User login
Welcome to Current Psychiatry, a leading source of information, online and in print, for practitioners of psychiatry and its related subspecialties, including addiction psychiatry, child and adolescent psychiatry, and geriatric psychiatry. This Web site contains evidence-based reviews of the prevention, diagnosis, and treatment of mental illness and psychological disorders; case reports; updates on psychopharmacology; news about the specialty of psychiatry; pearls for practice; and other topics of interest and use to this audience.
Dear Drupal User: You're seeing this because you're logged in to Drupal, and not redirected to MDedge.com/psychiatry.
Depression
adolescent depression
adolescent major depressive disorder
adolescent schizophrenia
adolescent with major depressive disorder
animals
autism
baby
brexpiprazole
child
child bipolar
child depression
child schizophrenia
children with bipolar disorder
children with depression
children with major depressive disorder
compulsive behaviors
cure
elderly bipolar
elderly depression
elderly major depressive disorder
elderly schizophrenia
elderly with dementia
first break
first episode
gambling
gaming
geriatric depression
geriatric major depressive disorder
geriatric schizophrenia
infant
kid
major depressive disorder
major depressive disorder in adolescents
major depressive disorder in children
parenting
pediatric
pediatric bipolar
pediatric depression
pediatric major depressive disorder
pediatric schizophrenia
pregnancy
pregnant
rexulti
skin care
teen
wine
section[contains(@class, 'nav-hidden')]
footer[@id='footer']
div[contains(@class, 'pane-pub-article-current-psychiatry')]
div[contains(@class, 'pane-pub-home-current-psychiatry')]
div[contains(@class, 'pane-pub-topic-current-psychiatry')]
div[contains(@class, 'panel-panel-inner')]
div[contains(@class, 'pane-node-field-article-topics')]
section[contains(@class, 'footer-nav-section-wrapper')]
Key steps to take when a patient commits suicide
The suicide of a patient is a relatively frequent occurrence in psychiatry. As many as 68% of consultant psychiatrists acknowledge the loss of a patient to suicide.1 Conservative estimates are that as many as 54% of psychiatry resident trainees experience patient suicide.2
Up to 57% of psychiatrists who have experienced a patient’s suicide have developed symptoms of posttraumatic stress disorder.3 There are steps you can take personally, with your staff, and with the patient’s family to mitigate social,
ethical, and legal consequences of a patient committing suicide, and to improve risk management.
Steps to take for yourself
1. In an inpatient psychiatric facility, be aware of standard operating procedures after a suicide; inform only an immediate supervisor if you learn of a suicide. In a group practice, inform the owner of the practice and receive advice on how to proceed. Do not contact the coroner’s office, the police, the deceased’s family, or legal counsel until advised to do so by a direct supervisor.
2. Be prepared to work with the coroner’s or medical examiner’s office. Write a detailed note summarizing the patient’s clinical history before the suicide; describe the clinical team’s work with the patient, the treatment plan, and an estimate of suicide risk.
3. Contact a trusted colleague or mentor; seeking formal and informal support from colleagues has shown to be helpful in coping with patient suicide.4 Group
support helps diminish feelings of pain and loneliness and helps one regain a sense of empowerment and willingness to treat other suicidal patients.
4. If possible, attend the patient’s funeral. This gesture often is welcomed by the family and facilitates the grieving process. Attending the funeral is not an admission of responsibility for the suicide.
5. Participate in the audit process (ie, what went wrong?, Could something have been done differently?).
Steps to take with the patient’s family
1. Once standard operating procedure allows, and, preferably within 24 hours of the suicide, contact the patient’s family to express your grief; give the family an opportunity to ask questions. Early communication and support reduces anger displaced on the psychiatrist. Initial contact can be used to provide support and as an opportunity to share and communicate.
2. When speaking with the family, discuss treatment efforts and emphasize that all realistic efforts were made to help the patient. Let family members vent their anger and hostility; the grieving process is hard, complex, and painful when a loved one has committed suicide.
3. Support the family’s decisions about mourning rituals specific to their culture and needs; involving the clergy early on can be helpful. Discussing the autopsy report with the family can be another way to show support.
4. Continue to offer support through stressful times, such as anniversaries and birthdays.
Steps to take with staff
1. Make staff aware of the death as a group; encourage them to attend funeral services.
2. Avoid placing blame; encourage group support and venting of emotions.
3. Be available to the staff so that they can share feelings of hurt and disappointment with you.
4. Maintain the schedule on unit, restoring a sense of stability and normalcy.
5. A so-called psychological autopsy exercise is recommended, in which you can emphasize the learning experience and focus on improvements4 that can help formulate policy reforms for providing better care.
Steps to improve risk management
1. If you work in a hospital, immediately contact the risk management team.
2. Seek legal counsel as soon as possible and involve counsel at all stages.
3. Notify your malpractice insurance carrier.
4. Complete the patient’s medical record and describe the facts as they occurred. Date the records accurately with clarification on notes entered after the suicide. Avoid drawing conclusions. Do not apologize for, or justify, your treatment decisions.
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Alexander DA, Klein S, Gray NM, et al. Suicide by patients: questionnaire study of its effect on consultant psychiatrists. BMJ. 2000;320(7249):1571-1574.
2. Courtenay KP, Stephens JP. The experience of patient suicide among trainees in psychiatry. The Psychiatrist. 2001;25:51-52.
3. Chemtob CM, Hamada RS, Bauer G, et al. Patients’ suicides: frequency and impact on psychiatrists. Am J Psychiatry. 1988;145(2):224-228.
4. Kaye NS, Soreff SM. The psychiatrist’s role, responses, and responsibilities when a patient commits suicide. Am J Psychiatry. 1991;148(6):739-743.
The suicide of a patient is a relatively frequent occurrence in psychiatry. As many as 68% of consultant psychiatrists acknowledge the loss of a patient to suicide.1 Conservative estimates are that as many as 54% of psychiatry resident trainees experience patient suicide.2
Up to 57% of psychiatrists who have experienced a patient’s suicide have developed symptoms of posttraumatic stress disorder.3 There are steps you can take personally, with your staff, and with the patient’s family to mitigate social,
ethical, and legal consequences of a patient committing suicide, and to improve risk management.
Steps to take for yourself
1. In an inpatient psychiatric facility, be aware of standard operating procedures after a suicide; inform only an immediate supervisor if you learn of a suicide. In a group practice, inform the owner of the practice and receive advice on how to proceed. Do not contact the coroner’s office, the police, the deceased’s family, or legal counsel until advised to do so by a direct supervisor.
2. Be prepared to work with the coroner’s or medical examiner’s office. Write a detailed note summarizing the patient’s clinical history before the suicide; describe the clinical team’s work with the patient, the treatment plan, and an estimate of suicide risk.
3. Contact a trusted colleague or mentor; seeking formal and informal support from colleagues has shown to be helpful in coping with patient suicide.4 Group
support helps diminish feelings of pain and loneliness and helps one regain a sense of empowerment and willingness to treat other suicidal patients.
4. If possible, attend the patient’s funeral. This gesture often is welcomed by the family and facilitates the grieving process. Attending the funeral is not an admission of responsibility for the suicide.
5. Participate in the audit process (ie, what went wrong?, Could something have been done differently?).
Steps to take with the patient’s family
1. Once standard operating procedure allows, and, preferably within 24 hours of the suicide, contact the patient’s family to express your grief; give the family an opportunity to ask questions. Early communication and support reduces anger displaced on the psychiatrist. Initial contact can be used to provide support and as an opportunity to share and communicate.
2. When speaking with the family, discuss treatment efforts and emphasize that all realistic efforts were made to help the patient. Let family members vent their anger and hostility; the grieving process is hard, complex, and painful when a loved one has committed suicide.
3. Support the family’s decisions about mourning rituals specific to their culture and needs; involving the clergy early on can be helpful. Discussing the autopsy report with the family can be another way to show support.
4. Continue to offer support through stressful times, such as anniversaries and birthdays.
Steps to take with staff
1. Make staff aware of the death as a group; encourage them to attend funeral services.
2. Avoid placing blame; encourage group support and venting of emotions.
3. Be available to the staff so that they can share feelings of hurt and disappointment with you.
4. Maintain the schedule on unit, restoring a sense of stability and normalcy.
5. A so-called psychological autopsy exercise is recommended, in which you can emphasize the learning experience and focus on improvements4 that can help formulate policy reforms for providing better care.
Steps to improve risk management
1. If you work in a hospital, immediately contact the risk management team.
2. Seek legal counsel as soon as possible and involve counsel at all stages.
3. Notify your malpractice insurance carrier.
4. Complete the patient’s medical record and describe the facts as they occurred. Date the records accurately with clarification on notes entered after the suicide. Avoid drawing conclusions. Do not apologize for, or justify, your treatment decisions.
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
The suicide of a patient is a relatively frequent occurrence in psychiatry. As many as 68% of consultant psychiatrists acknowledge the loss of a patient to suicide.1 Conservative estimates are that as many as 54% of psychiatry resident trainees experience patient suicide.2
Up to 57% of psychiatrists who have experienced a patient’s suicide have developed symptoms of posttraumatic stress disorder.3 There are steps you can take personally, with your staff, and with the patient’s family to mitigate social,
ethical, and legal consequences of a patient committing suicide, and to improve risk management.
Steps to take for yourself
1. In an inpatient psychiatric facility, be aware of standard operating procedures after a suicide; inform only an immediate supervisor if you learn of a suicide. In a group practice, inform the owner of the practice and receive advice on how to proceed. Do not contact the coroner’s office, the police, the deceased’s family, or legal counsel until advised to do so by a direct supervisor.
2. Be prepared to work with the coroner’s or medical examiner’s office. Write a detailed note summarizing the patient’s clinical history before the suicide; describe the clinical team’s work with the patient, the treatment plan, and an estimate of suicide risk.
3. Contact a trusted colleague or mentor; seeking formal and informal support from colleagues has shown to be helpful in coping with patient suicide.4 Group
support helps diminish feelings of pain and loneliness and helps one regain a sense of empowerment and willingness to treat other suicidal patients.
4. If possible, attend the patient’s funeral. This gesture often is welcomed by the family and facilitates the grieving process. Attending the funeral is not an admission of responsibility for the suicide.
5. Participate in the audit process (ie, what went wrong?, Could something have been done differently?).
Steps to take with the patient’s family
1. Once standard operating procedure allows, and, preferably within 24 hours of the suicide, contact the patient’s family to express your grief; give the family an opportunity to ask questions. Early communication and support reduces anger displaced on the psychiatrist. Initial contact can be used to provide support and as an opportunity to share and communicate.
2. When speaking with the family, discuss treatment efforts and emphasize that all realistic efforts were made to help the patient. Let family members vent their anger and hostility; the grieving process is hard, complex, and painful when a loved one has committed suicide.
3. Support the family’s decisions about mourning rituals specific to their culture and needs; involving the clergy early on can be helpful. Discussing the autopsy report with the family can be another way to show support.
4. Continue to offer support through stressful times, such as anniversaries and birthdays.
Steps to take with staff
1. Make staff aware of the death as a group; encourage them to attend funeral services.
2. Avoid placing blame; encourage group support and venting of emotions.
3. Be available to the staff so that they can share feelings of hurt and disappointment with you.
4. Maintain the schedule on unit, restoring a sense of stability and normalcy.
5. A so-called psychological autopsy exercise is recommended, in which you can emphasize the learning experience and focus on improvements4 that can help formulate policy reforms for providing better care.
Steps to improve risk management
1. If you work in a hospital, immediately contact the risk management team.
2. Seek legal counsel as soon as possible and involve counsel at all stages.
3. Notify your malpractice insurance carrier.
4. Complete the patient’s medical record and describe the facts as they occurred. Date the records accurately with clarification on notes entered after the suicide. Avoid drawing conclusions. Do not apologize for, or justify, your treatment decisions.
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Alexander DA, Klein S, Gray NM, et al. Suicide by patients: questionnaire study of its effect on consultant psychiatrists. BMJ. 2000;320(7249):1571-1574.
2. Courtenay KP, Stephens JP. The experience of patient suicide among trainees in psychiatry. The Psychiatrist. 2001;25:51-52.
3. Chemtob CM, Hamada RS, Bauer G, et al. Patients’ suicides: frequency and impact on psychiatrists. Am J Psychiatry. 1988;145(2):224-228.
4. Kaye NS, Soreff SM. The psychiatrist’s role, responses, and responsibilities when a patient commits suicide. Am J Psychiatry. 1991;148(6):739-743.
1. Alexander DA, Klein S, Gray NM, et al. Suicide by patients: questionnaire study of its effect on consultant psychiatrists. BMJ. 2000;320(7249):1571-1574.
2. Courtenay KP, Stephens JP. The experience of patient suicide among trainees in psychiatry. The Psychiatrist. 2001;25:51-52.
3. Chemtob CM, Hamada RS, Bauer G, et al. Patients’ suicides: frequency and impact on psychiatrists. Am J Psychiatry. 1988;145(2):224-228.
4. Kaye NS, Soreff SM. The psychiatrist’s role, responses, and responsibilities when a patient commits suicide. Am J Psychiatry. 1991;148(6):739-743.
Confused, cold, and lethargic
CASE Confused and cold
Ms. K, age 48, is brought to the emergency department (ED) by her husband because she has become increasingly lethargic over the past 2 weeks and cannot attend to activities of daily living. She is incontinent of stool and poorly responsive.
Ms. K’s husband reports that lethargy culminated in his wife sleeping 30 continuous hours. She has a history of a ruptured cerebral arteriovenous malformation (AVM) complicated by a secondary infarct 7 years ago, with residual symptoms of frontal lobe syndrome. Until 2 weeks ago, however, she was in her usual state of health.
Symptoms have included depression, mood lability, impulsivity, disinhibition, poor focus, and apathy. An outpatient psychiatrist has managed these symptoms with antidepressants and atypical antipsychotics.
When Ms. K arrives in the ED, she is taking citalopram, 30 mg/d, and paliperidone,
6 mg/d. Her psychiatrist started paliperidone 2 months ago, increasing the dosage to 6 mg/d 6 weeks before presentation because of worsening mood lability, disinhibition, and paranoia regarding her caregivers. Her husband denies any other medication changes or exposure to environmental toxins.
In the ED, Ms. K is confused and oriented only to person. Vital signs are: pulse 46 bpm; blood pressure, 66/51 mm Hg; respirations, 12/min; and temperature, 29.9ºC (85.8ºF) via bladder probe.
a) major depressive disorder, severe, with catatonic features
b) exposure to cold
c) hypothyroidism
d) drug-induced hypothermia
e) stroke
f) sepsis
g) delirium
The authors’ observations
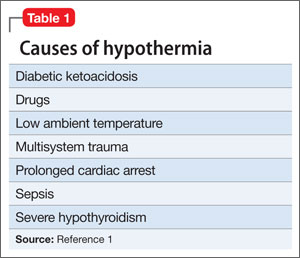
Hypothermia is core body temperature <35ºC (95ºF).1 It often is caused by exposure to low ambient temperature (Table 1),1 but Ms. K’s husband denied that she had been exposed to cold. Because of Ms. K’s neurologic history, stroke was high on the differential diagnosis, but physical examination did not reveal evidence of focal dysfunction and was significant only for altered mental status.
Ms. K had no posturing, rigidity, negativism, or excessive motor activity that would suggest catatonia. Before she became lethargic, her husband had not noted any deterioration of mood, although she did exhibit other behavioral changes that prompted her outpatient psychiatrist to increase the dosage of paliperidone. Although Ms. K began experiencing persecutory delusions—she believed that her caregivers were trying to harm her—she and her family denied perceptual disturbances. On examination, she did not appear responsive to auditory or visual hallucinations.
Frontal lobe syndrome is defined as a set of changes in the cognitive, behavioral, or emotional domains, often leading to disturbed affect, alteration of attention, aphasia, perseveration, disinhibition, and personality changes.2 These symptoms are not specific to lesions in the frontal lobes but can arise from lesions anywhere in the frontal-striatal-thalamic circuit.3 Causes include traumatic brain injury, neurodegenerative disorders, cerebrovascular disease, tumors, and aging.2 Recommended treatment incorporates psychosocial interventions with drug treatment to target specific symptoms. Medications reported to be effective include typical and atypical antipsychotics to target aggression and agitation; benzodiazepines to reduce arousal; antidepressants for mood symptoms, dopamine agonists (eg, bromocriptine) to decrease apathy, and mood stabilizers to target mood lability.4
Before her AVM rupture, review of Ms. K’s psychiatric history revealed no psychiatric symptoms or impaired functioning. When hospitalized for the AVM repair, she was started on sertraline. She began seeing a psychiatrist 2 years later because of increased agitation and behavioral disturbances, and aripiprazole was added. Persistent agitation prompted a trial of divalproex sodium, which was discontinued because of slurred speech and increased distractibility. Aripiprazole was tapered and replaced with paliperidone because of poor response. Citalopram was initiated 1 year before she presented to the ED.
a) brain MRI
b) infectious evaluation (lumbar puncture with analysis of cerebrospinal fluid, complete blood count, blood cultures, chest radiographs)
c) endocrine panel
d) urine toxicology screen
EVALUATION Hypothermia
Laboratory tests reveal multiple abnormalities, including thrombocytopenia (platelet level, 53 ×103/μL), altered coagulation (partial thromboplastin time, 55.6 s), elevated levels of hepatic transaminases (aspartate aminotransferase, 168 U/L; alanine aminotransferase, 357 U/L), and increased alkaline phosphatase (206 U/L). Other mild metabolic disturbances include: sodium, 149 mEq/L; CO2, 33 mEq/L; and blood urea nitrogen, 24 mg/dL.
These laboratory values are consistent with complications of hypothermia.1
ECG reveals sinus bradycardia (40 bpm) and Osborn waves (additional deflection at the end of the QRS complex), which are seen often in hypothermia.1 Head CT and brain MRI show chronic changes after Ms. K’s right temporoparietal AVM rupture, but no acute abnormality. Urinalysis, blood cultures, and chest radiographs are negative for infection. Urine toxicology screen is negative. Results of thyroid function tests and pituitary hormones studies are significant only for hyperprolactinemia of 155.7 ng/mL, a known adverse effect of antipsychotics.5
Ms. K is admitted and rewarmed passively and with warm IV fluids; by day 10 of hospitalization, temperature is stable (>35.1ºC [95.2ºF]). Thrombocytopenia, transaminitis, and altered mental status resolve.
Ms. K’s oral medications, including citalopram and paliperidone, have been held since admission because of her altered mental status. The psychiatry service is consulted to evaluate whether her presentation could be related to her change of medication.
A literature search reveals no report of paliperidone-induced hypothermia, but we consider it a possible explanation for Ms. K’s presentation. Lamotrigine (titrated to 50 mg/d), a benzodiazepine (oral lorazepam as needed), and discontinuing antipsychotics are recommended. After she returns to her baseline functioning, Ms. K is discharged to a skilled nursing facility.
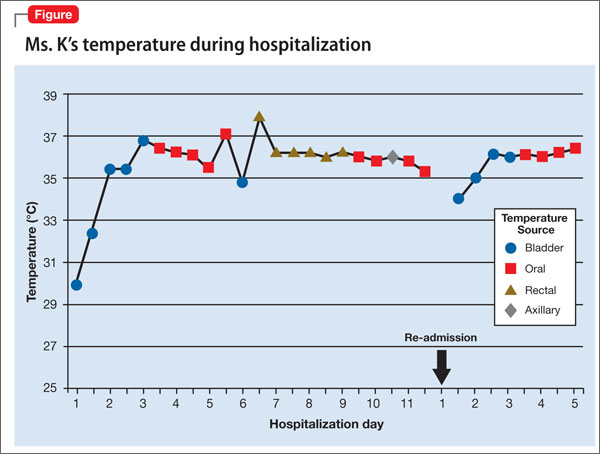
Ms. K presents to the ED 2 days after discharge with altered mental status. Vital signs are: blood pressure, 90/55 mm Hg; pulse, 59 bpm; respiratory rate, 14/min; and temperature, 34.4ºC (93.9ºF) via bladder probe (Figure). Laboratory tests were significant for hepatic transaminitis (aspartate aminotransferase, 75 U/L; alanine aminotransferase, 122 U/L) and elevated alkaline phosphatase (226 U/L). A review of records from the nursing facility revealed that Ms. K was receiving paliperidone because of an error in the discharge summary, which recommended restarting all prior medications.
The authors’ observations
The Naranjo Causality Scale,6 which categorizes the probability that an adverse event is related to a drug (based on several variables, including timing of the drug administration with the onset of event, drug dosage and levels, response relationships to a drug, including re-challenge when possible, and previous patient experience with the medication), often is used to evaluate whether an adverse clinical event has been caused by a drug (Table 2). We applied the Scale to Ms. K’s case, which revealed a score of 7—indicating a probable adverse drug reaction. The sequence of events in Ms. K’s case that led to a paliperidone challenge-dechallenge-rechallenge, and the resulting hypothermia, are, we concluded, evidence of an adverse drug reaction.

Using the World Health Organization database for adverse drug reactions, van Marum et al7 found 480 reports hypothermia with antipsychotics as of 2007 (compared with 524 reports of hyperthermia in the same period); 55% involved atypical antipsychotics, mainly risperidone. There are no case reports of paliperidone-induced hypothermia; however, several reports of hypothermia have been attributed to risperidone, and paliperidone is the primary active metabolite of risperidone.5
To identify risk factors for hypothermia with antipsychotic use, van Marum et al7 performed a literature search for case reports of antipsychotic-induced hypothermia, which revealed no association with age or sex. The most common diagnosis in cases of antipsychotic-induced hypothermia was schizophrenia (51%). In 73% of the cases, hypothermia followed the start or dosage increase of the antipsychotic. These observations have been noted in case reports and case series of hypothermia associated with antipsychotic use.8-12
Mechanism of action
One proposed mechanism for antipsychotic-induced hypothermia includes preferential 5-HT2A receptor antagonism over D2 receptor antagonism.7,12 It has been believed that, under normal conditions, the action of dopamine to reduce body temperature and the action of serotonin to elevate it are in balance.9
Another possible mechanism is peripheral á2-adrenergic blockade, which might increase the hypothermic effect by inhibiting peripheral responses to cooling, such as vasoconstriction and shivering.7,8 Boschi et al13 found that antipsychotics cause hypothermia in rats when the drug is administered intraperitoneally but not when given intrathecally. Perhaps for these reasons, in the early 1950s, before its psychotropic properties were known, chlorpromazine was used during surgery to induce artificial hibernation and suppress the body’s response to cooling.7 The therapeutic activity of paliperidone is mediated though a D2, 5-HT2A, and á2-receptor antagonism5; these mechanisms could, therefore, be contributing to Ms. K’s hypothermia.
Patients with preexisting brain damage— such as Ms. K—might be at increased risk of antipsychotic-induced hypothermia.7,8 This includes focal damage to central thermoregulatory centers, such as the pre-optic anterior hypothalamic region,14 and more diffuse damage seen in patients with cognitive impairment or a seizure disorder.8
Studies of people with schizophrenia show a decrease in core temperature after administration of an antipsychotic,15 raising the possibility of an impairment of baseline thermoregulatory control. Such thermal dysregulation in patients with schizophrenia might be explained by changes in neurotensin levels.7
The neuropeptide neurotensin has been implicated in the regulation of prolactin release and interacts to a significant degree with the dopaminergic system.16 When administered to animals, neurotensin suppresses heat production and increases heat loss.17 The neurotensin level in CSF was found to be lower in non-medicated patients with schizophrenia than in healthy controls, with an inverse correlation between the severity of symptoms and the neurotensin level.18
Additionally, persons with schizophrenia might be at increased risk of developing hypothermia when exposed to a low environmental temperature.7,8 Kudoh et al19 investigated temperature regulation during anesthesia in patients with chronic (≥7 years) schizophrenia receiving antipsychotics, and compared findings against what was seen in controls. The team reported that patients with schizophrenia had significantly lower intraoperative temperatures.
A published analysis of cases and studies of antipsychotic-induced hypothermia describes the combination of drug variables, patient variables, and environmental variables that contribute to thermal dysregulation (Table 3).7-12,15 The recommendation for practitioners is that, when considering an antipsychotic for a patient at high risk of thermal dysregulation, your choice of an agent should take that risk into account, especially when that drug is one that has comparatively stronger serotonergic and peripheral á-adrenergic effects. You should monitor patients closely for hypothermia after starting and when increasing the dosage of the drug. In patients with schizophrenia who might have a problem with baseline thermoregulation, advise them to take measures to counteract their increased susceptibility to low ambient temperatures.

OUTCOME Readmission
Ms. K was readmitted, rewarmed, and discharged to a skilled nursing facility 4 days later, after baseline function returned to normal and temperature stabilized. Paliperidone is now listed in her electronic medical record as “drug intolerance.”
This case also highlights the importance of adequate medication reconciliation at
admission and discharge, especially when using an electronic medical record system, because what might otherwise be considered a minor mistake can have devastating consequences.
Bottom Line
Thermal dysregulation—hyperthermia and hypothermia—can occur secondary to an antipsychotic. Determining whether a patient is at increased risk of either of these adverse effects is important when deciding to use antipsychotics. Recognizing agents that can cause hypothermia is essential, because management requires prompt discontinuation of the offending drug.
Related Resource
- Espay AJ, et al. Frontal lobe syndromes. http://emedicine.medscape.com/article/1135866-overview. Updated September 17, 2012. Accessed November 3, 2012.
Drug Brand Names
Aripiprazole • Abilify Lamotrigine • Lamictal
Bromocriptine • Parlodel Lorazepam • Ativan
Chlorpromazine • Thorazine Paliperidone • Invega
Citalopram • Celexa Risperidone • Risperdal
Clozapine • Clozaril Sertraline • Zoloft
Divalproex sodium • Depakote Thioridazine • Mellaril
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Aslam AF, Aslam AK, Vasavada BC, et al. Hypothermia: evaluation, electrocardiographic manifestations, and management. Am J Med. 2006;119(4):297-301.
2. Hanna-Pladdy B. Dysexecutive syndromes in neurologic disease. J Neurol Phys Ther. 2007;31(3):119-127.
3. Salloway SP. Diagnosis and treatment of patients with “frontal lobe” syndromes. J Neuropsychiatry Clin Neurosci. 1994;6(4):388-398.
4. Campbell JJ, Duffy JD, Salloway SP. Treatment strategies for patients with dysexecutive syndromes. In: Salloway SP, Malloy PF, Duffy JD, eds. The frontal lobes and neuropsychiatric illness. Washington, DC: American Psychiatric Press; 2001:153-163.
5. Stahl SM. Essential psychopharmacology: neuroscientific basis and practical applications. 3rd ed. New York, NY: Cambridge University Press; 2000:336.
6. Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30(2):239-245.
7. van Marum RJ, Wegewijs MA, Loonen AJM, et al. Hypothermia following antipsychotic drug use. Eur J Clin Pharmacol. 2007;63(6):627-631.
8. Kreuzer P, Landgrebe M, Wittmann M, et al. Hypothermia associated with antipsychotic drug use: a clinical case series and review of current literature. J Clin Pharmacol. 2012;52(7)1090-1097.
9. Hung CF, Huang TY, Lin PY. Hypothermia and rhabdomyolysis following olanzapine injection in an adolescent with schizophreniform disorder. Gen Hosp Psychiatry. 2009;31(4):376-378.
10. Razaq M, Samma M. A case of risperidone-induced hypothermia. Am J Ther. 2004;11(3):229-230.
11. Schwaninger M, Weisbrod M, Schwab S, et al. Hypothermia induced by atypical neuroleptics. Clin Neuropharmacol. 1998;21(6):344-346.
12. Bookstaver PB, Miller AD. Possible long-acting risperidone-induced hypothermia precipitating phenytoin toxicity in an elderly patient. J Clin Pharm Ther. 2011; 36(3):426-429.
13. Boschi G, Launay N, Rips R. Neuroleptic-induced hypothermia in mice: lack of evidence for a central mechanism. Br J Pharmacol. 1987;90(4):745-751.
14. Sessler DI. Thermoregulatory defense mechanisms. Crit Care Med. 2009;37(suppl 7):S203-S210.
15. Shiloh R, Weizman A, Epstein Y, et al. Abnormal thermoregulation in drug-free male schizophrenia patients. Eur Neuropsychopharmacol. 2001;11(4):285-288.
16. McCann SM, Vijayan E. Control of anterior pituitary hormone secretion by neurotensin. Ann N Y Acad Sci. 1992; 668:287-297.
17. Chandra A, Chou HC, Chang C, et al. Effecst of intraventricular administration of neurotensin and somatostatin on thermoregulation in the rat. Neuropharmacology. 1981;20(7):715-718.
18. Sharma RP, Janicak PG, Bissette G, et al. CSF neurotensin concentrations and antipsychotic treatment in schizophrenia and schizoaffective disorder. Am J Psychiatry. 1997; 154(7):1019-1021.
19. Kudoh A, Takase H, Takazawa T. Chronic treatment with antipsychotics enhances intraoperative core hypothermia. Anesth Analg. 2004;98(1):111-115.
CASE Confused and cold
Ms. K, age 48, is brought to the emergency department (ED) by her husband because she has become increasingly lethargic over the past 2 weeks and cannot attend to activities of daily living. She is incontinent of stool and poorly responsive.
Ms. K’s husband reports that lethargy culminated in his wife sleeping 30 continuous hours. She has a history of a ruptured cerebral arteriovenous malformation (AVM) complicated by a secondary infarct 7 years ago, with residual symptoms of frontal lobe syndrome. Until 2 weeks ago, however, she was in her usual state of health.
Symptoms have included depression, mood lability, impulsivity, disinhibition, poor focus, and apathy. An outpatient psychiatrist has managed these symptoms with antidepressants and atypical antipsychotics.
When Ms. K arrives in the ED, she is taking citalopram, 30 mg/d, and paliperidone,
6 mg/d. Her psychiatrist started paliperidone 2 months ago, increasing the dosage to 6 mg/d 6 weeks before presentation because of worsening mood lability, disinhibition, and paranoia regarding her caregivers. Her husband denies any other medication changes or exposure to environmental toxins.
In the ED, Ms. K is confused and oriented only to person. Vital signs are: pulse 46 bpm; blood pressure, 66/51 mm Hg; respirations, 12/min; and temperature, 29.9ºC (85.8ºF) via bladder probe.
a) major depressive disorder, severe, with catatonic features
b) exposure to cold
c) hypothyroidism
d) drug-induced hypothermia
e) stroke
f) sepsis
g) delirium
The authors’ observations
Hypothermia is core body temperature <35ºC (95ºF).1 It often is caused by exposure to low ambient temperature (Table 1),1 but Ms. K’s husband denied that she had been exposed to cold. Because of Ms. K’s neurologic history, stroke was high on the differential diagnosis, but physical examination did not reveal evidence of focal dysfunction and was significant only for altered mental status.
Ms. K had no posturing, rigidity, negativism, or excessive motor activity that would suggest catatonia. Before she became lethargic, her husband had not noted any deterioration of mood, although she did exhibit other behavioral changes that prompted her outpatient psychiatrist to increase the dosage of paliperidone. Although Ms. K began experiencing persecutory delusions—she believed that her caregivers were trying to harm her—she and her family denied perceptual disturbances. On examination, she did not appear responsive to auditory or visual hallucinations.
Frontal lobe syndrome is defined as a set of changes in the cognitive, behavioral, or emotional domains, often leading to disturbed affect, alteration of attention, aphasia, perseveration, disinhibition, and personality changes.2 These symptoms are not specific to lesions in the frontal lobes but can arise from lesions anywhere in the frontal-striatal-thalamic circuit.3 Causes include traumatic brain injury, neurodegenerative disorders, cerebrovascular disease, tumors, and aging.2 Recommended treatment incorporates psychosocial interventions with drug treatment to target specific symptoms. Medications reported to be effective include typical and atypical antipsychotics to target aggression and agitation; benzodiazepines to reduce arousal; antidepressants for mood symptoms, dopamine agonists (eg, bromocriptine) to decrease apathy, and mood stabilizers to target mood lability.4
Before her AVM rupture, review of Ms. K’s psychiatric history revealed no psychiatric symptoms or impaired functioning. When hospitalized for the AVM repair, she was started on sertraline. She began seeing a psychiatrist 2 years later because of increased agitation and behavioral disturbances, and aripiprazole was added. Persistent agitation prompted a trial of divalproex sodium, which was discontinued because of slurred speech and increased distractibility. Aripiprazole was tapered and replaced with paliperidone because of poor response. Citalopram was initiated 1 year before she presented to the ED.
a) brain MRI
b) infectious evaluation (lumbar puncture with analysis of cerebrospinal fluid, complete blood count, blood cultures, chest radiographs)
c) endocrine panel
d) urine toxicology screen
EVALUATION Hypothermia
Laboratory tests reveal multiple abnormalities, including thrombocytopenia (platelet level, 53 ×103/μL), altered coagulation (partial thromboplastin time, 55.6 s), elevated levels of hepatic transaminases (aspartate aminotransferase, 168 U/L; alanine aminotransferase, 357 U/L), and increased alkaline phosphatase (206 U/L). Other mild metabolic disturbances include: sodium, 149 mEq/L; CO2, 33 mEq/L; and blood urea nitrogen, 24 mg/dL.
These laboratory values are consistent with complications of hypothermia.1
ECG reveals sinus bradycardia (40 bpm) and Osborn waves (additional deflection at the end of the QRS complex), which are seen often in hypothermia.1 Head CT and brain MRI show chronic changes after Ms. K’s right temporoparietal AVM rupture, but no acute abnormality. Urinalysis, blood cultures, and chest radiographs are negative for infection. Urine toxicology screen is negative. Results of thyroid function tests and pituitary hormones studies are significant only for hyperprolactinemia of 155.7 ng/mL, a known adverse effect of antipsychotics.5
Ms. K is admitted and rewarmed passively and with warm IV fluids; by day 10 of hospitalization, temperature is stable (>35.1ºC [95.2ºF]). Thrombocytopenia, transaminitis, and altered mental status resolve.
Ms. K’s oral medications, including citalopram and paliperidone, have been held since admission because of her altered mental status. The psychiatry service is consulted to evaluate whether her presentation could be related to her change of medication.
A literature search reveals no report of paliperidone-induced hypothermia, but we consider it a possible explanation for Ms. K’s presentation. Lamotrigine (titrated to 50 mg/d), a benzodiazepine (oral lorazepam as needed), and discontinuing antipsychotics are recommended. After she returns to her baseline functioning, Ms. K is discharged to a skilled nursing facility.
Ms. K presents to the ED 2 days after discharge with altered mental status. Vital signs are: blood pressure, 90/55 mm Hg; pulse, 59 bpm; respiratory rate, 14/min; and temperature, 34.4ºC (93.9ºF) via bladder probe (Figure). Laboratory tests were significant for hepatic transaminitis (aspartate aminotransferase, 75 U/L; alanine aminotransferase, 122 U/L) and elevated alkaline phosphatase (226 U/L). A review of records from the nursing facility revealed that Ms. K was receiving paliperidone because of an error in the discharge summary, which recommended restarting all prior medications.
The authors’ observations
The Naranjo Causality Scale,6 which categorizes the probability that an adverse event is related to a drug (based on several variables, including timing of the drug administration with the onset of event, drug dosage and levels, response relationships to a drug, including re-challenge when possible, and previous patient experience with the medication), often is used to evaluate whether an adverse clinical event has been caused by a drug (Table 2). We applied the Scale to Ms. K’s case, which revealed a score of 7—indicating a probable adverse drug reaction. The sequence of events in Ms. K’s case that led to a paliperidone challenge-dechallenge-rechallenge, and the resulting hypothermia, are, we concluded, evidence of an adverse drug reaction.
Using the World Health Organization database for adverse drug reactions, van Marum et al7 found 480 reports hypothermia with antipsychotics as of 2007 (compared with 524 reports of hyperthermia in the same period); 55% involved atypical antipsychotics, mainly risperidone. There are no case reports of paliperidone-induced hypothermia; however, several reports of hypothermia have been attributed to risperidone, and paliperidone is the primary active metabolite of risperidone.5
To identify risk factors for hypothermia with antipsychotic use, van Marum et al7 performed a literature search for case reports of antipsychotic-induced hypothermia, which revealed no association with age or sex. The most common diagnosis in cases of antipsychotic-induced hypothermia was schizophrenia (51%). In 73% of the cases, hypothermia followed the start or dosage increase of the antipsychotic. These observations have been noted in case reports and case series of hypothermia associated with antipsychotic use.8-12
Mechanism of action
One proposed mechanism for antipsychotic-induced hypothermia includes preferential 5-HT2A receptor antagonism over D2 receptor antagonism.7,12 It has been believed that, under normal conditions, the action of dopamine to reduce body temperature and the action of serotonin to elevate it are in balance.9
Another possible mechanism is peripheral á2-adrenergic blockade, which might increase the hypothermic effect by inhibiting peripheral responses to cooling, such as vasoconstriction and shivering.7,8 Boschi et al13 found that antipsychotics cause hypothermia in rats when the drug is administered intraperitoneally but not when given intrathecally. Perhaps for these reasons, in the early 1950s, before its psychotropic properties were known, chlorpromazine was used during surgery to induce artificial hibernation and suppress the body’s response to cooling.7 The therapeutic activity of paliperidone is mediated though a D2, 5-HT2A, and á2-receptor antagonism5; these mechanisms could, therefore, be contributing to Ms. K’s hypothermia.
Patients with preexisting brain damage— such as Ms. K—might be at increased risk of antipsychotic-induced hypothermia.7,8 This includes focal damage to central thermoregulatory centers, such as the pre-optic anterior hypothalamic region,14 and more diffuse damage seen in patients with cognitive impairment or a seizure disorder.8
Studies of people with schizophrenia show a decrease in core temperature after administration of an antipsychotic,15 raising the possibility of an impairment of baseline thermoregulatory control. Such thermal dysregulation in patients with schizophrenia might be explained by changes in neurotensin levels.7
The neuropeptide neurotensin has been implicated in the regulation of prolactin release and interacts to a significant degree with the dopaminergic system.16 When administered to animals, neurotensin suppresses heat production and increases heat loss.17 The neurotensin level in CSF was found to be lower in non-medicated patients with schizophrenia than in healthy controls, with an inverse correlation between the severity of symptoms and the neurotensin level.18
Additionally, persons with schizophrenia might be at increased risk of developing hypothermia when exposed to a low environmental temperature.7,8 Kudoh et al19 investigated temperature regulation during anesthesia in patients with chronic (≥7 years) schizophrenia receiving antipsychotics, and compared findings against what was seen in controls. The team reported that patients with schizophrenia had significantly lower intraoperative temperatures.
A published analysis of cases and studies of antipsychotic-induced hypothermia describes the combination of drug variables, patient variables, and environmental variables that contribute to thermal dysregulation (Table 3).7-12,15 The recommendation for practitioners is that, when considering an antipsychotic for a patient at high risk of thermal dysregulation, your choice of an agent should take that risk into account, especially when that drug is one that has comparatively stronger serotonergic and peripheral á-adrenergic effects. You should monitor patients closely for hypothermia after starting and when increasing the dosage of the drug. In patients with schizophrenia who might have a problem with baseline thermoregulation, advise them to take measures to counteract their increased susceptibility to low ambient temperatures.
OUTCOME Readmission
Ms. K was readmitted, rewarmed, and discharged to a skilled nursing facility 4 days later, after baseline function returned to normal and temperature stabilized. Paliperidone is now listed in her electronic medical record as “drug intolerance.”
This case also highlights the importance of adequate medication reconciliation at
admission and discharge, especially when using an electronic medical record system, because what might otherwise be considered a minor mistake can have devastating consequences.
Bottom Line
Thermal dysregulation—hyperthermia and hypothermia—can occur secondary to an antipsychotic. Determining whether a patient is at increased risk of either of these adverse effects is important when deciding to use antipsychotics. Recognizing agents that can cause hypothermia is essential, because management requires prompt discontinuation of the offending drug.
Related Resource
- Espay AJ, et al. Frontal lobe syndromes. http://emedicine.medscape.com/article/1135866-overview. Updated September 17, 2012. Accessed November 3, 2012.
Drug Brand Names
Aripiprazole • Abilify Lamotrigine • Lamictal
Bromocriptine • Parlodel Lorazepam • Ativan
Chlorpromazine • Thorazine Paliperidone • Invega
Citalopram • Celexa Risperidone • Risperdal
Clozapine • Clozaril Sertraline • Zoloft
Divalproex sodium • Depakote Thioridazine • Mellaril
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
CASE Confused and cold
Ms. K, age 48, is brought to the emergency department (ED) by her husband because she has become increasingly lethargic over the past 2 weeks and cannot attend to activities of daily living. She is incontinent of stool and poorly responsive.
Ms. K’s husband reports that lethargy culminated in his wife sleeping 30 continuous hours. She has a history of a ruptured cerebral arteriovenous malformation (AVM) complicated by a secondary infarct 7 years ago, with residual symptoms of frontal lobe syndrome. Until 2 weeks ago, however, she was in her usual state of health.
Symptoms have included depression, mood lability, impulsivity, disinhibition, poor focus, and apathy. An outpatient psychiatrist has managed these symptoms with antidepressants and atypical antipsychotics.
When Ms. K arrives in the ED, she is taking citalopram, 30 mg/d, and paliperidone,
6 mg/d. Her psychiatrist started paliperidone 2 months ago, increasing the dosage to 6 mg/d 6 weeks before presentation because of worsening mood lability, disinhibition, and paranoia regarding her caregivers. Her husband denies any other medication changes or exposure to environmental toxins.
In the ED, Ms. K is confused and oriented only to person. Vital signs are: pulse 46 bpm; blood pressure, 66/51 mm Hg; respirations, 12/min; and temperature, 29.9ºC (85.8ºF) via bladder probe.
a) major depressive disorder, severe, with catatonic features
b) exposure to cold
c) hypothyroidism
d) drug-induced hypothermia
e) stroke
f) sepsis
g) delirium
The authors’ observations
Hypothermia is core body temperature <35ºC (95ºF).1 It often is caused by exposure to low ambient temperature (Table 1),1 but Ms. K’s husband denied that she had been exposed to cold. Because of Ms. K’s neurologic history, stroke was high on the differential diagnosis, but physical examination did not reveal evidence of focal dysfunction and was significant only for altered mental status.
Ms. K had no posturing, rigidity, negativism, or excessive motor activity that would suggest catatonia. Before she became lethargic, her husband had not noted any deterioration of mood, although she did exhibit other behavioral changes that prompted her outpatient psychiatrist to increase the dosage of paliperidone. Although Ms. K began experiencing persecutory delusions—she believed that her caregivers were trying to harm her—she and her family denied perceptual disturbances. On examination, she did not appear responsive to auditory or visual hallucinations.
Frontal lobe syndrome is defined as a set of changes in the cognitive, behavioral, or emotional domains, often leading to disturbed affect, alteration of attention, aphasia, perseveration, disinhibition, and personality changes.2 These symptoms are not specific to lesions in the frontal lobes but can arise from lesions anywhere in the frontal-striatal-thalamic circuit.3 Causes include traumatic brain injury, neurodegenerative disorders, cerebrovascular disease, tumors, and aging.2 Recommended treatment incorporates psychosocial interventions with drug treatment to target specific symptoms. Medications reported to be effective include typical and atypical antipsychotics to target aggression and agitation; benzodiazepines to reduce arousal; antidepressants for mood symptoms, dopamine agonists (eg, bromocriptine) to decrease apathy, and mood stabilizers to target mood lability.4
Before her AVM rupture, review of Ms. K’s psychiatric history revealed no psychiatric symptoms or impaired functioning. When hospitalized for the AVM repair, she was started on sertraline. She began seeing a psychiatrist 2 years later because of increased agitation and behavioral disturbances, and aripiprazole was added. Persistent agitation prompted a trial of divalproex sodium, which was discontinued because of slurred speech and increased distractibility. Aripiprazole was tapered and replaced with paliperidone because of poor response. Citalopram was initiated 1 year before she presented to the ED.
a) brain MRI
b) infectious evaluation (lumbar puncture with analysis of cerebrospinal fluid, complete blood count, blood cultures, chest radiographs)
c) endocrine panel
d) urine toxicology screen
EVALUATION Hypothermia
Laboratory tests reveal multiple abnormalities, including thrombocytopenia (platelet level, 53 ×103/μL), altered coagulation (partial thromboplastin time, 55.6 s), elevated levels of hepatic transaminases (aspartate aminotransferase, 168 U/L; alanine aminotransferase, 357 U/L), and increased alkaline phosphatase (206 U/L). Other mild metabolic disturbances include: sodium, 149 mEq/L; CO2, 33 mEq/L; and blood urea nitrogen, 24 mg/dL.
These laboratory values are consistent with complications of hypothermia.1
ECG reveals sinus bradycardia (40 bpm) and Osborn waves (additional deflection at the end of the QRS complex), which are seen often in hypothermia.1 Head CT and brain MRI show chronic changes after Ms. K’s right temporoparietal AVM rupture, but no acute abnormality. Urinalysis, blood cultures, and chest radiographs are negative for infection. Urine toxicology screen is negative. Results of thyroid function tests and pituitary hormones studies are significant only for hyperprolactinemia of 155.7 ng/mL, a known adverse effect of antipsychotics.5
Ms. K is admitted and rewarmed passively and with warm IV fluids; by day 10 of hospitalization, temperature is stable (>35.1ºC [95.2ºF]). Thrombocytopenia, transaminitis, and altered mental status resolve.
Ms. K’s oral medications, including citalopram and paliperidone, have been held since admission because of her altered mental status. The psychiatry service is consulted to evaluate whether her presentation could be related to her change of medication.
A literature search reveals no report of paliperidone-induced hypothermia, but we consider it a possible explanation for Ms. K’s presentation. Lamotrigine (titrated to 50 mg/d), a benzodiazepine (oral lorazepam as needed), and discontinuing antipsychotics are recommended. After she returns to her baseline functioning, Ms. K is discharged to a skilled nursing facility.
Ms. K presents to the ED 2 days after discharge with altered mental status. Vital signs are: blood pressure, 90/55 mm Hg; pulse, 59 bpm; respiratory rate, 14/min; and temperature, 34.4ºC (93.9ºF) via bladder probe (Figure). Laboratory tests were significant for hepatic transaminitis (aspartate aminotransferase, 75 U/L; alanine aminotransferase, 122 U/L) and elevated alkaline phosphatase (226 U/L). A review of records from the nursing facility revealed that Ms. K was receiving paliperidone because of an error in the discharge summary, which recommended restarting all prior medications.
The authors’ observations
The Naranjo Causality Scale,6 which categorizes the probability that an adverse event is related to a drug (based on several variables, including timing of the drug administration with the onset of event, drug dosage and levels, response relationships to a drug, including re-challenge when possible, and previous patient experience with the medication), often is used to evaluate whether an adverse clinical event has been caused by a drug (Table 2). We applied the Scale to Ms. K’s case, which revealed a score of 7—indicating a probable adverse drug reaction. The sequence of events in Ms. K’s case that led to a paliperidone challenge-dechallenge-rechallenge, and the resulting hypothermia, are, we concluded, evidence of an adverse drug reaction.
Using the World Health Organization database for adverse drug reactions, van Marum et al7 found 480 reports hypothermia with antipsychotics as of 2007 (compared with 524 reports of hyperthermia in the same period); 55% involved atypical antipsychotics, mainly risperidone. There are no case reports of paliperidone-induced hypothermia; however, several reports of hypothermia have been attributed to risperidone, and paliperidone is the primary active metabolite of risperidone.5
To identify risk factors for hypothermia with antipsychotic use, van Marum et al7 performed a literature search for case reports of antipsychotic-induced hypothermia, which revealed no association with age or sex. The most common diagnosis in cases of antipsychotic-induced hypothermia was schizophrenia (51%). In 73% of the cases, hypothermia followed the start or dosage increase of the antipsychotic. These observations have been noted in case reports and case series of hypothermia associated with antipsychotic use.8-12
Mechanism of action
One proposed mechanism for antipsychotic-induced hypothermia includes preferential 5-HT2A receptor antagonism over D2 receptor antagonism.7,12 It has been believed that, under normal conditions, the action of dopamine to reduce body temperature and the action of serotonin to elevate it are in balance.9
Another possible mechanism is peripheral á2-adrenergic blockade, which might increase the hypothermic effect by inhibiting peripheral responses to cooling, such as vasoconstriction and shivering.7,8 Boschi et al13 found that antipsychotics cause hypothermia in rats when the drug is administered intraperitoneally but not when given intrathecally. Perhaps for these reasons, in the early 1950s, before its psychotropic properties were known, chlorpromazine was used during surgery to induce artificial hibernation and suppress the body’s response to cooling.7 The therapeutic activity of paliperidone is mediated though a D2, 5-HT2A, and á2-receptor antagonism5; these mechanisms could, therefore, be contributing to Ms. K’s hypothermia.
Patients with preexisting brain damage— such as Ms. K—might be at increased risk of antipsychotic-induced hypothermia.7,8 This includes focal damage to central thermoregulatory centers, such as the pre-optic anterior hypothalamic region,14 and more diffuse damage seen in patients with cognitive impairment or a seizure disorder.8
Studies of people with schizophrenia show a decrease in core temperature after administration of an antipsychotic,15 raising the possibility of an impairment of baseline thermoregulatory control. Such thermal dysregulation in patients with schizophrenia might be explained by changes in neurotensin levels.7
The neuropeptide neurotensin has been implicated in the regulation of prolactin release and interacts to a significant degree with the dopaminergic system.16 When administered to animals, neurotensin suppresses heat production and increases heat loss.17 The neurotensin level in CSF was found to be lower in non-medicated patients with schizophrenia than in healthy controls, with an inverse correlation between the severity of symptoms and the neurotensin level.18
Additionally, persons with schizophrenia might be at increased risk of developing hypothermia when exposed to a low environmental temperature.7,8 Kudoh et al19 investigated temperature regulation during anesthesia in patients with chronic (≥7 years) schizophrenia receiving antipsychotics, and compared findings against what was seen in controls. The team reported that patients with schizophrenia had significantly lower intraoperative temperatures.
A published analysis of cases and studies of antipsychotic-induced hypothermia describes the combination of drug variables, patient variables, and environmental variables that contribute to thermal dysregulation (Table 3).7-12,15 The recommendation for practitioners is that, when considering an antipsychotic for a patient at high risk of thermal dysregulation, your choice of an agent should take that risk into account, especially when that drug is one that has comparatively stronger serotonergic and peripheral á-adrenergic effects. You should monitor patients closely for hypothermia after starting and when increasing the dosage of the drug. In patients with schizophrenia who might have a problem with baseline thermoregulation, advise them to take measures to counteract their increased susceptibility to low ambient temperatures.
OUTCOME Readmission
Ms. K was readmitted, rewarmed, and discharged to a skilled nursing facility 4 days later, after baseline function returned to normal and temperature stabilized. Paliperidone is now listed in her electronic medical record as “drug intolerance.”
This case also highlights the importance of adequate medication reconciliation at
admission and discharge, especially when using an electronic medical record system, because what might otherwise be considered a minor mistake can have devastating consequences.
Bottom Line
Thermal dysregulation—hyperthermia and hypothermia—can occur secondary to an antipsychotic. Determining whether a patient is at increased risk of either of these adverse effects is important when deciding to use antipsychotics. Recognizing agents that can cause hypothermia is essential, because management requires prompt discontinuation of the offending drug.
Related Resource
- Espay AJ, et al. Frontal lobe syndromes. http://emedicine.medscape.com/article/1135866-overview. Updated September 17, 2012. Accessed November 3, 2012.
Drug Brand Names
Aripiprazole • Abilify Lamotrigine • Lamictal
Bromocriptine • Parlodel Lorazepam • Ativan
Chlorpromazine • Thorazine Paliperidone • Invega
Citalopram • Celexa Risperidone • Risperdal
Clozapine • Clozaril Sertraline • Zoloft
Divalproex sodium • Depakote Thioridazine • Mellaril
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Aslam AF, Aslam AK, Vasavada BC, et al. Hypothermia: evaluation, electrocardiographic manifestations, and management. Am J Med. 2006;119(4):297-301.
2. Hanna-Pladdy B. Dysexecutive syndromes in neurologic disease. J Neurol Phys Ther. 2007;31(3):119-127.
3. Salloway SP. Diagnosis and treatment of patients with “frontal lobe” syndromes. J Neuropsychiatry Clin Neurosci. 1994;6(4):388-398.
4. Campbell JJ, Duffy JD, Salloway SP. Treatment strategies for patients with dysexecutive syndromes. In: Salloway SP, Malloy PF, Duffy JD, eds. The frontal lobes and neuropsychiatric illness. Washington, DC: American Psychiatric Press; 2001:153-163.
5. Stahl SM. Essential psychopharmacology: neuroscientific basis and practical applications. 3rd ed. New York, NY: Cambridge University Press; 2000:336.
6. Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30(2):239-245.
7. van Marum RJ, Wegewijs MA, Loonen AJM, et al. Hypothermia following antipsychotic drug use. Eur J Clin Pharmacol. 2007;63(6):627-631.
8. Kreuzer P, Landgrebe M, Wittmann M, et al. Hypothermia associated with antipsychotic drug use: a clinical case series and review of current literature. J Clin Pharmacol. 2012;52(7)1090-1097.
9. Hung CF, Huang TY, Lin PY. Hypothermia and rhabdomyolysis following olanzapine injection in an adolescent with schizophreniform disorder. Gen Hosp Psychiatry. 2009;31(4):376-378.
10. Razaq M, Samma M. A case of risperidone-induced hypothermia. Am J Ther. 2004;11(3):229-230.
11. Schwaninger M, Weisbrod M, Schwab S, et al. Hypothermia induced by atypical neuroleptics. Clin Neuropharmacol. 1998;21(6):344-346.
12. Bookstaver PB, Miller AD. Possible long-acting risperidone-induced hypothermia precipitating phenytoin toxicity in an elderly patient. J Clin Pharm Ther. 2011; 36(3):426-429.
13. Boschi G, Launay N, Rips R. Neuroleptic-induced hypothermia in mice: lack of evidence for a central mechanism. Br J Pharmacol. 1987;90(4):745-751.
14. Sessler DI. Thermoregulatory defense mechanisms. Crit Care Med. 2009;37(suppl 7):S203-S210.
15. Shiloh R, Weizman A, Epstein Y, et al. Abnormal thermoregulation in drug-free male schizophrenia patients. Eur Neuropsychopharmacol. 2001;11(4):285-288.
16. McCann SM, Vijayan E. Control of anterior pituitary hormone secretion by neurotensin. Ann N Y Acad Sci. 1992; 668:287-297.
17. Chandra A, Chou HC, Chang C, et al. Effecst of intraventricular administration of neurotensin and somatostatin on thermoregulation in the rat. Neuropharmacology. 1981;20(7):715-718.
18. Sharma RP, Janicak PG, Bissette G, et al. CSF neurotensin concentrations and antipsychotic treatment in schizophrenia and schizoaffective disorder. Am J Psychiatry. 1997; 154(7):1019-1021.
19. Kudoh A, Takase H, Takazawa T. Chronic treatment with antipsychotics enhances intraoperative core hypothermia. Anesth Analg. 2004;98(1):111-115.
1. Aslam AF, Aslam AK, Vasavada BC, et al. Hypothermia: evaluation, electrocardiographic manifestations, and management. Am J Med. 2006;119(4):297-301.
2. Hanna-Pladdy B. Dysexecutive syndromes in neurologic disease. J Neurol Phys Ther. 2007;31(3):119-127.
3. Salloway SP. Diagnosis and treatment of patients with “frontal lobe” syndromes. J Neuropsychiatry Clin Neurosci. 1994;6(4):388-398.
4. Campbell JJ, Duffy JD, Salloway SP. Treatment strategies for patients with dysexecutive syndromes. In: Salloway SP, Malloy PF, Duffy JD, eds. The frontal lobes and neuropsychiatric illness. Washington, DC: American Psychiatric Press; 2001:153-163.
5. Stahl SM. Essential psychopharmacology: neuroscientific basis and practical applications. 3rd ed. New York, NY: Cambridge University Press; 2000:336.
6. Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30(2):239-245.
7. van Marum RJ, Wegewijs MA, Loonen AJM, et al. Hypothermia following antipsychotic drug use. Eur J Clin Pharmacol. 2007;63(6):627-631.
8. Kreuzer P, Landgrebe M, Wittmann M, et al. Hypothermia associated with antipsychotic drug use: a clinical case series and review of current literature. J Clin Pharmacol. 2012;52(7)1090-1097.
9. Hung CF, Huang TY, Lin PY. Hypothermia and rhabdomyolysis following olanzapine injection in an adolescent with schizophreniform disorder. Gen Hosp Psychiatry. 2009;31(4):376-378.
10. Razaq M, Samma M. A case of risperidone-induced hypothermia. Am J Ther. 2004;11(3):229-230.
11. Schwaninger M, Weisbrod M, Schwab S, et al. Hypothermia induced by atypical neuroleptics. Clin Neuropharmacol. 1998;21(6):344-346.
12. Bookstaver PB, Miller AD. Possible long-acting risperidone-induced hypothermia precipitating phenytoin toxicity in an elderly patient. J Clin Pharm Ther. 2011; 36(3):426-429.
13. Boschi G, Launay N, Rips R. Neuroleptic-induced hypothermia in mice: lack of evidence for a central mechanism. Br J Pharmacol. 1987;90(4):745-751.
14. Sessler DI. Thermoregulatory defense mechanisms. Crit Care Med. 2009;37(suppl 7):S203-S210.
15. Shiloh R, Weizman A, Epstein Y, et al. Abnormal thermoregulation in drug-free male schizophrenia patients. Eur Neuropsychopharmacol. 2001;11(4):285-288.
16. McCann SM, Vijayan E. Control of anterior pituitary hormone secretion by neurotensin. Ann N Y Acad Sci. 1992; 668:287-297.
17. Chandra A, Chou HC, Chang C, et al. Effecst of intraventricular administration of neurotensin and somatostatin on thermoregulation in the rat. Neuropharmacology. 1981;20(7):715-718.
18. Sharma RP, Janicak PG, Bissette G, et al. CSF neurotensin concentrations and antipsychotic treatment in schizophrenia and schizoaffective disorder. Am J Psychiatry. 1997; 154(7):1019-1021.
19. Kudoh A, Takase H, Takazawa T. Chronic treatment with antipsychotics enhances intraoperative core hypothermia. Anesth Analg. 2004;98(1):111-115.
Vortioxetine for major depressive disorder
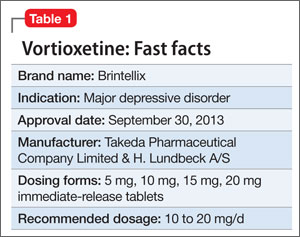
Vortioxetine is FDA-approved to treat major depressive disorder (MDD) (Table 1), having shown efficacy in relieving depressive symptoms in clinical trials.1 Vortioxetine’s mechanism of action enhances CNS serotonergic activity through inhibiting serotonin (5-HT) reuptake, agonizing the 5-HT1A receptor, partially agonizing the 5-HT1B receptor, and antagonizing the 5-HT3, 5-HT1D, and 5-HT7 receptors.
Clinical implications
It is hypothesized that depression is a heterogeneous disease caused by dysregulation of serotonin, norepinephrine, and dopamine, subsequently producing mood and neurovegetative symptoms of depression. Preclinical, in vivo studies indicate that vortioxetine enhances levels of serotonin, norepinephrine, dopamine, acetylcholine, and histamine in specific areas of the brain with the ability to improve depressive symptoms. Vortioxetine’s multimodal activity can be a useful alternative to other serotonergic antidepressants for some patients who are partial responders or non-responders to other treatment options. In addition, vortioxetine appears to have minimal effect on weight2 and sexual function—the latter being dose-dependent.3
How does it work?
Vortioxetine differs from other antidepressants in its multimodal activity (ie, affecting G-protein mode receptors, ion channel mode receptors, and neurotransmitter transporters). It inhibits the serotonin transporter (Ki = 1.6 nM), causing subsequent inhibition of serotonin reuptake into presynaptic neurons as well as selectively acting on the other subtypes of serotonergic receptors; however, activity on the norepinephrine transporter (Ki = 113 nM) and dopamine transporter (Ki > 1000 nM) is minimal. It is believed that mood-regulating effects of vortioxetine are caused by inhibition of serotonin reuptake, prolonged availability of serotonin to the postsynaptic neurons, its agonist activity on the 5-HT1A receptor (Ki = 15 nM), and partial agonist activity on the 5-HT1B receptor (Ki = 33 nM). Vortioxetine has strong affinity for the 5-HT3 receptor (Ki = 3.7 nM), which plays a role in modulation of centrally mediated nausea and vomiting. Positron emission tomography studies in humans determined that the occupancy of 5-HT transporter was 50% at 5 mg/d, 65% at 10 mg/d, and 80% at 20 mg/d.1,4 Human studies did not show that vortioxetine causes QTc prolongation.
Pharmacokinetics
Therapeutic activity of vortioxetine is thought to be due to the parent drug. It has a half-life of approximately 66 hours, and achieves steady state in 13.5 to 19 days. Bioavailability of vortioxetine is 75%; absorption does not depend on food; and 98% of drug is bound on plasma proteins.
Vortioxetine has linear pharmacokinetics, with maximum plasma concentration 7 to 11 hours after ingestion. The medication is metabolized primarily by oxidation through cytochrome P (CYP) 450: CYP2D6 (primary), CYP 3A4/5, CYP 2C19, CYP 2C9, CYP2A6, CYP2C8, and CYP2B6 with subsequent glucuronidation. This predisposes vortioxetine to potential pharmacokinetic drug-drug interaction warranting dose adjustment consideration when vortioxetine is coadministered with compounds inhibiting CYP2D6 or inducing CYP3A4 for ≥14 days, or for patients identified as poor 2D6 metabolizers.
In addition, coadministration of vortioxetine with serotonergic medications such as triptans, other antidepressants, and tramadol can cause potentially life-threatening serotonin syndrome, characterized by mental status changes, autonomic instability, neuromuscular aberrations, and GI symptoms. Concomitant use of vortioxetine and a nonsteroidal anti-inflammatory drug, aspirin, or warfarin can result in abnormal bleeding. Coadministration of vortioxetine with another highly protein-bound drug may increase or decrease the free concentration of either drug depending on the binding affinity of the drug for the protein.
Efficacy
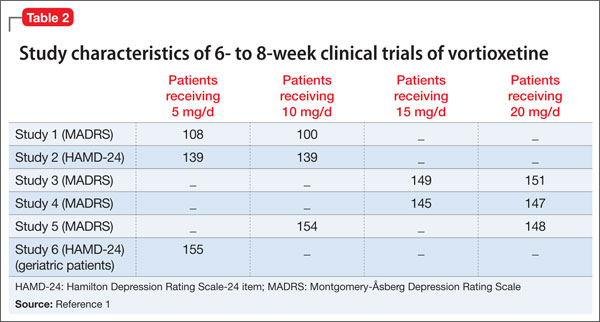
Vortioxetine reduced depressive symptoms in 6 positive, 6- to 8-week, double-blind, placebo controlled and randomized studies and 1 maintenance study.1 Subjects were adults (Studies 1 to 5) and geriatric patients from age 64 to 88 who had ≥1 depressive episode before age 60 (Study 6). All met DSM-IV-TR criteria for MDD. Subjects with cognitive impairment scoring <24 on the Mini-Mental Status Examination and children were excluded. Depending on the study, response to the treatment was primarily measured on the Montgomery-Åsberg Depression Rating Scale (MADRS) or Hamilton Depression Rating Scale (HAM-D).
See Table 2 for a description of the positive studies, including dosages. In all studies, vortioxetine was superior to placebo at least one dosage for treating depression. In the 6- to 8-week placebo-controlled studies, an effect of vortioxetine based on the primary efficacy measure was generally observed starting at Week 2; that effect increased in subsequent weeks with the full antidepressant effect of vortioxetine generally not seen until study Week 4 or later.1
The maintenance treatment study included 639 patients who met DSM-IV-TR criteria for MDD. This study lasted for as long as 64 weeks. The first 12-week period was open-label, during which patients were treated with vortioxetine, 5 mg/d or 10 mg/d, with a possibility to adjust the dosage in the first 8 weeks. By the end of Week 12, 396 subjects achieved remission (MADRS <10), 75% of whom were taking vortioxetine, 10 mg/d. These patients were then randomly assigned to placebo or the dosage of vortioxetine to which they had responded, and continued the study for as long as 64 weeks. Time to relapse (MADRS total score ≥22) or an insufficient therapeutic response (as judged by the investigator) was the primary efficacy outcome, and demonstrated that vortioxetine was superior to placebo.
Tolerability
The tolerability of vortioxetine is comparable with other serotonergic antidepressants. In pooled analysis of pre-marketing studies, 5% to 8% of patients receiving vortioxetine (5 to 20 mg/d) discontinued treatment because of adverse effects (AEs), compared with 4% in the placebo group. Nausea was the most commonly reported AE leading to discontinuation and appeared to be dose dependent.
AEs, such as nausea, constipation, and vomiting, most commonly occurred in the first week of treatment, with a median duration of 2 weeks.5 In the 6- to 8-week trials, the most common AEs were nausea, constipation, and vomiting. In longer trials (24 to 64 weeks), the most common AE was nausea.
In 6- to 8-week placebo-controlled studies, vortioxetine was not associated with any clinically significant effect on vital signs or laboratory values in hematology, urinalysis, or serum chemistry (except sodium). Hyponatremia, the result of the syndrome of inappropriate antidiuretic hormone secretion (SIADH), has occurred. The risk of developing SIADH and resultant hyponatremia is greater in geriatric patients and patients taking a diuretic.
Abruptly discontinuing vortioxetine can cause transient withdrawal symptoms, including headache and muscle tension, especially at a higher dosage (15 to 20 mg/d). Gradual tapering can reduce withdrawal symptoms.
Specific clinical issues
All antidepressants have a “black-box” warning about the potential for clinical worsening and increased suicidality early in treatment. Closely monitor patients for suicidal ideation and behaviors during the first months of treatment and with dosage changes.
Vortioxetine is categorized as pregnancy category C. Newborns exposed to a selective serotonin reuptake inhibitor (SSRI) in pregnancy may have an increased risk of persistent pulmonary hypertension during the neonatal period. When taken during the third trimester of pregnancy, SSRIs and serotonin-norepinephrine reuptake inhibitors can cause serious neonatal complications, including respiratory distress, cyanosis, apnea, and seizures, which may require longer hospitalization, respiratory support, or tube feeding for the infant. Consider risks and benefits of third-trimester use of an antidepressant. It is not known if vortioxetine is present in human breast milk.
Clinical studies on vortioxetine in pediatric patients have not been conducted.
No dosage adjustment is recommended on the basis of age for geriatric patients. No dose adjustment of vortioxetine is necessary on the basis of race, sex, ethnicity, renal function, or mild to moderate hepatic impairment. See Table 3 for practice points when prescribing vortioxetine. See Table 4 for contraindications to vortioxetine.


Dosing
The recommended starting dosage is 10 mg, administered orally once daily without regard to meals. Dosage should then be increased to 20 mg/d, as clinically warranted and tolerated. Consider a dosage decrease to 5 mg/d in patients who do not tolerate higher dosages or require drug adjustment because of drug-drug interaction or poor 2D6 metabolizer status.
Bottom Line
FDA-approved for major depressive disorder in adults, vortioxetine reduced depressive symptoms in 6 positive, double-blind, placebo-controlled, and randomized studies. The multimodal activity of vortioxeine can be a useful alternative to serotonergic antidepressants for some patients who are partial responders or nonresponders. Tolerability is comparable with other serotonergic antidepressants.
Related Resources
- Alam MY, Jacobsen PL, Chen Y, et al. Safety, tolerability, and efficacy of vortioxetine (Lu AA21004) in major depressive disorder: results of an open-label, flexible-dose, 52-week extension study. Int Clin Psychopharmacol. 2014; 29(1):36-44.
- Mahableshwarkar AR, Jacobsen PL, Chen Y. A randomized, double-blind trial of 2.5 mg and 5 mg vortioxetine (Lu AA21004) versus placebo for 8 weeks in adults with major depressive disorder. Curr Med Res Opin. 2013;29(3):217-226.
Drug Brand Names
Linezolid • Zyvox Vortioxetine • Brintellix
Methylene blue • Urolene Blue Warfarin • Coumadin
Tramadol • Ultram
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Vortioxetine [package insert]. Deerfield, IL: Takeda Pharmaceuticals America, Inc.; 2013.
2. Serretti A, Mandelli L. Antidepressants and body weight: a comprehensive review and meta-analysis. J Clin Psychiatry. 2010;71(10):1259-1272.
3. Serretti A, Chiesa A. Treatment-emergent sexual dysfunction related to antidepressants: a comprehensive review and meta-analysis. J Clin Psychopharmacol. 2009; 29(3):259-266.
4. Chen G, Lee R, Højer A, et al. Pharmacokinetic drug interactions involving vortioxetine (LU AA 21004), a multimodal antidepressant. Clin Drug Invetig. 2013; 33(10):727-736.
5. Citrome L. Vortioxetine for major depressive disorder: a systematic review of the efficacy and safety profile for this newly approved antidepressant—what is the number needed to treat, number needed to harm and likelihood to be helped or harmed? Int J Clin Prac. 2014;68(1):60-82.
Vortioxetine is FDA-approved to treat major depressive disorder (MDD) (Table 1), having shown efficacy in relieving depressive symptoms in clinical trials.1 Vortioxetine’s mechanism of action enhances CNS serotonergic activity through inhibiting serotonin (5-HT) reuptake, agonizing the 5-HT1A receptor, partially agonizing the 5-HT1B receptor, and antagonizing the 5-HT3, 5-HT1D, and 5-HT7 receptors.
Clinical implications
It is hypothesized that depression is a heterogeneous disease caused by dysregulation of serotonin, norepinephrine, and dopamine, subsequently producing mood and neurovegetative symptoms of depression. Preclinical, in vivo studies indicate that vortioxetine enhances levels of serotonin, norepinephrine, dopamine, acetylcholine, and histamine in specific areas of the brain with the ability to improve depressive symptoms. Vortioxetine’s multimodal activity can be a useful alternative to other serotonergic antidepressants for some patients who are partial responders or non-responders to other treatment options. In addition, vortioxetine appears to have minimal effect on weight2 and sexual function—the latter being dose-dependent.3
How does it work?
Vortioxetine differs from other antidepressants in its multimodal activity (ie, affecting G-protein mode receptors, ion channel mode receptors, and neurotransmitter transporters). It inhibits the serotonin transporter (Ki = 1.6 nM), causing subsequent inhibition of serotonin reuptake into presynaptic neurons as well as selectively acting on the other subtypes of serotonergic receptors; however, activity on the norepinephrine transporter (Ki = 113 nM) and dopamine transporter (Ki > 1000 nM) is minimal. It is believed that mood-regulating effects of vortioxetine are caused by inhibition of serotonin reuptake, prolonged availability of serotonin to the postsynaptic neurons, its agonist activity on the 5-HT1A receptor (Ki = 15 nM), and partial agonist activity on the 5-HT1B receptor (Ki = 33 nM). Vortioxetine has strong affinity for the 5-HT3 receptor (Ki = 3.7 nM), which plays a role in modulation of centrally mediated nausea and vomiting. Positron emission tomography studies in humans determined that the occupancy of 5-HT transporter was 50% at 5 mg/d, 65% at 10 mg/d, and 80% at 20 mg/d.1,4 Human studies did not show that vortioxetine causes QTc prolongation.
Pharmacokinetics
Therapeutic activity of vortioxetine is thought to be due to the parent drug. It has a half-life of approximately 66 hours, and achieves steady state in 13.5 to 19 days. Bioavailability of vortioxetine is 75%; absorption does not depend on food; and 98% of drug is bound on plasma proteins.
Vortioxetine has linear pharmacokinetics, with maximum plasma concentration 7 to 11 hours after ingestion. The medication is metabolized primarily by oxidation through cytochrome P (CYP) 450: CYP2D6 (primary), CYP 3A4/5, CYP 2C19, CYP 2C9, CYP2A6, CYP2C8, and CYP2B6 with subsequent glucuronidation. This predisposes vortioxetine to potential pharmacokinetic drug-drug interaction warranting dose adjustment consideration when vortioxetine is coadministered with compounds inhibiting CYP2D6 or inducing CYP3A4 for ≥14 days, or for patients identified as poor 2D6 metabolizers.
In addition, coadministration of vortioxetine with serotonergic medications such as triptans, other antidepressants, and tramadol can cause potentially life-threatening serotonin syndrome, characterized by mental status changes, autonomic instability, neuromuscular aberrations, and GI symptoms. Concomitant use of vortioxetine and a nonsteroidal anti-inflammatory drug, aspirin, or warfarin can result in abnormal bleeding. Coadministration of vortioxetine with another highly protein-bound drug may increase or decrease the free concentration of either drug depending on the binding affinity of the drug for the protein.
Efficacy
Vortioxetine reduced depressive symptoms in 6 positive, 6- to 8-week, double-blind, placebo controlled and randomized studies and 1 maintenance study.1 Subjects were adults (Studies 1 to 5) and geriatric patients from age 64 to 88 who had ≥1 depressive episode before age 60 (Study 6). All met DSM-IV-TR criteria for MDD. Subjects with cognitive impairment scoring <24 on the Mini-Mental Status Examination and children were excluded. Depending on the study, response to the treatment was primarily measured on the Montgomery-Åsberg Depression Rating Scale (MADRS) or Hamilton Depression Rating Scale (HAM-D).
See Table 2 for a description of the positive studies, including dosages. In all studies, vortioxetine was superior to placebo at least one dosage for treating depression. In the 6- to 8-week placebo-controlled studies, an effect of vortioxetine based on the primary efficacy measure was generally observed starting at Week 2; that effect increased in subsequent weeks with the full antidepressant effect of vortioxetine generally not seen until study Week 4 or later.1
The maintenance treatment study included 639 patients who met DSM-IV-TR criteria for MDD. This study lasted for as long as 64 weeks. The first 12-week period was open-label, during which patients were treated with vortioxetine, 5 mg/d or 10 mg/d, with a possibility to adjust the dosage in the first 8 weeks. By the end of Week 12, 396 subjects achieved remission (MADRS <10), 75% of whom were taking vortioxetine, 10 mg/d. These patients were then randomly assigned to placebo or the dosage of vortioxetine to which they had responded, and continued the study for as long as 64 weeks. Time to relapse (MADRS total score ≥22) or an insufficient therapeutic response (as judged by the investigator) was the primary efficacy outcome, and demonstrated that vortioxetine was superior to placebo.
Tolerability
The tolerability of vortioxetine is comparable with other serotonergic antidepressants. In pooled analysis of pre-marketing studies, 5% to 8% of patients receiving vortioxetine (5 to 20 mg/d) discontinued treatment because of adverse effects (AEs), compared with 4% in the placebo group. Nausea was the most commonly reported AE leading to discontinuation and appeared to be dose dependent.
AEs, such as nausea, constipation, and vomiting, most commonly occurred in the first week of treatment, with a median duration of 2 weeks.5 In the 6- to 8-week trials, the most common AEs were nausea, constipation, and vomiting. In longer trials (24 to 64 weeks), the most common AE was nausea.
In 6- to 8-week placebo-controlled studies, vortioxetine was not associated with any clinically significant effect on vital signs or laboratory values in hematology, urinalysis, or serum chemistry (except sodium). Hyponatremia, the result of the syndrome of inappropriate antidiuretic hormone secretion (SIADH), has occurred. The risk of developing SIADH and resultant hyponatremia is greater in geriatric patients and patients taking a diuretic.
Abruptly discontinuing vortioxetine can cause transient withdrawal symptoms, including headache and muscle tension, especially at a higher dosage (15 to 20 mg/d). Gradual tapering can reduce withdrawal symptoms.
Specific clinical issues
All antidepressants have a “black-box” warning about the potential for clinical worsening and increased suicidality early in treatment. Closely monitor patients for suicidal ideation and behaviors during the first months of treatment and with dosage changes.
Vortioxetine is categorized as pregnancy category C. Newborns exposed to a selective serotonin reuptake inhibitor (SSRI) in pregnancy may have an increased risk of persistent pulmonary hypertension during the neonatal period. When taken during the third trimester of pregnancy, SSRIs and serotonin-norepinephrine reuptake inhibitors can cause serious neonatal complications, including respiratory distress, cyanosis, apnea, and seizures, which may require longer hospitalization, respiratory support, or tube feeding for the infant. Consider risks and benefits of third-trimester use of an antidepressant. It is not known if vortioxetine is present in human breast milk.
Clinical studies on vortioxetine in pediatric patients have not been conducted.
No dosage adjustment is recommended on the basis of age for geriatric patients. No dose adjustment of vortioxetine is necessary on the basis of race, sex, ethnicity, renal function, or mild to moderate hepatic impairment. See Table 3 for practice points when prescribing vortioxetine. See Table 4 for contraindications to vortioxetine.
Dosing
The recommended starting dosage is 10 mg, administered orally once daily without regard to meals. Dosage should then be increased to 20 mg/d, as clinically warranted and tolerated. Consider a dosage decrease to 5 mg/d in patients who do not tolerate higher dosages or require drug adjustment because of drug-drug interaction or poor 2D6 metabolizer status.
Bottom Line
FDA-approved for major depressive disorder in adults, vortioxetine reduced depressive symptoms in 6 positive, double-blind, placebo-controlled, and randomized studies. The multimodal activity of vortioxeine can be a useful alternative to serotonergic antidepressants for some patients who are partial responders or nonresponders. Tolerability is comparable with other serotonergic antidepressants.
Related Resources
- Alam MY, Jacobsen PL, Chen Y, et al. Safety, tolerability, and efficacy of vortioxetine (Lu AA21004) in major depressive disorder: results of an open-label, flexible-dose, 52-week extension study. Int Clin Psychopharmacol. 2014; 29(1):36-44.
- Mahableshwarkar AR, Jacobsen PL, Chen Y. A randomized, double-blind trial of 2.5 mg and 5 mg vortioxetine (Lu AA21004) versus placebo for 8 weeks in adults with major depressive disorder. Curr Med Res Opin. 2013;29(3):217-226.
Drug Brand Names
Linezolid • Zyvox Vortioxetine • Brintellix
Methylene blue • Urolene Blue Warfarin • Coumadin
Tramadol • Ultram
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Vortioxetine is FDA-approved to treat major depressive disorder (MDD) (Table 1), having shown efficacy in relieving depressive symptoms in clinical trials.1 Vortioxetine’s mechanism of action enhances CNS serotonergic activity through inhibiting serotonin (5-HT) reuptake, agonizing the 5-HT1A receptor, partially agonizing the 5-HT1B receptor, and antagonizing the 5-HT3, 5-HT1D, and 5-HT7 receptors.
Clinical implications
It is hypothesized that depression is a heterogeneous disease caused by dysregulation of serotonin, norepinephrine, and dopamine, subsequently producing mood and neurovegetative symptoms of depression. Preclinical, in vivo studies indicate that vortioxetine enhances levels of serotonin, norepinephrine, dopamine, acetylcholine, and histamine in specific areas of the brain with the ability to improve depressive symptoms. Vortioxetine’s multimodal activity can be a useful alternative to other serotonergic antidepressants for some patients who are partial responders or non-responders to other treatment options. In addition, vortioxetine appears to have minimal effect on weight2 and sexual function—the latter being dose-dependent.3
How does it work?
Vortioxetine differs from other antidepressants in its multimodal activity (ie, affecting G-protein mode receptors, ion channel mode receptors, and neurotransmitter transporters). It inhibits the serotonin transporter (Ki = 1.6 nM), causing subsequent inhibition of serotonin reuptake into presynaptic neurons as well as selectively acting on the other subtypes of serotonergic receptors; however, activity on the norepinephrine transporter (Ki = 113 nM) and dopamine transporter (Ki > 1000 nM) is minimal. It is believed that mood-regulating effects of vortioxetine are caused by inhibition of serotonin reuptake, prolonged availability of serotonin to the postsynaptic neurons, its agonist activity on the 5-HT1A receptor (Ki = 15 nM), and partial agonist activity on the 5-HT1B receptor (Ki = 33 nM). Vortioxetine has strong affinity for the 5-HT3 receptor (Ki = 3.7 nM), which plays a role in modulation of centrally mediated nausea and vomiting. Positron emission tomography studies in humans determined that the occupancy of 5-HT transporter was 50% at 5 mg/d, 65% at 10 mg/d, and 80% at 20 mg/d.1,4 Human studies did not show that vortioxetine causes QTc prolongation.
Pharmacokinetics
Therapeutic activity of vortioxetine is thought to be due to the parent drug. It has a half-life of approximately 66 hours, and achieves steady state in 13.5 to 19 days. Bioavailability of vortioxetine is 75%; absorption does not depend on food; and 98% of drug is bound on plasma proteins.
Vortioxetine has linear pharmacokinetics, with maximum plasma concentration 7 to 11 hours after ingestion. The medication is metabolized primarily by oxidation through cytochrome P (CYP) 450: CYP2D6 (primary), CYP 3A4/5, CYP 2C19, CYP 2C9, CYP2A6, CYP2C8, and CYP2B6 with subsequent glucuronidation. This predisposes vortioxetine to potential pharmacokinetic drug-drug interaction warranting dose adjustment consideration when vortioxetine is coadministered with compounds inhibiting CYP2D6 or inducing CYP3A4 for ≥14 days, or for patients identified as poor 2D6 metabolizers.
In addition, coadministration of vortioxetine with serotonergic medications such as triptans, other antidepressants, and tramadol can cause potentially life-threatening serotonin syndrome, characterized by mental status changes, autonomic instability, neuromuscular aberrations, and GI symptoms. Concomitant use of vortioxetine and a nonsteroidal anti-inflammatory drug, aspirin, or warfarin can result in abnormal bleeding. Coadministration of vortioxetine with another highly protein-bound drug may increase or decrease the free concentration of either drug depending on the binding affinity of the drug for the protein.
Efficacy
Vortioxetine reduced depressive symptoms in 6 positive, 6- to 8-week, double-blind, placebo controlled and randomized studies and 1 maintenance study.1 Subjects were adults (Studies 1 to 5) and geriatric patients from age 64 to 88 who had ≥1 depressive episode before age 60 (Study 6). All met DSM-IV-TR criteria for MDD. Subjects with cognitive impairment scoring <24 on the Mini-Mental Status Examination and children were excluded. Depending on the study, response to the treatment was primarily measured on the Montgomery-Åsberg Depression Rating Scale (MADRS) or Hamilton Depression Rating Scale (HAM-D).
See Table 2 for a description of the positive studies, including dosages. In all studies, vortioxetine was superior to placebo at least one dosage for treating depression. In the 6- to 8-week placebo-controlled studies, an effect of vortioxetine based on the primary efficacy measure was generally observed starting at Week 2; that effect increased in subsequent weeks with the full antidepressant effect of vortioxetine generally not seen until study Week 4 or later.1
The maintenance treatment study included 639 patients who met DSM-IV-TR criteria for MDD. This study lasted for as long as 64 weeks. The first 12-week period was open-label, during which patients were treated with vortioxetine, 5 mg/d or 10 mg/d, with a possibility to adjust the dosage in the first 8 weeks. By the end of Week 12, 396 subjects achieved remission (MADRS <10), 75% of whom were taking vortioxetine, 10 mg/d. These patients were then randomly assigned to placebo or the dosage of vortioxetine to which they had responded, and continued the study for as long as 64 weeks. Time to relapse (MADRS total score ≥22) or an insufficient therapeutic response (as judged by the investigator) was the primary efficacy outcome, and demonstrated that vortioxetine was superior to placebo.
Tolerability
The tolerability of vortioxetine is comparable with other serotonergic antidepressants. In pooled analysis of pre-marketing studies, 5% to 8% of patients receiving vortioxetine (5 to 20 mg/d) discontinued treatment because of adverse effects (AEs), compared with 4% in the placebo group. Nausea was the most commonly reported AE leading to discontinuation and appeared to be dose dependent.
AEs, such as nausea, constipation, and vomiting, most commonly occurred in the first week of treatment, with a median duration of 2 weeks.5 In the 6- to 8-week trials, the most common AEs were nausea, constipation, and vomiting. In longer trials (24 to 64 weeks), the most common AE was nausea.
In 6- to 8-week placebo-controlled studies, vortioxetine was not associated with any clinically significant effect on vital signs or laboratory values in hematology, urinalysis, or serum chemistry (except sodium). Hyponatremia, the result of the syndrome of inappropriate antidiuretic hormone secretion (SIADH), has occurred. The risk of developing SIADH and resultant hyponatremia is greater in geriatric patients and patients taking a diuretic.
Abruptly discontinuing vortioxetine can cause transient withdrawal symptoms, including headache and muscle tension, especially at a higher dosage (15 to 20 mg/d). Gradual tapering can reduce withdrawal symptoms.
Specific clinical issues
All antidepressants have a “black-box” warning about the potential for clinical worsening and increased suicidality early in treatment. Closely monitor patients for suicidal ideation and behaviors during the first months of treatment and with dosage changes.
Vortioxetine is categorized as pregnancy category C. Newborns exposed to a selective serotonin reuptake inhibitor (SSRI) in pregnancy may have an increased risk of persistent pulmonary hypertension during the neonatal period. When taken during the third trimester of pregnancy, SSRIs and serotonin-norepinephrine reuptake inhibitors can cause serious neonatal complications, including respiratory distress, cyanosis, apnea, and seizures, which may require longer hospitalization, respiratory support, or tube feeding for the infant. Consider risks and benefits of third-trimester use of an antidepressant. It is not known if vortioxetine is present in human breast milk.
Clinical studies on vortioxetine in pediatric patients have not been conducted.
No dosage adjustment is recommended on the basis of age for geriatric patients. No dose adjustment of vortioxetine is necessary on the basis of race, sex, ethnicity, renal function, or mild to moderate hepatic impairment. See Table 3 for practice points when prescribing vortioxetine. See Table 4 for contraindications to vortioxetine.
Dosing
The recommended starting dosage is 10 mg, administered orally once daily without regard to meals. Dosage should then be increased to 20 mg/d, as clinically warranted and tolerated. Consider a dosage decrease to 5 mg/d in patients who do not tolerate higher dosages or require drug adjustment because of drug-drug interaction or poor 2D6 metabolizer status.
Bottom Line
FDA-approved for major depressive disorder in adults, vortioxetine reduced depressive symptoms in 6 positive, double-blind, placebo-controlled, and randomized studies. The multimodal activity of vortioxeine can be a useful alternative to serotonergic antidepressants for some patients who are partial responders or nonresponders. Tolerability is comparable with other serotonergic antidepressants.
Related Resources
- Alam MY, Jacobsen PL, Chen Y, et al. Safety, tolerability, and efficacy of vortioxetine (Lu AA21004) in major depressive disorder: results of an open-label, flexible-dose, 52-week extension study. Int Clin Psychopharmacol. 2014; 29(1):36-44.
- Mahableshwarkar AR, Jacobsen PL, Chen Y. A randomized, double-blind trial of 2.5 mg and 5 mg vortioxetine (Lu AA21004) versus placebo for 8 weeks in adults with major depressive disorder. Curr Med Res Opin. 2013;29(3):217-226.
Drug Brand Names
Linezolid • Zyvox Vortioxetine • Brintellix
Methylene blue • Urolene Blue Warfarin • Coumadin
Tramadol • Ultram
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Vortioxetine [package insert]. Deerfield, IL: Takeda Pharmaceuticals America, Inc.; 2013.
2. Serretti A, Mandelli L. Antidepressants and body weight: a comprehensive review and meta-analysis. J Clin Psychiatry. 2010;71(10):1259-1272.
3. Serretti A, Chiesa A. Treatment-emergent sexual dysfunction related to antidepressants: a comprehensive review and meta-analysis. J Clin Psychopharmacol. 2009; 29(3):259-266.
4. Chen G, Lee R, Højer A, et al. Pharmacokinetic drug interactions involving vortioxetine (LU AA 21004), a multimodal antidepressant. Clin Drug Invetig. 2013; 33(10):727-736.
5. Citrome L. Vortioxetine for major depressive disorder: a systematic review of the efficacy and safety profile for this newly approved antidepressant—what is the number needed to treat, number needed to harm and likelihood to be helped or harmed? Int J Clin Prac. 2014;68(1):60-82.
1. Vortioxetine [package insert]. Deerfield, IL: Takeda Pharmaceuticals America, Inc.; 2013.
2. Serretti A, Mandelli L. Antidepressants and body weight: a comprehensive review and meta-analysis. J Clin Psychiatry. 2010;71(10):1259-1272.
3. Serretti A, Chiesa A. Treatment-emergent sexual dysfunction related to antidepressants: a comprehensive review and meta-analysis. J Clin Psychopharmacol. 2009; 29(3):259-266.
4. Chen G, Lee R, Højer A, et al. Pharmacokinetic drug interactions involving vortioxetine (LU AA 21004), a multimodal antidepressant. Clin Drug Invetig. 2013; 33(10):727-736.
5. Citrome L. Vortioxetine for major depressive disorder: a systematic review of the efficacy and safety profile for this newly approved antidepressant—what is the number needed to treat, number needed to harm and likelihood to be helped or harmed? Int J Clin Prac. 2014;68(1):60-82.
Awakening to the dangers of obstructive sleep apnea
Estimates are that 50 to 70 million Americans suffer from a chronic disorder of sleep and wakefulness, hindering daily functioning and affecting health.1 Psychiatric illness is common among people who have a sleep disorder. The relationship between psychiatric illness and sleep disorders is bidirectional: People with mental illness often have sleep complaints, and a primary sleep disorder often results in neuropsychiatric complications.
What is obstructive sleep apnea?
The most common type of sleep-disordered breathing, obstructive sleep apnea (OSA) is characterized by frequent cessations of breathing during sleep because of an obstruction of the upper airway. The obstruction occurs secondary to inadequate motor tone of the tongue or airway dilator muscles, or both.1 In addition, many people with OSA have central apneic episodes, in which breathing stops temporarily without airway blockage or respiratory effort.2
The prevalence of OSA is growing as obesity in the United States increases. Risk factors for OSA include obesity, a craniofacial abnormality, an upper-airway abnormality, heredity, smoking, and nasal congestion. OSA plays a role in causing and exacerbating medical illness in people with severe and persistent mental illness, contributing to a significantly shortened life span. Attending to the general health of people who suffer from severe mental illness—including effective treatment of illnesses such as OSA—is crucial.3
Clinical features of OSA
OSA is characterized by hypopnea (a decrease in breathing during sleep) or apnea (an actual pause in breathing). Pauses in breathing during sleep of at least 10 seconds, with obstruction of oronasal airflow despite continuous chest and abdominal movements, are referred to as obstructive apneas. These pauses are associated with a decrease in oxygen saturation or arousal from sleep, or both.1
Primary features of OSA include sleep fragmentation accompanied by nocturnal hypoxemia and hypercapnia, with resulting excessive daytime sleepiness, mood problems, and poor neurocognitive performance (Table 1). OSA often causes potentially serious organ system dysfunction, including adverse cardiovascular and metabolic effects. Studies have suggested that executive dysfunction can be a feature of OSA, which is thought to be related to prefrontal lobe dysfunction caused by intermittent hypoxia. All of these conditions can contribute significantly to decreased quality of life.1

The prevalence of OSA in the general population is approximately 20% when the condition is defined as an apnea-hypopnea index >5 events an hour. The index is the number of apnea and hypopnea episodes that occur during 1 hour of sleep.4
OSA and psychiatric illness
Psychiatric disorders often are comorbid with OSA. These include depression, anxiety, bipolar disorder, schizophrenia, posttraumatic stress disorder (PTSD), panic disorder, and substance use disorder.
Depression. Several studies have documented that OSA and depressive disorder often are comorbid. Many symptoms are common to both, including fatigue, daytime sleepiness, poor concentration, irritability, and weight gain (Figure), although some core symptoms of depression (eg, sadness, anhedonia, guilt, and agitation) are clearly distinguishable from symptoms of OSA. The current recommendation is that a mood disorder should be considered secondary to OSA, and treated accordingly.5
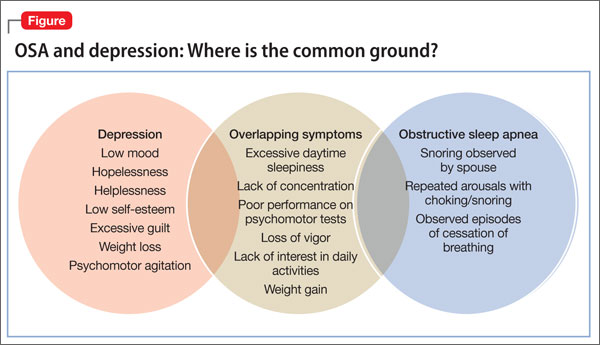
Anxiety. OSA also has been linked to anxiety and nocturnal panic attacks. Frequent awakening due to choking from breathing cessation might play a role in the development of anxiety in patients with OSA, although the association is unproven. Studies have shown a correlation between anxiety disorders and excessive daytime sleepiness, one of the core symptoms of OSA.6 OSA is highly prevalent among combat veterans who have PTSD and complain of being overly vigilant at night; experiencing nightmares and frequent awakening; and having non-restorative sleep.7 Anecdotal reports suggest an association between OSA and bipolar disorder: namely, that continuous positive airway pressure (CPAP) treatment (see “How is OSA treated?,” below) might switch depressed patients to mania.8
Schizophrenia. A strong association exists between OSA and schizophrenia. In a study,9 an OSA diagnosis was made 6 times more often in patients with schizophrenia than in patients with other psychiatric illnesses. Obesity, male sex, and chronic antipsychotic administration were risk factors for OSA in patients with
schizophrenia.9 OSA might be underdiagnosed in patients with schizophrenia because excessive daytime sleepiness, the most common daytime symptom of OSA, can be misattributed as a negative symptom of the disease or a side effect of pharmacotherapy.
OSA and medical illness
OSA can be comorbid with several medical conditions (Table 2). Sleep research in the past 15 years has demonstrated that chronic sleep deprivation has multiple untoward health consequences apart from excessive daytime sleepiness.10 Recent research suggests that chronic sleep loss (<7 hours a night), including sleep loss secondary to OSA, has wide-ranging effects on the cardiovascular, endocrine, immune, and nervous systems, including:
• obesity (adults and children)
• diabetes mellitus and impaired glucose tolerance
• cardiovascular disease and hypertension.

Obesity is one of the primary and more modifiable risk factors for OSA (Box). Studies suggest that reducing the severity of obesity would likely benefit people with a sleep disorder, and that treating sleep deprivation and sleep disorders might benefit persons with obesity.12 Chronic sleep loss can have a deleterious influence on appetite regulation through effects on 2 hormones, leptin and ghrelin, that play a major role in appetite regulation. Chronic sleep loss causes and perpetuates obesity through its interplay with these, and other, hormones.12

Diabetes. The link between obesity and diabetes is well-established, as is the long-term morbidity and mortality of these 2 diseases.13 Evidence shows that OSA is associated with impaired glucose tolerance and an increased risk of diabetes.14
Cardiovascular disease. OSA has a strong association with cardiovascular disease, including systemic hypertension, possibly myocardial infarction, congestive heart failure, and stroke.15 Institution of appropriate treatment for OSA including CPAP can minimize or reverse many of these effects.16
Making an OSA diagnosis
A diagnostic polysomnogram (PSG), or sleep study, is the standard test when OSA is suspected. It is performed most often at an attended sleep laboratory. Typically, a PSG measures several physiologic measures, including, but not limited to:
• airflow through mouth and nose
• stages of sleep (by means of electroencephalography channels)
• thoracic and abdominal movements (to assess effort of breathing)
• muscle activity of the chin
• oxyhemoglobin saturation (to monitor variability in oxygen saturation [SaO2] during OSA events).
Portable diagnostic instruments can provide reliable information when a patient cannot be studied in a laboratory. Assessments available on portable instruments include cardiopulmonary monitoring of respiration only; PSG; and peripheral arterial tonometry, which measures autonomic manifestations of respiratory obstructive events.17,18
The severity of OSA is established by the apnea/hypopnea index, which measures the number of apneas and hypopneas per hour of sleep.
How is OSA treated?
CPAP is still the gold standard for treating OSA. CPAP provides a pneumatic splint for the upper airway by administering positive pressure through a nasal or oronasal mask. CPAP distinctly improves daytime sleepiness.19,20
Pressure is determined initially by titration during PSG, although a number of automated CPAP machines are available in which pressure is adjusted based on the machine’s response to airflow obstruction. Advantages of using PSG to titrate CPAP are direct observation to control mask leak and the ability to observe the effects of body position and sleep stage and clearly distinguish periods of sleep from wakefulness.
Regrettably, adherence to a nightly regimen of CPAP is less than ideal for several reasons, including claustrophobia, interface failure, and other motivational variables. Some patients who experience claustrophobia can use desensitization techniques; others are, ultimately, unable to use the mask.
Oral appliances. A patient who has mild or moderate OSA but who cannot use the CPAP mask might be a good candidate for an oral appliance. These appliances, which hold the mandible in an advanced position during the night, can be effective in such cases.
CPAP autotitration changes the treatment pressure based on feedback from such patient measures as airflow and airway resistance. Autotitrating devices might have a role in beginning treatment in patients with OSA by means of a portable sleep study, in which CPAP titration is not performed. In addition, autotitrating offers the possibility of changing pressure over time—such as with changes in position during the night or over the longer term in response to weight loss or gain.
Surgery. In patients who are unable to use CPAP, surgery might be indicated to relieve an anatomical obstruction, such as adenotonsillar hypertrophy or other type of mass lesion.
Sleep positioning. A patient who demonstrates OSA exclusively while sleeping supine might benefit from being trained to sleep on either side only or arranging pillows so that he can only sleep on his side.
In conclusion
OSA is common and easily treatable. It coexists with, and exacerbates, medical and psychiatric illness. Treating OSA concomitantly with comorbid medical and psychiatric illness is essential to achieve full symptom remission and prevent associated long-term consequences of both medical and psychiatric illness.
BOTTOM LINE
Obstructive sleep apnea (OSA) and psychiatric illness, especially depression, often co-exist. Screen depressed patients—especially those with risk factors for OSA, such as obesity, smoking, and an upper-airway abnormality—for a sleep disorder. This is especially important if a patient complains of daytime somnolence, fatigue, cognitive problems, poor concentration, or weight gain. For optimal results, treat comorbid psychiatric illness and OSA concurrently; the same is true for other sleep disorders.
Related Resources
- Babson KA, Del Re AC, Bonn-Miller MO, et al. The comorbidity of sleep apnea and mood, anxiety, and substance use disorders among obese military veterans within the Veterans Health Administration. J Clin Sleep Med. 2013; 9(12):1253-1258.
- Karkoulias K, Lykouras D, Sampsonas F, et al. The impact of obstructive sleep apnea syndrome severity on physical performance and mental health. The use of SF-36 questionnaire in sleep apnea. Eur Rev Med Pharmacol Sci. 2013;17(4):531-536.
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Acknowledgment
Dr. Muhammad Awais Aftab, psychiatry resident at Hamad Medical Corporation, Doha, Qatar, and Umair Amin, final year MBBS student at King Edward Medical University, Lahore, Pakistan, assisted with development of the manuscript of this article.
1. Institute of Medicine. Sleep disorders and sleep deprivation: an unmet public health problem. Washington, DC: The National Academies Press; 2006:20.
2. Badr MS. Central sleep apnea. Prim Care. 2005;32(2):361-374.
3. Freedland KE, Carney RM, Hayano J, et al. Effect of obstructive sleep apnea on response to cognitive behavior therapy for depression after an acute myocardial infarction. J Psychosom Res. 2012;72(4):276-281.
4. Punjabi NM. The epidemiology of adult obstructive sleep apnea. Proc Am Thorac Soc. 2008;5(2):136-143.
5. El-Sherbini AM, Bediwy AS, El-Mitwalli A. Association between obstructive sleep apnea (OSA) and depression and the effect of continuous positive airway pressure (CPAP) treatment. Neuropsychiatr Dis Treat. 2011;7:715-721.
6. Hasler G, Buysse DJ, Gamma A, et al. Excessive daytime sleepiness in young adults: a 20-year prospective community study. J Clin Psychiatry. 2005;66(4):521-529.
7. Yesavage JA, Kinoshita LM, Kimball T, et al. Sleep-disordered breathing in Vietnam veterans with posttraumatic stress disorder. Am J Geriatr Psychiatry. 2012;20(3):199-204.
8. Plante D, Winkelman J. Sleep disturbance in bipolar disorder: therapeutic implications. Am J Psychiatry. 2008; 165(7):830-843.
9. Winkelman J. Schizophrenia, obesity, and obstructive sleep apnea. J Clin Psychiatry. 2001;62(1):8-11.
10. Partinen M, Hublin C. Epidemiology of sleep disorders. Philadelphia, PA: Elsevier Saunders; 2005.
11. Valderas JM, Starfield B, Sibbald B, et al. Defining comorbidity: implications for understanding health and health services. Ann Fam Med. 2009;7(4):357-363.
12. Romero-Corral A, Caples SM, Lopez-Jimenez F, et al. Interactions between obesity and obstructive sleep apnea: implications for treatment. Chest. 2010;137(3):711-719.
13. Villareal DT, Apovian CM, Kushner RF, et al. Obesity in older adults: technical review and position statement of the American Society for Nutrition and NAASO, The Obesity Society. Obes Res. 2005;13(11):1849-1863.
14. Pamidi S, Aronsohn RS, Tasali E. Obstructive sleep apnea: role in the risk and severity of diabetes. Best Pract Res Clin Endocrinol Metab. 2010;24(5):703-715.
15. Malhotra A, Loscalzo J. Sleep and cardiovascular disease: an overview. Prog Cardiovasc Dis. 2009;51(4):279-284.
16. Bradley TD, Floras JS. Obstructive sleep apnoea and its cardiovascular consequences. Lancet. 2009;373(9657):82-93.
17. Chesson A, Berry R, Pack A. Practice parameters for the use of portable monitoring devices in the investigation of suspected obstructive sleep apnea in adults. Sleep. 2003; 26(7):907-913.
18. Pittman S, Ayas N, MacDonald M, et al. Using a wrist-worn device based on peripheral arterial tonometry to diagnose obstructive sleep apnea: in-laboratory and ambulatory validation. Sleep. 2004;27(1):923-933.
19. Ballester E, Badia J, Hernandez L, et al. Evidence of the effectiveness of continuous positive airway pressure in the treatment of sleep apnea/hypopnea syndrome. Am J Respir Crit Care Med. 1999;159:495-501.
20. Jenkinson D, Davies J, Mullins R, et al. Comparison of therapeutic and subtherapeutic nasal continuous positive airway pressure for obstructive sleep apnoea: a randomised prospective parallel trial. Lancet. 1999;353:2100-2105.
Estimates are that 50 to 70 million Americans suffer from a chronic disorder of sleep and wakefulness, hindering daily functioning and affecting health.1 Psychiatric illness is common among people who have a sleep disorder. The relationship between psychiatric illness and sleep disorders is bidirectional: People with mental illness often have sleep complaints, and a primary sleep disorder often results in neuropsychiatric complications.
What is obstructive sleep apnea?
The most common type of sleep-disordered breathing, obstructive sleep apnea (OSA) is characterized by frequent cessations of breathing during sleep because of an obstruction of the upper airway. The obstruction occurs secondary to inadequate motor tone of the tongue or airway dilator muscles, or both.1 In addition, many people with OSA have central apneic episodes, in which breathing stops temporarily without airway blockage or respiratory effort.2
The prevalence of OSA is growing as obesity in the United States increases. Risk factors for OSA include obesity, a craniofacial abnormality, an upper-airway abnormality, heredity, smoking, and nasal congestion. OSA plays a role in causing and exacerbating medical illness in people with severe and persistent mental illness, contributing to a significantly shortened life span. Attending to the general health of people who suffer from severe mental illness—including effective treatment of illnesses such as OSA—is crucial.3
Clinical features of OSA
OSA is characterized by hypopnea (a decrease in breathing during sleep) or apnea (an actual pause in breathing). Pauses in breathing during sleep of at least 10 seconds, with obstruction of oronasal airflow despite continuous chest and abdominal movements, are referred to as obstructive apneas. These pauses are associated with a decrease in oxygen saturation or arousal from sleep, or both.1
Primary features of OSA include sleep fragmentation accompanied by nocturnal hypoxemia and hypercapnia, with resulting excessive daytime sleepiness, mood problems, and poor neurocognitive performance (Table 1). OSA often causes potentially serious organ system dysfunction, including adverse cardiovascular and metabolic effects. Studies have suggested that executive dysfunction can be a feature of OSA, which is thought to be related to prefrontal lobe dysfunction caused by intermittent hypoxia. All of these conditions can contribute significantly to decreased quality of life.1
The prevalence of OSA in the general population is approximately 20% when the condition is defined as an apnea-hypopnea index >5 events an hour. The index is the number of apnea and hypopnea episodes that occur during 1 hour of sleep.4
OSA and psychiatric illness
Psychiatric disorders often are comorbid with OSA. These include depression, anxiety, bipolar disorder, schizophrenia, posttraumatic stress disorder (PTSD), panic disorder, and substance use disorder.
Depression. Several studies have documented that OSA and depressive disorder often are comorbid. Many symptoms are common to both, including fatigue, daytime sleepiness, poor concentration, irritability, and weight gain (Figure), although some core symptoms of depression (eg, sadness, anhedonia, guilt, and agitation) are clearly distinguishable from symptoms of OSA. The current recommendation is that a mood disorder should be considered secondary to OSA, and treated accordingly.5
Anxiety. OSA also has been linked to anxiety and nocturnal panic attacks. Frequent awakening due to choking from breathing cessation might play a role in the development of anxiety in patients with OSA, although the association is unproven. Studies have shown a correlation between anxiety disorders and excessive daytime sleepiness, one of the core symptoms of OSA.6 OSA is highly prevalent among combat veterans who have PTSD and complain of being overly vigilant at night; experiencing nightmares and frequent awakening; and having non-restorative sleep.7 Anecdotal reports suggest an association between OSA and bipolar disorder: namely, that continuous positive airway pressure (CPAP) treatment (see “How is OSA treated?,” below) might switch depressed patients to mania.8
Schizophrenia. A strong association exists between OSA and schizophrenia. In a study,9 an OSA diagnosis was made 6 times more often in patients with schizophrenia than in patients with other psychiatric illnesses. Obesity, male sex, and chronic antipsychotic administration were risk factors for OSA in patients with
schizophrenia.9 OSA might be underdiagnosed in patients with schizophrenia because excessive daytime sleepiness, the most common daytime symptom of OSA, can be misattributed as a negative symptom of the disease or a side effect of pharmacotherapy.
OSA and medical illness
OSA can be comorbid with several medical conditions (Table 2). Sleep research in the past 15 years has demonstrated that chronic sleep deprivation has multiple untoward health consequences apart from excessive daytime sleepiness.10 Recent research suggests that chronic sleep loss (<7 hours a night), including sleep loss secondary to OSA, has wide-ranging effects on the cardiovascular, endocrine, immune, and nervous systems, including:
• obesity (adults and children)
• diabetes mellitus and impaired glucose tolerance
• cardiovascular disease and hypertension.
Obesity is one of the primary and more modifiable risk factors for OSA (Box). Studies suggest that reducing the severity of obesity would likely benefit people with a sleep disorder, and that treating sleep deprivation and sleep disorders might benefit persons with obesity.12 Chronic sleep loss can have a deleterious influence on appetite regulation through effects on 2 hormones, leptin and ghrelin, that play a major role in appetite regulation. Chronic sleep loss causes and perpetuates obesity through its interplay with these, and other, hormones.12
Diabetes. The link between obesity and diabetes is well-established, as is the long-term morbidity and mortality of these 2 diseases.13 Evidence shows that OSA is associated with impaired glucose tolerance and an increased risk of diabetes.14
Cardiovascular disease. OSA has a strong association with cardiovascular disease, including systemic hypertension, possibly myocardial infarction, congestive heart failure, and stroke.15 Institution of appropriate treatment for OSA including CPAP can minimize or reverse many of these effects.16
Making an OSA diagnosis
A diagnostic polysomnogram (PSG), or sleep study, is the standard test when OSA is suspected. It is performed most often at an attended sleep laboratory. Typically, a PSG measures several physiologic measures, including, but not limited to:
• airflow through mouth and nose
• stages of sleep (by means of electroencephalography channels)
• thoracic and abdominal movements (to assess effort of breathing)
• muscle activity of the chin
• oxyhemoglobin saturation (to monitor variability in oxygen saturation [SaO2] during OSA events).
Portable diagnostic instruments can provide reliable information when a patient cannot be studied in a laboratory. Assessments available on portable instruments include cardiopulmonary monitoring of respiration only; PSG; and peripheral arterial tonometry, which measures autonomic manifestations of respiratory obstructive events.17,18
The severity of OSA is established by the apnea/hypopnea index, which measures the number of apneas and hypopneas per hour of sleep.
How is OSA treated?
CPAP is still the gold standard for treating OSA. CPAP provides a pneumatic splint for the upper airway by administering positive pressure through a nasal or oronasal mask. CPAP distinctly improves daytime sleepiness.19,20
Pressure is determined initially by titration during PSG, although a number of automated CPAP machines are available in which pressure is adjusted based on the machine’s response to airflow obstruction. Advantages of using PSG to titrate CPAP are direct observation to control mask leak and the ability to observe the effects of body position and sleep stage and clearly distinguish periods of sleep from wakefulness.
Regrettably, adherence to a nightly regimen of CPAP is less than ideal for several reasons, including claustrophobia, interface failure, and other motivational variables. Some patients who experience claustrophobia can use desensitization techniques; others are, ultimately, unable to use the mask.
Oral appliances. A patient who has mild or moderate OSA but who cannot use the CPAP mask might be a good candidate for an oral appliance. These appliances, which hold the mandible in an advanced position during the night, can be effective in such cases.
CPAP autotitration changes the treatment pressure based on feedback from such patient measures as airflow and airway resistance. Autotitrating devices might have a role in beginning treatment in patients with OSA by means of a portable sleep study, in which CPAP titration is not performed. In addition, autotitrating offers the possibility of changing pressure over time—such as with changes in position during the night or over the longer term in response to weight loss or gain.
Surgery. In patients who are unable to use CPAP, surgery might be indicated to relieve an anatomical obstruction, such as adenotonsillar hypertrophy or other type of mass lesion.
Sleep positioning. A patient who demonstrates OSA exclusively while sleeping supine might benefit from being trained to sleep on either side only or arranging pillows so that he can only sleep on his side.
In conclusion
OSA is common and easily treatable. It coexists with, and exacerbates, medical and psychiatric illness. Treating OSA concomitantly with comorbid medical and psychiatric illness is essential to achieve full symptom remission and prevent associated long-term consequences of both medical and psychiatric illness.
BOTTOM LINE
Obstructive sleep apnea (OSA) and psychiatric illness, especially depression, often co-exist. Screen depressed patients—especially those with risk factors for OSA, such as obesity, smoking, and an upper-airway abnormality—for a sleep disorder. This is especially important if a patient complains of daytime somnolence, fatigue, cognitive problems, poor concentration, or weight gain. For optimal results, treat comorbid psychiatric illness and OSA concurrently; the same is true for other sleep disorders.
Related Resources
- Babson KA, Del Re AC, Bonn-Miller MO, et al. The comorbidity of sleep apnea and mood, anxiety, and substance use disorders among obese military veterans within the Veterans Health Administration. J Clin Sleep Med. 2013; 9(12):1253-1258.
- Karkoulias K, Lykouras D, Sampsonas F, et al. The impact of obstructive sleep apnea syndrome severity on physical performance and mental health. The use of SF-36 questionnaire in sleep apnea. Eur Rev Med Pharmacol Sci. 2013;17(4):531-536.
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Acknowledgment
Dr. Muhammad Awais Aftab, psychiatry resident at Hamad Medical Corporation, Doha, Qatar, and Umair Amin, final year MBBS student at King Edward Medical University, Lahore, Pakistan, assisted with development of the manuscript of this article.
Estimates are that 50 to 70 million Americans suffer from a chronic disorder of sleep and wakefulness, hindering daily functioning and affecting health.1 Psychiatric illness is common among people who have a sleep disorder. The relationship between psychiatric illness and sleep disorders is bidirectional: People with mental illness often have sleep complaints, and a primary sleep disorder often results in neuropsychiatric complications.
What is obstructive sleep apnea?
The most common type of sleep-disordered breathing, obstructive sleep apnea (OSA) is characterized by frequent cessations of breathing during sleep because of an obstruction of the upper airway. The obstruction occurs secondary to inadequate motor tone of the tongue or airway dilator muscles, or both.1 In addition, many people with OSA have central apneic episodes, in which breathing stops temporarily without airway blockage or respiratory effort.2
The prevalence of OSA is growing as obesity in the United States increases. Risk factors for OSA include obesity, a craniofacial abnormality, an upper-airway abnormality, heredity, smoking, and nasal congestion. OSA plays a role in causing and exacerbating medical illness in people with severe and persistent mental illness, contributing to a significantly shortened life span. Attending to the general health of people who suffer from severe mental illness—including effective treatment of illnesses such as OSA—is crucial.3
Clinical features of OSA
OSA is characterized by hypopnea (a decrease in breathing during sleep) or apnea (an actual pause in breathing). Pauses in breathing during sleep of at least 10 seconds, with obstruction of oronasal airflow despite continuous chest and abdominal movements, are referred to as obstructive apneas. These pauses are associated with a decrease in oxygen saturation or arousal from sleep, or both.1
Primary features of OSA include sleep fragmentation accompanied by nocturnal hypoxemia and hypercapnia, with resulting excessive daytime sleepiness, mood problems, and poor neurocognitive performance (Table 1). OSA often causes potentially serious organ system dysfunction, including adverse cardiovascular and metabolic effects. Studies have suggested that executive dysfunction can be a feature of OSA, which is thought to be related to prefrontal lobe dysfunction caused by intermittent hypoxia. All of these conditions can contribute significantly to decreased quality of life.1
The prevalence of OSA in the general population is approximately 20% when the condition is defined as an apnea-hypopnea index >5 events an hour. The index is the number of apnea and hypopnea episodes that occur during 1 hour of sleep.4
OSA and psychiatric illness
Psychiatric disorders often are comorbid with OSA. These include depression, anxiety, bipolar disorder, schizophrenia, posttraumatic stress disorder (PTSD), panic disorder, and substance use disorder.
Depression. Several studies have documented that OSA and depressive disorder often are comorbid. Many symptoms are common to both, including fatigue, daytime sleepiness, poor concentration, irritability, and weight gain (Figure), although some core symptoms of depression (eg, sadness, anhedonia, guilt, and agitation) are clearly distinguishable from symptoms of OSA. The current recommendation is that a mood disorder should be considered secondary to OSA, and treated accordingly.5
Anxiety. OSA also has been linked to anxiety and nocturnal panic attacks. Frequent awakening due to choking from breathing cessation might play a role in the development of anxiety in patients with OSA, although the association is unproven. Studies have shown a correlation between anxiety disorders and excessive daytime sleepiness, one of the core symptoms of OSA.6 OSA is highly prevalent among combat veterans who have PTSD and complain of being overly vigilant at night; experiencing nightmares and frequent awakening; and having non-restorative sleep.7 Anecdotal reports suggest an association between OSA and bipolar disorder: namely, that continuous positive airway pressure (CPAP) treatment (see “How is OSA treated?,” below) might switch depressed patients to mania.8
Schizophrenia. A strong association exists between OSA and schizophrenia. In a study,9 an OSA diagnosis was made 6 times more often in patients with schizophrenia than in patients with other psychiatric illnesses. Obesity, male sex, and chronic antipsychotic administration were risk factors for OSA in patients with
schizophrenia.9 OSA might be underdiagnosed in patients with schizophrenia because excessive daytime sleepiness, the most common daytime symptom of OSA, can be misattributed as a negative symptom of the disease or a side effect of pharmacotherapy.
OSA and medical illness
OSA can be comorbid with several medical conditions (Table 2). Sleep research in the past 15 years has demonstrated that chronic sleep deprivation has multiple untoward health consequences apart from excessive daytime sleepiness.10 Recent research suggests that chronic sleep loss (<7 hours a night), including sleep loss secondary to OSA, has wide-ranging effects on the cardiovascular, endocrine, immune, and nervous systems, including:
• obesity (adults and children)
• diabetes mellitus and impaired glucose tolerance
• cardiovascular disease and hypertension.
Obesity is one of the primary and more modifiable risk factors for OSA (Box). Studies suggest that reducing the severity of obesity would likely benefit people with a sleep disorder, and that treating sleep deprivation and sleep disorders might benefit persons with obesity.12 Chronic sleep loss can have a deleterious influence on appetite regulation through effects on 2 hormones, leptin and ghrelin, that play a major role in appetite regulation. Chronic sleep loss causes and perpetuates obesity through its interplay with these, and other, hormones.12
Diabetes. The link between obesity and diabetes is well-established, as is the long-term morbidity and mortality of these 2 diseases.13 Evidence shows that OSA is associated with impaired glucose tolerance and an increased risk of diabetes.14
Cardiovascular disease. OSA has a strong association with cardiovascular disease, including systemic hypertension, possibly myocardial infarction, congestive heart failure, and stroke.15 Institution of appropriate treatment for OSA including CPAP can minimize or reverse many of these effects.16
Making an OSA diagnosis
A diagnostic polysomnogram (PSG), or sleep study, is the standard test when OSA is suspected. It is performed most often at an attended sleep laboratory. Typically, a PSG measures several physiologic measures, including, but not limited to:
• airflow through mouth and nose
• stages of sleep (by means of electroencephalography channels)
• thoracic and abdominal movements (to assess effort of breathing)
• muscle activity of the chin
• oxyhemoglobin saturation (to monitor variability in oxygen saturation [SaO2] during OSA events).
Portable diagnostic instruments can provide reliable information when a patient cannot be studied in a laboratory. Assessments available on portable instruments include cardiopulmonary monitoring of respiration only; PSG; and peripheral arterial tonometry, which measures autonomic manifestations of respiratory obstructive events.17,18
The severity of OSA is established by the apnea/hypopnea index, which measures the number of apneas and hypopneas per hour of sleep.
How is OSA treated?
CPAP is still the gold standard for treating OSA. CPAP provides a pneumatic splint for the upper airway by administering positive pressure through a nasal or oronasal mask. CPAP distinctly improves daytime sleepiness.19,20
Pressure is determined initially by titration during PSG, although a number of automated CPAP machines are available in which pressure is adjusted based on the machine’s response to airflow obstruction. Advantages of using PSG to titrate CPAP are direct observation to control mask leak and the ability to observe the effects of body position and sleep stage and clearly distinguish periods of sleep from wakefulness.
Regrettably, adherence to a nightly regimen of CPAP is less than ideal for several reasons, including claustrophobia, interface failure, and other motivational variables. Some patients who experience claustrophobia can use desensitization techniques; others are, ultimately, unable to use the mask.
Oral appliances. A patient who has mild or moderate OSA but who cannot use the CPAP mask might be a good candidate for an oral appliance. These appliances, which hold the mandible in an advanced position during the night, can be effective in such cases.
CPAP autotitration changes the treatment pressure based on feedback from such patient measures as airflow and airway resistance. Autotitrating devices might have a role in beginning treatment in patients with OSA by means of a portable sleep study, in which CPAP titration is not performed. In addition, autotitrating offers the possibility of changing pressure over time—such as with changes in position during the night or over the longer term in response to weight loss or gain.
Surgery. In patients who are unable to use CPAP, surgery might be indicated to relieve an anatomical obstruction, such as adenotonsillar hypertrophy or other type of mass lesion.
Sleep positioning. A patient who demonstrates OSA exclusively while sleeping supine might benefit from being trained to sleep on either side only or arranging pillows so that he can only sleep on his side.
In conclusion
OSA is common and easily treatable. It coexists with, and exacerbates, medical and psychiatric illness. Treating OSA concomitantly with comorbid medical and psychiatric illness is essential to achieve full symptom remission and prevent associated long-term consequences of both medical and psychiatric illness.
BOTTOM LINE
Obstructive sleep apnea (OSA) and psychiatric illness, especially depression, often co-exist. Screen depressed patients—especially those with risk factors for OSA, such as obesity, smoking, and an upper-airway abnormality—for a sleep disorder. This is especially important if a patient complains of daytime somnolence, fatigue, cognitive problems, poor concentration, or weight gain. For optimal results, treat comorbid psychiatric illness and OSA concurrently; the same is true for other sleep disorders.
Related Resources
- Babson KA, Del Re AC, Bonn-Miller MO, et al. The comorbidity of sleep apnea and mood, anxiety, and substance use disorders among obese military veterans within the Veterans Health Administration. J Clin Sleep Med. 2013; 9(12):1253-1258.
- Karkoulias K, Lykouras D, Sampsonas F, et al. The impact of obstructive sleep apnea syndrome severity on physical performance and mental health. The use of SF-36 questionnaire in sleep apnea. Eur Rev Med Pharmacol Sci. 2013;17(4):531-536.
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Acknowledgment
Dr. Muhammad Awais Aftab, psychiatry resident at Hamad Medical Corporation, Doha, Qatar, and Umair Amin, final year MBBS student at King Edward Medical University, Lahore, Pakistan, assisted with development of the manuscript of this article.
1. Institute of Medicine. Sleep disorders and sleep deprivation: an unmet public health problem. Washington, DC: The National Academies Press; 2006:20.
2. Badr MS. Central sleep apnea. Prim Care. 2005;32(2):361-374.
3. Freedland KE, Carney RM, Hayano J, et al. Effect of obstructive sleep apnea on response to cognitive behavior therapy for depression after an acute myocardial infarction. J Psychosom Res. 2012;72(4):276-281.
4. Punjabi NM. The epidemiology of adult obstructive sleep apnea. Proc Am Thorac Soc. 2008;5(2):136-143.
5. El-Sherbini AM, Bediwy AS, El-Mitwalli A. Association between obstructive sleep apnea (OSA) and depression and the effect of continuous positive airway pressure (CPAP) treatment. Neuropsychiatr Dis Treat. 2011;7:715-721.
6. Hasler G, Buysse DJ, Gamma A, et al. Excessive daytime sleepiness in young adults: a 20-year prospective community study. J Clin Psychiatry. 2005;66(4):521-529.
7. Yesavage JA, Kinoshita LM, Kimball T, et al. Sleep-disordered breathing in Vietnam veterans with posttraumatic stress disorder. Am J Geriatr Psychiatry. 2012;20(3):199-204.
8. Plante D, Winkelman J. Sleep disturbance in bipolar disorder: therapeutic implications. Am J Psychiatry. 2008; 165(7):830-843.
9. Winkelman J. Schizophrenia, obesity, and obstructive sleep apnea. J Clin Psychiatry. 2001;62(1):8-11.
10. Partinen M, Hublin C. Epidemiology of sleep disorders. Philadelphia, PA: Elsevier Saunders; 2005.
11. Valderas JM, Starfield B, Sibbald B, et al. Defining comorbidity: implications for understanding health and health services. Ann Fam Med. 2009;7(4):357-363.
12. Romero-Corral A, Caples SM, Lopez-Jimenez F, et al. Interactions between obesity and obstructive sleep apnea: implications for treatment. Chest. 2010;137(3):711-719.
13. Villareal DT, Apovian CM, Kushner RF, et al. Obesity in older adults: technical review and position statement of the American Society for Nutrition and NAASO, The Obesity Society. Obes Res. 2005;13(11):1849-1863.
14. Pamidi S, Aronsohn RS, Tasali E. Obstructive sleep apnea: role in the risk and severity of diabetes. Best Pract Res Clin Endocrinol Metab. 2010;24(5):703-715.
15. Malhotra A, Loscalzo J. Sleep and cardiovascular disease: an overview. Prog Cardiovasc Dis. 2009;51(4):279-284.
16. Bradley TD, Floras JS. Obstructive sleep apnoea and its cardiovascular consequences. Lancet. 2009;373(9657):82-93.
17. Chesson A, Berry R, Pack A. Practice parameters for the use of portable monitoring devices in the investigation of suspected obstructive sleep apnea in adults. Sleep. 2003; 26(7):907-913.
18. Pittman S, Ayas N, MacDonald M, et al. Using a wrist-worn device based on peripheral arterial tonometry to diagnose obstructive sleep apnea: in-laboratory and ambulatory validation. Sleep. 2004;27(1):923-933.
19. Ballester E, Badia J, Hernandez L, et al. Evidence of the effectiveness of continuous positive airway pressure in the treatment of sleep apnea/hypopnea syndrome. Am J Respir Crit Care Med. 1999;159:495-501.
20. Jenkinson D, Davies J, Mullins R, et al. Comparison of therapeutic and subtherapeutic nasal continuous positive airway pressure for obstructive sleep apnoea: a randomised prospective parallel trial. Lancet. 1999;353:2100-2105.
1. Institute of Medicine. Sleep disorders and sleep deprivation: an unmet public health problem. Washington, DC: The National Academies Press; 2006:20.
2. Badr MS. Central sleep apnea. Prim Care. 2005;32(2):361-374.
3. Freedland KE, Carney RM, Hayano J, et al. Effect of obstructive sleep apnea on response to cognitive behavior therapy for depression after an acute myocardial infarction. J Psychosom Res. 2012;72(4):276-281.
4. Punjabi NM. The epidemiology of adult obstructive sleep apnea. Proc Am Thorac Soc. 2008;5(2):136-143.
5. El-Sherbini AM, Bediwy AS, El-Mitwalli A. Association between obstructive sleep apnea (OSA) and depression and the effect of continuous positive airway pressure (CPAP) treatment. Neuropsychiatr Dis Treat. 2011;7:715-721.
6. Hasler G, Buysse DJ, Gamma A, et al. Excessive daytime sleepiness in young adults: a 20-year prospective community study. J Clin Psychiatry. 2005;66(4):521-529.
7. Yesavage JA, Kinoshita LM, Kimball T, et al. Sleep-disordered breathing in Vietnam veterans with posttraumatic stress disorder. Am J Geriatr Psychiatry. 2012;20(3):199-204.
8. Plante D, Winkelman J. Sleep disturbance in bipolar disorder: therapeutic implications. Am J Psychiatry. 2008; 165(7):830-843.
9. Winkelman J. Schizophrenia, obesity, and obstructive sleep apnea. J Clin Psychiatry. 2001;62(1):8-11.
10. Partinen M, Hublin C. Epidemiology of sleep disorders. Philadelphia, PA: Elsevier Saunders; 2005.
11. Valderas JM, Starfield B, Sibbald B, et al. Defining comorbidity: implications for understanding health and health services. Ann Fam Med. 2009;7(4):357-363.
12. Romero-Corral A, Caples SM, Lopez-Jimenez F, et al. Interactions between obesity and obstructive sleep apnea: implications for treatment. Chest. 2010;137(3):711-719.
13. Villareal DT, Apovian CM, Kushner RF, et al. Obesity in older adults: technical review and position statement of the American Society for Nutrition and NAASO, The Obesity Society. Obes Res. 2005;13(11):1849-1863.
14. Pamidi S, Aronsohn RS, Tasali E. Obstructive sleep apnea: role in the risk and severity of diabetes. Best Pract Res Clin Endocrinol Metab. 2010;24(5):703-715.
15. Malhotra A, Loscalzo J. Sleep and cardiovascular disease: an overview. Prog Cardiovasc Dis. 2009;51(4):279-284.
16. Bradley TD, Floras JS. Obstructive sleep apnoea and its cardiovascular consequences. Lancet. 2009;373(9657):82-93.
17. Chesson A, Berry R, Pack A. Practice parameters for the use of portable monitoring devices in the investigation of suspected obstructive sleep apnea in adults. Sleep. 2003; 26(7):907-913.
18. Pittman S, Ayas N, MacDonald M, et al. Using a wrist-worn device based on peripheral arterial tonometry to diagnose obstructive sleep apnea: in-laboratory and ambulatory validation. Sleep. 2004;27(1):923-933.
19. Ballester E, Badia J, Hernandez L, et al. Evidence of the effectiveness of continuous positive airway pressure in the treatment of sleep apnea/hypopnea syndrome. Am J Respir Crit Care Med. 1999;159:495-501.
20. Jenkinson D, Davies J, Mullins R, et al. Comparison of therapeutic and subtherapeutic nasal continuous positive airway pressure for obstructive sleep apnoea: a randomised prospective parallel trial. Lancet. 1999;353:2100-2105.

Dissecting melancholia with evidence-based biomarker tools
For more than 50 years, depression has been studied, and understood, as a deficiency of specific neurotransmitters in the brain—namely dopamine, norepinephrine, and serotonin. Treatments for depression have been engineered to increase the release, or block the degradation, of these neurotransmitters within the synaptic cleft. Although a large body of evidence supports involvement of dopamine, norepinephrine, and serotonin in the pathophysiology of depression, the observation that pharmacotherapy is able to induce remission only in <50% of patients1 has prompted researchers to look beyond neurotransmitters for an understanding of depressive disorders (Table 1).

Today, theories of depression focus more on differences in neuron density in various regions of the brain; the effect of stress on neurogenesis and neuronal cell apoptosis; alterations in feedback pathways connecting the pre-frontal cortex to the limbic system; and the role of proinflammatory mediators evoked during the stress response (Box,2,3). These theories should not be viewed as separate entities because they are highly interconnected. Integrating them provides for a more expansive understanding of the pathophysiology of depression and biomarkers that are involved (Table 2).


In this article, we:
- integrate the large body of evidence supporting the contribution of the above variables to the onset and persistence of depression
- propose a possible risk stratification model
- explore possibilities for treatment.
The stress response: How does it affect the brain?
Stress initiates a cascade of events in the brain and peripheral systems that enable an organism to cope with, and adapt to, new and challenging situations. That is why physiologic and behavioral responses to stress generally are considered beneficial to survival.
When stress is maintained for a long period, both brain and body are harmed because target cells undergo prolonged exposure to physiologic stress mediators. For example, Woolley and Gould4 exposed rats to varying durations of glucocorticoids and observed that treating animals with corticosterone injection for 21 days induced neuronal atrophy in the hippocampus and prefrontal cortex and increased release of proinflammatory cytokines from astrocytes within the limbic system. Stressful experiences are believed to be closely associated with development of psychological alterations and, thus, neuropsychiatric disorders.5 To go further: Chronic stress is believed to be the leading cause of depression.
When the brain perceives an external threat, the stress response is called into action. The amygdala, part of the primitive limbic system, is the primary area of the brain responsible for triggering the stress response,6 signaling the hypothalamus to release corticotropin-releasing hormone (CRH) to the anterior pituitary gland, which, in turn releases adrenocorticotropic hormone to the adrenal glands (Figure 1).7 The adrenal glands are responsible for releasing glucocorticoids, which, because of their lipophilic nature, can cross the blood-brain barrier and are found in higher levels in the cerebrospinal fluid (CSF) of depressed persons.7
Once in the brain, glucocorticoids can be irreversibly degraded in the cytosol by the enzyme 11-β hydroxysteroid dehydrogenase type 2, a potential target for treating depression, or can bind to the glucocorticoid receptor (GR). Results of a research study of the role of cortisol in suppression of proinflammatory cytokine signaling activity in rainbow trout hepatocytes suggest a negative feedback loop for GR gene regulation during stress.8
Because this auto-regulation is a crucial step in the physiological stress response, the idea of the GR as an important biomarker in depression has gained popularity. In humans, when the GR binds to glucocorticoids that are released from the adrenal cortex during the stress response, the activated GR-cortisol complex represses expression of proinflammatory proteins in astrocytes and microglial cells and in all cells in the periphery before they are transcribed into proteins.9 The GR also has been shown to modulate neurogenesis.8 Repeated stress that persists over a long period leads to GR resistance, thereby reducing inhibition of production of proinflammatory cytokines.
Exposure to stress for >21 days leads to overactivity of the HPA axis and GR resistance,10 which decreases suppression of proinflammatory cytokines. There is evidence that proinflammatory cytokines, tumor necrosis factor-α, and interleukin-6 further induce GR receptor resistance by preventing the cortisol-GR receptor complex from entering cell nuclei and decreasing binding to DNA within the nuclei.11 Dexamethasone, a GR agonist, has been implicated in research studies for potential re-regulation of the HPA axis in depressed persons.12
Nerve cell death in the hippocampus
Studies showing reduced hippocampal volume in unipolar depression and a correlation between the number of episodes and a consequence of untreated depression and studies suggesting that treatment can stop or reduce shrinkage,13 and recent findings of rapid neurogenesis in hippocampi in response to ketamine, brings our focus to hippocampus in depression.
The greatest density of GRs is found in the hippocampus, which is closely associated with the limbic system.7 Therefore, the hippocampus is sensitive to increases in glucocorticoids in the brain and plays a crucial role in regulation of the HPA axis.
Evidence shows that in chronic stress exposure (≥21 days), nerve cells in the hippocampus begin to atrophy and can no longer provide negative feedback inhibition to the hypothalamus, causing HPA axis dysregulation and uncontrolled release of glucocorticoids into the bloodstream and CSF.2 In patients with Cushing syndrome, who produce abnormally high levels of glucocorticoid, the incidence of depression is as high as 50%.14 Similarly, patients treated with glucocorticoids such as prednisone often experience psychiatric symptoms, the most common being depression. Gould found that partial adrenalectomy increased hippocampal neurogenesis in rat brains, indicating the beneficial effect of stress hormone antagonism.4 CRH antagonists are being looked at as a promising and less invasive treatment option for depression.
Focus has been diverted to the role of the hippocampus in depression because of its ability to regenerate throughout adulthood, leading potentially to a re-regulation of the HPA axis and subsiding of the stress response, which is universally believed to be the primary precipitating factor in depression onset. Rats require 10 to 21 days of rest to recover from the effects of chronic (21 days) administration of glucocorticoids.15 If this proves to be a directly proportional relationship, then rats would need an estimated 120 days to recover from 6 months of constant glucocorticoid exposure. Considering that the same is true for humans, current depression treatment programs, which average 6 weeks, are not long enough for adequate recovery.
Antidepressants such as selective serotonin reuptake inhibitors, serotonin-norepinephrine reuptake inhibitors, and tricyclics stimulate neurogenesis in the hippocampus via increases in brain-derived neurotrophic factor (BDNF), suggesting that these neurotransmitters play an important role depression.16
Repetitive transcranial magnetic stimulation (rTMS), a noninvasive neuromodulation therapy approved to treat major depression, delivers brief magnetic pulses to the limbic structures. Treatment facilitates focal stimulation, rapidly applying electrical charges to the cortical neurons. TMS targets prefrontal circuits of the brain that are underactive during depressive episodes. Recent animal studies have suggested that bromodeoxyuridine (BrdU)-positive cells (newborn cells) are increased significantly in the dentate gyrus, in turn suggesting that hippocampal neurogenesis might be involved in the antidepressant effects of chronic rTMS.17 Although the underlying therapeutic mechanisms of rTMS treatment of depression remain unclear, it appears that hippocampal neurogenesis might be required to produce the effects of antidepressant treatments, including drugs and electroconvulsive therapy.17
Selective ‘shunting’ of energy occurs during the stress response
Hormones released from the adrenal glands during stress divert glucose to exercising muscles and the brain’s limbic system, which are involved in the fight-or-flight response.18 However, metabolic functions and areas of the brain that are not involved in the stress response, such as the cerebral cortex and hippocampus, are deprived of energy as a consequence of this innate selective shunting (Figure 2).19
Positron-emission tomography (PET) scanning of the resting brain shows that components of the cerebral cortex (prefrontal cortex, hippocampus, striatum) and areas connecting the cerebral cortex to the limbic system exhibit the most energy consumption in the brain during rest (Figure 3).20 PET studies also show that neuronal connections within these energy-demanding areas atrophy more rapidly than in any other area of the brain when their energy supply is reduced or cut off.6
When the supply of oxygen and glucose to certain areas of the brain is reduced—such as in traumatic brain injury or stroke—the excitatory neurotransmitter glutamate accumulates in extracellular fluid and causes nerve-cell death.21 When a conditioned stimulus is presented during fear acquisition, functional magnetic resonance imaging (fMRI) studies of fear-conditioning have consistently reported, in the prefrontal cortex:
- a decrease in the blood oxygen level-dependent signal, below resting baseline
- a reduction in blood flow (Figure 4).22
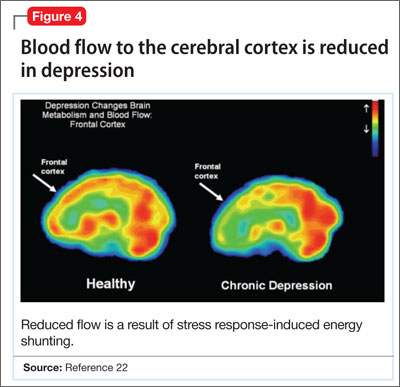
This discovery adds to evidence that demonstrates a decrease in gray-matter density in the frontal lobes as a result of glutaminergic toxicity (Figure 5).

Activation of L-glutamate, believed to play a significant role in depression and other neuropsychiatric disorders, triggers calcium-dependent intracellular responses that “excite cells to death,” so to speak—thereby causing nerve-cell apoptosis and a reduction in synaptic connections between different areas of the brain responsible for learning and memory.23 Malfunction of these synaptic connections is thought to be partially responsible for depression and other psychiatric disorders.
Excessive activation of N-methyl-d-asparate (NMDA) receptors is thought to be the underlying mechanism that leads to neuronal cell death in glutaminergic toxicity. Therefore, NMDA receptor proteins have become a target in treating neurodegenerative psychiatric illnesses. There is more than one type of NMDA receptor; some of them are excitatory, others are inhibitory. Four compounds have presented as therapeutic candidates for inhibition of NMDA receptor functioning and treatment of depression: those that inhibit glutamate binding, those that block the ion channel, and those that inhibit receptor binding to the terminal regulatory domain.24
Regrettably, these chemical compounds are not receptor-selective, but small structural modifications of these NMDA receptors have been found and lead to significant changes in potency and selectivity. This should serve as a unique starting point for developing highly specific NMDA receptor modulator agents for a variety of neuropsychiatric and neurological conditions. GLYX-13, a derivative of ketamine (an NMDA receptor antagonist), has been implicated for use in treating depression. It has been tested on 2 large phase-II study groups.25
Neuronal circuitry of depression is altered by prolonged stress
Symptoms of depression can be explained by the anatomical circuit shown in Figure 6.15,20 Impaired concentration, diminished ability to process new information, and decline in memory function are associated with decreased nerve density in the hippocampus, which plays a key role in learning, memory, and encoding of emotionally relevant data into memory.26 The hippocampus interacts with the amygdala to provide input about the context in which stimuli occur.
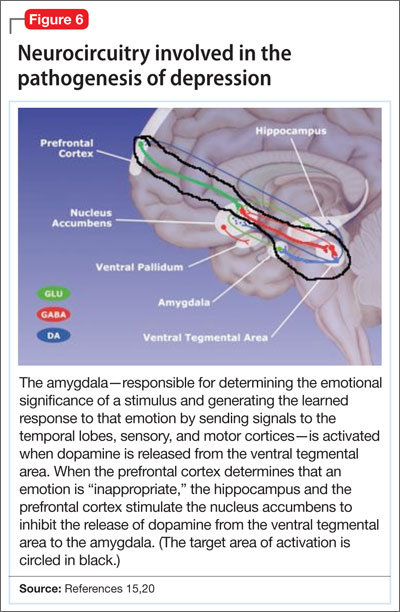
Depressed people often demonstrate impulsivity and have difficulty controlling expression of emotions—traits that are attributed to increased neuronal density in the amygdala and insula, which has been illustrated in PET scans and voxel-based morphometry in depressed patients.27 These brain areas are implicated in subjective emotional experience, processing of emotional reactions, and impulsive decision-making. The amygdala is normally highly regulated by the prefrontal cortex, which uses rational judgment to interpret stimuli and regulate the expression of emotion.
A study involving a facial expression processing task demonstrated reduced connectivity between the amygdala and prefrontal cortex and increased functional connectivity among the amygdala, hippocampus, and caudate-putamen in depressed patients.24 And in a study that measured white matter conduction in various brain areas in depressed patients, the greatest reduction was found in areas connecting the limbic system to the prefrontal cortex and hippocampus—believed to be caused by stress response-induced ischemic glutaminergic neuroapoptosis.21 Such neuroapoptosis might lead to irrational interpretation of stimuli, unchecked expression of emotion, and impulsive thoughts and behavior that are often present in depression and other mood disorders.
Deep brain stimulation (DBS), in which electrodes are implanted in the brain, has proved effective at increasing synaptic connections between the prefrontal cortex and the limbic system when electrodes are placed appropriately.28 Patients with refractory depression who are treated with DBS show increased gray-matter density and functional activity in the prefrontal cortex, hippocampus, and fronto-limbic connections.29 DBS also increases neurotransmission of dopamine, serotonin, and norepinephrine within the fronto-limbic circuitry.30
Identifying risk factors for depression
Genetic risk factors. Forty percent of patients with depression have a first-degree relative with depression, suggesting a strong genetic component.10 Inherited differences in hippocampal volume, synaptic connections between the prefrontal cortex and amygdala, γ-aminobutyric acid (GABA)/glutamate balance, BDNF neurotransmitter receptors, and anatomic positioning of the limbic system in relation to other brain structures might account for the heritability of psychiatric disorders such as depression.
Evidence has been consistent that hippocampal volume is diminished in the brain of depressed persons. However, there is no prospective cohort study to determine whether people who have lower gray-matter hippocampal density or volume, or both, before depression onset develop symptoms later in life. There also is no study to determine the percentage of people who have lower-than-average hippocampal gray-matter density or volume and who have a first-degree relative with depression. Such studies would yield valuable information about anatomic variables that increase the risk of depression.
It has been proposed that low GABA function is an inherited biomarker for depression. Bjork and co-workers found a lower plasma level of GABA in depressed subjects and in their first-degree relatives, confirming that GABAergic tone might be under genetic control.11 Genetic loci studies in mice have linked depressive-like behavior to GABAergic loci on chromosomes 8 and 11, encoding alpha 1, alpha 6, and gamma subunits of GABAA receptors.23
A recent study in humans showed that severe, treatment-resistant depression with anxiety was linked to a mutation in the B1 subunit of the GABAA receptor. Positive genetic associations were found between polymorphism in human GABAA receptor subunit genes.11
GABA metabolizing enzymes also can be considered biological modifiers of depression. For example:
- GABA uptake and metabolism is controlled by the enzyme glutamic acid decarboxylase (GAD); depression has been found to be associated with a polymorphism in the GAD67 gene encoding an isoform of GAD.11
- GABA transaminase (GABA-T) is another key enzyme in GABA turnover.31 It catabolizes GABA.
We can conclude that, to a high degree, depression depends on GABA production and metabolism.
A variant in the human BDNF gene, in which valine is substituted for methionine in position 66 of the pro-domain of the BDNF protein, is associated with
- a decrease in the production of BDNF
- increased susceptibility to neuropsychiatric disorders, including depression, anxiety disorder, and bipolar disorder (Figure 7).32
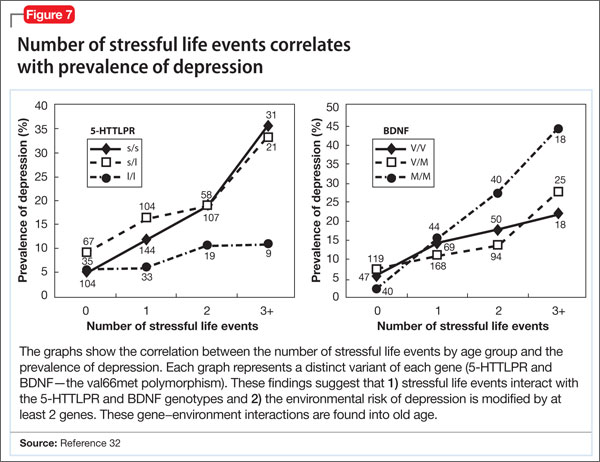
People with the MM allele have been found to have a small hippocampal neuronal density and poor hippocampus-dependent memory function in neuroimaging studies.23 They also displayed diminished ventromedial prefrontal cortex volume and presented with aversive memory extinction deficit (ie, “holding grudges”).
Another neurotrophic factor, vascular endothelial growth factor (VEGF), is a survival factor for endothelial cells and neurons and a modulator of synaptic transmission. Understanding the molecular and cellular specificity of antidepressant-induced VEGF will be critical to determine its potential as a therapeutic target in depression.33 Delineating the relationship between VEGF and depression has, ultimately, the potential to shed light on the still elusive neural mechanisms that underlie the pathophysiology of depression and the mechanisms by which antidepressants exert their effects.34
Genetic polymorphisms in monoamine receptors (5-HT2A), transporters (SERTPR, 5-HTTLPR, STin2, rs25531, SLC6A4), and regulatory enzymes should not be overlooked.35 There is reproducible evidence that variability in these polymorphisms are associated with variability in:
- vulnerability to depression
- the response to treatment with existing antidepressant medications.1
Most studies that look at changes in neuronal circuitry focus on the integrity of synaptic connections between the frontal cortex and limbic system; few of them have closely examined the importance of the anatomic proximity of the 2 regions. It might be that having an amygdala that is relatively closer to the frontal cortex and the hippocampus reduces a person’s risk of depression, and vice versa. This association needs to be investigated further with imaging studies.
Environmental risk factors. The brain is thought to be plastic until age 30.5 Plasticity diminishes with age after age 7—except for the hippocampus, which can regenerate throughout life.36 Early life experiences play an important role in forming synaptic connections between the frontal cortex and the limbic system, through a process known as fear conditioning.
Children learn early in life which stimuli are to be perceived as threatening or aversive and how to respond to best preserves their safety and internal sense of well-being. Those who grow up in a hostile environment learn to perceive more stimuli as threatening than children who grow up in a nurturing environment.32 It is possible that the amygdala is larger in children who grow up in less-than-ideal circumstances because this region is constantly being recruited—at the expense of the more rational frontal cortex.
Evidence suggests that these conditions reduce hippocampal neurogenesis37:
- increasing age
- substance abuse (opiates and methamphetamines)
- inadequate housing
- minimal physical activity
- little opportunity for social stimulation
- minimal learning experience.
Bottom Line
Depression has been understood as a neurotransmitter deficiency in the brain; treatments were engineered to increase release, or block degradation, of those neurotransmitters. Novel theories—all interconnected—of the neuroanatomical pathophysiology of depression focus more on differences in neuron density in the brain; effects of stress on neurogenesis and neuronal cell apoptosis; alterations in feedback pathways connecting the pre-frontal cortex to the limbic system; and the role of pro-inflammatory mediators evoked during the stress response.
Related Resources
- Fuchs E. Neurogenesis in the adult brain: is there an association with mental disorders? Eur Arch Psychiatry Clin Neurosci. 2007;257(5):247-249.
- Videbech P, Ravnkilde B. Hippocampal volume and depression: a meta-analysis of MRI studies. Am J Psychiatry. 2004; 161(11):1957-1966.
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Acknowledgement
Anita Rao, second-year medical student, Stritch School of Medicine, Loyola University, Chicago, Illinois, assisted in the preparation of this manuscript.
1. Eley TC, Sugden K, Corsico A, et al. Gene-environment interaction analysis of serotonin system markers with adolescent depression. Mol Psychiatry. 2004;9(10):908-915.
2. Haber SN, Rauch SL. Neurocircuitry: a window into the networks underlying neuropsychiatric disease. Neuropsychopharmacology. 2010;35(1):1-3.
3. Frodl T, Bokde AL, Scheuerecker J, et al. Functional connectivity bias of the orbitofrontal cortex in drug-free patients with major depression. Biol Psychiatry. 2010; 67(2):161-167.
4. Woolley CS, Gould E, McEwen BS. Exposure to excess glucocorticoids alters dendritic morphology of adult hippocampal pyramidal neurons. Brain Res. 1990;531(1-2): 225-231.
5. Heim C, Nemeroff CB. The impact of early adverse experiences on brain systems involved in the pathophysiology of anxiety and affective disorders. Biol Psychiatry. 1999;46(11):1509-1522.
6. Isgor C, Kabbaj M, Akil H, et al. Delayed effects of chronic variable stress during peripubertal-juvenile period on hippocampal morphology and on cognitive and stress axis functions in rats. Hippocampus. 2004;14(5):636-648.
7. De Kloet ER, Vreugdenhil E, Oitzl MS, et al. Brain corticosteroid receptor balance in health and disease. Endocr Rev. 1998;19(3):269-301.
8. Philip AM, Kim SD, Vijayan MM. Cortisol modulates the expression of cytokines and suppressors of cytokine signaling (SOCS) in rainbow trout hepatocytes. Dev Comp Immunol. 2012;38(2):360-367.
9. Coplan JD, Lydiard RB. Brain circuits in panic disorder. Biol Psychiatry. 1998;44(12):1264-1276.
10. Anisman H, Merali Z. Cytokines, stress and depressive illness: brain-immune interactions. Ann Med. 2003;35(1):2-11.
11. Crowley JJ, Lucki I. Opportunities to discover genes regulating depression and antidepressant response from rodent behavioral genetics. Curr Pharm Des. 2005;11(2):157-169.
12. Covington HE 3rd, Vialou V, Nestler EJ. From synapse to nucleus: novel targets for treating depression. Neuropharmacology. 2010;58(4-5):683-693.
13. Videbech P, Ravnkilde B. Hippocampal volume and depression: a meta-analysis of MRI studies. Am J Psychiatry. 2004;161(11):1957-1966.
14. Sandi C. Stress, cognitive impairment and cell adhesion molecules. Nat Rev Neurosci. 2004;5(12):917-930.
15. Hartley CA, Phelps EA. Changing fear: the neurocircuitry of emotion regulation. Neuropsychopharmacology. 2010;35(1): 136-146.
16. Kim DK, Lim SW, Lee S, et al. Serotonin transporter gene polymorphism and antidepressant response. Neuroreport. 2000;11(1):215-219.
17. Ueyama E, Ukai S, Ogawa A, et al, Chronic repetitive transcranial magnetic stimulation increases hippocampal neurogenesis in rats. Psychiatry Clin Neurosci. 2011; 65(1):77-81.
18. Irwin W, Anderle MJ, Abercrombie HC, et al. Amygdalar interhemispheric functional connectivity differs between the non-depressed and depressed human brain. Neuroimage. 2004;21(2):674-686.
19. McEwen BS. Physiology and neurobiology of stress and adaptation: central role of the brain. Physiol Rev. 2007; 87(3):873-904.
20. Gusnard DA, Raichle ME, Raichle ME. Searching for a baseline: functional imaging and the resting human brain. Nat Rev Neurosci. 2001;2(10):685-694.
21. Hulsebosch CE, Hains BC, Crown ED, et al. Mechanisms of chronic central neuropathic pain after spinal cord injury. Brain Res Rev. 2009;60(1):202-213.
22. Gottfried JA, Dolan RJ. Human orbitofrontal cortex mediates extinction learning while accessing conditioned representations of value. Nat Neurosci. 2004;7(10):1144-1152.
23 Arnone D, McKie S, Elliott R, et al. State-dependent changes in hippocampal grey matter in depression. Mol Psychiatry. 2012;1(8):1359-4184.
24. Brunoni AR, Lopes M, Fregni F. A systematic review and meta-analysis of clinical studies on major depression and BDNF levels: implications for the role of neuroplasticity in depression. Int J Neuropsychopharmacol. 2008;11(8):1169-1180.
25. Maeng S, Zarate CA Jr. The role of glutamate in mood disorders: results from the ketamine in major depression study and the presumed cellular mechanism underlying its antidepressant effects. Curr Psychiatry Rep. 2007;9(6):467-474.
26. Vaidya VA, Fernandes K, Jha S. Regulation of adult hippocampal neurogenesis: relevance to depression. Expert Rev Neurother. 2007;7(7):853-864.
27. Lisiecka DM, Carballedo A, Fagan AJ, et al. Altered inhibition of negative emotions in subjects at family risk of major depressive disorder. J Psychiatr Res. 2012;46(2):181-188.
28. Mayberg HS, Lozano AM, Voon V, et al. Deep brain stimulation for treatment-resistant depression. Neuron. 2005;45(5):651-660.
29. Levkovitz Y, Harel EV, Roth Y, et al. Deep transcranial magnetic stimulation over the prefrontal cortex: evaluation of antidepressant and cognitive effects in depressive patients. Brain Stimul. 2009;2(4):188-200.
30. Schlaepfer TE, Lieb K. Deep brain stimulation for treatment of refractory depression. Lancet. 2005;366(9495):1420-1422.
31. Astrup, J. Energy-requiring cell functions in the ischemic brain. Their critical supply and possible inhibition in protective therapy. J Neurosurg. 1982;56(4):482-497.
32. Fletcher JM. Childhood mistreatment and adolescent and young adult depression. Soc Sci Med. 2009;68(5):799-806.
33. Warner-Schmidt JL, Duman R. VEGF as a potential target for therapeutic intervention in depression. Curr Opin Pharmacol. 2008;8(1):14-19.
34. Clark-Raymond A, Halaris A. VEGF and depression: a comprehensive assessment of clinical data. J Psychiatr Res. 2013;47(8):1080-1087.
35. Alonso R, Griebel G, Pavone G, et al. Blockade of CRF(1) or V(1b) receptors reverses stress-induced suppression of neurogenesis in a mouse model of depression. Mol Psychiatry. 2004;9(3):278-286.
36. Thomas RM, Peterson DA. A neurogenic theory of depression gains momentum. Mol Interv. 2003;3(8):441-444.
37. Jacobs BL. Adult brain neurogenesis and depression. Brain Behav Immun. 2002;16(5):602-609.
For more than 50 years, depression has been studied, and understood, as a deficiency of specific neurotransmitters in the brain—namely dopamine, norepinephrine, and serotonin. Treatments for depression have been engineered to increase the release, or block the degradation, of these neurotransmitters within the synaptic cleft. Although a large body of evidence supports involvement of dopamine, norepinephrine, and serotonin in the pathophysiology of depression, the observation that pharmacotherapy is able to induce remission only in <50% of patients1 has prompted researchers to look beyond neurotransmitters for an understanding of depressive disorders (Table 1).
Today, theories of depression focus more on differences in neuron density in various regions of the brain; the effect of stress on neurogenesis and neuronal cell apoptosis; alterations in feedback pathways connecting the pre-frontal cortex to the limbic system; and the role of proinflammatory mediators evoked during the stress response (Box,2,3). These theories should not be viewed as separate entities because they are highly interconnected. Integrating them provides for a more expansive understanding of the pathophysiology of depression and biomarkers that are involved (Table 2).
In this article, we:
- integrate the large body of evidence supporting the contribution of the above variables to the onset and persistence of depression
- propose a possible risk stratification model
- explore possibilities for treatment.
The stress response: How does it affect the brain?
Stress initiates a cascade of events in the brain and peripheral systems that enable an organism to cope with, and adapt to, new and challenging situations. That is why physiologic and behavioral responses to stress generally are considered beneficial to survival.
When stress is maintained for a long period, both brain and body are harmed because target cells undergo prolonged exposure to physiologic stress mediators. For example, Woolley and Gould4 exposed rats to varying durations of glucocorticoids and observed that treating animals with corticosterone injection for 21 days induced neuronal atrophy in the hippocampus and prefrontal cortex and increased release of proinflammatory cytokines from astrocytes within the limbic system. Stressful experiences are believed to be closely associated with development of psychological alterations and, thus, neuropsychiatric disorders.5 To go further: Chronic stress is believed to be the leading cause of depression.
When the brain perceives an external threat, the stress response is called into action. The amygdala, part of the primitive limbic system, is the primary area of the brain responsible for triggering the stress response,6 signaling the hypothalamus to release corticotropin-releasing hormone (CRH) to the anterior pituitary gland, which, in turn releases adrenocorticotropic hormone to the adrenal glands (Figure 1).7 The adrenal glands are responsible for releasing glucocorticoids, which, because of their lipophilic nature, can cross the blood-brain barrier and are found in higher levels in the cerebrospinal fluid (CSF) of depressed persons.7
Once in the brain, glucocorticoids can be irreversibly degraded in the cytosol by the enzyme 11-β hydroxysteroid dehydrogenase type 2, a potential target for treating depression, or can bind to the glucocorticoid receptor (GR). Results of a research study of the role of cortisol in suppression of proinflammatory cytokine signaling activity in rainbow trout hepatocytes suggest a negative feedback loop for GR gene regulation during stress.8
Because this auto-regulation is a crucial step in the physiological stress response, the idea of the GR as an important biomarker in depression has gained popularity. In humans, when the GR binds to glucocorticoids that are released from the adrenal cortex during the stress response, the activated GR-cortisol complex represses expression of proinflammatory proteins in astrocytes and microglial cells and in all cells in the periphery before they are transcribed into proteins.9 The GR also has been shown to modulate neurogenesis.8 Repeated stress that persists over a long period leads to GR resistance, thereby reducing inhibition of production of proinflammatory cytokines.
Exposure to stress for >21 days leads to overactivity of the HPA axis and GR resistance,10 which decreases suppression of proinflammatory cytokines. There is evidence that proinflammatory cytokines, tumor necrosis factor-α, and interleukin-6 further induce GR receptor resistance by preventing the cortisol-GR receptor complex from entering cell nuclei and decreasing binding to DNA within the nuclei.11 Dexamethasone, a GR agonist, has been implicated in research studies for potential re-regulation of the HPA axis in depressed persons.12
Nerve cell death in the hippocampus
Studies showing reduced hippocampal volume in unipolar depression and a correlation between the number of episodes and a consequence of untreated depression and studies suggesting that treatment can stop or reduce shrinkage,13 and recent findings of rapid neurogenesis in hippocampi in response to ketamine, brings our focus to hippocampus in depression.
The greatest density of GRs is found in the hippocampus, which is closely associated with the limbic system.7 Therefore, the hippocampus is sensitive to increases in glucocorticoids in the brain and plays a crucial role in regulation of the HPA axis.
Evidence shows that in chronic stress exposure (≥21 days), nerve cells in the hippocampus begin to atrophy and can no longer provide negative feedback inhibition to the hypothalamus, causing HPA axis dysregulation and uncontrolled release of glucocorticoids into the bloodstream and CSF.2 In patients with Cushing syndrome, who produce abnormally high levels of glucocorticoid, the incidence of depression is as high as 50%.14 Similarly, patients treated with glucocorticoids such as prednisone often experience psychiatric symptoms, the most common being depression. Gould found that partial adrenalectomy increased hippocampal neurogenesis in rat brains, indicating the beneficial effect of stress hormone antagonism.4 CRH antagonists are being looked at as a promising and less invasive treatment option for depression.
Focus has been diverted to the role of the hippocampus in depression because of its ability to regenerate throughout adulthood, leading potentially to a re-regulation of the HPA axis and subsiding of the stress response, which is universally believed to be the primary precipitating factor in depression onset. Rats require 10 to 21 days of rest to recover from the effects of chronic (21 days) administration of glucocorticoids.15 If this proves to be a directly proportional relationship, then rats would need an estimated 120 days to recover from 6 months of constant glucocorticoid exposure. Considering that the same is true for humans, current depression treatment programs, which average 6 weeks, are not long enough for adequate recovery.
Antidepressants such as selective serotonin reuptake inhibitors, serotonin-norepinephrine reuptake inhibitors, and tricyclics stimulate neurogenesis in the hippocampus via increases in brain-derived neurotrophic factor (BDNF), suggesting that these neurotransmitters play an important role depression.16
Repetitive transcranial magnetic stimulation (rTMS), a noninvasive neuromodulation therapy approved to treat major depression, delivers brief magnetic pulses to the limbic structures. Treatment facilitates focal stimulation, rapidly applying electrical charges to the cortical neurons. TMS targets prefrontal circuits of the brain that are underactive during depressive episodes. Recent animal studies have suggested that bromodeoxyuridine (BrdU)-positive cells (newborn cells) are increased significantly in the dentate gyrus, in turn suggesting that hippocampal neurogenesis might be involved in the antidepressant effects of chronic rTMS.17 Although the underlying therapeutic mechanisms of rTMS treatment of depression remain unclear, it appears that hippocampal neurogenesis might be required to produce the effects of antidepressant treatments, including drugs and electroconvulsive therapy.17
Selective ‘shunting’ of energy occurs during the stress response
Hormones released from the adrenal glands during stress divert glucose to exercising muscles and the brain’s limbic system, which are involved in the fight-or-flight response.18 However, metabolic functions and areas of the brain that are not involved in the stress response, such as the cerebral cortex and hippocampus, are deprived of energy as a consequence of this innate selective shunting (Figure 2).19
Positron-emission tomography (PET) scanning of the resting brain shows that components of the cerebral cortex (prefrontal cortex, hippocampus, striatum) and areas connecting the cerebral cortex to the limbic system exhibit the most energy consumption in the brain during rest (Figure 3).20 PET studies also show that neuronal connections within these energy-demanding areas atrophy more rapidly than in any other area of the brain when their energy supply is reduced or cut off.6
When the supply of oxygen and glucose to certain areas of the brain is reduced—such as in traumatic brain injury or stroke—the excitatory neurotransmitter glutamate accumulates in extracellular fluid and causes nerve-cell death.21 When a conditioned stimulus is presented during fear acquisition, functional magnetic resonance imaging (fMRI) studies of fear-conditioning have consistently reported, in the prefrontal cortex:
- a decrease in the blood oxygen level-dependent signal, below resting baseline
- a reduction in blood flow (Figure 4).22
This discovery adds to evidence that demonstrates a decrease in gray-matter density in the frontal lobes as a result of glutaminergic toxicity (Figure 5).
Activation of L-glutamate, believed to play a significant role in depression and other neuropsychiatric disorders, triggers calcium-dependent intracellular responses that “excite cells to death,” so to speak—thereby causing nerve-cell apoptosis and a reduction in synaptic connections between different areas of the brain responsible for learning and memory.23 Malfunction of these synaptic connections is thought to be partially responsible for depression and other psychiatric disorders.
Excessive activation of N-methyl-d-asparate (NMDA) receptors is thought to be the underlying mechanism that leads to neuronal cell death in glutaminergic toxicity. Therefore, NMDA receptor proteins have become a target in treating neurodegenerative psychiatric illnesses. There is more than one type of NMDA receptor; some of them are excitatory, others are inhibitory. Four compounds have presented as therapeutic candidates for inhibition of NMDA receptor functioning and treatment of depression: those that inhibit glutamate binding, those that block the ion channel, and those that inhibit receptor binding to the terminal regulatory domain.24
Regrettably, these chemical compounds are not receptor-selective, but small structural modifications of these NMDA receptors have been found and lead to significant changes in potency and selectivity. This should serve as a unique starting point for developing highly specific NMDA receptor modulator agents for a variety of neuropsychiatric and neurological conditions. GLYX-13, a derivative of ketamine (an NMDA receptor antagonist), has been implicated for use in treating depression. It has been tested on 2 large phase-II study groups.25
Neuronal circuitry of depression is altered by prolonged stress
Symptoms of depression can be explained by the anatomical circuit shown in Figure 6.15,20 Impaired concentration, diminished ability to process new information, and decline in memory function are associated with decreased nerve density in the hippocampus, which plays a key role in learning, memory, and encoding of emotionally relevant data into memory.26 The hippocampus interacts with the amygdala to provide input about the context in which stimuli occur.
Depressed people often demonstrate impulsivity and have difficulty controlling expression of emotions—traits that are attributed to increased neuronal density in the amygdala and insula, which has been illustrated in PET scans and voxel-based morphometry in depressed patients.27 These brain areas are implicated in subjective emotional experience, processing of emotional reactions, and impulsive decision-making. The amygdala is normally highly regulated by the prefrontal cortex, which uses rational judgment to interpret stimuli and regulate the expression of emotion.
A study involving a facial expression processing task demonstrated reduced connectivity between the amygdala and prefrontal cortex and increased functional connectivity among the amygdala, hippocampus, and caudate-putamen in depressed patients.24 And in a study that measured white matter conduction in various brain areas in depressed patients, the greatest reduction was found in areas connecting the limbic system to the prefrontal cortex and hippocampus—believed to be caused by stress response-induced ischemic glutaminergic neuroapoptosis.21 Such neuroapoptosis might lead to irrational interpretation of stimuli, unchecked expression of emotion, and impulsive thoughts and behavior that are often present in depression and other mood disorders.
Deep brain stimulation (DBS), in which electrodes are implanted in the brain, has proved effective at increasing synaptic connections between the prefrontal cortex and the limbic system when electrodes are placed appropriately.28 Patients with refractory depression who are treated with DBS show increased gray-matter density and functional activity in the prefrontal cortex, hippocampus, and fronto-limbic connections.29 DBS also increases neurotransmission of dopamine, serotonin, and norepinephrine within the fronto-limbic circuitry.30
Identifying risk factors for depression
Genetic risk factors. Forty percent of patients with depression have a first-degree relative with depression, suggesting a strong genetic component.10 Inherited differences in hippocampal volume, synaptic connections between the prefrontal cortex and amygdala, γ-aminobutyric acid (GABA)/glutamate balance, BDNF neurotransmitter receptors, and anatomic positioning of the limbic system in relation to other brain structures might account for the heritability of psychiatric disorders such as depression.
Evidence has been consistent that hippocampal volume is diminished in the brain of depressed persons. However, there is no prospective cohort study to determine whether people who have lower gray-matter hippocampal density or volume, or both, before depression onset develop symptoms later in life. There also is no study to determine the percentage of people who have lower-than-average hippocampal gray-matter density or volume and who have a first-degree relative with depression. Such studies would yield valuable information about anatomic variables that increase the risk of depression.
It has been proposed that low GABA function is an inherited biomarker for depression. Bjork and co-workers found a lower plasma level of GABA in depressed subjects and in their first-degree relatives, confirming that GABAergic tone might be under genetic control.11 Genetic loci studies in mice have linked depressive-like behavior to GABAergic loci on chromosomes 8 and 11, encoding alpha 1, alpha 6, and gamma subunits of GABAA receptors.23
A recent study in humans showed that severe, treatment-resistant depression with anxiety was linked to a mutation in the B1 subunit of the GABAA receptor. Positive genetic associations were found between polymorphism in human GABAA receptor subunit genes.11
GABA metabolizing enzymes also can be considered biological modifiers of depression. For example:
- GABA uptake and metabolism is controlled by the enzyme glutamic acid decarboxylase (GAD); depression has been found to be associated with a polymorphism in the GAD67 gene encoding an isoform of GAD.11
- GABA transaminase (GABA-T) is another key enzyme in GABA turnover.31 It catabolizes GABA.
We can conclude that, to a high degree, depression depends on GABA production and metabolism.
A variant in the human BDNF gene, in which valine is substituted for methionine in position 66 of the pro-domain of the BDNF protein, is associated with
- a decrease in the production of BDNF
- increased susceptibility to neuropsychiatric disorders, including depression, anxiety disorder, and bipolar disorder (Figure 7).32
People with the MM allele have been found to have a small hippocampal neuronal density and poor hippocampus-dependent memory function in neuroimaging studies.23 They also displayed diminished ventromedial prefrontal cortex volume and presented with aversive memory extinction deficit (ie, “holding grudges”).
Another neurotrophic factor, vascular endothelial growth factor (VEGF), is a survival factor for endothelial cells and neurons and a modulator of synaptic transmission. Understanding the molecular and cellular specificity of antidepressant-induced VEGF will be critical to determine its potential as a therapeutic target in depression.33 Delineating the relationship between VEGF and depression has, ultimately, the potential to shed light on the still elusive neural mechanisms that underlie the pathophysiology of depression and the mechanisms by which antidepressants exert their effects.34
Genetic polymorphisms in monoamine receptors (5-HT2A), transporters (SERTPR, 5-HTTLPR, STin2, rs25531, SLC6A4), and regulatory enzymes should not be overlooked.35 There is reproducible evidence that variability in these polymorphisms are associated with variability in:
- vulnerability to depression
- the response to treatment with existing antidepressant medications.1
Most studies that look at changes in neuronal circuitry focus on the integrity of synaptic connections between the frontal cortex and limbic system; few of them have closely examined the importance of the anatomic proximity of the 2 regions. It might be that having an amygdala that is relatively closer to the frontal cortex and the hippocampus reduces a person’s risk of depression, and vice versa. This association needs to be investigated further with imaging studies.
Environmental risk factors. The brain is thought to be plastic until age 30.5 Plasticity diminishes with age after age 7—except for the hippocampus, which can regenerate throughout life.36 Early life experiences play an important role in forming synaptic connections between the frontal cortex and the limbic system, through a process known as fear conditioning.
Children learn early in life which stimuli are to be perceived as threatening or aversive and how to respond to best preserves their safety and internal sense of well-being. Those who grow up in a hostile environment learn to perceive more stimuli as threatening than children who grow up in a nurturing environment.32 It is possible that the amygdala is larger in children who grow up in less-than-ideal circumstances because this region is constantly being recruited—at the expense of the more rational frontal cortex.
Evidence suggests that these conditions reduce hippocampal neurogenesis37:
- increasing age
- substance abuse (opiates and methamphetamines)
- inadequate housing
- minimal physical activity
- little opportunity for social stimulation
- minimal learning experience.
Bottom Line
Depression has been understood as a neurotransmitter deficiency in the brain; treatments were engineered to increase release, or block degradation, of those neurotransmitters. Novel theories—all interconnected—of the neuroanatomical pathophysiology of depression focus more on differences in neuron density in the brain; effects of stress on neurogenesis and neuronal cell apoptosis; alterations in feedback pathways connecting the pre-frontal cortex to the limbic system; and the role of pro-inflammatory mediators evoked during the stress response.
Related Resources
- Fuchs E. Neurogenesis in the adult brain: is there an association with mental disorders? Eur Arch Psychiatry Clin Neurosci. 2007;257(5):247-249.
- Videbech P, Ravnkilde B. Hippocampal volume and depression: a meta-analysis of MRI studies. Am J Psychiatry. 2004; 161(11):1957-1966.
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Acknowledgement
Anita Rao, second-year medical student, Stritch School of Medicine, Loyola University, Chicago, Illinois, assisted in the preparation of this manuscript.
For more than 50 years, depression has been studied, and understood, as a deficiency of specific neurotransmitters in the brain—namely dopamine, norepinephrine, and serotonin. Treatments for depression have been engineered to increase the release, or block the degradation, of these neurotransmitters within the synaptic cleft. Although a large body of evidence supports involvement of dopamine, norepinephrine, and serotonin in the pathophysiology of depression, the observation that pharmacotherapy is able to induce remission only in <50% of patients1 has prompted researchers to look beyond neurotransmitters for an understanding of depressive disorders (Table 1).
Today, theories of depression focus more on differences in neuron density in various regions of the brain; the effect of stress on neurogenesis and neuronal cell apoptosis; alterations in feedback pathways connecting the pre-frontal cortex to the limbic system; and the role of proinflammatory mediators evoked during the stress response (Box,2,3). These theories should not be viewed as separate entities because they are highly interconnected. Integrating them provides for a more expansive understanding of the pathophysiology of depression and biomarkers that are involved (Table 2).
In this article, we:
- integrate the large body of evidence supporting the contribution of the above variables to the onset and persistence of depression
- propose a possible risk stratification model
- explore possibilities for treatment.
The stress response: How does it affect the brain?
Stress initiates a cascade of events in the brain and peripheral systems that enable an organism to cope with, and adapt to, new and challenging situations. That is why physiologic and behavioral responses to stress generally are considered beneficial to survival.
When stress is maintained for a long period, both brain and body are harmed because target cells undergo prolonged exposure to physiologic stress mediators. For example, Woolley and Gould4 exposed rats to varying durations of glucocorticoids and observed that treating animals with corticosterone injection for 21 days induced neuronal atrophy in the hippocampus and prefrontal cortex and increased release of proinflammatory cytokines from astrocytes within the limbic system. Stressful experiences are believed to be closely associated with development of psychological alterations and, thus, neuropsychiatric disorders.5 To go further: Chronic stress is believed to be the leading cause of depression.
When the brain perceives an external threat, the stress response is called into action. The amygdala, part of the primitive limbic system, is the primary area of the brain responsible for triggering the stress response,6 signaling the hypothalamus to release corticotropin-releasing hormone (CRH) to the anterior pituitary gland, which, in turn releases adrenocorticotropic hormone to the adrenal glands (Figure 1).7 The adrenal glands are responsible for releasing glucocorticoids, which, because of their lipophilic nature, can cross the blood-brain barrier and are found in higher levels in the cerebrospinal fluid (CSF) of depressed persons.7
Once in the brain, glucocorticoids can be irreversibly degraded in the cytosol by the enzyme 11-β hydroxysteroid dehydrogenase type 2, a potential target for treating depression, or can bind to the glucocorticoid receptor (GR). Results of a research study of the role of cortisol in suppression of proinflammatory cytokine signaling activity in rainbow trout hepatocytes suggest a negative feedback loop for GR gene regulation during stress.8
Because this auto-regulation is a crucial step in the physiological stress response, the idea of the GR as an important biomarker in depression has gained popularity. In humans, when the GR binds to glucocorticoids that are released from the adrenal cortex during the stress response, the activated GR-cortisol complex represses expression of proinflammatory proteins in astrocytes and microglial cells and in all cells in the periphery before they are transcribed into proteins.9 The GR also has been shown to modulate neurogenesis.8 Repeated stress that persists over a long period leads to GR resistance, thereby reducing inhibition of production of proinflammatory cytokines.
Exposure to stress for >21 days leads to overactivity of the HPA axis and GR resistance,10 which decreases suppression of proinflammatory cytokines. There is evidence that proinflammatory cytokines, tumor necrosis factor-α, and interleukin-6 further induce GR receptor resistance by preventing the cortisol-GR receptor complex from entering cell nuclei and decreasing binding to DNA within the nuclei.11 Dexamethasone, a GR agonist, has been implicated in research studies for potential re-regulation of the HPA axis in depressed persons.12
Nerve cell death in the hippocampus
Studies showing reduced hippocampal volume in unipolar depression and a correlation between the number of episodes and a consequence of untreated depression and studies suggesting that treatment can stop or reduce shrinkage,13 and recent findings of rapid neurogenesis in hippocampi in response to ketamine, brings our focus to hippocampus in depression.
The greatest density of GRs is found in the hippocampus, which is closely associated with the limbic system.7 Therefore, the hippocampus is sensitive to increases in glucocorticoids in the brain and plays a crucial role in regulation of the HPA axis.
Evidence shows that in chronic stress exposure (≥21 days), nerve cells in the hippocampus begin to atrophy and can no longer provide negative feedback inhibition to the hypothalamus, causing HPA axis dysregulation and uncontrolled release of glucocorticoids into the bloodstream and CSF.2 In patients with Cushing syndrome, who produce abnormally high levels of glucocorticoid, the incidence of depression is as high as 50%.14 Similarly, patients treated with glucocorticoids such as prednisone often experience psychiatric symptoms, the most common being depression. Gould found that partial adrenalectomy increased hippocampal neurogenesis in rat brains, indicating the beneficial effect of stress hormone antagonism.4 CRH antagonists are being looked at as a promising and less invasive treatment option for depression.
Focus has been diverted to the role of the hippocampus in depression because of its ability to regenerate throughout adulthood, leading potentially to a re-regulation of the HPA axis and subsiding of the stress response, which is universally believed to be the primary precipitating factor in depression onset. Rats require 10 to 21 days of rest to recover from the effects of chronic (21 days) administration of glucocorticoids.15 If this proves to be a directly proportional relationship, then rats would need an estimated 120 days to recover from 6 months of constant glucocorticoid exposure. Considering that the same is true for humans, current depression treatment programs, which average 6 weeks, are not long enough for adequate recovery.
Antidepressants such as selective serotonin reuptake inhibitors, serotonin-norepinephrine reuptake inhibitors, and tricyclics stimulate neurogenesis in the hippocampus via increases in brain-derived neurotrophic factor (BDNF), suggesting that these neurotransmitters play an important role depression.16
Repetitive transcranial magnetic stimulation (rTMS), a noninvasive neuromodulation therapy approved to treat major depression, delivers brief magnetic pulses to the limbic structures. Treatment facilitates focal stimulation, rapidly applying electrical charges to the cortical neurons. TMS targets prefrontal circuits of the brain that are underactive during depressive episodes. Recent animal studies have suggested that bromodeoxyuridine (BrdU)-positive cells (newborn cells) are increased significantly in the dentate gyrus, in turn suggesting that hippocampal neurogenesis might be involved in the antidepressant effects of chronic rTMS.17 Although the underlying therapeutic mechanisms of rTMS treatment of depression remain unclear, it appears that hippocampal neurogenesis might be required to produce the effects of antidepressant treatments, including drugs and electroconvulsive therapy.17
Selective ‘shunting’ of energy occurs during the stress response
Hormones released from the adrenal glands during stress divert glucose to exercising muscles and the brain’s limbic system, which are involved in the fight-or-flight response.18 However, metabolic functions and areas of the brain that are not involved in the stress response, such as the cerebral cortex and hippocampus, are deprived of energy as a consequence of this innate selective shunting (Figure 2).19
Positron-emission tomography (PET) scanning of the resting brain shows that components of the cerebral cortex (prefrontal cortex, hippocampus, striatum) and areas connecting the cerebral cortex to the limbic system exhibit the most energy consumption in the brain during rest (Figure 3).20 PET studies also show that neuronal connections within these energy-demanding areas atrophy more rapidly than in any other area of the brain when their energy supply is reduced or cut off.6
When the supply of oxygen and glucose to certain areas of the brain is reduced—such as in traumatic brain injury or stroke—the excitatory neurotransmitter glutamate accumulates in extracellular fluid and causes nerve-cell death.21 When a conditioned stimulus is presented during fear acquisition, functional magnetic resonance imaging (fMRI) studies of fear-conditioning have consistently reported, in the prefrontal cortex:
- a decrease in the blood oxygen level-dependent signal, below resting baseline
- a reduction in blood flow (Figure 4).22
This discovery adds to evidence that demonstrates a decrease in gray-matter density in the frontal lobes as a result of glutaminergic toxicity (Figure 5).
Activation of L-glutamate, believed to play a significant role in depression and other neuropsychiatric disorders, triggers calcium-dependent intracellular responses that “excite cells to death,” so to speak—thereby causing nerve-cell apoptosis and a reduction in synaptic connections between different areas of the brain responsible for learning and memory.23 Malfunction of these synaptic connections is thought to be partially responsible for depression and other psychiatric disorders.
Excessive activation of N-methyl-d-asparate (NMDA) receptors is thought to be the underlying mechanism that leads to neuronal cell death in glutaminergic toxicity. Therefore, NMDA receptor proteins have become a target in treating neurodegenerative psychiatric illnesses. There is more than one type of NMDA receptor; some of them are excitatory, others are inhibitory. Four compounds have presented as therapeutic candidates for inhibition of NMDA receptor functioning and treatment of depression: those that inhibit glutamate binding, those that block the ion channel, and those that inhibit receptor binding to the terminal regulatory domain.24
Regrettably, these chemical compounds are not receptor-selective, but small structural modifications of these NMDA receptors have been found and lead to significant changes in potency and selectivity. This should serve as a unique starting point for developing highly specific NMDA receptor modulator agents for a variety of neuropsychiatric and neurological conditions. GLYX-13, a derivative of ketamine (an NMDA receptor antagonist), has been implicated for use in treating depression. It has been tested on 2 large phase-II study groups.25
Neuronal circuitry of depression is altered by prolonged stress
Symptoms of depression can be explained by the anatomical circuit shown in Figure 6.15,20 Impaired concentration, diminished ability to process new information, and decline in memory function are associated with decreased nerve density in the hippocampus, which plays a key role in learning, memory, and encoding of emotionally relevant data into memory.26 The hippocampus interacts with the amygdala to provide input about the context in which stimuli occur.
Depressed people often demonstrate impulsivity and have difficulty controlling expression of emotions—traits that are attributed to increased neuronal density in the amygdala and insula, which has been illustrated in PET scans and voxel-based morphometry in depressed patients.27 These brain areas are implicated in subjective emotional experience, processing of emotional reactions, and impulsive decision-making. The amygdala is normally highly regulated by the prefrontal cortex, which uses rational judgment to interpret stimuli and regulate the expression of emotion.
A study involving a facial expression processing task demonstrated reduced connectivity between the amygdala and prefrontal cortex and increased functional connectivity among the amygdala, hippocampus, and caudate-putamen in depressed patients.24 And in a study that measured white matter conduction in various brain areas in depressed patients, the greatest reduction was found in areas connecting the limbic system to the prefrontal cortex and hippocampus—believed to be caused by stress response-induced ischemic glutaminergic neuroapoptosis.21 Such neuroapoptosis might lead to irrational interpretation of stimuli, unchecked expression of emotion, and impulsive thoughts and behavior that are often present in depression and other mood disorders.
Deep brain stimulation (DBS), in which electrodes are implanted in the brain, has proved effective at increasing synaptic connections between the prefrontal cortex and the limbic system when electrodes are placed appropriately.28 Patients with refractory depression who are treated with DBS show increased gray-matter density and functional activity in the prefrontal cortex, hippocampus, and fronto-limbic connections.29 DBS also increases neurotransmission of dopamine, serotonin, and norepinephrine within the fronto-limbic circuitry.30
Identifying risk factors for depression
Genetic risk factors. Forty percent of patients with depression have a first-degree relative with depression, suggesting a strong genetic component.10 Inherited differences in hippocampal volume, synaptic connections between the prefrontal cortex and amygdala, γ-aminobutyric acid (GABA)/glutamate balance, BDNF neurotransmitter receptors, and anatomic positioning of the limbic system in relation to other brain structures might account for the heritability of psychiatric disorders such as depression.
Evidence has been consistent that hippocampal volume is diminished in the brain of depressed persons. However, there is no prospective cohort study to determine whether people who have lower gray-matter hippocampal density or volume, or both, before depression onset develop symptoms later in life. There also is no study to determine the percentage of people who have lower-than-average hippocampal gray-matter density or volume and who have a first-degree relative with depression. Such studies would yield valuable information about anatomic variables that increase the risk of depression.
It has been proposed that low GABA function is an inherited biomarker for depression. Bjork and co-workers found a lower plasma level of GABA in depressed subjects and in their first-degree relatives, confirming that GABAergic tone might be under genetic control.11 Genetic loci studies in mice have linked depressive-like behavior to GABAergic loci on chromosomes 8 and 11, encoding alpha 1, alpha 6, and gamma subunits of GABAA receptors.23
A recent study in humans showed that severe, treatment-resistant depression with anxiety was linked to a mutation in the B1 subunit of the GABAA receptor. Positive genetic associations were found between polymorphism in human GABAA receptor subunit genes.11
GABA metabolizing enzymes also can be considered biological modifiers of depression. For example:
- GABA uptake and metabolism is controlled by the enzyme glutamic acid decarboxylase (GAD); depression has been found to be associated with a polymorphism in the GAD67 gene encoding an isoform of GAD.11
- GABA transaminase (GABA-T) is another key enzyme in GABA turnover.31 It catabolizes GABA.
We can conclude that, to a high degree, depression depends on GABA production and metabolism.
A variant in the human BDNF gene, in which valine is substituted for methionine in position 66 of the pro-domain of the BDNF protein, is associated with
- a decrease in the production of BDNF
- increased susceptibility to neuropsychiatric disorders, including depression, anxiety disorder, and bipolar disorder (Figure 7).32
People with the MM allele have been found to have a small hippocampal neuronal density and poor hippocampus-dependent memory function in neuroimaging studies.23 They also displayed diminished ventromedial prefrontal cortex volume and presented with aversive memory extinction deficit (ie, “holding grudges”).
Another neurotrophic factor, vascular endothelial growth factor (VEGF), is a survival factor for endothelial cells and neurons and a modulator of synaptic transmission. Understanding the molecular and cellular specificity of antidepressant-induced VEGF will be critical to determine its potential as a therapeutic target in depression.33 Delineating the relationship between VEGF and depression has, ultimately, the potential to shed light on the still elusive neural mechanisms that underlie the pathophysiology of depression and the mechanisms by which antidepressants exert their effects.34
Genetic polymorphisms in monoamine receptors (5-HT2A), transporters (SERTPR, 5-HTTLPR, STin2, rs25531, SLC6A4), and regulatory enzymes should not be overlooked.35 There is reproducible evidence that variability in these polymorphisms are associated with variability in:
- vulnerability to depression
- the response to treatment with existing antidepressant medications.1
Most studies that look at changes in neuronal circuitry focus on the integrity of synaptic connections between the frontal cortex and limbic system; few of them have closely examined the importance of the anatomic proximity of the 2 regions. It might be that having an amygdala that is relatively closer to the frontal cortex and the hippocampus reduces a person’s risk of depression, and vice versa. This association needs to be investigated further with imaging studies.
Environmental risk factors. The brain is thought to be plastic until age 30.5 Plasticity diminishes with age after age 7—except for the hippocampus, which can regenerate throughout life.36 Early life experiences play an important role in forming synaptic connections between the frontal cortex and the limbic system, through a process known as fear conditioning.
Children learn early in life which stimuli are to be perceived as threatening or aversive and how to respond to best preserves their safety and internal sense of well-being. Those who grow up in a hostile environment learn to perceive more stimuli as threatening than children who grow up in a nurturing environment.32 It is possible that the amygdala is larger in children who grow up in less-than-ideal circumstances because this region is constantly being recruited—at the expense of the more rational frontal cortex.
Evidence suggests that these conditions reduce hippocampal neurogenesis37:
- increasing age
- substance abuse (opiates and methamphetamines)
- inadequate housing
- minimal physical activity
- little opportunity for social stimulation
- minimal learning experience.
Bottom Line
Depression has been understood as a neurotransmitter deficiency in the brain; treatments were engineered to increase release, or block degradation, of those neurotransmitters. Novel theories—all interconnected—of the neuroanatomical pathophysiology of depression focus more on differences in neuron density in the brain; effects of stress on neurogenesis and neuronal cell apoptosis; alterations in feedback pathways connecting the pre-frontal cortex to the limbic system; and the role of pro-inflammatory mediators evoked during the stress response.
Related Resources
- Fuchs E. Neurogenesis in the adult brain: is there an association with mental disorders? Eur Arch Psychiatry Clin Neurosci. 2007;257(5):247-249.
- Videbech P, Ravnkilde B. Hippocampal volume and depression: a meta-analysis of MRI studies. Am J Psychiatry. 2004; 161(11):1957-1966.
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Acknowledgement
Anita Rao, second-year medical student, Stritch School of Medicine, Loyola University, Chicago, Illinois, assisted in the preparation of this manuscript.
1. Eley TC, Sugden K, Corsico A, et al. Gene-environment interaction analysis of serotonin system markers with adolescent depression. Mol Psychiatry. 2004;9(10):908-915.
2. Haber SN, Rauch SL. Neurocircuitry: a window into the networks underlying neuropsychiatric disease. Neuropsychopharmacology. 2010;35(1):1-3.
3. Frodl T, Bokde AL, Scheuerecker J, et al. Functional connectivity bias of the orbitofrontal cortex in drug-free patients with major depression. Biol Psychiatry. 2010; 67(2):161-167.
4. Woolley CS, Gould E, McEwen BS. Exposure to excess glucocorticoids alters dendritic morphology of adult hippocampal pyramidal neurons. Brain Res. 1990;531(1-2): 225-231.
5. Heim C, Nemeroff CB. The impact of early adverse experiences on brain systems involved in the pathophysiology of anxiety and affective disorders. Biol Psychiatry. 1999;46(11):1509-1522.
6. Isgor C, Kabbaj M, Akil H, et al. Delayed effects of chronic variable stress during peripubertal-juvenile period on hippocampal morphology and on cognitive and stress axis functions in rats. Hippocampus. 2004;14(5):636-648.
7. De Kloet ER, Vreugdenhil E, Oitzl MS, et al. Brain corticosteroid receptor balance in health and disease. Endocr Rev. 1998;19(3):269-301.
8. Philip AM, Kim SD, Vijayan MM. Cortisol modulates the expression of cytokines and suppressors of cytokine signaling (SOCS) in rainbow trout hepatocytes. Dev Comp Immunol. 2012;38(2):360-367.
9. Coplan JD, Lydiard RB. Brain circuits in panic disorder. Biol Psychiatry. 1998;44(12):1264-1276.
10. Anisman H, Merali Z. Cytokines, stress and depressive illness: brain-immune interactions. Ann Med. 2003;35(1):2-11.
11. Crowley JJ, Lucki I. Opportunities to discover genes regulating depression and antidepressant response from rodent behavioral genetics. Curr Pharm Des. 2005;11(2):157-169.
12. Covington HE 3rd, Vialou V, Nestler EJ. From synapse to nucleus: novel targets for treating depression. Neuropharmacology. 2010;58(4-5):683-693.
13. Videbech P, Ravnkilde B. Hippocampal volume and depression: a meta-analysis of MRI studies. Am J Psychiatry. 2004;161(11):1957-1966.
14. Sandi C. Stress, cognitive impairment and cell adhesion molecules. Nat Rev Neurosci. 2004;5(12):917-930.
15. Hartley CA, Phelps EA. Changing fear: the neurocircuitry of emotion regulation. Neuropsychopharmacology. 2010;35(1): 136-146.
16. Kim DK, Lim SW, Lee S, et al. Serotonin transporter gene polymorphism and antidepressant response. Neuroreport. 2000;11(1):215-219.
17. Ueyama E, Ukai S, Ogawa A, et al, Chronic repetitive transcranial magnetic stimulation increases hippocampal neurogenesis in rats. Psychiatry Clin Neurosci. 2011; 65(1):77-81.
18. Irwin W, Anderle MJ, Abercrombie HC, et al. Amygdalar interhemispheric functional connectivity differs between the non-depressed and depressed human brain. Neuroimage. 2004;21(2):674-686.
19. McEwen BS. Physiology and neurobiology of stress and adaptation: central role of the brain. Physiol Rev. 2007; 87(3):873-904.
20. Gusnard DA, Raichle ME, Raichle ME. Searching for a baseline: functional imaging and the resting human brain. Nat Rev Neurosci. 2001;2(10):685-694.
21. Hulsebosch CE, Hains BC, Crown ED, et al. Mechanisms of chronic central neuropathic pain after spinal cord injury. Brain Res Rev. 2009;60(1):202-213.
22. Gottfried JA, Dolan RJ. Human orbitofrontal cortex mediates extinction learning while accessing conditioned representations of value. Nat Neurosci. 2004;7(10):1144-1152.
23 Arnone D, McKie S, Elliott R, et al. State-dependent changes in hippocampal grey matter in depression. Mol Psychiatry. 2012;1(8):1359-4184.
24. Brunoni AR, Lopes M, Fregni F. A systematic review and meta-analysis of clinical studies on major depression and BDNF levels: implications for the role of neuroplasticity in depression. Int J Neuropsychopharmacol. 2008;11(8):1169-1180.
25. Maeng S, Zarate CA Jr. The role of glutamate in mood disorders: results from the ketamine in major depression study and the presumed cellular mechanism underlying its antidepressant effects. Curr Psychiatry Rep. 2007;9(6):467-474.
26. Vaidya VA, Fernandes K, Jha S. Regulation of adult hippocampal neurogenesis: relevance to depression. Expert Rev Neurother. 2007;7(7):853-864.
27. Lisiecka DM, Carballedo A, Fagan AJ, et al. Altered inhibition of negative emotions in subjects at family risk of major depressive disorder. J Psychiatr Res. 2012;46(2):181-188.
28. Mayberg HS, Lozano AM, Voon V, et al. Deep brain stimulation for treatment-resistant depression. Neuron. 2005;45(5):651-660.
29. Levkovitz Y, Harel EV, Roth Y, et al. Deep transcranial magnetic stimulation over the prefrontal cortex: evaluation of antidepressant and cognitive effects in depressive patients. Brain Stimul. 2009;2(4):188-200.
30. Schlaepfer TE, Lieb K. Deep brain stimulation for treatment of refractory depression. Lancet. 2005;366(9495):1420-1422.
31. Astrup, J. Energy-requiring cell functions in the ischemic brain. Their critical supply and possible inhibition in protective therapy. J Neurosurg. 1982;56(4):482-497.
32. Fletcher JM. Childhood mistreatment and adolescent and young adult depression. Soc Sci Med. 2009;68(5):799-806.
33. Warner-Schmidt JL, Duman R. VEGF as a potential target for therapeutic intervention in depression. Curr Opin Pharmacol. 2008;8(1):14-19.
34. Clark-Raymond A, Halaris A. VEGF and depression: a comprehensive assessment of clinical data. J Psychiatr Res. 2013;47(8):1080-1087.
35. Alonso R, Griebel G, Pavone G, et al. Blockade of CRF(1) or V(1b) receptors reverses stress-induced suppression of neurogenesis in a mouse model of depression. Mol Psychiatry. 2004;9(3):278-286.
36. Thomas RM, Peterson DA. A neurogenic theory of depression gains momentum. Mol Interv. 2003;3(8):441-444.
37. Jacobs BL. Adult brain neurogenesis and depression. Brain Behav Immun. 2002;16(5):602-609.
1. Eley TC, Sugden K, Corsico A, et al. Gene-environment interaction analysis of serotonin system markers with adolescent depression. Mol Psychiatry. 2004;9(10):908-915.
2. Haber SN, Rauch SL. Neurocircuitry: a window into the networks underlying neuropsychiatric disease. Neuropsychopharmacology. 2010;35(1):1-3.
3. Frodl T, Bokde AL, Scheuerecker J, et al. Functional connectivity bias of the orbitofrontal cortex in drug-free patients with major depression. Biol Psychiatry. 2010; 67(2):161-167.
4. Woolley CS, Gould E, McEwen BS. Exposure to excess glucocorticoids alters dendritic morphology of adult hippocampal pyramidal neurons. Brain Res. 1990;531(1-2): 225-231.
5. Heim C, Nemeroff CB. The impact of early adverse experiences on brain systems involved in the pathophysiology of anxiety and affective disorders. Biol Psychiatry. 1999;46(11):1509-1522.
6. Isgor C, Kabbaj M, Akil H, et al. Delayed effects of chronic variable stress during peripubertal-juvenile period on hippocampal morphology and on cognitive and stress axis functions in rats. Hippocampus. 2004;14(5):636-648.
7. De Kloet ER, Vreugdenhil E, Oitzl MS, et al. Brain corticosteroid receptor balance in health and disease. Endocr Rev. 1998;19(3):269-301.
8. Philip AM, Kim SD, Vijayan MM. Cortisol modulates the expression of cytokines and suppressors of cytokine signaling (SOCS) in rainbow trout hepatocytes. Dev Comp Immunol. 2012;38(2):360-367.
9. Coplan JD, Lydiard RB. Brain circuits in panic disorder. Biol Psychiatry. 1998;44(12):1264-1276.
10. Anisman H, Merali Z. Cytokines, stress and depressive illness: brain-immune interactions. Ann Med. 2003;35(1):2-11.
11. Crowley JJ, Lucki I. Opportunities to discover genes regulating depression and antidepressant response from rodent behavioral genetics. Curr Pharm Des. 2005;11(2):157-169.
12. Covington HE 3rd, Vialou V, Nestler EJ. From synapse to nucleus: novel targets for treating depression. Neuropharmacology. 2010;58(4-5):683-693.
13. Videbech P, Ravnkilde B. Hippocampal volume and depression: a meta-analysis of MRI studies. Am J Psychiatry. 2004;161(11):1957-1966.
14. Sandi C. Stress, cognitive impairment and cell adhesion molecules. Nat Rev Neurosci. 2004;5(12):917-930.
15. Hartley CA, Phelps EA. Changing fear: the neurocircuitry of emotion regulation. Neuropsychopharmacology. 2010;35(1): 136-146.
16. Kim DK, Lim SW, Lee S, et al. Serotonin transporter gene polymorphism and antidepressant response. Neuroreport. 2000;11(1):215-219.
17. Ueyama E, Ukai S, Ogawa A, et al, Chronic repetitive transcranial magnetic stimulation increases hippocampal neurogenesis in rats. Psychiatry Clin Neurosci. 2011; 65(1):77-81.
18. Irwin W, Anderle MJ, Abercrombie HC, et al. Amygdalar interhemispheric functional connectivity differs between the non-depressed and depressed human brain. Neuroimage. 2004;21(2):674-686.
19. McEwen BS. Physiology and neurobiology of stress and adaptation: central role of the brain. Physiol Rev. 2007; 87(3):873-904.
20. Gusnard DA, Raichle ME, Raichle ME. Searching for a baseline: functional imaging and the resting human brain. Nat Rev Neurosci. 2001;2(10):685-694.
21. Hulsebosch CE, Hains BC, Crown ED, et al. Mechanisms of chronic central neuropathic pain after spinal cord injury. Brain Res Rev. 2009;60(1):202-213.
22. Gottfried JA, Dolan RJ. Human orbitofrontal cortex mediates extinction learning while accessing conditioned representations of value. Nat Neurosci. 2004;7(10):1144-1152.
23 Arnone D, McKie S, Elliott R, et al. State-dependent changes in hippocampal grey matter in depression. Mol Psychiatry. 2012;1(8):1359-4184.
24. Brunoni AR, Lopes M, Fregni F. A systematic review and meta-analysis of clinical studies on major depression and BDNF levels: implications for the role of neuroplasticity in depression. Int J Neuropsychopharmacol. 2008;11(8):1169-1180.
25. Maeng S, Zarate CA Jr. The role of glutamate in mood disorders: results from the ketamine in major depression study and the presumed cellular mechanism underlying its antidepressant effects. Curr Psychiatry Rep. 2007;9(6):467-474.
26. Vaidya VA, Fernandes K, Jha S. Regulation of adult hippocampal neurogenesis: relevance to depression. Expert Rev Neurother. 2007;7(7):853-864.
27. Lisiecka DM, Carballedo A, Fagan AJ, et al. Altered inhibition of negative emotions in subjects at family risk of major depressive disorder. J Psychiatr Res. 2012;46(2):181-188.
28. Mayberg HS, Lozano AM, Voon V, et al. Deep brain stimulation for treatment-resistant depression. Neuron. 2005;45(5):651-660.
29. Levkovitz Y, Harel EV, Roth Y, et al. Deep transcranial magnetic stimulation over the prefrontal cortex: evaluation of antidepressant and cognitive effects in depressive patients. Brain Stimul. 2009;2(4):188-200.
30. Schlaepfer TE, Lieb K. Deep brain stimulation for treatment of refractory depression. Lancet. 2005;366(9495):1420-1422.
31. Astrup, J. Energy-requiring cell functions in the ischemic brain. Their critical supply and possible inhibition in protective therapy. J Neurosurg. 1982;56(4):482-497.
32. Fletcher JM. Childhood mistreatment and adolescent and young adult depression. Soc Sci Med. 2009;68(5):799-806.
33. Warner-Schmidt JL, Duman R. VEGF as a potential target for therapeutic intervention in depression. Curr Opin Pharmacol. 2008;8(1):14-19.
34. Clark-Raymond A, Halaris A. VEGF and depression: a comprehensive assessment of clinical data. J Psychiatr Res. 2013;47(8):1080-1087.
35. Alonso R, Griebel G, Pavone G, et al. Blockade of CRF(1) or V(1b) receptors reverses stress-induced suppression of neurogenesis in a mouse model of depression. Mol Psychiatry. 2004;9(3):278-286.
36. Thomas RM, Peterson DA. A neurogenic theory of depression gains momentum. Mol Interv. 2003;3(8):441-444.
37. Jacobs BL. Adult brain neurogenesis and depression. Brain Behav Immun. 2002;16(5):602-609.

Recovery on higher ground: Spirituality in the treatment of substance abuse
Mr. W, age 45, is a divorced Army veteran living on the street who has entered alcohol treatment for the sixth time. He has never stayed sober for longer than 1 month after each of his previous treatment episodes.
On a typical day, Mr. W drinks 40 oz of beer, a pint of vodka, and other alcoholic beverages when available. Although he has used drugs, he reports that he did so only to augment the effects of alcohol. Before entering rehab, Mr. W worked for a television network for 16 years and was promoted to associate vice president. He lost that job as a result of drinking.
Mr. W comes from a large Irish Catholic family and has sustained an active religious faith, going to Mass 2 or 3 times a month. Throughout the interview, he appears introspective and describes frequent periods of “going inside” of himself to “rehash” things. He states that he has never been satisfied with his spiritual life and has been unable to “quiet the hunger inside.”
He describes his benders as “mini-retreats” and comments that focusing on the condensation droplets on a glass of beer is nearly a “sacramental experience” for him. He states: “My drinking is a spiritual thing for me. I believe that every time I drink I am on a spiritual search. I believe this with all my heart. I have this emptiness inside of me and alcohol would temporarily fill the enormous hole in my insides. Just for a short period of time, I would feel at peace and connected to others, and maybe even to God.”
Mr. W observes that “Every time I relapse it’s because I’m going through a spiritual withdrawal. Booze filled the void inside of me and now the void is back again. Physically and psychologically I’m fine. I’m just empty inside and when I can’t stand it any longer, I drink again.”
Few patients can so directly articulate the role they feel that spirituality plays in their substance use disorder. It is important for clinicians to be aware of the dynamics of spirituality and religion in the cause, maintenance, and treatment of substance misuse problems.
In this article, we discuss how spirituality can be assessed and suggest ethical and clinical practice concerns that we believe may support treatment of substance use disorder. We do not advocate incorporating spiritual interventions into clinical practice for patients who are uncomfortable with doing so, nor do we feel that consideration of and respect for patients’ spirituality precludes evidence-based pharmaceutical and behavioral treatment strategies. We believe, however, that addressing these issues can enhance treatment adherence in select patients.
Defining spirituality
Although religion and spirituality are related concepts, they differ.
- Religion has been defined as “an organized system of beliefs, practices, rituals, and symbols through which ones’ relationship to God or others is nurtured and exercised.”1
- Spirituality is more complex and multifaceted. Reflecting his extensive review of articles on spirituality and addiction, Cook2 proposed the following definition:
Spirituality is a distinctive, potentially creative, and universal dimension of
human experience arising within inner subjective awareness of individuals and within communities, social groups, and traditions. It may be experienced as relationship with that which is intimately “inner,” immanent, and personal, within the self and others, and/or as relationship with that which is wholly “other,” transcendent and beyond the self. It is experienced as being of fundamental or ultimate importance and is therefore concerned with matters of meaning and purpose in life, truth, and values.
In some religions, any use of alcohol or drugs is forbidden; in most religions, abuse of these substances violates norms. Those who misuse a substance also might be alienated from their religious and social support community. People struggling with addiction might feel they are compromising their spiritual values directly through the action itself and indirectly because of the harm their substance misuse causes to close friends and family. Misuse of substances also might be an attempt to “fill the void,” as Mr. W described it, of a spiritual longing or a consequence of doubts about meaning, purpose in life, and God.
Perhaps it isn’t surprising that, among psychiatric disorders, substance abuse problems seem to be most associated with spiritual intervention—especially Alcoholics Anonymous (AA), Narcotics Anonymous (NA), and Cocaine Anonymous (CA). Many of the 12 steps contain references to God and spirituality. Although much of the evidence supporting the effectiveness of the spiritual components of AA/NA/CA is correlational,3,4 the resonance that many persons who are recovering from substance abuse find in the 12-step model suggests that spiritual issues are relevant to understanding patients’ viewpoints and for planning treatment.
Religion and spirituality in risk and recovery
Although alcohol and drugs can have positive religious significance (eg, wine in the Christian Eucharist or in the Hindu practice of Ayurvedic medicine; peyote in some indigenous American religious rituals), substance use and abuse are less prevalent among persons who identify themselves as highly religious or spiritual.5 Studies indicate that alcohol use and abuse are less prevalent among persons who identify as Jewish, Muslim, or conservative Protestant, compared with those who are Catholic or liberal Protestant.6
Research suggests that consideration and accommodation of religious and spiritual practices in the recovery process is effective7-9—and preferred—by many patients.10 Psychiatrists do not need to and, in many cases, should not deliver overtly spiritual treatments to patients recovering from substance abuse. Spirituality and religion are complex issues largely outside the expertise of psychiatry; it would be naïve to consider spirituality and religion as universally beneficial elements for all patients recovering from substance abuse. Instead, psychiatrists should be equipped to:
- assess for religion and spirituality in patients
- be aware of, and supportive of, resources for integrating spirituality into treatment (eg, clergy and hospital chaplains, local AA/NA/CA groups, community religious organizations, spirituality groups).
Assessment
Post and colleagues11 note: “When patients feel that their spiritual needs are neglected in standard clinical environments, many of them may be driven away from effective medical treatment.” This is of particular concern when working with persons who have a substance use disorder—among whom, regrettably, only a minority avail themselves of professional care. You should carefully gauge the importance of these dimensions in how patients understand their disorder and the components of treatment they think their recovery process should involve.
Asking about the religious and spiritual aspects of patients’ lives shows respect for their views and facilitates a therapeutic alliance by recognizing their autonomy in treatment. Pargament and Lomax12 state: “Religion speaks to highly sensitive issues that lie at the core of the individual’s identity, commitments, values and world view. Patients are unlikely to engage in a conversation about the deepest side of themselves unless their psychiatrist demonstrates an openness to, interest in, and appreciation of the patient’s religiousness.” This exploration process can suggest natural environmental support systems available to complement recovery efforts and can indicate whether consultation with a clergyperson knowledgeable about and sensitive to their particular religious tradition is appropriate.
Assessment of spirituality and religious practice usually should occur during the initial clinical interview. Examples of revealing interview questions are listed in Box 1.

The Table13-20 lists well-researched psychometric measures that can provide clues to aspects of your patient’s spirituality. These scales yield quantitative results, yet it may be more useful to review with the patient his (her) responses to the items and to pursue issues further based on the responses.
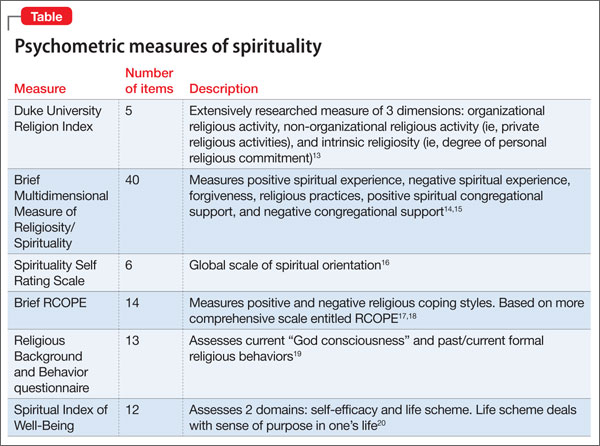
In discussing responses to questions about their spirituality and religious practices, some patients might ask about your views and whether you agree with their views. You could respond with a direct, concise answer, but refocusing the discussion on why the topic of spirituality is important to understanding one’s life and choices might be more therapeutic. This also might be a good time to remind patients that the treatment plan should reflect their view of the role of religion and spirituality in their life.
Of course, some patients are disinclined to discuss spirituality or religion, or prefer that it not be considered in treatment. This is clearly a matter of patient choice, but be aware that the patient may change his (her) mind as the recovery process continues and that the topic can be revisited if desired.
Recommendations for practice
Give careful consideration to ethical and clinical practice issues related to spiritual components in the recovery process. Plante21 and Meador and Koenig1 addressed several relevant ethical principles in considering spirituality in, respectively, psychological and psychiatric practice. Delaney and co-workers22 also presented similar principles, specifically within the context of substance use treatment. These can be summarized as a series of recommendations:
- Although psychiatrists as a group typically have a lower rate of conventional spirituality and religious practice than many of their patients,23-25 it is important not to ignore the patient’s perspective and show respect for the patient’s spiritual needs. However, if the psychiatrist has strong personal religious beliefs, he (she) must carefully guard against proselytizing or exerting undue, unintentional influence.
- Given the variety of religious traditions in the United States, understanding their particular features, as with other issues of cultural diversity, requires competence and sensitivity. Your work requires study and consultation with knowledgeable peers and experts in various faith traditions. Clergy and clinically trained chaplains, in particular, can conduct more comprehensive spiritual assessments that can yield additional treatment-relevant information.
- Maintaining professional boundaries is critical when dealing with religious and spiritual aspects of the patient’s life and thinking. Keep in mind issues of transference and the authoritative influence of the psychiatrist. The patient should understand the difference between services offered by a mental health professional and those given by a spiritual counselor or member of the clergy. Spiritual and religious issues, such as addressing concerns over guilt and sin and a relationship with God, should be referred to an appropriate clergyperson.
- Spiritual interventions introduced into the therapeutic process always should derive from the patient’s perspective and value system; they should not be imposed from an external source.
- Referral to AA often will help alcohol-dependent patients. The heart of AA’s philosophy is that addiction should be seen as a spiritual problem and that genuine recovery requires a profound spiritual awakening (Box 2). AA, as well as interventions inspired by it (eg, NA), are based on peer support, are readily available, and free. Although there is a dearth of controlled research demonstrating the efficacy of AA compared with other interventions, many recovering alcoholics credit these 12-step programs with their having maintained sobriety and adopting a positive lifestyle.

Recent research has identified specific components of AA participation that seem to be helpful.26 These include activities that are spiritual in nature and other generally active components of substance abuse care. Patient preference should be respected when encouraging AA involvement. For patients who are uncomfortable with AA—especially with its emphasis on spirituality—alternative peer support groups are available, such as SMART Recovery.27,28 If the patient adopts AA’s philosophy, it might be helpful for you to employ the language of AA and its constructs when talking with him (her).
Useful strategies on how therapists can encourage AA participation and integrate mutual help groups into treatment planning are described by Nowinski.25 Some AA members believe that use of medication is antithetical to the recovery process, but this is not the position of AA29; using FDA-approved medications, such as naltrexone and acamprosate, is evidence-based and often should be a part of the treatment regimen for alcohol dependence.
Bottom Line
Awareness of, and sensitivity to, the religious and spiritual characteristics of patients with substance use disorder can enhance clinical rapport, inform development of individualized treatment plans, and suggest strategies, such as professional consultation, that might increase the prospects for successful treatment.
Related Resources
- Galanter M. The concept of spirituality in relation to addiction recovery and general psychiatry. Recent Dev Alcohol. 2008;18:125-140.
- Monod S, Brennan M, Rochat E, et al. Instruments measuring spirituality in clinical research: a systematic review. J Gen Intern Med. 2011;26(11):1345-1357.
Drug Brand Names
Acamprosate • Campral
Naltrexone • ReVia, Vivitrol
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Featured Audio
John P. Allen, PhD, MPA, discusses whether spirituality plays a different role in treating substance use disorder than it might in treating other psychiatric illnesses. Dr. Allen works at the Department of Veterans Affairs, Mid-Atlantic Mental Illness Research, Education and Clinical Center, Division of Addictions Research and Treatment, Department of Psychiatry and Behavioral Sciences, Duke University School of Medicine, Durham, North Carolina.
1. Meador KG, Koenig HG. Spirituality and religion in psychiatry practice: parameters and implications. Psychiatr Ann. 2000;30(8):549-555.
2. Cook CC. Addiction and spirituality. Addiction. 2004;99(5):539-551.
3. Kelly J, Stout RL, Magill M, et al. Spirituality in recovery: a lagged mediational analysis of alcoholics anonymous’ principal theoretical mechanism of behavior change. Alcohol Clin Exp Res. 2011;35(3):454-463.
4. Zemore SE. A role for spiritual change in the benefits of 12-step involvement. Alcohol Clin Exp Res. 2007;31(10 suppl):76s-79s.
5. Kendler KS, Liu XQ, Gardner CO, et al. Dimensions of religiosity and their relationship to lifetime psychiatric and substance use disorders. Am J Psychiatry. 2003;160(3):496-503.
6. Koenig HG, King D, Carson VB. Handbook of religion and health, 2nd ed. New York, NY: Oxford University Press; 2012.
7. Carter TM. The effects of spiritual practices on recovery from substance abuse. J Psychiatr Ment Health Nurs. 1998; 5(5):409-413.
8. Conner BT, Anglin MD, Annon J, et al. Effect of religiosity and spirituality on drug treatment outcomes. J Behav Health Serv Res. 2009;36(2):189-198.
9. Robinson EA, Krentzman AR, Webb JR, et al. Six-month changes in spirituality and religiousness in alcoholics predict drinking outcomes at nine months. J Stud Alcohol Drugs. 2011;72(4):660-668.
10. Heinz AJ, Disney ER, Epstein DH, et al. A focus-group study on spirituality and substance-user treatment. Subst Use Misuse. 2010;45(1-2):134-153.
11. Post SG, Puchalski CM, Larson DB. Physicians and patient spirituality: professional boundaries, competency, and ethics. Ann Intern Med. 2000;132(7):578-583.
12. Pargament KI, Lomax JW. Understanding and addressing religion among people with mental illness. World Psychiatry. 2013;12(1):26-32.
13. Koenig HG, Büssing A. The Duke University Religion Index (DUREL): a five-item measure for use in epidemiological studies. Religions. 2010;1(1):78-85.
14. Brief Multidimensional Measure of Religiousness/Spirituality: 1999. http://www.primarycarecore.org/PDF/137.pdf. Accessed January 9, 2014.
15. Johnstone B, Yoon DP, Franklin KL, et al. Re-conceptualizing the factor structure of the Brief Multidimensional Measure of Religiousness/Spirituality. J Relig Health. 2009;48(2):146-163.
16. Galanter M, Dermatis H, Bunt G, et al. Assessment of spirituality and its relevance to addiction treatment. J Subst Abuse Treat. 2007;33(3):257-264.
17. Pargament K, Feuille M, Burdzy D. The Brief RCOPE: current psychometric status of a short measure of religious coping. Religions. 2011;2(1):51-76.
18. Afterdeployment.org. Spirituality assessment. http://www.afterdeployment.org/sites/default/files/pdfs/assessment-tools/spirituality-assessment.pdf. Accessed January 9, 2014.
19. Connors GJ, Tonigan JS, Miller WR. A measure of religious background and behavior for use in behavior change research. Psychol Addict Behav. 1996;10(2):90-96.
20. Daaleman TP, Frey BB. The Spirituality Index of Well-Being: a new Instrument for health-related quality-of-life research. Ann Fam Med. 2004;2(5):499-503.
21. Plante TG. Integrating spirituality and psychotherapy: ethical issues and principles to consider. J Clin Psychol. 2007;63(9):891-902.
22. Delaney HD, Forcehimes AA, Campbell WP, et al. Integrating spirituality into alcohol treatment. J Clin Psychol. 2009;65(2):185-198.
23. Shafranske EP. Religious involvement and professional practices of psychiatrists and other mental health professionals. Psychiatr Ann. 2000;30(8):525-532.
24. Curlin FA, Lantos JD, Roach CJ, et al. Religious characteristics of U.S. physicians: a national survey. Gen Intern Med. 2005;20(7):629-634.
25. Curlin FA, Odell SV, Lawrence RE, et al. The relationship between psychiatry and religion among U.S. physicians. Psychiatr Serv. 2007;58(9):1193-1198.
26. Kelly JF, Hoeppner B, Stout RL, et al. Determining the relative importance of the mechanisms of behavior change within Alcoholics Anonymous: a multiple mediator analysis. Addiction. 2012;107(2):289-299.
27. Nowinski J. Self-help groups for addictions. In: McCrady BS, Epstein EE, eds. Addictions. New York, NY: Oxford University Press; 1999:328-346.
28. SMART recovery: self-management and recovery training. http://www.smartrecovery.org. Accessed January 9, 2014.
29. Alcoholics Anonymous World Services. The AA member—medication & other drugs. 2nd ed. New York, NY: Alcoholics Anonymous World Services; 2011.
Mr. W, age 45, is a divorced Army veteran living on the street who has entered alcohol treatment for the sixth time. He has never stayed sober for longer than 1 month after each of his previous treatment episodes.
On a typical day, Mr. W drinks 40 oz of beer, a pint of vodka, and other alcoholic beverages when available. Although he has used drugs, he reports that he did so only to augment the effects of alcohol. Before entering rehab, Mr. W worked for a television network for 16 years and was promoted to associate vice president. He lost that job as a result of drinking.
Mr. W comes from a large Irish Catholic family and has sustained an active religious faith, going to Mass 2 or 3 times a month. Throughout the interview, he appears introspective and describes frequent periods of “going inside” of himself to “rehash” things. He states that he has never been satisfied with his spiritual life and has been unable to “quiet the hunger inside.”
He describes his benders as “mini-retreats” and comments that focusing on the condensation droplets on a glass of beer is nearly a “sacramental experience” for him. He states: “My drinking is a spiritual thing for me. I believe that every time I drink I am on a spiritual search. I believe this with all my heart. I have this emptiness inside of me and alcohol would temporarily fill the enormous hole in my insides. Just for a short period of time, I would feel at peace and connected to others, and maybe even to God.”
Mr. W observes that “Every time I relapse it’s because I’m going through a spiritual withdrawal. Booze filled the void inside of me and now the void is back again. Physically and psychologically I’m fine. I’m just empty inside and when I can’t stand it any longer, I drink again.”
Few patients can so directly articulate the role they feel that spirituality plays in their substance use disorder. It is important for clinicians to be aware of the dynamics of spirituality and religion in the cause, maintenance, and treatment of substance misuse problems.
In this article, we discuss how spirituality can be assessed and suggest ethical and clinical practice concerns that we believe may support treatment of substance use disorder. We do not advocate incorporating spiritual interventions into clinical practice for patients who are uncomfortable with doing so, nor do we feel that consideration of and respect for patients’ spirituality precludes evidence-based pharmaceutical and behavioral treatment strategies. We believe, however, that addressing these issues can enhance treatment adherence in select patients.
Defining spirituality
Although religion and spirituality are related concepts, they differ.
- Religion has been defined as “an organized system of beliefs, practices, rituals, and symbols through which ones’ relationship to God or others is nurtured and exercised.”1
- Spirituality is more complex and multifaceted. Reflecting his extensive review of articles on spirituality and addiction, Cook2 proposed the following definition:
Spirituality is a distinctive, potentially creative, and universal dimension of
human experience arising within inner subjective awareness of individuals and within communities, social groups, and traditions. It may be experienced as relationship with that which is intimately “inner,” immanent, and personal, within the self and others, and/or as relationship with that which is wholly “other,” transcendent and beyond the self. It is experienced as being of fundamental or ultimate importance and is therefore concerned with matters of meaning and purpose in life, truth, and values.
In some religions, any use of alcohol or drugs is forbidden; in most religions, abuse of these substances violates norms. Those who misuse a substance also might be alienated from their religious and social support community. People struggling with addiction might feel they are compromising their spiritual values directly through the action itself and indirectly because of the harm their substance misuse causes to close friends and family. Misuse of substances also might be an attempt to “fill the void,” as Mr. W described it, of a spiritual longing or a consequence of doubts about meaning, purpose in life, and God.
Perhaps it isn’t surprising that, among psychiatric disorders, substance abuse problems seem to be most associated with spiritual intervention—especially Alcoholics Anonymous (AA), Narcotics Anonymous (NA), and Cocaine Anonymous (CA). Many of the 12 steps contain references to God and spirituality. Although much of the evidence supporting the effectiveness of the spiritual components of AA/NA/CA is correlational,3,4 the resonance that many persons who are recovering from substance abuse find in the 12-step model suggests that spiritual issues are relevant to understanding patients’ viewpoints and for planning treatment.
Religion and spirituality in risk and recovery
Although alcohol and drugs can have positive religious significance (eg, wine in the Christian Eucharist or in the Hindu practice of Ayurvedic medicine; peyote in some indigenous American religious rituals), substance use and abuse are less prevalent among persons who identify themselves as highly religious or spiritual.5 Studies indicate that alcohol use and abuse are less prevalent among persons who identify as Jewish, Muslim, or conservative Protestant, compared with those who are Catholic or liberal Protestant.6
Research suggests that consideration and accommodation of religious and spiritual practices in the recovery process is effective7-9—and preferred—by many patients.10 Psychiatrists do not need to and, in many cases, should not deliver overtly spiritual treatments to patients recovering from substance abuse. Spirituality and religion are complex issues largely outside the expertise of psychiatry; it would be naïve to consider spirituality and religion as universally beneficial elements for all patients recovering from substance abuse. Instead, psychiatrists should be equipped to:
- assess for religion and spirituality in patients
- be aware of, and supportive of, resources for integrating spirituality into treatment (eg, clergy and hospital chaplains, local AA/NA/CA groups, community religious organizations, spirituality groups).
Assessment
Post and colleagues11 note: “When patients feel that their spiritual needs are neglected in standard clinical environments, many of them may be driven away from effective medical treatment.” This is of particular concern when working with persons who have a substance use disorder—among whom, regrettably, only a minority avail themselves of professional care. You should carefully gauge the importance of these dimensions in how patients understand their disorder and the components of treatment they think their recovery process should involve.
Asking about the religious and spiritual aspects of patients’ lives shows respect for their views and facilitates a therapeutic alliance by recognizing their autonomy in treatment. Pargament and Lomax12 state: “Religion speaks to highly sensitive issues that lie at the core of the individual’s identity, commitments, values and world view. Patients are unlikely to engage in a conversation about the deepest side of themselves unless their psychiatrist demonstrates an openness to, interest in, and appreciation of the patient’s religiousness.” This exploration process can suggest natural environmental support systems available to complement recovery efforts and can indicate whether consultation with a clergyperson knowledgeable about and sensitive to their particular religious tradition is appropriate.
Assessment of spirituality and religious practice usually should occur during the initial clinical interview. Examples of revealing interview questions are listed in Box 1.
The Table13-20 lists well-researched psychometric measures that can provide clues to aspects of your patient’s spirituality. These scales yield quantitative results, yet it may be more useful to review with the patient his (her) responses to the items and to pursue issues further based on the responses.
In discussing responses to questions about their spirituality and religious practices, some patients might ask about your views and whether you agree with their views. You could respond with a direct, concise answer, but refocusing the discussion on why the topic of spirituality is important to understanding one’s life and choices might be more therapeutic. This also might be a good time to remind patients that the treatment plan should reflect their view of the role of religion and spirituality in their life.
Of course, some patients are disinclined to discuss spirituality or religion, or prefer that it not be considered in treatment. This is clearly a matter of patient choice, but be aware that the patient may change his (her) mind as the recovery process continues and that the topic can be revisited if desired.
Recommendations for practice
Give careful consideration to ethical and clinical practice issues related to spiritual components in the recovery process. Plante21 and Meador and Koenig1 addressed several relevant ethical principles in considering spirituality in, respectively, psychological and psychiatric practice. Delaney and co-workers22 also presented similar principles, specifically within the context of substance use treatment. These can be summarized as a series of recommendations:
- Although psychiatrists as a group typically have a lower rate of conventional spirituality and religious practice than many of their patients,23-25 it is important not to ignore the patient’s perspective and show respect for the patient’s spiritual needs. However, if the psychiatrist has strong personal religious beliefs, he (she) must carefully guard against proselytizing or exerting undue, unintentional influence.
- Given the variety of religious traditions in the United States, understanding their particular features, as with other issues of cultural diversity, requires competence and sensitivity. Your work requires study and consultation with knowledgeable peers and experts in various faith traditions. Clergy and clinically trained chaplains, in particular, can conduct more comprehensive spiritual assessments that can yield additional treatment-relevant information.
- Maintaining professional boundaries is critical when dealing with religious and spiritual aspects of the patient’s life and thinking. Keep in mind issues of transference and the authoritative influence of the psychiatrist. The patient should understand the difference between services offered by a mental health professional and those given by a spiritual counselor or member of the clergy. Spiritual and religious issues, such as addressing concerns over guilt and sin and a relationship with God, should be referred to an appropriate clergyperson.
- Spiritual interventions introduced into the therapeutic process always should derive from the patient’s perspective and value system; they should not be imposed from an external source.
- Referral to AA often will help alcohol-dependent patients. The heart of AA’s philosophy is that addiction should be seen as a spiritual problem and that genuine recovery requires a profound spiritual awakening (Box 2). AA, as well as interventions inspired by it (eg, NA), are based on peer support, are readily available, and free. Although there is a dearth of controlled research demonstrating the efficacy of AA compared with other interventions, many recovering alcoholics credit these 12-step programs with their having maintained sobriety and adopting a positive lifestyle.
Recent research has identified specific components of AA participation that seem to be helpful.26 These include activities that are spiritual in nature and other generally active components of substance abuse care. Patient preference should be respected when encouraging AA involvement. For patients who are uncomfortable with AA—especially with its emphasis on spirituality—alternative peer support groups are available, such as SMART Recovery.27,28 If the patient adopts AA’s philosophy, it might be helpful for you to employ the language of AA and its constructs when talking with him (her).
Useful strategies on how therapists can encourage AA participation and integrate mutual help groups into treatment planning are described by Nowinski.25 Some AA members believe that use of medication is antithetical to the recovery process, but this is not the position of AA29; using FDA-approved medications, such as naltrexone and acamprosate, is evidence-based and often should be a part of the treatment regimen for alcohol dependence.
Bottom Line
Awareness of, and sensitivity to, the religious and spiritual characteristics of patients with substance use disorder can enhance clinical rapport, inform development of individualized treatment plans, and suggest strategies, such as professional consultation, that might increase the prospects for successful treatment.
Related Resources
- Galanter M. The concept of spirituality in relation to addiction recovery and general psychiatry. Recent Dev Alcohol. 2008;18:125-140.
- Monod S, Brennan M, Rochat E, et al. Instruments measuring spirituality in clinical research: a systematic review. J Gen Intern Med. 2011;26(11):1345-1357.
Drug Brand Names
Acamprosate • Campral
Naltrexone • ReVia, Vivitrol
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Featured Audio
John P. Allen, PhD, MPA, discusses whether spirituality plays a different role in treating substance use disorder than it might in treating other psychiatric illnesses. Dr. Allen works at the Department of Veterans Affairs, Mid-Atlantic Mental Illness Research, Education and Clinical Center, Division of Addictions Research and Treatment, Department of Psychiatry and Behavioral Sciences, Duke University School of Medicine, Durham, North Carolina.
Mr. W, age 45, is a divorced Army veteran living on the street who has entered alcohol treatment for the sixth time. He has never stayed sober for longer than 1 month after each of his previous treatment episodes.
On a typical day, Mr. W drinks 40 oz of beer, a pint of vodka, and other alcoholic beverages when available. Although he has used drugs, he reports that he did so only to augment the effects of alcohol. Before entering rehab, Mr. W worked for a television network for 16 years and was promoted to associate vice president. He lost that job as a result of drinking.
Mr. W comes from a large Irish Catholic family and has sustained an active religious faith, going to Mass 2 or 3 times a month. Throughout the interview, he appears introspective and describes frequent periods of “going inside” of himself to “rehash” things. He states that he has never been satisfied with his spiritual life and has been unable to “quiet the hunger inside.”
He describes his benders as “mini-retreats” and comments that focusing on the condensation droplets on a glass of beer is nearly a “sacramental experience” for him. He states: “My drinking is a spiritual thing for me. I believe that every time I drink I am on a spiritual search. I believe this with all my heart. I have this emptiness inside of me and alcohol would temporarily fill the enormous hole in my insides. Just for a short period of time, I would feel at peace and connected to others, and maybe even to God.”
Mr. W observes that “Every time I relapse it’s because I’m going through a spiritual withdrawal. Booze filled the void inside of me and now the void is back again. Physically and psychologically I’m fine. I’m just empty inside and when I can’t stand it any longer, I drink again.”
Few patients can so directly articulate the role they feel that spirituality plays in their substance use disorder. It is important for clinicians to be aware of the dynamics of spirituality and religion in the cause, maintenance, and treatment of substance misuse problems.
In this article, we discuss how spirituality can be assessed and suggest ethical and clinical practice concerns that we believe may support treatment of substance use disorder. We do not advocate incorporating spiritual interventions into clinical practice for patients who are uncomfortable with doing so, nor do we feel that consideration of and respect for patients’ spirituality precludes evidence-based pharmaceutical and behavioral treatment strategies. We believe, however, that addressing these issues can enhance treatment adherence in select patients.
Defining spirituality
Although religion and spirituality are related concepts, they differ.
- Religion has been defined as “an organized system of beliefs, practices, rituals, and symbols through which ones’ relationship to God or others is nurtured and exercised.”1
- Spirituality is more complex and multifaceted. Reflecting his extensive review of articles on spirituality and addiction, Cook2 proposed the following definition:
Spirituality is a distinctive, potentially creative, and universal dimension of
human experience arising within inner subjective awareness of individuals and within communities, social groups, and traditions. It may be experienced as relationship with that which is intimately “inner,” immanent, and personal, within the self and others, and/or as relationship with that which is wholly “other,” transcendent and beyond the self. It is experienced as being of fundamental or ultimate importance and is therefore concerned with matters of meaning and purpose in life, truth, and values.
In some religions, any use of alcohol or drugs is forbidden; in most religions, abuse of these substances violates norms. Those who misuse a substance also might be alienated from their religious and social support community. People struggling with addiction might feel they are compromising their spiritual values directly through the action itself and indirectly because of the harm their substance misuse causes to close friends and family. Misuse of substances also might be an attempt to “fill the void,” as Mr. W described it, of a spiritual longing or a consequence of doubts about meaning, purpose in life, and God.
Perhaps it isn’t surprising that, among psychiatric disorders, substance abuse problems seem to be most associated with spiritual intervention—especially Alcoholics Anonymous (AA), Narcotics Anonymous (NA), and Cocaine Anonymous (CA). Many of the 12 steps contain references to God and spirituality. Although much of the evidence supporting the effectiveness of the spiritual components of AA/NA/CA is correlational,3,4 the resonance that many persons who are recovering from substance abuse find in the 12-step model suggests that spiritual issues are relevant to understanding patients’ viewpoints and for planning treatment.
Religion and spirituality in risk and recovery
Although alcohol and drugs can have positive religious significance (eg, wine in the Christian Eucharist or in the Hindu practice of Ayurvedic medicine; peyote in some indigenous American religious rituals), substance use and abuse are less prevalent among persons who identify themselves as highly religious or spiritual.5 Studies indicate that alcohol use and abuse are less prevalent among persons who identify as Jewish, Muslim, or conservative Protestant, compared with those who are Catholic or liberal Protestant.6
Research suggests that consideration and accommodation of religious and spiritual practices in the recovery process is effective7-9—and preferred—by many patients.10 Psychiatrists do not need to and, in many cases, should not deliver overtly spiritual treatments to patients recovering from substance abuse. Spirituality and religion are complex issues largely outside the expertise of psychiatry; it would be naïve to consider spirituality and religion as universally beneficial elements for all patients recovering from substance abuse. Instead, psychiatrists should be equipped to:
- assess for religion and spirituality in patients
- be aware of, and supportive of, resources for integrating spirituality into treatment (eg, clergy and hospital chaplains, local AA/NA/CA groups, community religious organizations, spirituality groups).
Assessment
Post and colleagues11 note: “When patients feel that their spiritual needs are neglected in standard clinical environments, many of them may be driven away from effective medical treatment.” This is of particular concern when working with persons who have a substance use disorder—among whom, regrettably, only a minority avail themselves of professional care. You should carefully gauge the importance of these dimensions in how patients understand their disorder and the components of treatment they think their recovery process should involve.
Asking about the religious and spiritual aspects of patients’ lives shows respect for their views and facilitates a therapeutic alliance by recognizing their autonomy in treatment. Pargament and Lomax12 state: “Religion speaks to highly sensitive issues that lie at the core of the individual’s identity, commitments, values and world view. Patients are unlikely to engage in a conversation about the deepest side of themselves unless their psychiatrist demonstrates an openness to, interest in, and appreciation of the patient’s religiousness.” This exploration process can suggest natural environmental support systems available to complement recovery efforts and can indicate whether consultation with a clergyperson knowledgeable about and sensitive to their particular religious tradition is appropriate.
Assessment of spirituality and religious practice usually should occur during the initial clinical interview. Examples of revealing interview questions are listed in Box 1.
The Table13-20 lists well-researched psychometric measures that can provide clues to aspects of your patient’s spirituality. These scales yield quantitative results, yet it may be more useful to review with the patient his (her) responses to the items and to pursue issues further based on the responses.
In discussing responses to questions about their spirituality and religious practices, some patients might ask about your views and whether you agree with their views. You could respond with a direct, concise answer, but refocusing the discussion on why the topic of spirituality is important to understanding one’s life and choices might be more therapeutic. This also might be a good time to remind patients that the treatment plan should reflect their view of the role of religion and spirituality in their life.
Of course, some patients are disinclined to discuss spirituality or religion, or prefer that it not be considered in treatment. This is clearly a matter of patient choice, but be aware that the patient may change his (her) mind as the recovery process continues and that the topic can be revisited if desired.
Recommendations for practice
Give careful consideration to ethical and clinical practice issues related to spiritual components in the recovery process. Plante21 and Meador and Koenig1 addressed several relevant ethical principles in considering spirituality in, respectively, psychological and psychiatric practice. Delaney and co-workers22 also presented similar principles, specifically within the context of substance use treatment. These can be summarized as a series of recommendations:
- Although psychiatrists as a group typically have a lower rate of conventional spirituality and religious practice than many of their patients,23-25 it is important not to ignore the patient’s perspective and show respect for the patient’s spiritual needs. However, if the psychiatrist has strong personal religious beliefs, he (she) must carefully guard against proselytizing or exerting undue, unintentional influence.
- Given the variety of religious traditions in the United States, understanding their particular features, as with other issues of cultural diversity, requires competence and sensitivity. Your work requires study and consultation with knowledgeable peers and experts in various faith traditions. Clergy and clinically trained chaplains, in particular, can conduct more comprehensive spiritual assessments that can yield additional treatment-relevant information.
- Maintaining professional boundaries is critical when dealing with religious and spiritual aspects of the patient’s life and thinking. Keep in mind issues of transference and the authoritative influence of the psychiatrist. The patient should understand the difference between services offered by a mental health professional and those given by a spiritual counselor or member of the clergy. Spiritual and religious issues, such as addressing concerns over guilt and sin and a relationship with God, should be referred to an appropriate clergyperson.
- Spiritual interventions introduced into the therapeutic process always should derive from the patient’s perspective and value system; they should not be imposed from an external source.
- Referral to AA often will help alcohol-dependent patients. The heart of AA’s philosophy is that addiction should be seen as a spiritual problem and that genuine recovery requires a profound spiritual awakening (Box 2). AA, as well as interventions inspired by it (eg, NA), are based on peer support, are readily available, and free. Although there is a dearth of controlled research demonstrating the efficacy of AA compared with other interventions, many recovering alcoholics credit these 12-step programs with their having maintained sobriety and adopting a positive lifestyle.
Recent research has identified specific components of AA participation that seem to be helpful.26 These include activities that are spiritual in nature and other generally active components of substance abuse care. Patient preference should be respected when encouraging AA involvement. For patients who are uncomfortable with AA—especially with its emphasis on spirituality—alternative peer support groups are available, such as SMART Recovery.27,28 If the patient adopts AA’s philosophy, it might be helpful for you to employ the language of AA and its constructs when talking with him (her).
Useful strategies on how therapists can encourage AA participation and integrate mutual help groups into treatment planning are described by Nowinski.25 Some AA members believe that use of medication is antithetical to the recovery process, but this is not the position of AA29; using FDA-approved medications, such as naltrexone and acamprosate, is evidence-based and often should be a part of the treatment regimen for alcohol dependence.
Bottom Line
Awareness of, and sensitivity to, the religious and spiritual characteristics of patients with substance use disorder can enhance clinical rapport, inform development of individualized treatment plans, and suggest strategies, such as professional consultation, that might increase the prospects for successful treatment.
Related Resources
- Galanter M. The concept of spirituality in relation to addiction recovery and general psychiatry. Recent Dev Alcohol. 2008;18:125-140.
- Monod S, Brennan M, Rochat E, et al. Instruments measuring spirituality in clinical research: a systematic review. J Gen Intern Med. 2011;26(11):1345-1357.
Drug Brand Names
Acamprosate • Campral
Naltrexone • ReVia, Vivitrol
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Featured Audio
John P. Allen, PhD, MPA, discusses whether spirituality plays a different role in treating substance use disorder than it might in treating other psychiatric illnesses. Dr. Allen works at the Department of Veterans Affairs, Mid-Atlantic Mental Illness Research, Education and Clinical Center, Division of Addictions Research and Treatment, Department of Psychiatry and Behavioral Sciences, Duke University School of Medicine, Durham, North Carolina.
1. Meador KG, Koenig HG. Spirituality and religion in psychiatry practice: parameters and implications. Psychiatr Ann. 2000;30(8):549-555.
2. Cook CC. Addiction and spirituality. Addiction. 2004;99(5):539-551.
3. Kelly J, Stout RL, Magill M, et al. Spirituality in recovery: a lagged mediational analysis of alcoholics anonymous’ principal theoretical mechanism of behavior change. Alcohol Clin Exp Res. 2011;35(3):454-463.
4. Zemore SE. A role for spiritual change in the benefits of 12-step involvement. Alcohol Clin Exp Res. 2007;31(10 suppl):76s-79s.
5. Kendler KS, Liu XQ, Gardner CO, et al. Dimensions of religiosity and their relationship to lifetime psychiatric and substance use disorders. Am J Psychiatry. 2003;160(3):496-503.
6. Koenig HG, King D, Carson VB. Handbook of religion and health, 2nd ed. New York, NY: Oxford University Press; 2012.
7. Carter TM. The effects of spiritual practices on recovery from substance abuse. J Psychiatr Ment Health Nurs. 1998; 5(5):409-413.
8. Conner BT, Anglin MD, Annon J, et al. Effect of religiosity and spirituality on drug treatment outcomes. J Behav Health Serv Res. 2009;36(2):189-198.
9. Robinson EA, Krentzman AR, Webb JR, et al. Six-month changes in spirituality and religiousness in alcoholics predict drinking outcomes at nine months. J Stud Alcohol Drugs. 2011;72(4):660-668.
10. Heinz AJ, Disney ER, Epstein DH, et al. A focus-group study on spirituality and substance-user treatment. Subst Use Misuse. 2010;45(1-2):134-153.
11. Post SG, Puchalski CM, Larson DB. Physicians and patient spirituality: professional boundaries, competency, and ethics. Ann Intern Med. 2000;132(7):578-583.
12. Pargament KI, Lomax JW. Understanding and addressing religion among people with mental illness. World Psychiatry. 2013;12(1):26-32.
13. Koenig HG, Büssing A. The Duke University Religion Index (DUREL): a five-item measure for use in epidemiological studies. Religions. 2010;1(1):78-85.
14. Brief Multidimensional Measure of Religiousness/Spirituality: 1999. http://www.primarycarecore.org/PDF/137.pdf. Accessed January 9, 2014.
15. Johnstone B, Yoon DP, Franklin KL, et al. Re-conceptualizing the factor structure of the Brief Multidimensional Measure of Religiousness/Spirituality. J Relig Health. 2009;48(2):146-163.
16. Galanter M, Dermatis H, Bunt G, et al. Assessment of spirituality and its relevance to addiction treatment. J Subst Abuse Treat. 2007;33(3):257-264.
17. Pargament K, Feuille M, Burdzy D. The Brief RCOPE: current psychometric status of a short measure of religious coping. Religions. 2011;2(1):51-76.
18. Afterdeployment.org. Spirituality assessment. http://www.afterdeployment.org/sites/default/files/pdfs/assessment-tools/spirituality-assessment.pdf. Accessed January 9, 2014.
19. Connors GJ, Tonigan JS, Miller WR. A measure of religious background and behavior for use in behavior change research. Psychol Addict Behav. 1996;10(2):90-96.
20. Daaleman TP, Frey BB. The Spirituality Index of Well-Being: a new Instrument for health-related quality-of-life research. Ann Fam Med. 2004;2(5):499-503.
21. Plante TG. Integrating spirituality and psychotherapy: ethical issues and principles to consider. J Clin Psychol. 2007;63(9):891-902.
22. Delaney HD, Forcehimes AA, Campbell WP, et al. Integrating spirituality into alcohol treatment. J Clin Psychol. 2009;65(2):185-198.
23. Shafranske EP. Religious involvement and professional practices of psychiatrists and other mental health professionals. Psychiatr Ann. 2000;30(8):525-532.
24. Curlin FA, Lantos JD, Roach CJ, et al. Religious characteristics of U.S. physicians: a national survey. Gen Intern Med. 2005;20(7):629-634.
25. Curlin FA, Odell SV, Lawrence RE, et al. The relationship between psychiatry and religion among U.S. physicians. Psychiatr Serv. 2007;58(9):1193-1198.
26. Kelly JF, Hoeppner B, Stout RL, et al. Determining the relative importance of the mechanisms of behavior change within Alcoholics Anonymous: a multiple mediator analysis. Addiction. 2012;107(2):289-299.
27. Nowinski J. Self-help groups for addictions. In: McCrady BS, Epstein EE, eds. Addictions. New York, NY: Oxford University Press; 1999:328-346.
28. SMART recovery: self-management and recovery training. http://www.smartrecovery.org. Accessed January 9, 2014.
29. Alcoholics Anonymous World Services. The AA member—medication & other drugs. 2nd ed. New York, NY: Alcoholics Anonymous World Services; 2011.
1. Meador KG, Koenig HG. Spirituality and religion in psychiatry practice: parameters and implications. Psychiatr Ann. 2000;30(8):549-555.
2. Cook CC. Addiction and spirituality. Addiction. 2004;99(5):539-551.
3. Kelly J, Stout RL, Magill M, et al. Spirituality in recovery: a lagged mediational analysis of alcoholics anonymous’ principal theoretical mechanism of behavior change. Alcohol Clin Exp Res. 2011;35(3):454-463.
4. Zemore SE. A role for spiritual change in the benefits of 12-step involvement. Alcohol Clin Exp Res. 2007;31(10 suppl):76s-79s.
5. Kendler KS, Liu XQ, Gardner CO, et al. Dimensions of religiosity and their relationship to lifetime psychiatric and substance use disorders. Am J Psychiatry. 2003;160(3):496-503.
6. Koenig HG, King D, Carson VB. Handbook of religion and health, 2nd ed. New York, NY: Oxford University Press; 2012.
7. Carter TM. The effects of spiritual practices on recovery from substance abuse. J Psychiatr Ment Health Nurs. 1998; 5(5):409-413.
8. Conner BT, Anglin MD, Annon J, et al. Effect of religiosity and spirituality on drug treatment outcomes. J Behav Health Serv Res. 2009;36(2):189-198.
9. Robinson EA, Krentzman AR, Webb JR, et al. Six-month changes in spirituality and religiousness in alcoholics predict drinking outcomes at nine months. J Stud Alcohol Drugs. 2011;72(4):660-668.
10. Heinz AJ, Disney ER, Epstein DH, et al. A focus-group study on spirituality and substance-user treatment. Subst Use Misuse. 2010;45(1-2):134-153.
11. Post SG, Puchalski CM, Larson DB. Physicians and patient spirituality: professional boundaries, competency, and ethics. Ann Intern Med. 2000;132(7):578-583.
12. Pargament KI, Lomax JW. Understanding and addressing religion among people with mental illness. World Psychiatry. 2013;12(1):26-32.
13. Koenig HG, Büssing A. The Duke University Religion Index (DUREL): a five-item measure for use in epidemiological studies. Religions. 2010;1(1):78-85.
14. Brief Multidimensional Measure of Religiousness/Spirituality: 1999. http://www.primarycarecore.org/PDF/137.pdf. Accessed January 9, 2014.
15. Johnstone B, Yoon DP, Franklin KL, et al. Re-conceptualizing the factor structure of the Brief Multidimensional Measure of Religiousness/Spirituality. J Relig Health. 2009;48(2):146-163.
16. Galanter M, Dermatis H, Bunt G, et al. Assessment of spirituality and its relevance to addiction treatment. J Subst Abuse Treat. 2007;33(3):257-264.
17. Pargament K, Feuille M, Burdzy D. The Brief RCOPE: current psychometric status of a short measure of religious coping. Religions. 2011;2(1):51-76.
18. Afterdeployment.org. Spirituality assessment. http://www.afterdeployment.org/sites/default/files/pdfs/assessment-tools/spirituality-assessment.pdf. Accessed January 9, 2014.
19. Connors GJ, Tonigan JS, Miller WR. A measure of religious background and behavior for use in behavior change research. Psychol Addict Behav. 1996;10(2):90-96.
20. Daaleman TP, Frey BB. The Spirituality Index of Well-Being: a new Instrument for health-related quality-of-life research. Ann Fam Med. 2004;2(5):499-503.
21. Plante TG. Integrating spirituality and psychotherapy: ethical issues and principles to consider. J Clin Psychol. 2007;63(9):891-902.
22. Delaney HD, Forcehimes AA, Campbell WP, et al. Integrating spirituality into alcohol treatment. J Clin Psychol. 2009;65(2):185-198.
23. Shafranske EP. Religious involvement and professional practices of psychiatrists and other mental health professionals. Psychiatr Ann. 2000;30(8):525-532.
24. Curlin FA, Lantos JD, Roach CJ, et al. Religious characteristics of U.S. physicians: a national survey. Gen Intern Med. 2005;20(7):629-634.
25. Curlin FA, Odell SV, Lawrence RE, et al. The relationship between psychiatry and religion among U.S. physicians. Psychiatr Serv. 2007;58(9):1193-1198.
26. Kelly JF, Hoeppner B, Stout RL, et al. Determining the relative importance of the mechanisms of behavior change within Alcoholics Anonymous: a multiple mediator analysis. Addiction. 2012;107(2):289-299.
27. Nowinski J. Self-help groups for addictions. In: McCrady BS, Epstein EE, eds. Addictions. New York, NY: Oxford University Press; 1999:328-346.
28. SMART recovery: self-management and recovery training. http://www.smartrecovery.org. Accessed January 9, 2014.
29. Alcoholics Anonymous World Services. The AA member—medication & other drugs. 2nd ed. New York, NY: Alcoholics Anonymous World Services; 2011.

Psychiatry’s future shock
It might seem business as usual in clinical psychiatry, but major transformative changes in the scientific foundation of the specialty are taking place. The “neuroscientification” of psychiatry, ongoing for more than 3 decades, is now approaching a tipping point: The specialty is on the verge of an unprecedented denouement of the old tenets and assumptions. Just as smartphones have made a 25-volume encyclopedia set obsolete, coming changes in scientific psychiatry will be fully disruptive to your father’s practice of psychiatry.
Many psychiatrists still practice like it’s 1999
That situation will change, soon—as surely as medieval times gave way to the Renaissance. Psychiatry of the future will be drastically different once new models of objective diagnostic tests and physiologically specific interventions emerge from fast-moving discoveries of the molecular biology of the mind and its pathologies.
Most psychiatric practitioners do not regularly read neuroscience journals that describe the accelerating progress in molecular psychiatry, where the text is replete with an alphabet soup of terminology that one day will permeate the medical practice of the new psychiatry.
Fuzzy ambiguities will be cleared away
There are many reasons to be optimistic that transcendent scientific transformations will sweep away the fuzzy biologic, diagnostic, and therapeutic ambiguities that have plagued psychiatry for so long—plagued us because of the herculean challenges of investigating the divinely complex brain and its gloriously enigmatic mind. New methods and tools for exploration and paradigmatic shifts in conceptualizing the etiopathogenesis of psychiatric brain disorders are rapidly leading to a discarding of many simplistic, even primitive, notions that have guided psychiatry over the past century. Psychopharmacological breakthroughs of the past 50 years, which, admittedly, have yet to cure or eliminate disabilities associated with major psychiatric disorders, are only a prologue to the coming revolution in neuropsychiatry, in which prevention, not just intervention, will change everything. Curing deteriorative brain disorders will be a reality once that revolution in neuroscience enters its propitious translational phase.
The Table presents a sampling of scientific progress that is setting the stage for disruptive technologies and probes that will lead to far more advanced diagnosis, prevention, and treatment of neuropsychiatric diseases.
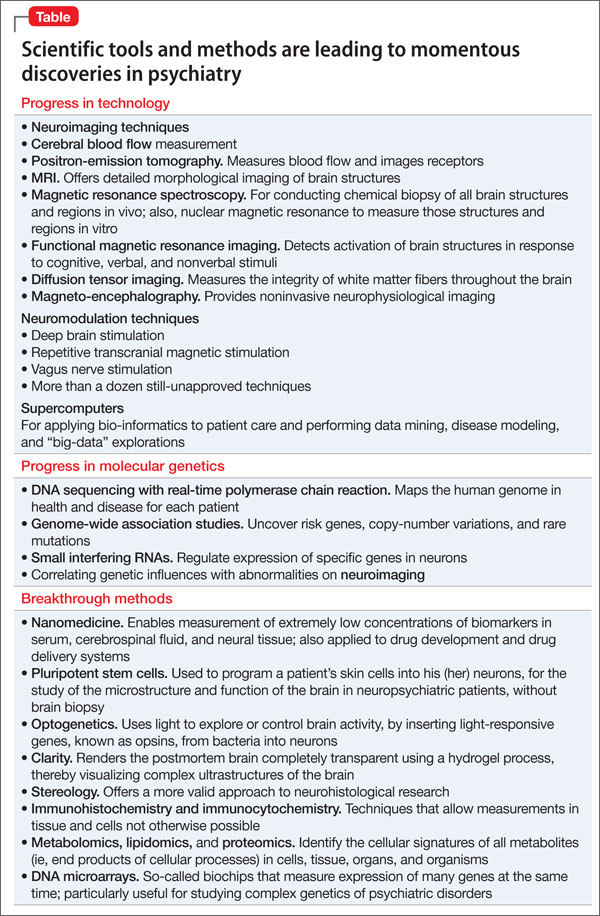
Prepare for psychiatry’s future shock!
We psychiatrists must keep up, regularly reading the latest literature to learn about the latest advances and to make sure we are familiar with the emerging language of psychiatric neuroscience. Instead of remaining fondly attached to ancient constructs such as id, ego, superego, and defense mechanisms, we should be thinking about the default mode network, seeking to understand the connectome, the envirome, the metabolome, and the proteome; microglial activation, inflammatory markers, IL-6, TNF alpha, oxidative and nitrosative stress, and physiologic vs pathologic apoptosis; BDNF, FGF, VEGF, MIF, GFAP, and S100B; neuroplasticity and dendritic spines; and genes such as CLOCK, NOTCH3, and Met-to-Val mutations—and so on.
Those of us who do not adapt to swift transition of knowledge might suffer the fate of clinical dinosaurs, as the massive asteroid of neuroscientific advances smashes into the placid landscape of psychiatry. As Alvin Toffler, author of Future Shock, proposed, the illiterates of the future will not be the people who cannot read or write. They will be the ones who fail to learn.
It might seem business as usual in clinical psychiatry, but major transformative changes in the scientific foundation of the specialty are taking place. The “neuroscientification” of psychiatry, ongoing for more than 3 decades, is now approaching a tipping point: The specialty is on the verge of an unprecedented denouement of the old tenets and assumptions. Just as smartphones have made a 25-volume encyclopedia set obsolete, coming changes in scientific psychiatry will be fully disruptive to your father’s practice of psychiatry.
Many psychiatrists still practice like it’s 1999
That situation will change, soon—as surely as medieval times gave way to the Renaissance. Psychiatry of the future will be drastically different once new models of objective diagnostic tests and physiologically specific interventions emerge from fast-moving discoveries of the molecular biology of the mind and its pathologies.
Most psychiatric practitioners do not regularly read neuroscience journals that describe the accelerating progress in molecular psychiatry, where the text is replete with an alphabet soup of terminology that one day will permeate the medical practice of the new psychiatry.
Fuzzy ambiguities will be cleared away
There are many reasons to be optimistic that transcendent scientific transformations will sweep away the fuzzy biologic, diagnostic, and therapeutic ambiguities that have plagued psychiatry for so long—plagued us because of the herculean challenges of investigating the divinely complex brain and its gloriously enigmatic mind. New methods and tools for exploration and paradigmatic shifts in conceptualizing the etiopathogenesis of psychiatric brain disorders are rapidly leading to a discarding of many simplistic, even primitive, notions that have guided psychiatry over the past century. Psychopharmacological breakthroughs of the past 50 years, which, admittedly, have yet to cure or eliminate disabilities associated with major psychiatric disorders, are only a prologue to the coming revolution in neuropsychiatry, in which prevention, not just intervention, will change everything. Curing deteriorative brain disorders will be a reality once that revolution in neuroscience enters its propitious translational phase.
The Table presents a sampling of scientific progress that is setting the stage for disruptive technologies and probes that will lead to far more advanced diagnosis, prevention, and treatment of neuropsychiatric diseases.
Prepare for psychiatry’s future shock!
We psychiatrists must keep up, regularly reading the latest literature to learn about the latest advances and to make sure we are familiar with the emerging language of psychiatric neuroscience. Instead of remaining fondly attached to ancient constructs such as id, ego, superego, and defense mechanisms, we should be thinking about the default mode network, seeking to understand the connectome, the envirome, the metabolome, and the proteome; microglial activation, inflammatory markers, IL-6, TNF alpha, oxidative and nitrosative stress, and physiologic vs pathologic apoptosis; BDNF, FGF, VEGF, MIF, GFAP, and S100B; neuroplasticity and dendritic spines; and genes such as CLOCK, NOTCH3, and Met-to-Val mutations—and so on.
Those of us who do not adapt to swift transition of knowledge might suffer the fate of clinical dinosaurs, as the massive asteroid of neuroscientific advances smashes into the placid landscape of psychiatry. As Alvin Toffler, author of Future Shock, proposed, the illiterates of the future will not be the people who cannot read or write. They will be the ones who fail to learn.
It might seem business as usual in clinical psychiatry, but major transformative changes in the scientific foundation of the specialty are taking place. The “neuroscientification” of psychiatry, ongoing for more than 3 decades, is now approaching a tipping point: The specialty is on the verge of an unprecedented denouement of the old tenets and assumptions. Just as smartphones have made a 25-volume encyclopedia set obsolete, coming changes in scientific psychiatry will be fully disruptive to your father’s practice of psychiatry.
Many psychiatrists still practice like it’s 1999
That situation will change, soon—as surely as medieval times gave way to the Renaissance. Psychiatry of the future will be drastically different once new models of objective diagnostic tests and physiologically specific interventions emerge from fast-moving discoveries of the molecular biology of the mind and its pathologies.
Most psychiatric practitioners do not regularly read neuroscience journals that describe the accelerating progress in molecular psychiatry, where the text is replete with an alphabet soup of terminology that one day will permeate the medical practice of the new psychiatry.
Fuzzy ambiguities will be cleared away
There are many reasons to be optimistic that transcendent scientific transformations will sweep away the fuzzy biologic, diagnostic, and therapeutic ambiguities that have plagued psychiatry for so long—plagued us because of the herculean challenges of investigating the divinely complex brain and its gloriously enigmatic mind. New methods and tools for exploration and paradigmatic shifts in conceptualizing the etiopathogenesis of psychiatric brain disorders are rapidly leading to a discarding of many simplistic, even primitive, notions that have guided psychiatry over the past century. Psychopharmacological breakthroughs of the past 50 years, which, admittedly, have yet to cure or eliminate disabilities associated with major psychiatric disorders, are only a prologue to the coming revolution in neuropsychiatry, in which prevention, not just intervention, will change everything. Curing deteriorative brain disorders will be a reality once that revolution in neuroscience enters its propitious translational phase.
The Table presents a sampling of scientific progress that is setting the stage for disruptive technologies and probes that will lead to far more advanced diagnosis, prevention, and treatment of neuropsychiatric diseases.
Prepare for psychiatry’s future shock!
We psychiatrists must keep up, regularly reading the latest literature to learn about the latest advances and to make sure we are familiar with the emerging language of psychiatric neuroscience. Instead of remaining fondly attached to ancient constructs such as id, ego, superego, and defense mechanisms, we should be thinking about the default mode network, seeking to understand the connectome, the envirome, the metabolome, and the proteome; microglial activation, inflammatory markers, IL-6, TNF alpha, oxidative and nitrosative stress, and physiologic vs pathologic apoptosis; BDNF, FGF, VEGF, MIF, GFAP, and S100B; neuroplasticity and dendritic spines; and genes such as CLOCK, NOTCH3, and Met-to-Val mutations—and so on.
Those of us who do not adapt to swift transition of knowledge might suffer the fate of clinical dinosaurs, as the massive asteroid of neuroscientific advances smashes into the placid landscape of psychiatry. As Alvin Toffler, author of Future Shock, proposed, the illiterates of the future will not be the people who cannot read or write. They will be the ones who fail to learn.
Apply your psychiatric skills to managing rheumatoid arthritis
Joint disease is the most common cause of disability and the source of considerable psychological distress. In the United States, 50 million adults complain of joint pain; in 2007, 1.5 million people suffered from rheumatoid arthritis (RA). A chronic inflammatory autoimmune disease of joints, RA can involve almost all organs.1
The link to mental illness
Mental illness in RA patients often is underdiagnosed and undertreated. These missed opportunities contribute to poor compliance with medical therapy, suboptimal therapeutic response, greater disability, and diminished quality of life.2
Limited mobility, chronic pain, sleep disturbance, fatigue, and immunological factors predispose RA patients to depression and anxiety.3 The proinflammatory cytokines, tumor necrosis factor-α (TNF-α), interleukin 1 (IL-1), IL-6, and interferon-g have a role in inducing affective symptoms. There also is a relationship between an elevated IL-17 level and anxiety.
Research substantiates a relationship between RA and depression.3 The prevalence of affective illness is approximately 6% among the general population, and 13% to 30% among RA patients.4 In arthritic populations, 52% exhibit depression and anxiety; joint discomfort contributes to insomnia in 25% to 42% of cases.4
Arthritic pain persists despite suppressed inflammation, which suggests involvement of the CNS.5 Increased levels of IL-6 and TNF-α can cause insomnia and affect pain perception.6 Decreased conditioned pain modulation, a lower pain threshold, and pressure pain intolerance lead to increased pain awareness and heightened discomfort.
How can you help your patient who has RA?
Because the focus of care in RA is on the disease’s physical attributes, psychiatric symptoms sometimes receive less attention.7 And because arthritic symptoms overlap with anorexia, weight loss, fatigue, pain, and insomnia, affective illness can go unrecognized.
Depression rating scales can overestimate affective illness, but a history and follow-up questionnaire can facilitate an accurate diagnosis of depression and help determine the need for, and type of, intervention.
Selective serotonin reuptake inhibitors (SSRIs) are considered first‐line treatment of depression associated with RA.7 Although SSRIs for RA can be administered to the maximum recommended dosage, titration is advised in accordance with patient response and tolerance.
Tricyclic antidepressants are not as well tolerated in RA, especially in older patients; however, they have more of an analgesic effect, even at lower dosages.
Joint disease activity and mood are associated with sleep disturbance, and vice versa.5 Insomnia calls for patient education about sleep hygiene, avoiding caffeine and other stimulants, and an individualized appraisal of options for pharmacotherapy.
Alleviating RA pain is important for psychosocial health.8 Although the medical team’s emphasis should be on controlling inflammation to minimize joint damage and pain, be sure to address your RA patients’ mood symptoms to improve the quality of their life.
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Centers for Disease Control and Prevention. Arthritis-related statistics. http://www.cdc.gov/arthritis/data_statistics/arthritis_related_stats.htm. Updated August 1, 2011. Accessed January 4, 2013.
2. Shih M, Hootman JM, Strine TW, et al. Serious psychological distress in U.S. adults with arthritis. J Gen Intern Med. 2006;21(11):1160-1166.
3. Sato E, Nishimura K, Nakajima A, et al. Major depressive disorder in patients with rheumatoid arthritis. Mod Rheumatol. 2013;23(2):237-244.
4. Wolfe F, Michaud K, Li T. Sleep disturbance in patients with rheumatoid arthritis: evaluation by medical outcomes study and visual analog sleep scales. J Rheumatol. 2006;33(10):1942-1951.
5. Fragiadaki K, Tektonidou MG, Konsta M, et al. Sleep disturbances and interleukin 6 receptor inhibition in rheumatoid arthritis. J Rheumatol. 2012;39(1):60-62.
6. Lee YC, Lu B, Edwards RR, et al. The role of sleep problems in central pain processing in rheumatoid arthritis. Arthritis Rheum. 2013;65(1):59-68.
7. Dickens C, Creed F. The burden of depression in patients with rheumatoid arthritis. Rheumatology (Oxford). 2001; 40(12):1327-1330.
8. Courvoisier DS, Agoritsas T, Glauser J, et al. Pain as an important predictor of psychosocial health in patients with rheumatoid arthritis. Arthritis Care Res (Hoboken). 2012;64(2):190-196.
Joint disease is the most common cause of disability and the source of considerable psychological distress. In the United States, 50 million adults complain of joint pain; in 2007, 1.5 million people suffered from rheumatoid arthritis (RA). A chronic inflammatory autoimmune disease of joints, RA can involve almost all organs.1
The link to mental illness
Mental illness in RA patients often is underdiagnosed and undertreated. These missed opportunities contribute to poor compliance with medical therapy, suboptimal therapeutic response, greater disability, and diminished quality of life.2
Limited mobility, chronic pain, sleep disturbance, fatigue, and immunological factors predispose RA patients to depression and anxiety.3 The proinflammatory cytokines, tumor necrosis factor-α (TNF-α), interleukin 1 (IL-1), IL-6, and interferon-g have a role in inducing affective symptoms. There also is a relationship between an elevated IL-17 level and anxiety.
Research substantiates a relationship between RA and depression.3 The prevalence of affective illness is approximately 6% among the general population, and 13% to 30% among RA patients.4 In arthritic populations, 52% exhibit depression and anxiety; joint discomfort contributes to insomnia in 25% to 42% of cases.4
Arthritic pain persists despite suppressed inflammation, which suggests involvement of the CNS.5 Increased levels of IL-6 and TNF-α can cause insomnia and affect pain perception.6 Decreased conditioned pain modulation, a lower pain threshold, and pressure pain intolerance lead to increased pain awareness and heightened discomfort.
How can you help your patient who has RA?
Because the focus of care in RA is on the disease’s physical attributes, psychiatric symptoms sometimes receive less attention.7 And because arthritic symptoms overlap with anorexia, weight loss, fatigue, pain, and insomnia, affective illness can go unrecognized.
Depression rating scales can overestimate affective illness, but a history and follow-up questionnaire can facilitate an accurate diagnosis of depression and help determine the need for, and type of, intervention.
Selective serotonin reuptake inhibitors (SSRIs) are considered first‐line treatment of depression associated with RA.7 Although SSRIs for RA can be administered to the maximum recommended dosage, titration is advised in accordance with patient response and tolerance.
Tricyclic antidepressants are not as well tolerated in RA, especially in older patients; however, they have more of an analgesic effect, even at lower dosages.
Joint disease activity and mood are associated with sleep disturbance, and vice versa.5 Insomnia calls for patient education about sleep hygiene, avoiding caffeine and other stimulants, and an individualized appraisal of options for pharmacotherapy.
Alleviating RA pain is important for psychosocial health.8 Although the medical team’s emphasis should be on controlling inflammation to minimize joint damage and pain, be sure to address your RA patients’ mood symptoms to improve the quality of their life.
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Joint disease is the most common cause of disability and the source of considerable psychological distress. In the United States, 50 million adults complain of joint pain; in 2007, 1.5 million people suffered from rheumatoid arthritis (RA). A chronic inflammatory autoimmune disease of joints, RA can involve almost all organs.1
The link to mental illness
Mental illness in RA patients often is underdiagnosed and undertreated. These missed opportunities contribute to poor compliance with medical therapy, suboptimal therapeutic response, greater disability, and diminished quality of life.2
Limited mobility, chronic pain, sleep disturbance, fatigue, and immunological factors predispose RA patients to depression and anxiety.3 The proinflammatory cytokines, tumor necrosis factor-α (TNF-α), interleukin 1 (IL-1), IL-6, and interferon-g have a role in inducing affective symptoms. There also is a relationship between an elevated IL-17 level and anxiety.
Research substantiates a relationship between RA and depression.3 The prevalence of affective illness is approximately 6% among the general population, and 13% to 30% among RA patients.4 In arthritic populations, 52% exhibit depression and anxiety; joint discomfort contributes to insomnia in 25% to 42% of cases.4
Arthritic pain persists despite suppressed inflammation, which suggests involvement of the CNS.5 Increased levels of IL-6 and TNF-α can cause insomnia and affect pain perception.6 Decreased conditioned pain modulation, a lower pain threshold, and pressure pain intolerance lead to increased pain awareness and heightened discomfort.
How can you help your patient who has RA?
Because the focus of care in RA is on the disease’s physical attributes, psychiatric symptoms sometimes receive less attention.7 And because arthritic symptoms overlap with anorexia, weight loss, fatigue, pain, and insomnia, affective illness can go unrecognized.
Depression rating scales can overestimate affective illness, but a history and follow-up questionnaire can facilitate an accurate diagnosis of depression and help determine the need for, and type of, intervention.
Selective serotonin reuptake inhibitors (SSRIs) are considered first‐line treatment of depression associated with RA.7 Although SSRIs for RA can be administered to the maximum recommended dosage, titration is advised in accordance with patient response and tolerance.
Tricyclic antidepressants are not as well tolerated in RA, especially in older patients; however, they have more of an analgesic effect, even at lower dosages.
Joint disease activity and mood are associated with sleep disturbance, and vice versa.5 Insomnia calls for patient education about sleep hygiene, avoiding caffeine and other stimulants, and an individualized appraisal of options for pharmacotherapy.
Alleviating RA pain is important for psychosocial health.8 Although the medical team’s emphasis should be on controlling inflammation to minimize joint damage and pain, be sure to address your RA patients’ mood symptoms to improve the quality of their life.
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Centers for Disease Control and Prevention. Arthritis-related statistics. http://www.cdc.gov/arthritis/data_statistics/arthritis_related_stats.htm. Updated August 1, 2011. Accessed January 4, 2013.
2. Shih M, Hootman JM, Strine TW, et al. Serious psychological distress in U.S. adults with arthritis. J Gen Intern Med. 2006;21(11):1160-1166.
3. Sato E, Nishimura K, Nakajima A, et al. Major depressive disorder in patients with rheumatoid arthritis. Mod Rheumatol. 2013;23(2):237-244.
4. Wolfe F, Michaud K, Li T. Sleep disturbance in patients with rheumatoid arthritis: evaluation by medical outcomes study and visual analog sleep scales. J Rheumatol. 2006;33(10):1942-1951.
5. Fragiadaki K, Tektonidou MG, Konsta M, et al. Sleep disturbances and interleukin 6 receptor inhibition in rheumatoid arthritis. J Rheumatol. 2012;39(1):60-62.
6. Lee YC, Lu B, Edwards RR, et al. The role of sleep problems in central pain processing in rheumatoid arthritis. Arthritis Rheum. 2013;65(1):59-68.
7. Dickens C, Creed F. The burden of depression in patients with rheumatoid arthritis. Rheumatology (Oxford). 2001; 40(12):1327-1330.
8. Courvoisier DS, Agoritsas T, Glauser J, et al. Pain as an important predictor of psychosocial health in patients with rheumatoid arthritis. Arthritis Care Res (Hoboken). 2012;64(2):190-196.
1. Centers for Disease Control and Prevention. Arthritis-related statistics. http://www.cdc.gov/arthritis/data_statistics/arthritis_related_stats.htm. Updated August 1, 2011. Accessed January 4, 2013.
2. Shih M, Hootman JM, Strine TW, et al. Serious psychological distress in U.S. adults with arthritis. J Gen Intern Med. 2006;21(11):1160-1166.
3. Sato E, Nishimura K, Nakajima A, et al. Major depressive disorder in patients with rheumatoid arthritis. Mod Rheumatol. 2013;23(2):237-244.
4. Wolfe F, Michaud K, Li T. Sleep disturbance in patients with rheumatoid arthritis: evaluation by medical outcomes study and visual analog sleep scales. J Rheumatol. 2006;33(10):1942-1951.
5. Fragiadaki K, Tektonidou MG, Konsta M, et al. Sleep disturbances and interleukin 6 receptor inhibition in rheumatoid arthritis. J Rheumatol. 2012;39(1):60-62.
6. Lee YC, Lu B, Edwards RR, et al. The role of sleep problems in central pain processing in rheumatoid arthritis. Arthritis Rheum. 2013;65(1):59-68.
7. Dickens C, Creed F. The burden of depression in patients with rheumatoid arthritis. Rheumatology (Oxford). 2001; 40(12):1327-1330.
8. Courvoisier DS, Agoritsas T, Glauser J, et al. Pain as an important predictor of psychosocial health in patients with rheumatoid arthritis. Arthritis Care Res (Hoboken). 2012;64(2):190-196.
Spirituality and substance abuse

Making a practical case for marrying psychiatry and neurology
Historically and recently, leaders within psychiatry have expressed disdain over the public’s misunderstanding of the specialty.1 There are many factors—cultural and sociopolitical influences, for example—that contribute to a generalized suspicion of the intent and the abilities of psychiatry. Few observers, however, have focused on how a lack of cohesion within the discipline might be an important, underappreciated influence in the misconceptions and mistrust.
One way to view the recent publication of the DSM-5 is as further positive application of evidence-based medicine and an indicator of the flexible, progressive adaptability of psychiatry. Indeed, Gawande has demonstrated the benefit of implementing a high degree of standardization in terms of maximizing economic efficiency and minimizing medical error.2
Yet critics of psychiatry use the DSM-5 to substantiate their claim that the field is still murky and unsure of itself. Major changes in classification and diagnostic criteria might support a Szaszian fallacy that we somehow create mental illness and simply fit individuals into the framework at our whim. In the midst of what is, at best, lateral movement in psychiatry, the extremism of critics of the specialty, such as Peter Breggin, might gain undeserved credence. Furthermore, the merits of these critics’ arguments remain largely unchallenged in the public arena.
It is worth noting 2 additional factors within psychiatry that contribute to its stagnation:
- Knowledge and practice are grossly misaligned. What practitioners know and what they do are quite different, and the best way to treat mental illness often takes a back seat to tradition or convenience. Consider neuroimaging, which has illustrated structural and functional changes in the brain that have contributed to the phenomenology of schizophrenia. Schizophrenia is considered a clinical diagnosis, but the value of imaging in predicting prognosis, progression, response to treatment, etc. is well known. Yet neuroimaging is underutilized and the cost-benefit analysis of this modality remains unexplored. Likewise, cognitive testing, an important tool in the diagnosis and prognosis of schizophrenia, is not standard practice. These are good reasons why psychiatry shouldn’t shy from the push toward medicalization: Incorporating imaging and genetic analysis into practice will go a long way toward building legitimacy.
- Mental illness is stigmatized within. The stigma of mental illness that psychiatry must overcome is rooted in ignorance and misunderstanding. However, psychiatry itself has done little to eliminate the stigma of mental illness among its practitioners. This is apparent in the punitive, non-progressive nature of most state programs for impaired physicians.3 This type of “individual discrimination” described by Carl Hart4 undoubtedly permeates the residency match and ranking process, even in psychiatry. How can any headway be made in curbing societal intolerance of, say, addiction when it thrives in the academic environment?
A marriage that will dispel ignorance
In light of the continued undervaluation and ignorance of psychiatry, we can start by heeding the Buddhist teaching that change must come from within. To undertake change means to consolidate information and begin to change the inner workings, practices, and structure of the field itself. It means taking seriously the Research Domain Criteria outlined by Thomas Insel, MD, Director of the National Institute of Mental Health.5
It is increasingly apparent that psychiatry and neurology are inseparable.6 Why is there still reluctance to collaborate between the specialties? Why are these 2 fields’ research efforts still relatively distinct from one another, and not being built upon what is already known?
Based on current knowledge, sophisticated proponents of neuropsychiatry aren’t being unreasonable in their desire to push for an elevated status. If the field is to move in the most constructive direction, we should encourage a marriage—a fusion—of psychiatry and neurology. We shouldn’t be satisfied with connecting the specialties in theory and discussion; we should seek a structural unison of departments, journals, teaching, texts, research efforts, and fellowship options and accreditations.
Disclosure
Dr. Siragusa reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Nawkova L, Nawka A, Adamkova T, et al. The picture of mental health/illness in the printed media in three Central European countries. J Health Commun. 2012;17(1):22-40.
2. Gawande A. The checklist manifesto: how to get things right. New York, NY: Henry Holt and Company; 2009.
3. Chander K. Licensing boards and the stigma of mental illness. JAMA. 1999;281(7):606-607.
4. Hart C. High price: a neuroscientist’s journey of self-discovery that challenges everything you know about drugs and society. New York, NY: Harper Collins; 2013.
5. National Institute of Mental Health. Research Domain Criteria (RDoC). http://www.nimh.nih.gov/research-priorities/rdoc/index.shtml. Accessed December 18, 2013.
6. Nasrallah HA. Let’s tear down the silos and reunify psychiatry and neurology! Current Psychiatry. 2013;12(8):8-9.
Historically and recently, leaders within psychiatry have expressed disdain over the public’s misunderstanding of the specialty.1 There are many factors—cultural and sociopolitical influences, for example—that contribute to a generalized suspicion of the intent and the abilities of psychiatry. Few observers, however, have focused on how a lack of cohesion within the discipline might be an important, underappreciated influence in the misconceptions and mistrust.
One way to view the recent publication of the DSM-5 is as further positive application of evidence-based medicine and an indicator of the flexible, progressive adaptability of psychiatry. Indeed, Gawande has demonstrated the benefit of implementing a high degree of standardization in terms of maximizing economic efficiency and minimizing medical error.2
Yet critics of psychiatry use the DSM-5 to substantiate their claim that the field is still murky and unsure of itself. Major changes in classification and diagnostic criteria might support a Szaszian fallacy that we somehow create mental illness and simply fit individuals into the framework at our whim. In the midst of what is, at best, lateral movement in psychiatry, the extremism of critics of the specialty, such as Peter Breggin, might gain undeserved credence. Furthermore, the merits of these critics’ arguments remain largely unchallenged in the public arena.
It is worth noting 2 additional factors within psychiatry that contribute to its stagnation:
- Knowledge and practice are grossly misaligned. What practitioners know and what they do are quite different, and the best way to treat mental illness often takes a back seat to tradition or convenience. Consider neuroimaging, which has illustrated structural and functional changes in the brain that have contributed to the phenomenology of schizophrenia. Schizophrenia is considered a clinical diagnosis, but the value of imaging in predicting prognosis, progression, response to treatment, etc. is well known. Yet neuroimaging is underutilized and the cost-benefit analysis of this modality remains unexplored. Likewise, cognitive testing, an important tool in the diagnosis and prognosis of schizophrenia, is not standard practice. These are good reasons why psychiatry shouldn’t shy from the push toward medicalization: Incorporating imaging and genetic analysis into practice will go a long way toward building legitimacy.
- Mental illness is stigmatized within. The stigma of mental illness that psychiatry must overcome is rooted in ignorance and misunderstanding. However, psychiatry itself has done little to eliminate the stigma of mental illness among its practitioners. This is apparent in the punitive, non-progressive nature of most state programs for impaired physicians.3 This type of “individual discrimination” described by Carl Hart4 undoubtedly permeates the residency match and ranking process, even in psychiatry. How can any headway be made in curbing societal intolerance of, say, addiction when it thrives in the academic environment?
A marriage that will dispel ignorance
In light of the continued undervaluation and ignorance of psychiatry, we can start by heeding the Buddhist teaching that change must come from within. To undertake change means to consolidate information and begin to change the inner workings, practices, and structure of the field itself. It means taking seriously the Research Domain Criteria outlined by Thomas Insel, MD, Director of the National Institute of Mental Health.5
It is increasingly apparent that psychiatry and neurology are inseparable.6 Why is there still reluctance to collaborate between the specialties? Why are these 2 fields’ research efforts still relatively distinct from one another, and not being built upon what is already known?
Based on current knowledge, sophisticated proponents of neuropsychiatry aren’t being unreasonable in their desire to push for an elevated status. If the field is to move in the most constructive direction, we should encourage a marriage—a fusion—of psychiatry and neurology. We shouldn’t be satisfied with connecting the specialties in theory and discussion; we should seek a structural unison of departments, journals, teaching, texts, research efforts, and fellowship options and accreditations.
Disclosure
Dr. Siragusa reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Historically and recently, leaders within psychiatry have expressed disdain over the public’s misunderstanding of the specialty.1 There are many factors—cultural and sociopolitical influences, for example—that contribute to a generalized suspicion of the intent and the abilities of psychiatry. Few observers, however, have focused on how a lack of cohesion within the discipline might be an important, underappreciated influence in the misconceptions and mistrust.
One way to view the recent publication of the DSM-5 is as further positive application of evidence-based medicine and an indicator of the flexible, progressive adaptability of psychiatry. Indeed, Gawande has demonstrated the benefit of implementing a high degree of standardization in terms of maximizing economic efficiency and minimizing medical error.2
Yet critics of psychiatry use the DSM-5 to substantiate their claim that the field is still murky and unsure of itself. Major changes in classification and diagnostic criteria might support a Szaszian fallacy that we somehow create mental illness and simply fit individuals into the framework at our whim. In the midst of what is, at best, lateral movement in psychiatry, the extremism of critics of the specialty, such as Peter Breggin, might gain undeserved credence. Furthermore, the merits of these critics’ arguments remain largely unchallenged in the public arena.
It is worth noting 2 additional factors within psychiatry that contribute to its stagnation:
- Knowledge and practice are grossly misaligned. What practitioners know and what they do are quite different, and the best way to treat mental illness often takes a back seat to tradition or convenience. Consider neuroimaging, which has illustrated structural and functional changes in the brain that have contributed to the phenomenology of schizophrenia. Schizophrenia is considered a clinical diagnosis, but the value of imaging in predicting prognosis, progression, response to treatment, etc. is well known. Yet neuroimaging is underutilized and the cost-benefit analysis of this modality remains unexplored. Likewise, cognitive testing, an important tool in the diagnosis and prognosis of schizophrenia, is not standard practice. These are good reasons why psychiatry shouldn’t shy from the push toward medicalization: Incorporating imaging and genetic analysis into practice will go a long way toward building legitimacy.
- Mental illness is stigmatized within. The stigma of mental illness that psychiatry must overcome is rooted in ignorance and misunderstanding. However, psychiatry itself has done little to eliminate the stigma of mental illness among its practitioners. This is apparent in the punitive, non-progressive nature of most state programs for impaired physicians.3 This type of “individual discrimination” described by Carl Hart4 undoubtedly permeates the residency match and ranking process, even in psychiatry. How can any headway be made in curbing societal intolerance of, say, addiction when it thrives in the academic environment?
A marriage that will dispel ignorance
In light of the continued undervaluation and ignorance of psychiatry, we can start by heeding the Buddhist teaching that change must come from within. To undertake change means to consolidate information and begin to change the inner workings, practices, and structure of the field itself. It means taking seriously the Research Domain Criteria outlined by Thomas Insel, MD, Director of the National Institute of Mental Health.5
It is increasingly apparent that psychiatry and neurology are inseparable.6 Why is there still reluctance to collaborate between the specialties? Why are these 2 fields’ research efforts still relatively distinct from one another, and not being built upon what is already known?
Based on current knowledge, sophisticated proponents of neuropsychiatry aren’t being unreasonable in their desire to push for an elevated status. If the field is to move in the most constructive direction, we should encourage a marriage—a fusion—of psychiatry and neurology. We shouldn’t be satisfied with connecting the specialties in theory and discussion; we should seek a structural unison of departments, journals, teaching, texts, research efforts, and fellowship options and accreditations.
Disclosure
Dr. Siragusa reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Nawkova L, Nawka A, Adamkova T, et al. The picture of mental health/illness in the printed media in three Central European countries. J Health Commun. 2012;17(1):22-40.
2. Gawande A. The checklist manifesto: how to get things right. New York, NY: Henry Holt and Company; 2009.
3. Chander K. Licensing boards and the stigma of mental illness. JAMA. 1999;281(7):606-607.
4. Hart C. High price: a neuroscientist’s journey of self-discovery that challenges everything you know about drugs and society. New York, NY: Harper Collins; 2013.
5. National Institute of Mental Health. Research Domain Criteria (RDoC). http://www.nimh.nih.gov/research-priorities/rdoc/index.shtml. Accessed December 18, 2013.
6. Nasrallah HA. Let’s tear down the silos and reunify psychiatry and neurology! Current Psychiatry. 2013;12(8):8-9.
1. Nawkova L, Nawka A, Adamkova T, et al. The picture of mental health/illness in the printed media in three Central European countries. J Health Commun. 2012;17(1):22-40.
2. Gawande A. The checklist manifesto: how to get things right. New York, NY: Henry Holt and Company; 2009.
3. Chander K. Licensing boards and the stigma of mental illness. JAMA. 1999;281(7):606-607.
4. Hart C. High price: a neuroscientist’s journey of self-discovery that challenges everything you know about drugs and society. New York, NY: Harper Collins; 2013.
5. National Institute of Mental Health. Research Domain Criteria (RDoC). http://www.nimh.nih.gov/research-priorities/rdoc/index.shtml. Accessed December 18, 2013.
6. Nasrallah HA. Let’s tear down the silos and reunify psychiatry and neurology! Current Psychiatry. 2013;12(8):8-9.