User login
Welcome to Current Psychiatry, a leading source of information, online and in print, for practitioners of psychiatry and its related subspecialties, including addiction psychiatry, child and adolescent psychiatry, and geriatric psychiatry. This Web site contains evidence-based reviews of the prevention, diagnosis, and treatment of mental illness and psychological disorders; case reports; updates on psychopharmacology; news about the specialty of psychiatry; pearls for practice; and other topics of interest and use to this audience.
Dear Drupal User: You're seeing this because you're logged in to Drupal, and not redirected to MDedge.com/psychiatry.
Depression
adolescent depression
adolescent major depressive disorder
adolescent schizophrenia
adolescent with major depressive disorder
animals
autism
baby
brexpiprazole
child
child bipolar
child depression
child schizophrenia
children with bipolar disorder
children with depression
children with major depressive disorder
compulsive behaviors
cure
elderly bipolar
elderly depression
elderly major depressive disorder
elderly schizophrenia
elderly with dementia
first break
first episode
gambling
gaming
geriatric depression
geriatric major depressive disorder
geriatric schizophrenia
infant
kid
major depressive disorder
major depressive disorder in adolescents
major depressive disorder in children
parenting
pediatric
pediatric bipolar
pediatric depression
pediatric major depressive disorder
pediatric schizophrenia
pregnancy
pregnant
rexulti
skin care
teen
wine
section[contains(@class, 'nav-hidden')]
footer[@id='footer']
div[contains(@class, 'pane-pub-article-current-psychiatry')]
div[contains(@class, 'pane-pub-home-current-psychiatry')]
div[contains(@class, 'pane-pub-topic-current-psychiatry')]
div[contains(@class, 'panel-panel-inner')]
div[contains(@class, 'pane-node-field-article-topics')]
section[contains(@class, 'footer-nav-section-wrapper')]
Psychological testing in psychiatric practice
Drs. C. Don Morgan and John Bober addressed the important topic of psychological testing in psychiatric practice (“Psychological testing: Use do-it-yourself tools or refer?,” Current Psychiatry, June 2005).
I would like to add some points that were not emphasized in the article:
- Psychological testing should never be used alone to diagnose or rule out a psychiatric disorder.
- Psychological testing can help screen for occult psychiatric disorders and confirm psychiatric diagnoses.
- Some psychological tests, such as the Minnesota Multiphasic Personality Inventory (MMPI-2), are vulnerable to practice effects and require time between subsequent administrations for results to be valid.
- Psychological tests should only be given, scored, and interpreted by properly trained individuals.
Michael Menaster, MD
San Francisco, CA
I, too, would like to add the following in reference to the article by Drs. Morgan and Bober:
While this article refers to use of board-certified neuropsychologists, most psychologists who offer neuropsychological testing are not certified by a recognized credentialing organization. The current definition of a clinical neuropsychologist, published by the National Academy of Neuropsychology, does not require psychologists to be board-certified to offer neuropsychological testing. Practitioners, however, must meet specific criteria to be called a clinical neuropsychologist.
Forensic assessment, which Drs. Morgan and Bober mention in passing, is another important application of psychological tests. Numerous well-standardized screening instruments and in-depth measures can be used to assess criminal responsibility or adjudicative competence and to gauge other features.1
Few psychologists are competent to test patients across a broad age range. In general, children and adolescents should be referred to pediatric psychologists, and patients age >18 should see psychologists with expertise in adult testing. Many pediatric psychologists limit their practice to school-age children, so when referring a preschool-age child, look for practitioners who routinely test this age group.
Also, many psychologists who evaluate adults have little or no experience or training in testing elderly patients, so psychiatrists should seek clinicians who are well-versed in geriatric assessment.
Managed care companies usually authorize psychological testing when a known or suspected medical cause contributes to mental status change. However, authorization requests for other cases are sometimes denied, or the approved assessment period is limited. In these cases, the psychiatrist and testing psychologist must collaborate closely to provide clear rationales for the proposed assessment.
Jerrold Pollak, PhD
Coordinator, program in medical and forensic neuropsychology
Seacoast Mental Health Center, Portsmouth, NH
Drs. Morgan and Bober respond
While tests of cognitive functions such as memory, intelligence, and achievement are susceptible to practice effects, the MMPI-2 is so lengthy (567 items) that it would be difficult to remember how one responded to individual items. The test, however, is state-dependent, meaning that situational stressors can influence test results.
These points aside, we agree with Dr. Menaster’s comments. We also thank Dr. Pollak for his useful and important thoughts.
C. Don Morgan, PhD
Associate professor
John F. Bober, MD
Assistant professor and residency program director
Department of psychiatry and behavioral sciences
University of Kansas School of Medicine
Wichita
Drs. C. Don Morgan and John Bober addressed the important topic of psychological testing in psychiatric practice (“Psychological testing: Use do-it-yourself tools or refer?,” Current Psychiatry, June 2005).
I would like to add some points that were not emphasized in the article:
- Psychological testing should never be used alone to diagnose or rule out a psychiatric disorder.
- Psychological testing can help screen for occult psychiatric disorders and confirm psychiatric diagnoses.
- Some psychological tests, such as the Minnesota Multiphasic Personality Inventory (MMPI-2), are vulnerable to practice effects and require time between subsequent administrations for results to be valid.
- Psychological tests should only be given, scored, and interpreted by properly trained individuals.
Michael Menaster, MD
San Francisco, CA
I, too, would like to add the following in reference to the article by Drs. Morgan and Bober:
While this article refers to use of board-certified neuropsychologists, most psychologists who offer neuropsychological testing are not certified by a recognized credentialing organization. The current definition of a clinical neuropsychologist, published by the National Academy of Neuropsychology, does not require psychologists to be board-certified to offer neuropsychological testing. Practitioners, however, must meet specific criteria to be called a clinical neuropsychologist.
Forensic assessment, which Drs. Morgan and Bober mention in passing, is another important application of psychological tests. Numerous well-standardized screening instruments and in-depth measures can be used to assess criminal responsibility or adjudicative competence and to gauge other features.1
Few psychologists are competent to test patients across a broad age range. In general, children and adolescents should be referred to pediatric psychologists, and patients age >18 should see psychologists with expertise in adult testing. Many pediatric psychologists limit their practice to school-age children, so when referring a preschool-age child, look for practitioners who routinely test this age group.
Also, many psychologists who evaluate adults have little or no experience or training in testing elderly patients, so psychiatrists should seek clinicians who are well-versed in geriatric assessment.
Managed care companies usually authorize psychological testing when a known or suspected medical cause contributes to mental status change. However, authorization requests for other cases are sometimes denied, or the approved assessment period is limited. In these cases, the psychiatrist and testing psychologist must collaborate closely to provide clear rationales for the proposed assessment.
Jerrold Pollak, PhD
Coordinator, program in medical and forensic neuropsychology
Seacoast Mental Health Center, Portsmouth, NH
Drs. Morgan and Bober respond
While tests of cognitive functions such as memory, intelligence, and achievement are susceptible to practice effects, the MMPI-2 is so lengthy (567 items) that it would be difficult to remember how one responded to individual items. The test, however, is state-dependent, meaning that situational stressors can influence test results.
These points aside, we agree with Dr. Menaster’s comments. We also thank Dr. Pollak for his useful and important thoughts.
C. Don Morgan, PhD
Associate professor
John F. Bober, MD
Assistant professor and residency program director
Department of psychiatry and behavioral sciences
University of Kansas School of Medicine
Wichita
Drs. C. Don Morgan and John Bober addressed the important topic of psychological testing in psychiatric practice (“Psychological testing: Use do-it-yourself tools or refer?,” Current Psychiatry, June 2005).
I would like to add some points that were not emphasized in the article:
- Psychological testing should never be used alone to diagnose or rule out a psychiatric disorder.
- Psychological testing can help screen for occult psychiatric disorders and confirm psychiatric diagnoses.
- Some psychological tests, such as the Minnesota Multiphasic Personality Inventory (MMPI-2), are vulnerable to practice effects and require time between subsequent administrations for results to be valid.
- Psychological tests should only be given, scored, and interpreted by properly trained individuals.
Michael Menaster, MD
San Francisco, CA
I, too, would like to add the following in reference to the article by Drs. Morgan and Bober:
While this article refers to use of board-certified neuropsychologists, most psychologists who offer neuropsychological testing are not certified by a recognized credentialing organization. The current definition of a clinical neuropsychologist, published by the National Academy of Neuropsychology, does not require psychologists to be board-certified to offer neuropsychological testing. Practitioners, however, must meet specific criteria to be called a clinical neuropsychologist.
Forensic assessment, which Drs. Morgan and Bober mention in passing, is another important application of psychological tests. Numerous well-standardized screening instruments and in-depth measures can be used to assess criminal responsibility or adjudicative competence and to gauge other features.1
Few psychologists are competent to test patients across a broad age range. In general, children and adolescents should be referred to pediatric psychologists, and patients age >18 should see psychologists with expertise in adult testing. Many pediatric psychologists limit their practice to school-age children, so when referring a preschool-age child, look for practitioners who routinely test this age group.
Also, many psychologists who evaluate adults have little or no experience or training in testing elderly patients, so psychiatrists should seek clinicians who are well-versed in geriatric assessment.
Managed care companies usually authorize psychological testing when a known or suspected medical cause contributes to mental status change. However, authorization requests for other cases are sometimes denied, or the approved assessment period is limited. In these cases, the psychiatrist and testing psychologist must collaborate closely to provide clear rationales for the proposed assessment.
Jerrold Pollak, PhD
Coordinator, program in medical and forensic neuropsychology
Seacoast Mental Health Center, Portsmouth, NH
Drs. Morgan and Bober respond
While tests of cognitive functions such as memory, intelligence, and achievement are susceptible to practice effects, the MMPI-2 is so lengthy (567 items) that it would be difficult to remember how one responded to individual items. The test, however, is state-dependent, meaning that situational stressors can influence test results.
These points aside, we agree with Dr. Menaster’s comments. We also thank Dr. Pollak for his useful and important thoughts.
C. Don Morgan, PhD
Associate professor
John F. Bober, MD
Assistant professor and residency program director
Department of psychiatry and behavioral sciences
University of Kansas School of Medicine
Wichita
It’s August; why aren’t you on vacation?
A persistent myth about psychiatrists is that we all take vacations in August:
- Judith Rossner wrote August, a successful novel based on the premise that all the psychiatrists left town that month, leaving their patients to their own devices.
- Bill Murray and Richard Dreyfuss starred in the film What About Bob?, in which a patient was so distressed to be left alone that he followed his psychiatrist on his August vacation.
- John Katzenbach wrote The Analyst, in which a patient plots to kill his psychiatrist/analyst for going on vacation in August.
This August vacation theme seems “anti-psychiatry” to me. It suggests we belong to some cult (Freud took August vacations, so the rest of us do, too), or that we do not care about our patients (or at least fail to provide adequate cross-coverage).
One can test the hypothesis that psychiatrists—compared with the general population—are more likely to take August vacations. Using vacation data from my university department in Cincinnati, I find no evidence that psychiatrists take August vacations more than other physicians do. This small study awaits confirmation by larger-scale epidemiologic studies.
If you’re reading this issue of Current Psychiatry in August, you are probably not on vacation. If it’s any consolation, most of your colleagues (at least here in Cincinnati) are not on vacation, either.
![]()

James Randolph Hillard, MD
Editor-in-Chief
James Randolph Hillard, MD
Editor-in-Chief
James Randolph Hillard, MD
Editor-in-Chief
A persistent myth about psychiatrists is that we all take vacations in August:
- Judith Rossner wrote August, a successful novel based on the premise that all the psychiatrists left town that month, leaving their patients to their own devices.
- Bill Murray and Richard Dreyfuss starred in the film What About Bob?, in which a patient was so distressed to be left alone that he followed his psychiatrist on his August vacation.
- John Katzenbach wrote The Analyst, in which a patient plots to kill his psychiatrist/analyst for going on vacation in August.
This August vacation theme seems “anti-psychiatry” to me. It suggests we belong to some cult (Freud took August vacations, so the rest of us do, too), or that we do not care about our patients (or at least fail to provide adequate cross-coverage).
One can test the hypothesis that psychiatrists—compared with the general population—are more likely to take August vacations. Using vacation data from my university department in Cincinnati, I find no evidence that psychiatrists take August vacations more than other physicians do. This small study awaits confirmation by larger-scale epidemiologic studies.
If you’re reading this issue of Current Psychiatry in August, you are probably not on vacation. If it’s any consolation, most of your colleagues (at least here in Cincinnati) are not on vacation, either.
![]()
A persistent myth about psychiatrists is that we all take vacations in August:
- Judith Rossner wrote August, a successful novel based on the premise that all the psychiatrists left town that month, leaving their patients to their own devices.
- Bill Murray and Richard Dreyfuss starred in the film What About Bob?, in which a patient was so distressed to be left alone that he followed his psychiatrist on his August vacation.
- John Katzenbach wrote The Analyst, in which a patient plots to kill his psychiatrist/analyst for going on vacation in August.
This August vacation theme seems “anti-psychiatry” to me. It suggests we belong to some cult (Freud took August vacations, so the rest of us do, too), or that we do not care about our patients (or at least fail to provide adequate cross-coverage).
One can test the hypothesis that psychiatrists—compared with the general population—are more likely to take August vacations. Using vacation data from my university department in Cincinnati, I find no evidence that psychiatrists take August vacations more than other physicians do. This small study awaits confirmation by larger-scale epidemiologic studies.
If you’re reading this issue of Current Psychiatry in August, you are probably not on vacation. If it’s any consolation, most of your colleagues (at least here in Cincinnati) are not on vacation, either.
![]()
When depression treatment goes nowhere
History: losing his ‘drive’
Mr. D, age 49, has been treated for major depressive disorder for approximately 1 year but reports only occasional minor symptom improvement. At presentation, he had been irritable and lethargic for about 2 weeks and had increased appetite, decreased concentration, and trouble falling asleep at night.
A once-gregarious family man, Mr. D had become apathetic and too tired to enjoy socializing. He denied suicidal thoughts or feelings of worthlessness and hopelessness but feared his fatigue was interfering with his job as a truck driver. He tired after driving only a few hours.
Mr. D had been diagnosed with sleep apnea when he was younger but had no other medical history. He said his erratic work schedule kept him from using his continuous positive airway pressure (CPAP) machine regularly. He was taking no medications and had not seen a primary care physician for more than 2 years because of lack of coverage. He denied past or current substance abuse.
The patient weighed 280 lbs at intake. His body mass index (BMI) was 37.5, indicating clinical obesity.
Because Mr. D lacked health insurance, we enrolled him 1 year ago in a free depression study at a psychiatric outpatient clinic. At intake, he said numerous life stresses—particularly the recent death of his brother in a motor vehicle accident—had left him feeling depressed.
We started Mr. D on citalopram, 20 mg/d, which was the study protocol. Two weeks later, he complained of dry mouth and sedation with minimal symptom improvement. We stopped citalopram and started sertraline, 25 mg/d.
Two weeks later, Mr. D again complained he had “no energy” and was “sleeping all day.” We titrated sertraline to 200 mg/d over 2 months, but his excessive tiredness, increased appetite, and decreased motivation persisted. Mr. D needed routine laboratory tests, so we referred him to a local clinic that charges on a sliding scale. He did not complete the tests, however, for fear of incurring medical expenses.
We tried to improve Mr. D’s mood symptoms by adding lithium—225 mg/d titrated to 675 mg/d over 7 weeks—but his depression and fatigue kept worsening. We tapered him off lithium and sertraline and switched to the monoamine oxidase inhibitor tranylcypromine, 30 mg/d, which was also part of the study protocol. We warned him not to eat pizza, fermented dry sausages, or other foods that could interact adversely with tranylcypromine. After 4 weeks, Mr. D stopped taking the agent, saying he could not follow the dietary restrictions while on the road.
We released Mr. D from the study because of nonresponse. Bupropion, started at 100 mg bid and titrated to 300 mg each morning and 150 mg nightly across 5 months, did not resolve his fatigue. He also started having agitation and “anger problems,” often getting into shouting matches over his CBradio with other truck drivers. We started quetiapine, 25 mg bid, hoping the low dose would calm his mood.
Until now, Mr. D has ignored our requests to undergo routine laboratory testing. We referred him to the local clinic four times over the past year but he has not complied, citing lack of health insurance and financial concerns.
The authors’ observations
Although Mr. D’s symptoms (constantly depressed mood, loss of interest in usual activities) clearly suggest treatment-resistant major depressive disorder, an underlying medical disorder cannot be ruled out, yet he refuses to get needed tests.
Medical comorbidities are more prevalent in patients with mental illness than in the general population.1 As many as 43% of patients referred to some psychiatry clinics have medical disorders, and almost one-half the diagnoses were missed by the referring physician.2
Compared to patients without psychiatric diagnoses, those with mental illness have more difficulty gaining access to medical care and are less likely to receive and follow guidelines for preventive care. Mental illness symptoms often compromise one’s ability to seek health care or follow a doctor’s orders. For example, a psychotic person may be overly suspicious of doctors, whereas someone with anxiety may seek care inappropriately.3,4 Also, some studies estimate that 1 in 5 persons with mental illness are uninsured.1,5,6
Mr. D denies substance abuse, but primary care and behavioral health clinicians often miss substance use disorders.7 Accuracy of substance abuse self-reports varies widely; some studies report high accuracy, whereas almost 33% of patients in other studies do not disclose substance abuse.8
Testing: stimulating findings
At his next visit, Mr. D reports worsening thirst and increased urination and complains of increased appetite, easy bruising, excessive sleepiness, and apathy. He also reveals that for 2 months he has been taking 2 to 3 fat-burning stimulant capsules a day to stay awake while driving.
Alarmed by his elevated blood pressure (177/99 mm Hg) and worsening physical symptoms, Mr. D finally consents to baseline laboratory testing. Blood glucose is 306 mg/dL (normal 70 to 110 mg/dL), and glycosylated hemoglobin is 12% (normal
Mr. D, who now weighs 270 lbs, is diagnosed as having hypertension and type 2 diabetes mellitus. Clinic doctors start him on metformin, 500 mg bid titrated to 1,000 mg bid, and glyburide, 5 mg/d, to control his glucose, and lisinopril, 10 mg/d, to control his hypertension, reduce cardiovascular risk, and preserve renal function. Clinicians also order Mr. D to follow an 1,800-calorie, American Diabetes Association-approved diet. We stop quetiapine and bupropion.
Mr. D’s diabetes and hypertension diagnosis, combined with his habitus and history of easy bruising, suggest Cushing’s syndrome. Doctors rule out this disorder based on a 24-hour free cortisol reading of 59 mg/L and normal dexamethasone suppression. Lab findings suggest he is not taking stimulants away from work.
The authors’ observations
Ideally, Mr. D should have undergone laboratory testing after the initial intake visit, before psychotropics were started. Routine vital signs also should have been taken.
Symptoms of major depressive disorder and early type 2 diabetes are strikingly similar (Table 1). For example, early diabetes symptoms such as fatigue can mimic depression or other medical problems. In one study of 69 diabetic patients who were referred by their primary care doctors to a psychiatric clinic, 57 had not been diagnosed as having diabetes before referral.9
Aside from its medical complications, diabetes also doubles the risk of comorbid depression, which can alter diabetes’ course and outcome.10
Earlier laboratory testing could have uncovered Mr. D’s comorbid stimulant abuse, which also can mimic depression and complicate its treatment.11 Signs of amphetamine withdrawal—such as dysphoric mood, fatigue, insomnia or hypersomnia, increased appetite, and psychomotor retardation—can be mistaken for depression (Table 1).
Patients with Cushing’s syndrome may present with nonspecific complaints of fatigue, decreased energy, apathy, depressed mood, and hypersomnia. A 24-hour free cortisol reading and dexamethasone suppression testing can differentiate Cushing’s syndrome from depression.
Costly, unnecessary care. Missing a medical cause of apparent psychiatric symptoms can lead to unnecessary treatment and needless expense. A complete metabolic profile and urine drug screen—approximately $60—could have saved the nearly $5,000 spent on treating Mr. D’s “resistant” depression ( Table 2).
Psychiatrists need to watch for potential medical problems and for cormorbidities associated with mental illness. Patients with frequent mental distress—defined as ≥ 14 mentally unhealthy days within 30 days—were found to be more likely to smoke, drink heavily, and be physically inactive and obese than were mentally healthy persons. Mentally distressed patients also were more likely to lack health care coverage and to engage in multiple adverse behaviors, increasing their risk for mental and physical illness.12
Ensuring proper medical care. Based on our experience with Mr. D, routine vital signs—including BMI, weight, blood pressure, and pulse rate—should be recorded at each visit. At intake, we recommend that psychiatrists:
- find out when the patient last saw a primary health provider other than in the emergency room, and whether the patient is receiving preventive medical care
- assess for unhealthy lifestyle habits (smoking, drug use, poor diet) or family history of serious medical illnesses.
Educate patients about the interplay between physical and mental illness to help them understand the importance of seeing a primary care doctor. Finally, be familiar with local indigent health clinics and their fee scales.
Table 1
Medical symptoms that mimic depression
| Symptom | Amphetamine withdrawal | Cushing’s syndrome | Diabetes |
|---|---|---|---|
| Anxiety | × | ||
| Dysphoric mood | × | ||
| Fatigue | × | × | × |
| Hypersomnia | × | ||
| Increased appetite | × | × | |
| Insomnia | × | ||
| Irritability | × | × | |
| Muscle aches and cramps | × | ||
| Psychomotor retardation | × | ||
| Vivid, unpleasant dreams | × | ||
| Weakness | × | ||
| Weight gain or loss | × |
The cost of treating Mr. D’s ‘resistant depression’
| Medication/dosage | Start date | Stop date | Approximate cost |
|---|---|---|---|
| Citalopram, 20 mg/d | 10/3/03 | 10/24/03 | $58.50 |
| Sertraline, 25 to 200 mg/d | 10/24/03 | 12/24/03 | $283.00 |
| Sertraline 150 mg/d, with lithium, 225 to 675 mg/d | 12/24/03 | 2/6/04 | $343.00 |
| Tranylcypromine, 10 mg each morning, 20 mg at bedtime | 2/27/04 | 4/20/04 | $322.00 |
| Bupropion (sustained release) up to 450 mg/d | 5/7/04 | 8/30/04 | $372.00 |
| Bupropion (sustained release), 450 mg/d, plus quetiapine, 25 mg/d | 8/30/04 | 11/8/04 | $554.00 |
| Total cost of psychotropics | $1,932.50 | ||
| Total cost of office visits ($95 X 30 visits) | $2,850.00 | ||
| TOTAL COST OF TREATMENT | $4,782.50 | ||
| Source: Walgreens Co. retail prices in Wichita, KS | |||
Follow-up: 30 lbs in 4 months
Mr. D has lost >30 lbs over 4 months, and his blood pressure and serum glucose are normal. BMI is now 32, in the lower range of clinical obesity. He feels more energetic and active, no longer reports excessive sedation and apathy, and has stopped taking stimulants. His depressive symptoms have remitted.
Related resources
- WrongDiagnosis.com. Information on differential diagnosis of medical and psychiatric problems. www.wrongdiagnosis.com.
- Mauksch LB, Tucker SM, Katon WJ, et al. Mental illness, functional impairment, and patient preferences for collaborative care in an uninsured, primary care population. J Fam Pract 2001;50:41-7.
- Glied S, Little SE. The uninsured and the benefits of medical progress. Health Aff (Millwood) 2003;22:210-9.
- Bupropion • Wellbutrin
- Citalopram • Celexa
- Dexamethasone • Ciprodex, others
- Glucophage • Metformin
- Glyburide • DiaBeta, others
- Lisinopril • Prinivil, Zestril
- Lithium • Eskalith, others
- Quetiapine • Seroquel
- Sertraline • Zoloft
- Tranylcypromine •Parnate
Dr. Khan is a speaker for Wyeth Pharmaceuticals.
Dr. Grimsley reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1 McAlpine DD, Mechanic D. Utilization of specialty mental health care among persons with severe mental illness: the roles of demographics, need, insurance, and risk. Health Serv Res 2000;35(1 Pt 2):277-92.
2 Rosse RB, Deutsch LH, Deutsch SI. Medical assessment and laboratory testing in psychiatry. In: Sadock BJ, Sadock VA (eds). Kaplan & Sadock’ s comprehensive textbook of psychiatry (7th ed), Vol 1. Baltimore: Lippincott Williams & Wilkins; 2000:732.
3 Rubin AS, Littenberg B, Ross R, et al. Effects on processes and costs of care associated with the addition of an internist to an inpatient psychiatry team. Psychiatr Serv 2005;56:463-7.
4 Salsberry PJ, Chipps E, Kennedy C. Use of general medical services among Medicaid patients with severe and persistent mental illness. Psychiatr Serv 2005;56:458-62.
5 McAlpine DD, Mechanic D. Datapoints: payer source for emergency room visits by persons with psychiatric disorders. Psychiatr Serv 2002;53:14.-
6 Yanos PT, Lu W, Minsky S, Kiely GL. Correlates of health insurance among persons with schizophrenia in a statewide behavioral health care system. Psychiatr Serv 2004;55:79-82.
7 Brown GS, Hermann R, Jones E, Wu J. Using self-report to improve substance abuse risk assessment in behavioral health care. Jt Comm J Qual Saf 2004;30:448-54.
8 Tassiopoulos K, Bernstein J, Heeren T, et al. Hair testing and self-report of cocaine use by heroin users. Addiction 2004;99:590-7.
9 Katon WJ, Lin EH, Russo J, et al. Cardiac risk factors in patients with diabetes mellitus and major depression. J Gen Intern Med 2004;19:1192-9.
10 Lustman PJ, Clouse RE. Depression in diabetic patients: the relationship between mood and glycemic control. J Diabetes Complications 2005;19:113-22.
11 Mallin R, Slott K, Tumblin M, Hunter M. Detection of substance use disorders in patients presenting with depression. Subst Abus 2002;23:115-20.
12. Strine TW, Balluz L, Chapman DP, et al. Risk behaviors and healthcare coverage among adults by frequent mental distress status, 2001. Am J Prev Med 2004;26:213-6.
History: losing his ‘drive’
Mr. D, age 49, has been treated for major depressive disorder for approximately 1 year but reports only occasional minor symptom improvement. At presentation, he had been irritable and lethargic for about 2 weeks and had increased appetite, decreased concentration, and trouble falling asleep at night.
A once-gregarious family man, Mr. D had become apathetic and too tired to enjoy socializing. He denied suicidal thoughts or feelings of worthlessness and hopelessness but feared his fatigue was interfering with his job as a truck driver. He tired after driving only a few hours.
Mr. D had been diagnosed with sleep apnea when he was younger but had no other medical history. He said his erratic work schedule kept him from using his continuous positive airway pressure (CPAP) machine regularly. He was taking no medications and had not seen a primary care physician for more than 2 years because of lack of coverage. He denied past or current substance abuse.
The patient weighed 280 lbs at intake. His body mass index (BMI) was 37.5, indicating clinical obesity.
Because Mr. D lacked health insurance, we enrolled him 1 year ago in a free depression study at a psychiatric outpatient clinic. At intake, he said numerous life stresses—particularly the recent death of his brother in a motor vehicle accident—had left him feeling depressed.
We started Mr. D on citalopram, 20 mg/d, which was the study protocol. Two weeks later, he complained of dry mouth and sedation with minimal symptom improvement. We stopped citalopram and started sertraline, 25 mg/d.
Two weeks later, Mr. D again complained he had “no energy” and was “sleeping all day.” We titrated sertraline to 200 mg/d over 2 months, but his excessive tiredness, increased appetite, and decreased motivation persisted. Mr. D needed routine laboratory tests, so we referred him to a local clinic that charges on a sliding scale. He did not complete the tests, however, for fear of incurring medical expenses.
We tried to improve Mr. D’s mood symptoms by adding lithium—225 mg/d titrated to 675 mg/d over 7 weeks—but his depression and fatigue kept worsening. We tapered him off lithium and sertraline and switched to the monoamine oxidase inhibitor tranylcypromine, 30 mg/d, which was also part of the study protocol. We warned him not to eat pizza, fermented dry sausages, or other foods that could interact adversely with tranylcypromine. After 4 weeks, Mr. D stopped taking the agent, saying he could not follow the dietary restrictions while on the road.
We released Mr. D from the study because of nonresponse. Bupropion, started at 100 mg bid and titrated to 300 mg each morning and 150 mg nightly across 5 months, did not resolve his fatigue. He also started having agitation and “anger problems,” often getting into shouting matches over his CBradio with other truck drivers. We started quetiapine, 25 mg bid, hoping the low dose would calm his mood.
Until now, Mr. D has ignored our requests to undergo routine laboratory testing. We referred him to the local clinic four times over the past year but he has not complied, citing lack of health insurance and financial concerns.
The authors’ observations
Although Mr. D’s symptoms (constantly depressed mood, loss of interest in usual activities) clearly suggest treatment-resistant major depressive disorder, an underlying medical disorder cannot be ruled out, yet he refuses to get needed tests.
Medical comorbidities are more prevalent in patients with mental illness than in the general population.1 As many as 43% of patients referred to some psychiatry clinics have medical disorders, and almost one-half the diagnoses were missed by the referring physician.2
Compared to patients without psychiatric diagnoses, those with mental illness have more difficulty gaining access to medical care and are less likely to receive and follow guidelines for preventive care. Mental illness symptoms often compromise one’s ability to seek health care or follow a doctor’s orders. For example, a psychotic person may be overly suspicious of doctors, whereas someone with anxiety may seek care inappropriately.3,4 Also, some studies estimate that 1 in 5 persons with mental illness are uninsured.1,5,6
Mr. D denies substance abuse, but primary care and behavioral health clinicians often miss substance use disorders.7 Accuracy of substance abuse self-reports varies widely; some studies report high accuracy, whereas almost 33% of patients in other studies do not disclose substance abuse.8
Testing: stimulating findings
At his next visit, Mr. D reports worsening thirst and increased urination and complains of increased appetite, easy bruising, excessive sleepiness, and apathy. He also reveals that for 2 months he has been taking 2 to 3 fat-burning stimulant capsules a day to stay awake while driving.
Alarmed by his elevated blood pressure (177/99 mm Hg) and worsening physical symptoms, Mr. D finally consents to baseline laboratory testing. Blood glucose is 306 mg/dL (normal 70 to 110 mg/dL), and glycosylated hemoglobin is 12% (normal
Mr. D, who now weighs 270 lbs, is diagnosed as having hypertension and type 2 diabetes mellitus. Clinic doctors start him on metformin, 500 mg bid titrated to 1,000 mg bid, and glyburide, 5 mg/d, to control his glucose, and lisinopril, 10 mg/d, to control his hypertension, reduce cardiovascular risk, and preserve renal function. Clinicians also order Mr. D to follow an 1,800-calorie, American Diabetes Association-approved diet. We stop quetiapine and bupropion.
Mr. D’s diabetes and hypertension diagnosis, combined with his habitus and history of easy bruising, suggest Cushing’s syndrome. Doctors rule out this disorder based on a 24-hour free cortisol reading of 59 mg/L and normal dexamethasone suppression. Lab findings suggest he is not taking stimulants away from work.
The authors’ observations
Ideally, Mr. D should have undergone laboratory testing after the initial intake visit, before psychotropics were started. Routine vital signs also should have been taken.
Symptoms of major depressive disorder and early type 2 diabetes are strikingly similar (Table 1). For example, early diabetes symptoms such as fatigue can mimic depression or other medical problems. In one study of 69 diabetic patients who were referred by their primary care doctors to a psychiatric clinic, 57 had not been diagnosed as having diabetes before referral.9
Aside from its medical complications, diabetes also doubles the risk of comorbid depression, which can alter diabetes’ course and outcome.10
Earlier laboratory testing could have uncovered Mr. D’s comorbid stimulant abuse, which also can mimic depression and complicate its treatment.11 Signs of amphetamine withdrawal—such as dysphoric mood, fatigue, insomnia or hypersomnia, increased appetite, and psychomotor retardation—can be mistaken for depression (Table 1).
Patients with Cushing’s syndrome may present with nonspecific complaints of fatigue, decreased energy, apathy, depressed mood, and hypersomnia. A 24-hour free cortisol reading and dexamethasone suppression testing can differentiate Cushing’s syndrome from depression.
Costly, unnecessary care. Missing a medical cause of apparent psychiatric symptoms can lead to unnecessary treatment and needless expense. A complete metabolic profile and urine drug screen—approximately $60—could have saved the nearly $5,000 spent on treating Mr. D’s “resistant” depression ( Table 2).
Psychiatrists need to watch for potential medical problems and for cormorbidities associated with mental illness. Patients with frequent mental distress—defined as ≥ 14 mentally unhealthy days within 30 days—were found to be more likely to smoke, drink heavily, and be physically inactive and obese than were mentally healthy persons. Mentally distressed patients also were more likely to lack health care coverage and to engage in multiple adverse behaviors, increasing their risk for mental and physical illness.12
Ensuring proper medical care. Based on our experience with Mr. D, routine vital signs—including BMI, weight, blood pressure, and pulse rate—should be recorded at each visit. At intake, we recommend that psychiatrists:
- find out when the patient last saw a primary health provider other than in the emergency room, and whether the patient is receiving preventive medical care
- assess for unhealthy lifestyle habits (smoking, drug use, poor diet) or family history of serious medical illnesses.
Educate patients about the interplay between physical and mental illness to help them understand the importance of seeing a primary care doctor. Finally, be familiar with local indigent health clinics and their fee scales.
Table 1
Medical symptoms that mimic depression
| Symptom | Amphetamine withdrawal | Cushing’s syndrome | Diabetes |
|---|---|---|---|
| Anxiety | × | ||
| Dysphoric mood | × | ||
| Fatigue | × | × | × |
| Hypersomnia | × | ||
| Increased appetite | × | × | |
| Insomnia | × | ||
| Irritability | × | × | |
| Muscle aches and cramps | × | ||
| Psychomotor retardation | × | ||
| Vivid, unpleasant dreams | × | ||
| Weakness | × | ||
| Weight gain or loss | × |
The cost of treating Mr. D’s ‘resistant depression’
| Medication/dosage | Start date | Stop date | Approximate cost |
|---|---|---|---|
| Citalopram, 20 mg/d | 10/3/03 | 10/24/03 | $58.50 |
| Sertraline, 25 to 200 mg/d | 10/24/03 | 12/24/03 | $283.00 |
| Sertraline 150 mg/d, with lithium, 225 to 675 mg/d | 12/24/03 | 2/6/04 | $343.00 |
| Tranylcypromine, 10 mg each morning, 20 mg at bedtime | 2/27/04 | 4/20/04 | $322.00 |
| Bupropion (sustained release) up to 450 mg/d | 5/7/04 | 8/30/04 | $372.00 |
| Bupropion (sustained release), 450 mg/d, plus quetiapine, 25 mg/d | 8/30/04 | 11/8/04 | $554.00 |
| Total cost of psychotropics | $1,932.50 | ||
| Total cost of office visits ($95 X 30 visits) | $2,850.00 | ||
| TOTAL COST OF TREATMENT | $4,782.50 | ||
| Source: Walgreens Co. retail prices in Wichita, KS | |||
Follow-up: 30 lbs in 4 months
Mr. D has lost >30 lbs over 4 months, and his blood pressure and serum glucose are normal. BMI is now 32, in the lower range of clinical obesity. He feels more energetic and active, no longer reports excessive sedation and apathy, and has stopped taking stimulants. His depressive symptoms have remitted.
Related resources
- WrongDiagnosis.com. Information on differential diagnosis of medical and psychiatric problems. www.wrongdiagnosis.com.
- Mauksch LB, Tucker SM, Katon WJ, et al. Mental illness, functional impairment, and patient preferences for collaborative care in an uninsured, primary care population. J Fam Pract 2001;50:41-7.
- Glied S, Little SE. The uninsured and the benefits of medical progress. Health Aff (Millwood) 2003;22:210-9.
- Bupropion • Wellbutrin
- Citalopram • Celexa
- Dexamethasone • Ciprodex, others
- Glucophage • Metformin
- Glyburide • DiaBeta, others
- Lisinopril • Prinivil, Zestril
- Lithium • Eskalith, others
- Quetiapine • Seroquel
- Sertraline • Zoloft
- Tranylcypromine •Parnate
Dr. Khan is a speaker for Wyeth Pharmaceuticals.
Dr. Grimsley reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
History: losing his ‘drive’
Mr. D, age 49, has been treated for major depressive disorder for approximately 1 year but reports only occasional minor symptom improvement. At presentation, he had been irritable and lethargic for about 2 weeks and had increased appetite, decreased concentration, and trouble falling asleep at night.
A once-gregarious family man, Mr. D had become apathetic and too tired to enjoy socializing. He denied suicidal thoughts or feelings of worthlessness and hopelessness but feared his fatigue was interfering with his job as a truck driver. He tired after driving only a few hours.
Mr. D had been diagnosed with sleep apnea when he was younger but had no other medical history. He said his erratic work schedule kept him from using his continuous positive airway pressure (CPAP) machine regularly. He was taking no medications and had not seen a primary care physician for more than 2 years because of lack of coverage. He denied past or current substance abuse.
The patient weighed 280 lbs at intake. His body mass index (BMI) was 37.5, indicating clinical obesity.
Because Mr. D lacked health insurance, we enrolled him 1 year ago in a free depression study at a psychiatric outpatient clinic. At intake, he said numerous life stresses—particularly the recent death of his brother in a motor vehicle accident—had left him feeling depressed.
We started Mr. D on citalopram, 20 mg/d, which was the study protocol. Two weeks later, he complained of dry mouth and sedation with minimal symptom improvement. We stopped citalopram and started sertraline, 25 mg/d.
Two weeks later, Mr. D again complained he had “no energy” and was “sleeping all day.” We titrated sertraline to 200 mg/d over 2 months, but his excessive tiredness, increased appetite, and decreased motivation persisted. Mr. D needed routine laboratory tests, so we referred him to a local clinic that charges on a sliding scale. He did not complete the tests, however, for fear of incurring medical expenses.
We tried to improve Mr. D’s mood symptoms by adding lithium—225 mg/d titrated to 675 mg/d over 7 weeks—but his depression and fatigue kept worsening. We tapered him off lithium and sertraline and switched to the monoamine oxidase inhibitor tranylcypromine, 30 mg/d, which was also part of the study protocol. We warned him not to eat pizza, fermented dry sausages, or other foods that could interact adversely with tranylcypromine. After 4 weeks, Mr. D stopped taking the agent, saying he could not follow the dietary restrictions while on the road.
We released Mr. D from the study because of nonresponse. Bupropion, started at 100 mg bid and titrated to 300 mg each morning and 150 mg nightly across 5 months, did not resolve his fatigue. He also started having agitation and “anger problems,” often getting into shouting matches over his CBradio with other truck drivers. We started quetiapine, 25 mg bid, hoping the low dose would calm his mood.
Until now, Mr. D has ignored our requests to undergo routine laboratory testing. We referred him to the local clinic four times over the past year but he has not complied, citing lack of health insurance and financial concerns.
The authors’ observations
Although Mr. D’s symptoms (constantly depressed mood, loss of interest in usual activities) clearly suggest treatment-resistant major depressive disorder, an underlying medical disorder cannot be ruled out, yet he refuses to get needed tests.
Medical comorbidities are more prevalent in patients with mental illness than in the general population.1 As many as 43% of patients referred to some psychiatry clinics have medical disorders, and almost one-half the diagnoses were missed by the referring physician.2
Compared to patients without psychiatric diagnoses, those with mental illness have more difficulty gaining access to medical care and are less likely to receive and follow guidelines for preventive care. Mental illness symptoms often compromise one’s ability to seek health care or follow a doctor’s orders. For example, a psychotic person may be overly suspicious of doctors, whereas someone with anxiety may seek care inappropriately.3,4 Also, some studies estimate that 1 in 5 persons with mental illness are uninsured.1,5,6
Mr. D denies substance abuse, but primary care and behavioral health clinicians often miss substance use disorders.7 Accuracy of substance abuse self-reports varies widely; some studies report high accuracy, whereas almost 33% of patients in other studies do not disclose substance abuse.8
Testing: stimulating findings
At his next visit, Mr. D reports worsening thirst and increased urination and complains of increased appetite, easy bruising, excessive sleepiness, and apathy. He also reveals that for 2 months he has been taking 2 to 3 fat-burning stimulant capsules a day to stay awake while driving.
Alarmed by his elevated blood pressure (177/99 mm Hg) and worsening physical symptoms, Mr. D finally consents to baseline laboratory testing. Blood glucose is 306 mg/dL (normal 70 to 110 mg/dL), and glycosylated hemoglobin is 12% (normal
Mr. D, who now weighs 270 lbs, is diagnosed as having hypertension and type 2 diabetes mellitus. Clinic doctors start him on metformin, 500 mg bid titrated to 1,000 mg bid, and glyburide, 5 mg/d, to control his glucose, and lisinopril, 10 mg/d, to control his hypertension, reduce cardiovascular risk, and preserve renal function. Clinicians also order Mr. D to follow an 1,800-calorie, American Diabetes Association-approved diet. We stop quetiapine and bupropion.
Mr. D’s diabetes and hypertension diagnosis, combined with his habitus and history of easy bruising, suggest Cushing’s syndrome. Doctors rule out this disorder based on a 24-hour free cortisol reading of 59 mg/L and normal dexamethasone suppression. Lab findings suggest he is not taking stimulants away from work.
The authors’ observations
Ideally, Mr. D should have undergone laboratory testing after the initial intake visit, before psychotropics were started. Routine vital signs also should have been taken.
Symptoms of major depressive disorder and early type 2 diabetes are strikingly similar (Table 1). For example, early diabetes symptoms such as fatigue can mimic depression or other medical problems. In one study of 69 diabetic patients who were referred by their primary care doctors to a psychiatric clinic, 57 had not been diagnosed as having diabetes before referral.9
Aside from its medical complications, diabetes also doubles the risk of comorbid depression, which can alter diabetes’ course and outcome.10
Earlier laboratory testing could have uncovered Mr. D’s comorbid stimulant abuse, which also can mimic depression and complicate its treatment.11 Signs of amphetamine withdrawal—such as dysphoric mood, fatigue, insomnia or hypersomnia, increased appetite, and psychomotor retardation—can be mistaken for depression (Table 1).
Patients with Cushing’s syndrome may present with nonspecific complaints of fatigue, decreased energy, apathy, depressed mood, and hypersomnia. A 24-hour free cortisol reading and dexamethasone suppression testing can differentiate Cushing’s syndrome from depression.
Costly, unnecessary care. Missing a medical cause of apparent psychiatric symptoms can lead to unnecessary treatment and needless expense. A complete metabolic profile and urine drug screen—approximately $60—could have saved the nearly $5,000 spent on treating Mr. D’s “resistant” depression ( Table 2).
Psychiatrists need to watch for potential medical problems and for cormorbidities associated with mental illness. Patients with frequent mental distress—defined as ≥ 14 mentally unhealthy days within 30 days—were found to be more likely to smoke, drink heavily, and be physically inactive and obese than were mentally healthy persons. Mentally distressed patients also were more likely to lack health care coverage and to engage in multiple adverse behaviors, increasing their risk for mental and physical illness.12
Ensuring proper medical care. Based on our experience with Mr. D, routine vital signs—including BMI, weight, blood pressure, and pulse rate—should be recorded at each visit. At intake, we recommend that psychiatrists:
- find out when the patient last saw a primary health provider other than in the emergency room, and whether the patient is receiving preventive medical care
- assess for unhealthy lifestyle habits (smoking, drug use, poor diet) or family history of serious medical illnesses.
Educate patients about the interplay between physical and mental illness to help them understand the importance of seeing a primary care doctor. Finally, be familiar with local indigent health clinics and their fee scales.
Table 1
Medical symptoms that mimic depression
| Symptom | Amphetamine withdrawal | Cushing’s syndrome | Diabetes |
|---|---|---|---|
| Anxiety | × | ||
| Dysphoric mood | × | ||
| Fatigue | × | × | × |
| Hypersomnia | × | ||
| Increased appetite | × | × | |
| Insomnia | × | ||
| Irritability | × | × | |
| Muscle aches and cramps | × | ||
| Psychomotor retardation | × | ||
| Vivid, unpleasant dreams | × | ||
| Weakness | × | ||
| Weight gain or loss | × |
The cost of treating Mr. D’s ‘resistant depression’
| Medication/dosage | Start date | Stop date | Approximate cost |
|---|---|---|---|
| Citalopram, 20 mg/d | 10/3/03 | 10/24/03 | $58.50 |
| Sertraline, 25 to 200 mg/d | 10/24/03 | 12/24/03 | $283.00 |
| Sertraline 150 mg/d, with lithium, 225 to 675 mg/d | 12/24/03 | 2/6/04 | $343.00 |
| Tranylcypromine, 10 mg each morning, 20 mg at bedtime | 2/27/04 | 4/20/04 | $322.00 |
| Bupropion (sustained release) up to 450 mg/d | 5/7/04 | 8/30/04 | $372.00 |
| Bupropion (sustained release), 450 mg/d, plus quetiapine, 25 mg/d | 8/30/04 | 11/8/04 | $554.00 |
| Total cost of psychotropics | $1,932.50 | ||
| Total cost of office visits ($95 X 30 visits) | $2,850.00 | ||
| TOTAL COST OF TREATMENT | $4,782.50 | ||
| Source: Walgreens Co. retail prices in Wichita, KS | |||
Follow-up: 30 lbs in 4 months
Mr. D has lost >30 lbs over 4 months, and his blood pressure and serum glucose are normal. BMI is now 32, in the lower range of clinical obesity. He feels more energetic and active, no longer reports excessive sedation and apathy, and has stopped taking stimulants. His depressive symptoms have remitted.
Related resources
- WrongDiagnosis.com. Information on differential diagnosis of medical and psychiatric problems. www.wrongdiagnosis.com.
- Mauksch LB, Tucker SM, Katon WJ, et al. Mental illness, functional impairment, and patient preferences for collaborative care in an uninsured, primary care population. J Fam Pract 2001;50:41-7.
- Glied S, Little SE. The uninsured and the benefits of medical progress. Health Aff (Millwood) 2003;22:210-9.
- Bupropion • Wellbutrin
- Citalopram • Celexa
- Dexamethasone • Ciprodex, others
- Glucophage • Metformin
- Glyburide • DiaBeta, others
- Lisinopril • Prinivil, Zestril
- Lithium • Eskalith, others
- Quetiapine • Seroquel
- Sertraline • Zoloft
- Tranylcypromine •Parnate
Dr. Khan is a speaker for Wyeth Pharmaceuticals.
Dr. Grimsley reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1 McAlpine DD, Mechanic D. Utilization of specialty mental health care among persons with severe mental illness: the roles of demographics, need, insurance, and risk. Health Serv Res 2000;35(1 Pt 2):277-92.
2 Rosse RB, Deutsch LH, Deutsch SI. Medical assessment and laboratory testing in psychiatry. In: Sadock BJ, Sadock VA (eds). Kaplan & Sadock’ s comprehensive textbook of psychiatry (7th ed), Vol 1. Baltimore: Lippincott Williams & Wilkins; 2000:732.
3 Rubin AS, Littenberg B, Ross R, et al. Effects on processes and costs of care associated with the addition of an internist to an inpatient psychiatry team. Psychiatr Serv 2005;56:463-7.
4 Salsberry PJ, Chipps E, Kennedy C. Use of general medical services among Medicaid patients with severe and persistent mental illness. Psychiatr Serv 2005;56:458-62.
5 McAlpine DD, Mechanic D. Datapoints: payer source for emergency room visits by persons with psychiatric disorders. Psychiatr Serv 2002;53:14.-
6 Yanos PT, Lu W, Minsky S, Kiely GL. Correlates of health insurance among persons with schizophrenia in a statewide behavioral health care system. Psychiatr Serv 2004;55:79-82.
7 Brown GS, Hermann R, Jones E, Wu J. Using self-report to improve substance abuse risk assessment in behavioral health care. Jt Comm J Qual Saf 2004;30:448-54.
8 Tassiopoulos K, Bernstein J, Heeren T, et al. Hair testing and self-report of cocaine use by heroin users. Addiction 2004;99:590-7.
9 Katon WJ, Lin EH, Russo J, et al. Cardiac risk factors in patients with diabetes mellitus and major depression. J Gen Intern Med 2004;19:1192-9.
10 Lustman PJ, Clouse RE. Depression in diabetic patients: the relationship between mood and glycemic control. J Diabetes Complications 2005;19:113-22.
11 Mallin R, Slott K, Tumblin M, Hunter M. Detection of substance use disorders in patients presenting with depression. Subst Abus 2002;23:115-20.
12. Strine TW, Balluz L, Chapman DP, et al. Risk behaviors and healthcare coverage among adults by frequent mental distress status, 2001. Am J Prev Med 2004;26:213-6.
1 McAlpine DD, Mechanic D. Utilization of specialty mental health care among persons with severe mental illness: the roles of demographics, need, insurance, and risk. Health Serv Res 2000;35(1 Pt 2):277-92.
2 Rosse RB, Deutsch LH, Deutsch SI. Medical assessment and laboratory testing in psychiatry. In: Sadock BJ, Sadock VA (eds). Kaplan & Sadock’ s comprehensive textbook of psychiatry (7th ed), Vol 1. Baltimore: Lippincott Williams & Wilkins; 2000:732.
3 Rubin AS, Littenberg B, Ross R, et al. Effects on processes and costs of care associated with the addition of an internist to an inpatient psychiatry team. Psychiatr Serv 2005;56:463-7.
4 Salsberry PJ, Chipps E, Kennedy C. Use of general medical services among Medicaid patients with severe and persistent mental illness. Psychiatr Serv 2005;56:458-62.
5 McAlpine DD, Mechanic D. Datapoints: payer source for emergency room visits by persons with psychiatric disorders. Psychiatr Serv 2002;53:14.-
6 Yanos PT, Lu W, Minsky S, Kiely GL. Correlates of health insurance among persons with schizophrenia in a statewide behavioral health care system. Psychiatr Serv 2004;55:79-82.
7 Brown GS, Hermann R, Jones E, Wu J. Using self-report to improve substance abuse risk assessment in behavioral health care. Jt Comm J Qual Saf 2004;30:448-54.
8 Tassiopoulos K, Bernstein J, Heeren T, et al. Hair testing and self-report of cocaine use by heroin users. Addiction 2004;99:590-7.
9 Katon WJ, Lin EH, Russo J, et al. Cardiac risk factors in patients with diabetes mellitus and major depression. J Gen Intern Med 2004;19:1192-9.
10 Lustman PJ, Clouse RE. Depression in diabetic patients: the relationship between mood and glycemic control. J Diabetes Complications 2005;19:113-22.
11 Mallin R, Slott K, Tumblin M, Hunter M. Detection of substance use disorders in patients presenting with depression. Subst Abus 2002;23:115-20.
12. Strine TW, Balluz L, Chapman DP, et al. Risk behaviors and healthcare coverage among adults by frequent mental distress status, 2001. Am J Prev Med 2004;26:213-6.
Counseling trauma victims: 4 brief therapies meet the test
Therapists once believed trauma survivors required years of treatment, yet we now know that relatively brief cognitive-behavioral interventions can yield long-term gains in psychosocial and psychological function.1 Many psychiatric patients meet diagnostic criteria for posttraumatic stress disorder (PTSD), including:
- 33% of women experiencing sexual assault2
- 30% of male war veterans3
- 30% of the 5 million U.S. children exposed to trauma each year4(Box).5
We offer recommendations on how to prepare traumatized adults and children for cognitive-behavioral therapy (CBT) and discuss four tested models—prolonged exposure (PE), cognitive processing therapy (CPT), eye movement desensitization and reprocessing (EMDR), and stress inoculation training (SIT)—that psychiatrists may find effective when treating PTSD.
Exposure therapy with children is usually more gradual than with adults, and the child is first taught relaxation techniques to use while recalling traumatic experiences. Although re-exposing children to traumatic events may seem harsh, exposure-based cognitive-behavioral therapy (CBT) appears to be most effective when trauma memories or reminders are most distressing to the child.
As with adults, CBT with children typically includes:
- exposure
- identifying and challenging unhealthy or distorted trauma-related thoughts
- teaching anxiety management techniques such as relaxation or assertiveness training.
In initial studies, CBT has been found safe and effective for treating posttraumatic stress disorder (PTSD) in children and adolescents.17 Through therapy, they can learn not to be afraid of their memories and can develop healthier, more-appropriate thoughts about the trauma. Children with uncomplicated PTSD—without severe, long-term physical injury—typically receive 12 to 20 CBT sessions. More sessions are needed for complex cases, such as when the trauma perpetrator was an integral family member.
Comorbid conditions—such as conduct disorder, attention-deficit/hyperactivity disorder, or depression—may need to be treated before PTSD or concurrently, using medication or other interventions.
Preparing trauma patients for CBT
Before starting CBT, evaluate patients thoroughly to determine if they meet DSM-IV-TR full or subthreshold criteria (
Not all patients are ready to confront their traumas when they arrive for psychiatric evaluation. For example:
- For a domestic violence victim, the therapist’s priority is to help begin safety planning and to address trauma after the patient is out of danger.
- Patients with poor coping skills and little social support often find it difficult to begin trauma treatment. For them, focus on building skills to offset the distress that accompanies trauma therapy.
- Patients with PTSD and substance abuse may benefit more from CBT if the therapist first addresses the substance dependence.
CBT core concepts
CBT therapists typically help patients identify and evaluate disruptive cognitions, which helps them challenge and modify emotions, thoughts, and behaviors related to traumatic experience(s). Other CBT components include:
- educating patients about PTSD
- exposing them to the traumatic material
- challenging and modifying their disruptive thoughts.
International Society for Traumatic Stress Studies (ISTSS) practice guidelines for PTSD8 include assessment and treatment suggestions (see Related resources). Whatever the model, CBT appears help patients manage their distress, not only during treatment but up to 5 years after completing therapy.9
Which CBT? Comparison studies have shown all four CBT interventions to be effective in treating PTSD, although initial trend data suggest that patients with:
- fear-based PTSD may do better with PE or EMDR
- PTSD-related guilt, anger, or other cognitive distortions may benefit more from CPT.
If you refer a patient, make sure the therapist is trained in CBT interventions and in working with trauma patients. To be effective, the therapist must be skilled in handling trauma processing work, suicidal thoughts/intent, and comorbid personality disorders.
Prolonged exposure
PE (Table 1) is typically conducted in 9 to 12 sessions lasting 90 minutes each and has been used to treat PTSD after sexual assault, combat, sexual abuse, and natural disasters. Although frequently offered in individual sessions, group PE has also been found to be effective.10
After educating the patient about PTSD and the treatment rationale, the therapist repeatedly asks the patient to describe the traumatic event as if it were occurring. During 45 to 60 minutes of this exposure, the therapist frequently asks the patient to rate his or her distress. This identifies “hot spots” in the account that need to be repeated. The therapist does not necessarily challenge distorted cognitions about the event (such as “I am to blame for the rape” or “No one can be trusted”).
Researchers hypothesize that exposing a PTSD patient to traumatic memories engages his or her brain’s pathologic “fear network,” which triggers an excessive fear response to non-threatening stimuli. Continued exposure allows the patient to habituate to this network, with subsequent extinction of fear and anxiety reactions. Foa et al11 found that mentally re-experiencing a traumatic event helps patients organize memory cues about it, which encourages cognitive restructuring of the trauma.
PE has been shown to enhance the trauma survivor’s self-control and personal competence and to decrease generalization of fear to non-assault stimuli.12 For example, many combat veterans report fear of situations—such as going to the beach or into the woods—that bring back memories of traumatic events. Their fears may keep them from enjoying a walk in the park or family vacations.
Through in vivo exposure, these patients can face associations between environmental cues and their trauma. As they learn to modify the fears associated with these cues, their personal and social functioning improves.
PE can be successful for those who complete therapy, but it has a relatively high drop-out rate, reported as 8%13 to 41%.14 The pain of continually reliving a traumatic event probably causes many patients to quit. To reduce drop-out rates, many therapists combine PE with cognitive restructuring or other techniques that help build patients’ coping skills.
Table 1
Using prolonged exposure therapy to treat PTSD, session by session
| Session | Content |
|---|---|
| 1 | Education |
| Treatment rationale | |
| Review of PTSD symptom response | |
| Introduce breathing retraining | |
| 2 | Review handout, ‘Common reactions to trauma’ |
| Introduce Subjective Units of Distress | |
| Create fear hierarchy for in vivo exposures | |
| 3 | Provide rationale for imaginal exposure |
| Conduct imaginal exposure | |
| Assign in vivo exposure homework | |
| 4 to 8 | Conduct imaginal exposure |
| Discuss in vivo exposures | |
| 9 or 9 to 12 | Conduct imaginal exposure |
| Suggest continued in vivo exercises | |
| Termination | |
| Source: Foa EB, Rothbaum BO. Treating the trauma of rape: cognitive behavioral therapy for PTSD. New York: Guilford Press, 1998. | |
Cognitive processing therapy
CPT (Table 2) was created as a protocol to treat PTSD and related symptoms in rape survivors.7 Sessions can be group, individual, or combined, depending on the needs and resources of the patients and clinic.
Originally, CPT contained 12 weekly sessions, although versions up to 17 weeks have been developed for adult survivors of child sexual abuse, domestic violence survivors, and war veterans.15 Sessions can be added or adapted to address each population’s type of traumatic experience (such as developmental impairment of sexual abuse survivors).
CPT is based on information processing theory, which suggests that as people access a traumatic memory, they experience and extinguish emotions attached to the event. Guided by the therapist, the patient identifies and challenges distortions the trauma created in three cognition domains: the self, others, and the world. Patients learn to change or replace these cognitive distortions—which therapists often call “stuck points” or “rules”—with more-adaptive, healthier beliefs.
Common byproducts of trauma are feeling out of control or hopeless. Thus, CPT focuses on personal safety, trust, power/control, esteem, and intimacy within each of the three domains. Modules on assertiveness, communication, and social support can also be added.
Although CPT is being adapted for populations other than rape survivors, comparison studies are needed to determine if it is as effective as other CBT therapies for these groups.
Table 2
Using cognitive processing therapy to treat PTSD, session by session
| Session | Content |
|---|---|
| 1 | Education |
| Review of symptoms | |
| Introduce ‘stuck points’/rules | |
| Write impact of event statement (IES) | |
| 2 | Review IES |
| Identify stuck points | |
| Introduce A-B-C sheets | |
| 3 | Review A-B-C sheets |
| Assign writing of traumatic account | |
| 4 | Read traumatic account |
| Identify stuck points | |
| Rewrite the account | |
| 5 | Read rewritten account |
| Identify stuck points | |
| Introduce challenging questions sheet (CQS) | |
| Assign writing of next-most traumatic incident and CQS | |
| 6 | Review CQS |
| Assign review of faulty thinking patterns (FTP) | |
| 7 | Review FTP |
| Assign safety module and challenging beliefs worksheets (CBW) on safety | |
| 8 | Review CBWs on safety |
| Assign module on trust | |
| 9 | Review CBWs on trust |
| Assign module on power/control | |
| 10 | Review CBWs on power/control |
| Assign module on esteem | |
| 11 | Review CBWs on esteem |
| Assign module on intimacy | |
| Rewrite IES | |
| 12 | Review CBWs on intimacy |
| Read both impact statements | |
| Address remaining areas of concern | |
| Termination | |
| Source: Resick PA, Schnicke MK Cognitive processing therapy for rape victims: a treatment manual. Newbury Park, CA: Sage, 1993. | |
Eye movement desensitization and reprocessing
Like other PTSD treatments, EMDR is based on an “accelerated information-processing” model.16 Because it also incorporates dissociation and nonverbal representation of traumas (such as visual memories), EMDR is often classified as a cognitive treatment, although ISTSS practice guidelines8 present it as a separate category.
EMDR protocols call for the trauma patient to watch rapid, rhythmic movements of the therapist’s hand or a set of lights to distract attention from the stress he or she feels when visualizing the traumatic event. The original technique—developed by Francine Shapiro, PhD—is based on the observation that persons with PTSD often have disrupted rapid eye-movement sleep. In theory, inducing eye movements inhibits stress, allowing patients to more freely access their memory networks and process disturbances. Subsequently, Dr. Shapiro has suggested that using other auditory cues or hand taps may be as effective as eye movements.16
EMDR is often conducted in 12 to 15 sessions, although some studies report positive changes after 3 to 6 sessions. After obtaining a patient history, establishing rapport, and explaining the treatment, the therapist asks the patient to identify:
- visual images of the trauma
- his or her affective and physiologic responses to the trauma
- negative self-representations the trauma created
- positive, alternate self-representations.
EMDR has been effective in treating male war veterans, rape victims, and other trauma groups.17 Initial dismantling studies suggest that eye movements (or other distracting cues) might not be essential for trauma reprocessing, calling into question the mechanisms thought to create change in EMDR. Studies with larger samples comparing EMDR with other CBT models are needed to assess EMDR’s efficacy for trauma survivors.17
Stress inoculation training
SIT was designed by Meichenbaum18 (Table 3) to treat anxiety and stress and was adapted for use with trauma survivors. It appears most effective in relieving fear, anxiety, and depressive symptoms associated with traumatic experiences. SIT includes education, muscle relaxation training, breathing retraining, covert modeling, role-playing, guided self-dialog, and thought stopping. Therapists often teach these skills to patients in modules that build on each other.
For example, a patient might receive relaxation training while role-playing a difficult scenario she may face in the future. This helps her learn to remain calm in anxiety-provoking situations.
Unlike PE, SIT does not directly ask patients to recount their traumatic memories, although exposure may be indirect (such as during role-playing exercises). Its purpose is to give patients new skills to manage their anxiety, which in turn decreases PTSD symptoms.
Studies suggest that PE is more effective than SIT alone or SIT/PE combined.13 Thus, instead of using SIT as a trauma-focused treatment, some therapists find it useful to help patients gain coping skills before beginning other trauma treatments.
Table 3
Where to learn more about cognitive therapies for PTSD
| CBT model | PTSD related to… | Resources |
|---|---|---|
| Prolonged exposure | Combat experience, sexual assault, childhood abuse, motor vehicle accidents | Foa EB, Rothbaum BO. Treating the trauma of rape: Cognitive-behavioral therapy for PTSD. New York: Guilford Press; 1998 |
| Cognitive processing | Sexual assault, childhood abuse, incarceration (of adolescents) | Resick P, Schnicke M. Cognitive processing therapy for rape victims: a treatment manual. Newbury Park, CA: Sage Publications; 1996 |
| EMDR | Combat experience, sexual assault, civilian disasters (for children or adults) | Shapiro F. Eye movement desensitization and reprocessing: basic principles, protocols, and procedures (2nd ed). New York: Guilford Press; 2001 |
| EMDR Institute, Inc. Available at: http://www.emdr.com | ||
| Stress inoculation training | Sexual and physical assault, motor vehicle accidents | Meichenbaum D. Stress inoculation training for coping with stressors. Available at: http://www.apa.org/divisions/div12/rev_est/sit_stress.html |
| EMDR: Eye movement desensitization and reprocessing | ||
- International Society for Traumatic Stress Studies. www.istss.org.
- Foa EB, Keane TM, Friedman MJ. Effective treatments for PTSD. New York: Guilford Press; 2000.
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Rothbaum BO, Meadows EA, Resick PA, Foy DW. Cognitive-behavioral therapy. In: Foa E, Keane T, Friedman M (eds). Effective treatments for PTSD. New York: Guilford Press; 2000.
2. Kilpatrick D, Edmonds CN, Seymour AK. Rape in America: A report to the nation. Arlington, VA: National Victims Center; 1992.
3. Kulka RA, Schlenger WE, Fairbank JA, et al. Trauma and the Vietnam War generation: report of findings from the National Vietnam Veterans Readjustment Study. New York: Brunner/Mazel; 1990.
4. Pfefferbaum B. Posttraumatic stress disorder in children: a review of the past 10 years. J Am Acad Child Adolesc Psychiatry 1997;36(11):1503-11.
5. Deblinger E, McLeer SV, Henry D. Cognitive behavioral treatment for sexually abused children suffering post-traumatic stress. J Am Acad Child Adolesc Psychiatry 1990;5:747-52.
6. Najavits LM. Seeking Safety: a treatment manual for PTSD and substance abuse. New York: Guilford Press; 2002.
7. Resick PA, Nishith P, Weaver TL, et al. A comparison of cognitive processing therapy, prolonged exposure and a waiting condition for the treatment of posttraumatic stress disorder in female rape victims. J Consult Clin Psychol 2002;70:867-79.
8. Foa EB, Keane TM. Friedman MJ (eds). Effective treatments for PTSD. New York: Guilford Press; 2000.
9. Tarrier N, Sommerfield C. Treatment of chronic PTSD by cognitive therapy and exposure: 5-year follow-up. Behavior Ther 2004;35(2):231-46.
10. Foa EB, Rauch SA. Cognitive changes during prolonged exposure versus prolonged exposure plus cognitive restructuring in female assault survivors with posttraumatic stress disorder. J Consult Clin Psychol 2004;72(5):879-84.
11. Foa EB, Riggs DS, Massie ED, Yarczower M. The impact of fear activation and anger on the efficacy of exposure treatment for PTSD. Behav Ther 1995;26:487-99.
12. Foa EB, Rothbaum EO, Riggs D, Murdock T. Treatment of PTSD in rape victims: a comparison between cognitive-behavioral procedures and counseling. J Consult Clin Psychol 1991;59:715-23.
13. Foa EB, Dancu CV, Hembree EA, Jaycox LH, et al. A comparison of exposure therapy, stress inoculation training, and their combination for reducing posttraumatic stress disorder in female assault victims. J Consult Clin Psychol 1999;67:194-200.
14. McDonagh A, Friedman M, McHugo G, et al. Randomized trial of cognitive-behavioral therapy for chronic posttraumatic stress disorder in adult female survivors of childhood sexual abuse. J Consult Clin Psychol 2005;73:515-24.
15. Chard K. An evaluation of cognitive processing therapy for the treatment of posttraumatic stress disorder related to childhood sexual abuse. J Consult Clin Psychol. (in press).
16. Shapiro F. Eye movement desensitization and reprocessing: basic principles, protocols and procedures (2nd ed). New York: Guilford Press; 2001.
17. Chemtob CM, Tolin DF, van der Kolk BA, Pitman RK. Eye movement desensitization and reprocessing. In: Foa E, Keane T, Friedman M (eds). Effective treatments for PTSD. New York: Guilford Press; 2000.
18. Meichenbaum D. Cognitive-behavior modification: An integrative approach. New York: Plenum Press; 1977.
Therapists once believed trauma survivors required years of treatment, yet we now know that relatively brief cognitive-behavioral interventions can yield long-term gains in psychosocial and psychological function.1 Many psychiatric patients meet diagnostic criteria for posttraumatic stress disorder (PTSD), including:
- 33% of women experiencing sexual assault2
- 30% of male war veterans3
- 30% of the 5 million U.S. children exposed to trauma each year4(Box).5
We offer recommendations on how to prepare traumatized adults and children for cognitive-behavioral therapy (CBT) and discuss four tested models—prolonged exposure (PE), cognitive processing therapy (CPT), eye movement desensitization and reprocessing (EMDR), and stress inoculation training (SIT)—that psychiatrists may find effective when treating PTSD.
Exposure therapy with children is usually more gradual than with adults, and the child is first taught relaxation techniques to use while recalling traumatic experiences. Although re-exposing children to traumatic events may seem harsh, exposure-based cognitive-behavioral therapy (CBT) appears to be most effective when trauma memories or reminders are most distressing to the child.
As with adults, CBT with children typically includes:
- exposure
- identifying and challenging unhealthy or distorted trauma-related thoughts
- teaching anxiety management techniques such as relaxation or assertiveness training.
In initial studies, CBT has been found safe and effective for treating posttraumatic stress disorder (PTSD) in children and adolescents.17 Through therapy, they can learn not to be afraid of their memories and can develop healthier, more-appropriate thoughts about the trauma. Children with uncomplicated PTSD—without severe, long-term physical injury—typically receive 12 to 20 CBT sessions. More sessions are needed for complex cases, such as when the trauma perpetrator was an integral family member.
Comorbid conditions—such as conduct disorder, attention-deficit/hyperactivity disorder, or depression—may need to be treated before PTSD or concurrently, using medication or other interventions.
Preparing trauma patients for CBT
Before starting CBT, evaluate patients thoroughly to determine if they meet DSM-IV-TR full or subthreshold criteria (
Not all patients are ready to confront their traumas when they arrive for psychiatric evaluation. For example:
- For a domestic violence victim, the therapist’s priority is to help begin safety planning and to address trauma after the patient is out of danger.
- Patients with poor coping skills and little social support often find it difficult to begin trauma treatment. For them, focus on building skills to offset the distress that accompanies trauma therapy.
- Patients with PTSD and substance abuse may benefit more from CBT if the therapist first addresses the substance dependence.
CBT core concepts
CBT therapists typically help patients identify and evaluate disruptive cognitions, which helps them challenge and modify emotions, thoughts, and behaviors related to traumatic experience(s). Other CBT components include:
- educating patients about PTSD
- exposing them to the traumatic material
- challenging and modifying their disruptive thoughts.
International Society for Traumatic Stress Studies (ISTSS) practice guidelines for PTSD8 include assessment and treatment suggestions (see Related resources). Whatever the model, CBT appears help patients manage their distress, not only during treatment but up to 5 years after completing therapy.9
Which CBT? Comparison studies have shown all four CBT interventions to be effective in treating PTSD, although initial trend data suggest that patients with:
- fear-based PTSD may do better with PE or EMDR
- PTSD-related guilt, anger, or other cognitive distortions may benefit more from CPT.
If you refer a patient, make sure the therapist is trained in CBT interventions and in working with trauma patients. To be effective, the therapist must be skilled in handling trauma processing work, suicidal thoughts/intent, and comorbid personality disorders.
Prolonged exposure
PE (Table 1) is typically conducted in 9 to 12 sessions lasting 90 minutes each and has been used to treat PTSD after sexual assault, combat, sexual abuse, and natural disasters. Although frequently offered in individual sessions, group PE has also been found to be effective.10
After educating the patient about PTSD and the treatment rationale, the therapist repeatedly asks the patient to describe the traumatic event as if it were occurring. During 45 to 60 minutes of this exposure, the therapist frequently asks the patient to rate his or her distress. This identifies “hot spots” in the account that need to be repeated. The therapist does not necessarily challenge distorted cognitions about the event (such as “I am to blame for the rape” or “No one can be trusted”).
Researchers hypothesize that exposing a PTSD patient to traumatic memories engages his or her brain’s pathologic “fear network,” which triggers an excessive fear response to non-threatening stimuli. Continued exposure allows the patient to habituate to this network, with subsequent extinction of fear and anxiety reactions. Foa et al11 found that mentally re-experiencing a traumatic event helps patients organize memory cues about it, which encourages cognitive restructuring of the trauma.
PE has been shown to enhance the trauma survivor’s self-control and personal competence and to decrease generalization of fear to non-assault stimuli.12 For example, many combat veterans report fear of situations—such as going to the beach or into the woods—that bring back memories of traumatic events. Their fears may keep them from enjoying a walk in the park or family vacations.
Through in vivo exposure, these patients can face associations between environmental cues and their trauma. As they learn to modify the fears associated with these cues, their personal and social functioning improves.
PE can be successful for those who complete therapy, but it has a relatively high drop-out rate, reported as 8%13 to 41%.14 The pain of continually reliving a traumatic event probably causes many patients to quit. To reduce drop-out rates, many therapists combine PE with cognitive restructuring or other techniques that help build patients’ coping skills.
Table 1
Using prolonged exposure therapy to treat PTSD, session by session
| Session | Content |
|---|---|
| 1 | Education |
| Treatment rationale | |
| Review of PTSD symptom response | |
| Introduce breathing retraining | |
| 2 | Review handout, ‘Common reactions to trauma’ |
| Introduce Subjective Units of Distress | |
| Create fear hierarchy for in vivo exposures | |
| 3 | Provide rationale for imaginal exposure |
| Conduct imaginal exposure | |
| Assign in vivo exposure homework | |
| 4 to 8 | Conduct imaginal exposure |
| Discuss in vivo exposures | |
| 9 or 9 to 12 | Conduct imaginal exposure |
| Suggest continued in vivo exercises | |
| Termination | |
| Source: Foa EB, Rothbaum BO. Treating the trauma of rape: cognitive behavioral therapy for PTSD. New York: Guilford Press, 1998. | |
Cognitive processing therapy
CPT (Table 2) was created as a protocol to treat PTSD and related symptoms in rape survivors.7 Sessions can be group, individual, or combined, depending on the needs and resources of the patients and clinic.
Originally, CPT contained 12 weekly sessions, although versions up to 17 weeks have been developed for adult survivors of child sexual abuse, domestic violence survivors, and war veterans.15 Sessions can be added or adapted to address each population’s type of traumatic experience (such as developmental impairment of sexual abuse survivors).
CPT is based on information processing theory, which suggests that as people access a traumatic memory, they experience and extinguish emotions attached to the event. Guided by the therapist, the patient identifies and challenges distortions the trauma created in three cognition domains: the self, others, and the world. Patients learn to change or replace these cognitive distortions—which therapists often call “stuck points” or “rules”—with more-adaptive, healthier beliefs.
Common byproducts of trauma are feeling out of control or hopeless. Thus, CPT focuses on personal safety, trust, power/control, esteem, and intimacy within each of the three domains. Modules on assertiveness, communication, and social support can also be added.
Although CPT is being adapted for populations other than rape survivors, comparison studies are needed to determine if it is as effective as other CBT therapies for these groups.
Table 2
Using cognitive processing therapy to treat PTSD, session by session
| Session | Content |
|---|---|
| 1 | Education |
| Review of symptoms | |
| Introduce ‘stuck points’/rules | |
| Write impact of event statement (IES) | |
| 2 | Review IES |
| Identify stuck points | |
| Introduce A-B-C sheets | |
| 3 | Review A-B-C sheets |
| Assign writing of traumatic account | |
| 4 | Read traumatic account |
| Identify stuck points | |
| Rewrite the account | |
| 5 | Read rewritten account |
| Identify stuck points | |
| Introduce challenging questions sheet (CQS) | |
| Assign writing of next-most traumatic incident and CQS | |
| 6 | Review CQS |
| Assign review of faulty thinking patterns (FTP) | |
| 7 | Review FTP |
| Assign safety module and challenging beliefs worksheets (CBW) on safety | |
| 8 | Review CBWs on safety |
| Assign module on trust | |
| 9 | Review CBWs on trust |
| Assign module on power/control | |
| 10 | Review CBWs on power/control |
| Assign module on esteem | |
| 11 | Review CBWs on esteem |
| Assign module on intimacy | |
| Rewrite IES | |
| 12 | Review CBWs on intimacy |
| Read both impact statements | |
| Address remaining areas of concern | |
| Termination | |
| Source: Resick PA, Schnicke MK Cognitive processing therapy for rape victims: a treatment manual. Newbury Park, CA: Sage, 1993. | |
Eye movement desensitization and reprocessing
Like other PTSD treatments, EMDR is based on an “accelerated information-processing” model.16 Because it also incorporates dissociation and nonverbal representation of traumas (such as visual memories), EMDR is often classified as a cognitive treatment, although ISTSS practice guidelines8 present it as a separate category.
EMDR protocols call for the trauma patient to watch rapid, rhythmic movements of the therapist’s hand or a set of lights to distract attention from the stress he or she feels when visualizing the traumatic event. The original technique—developed by Francine Shapiro, PhD—is based on the observation that persons with PTSD often have disrupted rapid eye-movement sleep. In theory, inducing eye movements inhibits stress, allowing patients to more freely access their memory networks and process disturbances. Subsequently, Dr. Shapiro has suggested that using other auditory cues or hand taps may be as effective as eye movements.16
EMDR is often conducted in 12 to 15 sessions, although some studies report positive changes after 3 to 6 sessions. After obtaining a patient history, establishing rapport, and explaining the treatment, the therapist asks the patient to identify:
- visual images of the trauma
- his or her affective and physiologic responses to the trauma
- negative self-representations the trauma created
- positive, alternate self-representations.
EMDR has been effective in treating male war veterans, rape victims, and other trauma groups.17 Initial dismantling studies suggest that eye movements (or other distracting cues) might not be essential for trauma reprocessing, calling into question the mechanisms thought to create change in EMDR. Studies with larger samples comparing EMDR with other CBT models are needed to assess EMDR’s efficacy for trauma survivors.17
Stress inoculation training
SIT was designed by Meichenbaum18 (Table 3) to treat anxiety and stress and was adapted for use with trauma survivors. It appears most effective in relieving fear, anxiety, and depressive symptoms associated with traumatic experiences. SIT includes education, muscle relaxation training, breathing retraining, covert modeling, role-playing, guided self-dialog, and thought stopping. Therapists often teach these skills to patients in modules that build on each other.
For example, a patient might receive relaxation training while role-playing a difficult scenario she may face in the future. This helps her learn to remain calm in anxiety-provoking situations.
Unlike PE, SIT does not directly ask patients to recount their traumatic memories, although exposure may be indirect (such as during role-playing exercises). Its purpose is to give patients new skills to manage their anxiety, which in turn decreases PTSD symptoms.
Studies suggest that PE is more effective than SIT alone or SIT/PE combined.13 Thus, instead of using SIT as a trauma-focused treatment, some therapists find it useful to help patients gain coping skills before beginning other trauma treatments.
Table 3
Where to learn more about cognitive therapies for PTSD
| CBT model | PTSD related to… | Resources |
|---|---|---|
| Prolonged exposure | Combat experience, sexual assault, childhood abuse, motor vehicle accidents | Foa EB, Rothbaum BO. Treating the trauma of rape: Cognitive-behavioral therapy for PTSD. New York: Guilford Press; 1998 |
| Cognitive processing | Sexual assault, childhood abuse, incarceration (of adolescents) | Resick P, Schnicke M. Cognitive processing therapy for rape victims: a treatment manual. Newbury Park, CA: Sage Publications; 1996 |
| EMDR | Combat experience, sexual assault, civilian disasters (for children or adults) | Shapiro F. Eye movement desensitization and reprocessing: basic principles, protocols, and procedures (2nd ed). New York: Guilford Press; 2001 |
| EMDR Institute, Inc. Available at: http://www.emdr.com | ||
| Stress inoculation training | Sexual and physical assault, motor vehicle accidents | Meichenbaum D. Stress inoculation training for coping with stressors. Available at: http://www.apa.org/divisions/div12/rev_est/sit_stress.html |
| EMDR: Eye movement desensitization and reprocessing | ||
- International Society for Traumatic Stress Studies. www.istss.org.
- Foa EB, Keane TM, Friedman MJ. Effective treatments for PTSD. New York: Guilford Press; 2000.
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Therapists once believed trauma survivors required years of treatment, yet we now know that relatively brief cognitive-behavioral interventions can yield long-term gains in psychosocial and psychological function.1 Many psychiatric patients meet diagnostic criteria for posttraumatic stress disorder (PTSD), including:
- 33% of women experiencing sexual assault2
- 30% of male war veterans3
- 30% of the 5 million U.S. children exposed to trauma each year4(Box).5
We offer recommendations on how to prepare traumatized adults and children for cognitive-behavioral therapy (CBT) and discuss four tested models—prolonged exposure (PE), cognitive processing therapy (CPT), eye movement desensitization and reprocessing (EMDR), and stress inoculation training (SIT)—that psychiatrists may find effective when treating PTSD.
Exposure therapy with children is usually more gradual than with adults, and the child is first taught relaxation techniques to use while recalling traumatic experiences. Although re-exposing children to traumatic events may seem harsh, exposure-based cognitive-behavioral therapy (CBT) appears to be most effective when trauma memories or reminders are most distressing to the child.
As with adults, CBT with children typically includes:
- exposure
- identifying and challenging unhealthy or distorted trauma-related thoughts
- teaching anxiety management techniques such as relaxation or assertiveness training.
In initial studies, CBT has been found safe and effective for treating posttraumatic stress disorder (PTSD) in children and adolescents.17 Through therapy, they can learn not to be afraid of their memories and can develop healthier, more-appropriate thoughts about the trauma. Children with uncomplicated PTSD—without severe, long-term physical injury—typically receive 12 to 20 CBT sessions. More sessions are needed for complex cases, such as when the trauma perpetrator was an integral family member.
Comorbid conditions—such as conduct disorder, attention-deficit/hyperactivity disorder, or depression—may need to be treated before PTSD or concurrently, using medication or other interventions.
Preparing trauma patients for CBT
Before starting CBT, evaluate patients thoroughly to determine if they meet DSM-IV-TR full or subthreshold criteria (
Not all patients are ready to confront their traumas when they arrive for psychiatric evaluation. For example:
- For a domestic violence victim, the therapist’s priority is to help begin safety planning and to address trauma after the patient is out of danger.
- Patients with poor coping skills and little social support often find it difficult to begin trauma treatment. For them, focus on building skills to offset the distress that accompanies trauma therapy.
- Patients with PTSD and substance abuse may benefit more from CBT if the therapist first addresses the substance dependence.
CBT core concepts
CBT therapists typically help patients identify and evaluate disruptive cognitions, which helps them challenge and modify emotions, thoughts, and behaviors related to traumatic experience(s). Other CBT components include:
- educating patients about PTSD
- exposing them to the traumatic material
- challenging and modifying their disruptive thoughts.
International Society for Traumatic Stress Studies (ISTSS) practice guidelines for PTSD8 include assessment and treatment suggestions (see Related resources). Whatever the model, CBT appears help patients manage their distress, not only during treatment but up to 5 years after completing therapy.9
Which CBT? Comparison studies have shown all four CBT interventions to be effective in treating PTSD, although initial trend data suggest that patients with:
- fear-based PTSD may do better with PE or EMDR
- PTSD-related guilt, anger, or other cognitive distortions may benefit more from CPT.
If you refer a patient, make sure the therapist is trained in CBT interventions and in working with trauma patients. To be effective, the therapist must be skilled in handling trauma processing work, suicidal thoughts/intent, and comorbid personality disorders.
Prolonged exposure
PE (Table 1) is typically conducted in 9 to 12 sessions lasting 90 minutes each and has been used to treat PTSD after sexual assault, combat, sexual abuse, and natural disasters. Although frequently offered in individual sessions, group PE has also been found to be effective.10
After educating the patient about PTSD and the treatment rationale, the therapist repeatedly asks the patient to describe the traumatic event as if it were occurring. During 45 to 60 minutes of this exposure, the therapist frequently asks the patient to rate his or her distress. This identifies “hot spots” in the account that need to be repeated. The therapist does not necessarily challenge distorted cognitions about the event (such as “I am to blame for the rape” or “No one can be trusted”).
Researchers hypothesize that exposing a PTSD patient to traumatic memories engages his or her brain’s pathologic “fear network,” which triggers an excessive fear response to non-threatening stimuli. Continued exposure allows the patient to habituate to this network, with subsequent extinction of fear and anxiety reactions. Foa et al11 found that mentally re-experiencing a traumatic event helps patients organize memory cues about it, which encourages cognitive restructuring of the trauma.
PE has been shown to enhance the trauma survivor’s self-control and personal competence and to decrease generalization of fear to non-assault stimuli.12 For example, many combat veterans report fear of situations—such as going to the beach or into the woods—that bring back memories of traumatic events. Their fears may keep them from enjoying a walk in the park or family vacations.
Through in vivo exposure, these patients can face associations between environmental cues and their trauma. As they learn to modify the fears associated with these cues, their personal and social functioning improves.
PE can be successful for those who complete therapy, but it has a relatively high drop-out rate, reported as 8%13 to 41%.14 The pain of continually reliving a traumatic event probably causes many patients to quit. To reduce drop-out rates, many therapists combine PE with cognitive restructuring or other techniques that help build patients’ coping skills.
Table 1
Using prolonged exposure therapy to treat PTSD, session by session
| Session | Content |
|---|---|
| 1 | Education |
| Treatment rationale | |
| Review of PTSD symptom response | |
| Introduce breathing retraining | |
| 2 | Review handout, ‘Common reactions to trauma’ |
| Introduce Subjective Units of Distress | |
| Create fear hierarchy for in vivo exposures | |
| 3 | Provide rationale for imaginal exposure |
| Conduct imaginal exposure | |
| Assign in vivo exposure homework | |
| 4 to 8 | Conduct imaginal exposure |
| Discuss in vivo exposures | |
| 9 or 9 to 12 | Conduct imaginal exposure |
| Suggest continued in vivo exercises | |
| Termination | |
| Source: Foa EB, Rothbaum BO. Treating the trauma of rape: cognitive behavioral therapy for PTSD. New York: Guilford Press, 1998. | |
Cognitive processing therapy
CPT (Table 2) was created as a protocol to treat PTSD and related symptoms in rape survivors.7 Sessions can be group, individual, or combined, depending on the needs and resources of the patients and clinic.
Originally, CPT contained 12 weekly sessions, although versions up to 17 weeks have been developed for adult survivors of child sexual abuse, domestic violence survivors, and war veterans.15 Sessions can be added or adapted to address each population’s type of traumatic experience (such as developmental impairment of sexual abuse survivors).
CPT is based on information processing theory, which suggests that as people access a traumatic memory, they experience and extinguish emotions attached to the event. Guided by the therapist, the patient identifies and challenges distortions the trauma created in three cognition domains: the self, others, and the world. Patients learn to change or replace these cognitive distortions—which therapists often call “stuck points” or “rules”—with more-adaptive, healthier beliefs.
Common byproducts of trauma are feeling out of control or hopeless. Thus, CPT focuses on personal safety, trust, power/control, esteem, and intimacy within each of the three domains. Modules on assertiveness, communication, and social support can also be added.
Although CPT is being adapted for populations other than rape survivors, comparison studies are needed to determine if it is as effective as other CBT therapies for these groups.
Table 2
Using cognitive processing therapy to treat PTSD, session by session
| Session | Content |
|---|---|
| 1 | Education |
| Review of symptoms | |
| Introduce ‘stuck points’/rules | |
| Write impact of event statement (IES) | |
| 2 | Review IES |
| Identify stuck points | |
| Introduce A-B-C sheets | |
| 3 | Review A-B-C sheets |
| Assign writing of traumatic account | |
| 4 | Read traumatic account |
| Identify stuck points | |
| Rewrite the account | |
| 5 | Read rewritten account |
| Identify stuck points | |
| Introduce challenging questions sheet (CQS) | |
| Assign writing of next-most traumatic incident and CQS | |
| 6 | Review CQS |
| Assign review of faulty thinking patterns (FTP) | |
| 7 | Review FTP |
| Assign safety module and challenging beliefs worksheets (CBW) on safety | |
| 8 | Review CBWs on safety |
| Assign module on trust | |
| 9 | Review CBWs on trust |
| Assign module on power/control | |
| 10 | Review CBWs on power/control |
| Assign module on esteem | |
| 11 | Review CBWs on esteem |
| Assign module on intimacy | |
| Rewrite IES | |
| 12 | Review CBWs on intimacy |
| Read both impact statements | |
| Address remaining areas of concern | |
| Termination | |
| Source: Resick PA, Schnicke MK Cognitive processing therapy for rape victims: a treatment manual. Newbury Park, CA: Sage, 1993. | |
Eye movement desensitization and reprocessing
Like other PTSD treatments, EMDR is based on an “accelerated information-processing” model.16 Because it also incorporates dissociation and nonverbal representation of traumas (such as visual memories), EMDR is often classified as a cognitive treatment, although ISTSS practice guidelines8 present it as a separate category.
EMDR protocols call for the trauma patient to watch rapid, rhythmic movements of the therapist’s hand or a set of lights to distract attention from the stress he or she feels when visualizing the traumatic event. The original technique—developed by Francine Shapiro, PhD—is based on the observation that persons with PTSD often have disrupted rapid eye-movement sleep. In theory, inducing eye movements inhibits stress, allowing patients to more freely access their memory networks and process disturbances. Subsequently, Dr. Shapiro has suggested that using other auditory cues or hand taps may be as effective as eye movements.16
EMDR is often conducted in 12 to 15 sessions, although some studies report positive changes after 3 to 6 sessions. After obtaining a patient history, establishing rapport, and explaining the treatment, the therapist asks the patient to identify:
- visual images of the trauma
- his or her affective and physiologic responses to the trauma
- negative self-representations the trauma created
- positive, alternate self-representations.
EMDR has been effective in treating male war veterans, rape victims, and other trauma groups.17 Initial dismantling studies suggest that eye movements (or other distracting cues) might not be essential for trauma reprocessing, calling into question the mechanisms thought to create change in EMDR. Studies with larger samples comparing EMDR with other CBT models are needed to assess EMDR’s efficacy for trauma survivors.17
Stress inoculation training
SIT was designed by Meichenbaum18 (Table 3) to treat anxiety and stress and was adapted for use with trauma survivors. It appears most effective in relieving fear, anxiety, and depressive symptoms associated with traumatic experiences. SIT includes education, muscle relaxation training, breathing retraining, covert modeling, role-playing, guided self-dialog, and thought stopping. Therapists often teach these skills to patients in modules that build on each other.
For example, a patient might receive relaxation training while role-playing a difficult scenario she may face in the future. This helps her learn to remain calm in anxiety-provoking situations.
Unlike PE, SIT does not directly ask patients to recount their traumatic memories, although exposure may be indirect (such as during role-playing exercises). Its purpose is to give patients new skills to manage their anxiety, which in turn decreases PTSD symptoms.
Studies suggest that PE is more effective than SIT alone or SIT/PE combined.13 Thus, instead of using SIT as a trauma-focused treatment, some therapists find it useful to help patients gain coping skills before beginning other trauma treatments.
Table 3
Where to learn more about cognitive therapies for PTSD
| CBT model | PTSD related to… | Resources |
|---|---|---|
| Prolonged exposure | Combat experience, sexual assault, childhood abuse, motor vehicle accidents | Foa EB, Rothbaum BO. Treating the trauma of rape: Cognitive-behavioral therapy for PTSD. New York: Guilford Press; 1998 |
| Cognitive processing | Sexual assault, childhood abuse, incarceration (of adolescents) | Resick P, Schnicke M. Cognitive processing therapy for rape victims: a treatment manual. Newbury Park, CA: Sage Publications; 1996 |
| EMDR | Combat experience, sexual assault, civilian disasters (for children or adults) | Shapiro F. Eye movement desensitization and reprocessing: basic principles, protocols, and procedures (2nd ed). New York: Guilford Press; 2001 |
| EMDR Institute, Inc. Available at: http://www.emdr.com | ||
| Stress inoculation training | Sexual and physical assault, motor vehicle accidents | Meichenbaum D. Stress inoculation training for coping with stressors. Available at: http://www.apa.org/divisions/div12/rev_est/sit_stress.html |
| EMDR: Eye movement desensitization and reprocessing | ||
- International Society for Traumatic Stress Studies. www.istss.org.
- Foa EB, Keane TM, Friedman MJ. Effective treatments for PTSD. New York: Guilford Press; 2000.
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Rothbaum BO, Meadows EA, Resick PA, Foy DW. Cognitive-behavioral therapy. In: Foa E, Keane T, Friedman M (eds). Effective treatments for PTSD. New York: Guilford Press; 2000.
2. Kilpatrick D, Edmonds CN, Seymour AK. Rape in America: A report to the nation. Arlington, VA: National Victims Center; 1992.
3. Kulka RA, Schlenger WE, Fairbank JA, et al. Trauma and the Vietnam War generation: report of findings from the National Vietnam Veterans Readjustment Study. New York: Brunner/Mazel; 1990.
4. Pfefferbaum B. Posttraumatic stress disorder in children: a review of the past 10 years. J Am Acad Child Adolesc Psychiatry 1997;36(11):1503-11.
5. Deblinger E, McLeer SV, Henry D. Cognitive behavioral treatment for sexually abused children suffering post-traumatic stress. J Am Acad Child Adolesc Psychiatry 1990;5:747-52.
6. Najavits LM. Seeking Safety: a treatment manual for PTSD and substance abuse. New York: Guilford Press; 2002.
7. Resick PA, Nishith P, Weaver TL, et al. A comparison of cognitive processing therapy, prolonged exposure and a waiting condition for the treatment of posttraumatic stress disorder in female rape victims. J Consult Clin Psychol 2002;70:867-79.
8. Foa EB, Keane TM. Friedman MJ (eds). Effective treatments for PTSD. New York: Guilford Press; 2000.
9. Tarrier N, Sommerfield C. Treatment of chronic PTSD by cognitive therapy and exposure: 5-year follow-up. Behavior Ther 2004;35(2):231-46.
10. Foa EB, Rauch SA. Cognitive changes during prolonged exposure versus prolonged exposure plus cognitive restructuring in female assault survivors with posttraumatic stress disorder. J Consult Clin Psychol 2004;72(5):879-84.
11. Foa EB, Riggs DS, Massie ED, Yarczower M. The impact of fear activation and anger on the efficacy of exposure treatment for PTSD. Behav Ther 1995;26:487-99.
12. Foa EB, Rothbaum EO, Riggs D, Murdock T. Treatment of PTSD in rape victims: a comparison between cognitive-behavioral procedures and counseling. J Consult Clin Psychol 1991;59:715-23.
13. Foa EB, Dancu CV, Hembree EA, Jaycox LH, et al. A comparison of exposure therapy, stress inoculation training, and their combination for reducing posttraumatic stress disorder in female assault victims. J Consult Clin Psychol 1999;67:194-200.
14. McDonagh A, Friedman M, McHugo G, et al. Randomized trial of cognitive-behavioral therapy for chronic posttraumatic stress disorder in adult female survivors of childhood sexual abuse. J Consult Clin Psychol 2005;73:515-24.
15. Chard K. An evaluation of cognitive processing therapy for the treatment of posttraumatic stress disorder related to childhood sexual abuse. J Consult Clin Psychol. (in press).
16. Shapiro F. Eye movement desensitization and reprocessing: basic principles, protocols and procedures (2nd ed). New York: Guilford Press; 2001.
17. Chemtob CM, Tolin DF, van der Kolk BA, Pitman RK. Eye movement desensitization and reprocessing. In: Foa E, Keane T, Friedman M (eds). Effective treatments for PTSD. New York: Guilford Press; 2000.
18. Meichenbaum D. Cognitive-behavior modification: An integrative approach. New York: Plenum Press; 1977.
1. Rothbaum BO, Meadows EA, Resick PA, Foy DW. Cognitive-behavioral therapy. In: Foa E, Keane T, Friedman M (eds). Effective treatments for PTSD. New York: Guilford Press; 2000.
2. Kilpatrick D, Edmonds CN, Seymour AK. Rape in America: A report to the nation. Arlington, VA: National Victims Center; 1992.
3. Kulka RA, Schlenger WE, Fairbank JA, et al. Trauma and the Vietnam War generation: report of findings from the National Vietnam Veterans Readjustment Study. New York: Brunner/Mazel; 1990.
4. Pfefferbaum B. Posttraumatic stress disorder in children: a review of the past 10 years. J Am Acad Child Adolesc Psychiatry 1997;36(11):1503-11.
5. Deblinger E, McLeer SV, Henry D. Cognitive behavioral treatment for sexually abused children suffering post-traumatic stress. J Am Acad Child Adolesc Psychiatry 1990;5:747-52.
6. Najavits LM. Seeking Safety: a treatment manual for PTSD and substance abuse. New York: Guilford Press; 2002.
7. Resick PA, Nishith P, Weaver TL, et al. A comparison of cognitive processing therapy, prolonged exposure and a waiting condition for the treatment of posttraumatic stress disorder in female rape victims. J Consult Clin Psychol 2002;70:867-79.
8. Foa EB, Keane TM. Friedman MJ (eds). Effective treatments for PTSD. New York: Guilford Press; 2000.
9. Tarrier N, Sommerfield C. Treatment of chronic PTSD by cognitive therapy and exposure: 5-year follow-up. Behavior Ther 2004;35(2):231-46.
10. Foa EB, Rauch SA. Cognitive changes during prolonged exposure versus prolonged exposure plus cognitive restructuring in female assault survivors with posttraumatic stress disorder. J Consult Clin Psychol 2004;72(5):879-84.
11. Foa EB, Riggs DS, Massie ED, Yarczower M. The impact of fear activation and anger on the efficacy of exposure treatment for PTSD. Behav Ther 1995;26:487-99.
12. Foa EB, Rothbaum EO, Riggs D, Murdock T. Treatment of PTSD in rape victims: a comparison between cognitive-behavioral procedures and counseling. J Consult Clin Psychol 1991;59:715-23.
13. Foa EB, Dancu CV, Hembree EA, Jaycox LH, et al. A comparison of exposure therapy, stress inoculation training, and their combination for reducing posttraumatic stress disorder in female assault victims. J Consult Clin Psychol 1999;67:194-200.
14. McDonagh A, Friedman M, McHugo G, et al. Randomized trial of cognitive-behavioral therapy for chronic posttraumatic stress disorder in adult female survivors of childhood sexual abuse. J Consult Clin Psychol 2005;73:515-24.
15. Chard K. An evaluation of cognitive processing therapy for the treatment of posttraumatic stress disorder related to childhood sexual abuse. J Consult Clin Psychol. (in press).
16. Shapiro F. Eye movement desensitization and reprocessing: basic principles, protocols and procedures (2nd ed). New York: Guilford Press; 2001.
17. Chemtob CM, Tolin DF, van der Kolk BA, Pitman RK. Eye movement desensitization and reprocessing. In: Foa E, Keane T, Friedman M (eds). Effective treatments for PTSD. New York: Guilford Press; 2000.
18. Meichenbaum D. Cognitive-behavior modification: An integrative approach. New York: Plenum Press; 1977.
Are anticonvulsants safe for pediatric bipolar disorder?
Are anticonvulsants safe and effective mood stabilizers for children and adolescents with bipolar disorder? The answer is unclear because most bipolar disorder treatment trials have included adults only, and clinicians are desperate for data.1
To help you care for young patients, we report what is known about the potential benefits and risks of using mood stabilizers and anticonvulsants in bipolar youth. We base our dosing, target serum level, and monitoring recommendations on clinical experience and the limited published evidence.
AGENTS OF CHOICE?
Bipolar disorder’s “atypical” presentation in children—often more irritability and explosiveness than euphoria—can complicate diagnosis. Bipolar children and adolescents often have comorbid attention-deficit/hyperactivity disorder (ADHD), other disruptive behavior disorders, or anxiety disorders. Thus, comorbidities and presenting symptoms often dictate medication choice.
An expert consensus guideline acknowledges that more evidence on pediatric bipolar disorder is needed. In the meantime, the guideline suggests trying valproate or lithium first to treat nonpsychotic mania in pediatric bipolar patients.1 It also recommends three atypical antipsychotics— olanzapine, quetiapine, and risperidone—as potential first-line treatments. Valproate and lithium may be preferred because of atypicals’ risk of weight gain and metabolic syndrome.
Trying other anticonvulsants may be justified for bipolar youths who are not functioning well with first-line agents. Lamotrigine, for example, has antidepressant and antimanic effects.2 When you try anticonvulsants that lack double-blind, placebo-controlled trials, we recommend that you:
- obtain consent from the parents and child
- monitor carefully for side effects.
LITHIUM: STRONGEST EVIDENCE
Lithium is one of the most well-studied medications for pediatric bipolar disorder and the only mood stabilizer FDA-approved for children and adolescents (Table 1).3 Although approved for ages 12 and older, lithium has been used in younger children in practice and in clinical trials.
Table 1
FDA-approval status of medications used to treat bipolar disorder
| Medication | Indications for adults | Indications for children |
|---|---|---|
| Carbamazepine | Acute manic episode and acute mixed episode | Not approved |
| Lamotrigine | Maintenance therapy | Not approved |
| Lithium | Acute manic episode and maintenance therapy | Age ≥ 12 years |
| Oxcarbazepine | Not approved | Not approved |
| Topiramate | Not approved | Not approved |
| Valproate | Acute manic episode | Not approved |
| Source: Reference 3 | ||
In the only double-blind, placebo-controlled trial of lithium in adolescents with bipolar disorder, some subjects had secondary substance dependency disorders.7 For 6 weeks, 25 outpatient adolescents received lithium (13 patients) or placebo (12 patients). Lithium was effective in treating bipolar and substance dependency symptoms, with significantly improved clinical global assessment scores and decreased positive urine assays for drugs. Little difference was seen in mood item scores on the Schedule for Affective Disorders and Schizophrenia, child version (KSADS-1986), whether patients were taking lithium or placebo.
Pediatric dosing. For bipolar patients ages 6 to 12, use the child’s weight to determine lithium dosage (Table 2).8 Maintain serum levels between 0.8 and 1.2 mEq/L,9 and check them frequently when starting therapy.10 After mood stabilization, check levels every 1 to 3 months or when you suspect noncompliance. Obtain renal and thyroid function values at baseline and every 4 to 6 months.
Table 2
Guide to dosing lithium for prepubertal school-aged children*
| Doses (mg) | ||||
|---|---|---|---|---|
| Child’s weight (kg) | 8 AM | 12 PM | 6 PM | Total daily |
| 150 | 150 | 300 | 600 | |
| 25 to 40 | 300 | 300 | 300 | 900 |
| 40 to 50 | 300 | 300 | 600 | 1,200 |
| 50 to 60 | 600 | 300 | 600 | 1,500 |
| * Maintain specified dose at least 5 days, drawing serum levels 12 hrs after the last lithium dose until two consecutive levels appear in the therapeutic range (0.6 to 1.2 mEq/L). Dose may then be adjusted based on serum level, side effects, or clinical response. Do not exceed 1.4 mEq/L. | ||||
| Source: Reference 8 | ||||
Consider teratogenicity when choosing mood stabilizers for bipolar adolescent girls who may be sexually active. Lithium, valproate, and carbamazepine are labeled pregnancy category D because of their potential to cause birth defects.
Lithium treatment has been associated with increased risk of cardiac defects, specifically Ebstein’s anomaly (malformation of the tricuspid valve). Its incidence in children of women who used lithium during pregnancy is estimated to be 1:1,000 (0.10%) to 2:1,000 (0.05%)— 20 to 40 times the rate in the general population.12
Valproate.Results from the North American Antiepileptic Drug (AED) Pregnancy Registry showed a 10.7% rate of major congenital malformations (MCM)— including neural tube defects (spina bifida) and cardiac defects (pulmonary atresia)—in children of women who used valproate during pregnancy. The rate of births with MCMs in the general population is 2.9%.13
Carbamazepine.Data from the Australian Pregnancy Registry showed no significant increase in malformation rates in infants of carbamazepine users compared with those of women receiving no antiepileptics.14 Other studies, however, have linked carbamazepine with an increased risk of craniofacial defects (11%), neural tube defects (0.5 to 1%), and cardiac malformations.12
Lamotrigine.The teratogenic effects of the newer anticonvulsants are unclear. An 11-year study of lamotrigine15 found MCM risk after first-trimester exposure to lamotrigine to be similar to the general population’s MCM risk.
Combination therapy.Teratogenic risk appears to increase when multiple antiepileptic drugs are used (9.9% risk in polytherapy vs 6.2% in monotherapy).16
VALPROATE: OPEN-LABEL TRIALS ONLY
Efficacy. No double-blind, placebo-controlled study has shown valproate to be effective in treating bipolar disorder in children and adolescents. When used as monotherapy in open-label studies, valproate has produced response rates of:
- 53% in a 6-week, randomized, open-label trial in which 42 outpatients (mean age 11.4 years) with bipolar disorder type I or II received lithium, divalproex sodium, or carbamazepine9
- 61% in an open-label study of 40 patients ages 7 to 19 with a manic, hypomanic, or mixed episode who received divalproex for 2 to 8 weeks17
- 80% in an 8-week open-label trial of 40 patients ages 6 to 17 with bipolar disorder type I (77.5%) or type II (22.5%) and a Young Mania Rating Scale (YMRS)score ≥ 14.18
Safety: Black-box warnings. Valproate therapy carries risks of hepatic failure, pancreatitis, and birth defects. Monitor blood counts and hepatic enzymes throughout therapy (Table 3).3 Rare yet potentially fatal hepatic toxicity appears to occur most often in children age 21 Other studies suggest:
- an association with congenital malformations, including spina bifida and pulmonary atresia, in children exposed to valproate in utero6
- a link between valproate and hyperammonemic encephalopathy, especially in patients with urea cycle disorders22
- potential for benign thrombocytopenia23
- increased incidence of polycystic ovary syndrome—ovarian cysts, hyperandrogenism, chronic anovulation—in peripubertal mentally retarded women treated with valproate for seizure disorders.24
Table 3
Mood stabilizers’ side effects and recommended monitoring
| Medication | Major side effects | Monitoring |
|---|---|---|
| Carbamazepine | Allergic skin rash, drowsiness, blood dyscrasias, diplopia | CBC with reticulocytes, iron, LFTs, urinalysis, BUN, TFTs, sodium, serum carbamazepine levels |
| Lamotrigine | Stevens-Johnson syndrome, headache, dizziness, ataxia, somnolence, nausea, diplopia, blurred vision, rhinitis | No serum monitoring recommended |
| Lithium | Polyuria, polydipsia, nausea, diarrhea, tininecleatremor, enuresis, fatigue, ataxia, leukocytosis, malaise, cardiac arrhythmias, weight gain | BUN/creatinine, crearance, TFTs, calcium/phosphorus, ECG, serum lithium levels every 1 to 3 months once stabilized |
| Oxcarbazepine | Dizziness, somnolence/fatigue, ataxia/gait disturbance, vertigo, headache, tremor, rash, hyponatremia, hypersensitivity reaction, GI symptoms, diplopia | Sodium levels (particularly ifirst 3 months) |
| Topiramate | Hyperchloremic metabolic acidosis, oligohydrosis and hyperthermia, acute myopia, somnolence/fatigue, nausea, anorexia/weight loss, paresthesia, tremor, difficulty concentrating | BUN/creatinine, sodium bicarbonate |
| Valproate | Irritability/restlessness, ataxia, headache, weight gain, hyperammonemic encephalopathy, alopecia, GI upset, pancreatitis,sedation, thrombocytopenia, liver failure, polycystic ovaries/hyperandrogenism, teratogenic effects,rash | Ammonia, LFTs, bilirubin, CBC with platelets, serum valproate levels |
| BUN: blood urea nitrogen; CBC: complete blood count; ECG: electrocardiography; LFT: liver function tests; TFTs: thyroid function tests | ||
| Note: Bolded items included in black-box warnings | ||
| Source: Reference 3 | ||
CARBAMAZEPINE: DRUG INTERACTION RISK
Carbamazepine is used less often than lithium or divalproex for bipolar disorder. It tends to be used adjunctively when lithium alone is ineffective.
Efficacy. In an open-label study,9 42 patients ages 8 to 18 with bipolar disorder type I or II were randomly assigned to lithium, divalproex sodium, or carbamazepine monotherapy for 6 weeks. Response rates—measured as a ≥ 50% change from baseline in YMRS scores—were 53% with divalproex, 38% with lithium, and 38% with carbamazepine.
A retrospective review of 44 hospitalized bipolar patients ages 5 to 12 treated for at least 7 days with lithium, valproate, or carbamazepine reported higher (ie, worse) Clinical Global Impression of Improvement scores with carbamazepine.27 Small sample sizes, particularly in the carbamazepine group, limited this naturalistic study.
Safety: Black-box warnings. Carbamazepine’s hematologic “black box” warns of increased risk of aplastic anemia, agranulocytosis, leukopenia, and thrombocytopenia. Risks associated with carbamazepine have been estimated at:
- aplastic anemia: 5.1/million patient years
- agranulocytosis: 1.4/million patient years.28
Body weight. Carbamazepine is not associated with significant weight gain, which could be clinically important for some patients.
Drug interactions. Carbamazepine activates the cytochrome P-450 liver enzyme system, increasing the metabolism of many medications and decreasing their blood levels. Consider monitoring serum levels when using carbamazepine with valproate, imipramine, corticosteroids, warfarin, oral contraceptives, and some antibiotics. Because carbamazepine induces its own metabolism, you might need to increase its dosage if its effects appear to be waning.3
Carbamazepine and tricyclic antidepressants may show cross-sensitivity because of structural similarity. Do not use monoamine oxidase inhibitors with carbamazepine; discontinue them at least 14 days before starting carbamazepine.3
OXCARBAZEPINE: FEWER INTERACTIONS
Oxcarbazepine has similar efficacy to carbamazepine but less side effect risk and does not require plasma level monitoring. A weaker inducer of CYP-450, it causes fewer clinically important drug-drug interactions and may be useful for patients who respond to carbamazepine but cannot tolerate its side effects.30
Efficacy. Case studies31,32 have been encouraging, but no published, double-blind, placebo-controlled studies support using oxcarbazepine in bipolar children and adolescents.
Safety. Oxcarbazepine appears to be generally well-tolerated but can cause potentially serious reactions—including hyponatremia.33 Somnolence, emesis, and ataxia are the most common side effects in pediatric patients.3
Hyponatremia —plasma sodium 125 mEq/L—occurs in 2.5% of adults taking oxcarbazepine3 and has been reported in a similar percentage of children.34 This potentially severe reaction—characterized by nausea, lethargy, malaise, headache, confusion, decreased seizure threshold, or simply decreased serum sodium35—is usually noted within the first 12 weeks of therapy. The risk increases with concomitant use of other sodium-altering drugs, such as antidepressants or antipsychotics.36
Evaluate serum sodium when starting oxcarbazepine, periodically in the first 3 months, and if symptoms occur.34,36 For sodium levels of 125 to 130 mEq/L, obtain repeat measurements to confirm that hyponatremia is not worsening. Intervention is often required when levels fall below 125 mEq/L.36
Other serious adverse reactions include Stevens-Johnson syndrome, toxic epidermal necrolysis, and hypersensitivity reactions; 25% to 30% of patients with hypersensitivity to carbamazepine also will react to oxcarbazepine.33
Contraceptive concerns. Oxcarbazepine may reduce contraceptive efficacy by altering estrogen and progesterone plasma concentrations.37 Consider other birth control methods for sexuallyactive bipolar adolescent girls.
LAMOTRIGINE
Neurologists often use lamotrigine for children with atypical seizure disorders, but no controlled data exist on the drug’s efficacy and safety in youths with bipolar disorder.
Efficacy. In a prospective, open-label study,38 13 adolescents with type I bipolar disorder received lamotrigine, 200 to 400 mg/d. After 12 weeks (mean dosage 241 mg/d), their symptoms had improved as shown by these mean scores:
- Montgomery-Asberg Depression Rating Scale: from 21 at baseline to 4 at endpoint
- Clinical Global Impressions–Severity of Illness scale: from 4 to 1
- Children’s Depression Rating Scale (CDRS-R): from 74 to 40
- YMRS: from 20 to 6.
Safety: Severe rash. An age-related association with Stevens-Johnson syndrome may limit pediatric use of lamotrigine. Severe and potentially life-threatening rashes have been reported in 0.8% of children treated with lamotrigine.40 Discontinue lamotrigine if a rash develops, unless it clearly is not drug-related. Three factors that increase rash risk include:
- co-administering lamotrigine with valproate
- higher-than-recommended initial dosages
- rapid dose titration.41
Pediatric dosing. We find no published studies of efficacious dosages and plasma levels of lamotrigine in pediatric bipolar disorder (Table 4).3,9,17 Based on our clinical experience, we recommend starting lamotrigine at 1 to 5 mg/kg/day (1 to 3 mg/kg/day if given with valproate) divided into two daily doses. Watch for rash or skin disorders. Do not exceed the recommended daily dosage by 200 mg in children age
Table 4
Using anticonvulsants in pediatric bipolar disorder patients
| Drug | Recommended dosage | Target serum level |
|---|---|---|
| Carbamazepine | Age 6 to 12: 20 to 30 mg/kg/d | ≥ 7.0 μg/L |
| Age >12: 400 to 1,200 mg/d | ||
| Lamotrigine* | Unknown | Unknown |
| Oxcarbazepine | 11 to 16 mg/kg/d (as adjunct) | Unknown |
| Topiramate* | Unknown | Unknown |
| Valproate | 15 to 20 mg/kg/d | 45 to 125 μg/mL (trough) |
| 85 to 110 μg/mL (target) | ||
| * No published studies found for efficacious dosage and plasma levels in pediatric bipolar disorder. | ||
| Dosage supported by case reports only; no studies found examining efficacious plasma levels. | ||
| Source: References 3,9, and 17. | ||
TOPIRAMATE: LIMITED INFORMATION
Efficacy. Little is known about using topiramate in children and adolescents. A retrospective chart review42 of 26 patients with bipolar disorder type I (n=23) or II (n=3) showed adjunctive topiramate to be effective, with response rates of 73% for mania and 62% overall. Topiramate was well tolerated, and no serious events were reported.
A randomized, controlled trial of topiramate for acute mania in youths with type I bipolar disorder43 was recently halted because of lack of efficacy in adul trials. Preliminary data from 56 of the pediatric patients—analyzed before the study was halted—showed improved YMRS scores. Although results were not statistically significant, the authors suggest topiramate might be effective in treating children and adolescents with bipolar disorder.
Safety: FDA warning. Decreased sodium bicarbonate leading to hyperchloremic metabolic acidosis has been reported in youths treated with topiramate for seizure disorder,44 leading to an FDA warning to prescribers.3 Although no monitoring guidelines exist, we recommend baseline and periodic serum bicarbonate measurements and acidbase evaluations during topiramate treatment, especially when adding other antiepileptics.44
Other rare but serious reactions include:
- impaired sweat production and resultant hyperthermia45
- ophthalmologic symptoms characterized by secondary acute angle closure glaucoma and acute myacute myopia (usually within 1 month of starting treatment)46
- sedation and cognitive difficulties.47
Cognitive effects? Reports of “word finding difficulties” with topiramate47 may suggest cognitive effects. Thus, be very cautious about using this medication in children and adolescents.
Related resources
- Kowatch R, Fristad M, Birmaher B, et al. Treatment guidelines for children and adolescents with bipolar disorder. J Am Acad Child Adolesc Psychiatry. 2005;44(3):213-35.
- Yonkers K, Wisner K, Stowe Z, et al. Management of bipolar disorder during pregnancy and the postpartum period. Am J Psychiatry 2004;161(4):608-20.
- American Academy of Child and Adolescent Psychiatry. Treatment guidelines for childhood psychiatric disorders. www.aacap.org
Dr. Kloos, Dr. Hitchcock, and Dr. Ronald Weller report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Elizabeth Weller has been a consultant to or received research grants from GlaxoSmithKline, Johnson & Johnson, Novartis Pharmaceuticals Corp., Abbott Laboratories, and Shire Pharmaceuticals.
Drug brand names
- Carbamazepine • Tegretol
- Divalproex • Depakote
- Lamotrigine • Lamictal
- Lithium • Eskalith, Lithobid, others
- Oxcarbazepine • Trileptal
- Topiramate • Topamax
1. Kowatch R, Fristad M, Birmaher B, et al. Treatment guidelines for children and adolescents with bipolar disorder. J Am Acad Child Adolesc Psychiatry 2005;44(3):213-35
2. Wang P, Ketter T, Becker O, et al. New anticonvulsant medication uses in bipolar disorder. CNS Spectrums 2003;8(12):930-47.
3. Prescribing information. Physicians’ Desk Reference (59th ed.) Montvale, NJ: Thomson Healthcare, 2005.
4. Kafantaris V, Coletti D, Dicker R, et al. Lithium treatment of acute mania in adolescents: a large open trial. J Am Acad Child Adolesc Psychiatry 2003;42(9):1038-45.
5. Kafantaris V, Coletti D, Dicker R, et al. Lithium treatment of acute mania in adolescents: a placebo-controlled discontinuation study. J Am Acad Child Adolesc Psychiatry 2004;43(8):984-93.
6. Strober M, DeAntonio M, Schmidt-Lackner S, et al. Early childhood attention deficit hyperactivity disorder predicts poorer response to acute lithium therapy in adolescent mania. J Affect Disord 1998;51(2):145-51.
7. Geller B, Cooper T, Zimerman B, et al. Lithium for prepubertal depressed children with family history predictors of future bipolarity: a double-blind, placebo-controlled study. J Affect Disord 1998;51(2):165-75.
8. Weller E, Weller R, Fristad M. Lithium dosage guide for prepubertal children: a preliminary report. J Am Acad Child Adolesc Psychiatry 1986;25(1):92-5.
9. Kowatch R, Suppes T, Carmody T, et al. Effect size of lithium, divalproex sodium, and carbamazepine in children and adolescents with bipolar disorder. J Am Acad Child Adolesc Psychiatry 2000;39(6):713-20.
10. Hagino O, Weller E, Weller R, et al. Untoward effects of lithium treatment in children aged four through six years. J Am Acad Child Adolesc Psychiatry 1995;34(12):1584-90.
11. Hagino O, Weller E, Weller R, Fristad M. Comparison of lithium dosage methods for preschool- and early school-age children. J Am Acad Child Adolesc Psychiatry 1995;37(1):60-5.
12. Yonkers K, Wisner K, Stowe Z, et al. Management of bipolar disorder during pregnancy and the postpartum period. Am J Psychiatry 2004;161(4):608-20.
13. Holmes L. The North American antiepileptic drug pregnancy registry: a seven-year experience. (paper presentation). New Orleans: American Epilepsy Society annual meeting, 2004.
14. Vajda F, Lander C, Cook M, et al. Antiepileptic medication in pregnancy: the Australian Pregnancy Register: 52 months data (poster presentation). New Orleans: American Epilepsy Society annual meeting, 2004.
15. Messenheimer J, Tennis P, Cunnington M. Eleven-year interim results of an international observational study of pregnancy outcomes following exposure to lamotrigine (poster presentation). New Orleans: American Epilepsy Society annual meeting, 2004.
16. Torbjorn T, Battino D, Bonizzoni E, et al. Eurap: an international registry of antiepileptic drugs and pregnancy (poster presentation). New Orleans: American Epilepsy Society annual meeting, 2004.
17. Wagner K, Weller E, Carlson G, et al. An open-label trial of divalproex in children and adolescents with bipolar disorder. J Am Acad Child Adolesc Psychiatry 2002;41(10):1224-30.
18. Scheffer RE, Kowatch RA, Carmody T, Rush AJ. Randomized, placebo-controlled trial of mixed amphetamine salts for symptoms of comorbid ADHD in pediatric bipolar disorder after mood stabilization with divalproex sodium. Am J Psychiatry 2005;162(1):58-64.
19. Findling R, McNamara N, Gracious B, et al. Combination lithium and divalproex sodium in pediatric bipolarity. J Am Acad Child Adolesc Psychiatry 2003;42(8):895-901.
20. DelBello M, Adler C, Strakowski S. Divalproex for the treatment of aggression associated with adolescent mania. J Child Adolesc Psychopharmacol 2004;14(2):325-8.
21. Anderson G. Children versus adults: pharmokinetic and adverseeffect differences. Epilepsia 2002;43(suppl 3):53-9.
22. Yehya N, Saldarini C, Koski M, et al. Valproate-induced hyperammonemic encephalopathy. J Am Acad Child Adolesc Psychiatry 2004;43(8):926-7.
23. Verrotti A, Greco R, Matera V, et al. Platelet count and function in children receiving sodium valproate. Pediatr Neurol 1999;21(3):611-14.
24. Isojarvi J, Tauboll E, Pakarinen A, et al. Altered ovarian function and cardiovascular risk factors in valproate-treated women. Am J Med 2001;111(4):290-6.
25. Bowden C, Calabrese J, McElroy S, et al. A randomized, placebocontrolled 12-month trial of divalproex and lithium in treatment of outpatients with bipolar I disorder. Arch Gen Psychiatry 2000;57(5):481-9.
26. Findling R, Gracious B, McNamara N, et al. Rapid, continuous cycling and psychiatric co-morbidity in pediatric bipolar I disorder. Bipolar Disord 2001;3(4):202-10.
27. Davanzo P, Gunderson B, Belin T, et al. Mood stabilizers in hospitalized children with bipolar disorder: a retrospective review. Psychiatry Clin Neurosci 2003;57(5):504-10.
28. Pellock J. Carbamazepine side effects in children and adults. Epilepsia 1987;28(suppl 3):64-70.
29. Sobotka J, Alexander B, Cook B. A review of carbamazepine’s hematologic reactions and monitoring. DICP 1990;24(12):1214-19.
30. Hellewell J. Oxcarbazepine (Trileptal) in the treatment of bipolar disorders: a review of efficacy and tolerability. J Affect Disord 2002;72(supp 1):23-34.
31. Davanzo P, Nikore V, Yehya N, Stevenson L. Oxcarbazepine treatment of juvenile-onset bipolar disorder. J Child Adolesc Psychopharmacol 2004;14(3):344-5.
32. Teitelbaum M. Oxcarbazepine in bipolar disorder. J Am Acad Child Adolesc Psychiatry 2001;40(9):993-4.
33. Dietrich D, Kropp S, Emrich H. Oxcarbazepine in affective and schizoaffective disorders. Pharmacopsychiatry 2001;34(6):242-50.
34. Holtmann M, Krause M, Opp J, et al. Oxcarbazepine-induced hyponatremia and the regulation of serum sodium after replacing carbamazepine with oxcarbazepine in children. Neuropediatrics 2002;33(6):298-300.
35. Prescribing information for oxcarbazepine (Trileptal) Novartis Pharmaceuticals Corp. 2005. Available at: http://www.pharma.us. novartis.com/product/pi/pdf/trileptal.pdf. Accessed June 26, 2005.
36. Asconape J. Some common issues in the use of antiepileptic drugs. Semin Neurol 2002;22(1):27-39.
37. Fattore C, Cipolla G, Gatti G, et al. Induction of ethinylestradiol and levonorgestrel metabolism by oxcarbazepine in healthy women. Epilepsia 1999;40(6):783-7.
38. Swope G, Hoopes S, Amy L, et al. An open-label study of lamotrigine in adolescents with bipolar mood disorder (poster presentation). New York: American Psychiatric Association annual meeting, 2004.
39. Saxena K, Howe M, Chang K. Lamotrigine adjunct or monotherapy for adolescent bipolar depression or mixed mania (poster presentation). Washington, DC: American Academy of Child and Adolescent Psychiatry annual meeting, 2004.
40. Prescribing information for lamotrigine (Lamictal). GlaxoSmith Kline 2004. Available at: http://us.gsk.com/products/assets/us_ lamictal.pdf. Accessed June 2, 2005.
41. Messenheimer J, Mullens E, Giorgi L, Young F. Safety review of adult clinical trial experience with lamotrigine. Drug Safety 1998;18(4):281-96.
42. DelBello M, Kowatch R, Warner J, et al. Adjunctive topiramate treatment for pediatric bipolar disorder: a retrospective chart review. J Child Adolesc Psychopharmacol 2002;12(4):323-30.
43. DelBello M, Kushner S, Wang D, et al. Topiramate for acute mania in children and adolescents with bipolar I disorder (abstract). New York: American Psychiatric Association annual meeting, 2004.
44. Philippi H, Boor R, Reitter B. Topiramate and metabolic acidosis in infants and toddlers. Epilepsia 2002;43(7):744-7.
45. Arcas J, Ferrer T, Roche M, et al. Hypohidrosis related to the administration of topiramate to children. Epilepsia 2001;42(10):1363-5.
46. Davanzo P, Cantwell E, Kleiner J, et al. Cognitive changes during topiramate therapy. J Am Acad Child Adolesc Psychiatry 2001;40(3):262-3.
47. McIntyre R, Mancini D, McCann S, et al. Topiramate versus bupropion SR when added to mood stabilizer therapy for the depressive phase of bipolar disorder: a preliminary single-blind study. Bipolar Disord 2002;4(3):207-13.
Are anticonvulsants safe and effective mood stabilizers for children and adolescents with bipolar disorder? The answer is unclear because most bipolar disorder treatment trials have included adults only, and clinicians are desperate for data.1
To help you care for young patients, we report what is known about the potential benefits and risks of using mood stabilizers and anticonvulsants in bipolar youth. We base our dosing, target serum level, and monitoring recommendations on clinical experience and the limited published evidence.
AGENTS OF CHOICE?
Bipolar disorder’s “atypical” presentation in children—often more irritability and explosiveness than euphoria—can complicate diagnosis. Bipolar children and adolescents often have comorbid attention-deficit/hyperactivity disorder (ADHD), other disruptive behavior disorders, or anxiety disorders. Thus, comorbidities and presenting symptoms often dictate medication choice.
An expert consensus guideline acknowledges that more evidence on pediatric bipolar disorder is needed. In the meantime, the guideline suggests trying valproate or lithium first to treat nonpsychotic mania in pediatric bipolar patients.1 It also recommends three atypical antipsychotics— olanzapine, quetiapine, and risperidone—as potential first-line treatments. Valproate and lithium may be preferred because of atypicals’ risk of weight gain and metabolic syndrome.
Trying other anticonvulsants may be justified for bipolar youths who are not functioning well with first-line agents. Lamotrigine, for example, has antidepressant and antimanic effects.2 When you try anticonvulsants that lack double-blind, placebo-controlled trials, we recommend that you:
- obtain consent from the parents and child
- monitor carefully for side effects.
LITHIUM: STRONGEST EVIDENCE
Lithium is one of the most well-studied medications for pediatric bipolar disorder and the only mood stabilizer FDA-approved for children and adolescents (Table 1).3 Although approved for ages 12 and older, lithium has been used in younger children in practice and in clinical trials.
Table 1
FDA-approval status of medications used to treat bipolar disorder
| Medication | Indications for adults | Indications for children |
|---|---|---|
| Carbamazepine | Acute manic episode and acute mixed episode | Not approved |
| Lamotrigine | Maintenance therapy | Not approved |
| Lithium | Acute manic episode and maintenance therapy | Age ≥ 12 years |
| Oxcarbazepine | Not approved | Not approved |
| Topiramate | Not approved | Not approved |
| Valproate | Acute manic episode | Not approved |
| Source: Reference 3 | ||
In the only double-blind, placebo-controlled trial of lithium in adolescents with bipolar disorder, some subjects had secondary substance dependency disorders.7 For 6 weeks, 25 outpatient adolescents received lithium (13 patients) or placebo (12 patients). Lithium was effective in treating bipolar and substance dependency symptoms, with significantly improved clinical global assessment scores and decreased positive urine assays for drugs. Little difference was seen in mood item scores on the Schedule for Affective Disorders and Schizophrenia, child version (KSADS-1986), whether patients were taking lithium or placebo.
Pediatric dosing. For bipolar patients ages 6 to 12, use the child’s weight to determine lithium dosage (Table 2).8 Maintain serum levels between 0.8 and 1.2 mEq/L,9 and check them frequently when starting therapy.10 After mood stabilization, check levels every 1 to 3 months or when you suspect noncompliance. Obtain renal and thyroid function values at baseline and every 4 to 6 months.
Table 2
Guide to dosing lithium for prepubertal school-aged children*
| Doses (mg) | ||||
|---|---|---|---|---|
| Child’s weight (kg) | 8 AM | 12 PM | 6 PM | Total daily |
| 150 | 150 | 300 | 600 | |
| 25 to 40 | 300 | 300 | 300 | 900 |
| 40 to 50 | 300 | 300 | 600 | 1,200 |
| 50 to 60 | 600 | 300 | 600 | 1,500 |
| * Maintain specified dose at least 5 days, drawing serum levels 12 hrs after the last lithium dose until two consecutive levels appear in the therapeutic range (0.6 to 1.2 mEq/L). Dose may then be adjusted based on serum level, side effects, or clinical response. Do not exceed 1.4 mEq/L. | ||||
| Source: Reference 8 | ||||
Consider teratogenicity when choosing mood stabilizers for bipolar adolescent girls who may be sexually active. Lithium, valproate, and carbamazepine are labeled pregnancy category D because of their potential to cause birth defects.
Lithium treatment has been associated with increased risk of cardiac defects, specifically Ebstein’s anomaly (malformation of the tricuspid valve). Its incidence in children of women who used lithium during pregnancy is estimated to be 1:1,000 (0.10%) to 2:1,000 (0.05%)— 20 to 40 times the rate in the general population.12
Valproate.Results from the North American Antiepileptic Drug (AED) Pregnancy Registry showed a 10.7% rate of major congenital malformations (MCM)— including neural tube defects (spina bifida) and cardiac defects (pulmonary atresia)—in children of women who used valproate during pregnancy. The rate of births with MCMs in the general population is 2.9%.13
Carbamazepine.Data from the Australian Pregnancy Registry showed no significant increase in malformation rates in infants of carbamazepine users compared with those of women receiving no antiepileptics.14 Other studies, however, have linked carbamazepine with an increased risk of craniofacial defects (11%), neural tube defects (0.5 to 1%), and cardiac malformations.12
Lamotrigine.The teratogenic effects of the newer anticonvulsants are unclear. An 11-year study of lamotrigine15 found MCM risk after first-trimester exposure to lamotrigine to be similar to the general population’s MCM risk.
Combination therapy.Teratogenic risk appears to increase when multiple antiepileptic drugs are used (9.9% risk in polytherapy vs 6.2% in monotherapy).16
VALPROATE: OPEN-LABEL TRIALS ONLY
Efficacy. No double-blind, placebo-controlled study has shown valproate to be effective in treating bipolar disorder in children and adolescents. When used as monotherapy in open-label studies, valproate has produced response rates of:
- 53% in a 6-week, randomized, open-label trial in which 42 outpatients (mean age 11.4 years) with bipolar disorder type I or II received lithium, divalproex sodium, or carbamazepine9
- 61% in an open-label study of 40 patients ages 7 to 19 with a manic, hypomanic, or mixed episode who received divalproex for 2 to 8 weeks17
- 80% in an 8-week open-label trial of 40 patients ages 6 to 17 with bipolar disorder type I (77.5%) or type II (22.5%) and a Young Mania Rating Scale (YMRS)score ≥ 14.18
Safety: Black-box warnings. Valproate therapy carries risks of hepatic failure, pancreatitis, and birth defects. Monitor blood counts and hepatic enzymes throughout therapy (Table 3).3 Rare yet potentially fatal hepatic toxicity appears to occur most often in children age 21 Other studies suggest:
- an association with congenital malformations, including spina bifida and pulmonary atresia, in children exposed to valproate in utero6
- a link between valproate and hyperammonemic encephalopathy, especially in patients with urea cycle disorders22
- potential for benign thrombocytopenia23
- increased incidence of polycystic ovary syndrome—ovarian cysts, hyperandrogenism, chronic anovulation—in peripubertal mentally retarded women treated with valproate for seizure disorders.24
Table 3
Mood stabilizers’ side effects and recommended monitoring
| Medication | Major side effects | Monitoring |
|---|---|---|
| Carbamazepine | Allergic skin rash, drowsiness, blood dyscrasias, diplopia | CBC with reticulocytes, iron, LFTs, urinalysis, BUN, TFTs, sodium, serum carbamazepine levels |
| Lamotrigine | Stevens-Johnson syndrome, headache, dizziness, ataxia, somnolence, nausea, diplopia, blurred vision, rhinitis | No serum monitoring recommended |
| Lithium | Polyuria, polydipsia, nausea, diarrhea, tininecleatremor, enuresis, fatigue, ataxia, leukocytosis, malaise, cardiac arrhythmias, weight gain | BUN/creatinine, crearance, TFTs, calcium/phosphorus, ECG, serum lithium levels every 1 to 3 months once stabilized |
| Oxcarbazepine | Dizziness, somnolence/fatigue, ataxia/gait disturbance, vertigo, headache, tremor, rash, hyponatremia, hypersensitivity reaction, GI symptoms, diplopia | Sodium levels (particularly ifirst 3 months) |
| Topiramate | Hyperchloremic metabolic acidosis, oligohydrosis and hyperthermia, acute myopia, somnolence/fatigue, nausea, anorexia/weight loss, paresthesia, tremor, difficulty concentrating | BUN/creatinine, sodium bicarbonate |
| Valproate | Irritability/restlessness, ataxia, headache, weight gain, hyperammonemic encephalopathy, alopecia, GI upset, pancreatitis,sedation, thrombocytopenia, liver failure, polycystic ovaries/hyperandrogenism, teratogenic effects,rash | Ammonia, LFTs, bilirubin, CBC with platelets, serum valproate levels |
| BUN: blood urea nitrogen; CBC: complete blood count; ECG: electrocardiography; LFT: liver function tests; TFTs: thyroid function tests | ||
| Note: Bolded items included in black-box warnings | ||
| Source: Reference 3 | ||
CARBAMAZEPINE: DRUG INTERACTION RISK
Carbamazepine is used less often than lithium or divalproex for bipolar disorder. It tends to be used adjunctively when lithium alone is ineffective.
Efficacy. In an open-label study,9 42 patients ages 8 to 18 with bipolar disorder type I or II were randomly assigned to lithium, divalproex sodium, or carbamazepine monotherapy for 6 weeks. Response rates—measured as a ≥ 50% change from baseline in YMRS scores—were 53% with divalproex, 38% with lithium, and 38% with carbamazepine.
A retrospective review of 44 hospitalized bipolar patients ages 5 to 12 treated for at least 7 days with lithium, valproate, or carbamazepine reported higher (ie, worse) Clinical Global Impression of Improvement scores with carbamazepine.27 Small sample sizes, particularly in the carbamazepine group, limited this naturalistic study.
Safety: Black-box warnings. Carbamazepine’s hematologic “black box” warns of increased risk of aplastic anemia, agranulocytosis, leukopenia, and thrombocytopenia. Risks associated with carbamazepine have been estimated at:
- aplastic anemia: 5.1/million patient years
- agranulocytosis: 1.4/million patient years.28
Body weight. Carbamazepine is not associated with significant weight gain, which could be clinically important for some patients.
Drug interactions. Carbamazepine activates the cytochrome P-450 liver enzyme system, increasing the metabolism of many medications and decreasing their blood levels. Consider monitoring serum levels when using carbamazepine with valproate, imipramine, corticosteroids, warfarin, oral contraceptives, and some antibiotics. Because carbamazepine induces its own metabolism, you might need to increase its dosage if its effects appear to be waning.3
Carbamazepine and tricyclic antidepressants may show cross-sensitivity because of structural similarity. Do not use monoamine oxidase inhibitors with carbamazepine; discontinue them at least 14 days before starting carbamazepine.3
OXCARBAZEPINE: FEWER INTERACTIONS
Oxcarbazepine has similar efficacy to carbamazepine but less side effect risk and does not require plasma level monitoring. A weaker inducer of CYP-450, it causes fewer clinically important drug-drug interactions and may be useful for patients who respond to carbamazepine but cannot tolerate its side effects.30
Efficacy. Case studies31,32 have been encouraging, but no published, double-blind, placebo-controlled studies support using oxcarbazepine in bipolar children and adolescents.
Safety. Oxcarbazepine appears to be generally well-tolerated but can cause potentially serious reactions—including hyponatremia.33 Somnolence, emesis, and ataxia are the most common side effects in pediatric patients.3
Hyponatremia —plasma sodium 125 mEq/L—occurs in 2.5% of adults taking oxcarbazepine3 and has been reported in a similar percentage of children.34 This potentially severe reaction—characterized by nausea, lethargy, malaise, headache, confusion, decreased seizure threshold, or simply decreased serum sodium35—is usually noted within the first 12 weeks of therapy. The risk increases with concomitant use of other sodium-altering drugs, such as antidepressants or antipsychotics.36
Evaluate serum sodium when starting oxcarbazepine, periodically in the first 3 months, and if symptoms occur.34,36 For sodium levels of 125 to 130 mEq/L, obtain repeat measurements to confirm that hyponatremia is not worsening. Intervention is often required when levels fall below 125 mEq/L.36
Other serious adverse reactions include Stevens-Johnson syndrome, toxic epidermal necrolysis, and hypersensitivity reactions; 25% to 30% of patients with hypersensitivity to carbamazepine also will react to oxcarbazepine.33
Contraceptive concerns. Oxcarbazepine may reduce contraceptive efficacy by altering estrogen and progesterone plasma concentrations.37 Consider other birth control methods for sexuallyactive bipolar adolescent girls.
LAMOTRIGINE
Neurologists often use lamotrigine for children with atypical seizure disorders, but no controlled data exist on the drug’s efficacy and safety in youths with bipolar disorder.
Efficacy. In a prospective, open-label study,38 13 adolescents with type I bipolar disorder received lamotrigine, 200 to 400 mg/d. After 12 weeks (mean dosage 241 mg/d), their symptoms had improved as shown by these mean scores:
- Montgomery-Asberg Depression Rating Scale: from 21 at baseline to 4 at endpoint
- Clinical Global Impressions–Severity of Illness scale: from 4 to 1
- Children’s Depression Rating Scale (CDRS-R): from 74 to 40
- YMRS: from 20 to 6.
Safety: Severe rash. An age-related association with Stevens-Johnson syndrome may limit pediatric use of lamotrigine. Severe and potentially life-threatening rashes have been reported in 0.8% of children treated with lamotrigine.40 Discontinue lamotrigine if a rash develops, unless it clearly is not drug-related. Three factors that increase rash risk include:
- co-administering lamotrigine with valproate
- higher-than-recommended initial dosages
- rapid dose titration.41
Pediatric dosing. We find no published studies of efficacious dosages and plasma levels of lamotrigine in pediatric bipolar disorder (Table 4).3,9,17 Based on our clinical experience, we recommend starting lamotrigine at 1 to 5 mg/kg/day (1 to 3 mg/kg/day if given with valproate) divided into two daily doses. Watch for rash or skin disorders. Do not exceed the recommended daily dosage by 200 mg in children age
Table 4
Using anticonvulsants in pediatric bipolar disorder patients
| Drug | Recommended dosage | Target serum level |
|---|---|---|
| Carbamazepine | Age 6 to 12: 20 to 30 mg/kg/d | ≥ 7.0 μg/L |
| Age >12: 400 to 1,200 mg/d | ||
| Lamotrigine* | Unknown | Unknown |
| Oxcarbazepine | 11 to 16 mg/kg/d (as adjunct) | Unknown |
| Topiramate* | Unknown | Unknown |
| Valproate | 15 to 20 mg/kg/d | 45 to 125 μg/mL (trough) |
| 85 to 110 μg/mL (target) | ||
| * No published studies found for efficacious dosage and plasma levels in pediatric bipolar disorder. | ||
| Dosage supported by case reports only; no studies found examining efficacious plasma levels. | ||
| Source: References 3,9, and 17. | ||
TOPIRAMATE: LIMITED INFORMATION
Efficacy. Little is known about using topiramate in children and adolescents. A retrospective chart review42 of 26 patients with bipolar disorder type I (n=23) or II (n=3) showed adjunctive topiramate to be effective, with response rates of 73% for mania and 62% overall. Topiramate was well tolerated, and no serious events were reported.
A randomized, controlled trial of topiramate for acute mania in youths with type I bipolar disorder43 was recently halted because of lack of efficacy in adul trials. Preliminary data from 56 of the pediatric patients—analyzed before the study was halted—showed improved YMRS scores. Although results were not statistically significant, the authors suggest topiramate might be effective in treating children and adolescents with bipolar disorder.
Safety: FDA warning. Decreased sodium bicarbonate leading to hyperchloremic metabolic acidosis has been reported in youths treated with topiramate for seizure disorder,44 leading to an FDA warning to prescribers.3 Although no monitoring guidelines exist, we recommend baseline and periodic serum bicarbonate measurements and acidbase evaluations during topiramate treatment, especially when adding other antiepileptics.44
Other rare but serious reactions include:
- impaired sweat production and resultant hyperthermia45
- ophthalmologic symptoms characterized by secondary acute angle closure glaucoma and acute myacute myopia (usually within 1 month of starting treatment)46
- sedation and cognitive difficulties.47
Cognitive effects? Reports of “word finding difficulties” with topiramate47 may suggest cognitive effects. Thus, be very cautious about using this medication in children and adolescents.
Related resources
- Kowatch R, Fristad M, Birmaher B, et al. Treatment guidelines for children and adolescents with bipolar disorder. J Am Acad Child Adolesc Psychiatry. 2005;44(3):213-35.
- Yonkers K, Wisner K, Stowe Z, et al. Management of bipolar disorder during pregnancy and the postpartum period. Am J Psychiatry 2004;161(4):608-20.
- American Academy of Child and Adolescent Psychiatry. Treatment guidelines for childhood psychiatric disorders. www.aacap.org
Dr. Kloos, Dr. Hitchcock, and Dr. Ronald Weller report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Elizabeth Weller has been a consultant to or received research grants from GlaxoSmithKline, Johnson & Johnson, Novartis Pharmaceuticals Corp., Abbott Laboratories, and Shire Pharmaceuticals.
Drug brand names
- Carbamazepine • Tegretol
- Divalproex • Depakote
- Lamotrigine • Lamictal
- Lithium • Eskalith, Lithobid, others
- Oxcarbazepine • Trileptal
- Topiramate • Topamax
Are anticonvulsants safe and effective mood stabilizers for children and adolescents with bipolar disorder? The answer is unclear because most bipolar disorder treatment trials have included adults only, and clinicians are desperate for data.1
To help you care for young patients, we report what is known about the potential benefits and risks of using mood stabilizers and anticonvulsants in bipolar youth. We base our dosing, target serum level, and monitoring recommendations on clinical experience and the limited published evidence.
AGENTS OF CHOICE?
Bipolar disorder’s “atypical” presentation in children—often more irritability and explosiveness than euphoria—can complicate diagnosis. Bipolar children and adolescents often have comorbid attention-deficit/hyperactivity disorder (ADHD), other disruptive behavior disorders, or anxiety disorders. Thus, comorbidities and presenting symptoms often dictate medication choice.
An expert consensus guideline acknowledges that more evidence on pediatric bipolar disorder is needed. In the meantime, the guideline suggests trying valproate or lithium first to treat nonpsychotic mania in pediatric bipolar patients.1 It also recommends three atypical antipsychotics— olanzapine, quetiapine, and risperidone—as potential first-line treatments. Valproate and lithium may be preferred because of atypicals’ risk of weight gain and metabolic syndrome.
Trying other anticonvulsants may be justified for bipolar youths who are not functioning well with first-line agents. Lamotrigine, for example, has antidepressant and antimanic effects.2 When you try anticonvulsants that lack double-blind, placebo-controlled trials, we recommend that you:
- obtain consent from the parents and child
- monitor carefully for side effects.
LITHIUM: STRONGEST EVIDENCE
Lithium is one of the most well-studied medications for pediatric bipolar disorder and the only mood stabilizer FDA-approved for children and adolescents (Table 1).3 Although approved for ages 12 and older, lithium has been used in younger children in practice and in clinical trials.
Table 1
FDA-approval status of medications used to treat bipolar disorder
| Medication | Indications for adults | Indications for children |
|---|---|---|
| Carbamazepine | Acute manic episode and acute mixed episode | Not approved |
| Lamotrigine | Maintenance therapy | Not approved |
| Lithium | Acute manic episode and maintenance therapy | Age ≥ 12 years |
| Oxcarbazepine | Not approved | Not approved |
| Topiramate | Not approved | Not approved |
| Valproate | Acute manic episode | Not approved |
| Source: Reference 3 | ||
In the only double-blind, placebo-controlled trial of lithium in adolescents with bipolar disorder, some subjects had secondary substance dependency disorders.7 For 6 weeks, 25 outpatient adolescents received lithium (13 patients) or placebo (12 patients). Lithium was effective in treating bipolar and substance dependency symptoms, with significantly improved clinical global assessment scores and decreased positive urine assays for drugs. Little difference was seen in mood item scores on the Schedule for Affective Disorders and Schizophrenia, child version (KSADS-1986), whether patients were taking lithium or placebo.
Pediatric dosing. For bipolar patients ages 6 to 12, use the child’s weight to determine lithium dosage (Table 2).8 Maintain serum levels between 0.8 and 1.2 mEq/L,9 and check them frequently when starting therapy.10 After mood stabilization, check levels every 1 to 3 months or when you suspect noncompliance. Obtain renal and thyroid function values at baseline and every 4 to 6 months.
Table 2
Guide to dosing lithium for prepubertal school-aged children*
| Doses (mg) | ||||
|---|---|---|---|---|
| Child’s weight (kg) | 8 AM | 12 PM | 6 PM | Total daily |
| 150 | 150 | 300 | 600 | |
| 25 to 40 | 300 | 300 | 300 | 900 |
| 40 to 50 | 300 | 300 | 600 | 1,200 |
| 50 to 60 | 600 | 300 | 600 | 1,500 |
| * Maintain specified dose at least 5 days, drawing serum levels 12 hrs after the last lithium dose until two consecutive levels appear in the therapeutic range (0.6 to 1.2 mEq/L). Dose may then be adjusted based on serum level, side effects, or clinical response. Do not exceed 1.4 mEq/L. | ||||
| Source: Reference 8 | ||||
Consider teratogenicity when choosing mood stabilizers for bipolar adolescent girls who may be sexually active. Lithium, valproate, and carbamazepine are labeled pregnancy category D because of their potential to cause birth defects.
Lithium treatment has been associated with increased risk of cardiac defects, specifically Ebstein’s anomaly (malformation of the tricuspid valve). Its incidence in children of women who used lithium during pregnancy is estimated to be 1:1,000 (0.10%) to 2:1,000 (0.05%)— 20 to 40 times the rate in the general population.12
Valproate.Results from the North American Antiepileptic Drug (AED) Pregnancy Registry showed a 10.7% rate of major congenital malformations (MCM)— including neural tube defects (spina bifida) and cardiac defects (pulmonary atresia)—in children of women who used valproate during pregnancy. The rate of births with MCMs in the general population is 2.9%.13
Carbamazepine.Data from the Australian Pregnancy Registry showed no significant increase in malformation rates in infants of carbamazepine users compared with those of women receiving no antiepileptics.14 Other studies, however, have linked carbamazepine with an increased risk of craniofacial defects (11%), neural tube defects (0.5 to 1%), and cardiac malformations.12
Lamotrigine.The teratogenic effects of the newer anticonvulsants are unclear. An 11-year study of lamotrigine15 found MCM risk after first-trimester exposure to lamotrigine to be similar to the general population’s MCM risk.
Combination therapy.Teratogenic risk appears to increase when multiple antiepileptic drugs are used (9.9% risk in polytherapy vs 6.2% in monotherapy).16
VALPROATE: OPEN-LABEL TRIALS ONLY
Efficacy. No double-blind, placebo-controlled study has shown valproate to be effective in treating bipolar disorder in children and adolescents. When used as monotherapy in open-label studies, valproate has produced response rates of:
- 53% in a 6-week, randomized, open-label trial in which 42 outpatients (mean age 11.4 years) with bipolar disorder type I or II received lithium, divalproex sodium, or carbamazepine9
- 61% in an open-label study of 40 patients ages 7 to 19 with a manic, hypomanic, or mixed episode who received divalproex for 2 to 8 weeks17
- 80% in an 8-week open-label trial of 40 patients ages 6 to 17 with bipolar disorder type I (77.5%) or type II (22.5%) and a Young Mania Rating Scale (YMRS)score ≥ 14.18
Safety: Black-box warnings. Valproate therapy carries risks of hepatic failure, pancreatitis, and birth defects. Monitor blood counts and hepatic enzymes throughout therapy (Table 3).3 Rare yet potentially fatal hepatic toxicity appears to occur most often in children age 21 Other studies suggest:
- an association with congenital malformations, including spina bifida and pulmonary atresia, in children exposed to valproate in utero6
- a link between valproate and hyperammonemic encephalopathy, especially in patients with urea cycle disorders22
- potential for benign thrombocytopenia23
- increased incidence of polycystic ovary syndrome—ovarian cysts, hyperandrogenism, chronic anovulation—in peripubertal mentally retarded women treated with valproate for seizure disorders.24
Table 3
Mood stabilizers’ side effects and recommended monitoring
| Medication | Major side effects | Monitoring |
|---|---|---|
| Carbamazepine | Allergic skin rash, drowsiness, blood dyscrasias, diplopia | CBC with reticulocytes, iron, LFTs, urinalysis, BUN, TFTs, sodium, serum carbamazepine levels |
| Lamotrigine | Stevens-Johnson syndrome, headache, dizziness, ataxia, somnolence, nausea, diplopia, blurred vision, rhinitis | No serum monitoring recommended |
| Lithium | Polyuria, polydipsia, nausea, diarrhea, tininecleatremor, enuresis, fatigue, ataxia, leukocytosis, malaise, cardiac arrhythmias, weight gain | BUN/creatinine, crearance, TFTs, calcium/phosphorus, ECG, serum lithium levels every 1 to 3 months once stabilized |
| Oxcarbazepine | Dizziness, somnolence/fatigue, ataxia/gait disturbance, vertigo, headache, tremor, rash, hyponatremia, hypersensitivity reaction, GI symptoms, diplopia | Sodium levels (particularly ifirst 3 months) |
| Topiramate | Hyperchloremic metabolic acidosis, oligohydrosis and hyperthermia, acute myopia, somnolence/fatigue, nausea, anorexia/weight loss, paresthesia, tremor, difficulty concentrating | BUN/creatinine, sodium bicarbonate |
| Valproate | Irritability/restlessness, ataxia, headache, weight gain, hyperammonemic encephalopathy, alopecia, GI upset, pancreatitis,sedation, thrombocytopenia, liver failure, polycystic ovaries/hyperandrogenism, teratogenic effects,rash | Ammonia, LFTs, bilirubin, CBC with platelets, serum valproate levels |
| BUN: blood urea nitrogen; CBC: complete blood count; ECG: electrocardiography; LFT: liver function tests; TFTs: thyroid function tests | ||
| Note: Bolded items included in black-box warnings | ||
| Source: Reference 3 | ||
CARBAMAZEPINE: DRUG INTERACTION RISK
Carbamazepine is used less often than lithium or divalproex for bipolar disorder. It tends to be used adjunctively when lithium alone is ineffective.
Efficacy. In an open-label study,9 42 patients ages 8 to 18 with bipolar disorder type I or II were randomly assigned to lithium, divalproex sodium, or carbamazepine monotherapy for 6 weeks. Response rates—measured as a ≥ 50% change from baseline in YMRS scores—were 53% with divalproex, 38% with lithium, and 38% with carbamazepine.
A retrospective review of 44 hospitalized bipolar patients ages 5 to 12 treated for at least 7 days with lithium, valproate, or carbamazepine reported higher (ie, worse) Clinical Global Impression of Improvement scores with carbamazepine.27 Small sample sizes, particularly in the carbamazepine group, limited this naturalistic study.
Safety: Black-box warnings. Carbamazepine’s hematologic “black box” warns of increased risk of aplastic anemia, agranulocytosis, leukopenia, and thrombocytopenia. Risks associated with carbamazepine have been estimated at:
- aplastic anemia: 5.1/million patient years
- agranulocytosis: 1.4/million patient years.28
Body weight. Carbamazepine is not associated with significant weight gain, which could be clinically important for some patients.
Drug interactions. Carbamazepine activates the cytochrome P-450 liver enzyme system, increasing the metabolism of many medications and decreasing their blood levels. Consider monitoring serum levels when using carbamazepine with valproate, imipramine, corticosteroids, warfarin, oral contraceptives, and some antibiotics. Because carbamazepine induces its own metabolism, you might need to increase its dosage if its effects appear to be waning.3
Carbamazepine and tricyclic antidepressants may show cross-sensitivity because of structural similarity. Do not use monoamine oxidase inhibitors with carbamazepine; discontinue them at least 14 days before starting carbamazepine.3
OXCARBAZEPINE: FEWER INTERACTIONS
Oxcarbazepine has similar efficacy to carbamazepine but less side effect risk and does not require plasma level monitoring. A weaker inducer of CYP-450, it causes fewer clinically important drug-drug interactions and may be useful for patients who respond to carbamazepine but cannot tolerate its side effects.30
Efficacy. Case studies31,32 have been encouraging, but no published, double-blind, placebo-controlled studies support using oxcarbazepine in bipolar children and adolescents.
Safety. Oxcarbazepine appears to be generally well-tolerated but can cause potentially serious reactions—including hyponatremia.33 Somnolence, emesis, and ataxia are the most common side effects in pediatric patients.3
Hyponatremia —plasma sodium 125 mEq/L—occurs in 2.5% of adults taking oxcarbazepine3 and has been reported in a similar percentage of children.34 This potentially severe reaction—characterized by nausea, lethargy, malaise, headache, confusion, decreased seizure threshold, or simply decreased serum sodium35—is usually noted within the first 12 weeks of therapy. The risk increases with concomitant use of other sodium-altering drugs, such as antidepressants or antipsychotics.36
Evaluate serum sodium when starting oxcarbazepine, periodically in the first 3 months, and if symptoms occur.34,36 For sodium levels of 125 to 130 mEq/L, obtain repeat measurements to confirm that hyponatremia is not worsening. Intervention is often required when levels fall below 125 mEq/L.36
Other serious adverse reactions include Stevens-Johnson syndrome, toxic epidermal necrolysis, and hypersensitivity reactions; 25% to 30% of patients with hypersensitivity to carbamazepine also will react to oxcarbazepine.33
Contraceptive concerns. Oxcarbazepine may reduce contraceptive efficacy by altering estrogen and progesterone plasma concentrations.37 Consider other birth control methods for sexuallyactive bipolar adolescent girls.
LAMOTRIGINE
Neurologists often use lamotrigine for children with atypical seizure disorders, but no controlled data exist on the drug’s efficacy and safety in youths with bipolar disorder.
Efficacy. In a prospective, open-label study,38 13 adolescents with type I bipolar disorder received lamotrigine, 200 to 400 mg/d. After 12 weeks (mean dosage 241 mg/d), their symptoms had improved as shown by these mean scores:
- Montgomery-Asberg Depression Rating Scale: from 21 at baseline to 4 at endpoint
- Clinical Global Impressions–Severity of Illness scale: from 4 to 1
- Children’s Depression Rating Scale (CDRS-R): from 74 to 40
- YMRS: from 20 to 6.
Safety: Severe rash. An age-related association with Stevens-Johnson syndrome may limit pediatric use of lamotrigine. Severe and potentially life-threatening rashes have been reported in 0.8% of children treated with lamotrigine.40 Discontinue lamotrigine if a rash develops, unless it clearly is not drug-related. Three factors that increase rash risk include:
- co-administering lamotrigine with valproate
- higher-than-recommended initial dosages
- rapid dose titration.41
Pediatric dosing. We find no published studies of efficacious dosages and plasma levels of lamotrigine in pediatric bipolar disorder (Table 4).3,9,17 Based on our clinical experience, we recommend starting lamotrigine at 1 to 5 mg/kg/day (1 to 3 mg/kg/day if given with valproate) divided into two daily doses. Watch for rash or skin disorders. Do not exceed the recommended daily dosage by 200 mg in children age
Table 4
Using anticonvulsants in pediatric bipolar disorder patients
| Drug | Recommended dosage | Target serum level |
|---|---|---|
| Carbamazepine | Age 6 to 12: 20 to 30 mg/kg/d | ≥ 7.0 μg/L |
| Age >12: 400 to 1,200 mg/d | ||
| Lamotrigine* | Unknown | Unknown |
| Oxcarbazepine | 11 to 16 mg/kg/d (as adjunct) | Unknown |
| Topiramate* | Unknown | Unknown |
| Valproate | 15 to 20 mg/kg/d | 45 to 125 μg/mL (trough) |
| 85 to 110 μg/mL (target) | ||
| * No published studies found for efficacious dosage and plasma levels in pediatric bipolar disorder. | ||
| Dosage supported by case reports only; no studies found examining efficacious plasma levels. | ||
| Source: References 3,9, and 17. | ||
TOPIRAMATE: LIMITED INFORMATION
Efficacy. Little is known about using topiramate in children and adolescents. A retrospective chart review42 of 26 patients with bipolar disorder type I (n=23) or II (n=3) showed adjunctive topiramate to be effective, with response rates of 73% for mania and 62% overall. Topiramate was well tolerated, and no serious events were reported.
A randomized, controlled trial of topiramate for acute mania in youths with type I bipolar disorder43 was recently halted because of lack of efficacy in adul trials. Preliminary data from 56 of the pediatric patients—analyzed before the study was halted—showed improved YMRS scores. Although results were not statistically significant, the authors suggest topiramate might be effective in treating children and adolescents with bipolar disorder.
Safety: FDA warning. Decreased sodium bicarbonate leading to hyperchloremic metabolic acidosis has been reported in youths treated with topiramate for seizure disorder,44 leading to an FDA warning to prescribers.3 Although no monitoring guidelines exist, we recommend baseline and periodic serum bicarbonate measurements and acidbase evaluations during topiramate treatment, especially when adding other antiepileptics.44
Other rare but serious reactions include:
- impaired sweat production and resultant hyperthermia45
- ophthalmologic symptoms characterized by secondary acute angle closure glaucoma and acute myacute myopia (usually within 1 month of starting treatment)46
- sedation and cognitive difficulties.47
Cognitive effects? Reports of “word finding difficulties” with topiramate47 may suggest cognitive effects. Thus, be very cautious about using this medication in children and adolescents.
Related resources
- Kowatch R, Fristad M, Birmaher B, et al. Treatment guidelines for children and adolescents with bipolar disorder. J Am Acad Child Adolesc Psychiatry. 2005;44(3):213-35.
- Yonkers K, Wisner K, Stowe Z, et al. Management of bipolar disorder during pregnancy and the postpartum period. Am J Psychiatry 2004;161(4):608-20.
- American Academy of Child and Adolescent Psychiatry. Treatment guidelines for childhood psychiatric disorders. www.aacap.org
Dr. Kloos, Dr. Hitchcock, and Dr. Ronald Weller report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Elizabeth Weller has been a consultant to or received research grants from GlaxoSmithKline, Johnson & Johnson, Novartis Pharmaceuticals Corp., Abbott Laboratories, and Shire Pharmaceuticals.
Drug brand names
- Carbamazepine • Tegretol
- Divalproex • Depakote
- Lamotrigine • Lamictal
- Lithium • Eskalith, Lithobid, others
- Oxcarbazepine • Trileptal
- Topiramate • Topamax
1. Kowatch R, Fristad M, Birmaher B, et al. Treatment guidelines for children and adolescents with bipolar disorder. J Am Acad Child Adolesc Psychiatry 2005;44(3):213-35
2. Wang P, Ketter T, Becker O, et al. New anticonvulsant medication uses in bipolar disorder. CNS Spectrums 2003;8(12):930-47.
3. Prescribing information. Physicians’ Desk Reference (59th ed.) Montvale, NJ: Thomson Healthcare, 2005.
4. Kafantaris V, Coletti D, Dicker R, et al. Lithium treatment of acute mania in adolescents: a large open trial. J Am Acad Child Adolesc Psychiatry 2003;42(9):1038-45.
5. Kafantaris V, Coletti D, Dicker R, et al. Lithium treatment of acute mania in adolescents: a placebo-controlled discontinuation study. J Am Acad Child Adolesc Psychiatry 2004;43(8):984-93.
6. Strober M, DeAntonio M, Schmidt-Lackner S, et al. Early childhood attention deficit hyperactivity disorder predicts poorer response to acute lithium therapy in adolescent mania. J Affect Disord 1998;51(2):145-51.
7. Geller B, Cooper T, Zimerman B, et al. Lithium for prepubertal depressed children with family history predictors of future bipolarity: a double-blind, placebo-controlled study. J Affect Disord 1998;51(2):165-75.
8. Weller E, Weller R, Fristad M. Lithium dosage guide for prepubertal children: a preliminary report. J Am Acad Child Adolesc Psychiatry 1986;25(1):92-5.
9. Kowatch R, Suppes T, Carmody T, et al. Effect size of lithium, divalproex sodium, and carbamazepine in children and adolescents with bipolar disorder. J Am Acad Child Adolesc Psychiatry 2000;39(6):713-20.
10. Hagino O, Weller E, Weller R, et al. Untoward effects of lithium treatment in children aged four through six years. J Am Acad Child Adolesc Psychiatry 1995;34(12):1584-90.
11. Hagino O, Weller E, Weller R, Fristad M. Comparison of lithium dosage methods for preschool- and early school-age children. J Am Acad Child Adolesc Psychiatry 1995;37(1):60-5.
12. Yonkers K, Wisner K, Stowe Z, et al. Management of bipolar disorder during pregnancy and the postpartum period. Am J Psychiatry 2004;161(4):608-20.
13. Holmes L. The North American antiepileptic drug pregnancy registry: a seven-year experience. (paper presentation). New Orleans: American Epilepsy Society annual meeting, 2004.
14. Vajda F, Lander C, Cook M, et al. Antiepileptic medication in pregnancy: the Australian Pregnancy Register: 52 months data (poster presentation). New Orleans: American Epilepsy Society annual meeting, 2004.
15. Messenheimer J, Tennis P, Cunnington M. Eleven-year interim results of an international observational study of pregnancy outcomes following exposure to lamotrigine (poster presentation). New Orleans: American Epilepsy Society annual meeting, 2004.
16. Torbjorn T, Battino D, Bonizzoni E, et al. Eurap: an international registry of antiepileptic drugs and pregnancy (poster presentation). New Orleans: American Epilepsy Society annual meeting, 2004.
17. Wagner K, Weller E, Carlson G, et al. An open-label trial of divalproex in children and adolescents with bipolar disorder. J Am Acad Child Adolesc Psychiatry 2002;41(10):1224-30.
18. Scheffer RE, Kowatch RA, Carmody T, Rush AJ. Randomized, placebo-controlled trial of mixed amphetamine salts for symptoms of comorbid ADHD in pediatric bipolar disorder after mood stabilization with divalproex sodium. Am J Psychiatry 2005;162(1):58-64.
19. Findling R, McNamara N, Gracious B, et al. Combination lithium and divalproex sodium in pediatric bipolarity. J Am Acad Child Adolesc Psychiatry 2003;42(8):895-901.
20. DelBello M, Adler C, Strakowski S. Divalproex for the treatment of aggression associated with adolescent mania. J Child Adolesc Psychopharmacol 2004;14(2):325-8.
21. Anderson G. Children versus adults: pharmokinetic and adverseeffect differences. Epilepsia 2002;43(suppl 3):53-9.
22. Yehya N, Saldarini C, Koski M, et al. Valproate-induced hyperammonemic encephalopathy. J Am Acad Child Adolesc Psychiatry 2004;43(8):926-7.
23. Verrotti A, Greco R, Matera V, et al. Platelet count and function in children receiving sodium valproate. Pediatr Neurol 1999;21(3):611-14.
24. Isojarvi J, Tauboll E, Pakarinen A, et al. Altered ovarian function and cardiovascular risk factors in valproate-treated women. Am J Med 2001;111(4):290-6.
25. Bowden C, Calabrese J, McElroy S, et al. A randomized, placebocontrolled 12-month trial of divalproex and lithium in treatment of outpatients with bipolar I disorder. Arch Gen Psychiatry 2000;57(5):481-9.
26. Findling R, Gracious B, McNamara N, et al. Rapid, continuous cycling and psychiatric co-morbidity in pediatric bipolar I disorder. Bipolar Disord 2001;3(4):202-10.
27. Davanzo P, Gunderson B, Belin T, et al. Mood stabilizers in hospitalized children with bipolar disorder: a retrospective review. Psychiatry Clin Neurosci 2003;57(5):504-10.
28. Pellock J. Carbamazepine side effects in children and adults. Epilepsia 1987;28(suppl 3):64-70.
29. Sobotka J, Alexander B, Cook B. A review of carbamazepine’s hematologic reactions and monitoring. DICP 1990;24(12):1214-19.
30. Hellewell J. Oxcarbazepine (Trileptal) in the treatment of bipolar disorders: a review of efficacy and tolerability. J Affect Disord 2002;72(supp 1):23-34.
31. Davanzo P, Nikore V, Yehya N, Stevenson L. Oxcarbazepine treatment of juvenile-onset bipolar disorder. J Child Adolesc Psychopharmacol 2004;14(3):344-5.
32. Teitelbaum M. Oxcarbazepine in bipolar disorder. J Am Acad Child Adolesc Psychiatry 2001;40(9):993-4.
33. Dietrich D, Kropp S, Emrich H. Oxcarbazepine in affective and schizoaffective disorders. Pharmacopsychiatry 2001;34(6):242-50.
34. Holtmann M, Krause M, Opp J, et al. Oxcarbazepine-induced hyponatremia and the regulation of serum sodium after replacing carbamazepine with oxcarbazepine in children. Neuropediatrics 2002;33(6):298-300.
35. Prescribing information for oxcarbazepine (Trileptal) Novartis Pharmaceuticals Corp. 2005. Available at: http://www.pharma.us. novartis.com/product/pi/pdf/trileptal.pdf. Accessed June 26, 2005.
36. Asconape J. Some common issues in the use of antiepileptic drugs. Semin Neurol 2002;22(1):27-39.
37. Fattore C, Cipolla G, Gatti G, et al. Induction of ethinylestradiol and levonorgestrel metabolism by oxcarbazepine in healthy women. Epilepsia 1999;40(6):783-7.
38. Swope G, Hoopes S, Amy L, et al. An open-label study of lamotrigine in adolescents with bipolar mood disorder (poster presentation). New York: American Psychiatric Association annual meeting, 2004.
39. Saxena K, Howe M, Chang K. Lamotrigine adjunct or monotherapy for adolescent bipolar depression or mixed mania (poster presentation). Washington, DC: American Academy of Child and Adolescent Psychiatry annual meeting, 2004.
40. Prescribing information for lamotrigine (Lamictal). GlaxoSmith Kline 2004. Available at: http://us.gsk.com/products/assets/us_ lamictal.pdf. Accessed June 2, 2005.
41. Messenheimer J, Mullens E, Giorgi L, Young F. Safety review of adult clinical trial experience with lamotrigine. Drug Safety 1998;18(4):281-96.
42. DelBello M, Kowatch R, Warner J, et al. Adjunctive topiramate treatment for pediatric bipolar disorder: a retrospective chart review. J Child Adolesc Psychopharmacol 2002;12(4):323-30.
43. DelBello M, Kushner S, Wang D, et al. Topiramate for acute mania in children and adolescents with bipolar I disorder (abstract). New York: American Psychiatric Association annual meeting, 2004.
44. Philippi H, Boor R, Reitter B. Topiramate and metabolic acidosis in infants and toddlers. Epilepsia 2002;43(7):744-7.
45. Arcas J, Ferrer T, Roche M, et al. Hypohidrosis related to the administration of topiramate to children. Epilepsia 2001;42(10):1363-5.
46. Davanzo P, Cantwell E, Kleiner J, et al. Cognitive changes during topiramate therapy. J Am Acad Child Adolesc Psychiatry 2001;40(3):262-3.
47. McIntyre R, Mancini D, McCann S, et al. Topiramate versus bupropion SR when added to mood stabilizer therapy for the depressive phase of bipolar disorder: a preliminary single-blind study. Bipolar Disord 2002;4(3):207-13.
1. Kowatch R, Fristad M, Birmaher B, et al. Treatment guidelines for children and adolescents with bipolar disorder. J Am Acad Child Adolesc Psychiatry 2005;44(3):213-35
2. Wang P, Ketter T, Becker O, et al. New anticonvulsant medication uses in bipolar disorder. CNS Spectrums 2003;8(12):930-47.
3. Prescribing information. Physicians’ Desk Reference (59th ed.) Montvale, NJ: Thomson Healthcare, 2005.
4. Kafantaris V, Coletti D, Dicker R, et al. Lithium treatment of acute mania in adolescents: a large open trial. J Am Acad Child Adolesc Psychiatry 2003;42(9):1038-45.
5. Kafantaris V, Coletti D, Dicker R, et al. Lithium treatment of acute mania in adolescents: a placebo-controlled discontinuation study. J Am Acad Child Adolesc Psychiatry 2004;43(8):984-93.
6. Strober M, DeAntonio M, Schmidt-Lackner S, et al. Early childhood attention deficit hyperactivity disorder predicts poorer response to acute lithium therapy in adolescent mania. J Affect Disord 1998;51(2):145-51.
7. Geller B, Cooper T, Zimerman B, et al. Lithium for prepubertal depressed children with family history predictors of future bipolarity: a double-blind, placebo-controlled study. J Affect Disord 1998;51(2):165-75.
8. Weller E, Weller R, Fristad M. Lithium dosage guide for prepubertal children: a preliminary report. J Am Acad Child Adolesc Psychiatry 1986;25(1):92-5.
9. Kowatch R, Suppes T, Carmody T, et al. Effect size of lithium, divalproex sodium, and carbamazepine in children and adolescents with bipolar disorder. J Am Acad Child Adolesc Psychiatry 2000;39(6):713-20.
10. Hagino O, Weller E, Weller R, et al. Untoward effects of lithium treatment in children aged four through six years. J Am Acad Child Adolesc Psychiatry 1995;34(12):1584-90.
11. Hagino O, Weller E, Weller R, Fristad M. Comparison of lithium dosage methods for preschool- and early school-age children. J Am Acad Child Adolesc Psychiatry 1995;37(1):60-5.
12. Yonkers K, Wisner K, Stowe Z, et al. Management of bipolar disorder during pregnancy and the postpartum period. Am J Psychiatry 2004;161(4):608-20.
13. Holmes L. The North American antiepileptic drug pregnancy registry: a seven-year experience. (paper presentation). New Orleans: American Epilepsy Society annual meeting, 2004.
14. Vajda F, Lander C, Cook M, et al. Antiepileptic medication in pregnancy: the Australian Pregnancy Register: 52 months data (poster presentation). New Orleans: American Epilepsy Society annual meeting, 2004.
15. Messenheimer J, Tennis P, Cunnington M. Eleven-year interim results of an international observational study of pregnancy outcomes following exposure to lamotrigine (poster presentation). New Orleans: American Epilepsy Society annual meeting, 2004.
16. Torbjorn T, Battino D, Bonizzoni E, et al. Eurap: an international registry of antiepileptic drugs and pregnancy (poster presentation). New Orleans: American Epilepsy Society annual meeting, 2004.
17. Wagner K, Weller E, Carlson G, et al. An open-label trial of divalproex in children and adolescents with bipolar disorder. J Am Acad Child Adolesc Psychiatry 2002;41(10):1224-30.
18. Scheffer RE, Kowatch RA, Carmody T, Rush AJ. Randomized, placebo-controlled trial of mixed amphetamine salts for symptoms of comorbid ADHD in pediatric bipolar disorder after mood stabilization with divalproex sodium. Am J Psychiatry 2005;162(1):58-64.
19. Findling R, McNamara N, Gracious B, et al. Combination lithium and divalproex sodium in pediatric bipolarity. J Am Acad Child Adolesc Psychiatry 2003;42(8):895-901.
20. DelBello M, Adler C, Strakowski S. Divalproex for the treatment of aggression associated with adolescent mania. J Child Adolesc Psychopharmacol 2004;14(2):325-8.
21. Anderson G. Children versus adults: pharmokinetic and adverseeffect differences. Epilepsia 2002;43(suppl 3):53-9.
22. Yehya N, Saldarini C, Koski M, et al. Valproate-induced hyperammonemic encephalopathy. J Am Acad Child Adolesc Psychiatry 2004;43(8):926-7.
23. Verrotti A, Greco R, Matera V, et al. Platelet count and function in children receiving sodium valproate. Pediatr Neurol 1999;21(3):611-14.
24. Isojarvi J, Tauboll E, Pakarinen A, et al. Altered ovarian function and cardiovascular risk factors in valproate-treated women. Am J Med 2001;111(4):290-6.
25. Bowden C, Calabrese J, McElroy S, et al. A randomized, placebocontrolled 12-month trial of divalproex and lithium in treatment of outpatients with bipolar I disorder. Arch Gen Psychiatry 2000;57(5):481-9.
26. Findling R, Gracious B, McNamara N, et al. Rapid, continuous cycling and psychiatric co-morbidity in pediatric bipolar I disorder. Bipolar Disord 2001;3(4):202-10.
27. Davanzo P, Gunderson B, Belin T, et al. Mood stabilizers in hospitalized children with bipolar disorder: a retrospective review. Psychiatry Clin Neurosci 2003;57(5):504-10.
28. Pellock J. Carbamazepine side effects in children and adults. Epilepsia 1987;28(suppl 3):64-70.
29. Sobotka J, Alexander B, Cook B. A review of carbamazepine’s hematologic reactions and monitoring. DICP 1990;24(12):1214-19.
30. Hellewell J. Oxcarbazepine (Trileptal) in the treatment of bipolar disorders: a review of efficacy and tolerability. J Affect Disord 2002;72(supp 1):23-34.
31. Davanzo P, Nikore V, Yehya N, Stevenson L. Oxcarbazepine treatment of juvenile-onset bipolar disorder. J Child Adolesc Psychopharmacol 2004;14(3):344-5.
32. Teitelbaum M. Oxcarbazepine in bipolar disorder. J Am Acad Child Adolesc Psychiatry 2001;40(9):993-4.
33. Dietrich D, Kropp S, Emrich H. Oxcarbazepine in affective and schizoaffective disorders. Pharmacopsychiatry 2001;34(6):242-50.
34. Holtmann M, Krause M, Opp J, et al. Oxcarbazepine-induced hyponatremia and the regulation of serum sodium after replacing carbamazepine with oxcarbazepine in children. Neuropediatrics 2002;33(6):298-300.
35. Prescribing information for oxcarbazepine (Trileptal) Novartis Pharmaceuticals Corp. 2005. Available at: http://www.pharma.us. novartis.com/product/pi/pdf/trileptal.pdf. Accessed June 26, 2005.
36. Asconape J. Some common issues in the use of antiepileptic drugs. Semin Neurol 2002;22(1):27-39.
37. Fattore C, Cipolla G, Gatti G, et al. Induction of ethinylestradiol and levonorgestrel metabolism by oxcarbazepine in healthy women. Epilepsia 1999;40(6):783-7.
38. Swope G, Hoopes S, Amy L, et al. An open-label study of lamotrigine in adolescents with bipolar mood disorder (poster presentation). New York: American Psychiatric Association annual meeting, 2004.
39. Saxena K, Howe M, Chang K. Lamotrigine adjunct or monotherapy for adolescent bipolar depression or mixed mania (poster presentation). Washington, DC: American Academy of Child and Adolescent Psychiatry annual meeting, 2004.
40. Prescribing information for lamotrigine (Lamictal). GlaxoSmith Kline 2004. Available at: http://us.gsk.com/products/assets/us_ lamictal.pdf. Accessed June 2, 2005.
41. Messenheimer J, Mullens E, Giorgi L, Young F. Safety review of adult clinical trial experience with lamotrigine. Drug Safety 1998;18(4):281-96.
42. DelBello M, Kowatch R, Warner J, et al. Adjunctive topiramate treatment for pediatric bipolar disorder: a retrospective chart review. J Child Adolesc Psychopharmacol 2002;12(4):323-30.
43. DelBello M, Kushner S, Wang D, et al. Topiramate for acute mania in children and adolescents with bipolar I disorder (abstract). New York: American Psychiatric Association annual meeting, 2004.
44. Philippi H, Boor R, Reitter B. Topiramate and metabolic acidosis in infants and toddlers. Epilepsia 2002;43(7):744-7.
45. Arcas J, Ferrer T, Roche M, et al. Hypohidrosis related to the administration of topiramate to children. Epilepsia 2001;42(10):1363-5.
46. Davanzo P, Cantwell E, Kleiner J, et al. Cognitive changes during topiramate therapy. J Am Acad Child Adolesc Psychiatry 2001;40(3):262-3.
47. McIntyre R, Mancini D, McCann S, et al. Topiramate versus bupropion SR when added to mood stabilizer therapy for the depressive phase of bipolar disorder: a preliminary single-blind study. Bipolar Disord 2002;4(3):207-13.
Antidepressants for bipolar depression: Tips to stay out of trouble
In clinical practice, 50% to 80% of bipolar patients receive long-term antidepressants,1 although potential benefits probably outweigh risks in 20% to 40%. This gap suggests that psychiatrists could do more to stay out of trouble when prescribing antidepressants for patients with bipolar depression.
Antidepressants have not shown efficacy in long-term treatment, and evidence of their effectiveness in acute bipolar depression is limited. They appear to pose greater risk of switching and mood destabilization for some patients and certain types of bipolar illness, and some antidepressant classes are more worrisome than others.
Because carefully analyzing risks and benefits is essential when considering antidepressants for a patient with bipolar illness, this article clarifies that delicate balance and offers evidence-based recommendations for using antidepressants in bipolar depression.
ACUTE THERAPY
Clinical trials support antidepressants as the treatment of choice for unipolar depression, but less evidence supports efficacy and safety in acute bipolar depression. Depressive episodes predominate in bipolar disorder, with chronic subsyndromal symptoms being most characteristic.2,3 Compared with mania or hypomania, depressive episodes:
- last longer and are more frequent
- contribute to greater morbidity and mortality
- pose a greater treatment challenge.
Antidepressants have shown benefit in multiple double-blind, bipolar depression trials and were as effective as mood stabilizers in one small study.4 Even so, no trials have found them more effective than mood stabilizers in acute bipolar depression.
Controlled trials. Two randomized, double-blind, placebo-controlled trials have examined antidepressant use in bipolar depression.5,6 The larger and better designed—a prospective 10-week study by Nemeroff et al6—examined 117 outpatients with type I bipolar disorder.
Subjects who had been taking lithium (serum levels 0.5 to 1.2 mEq/L) for ≥6 weeks and were experiencing moderate breakthrough depression then received paroxetine (mean dosage 32.6 mg/d), imipramine (mean dosage 166.7 mg/d), or placebo. Therapeutic response was defined as ≤7 on the Hamilton Rating Scale for Depression (HRSD) or ≤2 on the Clinical Global Impression (CGI) scale—normally considered criteria for depressive remission.
The authors hoped to show a statistically significant medication-placebo difference, but the antidepressants’ effects were similar to those of placebo. Thus, adding antidepressants to lithium conferred no added benefit, though the small sample size may have created a false negative.
Interestingly, a post-hoc analysis found different treatment outcomes when patients were separated into two groups by lithium serum levels:
- low therapeutic (≤0.8 mEq/L)
- high therapeutic (>0.8 mEq/L).
Adding antidepressants significantly reduced HRSD scores compared with placebo in the low lithium group but not in the high lithium group. Thus, therapeutic lithium levels may have moderate antidepressant effects, and adding antidepressants may help patients who cannot tolerate therapeutic lithium levels.
MAINTENANCE THERAPY
Antidepressants may have modest efficacy in acute bipolar depression, but they have not shown benefit—with or without mood stabilizers—in 7 studies of bipolar depression maintenance therapy. Most were double-blind, long-term trials comparing tricyclic antidepressants (TCAs) with lithium or adding TCAs to lithium; 3 were placebo-controlled.7 Antidepressants were not more effective than mood stabilizers such as lithium or lamotrigine in preventing bipolar depression.
Type II patients. For depression in type II bipolar disorder, the only data on using antidepressants as acute or maintenance therapy come from post-hoc analyses of unipolar depression trials and retrospective assessments of “manic switches.” No specific mania rating scales have been used.8,9
Long-term antidepressants. Two naturalistic studies by Altshuler et al10,11 explored continuing antidepressants as bipolar depression maintenance treatment. The larger trial11 included 84 patients (most with type I bipolar disorder) who experienced breakthrough depression while taking a mood stabilizer. This subset (15%) of the Stanley Foundation Bipolar Network had tolerated antidepressants at least 2 months without switching into hypomania/mania and remained in remission at least 6 weeks. None were rapid cyclers.
With counseling from clinicians, patients chose to continue or discontinue taking antidepressants. Relapse rates after 1 year were 70% in patients who stopped antidepressants after <6 months, compared with 24% in those who continued taking them for 1 year. The authors concluded that bipolar patients may benefit from staying on antidepressants at least 6 months and perhaps 12 or more months after depressive remission.
Keep in mind, however, that these findings may not apply to all bipolar patients. This study pertains to a minority of robust responders—none of whom were rapid cyclers—who tolerated the medication well and were not randomly assigned to continue or discontinue antidepressants. Other evidence suggests that depressed bipolar patients are three times more likely than unipolar patients (54% vs 16%) to develop tolerance to antidepressants.12
ANTIDEPRESSANT RISKS
Risks of using antidepressants in bipolar patients include acute switches into hypo/mania, usually within 8 weeks of starting an antidepressant, and new-onset mood destabilization—with cycle acceleration or rapid cycling—or worsening of pre-existing rapid cycling (Table 1).1
Table 1
Switches vs destabilization: Defining antidepressant risks
| Risk | Definition |
|---|---|
| Acute switches to hypomania/mania | ≤8 weeks by convention, unless dosage is increased |
| Mood destabilization | |
| Cycle acceleration | Increase of ≥2 mood episodes while taking antidepressants, compared with a similar exposure time before treatment |
| Rapid cycling | ≥4mood episodes in previous 12 months (new-onset or exacerbation of baseline pattern), according to DSM-IV-TR |
| Source: Reference 1 | |
Switching risk. Some researchers have reported antidepressant-induced switches to be milder and more brief than spontaneous hypo/manias,13 whereas others have observed more-severe mixed14 and even psychotic episodes. Risk factors that may predispose patients to switching include:
- personal or family history of switches or mood destabilization
- family history of bipolar disorder
- exposure to multiple antidepressant trials
- history of substance abuse or dependence
- early onset (age <25) and/or treatment of mood symptoms.15,16
True switch rates are difficult to estimate because clinical trials have used different switching definitions, durations, antidepressants (with or without mood stabilizers, and with different mood stabilizers), and cohorts (often excluding rapid cyclers). Except for the Nemeroff et al study,6 no prospective, double-blind, placebo-controlled studies have examined switch rates, and even this study was not large enough to detect statistically significant differences.
Thus we must rely on naturalistic evidence that is less rigorous but more applicable to clinical practice. This literature reveals switch rates of:
- 30% to 60% with TCAs and monoamine oxidase inhibitors (MAOIs)
- 15% to 27% with selective serotonin reuptake inhibitors (SSRIs), bupropion, and venlafaxine.
Average switch rates are thought to be approximately 40% with TCAs/MAOIs and 20% with the newer antidepressants.1 Preliminary data associate venlafaxine with higher switch rates than SSRIs or bupropion, so perhaps antidepressants with some noradrenergic effects (including TCAs) facilitate the switching phenomenon.17
Mood destabilization. Three randomized, controlled trials suggest that antidepressants—especially TCAs—increase the risk of cycle acceleration or rapid cycling in bipolar patients. The best designed study—sponsored by the National Institute of Mental Health—was a 10-year, prospective, double-blind trial of 51 rapid-cycling patients. The trial’s on-off-on design showed that 20% of these patients developed rapid cycling as a direct result of taking TCAs.18
Unfortunately, most randomized, controlled trials are not designed to show a relationship between antidepressants and mood destabilization. Observational literature is mixed but suggests that antidepressant use is associated with rapid cycling. Most evidence supports a relationship between antidepressants and long-term mood destabilization—especially cycle acceleration, which is believed to occur in approximately 20% of patients using TCAs or SSRIs.1
Are mood stabilizers protective? Some studies suggest that mood stabilizers may help protect against switches. Most of the evidence—using lithium and TCAs—suggests a 50% drop in switch rates when patients receive mood stabilizers with antidepressants. In one study, lithium was more protective than anticonvulsants for SSRI-induced mania, but the difference was not statistically significant.19
Because study data variability, we don’t know if some mood stabilizers are more effective than others in preventing antidepressant-related switching. This variability is likely caused by:
- medication-specific factors (such as higher switch rates with TCAs and possibly dual-reuptake inhibitors than with SSRIs)
- illness-specific factors (such as rapid cycling and cycle pattern)
- patient-specific factors, already described. Mood stabilizers appear to be more protective against switching than against mood destabilization, in which their effects are less clear (Table 2).15
Table 2
Frequency of switching or mood destabilization with antidepressants
| Bipolar risk | Causative agents | Frequency | Mood stabilizer effect |
|---|---|---|---|
| Acute switch | TCAs, MAOIs | ~ 40% | Variably protective; apparent partial risk reduction ~ 50% |
| SSRIs, bupropion, venlafaxine | ~ 20% | ||
| Mood destabilization | TCAs, SSRIs | ~ 20% | Not as clearly protective against mood destabilization as against acute switching |
| TCAs: tricyclic antidepressants; MAOIs: monoamine oxidase inhibitors; SSRIs: selective serotonin reuptake inhibitors | |||
| Source: References 1, 15 | |||
TREATMENT RECOMMENDATIONS
How does a clinician decide which bipolar depressed patients should receive antidepressants?
The first step in treating bipolar depression (Algorithm) is to provide optimal dosages of the patient’s mood stabilizers. Consensus guidelines20 suggest lithium or lamotrigine as first-line treatments for bipolar depression. Evidence also shows efficacy for atypical antipsychotics, including the olanzapine/fluoxetine combination (OFC)—FDA-approved for acute bipolar depression21—and quetiapine monotherapy.22 Dosages vary, but suggested ranges include:
- lithium: 0.6 to 1.2 mEq/L; aim for approximately 0.8 mEq/L, but some data suggest 0.6 to 0.7 mEq/L may be sufficient
- lamotrigine: 50 to 250 mg/d (the higher dosage is based on maintenance studies)
- OFC: 6 to 12 mg olanzapine/25 to 50 mg fluoxetine
- quetiapine: 300 to 600 mg/d.
The next step is an antidepressant risk/benefit analysis, weighing the considerable risks of switching/mood destabilization with the patient’s depressive illness severity, type of bipolar disorder (such as rapid cycling), and cycle pattern.
Algorithm Recommended treatment of bipolar depression
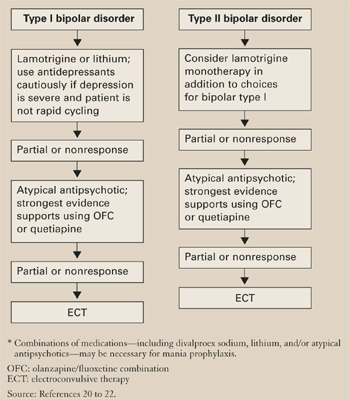
Cycle patterns. In a naturalistic study, Macqueen et al23 used life chart data for 42 bipolar patients to assess how the mood state preceding a prospectively observed depressive episode affected treatment response:
- A euthymic mood state in the previous 2 months represented a uniphasic pattern and an isolated depressive episode.
- A preceding hypomanic/manic mood state indicated a biphasic pattern.
Approximately 60% of bipolar patients show a biphasic pattern, although the episode sequence is usually depression-hypomania/mania rather than hypomania/mania-depression. These authors included patients whose breakthrough depressive episodes were treated with an antidepressant or a putative mood stabilizer but not an atypical antipsychotic.
In patients treated with an antidepressant, the response-to-switch ratio was 10:1 for those previously euthymic, compared with a less beneficial 0.75:1 in previously hypomanic/manic patients. This small study suggests that a patient’s cycle pattern may help you decide whether to use an antidepressant for bipolar depression.
How to use antidepressants. As described, some depressed bipolar patients are better candidates for antidepressant therapy than others (Table 3).
Table 3
Antidepressants for bipolar depression? Consider ‘ideal patient’ traits
| Severe depression refractory to optimal doses of ≥1 mood stabilizers |
| Uniphasic cycle pattern |
| Not rapid cycling |
| No history of switching or mood destabilization |
| No comorbid substance abuse |
Use antidepressants cautiously and conservatively in a minority of bipolar patients (approximately 20% to 40%) and usually for short periods (discussed below). SSRIs or bupropion are first-line agents because:
- they appear to be relatively less likely to cause switching than other antidepressant classes
- controlled trials have examined these antidepressants in bipolar depression.
Depressed patients with very mild, nonrapid-cycling, bipolar II disorder and no more than three previous hypomanic episodes might be candidates for antidepressant monotherapy. In other bipolar patients, always use at least one mood stabilizer if you decide to use an antidepressant.
TREATMENT DURATION
No randomized, controlled trial has examined what duration of antidepressant treatment may be optimum for bipolar depression, but consensus guidelines recommend:
- approximately 3 to 7 months, depending on depression severity
- approximately one-half that duration (2 to 4 months) for rapid-cycling bipolar disorder.20
Because of the switching risk, one could also argue for a shorter treatment duration in patients with a biphasic cycle pattern—especially with an episode sequence of depression to hypomania/mania to euthymia.
Ideally, patients would stay on antidepressants no longer than the natural course of their depression (usually 2 to 6 months in bipolar depression), although it could be shorter in rapid cyclers. Approximately 15% to 20% of patients may have a robust initial response to antidepressants and need to be maintained on these medications, especially after several tapers and relapses have failed.
Related resources
- Bipolar Clinic and Research Program. Massachusetts General Hospital. Includes tools for clinicians and the clinical site for the Systematic Treatment Enhancement Program for Bipolar Disorder (STEP-BD). www.manicdepressive.org.
- Goodwin FK, Jamison KR. Manic-depressive illness. New York: Oxford University Press, 1990.
Drug brand names
- Bupropion • Wellbutrin
- Imipramine • Tofranil
- Lamotrigine • Lamictal
- Lithium • Lithobid, others
- Olanzapine/fluoxetine • Symbyax
- Paroxetine • Paxil
- Quetiapine • Seroquel
- Venlafaxine • Effexor
Disclosures
Dr. Altman is a speaker for Forest Pharmaceuticals, Janssen Pharmaceutica, AstraZeneca Pharmaceuticals, and Abbott Laboratories.
1. Ghaemi SN, Hsu DJ, Soldani F, et al. Antidepressants in bipolar disorder: the case for caution. Bipolar Disorders 2003;5:421-33.
2. Judd LL. The long-term natural history of the weekly symptomatic status of bipolar I disorder. Arch Gen Psychiatry 2002;59:530-7.
3. Judd LL, Akiskal HS, Schettler PJ, et al. A prospective investigation of the natural history of the long-term weekly symptomatic status of bipolar II disorder. Arch Gen Psychiatry 2003;60(3):261-9.
4. Young LT, Joffe RT, Robb JC, et al. Double-blind comparison of addition of a second mood stabilizer versus an antidepressant to an initial mood stabilizer for treatment of patients with bipolar depression. Am J Psychiatry 2000;157:124-6.
5. Cohn JB, Collins G, Ashbrook E, et al. A comparison of fluoxetine, imipramine and placebo in patients with bipolar depressive disorder. Int Clin Psychopharmacol 1989;4(4):313-22.
6. Nemeroff CB, Evans DL, Gyulai L, et al. Double-blind, placebo-controlled comparison of imipramine and paroxetine in the treatment of bipolar depression. Am J Psychiatry 2001;158(6):906-12.
7. Ghaemi SN, Lenox MS, Baldessarini RJ. Effectiveness and safety of long-term antidepressant treatment in bipolar disorder. J Clin Psychiatry 2001;62(7):565-9.
8. Amsterdam JD, Garcia-Espana F, Fawcett J, et al. Efficacy and safety of fluoxetine in treating bipolar II major depressive episode. J Clin Psychopharmacol 1998;18:435-40.
9. Amsterdam JD, Garcia-Espana F. Venlafaxine monotherapy in women with bipolar II and unipolar major depression. J Affect Disord 2000;59:225-9.
10. Altshuler L, Kiriakos L, Calcagno J, et al. Impact of antidepressant discontinuation versus antidepressant continuation at 1-year risk for relapse of bipolar depression: a retrospective chart review. J Clin Psychiatry 2001;62:612-16.
11. Altshuler L, Suppes T, Black D, et al. Impact of antidepressant discontinuation after acute bipolar depression remission on rates of depressive relapse at 1-year follow-up. Am J Psychiatry 2003;160:1252-62.
12. Ghaemi SN, Rosenquist KJ, Ko JY, et al. Antidepressant treatment in bipolar versus unipolar depression. Am J Psychiatry 2004;161:163-5.
13. Stoll AL, Mayer PV, Kolbrener M, et al. Antidepressant-associated mania: a controlled comparison with spontaneous mania. Am J Psychiatry 1994;151:1642-5.
14. Zubieta JK, Demitrack MA. Possible bupropion precipitation of mania and mixed affective state. J Clin Psychopharmacol 1991;11(5):327-8.
15. Goldberg JF, Truman CJ. Antidepressant-induced mania: an overview of current controversies. Bipolar Disorders 2003;5:407-20.
16. Goldberg JF, Whiteside JE. The association between substance abuse and antidepressant-induced mania in bipolar disorder: a preliminary study. J Clin Psychiatry 2002;63:791-5.
17. Post RM, Leverich GS, Nolen WA, et al. A re-evaluation of the role of antidepressants in the treatment of bipolar depression: data from the Stanley Foundation Bipolar Network. Bipolar Disorders 2003;5:396-406.
18. Wehr TA, Sack DA, Rosenthal NE, et al. Rapid cycling affective disorder: contributing factors and treatment responses in 51 patients. Am J Psychiatry 1988;145:179-84.
19. Henry C, Sorbara F, Lacoste J, et al. Antidepressant-induced mania in bipolar patients: identification of risk factors. J Clin Psychiatry 2001;62:249-55.
20. Keck PE, Jr, Perlis RH, Otto MW, et al. The Expert Consensus Guideline Series: Treatment of bipolar disorder 2004. Postgrad Med 2004;1-120.
21. Tohen M, Vieta E, Calabrese J, et al. Efficacy of olanzapine and olanzapine-fluoxetine combination in the treatment of bipolar I depression. Arch Gen Psychiatry 2003;60:1079-88.
22. Calabrese JR. Quetiapine BOLDER study [presentation]. New York: American Psychiatric Association annual meeting, 2004.
23. MacQueen GM, Young LT, Marriott M, et al. Previous mood state predicts response and switch rates in patients with bipolar depression. Acta Psychiatr Scand 2002;105:414-18.
In clinical practice, 50% to 80% of bipolar patients receive long-term antidepressants,1 although potential benefits probably outweigh risks in 20% to 40%. This gap suggests that psychiatrists could do more to stay out of trouble when prescribing antidepressants for patients with bipolar depression.
Antidepressants have not shown efficacy in long-term treatment, and evidence of their effectiveness in acute bipolar depression is limited. They appear to pose greater risk of switching and mood destabilization for some patients and certain types of bipolar illness, and some antidepressant classes are more worrisome than others.
Because carefully analyzing risks and benefits is essential when considering antidepressants for a patient with bipolar illness, this article clarifies that delicate balance and offers evidence-based recommendations for using antidepressants in bipolar depression.
ACUTE THERAPY
Clinical trials support antidepressants as the treatment of choice for unipolar depression, but less evidence supports efficacy and safety in acute bipolar depression. Depressive episodes predominate in bipolar disorder, with chronic subsyndromal symptoms being most characteristic.2,3 Compared with mania or hypomania, depressive episodes:
- last longer and are more frequent
- contribute to greater morbidity and mortality
- pose a greater treatment challenge.
Antidepressants have shown benefit in multiple double-blind, bipolar depression trials and were as effective as mood stabilizers in one small study.4 Even so, no trials have found them more effective than mood stabilizers in acute bipolar depression.
Controlled trials. Two randomized, double-blind, placebo-controlled trials have examined antidepressant use in bipolar depression.5,6 The larger and better designed—a prospective 10-week study by Nemeroff et al6—examined 117 outpatients with type I bipolar disorder.
Subjects who had been taking lithium (serum levels 0.5 to 1.2 mEq/L) for ≥6 weeks and were experiencing moderate breakthrough depression then received paroxetine (mean dosage 32.6 mg/d), imipramine (mean dosage 166.7 mg/d), or placebo. Therapeutic response was defined as ≤7 on the Hamilton Rating Scale for Depression (HRSD) or ≤2 on the Clinical Global Impression (CGI) scale—normally considered criteria for depressive remission.
The authors hoped to show a statistically significant medication-placebo difference, but the antidepressants’ effects were similar to those of placebo. Thus, adding antidepressants to lithium conferred no added benefit, though the small sample size may have created a false negative.
Interestingly, a post-hoc analysis found different treatment outcomes when patients were separated into two groups by lithium serum levels:
- low therapeutic (≤0.8 mEq/L)
- high therapeutic (>0.8 mEq/L).
Adding antidepressants significantly reduced HRSD scores compared with placebo in the low lithium group but not in the high lithium group. Thus, therapeutic lithium levels may have moderate antidepressant effects, and adding antidepressants may help patients who cannot tolerate therapeutic lithium levels.
MAINTENANCE THERAPY
Antidepressants may have modest efficacy in acute bipolar depression, but they have not shown benefit—with or without mood stabilizers—in 7 studies of bipolar depression maintenance therapy. Most were double-blind, long-term trials comparing tricyclic antidepressants (TCAs) with lithium or adding TCAs to lithium; 3 were placebo-controlled.7 Antidepressants were not more effective than mood stabilizers such as lithium or lamotrigine in preventing bipolar depression.
Type II patients. For depression in type II bipolar disorder, the only data on using antidepressants as acute or maintenance therapy come from post-hoc analyses of unipolar depression trials and retrospective assessments of “manic switches.” No specific mania rating scales have been used.8,9
Long-term antidepressants. Two naturalistic studies by Altshuler et al10,11 explored continuing antidepressants as bipolar depression maintenance treatment. The larger trial11 included 84 patients (most with type I bipolar disorder) who experienced breakthrough depression while taking a mood stabilizer. This subset (15%) of the Stanley Foundation Bipolar Network had tolerated antidepressants at least 2 months without switching into hypomania/mania and remained in remission at least 6 weeks. None were rapid cyclers.
With counseling from clinicians, patients chose to continue or discontinue taking antidepressants. Relapse rates after 1 year were 70% in patients who stopped antidepressants after <6 months, compared with 24% in those who continued taking them for 1 year. The authors concluded that bipolar patients may benefit from staying on antidepressants at least 6 months and perhaps 12 or more months after depressive remission.
Keep in mind, however, that these findings may not apply to all bipolar patients. This study pertains to a minority of robust responders—none of whom were rapid cyclers—who tolerated the medication well and were not randomly assigned to continue or discontinue antidepressants. Other evidence suggests that depressed bipolar patients are three times more likely than unipolar patients (54% vs 16%) to develop tolerance to antidepressants.12
ANTIDEPRESSANT RISKS
Risks of using antidepressants in bipolar patients include acute switches into hypo/mania, usually within 8 weeks of starting an antidepressant, and new-onset mood destabilization—with cycle acceleration or rapid cycling—or worsening of pre-existing rapid cycling (Table 1).1
Table 1
Switches vs destabilization: Defining antidepressant risks
| Risk | Definition |
|---|---|
| Acute switches to hypomania/mania | ≤8 weeks by convention, unless dosage is increased |
| Mood destabilization | |
| Cycle acceleration | Increase of ≥2 mood episodes while taking antidepressants, compared with a similar exposure time before treatment |
| Rapid cycling | ≥4mood episodes in previous 12 months (new-onset or exacerbation of baseline pattern), according to DSM-IV-TR |
| Source: Reference 1 | |
Switching risk. Some researchers have reported antidepressant-induced switches to be milder and more brief than spontaneous hypo/manias,13 whereas others have observed more-severe mixed14 and even psychotic episodes. Risk factors that may predispose patients to switching include:
- personal or family history of switches or mood destabilization
- family history of bipolar disorder
- exposure to multiple antidepressant trials
- history of substance abuse or dependence
- early onset (age <25) and/or treatment of mood symptoms.15,16
True switch rates are difficult to estimate because clinical trials have used different switching definitions, durations, antidepressants (with or without mood stabilizers, and with different mood stabilizers), and cohorts (often excluding rapid cyclers). Except for the Nemeroff et al study,6 no prospective, double-blind, placebo-controlled studies have examined switch rates, and even this study was not large enough to detect statistically significant differences.
Thus we must rely on naturalistic evidence that is less rigorous but more applicable to clinical practice. This literature reveals switch rates of:
- 30% to 60% with TCAs and monoamine oxidase inhibitors (MAOIs)
- 15% to 27% with selective serotonin reuptake inhibitors (SSRIs), bupropion, and venlafaxine.
Average switch rates are thought to be approximately 40% with TCAs/MAOIs and 20% with the newer antidepressants.1 Preliminary data associate venlafaxine with higher switch rates than SSRIs or bupropion, so perhaps antidepressants with some noradrenergic effects (including TCAs) facilitate the switching phenomenon.17
Mood destabilization. Three randomized, controlled trials suggest that antidepressants—especially TCAs—increase the risk of cycle acceleration or rapid cycling in bipolar patients. The best designed study—sponsored by the National Institute of Mental Health—was a 10-year, prospective, double-blind trial of 51 rapid-cycling patients. The trial’s on-off-on design showed that 20% of these patients developed rapid cycling as a direct result of taking TCAs.18
Unfortunately, most randomized, controlled trials are not designed to show a relationship between antidepressants and mood destabilization. Observational literature is mixed but suggests that antidepressant use is associated with rapid cycling. Most evidence supports a relationship between antidepressants and long-term mood destabilization—especially cycle acceleration, which is believed to occur in approximately 20% of patients using TCAs or SSRIs.1
Are mood stabilizers protective? Some studies suggest that mood stabilizers may help protect against switches. Most of the evidence—using lithium and TCAs—suggests a 50% drop in switch rates when patients receive mood stabilizers with antidepressants. In one study, lithium was more protective than anticonvulsants for SSRI-induced mania, but the difference was not statistically significant.19
Because study data variability, we don’t know if some mood stabilizers are more effective than others in preventing antidepressant-related switching. This variability is likely caused by:
- medication-specific factors (such as higher switch rates with TCAs and possibly dual-reuptake inhibitors than with SSRIs)
- illness-specific factors (such as rapid cycling and cycle pattern)
- patient-specific factors, already described. Mood stabilizers appear to be more protective against switching than against mood destabilization, in which their effects are less clear (Table 2).15
Table 2
Frequency of switching or mood destabilization with antidepressants
| Bipolar risk | Causative agents | Frequency | Mood stabilizer effect |
|---|---|---|---|
| Acute switch | TCAs, MAOIs | ~ 40% | Variably protective; apparent partial risk reduction ~ 50% |
| SSRIs, bupropion, venlafaxine | ~ 20% | ||
| Mood destabilization | TCAs, SSRIs | ~ 20% | Not as clearly protective against mood destabilization as against acute switching |
| TCAs: tricyclic antidepressants; MAOIs: monoamine oxidase inhibitors; SSRIs: selective serotonin reuptake inhibitors | |||
| Source: References 1, 15 | |||
TREATMENT RECOMMENDATIONS
How does a clinician decide which bipolar depressed patients should receive antidepressants?
The first step in treating bipolar depression (Algorithm) is to provide optimal dosages of the patient’s mood stabilizers. Consensus guidelines20 suggest lithium or lamotrigine as first-line treatments for bipolar depression. Evidence also shows efficacy for atypical antipsychotics, including the olanzapine/fluoxetine combination (OFC)—FDA-approved for acute bipolar depression21—and quetiapine monotherapy.22 Dosages vary, but suggested ranges include:
- lithium: 0.6 to 1.2 mEq/L; aim for approximately 0.8 mEq/L, but some data suggest 0.6 to 0.7 mEq/L may be sufficient
- lamotrigine: 50 to 250 mg/d (the higher dosage is based on maintenance studies)
- OFC: 6 to 12 mg olanzapine/25 to 50 mg fluoxetine
- quetiapine: 300 to 600 mg/d.
The next step is an antidepressant risk/benefit analysis, weighing the considerable risks of switching/mood destabilization with the patient’s depressive illness severity, type of bipolar disorder (such as rapid cycling), and cycle pattern.
Algorithm Recommended treatment of bipolar depression
Cycle patterns. In a naturalistic study, Macqueen et al23 used life chart data for 42 bipolar patients to assess how the mood state preceding a prospectively observed depressive episode affected treatment response:
- A euthymic mood state in the previous 2 months represented a uniphasic pattern and an isolated depressive episode.
- A preceding hypomanic/manic mood state indicated a biphasic pattern.
Approximately 60% of bipolar patients show a biphasic pattern, although the episode sequence is usually depression-hypomania/mania rather than hypomania/mania-depression. These authors included patients whose breakthrough depressive episodes were treated with an antidepressant or a putative mood stabilizer but not an atypical antipsychotic.
In patients treated with an antidepressant, the response-to-switch ratio was 10:1 for those previously euthymic, compared with a less beneficial 0.75:1 in previously hypomanic/manic patients. This small study suggests that a patient’s cycle pattern may help you decide whether to use an antidepressant for bipolar depression.
How to use antidepressants. As described, some depressed bipolar patients are better candidates for antidepressant therapy than others (Table 3).
Table 3
Antidepressants for bipolar depression? Consider ‘ideal patient’ traits
| Severe depression refractory to optimal doses of ≥1 mood stabilizers |
| Uniphasic cycle pattern |
| Not rapid cycling |
| No history of switching or mood destabilization |
| No comorbid substance abuse |
Use antidepressants cautiously and conservatively in a minority of bipolar patients (approximately 20% to 40%) and usually for short periods (discussed below). SSRIs or bupropion are first-line agents because:
- they appear to be relatively less likely to cause switching than other antidepressant classes
- controlled trials have examined these antidepressants in bipolar depression.
Depressed patients with very mild, nonrapid-cycling, bipolar II disorder and no more than three previous hypomanic episodes might be candidates for antidepressant monotherapy. In other bipolar patients, always use at least one mood stabilizer if you decide to use an antidepressant.
TREATMENT DURATION
No randomized, controlled trial has examined what duration of antidepressant treatment may be optimum for bipolar depression, but consensus guidelines recommend:
- approximately 3 to 7 months, depending on depression severity
- approximately one-half that duration (2 to 4 months) for rapid-cycling bipolar disorder.20
Because of the switching risk, one could also argue for a shorter treatment duration in patients with a biphasic cycle pattern—especially with an episode sequence of depression to hypomania/mania to euthymia.
Ideally, patients would stay on antidepressants no longer than the natural course of their depression (usually 2 to 6 months in bipolar depression), although it could be shorter in rapid cyclers. Approximately 15% to 20% of patients may have a robust initial response to antidepressants and need to be maintained on these medications, especially after several tapers and relapses have failed.
Related resources
- Bipolar Clinic and Research Program. Massachusetts General Hospital. Includes tools for clinicians and the clinical site for the Systematic Treatment Enhancement Program for Bipolar Disorder (STEP-BD). www.manicdepressive.org.
- Goodwin FK, Jamison KR. Manic-depressive illness. New York: Oxford University Press, 1990.
Drug brand names
- Bupropion • Wellbutrin
- Imipramine • Tofranil
- Lamotrigine • Lamictal
- Lithium • Lithobid, others
- Olanzapine/fluoxetine • Symbyax
- Paroxetine • Paxil
- Quetiapine • Seroquel
- Venlafaxine • Effexor
Disclosures
Dr. Altman is a speaker for Forest Pharmaceuticals, Janssen Pharmaceutica, AstraZeneca Pharmaceuticals, and Abbott Laboratories.
In clinical practice, 50% to 80% of bipolar patients receive long-term antidepressants,1 although potential benefits probably outweigh risks in 20% to 40%. This gap suggests that psychiatrists could do more to stay out of trouble when prescribing antidepressants for patients with bipolar depression.
Antidepressants have not shown efficacy in long-term treatment, and evidence of their effectiveness in acute bipolar depression is limited. They appear to pose greater risk of switching and mood destabilization for some patients and certain types of bipolar illness, and some antidepressant classes are more worrisome than others.
Because carefully analyzing risks and benefits is essential when considering antidepressants for a patient with bipolar illness, this article clarifies that delicate balance and offers evidence-based recommendations for using antidepressants in bipolar depression.
ACUTE THERAPY
Clinical trials support antidepressants as the treatment of choice for unipolar depression, but less evidence supports efficacy and safety in acute bipolar depression. Depressive episodes predominate in bipolar disorder, with chronic subsyndromal symptoms being most characteristic.2,3 Compared with mania or hypomania, depressive episodes:
- last longer and are more frequent
- contribute to greater morbidity and mortality
- pose a greater treatment challenge.
Antidepressants have shown benefit in multiple double-blind, bipolar depression trials and were as effective as mood stabilizers in one small study.4 Even so, no trials have found them more effective than mood stabilizers in acute bipolar depression.
Controlled trials. Two randomized, double-blind, placebo-controlled trials have examined antidepressant use in bipolar depression.5,6 The larger and better designed—a prospective 10-week study by Nemeroff et al6—examined 117 outpatients with type I bipolar disorder.
Subjects who had been taking lithium (serum levels 0.5 to 1.2 mEq/L) for ≥6 weeks and were experiencing moderate breakthrough depression then received paroxetine (mean dosage 32.6 mg/d), imipramine (mean dosage 166.7 mg/d), or placebo. Therapeutic response was defined as ≤7 on the Hamilton Rating Scale for Depression (HRSD) or ≤2 on the Clinical Global Impression (CGI) scale—normally considered criteria for depressive remission.
The authors hoped to show a statistically significant medication-placebo difference, but the antidepressants’ effects were similar to those of placebo. Thus, adding antidepressants to lithium conferred no added benefit, though the small sample size may have created a false negative.
Interestingly, a post-hoc analysis found different treatment outcomes when patients were separated into two groups by lithium serum levels:
- low therapeutic (≤0.8 mEq/L)
- high therapeutic (>0.8 mEq/L).
Adding antidepressants significantly reduced HRSD scores compared with placebo in the low lithium group but not in the high lithium group. Thus, therapeutic lithium levels may have moderate antidepressant effects, and adding antidepressants may help patients who cannot tolerate therapeutic lithium levels.
MAINTENANCE THERAPY
Antidepressants may have modest efficacy in acute bipolar depression, but they have not shown benefit—with or without mood stabilizers—in 7 studies of bipolar depression maintenance therapy. Most were double-blind, long-term trials comparing tricyclic antidepressants (TCAs) with lithium or adding TCAs to lithium; 3 were placebo-controlled.7 Antidepressants were not more effective than mood stabilizers such as lithium or lamotrigine in preventing bipolar depression.
Type II patients. For depression in type II bipolar disorder, the only data on using antidepressants as acute or maintenance therapy come from post-hoc analyses of unipolar depression trials and retrospective assessments of “manic switches.” No specific mania rating scales have been used.8,9
Long-term antidepressants. Two naturalistic studies by Altshuler et al10,11 explored continuing antidepressants as bipolar depression maintenance treatment. The larger trial11 included 84 patients (most with type I bipolar disorder) who experienced breakthrough depression while taking a mood stabilizer. This subset (15%) of the Stanley Foundation Bipolar Network had tolerated antidepressants at least 2 months without switching into hypomania/mania and remained in remission at least 6 weeks. None were rapid cyclers.
With counseling from clinicians, patients chose to continue or discontinue taking antidepressants. Relapse rates after 1 year were 70% in patients who stopped antidepressants after <6 months, compared with 24% in those who continued taking them for 1 year. The authors concluded that bipolar patients may benefit from staying on antidepressants at least 6 months and perhaps 12 or more months after depressive remission.
Keep in mind, however, that these findings may not apply to all bipolar patients. This study pertains to a minority of robust responders—none of whom were rapid cyclers—who tolerated the medication well and were not randomly assigned to continue or discontinue antidepressants. Other evidence suggests that depressed bipolar patients are three times more likely than unipolar patients (54% vs 16%) to develop tolerance to antidepressants.12
ANTIDEPRESSANT RISKS
Risks of using antidepressants in bipolar patients include acute switches into hypo/mania, usually within 8 weeks of starting an antidepressant, and new-onset mood destabilization—with cycle acceleration or rapid cycling—or worsening of pre-existing rapid cycling (Table 1).1
Table 1
Switches vs destabilization: Defining antidepressant risks
| Risk | Definition |
|---|---|
| Acute switches to hypomania/mania | ≤8 weeks by convention, unless dosage is increased |
| Mood destabilization | |
| Cycle acceleration | Increase of ≥2 mood episodes while taking antidepressants, compared with a similar exposure time before treatment |
| Rapid cycling | ≥4mood episodes in previous 12 months (new-onset or exacerbation of baseline pattern), according to DSM-IV-TR |
| Source: Reference 1 | |
Switching risk. Some researchers have reported antidepressant-induced switches to be milder and more brief than spontaneous hypo/manias,13 whereas others have observed more-severe mixed14 and even psychotic episodes. Risk factors that may predispose patients to switching include:
- personal or family history of switches or mood destabilization
- family history of bipolar disorder
- exposure to multiple antidepressant trials
- history of substance abuse or dependence
- early onset (age <25) and/or treatment of mood symptoms.15,16
True switch rates are difficult to estimate because clinical trials have used different switching definitions, durations, antidepressants (with or without mood stabilizers, and with different mood stabilizers), and cohorts (often excluding rapid cyclers). Except for the Nemeroff et al study,6 no prospective, double-blind, placebo-controlled studies have examined switch rates, and even this study was not large enough to detect statistically significant differences.
Thus we must rely on naturalistic evidence that is less rigorous but more applicable to clinical practice. This literature reveals switch rates of:
- 30% to 60% with TCAs and monoamine oxidase inhibitors (MAOIs)
- 15% to 27% with selective serotonin reuptake inhibitors (SSRIs), bupropion, and venlafaxine.
Average switch rates are thought to be approximately 40% with TCAs/MAOIs and 20% with the newer antidepressants.1 Preliminary data associate venlafaxine with higher switch rates than SSRIs or bupropion, so perhaps antidepressants with some noradrenergic effects (including TCAs) facilitate the switching phenomenon.17
Mood destabilization. Three randomized, controlled trials suggest that antidepressants—especially TCAs—increase the risk of cycle acceleration or rapid cycling in bipolar patients. The best designed study—sponsored by the National Institute of Mental Health—was a 10-year, prospective, double-blind trial of 51 rapid-cycling patients. The trial’s on-off-on design showed that 20% of these patients developed rapid cycling as a direct result of taking TCAs.18
Unfortunately, most randomized, controlled trials are not designed to show a relationship between antidepressants and mood destabilization. Observational literature is mixed but suggests that antidepressant use is associated with rapid cycling. Most evidence supports a relationship between antidepressants and long-term mood destabilization—especially cycle acceleration, which is believed to occur in approximately 20% of patients using TCAs or SSRIs.1
Are mood stabilizers protective? Some studies suggest that mood stabilizers may help protect against switches. Most of the evidence—using lithium and TCAs—suggests a 50% drop in switch rates when patients receive mood stabilizers with antidepressants. In one study, lithium was more protective than anticonvulsants for SSRI-induced mania, but the difference was not statistically significant.19
Because study data variability, we don’t know if some mood stabilizers are more effective than others in preventing antidepressant-related switching. This variability is likely caused by:
- medication-specific factors (such as higher switch rates with TCAs and possibly dual-reuptake inhibitors than with SSRIs)
- illness-specific factors (such as rapid cycling and cycle pattern)
- patient-specific factors, already described. Mood stabilizers appear to be more protective against switching than against mood destabilization, in which their effects are less clear (Table 2).15
Table 2
Frequency of switching or mood destabilization with antidepressants
| Bipolar risk | Causative agents | Frequency | Mood stabilizer effect |
|---|---|---|---|
| Acute switch | TCAs, MAOIs | ~ 40% | Variably protective; apparent partial risk reduction ~ 50% |
| SSRIs, bupropion, venlafaxine | ~ 20% | ||
| Mood destabilization | TCAs, SSRIs | ~ 20% | Not as clearly protective against mood destabilization as against acute switching |
| TCAs: tricyclic antidepressants; MAOIs: monoamine oxidase inhibitors; SSRIs: selective serotonin reuptake inhibitors | |||
| Source: References 1, 15 | |||
TREATMENT RECOMMENDATIONS
How does a clinician decide which bipolar depressed patients should receive antidepressants?
The first step in treating bipolar depression (Algorithm) is to provide optimal dosages of the patient’s mood stabilizers. Consensus guidelines20 suggest lithium or lamotrigine as first-line treatments for bipolar depression. Evidence also shows efficacy for atypical antipsychotics, including the olanzapine/fluoxetine combination (OFC)—FDA-approved for acute bipolar depression21—and quetiapine monotherapy.22 Dosages vary, but suggested ranges include:
- lithium: 0.6 to 1.2 mEq/L; aim for approximately 0.8 mEq/L, but some data suggest 0.6 to 0.7 mEq/L may be sufficient
- lamotrigine: 50 to 250 mg/d (the higher dosage is based on maintenance studies)
- OFC: 6 to 12 mg olanzapine/25 to 50 mg fluoxetine
- quetiapine: 300 to 600 mg/d.
The next step is an antidepressant risk/benefit analysis, weighing the considerable risks of switching/mood destabilization with the patient’s depressive illness severity, type of bipolar disorder (such as rapid cycling), and cycle pattern.
Algorithm Recommended treatment of bipolar depression
Cycle patterns. In a naturalistic study, Macqueen et al23 used life chart data for 42 bipolar patients to assess how the mood state preceding a prospectively observed depressive episode affected treatment response:
- A euthymic mood state in the previous 2 months represented a uniphasic pattern and an isolated depressive episode.
- A preceding hypomanic/manic mood state indicated a biphasic pattern.
Approximately 60% of bipolar patients show a biphasic pattern, although the episode sequence is usually depression-hypomania/mania rather than hypomania/mania-depression. These authors included patients whose breakthrough depressive episodes were treated with an antidepressant or a putative mood stabilizer but not an atypical antipsychotic.
In patients treated with an antidepressant, the response-to-switch ratio was 10:1 for those previously euthymic, compared with a less beneficial 0.75:1 in previously hypomanic/manic patients. This small study suggests that a patient’s cycle pattern may help you decide whether to use an antidepressant for bipolar depression.
How to use antidepressants. As described, some depressed bipolar patients are better candidates for antidepressant therapy than others (Table 3).
Table 3
Antidepressants for bipolar depression? Consider ‘ideal patient’ traits
| Severe depression refractory to optimal doses of ≥1 mood stabilizers |
| Uniphasic cycle pattern |
| Not rapid cycling |
| No history of switching or mood destabilization |
| No comorbid substance abuse |
Use antidepressants cautiously and conservatively in a minority of bipolar patients (approximately 20% to 40%) and usually for short periods (discussed below). SSRIs or bupropion are first-line agents because:
- they appear to be relatively less likely to cause switching than other antidepressant classes
- controlled trials have examined these antidepressants in bipolar depression.
Depressed patients with very mild, nonrapid-cycling, bipolar II disorder and no more than three previous hypomanic episodes might be candidates for antidepressant monotherapy. In other bipolar patients, always use at least one mood stabilizer if you decide to use an antidepressant.
TREATMENT DURATION
No randomized, controlled trial has examined what duration of antidepressant treatment may be optimum for bipolar depression, but consensus guidelines recommend:
- approximately 3 to 7 months, depending on depression severity
- approximately one-half that duration (2 to 4 months) for rapid-cycling bipolar disorder.20
Because of the switching risk, one could also argue for a shorter treatment duration in patients with a biphasic cycle pattern—especially with an episode sequence of depression to hypomania/mania to euthymia.
Ideally, patients would stay on antidepressants no longer than the natural course of their depression (usually 2 to 6 months in bipolar depression), although it could be shorter in rapid cyclers. Approximately 15% to 20% of patients may have a robust initial response to antidepressants and need to be maintained on these medications, especially after several tapers and relapses have failed.
Related resources
- Bipolar Clinic and Research Program. Massachusetts General Hospital. Includes tools for clinicians and the clinical site for the Systematic Treatment Enhancement Program for Bipolar Disorder (STEP-BD). www.manicdepressive.org.
- Goodwin FK, Jamison KR. Manic-depressive illness. New York: Oxford University Press, 1990.
Drug brand names
- Bupropion • Wellbutrin
- Imipramine • Tofranil
- Lamotrigine • Lamictal
- Lithium • Lithobid, others
- Olanzapine/fluoxetine • Symbyax
- Paroxetine • Paxil
- Quetiapine • Seroquel
- Venlafaxine • Effexor
Disclosures
Dr. Altman is a speaker for Forest Pharmaceuticals, Janssen Pharmaceutica, AstraZeneca Pharmaceuticals, and Abbott Laboratories.
1. Ghaemi SN, Hsu DJ, Soldani F, et al. Antidepressants in bipolar disorder: the case for caution. Bipolar Disorders 2003;5:421-33.
2. Judd LL. The long-term natural history of the weekly symptomatic status of bipolar I disorder. Arch Gen Psychiatry 2002;59:530-7.
3. Judd LL, Akiskal HS, Schettler PJ, et al. A prospective investigation of the natural history of the long-term weekly symptomatic status of bipolar II disorder. Arch Gen Psychiatry 2003;60(3):261-9.
4. Young LT, Joffe RT, Robb JC, et al. Double-blind comparison of addition of a second mood stabilizer versus an antidepressant to an initial mood stabilizer for treatment of patients with bipolar depression. Am J Psychiatry 2000;157:124-6.
5. Cohn JB, Collins G, Ashbrook E, et al. A comparison of fluoxetine, imipramine and placebo in patients with bipolar depressive disorder. Int Clin Psychopharmacol 1989;4(4):313-22.
6. Nemeroff CB, Evans DL, Gyulai L, et al. Double-blind, placebo-controlled comparison of imipramine and paroxetine in the treatment of bipolar depression. Am J Psychiatry 2001;158(6):906-12.
7. Ghaemi SN, Lenox MS, Baldessarini RJ. Effectiveness and safety of long-term antidepressant treatment in bipolar disorder. J Clin Psychiatry 2001;62(7):565-9.
8. Amsterdam JD, Garcia-Espana F, Fawcett J, et al. Efficacy and safety of fluoxetine in treating bipolar II major depressive episode. J Clin Psychopharmacol 1998;18:435-40.
9. Amsterdam JD, Garcia-Espana F. Venlafaxine monotherapy in women with bipolar II and unipolar major depression. J Affect Disord 2000;59:225-9.
10. Altshuler L, Kiriakos L, Calcagno J, et al. Impact of antidepressant discontinuation versus antidepressant continuation at 1-year risk for relapse of bipolar depression: a retrospective chart review. J Clin Psychiatry 2001;62:612-16.
11. Altshuler L, Suppes T, Black D, et al. Impact of antidepressant discontinuation after acute bipolar depression remission on rates of depressive relapse at 1-year follow-up. Am J Psychiatry 2003;160:1252-62.
12. Ghaemi SN, Rosenquist KJ, Ko JY, et al. Antidepressant treatment in bipolar versus unipolar depression. Am J Psychiatry 2004;161:163-5.
13. Stoll AL, Mayer PV, Kolbrener M, et al. Antidepressant-associated mania: a controlled comparison with spontaneous mania. Am J Psychiatry 1994;151:1642-5.
14. Zubieta JK, Demitrack MA. Possible bupropion precipitation of mania and mixed affective state. J Clin Psychopharmacol 1991;11(5):327-8.
15. Goldberg JF, Truman CJ. Antidepressant-induced mania: an overview of current controversies. Bipolar Disorders 2003;5:407-20.
16. Goldberg JF, Whiteside JE. The association between substance abuse and antidepressant-induced mania in bipolar disorder: a preliminary study. J Clin Psychiatry 2002;63:791-5.
17. Post RM, Leverich GS, Nolen WA, et al. A re-evaluation of the role of antidepressants in the treatment of bipolar depression: data from the Stanley Foundation Bipolar Network. Bipolar Disorders 2003;5:396-406.
18. Wehr TA, Sack DA, Rosenthal NE, et al. Rapid cycling affective disorder: contributing factors and treatment responses in 51 patients. Am J Psychiatry 1988;145:179-84.
19. Henry C, Sorbara F, Lacoste J, et al. Antidepressant-induced mania in bipolar patients: identification of risk factors. J Clin Psychiatry 2001;62:249-55.
20. Keck PE, Jr, Perlis RH, Otto MW, et al. The Expert Consensus Guideline Series: Treatment of bipolar disorder 2004. Postgrad Med 2004;1-120.
21. Tohen M, Vieta E, Calabrese J, et al. Efficacy of olanzapine and olanzapine-fluoxetine combination in the treatment of bipolar I depression. Arch Gen Psychiatry 2003;60:1079-88.
22. Calabrese JR. Quetiapine BOLDER study [presentation]. New York: American Psychiatric Association annual meeting, 2004.
23. MacQueen GM, Young LT, Marriott M, et al. Previous mood state predicts response and switch rates in patients with bipolar depression. Acta Psychiatr Scand 2002;105:414-18.
1. Ghaemi SN, Hsu DJ, Soldani F, et al. Antidepressants in bipolar disorder: the case for caution. Bipolar Disorders 2003;5:421-33.
2. Judd LL. The long-term natural history of the weekly symptomatic status of bipolar I disorder. Arch Gen Psychiatry 2002;59:530-7.
3. Judd LL, Akiskal HS, Schettler PJ, et al. A prospective investigation of the natural history of the long-term weekly symptomatic status of bipolar II disorder. Arch Gen Psychiatry 2003;60(3):261-9.
4. Young LT, Joffe RT, Robb JC, et al. Double-blind comparison of addition of a second mood stabilizer versus an antidepressant to an initial mood stabilizer for treatment of patients with bipolar depression. Am J Psychiatry 2000;157:124-6.
5. Cohn JB, Collins G, Ashbrook E, et al. A comparison of fluoxetine, imipramine and placebo in patients with bipolar depressive disorder. Int Clin Psychopharmacol 1989;4(4):313-22.
6. Nemeroff CB, Evans DL, Gyulai L, et al. Double-blind, placebo-controlled comparison of imipramine and paroxetine in the treatment of bipolar depression. Am J Psychiatry 2001;158(6):906-12.
7. Ghaemi SN, Lenox MS, Baldessarini RJ. Effectiveness and safety of long-term antidepressant treatment in bipolar disorder. J Clin Psychiatry 2001;62(7):565-9.
8. Amsterdam JD, Garcia-Espana F, Fawcett J, et al. Efficacy and safety of fluoxetine in treating bipolar II major depressive episode. J Clin Psychopharmacol 1998;18:435-40.
9. Amsterdam JD, Garcia-Espana F. Venlafaxine monotherapy in women with bipolar II and unipolar major depression. J Affect Disord 2000;59:225-9.
10. Altshuler L, Kiriakos L, Calcagno J, et al. Impact of antidepressant discontinuation versus antidepressant continuation at 1-year risk for relapse of bipolar depression: a retrospective chart review. J Clin Psychiatry 2001;62:612-16.
11. Altshuler L, Suppes T, Black D, et al. Impact of antidepressant discontinuation after acute bipolar depression remission on rates of depressive relapse at 1-year follow-up. Am J Psychiatry 2003;160:1252-62.
12. Ghaemi SN, Rosenquist KJ, Ko JY, et al. Antidepressant treatment in bipolar versus unipolar depression. Am J Psychiatry 2004;161:163-5.
13. Stoll AL, Mayer PV, Kolbrener M, et al. Antidepressant-associated mania: a controlled comparison with spontaneous mania. Am J Psychiatry 1994;151:1642-5.
14. Zubieta JK, Demitrack MA. Possible bupropion precipitation of mania and mixed affective state. J Clin Psychopharmacol 1991;11(5):327-8.
15. Goldberg JF, Truman CJ. Antidepressant-induced mania: an overview of current controversies. Bipolar Disorders 2003;5:407-20.
16. Goldberg JF, Whiteside JE. The association between substance abuse and antidepressant-induced mania in bipolar disorder: a preliminary study. J Clin Psychiatry 2002;63:791-5.
17. Post RM, Leverich GS, Nolen WA, et al. A re-evaluation of the role of antidepressants in the treatment of bipolar depression: data from the Stanley Foundation Bipolar Network. Bipolar Disorders 2003;5:396-406.
18. Wehr TA, Sack DA, Rosenthal NE, et al. Rapid cycling affective disorder: contributing factors and treatment responses in 51 patients. Am J Psychiatry 1988;145:179-84.
19. Henry C, Sorbara F, Lacoste J, et al. Antidepressant-induced mania in bipolar patients: identification of risk factors. J Clin Psychiatry 2001;62:249-55.
20. Keck PE, Jr, Perlis RH, Otto MW, et al. The Expert Consensus Guideline Series: Treatment of bipolar disorder 2004. Postgrad Med 2004;1-120.
21. Tohen M, Vieta E, Calabrese J, et al. Efficacy of olanzapine and olanzapine-fluoxetine combination in the treatment of bipolar I depression. Arch Gen Psychiatry 2003;60:1079-88.
22. Calabrese JR. Quetiapine BOLDER study [presentation]. New York: American Psychiatric Association annual meeting, 2004.
23. MacQueen GM, Young LT, Marriott M, et al. Previous mood state predicts response and switch rates in patients with bipolar depression. Acta Psychiatr Scand 2002;105:414-18.
Speech recognition programs
Speech recognition technology—once too slow and inaccurate for clinical practice—is increasingly helping psychiatrists record patient notes, dictate letters and lengthy reports, and operate their computers.
Is speech recognition right for your practice? This article will help you decide by reviewing available programs and offering insights on choosing one for your practice.
Speaking of progress
Clinicians in radiology and pathology were among the first to use limited programs that employed voice commands and short phrases. Shortcuts that told the computer to type boilerplate passages have been available for more than 25 years. Early speech recognition programs required users to speak with a stilted voice and pauses between words.
By 1998, several “continuous speech” systems were available, but most were not suitable for a solo or small group psychiatric practice. Some included diagnosis-specific report templates that could be customized for initial assessment and progress notes.1 These programs were useful for dictating short memos or e-mails but not for composing longer documents because of awkward editing and difficulties with punctuation, formatting, and on-screen navigation.2
Today’s programs recognize natural speech without pauses between words. As the clinician speaks into a microphone at a normal pace, text is typed at speeds approaching 160 words per minute with 90% to 98% accuracy. New versions of some programs save the dictation in audio files, allowing an assistant to listen to the dictation later and edit the transcription. These programs also eliminate superfluous utterances and perform macro commands, including handling e-mail.
Newer speech recognition products are much easier to use than older programs. They can be mastered within days and are cost-effective for solo or small group practices. Single-user programs range in cost from free (included with the Windows XP or newer Macintosh operating systems), to approximately $175 for ViaVoice Professional, to about $800 for the highly recommended Dragon Naturally Speaking Version 8.0.3
How to get started
Speech recognition programs require a powerful computer with processor (microchip) speed >1 gigahertz, random access memory (RAM) ≥ 512 megabytes, and a platform no older than Windows 2000 or Mac OS X Version 10.1.
After the software is installed, some clicking and keystroking may still be necessary. Learning when to talk or type can help users increase efficiency and prevent repetitive strain injury.
Critical factors for successful use include user motivation and training (or consultation with a reseller), a specialized vocabulary and language model, and a high-quality sound card and microphone (the most sophisticated hardware available is recommended, and this usually must be purchased separately).
Most speech recognition programs allow different users to train on the same computer. Users can dictate directly into word-processing applications, and some products allow dictation into other office programs.
What’s available
Dragon Naturally Speaking Professional Medical Solutions Version 8.0 (www.dragontalk.com/DNS_MED_ PRO.htm) is widely recognized for its performance, high accuracy, and easy user interface. Users can dictate directly into a PC for immediate transcription or into selected digital recorders or personal digital assistants for transcription later. A file of your recorded speech can be saved along with the computer-transcribed document for future proofreading.
The program can process previously completed reports to customize word-use patterns and build a personalized vocabulary. The optional but useful medical vocabulary includes many terms unique to psychiatry and psychology.
Dragon responds to voice commands and macros and features online training and user guides. It works in most Windows-based applications but is not available for Macintosh.4
IBM’s ViaVoice Release 10 (uk.scansoft.com/viavoice) is available in six languages and multiple levels and comes in versions for Windows and Macintosh platforms (also optimized for G4). Its manufacturer offers support for selected digital handheld recorders.
ViaVoice can analyze previous documents and save recorded dictation, is strong on voice navigation, and recognizes file names and tool bar buttons. Users can open a file by speaking its name or activate a command by saying it.
There are some drawbacks, however. Specialized medical vocabularies must be obtained from outside the company, creating additional technical obstacles or requiring developer assistance. Also, several reviewers do not consider ViaVoice as robust, accurate, or fast as Dragon for lengthy or medical dictation.5
Philips SpeechMagic. (www.speech.philips.com) Philips has developed sophisticated tools for document creation, transcription, and commands that integrate with larger information systems. The products are network-based and scalable, essentially designed for large groups or medical centers. The programs cannot be purchased from Philips but are installed by its distributors and software vendors.
Microsoft Office XP (http://office.microsoft.com) includes an alternative user input speech recognition feature within the operating system that offers dictation and voice command modes. It works with any office program and offers a “taste” of speech recognition, but with extremely limited function. It requires awkward switching between dictation and commands, does not filter out extraneous noises, and has no specialized medical vocabulary.
Apple Speech Recognition (www.apple.com/macosx/features/speech/), included in Mac OS X, is rudimentary and is appropriate primarily for controlling a computer by voice commands. It requires no training and can convert English text to spoken words.
The future
Speech recognition programs that can be integrated with telephones, wireless phones, and tablet screens are in development. Microsoft has released speech-control software for Pocket PC devices that run on Windows Mobile 2003 and recognition software for navigating the Web.
Before long, personal digital assistants with built-in speech-recognition technology may respond to spoken questions or commands with a computer-synthesized voice, thus making a clumsy stylus or keypad outdated.
Related resources
- Huang MP, Alessi NE. The Internet and the future of psychiatry. Am J Psychiatry 1996;153:861-9.
- Fulton S. Chart comparing features of Windows-based continuous speech programs. www.out-loud.com/features.html.
- Taintor Z. Computers, the patient, and the psychiatrist. In Dickstein LJ, Riba MB, Oldham JM. Review of psychiatry. Vol 16. Washington, DC: American Psychiatric Press; 1997.
Disclosure
Dr. Green reports no financial relationship with any company whose products are mentioned in this article. The opinions expressed by Dr. Green are his and do not necessarily reflect those of Current Psychiatry.
Acknowledgment
The author thanks Dan Newman, author of several books and video guides on speech recognition, and Len Zullo, chief executive officer, Assistive Technologies Inc., for their help with researching this article and personal communication regarding current product features and comparisons.
1. Leipsic JS. Computer speech recognition in psychiatry. Psychiatric Times 1998;15(8):54-6.
2. Miastkowski S. Can we talk? PC World January 1999;127-36.
3. Manes S. Speech! Speech! Forbes February 28, 2005. Available at: http://www.forbes.com/forbes/2005/0228/054_print.html. Accessed June 21, 2005.
4. Dragon Naturally Speaking Professional Users Guide Version 6. Burlington, MA: ScanSoft 2002;1-25,125-40,185-8.
5. Newman D. Talk to your computer: speech recognition made easy. Berkeley CA: Waveside Publishing; 2000;9-41,122-3.
Dr. Green is a distinguished fellow, American Psychiatric Association, and chair of information services, San Diego Psychiatric Society. Psyber Psychiatry, edited by John S. Luo, MD, is published monthly at www.currentpsychiatry.com.
Speech recognition technology—once too slow and inaccurate for clinical practice—is increasingly helping psychiatrists record patient notes, dictate letters and lengthy reports, and operate their computers.
Is speech recognition right for your practice? This article will help you decide by reviewing available programs and offering insights on choosing one for your practice.
Speaking of progress
Clinicians in radiology and pathology were among the first to use limited programs that employed voice commands and short phrases. Shortcuts that told the computer to type boilerplate passages have been available for more than 25 years. Early speech recognition programs required users to speak with a stilted voice and pauses between words.
By 1998, several “continuous speech” systems were available, but most were not suitable for a solo or small group psychiatric practice. Some included diagnosis-specific report templates that could be customized for initial assessment and progress notes.1 These programs were useful for dictating short memos or e-mails but not for composing longer documents because of awkward editing and difficulties with punctuation, formatting, and on-screen navigation.2
Today’s programs recognize natural speech without pauses between words. As the clinician speaks into a microphone at a normal pace, text is typed at speeds approaching 160 words per minute with 90% to 98% accuracy. New versions of some programs save the dictation in audio files, allowing an assistant to listen to the dictation later and edit the transcription. These programs also eliminate superfluous utterances and perform macro commands, including handling e-mail.
Newer speech recognition products are much easier to use than older programs. They can be mastered within days and are cost-effective for solo or small group practices. Single-user programs range in cost from free (included with the Windows XP or newer Macintosh operating systems), to approximately $175 for ViaVoice Professional, to about $800 for the highly recommended Dragon Naturally Speaking Version 8.0.3
How to get started
Speech recognition programs require a powerful computer with processor (microchip) speed >1 gigahertz, random access memory (RAM) ≥ 512 megabytes, and a platform no older than Windows 2000 or Mac OS X Version 10.1.
After the software is installed, some clicking and keystroking may still be necessary. Learning when to talk or type can help users increase efficiency and prevent repetitive strain injury.
Critical factors for successful use include user motivation and training (or consultation with a reseller), a specialized vocabulary and language model, and a high-quality sound card and microphone (the most sophisticated hardware available is recommended, and this usually must be purchased separately).
Most speech recognition programs allow different users to train on the same computer. Users can dictate directly into word-processing applications, and some products allow dictation into other office programs.
What’s available
Dragon Naturally Speaking Professional Medical Solutions Version 8.0 (www.dragontalk.com/DNS_MED_ PRO.htm) is widely recognized for its performance, high accuracy, and easy user interface. Users can dictate directly into a PC for immediate transcription or into selected digital recorders or personal digital assistants for transcription later. A file of your recorded speech can be saved along with the computer-transcribed document for future proofreading.
The program can process previously completed reports to customize word-use patterns and build a personalized vocabulary. The optional but useful medical vocabulary includes many terms unique to psychiatry and psychology.
Dragon responds to voice commands and macros and features online training and user guides. It works in most Windows-based applications but is not available for Macintosh.4
IBM’s ViaVoice Release 10 (uk.scansoft.com/viavoice) is available in six languages and multiple levels and comes in versions for Windows and Macintosh platforms (also optimized for G4). Its manufacturer offers support for selected digital handheld recorders.
ViaVoice can analyze previous documents and save recorded dictation, is strong on voice navigation, and recognizes file names and tool bar buttons. Users can open a file by speaking its name or activate a command by saying it.
There are some drawbacks, however. Specialized medical vocabularies must be obtained from outside the company, creating additional technical obstacles or requiring developer assistance. Also, several reviewers do not consider ViaVoice as robust, accurate, or fast as Dragon for lengthy or medical dictation.5
Philips SpeechMagic. (www.speech.philips.com) Philips has developed sophisticated tools for document creation, transcription, and commands that integrate with larger information systems. The products are network-based and scalable, essentially designed for large groups or medical centers. The programs cannot be purchased from Philips but are installed by its distributors and software vendors.
Microsoft Office XP (http://office.microsoft.com) includes an alternative user input speech recognition feature within the operating system that offers dictation and voice command modes. It works with any office program and offers a “taste” of speech recognition, but with extremely limited function. It requires awkward switching between dictation and commands, does not filter out extraneous noises, and has no specialized medical vocabulary.
Apple Speech Recognition (www.apple.com/macosx/features/speech/), included in Mac OS X, is rudimentary and is appropriate primarily for controlling a computer by voice commands. It requires no training and can convert English text to spoken words.
The future
Speech recognition programs that can be integrated with telephones, wireless phones, and tablet screens are in development. Microsoft has released speech-control software for Pocket PC devices that run on Windows Mobile 2003 and recognition software for navigating the Web.
Before long, personal digital assistants with built-in speech-recognition technology may respond to spoken questions or commands with a computer-synthesized voice, thus making a clumsy stylus or keypad outdated.
Related resources
- Huang MP, Alessi NE. The Internet and the future of psychiatry. Am J Psychiatry 1996;153:861-9.
- Fulton S. Chart comparing features of Windows-based continuous speech programs. www.out-loud.com/features.html.
- Taintor Z. Computers, the patient, and the psychiatrist. In Dickstein LJ, Riba MB, Oldham JM. Review of psychiatry. Vol 16. Washington, DC: American Psychiatric Press; 1997.
Disclosure
Dr. Green reports no financial relationship with any company whose products are mentioned in this article. The opinions expressed by Dr. Green are his and do not necessarily reflect those of Current Psychiatry.
Acknowledgment
The author thanks Dan Newman, author of several books and video guides on speech recognition, and Len Zullo, chief executive officer, Assistive Technologies Inc., for their help with researching this article and personal communication regarding current product features and comparisons.
Speech recognition technology—once too slow and inaccurate for clinical practice—is increasingly helping psychiatrists record patient notes, dictate letters and lengthy reports, and operate their computers.
Is speech recognition right for your practice? This article will help you decide by reviewing available programs and offering insights on choosing one for your practice.
Speaking of progress
Clinicians in radiology and pathology were among the first to use limited programs that employed voice commands and short phrases. Shortcuts that told the computer to type boilerplate passages have been available for more than 25 years. Early speech recognition programs required users to speak with a stilted voice and pauses between words.
By 1998, several “continuous speech” systems were available, but most were not suitable for a solo or small group psychiatric practice. Some included diagnosis-specific report templates that could be customized for initial assessment and progress notes.1 These programs were useful for dictating short memos or e-mails but not for composing longer documents because of awkward editing and difficulties with punctuation, formatting, and on-screen navigation.2
Today’s programs recognize natural speech without pauses between words. As the clinician speaks into a microphone at a normal pace, text is typed at speeds approaching 160 words per minute with 90% to 98% accuracy. New versions of some programs save the dictation in audio files, allowing an assistant to listen to the dictation later and edit the transcription. These programs also eliminate superfluous utterances and perform macro commands, including handling e-mail.
Newer speech recognition products are much easier to use than older programs. They can be mastered within days and are cost-effective for solo or small group practices. Single-user programs range in cost from free (included with the Windows XP or newer Macintosh operating systems), to approximately $175 for ViaVoice Professional, to about $800 for the highly recommended Dragon Naturally Speaking Version 8.0.3
How to get started
Speech recognition programs require a powerful computer with processor (microchip) speed >1 gigahertz, random access memory (RAM) ≥ 512 megabytes, and a platform no older than Windows 2000 or Mac OS X Version 10.1.
After the software is installed, some clicking and keystroking may still be necessary. Learning when to talk or type can help users increase efficiency and prevent repetitive strain injury.
Critical factors for successful use include user motivation and training (or consultation with a reseller), a specialized vocabulary and language model, and a high-quality sound card and microphone (the most sophisticated hardware available is recommended, and this usually must be purchased separately).
Most speech recognition programs allow different users to train on the same computer. Users can dictate directly into word-processing applications, and some products allow dictation into other office programs.
What’s available
Dragon Naturally Speaking Professional Medical Solutions Version 8.0 (www.dragontalk.com/DNS_MED_ PRO.htm) is widely recognized for its performance, high accuracy, and easy user interface. Users can dictate directly into a PC for immediate transcription or into selected digital recorders or personal digital assistants for transcription later. A file of your recorded speech can be saved along with the computer-transcribed document for future proofreading.
The program can process previously completed reports to customize word-use patterns and build a personalized vocabulary. The optional but useful medical vocabulary includes many terms unique to psychiatry and psychology.
Dragon responds to voice commands and macros and features online training and user guides. It works in most Windows-based applications but is not available for Macintosh.4
IBM’s ViaVoice Release 10 (uk.scansoft.com/viavoice) is available in six languages and multiple levels and comes in versions for Windows and Macintosh platforms (also optimized for G4). Its manufacturer offers support for selected digital handheld recorders.
ViaVoice can analyze previous documents and save recorded dictation, is strong on voice navigation, and recognizes file names and tool bar buttons. Users can open a file by speaking its name or activate a command by saying it.
There are some drawbacks, however. Specialized medical vocabularies must be obtained from outside the company, creating additional technical obstacles or requiring developer assistance. Also, several reviewers do not consider ViaVoice as robust, accurate, or fast as Dragon for lengthy or medical dictation.5
Philips SpeechMagic. (www.speech.philips.com) Philips has developed sophisticated tools for document creation, transcription, and commands that integrate with larger information systems. The products are network-based and scalable, essentially designed for large groups or medical centers. The programs cannot be purchased from Philips but are installed by its distributors and software vendors.
Microsoft Office XP (http://office.microsoft.com) includes an alternative user input speech recognition feature within the operating system that offers dictation and voice command modes. It works with any office program and offers a “taste” of speech recognition, but with extremely limited function. It requires awkward switching between dictation and commands, does not filter out extraneous noises, and has no specialized medical vocabulary.
Apple Speech Recognition (www.apple.com/macosx/features/speech/), included in Mac OS X, is rudimentary and is appropriate primarily for controlling a computer by voice commands. It requires no training and can convert English text to spoken words.
The future
Speech recognition programs that can be integrated with telephones, wireless phones, and tablet screens are in development. Microsoft has released speech-control software for Pocket PC devices that run on Windows Mobile 2003 and recognition software for navigating the Web.
Before long, personal digital assistants with built-in speech-recognition technology may respond to spoken questions or commands with a computer-synthesized voice, thus making a clumsy stylus or keypad outdated.
Related resources
- Huang MP, Alessi NE. The Internet and the future of psychiatry. Am J Psychiatry 1996;153:861-9.
- Fulton S. Chart comparing features of Windows-based continuous speech programs. www.out-loud.com/features.html.
- Taintor Z. Computers, the patient, and the psychiatrist. In Dickstein LJ, Riba MB, Oldham JM. Review of psychiatry. Vol 16. Washington, DC: American Psychiatric Press; 1997.
Disclosure
Dr. Green reports no financial relationship with any company whose products are mentioned in this article. The opinions expressed by Dr. Green are his and do not necessarily reflect those of Current Psychiatry.
Acknowledgment
The author thanks Dan Newman, author of several books and video guides on speech recognition, and Len Zullo, chief executive officer, Assistive Technologies Inc., for their help with researching this article and personal communication regarding current product features and comparisons.
1. Leipsic JS. Computer speech recognition in psychiatry. Psychiatric Times 1998;15(8):54-6.
2. Miastkowski S. Can we talk? PC World January 1999;127-36.
3. Manes S. Speech! Speech! Forbes February 28, 2005. Available at: http://www.forbes.com/forbes/2005/0228/054_print.html. Accessed June 21, 2005.
4. Dragon Naturally Speaking Professional Users Guide Version 6. Burlington, MA: ScanSoft 2002;1-25,125-40,185-8.
5. Newman D. Talk to your computer: speech recognition made easy. Berkeley CA: Waveside Publishing; 2000;9-41,122-3.
Dr. Green is a distinguished fellow, American Psychiatric Association, and chair of information services, San Diego Psychiatric Society. Psyber Psychiatry, edited by John S. Luo, MD, is published monthly at www.currentpsychiatry.com.
1. Leipsic JS. Computer speech recognition in psychiatry. Psychiatric Times 1998;15(8):54-6.
2. Miastkowski S. Can we talk? PC World January 1999;127-36.
3. Manes S. Speech! Speech! Forbes February 28, 2005. Available at: http://www.forbes.com/forbes/2005/0228/054_print.html. Accessed June 21, 2005.
4. Dragon Naturally Speaking Professional Users Guide Version 6. Burlington, MA: ScanSoft 2002;1-25,125-40,185-8.
5. Newman D. Talk to your computer: speech recognition made easy. Berkeley CA: Waveside Publishing; 2000;9-41,122-3.
Dr. Green is a distinguished fellow, American Psychiatric Association, and chair of information services, San Diego Psychiatric Society. Psyber Psychiatry, edited by John S. Luo, MD, is published monthly at www.currentpsychiatry.com.
Pharmacogenomic DNA chip
Genotyping for cytochrome (CYP) P-450 gene variations can identify patients who will not benefit from, or may react badly to, some psychotropics.1 Psychiatrists can then more accurately tailor initial dosages to improve response and prevent adverse reactions.
An FDA-approved pharmacogenomic diagnostic DNA chip is expected to be available to clinical laboratories this month (Table 1). The chip provides an accurate genotype for two drug-metabolizing enzymes—2D6 and 2C19.
Table 1
Pharmacogenomic DNA chip: Fast facts
| Brand name: |
| AmpliChip CYP 450 Test |
| FDA-approved indication: |
| Genotyping patients |
| Manufacturer: |
| Roche Diagnostics |
| Estimated availability: |
| July 2005 |
| Recommended use: |
| Determining cytochrome P-450 2D6 and 2C19 gene variations in patients before prescribing a psychotropic metabolized through these pathways. |
| Laboratories that process AmpliChip results: |
| Labcore, Mayo Medical Laboratories, Quest Diagnostics |
Genotyping’S Role in Psychiatry
CYP 2D6 and 2C19 enzymes help metabolize many commonly prescribed psychotropics, including:
- fluoxetine, paroxetine, and venlafaxine, which are among the psychotropics primarily metabolized by the cytochrome P-450 2D6 enzyme (Table 2).
- amitriptyline and citalopram, which are among the psychotropics metabolized in part by 2C19 (Table 3).
The chip can identify patients who are genetically predisposed to abnormal metabolism of 2D6 and 2C19 substrates. This information can help psychiatrists improve response for ultrarapid metabolizers and minimize adverse effects experienced by poor metabolizers of these substrates.
For example, if the patient is an ultrarapid metabolizer of 2D6 and/or 2C19 substrates, the psychiatrist can:
- exceed the recommended dosage to reach adequate serum levels
- or choose an antidepressant not primarily metabolized by either enzyme.
For a poor metabolizer of 2D6 and/or 2C19 substrates, the psychiatrist can:
- choose an antidepressant metabolized by a different enzyme
- or prescribe 2D6 and 2C19 substrates at very low dosages.
For example, some poor metabolizers of 2D6 substrates have been successfully treated with fluoxetine, 2 to 5 mg/d.2,3 This approach can help avoid side effects and potentially save the patient money. To prevent prescription errors, make sure the pharmacist understands your rationale for lower-than-recommended dosages.
Patients who are poor metabolizers of 2C19 and extensive metabolizers of 2D6 substrates can probably tolerate citalopram and amitriptyline dosages at the low end of the therapeutic range. Watch for high serum levels of either or both drugs if both enzyme systems are inactive.
Table 2
Evidence suggests these drugs are predominantly metabolized by the 2D6 enzyme*
| Antidepressants | Antipsychotics | Stimulants |
|---|---|---|
| Desipramine | Fluphenazine | Atomoxetine |
| Fluoxetine | Perphenazine | |
| Nortriptyline | Risperidone | |
| Paroxetine | Thioridazine | |
| Venlafaxine | ||
| *Use caution when prescribing these agents to patients who are poor 2D6 metabolizers. | ||
Table 3
Evidence suggests these drugs are predominantly metabolized by the 2C19 enzyme*
| Antidepressants | |
|---|---|
| Diazepam | Citalopram |
| Clomipramine | Escitalopram |
| Imipramine | Sertraline |
| Benzodiazepines | |
| Amitriptyline | |
| *Use caution when prescribing these agents to patients who are poor 2C19 metabolizers. | |
Pharmacogenomic Chip’s Accuracy
The 2D6 gene has more than 100 variations, many of which are very rare mutations. The pharmacogenomic DNA chip can detect 27 of these variants, allowing the chip to accurately genotype most patients. By contrast, early 2D6 genotyping techniques identified only four or five variants, resulting in too many false negatives for clinical use.4
The chip also can identify the normal form of the 2C19 gene and two of its variants. Both variants produce an inactive 2C19 enzyme form that is ineffective in metabolizing 2C19 substrates.
Clinical Use
When should a psychiatrist obtain 2D6 and 2C19 genotypes?
First, understand that the pharmacogenomic chip does not predict which medications will produce a therapeutic response. Gene chips that predict response are in development but probably will not be available before 2008.
The chip, however, can identify the relatively few ultrarapid metabolizers who will not benefit from 2D6 or 2C19 substrate medications at normal dosages, as well as “poor metabolizers” of these substrates.1 The approximately 1% of whites in the United States who have ≥3 copies of the 2D6 gene metabolize 2D6 substrates very rapidly and will not respond to recommended dosages. About 10% of whites in the United States metabolize 2D6 or 2C19 substrates poorly and face increased risk of adverse reactions from these medications.
There is some evidence that the prevalence of these genetic variations differ among ethnicities. Approximately 15% of Saudi Arabians and 20% of Ethiopians are ultrarapid metabolizers of 2D6 and 2C19 substrates.5,6
The most common 2D6 poor metabolizer allele (*4) has been found in 12% to 21% of whites, whereas 23% to 32% of Asians and 13% of whites have the most common 2C19 poor metabolizer allele (*2).6-10 Prevalence of poor 2D6 and/or 2C19 metabolism among African Americans, Hispanics, and Native Americans has not been established.
Clinical Practicality
Clinicians’ unfamiliarity with genotyping and cost concerns pose potential barriers to the test’s use.
Clinician knowledge. Pharmacogenomic 2D6 and 2C19 tests will soon be offered nationwide at reference laboratories such as Quest Diagnostics, Labcore, and Mayo Medical Laboratories. The psychiatrist can call the lab for instructions, then send a blood sample and receive results by mail within 2 to 3 days.
While I believe the test’s usefulness will soon be widely understood, courses are available to help clinicians learn about genetic testing. Mayo Clinic College of Medicine (http://www.mayo. edu/cme/genomics.html) offers an annual week-long CME course in August. The American Psychiatric Association, as part of its May 2006 annual meeting, will offer a similar half-day course led by Mayo Clinic psychiatrists.
Cost. The exact cost of using the pharmacogenomic chip varies, as each laboratory sets fees for genotyping. Even so, genotyping could offer enormous cost savings by preventing failed medication trials and reducing the need for more-intensive psychiatric care. Furthermore, many insurance companies cover genotype testing.
Related resources
- Pharmacogenomic diagnostic DNA chip product information. www.rochediagnostics.com/products_services/amplichip_cyp450.html.
- Kirchheiner J, Borsen K, Dahl ML, et al. CYP2D6 and CYP2C19 genotype-based dose recommendations for antidepressants: a first step towards subpopulation-specific dosages. Acta Psychiatr Scand 2001;103(3):173-92.
Drug brand names
- Amitriptyline • Elavil
- Atomoxetine • Strattera
- Citalopram • Celexa
- Clomipramine • Anafranil
- Desipramine • Norpramin
- Diazepam • Valium
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Fluphenazine • Prolixin
- Nortriptyline • Pamelor
- Paroxetine • Paxil
- Perphenazine • Trilafon
- Risperidone • Risperdal
Disclosure
Dr. Mrazek is a consultant to Predix Pharmaceuticals.
1. Mrazek DA. New tool: genotyping makes prescribing safer, more effective. Current Psychiatry 2004;3(9):11-23.
2. Kirchheiner J, Borsen K, Dahl ML, et al. CYP2D6 and CYP2C19 genotype-based dose recommendations for antidepressants: a first step towards subpopulation-specific dosages. Acta Psychiatr Scand 2001;103:173-92.
3. Kirchheiner J, Nickchen K, Bauer M, et al. Pharmacogenetics of antidepressants and antipsychotics: the contribution of allelic variations to the phenotype of drug response. Mol Psychiatry 2004;9:442-73.
4. Chou WH, Yan FX, Robbins-Weilert DK, et al. Comparison of two CYP2D6 genotyping methods and assessment of genotype-phenotype relationships. Clin Chem 2003;49:542-51.
5. Ingelman-Sundberg M, Oscarson M, McLellan RA. Polymorphic human cytochrome P450 enzymes: an opportunity for individualized drug treatment. Trends Pharmacol Sci 1999;20:342-9.
6. Phillips KA, Veenstra DL, Oren E, et al. Potential role of pharmacogenomics in reducing adverse drug reactions: a systematic review. JAMA 2001;286:2270-9.
7. Ingelman-Sundberg M. Pharmacogenetics: an opportunity for a safer and more efficient pharmacotherapy. J Intern Med 2001;250:186-200.
8. Mizutani T. PM frequencies of major CYPs in Asians and Caucasians. Drug Metab Rev 2003;35:99-106.
9. Griese EU, Ilett KF, Kitteringham NR, et al. Allele and genotype frequencies of polymorphic cytochromes P450 2D6, 2C19, and 2E1 in aborigines from western Australia. Pharmacogenetics 2001;11:69-76.
10. Sachse C, Brockmoller J, Bauer S, Roots I. Cytochrome P450 2D6 variants in a Caucasian population: allele frequencies and phenotypic consequences. Am J Hum Genet 1997;60:284-95.
Genotyping for cytochrome (CYP) P-450 gene variations can identify patients who will not benefit from, or may react badly to, some psychotropics.1 Psychiatrists can then more accurately tailor initial dosages to improve response and prevent adverse reactions.
An FDA-approved pharmacogenomic diagnostic DNA chip is expected to be available to clinical laboratories this month (Table 1). The chip provides an accurate genotype for two drug-metabolizing enzymes—2D6 and 2C19.
Table 1
Pharmacogenomic DNA chip: Fast facts
| Brand name: |
| AmpliChip CYP 450 Test |
| FDA-approved indication: |
| Genotyping patients |
| Manufacturer: |
| Roche Diagnostics |
| Estimated availability: |
| July 2005 |
| Recommended use: |
| Determining cytochrome P-450 2D6 and 2C19 gene variations in patients before prescribing a psychotropic metabolized through these pathways. |
| Laboratories that process AmpliChip results: |
| Labcore, Mayo Medical Laboratories, Quest Diagnostics |
Genotyping’S Role in Psychiatry
CYP 2D6 and 2C19 enzymes help metabolize many commonly prescribed psychotropics, including:
- fluoxetine, paroxetine, and venlafaxine, which are among the psychotropics primarily metabolized by the cytochrome P-450 2D6 enzyme (Table 2).
- amitriptyline and citalopram, which are among the psychotropics metabolized in part by 2C19 (Table 3).
The chip can identify patients who are genetically predisposed to abnormal metabolism of 2D6 and 2C19 substrates. This information can help psychiatrists improve response for ultrarapid metabolizers and minimize adverse effects experienced by poor metabolizers of these substrates.
For example, if the patient is an ultrarapid metabolizer of 2D6 and/or 2C19 substrates, the psychiatrist can:
- exceed the recommended dosage to reach adequate serum levels
- or choose an antidepressant not primarily metabolized by either enzyme.
For a poor metabolizer of 2D6 and/or 2C19 substrates, the psychiatrist can:
- choose an antidepressant metabolized by a different enzyme
- or prescribe 2D6 and 2C19 substrates at very low dosages.
For example, some poor metabolizers of 2D6 substrates have been successfully treated with fluoxetine, 2 to 5 mg/d.2,3 This approach can help avoid side effects and potentially save the patient money. To prevent prescription errors, make sure the pharmacist understands your rationale for lower-than-recommended dosages.
Patients who are poor metabolizers of 2C19 and extensive metabolizers of 2D6 substrates can probably tolerate citalopram and amitriptyline dosages at the low end of the therapeutic range. Watch for high serum levels of either or both drugs if both enzyme systems are inactive.
Table 2
Evidence suggests these drugs are predominantly metabolized by the 2D6 enzyme*
| Antidepressants | Antipsychotics | Stimulants |
|---|---|---|
| Desipramine | Fluphenazine | Atomoxetine |
| Fluoxetine | Perphenazine | |
| Nortriptyline | Risperidone | |
| Paroxetine | Thioridazine | |
| Venlafaxine | ||
| *Use caution when prescribing these agents to patients who are poor 2D6 metabolizers. | ||
Table 3
Evidence suggests these drugs are predominantly metabolized by the 2C19 enzyme*
| Antidepressants | |
|---|---|
| Diazepam | Citalopram |
| Clomipramine | Escitalopram |
| Imipramine | Sertraline |
| Benzodiazepines | |
| Amitriptyline | |
| *Use caution when prescribing these agents to patients who are poor 2C19 metabolizers. | |
Pharmacogenomic Chip’s Accuracy
The 2D6 gene has more than 100 variations, many of which are very rare mutations. The pharmacogenomic DNA chip can detect 27 of these variants, allowing the chip to accurately genotype most patients. By contrast, early 2D6 genotyping techniques identified only four or five variants, resulting in too many false negatives for clinical use.4
The chip also can identify the normal form of the 2C19 gene and two of its variants. Both variants produce an inactive 2C19 enzyme form that is ineffective in metabolizing 2C19 substrates.
Clinical Use
When should a psychiatrist obtain 2D6 and 2C19 genotypes?
First, understand that the pharmacogenomic chip does not predict which medications will produce a therapeutic response. Gene chips that predict response are in development but probably will not be available before 2008.
The chip, however, can identify the relatively few ultrarapid metabolizers who will not benefit from 2D6 or 2C19 substrate medications at normal dosages, as well as “poor metabolizers” of these substrates.1 The approximately 1% of whites in the United States who have ≥3 copies of the 2D6 gene metabolize 2D6 substrates very rapidly and will not respond to recommended dosages. About 10% of whites in the United States metabolize 2D6 or 2C19 substrates poorly and face increased risk of adverse reactions from these medications.
There is some evidence that the prevalence of these genetic variations differ among ethnicities. Approximately 15% of Saudi Arabians and 20% of Ethiopians are ultrarapid metabolizers of 2D6 and 2C19 substrates.5,6
The most common 2D6 poor metabolizer allele (*4) has been found in 12% to 21% of whites, whereas 23% to 32% of Asians and 13% of whites have the most common 2C19 poor metabolizer allele (*2).6-10 Prevalence of poor 2D6 and/or 2C19 metabolism among African Americans, Hispanics, and Native Americans has not been established.
Clinical Practicality
Clinicians’ unfamiliarity with genotyping and cost concerns pose potential barriers to the test’s use.
Clinician knowledge. Pharmacogenomic 2D6 and 2C19 tests will soon be offered nationwide at reference laboratories such as Quest Diagnostics, Labcore, and Mayo Medical Laboratories. The psychiatrist can call the lab for instructions, then send a blood sample and receive results by mail within 2 to 3 days.
While I believe the test’s usefulness will soon be widely understood, courses are available to help clinicians learn about genetic testing. Mayo Clinic College of Medicine (http://www.mayo. edu/cme/genomics.html) offers an annual week-long CME course in August. The American Psychiatric Association, as part of its May 2006 annual meeting, will offer a similar half-day course led by Mayo Clinic psychiatrists.
Cost. The exact cost of using the pharmacogenomic chip varies, as each laboratory sets fees for genotyping. Even so, genotyping could offer enormous cost savings by preventing failed medication trials and reducing the need for more-intensive psychiatric care. Furthermore, many insurance companies cover genotype testing.
Related resources
- Pharmacogenomic diagnostic DNA chip product information. www.rochediagnostics.com/products_services/amplichip_cyp450.html.
- Kirchheiner J, Borsen K, Dahl ML, et al. CYP2D6 and CYP2C19 genotype-based dose recommendations for antidepressants: a first step towards subpopulation-specific dosages. Acta Psychiatr Scand 2001;103(3):173-92.
Drug brand names
- Amitriptyline • Elavil
- Atomoxetine • Strattera
- Citalopram • Celexa
- Clomipramine • Anafranil
- Desipramine • Norpramin
- Diazepam • Valium
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Fluphenazine • Prolixin
- Nortriptyline • Pamelor
- Paroxetine • Paxil
- Perphenazine • Trilafon
- Risperidone • Risperdal
Disclosure
Dr. Mrazek is a consultant to Predix Pharmaceuticals.
Genotyping for cytochrome (CYP) P-450 gene variations can identify patients who will not benefit from, or may react badly to, some psychotropics.1 Psychiatrists can then more accurately tailor initial dosages to improve response and prevent adverse reactions.
An FDA-approved pharmacogenomic diagnostic DNA chip is expected to be available to clinical laboratories this month (Table 1). The chip provides an accurate genotype for two drug-metabolizing enzymes—2D6 and 2C19.
Table 1
Pharmacogenomic DNA chip: Fast facts
| Brand name: |
| AmpliChip CYP 450 Test |
| FDA-approved indication: |
| Genotyping patients |
| Manufacturer: |
| Roche Diagnostics |
| Estimated availability: |
| July 2005 |
| Recommended use: |
| Determining cytochrome P-450 2D6 and 2C19 gene variations in patients before prescribing a psychotropic metabolized through these pathways. |
| Laboratories that process AmpliChip results: |
| Labcore, Mayo Medical Laboratories, Quest Diagnostics |
Genotyping’S Role in Psychiatry
CYP 2D6 and 2C19 enzymes help metabolize many commonly prescribed psychotropics, including:
- fluoxetine, paroxetine, and venlafaxine, which are among the psychotropics primarily metabolized by the cytochrome P-450 2D6 enzyme (Table 2).
- amitriptyline and citalopram, which are among the psychotropics metabolized in part by 2C19 (Table 3).
The chip can identify patients who are genetically predisposed to abnormal metabolism of 2D6 and 2C19 substrates. This information can help psychiatrists improve response for ultrarapid metabolizers and minimize adverse effects experienced by poor metabolizers of these substrates.
For example, if the patient is an ultrarapid metabolizer of 2D6 and/or 2C19 substrates, the psychiatrist can:
- exceed the recommended dosage to reach adequate serum levels
- or choose an antidepressant not primarily metabolized by either enzyme.
For a poor metabolizer of 2D6 and/or 2C19 substrates, the psychiatrist can:
- choose an antidepressant metabolized by a different enzyme
- or prescribe 2D6 and 2C19 substrates at very low dosages.
For example, some poor metabolizers of 2D6 substrates have been successfully treated with fluoxetine, 2 to 5 mg/d.2,3 This approach can help avoid side effects and potentially save the patient money. To prevent prescription errors, make sure the pharmacist understands your rationale for lower-than-recommended dosages.
Patients who are poor metabolizers of 2C19 and extensive metabolizers of 2D6 substrates can probably tolerate citalopram and amitriptyline dosages at the low end of the therapeutic range. Watch for high serum levels of either or both drugs if both enzyme systems are inactive.
Table 2
Evidence suggests these drugs are predominantly metabolized by the 2D6 enzyme*
| Antidepressants | Antipsychotics | Stimulants |
|---|---|---|
| Desipramine | Fluphenazine | Atomoxetine |
| Fluoxetine | Perphenazine | |
| Nortriptyline | Risperidone | |
| Paroxetine | Thioridazine | |
| Venlafaxine | ||
| *Use caution when prescribing these agents to patients who are poor 2D6 metabolizers. | ||
Table 3
Evidence suggests these drugs are predominantly metabolized by the 2C19 enzyme*
| Antidepressants | |
|---|---|
| Diazepam | Citalopram |
| Clomipramine | Escitalopram |
| Imipramine | Sertraline |
| Benzodiazepines | |
| Amitriptyline | |
| *Use caution when prescribing these agents to patients who are poor 2C19 metabolizers. | |
Pharmacogenomic Chip’s Accuracy
The 2D6 gene has more than 100 variations, many of which are very rare mutations. The pharmacogenomic DNA chip can detect 27 of these variants, allowing the chip to accurately genotype most patients. By contrast, early 2D6 genotyping techniques identified only four or five variants, resulting in too many false negatives for clinical use.4
The chip also can identify the normal form of the 2C19 gene and two of its variants. Both variants produce an inactive 2C19 enzyme form that is ineffective in metabolizing 2C19 substrates.
Clinical Use
When should a psychiatrist obtain 2D6 and 2C19 genotypes?
First, understand that the pharmacogenomic chip does not predict which medications will produce a therapeutic response. Gene chips that predict response are in development but probably will not be available before 2008.
The chip, however, can identify the relatively few ultrarapid metabolizers who will not benefit from 2D6 or 2C19 substrate medications at normal dosages, as well as “poor metabolizers” of these substrates.1 The approximately 1% of whites in the United States who have ≥3 copies of the 2D6 gene metabolize 2D6 substrates very rapidly and will not respond to recommended dosages. About 10% of whites in the United States metabolize 2D6 or 2C19 substrates poorly and face increased risk of adverse reactions from these medications.
There is some evidence that the prevalence of these genetic variations differ among ethnicities. Approximately 15% of Saudi Arabians and 20% of Ethiopians are ultrarapid metabolizers of 2D6 and 2C19 substrates.5,6
The most common 2D6 poor metabolizer allele (*4) has been found in 12% to 21% of whites, whereas 23% to 32% of Asians and 13% of whites have the most common 2C19 poor metabolizer allele (*2).6-10 Prevalence of poor 2D6 and/or 2C19 metabolism among African Americans, Hispanics, and Native Americans has not been established.
Clinical Practicality
Clinicians’ unfamiliarity with genotyping and cost concerns pose potential barriers to the test’s use.
Clinician knowledge. Pharmacogenomic 2D6 and 2C19 tests will soon be offered nationwide at reference laboratories such as Quest Diagnostics, Labcore, and Mayo Medical Laboratories. The psychiatrist can call the lab for instructions, then send a blood sample and receive results by mail within 2 to 3 days.
While I believe the test’s usefulness will soon be widely understood, courses are available to help clinicians learn about genetic testing. Mayo Clinic College of Medicine (http://www.mayo. edu/cme/genomics.html) offers an annual week-long CME course in August. The American Psychiatric Association, as part of its May 2006 annual meeting, will offer a similar half-day course led by Mayo Clinic psychiatrists.
Cost. The exact cost of using the pharmacogenomic chip varies, as each laboratory sets fees for genotyping. Even so, genotyping could offer enormous cost savings by preventing failed medication trials and reducing the need for more-intensive psychiatric care. Furthermore, many insurance companies cover genotype testing.
Related resources
- Pharmacogenomic diagnostic DNA chip product information. www.rochediagnostics.com/products_services/amplichip_cyp450.html.
- Kirchheiner J, Borsen K, Dahl ML, et al. CYP2D6 and CYP2C19 genotype-based dose recommendations for antidepressants: a first step towards subpopulation-specific dosages. Acta Psychiatr Scand 2001;103(3):173-92.
Drug brand names
- Amitriptyline • Elavil
- Atomoxetine • Strattera
- Citalopram • Celexa
- Clomipramine • Anafranil
- Desipramine • Norpramin
- Diazepam • Valium
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Fluphenazine • Prolixin
- Nortriptyline • Pamelor
- Paroxetine • Paxil
- Perphenazine • Trilafon
- Risperidone • Risperdal
Disclosure
Dr. Mrazek is a consultant to Predix Pharmaceuticals.
1. Mrazek DA. New tool: genotyping makes prescribing safer, more effective. Current Psychiatry 2004;3(9):11-23.
2. Kirchheiner J, Borsen K, Dahl ML, et al. CYP2D6 and CYP2C19 genotype-based dose recommendations for antidepressants: a first step towards subpopulation-specific dosages. Acta Psychiatr Scand 2001;103:173-92.
3. Kirchheiner J, Nickchen K, Bauer M, et al. Pharmacogenetics of antidepressants and antipsychotics: the contribution of allelic variations to the phenotype of drug response. Mol Psychiatry 2004;9:442-73.
4. Chou WH, Yan FX, Robbins-Weilert DK, et al. Comparison of two CYP2D6 genotyping methods and assessment of genotype-phenotype relationships. Clin Chem 2003;49:542-51.
5. Ingelman-Sundberg M, Oscarson M, McLellan RA. Polymorphic human cytochrome P450 enzymes: an opportunity for individualized drug treatment. Trends Pharmacol Sci 1999;20:342-9.
6. Phillips KA, Veenstra DL, Oren E, et al. Potential role of pharmacogenomics in reducing adverse drug reactions: a systematic review. JAMA 2001;286:2270-9.
7. Ingelman-Sundberg M. Pharmacogenetics: an opportunity for a safer and more efficient pharmacotherapy. J Intern Med 2001;250:186-200.
8. Mizutani T. PM frequencies of major CYPs in Asians and Caucasians. Drug Metab Rev 2003;35:99-106.
9. Griese EU, Ilett KF, Kitteringham NR, et al. Allele and genotype frequencies of polymorphic cytochromes P450 2D6, 2C19, and 2E1 in aborigines from western Australia. Pharmacogenetics 2001;11:69-76.
10. Sachse C, Brockmoller J, Bauer S, Roots I. Cytochrome P450 2D6 variants in a Caucasian population: allele frequencies and phenotypic consequences. Am J Hum Genet 1997;60:284-95.
1. Mrazek DA. New tool: genotyping makes prescribing safer, more effective. Current Psychiatry 2004;3(9):11-23.
2. Kirchheiner J, Borsen K, Dahl ML, et al. CYP2D6 and CYP2C19 genotype-based dose recommendations for antidepressants: a first step towards subpopulation-specific dosages. Acta Psychiatr Scand 2001;103:173-92.
3. Kirchheiner J, Nickchen K, Bauer M, et al. Pharmacogenetics of antidepressants and antipsychotics: the contribution of allelic variations to the phenotype of drug response. Mol Psychiatry 2004;9:442-73.
4. Chou WH, Yan FX, Robbins-Weilert DK, et al. Comparison of two CYP2D6 genotyping methods and assessment of genotype-phenotype relationships. Clin Chem 2003;49:542-51.
5. Ingelman-Sundberg M, Oscarson M, McLellan RA. Polymorphic human cytochrome P450 enzymes: an opportunity for individualized drug treatment. Trends Pharmacol Sci 1999;20:342-9.
6. Phillips KA, Veenstra DL, Oren E, et al. Potential role of pharmacogenomics in reducing adverse drug reactions: a systematic review. JAMA 2001;286:2270-9.
7. Ingelman-Sundberg M. Pharmacogenetics: an opportunity for a safer and more efficient pharmacotherapy. J Intern Med 2001;250:186-200.
8. Mizutani T. PM frequencies of major CYPs in Asians and Caucasians. Drug Metab Rev 2003;35:99-106.
9. Griese EU, Ilett KF, Kitteringham NR, et al. Allele and genotype frequencies of polymorphic cytochromes P450 2D6, 2C19, and 2E1 in aborigines from western Australia. Pharmacogenetics 2001;11:69-76.
10. Sachse C, Brockmoller J, Bauer S, Roots I. Cytochrome P450 2D6 variants in a Caucasian population: allele frequencies and phenotypic consequences. Am J Hum Genet 1997;60:284-95.
Identify neuroleptic malignant syndrome with FEVER
Neuroleptic malignant syndrome (NMS) is an uncommon but by no means rare side effect of antipsychotics and other dopamine-blocking agents. This life-threatening form of drug-induced hyperthermia can be disastrous if missed, as initial treatment is based squarely on discontinuing the offending agent (see Malpractice Verdicts).
The mnemonic FEVER can help identify clinical and laboratory NMS markers in patients who exhibit mental and neurologic deterioration while taking antipsychotics or dopaminergic antagonists.
Fever. Hyperthermia is often considered NMS’ hallmark and distinguishes it from other acute neuropsychiatric disorders. However, recent research suggests hyperthermia may be a late sign of NMS and predicts a fulminant state.1 Although it is never too late to intervene, do not wait until fever develops to start treatment if you suspect an evolving NMS.
Encephalopathy. Patients may abruptly and unexpectedly become confused, obtunded, and disoriented during early or prodomal NMS stages. Such mental status changes likely result from multiple causative mechanisms in the brain that promote a clouding of consciousness.
Vital sign instability. Autonomic instability symptoms—such as tachycardia, tachypnea, and/or labile blood pressure readings—are common.
Enzyme elevation. Extreme creatinine phosphokinase (CPK) elevations because of rhabdomyolysis can indicate NMS. Serum CPKs can sometimes be as high as 2,000 times normal.1 Rhabdomyolysis, caused by muscular rigidity (see below), can help distinguish NMS from other hyperthermic toxidromes such as serotonin syndrome and anticholinergic toxicity.
Rigidity. Generalized muscle rigidity—frequently described as “lead-pipe” in the literature—is an early and easily identified clinical sign.
Although no data clearly substantiate a temporal pattern of NMS, evidence suggests that symptoms progress sequentially. Mental status changes, muscle rigidity, and autonomic instability may appear first, with hyperthermia developing later.2 Recognizing the syndrome early and promptly discontinuing the neuroleptic agent can avert a medical crisis.3
1. Mann SC, Caroff SN, Keck PE, Lazarus A. Neuroleptic malignant syndrome and related conditions (2nd ed). Washington, DC: American Psychiatric Press, 2003.
2. Velamoor VR, Norman RM, Caroff SN, et al. Progression of symptoms in neuroleptic malignant syndrome. J Nerv Ment Dis 1994;182(3):168-73.
3. Christensen RC. Recognition and management of neuroleptic malignant syndrome. Primary Psychiatry 2004;11(2):20-31.
Dr. Christensen is associate professor of psychiatry, University of Florida College Medicine, Jacksonville, and director of the university’s community psychiatry program.
Neuroleptic malignant syndrome (NMS) is an uncommon but by no means rare side effect of antipsychotics and other dopamine-blocking agents. This life-threatening form of drug-induced hyperthermia can be disastrous if missed, as initial treatment is based squarely on discontinuing the offending agent (see Malpractice Verdicts).
The mnemonic FEVER can help identify clinical and laboratory NMS markers in patients who exhibit mental and neurologic deterioration while taking antipsychotics or dopaminergic antagonists.
Fever. Hyperthermia is often considered NMS’ hallmark and distinguishes it from other acute neuropsychiatric disorders. However, recent research suggests hyperthermia may be a late sign of NMS and predicts a fulminant state.1 Although it is never too late to intervene, do not wait until fever develops to start treatment if you suspect an evolving NMS.
Encephalopathy. Patients may abruptly and unexpectedly become confused, obtunded, and disoriented during early or prodomal NMS stages. Such mental status changes likely result from multiple causative mechanisms in the brain that promote a clouding of consciousness.
Vital sign instability. Autonomic instability symptoms—such as tachycardia, tachypnea, and/or labile blood pressure readings—are common.
Enzyme elevation. Extreme creatinine phosphokinase (CPK) elevations because of rhabdomyolysis can indicate NMS. Serum CPKs can sometimes be as high as 2,000 times normal.1 Rhabdomyolysis, caused by muscular rigidity (see below), can help distinguish NMS from other hyperthermic toxidromes such as serotonin syndrome and anticholinergic toxicity.
Rigidity. Generalized muscle rigidity—frequently described as “lead-pipe” in the literature—is an early and easily identified clinical sign.
Although no data clearly substantiate a temporal pattern of NMS, evidence suggests that symptoms progress sequentially. Mental status changes, muscle rigidity, and autonomic instability may appear first, with hyperthermia developing later.2 Recognizing the syndrome early and promptly discontinuing the neuroleptic agent can avert a medical crisis.3
Neuroleptic malignant syndrome (NMS) is an uncommon but by no means rare side effect of antipsychotics and other dopamine-blocking agents. This life-threatening form of drug-induced hyperthermia can be disastrous if missed, as initial treatment is based squarely on discontinuing the offending agent (see Malpractice Verdicts).
The mnemonic FEVER can help identify clinical and laboratory NMS markers in patients who exhibit mental and neurologic deterioration while taking antipsychotics or dopaminergic antagonists.
Fever. Hyperthermia is often considered NMS’ hallmark and distinguishes it from other acute neuropsychiatric disorders. However, recent research suggests hyperthermia may be a late sign of NMS and predicts a fulminant state.1 Although it is never too late to intervene, do not wait until fever develops to start treatment if you suspect an evolving NMS.
Encephalopathy. Patients may abruptly and unexpectedly become confused, obtunded, and disoriented during early or prodomal NMS stages. Such mental status changes likely result from multiple causative mechanisms in the brain that promote a clouding of consciousness.
Vital sign instability. Autonomic instability symptoms—such as tachycardia, tachypnea, and/or labile blood pressure readings—are common.
Enzyme elevation. Extreme creatinine phosphokinase (CPK) elevations because of rhabdomyolysis can indicate NMS. Serum CPKs can sometimes be as high as 2,000 times normal.1 Rhabdomyolysis, caused by muscular rigidity (see below), can help distinguish NMS from other hyperthermic toxidromes such as serotonin syndrome and anticholinergic toxicity.
Rigidity. Generalized muscle rigidity—frequently described as “lead-pipe” in the literature—is an early and easily identified clinical sign.
Although no data clearly substantiate a temporal pattern of NMS, evidence suggests that symptoms progress sequentially. Mental status changes, muscle rigidity, and autonomic instability may appear first, with hyperthermia developing later.2 Recognizing the syndrome early and promptly discontinuing the neuroleptic agent can avert a medical crisis.3
1. Mann SC, Caroff SN, Keck PE, Lazarus A. Neuroleptic malignant syndrome and related conditions (2nd ed). Washington, DC: American Psychiatric Press, 2003.
2. Velamoor VR, Norman RM, Caroff SN, et al. Progression of symptoms in neuroleptic malignant syndrome. J Nerv Ment Dis 1994;182(3):168-73.
3. Christensen RC. Recognition and management of neuroleptic malignant syndrome. Primary Psychiatry 2004;11(2):20-31.
Dr. Christensen is associate professor of psychiatry, University of Florida College Medicine, Jacksonville, and director of the university’s community psychiatry program.
1. Mann SC, Caroff SN, Keck PE, Lazarus A. Neuroleptic malignant syndrome and related conditions (2nd ed). Washington, DC: American Psychiatric Press, 2003.
2. Velamoor VR, Norman RM, Caroff SN, et al. Progression of symptoms in neuroleptic malignant syndrome. J Nerv Ment Dis 1994;182(3):168-73.
3. Christensen RC. Recognition and management of neuroleptic malignant syndrome. Primary Psychiatry 2004;11(2):20-31.
Dr. Christensen is associate professor of psychiatry, University of Florida College Medicine, Jacksonville, and director of the university’s community psychiatry program.
Reduce appetite suppression, insomnia in ADHD treatment
Appetite suppression and insomnia—both common, dose-related side effects of psychostimulants—can jeopardize treatment adherence for patients with attention-deficit/hyperactivity disorder (ADHD). The following strategies can minimize these effects.
First, wait and see
For most patients, the optimal psychostimulant dosage produces few or no side effects. Those that occur are usually minor, transient, and disappear as patients develop tolerance within days of starting medication.
The two most commonly used stimulants—methylphenidate and amphetamine—cause similar side effects.1 No evidence suggests either is more effective or less tolerable than the other.
Fine-tune psychostimulants to the lowest dosage that produces maximum benefit and minimum side effects. If side effects persist beyond 7 to 10 days, the dosage is probably too high or the patient is taking another stimulating medication. Before you attribute insomnia or appetite suppression to psychostimulants, ask the patient if he or she is using a decongestant, caffeine, diet pills, systemic corticosteroids, systemic albuterol, or theophylline.
Countering appetite suppression
Approximately one-third of adult and pediatric ADHD patients report appetite suppression at therapeutic psychostimulant dosages, but in most patients this effect is transient or clinically insignificant. If a child taking psychostimulants is not eating or gaining weight appropriately:
- suggest that parents plan mealtimes before the patient’s next dose or give high-calorie snacks throughout the day. (This strategy, although recommended by the American Academy of Pediatrics, can be cumbersome and has limited long-term efficacy.)
- switch from amphetamine to methylphenidate or vice versa.
- add the antihistamine cyproheptadine, 4 mg, with morning and evening meals
- add mirtazapine, one-half of a 15-mg tablet at bedtime to stimulate appetite and initiate sleep.
If none of these interventions work, recommend drug holidays from ADHD medications as a last resort when impairment is lowest, such as during weekends, holidays, or summers.
Curbing insomnia
About 20% of prepubertal children and 75% to 80% of adults have difficulty falling asleep while taking ADHD medications.2 For many patients it is not the medications but the mental and physical restlessness of ADHD that disturbs sleep. Take a careful baseline sleep history before starting psychostimulants to help you determine later if they are causing insomnia.
Avoid benzodiazepines, which may promote tolerance and dependence. I discourage using any hypnotic to treat insomnia that occurs as a side effect. Also avoid antihistamines (Benedryl, trazodone) that may leave the patient sedated the next day.
Try a trial nap. After fine-tuning the psychostimulant to the lowest optimal dosage, ask the patient to test his ability to sleep while on that dose by taking an afternoon nap. Most patients discover they can sleep well, proving to both patient and doctor that ADHD medications usually help sleep initiation or are sleep-neutral. A successful nap can ease a patient’s fear that her medication will keep her awake.
Even the longest extended-release psychostimulant formulations do not last the 14 to 16 hours of a typical waking day. This no-risk trial nap reassures patients that they can take supplemental doses as prescribed to assist them through even the longest workdays, without fear of sleep disruption.
Time-release formulations smooth the abrupt kinetics and rebound activation seen with immediate-release psychostimulants. But for patients taking immediate-release formulations, reducing the day’s last dose or taking the last dose earlier can often prevent medication-associated insomnia.
If insomnia persists, try:
- melatonin, 0.5 to 1.0 mg, at bedtime, 1 hour before bedtime, at sunset, or 6 hours before anticipated bedtime. I try to mimic the natural release of melatonin triggered by sunset, but no definitive data prove the most effective dosing time.
- alpha agonists such as clonidine, 0.1 to 0.2 mg at bedtime, or guanfacine, 1 to 2 mg at bedtime. These agents have proven efficacy for treating hyperactivity and sleep disturbances without causing tolerance but may be associated with nightmares in some children.3
- mirtazapine, one-half of a 15-mg tablet at bedtime.
1. Greenhill LL, Abikoff HB, Arnold LE, et al. Medication treatment strategies in the MTA study: relevance to clinicians and researchers. J Am Acad Child Adolesc Psychiatry 1996;35(10):1304-13.
2. Corkum P, Tannock R, Moldofsky H. Sleep disturbances in children with attention deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry 1998;37:637-46.
3. Biederman J, Spencer T, Wilens T. Evidence-based pharmacotherapy for attention-deficit hyperactivity disorder. Int J Neuropsycho-pharmacol 2004;7(1):77-97.
Dr. Dodson is a board-certified psychiatrist specializing in adult ADHD. He is director of the Attention Disorders Treatment Center, Denver, CO.
Appetite suppression and insomnia—both common, dose-related side effects of psychostimulants—can jeopardize treatment adherence for patients with attention-deficit/hyperactivity disorder (ADHD). The following strategies can minimize these effects.
First, wait and see
For most patients, the optimal psychostimulant dosage produces few or no side effects. Those that occur are usually minor, transient, and disappear as patients develop tolerance within days of starting medication.
The two most commonly used stimulants—methylphenidate and amphetamine—cause similar side effects.1 No evidence suggests either is more effective or less tolerable than the other.
Fine-tune psychostimulants to the lowest dosage that produces maximum benefit and minimum side effects. If side effects persist beyond 7 to 10 days, the dosage is probably too high or the patient is taking another stimulating medication. Before you attribute insomnia or appetite suppression to psychostimulants, ask the patient if he or she is using a decongestant, caffeine, diet pills, systemic corticosteroids, systemic albuterol, or theophylline.
Countering appetite suppression
Approximately one-third of adult and pediatric ADHD patients report appetite suppression at therapeutic psychostimulant dosages, but in most patients this effect is transient or clinically insignificant. If a child taking psychostimulants is not eating or gaining weight appropriately:
- suggest that parents plan mealtimes before the patient’s next dose or give high-calorie snacks throughout the day. (This strategy, although recommended by the American Academy of Pediatrics, can be cumbersome and has limited long-term efficacy.)
- switch from amphetamine to methylphenidate or vice versa.
- add the antihistamine cyproheptadine, 4 mg, with morning and evening meals
- add mirtazapine, one-half of a 15-mg tablet at bedtime to stimulate appetite and initiate sleep.
If none of these interventions work, recommend drug holidays from ADHD medications as a last resort when impairment is lowest, such as during weekends, holidays, or summers.
Curbing insomnia
About 20% of prepubertal children and 75% to 80% of adults have difficulty falling asleep while taking ADHD medications.2 For many patients it is not the medications but the mental and physical restlessness of ADHD that disturbs sleep. Take a careful baseline sleep history before starting psychostimulants to help you determine later if they are causing insomnia.
Avoid benzodiazepines, which may promote tolerance and dependence. I discourage using any hypnotic to treat insomnia that occurs as a side effect. Also avoid antihistamines (Benedryl, trazodone) that may leave the patient sedated the next day.
Try a trial nap. After fine-tuning the psychostimulant to the lowest optimal dosage, ask the patient to test his ability to sleep while on that dose by taking an afternoon nap. Most patients discover they can sleep well, proving to both patient and doctor that ADHD medications usually help sleep initiation or are sleep-neutral. A successful nap can ease a patient’s fear that her medication will keep her awake.
Even the longest extended-release psychostimulant formulations do not last the 14 to 16 hours of a typical waking day. This no-risk trial nap reassures patients that they can take supplemental doses as prescribed to assist them through even the longest workdays, without fear of sleep disruption.
Time-release formulations smooth the abrupt kinetics and rebound activation seen with immediate-release psychostimulants. But for patients taking immediate-release formulations, reducing the day’s last dose or taking the last dose earlier can often prevent medication-associated insomnia.
If insomnia persists, try:
- melatonin, 0.5 to 1.0 mg, at bedtime, 1 hour before bedtime, at sunset, or 6 hours before anticipated bedtime. I try to mimic the natural release of melatonin triggered by sunset, but no definitive data prove the most effective dosing time.
- alpha agonists such as clonidine, 0.1 to 0.2 mg at bedtime, or guanfacine, 1 to 2 mg at bedtime. These agents have proven efficacy for treating hyperactivity and sleep disturbances without causing tolerance but may be associated with nightmares in some children.3
- mirtazapine, one-half of a 15-mg tablet at bedtime.
Appetite suppression and insomnia—both common, dose-related side effects of psychostimulants—can jeopardize treatment adherence for patients with attention-deficit/hyperactivity disorder (ADHD). The following strategies can minimize these effects.
First, wait and see
For most patients, the optimal psychostimulant dosage produces few or no side effects. Those that occur are usually minor, transient, and disappear as patients develop tolerance within days of starting medication.
The two most commonly used stimulants—methylphenidate and amphetamine—cause similar side effects.1 No evidence suggests either is more effective or less tolerable than the other.
Fine-tune psychostimulants to the lowest dosage that produces maximum benefit and minimum side effects. If side effects persist beyond 7 to 10 days, the dosage is probably too high or the patient is taking another stimulating medication. Before you attribute insomnia or appetite suppression to psychostimulants, ask the patient if he or she is using a decongestant, caffeine, diet pills, systemic corticosteroids, systemic albuterol, or theophylline.
Countering appetite suppression
Approximately one-third of adult and pediatric ADHD patients report appetite suppression at therapeutic psychostimulant dosages, but in most patients this effect is transient or clinically insignificant. If a child taking psychostimulants is not eating or gaining weight appropriately:
- suggest that parents plan mealtimes before the patient’s next dose or give high-calorie snacks throughout the day. (This strategy, although recommended by the American Academy of Pediatrics, can be cumbersome and has limited long-term efficacy.)
- switch from amphetamine to methylphenidate or vice versa.
- add the antihistamine cyproheptadine, 4 mg, with morning and evening meals
- add mirtazapine, one-half of a 15-mg tablet at bedtime to stimulate appetite and initiate sleep.
If none of these interventions work, recommend drug holidays from ADHD medications as a last resort when impairment is lowest, such as during weekends, holidays, or summers.
Curbing insomnia
About 20% of prepubertal children and 75% to 80% of adults have difficulty falling asleep while taking ADHD medications.2 For many patients it is not the medications but the mental and physical restlessness of ADHD that disturbs sleep. Take a careful baseline sleep history before starting psychostimulants to help you determine later if they are causing insomnia.
Avoid benzodiazepines, which may promote tolerance and dependence. I discourage using any hypnotic to treat insomnia that occurs as a side effect. Also avoid antihistamines (Benedryl, trazodone) that may leave the patient sedated the next day.
Try a trial nap. After fine-tuning the psychostimulant to the lowest optimal dosage, ask the patient to test his ability to sleep while on that dose by taking an afternoon nap. Most patients discover they can sleep well, proving to both patient and doctor that ADHD medications usually help sleep initiation or are sleep-neutral. A successful nap can ease a patient’s fear that her medication will keep her awake.
Even the longest extended-release psychostimulant formulations do not last the 14 to 16 hours of a typical waking day. This no-risk trial nap reassures patients that they can take supplemental doses as prescribed to assist them through even the longest workdays, without fear of sleep disruption.
Time-release formulations smooth the abrupt kinetics and rebound activation seen with immediate-release psychostimulants. But for patients taking immediate-release formulations, reducing the day’s last dose or taking the last dose earlier can often prevent medication-associated insomnia.
If insomnia persists, try:
- melatonin, 0.5 to 1.0 mg, at bedtime, 1 hour before bedtime, at sunset, or 6 hours before anticipated bedtime. I try to mimic the natural release of melatonin triggered by sunset, but no definitive data prove the most effective dosing time.
- alpha agonists such as clonidine, 0.1 to 0.2 mg at bedtime, or guanfacine, 1 to 2 mg at bedtime. These agents have proven efficacy for treating hyperactivity and sleep disturbances without causing tolerance but may be associated with nightmares in some children.3
- mirtazapine, one-half of a 15-mg tablet at bedtime.
1. Greenhill LL, Abikoff HB, Arnold LE, et al. Medication treatment strategies in the MTA study: relevance to clinicians and researchers. J Am Acad Child Adolesc Psychiatry 1996;35(10):1304-13.
2. Corkum P, Tannock R, Moldofsky H. Sleep disturbances in children with attention deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry 1998;37:637-46.
3. Biederman J, Spencer T, Wilens T. Evidence-based pharmacotherapy for attention-deficit hyperactivity disorder. Int J Neuropsycho-pharmacol 2004;7(1):77-97.
Dr. Dodson is a board-certified psychiatrist specializing in adult ADHD. He is director of the Attention Disorders Treatment Center, Denver, CO.
1. Greenhill LL, Abikoff HB, Arnold LE, et al. Medication treatment strategies in the MTA study: relevance to clinicians and researchers. J Am Acad Child Adolesc Psychiatry 1996;35(10):1304-13.
2. Corkum P, Tannock R, Moldofsky H. Sleep disturbances in children with attention deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry 1998;37:637-46.
3. Biederman J, Spencer T, Wilens T. Evidence-based pharmacotherapy for attention-deficit hyperactivity disorder. Int J Neuropsycho-pharmacol 2004;7(1):77-97.
Dr. Dodson is a board-certified psychiatrist specializing in adult ADHD. He is director of the Attention Disorders Treatment Center, Denver, CO.