User login
Welcome to Current Psychiatry, a leading source of information, online and in print, for practitioners of psychiatry and its related subspecialties, including addiction psychiatry, child and adolescent psychiatry, and geriatric psychiatry. This Web site contains evidence-based reviews of the prevention, diagnosis, and treatment of mental illness and psychological disorders; case reports; updates on psychopharmacology; news about the specialty of psychiatry; pearls for practice; and other topics of interest and use to this audience.
Dear Drupal User: You're seeing this because you're logged in to Drupal, and not redirected to MDedge.com/psychiatry.
Depression
adolescent depression
adolescent major depressive disorder
adolescent schizophrenia
adolescent with major depressive disorder
animals
autism
baby
brexpiprazole
child
child bipolar
child depression
child schizophrenia
children with bipolar disorder
children with depression
children with major depressive disorder
compulsive behaviors
cure
elderly bipolar
elderly depression
elderly major depressive disorder
elderly schizophrenia
elderly with dementia
first break
first episode
gambling
gaming
geriatric depression
geriatric major depressive disorder
geriatric schizophrenia
infant
kid
major depressive disorder
major depressive disorder in adolescents
major depressive disorder in children
parenting
pediatric
pediatric bipolar
pediatric depression
pediatric major depressive disorder
pediatric schizophrenia
pregnancy
pregnant
rexulti
skin care
teen
wine
section[contains(@class, 'nav-hidden')]
footer[@id='footer']
div[contains(@class, 'pane-pub-article-current-psychiatry')]
div[contains(@class, 'pane-pub-home-current-psychiatry')]
div[contains(@class, 'pane-pub-topic-current-psychiatry')]
div[contains(@class, 'panel-panel-inner')]
div[contains(@class, 'pane-node-field-article-topics')]
section[contains(@class, 'footer-nav-section-wrapper')]
Duloxetine: Dual-action antidepressant
Depression’s remission rates remain low,1 and its common somatic symptoms (aches and pains, headaches, backaches) often complicate diagnosis and treatment.2
Duloxetine, recently FDA-approved for treating major depression Table 1, has shown efficacy against depression’s emotional and somatic symptoms in clinical trials.
HOW IT WORKS
Duloxetine inhibits both serotonin and norepinephrine reuptake. Researchers suggest that antidepressants exhibiting this dual action may be more effective and act faster than single-action selective serotonin reuptake inhibitors.3,4 Newer dual-action antidepressants also are more tolerable than dual-action tricyclic antidepressants.
Table 1
Duloxetine: Fast facts
| Drug brand name: Cymbalta |
| Class Serotonin and norepinephrine reuptake inhibitor |
| FDA-approved indication: Treatment of major depressive episodes |
| Approval date: August 3 2004 |
| Manufacturer: Eli Lilly and Co. |
| Dosing form: 20 mg, 30 mg, 60 mg capsules |
| Recommended dosage: 40 to 60 mg/d |
| Maximum dosage(studied in major depression): 120 mg/d |
Researchers have seen synergism between serotonergic and noradrenergic pain modulation at the spinal cord,4,5 suggesting that dual-action antidepressants may ameliorate major depression’s somatic symptoms.
Table 2
Plasma levels of these agents may affect—or be affected by— duloxetine coadministration
| CYP 2D6 substrates |
| Amitriptyline |
| Beta blockers Propranolol, metoprolol, timolol |
| Desipramine |
| Fluoxetine |
| Fluvoxamine |
| Haloperidol |
| Nortriptyline |
| Risperidone |
| Thioridazine |
| Type 1C antiarrhythmics Propafenone, flecainide |
| Venlafaxine |
| CYP 2D6 inhibitors |
| Cimetidine |
| Fluoxetine |
| Haloperidol |
| Paroxetine |
| Quinidine |
| CYP 1A2 inhibitors |
| Cimetidine |
| Ciprofloxacin |
| Enoxacin |
| Source: Reference 7 |
PHARMACOKINETICS
Despite its 12-hour plasma half-life, duloxetine has shown efficacy in clinical trials when given once daily. Mean plasma clearance is approximately 101 L/hr, with a mean volume of distribution of about 1640 L, meaning that duloxetine is distributed throughout the body.
The agent is more than 90% protein bound; thus, giving duloxetine concomitantly with another highly protein-bound agent could increase the side-effect risk of either drug.
Food does not alter duloxetine’s absorption but delays maximum concentration by about 4 hours Duloxetine may be taken before or after meals, though taking it after meals could reduce the risk of nausea—a common early side effect.
Duloxetine is metabolized by the 2D6 and 1A2 isoenzymes of the cytochrome P-450 system. It inhibits the CYP 2D6 isoenzyme but to a lesser extent than fluoxetine does.6 Co-administering duloxetine with a CYP 2D6 substrate or inhibitor or a CYP 1A2 inhibitor Table 27 could elevate plasma levels of duloxetine or the other agent, possibly increasing adverse effects.
EFFICACY
In an 8-week, placebo-controlled trial, Goldstein et al8 compared fluoxetine, 20 mg/d, and duloxetine, 40 mg/d titrated to 120 mg/d over 3 weeks, in 173 patients with major depressive disorder. Participants’ scores at baseline were 15 on the 17-item Hamilton Rating Scale for Depression (HAM-D-17) and 4 on the Clinical Global Improvement-Severity scale. Estimated probability of remission was 56% with duloxetine, 30% with fluoxetine, and 32% with placebo, with remission defined as achieving a HAM-D-17 score 7.
In two prospective, double-blind, placebocontrolled trials of 512 patients with major depression,9,10 duloxetine, 60 mg/d, reduced body, back, and shoulder pain based on Visual Analog Scale (VAS) scores; pre- and posttreatment VAS scores were not listed in the published studies. Estimated probability of remission in these two studies was 44% and 43% among patients taking duloxetine vs 16% and 28% in the placebo groups. Remission again was defined as HAM-D-17 score 7.
TOLERABILITY
In the two double-blind studies just mentioned,9,10 Detke et al reported adverse event-related drop out rates of 12.5% and 13.8% for duloxetine, 60 mg/d, vs 4.3% and 2.3% for placebo. Nausea, insomnia, headaches, somnolence, dry mouth, and sweating were most frequently reported.
Dizziness. Mild dizziness was reported in 11.3% of patients who abruptly stopped duloxetine after 9 weeks.9
Headaches. In one comparator trial,8 fewer headaches (20%) were reported among patients taking duloxetine, 40 to 120 mg/d, vs those taking fluoxetine, 20 mg/d (33.3%), or placebo (31.4%).
Hypertension. Detke et al10 found no statistical separation in systolic and diastolic blood pressures between the duloxetine (60 mg/d) and placebo groups. Likewise, Goldstein et al8 found a similar incidence of hypertension among patients taking duloxetine, 40 to 120 mg/d, or placebo. In clinical trials,11 duloxetine increased blood pressure by a mean of 2.0 mm Hg (systolic) and 0.5 mm Hg (diastolic).
As with several other noradrenergic medications, FDA recommends that clinicians check blood pressures before starting duloxetine therapy and periodically thereafter.
Duloxetine has not been studied in persons with poorly controlled hypertension.
Nausea. Mild to moderate nausea was the most common adverse event in one study;9 the effect dissipated after a median of 7 days. One patient reported severe nausea, and 1 patient out of 123 stopped the medication because of nausea.
Sexual dysfunction. Using the Arizona Sexual Experiences Scale (ASEX), Goldstein et al8 prospectively assessed sexual function in 70 men and women taking duloxetine or placebo. No statistical difference was seen between the two groups from baseline to endpoint.
In another study,12 duloxetine showed worsening only in ASEX item 4 (“How easily can you reach an orgasm?”), indicating some adverse sexual effects in men. No such differences were found in women. Duloxetine’s effect on sexual function needs to be studied further.
Duloxetine is an FDA Use-in-Pregnancy category C medication, meaning that risk to the fetus has not been ruled out. The agent is contraindicated in patients taking monoamine oxidase inhibitors and in those with narrow-angle glaucoma. FDA recommends not using duloxetine in patients with hepatic insufficiency, endstage renal disease, and substantial alcohol use.
DOSING STRATEGIES
Duloxetine, 40 to 120 mg/d, appears to be safe and effective for most adults.8-10 FDA recommends starting at 40 mg/d (20 mg bid) to 60 mg/d (once-daily or 30 mg bid) with no regard to meals. Dosages >60 mg/d have not shown additional benefit. Age and tolerability should drive initial dosing and titration. Side-effect incidence has not been directly compared at 60, 90, and 120 mg/d.
CLINICAL IMPLICATIONS
In clinical trials, duloxetine has shown a high estimated probability of remission in major depression and has shown efficacy against depression’s physical and emotional symptoms. Based on efficacy and safety data, duloxetine appears to be a first-line treatment option for major depression.
Related resources
- Gray GE. Concise guide to evidence-based psychiatry. Washington, DC: American Psychiatric Publishing, 2004.
- Schatzberg AF, Nemeroff CB (eds). Textbook of psychopharmacology (3rd ed). Washington, DC: American Psychiatric Publishing, 2004.
Drug brand names
- Amitriptyline • Elavil
- Cimetidine • Tagamet
- Ciprofloxacin • Cipro
- Desipramine • Norpramin
- Duloxetine • Cymbalta
- Enoxacin • Penetrex
- Fluoxetine • Prozac
- Flecainide • Tambocor
- Fluvoxamine • Luvox
- Haloperidol • Haldol
- Metoprolol • Toprol
- Nortriptyline • Pamelor
- Paroxetine • Paxil
- Propafenone • Rythmol
- Propranolol • Inderal
- Risperidone • Risperdal
- Thioridazine • Mellaril
- Timolol • Blocadren, others
- Venlafaxine • Effexor
Disclosure
Dr. Rakesh Jain receives research grants from Eli Lilly and Co., Forest Pharmaceuticals, GlaxoSmithKline, Merck and Co., Organon, Pfizer Inc., and Sepracor. He is a consultant to and speaker for Eli Lilly and Co., and is a speaker for GlaxoSmithKline, Pfizer Inc., and Wyeth Pharmaceuticals.
Dr. Shailesh Jain reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of compefting products.
1. Thase ME, Entsuah AR, Rudolph RL. Remission rates during treatment with venlafaxine or serotonin reuptake inhibitors. Br J Psychiatry 2001;178:234-41.
2. Fava M. The role of the serotonergic and noradrenergic neurotransmitter systems in the treatment of psychological and physical symptoms of depression. J Clin Psychiatry 2003;64(suppl 13):26-9.
3. Tran PV, Bymaster FP, McNamara RK, Potter WZ. Dual monoamine modulation for improved treatment of major depressive disorder. J Clin Psychopharmacol 2003;23:78-86.
4. Ansari A. The efficacy of newer antidepressants in the treatment of chronic pain: a review of current literature. Harv Rev Psychiatry 2000;7:257-77.
5. Willis WD, Westlund KN. Neuroanatomy of the pain system and the pathways that modulate pain. J Clin Neurophysiol 1997;14:2-31.
6. Skinner MH, Kuan HY, Pan A, et al. Duloxetine is both an inhibitor and a substrate of cytochrome P4502D6 in healthy volunteers. Clin Pharmacol Ther 2003;73:170-7.
7. Sadock BJ, Sadock VA. Kaplan and Sadock's pocket handbook of psychotropic drug treatment (3rd ed). Baltimore, MD: Lippincott Williams and Wilkins, 2001.
8. Goldstein DJ, Mallinckrodt C, Lu Y, Demitrack M. Duloxetine in the treatment of major depression disorder: a double-blind clinical trial. J Clin Psychiatry 2002;63:225-31.
9. Detke MJ, Lu Y, Goldstein DJ, et al. Duloxetine 60 mg once-daily for major depressive disorder: a randomized double-blind placebocontrolled trial. J Clin Psychiatry 2002;63:308-15.
10. Detke MJ, Lu Y, Goldstein DJ, et al. Duloxetine 60 mg once daily dosing versus placebo in the treatment of major depression. J Psychiatr Res 2002;36:383-90.
11. Cymbalta prescribing information Eli Lilly and Co 2004.
12. Goldstein DJ, Lu Y, Detke MJ, et al. Duloxetine in the treatment of depression: a double-blind placebo-controlled comparison with paroxetine. J Clin Psychopharmacol 2004;24:389-99.
Depression’s remission rates remain low,1 and its common somatic symptoms (aches and pains, headaches, backaches) often complicate diagnosis and treatment.2
Duloxetine, recently FDA-approved for treating major depression Table 1, has shown efficacy against depression’s emotional and somatic symptoms in clinical trials.
HOW IT WORKS
Duloxetine inhibits both serotonin and norepinephrine reuptake. Researchers suggest that antidepressants exhibiting this dual action may be more effective and act faster than single-action selective serotonin reuptake inhibitors.3,4 Newer dual-action antidepressants also are more tolerable than dual-action tricyclic antidepressants.
Table 1
Duloxetine: Fast facts
| Drug brand name: Cymbalta |
| Class Serotonin and norepinephrine reuptake inhibitor |
| FDA-approved indication: Treatment of major depressive episodes |
| Approval date: August 3 2004 |
| Manufacturer: Eli Lilly and Co. |
| Dosing form: 20 mg, 30 mg, 60 mg capsules |
| Recommended dosage: 40 to 60 mg/d |
| Maximum dosage(studied in major depression): 120 mg/d |
Researchers have seen synergism between serotonergic and noradrenergic pain modulation at the spinal cord,4,5 suggesting that dual-action antidepressants may ameliorate major depression’s somatic symptoms.
Table 2
Plasma levels of these agents may affect—or be affected by— duloxetine coadministration
| CYP 2D6 substrates |
| Amitriptyline |
| Beta blockers Propranolol, metoprolol, timolol |
| Desipramine |
| Fluoxetine |
| Fluvoxamine |
| Haloperidol |
| Nortriptyline |
| Risperidone |
| Thioridazine |
| Type 1C antiarrhythmics Propafenone, flecainide |
| Venlafaxine |
| CYP 2D6 inhibitors |
| Cimetidine |
| Fluoxetine |
| Haloperidol |
| Paroxetine |
| Quinidine |
| CYP 1A2 inhibitors |
| Cimetidine |
| Ciprofloxacin |
| Enoxacin |
| Source: Reference 7 |
PHARMACOKINETICS
Despite its 12-hour plasma half-life, duloxetine has shown efficacy in clinical trials when given once daily. Mean plasma clearance is approximately 101 L/hr, with a mean volume of distribution of about 1640 L, meaning that duloxetine is distributed throughout the body.
The agent is more than 90% protein bound; thus, giving duloxetine concomitantly with another highly protein-bound agent could increase the side-effect risk of either drug.
Food does not alter duloxetine’s absorption but delays maximum concentration by about 4 hours Duloxetine may be taken before or after meals, though taking it after meals could reduce the risk of nausea—a common early side effect.
Duloxetine is metabolized by the 2D6 and 1A2 isoenzymes of the cytochrome P-450 system. It inhibits the CYP 2D6 isoenzyme but to a lesser extent than fluoxetine does.6 Co-administering duloxetine with a CYP 2D6 substrate or inhibitor or a CYP 1A2 inhibitor Table 27 could elevate plasma levels of duloxetine or the other agent, possibly increasing adverse effects.
EFFICACY
In an 8-week, placebo-controlled trial, Goldstein et al8 compared fluoxetine, 20 mg/d, and duloxetine, 40 mg/d titrated to 120 mg/d over 3 weeks, in 173 patients with major depressive disorder. Participants’ scores at baseline were 15 on the 17-item Hamilton Rating Scale for Depression (HAM-D-17) and 4 on the Clinical Global Improvement-Severity scale. Estimated probability of remission was 56% with duloxetine, 30% with fluoxetine, and 32% with placebo, with remission defined as achieving a HAM-D-17 score 7.
In two prospective, double-blind, placebocontrolled trials of 512 patients with major depression,9,10 duloxetine, 60 mg/d, reduced body, back, and shoulder pain based on Visual Analog Scale (VAS) scores; pre- and posttreatment VAS scores were not listed in the published studies. Estimated probability of remission in these two studies was 44% and 43% among patients taking duloxetine vs 16% and 28% in the placebo groups. Remission again was defined as HAM-D-17 score 7.
TOLERABILITY
In the two double-blind studies just mentioned,9,10 Detke et al reported adverse event-related drop out rates of 12.5% and 13.8% for duloxetine, 60 mg/d, vs 4.3% and 2.3% for placebo. Nausea, insomnia, headaches, somnolence, dry mouth, and sweating were most frequently reported.
Dizziness. Mild dizziness was reported in 11.3% of patients who abruptly stopped duloxetine after 9 weeks.9
Headaches. In one comparator trial,8 fewer headaches (20%) were reported among patients taking duloxetine, 40 to 120 mg/d, vs those taking fluoxetine, 20 mg/d (33.3%), or placebo (31.4%).
Hypertension. Detke et al10 found no statistical separation in systolic and diastolic blood pressures between the duloxetine (60 mg/d) and placebo groups. Likewise, Goldstein et al8 found a similar incidence of hypertension among patients taking duloxetine, 40 to 120 mg/d, or placebo. In clinical trials,11 duloxetine increased blood pressure by a mean of 2.0 mm Hg (systolic) and 0.5 mm Hg (diastolic).
As with several other noradrenergic medications, FDA recommends that clinicians check blood pressures before starting duloxetine therapy and periodically thereafter.
Duloxetine has not been studied in persons with poorly controlled hypertension.
Nausea. Mild to moderate nausea was the most common adverse event in one study;9 the effect dissipated after a median of 7 days. One patient reported severe nausea, and 1 patient out of 123 stopped the medication because of nausea.
Sexual dysfunction. Using the Arizona Sexual Experiences Scale (ASEX), Goldstein et al8 prospectively assessed sexual function in 70 men and women taking duloxetine or placebo. No statistical difference was seen between the two groups from baseline to endpoint.
In another study,12 duloxetine showed worsening only in ASEX item 4 (“How easily can you reach an orgasm?”), indicating some adverse sexual effects in men. No such differences were found in women. Duloxetine’s effect on sexual function needs to be studied further.
Duloxetine is an FDA Use-in-Pregnancy category C medication, meaning that risk to the fetus has not been ruled out. The agent is contraindicated in patients taking monoamine oxidase inhibitors and in those with narrow-angle glaucoma. FDA recommends not using duloxetine in patients with hepatic insufficiency, endstage renal disease, and substantial alcohol use.
DOSING STRATEGIES
Duloxetine, 40 to 120 mg/d, appears to be safe and effective for most adults.8-10 FDA recommends starting at 40 mg/d (20 mg bid) to 60 mg/d (once-daily or 30 mg bid) with no regard to meals. Dosages >60 mg/d have not shown additional benefit. Age and tolerability should drive initial dosing and titration. Side-effect incidence has not been directly compared at 60, 90, and 120 mg/d.
CLINICAL IMPLICATIONS
In clinical trials, duloxetine has shown a high estimated probability of remission in major depression and has shown efficacy against depression’s physical and emotional symptoms. Based on efficacy and safety data, duloxetine appears to be a first-line treatment option for major depression.
Related resources
- Gray GE. Concise guide to evidence-based psychiatry. Washington, DC: American Psychiatric Publishing, 2004.
- Schatzberg AF, Nemeroff CB (eds). Textbook of psychopharmacology (3rd ed). Washington, DC: American Psychiatric Publishing, 2004.
Drug brand names
- Amitriptyline • Elavil
- Cimetidine • Tagamet
- Ciprofloxacin • Cipro
- Desipramine • Norpramin
- Duloxetine • Cymbalta
- Enoxacin • Penetrex
- Fluoxetine • Prozac
- Flecainide • Tambocor
- Fluvoxamine • Luvox
- Haloperidol • Haldol
- Metoprolol • Toprol
- Nortriptyline • Pamelor
- Paroxetine • Paxil
- Propafenone • Rythmol
- Propranolol • Inderal
- Risperidone • Risperdal
- Thioridazine • Mellaril
- Timolol • Blocadren, others
- Venlafaxine • Effexor
Disclosure
Dr. Rakesh Jain receives research grants from Eli Lilly and Co., Forest Pharmaceuticals, GlaxoSmithKline, Merck and Co., Organon, Pfizer Inc., and Sepracor. He is a consultant to and speaker for Eli Lilly and Co., and is a speaker for GlaxoSmithKline, Pfizer Inc., and Wyeth Pharmaceuticals.
Dr. Shailesh Jain reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of compefting products.
Depression’s remission rates remain low,1 and its common somatic symptoms (aches and pains, headaches, backaches) often complicate diagnosis and treatment.2
Duloxetine, recently FDA-approved for treating major depression Table 1, has shown efficacy against depression’s emotional and somatic symptoms in clinical trials.
HOW IT WORKS
Duloxetine inhibits both serotonin and norepinephrine reuptake. Researchers suggest that antidepressants exhibiting this dual action may be more effective and act faster than single-action selective serotonin reuptake inhibitors.3,4 Newer dual-action antidepressants also are more tolerable than dual-action tricyclic antidepressants.
Table 1
Duloxetine: Fast facts
| Drug brand name: Cymbalta |
| Class Serotonin and norepinephrine reuptake inhibitor |
| FDA-approved indication: Treatment of major depressive episodes |
| Approval date: August 3 2004 |
| Manufacturer: Eli Lilly and Co. |
| Dosing form: 20 mg, 30 mg, 60 mg capsules |
| Recommended dosage: 40 to 60 mg/d |
| Maximum dosage(studied in major depression): 120 mg/d |
Researchers have seen synergism between serotonergic and noradrenergic pain modulation at the spinal cord,4,5 suggesting that dual-action antidepressants may ameliorate major depression’s somatic symptoms.
Table 2
Plasma levels of these agents may affect—or be affected by— duloxetine coadministration
| CYP 2D6 substrates |
| Amitriptyline |
| Beta blockers Propranolol, metoprolol, timolol |
| Desipramine |
| Fluoxetine |
| Fluvoxamine |
| Haloperidol |
| Nortriptyline |
| Risperidone |
| Thioridazine |
| Type 1C antiarrhythmics Propafenone, flecainide |
| Venlafaxine |
| CYP 2D6 inhibitors |
| Cimetidine |
| Fluoxetine |
| Haloperidol |
| Paroxetine |
| Quinidine |
| CYP 1A2 inhibitors |
| Cimetidine |
| Ciprofloxacin |
| Enoxacin |
| Source: Reference 7 |
PHARMACOKINETICS
Despite its 12-hour plasma half-life, duloxetine has shown efficacy in clinical trials when given once daily. Mean plasma clearance is approximately 101 L/hr, with a mean volume of distribution of about 1640 L, meaning that duloxetine is distributed throughout the body.
The agent is more than 90% protein bound; thus, giving duloxetine concomitantly with another highly protein-bound agent could increase the side-effect risk of either drug.
Food does not alter duloxetine’s absorption but delays maximum concentration by about 4 hours Duloxetine may be taken before or after meals, though taking it after meals could reduce the risk of nausea—a common early side effect.
Duloxetine is metabolized by the 2D6 and 1A2 isoenzymes of the cytochrome P-450 system. It inhibits the CYP 2D6 isoenzyme but to a lesser extent than fluoxetine does.6 Co-administering duloxetine with a CYP 2D6 substrate or inhibitor or a CYP 1A2 inhibitor Table 27 could elevate plasma levels of duloxetine or the other agent, possibly increasing adverse effects.
EFFICACY
In an 8-week, placebo-controlled trial, Goldstein et al8 compared fluoxetine, 20 mg/d, and duloxetine, 40 mg/d titrated to 120 mg/d over 3 weeks, in 173 patients with major depressive disorder. Participants’ scores at baseline were 15 on the 17-item Hamilton Rating Scale for Depression (HAM-D-17) and 4 on the Clinical Global Improvement-Severity scale. Estimated probability of remission was 56% with duloxetine, 30% with fluoxetine, and 32% with placebo, with remission defined as achieving a HAM-D-17 score 7.
In two prospective, double-blind, placebocontrolled trials of 512 patients with major depression,9,10 duloxetine, 60 mg/d, reduced body, back, and shoulder pain based on Visual Analog Scale (VAS) scores; pre- and posttreatment VAS scores were not listed in the published studies. Estimated probability of remission in these two studies was 44% and 43% among patients taking duloxetine vs 16% and 28% in the placebo groups. Remission again was defined as HAM-D-17 score 7.
TOLERABILITY
In the two double-blind studies just mentioned,9,10 Detke et al reported adverse event-related drop out rates of 12.5% and 13.8% for duloxetine, 60 mg/d, vs 4.3% and 2.3% for placebo. Nausea, insomnia, headaches, somnolence, dry mouth, and sweating were most frequently reported.
Dizziness. Mild dizziness was reported in 11.3% of patients who abruptly stopped duloxetine after 9 weeks.9
Headaches. In one comparator trial,8 fewer headaches (20%) were reported among patients taking duloxetine, 40 to 120 mg/d, vs those taking fluoxetine, 20 mg/d (33.3%), or placebo (31.4%).
Hypertension. Detke et al10 found no statistical separation in systolic and diastolic blood pressures between the duloxetine (60 mg/d) and placebo groups. Likewise, Goldstein et al8 found a similar incidence of hypertension among patients taking duloxetine, 40 to 120 mg/d, or placebo. In clinical trials,11 duloxetine increased blood pressure by a mean of 2.0 mm Hg (systolic) and 0.5 mm Hg (diastolic).
As with several other noradrenergic medications, FDA recommends that clinicians check blood pressures before starting duloxetine therapy and periodically thereafter.
Duloxetine has not been studied in persons with poorly controlled hypertension.
Nausea. Mild to moderate nausea was the most common adverse event in one study;9 the effect dissipated after a median of 7 days. One patient reported severe nausea, and 1 patient out of 123 stopped the medication because of nausea.
Sexual dysfunction. Using the Arizona Sexual Experiences Scale (ASEX), Goldstein et al8 prospectively assessed sexual function in 70 men and women taking duloxetine or placebo. No statistical difference was seen between the two groups from baseline to endpoint.
In another study,12 duloxetine showed worsening only in ASEX item 4 (“How easily can you reach an orgasm?”), indicating some adverse sexual effects in men. No such differences were found in women. Duloxetine’s effect on sexual function needs to be studied further.
Duloxetine is an FDA Use-in-Pregnancy category C medication, meaning that risk to the fetus has not been ruled out. The agent is contraindicated in patients taking monoamine oxidase inhibitors and in those with narrow-angle glaucoma. FDA recommends not using duloxetine in patients with hepatic insufficiency, endstage renal disease, and substantial alcohol use.
DOSING STRATEGIES
Duloxetine, 40 to 120 mg/d, appears to be safe and effective for most adults.8-10 FDA recommends starting at 40 mg/d (20 mg bid) to 60 mg/d (once-daily or 30 mg bid) with no regard to meals. Dosages >60 mg/d have not shown additional benefit. Age and tolerability should drive initial dosing and titration. Side-effect incidence has not been directly compared at 60, 90, and 120 mg/d.
CLINICAL IMPLICATIONS
In clinical trials, duloxetine has shown a high estimated probability of remission in major depression and has shown efficacy against depression’s physical and emotional symptoms. Based on efficacy and safety data, duloxetine appears to be a first-line treatment option for major depression.
Related resources
- Gray GE. Concise guide to evidence-based psychiatry. Washington, DC: American Psychiatric Publishing, 2004.
- Schatzberg AF, Nemeroff CB (eds). Textbook of psychopharmacology (3rd ed). Washington, DC: American Psychiatric Publishing, 2004.
Drug brand names
- Amitriptyline • Elavil
- Cimetidine • Tagamet
- Ciprofloxacin • Cipro
- Desipramine • Norpramin
- Duloxetine • Cymbalta
- Enoxacin • Penetrex
- Fluoxetine • Prozac
- Flecainide • Tambocor
- Fluvoxamine • Luvox
- Haloperidol • Haldol
- Metoprolol • Toprol
- Nortriptyline • Pamelor
- Paroxetine • Paxil
- Propafenone • Rythmol
- Propranolol • Inderal
- Risperidone • Risperdal
- Thioridazine • Mellaril
- Timolol • Blocadren, others
- Venlafaxine • Effexor
Disclosure
Dr. Rakesh Jain receives research grants from Eli Lilly and Co., Forest Pharmaceuticals, GlaxoSmithKline, Merck and Co., Organon, Pfizer Inc., and Sepracor. He is a consultant to and speaker for Eli Lilly and Co., and is a speaker for GlaxoSmithKline, Pfizer Inc., and Wyeth Pharmaceuticals.
Dr. Shailesh Jain reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of compefting products.
1. Thase ME, Entsuah AR, Rudolph RL. Remission rates during treatment with venlafaxine or serotonin reuptake inhibitors. Br J Psychiatry 2001;178:234-41.
2. Fava M. The role of the serotonergic and noradrenergic neurotransmitter systems in the treatment of psychological and physical symptoms of depression. J Clin Psychiatry 2003;64(suppl 13):26-9.
3. Tran PV, Bymaster FP, McNamara RK, Potter WZ. Dual monoamine modulation for improved treatment of major depressive disorder. J Clin Psychopharmacol 2003;23:78-86.
4. Ansari A. The efficacy of newer antidepressants in the treatment of chronic pain: a review of current literature. Harv Rev Psychiatry 2000;7:257-77.
5. Willis WD, Westlund KN. Neuroanatomy of the pain system and the pathways that modulate pain. J Clin Neurophysiol 1997;14:2-31.
6. Skinner MH, Kuan HY, Pan A, et al. Duloxetine is both an inhibitor and a substrate of cytochrome P4502D6 in healthy volunteers. Clin Pharmacol Ther 2003;73:170-7.
7. Sadock BJ, Sadock VA. Kaplan and Sadock's pocket handbook of psychotropic drug treatment (3rd ed). Baltimore, MD: Lippincott Williams and Wilkins, 2001.
8. Goldstein DJ, Mallinckrodt C, Lu Y, Demitrack M. Duloxetine in the treatment of major depression disorder: a double-blind clinical trial. J Clin Psychiatry 2002;63:225-31.
9. Detke MJ, Lu Y, Goldstein DJ, et al. Duloxetine 60 mg once-daily for major depressive disorder: a randomized double-blind placebocontrolled trial. J Clin Psychiatry 2002;63:308-15.
10. Detke MJ, Lu Y, Goldstein DJ, et al. Duloxetine 60 mg once daily dosing versus placebo in the treatment of major depression. J Psychiatr Res 2002;36:383-90.
11. Cymbalta prescribing information Eli Lilly and Co 2004.
12. Goldstein DJ, Lu Y, Detke MJ, et al. Duloxetine in the treatment of depression: a double-blind placebo-controlled comparison with paroxetine. J Clin Psychopharmacol 2004;24:389-99.
1. Thase ME, Entsuah AR, Rudolph RL. Remission rates during treatment with venlafaxine or serotonin reuptake inhibitors. Br J Psychiatry 2001;178:234-41.
2. Fava M. The role of the serotonergic and noradrenergic neurotransmitter systems in the treatment of psychological and physical symptoms of depression. J Clin Psychiatry 2003;64(suppl 13):26-9.
3. Tran PV, Bymaster FP, McNamara RK, Potter WZ. Dual monoamine modulation for improved treatment of major depressive disorder. J Clin Psychopharmacol 2003;23:78-86.
4. Ansari A. The efficacy of newer antidepressants in the treatment of chronic pain: a review of current literature. Harv Rev Psychiatry 2000;7:257-77.
5. Willis WD, Westlund KN. Neuroanatomy of the pain system and the pathways that modulate pain. J Clin Neurophysiol 1997;14:2-31.
6. Skinner MH, Kuan HY, Pan A, et al. Duloxetine is both an inhibitor and a substrate of cytochrome P4502D6 in healthy volunteers. Clin Pharmacol Ther 2003;73:170-7.
7. Sadock BJ, Sadock VA. Kaplan and Sadock's pocket handbook of psychotropic drug treatment (3rd ed). Baltimore, MD: Lippincott Williams and Wilkins, 2001.
8. Goldstein DJ, Mallinckrodt C, Lu Y, Demitrack M. Duloxetine in the treatment of major depression disorder: a double-blind clinical trial. J Clin Psychiatry 2002;63:225-31.
9. Detke MJ, Lu Y, Goldstein DJ, et al. Duloxetine 60 mg once-daily for major depressive disorder: a randomized double-blind placebocontrolled trial. J Clin Psychiatry 2002;63:308-15.
10. Detke MJ, Lu Y, Goldstein DJ, et al. Duloxetine 60 mg once daily dosing versus placebo in the treatment of major depression. J Psychiatr Res 2002;36:383-90.
11. Cymbalta prescribing information Eli Lilly and Co 2004.
12. Goldstein DJ, Lu Y, Detke MJ, et al. Duloxetine in the treatment of depression: a double-blind placebo-controlled comparison with paroxetine. J Clin Psychopharmacol 2004;24:389-99.
Spotting a silent killer
CASE 1: BEWARE ‘OLD MAN KIPLING’
Mrs. A, age 87, has Alzheimer’s disease. About 1 month before presentation, she entered a nursing home because of increasing agitation, paranoia, auditory and visual hallucinations, and decreased ability to care for herself. Her doctor started risperidone, 0.5 mg bid, to treat her agitation and psychosis.
Two days later, Mrs. A barricaded herself in her room. She told staff that “Old Man Kipling” was trying to break in, steal her money, and kill her and her son. She was sent to the emergency room; psychiatric consultation was ordered.
Mrs. A also has hypertension, renal cell carcinoma, anemia, and chronic renal failure. She had seen a psychiatrist for worsening cognitive function but has no other psychiatric history. Brain CT without contrast revealed generalized atrophy with no acute cerebral events. Workup showed decreased potassium (3.1 mEq/L), which returned to normal after Mrs. A was given potassium chloride, 20 mEq/d for 5 days. Other lab results were normal. Hydrochlorothiazide, 25 mg/d for hypertension, was stopped to prevent potassium depletion. No neurologic deficits were found.
Upon admission to the geriatric psychiatry unit, Mrs. A was paranoid and agitated. She talked to an imaginary person, continued to fear “Old Man Kipling,” and again tried to barricade herself.
ECG at admission—done because of Mrs. A’s age, cardiac history, and hydrochlorothiazide use—showed a corrected QT (QTc) interval of 494 msec, nearly 50 msec above the high-normal range for women. The interval was 460 msec at baseline (before risperidone treatment). Mrs. A was switched to olanzapine, 5 mg at bedtime, but her QTc intervals stayed between 494 and 495 msec, and her psychotic symptoms continued unabated.
Table 1
Mean antipsychotic-induced QTc interval change from baseline to steady state
| Antipsychotic | Mean QTc interval change |
|---|---|
| Haloperidol | 4.7 msec |
| Olanzapine | 6.4 msec |
| Risperidone | 10.0 msec |
| Quetiapine | 14.5 msec |
| Ziprasidone | 20.6 msec |
| Thioridazine | 35.8 msec |
| Source: reference 2. | |
The authors’ observations
Antipsychotics, used to treat behavioral disturbances in older patients, can prolong QTc intervals. Although often asymptomatic, a prolonged interval can lead to torsade de pointes, a polymorphic ventricular arrhythmia that can progress to ventricular fibrillation and cause sudden death.
Reilly et al1 suggest that antipsychotic-induced QTc prolongation may be dose-dependent. Age >65 is also a risk factor.
Start low and go slow when prescribing antipsychotics to patients with QTc intervals 450 msec. If prolonged intervals persist, switch antipsychotics and consult a cardiologist to help manage the patient’s care.
Switching agents will not entirely eliminate the risk, however. Mrs. A’s QTc interval remained elevated despite the switch to olanzapine, which is less likely than most antipsychotics to increase the interval.
Among mostly healthy men, haloperidol was shown to cause a lower mean QTc interval increase than other antipsychotics (Table 1), although QTc prolongations >60 msec were reported in 4% of those who took haloperidol.2 The agent also may cause tardive dyskinesia, and that risk is multiplied in patients >age 65.3 For Mrs. A, however, persistent psychosis and declining function outweighed the risks.
With haloperidol, start low and titrate slowly to reduce the risk of extrapyramidal symptoms (EPS). Decrease the dosage if involuntary movements develop. If a haloperidol decrease would lead to decompensation, add an anticholinergic agent such as benztropine, but be careful because anticholinergics can worsen cognitive function.
Test for involuntary movements before starting an antipsychotic. Retest every 4 to 6 months, when changing dosages or switching antipsychotics, and when patients complain of EPS.
CASE 1 CONTINUED: GOODBYE MR. KIPLING
Mrs. A was switched to haloperidol, 0.5 mg bid titrated over 3 weeks to 2 mg every morning and 3 mg nightly. Daily ECGs across 10 days showed QTc intervals 467 msec. Abnormal Involuntary Movement Scale testing showed no EPS. Her blood pressure was stable, ranging from 110 to 130 mm Hg (systolic) and 70 to 80 mm Hg (diastolic).
The patient became calmer and her paranoid delusions and hallucinations disappeared. Her Folstein Mini-Mental Status Examination score during her third and final week of hospitalization was 16, indicating moderate dementia. She was discharged to her son’s care; outpatient psychiatric care was also arranged. The psychiatrist started donepezil, 5 mg/d titrated to 10 mg/d after 6 weeks, to treat her memory impairments.
More than 1 year later, Mrs. A lives at home with her son. She has not needed psychiatric hospitalization. Her primary care physician monitors her cardiac health.
CASE 2: SUICIDALITY AND SEXUAL BEHAVIOR
Mr. B, age 50, has battled schizoaffective disorder for more than 30 years. Upon presenting to the ER, he told clinicians he planned to jump from his seventh-floor apartment after arguing with his neighbor.
The patient had been taking gabapentin, 300 mg bid; olanzapine, 10 mg at bedtime; citalopram, 20 mg/d; clonazepam, 1 mg at bedtime for panic symptoms; atorvastatin, 10 mg/d for hyperlipidemia; and esomeprazole, 40 mg/d, for ongoing GI problems. He also has bradycardia.
Electrolyte and magnesium levels, thyroid function, and liver function tests were normal. Potassium was 3.9 mEq/L, indicating possible deficiency. Toxicity screen was negative, ruling out substance abuse or medication overdose. Baseline ECG—ordered because of Mr. B’s bradycardia—showed a QTc interval of 519 msec (almost 80 msec above high-normal for men) and a heart rate of 50 bpm.
The cardiology team found that 1 year before, while being examined for suspected syncope, Mr. B had a prolonged QTc interval that resolved after olanzapine was stopped. Acting on cardiology’s advice, the psychiatrist stopped olanzapine and clonazepam, continued gabapentin, 300 mg/d, and added lorazepam, 1 mg as needed for agitation.
Within 48 hours, Mr. B’s QTc interval decreased to 400 msec. Gabapentin and lorazepam were continued. He received potassium chloride, 40 mEq qid for 4 days, and within 2 days potassium was normal (4.4 to 4.8 mEq/L). Magnesium also was monitored.
Over the next few days, Mr. B decompensated. He exposed himself, requested sexual favors from staff, and became agitated. Staff reported that he was responding to internal stimuli and had pressured speech and flight of ideas.
After consulting cardiology, the psychiatrist restarted olanzapine, 10 mg/d, and lorazepam, 1 mg bid. Daily ECGs were ordered. After two olanzapine doses, Mr. B’s QTc interval rose to 550 msec. The psychiatrist stopped all psychotropics except lorazepam, which was increased to 2 mg bid. When Mr. B became more agitated, throwing himself to the floor and hitting himself, he was isolated for his safety.
The authors’ observations
For years, olanzapine abated Mr. B’s mood and psychotic symptoms, and until the previous year significant QTc prolongation had not been detected. Other risk factors—such as electrolyte imbalance and change in olanzapine metabolism—were ruled out.
Mr. B’s chart indicated that he had responded well to haloperidol during a prior hospitalization. Divalproex, which has little effect on QTc interval, was also considered to control his mood.
CASE 2 CONTINUED: DRUG TRIALS
Eight days after Mr. B was hospitalized, the psychiatrist added divalproex, 250 mg tid titrated over 4 days to 1,000 mg/d. Mr. B became less manic but remained psychotic and disorganized. Lorazepam was increased to 2 mg tid and 3 mg at bedtime. His QTc interval now averaged 400 msec.
Loxapine, 10 mg tid, was added but then quickly discontinued after Mr. B’s QTc interval approached 500 msec.
Table 2
QTc interval ranges in men and women
| Range | Men (msec) | Women (msec) |
|---|---|---|
| Normal | <430 | <450 |
| Borderline | 431-450 | 451-470 |
| Source: reference 8. | ||
The following week, after consulting cardiology, the psychiatrist started haloperidol, 2 mg tid, and added benztropine, 1 mg for dystonia as needed. The next day, Mr. B’s QTc interval was 402 msec.
Medications were readjusted gradually. Gabapentin was restarted and increased to 600 mg tid, lorazepam was decreased to 1 mg tid, and divalproex was increased to 500 mg tid with no major QTc change.
Haloperidol was titrated to 5 mg bid, but the interval increased to 549 msec, then fell below 500 msec after haloperidol was readjusted to 2 mg bid.
Over the next 2 weeks, Mr. B’s mood and psychotic symptoms gradually improved. He was discharged after 27 days, at which point his QTc interval ranged between 360 and 409 msec. He was told to continue his medications.
The authors’ observations
Many factors other than antipsychotic use can lengthen QTc interval. Patients with major psychiatric disorders tend to have more risk factors compared with the general population.4
Serial or signal-averaged ECGs are the most accurate ways to monitor QTc intervals.5 Obtain a baseline ECG before starting an antipsychotic for patients with one or more risk factors:
Age >65. Older persons without coronary artery disease (CAD) have longer QTc intervals than do younger patients in similar health.6
Drug-drug interactions—common among the elderly—can further prolong the interval. Decreased drug metabolism also raises drug plasma levels and increases QTc prolongation risk.
Cardiac diseases. CAD, cardiac arrhythmias, and congestive heart failure are serious risk factors, particularly for older patients. Watch for pre-existing heart disease—which heightens risk of conduction defects—and family history of cardiac disease, syncope, or sudden death.
CNS diseases. Stroke, tumors, and brain infections can cause autonomic dysfunction and electrolyte imbalances.
Electrolyte imbalance. Hypokalemia and hypomagnesemia can prolong the interval.7 Take complaints of diarrhea or frequent vomiting seriously, and refer patients with renal disease or who are using diuretics for an ECG. Regularly test for electrolytes, especially potassium and magnesium.
Endocrine diseases. Diabetes, hypothyroidism, and pituitary insufficiency can cause electrolyte abnormalities.
Female sex. QTc intervals are on average 20 msec longer in women <age>Table 2)8 and are prolonged during the first half of the menstrual cycle. Androgen may shorten intervals in men. Women account for about 70% of drug-induced torsade de pointes cases.9
</age>
Medications. Antipsychotics, tricyclics, and antihistamines can prolong the interval alone or when combined with drugs that inhibit their metabolism. Concomitant use of agents that inhibit cytochrome P-450 enzyme systems may elevate serum concentrations of the interval-prolonging medication,4 as can decreased CYP 2D6 activity.10 Check plasma drug levels in patients who exhibit side effects.
Also check for congenital long QTc interval, autonomic CNS abnormalities, and overdose of a prescribed psychotropic.
Check ECGs every 2 days for inpatients and at every visit for outpatients taking antipsychotics. Frequent testing is crucial for elderly patients with multiple cardiac risk factors who are taking medications likely to increase the interval. Repeat ECGs if the patient reports lightheadedness or palpitations.
QTc interval prolongation is minimal in healthy young adults taking antipsychotics, so order ECGs only when symptoms arise. A baseline ECG is advisable but not necessary.
Order a cardiology consult and immediate ECG when the QTc interval exceeds 500 msec11 or if the patient exhibits arrhythmia symptoms (palpitation chest pain, dizziness, presyncope, syncope). Work with the cardiologist to manage medication.
Related resources
- University of Arizona Center for Education and Research on Therapeutics. Drugs that prolong the QT interval. http://www.qtdrugs.org/medical-pros/drug-lists/drug-lists.htm
- Glassman AH, Bigger JT Jr. Antipsychotic drugs: prolonged QTc interval, torsade de pointes, and sudden death. Am J Psychiatry 2001;158:1774-82.
Drug brand names
- Atorvastatin • Lipitor
- Benztropine • Cogentin
- Citalopram • Celexa
- Clonazepam • Klonopin
- Divalproex • Depakote
- Donepezil • Aricept
- Esomeprazole • Nexium
- Gabapentin • Neurontin
- Haloperidol • Haldol
- Hydrochlorothiazide • Atacand, others
- Lorazepam • Ativan
- Loxapine • Loxitane
- Olanzapine • Zyprexa
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Thioridazine • Mellaril
- Ziprasidone •Geodon
Disclosure
Dr. Tampi receives research support from the division of state, community, and public health, bureau of health professions, Health Resources and Services Administration, Department of Health and Human Services.
Dr. Ruedrich receives grants from Pfizer Inc. and Eisai Inc., and is a consultant to Abbott Laboratories.
The other authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Reilly JG, Ayis SA, Ferrier IN, et al. QTc-interval abnormalities and psychotropic drug therapy in psychiatric patients. Lancet. 2000;355:1048-52.
2. U.S. Food and Drug Administration. Center for Drug Evaluation and Research, Psychopharmacological Drugs Advisory Committee. Meeting transcript for approval of Zeldox (ziprasidone), July 19, 2000. Available at: http://www.fda.gov/ohrms/docket/ac/00/transcripts/3619tla.pdf, 3619tlb.pdf. and 3619tlc.pdf.
3. Jeste DV, Caligiuri MP, Paulsen JS, et al. Risk of tardive dyskinesia in older patients. A prospective longitudinal study of 266 outpatients. Arch Gen Psychiatry. 1995;52:756-65.
4. Fayek M, Kingsbury SJ, Zada J, Simpson GM. Psychopharmacology: cardiac effects of antipsychotic medication. Psychiatr Serv. 2001;52:607-9.
5. Baker B, Dorian P, Sandor C, et al. Electrocardiographic effects of fluoxetine and doxepine in patients with major depression. J Clin Psychopharmacol. 1997;17:15-21.
6. Khan SP, Dhalvani S, Vieweg WVR, et al. Electrocardiographic QT interval in geropsychiatric inpatient population: a preliminary study. Med Psychiatr. 1998;1:71-4.
7. Crompton SJ, Lux RL, Ramsey MR, et al. Genetically defined therapy of inherited long-QT syndrome: correction of abnormal repolarization by potassium. Circulation. 1996;94:1018-22.
8. Piepho RW. Cardiovascular effects of antipsychotics used in bipolar illness. J Clin Psychiatry. 2002;63[suppl 4]:20-3.
9. Drici MD, Clement N. Is gender a risk factor for adverse drug reaction? The example of drug-induced long QT syndrome. Drug Saf. 2001;24(8):575-85
10. Francis PD. Effects of psychotropic medications on the pediatric electrocardiogram and recommendations for monitoring. Curr Opin Ped. 2002;14:224-30.
11. Bednar MM, Harrigan EP, Anziano RJ, et al. The QT interval. Prog Cardiovasc Dis. 2001;43:1-45.
CASE 1: BEWARE ‘OLD MAN KIPLING’
Mrs. A, age 87, has Alzheimer’s disease. About 1 month before presentation, she entered a nursing home because of increasing agitation, paranoia, auditory and visual hallucinations, and decreased ability to care for herself. Her doctor started risperidone, 0.5 mg bid, to treat her agitation and psychosis.
Two days later, Mrs. A barricaded herself in her room. She told staff that “Old Man Kipling” was trying to break in, steal her money, and kill her and her son. She was sent to the emergency room; psychiatric consultation was ordered.
Mrs. A also has hypertension, renal cell carcinoma, anemia, and chronic renal failure. She had seen a psychiatrist for worsening cognitive function but has no other psychiatric history. Brain CT without contrast revealed generalized atrophy with no acute cerebral events. Workup showed decreased potassium (3.1 mEq/L), which returned to normal after Mrs. A was given potassium chloride, 20 mEq/d for 5 days. Other lab results were normal. Hydrochlorothiazide, 25 mg/d for hypertension, was stopped to prevent potassium depletion. No neurologic deficits were found.
Upon admission to the geriatric psychiatry unit, Mrs. A was paranoid and agitated. She talked to an imaginary person, continued to fear “Old Man Kipling,” and again tried to barricade herself.
ECG at admission—done because of Mrs. A’s age, cardiac history, and hydrochlorothiazide use—showed a corrected QT (QTc) interval of 494 msec, nearly 50 msec above the high-normal range for women. The interval was 460 msec at baseline (before risperidone treatment). Mrs. A was switched to olanzapine, 5 mg at bedtime, but her QTc intervals stayed between 494 and 495 msec, and her psychotic symptoms continued unabated.
Table 1
Mean antipsychotic-induced QTc interval change from baseline to steady state
| Antipsychotic | Mean QTc interval change |
|---|---|
| Haloperidol | 4.7 msec |
| Olanzapine | 6.4 msec |
| Risperidone | 10.0 msec |
| Quetiapine | 14.5 msec |
| Ziprasidone | 20.6 msec |
| Thioridazine | 35.8 msec |
| Source: reference 2. | |
The authors’ observations
Antipsychotics, used to treat behavioral disturbances in older patients, can prolong QTc intervals. Although often asymptomatic, a prolonged interval can lead to torsade de pointes, a polymorphic ventricular arrhythmia that can progress to ventricular fibrillation and cause sudden death.
Reilly et al1 suggest that antipsychotic-induced QTc prolongation may be dose-dependent. Age >65 is also a risk factor.
Start low and go slow when prescribing antipsychotics to patients with QTc intervals 450 msec. If prolonged intervals persist, switch antipsychotics and consult a cardiologist to help manage the patient’s care.
Switching agents will not entirely eliminate the risk, however. Mrs. A’s QTc interval remained elevated despite the switch to olanzapine, which is less likely than most antipsychotics to increase the interval.
Among mostly healthy men, haloperidol was shown to cause a lower mean QTc interval increase than other antipsychotics (Table 1), although QTc prolongations >60 msec were reported in 4% of those who took haloperidol.2 The agent also may cause tardive dyskinesia, and that risk is multiplied in patients >age 65.3 For Mrs. A, however, persistent psychosis and declining function outweighed the risks.
With haloperidol, start low and titrate slowly to reduce the risk of extrapyramidal symptoms (EPS). Decrease the dosage if involuntary movements develop. If a haloperidol decrease would lead to decompensation, add an anticholinergic agent such as benztropine, but be careful because anticholinergics can worsen cognitive function.
Test for involuntary movements before starting an antipsychotic. Retest every 4 to 6 months, when changing dosages or switching antipsychotics, and when patients complain of EPS.
CASE 1 CONTINUED: GOODBYE MR. KIPLING
Mrs. A was switched to haloperidol, 0.5 mg bid titrated over 3 weeks to 2 mg every morning and 3 mg nightly. Daily ECGs across 10 days showed QTc intervals 467 msec. Abnormal Involuntary Movement Scale testing showed no EPS. Her blood pressure was stable, ranging from 110 to 130 mm Hg (systolic) and 70 to 80 mm Hg (diastolic).
The patient became calmer and her paranoid delusions and hallucinations disappeared. Her Folstein Mini-Mental Status Examination score during her third and final week of hospitalization was 16, indicating moderate dementia. She was discharged to her son’s care; outpatient psychiatric care was also arranged. The psychiatrist started donepezil, 5 mg/d titrated to 10 mg/d after 6 weeks, to treat her memory impairments.
More than 1 year later, Mrs. A lives at home with her son. She has not needed psychiatric hospitalization. Her primary care physician monitors her cardiac health.
CASE 2: SUICIDALITY AND SEXUAL BEHAVIOR
Mr. B, age 50, has battled schizoaffective disorder for more than 30 years. Upon presenting to the ER, he told clinicians he planned to jump from his seventh-floor apartment after arguing with his neighbor.
The patient had been taking gabapentin, 300 mg bid; olanzapine, 10 mg at bedtime; citalopram, 20 mg/d; clonazepam, 1 mg at bedtime for panic symptoms; atorvastatin, 10 mg/d for hyperlipidemia; and esomeprazole, 40 mg/d, for ongoing GI problems. He also has bradycardia.
Electrolyte and magnesium levels, thyroid function, and liver function tests were normal. Potassium was 3.9 mEq/L, indicating possible deficiency. Toxicity screen was negative, ruling out substance abuse or medication overdose. Baseline ECG—ordered because of Mr. B’s bradycardia—showed a QTc interval of 519 msec (almost 80 msec above high-normal for men) and a heart rate of 50 bpm.
The cardiology team found that 1 year before, while being examined for suspected syncope, Mr. B had a prolonged QTc interval that resolved after olanzapine was stopped. Acting on cardiology’s advice, the psychiatrist stopped olanzapine and clonazepam, continued gabapentin, 300 mg/d, and added lorazepam, 1 mg as needed for agitation.
Within 48 hours, Mr. B’s QTc interval decreased to 400 msec. Gabapentin and lorazepam were continued. He received potassium chloride, 40 mEq qid for 4 days, and within 2 days potassium was normal (4.4 to 4.8 mEq/L). Magnesium also was monitored.
Over the next few days, Mr. B decompensated. He exposed himself, requested sexual favors from staff, and became agitated. Staff reported that he was responding to internal stimuli and had pressured speech and flight of ideas.
After consulting cardiology, the psychiatrist restarted olanzapine, 10 mg/d, and lorazepam, 1 mg bid. Daily ECGs were ordered. After two olanzapine doses, Mr. B’s QTc interval rose to 550 msec. The psychiatrist stopped all psychotropics except lorazepam, which was increased to 2 mg bid. When Mr. B became more agitated, throwing himself to the floor and hitting himself, he was isolated for his safety.
The authors’ observations
For years, olanzapine abated Mr. B’s mood and psychotic symptoms, and until the previous year significant QTc prolongation had not been detected. Other risk factors—such as electrolyte imbalance and change in olanzapine metabolism—were ruled out.
Mr. B’s chart indicated that he had responded well to haloperidol during a prior hospitalization. Divalproex, which has little effect on QTc interval, was also considered to control his mood.
CASE 2 CONTINUED: DRUG TRIALS
Eight days after Mr. B was hospitalized, the psychiatrist added divalproex, 250 mg tid titrated over 4 days to 1,000 mg/d. Mr. B became less manic but remained psychotic and disorganized. Lorazepam was increased to 2 mg tid and 3 mg at bedtime. His QTc interval now averaged 400 msec.
Loxapine, 10 mg tid, was added but then quickly discontinued after Mr. B’s QTc interval approached 500 msec.
Table 2
QTc interval ranges in men and women
| Range | Men (msec) | Women (msec) |
|---|---|---|
| Normal | <430 | <450 |
| Borderline | 431-450 | 451-470 |
| Source: reference 8. | ||
The following week, after consulting cardiology, the psychiatrist started haloperidol, 2 mg tid, and added benztropine, 1 mg for dystonia as needed. The next day, Mr. B’s QTc interval was 402 msec.
Medications were readjusted gradually. Gabapentin was restarted and increased to 600 mg tid, lorazepam was decreased to 1 mg tid, and divalproex was increased to 500 mg tid with no major QTc change.
Haloperidol was titrated to 5 mg bid, but the interval increased to 549 msec, then fell below 500 msec after haloperidol was readjusted to 2 mg bid.
Over the next 2 weeks, Mr. B’s mood and psychotic symptoms gradually improved. He was discharged after 27 days, at which point his QTc interval ranged between 360 and 409 msec. He was told to continue his medications.
The authors’ observations
Many factors other than antipsychotic use can lengthen QTc interval. Patients with major psychiatric disorders tend to have more risk factors compared with the general population.4
Serial or signal-averaged ECGs are the most accurate ways to monitor QTc intervals.5 Obtain a baseline ECG before starting an antipsychotic for patients with one or more risk factors:
Age >65. Older persons without coronary artery disease (CAD) have longer QTc intervals than do younger patients in similar health.6
Drug-drug interactions—common among the elderly—can further prolong the interval. Decreased drug metabolism also raises drug plasma levels and increases QTc prolongation risk.
Cardiac diseases. CAD, cardiac arrhythmias, and congestive heart failure are serious risk factors, particularly for older patients. Watch for pre-existing heart disease—which heightens risk of conduction defects—and family history of cardiac disease, syncope, or sudden death.
CNS diseases. Stroke, tumors, and brain infections can cause autonomic dysfunction and electrolyte imbalances.
Electrolyte imbalance. Hypokalemia and hypomagnesemia can prolong the interval.7 Take complaints of diarrhea or frequent vomiting seriously, and refer patients with renal disease or who are using diuretics for an ECG. Regularly test for electrolytes, especially potassium and magnesium.
Endocrine diseases. Diabetes, hypothyroidism, and pituitary insufficiency can cause electrolyte abnormalities.
Female sex. QTc intervals are on average 20 msec longer in women <age>Table 2)8 and are prolonged during the first half of the menstrual cycle. Androgen may shorten intervals in men. Women account for about 70% of drug-induced torsade de pointes cases.9
</age>
Medications. Antipsychotics, tricyclics, and antihistamines can prolong the interval alone or when combined with drugs that inhibit their metabolism. Concomitant use of agents that inhibit cytochrome P-450 enzyme systems may elevate serum concentrations of the interval-prolonging medication,4 as can decreased CYP 2D6 activity.10 Check plasma drug levels in patients who exhibit side effects.
Also check for congenital long QTc interval, autonomic CNS abnormalities, and overdose of a prescribed psychotropic.
Check ECGs every 2 days for inpatients and at every visit for outpatients taking antipsychotics. Frequent testing is crucial for elderly patients with multiple cardiac risk factors who are taking medications likely to increase the interval. Repeat ECGs if the patient reports lightheadedness or palpitations.
QTc interval prolongation is minimal in healthy young adults taking antipsychotics, so order ECGs only when symptoms arise. A baseline ECG is advisable but not necessary.
Order a cardiology consult and immediate ECG when the QTc interval exceeds 500 msec11 or if the patient exhibits arrhythmia symptoms (palpitation chest pain, dizziness, presyncope, syncope). Work with the cardiologist to manage medication.
Related resources
- University of Arizona Center for Education and Research on Therapeutics. Drugs that prolong the QT interval. http://www.qtdrugs.org/medical-pros/drug-lists/drug-lists.htm
- Glassman AH, Bigger JT Jr. Antipsychotic drugs: prolonged QTc interval, torsade de pointes, and sudden death. Am J Psychiatry 2001;158:1774-82.
Drug brand names
- Atorvastatin • Lipitor
- Benztropine • Cogentin
- Citalopram • Celexa
- Clonazepam • Klonopin
- Divalproex • Depakote
- Donepezil • Aricept
- Esomeprazole • Nexium
- Gabapentin • Neurontin
- Haloperidol • Haldol
- Hydrochlorothiazide • Atacand, others
- Lorazepam • Ativan
- Loxapine • Loxitane
- Olanzapine • Zyprexa
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Thioridazine • Mellaril
- Ziprasidone •Geodon
Disclosure
Dr. Tampi receives research support from the division of state, community, and public health, bureau of health professions, Health Resources and Services Administration, Department of Health and Human Services.
Dr. Ruedrich receives grants from Pfizer Inc. and Eisai Inc., and is a consultant to Abbott Laboratories.
The other authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
CASE 1: BEWARE ‘OLD MAN KIPLING’
Mrs. A, age 87, has Alzheimer’s disease. About 1 month before presentation, she entered a nursing home because of increasing agitation, paranoia, auditory and visual hallucinations, and decreased ability to care for herself. Her doctor started risperidone, 0.5 mg bid, to treat her agitation and psychosis.
Two days later, Mrs. A barricaded herself in her room. She told staff that “Old Man Kipling” was trying to break in, steal her money, and kill her and her son. She was sent to the emergency room; psychiatric consultation was ordered.
Mrs. A also has hypertension, renal cell carcinoma, anemia, and chronic renal failure. She had seen a psychiatrist for worsening cognitive function but has no other psychiatric history. Brain CT without contrast revealed generalized atrophy with no acute cerebral events. Workup showed decreased potassium (3.1 mEq/L), which returned to normal after Mrs. A was given potassium chloride, 20 mEq/d for 5 days. Other lab results were normal. Hydrochlorothiazide, 25 mg/d for hypertension, was stopped to prevent potassium depletion. No neurologic deficits were found.
Upon admission to the geriatric psychiatry unit, Mrs. A was paranoid and agitated. She talked to an imaginary person, continued to fear “Old Man Kipling,” and again tried to barricade herself.
ECG at admission—done because of Mrs. A’s age, cardiac history, and hydrochlorothiazide use—showed a corrected QT (QTc) interval of 494 msec, nearly 50 msec above the high-normal range for women. The interval was 460 msec at baseline (before risperidone treatment). Mrs. A was switched to olanzapine, 5 mg at bedtime, but her QTc intervals stayed between 494 and 495 msec, and her psychotic symptoms continued unabated.
Table 1
Mean antipsychotic-induced QTc interval change from baseline to steady state
| Antipsychotic | Mean QTc interval change |
|---|---|
| Haloperidol | 4.7 msec |
| Olanzapine | 6.4 msec |
| Risperidone | 10.0 msec |
| Quetiapine | 14.5 msec |
| Ziprasidone | 20.6 msec |
| Thioridazine | 35.8 msec |
| Source: reference 2. | |
The authors’ observations
Antipsychotics, used to treat behavioral disturbances in older patients, can prolong QTc intervals. Although often asymptomatic, a prolonged interval can lead to torsade de pointes, a polymorphic ventricular arrhythmia that can progress to ventricular fibrillation and cause sudden death.
Reilly et al1 suggest that antipsychotic-induced QTc prolongation may be dose-dependent. Age >65 is also a risk factor.
Start low and go slow when prescribing antipsychotics to patients with QTc intervals 450 msec. If prolonged intervals persist, switch antipsychotics and consult a cardiologist to help manage the patient’s care.
Switching agents will not entirely eliminate the risk, however. Mrs. A’s QTc interval remained elevated despite the switch to olanzapine, which is less likely than most antipsychotics to increase the interval.
Among mostly healthy men, haloperidol was shown to cause a lower mean QTc interval increase than other antipsychotics (Table 1), although QTc prolongations >60 msec were reported in 4% of those who took haloperidol.2 The agent also may cause tardive dyskinesia, and that risk is multiplied in patients >age 65.3 For Mrs. A, however, persistent psychosis and declining function outweighed the risks.
With haloperidol, start low and titrate slowly to reduce the risk of extrapyramidal symptoms (EPS). Decrease the dosage if involuntary movements develop. If a haloperidol decrease would lead to decompensation, add an anticholinergic agent such as benztropine, but be careful because anticholinergics can worsen cognitive function.
Test for involuntary movements before starting an antipsychotic. Retest every 4 to 6 months, when changing dosages or switching antipsychotics, and when patients complain of EPS.
CASE 1 CONTINUED: GOODBYE MR. KIPLING
Mrs. A was switched to haloperidol, 0.5 mg bid titrated over 3 weeks to 2 mg every morning and 3 mg nightly. Daily ECGs across 10 days showed QTc intervals 467 msec. Abnormal Involuntary Movement Scale testing showed no EPS. Her blood pressure was stable, ranging from 110 to 130 mm Hg (systolic) and 70 to 80 mm Hg (diastolic).
The patient became calmer and her paranoid delusions and hallucinations disappeared. Her Folstein Mini-Mental Status Examination score during her third and final week of hospitalization was 16, indicating moderate dementia. She was discharged to her son’s care; outpatient psychiatric care was also arranged. The psychiatrist started donepezil, 5 mg/d titrated to 10 mg/d after 6 weeks, to treat her memory impairments.
More than 1 year later, Mrs. A lives at home with her son. She has not needed psychiatric hospitalization. Her primary care physician monitors her cardiac health.
CASE 2: SUICIDALITY AND SEXUAL BEHAVIOR
Mr. B, age 50, has battled schizoaffective disorder for more than 30 years. Upon presenting to the ER, he told clinicians he planned to jump from his seventh-floor apartment after arguing with his neighbor.
The patient had been taking gabapentin, 300 mg bid; olanzapine, 10 mg at bedtime; citalopram, 20 mg/d; clonazepam, 1 mg at bedtime for panic symptoms; atorvastatin, 10 mg/d for hyperlipidemia; and esomeprazole, 40 mg/d, for ongoing GI problems. He also has bradycardia.
Electrolyte and magnesium levels, thyroid function, and liver function tests were normal. Potassium was 3.9 mEq/L, indicating possible deficiency. Toxicity screen was negative, ruling out substance abuse or medication overdose. Baseline ECG—ordered because of Mr. B’s bradycardia—showed a QTc interval of 519 msec (almost 80 msec above high-normal for men) and a heart rate of 50 bpm.
The cardiology team found that 1 year before, while being examined for suspected syncope, Mr. B had a prolonged QTc interval that resolved after olanzapine was stopped. Acting on cardiology’s advice, the psychiatrist stopped olanzapine and clonazepam, continued gabapentin, 300 mg/d, and added lorazepam, 1 mg as needed for agitation.
Within 48 hours, Mr. B’s QTc interval decreased to 400 msec. Gabapentin and lorazepam were continued. He received potassium chloride, 40 mEq qid for 4 days, and within 2 days potassium was normal (4.4 to 4.8 mEq/L). Magnesium also was monitored.
Over the next few days, Mr. B decompensated. He exposed himself, requested sexual favors from staff, and became agitated. Staff reported that he was responding to internal stimuli and had pressured speech and flight of ideas.
After consulting cardiology, the psychiatrist restarted olanzapine, 10 mg/d, and lorazepam, 1 mg bid. Daily ECGs were ordered. After two olanzapine doses, Mr. B’s QTc interval rose to 550 msec. The psychiatrist stopped all psychotropics except lorazepam, which was increased to 2 mg bid. When Mr. B became more agitated, throwing himself to the floor and hitting himself, he was isolated for his safety.
The authors’ observations
For years, olanzapine abated Mr. B’s mood and psychotic symptoms, and until the previous year significant QTc prolongation had not been detected. Other risk factors—such as electrolyte imbalance and change in olanzapine metabolism—were ruled out.
Mr. B’s chart indicated that he had responded well to haloperidol during a prior hospitalization. Divalproex, which has little effect on QTc interval, was also considered to control his mood.
CASE 2 CONTINUED: DRUG TRIALS
Eight days after Mr. B was hospitalized, the psychiatrist added divalproex, 250 mg tid titrated over 4 days to 1,000 mg/d. Mr. B became less manic but remained psychotic and disorganized. Lorazepam was increased to 2 mg tid and 3 mg at bedtime. His QTc interval now averaged 400 msec.
Loxapine, 10 mg tid, was added but then quickly discontinued after Mr. B’s QTc interval approached 500 msec.
Table 2
QTc interval ranges in men and women
| Range | Men (msec) | Women (msec) |
|---|---|---|
| Normal | <430 | <450 |
| Borderline | 431-450 | 451-470 |
| Source: reference 8. | ||
The following week, after consulting cardiology, the psychiatrist started haloperidol, 2 mg tid, and added benztropine, 1 mg for dystonia as needed. The next day, Mr. B’s QTc interval was 402 msec.
Medications were readjusted gradually. Gabapentin was restarted and increased to 600 mg tid, lorazepam was decreased to 1 mg tid, and divalproex was increased to 500 mg tid with no major QTc change.
Haloperidol was titrated to 5 mg bid, but the interval increased to 549 msec, then fell below 500 msec after haloperidol was readjusted to 2 mg bid.
Over the next 2 weeks, Mr. B’s mood and psychotic symptoms gradually improved. He was discharged after 27 days, at which point his QTc interval ranged between 360 and 409 msec. He was told to continue his medications.
The authors’ observations
Many factors other than antipsychotic use can lengthen QTc interval. Patients with major psychiatric disorders tend to have more risk factors compared with the general population.4
Serial or signal-averaged ECGs are the most accurate ways to monitor QTc intervals.5 Obtain a baseline ECG before starting an antipsychotic for patients with one or more risk factors:
Age >65. Older persons without coronary artery disease (CAD) have longer QTc intervals than do younger patients in similar health.6
Drug-drug interactions—common among the elderly—can further prolong the interval. Decreased drug metabolism also raises drug plasma levels and increases QTc prolongation risk.
Cardiac diseases. CAD, cardiac arrhythmias, and congestive heart failure are serious risk factors, particularly for older patients. Watch for pre-existing heart disease—which heightens risk of conduction defects—and family history of cardiac disease, syncope, or sudden death.
CNS diseases. Stroke, tumors, and brain infections can cause autonomic dysfunction and electrolyte imbalances.
Electrolyte imbalance. Hypokalemia and hypomagnesemia can prolong the interval.7 Take complaints of diarrhea or frequent vomiting seriously, and refer patients with renal disease or who are using diuretics for an ECG. Regularly test for electrolytes, especially potassium and magnesium.
Endocrine diseases. Diabetes, hypothyroidism, and pituitary insufficiency can cause electrolyte abnormalities.
Female sex. QTc intervals are on average 20 msec longer in women <age>Table 2)8 and are prolonged during the first half of the menstrual cycle. Androgen may shorten intervals in men. Women account for about 70% of drug-induced torsade de pointes cases.9
</age>
Medications. Antipsychotics, tricyclics, and antihistamines can prolong the interval alone or when combined with drugs that inhibit their metabolism. Concomitant use of agents that inhibit cytochrome P-450 enzyme systems may elevate serum concentrations of the interval-prolonging medication,4 as can decreased CYP 2D6 activity.10 Check plasma drug levels in patients who exhibit side effects.
Also check for congenital long QTc interval, autonomic CNS abnormalities, and overdose of a prescribed psychotropic.
Check ECGs every 2 days for inpatients and at every visit for outpatients taking antipsychotics. Frequent testing is crucial for elderly patients with multiple cardiac risk factors who are taking medications likely to increase the interval. Repeat ECGs if the patient reports lightheadedness or palpitations.
QTc interval prolongation is minimal in healthy young adults taking antipsychotics, so order ECGs only when symptoms arise. A baseline ECG is advisable but not necessary.
Order a cardiology consult and immediate ECG when the QTc interval exceeds 500 msec11 or if the patient exhibits arrhythmia symptoms (palpitation chest pain, dizziness, presyncope, syncope). Work with the cardiologist to manage medication.
Related resources
- University of Arizona Center for Education and Research on Therapeutics. Drugs that prolong the QT interval. http://www.qtdrugs.org/medical-pros/drug-lists/drug-lists.htm
- Glassman AH, Bigger JT Jr. Antipsychotic drugs: prolonged QTc interval, torsade de pointes, and sudden death. Am J Psychiatry 2001;158:1774-82.
Drug brand names
- Atorvastatin • Lipitor
- Benztropine • Cogentin
- Citalopram • Celexa
- Clonazepam • Klonopin
- Divalproex • Depakote
- Donepezil • Aricept
- Esomeprazole • Nexium
- Gabapentin • Neurontin
- Haloperidol • Haldol
- Hydrochlorothiazide • Atacand, others
- Lorazepam • Ativan
- Loxapine • Loxitane
- Olanzapine • Zyprexa
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Thioridazine • Mellaril
- Ziprasidone •Geodon
Disclosure
Dr. Tampi receives research support from the division of state, community, and public health, bureau of health professions, Health Resources and Services Administration, Department of Health and Human Services.
Dr. Ruedrich receives grants from Pfizer Inc. and Eisai Inc., and is a consultant to Abbott Laboratories.
The other authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Reilly JG, Ayis SA, Ferrier IN, et al. QTc-interval abnormalities and psychotropic drug therapy in psychiatric patients. Lancet. 2000;355:1048-52.
2. U.S. Food and Drug Administration. Center for Drug Evaluation and Research, Psychopharmacological Drugs Advisory Committee. Meeting transcript for approval of Zeldox (ziprasidone), July 19, 2000. Available at: http://www.fda.gov/ohrms/docket/ac/00/transcripts/3619tla.pdf, 3619tlb.pdf. and 3619tlc.pdf.
3. Jeste DV, Caligiuri MP, Paulsen JS, et al. Risk of tardive dyskinesia in older patients. A prospective longitudinal study of 266 outpatients. Arch Gen Psychiatry. 1995;52:756-65.
4. Fayek M, Kingsbury SJ, Zada J, Simpson GM. Psychopharmacology: cardiac effects of antipsychotic medication. Psychiatr Serv. 2001;52:607-9.
5. Baker B, Dorian P, Sandor C, et al. Electrocardiographic effects of fluoxetine and doxepine in patients with major depression. J Clin Psychopharmacol. 1997;17:15-21.
6. Khan SP, Dhalvani S, Vieweg WVR, et al. Electrocardiographic QT interval in geropsychiatric inpatient population: a preliminary study. Med Psychiatr. 1998;1:71-4.
7. Crompton SJ, Lux RL, Ramsey MR, et al. Genetically defined therapy of inherited long-QT syndrome: correction of abnormal repolarization by potassium. Circulation. 1996;94:1018-22.
8. Piepho RW. Cardiovascular effects of antipsychotics used in bipolar illness. J Clin Psychiatry. 2002;63[suppl 4]:20-3.
9. Drici MD, Clement N. Is gender a risk factor for adverse drug reaction? The example of drug-induced long QT syndrome. Drug Saf. 2001;24(8):575-85
10. Francis PD. Effects of psychotropic medications on the pediatric electrocardiogram and recommendations for monitoring. Curr Opin Ped. 2002;14:224-30.
11. Bednar MM, Harrigan EP, Anziano RJ, et al. The QT interval. Prog Cardiovasc Dis. 2001;43:1-45.
1. Reilly JG, Ayis SA, Ferrier IN, et al. QTc-interval abnormalities and psychotropic drug therapy in psychiatric patients. Lancet. 2000;355:1048-52.
2. U.S. Food and Drug Administration. Center for Drug Evaluation and Research, Psychopharmacological Drugs Advisory Committee. Meeting transcript for approval of Zeldox (ziprasidone), July 19, 2000. Available at: http://www.fda.gov/ohrms/docket/ac/00/transcripts/3619tla.pdf, 3619tlb.pdf. and 3619tlc.pdf.
3. Jeste DV, Caligiuri MP, Paulsen JS, et al. Risk of tardive dyskinesia in older patients. A prospective longitudinal study of 266 outpatients. Arch Gen Psychiatry. 1995;52:756-65.
4. Fayek M, Kingsbury SJ, Zada J, Simpson GM. Psychopharmacology: cardiac effects of antipsychotic medication. Psychiatr Serv. 2001;52:607-9.
5. Baker B, Dorian P, Sandor C, et al. Electrocardiographic effects of fluoxetine and doxepine in patients with major depression. J Clin Psychopharmacol. 1997;17:15-21.
6. Khan SP, Dhalvani S, Vieweg WVR, et al. Electrocardiographic QT interval in geropsychiatric inpatient population: a preliminary study. Med Psychiatr. 1998;1:71-4.
7. Crompton SJ, Lux RL, Ramsey MR, et al. Genetically defined therapy of inherited long-QT syndrome: correction of abnormal repolarization by potassium. Circulation. 1996;94:1018-22.
8. Piepho RW. Cardiovascular effects of antipsychotics used in bipolar illness. J Clin Psychiatry. 2002;63[suppl 4]:20-3.
9. Drici MD, Clement N. Is gender a risk factor for adverse drug reaction? The example of drug-induced long QT syndrome. Drug Saf. 2001;24(8):575-85
10. Francis PD. Effects of psychotropic medications on the pediatric electrocardiogram and recommendations for monitoring. Curr Opin Ped. 2002;14:224-30.
11. Bednar MM, Harrigan EP, Anziano RJ, et al. The QT interval. Prog Cardiovasc Dis. 2001;43:1-45.
How to help nicotine-dependent adolescents quit smoking
Many adolescent psychiatric patients who smoke are not getting the help they need to quit. When we asked 120 teen inpatients if they smoked and then checked their charts, we found only 6 of 47 smokers had been diagnosed as nicotine-dependent.1
Adolescents who cannot quit on their own may benefit from smoking cessation therapies. Based on evidence and our experience, we offer a practical approach to treating nicotine dependence in adolescents, using drug and behavioral therapies.
PSYCHIATRIC COMORBIDITY
Psychiatric comorbidity is highly associated with cigarette smoking in adults and adolescents. In the United States:
- 44% of cigarettes smoked are sold to someone with a mental illness.2
- Persons with mental illness are 2.7 times more likely to smoke than are those without mental illness.2
- Most smokers start before age 18,3 and starting before age 13 is linked to psychopathology in later adolescence.4
Table
Smoking likelihood by age and comorbidity among adolescent psychiatric inpatients
| Significant variable | Logistic regression odds ratio | 95% confidence interval | Significance (P value) |
|---|---|---|---|
| Age | 1.30 | 1.03, 1.64 | 0.03 |
| Depressive disorders | 4.02 | 1.267, 12.734 | 0.018 |
| Conduct disorder | 12.96 | 1.678, 100.07 | 0.014 |
| Cannabis use disorder | 24.60 | 3.7, 163.42 | 0.0009 |
| Source: Data from 120 patients admitted to an inpatient child and adolescent psychiatry program. | |||
| Adapted with permission from reference 1. | |||
Disruptive behavior disorders in adolescent smokers include oppositional defiant disorder, conduct disorder, and attention-deficit/hyperactivity disorder (ADHD). Among psychiatric disorders, conduct disorder has the strongest association with smoking in adolescents.1 ADHD is associated with smoking and perhaps with increased difficulty in quitting.5,6
Mood disorders. Major depressive disorders have a strong, consistent, bidirectional association with smoking in the young. Depression may lead to smoking, and smoking to depression.7
Substance use disorders. Alcohol use disorders are strongly associated with smoking among adolescents, and the association is both bidirectional and dosedependent.8 Cannabis use disorder is also associated with cigarette smoking among adolescents (Table).9
Anxiety disorders. Evidence is emerging that anxiety disorders—especially social phobia—may be linked to smoking among adolescents.10
Nicotine withdrawal symptoms—irritability, anxiety, decreased concentration, increased appetite, craving for cigarettes—can mimic those of other psychiatric disorders. Adolescent smokers admitted to locked psychiatric units may experience withdrawal symptoms that require nicotine replacement treatment (Box).
Effect on quit rates. Psychiatric comorbidity may reduce quit rates during smoking cessation treatment.6 When smokers are trying to quit, watch for remission, worsening, or emergence of psychiatric conditions.
ASSESSING ADOLESCENT SMOKING
Adolescents with psychiatric diagnoses can be assessed for nicotine dependence—and vice versa—although accurately gauging their smoking habits is more difficult than in adults. For example:
- Rating scales for nicotine dependence severity—such as the modified Fagerstrom Tolerance Questionnaire11—lack standard cutoff scores for adolescents.
- Unlike adults, many adolescents cannot reliably report use in “packs per day” because the number of cigarettes they smoke varies widely from day to day.
Biological markers commonly used to assess smoking in adults include expired-air carbon monoxide (CO), cotinine (nicotine metabolite), and thiocynate levels. Preliminary evidence indicates that cotinine may be a more sensitive and specific biological marker for smoking among adolescents than CO levels.12 Thiocynate has not been evaluated as a marker for smoking in adolescents.
CO levels typically reflect smoking in the previous few hours, whereas the half-life of cotinine is longer (1 day or more). Also, factors such as environmental pollution or marijuana use can inflate CO levels. Thus, cotinine levels have greater accuracy and specificity, reflecting only the amount of nicotine consumed.
Unfortunately, most laboratories do not measure cotinine levels, and the expired-air CO test (CO Breathalyzer) is relatively expensive for most clinicians. Commercially available single-use cotinine test kits are modestly priced and provide semi-quantitative (a range instead of an exact number) urine cotinine levels. These tests, however, might not be covered by third-party insurers.
Until cotinine testing becomes widely available, we recommend a combination of self-report and expired-air CO level to monitor abstinence.
Self-report monitoring. Most clinicians rely on self-report rate of smoking among adolescents, as no screening assessment has been validated in this age group. As initial prompts, we recommend asking all adolescents if they smoke cigarettes, if they smoke regularly, and if they smoke daily.
We recommend using the “time line follow-back” method13 to monitor the self-reported smoking rate. Begin by providing the patient with a 30-day calendar, starting backwards from the day of assessment. Cite anchor points, such as special holidays and school or family events, to help the patient recall his or her cigarette use. Then have the patient fill in the number of cigarettes smoked each day for 30 days.
This assessment method appears more reliable than asking an adolescent “how many cigarettes do you smoke per day?”. After the initial time line follow-back assessment, encourage adolescent smokers to keep a daily diary of how many cigarettes they smoke, and monitor the diary at each visit.
Beth, age 15, was admitted overnight to an inpatient psychiatric unit after running away from home and being taken into police custody. Her primary diagnosis was conduct disorder.
At morning rounds, the nurse reported that Beth was very irritable, had threatened the staff, and had been moved to seclusion. During routine examination, the psychiatrist discovered that Beth was a half-pack/day smoker and “really” wanted a cigarette. The psychiatrist told her hospital policy did not allow smoking, but she could try a transdermal nicotine patch (TNP) to help reduce her nicotine withdrawal symptoms. She agreed and received a 14 mg/d nicotine patch.
Beth’s irritability improved substantially with TNP, and she moved back to her regular room within 2 hours without incident.
We have found daily smoking to be a good indicator of nicotine dependence, and anyone who smokes daily would receive significant health benefits from quitting. Hence, any daily smoker who wants to quit, regardless of DSM-IV nicotine dependence status, is a candidate for treatment.
BEHAVIORAL THERAPY
Unlike adults, adolescents usually lack smoking-related medical consequences, such as heart or lung disease. Even so, most adolescent smokers report that they would like to quit but face barriers such as:
- having to inform parents they smoke
- not knowing how to get help for smoking cessation
- lack of transportation for treatment
- lack of third-party reimbursement for smoking cessation treatment.
To help adolescents, we recommend following the U.S. Public Health Service guideline for smoking cessation.14 At least provide and discuss smoking cessation brochures developed specifically for adolescents. For example, one Centers for Disease Control and Prevention brochure describes what symptoms to expect when quitting, how to cope with craving, and other topics (see Related resources).
To manage peer pressure, we counsel teens to let their friends know they are trying to quit so that friends do not smoke in front of them. If that does not work, we ask patients to avoid being around friends who smoke at least for the first 2 weeks and preferably 2 months.
Many states have free telephone quit lines that provide support and advice on how to stop smoking. Several Web sites also are available for smokers (including adolescents) wanting to quit (see Related resources).
PHARMACOLOGIC TREATMENT
For adults, first-line FDA-approved medications for smoking cessation include nicotine replacement therapies (NRT)in transdermal, gum, inhaler, and lozenge forms and sustained-release bupropion. Nortriptyline, doxapine, and clonidine have shown effectiveness for smoking cessation but are not FDA-approved for this indication.15 Selegiline and mecamylamine have shown initial efficacy and are being examined in larger clinical trials.
For adolescents, little is known about what medications might help them stop smoking. Nicotine replacement therapies and bupropion SR have been most explored in adolescent smokers. The effect of psychiatric comorbidity on the quit rate is not well-studied in adolescents.
The transdermal nicotine patch (TNP) has shown modest results in preliminary trials among adolescents. One study found 11% abstinence at 6 weeks,16 and another found a <5% quit rate.17 A third study reported an 18% abstinence rate with a combination of TNP and contingency management therapy.18 Discussion of contingency management and other behavioral therapies is beyond the scope of this article.
A recent study comparing TNP, nicotine gum, and placebo in adolescent smokers found the lowest drop-out rate and highest compliance among the TNP group. Three-month abstinence rates were 17.6% for TNP, 6.5% for nicotine gum, and 2.5% for placebo. The difference between the TNP and placebo groups’ abstinence rates was statistically significant.19
Bupropion SR. In an open-label pilot study, our group treated 16 adolescent smokers weighing >90 lbs with bupropion SR, 150 mg bid. Average age was 18, and two-thirds of patients had ADHD. The endpoint abstinence rate—as measured by self-report and CO levels—was 31%, which is similar to rates reported in adult smokers treated with this dosage of bupropion SR.20
The adolescents did not gain weight during the study, which may be important to this age group. Reported side effects were similar to those in adults, with one adolescent reporting an allergic reaction (urticaria). We are conducting a larger follow-up study using bupropion SR with and without behavioral therapy.
A PRACTICAL CLINICAL APPROACH
Smoking behavior. For treatment, we propose two categories of adolescent smokers: regular (daily) and nonregular (nondaily) (see Algorithm). We recognize that many nondaily smokers smoke frequently and may benefit from aggressive treatment. However, we propose this two-track approach as a starting point because of limited data and medication risks, such as possible seizures with bupropion SR. We suggest:
- using behavioral therapy and patient education as first-line treatment for nonregular adolescent smokers
- using medication and behavioral therapy as first-line treatment for regular smokers and medication as second-line treatment for nonregular smokers who do not respond to behavior therapy/patient education.
Algorithm Suggested smoking cessation approaches for adolescents
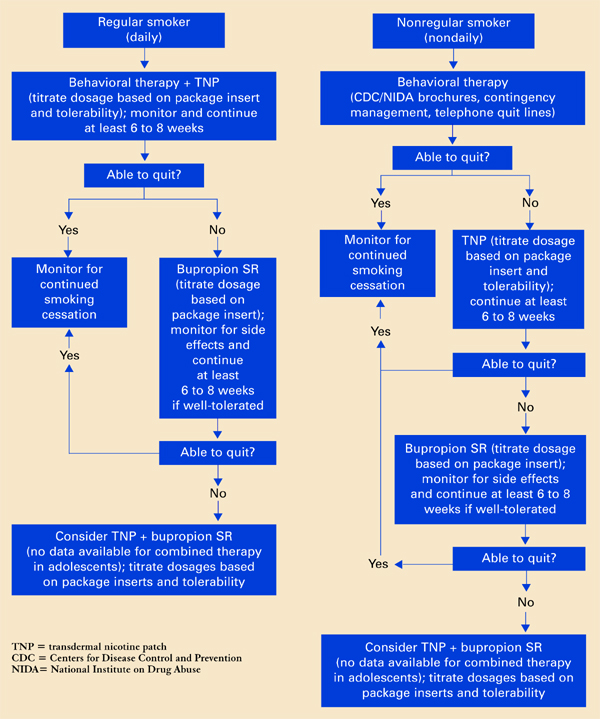
Offer a treatment for at least 6 to 8 weeks before considering a change in therapy. One definition of initial success is no tobacco use in a 7-day period by self-report and biological verification (such as CO levels).
Behavioral therapy is relatively low-risk and helps many adult smokers. Despite a lack of evidence, some sort of behavioral therapy in combination with pharmacologic therapy might also help adolescent smokers.
When adolescents get disheartened by a slip or relapse to smoking, be patient and encourage them to try again. Inform them that smokers often require multiple attempts before they can quit completely.
Medication. Based on the limited published evidence, we consider TNP and bupropion SR first-line medications for adolescent smokers who want to quit.
For adult smokers, clinicians often combine medication and NRT to increase success rates.15 No data suggest that combining TNP and bupropion SR may be more effective than monotherapy in adolescents, but the combination might help those who do not respond to either agent alone.
We recommend starting bupropion SR treatment at least 1 week before the patient’s quit date. Titrate the dosage based on the package insert and patient tolerance.
Start NRT according to package instructions, and titrate dosages based on response:
- increase if the patient reports substantial craving and withdrawal symptoms, such as irritability and anxiety.
- decrease in case of toxicity (such as nausea).
In our experience, adolescent smokers require slightly lower NRT dosages than adults, although this varies among individuals.
- Centers for Disease Control and Prevention. Tobacco Information and Prevention Source (TIPS). www.cdc.gov/tobacco/quit/iquit.htm
- National Institute of Drug Abuse. NIDA for Teens. The Science Behind Drug Abuse. http://teens.drugabuse.gov/index.asp
- Society for Research on Nicotine and Tobacco. www.srnt.org
Drug brand names
- Bupropion SR • Zyban
- Clonidine • Catapres
- Doxapine • Sinequan
- Mecamylamine • Inversine
- Nortriptyline • Pamelor
- Selegiline • Eldepryl
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Upadhyaya HP, Brady KT, Wharton M, Liao J. Psychiatric disorders and cigarette smoking among child and adolescent psychiatry inpatients. Am J Addict 2003;12:144-52.
2. Lasser K, Boyd JW, Woolhandler S, et al. Smoking and mental illness. A population-based prevalence study. JAMA 2000;284(20):2606-10.
3. Johnston LD, O’Malley PM, Bachman JG. Teen smoking continues to decline in 2003, but declines are slowing. Ann Arbor, MI: University of Michigan News and Information Services, Dec. 19, 2003. Available at: www.monitoringthefuture.org/press.html. Accessed 08/13/04.
4. Upadhyaya HP, Deas D, Brady KT, Kruesi M. Cigarette smoking and psychiatric comorbidity in children and adolescents. J Am Acad Child Adolesc Psychiatry 2002;41(11):1294-1305.
5. Molina BS, Pelham WE. Childhood predictors of adolescent substance use in a longitudinal study of children with ADHD. J Abnorm Psychol 2003;112(3):497-507.
6. Pomerleau OF, Downey KK, Stelson FW, Pomerleau CS. Cigarette smoking in adult patients diagnosed with attention-deficit/hyperactivity disorder. J Subst Abuse 1995;7:373-8.
7. Dierker LC, Avenevoli S, Merikangas KR, et al. Association between psychiatric disorders and the progression of tobacco use behaviors. J Am Acad Child Adolesc Psychiatry 2001;40(10):1159-67.
8. Zacny J. Behavioral aspects of alcohol-tobacco interactions. Recent Dev Alcohol 1990;8:205-19.
9. Rohde P, Lewinsohn P, Kahler C, et al. Natural course of alcohol use disorders from adolescence to young adulthood. J Am Acad Child Adolesc Psychiatry 2001;40(1):83-90.
10. Sonntag H, Wittchen HU, Hofler M, et al. Are social fears and DSM-IV social anxiety disorder associated with smoking and nicotine dependence in adolescents and young adults? Eur Psychiatry 2000;15:67-74.
11. Prokhorov AV, Pallonen UE, Fava JL, et al. Measuring nicotine dependence among high-risk adolescent smokers. Addict Behav 1996;21:117-27.
12. McDonald P, Colwell B, Backinger CL, et al. Better practices for youth tobacco cessation: evidence of review panel. Am J Health Behav 2003;27(suppl 2):S144-S158.
13. Sobell LC, Sobell MB, Leo GI, Cancilla A. Reliability of a timeline method: assessing normal drinkers’ reports of recent drinking and a comparative evaluation across several populations. Br J Addict 1988;83(4):393-402.
14. Fiore M, Bailey W, Cohen S. Treating tobacco use and dependence: Clinical practice guideline. Rockville, MD: US Public Health Service, 2000.
15. George TP, O’Malley SS. Current pharmacological treatments for nicotine dependence. Trends Pharmacol Sci 2004;25(1):42-8.
16. Hurt RD, Croghan GA, Beede SD, et al. Nicotine patch therapy in 101 adolescent smokers. Efficacy, withdrawal symptom relief, and carbon monoxide and plasma cotinine levels. Arch Pediatr Adolesc Med 2000;154:31-7.
17. Smith TA, House RF, Jr, Croghan IT, et al. Nicotine patch therapy in adolescent smokers. Pediatrics 1996;98:659-67.
18. Hanson K, Allen S, Jensen S, Hatsukami D. Treatment of adolescent smokers with the nicotine patch. Nicotine Tob Res 2003;5(4):515-26.
19. Moolchan ET. Efficacy of the nicotine patch and gum for the treatment of adolescent tobacco dependence. Scottsdale, AZ: Society for Research on Nicotine and Tobacco Research annual meeting, 2004.
20. Upadhyaya HP, Brady KT, Wang W. Bupropion SR in adolescents with comorbid ADHD and nicotine dependence: a pilot study. J Am Acad Child Adolesc Psychiatry 2004;43(2):199-205.
Many adolescent psychiatric patients who smoke are not getting the help they need to quit. When we asked 120 teen inpatients if they smoked and then checked their charts, we found only 6 of 47 smokers had been diagnosed as nicotine-dependent.1
Adolescents who cannot quit on their own may benefit from smoking cessation therapies. Based on evidence and our experience, we offer a practical approach to treating nicotine dependence in adolescents, using drug and behavioral therapies.
PSYCHIATRIC COMORBIDITY
Psychiatric comorbidity is highly associated with cigarette smoking in adults and adolescents. In the United States:
- 44% of cigarettes smoked are sold to someone with a mental illness.2
- Persons with mental illness are 2.7 times more likely to smoke than are those without mental illness.2
- Most smokers start before age 18,3 and starting before age 13 is linked to psychopathology in later adolescence.4
Table
Smoking likelihood by age and comorbidity among adolescent psychiatric inpatients
| Significant variable | Logistic regression odds ratio | 95% confidence interval | Significance (P value) |
|---|---|---|---|
| Age | 1.30 | 1.03, 1.64 | 0.03 |
| Depressive disorders | 4.02 | 1.267, 12.734 | 0.018 |
| Conduct disorder | 12.96 | 1.678, 100.07 | 0.014 |
| Cannabis use disorder | 24.60 | 3.7, 163.42 | 0.0009 |
| Source: Data from 120 patients admitted to an inpatient child and adolescent psychiatry program. | |||
| Adapted with permission from reference 1. | |||
Disruptive behavior disorders in adolescent smokers include oppositional defiant disorder, conduct disorder, and attention-deficit/hyperactivity disorder (ADHD). Among psychiatric disorders, conduct disorder has the strongest association with smoking in adolescents.1 ADHD is associated with smoking and perhaps with increased difficulty in quitting.5,6
Mood disorders. Major depressive disorders have a strong, consistent, bidirectional association with smoking in the young. Depression may lead to smoking, and smoking to depression.7
Substance use disorders. Alcohol use disorders are strongly associated with smoking among adolescents, and the association is both bidirectional and dosedependent.8 Cannabis use disorder is also associated with cigarette smoking among adolescents (Table).9
Anxiety disorders. Evidence is emerging that anxiety disorders—especially social phobia—may be linked to smoking among adolescents.10
Nicotine withdrawal symptoms—irritability, anxiety, decreased concentration, increased appetite, craving for cigarettes—can mimic those of other psychiatric disorders. Adolescent smokers admitted to locked psychiatric units may experience withdrawal symptoms that require nicotine replacement treatment (Box).
Effect on quit rates. Psychiatric comorbidity may reduce quit rates during smoking cessation treatment.6 When smokers are trying to quit, watch for remission, worsening, or emergence of psychiatric conditions.
ASSESSING ADOLESCENT SMOKING
Adolescents with psychiatric diagnoses can be assessed for nicotine dependence—and vice versa—although accurately gauging their smoking habits is more difficult than in adults. For example:
- Rating scales for nicotine dependence severity—such as the modified Fagerstrom Tolerance Questionnaire11—lack standard cutoff scores for adolescents.
- Unlike adults, many adolescents cannot reliably report use in “packs per day” because the number of cigarettes they smoke varies widely from day to day.
Biological markers commonly used to assess smoking in adults include expired-air carbon monoxide (CO), cotinine (nicotine metabolite), and thiocynate levels. Preliminary evidence indicates that cotinine may be a more sensitive and specific biological marker for smoking among adolescents than CO levels.12 Thiocynate has not been evaluated as a marker for smoking in adolescents.
CO levels typically reflect smoking in the previous few hours, whereas the half-life of cotinine is longer (1 day or more). Also, factors such as environmental pollution or marijuana use can inflate CO levels. Thus, cotinine levels have greater accuracy and specificity, reflecting only the amount of nicotine consumed.
Unfortunately, most laboratories do not measure cotinine levels, and the expired-air CO test (CO Breathalyzer) is relatively expensive for most clinicians. Commercially available single-use cotinine test kits are modestly priced and provide semi-quantitative (a range instead of an exact number) urine cotinine levels. These tests, however, might not be covered by third-party insurers.
Until cotinine testing becomes widely available, we recommend a combination of self-report and expired-air CO level to monitor abstinence.
Self-report monitoring. Most clinicians rely on self-report rate of smoking among adolescents, as no screening assessment has been validated in this age group. As initial prompts, we recommend asking all adolescents if they smoke cigarettes, if they smoke regularly, and if they smoke daily.
We recommend using the “time line follow-back” method13 to monitor the self-reported smoking rate. Begin by providing the patient with a 30-day calendar, starting backwards from the day of assessment. Cite anchor points, such as special holidays and school or family events, to help the patient recall his or her cigarette use. Then have the patient fill in the number of cigarettes smoked each day for 30 days.
This assessment method appears more reliable than asking an adolescent “how many cigarettes do you smoke per day?”. After the initial time line follow-back assessment, encourage adolescent smokers to keep a daily diary of how many cigarettes they smoke, and monitor the diary at each visit.
Beth, age 15, was admitted overnight to an inpatient psychiatric unit after running away from home and being taken into police custody. Her primary diagnosis was conduct disorder.
At morning rounds, the nurse reported that Beth was very irritable, had threatened the staff, and had been moved to seclusion. During routine examination, the psychiatrist discovered that Beth was a half-pack/day smoker and “really” wanted a cigarette. The psychiatrist told her hospital policy did not allow smoking, but she could try a transdermal nicotine patch (TNP) to help reduce her nicotine withdrawal symptoms. She agreed and received a 14 mg/d nicotine patch.
Beth’s irritability improved substantially with TNP, and she moved back to her regular room within 2 hours without incident.
We have found daily smoking to be a good indicator of nicotine dependence, and anyone who smokes daily would receive significant health benefits from quitting. Hence, any daily smoker who wants to quit, regardless of DSM-IV nicotine dependence status, is a candidate for treatment.
BEHAVIORAL THERAPY
Unlike adults, adolescents usually lack smoking-related medical consequences, such as heart or lung disease. Even so, most adolescent smokers report that they would like to quit but face barriers such as:
- having to inform parents they smoke
- not knowing how to get help for smoking cessation
- lack of transportation for treatment
- lack of third-party reimbursement for smoking cessation treatment.
To help adolescents, we recommend following the U.S. Public Health Service guideline for smoking cessation.14 At least provide and discuss smoking cessation brochures developed specifically for adolescents. For example, one Centers for Disease Control and Prevention brochure describes what symptoms to expect when quitting, how to cope with craving, and other topics (see Related resources).
To manage peer pressure, we counsel teens to let their friends know they are trying to quit so that friends do not smoke in front of them. If that does not work, we ask patients to avoid being around friends who smoke at least for the first 2 weeks and preferably 2 months.
Many states have free telephone quit lines that provide support and advice on how to stop smoking. Several Web sites also are available for smokers (including adolescents) wanting to quit (see Related resources).
PHARMACOLOGIC TREATMENT
For adults, first-line FDA-approved medications for smoking cessation include nicotine replacement therapies (NRT)in transdermal, gum, inhaler, and lozenge forms and sustained-release bupropion. Nortriptyline, doxapine, and clonidine have shown effectiveness for smoking cessation but are not FDA-approved for this indication.15 Selegiline and mecamylamine have shown initial efficacy and are being examined in larger clinical trials.
For adolescents, little is known about what medications might help them stop smoking. Nicotine replacement therapies and bupropion SR have been most explored in adolescent smokers. The effect of psychiatric comorbidity on the quit rate is not well-studied in adolescents.
The transdermal nicotine patch (TNP) has shown modest results in preliminary trials among adolescents. One study found 11% abstinence at 6 weeks,16 and another found a <5% quit rate.17 A third study reported an 18% abstinence rate with a combination of TNP and contingency management therapy.18 Discussion of contingency management and other behavioral therapies is beyond the scope of this article.
A recent study comparing TNP, nicotine gum, and placebo in adolescent smokers found the lowest drop-out rate and highest compliance among the TNP group. Three-month abstinence rates were 17.6% for TNP, 6.5% for nicotine gum, and 2.5% for placebo. The difference between the TNP and placebo groups’ abstinence rates was statistically significant.19
Bupropion SR. In an open-label pilot study, our group treated 16 adolescent smokers weighing >90 lbs with bupropion SR, 150 mg bid. Average age was 18, and two-thirds of patients had ADHD. The endpoint abstinence rate—as measured by self-report and CO levels—was 31%, which is similar to rates reported in adult smokers treated with this dosage of bupropion SR.20
The adolescents did not gain weight during the study, which may be important to this age group. Reported side effects were similar to those in adults, with one adolescent reporting an allergic reaction (urticaria). We are conducting a larger follow-up study using bupropion SR with and without behavioral therapy.
A PRACTICAL CLINICAL APPROACH
Smoking behavior. For treatment, we propose two categories of adolescent smokers: regular (daily) and nonregular (nondaily) (see Algorithm). We recognize that many nondaily smokers smoke frequently and may benefit from aggressive treatment. However, we propose this two-track approach as a starting point because of limited data and medication risks, such as possible seizures with bupropion SR. We suggest:
- using behavioral therapy and patient education as first-line treatment for nonregular adolescent smokers
- using medication and behavioral therapy as first-line treatment for regular smokers and medication as second-line treatment for nonregular smokers who do not respond to behavior therapy/patient education.
Algorithm Suggested smoking cessation approaches for adolescents
Offer a treatment for at least 6 to 8 weeks before considering a change in therapy. One definition of initial success is no tobacco use in a 7-day period by self-report and biological verification (such as CO levels).
Behavioral therapy is relatively low-risk and helps many adult smokers. Despite a lack of evidence, some sort of behavioral therapy in combination with pharmacologic therapy might also help adolescent smokers.
When adolescents get disheartened by a slip or relapse to smoking, be patient and encourage them to try again. Inform them that smokers often require multiple attempts before they can quit completely.
Medication. Based on the limited published evidence, we consider TNP and bupropion SR first-line medications for adolescent smokers who want to quit.
For adult smokers, clinicians often combine medication and NRT to increase success rates.15 No data suggest that combining TNP and bupropion SR may be more effective than monotherapy in adolescents, but the combination might help those who do not respond to either agent alone.
We recommend starting bupropion SR treatment at least 1 week before the patient’s quit date. Titrate the dosage based on the package insert and patient tolerance.
Start NRT according to package instructions, and titrate dosages based on response:
- increase if the patient reports substantial craving and withdrawal symptoms, such as irritability and anxiety.
- decrease in case of toxicity (such as nausea).
In our experience, adolescent smokers require slightly lower NRT dosages than adults, although this varies among individuals.
- Centers for Disease Control and Prevention. Tobacco Information and Prevention Source (TIPS). www.cdc.gov/tobacco/quit/iquit.htm
- National Institute of Drug Abuse. NIDA for Teens. The Science Behind Drug Abuse. http://teens.drugabuse.gov/index.asp
- Society for Research on Nicotine and Tobacco. www.srnt.org
Drug brand names
- Bupropion SR • Zyban
- Clonidine • Catapres
- Doxapine • Sinequan
- Mecamylamine • Inversine
- Nortriptyline • Pamelor
- Selegiline • Eldepryl
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Many adolescent psychiatric patients who smoke are not getting the help they need to quit. When we asked 120 teen inpatients if they smoked and then checked their charts, we found only 6 of 47 smokers had been diagnosed as nicotine-dependent.1
Adolescents who cannot quit on their own may benefit from smoking cessation therapies. Based on evidence and our experience, we offer a practical approach to treating nicotine dependence in adolescents, using drug and behavioral therapies.
PSYCHIATRIC COMORBIDITY
Psychiatric comorbidity is highly associated with cigarette smoking in adults and adolescents. In the United States:
- 44% of cigarettes smoked are sold to someone with a mental illness.2
- Persons with mental illness are 2.7 times more likely to smoke than are those without mental illness.2
- Most smokers start before age 18,3 and starting before age 13 is linked to psychopathology in later adolescence.4
Table
Smoking likelihood by age and comorbidity among adolescent psychiatric inpatients
| Significant variable | Logistic regression odds ratio | 95% confidence interval | Significance (P value) |
|---|---|---|---|
| Age | 1.30 | 1.03, 1.64 | 0.03 |
| Depressive disorders | 4.02 | 1.267, 12.734 | 0.018 |
| Conduct disorder | 12.96 | 1.678, 100.07 | 0.014 |
| Cannabis use disorder | 24.60 | 3.7, 163.42 | 0.0009 |
| Source: Data from 120 patients admitted to an inpatient child and adolescent psychiatry program. | |||
| Adapted with permission from reference 1. | |||
Disruptive behavior disorders in adolescent smokers include oppositional defiant disorder, conduct disorder, and attention-deficit/hyperactivity disorder (ADHD). Among psychiatric disorders, conduct disorder has the strongest association with smoking in adolescents.1 ADHD is associated with smoking and perhaps with increased difficulty in quitting.5,6
Mood disorders. Major depressive disorders have a strong, consistent, bidirectional association with smoking in the young. Depression may lead to smoking, and smoking to depression.7
Substance use disorders. Alcohol use disorders are strongly associated with smoking among adolescents, and the association is both bidirectional and dosedependent.8 Cannabis use disorder is also associated with cigarette smoking among adolescents (Table).9
Anxiety disorders. Evidence is emerging that anxiety disorders—especially social phobia—may be linked to smoking among adolescents.10
Nicotine withdrawal symptoms—irritability, anxiety, decreased concentration, increased appetite, craving for cigarettes—can mimic those of other psychiatric disorders. Adolescent smokers admitted to locked psychiatric units may experience withdrawal symptoms that require nicotine replacement treatment (Box).
Effect on quit rates. Psychiatric comorbidity may reduce quit rates during smoking cessation treatment.6 When smokers are trying to quit, watch for remission, worsening, or emergence of psychiatric conditions.
ASSESSING ADOLESCENT SMOKING
Adolescents with psychiatric diagnoses can be assessed for nicotine dependence—and vice versa—although accurately gauging their smoking habits is more difficult than in adults. For example:
- Rating scales for nicotine dependence severity—such as the modified Fagerstrom Tolerance Questionnaire11—lack standard cutoff scores for adolescents.
- Unlike adults, many adolescents cannot reliably report use in “packs per day” because the number of cigarettes they smoke varies widely from day to day.
Biological markers commonly used to assess smoking in adults include expired-air carbon monoxide (CO), cotinine (nicotine metabolite), and thiocynate levels. Preliminary evidence indicates that cotinine may be a more sensitive and specific biological marker for smoking among adolescents than CO levels.12 Thiocynate has not been evaluated as a marker for smoking in adolescents.
CO levels typically reflect smoking in the previous few hours, whereas the half-life of cotinine is longer (1 day or more). Also, factors such as environmental pollution or marijuana use can inflate CO levels. Thus, cotinine levels have greater accuracy and specificity, reflecting only the amount of nicotine consumed.
Unfortunately, most laboratories do not measure cotinine levels, and the expired-air CO test (CO Breathalyzer) is relatively expensive for most clinicians. Commercially available single-use cotinine test kits are modestly priced and provide semi-quantitative (a range instead of an exact number) urine cotinine levels. These tests, however, might not be covered by third-party insurers.
Until cotinine testing becomes widely available, we recommend a combination of self-report and expired-air CO level to monitor abstinence.
Self-report monitoring. Most clinicians rely on self-report rate of smoking among adolescents, as no screening assessment has been validated in this age group. As initial prompts, we recommend asking all adolescents if they smoke cigarettes, if they smoke regularly, and if they smoke daily.
We recommend using the “time line follow-back” method13 to monitor the self-reported smoking rate. Begin by providing the patient with a 30-day calendar, starting backwards from the day of assessment. Cite anchor points, such as special holidays and school or family events, to help the patient recall his or her cigarette use. Then have the patient fill in the number of cigarettes smoked each day for 30 days.
This assessment method appears more reliable than asking an adolescent “how many cigarettes do you smoke per day?”. After the initial time line follow-back assessment, encourage adolescent smokers to keep a daily diary of how many cigarettes they smoke, and monitor the diary at each visit.
Beth, age 15, was admitted overnight to an inpatient psychiatric unit after running away from home and being taken into police custody. Her primary diagnosis was conduct disorder.
At morning rounds, the nurse reported that Beth was very irritable, had threatened the staff, and had been moved to seclusion. During routine examination, the psychiatrist discovered that Beth was a half-pack/day smoker and “really” wanted a cigarette. The psychiatrist told her hospital policy did not allow smoking, but she could try a transdermal nicotine patch (TNP) to help reduce her nicotine withdrawal symptoms. She agreed and received a 14 mg/d nicotine patch.
Beth’s irritability improved substantially with TNP, and she moved back to her regular room within 2 hours without incident.
We have found daily smoking to be a good indicator of nicotine dependence, and anyone who smokes daily would receive significant health benefits from quitting. Hence, any daily smoker who wants to quit, regardless of DSM-IV nicotine dependence status, is a candidate for treatment.
BEHAVIORAL THERAPY
Unlike adults, adolescents usually lack smoking-related medical consequences, such as heart or lung disease. Even so, most adolescent smokers report that they would like to quit but face barriers such as:
- having to inform parents they smoke
- not knowing how to get help for smoking cessation
- lack of transportation for treatment
- lack of third-party reimbursement for smoking cessation treatment.
To help adolescents, we recommend following the U.S. Public Health Service guideline for smoking cessation.14 At least provide and discuss smoking cessation brochures developed specifically for adolescents. For example, one Centers for Disease Control and Prevention brochure describes what symptoms to expect when quitting, how to cope with craving, and other topics (see Related resources).
To manage peer pressure, we counsel teens to let their friends know they are trying to quit so that friends do not smoke in front of them. If that does not work, we ask patients to avoid being around friends who smoke at least for the first 2 weeks and preferably 2 months.
Many states have free telephone quit lines that provide support and advice on how to stop smoking. Several Web sites also are available for smokers (including adolescents) wanting to quit (see Related resources).
PHARMACOLOGIC TREATMENT
For adults, first-line FDA-approved medications for smoking cessation include nicotine replacement therapies (NRT)in transdermal, gum, inhaler, and lozenge forms and sustained-release bupropion. Nortriptyline, doxapine, and clonidine have shown effectiveness for smoking cessation but are not FDA-approved for this indication.15 Selegiline and mecamylamine have shown initial efficacy and are being examined in larger clinical trials.
For adolescents, little is known about what medications might help them stop smoking. Nicotine replacement therapies and bupropion SR have been most explored in adolescent smokers. The effect of psychiatric comorbidity on the quit rate is not well-studied in adolescents.
The transdermal nicotine patch (TNP) has shown modest results in preliminary trials among adolescents. One study found 11% abstinence at 6 weeks,16 and another found a <5% quit rate.17 A third study reported an 18% abstinence rate with a combination of TNP and contingency management therapy.18 Discussion of contingency management and other behavioral therapies is beyond the scope of this article.
A recent study comparing TNP, nicotine gum, and placebo in adolescent smokers found the lowest drop-out rate and highest compliance among the TNP group. Three-month abstinence rates were 17.6% for TNP, 6.5% for nicotine gum, and 2.5% for placebo. The difference between the TNP and placebo groups’ abstinence rates was statistically significant.19
Bupropion SR. In an open-label pilot study, our group treated 16 adolescent smokers weighing >90 lbs with bupropion SR, 150 mg bid. Average age was 18, and two-thirds of patients had ADHD. The endpoint abstinence rate—as measured by self-report and CO levels—was 31%, which is similar to rates reported in adult smokers treated with this dosage of bupropion SR.20
The adolescents did not gain weight during the study, which may be important to this age group. Reported side effects were similar to those in adults, with one adolescent reporting an allergic reaction (urticaria). We are conducting a larger follow-up study using bupropion SR with and without behavioral therapy.
A PRACTICAL CLINICAL APPROACH
Smoking behavior. For treatment, we propose two categories of adolescent smokers: regular (daily) and nonregular (nondaily) (see Algorithm). We recognize that many nondaily smokers smoke frequently and may benefit from aggressive treatment. However, we propose this two-track approach as a starting point because of limited data and medication risks, such as possible seizures with bupropion SR. We suggest:
- using behavioral therapy and patient education as first-line treatment for nonregular adolescent smokers
- using medication and behavioral therapy as first-line treatment for regular smokers and medication as second-line treatment for nonregular smokers who do not respond to behavior therapy/patient education.
Algorithm Suggested smoking cessation approaches for adolescents
Offer a treatment for at least 6 to 8 weeks before considering a change in therapy. One definition of initial success is no tobacco use in a 7-day period by self-report and biological verification (such as CO levels).
Behavioral therapy is relatively low-risk and helps many adult smokers. Despite a lack of evidence, some sort of behavioral therapy in combination with pharmacologic therapy might also help adolescent smokers.
When adolescents get disheartened by a slip or relapse to smoking, be patient and encourage them to try again. Inform them that smokers often require multiple attempts before they can quit completely.
Medication. Based on the limited published evidence, we consider TNP and bupropion SR first-line medications for adolescent smokers who want to quit.
For adult smokers, clinicians often combine medication and NRT to increase success rates.15 No data suggest that combining TNP and bupropion SR may be more effective than monotherapy in adolescents, but the combination might help those who do not respond to either agent alone.
We recommend starting bupropion SR treatment at least 1 week before the patient’s quit date. Titrate the dosage based on the package insert and patient tolerance.
Start NRT according to package instructions, and titrate dosages based on response:
- increase if the patient reports substantial craving and withdrawal symptoms, such as irritability and anxiety.
- decrease in case of toxicity (such as nausea).
In our experience, adolescent smokers require slightly lower NRT dosages than adults, although this varies among individuals.
- Centers for Disease Control and Prevention. Tobacco Information and Prevention Source (TIPS). www.cdc.gov/tobacco/quit/iquit.htm
- National Institute of Drug Abuse. NIDA for Teens. The Science Behind Drug Abuse. http://teens.drugabuse.gov/index.asp
- Society for Research on Nicotine and Tobacco. www.srnt.org
Drug brand names
- Bupropion SR • Zyban
- Clonidine • Catapres
- Doxapine • Sinequan
- Mecamylamine • Inversine
- Nortriptyline • Pamelor
- Selegiline • Eldepryl
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Upadhyaya HP, Brady KT, Wharton M, Liao J. Psychiatric disorders and cigarette smoking among child and adolescent psychiatry inpatients. Am J Addict 2003;12:144-52.
2. Lasser K, Boyd JW, Woolhandler S, et al. Smoking and mental illness. A population-based prevalence study. JAMA 2000;284(20):2606-10.
3. Johnston LD, O’Malley PM, Bachman JG. Teen smoking continues to decline in 2003, but declines are slowing. Ann Arbor, MI: University of Michigan News and Information Services, Dec. 19, 2003. Available at: www.monitoringthefuture.org/press.html. Accessed 08/13/04.
4. Upadhyaya HP, Deas D, Brady KT, Kruesi M. Cigarette smoking and psychiatric comorbidity in children and adolescents. J Am Acad Child Adolesc Psychiatry 2002;41(11):1294-1305.
5. Molina BS, Pelham WE. Childhood predictors of adolescent substance use in a longitudinal study of children with ADHD. J Abnorm Psychol 2003;112(3):497-507.
6. Pomerleau OF, Downey KK, Stelson FW, Pomerleau CS. Cigarette smoking in adult patients diagnosed with attention-deficit/hyperactivity disorder. J Subst Abuse 1995;7:373-8.
7. Dierker LC, Avenevoli S, Merikangas KR, et al. Association between psychiatric disorders and the progression of tobacco use behaviors. J Am Acad Child Adolesc Psychiatry 2001;40(10):1159-67.
8. Zacny J. Behavioral aspects of alcohol-tobacco interactions. Recent Dev Alcohol 1990;8:205-19.
9. Rohde P, Lewinsohn P, Kahler C, et al. Natural course of alcohol use disorders from adolescence to young adulthood. J Am Acad Child Adolesc Psychiatry 2001;40(1):83-90.
10. Sonntag H, Wittchen HU, Hofler M, et al. Are social fears and DSM-IV social anxiety disorder associated with smoking and nicotine dependence in adolescents and young adults? Eur Psychiatry 2000;15:67-74.
11. Prokhorov AV, Pallonen UE, Fava JL, et al. Measuring nicotine dependence among high-risk adolescent smokers. Addict Behav 1996;21:117-27.
12. McDonald P, Colwell B, Backinger CL, et al. Better practices for youth tobacco cessation: evidence of review panel. Am J Health Behav 2003;27(suppl 2):S144-S158.
13. Sobell LC, Sobell MB, Leo GI, Cancilla A. Reliability of a timeline method: assessing normal drinkers’ reports of recent drinking and a comparative evaluation across several populations. Br J Addict 1988;83(4):393-402.
14. Fiore M, Bailey W, Cohen S. Treating tobacco use and dependence: Clinical practice guideline. Rockville, MD: US Public Health Service, 2000.
15. George TP, O’Malley SS. Current pharmacological treatments for nicotine dependence. Trends Pharmacol Sci 2004;25(1):42-8.
16. Hurt RD, Croghan GA, Beede SD, et al. Nicotine patch therapy in 101 adolescent smokers. Efficacy, withdrawal symptom relief, and carbon monoxide and plasma cotinine levels. Arch Pediatr Adolesc Med 2000;154:31-7.
17. Smith TA, House RF, Jr, Croghan IT, et al. Nicotine patch therapy in adolescent smokers. Pediatrics 1996;98:659-67.
18. Hanson K, Allen S, Jensen S, Hatsukami D. Treatment of adolescent smokers with the nicotine patch. Nicotine Tob Res 2003;5(4):515-26.
19. Moolchan ET. Efficacy of the nicotine patch and gum for the treatment of adolescent tobacco dependence. Scottsdale, AZ: Society for Research on Nicotine and Tobacco Research annual meeting, 2004.
20. Upadhyaya HP, Brady KT, Wang W. Bupropion SR in adolescents with comorbid ADHD and nicotine dependence: a pilot study. J Am Acad Child Adolesc Psychiatry 2004;43(2):199-205.
1. Upadhyaya HP, Brady KT, Wharton M, Liao J. Psychiatric disorders and cigarette smoking among child and adolescent psychiatry inpatients. Am J Addict 2003;12:144-52.
2. Lasser K, Boyd JW, Woolhandler S, et al. Smoking and mental illness. A population-based prevalence study. JAMA 2000;284(20):2606-10.
3. Johnston LD, O’Malley PM, Bachman JG. Teen smoking continues to decline in 2003, but declines are slowing. Ann Arbor, MI: University of Michigan News and Information Services, Dec. 19, 2003. Available at: www.monitoringthefuture.org/press.html. Accessed 08/13/04.
4. Upadhyaya HP, Deas D, Brady KT, Kruesi M. Cigarette smoking and psychiatric comorbidity in children and adolescents. J Am Acad Child Adolesc Psychiatry 2002;41(11):1294-1305.
5. Molina BS, Pelham WE. Childhood predictors of adolescent substance use in a longitudinal study of children with ADHD. J Abnorm Psychol 2003;112(3):497-507.
6. Pomerleau OF, Downey KK, Stelson FW, Pomerleau CS. Cigarette smoking in adult patients diagnosed with attention-deficit/hyperactivity disorder. J Subst Abuse 1995;7:373-8.
7. Dierker LC, Avenevoli S, Merikangas KR, et al. Association between psychiatric disorders and the progression of tobacco use behaviors. J Am Acad Child Adolesc Psychiatry 2001;40(10):1159-67.
8. Zacny J. Behavioral aspects of alcohol-tobacco interactions. Recent Dev Alcohol 1990;8:205-19.
9. Rohde P, Lewinsohn P, Kahler C, et al. Natural course of alcohol use disorders from adolescence to young adulthood. J Am Acad Child Adolesc Psychiatry 2001;40(1):83-90.
10. Sonntag H, Wittchen HU, Hofler M, et al. Are social fears and DSM-IV social anxiety disorder associated with smoking and nicotine dependence in adolescents and young adults? Eur Psychiatry 2000;15:67-74.
11. Prokhorov AV, Pallonen UE, Fava JL, et al. Measuring nicotine dependence among high-risk adolescent smokers. Addict Behav 1996;21:117-27.
12. McDonald P, Colwell B, Backinger CL, et al. Better practices for youth tobacco cessation: evidence of review panel. Am J Health Behav 2003;27(suppl 2):S144-S158.
13. Sobell LC, Sobell MB, Leo GI, Cancilla A. Reliability of a timeline method: assessing normal drinkers’ reports of recent drinking and a comparative evaluation across several populations. Br J Addict 1988;83(4):393-402.
14. Fiore M, Bailey W, Cohen S. Treating tobacco use and dependence: Clinical practice guideline. Rockville, MD: US Public Health Service, 2000.
15. George TP, O’Malley SS. Current pharmacological treatments for nicotine dependence. Trends Pharmacol Sci 2004;25(1):42-8.
16. Hurt RD, Croghan GA, Beede SD, et al. Nicotine patch therapy in 101 adolescent smokers. Efficacy, withdrawal symptom relief, and carbon monoxide and plasma cotinine levels. Arch Pediatr Adolesc Med 2000;154:31-7.
17. Smith TA, House RF, Jr, Croghan IT, et al. Nicotine patch therapy in adolescent smokers. Pediatrics 1996;98:659-67.
18. Hanson K, Allen S, Jensen S, Hatsukami D. Treatment of adolescent smokers with the nicotine patch. Nicotine Tob Res 2003;5(4):515-26.
19. Moolchan ET. Efficacy of the nicotine patch and gum for the treatment of adolescent tobacco dependence. Scottsdale, AZ: Society for Research on Nicotine and Tobacco Research annual meeting, 2004.
20. Upadhyaya HP, Brady KT, Wang W. Bupropion SR in adolescents with comorbid ADHD and nicotine dependence: a pilot study. J Am Acad Child Adolesc Psychiatry 2004;43(2):199-205.
Acute bipolar mania: Aggressive initial dosing provides faster symptom relief
Evidence on aggressive initial dosing of mood stabilizers and antipsychotics is changing the way acute bipolar mania is treated. To help you apply this information, we searched the literature and meeting abstracts for aggressive strategies tested to date. This article discusses how to:
- identify patients who may benefit from loading or aggressive initial dosing
- calculate mood stabilizer dosages
- dose each atypical antipsychotic
- manage potential antipsychotic side effects during maintenance therapy.
PATIENT SELECTION
Rapidly achieving therapeutic blood levels relieves patient suffering faster than standard titration. It quickly calms the hyperactivity, impulsivity, tension, hostility, and uncooperativeness that distress patients and increase the risk of harm to themselves and others.
The challenge of using higher dosages is to minimize side effects. Loading is not a one-size-fits-all approach, as antimanic drugs’ unique pharmacokinetic and pharmacotherapeutic properties influence how each agent is used.
An interesting body of literature advocates using mood stabilizers plus antipsychotics to treat mania, suggesting greater efficacy than with either agent alone.16 This strategy raises important questions, such as:
- Can two drugs be loaded simultaneously?
- Can patients taking mood stabilizers be treated with antipsychotic loading, and can those taking antipsychotics receive loading dosages of mood stabilizers?
- Would “double loading” improve bipolar mania treatment?
Answers to these questions are needed because of increased demands on clinicians to control hospital costs by rapidly and effectively treating patients with bipolar mania.
Loading doses cannot be standardized but are calculated by multiplying target steady-state plasma concentration by volume of distribution. We suggest aggressive initial schedules for divalproex sodium and atypical antipsychotics in this article with the understanding that practitioners will adjust them based on each patient’s tolerance and response.
Hospitalization. Patients with acute bipolar mania should be supervised closely in the hospital during loading or aggressive initial dosing. Monitor for cardiovascular changes, neurologic disturbances, sensorium changes, and response.
Precautions. Not all patients are candidates for aggressive initial dosing. Contraindications include age <18 or >65 years, pregnancy, breast-feeding, medical illness, and known sensitivity to the medication being given.
Higher-than-usual dosing increases the risk of excessive drug concentrations in sensitive individuals—such as those with a history of sensitivity to lower dosages of similar medications—and toxic levels of drugs with long half-lives can persist. When in doubt, consider giving a smaller amount of the loading dose early in the day, followed later by a larger amount.
DRUG SELECTION
Loading and aggressive initial dosing strategies for bipolar mania were first advanced for divalproex sodium.1 Investigators then examined loading strategies for lithium and carbamazepine, as well as the antipsychotics olanzapine, risperidone, quetiapine, ziprasidone, and aripiprazole, which are known to have antimanic properties.
Olanzapine, quetiapine, and risperidone are FDA-approved for short-term treatment of acute manic episodes associated with bipolar I disorder, and similar indications were being considered for aripiprazole and ziprasidone as this article went to press. We discuss evidence on loading and aggressive initial dosing strategies for each agent.
No studies have compared one loading strategy with another. Thus, when we choose drugs for loading, we consider what each patient needs, available formulations, tolerability, and efficacy for long-term stabilization and maintenance treatment.
LITHIUM
Lithium loading targets the therapeutic range (<1.4 mEq/L), without crossing the toxic threshold (>1.5 mEq/L). Lithium loading has shown antimanic effects, although using >30 mg/kg/d causes severe nausea and vomiting.
Moscovich et al2 reported a case series of 9 adults with acute mania who received lithium loading dosages of 4,050 mg/d. Patients tolerated lithium well at plasma drug levels of approximately 1.2 to 1.4 mEq/L. Their manic symptoms declined significantly within 4 to 5 days, as measured by Clinical Global Impression (CGI) severity of illness, Biegel-Murphy Mania State Rating Scale, and Brief Psychiatric Rating Scale scores.
Table 1
How to calculate divalproex loading for acute bipolar mania*
| Days 1 and 2 |
| Patient weight in pounds x 15 = dosage (mg/d) (Example: 150 lbs x 15 = 1,750 mg/d) |
| Days 3 to 10 |
| Patient weight in pounds x 10 = dosage (mg/d) (Example: 150 lbs. x 10 = 1,500 mg/d) |
| To avoid splitting tablets, make dosage divisible by 125 (round up for young adults, round down for older adults). Divide into bid or tid doses for improved tolerability. |
| Days 4 and 7 |
| Draw blood to monitor valproic acid levels and for other values such as liver function studies. |
| * For use with oral divalproex sodium (delayed-release or extended-release formulations). Do not use valproic acid preparations, as loading is unlikely to be well tolerated. |
| Source: Reference 5 |
Kook et al3 attempted a 30-mg/kg loading dose of slow-release lithium carbonate in 38 patients to evaluate the safety of achieving a therapeutic level in 12 hours. No patient experienced adverse effects during loading or in the 12 hours after loading was completed.
Patients who develop a manic episode while taking lithium pose a therapeutic dilemma. If they stop taking lithium—especially abruptly—manic symptoms can return in as few as 2 days. For these patients, consider increasing the usual dosage by 50% to 100% on the first day of mania treatment,4 then continue a dosage that maintains efficacy and achieves a therapeutic blood level.
To avoid GI upset, avoid any single dose of >1,500 mg; if a greater dosage is needed, divide it for improved tolerance. Obtain lithium blood levels at baseline, after 4 days, and thereafter as clinically relevant to monitor for drug interactions that may affect serum levels.
Side effects—GI irritation, tremor, muscle weakness, thirst, polyuria—are common in the first week, and weight gain may occur after prolonged treatment.
DIVALPROEX
In a study by Keck et al1 of 19 adults with acute bipolar mania, aggressive initial dosing of divalproex sodium, 20 mg/kg/d, was well-tolerated and shortened hospitalization. Side effects including sedation and nausea occurred at a rate similar to that seen with more-gradual titration.
To calculate the initial dose, the authors used a conversion factor of 20 mg/kg/d or added a zero to the patient’s weight in pounds. This amount was given in single or divided doses the first day, then continued for 4 to 7 days. Blood levels were measured on day 4, and all 15 patients who completed the study achieved serum valproate levels >50 mcg/mL.
In a double-blind study by Hirschfeld et al,5 59 adults with Young Mania Rating Scale (YMRS) scores >14 were randomly assigned to receive divalproex oral loading; divalproex nonloading (250 mg tid on days 1 and 2, followed by standard titration on days 3 to 10); or lithium carbonate (300 mg tid, followed by standard titration on days 3 to 10).
Divalproex loading—30 mg/kg/d on days 1 and 2, followed by 20 mg/kg on days 3 to 10— yielded valproate levels >50 mcg/mL in 84% of patients by day 3. Rapid loading appeared to increase antimanic efficacy, as measured by YMRS endpoint scores, without increasing adverse effects. For Table 1, we converted this study’s metric values to patient weight in pounds.
Side effects. Divalproex can inhibit metabolism of other drugs (including anticonvulsants such as lamotrigine) and increase their blood levels. Side effects such as sedation, alopecia, abdominal pain, diarrhea, and tremor may require observation and treatment. Pancreatitis, hepatitis, and allergic reactions are rare but may require discontinuation.
Recommendation. Aggressive initial dosing and loading for patients with acute mania has been reported with enteric-coated delayed-release and extended-release6 divalproex sodium tablets. With identical dosages, the extended-release form produces 11% lower serum levels than the delayed-release form.7
Aggressive dosing of oral valproic acid preparations is not recommended and is likely to be poorly tolerated.8 A pilot study evaluating IV valproate loading in acute mania found no changes in mania in the 2 hours that followed loading.9
CARBAMAZEPINE
Carbamazepine is used off-label to treat mania and mixed phases of bipolar disorder.4,9,10 Excessive absorption rates are associated with dizziness, ataxia, and nausea. Side effects may occur when the plasma level is therapeutic for epilepsy (4 to 12 mcg/mL).11
In mania, the relationship between carbamazepine levels and clinical response is not always clear. Lerer et al12 found a correlation between acute mania response and a serum level of 8.8 mcg/mL (range 4.7 to 14.0 mcg/mL).
Patients with acute mania may require 600 to 1,600 mg/d. Carbamazepine is available in immediate-release and controlled-release preparations. Because generic preparations might not be bio-equivalent,13 use the same formulation throughout treatment to maintain consistent serum levels.
Enzymatic auto-induction—in which carbamazepine gradually increases the activity of its metabolic enzyme—is likely to occur at 3 to 5 weeks of administration, often after hospital discharge. An aggressive initial rescue adjustment can be used if a patient develops mania after having been stabilized on carbamazepine for >6 weeks. Because these patients are past the point of auto-induction, a target blood level of 8.8 mcg/mL can be used and the dosage adjusted proportionally.
For example, if mania recurs in a patient who is stabilized on carbamazepine, 400 mg/d (plasma level 4.5 mcg/mL), carbamazepine can be loaded up to 800 mg/d. Measure the plasma level within 4 days; the target level would be 9.0 mcg/mL (2 times 4.5 mcg/mL), which closely approximates steady state and sets up a ratio to reach the target level of 8.8 mcg/mL.
Side effects include ataxia, diplopia, and dizziness. Complete blood counts, liver function studies, plasma levels, and serum chemistries require regular monitoring.
Recommendation. Carbamazepine is not recommended for oral loading in patients who have never taken it or for those with hyponatremia, hepatic dysfunction, or history of intolerance or agranulocytosis.
OLANZAPINE
Olanzapine has pharmacologic properties favorable for loading.4 The recommended dosage for acute mania is 15 mg/d with standard titration; Karagianis et al14 showed that initial loading doses of >20 mg/d resulted in good control of agitated patients with psychosis. Side effects were uncommon, with sedation occurring in 14% of patients in this case series. The loading dose reduced agitation more effectively than did dosages <20 mg/d given to similar patients.
A multicenter study of 148 acutely agitated inpatients with a variety of psychiatric disorders compared olanzapine rapid initial dose escalation with usual clinical practice.15 Mean aggressive dosages were 28.8 mg/d on day 1, 30.3 mg/d on day 2, and 16.1 mg/d on day 5. Usual-practice dosages were 10 mg/d, plus lorazepam, 0 to 4 mg/d for the first 2 days and 0 to 2 mg/d on days 3 to 4. Based on Positive and Negative Syndrome Scale excited component subscale scores, olanzapine loading controlled agitation more effectively than did usual practice, with similar side-effect rates.
IM olanzapine or the orally dissolving form are bioequivalent to the tablets and may be used for acute agitation associated with bipolar mania in certain clinical settings.14
Table 2
Suggested antipsychotic loading for acute bipolar mania and mixed states*
| Drug | Day(s) | Aggressive initial dosing schedule | Comment |
|---|---|---|---|
| Aripiprazole24,25 | 1 2 to 3 4+ | 30 mg once daily with food 30 mg/d with food Reduce dosage by 10 to 15 mg/d, based on tolerance and response | Nausea and vomiting may occur in first few days; adjust dosage based on tolerance and response |
| Olanzapine14,15 | 1 and 2 3 and 4 5 to 10 | 40 mg in single or divided dosage 20 to 30 mg at bedtime 15 mg once daily (may reduce to 5, 7.5, or 10 mg/d) | Adjust dosage based on tolerance and response; oral or IM formulations may be used |
| Quetiapine19,21 | 1 2 and 3 4 to 10 | 100 mg upon admission and 100 mg at bedtime 100 mg bid (or tid to qid) plus 100 to 200 mg at bedtime 200 mg bid plus 200 to 300 mg at bedtime; may adjust to 400 to 800 mg/d) | Adjust dosage based on tolerance and response |
| Risperidone16,18 | 1 2 3 and 4 | 3 mg in single or divided dosage 4 mg in single or divided dosage 5 mg in single or divided dosage | Adjust dosage by 1 mg up or down, based on tolerance and response; use tablet, rapid-dissolving tablet, or liquid form, but not long-acting IM form |
| Ziprasidone22,23 | 1 2 | 20 mg IM in single dose (may repeat for severe agitation) or 40 mg po bid with food 60 to 80 mg po bid with food | Adjust dosage based on tolerance and response |
| * For hospitalized or partially hospitalized patients, ages18. Not recommended for patients who are pregnant, breastfeeding, medically ill, age >65, or with known sensitivity to the antipsychotic being given. | |||
RISPERIDONE
Sachs et al16 studied 156 inpatients who developed an acute manic or mixed episode while receiving lithium or divalproex. These patients were randomly assigned to begin adjunctive risperidone, 2 mg/d, haloperidol, or placebo. Dosing was flexible, increasing or decreasing by 1 mg/d. Risperidone’s mean modal dosage was 3.8 mg/d across 3 weeks, with mean exposure of 17 days. Risperidone plus a mood stabilizer was more effective than a mood stabilizer alone, and the combination provided rapid, well-tolerated control of manic symptoms.
In a double-blind trial, Hirschfeld et al17 randomly assigned 279 patients with acute bipolar mania to risperidone, 1 to 6 mg/d, or placebo for 3 weeks. As early as day 3, YMRS scores were reduced significantly more with risperidone than with placebo. Somnolence was the most common side effect, and mean modal dosage was 4.1 mg/d.
Table 3
Screening schedule for antipsychotic side effects during bipolar maintenance treatment
| Baseline | |
| Side effect | Recommended screening |
| Weight gain | Weight and body mass index (BMI) monthly for first 3 months; waist circumference |
| Hypertension | Blood pressure |
| Hyperglycemia | Fasting plasma glucose, with glycosylated hemoglobin (Hb A 1c ) if hyperglycemia is detected |
| Hyperlipidemia | Fasting lipid profile |
| Tardive dyskinesia | Abnormal Involuntary Movement Scale (AIMS) or other screen |
| Ophthalmic changes | Ophthalmologic examination for patients taking quetiapine and for all with diabetes mellitus |
| Follow-up schedules | |
| 3 months | |
| Weight and BMI | |
| Blood pressure | |
| Fasting plasma glucose, with Hb A 1c if hyperglycemia is detected; Hb A 1c values may be used to measure interval changes in glucose tolerance | |
| Fasting lipid profile | |
| 6 months | |
| AIMS or other tardive dyskinesia screen | |
| Ophthalmologic examination | |
| Source: Adapted from reference 26. | |
In a multicenter, randomized, double-blind, placebo-controlled study of patients with acute bipolar mania, Khanna et al18 assigned patients to receive risperidone monotherapy (mean modal dosage 5.6 mg/d) or placebo for 3 weeks. Mania scores of patients receiving risperidone were significantly lower at weeks 1 and 2, compared with the placebo group. Risperidone was well-tolerated, with no unexpected adverse events.
Recommendation. Because of a risk of extrapyramidal symptoms (EPS) and orthostatic hypotension, initial risperidone loading dosages >4 mg on day 1 are not recommended.
QUETIAPINE
Quetiapine has shown antimanic efficacy as monotherapy and as adjunctive therapy to mood stabilizers.19,20 The effective dosage was a mean 600 mg/d (range 400 to 800 mg/d) in monotherapy and adjunctive treatment. These studies achieved 400 mg/d within the first 4 days (100 mg on day 1, 200 mg on day 2, 300 mg on day 3, and 400 mg on day 4).
These data combined with revised prescribing information suggest aggressive initial dose escalation of quetiapine within the first 4 days for selected patients. A titration study in patients with schizophrenia used a more-rapid escalation rate of 400 mg within 2 days.21 Dizziness, orthostatic hypotension, and sedation were not more frequent in this high-dose group than in the two lower-dose titration groups. In our experience, 200 to 400 mg can be given the first day of treatment.
ZIPRASIDONE
In a randomized, double-blind, controlled trial, Keck et al22 assigned 210 patients with a manic or mixed bipolar episode to 3 weeks of ziprasidone, 40 to 80 mg bid, or placebo. Ziprasidone produced rapid, sustained improvement in manic symptoms on all primary and most secondary efficacy measures, such as the YMRS and CGI.
Significant improvements seen within 2 days were maintained. Ziprasidone was well tolerated and was associated with a low EPS rate; neither weight gain nor clinically significant changes in vital signs were seen.
IM ziprasidone, which is approved for use in schizophrenia, may also have efficacy in bipolar mania.23 The recommended dose of 20 mg IM is equivalent to 120 to 160 mg orally, so a single injection may reach the target antimanic dosage.
Recommendation. Ziprasidone could be an option for aggressive initial dosing for a patient who has previously received ziprasidone IM and is not at risk for QTc prolongation.
ARIPIPRAZOLE
In a randomized controlled trial, Keck et al24 assigned 262 patients with acute bipolar mania or mixed states to aripiprazole, 30 mg/d, or placebo for 3 weeks. By day 4, manic symptoms were improved significantly more in patients receiving aripiprazole, and discontinuation rates were similar.
Similarly, in a randomized, controlled multi-center study, Sachs et al25 used 30 mg/d in 272 patients with bipolar mania or mixed states. Compared with placebo, aripiprazole produced significant improvement by day 4, with similar discontinuation rates.
Recommendation. Aggressive initial dosing of aripiprazole could be useful for a patient who does not require an IM or rapidly dissolvable medication.
Table 4
Suggested response to metabolic changes during bipolar maintenance therapy
| Metabolic change | Therapeutic action |
|---|---|
| ≥5% increase in total body weight | Consider weight-reduction strategies or medication adjustment |
| Fasting glucose: ≥126 mg/mL ≥300 mg/mL or ≤60 mg/mL | Consider evaluation for diabetes mellitus Seek immediate consultation |
| Total cholesterol ≥200 mg/dL or triglycerides ≥165 mg/dL | Consider lipid-lowering with dietary and/or medication changes |
| Source: Adapted from reference 26. | |
MAINTENANCE THERAPY
Ideally, if a medication stabilizes a patient’s acute bipolar mania, that medication is continued for further stabilization and maintenance. Aggressive initial dosing befits this approach because it establishes a therapeutic blood level and usually reveals any side effects within days. Moreover, patients often prefer to continue the medication that provided relief when they felt most distressed.
Weight gain. Long-term use of atypical antipsychotics may be associated with weight gain, dyslipidemia, and the development of metabolic syndromes and diabetes mellitus. Weight gain risk may be further elevated in patients taking both antipsychotics and lithium or valproic acid.26 When atypical antipsychotics are used for bipolar maintenance therapy, the American Diabetes Association and American Psychiatric Association recommend close monitoring (Tables 3 and 4).
Abnormal movements. Though tardive dyskinesia risk is very low with atypical antipsychotics, we recommend screening during the first year of treatment. The development of diabetes mellitus may precipitate or worsen abnormal movements.
Related resources
- American Diabetes Association. www.diabetes.org
- American Obesity Association. www.obesity.org
- Expert Consensus Treatment Guidelines for Bipolar Disorder: A Guide for Patients and Families. Task Force for the APA Practice Guideline for the Treatment of Patients with Bipolar Disorder. www.psychguides.com/pfg3.php
Drug brand names
- Aripiprazole • Abilify
- Carbamazepine • Tegretol
- Divalproex sodium • Depakote
- Haloperidol • Haldol
- Olanzapine • Zyprexa
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Valproic acid • Depakene
- Ziprasidone • Geodon
Disclosures
Dr. Carroll is a consultant to Abbott Laboratories, Bristol-Myers Squibb Co., AstraZeneca Pharmaceuticals, Eli Lilly and Co., Janssen Pharmaceutica, and Pfizer Inc.
Dr. Fawver is a consultant to Eli Lilly and Co. and Pfizer Inc.
Dr. Thalassinos is a consultant for AstraZeneca Pharamaceuticals, Eli Lilly and Co., and Pfizer Inc.
Acknowledgment
The authors thank Donald R. Schmitt, PharmD, Christopher Thomas, PharmD, and Tina Fore, Library Service, Chillicothe VA Medical Center, for their help in identifying articles used in this review.
1. Keck PE, Jr, McElroy SL, Tugrul KC, et al. Valproate oral loading in the treatment of acute mania. J Clin Psychiatry. 1993;54:305-8.
2. Moscovich DG, Shapira B, Lerer B, et al. Rapid lithiumization in acute manic patients. Hum Psychopharmacol. 1992;7:343-5.
3. Kook KA, Stimmel GL, Wilkins JN, et al. Accuracy and safety of a priori lithium loading. J Clin Psychiatry. 1985;46:49-51.
4. Carroll BT, Thalassinos A, Fawver JD. Loading strategies in acute mania. CNS Spectrums. 2001;6:919-30.
5. Hirschfeld RMA, Allen MH, McEvoy J, et al. Safety and tolerability of oral loading divalproex sodium in acutely manic bipolar patients. J Clin Psychiatry. 1999;60:815-18.
6. Miller BP, et al. Oral loading of extended-release divalproex in acute mania (presentation). New Orleans: American Psychiatric Association annual meeting, 2001.
7. Thibault M, Blume WT, Saint-Hilaire JM, et al. Divalproex extended-release versus the original divalproex tablet: results of a randomized, crossover study of well-controlled epileptic patients with primary generalized seizures. Epilepsy Res. 2002;50:243-9.
8. Hirschfeld RMA, Baker JD, Wozniak P, et al. The safety and early efficacy of oral-loaded divalproex versus standard-titration divalproex, lithium, olanzapine, and placebo in the treatment of acute mania associated with bipolar disorder. J Clin Psychiatry. 2003;64:841-6.
9. Phrolov K, Applebaum J, Levine J, et al. Single-dose intravenous valproate in acute mania. J Clin Psychiatry. 2004;65:68-70.
10. Keck PE, Jr, McElroy SL, Bennet TA. Pharmacologic loading in the treatment of acute mania. Bipolar Disord. 2000;2:42-6.
11. Dunn RT, Frye MS, Tim KA, et al. The efficacy and use of anticonvulsants in mood disorders. Clin Neuropharmacol. 1998;21:215-35.
12. Lerer B, Moore N, Meyendor E, et al. Carbamazepine versus lithium in mania a double-blind study. J Clin Psychiatry. 1987;48:89-93.
13. Brown B. The use of generic mood stabilizers: carbamazepine (monograph). J Clin Psychiatry. 1997;15(4):11-17.
14. Karagianis JL, Dawe IC, Thakur A, et al. Rapid tranquilization with olanzapine in acute psychosis: a case series. J Clin Psychiatry. 2001;62(suppl 2):12-16.
15. Baker RW, Kinon BJ, Maguire GA, et al. Effectiveness of rapid initial dose escalation of up to 40 milligrams per day of oral olanzapine in acute agitation. J Clin Psychopharmacol. 2003;23(4):342-8.
16. Sachs GS, Grossman F, Ghaemi SN, et al. Combination of mood stabilizer with risperidone or haloperidol for the treatment of acute mania: a double-blind, placebo controlled comparison of efficacy and safety. Am J Psychiatry. 2002;159:1146-54.
17. Hirschfeld RMA, Keck PE, Jr, Karcher K, et al. Rapid antimanic effect of risperidone monotherapy: a 3-week multicenter, randomized, double-blind, placebo-controlled trial. Am J Psychiatry. 2004;161:1057-65.
18. Khanna S, Vieta E, Lyons B, et al. Risperidone monotherapy in acute bipolar mania (abstract P219). Pittsburgh: Fifth International Conference on Bipolar Disorder, 2003.
19. Jones MW, Huizar K, et al. Quetiapine monotherapy for acute mania associated with bipolar disorder (poster presentation). San Francisco: American Psychiatric Association annual meeting, 2003.
20. Sachs G, Mullen JA, Devine NA, Sweitzer DE. Quetiapine versus placebo as adjunct to mood stabilizer for the treatment of acute mania (abstract). Bipolar Disord. 2002;4(suppl 1):133.-
21. Smith MA, McCoy R, Hamer J, Brecher M. Optimal titration for quetiapine (poster presentation): Boca Raton, FL: National Clinical Drug Evaluation Unit annual meeting, 2002.
22. Keck PE, Jr, Versiani M, Potkin S, et al. Ziprasidone in the treatment of acute bipolar mania: a three-week, placebo-controlled, double-blind, randomized trial. Am J Psychiatry. 2003;160:741-8.
23. Holtzheimer PE, Neumaier JF. Treatment of acute mania. CNS Spectrums. 2003;8(12):917-28.
24. Keck PE, Jr, Marcus R, Tourkodimitris S, et al. A placebo-controlled, double-blind study of the efficacy and safety of aripiprazole in patients with acute bipolar mania. Am J Psychiatry. 2003;160(9):1651-8.
25. Sachs G, Sanchez R, Marcus R, et al. Aripiprazole vs placebo with an acute manic or mixed episode. New York: American Psychiatric Association annual meeting, 2004.
26. American Diabetes Association, American Psychiatric Association, American Association of Clinical Endocrinologists, North American Association for the Study of Obesity. Consensus Development Conference on Antipsychotic Drugs and Obesity and Diabetes. Diabetes Care. 2004;27:596-601.
Evidence on aggressive initial dosing of mood stabilizers and antipsychotics is changing the way acute bipolar mania is treated. To help you apply this information, we searched the literature and meeting abstracts for aggressive strategies tested to date. This article discusses how to:
- identify patients who may benefit from loading or aggressive initial dosing
- calculate mood stabilizer dosages
- dose each atypical antipsychotic
- manage potential antipsychotic side effects during maintenance therapy.
PATIENT SELECTION
Rapidly achieving therapeutic blood levels relieves patient suffering faster than standard titration. It quickly calms the hyperactivity, impulsivity, tension, hostility, and uncooperativeness that distress patients and increase the risk of harm to themselves and others.
The challenge of using higher dosages is to minimize side effects. Loading is not a one-size-fits-all approach, as antimanic drugs’ unique pharmacokinetic and pharmacotherapeutic properties influence how each agent is used.
An interesting body of literature advocates using mood stabilizers plus antipsychotics to treat mania, suggesting greater efficacy than with either agent alone.16 This strategy raises important questions, such as:
- Can two drugs be loaded simultaneously?
- Can patients taking mood stabilizers be treated with antipsychotic loading, and can those taking antipsychotics receive loading dosages of mood stabilizers?
- Would “double loading” improve bipolar mania treatment?
Answers to these questions are needed because of increased demands on clinicians to control hospital costs by rapidly and effectively treating patients with bipolar mania.
Loading doses cannot be standardized but are calculated by multiplying target steady-state plasma concentration by volume of distribution. We suggest aggressive initial schedules for divalproex sodium and atypical antipsychotics in this article with the understanding that practitioners will adjust them based on each patient’s tolerance and response.
Hospitalization. Patients with acute bipolar mania should be supervised closely in the hospital during loading or aggressive initial dosing. Monitor for cardiovascular changes, neurologic disturbances, sensorium changes, and response.
Precautions. Not all patients are candidates for aggressive initial dosing. Contraindications include age <18 or >65 years, pregnancy, breast-feeding, medical illness, and known sensitivity to the medication being given.
Higher-than-usual dosing increases the risk of excessive drug concentrations in sensitive individuals—such as those with a history of sensitivity to lower dosages of similar medications—and toxic levels of drugs with long half-lives can persist. When in doubt, consider giving a smaller amount of the loading dose early in the day, followed later by a larger amount.
DRUG SELECTION
Loading and aggressive initial dosing strategies for bipolar mania were first advanced for divalproex sodium.1 Investigators then examined loading strategies for lithium and carbamazepine, as well as the antipsychotics olanzapine, risperidone, quetiapine, ziprasidone, and aripiprazole, which are known to have antimanic properties.
Olanzapine, quetiapine, and risperidone are FDA-approved for short-term treatment of acute manic episodes associated with bipolar I disorder, and similar indications were being considered for aripiprazole and ziprasidone as this article went to press. We discuss evidence on loading and aggressive initial dosing strategies for each agent.
No studies have compared one loading strategy with another. Thus, when we choose drugs for loading, we consider what each patient needs, available formulations, tolerability, and efficacy for long-term stabilization and maintenance treatment.
LITHIUM
Lithium loading targets the therapeutic range (<1.4 mEq/L), without crossing the toxic threshold (>1.5 mEq/L). Lithium loading has shown antimanic effects, although using >30 mg/kg/d causes severe nausea and vomiting.
Moscovich et al2 reported a case series of 9 adults with acute mania who received lithium loading dosages of 4,050 mg/d. Patients tolerated lithium well at plasma drug levels of approximately 1.2 to 1.4 mEq/L. Their manic symptoms declined significantly within 4 to 5 days, as measured by Clinical Global Impression (CGI) severity of illness, Biegel-Murphy Mania State Rating Scale, and Brief Psychiatric Rating Scale scores.
Table 1
How to calculate divalproex loading for acute bipolar mania*
| Days 1 and 2 |
| Patient weight in pounds x 15 = dosage (mg/d) (Example: 150 lbs x 15 = 1,750 mg/d) |
| Days 3 to 10 |
| Patient weight in pounds x 10 = dosage (mg/d) (Example: 150 lbs. x 10 = 1,500 mg/d) |
| To avoid splitting tablets, make dosage divisible by 125 (round up for young adults, round down for older adults). Divide into bid or tid doses for improved tolerability. |
| Days 4 and 7 |
| Draw blood to monitor valproic acid levels and for other values such as liver function studies. |
| * For use with oral divalproex sodium (delayed-release or extended-release formulations). Do not use valproic acid preparations, as loading is unlikely to be well tolerated. |
| Source: Reference 5 |
Kook et al3 attempted a 30-mg/kg loading dose of slow-release lithium carbonate in 38 patients to evaluate the safety of achieving a therapeutic level in 12 hours. No patient experienced adverse effects during loading or in the 12 hours after loading was completed.
Patients who develop a manic episode while taking lithium pose a therapeutic dilemma. If they stop taking lithium—especially abruptly—manic symptoms can return in as few as 2 days. For these patients, consider increasing the usual dosage by 50% to 100% on the first day of mania treatment,4 then continue a dosage that maintains efficacy and achieves a therapeutic blood level.
To avoid GI upset, avoid any single dose of >1,500 mg; if a greater dosage is needed, divide it for improved tolerance. Obtain lithium blood levels at baseline, after 4 days, and thereafter as clinically relevant to monitor for drug interactions that may affect serum levels.
Side effects—GI irritation, tremor, muscle weakness, thirst, polyuria—are common in the first week, and weight gain may occur after prolonged treatment.
DIVALPROEX
In a study by Keck et al1 of 19 adults with acute bipolar mania, aggressive initial dosing of divalproex sodium, 20 mg/kg/d, was well-tolerated and shortened hospitalization. Side effects including sedation and nausea occurred at a rate similar to that seen with more-gradual titration.
To calculate the initial dose, the authors used a conversion factor of 20 mg/kg/d or added a zero to the patient’s weight in pounds. This amount was given in single or divided doses the first day, then continued for 4 to 7 days. Blood levels were measured on day 4, and all 15 patients who completed the study achieved serum valproate levels >50 mcg/mL.
In a double-blind study by Hirschfeld et al,5 59 adults with Young Mania Rating Scale (YMRS) scores >14 were randomly assigned to receive divalproex oral loading; divalproex nonloading (250 mg tid on days 1 and 2, followed by standard titration on days 3 to 10); or lithium carbonate (300 mg tid, followed by standard titration on days 3 to 10).
Divalproex loading—30 mg/kg/d on days 1 and 2, followed by 20 mg/kg on days 3 to 10— yielded valproate levels >50 mcg/mL in 84% of patients by day 3. Rapid loading appeared to increase antimanic efficacy, as measured by YMRS endpoint scores, without increasing adverse effects. For Table 1, we converted this study’s metric values to patient weight in pounds.
Side effects. Divalproex can inhibit metabolism of other drugs (including anticonvulsants such as lamotrigine) and increase their blood levels. Side effects such as sedation, alopecia, abdominal pain, diarrhea, and tremor may require observation and treatment. Pancreatitis, hepatitis, and allergic reactions are rare but may require discontinuation.
Recommendation. Aggressive initial dosing and loading for patients with acute mania has been reported with enteric-coated delayed-release and extended-release6 divalproex sodium tablets. With identical dosages, the extended-release form produces 11% lower serum levels than the delayed-release form.7
Aggressive dosing of oral valproic acid preparations is not recommended and is likely to be poorly tolerated.8 A pilot study evaluating IV valproate loading in acute mania found no changes in mania in the 2 hours that followed loading.9
CARBAMAZEPINE
Carbamazepine is used off-label to treat mania and mixed phases of bipolar disorder.4,9,10 Excessive absorption rates are associated with dizziness, ataxia, and nausea. Side effects may occur when the plasma level is therapeutic for epilepsy (4 to 12 mcg/mL).11
In mania, the relationship between carbamazepine levels and clinical response is not always clear. Lerer et al12 found a correlation between acute mania response and a serum level of 8.8 mcg/mL (range 4.7 to 14.0 mcg/mL).
Patients with acute mania may require 600 to 1,600 mg/d. Carbamazepine is available in immediate-release and controlled-release preparations. Because generic preparations might not be bio-equivalent,13 use the same formulation throughout treatment to maintain consistent serum levels.
Enzymatic auto-induction—in which carbamazepine gradually increases the activity of its metabolic enzyme—is likely to occur at 3 to 5 weeks of administration, often after hospital discharge. An aggressive initial rescue adjustment can be used if a patient develops mania after having been stabilized on carbamazepine for >6 weeks. Because these patients are past the point of auto-induction, a target blood level of 8.8 mcg/mL can be used and the dosage adjusted proportionally.
For example, if mania recurs in a patient who is stabilized on carbamazepine, 400 mg/d (plasma level 4.5 mcg/mL), carbamazepine can be loaded up to 800 mg/d. Measure the plasma level within 4 days; the target level would be 9.0 mcg/mL (2 times 4.5 mcg/mL), which closely approximates steady state and sets up a ratio to reach the target level of 8.8 mcg/mL.
Side effects include ataxia, diplopia, and dizziness. Complete blood counts, liver function studies, plasma levels, and serum chemistries require regular monitoring.
Recommendation. Carbamazepine is not recommended for oral loading in patients who have never taken it or for those with hyponatremia, hepatic dysfunction, or history of intolerance or agranulocytosis.
OLANZAPINE
Olanzapine has pharmacologic properties favorable for loading.4 The recommended dosage for acute mania is 15 mg/d with standard titration; Karagianis et al14 showed that initial loading doses of >20 mg/d resulted in good control of agitated patients with psychosis. Side effects were uncommon, with sedation occurring in 14% of patients in this case series. The loading dose reduced agitation more effectively than did dosages <20 mg/d given to similar patients.
A multicenter study of 148 acutely agitated inpatients with a variety of psychiatric disorders compared olanzapine rapid initial dose escalation with usual clinical practice.15 Mean aggressive dosages were 28.8 mg/d on day 1, 30.3 mg/d on day 2, and 16.1 mg/d on day 5. Usual-practice dosages were 10 mg/d, plus lorazepam, 0 to 4 mg/d for the first 2 days and 0 to 2 mg/d on days 3 to 4. Based on Positive and Negative Syndrome Scale excited component subscale scores, olanzapine loading controlled agitation more effectively than did usual practice, with similar side-effect rates.
IM olanzapine or the orally dissolving form are bioequivalent to the tablets and may be used for acute agitation associated with bipolar mania in certain clinical settings.14
Table 2
Suggested antipsychotic loading for acute bipolar mania and mixed states*
| Drug | Day(s) | Aggressive initial dosing schedule | Comment |
|---|---|---|---|
| Aripiprazole24,25 | 1 2 to 3 4+ | 30 mg once daily with food 30 mg/d with food Reduce dosage by 10 to 15 mg/d, based on tolerance and response | Nausea and vomiting may occur in first few days; adjust dosage based on tolerance and response |
| Olanzapine14,15 | 1 and 2 3 and 4 5 to 10 | 40 mg in single or divided dosage 20 to 30 mg at bedtime 15 mg once daily (may reduce to 5, 7.5, or 10 mg/d) | Adjust dosage based on tolerance and response; oral or IM formulations may be used |
| Quetiapine19,21 | 1 2 and 3 4 to 10 | 100 mg upon admission and 100 mg at bedtime 100 mg bid (or tid to qid) plus 100 to 200 mg at bedtime 200 mg bid plus 200 to 300 mg at bedtime; may adjust to 400 to 800 mg/d) | Adjust dosage based on tolerance and response |
| Risperidone16,18 | 1 2 3 and 4 | 3 mg in single or divided dosage 4 mg in single or divided dosage 5 mg in single or divided dosage | Adjust dosage by 1 mg up or down, based on tolerance and response; use tablet, rapid-dissolving tablet, or liquid form, but not long-acting IM form |
| Ziprasidone22,23 | 1 2 | 20 mg IM in single dose (may repeat for severe agitation) or 40 mg po bid with food 60 to 80 mg po bid with food | Adjust dosage based on tolerance and response |
| * For hospitalized or partially hospitalized patients, ages18. Not recommended for patients who are pregnant, breastfeeding, medically ill, age >65, or with known sensitivity to the antipsychotic being given. | |||
RISPERIDONE
Sachs et al16 studied 156 inpatients who developed an acute manic or mixed episode while receiving lithium or divalproex. These patients were randomly assigned to begin adjunctive risperidone, 2 mg/d, haloperidol, or placebo. Dosing was flexible, increasing or decreasing by 1 mg/d. Risperidone’s mean modal dosage was 3.8 mg/d across 3 weeks, with mean exposure of 17 days. Risperidone plus a mood stabilizer was more effective than a mood stabilizer alone, and the combination provided rapid, well-tolerated control of manic symptoms.
In a double-blind trial, Hirschfeld et al17 randomly assigned 279 patients with acute bipolar mania to risperidone, 1 to 6 mg/d, or placebo for 3 weeks. As early as day 3, YMRS scores were reduced significantly more with risperidone than with placebo. Somnolence was the most common side effect, and mean modal dosage was 4.1 mg/d.
Table 3
Screening schedule for antipsychotic side effects during bipolar maintenance treatment
| Baseline | |
| Side effect | Recommended screening |
| Weight gain | Weight and body mass index (BMI) monthly for first 3 months; waist circumference |
| Hypertension | Blood pressure |
| Hyperglycemia | Fasting plasma glucose, with glycosylated hemoglobin (Hb A 1c ) if hyperglycemia is detected |
| Hyperlipidemia | Fasting lipid profile |
| Tardive dyskinesia | Abnormal Involuntary Movement Scale (AIMS) or other screen |
| Ophthalmic changes | Ophthalmologic examination for patients taking quetiapine and for all with diabetes mellitus |
| Follow-up schedules | |
| 3 months | |
| Weight and BMI | |
| Blood pressure | |
| Fasting plasma glucose, with Hb A 1c if hyperglycemia is detected; Hb A 1c values may be used to measure interval changes in glucose tolerance | |
| Fasting lipid profile | |
| 6 months | |
| AIMS or other tardive dyskinesia screen | |
| Ophthalmologic examination | |
| Source: Adapted from reference 26. | |
In a multicenter, randomized, double-blind, placebo-controlled study of patients with acute bipolar mania, Khanna et al18 assigned patients to receive risperidone monotherapy (mean modal dosage 5.6 mg/d) or placebo for 3 weeks. Mania scores of patients receiving risperidone were significantly lower at weeks 1 and 2, compared with the placebo group. Risperidone was well-tolerated, with no unexpected adverse events.
Recommendation. Because of a risk of extrapyramidal symptoms (EPS) and orthostatic hypotension, initial risperidone loading dosages >4 mg on day 1 are not recommended.
QUETIAPINE
Quetiapine has shown antimanic efficacy as monotherapy and as adjunctive therapy to mood stabilizers.19,20 The effective dosage was a mean 600 mg/d (range 400 to 800 mg/d) in monotherapy and adjunctive treatment. These studies achieved 400 mg/d within the first 4 days (100 mg on day 1, 200 mg on day 2, 300 mg on day 3, and 400 mg on day 4).
These data combined with revised prescribing information suggest aggressive initial dose escalation of quetiapine within the first 4 days for selected patients. A titration study in patients with schizophrenia used a more-rapid escalation rate of 400 mg within 2 days.21 Dizziness, orthostatic hypotension, and sedation were not more frequent in this high-dose group than in the two lower-dose titration groups. In our experience, 200 to 400 mg can be given the first day of treatment.
ZIPRASIDONE
In a randomized, double-blind, controlled trial, Keck et al22 assigned 210 patients with a manic or mixed bipolar episode to 3 weeks of ziprasidone, 40 to 80 mg bid, or placebo. Ziprasidone produced rapid, sustained improvement in manic symptoms on all primary and most secondary efficacy measures, such as the YMRS and CGI.
Significant improvements seen within 2 days were maintained. Ziprasidone was well tolerated and was associated with a low EPS rate; neither weight gain nor clinically significant changes in vital signs were seen.
IM ziprasidone, which is approved for use in schizophrenia, may also have efficacy in bipolar mania.23 The recommended dose of 20 mg IM is equivalent to 120 to 160 mg orally, so a single injection may reach the target antimanic dosage.
Recommendation. Ziprasidone could be an option for aggressive initial dosing for a patient who has previously received ziprasidone IM and is not at risk for QTc prolongation.
ARIPIPRAZOLE
In a randomized controlled trial, Keck et al24 assigned 262 patients with acute bipolar mania or mixed states to aripiprazole, 30 mg/d, or placebo for 3 weeks. By day 4, manic symptoms were improved significantly more in patients receiving aripiprazole, and discontinuation rates were similar.
Similarly, in a randomized, controlled multi-center study, Sachs et al25 used 30 mg/d in 272 patients with bipolar mania or mixed states. Compared with placebo, aripiprazole produced significant improvement by day 4, with similar discontinuation rates.
Recommendation. Aggressive initial dosing of aripiprazole could be useful for a patient who does not require an IM or rapidly dissolvable medication.
Table 4
Suggested response to metabolic changes during bipolar maintenance therapy
| Metabolic change | Therapeutic action |
|---|---|
| ≥5% increase in total body weight | Consider weight-reduction strategies or medication adjustment |
| Fasting glucose: ≥126 mg/mL ≥300 mg/mL or ≤60 mg/mL | Consider evaluation for diabetes mellitus Seek immediate consultation |
| Total cholesterol ≥200 mg/dL or triglycerides ≥165 mg/dL | Consider lipid-lowering with dietary and/or medication changes |
| Source: Adapted from reference 26. | |
MAINTENANCE THERAPY
Ideally, if a medication stabilizes a patient’s acute bipolar mania, that medication is continued for further stabilization and maintenance. Aggressive initial dosing befits this approach because it establishes a therapeutic blood level and usually reveals any side effects within days. Moreover, patients often prefer to continue the medication that provided relief when they felt most distressed.
Weight gain. Long-term use of atypical antipsychotics may be associated with weight gain, dyslipidemia, and the development of metabolic syndromes and diabetes mellitus. Weight gain risk may be further elevated in patients taking both antipsychotics and lithium or valproic acid.26 When atypical antipsychotics are used for bipolar maintenance therapy, the American Diabetes Association and American Psychiatric Association recommend close monitoring (Tables 3 and 4).
Abnormal movements. Though tardive dyskinesia risk is very low with atypical antipsychotics, we recommend screening during the first year of treatment. The development of diabetes mellitus may precipitate or worsen abnormal movements.
Related resources
- American Diabetes Association. www.diabetes.org
- American Obesity Association. www.obesity.org
- Expert Consensus Treatment Guidelines for Bipolar Disorder: A Guide for Patients and Families. Task Force for the APA Practice Guideline for the Treatment of Patients with Bipolar Disorder. www.psychguides.com/pfg3.php
Drug brand names
- Aripiprazole • Abilify
- Carbamazepine • Tegretol
- Divalproex sodium • Depakote
- Haloperidol • Haldol
- Olanzapine • Zyprexa
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Valproic acid • Depakene
- Ziprasidone • Geodon
Disclosures
Dr. Carroll is a consultant to Abbott Laboratories, Bristol-Myers Squibb Co., AstraZeneca Pharmaceuticals, Eli Lilly and Co., Janssen Pharmaceutica, and Pfizer Inc.
Dr. Fawver is a consultant to Eli Lilly and Co. and Pfizer Inc.
Dr. Thalassinos is a consultant for AstraZeneca Pharamaceuticals, Eli Lilly and Co., and Pfizer Inc.
Acknowledgment
The authors thank Donald R. Schmitt, PharmD, Christopher Thomas, PharmD, and Tina Fore, Library Service, Chillicothe VA Medical Center, for their help in identifying articles used in this review.
Evidence on aggressive initial dosing of mood stabilizers and antipsychotics is changing the way acute bipolar mania is treated. To help you apply this information, we searched the literature and meeting abstracts for aggressive strategies tested to date. This article discusses how to:
- identify patients who may benefit from loading or aggressive initial dosing
- calculate mood stabilizer dosages
- dose each atypical antipsychotic
- manage potential antipsychotic side effects during maintenance therapy.
PATIENT SELECTION
Rapidly achieving therapeutic blood levels relieves patient suffering faster than standard titration. It quickly calms the hyperactivity, impulsivity, tension, hostility, and uncooperativeness that distress patients and increase the risk of harm to themselves and others.
The challenge of using higher dosages is to minimize side effects. Loading is not a one-size-fits-all approach, as antimanic drugs’ unique pharmacokinetic and pharmacotherapeutic properties influence how each agent is used.
An interesting body of literature advocates using mood stabilizers plus antipsychotics to treat mania, suggesting greater efficacy than with either agent alone.16 This strategy raises important questions, such as:
- Can two drugs be loaded simultaneously?
- Can patients taking mood stabilizers be treated with antipsychotic loading, and can those taking antipsychotics receive loading dosages of mood stabilizers?
- Would “double loading” improve bipolar mania treatment?
Answers to these questions are needed because of increased demands on clinicians to control hospital costs by rapidly and effectively treating patients with bipolar mania.
Loading doses cannot be standardized but are calculated by multiplying target steady-state plasma concentration by volume of distribution. We suggest aggressive initial schedules for divalproex sodium and atypical antipsychotics in this article with the understanding that practitioners will adjust them based on each patient’s tolerance and response.
Hospitalization. Patients with acute bipolar mania should be supervised closely in the hospital during loading or aggressive initial dosing. Monitor for cardiovascular changes, neurologic disturbances, sensorium changes, and response.
Precautions. Not all patients are candidates for aggressive initial dosing. Contraindications include age <18 or >65 years, pregnancy, breast-feeding, medical illness, and known sensitivity to the medication being given.
Higher-than-usual dosing increases the risk of excessive drug concentrations in sensitive individuals—such as those with a history of sensitivity to lower dosages of similar medications—and toxic levels of drugs with long half-lives can persist. When in doubt, consider giving a smaller amount of the loading dose early in the day, followed later by a larger amount.
DRUG SELECTION
Loading and aggressive initial dosing strategies for bipolar mania were first advanced for divalproex sodium.1 Investigators then examined loading strategies for lithium and carbamazepine, as well as the antipsychotics olanzapine, risperidone, quetiapine, ziprasidone, and aripiprazole, which are known to have antimanic properties.
Olanzapine, quetiapine, and risperidone are FDA-approved for short-term treatment of acute manic episodes associated with bipolar I disorder, and similar indications were being considered for aripiprazole and ziprasidone as this article went to press. We discuss evidence on loading and aggressive initial dosing strategies for each agent.
No studies have compared one loading strategy with another. Thus, when we choose drugs for loading, we consider what each patient needs, available formulations, tolerability, and efficacy for long-term stabilization and maintenance treatment.
LITHIUM
Lithium loading targets the therapeutic range (<1.4 mEq/L), without crossing the toxic threshold (>1.5 mEq/L). Lithium loading has shown antimanic effects, although using >30 mg/kg/d causes severe nausea and vomiting.
Moscovich et al2 reported a case series of 9 adults with acute mania who received lithium loading dosages of 4,050 mg/d. Patients tolerated lithium well at plasma drug levels of approximately 1.2 to 1.4 mEq/L. Their manic symptoms declined significantly within 4 to 5 days, as measured by Clinical Global Impression (CGI) severity of illness, Biegel-Murphy Mania State Rating Scale, and Brief Psychiatric Rating Scale scores.
Table 1
How to calculate divalproex loading for acute bipolar mania*
| Days 1 and 2 |
| Patient weight in pounds x 15 = dosage (mg/d) (Example: 150 lbs x 15 = 1,750 mg/d) |
| Days 3 to 10 |
| Patient weight in pounds x 10 = dosage (mg/d) (Example: 150 lbs. x 10 = 1,500 mg/d) |
| To avoid splitting tablets, make dosage divisible by 125 (round up for young adults, round down for older adults). Divide into bid or tid doses for improved tolerability. |
| Days 4 and 7 |
| Draw blood to monitor valproic acid levels and for other values such as liver function studies. |
| * For use with oral divalproex sodium (delayed-release or extended-release formulations). Do not use valproic acid preparations, as loading is unlikely to be well tolerated. |
| Source: Reference 5 |
Kook et al3 attempted a 30-mg/kg loading dose of slow-release lithium carbonate in 38 patients to evaluate the safety of achieving a therapeutic level in 12 hours. No patient experienced adverse effects during loading or in the 12 hours after loading was completed.
Patients who develop a manic episode while taking lithium pose a therapeutic dilemma. If they stop taking lithium—especially abruptly—manic symptoms can return in as few as 2 days. For these patients, consider increasing the usual dosage by 50% to 100% on the first day of mania treatment,4 then continue a dosage that maintains efficacy and achieves a therapeutic blood level.
To avoid GI upset, avoid any single dose of >1,500 mg; if a greater dosage is needed, divide it for improved tolerance. Obtain lithium blood levels at baseline, after 4 days, and thereafter as clinically relevant to monitor for drug interactions that may affect serum levels.
Side effects—GI irritation, tremor, muscle weakness, thirst, polyuria—are common in the first week, and weight gain may occur after prolonged treatment.
DIVALPROEX
In a study by Keck et al1 of 19 adults with acute bipolar mania, aggressive initial dosing of divalproex sodium, 20 mg/kg/d, was well-tolerated and shortened hospitalization. Side effects including sedation and nausea occurred at a rate similar to that seen with more-gradual titration.
To calculate the initial dose, the authors used a conversion factor of 20 mg/kg/d or added a zero to the patient’s weight in pounds. This amount was given in single or divided doses the first day, then continued for 4 to 7 days. Blood levels were measured on day 4, and all 15 patients who completed the study achieved serum valproate levels >50 mcg/mL.
In a double-blind study by Hirschfeld et al,5 59 adults with Young Mania Rating Scale (YMRS) scores >14 were randomly assigned to receive divalproex oral loading; divalproex nonloading (250 mg tid on days 1 and 2, followed by standard titration on days 3 to 10); or lithium carbonate (300 mg tid, followed by standard titration on days 3 to 10).
Divalproex loading—30 mg/kg/d on days 1 and 2, followed by 20 mg/kg on days 3 to 10— yielded valproate levels >50 mcg/mL in 84% of patients by day 3. Rapid loading appeared to increase antimanic efficacy, as measured by YMRS endpoint scores, without increasing adverse effects. For Table 1, we converted this study’s metric values to patient weight in pounds.
Side effects. Divalproex can inhibit metabolism of other drugs (including anticonvulsants such as lamotrigine) and increase their blood levels. Side effects such as sedation, alopecia, abdominal pain, diarrhea, and tremor may require observation and treatment. Pancreatitis, hepatitis, and allergic reactions are rare but may require discontinuation.
Recommendation. Aggressive initial dosing and loading for patients with acute mania has been reported with enteric-coated delayed-release and extended-release6 divalproex sodium tablets. With identical dosages, the extended-release form produces 11% lower serum levels than the delayed-release form.7
Aggressive dosing of oral valproic acid preparations is not recommended and is likely to be poorly tolerated.8 A pilot study evaluating IV valproate loading in acute mania found no changes in mania in the 2 hours that followed loading.9
CARBAMAZEPINE
Carbamazepine is used off-label to treat mania and mixed phases of bipolar disorder.4,9,10 Excessive absorption rates are associated with dizziness, ataxia, and nausea. Side effects may occur when the plasma level is therapeutic for epilepsy (4 to 12 mcg/mL).11
In mania, the relationship between carbamazepine levels and clinical response is not always clear. Lerer et al12 found a correlation between acute mania response and a serum level of 8.8 mcg/mL (range 4.7 to 14.0 mcg/mL).
Patients with acute mania may require 600 to 1,600 mg/d. Carbamazepine is available in immediate-release and controlled-release preparations. Because generic preparations might not be bio-equivalent,13 use the same formulation throughout treatment to maintain consistent serum levels.
Enzymatic auto-induction—in which carbamazepine gradually increases the activity of its metabolic enzyme—is likely to occur at 3 to 5 weeks of administration, often after hospital discharge. An aggressive initial rescue adjustment can be used if a patient develops mania after having been stabilized on carbamazepine for >6 weeks. Because these patients are past the point of auto-induction, a target blood level of 8.8 mcg/mL can be used and the dosage adjusted proportionally.
For example, if mania recurs in a patient who is stabilized on carbamazepine, 400 mg/d (plasma level 4.5 mcg/mL), carbamazepine can be loaded up to 800 mg/d. Measure the plasma level within 4 days; the target level would be 9.0 mcg/mL (2 times 4.5 mcg/mL), which closely approximates steady state and sets up a ratio to reach the target level of 8.8 mcg/mL.
Side effects include ataxia, diplopia, and dizziness. Complete blood counts, liver function studies, plasma levels, and serum chemistries require regular monitoring.
Recommendation. Carbamazepine is not recommended for oral loading in patients who have never taken it or for those with hyponatremia, hepatic dysfunction, or history of intolerance or agranulocytosis.
OLANZAPINE
Olanzapine has pharmacologic properties favorable for loading.4 The recommended dosage for acute mania is 15 mg/d with standard titration; Karagianis et al14 showed that initial loading doses of >20 mg/d resulted in good control of agitated patients with psychosis. Side effects were uncommon, with sedation occurring in 14% of patients in this case series. The loading dose reduced agitation more effectively than did dosages <20 mg/d given to similar patients.
A multicenter study of 148 acutely agitated inpatients with a variety of psychiatric disorders compared olanzapine rapid initial dose escalation with usual clinical practice.15 Mean aggressive dosages were 28.8 mg/d on day 1, 30.3 mg/d on day 2, and 16.1 mg/d on day 5. Usual-practice dosages were 10 mg/d, plus lorazepam, 0 to 4 mg/d for the first 2 days and 0 to 2 mg/d on days 3 to 4. Based on Positive and Negative Syndrome Scale excited component subscale scores, olanzapine loading controlled agitation more effectively than did usual practice, with similar side-effect rates.
IM olanzapine or the orally dissolving form are bioequivalent to the tablets and may be used for acute agitation associated with bipolar mania in certain clinical settings.14
Table 2
Suggested antipsychotic loading for acute bipolar mania and mixed states*
| Drug | Day(s) | Aggressive initial dosing schedule | Comment |
|---|---|---|---|
| Aripiprazole24,25 | 1 2 to 3 4+ | 30 mg once daily with food 30 mg/d with food Reduce dosage by 10 to 15 mg/d, based on tolerance and response | Nausea and vomiting may occur in first few days; adjust dosage based on tolerance and response |
| Olanzapine14,15 | 1 and 2 3 and 4 5 to 10 | 40 mg in single or divided dosage 20 to 30 mg at bedtime 15 mg once daily (may reduce to 5, 7.5, or 10 mg/d) | Adjust dosage based on tolerance and response; oral or IM formulations may be used |
| Quetiapine19,21 | 1 2 and 3 4 to 10 | 100 mg upon admission and 100 mg at bedtime 100 mg bid (or tid to qid) plus 100 to 200 mg at bedtime 200 mg bid plus 200 to 300 mg at bedtime; may adjust to 400 to 800 mg/d) | Adjust dosage based on tolerance and response |
| Risperidone16,18 | 1 2 3 and 4 | 3 mg in single or divided dosage 4 mg in single or divided dosage 5 mg in single or divided dosage | Adjust dosage by 1 mg up or down, based on tolerance and response; use tablet, rapid-dissolving tablet, or liquid form, but not long-acting IM form |
| Ziprasidone22,23 | 1 2 | 20 mg IM in single dose (may repeat for severe agitation) or 40 mg po bid with food 60 to 80 mg po bid with food | Adjust dosage based on tolerance and response |
| * For hospitalized or partially hospitalized patients, ages18. Not recommended for patients who are pregnant, breastfeeding, medically ill, age >65, or with known sensitivity to the antipsychotic being given. | |||
RISPERIDONE
Sachs et al16 studied 156 inpatients who developed an acute manic or mixed episode while receiving lithium or divalproex. These patients were randomly assigned to begin adjunctive risperidone, 2 mg/d, haloperidol, or placebo. Dosing was flexible, increasing or decreasing by 1 mg/d. Risperidone’s mean modal dosage was 3.8 mg/d across 3 weeks, with mean exposure of 17 days. Risperidone plus a mood stabilizer was more effective than a mood stabilizer alone, and the combination provided rapid, well-tolerated control of manic symptoms.
In a double-blind trial, Hirschfeld et al17 randomly assigned 279 patients with acute bipolar mania to risperidone, 1 to 6 mg/d, or placebo for 3 weeks. As early as day 3, YMRS scores were reduced significantly more with risperidone than with placebo. Somnolence was the most common side effect, and mean modal dosage was 4.1 mg/d.
Table 3
Screening schedule for antipsychotic side effects during bipolar maintenance treatment
| Baseline | |
| Side effect | Recommended screening |
| Weight gain | Weight and body mass index (BMI) monthly for first 3 months; waist circumference |
| Hypertension | Blood pressure |
| Hyperglycemia | Fasting plasma glucose, with glycosylated hemoglobin (Hb A 1c ) if hyperglycemia is detected |
| Hyperlipidemia | Fasting lipid profile |
| Tardive dyskinesia | Abnormal Involuntary Movement Scale (AIMS) or other screen |
| Ophthalmic changes | Ophthalmologic examination for patients taking quetiapine and for all with diabetes mellitus |
| Follow-up schedules | |
| 3 months | |
| Weight and BMI | |
| Blood pressure | |
| Fasting plasma glucose, with Hb A 1c if hyperglycemia is detected; Hb A 1c values may be used to measure interval changes in glucose tolerance | |
| Fasting lipid profile | |
| 6 months | |
| AIMS or other tardive dyskinesia screen | |
| Ophthalmologic examination | |
| Source: Adapted from reference 26. | |
In a multicenter, randomized, double-blind, placebo-controlled study of patients with acute bipolar mania, Khanna et al18 assigned patients to receive risperidone monotherapy (mean modal dosage 5.6 mg/d) or placebo for 3 weeks. Mania scores of patients receiving risperidone were significantly lower at weeks 1 and 2, compared with the placebo group. Risperidone was well-tolerated, with no unexpected adverse events.
Recommendation. Because of a risk of extrapyramidal symptoms (EPS) and orthostatic hypotension, initial risperidone loading dosages >4 mg on day 1 are not recommended.
QUETIAPINE
Quetiapine has shown antimanic efficacy as monotherapy and as adjunctive therapy to mood stabilizers.19,20 The effective dosage was a mean 600 mg/d (range 400 to 800 mg/d) in monotherapy and adjunctive treatment. These studies achieved 400 mg/d within the first 4 days (100 mg on day 1, 200 mg on day 2, 300 mg on day 3, and 400 mg on day 4).
These data combined with revised prescribing information suggest aggressive initial dose escalation of quetiapine within the first 4 days for selected patients. A titration study in patients with schizophrenia used a more-rapid escalation rate of 400 mg within 2 days.21 Dizziness, orthostatic hypotension, and sedation were not more frequent in this high-dose group than in the two lower-dose titration groups. In our experience, 200 to 400 mg can be given the first day of treatment.
ZIPRASIDONE
In a randomized, double-blind, controlled trial, Keck et al22 assigned 210 patients with a manic or mixed bipolar episode to 3 weeks of ziprasidone, 40 to 80 mg bid, or placebo. Ziprasidone produced rapid, sustained improvement in manic symptoms on all primary and most secondary efficacy measures, such as the YMRS and CGI.
Significant improvements seen within 2 days were maintained. Ziprasidone was well tolerated and was associated with a low EPS rate; neither weight gain nor clinically significant changes in vital signs were seen.
IM ziprasidone, which is approved for use in schizophrenia, may also have efficacy in bipolar mania.23 The recommended dose of 20 mg IM is equivalent to 120 to 160 mg orally, so a single injection may reach the target antimanic dosage.
Recommendation. Ziprasidone could be an option for aggressive initial dosing for a patient who has previously received ziprasidone IM and is not at risk for QTc prolongation.
ARIPIPRAZOLE
In a randomized controlled trial, Keck et al24 assigned 262 patients with acute bipolar mania or mixed states to aripiprazole, 30 mg/d, or placebo for 3 weeks. By day 4, manic symptoms were improved significantly more in patients receiving aripiprazole, and discontinuation rates were similar.
Similarly, in a randomized, controlled multi-center study, Sachs et al25 used 30 mg/d in 272 patients with bipolar mania or mixed states. Compared with placebo, aripiprazole produced significant improvement by day 4, with similar discontinuation rates.
Recommendation. Aggressive initial dosing of aripiprazole could be useful for a patient who does not require an IM or rapidly dissolvable medication.
Table 4
Suggested response to metabolic changes during bipolar maintenance therapy
| Metabolic change | Therapeutic action |
|---|---|
| ≥5% increase in total body weight | Consider weight-reduction strategies or medication adjustment |
| Fasting glucose: ≥126 mg/mL ≥300 mg/mL or ≤60 mg/mL | Consider evaluation for diabetes mellitus Seek immediate consultation |
| Total cholesterol ≥200 mg/dL or triglycerides ≥165 mg/dL | Consider lipid-lowering with dietary and/or medication changes |
| Source: Adapted from reference 26. | |
MAINTENANCE THERAPY
Ideally, if a medication stabilizes a patient’s acute bipolar mania, that medication is continued for further stabilization and maintenance. Aggressive initial dosing befits this approach because it establishes a therapeutic blood level and usually reveals any side effects within days. Moreover, patients often prefer to continue the medication that provided relief when they felt most distressed.
Weight gain. Long-term use of atypical antipsychotics may be associated with weight gain, dyslipidemia, and the development of metabolic syndromes and diabetes mellitus. Weight gain risk may be further elevated in patients taking both antipsychotics and lithium or valproic acid.26 When atypical antipsychotics are used for bipolar maintenance therapy, the American Diabetes Association and American Psychiatric Association recommend close monitoring (Tables 3 and 4).
Abnormal movements. Though tardive dyskinesia risk is very low with atypical antipsychotics, we recommend screening during the first year of treatment. The development of diabetes mellitus may precipitate or worsen abnormal movements.
Related resources
- American Diabetes Association. www.diabetes.org
- American Obesity Association. www.obesity.org
- Expert Consensus Treatment Guidelines for Bipolar Disorder: A Guide for Patients and Families. Task Force for the APA Practice Guideline for the Treatment of Patients with Bipolar Disorder. www.psychguides.com/pfg3.php
Drug brand names
- Aripiprazole • Abilify
- Carbamazepine • Tegretol
- Divalproex sodium • Depakote
- Haloperidol • Haldol
- Olanzapine • Zyprexa
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Valproic acid • Depakene
- Ziprasidone • Geodon
Disclosures
Dr. Carroll is a consultant to Abbott Laboratories, Bristol-Myers Squibb Co., AstraZeneca Pharmaceuticals, Eli Lilly and Co., Janssen Pharmaceutica, and Pfizer Inc.
Dr. Fawver is a consultant to Eli Lilly and Co. and Pfizer Inc.
Dr. Thalassinos is a consultant for AstraZeneca Pharamaceuticals, Eli Lilly and Co., and Pfizer Inc.
Acknowledgment
The authors thank Donald R. Schmitt, PharmD, Christopher Thomas, PharmD, and Tina Fore, Library Service, Chillicothe VA Medical Center, for their help in identifying articles used in this review.
1. Keck PE, Jr, McElroy SL, Tugrul KC, et al. Valproate oral loading in the treatment of acute mania. J Clin Psychiatry. 1993;54:305-8.
2. Moscovich DG, Shapira B, Lerer B, et al. Rapid lithiumization in acute manic patients. Hum Psychopharmacol. 1992;7:343-5.
3. Kook KA, Stimmel GL, Wilkins JN, et al. Accuracy and safety of a priori lithium loading. J Clin Psychiatry. 1985;46:49-51.
4. Carroll BT, Thalassinos A, Fawver JD. Loading strategies in acute mania. CNS Spectrums. 2001;6:919-30.
5. Hirschfeld RMA, Allen MH, McEvoy J, et al. Safety and tolerability of oral loading divalproex sodium in acutely manic bipolar patients. J Clin Psychiatry. 1999;60:815-18.
6. Miller BP, et al. Oral loading of extended-release divalproex in acute mania (presentation). New Orleans: American Psychiatric Association annual meeting, 2001.
7. Thibault M, Blume WT, Saint-Hilaire JM, et al. Divalproex extended-release versus the original divalproex tablet: results of a randomized, crossover study of well-controlled epileptic patients with primary generalized seizures. Epilepsy Res. 2002;50:243-9.
8. Hirschfeld RMA, Baker JD, Wozniak P, et al. The safety and early efficacy of oral-loaded divalproex versus standard-titration divalproex, lithium, olanzapine, and placebo in the treatment of acute mania associated with bipolar disorder. J Clin Psychiatry. 2003;64:841-6.
9. Phrolov K, Applebaum J, Levine J, et al. Single-dose intravenous valproate in acute mania. J Clin Psychiatry. 2004;65:68-70.
10. Keck PE, Jr, McElroy SL, Bennet TA. Pharmacologic loading in the treatment of acute mania. Bipolar Disord. 2000;2:42-6.
11. Dunn RT, Frye MS, Tim KA, et al. The efficacy and use of anticonvulsants in mood disorders. Clin Neuropharmacol. 1998;21:215-35.
12. Lerer B, Moore N, Meyendor E, et al. Carbamazepine versus lithium in mania a double-blind study. J Clin Psychiatry. 1987;48:89-93.
13. Brown B. The use of generic mood stabilizers: carbamazepine (monograph). J Clin Psychiatry. 1997;15(4):11-17.
14. Karagianis JL, Dawe IC, Thakur A, et al. Rapid tranquilization with olanzapine in acute psychosis: a case series. J Clin Psychiatry. 2001;62(suppl 2):12-16.
15. Baker RW, Kinon BJ, Maguire GA, et al. Effectiveness of rapid initial dose escalation of up to 40 milligrams per day of oral olanzapine in acute agitation. J Clin Psychopharmacol. 2003;23(4):342-8.
16. Sachs GS, Grossman F, Ghaemi SN, et al. Combination of mood stabilizer with risperidone or haloperidol for the treatment of acute mania: a double-blind, placebo controlled comparison of efficacy and safety. Am J Psychiatry. 2002;159:1146-54.
17. Hirschfeld RMA, Keck PE, Jr, Karcher K, et al. Rapid antimanic effect of risperidone monotherapy: a 3-week multicenter, randomized, double-blind, placebo-controlled trial. Am J Psychiatry. 2004;161:1057-65.
18. Khanna S, Vieta E, Lyons B, et al. Risperidone monotherapy in acute bipolar mania (abstract P219). Pittsburgh: Fifth International Conference on Bipolar Disorder, 2003.
19. Jones MW, Huizar K, et al. Quetiapine monotherapy for acute mania associated with bipolar disorder (poster presentation). San Francisco: American Psychiatric Association annual meeting, 2003.
20. Sachs G, Mullen JA, Devine NA, Sweitzer DE. Quetiapine versus placebo as adjunct to mood stabilizer for the treatment of acute mania (abstract). Bipolar Disord. 2002;4(suppl 1):133.-
21. Smith MA, McCoy R, Hamer J, Brecher M. Optimal titration for quetiapine (poster presentation): Boca Raton, FL: National Clinical Drug Evaluation Unit annual meeting, 2002.
22. Keck PE, Jr, Versiani M, Potkin S, et al. Ziprasidone in the treatment of acute bipolar mania: a three-week, placebo-controlled, double-blind, randomized trial. Am J Psychiatry. 2003;160:741-8.
23. Holtzheimer PE, Neumaier JF. Treatment of acute mania. CNS Spectrums. 2003;8(12):917-28.
24. Keck PE, Jr, Marcus R, Tourkodimitris S, et al. A placebo-controlled, double-blind study of the efficacy and safety of aripiprazole in patients with acute bipolar mania. Am J Psychiatry. 2003;160(9):1651-8.
25. Sachs G, Sanchez R, Marcus R, et al. Aripiprazole vs placebo with an acute manic or mixed episode. New York: American Psychiatric Association annual meeting, 2004.
26. American Diabetes Association, American Psychiatric Association, American Association of Clinical Endocrinologists, North American Association for the Study of Obesity. Consensus Development Conference on Antipsychotic Drugs and Obesity and Diabetes. Diabetes Care. 2004;27:596-601.
1. Keck PE, Jr, McElroy SL, Tugrul KC, et al. Valproate oral loading in the treatment of acute mania. J Clin Psychiatry. 1993;54:305-8.
2. Moscovich DG, Shapira B, Lerer B, et al. Rapid lithiumization in acute manic patients. Hum Psychopharmacol. 1992;7:343-5.
3. Kook KA, Stimmel GL, Wilkins JN, et al. Accuracy and safety of a priori lithium loading. J Clin Psychiatry. 1985;46:49-51.
4. Carroll BT, Thalassinos A, Fawver JD. Loading strategies in acute mania. CNS Spectrums. 2001;6:919-30.
5. Hirschfeld RMA, Allen MH, McEvoy J, et al. Safety and tolerability of oral loading divalproex sodium in acutely manic bipolar patients. J Clin Psychiatry. 1999;60:815-18.
6. Miller BP, et al. Oral loading of extended-release divalproex in acute mania (presentation). New Orleans: American Psychiatric Association annual meeting, 2001.
7. Thibault M, Blume WT, Saint-Hilaire JM, et al. Divalproex extended-release versus the original divalproex tablet: results of a randomized, crossover study of well-controlled epileptic patients with primary generalized seizures. Epilepsy Res. 2002;50:243-9.
8. Hirschfeld RMA, Baker JD, Wozniak P, et al. The safety and early efficacy of oral-loaded divalproex versus standard-titration divalproex, lithium, olanzapine, and placebo in the treatment of acute mania associated with bipolar disorder. J Clin Psychiatry. 2003;64:841-6.
9. Phrolov K, Applebaum J, Levine J, et al. Single-dose intravenous valproate in acute mania. J Clin Psychiatry. 2004;65:68-70.
10. Keck PE, Jr, McElroy SL, Bennet TA. Pharmacologic loading in the treatment of acute mania. Bipolar Disord. 2000;2:42-6.
11. Dunn RT, Frye MS, Tim KA, et al. The efficacy and use of anticonvulsants in mood disorders. Clin Neuropharmacol. 1998;21:215-35.
12. Lerer B, Moore N, Meyendor E, et al. Carbamazepine versus lithium in mania a double-blind study. J Clin Psychiatry. 1987;48:89-93.
13. Brown B. The use of generic mood stabilizers: carbamazepine (monograph). J Clin Psychiatry. 1997;15(4):11-17.
14. Karagianis JL, Dawe IC, Thakur A, et al. Rapid tranquilization with olanzapine in acute psychosis: a case series. J Clin Psychiatry. 2001;62(suppl 2):12-16.
15. Baker RW, Kinon BJ, Maguire GA, et al. Effectiveness of rapid initial dose escalation of up to 40 milligrams per day of oral olanzapine in acute agitation. J Clin Psychopharmacol. 2003;23(4):342-8.
16. Sachs GS, Grossman F, Ghaemi SN, et al. Combination of mood stabilizer with risperidone or haloperidol for the treatment of acute mania: a double-blind, placebo controlled comparison of efficacy and safety. Am J Psychiatry. 2002;159:1146-54.
17. Hirschfeld RMA, Keck PE, Jr, Karcher K, et al. Rapid antimanic effect of risperidone monotherapy: a 3-week multicenter, randomized, double-blind, placebo-controlled trial. Am J Psychiatry. 2004;161:1057-65.
18. Khanna S, Vieta E, Lyons B, et al. Risperidone monotherapy in acute bipolar mania (abstract P219). Pittsburgh: Fifth International Conference on Bipolar Disorder, 2003.
19. Jones MW, Huizar K, et al. Quetiapine monotherapy for acute mania associated with bipolar disorder (poster presentation). San Francisco: American Psychiatric Association annual meeting, 2003.
20. Sachs G, Mullen JA, Devine NA, Sweitzer DE. Quetiapine versus placebo as adjunct to mood stabilizer for the treatment of acute mania (abstract). Bipolar Disord. 2002;4(suppl 1):133.-
21. Smith MA, McCoy R, Hamer J, Brecher M. Optimal titration for quetiapine (poster presentation): Boca Raton, FL: National Clinical Drug Evaluation Unit annual meeting, 2002.
22. Keck PE, Jr, Versiani M, Potkin S, et al. Ziprasidone in the treatment of acute bipolar mania: a three-week, placebo-controlled, double-blind, randomized trial. Am J Psychiatry. 2003;160:741-8.
23. Holtzheimer PE, Neumaier JF. Treatment of acute mania. CNS Spectrums. 2003;8(12):917-28.
24. Keck PE, Jr, Marcus R, Tourkodimitris S, et al. A placebo-controlled, double-blind study of the efficacy and safety of aripiprazole in patients with acute bipolar mania. Am J Psychiatry. 2003;160(9):1651-8.
25. Sachs G, Sanchez R, Marcus R, et al. Aripiprazole vs placebo with an acute manic or mixed episode. New York: American Psychiatric Association annual meeting, 2004.
26. American Diabetes Association, American Psychiatric Association, American Association of Clinical Endocrinologists, North American Association for the Study of Obesity. Consensus Development Conference on Antipsychotic Drugs and Obesity and Diabetes. Diabetes Care. 2004;27:596-601.
New tool: Genotyping makes prescribing safer, more effective
Genotyping for cytochrome P-450 2D6 gene variations is emerging as a valuable clinical tool to help psychiatrists identify patients who:
Genetic variation has long been known to influence how individuals metabolize drugs, but only recently could we apply this information.5 Many academic medical centers and the two largest U.S. reference laboratories offer 2D6 testing at costs of $200 to $500.
Before long, psychiatrists may adopt routine genotyping before prescribing 2D6 substrate medications. This article and four vignettes illustrate the clinical benefits of psychiatric pharmacogenomics and suggest when prospective genotyping could help you select and dose medications.
Table 1
Drugs metabolized by the 2D6 enzyme*
| Antidepressants | Antipsychotics | Stimulants | Other medications |
|---|---|---|---|
| Desipramine | Fluphenazine | Atomoxetine | Codeine |
| Fluoxetine | Perphenazine | Dextromethorphan | |
| Nortriptyline | Risperidone | Oxycodone | |
| Paroxetine | Thioridazine | ||
| Venlafaxine | |||
| *Evidence suggests that these medications are predominantly metabolized by the 2D6 enzyme. | |||
| Be careful when prescribing these agents to patients who are poor 2D6 metabolizers. | |||
Why test for the 2D6 gene?
The 2D6 gene codes for the 2D6 enzyme, the primary enzyme required to metabolize many psychotropics Table 1.
Genetic variations. A common variation in a gene is frequently called an allele. More than 100 2D6 gene variations have been described. Consequently, the 2D6 gene’s enzyme activity also varies widely. Most mutations decrease the enzyme’s activity, but some polymorphisms change the gene’s promoter region, which can lead to upregulation and increased enzyme production.
Each 2D6 gene variation has been labeled with a standardized abbreviation (Table 2):
- *1 refers to the “normal” gene
- *2 stands for several variants with different activity levels.
- *3, *4, *6, *7, *8, *9, *10, *11, *12, *14, and *17 code for proteins with little or no activity.
- *5 indicates that the gene is deleted, and no enzyme can be produced.
Multiple copies. Another characteristic of the 2D6 gene is its unusually high propensity to accumulate in multiple copies on the 22nd chromosome. As many as 13 copies of the 2D6 gene have been shown on a single chromosome. Given that each gene can code for the 2D6 enzyme, patients with multiple copies can metabolize 2D6 substrate medications very rapidly.
Nonpsychiatric drugs. The 2D6 enzyme is also involved in metabolizing many nonpsychiatric drugs. To produce analgesia, for example, the 2D6 enzyme must metabolize the prodrug codeine to morphine. Thus, individuals with no 2D6 enzyme activity experience no analgesia with codeine. Approximately 7% of Caucasians metabolize codeine poorly. Conversely, individuals with multiple 2D6 gene copies metabolize codeine to morphine very rapidly, with potential for acute mental status changes, including psychosis.
4 metabolizer types. Based on variation in individual 2D6 genotype, a patient is usually categorized as being an ultrarapid, extensive, intermediate, or poor metabolizer (Table 3). The following case vignettes of patients in each category illustrate the clinical benefits of 2D6 genotyping.
Ultrarapid metabolizer: Extra 2D6 copies
Abdul, 49, is an Ethiopian businessman engaged in international commerce. While in the United States, he underwent a routine wisdom-tooth extraction and was treated with acetaminophen and codeine. Despite having no psychiatric history, he began to experience extreme discomfort and flashing visual hallucinations within 24 hours of taking two codeine doses. The oral surgeon instructed him to discontinue codeine, and his symptoms resolved within 24 hours.
Because of this experience, Abdul underwent genotyping for the 2D6 gene. He was found to have five active copies on one 22nd chromosome and no copies on the other (Figure 1). This genotype is unusual in western European populations but common in North Africa. Abdul then received alternate analgesics; psychiatric symptoms did not recur.
A patient such as Abdul, with multiple copies of a functional 2D6 gene, is an ultrarapid metabolizer. The 22nd chromosome—where the 2D6 gene is located—is short and contains areas of high homology. As a result, uneven crossover events occur more frequently during meiosis than is typical of larger chromosomes. Uneven crossover results in one gamete with two copies of the 2D6 gene and the other gamete with none.
2D6 enzyme activity is not essential for survival, which raises fascinating questions about this gene’s evolutionary importance. In certain geographic regions, many individuals have multiple copies of the gene. In Ethiopia—the country with the highest documented number of ultrarapid metabolizers—more than 25% of the population has one chromosome with multiple copies of the 2D6 gene.6 Because these copies produce an increased amount of 2D6 active enzyme, normal doses of 2D6 substrate medications do not benefit these individuals.
Table 2
How common 2D6 gene variations (alleles) affect 2D6 enzyme activity
| Allele label | 2D6 enzyme activity | Allele frequency (%)† |
|---|---|---|
| *1 | Normal | 37 |
| *2 | Decreased | 3.3 |
| *2P | Modestly increased | 6 |
| *3 | None | 1 |
| *4 | None | 18 |
| *5 | None (gene deletion) | 4 |
| *6 | None | 1 |
| *7 | None | <1 |
| *9 | Decreased | 3 |
| *10 | Decreased | 2 |
| *11 | None | 0 |
| *12 | None | <1 |
| *14 | Decreased | <1 |
| *17 | Decreased | <1 |
| †In Caucasian populations | ||
Table 3
Four ways patients respond to 2D6 substrate drugs
| Category | Patient characteristics | % of Caucasian population |
|---|---|---|
| Ultrarapid | Metabolize 2D6 medications rapidly resulting in poor response | 1 to 2 |
| Extensive | Metabolize 2D6 medications at a normal rate | 73 to 82 |
| Intermediate | Metabolize 2D6 medications at a slower-than-normal rate | 10 to 15 |
| Poor | Metabolize 2D6 medications very slowly with increased risk of side effects | 7 to 10 |
When treating ultrarapid metabolizers one strategy is to increase the dosage to obtain a therapeutic effect Because some substrates have complex metabolic pathways, however, high concentrations of secondary or tertiary metabolites can accumulate. Thus, when a substance’s metabolic pathway is not well-documented, a more cautious approach is to choose a medication metabolized by another pathway.
Figure 1 Genotypes and metabolizer categories of 4 illustrative patients
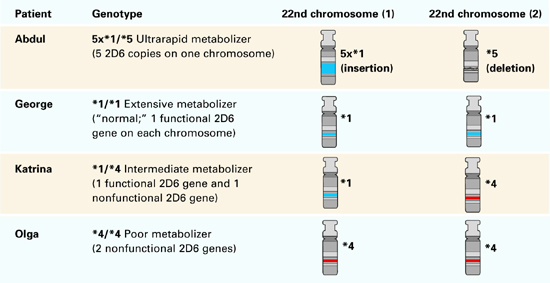
Extensive metabolizer: The ‘norm’
George, a 31-year-old Ethiopian architect, is Abdul’s second cousin. He developed acute depression with intense suicidal ideation and sought psychiatric consultation. He had no history of atypical drug reactions, but—because of his ethnic background—his psychiatrist was concerned that George might be a rapid metabolizer.
2D6 genotyping showed that George’s genotype was *1/*1, which meant he had two functional 2D6 copies (Figure 1). This genotype suggests that he could tolerate many antidepressants. The psychiatrist concluded—with some confidence—that George would not experience adverse effects or low serum levels when prescribed fluoxetine at usual dosages.
Extensive metabolizers have two normal 2D6 gene copies and can produce adequate active 2D6 enzyme Patients with this genotype—common in Caucasians—are generally said to have “normal” 2D6 metabolism. This means they metabolize 2D6 substrate medications at a rate within the recommended dosage ranges determined from North American or European pharmacokinetic studies.
Intermediate metabolizer: Mixed message
Katrina, 27, represents the government of her native Sweden in trade agreements. When she developed depressive symptoms (insomnia, sense of hopelessness), Katrina saw her psychiatrist. She reported that her family has a history of adverse reactions to multiple medications, but she had tolerated most medications. In fact, she had twice been successfully treated with relatively high doses of codeine.
Her psychiatrist suspected she was an intermediate 2D6 metabolizer and ordered testing. Her genotype was *1/*4, with one normal copy and one that produced no functional 2D6 enzyme (Figure 1).
Based on her clinical history and this genotypic information, the psychiatrist prescribed sertraline—metabolized by both 2D6 and 3A4 enzymes— at 50 mg/d. Because Katrina metabolized sertraline at a slower-than-usual rate, she developed a therapeutic blood level and responded well to this low dosage.
Intermediate metabolizers have a chromosome with one functional 2D6 gene copy. The other chromosome has either a copy with a defective functional polymorphism or a deletion of the gene. These patients usually tolerate 2D6 substrate drugs in low dosages.
Poor metabolizer: ‘medication-sensitive’
Olga, Katrina’s mother, has always lived in northern Sweden. She has no psychiatric history except for one psychotic episode that required hospitalization.
Her psychotic illness began on the summer solstice, during an all-night celebration. In addition to using unspecified recreational drugs, she took three 20-mg capsules of fluoxetine that her friend told her would make her feel high. She instead developed acute mania and dramatic paranoid delusions.
Figure 2 Possible genotypes of Brad, son of Abdul and Katrina
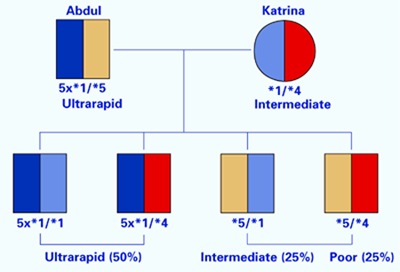
Olga was hospitalized and treated with moderate doses of haloperidol that precipitated an acute dystonic reaction. She was subsequently given ben-ztropine, and her extrapyramidal symptoms resolved. After discharge, she was treated with haloperidol and benztropine for 2 years, after which she spontaneously discontinued these drugs against medical advice. Her psychotic illness has not recurred.
Knowing her own genotype, Katrina understood that her mother had a 50% probability of having one copy of the 2D6 *4 allele. Given her mother’s history of medication intolerance, Katrina believed that her mother’s psychiatric illness might have been related to a drug reaction. She persuaded her mother to send a blood sample to a laboratory in Stockholm.
Olga’s genotype was *4/*4, indicating that she would be unlikely to tolerate even moderate doses of 2D6 substrate medications (Figure 1). Given her complete recovery and continued good health without medication, the most probable retrospective diagnosis was drug-induced psychosis. Her 2-year neuroleptic treatment probably was unnecessary.
Figure 3 Genogram for Brad, son of Abdul and Katrina
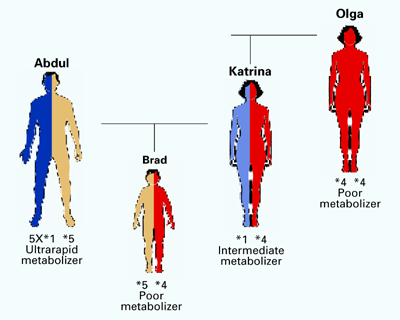
Poor metabolizers without a functional 2D6 gene copy have low tolerance for many medications and often become labeled as “medication sensitive.” When genotyping reveals that an individual is a poor metabolizer, prescribing medications that do not require 2D6 metabolism is usually prudent.
In rare cases, poor metabolizers have died from normal doses of 2D6 substrate medications.7 Far more commonly, however, they spontaneously discontinue taking these drugs because of adverse side effects.
Benefits of prospective testing
When used in clinical practice, pharmacogenomic testing’s two goals are to identify:
- ultrarapid metabolizers, who will not benefit from a medication
- poor metabolizers, who likely will have adverse responses to a medication.
The following case demonstrates the benefit of prospective 2D6 genotyping:
Brad, age 14, is the son of Abdul and Katrina, whose genotypes have been described. Brad developed a serious depression that was similar in severity and onset to an illness his mother experienced as a teen.
Brad’s parents want him to get the maximum benefit from psychopharmacologic treatment while avoiding distressing side effects. He had been healthy and had received no prescriptions other than antibiotics in the past.
How would you proceed? Without knowing Brad’s parents’ genotypes, you might reason that Brad would resemble one of them in drug response. However, when you review each parent’s genotype, you realize four scenarios are possible (Figure 2):
- Brad has a high likelihood of being an ultrarapid metabolizer because he has a 50% chance of inheriting a chromosome with five copies of the 2D6 gene from his father. He inherited the *1 or *4 form from his mother, but the effect of either will be clinically irrelevant.
- If Brad inherited the chromosome with the deletion from his father and the *1 form from his mother, he would be an intermediate metabolizer, as is his mother.
- If he inherited the chromosome with the deletion from his father and the *4 form from his mother, he would be a poor metabolizer like his grandmother, Olga. He would be at substantial risk for adverse reactions (such as intense headaches or vomiting) to 2D6 substrate medications.
On testing, Brad was found to be a poor metabolizer (Figure 3) The psychiatrist prescribed bupropion, which is metabolized by the 2B6 enzyme rather than the 2D6 enzyme.
Conclusion. To introduce the concept of genotypic testing, this review has focused on simple illustrations of variations in a single gene. However, many genes in the P-450 family play important roles in metabolizing psychotropics. In the future, genotyping of panels of these genes will likely provide more-specific guidance than can be achieved by simply testing one gene at a time.
Related resources
- Lerer B (ed). Pharmacogenetics of psychotropic drugs. Cambridge, UK: Cambridge University Press, 2002.
- Kirchheiner J, Borsen K, Dahl ML, et al. CYP2D6 and CYP2C19 genotype-based dose recommendations for antidepressants: a first step towards subpopulation-specific dosages. Acta Psychiatr Scand 2001;103(3):173-92.
- Indiana University School of Medicine, Division of Clinical Pharmacology. Drug Interactions—Defining Genetic Influences on Pharmacologic Responses. http://medicine.iupui.edu/flockhart.
Drug brand names
- Acetaminophen w/codeine phosphate • Tylenol w/codeine
- Atomoxetine • Strattera
- Benztropine mesylate • Cogentin
- Bupropion • Wellbutrin
- Desipramine • Norpramin
- Fluoxetine • Prozac
- Fluphenazine • Prolixin
- Haloperidol • Haldol
- Nortriptyline • Aventyl, Pamelor
- Oxycodone • Oxycontin
- Paroxetine • Paxil
- Perphenazine • Trilafon
- Risperidone • Risperdal
- Sertraline • Zoloft
- Thioridazine • Mellaril
- Venlafaxine • Effexor
Disclosure
Dr. Mrazek reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Mrazek DA. Clinical genomic testing. In: Wiener J, Dulcan M (eds). Textbook of child and adolescent psychiatry (3rd ed). Washington, DC: American Psychiatric Publishing, Inc., 2001;193-203.
2. Mrazek DA. Pharmacogenomic screening for depressed children and adolescents (scientific proceedings). Miami Beach, FL: American Academy of Child and Adolescent Psychiatry annual meeting, 2003;159.-
3. Phillips KA, Veenstra DL, Oren E, et al. Potential role of pharmacogenomics in reducing adverse drug reactions. JAMA 2001;286(18):2270-9.
4. Shi MM, Mehrens D, Dacus K. Pharmacogenomics: Changing the health care paradigm. Modern Drug Discovery 2001;4(7):27-32.
5. Kirchheiner J, Brosen K, Dahl ML, et al. CYP2D6 and CYP2C19 genotype-based dose recommendations for antidepressants: a first step towards subpopulation-specific dosages. Acta Psychiatr Scand 2001;104:173-92.
6. Masimirembwa CM, Hasler JA. Genetic polymorphism of drug metabolising enzymes in African populations: implications for the use of neuroleptics and antidepressants. Brain Res Bull 1997;44(5):561-71.
7. Sallee FR, DeVane CL, Ferrell RE. Fluoxetine-related death in a child with cytochrome P-450 2D6 genetic deficiency. J Child Adolesc Psychopharmacol 2000;10(1):27-34.
8. Gaedigk A, Gotschall RR, Forbes NS, et al. Optimization of cytochrome P4502D6 (CYP2D6) phenotype assignment using a genotyping algorithm based on allele frequency data. Pharmacogenetics 1999;9(6):669-82.
Genotyping for cytochrome P-450 2D6 gene variations is emerging as a valuable clinical tool to help psychiatrists identify patients who:
Genetic variation has long been known to influence how individuals metabolize drugs, but only recently could we apply this information.5 Many academic medical centers and the two largest U.S. reference laboratories offer 2D6 testing at costs of $200 to $500.
Before long, psychiatrists may adopt routine genotyping before prescribing 2D6 substrate medications. This article and four vignettes illustrate the clinical benefits of psychiatric pharmacogenomics and suggest when prospective genotyping could help you select and dose medications.
Table 1
Drugs metabolized by the 2D6 enzyme*
| Antidepressants | Antipsychotics | Stimulants | Other medications |
|---|---|---|---|
| Desipramine | Fluphenazine | Atomoxetine | Codeine |
| Fluoxetine | Perphenazine | Dextromethorphan | |
| Nortriptyline | Risperidone | Oxycodone | |
| Paroxetine | Thioridazine | ||
| Venlafaxine | |||
| *Evidence suggests that these medications are predominantly metabolized by the 2D6 enzyme. | |||
| Be careful when prescribing these agents to patients who are poor 2D6 metabolizers. | |||
Why test for the 2D6 gene?
The 2D6 gene codes for the 2D6 enzyme, the primary enzyme required to metabolize many psychotropics Table 1.
Genetic variations. A common variation in a gene is frequently called an allele. More than 100 2D6 gene variations have been described. Consequently, the 2D6 gene’s enzyme activity also varies widely. Most mutations decrease the enzyme’s activity, but some polymorphisms change the gene’s promoter region, which can lead to upregulation and increased enzyme production.
Each 2D6 gene variation has been labeled with a standardized abbreviation (Table 2):
- *1 refers to the “normal” gene
- *2 stands for several variants with different activity levels.
- *3, *4, *6, *7, *8, *9, *10, *11, *12, *14, and *17 code for proteins with little or no activity.
- *5 indicates that the gene is deleted, and no enzyme can be produced.
Multiple copies. Another characteristic of the 2D6 gene is its unusually high propensity to accumulate in multiple copies on the 22nd chromosome. As many as 13 copies of the 2D6 gene have been shown on a single chromosome. Given that each gene can code for the 2D6 enzyme, patients with multiple copies can metabolize 2D6 substrate medications very rapidly.
Nonpsychiatric drugs. The 2D6 enzyme is also involved in metabolizing many nonpsychiatric drugs. To produce analgesia, for example, the 2D6 enzyme must metabolize the prodrug codeine to morphine. Thus, individuals with no 2D6 enzyme activity experience no analgesia with codeine. Approximately 7% of Caucasians metabolize codeine poorly. Conversely, individuals with multiple 2D6 gene copies metabolize codeine to morphine very rapidly, with potential for acute mental status changes, including psychosis.
4 metabolizer types. Based on variation in individual 2D6 genotype, a patient is usually categorized as being an ultrarapid, extensive, intermediate, or poor metabolizer (Table 3). The following case vignettes of patients in each category illustrate the clinical benefits of 2D6 genotyping.
Ultrarapid metabolizer: Extra 2D6 copies
Abdul, 49, is an Ethiopian businessman engaged in international commerce. While in the United States, he underwent a routine wisdom-tooth extraction and was treated with acetaminophen and codeine. Despite having no psychiatric history, he began to experience extreme discomfort and flashing visual hallucinations within 24 hours of taking two codeine doses. The oral surgeon instructed him to discontinue codeine, and his symptoms resolved within 24 hours.
Because of this experience, Abdul underwent genotyping for the 2D6 gene. He was found to have five active copies on one 22nd chromosome and no copies on the other (Figure 1). This genotype is unusual in western European populations but common in North Africa. Abdul then received alternate analgesics; psychiatric symptoms did not recur.
A patient such as Abdul, with multiple copies of a functional 2D6 gene, is an ultrarapid metabolizer. The 22nd chromosome—where the 2D6 gene is located—is short and contains areas of high homology. As a result, uneven crossover events occur more frequently during meiosis than is typical of larger chromosomes. Uneven crossover results in one gamete with two copies of the 2D6 gene and the other gamete with none.
2D6 enzyme activity is not essential for survival, which raises fascinating questions about this gene’s evolutionary importance. In certain geographic regions, many individuals have multiple copies of the gene. In Ethiopia—the country with the highest documented number of ultrarapid metabolizers—more than 25% of the population has one chromosome with multiple copies of the 2D6 gene.6 Because these copies produce an increased amount of 2D6 active enzyme, normal doses of 2D6 substrate medications do not benefit these individuals.
Table 2
How common 2D6 gene variations (alleles) affect 2D6 enzyme activity
| Allele label | 2D6 enzyme activity | Allele frequency (%)† |
|---|---|---|
| *1 | Normal | 37 |
| *2 | Decreased | 3.3 |
| *2P | Modestly increased | 6 |
| *3 | None | 1 |
| *4 | None | 18 |
| *5 | None (gene deletion) | 4 |
| *6 | None | 1 |
| *7 | None | <1 |
| *9 | Decreased | 3 |
| *10 | Decreased | 2 |
| *11 | None | 0 |
| *12 | None | <1 |
| *14 | Decreased | <1 |
| *17 | Decreased | <1 |
| †In Caucasian populations | ||
Table 3
Four ways patients respond to 2D6 substrate drugs
| Category | Patient characteristics | % of Caucasian population |
|---|---|---|
| Ultrarapid | Metabolize 2D6 medications rapidly resulting in poor response | 1 to 2 |
| Extensive | Metabolize 2D6 medications at a normal rate | 73 to 82 |
| Intermediate | Metabolize 2D6 medications at a slower-than-normal rate | 10 to 15 |
| Poor | Metabolize 2D6 medications very slowly with increased risk of side effects | 7 to 10 |
When treating ultrarapid metabolizers one strategy is to increase the dosage to obtain a therapeutic effect Because some substrates have complex metabolic pathways, however, high concentrations of secondary or tertiary metabolites can accumulate. Thus, when a substance’s metabolic pathway is not well-documented, a more cautious approach is to choose a medication metabolized by another pathway.
Figure 1 Genotypes and metabolizer categories of 4 illustrative patients
Extensive metabolizer: The ‘norm’
George, a 31-year-old Ethiopian architect, is Abdul’s second cousin. He developed acute depression with intense suicidal ideation and sought psychiatric consultation. He had no history of atypical drug reactions, but—because of his ethnic background—his psychiatrist was concerned that George might be a rapid metabolizer.
2D6 genotyping showed that George’s genotype was *1/*1, which meant he had two functional 2D6 copies (Figure 1). This genotype suggests that he could tolerate many antidepressants. The psychiatrist concluded—with some confidence—that George would not experience adverse effects or low serum levels when prescribed fluoxetine at usual dosages.
Extensive metabolizers have two normal 2D6 gene copies and can produce adequate active 2D6 enzyme Patients with this genotype—common in Caucasians—are generally said to have “normal” 2D6 metabolism. This means they metabolize 2D6 substrate medications at a rate within the recommended dosage ranges determined from North American or European pharmacokinetic studies.
Intermediate metabolizer: Mixed message
Katrina, 27, represents the government of her native Sweden in trade agreements. When she developed depressive symptoms (insomnia, sense of hopelessness), Katrina saw her psychiatrist. She reported that her family has a history of adverse reactions to multiple medications, but she had tolerated most medications. In fact, she had twice been successfully treated with relatively high doses of codeine.
Her psychiatrist suspected she was an intermediate 2D6 metabolizer and ordered testing. Her genotype was *1/*4, with one normal copy and one that produced no functional 2D6 enzyme (Figure 1).
Based on her clinical history and this genotypic information, the psychiatrist prescribed sertraline—metabolized by both 2D6 and 3A4 enzymes— at 50 mg/d. Because Katrina metabolized sertraline at a slower-than-usual rate, she developed a therapeutic blood level and responded well to this low dosage.
Intermediate metabolizers have a chromosome with one functional 2D6 gene copy. The other chromosome has either a copy with a defective functional polymorphism or a deletion of the gene. These patients usually tolerate 2D6 substrate drugs in low dosages.
Poor metabolizer: ‘medication-sensitive’
Olga, Katrina’s mother, has always lived in northern Sweden. She has no psychiatric history except for one psychotic episode that required hospitalization.
Her psychotic illness began on the summer solstice, during an all-night celebration. In addition to using unspecified recreational drugs, she took three 20-mg capsules of fluoxetine that her friend told her would make her feel high. She instead developed acute mania and dramatic paranoid delusions.
Figure 2 Possible genotypes of Brad, son of Abdul and Katrina
Olga was hospitalized and treated with moderate doses of haloperidol that precipitated an acute dystonic reaction. She was subsequently given ben-ztropine, and her extrapyramidal symptoms resolved. After discharge, she was treated with haloperidol and benztropine for 2 years, after which she spontaneously discontinued these drugs against medical advice. Her psychotic illness has not recurred.
Knowing her own genotype, Katrina understood that her mother had a 50% probability of having one copy of the 2D6 *4 allele. Given her mother’s history of medication intolerance, Katrina believed that her mother’s psychiatric illness might have been related to a drug reaction. She persuaded her mother to send a blood sample to a laboratory in Stockholm.
Olga’s genotype was *4/*4, indicating that she would be unlikely to tolerate even moderate doses of 2D6 substrate medications (Figure 1). Given her complete recovery and continued good health without medication, the most probable retrospective diagnosis was drug-induced psychosis. Her 2-year neuroleptic treatment probably was unnecessary.
Figure 3 Genogram for Brad, son of Abdul and Katrina
Poor metabolizers without a functional 2D6 gene copy have low tolerance for many medications and often become labeled as “medication sensitive.” When genotyping reveals that an individual is a poor metabolizer, prescribing medications that do not require 2D6 metabolism is usually prudent.
In rare cases, poor metabolizers have died from normal doses of 2D6 substrate medications.7 Far more commonly, however, they spontaneously discontinue taking these drugs because of adverse side effects.
Benefits of prospective testing
When used in clinical practice, pharmacogenomic testing’s two goals are to identify:
- ultrarapid metabolizers, who will not benefit from a medication
- poor metabolizers, who likely will have adverse responses to a medication.
The following case demonstrates the benefit of prospective 2D6 genotyping:
Brad, age 14, is the son of Abdul and Katrina, whose genotypes have been described. Brad developed a serious depression that was similar in severity and onset to an illness his mother experienced as a teen.
Brad’s parents want him to get the maximum benefit from psychopharmacologic treatment while avoiding distressing side effects. He had been healthy and had received no prescriptions other than antibiotics in the past.
How would you proceed? Without knowing Brad’s parents’ genotypes, you might reason that Brad would resemble one of them in drug response. However, when you review each parent’s genotype, you realize four scenarios are possible (Figure 2):
- Brad has a high likelihood of being an ultrarapid metabolizer because he has a 50% chance of inheriting a chromosome with five copies of the 2D6 gene from his father. He inherited the *1 or *4 form from his mother, but the effect of either will be clinically irrelevant.
- If Brad inherited the chromosome with the deletion from his father and the *1 form from his mother, he would be an intermediate metabolizer, as is his mother.
- If he inherited the chromosome with the deletion from his father and the *4 form from his mother, he would be a poor metabolizer like his grandmother, Olga. He would be at substantial risk for adverse reactions (such as intense headaches or vomiting) to 2D6 substrate medications.
On testing, Brad was found to be a poor metabolizer (Figure 3) The psychiatrist prescribed bupropion, which is metabolized by the 2B6 enzyme rather than the 2D6 enzyme.
Conclusion. To introduce the concept of genotypic testing, this review has focused on simple illustrations of variations in a single gene. However, many genes in the P-450 family play important roles in metabolizing psychotropics. In the future, genotyping of panels of these genes will likely provide more-specific guidance than can be achieved by simply testing one gene at a time.
Related resources
- Lerer B (ed). Pharmacogenetics of psychotropic drugs. Cambridge, UK: Cambridge University Press, 2002.
- Kirchheiner J, Borsen K, Dahl ML, et al. CYP2D6 and CYP2C19 genotype-based dose recommendations for antidepressants: a first step towards subpopulation-specific dosages. Acta Psychiatr Scand 2001;103(3):173-92.
- Indiana University School of Medicine, Division of Clinical Pharmacology. Drug Interactions—Defining Genetic Influences on Pharmacologic Responses. http://medicine.iupui.edu/flockhart.
Drug brand names
- Acetaminophen w/codeine phosphate • Tylenol w/codeine
- Atomoxetine • Strattera
- Benztropine mesylate • Cogentin
- Bupropion • Wellbutrin
- Desipramine • Norpramin
- Fluoxetine • Prozac
- Fluphenazine • Prolixin
- Haloperidol • Haldol
- Nortriptyline • Aventyl, Pamelor
- Oxycodone • Oxycontin
- Paroxetine • Paxil
- Perphenazine • Trilafon
- Risperidone • Risperdal
- Sertraline • Zoloft
- Thioridazine • Mellaril
- Venlafaxine • Effexor
Disclosure
Dr. Mrazek reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Genotyping for cytochrome P-450 2D6 gene variations is emerging as a valuable clinical tool to help psychiatrists identify patients who:
Genetic variation has long been known to influence how individuals metabolize drugs, but only recently could we apply this information.5 Many academic medical centers and the two largest U.S. reference laboratories offer 2D6 testing at costs of $200 to $500.
Before long, psychiatrists may adopt routine genotyping before prescribing 2D6 substrate medications. This article and four vignettes illustrate the clinical benefits of psychiatric pharmacogenomics and suggest when prospective genotyping could help you select and dose medications.
Table 1
Drugs metabolized by the 2D6 enzyme*
| Antidepressants | Antipsychotics | Stimulants | Other medications |
|---|---|---|---|
| Desipramine | Fluphenazine | Atomoxetine | Codeine |
| Fluoxetine | Perphenazine | Dextromethorphan | |
| Nortriptyline | Risperidone | Oxycodone | |
| Paroxetine | Thioridazine | ||
| Venlafaxine | |||
| *Evidence suggests that these medications are predominantly metabolized by the 2D6 enzyme. | |||
| Be careful when prescribing these agents to patients who are poor 2D6 metabolizers. | |||
Why test for the 2D6 gene?
The 2D6 gene codes for the 2D6 enzyme, the primary enzyme required to metabolize many psychotropics Table 1.
Genetic variations. A common variation in a gene is frequently called an allele. More than 100 2D6 gene variations have been described. Consequently, the 2D6 gene’s enzyme activity also varies widely. Most mutations decrease the enzyme’s activity, but some polymorphisms change the gene’s promoter region, which can lead to upregulation and increased enzyme production.
Each 2D6 gene variation has been labeled with a standardized abbreviation (Table 2):
- *1 refers to the “normal” gene
- *2 stands for several variants with different activity levels.
- *3, *4, *6, *7, *8, *9, *10, *11, *12, *14, and *17 code for proteins with little or no activity.
- *5 indicates that the gene is deleted, and no enzyme can be produced.
Multiple copies. Another characteristic of the 2D6 gene is its unusually high propensity to accumulate in multiple copies on the 22nd chromosome. As many as 13 copies of the 2D6 gene have been shown on a single chromosome. Given that each gene can code for the 2D6 enzyme, patients with multiple copies can metabolize 2D6 substrate medications very rapidly.
Nonpsychiatric drugs. The 2D6 enzyme is also involved in metabolizing many nonpsychiatric drugs. To produce analgesia, for example, the 2D6 enzyme must metabolize the prodrug codeine to morphine. Thus, individuals with no 2D6 enzyme activity experience no analgesia with codeine. Approximately 7% of Caucasians metabolize codeine poorly. Conversely, individuals with multiple 2D6 gene copies metabolize codeine to morphine very rapidly, with potential for acute mental status changes, including psychosis.
4 metabolizer types. Based on variation in individual 2D6 genotype, a patient is usually categorized as being an ultrarapid, extensive, intermediate, or poor metabolizer (Table 3). The following case vignettes of patients in each category illustrate the clinical benefits of 2D6 genotyping.
Ultrarapid metabolizer: Extra 2D6 copies
Abdul, 49, is an Ethiopian businessman engaged in international commerce. While in the United States, he underwent a routine wisdom-tooth extraction and was treated with acetaminophen and codeine. Despite having no psychiatric history, he began to experience extreme discomfort and flashing visual hallucinations within 24 hours of taking two codeine doses. The oral surgeon instructed him to discontinue codeine, and his symptoms resolved within 24 hours.
Because of this experience, Abdul underwent genotyping for the 2D6 gene. He was found to have five active copies on one 22nd chromosome and no copies on the other (Figure 1). This genotype is unusual in western European populations but common in North Africa. Abdul then received alternate analgesics; psychiatric symptoms did not recur.
A patient such as Abdul, with multiple copies of a functional 2D6 gene, is an ultrarapid metabolizer. The 22nd chromosome—where the 2D6 gene is located—is short and contains areas of high homology. As a result, uneven crossover events occur more frequently during meiosis than is typical of larger chromosomes. Uneven crossover results in one gamete with two copies of the 2D6 gene and the other gamete with none.
2D6 enzyme activity is not essential for survival, which raises fascinating questions about this gene’s evolutionary importance. In certain geographic regions, many individuals have multiple copies of the gene. In Ethiopia—the country with the highest documented number of ultrarapid metabolizers—more than 25% of the population has one chromosome with multiple copies of the 2D6 gene.6 Because these copies produce an increased amount of 2D6 active enzyme, normal doses of 2D6 substrate medications do not benefit these individuals.
Table 2
How common 2D6 gene variations (alleles) affect 2D6 enzyme activity
| Allele label | 2D6 enzyme activity | Allele frequency (%)† |
|---|---|---|
| *1 | Normal | 37 |
| *2 | Decreased | 3.3 |
| *2P | Modestly increased | 6 |
| *3 | None | 1 |
| *4 | None | 18 |
| *5 | None (gene deletion) | 4 |
| *6 | None | 1 |
| *7 | None | <1 |
| *9 | Decreased | 3 |
| *10 | Decreased | 2 |
| *11 | None | 0 |
| *12 | None | <1 |
| *14 | Decreased | <1 |
| *17 | Decreased | <1 |
| †In Caucasian populations | ||
Table 3
Four ways patients respond to 2D6 substrate drugs
| Category | Patient characteristics | % of Caucasian population |
|---|---|---|
| Ultrarapid | Metabolize 2D6 medications rapidly resulting in poor response | 1 to 2 |
| Extensive | Metabolize 2D6 medications at a normal rate | 73 to 82 |
| Intermediate | Metabolize 2D6 medications at a slower-than-normal rate | 10 to 15 |
| Poor | Metabolize 2D6 medications very slowly with increased risk of side effects | 7 to 10 |
When treating ultrarapid metabolizers one strategy is to increase the dosage to obtain a therapeutic effect Because some substrates have complex metabolic pathways, however, high concentrations of secondary or tertiary metabolites can accumulate. Thus, when a substance’s metabolic pathway is not well-documented, a more cautious approach is to choose a medication metabolized by another pathway.
Figure 1 Genotypes and metabolizer categories of 4 illustrative patients
Extensive metabolizer: The ‘norm’
George, a 31-year-old Ethiopian architect, is Abdul’s second cousin. He developed acute depression with intense suicidal ideation and sought psychiatric consultation. He had no history of atypical drug reactions, but—because of his ethnic background—his psychiatrist was concerned that George might be a rapid metabolizer.
2D6 genotyping showed that George’s genotype was *1/*1, which meant he had two functional 2D6 copies (Figure 1). This genotype suggests that he could tolerate many antidepressants. The psychiatrist concluded—with some confidence—that George would not experience adverse effects or low serum levels when prescribed fluoxetine at usual dosages.
Extensive metabolizers have two normal 2D6 gene copies and can produce adequate active 2D6 enzyme Patients with this genotype—common in Caucasians—are generally said to have “normal” 2D6 metabolism. This means they metabolize 2D6 substrate medications at a rate within the recommended dosage ranges determined from North American or European pharmacokinetic studies.
Intermediate metabolizer: Mixed message
Katrina, 27, represents the government of her native Sweden in trade agreements. When she developed depressive symptoms (insomnia, sense of hopelessness), Katrina saw her psychiatrist. She reported that her family has a history of adverse reactions to multiple medications, but she had tolerated most medications. In fact, she had twice been successfully treated with relatively high doses of codeine.
Her psychiatrist suspected she was an intermediate 2D6 metabolizer and ordered testing. Her genotype was *1/*4, with one normal copy and one that produced no functional 2D6 enzyme (Figure 1).
Based on her clinical history and this genotypic information, the psychiatrist prescribed sertraline—metabolized by both 2D6 and 3A4 enzymes— at 50 mg/d. Because Katrina metabolized sertraline at a slower-than-usual rate, she developed a therapeutic blood level and responded well to this low dosage.
Intermediate metabolizers have a chromosome with one functional 2D6 gene copy. The other chromosome has either a copy with a defective functional polymorphism or a deletion of the gene. These patients usually tolerate 2D6 substrate drugs in low dosages.
Poor metabolizer: ‘medication-sensitive’
Olga, Katrina’s mother, has always lived in northern Sweden. She has no psychiatric history except for one psychotic episode that required hospitalization.
Her psychotic illness began on the summer solstice, during an all-night celebration. In addition to using unspecified recreational drugs, she took three 20-mg capsules of fluoxetine that her friend told her would make her feel high. She instead developed acute mania and dramatic paranoid delusions.
Figure 2 Possible genotypes of Brad, son of Abdul and Katrina
Olga was hospitalized and treated with moderate doses of haloperidol that precipitated an acute dystonic reaction. She was subsequently given ben-ztropine, and her extrapyramidal symptoms resolved. After discharge, she was treated with haloperidol and benztropine for 2 years, after which she spontaneously discontinued these drugs against medical advice. Her psychotic illness has not recurred.
Knowing her own genotype, Katrina understood that her mother had a 50% probability of having one copy of the 2D6 *4 allele. Given her mother’s history of medication intolerance, Katrina believed that her mother’s psychiatric illness might have been related to a drug reaction. She persuaded her mother to send a blood sample to a laboratory in Stockholm.
Olga’s genotype was *4/*4, indicating that she would be unlikely to tolerate even moderate doses of 2D6 substrate medications (Figure 1). Given her complete recovery and continued good health without medication, the most probable retrospective diagnosis was drug-induced psychosis. Her 2-year neuroleptic treatment probably was unnecessary.
Figure 3 Genogram for Brad, son of Abdul and Katrina
Poor metabolizers without a functional 2D6 gene copy have low tolerance for many medications and often become labeled as “medication sensitive.” When genotyping reveals that an individual is a poor metabolizer, prescribing medications that do not require 2D6 metabolism is usually prudent.
In rare cases, poor metabolizers have died from normal doses of 2D6 substrate medications.7 Far more commonly, however, they spontaneously discontinue taking these drugs because of adverse side effects.
Benefits of prospective testing
When used in clinical practice, pharmacogenomic testing’s two goals are to identify:
- ultrarapid metabolizers, who will not benefit from a medication
- poor metabolizers, who likely will have adverse responses to a medication.
The following case demonstrates the benefit of prospective 2D6 genotyping:
Brad, age 14, is the son of Abdul and Katrina, whose genotypes have been described. Brad developed a serious depression that was similar in severity and onset to an illness his mother experienced as a teen.
Brad’s parents want him to get the maximum benefit from psychopharmacologic treatment while avoiding distressing side effects. He had been healthy and had received no prescriptions other than antibiotics in the past.
How would you proceed? Without knowing Brad’s parents’ genotypes, you might reason that Brad would resemble one of them in drug response. However, when you review each parent’s genotype, you realize four scenarios are possible (Figure 2):
- Brad has a high likelihood of being an ultrarapid metabolizer because he has a 50% chance of inheriting a chromosome with five copies of the 2D6 gene from his father. He inherited the *1 or *4 form from his mother, but the effect of either will be clinically irrelevant.
- If Brad inherited the chromosome with the deletion from his father and the *1 form from his mother, he would be an intermediate metabolizer, as is his mother.
- If he inherited the chromosome with the deletion from his father and the *4 form from his mother, he would be a poor metabolizer like his grandmother, Olga. He would be at substantial risk for adverse reactions (such as intense headaches or vomiting) to 2D6 substrate medications.
On testing, Brad was found to be a poor metabolizer (Figure 3) The psychiatrist prescribed bupropion, which is metabolized by the 2B6 enzyme rather than the 2D6 enzyme.
Conclusion. To introduce the concept of genotypic testing, this review has focused on simple illustrations of variations in a single gene. However, many genes in the P-450 family play important roles in metabolizing psychotropics. In the future, genotyping of panels of these genes will likely provide more-specific guidance than can be achieved by simply testing one gene at a time.
Related resources
- Lerer B (ed). Pharmacogenetics of psychotropic drugs. Cambridge, UK: Cambridge University Press, 2002.
- Kirchheiner J, Borsen K, Dahl ML, et al. CYP2D6 and CYP2C19 genotype-based dose recommendations for antidepressants: a first step towards subpopulation-specific dosages. Acta Psychiatr Scand 2001;103(3):173-92.
- Indiana University School of Medicine, Division of Clinical Pharmacology. Drug Interactions—Defining Genetic Influences on Pharmacologic Responses. http://medicine.iupui.edu/flockhart.
Drug brand names
- Acetaminophen w/codeine phosphate • Tylenol w/codeine
- Atomoxetine • Strattera
- Benztropine mesylate • Cogentin
- Bupropion • Wellbutrin
- Desipramine • Norpramin
- Fluoxetine • Prozac
- Fluphenazine • Prolixin
- Haloperidol • Haldol
- Nortriptyline • Aventyl, Pamelor
- Oxycodone • Oxycontin
- Paroxetine • Paxil
- Perphenazine • Trilafon
- Risperidone • Risperdal
- Sertraline • Zoloft
- Thioridazine • Mellaril
- Venlafaxine • Effexor
Disclosure
Dr. Mrazek reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Mrazek DA. Clinical genomic testing. In: Wiener J, Dulcan M (eds). Textbook of child and adolescent psychiatry (3rd ed). Washington, DC: American Psychiatric Publishing, Inc., 2001;193-203.
2. Mrazek DA. Pharmacogenomic screening for depressed children and adolescents (scientific proceedings). Miami Beach, FL: American Academy of Child and Adolescent Psychiatry annual meeting, 2003;159.-
3. Phillips KA, Veenstra DL, Oren E, et al. Potential role of pharmacogenomics in reducing adverse drug reactions. JAMA 2001;286(18):2270-9.
4. Shi MM, Mehrens D, Dacus K. Pharmacogenomics: Changing the health care paradigm. Modern Drug Discovery 2001;4(7):27-32.
5. Kirchheiner J, Brosen K, Dahl ML, et al. CYP2D6 and CYP2C19 genotype-based dose recommendations for antidepressants: a first step towards subpopulation-specific dosages. Acta Psychiatr Scand 2001;104:173-92.
6. Masimirembwa CM, Hasler JA. Genetic polymorphism of drug metabolising enzymes in African populations: implications for the use of neuroleptics and antidepressants. Brain Res Bull 1997;44(5):561-71.
7. Sallee FR, DeVane CL, Ferrell RE. Fluoxetine-related death in a child with cytochrome P-450 2D6 genetic deficiency. J Child Adolesc Psychopharmacol 2000;10(1):27-34.
8. Gaedigk A, Gotschall RR, Forbes NS, et al. Optimization of cytochrome P4502D6 (CYP2D6) phenotype assignment using a genotyping algorithm based on allele frequency data. Pharmacogenetics 1999;9(6):669-82.
1. Mrazek DA. Clinical genomic testing. In: Wiener J, Dulcan M (eds). Textbook of child and adolescent psychiatry (3rd ed). Washington, DC: American Psychiatric Publishing, Inc., 2001;193-203.
2. Mrazek DA. Pharmacogenomic screening for depressed children and adolescents (scientific proceedings). Miami Beach, FL: American Academy of Child and Adolescent Psychiatry annual meeting, 2003;159.-
3. Phillips KA, Veenstra DL, Oren E, et al. Potential role of pharmacogenomics in reducing adverse drug reactions. JAMA 2001;286(18):2270-9.
4. Shi MM, Mehrens D, Dacus K. Pharmacogenomics: Changing the health care paradigm. Modern Drug Discovery 2001;4(7):27-32.
5. Kirchheiner J, Brosen K, Dahl ML, et al. CYP2D6 and CYP2C19 genotype-based dose recommendations for antidepressants: a first step towards subpopulation-specific dosages. Acta Psychiatr Scand 2001;104:173-92.
6. Masimirembwa CM, Hasler JA. Genetic polymorphism of drug metabolising enzymes in African populations: implications for the use of neuroleptics and antidepressants. Brain Res Bull 1997;44(5):561-71.
7. Sallee FR, DeVane CL, Ferrell RE. Fluoxetine-related death in a child with cytochrome P-450 2D6 genetic deficiency. J Child Adolesc Psychopharmacol 2000;10(1):27-34.
8. Gaedigk A, Gotschall RR, Forbes NS, et al. Optimization of cytochrome P4502D6 (CYP2D6) phenotype assignment using a genotyping algorithm based on allele frequency data. Pharmacogenetics 1999;9(6):669-82.
Is depression neurochemical or neurodegenerative?
A relative lack of important neurotransmitters is popularly believed to cause depression. This “monoamine hypothesis” makes sense: We give patients “chemicals,” and many depression symptoms improve. Just because an antidepressant works, however, does not mean that a “chemical imbalance” is causing the depression.
For one, 40 years of research has not consistently found diminished neurotransmitters or their metabolites in depressed persons. Also, the monoamine hypothesis fails to explain why clinical improvement can be delayed for weeks, even though antidepressants rapidly increase extracellular serotonin and norepinephrine.
Figure 1 How neural cells differentiate in the brain
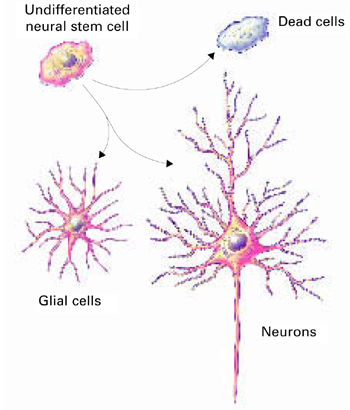
In neurogenesis, undifferentiated neural stem cells proliferate and migrate in the brain. Approximately one-half develop into useful cells, such as neurons and glial cells, and one-half die.
Source: Ilustration for CURRENTPSYCHIATRY by Maura Flynn
Increasing evidence suggests that depression may be a subtle neurodegenerative disorder. Postmortem and imaging studies have consistently found atrophy or neuron loss in the prefrontal cortices and hippocampi of depressed patients.1 Some studies suggest that antidepressants prevent the atrophy.2
Figure 2 Neural cell development may explain delayed antidepressant effect
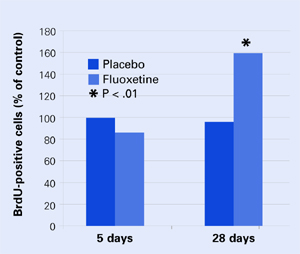
In mouse brains, fluoxetine—but not placebo—increased the number of recently developed neuralcells (as shown by the injected marker 5-bromo-2’- deoxyuridine [BrdU]), but only after 28 days.
Source: Adapted from reference 4.
NEW VIEWS ON NEUROGENESIS
Until recently, the brain was believed incapable of generating nerve cells. We were thought to be born with our entire allotment of brain cells and we could only lose them because of age, trauma, or toxins. Within the past 5 years, however, it has become clear that human neuronal stem cells are capable of neurogenesis (Figure 1).3
Neurogenesis is an ongoing process in the brain, and depression may result from a relative decrease in new neuron development. Effective depression treatments may work by stimulating neurogenesis, and Santarelli et al4 offer compelling support for this idea.
A group of mice was treated orally with fluoxetine or placebo. Several from each group were sacrificed after 5 days and others at 28 days. Twenty-four hours before being sacrificed, each mouse was injected with 5-bromo-2’-deoxyuridine (BrdU), which is incorporated into DNA and serves as a marker for recently developed nerve cells.
None of the mice at 5 days showed any change in BrdU-positive cells, and only the mice who received fluoxetine showed an increase in new cells after 28 days Figure 2. These results correlated with a greater willingness after 28 days by those mice on fluoxetine to venture into open, lighted areas—a behavioral change that was not seen at 5 days or in the placebo group.
This study shows that an antidepressant can increase development of new neurons and does so in a time course similar to the onset of efficacy seen in human clinical trials.
CHEMICAL OR CELLULAR?
One can speculate that insufficient neurogenesis is a possible cause of depression and that effective depression treatments may reverse that problem. Supporting this concept are other studies showing that lithium and electroconvulsive therapy—well-known treatments for depression—also increase neurogenesis.5
All this evidence makes depression look more like a cellular than a chemical imbalance.
1. Manji HK, Quiroz JA, Sporn J, et al. Enhancing neuronal plasticity and cellular resilience to develop novel, improved therapeutics for difficult-to-treat depression. Biol Psychiatry 2003;53:707-42.
2. Sheline YI, Gado MH, Kraemer HC. Untreated depression and hippocampal volume loss. Am J Psychiatry 2003;160:1516-8.
3. Gage FH. Brain, repair yourself. Sci Am 2003;289:46-53.
4. Santarelli L, Saxe M, Gross C, et al. Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science 2003;301:805-9.
5. Kempermann G, Kronenberg G. Depressed new neurons—adult hippocampal neurogenesis and a cellular plasticity hypothesis of major depression. Biol Psychiatry 2003;54:499-503.
A relative lack of important neurotransmitters is popularly believed to cause depression. This “monoamine hypothesis” makes sense: We give patients “chemicals,” and many depression symptoms improve. Just because an antidepressant works, however, does not mean that a “chemical imbalance” is causing the depression.
For one, 40 years of research has not consistently found diminished neurotransmitters or their metabolites in depressed persons. Also, the monoamine hypothesis fails to explain why clinical improvement can be delayed for weeks, even though antidepressants rapidly increase extracellular serotonin and norepinephrine.
Figure 1 How neural cells differentiate in the brain
In neurogenesis, undifferentiated neural stem cells proliferate and migrate in the brain. Approximately one-half develop into useful cells, such as neurons and glial cells, and one-half die.
Source: Ilustration for CURRENTPSYCHIATRY by Maura Flynn
Increasing evidence suggests that depression may be a subtle neurodegenerative disorder. Postmortem and imaging studies have consistently found atrophy or neuron loss in the prefrontal cortices and hippocampi of depressed patients.1 Some studies suggest that antidepressants prevent the atrophy.2
Figure 2 Neural cell development may explain delayed antidepressant effect
In mouse brains, fluoxetine—but not placebo—increased the number of recently developed neuralcells (as shown by the injected marker 5-bromo-2’- deoxyuridine [BrdU]), but only after 28 days.
Source: Adapted from reference 4.
NEW VIEWS ON NEUROGENESIS
Until recently, the brain was believed incapable of generating nerve cells. We were thought to be born with our entire allotment of brain cells and we could only lose them because of age, trauma, or toxins. Within the past 5 years, however, it has become clear that human neuronal stem cells are capable of neurogenesis (Figure 1).3
Neurogenesis is an ongoing process in the brain, and depression may result from a relative decrease in new neuron development. Effective depression treatments may work by stimulating neurogenesis, and Santarelli et al4 offer compelling support for this idea.
A group of mice was treated orally with fluoxetine or placebo. Several from each group were sacrificed after 5 days and others at 28 days. Twenty-four hours before being sacrificed, each mouse was injected with 5-bromo-2’-deoxyuridine (BrdU), which is incorporated into DNA and serves as a marker for recently developed nerve cells.
None of the mice at 5 days showed any change in BrdU-positive cells, and only the mice who received fluoxetine showed an increase in new cells after 28 days Figure 2. These results correlated with a greater willingness after 28 days by those mice on fluoxetine to venture into open, lighted areas—a behavioral change that was not seen at 5 days or in the placebo group.
This study shows that an antidepressant can increase development of new neurons and does so in a time course similar to the onset of efficacy seen in human clinical trials.
CHEMICAL OR CELLULAR?
One can speculate that insufficient neurogenesis is a possible cause of depression and that effective depression treatments may reverse that problem. Supporting this concept are other studies showing that lithium and electroconvulsive therapy—well-known treatments for depression—also increase neurogenesis.5
All this evidence makes depression look more like a cellular than a chemical imbalance.
A relative lack of important neurotransmitters is popularly believed to cause depression. This “monoamine hypothesis” makes sense: We give patients “chemicals,” and many depression symptoms improve. Just because an antidepressant works, however, does not mean that a “chemical imbalance” is causing the depression.
For one, 40 years of research has not consistently found diminished neurotransmitters or their metabolites in depressed persons. Also, the monoamine hypothesis fails to explain why clinical improvement can be delayed for weeks, even though antidepressants rapidly increase extracellular serotonin and norepinephrine.
Figure 1 How neural cells differentiate in the brain
In neurogenesis, undifferentiated neural stem cells proliferate and migrate in the brain. Approximately one-half develop into useful cells, such as neurons and glial cells, and one-half die.
Source: Ilustration for CURRENTPSYCHIATRY by Maura Flynn
Increasing evidence suggests that depression may be a subtle neurodegenerative disorder. Postmortem and imaging studies have consistently found atrophy or neuron loss in the prefrontal cortices and hippocampi of depressed patients.1 Some studies suggest that antidepressants prevent the atrophy.2
Figure 2 Neural cell development may explain delayed antidepressant effect
In mouse brains, fluoxetine—but not placebo—increased the number of recently developed neuralcells (as shown by the injected marker 5-bromo-2’- deoxyuridine [BrdU]), but only after 28 days.
Source: Adapted from reference 4.
NEW VIEWS ON NEUROGENESIS
Until recently, the brain was believed incapable of generating nerve cells. We were thought to be born with our entire allotment of brain cells and we could only lose them because of age, trauma, or toxins. Within the past 5 years, however, it has become clear that human neuronal stem cells are capable of neurogenesis (Figure 1).3
Neurogenesis is an ongoing process in the brain, and depression may result from a relative decrease in new neuron development. Effective depression treatments may work by stimulating neurogenesis, and Santarelli et al4 offer compelling support for this idea.
A group of mice was treated orally with fluoxetine or placebo. Several from each group were sacrificed after 5 days and others at 28 days. Twenty-four hours before being sacrificed, each mouse was injected with 5-bromo-2’-deoxyuridine (BrdU), which is incorporated into DNA and serves as a marker for recently developed nerve cells.
None of the mice at 5 days showed any change in BrdU-positive cells, and only the mice who received fluoxetine showed an increase in new cells after 28 days Figure 2. These results correlated with a greater willingness after 28 days by those mice on fluoxetine to venture into open, lighted areas—a behavioral change that was not seen at 5 days or in the placebo group.
This study shows that an antidepressant can increase development of new neurons and does so in a time course similar to the onset of efficacy seen in human clinical trials.
CHEMICAL OR CELLULAR?
One can speculate that insufficient neurogenesis is a possible cause of depression and that effective depression treatments may reverse that problem. Supporting this concept are other studies showing that lithium and electroconvulsive therapy—well-known treatments for depression—also increase neurogenesis.5
All this evidence makes depression look more like a cellular than a chemical imbalance.
1. Manji HK, Quiroz JA, Sporn J, et al. Enhancing neuronal plasticity and cellular resilience to develop novel, improved therapeutics for difficult-to-treat depression. Biol Psychiatry 2003;53:707-42.
2. Sheline YI, Gado MH, Kraemer HC. Untreated depression and hippocampal volume loss. Am J Psychiatry 2003;160:1516-8.
3. Gage FH. Brain, repair yourself. Sci Am 2003;289:46-53.
4. Santarelli L, Saxe M, Gross C, et al. Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science 2003;301:805-9.
5. Kempermann G, Kronenberg G. Depressed new neurons—adult hippocampal neurogenesis and a cellular plasticity hypothesis of major depression. Biol Psychiatry 2003;54:499-503.
1. Manji HK, Quiroz JA, Sporn J, et al. Enhancing neuronal plasticity and cellular resilience to develop novel, improved therapeutics for difficult-to-treat depression. Biol Psychiatry 2003;53:707-42.
2. Sheline YI, Gado MH, Kraemer HC. Untreated depression and hippocampal volume loss. Am J Psychiatry 2003;160:1516-8.
3. Gage FH. Brain, repair yourself. Sci Am 2003;289:46-53.
4. Santarelli L, Saxe M, Gross C, et al. Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science 2003;301:805-9.
5. Kempermann G, Kronenberg G. Depressed new neurons—adult hippocampal neurogenesis and a cellular plasticity hypothesis of major depression. Biol Psychiatry 2003;54:499-503.
Assessing suicide risk
“Suicide risk assessment: Questions that reveal what you really need to know” (Current Psychiatry, July 2004) points out the critical information psychiatrists need to obtain from the patient and from the suicide gesture’s circumstances to determine the true risk.
The article by Dr. Rajnish Mago and colleagues was to-the-point and based on real issues. Thank you for publishing such a useful article.
Maria S. Arrubla, MD
Veterans Administration Medical Center
Leeds, MA
“Suicide risk assessment: Questions that reveal what you really need to know” (Current Psychiatry, July 2004) points out the critical information psychiatrists need to obtain from the patient and from the suicide gesture’s circumstances to determine the true risk.
The article by Dr. Rajnish Mago and colleagues was to-the-point and based on real issues. Thank you for publishing such a useful article.
Maria S. Arrubla, MD
Veterans Administration Medical Center
Leeds, MA
“Suicide risk assessment: Questions that reveal what you really need to know” (Current Psychiatry, July 2004) points out the critical information psychiatrists need to obtain from the patient and from the suicide gesture’s circumstances to determine the true risk.
The article by Dr. Rajnish Mago and colleagues was to-the-point and based on real issues. Thank you for publishing such a useful article.
Maria S. Arrubla, MD
Veterans Administration Medical Center
Leeds, MA
Promiscuous prairie voles
I enjoyed “Understanding the ‘joy’ of aggression.”
Does Dr. Higgins know about investigations of oxycodone’s effect on bonding between people? About 2 years ago, a public radio program addressed brain oxycodone in prairie voles. One vole that mated for life had high levels of brain oxycodone. Nonmonogamous prairie voles had low oxycodone levels.
It would be interesting to see whether aggression alters oxycodone levels, or if dosing before the animals become angry changes the pleasure response in the brain.
George Hilton, MD
Great Bay Mental Health Associates
Dover, NH
Dr. Higgins responds
Yes, there is an association between oxycodone and bonding.
The more-interesting neuropeptide—on which data exist relevant to aggression—is vasopressin. Both oxycodone and vasopressin are produced by the hypothalamus.
Recent research has associated vasopressin with bonding1 and has shown that the solitary, promiscuous vole became monogamous when investigators increased receptors for vasopressin in the rodent’s ventral forebrain.
Of relevance to aggression is an article showing an association between CSF vasopressin and violence.2 The researchers found that more vasopressin was associated with more violence.
Both studies show an important link between neuropeptides and social affiliation—or lack thereof.
Edmund S. Higgins, MD
Clinical associate professor of family medicine and psychiatry
Medical University of South Carolina, Charleston
- Lim MM, Wang Z, Olazabal DE, et al. Enhanced partner preference in a promiscuous species by manipulating the expression of a single gene. Nature 2004;429:754–7.
- Coccaro EF, Kavoussi RJ, Hauger RL, et al. Cerebrospinal fluid vasopressin levels correlate with aggression and serotonin function in personality-disordered subjects. Arch Gen Psych 1998;55:708–14.
I enjoyed “Understanding the ‘joy’ of aggression.”
Does Dr. Higgins know about investigations of oxycodone’s effect on bonding between people? About 2 years ago, a public radio program addressed brain oxycodone in prairie voles. One vole that mated for life had high levels of brain oxycodone. Nonmonogamous prairie voles had low oxycodone levels.
It would be interesting to see whether aggression alters oxycodone levels, or if dosing before the animals become angry changes the pleasure response in the brain.
George Hilton, MD
Great Bay Mental Health Associates
Dover, NH
Dr. Higgins responds
Yes, there is an association between oxycodone and bonding.
The more-interesting neuropeptide—on which data exist relevant to aggression—is vasopressin. Both oxycodone and vasopressin are produced by the hypothalamus.
Recent research has associated vasopressin with bonding1 and has shown that the solitary, promiscuous vole became monogamous when investigators increased receptors for vasopressin in the rodent’s ventral forebrain.
Of relevance to aggression is an article showing an association between CSF vasopressin and violence.2 The researchers found that more vasopressin was associated with more violence.
Both studies show an important link between neuropeptides and social affiliation—or lack thereof.
Edmund S. Higgins, MD
Clinical associate professor of family medicine and psychiatry
Medical University of South Carolina, Charleston
- Lim MM, Wang Z, Olazabal DE, et al. Enhanced partner preference in a promiscuous species by manipulating the expression of a single gene. Nature 2004;429:754–7.
- Coccaro EF, Kavoussi RJ, Hauger RL, et al. Cerebrospinal fluid vasopressin levels correlate with aggression and serotonin function in personality-disordered subjects. Arch Gen Psych 1998;55:708–14.
I enjoyed “Understanding the ‘joy’ of aggression.”
Does Dr. Higgins know about investigations of oxycodone’s effect on bonding between people? About 2 years ago, a public radio program addressed brain oxycodone in prairie voles. One vole that mated for life had high levels of brain oxycodone. Nonmonogamous prairie voles had low oxycodone levels.
It would be interesting to see whether aggression alters oxycodone levels, or if dosing before the animals become angry changes the pleasure response in the brain.
George Hilton, MD
Great Bay Mental Health Associates
Dover, NH
Dr. Higgins responds
Yes, there is an association between oxycodone and bonding.
The more-interesting neuropeptide—on which data exist relevant to aggression—is vasopressin. Both oxycodone and vasopressin are produced by the hypothalamus.
Recent research has associated vasopressin with bonding1 and has shown that the solitary, promiscuous vole became monogamous when investigators increased receptors for vasopressin in the rodent’s ventral forebrain.
Of relevance to aggression is an article showing an association between CSF vasopressin and violence.2 The researchers found that more vasopressin was associated with more violence.
Both studies show an important link between neuropeptides and social affiliation—or lack thereof.
Edmund S. Higgins, MD
Clinical associate professor of family medicine and psychiatry
Medical University of South Carolina, Charleston
- Lim MM, Wang Z, Olazabal DE, et al. Enhanced partner preference in a promiscuous species by manipulating the expression of a single gene. Nature 2004;429:754–7.
- Coccaro EF, Kavoussi RJ, Hauger RL, et al. Cerebrospinal fluid vasopressin levels correlate with aggression and serotonin function in personality-disordered subjects. Arch Gen Psych 1998;55:708–14.
Why some of us love music
As a psychiatrist and music lover, I wonder if Dr. Higgins knows of any quality research that addresses why some of us love music.
I’ve talked with a couple of musicians and psychiatrists about this, and their reactions have been blank stares or even a little resentment. I think music is just assumed to be exciting, and that’s that. But why?
Arnold Knepfer, MD
Corte Madera, CA
Dr Higgins responds
I believe that a little squirt of dopamine at the nucleus accumbens reinforces any activity we enjoy.
After comparing PET scans, Jeffries et al1 found relative increased activity at the nucleus accumbens and other areas when the subjects were singing familiar songs compared to when they were speaking.
This may explain why some music is so pleasurable. I imagine the music we love gives a bigger squirt than the stuff they play on AM radio.
What is the survival value of this? I would speculate that music—commonly a group activity—enhances social connectedness and hence protection, but that’s just a guess.
- Jeffries KJ, Fritz JB, Braun AR. Words in melody: an H(2)15O PET study of brain activation during singing and speaking. Neuroreport 2003;14:749–54.
As a psychiatrist and music lover, I wonder if Dr. Higgins knows of any quality research that addresses why some of us love music.
I’ve talked with a couple of musicians and psychiatrists about this, and their reactions have been blank stares or even a little resentment. I think music is just assumed to be exciting, and that’s that. But why?
Arnold Knepfer, MD
Corte Madera, CA
Dr Higgins responds
I believe that a little squirt of dopamine at the nucleus accumbens reinforces any activity we enjoy.
After comparing PET scans, Jeffries et al1 found relative increased activity at the nucleus accumbens and other areas when the subjects were singing familiar songs compared to when they were speaking.
This may explain why some music is so pleasurable. I imagine the music we love gives a bigger squirt than the stuff they play on AM radio.
What is the survival value of this? I would speculate that music—commonly a group activity—enhances social connectedness and hence protection, but that’s just a guess.
- Jeffries KJ, Fritz JB, Braun AR. Words in melody: an H(2)15O PET study of brain activation during singing and speaking. Neuroreport 2003;14:749–54.
As a psychiatrist and music lover, I wonder if Dr. Higgins knows of any quality research that addresses why some of us love music.
I’ve talked with a couple of musicians and psychiatrists about this, and their reactions have been blank stares or even a little resentment. I think music is just assumed to be exciting, and that’s that. But why?
Arnold Knepfer, MD
Corte Madera, CA
Dr Higgins responds
I believe that a little squirt of dopamine at the nucleus accumbens reinforces any activity we enjoy.
After comparing PET scans, Jeffries et al1 found relative increased activity at the nucleus accumbens and other areas when the subjects were singing familiar songs compared to when they were speaking.
This may explain why some music is so pleasurable. I imagine the music we love gives a bigger squirt than the stuff they play on AM radio.
What is the survival value of this? I would speculate that music—commonly a group activity—enhances social connectedness and hence protection, but that’s just a guess.
- Jeffries KJ, Fritz JB, Braun AR. Words in melody: an H(2)15O PET study of brain activation during singing and speaking. Neuroreport 2003;14:749–54.
Ethics and patient privacy
I was dismayed that in his article “Understanding the ‘joy’ of aggression” (Neuroscience News, Current Psychiatry, July 2004), Dr. Edmund Higgins discussed the pathology of the specific individuals shown in the photo that accompanied the article. Publicly attributing pathology to people (in this case, U.S. soldiers at Iraq’s Abu Ghraib prison) is inconsistent with our historical role as doctors who are sympathetic to our patients and respect their privacy.
As physicians, we encourage patients to share with us their problems, secure in knowing that they have our sympathy, that we advocate for them, and that we apply on their behalf our understanding and professional knowledge. These professional values are worth maintaining.
Whatever our feelings about atrocities committed by soldiers, individuals should be able to expect these values from medical doctors. The American Psychiatric Association’s Principles of Medical Ethics with Annotations Especially Applicable to Psychiatry addresses this issue:
“Psychiatrists occasionally are asked for an opinion about an individual in the light of public attention, or who has disclosed information about himself or herself through the media. In such circumstances, a psychiatrist may share with the public his/her expertise about psychiatric issues in general. However, it is unethical for a psychiatrist to offer a professional opinion unless he/she has conducted an examination and has been granted proper authorization for such a statement (1998 edition, section 7-3).”
Alan Lipschitz, MD
North Wales, PA
Dr. Higgins responds
Dr. Lipschitz raises an excellent point—one we must always keep in mind in these times of high media attention. His point does not apply to my article, however.
No individual was singled out nor were any names included. Likewise, no psychiatric diagnosis was made nor was any treatment suggested for any individual. The Abu Ghraib prison situation was raised simply as an example to speculate about brain activity during aggressive acts in general.
I was dismayed that in his article “Understanding the ‘joy’ of aggression” (Neuroscience News, Current Psychiatry, July 2004), Dr. Edmund Higgins discussed the pathology of the specific individuals shown in the photo that accompanied the article. Publicly attributing pathology to people (in this case, U.S. soldiers at Iraq’s Abu Ghraib prison) is inconsistent with our historical role as doctors who are sympathetic to our patients and respect their privacy.
As physicians, we encourage patients to share with us their problems, secure in knowing that they have our sympathy, that we advocate for them, and that we apply on their behalf our understanding and professional knowledge. These professional values are worth maintaining.
Whatever our feelings about atrocities committed by soldiers, individuals should be able to expect these values from medical doctors. The American Psychiatric Association’s Principles of Medical Ethics with Annotations Especially Applicable to Psychiatry addresses this issue:
“Psychiatrists occasionally are asked for an opinion about an individual in the light of public attention, or who has disclosed information about himself or herself through the media. In such circumstances, a psychiatrist may share with the public his/her expertise about psychiatric issues in general. However, it is unethical for a psychiatrist to offer a professional opinion unless he/she has conducted an examination and has been granted proper authorization for such a statement (1998 edition, section 7-3).”
Alan Lipschitz, MD
North Wales, PA
Dr. Higgins responds
Dr. Lipschitz raises an excellent point—one we must always keep in mind in these times of high media attention. His point does not apply to my article, however.
No individual was singled out nor were any names included. Likewise, no psychiatric diagnosis was made nor was any treatment suggested for any individual. The Abu Ghraib prison situation was raised simply as an example to speculate about brain activity during aggressive acts in general.
I was dismayed that in his article “Understanding the ‘joy’ of aggression” (Neuroscience News, Current Psychiatry, July 2004), Dr. Edmund Higgins discussed the pathology of the specific individuals shown in the photo that accompanied the article. Publicly attributing pathology to people (in this case, U.S. soldiers at Iraq’s Abu Ghraib prison) is inconsistent with our historical role as doctors who are sympathetic to our patients and respect their privacy.
As physicians, we encourage patients to share with us their problems, secure in knowing that they have our sympathy, that we advocate for them, and that we apply on their behalf our understanding and professional knowledge. These professional values are worth maintaining.
Whatever our feelings about atrocities committed by soldiers, individuals should be able to expect these values from medical doctors. The American Psychiatric Association’s Principles of Medical Ethics with Annotations Especially Applicable to Psychiatry addresses this issue:
“Psychiatrists occasionally are asked for an opinion about an individual in the light of public attention, or who has disclosed information about himself or herself through the media. In such circumstances, a psychiatrist may share with the public his/her expertise about psychiatric issues in general. However, it is unethical for a psychiatrist to offer a professional opinion unless he/she has conducted an examination and has been granted proper authorization for such a statement (1998 edition, section 7-3).”
Alan Lipschitz, MD
North Wales, PA
Dr. Higgins responds
Dr. Lipschitz raises an excellent point—one we must always keep in mind in these times of high media attention. His point does not apply to my article, however.
No individual was singled out nor were any names included. Likewise, no psychiatric diagnosis was made nor was any treatment suggested for any individual. The Abu Ghraib prison situation was raised simply as an example to speculate about brain activity during aggressive acts in general.