User login
The Official Newspaper of the American Association for Thoracic Surgery
FDA approves biosimilar to bevacizumab
The Food and Drug Administration has approved a biosimilar to bevacizumab (Avastin) for the treatment of certain colorectal, lung, brain, kidney, and cervical cancers.
Bevacizumab-awwb is the first biosimilar approved in the United States for the treatment of cancer, the FDA said in a press release.
Approval is based on structural and functional characterization, animal study data, human pharmacokinetic and pharmacodynamics data, clinical immunogenicity data, and other clinical safety and effectiveness data that demonstrate bevacizumab-awwb is biosimilar to bevacizumab, the FDA said.
• Metastatic colorectal cancer, in combination with intravenous 5-fluorouracil-based chemotherapy for first- or second-line treatment.
• Metastatic colorectal cancer, in combination with fluoropyrimidine-irinotecan–based or fluoropyrimidine-oxaliplatin–based chemotherapy for the second-line treatment of patients who have progressed on a first-line bevacizumab product–containing regimen.
• Non-squamous non–small cell lung cancer, in combination with carboplatin and paclitaxel for first line treatment of unresectable, locally advanced, recurrent, or metastatic disease.
• Glioblastoma with progressive disease following prior therapy, based on improvement in objective response rate.
• Metastatic renal cell carcinoma, in combination with interferon alfa.
• Cervical cancer that is persistent, recurrent, or metastatic, in combination with paclitaxel and cisplatin or paclitaxel and topotecan.
Common expected side effects of the biosimilar include epistaxis, headache, hypertension, rhinitis, proteinuria, taste alteration, dry skin, hemorrhage, lacrimation disorder, back pain, and exfoliative dermatitis.
Serious expected side effects include perforation or fistula, arterial and venous thromboembolic events, hypertension, posterior reversible encephalopathy syndrome, proteinuria, infusion-related reactions, and ovarian failure. Women who are pregnant should not take bevacizumab-awwb.
The biosimilar to bevacizumab carries a similar boxed warning regarding the increased risk of gastrointestinal perforations; surgery and wound healing complications; and severe or fatal pulmonary, gastrointestinal, central nervous system, and vaginal hemorrhage.
The biosimilar approval was granted to Amgen, which will market the drug under the trade name Mvasi.
The Food and Drug Administration has approved a biosimilar to bevacizumab (Avastin) for the treatment of certain colorectal, lung, brain, kidney, and cervical cancers.
Bevacizumab-awwb is the first biosimilar approved in the United States for the treatment of cancer, the FDA said in a press release.
Approval is based on structural and functional characterization, animal study data, human pharmacokinetic and pharmacodynamics data, clinical immunogenicity data, and other clinical safety and effectiveness data that demonstrate bevacizumab-awwb is biosimilar to bevacizumab, the FDA said.
• Metastatic colorectal cancer, in combination with intravenous 5-fluorouracil-based chemotherapy for first- or second-line treatment.
• Metastatic colorectal cancer, in combination with fluoropyrimidine-irinotecan–based or fluoropyrimidine-oxaliplatin–based chemotherapy for the second-line treatment of patients who have progressed on a first-line bevacizumab product–containing regimen.
• Non-squamous non–small cell lung cancer, in combination with carboplatin and paclitaxel for first line treatment of unresectable, locally advanced, recurrent, or metastatic disease.
• Glioblastoma with progressive disease following prior therapy, based on improvement in objective response rate.
• Metastatic renal cell carcinoma, in combination with interferon alfa.
• Cervical cancer that is persistent, recurrent, or metastatic, in combination with paclitaxel and cisplatin or paclitaxel and topotecan.
Common expected side effects of the biosimilar include epistaxis, headache, hypertension, rhinitis, proteinuria, taste alteration, dry skin, hemorrhage, lacrimation disorder, back pain, and exfoliative dermatitis.
Serious expected side effects include perforation or fistula, arterial and venous thromboembolic events, hypertension, posterior reversible encephalopathy syndrome, proteinuria, infusion-related reactions, and ovarian failure. Women who are pregnant should not take bevacizumab-awwb.
The biosimilar to bevacizumab carries a similar boxed warning regarding the increased risk of gastrointestinal perforations; surgery and wound healing complications; and severe or fatal pulmonary, gastrointestinal, central nervous system, and vaginal hemorrhage.
The biosimilar approval was granted to Amgen, which will market the drug under the trade name Mvasi.
The Food and Drug Administration has approved a biosimilar to bevacizumab (Avastin) for the treatment of certain colorectal, lung, brain, kidney, and cervical cancers.
Bevacizumab-awwb is the first biosimilar approved in the United States for the treatment of cancer, the FDA said in a press release.
Approval is based on structural and functional characterization, animal study data, human pharmacokinetic and pharmacodynamics data, clinical immunogenicity data, and other clinical safety and effectiveness data that demonstrate bevacizumab-awwb is biosimilar to bevacizumab, the FDA said.
• Metastatic colorectal cancer, in combination with intravenous 5-fluorouracil-based chemotherapy for first- or second-line treatment.
• Metastatic colorectal cancer, in combination with fluoropyrimidine-irinotecan–based or fluoropyrimidine-oxaliplatin–based chemotherapy for the second-line treatment of patients who have progressed on a first-line bevacizumab product–containing regimen.
• Non-squamous non–small cell lung cancer, in combination with carboplatin and paclitaxel for first line treatment of unresectable, locally advanced, recurrent, or metastatic disease.
• Glioblastoma with progressive disease following prior therapy, based on improvement in objective response rate.
• Metastatic renal cell carcinoma, in combination with interferon alfa.
• Cervical cancer that is persistent, recurrent, or metastatic, in combination with paclitaxel and cisplatin or paclitaxel and topotecan.
Common expected side effects of the biosimilar include epistaxis, headache, hypertension, rhinitis, proteinuria, taste alteration, dry skin, hemorrhage, lacrimation disorder, back pain, and exfoliative dermatitis.
Serious expected side effects include perforation or fistula, arterial and venous thromboembolic events, hypertension, posterior reversible encephalopathy syndrome, proteinuria, infusion-related reactions, and ovarian failure. Women who are pregnant should not take bevacizumab-awwb.
The biosimilar to bevacizumab carries a similar boxed warning regarding the increased risk of gastrointestinal perforations; surgery and wound healing complications; and severe or fatal pulmonary, gastrointestinal, central nervous system, and vaginal hemorrhage.
The biosimilar approval was granted to Amgen, which will market the drug under the trade name Mvasi.
New data prompt update to ACC guidance on nonstatin LDL lowering
The American College of Cardiology Task Force on Expert Consensus Decision Pathways has released a “focused update” for the 2016 ACC Expert Consensus Decision Pathway (ECDP) on the role of nonstatin therapies for LDL-cholesterol lowering in the management of atherosclerotic cardiovascular disease (ASCVD) risk.
The update was deemed by the ECDP writing committee to be desirable given the additional evidence and perspectives that have emerged since the publication of the 2016 version, particularly with respect to the efficacy and safety of proprotein convertase subtilisin/kexin 9 (PCSK9) inhibitors for the secondary prevention of ASCVD, as well as the best use of ezetimibe in addition to statin therapy after acute coronary syndrome.

The ECDP algorithms endorse the four evidence-based statin benefit groups identified in the 2013 guidelines (adults aged 21 and older with clinical ASCVD, adults aged 21 and older with LDL-C of 190 mg/dL or greater, adults aged 40-75 years without ASCVD but with diabetes and with LDL-C of 70-189 mg/dL, and adults aged 40-75 without ASCVD or diabetes but with LDL-C of 70-189 mg/dL and an estimated 10-year risk for ASCVD of 7.5% or greater) and assume that the patient is currently taking or has attempted to take a statin, they noted.
Among the changes in the 2017 focused update are:
- Consideration of new randomized clinical trial data for the PCSK9 inhibitors evolocumab and bococizumab. Namely, they included results from the cardiovascular outcomes trials FOURIER (Further Cardiovascular Outcomes Research with PCSK9 Inhibition in Subjects with Elevated Risk) and SPIRE-1 and SPIRE-2 (Studies of PCSK9 Inhibition and the Reduction of Vascular Events), which were published in early 2017.
- An adjustment in the ECDP algorithms with respect to thresholds for consideration of net ASCVD risk reduction. The 2016 ECDP thresholds for risk reduction benefit were percent reduction in LDL-C with consideration of absolute LDL-C level in patients with clinical ASCVD, baseline LDL-C of 190 mg/dL or greater, and primary prevention. In patients with diabetes with or without clinical ASCVD, clinicians were allowed to consider absolute LDL-C and/or non-HDL-cholesterol levels. In the 2017 ECDP update, the thresholds are percent reduction in LDL-C with consideration of absolute LDL-C or non-HDL-C levels for patients in each of the four statin benefit groups. This change was based on the inclusion criteria of the FOURIER trial, the ongoing ODYSSEY Outcomes trial (Evaluation of Cardiovascular Outcomes after an Acute Coronary Syndrome During Treatment with Alirocumab), and the SPIRE-2 trial, all of which included non-HDL-C thresholds. “In alignment with these inclusion criteria, the 2017 Focused Update includes both LDL-C and non-HDL-C thresholds for evaluation of net ASCVD risk-reduction benefit when considering the addition of nonstatin therapies for patients in each of the four statin benefit groups” the update explained.
- An expansion of the threshold for consideration of net ASCVD risk-reduction benefit from a reduction of LDL C of at least 50%, as well as consideration of LDL-C less than 70 mg/dL or non-HDL-C less than 100 mg/dL for all patients (that is, both those with and those without comorbidities) who have clinical ASCVD and baseline LDL-C of 70-189 mg/dL. The 2016 ECDP had different thresholds for those with versus those without comorbidities. This change was based on findings from the FOURIER trial and the IMPROVE-IT (Improved Reduction of Outcomes: Vytorin Efficacy International Trial).“Based on consideration of all available evidence, the consensus of the writing committee members is that lower LDL-C levels are safe and optimal in patients with clinical ASCVD due to [their] increased risk of recurrent events,” they said.
- An expanded recommendation on the use of ezetimibe and PCSK9 inhibition. The 2016 ECDP stated that “if a decision is made to proceed with the addition of nonstatin therapy to maximally tolerated statin therapy, it is reasonable to consider the addition of ezetimibe as the initial agent and a PCSK9 inhibitor as the second agent.” However, based on the FOURIER findings, the ongoing ODYSSEY Outcomes trial, and the IMPROVE-IT trial, the 2017 Focused Update states that, if such a decision is made in patients with clinical ASCVD with comorbidities and baseline LDL-C of 70-189 mg/dL, it is reasonable to weigh the addition of either ezetimibe or a PCSK9 inhibitor in light of “considerations of the additional percent LDL-C reduction desired, patient preferences, costs, route of administration, and other factors.” The update also spells out considerations that may favor the initial choice of ezetimibe versus a PCSK9 inhibitor (such as requiring less than 25% additional lowering of LDL-C, an age of over 75 years, cost, and other patient factors and preferences) .
- Additional factors, based on the FOURIER trial results and inclusion criteria, that may be considered for the identification of higher-risk patients with clinical ASCVD. The 2016 ECDP included on this list diabetes, a recent ASCVD event, an ASCVD event while already taking a statin, poorly controlled other major ASCVD risk factors, elevated lipoprotein, chronic kidney disease, symptomatic heart failure, maintenance hemodialysis, and baseline LDL-C of at least 190 mg/dL not due to secondary causes. The 2017 update added being 65 years or older, prior MI or nonhemorrhagic stroke, current daily cigarette smoking, symptomatic peripheral artery disease with prior MI or stroke, history of non-MI related coronary revascularization, residual coronary artery disease with at least 40% stenosis in at least two large vessels, HDL-C less than 40 mg/dL for men and less than 50 mg/dL for women, high-sensitivity C-reactive protein greater than 2 mg/L, and metabolic syndrome.
The content of the full ECDP has been changed in accordance with these updates and now includes more extensive and detailed guidance for decision making – both in the text and in treatment algorithms.
Aspects that remain unchanged include the decision pathways and algorithms for the use of ezetimibe or PCSK9 inhibitors in primary prevention patients with LDL-C less than 190 mg/dL or in those without ASCVD and LDL-C of 190 mg/dL or greater unattributable to secondary causes.
In addition to other changes made for the purpose of clarification and consistency, recommendations regarding bile acid–sequestrant use were downgraded; these are now only recommended as optional secondary agents for consideration in patients who cannot tolerate ezetimibe.
“[These] recommendations attempt to provide practical guidance for clinicians and patients regarding the use of nonstatin therapies to further reduce ASCVD risk in situations not covered by the guideline until such time as the scientific evidence base expands and cardiovascular outcomes trials are completed with new agents for ASCVD risk reduction,” the committee concluded.
Dr. Lloyd-Jones reported having no disclosures.
The American College of Cardiology Task Force on Expert Consensus Decision Pathways has released a “focused update” for the 2016 ACC Expert Consensus Decision Pathway (ECDP) on the role of nonstatin therapies for LDL-cholesterol lowering in the management of atherosclerotic cardiovascular disease (ASCVD) risk.
The update was deemed by the ECDP writing committee to be desirable given the additional evidence and perspectives that have emerged since the publication of the 2016 version, particularly with respect to the efficacy and safety of proprotein convertase subtilisin/kexin 9 (PCSK9) inhibitors for the secondary prevention of ASCVD, as well as the best use of ezetimibe in addition to statin therapy after acute coronary syndrome.
The ECDP algorithms endorse the four evidence-based statin benefit groups identified in the 2013 guidelines (adults aged 21 and older with clinical ASCVD, adults aged 21 and older with LDL-C of 190 mg/dL or greater, adults aged 40-75 years without ASCVD but with diabetes and with LDL-C of 70-189 mg/dL, and adults aged 40-75 without ASCVD or diabetes but with LDL-C of 70-189 mg/dL and an estimated 10-year risk for ASCVD of 7.5% or greater) and assume that the patient is currently taking or has attempted to take a statin, they noted.
Among the changes in the 2017 focused update are:
- Consideration of new randomized clinical trial data for the PCSK9 inhibitors evolocumab and bococizumab. Namely, they included results from the cardiovascular outcomes trials FOURIER (Further Cardiovascular Outcomes Research with PCSK9 Inhibition in Subjects with Elevated Risk) and SPIRE-1 and SPIRE-2 (Studies of PCSK9 Inhibition and the Reduction of Vascular Events), which were published in early 2017.
- An adjustment in the ECDP algorithms with respect to thresholds for consideration of net ASCVD risk reduction. The 2016 ECDP thresholds for risk reduction benefit were percent reduction in LDL-C with consideration of absolute LDL-C level in patients with clinical ASCVD, baseline LDL-C of 190 mg/dL or greater, and primary prevention. In patients with diabetes with or without clinical ASCVD, clinicians were allowed to consider absolute LDL-C and/or non-HDL-cholesterol levels. In the 2017 ECDP update, the thresholds are percent reduction in LDL-C with consideration of absolute LDL-C or non-HDL-C levels for patients in each of the four statin benefit groups. This change was based on the inclusion criteria of the FOURIER trial, the ongoing ODYSSEY Outcomes trial (Evaluation of Cardiovascular Outcomes after an Acute Coronary Syndrome During Treatment with Alirocumab), and the SPIRE-2 trial, all of which included non-HDL-C thresholds. “In alignment with these inclusion criteria, the 2017 Focused Update includes both LDL-C and non-HDL-C thresholds for evaluation of net ASCVD risk-reduction benefit when considering the addition of nonstatin therapies for patients in each of the four statin benefit groups” the update explained.
- An expansion of the threshold for consideration of net ASCVD risk-reduction benefit from a reduction of LDL C of at least 50%, as well as consideration of LDL-C less than 70 mg/dL or non-HDL-C less than 100 mg/dL for all patients (that is, both those with and those without comorbidities) who have clinical ASCVD and baseline LDL-C of 70-189 mg/dL. The 2016 ECDP had different thresholds for those with versus those without comorbidities. This change was based on findings from the FOURIER trial and the IMPROVE-IT (Improved Reduction of Outcomes: Vytorin Efficacy International Trial).“Based on consideration of all available evidence, the consensus of the writing committee members is that lower LDL-C levels are safe and optimal in patients with clinical ASCVD due to [their] increased risk of recurrent events,” they said.
- An expanded recommendation on the use of ezetimibe and PCSK9 inhibition. The 2016 ECDP stated that “if a decision is made to proceed with the addition of nonstatin therapy to maximally tolerated statin therapy, it is reasonable to consider the addition of ezetimibe as the initial agent and a PCSK9 inhibitor as the second agent.” However, based on the FOURIER findings, the ongoing ODYSSEY Outcomes trial, and the IMPROVE-IT trial, the 2017 Focused Update states that, if such a decision is made in patients with clinical ASCVD with comorbidities and baseline LDL-C of 70-189 mg/dL, it is reasonable to weigh the addition of either ezetimibe or a PCSK9 inhibitor in light of “considerations of the additional percent LDL-C reduction desired, patient preferences, costs, route of administration, and other factors.” The update also spells out considerations that may favor the initial choice of ezetimibe versus a PCSK9 inhibitor (such as requiring less than 25% additional lowering of LDL-C, an age of over 75 years, cost, and other patient factors and preferences) .
- Additional factors, based on the FOURIER trial results and inclusion criteria, that may be considered for the identification of higher-risk patients with clinical ASCVD. The 2016 ECDP included on this list diabetes, a recent ASCVD event, an ASCVD event while already taking a statin, poorly controlled other major ASCVD risk factors, elevated lipoprotein, chronic kidney disease, symptomatic heart failure, maintenance hemodialysis, and baseline LDL-C of at least 190 mg/dL not due to secondary causes. The 2017 update added being 65 years or older, prior MI or nonhemorrhagic stroke, current daily cigarette smoking, symptomatic peripheral artery disease with prior MI or stroke, history of non-MI related coronary revascularization, residual coronary artery disease with at least 40% stenosis in at least two large vessels, HDL-C less than 40 mg/dL for men and less than 50 mg/dL for women, high-sensitivity C-reactive protein greater than 2 mg/L, and metabolic syndrome.
The content of the full ECDP has been changed in accordance with these updates and now includes more extensive and detailed guidance for decision making – both in the text and in treatment algorithms.
Aspects that remain unchanged include the decision pathways and algorithms for the use of ezetimibe or PCSK9 inhibitors in primary prevention patients with LDL-C less than 190 mg/dL or in those without ASCVD and LDL-C of 190 mg/dL or greater unattributable to secondary causes.
In addition to other changes made for the purpose of clarification and consistency, recommendations regarding bile acid–sequestrant use were downgraded; these are now only recommended as optional secondary agents for consideration in patients who cannot tolerate ezetimibe.
“[These] recommendations attempt to provide practical guidance for clinicians and patients regarding the use of nonstatin therapies to further reduce ASCVD risk in situations not covered by the guideline until such time as the scientific evidence base expands and cardiovascular outcomes trials are completed with new agents for ASCVD risk reduction,” the committee concluded.
Dr. Lloyd-Jones reported having no disclosures.
The American College of Cardiology Task Force on Expert Consensus Decision Pathways has released a “focused update” for the 2016 ACC Expert Consensus Decision Pathway (ECDP) on the role of nonstatin therapies for LDL-cholesterol lowering in the management of atherosclerotic cardiovascular disease (ASCVD) risk.
The update was deemed by the ECDP writing committee to be desirable given the additional evidence and perspectives that have emerged since the publication of the 2016 version, particularly with respect to the efficacy and safety of proprotein convertase subtilisin/kexin 9 (PCSK9) inhibitors for the secondary prevention of ASCVD, as well as the best use of ezetimibe in addition to statin therapy after acute coronary syndrome.
The ECDP algorithms endorse the four evidence-based statin benefit groups identified in the 2013 guidelines (adults aged 21 and older with clinical ASCVD, adults aged 21 and older with LDL-C of 190 mg/dL or greater, adults aged 40-75 years without ASCVD but with diabetes and with LDL-C of 70-189 mg/dL, and adults aged 40-75 without ASCVD or diabetes but with LDL-C of 70-189 mg/dL and an estimated 10-year risk for ASCVD of 7.5% or greater) and assume that the patient is currently taking or has attempted to take a statin, they noted.
Among the changes in the 2017 focused update are:
- Consideration of new randomized clinical trial data for the PCSK9 inhibitors evolocumab and bococizumab. Namely, they included results from the cardiovascular outcomes trials FOURIER (Further Cardiovascular Outcomes Research with PCSK9 Inhibition in Subjects with Elevated Risk) and SPIRE-1 and SPIRE-2 (Studies of PCSK9 Inhibition and the Reduction of Vascular Events), which were published in early 2017.
- An adjustment in the ECDP algorithms with respect to thresholds for consideration of net ASCVD risk reduction. The 2016 ECDP thresholds for risk reduction benefit were percent reduction in LDL-C with consideration of absolute LDL-C level in patients with clinical ASCVD, baseline LDL-C of 190 mg/dL or greater, and primary prevention. In patients with diabetes with or without clinical ASCVD, clinicians were allowed to consider absolute LDL-C and/or non-HDL-cholesterol levels. In the 2017 ECDP update, the thresholds are percent reduction in LDL-C with consideration of absolute LDL-C or non-HDL-C levels for patients in each of the four statin benefit groups. This change was based on the inclusion criteria of the FOURIER trial, the ongoing ODYSSEY Outcomes trial (Evaluation of Cardiovascular Outcomes after an Acute Coronary Syndrome During Treatment with Alirocumab), and the SPIRE-2 trial, all of which included non-HDL-C thresholds. “In alignment with these inclusion criteria, the 2017 Focused Update includes both LDL-C and non-HDL-C thresholds for evaluation of net ASCVD risk-reduction benefit when considering the addition of nonstatin therapies for patients in each of the four statin benefit groups” the update explained.
- An expansion of the threshold for consideration of net ASCVD risk-reduction benefit from a reduction of LDL C of at least 50%, as well as consideration of LDL-C less than 70 mg/dL or non-HDL-C less than 100 mg/dL for all patients (that is, both those with and those without comorbidities) who have clinical ASCVD and baseline LDL-C of 70-189 mg/dL. The 2016 ECDP had different thresholds for those with versus those without comorbidities. This change was based on findings from the FOURIER trial and the IMPROVE-IT (Improved Reduction of Outcomes: Vytorin Efficacy International Trial).“Based on consideration of all available evidence, the consensus of the writing committee members is that lower LDL-C levels are safe and optimal in patients with clinical ASCVD due to [their] increased risk of recurrent events,” they said.
- An expanded recommendation on the use of ezetimibe and PCSK9 inhibition. The 2016 ECDP stated that “if a decision is made to proceed with the addition of nonstatin therapy to maximally tolerated statin therapy, it is reasonable to consider the addition of ezetimibe as the initial agent and a PCSK9 inhibitor as the second agent.” However, based on the FOURIER findings, the ongoing ODYSSEY Outcomes trial, and the IMPROVE-IT trial, the 2017 Focused Update states that, if such a decision is made in patients with clinical ASCVD with comorbidities and baseline LDL-C of 70-189 mg/dL, it is reasonable to weigh the addition of either ezetimibe or a PCSK9 inhibitor in light of “considerations of the additional percent LDL-C reduction desired, patient preferences, costs, route of administration, and other factors.” The update also spells out considerations that may favor the initial choice of ezetimibe versus a PCSK9 inhibitor (such as requiring less than 25% additional lowering of LDL-C, an age of over 75 years, cost, and other patient factors and preferences) .
- Additional factors, based on the FOURIER trial results and inclusion criteria, that may be considered for the identification of higher-risk patients with clinical ASCVD. The 2016 ECDP included on this list diabetes, a recent ASCVD event, an ASCVD event while already taking a statin, poorly controlled other major ASCVD risk factors, elevated lipoprotein, chronic kidney disease, symptomatic heart failure, maintenance hemodialysis, and baseline LDL-C of at least 190 mg/dL not due to secondary causes. The 2017 update added being 65 years or older, prior MI or nonhemorrhagic stroke, current daily cigarette smoking, symptomatic peripheral artery disease with prior MI or stroke, history of non-MI related coronary revascularization, residual coronary artery disease with at least 40% stenosis in at least two large vessels, HDL-C less than 40 mg/dL for men and less than 50 mg/dL for women, high-sensitivity C-reactive protein greater than 2 mg/L, and metabolic syndrome.
The content of the full ECDP has been changed in accordance with these updates and now includes more extensive and detailed guidance for decision making – both in the text and in treatment algorithms.
Aspects that remain unchanged include the decision pathways and algorithms for the use of ezetimibe or PCSK9 inhibitors in primary prevention patients with LDL-C less than 190 mg/dL or in those without ASCVD and LDL-C of 190 mg/dL or greater unattributable to secondary causes.
In addition to other changes made for the purpose of clarification and consistency, recommendations regarding bile acid–sequestrant use were downgraded; these are now only recommended as optional secondary agents for consideration in patients who cannot tolerate ezetimibe.
“[These] recommendations attempt to provide practical guidance for clinicians and patients regarding the use of nonstatin therapies to further reduce ASCVD risk in situations not covered by the guideline until such time as the scientific evidence base expands and cardiovascular outcomes trials are completed with new agents for ASCVD risk reduction,” the committee concluded.
Dr. Lloyd-Jones reported having no disclosures.
FROM JOURNAL OF THE AMERICAN COLLEGE OF CARDIOLOGY
FDA moves to guard against abuse of ‘orphan drug’ program
The Food and Drug Administration is changing the way it approves orphan drugs after revelations that drugmakers may be abusing a law intended to help patients with rare diseases.
In a blog post Sept. 12, FDA Commissioner Scott Gottlieb, MD, said he wants to ensure financial incentives are granted “in a way that’s consistent with the manner Congress intended” when the Orphan Drug Act was passed in 1983. That legislation gave drugmakers a package of incentives, including tax credits, user-fee waivers and 7 years of market exclusivity if they developed medicines for rare diseases.
A Kaiser Health News investigation published in January 2017 found many drugs that now have orphan status aren’t entirely new. Of about 450 drugs that have won orphan approval since 1983, more than 70 were drugs first approved by the FDA for mass-market use. Those include rosuvastatin (Crestor), aripiprazole (Abilify), and adalimumab (Humira), the world’s best-selling drug.
Dr. Gottlieb announced plans to close a loophole that allows manufacturers to skip pediatric testing requirements when developing a common-disease drug for orphan use in children. He also signaled that bigger changes are being considered, announcing a public meeting to explore issues raised by scientific advances, such as the increase in precision medicine and biologics.
“We need to make sure our policies take notice of all of these new challenges and opportunities,” he wrote. Dr. Gottlieb, through his agency, declined multiple requests for interviews.
Over the years, drugmakers have fueled a boom in orphan drugs, which often carry six-figure price tags. Nearly half of the new drugs approved by the FDA are now for rare diseases – even though many of them also treat and are marketed for common diseases.
Dr. Gottlieb became commissioner in May, a few months after three key Republican senators called for a federal investigation into potential abuses of the Orphan Drug Act, and the Government Accountability Office agreed to investigate.
The GAO has yet to begin its investigation, saying it doesn’t expect to start work until late this year, when staff is available. Regardless, in late June, Dr. Gottlieb announced what would be the first in a series of updates that shift the way the FDA handles orphan drugs.
Those include:
- Eliminating a backlog in drug applications for orphan designation or status. Getting a designation is a critical first step if a company wants to win orphan incentives once the drug is approved for treatment use. And, much like the rise in approvals, the requests by companies to get drugs designated with orphan status has also skyrocketed. Dr. Gottlieb said in June that he wanted to get rid of the backlog; his blog post noted the effort was complete. About half of the 200 applications from drugmakers won orphan status.
- Mandating that drugmakers prove their medicine is clinically superior before getting the market exclusivity that comes with orphan drug status. The agency had lost a lawsuit in which a company said it was owed the exclusivity period regardless of whether its medicine was better. And two more lawsuits had been filed by Eagle Pharmaceuticals and United Therapeutics. The FDA Reauthorization Act, which passed in August, made it law that a drug has to be clinically superior to get the incentives.
- Closing the loophole for pediatric orphan drugs by requiring all drugs approved for common adult diseases, like inflammatory bowel disease, undergo pediatric testing when getting approval as a pediatric orphan drug. Pediatric testing is not required for orphan drugs, and last month Congress mandated that orphan drugs for cancer be tested for children. Still, the American Academy of Pediatrics celebrated the proposed change but warned it was only a first step. Bridgette Jones, MD, chair of American Academy of Pediatrics Committee on Drugs, said Sept. 12 that orphan drugs are “still mostly exempt from pediatric study requirements … children deserve access to safe, effective medications.”
Martin Makary, MD, who wrote a critical 2015 paper on orphan approvals, said the changes at the agency indicate that Dr. Gottlieb seems “concerned about all the right things. The government does a lot of lip service in general. This is not lip service.”
The restructuring has been swift in some ways.
Sandra Heibel, PhD, a senior consultant at Haffner Associates, a firm that helps companies submit orphan drug applications, noted that the approval process for designations definitely sped up over the summer, and “we are absolutely getting responses from the FDA back in 90 days. That has come through.”
Other changes to the agency, though, will evolve slowly. For example, the orphan drug office has begun reaching across the FDA’s divisions for help in reviewing drugs. In May, the FDA’s orphan reviews began to work with the office of pediatric therapeutics to review pediatric applications – ideally increasing the expertise applied when considering a company’s request for orphan drug use in children.
In an interview, FDA confirmed that Dr. Gottlieb’s orphan modernization plan is part of a larger effort to increase competition and decrease drug prices. One focus is on targeted drugs – especially those that affect rare diseases or diseases for which there is no effective therapy, the agency said.
“Such drugs present some of the biggest opportunities in medicine to treat and cure debilitating and very costly diseases,” the agency stated.
Kaiser Health News is a national health policy news service that is part of the nonpartisan Henry J. Kaiser Family Foundation.
The Food and Drug Administration is changing the way it approves orphan drugs after revelations that drugmakers may be abusing a law intended to help patients with rare diseases.
In a blog post Sept. 12, FDA Commissioner Scott Gottlieb, MD, said he wants to ensure financial incentives are granted “in a way that’s consistent with the manner Congress intended” when the Orphan Drug Act was passed in 1983. That legislation gave drugmakers a package of incentives, including tax credits, user-fee waivers and 7 years of market exclusivity if they developed medicines for rare diseases.
A Kaiser Health News investigation published in January 2017 found many drugs that now have orphan status aren’t entirely new. Of about 450 drugs that have won orphan approval since 1983, more than 70 were drugs first approved by the FDA for mass-market use. Those include rosuvastatin (Crestor), aripiprazole (Abilify), and adalimumab (Humira), the world’s best-selling drug.
Dr. Gottlieb announced plans to close a loophole that allows manufacturers to skip pediatric testing requirements when developing a common-disease drug for orphan use in children. He also signaled that bigger changes are being considered, announcing a public meeting to explore issues raised by scientific advances, such as the increase in precision medicine and biologics.
“We need to make sure our policies take notice of all of these new challenges and opportunities,” he wrote. Dr. Gottlieb, through his agency, declined multiple requests for interviews.
Over the years, drugmakers have fueled a boom in orphan drugs, which often carry six-figure price tags. Nearly half of the new drugs approved by the FDA are now for rare diseases – even though many of them also treat and are marketed for common diseases.
Dr. Gottlieb became commissioner in May, a few months after three key Republican senators called for a federal investigation into potential abuses of the Orphan Drug Act, and the Government Accountability Office agreed to investigate.
The GAO has yet to begin its investigation, saying it doesn’t expect to start work until late this year, when staff is available. Regardless, in late June, Dr. Gottlieb announced what would be the first in a series of updates that shift the way the FDA handles orphan drugs.
Those include:
- Eliminating a backlog in drug applications for orphan designation or status. Getting a designation is a critical first step if a company wants to win orphan incentives once the drug is approved for treatment use. And, much like the rise in approvals, the requests by companies to get drugs designated with orphan status has also skyrocketed. Dr. Gottlieb said in June that he wanted to get rid of the backlog; his blog post noted the effort was complete. About half of the 200 applications from drugmakers won orphan status.
- Mandating that drugmakers prove their medicine is clinically superior before getting the market exclusivity that comes with orphan drug status. The agency had lost a lawsuit in which a company said it was owed the exclusivity period regardless of whether its medicine was better. And two more lawsuits had been filed by Eagle Pharmaceuticals and United Therapeutics. The FDA Reauthorization Act, which passed in August, made it law that a drug has to be clinically superior to get the incentives.
- Closing the loophole for pediatric orphan drugs by requiring all drugs approved for common adult diseases, like inflammatory bowel disease, undergo pediatric testing when getting approval as a pediatric orphan drug. Pediatric testing is not required for orphan drugs, and last month Congress mandated that orphan drugs for cancer be tested for children. Still, the American Academy of Pediatrics celebrated the proposed change but warned it was only a first step. Bridgette Jones, MD, chair of American Academy of Pediatrics Committee on Drugs, said Sept. 12 that orphan drugs are “still mostly exempt from pediatric study requirements … children deserve access to safe, effective medications.”
Martin Makary, MD, who wrote a critical 2015 paper on orphan approvals, said the changes at the agency indicate that Dr. Gottlieb seems “concerned about all the right things. The government does a lot of lip service in general. This is not lip service.”
The restructuring has been swift in some ways.
Sandra Heibel, PhD, a senior consultant at Haffner Associates, a firm that helps companies submit orphan drug applications, noted that the approval process for designations definitely sped up over the summer, and “we are absolutely getting responses from the FDA back in 90 days. That has come through.”
Other changes to the agency, though, will evolve slowly. For example, the orphan drug office has begun reaching across the FDA’s divisions for help in reviewing drugs. In May, the FDA’s orphan reviews began to work with the office of pediatric therapeutics to review pediatric applications – ideally increasing the expertise applied when considering a company’s request for orphan drug use in children.
In an interview, FDA confirmed that Dr. Gottlieb’s orphan modernization plan is part of a larger effort to increase competition and decrease drug prices. One focus is on targeted drugs – especially those that affect rare diseases or diseases for which there is no effective therapy, the agency said.
“Such drugs present some of the biggest opportunities in medicine to treat and cure debilitating and very costly diseases,” the agency stated.
Kaiser Health News is a national health policy news service that is part of the nonpartisan Henry J. Kaiser Family Foundation.
The Food and Drug Administration is changing the way it approves orphan drugs after revelations that drugmakers may be abusing a law intended to help patients with rare diseases.
In a blog post Sept. 12, FDA Commissioner Scott Gottlieb, MD, said he wants to ensure financial incentives are granted “in a way that’s consistent with the manner Congress intended” when the Orphan Drug Act was passed in 1983. That legislation gave drugmakers a package of incentives, including tax credits, user-fee waivers and 7 years of market exclusivity if they developed medicines for rare diseases.
A Kaiser Health News investigation published in January 2017 found many drugs that now have orphan status aren’t entirely new. Of about 450 drugs that have won orphan approval since 1983, more than 70 were drugs first approved by the FDA for mass-market use. Those include rosuvastatin (Crestor), aripiprazole (Abilify), and adalimumab (Humira), the world’s best-selling drug.
Dr. Gottlieb announced plans to close a loophole that allows manufacturers to skip pediatric testing requirements when developing a common-disease drug for orphan use in children. He also signaled that bigger changes are being considered, announcing a public meeting to explore issues raised by scientific advances, such as the increase in precision medicine and biologics.
“We need to make sure our policies take notice of all of these new challenges and opportunities,” he wrote. Dr. Gottlieb, through his agency, declined multiple requests for interviews.
Over the years, drugmakers have fueled a boom in orphan drugs, which often carry six-figure price tags. Nearly half of the new drugs approved by the FDA are now for rare diseases – even though many of them also treat and are marketed for common diseases.
Dr. Gottlieb became commissioner in May, a few months after three key Republican senators called for a federal investigation into potential abuses of the Orphan Drug Act, and the Government Accountability Office agreed to investigate.
The GAO has yet to begin its investigation, saying it doesn’t expect to start work until late this year, when staff is available. Regardless, in late June, Dr. Gottlieb announced what would be the first in a series of updates that shift the way the FDA handles orphan drugs.
Those include:
- Eliminating a backlog in drug applications for orphan designation or status. Getting a designation is a critical first step if a company wants to win orphan incentives once the drug is approved for treatment use. And, much like the rise in approvals, the requests by companies to get drugs designated with orphan status has also skyrocketed. Dr. Gottlieb said in June that he wanted to get rid of the backlog; his blog post noted the effort was complete. About half of the 200 applications from drugmakers won orphan status.
- Mandating that drugmakers prove their medicine is clinically superior before getting the market exclusivity that comes with orphan drug status. The agency had lost a lawsuit in which a company said it was owed the exclusivity period regardless of whether its medicine was better. And two more lawsuits had been filed by Eagle Pharmaceuticals and United Therapeutics. The FDA Reauthorization Act, which passed in August, made it law that a drug has to be clinically superior to get the incentives.
- Closing the loophole for pediatric orphan drugs by requiring all drugs approved for common adult diseases, like inflammatory bowel disease, undergo pediatric testing when getting approval as a pediatric orphan drug. Pediatric testing is not required for orphan drugs, and last month Congress mandated that orphan drugs for cancer be tested for children. Still, the American Academy of Pediatrics celebrated the proposed change but warned it was only a first step. Bridgette Jones, MD, chair of American Academy of Pediatrics Committee on Drugs, said Sept. 12 that orphan drugs are “still mostly exempt from pediatric study requirements … children deserve access to safe, effective medications.”
Martin Makary, MD, who wrote a critical 2015 paper on orphan approvals, said the changes at the agency indicate that Dr. Gottlieb seems “concerned about all the right things. The government does a lot of lip service in general. This is not lip service.”
The restructuring has been swift in some ways.
Sandra Heibel, PhD, a senior consultant at Haffner Associates, a firm that helps companies submit orphan drug applications, noted that the approval process for designations definitely sped up over the summer, and “we are absolutely getting responses from the FDA back in 90 days. That has come through.”
Other changes to the agency, though, will evolve slowly. For example, the orphan drug office has begun reaching across the FDA’s divisions for help in reviewing drugs. In May, the FDA’s orphan reviews began to work with the office of pediatric therapeutics to review pediatric applications – ideally increasing the expertise applied when considering a company’s request for orphan drug use in children.
In an interview, FDA confirmed that Dr. Gottlieb’s orphan modernization plan is part of a larger effort to increase competition and decrease drug prices. One focus is on targeted drugs – especially those that affect rare diseases or diseases for which there is no effective therapy, the agency said.
“Such drugs present some of the biggest opportunities in medicine to treat and cure debilitating and very costly diseases,” the agency stated.
Kaiser Health News is a national health policy news service that is part of the nonpartisan Henry J. Kaiser Family Foundation.
Uninsured rate falls to record low of 8.8%
Three years after the Affordable Care Act’s coverage expansion took effect, the number of Americans without health insurance fell to 28.1 million in 2016, down from 29 million in 2015, according to a federal report released Sept. 12.
The latest numbers from the U.S. Census Bureau showed the nation’s uninsured rate dropped to 8.8%. It had been 9.1% in 2015.
Both the overall number of uninsured and the percentage are record lows.
The latest figures from the Census Bureau effectively close the book on President Barack Obama’s record on lowering the number of uninsured. He made that a linchpin of his 2008 campaign, and his administration’s effort to overhaul the nation’s health system through the ACA focused on expanding coverage.
When Mr. Obama took office in 2009, during the worst economic recession since the Great Depression, more than 50 million Americans were uninsured, or nearly 17% of the population.
The number of uninsured has fallen from 42 million in 2013 – before the ACA in 2014 allowed states to expand Medicaid, the federal-state program that provides coverage to low-income people, and provided federal subsidies to help lower- and middle-income Americans buy coverage on the insurance marketplaces. The decline also reflected the improving economy, which has put more Americans in jobs that offer health coverage.
The dramatic drop in the uninsured over the past few years played a major role in the congressional debate over the summer about whether to replace the 2010 health law. Advocates pleaded with the Republican-controlled Congress not to take steps to reverse the gains in coverage.
The Census Bureau numbers are considered the gold standard for tracking who has insurance because the survey samples are so large.
The uninsured rate has fallen in all 50 states and the District of Columbia since 2013, although the rate has been lower among the 31 states that expanded Medicaid as part of the health law. The lowest uninsured rate last year was 2.5% in Massachusetts, and the highest was 16.6% in Texas, the Census Bureau reported. States that expanded Medicaid had an average uninsured rate of 6.5%, compared with an 11.7% average among states that did not expand.
More than half of Americans – 55.7% – get health insurance through their jobs. But government coverage is becoming more common. Medicaid now covers more than 19% of the population and Medicare, nearly 17%.
Kaiser Health News is a national health policy news service that is part of the nonpartisan Henry J. Kaiser Family Foundation.
Three years after the Affordable Care Act’s coverage expansion took effect, the number of Americans without health insurance fell to 28.1 million in 2016, down from 29 million in 2015, according to a federal report released Sept. 12.
The latest numbers from the U.S. Census Bureau showed the nation’s uninsured rate dropped to 8.8%. It had been 9.1% in 2015.
Both the overall number of uninsured and the percentage are record lows.
The latest figures from the Census Bureau effectively close the book on President Barack Obama’s record on lowering the number of uninsured. He made that a linchpin of his 2008 campaign, and his administration’s effort to overhaul the nation’s health system through the ACA focused on expanding coverage.
When Mr. Obama took office in 2009, during the worst economic recession since the Great Depression, more than 50 million Americans were uninsured, or nearly 17% of the population.
The number of uninsured has fallen from 42 million in 2013 – before the ACA in 2014 allowed states to expand Medicaid, the federal-state program that provides coverage to low-income people, and provided federal subsidies to help lower- and middle-income Americans buy coverage on the insurance marketplaces. The decline also reflected the improving economy, which has put more Americans in jobs that offer health coverage.
The dramatic drop in the uninsured over the past few years played a major role in the congressional debate over the summer about whether to replace the 2010 health law. Advocates pleaded with the Republican-controlled Congress not to take steps to reverse the gains in coverage.
The Census Bureau numbers are considered the gold standard for tracking who has insurance because the survey samples are so large.
The uninsured rate has fallen in all 50 states and the District of Columbia since 2013, although the rate has been lower among the 31 states that expanded Medicaid as part of the health law. The lowest uninsured rate last year was 2.5% in Massachusetts, and the highest was 16.6% in Texas, the Census Bureau reported. States that expanded Medicaid had an average uninsured rate of 6.5%, compared with an 11.7% average among states that did not expand.
More than half of Americans – 55.7% – get health insurance through their jobs. But government coverage is becoming more common. Medicaid now covers more than 19% of the population and Medicare, nearly 17%.
Kaiser Health News is a national health policy news service that is part of the nonpartisan Henry J. Kaiser Family Foundation.
Three years after the Affordable Care Act’s coverage expansion took effect, the number of Americans without health insurance fell to 28.1 million in 2016, down from 29 million in 2015, according to a federal report released Sept. 12.
The latest numbers from the U.S. Census Bureau showed the nation’s uninsured rate dropped to 8.8%. It had been 9.1% in 2015.
Both the overall number of uninsured and the percentage are record lows.
The latest figures from the Census Bureau effectively close the book on President Barack Obama’s record on lowering the number of uninsured. He made that a linchpin of his 2008 campaign, and his administration’s effort to overhaul the nation’s health system through the ACA focused on expanding coverage.
When Mr. Obama took office in 2009, during the worst economic recession since the Great Depression, more than 50 million Americans were uninsured, or nearly 17% of the population.
The number of uninsured has fallen from 42 million in 2013 – before the ACA in 2014 allowed states to expand Medicaid, the federal-state program that provides coverage to low-income people, and provided federal subsidies to help lower- and middle-income Americans buy coverage on the insurance marketplaces. The decline also reflected the improving economy, which has put more Americans in jobs that offer health coverage.
The dramatic drop in the uninsured over the past few years played a major role in the congressional debate over the summer about whether to replace the 2010 health law. Advocates pleaded with the Republican-controlled Congress not to take steps to reverse the gains in coverage.
The Census Bureau numbers are considered the gold standard for tracking who has insurance because the survey samples are so large.
The uninsured rate has fallen in all 50 states and the District of Columbia since 2013, although the rate has been lower among the 31 states that expanded Medicaid as part of the health law. The lowest uninsured rate last year was 2.5% in Massachusetts, and the highest was 16.6% in Texas, the Census Bureau reported. States that expanded Medicaid had an average uninsured rate of 6.5%, compared with an 11.7% average among states that did not expand.
More than half of Americans – 55.7% – get health insurance through their jobs. But government coverage is becoming more common. Medicaid now covers more than 19% of the population and Medicare, nearly 17%.
Kaiser Health News is a national health policy news service that is part of the nonpartisan Henry J. Kaiser Family Foundation.
BIMA’s benefits extend to high-risk CABG patients
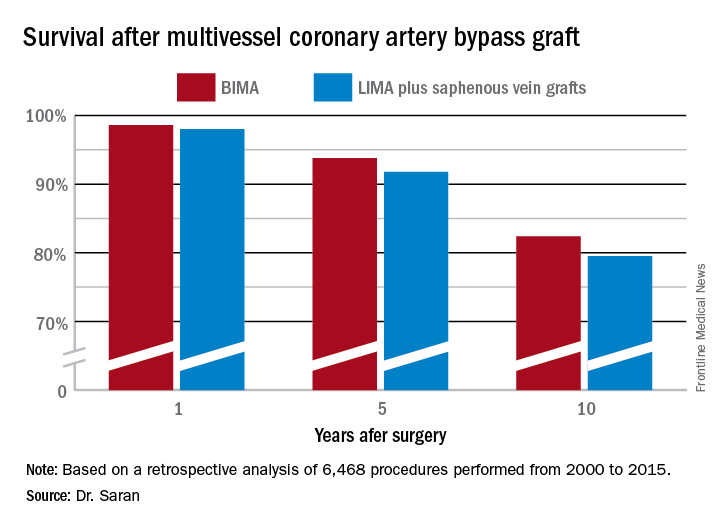
COLORADO SPRINGS – The survival advantage of bilateral internal over left internal mammary artery grafts persists even among multivessel CABG patients perceived to be at high surgical risk, Nishant Saran, MD, reported at the annual meeting of the Western Thoracic Surgical Association.
Many surgeons hesitate to perform bilateral internal mammary artery (BIMA) grafting in high-risk patients on the presumption that BIMA might not benefit them. It’s a concern that appears to be without merit, however, based on a retrospective analysis of the 6,468 multivessel CABG procedures performed at the Mayo Clinic during 2000-2015, said Dr. Saran of the Mayo Clinic in Rochester, Minn.

The BIMA patients were as a whole significantly younger, primarily men, and less likely to have diabetes or to be obese than the LIMA patients. Also, LIMA patients were fourfold more likely to have baseline heart failure, twice as likely to have a history of stroke, and had a twofold greater prevalence of chronic lung disease.
“The unmatched comparison shows the clear treatment selection bias we have: BIMA goes to the healthier patients,” Dr. Saran observed.
But is that bias justified? To find out, he and his coinvestigators performed extensive propensity score matching using several dozen baseline variables in order to identify 1,011 closely matched patient pairs. In this propensity score-matched analysis, 5- and 10-year survival rates were significantly better in the BIMA group. The gap between the two survival curves widened after about 7 years and continued to expand steadily through year 10. Incision time averaged 298 minutes in the BIMA group and 254 minutes in the propensity-matched LIMA group.
Discussant Eric J. Lehr, MD, a cardiac surgeon at Swedish Medical Center in Seattle, noted that the impressive survival benefit for BIMA in the retrospective Mayo Clinic study came at what he termed “a modest cost”: a doubled incidence of sternal site infections, from 1.4% in the LIMA group to 3% with BIMA. Importantly, though, there was no significant difference in the more serious deep sternal wound infections.
He agreed with Dr. Saran that BIMA is seriously underutilized, noting that only one cardiothoracic surgery program in the state of Washington uses BIMA more than 10% of the time in multivessel CABG.
Dr. Lehr then posed a provocative question: “Should BIMA grafting be considered a quality metric in coronary revascularization surgery, despite the small increase in sternal site infections, even though sternal wound infections have been declared a ‘never’ event and are tied to reimbursement?”
“I think BIMA should be a gold standard,” Dr. Saran replied. “The first thing that a cardiac surgeon should always think of when a patient is going to have CABG is ‘BIMA first,’ and only then look into reasons for not doing it. But I guess in current real-world practice, things are different.”
Howard K. Song, MD, commented, “I think a study like this doesn’t necessarily show that every surgeon should be using BIMA liberally, it shows that surgeons in your practice who do that have excellent outcomes.”
Dr. Song, professor of surgery and chief of the division of cardiothoracic surgery at Oregon Health and Science University, Portland, added that he believes extensive use of BIMA is actually a surrogate marker for a highly skilled subspecialist who would be expected to have very good outcomes as a matter of course.
“That may be one way of looking at it; however, I do think that even very skilled surgeons still have an inherent resistance to doing BIMA,” Dr. Saran responded.
“In the current era, the surgeon is pressured to achieve improved short-term outcomes and improved OR turnover times. An extra half hour for BIMA tends to push the surgeon away,” he added.
Dr. Saran reported having no financial conflicts of interest.
COLORADO SPRINGS – The survival advantage of bilateral internal over left internal mammary artery grafts persists even among multivessel CABG patients perceived to be at high surgical risk, Nishant Saran, MD, reported at the annual meeting of the Western Thoracic Surgical Association.
Many surgeons hesitate to perform bilateral internal mammary artery (BIMA) grafting in high-risk patients on the presumption that BIMA might not benefit them. It’s a concern that appears to be without merit, however, based on a retrospective analysis of the 6,468 multivessel CABG procedures performed at the Mayo Clinic during 2000-2015, said Dr. Saran of the Mayo Clinic in Rochester, Minn.
The BIMA patients were as a whole significantly younger, primarily men, and less likely to have diabetes or to be obese than the LIMA patients. Also, LIMA patients were fourfold more likely to have baseline heart failure, twice as likely to have a history of stroke, and had a twofold greater prevalence of chronic lung disease.
“The unmatched comparison shows the clear treatment selection bias we have: BIMA goes to the healthier patients,” Dr. Saran observed.
But is that bias justified? To find out, he and his coinvestigators performed extensive propensity score matching using several dozen baseline variables in order to identify 1,011 closely matched patient pairs. In this propensity score-matched analysis, 5- and 10-year survival rates were significantly better in the BIMA group. The gap between the two survival curves widened after about 7 years and continued to expand steadily through year 10. Incision time averaged 298 minutes in the BIMA group and 254 minutes in the propensity-matched LIMA group.
Discussant Eric J. Lehr, MD, a cardiac surgeon at Swedish Medical Center in Seattle, noted that the impressive survival benefit for BIMA in the retrospective Mayo Clinic study came at what he termed “a modest cost”: a doubled incidence of sternal site infections, from 1.4% in the LIMA group to 3% with BIMA. Importantly, though, there was no significant difference in the more serious deep sternal wound infections.
He agreed with Dr. Saran that BIMA is seriously underutilized, noting that only one cardiothoracic surgery program in the state of Washington uses BIMA more than 10% of the time in multivessel CABG.
Dr. Lehr then posed a provocative question: “Should BIMA grafting be considered a quality metric in coronary revascularization surgery, despite the small increase in sternal site infections, even though sternal wound infections have been declared a ‘never’ event and are tied to reimbursement?”
“I think BIMA should be a gold standard,” Dr. Saran replied. “The first thing that a cardiac surgeon should always think of when a patient is going to have CABG is ‘BIMA first,’ and only then look into reasons for not doing it. But I guess in current real-world practice, things are different.”
Howard K. Song, MD, commented, “I think a study like this doesn’t necessarily show that every surgeon should be using BIMA liberally, it shows that surgeons in your practice who do that have excellent outcomes.”
Dr. Song, professor of surgery and chief of the division of cardiothoracic surgery at Oregon Health and Science University, Portland, added that he believes extensive use of BIMA is actually a surrogate marker for a highly skilled subspecialist who would be expected to have very good outcomes as a matter of course.
“That may be one way of looking at it; however, I do think that even very skilled surgeons still have an inherent resistance to doing BIMA,” Dr. Saran responded.
“In the current era, the surgeon is pressured to achieve improved short-term outcomes and improved OR turnover times. An extra half hour for BIMA tends to push the surgeon away,” he added.
Dr. Saran reported having no financial conflicts of interest.
COLORADO SPRINGS – The survival advantage of bilateral internal over left internal mammary artery grafts persists even among multivessel CABG patients perceived to be at high surgical risk, Nishant Saran, MD, reported at the annual meeting of the Western Thoracic Surgical Association.
Many surgeons hesitate to perform bilateral internal mammary artery (BIMA) grafting in high-risk patients on the presumption that BIMA might not benefit them. It’s a concern that appears to be without merit, however, based on a retrospective analysis of the 6,468 multivessel CABG procedures performed at the Mayo Clinic during 2000-2015, said Dr. Saran of the Mayo Clinic in Rochester, Minn.
The BIMA patients were as a whole significantly younger, primarily men, and less likely to have diabetes or to be obese than the LIMA patients. Also, LIMA patients were fourfold more likely to have baseline heart failure, twice as likely to have a history of stroke, and had a twofold greater prevalence of chronic lung disease.
“The unmatched comparison shows the clear treatment selection bias we have: BIMA goes to the healthier patients,” Dr. Saran observed.
But is that bias justified? To find out, he and his coinvestigators performed extensive propensity score matching using several dozen baseline variables in order to identify 1,011 closely matched patient pairs. In this propensity score-matched analysis, 5- and 10-year survival rates were significantly better in the BIMA group. The gap between the two survival curves widened after about 7 years and continued to expand steadily through year 10. Incision time averaged 298 minutes in the BIMA group and 254 minutes in the propensity-matched LIMA group.
Discussant Eric J. Lehr, MD, a cardiac surgeon at Swedish Medical Center in Seattle, noted that the impressive survival benefit for BIMA in the retrospective Mayo Clinic study came at what he termed “a modest cost”: a doubled incidence of sternal site infections, from 1.4% in the LIMA group to 3% with BIMA. Importantly, though, there was no significant difference in the more serious deep sternal wound infections.
He agreed with Dr. Saran that BIMA is seriously underutilized, noting that only one cardiothoracic surgery program in the state of Washington uses BIMA more than 10% of the time in multivessel CABG.
Dr. Lehr then posed a provocative question: “Should BIMA grafting be considered a quality metric in coronary revascularization surgery, despite the small increase in sternal site infections, even though sternal wound infections have been declared a ‘never’ event and are tied to reimbursement?”
“I think BIMA should be a gold standard,” Dr. Saran replied. “The first thing that a cardiac surgeon should always think of when a patient is going to have CABG is ‘BIMA first,’ and only then look into reasons for not doing it. But I guess in current real-world practice, things are different.”
Howard K. Song, MD, commented, “I think a study like this doesn’t necessarily show that every surgeon should be using BIMA liberally, it shows that surgeons in your practice who do that have excellent outcomes.”
Dr. Song, professor of surgery and chief of the division of cardiothoracic surgery at Oregon Health and Science University, Portland, added that he believes extensive use of BIMA is actually a surrogate marker for a highly skilled subspecialist who would be expected to have very good outcomes as a matter of course.
“That may be one way of looking at it; however, I do think that even very skilled surgeons still have an inherent resistance to doing BIMA,” Dr. Saran responded.
“In the current era, the surgeon is pressured to achieve improved short-term outcomes and improved OR turnover times. An extra half hour for BIMA tends to push the surgeon away,” he added.
Dr. Saran reported having no financial conflicts of interest.
AT THE WTSA ANNUAL MEETING
Key clinical point:
Major finding: Ten-year survival following multivessel CABG using bilateral internal mammary artery grafting was 82.4%, significantly better than the 79.5% rate with left internal mammary artery grafting plus saphenous vein grafts.
Data source: This retrospective observational single-center included 6,468 patients who underwent multivessel CABG during 2000-2015.
Disclosures: Dr. Saran reported having no financial conflicts of interest.
Metabolically healthy obese still at elevated cardiovascular risk
Obese individuals with no metabolic abnormalities, such as dyslipidemia, high blood pressure, or high blood sugar levels, still have a higher risk of cardiovascular disease than do metabolically healthy people of normal weight, new data suggests.
“Our study robustly challenges the assertion that MHO [metabolically healthy obese] is a benign condition and adds to the evidence base that MHO is a high-risk state for future CVD events,” wrote Rishi Caleyachetty, MD, of the University of Birmingham, England, and his coauthors online (J Am Coll Cardiol. 2017, Sep 11. doi. 10.1016/j.jacc.2017.07.763).
Dr. Caleyachetty and his associates reported findings from a population-based study using the electronic health records of nearly 3.5 million individuals aged 18 years or older who were free of cardiovascular disease at baseline.
Overall, 15% of the population were classified as being metabolically healthy obese, meaning that they had a body mass index (BMI) of at least 30 kg/m2 with no sign of diabetes, hypertension, or hyperlipidemia, and 26% were overweight with no metabolic abnormalities. Despite their lack of metabolic disease, these obese individuals still had a significant 49% higher risk of coronary heart disease, 7% higher risk of cerebrovascular disease, and 96% higher risk of heart failure, compared with normal-weight individuals with no metabolic disease, after researchers adjusted for age, sex, smoking status, and social deprivation.
Individuals who were overweight but metabolically healthy had a 30% increased risk of ischemic heart disease, 11% increased risk of heart failure, and the same risk of cerebrovascular disease as normal-weight, healthy individuals.
They also saw an increasing risk of ischemic heart disease, cerebrovascular disease, heart failure, and peripheral vascular disease with each additional metabolic abnormality, even among underweight and normal-weight individuals, and suggested that a focus on screening overweight and obese individuals only could miss metabolic abnormalities in many patients.
Overweight and obese individuals without metabolic disease had a significantly lower risk of peripheral vascular disease, compared with healthy normal-weight individuals. The authors said this was a surprising finding but suggested cigarette smoking could be a confounding factor, as this is associated with both peripheral vascular disease and lower BMI.
“In sensitivity analyses restricted to individuals who were obese with no metabolic abnormalities and reported never smoking cigarettes, risk for PVD [peripheral vascular disease] was increased, compared [with] normal-weight individuals with no metabolic abnormalities,” Dr. Caleyachetty and his coinvestigators wrote.
Over the mean follow-up of 5.4 years, 5.6% of initially metabolically healthy obese individuals developed diabetes, 11.5% developed hyperlipidemia and 10.5% developed hypertension. In contrast, among the metabolically healthy overweight individuals at baseline, 1.9% developed diabetes, 9.4% developed hyperlipidemia, and 7.2% developed hypertension.
While the analysis adjusted for sex, the authors did note that women who were overweight or obese but metabolically healthy had stronger positive associations than did males with cerebrovascular disease and heart failure.
“Clinicians need to be aware that individuals who would otherwise be considered nonobese, based on a normal BMI, can have metabolic abnormalities, and therefore also be at high risk for CVD events,” the investigators concluded.
No conflicts of interest were declared.
Recently, studies have consistently placed metabolically healthy obese individuals between metabolically healthy lean and metabolically unhealthy obese individuals in terms of cardiovascular disease risk, occult cardiac dysfunction, and type 2 diabetes. Thus, either metabolic dysfunction or elevated body mass index appears to increases CVD risk factors.
Often, one or two metabolic risk factors in normal weight individuals are dismissed as unimportant because they are of healthy weight; however, these data suggest that the normal-weight group is at similar risk, compared with overweight, and at times, obese individuals, when metabolic abnormalities are present. The study not only definitively counters the concept of metabolically benign obesity but also demonstrates great risk to normal weight individuals if metabolic dysfunction is present. Thus, we would suggest an increased need for screening in the normal-weight population.
Jennifer W. Bea, PhD, is from the Collaboratory for Metabolic Disease Prevention and Treatment in Tucson, Ariz., and Nancy K. Sweitzer, MD, is chief of the division of cardiology at the Sarver Heart Center. These comments are taken from an accompanying editorial (J Am Coll Cardiol. 2017 Sep 19;70:1438-40. doi. org/10.1016/j.jacc.2017.07.742). No conflicts of interest were declared.
Recently, studies have consistently placed metabolically healthy obese individuals between metabolically healthy lean and metabolically unhealthy obese individuals in terms of cardiovascular disease risk, occult cardiac dysfunction, and type 2 diabetes. Thus, either metabolic dysfunction or elevated body mass index appears to increases CVD risk factors.
Often, one or two metabolic risk factors in normal weight individuals are dismissed as unimportant because they are of healthy weight; however, these data suggest that the normal-weight group is at similar risk, compared with overweight, and at times, obese individuals, when metabolic abnormalities are present. The study not only definitively counters the concept of metabolically benign obesity but also demonstrates great risk to normal weight individuals if metabolic dysfunction is present. Thus, we would suggest an increased need for screening in the normal-weight population.
Jennifer W. Bea, PhD, is from the Collaboratory for Metabolic Disease Prevention and Treatment in Tucson, Ariz., and Nancy K. Sweitzer, MD, is chief of the division of cardiology at the Sarver Heart Center. These comments are taken from an accompanying editorial (J Am Coll Cardiol. 2017 Sep 19;70:1438-40. doi. org/10.1016/j.jacc.2017.07.742). No conflicts of interest were declared.
Recently, studies have consistently placed metabolically healthy obese individuals between metabolically healthy lean and metabolically unhealthy obese individuals in terms of cardiovascular disease risk, occult cardiac dysfunction, and type 2 diabetes. Thus, either metabolic dysfunction or elevated body mass index appears to increases CVD risk factors.
Often, one or two metabolic risk factors in normal weight individuals are dismissed as unimportant because they are of healthy weight; however, these data suggest that the normal-weight group is at similar risk, compared with overweight, and at times, obese individuals, when metabolic abnormalities are present. The study not only definitively counters the concept of metabolically benign obesity but also demonstrates great risk to normal weight individuals if metabolic dysfunction is present. Thus, we would suggest an increased need for screening in the normal-weight population.
Jennifer W. Bea, PhD, is from the Collaboratory for Metabolic Disease Prevention and Treatment in Tucson, Ariz., and Nancy K. Sweitzer, MD, is chief of the division of cardiology at the Sarver Heart Center. These comments are taken from an accompanying editorial (J Am Coll Cardiol. 2017 Sep 19;70:1438-40. doi. org/10.1016/j.jacc.2017.07.742). No conflicts of interest were declared.
Obese individuals with no metabolic abnormalities, such as dyslipidemia, high blood pressure, or high blood sugar levels, still have a higher risk of cardiovascular disease than do metabolically healthy people of normal weight, new data suggests.
“Our study robustly challenges the assertion that MHO [metabolically healthy obese] is a benign condition and adds to the evidence base that MHO is a high-risk state for future CVD events,” wrote Rishi Caleyachetty, MD, of the University of Birmingham, England, and his coauthors online (J Am Coll Cardiol. 2017, Sep 11. doi. 10.1016/j.jacc.2017.07.763).
Dr. Caleyachetty and his associates reported findings from a population-based study using the electronic health records of nearly 3.5 million individuals aged 18 years or older who were free of cardiovascular disease at baseline.
Overall, 15% of the population were classified as being metabolically healthy obese, meaning that they had a body mass index (BMI) of at least 30 kg/m2 with no sign of diabetes, hypertension, or hyperlipidemia, and 26% were overweight with no metabolic abnormalities. Despite their lack of metabolic disease, these obese individuals still had a significant 49% higher risk of coronary heart disease, 7% higher risk of cerebrovascular disease, and 96% higher risk of heart failure, compared with normal-weight individuals with no metabolic disease, after researchers adjusted for age, sex, smoking status, and social deprivation.
Individuals who were overweight but metabolically healthy had a 30% increased risk of ischemic heart disease, 11% increased risk of heart failure, and the same risk of cerebrovascular disease as normal-weight, healthy individuals.
They also saw an increasing risk of ischemic heart disease, cerebrovascular disease, heart failure, and peripheral vascular disease with each additional metabolic abnormality, even among underweight and normal-weight individuals, and suggested that a focus on screening overweight and obese individuals only could miss metabolic abnormalities in many patients.
Overweight and obese individuals without metabolic disease had a significantly lower risk of peripheral vascular disease, compared with healthy normal-weight individuals. The authors said this was a surprising finding but suggested cigarette smoking could be a confounding factor, as this is associated with both peripheral vascular disease and lower BMI.
“In sensitivity analyses restricted to individuals who were obese with no metabolic abnormalities and reported never smoking cigarettes, risk for PVD [peripheral vascular disease] was increased, compared [with] normal-weight individuals with no metabolic abnormalities,” Dr. Caleyachetty and his coinvestigators wrote.
Over the mean follow-up of 5.4 years, 5.6% of initially metabolically healthy obese individuals developed diabetes, 11.5% developed hyperlipidemia and 10.5% developed hypertension. In contrast, among the metabolically healthy overweight individuals at baseline, 1.9% developed diabetes, 9.4% developed hyperlipidemia, and 7.2% developed hypertension.
While the analysis adjusted for sex, the authors did note that women who were overweight or obese but metabolically healthy had stronger positive associations than did males with cerebrovascular disease and heart failure.
“Clinicians need to be aware that individuals who would otherwise be considered nonobese, based on a normal BMI, can have metabolic abnormalities, and therefore also be at high risk for CVD events,” the investigators concluded.
No conflicts of interest were declared.
Obese individuals with no metabolic abnormalities, such as dyslipidemia, high blood pressure, or high blood sugar levels, still have a higher risk of cardiovascular disease than do metabolically healthy people of normal weight, new data suggests.
“Our study robustly challenges the assertion that MHO [metabolically healthy obese] is a benign condition and adds to the evidence base that MHO is a high-risk state for future CVD events,” wrote Rishi Caleyachetty, MD, of the University of Birmingham, England, and his coauthors online (J Am Coll Cardiol. 2017, Sep 11. doi. 10.1016/j.jacc.2017.07.763).
Dr. Caleyachetty and his associates reported findings from a population-based study using the electronic health records of nearly 3.5 million individuals aged 18 years or older who were free of cardiovascular disease at baseline.
Overall, 15% of the population were classified as being metabolically healthy obese, meaning that they had a body mass index (BMI) of at least 30 kg/m2 with no sign of diabetes, hypertension, or hyperlipidemia, and 26% were overweight with no metabolic abnormalities. Despite their lack of metabolic disease, these obese individuals still had a significant 49% higher risk of coronary heart disease, 7% higher risk of cerebrovascular disease, and 96% higher risk of heart failure, compared with normal-weight individuals with no metabolic disease, after researchers adjusted for age, sex, smoking status, and social deprivation.
Individuals who were overweight but metabolically healthy had a 30% increased risk of ischemic heart disease, 11% increased risk of heart failure, and the same risk of cerebrovascular disease as normal-weight, healthy individuals.
They also saw an increasing risk of ischemic heart disease, cerebrovascular disease, heart failure, and peripheral vascular disease with each additional metabolic abnormality, even among underweight and normal-weight individuals, and suggested that a focus on screening overweight and obese individuals only could miss metabolic abnormalities in many patients.
Overweight and obese individuals without metabolic disease had a significantly lower risk of peripheral vascular disease, compared with healthy normal-weight individuals. The authors said this was a surprising finding but suggested cigarette smoking could be a confounding factor, as this is associated with both peripheral vascular disease and lower BMI.
“In sensitivity analyses restricted to individuals who were obese with no metabolic abnormalities and reported never smoking cigarettes, risk for PVD [peripheral vascular disease] was increased, compared [with] normal-weight individuals with no metabolic abnormalities,” Dr. Caleyachetty and his coinvestigators wrote.
Over the mean follow-up of 5.4 years, 5.6% of initially metabolically healthy obese individuals developed diabetes, 11.5% developed hyperlipidemia and 10.5% developed hypertension. In contrast, among the metabolically healthy overweight individuals at baseline, 1.9% developed diabetes, 9.4% developed hyperlipidemia, and 7.2% developed hypertension.
While the analysis adjusted for sex, the authors did note that women who were overweight or obese but metabolically healthy had stronger positive associations than did males with cerebrovascular disease and heart failure.
“Clinicians need to be aware that individuals who would otherwise be considered nonobese, based on a normal BMI, can have metabolic abnormalities, and therefore also be at high risk for CVD events,” the investigators concluded.
No conflicts of interest were declared.
FROM THE JOURNAL OF THE AMERICAN COLLEGE OF CARDIOLOGY
Key clinical point: Obese individuals without any sign of metabolic disease, such as hypertension or dyslipidemia, still have a greater risk of cardiovascular disease than do normal weight individuals.
Major finding: Metabolically healthy obese individuals have a 49% higher risk of coronary heart disease and 96% higher risk of heart failure than metabolically healthy normal weight individuals.
Data source: Population-based electronic health record study of 3.5 million people.
Disclosures: No conflicts of interest were declared.
PACIFIC: Durvalumab extends PFS in stage 3 NSCLC
MADRID – For patients with locally advanced, unresectable non-small cell lung cancer, consolidation therapy with the anti-programmed death ligand 1 (PD-L1) inhibitor durvalumab after chemoradiation was associated with significantly better progression-free survival (PFS) than placebo, results of an interim analysis of the phase 3 PACIFIC trial showed.
Among 713 patients with stage III NSCLC treated with definitive chemoradiotherapy, the median PFS from randomization was 16.8 months for patients assigned to durvalumab compared with 5.6 months for patients assigned to placebo, reported Luis Paz-Ares, MD, from the University of Madrid, Spain.
“Overall, we think durvalumab is a promising option for patients with stage III non-small cell lung cancer treated with chemoradiation,” he said at a briefing at the European Society of Medical Oncology (ESMO) Congress.
Approximately 25% to 30% of patients with NSCLC have locally advanced disease at the time of presentation. Patients with unresectable disease and good performance status are treated with chemoradiotheraoy consisting of a platinum doublet with concurrent radiation, but median PFS in these patients is generally short, on the order of 8 months. The 5-year overall survival (OS) rate is 15%, he said.
Given the lack of major advances in the care of patients with stage 3 disease, investigators have been looking to newer therapies such as immune checkpoint inhibitors to see whether they could improve outcomes.
PACIFIC is a phase 3 trial in which patients with stage III NSCLC who did not have disease progression after a minimum of two cycles of platinum-based chemotherapy were assigned on a 2:1 basis to receive intravenous durvalumab 10 mg/kg or placebo every 2 weeks for up to 12 months. Patients were stratified by age, sex, and smoking history.
Dr. Paz-Ares presented PFS results from the interim analysis, planned when about 367 events had occurred. Data on the co-primary endpoint of OS were not mature at the time of the data cutoff.
Median PFS from randomization according to blinded independent central review for 476 patients treated with durvalumab was 16.8 months, compared with 5.6 months for 237 patients who received the placebo. This translated into a stratified hazard ratio (HR) of 0.52 (P less than .0001).
The respective PFS rates for durvalumab and placebo, were 59.9% vs. 35.3% at 12 months, and 44.2% vs. 27% at 18 months.
Among 443 patients on durvalumab and 213 on placebo who were evaluable for objective responses (OR), the respective OR rates were 28.4% vs. 16.
There were 6 complete responses (CR), 120 partial responses (PR), and 233 cases of stable disease in the durvalumab arm, compared with one CR, 33 PR, and 119 cases of stable disease in the placebo arm. In the durvalumab arm, 73 patients (16.5%) had progressive disease, compared with 59 patients (27.7%) in the placebo arm.
The median duration of response was not reached in the durvalumab arm, compared with 13.8 months in the placebo arm.
The PD-L1 inhibitor was also associated with a lower incidence of any new lesion among the intention-to-treat population (20.4% vs. 32.1%).
Grade 3 or 4 toxicities from any cause were slightly higher with durvalumab, at 29.9% vs. 26.1%. Events leading to discontinuation were also higher with the active drug, at 15.4% vs. 9.8% for placebo.
There were 21 deaths (4.4%) of patients treated with durvalumab, and 13 deaths (5.6%) of patients treated with placebo.
“In my opinion, this is a very well-designed study, and the results are very promising,” commented Enriqueta Felip, MD, who was not involved in the PACIFIC trial.
“We need to wait for the overall survival results, but in my opinion this is a very valuable trial in a group of patients [for whom] we need new strategies,” she said.
Dr. Felip was invited to the briefing as an independent commentator.
Results of the interim analysis of the PACIFIC trial were also published online in the New England Journal of Medicine.
The PACIFIC trial was funded by AstraZeneca. Dr. Paz-Ares has received consultancy fees from the company. Dr. Felip disclosed financial relationships with multiple companies not including AstraZeneca.
MADRID – For patients with locally advanced, unresectable non-small cell lung cancer, consolidation therapy with the anti-programmed death ligand 1 (PD-L1) inhibitor durvalumab after chemoradiation was associated with significantly better progression-free survival (PFS) than placebo, results of an interim analysis of the phase 3 PACIFIC trial showed.
Among 713 patients with stage III NSCLC treated with definitive chemoradiotherapy, the median PFS from randomization was 16.8 months for patients assigned to durvalumab compared with 5.6 months for patients assigned to placebo, reported Luis Paz-Ares, MD, from the University of Madrid, Spain.
“Overall, we think durvalumab is a promising option for patients with stage III non-small cell lung cancer treated with chemoradiation,” he said at a briefing at the European Society of Medical Oncology (ESMO) Congress.
Approximately 25% to 30% of patients with NSCLC have locally advanced disease at the time of presentation. Patients with unresectable disease and good performance status are treated with chemoradiotheraoy consisting of a platinum doublet with concurrent radiation, but median PFS in these patients is generally short, on the order of 8 months. The 5-year overall survival (OS) rate is 15%, he said.
Given the lack of major advances in the care of patients with stage 3 disease, investigators have been looking to newer therapies such as immune checkpoint inhibitors to see whether they could improve outcomes.
PACIFIC is a phase 3 trial in which patients with stage III NSCLC who did not have disease progression after a minimum of two cycles of platinum-based chemotherapy were assigned on a 2:1 basis to receive intravenous durvalumab 10 mg/kg or placebo every 2 weeks for up to 12 months. Patients were stratified by age, sex, and smoking history.
Dr. Paz-Ares presented PFS results from the interim analysis, planned when about 367 events had occurred. Data on the co-primary endpoint of OS were not mature at the time of the data cutoff.
Median PFS from randomization according to blinded independent central review for 476 patients treated with durvalumab was 16.8 months, compared with 5.6 months for 237 patients who received the placebo. This translated into a stratified hazard ratio (HR) of 0.52 (P less than .0001).
The respective PFS rates for durvalumab and placebo, were 59.9% vs. 35.3% at 12 months, and 44.2% vs. 27% at 18 months.
Among 443 patients on durvalumab and 213 on placebo who were evaluable for objective responses (OR), the respective OR rates were 28.4% vs. 16.
There were 6 complete responses (CR), 120 partial responses (PR), and 233 cases of stable disease in the durvalumab arm, compared with one CR, 33 PR, and 119 cases of stable disease in the placebo arm. In the durvalumab arm, 73 patients (16.5%) had progressive disease, compared with 59 patients (27.7%) in the placebo arm.
The median duration of response was not reached in the durvalumab arm, compared with 13.8 months in the placebo arm.
The PD-L1 inhibitor was also associated with a lower incidence of any new lesion among the intention-to-treat population (20.4% vs. 32.1%).
Grade 3 or 4 toxicities from any cause were slightly higher with durvalumab, at 29.9% vs. 26.1%. Events leading to discontinuation were also higher with the active drug, at 15.4% vs. 9.8% for placebo.
There were 21 deaths (4.4%) of patients treated with durvalumab, and 13 deaths (5.6%) of patients treated with placebo.
“In my opinion, this is a very well-designed study, and the results are very promising,” commented Enriqueta Felip, MD, who was not involved in the PACIFIC trial.
“We need to wait for the overall survival results, but in my opinion this is a very valuable trial in a group of patients [for whom] we need new strategies,” she said.
Dr. Felip was invited to the briefing as an independent commentator.
Results of the interim analysis of the PACIFIC trial were also published online in the New England Journal of Medicine.
The PACIFIC trial was funded by AstraZeneca. Dr. Paz-Ares has received consultancy fees from the company. Dr. Felip disclosed financial relationships with multiple companies not including AstraZeneca.
MADRID – For patients with locally advanced, unresectable non-small cell lung cancer, consolidation therapy with the anti-programmed death ligand 1 (PD-L1) inhibitor durvalumab after chemoradiation was associated with significantly better progression-free survival (PFS) than placebo, results of an interim analysis of the phase 3 PACIFIC trial showed.
Among 713 patients with stage III NSCLC treated with definitive chemoradiotherapy, the median PFS from randomization was 16.8 months for patients assigned to durvalumab compared with 5.6 months for patients assigned to placebo, reported Luis Paz-Ares, MD, from the University of Madrid, Spain.
“Overall, we think durvalumab is a promising option for patients with stage III non-small cell lung cancer treated with chemoradiation,” he said at a briefing at the European Society of Medical Oncology (ESMO) Congress.
Approximately 25% to 30% of patients with NSCLC have locally advanced disease at the time of presentation. Patients with unresectable disease and good performance status are treated with chemoradiotheraoy consisting of a platinum doublet with concurrent radiation, but median PFS in these patients is generally short, on the order of 8 months. The 5-year overall survival (OS) rate is 15%, he said.
Given the lack of major advances in the care of patients with stage 3 disease, investigators have been looking to newer therapies such as immune checkpoint inhibitors to see whether they could improve outcomes.
PACIFIC is a phase 3 trial in which patients with stage III NSCLC who did not have disease progression after a minimum of two cycles of platinum-based chemotherapy were assigned on a 2:1 basis to receive intravenous durvalumab 10 mg/kg or placebo every 2 weeks for up to 12 months. Patients were stratified by age, sex, and smoking history.
Dr. Paz-Ares presented PFS results from the interim analysis, planned when about 367 events had occurred. Data on the co-primary endpoint of OS were not mature at the time of the data cutoff.
Median PFS from randomization according to blinded independent central review for 476 patients treated with durvalumab was 16.8 months, compared with 5.6 months for 237 patients who received the placebo. This translated into a stratified hazard ratio (HR) of 0.52 (P less than .0001).
The respective PFS rates for durvalumab and placebo, were 59.9% vs. 35.3% at 12 months, and 44.2% vs. 27% at 18 months.
Among 443 patients on durvalumab and 213 on placebo who were evaluable for objective responses (OR), the respective OR rates were 28.4% vs. 16.
There were 6 complete responses (CR), 120 partial responses (PR), and 233 cases of stable disease in the durvalumab arm, compared with one CR, 33 PR, and 119 cases of stable disease in the placebo arm. In the durvalumab arm, 73 patients (16.5%) had progressive disease, compared with 59 patients (27.7%) in the placebo arm.
The median duration of response was not reached in the durvalumab arm, compared with 13.8 months in the placebo arm.
The PD-L1 inhibitor was also associated with a lower incidence of any new lesion among the intention-to-treat population (20.4% vs. 32.1%).
Grade 3 or 4 toxicities from any cause were slightly higher with durvalumab, at 29.9% vs. 26.1%. Events leading to discontinuation were also higher with the active drug, at 15.4% vs. 9.8% for placebo.
There were 21 deaths (4.4%) of patients treated with durvalumab, and 13 deaths (5.6%) of patients treated with placebo.
“In my opinion, this is a very well-designed study, and the results are very promising,” commented Enriqueta Felip, MD, who was not involved in the PACIFIC trial.
“We need to wait for the overall survival results, but in my opinion this is a very valuable trial in a group of patients [for whom] we need new strategies,” she said.
Dr. Felip was invited to the briefing as an independent commentator.
Results of the interim analysis of the PACIFIC trial were also published online in the New England Journal of Medicine.
The PACIFIC trial was funded by AstraZeneca. Dr. Paz-Ares has received consultancy fees from the company. Dr. Felip disclosed financial relationships with multiple companies not including AstraZeneca.
AT ESMO 2017
Minimally invasive esophagectomy may mean less major morbidity
COLORADO SPRINGS – Minimally invasive esophagectomy was associated with a significantly lower rate of postoperative major morbidity as well as a mean 1-day briefer length of stay than open esophagectomy in a propensity-matched analysis of the real-world American College of Surgeons-National Quality Improvement Program database, Mark F. Berry, MD, reported at the annual meeting of the Western Thoracic Surgical Association.
However, both of the study’s discussants questioned whether the reported modest absolute reduction in major morbidity was really attributable to the minimally invasive approach or could instead have resulted from one of several potential confounders that couldn’t be fully adjusted for, given inherent limitations of the ACS-NSQIP database.
“There was a statistically significant difference in morbidity,” replied Dr. Berry of Stanford (Calif.) University. “It was a 4% absolute difference, which I think is probably clinically meaningful, but certainly it’s not really, really dramatic.”

“What I think we found is that it’s safe to do a minimally invasive esophagectomy and safe for people to introduce it into their practice. But it’s not necessarily something that’s a game changer, unlike what’s been seen with minimally invasive approaches for some other things,” said Dr. Berry, who added that he didn’t wish to overstate the importance of the observed difference in morbidity.
Studies from high-volume centers show that minimally-invasive esophagectomy (MIE) reduces length of stay, postoperative major morbidity, and features equivalent or even slightly lower mortality than traditional open esophagectomy, the generalizability of these findings beyond such centers is questionable. That’s why Dr. Berry and his coinvestigators turned to the ACS-NSQIP database, which includes all esophagectomies performed for esophageal cancer at roughly 700 U.S. hospitals, not just those done by board-certified thoracic surgeons.
He presented a retrospective cohort study of 3,901 esophagectomy patients during 2005-2013 who met study criteria, 16.4% of whom had MIE. The use of this approach increased steadily from 6.5% of all esophagectomies in 2005 to 22.3% in 2013. A propensity-matched analysis designed to neutralize potentially confounding differences included 638 MIE and 1,914 open esophagectomy patients.
The primary outcome was the 30-day rate of composite major morbidity in the realms of various wound, respiratory, renal, and cardiovascular complications. The rate was 36.1% in the MIE group and 40.5% with open esophagectomy in the propensity-matched analysis, an absolute risk reduction of 4.4% and a relative risk reduction of 17%. Although rates were consistently slightly lower in each of the categories of major morbidity, those individual differences didn’t achieve statistical significance. The difference in major morbidity became significant only when major morbidity was considered as a whole.
Mean length of stay was 9 days with MIE and 10 days with open surgery.
There was no significant difference between the two study groups in 30-day rates of readmission, reoperation, or mortality.
Discussant Donald E. Low said “esophagectomy is being analysed regarding its place in all sorts of presentations, stages, and situations, so the aspect of making sure that we’re delivering the services as efficiently as possible is going to become more important, not less important.”
That being said, he noted that there is no specific CPT code for MIE. That raises the possibility of an uncertain amount of procedural misclassification in the ACS-NSQIP database.
Also, the only significant difference in major morbidity between the two study groups was in the subcategory of intra- or postoperative bleeding requiring transfusion, which occurred in 10.8% of the MIE and 16.7% of the open esophagectomy groups, observed Dr. Low, director of the Esophageal Center of Excellence at Virginia Mason Medical Center, Seattle.
“Some of us believe that blood utilization and transfusion requirement is really a quality measure and not a complication,” the surgeon said. And if that outcome is excluded from consideration, then there is no significant difference in major morbidity.
Discussant Douglas E. Wood, MD, professor and chair of the department of surgery at the University of Washington, Seattle, took the opportunity to share a self-described “pet peeve” about analyses of national surgical databases: these databases typically don’t contain key details necessary to correct for provider and hospital characteristics.
“The small differences that you demonstrate could easily have been completely driven by providers who choose to do minimally invasive esophagectomy and are in higher-volume, more specialized centers,” he said. “I’m not convinced of your conclusion that MIE produces less morbidity based on a 4% difference and no analysis of provider characteristics.”
COLORADO SPRINGS – Minimally invasive esophagectomy was associated with a significantly lower rate of postoperative major morbidity as well as a mean 1-day briefer length of stay than open esophagectomy in a propensity-matched analysis of the real-world American College of Surgeons-National Quality Improvement Program database, Mark F. Berry, MD, reported at the annual meeting of the Western Thoracic Surgical Association.
However, both of the study’s discussants questioned whether the reported modest absolute reduction in major morbidity was really attributable to the minimally invasive approach or could instead have resulted from one of several potential confounders that couldn’t be fully adjusted for, given inherent limitations of the ACS-NSQIP database.
“There was a statistically significant difference in morbidity,” replied Dr. Berry of Stanford (Calif.) University. “It was a 4% absolute difference, which I think is probably clinically meaningful, but certainly it’s not really, really dramatic.”
“What I think we found is that it’s safe to do a minimally invasive esophagectomy and safe for people to introduce it into their practice. But it’s not necessarily something that’s a game changer, unlike what’s been seen with minimally invasive approaches for some other things,” said Dr. Berry, who added that he didn’t wish to overstate the importance of the observed difference in morbidity.
Studies from high-volume centers show that minimally-invasive esophagectomy (MIE) reduces length of stay, postoperative major morbidity, and features equivalent or even slightly lower mortality than traditional open esophagectomy, the generalizability of these findings beyond such centers is questionable. That’s why Dr. Berry and his coinvestigators turned to the ACS-NSQIP database, which includes all esophagectomies performed for esophageal cancer at roughly 700 U.S. hospitals, not just those done by board-certified thoracic surgeons.
He presented a retrospective cohort study of 3,901 esophagectomy patients during 2005-2013 who met study criteria, 16.4% of whom had MIE. The use of this approach increased steadily from 6.5% of all esophagectomies in 2005 to 22.3% in 2013. A propensity-matched analysis designed to neutralize potentially confounding differences included 638 MIE and 1,914 open esophagectomy patients.
The primary outcome was the 30-day rate of composite major morbidity in the realms of various wound, respiratory, renal, and cardiovascular complications. The rate was 36.1% in the MIE group and 40.5% with open esophagectomy in the propensity-matched analysis, an absolute risk reduction of 4.4% and a relative risk reduction of 17%. Although rates were consistently slightly lower in each of the categories of major morbidity, those individual differences didn’t achieve statistical significance. The difference in major morbidity became significant only when major morbidity was considered as a whole.
Mean length of stay was 9 days with MIE and 10 days with open surgery.
There was no significant difference between the two study groups in 30-day rates of readmission, reoperation, or mortality.
Discussant Donald E. Low said “esophagectomy is being analysed regarding its place in all sorts of presentations, stages, and situations, so the aspect of making sure that we’re delivering the services as efficiently as possible is going to become more important, not less important.”
That being said, he noted that there is no specific CPT code for MIE. That raises the possibility of an uncertain amount of procedural misclassification in the ACS-NSQIP database.
Also, the only significant difference in major morbidity between the two study groups was in the subcategory of intra- or postoperative bleeding requiring transfusion, which occurred in 10.8% of the MIE and 16.7% of the open esophagectomy groups, observed Dr. Low, director of the Esophageal Center of Excellence at Virginia Mason Medical Center, Seattle.
“Some of us believe that blood utilization and transfusion requirement is really a quality measure and not a complication,” the surgeon said. And if that outcome is excluded from consideration, then there is no significant difference in major morbidity.
Discussant Douglas E. Wood, MD, professor and chair of the department of surgery at the University of Washington, Seattle, took the opportunity to share a self-described “pet peeve” about analyses of national surgical databases: these databases typically don’t contain key details necessary to correct for provider and hospital characteristics.
“The small differences that you demonstrate could easily have been completely driven by providers who choose to do minimally invasive esophagectomy and are in higher-volume, more specialized centers,” he said. “I’m not convinced of your conclusion that MIE produces less morbidity based on a 4% difference and no analysis of provider characteristics.”
COLORADO SPRINGS – Minimally invasive esophagectomy was associated with a significantly lower rate of postoperative major morbidity as well as a mean 1-day briefer length of stay than open esophagectomy in a propensity-matched analysis of the real-world American College of Surgeons-National Quality Improvement Program database, Mark F. Berry, MD, reported at the annual meeting of the Western Thoracic Surgical Association.
However, both of the study’s discussants questioned whether the reported modest absolute reduction in major morbidity was really attributable to the minimally invasive approach or could instead have resulted from one of several potential confounders that couldn’t be fully adjusted for, given inherent limitations of the ACS-NSQIP database.
“There was a statistically significant difference in morbidity,” replied Dr. Berry of Stanford (Calif.) University. “It was a 4% absolute difference, which I think is probably clinically meaningful, but certainly it’s not really, really dramatic.”
“What I think we found is that it’s safe to do a minimally invasive esophagectomy and safe for people to introduce it into their practice. But it’s not necessarily something that’s a game changer, unlike what’s been seen with minimally invasive approaches for some other things,” said Dr. Berry, who added that he didn’t wish to overstate the importance of the observed difference in morbidity.
Studies from high-volume centers show that minimally-invasive esophagectomy (MIE) reduces length of stay, postoperative major morbidity, and features equivalent or even slightly lower mortality than traditional open esophagectomy, the generalizability of these findings beyond such centers is questionable. That’s why Dr. Berry and his coinvestigators turned to the ACS-NSQIP database, which includes all esophagectomies performed for esophageal cancer at roughly 700 U.S. hospitals, not just those done by board-certified thoracic surgeons.
He presented a retrospective cohort study of 3,901 esophagectomy patients during 2005-2013 who met study criteria, 16.4% of whom had MIE. The use of this approach increased steadily from 6.5% of all esophagectomies in 2005 to 22.3% in 2013. A propensity-matched analysis designed to neutralize potentially confounding differences included 638 MIE and 1,914 open esophagectomy patients.
The primary outcome was the 30-day rate of composite major morbidity in the realms of various wound, respiratory, renal, and cardiovascular complications. The rate was 36.1% in the MIE group and 40.5% with open esophagectomy in the propensity-matched analysis, an absolute risk reduction of 4.4% and a relative risk reduction of 17%. Although rates were consistently slightly lower in each of the categories of major morbidity, those individual differences didn’t achieve statistical significance. The difference in major morbidity became significant only when major morbidity was considered as a whole.
Mean length of stay was 9 days with MIE and 10 days with open surgery.
There was no significant difference between the two study groups in 30-day rates of readmission, reoperation, or mortality.
Discussant Donald E. Low said “esophagectomy is being analysed regarding its place in all sorts of presentations, stages, and situations, so the aspect of making sure that we’re delivering the services as efficiently as possible is going to become more important, not less important.”
That being said, he noted that there is no specific CPT code for MIE. That raises the possibility of an uncertain amount of procedural misclassification in the ACS-NSQIP database.
Also, the only significant difference in major morbidity between the two study groups was in the subcategory of intra- or postoperative bleeding requiring transfusion, which occurred in 10.8% of the MIE and 16.7% of the open esophagectomy groups, observed Dr. Low, director of the Esophageal Center of Excellence at Virginia Mason Medical Center, Seattle.
“Some of us believe that blood utilization and transfusion requirement is really a quality measure and not a complication,” the surgeon said. And if that outcome is excluded from consideration, then there is no significant difference in major morbidity.
Discussant Douglas E. Wood, MD, professor and chair of the department of surgery at the University of Washington, Seattle, took the opportunity to share a self-described “pet peeve” about analyses of national surgical databases: these databases typically don’t contain key details necessary to correct for provider and hospital characteristics.
“The small differences that you demonstrate could easily have been completely driven by providers who choose to do minimally invasive esophagectomy and are in higher-volume, more specialized centers,” he said. “I’m not convinced of your conclusion that MIE produces less morbidity based on a 4% difference and no analysis of provider characteristics.”
AT WTSA 2017
Key clinical point:
Major finding: The 30-day rate of major morbidity was 36.1% in patients who underwent minimally invasive esophagectomy, significantly lower than the 40.5% rate with open esophagectomy in a propensity-matched analysis.
Data source: This retrospective cohort study included 3,901 patients who underwent esophagectomy for esophageal cancer as recorded in the American College of Surgeons-National Quality Improvement Program database for 2005-2013.
Disclosures: The study presenter reported having no financial conflicts of interest.
Absorb bioresorbable scaffold folds
The first bioresorbable vascular scaffold to reach clinical practice is no longer available because of poor sales, following nearly a year of bad news for the innovative device.
"We took this decision for commercial reasons, not for safety," Kristina Becker, director of communications at Abbott's Vascular business. Abbott announced that sales of the Absorb scaffold would cease worldwide as of Sept. 14, 2017, adding that the company would continue to follow patients in existing clinical trials according to their protocols.
The device was approved by the Food and Drug Administration in July 2016, but soon after, long-term trial results took an ominous turn. In March 2017, the agency issued a safety alert regarding the Absorb scaffold after release of the 2-year data from the 2,008-patient ABSORB III trial showing a significantly higher rate of target-lesion failure than with the everolimus-eluting metallic Xience stent, also marketed by Abbott.
Meanwhile, 3-year results of the ABSORB II trial, reported last October, revealed a 1% per year rate of late stent thrombosis during both the second and third years following Absorb placement in coronary arteries, the period when the Absorb scaffold was in the process of disappearing. More bad news came in July, when device thrombosis occurred nearly four times more frequently in recipients of the Absorb scaffold than with the Xience stent during 2 years of prospective follow-up in the randomized, investigator-initiated AIDA trial.
Abbott is not giving up on the idea of nonpermanent stents. “We recognize that [bioresorbable scaffold] technologies offer patients the possibility of life without permanent metallic implants, and we will continue to work on a next generation device while monitoring long-term outcomes after stent resorption in current Absorb trials,” the announcement said.
This article was updated 9/8/17.
The first bioresorbable vascular scaffold to reach clinical practice is no longer available because of poor sales, following nearly a year of bad news for the innovative device.
"We took this decision for commercial reasons, not for safety," Kristina Becker, director of communications at Abbott's Vascular business. Abbott announced that sales of the Absorb scaffold would cease worldwide as of Sept. 14, 2017, adding that the company would continue to follow patients in existing clinical trials according to their protocols.
The device was approved by the Food and Drug Administration in July 2016, but soon after, long-term trial results took an ominous turn. In March 2017, the agency issued a safety alert regarding the Absorb scaffold after release of the 2-year data from the 2,008-patient ABSORB III trial showing a significantly higher rate of target-lesion failure than with the everolimus-eluting metallic Xience stent, also marketed by Abbott.
Meanwhile, 3-year results of the ABSORB II trial, reported last October, revealed a 1% per year rate of late stent thrombosis during both the second and third years following Absorb placement in coronary arteries, the period when the Absorb scaffold was in the process of disappearing. More bad news came in July, when device thrombosis occurred nearly four times more frequently in recipients of the Absorb scaffold than with the Xience stent during 2 years of prospective follow-up in the randomized, investigator-initiated AIDA trial.
Abbott is not giving up on the idea of nonpermanent stents. “We recognize that [bioresorbable scaffold] technologies offer patients the possibility of life without permanent metallic implants, and we will continue to work on a next generation device while monitoring long-term outcomes after stent resorption in current Absorb trials,” the announcement said.
This article was updated 9/8/17.
The first bioresorbable vascular scaffold to reach clinical practice is no longer available because of poor sales, following nearly a year of bad news for the innovative device.
"We took this decision for commercial reasons, not for safety," Kristina Becker, director of communications at Abbott's Vascular business. Abbott announced that sales of the Absorb scaffold would cease worldwide as of Sept. 14, 2017, adding that the company would continue to follow patients in existing clinical trials according to their protocols.
The device was approved by the Food and Drug Administration in July 2016, but soon after, long-term trial results took an ominous turn. In March 2017, the agency issued a safety alert regarding the Absorb scaffold after release of the 2-year data from the 2,008-patient ABSORB III trial showing a significantly higher rate of target-lesion failure than with the everolimus-eluting metallic Xience stent, also marketed by Abbott.
Meanwhile, 3-year results of the ABSORB II trial, reported last October, revealed a 1% per year rate of late stent thrombosis during both the second and third years following Absorb placement in coronary arteries, the period when the Absorb scaffold was in the process of disappearing. More bad news came in July, when device thrombosis occurred nearly four times more frequently in recipients of the Absorb scaffold than with the Xience stent during 2 years of prospective follow-up in the randomized, investigator-initiated AIDA trial.
Abbott is not giving up on the idea of nonpermanent stents. “We recognize that [bioresorbable scaffold] technologies offer patients the possibility of life without permanent metallic implants, and we will continue to work on a next generation device while monitoring long-term outcomes after stent resorption in current Absorb trials,” the announcement said.
This article was updated 9/8/17.
Making Practice Perfect Download
Please click either of the links below to download your free eBook