User login
What Are the Best Therapeutic Options for Parkinson’s Disease?
Levodopa remains the most effective treatment, and techniques for deep brain stimulation are improving.
LOS ANGELES—Physicians who treat patients with Parkinson’s disease have many decisions to make based on therapeutic efficacy and desired outcomes. At the 70th Annual Meeting of the American Academy of Neurology, Melissa J. Nirenberg, MD, PhD, outlined the current landscape of Parkinson’s disease therapeutics, including data about symptom control, timing of treatment, and new therapies.

Initial Therapy: No Benefit to Levodopa Sparing
Levodopa, along with dopamine agonists and monoamine oxidase B (MAO-B) inhibitors, has Level A evidence as initial symptomatic therapy for Parkinson’s disease. “There is no question that levodopa is the most effective treatment for Parkinson’s disease,” said Dr. Nirenberg, Chief Medical Officer of the New York Stem Cell Foundation Research Institute and Adjunct Professor of Neurology at NYU Langone Health in New York City. “However, after people have been taking levodopa for a number of years, its therapeutic effect lasts for shorter periods of time, and patients spend an increasing amount of time in the off state, rather than in the on state.”
In addition to this wearing-off effect, levodopa treatment is associated with dyskinesias. This association and the wearing-off effect have prompted many physicians to adopt levodopa-sparing strategies, such as using dopamine agonists as initial treatment. However, dopamine agonists have other serious side effects, and research shows that in the long run, starting with a dopamine agonist does not improve outcomes.
In one study, data were compared between a large cohort of patients with Parkinson’s disease in Ghana, where levodopa therapy was initiated after a mean of 4.2 years’ disease duration, and patients with Parkinson’s disease in Italy, where levodopa was initiated at a mean of 2.4 years’ disease duration. “Disease duration and medication dosage, rather than the duration of levodopa therapy, affected the likelihood of dyskinesia,” Dr. Nirenberg said. “When you start levodopa late, you miss the honeymoon period,” she said, referring to the period during which patients experience the benefits of levodopa therapy before developing motor complications. “Simply put, levodopa as initial treatment works better [and] has fewer short- and long-term adverse effects [than dopamine agonists].”
Other Therapies
Dopamine agonists are highly efficacious as add-on treatment, but they also can have serious adverse effects. “Neurogenic orthostatic hypotension, psychosis, and sleepiness are adverse effects that are worse with dopamine agonists than with levodopa,” Dr. Nirenberg noted. “Another common adverse effect associated with dopamine agonists is impulse control disorders—pathologic gambling, compulsive eating, compulsive shopping, and hypersexuality.”
MAO-B inhibitors are also commonly used, well-tolerated medications that can be administered alone or in combination with levodopa or other medications. Of these drugs, selegiline and rasagiline can be used as monotherapy, Dr. Nirenberg noted, but a newer MAO-B inhibitor, safinamide, is not effective as monotherapy and should only be used as an adjunctive therapy with levodopa.
Extended release (ER) carbidopa–levodopa capsules, which contain immediate-release and ER beads to provide initial and extended levodopa plasma concentrations, have been effective in reducing wearing off between doses of levodopa, but conversion to this formulation from immediate-release levodopa is not straightforward. Rather than using the suggested conversion table in the package insert, neurologists might try the approach suggested by investigators who participated in the original clinical trials, said Dr. Nirenberg. Extended-release “capsules can be twisted open, and the beads poured into applesauce for people who have trouble swallowing,” she added.
Amantadine and anticholinergics are second-line medications that can be used as initial or adjunctive therapy. “They are weaker than the first-line drugs and have unfavorable adverse-effect profiles,” said Dr. Nirenberg. Amantadine, of which a newly approved extended-release formulation is available, can reduce dyskinesias.
New and Investigational Treatments
Deep brain stimulation (DBS) techniques are advancing, said Dr. Nirenberg. With DBS, a device implanted in the chest sends electrical pulses to electrodes inserted into targeted areas of the brain. “Recent studies are looking at closed-loop systems that provide direct feedback from the brain to the pacemaker so that stimulation is adjusted in real time.”
Continuous enteral infusion of carbidopa–levodopa intestinal gel over 16 hours via percutaneous endoscopic gastrojejunostomy is an option for people for whom DBS is being considered, but who have contraindications such as cognitive impairment or psychosis. “This [treatment] should only be prescribed to someone who has a good caregiver, because the pump has to be flushed often, removed before bathing, and checked to make sure there are no hardware problems or infections associated with its use.”
Droxidopa, a synthetic amino acid precursor of noradrenaline, received orphan-product designation for treatment of
Pimavanserin, a first-in-class drug approved in 2016 to treat hallucinations and delusions associated with Parkinson’s disease psychosis, is an atypical antipsychotic with a serotonergic mechanism of action. While the prospect of having such a treatment option initially generated excitement in the medical community, there have been recent concerns about adverse events in patients taking pimavanserin, including deaths, falls, insomnia, and nausea, in addition to continued hallucinations.
Focused ultrasound is approved for essential tremor and is investigational for Parkinson’s disease, Dr. Nirenberg noted. During the procedure, which can be performed on an outpatient basis, focused beams of ultrasonic energy are trained on targets deep in the brain to destroy diseased tissue without damaging surrounding normal tissue. Because of the lack of long-term follow-up of these patients, neurologists “do not know where this ultimately will fit in with Parkinson’s disease management,” said Dr. Nirenberg. Focused ultrasound is mainly being investigated as unilateral treatment because of concerns about the safety of bilateral ablative therapy.
To date, research on oral cannabinoids has not shown evidence of benefit for Parkinson’s disease, said Dr. Nirenberg. Neurologists have concerns about potential drug interactions and side effects such as imbalance, falls, cognitive impairment, and psychosis, which are of particular concern in people with Parkinson’s disease.
—Adriene Marshall
Cilia R, Akpalu A, Sarfo FS, et al. The modern pre-levodopa era of Parkinson’s disease: insights into motor complications from sub-Saharan Africa. Brain. 2014;137(Pt 10):2731-2742.
Tetrud J, Nausieda P, Kreitzman D, et al. Conversion to carbidopa and levodopa extended-release (IPX066) followed by its extended use in patients previously taking controlled-release carbidopa-levodopa for advanced Parkinson’s disease. J Neurol Sci. 2017;373:116-123.
Levodopa remains the most effective treatment, and techniques for deep brain stimulation are improving.
Levodopa remains the most effective treatment, and techniques for deep brain stimulation are improving.
LOS ANGELES—Physicians who treat patients with Parkinson’s disease have many decisions to make based on therapeutic efficacy and desired outcomes. At the 70th Annual Meeting of the American Academy of Neurology, Melissa J. Nirenberg, MD, PhD, outlined the current landscape of Parkinson’s disease therapeutics, including data about symptom control, timing of treatment, and new therapies.
Initial Therapy: No Benefit to Levodopa Sparing
Levodopa, along with dopamine agonists and monoamine oxidase B (MAO-B) inhibitors, has Level A evidence as initial symptomatic therapy for Parkinson’s disease. “There is no question that levodopa is the most effective treatment for Parkinson’s disease,” said Dr. Nirenberg, Chief Medical Officer of the New York Stem Cell Foundation Research Institute and Adjunct Professor of Neurology at NYU Langone Health in New York City. “However, after people have been taking levodopa for a number of years, its therapeutic effect lasts for shorter periods of time, and patients spend an increasing amount of time in the off state, rather than in the on state.”
In addition to this wearing-off effect, levodopa treatment is associated with dyskinesias. This association and the wearing-off effect have prompted many physicians to adopt levodopa-sparing strategies, such as using dopamine agonists as initial treatment. However, dopamine agonists have other serious side effects, and research shows that in the long run, starting with a dopamine agonist does not improve outcomes.
In one study, data were compared between a large cohort of patients with Parkinson’s disease in Ghana, where levodopa therapy was initiated after a mean of 4.2 years’ disease duration, and patients with Parkinson’s disease in Italy, where levodopa was initiated at a mean of 2.4 years’ disease duration. “Disease duration and medication dosage, rather than the duration of levodopa therapy, affected the likelihood of dyskinesia,” Dr. Nirenberg said. “When you start levodopa late, you miss the honeymoon period,” she said, referring to the period during which patients experience the benefits of levodopa therapy before developing motor complications. “Simply put, levodopa as initial treatment works better [and] has fewer short- and long-term adverse effects [than dopamine agonists].”
Other Therapies
Dopamine agonists are highly efficacious as add-on treatment, but they also can have serious adverse effects. “Neurogenic orthostatic hypotension, psychosis, and sleepiness are adverse effects that are worse with dopamine agonists than with levodopa,” Dr. Nirenberg noted. “Another common adverse effect associated with dopamine agonists is impulse control disorders—pathologic gambling, compulsive eating, compulsive shopping, and hypersexuality.”
MAO-B inhibitors are also commonly used, well-tolerated medications that can be administered alone or in combination with levodopa or other medications. Of these drugs, selegiline and rasagiline can be used as monotherapy, Dr. Nirenberg noted, but a newer MAO-B inhibitor, safinamide, is not effective as monotherapy and should only be used as an adjunctive therapy with levodopa.
Extended release (ER) carbidopa–levodopa capsules, which contain immediate-release and ER beads to provide initial and extended levodopa plasma concentrations, have been effective in reducing wearing off between doses of levodopa, but conversion to this formulation from immediate-release levodopa is not straightforward. Rather than using the suggested conversion table in the package insert, neurologists might try the approach suggested by investigators who participated in the original clinical trials, said Dr. Nirenberg. Extended-release “capsules can be twisted open, and the beads poured into applesauce for people who have trouble swallowing,” she added.
Amantadine and anticholinergics are second-line medications that can be used as initial or adjunctive therapy. “They are weaker than the first-line drugs and have unfavorable adverse-effect profiles,” said Dr. Nirenberg. Amantadine, of which a newly approved extended-release formulation is available, can reduce dyskinesias.
New and Investigational Treatments
Deep brain stimulation (DBS) techniques are advancing, said Dr. Nirenberg. With DBS, a device implanted in the chest sends electrical pulses to electrodes inserted into targeted areas of the brain. “Recent studies are looking at closed-loop systems that provide direct feedback from the brain to the pacemaker so that stimulation is adjusted in real time.”
Continuous enteral infusion of carbidopa–levodopa intestinal gel over 16 hours via percutaneous endoscopic gastrojejunostomy is an option for people for whom DBS is being considered, but who have contraindications such as cognitive impairment or psychosis. “This [treatment] should only be prescribed to someone who has a good caregiver, because the pump has to be flushed often, removed before bathing, and checked to make sure there are no hardware problems or infections associated with its use.”
Droxidopa, a synthetic amino acid precursor of noradrenaline, received orphan-product designation for treatment of
Pimavanserin, a first-in-class drug approved in 2016 to treat hallucinations and delusions associated with Parkinson’s disease psychosis, is an atypical antipsychotic with a serotonergic mechanism of action. While the prospect of having such a treatment option initially generated excitement in the medical community, there have been recent concerns about adverse events in patients taking pimavanserin, including deaths, falls, insomnia, and nausea, in addition to continued hallucinations.
Focused ultrasound is approved for essential tremor and is investigational for Parkinson’s disease, Dr. Nirenberg noted. During the procedure, which can be performed on an outpatient basis, focused beams of ultrasonic energy are trained on targets deep in the brain to destroy diseased tissue without damaging surrounding normal tissue. Because of the lack of long-term follow-up of these patients, neurologists “do not know where this ultimately will fit in with Parkinson’s disease management,” said Dr. Nirenberg. Focused ultrasound is mainly being investigated as unilateral treatment because of concerns about the safety of bilateral ablative therapy.
To date, research on oral cannabinoids has not shown evidence of benefit for Parkinson’s disease, said Dr. Nirenberg. Neurologists have concerns about potential drug interactions and side effects such as imbalance, falls, cognitive impairment, and psychosis, which are of particular concern in people with Parkinson’s disease.
—Adriene Marshall
Cilia R, Akpalu A, Sarfo FS, et al. The modern pre-levodopa era of Parkinson’s disease: insights into motor complications from sub-Saharan Africa. Brain. 2014;137(Pt 10):2731-2742.
Tetrud J, Nausieda P, Kreitzman D, et al. Conversion to carbidopa and levodopa extended-release (IPX066) followed by its extended use in patients previously taking controlled-release carbidopa-levodopa for advanced Parkinson’s disease. J Neurol Sci. 2017;373:116-123.
LOS ANGELES—Physicians who treat patients with Parkinson’s disease have many decisions to make based on therapeutic efficacy and desired outcomes. At the 70th Annual Meeting of the American Academy of Neurology, Melissa J. Nirenberg, MD, PhD, outlined the current landscape of Parkinson’s disease therapeutics, including data about symptom control, timing of treatment, and new therapies.
Initial Therapy: No Benefit to Levodopa Sparing
Levodopa, along with dopamine agonists and monoamine oxidase B (MAO-B) inhibitors, has Level A evidence as initial symptomatic therapy for Parkinson’s disease. “There is no question that levodopa is the most effective treatment for Parkinson’s disease,” said Dr. Nirenberg, Chief Medical Officer of the New York Stem Cell Foundation Research Institute and Adjunct Professor of Neurology at NYU Langone Health in New York City. “However, after people have been taking levodopa for a number of years, its therapeutic effect lasts for shorter periods of time, and patients spend an increasing amount of time in the off state, rather than in the on state.”
In addition to this wearing-off effect, levodopa treatment is associated with dyskinesias. This association and the wearing-off effect have prompted many physicians to adopt levodopa-sparing strategies, such as using dopamine agonists as initial treatment. However, dopamine agonists have other serious side effects, and research shows that in the long run, starting with a dopamine agonist does not improve outcomes.
In one study, data were compared between a large cohort of patients with Parkinson’s disease in Ghana, where levodopa therapy was initiated after a mean of 4.2 years’ disease duration, and patients with Parkinson’s disease in Italy, where levodopa was initiated at a mean of 2.4 years’ disease duration. “Disease duration and medication dosage, rather than the duration of levodopa therapy, affected the likelihood of dyskinesia,” Dr. Nirenberg said. “When you start levodopa late, you miss the honeymoon period,” she said, referring to the period during which patients experience the benefits of levodopa therapy before developing motor complications. “Simply put, levodopa as initial treatment works better [and] has fewer short- and long-term adverse effects [than dopamine agonists].”
Other Therapies
Dopamine agonists are highly efficacious as add-on treatment, but they also can have serious adverse effects. “Neurogenic orthostatic hypotension, psychosis, and sleepiness are adverse effects that are worse with dopamine agonists than with levodopa,” Dr. Nirenberg noted. “Another common adverse effect associated with dopamine agonists is impulse control disorders—pathologic gambling, compulsive eating, compulsive shopping, and hypersexuality.”
MAO-B inhibitors are also commonly used, well-tolerated medications that can be administered alone or in combination with levodopa or other medications. Of these drugs, selegiline and rasagiline can be used as monotherapy, Dr. Nirenberg noted, but a newer MAO-B inhibitor, safinamide, is not effective as monotherapy and should only be used as an adjunctive therapy with levodopa.
Extended release (ER) carbidopa–levodopa capsules, which contain immediate-release and ER beads to provide initial and extended levodopa plasma concentrations, have been effective in reducing wearing off between doses of levodopa, but conversion to this formulation from immediate-release levodopa is not straightforward. Rather than using the suggested conversion table in the package insert, neurologists might try the approach suggested by investigators who participated in the original clinical trials, said Dr. Nirenberg. Extended-release “capsules can be twisted open, and the beads poured into applesauce for people who have trouble swallowing,” she added.
Amantadine and anticholinergics are second-line medications that can be used as initial or adjunctive therapy. “They are weaker than the first-line drugs and have unfavorable adverse-effect profiles,” said Dr. Nirenberg. Amantadine, of which a newly approved extended-release formulation is available, can reduce dyskinesias.
New and Investigational Treatments
Deep brain stimulation (DBS) techniques are advancing, said Dr. Nirenberg. With DBS, a device implanted in the chest sends electrical pulses to electrodes inserted into targeted areas of the brain. “Recent studies are looking at closed-loop systems that provide direct feedback from the brain to the pacemaker so that stimulation is adjusted in real time.”
Continuous enteral infusion of carbidopa–levodopa intestinal gel over 16 hours via percutaneous endoscopic gastrojejunostomy is an option for people for whom DBS is being considered, but who have contraindications such as cognitive impairment or psychosis. “This [treatment] should only be prescribed to someone who has a good caregiver, because the pump has to be flushed often, removed before bathing, and checked to make sure there are no hardware problems or infections associated with its use.”
Droxidopa, a synthetic amino acid precursor of noradrenaline, received orphan-product designation for treatment of
Pimavanserin, a first-in-class drug approved in 2016 to treat hallucinations and delusions associated with Parkinson’s disease psychosis, is an atypical antipsychotic with a serotonergic mechanism of action. While the prospect of having such a treatment option initially generated excitement in the medical community, there have been recent concerns about adverse events in patients taking pimavanserin, including deaths, falls, insomnia, and nausea, in addition to continued hallucinations.
Focused ultrasound is approved for essential tremor and is investigational for Parkinson’s disease, Dr. Nirenberg noted. During the procedure, which can be performed on an outpatient basis, focused beams of ultrasonic energy are trained on targets deep in the brain to destroy diseased tissue without damaging surrounding normal tissue. Because of the lack of long-term follow-up of these patients, neurologists “do not know where this ultimately will fit in with Parkinson’s disease management,” said Dr. Nirenberg. Focused ultrasound is mainly being investigated as unilateral treatment because of concerns about the safety of bilateral ablative therapy.
To date, research on oral cannabinoids has not shown evidence of benefit for Parkinson’s disease, said Dr. Nirenberg. Neurologists have concerns about potential drug interactions and side effects such as imbalance, falls, cognitive impairment, and psychosis, which are of particular concern in people with Parkinson’s disease.
—Adriene Marshall
Cilia R, Akpalu A, Sarfo FS, et al. The modern pre-levodopa era of Parkinson’s disease: insights into motor complications from sub-Saharan Africa. Brain. 2014;137(Pt 10):2731-2742.
Tetrud J, Nausieda P, Kreitzman D, et al. Conversion to carbidopa and levodopa extended-release (IPX066) followed by its extended use in patients previously taking controlled-release carbidopa-levodopa for advanced Parkinson’s disease. J Neurol Sci. 2017;373:116-123.
Eptinezumab May Reduce Migraine Frequency in Patients With Chronic and Episodic Migraine
Infusion of the anti-CGRP monoclonal antibody significantly reduces monthly migraine days, versus placebo.
LOS ANGELES—Among patients with chronic migraine, IV infusion of eptinezumab significantly reduces the average number of migraine days per
Eptinezumab (formerly known as ALD403) is a humanized monoclonal antibody that inhibits calcitonin gene-related peptide (CGRP). It is designed to be administered quarterly via IV infusion. Eptinezumab previously was found to be effective and well tolerated in phase II studies in episodic and chronic migraine and in a phase III trial in episodic migraine. Alder BioPharmaceuticals, based in Bothell, Washington, is developing the therapy.
Treatment-emergent adverse events in phase III studies were similar for eptinezumab and placebo, and the safety profile was consistent with that in prior studies, researchers said.
The PROMISE Clinical Trial Program
The Prevention of Migraine via Intravenous ALD403 Safety and Efficacy (PROMISE) clinical trial program includes phase III, randomized, double-blind, placebo-controlled trials of eptinezumab for chronic migraine prevention (PROMISE-2) and episodic migraine prevention (PROMISE-1).
In the PROMISE-2 trial, 1,072 patients were randomized to receive eptinezumab (300 mg), eptinezumab (100 mg), or placebo administered by IV infusion once every 12 weeks. Eligible patients had at least 15 headache days per month, of which at least eight met criteria for migraine. The primary end point was the mean change from baseline in monthly migraine days over the 12-week treatment period. Secondary end points included reduction in migraine prevalence on Day 1 and the proportion of patients with reductions of at least 50% and 75%.
In the PROMISE-1 trial, 888 patients were randomized to receive eptinezumab (300 mg), eptinezumab (100 mg), eptinezumab (30 mg), or placebo administered by IV infusion once every 12 weeks. Eligible patients had 14 or fewer headache days per month, of which at least four met the criteria for migraine. The primary end point was the mean change from baseline in monthly migraine days over the 12-week treatment period.
PROMISE-2
Patients in PROMISE-2 had an average of about 20 headache days per month, including 16 migraine days, at baseline, said Richard B. Lipton, MD, Edwin S. Lowe Chair in Neurology at Albert Einstein College of Medicine in New York. Patients’ mean age was about 40, mean BMI was 26, and about 88% were female.

Patients had had migraine for about 18 years on average, and mean duration of chronic migraine was about 12 years.
During the 12 weeks after treatment, the mean change from baseline in monthly migraine days was –7.7 for patients who received the 100-mg dose of eptinezumab and –8.2 for patients who received the 300-mg dose of eptinezumab, compared with –5.6 for patients who received placebo. The difference was statistically significant for both active treatment groups versus placebo.
Secondary end points significantly favored eptinezumab. The percentage of patients with at least a 75% reduction in migraine days was 15.0% for placebo, 26.7% for the 100-mg dose of eptinezumab, and 33.1% for the 300-mg dose of eptinezumab. The percentage of patients with at least a 50% reduction in migraine days was 39.3% for placebo, 57.6% for the 100-mg dose of eptinezumab, and 61.4% for the 300-mg dose of eptinezumab.
On Day 1 post infusion, the percentage of patients with migraine decreased by 51% and 52% in the 100-mg and 300-mg treatment arms, respectively, compared with a decrease of 27% in the placebo arm.
Adverse event rates among eptinezumab-treated subjects were similar to those in placebo-treated subjects. The most commonly reported adverse events for eptinezumab were nasopharyngitis (6.3%), upper respiratory infection (4.0%), and nausea (3.4%).
Treatment-emergent adverse events occurred in approximately 40% of all treatment arms (39% in the placebo group, 38% in the 100-mg group, and 44% in the 300-mg group). Serious adverse events were rare, occurred in approximately equal rates in all treatment arms, and were considered unrelated to study drug.
PROMISE-1
Patients in PROMISE-1 had a mean age of about 40, and about 85% were female. They had about 8.5 migraine days per month on average, said Stephen D. Silberstein, MD, Professor of Neurology and Director of the Jefferson Headache Center at Thomas Jefferson University in Philadelphia.
During Weeks 1–12, mean change in monthly migraine days was significantly greater among patients who received eptinezumab (–4.0 with the 30-mg dose, –3.9 with the 100-mg dose, and –4.3 with the 300-mg dose), compared with patients who received placebo (–3.2).
One-year data indicated that the percentage of participants in the trial with 75% and 50% reductions in migraine increased over time. “With each subsequent infusion, there seems to be a cumulative increase in response,” Dr. Silberstein said. At the end of the trial, the proportion of patients in the 300-mg dose group with at least a 75% reduction in migraine days was 52%, whereas the proportion at Month 3 was 37%. The long-term results are encouraging, said Dr. Silberstein.
In PROMISE-1, the most commonly reported adverse events among treated patients were upper respiratory infection (10.3%), nasopharyngitis (6.6%), and sinusitis (3.6%).
—Jake Remaly
Infusion of the anti-CGRP monoclonal antibody significantly reduces monthly migraine days, versus placebo.
Infusion of the anti-CGRP monoclonal antibody significantly reduces monthly migraine days, versus placebo.
LOS ANGELES—Among patients with chronic migraine, IV infusion of eptinezumab significantly reduces the average number of migraine days per
Eptinezumab (formerly known as ALD403) is a humanized monoclonal antibody that inhibits calcitonin gene-related peptide (CGRP). It is designed to be administered quarterly via IV infusion. Eptinezumab previously was found to be effective and well tolerated in phase II studies in episodic and chronic migraine and in a phase III trial in episodic migraine. Alder BioPharmaceuticals, based in Bothell, Washington, is developing the therapy.
Treatment-emergent adverse events in phase III studies were similar for eptinezumab and placebo, and the safety profile was consistent with that in prior studies, researchers said.
The PROMISE Clinical Trial Program
The Prevention of Migraine via Intravenous ALD403 Safety and Efficacy (PROMISE) clinical trial program includes phase III, randomized, double-blind, placebo-controlled trials of eptinezumab for chronic migraine prevention (PROMISE-2) and episodic migraine prevention (PROMISE-1).
In the PROMISE-2 trial, 1,072 patients were randomized to receive eptinezumab (300 mg), eptinezumab (100 mg), or placebo administered by IV infusion once every 12 weeks. Eligible patients had at least 15 headache days per month, of which at least eight met criteria for migraine. The primary end point was the mean change from baseline in monthly migraine days over the 12-week treatment period. Secondary end points included reduction in migraine prevalence on Day 1 and the proportion of patients with reductions of at least 50% and 75%.
In the PROMISE-1 trial, 888 patients were randomized to receive eptinezumab (300 mg), eptinezumab (100 mg), eptinezumab (30 mg), or placebo administered by IV infusion once every 12 weeks. Eligible patients had 14 or fewer headache days per month, of which at least four met the criteria for migraine. The primary end point was the mean change from baseline in monthly migraine days over the 12-week treatment period.
PROMISE-2
Patients in PROMISE-2 had an average of about 20 headache days per month, including 16 migraine days, at baseline, said Richard B. Lipton, MD, Edwin S. Lowe Chair in Neurology at Albert Einstein College of Medicine in New York. Patients’ mean age was about 40, mean BMI was 26, and about 88% were female.
Patients had had migraine for about 18 years on average, and mean duration of chronic migraine was about 12 years.
During the 12 weeks after treatment, the mean change from baseline in monthly migraine days was –7.7 for patients who received the 100-mg dose of eptinezumab and –8.2 for patients who received the 300-mg dose of eptinezumab, compared with –5.6 for patients who received placebo. The difference was statistically significant for both active treatment groups versus placebo.
Secondary end points significantly favored eptinezumab. The percentage of patients with at least a 75% reduction in migraine days was 15.0% for placebo, 26.7% for the 100-mg dose of eptinezumab, and 33.1% for the 300-mg dose of eptinezumab. The percentage of patients with at least a 50% reduction in migraine days was 39.3% for placebo, 57.6% for the 100-mg dose of eptinezumab, and 61.4% for the 300-mg dose of eptinezumab.
On Day 1 post infusion, the percentage of patients with migraine decreased by 51% and 52% in the 100-mg and 300-mg treatment arms, respectively, compared with a decrease of 27% in the placebo arm.
Adverse event rates among eptinezumab-treated subjects were similar to those in placebo-treated subjects. The most commonly reported adverse events for eptinezumab were nasopharyngitis (6.3%), upper respiratory infection (4.0%), and nausea (3.4%).
Treatment-emergent adverse events occurred in approximately 40% of all treatment arms (39% in the placebo group, 38% in the 100-mg group, and 44% in the 300-mg group). Serious adverse events were rare, occurred in approximately equal rates in all treatment arms, and were considered unrelated to study drug.
PROMISE-1
Patients in PROMISE-1 had a mean age of about 40, and about 85% were female. They had about 8.5 migraine days per month on average, said Stephen D. Silberstein, MD, Professor of Neurology and Director of the Jefferson Headache Center at Thomas Jefferson University in Philadelphia.
During Weeks 1–12, mean change in monthly migraine days was significantly greater among patients who received eptinezumab (–4.0 with the 30-mg dose, –3.9 with the 100-mg dose, and –4.3 with the 300-mg dose), compared with patients who received placebo (–3.2).
One-year data indicated that the percentage of participants in the trial with 75% and 50% reductions in migraine increased over time. “With each subsequent infusion, there seems to be a cumulative increase in response,” Dr. Silberstein said. At the end of the trial, the proportion of patients in the 300-mg dose group with at least a 75% reduction in migraine days was 52%, whereas the proportion at Month 3 was 37%. The long-term results are encouraging, said Dr. Silberstein.
In PROMISE-1, the most commonly reported adverse events among treated patients were upper respiratory infection (10.3%), nasopharyngitis (6.6%), and sinusitis (3.6%).
—Jake Remaly
LOS ANGELES—Among patients with chronic migraine, IV infusion of eptinezumab significantly reduces the average number of migraine days per
Eptinezumab (formerly known as ALD403) is a humanized monoclonal antibody that inhibits calcitonin gene-related peptide (CGRP). It is designed to be administered quarterly via IV infusion. Eptinezumab previously was found to be effective and well tolerated in phase II studies in episodic and chronic migraine and in a phase III trial in episodic migraine. Alder BioPharmaceuticals, based in Bothell, Washington, is developing the therapy.
Treatment-emergent adverse events in phase III studies were similar for eptinezumab and placebo, and the safety profile was consistent with that in prior studies, researchers said.
The PROMISE Clinical Trial Program
The Prevention of Migraine via Intravenous ALD403 Safety and Efficacy (PROMISE) clinical trial program includes phase III, randomized, double-blind, placebo-controlled trials of eptinezumab for chronic migraine prevention (PROMISE-2) and episodic migraine prevention (PROMISE-1).
In the PROMISE-2 trial, 1,072 patients were randomized to receive eptinezumab (300 mg), eptinezumab (100 mg), or placebo administered by IV infusion once every 12 weeks. Eligible patients had at least 15 headache days per month, of which at least eight met criteria for migraine. The primary end point was the mean change from baseline in monthly migraine days over the 12-week treatment period. Secondary end points included reduction in migraine prevalence on Day 1 and the proportion of patients with reductions of at least 50% and 75%.
In the PROMISE-1 trial, 888 patients were randomized to receive eptinezumab (300 mg), eptinezumab (100 mg), eptinezumab (30 mg), or placebo administered by IV infusion once every 12 weeks. Eligible patients had 14 or fewer headache days per month, of which at least four met the criteria for migraine. The primary end point was the mean change from baseline in monthly migraine days over the 12-week treatment period.
PROMISE-2
Patients in PROMISE-2 had an average of about 20 headache days per month, including 16 migraine days, at baseline, said Richard B. Lipton, MD, Edwin S. Lowe Chair in Neurology at Albert Einstein College of Medicine in New York. Patients’ mean age was about 40, mean BMI was 26, and about 88% were female.
Patients had had migraine for about 18 years on average, and mean duration of chronic migraine was about 12 years.
During the 12 weeks after treatment, the mean change from baseline in monthly migraine days was –7.7 for patients who received the 100-mg dose of eptinezumab and –8.2 for patients who received the 300-mg dose of eptinezumab, compared with –5.6 for patients who received placebo. The difference was statistically significant for both active treatment groups versus placebo.
Secondary end points significantly favored eptinezumab. The percentage of patients with at least a 75% reduction in migraine days was 15.0% for placebo, 26.7% for the 100-mg dose of eptinezumab, and 33.1% for the 300-mg dose of eptinezumab. The percentage of patients with at least a 50% reduction in migraine days was 39.3% for placebo, 57.6% for the 100-mg dose of eptinezumab, and 61.4% for the 300-mg dose of eptinezumab.
On Day 1 post infusion, the percentage of patients with migraine decreased by 51% and 52% in the 100-mg and 300-mg treatment arms, respectively, compared with a decrease of 27% in the placebo arm.
Adverse event rates among eptinezumab-treated subjects were similar to those in placebo-treated subjects. The most commonly reported adverse events for eptinezumab were nasopharyngitis (6.3%), upper respiratory infection (4.0%), and nausea (3.4%).
Treatment-emergent adverse events occurred in approximately 40% of all treatment arms (39% in the placebo group, 38% in the 100-mg group, and 44% in the 300-mg group). Serious adverse events were rare, occurred in approximately equal rates in all treatment arms, and were considered unrelated to study drug.
PROMISE-1
Patients in PROMISE-1 had a mean age of about 40, and about 85% were female. They had about 8.5 migraine days per month on average, said Stephen D. Silberstein, MD, Professor of Neurology and Director of the Jefferson Headache Center at Thomas Jefferson University in Philadelphia.
During Weeks 1–12, mean change in monthly migraine days was significantly greater among patients who received eptinezumab (–4.0 with the 30-mg dose, –3.9 with the 100-mg dose, and –4.3 with the 300-mg dose), compared with patients who received placebo (–3.2).
One-year data indicated that the percentage of participants in the trial with 75% and 50% reductions in migraine increased over time. “With each subsequent infusion, there seems to be a cumulative increase in response,” Dr. Silberstein said. At the end of the trial, the proportion of patients in the 300-mg dose group with at least a 75% reduction in migraine days was 52%, whereas the proportion at Month 3 was 37%. The long-term results are encouraging, said Dr. Silberstein.
In PROMISE-1, the most commonly reported adverse events among treated patients were upper respiratory infection (10.3%), nasopharyngitis (6.6%), and sinusitis (3.6%).
—Jake Remaly
Methamphetamine use climbing among opioid users
SAN DIEGO – As the deadly opioid epidemic continues, a new study suggests that a fast-rising number of users are turning to another drug of abuse – methamphetamine. In some cases, a researcher says, their co-use is reminiscent of the fad for “speedball” mixtures of cocaine and heroin.
During 2011-2017, the percentage of surveyed opioid users seeking treatment who reported also using methamphetamine over the past month skyrocketed from 19% to 34%, researchers reported at the 2018 annual meeting of the College on Problems of Drug Dependence.

Use of crystal meth specifically went up by 82% and the use of prescription stimulants rose by 15%. By contrast, use of marijuana went up by just 6%, while the use of muscle relaxants and prescription sleep drugs fell by more than half.
The findings matter, because the use of multiple illicit drugs is even more dangerous than one alone, said study coauthor and doctoral candidate Matthew S. Ellis, of Washington University in St. Louis, in an interview. “Illicit opioids carry their own serious risks such as unknown purity, not knowing if heroin is laced with fentanyl, or inexperience of users who are used to clearly marked prescription pills. Add in a secondary drug, also often used in non-oral ways, and your risk for overdose is going to significantly increase.”
The rising use of methamphetamine, which comes in such forms as crystal meth, has been overshadowed by news about the opioid epidemic. Still, as a 2018 Lancet report put it, “while the opioid crisis has exploded, the lull in the methamphetamine epidemic has quietly and swiftly reversed course, now accounting for 11% of the total number of overdose deaths.”
In regard to co-use of opioids and methamphetamines, the report said, “in states including Wisconsin and Oregon, new patterns suggest they are beginning to overlap as increasing numbers of people use both drugs” (Lancet. 2018 Feb. 24;391[10122]:713).
Meanwhile, the New York Times published a story in February 2018 headlined “Meth, the forgotten killer, is back. And it’s everywhere.” It noted that meth overdose deaths in Oregon outpace those from opioids and added: “At the United States border, agents are seizing 10-20 times the amounts they did a decade ago. Methamphetamine, experts say, has never been purer, cheaper, or more lethal.”
Overall, there’s little known about co-use of opioids and methamphetamines, said study lead author Mr. Ellis. “The reason for this is that opioid use patterns and populations of users have drastically changed in the past 20 years, and continue to do so,” he said. “Methamphetamine is becoming increasingly available at the same time that heroin and illicit fentanyl are as well. Reports suggest that the United States has shifted from a market of home-grown methamphetamine to that manufactured and sent from other countries, creating a broader market than previously seen.”
For the new study, Mr. Ellis and his colleagues examined statistics from a U.S. surveillance program of opioid users entering substance abuse programs. They focused on 13,521 participants in 47 states during 2011-2017.
Of 12 drug classes examined, only co-use of methamphetamine rose significantly over the 6-year period, Mr. Ellis said.
Among demographic and geographic groups, the researchers saw the largest increases in co-use of the two drugs in the West, Northeast, and Midwest regions, in rural and suburban areas, among groups aged 18-44 years, and among whites.
Why is co-use among opioid users increasing? “We have begun to do some qualitative work with a number of participants suggesting they use opioids and methamphetamine to balance each other out,” Mr. Ellis said. “So an addict can use opioids, but if they need to go to work, they can reinvigorate themselves with methamphetamine.”
Mr. Ellis said “this is not necessarily a new trend,” noting that the co-use of the drugs is akin to the “speedball” – a mixture of cocaine and heroin designed to blend their opposite modes of action.
However, Mr. Ellis said, “ The increases in production and spread of illicit opioids and methamphetamine into an existing market of those previously using prescription opioids was a perfect storm for these two drugs to be a problem, both separately and together.”
He said researchers also are finding that “if methamphetamine is the only thing an opioid addict can find, they will use it to stave off withdrawals as well.”
Indeed, National Public Radio reported in June 2018 that “as opioids are becoming harder to obtain, more and more users are turning to cheap methamphetamine” in Ohio’s tiny Vinton County, near Columbus.
Moving forward, Mr. Ellis said, “we cannot treat substance use in a silo of a single drug. If we attempt to treat opioid abusers by simply treating their opioid abuse – and not other drugs – then we have less of a chance of success. More of a focus needs to be put on the fact that the vast majority of opioid abusers are polysubstance users.”
The study is funded by the RADARS (Researched Abuse, Diversion and Addiction-Related Surveillance) System, an independent, nonprofit postmarketing surveillance system supported by subscription fees from pharmaceutical manufacturers that use RADARS data to track medication use and meet regulatory obligations. The study authors report no relevant disclosures.
SAN DIEGO – As the deadly opioid epidemic continues, a new study suggests that a fast-rising number of users are turning to another drug of abuse – methamphetamine. In some cases, a researcher says, their co-use is reminiscent of the fad for “speedball” mixtures of cocaine and heroin.
During 2011-2017, the percentage of surveyed opioid users seeking treatment who reported also using methamphetamine over the past month skyrocketed from 19% to 34%, researchers reported at the 2018 annual meeting of the College on Problems of Drug Dependence.
Use of crystal meth specifically went up by 82% and the use of prescription stimulants rose by 15%. By contrast, use of marijuana went up by just 6%, while the use of muscle relaxants and prescription sleep drugs fell by more than half.
The findings matter, because the use of multiple illicit drugs is even more dangerous than one alone, said study coauthor and doctoral candidate Matthew S. Ellis, of Washington University in St. Louis, in an interview. “Illicit opioids carry their own serious risks such as unknown purity, not knowing if heroin is laced with fentanyl, or inexperience of users who are used to clearly marked prescription pills. Add in a secondary drug, also often used in non-oral ways, and your risk for overdose is going to significantly increase.”
The rising use of methamphetamine, which comes in such forms as crystal meth, has been overshadowed by news about the opioid epidemic. Still, as a 2018 Lancet report put it, “while the opioid crisis has exploded, the lull in the methamphetamine epidemic has quietly and swiftly reversed course, now accounting for 11% of the total number of overdose deaths.”
In regard to co-use of opioids and methamphetamines, the report said, “in states including Wisconsin and Oregon, new patterns suggest they are beginning to overlap as increasing numbers of people use both drugs” (Lancet. 2018 Feb. 24;391[10122]:713).
Meanwhile, the New York Times published a story in February 2018 headlined “Meth, the forgotten killer, is back. And it’s everywhere.” It noted that meth overdose deaths in Oregon outpace those from opioids and added: “At the United States border, agents are seizing 10-20 times the amounts they did a decade ago. Methamphetamine, experts say, has never been purer, cheaper, or more lethal.”
Overall, there’s little known about co-use of opioids and methamphetamines, said study lead author Mr. Ellis. “The reason for this is that opioid use patterns and populations of users have drastically changed in the past 20 years, and continue to do so,” he said. “Methamphetamine is becoming increasingly available at the same time that heroin and illicit fentanyl are as well. Reports suggest that the United States has shifted from a market of home-grown methamphetamine to that manufactured and sent from other countries, creating a broader market than previously seen.”
For the new study, Mr. Ellis and his colleagues examined statistics from a U.S. surveillance program of opioid users entering substance abuse programs. They focused on 13,521 participants in 47 states during 2011-2017.
Of 12 drug classes examined, only co-use of methamphetamine rose significantly over the 6-year period, Mr. Ellis said.
Among demographic and geographic groups, the researchers saw the largest increases in co-use of the two drugs in the West, Northeast, and Midwest regions, in rural and suburban areas, among groups aged 18-44 years, and among whites.
Why is co-use among opioid users increasing? “We have begun to do some qualitative work with a number of participants suggesting they use opioids and methamphetamine to balance each other out,” Mr. Ellis said. “So an addict can use opioids, but if they need to go to work, they can reinvigorate themselves with methamphetamine.”
Mr. Ellis said “this is not necessarily a new trend,” noting that the co-use of the drugs is akin to the “speedball” – a mixture of cocaine and heroin designed to blend their opposite modes of action.
However, Mr. Ellis said, “ The increases in production and spread of illicit opioids and methamphetamine into an existing market of those previously using prescription opioids was a perfect storm for these two drugs to be a problem, both separately and together.”
He said researchers also are finding that “if methamphetamine is the only thing an opioid addict can find, they will use it to stave off withdrawals as well.”
Indeed, National Public Radio reported in June 2018 that “as opioids are becoming harder to obtain, more and more users are turning to cheap methamphetamine” in Ohio’s tiny Vinton County, near Columbus.
Moving forward, Mr. Ellis said, “we cannot treat substance use in a silo of a single drug. If we attempt to treat opioid abusers by simply treating their opioid abuse – and not other drugs – then we have less of a chance of success. More of a focus needs to be put on the fact that the vast majority of opioid abusers are polysubstance users.”
The study is funded by the RADARS (Researched Abuse, Diversion and Addiction-Related Surveillance) System, an independent, nonprofit postmarketing surveillance system supported by subscription fees from pharmaceutical manufacturers that use RADARS data to track medication use and meet regulatory obligations. The study authors report no relevant disclosures.
SAN DIEGO – As the deadly opioid epidemic continues, a new study suggests that a fast-rising number of users are turning to another drug of abuse – methamphetamine. In some cases, a researcher says, their co-use is reminiscent of the fad for “speedball” mixtures of cocaine and heroin.
During 2011-2017, the percentage of surveyed opioid users seeking treatment who reported also using methamphetamine over the past month skyrocketed from 19% to 34%, researchers reported at the 2018 annual meeting of the College on Problems of Drug Dependence.
Use of crystal meth specifically went up by 82% and the use of prescription stimulants rose by 15%. By contrast, use of marijuana went up by just 6%, while the use of muscle relaxants and prescription sleep drugs fell by more than half.
The findings matter, because the use of multiple illicit drugs is even more dangerous than one alone, said study coauthor and doctoral candidate Matthew S. Ellis, of Washington University in St. Louis, in an interview. “Illicit opioids carry their own serious risks such as unknown purity, not knowing if heroin is laced with fentanyl, or inexperience of users who are used to clearly marked prescription pills. Add in a secondary drug, also often used in non-oral ways, and your risk for overdose is going to significantly increase.”
The rising use of methamphetamine, which comes in such forms as crystal meth, has been overshadowed by news about the opioid epidemic. Still, as a 2018 Lancet report put it, “while the opioid crisis has exploded, the lull in the methamphetamine epidemic has quietly and swiftly reversed course, now accounting for 11% of the total number of overdose deaths.”
In regard to co-use of opioids and methamphetamines, the report said, “in states including Wisconsin and Oregon, new patterns suggest they are beginning to overlap as increasing numbers of people use both drugs” (Lancet. 2018 Feb. 24;391[10122]:713).
Meanwhile, the New York Times published a story in February 2018 headlined “Meth, the forgotten killer, is back. And it’s everywhere.” It noted that meth overdose deaths in Oregon outpace those from opioids and added: “At the United States border, agents are seizing 10-20 times the amounts they did a decade ago. Methamphetamine, experts say, has never been purer, cheaper, or more lethal.”
Overall, there’s little known about co-use of opioids and methamphetamines, said study lead author Mr. Ellis. “The reason for this is that opioid use patterns and populations of users have drastically changed in the past 20 years, and continue to do so,” he said. “Methamphetamine is becoming increasingly available at the same time that heroin and illicit fentanyl are as well. Reports suggest that the United States has shifted from a market of home-grown methamphetamine to that manufactured and sent from other countries, creating a broader market than previously seen.”
For the new study, Mr. Ellis and his colleagues examined statistics from a U.S. surveillance program of opioid users entering substance abuse programs. They focused on 13,521 participants in 47 states during 2011-2017.
Of 12 drug classes examined, only co-use of methamphetamine rose significantly over the 6-year period, Mr. Ellis said.
Among demographic and geographic groups, the researchers saw the largest increases in co-use of the two drugs in the West, Northeast, and Midwest regions, in rural and suburban areas, among groups aged 18-44 years, and among whites.
Why is co-use among opioid users increasing? “We have begun to do some qualitative work with a number of participants suggesting they use opioids and methamphetamine to balance each other out,” Mr. Ellis said. “So an addict can use opioids, but if they need to go to work, they can reinvigorate themselves with methamphetamine.”
Mr. Ellis said “this is not necessarily a new trend,” noting that the co-use of the drugs is akin to the “speedball” – a mixture of cocaine and heroin designed to blend their opposite modes of action.
However, Mr. Ellis said, “ The increases in production and spread of illicit opioids and methamphetamine into an existing market of those previously using prescription opioids was a perfect storm for these two drugs to be a problem, both separately and together.”
He said researchers also are finding that “if methamphetamine is the only thing an opioid addict can find, they will use it to stave off withdrawals as well.”
Indeed, National Public Radio reported in June 2018 that “as opioids are becoming harder to obtain, more and more users are turning to cheap methamphetamine” in Ohio’s tiny Vinton County, near Columbus.
Moving forward, Mr. Ellis said, “we cannot treat substance use in a silo of a single drug. If we attempt to treat opioid abusers by simply treating their opioid abuse – and not other drugs – then we have less of a chance of success. More of a focus needs to be put on the fact that the vast majority of opioid abusers are polysubstance users.”
The study is funded by the RADARS (Researched Abuse, Diversion and Addiction-Related Surveillance) System, an independent, nonprofit postmarketing surveillance system supported by subscription fees from pharmaceutical manufacturers that use RADARS data to track medication use and meet regulatory obligations. The study authors report no relevant disclosures.
REPORTING FROM CPDD 2018
Key clinical point: The percentage of opioid users who also use methamphetamine is on the rise.
Major finding: During 2011-2017, the percentage of opioid users reporting methamphetamine use over the past month grew from 19% to 34%.
Study details: Analysis of 2011-2017 data from 13,521 opioid-using participants entering substance abuse programs.
Disclosures: The study is funded by the RADARS System, an independent, nonprofit postmarketing surveillance system supported by subscription fees from pharmaceutical manufacturers that use RADARS data to track medication use and meet regulatory obligations. The study authors report no relevant disclosures.
Pain remains after rheumatoid arthritis diagnosis
AMSTERDAM – according to data from a large prospective study reported at the European Congress of Rheumatology.
At enrollment, 64% of patients still had remaining pain, and at 1 year, 24% had remaining pain, according to data from the Canadian Early Arthritis Cohort (CATCH).

Dr. Bykerk of the University of Toronto and the Hospital for Special Surgery, New York, presented the research on behalf of Yvonne C. Lee, MD, of Northwestern University, Chicago, and noted that they had previously published data showing the incidence of widespread pain was highest within the first year of an RA diagnosis and then plateaued (Ann Rheum Dis. 2013;72[6];949-54). This plateau could have been related to the control of inflammation, Dr. Bykerk observed.
“We think the first 12 months are really a critical window during which time acute inflammatory pain may transition, if you will, to chronic noninflammatory pain,” Dr. Bykerk said.
The aim of their current study was to look at the evolution of pain in the year following an RA diagnosis and to see whether there were any predictive factors for remaining pain and widespread pain. The latter might previously have been described as fibromyalgia, Dr. Bykerk said, and was defined as a difference between the tender and swollen 28-joint counts of at least seven points. Remaining pain was defined as a score of more than 4 on a pain intensity numerical rating scale.
Data on 1,270 patients who were enrolled in CATCH across 17 sites in Canada during 2007-2017 were used. The majority of the sample was female (73%) and white (82%) and had been experiencing symptoms for a median of 6 months. The mean age of patients was 54 years.
At enrollment, patients had a mean number of two comorbidities, with up to 12% having another condition that might contribute to pain, such as osteoarthritis (12%), a back or spinal problem (10%), or fibromyalgia (9%).
Most (72%) patients were being treated with methotrexate initially, either alone (29%) or in combination with other drugs, and almost two-thirds were using steroids, either orally (29%) or parenterally (32%). A small percentage (2%) were treated with biologics, but this is unusual until almost 12 months, Dr. Bykerk observed.
The baseline predictors for remaining pain at 1 year were high baseline pain (odds ratio, 2.1), sleep difficulties (OR, 2.2), and disability measured by the Health Assessment Questionnaire Disability Index (OR, 1.5). The latter was the strongest predictor for lingering widespread pain. The number of comorbidities were also predictive of lingering pain, though this was not a significant difference.
Assessing and considering treatment for these predictors may be important for improving persisting pain, Dr. Bykerk suggested. “In future we’d like to validate our findings with other validated questionnaires; we have a depression questionnaire now being used in the study, and we want to understand what subgroups of patients have different trajectories of pain.”
CATCH is currently funded via unrestricted research grants from Amgen, Pfizer, Bristol-Myers Squibb, Medexus, Eli Lilly, Merck, and Sandoz Canada Pharmaceuticals. Dr. Bykerk had no personal financial disclosures.
SOURCE: Lee YC et al. EULAR 2018 Congress.Abstract OP0349.
AMSTERDAM – according to data from a large prospective study reported at the European Congress of Rheumatology.
At enrollment, 64% of patients still had remaining pain, and at 1 year, 24% had remaining pain, according to data from the Canadian Early Arthritis Cohort (CATCH).
Dr. Bykerk of the University of Toronto and the Hospital for Special Surgery, New York, presented the research on behalf of Yvonne C. Lee, MD, of Northwestern University, Chicago, and noted that they had previously published data showing the incidence of widespread pain was highest within the first year of an RA diagnosis and then plateaued (Ann Rheum Dis. 2013;72[6];949-54). This plateau could have been related to the control of inflammation, Dr. Bykerk observed.
“We think the first 12 months are really a critical window during which time acute inflammatory pain may transition, if you will, to chronic noninflammatory pain,” Dr. Bykerk said.
The aim of their current study was to look at the evolution of pain in the year following an RA diagnosis and to see whether there were any predictive factors for remaining pain and widespread pain. The latter might previously have been described as fibromyalgia, Dr. Bykerk said, and was defined as a difference between the tender and swollen 28-joint counts of at least seven points. Remaining pain was defined as a score of more than 4 on a pain intensity numerical rating scale.
Data on 1,270 patients who were enrolled in CATCH across 17 sites in Canada during 2007-2017 were used. The majority of the sample was female (73%) and white (82%) and had been experiencing symptoms for a median of 6 months. The mean age of patients was 54 years.
At enrollment, patients had a mean number of two comorbidities, with up to 12% having another condition that might contribute to pain, such as osteoarthritis (12%), a back or spinal problem (10%), or fibromyalgia (9%).
Most (72%) patients were being treated with methotrexate initially, either alone (29%) or in combination with other drugs, and almost two-thirds were using steroids, either orally (29%) or parenterally (32%). A small percentage (2%) were treated with biologics, but this is unusual until almost 12 months, Dr. Bykerk observed.
The baseline predictors for remaining pain at 1 year were high baseline pain (odds ratio, 2.1), sleep difficulties (OR, 2.2), and disability measured by the Health Assessment Questionnaire Disability Index (OR, 1.5). The latter was the strongest predictor for lingering widespread pain. The number of comorbidities were also predictive of lingering pain, though this was not a significant difference.
Assessing and considering treatment for these predictors may be important for improving persisting pain, Dr. Bykerk suggested. “In future we’d like to validate our findings with other validated questionnaires; we have a depression questionnaire now being used in the study, and we want to understand what subgroups of patients have different trajectories of pain.”
CATCH is currently funded via unrestricted research grants from Amgen, Pfizer, Bristol-Myers Squibb, Medexus, Eli Lilly, Merck, and Sandoz Canada Pharmaceuticals. Dr. Bykerk had no personal financial disclosures.
SOURCE: Lee YC et al. EULAR 2018 Congress.Abstract OP0349.
AMSTERDAM – according to data from a large prospective study reported at the European Congress of Rheumatology.
At enrollment, 64% of patients still had remaining pain, and at 1 year, 24% had remaining pain, according to data from the Canadian Early Arthritis Cohort (CATCH).
Dr. Bykerk of the University of Toronto and the Hospital for Special Surgery, New York, presented the research on behalf of Yvonne C. Lee, MD, of Northwestern University, Chicago, and noted that they had previously published data showing the incidence of widespread pain was highest within the first year of an RA diagnosis and then plateaued (Ann Rheum Dis. 2013;72[6];949-54). This plateau could have been related to the control of inflammation, Dr. Bykerk observed.
“We think the first 12 months are really a critical window during which time acute inflammatory pain may transition, if you will, to chronic noninflammatory pain,” Dr. Bykerk said.
The aim of their current study was to look at the evolution of pain in the year following an RA diagnosis and to see whether there were any predictive factors for remaining pain and widespread pain. The latter might previously have been described as fibromyalgia, Dr. Bykerk said, and was defined as a difference between the tender and swollen 28-joint counts of at least seven points. Remaining pain was defined as a score of more than 4 on a pain intensity numerical rating scale.
Data on 1,270 patients who were enrolled in CATCH across 17 sites in Canada during 2007-2017 were used. The majority of the sample was female (73%) and white (82%) and had been experiencing symptoms for a median of 6 months. The mean age of patients was 54 years.
At enrollment, patients had a mean number of two comorbidities, with up to 12% having another condition that might contribute to pain, such as osteoarthritis (12%), a back or spinal problem (10%), or fibromyalgia (9%).
Most (72%) patients were being treated with methotrexate initially, either alone (29%) or in combination with other drugs, and almost two-thirds were using steroids, either orally (29%) or parenterally (32%). A small percentage (2%) were treated with biologics, but this is unusual until almost 12 months, Dr. Bykerk observed.
The baseline predictors for remaining pain at 1 year were high baseline pain (odds ratio, 2.1), sleep difficulties (OR, 2.2), and disability measured by the Health Assessment Questionnaire Disability Index (OR, 1.5). The latter was the strongest predictor for lingering widespread pain. The number of comorbidities were also predictive of lingering pain, though this was not a significant difference.
Assessing and considering treatment for these predictors may be important for improving persisting pain, Dr. Bykerk suggested. “In future we’d like to validate our findings with other validated questionnaires; we have a depression questionnaire now being used in the study, and we want to understand what subgroups of patients have different trajectories of pain.”
CATCH is currently funded via unrestricted research grants from Amgen, Pfizer, Bristol-Myers Squibb, Medexus, Eli Lilly, Merck, and Sandoz Canada Pharmaceuticals. Dr. Bykerk had no personal financial disclosures.
SOURCE: Lee YC et al. EULAR 2018 Congress.Abstract OP0349.
REPORTING FROM THE EULAR 2018 CONGRESS
Key clinical point: A substantial number of patients have pain 1 year after being diagnosed with rheumatoid arthritis.
Major finding: Remaining pain and widespread pain were a respective 64% and 9% at baseline and 9% and 5% at 1 year.
Study details: A prospective, observational, cohort of 1,270 patients with new-onset inflammatory arthritis.
Disclosures: Dr. Bykerk is the principle investigator of the Canadian Early Arthritis Cohort (CATCH). CATCH is currently funded via unrestricted research grants from Amgen, Pfizer, Bristol-Myers Squibb, Medexus, Eli Lilly, Merck, and Sandoz Canada Pharmaceuticals. Dr. Bykerk had no personal financial disclosures.
Source: Lee YC et al. EULAR 2018 Congress, Abstract OP0349.
Limited anakinra course often enough for systemic JIA if used first
AMSTERDAM – Rather than waiting for other drugs or immunotherapies to fail, an immediate up-front but time-limited course of an interleukin-1 receptor (IL-1R) antagonist induced rapid and sustained remissions in most children with systemic juvenile idiopathic arthritis (JIA), according to 5-year data presented at the European Congress of Rheumatology.
In the latest follow-up of a protocol first described in 2014, over 90% of patients still had inactive disease, 75% of whom were completely off therapy, reported Sebastiaan J. Vastert, MD, PhD, of the division of pediatrics at University Medical Center, Utrecht, the Netherlands.

“Translating this into clinical practice, you could say that there might be a window of opportunity early in systemic JIA in which the innate immune system is the major player and perhaps you could down-regulate this to control the inflammation,” Dr. Vastert explained.
Citing a series of experimental studies at his institution that suggest immune mediators change as systemic JIA evolves from an acute to a chronic phase, Dr. Vastert believes that early use of an anti–IL-1R therapy may alter the trajectory of systemic JIA, compared with when it goes untreated or is treated with conventional therapies.
In the original series reported in 2014 (Arthritis Rheumatol. 2014 Apr;66:1034-43), data were presented on 20 patients. All fulfilled the International League of Associations for Rheumatology criteria for systemic JIA. They were treated with anakinra after failing to respond to indomethacin but before receiving any other therapy, including corticosteroids, disease-modifying antirheumatic drugs, or other biologics.
In the protocol described in the initial publication, a stop-therapy strategy permitted treatment discontinuation after 3 months in those who met American College of Rheumatology criteria for 90% improvement (ACR Pedi 90) in JIA.
By 1 year, 73% of the patients had met criteria to stop therapy. Of 11 patients followed for 3 years, 10 met criteria for disease remission, 8 of whom were off medication. The remaining two continued to receive anti–IL-1R or another therapy.
The systemic JIA cohort at Dr. Vastert’s institution has now grown to 50 patients, of whom 42 patients have received first-line anakinra. Among the 25 patients who have been followed for at least 5 years, 72% have inactive disease as defined by ACR Pedi 90 criteria off therapy. Another 20% have inactive disease on therapy, which is anakinra or another biologic in most cases. The majority of patients have avoided corticosteroids completely.
Freedom from corticosteroids has been accompanied by high rates of satisfaction and has allowed patients to avoid adverse events associated with corticosteroids. For example, only one patient in this series has a growth curve more than two standard deviations below normal for age and gender, according to Dr. Vastert.
“This is just a single-center cohort study, but we now have 3 more years of data to be convinced of this concept,” Dr. Vastert said.
Another notable finding from this cohort: 12 patients have been enrolled that did not fulfill International League of Associations for Rheumatology criteria for systemic JIA because of the absence of joint involvement. Strongly suspected of having systemic JIA because of other clinical signs and features, these patients have also responded well to first-line anakinra therapy.
“Our data point to a classification [of systemic JIA] that does not include arthritis as a prerequisite for diagnosis,” said Dr. Vastert, who provided data suggesting that elevated levels of IL-18 might be among biomarkers that could be employed in a revised classification system.
The study was not funded by industry. Dr. Vastert reported receiving consulting fees from Novartis.
AMSTERDAM – Rather than waiting for other drugs or immunotherapies to fail, an immediate up-front but time-limited course of an interleukin-1 receptor (IL-1R) antagonist induced rapid and sustained remissions in most children with systemic juvenile idiopathic arthritis (JIA), according to 5-year data presented at the European Congress of Rheumatology.
In the latest follow-up of a protocol first described in 2014, over 90% of patients still had inactive disease, 75% of whom were completely off therapy, reported Sebastiaan J. Vastert, MD, PhD, of the division of pediatrics at University Medical Center, Utrecht, the Netherlands.
“Translating this into clinical practice, you could say that there might be a window of opportunity early in systemic JIA in which the innate immune system is the major player and perhaps you could down-regulate this to control the inflammation,” Dr. Vastert explained.
Citing a series of experimental studies at his institution that suggest immune mediators change as systemic JIA evolves from an acute to a chronic phase, Dr. Vastert believes that early use of an anti–IL-1R therapy may alter the trajectory of systemic JIA, compared with when it goes untreated or is treated with conventional therapies.
In the original series reported in 2014 (Arthritis Rheumatol. 2014 Apr;66:1034-43), data were presented on 20 patients. All fulfilled the International League of Associations for Rheumatology criteria for systemic JIA. They were treated with anakinra after failing to respond to indomethacin but before receiving any other therapy, including corticosteroids, disease-modifying antirheumatic drugs, or other biologics.
In the protocol described in the initial publication, a stop-therapy strategy permitted treatment discontinuation after 3 months in those who met American College of Rheumatology criteria for 90% improvement (ACR Pedi 90) in JIA.
By 1 year, 73% of the patients had met criteria to stop therapy. Of 11 patients followed for 3 years, 10 met criteria for disease remission, 8 of whom were off medication. The remaining two continued to receive anti–IL-1R or another therapy.
The systemic JIA cohort at Dr. Vastert’s institution has now grown to 50 patients, of whom 42 patients have received first-line anakinra. Among the 25 patients who have been followed for at least 5 years, 72% have inactive disease as defined by ACR Pedi 90 criteria off therapy. Another 20% have inactive disease on therapy, which is anakinra or another biologic in most cases. The majority of patients have avoided corticosteroids completely.
Freedom from corticosteroids has been accompanied by high rates of satisfaction and has allowed patients to avoid adverse events associated with corticosteroids. For example, only one patient in this series has a growth curve more than two standard deviations below normal for age and gender, according to Dr. Vastert.
“This is just a single-center cohort study, but we now have 3 more years of data to be convinced of this concept,” Dr. Vastert said.
Another notable finding from this cohort: 12 patients have been enrolled that did not fulfill International League of Associations for Rheumatology criteria for systemic JIA because of the absence of joint involvement. Strongly suspected of having systemic JIA because of other clinical signs and features, these patients have also responded well to first-line anakinra therapy.
“Our data point to a classification [of systemic JIA] that does not include arthritis as a prerequisite for diagnosis,” said Dr. Vastert, who provided data suggesting that elevated levels of IL-18 might be among biomarkers that could be employed in a revised classification system.
The study was not funded by industry. Dr. Vastert reported receiving consulting fees from Novartis.
AMSTERDAM – Rather than waiting for other drugs or immunotherapies to fail, an immediate up-front but time-limited course of an interleukin-1 receptor (IL-1R) antagonist induced rapid and sustained remissions in most children with systemic juvenile idiopathic arthritis (JIA), according to 5-year data presented at the European Congress of Rheumatology.
In the latest follow-up of a protocol first described in 2014, over 90% of patients still had inactive disease, 75% of whom were completely off therapy, reported Sebastiaan J. Vastert, MD, PhD, of the division of pediatrics at University Medical Center, Utrecht, the Netherlands.
“Translating this into clinical practice, you could say that there might be a window of opportunity early in systemic JIA in which the innate immune system is the major player and perhaps you could down-regulate this to control the inflammation,” Dr. Vastert explained.
Citing a series of experimental studies at his institution that suggest immune mediators change as systemic JIA evolves from an acute to a chronic phase, Dr. Vastert believes that early use of an anti–IL-1R therapy may alter the trajectory of systemic JIA, compared with when it goes untreated or is treated with conventional therapies.
In the original series reported in 2014 (Arthritis Rheumatol. 2014 Apr;66:1034-43), data were presented on 20 patients. All fulfilled the International League of Associations for Rheumatology criteria for systemic JIA. They were treated with anakinra after failing to respond to indomethacin but before receiving any other therapy, including corticosteroids, disease-modifying antirheumatic drugs, or other biologics.
In the protocol described in the initial publication, a stop-therapy strategy permitted treatment discontinuation after 3 months in those who met American College of Rheumatology criteria for 90% improvement (ACR Pedi 90) in JIA.
By 1 year, 73% of the patients had met criteria to stop therapy. Of 11 patients followed for 3 years, 10 met criteria for disease remission, 8 of whom were off medication. The remaining two continued to receive anti–IL-1R or another therapy.
The systemic JIA cohort at Dr. Vastert’s institution has now grown to 50 patients, of whom 42 patients have received first-line anakinra. Among the 25 patients who have been followed for at least 5 years, 72% have inactive disease as defined by ACR Pedi 90 criteria off therapy. Another 20% have inactive disease on therapy, which is anakinra or another biologic in most cases. The majority of patients have avoided corticosteroids completely.
Freedom from corticosteroids has been accompanied by high rates of satisfaction and has allowed patients to avoid adverse events associated with corticosteroids. For example, only one patient in this series has a growth curve more than two standard deviations below normal for age and gender, according to Dr. Vastert.
“This is just a single-center cohort study, but we now have 3 more years of data to be convinced of this concept,” Dr. Vastert said.
Another notable finding from this cohort: 12 patients have been enrolled that did not fulfill International League of Associations for Rheumatology criteria for systemic JIA because of the absence of joint involvement. Strongly suspected of having systemic JIA because of other clinical signs and features, these patients have also responded well to first-line anakinra therapy.
“Our data point to a classification [of systemic JIA] that does not include arthritis as a prerequisite for diagnosis,” said Dr. Vastert, who provided data suggesting that elevated levels of IL-18 might be among biomarkers that could be employed in a revised classification system.
The study was not funded by industry. Dr. Vastert reported receiving consulting fees from Novartis.
REPORTING FROM THE EULAR 2018 CONGRESS
Key clinical point: Based on long-term outcomes, first-line anakinra may change natural history of systemic juvenile idiopathic arthritis.
Major finding: In a 5-year follow-up of 25 patients with systemic JIA, 92% have inactive disease, most of whom are off therapy.
Study details: A prospective, single-center cohort registry.
Disclosures: The study was not funded by industry. Dr. Vastert reported receiving consulting fees from Novartis.
European Medicines Agency recommends CAR T-cell approvals
The European Medicines Agency (EMA) has recommended a handful of hematology medications for approval, including two chimeric antigen receptor (CAR) T-cell therapies.
All of the drugs must next be approved by the European Commission in order to be marketed to patients throughout Europe.
At the end of June, the EMA’s Committee for Medicinal Products for Human Use tisagenlecleucel (Kymriah) and axicabtagene ciloleucel (Yescarta).
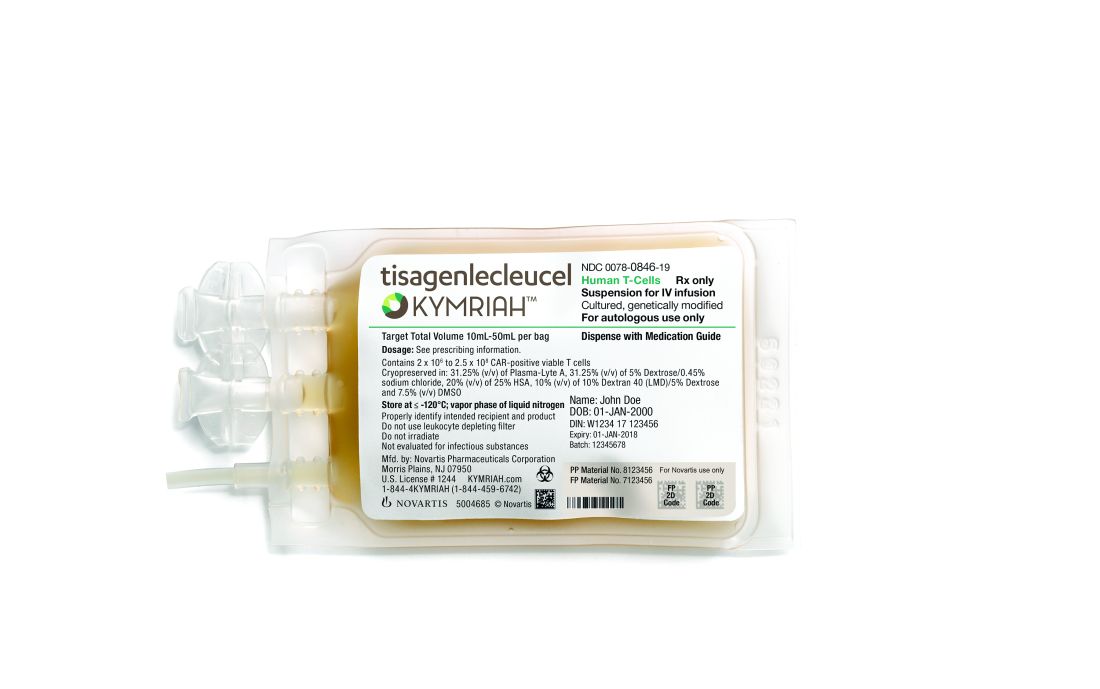
The EMA approval recommendations come with risk management measures to address the potential for cytokine release syndrome with both of these treatments. Drug makers must use a patient registry to monitor the long-term safety and efficacy of the therapies.
The EMA is also recommending approval of caplacizumab for acquired thrombotic thrombocytopenic purpura, vonicog alfa for the treatment of von Willebrand disease, and daunorubicin/cytarabine for the treatment of acute myeloid leukemia.
The European Medicines Agency (EMA) has recommended a handful of hematology medications for approval, including two chimeric antigen receptor (CAR) T-cell therapies.
All of the drugs must next be approved by the European Commission in order to be marketed to patients throughout Europe.
At the end of June, the EMA’s Committee for Medicinal Products for Human Use tisagenlecleucel (Kymriah) and axicabtagene ciloleucel (Yescarta).
The EMA approval recommendations come with risk management measures to address the potential for cytokine release syndrome with both of these treatments. Drug makers must use a patient registry to monitor the long-term safety and efficacy of the therapies.
The EMA is also recommending approval of caplacizumab for acquired thrombotic thrombocytopenic purpura, vonicog alfa for the treatment of von Willebrand disease, and daunorubicin/cytarabine for the treatment of acute myeloid leukemia.
The European Medicines Agency (EMA) has recommended a handful of hematology medications for approval, including two chimeric antigen receptor (CAR) T-cell therapies.
All of the drugs must next be approved by the European Commission in order to be marketed to patients throughout Europe.
At the end of June, the EMA’s Committee for Medicinal Products for Human Use tisagenlecleucel (Kymriah) and axicabtagene ciloleucel (Yescarta).
The EMA approval recommendations come with risk management measures to address the potential for cytokine release syndrome with both of these treatments. Drug makers must use a patient registry to monitor the long-term safety and efficacy of the therapies.
The EMA is also recommending approval of caplacizumab for acquired thrombotic thrombocytopenic purpura, vonicog alfa for the treatment of von Willebrand disease, and daunorubicin/cytarabine for the treatment of acute myeloid leukemia.
Bortezomib plus vorinostat shows modest response in MCL
but was less impressive among patients with diffuse large B-cell lymphoma (DLBCL).
Victor Yazbeck, MD, of the Massey Cancer Center at Virginia Commonwealth University in Richmond, and his colleagues reported the findings from the multicenter, nonrandomized, phase 2 trial with 65 treated patients. The trial included three cohorts: 22 patients with MCL and no prior treatment with bortezomib; 4 patients with MCL and prior treatment with bortezomib; and 39 patients with relapsed or refractory DLBCL and no prior bortezomib.
The best results were seen among MCL patients with no prior bortezomib treatment, with an overall response rate of 31.8% and a median progression-free survival (PFS) of 7.6 months. Responses were limited among the DLBCL cohort, which had an overall response rate of 7.7% and a median PFS of just 1.8 months. Among MCL patients who had received prior bortezomib treatment, there were no responses.
From a safety perspective, the combination treatment was well tolerated. The most common grade 3 and 4 hematologic toxicities were thrombocytopenia, lymphopenia, and neutropenia. There was one death among the DLBCL patients and it was unclear if it was related to treatment or progression of disease.
“Patients with MCL had a higher [overall response rate] compared to those with DLBCL, most likely due to the single-agent activity of bortezomib in MCL,” the researchers wrote. “Overall, the synergism previously demonstrated in preclinical models could not be confirmed.”
The study was supported by the Southeast Phase 2 Consortium and by a grant from the National Cancer Institute. Dr. Yazbeck reported having no financial disclosures. One of his coauthors is an employee of Amgen and owns Amgen stock. Another coauthor receives research support from Takeda, Celgene, Karyopharm Therapeutics, Bristol-Myers Squibb, Merck, and Signal Genetics.
SOURCE: Yazbeck V et al. Clin Lymphoma Myeloma Leuk. 2018 Jun 6. doi: 10.1016/j.clml.2018.05.023.
but was less impressive among patients with diffuse large B-cell lymphoma (DLBCL).
Victor Yazbeck, MD, of the Massey Cancer Center at Virginia Commonwealth University in Richmond, and his colleagues reported the findings from the multicenter, nonrandomized, phase 2 trial with 65 treated patients. The trial included three cohorts: 22 patients with MCL and no prior treatment with bortezomib; 4 patients with MCL and prior treatment with bortezomib; and 39 patients with relapsed or refractory DLBCL and no prior bortezomib.
The best results were seen among MCL patients with no prior bortezomib treatment, with an overall response rate of 31.8% and a median progression-free survival (PFS) of 7.6 months. Responses were limited among the DLBCL cohort, which had an overall response rate of 7.7% and a median PFS of just 1.8 months. Among MCL patients who had received prior bortezomib treatment, there were no responses.
From a safety perspective, the combination treatment was well tolerated. The most common grade 3 and 4 hematologic toxicities were thrombocytopenia, lymphopenia, and neutropenia. There was one death among the DLBCL patients and it was unclear if it was related to treatment or progression of disease.
“Patients with MCL had a higher [overall response rate] compared to those with DLBCL, most likely due to the single-agent activity of bortezomib in MCL,” the researchers wrote. “Overall, the synergism previously demonstrated in preclinical models could not be confirmed.”
The study was supported by the Southeast Phase 2 Consortium and by a grant from the National Cancer Institute. Dr. Yazbeck reported having no financial disclosures. One of his coauthors is an employee of Amgen and owns Amgen stock. Another coauthor receives research support from Takeda, Celgene, Karyopharm Therapeutics, Bristol-Myers Squibb, Merck, and Signal Genetics.
SOURCE: Yazbeck V et al. Clin Lymphoma Myeloma Leuk. 2018 Jun 6. doi: 10.1016/j.clml.2018.05.023.
but was less impressive among patients with diffuse large B-cell lymphoma (DLBCL).
Victor Yazbeck, MD, of the Massey Cancer Center at Virginia Commonwealth University in Richmond, and his colleagues reported the findings from the multicenter, nonrandomized, phase 2 trial with 65 treated patients. The trial included three cohorts: 22 patients with MCL and no prior treatment with bortezomib; 4 patients with MCL and prior treatment with bortezomib; and 39 patients with relapsed or refractory DLBCL and no prior bortezomib.
The best results were seen among MCL patients with no prior bortezomib treatment, with an overall response rate of 31.8% and a median progression-free survival (PFS) of 7.6 months. Responses were limited among the DLBCL cohort, which had an overall response rate of 7.7% and a median PFS of just 1.8 months. Among MCL patients who had received prior bortezomib treatment, there were no responses.
From a safety perspective, the combination treatment was well tolerated. The most common grade 3 and 4 hematologic toxicities were thrombocytopenia, lymphopenia, and neutropenia. There was one death among the DLBCL patients and it was unclear if it was related to treatment or progression of disease.
“Patients with MCL had a higher [overall response rate] compared to those with DLBCL, most likely due to the single-agent activity of bortezomib in MCL,” the researchers wrote. “Overall, the synergism previously demonstrated in preclinical models could not be confirmed.”
The study was supported by the Southeast Phase 2 Consortium and by a grant from the National Cancer Institute. Dr. Yazbeck reported having no financial disclosures. One of his coauthors is an employee of Amgen and owns Amgen stock. Another coauthor receives research support from Takeda, Celgene, Karyopharm Therapeutics, Bristol-Myers Squibb, Merck, and Signal Genetics.
SOURCE: Yazbeck V et al. Clin Lymphoma Myeloma Leuk. 2018 Jun 6. doi: 10.1016/j.clml.2018.05.023.
FROM CLINICAL LYMPHOMA, MYELOMA AND LEUKEMIA
Antidrug vaccines: A shot in the arm against addiction?
SAN DIEGO – Heroin, methamphetamine, and cocaine might not seem to have much in common with germs, but chemist Kim D. Janda, PhD, and his colleagues are trying to trick the human body into looking at them the same way: As invaders who must be conquered.
And what’s the best way to prepare the immune system for a fight? Train it through vaccination – the key to so many victories over viruses and bacteria.
For decades now, Dr. Janda, of the Scripps Research Institute in La Jolla, Calif., and other scientists have tried to develop a vaccine that would blunt the effects of substances of abuse, such as illicit drugs and even tobacco. The idea is to arm the immune system with antibodies that will prevent some or all drug components from breaking through the blood-brain barrier and producing psychoactive reactions.
In a follow-up interview after his presentation at the annual meeting of the College on Problems of Drug Dependence, Dr. Janda spoke about the history of research in this area, the potential workings of a vaccine, and the challenges of moving his own research forward.
Question: What’s the history of scientists’ efforts to develop a vaccine against drugs of abuse?
Answer: People looked at it back in the 1970s, but it didn’t really pan out. It was pretty dormant until we published research about a cocaine vaccine in 1995.
We worked on that for a while, then we looked at methamphetamine, Rohypnol, and opioids. There have been clinical trials for nicotine and cocaine, but they’ve failed.
Q: How are these vaccines expected to work?
A: The idea of the vaccines is that you don’t get high, so you won’t feel the effect of the drugs. They’ll never reach the pleasure centers that they’re supposed to.
Q: Is the idea that everyone would get a vaccine against, say, cocaine or heroin, like we routinely get vaccines against various diseases?
A: For an infectious disease like measles or mumps, you give vaccinations, and herd immunity develops. That’s not the case here. These work in a different manner, and they’re not going to be useful for people who don’t want to get off the drug. Instead, they’re going to be useful for people who want to obtain sobriety. If someone has a weak moment and tries to take the drug to get high, this would hopefully stop that from happening. It could also help overdoses in some cases, since you’re not getting that much drug into the central nervous system where it can cause cardiac arrest.
Q: How often would people be vaccinated?
A: These are vaccines that will be on board for a certain amount of time. We envision 3-4 shots over a 3-month period. You’d come back once a month for 3 months, and you’d be good for maybe 4-6 months.
Q: Where does your research stand now?
A: We’ve made a lot of advances with the opioids, and they’re good enough to put a vaccine in the clinic. Money is the issue. It’s always the issue. But we have to get up to the plate and take our swings – and find if we’ll strike out or get a hit.
SAN DIEGO – Heroin, methamphetamine, and cocaine might not seem to have much in common with germs, but chemist Kim D. Janda, PhD, and his colleagues are trying to trick the human body into looking at them the same way: As invaders who must be conquered.
And what’s the best way to prepare the immune system for a fight? Train it through vaccination – the key to so many victories over viruses and bacteria.
For decades now, Dr. Janda, of the Scripps Research Institute in La Jolla, Calif., and other scientists have tried to develop a vaccine that would blunt the effects of substances of abuse, such as illicit drugs and even tobacco. The idea is to arm the immune system with antibodies that will prevent some or all drug components from breaking through the blood-brain barrier and producing psychoactive reactions.
In a follow-up interview after his presentation at the annual meeting of the College on Problems of Drug Dependence, Dr. Janda spoke about the history of research in this area, the potential workings of a vaccine, and the challenges of moving his own research forward.
Question: What’s the history of scientists’ efforts to develop a vaccine against drugs of abuse?
Answer: People looked at it back in the 1970s, but it didn’t really pan out. It was pretty dormant until we published research about a cocaine vaccine in 1995.
We worked on that for a while, then we looked at methamphetamine, Rohypnol, and opioids. There have been clinical trials for nicotine and cocaine, but they’ve failed.
Q: How are these vaccines expected to work?
A: The idea of the vaccines is that you don’t get high, so you won’t feel the effect of the drugs. They’ll never reach the pleasure centers that they’re supposed to.
Q: Is the idea that everyone would get a vaccine against, say, cocaine or heroin, like we routinely get vaccines against various diseases?
A: For an infectious disease like measles or mumps, you give vaccinations, and herd immunity develops. That’s not the case here. These work in a different manner, and they’re not going to be useful for people who don’t want to get off the drug. Instead, they’re going to be useful for people who want to obtain sobriety. If someone has a weak moment and tries to take the drug to get high, this would hopefully stop that from happening. It could also help overdoses in some cases, since you’re not getting that much drug into the central nervous system where it can cause cardiac arrest.
Q: How often would people be vaccinated?
A: These are vaccines that will be on board for a certain amount of time. We envision 3-4 shots over a 3-month period. You’d come back once a month for 3 months, and you’d be good for maybe 4-6 months.
Q: Where does your research stand now?
A: We’ve made a lot of advances with the opioids, and they’re good enough to put a vaccine in the clinic. Money is the issue. It’s always the issue. But we have to get up to the plate and take our swings – and find if we’ll strike out or get a hit.
SAN DIEGO – Heroin, methamphetamine, and cocaine might not seem to have much in common with germs, but chemist Kim D. Janda, PhD, and his colleagues are trying to trick the human body into looking at them the same way: As invaders who must be conquered.
And what’s the best way to prepare the immune system for a fight? Train it through vaccination – the key to so many victories over viruses and bacteria.
For decades now, Dr. Janda, of the Scripps Research Institute in La Jolla, Calif., and other scientists have tried to develop a vaccine that would blunt the effects of substances of abuse, such as illicit drugs and even tobacco. The idea is to arm the immune system with antibodies that will prevent some or all drug components from breaking through the blood-brain barrier and producing psychoactive reactions.
In a follow-up interview after his presentation at the annual meeting of the College on Problems of Drug Dependence, Dr. Janda spoke about the history of research in this area, the potential workings of a vaccine, and the challenges of moving his own research forward.
Question: What’s the history of scientists’ efforts to develop a vaccine against drugs of abuse?
Answer: People looked at it back in the 1970s, but it didn’t really pan out. It was pretty dormant until we published research about a cocaine vaccine in 1995.
We worked on that for a while, then we looked at methamphetamine, Rohypnol, and opioids. There have been clinical trials for nicotine and cocaine, but they’ve failed.
Q: How are these vaccines expected to work?
A: The idea of the vaccines is that you don’t get high, so you won’t feel the effect of the drugs. They’ll never reach the pleasure centers that they’re supposed to.
Q: Is the idea that everyone would get a vaccine against, say, cocaine or heroin, like we routinely get vaccines against various diseases?
A: For an infectious disease like measles or mumps, you give vaccinations, and herd immunity develops. That’s not the case here. These work in a different manner, and they’re not going to be useful for people who don’t want to get off the drug. Instead, they’re going to be useful for people who want to obtain sobriety. If someone has a weak moment and tries to take the drug to get high, this would hopefully stop that from happening. It could also help overdoses in some cases, since you’re not getting that much drug into the central nervous system where it can cause cardiac arrest.
Q: How often would people be vaccinated?
A: These are vaccines that will be on board for a certain amount of time. We envision 3-4 shots over a 3-month period. You’d come back once a month for 3 months, and you’d be good for maybe 4-6 months.
Q: Where does your research stand now?
A: We’ve made a lot of advances with the opioids, and they’re good enough to put a vaccine in the clinic. Money is the issue. It’s always the issue. But we have to get up to the plate and take our swings – and find if we’ll strike out or get a hit.
REPORTING FROM CPDD 2018
High RA biologic drug levels linked with more infections
AMSTERDAM – Patients with rheumatoid arthritis who had a high serum level of biologic immunomodulatory drugs had a statistically significant 51% higher rate of infection during their first year on the drug, compared with RA patients who maintained usual or low serum levels of the same drugs, according to an analysis of 703 U.K. patients in a national database.
The results suggest that, once patients with rheumatoid arthritis go into remission on a higher dosage of biologic agents that produce a high serum level “dose tapering may lower their risk of infection,” Meghna Jani, MD, said at the European Congress of Rheumatology.

The study used data and specimens collected in two separate, prospective U.K. studies: the British Society for Rheumatology Biologics Register-RA, which had data from more than 20,000 U.K. patients with RA who started treatment with a biologic agent, and BRAGGSS (Biologics in Rheumatoid Arthritis and Genetics and Genomics Study Syndicate), a national prospective cohort of 3,000 RA patients who had serum specimens drawn at 3, 6, and 12 months after starting biologic drug treatment and tested by an immunoassay for the concentration of the drug each patient received.

Dr. Jani and her associates considered serum levels that exceeded the following thresholds to categorize patients as having a high drug level: 8 mcg/mL adalimumab, 25 mcg/mL certolizumab, 4 mcg/mL etanercept, 4 mcg/mL infliximab, and 4 mcg/mL tocilizumab. The patients averaged about 59 years old, about three-quarters were women, and they had been diagnosed with RA for approximately 5-7 years. About 22% were also on treatment with a steroid, and most patients had not received prior treatment with a biologic agent.
The researchers tallied 229 diagnosed infections in the subgroup with high serum levels of their biologic drug, and 63 infections in those with levels below this threshold. After adjustment for age, sex, methotrexate use, and disease activity score, patients with high serum levels of their biologic drug had a 51% higher rate of all infections than did patients with levels that fell below the high-level threshold, reported Dr. Jani, a rheumatologist at Manchester (England) University. Analysis of the accumulation of infections over the course of 1 year of follow-up showed that this difference in infection rates became apparent after about 2 months of exposure and then began to diverge more sharply after about 5 months of exposure.
The results also showed that the rate of serious infections – defined as those needing intravenous antibiotics, hospitalization, or resulting in death – were similar in the two subgroups. The types of infections and their relative frequencies were also roughly similar in the two subgroups. Lower respiratory infections were the most common infection in both subgroups, followed by infections of the upper respiratory tract, urinary tract, and skin as the next three most common infections in both subgroups.
SOURCE: Jani M et al. EULAR 2018 Congress, Abstract OP0229.
AMSTERDAM – Patients with rheumatoid arthritis who had a high serum level of biologic immunomodulatory drugs had a statistically significant 51% higher rate of infection during their first year on the drug, compared with RA patients who maintained usual or low serum levels of the same drugs, according to an analysis of 703 U.K. patients in a national database.
The results suggest that, once patients with rheumatoid arthritis go into remission on a higher dosage of biologic agents that produce a high serum level “dose tapering may lower their risk of infection,” Meghna Jani, MD, said at the European Congress of Rheumatology.
The study used data and specimens collected in two separate, prospective U.K. studies: the British Society for Rheumatology Biologics Register-RA, which had data from more than 20,000 U.K. patients with RA who started treatment with a biologic agent, and BRAGGSS (Biologics in Rheumatoid Arthritis and Genetics and Genomics Study Syndicate), a national prospective cohort of 3,000 RA patients who had serum specimens drawn at 3, 6, and 12 months after starting biologic drug treatment and tested by an immunoassay for the concentration of the drug each patient received.
Dr. Jani and her associates considered serum levels that exceeded the following thresholds to categorize patients as having a high drug level: 8 mcg/mL adalimumab, 25 mcg/mL certolizumab, 4 mcg/mL etanercept, 4 mcg/mL infliximab, and 4 mcg/mL tocilizumab. The patients averaged about 59 years old, about three-quarters were women, and they had been diagnosed with RA for approximately 5-7 years. About 22% were also on treatment with a steroid, and most patients had not received prior treatment with a biologic agent.
The researchers tallied 229 diagnosed infections in the subgroup with high serum levels of their biologic drug, and 63 infections in those with levels below this threshold. After adjustment for age, sex, methotrexate use, and disease activity score, patients with high serum levels of their biologic drug had a 51% higher rate of all infections than did patients with levels that fell below the high-level threshold, reported Dr. Jani, a rheumatologist at Manchester (England) University. Analysis of the accumulation of infections over the course of 1 year of follow-up showed that this difference in infection rates became apparent after about 2 months of exposure and then began to diverge more sharply after about 5 months of exposure.
The results also showed that the rate of serious infections – defined as those needing intravenous antibiotics, hospitalization, or resulting in death – were similar in the two subgroups. The types of infections and their relative frequencies were also roughly similar in the two subgroups. Lower respiratory infections were the most common infection in both subgroups, followed by infections of the upper respiratory tract, urinary tract, and skin as the next three most common infections in both subgroups.
SOURCE: Jani M et al. EULAR 2018 Congress, Abstract OP0229.
AMSTERDAM – Patients with rheumatoid arthritis who had a high serum level of biologic immunomodulatory drugs had a statistically significant 51% higher rate of infection during their first year on the drug, compared with RA patients who maintained usual or low serum levels of the same drugs, according to an analysis of 703 U.K. patients in a national database.
The results suggest that, once patients with rheumatoid arthritis go into remission on a higher dosage of biologic agents that produce a high serum level “dose tapering may lower their risk of infection,” Meghna Jani, MD, said at the European Congress of Rheumatology.
The study used data and specimens collected in two separate, prospective U.K. studies: the British Society for Rheumatology Biologics Register-RA, which had data from more than 20,000 U.K. patients with RA who started treatment with a biologic agent, and BRAGGSS (Biologics in Rheumatoid Arthritis and Genetics and Genomics Study Syndicate), a national prospective cohort of 3,000 RA patients who had serum specimens drawn at 3, 6, and 12 months after starting biologic drug treatment and tested by an immunoassay for the concentration of the drug each patient received.
Dr. Jani and her associates considered serum levels that exceeded the following thresholds to categorize patients as having a high drug level: 8 mcg/mL adalimumab, 25 mcg/mL certolizumab, 4 mcg/mL etanercept, 4 mcg/mL infliximab, and 4 mcg/mL tocilizumab. The patients averaged about 59 years old, about three-quarters were women, and they had been diagnosed with RA for approximately 5-7 years. About 22% were also on treatment with a steroid, and most patients had not received prior treatment with a biologic agent.
The researchers tallied 229 diagnosed infections in the subgroup with high serum levels of their biologic drug, and 63 infections in those with levels below this threshold. After adjustment for age, sex, methotrexate use, and disease activity score, patients with high serum levels of their biologic drug had a 51% higher rate of all infections than did patients with levels that fell below the high-level threshold, reported Dr. Jani, a rheumatologist at Manchester (England) University. Analysis of the accumulation of infections over the course of 1 year of follow-up showed that this difference in infection rates became apparent after about 2 months of exposure and then began to diverge more sharply after about 5 months of exposure.
The results also showed that the rate of serious infections – defined as those needing intravenous antibiotics, hospitalization, or resulting in death – were similar in the two subgroups. The types of infections and their relative frequencies were also roughly similar in the two subgroups. Lower respiratory infections were the most common infection in both subgroups, followed by infections of the upper respiratory tract, urinary tract, and skin as the next three most common infections in both subgroups.
SOURCE: Jani M et al. EULAR 2018 Congress, Abstract OP0229.
REPORTING FROM THE EULAR 2018 CONGRESS
Key clinical point:
Major finding: High serum levels of a biologic drug were linked with a 51% higher infection rate, compared with those who had normal or low levels.
Study details: Prospective data collected from 703 patients throughout the United Kingdom.
Disclosures: Dr. Jani had no relevant disclosures. Dr. Isaacs has been a consultant to several companies that market biologic drugs for treating rheumatoid arthritis.
Source: Jani M et al. EULAR 2018 Congress, Abstract OP0229.
What to do if you encounter Candida auris
Closely monitor patients for treatment failure
Candida auris is an emerging, often multidrug-resistant yeast that causes invasive infections (such as bloodstream, intra-abdominal) and is transmitted in health care settings. It is difficult to diagnose using traditional yeast identification methods. C. auris also has been found in noninvasive body sites and can colonize a person without causing active infection and hence permitting transmission of the pathogen between patients. These sites include skin, urine, external ear, wounds, and respiratory specimens.
This fungus was first described in 2009 in an ear-discharge culture from a patient in Japan. The first clinical cases were described in South Korea in 2011. An unknown pathogen before 2009, C. auris caused 4%-8% of candidemia in Indian ICUs during 2011-2012 and 38% of candidemia in one Kenyan hospital during 2010-2013. It has now spread across Asia and Europe, only to arrive in the United States in 2016.
As of Aug. 31, 2017, a total of 153 clinical cases of C. auris infection have been reported to CDC from 10 U.S. states; most have occurred in New York and New Jersey. An additional 143 patients have been found to be colonized with C. auris. Based on epidemiologic and molecular information, including whole genome sequencing, the Centers for Disease Control and Prevention infers that most U.S. cases likely resulted from local transmission of C. auris following previous introduction from other countries in Asia.

The majority of infections within the United States have been in blood streams. The reported all-cause mortality from these infections has been up to 60%. Most C. auris isolates in the United States have been resistant to at least one antifungal, most commonly fluconazole, and patients have developed resistance to echinocandin drugs while on treatment. Amphotericin B resistance also has been seen in about 30% of isolates.
In response to global reports and a large outbreak in a specialty hospital in the United Kingdom, the CDC issued its first advisory and clinical alert to health care facilities in June 2016. It is essential for hospitalist physicians to be aware of this emerging pathogen and also of the interventions needed to curb its spread, given they are the frontline warriors in the fight against hospital-acquired infections.
The first step in controlling C. auris is identification. C. auris can be misidentified when using traditional biochemical methods. They are most commonly misidentified as Candida haemulonii. Currently, accurate identification for C. auris can be performed by Vitek MS and matrix-assisted laser desorption/ionization time-of-flight using research use–only databases. Hospitalists should be aware of the diagnostic instruments used in their hospital laboratories and their ability to detect C. auris. Clinical laboratories should request testing of suspect C. auris isolates from their state or regional public health laboratory or the CDC. Laboratories should also consider reviewing historical microbiology records for suspect isolates (e.g., C. haemulonii) to identify missed cases of C. auris.
All cultures positive for Candida should be further speciated and antifungal susceptibilities should be reported as per new Infectious Diseases Society of America guidelines for candidiasis from 2016. As many clinical laboratories do not determine the species of Candida from noninvasive sites, C. auris colonization may go unrecognized and lead to transmission. About 54% of recognized U.S. clinical cases have been identified from blood cultures. The remaining patients with positive C. auris cultures, including those with recent hospitalizations abroad, have had the organism isolated from other body sites, including skin wounds, urine, respiratory specimens, bile fluid, and ears. Determining the species of Candida for isolates from these noninvasive sites in certain situations may allow for more rapid identification of C. auris and allow for timely implementation of targeted infection control measures to reduce transmission.
Patients have been persistently colonized with C. auris, posing long-term risk of transmission. Currently, data on effective decolonization methods are lacking. Patients with suspected or confirmed C. auris infection should be placed in a single room if possible and standard and contact precautions should be initiated and thorough environmental cleaning and disinfection of the patient care area should be undertaken. Using an Environmental Protection Agency–registered antimicrobial product active against Clostridium difficile for routine and terminal disinfection is recommended.
Implement contact tracing and testing to identify other patients colonized with C. auris. Review past microbiology records (at least for the preceding 1 year) for suspect or confirmed cases of C. auris at the institution. Set up enhanced surveillance for C. auris in the laboratory setting.
Echinocandin drugs are the first-line treatment for most invasive Candida infections, making resistance to this class of antifungal drugs particularly concerning. As of Sept. 15, 2017, at least five patients in the United States had echinocandin-resistant isolates. In one patient, resistance to echinocandin drugs developed while being treated with echinocandins.
Based on these findings, CDC is concerned that echinocandin-resistant C. auris could become more common. Patients with C. auris infection should be closely monitored for treatment failure, as indicated by persistently positive clinical cultures (lasting more than 5 days). Consultation with an infectious disease specialist is highly recommended.
Dr. Tirupathi is medical director, infectious diseases/HIV at Keystone Health, and chair, infection prevention, at Summit Health, both in Chambersburg, Pa. He is clinical assistant professor of medicine at Penn State University, Hershey.
Closely monitor patients for treatment failure
Closely monitor patients for treatment failure
Candida auris is an emerging, often multidrug-resistant yeast that causes invasive infections (such as bloodstream, intra-abdominal) and is transmitted in health care settings. It is difficult to diagnose using traditional yeast identification methods. C. auris also has been found in noninvasive body sites and can colonize a person without causing active infection and hence permitting transmission of the pathogen between patients. These sites include skin, urine, external ear, wounds, and respiratory specimens.
This fungus was first described in 2009 in an ear-discharge culture from a patient in Japan. The first clinical cases were described in South Korea in 2011. An unknown pathogen before 2009, C. auris caused 4%-8% of candidemia in Indian ICUs during 2011-2012 and 38% of candidemia in one Kenyan hospital during 2010-2013. It has now spread across Asia and Europe, only to arrive in the United States in 2016.
As of Aug. 31, 2017, a total of 153 clinical cases of C. auris infection have been reported to CDC from 10 U.S. states; most have occurred in New York and New Jersey. An additional 143 patients have been found to be colonized with C. auris. Based on epidemiologic and molecular information, including whole genome sequencing, the Centers for Disease Control and Prevention infers that most U.S. cases likely resulted from local transmission of C. auris following previous introduction from other countries in Asia.
The majority of infections within the United States have been in blood streams. The reported all-cause mortality from these infections has been up to 60%. Most C. auris isolates in the United States have been resistant to at least one antifungal, most commonly fluconazole, and patients have developed resistance to echinocandin drugs while on treatment. Amphotericin B resistance also has been seen in about 30% of isolates.
In response to global reports and a large outbreak in a specialty hospital in the United Kingdom, the CDC issued its first advisory and clinical alert to health care facilities in June 2016. It is essential for hospitalist physicians to be aware of this emerging pathogen and also of the interventions needed to curb its spread, given they are the frontline warriors in the fight against hospital-acquired infections.
The first step in controlling C. auris is identification. C. auris can be misidentified when using traditional biochemical methods. They are most commonly misidentified as Candida haemulonii. Currently, accurate identification for C. auris can be performed by Vitek MS and matrix-assisted laser desorption/ionization time-of-flight using research use–only databases. Hospitalists should be aware of the diagnostic instruments used in their hospital laboratories and their ability to detect C. auris. Clinical laboratories should request testing of suspect C. auris isolates from their state or regional public health laboratory or the CDC. Laboratories should also consider reviewing historical microbiology records for suspect isolates (e.g., C. haemulonii) to identify missed cases of C. auris.
All cultures positive for Candida should be further speciated and antifungal susceptibilities should be reported as per new Infectious Diseases Society of America guidelines for candidiasis from 2016. As many clinical laboratories do not determine the species of Candida from noninvasive sites, C. auris colonization may go unrecognized and lead to transmission. About 54% of recognized U.S. clinical cases have been identified from blood cultures. The remaining patients with positive C. auris cultures, including those with recent hospitalizations abroad, have had the organism isolated from other body sites, including skin wounds, urine, respiratory specimens, bile fluid, and ears. Determining the species of Candida for isolates from these noninvasive sites in certain situations may allow for more rapid identification of C. auris and allow for timely implementation of targeted infection control measures to reduce transmission.
Patients have been persistently colonized with C. auris, posing long-term risk of transmission. Currently, data on effective decolonization methods are lacking. Patients with suspected or confirmed C. auris infection should be placed in a single room if possible and standard and contact precautions should be initiated and thorough environmental cleaning and disinfection of the patient care area should be undertaken. Using an Environmental Protection Agency–registered antimicrobial product active against Clostridium difficile for routine and terminal disinfection is recommended.
Implement contact tracing and testing to identify other patients colonized with C. auris. Review past microbiology records (at least for the preceding 1 year) for suspect or confirmed cases of C. auris at the institution. Set up enhanced surveillance for C. auris in the laboratory setting.
Echinocandin drugs are the first-line treatment for most invasive Candida infections, making resistance to this class of antifungal drugs particularly concerning. As of Sept. 15, 2017, at least five patients in the United States had echinocandin-resistant isolates. In one patient, resistance to echinocandin drugs developed while being treated with echinocandins.
Based on these findings, CDC is concerned that echinocandin-resistant C. auris could become more common. Patients with C. auris infection should be closely monitored for treatment failure, as indicated by persistently positive clinical cultures (lasting more than 5 days). Consultation with an infectious disease specialist is highly recommended.
Dr. Tirupathi is medical director, infectious diseases/HIV at Keystone Health, and chair, infection prevention, at Summit Health, both in Chambersburg, Pa. He is clinical assistant professor of medicine at Penn State University, Hershey.
Candida auris is an emerging, often multidrug-resistant yeast that causes invasive infections (such as bloodstream, intra-abdominal) and is transmitted in health care settings. It is difficult to diagnose using traditional yeast identification methods. C. auris also has been found in noninvasive body sites and can colonize a person without causing active infection and hence permitting transmission of the pathogen between patients. These sites include skin, urine, external ear, wounds, and respiratory specimens.
This fungus was first described in 2009 in an ear-discharge culture from a patient in Japan. The first clinical cases were described in South Korea in 2011. An unknown pathogen before 2009, C. auris caused 4%-8% of candidemia in Indian ICUs during 2011-2012 and 38% of candidemia in one Kenyan hospital during 2010-2013. It has now spread across Asia and Europe, only to arrive in the United States in 2016.
As of Aug. 31, 2017, a total of 153 clinical cases of C. auris infection have been reported to CDC from 10 U.S. states; most have occurred in New York and New Jersey. An additional 143 patients have been found to be colonized with C. auris. Based on epidemiologic and molecular information, including whole genome sequencing, the Centers for Disease Control and Prevention infers that most U.S. cases likely resulted from local transmission of C. auris following previous introduction from other countries in Asia.
The majority of infections within the United States have been in blood streams. The reported all-cause mortality from these infections has been up to 60%. Most C. auris isolates in the United States have been resistant to at least one antifungal, most commonly fluconazole, and patients have developed resistance to echinocandin drugs while on treatment. Amphotericin B resistance also has been seen in about 30% of isolates.
In response to global reports and a large outbreak in a specialty hospital in the United Kingdom, the CDC issued its first advisory and clinical alert to health care facilities in June 2016. It is essential for hospitalist physicians to be aware of this emerging pathogen and also of the interventions needed to curb its spread, given they are the frontline warriors in the fight against hospital-acquired infections.
The first step in controlling C. auris is identification. C. auris can be misidentified when using traditional biochemical methods. They are most commonly misidentified as Candida haemulonii. Currently, accurate identification for C. auris can be performed by Vitek MS and matrix-assisted laser desorption/ionization time-of-flight using research use–only databases. Hospitalists should be aware of the diagnostic instruments used in their hospital laboratories and their ability to detect C. auris. Clinical laboratories should request testing of suspect C. auris isolates from their state or regional public health laboratory or the CDC. Laboratories should also consider reviewing historical microbiology records for suspect isolates (e.g., C. haemulonii) to identify missed cases of C. auris.
All cultures positive for Candida should be further speciated and antifungal susceptibilities should be reported as per new Infectious Diseases Society of America guidelines for candidiasis from 2016. As many clinical laboratories do not determine the species of Candida from noninvasive sites, C. auris colonization may go unrecognized and lead to transmission. About 54% of recognized U.S. clinical cases have been identified from blood cultures. The remaining patients with positive C. auris cultures, including those with recent hospitalizations abroad, have had the organism isolated from other body sites, including skin wounds, urine, respiratory specimens, bile fluid, and ears. Determining the species of Candida for isolates from these noninvasive sites in certain situations may allow for more rapid identification of C. auris and allow for timely implementation of targeted infection control measures to reduce transmission.
Patients have been persistently colonized with C. auris, posing long-term risk of transmission. Currently, data on effective decolonization methods are lacking. Patients with suspected or confirmed C. auris infection should be placed in a single room if possible and standard and contact precautions should be initiated and thorough environmental cleaning and disinfection of the patient care area should be undertaken. Using an Environmental Protection Agency–registered antimicrobial product active against Clostridium difficile for routine and terminal disinfection is recommended.
Implement contact tracing and testing to identify other patients colonized with C. auris. Review past microbiology records (at least for the preceding 1 year) for suspect or confirmed cases of C. auris at the institution. Set up enhanced surveillance for C. auris in the laboratory setting.
Echinocandin drugs are the first-line treatment for most invasive Candida infections, making resistance to this class of antifungal drugs particularly concerning. As of Sept. 15, 2017, at least five patients in the United States had echinocandin-resistant isolates. In one patient, resistance to echinocandin drugs developed while being treated with echinocandins.
Based on these findings, CDC is concerned that echinocandin-resistant C. auris could become more common. Patients with C. auris infection should be closely monitored for treatment failure, as indicated by persistently positive clinical cultures (lasting more than 5 days). Consultation with an infectious disease specialist is highly recommended.
Dr. Tirupathi is medical director, infectious diseases/HIV at Keystone Health, and chair, infection prevention, at Summit Health, both in Chambersburg, Pa. He is clinical assistant professor of medicine at Penn State University, Hershey.