User login
Breast Cancer Screening Improvement Initiative
The purpose of the initiative is to increase the percentage of eligible female veterans who receive biennial mammography to over 80% consistently at the Minneapolis VA (MVA) medical center.
The External Peer Review Program (EPRP) evaluates a number of charts each month to report quality of care on a variety of performance measures. Breast Cancer Screening is among those evaluated with the goal of a 77% completion rate. In 2012 the average EPRP showed MVA’s completion rate at 77%. It also highlighted that it was below target for 4 months. Clinical Reminders are relied upon for provider notification, however, the current process depends on clinicians going into individual charts to see and act on them. Oftentimes this is not done in an acceptable amount of time.
The Corporate Data Warehouse (CDW) stores clinical information from VistA. A pro-active process to address biennial mammography completion was reproduced using methods that query the CDW. Reports that evaluated all veterans, overdue or nearly due, for breast cancer screening were created. Lists of these patients were generated and sent to providers/PACT teams for evaluation and resolution. These “push” reports were distributed quarterly, starting in January 2014, and increased to monthly in 2015.
EPRP results are highly variable and depend on the sample selection for any given month. Therefore a rolling 12 month average, median, and standard deviation of the samples are reported along with actual monthly values.
The MVA breast cancer screening rate, as reported by EPRP, improved from a 12 month average of 80% to a 12 month average of 95% over 2 years. The variability/standard deviation was reduced by 50% as quality improved.
“Push” reports can help improve rates of early breast cancer detection by screening within the recommended period of time. This panel management tool can be created at any VA facility and can be extrapolated to other quality measures.
The purpose of the initiative is to increase the percentage of eligible female veterans who receive biennial mammography to over 80% consistently at the Minneapolis VA (MVA) medical center.
The External Peer Review Program (EPRP) evaluates a number of charts each month to report quality of care on a variety of performance measures. Breast Cancer Screening is among those evaluated with the goal of a 77% completion rate. In 2012 the average EPRP showed MVA’s completion rate at 77%. It also highlighted that it was below target for 4 months. Clinical Reminders are relied upon for provider notification, however, the current process depends on clinicians going into individual charts to see and act on them. Oftentimes this is not done in an acceptable amount of time.
The Corporate Data Warehouse (CDW) stores clinical information from VistA. A pro-active process to address biennial mammography completion was reproduced using methods that query the CDW. Reports that evaluated all veterans, overdue or nearly due, for breast cancer screening were created. Lists of these patients were generated and sent to providers/PACT teams for evaluation and resolution. These “push” reports were distributed quarterly, starting in January 2014, and increased to monthly in 2015.
EPRP results are highly variable and depend on the sample selection for any given month. Therefore a rolling 12 month average, median, and standard deviation of the samples are reported along with actual monthly values.
The MVA breast cancer screening rate, as reported by EPRP, improved from a 12 month average of 80% to a 12 month average of 95% over 2 years. The variability/standard deviation was reduced by 50% as quality improved.
“Push” reports can help improve rates of early breast cancer detection by screening within the recommended period of time. This panel management tool can be created at any VA facility and can be extrapolated to other quality measures.
The purpose of the initiative is to increase the percentage of eligible female veterans who receive biennial mammography to over 80% consistently at the Minneapolis VA (MVA) medical center.
The External Peer Review Program (EPRP) evaluates a number of charts each month to report quality of care on a variety of performance measures. Breast Cancer Screening is among those evaluated with the goal of a 77% completion rate. In 2012 the average EPRP showed MVA’s completion rate at 77%. It also highlighted that it was below target for 4 months. Clinical Reminders are relied upon for provider notification, however, the current process depends on clinicians going into individual charts to see and act on them. Oftentimes this is not done in an acceptable amount of time.
The Corporate Data Warehouse (CDW) stores clinical information from VistA. A pro-active process to address biennial mammography completion was reproduced using methods that query the CDW. Reports that evaluated all veterans, overdue or nearly due, for breast cancer screening were created. Lists of these patients were generated and sent to providers/PACT teams for evaluation and resolution. These “push” reports were distributed quarterly, starting in January 2014, and increased to monthly in 2015.
EPRP results are highly variable and depend on the sample selection for any given month. Therefore a rolling 12 month average, median, and standard deviation of the samples are reported along with actual monthly values.
The MVA breast cancer screening rate, as reported by EPRP, improved from a 12 month average of 80% to a 12 month average of 95% over 2 years. The variability/standard deviation was reduced by 50% as quality improved.
“Push” reports can help improve rates of early breast cancer detection by screening within the recommended period of time. This panel management tool can be created at any VA facility and can be extrapolated to other quality measures.
Evaluation of Acute Toxicities of Hypofractionated Radiotherapy Using Volumetric Arc Therapy
Purpose: Traditional radiation therapy for prostate cancer is given over 39 – 42 daily fractions. There have been increasing efforts to decrease the treatment time by using hypofractionated radiotherapy. The purpose of this retrospective study is to evaluate the acute toxicities (RTOG definition) in prostate cancer patients when using hypofractionated radiotherapy.
Methods: 42 patients were treated with 25 daily fractions from 2014 – 2015. Patient, tumor, and dosimetric factors (rectal and bladder min dose, max dose, mean dose, median dose, volume, V31, V50, as well as PTV max, min, mean, and median doses) were analyzed to find associations between acute (< 90 days) GU and GI toxicities.
Results: The median age was 68 with a median follow-up of 18 months. There were 2 low, 12 intermediate, and 18 high risk patients (NCCN criteria). Dose fractionations schemas used were 267 cGy (n = 6), 270 cGy (n = 14); 275 cGy (n = 17), and some high risk patients received a simultaneous integrated boost (SIB) of 300 cGy (n = 5) to a smaller volume of the prostate. 13 patients received pelvic irradiation via SIB at 200 cGy per fraction. 10 patients received no androgen deprivation therapy (ADT), 15 received short term ADT (≤ 6 months), and 17 received long term ADT (> 6 months). Grade 0 acute GU toxicity occurred in 12 patients (29%), grade 1 in 7 (17%), grade 2 in 22 (52%), and grade 3 in 1 (2%). Grade 0 acute GI toxicity occurred in 30 patients (71%), grade 1 in 5 (12%), grade 2 in 7 (17), and no grade 3 toxicity. There were no grade 4 or 5 toxicities. On univariate analysis, factors positively associated with acute GU toxicity were AUA score (P = .02) and PTV max dose (P = .04); acute GI were pelvic radiation (P = .04) and rectal min dose (P = .02). These factors were not significant on multivariate analysis.
Conclusion: Volumetric Arc Therapy based hypofractionated radiotherapy was well tolerated and is an acceptable treatment for prostate cancer patients. Larger and adequately powered studies are needed to validate these findings.
Purpose: Traditional radiation therapy for prostate cancer is given over 39 – 42 daily fractions. There have been increasing efforts to decrease the treatment time by using hypofractionated radiotherapy. The purpose of this retrospective study is to evaluate the acute toxicities (RTOG definition) in prostate cancer patients when using hypofractionated radiotherapy.
Methods: 42 patients were treated with 25 daily fractions from 2014 – 2015. Patient, tumor, and dosimetric factors (rectal and bladder min dose, max dose, mean dose, median dose, volume, V31, V50, as well as PTV max, min, mean, and median doses) were analyzed to find associations between acute (< 90 days) GU and GI toxicities.
Results: The median age was 68 with a median follow-up of 18 months. There were 2 low, 12 intermediate, and 18 high risk patients (NCCN criteria). Dose fractionations schemas used were 267 cGy (n = 6), 270 cGy (n = 14); 275 cGy (n = 17), and some high risk patients received a simultaneous integrated boost (SIB) of 300 cGy (n = 5) to a smaller volume of the prostate. 13 patients received pelvic irradiation via SIB at 200 cGy per fraction. 10 patients received no androgen deprivation therapy (ADT), 15 received short term ADT (≤ 6 months), and 17 received long term ADT (> 6 months). Grade 0 acute GU toxicity occurred in 12 patients (29%), grade 1 in 7 (17%), grade 2 in 22 (52%), and grade 3 in 1 (2%). Grade 0 acute GI toxicity occurred in 30 patients (71%), grade 1 in 5 (12%), grade 2 in 7 (17), and no grade 3 toxicity. There were no grade 4 or 5 toxicities. On univariate analysis, factors positively associated with acute GU toxicity were AUA score (P = .02) and PTV max dose (P = .04); acute GI were pelvic radiation (P = .04) and rectal min dose (P = .02). These factors were not significant on multivariate analysis.
Conclusion: Volumetric Arc Therapy based hypofractionated radiotherapy was well tolerated and is an acceptable treatment for prostate cancer patients. Larger and adequately powered studies are needed to validate these findings.
Purpose: Traditional radiation therapy for prostate cancer is given over 39 – 42 daily fractions. There have been increasing efforts to decrease the treatment time by using hypofractionated radiotherapy. The purpose of this retrospective study is to evaluate the acute toxicities (RTOG definition) in prostate cancer patients when using hypofractionated radiotherapy.
Methods: 42 patients were treated with 25 daily fractions from 2014 – 2015. Patient, tumor, and dosimetric factors (rectal and bladder min dose, max dose, mean dose, median dose, volume, V31, V50, as well as PTV max, min, mean, and median doses) were analyzed to find associations between acute (< 90 days) GU and GI toxicities.
Results: The median age was 68 with a median follow-up of 18 months. There were 2 low, 12 intermediate, and 18 high risk patients (NCCN criteria). Dose fractionations schemas used were 267 cGy (n = 6), 270 cGy (n = 14); 275 cGy (n = 17), and some high risk patients received a simultaneous integrated boost (SIB) of 300 cGy (n = 5) to a smaller volume of the prostate. 13 patients received pelvic irradiation via SIB at 200 cGy per fraction. 10 patients received no androgen deprivation therapy (ADT), 15 received short term ADT (≤ 6 months), and 17 received long term ADT (> 6 months). Grade 0 acute GU toxicity occurred in 12 patients (29%), grade 1 in 7 (17%), grade 2 in 22 (52%), and grade 3 in 1 (2%). Grade 0 acute GI toxicity occurred in 30 patients (71%), grade 1 in 5 (12%), grade 2 in 7 (17), and no grade 3 toxicity. There were no grade 4 or 5 toxicities. On univariate analysis, factors positively associated with acute GU toxicity were AUA score (P = .02) and PTV max dose (P = .04); acute GI were pelvic radiation (P = .04) and rectal min dose (P = .02). These factors were not significant on multivariate analysis.
Conclusion: Volumetric Arc Therapy based hypofractionated radiotherapy was well tolerated and is an acceptable treatment for prostate cancer patients. Larger and adequately powered studies are needed to validate these findings.
Distress Screening at the Central Arkansas Veterans Healthcare System Hematology/Oncology Clinics: A Quality Improvement Project
Purpose: Distress screening (DS) of cancer patients is likely to improve access to supportive care services and adherence to cancer treatment.
Background: We assessed DS at the CAVHS and the University of Arkansas for Medical Sciences by completing Quality Oncology and Practice Initiatives (QOPI) survey. At baseline, we identified assessment of distress screening in only 15% of patients compared to the national average of 50%.
Methods: Based on QOPI data, we performed 3 Plan-Do-Study-Act (PDSA) cycles. We implemented a DS template on computer patient record system (CPRS) at the CAVHS hematology-oncology clinic, validated the template after initial use, and modified it in each PDSA cycle.
Results: At baseline DS was identified in 18 out of 121 charts (15%) per QOPI survey results in 2013 Spring Round. During our first PDSA cycle, we decided to add DS templates to all electronic medical notes in CPRS. We validated the template in 20 patient charts. Thereafter, in the second PDSA cycle, we modified the template and included distress thermometer (DT). After this intervention, we noted distress screening in 27 of 100 charts from 14 providers. Out of these 27 patients, 4 had a distress score of 4 or greater; these patients were all referred to subspecialty services. We did a third PDSA cycle and DS improved to 46% (51 out of 111 charts) on QOPI 2015 Spring Round. Subsequently, we added daily reminders at staff meetings, weekly e-mail reminders, and visual DT in each clinic room to perform DS and improved DS to 55 screenings out of 100 charts audited (27% to 55%). In these patients, only 1 had score of 4 and 7 had scores of 3. Intervention was offered in 15 out of 55 (27.2%) patients including counseling and referral to subspecialty services.
Conclusion: Distress screening is important for identifying patients who need intervention. QOPI is an excellent method of evaluating compliance to distress screening and PDSA cycles are effective in improving compliance. We improved DS by more than 200% using QOPI, PDSA and other quality improvement methods.
Purpose: Distress screening (DS) of cancer patients is likely to improve access to supportive care services and adherence to cancer treatment.
Background: We assessed DS at the CAVHS and the University of Arkansas for Medical Sciences by completing Quality Oncology and Practice Initiatives (QOPI) survey. At baseline, we identified assessment of distress screening in only 15% of patients compared to the national average of 50%.
Methods: Based on QOPI data, we performed 3 Plan-Do-Study-Act (PDSA) cycles. We implemented a DS template on computer patient record system (CPRS) at the CAVHS hematology-oncology clinic, validated the template after initial use, and modified it in each PDSA cycle.
Results: At baseline DS was identified in 18 out of 121 charts (15%) per QOPI survey results in 2013 Spring Round. During our first PDSA cycle, we decided to add DS templates to all electronic medical notes in CPRS. We validated the template in 20 patient charts. Thereafter, in the second PDSA cycle, we modified the template and included distress thermometer (DT). After this intervention, we noted distress screening in 27 of 100 charts from 14 providers. Out of these 27 patients, 4 had a distress score of 4 or greater; these patients were all referred to subspecialty services. We did a third PDSA cycle and DS improved to 46% (51 out of 111 charts) on QOPI 2015 Spring Round. Subsequently, we added daily reminders at staff meetings, weekly e-mail reminders, and visual DT in each clinic room to perform DS and improved DS to 55 screenings out of 100 charts audited (27% to 55%). In these patients, only 1 had score of 4 and 7 had scores of 3. Intervention was offered in 15 out of 55 (27.2%) patients including counseling and referral to subspecialty services.
Conclusion: Distress screening is important for identifying patients who need intervention. QOPI is an excellent method of evaluating compliance to distress screening and PDSA cycles are effective in improving compliance. We improved DS by more than 200% using QOPI, PDSA and other quality improvement methods.
Purpose: Distress screening (DS) of cancer patients is likely to improve access to supportive care services and adherence to cancer treatment.
Background: We assessed DS at the CAVHS and the University of Arkansas for Medical Sciences by completing Quality Oncology and Practice Initiatives (QOPI) survey. At baseline, we identified assessment of distress screening in only 15% of patients compared to the national average of 50%.
Methods: Based on QOPI data, we performed 3 Plan-Do-Study-Act (PDSA) cycles. We implemented a DS template on computer patient record system (CPRS) at the CAVHS hematology-oncology clinic, validated the template after initial use, and modified it in each PDSA cycle.
Results: At baseline DS was identified in 18 out of 121 charts (15%) per QOPI survey results in 2013 Spring Round. During our first PDSA cycle, we decided to add DS templates to all electronic medical notes in CPRS. We validated the template in 20 patient charts. Thereafter, in the second PDSA cycle, we modified the template and included distress thermometer (DT). After this intervention, we noted distress screening in 27 of 100 charts from 14 providers. Out of these 27 patients, 4 had a distress score of 4 or greater; these patients were all referred to subspecialty services. We did a third PDSA cycle and DS improved to 46% (51 out of 111 charts) on QOPI 2015 Spring Round. Subsequently, we added daily reminders at staff meetings, weekly e-mail reminders, and visual DT in each clinic room to perform DS and improved DS to 55 screenings out of 100 charts audited (27% to 55%). In these patients, only 1 had score of 4 and 7 had scores of 3. Intervention was offered in 15 out of 55 (27.2%) patients including counseling and referral to subspecialty services.
Conclusion: Distress screening is important for identifying patients who need intervention. QOPI is an excellent method of evaluating compliance to distress screening and PDSA cycles are effective in improving compliance. We improved DS by more than 200% using QOPI, PDSA and other quality improvement methods.
Pediatric Hospital Medicine 2016 Wrap Up
Pediatric Hospital Medicine 2016, cosponsored by the American Academy of Pediatrics (AAP), the Academic Pediatric Association (APA), and the Society of Hospital Medicine (SHM), took place July 28–31 in Chicago. Didn’t make it? Here are all the news, research, and talking points you need to know.

Shape Your Brain to Avoid Burnout
Presenter: Lisa Zaoutis, MD, FHM
Amid the skyscrapers of the Windy City, Pediatric Hospital Medicine (PHM) 2016 swept into town, bringing with it the denizens of pediatric hospitalist programs across the country. Some 1,150 attendees, composed of hospitalists, PHM program leaders, and advanced-care practitioners, gathered to educate and inspire one another in the care of hospitalized children.
Lisa Zaoutis, MD, FHM, director of the pediatric residency program at The Children’s Hospital of Philadelphia, kicked off the conference with the opening plenary. Initially titled “North Star and Space,” she quickly changed the title to “Changing Our Minds.” Touching on the disconnect between positive experiences that bring physicians into pediatric hospital medicine and negative experiences that often drive behavior, she started with the beginning: the evolution of our brains.
“We are wired toward the negative,” Dr. Zaoutis said. “We are Teflon for positive experiences and Velcro for negative experiences.”
Delving deeper into neuroanatomy, Dr. Zaoutis spoke of “amygdala hijack,” where chronic stress inherent to the professional lives of pediatric hospitalists leads to anxiety responses that are faster, more robust, and more easily triggered.
But all is not lost, Dr. Zaoutis noted, as our brains are more plastic than previously known. The “neural Darwinism” of our brains, she said, leads to epigenetic intracellular changes, more sensitive synapses, improved blood flow, and even new cells as a result of experience-dependent neuroplasticity. For example, London taxi drivers have thicker white matter in their hippocampus as a result of learning London city streets, and mindfulness meditators have thicker gray matter in regions that control attention and self-insight.
Key Takeaways
The lesson for pediatric hospitalists, according Dr. Zaoutis, is that you can shape your brain for greater joy.
“Consciously choose activities” that counter our evolutionary negativity bias, Dr. Zaoutis said.
Here’s how to do it:
- Have a positive experience. (You can create one or retrieve a prior one.)
- Enrich it and install it by dwelling on it for at least 15–30 seconds.
- Absorb it into your body, which may require somatizing it. (Dr. Zaoutis presses her hand into her chest to aid in this.)
Further, spread this to your group by the old medical training technique of “see one, do one, teach one.” See if you can start your sign-out with the best thing that happened to you in the week. Most important, start with observing yourself.
Weijen Chang, MD, SFHM, is pediatric editor of The Hospitalist. He is associate clinical professor of medicine and pediatrics at the University of California, San Diego (UCSD) School of Medicine and a hospitalist at both UCSD Medical Center and Rady Children’s Hospital. Send comments and questions to [email protected].
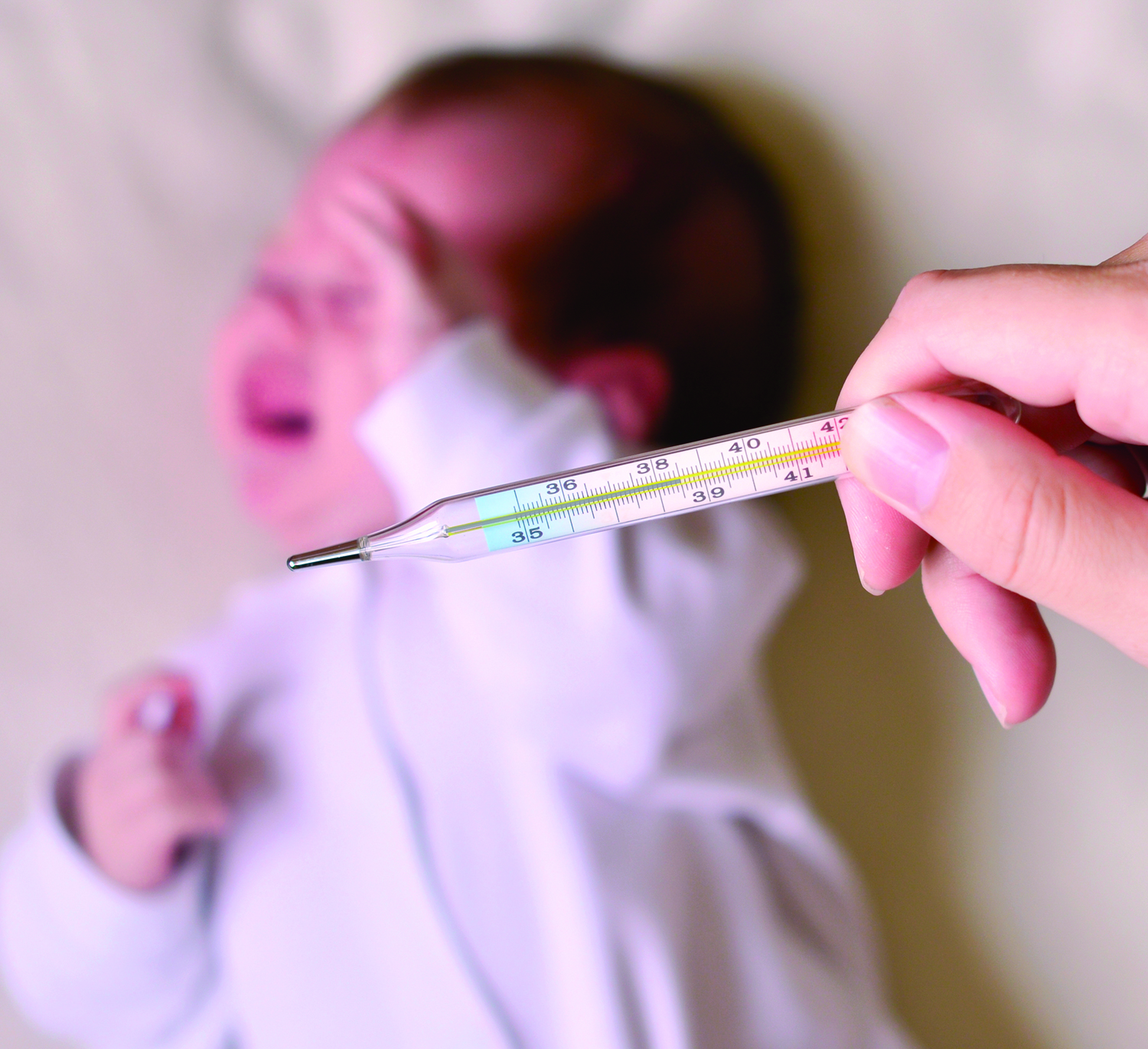
New AAP Guideline on Evaluating, Managing Febrile Infants
Presenter: Kenneth Roberts, MD
One of PHM16’s most highly attended sessions was an update on the anticipated AAP guidelines for febrile infants ages 7–90 days. The updated guidelines stress the need to separate individual components of serious bacterial infections (UTI, bacteremia, and meningitis) as the incidence and clinical course can vary greatly in this population.
The inclusion criteria for infants for this upcoming algorithm require an infant to be full-term (37–43 weeks’ gestation), aged 7–90 days, well-appearing, and presenting with a temperature of 38°C.
Exclusion criteria include perinatal/prenatal/neonatal maternal fever, infection, or antimicrobial treatment; the presence of any evident infection; being technology-dependent; and the presence of congenital anomalies.
The updated guideline will aim to stratify management by ages 7–28 days, 29–60 days, and 61–90 days to provide the most appropriate and directed treatment.
It will also include a role for inflammatory markers and allow for a “kinder, gentler” approach, including withholding certain treatments and procedures if infants are at low risk of infection. An active need for observation may be appropriate for certain infants as well. These guidelines should be tailored for individual patients to provide the best care possible while minimizing risk.
Key Takeaway
An updated AAP guideline algorithm for the management of well-appearing febrile infants ages 7–28 days, 29–60 days, and 60–90 days will be coming in the near future. It will help standardize care in this population but should not be used as a substitute for clinical judgment.
Chandani DeZure, MD, FAAP, is a pediatric hospitalist at Children’s National Health System and instructor of pediatrics at George Washington University School of Medicine & Health Sciences in Washington, D.C.
Promoting, Teaching Pediatric High-Value Care
Presenters: Lauren L. Walker, MD, FAAP; Alan Schroeder, MD, FAAP; Michael
Tchou, MD, FAAP; Jimmy Beck, MD, MEd; Lisa Herrmann, MD; Ricardo Quinonez, MD, FAAP
Pediatric hospitalists gathered to attend a fruitful discussion on not only how to change our way of thinking but also how to feed it forward to our trainees. The barriers to promoting and teaching high-value care are plenty and essentially universal to academic and community sites: We have had no formal teaching, there is cultural resistance, and there is lack of transparency on costs and charges.
The questions we perhaps should be asking ourselves, our trainees, and our families are:
- “What matters?” instead of “What’s the matter?”
- “Does that test benefit the patient? What are the harms of the test?” instead of “Will that test change our management?”
There is still a long way to go to move the pendulum to the side of value-based practice and teaching. There is still controversy on how and whether cost should be discussed with the family. Cost is more than just monetary value; family anxiety and patient harm may resonate more with families as we perfect our skills in shared decision making.
Key Takeaway
This serves as an exciting time to unite and better our understanding about why we do what we do and deliberately think about downstream effects. High-value care curriculum for medical students, residents, fellows, and even faculty is an area ripe for further research.
Akshata Hopkins, MD, FAAP, is an academic hospitalist at Johns Hopkins All Children’s Hospital in St. Petersburg, Fla.

How to Design, Improve Educational Programs at Community Hospitals
Presenters: Christopher Russo, MD, FAAP; Laura Hodo, MD, and Lauren Wilson, MD
One session at PHM16 focused on ways to design and improve education within community hospital settings. It was done via a didactic session, breakout groups, and an electronic assessment tool. Facilitators included the workshop leaders and co-leaders along with current PHM fellows and educators from community and academic settings.
During the didactic session, a general background of the importance of education during times of increasing academic and community site affiliations was discussed. This included the strengths of community hospitals for learners such as “appropriate learner autonomy,” “exposure to different career paths,” and “transfer decision making.”
Some of the challenges discussed in regard to developing an educational structure in community settings included:
1. Logistics
- Making the case for education
- Legal framework (e.g., affiliation agreements, liability)
- Finances (e.g., GME funding)
- Paperwork burden (e.g., licensing, credentialing)
2. Learning environment
- Complementing clinical work with materials
- Autonomy/supervision balancing
- Developing clinical teachers
The didactic session also reviewed the six steps for curriculum development: general needs assessment, targeted needs assessment, goals and objectives, educational strategies, implementation, and evaluation/feedback. Each of these was described in further detail with relevant examples.
Groups were broken into small groups based on four learner types: medical students, family medicine residents, pediatric residents, and PHM fellows. Within each group, a “program development matrix” was distributed to assess the support from leadership and logistics within each setting. Each one of these was separated into subgroups such as credentialing, financial support, housing/travel, and preceptor recruitment.
A separate “curriculum development matrix” was used during breakout groups that focused on curriculum development. This matrix was broken into three areas: educational strategies, implementation, and evaluation/feedback. These were further broken down into subgroups such as content, identifying resources, and remediation planning. The group was asked to determine short- and long-term goals with action steps for both of these matrix subgroups.
Key Takeaway
Overall, the session presented a structured way of assessing the educational environment for learners in community settings. It gave tangible tools for sites that wish to develop or improve their current educational framework.
Francisco Alvarez, MD, FAAP, is a pediatric hospitalist and director of the Children’s National Health System Community Hospital Services in Washington, D.C.
Tips on Meeting Needs of Children with a Medical Complexity
Presenters: Mary L. Ehlenbach, MD, FAAP; Megan Z. Cardoso, MD, FAAP, and Christina Kleier, ARNP, PNP
This session at PHM16 was focused on logistical tips on how to build a pediatric complex-care program. Presenters opened with a discussion on how to define children with medical complexity. This involved reviewing different methods, including using research-based aggregation of ICD-10 codes, relying on referral from both families and other providers, and identifying patients by consumption of hospital resources. The presentation continued by highlighting that although medically complex children make up only a small percentage of the overall population of children, they account for about one-third of healthcare spending. Because of advances in technology and medicine, this group of children is growing in numbers. It currently makes up about 10% of all pediatric admissions.
Key Takeaways
1. Children with medical complexity are a growing population on which a large proportion of healthcare resources are utilized. A program dedicated to serving the needs of this population may be helpful in reducing costs and improving the patient and family experience during hospitalizations.
2. When working to initiate a complex-care program:
- Set clear guidelines about which children the program is intended to serve and in what capacity it will function.
- Ensure the team composition is sustainable and meets the needs of the patients.
- Aggregate data about if the program is helping. This may be difficult to quantify since these are mostly qualitative measures.
- Include team members who are nonclinical to aid in improving hospital revenue and highlighting program benefits to the institution.
Margaret Rush, MD, is a hospitalist fellow at Children’s National Medical Center in Washington, D.C.
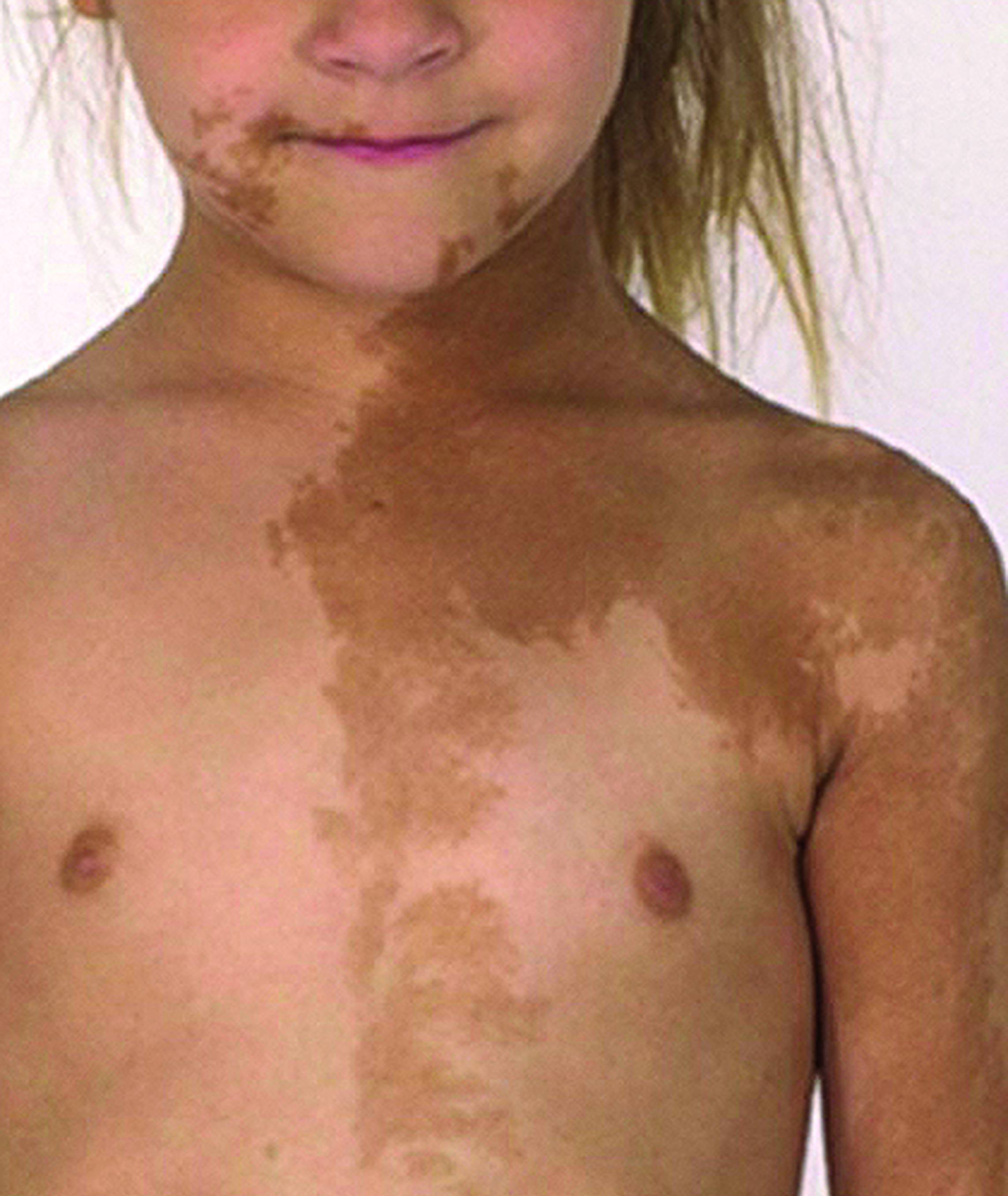
A Picture Is Worth a Thousand Words
Presenter: Kenneth Roberts, MD
PHM16’s “Visual Diagnosis: Signs and Why They Matter” session was a review of case presentations in which visual clues were vital to establishing a diagnosis. Though much of the content was presented with pictures, the emphasis was placed on the importance of correct diagnosis to avoid both misdiagnoses or overdiagnoses and the potential harm that may result from inappropriate treatment. This may also translate into poor utilization of resources and significant financial burden that can result from the unnecessary hospitalization of a patient.
Many of the presented cases highlighted examples in which there was extensive workup, hospitalization, subspecialty evaluation, and even incorrect treatment of patients.
In other instances, such as with Henoch-Schonlein purpura, Waardenburg syndrome, or McCune-Albright syndrome, the correct diagnosis was necessary to help guide management and future treatment, including subspecialty evaluation.
Key Takeaway
Many diseases with visual presentations will have a benign course and require no treatment. Acknowledging this is important in providing reassurance to a family that may be very anxious over the physical appearance of their child.
This session underscores the need for experience and exposure to various signs not only with rare medical conditions but also in common illnesses such as Kawasaki disease and scarlet fever that may present similarly.
Chandani DeZure, MD, FAAP, is a pediatric hospitalist at Children’s National Health System and instructor of pediatrics at George Washington University School of Medicine & Health Sciences in Washington, D.C.
Pediatric Hospital Medicine 2016, cosponsored by the American Academy of Pediatrics (AAP), the Academic Pediatric Association (APA), and the Society of Hospital Medicine (SHM), took place July 28–31 in Chicago. Didn’t make it? Here are all the news, research, and talking points you need to know.
Shape Your Brain to Avoid Burnout
Presenter: Lisa Zaoutis, MD, FHM
Amid the skyscrapers of the Windy City, Pediatric Hospital Medicine (PHM) 2016 swept into town, bringing with it the denizens of pediatric hospitalist programs across the country. Some 1,150 attendees, composed of hospitalists, PHM program leaders, and advanced-care practitioners, gathered to educate and inspire one another in the care of hospitalized children.
Lisa Zaoutis, MD, FHM, director of the pediatric residency program at The Children’s Hospital of Philadelphia, kicked off the conference with the opening plenary. Initially titled “North Star and Space,” she quickly changed the title to “Changing Our Minds.” Touching on the disconnect between positive experiences that bring physicians into pediatric hospital medicine and negative experiences that often drive behavior, she started with the beginning: the evolution of our brains.
“We are wired toward the negative,” Dr. Zaoutis said. “We are Teflon for positive experiences and Velcro for negative experiences.”
Delving deeper into neuroanatomy, Dr. Zaoutis spoke of “amygdala hijack,” where chronic stress inherent to the professional lives of pediatric hospitalists leads to anxiety responses that are faster, more robust, and more easily triggered.
But all is not lost, Dr. Zaoutis noted, as our brains are more plastic than previously known. The “neural Darwinism” of our brains, she said, leads to epigenetic intracellular changes, more sensitive synapses, improved blood flow, and even new cells as a result of experience-dependent neuroplasticity. For example, London taxi drivers have thicker white matter in their hippocampus as a result of learning London city streets, and mindfulness meditators have thicker gray matter in regions that control attention and self-insight.
Key Takeaways
The lesson for pediatric hospitalists, according Dr. Zaoutis, is that you can shape your brain for greater joy.
“Consciously choose activities” that counter our evolutionary negativity bias, Dr. Zaoutis said.
Here’s how to do it:
- Have a positive experience. (You can create one or retrieve a prior one.)
- Enrich it and install it by dwelling on it for at least 15–30 seconds.
- Absorb it into your body, which may require somatizing it. (Dr. Zaoutis presses her hand into her chest to aid in this.)
Further, spread this to your group by the old medical training technique of “see one, do one, teach one.” See if you can start your sign-out with the best thing that happened to you in the week. Most important, start with observing yourself.
Weijen Chang, MD, SFHM, is pediatric editor of The Hospitalist. He is associate clinical professor of medicine and pediatrics at the University of California, San Diego (UCSD) School of Medicine and a hospitalist at both UCSD Medical Center and Rady Children’s Hospital. Send comments and questions to [email protected].
New AAP Guideline on Evaluating, Managing Febrile Infants
Presenter: Kenneth Roberts, MD
One of PHM16’s most highly attended sessions was an update on the anticipated AAP guidelines for febrile infants ages 7–90 days. The updated guidelines stress the need to separate individual components of serious bacterial infections (UTI, bacteremia, and meningitis) as the incidence and clinical course can vary greatly in this population.
The inclusion criteria for infants for this upcoming algorithm require an infant to be full-term (37–43 weeks’ gestation), aged 7–90 days, well-appearing, and presenting with a temperature of 38°C.
Exclusion criteria include perinatal/prenatal/neonatal maternal fever, infection, or antimicrobial treatment; the presence of any evident infection; being technology-dependent; and the presence of congenital anomalies.
The updated guideline will aim to stratify management by ages 7–28 days, 29–60 days, and 61–90 days to provide the most appropriate and directed treatment.
It will also include a role for inflammatory markers and allow for a “kinder, gentler” approach, including withholding certain treatments and procedures if infants are at low risk of infection. An active need for observation may be appropriate for certain infants as well. These guidelines should be tailored for individual patients to provide the best care possible while minimizing risk.
Key Takeaway
An updated AAP guideline algorithm for the management of well-appearing febrile infants ages 7–28 days, 29–60 days, and 60–90 days will be coming in the near future. It will help standardize care in this population but should not be used as a substitute for clinical judgment.
Chandani DeZure, MD, FAAP, is a pediatric hospitalist at Children’s National Health System and instructor of pediatrics at George Washington University School of Medicine & Health Sciences in Washington, D.C.
Promoting, Teaching Pediatric High-Value Care
Presenters: Lauren L. Walker, MD, FAAP; Alan Schroeder, MD, FAAP; Michael
Tchou, MD, FAAP; Jimmy Beck, MD, MEd; Lisa Herrmann, MD; Ricardo Quinonez, MD, FAAP
Pediatric hospitalists gathered to attend a fruitful discussion on not only how to change our way of thinking but also how to feed it forward to our trainees. The barriers to promoting and teaching high-value care are plenty and essentially universal to academic and community sites: We have had no formal teaching, there is cultural resistance, and there is lack of transparency on costs and charges.
The questions we perhaps should be asking ourselves, our trainees, and our families are:
- “What matters?” instead of “What’s the matter?”
- “Does that test benefit the patient? What are the harms of the test?” instead of “Will that test change our management?”
There is still a long way to go to move the pendulum to the side of value-based practice and teaching. There is still controversy on how and whether cost should be discussed with the family. Cost is more than just monetary value; family anxiety and patient harm may resonate more with families as we perfect our skills in shared decision making.
Key Takeaway
This serves as an exciting time to unite and better our understanding about why we do what we do and deliberately think about downstream effects. High-value care curriculum for medical students, residents, fellows, and even faculty is an area ripe for further research.
Akshata Hopkins, MD, FAAP, is an academic hospitalist at Johns Hopkins All Children’s Hospital in St. Petersburg, Fla.
How to Design, Improve Educational Programs at Community Hospitals
Presenters: Christopher Russo, MD, FAAP; Laura Hodo, MD, and Lauren Wilson, MD
One session at PHM16 focused on ways to design and improve education within community hospital settings. It was done via a didactic session, breakout groups, and an electronic assessment tool. Facilitators included the workshop leaders and co-leaders along with current PHM fellows and educators from community and academic settings.
During the didactic session, a general background of the importance of education during times of increasing academic and community site affiliations was discussed. This included the strengths of community hospitals for learners such as “appropriate learner autonomy,” “exposure to different career paths,” and “transfer decision making.”
Some of the challenges discussed in regard to developing an educational structure in community settings included:
1. Logistics
- Making the case for education
- Legal framework (e.g., affiliation agreements, liability)
- Finances (e.g., GME funding)
- Paperwork burden (e.g., licensing, credentialing)
2. Learning environment
- Complementing clinical work with materials
- Autonomy/supervision balancing
- Developing clinical teachers
The didactic session also reviewed the six steps for curriculum development: general needs assessment, targeted needs assessment, goals and objectives, educational strategies, implementation, and evaluation/feedback. Each of these was described in further detail with relevant examples.
Groups were broken into small groups based on four learner types: medical students, family medicine residents, pediatric residents, and PHM fellows. Within each group, a “program development matrix” was distributed to assess the support from leadership and logistics within each setting. Each one of these was separated into subgroups such as credentialing, financial support, housing/travel, and preceptor recruitment.
A separate “curriculum development matrix” was used during breakout groups that focused on curriculum development. This matrix was broken into three areas: educational strategies, implementation, and evaluation/feedback. These were further broken down into subgroups such as content, identifying resources, and remediation planning. The group was asked to determine short- and long-term goals with action steps for both of these matrix subgroups.
Key Takeaway
Overall, the session presented a structured way of assessing the educational environment for learners in community settings. It gave tangible tools for sites that wish to develop or improve their current educational framework.
Francisco Alvarez, MD, FAAP, is a pediatric hospitalist and director of the Children’s National Health System Community Hospital Services in Washington, D.C.
Tips on Meeting Needs of Children with a Medical Complexity
Presenters: Mary L. Ehlenbach, MD, FAAP; Megan Z. Cardoso, MD, FAAP, and Christina Kleier, ARNP, PNP
This session at PHM16 was focused on logistical tips on how to build a pediatric complex-care program. Presenters opened with a discussion on how to define children with medical complexity. This involved reviewing different methods, including using research-based aggregation of ICD-10 codes, relying on referral from both families and other providers, and identifying patients by consumption of hospital resources. The presentation continued by highlighting that although medically complex children make up only a small percentage of the overall population of children, they account for about one-third of healthcare spending. Because of advances in technology and medicine, this group of children is growing in numbers. It currently makes up about 10% of all pediatric admissions.
Key Takeaways
1. Children with medical complexity are a growing population on which a large proportion of healthcare resources are utilized. A program dedicated to serving the needs of this population may be helpful in reducing costs and improving the patient and family experience during hospitalizations.
2. When working to initiate a complex-care program:
- Set clear guidelines about which children the program is intended to serve and in what capacity it will function.
- Ensure the team composition is sustainable and meets the needs of the patients.
- Aggregate data about if the program is helping. This may be difficult to quantify since these are mostly qualitative measures.
- Include team members who are nonclinical to aid in improving hospital revenue and highlighting program benefits to the institution.
Margaret Rush, MD, is a hospitalist fellow at Children’s National Medical Center in Washington, D.C.
A Picture Is Worth a Thousand Words
Presenter: Kenneth Roberts, MD
PHM16’s “Visual Diagnosis: Signs and Why They Matter” session was a review of case presentations in which visual clues were vital to establishing a diagnosis. Though much of the content was presented with pictures, the emphasis was placed on the importance of correct diagnosis to avoid both misdiagnoses or overdiagnoses and the potential harm that may result from inappropriate treatment. This may also translate into poor utilization of resources and significant financial burden that can result from the unnecessary hospitalization of a patient.
Many of the presented cases highlighted examples in which there was extensive workup, hospitalization, subspecialty evaluation, and even incorrect treatment of patients.
In other instances, such as with Henoch-Schonlein purpura, Waardenburg syndrome, or McCune-Albright syndrome, the correct diagnosis was necessary to help guide management and future treatment, including subspecialty evaluation.
Key Takeaway
Many diseases with visual presentations will have a benign course and require no treatment. Acknowledging this is important in providing reassurance to a family that may be very anxious over the physical appearance of their child.
This session underscores the need for experience and exposure to various signs not only with rare medical conditions but also in common illnesses such as Kawasaki disease and scarlet fever that may present similarly.
Chandani DeZure, MD, FAAP, is a pediatric hospitalist at Children’s National Health System and instructor of pediatrics at George Washington University School of Medicine & Health Sciences in Washington, D.C.
Pediatric Hospital Medicine 2016, cosponsored by the American Academy of Pediatrics (AAP), the Academic Pediatric Association (APA), and the Society of Hospital Medicine (SHM), took place July 28–31 in Chicago. Didn’t make it? Here are all the news, research, and talking points you need to know.
Shape Your Brain to Avoid Burnout
Presenter: Lisa Zaoutis, MD, FHM
Amid the skyscrapers of the Windy City, Pediatric Hospital Medicine (PHM) 2016 swept into town, bringing with it the denizens of pediatric hospitalist programs across the country. Some 1,150 attendees, composed of hospitalists, PHM program leaders, and advanced-care practitioners, gathered to educate and inspire one another in the care of hospitalized children.
Lisa Zaoutis, MD, FHM, director of the pediatric residency program at The Children’s Hospital of Philadelphia, kicked off the conference with the opening plenary. Initially titled “North Star and Space,” she quickly changed the title to “Changing Our Minds.” Touching on the disconnect between positive experiences that bring physicians into pediatric hospital medicine and negative experiences that often drive behavior, she started with the beginning: the evolution of our brains.
“We are wired toward the negative,” Dr. Zaoutis said. “We are Teflon for positive experiences and Velcro for negative experiences.”
Delving deeper into neuroanatomy, Dr. Zaoutis spoke of “amygdala hijack,” where chronic stress inherent to the professional lives of pediatric hospitalists leads to anxiety responses that are faster, more robust, and more easily triggered.
But all is not lost, Dr. Zaoutis noted, as our brains are more plastic than previously known. The “neural Darwinism” of our brains, she said, leads to epigenetic intracellular changes, more sensitive synapses, improved blood flow, and even new cells as a result of experience-dependent neuroplasticity. For example, London taxi drivers have thicker white matter in their hippocampus as a result of learning London city streets, and mindfulness meditators have thicker gray matter in regions that control attention and self-insight.
Key Takeaways
The lesson for pediatric hospitalists, according Dr. Zaoutis, is that you can shape your brain for greater joy.
“Consciously choose activities” that counter our evolutionary negativity bias, Dr. Zaoutis said.
Here’s how to do it:
- Have a positive experience. (You can create one or retrieve a prior one.)
- Enrich it and install it by dwelling on it for at least 15–30 seconds.
- Absorb it into your body, which may require somatizing it. (Dr. Zaoutis presses her hand into her chest to aid in this.)
Further, spread this to your group by the old medical training technique of “see one, do one, teach one.” See if you can start your sign-out with the best thing that happened to you in the week. Most important, start with observing yourself.
Weijen Chang, MD, SFHM, is pediatric editor of The Hospitalist. He is associate clinical professor of medicine and pediatrics at the University of California, San Diego (UCSD) School of Medicine and a hospitalist at both UCSD Medical Center and Rady Children’s Hospital. Send comments and questions to [email protected].
New AAP Guideline on Evaluating, Managing Febrile Infants
Presenter: Kenneth Roberts, MD
One of PHM16’s most highly attended sessions was an update on the anticipated AAP guidelines for febrile infants ages 7–90 days. The updated guidelines stress the need to separate individual components of serious bacterial infections (UTI, bacteremia, and meningitis) as the incidence and clinical course can vary greatly in this population.
The inclusion criteria for infants for this upcoming algorithm require an infant to be full-term (37–43 weeks’ gestation), aged 7–90 days, well-appearing, and presenting with a temperature of 38°C.
Exclusion criteria include perinatal/prenatal/neonatal maternal fever, infection, or antimicrobial treatment; the presence of any evident infection; being technology-dependent; and the presence of congenital anomalies.
The updated guideline will aim to stratify management by ages 7–28 days, 29–60 days, and 61–90 days to provide the most appropriate and directed treatment.
It will also include a role for inflammatory markers and allow for a “kinder, gentler” approach, including withholding certain treatments and procedures if infants are at low risk of infection. An active need for observation may be appropriate for certain infants as well. These guidelines should be tailored for individual patients to provide the best care possible while minimizing risk.
Key Takeaway
An updated AAP guideline algorithm for the management of well-appearing febrile infants ages 7–28 days, 29–60 days, and 60–90 days will be coming in the near future. It will help standardize care in this population but should not be used as a substitute for clinical judgment.
Chandani DeZure, MD, FAAP, is a pediatric hospitalist at Children’s National Health System and instructor of pediatrics at George Washington University School of Medicine & Health Sciences in Washington, D.C.
Promoting, Teaching Pediatric High-Value Care
Presenters: Lauren L. Walker, MD, FAAP; Alan Schroeder, MD, FAAP; Michael
Tchou, MD, FAAP; Jimmy Beck, MD, MEd; Lisa Herrmann, MD; Ricardo Quinonez, MD, FAAP
Pediatric hospitalists gathered to attend a fruitful discussion on not only how to change our way of thinking but also how to feed it forward to our trainees. The barriers to promoting and teaching high-value care are plenty and essentially universal to academic and community sites: We have had no formal teaching, there is cultural resistance, and there is lack of transparency on costs and charges.
The questions we perhaps should be asking ourselves, our trainees, and our families are:
- “What matters?” instead of “What’s the matter?”
- “Does that test benefit the patient? What are the harms of the test?” instead of “Will that test change our management?”
There is still a long way to go to move the pendulum to the side of value-based practice and teaching. There is still controversy on how and whether cost should be discussed with the family. Cost is more than just monetary value; family anxiety and patient harm may resonate more with families as we perfect our skills in shared decision making.
Key Takeaway
This serves as an exciting time to unite and better our understanding about why we do what we do and deliberately think about downstream effects. High-value care curriculum for medical students, residents, fellows, and even faculty is an area ripe for further research.
Akshata Hopkins, MD, FAAP, is an academic hospitalist at Johns Hopkins All Children’s Hospital in St. Petersburg, Fla.
How to Design, Improve Educational Programs at Community Hospitals
Presenters: Christopher Russo, MD, FAAP; Laura Hodo, MD, and Lauren Wilson, MD
One session at PHM16 focused on ways to design and improve education within community hospital settings. It was done via a didactic session, breakout groups, and an electronic assessment tool. Facilitators included the workshop leaders and co-leaders along with current PHM fellows and educators from community and academic settings.
During the didactic session, a general background of the importance of education during times of increasing academic and community site affiliations was discussed. This included the strengths of community hospitals for learners such as “appropriate learner autonomy,” “exposure to different career paths,” and “transfer decision making.”
Some of the challenges discussed in regard to developing an educational structure in community settings included:
1. Logistics
- Making the case for education
- Legal framework (e.g., affiliation agreements, liability)
- Finances (e.g., GME funding)
- Paperwork burden (e.g., licensing, credentialing)
2. Learning environment
- Complementing clinical work with materials
- Autonomy/supervision balancing
- Developing clinical teachers
The didactic session also reviewed the six steps for curriculum development: general needs assessment, targeted needs assessment, goals and objectives, educational strategies, implementation, and evaluation/feedback. Each of these was described in further detail with relevant examples.
Groups were broken into small groups based on four learner types: medical students, family medicine residents, pediatric residents, and PHM fellows. Within each group, a “program development matrix” was distributed to assess the support from leadership and logistics within each setting. Each one of these was separated into subgroups such as credentialing, financial support, housing/travel, and preceptor recruitment.
A separate “curriculum development matrix” was used during breakout groups that focused on curriculum development. This matrix was broken into three areas: educational strategies, implementation, and evaluation/feedback. These were further broken down into subgroups such as content, identifying resources, and remediation planning. The group was asked to determine short- and long-term goals with action steps for both of these matrix subgroups.
Key Takeaway
Overall, the session presented a structured way of assessing the educational environment for learners in community settings. It gave tangible tools for sites that wish to develop or improve their current educational framework.
Francisco Alvarez, MD, FAAP, is a pediatric hospitalist and director of the Children’s National Health System Community Hospital Services in Washington, D.C.
Tips on Meeting Needs of Children with a Medical Complexity
Presenters: Mary L. Ehlenbach, MD, FAAP; Megan Z. Cardoso, MD, FAAP, and Christina Kleier, ARNP, PNP
This session at PHM16 was focused on logistical tips on how to build a pediatric complex-care program. Presenters opened with a discussion on how to define children with medical complexity. This involved reviewing different methods, including using research-based aggregation of ICD-10 codes, relying on referral from both families and other providers, and identifying patients by consumption of hospital resources. The presentation continued by highlighting that although medically complex children make up only a small percentage of the overall population of children, they account for about one-third of healthcare spending. Because of advances in technology and medicine, this group of children is growing in numbers. It currently makes up about 10% of all pediatric admissions.
Key Takeaways
1. Children with medical complexity are a growing population on which a large proportion of healthcare resources are utilized. A program dedicated to serving the needs of this population may be helpful in reducing costs and improving the patient and family experience during hospitalizations.
2. When working to initiate a complex-care program:
- Set clear guidelines about which children the program is intended to serve and in what capacity it will function.
- Ensure the team composition is sustainable and meets the needs of the patients.
- Aggregate data about if the program is helping. This may be difficult to quantify since these are mostly qualitative measures.
- Include team members who are nonclinical to aid in improving hospital revenue and highlighting program benefits to the institution.
Margaret Rush, MD, is a hospitalist fellow at Children’s National Medical Center in Washington, D.C.
A Picture Is Worth a Thousand Words
Presenter: Kenneth Roberts, MD
PHM16’s “Visual Diagnosis: Signs and Why They Matter” session was a review of case presentations in which visual clues were vital to establishing a diagnosis. Though much of the content was presented with pictures, the emphasis was placed on the importance of correct diagnosis to avoid both misdiagnoses or overdiagnoses and the potential harm that may result from inappropriate treatment. This may also translate into poor utilization of resources and significant financial burden that can result from the unnecessary hospitalization of a patient.
Many of the presented cases highlighted examples in which there was extensive workup, hospitalization, subspecialty evaluation, and even incorrect treatment of patients.
In other instances, such as with Henoch-Schonlein purpura, Waardenburg syndrome, or McCune-Albright syndrome, the correct diagnosis was necessary to help guide management and future treatment, including subspecialty evaluation.
Key Takeaway
Many diseases with visual presentations will have a benign course and require no treatment. Acknowledging this is important in providing reassurance to a family that may be very anxious over the physical appearance of their child.
This session underscores the need for experience and exposure to various signs not only with rare medical conditions but also in common illnesses such as Kawasaki disease and scarlet fever that may present similarly.
Chandani DeZure, MD, FAAP, is a pediatric hospitalist at Children’s National Health System and instructor of pediatrics at George Washington University School of Medicine & Health Sciences in Washington, D.C.
Docetaxel-Induced Stevens-Johnson Syndrome
Abstract: Docetaxel is a commonly used chemotherapeutic agent used in a variety of cancer treatment plans. We present a case of apparent docetaxel-induced Stevens-Johnson syndrome (SJS) in a patient recently treated with docetaxel for metastatic prostate cancer. This medication is not classically associated with the development of SJS.
A 63-year-old gentleman with a past medical history of hypertension, hyperlipidemia, and metastatic prostate adenocarcinoma to retroperitoneal lymph nodes, lung, and bone presented to the emergency room with a one-week history of a rash affecting his hands, feet, back, and chest. It developed into blisters that later ruptured. it was especially painful in the hands and feet. He also reported red eyes and difficulty eating for a week.
Vital Signs: Temp 36.4°C, 83/min, R.R 12/min, BP 121/60 mm Hg. Physical examination of the patient revealed a severe rash covering less than 30% of the body, oral ulcers, and conjunctival redness.
The patient’s cancer treatment regimen included active hormonal therapy with leuprolide and active chemotherapy with docetaxel. He had received 2 cycles of docetaxel therapy (75 mg/m2), with the last dose of docetaxel received 2 weeks prior to presentation.
Treatment of the patient included intravenous fluid replacement, prednisone, piperacillin/tazobactam, ondansetron, and morphine. The skin lesions were kept clean with regular dressing changes.
Because our searches for an established association between SJS and docetaxel were in vain, we elected to obtain a punch biopsy of the lesions to establish pathological evidence of our diagnosis. The punch biopsies were obtained from the edges of lesions on the left forearm and left medial foot. Light microscopy confirmed the diagnosis of SJS (clinical pics, pathology slides are available).
Discussion: Docetaxel is a widely used chemotherapeutic agent in the treatment of breast, lung, prostate and other cancers. The classically known side effects of docetaxel therapy include alopecia, pancytopenia, hepatotoxicity, nausea, vomiting, and diarrhea. A number of popular clinical pharmacology resources do not include Stevens-Johnson syndrome (SJS) as a known complication of docetaxel chemotherapy. However, the current case and the cases written by a handful of other clinicians may provide clinical evidence that docetaxel therapy is associated with the development of this potentially life-threatening dermatologic condition.
Abstract: Docetaxel is a commonly used chemotherapeutic agent used in a variety of cancer treatment plans. We present a case of apparent docetaxel-induced Stevens-Johnson syndrome (SJS) in a patient recently treated with docetaxel for metastatic prostate cancer. This medication is not classically associated with the development of SJS.
A 63-year-old gentleman with a past medical history of hypertension, hyperlipidemia, and metastatic prostate adenocarcinoma to retroperitoneal lymph nodes, lung, and bone presented to the emergency room with a one-week history of a rash affecting his hands, feet, back, and chest. It developed into blisters that later ruptured. it was especially painful in the hands and feet. He also reported red eyes and difficulty eating for a week.
Vital Signs: Temp 36.4°C, 83/min, R.R 12/min, BP 121/60 mm Hg. Physical examination of the patient revealed a severe rash covering less than 30% of the body, oral ulcers, and conjunctival redness.
The patient’s cancer treatment regimen included active hormonal therapy with leuprolide and active chemotherapy with docetaxel. He had received 2 cycles of docetaxel therapy (75 mg/m2), with the last dose of docetaxel received 2 weeks prior to presentation.
Treatment of the patient included intravenous fluid replacement, prednisone, piperacillin/tazobactam, ondansetron, and morphine. The skin lesions were kept clean with regular dressing changes.
Because our searches for an established association between SJS and docetaxel were in vain, we elected to obtain a punch biopsy of the lesions to establish pathological evidence of our diagnosis. The punch biopsies were obtained from the edges of lesions on the left forearm and left medial foot. Light microscopy confirmed the diagnosis of SJS (clinical pics, pathology slides are available).
Discussion: Docetaxel is a widely used chemotherapeutic agent in the treatment of breast, lung, prostate and other cancers. The classically known side effects of docetaxel therapy include alopecia, pancytopenia, hepatotoxicity, nausea, vomiting, and diarrhea. A number of popular clinical pharmacology resources do not include Stevens-Johnson syndrome (SJS) as a known complication of docetaxel chemotherapy. However, the current case and the cases written by a handful of other clinicians may provide clinical evidence that docetaxel therapy is associated with the development of this potentially life-threatening dermatologic condition.
Abstract: Docetaxel is a commonly used chemotherapeutic agent used in a variety of cancer treatment plans. We present a case of apparent docetaxel-induced Stevens-Johnson syndrome (SJS) in a patient recently treated with docetaxel for metastatic prostate cancer. This medication is not classically associated with the development of SJS.
A 63-year-old gentleman with a past medical history of hypertension, hyperlipidemia, and metastatic prostate adenocarcinoma to retroperitoneal lymph nodes, lung, and bone presented to the emergency room with a one-week history of a rash affecting his hands, feet, back, and chest. It developed into blisters that later ruptured. it was especially painful in the hands and feet. He also reported red eyes and difficulty eating for a week.
Vital Signs: Temp 36.4°C, 83/min, R.R 12/min, BP 121/60 mm Hg. Physical examination of the patient revealed a severe rash covering less than 30% of the body, oral ulcers, and conjunctival redness.
The patient’s cancer treatment regimen included active hormonal therapy with leuprolide and active chemotherapy with docetaxel. He had received 2 cycles of docetaxel therapy (75 mg/m2), with the last dose of docetaxel received 2 weeks prior to presentation.
Treatment of the patient included intravenous fluid replacement, prednisone, piperacillin/tazobactam, ondansetron, and morphine. The skin lesions were kept clean with regular dressing changes.
Because our searches for an established association between SJS and docetaxel were in vain, we elected to obtain a punch biopsy of the lesions to establish pathological evidence of our diagnosis. The punch biopsies were obtained from the edges of lesions on the left forearm and left medial foot. Light microscopy confirmed the diagnosis of SJS (clinical pics, pathology slides are available).
Discussion: Docetaxel is a widely used chemotherapeutic agent in the treatment of breast, lung, prostate and other cancers. The classically known side effects of docetaxel therapy include alopecia, pancytopenia, hepatotoxicity, nausea, vomiting, and diarrhea. A number of popular clinical pharmacology resources do not include Stevens-Johnson syndrome (SJS) as a known complication of docetaxel chemotherapy. However, the current case and the cases written by a handful of other clinicians may provide clinical evidence that docetaxel therapy is associated with the development of this potentially life-threatening dermatologic condition.
The Use of IVC Filters in Cancer Patients: A 15-Year Experience at a Single Veterans Affairs Medical Center
Background: Cancer and chemotherapy are both associated with an increased risk of thrombosis. Venous thromboembolism (VTE) is the leading cause of death in cancer patients. Inferior vena cava (IVC) filter use is recommended in patients with VTE and contraindication to anticoagulation (AC) or recurrent VTE despite treatment. The use of IVC filters in cancer patients outside the Veterans Affairs Medical Center (VAMC) has been described; no such study exists within the VAMC.
Purpose: Descriptive analysis of IVC filters use in cancer patients at the Washington DC VAMC.
Methods: Retrospective study utilizing data from the Washington DC VA Cancer Registry and the electronic health records (EHR). Current Procedural Terminology (CPT) codes for IVC filter placement were used to identify subjects in the cancer registry who received an IVC filter. Demographics, cancer date of diagnosis, VTE date of diagnosis, type of filter, indication, placement date, procedural complications, and AC medication use at time of filter placement were collected. Cancer subjects (n = 6,678) were identified from October 1999 – May 2015 and 64 patients met inclusion criteria.
Data Analysis: Percentages were calculated for the aforementioned data points.
Results: Characteristics of the 64 cancer patients with IVC filter placement include: 100% male, 75% black or African American, 15.6% white, 1.6% Hispanic or Latino, 7.8% unknown. The average age at cancer diagnosis was 65.3 years. Date of cancer diagnosis to VTE diagnosis is currently being analyzed.
VTE diagnosis with an immediate contraindication to AC medication led the list of IVC filter indications (59%). An additional 16% of patients received filters due to subsequent development of AC contraindication. The remaining patients (25%) received filters for prophylaxis, AC failure, or in combination with AC medications. For subjects whose filter type was captured from chart review, 51.8% were given permanent filters versus 48.1% who were given retrievable filters. Overall, there were no complications associated with the procedure.
Implications: Contraindication to immediate AC is the leading indication for IVC filter use in our cancer patients with VTE, consistent with current guidelines. Overall, rates of permanent versus retrievable filter use were similar.
Background: Cancer and chemotherapy are both associated with an increased risk of thrombosis. Venous thromboembolism (VTE) is the leading cause of death in cancer patients. Inferior vena cava (IVC) filter use is recommended in patients with VTE and contraindication to anticoagulation (AC) or recurrent VTE despite treatment. The use of IVC filters in cancer patients outside the Veterans Affairs Medical Center (VAMC) has been described; no such study exists within the VAMC.
Purpose: Descriptive analysis of IVC filters use in cancer patients at the Washington DC VAMC.
Methods: Retrospective study utilizing data from the Washington DC VA Cancer Registry and the electronic health records (EHR). Current Procedural Terminology (CPT) codes for IVC filter placement were used to identify subjects in the cancer registry who received an IVC filter. Demographics, cancer date of diagnosis, VTE date of diagnosis, type of filter, indication, placement date, procedural complications, and AC medication use at time of filter placement were collected. Cancer subjects (n = 6,678) were identified from October 1999 – May 2015 and 64 patients met inclusion criteria.
Data Analysis: Percentages were calculated for the aforementioned data points.
Results: Characteristics of the 64 cancer patients with IVC filter placement include: 100% male, 75% black or African American, 15.6% white, 1.6% Hispanic or Latino, 7.8% unknown. The average age at cancer diagnosis was 65.3 years. Date of cancer diagnosis to VTE diagnosis is currently being analyzed.
VTE diagnosis with an immediate contraindication to AC medication led the list of IVC filter indications (59%). An additional 16% of patients received filters due to subsequent development of AC contraindication. The remaining patients (25%) received filters for prophylaxis, AC failure, or in combination with AC medications. For subjects whose filter type was captured from chart review, 51.8% were given permanent filters versus 48.1% who were given retrievable filters. Overall, there were no complications associated with the procedure.
Implications: Contraindication to immediate AC is the leading indication for IVC filter use in our cancer patients with VTE, consistent with current guidelines. Overall, rates of permanent versus retrievable filter use were similar.
Background: Cancer and chemotherapy are both associated with an increased risk of thrombosis. Venous thromboembolism (VTE) is the leading cause of death in cancer patients. Inferior vena cava (IVC) filter use is recommended in patients with VTE and contraindication to anticoagulation (AC) or recurrent VTE despite treatment. The use of IVC filters in cancer patients outside the Veterans Affairs Medical Center (VAMC) has been described; no such study exists within the VAMC.
Purpose: Descriptive analysis of IVC filters use in cancer patients at the Washington DC VAMC.
Methods: Retrospective study utilizing data from the Washington DC VA Cancer Registry and the electronic health records (EHR). Current Procedural Terminology (CPT) codes for IVC filter placement were used to identify subjects in the cancer registry who received an IVC filter. Demographics, cancer date of diagnosis, VTE date of diagnosis, type of filter, indication, placement date, procedural complications, and AC medication use at time of filter placement were collected. Cancer subjects (n = 6,678) were identified from October 1999 – May 2015 and 64 patients met inclusion criteria.
Data Analysis: Percentages were calculated for the aforementioned data points.
Results: Characteristics of the 64 cancer patients with IVC filter placement include: 100% male, 75% black or African American, 15.6% white, 1.6% Hispanic or Latino, 7.8% unknown. The average age at cancer diagnosis was 65.3 years. Date of cancer diagnosis to VTE diagnosis is currently being analyzed.
VTE diagnosis with an immediate contraindication to AC medication led the list of IVC filter indications (59%). An additional 16% of patients received filters due to subsequent development of AC contraindication. The remaining patients (25%) received filters for prophylaxis, AC failure, or in combination with AC medications. For subjects whose filter type was captured from chart review, 51.8% were given permanent filters versus 48.1% who were given retrievable filters. Overall, there were no complications associated with the procedure.
Implications: Contraindication to immediate AC is the leading indication for IVC filter use in our cancer patients with VTE, consistent with current guidelines. Overall, rates of permanent versus retrievable filter use were similar.
Lean Six Sigma Applied to Tracking Head/Neck Cancer Patients
Purpose: The ENT Clinic will provide safe and quality care to its head/neck cancer (HNC) patients with optimal treatment interventions and cancer surveillance through regularly scheduled follow-up visits by preventing patients from being inadvertently lost to follow-up care.
Background/Problem: ~ 400,000 new cases of HNC are diagnosed and reported each year. A study reported a 47.3% disease recurrence in the first year post-treatment. HNC patients require frequent follow-up care due to the high percentage of potential disease recurrence.
Before activation of a HNC Cancer Surveillance Program a record review in 2012 showed 31.1% of HNC patients in the ENT clinic were lost to follow-up care when appointments were canceled or missed by patients and did not get a rescheduled appointment.
Methods: Vigorous Lean Six Sigma methodological tools were used to carefully assess the problem and to improve outcomes encompassing such tools as root-cause analysis, defining waste barriers, Plan, Do, Study, Act (PDSA).
In Phase I, an Excel spreadsheet was created to manually track and monitor HNC patients for cancer surveillance. Monthly reports thereafter proved that tracking HNC patients using an Excel spreadsheet was successful, and 100% of HNC patients had received follow-up appointments. However, the manual process of tracking HNC patients on an Excel spreadsheet was time consuming with limited functionality.
Phase II – A robust automated electronic identification system was implemented for tracking HNC patients which included additional features that far exceeded the capabilities of manual tracking.
Data Analysis: During the first 8 months of its operation (February 2014 – September 2014) 25 newly diagnosed HNC patients were identified electronically; patients that manual tracking might have missed.
Results: FY15 and FY16 targeted goal was achieved. 100% of HNC patient appointments were recaptured for cancer surveillance that otherwise might have been lost to follow-up using the automated electronic tracking system.
Implications: The automated HNC Dashboard has proved to be a vital tool providing improved access to care. It can be used and customized for tracking other cancer types.
Purpose: The ENT Clinic will provide safe and quality care to its head/neck cancer (HNC) patients with optimal treatment interventions and cancer surveillance through regularly scheduled follow-up visits by preventing patients from being inadvertently lost to follow-up care.
Background/Problem: ~ 400,000 new cases of HNC are diagnosed and reported each year. A study reported a 47.3% disease recurrence in the first year post-treatment. HNC patients require frequent follow-up care due to the high percentage of potential disease recurrence.
Before activation of a HNC Cancer Surveillance Program a record review in 2012 showed 31.1% of HNC patients in the ENT clinic were lost to follow-up care when appointments were canceled or missed by patients and did not get a rescheduled appointment.
Methods: Vigorous Lean Six Sigma methodological tools were used to carefully assess the problem and to improve outcomes encompassing such tools as root-cause analysis, defining waste barriers, Plan, Do, Study, Act (PDSA).
In Phase I, an Excel spreadsheet was created to manually track and monitor HNC patients for cancer surveillance. Monthly reports thereafter proved that tracking HNC patients using an Excel spreadsheet was successful, and 100% of HNC patients had received follow-up appointments. However, the manual process of tracking HNC patients on an Excel spreadsheet was time consuming with limited functionality.
Phase II – A robust automated electronic identification system was implemented for tracking HNC patients which included additional features that far exceeded the capabilities of manual tracking.
Data Analysis: During the first 8 months of its operation (February 2014 – September 2014) 25 newly diagnosed HNC patients were identified electronically; patients that manual tracking might have missed.
Results: FY15 and FY16 targeted goal was achieved. 100% of HNC patient appointments were recaptured for cancer surveillance that otherwise might have been lost to follow-up using the automated electronic tracking system.
Implications: The automated HNC Dashboard has proved to be a vital tool providing improved access to care. It can be used and customized for tracking other cancer types.
Purpose: The ENT Clinic will provide safe and quality care to its head/neck cancer (HNC) patients with optimal treatment interventions and cancer surveillance through regularly scheduled follow-up visits by preventing patients from being inadvertently lost to follow-up care.
Background/Problem: ~ 400,000 new cases of HNC are diagnosed and reported each year. A study reported a 47.3% disease recurrence in the first year post-treatment. HNC patients require frequent follow-up care due to the high percentage of potential disease recurrence.
Before activation of a HNC Cancer Surveillance Program a record review in 2012 showed 31.1% of HNC patients in the ENT clinic were lost to follow-up care when appointments were canceled or missed by patients and did not get a rescheduled appointment.
Methods: Vigorous Lean Six Sigma methodological tools were used to carefully assess the problem and to improve outcomes encompassing such tools as root-cause analysis, defining waste barriers, Plan, Do, Study, Act (PDSA).
In Phase I, an Excel spreadsheet was created to manually track and monitor HNC patients for cancer surveillance. Monthly reports thereafter proved that tracking HNC patients using an Excel spreadsheet was successful, and 100% of HNC patients had received follow-up appointments. However, the manual process of tracking HNC patients on an Excel spreadsheet was time consuming with limited functionality.
Phase II – A robust automated electronic identification system was implemented for tracking HNC patients which included additional features that far exceeded the capabilities of manual tracking.
Data Analysis: During the first 8 months of its operation (February 2014 – September 2014) 25 newly diagnosed HNC patients were identified electronically; patients that manual tracking might have missed.
Results: FY15 and FY16 targeted goal was achieved. 100% of HNC patient appointments were recaptured for cancer surveillance that otherwise might have been lost to follow-up using the automated electronic tracking system.
Implications: The automated HNC Dashboard has proved to be a vital tool providing improved access to care. It can be used and customized for tracking other cancer types.
Implementing Psychosocial Distress Screening at the VA Palo Alto Health Care System: Lessons Learned and Future Directions
Purpose: To create and evaluate psychosocial distress screening procedures for patients diagnosed with cancer at the VA Palo Alto Health Care System (VAPAHCS).
Relevant Background/Problem: A program development and evaluation project was conducted at VAPAHCS in order to implement psychosocial distress screening according to the Cancer Program Standards (Standard 3.2) of the American College of Surgeon’s Commission on Cancer.
Methods: Veterans recently diagnosed with cancer were screened for distress by a psychologist, social worker, or psychology trainee during one of their regularly scheduled oncology appointments using the National Comprehensive Cancer Network Distress Thermometer and Symptom Checklist. Data were abstracted from CPRS for distress screens conducted from January 2015-April 2016. The number of new cancer cases at VAPAHCS during the same time period was obtained from the cancer registry.
Data Analysis: The number of distress screens conducted was compared to cancer registry data of total new cases of cancer. Descriptive statistics were used to explore the level of distress and frequency of biopsychosocial symptoms endorsed by veterans in this sample.
Results: A total of 372 veterans completed distress screening during the program evaluation period. During the same time period, 920 veterans were newly diagnosed with cancer at VAPAHCS. Average level of distress was 3.3 (0 = no distress, 10 = extreme distress). Forty-one percent (n = 152) of veterans scored above the clinical cut-off for significant distress (4/10). The most commonly endorsed symptoms were fatigue (n = 97; 26.0%), worry (n = 93; 25.0%), pain (n = 92; 24.7%), sleep (n = 78; 21.0%), and skin dry/itchy (n = 78; 21.0%).
Implications: The discrepancy between number of new cases of cancer and number of distress screenings conducted during the program evaluation period suggests that modifications to current procedures are necessary to ensure that all cancer patients at VAPAHCS are screened for distress. Over 40% of veterans screened endorsed clinically significant levels of distress and over 20% of veterans endorsed problems with fatigue, worry, pain, sleep and/or skin issues. Future directions for care include introducing psychosocial interventions in the oncology clinic to reduce distress and cope with commonly reported symptoms/side effects.
Purpose: To create and evaluate psychosocial distress screening procedures for patients diagnosed with cancer at the VA Palo Alto Health Care System (VAPAHCS).
Relevant Background/Problem: A program development and evaluation project was conducted at VAPAHCS in order to implement psychosocial distress screening according to the Cancer Program Standards (Standard 3.2) of the American College of Surgeon’s Commission on Cancer.
Methods: Veterans recently diagnosed with cancer were screened for distress by a psychologist, social worker, or psychology trainee during one of their regularly scheduled oncology appointments using the National Comprehensive Cancer Network Distress Thermometer and Symptom Checklist. Data were abstracted from CPRS for distress screens conducted from January 2015-April 2016. The number of new cancer cases at VAPAHCS during the same time period was obtained from the cancer registry.
Data Analysis: The number of distress screens conducted was compared to cancer registry data of total new cases of cancer. Descriptive statistics were used to explore the level of distress and frequency of biopsychosocial symptoms endorsed by veterans in this sample.
Results: A total of 372 veterans completed distress screening during the program evaluation period. During the same time period, 920 veterans were newly diagnosed with cancer at VAPAHCS. Average level of distress was 3.3 (0 = no distress, 10 = extreme distress). Forty-one percent (n = 152) of veterans scored above the clinical cut-off for significant distress (4/10). The most commonly endorsed symptoms were fatigue (n = 97; 26.0%), worry (n = 93; 25.0%), pain (n = 92; 24.7%), sleep (n = 78; 21.0%), and skin dry/itchy (n = 78; 21.0%).
Implications: The discrepancy between number of new cases of cancer and number of distress screenings conducted during the program evaluation period suggests that modifications to current procedures are necessary to ensure that all cancer patients at VAPAHCS are screened for distress. Over 40% of veterans screened endorsed clinically significant levels of distress and over 20% of veterans endorsed problems with fatigue, worry, pain, sleep and/or skin issues. Future directions for care include introducing psychosocial interventions in the oncology clinic to reduce distress and cope with commonly reported symptoms/side effects.
Purpose: To create and evaluate psychosocial distress screening procedures for patients diagnosed with cancer at the VA Palo Alto Health Care System (VAPAHCS).
Relevant Background/Problem: A program development and evaluation project was conducted at VAPAHCS in order to implement psychosocial distress screening according to the Cancer Program Standards (Standard 3.2) of the American College of Surgeon’s Commission on Cancer.
Methods: Veterans recently diagnosed with cancer were screened for distress by a psychologist, social worker, or psychology trainee during one of their regularly scheduled oncology appointments using the National Comprehensive Cancer Network Distress Thermometer and Symptom Checklist. Data were abstracted from CPRS for distress screens conducted from January 2015-April 2016. The number of new cancer cases at VAPAHCS during the same time period was obtained from the cancer registry.
Data Analysis: The number of distress screens conducted was compared to cancer registry data of total new cases of cancer. Descriptive statistics were used to explore the level of distress and frequency of biopsychosocial symptoms endorsed by veterans in this sample.
Results: A total of 372 veterans completed distress screening during the program evaluation period. During the same time period, 920 veterans were newly diagnosed with cancer at VAPAHCS. Average level of distress was 3.3 (0 = no distress, 10 = extreme distress). Forty-one percent (n = 152) of veterans scored above the clinical cut-off for significant distress (4/10). The most commonly endorsed symptoms were fatigue (n = 97; 26.0%), worry (n = 93; 25.0%), pain (n = 92; 24.7%), sleep (n = 78; 21.0%), and skin dry/itchy (n = 78; 21.0%).
Implications: The discrepancy between number of new cases of cancer and number of distress screenings conducted during the program evaluation period suggests that modifications to current procedures are necessary to ensure that all cancer patients at VAPAHCS are screened for distress. Over 40% of veterans screened endorsed clinically significant levels of distress and over 20% of veterans endorsed problems with fatigue, worry, pain, sleep and/or skin issues. Future directions for care include introducing psychosocial interventions in the oncology clinic to reduce distress and cope with commonly reported symptoms/side effects.
Use of the Community Needs Assessment (CNA) to Identify Barriers and Improve Access to Cancer Care for Veterans
Purpose: To disseminate information regarding The American College of Surgeons Commission on Cancer (ACOS COC) requirements of Patient Navigation that can be used across VAMCs.
Background: The ACOS COC requires that each facility determine a patient navigation process. The process must focus on a barrier to care identified within a Community Needs Assessment (CNA) that is administered at least every 3 years. From the results of the CNA, a patient navigation process can be developed to address patient, provider, or system barriers to care. These results are also presented to the Cancer Committee (CC) to compile a report summarizing the findings and implementing a plan to improve the quality of cancer care delivered.
Methods: A CNA questionnaire was reviewed by an interdisciplinary group consisting of oncology social worker, oncology psychologist, medical oncologist, survivorship advanced practice nurse, 3 oncology nurse care coordinators and the Cancer Center Program Administrator. The questionnaire was formatted for ease of reading and comprehension. The group presented the CNA questionnaire to the CC for review and approval. The questionnaire was distributed and completed by Veteran’s at varying stages along the cancer trajectory.
Data Analysis: The questionnaire was distributed and completed by 50 Veterans from Feb 2014-Sept 2014. The questionnaires were distributed and collected in the chemotherapy infusion clinic, during outpatient clinic visits, and during the Louis Stokes Cleveland VAMC (LSCVAMC) annual Cancer Fair.
Results: The top rated concern was found to be travel. According the National Cancer Data Base (NCDB) generated in May 2015 shows that from the years of 2007-2012, 34% of Veterans receiving their care at the LSCVAMC traveled between 50-99 miles to receive their cancer care. The data were presented to the CC, and plans were made to further look at travel resources and community services available to our Veterans. A comprehensive report addressing resources was compiled and presented to the CC.
Implications: Identifying and breaking down barriers to transportation is vital to improving access to Veteran’s cancer care.
Purpose: To disseminate information regarding The American College of Surgeons Commission on Cancer (ACOS COC) requirements of Patient Navigation that can be used across VAMCs.
Background: The ACOS COC requires that each facility determine a patient navigation process. The process must focus on a barrier to care identified within a Community Needs Assessment (CNA) that is administered at least every 3 years. From the results of the CNA, a patient navigation process can be developed to address patient, provider, or system barriers to care. These results are also presented to the Cancer Committee (CC) to compile a report summarizing the findings and implementing a plan to improve the quality of cancer care delivered.
Methods: A CNA questionnaire was reviewed by an interdisciplinary group consisting of oncology social worker, oncology psychologist, medical oncologist, survivorship advanced practice nurse, 3 oncology nurse care coordinators and the Cancer Center Program Administrator. The questionnaire was formatted for ease of reading and comprehension. The group presented the CNA questionnaire to the CC for review and approval. The questionnaire was distributed and completed by Veteran’s at varying stages along the cancer trajectory.
Data Analysis: The questionnaire was distributed and completed by 50 Veterans from Feb 2014-Sept 2014. The questionnaires were distributed and collected in the chemotherapy infusion clinic, during outpatient clinic visits, and during the Louis Stokes Cleveland VAMC (LSCVAMC) annual Cancer Fair.
Results: The top rated concern was found to be travel. According the National Cancer Data Base (NCDB) generated in May 2015 shows that from the years of 2007-2012, 34% of Veterans receiving their care at the LSCVAMC traveled between 50-99 miles to receive their cancer care. The data were presented to the CC, and plans were made to further look at travel resources and community services available to our Veterans. A comprehensive report addressing resources was compiled and presented to the CC.
Implications: Identifying and breaking down barriers to transportation is vital to improving access to Veteran’s cancer care.
Purpose: To disseminate information regarding The American College of Surgeons Commission on Cancer (ACOS COC) requirements of Patient Navigation that can be used across VAMCs.
Background: The ACOS COC requires that each facility determine a patient navigation process. The process must focus on a barrier to care identified within a Community Needs Assessment (CNA) that is administered at least every 3 years. From the results of the CNA, a patient navigation process can be developed to address patient, provider, or system barriers to care. These results are also presented to the Cancer Committee (CC) to compile a report summarizing the findings and implementing a plan to improve the quality of cancer care delivered.
Methods: A CNA questionnaire was reviewed by an interdisciplinary group consisting of oncology social worker, oncology psychologist, medical oncologist, survivorship advanced practice nurse, 3 oncology nurse care coordinators and the Cancer Center Program Administrator. The questionnaire was formatted for ease of reading and comprehension. The group presented the CNA questionnaire to the CC for review and approval. The questionnaire was distributed and completed by Veteran’s at varying stages along the cancer trajectory.
Data Analysis: The questionnaire was distributed and completed by 50 Veterans from Feb 2014-Sept 2014. The questionnaires were distributed and collected in the chemotherapy infusion clinic, during outpatient clinic visits, and during the Louis Stokes Cleveland VAMC (LSCVAMC) annual Cancer Fair.
Results: The top rated concern was found to be travel. According the National Cancer Data Base (NCDB) generated in May 2015 shows that from the years of 2007-2012, 34% of Veterans receiving their care at the LSCVAMC traveled between 50-99 miles to receive their cancer care. The data were presented to the CC, and plans were made to further look at travel resources and community services available to our Veterans. A comprehensive report addressing resources was compiled and presented to the CC.
Implications: Identifying and breaking down barriers to transportation is vital to improving access to Veteran’s cancer care.
The VA Precision Oncology Research Program
The VA recently launched the Precision Oncology Program (POP), a clinical program that provides a turnkey operation for targeted sequencing of tumor samples and return of annotated results to the patient record. The Program recommends available clinical trials and consultative advice to clinicians and patients. We describe here the accompanying Research Program (Re-POP) that leverages the artifacts of the POP.
Lung cancer patients whose tumors are sequenced as part of the clinical POP will be given the opportunity to participate in the research Program. The goals of the Re-POP include: 1) creation of a network of VA sites to participate as a consortium in clinical trials; 2) provision of a research data repository containing information regarding tumor features including mutational status, patient demographics, and cancer treatments and outcomes; and 3) re-use of residual patient tumor tissue for expanded analysis.
Through collaborative efforts with research groups Re-POP will open large trials on a national level throughout the VA. Use of a centralized IRB and coordinating center (at Massachusetts Veterans Epidemiology Research and Information Center) will facilitate opening studies at any VA site that wishes to participate. The VA Cooperative Studies Program will support these activities. Preliminary data suggest that the Program can make available protocols offering either experimental or off-label therapies for approximately half of all nonsmall cell lung cancer patients.
The Re-POP data repository will reside at the NCI Genomic Data Commons and is available to both academic and industry researchers. Predictive analytics of these data support learning healthcare system activities (predicting individual patient outcomes) and the production of population-level generalizable knowledge.
Residual tissue and clinical data from Re-POP will be made available to researchers to identify new biomarkers that will enhance understanding of response to therapies as well as identify new therapeutic targets. An NCI-VA-DoD collaboration to study the value of proteomics in predicting response to targeted therapies in lung cancer was recently announced as part of the White House’s cancer moonshot initiative.
The VA recently launched the Precision Oncology Program (POP), a clinical program that provides a turnkey operation for targeted sequencing of tumor samples and return of annotated results to the patient record. The Program recommends available clinical trials and consultative advice to clinicians and patients. We describe here the accompanying Research Program (Re-POP) that leverages the artifacts of the POP.
Lung cancer patients whose tumors are sequenced as part of the clinical POP will be given the opportunity to participate in the research Program. The goals of the Re-POP include: 1) creation of a network of VA sites to participate as a consortium in clinical trials; 2) provision of a research data repository containing information regarding tumor features including mutational status, patient demographics, and cancer treatments and outcomes; and 3) re-use of residual patient tumor tissue for expanded analysis.
Through collaborative efforts with research groups Re-POP will open large trials on a national level throughout the VA. Use of a centralized IRB and coordinating center (at Massachusetts Veterans Epidemiology Research and Information Center) will facilitate opening studies at any VA site that wishes to participate. The VA Cooperative Studies Program will support these activities. Preliminary data suggest that the Program can make available protocols offering either experimental or off-label therapies for approximately half of all nonsmall cell lung cancer patients.
The Re-POP data repository will reside at the NCI Genomic Data Commons and is available to both academic and industry researchers. Predictive analytics of these data support learning healthcare system activities (predicting individual patient outcomes) and the production of population-level generalizable knowledge.
Residual tissue and clinical data from Re-POP will be made available to researchers to identify new biomarkers that will enhance understanding of response to therapies as well as identify new therapeutic targets. An NCI-VA-DoD collaboration to study the value of proteomics in predicting response to targeted therapies in lung cancer was recently announced as part of the White House’s cancer moonshot initiative.
The VA recently launched the Precision Oncology Program (POP), a clinical program that provides a turnkey operation for targeted sequencing of tumor samples and return of annotated results to the patient record. The Program recommends available clinical trials and consultative advice to clinicians and patients. We describe here the accompanying Research Program (Re-POP) that leverages the artifacts of the POP.
Lung cancer patients whose tumors are sequenced as part of the clinical POP will be given the opportunity to participate in the research Program. The goals of the Re-POP include: 1) creation of a network of VA sites to participate as a consortium in clinical trials; 2) provision of a research data repository containing information regarding tumor features including mutational status, patient demographics, and cancer treatments and outcomes; and 3) re-use of residual patient tumor tissue for expanded analysis.
Through collaborative efforts with research groups Re-POP will open large trials on a national level throughout the VA. Use of a centralized IRB and coordinating center (at Massachusetts Veterans Epidemiology Research and Information Center) will facilitate opening studies at any VA site that wishes to participate. The VA Cooperative Studies Program will support these activities. Preliminary data suggest that the Program can make available protocols offering either experimental or off-label therapies for approximately half of all nonsmall cell lung cancer patients.
The Re-POP data repository will reside at the NCI Genomic Data Commons and is available to both academic and industry researchers. Predictive analytics of these data support learning healthcare system activities (predicting individual patient outcomes) and the production of population-level generalizable knowledge.
Residual tissue and clinical data from Re-POP will be made available to researchers to identify new biomarkers that will enhance understanding of response to therapies as well as identify new therapeutic targets. An NCI-VA-DoD collaboration to study the value of proteomics in predicting response to targeted therapies in lung cancer was recently announced as part of the White House’s cancer moonshot initiative.