User login
Longer DAPT better for PAD, study suggests

Photo by Sage Ross
ROME—A subanalysis of the PRODIGY study suggests a longer duration of dual antiplatelet therapy (DAPT) improves outcomes after percutaneous coronary intervention (PCI) for patients with peripheral arterial disease (PAD).
Receiving long-term DAPT after PCI reduced the risk of atherothrombotic events and death in patients with PAD, without increasing the risk of actionable bleeding episodes.
However, patients without PAD fared better with short-term DAPT.
These results were presented at ESC Congress 2016 (abstract 5154) and published in JAMA Cardiology.
Marco Valgimigli, MD, PhD, of Bern University Hospital in Bern, Switzerland, and his colleagues performed this analysis of PRODIGY data.
The study included patients from tertiary care hospitals who had stable coronary artery disease or acute coronary syndromes, with or without concomitant PAD, and were undergoing PCI.
There were 246 patients with PAD—118 who were randomized to receive DAPT for 24 months after PCI and 128 who were randomized to DAPT for 6 months or less.
There were 1724 patients without PAD—869 who were randomized to receive DAPT for 24 months after PCI and 855 who were randomized to DAPT for 6 months or less.
The patients with PAD were older and more frequently underwent multivessel intervention. They were also more likely to have hypertension, type 1 or 2 diabetes, previous myocardial infarction, previous coronary artery bypass grafting, non-ST-segment elevation myocardial infarction, and more complex coronary artery disease.
At 30 days, patients with PAD were more often taking diuretics, and patients without PAD were more often taking beta-blockers and statins.
Patients with PAD who were randomized to long-term DAPT were younger, had a higher body mass index, and less frequently underwent PCI of the left main coronary artery than PAD patients randomized to short-term DAPT.
Having PAD was associated with a higher risk of death and ischemic events, with a hazard ratio (HR) of 2.80 (95% CI, 2.05-3.83; P<0.001).
Results
The primary efficacy endpoint of this study was a composite of death, myocardial infarction, and cerebrovascular accidents.
Among patients with PAD, those who received long-term DAPT had a lower risk of this endpoint than those who received short-term DAPT—16.1% and 27.3%, respectively. The HR was 0.54 (95% CI, 0.31-0.95; P=0.03).
Among patients without PAD, there was no significant difference in the incidence of the primary endpoint according to DAPT duration. It occurred in 9.3% of patients who received long-term DAPT and 7.4% of patients who received short-term DAPT. The HR was 1.28 (95% CI, 0.92-1.77; P=0.15).
The key safety endpoint was a composite of Bleeding Academic Research Consortium (BARC) type 2, 3, or 5 bleeding.
There was no significant difference in this endpoint according to DAPT duration for patients with PAD, but long-term DAPT was associated with a significant increase in this endpoint for patients without PAD.
Among patients with PAD, BARC type 2, 3, or 5 bleeding occurred in 5.2% of those receiving long-term DAPT and 6.9% of those receiving short-term DAPT. The HR was 0.77 (95% CI, 0.27-2.21; P=0.62).
Among patients without PAD, BARC type 2, 3, or 5 bleeding occurred in 8% of those receiving long-term DAPT and 3.1% of those receiving short-term DAPT. The HR was 2.61 (95% CI, 0.27-2.21; P<0.001).
The researchers said the apparent neutral effect of long-term DAPT on bleeding risk in PAD patients requires further evaluation in adequately powered studies, but this research suggests patients with PAD will benefit from prolonged DAPT after PCI. ![]()
Photo by Sage Ross
ROME—A subanalysis of the PRODIGY study suggests a longer duration of dual antiplatelet therapy (DAPT) improves outcomes after percutaneous coronary intervention (PCI) for patients with peripheral arterial disease (PAD).
Receiving long-term DAPT after PCI reduced the risk of atherothrombotic events and death in patients with PAD, without increasing the risk of actionable bleeding episodes.
However, patients without PAD fared better with short-term DAPT.
These results were presented at ESC Congress 2016 (abstract 5154) and published in JAMA Cardiology.
Marco Valgimigli, MD, PhD, of Bern University Hospital in Bern, Switzerland, and his colleagues performed this analysis of PRODIGY data.
The study included patients from tertiary care hospitals who had stable coronary artery disease or acute coronary syndromes, with or without concomitant PAD, and were undergoing PCI.
There were 246 patients with PAD—118 who were randomized to receive DAPT for 24 months after PCI and 128 who were randomized to DAPT for 6 months or less.
There were 1724 patients without PAD—869 who were randomized to receive DAPT for 24 months after PCI and 855 who were randomized to DAPT for 6 months or less.
The patients with PAD were older and more frequently underwent multivessel intervention. They were also more likely to have hypertension, type 1 or 2 diabetes, previous myocardial infarction, previous coronary artery bypass grafting, non-ST-segment elevation myocardial infarction, and more complex coronary artery disease.
At 30 days, patients with PAD were more often taking diuretics, and patients without PAD were more often taking beta-blockers and statins.
Patients with PAD who were randomized to long-term DAPT were younger, had a higher body mass index, and less frequently underwent PCI of the left main coronary artery than PAD patients randomized to short-term DAPT.
Having PAD was associated with a higher risk of death and ischemic events, with a hazard ratio (HR) of 2.80 (95% CI, 2.05-3.83; P<0.001).
Results
The primary efficacy endpoint of this study was a composite of death, myocardial infarction, and cerebrovascular accidents.
Among patients with PAD, those who received long-term DAPT had a lower risk of this endpoint than those who received short-term DAPT—16.1% and 27.3%, respectively. The HR was 0.54 (95% CI, 0.31-0.95; P=0.03).
Among patients without PAD, there was no significant difference in the incidence of the primary endpoint according to DAPT duration. It occurred in 9.3% of patients who received long-term DAPT and 7.4% of patients who received short-term DAPT. The HR was 1.28 (95% CI, 0.92-1.77; P=0.15).
The key safety endpoint was a composite of Bleeding Academic Research Consortium (BARC) type 2, 3, or 5 bleeding.
There was no significant difference in this endpoint according to DAPT duration for patients with PAD, but long-term DAPT was associated with a significant increase in this endpoint for patients without PAD.
Among patients with PAD, BARC type 2, 3, or 5 bleeding occurred in 5.2% of those receiving long-term DAPT and 6.9% of those receiving short-term DAPT. The HR was 0.77 (95% CI, 0.27-2.21; P=0.62).
Among patients without PAD, BARC type 2, 3, or 5 bleeding occurred in 8% of those receiving long-term DAPT and 3.1% of those receiving short-term DAPT. The HR was 2.61 (95% CI, 0.27-2.21; P<0.001).
The researchers said the apparent neutral effect of long-term DAPT on bleeding risk in PAD patients requires further evaluation in adequately powered studies, but this research suggests patients with PAD will benefit from prolonged DAPT after PCI. ![]()
Photo by Sage Ross
ROME—A subanalysis of the PRODIGY study suggests a longer duration of dual antiplatelet therapy (DAPT) improves outcomes after percutaneous coronary intervention (PCI) for patients with peripheral arterial disease (PAD).
Receiving long-term DAPT after PCI reduced the risk of atherothrombotic events and death in patients with PAD, without increasing the risk of actionable bleeding episodes.
However, patients without PAD fared better with short-term DAPT.
These results were presented at ESC Congress 2016 (abstract 5154) and published in JAMA Cardiology.
Marco Valgimigli, MD, PhD, of Bern University Hospital in Bern, Switzerland, and his colleagues performed this analysis of PRODIGY data.
The study included patients from tertiary care hospitals who had stable coronary artery disease or acute coronary syndromes, with or without concomitant PAD, and were undergoing PCI.
There were 246 patients with PAD—118 who were randomized to receive DAPT for 24 months after PCI and 128 who were randomized to DAPT for 6 months or less.
There were 1724 patients without PAD—869 who were randomized to receive DAPT for 24 months after PCI and 855 who were randomized to DAPT for 6 months or less.
The patients with PAD were older and more frequently underwent multivessel intervention. They were also more likely to have hypertension, type 1 or 2 diabetes, previous myocardial infarction, previous coronary artery bypass grafting, non-ST-segment elevation myocardial infarction, and more complex coronary artery disease.
At 30 days, patients with PAD were more often taking diuretics, and patients without PAD were more often taking beta-blockers and statins.
Patients with PAD who were randomized to long-term DAPT were younger, had a higher body mass index, and less frequently underwent PCI of the left main coronary artery than PAD patients randomized to short-term DAPT.
Having PAD was associated with a higher risk of death and ischemic events, with a hazard ratio (HR) of 2.80 (95% CI, 2.05-3.83; P<0.001).
Results
The primary efficacy endpoint of this study was a composite of death, myocardial infarction, and cerebrovascular accidents.
Among patients with PAD, those who received long-term DAPT had a lower risk of this endpoint than those who received short-term DAPT—16.1% and 27.3%, respectively. The HR was 0.54 (95% CI, 0.31-0.95; P=0.03).
Among patients without PAD, there was no significant difference in the incidence of the primary endpoint according to DAPT duration. It occurred in 9.3% of patients who received long-term DAPT and 7.4% of patients who received short-term DAPT. The HR was 1.28 (95% CI, 0.92-1.77; P=0.15).
The key safety endpoint was a composite of Bleeding Academic Research Consortium (BARC) type 2, 3, or 5 bleeding.
There was no significant difference in this endpoint according to DAPT duration for patients with PAD, but long-term DAPT was associated with a significant increase in this endpoint for patients without PAD.
Among patients with PAD, BARC type 2, 3, or 5 bleeding occurred in 5.2% of those receiving long-term DAPT and 6.9% of those receiving short-term DAPT. The HR was 0.77 (95% CI, 0.27-2.21; P=0.62).
Among patients without PAD, BARC type 2, 3, or 5 bleeding occurred in 8% of those receiving long-term DAPT and 3.1% of those receiving short-term DAPT. The HR was 2.61 (95% CI, 0.27-2.21; P<0.001).
The researchers said the apparent neutral effect of long-term DAPT on bleeding risk in PAD patients requires further evaluation in adequately powered studies, but this research suggests patients with PAD will benefit from prolonged DAPT after PCI. ![]()
MRI measurements reveal effects of sleep deprivation
Lack of sleep had a significant impact on brain responses to an attention task, and circadian rhythms played a role, according to functional magnetic resonance imaging data from 33 healthy adults. The findings were published online Aug. 11 in Science.
Despite the data showing that acute sleep loss impacts cognition, “human performance remains remarkably well preserved until wakefulness is extended into the biological night,” wrote Vincenzo Muto of the University of Liège, Belgium, and his colleagues (Science 2016;353:687-90. doi: 10.1126/science.aad2993).

Study participants stayed awake for 42 hours, beginning in the morning and covering 2 biological days, 1 biological night, and part of a second night. They periodically performed the psychomotor vigilance task (PVT), a visual reaction time task designed to measure attention; and an auditory n-back task, and the researchers collected functional and structural MRI data across 13 sessions. The average age of the participants (17 men and 16 women) was 21 years.
Overall, PVT performance was stable during the first day, but decreased significantly after sleep deprivation, then recovered during the second day, and returned to baseline after a period of recovery sleep, the researchers said.
Brain responses to the n-back task were “significantly modulated by a circadian oscillation, synchronous to the melatonin rhythm,” they noted. “This finding rules out a global task-independent circadian influence and suggest the influence of a local, region-specific task-dependent circadian signal,” they added.
Although more research is needed on how different cognitive tasks are affected by sleep deprivation, the findings may help in “understanding of the brain mechanisms underlying the maintenance of daytime cognitive performance and its deterioration, as observed in shift work, jet lag, sleep disorders, aging, and neurodegenerative diseases,” the researchers wrote.
They had no financial conflicts to disclose.
Lack of sleep had a significant impact on brain responses to an attention task, and circadian rhythms played a role, according to functional magnetic resonance imaging data from 33 healthy adults. The findings were published online Aug. 11 in Science.
Despite the data showing that acute sleep loss impacts cognition, “human performance remains remarkably well preserved until wakefulness is extended into the biological night,” wrote Vincenzo Muto of the University of Liège, Belgium, and his colleagues (Science 2016;353:687-90. doi: 10.1126/science.aad2993).
Study participants stayed awake for 42 hours, beginning in the morning and covering 2 biological days, 1 biological night, and part of a second night. They periodically performed the psychomotor vigilance task (PVT), a visual reaction time task designed to measure attention; and an auditory n-back task, and the researchers collected functional and structural MRI data across 13 sessions. The average age of the participants (17 men and 16 women) was 21 years.
Overall, PVT performance was stable during the first day, but decreased significantly after sleep deprivation, then recovered during the second day, and returned to baseline after a period of recovery sleep, the researchers said.
Brain responses to the n-back task were “significantly modulated by a circadian oscillation, synchronous to the melatonin rhythm,” they noted. “This finding rules out a global task-independent circadian influence and suggest the influence of a local, region-specific task-dependent circadian signal,” they added.
Although more research is needed on how different cognitive tasks are affected by sleep deprivation, the findings may help in “understanding of the brain mechanisms underlying the maintenance of daytime cognitive performance and its deterioration, as observed in shift work, jet lag, sleep disorders, aging, and neurodegenerative diseases,” the researchers wrote.
They had no financial conflicts to disclose.
Lack of sleep had a significant impact on brain responses to an attention task, and circadian rhythms played a role, according to functional magnetic resonance imaging data from 33 healthy adults. The findings were published online Aug. 11 in Science.
Despite the data showing that acute sleep loss impacts cognition, “human performance remains remarkably well preserved until wakefulness is extended into the biological night,” wrote Vincenzo Muto of the University of Liège, Belgium, and his colleagues (Science 2016;353:687-90. doi: 10.1126/science.aad2993).
Study participants stayed awake for 42 hours, beginning in the morning and covering 2 biological days, 1 biological night, and part of a second night. They periodically performed the psychomotor vigilance task (PVT), a visual reaction time task designed to measure attention; and an auditory n-back task, and the researchers collected functional and structural MRI data across 13 sessions. The average age of the participants (17 men and 16 women) was 21 years.
Overall, PVT performance was stable during the first day, but decreased significantly after sleep deprivation, then recovered during the second day, and returned to baseline after a period of recovery sleep, the researchers said.
Brain responses to the n-back task were “significantly modulated by a circadian oscillation, synchronous to the melatonin rhythm,” they noted. “This finding rules out a global task-independent circadian influence and suggest the influence of a local, region-specific task-dependent circadian signal,” they added.
Although more research is needed on how different cognitive tasks are affected by sleep deprivation, the findings may help in “understanding of the brain mechanisms underlying the maintenance of daytime cognitive performance and its deterioration, as observed in shift work, jet lag, sleep disorders, aging, and neurodegenerative diseases,” the researchers wrote.
They had no financial conflicts to disclose.
FROM SCIENCE
Key clinical point: Brain responses to sustained-attention tasks deteriorated with sleep deprivation and varied according to circadian rhythms, according to functional MRI data.
Major finding: MRI data collected over 42 hours of wakefulness and after recovery sleep showed a significant (P less than .05) impact of circadian rhythms on participants’ abilities to perform visual and auditory tasks.
Data source: A sleep study that used functional MRI to measure changes in brain response in 33 healthy adults.
Disclosures: The researchers had no financial conflicts to disclose.

FDA rule will pull many consumer antibacterial soaps from market
Over-the-counter consumer antiseptic wash products with active ingredients such as triclosan and triclocarban will be pulled from the market, following a final rule issued Sept. 2 by the Food and Drug Administration.
Companies will no longer be able to sell antibacterial washes with those ingredients, the FDA said, because manufacturers failed to show the ingredients are safe for long-term daily use and are better than plain soap and water at preventing illness and the spread of infections.

The final rule targets consumer antiseptic wash products containing 1 or more of 19 active ingredients, including the 2 most commonly used ingredients, triclosan and triclocarban. Companies have 1 year to comply with the new rule.
The FDA’s rule does not apply to hand sanitizers, wipes, or antibacterial products used in health care settings.
The agency has deferred for 1 year a decision on the continued use of three other ingredients in consumer wash products: benzalkonium chloride, benzethonium chloride, and chloroxylenol.
The FDA’s decision was driven in part by concerns about the risks posed by long-term exposure to such products, including bacterial resistance or hormonal effects.
“Consumers may think antibacterial washes are more effective at preventing the spread of germs, but we have no scientific evidence that they are any better than plain soap and water,” said Janet Woodcock, MD, director of the FDA’s Center for Drug Evaluation and Research, in a statement. “In fact, some data suggest that antibacterial ingredients may do more harm than good over the long term.”
Washing with plain soap and water remains one of the most important steps consumers can take to prevent illness and the spread of infection, the FDA advised. The agency also recommended use of alcohol-based hand sanitizer with at least 60% alcohol.
Read the full press release on the FDA website.
Over-the-counter consumer antiseptic wash products with active ingredients such as triclosan and triclocarban will be pulled from the market, following a final rule issued Sept. 2 by the Food and Drug Administration.
Companies will no longer be able to sell antibacterial washes with those ingredients, the FDA said, because manufacturers failed to show the ingredients are safe for long-term daily use and are better than plain soap and water at preventing illness and the spread of infections.
The final rule targets consumer antiseptic wash products containing 1 or more of 19 active ingredients, including the 2 most commonly used ingredients, triclosan and triclocarban. Companies have 1 year to comply with the new rule.
The FDA’s rule does not apply to hand sanitizers, wipes, or antibacterial products used in health care settings.
The agency has deferred for 1 year a decision on the continued use of three other ingredients in consumer wash products: benzalkonium chloride, benzethonium chloride, and chloroxylenol.
The FDA’s decision was driven in part by concerns about the risks posed by long-term exposure to such products, including bacterial resistance or hormonal effects.
“Consumers may think antibacterial washes are more effective at preventing the spread of germs, but we have no scientific evidence that they are any better than plain soap and water,” said Janet Woodcock, MD, director of the FDA’s Center for Drug Evaluation and Research, in a statement. “In fact, some data suggest that antibacterial ingredients may do more harm than good over the long term.”
Washing with plain soap and water remains one of the most important steps consumers can take to prevent illness and the spread of infection, the FDA advised. The agency also recommended use of alcohol-based hand sanitizer with at least 60% alcohol.
Read the full press release on the FDA website.
Over-the-counter consumer antiseptic wash products with active ingredients such as triclosan and triclocarban will be pulled from the market, following a final rule issued Sept. 2 by the Food and Drug Administration.
Companies will no longer be able to sell antibacterial washes with those ingredients, the FDA said, because manufacturers failed to show the ingredients are safe for long-term daily use and are better than plain soap and water at preventing illness and the spread of infections.
The final rule targets consumer antiseptic wash products containing 1 or more of 19 active ingredients, including the 2 most commonly used ingredients, triclosan and triclocarban. Companies have 1 year to comply with the new rule.
The FDA’s rule does not apply to hand sanitizers, wipes, or antibacterial products used in health care settings.
The agency has deferred for 1 year a decision on the continued use of three other ingredients in consumer wash products: benzalkonium chloride, benzethonium chloride, and chloroxylenol.
The FDA’s decision was driven in part by concerns about the risks posed by long-term exposure to such products, including bacterial resistance or hormonal effects.
“Consumers may think antibacterial washes are more effective at preventing the spread of germs, but we have no scientific evidence that they are any better than plain soap and water,” said Janet Woodcock, MD, director of the FDA’s Center for Drug Evaluation and Research, in a statement. “In fact, some data suggest that antibacterial ingredients may do more harm than good over the long term.”
Washing with plain soap and water remains one of the most important steps consumers can take to prevent illness and the spread of infection, the FDA advised. The agency also recommended use of alcohol-based hand sanitizer with at least 60% alcohol.
Read the full press release on the FDA website.

Treat bed bug bites with topical steroids
BOSTON – Preventing repeated occurrences of bed bug bites means eliminating infestations or – if the bites occur during travel – avoiding bringing the bugs home, according to Theodore Rosen, MD.
Bed bugs are “very, very, very hardy,” and can live months or maybe even years between blood meals, Dr. Rosen, professor of dermatology at Baylor College of Medicine, Houston, said at the American Academy of Dermatology summer meeting.
“They lay lots of eggs and have lots of offspring,” he said, adding that bed bugs have evolved and developed mutations that make them resistant to insecticides, and can survive up to a week at a continuous temperature of 10 degrees Fahrenheit.

Advise patients who will be traveling to check whether the area they are traveling to has known bed bug infestations. Recommend that they always check areas within 3 feet or so of hotel beds for evidence of bed bugs, he said.
Bed bugs like right angles, so they may be found on headboards, bed frames, mattress piping, drawers, and windows. They can also be found hiding under peeling paint, and or in the piping or under surfaces of furniture in hotel rooms.
Dr. Rosen is often asked by patients if he really looks for bed bugs when he travels. “I do look – and I’ve found them,” he said, noting that they can be found in the nicest of hotels. He found some lurking beneath the drawer of a nightstand in one luxury hotel.
Bugs can be seen, but droppings are another telltale sign of infestation; bed bug excrement stains fabrics, so if round brown splotches are seen on mattress or other fabric, bed bugs have been there and probably still are there, Dr. Rosen said.
“I pull up the sheets, look at the piping on the mattress, pull up the mattress and look at the frame, look at the coils, look at the night stand,” he said.
A pricey monitoring and trapping device called the NightWatch can be purchased to help in the event of an infestation. The device emits heated carbon dioxide and pheromones that attract and trap bed bugs, but the $300-$400 price tag can be prohibitive, he noted.
The best bet is to avoid bringing home bugs by keeping all bags and clothes off the floor. Bags should be kept on a dresser or stand, clothes should be hung.
If infestation occurs, let patients know that the bites aren’t dangerous. There is no convincing evidence that bed bugs transmit any diseases. Anemia with heavy infestation and repeated bites could theoretically be a concern, but other than itching and looking bad, bites are rarely an issue.
Bed bug bites usually respond well to topical steroid treatment, and only in rare cases are oral steroids or antihistamines needed. Home bites will recur if there is infestation, and the problem can be a source of depression and anxiety, which in some patients can be severe and lead to suicidal ideation.
Infestation should be addressed by heavy vacuuming and steaming, which kills bed bugs if he temperatures are high enough. Mattress encasement can also help, but insecticides are rarely effective. Freezing smaller objects can be tried, but bed bugs can withstand freezing temperatures for long periods of time, he said.
“If you bring bed bugs home, you have to treat the whole environment,” he said.
One treatment that has shown promise is ivermectin in the host. Studies suggest this approach kills some bugs, but it is not 100% effective and is probably best reserved for widespread infestation, such as in a nursing home, Dr. Rosen said.
He disclosed financial or other relationships with Anacor Pharmaceuticals; Innocutis; Sandoz, a Novartis company; and Valeant Pharmaceuticals International.
BOSTON – Preventing repeated occurrences of bed bug bites means eliminating infestations or – if the bites occur during travel – avoiding bringing the bugs home, according to Theodore Rosen, MD.
Bed bugs are “very, very, very hardy,” and can live months or maybe even years between blood meals, Dr. Rosen, professor of dermatology at Baylor College of Medicine, Houston, said at the American Academy of Dermatology summer meeting.
“They lay lots of eggs and have lots of offspring,” he said, adding that bed bugs have evolved and developed mutations that make them resistant to insecticides, and can survive up to a week at a continuous temperature of 10 degrees Fahrenheit.
Advise patients who will be traveling to check whether the area they are traveling to has known bed bug infestations. Recommend that they always check areas within 3 feet or so of hotel beds for evidence of bed bugs, he said.
Bed bugs like right angles, so they may be found on headboards, bed frames, mattress piping, drawers, and windows. They can also be found hiding under peeling paint, and or in the piping or under surfaces of furniture in hotel rooms.
Dr. Rosen is often asked by patients if he really looks for bed bugs when he travels. “I do look – and I’ve found them,” he said, noting that they can be found in the nicest of hotels. He found some lurking beneath the drawer of a nightstand in one luxury hotel.
Bugs can be seen, but droppings are another telltale sign of infestation; bed bug excrement stains fabrics, so if round brown splotches are seen on mattress or other fabric, bed bugs have been there and probably still are there, Dr. Rosen said.
“I pull up the sheets, look at the piping on the mattress, pull up the mattress and look at the frame, look at the coils, look at the night stand,” he said.
A pricey monitoring and trapping device called the NightWatch can be purchased to help in the event of an infestation. The device emits heated carbon dioxide and pheromones that attract and trap bed bugs, but the $300-$400 price tag can be prohibitive, he noted.
The best bet is to avoid bringing home bugs by keeping all bags and clothes off the floor. Bags should be kept on a dresser or stand, clothes should be hung.
If infestation occurs, let patients know that the bites aren’t dangerous. There is no convincing evidence that bed bugs transmit any diseases. Anemia with heavy infestation and repeated bites could theoretically be a concern, but other than itching and looking bad, bites are rarely an issue.
Bed bug bites usually respond well to topical steroid treatment, and only in rare cases are oral steroids or antihistamines needed. Home bites will recur if there is infestation, and the problem can be a source of depression and anxiety, which in some patients can be severe and lead to suicidal ideation.
Infestation should be addressed by heavy vacuuming and steaming, which kills bed bugs if he temperatures are high enough. Mattress encasement can also help, but insecticides are rarely effective. Freezing smaller objects can be tried, but bed bugs can withstand freezing temperatures for long periods of time, he said.
“If you bring bed bugs home, you have to treat the whole environment,” he said.
One treatment that has shown promise is ivermectin in the host. Studies suggest this approach kills some bugs, but it is not 100% effective and is probably best reserved for widespread infestation, such as in a nursing home, Dr. Rosen said.
He disclosed financial or other relationships with Anacor Pharmaceuticals; Innocutis; Sandoz, a Novartis company; and Valeant Pharmaceuticals International.
BOSTON – Preventing repeated occurrences of bed bug bites means eliminating infestations or – if the bites occur during travel – avoiding bringing the bugs home, according to Theodore Rosen, MD.
Bed bugs are “very, very, very hardy,” and can live months or maybe even years between blood meals, Dr. Rosen, professor of dermatology at Baylor College of Medicine, Houston, said at the American Academy of Dermatology summer meeting.
“They lay lots of eggs and have lots of offspring,” he said, adding that bed bugs have evolved and developed mutations that make them resistant to insecticides, and can survive up to a week at a continuous temperature of 10 degrees Fahrenheit.
Advise patients who will be traveling to check whether the area they are traveling to has known bed bug infestations. Recommend that they always check areas within 3 feet or so of hotel beds for evidence of bed bugs, he said.
Bed bugs like right angles, so they may be found on headboards, bed frames, mattress piping, drawers, and windows. They can also be found hiding under peeling paint, and or in the piping or under surfaces of furniture in hotel rooms.
Dr. Rosen is often asked by patients if he really looks for bed bugs when he travels. “I do look – and I’ve found them,” he said, noting that they can be found in the nicest of hotels. He found some lurking beneath the drawer of a nightstand in one luxury hotel.
Bugs can be seen, but droppings are another telltale sign of infestation; bed bug excrement stains fabrics, so if round brown splotches are seen on mattress or other fabric, bed bugs have been there and probably still are there, Dr. Rosen said.
“I pull up the sheets, look at the piping on the mattress, pull up the mattress and look at the frame, look at the coils, look at the night stand,” he said.
A pricey monitoring and trapping device called the NightWatch can be purchased to help in the event of an infestation. The device emits heated carbon dioxide and pheromones that attract and trap bed bugs, but the $300-$400 price tag can be prohibitive, he noted.
The best bet is to avoid bringing home bugs by keeping all bags and clothes off the floor. Bags should be kept on a dresser or stand, clothes should be hung.
If infestation occurs, let patients know that the bites aren’t dangerous. There is no convincing evidence that bed bugs transmit any diseases. Anemia with heavy infestation and repeated bites could theoretically be a concern, but other than itching and looking bad, bites are rarely an issue.
Bed bug bites usually respond well to topical steroid treatment, and only in rare cases are oral steroids or antihistamines needed. Home bites will recur if there is infestation, and the problem can be a source of depression and anxiety, which in some patients can be severe and lead to suicidal ideation.
Infestation should be addressed by heavy vacuuming and steaming, which kills bed bugs if he temperatures are high enough. Mattress encasement can also help, but insecticides are rarely effective. Freezing smaller objects can be tried, but bed bugs can withstand freezing temperatures for long periods of time, he said.
“If you bring bed bugs home, you have to treat the whole environment,” he said.
One treatment that has shown promise is ivermectin in the host. Studies suggest this approach kills some bugs, but it is not 100% effective and is probably best reserved for widespread infestation, such as in a nursing home, Dr. Rosen said.
He disclosed financial or other relationships with Anacor Pharmaceuticals; Innocutis; Sandoz, a Novartis company; and Valeant Pharmaceuticals International.
EXPERT ANALYSIS FROM AAD Summer Academy 2016

Another 199 pregnant women with Zika
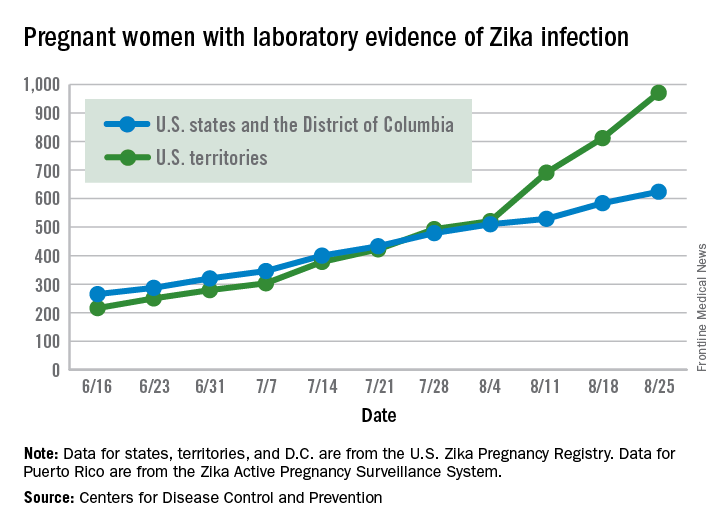
Zika virus shows no signs of slowing down, as the number of pregnant women with laboratory evidence of possible infection in the United States and its territories took its largest jump yet during the week ending Aug. 25, according to the Centers for Disease Control and Prevention.
There were 199 new cases of Zika that week: 159 in the U.S. territories and 40 in the 50 states and the District of Columbia. The previous high had been 189 for the week ending Aug. 11. Cases in pregnant women for 2016 so far number 971 in the territories and 624 in the states and D.C. – a total of 1,595, the CDC reported Sept. 1.
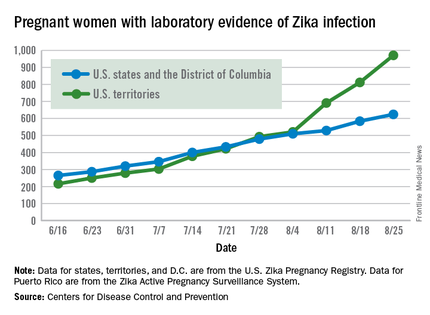
The number of poor outcomes among pregnant women with Zika virus infection did not change for the week ending Aug. 25. The number of live-born infants with Zika-related birth defects remained at 17 – 16 in the states/D.C. and 1 in the territories – and the number of pregnancy losses with birth defects was still 6 – 5 in the states/D.C. and 1 in the territories, the CDC said. State- or territorial-level data are not being reported to protect the privacy of affected women and children.
Among the entire U.S. population, 16,832 cases of Zika have been reported to the CDC Arboviral Disease Branch in 2015-2016, with 5,304 reported for the week ending Aug. 31 (Puerto Rico retroactively reported 5,000 cases that had been identified between June 4 and Aug. 6). The states/D.C. account for 2,722 of total cases, and the territories have reported 14,110 cases, of which Puerto Rico accounts for 13,791, the CDC noted.
The figures for states, territories, and D.C. reflect reporting to the U.S. Zika Pregnancy Registry; data for Puerto Rico are reported to the U.S. Zika Active Pregnancy Surveillance System.
Zika-related birth defects recorded by the CDC could include microcephaly, calcium deposits in the brain indicating possible brain damage, excess fluid in the brain cavities and surrounding the brain, absent or poorly formed brain structures, abnormal eye development, or other problems resulting from brain damage that affect nerves, muscles, and bones. The pregnancy losses encompass any miscarriage, stillbirth, and termination with evidence of birth defects.
Zika virus shows no signs of slowing down, as the number of pregnant women with laboratory evidence of possible infection in the United States and its territories took its largest jump yet during the week ending Aug. 25, according to the Centers for Disease Control and Prevention.
There were 199 new cases of Zika that week: 159 in the U.S. territories and 40 in the 50 states and the District of Columbia. The previous high had been 189 for the week ending Aug. 11. Cases in pregnant women for 2016 so far number 971 in the territories and 624 in the states and D.C. – a total of 1,595, the CDC reported Sept. 1.
The number of poor outcomes among pregnant women with Zika virus infection did not change for the week ending Aug. 25. The number of live-born infants with Zika-related birth defects remained at 17 – 16 in the states/D.C. and 1 in the territories – and the number of pregnancy losses with birth defects was still 6 – 5 in the states/D.C. and 1 in the territories, the CDC said. State- or territorial-level data are not being reported to protect the privacy of affected women and children.
Among the entire U.S. population, 16,832 cases of Zika have been reported to the CDC Arboviral Disease Branch in 2015-2016, with 5,304 reported for the week ending Aug. 31 (Puerto Rico retroactively reported 5,000 cases that had been identified between June 4 and Aug. 6). The states/D.C. account for 2,722 of total cases, and the territories have reported 14,110 cases, of which Puerto Rico accounts for 13,791, the CDC noted.
The figures for states, territories, and D.C. reflect reporting to the U.S. Zika Pregnancy Registry; data for Puerto Rico are reported to the U.S. Zika Active Pregnancy Surveillance System.
Zika-related birth defects recorded by the CDC could include microcephaly, calcium deposits in the brain indicating possible brain damage, excess fluid in the brain cavities and surrounding the brain, absent or poorly formed brain structures, abnormal eye development, or other problems resulting from brain damage that affect nerves, muscles, and bones. The pregnancy losses encompass any miscarriage, stillbirth, and termination with evidence of birth defects.
Zika virus shows no signs of slowing down, as the number of pregnant women with laboratory evidence of possible infection in the United States and its territories took its largest jump yet during the week ending Aug. 25, according to the Centers for Disease Control and Prevention.
There were 199 new cases of Zika that week: 159 in the U.S. territories and 40 in the 50 states and the District of Columbia. The previous high had been 189 for the week ending Aug. 11. Cases in pregnant women for 2016 so far number 971 in the territories and 624 in the states and D.C. – a total of 1,595, the CDC reported Sept. 1.
The number of poor outcomes among pregnant women with Zika virus infection did not change for the week ending Aug. 25. The number of live-born infants with Zika-related birth defects remained at 17 – 16 in the states/D.C. and 1 in the territories – and the number of pregnancy losses with birth defects was still 6 – 5 in the states/D.C. and 1 in the territories, the CDC said. State- or territorial-level data are not being reported to protect the privacy of affected women and children.
Among the entire U.S. population, 16,832 cases of Zika have been reported to the CDC Arboviral Disease Branch in 2015-2016, with 5,304 reported for the week ending Aug. 31 (Puerto Rico retroactively reported 5,000 cases that had been identified between June 4 and Aug. 6). The states/D.C. account for 2,722 of total cases, and the territories have reported 14,110 cases, of which Puerto Rico accounts for 13,791, the CDC noted.
The figures for states, territories, and D.C. reflect reporting to the U.S. Zika Pregnancy Registry; data for Puerto Rico are reported to the U.S. Zika Active Pregnancy Surveillance System.
Zika-related birth defects recorded by the CDC could include microcephaly, calcium deposits in the brain indicating possible brain damage, excess fluid in the brain cavities and surrounding the brain, absent or poorly formed brain structures, abnormal eye development, or other problems resulting from brain damage that affect nerves, muscles, and bones. The pregnancy losses encompass any miscarriage, stillbirth, and termination with evidence of birth defects.
Vitamin and Mineral Intake Is Inadequate for Most Americans: What Should We Advise Patients About Supplements?

This supplement examines the role of vitamin and mineral supplements in increasing nutrient intake and reducing nutrient deficiencies and inadequacies. Although research is needed to study the effects of dietary supplements on chronic disease outcomes, US health care providers need to know how to advise their patients about adding vitamins and minerals to their diets.
This supplement examines the role of vitamin and mineral supplements in increasing nutrient intake and reducing nutrient deficiencies and inadequacies. Although research is needed to study the effects of dietary supplements on chronic disease outcomes, US health care providers need to know how to advise their patients about adding vitamins and minerals to their diets.
This supplement examines the role of vitamin and mineral supplements in increasing nutrient intake and reducing nutrient deficiencies and inadequacies. Although research is needed to study the effects of dietary supplements on chronic disease outcomes, US health care providers need to know how to advise their patients about adding vitamins and minerals to their diets.

Robot-assisted laparoscopic surgery performed mostly by and for white males
BOSTON – Patients who receive robot-assisted laparoscopic surgery (RALS), an increasingly widespread facet of surgical medicine, tend to be higher income white males, according to an extensive new study presented at Minimally Invasive Surgery Week.
“We wanted to look at how the technology is rolling out ... and what some of those characteristics are that are occurring, not only with the types of patients that are picking up these surgeries but also who the surgeons are that are performing these surgeries,” the study’s lead investigator, Michael A. Palese, MD, of Mount Sinai Health System, New York, explained during a video interview.
A total of 63,725 RALS cases were included, all of which occurred during 2009-2015. In addition to affluent white males being the predominant recipients of this type of surgery, younger white male surgeons tended to be the ones more likely to perform RALS. Across specialties, RALS use has increased substantially over the study period, with the largest increases seen among cardiothoracic surgeons (from 197 cases, 3.1% of all cases per year, to 1,159, 8.7% of all cases). Among general surgeons, RALS use increased from 98 cases (3.2%) to 2,559 cases (19.1%), and for orthopedic surgeons, 55 (0.8%) to 985 (7.4%).
Dr. Palese discussed the genesis of the study, the importance of the study’s findings, and where he foresees RALS heading in the near future. He did not report any relevant financial disclosures.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
BOSTON – Patients who receive robot-assisted laparoscopic surgery (RALS), an increasingly widespread facet of surgical medicine, tend to be higher income white males, according to an extensive new study presented at Minimally Invasive Surgery Week.
“We wanted to look at how the technology is rolling out ... and what some of those characteristics are that are occurring, not only with the types of patients that are picking up these surgeries but also who the surgeons are that are performing these surgeries,” the study’s lead investigator, Michael A. Palese, MD, of Mount Sinai Health System, New York, explained during a video interview.
A total of 63,725 RALS cases were included, all of which occurred during 2009-2015. In addition to affluent white males being the predominant recipients of this type of surgery, younger white male surgeons tended to be the ones more likely to perform RALS. Across specialties, RALS use has increased substantially over the study period, with the largest increases seen among cardiothoracic surgeons (from 197 cases, 3.1% of all cases per year, to 1,159, 8.7% of all cases). Among general surgeons, RALS use increased from 98 cases (3.2%) to 2,559 cases (19.1%), and for orthopedic surgeons, 55 (0.8%) to 985 (7.4%).
Dr. Palese discussed the genesis of the study, the importance of the study’s findings, and where he foresees RALS heading in the near future. He did not report any relevant financial disclosures.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
BOSTON – Patients who receive robot-assisted laparoscopic surgery (RALS), an increasingly widespread facet of surgical medicine, tend to be higher income white males, according to an extensive new study presented at Minimally Invasive Surgery Week.
“We wanted to look at how the technology is rolling out ... and what some of those characteristics are that are occurring, not only with the types of patients that are picking up these surgeries but also who the surgeons are that are performing these surgeries,” the study’s lead investigator, Michael A. Palese, MD, of Mount Sinai Health System, New York, explained during a video interview.
A total of 63,725 RALS cases were included, all of which occurred during 2009-2015. In addition to affluent white males being the predominant recipients of this type of surgery, younger white male surgeons tended to be the ones more likely to perform RALS. Across specialties, RALS use has increased substantially over the study period, with the largest increases seen among cardiothoracic surgeons (from 197 cases, 3.1% of all cases per year, to 1,159, 8.7% of all cases). Among general surgeons, RALS use increased from 98 cases (3.2%) to 2,559 cases (19.1%), and for orthopedic surgeons, 55 (0.8%) to 985 (7.4%).
Dr. Palese discussed the genesis of the study, the importance of the study’s findings, and where he foresees RALS heading in the near future. He did not report any relevant financial disclosures.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
AT MINIMALLY INVASIVE SURGERY WEEK

Study supports extending docetaxel therapy in metastatic castration-resistant prostate cancer
Extending docetaxel chemotherapy significantly improved overall survival in patients with metastatic castration-resistant prostate cancer (mCRPC), regardless of whether they received lenalidomide, according to a retrospective analysis of 1,059 patients from a randomized, phase III trial.
“We found a robust and independent effect on overall survival by the number of docetaxel cycles administered in the setting of mCRPC,” wrote Ellen de Morree of Erasmus MC Cancer Institute (Rotterdam, the Netherlands) and her associates (JAMA Oncol. 2016 Aug 25. doi: 10.1001/jamaoncol.2016.3000).
The association was independent of performance status (Eastern Cooperative Oncology Group score) or baseline levels of lactate dehydrogenase level, hemoglobin, and albumin, they noted. “These data indicate that patients who appear to have clinical, radiological, or biochemical benefit by docetaxel should continue beyond 6 cycles as long as they tolerate their treatment well,” they concluded.
This study, the first to investigate the optimal number of docetaxel cycles in mCRPC, analyzed data from the multicenter Mainsail trial, in which patients received docetaxel, prednisone, and lenalidomide (DPL) or docetaxel, prednisone, and a placebo (DP) until they developed progressive disease or unacceptable adverse effects. Although dose intensity was similar between the two trial arms, DPL patients developed myelotoxicity with lenalidomide and therefore received a median of only six treatment cycles, while DP patients received a median of eight cycles. That difference enabled this analysis, the investigators noted.
Cumulative dose of docetaxel, duration of lenalidomide treatment, and allocated treatment regimen were significant predictors of overall survival in the univariate analysis. Overall survival was associated with treatment arm in a multivariable analysis that did not account for number of docetaxel cycles (hazard ratio, 1.6; 95% confidence interval, 1.2 to 2.1; P less than .001). But that changed after the addition of a number of docetaxel cycles to the model, the researchers said. In this final model, treatment with eight or more cycles of docetaxel led to substantially improved overall survival (hazard ratio, 1.9; P less than .001), regardless of lenalidomide treatment (HR, 1.1; 95% CI, 0.9 to 1.2; P = .4). Sensitivity analyses confirmed the association – patients who received more than 10 cycles of docetaxel had a median overall survival of 33 months (30-37 months), versus 27 months (24-30 months) with 8-10 cycles and about 23 months (18-27 months) with 5-7 cycles (P less than .001).
Extending docetaxel chemotherapy significantly improved overall survival in patients with metastatic castration-resistant prostate cancer (mCRPC), regardless of whether they received lenalidomide, according to a retrospective analysis of 1,059 patients from a randomized, phase III trial.
“We found a robust and independent effect on overall survival by the number of docetaxel cycles administered in the setting of mCRPC,” wrote Ellen de Morree of Erasmus MC Cancer Institute (Rotterdam, the Netherlands) and her associates (JAMA Oncol. 2016 Aug 25. doi: 10.1001/jamaoncol.2016.3000).
The association was independent of performance status (Eastern Cooperative Oncology Group score) or baseline levels of lactate dehydrogenase level, hemoglobin, and albumin, they noted. “These data indicate that patients who appear to have clinical, radiological, or biochemical benefit by docetaxel should continue beyond 6 cycles as long as they tolerate their treatment well,” they concluded.
This study, the first to investigate the optimal number of docetaxel cycles in mCRPC, analyzed data from the multicenter Mainsail trial, in which patients received docetaxel, prednisone, and lenalidomide (DPL) or docetaxel, prednisone, and a placebo (DP) until they developed progressive disease or unacceptable adverse effects. Although dose intensity was similar between the two trial arms, DPL patients developed myelotoxicity with lenalidomide and therefore received a median of only six treatment cycles, while DP patients received a median of eight cycles. That difference enabled this analysis, the investigators noted.
Cumulative dose of docetaxel, duration of lenalidomide treatment, and allocated treatment regimen were significant predictors of overall survival in the univariate analysis. Overall survival was associated with treatment arm in a multivariable analysis that did not account for number of docetaxel cycles (hazard ratio, 1.6; 95% confidence interval, 1.2 to 2.1; P less than .001). But that changed after the addition of a number of docetaxel cycles to the model, the researchers said. In this final model, treatment with eight or more cycles of docetaxel led to substantially improved overall survival (hazard ratio, 1.9; P less than .001), regardless of lenalidomide treatment (HR, 1.1; 95% CI, 0.9 to 1.2; P = .4). Sensitivity analyses confirmed the association – patients who received more than 10 cycles of docetaxel had a median overall survival of 33 months (30-37 months), versus 27 months (24-30 months) with 8-10 cycles and about 23 months (18-27 months) with 5-7 cycles (P less than .001).
Extending docetaxel chemotherapy significantly improved overall survival in patients with metastatic castration-resistant prostate cancer (mCRPC), regardless of whether they received lenalidomide, according to a retrospective analysis of 1,059 patients from a randomized, phase III trial.
“We found a robust and independent effect on overall survival by the number of docetaxel cycles administered in the setting of mCRPC,” wrote Ellen de Morree of Erasmus MC Cancer Institute (Rotterdam, the Netherlands) and her associates (JAMA Oncol. 2016 Aug 25. doi: 10.1001/jamaoncol.2016.3000).
The association was independent of performance status (Eastern Cooperative Oncology Group score) or baseline levels of lactate dehydrogenase level, hemoglobin, and albumin, they noted. “These data indicate that patients who appear to have clinical, radiological, or biochemical benefit by docetaxel should continue beyond 6 cycles as long as they tolerate their treatment well,” they concluded.
This study, the first to investigate the optimal number of docetaxel cycles in mCRPC, analyzed data from the multicenter Mainsail trial, in which patients received docetaxel, prednisone, and lenalidomide (DPL) or docetaxel, prednisone, and a placebo (DP) until they developed progressive disease or unacceptable adverse effects. Although dose intensity was similar between the two trial arms, DPL patients developed myelotoxicity with lenalidomide and therefore received a median of only six treatment cycles, while DP patients received a median of eight cycles. That difference enabled this analysis, the investigators noted.
Cumulative dose of docetaxel, duration of lenalidomide treatment, and allocated treatment regimen were significant predictors of overall survival in the univariate analysis. Overall survival was associated with treatment arm in a multivariable analysis that did not account for number of docetaxel cycles (hazard ratio, 1.6; 95% confidence interval, 1.2 to 2.1; P less than .001). But that changed after the addition of a number of docetaxel cycles to the model, the researchers said. In this final model, treatment with eight or more cycles of docetaxel led to substantially improved overall survival (hazard ratio, 1.9; P less than .001), regardless of lenalidomide treatment (HR, 1.1; 95% CI, 0.9 to 1.2; P = .4). Sensitivity analyses confirmed the association – patients who received more than 10 cycles of docetaxel had a median overall survival of 33 months (30-37 months), versus 27 months (24-30 months) with 8-10 cycles and about 23 months (18-27 months) with 5-7 cycles (P less than .001).
FROM JAMA ONCOLOGY
Key clinical point: The number of docetaxel cycles independently predicted overall survival in metastatic castration-resistant prostate cancer (mCRPC).
Major finding: Treatment with eight or more cycles of docetaxel led to improved overall survival (hazard ratio, 1.9; P less than .001) regardless of lenalidomide treatment.
Data source: A retrospective study of 1,059 patients from the Mainsail trial, a randomized phase III study that compared docetaxel, prednisone, and lenalidomide with docetaxel, prednisone, and placebo.
Disclosures: Celgene funded the study but had no role in its design, conduct, interpretation, or in manuscript preparation or review. Dr. Morree had no disclosures.
Amyloid pathology associated with neuropsychiatric symptoms in MCI
TORONTO – Patients with mild cognitive impairment have a greater likelihood of having neuropsychiatric symptoms if they test positive for amyloid pathology on PET imaging, according to a study of patients in the Alzheimer’s Disease Neuroimaging Initiative.
Amyloid-positive patients were significantly more likely to develop agitation, anxiety, apathy, and other symptoms over 4 years than were amyloid-negative patients, Naira Goukasian said at the Alzheimer’s Association International Conference 2016.
“In MCI [mild cognitive impairment], we found that amyloid pathology was a significant risk factor for developing these symptoms,” said Ms. Goukasian, a researcher at the University of California, Los Angeles.

She investigated the presence and development of neuropsychiatric symptoms in 1,077 subjects drawn from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) cohort. The cohort comprised 275 cognitively normal subjects, 100 with subjective memory complaint, 559 with MCI, and 143 with Alzheimer’s disease. As part of ADNI, all patients had baseline neurocognitive and neuropsychiatric testing, and florbetapir F 18 (Amyvid) scans to determine brain amyloid status. Neuropsychiatric symptoms were measured with the Neuropsychiatric Inventory (NPI) and the Neuropsychiatric Inventory Questionnaire (NPI-Q) at baseline and during every annual visit. Patients were followed for up to 4 years.
At baseline, amyloid pathology was associated with some neuropsychiatric symptomatology in every group except those with subjective memory complaints.
Amyloid-positive control subjects were significantly more likely to present with depression than were amyloid-negative controls. Amyloid-positive MCI patients were significantly more likely to present with anxiety when they had amyloid pathology than when they did not. Amyloid-positive dementia patients were significantly more likely to present with apathy than were amyloid-negative dementia patients.
There were no amyloid-dependent differences in neuropsychiatric symptoms among those with subjective memory complaints.
Over the 4-year follow-up period, no new neuropsychiatric symptoms developed in the control, subjective memory complaint, or dementia groups, whether they were amyloid positive or negative.
Amyloid-positive MCI patients, however, were significantly more likely to develop new symptoms than were amyloid-negative MCI patients, including delusions (13% vs. 2%), hallucinations (8% vs. 2%), anxiety (36% vs. 25%), apathy (38% vs. 22%), agitation (36% vs. 27%), disinhibition (24% vs. 15%), irritability (46% vs. 33%), and motor disturbances (18% vs. 9%).
Ms. Goukasian did not elaborate on the pathophysiologic relationship between amyloid and these symptoms. However, a 2015 study using a similar ADNI cohort localized some of them to specific amyloid-burdened brain regions (J Alzheimers Dis. 2016;49[2]:387-98).
The study by David Bensamoun, MD, and colleagues comprised 657 ADNI participants (230 controls, 308 MCI patients, and 119 Alzheimer’s patients).
In the entire group, Dr. Bensamoun, of the Regional Memory Center, Nice, France, found positive significant correlations between anxiety and global cerebral florbetapir F 18 uptake, as well as uptake in the frontal and cingulate regions. Irritability was associated with global florbetapir F 18 uptake and increased signal in the frontal, cingulate, and parietal regions.
In the MCI subgroup, there was an association between anxiety and frontal and global cerebral uptake. In the Alzheimer’s subgroup, there was an association between irritability and parietal uptake.
“Anxiety and irritability appear to be associated with greater amyloid deposition in the neurodegenerative process leading to Alzheimer’s,” the investigators said.
Ms. Goukasian had no financial disclosures.
On Twitter @alz_gal
TORONTO – Patients with mild cognitive impairment have a greater likelihood of having neuropsychiatric symptoms if they test positive for amyloid pathology on PET imaging, according to a study of patients in the Alzheimer’s Disease Neuroimaging Initiative.
Amyloid-positive patients were significantly more likely to develop agitation, anxiety, apathy, and other symptoms over 4 years than were amyloid-negative patients, Naira Goukasian said at the Alzheimer’s Association International Conference 2016.
“In MCI [mild cognitive impairment], we found that amyloid pathology was a significant risk factor for developing these symptoms,” said Ms. Goukasian, a researcher at the University of California, Los Angeles.
She investigated the presence and development of neuropsychiatric symptoms in 1,077 subjects drawn from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) cohort. The cohort comprised 275 cognitively normal subjects, 100 with subjective memory complaint, 559 with MCI, and 143 with Alzheimer’s disease. As part of ADNI, all patients had baseline neurocognitive and neuropsychiatric testing, and florbetapir F 18 (Amyvid) scans to determine brain amyloid status. Neuropsychiatric symptoms were measured with the Neuropsychiatric Inventory (NPI) and the Neuropsychiatric Inventory Questionnaire (NPI-Q) at baseline and during every annual visit. Patients were followed for up to 4 years.
At baseline, amyloid pathology was associated with some neuropsychiatric symptomatology in every group except those with subjective memory complaints.
Amyloid-positive control subjects were significantly more likely to present with depression than were amyloid-negative controls. Amyloid-positive MCI patients were significantly more likely to present with anxiety when they had amyloid pathology than when they did not. Amyloid-positive dementia patients were significantly more likely to present with apathy than were amyloid-negative dementia patients.
There were no amyloid-dependent differences in neuropsychiatric symptoms among those with subjective memory complaints.
Over the 4-year follow-up period, no new neuropsychiatric symptoms developed in the control, subjective memory complaint, or dementia groups, whether they were amyloid positive or negative.
Amyloid-positive MCI patients, however, were significantly more likely to develop new symptoms than were amyloid-negative MCI patients, including delusions (13% vs. 2%), hallucinations (8% vs. 2%), anxiety (36% vs. 25%), apathy (38% vs. 22%), agitation (36% vs. 27%), disinhibition (24% vs. 15%), irritability (46% vs. 33%), and motor disturbances (18% vs. 9%).
Ms. Goukasian did not elaborate on the pathophysiologic relationship between amyloid and these symptoms. However, a 2015 study using a similar ADNI cohort localized some of them to specific amyloid-burdened brain regions (J Alzheimers Dis. 2016;49[2]:387-98).
The study by David Bensamoun, MD, and colleagues comprised 657 ADNI participants (230 controls, 308 MCI patients, and 119 Alzheimer’s patients).
In the entire group, Dr. Bensamoun, of the Regional Memory Center, Nice, France, found positive significant correlations between anxiety and global cerebral florbetapir F 18 uptake, as well as uptake in the frontal and cingulate regions. Irritability was associated with global florbetapir F 18 uptake and increased signal in the frontal, cingulate, and parietal regions.
In the MCI subgroup, there was an association between anxiety and frontal and global cerebral uptake. In the Alzheimer’s subgroup, there was an association between irritability and parietal uptake.
“Anxiety and irritability appear to be associated with greater amyloid deposition in the neurodegenerative process leading to Alzheimer’s,” the investigators said.
Ms. Goukasian had no financial disclosures.
On Twitter @alz_gal
TORONTO – Patients with mild cognitive impairment have a greater likelihood of having neuropsychiatric symptoms if they test positive for amyloid pathology on PET imaging, according to a study of patients in the Alzheimer’s Disease Neuroimaging Initiative.
Amyloid-positive patients were significantly more likely to develop agitation, anxiety, apathy, and other symptoms over 4 years than were amyloid-negative patients, Naira Goukasian said at the Alzheimer’s Association International Conference 2016.
“In MCI [mild cognitive impairment], we found that amyloid pathology was a significant risk factor for developing these symptoms,” said Ms. Goukasian, a researcher at the University of California, Los Angeles.
She investigated the presence and development of neuropsychiatric symptoms in 1,077 subjects drawn from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) cohort. The cohort comprised 275 cognitively normal subjects, 100 with subjective memory complaint, 559 with MCI, and 143 with Alzheimer’s disease. As part of ADNI, all patients had baseline neurocognitive and neuropsychiatric testing, and florbetapir F 18 (Amyvid) scans to determine brain amyloid status. Neuropsychiatric symptoms were measured with the Neuropsychiatric Inventory (NPI) and the Neuropsychiatric Inventory Questionnaire (NPI-Q) at baseline and during every annual visit. Patients were followed for up to 4 years.
At baseline, amyloid pathology was associated with some neuropsychiatric symptomatology in every group except those with subjective memory complaints.
Amyloid-positive control subjects were significantly more likely to present with depression than were amyloid-negative controls. Amyloid-positive MCI patients were significantly more likely to present with anxiety when they had amyloid pathology than when they did not. Amyloid-positive dementia patients were significantly more likely to present with apathy than were amyloid-negative dementia patients.
There were no amyloid-dependent differences in neuropsychiatric symptoms among those with subjective memory complaints.
Over the 4-year follow-up period, no new neuropsychiatric symptoms developed in the control, subjective memory complaint, or dementia groups, whether they were amyloid positive or negative.
Amyloid-positive MCI patients, however, were significantly more likely to develop new symptoms than were amyloid-negative MCI patients, including delusions (13% vs. 2%), hallucinations (8% vs. 2%), anxiety (36% vs. 25%), apathy (38% vs. 22%), agitation (36% vs. 27%), disinhibition (24% vs. 15%), irritability (46% vs. 33%), and motor disturbances (18% vs. 9%).
Ms. Goukasian did not elaborate on the pathophysiologic relationship between amyloid and these symptoms. However, a 2015 study using a similar ADNI cohort localized some of them to specific amyloid-burdened brain regions (J Alzheimers Dis. 2016;49[2]:387-98).
The study by David Bensamoun, MD, and colleagues comprised 657 ADNI participants (230 controls, 308 MCI patients, and 119 Alzheimer’s patients).
In the entire group, Dr. Bensamoun, of the Regional Memory Center, Nice, France, found positive significant correlations between anxiety and global cerebral florbetapir F 18 uptake, as well as uptake in the frontal and cingulate regions. Irritability was associated with global florbetapir F 18 uptake and increased signal in the frontal, cingulate, and parietal regions.
In the MCI subgroup, there was an association between anxiety and frontal and global cerebral uptake. In the Alzheimer’s subgroup, there was an association between irritability and parietal uptake.
“Anxiety and irritability appear to be associated with greater amyloid deposition in the neurodegenerative process leading to Alzheimer’s,” the investigators said.
Ms. Goukasian had no financial disclosures.
On Twitter @alz_gal
AT AAIC 2016
Key clinical point: Amyloid pathology is a risk factor for neuropsychiatric symptoms in mild cognitive impairment.
Major finding: Amyloid-positive patients with MCI were more likely than were amyloid-negative patients to develop anxiety (36% vs. 25%), apathy (38% vs. 22%), agitation (36% vs. 27%), and other symptoms.
Data source: The study comprised 1,077 patients drawn from the Alzheimer’s Disease Neuroimaging Initiative.
Disclosures: Ms. Goukasian had no financial disclosures.

Presentation of Primary Ocular Melanoma in an Adult Male
Ocular melanoma is the most common primary intraocular malignancy and can often be fatal. It is relatively uncommon and presents in about 5.1 cases per million population per year. Oftentimes, the patient is asymptomatic at diagnosis and the presentation is highly variable. We present a case of ocular melanoma.
A 68-year-old man with a history of hypertension, osteoarthritis, and coronary artery disease came in after having worsening pain in multiple joints. Review of systems revealed worsening blurry vision and eye floaters. He denied eye pain or other associated complaints. He had no past history of any ocular pigmented lesions or history of skin cancer. Ophthalmology evaluation a few years earlier did not identify any abnormalities. Approximately 10 years prior to presentation, he did have LASIK surgery on both eyes. Subsequent ophthalmological evaluation showed an iris mass, elevated pressure, intra-retinal hemorrhages, and evidence of involvement in the choroid and conjunctivae. This was highly suspicious for iris melanoma of the right eye. He was started on intraocular pressure lowering medications and further workup was initiated. Biopsy confirmed the diagnoses of choroidal melanoma with an iris mass measuring 1 mm radially by 4 mm circumferentially. The mass extended posteriorly and involved well over half his iridocorneal angle resulting in very high intraocular pressure. A metastatic workup was done and was negative at the time. He underwent successful enucleation surgery with prostheses placement. Patient did well until about 1.5 years later when he was found to have multiple liver lesions suggestive of metastasis. This is currently being further evaluated.
No current guidelines exist for the screening of primary ocular melanoma as well as for screening for metastasis in those already diagnosed. Unfortunately, up to 50% of patients with ocular melanoma develop metastases. This case opens the discussion of needing current guidelines for screening and better surveillance in ocular melanomas. It highlights the importance of looking into screening using genomics and also developing targeted therapies, as well as focusing on immunotherapies for these cases.
Ocular melanoma is the most common primary intraocular malignancy and can often be fatal. It is relatively uncommon and presents in about 5.1 cases per million population per year. Oftentimes, the patient is asymptomatic at diagnosis and the presentation is highly variable. We present a case of ocular melanoma.
A 68-year-old man with a history of hypertension, osteoarthritis, and coronary artery disease came in after having worsening pain in multiple joints. Review of systems revealed worsening blurry vision and eye floaters. He denied eye pain or other associated complaints. He had no past history of any ocular pigmented lesions or history of skin cancer. Ophthalmology evaluation a few years earlier did not identify any abnormalities. Approximately 10 years prior to presentation, he did have LASIK surgery on both eyes. Subsequent ophthalmological evaluation showed an iris mass, elevated pressure, intra-retinal hemorrhages, and evidence of involvement in the choroid and conjunctivae. This was highly suspicious for iris melanoma of the right eye. He was started on intraocular pressure lowering medications and further workup was initiated. Biopsy confirmed the diagnoses of choroidal melanoma with an iris mass measuring 1 mm radially by 4 mm circumferentially. The mass extended posteriorly and involved well over half his iridocorneal angle resulting in very high intraocular pressure. A metastatic workup was done and was negative at the time. He underwent successful enucleation surgery with prostheses placement. Patient did well until about 1.5 years later when he was found to have multiple liver lesions suggestive of metastasis. This is currently being further evaluated.
No current guidelines exist for the screening of primary ocular melanoma as well as for screening for metastasis in those already diagnosed. Unfortunately, up to 50% of patients with ocular melanoma develop metastases. This case opens the discussion of needing current guidelines for screening and better surveillance in ocular melanomas. It highlights the importance of looking into screening using genomics and also developing targeted therapies, as well as focusing on immunotherapies for these cases.
Ocular melanoma is the most common primary intraocular malignancy and can often be fatal. It is relatively uncommon and presents in about 5.1 cases per million population per year. Oftentimes, the patient is asymptomatic at diagnosis and the presentation is highly variable. We present a case of ocular melanoma.
A 68-year-old man with a history of hypertension, osteoarthritis, and coronary artery disease came in after having worsening pain in multiple joints. Review of systems revealed worsening blurry vision and eye floaters. He denied eye pain or other associated complaints. He had no past history of any ocular pigmented lesions or history of skin cancer. Ophthalmology evaluation a few years earlier did not identify any abnormalities. Approximately 10 years prior to presentation, he did have LASIK surgery on both eyes. Subsequent ophthalmological evaluation showed an iris mass, elevated pressure, intra-retinal hemorrhages, and evidence of involvement in the choroid and conjunctivae. This was highly suspicious for iris melanoma of the right eye. He was started on intraocular pressure lowering medications and further workup was initiated. Biopsy confirmed the diagnoses of choroidal melanoma with an iris mass measuring 1 mm radially by 4 mm circumferentially. The mass extended posteriorly and involved well over half his iridocorneal angle resulting in very high intraocular pressure. A metastatic workup was done and was negative at the time. He underwent successful enucleation surgery with prostheses placement. Patient did well until about 1.5 years later when he was found to have multiple liver lesions suggestive of metastasis. This is currently being further evaluated.
No current guidelines exist for the screening of primary ocular melanoma as well as for screening for metastasis in those already diagnosed. Unfortunately, up to 50% of patients with ocular melanoma develop metastases. This case opens the discussion of needing current guidelines for screening and better surveillance in ocular melanomas. It highlights the importance of looking into screening using genomics and also developing targeted therapies, as well as focusing on immunotherapies for these cases.