User login
Esophageal Cancer: Current Diagnosis and Management
From the University of Utah School of Medicine, Salt Lake City, UT.
Abstract
- Objective: To review the evaluation, diagnosis, and management of patients with esophageal cancer.
- Methods: Review of the literature.
- Results: Esophageal adenocarcinoma and esophageal squamous cell carcinoma (SCC) are aggressive cancers with a poor prognosis. GERD is the most common cause of esophageal adenocarcinoma, whereas increased alcohol consumption and tobacco commonly lead to esophageal SCC. Diagnosis is made via esophagogastroduodenoscopy and biopsies, and endoscopic ultrasound is typically used for locoregional staging. The endoscopic treatment of dysphagia is complex and several treatment options are available. Patients with locally advanced esophageal cancers are usually treated with neoadjuvant chemoradiation in combination with surgery. Improvement of quality of life is a major goal in patients with unresectable disease.
- Conclusion: Esophageal cancer remains a commonly encountered clinical entity requiring multidisciplinary evaluation and treatment.
Esophageal cancer is an aggressive disease with an overall poor outcome. It is the eighth most common cancer and sixth most common cause of cancer-related death worldwide [1]. In 2012, there were an estimated 456,000 new diagnoses of esophageal cancer and 400,000 deaths worldwide [1]. In the United States alone, an estimated 18,170 cases of esophageal cancer will be diagnosed in 2014, with 15,450 expected deaths [2].
Esophageal cancer includes 2 distinct histologic diseases: esophageal adenocarcinoma and esophageal squamous cell carcinoma (SCC). Overall, esophageal adenocarcinoma has increased in incidence, while the incidence of SCC has decreased in the Western world due to long-term reductions in smoking and alcohol consumption and increased incidence of gastroesophageal reflux disease (GERD) and obesity [3,4]. Esophageal adenocarcinoma accounted for less than 15% of esophageal cancers in the early 1980s, but now represents more than 60% of all esophageal cancers in the United States [5]. Esophageal SCC is still more common in China, central Asia, sub-Saharan Africa, and India and among the African-American and Caucasian female population in the United States [3,5].
Etiology
Esophageal Adenocarcinoma
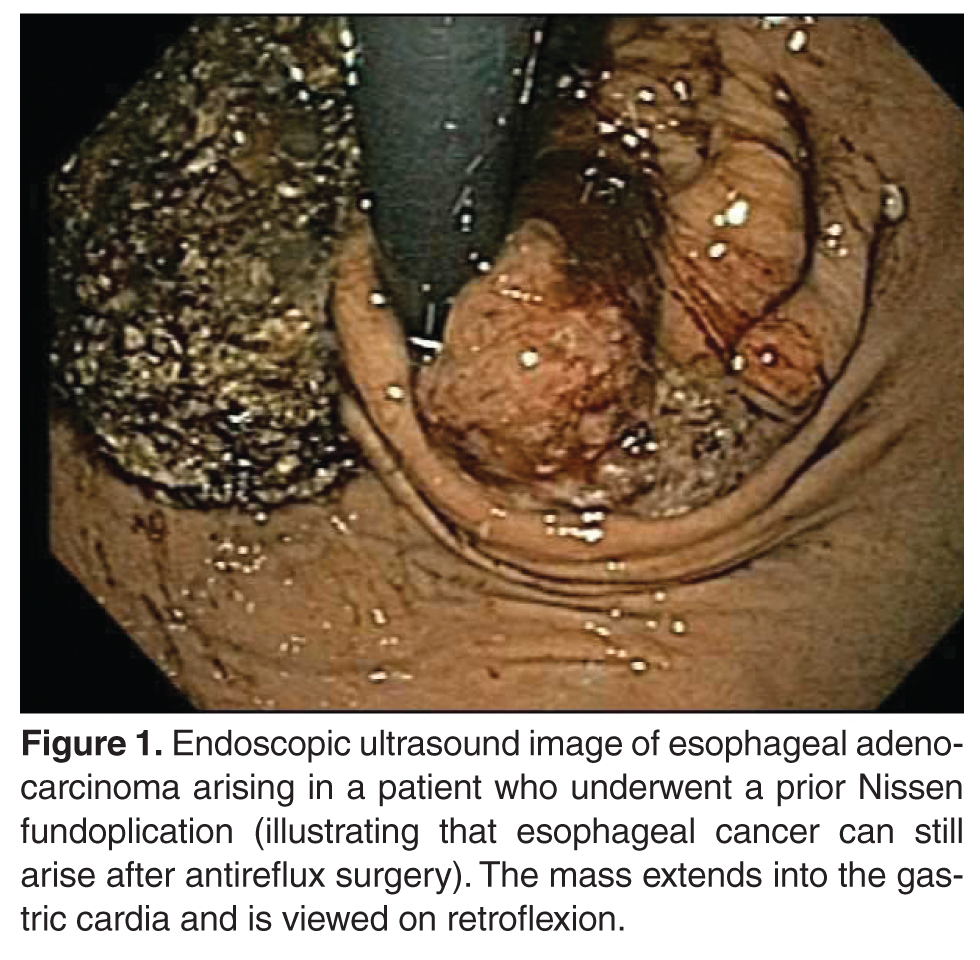
While GERD is the most common cause of esophageal adenocarcinoma, other important causes/risk factors have been identified such as male sex, Caucasian race, older age, and obesity [8,12].In a prospective study by Abnet et al, patients who had a body mass index (BMI) greater than 35 kg/m2 had a significantly increased risk of esophageal adenocarcinoma when compared to patients with a BMI of 18.5 to 25 kg/m2 (hazard ratio [HR], 2.27; 95% CI, 1.44 to 3.59) [13]. Similarly, a recent meta-analysis found that patients with a BMI of 30 kg/m2 or greater had a relative risk for esophageal adenocarcinoma of 2.71 (95% CI, 2.16 to 3.46) [14]. Despite the strong correlation, the etiology of esophageal adenocarcinoma is complex and cannot be fully explained by obesity trends [15].
Smoking is another important risk factor associated with the development of esophageal adenocarcinoma. A study from the Barrett’s and Esophageal Adenocarcinoma Consortium revealed strong associations with esophageal adenocarcinoma and cigarette smoking (OR, 1.96; 95% CI, 1.64 to 2.34) [16]. Furthermore, the study found a statistically significant dose-response association between cigarette smoking and esophageal adenocarcinoma (P < 0.001).
Finally, dietary intake of vegetables and fruits has been shown to reduce the risk of Barrett’s esophagus. In a case-control study, patients with a median intake of 8.3 servings per day of vegetables and fruits had a 73% lower risk of developing Barrett’s esophagus versus those with 2.0 servings per day (OR, 0.27; 95% CI, 0.15 to 0.50) [17]. Each additional serving of vegetables and fruit was associated with a 14% reduction of risk (OR, 0.86; 95% CI, 0.80 to 0.93).
Esophageal Squamous Cell Carcinoma
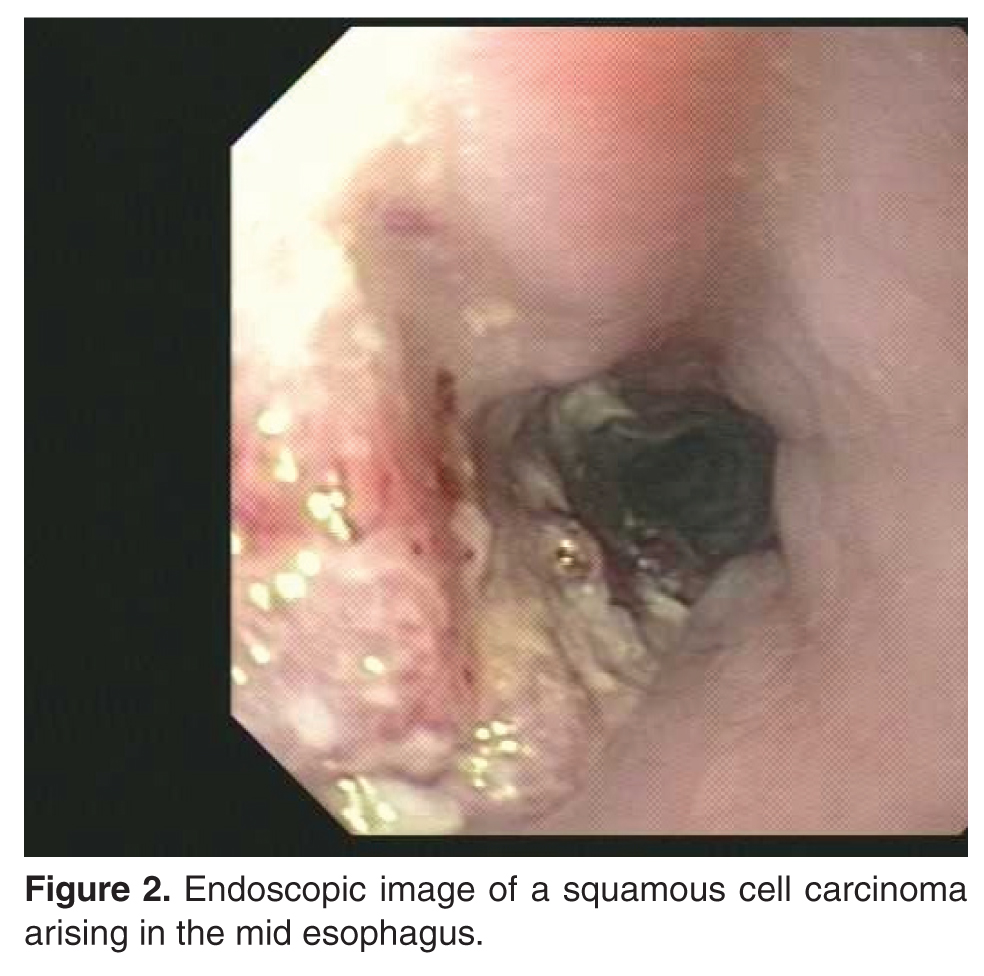
In the study by Freedman et al, when compared with nonsmokers, current cigarette smokers were at significantly increased risk for esophageal SCC (HR, 9.27; 95% CI, 4.04 to 21.29) [18].Smoking has a stronger correlation with esophageal SCC than with esophageal adenocarcinoma [20]. In current smokers, the risk for developing esophageal SCC increases approximately three- to sevenfold [20]. The duration and intensity of smoking has been shown to increase the risk of esophageal SCC as well [21]. Smoking cessation has been shown to reduce the risk of esophageal SCC, but data shows that former cigarette smokers still are at a significant risk [18,21]. In a population-based case-control study, the risk of esophageal SCC in ex-smokers remained elevated for up to 30 years (OR, 1.44; 95% CI, 0.82 to 2.52) [21].
There are only limited studies that have examined the relationship between esophageal SCC and smokeless tobacco and other smoking products. Despite the limited number of studies, smokeless tobacco has been associated with esophageal SCC [22]. In a 2012 study of patients from India, chewing nass (a mix of tobacco, ash, oil, lime, and coloring and flavoring agents) and smoking hookah were associated with an increased risk of developing esophageal SCC [23].
Other risk factors associated with esophageal SCC include poor oral hygiene, atrophic gastritis, caustic esophageal injuries, and achalasia (likely due to stasis of esophageal contents in the case of achalasia) [24–27].Dietary causes of esophageal SCC have also been implicated in many international studies. Foods containing N-nitroso compounds and diets with selenium and zinc mineral deficiencies have been found to be risk factors for esophageal SCC [20,28–30].Thermal injury to the esophageal mucosa caused by food and beverages served at high temperatures has been shown to increase the risk of esophageal cancer [31]. Also, as seen in esophageal adenocarcinoma, diets rich with vegetables and fruits have been associated with a reduced risk of esophageal SCC [32].
In a meta-analysis of 1813 esophageal cancer cases by Corley et al, the use of aspirin and nonsteroidal anti-inflammatory drugs (NSAIDs) was found to be protective against both esophageal SCC and esophageal adenocarcinoma [33]. The study found a dose-dependent effect in the protective association between aspirin/NSAID use and esophageal cancer. Frequent aspirin/NSAID use was associated with a 46% reduction of the odds for developing any esophageal cancer, whereas intermittent use provided an 18% reduction in the odds. However, any use of aspirin or NSAIDs offered some degree of protection against both esophageal SCC (OR, 0.58; 95% CI, 0.43 to 0.78) and esophageal adenocarcinoma (OR, 0.67; 95% CI, 0.51 to 0.87). The mechanism of the risk reduction with aspirin and NSAIDs is still unclear but may be associated with inhibition of the cyclooxygenase-2 enzyme and the reduction of inflammation [33–35].
Clinical Manifestations
Esophageal cancer commonly presents with dysphagia, weight loss, gastrointestinal reflux, and/or odynophagia. In a study by Daly et al, 74% of esophageal cancer patients reported dysphagia and 16.6% reported having odynophagia at the time of initial diagnosis [36]. Patients can have the sensation of food getting “stuck,” which initially can be overcome by careful chewing and/or dietary modification [37]. A history of trouble swallowing solid foods followed by difficulty with drinking liquids is frequently seen. Some patients complain of regurgitation of undigested foods, and approximately 20% of patients have reported having GERD symptoms [36,37]. Due to the complete or partial esophageal obstruction combined with tumor effects, patients with esophageal cancer often develop significant weight loss. In the study by Daly et al, 57.3% of patients reported weight loss at the time of their cancer diagnosis [36]. Weight loss of more than 10% body mass has been identified as an independent indicator for poor prognosis [36,38]. Pain, dyspnea, hoarseness, and cough occur less frequently but may reflect extensive cancer burden [39]. Some patients with advanced tumors have hematemesis from tumor erosion or have recurrent pneumonias due to tracheobronchial fistulas.
Hepatomegaly, pleural effusion, and lymphadenopathy, especially in Virchow’s node (left supraclavicular fossa), are physical examination findings suggestive of metastatic disease [39]. However, most patients with esophageal cancer will have unremarkable physical examination findings.
It should be noted that patients with early stage lesions (ie, stage T1 lesions) may have minimal or no symptoms, with lesions detected either incidentally or as part of endoscopic screening/surveillance programs.
Diagnostic Studies
For patients with suspected esophageal cancer, a barium swallow is an inexpensive and readily available diagnostic study [39]. A barium swallow may show a mass lesion and/or a stricture. If the barium swallow is suggestive of cancer, the diagnosis is usually confirmed via an esophagogastroduodenoscopy (EGD) and biopsies, although in practice many patients with dysphagia and/or a history suspicious for esophageal cancer will proceed directly to EGD [40]. Findings suspicious for cancer are routinely biopsied [39].Traditionally, the more biopsies obtained (up to 7), the higher the diagnostic yield of cancer [41]. The addition of brush cytology to biopsies has also been found to increase the diagnostic accuracy, although this is not widely performed [41].
Once the diagnosis of cancer is confirmed, a computed tomography (CT) scan of the chest, abdomen, and pelvis with intravenous (IV) contrast is usually the next step in the patient’s evaluation, primarily to detect distant metastasis and to look for peritumoral adenopathy [39]. However, in terms of locoregional tumor staging, CT scans are less sensitive and specific than endoscopic ultrasonography (EUS) [42]. Patients who do not have evidence of metastasis on CT scan typically undergo EUS for definitive locoregional staging.
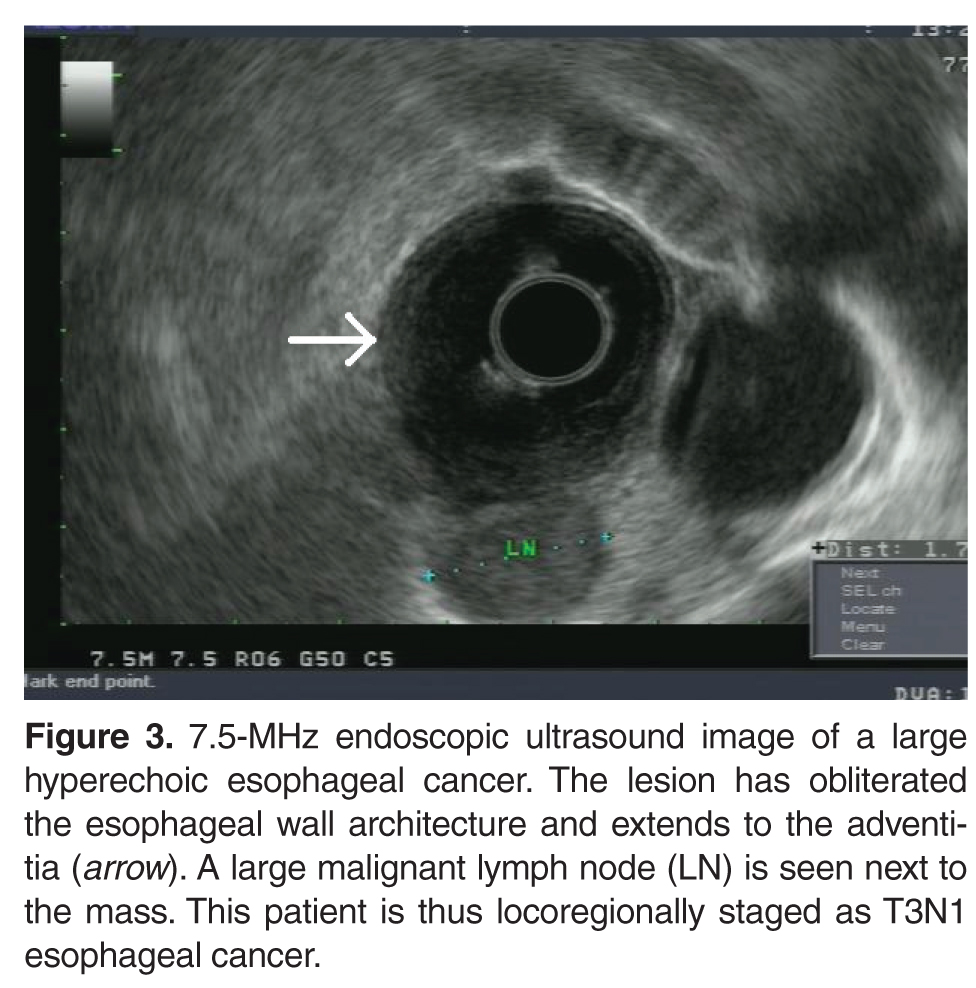
EUS does have its limitations. Between 25% and 36% of patients with esophageal carcinoma present with high-grade malignant strictures that do not allow passage of the scope, although if the exam can show malignant adenopathy and/or tumor extension through the muscularis propria, further evaluation is often of little additional benefit [46]. Dilation of malignant esophageal strictures to facilitate EUS is uncommon as there is a high risk of perforation (up to 24%) [47]. High-frequency (12.5 MHz) EUS mini-probes have been used to interrogate tumors with a very narrow lumen; however, the mini-probes are limited by the penetration depth of the transducer, which can lead to an incomplete locoregional tumor assessment [48]. EUS is not usually used for restaging after neoadjuvant therapy [49].
Endoscopic mucosal resection (EMR) is another technique for staging and treatment of superficial neoplasms (see Treatment section for more details). EMR is critical for distinguishing between T1a lesions (often candidates for definitive endoscopic therapy given the low likelihood of nodal involvement) versus T1b lesions (invasive to submucosa and more likely to prompt surgical esophagectomy with lymph node sampling). The distinction between T1a and T1b disease cannot be established as reliably by EUS when compared with EMR. The American Society for Gastrointestinal Endoscopy 2013 guidelines recommend EMR for the treatment and staging of nodular Barrett’s esophagus and suspected intramucosal adenocarcinoma [50].
Looking for distant metastasis, or M staging, is carried out with EUS, diagnostic laparoscopy/thoracoscopy, and CT and/or positron emission tomography (PET) scans. Despite the high accuracy of esophageal cancer staging with laparoscopy and thoracoscopy, these are invasive procedures and have generally been replaced by PET scan [39,51,52]. PET with 18F-fludeoxyglucose has been shown to significantly improve the detection rate of metastatic disease compared with the conventional staging methods (CT scan and EUS) [53]. In a prospective study, PET scans detected metastasis in 15% of patients who were thought to have localized cancer by conventional staging modalities [39,54].
Unlike several other cancers, tumor markers such as carbohydrate antigen (CA) 19-9, CA 125, and carcinoembryonic antigen (CEA) have low specificity and sensitivity in esophageal cancer and are not routinely obtained and/or followed [39,55].
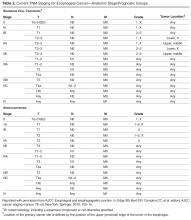
Staging

Treatment

Early Stage
Historically, patients with early stage esophageal cancer (those without evidence of deep invasion into the esophageal wall and no evidence of peritumoral malignant adenopathy or metastases, typically T1N0M0) were referred for esophagectomy [59]. Recent treatment trends suggest proportionately more patients with T1 disease are being treated endoscopically (up to 29% of patients) and proportionately fewer with esophagectomy [60]. EMR has emerged as a viable alternative treatment to esophagectomy when the lesion is staged T1aN0 (tumor invading the lamina propria or muscularis mucosae but not the submucosa) [3]. EMR is performed via several techniques, but most commonly as follows. First, saline is injected under the lesion to create a submucosal cushion, separating the lesion from the underlying muscularis propria. The actual endoscopic resection of the lesion is usually accomplished via snare electrocautery and the resected lesion is sent for pathologic analysis. Endoscopic caps and band ligation devices are available to facilitate removal of the lesion in one or more pieces [61].
In a retrospective cohort study by Prasad et al of 178 patients from 1998 to 2007, the cumulative mortality in the EMR group was comparable to that of the surgery group (17% vs. 20%, respectively, P = 0.75) [62]. Recurrent cancer was detected in 12% of EMR patients; however, all patients were successfully re-treated without affecting overall survival.
In another study of 742 patients, long-term survival in those with early esophageal cancer managed with endoscopic therapy was comparable to that in patients treated with surgical resection [63]. The median cancer-free survival in the endoscopic group was not significantly different from that in the surgical group (56 and 59 months, respectively, P = 0.41) The study found that the relative hazard for 1esophageal cancer–specific mortality in the endoscopic group did not differ from that of the surgical group (relative hazard, 0.89; 95% CI, 0.51 to 1.56; P = 0.68).
Locally Advanced Disease
Neoadjuvant Therapy
For patients with locally advanced cancer (ie, patients without distant metastases who have extension of the primary tumor into the deeper layers of the esophageal wall, including the muscularis propria and the adventitia with or without peritumoral malignant adenopathy, or T2 or T3 lesions with N0 or N1, N2, or N3 status, neoadjuvant therapy is the norm, although the optimal management remains controversial and treatment protocols vary around the world [3,62]. Most neoadjuvant therapy regimens in the United States combine chemotherapy and external beam radiation therapy.
Neoadjuvant treatment with chemoradiation has been found to be beneficial in all esophageal cancers [3,64]. A meta-analysis of 1209 patients found a significant survival benefit for preoperative chemoradiotherapy and, to a lesser extent, for chemotherapy when compared to surgery alone [65]. When comparing neoadjuvant chemoradiotherapy to surgery alone, there was a 19% decrease in the risk of death corresponding to a 13% absolute difference in 2-year survival in the neoadjuvant chemotherapy group. HR for all-cause mortality with neoadjuvant chemoradiotherapy versus surgery alone was 0.81 (95% CI, 0.70 to 0.93; P = 0.002). The benefits of neoadjuvant chemoradiotherapy were similar for both esophageal SCC and adenocarcinoma. The benefits of chemotherapy, however, were less than chemoradiotherapy. When comparing neoadjuvant chemotherapy to surgery alone, there was an absolute survival benefit of 7%.
Following neoadjuvant therapy, patients typically undergo restaging via cross-sectional imaging, most commonly PET/CT scans. If the patient is felt to have active residual disease and has not developed metastases or contraindications to surgery, esophagectomy is appropriate. Some data suggests that patients with esophageal SCC who have complete clinical response after chemoradiation can be observed closely rather than proceed to surgery [3,62,66]. However, the data concerning the usefulness of definitive chemoradiotherapy in esophageal adenocarcinoma is lacking at this time. In a retrospective study of nonmetastatic esophageal adenocarcinoma patients by Tougeron et al comparing surgical patients (± preoperative treatment) to definitive chemoradiotherapy patients, a complete resection was achieved in 92.5% of patients in the surgical group and a clinical complete response was observed in 49.4% of patients with definitive chemoradiotherapy [67]. The overall survival was 36.2 ± 2 months for the surgery group versus 16.5 ± 0.8 months for the definitive chemoradiotherapy group (P = 0.02).
Stenting Prior to Neoadjuvant Therapy
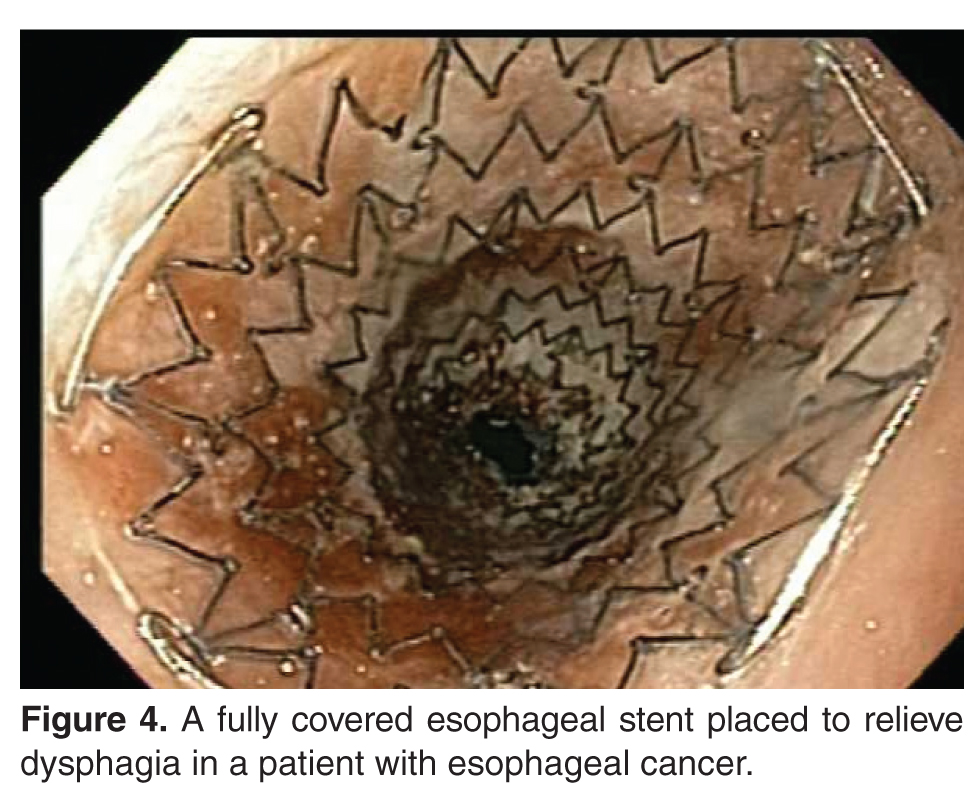
In a meta-analysis of 9 studies comprising 180 patients, placement of esophageal stents in patients with locally advanced esophageal cancer significantly improved dysphagia and allowed for oral nutrition during neoadjuvant therapy [69]. There was a substantial decrease in the dysphagia scores standard difference in means (SDM) of –0.81 (standard error, 0.15; 95% CI, –1.1 to –0.51), an increase in weight SDM of 0.591 (standard error, 0.434; 95% CI, –0.261 to 1.442), and an increase in serum albumin SDM of 0.35 (standard error, 0.271; 95% CI, –0.181 to 0.881). The overall procedural success rate was 95% (95% CI, 0.895 to 0.977). Major adverse events included stent migration in 32% of patients (95% CI, 0.258 to 0.395) and chest discomfort in 51.4% (95% CI, 0.206 to 0.812). However, it was believed that the stent migration may have been a sign of tumor response to neoadjuvant therapy.
In a prospective nonrandomized study of 13 patients with polyflex stents (polyester mesh stents covered in a silicone membrane) placed prior to neoadjuvant therapy, similar improvements with dysphagia scores were observed after stent placement [70]. In the study, the mean baseline dysphagia score at the time of stent placement was 3. Dysphagia scores were subsequently obtained at 1, 2, 3, and 4 weeks after stent placement and were 1.1, 0.8, 0.9, and 1.0, respectively (P = 0.005, P = 0.01, P = 0.02, and P = 0.008, respectively). There were no episodes of bleeding or esophageal perforation. Immediate complications from stenting included chest discomfort, seen in 12 of the 13 patients. Stent migration occurred at some point in 6 of 13 patients, although not all patients with a migrated stent required stent replacement. Again, it was thought that the stent migration could be a sign of tumor response to neoadjuvant therapy.
Surgery
Surgery is an essential part of treatment of esophageal cancer [3,71]. Transthoracic, transhiatal, and radical (en bloc) are the 3 different basic approaches for esophagectomy [3]. Because it does not require a thoracotomy, the transhiatal approach has a theoretical advantage of decreased morbidity and mortality, although several studies have shown no differences in outcome between the transthoracic and transhiatal approach [3,72,73]. In a study by Chang et al comparing the transhiatal to the transthoracic approach, the 5-year survival was higher for patients undergoing transhiatal versus transthoracic esophagectomy (30.5% vs. 22.7%, P = 0.02) [73]. However, after adjusting for differences in tumor stage and patient and provider factors the survival advantage was no longer statistically significant (adjusted HR for mortality, 0.95; 95% CI, 0.75 to 1.20).
Adjuvant Therapy
Despite the benefits of chemoradiation as a neoadjuvant treatment, the data for chemoradiation as adjuvant therapy after resection is lacking in most clinical situations [74].
Metastatic Disease
Between 25% and 40% of esophageal cancer patients will present with metastases to liver, bone, and lung or widespread nodal metastases [61].Improvement of quality of life is a major goal in patients with unresectable disease. Patients with nonsurgical esophageal cancer who have an estimated life expectancy of greater than a few weeks are recommended to have concurrent chemoradiotherapy as most patients have symptomatic obstructive disease and dysphagia [62]. A study by Harvey et al examined the palliative benefit of chemoradiotherapy on dysphagia versus toxicity in patients with invasive esophageal carcinoma [75]. The study found that treatment was well tolerated, with only 5% of patients failing to complete treatment. The study used the DeMeester (4-point) symptom scores for the assessment of dysphagia. The median baseline score at presentation was 2 (moderate: difficulty with soft food, predominately liquid diet). After chemoradiotherapy, 49% of patients were assessed as having a dysphagia score of 0 (no dysphagia). Of those patients who received chemoradiotherapy, 78% had an improvement of at least 1 grade in their DeMeester dysphagia, while only 14% of patients did not improve with therapy. The median survival for the study population was 7 months, with a 6% treatment-related mortality. Chemoradiation therapy as a primary treatment for dysphagia can take days to weeks to take effect, and can be associated with significant pain, usually from radiation esophagitis.
Other alternatives for palliation of nonresectable esophageal cancer include esophageal stenting with SEMS and brachytherapy. SEMS are effective and safe for palliation of dysphagia caused by primary esophageal tumors, postoperative cancer recurrence, esophagorespiratory fistulae, and tumors near the upper esophageal sphincter [76]. A study looking at the use of esophageal SEMS in cancer found that after SEMS placement, the dysphagia score improved from a mean of 3.6 to 1.6 (P < 0.001) [75]. The procedure was technically successful in 96% of the patients. In all cases, esophagorespiratory fistulas were occluded. Pain, reflux, and stent migration are the most common complications of esophageal SEMS.
In a study comparing single-dose brachytherapy versus SEMS, the SEMS group had quicker improvement of dysphagia symptoms than the brachytherapy group, but the long-term relief of dysphagia was better after brachytherapy [77]. In addition, SEMS placement had more complications than brachytherapy (33% vs. 21%, respectively; P = 0.02), which was mainly due to an increased incidence of late hemorrhage. However, brachytherapy and SEMS did not differ in terms of median survival (P = 0.23) or recurrent or persistent dysphagia (P = 0.81).
Tracheoesophageal fistulas may develop in the setting of a locally advanced tumor, or as a complication of RT or chemoradiotherapy. SEMS can also be used successfully in the palliation therapy for tracheoesophageal fistulas or post-esophagectomy anastomotic strictures [78].
Prognosis
The overall survival for patients with resectable esophageal cancer has improved significantly over the past 30 years; however, more than 50% of patients presenting with esophageal cancer will have unresectable or metastatic disease at the time of presentation [3,39,79].Prognosis is primarily TMN stage–dependent, as patients with early stage cancer limited to the mucosa are expected to have curable disease [3]. Poor prognostic predictors include advanced stage cancer, dysphagia, advanced age, large tumors, more than 10% loss in body mass, and malignant adenopathy [39,80–84].
In 2010, the American Joint Committee on Cancer/International Union against Cancer Staging system looked at the prognosis of 4627 patients who underwent esophagectomy alone without radiation or chemotherapy [3,56]. For stage Tis (tumor in situ or high-grade dysplasia) and 1A cancers, there was an approximate 80% 5-year risk-adjusted survival rate [3,56]. The survival rate was marginally better for esophageal adenocarcinoma than for esophageal SCC. With surgery alone, stage 1B disease had a 5-year survival of 62% with SCC and 64% with adenocarcinoma [3,56]. For patients with stage 2A cancer, the 5-year survival was 55% for SCC and 50% for adenocarcinoma as long as there was not nodal involvement [3,56]. If there was nodal involvement, the survival rate dropped to 40% for stage 2B cancer, 25% for stage 3A cancer, and 15% to 17% for stage 3B to 3C cancer [3,56]. As stated earlier, neoadjuvant chemoradiation helps improve outcomes when compared to surgery alone (see Neoadjuvant Therapy in the Treatment section). Thus, one would expect a slightly better prognosis with neoadjuvant therapy and surgery than the previously stated data for surgery alone. Unfortunately, patients with unresectable or metastatic disease at time of diagnosis have a poor prognosis, with a 1-year survival rate less than 20% [3].
Esophageal cancer patients who are treated successfully need to be followed closely because a majority of esophageal cancers will recur within 3 years of treatment [61]. For the first 3 years post treatment, patients should be followed every 3 to 6 months [61]. For 3 to 5 years after treatment, patients should be followed every 6 months and annually thereafter [61]. During each visit, patients should have a thorough history and physical exam and assessment of quality of life [61]. Laboratory studies and EGD are performed as clinically indicated [61]. The importance of intensive post-treatment endoscopic surveillance should be emphasized given a defined rate of disease recurrence. Additionally, radiographic imaging such as CT of the chest and abdomen with contrast or PET/CT may be needed for restaging purposes [61].
Conclusion
Esophageal adenocarcinoma and esophageal SCC are aggressive cancers with poor prognosis. Overall, esophageal adenocarcinoma has increased in incidence, while the incidence of SCC has decreased in the Western world. GERD is the most common cause of esophageal adenocarcinoma, whereas increased alcohol consumption and tobacco commonly lead to esophageal SCC.
For patients with suspected esophageal cancer, a barium swallow is an inexpensive initial diagnostic study that is usually followed up with EGD with biopsies if suggestive of cancer. Once cancer is confirmed, a CT scan with intravenous contrast is obtained to look for adenopathy and metastasis. Those who do not have evidence of metastasis on CT scan typically undergo EUS for definitive locoregional staging.
In the past, patients with early stage esophageal cancer were referred for esophagectomy, but recently EMR has emerged as a viable alternative. Patients with locally advanced esophageal cancers are usually treated with neoadjuvant chemoradiation in combination with surgery. In addition, several studies have showed that esophageal stenting prior to neoadjuvant treatment significantly improves patients’ dysphagia. Unfortunately, many patients still initially present with metastatic or nonresectable disease. Improvement of quality of life is a major goal in patients with unresectable disease. Chemoradiotherapy, esophageal stenting, and brachytherapy are options for improvement of quality of life. Further studies are still needed to evaluate current and new therapeutic guidelines for resectable and nonresectable disease.
Corresponding author: Douglas G. Adler, MD, 30N 1900E 4R118, Salt Lake City, UT 84132, [email protected].
Financial disclosures: None.
1. Ferlay J, Soerjomataram I, Ervik M, et al. GLOBOCAN 2012 v1.0, Cancer Incidence and Mortality Worldwide: IARC CancerBase No. 11 [Internet]. Lyon, France: International Agency for Research on Cancer; 2013. http://globocan.iarc.fr. Accessed on May 5, 2014.
2. Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin 2014;64:9–29.
3. Nieman DR, Peters JH. Treatment strategies for esophageal cancer. Gastroenterol Clin North Am 2013;42:187–97.
4. Shridhar R, Almhanna K, Meredith KL, et al. Radiation therapy and esophageal cancer. Cancer Control 2013;20:97–110.
5. Baquet CR, Commiskey P, Mack K, et al. Esophageal cancer epidemiology in blacks and whites: racial and gender disparities in incidence, mortality, survival rates and histology. J Natl Med Assoc 2005;97:1471–8.
6. Rubenstein JH, Taylor JB. Meta-analysis: the association of oesophagealadenocarcinoma with symptoms of gastro-oesophageal reflux. Aliment Pharmacol Ther 2010;32:1222–7.
7. Lagergren J, Bergström R, Lindgren A, Nyrén O. Symptomatic gastroesophageal reflux as a risk factor for esophageal adenocarcinoma. N Engl J Med 1999;340:825–31.
8. Pohl H, Wrobel K, Bojarski C, et al. Risk factors in the development of esophageal adenocarcinoma. Am J Gastroenterol 2013;108:200–7.
9. Coleman HG, Bhat SK, Murray LJ, et al. Symptoms and endoscopic features at Barrett’s esophagus diagnosis: implications for neoplastic progression risk. Am J Gastroenterol 2014;109:527–34.
10. Nguyen DM, El-Serag HB, Henderson L, et al. Medication usage and the risk of neoplasia in patients with Barrett’s esophagus. Clin Gastroenterol Hepatol 2009;7:1299–304.
11. Lagergren J, Ye W, Lagergren P, Lu Y. The risk of esophageal adenocarcinomaafter antireflux surgery. Gastroenterology 2010;138:1297–301.
12. el-Serag HB. The epidemic of esophageal adenocarcinoma. Gastroenterol Clin North Am 2002;31:421–40, viii.
13. Abnet CC, Freedman ND, Hollenbeck AR, et al. A prospective study of BMI and risk of oesophageal and gastric adenocarcinoma. Eur J Cancer 2008;44:465–71.
14. Turati F, Tramacere I, La Vecchia C, Negri E. A meta-analysis of body mass index and esophageal and gastric cardia adenocarcinoma. Ann Oncol 2013;24:609–17.
15. Kroep S, Lansdorp-Vogelaar I, Rubenstein JH, et al. Comparing trends in esophageal adenocarcinoma incidence and lifestyle factors between the United States, Spain, and the Netherlands. Am J Gastroenterol 2014;109:336–43.
16. Cook MB, Kamangar F, Whiteman DC, et al. Cigarette smoking and adenocarcinomas of the esophagus and esophagogastric junction: a pooled analysis from the international BEACON consortium. J Natl Cancer Inst 2010;102:1344–53.
17. Kubo A, Levin TR, Block G, et al. Dietary antioxidants, fruits, and vegetables and the risk of Barrett’s esophagus. Am J Gastroenterol 2008;103:1614–23.
18. Freedman ND, Abnet CC, Leitzmann MF, et al. A prospective study of tobacco, alcohol, and the risk of esophageal and gastric cancer subtypes. Am J Epidemiol 2007;165:1424–33.
19. Pandeya N, Williams G, Green AC, et al; Australian Cancer Study. Alcohol consumption and the risks of adenocarcinoma and squamous cell carcinoma of the esophagus. Gastroenterology 2009;136:1215–24, e1-2.
20. Kamangar F, Chow WH, Abnet CC, Dawsey SM. Environmental causes of esophageal cancer. Gastroenterol Clin North Am 2009;38:27–57, vii.
21. Pandeya N, Williams GM, Sadhegi S, et al. Associations of duration, intensity, and quantity of smoking with adenocarcinoma and squamous cell carcinoma of the esophagus. Am J Epidemiol 2008;168:105–14.
22. Boffetta P, Hecht S, Gray N, et al. Smokeless tobacco and cancer. Lancet Oncol 2008;9:667–75.
23. Dar NA, Bhat GA, Shah IA, eta l. Hookah smoking, nass chewing, and oesophageal squamous cell carcinoma in Kashmir, India. Br J Cancer 2012;107:1618–23.
24. Abnet CC, Kamangar F, Islami F, et al. Tooth loss and lack of regular oral hygiene are associated with higher risk of esophageal squamous cell carcinoma. Cancer Epidemiol Biomarkers Prev 2008;17:3062–8.
25. Islami F, Sheikhattari P, Ren JS, Kamangar F. Gastric atrophy and risk of oesophageal cancer and gastric cardia adenocarcinoma--a systematic review and meta-analysis. Ann Oncol 2011;22:754–60.
26. Appelqvist P, Salmo M. Lye corrosion carcinoma of the esophagus: a review of 63 cases. Cancer 1980;45:2655–8.
27. Sandler RS, Nyrén O, Ekbom A, et al. The risk of esophageal cancer in patients with achalasia. A population-based study. JAMA 1995;274:1359–62.
28. Lu SH, Montesano R, Zhang MS, et al. Relevance of N-nitrosamines to esophageal cancer in China. J Cell Physiol Suppl 1986;4:51–8.
29. Steevens J, van den Brandt PA, Goldbohm RA, Schouten LJ. Selenium status and the risk of esophageal and gastric cancer subtypes: the Netherlands cohort study. Gastroenterology 2010;138:1704–13.
30. Abnet CC, Lai B, Qiao YL, et al. Zinc concentration in esophageal biopsy specimens measured by x-ray fluorescence and esophageal cancer risk. J Natl Cancer Inst 2005;97:301–6.
31. Islami F, Boffetta P, Ren JS, et al. High-temperature beverages and foods and esophageal cancer risk--a systematic review. Int J Cancer 2009;125:491–524.
32. Liu J, Wang J, Leng Y, Lv C. Intake of fruit and vegetables and risk of esophageal squamous cell carcinoma: a meta-analysis of observational studies. Int J Cancer 2013;133:473–85.
33. Corley DA, Kerlikowske K, Verma R, Buffler P. Protective association of aspirin/NSAIDs and esophageal cancer: a systematic review and meta-analysis. Gastroenterology 2003;124:47–56.
34. Ratnasinghe D, Tangrea J, Roth MJ, et al. Expression of cyclooxygenase-2 in human squamous cell carcinoma of the esophagus; an immunohistochemical survey. Anticancer Res 1999;19:171–4.
35. Morris CD, Armstrong GR, Bigley G, et al. Cyclooxygenase-2 expression in the Barrett’s metaplasia-dysplasia-adenocarcinoma sequence. Am J Gastroenterol 2001;96:990–6.
36. Daly JM, Fry WA, Little AG, et al. Esophageal cancer: results of an American College of Surgeons Patient Care Evaluation Study. J Am Coll Surg 2000;190:562–72.
37. Gastrointestinal cancer. In: Fishman MC, Hoffman R, Klausner RD, Thaler MS, editors. Medicine. 5th ed. Baltimore: Lippincott Williams & Wilkins; 2002:423–6.
38. Fein R, Kelsen DP, Geller N, et al. Adenocarcinoma of the esophagus and gastroesophageal junction. Prognostic factors and results of therapy. Cancer 1985;56:2512–8.
39. Enzinger PC, Mayer RJ. Esophageal cancer. N Engl J Med 2003;349:2241–52.
40. Lightdale CJ. Esophageal cancer. Am J Gastroenterol 1999;94:20–9.
41. Graham DY, Schwartz JT, Cain GD, Gyorkey F. Prospective evaluation of biopsy number in the diagnosis of esophageal and gastric carcinoma. Gastroenterology 1982;82:228–31.
42. Lowe VJ, Booya F, Fletcher JG, et al. Comparison of positron emission tomography, computed tomography, and endoscopic ultrasound in the initial staging of patients with esophageal cancer. Mol Imaging Biol 2005;7:422–30.
43. Röesch T. Endoscopic ultrasonography: equipment and technique. Gastrointest Endosc Clin N Am 2005;15:13–31, vii.
44. Vazquez-Sequeiros E, Norton ID, Clain JE, et al. Impact of EUS-guided fine-needle aspiration on lymph node staging in patients with esophageal carcinoma. Gastrointest Endosc 2001;53:751–7.
45. Van Dam J. Endosonographic evaluation of the patient with esophageal cancer. Chest 1997;112(4 Suppl):184S–190S.
46. Catalano MF, Van Dam J, Sivak MV Jr. Malignant esophageal strictures: staging accuracy of endoscopic ultrasonography. Gastrointest Endosc 1995;41:535–9.
47. Van Dam J, Rice TW, Catalano MF, et al. High-grade malignant stricture is predictive of esophageal tumor stage. Risks of endosonographic evaluation. Cancer 1993;71:2910–7.
48. Hünerbein M, Ghadimi BM, Haensch W, Schlag PM. Transendoscopic ultrasound of esophageal and gastric cancer using miniaturized ultrasound catheter probes. Gastrointest Endosc 1998;48:371–5.
49. Zuccaro G Jr, Rice TW, Goldblum J, et al. Endoscopic ultrasound cannot determine suitability for esophagectomy after aggressive chemoradiotherapy for esophageal cancer. Am J Gastroenterol 1999;94:906–12.
50. ASGE Standards of Practice Committee, Evans JA, Early DS, Chandraskhara V, et al; American Society for Gastrointestinal Endoscopy. The role of endoscopy in the assessment and treatment of esophageal cancer. Gastrointest Endosc 2013;77:328–34.
51. Luketich JD, Schauer P, Landreneau R, et al. Minimally invasive surgical staging is superior to endoscopic ultrasound in detecting lymph node metastases in esophageal cancer. J Thorac Cardiovasc Surg 1997;114:817–21.
52. Krasna MJ, Flowers JL, Attar S, McLaughlin J. Combined thoracoscopic/laparoscopic staging of esophageal cancer. J Thorac Cardiovasc Surg 1996;111:800–6.
53. Flamen P, Lerut A, Van Cutsem E, et al. Utility of positron emission tomography for the staging of patients with potentially operable esophageal carcinoma. J Clin Oncol 2000;18:3202–10.
54. Downey RJ, Akhurst T, Ilson D, et al. Whole body 18FDG-PET and the response of esophageal cancer to induction therapy: results of a prospective trial. J Clin Oncol 2003;21:428–32.
55. Mealy K, Feely J, Reid I, et al. Tumour marker detection in oesophageal carcinoma. Eur J Surg Oncol 1996;22:505–7.
56. Rice TW, Rusch VW, Ishwaran H, Blackstone EH; Worldwide Esophageal Cancer Collaboration. Cancer of the esophagus and esophagogastric junction: data-driven staging for the seventh edition of the American Joint Committee on Cancer/International Union Against Cancer Cancer Staging Manuals. Cancer 2010;116:3763–73.
57. Edge SB, Byrd DR, Compton CC, et al, eds. AJCC cancer staging manual. 7th ed. New York: Springer-Verlag; 2009:103–5.
58. Rice TW, Rusch VW, Apperson-Hansen C, et al. Worldwide esophageal cancer collaboration. Dis Esophagus 2009;22:1–8.
59. Villaflor VM, Allaix ME, Minsky B, et al. Multidisciplinary approach for patients with esophageal cancer. World J Gastroenterol 2012;18:6737–46.
60. Ngamruengphong S, Wolfsen HC, Wallace MB. Survival of patients with superficial esophageal adenocarcinoma after endoscopic treatment vs surgery. Clin Gastroenterol Hepatol 2013;11:1424–9.e2.
61. Lin SH, Chang JY. Esophageal cancer: diagnosis and management. Chin J Cancer 2010;29:843–54.
62. Prasad GA, Wu TT, Wigle DA, et al. Endoscopic and surgical treatment of mucosal (T1a) esophageal adenocarcinoma in Barrett’s esophagus. Gastroenterology 2009;137:815–23.
63. Das A, Singh V, Fleischer DE, Sharma VK. A comparison of endoscopic treatment and surgery in early esophageal cancer: an analysis of surveillance epidemiology and end results data. Am J Gastroenterol 2008;103:1340–5.
64. Herskovic A, Martz K, al-Sarraf M, et al. Combined chemotherapy and radiotherapy compared with radiotherapy alone in patients with cancer of the esophagus. N Engl J Med 1992;326:1593–8.
65. Gebski V, Burmeister B, Smithers BM, et al; Australasian Gastro-Intestinal Trials Group. Survival benefits from neoadjuvant chemoradiotherapy or chemotherapy in oesophageal carcinoma: a meta-analysis. Lancet Oncol 2007;8:226–34.
66. Bedenne L, Michel P, Bouché O, et al. Chemoradiation followed by surgery compared with chemoradiation alone in squamous cancer of the esophagus: FFCD 9102. J Clin Oncol 2007;25:1160–8.
67. Tougeron D, Scotté M, Hamidou H, et al. Definitive chemoradiotherapy in patients with esophageal adenocarcinoma: an alternative to surgery? J Surg Oncol 2012;105: 761–6.
68. Siddiqui AA, Sarkar A, Beltz S, et al. Placement of fully covered self-expandable metal stents in patients with locally advanced esophageal cancer before neoadjuvant therapy. Gastrointest Endosc 2012;76:44–51.
69. Nagaraja V, Cox MR, Eslick GD. Safety and efficacy of esophageal stents preceding or during neoadjuvant chemotherapy for esophageal cancer: a systematic review and meta-analysis. J Gastrointest Oncol 2014;5:119–26.
70. Adler DG, Fang J, Wong R, et al. Placement of Polyflex stents in patients with locally advanced esophageal cancer is safe and improves dysphagia during neoadjuvant therapy. Gastrointest Endosc 2009;70:614–9.
71. Dubecz A, Sepesi B, Salvador R, et al. Surgical resection for locoregional esophageal cancer is underutilized in the United States. J Am Coll Surg 2010;211:754–61.
72. Hulscher JB, Tijssen JG, Obertop H, van Lanschot JJ. Transthoracic versus transhiatal resection for carcinoma of the esophagus: a meta-analysis. Ann Thorac Surg 2001;72:306–13.
73. Chang AC, Ji H, Birkmeyer NJ, et al. Outcomes after transhiatal and transthoracic esophagectomy for cancer. Ann Thorac Surg 2008;85:424–9.
74. Almhanna K, Shridhar R, Meredith KL. Neoadjuvant or adjuvant therapy for resectable esophageal cancer: is there a standard of care? Cancer Control 2013;20:89–96.
75. Harvey JA, Bessell JR, Beller E, et al. Chemoradiation therapy is effective for the palliative treatment of malignant dysphagia. Dis Esophagus 2004;17:260–5.
76. Siersema PD, Schrauwen SL, van Blankenstein M, et al; Rotterdam Esophageal Tumor Study Group. Self-expanding metal stents for complicated and recurrent esophagogastric cancer. Gastrointest Endosc 2001;54:579–86.
77. Homs MY, Steyerberg EW, Eijkenboom WM, et al. Single-dose brachytherapy versus metal stent placement for the palliation of dysphagia from oesophageal cancer: multicentre randomised trial. Lancet 2004;364(9444):1497–504.
78. van den Bongard HJ, Boot H, Baas P, Taal BG. The role of parallel stent insertion in patients with esophagorespiratory fistulas. Gastrointest Endosc 2002;55:110–5.
79. Siegel R, DeSantis C, Virgo K, et al. Cancer treatment and survivorship statistics, 2012. CA Cancer J Clin 2012;62:220–41.
80. Fein R, Kelsen DP, Geller N, et al. Adenocarcinoma of the esophagus and gastroesophageal junction. Prognostic factors and results of therapy. Cancer 1985;56:2512–8.
81. Mariette C, Maurel A, Fabre S, et al. [Preoperative prognostic factors for squamous cell carcinomas of the thoracic esophagus]. Gastroenterol Clin Biol 2001;25:468–72.
82. Swanson SJ, Batirel HF, Bueno R, et al. Transthoracic esophagectomy with radical mediastinal and abdominal lymph node dissection and cervical esophagogastrostomy for esophageal carcinoma. Ann Thorac Surg 2001;72:1918–24.
83. Urba SG, Orringer MB, Turrisi A, et al. Randomized trial of preoperative chemoradiation versus surgery alone in patients with locoregional esophageal carcinoma. J Clin Oncol 2001;19:305–13.
84. Hosch SB, Stoecklein NH, Pichlmeier U, et al. Esophageal cancer: the mode of lymphatic tumor cell spread and its prognostic significance. J Clin Oncol 2001;19:1970–5.
From the University of Utah School of Medicine, Salt Lake City, UT.
Abstract
- Objective: To review the evaluation, diagnosis, and management of patients with esophageal cancer.
- Methods: Review of the literature.
- Results: Esophageal adenocarcinoma and esophageal squamous cell carcinoma (SCC) are aggressive cancers with a poor prognosis. GERD is the most common cause of esophageal adenocarcinoma, whereas increased alcohol consumption and tobacco commonly lead to esophageal SCC. Diagnosis is made via esophagogastroduodenoscopy and biopsies, and endoscopic ultrasound is typically used for locoregional staging. The endoscopic treatment of dysphagia is complex and several treatment options are available. Patients with locally advanced esophageal cancers are usually treated with neoadjuvant chemoradiation in combination with surgery. Improvement of quality of life is a major goal in patients with unresectable disease.
- Conclusion: Esophageal cancer remains a commonly encountered clinical entity requiring multidisciplinary evaluation and treatment.
Esophageal cancer is an aggressive disease with an overall poor outcome. It is the eighth most common cancer and sixth most common cause of cancer-related death worldwide [1]. In 2012, there were an estimated 456,000 new diagnoses of esophageal cancer and 400,000 deaths worldwide [1]. In the United States alone, an estimated 18,170 cases of esophageal cancer will be diagnosed in 2014, with 15,450 expected deaths [2].
Esophageal cancer includes 2 distinct histologic diseases: esophageal adenocarcinoma and esophageal squamous cell carcinoma (SCC). Overall, esophageal adenocarcinoma has increased in incidence, while the incidence of SCC has decreased in the Western world due to long-term reductions in smoking and alcohol consumption and increased incidence of gastroesophageal reflux disease (GERD) and obesity [3,4]. Esophageal adenocarcinoma accounted for less than 15% of esophageal cancers in the early 1980s, but now represents more than 60% of all esophageal cancers in the United States [5]. Esophageal SCC is still more common in China, central Asia, sub-Saharan Africa, and India and among the African-American and Caucasian female population in the United States [3,5].
Etiology
Esophageal Adenocarcinoma
While GERD is the most common cause of esophageal adenocarcinoma, other important causes/risk factors have been identified such as male sex, Caucasian race, older age, and obesity [8,12].In a prospective study by Abnet et al, patients who had a body mass index (BMI) greater than 35 kg/m2 had a significantly increased risk of esophageal adenocarcinoma when compared to patients with a BMI of 18.5 to 25 kg/m2 (hazard ratio [HR], 2.27; 95% CI, 1.44 to 3.59) [13]. Similarly, a recent meta-analysis found that patients with a BMI of 30 kg/m2 or greater had a relative risk for esophageal adenocarcinoma of 2.71 (95% CI, 2.16 to 3.46) [14]. Despite the strong correlation, the etiology of esophageal adenocarcinoma is complex and cannot be fully explained by obesity trends [15].
Smoking is another important risk factor associated with the development of esophageal adenocarcinoma. A study from the Barrett’s and Esophageal Adenocarcinoma Consortium revealed strong associations with esophageal adenocarcinoma and cigarette smoking (OR, 1.96; 95% CI, 1.64 to 2.34) [16]. Furthermore, the study found a statistically significant dose-response association between cigarette smoking and esophageal adenocarcinoma (P < 0.001).
Finally, dietary intake of vegetables and fruits has been shown to reduce the risk of Barrett’s esophagus. In a case-control study, patients with a median intake of 8.3 servings per day of vegetables and fruits had a 73% lower risk of developing Barrett’s esophagus versus those with 2.0 servings per day (OR, 0.27; 95% CI, 0.15 to 0.50) [17]. Each additional serving of vegetables and fruit was associated with a 14% reduction of risk (OR, 0.86; 95% CI, 0.80 to 0.93).
Esophageal Squamous Cell Carcinoma
In the study by Freedman et al, when compared with nonsmokers, current cigarette smokers were at significantly increased risk for esophageal SCC (HR, 9.27; 95% CI, 4.04 to 21.29) [18].Smoking has a stronger correlation with esophageal SCC than with esophageal adenocarcinoma [20]. In current smokers, the risk for developing esophageal SCC increases approximately three- to sevenfold [20]. The duration and intensity of smoking has been shown to increase the risk of esophageal SCC as well [21]. Smoking cessation has been shown to reduce the risk of esophageal SCC, but data shows that former cigarette smokers still are at a significant risk [18,21]. In a population-based case-control study, the risk of esophageal SCC in ex-smokers remained elevated for up to 30 years (OR, 1.44; 95% CI, 0.82 to 2.52) [21].
There are only limited studies that have examined the relationship between esophageal SCC and smokeless tobacco and other smoking products. Despite the limited number of studies, smokeless tobacco has been associated with esophageal SCC [22]. In a 2012 study of patients from India, chewing nass (a mix of tobacco, ash, oil, lime, and coloring and flavoring agents) and smoking hookah were associated with an increased risk of developing esophageal SCC [23].
Other risk factors associated with esophageal SCC include poor oral hygiene, atrophic gastritis, caustic esophageal injuries, and achalasia (likely due to stasis of esophageal contents in the case of achalasia) [24–27].Dietary causes of esophageal SCC have also been implicated in many international studies. Foods containing N-nitroso compounds and diets with selenium and zinc mineral deficiencies have been found to be risk factors for esophageal SCC [20,28–30].Thermal injury to the esophageal mucosa caused by food and beverages served at high temperatures has been shown to increase the risk of esophageal cancer [31]. Also, as seen in esophageal adenocarcinoma, diets rich with vegetables and fruits have been associated with a reduced risk of esophageal SCC [32].
In a meta-analysis of 1813 esophageal cancer cases by Corley et al, the use of aspirin and nonsteroidal anti-inflammatory drugs (NSAIDs) was found to be protective against both esophageal SCC and esophageal adenocarcinoma [33]. The study found a dose-dependent effect in the protective association between aspirin/NSAID use and esophageal cancer. Frequent aspirin/NSAID use was associated with a 46% reduction of the odds for developing any esophageal cancer, whereas intermittent use provided an 18% reduction in the odds. However, any use of aspirin or NSAIDs offered some degree of protection against both esophageal SCC (OR, 0.58; 95% CI, 0.43 to 0.78) and esophageal adenocarcinoma (OR, 0.67; 95% CI, 0.51 to 0.87). The mechanism of the risk reduction with aspirin and NSAIDs is still unclear but may be associated with inhibition of the cyclooxygenase-2 enzyme and the reduction of inflammation [33–35].
Clinical Manifestations
Esophageal cancer commonly presents with dysphagia, weight loss, gastrointestinal reflux, and/or odynophagia. In a study by Daly et al, 74% of esophageal cancer patients reported dysphagia and 16.6% reported having odynophagia at the time of initial diagnosis [36]. Patients can have the sensation of food getting “stuck,” which initially can be overcome by careful chewing and/or dietary modification [37]. A history of trouble swallowing solid foods followed by difficulty with drinking liquids is frequently seen. Some patients complain of regurgitation of undigested foods, and approximately 20% of patients have reported having GERD symptoms [36,37]. Due to the complete or partial esophageal obstruction combined with tumor effects, patients with esophageal cancer often develop significant weight loss. In the study by Daly et al, 57.3% of patients reported weight loss at the time of their cancer diagnosis [36]. Weight loss of more than 10% body mass has been identified as an independent indicator for poor prognosis [36,38]. Pain, dyspnea, hoarseness, and cough occur less frequently but may reflect extensive cancer burden [39]. Some patients with advanced tumors have hematemesis from tumor erosion or have recurrent pneumonias due to tracheobronchial fistulas.
Hepatomegaly, pleural effusion, and lymphadenopathy, especially in Virchow’s node (left supraclavicular fossa), are physical examination findings suggestive of metastatic disease [39]. However, most patients with esophageal cancer will have unremarkable physical examination findings.
It should be noted that patients with early stage lesions (ie, stage T1 lesions) may have minimal or no symptoms, with lesions detected either incidentally or as part of endoscopic screening/surveillance programs.
Diagnostic Studies
For patients with suspected esophageal cancer, a barium swallow is an inexpensive and readily available diagnostic study [39]. A barium swallow may show a mass lesion and/or a stricture. If the barium swallow is suggestive of cancer, the diagnosis is usually confirmed via an esophagogastroduodenoscopy (EGD) and biopsies, although in practice many patients with dysphagia and/or a history suspicious for esophageal cancer will proceed directly to EGD [40]. Findings suspicious for cancer are routinely biopsied [39].Traditionally, the more biopsies obtained (up to 7), the higher the diagnostic yield of cancer [41]. The addition of brush cytology to biopsies has also been found to increase the diagnostic accuracy, although this is not widely performed [41].
Once the diagnosis of cancer is confirmed, a computed tomography (CT) scan of the chest, abdomen, and pelvis with intravenous (IV) contrast is usually the next step in the patient’s evaluation, primarily to detect distant metastasis and to look for peritumoral adenopathy [39]. However, in terms of locoregional tumor staging, CT scans are less sensitive and specific than endoscopic ultrasonography (EUS) [42]. Patients who do not have evidence of metastasis on CT scan typically undergo EUS for definitive locoregional staging.
EUS does have its limitations. Between 25% and 36% of patients with esophageal carcinoma present with high-grade malignant strictures that do not allow passage of the scope, although if the exam can show malignant adenopathy and/or tumor extension through the muscularis propria, further evaluation is often of little additional benefit [46]. Dilation of malignant esophageal strictures to facilitate EUS is uncommon as there is a high risk of perforation (up to 24%) [47]. High-frequency (12.5 MHz) EUS mini-probes have been used to interrogate tumors with a very narrow lumen; however, the mini-probes are limited by the penetration depth of the transducer, which can lead to an incomplete locoregional tumor assessment [48]. EUS is not usually used for restaging after neoadjuvant therapy [49].
Endoscopic mucosal resection (EMR) is another technique for staging and treatment of superficial neoplasms (see Treatment section for more details). EMR is critical for distinguishing between T1a lesions (often candidates for definitive endoscopic therapy given the low likelihood of nodal involvement) versus T1b lesions (invasive to submucosa and more likely to prompt surgical esophagectomy with lymph node sampling). The distinction between T1a and T1b disease cannot be established as reliably by EUS when compared with EMR. The American Society for Gastrointestinal Endoscopy 2013 guidelines recommend EMR for the treatment and staging of nodular Barrett’s esophagus and suspected intramucosal adenocarcinoma [50].
Looking for distant metastasis, or M staging, is carried out with EUS, diagnostic laparoscopy/thoracoscopy, and CT and/or positron emission tomography (PET) scans. Despite the high accuracy of esophageal cancer staging with laparoscopy and thoracoscopy, these are invasive procedures and have generally been replaced by PET scan [39,51,52]. PET with 18F-fludeoxyglucose has been shown to significantly improve the detection rate of metastatic disease compared with the conventional staging methods (CT scan and EUS) [53]. In a prospective study, PET scans detected metastasis in 15% of patients who were thought to have localized cancer by conventional staging modalities [39,54].
Unlike several other cancers, tumor markers such as carbohydrate antigen (CA) 19-9, CA 125, and carcinoembryonic antigen (CEA) have low specificity and sensitivity in esophageal cancer and are not routinely obtained and/or followed [39,55].
Staging
Treatment
Early Stage
Historically, patients with early stage esophageal cancer (those without evidence of deep invasion into the esophageal wall and no evidence of peritumoral malignant adenopathy or metastases, typically T1N0M0) were referred for esophagectomy [59]. Recent treatment trends suggest proportionately more patients with T1 disease are being treated endoscopically (up to 29% of patients) and proportionately fewer with esophagectomy [60]. EMR has emerged as a viable alternative treatment to esophagectomy when the lesion is staged T1aN0 (tumor invading the lamina propria or muscularis mucosae but not the submucosa) [3]. EMR is performed via several techniques, but most commonly as follows. First, saline is injected under the lesion to create a submucosal cushion, separating the lesion from the underlying muscularis propria. The actual endoscopic resection of the lesion is usually accomplished via snare electrocautery and the resected lesion is sent for pathologic analysis. Endoscopic caps and band ligation devices are available to facilitate removal of the lesion in one or more pieces [61].
In a retrospective cohort study by Prasad et al of 178 patients from 1998 to 2007, the cumulative mortality in the EMR group was comparable to that of the surgery group (17% vs. 20%, respectively, P = 0.75) [62]. Recurrent cancer was detected in 12% of EMR patients; however, all patients were successfully re-treated without affecting overall survival.
In another study of 742 patients, long-term survival in those with early esophageal cancer managed with endoscopic therapy was comparable to that in patients treated with surgical resection [63]. The median cancer-free survival in the endoscopic group was not significantly different from that in the surgical group (56 and 59 months, respectively, P = 0.41) The study found that the relative hazard for 1esophageal cancer–specific mortality in the endoscopic group did not differ from that of the surgical group (relative hazard, 0.89; 95% CI, 0.51 to 1.56; P = 0.68).
Locally Advanced Disease
Neoadjuvant Therapy
For patients with locally advanced cancer (ie, patients without distant metastases who have extension of the primary tumor into the deeper layers of the esophageal wall, including the muscularis propria and the adventitia with or without peritumoral malignant adenopathy, or T2 or T3 lesions with N0 or N1, N2, or N3 status, neoadjuvant therapy is the norm, although the optimal management remains controversial and treatment protocols vary around the world [3,62]. Most neoadjuvant therapy regimens in the United States combine chemotherapy and external beam radiation therapy.
Neoadjuvant treatment with chemoradiation has been found to be beneficial in all esophageal cancers [3,64]. A meta-analysis of 1209 patients found a significant survival benefit for preoperative chemoradiotherapy and, to a lesser extent, for chemotherapy when compared to surgery alone [65]. When comparing neoadjuvant chemoradiotherapy to surgery alone, there was a 19% decrease in the risk of death corresponding to a 13% absolute difference in 2-year survival in the neoadjuvant chemotherapy group. HR for all-cause mortality with neoadjuvant chemoradiotherapy versus surgery alone was 0.81 (95% CI, 0.70 to 0.93; P = 0.002). The benefits of neoadjuvant chemoradiotherapy were similar for both esophageal SCC and adenocarcinoma. The benefits of chemotherapy, however, were less than chemoradiotherapy. When comparing neoadjuvant chemotherapy to surgery alone, there was an absolute survival benefit of 7%.
Following neoadjuvant therapy, patients typically undergo restaging via cross-sectional imaging, most commonly PET/CT scans. If the patient is felt to have active residual disease and has not developed metastases or contraindications to surgery, esophagectomy is appropriate. Some data suggests that patients with esophageal SCC who have complete clinical response after chemoradiation can be observed closely rather than proceed to surgery [3,62,66]. However, the data concerning the usefulness of definitive chemoradiotherapy in esophageal adenocarcinoma is lacking at this time. In a retrospective study of nonmetastatic esophageal adenocarcinoma patients by Tougeron et al comparing surgical patients (± preoperative treatment) to definitive chemoradiotherapy patients, a complete resection was achieved in 92.5% of patients in the surgical group and a clinical complete response was observed in 49.4% of patients with definitive chemoradiotherapy [67]. The overall survival was 36.2 ± 2 months for the surgery group versus 16.5 ± 0.8 months for the definitive chemoradiotherapy group (P = 0.02).
Stenting Prior to Neoadjuvant Therapy
In a meta-analysis of 9 studies comprising 180 patients, placement of esophageal stents in patients with locally advanced esophageal cancer significantly improved dysphagia and allowed for oral nutrition during neoadjuvant therapy [69]. There was a substantial decrease in the dysphagia scores standard difference in means (SDM) of –0.81 (standard error, 0.15; 95% CI, –1.1 to –0.51), an increase in weight SDM of 0.591 (standard error, 0.434; 95% CI, –0.261 to 1.442), and an increase in serum albumin SDM of 0.35 (standard error, 0.271; 95% CI, –0.181 to 0.881). The overall procedural success rate was 95% (95% CI, 0.895 to 0.977). Major adverse events included stent migration in 32% of patients (95% CI, 0.258 to 0.395) and chest discomfort in 51.4% (95% CI, 0.206 to 0.812). However, it was believed that the stent migration may have been a sign of tumor response to neoadjuvant therapy.
In a prospective nonrandomized study of 13 patients with polyflex stents (polyester mesh stents covered in a silicone membrane) placed prior to neoadjuvant therapy, similar improvements with dysphagia scores were observed after stent placement [70]. In the study, the mean baseline dysphagia score at the time of stent placement was 3. Dysphagia scores were subsequently obtained at 1, 2, 3, and 4 weeks after stent placement and were 1.1, 0.8, 0.9, and 1.0, respectively (P = 0.005, P = 0.01, P = 0.02, and P = 0.008, respectively). There were no episodes of bleeding or esophageal perforation. Immediate complications from stenting included chest discomfort, seen in 12 of the 13 patients. Stent migration occurred at some point in 6 of 13 patients, although not all patients with a migrated stent required stent replacement. Again, it was thought that the stent migration could be a sign of tumor response to neoadjuvant therapy.
Surgery
Surgery is an essential part of treatment of esophageal cancer [3,71]. Transthoracic, transhiatal, and radical (en bloc) are the 3 different basic approaches for esophagectomy [3]. Because it does not require a thoracotomy, the transhiatal approach has a theoretical advantage of decreased morbidity and mortality, although several studies have shown no differences in outcome between the transthoracic and transhiatal approach [3,72,73]. In a study by Chang et al comparing the transhiatal to the transthoracic approach, the 5-year survival was higher for patients undergoing transhiatal versus transthoracic esophagectomy (30.5% vs. 22.7%, P = 0.02) [73]. However, after adjusting for differences in tumor stage and patient and provider factors the survival advantage was no longer statistically significant (adjusted HR for mortality, 0.95; 95% CI, 0.75 to 1.20).
Adjuvant Therapy
Despite the benefits of chemoradiation as a neoadjuvant treatment, the data for chemoradiation as adjuvant therapy after resection is lacking in most clinical situations [74].
Metastatic Disease
Between 25% and 40% of esophageal cancer patients will present with metastases to liver, bone, and lung or widespread nodal metastases [61].Improvement of quality of life is a major goal in patients with unresectable disease. Patients with nonsurgical esophageal cancer who have an estimated life expectancy of greater than a few weeks are recommended to have concurrent chemoradiotherapy as most patients have symptomatic obstructive disease and dysphagia [62]. A study by Harvey et al examined the palliative benefit of chemoradiotherapy on dysphagia versus toxicity in patients with invasive esophageal carcinoma [75]. The study found that treatment was well tolerated, with only 5% of patients failing to complete treatment. The study used the DeMeester (4-point) symptom scores for the assessment of dysphagia. The median baseline score at presentation was 2 (moderate: difficulty with soft food, predominately liquid diet). After chemoradiotherapy, 49% of patients were assessed as having a dysphagia score of 0 (no dysphagia). Of those patients who received chemoradiotherapy, 78% had an improvement of at least 1 grade in their DeMeester dysphagia, while only 14% of patients did not improve with therapy. The median survival for the study population was 7 months, with a 6% treatment-related mortality. Chemoradiation therapy as a primary treatment for dysphagia can take days to weeks to take effect, and can be associated with significant pain, usually from radiation esophagitis.
Other alternatives for palliation of nonresectable esophageal cancer include esophageal stenting with SEMS and brachytherapy. SEMS are effective and safe for palliation of dysphagia caused by primary esophageal tumors, postoperative cancer recurrence, esophagorespiratory fistulae, and tumors near the upper esophageal sphincter [76]. A study looking at the use of esophageal SEMS in cancer found that after SEMS placement, the dysphagia score improved from a mean of 3.6 to 1.6 (P < 0.001) [75]. The procedure was technically successful in 96% of the patients. In all cases, esophagorespiratory fistulas were occluded. Pain, reflux, and stent migration are the most common complications of esophageal SEMS.
In a study comparing single-dose brachytherapy versus SEMS, the SEMS group had quicker improvement of dysphagia symptoms than the brachytherapy group, but the long-term relief of dysphagia was better after brachytherapy [77]. In addition, SEMS placement had more complications than brachytherapy (33% vs. 21%, respectively; P = 0.02), which was mainly due to an increased incidence of late hemorrhage. However, brachytherapy and SEMS did not differ in terms of median survival (P = 0.23) or recurrent or persistent dysphagia (P = 0.81).
Tracheoesophageal fistulas may develop in the setting of a locally advanced tumor, or as a complication of RT or chemoradiotherapy. SEMS can also be used successfully in the palliation therapy for tracheoesophageal fistulas or post-esophagectomy anastomotic strictures [78].
Prognosis
The overall survival for patients with resectable esophageal cancer has improved significantly over the past 30 years; however, more than 50% of patients presenting with esophageal cancer will have unresectable or metastatic disease at the time of presentation [3,39,79].Prognosis is primarily TMN stage–dependent, as patients with early stage cancer limited to the mucosa are expected to have curable disease [3]. Poor prognostic predictors include advanced stage cancer, dysphagia, advanced age, large tumors, more than 10% loss in body mass, and malignant adenopathy [39,80–84].
In 2010, the American Joint Committee on Cancer/International Union against Cancer Staging system looked at the prognosis of 4627 patients who underwent esophagectomy alone without radiation or chemotherapy [3,56]. For stage Tis (tumor in situ or high-grade dysplasia) and 1A cancers, there was an approximate 80% 5-year risk-adjusted survival rate [3,56]. The survival rate was marginally better for esophageal adenocarcinoma than for esophageal SCC. With surgery alone, stage 1B disease had a 5-year survival of 62% with SCC and 64% with adenocarcinoma [3,56]. For patients with stage 2A cancer, the 5-year survival was 55% for SCC and 50% for adenocarcinoma as long as there was not nodal involvement [3,56]. If there was nodal involvement, the survival rate dropped to 40% for stage 2B cancer, 25% for stage 3A cancer, and 15% to 17% for stage 3B to 3C cancer [3,56]. As stated earlier, neoadjuvant chemoradiation helps improve outcomes when compared to surgery alone (see Neoadjuvant Therapy in the Treatment section). Thus, one would expect a slightly better prognosis with neoadjuvant therapy and surgery than the previously stated data for surgery alone. Unfortunately, patients with unresectable or metastatic disease at time of diagnosis have a poor prognosis, with a 1-year survival rate less than 20% [3].
Esophageal cancer patients who are treated successfully need to be followed closely because a majority of esophageal cancers will recur within 3 years of treatment [61]. For the first 3 years post treatment, patients should be followed every 3 to 6 months [61]. For 3 to 5 years after treatment, patients should be followed every 6 months and annually thereafter [61]. During each visit, patients should have a thorough history and physical exam and assessment of quality of life [61]. Laboratory studies and EGD are performed as clinically indicated [61]. The importance of intensive post-treatment endoscopic surveillance should be emphasized given a defined rate of disease recurrence. Additionally, radiographic imaging such as CT of the chest and abdomen with contrast or PET/CT may be needed for restaging purposes [61].
Conclusion
Esophageal adenocarcinoma and esophageal SCC are aggressive cancers with poor prognosis. Overall, esophageal adenocarcinoma has increased in incidence, while the incidence of SCC has decreased in the Western world. GERD is the most common cause of esophageal adenocarcinoma, whereas increased alcohol consumption and tobacco commonly lead to esophageal SCC.
For patients with suspected esophageal cancer, a barium swallow is an inexpensive initial diagnostic study that is usually followed up with EGD with biopsies if suggestive of cancer. Once cancer is confirmed, a CT scan with intravenous contrast is obtained to look for adenopathy and metastasis. Those who do not have evidence of metastasis on CT scan typically undergo EUS for definitive locoregional staging.
In the past, patients with early stage esophageal cancer were referred for esophagectomy, but recently EMR has emerged as a viable alternative. Patients with locally advanced esophageal cancers are usually treated with neoadjuvant chemoradiation in combination with surgery. In addition, several studies have showed that esophageal stenting prior to neoadjuvant treatment significantly improves patients’ dysphagia. Unfortunately, many patients still initially present with metastatic or nonresectable disease. Improvement of quality of life is a major goal in patients with unresectable disease. Chemoradiotherapy, esophageal stenting, and brachytherapy are options for improvement of quality of life. Further studies are still needed to evaluate current and new therapeutic guidelines for resectable and nonresectable disease.
Corresponding author: Douglas G. Adler, MD, 30N 1900E 4R118, Salt Lake City, UT 84132, [email protected].
Financial disclosures: None.
From the University of Utah School of Medicine, Salt Lake City, UT.
Abstract
- Objective: To review the evaluation, diagnosis, and management of patients with esophageal cancer.
- Methods: Review of the literature.
- Results: Esophageal adenocarcinoma and esophageal squamous cell carcinoma (SCC) are aggressive cancers with a poor prognosis. GERD is the most common cause of esophageal adenocarcinoma, whereas increased alcohol consumption and tobacco commonly lead to esophageal SCC. Diagnosis is made via esophagogastroduodenoscopy and biopsies, and endoscopic ultrasound is typically used for locoregional staging. The endoscopic treatment of dysphagia is complex and several treatment options are available. Patients with locally advanced esophageal cancers are usually treated with neoadjuvant chemoradiation in combination with surgery. Improvement of quality of life is a major goal in patients with unresectable disease.
- Conclusion: Esophageal cancer remains a commonly encountered clinical entity requiring multidisciplinary evaluation and treatment.
Esophageal cancer is an aggressive disease with an overall poor outcome. It is the eighth most common cancer and sixth most common cause of cancer-related death worldwide [1]. In 2012, there were an estimated 456,000 new diagnoses of esophageal cancer and 400,000 deaths worldwide [1]. In the United States alone, an estimated 18,170 cases of esophageal cancer will be diagnosed in 2014, with 15,450 expected deaths [2].
Esophageal cancer includes 2 distinct histologic diseases: esophageal adenocarcinoma and esophageal squamous cell carcinoma (SCC). Overall, esophageal adenocarcinoma has increased in incidence, while the incidence of SCC has decreased in the Western world due to long-term reductions in smoking and alcohol consumption and increased incidence of gastroesophageal reflux disease (GERD) and obesity [3,4]. Esophageal adenocarcinoma accounted for less than 15% of esophageal cancers in the early 1980s, but now represents more than 60% of all esophageal cancers in the United States [5]. Esophageal SCC is still more common in China, central Asia, sub-Saharan Africa, and India and among the African-American and Caucasian female population in the United States [3,5].
Etiology
Esophageal Adenocarcinoma
While GERD is the most common cause of esophageal adenocarcinoma, other important causes/risk factors have been identified such as male sex, Caucasian race, older age, and obesity [8,12].In a prospective study by Abnet et al, patients who had a body mass index (BMI) greater than 35 kg/m2 had a significantly increased risk of esophageal adenocarcinoma when compared to patients with a BMI of 18.5 to 25 kg/m2 (hazard ratio [HR], 2.27; 95% CI, 1.44 to 3.59) [13]. Similarly, a recent meta-analysis found that patients with a BMI of 30 kg/m2 or greater had a relative risk for esophageal adenocarcinoma of 2.71 (95% CI, 2.16 to 3.46) [14]. Despite the strong correlation, the etiology of esophageal adenocarcinoma is complex and cannot be fully explained by obesity trends [15].
Smoking is another important risk factor associated with the development of esophageal adenocarcinoma. A study from the Barrett’s and Esophageal Adenocarcinoma Consortium revealed strong associations with esophageal adenocarcinoma and cigarette smoking (OR, 1.96; 95% CI, 1.64 to 2.34) [16]. Furthermore, the study found a statistically significant dose-response association between cigarette smoking and esophageal adenocarcinoma (P < 0.001).
Finally, dietary intake of vegetables and fruits has been shown to reduce the risk of Barrett’s esophagus. In a case-control study, patients with a median intake of 8.3 servings per day of vegetables and fruits had a 73% lower risk of developing Barrett’s esophagus versus those with 2.0 servings per day (OR, 0.27; 95% CI, 0.15 to 0.50) [17]. Each additional serving of vegetables and fruit was associated with a 14% reduction of risk (OR, 0.86; 95% CI, 0.80 to 0.93).
Esophageal Squamous Cell Carcinoma
In the study by Freedman et al, when compared with nonsmokers, current cigarette smokers were at significantly increased risk for esophageal SCC (HR, 9.27; 95% CI, 4.04 to 21.29) [18].Smoking has a stronger correlation with esophageal SCC than with esophageal adenocarcinoma [20]. In current smokers, the risk for developing esophageal SCC increases approximately three- to sevenfold [20]. The duration and intensity of smoking has been shown to increase the risk of esophageal SCC as well [21]. Smoking cessation has been shown to reduce the risk of esophageal SCC, but data shows that former cigarette smokers still are at a significant risk [18,21]. In a population-based case-control study, the risk of esophageal SCC in ex-smokers remained elevated for up to 30 years (OR, 1.44; 95% CI, 0.82 to 2.52) [21].
There are only limited studies that have examined the relationship between esophageal SCC and smokeless tobacco and other smoking products. Despite the limited number of studies, smokeless tobacco has been associated with esophageal SCC [22]. In a 2012 study of patients from India, chewing nass (a mix of tobacco, ash, oil, lime, and coloring and flavoring agents) and smoking hookah were associated with an increased risk of developing esophageal SCC [23].
Other risk factors associated with esophageal SCC include poor oral hygiene, atrophic gastritis, caustic esophageal injuries, and achalasia (likely due to stasis of esophageal contents in the case of achalasia) [24–27].Dietary causes of esophageal SCC have also been implicated in many international studies. Foods containing N-nitroso compounds and diets with selenium and zinc mineral deficiencies have been found to be risk factors for esophageal SCC [20,28–30].Thermal injury to the esophageal mucosa caused by food and beverages served at high temperatures has been shown to increase the risk of esophageal cancer [31]. Also, as seen in esophageal adenocarcinoma, diets rich with vegetables and fruits have been associated with a reduced risk of esophageal SCC [32].
In a meta-analysis of 1813 esophageal cancer cases by Corley et al, the use of aspirin and nonsteroidal anti-inflammatory drugs (NSAIDs) was found to be protective against both esophageal SCC and esophageal adenocarcinoma [33]. The study found a dose-dependent effect in the protective association between aspirin/NSAID use and esophageal cancer. Frequent aspirin/NSAID use was associated with a 46% reduction of the odds for developing any esophageal cancer, whereas intermittent use provided an 18% reduction in the odds. However, any use of aspirin or NSAIDs offered some degree of protection against both esophageal SCC (OR, 0.58; 95% CI, 0.43 to 0.78) and esophageal adenocarcinoma (OR, 0.67; 95% CI, 0.51 to 0.87). The mechanism of the risk reduction with aspirin and NSAIDs is still unclear but may be associated with inhibition of the cyclooxygenase-2 enzyme and the reduction of inflammation [33–35].
Clinical Manifestations
Esophageal cancer commonly presents with dysphagia, weight loss, gastrointestinal reflux, and/or odynophagia. In a study by Daly et al, 74% of esophageal cancer patients reported dysphagia and 16.6% reported having odynophagia at the time of initial diagnosis [36]. Patients can have the sensation of food getting “stuck,” which initially can be overcome by careful chewing and/or dietary modification [37]. A history of trouble swallowing solid foods followed by difficulty with drinking liquids is frequently seen. Some patients complain of regurgitation of undigested foods, and approximately 20% of patients have reported having GERD symptoms [36,37]. Due to the complete or partial esophageal obstruction combined with tumor effects, patients with esophageal cancer often develop significant weight loss. In the study by Daly et al, 57.3% of patients reported weight loss at the time of their cancer diagnosis [36]. Weight loss of more than 10% body mass has been identified as an independent indicator for poor prognosis [36,38]. Pain, dyspnea, hoarseness, and cough occur less frequently but may reflect extensive cancer burden [39]. Some patients with advanced tumors have hematemesis from tumor erosion or have recurrent pneumonias due to tracheobronchial fistulas.
Hepatomegaly, pleural effusion, and lymphadenopathy, especially in Virchow’s node (left supraclavicular fossa), are physical examination findings suggestive of metastatic disease [39]. However, most patients with esophageal cancer will have unremarkable physical examination findings.
It should be noted that patients with early stage lesions (ie, stage T1 lesions) may have minimal or no symptoms, with lesions detected either incidentally or as part of endoscopic screening/surveillance programs.
Diagnostic Studies
For patients with suspected esophageal cancer, a barium swallow is an inexpensive and readily available diagnostic study [39]. A barium swallow may show a mass lesion and/or a stricture. If the barium swallow is suggestive of cancer, the diagnosis is usually confirmed via an esophagogastroduodenoscopy (EGD) and biopsies, although in practice many patients with dysphagia and/or a history suspicious for esophageal cancer will proceed directly to EGD [40]. Findings suspicious for cancer are routinely biopsied [39].Traditionally, the more biopsies obtained (up to 7), the higher the diagnostic yield of cancer [41]. The addition of brush cytology to biopsies has also been found to increase the diagnostic accuracy, although this is not widely performed [41].
Once the diagnosis of cancer is confirmed, a computed tomography (CT) scan of the chest, abdomen, and pelvis with intravenous (IV) contrast is usually the next step in the patient’s evaluation, primarily to detect distant metastasis and to look for peritumoral adenopathy [39]. However, in terms of locoregional tumor staging, CT scans are less sensitive and specific than endoscopic ultrasonography (EUS) [42]. Patients who do not have evidence of metastasis on CT scan typically undergo EUS for definitive locoregional staging.
EUS does have its limitations. Between 25% and 36% of patients with esophageal carcinoma present with high-grade malignant strictures that do not allow passage of the scope, although if the exam can show malignant adenopathy and/or tumor extension through the muscularis propria, further evaluation is often of little additional benefit [46]. Dilation of malignant esophageal strictures to facilitate EUS is uncommon as there is a high risk of perforation (up to 24%) [47]. High-frequency (12.5 MHz) EUS mini-probes have been used to interrogate tumors with a very narrow lumen; however, the mini-probes are limited by the penetration depth of the transducer, which can lead to an incomplete locoregional tumor assessment [48]. EUS is not usually used for restaging after neoadjuvant therapy [49].
Endoscopic mucosal resection (EMR) is another technique for staging and treatment of superficial neoplasms (see Treatment section for more details). EMR is critical for distinguishing between T1a lesions (often candidates for definitive endoscopic therapy given the low likelihood of nodal involvement) versus T1b lesions (invasive to submucosa and more likely to prompt surgical esophagectomy with lymph node sampling). The distinction between T1a and T1b disease cannot be established as reliably by EUS when compared with EMR. The American Society for Gastrointestinal Endoscopy 2013 guidelines recommend EMR for the treatment and staging of nodular Barrett’s esophagus and suspected intramucosal adenocarcinoma [50].
Looking for distant metastasis, or M staging, is carried out with EUS, diagnostic laparoscopy/thoracoscopy, and CT and/or positron emission tomography (PET) scans. Despite the high accuracy of esophageal cancer staging with laparoscopy and thoracoscopy, these are invasive procedures and have generally been replaced by PET scan [39,51,52]. PET with 18F-fludeoxyglucose has been shown to significantly improve the detection rate of metastatic disease compared with the conventional staging methods (CT scan and EUS) [53]. In a prospective study, PET scans detected metastasis in 15% of patients who were thought to have localized cancer by conventional staging modalities [39,54].
Unlike several other cancers, tumor markers such as carbohydrate antigen (CA) 19-9, CA 125, and carcinoembryonic antigen (CEA) have low specificity and sensitivity in esophageal cancer and are not routinely obtained and/or followed [39,55].
Staging
Treatment
Early Stage
Historically, patients with early stage esophageal cancer (those without evidence of deep invasion into the esophageal wall and no evidence of peritumoral malignant adenopathy or metastases, typically T1N0M0) were referred for esophagectomy [59]. Recent treatment trends suggest proportionately more patients with T1 disease are being treated endoscopically (up to 29% of patients) and proportionately fewer with esophagectomy [60]. EMR has emerged as a viable alternative treatment to esophagectomy when the lesion is staged T1aN0 (tumor invading the lamina propria or muscularis mucosae but not the submucosa) [3]. EMR is performed via several techniques, but most commonly as follows. First, saline is injected under the lesion to create a submucosal cushion, separating the lesion from the underlying muscularis propria. The actual endoscopic resection of the lesion is usually accomplished via snare electrocautery and the resected lesion is sent for pathologic analysis. Endoscopic caps and band ligation devices are available to facilitate removal of the lesion in one or more pieces [61].
In a retrospective cohort study by Prasad et al of 178 patients from 1998 to 2007, the cumulative mortality in the EMR group was comparable to that of the surgery group (17% vs. 20%, respectively, P = 0.75) [62]. Recurrent cancer was detected in 12% of EMR patients; however, all patients were successfully re-treated without affecting overall survival.
In another study of 742 patients, long-term survival in those with early esophageal cancer managed with endoscopic therapy was comparable to that in patients treated with surgical resection [63]. The median cancer-free survival in the endoscopic group was not significantly different from that in the surgical group (56 and 59 months, respectively, P = 0.41) The study found that the relative hazard for 1esophageal cancer–specific mortality in the endoscopic group did not differ from that of the surgical group (relative hazard, 0.89; 95% CI, 0.51 to 1.56; P = 0.68).
Locally Advanced Disease
Neoadjuvant Therapy
For patients with locally advanced cancer (ie, patients without distant metastases who have extension of the primary tumor into the deeper layers of the esophageal wall, including the muscularis propria and the adventitia with or without peritumoral malignant adenopathy, or T2 or T3 lesions with N0 or N1, N2, or N3 status, neoadjuvant therapy is the norm, although the optimal management remains controversial and treatment protocols vary around the world [3,62]. Most neoadjuvant therapy regimens in the United States combine chemotherapy and external beam radiation therapy.
Neoadjuvant treatment with chemoradiation has been found to be beneficial in all esophageal cancers [3,64]. A meta-analysis of 1209 patients found a significant survival benefit for preoperative chemoradiotherapy and, to a lesser extent, for chemotherapy when compared to surgery alone [65]. When comparing neoadjuvant chemoradiotherapy to surgery alone, there was a 19% decrease in the risk of death corresponding to a 13% absolute difference in 2-year survival in the neoadjuvant chemotherapy group. HR for all-cause mortality with neoadjuvant chemoradiotherapy versus surgery alone was 0.81 (95% CI, 0.70 to 0.93; P = 0.002). The benefits of neoadjuvant chemoradiotherapy were similar for both esophageal SCC and adenocarcinoma. The benefits of chemotherapy, however, were less than chemoradiotherapy. When comparing neoadjuvant chemotherapy to surgery alone, there was an absolute survival benefit of 7%.
Following neoadjuvant therapy, patients typically undergo restaging via cross-sectional imaging, most commonly PET/CT scans. If the patient is felt to have active residual disease and has not developed metastases or contraindications to surgery, esophagectomy is appropriate. Some data suggests that patients with esophageal SCC who have complete clinical response after chemoradiation can be observed closely rather than proceed to surgery [3,62,66]. However, the data concerning the usefulness of definitive chemoradiotherapy in esophageal adenocarcinoma is lacking at this time. In a retrospective study of nonmetastatic esophageal adenocarcinoma patients by Tougeron et al comparing surgical patients (± preoperative treatment) to definitive chemoradiotherapy patients, a complete resection was achieved in 92.5% of patients in the surgical group and a clinical complete response was observed in 49.4% of patients with definitive chemoradiotherapy [67]. The overall survival was 36.2 ± 2 months for the surgery group versus 16.5 ± 0.8 months for the definitive chemoradiotherapy group (P = 0.02).
Stenting Prior to Neoadjuvant Therapy
In a meta-analysis of 9 studies comprising 180 patients, placement of esophageal stents in patients with locally advanced esophageal cancer significantly improved dysphagia and allowed for oral nutrition during neoadjuvant therapy [69]. There was a substantial decrease in the dysphagia scores standard difference in means (SDM) of –0.81 (standard error, 0.15; 95% CI, –1.1 to –0.51), an increase in weight SDM of 0.591 (standard error, 0.434; 95% CI, –0.261 to 1.442), and an increase in serum albumin SDM of 0.35 (standard error, 0.271; 95% CI, –0.181 to 0.881). The overall procedural success rate was 95% (95% CI, 0.895 to 0.977). Major adverse events included stent migration in 32% of patients (95% CI, 0.258 to 0.395) and chest discomfort in 51.4% (95% CI, 0.206 to 0.812). However, it was believed that the stent migration may have been a sign of tumor response to neoadjuvant therapy.
In a prospective nonrandomized study of 13 patients with polyflex stents (polyester mesh stents covered in a silicone membrane) placed prior to neoadjuvant therapy, similar improvements with dysphagia scores were observed after stent placement [70]. In the study, the mean baseline dysphagia score at the time of stent placement was 3. Dysphagia scores were subsequently obtained at 1, 2, 3, and 4 weeks after stent placement and were 1.1, 0.8, 0.9, and 1.0, respectively (P = 0.005, P = 0.01, P = 0.02, and P = 0.008, respectively). There were no episodes of bleeding or esophageal perforation. Immediate complications from stenting included chest discomfort, seen in 12 of the 13 patients. Stent migration occurred at some point in 6 of 13 patients, although not all patients with a migrated stent required stent replacement. Again, it was thought that the stent migration could be a sign of tumor response to neoadjuvant therapy.
Surgery
Surgery is an essential part of treatment of esophageal cancer [3,71]. Transthoracic, transhiatal, and radical (en bloc) are the 3 different basic approaches for esophagectomy [3]. Because it does not require a thoracotomy, the transhiatal approach has a theoretical advantage of decreased morbidity and mortality, although several studies have shown no differences in outcome between the transthoracic and transhiatal approach [3,72,73]. In a study by Chang et al comparing the transhiatal to the transthoracic approach, the 5-year survival was higher for patients undergoing transhiatal versus transthoracic esophagectomy (30.5% vs. 22.7%, P = 0.02) [73]. However, after adjusting for differences in tumor stage and patient and provider factors the survival advantage was no longer statistically significant (adjusted HR for mortality, 0.95; 95% CI, 0.75 to 1.20).
Adjuvant Therapy
Despite the benefits of chemoradiation as a neoadjuvant treatment, the data for chemoradiation as adjuvant therapy after resection is lacking in most clinical situations [74].
Metastatic Disease
Between 25% and 40% of esophageal cancer patients will present with metastases to liver, bone, and lung or widespread nodal metastases [61].Improvement of quality of life is a major goal in patients with unresectable disease. Patients with nonsurgical esophageal cancer who have an estimated life expectancy of greater than a few weeks are recommended to have concurrent chemoradiotherapy as most patients have symptomatic obstructive disease and dysphagia [62]. A study by Harvey et al examined the palliative benefit of chemoradiotherapy on dysphagia versus toxicity in patients with invasive esophageal carcinoma [75]. The study found that treatment was well tolerated, with only 5% of patients failing to complete treatment. The study used the DeMeester (4-point) symptom scores for the assessment of dysphagia. The median baseline score at presentation was 2 (moderate: difficulty with soft food, predominately liquid diet). After chemoradiotherapy, 49% of patients were assessed as having a dysphagia score of 0 (no dysphagia). Of those patients who received chemoradiotherapy, 78% had an improvement of at least 1 grade in their DeMeester dysphagia, while only 14% of patients did not improve with therapy. The median survival for the study population was 7 months, with a 6% treatment-related mortality. Chemoradiation therapy as a primary treatment for dysphagia can take days to weeks to take effect, and can be associated with significant pain, usually from radiation esophagitis.
Other alternatives for palliation of nonresectable esophageal cancer include esophageal stenting with SEMS and brachytherapy. SEMS are effective and safe for palliation of dysphagia caused by primary esophageal tumors, postoperative cancer recurrence, esophagorespiratory fistulae, and tumors near the upper esophageal sphincter [76]. A study looking at the use of esophageal SEMS in cancer found that after SEMS placement, the dysphagia score improved from a mean of 3.6 to 1.6 (P < 0.001) [75]. The procedure was technically successful in 96% of the patients. In all cases, esophagorespiratory fistulas were occluded. Pain, reflux, and stent migration are the most common complications of esophageal SEMS.
In a study comparing single-dose brachytherapy versus SEMS, the SEMS group had quicker improvement of dysphagia symptoms than the brachytherapy group, but the long-term relief of dysphagia was better after brachytherapy [77]. In addition, SEMS placement had more complications than brachytherapy (33% vs. 21%, respectively; P = 0.02), which was mainly due to an increased incidence of late hemorrhage. However, brachytherapy and SEMS did not differ in terms of median survival (P = 0.23) or recurrent or persistent dysphagia (P = 0.81).
Tracheoesophageal fistulas may develop in the setting of a locally advanced tumor, or as a complication of RT or chemoradiotherapy. SEMS can also be used successfully in the palliation therapy for tracheoesophageal fistulas or post-esophagectomy anastomotic strictures [78].
Prognosis
The overall survival for patients with resectable esophageal cancer has improved significantly over the past 30 years; however, more than 50% of patients presenting with esophageal cancer will have unresectable or metastatic disease at the time of presentation [3,39,79].Prognosis is primarily TMN stage–dependent, as patients with early stage cancer limited to the mucosa are expected to have curable disease [3]. Poor prognostic predictors include advanced stage cancer, dysphagia, advanced age, large tumors, more than 10% loss in body mass, and malignant adenopathy [39,80–84].
In 2010, the American Joint Committee on Cancer/International Union against Cancer Staging system looked at the prognosis of 4627 patients who underwent esophagectomy alone without radiation or chemotherapy [3,56]. For stage Tis (tumor in situ or high-grade dysplasia) and 1A cancers, there was an approximate 80% 5-year risk-adjusted survival rate [3,56]. The survival rate was marginally better for esophageal adenocarcinoma than for esophageal SCC. With surgery alone, stage 1B disease had a 5-year survival of 62% with SCC and 64% with adenocarcinoma [3,56]. For patients with stage 2A cancer, the 5-year survival was 55% for SCC and 50% for adenocarcinoma as long as there was not nodal involvement [3,56]. If there was nodal involvement, the survival rate dropped to 40% for stage 2B cancer, 25% for stage 3A cancer, and 15% to 17% for stage 3B to 3C cancer [3,56]. As stated earlier, neoadjuvant chemoradiation helps improve outcomes when compared to surgery alone (see Neoadjuvant Therapy in the Treatment section). Thus, one would expect a slightly better prognosis with neoadjuvant therapy and surgery than the previously stated data for surgery alone. Unfortunately, patients with unresectable or metastatic disease at time of diagnosis have a poor prognosis, with a 1-year survival rate less than 20% [3].
Esophageal cancer patients who are treated successfully need to be followed closely because a majority of esophageal cancers will recur within 3 years of treatment [61]. For the first 3 years post treatment, patients should be followed every 3 to 6 months [61]. For 3 to 5 years after treatment, patients should be followed every 6 months and annually thereafter [61]. During each visit, patients should have a thorough history and physical exam and assessment of quality of life [61]. Laboratory studies and EGD are performed as clinically indicated [61]. The importance of intensive post-treatment endoscopic surveillance should be emphasized given a defined rate of disease recurrence. Additionally, radiographic imaging such as CT of the chest and abdomen with contrast or PET/CT may be needed for restaging purposes [61].
Conclusion
Esophageal adenocarcinoma and esophageal SCC are aggressive cancers with poor prognosis. Overall, esophageal adenocarcinoma has increased in incidence, while the incidence of SCC has decreased in the Western world. GERD is the most common cause of esophageal adenocarcinoma, whereas increased alcohol consumption and tobacco commonly lead to esophageal SCC.
For patients with suspected esophageal cancer, a barium swallow is an inexpensive initial diagnostic study that is usually followed up with EGD with biopsies if suggestive of cancer. Once cancer is confirmed, a CT scan with intravenous contrast is obtained to look for adenopathy and metastasis. Those who do not have evidence of metastasis on CT scan typically undergo EUS for definitive locoregional staging.
In the past, patients with early stage esophageal cancer were referred for esophagectomy, but recently EMR has emerged as a viable alternative. Patients with locally advanced esophageal cancers are usually treated with neoadjuvant chemoradiation in combination with surgery. In addition, several studies have showed that esophageal stenting prior to neoadjuvant treatment significantly improves patients’ dysphagia. Unfortunately, many patients still initially present with metastatic or nonresectable disease. Improvement of quality of life is a major goal in patients with unresectable disease. Chemoradiotherapy, esophageal stenting, and brachytherapy are options for improvement of quality of life. Further studies are still needed to evaluate current and new therapeutic guidelines for resectable and nonresectable disease.
Corresponding author: Douglas G. Adler, MD, 30N 1900E 4R118, Salt Lake City, UT 84132, [email protected].
Financial disclosures: None.
1. Ferlay J, Soerjomataram I, Ervik M, et al. GLOBOCAN 2012 v1.0, Cancer Incidence and Mortality Worldwide: IARC CancerBase No. 11 [Internet]. Lyon, France: International Agency for Research on Cancer; 2013. http://globocan.iarc.fr. Accessed on May 5, 2014.
2. Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin 2014;64:9–29.
3. Nieman DR, Peters JH. Treatment strategies for esophageal cancer. Gastroenterol Clin North Am 2013;42:187–97.
4. Shridhar R, Almhanna K, Meredith KL, et al. Radiation therapy and esophageal cancer. Cancer Control 2013;20:97–110.
5. Baquet CR, Commiskey P, Mack K, et al. Esophageal cancer epidemiology in blacks and whites: racial and gender disparities in incidence, mortality, survival rates and histology. J Natl Med Assoc 2005;97:1471–8.
6. Rubenstein JH, Taylor JB. Meta-analysis: the association of oesophagealadenocarcinoma with symptoms of gastro-oesophageal reflux. Aliment Pharmacol Ther 2010;32:1222–7.
7. Lagergren J, Bergström R, Lindgren A, Nyrén O. Symptomatic gastroesophageal reflux as a risk factor for esophageal adenocarcinoma. N Engl J Med 1999;340:825–31.
8. Pohl H, Wrobel K, Bojarski C, et al. Risk factors in the development of esophageal adenocarcinoma. Am J Gastroenterol 2013;108:200–7.
9. Coleman HG, Bhat SK, Murray LJ, et al. Symptoms and endoscopic features at Barrett’s esophagus diagnosis: implications for neoplastic progression risk. Am J Gastroenterol 2014;109:527–34.
10. Nguyen DM, El-Serag HB, Henderson L, et al. Medication usage and the risk of neoplasia in patients with Barrett’s esophagus. Clin Gastroenterol Hepatol 2009;7:1299–304.
11. Lagergren J, Ye W, Lagergren P, Lu Y. The risk of esophageal adenocarcinomaafter antireflux surgery. Gastroenterology 2010;138:1297–301.
12. el-Serag HB. The epidemic of esophageal adenocarcinoma. Gastroenterol Clin North Am 2002;31:421–40, viii.
13. Abnet CC, Freedman ND, Hollenbeck AR, et al. A prospective study of BMI and risk of oesophageal and gastric adenocarcinoma. Eur J Cancer 2008;44:465–71.
14. Turati F, Tramacere I, La Vecchia C, Negri E. A meta-analysis of body mass index and esophageal and gastric cardia adenocarcinoma. Ann Oncol 2013;24:609–17.
15. Kroep S, Lansdorp-Vogelaar I, Rubenstein JH, et al. Comparing trends in esophageal adenocarcinoma incidence and lifestyle factors between the United States, Spain, and the Netherlands. Am J Gastroenterol 2014;109:336–43.
16. Cook MB, Kamangar F, Whiteman DC, et al. Cigarette smoking and adenocarcinomas of the esophagus and esophagogastric junction: a pooled analysis from the international BEACON consortium. J Natl Cancer Inst 2010;102:1344–53.
17. Kubo A, Levin TR, Block G, et al. Dietary antioxidants, fruits, and vegetables and the risk of Barrett’s esophagus. Am J Gastroenterol 2008;103:1614–23.
18. Freedman ND, Abnet CC, Leitzmann MF, et al. A prospective study of tobacco, alcohol, and the risk of esophageal and gastric cancer subtypes. Am J Epidemiol 2007;165:1424–33.
19. Pandeya N, Williams G, Green AC, et al; Australian Cancer Study. Alcohol consumption and the risks of adenocarcinoma and squamous cell carcinoma of the esophagus. Gastroenterology 2009;136:1215–24, e1-2.
20. Kamangar F, Chow WH, Abnet CC, Dawsey SM. Environmental causes of esophageal cancer. Gastroenterol Clin North Am 2009;38:27–57, vii.
21. Pandeya N, Williams GM, Sadhegi S, et al. Associations of duration, intensity, and quantity of smoking with adenocarcinoma and squamous cell carcinoma of the esophagus. Am J Epidemiol 2008;168:105–14.
22. Boffetta P, Hecht S, Gray N, et al. Smokeless tobacco and cancer. Lancet Oncol 2008;9:667–75.
23. Dar NA, Bhat GA, Shah IA, eta l. Hookah smoking, nass chewing, and oesophageal squamous cell carcinoma in Kashmir, India. Br J Cancer 2012;107:1618–23.
24. Abnet CC, Kamangar F, Islami F, et al. Tooth loss and lack of regular oral hygiene are associated with higher risk of esophageal squamous cell carcinoma. Cancer Epidemiol Biomarkers Prev 2008;17:3062–8.
25. Islami F, Sheikhattari P, Ren JS, Kamangar F. Gastric atrophy and risk of oesophageal cancer and gastric cardia adenocarcinoma--a systematic review and meta-analysis. Ann Oncol 2011;22:754–60.
26. Appelqvist P, Salmo M. Lye corrosion carcinoma of the esophagus: a review of 63 cases. Cancer 1980;45:2655–8.
27. Sandler RS, Nyrén O, Ekbom A, et al. The risk of esophageal cancer in patients with achalasia. A population-based study. JAMA 1995;274:1359–62.
28. Lu SH, Montesano R, Zhang MS, et al. Relevance of N-nitrosamines to esophageal cancer in China. J Cell Physiol Suppl 1986;4:51–8.
29. Steevens J, van den Brandt PA, Goldbohm RA, Schouten LJ. Selenium status and the risk of esophageal and gastric cancer subtypes: the Netherlands cohort study. Gastroenterology 2010;138:1704–13.
30. Abnet CC, Lai B, Qiao YL, et al. Zinc concentration in esophageal biopsy specimens measured by x-ray fluorescence and esophageal cancer risk. J Natl Cancer Inst 2005;97:301–6.
31. Islami F, Boffetta P, Ren JS, et al. High-temperature beverages and foods and esophageal cancer risk--a systematic review. Int J Cancer 2009;125:491–524.
32. Liu J, Wang J, Leng Y, Lv C. Intake of fruit and vegetables and risk of esophageal squamous cell carcinoma: a meta-analysis of observational studies. Int J Cancer 2013;133:473–85.
33. Corley DA, Kerlikowske K, Verma R, Buffler P. Protective association of aspirin/NSAIDs and esophageal cancer: a systematic review and meta-analysis. Gastroenterology 2003;124:47–56.
34. Ratnasinghe D, Tangrea J, Roth MJ, et al. Expression of cyclooxygenase-2 in human squamous cell carcinoma of the esophagus; an immunohistochemical survey. Anticancer Res 1999;19:171–4.
35. Morris CD, Armstrong GR, Bigley G, et al. Cyclooxygenase-2 expression in the Barrett’s metaplasia-dysplasia-adenocarcinoma sequence. Am J Gastroenterol 2001;96:990–6.
36. Daly JM, Fry WA, Little AG, et al. Esophageal cancer: results of an American College of Surgeons Patient Care Evaluation Study. J Am Coll Surg 2000;190:562–72.
37. Gastrointestinal cancer. In: Fishman MC, Hoffman R, Klausner RD, Thaler MS, editors. Medicine. 5th ed. Baltimore: Lippincott Williams & Wilkins; 2002:423–6.
38. Fein R, Kelsen DP, Geller N, et al. Adenocarcinoma of the esophagus and gastroesophageal junction. Prognostic factors and results of therapy. Cancer 1985;56:2512–8.
39. Enzinger PC, Mayer RJ. Esophageal cancer. N Engl J Med 2003;349:2241–52.
40. Lightdale CJ. Esophageal cancer. Am J Gastroenterol 1999;94:20–9.
41. Graham DY, Schwartz JT, Cain GD, Gyorkey F. Prospective evaluation of biopsy number in the diagnosis of esophageal and gastric carcinoma. Gastroenterology 1982;82:228–31.
42. Lowe VJ, Booya F, Fletcher JG, et al. Comparison of positron emission tomography, computed tomography, and endoscopic ultrasound in the initial staging of patients with esophageal cancer. Mol Imaging Biol 2005;7:422–30.
43. Röesch T. Endoscopic ultrasonography: equipment and technique. Gastrointest Endosc Clin N Am 2005;15:13–31, vii.
44. Vazquez-Sequeiros E, Norton ID, Clain JE, et al. Impact of EUS-guided fine-needle aspiration on lymph node staging in patients with esophageal carcinoma. Gastrointest Endosc 2001;53:751–7.
45. Van Dam J. Endosonographic evaluation of the patient with esophageal cancer. Chest 1997;112(4 Suppl):184S–190S.
46. Catalano MF, Van Dam J, Sivak MV Jr. Malignant esophageal strictures: staging accuracy of endoscopic ultrasonography. Gastrointest Endosc 1995;41:535–9.
47. Van Dam J, Rice TW, Catalano MF, et al. High-grade malignant stricture is predictive of esophageal tumor stage. Risks of endosonographic evaluation. Cancer 1993;71:2910–7.
48. Hünerbein M, Ghadimi BM, Haensch W, Schlag PM. Transendoscopic ultrasound of esophageal and gastric cancer using miniaturized ultrasound catheter probes. Gastrointest Endosc 1998;48:371–5.
49. Zuccaro G Jr, Rice TW, Goldblum J, et al. Endoscopic ultrasound cannot determine suitability for esophagectomy after aggressive chemoradiotherapy for esophageal cancer. Am J Gastroenterol 1999;94:906–12.
50. ASGE Standards of Practice Committee, Evans JA, Early DS, Chandraskhara V, et al; American Society for Gastrointestinal Endoscopy. The role of endoscopy in the assessment and treatment of esophageal cancer. Gastrointest Endosc 2013;77:328–34.
51. Luketich JD, Schauer P, Landreneau R, et al. Minimally invasive surgical staging is superior to endoscopic ultrasound in detecting lymph node metastases in esophageal cancer. J Thorac Cardiovasc Surg 1997;114:817–21.
52. Krasna MJ, Flowers JL, Attar S, McLaughlin J. Combined thoracoscopic/laparoscopic staging of esophageal cancer. J Thorac Cardiovasc Surg 1996;111:800–6.
53. Flamen P, Lerut A, Van Cutsem E, et al. Utility of positron emission tomography for the staging of patients with potentially operable esophageal carcinoma. J Clin Oncol 2000;18:3202–10.
54. Downey RJ, Akhurst T, Ilson D, et al. Whole body 18FDG-PET and the response of esophageal cancer to induction therapy: results of a prospective trial. J Clin Oncol 2003;21:428–32.
55. Mealy K, Feely J, Reid I, et al. Tumour marker detection in oesophageal carcinoma. Eur J Surg Oncol 1996;22:505–7.
56. Rice TW, Rusch VW, Ishwaran H, Blackstone EH; Worldwide Esophageal Cancer Collaboration. Cancer of the esophagus and esophagogastric junction: data-driven staging for the seventh edition of the American Joint Committee on Cancer/International Union Against Cancer Cancer Staging Manuals. Cancer 2010;116:3763–73.
57. Edge SB, Byrd DR, Compton CC, et al, eds. AJCC cancer staging manual. 7th ed. New York: Springer-Verlag; 2009:103–5.
58. Rice TW, Rusch VW, Apperson-Hansen C, et al. Worldwide esophageal cancer collaboration. Dis Esophagus 2009;22:1–8.
59. Villaflor VM, Allaix ME, Minsky B, et al. Multidisciplinary approach for patients with esophageal cancer. World J Gastroenterol 2012;18:6737–46.
60. Ngamruengphong S, Wolfsen HC, Wallace MB. Survival of patients with superficial esophageal adenocarcinoma after endoscopic treatment vs surgery. Clin Gastroenterol Hepatol 2013;11:1424–9.e2.
61. Lin SH, Chang JY. Esophageal cancer: diagnosis and management. Chin J Cancer 2010;29:843–54.
62. Prasad GA, Wu TT, Wigle DA, et al. Endoscopic and surgical treatment of mucosal (T1a) esophageal adenocarcinoma in Barrett’s esophagus. Gastroenterology 2009;137:815–23.
63. Das A, Singh V, Fleischer DE, Sharma VK. A comparison of endoscopic treatment and surgery in early esophageal cancer: an analysis of surveillance epidemiology and end results data. Am J Gastroenterol 2008;103:1340–5.
64. Herskovic A, Martz K, al-Sarraf M, et al. Combined chemotherapy and radiotherapy compared with radiotherapy alone in patients with cancer of the esophagus. N Engl J Med 1992;326:1593–8.
65. Gebski V, Burmeister B, Smithers BM, et al; Australasian Gastro-Intestinal Trials Group. Survival benefits from neoadjuvant chemoradiotherapy or chemotherapy in oesophageal carcinoma: a meta-analysis. Lancet Oncol 2007;8:226–34.
66. Bedenne L, Michel P, Bouché O, et al. Chemoradiation followed by surgery compared with chemoradiation alone in squamous cancer of the esophagus: FFCD 9102. J Clin Oncol 2007;25:1160–8.
67. Tougeron D, Scotté M, Hamidou H, et al. Definitive chemoradiotherapy in patients with esophageal adenocarcinoma: an alternative to surgery? J Surg Oncol 2012;105: 761–6.
68. Siddiqui AA, Sarkar A, Beltz S, et al. Placement of fully covered self-expandable metal stents in patients with locally advanced esophageal cancer before neoadjuvant therapy. Gastrointest Endosc 2012;76:44–51.
69. Nagaraja V, Cox MR, Eslick GD. Safety and efficacy of esophageal stents preceding or during neoadjuvant chemotherapy for esophageal cancer: a systematic review and meta-analysis. J Gastrointest Oncol 2014;5:119–26.
70. Adler DG, Fang J, Wong R, et al. Placement of Polyflex stents in patients with locally advanced esophageal cancer is safe and improves dysphagia during neoadjuvant therapy. Gastrointest Endosc 2009;70:614–9.
71. Dubecz A, Sepesi B, Salvador R, et al. Surgical resection for locoregional esophageal cancer is underutilized in the United States. J Am Coll Surg 2010;211:754–61.
72. Hulscher JB, Tijssen JG, Obertop H, van Lanschot JJ. Transthoracic versus transhiatal resection for carcinoma of the esophagus: a meta-analysis. Ann Thorac Surg 2001;72:306–13.
73. Chang AC, Ji H, Birkmeyer NJ, et al. Outcomes after transhiatal and transthoracic esophagectomy for cancer. Ann Thorac Surg 2008;85:424–9.
74. Almhanna K, Shridhar R, Meredith KL. Neoadjuvant or adjuvant therapy for resectable esophageal cancer: is there a standard of care? Cancer Control 2013;20:89–96.
75. Harvey JA, Bessell JR, Beller E, et al. Chemoradiation therapy is effective for the palliative treatment of malignant dysphagia. Dis Esophagus 2004;17:260–5.
76. Siersema PD, Schrauwen SL, van Blankenstein M, et al; Rotterdam Esophageal Tumor Study Group. Self-expanding metal stents for complicated and recurrent esophagogastric cancer. Gastrointest Endosc 2001;54:579–86.
77. Homs MY, Steyerberg EW, Eijkenboom WM, et al. Single-dose brachytherapy versus metal stent placement for the palliation of dysphagia from oesophageal cancer: multicentre randomised trial. Lancet 2004;364(9444):1497–504.
78. van den Bongard HJ, Boot H, Baas P, Taal BG. The role of parallel stent insertion in patients with esophagorespiratory fistulas. Gastrointest Endosc 2002;55:110–5.
79. Siegel R, DeSantis C, Virgo K, et al. Cancer treatment and survivorship statistics, 2012. CA Cancer J Clin 2012;62:220–41.
80. Fein R, Kelsen DP, Geller N, et al. Adenocarcinoma of the esophagus and gastroesophageal junction. Prognostic factors and results of therapy. Cancer 1985;56:2512–8.
81. Mariette C, Maurel A, Fabre S, et al. [Preoperative prognostic factors for squamous cell carcinomas of the thoracic esophagus]. Gastroenterol Clin Biol 2001;25:468–72.
82. Swanson SJ, Batirel HF, Bueno R, et al. Transthoracic esophagectomy with radical mediastinal and abdominal lymph node dissection and cervical esophagogastrostomy for esophageal carcinoma. Ann Thorac Surg 2001;72:1918–24.
83. Urba SG, Orringer MB, Turrisi A, et al. Randomized trial of preoperative chemoradiation versus surgery alone in patients with locoregional esophageal carcinoma. J Clin Oncol 2001;19:305–13.
84. Hosch SB, Stoecklein NH, Pichlmeier U, et al. Esophageal cancer: the mode of lymphatic tumor cell spread and its prognostic significance. J Clin Oncol 2001;19:1970–5.
1. Ferlay J, Soerjomataram I, Ervik M, et al. GLOBOCAN 2012 v1.0, Cancer Incidence and Mortality Worldwide: IARC CancerBase No. 11 [Internet]. Lyon, France: International Agency for Research on Cancer; 2013. http://globocan.iarc.fr. Accessed on May 5, 2014.
2. Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin 2014;64:9–29.
3. Nieman DR, Peters JH. Treatment strategies for esophageal cancer. Gastroenterol Clin North Am 2013;42:187–97.
4. Shridhar R, Almhanna K, Meredith KL, et al. Radiation therapy and esophageal cancer. Cancer Control 2013;20:97–110.
5. Baquet CR, Commiskey P, Mack K, et al. Esophageal cancer epidemiology in blacks and whites: racial and gender disparities in incidence, mortality, survival rates and histology. J Natl Med Assoc 2005;97:1471–8.
6. Rubenstein JH, Taylor JB. Meta-analysis: the association of oesophagealadenocarcinoma with symptoms of gastro-oesophageal reflux. Aliment Pharmacol Ther 2010;32:1222–7.
7. Lagergren J, Bergström R, Lindgren A, Nyrén O. Symptomatic gastroesophageal reflux as a risk factor for esophageal adenocarcinoma. N Engl J Med 1999;340:825–31.
8. Pohl H, Wrobel K, Bojarski C, et al. Risk factors in the development of esophageal adenocarcinoma. Am J Gastroenterol 2013;108:200–7.
9. Coleman HG, Bhat SK, Murray LJ, et al. Symptoms and endoscopic features at Barrett’s esophagus diagnosis: implications for neoplastic progression risk. Am J Gastroenterol 2014;109:527–34.
10. Nguyen DM, El-Serag HB, Henderson L, et al. Medication usage and the risk of neoplasia in patients with Barrett’s esophagus. Clin Gastroenterol Hepatol 2009;7:1299–304.
11. Lagergren J, Ye W, Lagergren P, Lu Y. The risk of esophageal adenocarcinomaafter antireflux surgery. Gastroenterology 2010;138:1297–301.
12. el-Serag HB. The epidemic of esophageal adenocarcinoma. Gastroenterol Clin North Am 2002;31:421–40, viii.
13. Abnet CC, Freedman ND, Hollenbeck AR, et al. A prospective study of BMI and risk of oesophageal and gastric adenocarcinoma. Eur J Cancer 2008;44:465–71.
14. Turati F, Tramacere I, La Vecchia C, Negri E. A meta-analysis of body mass index and esophageal and gastric cardia adenocarcinoma. Ann Oncol 2013;24:609–17.
15. Kroep S, Lansdorp-Vogelaar I, Rubenstein JH, et al. Comparing trends in esophageal adenocarcinoma incidence and lifestyle factors between the United States, Spain, and the Netherlands. Am J Gastroenterol 2014;109:336–43.
16. Cook MB, Kamangar F, Whiteman DC, et al. Cigarette smoking and adenocarcinomas of the esophagus and esophagogastric junction: a pooled analysis from the international BEACON consortium. J Natl Cancer Inst 2010;102:1344–53.
17. Kubo A, Levin TR, Block G, et al. Dietary antioxidants, fruits, and vegetables and the risk of Barrett’s esophagus. Am J Gastroenterol 2008;103:1614–23.
18. Freedman ND, Abnet CC, Leitzmann MF, et al. A prospective study of tobacco, alcohol, and the risk of esophageal and gastric cancer subtypes. Am J Epidemiol 2007;165:1424–33.
19. Pandeya N, Williams G, Green AC, et al; Australian Cancer Study. Alcohol consumption and the risks of adenocarcinoma and squamous cell carcinoma of the esophagus. Gastroenterology 2009;136:1215–24, e1-2.
20. Kamangar F, Chow WH, Abnet CC, Dawsey SM. Environmental causes of esophageal cancer. Gastroenterol Clin North Am 2009;38:27–57, vii.
21. Pandeya N, Williams GM, Sadhegi S, et al. Associations of duration, intensity, and quantity of smoking with adenocarcinoma and squamous cell carcinoma of the esophagus. Am J Epidemiol 2008;168:105–14.
22. Boffetta P, Hecht S, Gray N, et al. Smokeless tobacco and cancer. Lancet Oncol 2008;9:667–75.
23. Dar NA, Bhat GA, Shah IA, eta l. Hookah smoking, nass chewing, and oesophageal squamous cell carcinoma in Kashmir, India. Br J Cancer 2012;107:1618–23.
24. Abnet CC, Kamangar F, Islami F, et al. Tooth loss and lack of regular oral hygiene are associated with higher risk of esophageal squamous cell carcinoma. Cancer Epidemiol Biomarkers Prev 2008;17:3062–8.
25. Islami F, Sheikhattari P, Ren JS, Kamangar F. Gastric atrophy and risk of oesophageal cancer and gastric cardia adenocarcinoma--a systematic review and meta-analysis. Ann Oncol 2011;22:754–60.
26. Appelqvist P, Salmo M. Lye corrosion carcinoma of the esophagus: a review of 63 cases. Cancer 1980;45:2655–8.
27. Sandler RS, Nyrén O, Ekbom A, et al. The risk of esophageal cancer in patients with achalasia. A population-based study. JAMA 1995;274:1359–62.
28. Lu SH, Montesano R, Zhang MS, et al. Relevance of N-nitrosamines to esophageal cancer in China. J Cell Physiol Suppl 1986;4:51–8.
29. Steevens J, van den Brandt PA, Goldbohm RA, Schouten LJ. Selenium status and the risk of esophageal and gastric cancer subtypes: the Netherlands cohort study. Gastroenterology 2010;138:1704–13.
30. Abnet CC, Lai B, Qiao YL, et al. Zinc concentration in esophageal biopsy specimens measured by x-ray fluorescence and esophageal cancer risk. J Natl Cancer Inst 2005;97:301–6.
31. Islami F, Boffetta P, Ren JS, et al. High-temperature beverages and foods and esophageal cancer risk--a systematic review. Int J Cancer 2009;125:491–524.
32. Liu J, Wang J, Leng Y, Lv C. Intake of fruit and vegetables and risk of esophageal squamous cell carcinoma: a meta-analysis of observational studies. Int J Cancer 2013;133:473–85.
33. Corley DA, Kerlikowske K, Verma R, Buffler P. Protective association of aspirin/NSAIDs and esophageal cancer: a systematic review and meta-analysis. Gastroenterology 2003;124:47–56.
34. Ratnasinghe D, Tangrea J, Roth MJ, et al. Expression of cyclooxygenase-2 in human squamous cell carcinoma of the esophagus; an immunohistochemical survey. Anticancer Res 1999;19:171–4.
35. Morris CD, Armstrong GR, Bigley G, et al. Cyclooxygenase-2 expression in the Barrett’s metaplasia-dysplasia-adenocarcinoma sequence. Am J Gastroenterol 2001;96:990–6.
36. Daly JM, Fry WA, Little AG, et al. Esophageal cancer: results of an American College of Surgeons Patient Care Evaluation Study. J Am Coll Surg 2000;190:562–72.
37. Gastrointestinal cancer. In: Fishman MC, Hoffman R, Klausner RD, Thaler MS, editors. Medicine. 5th ed. Baltimore: Lippincott Williams & Wilkins; 2002:423–6.
38. Fein R, Kelsen DP, Geller N, et al. Adenocarcinoma of the esophagus and gastroesophageal junction. Prognostic factors and results of therapy. Cancer 1985;56:2512–8.
39. Enzinger PC, Mayer RJ. Esophageal cancer. N Engl J Med 2003;349:2241–52.
40. Lightdale CJ. Esophageal cancer. Am J Gastroenterol 1999;94:20–9.
41. Graham DY, Schwartz JT, Cain GD, Gyorkey F. Prospective evaluation of biopsy number in the diagnosis of esophageal and gastric carcinoma. Gastroenterology 1982;82:228–31.
42. Lowe VJ, Booya F, Fletcher JG, et al. Comparison of positron emission tomography, computed tomography, and endoscopic ultrasound in the initial staging of patients with esophageal cancer. Mol Imaging Biol 2005;7:422–30.
43. Röesch T. Endoscopic ultrasonography: equipment and technique. Gastrointest Endosc Clin N Am 2005;15:13–31, vii.
44. Vazquez-Sequeiros E, Norton ID, Clain JE, et al. Impact of EUS-guided fine-needle aspiration on lymph node staging in patients with esophageal carcinoma. Gastrointest Endosc 2001;53:751–7.
45. Van Dam J. Endosonographic evaluation of the patient with esophageal cancer. Chest 1997;112(4 Suppl):184S–190S.
46. Catalano MF, Van Dam J, Sivak MV Jr. Malignant esophageal strictures: staging accuracy of endoscopic ultrasonography. Gastrointest Endosc 1995;41:535–9.
47. Van Dam J, Rice TW, Catalano MF, et al. High-grade malignant stricture is predictive of esophageal tumor stage. Risks of endosonographic evaluation. Cancer 1993;71:2910–7.
48. Hünerbein M, Ghadimi BM, Haensch W, Schlag PM. Transendoscopic ultrasound of esophageal and gastric cancer using miniaturized ultrasound catheter probes. Gastrointest Endosc 1998;48:371–5.
49. Zuccaro G Jr, Rice TW, Goldblum J, et al. Endoscopic ultrasound cannot determine suitability for esophagectomy after aggressive chemoradiotherapy for esophageal cancer. Am J Gastroenterol 1999;94:906–12.
50. ASGE Standards of Practice Committee, Evans JA, Early DS, Chandraskhara V, et al; American Society for Gastrointestinal Endoscopy. The role of endoscopy in the assessment and treatment of esophageal cancer. Gastrointest Endosc 2013;77:328–34.
51. Luketich JD, Schauer P, Landreneau R, et al. Minimally invasive surgical staging is superior to endoscopic ultrasound in detecting lymph node metastases in esophageal cancer. J Thorac Cardiovasc Surg 1997;114:817–21.
52. Krasna MJ, Flowers JL, Attar S, McLaughlin J. Combined thoracoscopic/laparoscopic staging of esophageal cancer. J Thorac Cardiovasc Surg 1996;111:800–6.
53. Flamen P, Lerut A, Van Cutsem E, et al. Utility of positron emission tomography for the staging of patients with potentially operable esophageal carcinoma. J Clin Oncol 2000;18:3202–10.
54. Downey RJ, Akhurst T, Ilson D, et al. Whole body 18FDG-PET and the response of esophageal cancer to induction therapy: results of a prospective trial. J Clin Oncol 2003;21:428–32.
55. Mealy K, Feely J, Reid I, et al. Tumour marker detection in oesophageal carcinoma. Eur J Surg Oncol 1996;22:505–7.
56. Rice TW, Rusch VW, Ishwaran H, Blackstone EH; Worldwide Esophageal Cancer Collaboration. Cancer of the esophagus and esophagogastric junction: data-driven staging for the seventh edition of the American Joint Committee on Cancer/International Union Against Cancer Cancer Staging Manuals. Cancer 2010;116:3763–73.
57. Edge SB, Byrd DR, Compton CC, et al, eds. AJCC cancer staging manual. 7th ed. New York: Springer-Verlag; 2009:103–5.
58. Rice TW, Rusch VW, Apperson-Hansen C, et al. Worldwide esophageal cancer collaboration. Dis Esophagus 2009;22:1–8.
59. Villaflor VM, Allaix ME, Minsky B, et al. Multidisciplinary approach for patients with esophageal cancer. World J Gastroenterol 2012;18:6737–46.
60. Ngamruengphong S, Wolfsen HC, Wallace MB. Survival of patients with superficial esophageal adenocarcinoma after endoscopic treatment vs surgery. Clin Gastroenterol Hepatol 2013;11:1424–9.e2.
61. Lin SH, Chang JY. Esophageal cancer: diagnosis and management. Chin J Cancer 2010;29:843–54.
62. Prasad GA, Wu TT, Wigle DA, et al. Endoscopic and surgical treatment of mucosal (T1a) esophageal adenocarcinoma in Barrett’s esophagus. Gastroenterology 2009;137:815–23.
63. Das A, Singh V, Fleischer DE, Sharma VK. A comparison of endoscopic treatment and surgery in early esophageal cancer: an analysis of surveillance epidemiology and end results data. Am J Gastroenterol 2008;103:1340–5.
64. Herskovic A, Martz K, al-Sarraf M, et al. Combined chemotherapy and radiotherapy compared with radiotherapy alone in patients with cancer of the esophagus. N Engl J Med 1992;326:1593–8.
65. Gebski V, Burmeister B, Smithers BM, et al; Australasian Gastro-Intestinal Trials Group. Survival benefits from neoadjuvant chemoradiotherapy or chemotherapy in oesophageal carcinoma: a meta-analysis. Lancet Oncol 2007;8:226–34.
66. Bedenne L, Michel P, Bouché O, et al. Chemoradiation followed by surgery compared with chemoradiation alone in squamous cancer of the esophagus: FFCD 9102. J Clin Oncol 2007;25:1160–8.
67. Tougeron D, Scotté M, Hamidou H, et al. Definitive chemoradiotherapy in patients with esophageal adenocarcinoma: an alternative to surgery? J Surg Oncol 2012;105: 761–6.
68. Siddiqui AA, Sarkar A, Beltz S, et al. Placement of fully covered self-expandable metal stents in patients with locally advanced esophageal cancer before neoadjuvant therapy. Gastrointest Endosc 2012;76:44–51.
69. Nagaraja V, Cox MR, Eslick GD. Safety and efficacy of esophageal stents preceding or during neoadjuvant chemotherapy for esophageal cancer: a systematic review and meta-analysis. J Gastrointest Oncol 2014;5:119–26.
70. Adler DG, Fang J, Wong R, et al. Placement of Polyflex stents in patients with locally advanced esophageal cancer is safe and improves dysphagia during neoadjuvant therapy. Gastrointest Endosc 2009;70:614–9.
71. Dubecz A, Sepesi B, Salvador R, et al. Surgical resection for locoregional esophageal cancer is underutilized in the United States. J Am Coll Surg 2010;211:754–61.
72. Hulscher JB, Tijssen JG, Obertop H, van Lanschot JJ. Transthoracic versus transhiatal resection for carcinoma of the esophagus: a meta-analysis. Ann Thorac Surg 2001;72:306–13.
73. Chang AC, Ji H, Birkmeyer NJ, et al. Outcomes after transhiatal and transthoracic esophagectomy for cancer. Ann Thorac Surg 2008;85:424–9.
74. Almhanna K, Shridhar R, Meredith KL. Neoadjuvant or adjuvant therapy for resectable esophageal cancer: is there a standard of care? Cancer Control 2013;20:89–96.
75. Harvey JA, Bessell JR, Beller E, et al. Chemoradiation therapy is effective for the palliative treatment of malignant dysphagia. Dis Esophagus 2004;17:260–5.
76. Siersema PD, Schrauwen SL, van Blankenstein M, et al; Rotterdam Esophageal Tumor Study Group. Self-expanding metal stents for complicated and recurrent esophagogastric cancer. Gastrointest Endosc 2001;54:579–86.
77. Homs MY, Steyerberg EW, Eijkenboom WM, et al. Single-dose brachytherapy versus metal stent placement for the palliation of dysphagia from oesophageal cancer: multicentre randomised trial. Lancet 2004;364(9444):1497–504.
78. van den Bongard HJ, Boot H, Baas P, Taal BG. The role of parallel stent insertion in patients with esophagorespiratory fistulas. Gastrointest Endosc 2002;55:110–5.
79. Siegel R, DeSantis C, Virgo K, et al. Cancer treatment and survivorship statistics, 2012. CA Cancer J Clin 2012;62:220–41.
80. Fein R, Kelsen DP, Geller N, et al. Adenocarcinoma of the esophagus and gastroesophageal junction. Prognostic factors and results of therapy. Cancer 1985;56:2512–8.
81. Mariette C, Maurel A, Fabre S, et al. [Preoperative prognostic factors for squamous cell carcinomas of the thoracic esophagus]. Gastroenterol Clin Biol 2001;25:468–72.
82. Swanson SJ, Batirel HF, Bueno R, et al. Transthoracic esophagectomy with radical mediastinal and abdominal lymph node dissection and cervical esophagogastrostomy for esophageal carcinoma. Ann Thorac Surg 2001;72:1918–24.
83. Urba SG, Orringer MB, Turrisi A, et al. Randomized trial of preoperative chemoradiation versus surgery alone in patients with locoregional esophageal carcinoma. J Clin Oncol 2001;19:305–13.
84. Hosch SB, Stoecklein NH, Pichlmeier U, et al. Esophageal cancer: the mode of lymphatic tumor cell spread and its prognostic significance. J Clin Oncol 2001;19:1970–5.
Powered toothbrushes really are better than manual ones at plaque control
Maintaining close collaborative relationships with my dental colleagues is one of the many benefits of my primary care practice. I never cease to be amazed by how much my dental colleagues know about medicine and how little I know about dentistry. But I do ask my patients how frequently they see a dentist because it is a powerful marker for what I am going to find during the oral examination.
Many of my patients seem to have trouble maintaining their native teeth. This is surprising to me given the abundance of options for dental care; and yet, not surprising when I remember that caries is the most prevalent disease worldwide. Oral health has a huge potential impact on overall health, and the control of dental plaque is the key to oral health. I typically do not recommend toothbrushes to my patients who have identified dental disease, but I may start doing this now that I understand more about toothbrushes.
Powered toothbrushes clean teeth through a variety of mechanisms: side-to-side action, counter oscillation, rotation oscillation, circular, ultrasonic, and ionic, just to name a few. They are more expensive than regular toothbrushes, but are they better for removing plaque?
An updated systematic review of the literature has been published comparing powered versus manual toothbrushing for the maintenance of oral health. Trials were selected if they evaluated at least 4 weeks of unsupervised toothbrushing. Fifty-one trials involving 4,624 participants provided data for the meta-analysis (Cochrane Database Syst. Rev. 2014;6:CD002281 [doi:10.1002/14651858.CD002281.pub3]).
Powered toothbrushes provide a statistically significant benefit, compared with manual toothbrushes, for the reduction of plaque in both the short (1-3 months; 11% reduction) and long term (longer than 3 months; 21% reduction) over manual toothbrushes. Powered toothbrushes also provide a statistically significant benefit in the short and long term for reduction in gingivitis. Most of the evidence is for rotation oscillation brushes.
So now I can give my patients a useful tip for maintaining oral health. Does improved plaque removal translate into general health benefits? We are uncertain, but it will certainly make for more enjoyable oral examinations.
Dr. Ebbert is a professor of medicine, a general internist at the Mayo Clinic in Rochester, Minn., and a diplomate of the American Board of Addiction Medicine. Dr. Ebbert reports no disclosures. The opinions expressed are his alone and should not be used to diagnose or treat any medical condition nor should they be used as a substitute for medical advice from a qualified, board-certified practicing clinician.
Maintaining close collaborative relationships with my dental colleagues is one of the many benefits of my primary care practice. I never cease to be amazed by how much my dental colleagues know about medicine and how little I know about dentistry. But I do ask my patients how frequently they see a dentist because it is a powerful marker for what I am going to find during the oral examination.
Many of my patients seem to have trouble maintaining their native teeth. This is surprising to me given the abundance of options for dental care; and yet, not surprising when I remember that caries is the most prevalent disease worldwide. Oral health has a huge potential impact on overall health, and the control of dental plaque is the key to oral health. I typically do not recommend toothbrushes to my patients who have identified dental disease, but I may start doing this now that I understand more about toothbrushes.
Powered toothbrushes clean teeth through a variety of mechanisms: side-to-side action, counter oscillation, rotation oscillation, circular, ultrasonic, and ionic, just to name a few. They are more expensive than regular toothbrushes, but are they better for removing plaque?
An updated systematic review of the literature has been published comparing powered versus manual toothbrushing for the maintenance of oral health. Trials were selected if they evaluated at least 4 weeks of unsupervised toothbrushing. Fifty-one trials involving 4,624 participants provided data for the meta-analysis (Cochrane Database Syst. Rev. 2014;6:CD002281 [doi:10.1002/14651858.CD002281.pub3]).
Powered toothbrushes provide a statistically significant benefit, compared with manual toothbrushes, for the reduction of plaque in both the short (1-3 months; 11% reduction) and long term (longer than 3 months; 21% reduction) over manual toothbrushes. Powered toothbrushes also provide a statistically significant benefit in the short and long term for reduction in gingivitis. Most of the evidence is for rotation oscillation brushes.
So now I can give my patients a useful tip for maintaining oral health. Does improved plaque removal translate into general health benefits? We are uncertain, but it will certainly make for more enjoyable oral examinations.
Dr. Ebbert is a professor of medicine, a general internist at the Mayo Clinic in Rochester, Minn., and a diplomate of the American Board of Addiction Medicine. Dr. Ebbert reports no disclosures. The opinions expressed are his alone and should not be used to diagnose or treat any medical condition nor should they be used as a substitute for medical advice from a qualified, board-certified practicing clinician.
Maintaining close collaborative relationships with my dental colleagues is one of the many benefits of my primary care practice. I never cease to be amazed by how much my dental colleagues know about medicine and how little I know about dentistry. But I do ask my patients how frequently they see a dentist because it is a powerful marker for what I am going to find during the oral examination.
Many of my patients seem to have trouble maintaining their native teeth. This is surprising to me given the abundance of options for dental care; and yet, not surprising when I remember that caries is the most prevalent disease worldwide. Oral health has a huge potential impact on overall health, and the control of dental plaque is the key to oral health. I typically do not recommend toothbrushes to my patients who have identified dental disease, but I may start doing this now that I understand more about toothbrushes.
Powered toothbrushes clean teeth through a variety of mechanisms: side-to-side action, counter oscillation, rotation oscillation, circular, ultrasonic, and ionic, just to name a few. They are more expensive than regular toothbrushes, but are they better for removing plaque?
An updated systematic review of the literature has been published comparing powered versus manual toothbrushing for the maintenance of oral health. Trials were selected if they evaluated at least 4 weeks of unsupervised toothbrushing. Fifty-one trials involving 4,624 participants provided data for the meta-analysis (Cochrane Database Syst. Rev. 2014;6:CD002281 [doi:10.1002/14651858.CD002281.pub3]).
Powered toothbrushes provide a statistically significant benefit, compared with manual toothbrushes, for the reduction of plaque in both the short (1-3 months; 11% reduction) and long term (longer than 3 months; 21% reduction) over manual toothbrushes. Powered toothbrushes also provide a statistically significant benefit in the short and long term for reduction in gingivitis. Most of the evidence is for rotation oscillation brushes.
So now I can give my patients a useful tip for maintaining oral health. Does improved plaque removal translate into general health benefits? We are uncertain, but it will certainly make for more enjoyable oral examinations.
Dr. Ebbert is a professor of medicine, a general internist at the Mayo Clinic in Rochester, Minn., and a diplomate of the American Board of Addiction Medicine. Dr. Ebbert reports no disclosures. The opinions expressed are his alone and should not be used to diagnose or treat any medical condition nor should they be used as a substitute for medical advice from a qualified, board-certified practicing clinician.

Pediatric Hospital Medicine 2014: Over-Diagnosis Is Harming Children
Presenters
Eric Coon, Ricardo Quinonez, Alan Schroeder
Summary
One of PHM2014’s first breakout sessions, coming on the heels of Dr. Meuthing’s opening talk on reducing serious safety events, focused on the topic of over-diagnosis in pediatric HM and its contribution to patient harm. The first key point is the distinction between over-diagnosis and mis-diagnosis. Over-diagnosis is the identification of an abnormality where detection will not benefit the patient. This is different from mis-diagnosis or incorrect diagnosis. Over-diagnosis has grown over the years due to several causes, including our fear of missing a diagnosis and increasing use of screening tests.
The speakers outlined many, varied drivers of over-diagnosis, including physicians’ unawareness of over-diagnosis, physicians’ discomfort with uncertainty, system incentives such as fee for service which reimburses or rewards increased testing, quality measures based on usage and testing, a perceived imperative to use testing and technology because it is available, and physicians’ inherent belief in technology and its results.
The classic example of over-diagnosis in pediatrics is asymptomatic urinary screening for neuroblastomas, where studies showed an increase in testing, an increase in diagnosis, but no change in mortality. A current example is children receiving head CT scans for minor head trauma that can lead to a diagnosis of small asymptomatic head bleeds or non-displaced skull fractures, which can then lead to PICU admissions, transfers to higher level centers, prophylactic administration of anti-seizure medications, and repeat CT scans.
From the patient perspective, over-diagnosis can lead to unnecessary hospitalizations, inappropriate medications and treatments, and increased patient or parental anxiety secondary being given a label of a diagnosis or disease.
Key Takeaway
The bottom line for physicians to consider when ordering a test is not just does the patient benefit from detection or diagnosis, but also what is the potential harm?
James O’Callaghan is a clinical assistant professor of pediatrics at the University of Washington and a member of Team Hospitalist.
Presenters
Eric Coon, Ricardo Quinonez, Alan Schroeder
Summary
One of PHM2014’s first breakout sessions, coming on the heels of Dr. Meuthing’s opening talk on reducing serious safety events, focused on the topic of over-diagnosis in pediatric HM and its contribution to patient harm. The first key point is the distinction between over-diagnosis and mis-diagnosis. Over-diagnosis is the identification of an abnormality where detection will not benefit the patient. This is different from mis-diagnosis or incorrect diagnosis. Over-diagnosis has grown over the years due to several causes, including our fear of missing a diagnosis and increasing use of screening tests.
The speakers outlined many, varied drivers of over-diagnosis, including physicians’ unawareness of over-diagnosis, physicians’ discomfort with uncertainty, system incentives such as fee for service which reimburses or rewards increased testing, quality measures based on usage and testing, a perceived imperative to use testing and technology because it is available, and physicians’ inherent belief in technology and its results.
The classic example of over-diagnosis in pediatrics is asymptomatic urinary screening for neuroblastomas, where studies showed an increase in testing, an increase in diagnosis, but no change in mortality. A current example is children receiving head CT scans for minor head trauma that can lead to a diagnosis of small asymptomatic head bleeds or non-displaced skull fractures, which can then lead to PICU admissions, transfers to higher level centers, prophylactic administration of anti-seizure medications, and repeat CT scans.
From the patient perspective, over-diagnosis can lead to unnecessary hospitalizations, inappropriate medications and treatments, and increased patient or parental anxiety secondary being given a label of a diagnosis or disease.
Key Takeaway
The bottom line for physicians to consider when ordering a test is not just does the patient benefit from detection or diagnosis, but also what is the potential harm?
James O’Callaghan is a clinical assistant professor of pediatrics at the University of Washington and a member of Team Hospitalist.
Presenters
Eric Coon, Ricardo Quinonez, Alan Schroeder
Summary
One of PHM2014’s first breakout sessions, coming on the heels of Dr. Meuthing’s opening talk on reducing serious safety events, focused on the topic of over-diagnosis in pediatric HM and its contribution to patient harm. The first key point is the distinction between over-diagnosis and mis-diagnosis. Over-diagnosis is the identification of an abnormality where detection will not benefit the patient. This is different from mis-diagnosis or incorrect diagnosis. Over-diagnosis has grown over the years due to several causes, including our fear of missing a diagnosis and increasing use of screening tests.
The speakers outlined many, varied drivers of over-diagnosis, including physicians’ unawareness of over-diagnosis, physicians’ discomfort with uncertainty, system incentives such as fee for service which reimburses or rewards increased testing, quality measures based on usage and testing, a perceived imperative to use testing and technology because it is available, and physicians’ inherent belief in technology and its results.
The classic example of over-diagnosis in pediatrics is asymptomatic urinary screening for neuroblastomas, where studies showed an increase in testing, an increase in diagnosis, but no change in mortality. A current example is children receiving head CT scans for minor head trauma that can lead to a diagnosis of small asymptomatic head bleeds or non-displaced skull fractures, which can then lead to PICU admissions, transfers to higher level centers, prophylactic administration of anti-seizure medications, and repeat CT scans.
From the patient perspective, over-diagnosis can lead to unnecessary hospitalizations, inappropriate medications and treatments, and increased patient or parental anxiety secondary being given a label of a diagnosis or disease.
Key Takeaway
The bottom line for physicians to consider when ordering a test is not just does the patient benefit from detection or diagnosis, but also what is the potential harm?
James O’Callaghan is a clinical assistant professor of pediatrics at the University of Washington and a member of Team Hospitalist.
Pediatric Hospital Medicine 2014: Building Blocks in the Evolution of a Successful Distributed Hospitalist Program
Presenters
Dan Hale, MD, FAAP, and Elisabeth Schainker, MD, FAAP, The Floating Hospital for Children at Tufts Medical Center, Boston
Summary
"Master the basics of a good hospitalist program and keep revisiting your core values, and you will continue to have a high-quality and sustainable program,” said Dr. Dan Hale at the PHM14 workshop “Building Blocks in the Evolution of a Successful Distributed Hospitalist Program.”
Dr. Elisabeth Schainker, chief of hospitalist medicine at The Floating Hospital for Children at Tufts Medical Center in Boston, and Dr. Hale, a hospitalist at The Floating Hospital and site director of the Lawrence General Hospital affiliated pediatric hospitalist program, allowed participants to share their experiences in program development.
This workshop reviewed the fundamentals that programs should review before starting and also periodically after established. Program changes should be made as needed. The workshop used an assessment tool to evaluate the basic elements of the participants’ programs. The February 2014 article “Key Principles and Characteristics of an Effective Hospital Medicine Group” in the Journal of Hospital Medicine were used as a starting point for program self-evaluation. These “building blocks” include:
• Establish the rationale for the program and include all stakeholders;
• Financial expectations; • Define scope of practice;
• Nursing and referral physician collaboration;
• Assess staffing and workload expectations;
• Referral base; and
• Basic code and emergency preparedness.
Ongoing program development elements of a program were discussed as well. These components help further integrate a hospitalist program with the hospital as a whole and help add value. These ongoing “building blocks” include:
• Communication and collaboration with other hospital departments (emergency, radiology, surgery, etc.);
• Newborn medicine care;
• Internal group clinical practice guidelines;
• Co-management of surgical or specialty patients;
• Transfers from other hospitals or continuing care from tertiary care centers;
• Pediatric code teams and rapid response teams;
• Advanced code and emergency preparedness and mock code training; and
• Nursing education.
These additive features may be different at each program. Not all of these components are applicable or needed at all hospitals. Thoughtful approaches and thorough planning can create synergy with other components of a program. The essentials of a successful distributed network of multiple hospitalist program site were also described. After assuring the fundamentals are present at each site, transparency and institutional alignment are imperative.
Key Takeaways
1. It is important to understand several fundamental elements of hospitalist programs and address goals before starting a program.
2. For existing programs, it is important to review the fundamentals periodically and provide program maintenance.
3. After a program is established and fundamentals are in place, other important advance practices can be added on. These include ongoing collaboration, advanced emergency planning, staff education, and clinical practice guidelines.
4. For a multiple site or distributed program, high level collaboration and transparency is essential.
Dr. Hale is a past member of Team Hospitalist and is a pediatric hospitalist at the Floating Hospital for Children at Tufts Medical Center in Boston.
Presenters
Dan Hale, MD, FAAP, and Elisabeth Schainker, MD, FAAP, The Floating Hospital for Children at Tufts Medical Center, Boston
Summary
"Master the basics of a good hospitalist program and keep revisiting your core values, and you will continue to have a high-quality and sustainable program,” said Dr. Dan Hale at the PHM14 workshop “Building Blocks in the Evolution of a Successful Distributed Hospitalist Program.”
Dr. Elisabeth Schainker, chief of hospitalist medicine at The Floating Hospital for Children at Tufts Medical Center in Boston, and Dr. Hale, a hospitalist at The Floating Hospital and site director of the Lawrence General Hospital affiliated pediatric hospitalist program, allowed participants to share their experiences in program development.
This workshop reviewed the fundamentals that programs should review before starting and also periodically after established. Program changes should be made as needed. The workshop used an assessment tool to evaluate the basic elements of the participants’ programs. The February 2014 article “Key Principles and Characteristics of an Effective Hospital Medicine Group” in the Journal of Hospital Medicine were used as a starting point for program self-evaluation. These “building blocks” include:
• Establish the rationale for the program and include all stakeholders;
• Financial expectations; • Define scope of practice;
• Nursing and referral physician collaboration;
• Assess staffing and workload expectations;
• Referral base; and
• Basic code and emergency preparedness.
Ongoing program development elements of a program were discussed as well. These components help further integrate a hospitalist program with the hospital as a whole and help add value. These ongoing “building blocks” include:
• Communication and collaboration with other hospital departments (emergency, radiology, surgery, etc.);
• Newborn medicine care;
• Internal group clinical practice guidelines;
• Co-management of surgical or specialty patients;
• Transfers from other hospitals or continuing care from tertiary care centers;
• Pediatric code teams and rapid response teams;
• Advanced code and emergency preparedness and mock code training; and
• Nursing education.
These additive features may be different at each program. Not all of these components are applicable or needed at all hospitals. Thoughtful approaches and thorough planning can create synergy with other components of a program. The essentials of a successful distributed network of multiple hospitalist program site were also described. After assuring the fundamentals are present at each site, transparency and institutional alignment are imperative.
Key Takeaways
1. It is important to understand several fundamental elements of hospitalist programs and address goals before starting a program.
2. For existing programs, it is important to review the fundamentals periodically and provide program maintenance.
3. After a program is established and fundamentals are in place, other important advance practices can be added on. These include ongoing collaboration, advanced emergency planning, staff education, and clinical practice guidelines.
4. For a multiple site or distributed program, high level collaboration and transparency is essential.
Dr. Hale is a past member of Team Hospitalist and is a pediatric hospitalist at the Floating Hospital for Children at Tufts Medical Center in Boston.
Presenters
Dan Hale, MD, FAAP, and Elisabeth Schainker, MD, FAAP, The Floating Hospital for Children at Tufts Medical Center, Boston
Summary
"Master the basics of a good hospitalist program and keep revisiting your core values, and you will continue to have a high-quality and sustainable program,” said Dr. Dan Hale at the PHM14 workshop “Building Blocks in the Evolution of a Successful Distributed Hospitalist Program.”
Dr. Elisabeth Schainker, chief of hospitalist medicine at The Floating Hospital for Children at Tufts Medical Center in Boston, and Dr. Hale, a hospitalist at The Floating Hospital and site director of the Lawrence General Hospital affiliated pediatric hospitalist program, allowed participants to share their experiences in program development.
This workshop reviewed the fundamentals that programs should review before starting and also periodically after established. Program changes should be made as needed. The workshop used an assessment tool to evaluate the basic elements of the participants’ programs. The February 2014 article “Key Principles and Characteristics of an Effective Hospital Medicine Group” in the Journal of Hospital Medicine were used as a starting point for program self-evaluation. These “building blocks” include:
• Establish the rationale for the program and include all stakeholders;
• Financial expectations; • Define scope of practice;
• Nursing and referral physician collaboration;
• Assess staffing and workload expectations;
• Referral base; and
• Basic code and emergency preparedness.
Ongoing program development elements of a program were discussed as well. These components help further integrate a hospitalist program with the hospital as a whole and help add value. These ongoing “building blocks” include:
• Communication and collaboration with other hospital departments (emergency, radiology, surgery, etc.);
• Newborn medicine care;
• Internal group clinical practice guidelines;
• Co-management of surgical or specialty patients;
• Transfers from other hospitals or continuing care from tertiary care centers;
• Pediatric code teams and rapid response teams;
• Advanced code and emergency preparedness and mock code training; and
• Nursing education.
These additive features may be different at each program. Not all of these components are applicable or needed at all hospitals. Thoughtful approaches and thorough planning can create synergy with other components of a program. The essentials of a successful distributed network of multiple hospitalist program site were also described. After assuring the fundamentals are present at each site, transparency and institutional alignment are imperative.
Key Takeaways
1. It is important to understand several fundamental elements of hospitalist programs and address goals before starting a program.
2. For existing programs, it is important to review the fundamentals periodically and provide program maintenance.
3. After a program is established and fundamentals are in place, other important advance practices can be added on. These include ongoing collaboration, advanced emergency planning, staff education, and clinical practice guidelines.
4. For a multiple site or distributed program, high level collaboration and transparency is essential.
Dr. Hale is a past member of Team Hospitalist and is a pediatric hospitalist at the Floating Hospital for Children at Tufts Medical Center in Boston.
Intergluteal Itching in Need of Relief
ANSWER
Admittedly, this is a bit of a trick question—but with a good teaching point to make. A course of oral fluconazole (choice “a”) is futile, since there’s no reason to think this problem is yeast-driven and since the patient has already demonstrated a lack of response to topical imidazoles.
Punch biopsy (choice “b”) would be a good choice, but not in this area, where it could quickly become a bigger problem than the one the patient presented with. Sutures would not likely hold the biopsy wound together, and resultant infection is all too likely.
A KOH test to detect fungal or yeast elements (choice “c”) is unlikely to shed any light on the problem, given the lack of response to antifungal creams. Finally, there’s no reason to suspect a bacterial origin, so oral antibiotics such as cephalexin (choice “d”) would be useless (and had already been tried unsuccessfully).
The correct answer is none of the above (choice “e”).
DISCUSSION
This case illustrates why dermatology seems so maddeningly difficult to the uninitiated. Any experienced derm provider would know the correct diagnosis, lichen sclerosus et atrophicus (LS&A), because it presents in such a distinctive way (in limited locations, predominantly in women) and because the differential is so limited. But if you’ve never heard of LS&A, you’re unlikely to diagnose it, let alone know how to treat it.
LS&A is an inflammatory condition of unknown origin that affects the upper epidermis. It can present in extragenital locations (particularly shoulders and legs) but is far more common in genital areas. As exhibited in this case, it presents with well-defined pigment loss, which is especially easy to see in patients with darker skin.
Although more commonly seen in women, LS&A can occur in men, usually manifesting on the penile glans and distal foreskin of uncircumcised patients. The dry atrophic changes seen on the glans can lead to stenosis of the urethral meatus and, proximally, to adhesions (phimosis) of the foreskin. (This condition was termed balanitis xerotica obliterans [BXO] long before its pathologic process was determined to be identical to LS&A’s. Tissue specimens obtained during circumcisions performed for chronic phimosis often yield evidence of BXO.)
In women, untreated chronic LS&A can lead to sclerotic changes in and around the urethra and labia minora and can cause introital stenosis. This case is a bit atypical; LS&A more often manifests in perivaginal and perirectal areas, where the intense hypopigmentation produces a classic “figure eight” appearance.
The differential includes lichen simplex chronicus, psoriasis, lichen planus, contact/irritant dermatitis, and seborrhea. Often, biopsy is necessary and appropriate to settle the issue, other factors being equal.
TREATMENT/PROGNOSIS
The patient was given a prescription for clobetasol 0.05% ointment for twice-daily application Monday through Friday (and no application for two consecutive days—in this case, the weekend—per week). Studies have established the efficacy and safety of this treatment regimen.
In a month or two, application can be reduced to once or twice a week to control the condition.
ANSWER
Admittedly, this is a bit of a trick question—but with a good teaching point to make. A course of oral fluconazole (choice “a”) is futile, since there’s no reason to think this problem is yeast-driven and since the patient has already demonstrated a lack of response to topical imidazoles.
Punch biopsy (choice “b”) would be a good choice, but not in this area, where it could quickly become a bigger problem than the one the patient presented with. Sutures would not likely hold the biopsy wound together, and resultant infection is all too likely.
A KOH test to detect fungal or yeast elements (choice “c”) is unlikely to shed any light on the problem, given the lack of response to antifungal creams. Finally, there’s no reason to suspect a bacterial origin, so oral antibiotics such as cephalexin (choice “d”) would be useless (and had already been tried unsuccessfully).
The correct answer is none of the above (choice “e”).
DISCUSSION
This case illustrates why dermatology seems so maddeningly difficult to the uninitiated. Any experienced derm provider would know the correct diagnosis, lichen sclerosus et atrophicus (LS&A), because it presents in such a distinctive way (in limited locations, predominantly in women) and because the differential is so limited. But if you’ve never heard of LS&A, you’re unlikely to diagnose it, let alone know how to treat it.
LS&A is an inflammatory condition of unknown origin that affects the upper epidermis. It can present in extragenital locations (particularly shoulders and legs) but is far more common in genital areas. As exhibited in this case, it presents with well-defined pigment loss, which is especially easy to see in patients with darker skin.
Although more commonly seen in women, LS&A can occur in men, usually manifesting on the penile glans and distal foreskin of uncircumcised patients. The dry atrophic changes seen on the glans can lead to stenosis of the urethral meatus and, proximally, to adhesions (phimosis) of the foreskin. (This condition was termed balanitis xerotica obliterans [BXO] long before its pathologic process was determined to be identical to LS&A’s. Tissue specimens obtained during circumcisions performed for chronic phimosis often yield evidence of BXO.)
In women, untreated chronic LS&A can lead to sclerotic changes in and around the urethra and labia minora and can cause introital stenosis. This case is a bit atypical; LS&A more often manifests in perivaginal and perirectal areas, where the intense hypopigmentation produces a classic “figure eight” appearance.
The differential includes lichen simplex chronicus, psoriasis, lichen planus, contact/irritant dermatitis, and seborrhea. Often, biopsy is necessary and appropriate to settle the issue, other factors being equal.
TREATMENT/PROGNOSIS
The patient was given a prescription for clobetasol 0.05% ointment for twice-daily application Monday through Friday (and no application for two consecutive days—in this case, the weekend—per week). Studies have established the efficacy and safety of this treatment regimen.
In a month or two, application can be reduced to once or twice a week to control the condition.
ANSWER
Admittedly, this is a bit of a trick question—but with a good teaching point to make. A course of oral fluconazole (choice “a”) is futile, since there’s no reason to think this problem is yeast-driven and since the patient has already demonstrated a lack of response to topical imidazoles.
Punch biopsy (choice “b”) would be a good choice, but not in this area, where it could quickly become a bigger problem than the one the patient presented with. Sutures would not likely hold the biopsy wound together, and resultant infection is all too likely.
A KOH test to detect fungal or yeast elements (choice “c”) is unlikely to shed any light on the problem, given the lack of response to antifungal creams. Finally, there’s no reason to suspect a bacterial origin, so oral antibiotics such as cephalexin (choice “d”) would be useless (and had already been tried unsuccessfully).
The correct answer is none of the above (choice “e”).
DISCUSSION
This case illustrates why dermatology seems so maddeningly difficult to the uninitiated. Any experienced derm provider would know the correct diagnosis, lichen sclerosus et atrophicus (LS&A), because it presents in such a distinctive way (in limited locations, predominantly in women) and because the differential is so limited. But if you’ve never heard of LS&A, you’re unlikely to diagnose it, let alone know how to treat it.
LS&A is an inflammatory condition of unknown origin that affects the upper epidermis. It can present in extragenital locations (particularly shoulders and legs) but is far more common in genital areas. As exhibited in this case, it presents with well-defined pigment loss, which is especially easy to see in patients with darker skin.
Although more commonly seen in women, LS&A can occur in men, usually manifesting on the penile glans and distal foreskin of uncircumcised patients. The dry atrophic changes seen on the glans can lead to stenosis of the urethral meatus and, proximally, to adhesions (phimosis) of the foreskin. (This condition was termed balanitis xerotica obliterans [BXO] long before its pathologic process was determined to be identical to LS&A’s. Tissue specimens obtained during circumcisions performed for chronic phimosis often yield evidence of BXO.)
In women, untreated chronic LS&A can lead to sclerotic changes in and around the urethra and labia minora and can cause introital stenosis. This case is a bit atypical; LS&A more often manifests in perivaginal and perirectal areas, where the intense hypopigmentation produces a classic “figure eight” appearance.
The differential includes lichen simplex chronicus, psoriasis, lichen planus, contact/irritant dermatitis, and seborrhea. Often, biopsy is necessary and appropriate to settle the issue, other factors being equal.
TREATMENT/PROGNOSIS
The patient was given a prescription for clobetasol 0.05% ointment for twice-daily application Monday through Friday (and no application for two consecutive days—in this case, the weekend—per week). Studies have established the efficacy and safety of this treatment regimen.
In a month or two, application can be reduced to once or twice a week to control the condition.

For almost a year, a 55-year-old African-American woman has experienced itchy skin changes in her perianal area. Treatment attempts with several topical creams—including clotrimazole, combination clotrimazole/betamethasone, and ketoconazole—have not helped. The patient has seen several primary care providers for the problem. All have told her that it was yeast-related, except the last clinician, who suspected psoriasis. When the topical medication prescribed by that provider did not yield a resolution, the patient decided to consult dermatology. Due to her lack of insurance, she had to wait four months to see a derm clinician, since her only option was a once-a-month free clinic in her community. Aside from mild hypertension, the patient claims to be in good health. Recent work-up indicated she does not have diabetes. She denies any family history of skin diseases, including psoriasis. She has had no previous complaints regarding her vaginal/perivaginal areas. The patient’s type V skin is free of notable changes except in the intergluteal and perianal areas. Specifically, no rash is noted on her extensor elbows or knees or in her scalp, and there are no changes in her fingernails. When the patient lies on her left side, extending her left leg and bringing her right knee toward her chest, the entire intergluteal and perianal areas can be visualized. Distinct loss of dark pigment is seen in the upper intergluteal/lower coccygeal areas. Closer inspection reveals that the pigment loss is complete, giving the affected skin a porcelain-like white appearance that also seems moderately atrophic. Palpation confirms this impression. No such changes are noted in the perianal or perineal areas. However, there is diffuse hyperpigmentation, as well as signs of mild chronic excoriation.

Disseminated Coccidioidomycosis of the Spine in an Immunocompetent Patient
Transforming vaginal hysterectomy: 7 solutions to the most daunting challenges
Vaginal hysterectomy is the preferred route to benign hysterectomy because it is associated with better outcomes and fewer complications than the laparoscopic and open abdominal approaches.1,2 Yet, despite superior patient outcomes and cost benefits, the rate of vaginal hysterectomy is declining.
According to the Nationwide Inpatient Sample, the use of vaginal hysterectomy declined from 24.8% in 1998 to 16.7% in 2010.3 In fact, more than 80% of surgeons in the United States now perform fewer than five vaginal procedures in a year.4
The increasing use of other minimally invasive routes, such as laparoscopy and robotics, indicates that most practicing surgeons and recent graduates are choosing these approaches over the vaginal route. In only 3 years, the rate of laparoscopy increased by 6% and robotics increased by almost 10%.3
Many surgeons assume that vaginal hysterectomy exists in a state of suspended animation, with nothing much changed in the way it has been performed over the past few decades. Further, vaginal surgery is difficult to teach and learn, given limitations in exposure and visualization, difficulty in securing hemostasis, and challenges in the removal of the large uterus and adnexae. As a result, vaginal hysterectomy often is thought, erroneously, to be indicated only in procedures involving a small and prolapsing uterus.
To increase the rate of vaginal hysterectomy, we can benefit from experience gained in laparoscopy and robotics—whether we are teachers or learners—while maintaining patient safety and containing costs.
In this article, I describe common challenges in vaginal hysterectomy and offer tools and techniques to overcome them:
- achieving and enhancing ergonomics, exposure, and visualization
- the need to work in a long vaginal vault
- the task of securing vascular and thick tissue pedicles when the introitus and vaginal vault are narrow.
The vaginal approach is less costly
Vaginal hysterectomy costs significantly less to perform than other approaches. At a tertiary referral center, vaginal hysterectomy costs approximately $7,000 to $18,000 per case less than laparoscopic, abdominal, and robotic hysterectomy.5 With declining use of vaginal hysterectomy and increasing use of more costly approaches, we face a health-care crisis.
Residents are inadequately trained to perform vaginal hysterectomy
Data reveal that not only are our recent graduates inadequately prepared to perform vaginal hysterectomy, but national health-care dollars and resources are depleted when surgeons choose to perform more costly approaches. As a result, many eligible patients end up deprived of the benefits of a single, concealed, and minimally invasive procedure.
The increase in laparoscopic and robotic approaches to hysterectomy has affected residency training. National case log reports from the Accreditation Council of Graduate Medical Education show that the number of vaginal hysterectomies performed by residents as “primary surgeons” decreased by 40%, from a mean of 35 cases in 2002 to 19 cases in 2012.6 A recent survey found that only 28% of graduating residents were “completely prepared” to perform a vaginal hysterectomy, compared with 58% for abdominal hysterectomy, 22% for laparoscopic hysterectomy, and 3% for the robotic approach.7
The rate of vaginal hysterectomy will continue to decline if we perform it in the same manner it was done 30 years ago. The current generation of practicing gynecologists and graduates is choosing to perform the procedure laparoscopically or robotically because of the advantages these technologies provide. It is time that we incorporate features from these minimally invasive approaches to streamline vaginal hysterectomy while maintaining patient safety and containing costs.
Challenges: Ergonomics, exposure, and visualization
In conventional vaginal surgery, the surgeon often is the person who has the best and, sometimes, the sole view. Two bedside assistants are required to hold retractors during the entire case, which can lead to fatigue and muscle strain. Poor lighting also can greatly limit visualization into the pelvic cavity.
Both laparoscopy and robotics provide a well-illuminated and magnified view, with three-dimensional images now available in both platforms. This view is projected to overhead monitors for the entire surgical team to see. Magnification of the pelvic anatomic structures and projection to an external monitor facilitate teaching and learning, better anticipation of the surgical and procedural needs, and overall patient safety.
From robotics, where ergonomics is exemplified, we also learn the importance of surgeon comfort during the procedure.
Solution #1: A self-retaining retractor
A self-retaining system such as the Magrina-Bookwalter vaginal retractor (Symmetry Surgical, Nashville, Tennessee) (FIGURE 1)
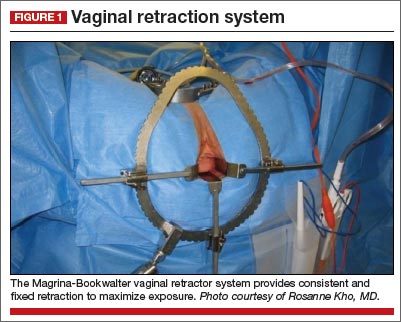
Solution #2: Seat the surgeon for an optimal view
With the patient in the lithotomy position and her legs in candy cane stirrups, the surgeon can be seated on a high chair so that the operative field is at the approximate level of the assistants’ view (FIGURE 2)
Solution #3: Illuminate the cavity
The deep pelvic cavity can be easily illuminated using a lighted suction tip, a flexible light source (as part of the cystoscopy set) held with a Babcock clamp (FIGURE 3), or a malleable illuminating mat taped to the retractor blades (such as Lightmat surgical illuminator, Lumitex, Inc., Strongsville, Ohio).
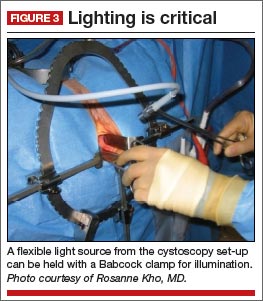
Solution #4: Project the image
Cameras attached to an overhead boom or operating room light handles (FIGURE 4) and an external telescope with integrated illumination, such as a standard cystoscope or VITOM Exoscope (Karl Storz, El Segundo, California) (FIGURE 5) provide both magnification and projection of the procedure to an overhead monitor.
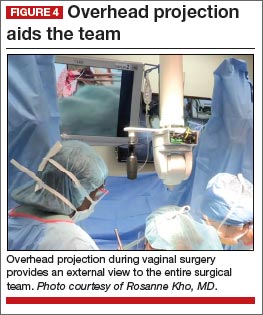
Glass technology (Google, Mountain View, California) also has been utilized in surgery and can be a good application of simultaneous projection and recording of the procedure to an external monitor (FIGURE 6). Google Glass is a wearable computer with an optical head-mounted display. The device, similar to eyeglasses, is voice-activated, thereby allowing the surgeon to record the procedure hands-free. Simultaneous projection to an external monitor allows the entire team in the operating room to be aware of the flow of the procedure.

Challenge: Working in a narrow vaginal vault
Without correct instrumentation, this challenge can be especially daunting. Laparoscopy and robotics have changed the way we perform pelvic surgery by providing advanced instrumentation.
Solution #5: Adapt your instruments
Modified vaginal instruments can be used to facilitate a case. Watch the accompanying VIDEO on the use of improved vaginal instruments during morcellation.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel |
| Click to enlarge >>> |
Among the instruments adaptable for vaginal surgery:
- curving, articulating instruments
- long, curved, and rounded knife handles, which allow for better ergonomics during prolonged morcellation
- modified long retractors and use of a single long vaginal pack provide retraction of loops of bowel and easy access to secure pedicles deep in the pelvis.
All of these instruments are available through Marina Medical in Sunrise, Florida.
Challenge: Securing vascular and thick tissue pediclesA narrow introitus and vaginal vault can be difficult to manage during vaginal surgery. Another challenge is a uterus that is large or deformed by multiple fibroids.
Solution #6: Vaginal incision
A simple superficial 2- to 3-cm incision on the distal posterior aspect of the vaginal wall can widen the introitus and vault to facilitate the procedure (FIGURE 7)
Solution #7: Vessel-sealing tools
The use of energy is integral to laparoscopy and robotics for dissection and securing vessels. In a meta-analysis that included seven randomized controlled trials, advanced vessel-sealing devices proved useful in vaginal surgery by decreasing blood loss and operative time.8
In the setting of a difficult vaginal hysterectomy with a narrow introitus and large uterus, the use of vessel-sealing technology allows the surgeon to skeletonize the uterine arteries while allowing progressive descensus to secure the upper pedicles.
In my experience, the use of an advanced vessel-sealing device, compared with traditional clamp-cut-tying technique, facilitated successful completion of vaginal hysterectomy in 650 patients with relative contraindications to the vaginal approach, such as nulliparity, a uterus weighing more than 250 g, and a history of cesarean delivery (Mayo Clinic data; yet unpublished).
We must change with the times
The rate of vaginal hysterectomy will continue to decline unless we modify our technique to incorporate new technology. The current generation of practicing gynecologists and recent graduates are choosing the laparoscopic and robotic approaches because of the advantages these technologies offer. It is time we incorporate relevant features from these minimally invasive approaches while maintaining patient safety and containing costs by performing vaginal hysterectomy whenever possible. A willingness to change and ability to think outside the usual box will help us train new generations of vaginal surgeons who can bring back vaginal hysterectomy as the preferred route to the benign hysterectomy.
WE WANT TO HEAR FROM YOU! Share your thoughts on this article. Send your Letter to the Editor to: [email protected]
1. Nieboer TE, Johnson N, Lethaby A, et al. Surgical approach to hysterectomy for benign gynaecological disease. Cochrane Database Syst Rev. 2009;(3):CD003677.
2. American College of Obstetricians and Gynecologists. ACOG Committee Opinion No. 444: Choosing the route of hysterectomy for benign disease. Obstet Gynecol. 2009;114(5):1156–1158.
3. Wright T, Herzog T, Tsul J, et al. Nationwide trends in inpatient hysterectomy in the United States. Obstet Gynecol. 2013:122(2):233–241.
4. Rogo-Gupta L, Lewyn S, Jum JH, et al. Effect of surgeon volume on outcomes and resource use for vaginal hysterectomy. Obstet Gynecol. 2010;116(6):1341–1347.
5. Wright KN, Jonsdottir GM, Jorgensen S, Shah N, Einarsson JI. Costs and outcomes of abdominal, vaginal, laparoscopic and robotic hysterectomies. JSLS. 2012;16(4):519–524.
6. Washburn EE, Cohen SL, Manoucherie E, Zurawin, RJ, Einarsson JI. Trends in reported residency surgical experience in hysterectomy [published online ahead of print June 4, 2014]. J Minim Invasive Gynecol. doi:10.1016/j.jmig.2014.05.005.
7. Burkett D, Horwitz J, Kennedy V, et al. Assessing current trends in resident hysterectomy training. Female Pelvic Med Reconstr Surg. 2011;17(5):210–214.
8. Kroft J, Selk K. Energy-based vessel sealing in vaginal hysterectomy. A systematic review and meta-analysis. Obstet Gynecol. 2011;118(5):1127–1136.

Rosanne M. Kho, MD
Dr. Kho is Assistant Professor, Director of the Urogynecology Program, and Co-Director of the MIGS Fellowship Program at Columbia University Medical Center in New York, New York.
The author reports that she is a consultant to Marina Medical and Symmetry Surgical.
Rosanne M. Kho, MD
Dr. Kho is Assistant Professor, Director of the Urogynecology Program, and Co-Director of the MIGS Fellowship Program at Columbia University Medical Center in New York, New York.
The author reports that she is a consultant to Marina Medical and Symmetry Surgical.
Rosanne M. Kho, MD
Dr. Kho is Assistant Professor, Director of the Urogynecology Program, and Co-Director of the MIGS Fellowship Program at Columbia University Medical Center in New York, New York.
The author reports that she is a consultant to Marina Medical and Symmetry Surgical.
Vaginal hysterectomy is the preferred route to benign hysterectomy because it is associated with better outcomes and fewer complications than the laparoscopic and open abdominal approaches.1,2 Yet, despite superior patient outcomes and cost benefits, the rate of vaginal hysterectomy is declining.
According to the Nationwide Inpatient Sample, the use of vaginal hysterectomy declined from 24.8% in 1998 to 16.7% in 2010.3 In fact, more than 80% of surgeons in the United States now perform fewer than five vaginal procedures in a year.4
The increasing use of other minimally invasive routes, such as laparoscopy and robotics, indicates that most practicing surgeons and recent graduates are choosing these approaches over the vaginal route. In only 3 years, the rate of laparoscopy increased by 6% and robotics increased by almost 10%.3
Many surgeons assume that vaginal hysterectomy exists in a state of suspended animation, with nothing much changed in the way it has been performed over the past few decades. Further, vaginal surgery is difficult to teach and learn, given limitations in exposure and visualization, difficulty in securing hemostasis, and challenges in the removal of the large uterus and adnexae. As a result, vaginal hysterectomy often is thought, erroneously, to be indicated only in procedures involving a small and prolapsing uterus.
To increase the rate of vaginal hysterectomy, we can benefit from experience gained in laparoscopy and robotics—whether we are teachers or learners—while maintaining patient safety and containing costs.
In this article, I describe common challenges in vaginal hysterectomy and offer tools and techniques to overcome them:
- achieving and enhancing ergonomics, exposure, and visualization
- the need to work in a long vaginal vault
- the task of securing vascular and thick tissue pedicles when the introitus and vaginal vault are narrow.
The vaginal approach is less costly
Vaginal hysterectomy costs significantly less to perform than other approaches. At a tertiary referral center, vaginal hysterectomy costs approximately $7,000 to $18,000 per case less than laparoscopic, abdominal, and robotic hysterectomy.5 With declining use of vaginal hysterectomy and increasing use of more costly approaches, we face a health-care crisis.
Residents are inadequately trained to perform vaginal hysterectomy
Data reveal that not only are our recent graduates inadequately prepared to perform vaginal hysterectomy, but national health-care dollars and resources are depleted when surgeons choose to perform more costly approaches. As a result, many eligible patients end up deprived of the benefits of a single, concealed, and minimally invasive procedure.
The increase in laparoscopic and robotic approaches to hysterectomy has affected residency training. National case log reports from the Accreditation Council of Graduate Medical Education show that the number of vaginal hysterectomies performed by residents as “primary surgeons” decreased by 40%, from a mean of 35 cases in 2002 to 19 cases in 2012.6 A recent survey found that only 28% of graduating residents were “completely prepared” to perform a vaginal hysterectomy, compared with 58% for abdominal hysterectomy, 22% for laparoscopic hysterectomy, and 3% for the robotic approach.7
The rate of vaginal hysterectomy will continue to decline if we perform it in the same manner it was done 30 years ago. The current generation of practicing gynecologists and graduates is choosing to perform the procedure laparoscopically or robotically because of the advantages these technologies provide. It is time that we incorporate features from these minimally invasive approaches to streamline vaginal hysterectomy while maintaining patient safety and containing costs.
Challenges: Ergonomics, exposure, and visualization
In conventional vaginal surgery, the surgeon often is the person who has the best and, sometimes, the sole view. Two bedside assistants are required to hold retractors during the entire case, which can lead to fatigue and muscle strain. Poor lighting also can greatly limit visualization into the pelvic cavity.
Both laparoscopy and robotics provide a well-illuminated and magnified view, with three-dimensional images now available in both platforms. This view is projected to overhead monitors for the entire surgical team to see. Magnification of the pelvic anatomic structures and projection to an external monitor facilitate teaching and learning, better anticipation of the surgical and procedural needs, and overall patient safety.
From robotics, where ergonomics is exemplified, we also learn the importance of surgeon comfort during the procedure.
Solution #1: A self-retaining retractor
A self-retaining system such as the Magrina-Bookwalter vaginal retractor (Symmetry Surgical, Nashville, Tennessee) (FIGURE 1)
Solution #2: Seat the surgeon for an optimal view
With the patient in the lithotomy position and her legs in candy cane stirrups, the surgeon can be seated on a high chair so that the operative field is at the approximate level of the assistants’ view (FIGURE 2)
Solution #3: Illuminate the cavity
The deep pelvic cavity can be easily illuminated using a lighted suction tip, a flexible light source (as part of the cystoscopy set) held with a Babcock clamp (FIGURE 3), or a malleable illuminating mat taped to the retractor blades (such as Lightmat surgical illuminator, Lumitex, Inc., Strongsville, Ohio).
Solution #4: Project the image
Cameras attached to an overhead boom or operating room light handles (FIGURE 4) and an external telescope with integrated illumination, such as a standard cystoscope or VITOM Exoscope (Karl Storz, El Segundo, California) (FIGURE 5) provide both magnification and projection of the procedure to an overhead monitor.
Glass technology (Google, Mountain View, California) also has been utilized in surgery and can be a good application of simultaneous projection and recording of the procedure to an external monitor (FIGURE 6). Google Glass is a wearable computer with an optical head-mounted display. The device, similar to eyeglasses, is voice-activated, thereby allowing the surgeon to record the procedure hands-free. Simultaneous projection to an external monitor allows the entire team in the operating room to be aware of the flow of the procedure.
Challenge: Working in a narrow vaginal vault
Without correct instrumentation, this challenge can be especially daunting. Laparoscopy and robotics have changed the way we perform pelvic surgery by providing advanced instrumentation.
Solution #5: Adapt your instruments
Modified vaginal instruments can be used to facilitate a case. Watch the accompanying VIDEO on the use of improved vaginal instruments during morcellation.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel |
| Click to enlarge >>> |
Among the instruments adaptable for vaginal surgery:
- curving, articulating instruments
- long, curved, and rounded knife handles, which allow for better ergonomics during prolonged morcellation
- modified long retractors and use of a single long vaginal pack provide retraction of loops of bowel and easy access to secure pedicles deep in the pelvis.
All of these instruments are available through Marina Medical in Sunrise, Florida.
Challenge: Securing vascular and thick tissue pediclesA narrow introitus and vaginal vault can be difficult to manage during vaginal surgery. Another challenge is a uterus that is large or deformed by multiple fibroids.
Solution #6: Vaginal incision
A simple superficial 2- to 3-cm incision on the distal posterior aspect of the vaginal wall can widen the introitus and vault to facilitate the procedure (FIGURE 7)
Solution #7: Vessel-sealing tools
The use of energy is integral to laparoscopy and robotics for dissection and securing vessels. In a meta-analysis that included seven randomized controlled trials, advanced vessel-sealing devices proved useful in vaginal surgery by decreasing blood loss and operative time.8
In the setting of a difficult vaginal hysterectomy with a narrow introitus and large uterus, the use of vessel-sealing technology allows the surgeon to skeletonize the uterine arteries while allowing progressive descensus to secure the upper pedicles.
In my experience, the use of an advanced vessel-sealing device, compared with traditional clamp-cut-tying technique, facilitated successful completion of vaginal hysterectomy in 650 patients with relative contraindications to the vaginal approach, such as nulliparity, a uterus weighing more than 250 g, and a history of cesarean delivery (Mayo Clinic data; yet unpublished).
We must change with the times
The rate of vaginal hysterectomy will continue to decline unless we modify our technique to incorporate new technology. The current generation of practicing gynecologists and recent graduates are choosing the laparoscopic and robotic approaches because of the advantages these technologies offer. It is time we incorporate relevant features from these minimally invasive approaches while maintaining patient safety and containing costs by performing vaginal hysterectomy whenever possible. A willingness to change and ability to think outside the usual box will help us train new generations of vaginal surgeons who can bring back vaginal hysterectomy as the preferred route to the benign hysterectomy.
WE WANT TO HEAR FROM YOU! Share your thoughts on this article. Send your Letter to the Editor to: [email protected]
Vaginal hysterectomy is the preferred route to benign hysterectomy because it is associated with better outcomes and fewer complications than the laparoscopic and open abdominal approaches.1,2 Yet, despite superior patient outcomes and cost benefits, the rate of vaginal hysterectomy is declining.
According to the Nationwide Inpatient Sample, the use of vaginal hysterectomy declined from 24.8% in 1998 to 16.7% in 2010.3 In fact, more than 80% of surgeons in the United States now perform fewer than five vaginal procedures in a year.4
The increasing use of other minimally invasive routes, such as laparoscopy and robotics, indicates that most practicing surgeons and recent graduates are choosing these approaches over the vaginal route. In only 3 years, the rate of laparoscopy increased by 6% and robotics increased by almost 10%.3
Many surgeons assume that vaginal hysterectomy exists in a state of suspended animation, with nothing much changed in the way it has been performed over the past few decades. Further, vaginal surgery is difficult to teach and learn, given limitations in exposure and visualization, difficulty in securing hemostasis, and challenges in the removal of the large uterus and adnexae. As a result, vaginal hysterectomy often is thought, erroneously, to be indicated only in procedures involving a small and prolapsing uterus.
To increase the rate of vaginal hysterectomy, we can benefit from experience gained in laparoscopy and robotics—whether we are teachers or learners—while maintaining patient safety and containing costs.
In this article, I describe common challenges in vaginal hysterectomy and offer tools and techniques to overcome them:
- achieving and enhancing ergonomics, exposure, and visualization
- the need to work in a long vaginal vault
- the task of securing vascular and thick tissue pedicles when the introitus and vaginal vault are narrow.
The vaginal approach is less costly
Vaginal hysterectomy costs significantly less to perform than other approaches. At a tertiary referral center, vaginal hysterectomy costs approximately $7,000 to $18,000 per case less than laparoscopic, abdominal, and robotic hysterectomy.5 With declining use of vaginal hysterectomy and increasing use of more costly approaches, we face a health-care crisis.
Residents are inadequately trained to perform vaginal hysterectomy
Data reveal that not only are our recent graduates inadequately prepared to perform vaginal hysterectomy, but national health-care dollars and resources are depleted when surgeons choose to perform more costly approaches. As a result, many eligible patients end up deprived of the benefits of a single, concealed, and minimally invasive procedure.
The increase in laparoscopic and robotic approaches to hysterectomy has affected residency training. National case log reports from the Accreditation Council of Graduate Medical Education show that the number of vaginal hysterectomies performed by residents as “primary surgeons” decreased by 40%, from a mean of 35 cases in 2002 to 19 cases in 2012.6 A recent survey found that only 28% of graduating residents were “completely prepared” to perform a vaginal hysterectomy, compared with 58% for abdominal hysterectomy, 22% for laparoscopic hysterectomy, and 3% for the robotic approach.7
The rate of vaginal hysterectomy will continue to decline if we perform it in the same manner it was done 30 years ago. The current generation of practicing gynecologists and graduates is choosing to perform the procedure laparoscopically or robotically because of the advantages these technologies provide. It is time that we incorporate features from these minimally invasive approaches to streamline vaginal hysterectomy while maintaining patient safety and containing costs.
Challenges: Ergonomics, exposure, and visualization
In conventional vaginal surgery, the surgeon often is the person who has the best and, sometimes, the sole view. Two bedside assistants are required to hold retractors during the entire case, which can lead to fatigue and muscle strain. Poor lighting also can greatly limit visualization into the pelvic cavity.
Both laparoscopy and robotics provide a well-illuminated and magnified view, with three-dimensional images now available in both platforms. This view is projected to overhead monitors for the entire surgical team to see. Magnification of the pelvic anatomic structures and projection to an external monitor facilitate teaching and learning, better anticipation of the surgical and procedural needs, and overall patient safety.
From robotics, where ergonomics is exemplified, we also learn the importance of surgeon comfort during the procedure.
Solution #1: A self-retaining retractor
A self-retaining system such as the Magrina-Bookwalter vaginal retractor (Symmetry Surgical, Nashville, Tennessee) (FIGURE 1)
Solution #2: Seat the surgeon for an optimal view
With the patient in the lithotomy position and her legs in candy cane stirrups, the surgeon can be seated on a high chair so that the operative field is at the approximate level of the assistants’ view (FIGURE 2)
Solution #3: Illuminate the cavity
The deep pelvic cavity can be easily illuminated using a lighted suction tip, a flexible light source (as part of the cystoscopy set) held with a Babcock clamp (FIGURE 3), or a malleable illuminating mat taped to the retractor blades (such as Lightmat surgical illuminator, Lumitex, Inc., Strongsville, Ohio).
Solution #4: Project the image
Cameras attached to an overhead boom or operating room light handles (FIGURE 4) and an external telescope with integrated illumination, such as a standard cystoscope or VITOM Exoscope (Karl Storz, El Segundo, California) (FIGURE 5) provide both magnification and projection of the procedure to an overhead monitor.
Glass technology (Google, Mountain View, California) also has been utilized in surgery and can be a good application of simultaneous projection and recording of the procedure to an external monitor (FIGURE 6). Google Glass is a wearable computer with an optical head-mounted display. The device, similar to eyeglasses, is voice-activated, thereby allowing the surgeon to record the procedure hands-free. Simultaneous projection to an external monitor allows the entire team in the operating room to be aware of the flow of the procedure.
Challenge: Working in a narrow vaginal vault
Without correct instrumentation, this challenge can be especially daunting. Laparoscopy and robotics have changed the way we perform pelvic surgery by providing advanced instrumentation.
Solution #5: Adapt your instruments
Modified vaginal instruments can be used to facilitate a case. Watch the accompanying VIDEO on the use of improved vaginal instruments during morcellation.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel |
| Click to enlarge >>> |
Among the instruments adaptable for vaginal surgery:
- curving, articulating instruments
- long, curved, and rounded knife handles, which allow for better ergonomics during prolonged morcellation
- modified long retractors and use of a single long vaginal pack provide retraction of loops of bowel and easy access to secure pedicles deep in the pelvis.
All of these instruments are available through Marina Medical in Sunrise, Florida.
Challenge: Securing vascular and thick tissue pediclesA narrow introitus and vaginal vault can be difficult to manage during vaginal surgery. Another challenge is a uterus that is large or deformed by multiple fibroids.
Solution #6: Vaginal incision
A simple superficial 2- to 3-cm incision on the distal posterior aspect of the vaginal wall can widen the introitus and vault to facilitate the procedure (FIGURE 7)
Solution #7: Vessel-sealing tools
The use of energy is integral to laparoscopy and robotics for dissection and securing vessels. In a meta-analysis that included seven randomized controlled trials, advanced vessel-sealing devices proved useful in vaginal surgery by decreasing blood loss and operative time.8
In the setting of a difficult vaginal hysterectomy with a narrow introitus and large uterus, the use of vessel-sealing technology allows the surgeon to skeletonize the uterine arteries while allowing progressive descensus to secure the upper pedicles.
In my experience, the use of an advanced vessel-sealing device, compared with traditional clamp-cut-tying technique, facilitated successful completion of vaginal hysterectomy in 650 patients with relative contraindications to the vaginal approach, such as nulliparity, a uterus weighing more than 250 g, and a history of cesarean delivery (Mayo Clinic data; yet unpublished).
We must change with the times
The rate of vaginal hysterectomy will continue to decline unless we modify our technique to incorporate new technology. The current generation of practicing gynecologists and recent graduates are choosing the laparoscopic and robotic approaches because of the advantages these technologies offer. It is time we incorporate relevant features from these minimally invasive approaches while maintaining patient safety and containing costs by performing vaginal hysterectomy whenever possible. A willingness to change and ability to think outside the usual box will help us train new generations of vaginal surgeons who can bring back vaginal hysterectomy as the preferred route to the benign hysterectomy.
WE WANT TO HEAR FROM YOU! Share your thoughts on this article. Send your Letter to the Editor to: [email protected]
1. Nieboer TE, Johnson N, Lethaby A, et al. Surgical approach to hysterectomy for benign gynaecological disease. Cochrane Database Syst Rev. 2009;(3):CD003677.
2. American College of Obstetricians and Gynecologists. ACOG Committee Opinion No. 444: Choosing the route of hysterectomy for benign disease. Obstet Gynecol. 2009;114(5):1156–1158.
3. Wright T, Herzog T, Tsul J, et al. Nationwide trends in inpatient hysterectomy in the United States. Obstet Gynecol. 2013:122(2):233–241.
4. Rogo-Gupta L, Lewyn S, Jum JH, et al. Effect of surgeon volume on outcomes and resource use for vaginal hysterectomy. Obstet Gynecol. 2010;116(6):1341–1347.
5. Wright KN, Jonsdottir GM, Jorgensen S, Shah N, Einarsson JI. Costs and outcomes of abdominal, vaginal, laparoscopic and robotic hysterectomies. JSLS. 2012;16(4):519–524.
6. Washburn EE, Cohen SL, Manoucherie E, Zurawin, RJ, Einarsson JI. Trends in reported residency surgical experience in hysterectomy [published online ahead of print June 4, 2014]. J Minim Invasive Gynecol. doi:10.1016/j.jmig.2014.05.005.
7. Burkett D, Horwitz J, Kennedy V, et al. Assessing current trends in resident hysterectomy training. Female Pelvic Med Reconstr Surg. 2011;17(5):210–214.
8. Kroft J, Selk K. Energy-based vessel sealing in vaginal hysterectomy. A systematic review and meta-analysis. Obstet Gynecol. 2011;118(5):1127–1136.
1. Nieboer TE, Johnson N, Lethaby A, et al. Surgical approach to hysterectomy for benign gynaecological disease. Cochrane Database Syst Rev. 2009;(3):CD003677.
2. American College of Obstetricians and Gynecologists. ACOG Committee Opinion No. 444: Choosing the route of hysterectomy for benign disease. Obstet Gynecol. 2009;114(5):1156–1158.
3. Wright T, Herzog T, Tsul J, et al. Nationwide trends in inpatient hysterectomy in the United States. Obstet Gynecol. 2013:122(2):233–241.
4. Rogo-Gupta L, Lewyn S, Jum JH, et al. Effect of surgeon volume on outcomes and resource use for vaginal hysterectomy. Obstet Gynecol. 2010;116(6):1341–1347.
5. Wright KN, Jonsdottir GM, Jorgensen S, Shah N, Einarsson JI. Costs and outcomes of abdominal, vaginal, laparoscopic and robotic hysterectomies. JSLS. 2012;16(4):519–524.
6. Washburn EE, Cohen SL, Manoucherie E, Zurawin, RJ, Einarsson JI. Trends in reported residency surgical experience in hysterectomy [published online ahead of print June 4, 2014]. J Minim Invasive Gynecol. doi:10.1016/j.jmig.2014.05.005.
7. Burkett D, Horwitz J, Kennedy V, et al. Assessing current trends in resident hysterectomy training. Female Pelvic Med Reconstr Surg. 2011;17(5):210–214.
8. Kroft J, Selk K. Energy-based vessel sealing in vaginal hysterectomy. A systematic review and meta-analysis. Obstet Gynecol. 2011;118(5):1127–1136.
![]()
Dr. Kho presents vaginal morcellation by hand, using advanced instrumentation
Key ways to differentiate a benign from a malignant adnexal mass
Click here to register for PAGS 2014 December 4 to 6 at the Bellagio in Las Vegas
More from PAGS 2013:
Lichen sclerosis: My approach to treatment
Michael Baggish, MD
Click here to register for PAGS 2014 December 4 to 6 at the Bellagio in Las Vegas
More from PAGS 2013:
Lichen sclerosis: My approach to treatment
Michael Baggish, MD
Click here to register for PAGS 2014 December 4 to 6 at the Bellagio in Las Vegas
More from PAGS 2013:
Lichen sclerosis: My approach to treatment
Michael Baggish, MD