User login
Thromboprophylaxis in Patients with HIV
Patients with human immunodeficiency virus (HIV) are at a 2‐ to 10‐fold greater risk for venous thromboembolism (VTE) compared with the general population.[1] Although antiphospholipid antibodies and protein S deficiency have often been cited as reasons for the thrombophilia associated with HIV, previous studies have also documented an increased risk of VTE with declining CD4+ cell count.[2, 3, 4, 5, 6, 7, 8] Worsening immune function places HIV patients at increased risk for opportunistic and nonopportunistic infections and malignancies, all independently associated with an increased risk of VTE.[5, 9, 10, 11, 12] Although increasing use of antiretroviral therapy has greatly decreased these sequelae, these complications of HIV infection are associated with an increased frequency of hospitalization.[13, 14, 15, 16] HIV infection and associated inflammation has been implicated in cardiovascular conditions such as cardiomyopathy, pulmonary hypertension, and myocardial infarction.[17, 18] Additionally, progression of HIV infection appears to influence T‐cell activation and differentiation in a manner that leads to early immunosenescence in infected individuals.[19, 20]
VTE prophylaxis is effective.[21] Virtually all efforts to decrease VTE have been focused on improving the prescription of prophylaxis with varying degrees of success.[22] These interventions have been employed with the tacit assumption that medication prescribed for inpatients will always be administered. However, at our institution, recent research has demonstrated that a significant proportion of prescribed thromboprophylaxis doses are not administered to hospitalized patients.[23] Refusal by the patient or a family member was the most commonly documented reason for dose nonadministration. In addition, the rate of thromboprophylaxis nonadministration varied greatly between nursing units with distinct patient populations. We hypothesized that nonadministration of VTE prophylaxis may be more common in patients with HIV, and this phenomenon may contribute to their increased risk for VTE.
The purpose of this study was to determine if the proportion of nonadministered thromboprophylaxis is greater among hospitalized patients with HIV and to characterize documented reasons for dose nonadministration.
METHODS
This study was conducted at The Johns Hopkins Hospital (JHH), a large, urban, academic medical center in Baltimore, Maryland. This single‐center retrospective cohort study utilized an existing dataset containing dose administration data extracted from an electronic medication administration record (eMAR). This dataset included information for all prescribed doses of thromboprophylaxis (heparin 5000 U subcutaneously every 8 or 12 hours, heparin 7500 U subcutaneously every 12 hours, enoxaparin 30 mg subcutaneously every 12 hours, or enoxaparin 40 mg subcutaneously daily) for patients hospitalized on medicine units at JHH from November 2007 to December 2008. This time period follows the implementation of an electronic order set for VTE prophylaxis.[24, 25] Data available for each dose included drug name, dose, frequency, patient demographics, and whether or not the dose was administered. Each dose not administered included a reason for nonadministration, which was chosen from a dropdown menu of responses on the eMAR by the nurse at the time the dose was due. A separate electronic report was obtained from an internal administrative database, which identified all patients within the dose administration dataset who had the International Classification of Diseases, 9th Revision code 042 (HIV diagnosis). A report identifying patient history numbers with matching diagnostic code for HIV was appended to the dose administration dataset using a relational database (Microsoft Access; Microsoft Corp., Redmond, WA) prior to analysis. The dose administration data were obtained previously for a separate analysis.[23] Approval for this study was granted from the institutional review board of Johns Hopkins Medicine.
Our analytic plan included comparisons between patients with and without HIV on a dose, patient, and unit level. As JHH operates a nursing unit dedicated to the inpatient care of patients with HIV, we included analyses of dose characteristics between this unit and other medicine units. It should be noted that patients without a diagnosis of HIV are sometimes cared for on this unit. Therefore, the electronic medical record for each patient without the diagnosis code for HIV hospitalized on this unit was reviewed to determine HIV status. An analysis was performed comparing visit identification numbers with diagnosis codes to identify potential seroconversions during the study period. Although we planned to compare nonadministration and documented refusal of doses on the unit level, a lack of patients with HIV on a number of units limited our ability to perform these analyses.
Statistical Analysis
The percent of doses not administered was calculated as the number of doses not administered divided by the number of doses prescribed. Likewise, the percent of prescribed doses documented as refused was calculated as the number of prescribed doses documented as refused divided by the number of doses prescribed. For each comparison, an odds ratio (OR) with 95% confidence interval (CI) was reported. Univariate and multivariate regression analyses were performed to assess the relationship between patient factors and dose nonadministration and documented refusal, respectively. Generalized estimating equations (GEE) using a logit link and an exchangeable correlation structure were used in these analyses. The GEE technique was used to account for within‐individual correlation of administration and documented refusal status.
Categorical data were compared using the two‐sided [2] test. Parametric and nonparametric continuous data were compared using the Student t test and Mann‐Whitney U test, respectively. A P value of <0.05 was considered statistically significant for all analyses. Analyses were performed using Minitab 15 (Minitab Inc., State College, PA) and Stata (StataCorp, College Station, TX).
RESULTS
During the 8‐month study period, 42,870 doses of thromboprophylaxis were prescribed during 4947 patient admissions to 13 individual medicine units. Overall, the diagnosis code for HIV was present in 12% of patient visits. The proportion of nonadministered doses per unit ranged from 6% to 27%, whereas the number of doses prescribed per unit ranged from 34 to 7301.
Patient characteristics were described on the visit level (Table 1). Patients with HIV were significantly younger, more often male and black, and had a longer length of stay compared with patients without HIV. Patients hospitalized on the HIV care unit had similar characteristics to the overall population of patients with HIV. It should be noted that not all patients cared for on this unit had a diagnosis of HIV, as patients from other medicine services are sometimes cared for in this location.
| Patients Without HIV | Patients With HIV | P | |
|---|---|---|---|
| |||
| Visits, n | 4,364 | 583 | N/A |
| Male, n (%) | 2,039 (47) | 370 (64) | <0.001 |
| Mean ageSD, y | 5618 | 469 | <0.001 |
| Race, n (%) | |||
| African American | 2,603 (60) | 522 (90) | <0.001 |
| Caucasian | 1,610 (37) | 53 (9) | <0.001 |
| Asian, Pacific Islander, other | 151 (4) | 8 (1) | 0.006 |
| Median length of stay (IQR), d | 3 (15) | 4 (27) | 0.002 |
| Marital status, n (%) | |||
| Single | 2,051 (47) | 471 (81) | <0.001 |
| Married | 1,405 (32) | 71 (12) | <0.001 |
| Widowed | 486 (11) | 10 (1) | <0.001 |
| Divorced | 402 (9) | 28 (5) | <0.001 |
| Separated | 33 (1) | 3 (1) | 0.607 |
| Unknown | 5 (0) | 0 (0) | 0.465 |
| Payor, n (%) | |||
| Medicare | 1,771 (41) | 133 (23) | <0.001 |
| Medicaid | 1,343 (31) | 392 (67) | <0.001 |
| Commercial | 1,181 (27) | 43 (7) | <0.001 |
| Other including self‐pay | 69 (1) | 15 (3) | 0.087 |
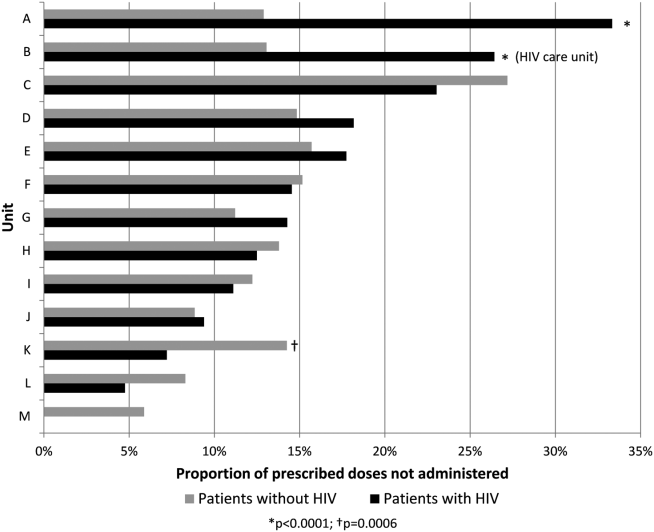
Overall, 17% of prescribed prophylaxis doses were not administered. A greater proportion of prescribed doses were not administered to patients with HIV compared with patients without HIV (23.5% vs 16.1%, OR: 1.59, 95% CI: 1.49‐1.70, P<0.001) (Table 2). Using a GEE and univariate regression, HIV diagnosis was associated with nonadministration of doses (OR: 1.37, 95% CI: 1.17‐1.60, P<0.001) (Table 3). Race, age, length of stay, and drug (heparin vs enoxaparin) were each associated with nonadministration. There was no significant association between nonadministration and sex, marital status, or payor. When stratified by nursing unit, there was substantial variation in the proportion of nonadministered doses between units. Within each unit, the proportion of doses not administered varied when stratified by HIV status. For example, on unit A, the proportion of doses not administered was greater for patients with HIV compared with patients without HIV (33.3% vs 12.9%, OR: 3.38, 95% CI: 2.61 to 4.37, P<0.001) (Figure 1). However, on unit K, the proportion of doses not administered to patients with HIV was 2‐fold less than in patients without HIV (7.2% vs 14.3%, OR: 0.47, 95% CI: 0.30‐0.74, P<0.001). Unit‐level analysis was not possible in regression models due to drastic imbalance in the prevalence of HIV across units. When comparing doses prescribed in the HIV care unit to all other medicine units, the proportion not administered (23.9% vs 16.3%, OR: 1.61, 95% CI: 1.49‐1.73, P<0.001) closely resembled the values seen when comparing patients with and without HIV hospital wide (23.5% vs 16.1%). However, when doses on the HIV care unit were stratified by HIV status, the doses not administered were 2‐fold greater, as a proportion, for patients with HIV compared with those without HIV (26.4% vs 13.1%, OR: 2.39, 95% CI: 1.93‐2.96, P<0.001).
| Doses Prescribed | Doses Not Administered (% of Doses Prescribed) | Doses Documented as Refused (% of All Doses Prescribed) | |
|---|---|---|---|
| |||
| All patients with HIV | 5,681 | 1,334 (23.5%)a | 935 (16.5%)a |
| All patients without HIV | 37,189 | 6,005 (16.1%) | 3,935 (10.6%) |
| HIV care unit | 4,452 | 1,063 (23.9%)a | 709 (15.9%)a |
| All other units | 38,418 | 6,276 (16.3%) | 4,161 (10.8%) |
| HIV care unit: patients with HIV | 3,602 | 952 (26.4%)a | 651 (18.1%)a |
| HIV care unit: patients without HIV | 850 | 111 (13.1%) | 58 (6.8%) |
| All other units: patients with HIV | 2,079 | 382 (18.4%)b | 284 (13.7%)a |
| All other units: patients without HIV | 36,339 | 5,894 (16.2%) | 3,877 (10.7%) |
| Nonadministered, n (%) | P | Documented as Refused, n (%) | P | |
|---|---|---|---|---|
| ||||
| Race | 0.001 | 0.072 | ||
| African American | 2,601 (17.8) | 1,708 (11.7) | ||
| Caucasian | 4,379 (16.4) | 2,922 (10.9) | ||
| Asian, Pacific Islander, other | 359 (23.4) | 240 (15.6) | ||
| HIV status | <0.001 | 0.002 | ||
| Negative | 6,005 (16.2) | 3,935 (10.6) | ||
| Positive | 1,344 (23.5) | 935 (16.5) | ||
| Age, y | <0.001 | <0.001 | ||
| 19 | 59 (20.6) | 44 (15.3) | ||
| 2029 | 1,260 (33.8) | 1,000 (26.8) | ||
| 3039 | 1,088 (28.1) | 845 (21.8) | ||
| 4049 | 1,628 (21.0) | 1,104 (14.2) | ||
| 5059 | 1,493 (16.1) | 953 (10.3) | ||
| 6069 | 900 (12.6) | 515 (7.2) | ||
| 7079 | 571 (9.6) | 250 (4.2) | ||
| 8089 | 252 (6.2) | 95 (2.3) | ||
| 90 | 88 (11.5) | 84 (8.4) | ||
| Sex | 0.372 | 0.919 | ||
| Male | 3,689 (17.3) | 2,392 (11.2) | ||
| Female | 3,650 (17.0) | 2,478 (11.5) | ||
| Drug | <0.001 | <0.001 | ||
| Heparin | 6,833 (18.4) | 4,515 (12.2) | ||
| Enoxaparin | 506 (8.9) | 355 (6.2) | ||
| Length of stay, d | <0.001 | <0.001 | ||
| 01 | 446 (24.3) | 282 (15.4) | ||
| 23 | 1,463 (19.4) | 971 (12.9) | ||
| 47 | 2,332 (18.9) | 1,620 (13.1) | ||
| 8 | 3,098 (14.6) | 1,997 (9.4) | ||
The results of the multivariate regression analyses with GEE are displayed in Table 4. HIV diagnosis, non‐African American race, and heparin (as compared with enoxaparin) were associated with increased likelihood of nonadministration. Increasing age and increasing length of stay were associated with decreased likelihood of nonadministration by a small but significant amount.
| OR of Nonadministration | 95% CI, P | OR of Documented Refusal | 95% CI, P | |
|---|---|---|---|---|
| ||||
| Race | ||||
| African American | 1.00 | Reference | 1.00 | Reference |
| Caucasian | 1.62 | 1.44‐1.81, <0.001 | 1.53 | 1.32‐1.77, <0.001 |
| Asian, Pacific Islander, Other | 1.54 | 1.19‐2.00, 0.001 | 1.48 | 1.07‐2.01, 0.019 |
| HIV status | ||||
| Negative | 1.00 | Reference | 1.00 | Reference |
| Positive | 1.21 | 1.001.45, 0.039 | 1.29 | 1.06‐1.56, 0.012 |
| Age, per year | 0.97 | 0.97‐0.98, <0.001 | 0.97 | 0.96‐0.97, <0.001 |
| Drug | ||||
| Heparin | 1.00 | Reference | 1.00 | Reference |
| Enoxaparin | 0.45 | 0.40‐0.51, <0.001 | 0.53 | 0.47‐0.61, <0.001 |
| Length of stay, per day | 0.991 | 0.987‐0.995, <0.001 | 0.989 | 0.983‐0.993, <0.001 |
The most commonly documented reason for nonadministration was refusal by the patient or family member (66% of all doses not administered). The second most common reason, patient condition not appropriate, accounted for an additional 10% of doses. Across all nursing units, the proportion of prescribed doses that were documented as refused was significantly greater for patients with HIV compared with patients without HIV (16.5% vs 10.6%, OR: 1.66, 95% CI: 1.54‐1.80, P<0.0001) (Table 2). Using the GEE and multivariate regression, HIV diagnosis, non‐African American race, and heparin were associated with increased risk of documented dose refusal. Age and length of stay were inversely related to the likelihood of documented dose refusal. When all administered doses were excluded from the analysis, the association between these variables and documented dose refusal were not as strong. Age and length of stay remained significantly inversely related; however, the other factors were no longer significantly positively associated with documented dose refusal.
Within the HIV care unit, the proportion of prescribed doses documented as refused was greater for patients with HIV compared with patients without HIV (18.1% vs 6.8%, OR: 3.01, 95% CI: 2.28‐3.99, P<0.0001). For all other medicine units, the proportion of nonadministered doses documented as refused was also greater for patients with HIV compared with patients without HIV (13.7% vs 10.7%, OR: 1.32, 95% CI: 1.16‐1.51, P<0.0001).
DISCUSSION
We have identified that nonadministration of thromboprophylaxis was more common among patients with HIV at our institution. Substantial variation in the proportion of doses not administered existed on the nursing unit level, as well as within each unit when stratified by HIV status. This disparity in dose administration was observed on the HIV care unit as well, as the proportion not administered was about 2‐fold greater for patients with HIV compared with those without HIV. Documented dose refusal appeared to account for the majority of nonadministered doses in our cohort. Our analysis also demonstrated that HIV diagnosis is significantly associated with both dose nonadministration and documented dose refusal at our institution.
Medication refusal is a well‐recognized phenomenon among hospitalized patients. A recent study of medication administration in hospitalized patients in the United Kingdom noted that refusal accounted for about 45% of omitted doses.[26] Fanikos et al. also found that documented refusal of doses contributed significantly to the overall number of VTE prophylaxis doses not administered to patients.[27] In our study, the proportion of nonadministered doses documented as refused by the patient or family member was significantly greater in patients with HIV than in patients without HIV across all units. Interestingly, the difference was greater on the HIV care unit when doses were stratified by HIV status. This observation leads us to hypothesize that specific hospital care environments may influence dose nonadministration and refusal rates among our patient population.
Based on regression analyses, increasing age and length of stay were associated with a decreased likelihood of any particular dose not being administered and with any particular dose being documented as refused. It is important to note that our GEE did not take into account date or time of each dose, and therefore we cannot make conclusions as to the likelihood of dose nonadministration or refusal of doses in relation to each other on a time scale. One cannot assume that a dose due later in a hospital course was more or less likely to be given than a dose due on the first hospital day. Although we did not expect these findings, one can hypothesize that patients who are older or have longer stays may be perceived to have more severe illness, and therefore greater need for prophylaxis, from nursing staff and others involved in their care. The associations were small but significant and warrant future investigation.
To our knowledge, this is the first investigation comparing the proportion of nonadministered doses of thromboprophylaxis between patients with and without HIV. Our data show that nonadministered doses and refused doses of thromboprophylaxis are more frequent among patients with HIV. In addition, we noted that nonadministration was more common on the dedicated HIV care unit compared with other units. We cannot currently offer a clear explanation for the disparity observed between units, and more specifically, within the HIV care unit. However, it is possible that a unique culture of care and provider‐specific factors may contribute.
Our study was limited by a number of factors. Seroconversion among patients during the study period was possible; however, our analysis revealed only 2 instances among nearly 4000 unique patients. A more significant limitation was the level of analysis allowed by the dataset. We examined dose characteristics on a dose and unit level, but the ability to analyze doses based on the prescriber and nurse level may have provided valuable insight into the specific reasons behind the observations presented here. Additionally, the specific unit assigned to a given dose in our dataset represented the discharge location for the corresponding patient, making it possible that some amount of nonadministered doses may be attributed to the incorrect unit. However, we do not believe that unit‐to‐unit transfers would be frequent enough to influence the overall results. In addition, we did not link nonadministration of thromboprophylaxis with VTE events, as these data were not present in the current dataset. Although this is a limitation of the current study, we believe that the notion that missed doses of thromboprophylaxis place patients at higher risk for VTE is plausible, as the efficacy of thromboprophylaxis is well established.[28, 29, 30] It is important to note that the reason for nonadministration selected by the nurse on the eMAR may not always represent the only reason or even the true reason for dose nonadministration. It is possible that dose refusal may be over‐represented in our sample, in part due to inaccurate documentation. Recent investigations at JHH have identified varying attitudes on the part of the patient and the nurse regarding thromboprophylaxis. A questionnaire and interview of patients showed a large knowledge gap regarding thromboprophylaxis, with many individuals unable to explain its role or significance in their medical care.[31] A common theme was also observed in a survey of nurses regarding VTE prophylaxis: doses were sometimes considered optional for reasons such as ambulation status, perceived severity of illness, or reason for hospitalization. Some nurses also reported that after an initial refused dose, they may continue to document subsequent doses as refused, sometimes without offering the dose to the patient.[32] As variation in practice was observed between individual nurses, it is also likely that the culture of care may vary between units, influencing thromboprophylaxis nonadministration rates as well as documentation of doses as refused. The dose‐level data used for the GEE analyses did not include date and time of administration, which limited the ability of the GEE to more completely account for autocorrelation.
To further investigate the findings of this and related studies, we intend to more closely analyze data at multiple levels with the goal of identifying an appropriate and feasible target for intervention. Additionally, further investigation should be performed with the goal of determining the relationship between decreased exposure to thromboprophylaxis and VTE. However, as patients with HIV appear to be at increased risk of VTE, ensuring that thromboprophylaxis is delivered appropriately and consistently should be an important goal for all who provide care to this population.
- , , , , . Venous thromboembolism in patients with HIV/AIDS: a case‐control study. J Acquir Immune Defic Syndr. 2008;48(3):310–314.
- , , . AIDS and thrombosis: retrospective study of 131 HIV‐infected patients. AIDS Patient Care STDS. 2001;15(6):311–320.
- , , , et al. HIV and risk of venous thromboembolism: a Danish nationwide population‐based cohort study. HIV Med. 2011;12(4):202–210.
- , , , . Epidemiology of thrombosis in HIV‐infected individuals. The adult/adolescent spectrum of HIV disease project. AIDS. 2000;14(3):321–324.
- , , . Thrombotic complications in patients infected with HIV in the era of highly active antiretroviral therapy: a case series. Clin Infect Dis. 2004;39(8):1214–1222.
- , , . Circulating coagulation inhibitors in the acquired immunodeficiency syndrome. Ann Intern Med. 1986;104(2):175–180.
- , . The pathogenesis of the antiphospholipid syndrome. N Engl J Med. 2013;368(11):1033–1044.
- , , , . Acquired protein C and protein S deficiency in HIV‐infected patients. Clin Appl Thromb Hemost. 2003;9(4):325–331.
- , , , et al. Antiphospholipid syndrome associated with cytomegalovirus infection: case report and review. Clin Infect Dis. 1997;24(2):197–200.
- , . Viral infections and antiphospholipid antibodies. Semin Arthritis Rheum. 2002;31(4):256–263.
- , . AIDS‐defining and non‐AIDS‐defining malignancies: cancer occurrence in the antiretroviral therapy era. Curr Opin Oncol. 2007;19(5):446–451.
- , , , , . Pathogenesis, clinical and laboratory aspects of thrombosis in cancer. J Thromb Thrombolysis. 2007;24(1):29–38.
- , , , et al. Patterns of diagnoses in hospital admissions in a multistate cohort of HIV‐positive adults in 2001. Med Care. 2005;43(9 suppl):III3–III14.
- , , , et al. Opportunistic infections as causes of death in HIV‐infected patients in the HAART era in France. Scand J Infect Dis. 2005;37(6‐7):482–487.
- , , , et al. Rates of hospitalizations and associated diagnoses in a large multisite cohort of HIV patients in the United States, 1994–2005. AIDS. 2008;22(11):1345–1354.
- , , . Hospitalizations for metabolic conditions, opportunistic infections, and injection drug use among HIV patients: trends between 1996 and 2000 in 12 states. J Acquir Immune Defic Syndr. 2005;40(5):609–616.
- , , , , , . Cardiovascular disease in HIV infection. Am Heart J. 2006;151:1147–1155.
- , , , et al. Epidemiological evidence for cardiovascular disease in HIV‐infected patients and relationship to highly active antiretroviral therapy. Circulation. 2008;118(2):e29–e35.
- , , , et al. Immune activation and CD8+ T‐cell differentiation towards senescence in HIV‐1 infection. PLoS Biol. 2004;2(2):E20.
- , , , , . CD4 T cell depletion is linked directly to immune activation in the pathogenesis of HIV‐1 and HIV‐2 but only indirectly to the viral load. J Immunol. 2002;169(6):3400–3406.
- , . Thromboprophylaxis in nonsurgical patients. Hematology Am Soc Hematol Educ Program. 2012;2012:631–637.
- , . Practices to prevent venous thromboembolism [published online ahead of print May 24, 2013]. BMJ Qual Saf. doi:10.1136/bmjqs‐2012‐001782.
- , , , et al. Patterns of non‐administration of ordered doses of venous thromboembolism prophylaxis: implications for novel intervention strategies. PLoS One. 2013;8(6):e66311.
- , , , et al. Lessons from The Johns Hopkins multi‐disciplinary venous thromboembolism (VTE) prevention collaborative. BMJ. 2012;344:e3935.
- , , , et al. Impact of a venous thromboembolism prophylaxis “smart order set”: improved compliance, fewer events [published online ahead of print April 4, 2013]. Am J Hematol. doi: 10.1002/ajh.23450.
- , , . Dose omissions in hospitalized patients in a UK hospital: an analysis of the relative contribution of adverse drug reactions. Drug Saf. 2012;35(8):677–683.
- , , , et al. Adherence to pharmacological thromboprophylaxis orders in hospitalized patients. Am J Med. 2010;123(6):536–541.
- , , , et al. A comparison of enoxaparin with placebo for the prevention of venous thromboembolism in acutely ill medical patients. Prophylaxis in medical patients with enoxaparin study group. N Engl J Med. 1999;341(11):793–800.
- , , , et al. Randomized, placebo‐controlled trial of dalteparin for the prevention of venous thromboembolism in acutely ill medical patients. Circulation. 2004;110(7):874–879.
- , , , et al. Efficacy and safety of fondaparinux for the prevention of venous thromboembolism in older acute medical patients: randomised placebo controlled trial. BMJ. 2006;332(7537):325–329.
- , , , et al. Patient perspectives on pharmacological venous thromboembolism prophylaxis at The Johns Hopkins Hospital. J Thromb Thrombolysis. 2013;35(3):416.
- , , , et al. Culture of care and documented patient refusal of pharmacologic venous thromboembolism prophylaxis. J Thromb Thrombolysis. 2011;31(3):367–400.
Patients with human immunodeficiency virus (HIV) are at a 2‐ to 10‐fold greater risk for venous thromboembolism (VTE) compared with the general population.[1] Although antiphospholipid antibodies and protein S deficiency have often been cited as reasons for the thrombophilia associated with HIV, previous studies have also documented an increased risk of VTE with declining CD4+ cell count.[2, 3, 4, 5, 6, 7, 8] Worsening immune function places HIV patients at increased risk for opportunistic and nonopportunistic infections and malignancies, all independently associated with an increased risk of VTE.[5, 9, 10, 11, 12] Although increasing use of antiretroviral therapy has greatly decreased these sequelae, these complications of HIV infection are associated with an increased frequency of hospitalization.[13, 14, 15, 16] HIV infection and associated inflammation has been implicated in cardiovascular conditions such as cardiomyopathy, pulmonary hypertension, and myocardial infarction.[17, 18] Additionally, progression of HIV infection appears to influence T‐cell activation and differentiation in a manner that leads to early immunosenescence in infected individuals.[19, 20]
VTE prophylaxis is effective.[21] Virtually all efforts to decrease VTE have been focused on improving the prescription of prophylaxis with varying degrees of success.[22] These interventions have been employed with the tacit assumption that medication prescribed for inpatients will always be administered. However, at our institution, recent research has demonstrated that a significant proportion of prescribed thromboprophylaxis doses are not administered to hospitalized patients.[23] Refusal by the patient or a family member was the most commonly documented reason for dose nonadministration. In addition, the rate of thromboprophylaxis nonadministration varied greatly between nursing units with distinct patient populations. We hypothesized that nonadministration of VTE prophylaxis may be more common in patients with HIV, and this phenomenon may contribute to their increased risk for VTE.
The purpose of this study was to determine if the proportion of nonadministered thromboprophylaxis is greater among hospitalized patients with HIV and to characterize documented reasons for dose nonadministration.
METHODS
This study was conducted at The Johns Hopkins Hospital (JHH), a large, urban, academic medical center in Baltimore, Maryland. This single‐center retrospective cohort study utilized an existing dataset containing dose administration data extracted from an electronic medication administration record (eMAR). This dataset included information for all prescribed doses of thromboprophylaxis (heparin 5000 U subcutaneously every 8 or 12 hours, heparin 7500 U subcutaneously every 12 hours, enoxaparin 30 mg subcutaneously every 12 hours, or enoxaparin 40 mg subcutaneously daily) for patients hospitalized on medicine units at JHH from November 2007 to December 2008. This time period follows the implementation of an electronic order set for VTE prophylaxis.[24, 25] Data available for each dose included drug name, dose, frequency, patient demographics, and whether or not the dose was administered. Each dose not administered included a reason for nonadministration, which was chosen from a dropdown menu of responses on the eMAR by the nurse at the time the dose was due. A separate electronic report was obtained from an internal administrative database, which identified all patients within the dose administration dataset who had the International Classification of Diseases, 9th Revision code 042 (HIV diagnosis). A report identifying patient history numbers with matching diagnostic code for HIV was appended to the dose administration dataset using a relational database (Microsoft Access; Microsoft Corp., Redmond, WA) prior to analysis. The dose administration data were obtained previously for a separate analysis.[23] Approval for this study was granted from the institutional review board of Johns Hopkins Medicine.
Our analytic plan included comparisons between patients with and without HIV on a dose, patient, and unit level. As JHH operates a nursing unit dedicated to the inpatient care of patients with HIV, we included analyses of dose characteristics between this unit and other medicine units. It should be noted that patients without a diagnosis of HIV are sometimes cared for on this unit. Therefore, the electronic medical record for each patient without the diagnosis code for HIV hospitalized on this unit was reviewed to determine HIV status. An analysis was performed comparing visit identification numbers with diagnosis codes to identify potential seroconversions during the study period. Although we planned to compare nonadministration and documented refusal of doses on the unit level, a lack of patients with HIV on a number of units limited our ability to perform these analyses.
Statistical Analysis
The percent of doses not administered was calculated as the number of doses not administered divided by the number of doses prescribed. Likewise, the percent of prescribed doses documented as refused was calculated as the number of prescribed doses documented as refused divided by the number of doses prescribed. For each comparison, an odds ratio (OR) with 95% confidence interval (CI) was reported. Univariate and multivariate regression analyses were performed to assess the relationship between patient factors and dose nonadministration and documented refusal, respectively. Generalized estimating equations (GEE) using a logit link and an exchangeable correlation structure were used in these analyses. The GEE technique was used to account for within‐individual correlation of administration and documented refusal status.
Categorical data were compared using the two‐sided [2] test. Parametric and nonparametric continuous data were compared using the Student t test and Mann‐Whitney U test, respectively. A P value of <0.05 was considered statistically significant for all analyses. Analyses were performed using Minitab 15 (Minitab Inc., State College, PA) and Stata (StataCorp, College Station, TX).
RESULTS
During the 8‐month study period, 42,870 doses of thromboprophylaxis were prescribed during 4947 patient admissions to 13 individual medicine units. Overall, the diagnosis code for HIV was present in 12% of patient visits. The proportion of nonadministered doses per unit ranged from 6% to 27%, whereas the number of doses prescribed per unit ranged from 34 to 7301.
Patient characteristics were described on the visit level (Table 1). Patients with HIV were significantly younger, more often male and black, and had a longer length of stay compared with patients without HIV. Patients hospitalized on the HIV care unit had similar characteristics to the overall population of patients with HIV. It should be noted that not all patients cared for on this unit had a diagnosis of HIV, as patients from other medicine services are sometimes cared for in this location.
| Patients Without HIV | Patients With HIV | P | |
|---|---|---|---|
| |||
| Visits, n | 4,364 | 583 | N/A |
| Male, n (%) | 2,039 (47) | 370 (64) | <0.001 |
| Mean ageSD, y | 5618 | 469 | <0.001 |
| Race, n (%) | |||
| African American | 2,603 (60) | 522 (90) | <0.001 |
| Caucasian | 1,610 (37) | 53 (9) | <0.001 |
| Asian, Pacific Islander, other | 151 (4) | 8 (1) | 0.006 |
| Median length of stay (IQR), d | 3 (15) | 4 (27) | 0.002 |
| Marital status, n (%) | |||
| Single | 2,051 (47) | 471 (81) | <0.001 |
| Married | 1,405 (32) | 71 (12) | <0.001 |
| Widowed | 486 (11) | 10 (1) | <0.001 |
| Divorced | 402 (9) | 28 (5) | <0.001 |
| Separated | 33 (1) | 3 (1) | 0.607 |
| Unknown | 5 (0) | 0 (0) | 0.465 |
| Payor, n (%) | |||
| Medicare | 1,771 (41) | 133 (23) | <0.001 |
| Medicaid | 1,343 (31) | 392 (67) | <0.001 |
| Commercial | 1,181 (27) | 43 (7) | <0.001 |
| Other including self‐pay | 69 (1) | 15 (3) | 0.087 |
Overall, 17% of prescribed prophylaxis doses were not administered. A greater proportion of prescribed doses were not administered to patients with HIV compared with patients without HIV (23.5% vs 16.1%, OR: 1.59, 95% CI: 1.49‐1.70, P<0.001) (Table 2). Using a GEE and univariate regression, HIV diagnosis was associated with nonadministration of doses (OR: 1.37, 95% CI: 1.17‐1.60, P<0.001) (Table 3). Race, age, length of stay, and drug (heparin vs enoxaparin) were each associated with nonadministration. There was no significant association between nonadministration and sex, marital status, or payor. When stratified by nursing unit, there was substantial variation in the proportion of nonadministered doses between units. Within each unit, the proportion of doses not administered varied when stratified by HIV status. For example, on unit A, the proportion of doses not administered was greater for patients with HIV compared with patients without HIV (33.3% vs 12.9%, OR: 3.38, 95% CI: 2.61 to 4.37, P<0.001) (Figure 1). However, on unit K, the proportion of doses not administered to patients with HIV was 2‐fold less than in patients without HIV (7.2% vs 14.3%, OR: 0.47, 95% CI: 0.30‐0.74, P<0.001). Unit‐level analysis was not possible in regression models due to drastic imbalance in the prevalence of HIV across units. When comparing doses prescribed in the HIV care unit to all other medicine units, the proportion not administered (23.9% vs 16.3%, OR: 1.61, 95% CI: 1.49‐1.73, P<0.001) closely resembled the values seen when comparing patients with and without HIV hospital wide (23.5% vs 16.1%). However, when doses on the HIV care unit were stratified by HIV status, the doses not administered were 2‐fold greater, as a proportion, for patients with HIV compared with those without HIV (26.4% vs 13.1%, OR: 2.39, 95% CI: 1.93‐2.96, P<0.001).
| Doses Prescribed | Doses Not Administered (% of Doses Prescribed) | Doses Documented as Refused (% of All Doses Prescribed) | |
|---|---|---|---|
| |||
| All patients with HIV | 5,681 | 1,334 (23.5%)a | 935 (16.5%)a |
| All patients without HIV | 37,189 | 6,005 (16.1%) | 3,935 (10.6%) |
| HIV care unit | 4,452 | 1,063 (23.9%)a | 709 (15.9%)a |
| All other units | 38,418 | 6,276 (16.3%) | 4,161 (10.8%) |
| HIV care unit: patients with HIV | 3,602 | 952 (26.4%)a | 651 (18.1%)a |
| HIV care unit: patients without HIV | 850 | 111 (13.1%) | 58 (6.8%) |
| All other units: patients with HIV | 2,079 | 382 (18.4%)b | 284 (13.7%)a |
| All other units: patients without HIV | 36,339 | 5,894 (16.2%) | 3,877 (10.7%) |
| Nonadministered, n (%) | P | Documented as Refused, n (%) | P | |
|---|---|---|---|---|
| ||||
| Race | 0.001 | 0.072 | ||
| African American | 2,601 (17.8) | 1,708 (11.7) | ||
| Caucasian | 4,379 (16.4) | 2,922 (10.9) | ||
| Asian, Pacific Islander, other | 359 (23.4) | 240 (15.6) | ||
| HIV status | <0.001 | 0.002 | ||
| Negative | 6,005 (16.2) | 3,935 (10.6) | ||
| Positive | 1,344 (23.5) | 935 (16.5) | ||
| Age, y | <0.001 | <0.001 | ||
| 19 | 59 (20.6) | 44 (15.3) | ||
| 2029 | 1,260 (33.8) | 1,000 (26.8) | ||
| 3039 | 1,088 (28.1) | 845 (21.8) | ||
| 4049 | 1,628 (21.0) | 1,104 (14.2) | ||
| 5059 | 1,493 (16.1) | 953 (10.3) | ||
| 6069 | 900 (12.6) | 515 (7.2) | ||
| 7079 | 571 (9.6) | 250 (4.2) | ||
| 8089 | 252 (6.2) | 95 (2.3) | ||
| 90 | 88 (11.5) | 84 (8.4) | ||
| Sex | 0.372 | 0.919 | ||
| Male | 3,689 (17.3) | 2,392 (11.2) | ||
| Female | 3,650 (17.0) | 2,478 (11.5) | ||
| Drug | <0.001 | <0.001 | ||
| Heparin | 6,833 (18.4) | 4,515 (12.2) | ||
| Enoxaparin | 506 (8.9) | 355 (6.2) | ||
| Length of stay, d | <0.001 | <0.001 | ||
| 01 | 446 (24.3) | 282 (15.4) | ||
| 23 | 1,463 (19.4) | 971 (12.9) | ||
| 47 | 2,332 (18.9) | 1,620 (13.1) | ||
| 8 | 3,098 (14.6) | 1,997 (9.4) | ||
The results of the multivariate regression analyses with GEE are displayed in Table 4. HIV diagnosis, non‐African American race, and heparin (as compared with enoxaparin) were associated with increased likelihood of nonadministration. Increasing age and increasing length of stay were associated with decreased likelihood of nonadministration by a small but significant amount.
| OR of Nonadministration | 95% CI, P | OR of Documented Refusal | 95% CI, P | |
|---|---|---|---|---|
| ||||
| Race | ||||
| African American | 1.00 | Reference | 1.00 | Reference |
| Caucasian | 1.62 | 1.44‐1.81, <0.001 | 1.53 | 1.32‐1.77, <0.001 |
| Asian, Pacific Islander, Other | 1.54 | 1.19‐2.00, 0.001 | 1.48 | 1.07‐2.01, 0.019 |
| HIV status | ||||
| Negative | 1.00 | Reference | 1.00 | Reference |
| Positive | 1.21 | 1.001.45, 0.039 | 1.29 | 1.06‐1.56, 0.012 |
| Age, per year | 0.97 | 0.97‐0.98, <0.001 | 0.97 | 0.96‐0.97, <0.001 |
| Drug | ||||
| Heparin | 1.00 | Reference | 1.00 | Reference |
| Enoxaparin | 0.45 | 0.40‐0.51, <0.001 | 0.53 | 0.47‐0.61, <0.001 |
| Length of stay, per day | 0.991 | 0.987‐0.995, <0.001 | 0.989 | 0.983‐0.993, <0.001 |
The most commonly documented reason for nonadministration was refusal by the patient or family member (66% of all doses not administered). The second most common reason, patient condition not appropriate, accounted for an additional 10% of doses. Across all nursing units, the proportion of prescribed doses that were documented as refused was significantly greater for patients with HIV compared with patients without HIV (16.5% vs 10.6%, OR: 1.66, 95% CI: 1.54‐1.80, P<0.0001) (Table 2). Using the GEE and multivariate regression, HIV diagnosis, non‐African American race, and heparin were associated with increased risk of documented dose refusal. Age and length of stay were inversely related to the likelihood of documented dose refusal. When all administered doses were excluded from the analysis, the association between these variables and documented dose refusal were not as strong. Age and length of stay remained significantly inversely related; however, the other factors were no longer significantly positively associated with documented dose refusal.
Within the HIV care unit, the proportion of prescribed doses documented as refused was greater for patients with HIV compared with patients without HIV (18.1% vs 6.8%, OR: 3.01, 95% CI: 2.28‐3.99, P<0.0001). For all other medicine units, the proportion of nonadministered doses documented as refused was also greater for patients with HIV compared with patients without HIV (13.7% vs 10.7%, OR: 1.32, 95% CI: 1.16‐1.51, P<0.0001).
DISCUSSION
We have identified that nonadministration of thromboprophylaxis was more common among patients with HIV at our institution. Substantial variation in the proportion of doses not administered existed on the nursing unit level, as well as within each unit when stratified by HIV status. This disparity in dose administration was observed on the HIV care unit as well, as the proportion not administered was about 2‐fold greater for patients with HIV compared with those without HIV. Documented dose refusal appeared to account for the majority of nonadministered doses in our cohort. Our analysis also demonstrated that HIV diagnosis is significantly associated with both dose nonadministration and documented dose refusal at our institution.
Medication refusal is a well‐recognized phenomenon among hospitalized patients. A recent study of medication administration in hospitalized patients in the United Kingdom noted that refusal accounted for about 45% of omitted doses.[26] Fanikos et al. also found that documented refusal of doses contributed significantly to the overall number of VTE prophylaxis doses not administered to patients.[27] In our study, the proportion of nonadministered doses documented as refused by the patient or family member was significantly greater in patients with HIV than in patients without HIV across all units. Interestingly, the difference was greater on the HIV care unit when doses were stratified by HIV status. This observation leads us to hypothesize that specific hospital care environments may influence dose nonadministration and refusal rates among our patient population.
Based on regression analyses, increasing age and length of stay were associated with a decreased likelihood of any particular dose not being administered and with any particular dose being documented as refused. It is important to note that our GEE did not take into account date or time of each dose, and therefore we cannot make conclusions as to the likelihood of dose nonadministration or refusal of doses in relation to each other on a time scale. One cannot assume that a dose due later in a hospital course was more or less likely to be given than a dose due on the first hospital day. Although we did not expect these findings, one can hypothesize that patients who are older or have longer stays may be perceived to have more severe illness, and therefore greater need for prophylaxis, from nursing staff and others involved in their care. The associations were small but significant and warrant future investigation.
To our knowledge, this is the first investigation comparing the proportion of nonadministered doses of thromboprophylaxis between patients with and without HIV. Our data show that nonadministered doses and refused doses of thromboprophylaxis are more frequent among patients with HIV. In addition, we noted that nonadministration was more common on the dedicated HIV care unit compared with other units. We cannot currently offer a clear explanation for the disparity observed between units, and more specifically, within the HIV care unit. However, it is possible that a unique culture of care and provider‐specific factors may contribute.
Our study was limited by a number of factors. Seroconversion among patients during the study period was possible; however, our analysis revealed only 2 instances among nearly 4000 unique patients. A more significant limitation was the level of analysis allowed by the dataset. We examined dose characteristics on a dose and unit level, but the ability to analyze doses based on the prescriber and nurse level may have provided valuable insight into the specific reasons behind the observations presented here. Additionally, the specific unit assigned to a given dose in our dataset represented the discharge location for the corresponding patient, making it possible that some amount of nonadministered doses may be attributed to the incorrect unit. However, we do not believe that unit‐to‐unit transfers would be frequent enough to influence the overall results. In addition, we did not link nonadministration of thromboprophylaxis with VTE events, as these data were not present in the current dataset. Although this is a limitation of the current study, we believe that the notion that missed doses of thromboprophylaxis place patients at higher risk for VTE is plausible, as the efficacy of thromboprophylaxis is well established.[28, 29, 30] It is important to note that the reason for nonadministration selected by the nurse on the eMAR may not always represent the only reason or even the true reason for dose nonadministration. It is possible that dose refusal may be over‐represented in our sample, in part due to inaccurate documentation. Recent investigations at JHH have identified varying attitudes on the part of the patient and the nurse regarding thromboprophylaxis. A questionnaire and interview of patients showed a large knowledge gap regarding thromboprophylaxis, with many individuals unable to explain its role or significance in their medical care.[31] A common theme was also observed in a survey of nurses regarding VTE prophylaxis: doses were sometimes considered optional for reasons such as ambulation status, perceived severity of illness, or reason for hospitalization. Some nurses also reported that after an initial refused dose, they may continue to document subsequent doses as refused, sometimes without offering the dose to the patient.[32] As variation in practice was observed between individual nurses, it is also likely that the culture of care may vary between units, influencing thromboprophylaxis nonadministration rates as well as documentation of doses as refused. The dose‐level data used for the GEE analyses did not include date and time of administration, which limited the ability of the GEE to more completely account for autocorrelation.
To further investigate the findings of this and related studies, we intend to more closely analyze data at multiple levels with the goal of identifying an appropriate and feasible target for intervention. Additionally, further investigation should be performed with the goal of determining the relationship between decreased exposure to thromboprophylaxis and VTE. However, as patients with HIV appear to be at increased risk of VTE, ensuring that thromboprophylaxis is delivered appropriately and consistently should be an important goal for all who provide care to this population.
Patients with human immunodeficiency virus (HIV) are at a 2‐ to 10‐fold greater risk for venous thromboembolism (VTE) compared with the general population.[1] Although antiphospholipid antibodies and protein S deficiency have often been cited as reasons for the thrombophilia associated with HIV, previous studies have also documented an increased risk of VTE with declining CD4+ cell count.[2, 3, 4, 5, 6, 7, 8] Worsening immune function places HIV patients at increased risk for opportunistic and nonopportunistic infections and malignancies, all independently associated with an increased risk of VTE.[5, 9, 10, 11, 12] Although increasing use of antiretroviral therapy has greatly decreased these sequelae, these complications of HIV infection are associated with an increased frequency of hospitalization.[13, 14, 15, 16] HIV infection and associated inflammation has been implicated in cardiovascular conditions such as cardiomyopathy, pulmonary hypertension, and myocardial infarction.[17, 18] Additionally, progression of HIV infection appears to influence T‐cell activation and differentiation in a manner that leads to early immunosenescence in infected individuals.[19, 20]
VTE prophylaxis is effective.[21] Virtually all efforts to decrease VTE have been focused on improving the prescription of prophylaxis with varying degrees of success.[22] These interventions have been employed with the tacit assumption that medication prescribed for inpatients will always be administered. However, at our institution, recent research has demonstrated that a significant proportion of prescribed thromboprophylaxis doses are not administered to hospitalized patients.[23] Refusal by the patient or a family member was the most commonly documented reason for dose nonadministration. In addition, the rate of thromboprophylaxis nonadministration varied greatly between nursing units with distinct patient populations. We hypothesized that nonadministration of VTE prophylaxis may be more common in patients with HIV, and this phenomenon may contribute to their increased risk for VTE.
The purpose of this study was to determine if the proportion of nonadministered thromboprophylaxis is greater among hospitalized patients with HIV and to characterize documented reasons for dose nonadministration.
METHODS
This study was conducted at The Johns Hopkins Hospital (JHH), a large, urban, academic medical center in Baltimore, Maryland. This single‐center retrospective cohort study utilized an existing dataset containing dose administration data extracted from an electronic medication administration record (eMAR). This dataset included information for all prescribed doses of thromboprophylaxis (heparin 5000 U subcutaneously every 8 or 12 hours, heparin 7500 U subcutaneously every 12 hours, enoxaparin 30 mg subcutaneously every 12 hours, or enoxaparin 40 mg subcutaneously daily) for patients hospitalized on medicine units at JHH from November 2007 to December 2008. This time period follows the implementation of an electronic order set for VTE prophylaxis.[24, 25] Data available for each dose included drug name, dose, frequency, patient demographics, and whether or not the dose was administered. Each dose not administered included a reason for nonadministration, which was chosen from a dropdown menu of responses on the eMAR by the nurse at the time the dose was due. A separate electronic report was obtained from an internal administrative database, which identified all patients within the dose administration dataset who had the International Classification of Diseases, 9th Revision code 042 (HIV diagnosis). A report identifying patient history numbers with matching diagnostic code for HIV was appended to the dose administration dataset using a relational database (Microsoft Access; Microsoft Corp., Redmond, WA) prior to analysis. The dose administration data were obtained previously for a separate analysis.[23] Approval for this study was granted from the institutional review board of Johns Hopkins Medicine.
Our analytic plan included comparisons between patients with and without HIV on a dose, patient, and unit level. As JHH operates a nursing unit dedicated to the inpatient care of patients with HIV, we included analyses of dose characteristics between this unit and other medicine units. It should be noted that patients without a diagnosis of HIV are sometimes cared for on this unit. Therefore, the electronic medical record for each patient without the diagnosis code for HIV hospitalized on this unit was reviewed to determine HIV status. An analysis was performed comparing visit identification numbers with diagnosis codes to identify potential seroconversions during the study period. Although we planned to compare nonadministration and documented refusal of doses on the unit level, a lack of patients with HIV on a number of units limited our ability to perform these analyses.
Statistical Analysis
The percent of doses not administered was calculated as the number of doses not administered divided by the number of doses prescribed. Likewise, the percent of prescribed doses documented as refused was calculated as the number of prescribed doses documented as refused divided by the number of doses prescribed. For each comparison, an odds ratio (OR) with 95% confidence interval (CI) was reported. Univariate and multivariate regression analyses were performed to assess the relationship between patient factors and dose nonadministration and documented refusal, respectively. Generalized estimating equations (GEE) using a logit link and an exchangeable correlation structure were used in these analyses. The GEE technique was used to account for within‐individual correlation of administration and documented refusal status.
Categorical data were compared using the two‐sided [2] test. Parametric and nonparametric continuous data were compared using the Student t test and Mann‐Whitney U test, respectively. A P value of <0.05 was considered statistically significant for all analyses. Analyses were performed using Minitab 15 (Minitab Inc., State College, PA) and Stata (StataCorp, College Station, TX).
RESULTS
During the 8‐month study period, 42,870 doses of thromboprophylaxis were prescribed during 4947 patient admissions to 13 individual medicine units. Overall, the diagnosis code for HIV was present in 12% of patient visits. The proportion of nonadministered doses per unit ranged from 6% to 27%, whereas the number of doses prescribed per unit ranged from 34 to 7301.
Patient characteristics were described on the visit level (Table 1). Patients with HIV were significantly younger, more often male and black, and had a longer length of stay compared with patients without HIV. Patients hospitalized on the HIV care unit had similar characteristics to the overall population of patients with HIV. It should be noted that not all patients cared for on this unit had a diagnosis of HIV, as patients from other medicine services are sometimes cared for in this location.
| Patients Without HIV | Patients With HIV | P | |
|---|---|---|---|
| |||
| Visits, n | 4,364 | 583 | N/A |
| Male, n (%) | 2,039 (47) | 370 (64) | <0.001 |
| Mean ageSD, y | 5618 | 469 | <0.001 |
| Race, n (%) | |||
| African American | 2,603 (60) | 522 (90) | <0.001 |
| Caucasian | 1,610 (37) | 53 (9) | <0.001 |
| Asian, Pacific Islander, other | 151 (4) | 8 (1) | 0.006 |
| Median length of stay (IQR), d | 3 (15) | 4 (27) | 0.002 |
| Marital status, n (%) | |||
| Single | 2,051 (47) | 471 (81) | <0.001 |
| Married | 1,405 (32) | 71 (12) | <0.001 |
| Widowed | 486 (11) | 10 (1) | <0.001 |
| Divorced | 402 (9) | 28 (5) | <0.001 |
| Separated | 33 (1) | 3 (1) | 0.607 |
| Unknown | 5 (0) | 0 (0) | 0.465 |
| Payor, n (%) | |||
| Medicare | 1,771 (41) | 133 (23) | <0.001 |
| Medicaid | 1,343 (31) | 392 (67) | <0.001 |
| Commercial | 1,181 (27) | 43 (7) | <0.001 |
| Other including self‐pay | 69 (1) | 15 (3) | 0.087 |
Overall, 17% of prescribed prophylaxis doses were not administered. A greater proportion of prescribed doses were not administered to patients with HIV compared with patients without HIV (23.5% vs 16.1%, OR: 1.59, 95% CI: 1.49‐1.70, P<0.001) (Table 2). Using a GEE and univariate regression, HIV diagnosis was associated with nonadministration of doses (OR: 1.37, 95% CI: 1.17‐1.60, P<0.001) (Table 3). Race, age, length of stay, and drug (heparin vs enoxaparin) were each associated with nonadministration. There was no significant association between nonadministration and sex, marital status, or payor. When stratified by nursing unit, there was substantial variation in the proportion of nonadministered doses between units. Within each unit, the proportion of doses not administered varied when stratified by HIV status. For example, on unit A, the proportion of doses not administered was greater for patients with HIV compared with patients without HIV (33.3% vs 12.9%, OR: 3.38, 95% CI: 2.61 to 4.37, P<0.001) (Figure 1). However, on unit K, the proportion of doses not administered to patients with HIV was 2‐fold less than in patients without HIV (7.2% vs 14.3%, OR: 0.47, 95% CI: 0.30‐0.74, P<0.001). Unit‐level analysis was not possible in regression models due to drastic imbalance in the prevalence of HIV across units. When comparing doses prescribed in the HIV care unit to all other medicine units, the proportion not administered (23.9% vs 16.3%, OR: 1.61, 95% CI: 1.49‐1.73, P<0.001) closely resembled the values seen when comparing patients with and without HIV hospital wide (23.5% vs 16.1%). However, when doses on the HIV care unit were stratified by HIV status, the doses not administered were 2‐fold greater, as a proportion, for patients with HIV compared with those without HIV (26.4% vs 13.1%, OR: 2.39, 95% CI: 1.93‐2.96, P<0.001).
| Doses Prescribed | Doses Not Administered (% of Doses Prescribed) | Doses Documented as Refused (% of All Doses Prescribed) | |
|---|---|---|---|
| |||
| All patients with HIV | 5,681 | 1,334 (23.5%)a | 935 (16.5%)a |
| All patients without HIV | 37,189 | 6,005 (16.1%) | 3,935 (10.6%) |
| HIV care unit | 4,452 | 1,063 (23.9%)a | 709 (15.9%)a |
| All other units | 38,418 | 6,276 (16.3%) | 4,161 (10.8%) |
| HIV care unit: patients with HIV | 3,602 | 952 (26.4%)a | 651 (18.1%)a |
| HIV care unit: patients without HIV | 850 | 111 (13.1%) | 58 (6.8%) |
| All other units: patients with HIV | 2,079 | 382 (18.4%)b | 284 (13.7%)a |
| All other units: patients without HIV | 36,339 | 5,894 (16.2%) | 3,877 (10.7%) |
| Nonadministered, n (%) | P | Documented as Refused, n (%) | P | |
|---|---|---|---|---|
| ||||
| Race | 0.001 | 0.072 | ||
| African American | 2,601 (17.8) | 1,708 (11.7) | ||
| Caucasian | 4,379 (16.4) | 2,922 (10.9) | ||
| Asian, Pacific Islander, other | 359 (23.4) | 240 (15.6) | ||
| HIV status | <0.001 | 0.002 | ||
| Negative | 6,005 (16.2) | 3,935 (10.6) | ||
| Positive | 1,344 (23.5) | 935 (16.5) | ||
| Age, y | <0.001 | <0.001 | ||
| 19 | 59 (20.6) | 44 (15.3) | ||
| 2029 | 1,260 (33.8) | 1,000 (26.8) | ||
| 3039 | 1,088 (28.1) | 845 (21.8) | ||
| 4049 | 1,628 (21.0) | 1,104 (14.2) | ||
| 5059 | 1,493 (16.1) | 953 (10.3) | ||
| 6069 | 900 (12.6) | 515 (7.2) | ||
| 7079 | 571 (9.6) | 250 (4.2) | ||
| 8089 | 252 (6.2) | 95 (2.3) | ||
| 90 | 88 (11.5) | 84 (8.4) | ||
| Sex | 0.372 | 0.919 | ||
| Male | 3,689 (17.3) | 2,392 (11.2) | ||
| Female | 3,650 (17.0) | 2,478 (11.5) | ||
| Drug | <0.001 | <0.001 | ||
| Heparin | 6,833 (18.4) | 4,515 (12.2) | ||
| Enoxaparin | 506 (8.9) | 355 (6.2) | ||
| Length of stay, d | <0.001 | <0.001 | ||
| 01 | 446 (24.3) | 282 (15.4) | ||
| 23 | 1,463 (19.4) | 971 (12.9) | ||
| 47 | 2,332 (18.9) | 1,620 (13.1) | ||
| 8 | 3,098 (14.6) | 1,997 (9.4) | ||
The results of the multivariate regression analyses with GEE are displayed in Table 4. HIV diagnosis, non‐African American race, and heparin (as compared with enoxaparin) were associated with increased likelihood of nonadministration. Increasing age and increasing length of stay were associated with decreased likelihood of nonadministration by a small but significant amount.
| OR of Nonadministration | 95% CI, P | OR of Documented Refusal | 95% CI, P | |
|---|---|---|---|---|
| ||||
| Race | ||||
| African American | 1.00 | Reference | 1.00 | Reference |
| Caucasian | 1.62 | 1.44‐1.81, <0.001 | 1.53 | 1.32‐1.77, <0.001 |
| Asian, Pacific Islander, Other | 1.54 | 1.19‐2.00, 0.001 | 1.48 | 1.07‐2.01, 0.019 |
| HIV status | ||||
| Negative | 1.00 | Reference | 1.00 | Reference |
| Positive | 1.21 | 1.001.45, 0.039 | 1.29 | 1.06‐1.56, 0.012 |
| Age, per year | 0.97 | 0.97‐0.98, <0.001 | 0.97 | 0.96‐0.97, <0.001 |
| Drug | ||||
| Heparin | 1.00 | Reference | 1.00 | Reference |
| Enoxaparin | 0.45 | 0.40‐0.51, <0.001 | 0.53 | 0.47‐0.61, <0.001 |
| Length of stay, per day | 0.991 | 0.987‐0.995, <0.001 | 0.989 | 0.983‐0.993, <0.001 |
The most commonly documented reason for nonadministration was refusal by the patient or family member (66% of all doses not administered). The second most common reason, patient condition not appropriate, accounted for an additional 10% of doses. Across all nursing units, the proportion of prescribed doses that were documented as refused was significantly greater for patients with HIV compared with patients without HIV (16.5% vs 10.6%, OR: 1.66, 95% CI: 1.54‐1.80, P<0.0001) (Table 2). Using the GEE and multivariate regression, HIV diagnosis, non‐African American race, and heparin were associated with increased risk of documented dose refusal. Age and length of stay were inversely related to the likelihood of documented dose refusal. When all administered doses were excluded from the analysis, the association between these variables and documented dose refusal were not as strong. Age and length of stay remained significantly inversely related; however, the other factors were no longer significantly positively associated with documented dose refusal.
Within the HIV care unit, the proportion of prescribed doses documented as refused was greater for patients with HIV compared with patients without HIV (18.1% vs 6.8%, OR: 3.01, 95% CI: 2.28‐3.99, P<0.0001). For all other medicine units, the proportion of nonadministered doses documented as refused was also greater for patients with HIV compared with patients without HIV (13.7% vs 10.7%, OR: 1.32, 95% CI: 1.16‐1.51, P<0.0001).
DISCUSSION
We have identified that nonadministration of thromboprophylaxis was more common among patients with HIV at our institution. Substantial variation in the proportion of doses not administered existed on the nursing unit level, as well as within each unit when stratified by HIV status. This disparity in dose administration was observed on the HIV care unit as well, as the proportion not administered was about 2‐fold greater for patients with HIV compared with those without HIV. Documented dose refusal appeared to account for the majority of nonadministered doses in our cohort. Our analysis also demonstrated that HIV diagnosis is significantly associated with both dose nonadministration and documented dose refusal at our institution.
Medication refusal is a well‐recognized phenomenon among hospitalized patients. A recent study of medication administration in hospitalized patients in the United Kingdom noted that refusal accounted for about 45% of omitted doses.[26] Fanikos et al. also found that documented refusal of doses contributed significantly to the overall number of VTE prophylaxis doses not administered to patients.[27] In our study, the proportion of nonadministered doses documented as refused by the patient or family member was significantly greater in patients with HIV than in patients without HIV across all units. Interestingly, the difference was greater on the HIV care unit when doses were stratified by HIV status. This observation leads us to hypothesize that specific hospital care environments may influence dose nonadministration and refusal rates among our patient population.
Based on regression analyses, increasing age and length of stay were associated with a decreased likelihood of any particular dose not being administered and with any particular dose being documented as refused. It is important to note that our GEE did not take into account date or time of each dose, and therefore we cannot make conclusions as to the likelihood of dose nonadministration or refusal of doses in relation to each other on a time scale. One cannot assume that a dose due later in a hospital course was more or less likely to be given than a dose due on the first hospital day. Although we did not expect these findings, one can hypothesize that patients who are older or have longer stays may be perceived to have more severe illness, and therefore greater need for prophylaxis, from nursing staff and others involved in their care. The associations were small but significant and warrant future investigation.
To our knowledge, this is the first investigation comparing the proportion of nonadministered doses of thromboprophylaxis between patients with and without HIV. Our data show that nonadministered doses and refused doses of thromboprophylaxis are more frequent among patients with HIV. In addition, we noted that nonadministration was more common on the dedicated HIV care unit compared with other units. We cannot currently offer a clear explanation for the disparity observed between units, and more specifically, within the HIV care unit. However, it is possible that a unique culture of care and provider‐specific factors may contribute.
Our study was limited by a number of factors. Seroconversion among patients during the study period was possible; however, our analysis revealed only 2 instances among nearly 4000 unique patients. A more significant limitation was the level of analysis allowed by the dataset. We examined dose characteristics on a dose and unit level, but the ability to analyze doses based on the prescriber and nurse level may have provided valuable insight into the specific reasons behind the observations presented here. Additionally, the specific unit assigned to a given dose in our dataset represented the discharge location for the corresponding patient, making it possible that some amount of nonadministered doses may be attributed to the incorrect unit. However, we do not believe that unit‐to‐unit transfers would be frequent enough to influence the overall results. In addition, we did not link nonadministration of thromboprophylaxis with VTE events, as these data were not present in the current dataset. Although this is a limitation of the current study, we believe that the notion that missed doses of thromboprophylaxis place patients at higher risk for VTE is plausible, as the efficacy of thromboprophylaxis is well established.[28, 29, 30] It is important to note that the reason for nonadministration selected by the nurse on the eMAR may not always represent the only reason or even the true reason for dose nonadministration. It is possible that dose refusal may be over‐represented in our sample, in part due to inaccurate documentation. Recent investigations at JHH have identified varying attitudes on the part of the patient and the nurse regarding thromboprophylaxis. A questionnaire and interview of patients showed a large knowledge gap regarding thromboprophylaxis, with many individuals unable to explain its role or significance in their medical care.[31] A common theme was also observed in a survey of nurses regarding VTE prophylaxis: doses were sometimes considered optional for reasons such as ambulation status, perceived severity of illness, or reason for hospitalization. Some nurses also reported that after an initial refused dose, they may continue to document subsequent doses as refused, sometimes without offering the dose to the patient.[32] As variation in practice was observed between individual nurses, it is also likely that the culture of care may vary between units, influencing thromboprophylaxis nonadministration rates as well as documentation of doses as refused. The dose‐level data used for the GEE analyses did not include date and time of administration, which limited the ability of the GEE to more completely account for autocorrelation.
To further investigate the findings of this and related studies, we intend to more closely analyze data at multiple levels with the goal of identifying an appropriate and feasible target for intervention. Additionally, further investigation should be performed with the goal of determining the relationship between decreased exposure to thromboprophylaxis and VTE. However, as patients with HIV appear to be at increased risk of VTE, ensuring that thromboprophylaxis is delivered appropriately and consistently should be an important goal for all who provide care to this population.
- , , , , . Venous thromboembolism in patients with HIV/AIDS: a case‐control study. J Acquir Immune Defic Syndr. 2008;48(3):310–314.
- , , . AIDS and thrombosis: retrospective study of 131 HIV‐infected patients. AIDS Patient Care STDS. 2001;15(6):311–320.
- , , , et al. HIV and risk of venous thromboembolism: a Danish nationwide population‐based cohort study. HIV Med. 2011;12(4):202–210.
- , , , . Epidemiology of thrombosis in HIV‐infected individuals. The adult/adolescent spectrum of HIV disease project. AIDS. 2000;14(3):321–324.
- , , . Thrombotic complications in patients infected with HIV in the era of highly active antiretroviral therapy: a case series. Clin Infect Dis. 2004;39(8):1214–1222.
- , , . Circulating coagulation inhibitors in the acquired immunodeficiency syndrome. Ann Intern Med. 1986;104(2):175–180.
- , . The pathogenesis of the antiphospholipid syndrome. N Engl J Med. 2013;368(11):1033–1044.
- , , , . Acquired protein C and protein S deficiency in HIV‐infected patients. Clin Appl Thromb Hemost. 2003;9(4):325–331.
- , , , et al. Antiphospholipid syndrome associated with cytomegalovirus infection: case report and review. Clin Infect Dis. 1997;24(2):197–200.
- , . Viral infections and antiphospholipid antibodies. Semin Arthritis Rheum. 2002;31(4):256–263.
- , . AIDS‐defining and non‐AIDS‐defining malignancies: cancer occurrence in the antiretroviral therapy era. Curr Opin Oncol. 2007;19(5):446–451.
- , , , , . Pathogenesis, clinical and laboratory aspects of thrombosis in cancer. J Thromb Thrombolysis. 2007;24(1):29–38.
- , , , et al. Patterns of diagnoses in hospital admissions in a multistate cohort of HIV‐positive adults in 2001. Med Care. 2005;43(9 suppl):III3–III14.
- , , , et al. Opportunistic infections as causes of death in HIV‐infected patients in the HAART era in France. Scand J Infect Dis. 2005;37(6‐7):482–487.
- , , , et al. Rates of hospitalizations and associated diagnoses in a large multisite cohort of HIV patients in the United States, 1994–2005. AIDS. 2008;22(11):1345–1354.
- , , . Hospitalizations for metabolic conditions, opportunistic infections, and injection drug use among HIV patients: trends between 1996 and 2000 in 12 states. J Acquir Immune Defic Syndr. 2005;40(5):609–616.
- , , , , , . Cardiovascular disease in HIV infection. Am Heart J. 2006;151:1147–1155.
- , , , et al. Epidemiological evidence for cardiovascular disease in HIV‐infected patients and relationship to highly active antiretroviral therapy. Circulation. 2008;118(2):e29–e35.
- , , , et al. Immune activation and CD8+ T‐cell differentiation towards senescence in HIV‐1 infection. PLoS Biol. 2004;2(2):E20.
- , , , , . CD4 T cell depletion is linked directly to immune activation in the pathogenesis of HIV‐1 and HIV‐2 but only indirectly to the viral load. J Immunol. 2002;169(6):3400–3406.
- , . Thromboprophylaxis in nonsurgical patients. Hematology Am Soc Hematol Educ Program. 2012;2012:631–637.
- , . Practices to prevent venous thromboembolism [published online ahead of print May 24, 2013]. BMJ Qual Saf. doi:10.1136/bmjqs‐2012‐001782.
- , , , et al. Patterns of non‐administration of ordered doses of venous thromboembolism prophylaxis: implications for novel intervention strategies. PLoS One. 2013;8(6):e66311.
- , , , et al. Lessons from The Johns Hopkins multi‐disciplinary venous thromboembolism (VTE) prevention collaborative. BMJ. 2012;344:e3935.
- , , , et al. Impact of a venous thromboembolism prophylaxis “smart order set”: improved compliance, fewer events [published online ahead of print April 4, 2013]. Am J Hematol. doi: 10.1002/ajh.23450.
- , , . Dose omissions in hospitalized patients in a UK hospital: an analysis of the relative contribution of adverse drug reactions. Drug Saf. 2012;35(8):677–683.
- , , , et al. Adherence to pharmacological thromboprophylaxis orders in hospitalized patients. Am J Med. 2010;123(6):536–541.
- , , , et al. A comparison of enoxaparin with placebo for the prevention of venous thromboembolism in acutely ill medical patients. Prophylaxis in medical patients with enoxaparin study group. N Engl J Med. 1999;341(11):793–800.
- , , , et al. Randomized, placebo‐controlled trial of dalteparin for the prevention of venous thromboembolism in acutely ill medical patients. Circulation. 2004;110(7):874–879.
- , , , et al. Efficacy and safety of fondaparinux for the prevention of venous thromboembolism in older acute medical patients: randomised placebo controlled trial. BMJ. 2006;332(7537):325–329.
- , , , et al. Patient perspectives on pharmacological venous thromboembolism prophylaxis at The Johns Hopkins Hospital. J Thromb Thrombolysis. 2013;35(3):416.
- , , , et al. Culture of care and documented patient refusal of pharmacologic venous thromboembolism prophylaxis. J Thromb Thrombolysis. 2011;31(3):367–400.
- , , , , . Venous thromboembolism in patients with HIV/AIDS: a case‐control study. J Acquir Immune Defic Syndr. 2008;48(3):310–314.
- , , . AIDS and thrombosis: retrospective study of 131 HIV‐infected patients. AIDS Patient Care STDS. 2001;15(6):311–320.
- , , , et al. HIV and risk of venous thromboembolism: a Danish nationwide population‐based cohort study. HIV Med. 2011;12(4):202–210.
- , , , . Epidemiology of thrombosis in HIV‐infected individuals. The adult/adolescent spectrum of HIV disease project. AIDS. 2000;14(3):321–324.
- , , . Thrombotic complications in patients infected with HIV in the era of highly active antiretroviral therapy: a case series. Clin Infect Dis. 2004;39(8):1214–1222.
- , , . Circulating coagulation inhibitors in the acquired immunodeficiency syndrome. Ann Intern Med. 1986;104(2):175–180.
- , . The pathogenesis of the antiphospholipid syndrome. N Engl J Med. 2013;368(11):1033–1044.
- , , , . Acquired protein C and protein S deficiency in HIV‐infected patients. Clin Appl Thromb Hemost. 2003;9(4):325–331.
- , , , et al. Antiphospholipid syndrome associated with cytomegalovirus infection: case report and review. Clin Infect Dis. 1997;24(2):197–200.
- , . Viral infections and antiphospholipid antibodies. Semin Arthritis Rheum. 2002;31(4):256–263.
- , . AIDS‐defining and non‐AIDS‐defining malignancies: cancer occurrence in the antiretroviral therapy era. Curr Opin Oncol. 2007;19(5):446–451.
- , , , , . Pathogenesis, clinical and laboratory aspects of thrombosis in cancer. J Thromb Thrombolysis. 2007;24(1):29–38.
- , , , et al. Patterns of diagnoses in hospital admissions in a multistate cohort of HIV‐positive adults in 2001. Med Care. 2005;43(9 suppl):III3–III14.
- , , , et al. Opportunistic infections as causes of death in HIV‐infected patients in the HAART era in France. Scand J Infect Dis. 2005;37(6‐7):482–487.
- , , , et al. Rates of hospitalizations and associated diagnoses in a large multisite cohort of HIV patients in the United States, 1994–2005. AIDS. 2008;22(11):1345–1354.
- , , . Hospitalizations for metabolic conditions, opportunistic infections, and injection drug use among HIV patients: trends between 1996 and 2000 in 12 states. J Acquir Immune Defic Syndr. 2005;40(5):609–616.
- , , , , , . Cardiovascular disease in HIV infection. Am Heart J. 2006;151:1147–1155.
- , , , et al. Epidemiological evidence for cardiovascular disease in HIV‐infected patients and relationship to highly active antiretroviral therapy. Circulation. 2008;118(2):e29–e35.
- , , , et al. Immune activation and CD8+ T‐cell differentiation towards senescence in HIV‐1 infection. PLoS Biol. 2004;2(2):E20.
- , , , , . CD4 T cell depletion is linked directly to immune activation in the pathogenesis of HIV‐1 and HIV‐2 but only indirectly to the viral load. J Immunol. 2002;169(6):3400–3406.
- , . Thromboprophylaxis in nonsurgical patients. Hematology Am Soc Hematol Educ Program. 2012;2012:631–637.
- , . Practices to prevent venous thromboembolism [published online ahead of print May 24, 2013]. BMJ Qual Saf. doi:10.1136/bmjqs‐2012‐001782.
- , , , et al. Patterns of non‐administration of ordered doses of venous thromboembolism prophylaxis: implications for novel intervention strategies. PLoS One. 2013;8(6):e66311.
- , , , et al. Lessons from The Johns Hopkins multi‐disciplinary venous thromboembolism (VTE) prevention collaborative. BMJ. 2012;344:e3935.
- , , , et al. Impact of a venous thromboembolism prophylaxis “smart order set”: improved compliance, fewer events [published online ahead of print April 4, 2013]. Am J Hematol. doi: 10.1002/ajh.23450.
- , , . Dose omissions in hospitalized patients in a UK hospital: an analysis of the relative contribution of adverse drug reactions. Drug Saf. 2012;35(8):677–683.
- , , , et al. Adherence to pharmacological thromboprophylaxis orders in hospitalized patients. Am J Med. 2010;123(6):536–541.
- , , , et al. A comparison of enoxaparin with placebo for the prevention of venous thromboembolism in acutely ill medical patients. Prophylaxis in medical patients with enoxaparin study group. N Engl J Med. 1999;341(11):793–800.
- , , , et al. Randomized, placebo‐controlled trial of dalteparin for the prevention of venous thromboembolism in acutely ill medical patients. Circulation. 2004;110(7):874–879.
- , , , et al. Efficacy and safety of fondaparinux for the prevention of venous thromboembolism in older acute medical patients: randomised placebo controlled trial. BMJ. 2006;332(7537):325–329.
- , , , et al. Patient perspectives on pharmacological venous thromboembolism prophylaxis at The Johns Hopkins Hospital. J Thromb Thrombolysis. 2013;35(3):416.
- , , , et al. Culture of care and documented patient refusal of pharmacologic venous thromboembolism prophylaxis. J Thromb Thrombolysis. 2011;31(3):367–400.
© 2014 Society of Hospital Medicine
Aging and Inpatient Demand
The number of older people in the United States is expected to increase, due to the aging of the post‐World War II baby boomers.[1] For example, those aged 65 years are expected to number 88.5 million in 2050, more than double the number in 2010 of 40.2 million. This demographic shift has raised concerns about future hospital capacity, but the scope of the problem has not been quantified.[2]
A recent analysis calculated the number and length of emergency department visits expected to occur based on the aging of the US population.[3] One finding was that hospital admissions would increase 23% faster than population growth. However, this considered only hospitalizations originating in the emergency department and did not consider all‐source hospitalizations. We obtained data on all‐source hospitalizations and applied them to the US Census Bureau's demographic projections for the future through 2050. This provides a base‐case estimate for how inpatient demand would change if all other influences remained equal. The goal was to isolate the effect of population age makeup on inpatient requirements while holding other influences constant.
METHODS
We used the method of actuarial life table adjustment as described previously.[3] To calculate age‐specific hospitalization rates, we estimated age‐specific hospitalization frequencies (counts) in the United States for 2011 from the Nationwide Inpatient Sample (NIS).[4] This is a stratified probability sample of US community hospitals, defined as all nonfederal, short term, general, and other specialty hospitals, excluding hospital units of institutions. Veterans hospitals and other federal facilities, short‐term rehabilitation hospitals, long‐term non‐acute care hospitals, psychiatric hospitals, and alcoholism/chemical dependency treatment facilities were excluded from NIS 2011. Of hospitals in the sample, 21% are government (nonfederal) owned.
We converted age‐specific hospitalization frequencies derived from this sample into rates by dividing each stratum‐specific admission count by the 2011 population count in each age stratum from the US Census Bureau.[5] The Census Bureau provides detailed predictions of the US population through 2050. Births, deaths, and net international migration are projected for each birth cohort. Using 2011 as the origin, we applied baseline age‐specific hospitalization rates stratum‐wise to the general population expected by the Census Bureau in future years. This gave us stratum‐specific hospitalization frequencies for each future year. We summed these to arrive at the aggregate anticipated hospitalization frequency in each year. For our main outcome measure, we calculated the ratio of change in hospitalization frequency to change in population, comparing each future year to the 2011 baseline. We also calculated aggregate inpatient days, using the same data sources and methods. Our institutional review board exempted this study from review. We used Stata 13.0 (StataCorp, College Station, TX), and Microsoft Excel (Microsoft, Redmond, WA) for all analyses.
RESULTS
Baseline data are displayed in Figure 1. In 2011, there were 0.23 hospitalizations per US resident aged 0 to 4 years, and 0.01 per resident aged 5 to 9 years. From this age forward, hospitalization rates increased steadily with advancing age, reaching 0.63 per resident aged 90 to 94 years. Length of stay also was generally associated with age, though there was a peak among older children.
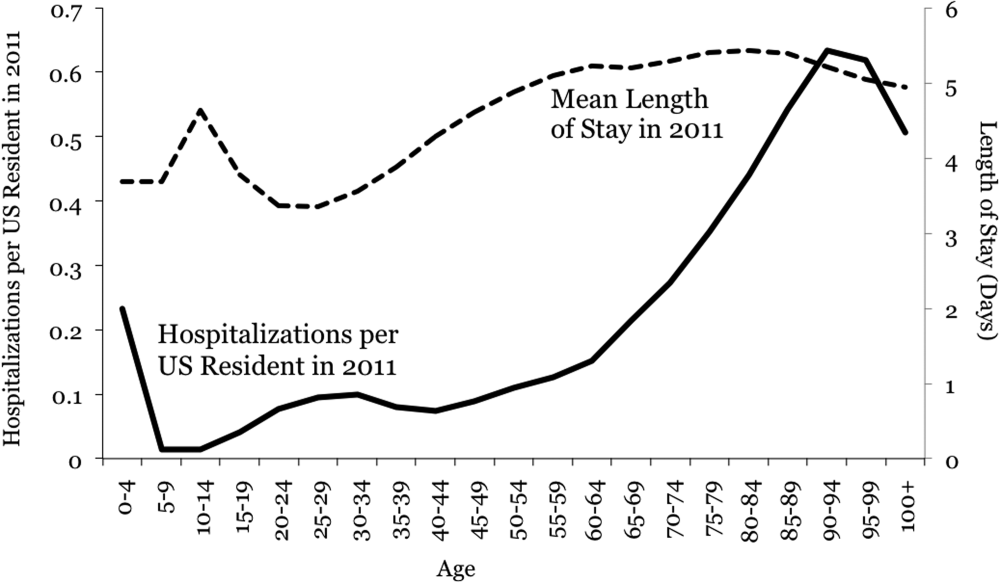
Projections through 2050 are shown in Table 1 and Figure 2. Table 1 displays the population projections of the US Census Bureau, which expects the US population to increase by 41% between now and 2050. Also shown in the table are our projections, which indicate that, all other things being equal, the annual number of inpatient admissions in the US will increase by 67%. The ratio of 67% to 41% is 1.18, meaning that the frequency of inpatient admissions will grow 18% more than population growth due to the aging of the population. The aggregate number of inpatient days will increase 22% more than population growth. Overall, inpatient capacity must expand by 72% to keep pace.
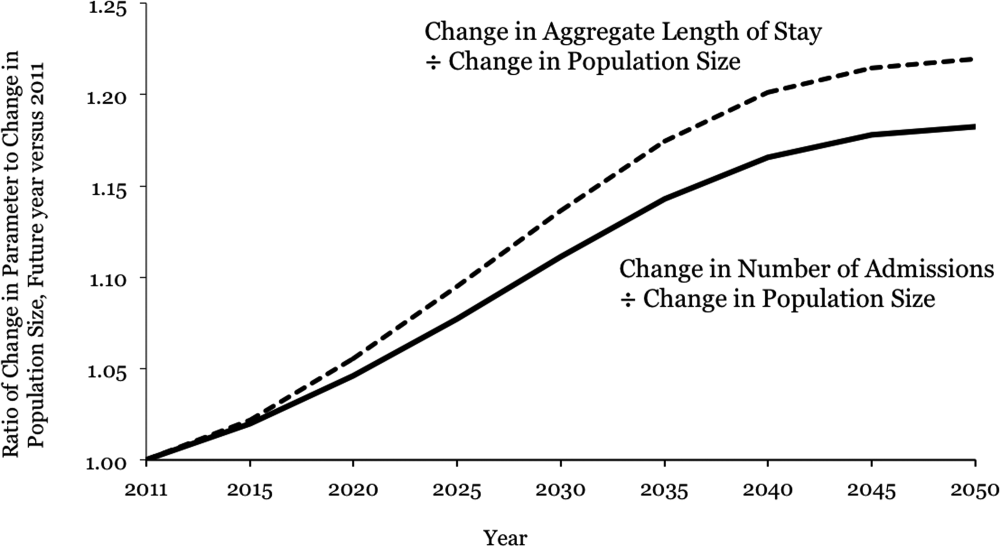
| Year | Population | Hospital Admissions | Aggregate Inpatient Days | Population: Ratio of Future Year to 2011 | Admissions: Ratio of Future Year to 2011 | Ratio of Admission Increase to Population Increase | Aggregate Inpatient Days: Ratio of Future Year to 2011 | Ratio of Increase in Inpatient Days to Population Increase |
|---|---|---|---|---|---|---|---|---|
| ||||||||
| 2011 | 311,591,917 | 38,560,751* | 177,501,515 | 1 | 1 | 1 | 1 | 1 |
| 2015 | 325,539,790 | 41,093,154 | 189,520,706 | 1.04 | 1.07 | 1.02 | 1.07 | 1.02 |
| 2020 | 341,386,665 | 44,196,669 | 205,205,962 | 1.10 | 1.15 | 1.05 | 1.16 | 1.06 |
| 2025 | 357,451,620 | 47,655,492 | 222,911,204 | 1.15 | 1.24 | 1.08 | 1.26 | 1.09 |
| 2030 | 373,503,674 | 51,365,441 | 241,852,384 | 1.20 | 1.33 | 1.11 | 1.36 | 1.14 |
| 2035 | 389,531,156 | 55,091,242 | 260,603,998 | 1.25 | 1.43 | 1.14 | 1.47 | 1.17 |
| 2040 | 405,655,295 | 58,524,016 | 277,530,732 | 1.30 | 1.52 | 1.17 | 1.56 | 1.20 |
| 2045 | 422,058,629 | 61,525,903 | 292,014,192 | 1.35 | 1.60 | 1.18 | 1.65 | 1.21 |
| 2050 | 439,010,253 | 64,249,181 | 304,945,179 | 1.41 | 1.67 | 1.18 | 1.72 | 1.22 |
DISCUSSION
Although US hospital capacity has fallen over the past 3 decades,[6, 7] our analysis suggests that demand for inpatient beds will increase 22% faster than population growth by 2050. The total projected demand increase is 72%, including that attributable to population growth and that attributable to population aging.
These are ceteris paribus projections, which reveal the changes in inpatient demand that would result if 2 conditions held: (1) the US Census Bureau's expectations for population makeup proved correct, and (2) age‐specific hospitalization rates and lengths of stay did not change. In reality, age‐specific hospitalization rates and lengths of stay could change. Examples of change drivers include epidemics, technology, and financial incentives provided by third‐party payers.[7] For example, if an epidemic of a new disease were to occur, age‐specific hospitalization rates could increase across all age groups. Our projections depict what would happen in the absence of any such change. This is useful because we do not know if changes in age‐specific hospitalization rates will occur, and whether there will be increases or decreases. Therefore, our projections should not be viewed as literal predictions, but rather as pieces of the puzzle, necessary but not sufficient elements of an understanding of what the future may hold for inpatient demand.
Clinicians, academics, and government agencies have an interest in understanding inpatient supply and demand on national and local levels. However, their ability to influence supply is limited by the fact that of all registered hospitals in the United States, only 22% are government owned.[1] As a result, decisions about hospital construction and closure are generally left to the free market.[6] Nonetheless, we bear responsibility for monitoring supply and demand, and government regulation of hospitals and reimbursement for inpatient care mean that the public is not entirely without influence. Thirty‐two percent of US residents have government‐issued health insurance.[8]
In the early 20th century, very little healthcare took place in the inpatient setting. However, by the 1970s, inpatient care accounted for a large part of healthcare, due largely to changes in technology and reimbursement. This trend reversed in the 1980s and 1990s, and hospitals closed.[7] In 1975, there were 5875 hospitals in the United States, and in 2000 there were 4915.[6] The number of staffed beds decreased from 942,000 to 826,000.[6] In parallel, likely due to changes in technology (ie, the nature of healthcare), total inpatient days in community hospitals decreased from 223 million in 1991 to 187 million in 2011.[9] On the other hand, increasing access to insurance under the Affordable Care Act could increase utilization, as seen when a 30% increase in hospital utilization occurred when people were enrolled in Oregon's Medicaid program.[10] Also, hospital utilization may increase if Medicare patients require more services.[11]
Actuarial life table analysis has been used to make forecasts related to healthcare supply and demand, though we are not aware of prior applications to the question of hospitalization. A prior study used actuarial life table adjustment to forecast demand for emergency department services.[3] These methods have also been used to forecast the influence of longevity upon healthcare expenditures[12, 13, 14] and to predict demand for specialty services.[15, 16] Of note, rather than reporting ratios of demand growth to population growth, another option would have been to derive a compound growth rate. We are not aware of a precedent for such methods in the prior published applications of actuarial life table analysis and felt that such inductive methods would complicate the interpretation of our results.
The main limitation of our investigation is its scope. We used actuarial life table adjustment to isolate the effect of population aging upon demand for inpatient hospitalizations. This method does not yield a comprehensive prediction of inpatient demand, but rather provides a robust estimate under the assumption that all other things remain equal. Another obvious limitation is that our analysis has a nationwide scope, and was not designed to account for variation from one locale to the next. However, these methods can be used by local health authorities.
CONCLUSIONS
The US Census Bureau expects the US population to increase by 41% over the next 4 decades, and the number of US residents aged 65 years to more than double. Our results indicate that, all other things being equal, this will cause the number of hospital admissions to increase 18% faster than population growth, and the aggregate number of inpatient days to increase 22% faster than population growth. Including both population growth and population aging, the total projected increase required for inpatient capacity is 72%. This is a base‐case, ceteris paribus analysis, and understanding how demand for inpatient services may change will require multiple perspectives. Increasing access to insurance, changing poverty rates, and changes in healthcare delivery and technology are other important factors. The present analysis provides a focused estimate of the influence upon demand for inpatient services due to expected changes in our population's age distribution.
- American Hospital Association. Fast facts on US hospitals, 2011. Available at: http://www.aha.org/research/rc/stat‐studies/fast‐facts.shtml. Accessed August 7, 2013.
- American Hospital Association. Cracks in the foundation: averting a crisis in America's hospitals. AHA 2002. Available at: http://www.aha.org/content/00–10/cracksreprint08‐02.pdf. Accessed August 4, 2013.
- , , , , . Population aging and emergency departments: visits will not increase, lengths‐of‐stay and hospitalizations will. Health Aff (Millwood). 2013;32(7):1306–1312.
- HCUP Nationwide Inpatient Sample (NIS). Healthcare Cost and Utilization Project (HCUP). 2011. Agency for Healthcare Research and Quality, Rockville, MD. Available at: http://www.hcup‐us.ahrq.gov/nisoverview.jsp. Accessed July 25, 2013.
- Bureau of the Census. Population Projections by Age, Sex, Race, and Hispanic Origin: July 1, 2000–2050. Washington, DC: The Bureau; 2008.
- , , , . Does U.S. hospital capacity need to be expanded? Health Aff (Millwood). 2003;22(6):40–54.
- . Decline in hospital utilization and cost inflation under managed care in California. JAMA. 1996;276(13):1060–1064.
- , , . Income, poverty, and health insurance coverage in the United States, 2011. US Census Bureau. Available at: http://www.census.gov/prod/2012pubs/p60–243.pdf. Published September 2012. Accessed August 7, 2013.
- American Hospital Association. Trendwatch. Table 3.1: trends in inpatient utilization in community hospitals, 1991–2011. Available at: http://www.aha.org/research/reports/tw/chartbook/2013/table3‐1.pdf. Accessed November 9, 2013.
- , , , et al. The Oregon health insurance experiment: evidence from the first year. Q J Econ. 2012;127(3):1057–1106.
- American Hospital Association. Trendwatch. Are Medicare patients getting sicker? Available at: http://www.aha.org/research/reports/tw/12dec‐tw‐ptacuity.pdf. Accessed November 9, 2013.
- , , . Longevity and Medicare expenditures. N Engl J Med. 1995;332(15):999–1003.
- , . The aging of America. Impact on health care costs. JAMA. 1990;263(17):2335–2340.
- , . The effect of longevity on spending for acute and long‐term care. N Engl J Med. 2000;342(19):1409–1415.
- , , , . Demographics and cardiology, 1950–2050. J Am Coll Cardiol. 2000;35(4):1067–1081.
- , , , . Population‐based analysis of inpatient vascular procedures and predicting future workload and implications for training. J Vasc Surg. 2012;55(5):1394–1399; discussion 1399–1400.
The number of older people in the United States is expected to increase, due to the aging of the post‐World War II baby boomers.[1] For example, those aged 65 years are expected to number 88.5 million in 2050, more than double the number in 2010 of 40.2 million. This demographic shift has raised concerns about future hospital capacity, but the scope of the problem has not been quantified.[2]
A recent analysis calculated the number and length of emergency department visits expected to occur based on the aging of the US population.[3] One finding was that hospital admissions would increase 23% faster than population growth. However, this considered only hospitalizations originating in the emergency department and did not consider all‐source hospitalizations. We obtained data on all‐source hospitalizations and applied them to the US Census Bureau's demographic projections for the future through 2050. This provides a base‐case estimate for how inpatient demand would change if all other influences remained equal. The goal was to isolate the effect of population age makeup on inpatient requirements while holding other influences constant.
METHODS
We used the method of actuarial life table adjustment as described previously.[3] To calculate age‐specific hospitalization rates, we estimated age‐specific hospitalization frequencies (counts) in the United States for 2011 from the Nationwide Inpatient Sample (NIS).[4] This is a stratified probability sample of US community hospitals, defined as all nonfederal, short term, general, and other specialty hospitals, excluding hospital units of institutions. Veterans hospitals and other federal facilities, short‐term rehabilitation hospitals, long‐term non‐acute care hospitals, psychiatric hospitals, and alcoholism/chemical dependency treatment facilities were excluded from NIS 2011. Of hospitals in the sample, 21% are government (nonfederal) owned.
We converted age‐specific hospitalization frequencies derived from this sample into rates by dividing each stratum‐specific admission count by the 2011 population count in each age stratum from the US Census Bureau.[5] The Census Bureau provides detailed predictions of the US population through 2050. Births, deaths, and net international migration are projected for each birth cohort. Using 2011 as the origin, we applied baseline age‐specific hospitalization rates stratum‐wise to the general population expected by the Census Bureau in future years. This gave us stratum‐specific hospitalization frequencies for each future year. We summed these to arrive at the aggregate anticipated hospitalization frequency in each year. For our main outcome measure, we calculated the ratio of change in hospitalization frequency to change in population, comparing each future year to the 2011 baseline. We also calculated aggregate inpatient days, using the same data sources and methods. Our institutional review board exempted this study from review. We used Stata 13.0 (StataCorp, College Station, TX), and Microsoft Excel (Microsoft, Redmond, WA) for all analyses.
RESULTS
Baseline data are displayed in Figure 1. In 2011, there were 0.23 hospitalizations per US resident aged 0 to 4 years, and 0.01 per resident aged 5 to 9 years. From this age forward, hospitalization rates increased steadily with advancing age, reaching 0.63 per resident aged 90 to 94 years. Length of stay also was generally associated with age, though there was a peak among older children.
Projections through 2050 are shown in Table 1 and Figure 2. Table 1 displays the population projections of the US Census Bureau, which expects the US population to increase by 41% between now and 2050. Also shown in the table are our projections, which indicate that, all other things being equal, the annual number of inpatient admissions in the US will increase by 67%. The ratio of 67% to 41% is 1.18, meaning that the frequency of inpatient admissions will grow 18% more than population growth due to the aging of the population. The aggregate number of inpatient days will increase 22% more than population growth. Overall, inpatient capacity must expand by 72% to keep pace.
| Year | Population | Hospital Admissions | Aggregate Inpatient Days | Population: Ratio of Future Year to 2011 | Admissions: Ratio of Future Year to 2011 | Ratio of Admission Increase to Population Increase | Aggregate Inpatient Days: Ratio of Future Year to 2011 | Ratio of Increase in Inpatient Days to Population Increase |
|---|---|---|---|---|---|---|---|---|
| ||||||||
| 2011 | 311,591,917 | 38,560,751* | 177,501,515 | 1 | 1 | 1 | 1 | 1 |
| 2015 | 325,539,790 | 41,093,154 | 189,520,706 | 1.04 | 1.07 | 1.02 | 1.07 | 1.02 |
| 2020 | 341,386,665 | 44,196,669 | 205,205,962 | 1.10 | 1.15 | 1.05 | 1.16 | 1.06 |
| 2025 | 357,451,620 | 47,655,492 | 222,911,204 | 1.15 | 1.24 | 1.08 | 1.26 | 1.09 |
| 2030 | 373,503,674 | 51,365,441 | 241,852,384 | 1.20 | 1.33 | 1.11 | 1.36 | 1.14 |
| 2035 | 389,531,156 | 55,091,242 | 260,603,998 | 1.25 | 1.43 | 1.14 | 1.47 | 1.17 |
| 2040 | 405,655,295 | 58,524,016 | 277,530,732 | 1.30 | 1.52 | 1.17 | 1.56 | 1.20 |
| 2045 | 422,058,629 | 61,525,903 | 292,014,192 | 1.35 | 1.60 | 1.18 | 1.65 | 1.21 |
| 2050 | 439,010,253 | 64,249,181 | 304,945,179 | 1.41 | 1.67 | 1.18 | 1.72 | 1.22 |
DISCUSSION
Although US hospital capacity has fallen over the past 3 decades,[6, 7] our analysis suggests that demand for inpatient beds will increase 22% faster than population growth by 2050. The total projected demand increase is 72%, including that attributable to population growth and that attributable to population aging.
These are ceteris paribus projections, which reveal the changes in inpatient demand that would result if 2 conditions held: (1) the US Census Bureau's expectations for population makeup proved correct, and (2) age‐specific hospitalization rates and lengths of stay did not change. In reality, age‐specific hospitalization rates and lengths of stay could change. Examples of change drivers include epidemics, technology, and financial incentives provided by third‐party payers.[7] For example, if an epidemic of a new disease were to occur, age‐specific hospitalization rates could increase across all age groups. Our projections depict what would happen in the absence of any such change. This is useful because we do not know if changes in age‐specific hospitalization rates will occur, and whether there will be increases or decreases. Therefore, our projections should not be viewed as literal predictions, but rather as pieces of the puzzle, necessary but not sufficient elements of an understanding of what the future may hold for inpatient demand.
Clinicians, academics, and government agencies have an interest in understanding inpatient supply and demand on national and local levels. However, their ability to influence supply is limited by the fact that of all registered hospitals in the United States, only 22% are government owned.[1] As a result, decisions about hospital construction and closure are generally left to the free market.[6] Nonetheless, we bear responsibility for monitoring supply and demand, and government regulation of hospitals and reimbursement for inpatient care mean that the public is not entirely without influence. Thirty‐two percent of US residents have government‐issued health insurance.[8]
In the early 20th century, very little healthcare took place in the inpatient setting. However, by the 1970s, inpatient care accounted for a large part of healthcare, due largely to changes in technology and reimbursement. This trend reversed in the 1980s and 1990s, and hospitals closed.[7] In 1975, there were 5875 hospitals in the United States, and in 2000 there were 4915.[6] The number of staffed beds decreased from 942,000 to 826,000.[6] In parallel, likely due to changes in technology (ie, the nature of healthcare), total inpatient days in community hospitals decreased from 223 million in 1991 to 187 million in 2011.[9] On the other hand, increasing access to insurance under the Affordable Care Act could increase utilization, as seen when a 30% increase in hospital utilization occurred when people were enrolled in Oregon's Medicaid program.[10] Also, hospital utilization may increase if Medicare patients require more services.[11]
Actuarial life table analysis has been used to make forecasts related to healthcare supply and demand, though we are not aware of prior applications to the question of hospitalization. A prior study used actuarial life table adjustment to forecast demand for emergency department services.[3] These methods have also been used to forecast the influence of longevity upon healthcare expenditures[12, 13, 14] and to predict demand for specialty services.[15, 16] Of note, rather than reporting ratios of demand growth to population growth, another option would have been to derive a compound growth rate. We are not aware of a precedent for such methods in the prior published applications of actuarial life table analysis and felt that such inductive methods would complicate the interpretation of our results.
The main limitation of our investigation is its scope. We used actuarial life table adjustment to isolate the effect of population aging upon demand for inpatient hospitalizations. This method does not yield a comprehensive prediction of inpatient demand, but rather provides a robust estimate under the assumption that all other things remain equal. Another obvious limitation is that our analysis has a nationwide scope, and was not designed to account for variation from one locale to the next. However, these methods can be used by local health authorities.
CONCLUSIONS
The US Census Bureau expects the US population to increase by 41% over the next 4 decades, and the number of US residents aged 65 years to more than double. Our results indicate that, all other things being equal, this will cause the number of hospital admissions to increase 18% faster than population growth, and the aggregate number of inpatient days to increase 22% faster than population growth. Including both population growth and population aging, the total projected increase required for inpatient capacity is 72%. This is a base‐case, ceteris paribus analysis, and understanding how demand for inpatient services may change will require multiple perspectives. Increasing access to insurance, changing poverty rates, and changes in healthcare delivery and technology are other important factors. The present analysis provides a focused estimate of the influence upon demand for inpatient services due to expected changes in our population's age distribution.
The number of older people in the United States is expected to increase, due to the aging of the post‐World War II baby boomers.[1] For example, those aged 65 years are expected to number 88.5 million in 2050, more than double the number in 2010 of 40.2 million. This demographic shift has raised concerns about future hospital capacity, but the scope of the problem has not been quantified.[2]
A recent analysis calculated the number and length of emergency department visits expected to occur based on the aging of the US population.[3] One finding was that hospital admissions would increase 23% faster than population growth. However, this considered only hospitalizations originating in the emergency department and did not consider all‐source hospitalizations. We obtained data on all‐source hospitalizations and applied them to the US Census Bureau's demographic projections for the future through 2050. This provides a base‐case estimate for how inpatient demand would change if all other influences remained equal. The goal was to isolate the effect of population age makeup on inpatient requirements while holding other influences constant.
METHODS
We used the method of actuarial life table adjustment as described previously.[3] To calculate age‐specific hospitalization rates, we estimated age‐specific hospitalization frequencies (counts) in the United States for 2011 from the Nationwide Inpatient Sample (NIS).[4] This is a stratified probability sample of US community hospitals, defined as all nonfederal, short term, general, and other specialty hospitals, excluding hospital units of institutions. Veterans hospitals and other federal facilities, short‐term rehabilitation hospitals, long‐term non‐acute care hospitals, psychiatric hospitals, and alcoholism/chemical dependency treatment facilities were excluded from NIS 2011. Of hospitals in the sample, 21% are government (nonfederal) owned.
We converted age‐specific hospitalization frequencies derived from this sample into rates by dividing each stratum‐specific admission count by the 2011 population count in each age stratum from the US Census Bureau.[5] The Census Bureau provides detailed predictions of the US population through 2050. Births, deaths, and net international migration are projected for each birth cohort. Using 2011 as the origin, we applied baseline age‐specific hospitalization rates stratum‐wise to the general population expected by the Census Bureau in future years. This gave us stratum‐specific hospitalization frequencies for each future year. We summed these to arrive at the aggregate anticipated hospitalization frequency in each year. For our main outcome measure, we calculated the ratio of change in hospitalization frequency to change in population, comparing each future year to the 2011 baseline. We also calculated aggregate inpatient days, using the same data sources and methods. Our institutional review board exempted this study from review. We used Stata 13.0 (StataCorp, College Station, TX), and Microsoft Excel (Microsoft, Redmond, WA) for all analyses.
RESULTS
Baseline data are displayed in Figure 1. In 2011, there were 0.23 hospitalizations per US resident aged 0 to 4 years, and 0.01 per resident aged 5 to 9 years. From this age forward, hospitalization rates increased steadily with advancing age, reaching 0.63 per resident aged 90 to 94 years. Length of stay also was generally associated with age, though there was a peak among older children.
Projections through 2050 are shown in Table 1 and Figure 2. Table 1 displays the population projections of the US Census Bureau, which expects the US population to increase by 41% between now and 2050. Also shown in the table are our projections, which indicate that, all other things being equal, the annual number of inpatient admissions in the US will increase by 67%. The ratio of 67% to 41% is 1.18, meaning that the frequency of inpatient admissions will grow 18% more than population growth due to the aging of the population. The aggregate number of inpatient days will increase 22% more than population growth. Overall, inpatient capacity must expand by 72% to keep pace.
| Year | Population | Hospital Admissions | Aggregate Inpatient Days | Population: Ratio of Future Year to 2011 | Admissions: Ratio of Future Year to 2011 | Ratio of Admission Increase to Population Increase | Aggregate Inpatient Days: Ratio of Future Year to 2011 | Ratio of Increase in Inpatient Days to Population Increase |
|---|---|---|---|---|---|---|---|---|
| ||||||||
| 2011 | 311,591,917 | 38,560,751* | 177,501,515 | 1 | 1 | 1 | 1 | 1 |
| 2015 | 325,539,790 | 41,093,154 | 189,520,706 | 1.04 | 1.07 | 1.02 | 1.07 | 1.02 |
| 2020 | 341,386,665 | 44,196,669 | 205,205,962 | 1.10 | 1.15 | 1.05 | 1.16 | 1.06 |
| 2025 | 357,451,620 | 47,655,492 | 222,911,204 | 1.15 | 1.24 | 1.08 | 1.26 | 1.09 |
| 2030 | 373,503,674 | 51,365,441 | 241,852,384 | 1.20 | 1.33 | 1.11 | 1.36 | 1.14 |
| 2035 | 389,531,156 | 55,091,242 | 260,603,998 | 1.25 | 1.43 | 1.14 | 1.47 | 1.17 |
| 2040 | 405,655,295 | 58,524,016 | 277,530,732 | 1.30 | 1.52 | 1.17 | 1.56 | 1.20 |
| 2045 | 422,058,629 | 61,525,903 | 292,014,192 | 1.35 | 1.60 | 1.18 | 1.65 | 1.21 |
| 2050 | 439,010,253 | 64,249,181 | 304,945,179 | 1.41 | 1.67 | 1.18 | 1.72 | 1.22 |
DISCUSSION
Although US hospital capacity has fallen over the past 3 decades,[6, 7] our analysis suggests that demand for inpatient beds will increase 22% faster than population growth by 2050. The total projected demand increase is 72%, including that attributable to population growth and that attributable to population aging.
These are ceteris paribus projections, which reveal the changes in inpatient demand that would result if 2 conditions held: (1) the US Census Bureau's expectations for population makeup proved correct, and (2) age‐specific hospitalization rates and lengths of stay did not change. In reality, age‐specific hospitalization rates and lengths of stay could change. Examples of change drivers include epidemics, technology, and financial incentives provided by third‐party payers.[7] For example, if an epidemic of a new disease were to occur, age‐specific hospitalization rates could increase across all age groups. Our projections depict what would happen in the absence of any such change. This is useful because we do not know if changes in age‐specific hospitalization rates will occur, and whether there will be increases or decreases. Therefore, our projections should not be viewed as literal predictions, but rather as pieces of the puzzle, necessary but not sufficient elements of an understanding of what the future may hold for inpatient demand.
Clinicians, academics, and government agencies have an interest in understanding inpatient supply and demand on national and local levels. However, their ability to influence supply is limited by the fact that of all registered hospitals in the United States, only 22% are government owned.[1] As a result, decisions about hospital construction and closure are generally left to the free market.[6] Nonetheless, we bear responsibility for monitoring supply and demand, and government regulation of hospitals and reimbursement for inpatient care mean that the public is not entirely without influence. Thirty‐two percent of US residents have government‐issued health insurance.[8]
In the early 20th century, very little healthcare took place in the inpatient setting. However, by the 1970s, inpatient care accounted for a large part of healthcare, due largely to changes in technology and reimbursement. This trend reversed in the 1980s and 1990s, and hospitals closed.[7] In 1975, there were 5875 hospitals in the United States, and in 2000 there were 4915.[6] The number of staffed beds decreased from 942,000 to 826,000.[6] In parallel, likely due to changes in technology (ie, the nature of healthcare), total inpatient days in community hospitals decreased from 223 million in 1991 to 187 million in 2011.[9] On the other hand, increasing access to insurance under the Affordable Care Act could increase utilization, as seen when a 30% increase in hospital utilization occurred when people were enrolled in Oregon's Medicaid program.[10] Also, hospital utilization may increase if Medicare patients require more services.[11]
Actuarial life table analysis has been used to make forecasts related to healthcare supply and demand, though we are not aware of prior applications to the question of hospitalization. A prior study used actuarial life table adjustment to forecast demand for emergency department services.[3] These methods have also been used to forecast the influence of longevity upon healthcare expenditures[12, 13, 14] and to predict demand for specialty services.[15, 16] Of note, rather than reporting ratios of demand growth to population growth, another option would have been to derive a compound growth rate. We are not aware of a precedent for such methods in the prior published applications of actuarial life table analysis and felt that such inductive methods would complicate the interpretation of our results.
The main limitation of our investigation is its scope. We used actuarial life table adjustment to isolate the effect of population aging upon demand for inpatient hospitalizations. This method does not yield a comprehensive prediction of inpatient demand, but rather provides a robust estimate under the assumption that all other things remain equal. Another obvious limitation is that our analysis has a nationwide scope, and was not designed to account for variation from one locale to the next. However, these methods can be used by local health authorities.
CONCLUSIONS
The US Census Bureau expects the US population to increase by 41% over the next 4 decades, and the number of US residents aged 65 years to more than double. Our results indicate that, all other things being equal, this will cause the number of hospital admissions to increase 18% faster than population growth, and the aggregate number of inpatient days to increase 22% faster than population growth. Including both population growth and population aging, the total projected increase required for inpatient capacity is 72%. This is a base‐case, ceteris paribus analysis, and understanding how demand for inpatient services may change will require multiple perspectives. Increasing access to insurance, changing poverty rates, and changes in healthcare delivery and technology are other important factors. The present analysis provides a focused estimate of the influence upon demand for inpatient services due to expected changes in our population's age distribution.
- American Hospital Association. Fast facts on US hospitals, 2011. Available at: http://www.aha.org/research/rc/stat‐studies/fast‐facts.shtml. Accessed August 7, 2013.
- American Hospital Association. Cracks in the foundation: averting a crisis in America's hospitals. AHA 2002. Available at: http://www.aha.org/content/00–10/cracksreprint08‐02.pdf. Accessed August 4, 2013.
- , , , , . Population aging and emergency departments: visits will not increase, lengths‐of‐stay and hospitalizations will. Health Aff (Millwood). 2013;32(7):1306–1312.
- HCUP Nationwide Inpatient Sample (NIS). Healthcare Cost and Utilization Project (HCUP). 2011. Agency for Healthcare Research and Quality, Rockville, MD. Available at: http://www.hcup‐us.ahrq.gov/nisoverview.jsp. Accessed July 25, 2013.
- Bureau of the Census. Population Projections by Age, Sex, Race, and Hispanic Origin: July 1, 2000–2050. Washington, DC: The Bureau; 2008.
- , , , . Does U.S. hospital capacity need to be expanded? Health Aff (Millwood). 2003;22(6):40–54.
- . Decline in hospital utilization and cost inflation under managed care in California. JAMA. 1996;276(13):1060–1064.
- , , . Income, poverty, and health insurance coverage in the United States, 2011. US Census Bureau. Available at: http://www.census.gov/prod/2012pubs/p60–243.pdf. Published September 2012. Accessed August 7, 2013.
- American Hospital Association. Trendwatch. Table 3.1: trends in inpatient utilization in community hospitals, 1991–2011. Available at: http://www.aha.org/research/reports/tw/chartbook/2013/table3‐1.pdf. Accessed November 9, 2013.
- , , , et al. The Oregon health insurance experiment: evidence from the first year. Q J Econ. 2012;127(3):1057–1106.
- American Hospital Association. Trendwatch. Are Medicare patients getting sicker? Available at: http://www.aha.org/research/reports/tw/12dec‐tw‐ptacuity.pdf. Accessed November 9, 2013.
- , , . Longevity and Medicare expenditures. N Engl J Med. 1995;332(15):999–1003.
- , . The aging of America. Impact on health care costs. JAMA. 1990;263(17):2335–2340.
- , . The effect of longevity on spending for acute and long‐term care. N Engl J Med. 2000;342(19):1409–1415.
- , , , . Demographics and cardiology, 1950–2050. J Am Coll Cardiol. 2000;35(4):1067–1081.
- , , , . Population‐based analysis of inpatient vascular procedures and predicting future workload and implications for training. J Vasc Surg. 2012;55(5):1394–1399; discussion 1399–1400.
- American Hospital Association. Fast facts on US hospitals, 2011. Available at: http://www.aha.org/research/rc/stat‐studies/fast‐facts.shtml. Accessed August 7, 2013.
- American Hospital Association. Cracks in the foundation: averting a crisis in America's hospitals. AHA 2002. Available at: http://www.aha.org/content/00–10/cracksreprint08‐02.pdf. Accessed August 4, 2013.
- , , , , . Population aging and emergency departments: visits will not increase, lengths‐of‐stay and hospitalizations will. Health Aff (Millwood). 2013;32(7):1306–1312.
- HCUP Nationwide Inpatient Sample (NIS). Healthcare Cost and Utilization Project (HCUP). 2011. Agency for Healthcare Research and Quality, Rockville, MD. Available at: http://www.hcup‐us.ahrq.gov/nisoverview.jsp. Accessed July 25, 2013.
- Bureau of the Census. Population Projections by Age, Sex, Race, and Hispanic Origin: July 1, 2000–2050. Washington, DC: The Bureau; 2008.
- , , , . Does U.S. hospital capacity need to be expanded? Health Aff (Millwood). 2003;22(6):40–54.
- . Decline in hospital utilization and cost inflation under managed care in California. JAMA. 1996;276(13):1060–1064.
- , , . Income, poverty, and health insurance coverage in the United States, 2011. US Census Bureau. Available at: http://www.census.gov/prod/2012pubs/p60–243.pdf. Published September 2012. Accessed August 7, 2013.
- American Hospital Association. Trendwatch. Table 3.1: trends in inpatient utilization in community hospitals, 1991–2011. Available at: http://www.aha.org/research/reports/tw/chartbook/2013/table3‐1.pdf. Accessed November 9, 2013.
- , , , et al. The Oregon health insurance experiment: evidence from the first year. Q J Econ. 2012;127(3):1057–1106.
- American Hospital Association. Trendwatch. Are Medicare patients getting sicker? Available at: http://www.aha.org/research/reports/tw/12dec‐tw‐ptacuity.pdf. Accessed November 9, 2013.
- , , . Longevity and Medicare expenditures. N Engl J Med. 1995;332(15):999–1003.
- , . The aging of America. Impact on health care costs. JAMA. 1990;263(17):2335–2340.
- , . The effect of longevity on spending for acute and long‐term care. N Engl J Med. 2000;342(19):1409–1415.
- , , , . Demographics and cardiology, 1950–2050. J Am Coll Cardiol. 2000;35(4):1067–1081.
- , , , . Population‐based analysis of inpatient vascular procedures and predicting future workload and implications for training. J Vasc Surg. 2012;55(5):1394–1399; discussion 1399–1400.
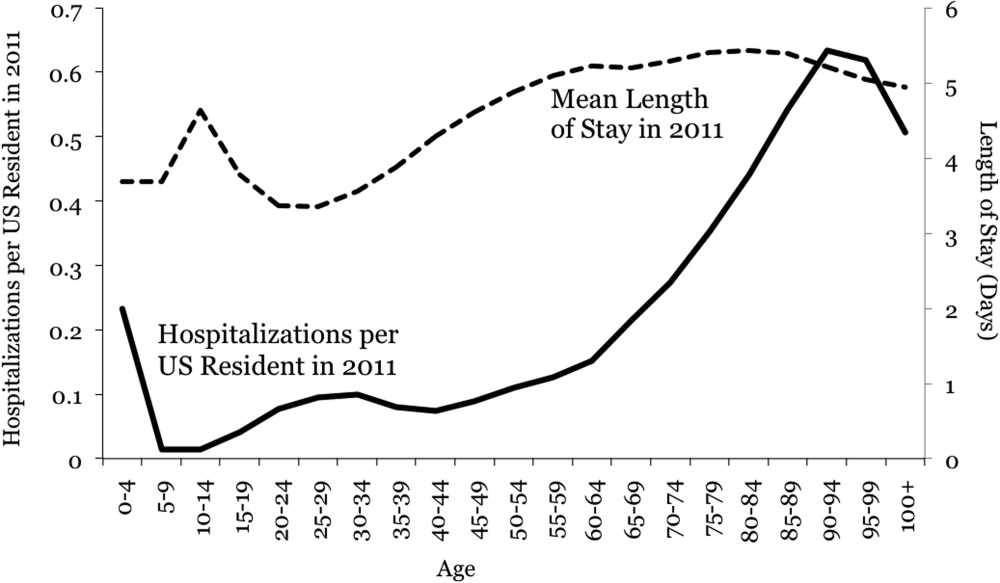
AAB Therapy Risks in Orthopedic Surgery
Patients presenting for surgery with angiotensin axis blockade (AAB) from therapy with either angiotensin‐converting enzyme inhibitors (ACEIs) or angiotensin receptor blockers (ARBs) experience an increased incidence of perioperative hypotension.[1, 2, 3, 4] Debate continues as to whether this hypotension results in any significant clinical sequelae. Some authors found that the use of an ACEI decreased the incidence of acute kidney injury (AKI),[5] mortality, and septicemia in cardiac and vascular surgical patients.[6] However, others found that in vascular and cardiac surgery there is increased mortality[7] as well as increased incidence of postoperative AKI.[8, 9, 10] A retrospective study of 10,000 coronary artery bypass graft patients found that ACEI was associated with increased inotropic support, AKI, mortality, and new onset atrial fibrillation.[11] In a meta‐analysis of 69,000 cardiothoracic surgery patients, the use of ACEIs/ARBs was associated with an increase in AKI and mortality.[12] AKI has also been demonstrated after lung resection surgery in patients receiving preoperative therapy with an ARB.[13]
Studies on noncardiac general surgery patients demonstrate that the use of AAB results in postinduction hypotension, but they fail to show an increased incidence in postoperative AKI.[14, 15] We propose, however, that major orthopedic surgery patients are a specific surgical cohort, like cardiac, vascular, and lung, who can develop operative hypotension and postoperative AKI when AAB is taken on the morning of surgery. To address this question we performed a retrospective study of 1154 patients undergoing either spinal fusion, total knee arthroplasty (TKA), or total hip arthroplasty (THA) during the 2010 calendar year in our academic medical center. We measured the incidence of postanesthesia induction hypotension, intraoperative hypotension, and postoperative AKI as it relates to the administration of AAB preoperatively.
MATERIALS AND METHODS
This study was a retrospective, observational investigation at a single, large academic hospital. The study design for chart review was approved by the institutional review board prior to data collection. Informed patient consent was not required for this retrospective study.
Patient Selection
We retrospectively reviewed the computerized chart and operating room electronic medical records of all patients who underwent elective major orthopedic surgery in the 2010 calendar year. We chose vertebral spine fusion, TKA, and THA as the 3 surgeries to represent major elective orthopedic surgery. Electronic query of the hospital database identified 1154 patients having undergone these surgeries in the year 2010. Nine hundred twenty‐two patients met inclusion criteria: 21 years old and evaluated in the preanesthesia clinic with documented vital signs and clearly defined preoperative medication recommendations. The policy in the preanesthesia clinic was to recommend taking the ACEI and ARB on the morning of surgery. All 922 patients were included in the analysis of the outcomes for induction hypotension and intraoperative hypotension. Of the 922 patients, 798 had the documented preoperative and postoperative creatinine values needed to define AKI. Therefore, only these 798 patients were included in the AKI outcome analysis. During the time of the study it was the practice at our medical center that all such surgeries were performed under general anesthesia.
Data Collection
Preanesthesia records were reviewed for patient demographics including age, body mass index (BMI), baseline blood pressure, diabetes mellitus (DM), coronary artery disease (CAD), hypertension (HTN), and congestive heart failure (CHF), as well as for therapy with ACEI or ARB, diuretics, ‐blockers, nonsteroidal anti‐inflammatory drugs (NSAIDs), and calcium channel blockers (CCB). The 4 statistically and clinically significant comorbidities were looked at individually as well as added together for a total sum of weighted comorbidity risk factors. The Anesthesia Electronic Record (Surginet Anesthesia, Kansas City, MO) was reviewed for each corresponding patient to determine the general anesthesia induction agent used and to assess the postinduction and intraoperative systolic blood pressures. Blood pressure was determined using an automated blood pressure cuff and automatically recorded at intervals of 5 minutes or less. Further, ephedrine, phenylephrine and vasopressin doses, estimated blood loss, blood transfusion requirements, and intravenous fluid administration (colloid and crystalloid) were noted. Preoperative (<30 days) and postoperative (within 24 hours after surgery) serum creatinine and hematocrit values were also recorded.
Outcome Measures
The primary outcome measures studied were:
- Postinduction hypotension (systolic blood pressure [SBP] 80 mm Hg for5 minutes) occurring within 30 minutes after anesthesia induction but before surgical incision.[16]
- Intraoperative hypotension (SBP 80 mm Hg for 10 minutes) occurring after surgical incision.[16]
- Postoperative AKI defined as an increase in serum creatinine 0.3 mg/dL or an increase of 50% from preoperative creatinine (Kidney Disease: Improving Global Outcomes Clinical Practice Guideline for Acute Kidney Injury)[17] within 24 hours postoperatively.
The secondary outcome measures were:
- Hospital length of stay (LOS).
- Two‐year mortality associated with the development of AKI.
Statistical Analysis
Categorical variables were summarized with frequencies and percentages, whereas continuous variables were summarized with means, standard deviations, medians, and quartiles. A [2] test or a Wilcoxon rank sum test was used to determine differences in preoperative and intraoperative characteristics between those patients with AAB and those patients without AAB. Logistic regression was used to determine the association between the main outcome variables (hypotension at anesthetic induction, hypotension during the operative procedure, and postoperative AKI) and the primary independent variable, AAB, as well as other preoperative and intraoperative characteristics. The significance and magnitude of the associations were quantified with percentages and odds ratios. Exact logistic regression was used as necessary when expected cell counts were too small for the usual asymptotic logistic regression to be valid. The statistically significant (P<0.05) variables resulting from this bivariate analysis as well as important clinically significant variables with known associations with the outcome variables were included as covariates in a multivariable logistic regression model for each outcome variable with AAB. By adjusting for these covariates, any potential and unwanted variation resulting from differences between the AAB groups in other preoperative or postoperative characteristics is removed from the association between each outcome variable and AAB. Variance inflation factor and tolerance statistics were used to test for multicollinearity between all independent variables before including them in the final models. The Hosmer and Lemeshow goodness‐of‐fit test was used to assess the fit of the final models. Logistic regression was used to test the association of AKI with mortality, whereas a Wilcoxon rank sum test was used to test the difference in mean/median LOS between AKI and non‐AKI groups.
RESULTS
Preoperative, Surgical, and Anesthesia Data
A total of 922 patients met inclusion criteria, of which 343 (37%) were receiving AAB with either an ACEI or ARB preoperatively. Preoperative characteristics are documented in Table 1. Patients receiving AAB were older (63.010.8 vs 57.313.9 years) and had a higher BMI (34.67.3 vs 31.97.7) than patients not receiving this therapy. They were also more likely to be receiving diuretics, ‐blockers, and CCBs as well as have DM, CHF, CAD, and HTN. These characteristics were included as covariates in a multivariable logistic regression model so that any confounding resulting differences caused by these variables were removed from the association between each outcome variable and AAB use. There was no difference in baseline SBP or diastolic blood pressures. There was no difference in the use of NSAIDs. Patients receiving AAB underwent a higher percentage of TKAs (56.0% vs 44.2%) and lower percentage of spine fusions (16.6% vs 24.4%) and THAs (27.4% vs 31.4%). Propofol was the most commonly used general anesthetic induction agent (78%). Anesthetic induction agent usage was not different across the groups.
| AAB (N=343) | Non‐AAB (N=579) | P Value* | |
|---|---|---|---|
| |||
| Patient demographics | |||
| Age (y), meanSD | 63.010.8 | 57.313.9 | <0.001 |
| BMI (kg/m2), meanSD | 34.67.3 | 31.87.7 | <0.001 |
| Baseline systolic BP (mm Hg), meanSD | 135.016.8 | 130.819.8 | 0.339 |
| Baseline diastolic BP (mm Hg), meanSD | 74.514.5 | 75.311.8 | 0.798 |
| Medications | |||
| Diuretic, % | 53.6 | 18.5 | <0.001 |
| ‐Blocker, % | 39.4 | 22.4 | <0.001 |
| Calcium channel blocker, % | 23.3 | 9.9 | <0.001 |
| Nonsteroidal anti‐inflammatory drug, % | 36.1 | 35.7 | 0.901 |
| Comorbidities | |||
| Diabetes mellitus, % | 32.9 | 9.5 | <0.001 |
| Congestive heart failure, % | 2.9 | 0.4 | 0.001 |
| Coronary artery disease, % | 20.1 | 9.5 | <0.001 |
| Hypertension, % | 95.3 | 36.8 | <0.001 |
| Total comorbidities, meanSD | 1.50.7 | 0.50.7 | <0.001 |
| Procedure type | |||
| Spinal fusions, % | 16.6 | 24.4 | 0.001 |
| Total knee arthroplasty, % | 56.0 | 44.2 | 0.001 |
| Total hip arthroplasty, % | 27.4 | 31.4 | 0.001 |
| Induction agents | |||
| Etomidate, % | 14.9 | 11.9 | 0.199 |
| Propofol, % | 77.6 | 78.3 | 0.801 |
| Methohexital, % | 7.3 | 9.2 | 0.329 |
| Other (gas, ketamine), % | 0.3 | 1.2 | 0.270 |
| Hematocrit | |||
| Preoperative (%), meanSD | 40.53.7 | 40.23.8 | 0.150 |
| Postoperative (%), meanSD | 34.04.6 | 33.74.3 | 0.511 |
| % Change, meanSD | 15.38.5 | 16.88.9 | 0.036 |
| Creatinine | |||
| Preoperative (mg/dL), meanSD | 0.960.41 | 0.850.23 | <0.001 |
| Postoperative (mg/dL), meanSD | 0.960.42 | 0.810.23 | <0.001 |
| % change, meanSD | 1.4829.22 | 4.1414.18 | 0.235 |
| Fluids | |||
| Estimated blood loss (mL), meanSD | 301.2340.0 | 356.9482.7 | 0.125 |
| Crystalloid (mL), meanSD | 2584.41401.6 | 2765.21487.2 | 0.036 |
| Colloid (mL), meanSD | 124.3322.4 | 151.0553.5 | 0.810 |
| Packed red blood cells (mL), meanSD | 78.5323.1 | 99.5452.0 | 0.613 |
| Vasopressors | |||
| Ephedrine (mg), meanSD | 11.715.0 | 8.013.0 | <0.001 |
| Phenylephrine (g), meanSD | 615.72210.9 | 687.13905.3 | 0.002 |
| Vasopressin (U), meanSD | 0.351.90 | 0.080.90 | <0.001 |
| Outcomes | |||
| Induction hypotension, % | 12.2 | 1.93 | 0.005 |
| Intraoperative hypotension, % | 26.0 | 20.9 | 0.078 |
| Acute kidney injury, % | 8.3 | 1.7 | <0.001 |
Estimated blood loss for the procedures was similar between the 2 groups (301.2340 vs 356.9482.7 mL) and similar colloid and packed red cell administration occurred. However, patients receiving AAB were administered less crystalloid infusion (2584.41401.6 vs 2765.21487.2 mL, P=0.036) and received less phenylephrine but higher ephedrine and vasopressin dosages as a group. Patients in both groups had similar preoperative and postoperative hematocrit concentrations. Average preoperative serum creatinine was higher in the AAB group than in the non‐AAB group (0.960.41 vs 0.850.23, P<0.001) and remained so postoperatively (0.960.42 vs 0.810.23, P<0.001).
Primary and Secondary Outcome Measures
Postinduction Hypotension
Therapy with AAB was associated with a greater incidence of postinduction hypotension (12.2% vs 6.7%, P=0.005). Using a multivariate logistic regression model adjusting for the effects of age, BMI, antihypertensive medications, comorbidities, and anesthetic induction agents, the use of AAB had a greater odds ratio (OR) of 1.93 (95% confidence interval [CI]: 1.10‐3.41, P=0.023) for developing postinduction hypotension (Table 2). A higher BMI had a lower OR for postinduction hypotension.
| Comparison | Hypotension at Induction, OR (95% CI), P Value | Intraoperative Hypotension, OR (95% CI), P Value | Acute Kidney Injury, OR (95% CI), P Value |
|---|---|---|---|
| |||
| AAB vs none, unadjusted | 1.93 (1.223.06), 0.005 | 1.33 (0.971.82), 0.078 | 5.40 (2.4112.06), <0.001 |
| AAB vs none, adjusted for covariates | 1.93 (1.103.41), 0.023 | 1.30 (0.851.97), 0.226 | 2.68 (1.086.69), 0.034 |
| AAB vs none, adjusted for covariates+hypotension at induction | N/A | N/A | 2.51 (1.06.32), 0.051 |
| AAB vs none, adjusted for covariates+intraoperative hypotension | N/A | N/A | 2.66 (1.066.64), 0.037 |
| AAB vs none, adjusted for covariates+any hypotension | N/A | N/A | 2.60 (1.046.51), 0.042 |
Postincision (Intraoperative) Hypotension
The incidence of postincision, intraoperative hypotension in patients receiving AAB (26.0%) was not statistically different (P=0.078) from those not receiving these agents (20.9%). Multivariate logistic regression demonstrated that preoperative hypertension (OR: 1.73, 95% CI: 1.05‐2.85, P=0.031) and THA were each independent risk factors for intraoperative hypotension. The other comorbidities of DM, CHF, CAD, and the individual antihypertensive agents were not found to have a strong influence on the outcome of intraoperative hypotension. The odds ratio of developing intraoperative hypotension during the procedure in patients receiving AAB was not statistically significant (OR: 1.30, 95% CI: 0.85‐1.97, P=0.226) from those not receiving this therapy preoperatively (Table 2).
AKI
There was a significantly higher incidence of AKI (26/313 [8.3%] vs 8/485 [1.7%], P<0.001) in patients receiving preoperative AAB. No patients required renal replacement therapy. Of those patients who developed postoperative AKI, 73% of the AAB group and 75% of the non‐AAB group had a normal glomerular filtration rate (GFR) (mL/min/1.73 m2, GFR >90 prior to surgery. Of both the AAB and non‐AAB groups, 20% to 25% were in stage 2 (GFR 6089) chronic kidney disease (CKD)[18] preoperatively. Only 2 patients in the AAB group began in stage 3 (GFR 3059) CKD. Fifty percent of both groups went from stage 1 kidney function to stage 3. For all others who developed AKI, the GFR rose by a single stage.
Multivariate logistic regression, controlling for statistically significant and clinically significant variables, demonstrated that AAB administered preoperatively was independently associated with a greater OR of 2.68 (95% CI: 1.08‐6.69, P=0.034) for developing AKI postoperatively than if AAB was not taken (Table 2). Higher BMI was a further independent risk factor for postoperative AKI; a 5‐unit increase in BMI revealed an OR of 1.58 (95% CI: 1.25‐1.99, P<0.001) for experiencing postoperative AKI. Although the AAB group had statistically significantly more comorbidities, in the final analysis only the presence of CAD trended to an association with the development of AKI (OR: 2.37, 95% CI: 1.005.60, P=0.050).
We wished to determine whether the increased risk of AKI associated with AAB therapy was explained by the associated postinduction or postincision, intraoperative hypotension experienced by the patients or independent of these effects. To do so we accounted for the development of either postinduction or intraoperative hypotension as a possible confounder in the multivariate analysis for the development of AKI (Table 2). Postinduction hypotension alone was not statistically associated with AKI (OR 2.04, 95% CI: 0.70‐6.0, P=0.193). However, intraoperative hypotension was found to be an independent risk factor for the development of AKI (OR: 2.62, 95% CI: 1.17‐5.84, P=0.019). When eliminating the effect of this intraoperative hypotension as a confounder, patients on AAB therapy continued to have a 2.66 OR for the development of AKI postoperatively (95% CI: 1.06‐6.64, P=0.037), independent of the development of intraoperative hypotension. When eliminating the effect of both postinduction and intraoperative hypotension (all hypotension Table 2), AAB therapy continued to have a statistically significant independent effect (OR: 2.60, 95% CI: 1.046.51, P=0.042) on developing postoperative AKI.
As secondary outcome measures, we looked at the development of AKI and its relationship to hospital LOS and mortality. The development of AKI was associated with a significantly greater mean length of hospital stay (5.76 days vs 3.28 days, P<0.001). Although 2‐year mortality was higher, 5.9% in the AKI group compared to 2.4% in the non‐AAB group, it was not statistically significant (P=0.211).
DISCUSSION
In this retrospective review of 922 patients presenting for major orthopedic surgery, we found that 343(37%) were receiving therapy with either ACEIs or ARBs. In such patients, we demonstrated a higher incidence of postinduction hypotension and an increased incidence of AKI. We further demonstrated that the development of AKI associated with AAB therapy was independent of hypotension occurring either postinduction or intraoperatively after incision.
Postinduction hypotension in patients receiving AAB was demonstrated to be 12.2% compared to 7.7% in patients not receiving this therapy. Hypotension after general anesthesia induction in patients receiving AAB is widely reported,[1, 14, 15] and ranges from 22% to 100%[19, 20] based on varying definitions of what constitutes hypotension. We chose an absolute value of a systolic blood pressure of 80 mm Hg occurring for 5 minutes as constituting significant hypotension.[16] Monk et al. reported an increased one year postnon‐cardiac surgery mortality risk of 1.036 times per minute of intraoperative hypotension, defining hypotension as a systolic blood pressure of <80 mm Hg.[21]
We further demonstrated that AAB therapy resulted in an 8.3% incidence of AKI versus 1.7% in non‐AAB patients (P<0.001). AKI was defined as an increase in serum creatinine of 0.3 mg/dL or a 50% increase in creatinine when pre‐ and postoperative values were compared.[17] A number of other investigators have identified AKI associated with AAB use in patients undergoing cardiac,[6, 11] vascular,[7, 10] and lung[13] surgery. Similarly, in the present study, in orthopedic patients, AAB remained a significant risk factor for developing AKI (OR: 2.68, P=0.034) independent of patient comorbidities and adjunct therapy (Table 2).
ACEIs and ARBs are prescribed to treat HTN, CHF, and improve renal function in diabetic and proteinuric nephropathy.[22] AAB therapy is prescribed for nephropathy because these medications decrease glomerular pressure by selective inhibition of angiotensin II mediated vasoconstriction of the efferent glomerular arteriole.[23] Normally, this is beneficial to patients and is associated with a decrease in serum creatinine concentration. However, during hypotension, when there is decreased renal perfusion, further decreases in intraglomerular pressure may occur, precipitating renal failure.[23] In addition, other factors may contribute to the development of AKI, as AAB has both tissue and systemic effects that extend beyond simply dilating the efferent glomerular arteriole. These include effects on the sympathetic nervous system, oxidative stress, and altering the release and synthesis of vasodilators such as bradykinin, nitric oxide, and prostacyclins[24] as well as effects through the release of aldosterone and arginine‐vasopressin.[25] These other factors might help explain the present study's findings that, when eliminating the effect of both postinduction and intraoperative hypotension, AAB therapy continued to have a statistically significant independent effect (OR: 2.60, 95% CI: 1.046.51, P=0.042) on developing postoperative AKI.
Although we demonstrated an association of AAB therapy with the development of hypotension after induction, we demonstrated only a trend in the development of postincisional, intraoperative hypotension (P=0.078). We defined intraoperative hypotension as a systolic BP <80 mm Hg for 10 minutes occurring after skin incision.[16] One must take into consideration, however, that a significant number of AAB patients were hypotensive during induction and received higher doses of ephedrine and vasopressin during the operative period. These patients may have been rescued from intraoperative hypotension by receiving vasopressor treatment at the outset. We did find that intraoperative hypotension was a significant, independent risk factor for AKI (OR: 2.62, P=0.019).
We looked further at the consequences of developing AKI. Patients who developed AKI had a significant greater mean length of hospital stay (5.76 days vs 3.28 days, P<0.001), which is consistent with other investigators' findings.[25, 26, 27] Although 2‐year mortality was higher at 5.88% in the AKI group compared to 2.38% in the non‐AAB group, this was not statistically significant (P=0.211). Other studies have shown that the development of AKI results in greater mortality.[26, 28]
The American College of Physicians (ACP) recommendations as of 2013 regarding the use of ACEIs and ARBs preoperatively is: uncertain, continue with caution, avoid hypovolemia. Potential for hypotension with induction of anesthesia and increased vasoconstrictor requirements and decreased responsiveness to pressors.[29] The ACP acknowledges that preoperative ACEIs and ARBs have the potential for postinduction hypotension and increased requirements for vasopressors. We have implemented recommendations at our preoperative anesthesia clinic to hold ACEIs and ARBs on the morning of surgery in patients with controlled blood pressure scheduled for spine fusion, and hip and knee arthroplasties. In accordance with ACP guidelines, other antihypertensives such as ‐blockers, calcium channel blockers, nitrates, and sympatholytics should be continued preoperatively and can be used perioperatively.
Limitations of the Study
There are several limitations to our study. This was a retrospective analysis over a fixed time period in one academic institution. Further, because of the retrospective nature, anesthesia and intraoperative (fluid and vasoconstrictor) management was not standardized. The definition of hypotension (SBP 80 mm Hg for 5 minutes after induction and 10 minutes after incision) may have been too stringent, so that more subtle decreases in blood pressure that could have impacted AKI might not have been captured to show statistical significance. Thus, our finding, that the development of AKI associated with preoperative AAB therapy may be independent of the occurrence of hypotension, must be interpreted with this in mind.
CONCLUSIONS
Patients who receive preoperative ACEI or ARB therapy and undergo major orthopedic surgery such as spinal fusion, and hip or knee arthroplasties experience a higher incidence of postinduction hypotension and AKI than those not receiving such therapy. The development of AKI in such patients is associated with a significantly prolonged length of hospital stay. Our findings suggest an association between preoperative ACEI/ARB use and moderate kidney injury following major orthopedic surgeries. However, a prospective, multicentered, randomized trial needs to be performed to confirm that withdrawal of AAB therapy preoperatively will decrease the incidence of AKI in patients undergoing major orthopedic procedures under general anesthesia. Future studies also need to determine the optimal time duration of withholding AAB therapy and the consequences on cardiac outcomes.
ACKNOWLEDGMENTS
Disclosures: Presented at the Society of Hospital Medicine National Meeting, May 18, 2013, National Harbor, Maryland; and the Society of General Internal Medicine Mid‐Atlantic Regional Meeting, March 1, 2013, Philadelphia, Pennsylvania. The authors report no conflicts of interest.
- . Management of hypotension associated with angiotensin‐axis blockade and general anesthesia administration. J Cardiothorac Vasc Anesth. 2013;27:156–167.
- , , , , , . Clinical consequences of withholding versus administering renin‐angiotensin‐aldosterone system antagonists in the preoperative period. J Hosp Med. 2008;3:319–325.
- , , , et al. Influence of chronic angiotensin‐converting enzyme inhibition on anesthetic induction. Anesthesiology. 1994;81(2):299–307.
- , , , , . Pressor responses to tracheal intubation after sublingual captopril. A pilot study. Anaesthesia. 1990;45(3):243–245.
- , , , , , , . Preoperative angiotensin‐converting enzyme inhibitors and acute kidney injury after coronary artery bypass grafting. Ann Thorac Surg. 2008;86(4):1160–1165.
- , , , et al. The effects of preoperative renin‐angiotensin system inhibitors on outcomes in patients undergoing cardiac surgery. J Cardiothorac Vasc Anesth. 2013;27(4):703–709.
- , , , . Renin‐angiotensin blockade is associated with increased mortality after vascular surgery. Can J Anaesth. 2010;57:736–744.
- , . , et al. Preoperative use of angiotensin‐converting enzyme inhibitors/angiotensin receptor blockers is associated with increased risk for acute kidney injury after cardiovascular surgery. Clin J Am Soc Nephrol. 2008;3(5):1266–1273.
- , , , et al.; TRIBE‐AKI Consortium. Preoperative angiotensin‐converting enzyme inhibitors and angiotensin receptor blocker use and acute kidney injury in patients undergoing cardiac surgery. Nephrol Dial Transplant. 2013;28(11):2787–2799.
- , , , et al. The chronic inhibition of angiotensin‐converting enzyme impairs postoperative renal function. Anesth Analg. 2001;93(5):1111–1115.
- , , , et al. Effects of angiotensin‐converting enzyme inhibitor therapy on clinical outcome in patients undergoing coronary artery bypass grafting. J Am Coll Cardiol. 2009;54:1778–1784.
- , , , , , . Acute kidney injury and death associated with renin angiotensin system blockade in cardiothoracic surgery: a meta‐analysis of observational studies. Am J Kidney Dis. 2013;63(6):1077–1086.
- , , . Acute kidney injury after lung resection surgery: incidence and perioperative risk factors. Anesth Analg. 2012;114:1256–1262.
- , , , et al. Angiotensin system inhibitors in a general surgical population. Anesth Analg. 2005;100:636–644, table of contents.
- , , , , . Chronic angiotensin‐converting enzyme inhibitor or angiotensin receptor blocker therapy combined with diuretic therapy is associated with increased episodes of hypotension in noncardiac surgery. J Cardiothorac Vasc Anesth. 2008;22:180–186.
- , , , , . Hemodynamic impact of dexmedetomidine administration in 15,656 noncardiac surgical cases. J Clin Anesth. 2012;24:212–220.
- Kidney Disease: Improving Global Outcomes (KDIGO). Acute Kidney Injury Work Group. KDIGO clinical practice guideline for acute kidney injury. Kidney Int Suppl. 2012;2:8.
- Kidney Disease: Improving Global Outcomes (KDIGO). Clincal practice guidelines for evaluation and management of chronic kidney disease. Kidney Int Suppl. 2013;3(1):8.
- , . Hemodynamic effects of anesthesia in patients with ischemic heart failure chronically treated with angiotensin‐converting enzyme inhibitors. Anesth Analg. 1997;84:945–949.
- , , , , . The hemodynamic effects of anesthetic induction in vascular surgical patients chronically treated with angiotensin II receptor antagonists. Anesth Analg. 1999;89:1388–1392.
- , , , . Anesthetic management and one‐year mortality after noncardiac surgery. Anesth Analg. 2005;100:4–10.
- , , , , . Use of angiotensin‐converting enzyme inhibitors and angiotensin receptor blockers in clinical practice. Expert Rev Cardiovasc Ther. 2012;10:159–166.
- , . An overview of drug‐induced acute kidney injury. Crit Care Med. 2008;36:S216–S223.
- , , , et al. Improvement of endothelial function by chronic angiotensin‐converting enzyme inhibition in heart failure: role of nitric oxide, prostanoids, oxidant stress, and bradykinin. Circulation. 2000;102:351–356.
- , , , . Renin‐angiotensin system antagonists in the perioperative setting: clinical consequences and recommendations for practice. Postgrad Med J. 2011;87:472–481.
- , , , , . Early postoperative statin therapy is associated with a lower incidence of acute kidney injury after cardiac surgery. J Cardiothorac Vasc Anesth. 2010;24:913–920.
- , , , , . Improved survival in acute kidney injury after cardiac surgery. Am J Kidney Dis. 2007;50:703–711.
- , , , . Determinants of postoperative acute kidney injury. Crit Care. 2009;13:R79.
- American College of Physicians. ACP Smart Medicine: Perioperative Medication Management. Tables: Perioperative Cardiovascular Medication Management. http://smartmedicine.acponline.org/content.aspx?gbosID=336. Accessed January 19, 2014.
Patients presenting for surgery with angiotensin axis blockade (AAB) from therapy with either angiotensin‐converting enzyme inhibitors (ACEIs) or angiotensin receptor blockers (ARBs) experience an increased incidence of perioperative hypotension.[1, 2, 3, 4] Debate continues as to whether this hypotension results in any significant clinical sequelae. Some authors found that the use of an ACEI decreased the incidence of acute kidney injury (AKI),[5] mortality, and septicemia in cardiac and vascular surgical patients.[6] However, others found that in vascular and cardiac surgery there is increased mortality[7] as well as increased incidence of postoperative AKI.[8, 9, 10] A retrospective study of 10,000 coronary artery bypass graft patients found that ACEI was associated with increased inotropic support, AKI, mortality, and new onset atrial fibrillation.[11] In a meta‐analysis of 69,000 cardiothoracic surgery patients, the use of ACEIs/ARBs was associated with an increase in AKI and mortality.[12] AKI has also been demonstrated after lung resection surgery in patients receiving preoperative therapy with an ARB.[13]
Studies on noncardiac general surgery patients demonstrate that the use of AAB results in postinduction hypotension, but they fail to show an increased incidence in postoperative AKI.[14, 15] We propose, however, that major orthopedic surgery patients are a specific surgical cohort, like cardiac, vascular, and lung, who can develop operative hypotension and postoperative AKI when AAB is taken on the morning of surgery. To address this question we performed a retrospective study of 1154 patients undergoing either spinal fusion, total knee arthroplasty (TKA), or total hip arthroplasty (THA) during the 2010 calendar year in our academic medical center. We measured the incidence of postanesthesia induction hypotension, intraoperative hypotension, and postoperative AKI as it relates to the administration of AAB preoperatively.
MATERIALS AND METHODS
This study was a retrospective, observational investigation at a single, large academic hospital. The study design for chart review was approved by the institutional review board prior to data collection. Informed patient consent was not required for this retrospective study.
Patient Selection
We retrospectively reviewed the computerized chart and operating room electronic medical records of all patients who underwent elective major orthopedic surgery in the 2010 calendar year. We chose vertebral spine fusion, TKA, and THA as the 3 surgeries to represent major elective orthopedic surgery. Electronic query of the hospital database identified 1154 patients having undergone these surgeries in the year 2010. Nine hundred twenty‐two patients met inclusion criteria: 21 years old and evaluated in the preanesthesia clinic with documented vital signs and clearly defined preoperative medication recommendations. The policy in the preanesthesia clinic was to recommend taking the ACEI and ARB on the morning of surgery. All 922 patients were included in the analysis of the outcomes for induction hypotension and intraoperative hypotension. Of the 922 patients, 798 had the documented preoperative and postoperative creatinine values needed to define AKI. Therefore, only these 798 patients were included in the AKI outcome analysis. During the time of the study it was the practice at our medical center that all such surgeries were performed under general anesthesia.
Data Collection
Preanesthesia records were reviewed for patient demographics including age, body mass index (BMI), baseline blood pressure, diabetes mellitus (DM), coronary artery disease (CAD), hypertension (HTN), and congestive heart failure (CHF), as well as for therapy with ACEI or ARB, diuretics, ‐blockers, nonsteroidal anti‐inflammatory drugs (NSAIDs), and calcium channel blockers (CCB). The 4 statistically and clinically significant comorbidities were looked at individually as well as added together for a total sum of weighted comorbidity risk factors. The Anesthesia Electronic Record (Surginet Anesthesia, Kansas City, MO) was reviewed for each corresponding patient to determine the general anesthesia induction agent used and to assess the postinduction and intraoperative systolic blood pressures. Blood pressure was determined using an automated blood pressure cuff and automatically recorded at intervals of 5 minutes or less. Further, ephedrine, phenylephrine and vasopressin doses, estimated blood loss, blood transfusion requirements, and intravenous fluid administration (colloid and crystalloid) were noted. Preoperative (<30 days) and postoperative (within 24 hours after surgery) serum creatinine and hematocrit values were also recorded.
Outcome Measures
The primary outcome measures studied were:
- Postinduction hypotension (systolic blood pressure [SBP] 80 mm Hg for5 minutes) occurring within 30 minutes after anesthesia induction but before surgical incision.[16]
- Intraoperative hypotension (SBP 80 mm Hg for 10 minutes) occurring after surgical incision.[16]
- Postoperative AKI defined as an increase in serum creatinine 0.3 mg/dL or an increase of 50% from preoperative creatinine (Kidney Disease: Improving Global Outcomes Clinical Practice Guideline for Acute Kidney Injury)[17] within 24 hours postoperatively.
The secondary outcome measures were:
- Hospital length of stay (LOS).
- Two‐year mortality associated with the development of AKI.
Statistical Analysis
Categorical variables were summarized with frequencies and percentages, whereas continuous variables were summarized with means, standard deviations, medians, and quartiles. A [2] test or a Wilcoxon rank sum test was used to determine differences in preoperative and intraoperative characteristics between those patients with AAB and those patients without AAB. Logistic regression was used to determine the association between the main outcome variables (hypotension at anesthetic induction, hypotension during the operative procedure, and postoperative AKI) and the primary independent variable, AAB, as well as other preoperative and intraoperative characteristics. The significance and magnitude of the associations were quantified with percentages and odds ratios. Exact logistic regression was used as necessary when expected cell counts were too small for the usual asymptotic logistic regression to be valid. The statistically significant (P<0.05) variables resulting from this bivariate analysis as well as important clinically significant variables with known associations with the outcome variables were included as covariates in a multivariable logistic regression model for each outcome variable with AAB. By adjusting for these covariates, any potential and unwanted variation resulting from differences between the AAB groups in other preoperative or postoperative characteristics is removed from the association between each outcome variable and AAB. Variance inflation factor and tolerance statistics were used to test for multicollinearity between all independent variables before including them in the final models. The Hosmer and Lemeshow goodness‐of‐fit test was used to assess the fit of the final models. Logistic regression was used to test the association of AKI with mortality, whereas a Wilcoxon rank sum test was used to test the difference in mean/median LOS between AKI and non‐AKI groups.
RESULTS
Preoperative, Surgical, and Anesthesia Data
A total of 922 patients met inclusion criteria, of which 343 (37%) were receiving AAB with either an ACEI or ARB preoperatively. Preoperative characteristics are documented in Table 1. Patients receiving AAB were older (63.010.8 vs 57.313.9 years) and had a higher BMI (34.67.3 vs 31.97.7) than patients not receiving this therapy. They were also more likely to be receiving diuretics, ‐blockers, and CCBs as well as have DM, CHF, CAD, and HTN. These characteristics were included as covariates in a multivariable logistic regression model so that any confounding resulting differences caused by these variables were removed from the association between each outcome variable and AAB use. There was no difference in baseline SBP or diastolic blood pressures. There was no difference in the use of NSAIDs. Patients receiving AAB underwent a higher percentage of TKAs (56.0% vs 44.2%) and lower percentage of spine fusions (16.6% vs 24.4%) and THAs (27.4% vs 31.4%). Propofol was the most commonly used general anesthetic induction agent (78%). Anesthetic induction agent usage was not different across the groups.
| AAB (N=343) | Non‐AAB (N=579) | P Value* | |
|---|---|---|---|
| |||
| Patient demographics | |||
| Age (y), meanSD | 63.010.8 | 57.313.9 | <0.001 |
| BMI (kg/m2), meanSD | 34.67.3 | 31.87.7 | <0.001 |
| Baseline systolic BP (mm Hg), meanSD | 135.016.8 | 130.819.8 | 0.339 |
| Baseline diastolic BP (mm Hg), meanSD | 74.514.5 | 75.311.8 | 0.798 |
| Medications | |||
| Diuretic, % | 53.6 | 18.5 | <0.001 |
| ‐Blocker, % | 39.4 | 22.4 | <0.001 |
| Calcium channel blocker, % | 23.3 | 9.9 | <0.001 |
| Nonsteroidal anti‐inflammatory drug, % | 36.1 | 35.7 | 0.901 |
| Comorbidities | |||
| Diabetes mellitus, % | 32.9 | 9.5 | <0.001 |
| Congestive heart failure, % | 2.9 | 0.4 | 0.001 |
| Coronary artery disease, % | 20.1 | 9.5 | <0.001 |
| Hypertension, % | 95.3 | 36.8 | <0.001 |
| Total comorbidities, meanSD | 1.50.7 | 0.50.7 | <0.001 |
| Procedure type | |||
| Spinal fusions, % | 16.6 | 24.4 | 0.001 |
| Total knee arthroplasty, % | 56.0 | 44.2 | 0.001 |
| Total hip arthroplasty, % | 27.4 | 31.4 | 0.001 |
| Induction agents | |||
| Etomidate, % | 14.9 | 11.9 | 0.199 |
| Propofol, % | 77.6 | 78.3 | 0.801 |
| Methohexital, % | 7.3 | 9.2 | 0.329 |
| Other (gas, ketamine), % | 0.3 | 1.2 | 0.270 |
| Hematocrit | |||
| Preoperative (%), meanSD | 40.53.7 | 40.23.8 | 0.150 |
| Postoperative (%), meanSD | 34.04.6 | 33.74.3 | 0.511 |
| % Change, meanSD | 15.38.5 | 16.88.9 | 0.036 |
| Creatinine | |||
| Preoperative (mg/dL), meanSD | 0.960.41 | 0.850.23 | <0.001 |
| Postoperative (mg/dL), meanSD | 0.960.42 | 0.810.23 | <0.001 |
| % change, meanSD | 1.4829.22 | 4.1414.18 | 0.235 |
| Fluids | |||
| Estimated blood loss (mL), meanSD | 301.2340.0 | 356.9482.7 | 0.125 |
| Crystalloid (mL), meanSD | 2584.41401.6 | 2765.21487.2 | 0.036 |
| Colloid (mL), meanSD | 124.3322.4 | 151.0553.5 | 0.810 |
| Packed red blood cells (mL), meanSD | 78.5323.1 | 99.5452.0 | 0.613 |
| Vasopressors | |||
| Ephedrine (mg), meanSD | 11.715.0 | 8.013.0 | <0.001 |
| Phenylephrine (g), meanSD | 615.72210.9 | 687.13905.3 | 0.002 |
| Vasopressin (U), meanSD | 0.351.90 | 0.080.90 | <0.001 |
| Outcomes | |||
| Induction hypotension, % | 12.2 | 1.93 | 0.005 |
| Intraoperative hypotension, % | 26.0 | 20.9 | 0.078 |
| Acute kidney injury, % | 8.3 | 1.7 | <0.001 |
Estimated blood loss for the procedures was similar between the 2 groups (301.2340 vs 356.9482.7 mL) and similar colloid and packed red cell administration occurred. However, patients receiving AAB were administered less crystalloid infusion (2584.41401.6 vs 2765.21487.2 mL, P=0.036) and received less phenylephrine but higher ephedrine and vasopressin dosages as a group. Patients in both groups had similar preoperative and postoperative hematocrit concentrations. Average preoperative serum creatinine was higher in the AAB group than in the non‐AAB group (0.960.41 vs 0.850.23, P<0.001) and remained so postoperatively (0.960.42 vs 0.810.23, P<0.001).
Primary and Secondary Outcome Measures
Postinduction Hypotension
Therapy with AAB was associated with a greater incidence of postinduction hypotension (12.2% vs 6.7%, P=0.005). Using a multivariate logistic regression model adjusting for the effects of age, BMI, antihypertensive medications, comorbidities, and anesthetic induction agents, the use of AAB had a greater odds ratio (OR) of 1.93 (95% confidence interval [CI]: 1.10‐3.41, P=0.023) for developing postinduction hypotension (Table 2). A higher BMI had a lower OR for postinduction hypotension.
| Comparison | Hypotension at Induction, OR (95% CI), P Value | Intraoperative Hypotension, OR (95% CI), P Value | Acute Kidney Injury, OR (95% CI), P Value |
|---|---|---|---|
| |||
| AAB vs none, unadjusted | 1.93 (1.223.06), 0.005 | 1.33 (0.971.82), 0.078 | 5.40 (2.4112.06), <0.001 |
| AAB vs none, adjusted for covariates | 1.93 (1.103.41), 0.023 | 1.30 (0.851.97), 0.226 | 2.68 (1.086.69), 0.034 |
| AAB vs none, adjusted for covariates+hypotension at induction | N/A | N/A | 2.51 (1.06.32), 0.051 |
| AAB vs none, adjusted for covariates+intraoperative hypotension | N/A | N/A | 2.66 (1.066.64), 0.037 |
| AAB vs none, adjusted for covariates+any hypotension | N/A | N/A | 2.60 (1.046.51), 0.042 |
Postincision (Intraoperative) Hypotension
The incidence of postincision, intraoperative hypotension in patients receiving AAB (26.0%) was not statistically different (P=0.078) from those not receiving these agents (20.9%). Multivariate logistic regression demonstrated that preoperative hypertension (OR: 1.73, 95% CI: 1.05‐2.85, P=0.031) and THA were each independent risk factors for intraoperative hypotension. The other comorbidities of DM, CHF, CAD, and the individual antihypertensive agents were not found to have a strong influence on the outcome of intraoperative hypotension. The odds ratio of developing intraoperative hypotension during the procedure in patients receiving AAB was not statistically significant (OR: 1.30, 95% CI: 0.85‐1.97, P=0.226) from those not receiving this therapy preoperatively (Table 2).
AKI
There was a significantly higher incidence of AKI (26/313 [8.3%] vs 8/485 [1.7%], P<0.001) in patients receiving preoperative AAB. No patients required renal replacement therapy. Of those patients who developed postoperative AKI, 73% of the AAB group and 75% of the non‐AAB group had a normal glomerular filtration rate (GFR) (mL/min/1.73 m2, GFR >90 prior to surgery. Of both the AAB and non‐AAB groups, 20% to 25% were in stage 2 (GFR 6089) chronic kidney disease (CKD)[18] preoperatively. Only 2 patients in the AAB group began in stage 3 (GFR 3059) CKD. Fifty percent of both groups went from stage 1 kidney function to stage 3. For all others who developed AKI, the GFR rose by a single stage.
Multivariate logistic regression, controlling for statistically significant and clinically significant variables, demonstrated that AAB administered preoperatively was independently associated with a greater OR of 2.68 (95% CI: 1.08‐6.69, P=0.034) for developing AKI postoperatively than if AAB was not taken (Table 2). Higher BMI was a further independent risk factor for postoperative AKI; a 5‐unit increase in BMI revealed an OR of 1.58 (95% CI: 1.25‐1.99, P<0.001) for experiencing postoperative AKI. Although the AAB group had statistically significantly more comorbidities, in the final analysis only the presence of CAD trended to an association with the development of AKI (OR: 2.37, 95% CI: 1.005.60, P=0.050).
We wished to determine whether the increased risk of AKI associated with AAB therapy was explained by the associated postinduction or postincision, intraoperative hypotension experienced by the patients or independent of these effects. To do so we accounted for the development of either postinduction or intraoperative hypotension as a possible confounder in the multivariate analysis for the development of AKI (Table 2). Postinduction hypotension alone was not statistically associated with AKI (OR 2.04, 95% CI: 0.70‐6.0, P=0.193). However, intraoperative hypotension was found to be an independent risk factor for the development of AKI (OR: 2.62, 95% CI: 1.17‐5.84, P=0.019). When eliminating the effect of this intraoperative hypotension as a confounder, patients on AAB therapy continued to have a 2.66 OR for the development of AKI postoperatively (95% CI: 1.06‐6.64, P=0.037), independent of the development of intraoperative hypotension. When eliminating the effect of both postinduction and intraoperative hypotension (all hypotension Table 2), AAB therapy continued to have a statistically significant independent effect (OR: 2.60, 95% CI: 1.046.51, P=0.042) on developing postoperative AKI.
As secondary outcome measures, we looked at the development of AKI and its relationship to hospital LOS and mortality. The development of AKI was associated with a significantly greater mean length of hospital stay (5.76 days vs 3.28 days, P<0.001). Although 2‐year mortality was higher, 5.9% in the AKI group compared to 2.4% in the non‐AAB group, it was not statistically significant (P=0.211).
DISCUSSION
In this retrospective review of 922 patients presenting for major orthopedic surgery, we found that 343(37%) were receiving therapy with either ACEIs or ARBs. In such patients, we demonstrated a higher incidence of postinduction hypotension and an increased incidence of AKI. We further demonstrated that the development of AKI associated with AAB therapy was independent of hypotension occurring either postinduction or intraoperatively after incision.
Postinduction hypotension in patients receiving AAB was demonstrated to be 12.2% compared to 7.7% in patients not receiving this therapy. Hypotension after general anesthesia induction in patients receiving AAB is widely reported,[1, 14, 15] and ranges from 22% to 100%[19, 20] based on varying definitions of what constitutes hypotension. We chose an absolute value of a systolic blood pressure of 80 mm Hg occurring for 5 minutes as constituting significant hypotension.[16] Monk et al. reported an increased one year postnon‐cardiac surgery mortality risk of 1.036 times per minute of intraoperative hypotension, defining hypotension as a systolic blood pressure of <80 mm Hg.[21]
We further demonstrated that AAB therapy resulted in an 8.3% incidence of AKI versus 1.7% in non‐AAB patients (P<0.001). AKI was defined as an increase in serum creatinine of 0.3 mg/dL or a 50% increase in creatinine when pre‐ and postoperative values were compared.[17] A number of other investigators have identified AKI associated with AAB use in patients undergoing cardiac,[6, 11] vascular,[7, 10] and lung[13] surgery. Similarly, in the present study, in orthopedic patients, AAB remained a significant risk factor for developing AKI (OR: 2.68, P=0.034) independent of patient comorbidities and adjunct therapy (Table 2).
ACEIs and ARBs are prescribed to treat HTN, CHF, and improve renal function in diabetic and proteinuric nephropathy.[22] AAB therapy is prescribed for nephropathy because these medications decrease glomerular pressure by selective inhibition of angiotensin II mediated vasoconstriction of the efferent glomerular arteriole.[23] Normally, this is beneficial to patients and is associated with a decrease in serum creatinine concentration. However, during hypotension, when there is decreased renal perfusion, further decreases in intraglomerular pressure may occur, precipitating renal failure.[23] In addition, other factors may contribute to the development of AKI, as AAB has both tissue and systemic effects that extend beyond simply dilating the efferent glomerular arteriole. These include effects on the sympathetic nervous system, oxidative stress, and altering the release and synthesis of vasodilators such as bradykinin, nitric oxide, and prostacyclins[24] as well as effects through the release of aldosterone and arginine‐vasopressin.[25] These other factors might help explain the present study's findings that, when eliminating the effect of both postinduction and intraoperative hypotension, AAB therapy continued to have a statistically significant independent effect (OR: 2.60, 95% CI: 1.046.51, P=0.042) on developing postoperative AKI.
Although we demonstrated an association of AAB therapy with the development of hypotension after induction, we demonstrated only a trend in the development of postincisional, intraoperative hypotension (P=0.078). We defined intraoperative hypotension as a systolic BP <80 mm Hg for 10 minutes occurring after skin incision.[16] One must take into consideration, however, that a significant number of AAB patients were hypotensive during induction and received higher doses of ephedrine and vasopressin during the operative period. These patients may have been rescued from intraoperative hypotension by receiving vasopressor treatment at the outset. We did find that intraoperative hypotension was a significant, independent risk factor for AKI (OR: 2.62, P=0.019).
We looked further at the consequences of developing AKI. Patients who developed AKI had a significant greater mean length of hospital stay (5.76 days vs 3.28 days, P<0.001), which is consistent with other investigators' findings.[25, 26, 27] Although 2‐year mortality was higher at 5.88% in the AKI group compared to 2.38% in the non‐AAB group, this was not statistically significant (P=0.211). Other studies have shown that the development of AKI results in greater mortality.[26, 28]
The American College of Physicians (ACP) recommendations as of 2013 regarding the use of ACEIs and ARBs preoperatively is: uncertain, continue with caution, avoid hypovolemia. Potential for hypotension with induction of anesthesia and increased vasoconstrictor requirements and decreased responsiveness to pressors.[29] The ACP acknowledges that preoperative ACEIs and ARBs have the potential for postinduction hypotension and increased requirements for vasopressors. We have implemented recommendations at our preoperative anesthesia clinic to hold ACEIs and ARBs on the morning of surgery in patients with controlled blood pressure scheduled for spine fusion, and hip and knee arthroplasties. In accordance with ACP guidelines, other antihypertensives such as ‐blockers, calcium channel blockers, nitrates, and sympatholytics should be continued preoperatively and can be used perioperatively.
Limitations of the Study
There are several limitations to our study. This was a retrospective analysis over a fixed time period in one academic institution. Further, because of the retrospective nature, anesthesia and intraoperative (fluid and vasoconstrictor) management was not standardized. The definition of hypotension (SBP 80 mm Hg for 5 minutes after induction and 10 minutes after incision) may have been too stringent, so that more subtle decreases in blood pressure that could have impacted AKI might not have been captured to show statistical significance. Thus, our finding, that the development of AKI associated with preoperative AAB therapy may be independent of the occurrence of hypotension, must be interpreted with this in mind.
CONCLUSIONS
Patients who receive preoperative ACEI or ARB therapy and undergo major orthopedic surgery such as spinal fusion, and hip or knee arthroplasties experience a higher incidence of postinduction hypotension and AKI than those not receiving such therapy. The development of AKI in such patients is associated with a significantly prolonged length of hospital stay. Our findings suggest an association between preoperative ACEI/ARB use and moderate kidney injury following major orthopedic surgeries. However, a prospective, multicentered, randomized trial needs to be performed to confirm that withdrawal of AAB therapy preoperatively will decrease the incidence of AKI in patients undergoing major orthopedic procedures under general anesthesia. Future studies also need to determine the optimal time duration of withholding AAB therapy and the consequences on cardiac outcomes.
ACKNOWLEDGMENTS
Disclosures: Presented at the Society of Hospital Medicine National Meeting, May 18, 2013, National Harbor, Maryland; and the Society of General Internal Medicine Mid‐Atlantic Regional Meeting, March 1, 2013, Philadelphia, Pennsylvania. The authors report no conflicts of interest.
Patients presenting for surgery with angiotensin axis blockade (AAB) from therapy with either angiotensin‐converting enzyme inhibitors (ACEIs) or angiotensin receptor blockers (ARBs) experience an increased incidence of perioperative hypotension.[1, 2, 3, 4] Debate continues as to whether this hypotension results in any significant clinical sequelae. Some authors found that the use of an ACEI decreased the incidence of acute kidney injury (AKI),[5] mortality, and septicemia in cardiac and vascular surgical patients.[6] However, others found that in vascular and cardiac surgery there is increased mortality[7] as well as increased incidence of postoperative AKI.[8, 9, 10] A retrospective study of 10,000 coronary artery bypass graft patients found that ACEI was associated with increased inotropic support, AKI, mortality, and new onset atrial fibrillation.[11] In a meta‐analysis of 69,000 cardiothoracic surgery patients, the use of ACEIs/ARBs was associated with an increase in AKI and mortality.[12] AKI has also been demonstrated after lung resection surgery in patients receiving preoperative therapy with an ARB.[13]
Studies on noncardiac general surgery patients demonstrate that the use of AAB results in postinduction hypotension, but they fail to show an increased incidence in postoperative AKI.[14, 15] We propose, however, that major orthopedic surgery patients are a specific surgical cohort, like cardiac, vascular, and lung, who can develop operative hypotension and postoperative AKI when AAB is taken on the morning of surgery. To address this question we performed a retrospective study of 1154 patients undergoing either spinal fusion, total knee arthroplasty (TKA), or total hip arthroplasty (THA) during the 2010 calendar year in our academic medical center. We measured the incidence of postanesthesia induction hypotension, intraoperative hypotension, and postoperative AKI as it relates to the administration of AAB preoperatively.
MATERIALS AND METHODS
This study was a retrospective, observational investigation at a single, large academic hospital. The study design for chart review was approved by the institutional review board prior to data collection. Informed patient consent was not required for this retrospective study.
Patient Selection
We retrospectively reviewed the computerized chart and operating room electronic medical records of all patients who underwent elective major orthopedic surgery in the 2010 calendar year. We chose vertebral spine fusion, TKA, and THA as the 3 surgeries to represent major elective orthopedic surgery. Electronic query of the hospital database identified 1154 patients having undergone these surgeries in the year 2010. Nine hundred twenty‐two patients met inclusion criteria: 21 years old and evaluated in the preanesthesia clinic with documented vital signs and clearly defined preoperative medication recommendations. The policy in the preanesthesia clinic was to recommend taking the ACEI and ARB on the morning of surgery. All 922 patients were included in the analysis of the outcomes for induction hypotension and intraoperative hypotension. Of the 922 patients, 798 had the documented preoperative and postoperative creatinine values needed to define AKI. Therefore, only these 798 patients were included in the AKI outcome analysis. During the time of the study it was the practice at our medical center that all such surgeries were performed under general anesthesia.
Data Collection
Preanesthesia records were reviewed for patient demographics including age, body mass index (BMI), baseline blood pressure, diabetes mellitus (DM), coronary artery disease (CAD), hypertension (HTN), and congestive heart failure (CHF), as well as for therapy with ACEI or ARB, diuretics, ‐blockers, nonsteroidal anti‐inflammatory drugs (NSAIDs), and calcium channel blockers (CCB). The 4 statistically and clinically significant comorbidities were looked at individually as well as added together for a total sum of weighted comorbidity risk factors. The Anesthesia Electronic Record (Surginet Anesthesia, Kansas City, MO) was reviewed for each corresponding patient to determine the general anesthesia induction agent used and to assess the postinduction and intraoperative systolic blood pressures. Blood pressure was determined using an automated blood pressure cuff and automatically recorded at intervals of 5 minutes or less. Further, ephedrine, phenylephrine and vasopressin doses, estimated blood loss, blood transfusion requirements, and intravenous fluid administration (colloid and crystalloid) were noted. Preoperative (<30 days) and postoperative (within 24 hours after surgery) serum creatinine and hematocrit values were also recorded.
Outcome Measures
The primary outcome measures studied were:
- Postinduction hypotension (systolic blood pressure [SBP] 80 mm Hg for5 minutes) occurring within 30 minutes after anesthesia induction but before surgical incision.[16]
- Intraoperative hypotension (SBP 80 mm Hg for 10 minutes) occurring after surgical incision.[16]
- Postoperative AKI defined as an increase in serum creatinine 0.3 mg/dL or an increase of 50% from preoperative creatinine (Kidney Disease: Improving Global Outcomes Clinical Practice Guideline for Acute Kidney Injury)[17] within 24 hours postoperatively.
The secondary outcome measures were:
- Hospital length of stay (LOS).
- Two‐year mortality associated with the development of AKI.
Statistical Analysis
Categorical variables were summarized with frequencies and percentages, whereas continuous variables were summarized with means, standard deviations, medians, and quartiles. A [2] test or a Wilcoxon rank sum test was used to determine differences in preoperative and intraoperative characteristics between those patients with AAB and those patients without AAB. Logistic regression was used to determine the association between the main outcome variables (hypotension at anesthetic induction, hypotension during the operative procedure, and postoperative AKI) and the primary independent variable, AAB, as well as other preoperative and intraoperative characteristics. The significance and magnitude of the associations were quantified with percentages and odds ratios. Exact logistic regression was used as necessary when expected cell counts were too small for the usual asymptotic logistic regression to be valid. The statistically significant (P<0.05) variables resulting from this bivariate analysis as well as important clinically significant variables with known associations with the outcome variables were included as covariates in a multivariable logistic regression model for each outcome variable with AAB. By adjusting for these covariates, any potential and unwanted variation resulting from differences between the AAB groups in other preoperative or postoperative characteristics is removed from the association between each outcome variable and AAB. Variance inflation factor and tolerance statistics were used to test for multicollinearity between all independent variables before including them in the final models. The Hosmer and Lemeshow goodness‐of‐fit test was used to assess the fit of the final models. Logistic regression was used to test the association of AKI with mortality, whereas a Wilcoxon rank sum test was used to test the difference in mean/median LOS between AKI and non‐AKI groups.
RESULTS
Preoperative, Surgical, and Anesthesia Data
A total of 922 patients met inclusion criteria, of which 343 (37%) were receiving AAB with either an ACEI or ARB preoperatively. Preoperative characteristics are documented in Table 1. Patients receiving AAB were older (63.010.8 vs 57.313.9 years) and had a higher BMI (34.67.3 vs 31.97.7) than patients not receiving this therapy. They were also more likely to be receiving diuretics, ‐blockers, and CCBs as well as have DM, CHF, CAD, and HTN. These characteristics were included as covariates in a multivariable logistic regression model so that any confounding resulting differences caused by these variables were removed from the association between each outcome variable and AAB use. There was no difference in baseline SBP or diastolic blood pressures. There was no difference in the use of NSAIDs. Patients receiving AAB underwent a higher percentage of TKAs (56.0% vs 44.2%) and lower percentage of spine fusions (16.6% vs 24.4%) and THAs (27.4% vs 31.4%). Propofol was the most commonly used general anesthetic induction agent (78%). Anesthetic induction agent usage was not different across the groups.
| AAB (N=343) | Non‐AAB (N=579) | P Value* | |
|---|---|---|---|
| |||
| Patient demographics | |||
| Age (y), meanSD | 63.010.8 | 57.313.9 | <0.001 |
| BMI (kg/m2), meanSD | 34.67.3 | 31.87.7 | <0.001 |
| Baseline systolic BP (mm Hg), meanSD | 135.016.8 | 130.819.8 | 0.339 |
| Baseline diastolic BP (mm Hg), meanSD | 74.514.5 | 75.311.8 | 0.798 |
| Medications | |||
| Diuretic, % | 53.6 | 18.5 | <0.001 |
| ‐Blocker, % | 39.4 | 22.4 | <0.001 |
| Calcium channel blocker, % | 23.3 | 9.9 | <0.001 |
| Nonsteroidal anti‐inflammatory drug, % | 36.1 | 35.7 | 0.901 |
| Comorbidities | |||
| Diabetes mellitus, % | 32.9 | 9.5 | <0.001 |
| Congestive heart failure, % | 2.9 | 0.4 | 0.001 |
| Coronary artery disease, % | 20.1 | 9.5 | <0.001 |
| Hypertension, % | 95.3 | 36.8 | <0.001 |
| Total comorbidities, meanSD | 1.50.7 | 0.50.7 | <0.001 |
| Procedure type | |||
| Spinal fusions, % | 16.6 | 24.4 | 0.001 |
| Total knee arthroplasty, % | 56.0 | 44.2 | 0.001 |
| Total hip arthroplasty, % | 27.4 | 31.4 | 0.001 |
| Induction agents | |||
| Etomidate, % | 14.9 | 11.9 | 0.199 |
| Propofol, % | 77.6 | 78.3 | 0.801 |
| Methohexital, % | 7.3 | 9.2 | 0.329 |
| Other (gas, ketamine), % | 0.3 | 1.2 | 0.270 |
| Hematocrit | |||
| Preoperative (%), meanSD | 40.53.7 | 40.23.8 | 0.150 |
| Postoperative (%), meanSD | 34.04.6 | 33.74.3 | 0.511 |
| % Change, meanSD | 15.38.5 | 16.88.9 | 0.036 |
| Creatinine | |||
| Preoperative (mg/dL), meanSD | 0.960.41 | 0.850.23 | <0.001 |
| Postoperative (mg/dL), meanSD | 0.960.42 | 0.810.23 | <0.001 |
| % change, meanSD | 1.4829.22 | 4.1414.18 | 0.235 |
| Fluids | |||
| Estimated blood loss (mL), meanSD | 301.2340.0 | 356.9482.7 | 0.125 |
| Crystalloid (mL), meanSD | 2584.41401.6 | 2765.21487.2 | 0.036 |
| Colloid (mL), meanSD | 124.3322.4 | 151.0553.5 | 0.810 |
| Packed red blood cells (mL), meanSD | 78.5323.1 | 99.5452.0 | 0.613 |
| Vasopressors | |||
| Ephedrine (mg), meanSD | 11.715.0 | 8.013.0 | <0.001 |
| Phenylephrine (g), meanSD | 615.72210.9 | 687.13905.3 | 0.002 |
| Vasopressin (U), meanSD | 0.351.90 | 0.080.90 | <0.001 |
| Outcomes | |||
| Induction hypotension, % | 12.2 | 1.93 | 0.005 |
| Intraoperative hypotension, % | 26.0 | 20.9 | 0.078 |
| Acute kidney injury, % | 8.3 | 1.7 | <0.001 |
Estimated blood loss for the procedures was similar between the 2 groups (301.2340 vs 356.9482.7 mL) and similar colloid and packed red cell administration occurred. However, patients receiving AAB were administered less crystalloid infusion (2584.41401.6 vs 2765.21487.2 mL, P=0.036) and received less phenylephrine but higher ephedrine and vasopressin dosages as a group. Patients in both groups had similar preoperative and postoperative hematocrit concentrations. Average preoperative serum creatinine was higher in the AAB group than in the non‐AAB group (0.960.41 vs 0.850.23, P<0.001) and remained so postoperatively (0.960.42 vs 0.810.23, P<0.001).
Primary and Secondary Outcome Measures
Postinduction Hypotension
Therapy with AAB was associated with a greater incidence of postinduction hypotension (12.2% vs 6.7%, P=0.005). Using a multivariate logistic regression model adjusting for the effects of age, BMI, antihypertensive medications, comorbidities, and anesthetic induction agents, the use of AAB had a greater odds ratio (OR) of 1.93 (95% confidence interval [CI]: 1.10‐3.41, P=0.023) for developing postinduction hypotension (Table 2). A higher BMI had a lower OR for postinduction hypotension.
| Comparison | Hypotension at Induction, OR (95% CI), P Value | Intraoperative Hypotension, OR (95% CI), P Value | Acute Kidney Injury, OR (95% CI), P Value |
|---|---|---|---|
| |||
| AAB vs none, unadjusted | 1.93 (1.223.06), 0.005 | 1.33 (0.971.82), 0.078 | 5.40 (2.4112.06), <0.001 |
| AAB vs none, adjusted for covariates | 1.93 (1.103.41), 0.023 | 1.30 (0.851.97), 0.226 | 2.68 (1.086.69), 0.034 |
| AAB vs none, adjusted for covariates+hypotension at induction | N/A | N/A | 2.51 (1.06.32), 0.051 |
| AAB vs none, adjusted for covariates+intraoperative hypotension | N/A | N/A | 2.66 (1.066.64), 0.037 |
| AAB vs none, adjusted for covariates+any hypotension | N/A | N/A | 2.60 (1.046.51), 0.042 |
Postincision (Intraoperative) Hypotension
The incidence of postincision, intraoperative hypotension in patients receiving AAB (26.0%) was not statistically different (P=0.078) from those not receiving these agents (20.9%). Multivariate logistic regression demonstrated that preoperative hypertension (OR: 1.73, 95% CI: 1.05‐2.85, P=0.031) and THA were each independent risk factors for intraoperative hypotension. The other comorbidities of DM, CHF, CAD, and the individual antihypertensive agents were not found to have a strong influence on the outcome of intraoperative hypotension. The odds ratio of developing intraoperative hypotension during the procedure in patients receiving AAB was not statistically significant (OR: 1.30, 95% CI: 0.85‐1.97, P=0.226) from those not receiving this therapy preoperatively (Table 2).
AKI
There was a significantly higher incidence of AKI (26/313 [8.3%] vs 8/485 [1.7%], P<0.001) in patients receiving preoperative AAB. No patients required renal replacement therapy. Of those patients who developed postoperative AKI, 73% of the AAB group and 75% of the non‐AAB group had a normal glomerular filtration rate (GFR) (mL/min/1.73 m2, GFR >90 prior to surgery. Of both the AAB and non‐AAB groups, 20% to 25% were in stage 2 (GFR 6089) chronic kidney disease (CKD)[18] preoperatively. Only 2 patients in the AAB group began in stage 3 (GFR 3059) CKD. Fifty percent of both groups went from stage 1 kidney function to stage 3. For all others who developed AKI, the GFR rose by a single stage.
Multivariate logistic regression, controlling for statistically significant and clinically significant variables, demonstrated that AAB administered preoperatively was independently associated with a greater OR of 2.68 (95% CI: 1.08‐6.69, P=0.034) for developing AKI postoperatively than if AAB was not taken (Table 2). Higher BMI was a further independent risk factor for postoperative AKI; a 5‐unit increase in BMI revealed an OR of 1.58 (95% CI: 1.25‐1.99, P<0.001) for experiencing postoperative AKI. Although the AAB group had statistically significantly more comorbidities, in the final analysis only the presence of CAD trended to an association with the development of AKI (OR: 2.37, 95% CI: 1.005.60, P=0.050).
We wished to determine whether the increased risk of AKI associated with AAB therapy was explained by the associated postinduction or postincision, intraoperative hypotension experienced by the patients or independent of these effects. To do so we accounted for the development of either postinduction or intraoperative hypotension as a possible confounder in the multivariate analysis for the development of AKI (Table 2). Postinduction hypotension alone was not statistically associated with AKI (OR 2.04, 95% CI: 0.70‐6.0, P=0.193). However, intraoperative hypotension was found to be an independent risk factor for the development of AKI (OR: 2.62, 95% CI: 1.17‐5.84, P=0.019). When eliminating the effect of this intraoperative hypotension as a confounder, patients on AAB therapy continued to have a 2.66 OR for the development of AKI postoperatively (95% CI: 1.06‐6.64, P=0.037), independent of the development of intraoperative hypotension. When eliminating the effect of both postinduction and intraoperative hypotension (all hypotension Table 2), AAB therapy continued to have a statistically significant independent effect (OR: 2.60, 95% CI: 1.046.51, P=0.042) on developing postoperative AKI.
As secondary outcome measures, we looked at the development of AKI and its relationship to hospital LOS and mortality. The development of AKI was associated with a significantly greater mean length of hospital stay (5.76 days vs 3.28 days, P<0.001). Although 2‐year mortality was higher, 5.9% in the AKI group compared to 2.4% in the non‐AAB group, it was not statistically significant (P=0.211).
DISCUSSION
In this retrospective review of 922 patients presenting for major orthopedic surgery, we found that 343(37%) were receiving therapy with either ACEIs or ARBs. In such patients, we demonstrated a higher incidence of postinduction hypotension and an increased incidence of AKI. We further demonstrated that the development of AKI associated with AAB therapy was independent of hypotension occurring either postinduction or intraoperatively after incision.
Postinduction hypotension in patients receiving AAB was demonstrated to be 12.2% compared to 7.7% in patients not receiving this therapy. Hypotension after general anesthesia induction in patients receiving AAB is widely reported,[1, 14, 15] and ranges from 22% to 100%[19, 20] based on varying definitions of what constitutes hypotension. We chose an absolute value of a systolic blood pressure of 80 mm Hg occurring for 5 minutes as constituting significant hypotension.[16] Monk et al. reported an increased one year postnon‐cardiac surgery mortality risk of 1.036 times per minute of intraoperative hypotension, defining hypotension as a systolic blood pressure of <80 mm Hg.[21]
We further demonstrated that AAB therapy resulted in an 8.3% incidence of AKI versus 1.7% in non‐AAB patients (P<0.001). AKI was defined as an increase in serum creatinine of 0.3 mg/dL or a 50% increase in creatinine when pre‐ and postoperative values were compared.[17] A number of other investigators have identified AKI associated with AAB use in patients undergoing cardiac,[6, 11] vascular,[7, 10] and lung[13] surgery. Similarly, in the present study, in orthopedic patients, AAB remained a significant risk factor for developing AKI (OR: 2.68, P=0.034) independent of patient comorbidities and adjunct therapy (Table 2).
ACEIs and ARBs are prescribed to treat HTN, CHF, and improve renal function in diabetic and proteinuric nephropathy.[22] AAB therapy is prescribed for nephropathy because these medications decrease glomerular pressure by selective inhibition of angiotensin II mediated vasoconstriction of the efferent glomerular arteriole.[23] Normally, this is beneficial to patients and is associated with a decrease in serum creatinine concentration. However, during hypotension, when there is decreased renal perfusion, further decreases in intraglomerular pressure may occur, precipitating renal failure.[23] In addition, other factors may contribute to the development of AKI, as AAB has both tissue and systemic effects that extend beyond simply dilating the efferent glomerular arteriole. These include effects on the sympathetic nervous system, oxidative stress, and altering the release and synthesis of vasodilators such as bradykinin, nitric oxide, and prostacyclins[24] as well as effects through the release of aldosterone and arginine‐vasopressin.[25] These other factors might help explain the present study's findings that, when eliminating the effect of both postinduction and intraoperative hypotension, AAB therapy continued to have a statistically significant independent effect (OR: 2.60, 95% CI: 1.046.51, P=0.042) on developing postoperative AKI.
Although we demonstrated an association of AAB therapy with the development of hypotension after induction, we demonstrated only a trend in the development of postincisional, intraoperative hypotension (P=0.078). We defined intraoperative hypotension as a systolic BP <80 mm Hg for 10 minutes occurring after skin incision.[16] One must take into consideration, however, that a significant number of AAB patients were hypotensive during induction and received higher doses of ephedrine and vasopressin during the operative period. These patients may have been rescued from intraoperative hypotension by receiving vasopressor treatment at the outset. We did find that intraoperative hypotension was a significant, independent risk factor for AKI (OR: 2.62, P=0.019).
We looked further at the consequences of developing AKI. Patients who developed AKI had a significant greater mean length of hospital stay (5.76 days vs 3.28 days, P<0.001), which is consistent with other investigators' findings.[25, 26, 27] Although 2‐year mortality was higher at 5.88% in the AKI group compared to 2.38% in the non‐AAB group, this was not statistically significant (P=0.211). Other studies have shown that the development of AKI results in greater mortality.[26, 28]
The American College of Physicians (ACP) recommendations as of 2013 regarding the use of ACEIs and ARBs preoperatively is: uncertain, continue with caution, avoid hypovolemia. Potential for hypotension with induction of anesthesia and increased vasoconstrictor requirements and decreased responsiveness to pressors.[29] The ACP acknowledges that preoperative ACEIs and ARBs have the potential for postinduction hypotension and increased requirements for vasopressors. We have implemented recommendations at our preoperative anesthesia clinic to hold ACEIs and ARBs on the morning of surgery in patients with controlled blood pressure scheduled for spine fusion, and hip and knee arthroplasties. In accordance with ACP guidelines, other antihypertensives such as ‐blockers, calcium channel blockers, nitrates, and sympatholytics should be continued preoperatively and can be used perioperatively.
Limitations of the Study
There are several limitations to our study. This was a retrospective analysis over a fixed time period in one academic institution. Further, because of the retrospective nature, anesthesia and intraoperative (fluid and vasoconstrictor) management was not standardized. The definition of hypotension (SBP 80 mm Hg for 5 minutes after induction and 10 minutes after incision) may have been too stringent, so that more subtle decreases in blood pressure that could have impacted AKI might not have been captured to show statistical significance. Thus, our finding, that the development of AKI associated with preoperative AAB therapy may be independent of the occurrence of hypotension, must be interpreted with this in mind.
CONCLUSIONS
Patients who receive preoperative ACEI or ARB therapy and undergo major orthopedic surgery such as spinal fusion, and hip or knee arthroplasties experience a higher incidence of postinduction hypotension and AKI than those not receiving such therapy. The development of AKI in such patients is associated with a significantly prolonged length of hospital stay. Our findings suggest an association between preoperative ACEI/ARB use and moderate kidney injury following major orthopedic surgeries. However, a prospective, multicentered, randomized trial needs to be performed to confirm that withdrawal of AAB therapy preoperatively will decrease the incidence of AKI in patients undergoing major orthopedic procedures under general anesthesia. Future studies also need to determine the optimal time duration of withholding AAB therapy and the consequences on cardiac outcomes.
ACKNOWLEDGMENTS
Disclosures: Presented at the Society of Hospital Medicine National Meeting, May 18, 2013, National Harbor, Maryland; and the Society of General Internal Medicine Mid‐Atlantic Regional Meeting, March 1, 2013, Philadelphia, Pennsylvania. The authors report no conflicts of interest.
- . Management of hypotension associated with angiotensin‐axis blockade and general anesthesia administration. J Cardiothorac Vasc Anesth. 2013;27:156–167.
- , , , , , . Clinical consequences of withholding versus administering renin‐angiotensin‐aldosterone system antagonists in the preoperative period. J Hosp Med. 2008;3:319–325.
- , , , et al. Influence of chronic angiotensin‐converting enzyme inhibition on anesthetic induction. Anesthesiology. 1994;81(2):299–307.
- , , , , . Pressor responses to tracheal intubation after sublingual captopril. A pilot study. Anaesthesia. 1990;45(3):243–245.
- , , , , , , . Preoperative angiotensin‐converting enzyme inhibitors and acute kidney injury after coronary artery bypass grafting. Ann Thorac Surg. 2008;86(4):1160–1165.
- , , , et al. The effects of preoperative renin‐angiotensin system inhibitors on outcomes in patients undergoing cardiac surgery. J Cardiothorac Vasc Anesth. 2013;27(4):703–709.
- , , , . Renin‐angiotensin blockade is associated with increased mortality after vascular surgery. Can J Anaesth. 2010;57:736–744.
- , . , et al. Preoperative use of angiotensin‐converting enzyme inhibitors/angiotensin receptor blockers is associated with increased risk for acute kidney injury after cardiovascular surgery. Clin J Am Soc Nephrol. 2008;3(5):1266–1273.
- , , , et al.; TRIBE‐AKI Consortium. Preoperative angiotensin‐converting enzyme inhibitors and angiotensin receptor blocker use and acute kidney injury in patients undergoing cardiac surgery. Nephrol Dial Transplant. 2013;28(11):2787–2799.
- , , , et al. The chronic inhibition of angiotensin‐converting enzyme impairs postoperative renal function. Anesth Analg. 2001;93(5):1111–1115.
- , , , et al. Effects of angiotensin‐converting enzyme inhibitor therapy on clinical outcome in patients undergoing coronary artery bypass grafting. J Am Coll Cardiol. 2009;54:1778–1784.
- , , , , , . Acute kidney injury and death associated with renin angiotensin system blockade in cardiothoracic surgery: a meta‐analysis of observational studies. Am J Kidney Dis. 2013;63(6):1077–1086.
- , , . Acute kidney injury after lung resection surgery: incidence and perioperative risk factors. Anesth Analg. 2012;114:1256–1262.
- , , , et al. Angiotensin system inhibitors in a general surgical population. Anesth Analg. 2005;100:636–644, table of contents.
- , , , , . Chronic angiotensin‐converting enzyme inhibitor or angiotensin receptor blocker therapy combined with diuretic therapy is associated with increased episodes of hypotension in noncardiac surgery. J Cardiothorac Vasc Anesth. 2008;22:180–186.
- , , , , . Hemodynamic impact of dexmedetomidine administration in 15,656 noncardiac surgical cases. J Clin Anesth. 2012;24:212–220.
- Kidney Disease: Improving Global Outcomes (KDIGO). Acute Kidney Injury Work Group. KDIGO clinical practice guideline for acute kidney injury. Kidney Int Suppl. 2012;2:8.
- Kidney Disease: Improving Global Outcomes (KDIGO). Clincal practice guidelines for evaluation and management of chronic kidney disease. Kidney Int Suppl. 2013;3(1):8.
- , . Hemodynamic effects of anesthesia in patients with ischemic heart failure chronically treated with angiotensin‐converting enzyme inhibitors. Anesth Analg. 1997;84:945–949.
- , , , , . The hemodynamic effects of anesthetic induction in vascular surgical patients chronically treated with angiotensin II receptor antagonists. Anesth Analg. 1999;89:1388–1392.
- , , , . Anesthetic management and one‐year mortality after noncardiac surgery. Anesth Analg. 2005;100:4–10.
- , , , , . Use of angiotensin‐converting enzyme inhibitors and angiotensin receptor blockers in clinical practice. Expert Rev Cardiovasc Ther. 2012;10:159–166.
- , . An overview of drug‐induced acute kidney injury. Crit Care Med. 2008;36:S216–S223.
- , , , et al. Improvement of endothelial function by chronic angiotensin‐converting enzyme inhibition in heart failure: role of nitric oxide, prostanoids, oxidant stress, and bradykinin. Circulation. 2000;102:351–356.
- , , , . Renin‐angiotensin system antagonists in the perioperative setting: clinical consequences and recommendations for practice. Postgrad Med J. 2011;87:472–481.
- , , , , . Early postoperative statin therapy is associated with a lower incidence of acute kidney injury after cardiac surgery. J Cardiothorac Vasc Anesth. 2010;24:913–920.
- , , , , . Improved survival in acute kidney injury after cardiac surgery. Am J Kidney Dis. 2007;50:703–711.
- , , , . Determinants of postoperative acute kidney injury. Crit Care. 2009;13:R79.
- American College of Physicians. ACP Smart Medicine: Perioperative Medication Management. Tables: Perioperative Cardiovascular Medication Management. http://smartmedicine.acponline.org/content.aspx?gbosID=336. Accessed January 19, 2014.
- . Management of hypotension associated with angiotensin‐axis blockade and general anesthesia administration. J Cardiothorac Vasc Anesth. 2013;27:156–167.
- , , , , , . Clinical consequences of withholding versus administering renin‐angiotensin‐aldosterone system antagonists in the preoperative period. J Hosp Med. 2008;3:319–325.
- , , , et al. Influence of chronic angiotensin‐converting enzyme inhibition on anesthetic induction. Anesthesiology. 1994;81(2):299–307.
- , , , , . Pressor responses to tracheal intubation after sublingual captopril. A pilot study. Anaesthesia. 1990;45(3):243–245.
- , , , , , , . Preoperative angiotensin‐converting enzyme inhibitors and acute kidney injury after coronary artery bypass grafting. Ann Thorac Surg. 2008;86(4):1160–1165.
- , , , et al. The effects of preoperative renin‐angiotensin system inhibitors on outcomes in patients undergoing cardiac surgery. J Cardiothorac Vasc Anesth. 2013;27(4):703–709.
- , , , . Renin‐angiotensin blockade is associated with increased mortality after vascular surgery. Can J Anaesth. 2010;57:736–744.
- , . , et al. Preoperative use of angiotensin‐converting enzyme inhibitors/angiotensin receptor blockers is associated with increased risk for acute kidney injury after cardiovascular surgery. Clin J Am Soc Nephrol. 2008;3(5):1266–1273.
- , , , et al.; TRIBE‐AKI Consortium. Preoperative angiotensin‐converting enzyme inhibitors and angiotensin receptor blocker use and acute kidney injury in patients undergoing cardiac surgery. Nephrol Dial Transplant. 2013;28(11):2787–2799.
- , , , et al. The chronic inhibition of angiotensin‐converting enzyme impairs postoperative renal function. Anesth Analg. 2001;93(5):1111–1115.
- , , , et al. Effects of angiotensin‐converting enzyme inhibitor therapy on clinical outcome in patients undergoing coronary artery bypass grafting. J Am Coll Cardiol. 2009;54:1778–1784.
- , , , , , . Acute kidney injury and death associated with renin angiotensin system blockade in cardiothoracic surgery: a meta‐analysis of observational studies. Am J Kidney Dis. 2013;63(6):1077–1086.
- , , . Acute kidney injury after lung resection surgery: incidence and perioperative risk factors. Anesth Analg. 2012;114:1256–1262.
- , , , et al. Angiotensin system inhibitors in a general surgical population. Anesth Analg. 2005;100:636–644, table of contents.
- , , , , . Chronic angiotensin‐converting enzyme inhibitor or angiotensin receptor blocker therapy combined with diuretic therapy is associated with increased episodes of hypotension in noncardiac surgery. J Cardiothorac Vasc Anesth. 2008;22:180–186.
- , , , , . Hemodynamic impact of dexmedetomidine administration in 15,656 noncardiac surgical cases. J Clin Anesth. 2012;24:212–220.
- Kidney Disease: Improving Global Outcomes (KDIGO). Acute Kidney Injury Work Group. KDIGO clinical practice guideline for acute kidney injury. Kidney Int Suppl. 2012;2:8.
- Kidney Disease: Improving Global Outcomes (KDIGO). Clincal practice guidelines for evaluation and management of chronic kidney disease. Kidney Int Suppl. 2013;3(1):8.
- , . Hemodynamic effects of anesthesia in patients with ischemic heart failure chronically treated with angiotensin‐converting enzyme inhibitors. Anesth Analg. 1997;84:945–949.
- , , , , . The hemodynamic effects of anesthetic induction in vascular surgical patients chronically treated with angiotensin II receptor antagonists. Anesth Analg. 1999;89:1388–1392.
- , , , . Anesthetic management and one‐year mortality after noncardiac surgery. Anesth Analg. 2005;100:4–10.
- , , , , . Use of angiotensin‐converting enzyme inhibitors and angiotensin receptor blockers in clinical practice. Expert Rev Cardiovasc Ther. 2012;10:159–166.
- , . An overview of drug‐induced acute kidney injury. Crit Care Med. 2008;36:S216–S223.
- , , , et al. Improvement of endothelial function by chronic angiotensin‐converting enzyme inhibition in heart failure: role of nitric oxide, prostanoids, oxidant stress, and bradykinin. Circulation. 2000;102:351–356.
- , , , . Renin‐angiotensin system antagonists in the perioperative setting: clinical consequences and recommendations for practice. Postgrad Med J. 2011;87:472–481.
- , , , , . Early postoperative statin therapy is associated with a lower incidence of acute kidney injury after cardiac surgery. J Cardiothorac Vasc Anesth. 2010;24:913–920.
- , , , , . Improved survival in acute kidney injury after cardiac surgery. Am J Kidney Dis. 2007;50:703–711.
- , , , . Determinants of postoperative acute kidney injury. Crit Care. 2009;13:R79.
- American College of Physicians. ACP Smart Medicine: Perioperative Medication Management. Tables: Perioperative Cardiovascular Medication Management. http://smartmedicine.acponline.org/content.aspx?gbosID=336. Accessed January 19, 2014.
© 2014 Society of Hospital Medicine
Obesity and Gynecologic Cancer
For the last decade, the obesity epidemic in the United States has been well recognized. In 2001, the surgeon general made a call to action to combat obesity. Despite this effort, obesity rates in the United States continued to rise, and in 2009-2010, more than one third (35.7%) of adults in the United States were classified as obese, according to the Centers for Disease Control and Prevention.
The definition of obesity relies on the body mass index. BMI is defined as a person’s weight in kilograms divided by the individual’s height in meters squared. Overweight is defined as a BMI of 25-29.9 kg/m2 and obesity as a BMI of greater than 30 kg/m2. Obesity has been further classified by the World Health Organization into class I (BMI, 30-34.9 kg/m2), class II (BMI, 35-39.9 kg/ m2), and class III (BMI, greater than 40 kg/ m2).
In the United States in 2013, there were approximately 50,000 new cases and more than 8,000 deaths from endometrial cancer (CA Cancer J. Clin. 2013;63:11-30). Rates of endometrial cancer have risen steadily along with the obesity epidemic. This is no surprise, as obesity has been linked to the development of endometrial cancer. It is believed that high levels of circulating estrogen created by adipose tissue convert androstenedione to estrone, and there is aromatization of androgens. For each 5-kg/m2 increase in BMI, there is an increased risk of development of endometrial cancer (relative risk, 1.59) (Lancet 2008;371:569). While many physicians realize the link between obesity and the hyperestrogenic state associated with endometrial cancers, increased BMI is also associated with an increased risk of ovarian cancer (odds ratio, 1.3) (Eur. J. Cancer 2007;43:690).

In addition to increasing the risk of developing gynecologic cancers, obesity also increases the risk of death from all gynecologic malignancies. In the Cancer Prevention Study II, a large prospective cohort study, a BMI greater than 35 was associated with increased mortality compared with normal weight in ovarian (RR, 1.51), endometrial (RR, 2.77), and cervical cancer (RR, 3.20) (N. Engl. J. Med. 2003;348:1625). The same study found that those with a BMI greater than 40 with endometrial cancer had a relative risk of death of 6.25.
The increased mortality seen in obese endometrial cancer patients is particularly striking, given the fact that these women are more likely to have less-aggressive histologies and earlier-stage cancers (Gynecol. Oncol. 2009;90:150-7; Gynecol. Oncol. 2009;114:121-7). This highlights the importance of weight loss and healthy lifestyle choices in this population. The American Cancer Society recommends focusing on healthy lifestyles in cancer survivors. Key recommendations include the maintenance of healthy weight or weight loss for the overweight/obese, physical activity with at least 30 minutes of moderate activity on 5 or more days per week, a healthy diet with at least five servings of fruits and vegetables per day with limited processed foods and red meats, and limited alcohol intake (CA Cancer J. Clin. 2012;62:243).
Practicing gynecologists should appreciate the increasing rates of endometrial cancer and remain highly suspicious of abnormal uterine bleeding in their obese patients. Early detection of cancers and modification of lifestyle remain the mainstay of improving outcomes in obese patients.
Dr. Gehrig is professor and director of gynecologic oncology at the University of North Carolina at Chapel Hill. Dr. Clark is a chief resident in the department of obstetrics and gynecology at the university. Dr. Gehrig and Dr. Clark have no relevant conflicts of interest.*
* This story was updated 1/27/2014
For the last decade, the obesity epidemic in the United States has been well recognized. In 2001, the surgeon general made a call to action to combat obesity. Despite this effort, obesity rates in the United States continued to rise, and in 2009-2010, more than one third (35.7%) of adults in the United States were classified as obese, according to the Centers for Disease Control and Prevention.
The definition of obesity relies on the body mass index. BMI is defined as a person’s weight in kilograms divided by the individual’s height in meters squared. Overweight is defined as a BMI of 25-29.9 kg/m2 and obesity as a BMI of greater than 30 kg/m2. Obesity has been further classified by the World Health Organization into class I (BMI, 30-34.9 kg/m2), class II (BMI, 35-39.9 kg/ m2), and class III (BMI, greater than 40 kg/ m2).
In the United States in 2013, there were approximately 50,000 new cases and more than 8,000 deaths from endometrial cancer (CA Cancer J. Clin. 2013;63:11-30). Rates of endometrial cancer have risen steadily along with the obesity epidemic. This is no surprise, as obesity has been linked to the development of endometrial cancer. It is believed that high levels of circulating estrogen created by adipose tissue convert androstenedione to estrone, and there is aromatization of androgens. For each 5-kg/m2 increase in BMI, there is an increased risk of development of endometrial cancer (relative risk, 1.59) (Lancet 2008;371:569). While many physicians realize the link between obesity and the hyperestrogenic state associated with endometrial cancers, increased BMI is also associated with an increased risk of ovarian cancer (odds ratio, 1.3) (Eur. J. Cancer 2007;43:690).
In addition to increasing the risk of developing gynecologic cancers, obesity also increases the risk of death from all gynecologic malignancies. In the Cancer Prevention Study II, a large prospective cohort study, a BMI greater than 35 was associated with increased mortality compared with normal weight in ovarian (RR, 1.51), endometrial (RR, 2.77), and cervical cancer (RR, 3.20) (N. Engl. J. Med. 2003;348:1625). The same study found that those with a BMI greater than 40 with endometrial cancer had a relative risk of death of 6.25.
The increased mortality seen in obese endometrial cancer patients is particularly striking, given the fact that these women are more likely to have less-aggressive histologies and earlier-stage cancers (Gynecol. Oncol. 2009;90:150-7; Gynecol. Oncol. 2009;114:121-7). This highlights the importance of weight loss and healthy lifestyle choices in this population. The American Cancer Society recommends focusing on healthy lifestyles in cancer survivors. Key recommendations include the maintenance of healthy weight or weight loss for the overweight/obese, physical activity with at least 30 minutes of moderate activity on 5 or more days per week, a healthy diet with at least five servings of fruits and vegetables per day with limited processed foods and red meats, and limited alcohol intake (CA Cancer J. Clin. 2012;62:243).
Practicing gynecologists should appreciate the increasing rates of endometrial cancer and remain highly suspicious of abnormal uterine bleeding in their obese patients. Early detection of cancers and modification of lifestyle remain the mainstay of improving outcomes in obese patients.
Dr. Gehrig is professor and director of gynecologic oncology at the University of North Carolina at Chapel Hill. Dr. Clark is a chief resident in the department of obstetrics and gynecology at the university. Dr. Gehrig and Dr. Clark have no relevant conflicts of interest.*
* This story was updated 1/27/2014
For the last decade, the obesity epidemic in the United States has been well recognized. In 2001, the surgeon general made a call to action to combat obesity. Despite this effort, obesity rates in the United States continued to rise, and in 2009-2010, more than one third (35.7%) of adults in the United States were classified as obese, according to the Centers for Disease Control and Prevention.
The definition of obesity relies on the body mass index. BMI is defined as a person’s weight in kilograms divided by the individual’s height in meters squared. Overweight is defined as a BMI of 25-29.9 kg/m2 and obesity as a BMI of greater than 30 kg/m2. Obesity has been further classified by the World Health Organization into class I (BMI, 30-34.9 kg/m2), class II (BMI, 35-39.9 kg/ m2), and class III (BMI, greater than 40 kg/ m2).
In the United States in 2013, there were approximately 50,000 new cases and more than 8,000 deaths from endometrial cancer (CA Cancer J. Clin. 2013;63:11-30). Rates of endometrial cancer have risen steadily along with the obesity epidemic. This is no surprise, as obesity has been linked to the development of endometrial cancer. It is believed that high levels of circulating estrogen created by adipose tissue convert androstenedione to estrone, and there is aromatization of androgens. For each 5-kg/m2 increase in BMI, there is an increased risk of development of endometrial cancer (relative risk, 1.59) (Lancet 2008;371:569). While many physicians realize the link between obesity and the hyperestrogenic state associated with endometrial cancers, increased BMI is also associated with an increased risk of ovarian cancer (odds ratio, 1.3) (Eur. J. Cancer 2007;43:690).
In addition to increasing the risk of developing gynecologic cancers, obesity also increases the risk of death from all gynecologic malignancies. In the Cancer Prevention Study II, a large prospective cohort study, a BMI greater than 35 was associated with increased mortality compared with normal weight in ovarian (RR, 1.51), endometrial (RR, 2.77), and cervical cancer (RR, 3.20) (N. Engl. J. Med. 2003;348:1625). The same study found that those with a BMI greater than 40 with endometrial cancer had a relative risk of death of 6.25.
The increased mortality seen in obese endometrial cancer patients is particularly striking, given the fact that these women are more likely to have less-aggressive histologies and earlier-stage cancers (Gynecol. Oncol. 2009;90:150-7; Gynecol. Oncol. 2009;114:121-7). This highlights the importance of weight loss and healthy lifestyle choices in this population. The American Cancer Society recommends focusing on healthy lifestyles in cancer survivors. Key recommendations include the maintenance of healthy weight or weight loss for the overweight/obese, physical activity with at least 30 minutes of moderate activity on 5 or more days per week, a healthy diet with at least five servings of fruits and vegetables per day with limited processed foods and red meats, and limited alcohol intake (CA Cancer J. Clin. 2012;62:243).
Practicing gynecologists should appreciate the increasing rates of endometrial cancer and remain highly suspicious of abnormal uterine bleeding in their obese patients. Early detection of cancers and modification of lifestyle remain the mainstay of improving outcomes in obese patients.
Dr. Gehrig is professor and director of gynecologic oncology at the University of North Carolina at Chapel Hill. Dr. Clark is a chief resident in the department of obstetrics and gynecology at the university. Dr. Gehrig and Dr. Clark have no relevant conflicts of interest.*
* This story was updated 1/27/2014

Group simulates blood vessel growth
Louis Heiser & Robert Ackland
Bioengineers say they’ve found a way to accurately predict blood vessel growth, and this finding has implications for cancers and other diseases.
The team discovered that tiny blood vessels grow better in the lab if the tissue surrounding them is less dense.
And this discovery allowed them to create a computer simulation that can accurately predict such growth.
“Better understanding of the processes that regulate the growth of blood vessels puts us in a position, ultimately, to develop new treatments for diseases related to blood vessel growth,” said study author Jeff Weiss, PhD, of the University of Utah in Salt Lake City.
Dr Weiss and his colleagues described their research in PLOS ONE.
Like some previous studies, the group’s research showed that capillaries grow, branch, and interconnect best when the density of the surrounding tissue, the extracellular matrix, is lower rather than higher. But unlike earlier research, Dr Weiss and his colleagues used pieces of real blood vessels from rats (rather than single cells).
Earlier work also focused on how the extracellular matrix, made mostly of collagen, sends chemical signals to promote capillary growth. The current study focused more on how the collagen’s mechanical or physical properties—specifically, the density or stiffness of the matrix—affect blood vessel growth.
Both the lab experiments and computer simulations showed that the denser or stiffer this collagen matrix, the more difficult it is for blood vessels to form a network necessary to supply blood to living tissue.
Growing blood vessels
To grow a network of blood vessels, the researchers extracted blood vessel fragments from the fat tissues of rats and suspended them in liquid. This extract contained 35,000 of those blood-vessel fragments per mL of solution.
The blood vessel fragments were grown in plastic plates with tiny mold-like wells filled with gel-like collagen as the extracellular matrix. The team cultured the fragments for 6 days with 3 densities of collagen: 2 mg, 3 mg, and 4 mg of collagen per mL of solution.
Vessels in the lower-density collagen grew and branched more, had fewer dead ends, and interconnected with each other better than the vessels growing in the higher-density collagen. These blood vessel networks mirrored those found in living mammals.
Simulating growth
The vessels grown in the lab provided data on total length of the vessels, the degree to which they connected into a network of vessels, and the number of vessels branches and dead ends.
And these data allowed the researchers to program a 3-D computer simulation that accurately predicted blood vessel network formation based on collagen matrix density.
“Now, we can answer all sorts of ‘what if’ questions about the geometry of these tissues, their shape, boundaries, initial densities, and mechanical properties,” Dr Weiss said. “We can use the computer to predict the influence that these factors have in the layout of a vascular network structure.”
The 3-D computer simulation also enabled the researchers to “conduct” experiments that couldn’t be done in the lab. One simulation showed blood vessels grow easily from denser toward less-dense collagen, but not the other way around.
A second simulation showed that vessels grew in collagen, except where a dense piece of collagen was placed in the center of less-dense collagen.
The third simulation showed that when researchers simulated 2 bands of less-dense collagen surrounded by bands of stiffer collagen, the nerve vessels grew along the bands of lower density.
Applications for cancer, other diseases
The researchers said these findings could ultimately be applied to aid the development of treatments for patients with cancer or diabetes, as well as patients who have had a heart attack and those who require tissue implants.
By better understanding the role that density of surrounding tissue plays in vessel formation, bioengineers could prepare “prevascularized” implantable tissues already equipped with blood vessels that match a patient’s blood vessel structure.
Prevascularized tissues might also help diabetes patients suffering from wounds that heal slowly—if at all—due to impaired blood microcirculation. Implanted skin grafts with their own blood vessels could stimulate blood flow to promote healing of diabetic ulcers.
Dr Weiss said he envisions prevascularized patches rehabilitating heart muscle that is damaged when a heart attack cuts off part of the heart’s oxygen supply, turning some of the heart into stiff scar tissue. A tissue patch implanted on the scar tissue could encourage blood vessel regrowth to repair the damaged, oxygen-deprived heart muscle.
As for cancer metastasis, most tumors begin as dense, blood-free masses. To grow and spread, the tumor tricks the body into fueling it with oxygenated blood vessels.
“The vessels grow in and then provide a pathway for the tumor to spread,” Dr Weiss noted. “This research will help us understand the physical parameters that control whether blood vessels reach the tumor.” ![]()
Louis Heiser & Robert Ackland
Bioengineers say they’ve found a way to accurately predict blood vessel growth, and this finding has implications for cancers and other diseases.
The team discovered that tiny blood vessels grow better in the lab if the tissue surrounding them is less dense.
And this discovery allowed them to create a computer simulation that can accurately predict such growth.
“Better understanding of the processes that regulate the growth of blood vessels puts us in a position, ultimately, to develop new treatments for diseases related to blood vessel growth,” said study author Jeff Weiss, PhD, of the University of Utah in Salt Lake City.
Dr Weiss and his colleagues described their research in PLOS ONE.
Like some previous studies, the group’s research showed that capillaries grow, branch, and interconnect best when the density of the surrounding tissue, the extracellular matrix, is lower rather than higher. But unlike earlier research, Dr Weiss and his colleagues used pieces of real blood vessels from rats (rather than single cells).
Earlier work also focused on how the extracellular matrix, made mostly of collagen, sends chemical signals to promote capillary growth. The current study focused more on how the collagen’s mechanical or physical properties—specifically, the density or stiffness of the matrix—affect blood vessel growth.
Both the lab experiments and computer simulations showed that the denser or stiffer this collagen matrix, the more difficult it is for blood vessels to form a network necessary to supply blood to living tissue.
Growing blood vessels
To grow a network of blood vessels, the researchers extracted blood vessel fragments from the fat tissues of rats and suspended them in liquid. This extract contained 35,000 of those blood-vessel fragments per mL of solution.
The blood vessel fragments were grown in plastic plates with tiny mold-like wells filled with gel-like collagen as the extracellular matrix. The team cultured the fragments for 6 days with 3 densities of collagen: 2 mg, 3 mg, and 4 mg of collagen per mL of solution.
Vessels in the lower-density collagen grew and branched more, had fewer dead ends, and interconnected with each other better than the vessels growing in the higher-density collagen. These blood vessel networks mirrored those found in living mammals.
Simulating growth
The vessels grown in the lab provided data on total length of the vessels, the degree to which they connected into a network of vessels, and the number of vessels branches and dead ends.
And these data allowed the researchers to program a 3-D computer simulation that accurately predicted blood vessel network formation based on collagen matrix density.
“Now, we can answer all sorts of ‘what if’ questions about the geometry of these tissues, their shape, boundaries, initial densities, and mechanical properties,” Dr Weiss said. “We can use the computer to predict the influence that these factors have in the layout of a vascular network structure.”
The 3-D computer simulation also enabled the researchers to “conduct” experiments that couldn’t be done in the lab. One simulation showed blood vessels grow easily from denser toward less-dense collagen, but not the other way around.
A second simulation showed that vessels grew in collagen, except where a dense piece of collagen was placed in the center of less-dense collagen.
The third simulation showed that when researchers simulated 2 bands of less-dense collagen surrounded by bands of stiffer collagen, the nerve vessels grew along the bands of lower density.
Applications for cancer, other diseases
The researchers said these findings could ultimately be applied to aid the development of treatments for patients with cancer or diabetes, as well as patients who have had a heart attack and those who require tissue implants.
By better understanding the role that density of surrounding tissue plays in vessel formation, bioengineers could prepare “prevascularized” implantable tissues already equipped with blood vessels that match a patient’s blood vessel structure.
Prevascularized tissues might also help diabetes patients suffering from wounds that heal slowly—if at all—due to impaired blood microcirculation. Implanted skin grafts with their own blood vessels could stimulate blood flow to promote healing of diabetic ulcers.
Dr Weiss said he envisions prevascularized patches rehabilitating heart muscle that is damaged when a heart attack cuts off part of the heart’s oxygen supply, turning some of the heart into stiff scar tissue. A tissue patch implanted on the scar tissue could encourage blood vessel regrowth to repair the damaged, oxygen-deprived heart muscle.
As for cancer metastasis, most tumors begin as dense, blood-free masses. To grow and spread, the tumor tricks the body into fueling it with oxygenated blood vessels.
“The vessels grow in and then provide a pathway for the tumor to spread,” Dr Weiss noted. “This research will help us understand the physical parameters that control whether blood vessels reach the tumor.” ![]()
Louis Heiser & Robert Ackland
Bioengineers say they’ve found a way to accurately predict blood vessel growth, and this finding has implications for cancers and other diseases.
The team discovered that tiny blood vessels grow better in the lab if the tissue surrounding them is less dense.
And this discovery allowed them to create a computer simulation that can accurately predict such growth.
“Better understanding of the processes that regulate the growth of blood vessels puts us in a position, ultimately, to develop new treatments for diseases related to blood vessel growth,” said study author Jeff Weiss, PhD, of the University of Utah in Salt Lake City.
Dr Weiss and his colleagues described their research in PLOS ONE.
Like some previous studies, the group’s research showed that capillaries grow, branch, and interconnect best when the density of the surrounding tissue, the extracellular matrix, is lower rather than higher. But unlike earlier research, Dr Weiss and his colleagues used pieces of real blood vessels from rats (rather than single cells).
Earlier work also focused on how the extracellular matrix, made mostly of collagen, sends chemical signals to promote capillary growth. The current study focused more on how the collagen’s mechanical or physical properties—specifically, the density or stiffness of the matrix—affect blood vessel growth.
Both the lab experiments and computer simulations showed that the denser or stiffer this collagen matrix, the more difficult it is for blood vessels to form a network necessary to supply blood to living tissue.
Growing blood vessels
To grow a network of blood vessels, the researchers extracted blood vessel fragments from the fat tissues of rats and suspended them in liquid. This extract contained 35,000 of those blood-vessel fragments per mL of solution.
The blood vessel fragments were grown in plastic plates with tiny mold-like wells filled with gel-like collagen as the extracellular matrix. The team cultured the fragments for 6 days with 3 densities of collagen: 2 mg, 3 mg, and 4 mg of collagen per mL of solution.
Vessels in the lower-density collagen grew and branched more, had fewer dead ends, and interconnected with each other better than the vessels growing in the higher-density collagen. These blood vessel networks mirrored those found in living mammals.
Simulating growth
The vessels grown in the lab provided data on total length of the vessels, the degree to which they connected into a network of vessels, and the number of vessels branches and dead ends.
And these data allowed the researchers to program a 3-D computer simulation that accurately predicted blood vessel network formation based on collagen matrix density.
“Now, we can answer all sorts of ‘what if’ questions about the geometry of these tissues, their shape, boundaries, initial densities, and mechanical properties,” Dr Weiss said. “We can use the computer to predict the influence that these factors have in the layout of a vascular network structure.”
The 3-D computer simulation also enabled the researchers to “conduct” experiments that couldn’t be done in the lab. One simulation showed blood vessels grow easily from denser toward less-dense collagen, but not the other way around.
A second simulation showed that vessels grew in collagen, except where a dense piece of collagen was placed in the center of less-dense collagen.
The third simulation showed that when researchers simulated 2 bands of less-dense collagen surrounded by bands of stiffer collagen, the nerve vessels grew along the bands of lower density.
Applications for cancer, other diseases
The researchers said these findings could ultimately be applied to aid the development of treatments for patients with cancer or diabetes, as well as patients who have had a heart attack and those who require tissue implants.
By better understanding the role that density of surrounding tissue plays in vessel formation, bioengineers could prepare “prevascularized” implantable tissues already equipped with blood vessels that match a patient’s blood vessel structure.
Prevascularized tissues might also help diabetes patients suffering from wounds that heal slowly—if at all—due to impaired blood microcirculation. Implanted skin grafts with their own blood vessels could stimulate blood flow to promote healing of diabetic ulcers.
Dr Weiss said he envisions prevascularized patches rehabilitating heart muscle that is damaged when a heart attack cuts off part of the heart’s oxygen supply, turning some of the heart into stiff scar tissue. A tissue patch implanted on the scar tissue could encourage blood vessel regrowth to repair the damaged, oxygen-deprived heart muscle.
As for cancer metastasis, most tumors begin as dense, blood-free masses. To grow and spread, the tumor tricks the body into fueling it with oxygenated blood vessels.
“The vessels grow in and then provide a pathway for the tumor to spread,” Dr Weiss noted. “This research will help us understand the physical parameters that control whether blood vessels reach the tumor.” ![]()
Adding idelalisib improves CLL treatment

The PI3K delta inhibitor idelalisib could turn chronic lymphocytic leukemia (CLL) into a highly treatable disease, according to the lead investigator of a phase 3 trial.
Results of the trial showed that adding idelalisib to treatment with rituximab can improve response and survival rates in patients with relapsed CLL.
In fact, the study was stopped early because idelalisib had a significant impact on progression-free survival.
“This study, and others we have conducted on idelalisib, demonstrates that we may no longer need to use chemotherapy in CLL,” said lead investigator Richard R. Furman, MD, of Weill Cornell Medical College in New York.
“Even if this cancer remains incurable, it now can be treated as if it was a chronic disease—with a pill, in the same way that high blood pressure is treated.”
Dr Furman and his colleagues reported the results of this study in The New England Journal of Medicine.
The trial was funded by Gilead, the makers of idelalisib. Dr Furman has served as an advisor for this company.
The study included 220 CLL patients who could not receive chemotherapy. Nearly two-thirds of the patients had advanced-stage disease. The median time from CLL diagnosis was 9 years, and patients had received a median of 3 previous treatments.
Most of the patients were 65 or older (78%). Forty percent had at least moderate renal dysfunction, and 35% had poor bone marrow function.
Half of the patients were randomized to receive idelalisib plus rituximab and the other half to rituximab plus placebo.
“It is remarkable how quickly idelalisib worked in this heavily treated group of patients, many of whom were resistant to chemotherapy,” Dr Furman said. “We saw responses within a week.”
Patients in the idelalisib arm had a much higher overall response rate than patients in the placebo arm—81% and 13%, respectively (P<0.001). But all responses were partial responses.
At 24 weeks, the rate of progression-free survival was 93% in the idelalisib arm and 46% in the placebo arm (P<0.001). The median progression-free survival was 5.5 months in the placebo arm and not reached in the idelalisib arm (P<0.001).
And at 12 months, the overall survival rate was 92% in the idelalisib arm and 80% in the placebo arm (P=0.02).
The difference in outcomes between the treatment groups prompted an independent data-monitoring committee to halt the study early, in October 2013, so that all participants could receive idelalisib.
Most adverse events (AEs), in either treatment group, were grade 2 or lower. The most common AEs in the idelalisib arm were pyrexia, fatigue, nausea, chills, and diarrhea. In the placebo arm, the most common AEs were infusion-related reactions, fatigue, cough, nausea, and dyspnea.
There were more serious AEs in the idelalisib arm than in the placebo arm—40% and 35%, respectively. The most frequent serious AEs were pneumonia, pyrexia, and febrile neutropenia (in both treatment arms).
Despite these events, the researchers considered idelalisib to be well-tolerated in this patient population.
“Having a treatment like idelalisib, which is highly effective and well-tolerated, and thus can generate responses in patients that are unable to tolerate treatment and unlikely to respond, indicates the potential for idelalisib in all patients,” Dr Furman said. ![]()
The PI3K delta inhibitor idelalisib could turn chronic lymphocytic leukemia (CLL) into a highly treatable disease, according to the lead investigator of a phase 3 trial.
Results of the trial showed that adding idelalisib to treatment with rituximab can improve response and survival rates in patients with relapsed CLL.
In fact, the study was stopped early because idelalisib had a significant impact on progression-free survival.
“This study, and others we have conducted on idelalisib, demonstrates that we may no longer need to use chemotherapy in CLL,” said lead investigator Richard R. Furman, MD, of Weill Cornell Medical College in New York.
“Even if this cancer remains incurable, it now can be treated as if it was a chronic disease—with a pill, in the same way that high blood pressure is treated.”
Dr Furman and his colleagues reported the results of this study in The New England Journal of Medicine.
The trial was funded by Gilead, the makers of idelalisib. Dr Furman has served as an advisor for this company.
The study included 220 CLL patients who could not receive chemotherapy. Nearly two-thirds of the patients had advanced-stage disease. The median time from CLL diagnosis was 9 years, and patients had received a median of 3 previous treatments.
Most of the patients were 65 or older (78%). Forty percent had at least moderate renal dysfunction, and 35% had poor bone marrow function.
Half of the patients were randomized to receive idelalisib plus rituximab and the other half to rituximab plus placebo.
“It is remarkable how quickly idelalisib worked in this heavily treated group of patients, many of whom were resistant to chemotherapy,” Dr Furman said. “We saw responses within a week.”
Patients in the idelalisib arm had a much higher overall response rate than patients in the placebo arm—81% and 13%, respectively (P<0.001). But all responses were partial responses.
At 24 weeks, the rate of progression-free survival was 93% in the idelalisib arm and 46% in the placebo arm (P<0.001). The median progression-free survival was 5.5 months in the placebo arm and not reached in the idelalisib arm (P<0.001).
And at 12 months, the overall survival rate was 92% in the idelalisib arm and 80% in the placebo arm (P=0.02).
The difference in outcomes between the treatment groups prompted an independent data-monitoring committee to halt the study early, in October 2013, so that all participants could receive idelalisib.
Most adverse events (AEs), in either treatment group, were grade 2 or lower. The most common AEs in the idelalisib arm were pyrexia, fatigue, nausea, chills, and diarrhea. In the placebo arm, the most common AEs were infusion-related reactions, fatigue, cough, nausea, and dyspnea.
There were more serious AEs in the idelalisib arm than in the placebo arm—40% and 35%, respectively. The most frequent serious AEs were pneumonia, pyrexia, and febrile neutropenia (in both treatment arms).
Despite these events, the researchers considered idelalisib to be well-tolerated in this patient population.
“Having a treatment like idelalisib, which is highly effective and well-tolerated, and thus can generate responses in patients that are unable to tolerate treatment and unlikely to respond, indicates the potential for idelalisib in all patients,” Dr Furman said. ![]()
The PI3K delta inhibitor idelalisib could turn chronic lymphocytic leukemia (CLL) into a highly treatable disease, according to the lead investigator of a phase 3 trial.
Results of the trial showed that adding idelalisib to treatment with rituximab can improve response and survival rates in patients with relapsed CLL.
In fact, the study was stopped early because idelalisib had a significant impact on progression-free survival.
“This study, and others we have conducted on idelalisib, demonstrates that we may no longer need to use chemotherapy in CLL,” said lead investigator Richard R. Furman, MD, of Weill Cornell Medical College in New York.
“Even if this cancer remains incurable, it now can be treated as if it was a chronic disease—with a pill, in the same way that high blood pressure is treated.”
Dr Furman and his colleagues reported the results of this study in The New England Journal of Medicine.
The trial was funded by Gilead, the makers of idelalisib. Dr Furman has served as an advisor for this company.
The study included 220 CLL patients who could not receive chemotherapy. Nearly two-thirds of the patients had advanced-stage disease. The median time from CLL diagnosis was 9 years, and patients had received a median of 3 previous treatments.
Most of the patients were 65 or older (78%). Forty percent had at least moderate renal dysfunction, and 35% had poor bone marrow function.
Half of the patients were randomized to receive idelalisib plus rituximab and the other half to rituximab plus placebo.
“It is remarkable how quickly idelalisib worked in this heavily treated group of patients, many of whom were resistant to chemotherapy,” Dr Furman said. “We saw responses within a week.”
Patients in the idelalisib arm had a much higher overall response rate than patients in the placebo arm—81% and 13%, respectively (P<0.001). But all responses were partial responses.
At 24 weeks, the rate of progression-free survival was 93% in the idelalisib arm and 46% in the placebo arm (P<0.001). The median progression-free survival was 5.5 months in the placebo arm and not reached in the idelalisib arm (P<0.001).
And at 12 months, the overall survival rate was 92% in the idelalisib arm and 80% in the placebo arm (P=0.02).
The difference in outcomes between the treatment groups prompted an independent data-monitoring committee to halt the study early, in October 2013, so that all participants could receive idelalisib.
Most adverse events (AEs), in either treatment group, were grade 2 or lower. The most common AEs in the idelalisib arm were pyrexia, fatigue, nausea, chills, and diarrhea. In the placebo arm, the most common AEs were infusion-related reactions, fatigue, cough, nausea, and dyspnea.
There were more serious AEs in the idelalisib arm than in the placebo arm—40% and 35%, respectively. The most frequent serious AEs were pneumonia, pyrexia, and febrile neutropenia (in both treatment arms).
Despite these events, the researchers considered idelalisib to be well-tolerated in this patient population.
“Having a treatment like idelalisib, which is highly effective and well-tolerated, and thus can generate responses in patients that are unable to tolerate treatment and unlikely to respond, indicates the potential for idelalisib in all patients,” Dr Furman said. ![]()
Study explains why FDA rejects new drug applications

Credit: Esther Dyson
New research suggests that drugs are often rejected by the US Food and Drug Administration (FDA), not because they are unsafe or ineffective, but because there is not enough evidence to determine the drugs’ safety and efficacy.
Investigators reviewed about 300 drug applications and discovered a number of reasons why drugs were denied approval on the first try.
The FDA cited issues with dosing, trial populations, study endpoints, and inconsistencies in data as reasons for denial.
Leonard V. Sacks, MBBCh, of the FDA in Silver Springs, Maryland, and his colleagues conducted this research and reported the results in JAMA.
The team reviewed marketing applications for all new molecular entities (NMEs; active ingredients never before marketed in the US) first submitted to the FDA between 2000 and 2012.
They used FDA correspondence and reviews to determine the scientific and regulatory reasons approvals were delayed or denied.
Of the 302 NME applications, 222 (73.5%) ultimately achieved marketing approval.
Half of all NMEs (151) were rejected on the first try, but 71 (47.0%) of these were approved following resubmission. The median time to approval was 435 days after the first action letter (range, 47-2374 days).
Drugs were denied approval for a number of reasons, including:
- Uncertainty about the optimal dose to maximize efficacy and minimize safety risks (15.9%)
- Inconsistent results for multiple predefined study endpoints (13.2%)
- Trial endpoints were unsatisfactory (13.2%)
- Inconsistencies in efficacy for portions of the study population (11.3%)
- The populations studied did not reflect the populations likely to use the drug (7.3%).
The investigators also found that the frequency of safety deficiencies was similar among never-approved drugs and drugs with delayed approval. However, efficacy deficiencies were significantly more frequent among the never-approved drugs than among those with delayed approvals.
There were 48 drugs with initial efficacy concerns, and only 31.3% of these were eventually approved, compared to 61.5% of the 39 drugs with safety concerns alone.
There were 20 drugs (13.2%) that, despite showing superiority to placebo, were considered to have inadequate efficacy compared with the standard of care.
The investigators said that, taken together, these findings suggest there is room for improvement in new drug applications. But if drug sponsors increase communication with the FDA, particularly with regard to study design, they could reduce delays in drug approval. ![]()
Credit: Esther Dyson
New research suggests that drugs are often rejected by the US Food and Drug Administration (FDA), not because they are unsafe or ineffective, but because there is not enough evidence to determine the drugs’ safety and efficacy.
Investigators reviewed about 300 drug applications and discovered a number of reasons why drugs were denied approval on the first try.
The FDA cited issues with dosing, trial populations, study endpoints, and inconsistencies in data as reasons for denial.
Leonard V. Sacks, MBBCh, of the FDA in Silver Springs, Maryland, and his colleagues conducted this research and reported the results in JAMA.
The team reviewed marketing applications for all new molecular entities (NMEs; active ingredients never before marketed in the US) first submitted to the FDA between 2000 and 2012.
They used FDA correspondence and reviews to determine the scientific and regulatory reasons approvals were delayed or denied.
Of the 302 NME applications, 222 (73.5%) ultimately achieved marketing approval.
Half of all NMEs (151) were rejected on the first try, but 71 (47.0%) of these were approved following resubmission. The median time to approval was 435 days after the first action letter (range, 47-2374 days).
Drugs were denied approval for a number of reasons, including:
- Uncertainty about the optimal dose to maximize efficacy and minimize safety risks (15.9%)
- Inconsistent results for multiple predefined study endpoints (13.2%)
- Trial endpoints were unsatisfactory (13.2%)
- Inconsistencies in efficacy for portions of the study population (11.3%)
- The populations studied did not reflect the populations likely to use the drug (7.3%).
The investigators also found that the frequency of safety deficiencies was similar among never-approved drugs and drugs with delayed approval. However, efficacy deficiencies were significantly more frequent among the never-approved drugs than among those with delayed approvals.
There were 48 drugs with initial efficacy concerns, and only 31.3% of these were eventually approved, compared to 61.5% of the 39 drugs with safety concerns alone.
There were 20 drugs (13.2%) that, despite showing superiority to placebo, were considered to have inadequate efficacy compared with the standard of care.
The investigators said that, taken together, these findings suggest there is room for improvement in new drug applications. But if drug sponsors increase communication with the FDA, particularly with regard to study design, they could reduce delays in drug approval. ![]()
Credit: Esther Dyson
New research suggests that drugs are often rejected by the US Food and Drug Administration (FDA), not because they are unsafe or ineffective, but because there is not enough evidence to determine the drugs’ safety and efficacy.
Investigators reviewed about 300 drug applications and discovered a number of reasons why drugs were denied approval on the first try.
The FDA cited issues with dosing, trial populations, study endpoints, and inconsistencies in data as reasons for denial.
Leonard V. Sacks, MBBCh, of the FDA in Silver Springs, Maryland, and his colleagues conducted this research and reported the results in JAMA.
The team reviewed marketing applications for all new molecular entities (NMEs; active ingredients never before marketed in the US) first submitted to the FDA between 2000 and 2012.
They used FDA correspondence and reviews to determine the scientific and regulatory reasons approvals were delayed or denied.
Of the 302 NME applications, 222 (73.5%) ultimately achieved marketing approval.
Half of all NMEs (151) were rejected on the first try, but 71 (47.0%) of these were approved following resubmission. The median time to approval was 435 days after the first action letter (range, 47-2374 days).
Drugs were denied approval for a number of reasons, including:
- Uncertainty about the optimal dose to maximize efficacy and minimize safety risks (15.9%)
- Inconsistent results for multiple predefined study endpoints (13.2%)
- Trial endpoints were unsatisfactory (13.2%)
- Inconsistencies in efficacy for portions of the study population (11.3%)
- The populations studied did not reflect the populations likely to use the drug (7.3%).
The investigators also found that the frequency of safety deficiencies was similar among never-approved drugs and drugs with delayed approval. However, efficacy deficiencies were significantly more frequent among the never-approved drugs than among those with delayed approvals.
There were 48 drugs with initial efficacy concerns, and only 31.3% of these were eventually approved, compared to 61.5% of the 39 drugs with safety concerns alone.
There were 20 drugs (13.2%) that, despite showing superiority to placebo, were considered to have inadequate efficacy compared with the standard of care.
The investigators said that, taken together, these findings suggest there is room for improvement in new drug applications. But if drug sponsors increase communication with the FDA, particularly with regard to study design, they could reduce delays in drug approval. ![]()
Decline in Healthcare Employment Unlikely to Hit Hospitalists
Hospitalists needn't worry much about federal data released this month that shows healthcare employment figures dropping for the first time in a decade. A recent report from the Bureau of Labor Statistics reflects the loss of 6,000 jobs in healthcare last December, and employment gains falling to 17,000 per month on average in 2013 compared with 27,000 per month in 2012.
"The overall decline in healthcare spending and employment will have a small effect on hospitalist growth," says Anupam Jena, MD, PhD, assistant professor of healthcare policy and medicine at Harvard Medical School, and an internist at Massachusetts General Hospital, both in Boston. "It certainly is true that the demand for hospital care has decreased over the last two decades and will continue to decrease as hospitals and providers become more incentivized to keep patients out of the hospital. But this effect is offset by the fact that hospitalists continue to account for larger and larger shares of all inpatient care in the U.S."
Dr. Jena says the fact that healthcare companies lost 6,000 positions last December—the first monthly loss since July 2003, according to CNN—isn't surprising, as it comes on the heels of a report from the Centers for Medicare and Medicaid Services that the rate of increases in healthcare spending has slowed over the past four years as well.
Dr. Jena says that, while healthcare reform under the Affordable Care Act may "trim the fat" by reducing the amount of lower-value health services and the number of providers that perform them, HM is still a budding field. As long as the specialty continues to demonstrate value via cost savings and reduced length of stay, that scenario isn’t likely to change, he adds.
"Although healthcare reforms will probably reduce hospitalizations, I expect the demand for hospitalists to continue to grow," Dr. Jena says. "There are still 30% of hospitalizations in the U.S. that are not covered by hospitalists, which means there is room to grow."
Visit our website for more information on healthcare economics.
Hospitalists needn't worry much about federal data released this month that shows healthcare employment figures dropping for the first time in a decade. A recent report from the Bureau of Labor Statistics reflects the loss of 6,000 jobs in healthcare last December, and employment gains falling to 17,000 per month on average in 2013 compared with 27,000 per month in 2012.
"The overall decline in healthcare spending and employment will have a small effect on hospitalist growth," says Anupam Jena, MD, PhD, assistant professor of healthcare policy and medicine at Harvard Medical School, and an internist at Massachusetts General Hospital, both in Boston. "It certainly is true that the demand for hospital care has decreased over the last two decades and will continue to decrease as hospitals and providers become more incentivized to keep patients out of the hospital. But this effect is offset by the fact that hospitalists continue to account for larger and larger shares of all inpatient care in the U.S."
Dr. Jena says the fact that healthcare companies lost 6,000 positions last December—the first monthly loss since July 2003, according to CNN—isn't surprising, as it comes on the heels of a report from the Centers for Medicare and Medicaid Services that the rate of increases in healthcare spending has slowed over the past four years as well.
Dr. Jena says that, while healthcare reform under the Affordable Care Act may "trim the fat" by reducing the amount of lower-value health services and the number of providers that perform them, HM is still a budding field. As long as the specialty continues to demonstrate value via cost savings and reduced length of stay, that scenario isn’t likely to change, he adds.
"Although healthcare reforms will probably reduce hospitalizations, I expect the demand for hospitalists to continue to grow," Dr. Jena says. "There are still 30% of hospitalizations in the U.S. that are not covered by hospitalists, which means there is room to grow."
Visit our website for more information on healthcare economics.
Hospitalists needn't worry much about federal data released this month that shows healthcare employment figures dropping for the first time in a decade. A recent report from the Bureau of Labor Statistics reflects the loss of 6,000 jobs in healthcare last December, and employment gains falling to 17,000 per month on average in 2013 compared with 27,000 per month in 2012.
"The overall decline in healthcare spending and employment will have a small effect on hospitalist growth," says Anupam Jena, MD, PhD, assistant professor of healthcare policy and medicine at Harvard Medical School, and an internist at Massachusetts General Hospital, both in Boston. "It certainly is true that the demand for hospital care has decreased over the last two decades and will continue to decrease as hospitals and providers become more incentivized to keep patients out of the hospital. But this effect is offset by the fact that hospitalists continue to account for larger and larger shares of all inpatient care in the U.S."
Dr. Jena says the fact that healthcare companies lost 6,000 positions last December—the first monthly loss since July 2003, according to CNN—isn't surprising, as it comes on the heels of a report from the Centers for Medicare and Medicaid Services that the rate of increases in healthcare spending has slowed over the past four years as well.
Dr. Jena says that, while healthcare reform under the Affordable Care Act may "trim the fat" by reducing the amount of lower-value health services and the number of providers that perform them, HM is still a budding field. As long as the specialty continues to demonstrate value via cost savings and reduced length of stay, that scenario isn’t likely to change, he adds.
"Although healthcare reforms will probably reduce hospitalizations, I expect the demand for hospitalists to continue to grow," Dr. Jena says. "There are still 30% of hospitalizations in the U.S. that are not covered by hospitalists, which means there is room to grow."
Visit our website for more information on healthcare economics.
Oral Proton Pump Inhibitors (PPIs) as Effective as IV PPIs in Peptic Ulcer Bleeding
Clinical question: In patients with peptic ulcer bleeding, are oral PPIs of equal benefit to intravenous PPIs?
Background: PPI therapy has been shown in several studies to reduce re-bleeding risk in patients when used adjunctively for peptic ulcer bleeding. In spite of this data, there is still uncertainty about the optimal dose and route of administration.
Study design: Meta-analysis of prospective, randomized control trials.
Setting: OVID database search in June 2012.
Synopsis: A literature search identified six prospective randomized control trials. Overall, 615 patients were included across the six trials. No significant difference in risk of re-bleeding was discovered between the two groups (8.6% oral vs. 9.3% IV, RR: 0.92, 95% CI: 0.56–1.5). Length of hospital stay was statistically significantly lower for oral PPIs (-0.74 day, 95% CI: -1.10 to -0.39 day).
Because these findings are based on a meta-analysis of studies with notable flaws—including lack of blinding—it is difficult to draw any definitive conclusions from this data. Hospitalists should use care before changing their practice patterns, given the risk of bias and need for further study.
Bottom line: Oral PPIs may reduce hospital length of stay without an increased risk of re-bleeding; however, further study with a well-powered, double-blind, randomized control trial is necessary.
Citation: Tsoi KK, Hirai HW, Sung JJ. Meta-analysis: Comparison of oral vs. intravenous proton pump inhibitors in patients with peptic ulcer bleeding. Aliment Pharmacol Ther. 2013;38(7):721-728.
Visit our website for more information on the use of proton pump inhibitors.
Clinical question: In patients with peptic ulcer bleeding, are oral PPIs of equal benefit to intravenous PPIs?
Background: PPI therapy has been shown in several studies to reduce re-bleeding risk in patients when used adjunctively for peptic ulcer bleeding. In spite of this data, there is still uncertainty about the optimal dose and route of administration.
Study design: Meta-analysis of prospective, randomized control trials.
Setting: OVID database search in June 2012.
Synopsis: A literature search identified six prospective randomized control trials. Overall, 615 patients were included across the six trials. No significant difference in risk of re-bleeding was discovered between the two groups (8.6% oral vs. 9.3% IV, RR: 0.92, 95% CI: 0.56–1.5). Length of hospital stay was statistically significantly lower for oral PPIs (-0.74 day, 95% CI: -1.10 to -0.39 day).
Because these findings are based on a meta-analysis of studies with notable flaws—including lack of blinding—it is difficult to draw any definitive conclusions from this data. Hospitalists should use care before changing their practice patterns, given the risk of bias and need for further study.
Bottom line: Oral PPIs may reduce hospital length of stay without an increased risk of re-bleeding; however, further study with a well-powered, double-blind, randomized control trial is necessary.
Citation: Tsoi KK, Hirai HW, Sung JJ. Meta-analysis: Comparison of oral vs. intravenous proton pump inhibitors in patients with peptic ulcer bleeding. Aliment Pharmacol Ther. 2013;38(7):721-728.
Visit our website for more information on the use of proton pump inhibitors.
Clinical question: In patients with peptic ulcer bleeding, are oral PPIs of equal benefit to intravenous PPIs?
Background: PPI therapy has been shown in several studies to reduce re-bleeding risk in patients when used adjunctively for peptic ulcer bleeding. In spite of this data, there is still uncertainty about the optimal dose and route of administration.
Study design: Meta-analysis of prospective, randomized control trials.
Setting: OVID database search in June 2012.
Synopsis: A literature search identified six prospective randomized control trials. Overall, 615 patients were included across the six trials. No significant difference in risk of re-bleeding was discovered between the two groups (8.6% oral vs. 9.3% IV, RR: 0.92, 95% CI: 0.56–1.5). Length of hospital stay was statistically significantly lower for oral PPIs (-0.74 day, 95% CI: -1.10 to -0.39 day).
Because these findings are based on a meta-analysis of studies with notable flaws—including lack of blinding—it is difficult to draw any definitive conclusions from this data. Hospitalists should use care before changing their practice patterns, given the risk of bias and need for further study.
Bottom line: Oral PPIs may reduce hospital length of stay without an increased risk of re-bleeding; however, further study with a well-powered, double-blind, randomized control trial is necessary.
Citation: Tsoi KK, Hirai HW, Sung JJ. Meta-analysis: Comparison of oral vs. intravenous proton pump inhibitors in patients with peptic ulcer bleeding. Aliment Pharmacol Ther. 2013;38(7):721-728.
Visit our website for more information on the use of proton pump inhibitors.
Storytelling
Tell me a story. Is there anyone who hasn’t uttered those four words? As humans we are hard wired to both tell and listen to stories. Indeed, professional storyteller Bill Harley, in a 2012 TEDx talk entitled, "Stories Out Loud," said that storytelling is "at the very center of what it means to be human."
This is why storytelling is a powerful marketing tool for you and your practice. In a noisy social media world, stories allow your voice to be heard.
Here are some reasons why you should be using storytelling to market your practice:
• A story is experiential – it shares an experience or observation.
• Stories help us make sense of our lives.
• They help you connect with your patients, build trust, and market your brand.
• They can capture your patients’ attention.
• They can inspire and appeal to emotions.
• Stories are easier to remember than facts and statistics.
• They feel authentic and help show the real you and real patients.
• Stories can be educational by putting difficult concepts into meaningful context.
• They can be an effective call to action for patients.
How powerful is storytelling? Do an Internet search of "patient stories," and the results will feature some of the country’s top hospitals such as the Mayo Clinic, Memorial Sloan-Kettering Cancer Center, and St. Jude's Children's Research Hospital. Take some time visiting these sites. Watch a few videos and ask yourself what makes them effective.
When it comes to marketing your practice, you can tell stories in person to patients, on video, in blog posts, or even in images on sites like Pinterest. Although videos can be done by a professional videographer using ambient lighting and music, they don’t have to be. You might decide to videotape a procedure with your video camera or upload patient stories using your iPhone.
Before using a story, remember to respect patients’ privacy and to get their consent appropriately if they are identifiable in a story. Make sure your story is short, relevant, and compelling and leaves the consumer with a better understanding of the issue.
Do you use stories in your practice? Do you have tips for physicians on how to use stories effectively in their practice? Please share them.
Dr. Benabio is a practicing dermatologist and physician director of health care transformation at Kaiser Permanente in San Diego. Connect with him on Twitter @Dermdoc or drop him a line at [email protected].
Tell me a story. Is there anyone who hasn’t uttered those four words? As humans we are hard wired to both tell and listen to stories. Indeed, professional storyteller Bill Harley, in a 2012 TEDx talk entitled, "Stories Out Loud," said that storytelling is "at the very center of what it means to be human."
This is why storytelling is a powerful marketing tool for you and your practice. In a noisy social media world, stories allow your voice to be heard.
Here are some reasons why you should be using storytelling to market your practice:
• A story is experiential – it shares an experience or observation.
• Stories help us make sense of our lives.
• They help you connect with your patients, build trust, and market your brand.
• They can capture your patients’ attention.
• They can inspire and appeal to emotions.
• Stories are easier to remember than facts and statistics.
• They feel authentic and help show the real you and real patients.
• Stories can be educational by putting difficult concepts into meaningful context.
• They can be an effective call to action for patients.
How powerful is storytelling? Do an Internet search of "patient stories," and the results will feature some of the country’s top hospitals such as the Mayo Clinic, Memorial Sloan-Kettering Cancer Center, and St. Jude's Children's Research Hospital. Take some time visiting these sites. Watch a few videos and ask yourself what makes them effective.
When it comes to marketing your practice, you can tell stories in person to patients, on video, in blog posts, or even in images on sites like Pinterest. Although videos can be done by a professional videographer using ambient lighting and music, they don’t have to be. You might decide to videotape a procedure with your video camera or upload patient stories using your iPhone.
Before using a story, remember to respect patients’ privacy and to get their consent appropriately if they are identifiable in a story. Make sure your story is short, relevant, and compelling and leaves the consumer with a better understanding of the issue.
Do you use stories in your practice? Do you have tips for physicians on how to use stories effectively in their practice? Please share them.
Dr. Benabio is a practicing dermatologist and physician director of health care transformation at Kaiser Permanente in San Diego. Connect with him on Twitter @Dermdoc or drop him a line at [email protected].
Tell me a story. Is there anyone who hasn’t uttered those four words? As humans we are hard wired to both tell and listen to stories. Indeed, professional storyteller Bill Harley, in a 2012 TEDx talk entitled, "Stories Out Loud," said that storytelling is "at the very center of what it means to be human."
This is why storytelling is a powerful marketing tool for you and your practice. In a noisy social media world, stories allow your voice to be heard.
Here are some reasons why you should be using storytelling to market your practice:
• A story is experiential – it shares an experience or observation.
• Stories help us make sense of our lives.
• They help you connect with your patients, build trust, and market your brand.
• They can capture your patients’ attention.
• They can inspire and appeal to emotions.
• Stories are easier to remember than facts and statistics.
• They feel authentic and help show the real you and real patients.
• Stories can be educational by putting difficult concepts into meaningful context.
• They can be an effective call to action for patients.
How powerful is storytelling? Do an Internet search of "patient stories," and the results will feature some of the country’s top hospitals such as the Mayo Clinic, Memorial Sloan-Kettering Cancer Center, and St. Jude's Children's Research Hospital. Take some time visiting these sites. Watch a few videos and ask yourself what makes them effective.
When it comes to marketing your practice, you can tell stories in person to patients, on video, in blog posts, or even in images on sites like Pinterest. Although videos can be done by a professional videographer using ambient lighting and music, they don’t have to be. You might decide to videotape a procedure with your video camera or upload patient stories using your iPhone.
Before using a story, remember to respect patients’ privacy and to get their consent appropriately if they are identifiable in a story. Make sure your story is short, relevant, and compelling and leaves the consumer with a better understanding of the issue.
Do you use stories in your practice? Do you have tips for physicians on how to use stories effectively in their practice? Please share them.
Dr. Benabio is a practicing dermatologist and physician director of health care transformation at Kaiser Permanente in San Diego. Connect with him on Twitter @Dermdoc or drop him a line at [email protected].
