User login
Renal denervation to treat resistant hypertension: Guarded optimism
Can a percutaneous catheter-based procedure effectively treat resistant hypertension?
Radiofrequency ablation of the renal sympathetic nerves is undergoing randomized controlled trials in patients who have resistant hypertension and other disorders that involve the sympathetic nervous system. Remarkably, the limited results available so far look good.
This article discusses the physiologic rationale for renal denervation, the evidence from studies in humans of the benefits, risks, and complications of the procedure, upcoming trials, and areas for future research.
DESPITE MANY TREATMENT OPTIONS, RESISTANT HYPERTENSION IS COMMON
Hypertension is a leading reason for visits to physicians in the United States and is associated with increased rates of cardiovascular disease and death.1,2 A variety of antihypertensive agents are available, and the percentage of people with hypertension whose blood pressure is under control has increased over the past 2 decades. Nevertheless, population-based studies show that the control rate remains suboptimal.3 Effective pharmacologic treatment may be limited by inadequate doses or inappropriate combinations of antihypertensive drugs, concurrent use of agents that raise the blood pressure, noncompliance with dietary restrictions, and side effects that result in poor compliance with drug therapy.
Resistant hypertension is defined as failure to achieve goal blood pressure in patients who are adhering to full tolerated doses of an appropriate three-drug regimen that includes a diuretic.1,4,5 If we use these criteria, many patients labelled as having resistant hypertension probably do not truly have it; instead, they are nonadherent to therapy or are on an inadequate or inappropriate regimen. Although the true prevalence of resistant hypertension is not clear, estimates from large clinical trials suggest that about 20% to 30% of hypertensive patients may meet the criteria for it.4 For the subset of patients who have truly resistant hypertension, nonpharmacologic treatments such as renal sympathetic denervation are an intriguing avenue.
SURGICAL SYMPATHETIC DENERVATION: TRIED AND ABANDONED IN THE 1950s
More than a half century ago, a surgical procedure, thoracolumbar sympathectomy (in which sympathetic nerve trunks and splanchnic nerves were removed), was sometimes performed to control blood pressure in patients with malignant hypertension. This was effective but caused debilitating side effects such as postural hypotension, erectile dysfunction, and syncope.
Smithwick and Thompson6 reported that, in 1,266 hypertensive patients who underwent this procedure and 467 medically treated controls, the 5-year mortality rates were 19% and 54%, respectively. Forty-five percent of those who survived the surgery had significantly lower blood pressure afterward, and the antihypertensive effect lasted 10 years or more.
The procedure fell out of favor due to the morbidity associated with this nonselective approach and to the increased availability of drug therapy.
THE SYMPATHETIC NERVOUS SYSTEM IS A DRIVER OF HYPERTENSION
A variety of evidence suggests that hyperactivation of the sympathetic nervous system plays a major role in initiating and maintaining hypertension. For example, drugs that inhibit the sympathetic drive at various levels have a blood-pressure-lowering effect. Further, direct intraneural recordings show a high level of sympathetic nerve activity in the muscles of hypertensive patients, who also have high levels of cardiac and renal norepinephrine “spillover”—ie, the amount of this neurotransmitter that escapes neuronal uptake and local metabolism and spills over into the circulation.7
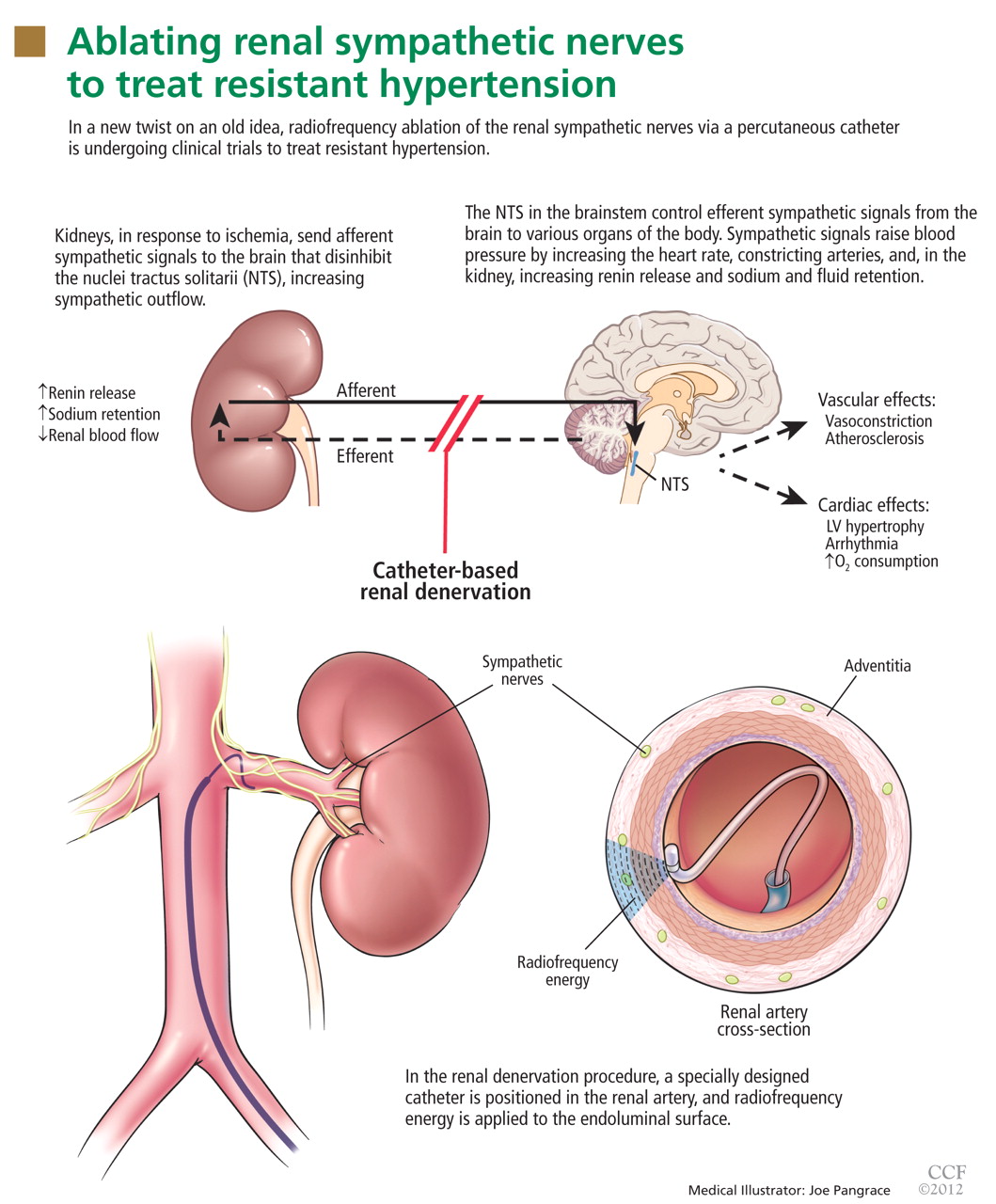
The kidneys are supplied with postganglionic sympathetic nerve fibers that end in the efferent and afferent renal arterioles, the juxtaglomerular apparatus, and the renal tubular system. Studies in animals and humans have shown that an increase in efferent signals (ie, from the brain to the kidney) leads to renal vasoconstriction and decreased renal blood flow, increased renin release, and sodium retention.8,9 Afferent signals (from the kidney to the central nervous system), which are increased in states of renal ischemia, renal parenchymal injury, and hypoxia, disinhibit the vasomotor center (the nuclei tractus solitarii) in the central nervous system, leading to increased efferent signals to the kidneys, heart, and peripheral blood vessels (Figure 1).10
Enhanced sympathetic activity in patients with hypertension may play a role in subsequent target-organ damage such as left ventricular hypertrophy, congestive heart failure, and progressive renal damage.11
Studies of renal denervation in animals, using surgical and chemical techniques, have further helped to establish the role of renal sympathetic nerves in hypertension.12,13
CATHETER-BASED RENAL DENERVATION
Renal sympathetic nerves run through the adventitia of the renal arteries in a mesh-like pattern.
In the renal denervation procedure, a specially designed catheter is inserted into a femoral artery and advanced into one of the renal arteries. There, radiofrequency energy is applied to the endoluminal surface according to a proprietary algorithm, thereby delivering thermal injury selectively to the renal sympathetic nerves without affecting the abdominal, pelvic, or lower-extremity nerves. The energy delivered is lower than that used for cardiac electrophysiologic procedures.
The nerves are not imaged or mapped before treatment. The procedure is performed on both sides, with four to six sites ablated in a longitudinal and rotational manner in 2-minute treatments at each site, to cover the full circumference (Figure 1).
In the United States, the device (Symplicity Renal Denervation System; Medtronic, Inc, Mountain View, CA) is available only for investigational use.
Below, we briefly review the studies of renal denervation to date. SYMPLICITY HTN-1 Symplicity HTN-1 was a proof-of-principle study in 45 patients with resistant hypertension (Table 1).14,15
Effect on blood pressure. Six months after renal denervation, blood pressure was significantly lower than at baseline (−22/−11 mm Hg, 95% confidence interval [CI] 10/5 mm Hg) in 26 patients available for follow-up. At 12 months, the difference from baseline was −27/−10 mm Hg (95% CI 16/11 mm Hg) in 9 patients available for follow-up (Table 2).14
Evidence of the durability of blood pressure reduction came from an expanded cohort of 153 patients followed for 2 years after denervation.16
Further follow-up data showed a sustained and significant blood pressure reduction through 3 years after denervation (unpublished results presented at the 2012 annual meeting of the American College of Cardiology). Notably, patients who were initially considered to be nonresponders (defined as failure of their blood pressure to go down by at least 10 mm Hg) were all reported to have a clinical response at 36 months.
Adverse events. In the initial and expanded cohorts combined, one patient suffered a renal artery dissection due to manipulation of the guiding catheter before the radiofrequency energy was delivered, and three patients developed a femoral pseudoaneurysm. No other long-term arterial complications were observed.
Comments. Limitations of this study included a small number of patients, no control group, and a primary outcome of a reduction in office blood pressure rather than in ambulatory blood pressure.
Additionally, although the authors concluded that there was no significant deterioration in renal function during the study period, we should note that in an additional follow-up period in this cohort, 10 patients with available 2-year data had a decrease in estimated glomerular filtration rate (eGFR) of −16.0 mL/min/1.73 m2. In 5 patients who did not have spironolactone (Aldactone) or another diuretic added after the first year of followup, a lesser but significant decrease (−7.8 mL/min/1.73 m2) was noted. The investigators surmised that denervation may enhance diuretic sensitivity, leading to prerenal azotemia in some patients.17
SYMPLICITY HTN-2
The Symplicity HTN-2 trial was a larger, randomized, efficacy study that built on the earlier results, providing additional evidence of therapeutic benefit.15
An international cohort of 106 patients with resistant hypertension, defined as systolic blood pressure of 160 mm Hg or higher (or ≥ 150 mm Hg in patients with type 2 diabetes) despite the use of three or more antihypertensive medications, were randomly assigned to undergo renal denervation with the Symplicity device (n = 52) or to continue their previous treatment with antihypertensive medications alone (n = 54). The primary effectiveness end point was the change in seated office blood pressure from baseline to 6 months (Table 1).
Effect on blood pressure. In the denervation group, at 6 months, office blood pressure had changed by a mean of −32/−12 mm Hg (standard deviation [SD] 23/11 mm Hg) compared with a mean change of 1/0 mm Hg (SD 21/10 mm Hg) in the control group. Fortyone (84%) of the 49 patients who underwent denervation had a decrease in systolic blood pressure of 10 mm Hg or more at 6 months compared with baseline values, while five (10%) had no decline in systolic blood pressure. Nineteen patients had a reduction in systolic pressure to less than 140 mm Hg in the denervation group.
A subset of patients (20 in the denervation group and 25 in the control group) underwent 24-hour ambulatory blood pressure monitoring at 6 months. This showed a similar though less pronounced fall in blood pressure in the denervation group and no change in the controls. A subanalysis that censored all data for patients whose medication was increased during the follow-up period showed a blood pressure reduction of −31/−12 mm Hg (SD 22/11 mm Hg) in the renal denervation group.
Adverse events. Procedure-related adverse events included a single femoral artery pseudoaneurysm, one case of postprocedural hypotension requiring a reduction in antihypertensive medications, and 7 (13%) of 52 patients who experienced intraprocedural bradycardia requiring atropine.
Effect on renal function. No significant difference was noted between groups in the mean change in renal function at 6 months, whether assessed by eGFR, serum creatinine level, or cystatin C level. At 6 months, no patient had a decrease of more than 50% in eGFR, although two patients who underwent renal denervation and three controls had more than a 25% decrease in eGFR.
At 6 months, the urine albumin-to-creatinine ratio had changed by a median of −3 mg/g (range −1,089 to 76) in 38 patients in the treatment group and by 1 mg/g (range −538 to 227) in 37 controls.
Most patients (88%) undergoing renal denervation underwent renal arterial imaging at 6 months, on which a single patient showed possible progression of an underlying atherosclerotic lesion that was unrelated to the procedure and that did not require intervention.
Denervation and the normal stress response. Whether renal denervation negatively affects the body’s physiologic response to stress that is normally mediated by sympathetic nerve activity was addressed in an extended investigation of Symplicity HTN-2 using cardiopulmonary exercise tests at baseline and 3 months after renal denervation.18 In the denervation group, blood pressure during exercise was significantly lower at 3 months than at baseline, but the heart rate increase at different levels of exercise was not affected. Additionally, the resting heart rate was lower and heart rate recovery after exercise improved after the procedure, particularly in patients without diabetes.
Comments. The Symplicity HTN-2 trial benefited from a randomized trial design and strict inclusion criteria of treatment resistance, but it still had notable limitations. A pretrial evaluation for causes of secondary hypertension or white-coat hypertension was not explicitly described. The control group did not undergo a sham procedure, and data analyzers were not masked to treatment assignment. Although not analyzed as a primary end point, the use of home-based and 24-hour ambulatory blood pressure assessment—measures important for determining white-coat hypertension—revealed substantial differences in blood pressure changes relative to office measurements. Because nearly all the patients (97%) were white, the generalizability of treatment results to black patients with resistant hypertension may be limited. Isolated diastolic hypertension (defined as diastolic pressure ≥ 90 mm Hg with systolic pressure < 140 mm Hg), which is more common in younger patients, was not studied.
DOES RENAL DENERVATION REDUCE SYMPATHETIC TONE?
A subgroup of 10 patients in the Symplicity HTN-1 trial whose mean 6-month office blood pressure was reduced by 22/12 mm Hg underwent assessment of renal norepinephrine spillover. A substantial (47%) reduction in renal norepinephrine spillover was noted 1 month after the procedure.14
The investigators additionally described a marked reduction in renal norepinephrine spillover from both kidneys in one patient, with a reduction of 48% from the left kidney and 75% from the right kidney 1 month after the procedure. Whole-body norepinephrine spillover in this patient was reduced by 42%. This effect was accompanied by a 50% decrease in plasma renin activity and by an increase in renal plasma flow. Aldosterone levels were not reported.19
Thus, the decrease in renal norepinephrine spillover suggests a reduction of renal efferent activity, and the decrease in total body norepinephrine spillover suggests a reduction in central sympathetic drive via the renal afferent pathway.
Microneurography in this same patient showed a gradual reduction in muscle sympathetic nerve activity to normal levels, from 56 bursts per minute at baseline to 41 at 30 days and 19 at 12 months).19 Decreased renin secretion, via circulating angiotensin II, may affect central sympathetic outflow as well.
Comments. While these findings address some of the underlying mechanisms, the small number of patients in whom these studies were done limits the generalizability of the results. The impact of the procedure on renal hemodynamics will need to be studied, including possible direct effects of the procedure, and whether there are differences in different study populations or differences based on blood pressure levels.
WHICH PATIENTS RESPOND BEST TO THIS PROCEDURE?
Although the Symplicity HTN-2 investigators report some predictors of increased reduction in blood pressure on multivariate analysis, including increased blood pressure at baseline and reduced heart rate at baseline, these are not specific enough to enable patient selection.
Interestingly, results from the expanded cohort of the Symplicity HTN-1 study found that patients on central sympatholytic agents such as clonidine had a greater reduction in blood pressure, although the reason for this is unclear.16 Identifying specific predictors of treatment success at baseline will be essential in future studies.
The earlier Symplicity trials and the ongoing Symplicity HTN-3 trial are in patients who have high blood pressure not responding to three or more antihypertensive drugs. The mean baseline systolic blood pressure in the Symplicity HTN-1 and HTN-2 trials was 178 mm Hg, and patients were taking an average of five antihypertensive drugs (Table 1). It is not known whether denervation will produce similar blood-pressure-lowering results across the spectrum of hypertension severity.
WHAT ARE THE LONG-TERM RESULTS OF DENERVATION?
Enthusiasm for the results from the Symplicity trials is tempered by concerns about the durability of the effects of the procedure, the need for better understanding of the impact of renal denervation on a wide array of pathophysiologic cascades leading to hypertension, and the effect on renal hemodynamics.
Antihypertensive efficacy has been reported to persist up to 2 years after the procedure,16 with recent unpublished data suggesting efficacy up to 3 years, but longer follow-up is needed to address whether these effects are finite.
Although reinnervation of afferent renal nerves has not been described, transplant models have shown anatomic regrowth of efferent nerves; the impact of this efferent reinnervation on blood pressure remains unclear. Experience from renal transplantation also shows that implanted kidneys that are “denervated” can still maintain fluid and electrolyte regulation.
Follow-up renal imaging in the Symplicity trials did not indicate renal artery stenosis at the sites of denervation in patients who underwent the procedure. Animal studies using the Symplicity catheter system showed renal nerve injury as evidenced by nerve fibrosis and thickened epineurium and perineurium, but no significant smooth muscle hyperplasia, arterial stenosis, or thrombosis by angiography or histology at 6 months.20
WHAT ARE THE RISKS?
Adverse effects that were noted in the short term are detailed under discussion of the trials and in Table 2.
Long-term adverse events in the Symplicity HTN-2 trial that required hospitalization were reported in five patients in the denervation group and three patients in the control group (Table 2). These included transient ischemic attacks, hypertensive crises, hypotensive episodes, angina, and nausea.
Renal function was maintained for the duration of both trials, and details regarding eGFR change have been described above under the discussion of the trials.
Diffuse visceral pain at the time of the procedure is reported as an expected occurrence, managed with intravenous analgesic medications.
DOES SYMPATHETIC DENERVATION HAVE A ROLE IN OTHER CONDITIONS?
Interestingly, other sympathetically driven diseases, such as diabetes mellitus and polycystic ovary syndrome, may prove to be targets for this therapy in the future.21
Mahfoud et al22 conducted a pilot study in 37 patients with resistant hypertension undergoing renal denervation and 13 control patients. Fasting glucose levels declined from 118 ± 3.4 mg/dL to 108 ± 3.8 mg/dL after 3 months in the intervention group (P = .039), compared with no change in the control group. Insulin and C-peptide levels were also lower in the intervention group. The reported improvement in glucose metabolism and insulin sensitivity suggests that the beneficial effects of this procedure may extend beyond blood pressure reduction.
Brandt et al23 reported regression of left ventricular hypertrophy and significantly improved cardiac functional parameters, including increase in ejection fraction and improved diastolic dysfunction, in a study of 46 patients who underwent renal denervation. This findings suggests a potential beneficial effect on cardiac remodeling.
Witkowski et al24 reported lowering of blood pressure in 10 patients with refractory hypertension and obstructive sleep apnea who underwent renal denervation, which was accompanied by improvement of sleep apnea severity.
Ukena et al25 reported reduction in ventricular tachyarrhythmias in two patients with congestive heart failure who had therapy-resistant electrical storm.
A recent pilot study in 15 patients with stage 3 and 4 chronic kidney disease (mean eGFR 31 mL/min/1.73 m2) showed significantly improved office blood pressure control up to 1 year, restoration of nocturnal dipping on 24-hour monitoring, as well as a nonsignificant trend towards increased hemoglobin levels and decreased proteinuria. No additional deterioration of renal function was reported in these patients (2 patients had renal function assessed up to 1 year).26
Thus, the benefits of this procedure may extend to other diseases that have a common underlying thread of elevated sympathetic activity, by targeting the “sympathorenal” axis.27
GUARDED OPTIMISM AND FUTURE DIRECTIONS
Given the well-known cardiovascular risks and health care costs associated with uncontrolled hypertension and the continued challenge that physicians face in managing it, novel therapies such as renal denervation may provide an adjunct to existing pharmacologic approaches.
While there is certainly cause for guarded optimism, especially with the striking blood pressure-lowering results seen in trials so far, it should be kept in mind that the mechanisms leading to the hypertensive response are complex and multifactorial, and further understanding of this therapy with long-term follow-up is needed. A comparison study with spironolactone, which is increasingly being used to treat resistant hypertension (in the absence of a diagnosis of primary aldosteronism)28,29 would help to further establish the role of this procedure.
Studies of carotid baroreceptor stimulation via an implantable device have shown sustained reduction in blood pressure in patients with resistant hypertension. A study comparing this technique with renal denervation for efficacy and safety end points could be considered in the future.30,31
The planned Symplicity HTN-3 study in the United States will be the largest trial to date, with a targeted randomization of more than 500 patients using strict enrollment criteria, including the use of maximally tolerated doses of diuretics and more focus on the use of ambulatory blood pressure monitoring and on the blinding of participants. This study will help further analysis of this technology in a more diverse population.32,33
Future studies should be designed to clarify pathophysiologic mechanisms, patient selection criteria, effects on target organ damage, and efficacy in patients with chronic kidney disease, obesity, congestive heart failure, and in less severe forms of hypertension.
A CALL FOR PARTICIPANTS IN A CLINICAL TRIAL
The Departments of Cardiology and Nephrology and Hypertension at Cleveland Clinic are currently enrolling patients in the Symplicity HTN-3 trial. For more information, please contact George Thomas, MD ([email protected]), or Mehdi Shishehbor, DO, MPH ([email protected]), or visit www.symplifybptrial.com.
- Chobanian AV, Bakris GL, Black HR, et al; National Heart, Lung, and Blood Institute Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure; National High Blood Pressure Education Program Coordinating Committee. The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. JAMA 2003; 289:2560–2572.
- Schappert SM, Rechtsteiner EA. Ambulatory medical care utilization estimates for 2007. National Center for Health Statistics. Vital Health Stat 13( 169) 2011. http://www.cdc.gov/nchs/data/series/sr_13/sr13_169.pdf. Accessed April 24, 2012.
- Egan BM, Zhao Y, Axon RN. US trends in prevalence, awareness, treatment, and control of hypertension, 1988–2008. JAMA 2010; 303:2043–2050.
- Persell SD. Prevalence of resistant hypertension in the United States, 2003–2008. Hypertension 2011; 57:1076–1080.
- Calhoun DA, Jones D, Textor S, et al; American Heart Association Professional Education Committee. Resistant hypertension: diagnosis, evaluation, and treatment: a scientific statement from the American Heart Association Professional Education Committee of the Council for High Blood Pressure Research. Circulation 2008; 117:e510–e526.
- Smithwick RH, Thompson JE. Splanchnicectomy for essential hypertension; results in 1,266 cases. J Am Med Assoc 1953; 152:1501–1504.
- Schlaich MP, Sobotka PA, Krum H, Whitbourn R, Walton A, Esler MD. Renal denervation as a therapeutic approach for hypertension: novel implications for an old concept. Hypertension 2009; 54:1195–1201.
- Zanchetti AS. Neural regulation of renin release: experimental evidence and clinical implications in arterial hypertension. Circulation 1977; 56:691–698.
- Kon V. Neural control of renal circulation. Miner Electrolyte Metab 1989; 15:33–43.
- Campese VM. Neurogenic factors and hypertension in renal disease. Kidney Int Suppl 2000; 75:S2–S6.
- Mancia G, Grassi G, Giannattasio C, Seravalle G. Sympathetic activation in the pathogenesis of hypertension and progression of organ damage. Hypertension 1999; 34:724–728.
- Campese VM, Ye S, Zhong H, Yanamadala V, Ye Z, Chiu J. Reactive oxygen species stimulate central and peripheral sympathetic nervous system activity. Am J Physiol Heart Circ Physiol 2004; 287:H695–H703.
- Katholi RE. Renal nerves in the pathogenesis of hypertension in experimental animals and humans. Am J Physiol 1983; 245:F1–F14.
- Krum H, Schlaich M, Whitbourn R, et al. Catheter-based renal sympathetic denervation for resistant hypertension: a multicentre safety and proof-of-principle cohort study. Lancet 2009; 373:1275–1281.
- Esler MD, Krum H, Sobotka PA, Schlaich MP, Schmieder RE, Böhm M; Symplicity HTN-2 Investigators. Renal sympathetic denervation in patients with treatmentresistant hypertension (The Symplicity HTN-2 Trial): a randomised controlled trial. Lancet 2010; 376:1903–1909.
- Symplicity HTN-1 Investigators. Catheter-based renal sympathetic denervation for resistant hypertension: durability of blood pressure reduction out to 24 months. Hypertension 2011; 57:911–917.
- Petidis K, Anyfanti P, Doumas M. Renal sympathetic denervation: renal function concerns. Hypertension 2011; 58:e19; author replye20.
- Ukena C, Mahfoud F, Kindermann I, et al. Cardiorespiratory response to exercise after renal sympathetic denervation in patients with resistant hypertension. J Am Coll Cardiol 2011; 58:1176–1182.
- Schlaich MP, Sobotka PA, Krum H, Lambert E, Esler MD. Renal sympathetic-nerve ablation for uncontrolled hypertension (letter). N Engl J Med 2009; 361:932–934.
- Rippy MK, Zarins D, Barman NC, Wu A, Duncan KL, Zarins CK. Catheter-based renal sympathetic denervation: chronic preclinical evidence for renal artery safety. Clin Res Cardiol 2011; 100:1095–1101.
- Schlaich MP, Straznicky N, Grima M, et al. Renal denervation: a potential new treatment modality for polycystic ovary syndrome? J Hypertens 2011; 29:991–996.
- Mahfoud F, Schlaich M, Kindermann I, et al. Effect of renal sympathetic denervation on glucose metabolism in patients with resistant hypertension: a pilot study. Circulation 2011; 123:1940–1946.
- Brandt MC, Mahfoud F, Reda S, et al. Renal sympathetic denervation reduces left ventricular hypertrophy and improves cardiac function in patients with resistant hypertension. J Am Coll Cardiol 2012; 59:901–909.
- Witkowski A, Prejbisz A, Florczak E, et al. Effects of renal sympathetic denervation on blood pressure, sleep apnea course, and glycemic control in patients with resistant hypertension and sleep apnea. Hypertension 2011; 58:559–565.
- Ukena C, Bauer A, Mahfoud F, et al. Renal sympathetic denervation for treatment of electrical storm: first-inman experience. Clin Res Cardiol 2012; 101:63–67.
- Herring D, Mahfoud F, Walton AS, et al. Renal denervation in moderate to severe CKD. J Am Soc Nephrol 2012; May 17[Epub ahead of print]
- Sobotka PA, Mahfoud F, Schlaich MP, Hoppe UC, Böhm M, Krum H. Sympatho-renal axis in chronic disease. Clin Res Cardiol 2011; 100:1049–1057.
- Chapman N, Dobson J, Wilson S, et al; Anglo-Scandinavian Cardiac Outcomes Trial Investigators. Effect of spironolactone on blood pressure in subjects with resistant hypertension. Hypertension 2007; 49:839–845.
- Nishizaka MK, Zaman MA, Calhoun DA. Efficacy of low-dose spironolactone in subjects with resistant hypertension. Am J Hypertens 2003; 16:925–930.
- Papademetriou V, Doumas M, Faselis C, et al. Carotid baroreceptor stimulation for the treatment of resistant hypertension. Int J Hypertens 2011; 2011:964394.
- Ng MM, Sica DA, Frishman WH. Rheos: an implantable carotid sinus stimulation device for the nonpharmacologic treatment of resistant hypertension. Cardiol Rev 2011; 19:52–57.
- US National Institutes of Health. Renal denervation in patients with uncontrolled hypertension (SYMPLICITY HTN-3). http://www.clinicaltrials.gov/ct2/show/NCT01418261. Accessed June 7, 2012.
- Kandzari DE, Bhatt DL, Sobotka PA, et al. Catheter-based renal denervation for resistant hypertension: rationale and design of the Symplicity HTN-3 trial. Clin Cardiol 2012 May 9. [Epub ahead of print]
Can a percutaneous catheter-based procedure effectively treat resistant hypertension?
Radiofrequency ablation of the renal sympathetic nerves is undergoing randomized controlled trials in patients who have resistant hypertension and other disorders that involve the sympathetic nervous system. Remarkably, the limited results available so far look good.
This article discusses the physiologic rationale for renal denervation, the evidence from studies in humans of the benefits, risks, and complications of the procedure, upcoming trials, and areas for future research.
DESPITE MANY TREATMENT OPTIONS, RESISTANT HYPERTENSION IS COMMON
Hypertension is a leading reason for visits to physicians in the United States and is associated with increased rates of cardiovascular disease and death.1,2 A variety of antihypertensive agents are available, and the percentage of people with hypertension whose blood pressure is under control has increased over the past 2 decades. Nevertheless, population-based studies show that the control rate remains suboptimal.3 Effective pharmacologic treatment may be limited by inadequate doses or inappropriate combinations of antihypertensive drugs, concurrent use of agents that raise the blood pressure, noncompliance with dietary restrictions, and side effects that result in poor compliance with drug therapy.
Resistant hypertension is defined as failure to achieve goal blood pressure in patients who are adhering to full tolerated doses of an appropriate three-drug regimen that includes a diuretic.1,4,5 If we use these criteria, many patients labelled as having resistant hypertension probably do not truly have it; instead, they are nonadherent to therapy or are on an inadequate or inappropriate regimen. Although the true prevalence of resistant hypertension is not clear, estimates from large clinical trials suggest that about 20% to 30% of hypertensive patients may meet the criteria for it.4 For the subset of patients who have truly resistant hypertension, nonpharmacologic treatments such as renal sympathetic denervation are an intriguing avenue.
SURGICAL SYMPATHETIC DENERVATION: TRIED AND ABANDONED IN THE 1950s
More than a half century ago, a surgical procedure, thoracolumbar sympathectomy (in which sympathetic nerve trunks and splanchnic nerves were removed), was sometimes performed to control blood pressure in patients with malignant hypertension. This was effective but caused debilitating side effects such as postural hypotension, erectile dysfunction, and syncope.
Smithwick and Thompson6 reported that, in 1,266 hypertensive patients who underwent this procedure and 467 medically treated controls, the 5-year mortality rates were 19% and 54%, respectively. Forty-five percent of those who survived the surgery had significantly lower blood pressure afterward, and the antihypertensive effect lasted 10 years or more.
The procedure fell out of favor due to the morbidity associated with this nonselective approach and to the increased availability of drug therapy.
THE SYMPATHETIC NERVOUS SYSTEM IS A DRIVER OF HYPERTENSION
A variety of evidence suggests that hyperactivation of the sympathetic nervous system plays a major role in initiating and maintaining hypertension. For example, drugs that inhibit the sympathetic drive at various levels have a blood-pressure-lowering effect. Further, direct intraneural recordings show a high level of sympathetic nerve activity in the muscles of hypertensive patients, who also have high levels of cardiac and renal norepinephrine “spillover”—ie, the amount of this neurotransmitter that escapes neuronal uptake and local metabolism and spills over into the circulation.7
The kidneys are supplied with postganglionic sympathetic nerve fibers that end in the efferent and afferent renal arterioles, the juxtaglomerular apparatus, and the renal tubular system. Studies in animals and humans have shown that an increase in efferent signals (ie, from the brain to the kidney) leads to renal vasoconstriction and decreased renal blood flow, increased renin release, and sodium retention.8,9 Afferent signals (from the kidney to the central nervous system), which are increased in states of renal ischemia, renal parenchymal injury, and hypoxia, disinhibit the vasomotor center (the nuclei tractus solitarii) in the central nervous system, leading to increased efferent signals to the kidneys, heart, and peripheral blood vessels (Figure 1).10
Enhanced sympathetic activity in patients with hypertension may play a role in subsequent target-organ damage such as left ventricular hypertrophy, congestive heart failure, and progressive renal damage.11
Studies of renal denervation in animals, using surgical and chemical techniques, have further helped to establish the role of renal sympathetic nerves in hypertension.12,13
CATHETER-BASED RENAL DENERVATION
Renal sympathetic nerves run through the adventitia of the renal arteries in a mesh-like pattern.
In the renal denervation procedure, a specially designed catheter is inserted into a femoral artery and advanced into one of the renal arteries. There, radiofrequency energy is applied to the endoluminal surface according to a proprietary algorithm, thereby delivering thermal injury selectively to the renal sympathetic nerves without affecting the abdominal, pelvic, or lower-extremity nerves. The energy delivered is lower than that used for cardiac electrophysiologic procedures.
The nerves are not imaged or mapped before treatment. The procedure is performed on both sides, with four to six sites ablated in a longitudinal and rotational manner in 2-minute treatments at each site, to cover the full circumference (Figure 1).
In the United States, the device (Symplicity Renal Denervation System; Medtronic, Inc, Mountain View, CA) is available only for investigational use.
Below, we briefly review the studies of renal denervation to date. SYMPLICITY HTN-1 Symplicity HTN-1 was a proof-of-principle study in 45 patients with resistant hypertension (Table 1).14,15
Effect on blood pressure. Six months after renal denervation, blood pressure was significantly lower than at baseline (−22/−11 mm Hg, 95% confidence interval [CI] 10/5 mm Hg) in 26 patients available for follow-up. At 12 months, the difference from baseline was −27/−10 mm Hg (95% CI 16/11 mm Hg) in 9 patients available for follow-up (Table 2).14
Evidence of the durability of blood pressure reduction came from an expanded cohort of 153 patients followed for 2 years after denervation.16
Further follow-up data showed a sustained and significant blood pressure reduction through 3 years after denervation (unpublished results presented at the 2012 annual meeting of the American College of Cardiology). Notably, patients who were initially considered to be nonresponders (defined as failure of their blood pressure to go down by at least 10 mm Hg) were all reported to have a clinical response at 36 months.
Adverse events. In the initial and expanded cohorts combined, one patient suffered a renal artery dissection due to manipulation of the guiding catheter before the radiofrequency energy was delivered, and three patients developed a femoral pseudoaneurysm. No other long-term arterial complications were observed.
Comments. Limitations of this study included a small number of patients, no control group, and a primary outcome of a reduction in office blood pressure rather than in ambulatory blood pressure.
Additionally, although the authors concluded that there was no significant deterioration in renal function during the study period, we should note that in an additional follow-up period in this cohort, 10 patients with available 2-year data had a decrease in estimated glomerular filtration rate (eGFR) of −16.0 mL/min/1.73 m2. In 5 patients who did not have spironolactone (Aldactone) or another diuretic added after the first year of followup, a lesser but significant decrease (−7.8 mL/min/1.73 m2) was noted. The investigators surmised that denervation may enhance diuretic sensitivity, leading to prerenal azotemia in some patients.17
SYMPLICITY HTN-2
The Symplicity HTN-2 trial was a larger, randomized, efficacy study that built on the earlier results, providing additional evidence of therapeutic benefit.15
An international cohort of 106 patients with resistant hypertension, defined as systolic blood pressure of 160 mm Hg or higher (or ≥ 150 mm Hg in patients with type 2 diabetes) despite the use of three or more antihypertensive medications, were randomly assigned to undergo renal denervation with the Symplicity device (n = 52) or to continue their previous treatment with antihypertensive medications alone (n = 54). The primary effectiveness end point was the change in seated office blood pressure from baseline to 6 months (Table 1).
Effect on blood pressure. In the denervation group, at 6 months, office blood pressure had changed by a mean of −32/−12 mm Hg (standard deviation [SD] 23/11 mm Hg) compared with a mean change of 1/0 mm Hg (SD 21/10 mm Hg) in the control group. Fortyone (84%) of the 49 patients who underwent denervation had a decrease in systolic blood pressure of 10 mm Hg or more at 6 months compared with baseline values, while five (10%) had no decline in systolic blood pressure. Nineteen patients had a reduction in systolic pressure to less than 140 mm Hg in the denervation group.
A subset of patients (20 in the denervation group and 25 in the control group) underwent 24-hour ambulatory blood pressure monitoring at 6 months. This showed a similar though less pronounced fall in blood pressure in the denervation group and no change in the controls. A subanalysis that censored all data for patients whose medication was increased during the follow-up period showed a blood pressure reduction of −31/−12 mm Hg (SD 22/11 mm Hg) in the renal denervation group.
Adverse events. Procedure-related adverse events included a single femoral artery pseudoaneurysm, one case of postprocedural hypotension requiring a reduction in antihypertensive medications, and 7 (13%) of 52 patients who experienced intraprocedural bradycardia requiring atropine.
Effect on renal function. No significant difference was noted between groups in the mean change in renal function at 6 months, whether assessed by eGFR, serum creatinine level, or cystatin C level. At 6 months, no patient had a decrease of more than 50% in eGFR, although two patients who underwent renal denervation and three controls had more than a 25% decrease in eGFR.
At 6 months, the urine albumin-to-creatinine ratio had changed by a median of −3 mg/g (range −1,089 to 76) in 38 patients in the treatment group and by 1 mg/g (range −538 to 227) in 37 controls.
Most patients (88%) undergoing renal denervation underwent renal arterial imaging at 6 months, on which a single patient showed possible progression of an underlying atherosclerotic lesion that was unrelated to the procedure and that did not require intervention.
Denervation and the normal stress response. Whether renal denervation negatively affects the body’s physiologic response to stress that is normally mediated by sympathetic nerve activity was addressed in an extended investigation of Symplicity HTN-2 using cardiopulmonary exercise tests at baseline and 3 months after renal denervation.18 In the denervation group, blood pressure during exercise was significantly lower at 3 months than at baseline, but the heart rate increase at different levels of exercise was not affected. Additionally, the resting heart rate was lower and heart rate recovery after exercise improved after the procedure, particularly in patients without diabetes.
Comments. The Symplicity HTN-2 trial benefited from a randomized trial design and strict inclusion criteria of treatment resistance, but it still had notable limitations. A pretrial evaluation for causes of secondary hypertension or white-coat hypertension was not explicitly described. The control group did not undergo a sham procedure, and data analyzers were not masked to treatment assignment. Although not analyzed as a primary end point, the use of home-based and 24-hour ambulatory blood pressure assessment—measures important for determining white-coat hypertension—revealed substantial differences in blood pressure changes relative to office measurements. Because nearly all the patients (97%) were white, the generalizability of treatment results to black patients with resistant hypertension may be limited. Isolated diastolic hypertension (defined as diastolic pressure ≥ 90 mm Hg with systolic pressure < 140 mm Hg), which is more common in younger patients, was not studied.
DOES RENAL DENERVATION REDUCE SYMPATHETIC TONE?
A subgroup of 10 patients in the Symplicity HTN-1 trial whose mean 6-month office blood pressure was reduced by 22/12 mm Hg underwent assessment of renal norepinephrine spillover. A substantial (47%) reduction in renal norepinephrine spillover was noted 1 month after the procedure.14
The investigators additionally described a marked reduction in renal norepinephrine spillover from both kidneys in one patient, with a reduction of 48% from the left kidney and 75% from the right kidney 1 month after the procedure. Whole-body norepinephrine spillover in this patient was reduced by 42%. This effect was accompanied by a 50% decrease in plasma renin activity and by an increase in renal plasma flow. Aldosterone levels were not reported.19
Thus, the decrease in renal norepinephrine spillover suggests a reduction of renal efferent activity, and the decrease in total body norepinephrine spillover suggests a reduction in central sympathetic drive via the renal afferent pathway.
Microneurography in this same patient showed a gradual reduction in muscle sympathetic nerve activity to normal levels, from 56 bursts per minute at baseline to 41 at 30 days and 19 at 12 months).19 Decreased renin secretion, via circulating angiotensin II, may affect central sympathetic outflow as well.
Comments. While these findings address some of the underlying mechanisms, the small number of patients in whom these studies were done limits the generalizability of the results. The impact of the procedure on renal hemodynamics will need to be studied, including possible direct effects of the procedure, and whether there are differences in different study populations or differences based on blood pressure levels.
WHICH PATIENTS RESPOND BEST TO THIS PROCEDURE?
Although the Symplicity HTN-2 investigators report some predictors of increased reduction in blood pressure on multivariate analysis, including increased blood pressure at baseline and reduced heart rate at baseline, these are not specific enough to enable patient selection.
Interestingly, results from the expanded cohort of the Symplicity HTN-1 study found that patients on central sympatholytic agents such as clonidine had a greater reduction in blood pressure, although the reason for this is unclear.16 Identifying specific predictors of treatment success at baseline will be essential in future studies.
The earlier Symplicity trials and the ongoing Symplicity HTN-3 trial are in patients who have high blood pressure not responding to three or more antihypertensive drugs. The mean baseline systolic blood pressure in the Symplicity HTN-1 and HTN-2 trials was 178 mm Hg, and patients were taking an average of five antihypertensive drugs (Table 1). It is not known whether denervation will produce similar blood-pressure-lowering results across the spectrum of hypertension severity.
WHAT ARE THE LONG-TERM RESULTS OF DENERVATION?
Enthusiasm for the results from the Symplicity trials is tempered by concerns about the durability of the effects of the procedure, the need for better understanding of the impact of renal denervation on a wide array of pathophysiologic cascades leading to hypertension, and the effect on renal hemodynamics.
Antihypertensive efficacy has been reported to persist up to 2 years after the procedure,16 with recent unpublished data suggesting efficacy up to 3 years, but longer follow-up is needed to address whether these effects are finite.
Although reinnervation of afferent renal nerves has not been described, transplant models have shown anatomic regrowth of efferent nerves; the impact of this efferent reinnervation on blood pressure remains unclear. Experience from renal transplantation also shows that implanted kidneys that are “denervated” can still maintain fluid and electrolyte regulation.
Follow-up renal imaging in the Symplicity trials did not indicate renal artery stenosis at the sites of denervation in patients who underwent the procedure. Animal studies using the Symplicity catheter system showed renal nerve injury as evidenced by nerve fibrosis and thickened epineurium and perineurium, but no significant smooth muscle hyperplasia, arterial stenosis, or thrombosis by angiography or histology at 6 months.20
WHAT ARE THE RISKS?
Adverse effects that were noted in the short term are detailed under discussion of the trials and in Table 2.
Long-term adverse events in the Symplicity HTN-2 trial that required hospitalization were reported in five patients in the denervation group and three patients in the control group (Table 2). These included transient ischemic attacks, hypertensive crises, hypotensive episodes, angina, and nausea.
Renal function was maintained for the duration of both trials, and details regarding eGFR change have been described above under the discussion of the trials.
Diffuse visceral pain at the time of the procedure is reported as an expected occurrence, managed with intravenous analgesic medications.
DOES SYMPATHETIC DENERVATION HAVE A ROLE IN OTHER CONDITIONS?
Interestingly, other sympathetically driven diseases, such as diabetes mellitus and polycystic ovary syndrome, may prove to be targets for this therapy in the future.21
Mahfoud et al22 conducted a pilot study in 37 patients with resistant hypertension undergoing renal denervation and 13 control patients. Fasting glucose levels declined from 118 ± 3.4 mg/dL to 108 ± 3.8 mg/dL after 3 months in the intervention group (P = .039), compared with no change in the control group. Insulin and C-peptide levels were also lower in the intervention group. The reported improvement in glucose metabolism and insulin sensitivity suggests that the beneficial effects of this procedure may extend beyond blood pressure reduction.
Brandt et al23 reported regression of left ventricular hypertrophy and significantly improved cardiac functional parameters, including increase in ejection fraction and improved diastolic dysfunction, in a study of 46 patients who underwent renal denervation. This findings suggests a potential beneficial effect on cardiac remodeling.
Witkowski et al24 reported lowering of blood pressure in 10 patients with refractory hypertension and obstructive sleep apnea who underwent renal denervation, which was accompanied by improvement of sleep apnea severity.
Ukena et al25 reported reduction in ventricular tachyarrhythmias in two patients with congestive heart failure who had therapy-resistant electrical storm.
A recent pilot study in 15 patients with stage 3 and 4 chronic kidney disease (mean eGFR 31 mL/min/1.73 m2) showed significantly improved office blood pressure control up to 1 year, restoration of nocturnal dipping on 24-hour monitoring, as well as a nonsignificant trend towards increased hemoglobin levels and decreased proteinuria. No additional deterioration of renal function was reported in these patients (2 patients had renal function assessed up to 1 year).26
Thus, the benefits of this procedure may extend to other diseases that have a common underlying thread of elevated sympathetic activity, by targeting the “sympathorenal” axis.27
GUARDED OPTIMISM AND FUTURE DIRECTIONS
Given the well-known cardiovascular risks and health care costs associated with uncontrolled hypertension and the continued challenge that physicians face in managing it, novel therapies such as renal denervation may provide an adjunct to existing pharmacologic approaches.
While there is certainly cause for guarded optimism, especially with the striking blood pressure-lowering results seen in trials so far, it should be kept in mind that the mechanisms leading to the hypertensive response are complex and multifactorial, and further understanding of this therapy with long-term follow-up is needed. A comparison study with spironolactone, which is increasingly being used to treat resistant hypertension (in the absence of a diagnosis of primary aldosteronism)28,29 would help to further establish the role of this procedure.
Studies of carotid baroreceptor stimulation via an implantable device have shown sustained reduction in blood pressure in patients with resistant hypertension. A study comparing this technique with renal denervation for efficacy and safety end points could be considered in the future.30,31
The planned Symplicity HTN-3 study in the United States will be the largest trial to date, with a targeted randomization of more than 500 patients using strict enrollment criteria, including the use of maximally tolerated doses of diuretics and more focus on the use of ambulatory blood pressure monitoring and on the blinding of participants. This study will help further analysis of this technology in a more diverse population.32,33
Future studies should be designed to clarify pathophysiologic mechanisms, patient selection criteria, effects on target organ damage, and efficacy in patients with chronic kidney disease, obesity, congestive heart failure, and in less severe forms of hypertension.
A CALL FOR PARTICIPANTS IN A CLINICAL TRIAL
The Departments of Cardiology and Nephrology and Hypertension at Cleveland Clinic are currently enrolling patients in the Symplicity HTN-3 trial. For more information, please contact George Thomas, MD ([email protected]), or Mehdi Shishehbor, DO, MPH ([email protected]), or visit www.symplifybptrial.com.
Can a percutaneous catheter-based procedure effectively treat resistant hypertension?
Radiofrequency ablation of the renal sympathetic nerves is undergoing randomized controlled trials in patients who have resistant hypertension and other disorders that involve the sympathetic nervous system. Remarkably, the limited results available so far look good.
This article discusses the physiologic rationale for renal denervation, the evidence from studies in humans of the benefits, risks, and complications of the procedure, upcoming trials, and areas for future research.
DESPITE MANY TREATMENT OPTIONS, RESISTANT HYPERTENSION IS COMMON
Hypertension is a leading reason for visits to physicians in the United States and is associated with increased rates of cardiovascular disease and death.1,2 A variety of antihypertensive agents are available, and the percentage of people with hypertension whose blood pressure is under control has increased over the past 2 decades. Nevertheless, population-based studies show that the control rate remains suboptimal.3 Effective pharmacologic treatment may be limited by inadequate doses or inappropriate combinations of antihypertensive drugs, concurrent use of agents that raise the blood pressure, noncompliance with dietary restrictions, and side effects that result in poor compliance with drug therapy.
Resistant hypertension is defined as failure to achieve goal blood pressure in patients who are adhering to full tolerated doses of an appropriate three-drug regimen that includes a diuretic.1,4,5 If we use these criteria, many patients labelled as having resistant hypertension probably do not truly have it; instead, they are nonadherent to therapy or are on an inadequate or inappropriate regimen. Although the true prevalence of resistant hypertension is not clear, estimates from large clinical trials suggest that about 20% to 30% of hypertensive patients may meet the criteria for it.4 For the subset of patients who have truly resistant hypertension, nonpharmacologic treatments such as renal sympathetic denervation are an intriguing avenue.
SURGICAL SYMPATHETIC DENERVATION: TRIED AND ABANDONED IN THE 1950s
More than a half century ago, a surgical procedure, thoracolumbar sympathectomy (in which sympathetic nerve trunks and splanchnic nerves were removed), was sometimes performed to control blood pressure in patients with malignant hypertension. This was effective but caused debilitating side effects such as postural hypotension, erectile dysfunction, and syncope.
Smithwick and Thompson6 reported that, in 1,266 hypertensive patients who underwent this procedure and 467 medically treated controls, the 5-year mortality rates were 19% and 54%, respectively. Forty-five percent of those who survived the surgery had significantly lower blood pressure afterward, and the antihypertensive effect lasted 10 years or more.
The procedure fell out of favor due to the morbidity associated with this nonselective approach and to the increased availability of drug therapy.
THE SYMPATHETIC NERVOUS SYSTEM IS A DRIVER OF HYPERTENSION
A variety of evidence suggests that hyperactivation of the sympathetic nervous system plays a major role in initiating and maintaining hypertension. For example, drugs that inhibit the sympathetic drive at various levels have a blood-pressure-lowering effect. Further, direct intraneural recordings show a high level of sympathetic nerve activity in the muscles of hypertensive patients, who also have high levels of cardiac and renal norepinephrine “spillover”—ie, the amount of this neurotransmitter that escapes neuronal uptake and local metabolism and spills over into the circulation.7
The kidneys are supplied with postganglionic sympathetic nerve fibers that end in the efferent and afferent renal arterioles, the juxtaglomerular apparatus, and the renal tubular system. Studies in animals and humans have shown that an increase in efferent signals (ie, from the brain to the kidney) leads to renal vasoconstriction and decreased renal blood flow, increased renin release, and sodium retention.8,9 Afferent signals (from the kidney to the central nervous system), which are increased in states of renal ischemia, renal parenchymal injury, and hypoxia, disinhibit the vasomotor center (the nuclei tractus solitarii) in the central nervous system, leading to increased efferent signals to the kidneys, heart, and peripheral blood vessels (Figure 1).10
Enhanced sympathetic activity in patients with hypertension may play a role in subsequent target-organ damage such as left ventricular hypertrophy, congestive heart failure, and progressive renal damage.11
Studies of renal denervation in animals, using surgical and chemical techniques, have further helped to establish the role of renal sympathetic nerves in hypertension.12,13
CATHETER-BASED RENAL DENERVATION
Renal sympathetic nerves run through the adventitia of the renal arteries in a mesh-like pattern.
In the renal denervation procedure, a specially designed catheter is inserted into a femoral artery and advanced into one of the renal arteries. There, radiofrequency energy is applied to the endoluminal surface according to a proprietary algorithm, thereby delivering thermal injury selectively to the renal sympathetic nerves without affecting the abdominal, pelvic, or lower-extremity nerves. The energy delivered is lower than that used for cardiac electrophysiologic procedures.
The nerves are not imaged or mapped before treatment. The procedure is performed on both sides, with four to six sites ablated in a longitudinal and rotational manner in 2-minute treatments at each site, to cover the full circumference (Figure 1).
In the United States, the device (Symplicity Renal Denervation System; Medtronic, Inc, Mountain View, CA) is available only for investigational use.
Below, we briefly review the studies of renal denervation to date. SYMPLICITY HTN-1 Symplicity HTN-1 was a proof-of-principle study in 45 patients with resistant hypertension (Table 1).14,15
Effect on blood pressure. Six months after renal denervation, blood pressure was significantly lower than at baseline (−22/−11 mm Hg, 95% confidence interval [CI] 10/5 mm Hg) in 26 patients available for follow-up. At 12 months, the difference from baseline was −27/−10 mm Hg (95% CI 16/11 mm Hg) in 9 patients available for follow-up (Table 2).14
Evidence of the durability of blood pressure reduction came from an expanded cohort of 153 patients followed for 2 years after denervation.16
Further follow-up data showed a sustained and significant blood pressure reduction through 3 years after denervation (unpublished results presented at the 2012 annual meeting of the American College of Cardiology). Notably, patients who were initially considered to be nonresponders (defined as failure of their blood pressure to go down by at least 10 mm Hg) were all reported to have a clinical response at 36 months.
Adverse events. In the initial and expanded cohorts combined, one patient suffered a renal artery dissection due to manipulation of the guiding catheter before the radiofrequency energy was delivered, and three patients developed a femoral pseudoaneurysm. No other long-term arterial complications were observed.
Comments. Limitations of this study included a small number of patients, no control group, and a primary outcome of a reduction in office blood pressure rather than in ambulatory blood pressure.
Additionally, although the authors concluded that there was no significant deterioration in renal function during the study period, we should note that in an additional follow-up period in this cohort, 10 patients with available 2-year data had a decrease in estimated glomerular filtration rate (eGFR) of −16.0 mL/min/1.73 m2. In 5 patients who did not have spironolactone (Aldactone) or another diuretic added after the first year of followup, a lesser but significant decrease (−7.8 mL/min/1.73 m2) was noted. The investigators surmised that denervation may enhance diuretic sensitivity, leading to prerenal azotemia in some patients.17
SYMPLICITY HTN-2
The Symplicity HTN-2 trial was a larger, randomized, efficacy study that built on the earlier results, providing additional evidence of therapeutic benefit.15
An international cohort of 106 patients with resistant hypertension, defined as systolic blood pressure of 160 mm Hg or higher (or ≥ 150 mm Hg in patients with type 2 diabetes) despite the use of three or more antihypertensive medications, were randomly assigned to undergo renal denervation with the Symplicity device (n = 52) or to continue their previous treatment with antihypertensive medications alone (n = 54). The primary effectiveness end point was the change in seated office blood pressure from baseline to 6 months (Table 1).
Effect on blood pressure. In the denervation group, at 6 months, office blood pressure had changed by a mean of −32/−12 mm Hg (standard deviation [SD] 23/11 mm Hg) compared with a mean change of 1/0 mm Hg (SD 21/10 mm Hg) in the control group. Fortyone (84%) of the 49 patients who underwent denervation had a decrease in systolic blood pressure of 10 mm Hg or more at 6 months compared with baseline values, while five (10%) had no decline in systolic blood pressure. Nineteen patients had a reduction in systolic pressure to less than 140 mm Hg in the denervation group.
A subset of patients (20 in the denervation group and 25 in the control group) underwent 24-hour ambulatory blood pressure monitoring at 6 months. This showed a similar though less pronounced fall in blood pressure in the denervation group and no change in the controls. A subanalysis that censored all data for patients whose medication was increased during the follow-up period showed a blood pressure reduction of −31/−12 mm Hg (SD 22/11 mm Hg) in the renal denervation group.
Adverse events. Procedure-related adverse events included a single femoral artery pseudoaneurysm, one case of postprocedural hypotension requiring a reduction in antihypertensive medications, and 7 (13%) of 52 patients who experienced intraprocedural bradycardia requiring atropine.
Effect on renal function. No significant difference was noted between groups in the mean change in renal function at 6 months, whether assessed by eGFR, serum creatinine level, or cystatin C level. At 6 months, no patient had a decrease of more than 50% in eGFR, although two patients who underwent renal denervation and three controls had more than a 25% decrease in eGFR.
At 6 months, the urine albumin-to-creatinine ratio had changed by a median of −3 mg/g (range −1,089 to 76) in 38 patients in the treatment group and by 1 mg/g (range −538 to 227) in 37 controls.
Most patients (88%) undergoing renal denervation underwent renal arterial imaging at 6 months, on which a single patient showed possible progression of an underlying atherosclerotic lesion that was unrelated to the procedure and that did not require intervention.
Denervation and the normal stress response. Whether renal denervation negatively affects the body’s physiologic response to stress that is normally mediated by sympathetic nerve activity was addressed in an extended investigation of Symplicity HTN-2 using cardiopulmonary exercise tests at baseline and 3 months after renal denervation.18 In the denervation group, blood pressure during exercise was significantly lower at 3 months than at baseline, but the heart rate increase at different levels of exercise was not affected. Additionally, the resting heart rate was lower and heart rate recovery after exercise improved after the procedure, particularly in patients without diabetes.
Comments. The Symplicity HTN-2 trial benefited from a randomized trial design and strict inclusion criteria of treatment resistance, but it still had notable limitations. A pretrial evaluation for causes of secondary hypertension or white-coat hypertension was not explicitly described. The control group did not undergo a sham procedure, and data analyzers were not masked to treatment assignment. Although not analyzed as a primary end point, the use of home-based and 24-hour ambulatory blood pressure assessment—measures important for determining white-coat hypertension—revealed substantial differences in blood pressure changes relative to office measurements. Because nearly all the patients (97%) were white, the generalizability of treatment results to black patients with resistant hypertension may be limited. Isolated diastolic hypertension (defined as diastolic pressure ≥ 90 mm Hg with systolic pressure < 140 mm Hg), which is more common in younger patients, was not studied.
DOES RENAL DENERVATION REDUCE SYMPATHETIC TONE?
A subgroup of 10 patients in the Symplicity HTN-1 trial whose mean 6-month office blood pressure was reduced by 22/12 mm Hg underwent assessment of renal norepinephrine spillover. A substantial (47%) reduction in renal norepinephrine spillover was noted 1 month after the procedure.14
The investigators additionally described a marked reduction in renal norepinephrine spillover from both kidneys in one patient, with a reduction of 48% from the left kidney and 75% from the right kidney 1 month after the procedure. Whole-body norepinephrine spillover in this patient was reduced by 42%. This effect was accompanied by a 50% decrease in plasma renin activity and by an increase in renal plasma flow. Aldosterone levels were not reported.19
Thus, the decrease in renal norepinephrine spillover suggests a reduction of renal efferent activity, and the decrease in total body norepinephrine spillover suggests a reduction in central sympathetic drive via the renal afferent pathway.
Microneurography in this same patient showed a gradual reduction in muscle sympathetic nerve activity to normal levels, from 56 bursts per minute at baseline to 41 at 30 days and 19 at 12 months).19 Decreased renin secretion, via circulating angiotensin II, may affect central sympathetic outflow as well.
Comments. While these findings address some of the underlying mechanisms, the small number of patients in whom these studies were done limits the generalizability of the results. The impact of the procedure on renal hemodynamics will need to be studied, including possible direct effects of the procedure, and whether there are differences in different study populations or differences based on blood pressure levels.
WHICH PATIENTS RESPOND BEST TO THIS PROCEDURE?
Although the Symplicity HTN-2 investigators report some predictors of increased reduction in blood pressure on multivariate analysis, including increased blood pressure at baseline and reduced heart rate at baseline, these are not specific enough to enable patient selection.
Interestingly, results from the expanded cohort of the Symplicity HTN-1 study found that patients on central sympatholytic agents such as clonidine had a greater reduction in blood pressure, although the reason for this is unclear.16 Identifying specific predictors of treatment success at baseline will be essential in future studies.
The earlier Symplicity trials and the ongoing Symplicity HTN-3 trial are in patients who have high blood pressure not responding to three or more antihypertensive drugs. The mean baseline systolic blood pressure in the Symplicity HTN-1 and HTN-2 trials was 178 mm Hg, and patients were taking an average of five antihypertensive drugs (Table 1). It is not known whether denervation will produce similar blood-pressure-lowering results across the spectrum of hypertension severity.
WHAT ARE THE LONG-TERM RESULTS OF DENERVATION?
Enthusiasm for the results from the Symplicity trials is tempered by concerns about the durability of the effects of the procedure, the need for better understanding of the impact of renal denervation on a wide array of pathophysiologic cascades leading to hypertension, and the effect on renal hemodynamics.
Antihypertensive efficacy has been reported to persist up to 2 years after the procedure,16 with recent unpublished data suggesting efficacy up to 3 years, but longer follow-up is needed to address whether these effects are finite.
Although reinnervation of afferent renal nerves has not been described, transplant models have shown anatomic regrowth of efferent nerves; the impact of this efferent reinnervation on blood pressure remains unclear. Experience from renal transplantation also shows that implanted kidneys that are “denervated” can still maintain fluid and electrolyte regulation.
Follow-up renal imaging in the Symplicity trials did not indicate renal artery stenosis at the sites of denervation in patients who underwent the procedure. Animal studies using the Symplicity catheter system showed renal nerve injury as evidenced by nerve fibrosis and thickened epineurium and perineurium, but no significant smooth muscle hyperplasia, arterial stenosis, or thrombosis by angiography or histology at 6 months.20
WHAT ARE THE RISKS?
Adverse effects that were noted in the short term are detailed under discussion of the trials and in Table 2.
Long-term adverse events in the Symplicity HTN-2 trial that required hospitalization were reported in five patients in the denervation group and three patients in the control group (Table 2). These included transient ischemic attacks, hypertensive crises, hypotensive episodes, angina, and nausea.
Renal function was maintained for the duration of both trials, and details regarding eGFR change have been described above under the discussion of the trials.
Diffuse visceral pain at the time of the procedure is reported as an expected occurrence, managed with intravenous analgesic medications.
DOES SYMPATHETIC DENERVATION HAVE A ROLE IN OTHER CONDITIONS?
Interestingly, other sympathetically driven diseases, such as diabetes mellitus and polycystic ovary syndrome, may prove to be targets for this therapy in the future.21
Mahfoud et al22 conducted a pilot study in 37 patients with resistant hypertension undergoing renal denervation and 13 control patients. Fasting glucose levels declined from 118 ± 3.4 mg/dL to 108 ± 3.8 mg/dL after 3 months in the intervention group (P = .039), compared with no change in the control group. Insulin and C-peptide levels were also lower in the intervention group. The reported improvement in glucose metabolism and insulin sensitivity suggests that the beneficial effects of this procedure may extend beyond blood pressure reduction.
Brandt et al23 reported regression of left ventricular hypertrophy and significantly improved cardiac functional parameters, including increase in ejection fraction and improved diastolic dysfunction, in a study of 46 patients who underwent renal denervation. This findings suggests a potential beneficial effect on cardiac remodeling.
Witkowski et al24 reported lowering of blood pressure in 10 patients with refractory hypertension and obstructive sleep apnea who underwent renal denervation, which was accompanied by improvement of sleep apnea severity.
Ukena et al25 reported reduction in ventricular tachyarrhythmias in two patients with congestive heart failure who had therapy-resistant electrical storm.
A recent pilot study in 15 patients with stage 3 and 4 chronic kidney disease (mean eGFR 31 mL/min/1.73 m2) showed significantly improved office blood pressure control up to 1 year, restoration of nocturnal dipping on 24-hour monitoring, as well as a nonsignificant trend towards increased hemoglobin levels and decreased proteinuria. No additional deterioration of renal function was reported in these patients (2 patients had renal function assessed up to 1 year).26
Thus, the benefits of this procedure may extend to other diseases that have a common underlying thread of elevated sympathetic activity, by targeting the “sympathorenal” axis.27
GUARDED OPTIMISM AND FUTURE DIRECTIONS
Given the well-known cardiovascular risks and health care costs associated with uncontrolled hypertension and the continued challenge that physicians face in managing it, novel therapies such as renal denervation may provide an adjunct to existing pharmacologic approaches.
While there is certainly cause for guarded optimism, especially with the striking blood pressure-lowering results seen in trials so far, it should be kept in mind that the mechanisms leading to the hypertensive response are complex and multifactorial, and further understanding of this therapy with long-term follow-up is needed. A comparison study with spironolactone, which is increasingly being used to treat resistant hypertension (in the absence of a diagnosis of primary aldosteronism)28,29 would help to further establish the role of this procedure.
Studies of carotid baroreceptor stimulation via an implantable device have shown sustained reduction in blood pressure in patients with resistant hypertension. A study comparing this technique with renal denervation for efficacy and safety end points could be considered in the future.30,31
The planned Symplicity HTN-3 study in the United States will be the largest trial to date, with a targeted randomization of more than 500 patients using strict enrollment criteria, including the use of maximally tolerated doses of diuretics and more focus on the use of ambulatory blood pressure monitoring and on the blinding of participants. This study will help further analysis of this technology in a more diverse population.32,33
Future studies should be designed to clarify pathophysiologic mechanisms, patient selection criteria, effects on target organ damage, and efficacy in patients with chronic kidney disease, obesity, congestive heart failure, and in less severe forms of hypertension.
A CALL FOR PARTICIPANTS IN A CLINICAL TRIAL
The Departments of Cardiology and Nephrology and Hypertension at Cleveland Clinic are currently enrolling patients in the Symplicity HTN-3 trial. For more information, please contact George Thomas, MD ([email protected]), or Mehdi Shishehbor, DO, MPH ([email protected]), or visit www.symplifybptrial.com.
- Chobanian AV, Bakris GL, Black HR, et al; National Heart, Lung, and Blood Institute Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure; National High Blood Pressure Education Program Coordinating Committee. The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. JAMA 2003; 289:2560–2572.
- Schappert SM, Rechtsteiner EA. Ambulatory medical care utilization estimates for 2007. National Center for Health Statistics. Vital Health Stat 13( 169) 2011. http://www.cdc.gov/nchs/data/series/sr_13/sr13_169.pdf. Accessed April 24, 2012.
- Egan BM, Zhao Y, Axon RN. US trends in prevalence, awareness, treatment, and control of hypertension, 1988–2008. JAMA 2010; 303:2043–2050.
- Persell SD. Prevalence of resistant hypertension in the United States, 2003–2008. Hypertension 2011; 57:1076–1080.
- Calhoun DA, Jones D, Textor S, et al; American Heart Association Professional Education Committee. Resistant hypertension: diagnosis, evaluation, and treatment: a scientific statement from the American Heart Association Professional Education Committee of the Council for High Blood Pressure Research. Circulation 2008; 117:e510–e526.
- Smithwick RH, Thompson JE. Splanchnicectomy for essential hypertension; results in 1,266 cases. J Am Med Assoc 1953; 152:1501–1504.
- Schlaich MP, Sobotka PA, Krum H, Whitbourn R, Walton A, Esler MD. Renal denervation as a therapeutic approach for hypertension: novel implications for an old concept. Hypertension 2009; 54:1195–1201.
- Zanchetti AS. Neural regulation of renin release: experimental evidence and clinical implications in arterial hypertension. Circulation 1977; 56:691–698.
- Kon V. Neural control of renal circulation. Miner Electrolyte Metab 1989; 15:33–43.
- Campese VM. Neurogenic factors and hypertension in renal disease. Kidney Int Suppl 2000; 75:S2–S6.
- Mancia G, Grassi G, Giannattasio C, Seravalle G. Sympathetic activation in the pathogenesis of hypertension and progression of organ damage. Hypertension 1999; 34:724–728.
- Campese VM, Ye S, Zhong H, Yanamadala V, Ye Z, Chiu J. Reactive oxygen species stimulate central and peripheral sympathetic nervous system activity. Am J Physiol Heart Circ Physiol 2004; 287:H695–H703.
- Katholi RE. Renal nerves in the pathogenesis of hypertension in experimental animals and humans. Am J Physiol 1983; 245:F1–F14.
- Krum H, Schlaich M, Whitbourn R, et al. Catheter-based renal sympathetic denervation for resistant hypertension: a multicentre safety and proof-of-principle cohort study. Lancet 2009; 373:1275–1281.
- Esler MD, Krum H, Sobotka PA, Schlaich MP, Schmieder RE, Böhm M; Symplicity HTN-2 Investigators. Renal sympathetic denervation in patients with treatmentresistant hypertension (The Symplicity HTN-2 Trial): a randomised controlled trial. Lancet 2010; 376:1903–1909.
- Symplicity HTN-1 Investigators. Catheter-based renal sympathetic denervation for resistant hypertension: durability of blood pressure reduction out to 24 months. Hypertension 2011; 57:911–917.
- Petidis K, Anyfanti P, Doumas M. Renal sympathetic denervation: renal function concerns. Hypertension 2011; 58:e19; author replye20.
- Ukena C, Mahfoud F, Kindermann I, et al. Cardiorespiratory response to exercise after renal sympathetic denervation in patients with resistant hypertension. J Am Coll Cardiol 2011; 58:1176–1182.
- Schlaich MP, Sobotka PA, Krum H, Lambert E, Esler MD. Renal sympathetic-nerve ablation for uncontrolled hypertension (letter). N Engl J Med 2009; 361:932–934.
- Rippy MK, Zarins D, Barman NC, Wu A, Duncan KL, Zarins CK. Catheter-based renal sympathetic denervation: chronic preclinical evidence for renal artery safety. Clin Res Cardiol 2011; 100:1095–1101.
- Schlaich MP, Straznicky N, Grima M, et al. Renal denervation: a potential new treatment modality for polycystic ovary syndrome? J Hypertens 2011; 29:991–996.
- Mahfoud F, Schlaich M, Kindermann I, et al. Effect of renal sympathetic denervation on glucose metabolism in patients with resistant hypertension: a pilot study. Circulation 2011; 123:1940–1946.
- Brandt MC, Mahfoud F, Reda S, et al. Renal sympathetic denervation reduces left ventricular hypertrophy and improves cardiac function in patients with resistant hypertension. J Am Coll Cardiol 2012; 59:901–909.
- Witkowski A, Prejbisz A, Florczak E, et al. Effects of renal sympathetic denervation on blood pressure, sleep apnea course, and glycemic control in patients with resistant hypertension and sleep apnea. Hypertension 2011; 58:559–565.
- Ukena C, Bauer A, Mahfoud F, et al. Renal sympathetic denervation for treatment of electrical storm: first-inman experience. Clin Res Cardiol 2012; 101:63–67.
- Herring D, Mahfoud F, Walton AS, et al. Renal denervation in moderate to severe CKD. J Am Soc Nephrol 2012; May 17[Epub ahead of print]
- Sobotka PA, Mahfoud F, Schlaich MP, Hoppe UC, Böhm M, Krum H. Sympatho-renal axis in chronic disease. Clin Res Cardiol 2011; 100:1049–1057.
- Chapman N, Dobson J, Wilson S, et al; Anglo-Scandinavian Cardiac Outcomes Trial Investigators. Effect of spironolactone on blood pressure in subjects with resistant hypertension. Hypertension 2007; 49:839–845.
- Nishizaka MK, Zaman MA, Calhoun DA. Efficacy of low-dose spironolactone in subjects with resistant hypertension. Am J Hypertens 2003; 16:925–930.
- Papademetriou V, Doumas M, Faselis C, et al. Carotid baroreceptor stimulation for the treatment of resistant hypertension. Int J Hypertens 2011; 2011:964394.
- Ng MM, Sica DA, Frishman WH. Rheos: an implantable carotid sinus stimulation device for the nonpharmacologic treatment of resistant hypertension. Cardiol Rev 2011; 19:52–57.
- US National Institutes of Health. Renal denervation in patients with uncontrolled hypertension (SYMPLICITY HTN-3). http://www.clinicaltrials.gov/ct2/show/NCT01418261. Accessed June 7, 2012.
- Kandzari DE, Bhatt DL, Sobotka PA, et al. Catheter-based renal denervation for resistant hypertension: rationale and design of the Symplicity HTN-3 trial. Clin Cardiol 2012 May 9. [Epub ahead of print]
- Chobanian AV, Bakris GL, Black HR, et al; National Heart, Lung, and Blood Institute Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure; National High Blood Pressure Education Program Coordinating Committee. The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. JAMA 2003; 289:2560–2572.
- Schappert SM, Rechtsteiner EA. Ambulatory medical care utilization estimates for 2007. National Center for Health Statistics. Vital Health Stat 13( 169) 2011. http://www.cdc.gov/nchs/data/series/sr_13/sr13_169.pdf. Accessed April 24, 2012.
- Egan BM, Zhao Y, Axon RN. US trends in prevalence, awareness, treatment, and control of hypertension, 1988–2008. JAMA 2010; 303:2043–2050.
- Persell SD. Prevalence of resistant hypertension in the United States, 2003–2008. Hypertension 2011; 57:1076–1080.
- Calhoun DA, Jones D, Textor S, et al; American Heart Association Professional Education Committee. Resistant hypertension: diagnosis, evaluation, and treatment: a scientific statement from the American Heart Association Professional Education Committee of the Council for High Blood Pressure Research. Circulation 2008; 117:e510–e526.
- Smithwick RH, Thompson JE. Splanchnicectomy for essential hypertension; results in 1,266 cases. J Am Med Assoc 1953; 152:1501–1504.
- Schlaich MP, Sobotka PA, Krum H, Whitbourn R, Walton A, Esler MD. Renal denervation as a therapeutic approach for hypertension: novel implications for an old concept. Hypertension 2009; 54:1195–1201.
- Zanchetti AS. Neural regulation of renin release: experimental evidence and clinical implications in arterial hypertension. Circulation 1977; 56:691–698.
- Kon V. Neural control of renal circulation. Miner Electrolyte Metab 1989; 15:33–43.
- Campese VM. Neurogenic factors and hypertension in renal disease. Kidney Int Suppl 2000; 75:S2–S6.
- Mancia G, Grassi G, Giannattasio C, Seravalle G. Sympathetic activation in the pathogenesis of hypertension and progression of organ damage. Hypertension 1999; 34:724–728.
- Campese VM, Ye S, Zhong H, Yanamadala V, Ye Z, Chiu J. Reactive oxygen species stimulate central and peripheral sympathetic nervous system activity. Am J Physiol Heart Circ Physiol 2004; 287:H695–H703.
- Katholi RE. Renal nerves in the pathogenesis of hypertension in experimental animals and humans. Am J Physiol 1983; 245:F1–F14.
- Krum H, Schlaich M, Whitbourn R, et al. Catheter-based renal sympathetic denervation for resistant hypertension: a multicentre safety and proof-of-principle cohort study. Lancet 2009; 373:1275–1281.
- Esler MD, Krum H, Sobotka PA, Schlaich MP, Schmieder RE, Böhm M; Symplicity HTN-2 Investigators. Renal sympathetic denervation in patients with treatmentresistant hypertension (The Symplicity HTN-2 Trial): a randomised controlled trial. Lancet 2010; 376:1903–1909.
- Symplicity HTN-1 Investigators. Catheter-based renal sympathetic denervation for resistant hypertension: durability of blood pressure reduction out to 24 months. Hypertension 2011; 57:911–917.
- Petidis K, Anyfanti P, Doumas M. Renal sympathetic denervation: renal function concerns. Hypertension 2011; 58:e19; author replye20.
- Ukena C, Mahfoud F, Kindermann I, et al. Cardiorespiratory response to exercise after renal sympathetic denervation in patients with resistant hypertension. J Am Coll Cardiol 2011; 58:1176–1182.
- Schlaich MP, Sobotka PA, Krum H, Lambert E, Esler MD. Renal sympathetic-nerve ablation for uncontrolled hypertension (letter). N Engl J Med 2009; 361:932–934.
- Rippy MK, Zarins D, Barman NC, Wu A, Duncan KL, Zarins CK. Catheter-based renal sympathetic denervation: chronic preclinical evidence for renal artery safety. Clin Res Cardiol 2011; 100:1095–1101.
- Schlaich MP, Straznicky N, Grima M, et al. Renal denervation: a potential new treatment modality for polycystic ovary syndrome? J Hypertens 2011; 29:991–996.
- Mahfoud F, Schlaich M, Kindermann I, et al. Effect of renal sympathetic denervation on glucose metabolism in patients with resistant hypertension: a pilot study. Circulation 2011; 123:1940–1946.
- Brandt MC, Mahfoud F, Reda S, et al. Renal sympathetic denervation reduces left ventricular hypertrophy and improves cardiac function in patients with resistant hypertension. J Am Coll Cardiol 2012; 59:901–909.
- Witkowski A, Prejbisz A, Florczak E, et al. Effects of renal sympathetic denervation on blood pressure, sleep apnea course, and glycemic control in patients with resistant hypertension and sleep apnea. Hypertension 2011; 58:559–565.
- Ukena C, Bauer A, Mahfoud F, et al. Renal sympathetic denervation for treatment of electrical storm: first-inman experience. Clin Res Cardiol 2012; 101:63–67.
- Herring D, Mahfoud F, Walton AS, et al. Renal denervation in moderate to severe CKD. J Am Soc Nephrol 2012; May 17[Epub ahead of print]
- Sobotka PA, Mahfoud F, Schlaich MP, Hoppe UC, Böhm M, Krum H. Sympatho-renal axis in chronic disease. Clin Res Cardiol 2011; 100:1049–1057.
- Chapman N, Dobson J, Wilson S, et al; Anglo-Scandinavian Cardiac Outcomes Trial Investigators. Effect of spironolactone on blood pressure in subjects with resistant hypertension. Hypertension 2007; 49:839–845.
- Nishizaka MK, Zaman MA, Calhoun DA. Efficacy of low-dose spironolactone in subjects with resistant hypertension. Am J Hypertens 2003; 16:925–930.
- Papademetriou V, Doumas M, Faselis C, et al. Carotid baroreceptor stimulation for the treatment of resistant hypertension. Int J Hypertens 2011; 2011:964394.
- Ng MM, Sica DA, Frishman WH. Rheos: an implantable carotid sinus stimulation device for the nonpharmacologic treatment of resistant hypertension. Cardiol Rev 2011; 19:52–57.
- US National Institutes of Health. Renal denervation in patients with uncontrolled hypertension (SYMPLICITY HTN-3). http://www.clinicaltrials.gov/ct2/show/NCT01418261. Accessed June 7, 2012.
- Kandzari DE, Bhatt DL, Sobotka PA, et al. Catheter-based renal denervation for resistant hypertension: rationale and design of the Symplicity HTN-3 trial. Clin Cardiol 2012 May 9. [Epub ahead of print]
KEY POINTS
- Renal sympathetic nerves help regulate volume and blood pressure as they innervate the renal tubules, blood vessels, and juxtaglomerular apparatus. They carry both afferent and efferent signals between the central nervous system and the kidneys.
- Surgical sympathectomy was done in the 1950s for malignant hypertension. It had lasting antihypertensive results but also caused severe procedure-related morbidity. A new percutaneous procedure for selective renal denervation offers the advantage of causing few major procedure-related adverse effects.
- Selective renal denervation decreases norepinephrine spillover and muscle sympathetic nerve activity, evidence that the procedure reduces sympathetic tone.
- The major clinical trials done so far have found that renal denervation lowers blood pressure significantly, and the reduction is sustained for at least 3 years.
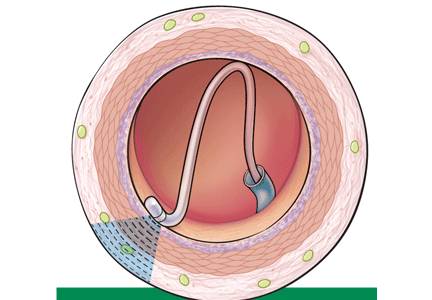
Correction: Anemia, leukocytosis, abdominal pain, flushing, and bone and skin lesion
In the June 2012 issue, on page 384 of the Clinical Picture article by Álvarez-Twose et al (Álvarez-Twose I, Vañó-Galván S, Sanchez-Muñoz L, Fernandez-Zapardiel S, Escribano L. The Clinical Picture: anemia, leukocytosis, abdominal pain, flushing, and bone and skin lesions. Cleve Clin J Med 2012; 79:384–386), Dr. Alvarez-Twose’s first name was spelled incorrectly. The correct spelling is Iván. This error has been corrected in the online version.
In the June 2012 issue, on page 384 of the Clinical Picture article by Álvarez-Twose et al (Álvarez-Twose I, Vañó-Galván S, Sanchez-Muñoz L, Fernandez-Zapardiel S, Escribano L. The Clinical Picture: anemia, leukocytosis, abdominal pain, flushing, and bone and skin lesions. Cleve Clin J Med 2012; 79:384–386), Dr. Alvarez-Twose’s first name was spelled incorrectly. The correct spelling is Iván. This error has been corrected in the online version.
In the June 2012 issue, on page 384 of the Clinical Picture article by Álvarez-Twose et al (Álvarez-Twose I, Vañó-Galván S, Sanchez-Muñoz L, Fernandez-Zapardiel S, Escribano L. The Clinical Picture: anemia, leukocytosis, abdominal pain, flushing, and bone and skin lesions. Cleve Clin J Med 2012; 79:384–386), Dr. Alvarez-Twose’s first name was spelled incorrectly. The correct spelling is Iván. This error has been corrected in the online version.
Septic Arthritis and Osteomyelitis Due to the Chromoblastomycosis Agent Fonsecaea pedrosoi
Lichen Planus: An Update and Review
A perplexing case of altered mental status
Discuss this article at www.facebook.com/CurrentPsychiatry
CASE: Agitated and paranoid
Mr. E, age 55, presents to the emergency department (ED) with a 2-week history of altered mental status (AMS). His wife reports, “He was normal one day and the next day he was not.” Mr. E also presents with sleep disturbance, decreased appetite and speech, and a 20-lb weight loss. His family reports no recent stressors or head trauma. Mr. E is agitated in the ED and receives a single dose of IV haloperidol, 5 mg. He exhibits paranoia and is afraid to get a CT scan. The medical team attempts a lumbar puncture (LP), but Mr. E does not cooperate.
His laboratory values are: potassium, 3.0 mEq/L; creatinine, 1.60 mg/dL; calcium, 10.6 mg/dL; thyroid-stimulating hormone, 0.177 IU/L; vitamin B12, >1500 pg/mL; folate, >20 ng/mL; and creatine kinase, 281 U/L. Urine drug screen is positive for benzodiazepines and opiates, neither of which was prescribed, and blood alcohol is negative.
Mr. E is admitted for further workup of AMS. His daughter-in-law states that Mr. E is an alcoholic and she is concerned that he may have mixed drugs and alcohol. The medical service starts Mr. E on empiric antimicrobials—vancomycin, ceftriaxone, and acyclovir—because of his AMS, and performs an LP to rule out intracranial pathology. His LP results are unremarkable.
Mr. E appears to be confused during psychiatric evaluation. He requests to be “hypnotized on a helicopter to find out what is wrong with me.” His wife states that Mr. E drank vodka daily but decreased his alcohol consumption after surgery 5 months ago. Before his current admission, he was drinking approximately half a glass of vodka every few days, according to his wife. Mr. E says he has no prior psychiatric admissions. During the mental status exam, his eye contact is poor, with latency of response to questions, thought blocking, and psychomotor retardation. He is alert, oriented to time, place, and person, and cooperative. He cannot concentrate or focus during the interview. He denies suicidal or homicidal ideation.
The authors’ observations
Mr. E appeared to be delirious, as evidenced by the sudden onset of waxing and waning changes in consciousness, attention deficits, and cognition. He also had a history of daily alcohol use and decreased his alcohol intake after a surgery 5 months ago, which puts him at risk for Wernicke’s encephalopathy.1-3 The type of surgery and whether he received adequate thiamine supplementation at that time was unclear. Because Mr. E is older, he has a higher risk of mortality and morbidity from delirium.4,5 We started Mr. E on quetiapine, 50 mg/d, for delirium and an IV lorazepam taper, starting at 2 mg every 8 hours, because the extent of his alcohol and benzodiazepine use was unclear—we weren’t sure how forthcoming he was about his alcohol use. He received IV thiamine supplementation followed by oral thiamine, 100 mg/d.
The authors’ recommendations
We requested a neurology consult, EEG, CSF cultures, and brain MRI (Table 1).6 EEG, chest radiography, thyroid scan, and CT scan were normal and MRI showed no acute intracranial process. However, there was a redemonstration of increased T1 signal seen within the bilateral basal ganglia and relative diminutive appearance to the bilateral mamillary bodies, which suggests possible liver disease and/or alcohol abuse. These findings were unchanged from an MRI Mr. E received 10 years ago, were consistent with his history of alcohol abuse, and may indicate an underlying predisposition to delirium. A CT scan of the abdomen showed hepatic cirrhosis with prominent, tortuous vessels of the upper abdomen, subtle ill-defined hypodensity of the anterior aspect of the liver, and an enlarged spleen.
Mr. E’s mental functioning did not improve with quetiapine and lorazepam. Further evaluation revealed a negative human immunodeficiency virus test and normal heavy metals, ammonia, ceruloplasmin, and thiamine. We suspected limbic encephalitis because of Mr. E’s memory problems and behavioral and psychiatric manifestations,7 but CSF was unremarkable and limbic encephalitis workup of CSF and paraneoplastic antibody panel were negative.
Mr. E’s primary care physician stated that at an appointment 1 month ago, Mr. E was alert, oriented, and conversational with normal thought processing. At that time he had presented with rectal bleeding, occult blood in his stool, and an unintentional 25-lb weight loss over 2 months. It was not clear if his weight loss was caused by poor nutrition—which is common among chronic alcoholics—or an occult disease process.
After 10 days, Mr. E was discharged home from the medicine service with no clear cause of his AMS.
Table 1
Suggested workup for altered mental status
| Complete blood count, basic metabolic profile, creatine kinase |
| Thyroid-stimulating hormone, thyroid scan |
| Vitamin B12, folate, thiamine |
| Blood alcohol, urine drug screen |
| Urine analysis and cultures |
| Lumbar puncture—CSF staining and cultures |
| Chest radiography |
| CT and MRI scan of brain |
| Electroencephalography |
| Neuropsychiatric testing |
| CSF: cerebrospinal fluid Source: Reference 6 |
EVALUATION: Worsening behavior
One week later, Mr. E presents to the ED with continued AMS and worsening behavior at home. Two days ago, he attempted to strangle his dog and cut himself with a knife. His paranoia was worsening and his oral intake continued to decrease. In the ED, Mr. E does not want a chest radiograph because, “I don’t like radiation contaminating my body”; his family stated that he had been suspicious of radiography all of his life. He receives empiric ceftriaxone because of a possible urinary tract infection. Urine culture is positive for Pseudomonas aeruginosa and he is switched to ciprofloxacin. Mr. E is admitted for further workup.
Mr. E’s mother states, “I think this change in behavior is related to my son drinking alcohol for 20 years. This is exactly how he acted when he was on drugs. I think he is having a flashback.” She also reports her son purchased multiple chemicals—the details of which are unclear—that he left lying around the house.
His wife says that after discharge a week ago, Mr. E was stable for 1 or 2 days and then “he started going downhill.” He became more paranoid and he started talking about cameras watching him in his house. Mr. E took quetiapine, 50 mg/d, for a few days, then refused because he thought there was something in the medication. Mr. E’s family feels that at times he is responding to internal stimuli. He makes statements about his DNA being changed and reports that he has 2 wives and the wife in the room was not the real one, which suggests Capgras syndrome. His wife provides a home medication list that includes vitamin B complex, vitamins B12, E, and C, a multivitamin, zinc, magnesium, fish oil, garlic, calcium, glucosamine, chondroitin, herbal supplements, and gingko. The psychiatry team recommends switching from quetiapine to olanzapine, 15 mg/d, because Mr. E was paranoid about taking quetiapine.
We determine that Mr. E does not have medical decision-making capacity.
Because his symptoms do not improve, Mr. E is transferred to the psychiatric intensive care unit. His mental status shows little change while there. Neuropsychiatric testing shows only “cognitive deficits.” He shows signs consistent with neurologic dysfunction in terms of stimulus-bound responding and perseveration, which is compatible with the bilateral basal ganglia lesion seen on MRI. However, some of his behaviors suggest psychiatric and motivationally driven or manipulative etiology. During this testing he was difficult to evaluate and needed to be convinced to engage. At times he was illogical and at other times he showed good focus and recall. It is difficult to draw more definitive conclusions and Mr. E is discharged home with minor improvement in his symptoms. He didn’t attend follow-up appointments. During a courtesy call a few months after his admission, his wife revealed that Mr. E had died after shooting himself. It is unclear if it was an accident or suicide.
The authors’ observations
Mr. E’s diagnosis remains unclear (for a summary of his clinical course, see Table 2). Although his initial presentation was consistent with delirium, the lack of an identifiable medical cause, prolonged time course, and lack of improvement with dopamine blocking agents suggest additional diagnoses such as Wernicke-Korsakoff syndrome, rapidly progressive dementia, or a substance-induced disorder. He displayed paranoia and bizarre delusions, which would suggest a thought disorder. However, he also had a history of substance use. A few months after we saw Mr. E, “bath salt” (methylenedioxypyrovalerone) abuse gained national attention. Patients with bath salt intoxication present with confusion, paranoia, and behavioral disturbances as well as a prolonged course.8
Mr. E’s CT and MRI scans, history of alcohol use, and cirrhosis also point to Wernicke-Korsakoff syndrome as an underlying diagnosis. It is unclear whether Mr. E experienced alcohol withdrawal and IV glucose without adequate thiamine replacement during a prior surgery. However, MRI findings were unchanged from a previous study 10 years ago.
It is puzzling whether Mr. E’s AMS was a first psychotic break, a result of drug and alcohol use, rapidly progressing dementia, or another neurologic problem that we have not identified. Our tentative diagnosis was Wernicke-Korsakoff syndrome because of his history of alcohol use and imaging findings.
Although we used a multidisciplinary team approach that included psychiatry, internal medicine, neurology, neuropsychology, and an aggressive and thorough workup, we could not establish a definitive diagnosis. Unsolved cases such as this can leave patients and clinicians frustrated and may lead to unfavorable outcomes. Additional resources such as a telephone call after the first missed appointment may have been warranted.
Table 2
Mr. E’s clinical course
| Symptoms | Treatment | |
|---|---|---|
| First ED visit | Agitation Confusion Sleep disturbance Decreased appetite and speech 20-lb weight loss | Empiric antimicrobials for possible meningitis Haloperidol for agitation Quetiapine for delirium Lorazepam taper Thiamine supplementation |
| Second ED visit | Violent behavior Worsening paranoia Responding to internal stimuli Mr. E believes he has 2 wives, but the wife in the room is not the real one, which suggests possible Capgras syndrome Cognitive deficits on mental status exam | Switch from ceftriaxone to ciprofloxacin for Pseudomonas aeruginosa Switch from quetiapine to olanzapine |
| ED: emergency department | ||
Related Resources
- Kaufman DM. Clinical neurology for psychiatrists. 6th ed. Philadelphia, PA: Saunders Elsevier; 2007.
- Sidhu KS, Balon R, Ajluni V, et al. Standard EEG and the difficult-to-assess mental status. Ann Clin Psychiatry. 2009;21(2):103-108.
Drug Brand Names
- Acyclovir • Zovirax
- Ceftriaxone • Rocephin
- Ciprofloxacin • Cipro
- Haloperidol • Haldol
- Lorazepam • Ativan
- Olanzapine • Zyprexa
- Quetiapine • Seroquel
- Thiamine • Betaxin
- Vancomycin • Vancocin
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Jiang W, Gagliardi JP, Raj YP, et al. Acute psychotic disorder after gastric bypass surgery: differential diagnosis and treatment. Am J Psychiatry. 2006;163(1):15-19.
2. Harrison RA, Vu T, Hunter AJ. Wernicke’s encephalopathy in a patient with schizophrenia. J Gen Intern Med. 2006;21(12):C8-C11.
3. Sechi GP, Serra A. Wernicke’s encephalopathy: new clinical settings and recent advances in diagnosis and treatment. Lancet Neurol. 2007;6(5):442-455.
4. American Psychiatric Association. Practice guidelines for the treatment of patients with delirium. Am J Psychiatry. 1999;156(5 suppl):1-20.
5. Sharon KI, Fearing MA, Marcantonio RA. Delirium. In: Halter JB Ouslander JG, Tinetti ME, et al, eds. Hazzard’s geriatric medicine and gerontology. 6th ed. New York, NY: McGraw-Hill Medical; 2009:647–658.
6. Sadock BJ, Sadock VA. Delirium dementia, and amnestic and other cognitive disorders. In: Sadock BJ, Kaplan HI, Sadock VA. Kaplan and Sadock’s synopsis of psychiatry: behavioral sciences/clinical psychiatry. Philadelphia, PA: Lippincott Williams and Wilkins; 2007:319–372.
7. Ahmad SA, Archer HA, Rice CM, et al. Seronegative limbic encephalitis: case report, literature review and proposed treatment algorithm. Pract Neurol. 2011;11(6):355-361.
8. Ross EA, Watson M, Goldberger B. “Bath salts” intoxication. N Engl J Med. 2011;365(10):967-968.
Discuss this article at www.facebook.com/CurrentPsychiatry
CASE: Agitated and paranoid
Mr. E, age 55, presents to the emergency department (ED) with a 2-week history of altered mental status (AMS). His wife reports, “He was normal one day and the next day he was not.” Mr. E also presents with sleep disturbance, decreased appetite and speech, and a 20-lb weight loss. His family reports no recent stressors or head trauma. Mr. E is agitated in the ED and receives a single dose of IV haloperidol, 5 mg. He exhibits paranoia and is afraid to get a CT scan. The medical team attempts a lumbar puncture (LP), but Mr. E does not cooperate.
His laboratory values are: potassium, 3.0 mEq/L; creatinine, 1.60 mg/dL; calcium, 10.6 mg/dL; thyroid-stimulating hormone, 0.177 IU/L; vitamin B12, >1500 pg/mL; folate, >20 ng/mL; and creatine kinase, 281 U/L. Urine drug screen is positive for benzodiazepines and opiates, neither of which was prescribed, and blood alcohol is negative.
Mr. E is admitted for further workup of AMS. His daughter-in-law states that Mr. E is an alcoholic and she is concerned that he may have mixed drugs and alcohol. The medical service starts Mr. E on empiric antimicrobials—vancomycin, ceftriaxone, and acyclovir—because of his AMS, and performs an LP to rule out intracranial pathology. His LP results are unremarkable.
Mr. E appears to be confused during psychiatric evaluation. He requests to be “hypnotized on a helicopter to find out what is wrong with me.” His wife states that Mr. E drank vodka daily but decreased his alcohol consumption after surgery 5 months ago. Before his current admission, he was drinking approximately half a glass of vodka every few days, according to his wife. Mr. E says he has no prior psychiatric admissions. During the mental status exam, his eye contact is poor, with latency of response to questions, thought blocking, and psychomotor retardation. He is alert, oriented to time, place, and person, and cooperative. He cannot concentrate or focus during the interview. He denies suicidal or homicidal ideation.
The authors’ observations
Mr. E appeared to be delirious, as evidenced by the sudden onset of waxing and waning changes in consciousness, attention deficits, and cognition. He also had a history of daily alcohol use and decreased his alcohol intake after a surgery 5 months ago, which puts him at risk for Wernicke’s encephalopathy.1-3 The type of surgery and whether he received adequate thiamine supplementation at that time was unclear. Because Mr. E is older, he has a higher risk of mortality and morbidity from delirium.4,5 We started Mr. E on quetiapine, 50 mg/d, for delirium and an IV lorazepam taper, starting at 2 mg every 8 hours, because the extent of his alcohol and benzodiazepine use was unclear—we weren’t sure how forthcoming he was about his alcohol use. He received IV thiamine supplementation followed by oral thiamine, 100 mg/d.
The authors’ recommendations
We requested a neurology consult, EEG, CSF cultures, and brain MRI (Table 1).6 EEG, chest radiography, thyroid scan, and CT scan were normal and MRI showed no acute intracranial process. However, there was a redemonstration of increased T1 signal seen within the bilateral basal ganglia and relative diminutive appearance to the bilateral mamillary bodies, which suggests possible liver disease and/or alcohol abuse. These findings were unchanged from an MRI Mr. E received 10 years ago, were consistent with his history of alcohol abuse, and may indicate an underlying predisposition to delirium. A CT scan of the abdomen showed hepatic cirrhosis with prominent, tortuous vessels of the upper abdomen, subtle ill-defined hypodensity of the anterior aspect of the liver, and an enlarged spleen.
Mr. E’s mental functioning did not improve with quetiapine and lorazepam. Further evaluation revealed a negative human immunodeficiency virus test and normal heavy metals, ammonia, ceruloplasmin, and thiamine. We suspected limbic encephalitis because of Mr. E’s memory problems and behavioral and psychiatric manifestations,7 but CSF was unremarkable and limbic encephalitis workup of CSF and paraneoplastic antibody panel were negative.
Mr. E’s primary care physician stated that at an appointment 1 month ago, Mr. E was alert, oriented, and conversational with normal thought processing. At that time he had presented with rectal bleeding, occult blood in his stool, and an unintentional 25-lb weight loss over 2 months. It was not clear if his weight loss was caused by poor nutrition—which is common among chronic alcoholics—or an occult disease process.
After 10 days, Mr. E was discharged home from the medicine service with no clear cause of his AMS.
Table 1
Suggested workup for altered mental status
| Complete blood count, basic metabolic profile, creatine kinase |
| Thyroid-stimulating hormone, thyroid scan |
| Vitamin B12, folate, thiamine |
| Blood alcohol, urine drug screen |
| Urine analysis and cultures |
| Lumbar puncture—CSF staining and cultures |
| Chest radiography |
| CT and MRI scan of brain |
| Electroencephalography |
| Neuropsychiatric testing |
| CSF: cerebrospinal fluid Source: Reference 6 |
EVALUATION: Worsening behavior
One week later, Mr. E presents to the ED with continued AMS and worsening behavior at home. Two days ago, he attempted to strangle his dog and cut himself with a knife. His paranoia was worsening and his oral intake continued to decrease. In the ED, Mr. E does not want a chest radiograph because, “I don’t like radiation contaminating my body”; his family stated that he had been suspicious of radiography all of his life. He receives empiric ceftriaxone because of a possible urinary tract infection. Urine culture is positive for Pseudomonas aeruginosa and he is switched to ciprofloxacin. Mr. E is admitted for further workup.
Mr. E’s mother states, “I think this change in behavior is related to my son drinking alcohol for 20 years. This is exactly how he acted when he was on drugs. I think he is having a flashback.” She also reports her son purchased multiple chemicals—the details of which are unclear—that he left lying around the house.
His wife says that after discharge a week ago, Mr. E was stable for 1 or 2 days and then “he started going downhill.” He became more paranoid and he started talking about cameras watching him in his house. Mr. E took quetiapine, 50 mg/d, for a few days, then refused because he thought there was something in the medication. Mr. E’s family feels that at times he is responding to internal stimuli. He makes statements about his DNA being changed and reports that he has 2 wives and the wife in the room was not the real one, which suggests Capgras syndrome. His wife provides a home medication list that includes vitamin B complex, vitamins B12, E, and C, a multivitamin, zinc, magnesium, fish oil, garlic, calcium, glucosamine, chondroitin, herbal supplements, and gingko. The psychiatry team recommends switching from quetiapine to olanzapine, 15 mg/d, because Mr. E was paranoid about taking quetiapine.
We determine that Mr. E does not have medical decision-making capacity.
Because his symptoms do not improve, Mr. E is transferred to the psychiatric intensive care unit. His mental status shows little change while there. Neuropsychiatric testing shows only “cognitive deficits.” He shows signs consistent with neurologic dysfunction in terms of stimulus-bound responding and perseveration, which is compatible with the bilateral basal ganglia lesion seen on MRI. However, some of his behaviors suggest psychiatric and motivationally driven or manipulative etiology. During this testing he was difficult to evaluate and needed to be convinced to engage. At times he was illogical and at other times he showed good focus and recall. It is difficult to draw more definitive conclusions and Mr. E is discharged home with minor improvement in his symptoms. He didn’t attend follow-up appointments. During a courtesy call a few months after his admission, his wife revealed that Mr. E had died after shooting himself. It is unclear if it was an accident or suicide.
The authors’ observations
Mr. E’s diagnosis remains unclear (for a summary of his clinical course, see Table 2). Although his initial presentation was consistent with delirium, the lack of an identifiable medical cause, prolonged time course, and lack of improvement with dopamine blocking agents suggest additional diagnoses such as Wernicke-Korsakoff syndrome, rapidly progressive dementia, or a substance-induced disorder. He displayed paranoia and bizarre delusions, which would suggest a thought disorder. However, he also had a history of substance use. A few months after we saw Mr. E, “bath salt” (methylenedioxypyrovalerone) abuse gained national attention. Patients with bath salt intoxication present with confusion, paranoia, and behavioral disturbances as well as a prolonged course.8
Mr. E’s CT and MRI scans, history of alcohol use, and cirrhosis also point to Wernicke-Korsakoff syndrome as an underlying diagnosis. It is unclear whether Mr. E experienced alcohol withdrawal and IV glucose without adequate thiamine replacement during a prior surgery. However, MRI findings were unchanged from a previous study 10 years ago.
It is puzzling whether Mr. E’s AMS was a first psychotic break, a result of drug and alcohol use, rapidly progressing dementia, or another neurologic problem that we have not identified. Our tentative diagnosis was Wernicke-Korsakoff syndrome because of his history of alcohol use and imaging findings.
Although we used a multidisciplinary team approach that included psychiatry, internal medicine, neurology, neuropsychology, and an aggressive and thorough workup, we could not establish a definitive diagnosis. Unsolved cases such as this can leave patients and clinicians frustrated and may lead to unfavorable outcomes. Additional resources such as a telephone call after the first missed appointment may have been warranted.
Table 2
Mr. E’s clinical course
| Symptoms | Treatment | |
|---|---|---|
| First ED visit | Agitation Confusion Sleep disturbance Decreased appetite and speech 20-lb weight loss | Empiric antimicrobials for possible meningitis Haloperidol for agitation Quetiapine for delirium Lorazepam taper Thiamine supplementation |
| Second ED visit | Violent behavior Worsening paranoia Responding to internal stimuli Mr. E believes he has 2 wives, but the wife in the room is not the real one, which suggests possible Capgras syndrome Cognitive deficits on mental status exam | Switch from ceftriaxone to ciprofloxacin for Pseudomonas aeruginosa Switch from quetiapine to olanzapine |
| ED: emergency department | ||
Related Resources
- Kaufman DM. Clinical neurology for psychiatrists. 6th ed. Philadelphia, PA: Saunders Elsevier; 2007.
- Sidhu KS, Balon R, Ajluni V, et al. Standard EEG and the difficult-to-assess mental status. Ann Clin Psychiatry. 2009;21(2):103-108.
Drug Brand Names
- Acyclovir • Zovirax
- Ceftriaxone • Rocephin
- Ciprofloxacin • Cipro
- Haloperidol • Haldol
- Lorazepam • Ativan
- Olanzapine • Zyprexa
- Quetiapine • Seroquel
- Thiamine • Betaxin
- Vancomycin • Vancocin
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Discuss this article at www.facebook.com/CurrentPsychiatry
CASE: Agitated and paranoid
Mr. E, age 55, presents to the emergency department (ED) with a 2-week history of altered mental status (AMS). His wife reports, “He was normal one day and the next day he was not.” Mr. E also presents with sleep disturbance, decreased appetite and speech, and a 20-lb weight loss. His family reports no recent stressors or head trauma. Mr. E is agitated in the ED and receives a single dose of IV haloperidol, 5 mg. He exhibits paranoia and is afraid to get a CT scan. The medical team attempts a lumbar puncture (LP), but Mr. E does not cooperate.
His laboratory values are: potassium, 3.0 mEq/L; creatinine, 1.60 mg/dL; calcium, 10.6 mg/dL; thyroid-stimulating hormone, 0.177 IU/L; vitamin B12, >1500 pg/mL; folate, >20 ng/mL; and creatine kinase, 281 U/L. Urine drug screen is positive for benzodiazepines and opiates, neither of which was prescribed, and blood alcohol is negative.
Mr. E is admitted for further workup of AMS. His daughter-in-law states that Mr. E is an alcoholic and she is concerned that he may have mixed drugs and alcohol. The medical service starts Mr. E on empiric antimicrobials—vancomycin, ceftriaxone, and acyclovir—because of his AMS, and performs an LP to rule out intracranial pathology. His LP results are unremarkable.
Mr. E appears to be confused during psychiatric evaluation. He requests to be “hypnotized on a helicopter to find out what is wrong with me.” His wife states that Mr. E drank vodka daily but decreased his alcohol consumption after surgery 5 months ago. Before his current admission, he was drinking approximately half a glass of vodka every few days, according to his wife. Mr. E says he has no prior psychiatric admissions. During the mental status exam, his eye contact is poor, with latency of response to questions, thought blocking, and psychomotor retardation. He is alert, oriented to time, place, and person, and cooperative. He cannot concentrate or focus during the interview. He denies suicidal or homicidal ideation.
The authors’ observations
Mr. E appeared to be delirious, as evidenced by the sudden onset of waxing and waning changes in consciousness, attention deficits, and cognition. He also had a history of daily alcohol use and decreased his alcohol intake after a surgery 5 months ago, which puts him at risk for Wernicke’s encephalopathy.1-3 The type of surgery and whether he received adequate thiamine supplementation at that time was unclear. Because Mr. E is older, he has a higher risk of mortality and morbidity from delirium.4,5 We started Mr. E on quetiapine, 50 mg/d, for delirium and an IV lorazepam taper, starting at 2 mg every 8 hours, because the extent of his alcohol and benzodiazepine use was unclear—we weren’t sure how forthcoming he was about his alcohol use. He received IV thiamine supplementation followed by oral thiamine, 100 mg/d.
The authors’ recommendations
We requested a neurology consult, EEG, CSF cultures, and brain MRI (Table 1).6 EEG, chest radiography, thyroid scan, and CT scan were normal and MRI showed no acute intracranial process. However, there was a redemonstration of increased T1 signal seen within the bilateral basal ganglia and relative diminutive appearance to the bilateral mamillary bodies, which suggests possible liver disease and/or alcohol abuse. These findings were unchanged from an MRI Mr. E received 10 years ago, were consistent with his history of alcohol abuse, and may indicate an underlying predisposition to delirium. A CT scan of the abdomen showed hepatic cirrhosis with prominent, tortuous vessels of the upper abdomen, subtle ill-defined hypodensity of the anterior aspect of the liver, and an enlarged spleen.
Mr. E’s mental functioning did not improve with quetiapine and lorazepam. Further evaluation revealed a negative human immunodeficiency virus test and normal heavy metals, ammonia, ceruloplasmin, and thiamine. We suspected limbic encephalitis because of Mr. E’s memory problems and behavioral and psychiatric manifestations,7 but CSF was unremarkable and limbic encephalitis workup of CSF and paraneoplastic antibody panel were negative.
Mr. E’s primary care physician stated that at an appointment 1 month ago, Mr. E was alert, oriented, and conversational with normal thought processing. At that time he had presented with rectal bleeding, occult blood in his stool, and an unintentional 25-lb weight loss over 2 months. It was not clear if his weight loss was caused by poor nutrition—which is common among chronic alcoholics—or an occult disease process.
After 10 days, Mr. E was discharged home from the medicine service with no clear cause of his AMS.
Table 1
Suggested workup for altered mental status
| Complete blood count, basic metabolic profile, creatine kinase |
| Thyroid-stimulating hormone, thyroid scan |
| Vitamin B12, folate, thiamine |
| Blood alcohol, urine drug screen |
| Urine analysis and cultures |
| Lumbar puncture—CSF staining and cultures |
| Chest radiography |
| CT and MRI scan of brain |
| Electroencephalography |
| Neuropsychiatric testing |
| CSF: cerebrospinal fluid Source: Reference 6 |
EVALUATION: Worsening behavior
One week later, Mr. E presents to the ED with continued AMS and worsening behavior at home. Two days ago, he attempted to strangle his dog and cut himself with a knife. His paranoia was worsening and his oral intake continued to decrease. In the ED, Mr. E does not want a chest radiograph because, “I don’t like radiation contaminating my body”; his family stated that he had been suspicious of radiography all of his life. He receives empiric ceftriaxone because of a possible urinary tract infection. Urine culture is positive for Pseudomonas aeruginosa and he is switched to ciprofloxacin. Mr. E is admitted for further workup.
Mr. E’s mother states, “I think this change in behavior is related to my son drinking alcohol for 20 years. This is exactly how he acted when he was on drugs. I think he is having a flashback.” She also reports her son purchased multiple chemicals—the details of which are unclear—that he left lying around the house.
His wife says that after discharge a week ago, Mr. E was stable for 1 or 2 days and then “he started going downhill.” He became more paranoid and he started talking about cameras watching him in his house. Mr. E took quetiapine, 50 mg/d, for a few days, then refused because he thought there was something in the medication. Mr. E’s family feels that at times he is responding to internal stimuli. He makes statements about his DNA being changed and reports that he has 2 wives and the wife in the room was not the real one, which suggests Capgras syndrome. His wife provides a home medication list that includes vitamin B complex, vitamins B12, E, and C, a multivitamin, zinc, magnesium, fish oil, garlic, calcium, glucosamine, chondroitin, herbal supplements, and gingko. The psychiatry team recommends switching from quetiapine to olanzapine, 15 mg/d, because Mr. E was paranoid about taking quetiapine.
We determine that Mr. E does not have medical decision-making capacity.
Because his symptoms do not improve, Mr. E is transferred to the psychiatric intensive care unit. His mental status shows little change while there. Neuropsychiatric testing shows only “cognitive deficits.” He shows signs consistent with neurologic dysfunction in terms of stimulus-bound responding and perseveration, which is compatible with the bilateral basal ganglia lesion seen on MRI. However, some of his behaviors suggest psychiatric and motivationally driven or manipulative etiology. During this testing he was difficult to evaluate and needed to be convinced to engage. At times he was illogical and at other times he showed good focus and recall. It is difficult to draw more definitive conclusions and Mr. E is discharged home with minor improvement in his symptoms. He didn’t attend follow-up appointments. During a courtesy call a few months after his admission, his wife revealed that Mr. E had died after shooting himself. It is unclear if it was an accident or suicide.
The authors’ observations
Mr. E’s diagnosis remains unclear (for a summary of his clinical course, see Table 2). Although his initial presentation was consistent with delirium, the lack of an identifiable medical cause, prolonged time course, and lack of improvement with dopamine blocking agents suggest additional diagnoses such as Wernicke-Korsakoff syndrome, rapidly progressive dementia, or a substance-induced disorder. He displayed paranoia and bizarre delusions, which would suggest a thought disorder. However, he also had a history of substance use. A few months after we saw Mr. E, “bath salt” (methylenedioxypyrovalerone) abuse gained national attention. Patients with bath salt intoxication present with confusion, paranoia, and behavioral disturbances as well as a prolonged course.8
Mr. E’s CT and MRI scans, history of alcohol use, and cirrhosis also point to Wernicke-Korsakoff syndrome as an underlying diagnosis. It is unclear whether Mr. E experienced alcohol withdrawal and IV glucose without adequate thiamine replacement during a prior surgery. However, MRI findings were unchanged from a previous study 10 years ago.
It is puzzling whether Mr. E’s AMS was a first psychotic break, a result of drug and alcohol use, rapidly progressing dementia, or another neurologic problem that we have not identified. Our tentative diagnosis was Wernicke-Korsakoff syndrome because of his history of alcohol use and imaging findings.
Although we used a multidisciplinary team approach that included psychiatry, internal medicine, neurology, neuropsychology, and an aggressive and thorough workup, we could not establish a definitive diagnosis. Unsolved cases such as this can leave patients and clinicians frustrated and may lead to unfavorable outcomes. Additional resources such as a telephone call after the first missed appointment may have been warranted.
Table 2
Mr. E’s clinical course
| Symptoms | Treatment | |
|---|---|---|
| First ED visit | Agitation Confusion Sleep disturbance Decreased appetite and speech 20-lb weight loss | Empiric antimicrobials for possible meningitis Haloperidol for agitation Quetiapine for delirium Lorazepam taper Thiamine supplementation |
| Second ED visit | Violent behavior Worsening paranoia Responding to internal stimuli Mr. E believes he has 2 wives, but the wife in the room is not the real one, which suggests possible Capgras syndrome Cognitive deficits on mental status exam | Switch from ceftriaxone to ciprofloxacin for Pseudomonas aeruginosa Switch from quetiapine to olanzapine |
| ED: emergency department | ||
Related Resources
- Kaufman DM. Clinical neurology for psychiatrists. 6th ed. Philadelphia, PA: Saunders Elsevier; 2007.
- Sidhu KS, Balon R, Ajluni V, et al. Standard EEG and the difficult-to-assess mental status. Ann Clin Psychiatry. 2009;21(2):103-108.
Drug Brand Names
- Acyclovir • Zovirax
- Ceftriaxone • Rocephin
- Ciprofloxacin • Cipro
- Haloperidol • Haldol
- Lorazepam • Ativan
- Olanzapine • Zyprexa
- Quetiapine • Seroquel
- Thiamine • Betaxin
- Vancomycin • Vancocin
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Jiang W, Gagliardi JP, Raj YP, et al. Acute psychotic disorder after gastric bypass surgery: differential diagnosis and treatment. Am J Psychiatry. 2006;163(1):15-19.
2. Harrison RA, Vu T, Hunter AJ. Wernicke’s encephalopathy in a patient with schizophrenia. J Gen Intern Med. 2006;21(12):C8-C11.
3. Sechi GP, Serra A. Wernicke’s encephalopathy: new clinical settings and recent advances in diagnosis and treatment. Lancet Neurol. 2007;6(5):442-455.
4. American Psychiatric Association. Practice guidelines for the treatment of patients with delirium. Am J Psychiatry. 1999;156(5 suppl):1-20.
5. Sharon KI, Fearing MA, Marcantonio RA. Delirium. In: Halter JB Ouslander JG, Tinetti ME, et al, eds. Hazzard’s geriatric medicine and gerontology. 6th ed. New York, NY: McGraw-Hill Medical; 2009:647–658.
6. Sadock BJ, Sadock VA. Delirium dementia, and amnestic and other cognitive disorders. In: Sadock BJ, Kaplan HI, Sadock VA. Kaplan and Sadock’s synopsis of psychiatry: behavioral sciences/clinical psychiatry. Philadelphia, PA: Lippincott Williams and Wilkins; 2007:319–372.
7. Ahmad SA, Archer HA, Rice CM, et al. Seronegative limbic encephalitis: case report, literature review and proposed treatment algorithm. Pract Neurol. 2011;11(6):355-361.
8. Ross EA, Watson M, Goldberger B. “Bath salts” intoxication. N Engl J Med. 2011;365(10):967-968.
1. Jiang W, Gagliardi JP, Raj YP, et al. Acute psychotic disorder after gastric bypass surgery: differential diagnosis and treatment. Am J Psychiatry. 2006;163(1):15-19.
2. Harrison RA, Vu T, Hunter AJ. Wernicke’s encephalopathy in a patient with schizophrenia. J Gen Intern Med. 2006;21(12):C8-C11.
3. Sechi GP, Serra A. Wernicke’s encephalopathy: new clinical settings and recent advances in diagnosis and treatment. Lancet Neurol. 2007;6(5):442-455.
4. American Psychiatric Association. Practice guidelines for the treatment of patients with delirium. Am J Psychiatry. 1999;156(5 suppl):1-20.
5. Sharon KI, Fearing MA, Marcantonio RA. Delirium. In: Halter JB Ouslander JG, Tinetti ME, et al, eds. Hazzard’s geriatric medicine and gerontology. 6th ed. New York, NY: McGraw-Hill Medical; 2009:647–658.
6. Sadock BJ, Sadock VA. Delirium dementia, and amnestic and other cognitive disorders. In: Sadock BJ, Kaplan HI, Sadock VA. Kaplan and Sadock’s synopsis of psychiatry: behavioral sciences/clinical psychiatry. Philadelphia, PA: Lippincott Williams and Wilkins; 2007:319–372.
7. Ahmad SA, Archer HA, Rice CM, et al. Seronegative limbic encephalitis: case report, literature review and proposed treatment algorithm. Pract Neurol. 2011;11(6):355-361.
8. Ross EA, Watson M, Goldberger B. “Bath salts” intoxication. N Engl J Med. 2011;365(10):967-968.
New mother dies from PE; 7 cases of bowel injury … and more
A 39-YEAR-OLD WOMAN’S SECOND CHILD WAS BORN by cesarean delivery. The mother died the next day from a pulmonary embolism.
ESTATE’S CLAIM Physicians and nurses at the hospital were negligent in failing to recognize the mother’s risk factors for pulmonary embolism, including obesity, being over age 35, and hypertension. They failed to ensure that compression boots were in place and working prior to delivery. Although orders had been given for the woman to walk within 8 hours of delivery, she did not get out of bed and walk for 24 hours after delivery.
DEFENDANTS’ DEFENSE The case was settled before trial.
VERDICT A $3.5 million Illinois settlement was reached.
Woman told “Biopsy isn’t urgent”
TWO MONTHS AFTER HER INITIAL VISIT, a 58-year-old woman returned to the gynecologist with vaginal bleeding. In March 2004, ultrasonography (US) showed slight thickening of the endometrial lining and a “pin dot” described as being a prepolyp. Vaginal bleeding was determined to be due to thinning of the vaginal wall with menopause.
The patient reported daily vaginal bleeding when she saw the gynecologist in January 2005. A new, large, rounded, solid mass within the endometrial cavity consistent with a large endometrial polyp was seen on US. The radiologist recommended hysteroscopic biopsy with excision, but the gynecologist told the patient it was not urgent.
In March 2005, hysteroscopy confirmed carcinosarcoma of the uterus. The patient underwent a hysterectomy followed by pelvic radiation and brachytherapy.
Eight months later, metastasis was found in the lungs; she died in October 2006.
ESTATE’S CLAIM The gynecologist failed to react when the patient first reported vaginal bleeding. An earlier diagnosis could have prevented her death.
PHYSICIAN’S DEFENSE The case was settled before trial.
VERDICT An $820,000 Massachusetts settlement was reached.
US report misses fetal abnormalities
A PREGNANT WOMAN UNDERWENT US. The preliminary report indicated echogenic cardiac focus and unilateral pyelectasis. Twenty-five days later, the mother underwent a level II US. A radiologist wrote that fetal anatomy was normal in both reports. The mother had two additional sonograms, with no reported abnormality.
The baby was born with aplasia and hypoplasia with both arms absent below a short humerus, an absent left leg, and a shortened right leg with a remnant foot and three small toes.
PARENTS’ CLAIM The radiologist’s US reports failed to accurately describe the fetal anatomy, depriving the parents of the chance to terminate the pregnancy.
DEFENDANTS’ DEFENSE Proper treatment was given.
VERDICT A $4.5 million Florida verdict was returned. Fault was assigned to the radiologist (85%) and the level II technologist (15%).
Forceps delivery injures mother’s pelvic floor
DURING A TRIAL OF LABOR, a 34-year-old woman experienced deep transverse arrest and lack of progress due to pelvic restriction. The ObGyn proceeded to deliver the baby vaginally using forceps, which caused pelvic floor injuries to the mother.
Several months later, she underwent corrective repair surgery for pelvic floor prolapse. She has continuing vaginal and rectal pain and dysfunction.
PATIENT’S CLAIM A cesarean delivery should have been performed as soon as pelvic restriction was found. The injuries reduce the woman’s chances of having another child.
PHYSICIAN’S DEFENSE A trial of labor was proper. The patient’s continuing fertility problems are related to chronic yeast infections and prescription birth control.
VERDICT A $1,716,469 Illinois verdict was returned, which included $484,000 to the patient’s husband for loss of consortium.
7 CASES OF INJURED BOWEL
1 Woman dies from bowel injury
DURING A SLING PROCEDURE for vaginal prolapse, a 50-year-old woman required a transfusion. The next day, she was nauseated and constipated. A day later, she went to the ED with shortness of breath and chest and abdominal pain. Her symptoms persisted for 8 days before an injury to her transverse colon was found during exploratory surgery. She suffered massive organ failure caused by sepsis and died 3 weeks after the initial surgery.
ESTATE’S CLAIM The gynecologist should have investigated why she needed a transfusion during surgery. He should have reacted earlier to her postsurgical complaints.
PHYSICIAN’S DEFENSE Bowel injury is a known risk of the procedure. The patient suffered multiple strokes after being readmitted to the hospital.
VERDICT A $2.4 million South Carolina verdict was returned.
2 Colostomy, coma after hysterectomy
DUE TO FIBROID TUMORS and pelvic pain, a 39-year-old woman’s ObGyn suggested laparoscopic-assisted vaginal hysterectomy. A third-year resident performed most of the procedure. The ObGyn’s associate covered postsurgical care.
When the patient reported increasing pain and rectal bleeding, an exploratory laparotomy was performed 3 days after surgery. Bowel and ureter injuries were repaired and a permanent colostomy was created. The patient developed septic shock with multiple organ failure, and was placed in a chemically induced coma for 3 weeks, after which she had to relearn to walk, talk, and care for herself.
PATIENT’S CLAIM The ObGyn was negligent in performing the surgery. He failed to obtain consent for the resident’s participation. The associate failed to respond to her declining postoperative condition in a timely manner.
DEFENDANTS’ DEFENSE Surgery was properly performed and postoperative care was appropriate. The bowel injury was a thermal or pressure necrosis that occurred 3 days after surgery. Two different consent forms signed by the patient included notification that a resident might assist; the resident was introduced to the patient prior to surgery. The patient’s injury claims were exaggerated; her future medical bills would be limited to colostomy supplies.
VERDICT A $1,926,069 Texas verdict was returned.
3 Were physicians qualified on robot?
A 48-YEAR-OLD WOMAN UNDERWENT robotic-assisted total hysterectomy and oophorectomy for uterine fibroids and cysts. During surgery, the physicians realized that the sigmoid colon had been perforated. A general surgeon repaired the injury with a loop ileostomy, which was successfully reversed 3 months later. The patient continues to have constipation, with occasional bleeding, pain, and burning.
PATIENT’S CLAIM The risks of robotic surgery were never fully explained to her. Failure to properly visualize her internal organs led to the injury; the extent of damage exceeded what is considered “acceptable risk” of the procedure. The physicians had little experience and training in robotic surgery.
PHYSICIANS’ DEFENSE The case was settled before trial.
VERDICT A $350,000 Massachusetts settlement was reached.
4 Adhesions limit view of bowel
A 76-YEAR-OLD WOMAN UNDERWENT surgical removal of an ovarian cyst. The ObGyn attempted a laparoscopic procedure but converted to laparotomy when extensive adhesions were encountered. The next morning, the patient discovered that her navel was discharging fecal matter. Exploratory surgery determined that the bowel had been perforated. She required additional surgery and had a long recovery.
PATIENT’S CLAIM The ObGyn was negligent in failing to diagnose and treat bowel perforation in a timely manner. An intraoperative bowel inspection should have occurred due to the likelihood of a bowel injury related to the adhesions.
PHYSICIAN’S DEFENSE Adhesions restricted inspection of every area.
VERDICT A $225,000 New York settlement was reached.
5 Skydiver’s ongoing postop pain
AFTER REPORTING DYSMENORRHEA and menometrorrhagia, a 34-year-old woman underwent dilatation and curettage, thermal endometrial ablation, and diagnostic laparoscopy. A day later, she reported increasing pain. The ObGyn’s examination revealed minimal abdominal distension, sluggish bowel sounds, and some guarding, with no rebound tenderness or acute distress. US showed a 3-cm pocket of fluid in the abdomen. Two hours later, an exam revealed a soft abdomen and normal bowel sounds. She was sent home with instructions to return the next day or, if her condition worsened, to go to the ED.
Her husband called the next day to report she was feeling better. The patient woke the following morning with massive distension, worse pain, and severe shortness of breath. At the ED, a CT scan revealed a large amount of abdominal fluid. During emergency laparotomy, an injury was found in the jejunum, necessitating a 3-inch resection.
PATIENT’S CLAIM The ObGyn was negligent in not treating her postoperative symptoms in a more proactive manner. Adhesions developed from peritonitis, leading to chronic abdominal pain. Several operations were required.
PHYSICIAN’S DEFENSE Bowel injury is a known complication of the procedure. There was no indication during surgery or at the office visit that the jejunum was injured. Adhesions were not the cause of the patient’s ongoing pain; very few adhesions were found during subsequent operations. The woman was an avid skydiver who had completed 200 jumps since her initial surgery.
VERDICT An Illinois defense verdict was returned.
6 Bowel injury at laparoscopy
WHEN THE GYNECOLOGIST recognized a bowel injury during laparoscopic salpingectomy, he called a general surgeon, who repaired three areas of bowel. The patient was released 2 days after surgery. She called the gynecologist 2 days later to report fever and vaginal bleeding. She was told to come to the office, but she cancelled when the fever subsided. The next day, she went to the ED, where sepsis was diagnosed. She was flown to another hospital for surgery. A 1-cm small-bowel perforation was found in an area of earlier repair because a suture had been disrupted. A temporary colostomy was reversed 3 months later.
PATIENT’S CLAIM The gynecologist was negligent in performing laparoscopic salpingectomy. The patient should not have been discharged because her white blood cell count and heart rate were elevated.
DEFENDANTS’ DEFENSE Performance of a laparoscopic procedure was proper. Discharge was reasonable, as there was only a potential for complications with no evident problems.
VERDICT An Missouri defense verdict was returned.
7 Was treatment of abscess delayed?
A 49-YEAR-OLD WOMAN with menorrhagia underwent cryoablation. Two weeks later, she went to the ED with pain and constipation. Following CT scans and US, she was found to have a tubo-ovarian abscess. After an enema and subsequent bowel movement, her pain improved. She was discharged with instructions to follow-up with her gynecologist. Six days later, the gynecologist prescribed triple antibiotics, analgesics, and weekly visits for the abscess. Two weeks later, she reported unbearable pain and was sent to the ED. She was found to have a microperforation of the sigmoid colon and multiple gynecologic pathologies, including myomata, right serous cystadenoma, and left tubo-ovarian complex suggestive of endometriosis. Hysterectomy and colostomy were performed; the colostomy was reversed several months later.
PATIENT’S CLAIM She should have been hospitalized when the abscess was found so that the infection could be treated properly. She alleged lack of informed consent for the cryoablation.
PHYSICIAN’S DEFENSE Hospitalization was unnecessary; the patient had initially improved, and the outcome would not have changed with intravenous antibiotics. The patient was fully informed of the risks of the procedure.
VERDICT A Pennsylvania defense verdict was returned.
These cases were selected by the editors of OBG Management from Medical Malpractice Verdicts, Settlements & Experts, with permission of the editor, Lewis Laska (www.verdictslaska.com). The information available to the editors about the cases presented here is sometimes incomplete. Moreover, the cases may or may not have merit. Nevertheless, these cases represent the types of clinical situations that typically result in litigation and are meant to illustrate nationwide variation in jury verdicts and awards.
We want to hear from you! Tell us what you think.
A 39-YEAR-OLD WOMAN’S SECOND CHILD WAS BORN by cesarean delivery. The mother died the next day from a pulmonary embolism.
ESTATE’S CLAIM Physicians and nurses at the hospital were negligent in failing to recognize the mother’s risk factors for pulmonary embolism, including obesity, being over age 35, and hypertension. They failed to ensure that compression boots were in place and working prior to delivery. Although orders had been given for the woman to walk within 8 hours of delivery, she did not get out of bed and walk for 24 hours after delivery.
DEFENDANTS’ DEFENSE The case was settled before trial.
VERDICT A $3.5 million Illinois settlement was reached.
Woman told “Biopsy isn’t urgent”
TWO MONTHS AFTER HER INITIAL VISIT, a 58-year-old woman returned to the gynecologist with vaginal bleeding. In March 2004, ultrasonography (US) showed slight thickening of the endometrial lining and a “pin dot” described as being a prepolyp. Vaginal bleeding was determined to be due to thinning of the vaginal wall with menopause.
The patient reported daily vaginal bleeding when she saw the gynecologist in January 2005. A new, large, rounded, solid mass within the endometrial cavity consistent with a large endometrial polyp was seen on US. The radiologist recommended hysteroscopic biopsy with excision, but the gynecologist told the patient it was not urgent.
In March 2005, hysteroscopy confirmed carcinosarcoma of the uterus. The patient underwent a hysterectomy followed by pelvic radiation and brachytherapy.
Eight months later, metastasis was found in the lungs; she died in October 2006.
ESTATE’S CLAIM The gynecologist failed to react when the patient first reported vaginal bleeding. An earlier diagnosis could have prevented her death.
PHYSICIAN’S DEFENSE The case was settled before trial.
VERDICT An $820,000 Massachusetts settlement was reached.
US report misses fetal abnormalities
A PREGNANT WOMAN UNDERWENT US. The preliminary report indicated echogenic cardiac focus and unilateral pyelectasis. Twenty-five days later, the mother underwent a level II US. A radiologist wrote that fetal anatomy was normal in both reports. The mother had two additional sonograms, with no reported abnormality.
The baby was born with aplasia and hypoplasia with both arms absent below a short humerus, an absent left leg, and a shortened right leg with a remnant foot and three small toes.
PARENTS’ CLAIM The radiologist’s US reports failed to accurately describe the fetal anatomy, depriving the parents of the chance to terminate the pregnancy.
DEFENDANTS’ DEFENSE Proper treatment was given.
VERDICT A $4.5 million Florida verdict was returned. Fault was assigned to the radiologist (85%) and the level II technologist (15%).
Forceps delivery injures mother’s pelvic floor
DURING A TRIAL OF LABOR, a 34-year-old woman experienced deep transverse arrest and lack of progress due to pelvic restriction. The ObGyn proceeded to deliver the baby vaginally using forceps, which caused pelvic floor injuries to the mother.
Several months later, she underwent corrective repair surgery for pelvic floor prolapse. She has continuing vaginal and rectal pain and dysfunction.
PATIENT’S CLAIM A cesarean delivery should have been performed as soon as pelvic restriction was found. The injuries reduce the woman’s chances of having another child.
PHYSICIAN’S DEFENSE A trial of labor was proper. The patient’s continuing fertility problems are related to chronic yeast infections and prescription birth control.
VERDICT A $1,716,469 Illinois verdict was returned, which included $484,000 to the patient’s husband for loss of consortium.
7 CASES OF INJURED BOWEL
1 Woman dies from bowel injury
DURING A SLING PROCEDURE for vaginal prolapse, a 50-year-old woman required a transfusion. The next day, she was nauseated and constipated. A day later, she went to the ED with shortness of breath and chest and abdominal pain. Her symptoms persisted for 8 days before an injury to her transverse colon was found during exploratory surgery. She suffered massive organ failure caused by sepsis and died 3 weeks after the initial surgery.
ESTATE’S CLAIM The gynecologist should have investigated why she needed a transfusion during surgery. He should have reacted earlier to her postsurgical complaints.
PHYSICIAN’S DEFENSE Bowel injury is a known risk of the procedure. The patient suffered multiple strokes after being readmitted to the hospital.
VERDICT A $2.4 million South Carolina verdict was returned.
2 Colostomy, coma after hysterectomy
DUE TO FIBROID TUMORS and pelvic pain, a 39-year-old woman’s ObGyn suggested laparoscopic-assisted vaginal hysterectomy. A third-year resident performed most of the procedure. The ObGyn’s associate covered postsurgical care.
When the patient reported increasing pain and rectal bleeding, an exploratory laparotomy was performed 3 days after surgery. Bowel and ureter injuries were repaired and a permanent colostomy was created. The patient developed septic shock with multiple organ failure, and was placed in a chemically induced coma for 3 weeks, after which she had to relearn to walk, talk, and care for herself.
PATIENT’S CLAIM The ObGyn was negligent in performing the surgery. He failed to obtain consent for the resident’s participation. The associate failed to respond to her declining postoperative condition in a timely manner.
DEFENDANTS’ DEFENSE Surgery was properly performed and postoperative care was appropriate. The bowel injury was a thermal or pressure necrosis that occurred 3 days after surgery. Two different consent forms signed by the patient included notification that a resident might assist; the resident was introduced to the patient prior to surgery. The patient’s injury claims were exaggerated; her future medical bills would be limited to colostomy supplies.
VERDICT A $1,926,069 Texas verdict was returned.
3 Were physicians qualified on robot?
A 48-YEAR-OLD WOMAN UNDERWENT robotic-assisted total hysterectomy and oophorectomy for uterine fibroids and cysts. During surgery, the physicians realized that the sigmoid colon had been perforated. A general surgeon repaired the injury with a loop ileostomy, which was successfully reversed 3 months later. The patient continues to have constipation, with occasional bleeding, pain, and burning.
PATIENT’S CLAIM The risks of robotic surgery were never fully explained to her. Failure to properly visualize her internal organs led to the injury; the extent of damage exceeded what is considered “acceptable risk” of the procedure. The physicians had little experience and training in robotic surgery.
PHYSICIANS’ DEFENSE The case was settled before trial.
VERDICT A $350,000 Massachusetts settlement was reached.
4 Adhesions limit view of bowel
A 76-YEAR-OLD WOMAN UNDERWENT surgical removal of an ovarian cyst. The ObGyn attempted a laparoscopic procedure but converted to laparotomy when extensive adhesions were encountered. The next morning, the patient discovered that her navel was discharging fecal matter. Exploratory surgery determined that the bowel had been perforated. She required additional surgery and had a long recovery.
PATIENT’S CLAIM The ObGyn was negligent in failing to diagnose and treat bowel perforation in a timely manner. An intraoperative bowel inspection should have occurred due to the likelihood of a bowel injury related to the adhesions.
PHYSICIAN’S DEFENSE Adhesions restricted inspection of every area.
VERDICT A $225,000 New York settlement was reached.
5 Skydiver’s ongoing postop pain
AFTER REPORTING DYSMENORRHEA and menometrorrhagia, a 34-year-old woman underwent dilatation and curettage, thermal endometrial ablation, and diagnostic laparoscopy. A day later, she reported increasing pain. The ObGyn’s examination revealed minimal abdominal distension, sluggish bowel sounds, and some guarding, with no rebound tenderness or acute distress. US showed a 3-cm pocket of fluid in the abdomen. Two hours later, an exam revealed a soft abdomen and normal bowel sounds. She was sent home with instructions to return the next day or, if her condition worsened, to go to the ED.
Her husband called the next day to report she was feeling better. The patient woke the following morning with massive distension, worse pain, and severe shortness of breath. At the ED, a CT scan revealed a large amount of abdominal fluid. During emergency laparotomy, an injury was found in the jejunum, necessitating a 3-inch resection.
PATIENT’S CLAIM The ObGyn was negligent in not treating her postoperative symptoms in a more proactive manner. Adhesions developed from peritonitis, leading to chronic abdominal pain. Several operations were required.
PHYSICIAN’S DEFENSE Bowel injury is a known complication of the procedure. There was no indication during surgery or at the office visit that the jejunum was injured. Adhesions were not the cause of the patient’s ongoing pain; very few adhesions were found during subsequent operations. The woman was an avid skydiver who had completed 200 jumps since her initial surgery.
VERDICT An Illinois defense verdict was returned.
6 Bowel injury at laparoscopy
WHEN THE GYNECOLOGIST recognized a bowel injury during laparoscopic salpingectomy, he called a general surgeon, who repaired three areas of bowel. The patient was released 2 days after surgery. She called the gynecologist 2 days later to report fever and vaginal bleeding. She was told to come to the office, but she cancelled when the fever subsided. The next day, she went to the ED, where sepsis was diagnosed. She was flown to another hospital for surgery. A 1-cm small-bowel perforation was found in an area of earlier repair because a suture had been disrupted. A temporary colostomy was reversed 3 months later.
PATIENT’S CLAIM The gynecologist was negligent in performing laparoscopic salpingectomy. The patient should not have been discharged because her white blood cell count and heart rate were elevated.
DEFENDANTS’ DEFENSE Performance of a laparoscopic procedure was proper. Discharge was reasonable, as there was only a potential for complications with no evident problems.
VERDICT An Missouri defense verdict was returned.
7 Was treatment of abscess delayed?
A 49-YEAR-OLD WOMAN with menorrhagia underwent cryoablation. Two weeks later, she went to the ED with pain and constipation. Following CT scans and US, she was found to have a tubo-ovarian abscess. After an enema and subsequent bowel movement, her pain improved. She was discharged with instructions to follow-up with her gynecologist. Six days later, the gynecologist prescribed triple antibiotics, analgesics, and weekly visits for the abscess. Two weeks later, she reported unbearable pain and was sent to the ED. She was found to have a microperforation of the sigmoid colon and multiple gynecologic pathologies, including myomata, right serous cystadenoma, and left tubo-ovarian complex suggestive of endometriosis. Hysterectomy and colostomy were performed; the colostomy was reversed several months later.
PATIENT’S CLAIM She should have been hospitalized when the abscess was found so that the infection could be treated properly. She alleged lack of informed consent for the cryoablation.
PHYSICIAN’S DEFENSE Hospitalization was unnecessary; the patient had initially improved, and the outcome would not have changed with intravenous antibiotics. The patient was fully informed of the risks of the procedure.
VERDICT A Pennsylvania defense verdict was returned.
A 39-YEAR-OLD WOMAN’S SECOND CHILD WAS BORN by cesarean delivery. The mother died the next day from a pulmonary embolism.
ESTATE’S CLAIM Physicians and nurses at the hospital were negligent in failing to recognize the mother’s risk factors for pulmonary embolism, including obesity, being over age 35, and hypertension. They failed to ensure that compression boots were in place and working prior to delivery. Although orders had been given for the woman to walk within 8 hours of delivery, she did not get out of bed and walk for 24 hours after delivery.
DEFENDANTS’ DEFENSE The case was settled before trial.
VERDICT A $3.5 million Illinois settlement was reached.
Woman told “Biopsy isn’t urgent”
TWO MONTHS AFTER HER INITIAL VISIT, a 58-year-old woman returned to the gynecologist with vaginal bleeding. In March 2004, ultrasonography (US) showed slight thickening of the endometrial lining and a “pin dot” described as being a prepolyp. Vaginal bleeding was determined to be due to thinning of the vaginal wall with menopause.
The patient reported daily vaginal bleeding when she saw the gynecologist in January 2005. A new, large, rounded, solid mass within the endometrial cavity consistent with a large endometrial polyp was seen on US. The radiologist recommended hysteroscopic biopsy with excision, but the gynecologist told the patient it was not urgent.
In March 2005, hysteroscopy confirmed carcinosarcoma of the uterus. The patient underwent a hysterectomy followed by pelvic radiation and brachytherapy.
Eight months later, metastasis was found in the lungs; she died in October 2006.
ESTATE’S CLAIM The gynecologist failed to react when the patient first reported vaginal bleeding. An earlier diagnosis could have prevented her death.
PHYSICIAN’S DEFENSE The case was settled before trial.
VERDICT An $820,000 Massachusetts settlement was reached.
US report misses fetal abnormalities
A PREGNANT WOMAN UNDERWENT US. The preliminary report indicated echogenic cardiac focus and unilateral pyelectasis. Twenty-five days later, the mother underwent a level II US. A radiologist wrote that fetal anatomy was normal in both reports. The mother had two additional sonograms, with no reported abnormality.
The baby was born with aplasia and hypoplasia with both arms absent below a short humerus, an absent left leg, and a shortened right leg with a remnant foot and three small toes.
PARENTS’ CLAIM The radiologist’s US reports failed to accurately describe the fetal anatomy, depriving the parents of the chance to terminate the pregnancy.
DEFENDANTS’ DEFENSE Proper treatment was given.
VERDICT A $4.5 million Florida verdict was returned. Fault was assigned to the radiologist (85%) and the level II technologist (15%).
Forceps delivery injures mother’s pelvic floor
DURING A TRIAL OF LABOR, a 34-year-old woman experienced deep transverse arrest and lack of progress due to pelvic restriction. The ObGyn proceeded to deliver the baby vaginally using forceps, which caused pelvic floor injuries to the mother.
Several months later, she underwent corrective repair surgery for pelvic floor prolapse. She has continuing vaginal and rectal pain and dysfunction.
PATIENT’S CLAIM A cesarean delivery should have been performed as soon as pelvic restriction was found. The injuries reduce the woman’s chances of having another child.
PHYSICIAN’S DEFENSE A trial of labor was proper. The patient’s continuing fertility problems are related to chronic yeast infections and prescription birth control.
VERDICT A $1,716,469 Illinois verdict was returned, which included $484,000 to the patient’s husband for loss of consortium.
7 CASES OF INJURED BOWEL
1 Woman dies from bowel injury
DURING A SLING PROCEDURE for vaginal prolapse, a 50-year-old woman required a transfusion. The next day, she was nauseated and constipated. A day later, she went to the ED with shortness of breath and chest and abdominal pain. Her symptoms persisted for 8 days before an injury to her transverse colon was found during exploratory surgery. She suffered massive organ failure caused by sepsis and died 3 weeks after the initial surgery.
ESTATE’S CLAIM The gynecologist should have investigated why she needed a transfusion during surgery. He should have reacted earlier to her postsurgical complaints.
PHYSICIAN’S DEFENSE Bowel injury is a known risk of the procedure. The patient suffered multiple strokes after being readmitted to the hospital.
VERDICT A $2.4 million South Carolina verdict was returned.
2 Colostomy, coma after hysterectomy
DUE TO FIBROID TUMORS and pelvic pain, a 39-year-old woman’s ObGyn suggested laparoscopic-assisted vaginal hysterectomy. A third-year resident performed most of the procedure. The ObGyn’s associate covered postsurgical care.
When the patient reported increasing pain and rectal bleeding, an exploratory laparotomy was performed 3 days after surgery. Bowel and ureter injuries were repaired and a permanent colostomy was created. The patient developed septic shock with multiple organ failure, and was placed in a chemically induced coma for 3 weeks, after which she had to relearn to walk, talk, and care for herself.
PATIENT’S CLAIM The ObGyn was negligent in performing the surgery. He failed to obtain consent for the resident’s participation. The associate failed to respond to her declining postoperative condition in a timely manner.
DEFENDANTS’ DEFENSE Surgery was properly performed and postoperative care was appropriate. The bowel injury was a thermal or pressure necrosis that occurred 3 days after surgery. Two different consent forms signed by the patient included notification that a resident might assist; the resident was introduced to the patient prior to surgery. The patient’s injury claims were exaggerated; her future medical bills would be limited to colostomy supplies.
VERDICT A $1,926,069 Texas verdict was returned.
3 Were physicians qualified on robot?
A 48-YEAR-OLD WOMAN UNDERWENT robotic-assisted total hysterectomy and oophorectomy for uterine fibroids and cysts. During surgery, the physicians realized that the sigmoid colon had been perforated. A general surgeon repaired the injury with a loop ileostomy, which was successfully reversed 3 months later. The patient continues to have constipation, with occasional bleeding, pain, and burning.
PATIENT’S CLAIM The risks of robotic surgery were never fully explained to her. Failure to properly visualize her internal organs led to the injury; the extent of damage exceeded what is considered “acceptable risk” of the procedure. The physicians had little experience and training in robotic surgery.
PHYSICIANS’ DEFENSE The case was settled before trial.
VERDICT A $350,000 Massachusetts settlement was reached.
4 Adhesions limit view of bowel
A 76-YEAR-OLD WOMAN UNDERWENT surgical removal of an ovarian cyst. The ObGyn attempted a laparoscopic procedure but converted to laparotomy when extensive adhesions were encountered. The next morning, the patient discovered that her navel was discharging fecal matter. Exploratory surgery determined that the bowel had been perforated. She required additional surgery and had a long recovery.
PATIENT’S CLAIM The ObGyn was negligent in failing to diagnose and treat bowel perforation in a timely manner. An intraoperative bowel inspection should have occurred due to the likelihood of a bowel injury related to the adhesions.
PHYSICIAN’S DEFENSE Adhesions restricted inspection of every area.
VERDICT A $225,000 New York settlement was reached.
5 Skydiver’s ongoing postop pain
AFTER REPORTING DYSMENORRHEA and menometrorrhagia, a 34-year-old woman underwent dilatation and curettage, thermal endometrial ablation, and diagnostic laparoscopy. A day later, she reported increasing pain. The ObGyn’s examination revealed minimal abdominal distension, sluggish bowel sounds, and some guarding, with no rebound tenderness or acute distress. US showed a 3-cm pocket of fluid in the abdomen. Two hours later, an exam revealed a soft abdomen and normal bowel sounds. She was sent home with instructions to return the next day or, if her condition worsened, to go to the ED.
Her husband called the next day to report she was feeling better. The patient woke the following morning with massive distension, worse pain, and severe shortness of breath. At the ED, a CT scan revealed a large amount of abdominal fluid. During emergency laparotomy, an injury was found in the jejunum, necessitating a 3-inch resection.
PATIENT’S CLAIM The ObGyn was negligent in not treating her postoperative symptoms in a more proactive manner. Adhesions developed from peritonitis, leading to chronic abdominal pain. Several operations were required.
PHYSICIAN’S DEFENSE Bowel injury is a known complication of the procedure. There was no indication during surgery or at the office visit that the jejunum was injured. Adhesions were not the cause of the patient’s ongoing pain; very few adhesions were found during subsequent operations. The woman was an avid skydiver who had completed 200 jumps since her initial surgery.
VERDICT An Illinois defense verdict was returned.
6 Bowel injury at laparoscopy
WHEN THE GYNECOLOGIST recognized a bowel injury during laparoscopic salpingectomy, he called a general surgeon, who repaired three areas of bowel. The patient was released 2 days after surgery. She called the gynecologist 2 days later to report fever and vaginal bleeding. She was told to come to the office, but she cancelled when the fever subsided. The next day, she went to the ED, where sepsis was diagnosed. She was flown to another hospital for surgery. A 1-cm small-bowel perforation was found in an area of earlier repair because a suture had been disrupted. A temporary colostomy was reversed 3 months later.
PATIENT’S CLAIM The gynecologist was negligent in performing laparoscopic salpingectomy. The patient should not have been discharged because her white blood cell count and heart rate were elevated.
DEFENDANTS’ DEFENSE Performance of a laparoscopic procedure was proper. Discharge was reasonable, as there was only a potential for complications with no evident problems.
VERDICT An Missouri defense verdict was returned.
7 Was treatment of abscess delayed?
A 49-YEAR-OLD WOMAN with menorrhagia underwent cryoablation. Two weeks later, she went to the ED with pain and constipation. Following CT scans and US, she was found to have a tubo-ovarian abscess. After an enema and subsequent bowel movement, her pain improved. She was discharged with instructions to follow-up with her gynecologist. Six days later, the gynecologist prescribed triple antibiotics, analgesics, and weekly visits for the abscess. Two weeks later, she reported unbearable pain and was sent to the ED. She was found to have a microperforation of the sigmoid colon and multiple gynecologic pathologies, including myomata, right serous cystadenoma, and left tubo-ovarian complex suggestive of endometriosis. Hysterectomy and colostomy were performed; the colostomy was reversed several months later.
PATIENT’S CLAIM She should have been hospitalized when the abscess was found so that the infection could be treated properly. She alleged lack of informed consent for the cryoablation.
PHYSICIAN’S DEFENSE Hospitalization was unnecessary; the patient had initially improved, and the outcome would not have changed with intravenous antibiotics. The patient was fully informed of the risks of the procedure.
VERDICT A Pennsylvania defense verdict was returned.
These cases were selected by the editors of OBG Management from Medical Malpractice Verdicts, Settlements & Experts, with permission of the editor, Lewis Laska (www.verdictslaska.com). The information available to the editors about the cases presented here is sometimes incomplete. Moreover, the cases may or may not have merit. Nevertheless, these cases represent the types of clinical situations that typically result in litigation and are meant to illustrate nationwide variation in jury verdicts and awards.
We want to hear from you! Tell us what you think.
These cases were selected by the editors of OBG Management from Medical Malpractice Verdicts, Settlements & Experts, with permission of the editor, Lewis Laska (www.verdictslaska.com). The information available to the editors about the cases presented here is sometimes incomplete. Moreover, the cases may or may not have merit. Nevertheless, these cases represent the types of clinical situations that typically result in litigation and are meant to illustrate nationwide variation in jury verdicts and awards.
We want to hear from you! Tell us what you think.
Gallbladder surgery uncovers something more...Diagnosis minus treatment equals catastrophe...more
Gallbladder surgery uncovers something more
ABDOMINAL PAIN prompted a 46-year-old woman to seek treatment at a local medical center, where she had minor therapy. She returned to the hospital repeatedly over the next 3 years and received various treatments for abdominal pain, culminating in the removal of her gallbladder.
During the procedure, the surgeon found an ovarian tumor that turned out to be stage III cancer. The patient underwent oophorectomy and several courses of chemotherapy.
PLAINTIFF’S CLAIM The cancer should have been diagnosed at any of the patient’s previous examinations.
THE DEFENSE The patient’s symptoms were vague; ovarian cancer is often diagnosed at a late stage.
VERDICT $160,000 New York settlement.
COMMENT It never ceases to amaze that we’re held to such high (irrational?) standards whenever cancer is diagnosed. Although pertinent details of this case—such as the size of the tumor and frequency of pelvic exams—aren’t provided, it goes to show you that lawyers will do what lawyers do.
Diagnosis minus treatment equals catastrophe
A SWOLLEN, PAINFUL LEFT KNEE led a 65-year-old man to go to the emergency department (ED). The physician who examined his knee prescribed acetaminophen and hydrocodone and naproxen and sent the patient home with instructions to apply ice and heat.
The patient went back to the ED 2 days later because the knee was still swollen and painful. He was told to keep taking the prescribed medications and to follow up with a doctor at a local practice, who examined the patient later that day. The doctor aspirated brown, pus-filled material from the knee and diagnosed sepsis in the knee joint. He told the patient to drive to his family physician’s office, about 70 miles away, for treatment. The patient was carried back to his car and made the drive slowly.
By the time he arrived at his doctor’s office, the patient was in shock and kidney failure and breathing with difficulty. He was put on a ventilator and given antibiotics. He died several days later from septic shock and multiple organ failure.
PLAINTIFF’S CLAIM If the patient had been given antibiotics during his first or second examination, he would have lived.
THE DEFENSE No information about the defense is available.
VERDICT $10.9 million South Carolina verdict.
COMMENT It’s horrible enough that this patient wasn’t diagnosed promptly, but unfathomable that he was sent on his way without treatment!
Circumcision proceeds without consent
AFTER THE BIRTH OF A HEALTHY BABY BOY, a nurse presented the baby’s mother with a consent form for circumcision, which she didn’t sign. Before the birth, the parents had told the child’s pediatrician—who had also been the pediatrician for the mother’s 2 brothers and her oldest son—that they didn’t want their baby circumcised if it was a boy. Despite a lack of consent, the pediatrician circumcised the infant, without incident, the day after his birth. The parents were outraged.
PLAINTIFF’S CLAIM Because the pediatrician had cared for other male members of the family, he should have been aware of the family’s wishes regarding circumcision. The Gomco clamp method used to circumcise the baby caused pain throughout the 25-minute procedure, and the child suffered pain for 2 weeks while his penis healed. The baby, who had been calm before the surgery, became fussy afterwards and remained so for a year. He has a greater risk of developing some health problems because of the circumcision.
THE DEFENSE The circumcision was performed because the hospital staff erred in not following the hospital’s protocol. The procedure was done properly and without complications; the baby suffered no injuries from it. Remaining uncircumcised has no benefit; because circumcision lowers the risk of urinary tract and foreskin infections, as well as other illnesses, the child would be healthier than uncircumcised boys.
VERDICT Indiana defense verdict for the pediatrician. (The hospital reached a confidential settlement with the parents before trial.)
COMMENT It still astounds when wrong side surgeries occur—and how about this example of a circumcision without consent?! This is why checklists are so important. Obviously, appropriate informed consent should precede any procedure.
A drug adverse effect—that wasn’t
A 68-YEAR-OLD WOMAN went to her physician complaining of gastrointestinal discomfort. The doctor surmised that medication prescribed for hypertension was causing the discomfort and changed the medication. He recommended a follow-up visit in 2 weeks.
Three days later, the patient returned to the clinic complaining of abdominal pain. A physician assistant made the same diagnosis as the physician.
The patient went to the hospital 4 days later because the pain had increased. She was found to have a ruptured appendix and underwent an appendectomy. After surgery, the patient experienced residual pain.
PLAINTIFF’S CLAIM The physician and physician assistant were negligent in failing to diagnose appendicitis promptly. The case proceeded to trial against the physician assistant and the clinic.
THE DEFENSE The patient was properly evaluated and didn’t have symptoms suggesting appendicitis. Diagnostic tests weren’t necessary because the second visit was a follow-up examination.
VERDICT $150,000 New York verdict.
COMMENT Thoroughly documenting the history and physical examination is key to avoiding malpractice claims.
Antibiotics prescribed by phone can’t substitute for office visit
THREE DAYS OF FATIGUE AND A 103°F FEVER in a 42-year-old man prompted his wife to call his primary care physician. She discussed the symptoms with a nurse, who told her the doctor didn’t have an opening to see her husband. Instead, the physician called in a prescription for antibiotics because the symptoms resembled ones the patient had had about 8 months earlier that cleared up with antibiotics.
The patient felt well enough to pick up the antibiotics and the couple’s 2 children from preschool. When he got home, he took the antibiotics and went to bed. His wife found him dead that evening. The cause was determined to be cardiac arrest from myocarditis.
PLAINTIFF’S CLAIM Based on the reported symptoms, the doctor should have seen the patient immediately and referred him to an emergency department, where myocarditis would have been diagnosed and lifesaving treatment could have been started.
THE DEFENSE The patient’s wife didn’t properly describe all the symptoms to the nurse when she called. If she had, the doctor’s office would have scheduled an immediate appointment. In any case, myocarditis is difficult to diagnose; a pathologist’s findings indicated that the patient had focal myocarditis only in the right ventricle, which would have caused no symptoms detectable by a physical examination or electrocardiogram.
VERDICT $220,255 New Jersey judgment. The jury returned a $1 million verdict on a finding that the doctor was 20% responsible for the damages and the patient’s pre-existing condition was 80% responsible.
COMMENT I’m increasingly alarmed by the trend to find clinicians partially responsible for damages. A 20% share of liability added up to more than $200,000 in this case.
Gallbladder surgery uncovers something more
ABDOMINAL PAIN prompted a 46-year-old woman to seek treatment at a local medical center, where she had minor therapy. She returned to the hospital repeatedly over the next 3 years and received various treatments for abdominal pain, culminating in the removal of her gallbladder.
During the procedure, the surgeon found an ovarian tumor that turned out to be stage III cancer. The patient underwent oophorectomy and several courses of chemotherapy.
PLAINTIFF’S CLAIM The cancer should have been diagnosed at any of the patient’s previous examinations.
THE DEFENSE The patient’s symptoms were vague; ovarian cancer is often diagnosed at a late stage.
VERDICT $160,000 New York settlement.
COMMENT It never ceases to amaze that we’re held to such high (irrational?) standards whenever cancer is diagnosed. Although pertinent details of this case—such as the size of the tumor and frequency of pelvic exams—aren’t provided, it goes to show you that lawyers will do what lawyers do.
Diagnosis minus treatment equals catastrophe
A SWOLLEN, PAINFUL LEFT KNEE led a 65-year-old man to go to the emergency department (ED). The physician who examined his knee prescribed acetaminophen and hydrocodone and naproxen and sent the patient home with instructions to apply ice and heat.
The patient went back to the ED 2 days later because the knee was still swollen and painful. He was told to keep taking the prescribed medications and to follow up with a doctor at a local practice, who examined the patient later that day. The doctor aspirated brown, pus-filled material from the knee and diagnosed sepsis in the knee joint. He told the patient to drive to his family physician’s office, about 70 miles away, for treatment. The patient was carried back to his car and made the drive slowly.
By the time he arrived at his doctor’s office, the patient was in shock and kidney failure and breathing with difficulty. He was put on a ventilator and given antibiotics. He died several days later from septic shock and multiple organ failure.
PLAINTIFF’S CLAIM If the patient had been given antibiotics during his first or second examination, he would have lived.
THE DEFENSE No information about the defense is available.
VERDICT $10.9 million South Carolina verdict.
COMMENT It’s horrible enough that this patient wasn’t diagnosed promptly, but unfathomable that he was sent on his way without treatment!
Circumcision proceeds without consent
AFTER THE BIRTH OF A HEALTHY BABY BOY, a nurse presented the baby’s mother with a consent form for circumcision, which she didn’t sign. Before the birth, the parents had told the child’s pediatrician—who had also been the pediatrician for the mother’s 2 brothers and her oldest son—that they didn’t want their baby circumcised if it was a boy. Despite a lack of consent, the pediatrician circumcised the infant, without incident, the day after his birth. The parents were outraged.
PLAINTIFF’S CLAIM Because the pediatrician had cared for other male members of the family, he should have been aware of the family’s wishes regarding circumcision. The Gomco clamp method used to circumcise the baby caused pain throughout the 25-minute procedure, and the child suffered pain for 2 weeks while his penis healed. The baby, who had been calm before the surgery, became fussy afterwards and remained so for a year. He has a greater risk of developing some health problems because of the circumcision.
THE DEFENSE The circumcision was performed because the hospital staff erred in not following the hospital’s protocol. The procedure was done properly and without complications; the baby suffered no injuries from it. Remaining uncircumcised has no benefit; because circumcision lowers the risk of urinary tract and foreskin infections, as well as other illnesses, the child would be healthier than uncircumcised boys.
VERDICT Indiana defense verdict for the pediatrician. (The hospital reached a confidential settlement with the parents before trial.)
COMMENT It still astounds when wrong side surgeries occur—and how about this example of a circumcision without consent?! This is why checklists are so important. Obviously, appropriate informed consent should precede any procedure.
A drug adverse effect—that wasn’t
A 68-YEAR-OLD WOMAN went to her physician complaining of gastrointestinal discomfort. The doctor surmised that medication prescribed for hypertension was causing the discomfort and changed the medication. He recommended a follow-up visit in 2 weeks.
Three days later, the patient returned to the clinic complaining of abdominal pain. A physician assistant made the same diagnosis as the physician.
The patient went to the hospital 4 days later because the pain had increased. She was found to have a ruptured appendix and underwent an appendectomy. After surgery, the patient experienced residual pain.
PLAINTIFF’S CLAIM The physician and physician assistant were negligent in failing to diagnose appendicitis promptly. The case proceeded to trial against the physician assistant and the clinic.
THE DEFENSE The patient was properly evaluated and didn’t have symptoms suggesting appendicitis. Diagnostic tests weren’t necessary because the second visit was a follow-up examination.
VERDICT $150,000 New York verdict.
COMMENT Thoroughly documenting the history and physical examination is key to avoiding malpractice claims.
Antibiotics prescribed by phone can’t substitute for office visit
THREE DAYS OF FATIGUE AND A 103°F FEVER in a 42-year-old man prompted his wife to call his primary care physician. She discussed the symptoms with a nurse, who told her the doctor didn’t have an opening to see her husband. Instead, the physician called in a prescription for antibiotics because the symptoms resembled ones the patient had had about 8 months earlier that cleared up with antibiotics.
The patient felt well enough to pick up the antibiotics and the couple’s 2 children from preschool. When he got home, he took the antibiotics and went to bed. His wife found him dead that evening. The cause was determined to be cardiac arrest from myocarditis.
PLAINTIFF’S CLAIM Based on the reported symptoms, the doctor should have seen the patient immediately and referred him to an emergency department, where myocarditis would have been diagnosed and lifesaving treatment could have been started.
THE DEFENSE The patient’s wife didn’t properly describe all the symptoms to the nurse when she called. If she had, the doctor’s office would have scheduled an immediate appointment. In any case, myocarditis is difficult to diagnose; a pathologist’s findings indicated that the patient had focal myocarditis only in the right ventricle, which would have caused no symptoms detectable by a physical examination or electrocardiogram.
VERDICT $220,255 New Jersey judgment. The jury returned a $1 million verdict on a finding that the doctor was 20% responsible for the damages and the patient’s pre-existing condition was 80% responsible.
COMMENT I’m increasingly alarmed by the trend to find clinicians partially responsible for damages. A 20% share of liability added up to more than $200,000 in this case.
Gallbladder surgery uncovers something more
ABDOMINAL PAIN prompted a 46-year-old woman to seek treatment at a local medical center, where she had minor therapy. She returned to the hospital repeatedly over the next 3 years and received various treatments for abdominal pain, culminating in the removal of her gallbladder.
During the procedure, the surgeon found an ovarian tumor that turned out to be stage III cancer. The patient underwent oophorectomy and several courses of chemotherapy.
PLAINTIFF’S CLAIM The cancer should have been diagnosed at any of the patient’s previous examinations.
THE DEFENSE The patient’s symptoms were vague; ovarian cancer is often diagnosed at a late stage.
VERDICT $160,000 New York settlement.
COMMENT It never ceases to amaze that we’re held to such high (irrational?) standards whenever cancer is diagnosed. Although pertinent details of this case—such as the size of the tumor and frequency of pelvic exams—aren’t provided, it goes to show you that lawyers will do what lawyers do.
Diagnosis minus treatment equals catastrophe
A SWOLLEN, PAINFUL LEFT KNEE led a 65-year-old man to go to the emergency department (ED). The physician who examined his knee prescribed acetaminophen and hydrocodone and naproxen and sent the patient home with instructions to apply ice and heat.
The patient went back to the ED 2 days later because the knee was still swollen and painful. He was told to keep taking the prescribed medications and to follow up with a doctor at a local practice, who examined the patient later that day. The doctor aspirated brown, pus-filled material from the knee and diagnosed sepsis in the knee joint. He told the patient to drive to his family physician’s office, about 70 miles away, for treatment. The patient was carried back to his car and made the drive slowly.
By the time he arrived at his doctor’s office, the patient was in shock and kidney failure and breathing with difficulty. He was put on a ventilator and given antibiotics. He died several days later from septic shock and multiple organ failure.
PLAINTIFF’S CLAIM If the patient had been given antibiotics during his first or second examination, he would have lived.
THE DEFENSE No information about the defense is available.
VERDICT $10.9 million South Carolina verdict.
COMMENT It’s horrible enough that this patient wasn’t diagnosed promptly, but unfathomable that he was sent on his way without treatment!
Circumcision proceeds without consent
AFTER THE BIRTH OF A HEALTHY BABY BOY, a nurse presented the baby’s mother with a consent form for circumcision, which she didn’t sign. Before the birth, the parents had told the child’s pediatrician—who had also been the pediatrician for the mother’s 2 brothers and her oldest son—that they didn’t want their baby circumcised if it was a boy. Despite a lack of consent, the pediatrician circumcised the infant, without incident, the day after his birth. The parents were outraged.
PLAINTIFF’S CLAIM Because the pediatrician had cared for other male members of the family, he should have been aware of the family’s wishes regarding circumcision. The Gomco clamp method used to circumcise the baby caused pain throughout the 25-minute procedure, and the child suffered pain for 2 weeks while his penis healed. The baby, who had been calm before the surgery, became fussy afterwards and remained so for a year. He has a greater risk of developing some health problems because of the circumcision.
THE DEFENSE The circumcision was performed because the hospital staff erred in not following the hospital’s protocol. The procedure was done properly and without complications; the baby suffered no injuries from it. Remaining uncircumcised has no benefit; because circumcision lowers the risk of urinary tract and foreskin infections, as well as other illnesses, the child would be healthier than uncircumcised boys.
VERDICT Indiana defense verdict for the pediatrician. (The hospital reached a confidential settlement with the parents before trial.)
COMMENT It still astounds when wrong side surgeries occur—and how about this example of a circumcision without consent?! This is why checklists are so important. Obviously, appropriate informed consent should precede any procedure.
A drug adverse effect—that wasn’t
A 68-YEAR-OLD WOMAN went to her physician complaining of gastrointestinal discomfort. The doctor surmised that medication prescribed for hypertension was causing the discomfort and changed the medication. He recommended a follow-up visit in 2 weeks.
Three days later, the patient returned to the clinic complaining of abdominal pain. A physician assistant made the same diagnosis as the physician.
The patient went to the hospital 4 days later because the pain had increased. She was found to have a ruptured appendix and underwent an appendectomy. After surgery, the patient experienced residual pain.
PLAINTIFF’S CLAIM The physician and physician assistant were negligent in failing to diagnose appendicitis promptly. The case proceeded to trial against the physician assistant and the clinic.
THE DEFENSE The patient was properly evaluated and didn’t have symptoms suggesting appendicitis. Diagnostic tests weren’t necessary because the second visit was a follow-up examination.
VERDICT $150,000 New York verdict.
COMMENT Thoroughly documenting the history and physical examination is key to avoiding malpractice claims.
Antibiotics prescribed by phone can’t substitute for office visit
THREE DAYS OF FATIGUE AND A 103°F FEVER in a 42-year-old man prompted his wife to call his primary care physician. She discussed the symptoms with a nurse, who told her the doctor didn’t have an opening to see her husband. Instead, the physician called in a prescription for antibiotics because the symptoms resembled ones the patient had had about 8 months earlier that cleared up with antibiotics.
The patient felt well enough to pick up the antibiotics and the couple’s 2 children from preschool. When he got home, he took the antibiotics and went to bed. His wife found him dead that evening. The cause was determined to be cardiac arrest from myocarditis.
PLAINTIFF’S CLAIM Based on the reported symptoms, the doctor should have seen the patient immediately and referred him to an emergency department, where myocarditis would have been diagnosed and lifesaving treatment could have been started.
THE DEFENSE The patient’s wife didn’t properly describe all the symptoms to the nurse when she called. If she had, the doctor’s office would have scheduled an immediate appointment. In any case, myocarditis is difficult to diagnose; a pathologist’s findings indicated that the patient had focal myocarditis only in the right ventricle, which would have caused no symptoms detectable by a physical examination or electrocardiogram.
VERDICT $220,255 New Jersey judgment. The jury returned a $1 million verdict on a finding that the doctor was 20% responsible for the damages and the patient’s pre-existing condition was 80% responsible.
COMMENT I’m increasingly alarmed by the trend to find clinicians partially responsible for damages. A 20% share of liability added up to more than $200,000 in this case.
The shrinking case for saw palmetto
Advise men with benign prostatic hyperplasia (BPH) not to take saw palmetto for urinary symptoms. Explain that it has not been found to alleviate symptoms, even at triple the standard dose.1
A: Based on evidence from a high-quality randomized controlled trial (RCT)1 and a 2009 meta-analysis.2
1. Barry MJ, Meleth S, Lee JY, et al. Effect of increasing doses of saw palmetto extract on lower urinary tract symptoms: a randomized trial. JAMA. 2011;306:1344-1351.
ILLUSTRATIVE CASE
A 66-year-old man comes to your office complaining of urinary frequency and straining to begin urination. He was recently diagnosed with BPh by a urologist, but is hesitant to begin taking a prescription drug. The patient, who is on a fixed income, asks you if saw palmetto extract might relieve his urinary symptoms. What should you tell him?
Roughly 40% of American men older than 60 years and nearly 90% of men older than 80 suffer from BPH and the troublesome lower urinary tract symptoms (LUTS) that it causes.3 Established medical and surgical options, as well as over-the-counter (OTC) plant-based products, are used for symptom relief. The OTC remedy most commonly used for BPH is Serenoa repens, derived from the saw palmetto dwarf palm tree. In a 2007 survey, 1.6 million US adults reported using saw palmetto extract, often as a treatment for BPH, in the 30 days prior to the survey.4
Until now, more questions than answers
As a family physician, you undoubtedly have many patients who are taking or considering taking saw palmetto for relief of BPH symptoms. The significant adverse effects of alpha-blockers and 5-alpha-reductase inhibitors, which are typically prescribed for LUTS—including decreased libido and dizziness—may help account for their interest in this alternative treatment.5,6
Until recently, evidence of saw palmetto’s efficacy has been limited and conflicting, despite widespread use of the extract. That has left many of us wondering whether we should recommend that men with BPH try saw palmetto despite the limited evidence; whether it is effective for some, but not all, BPH symptoms; and whether an increase in dose would increase its efficacy.
A 2002 Cochrane meta-analysis of 21 trials of saw palmetto extract for LUTS reported reduced nocturia, improved self-reported symptoms, and increased peak uroflow compared with placebo, without significant adverse effects.7 An updated Cochrane review published in 2009 included several more rigorous trials—and had very different results: This meta-analysis, which was based on 30 trials, found a reduction in nocturia, but failed to show improvement in other self-reported symptoms or peak uroflow.2
The largest trial included in the 2009 review was the Saw Palmetto Treatment for Enlarged Prostates (STEP) study,8 a one-year study with 225 participants. Its findings: no improvement in the treatment group compared with the placebo group in symptom scores or any secondary endpoints, and no important toxicity.8 Of note, the STEP study and most trials included in the 2009 Cochrane review used the standard saw palmetto extract dose of 160 mg twice daily.1,2
STUDY SUMMARY: Saw palmetto is ineffective, even at triple the dose
Barry et al conducted a 72-week double-blind, multicenter placebo-controlled trial to assess the effect of double (640 mg/d) and triple (960 mg/d) the standard dose of saw palmetto extract on BPH symptoms.1 The study included 369 men with moderate LUTS who had not recently received treatment for BPH. Exclusion criteria included a history of invasive BPH treatment, recent treatment with either an alpha-blocker or a 5-alpha-reductase inhibitor; recent phytotherapy, including saw palmetto; and a history of prostate or bladder cancer. Participants were randomized to receive either saw palmetto extract or an identical-looking placebo gel cap. Doses started at 320 mg/d and were increased to 640 mg/d at 24 weeks and 960 mg/d at 48 weeks.
The primary outcome was the change in the American Urological Association Symptom Index (AUASI) score from baseline to 72 weeks. AUASI, a scale of 0 to 35 in which higher numbers represent increased symptoms, is the same scoring tool used in both the Cochrane review and the STEP trial. Secondary measures included other symptom scales, peak uroflow, and poststudy satisfaction. The treatment and placebo groups had statistically identical baseline characteristics, and the sample size was large enough to detect clinically significant differences.
The AUASI score decreased by a mean of 2.20 points (95% confidence interval [CI], -3.04 to -0.36) in the group that received saw palmetto and by 2.99 points (95% CI, -3.81 to -2.17) in the placebo group—a mean difference of 0.79 in favor of the placebo group (P=.91). The proportion of participants achieving a 3-point reduction in AUASI score was statistically similar between the 2 groups (P=0.66). There was no significant dose response difference between the 2 groups, and saw palmetto proved to be no better than placebo for any of the secondary outcomes.
Subgroup analysis did not reveal any results that differed from the main outcomes. The only adverse events that were significantly different between the 2 groups related to physical injury or trauma, which were unlikely to be due to the intervention.1
By using the same symptom scoring system (AUASI) as many studies in the previous Cochrane reviews, Barry et al were able to compare their findings with those of other high-quality studies with similar methodologies and outcome measures. Despite using an even higher dose of the extract, the results of this trial are remarkably consistent with previous conclusions: Saw palmetto is not an effective treatment for symptoms associated with BPH. Moreover, this trial had a broad base of participants similar to the population in a primary care practice, including patients who would typically choose a natural remedy for LUTS.1
WHAT’S NEW: We now have answers to queries about saw palmetto
This trial is the first to compare higher doses of saw palmetto with placebo to assess a dosage threshold for effectiveness. While the study found no evidence of saw palmetto toxicity even at these higher doses, the extract did not outperform placebo for any measured outcome.1
This high-quality study confirmed the recent series of rigorous studies with negative outcomes by showing that the use of a standard dosage was not a study limitation and that saw palmetto extract is not effective for treating LUTS at any dosage. This trial should substantially affect future guideline recommendations that were limited by methodological concerns in the past.9-11
CAVEATS: In theory, individual preparations could work differently
It is not possible to be absolutely certain that these findings apply to all saw palmetto extract preparations, given the unknown active ingredients and unknown mechanism of action. However, the researchers used a high-quality preparation (a proprietary lipidic ethanolic extract) of saw palmetto at higher doses than the STEP trial and came to a similar conclusion, making it highly unlikely that another preparation would perform differently.
CHALLENGES TO IMPLEMENTATION: There are none
We see no challenges to implementation of this recommendation.
Acknowledgement
The PURLs Surveillance System is supported in part by Grant Number UL1RR024999 from the National Center for Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
1. Barry MJ, Meleth S, Lee JY, et al. Effect of increasing doses of saw palmetto extract on lower urinary tract symptoms: a randomized trial. JAMA. 2011;306:1344-1351.
2. Tacklind J, MacDonald R, Rutks I, et al. Serenoa repens for benign prostatic hyperplasia. Cochrane Database Syst Rev. 2009;(2):CD001423.-
3. Roehrborn CG, McConnell JD. Etiology, Pathophysiology, Epidemiology, and Natural History of Benign Prostatic Hyperplasia. 8th ed. Philadelphia, Pa: Campbell’s Urology; 2002.
4. Barnes PM, Bloom B, Nahin RL. Complementary and alternative medicine use among adults and children: United States, 2007. Natl Health Stat Report. 2008;(12):1-23.
5. Traish AM, Hassani J, Guay AT, et al. Adverse side effects of 5-alpha-reductase inhibitors therapy: persistent diminished libido and erectile dysfunction and depression in a subset of patients. J Sex Med. 2011;8:872-884.
6. Clifford GM, Farmer RD. Medical therapy for benign prostatic hyperplasia: a review of the literature. Eur Urol. 2000;38:2-19.
7. Wilt T, Ishani A, MacDonald R. Serenoa repens for benign prostatic hyperplasia. Cochrane Database Syst Rev. 2002;(3):CD001423.-
8. Bent S, Kane C, Shinohara K, et al. Saw palmetto for benign prostatic hyperplasia. N Engl J Med. 2006;354:557-566.
9. Benign prostatic hyperplasia: treatment {saw palmetto}. In: DynaMed. Available at: http://www.DynamicMedical.com. Accessed April 16, 2012.
10. Saper R. Clinical use of saw palmetto {saw palmetto}. In: Basow DS, ed. UpToDate [database online]. Waltham, Mass: UpToDate 2012. Available at: http://www.uptodate.com. Accessed April 16, 2012.
11. Agency for Healthcare Research and Quality. Guidelines on the treatment of non-neurogenic male LUTS. Available at: http://www.guideline.gov/content.aspx?id=34066. Accessed June 17, 2012.
Advise men with benign prostatic hyperplasia (BPH) not to take saw palmetto for urinary symptoms. Explain that it has not been found to alleviate symptoms, even at triple the standard dose.1
A: Based on evidence from a high-quality randomized controlled trial (RCT)1 and a 2009 meta-analysis.2
1. Barry MJ, Meleth S, Lee JY, et al. Effect of increasing doses of saw palmetto extract on lower urinary tract symptoms: a randomized trial. JAMA. 2011;306:1344-1351.
ILLUSTRATIVE CASE
A 66-year-old man comes to your office complaining of urinary frequency and straining to begin urination. He was recently diagnosed with BPh by a urologist, but is hesitant to begin taking a prescription drug. The patient, who is on a fixed income, asks you if saw palmetto extract might relieve his urinary symptoms. What should you tell him?
Roughly 40% of American men older than 60 years and nearly 90% of men older than 80 suffer from BPH and the troublesome lower urinary tract symptoms (LUTS) that it causes.3 Established medical and surgical options, as well as over-the-counter (OTC) plant-based products, are used for symptom relief. The OTC remedy most commonly used for BPH is Serenoa repens, derived from the saw palmetto dwarf palm tree. In a 2007 survey, 1.6 million US adults reported using saw palmetto extract, often as a treatment for BPH, in the 30 days prior to the survey.4
Until now, more questions than answers
As a family physician, you undoubtedly have many patients who are taking or considering taking saw palmetto for relief of BPH symptoms. The significant adverse effects of alpha-blockers and 5-alpha-reductase inhibitors, which are typically prescribed for LUTS—including decreased libido and dizziness—may help account for their interest in this alternative treatment.5,6
Until recently, evidence of saw palmetto’s efficacy has been limited and conflicting, despite widespread use of the extract. That has left many of us wondering whether we should recommend that men with BPH try saw palmetto despite the limited evidence; whether it is effective for some, but not all, BPH symptoms; and whether an increase in dose would increase its efficacy.
A 2002 Cochrane meta-analysis of 21 trials of saw palmetto extract for LUTS reported reduced nocturia, improved self-reported symptoms, and increased peak uroflow compared with placebo, without significant adverse effects.7 An updated Cochrane review published in 2009 included several more rigorous trials—and had very different results: This meta-analysis, which was based on 30 trials, found a reduction in nocturia, but failed to show improvement in other self-reported symptoms or peak uroflow.2
The largest trial included in the 2009 review was the Saw Palmetto Treatment for Enlarged Prostates (STEP) study,8 a one-year study with 225 participants. Its findings: no improvement in the treatment group compared with the placebo group in symptom scores or any secondary endpoints, and no important toxicity.8 Of note, the STEP study and most trials included in the 2009 Cochrane review used the standard saw palmetto extract dose of 160 mg twice daily.1,2
STUDY SUMMARY: Saw palmetto is ineffective, even at triple the dose
Barry et al conducted a 72-week double-blind, multicenter placebo-controlled trial to assess the effect of double (640 mg/d) and triple (960 mg/d) the standard dose of saw palmetto extract on BPH symptoms.1 The study included 369 men with moderate LUTS who had not recently received treatment for BPH. Exclusion criteria included a history of invasive BPH treatment, recent treatment with either an alpha-blocker or a 5-alpha-reductase inhibitor; recent phytotherapy, including saw palmetto; and a history of prostate or bladder cancer. Participants were randomized to receive either saw palmetto extract or an identical-looking placebo gel cap. Doses started at 320 mg/d and were increased to 640 mg/d at 24 weeks and 960 mg/d at 48 weeks.
The primary outcome was the change in the American Urological Association Symptom Index (AUASI) score from baseline to 72 weeks. AUASI, a scale of 0 to 35 in which higher numbers represent increased symptoms, is the same scoring tool used in both the Cochrane review and the STEP trial. Secondary measures included other symptom scales, peak uroflow, and poststudy satisfaction. The treatment and placebo groups had statistically identical baseline characteristics, and the sample size was large enough to detect clinically significant differences.
The AUASI score decreased by a mean of 2.20 points (95% confidence interval [CI], -3.04 to -0.36) in the group that received saw palmetto and by 2.99 points (95% CI, -3.81 to -2.17) in the placebo group—a mean difference of 0.79 in favor of the placebo group (P=.91). The proportion of participants achieving a 3-point reduction in AUASI score was statistically similar between the 2 groups (P=0.66). There was no significant dose response difference between the 2 groups, and saw palmetto proved to be no better than placebo for any of the secondary outcomes.
Subgroup analysis did not reveal any results that differed from the main outcomes. The only adverse events that were significantly different between the 2 groups related to physical injury or trauma, which were unlikely to be due to the intervention.1
By using the same symptom scoring system (AUASI) as many studies in the previous Cochrane reviews, Barry et al were able to compare their findings with those of other high-quality studies with similar methodologies and outcome measures. Despite using an even higher dose of the extract, the results of this trial are remarkably consistent with previous conclusions: Saw palmetto is not an effective treatment for symptoms associated with BPH. Moreover, this trial had a broad base of participants similar to the population in a primary care practice, including patients who would typically choose a natural remedy for LUTS.1
WHAT’S NEW: We now have answers to queries about saw palmetto
This trial is the first to compare higher doses of saw palmetto with placebo to assess a dosage threshold for effectiveness. While the study found no evidence of saw palmetto toxicity even at these higher doses, the extract did not outperform placebo for any measured outcome.1
This high-quality study confirmed the recent series of rigorous studies with negative outcomes by showing that the use of a standard dosage was not a study limitation and that saw palmetto extract is not effective for treating LUTS at any dosage. This trial should substantially affect future guideline recommendations that were limited by methodological concerns in the past.9-11
CAVEATS: In theory, individual preparations could work differently
It is not possible to be absolutely certain that these findings apply to all saw palmetto extract preparations, given the unknown active ingredients and unknown mechanism of action. However, the researchers used a high-quality preparation (a proprietary lipidic ethanolic extract) of saw palmetto at higher doses than the STEP trial and came to a similar conclusion, making it highly unlikely that another preparation would perform differently.
CHALLENGES TO IMPLEMENTATION: There are none
We see no challenges to implementation of this recommendation.
Acknowledgement
The PURLs Surveillance System is supported in part by Grant Number UL1RR024999 from the National Center for Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
Advise men with benign prostatic hyperplasia (BPH) not to take saw palmetto for urinary symptoms. Explain that it has not been found to alleviate symptoms, even at triple the standard dose.1
A: Based on evidence from a high-quality randomized controlled trial (RCT)1 and a 2009 meta-analysis.2
1. Barry MJ, Meleth S, Lee JY, et al. Effect of increasing doses of saw palmetto extract on lower urinary tract symptoms: a randomized trial. JAMA. 2011;306:1344-1351.
ILLUSTRATIVE CASE
A 66-year-old man comes to your office complaining of urinary frequency and straining to begin urination. He was recently diagnosed with BPh by a urologist, but is hesitant to begin taking a prescription drug. The patient, who is on a fixed income, asks you if saw palmetto extract might relieve his urinary symptoms. What should you tell him?
Roughly 40% of American men older than 60 years and nearly 90% of men older than 80 suffer from BPH and the troublesome lower urinary tract symptoms (LUTS) that it causes.3 Established medical and surgical options, as well as over-the-counter (OTC) plant-based products, are used for symptom relief. The OTC remedy most commonly used for BPH is Serenoa repens, derived from the saw palmetto dwarf palm tree. In a 2007 survey, 1.6 million US adults reported using saw palmetto extract, often as a treatment for BPH, in the 30 days prior to the survey.4
Until now, more questions than answers
As a family physician, you undoubtedly have many patients who are taking or considering taking saw palmetto for relief of BPH symptoms. The significant adverse effects of alpha-blockers and 5-alpha-reductase inhibitors, which are typically prescribed for LUTS—including decreased libido and dizziness—may help account for their interest in this alternative treatment.5,6
Until recently, evidence of saw palmetto’s efficacy has been limited and conflicting, despite widespread use of the extract. That has left many of us wondering whether we should recommend that men with BPH try saw palmetto despite the limited evidence; whether it is effective for some, but not all, BPH symptoms; and whether an increase in dose would increase its efficacy.
A 2002 Cochrane meta-analysis of 21 trials of saw palmetto extract for LUTS reported reduced nocturia, improved self-reported symptoms, and increased peak uroflow compared with placebo, without significant adverse effects.7 An updated Cochrane review published in 2009 included several more rigorous trials—and had very different results: This meta-analysis, which was based on 30 trials, found a reduction in nocturia, but failed to show improvement in other self-reported symptoms or peak uroflow.2
The largest trial included in the 2009 review was the Saw Palmetto Treatment for Enlarged Prostates (STEP) study,8 a one-year study with 225 participants. Its findings: no improvement in the treatment group compared with the placebo group in symptom scores or any secondary endpoints, and no important toxicity.8 Of note, the STEP study and most trials included in the 2009 Cochrane review used the standard saw palmetto extract dose of 160 mg twice daily.1,2
STUDY SUMMARY: Saw palmetto is ineffective, even at triple the dose
Barry et al conducted a 72-week double-blind, multicenter placebo-controlled trial to assess the effect of double (640 mg/d) and triple (960 mg/d) the standard dose of saw palmetto extract on BPH symptoms.1 The study included 369 men with moderate LUTS who had not recently received treatment for BPH. Exclusion criteria included a history of invasive BPH treatment, recent treatment with either an alpha-blocker or a 5-alpha-reductase inhibitor; recent phytotherapy, including saw palmetto; and a history of prostate or bladder cancer. Participants were randomized to receive either saw palmetto extract or an identical-looking placebo gel cap. Doses started at 320 mg/d and were increased to 640 mg/d at 24 weeks and 960 mg/d at 48 weeks.
The primary outcome was the change in the American Urological Association Symptom Index (AUASI) score from baseline to 72 weeks. AUASI, a scale of 0 to 35 in which higher numbers represent increased symptoms, is the same scoring tool used in both the Cochrane review and the STEP trial. Secondary measures included other symptom scales, peak uroflow, and poststudy satisfaction. The treatment and placebo groups had statistically identical baseline characteristics, and the sample size was large enough to detect clinically significant differences.
The AUASI score decreased by a mean of 2.20 points (95% confidence interval [CI], -3.04 to -0.36) in the group that received saw palmetto and by 2.99 points (95% CI, -3.81 to -2.17) in the placebo group—a mean difference of 0.79 in favor of the placebo group (P=.91). The proportion of participants achieving a 3-point reduction in AUASI score was statistically similar between the 2 groups (P=0.66). There was no significant dose response difference between the 2 groups, and saw palmetto proved to be no better than placebo for any of the secondary outcomes.
Subgroup analysis did not reveal any results that differed from the main outcomes. The only adverse events that were significantly different between the 2 groups related to physical injury or trauma, which were unlikely to be due to the intervention.1
By using the same symptom scoring system (AUASI) as many studies in the previous Cochrane reviews, Barry et al were able to compare their findings with those of other high-quality studies with similar methodologies and outcome measures. Despite using an even higher dose of the extract, the results of this trial are remarkably consistent with previous conclusions: Saw palmetto is not an effective treatment for symptoms associated with BPH. Moreover, this trial had a broad base of participants similar to the population in a primary care practice, including patients who would typically choose a natural remedy for LUTS.1
WHAT’S NEW: We now have answers to queries about saw palmetto
This trial is the first to compare higher doses of saw palmetto with placebo to assess a dosage threshold for effectiveness. While the study found no evidence of saw palmetto toxicity even at these higher doses, the extract did not outperform placebo for any measured outcome.1
This high-quality study confirmed the recent series of rigorous studies with negative outcomes by showing that the use of a standard dosage was not a study limitation and that saw palmetto extract is not effective for treating LUTS at any dosage. This trial should substantially affect future guideline recommendations that were limited by methodological concerns in the past.9-11
CAVEATS: In theory, individual preparations could work differently
It is not possible to be absolutely certain that these findings apply to all saw palmetto extract preparations, given the unknown active ingredients and unknown mechanism of action. However, the researchers used a high-quality preparation (a proprietary lipidic ethanolic extract) of saw palmetto at higher doses than the STEP trial and came to a similar conclusion, making it highly unlikely that another preparation would perform differently.
CHALLENGES TO IMPLEMENTATION: There are none
We see no challenges to implementation of this recommendation.
Acknowledgement
The PURLs Surveillance System is supported in part by Grant Number UL1RR024999 from the National Center for Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
1. Barry MJ, Meleth S, Lee JY, et al. Effect of increasing doses of saw palmetto extract on lower urinary tract symptoms: a randomized trial. JAMA. 2011;306:1344-1351.
2. Tacklind J, MacDonald R, Rutks I, et al. Serenoa repens for benign prostatic hyperplasia. Cochrane Database Syst Rev. 2009;(2):CD001423.-
3. Roehrborn CG, McConnell JD. Etiology, Pathophysiology, Epidemiology, and Natural History of Benign Prostatic Hyperplasia. 8th ed. Philadelphia, Pa: Campbell’s Urology; 2002.
4. Barnes PM, Bloom B, Nahin RL. Complementary and alternative medicine use among adults and children: United States, 2007. Natl Health Stat Report. 2008;(12):1-23.
5. Traish AM, Hassani J, Guay AT, et al. Adverse side effects of 5-alpha-reductase inhibitors therapy: persistent diminished libido and erectile dysfunction and depression in a subset of patients. J Sex Med. 2011;8:872-884.
6. Clifford GM, Farmer RD. Medical therapy for benign prostatic hyperplasia: a review of the literature. Eur Urol. 2000;38:2-19.
7. Wilt T, Ishani A, MacDonald R. Serenoa repens for benign prostatic hyperplasia. Cochrane Database Syst Rev. 2002;(3):CD001423.-
8. Bent S, Kane C, Shinohara K, et al. Saw palmetto for benign prostatic hyperplasia. N Engl J Med. 2006;354:557-566.
9. Benign prostatic hyperplasia: treatment {saw palmetto}. In: DynaMed. Available at: http://www.DynamicMedical.com. Accessed April 16, 2012.
10. Saper R. Clinical use of saw palmetto {saw palmetto}. In: Basow DS, ed. UpToDate [database online]. Waltham, Mass: UpToDate 2012. Available at: http://www.uptodate.com. Accessed April 16, 2012.
11. Agency for Healthcare Research and Quality. Guidelines on the treatment of non-neurogenic male LUTS. Available at: http://www.guideline.gov/content.aspx?id=34066. Accessed June 17, 2012.
1. Barry MJ, Meleth S, Lee JY, et al. Effect of increasing doses of saw palmetto extract on lower urinary tract symptoms: a randomized trial. JAMA. 2011;306:1344-1351.
2. Tacklind J, MacDonald R, Rutks I, et al. Serenoa repens for benign prostatic hyperplasia. Cochrane Database Syst Rev. 2009;(2):CD001423.-
3. Roehrborn CG, McConnell JD. Etiology, Pathophysiology, Epidemiology, and Natural History of Benign Prostatic Hyperplasia. 8th ed. Philadelphia, Pa: Campbell’s Urology; 2002.
4. Barnes PM, Bloom B, Nahin RL. Complementary and alternative medicine use among adults and children: United States, 2007. Natl Health Stat Report. 2008;(12):1-23.
5. Traish AM, Hassani J, Guay AT, et al. Adverse side effects of 5-alpha-reductase inhibitors therapy: persistent diminished libido and erectile dysfunction and depression in a subset of patients. J Sex Med. 2011;8:872-884.
6. Clifford GM, Farmer RD. Medical therapy for benign prostatic hyperplasia: a review of the literature. Eur Urol. 2000;38:2-19.
7. Wilt T, Ishani A, MacDonald R. Serenoa repens for benign prostatic hyperplasia. Cochrane Database Syst Rev. 2002;(3):CD001423.-
8. Bent S, Kane C, Shinohara K, et al. Saw palmetto for benign prostatic hyperplasia. N Engl J Med. 2006;354:557-566.
9. Benign prostatic hyperplasia: treatment {saw palmetto}. In: DynaMed. Available at: http://www.DynamicMedical.com. Accessed April 16, 2012.
10. Saper R. Clinical use of saw palmetto {saw palmetto}. In: Basow DS, ed. UpToDate [database online]. Waltham, Mass: UpToDate 2012. Available at: http://www.uptodate.com. Accessed April 16, 2012.
11. Agency for Healthcare Research and Quality. Guidelines on the treatment of non-neurogenic male LUTS. Available at: http://www.guideline.gov/content.aspx?id=34066. Accessed June 17, 2012.
Copyright © 2012 The Family Physicians Inquiries Network. All rights reserved.
Does anal cancer screening reduce morbidity and mortality in men who have sex with men?
IT’S UNCLEAR whether anal cancer screening benefits men who have sex with men because high-quality studies on this subject are lacking. In the absence of high-quality data, anal pap smears aren’t recommended for routine screening of men who have sex with men (strength of recommendation: C, expert opinion).
Evidence summary
The National Cancer Institute reports an annual anal cancer incidence of 1.6 per 100,000 and as of November of last year, expected that 5820 men and women would receive the diagnosis in 2011.1 The 5-year survival rate is 64.9%. For men who have sex with men, the incidence ranges from 35 to 100 per 100,000, with a higher incidence in HIV-positive men.2
Men who have sex with men also have a higher prevalence of human papillomavirus (HPV) than the general population.3,4 HPV is the most common cause of anal squamous intraepithelial lesions. In theory, screening for anal cancer may reduce morbidity and mortality by identifying and treating anal cancer precursors, much as screening has done for cervical cancer.
Small studies suggest that screening may be effective
One study has demonstrated that anal pap smears are potentially effective as a screening tool for detecting anal intraepithelial neoplasia.5 The study was limited by small sample size and failure to address patient-centered outcomes, however. It included only 395 subjects, most of whom (54%) were HIV positive. Additional studies evaluated 265 HIV-positive men and 658 men, of whom 407 were HIV positive, with similar findings.6,7
But a larger study shows no impact
The largest study to date, which included 5083 HIV-positive patients (contributing 13,411 patient-years), didn’t demonstrate a decrease in invasive anal carcinoma during the screening period.8 The difference in HPV prevalence between HIV-positive and HIV-negative men who have sex with men (96% vs 58.9%; P<.001) limits the ability to generalize the conclusions of this study to all men who have sex with men.9
Recommendations
No consensus guidelines exist on screening for anal cancer in men who have sex with men, regardless of HIV status.
The New York State Department of Health recommends baseline cytology and annual anal cancer screening for all HIV-positive men who have sex with men.
Based on the high prevalence of HPV in the HIV-positive population, some experts suggest anal cancer screening for HIV-positive men who have sex with men.10
1. National Cancer Institute. SEER stat fact sheets: anal cancer. November 10, 2011. Available at: http://seer.cancer.gov/statfacts/html/anus.html. Accessed February 24, 2012.
2. Altekruse SF, Kosary CL, Krapcho M, et al. eds. SEER cancer statistics review, 1975-2007. Available at: http://seer.cancer.gov/csr/1975_2007. Accessed February 24, 2012.
3. Chin-Hong PV, Vittinghoff E, Cranston RD, et al. Age-specific prevalence of anal human papillomavirus infection in HIV-negative sexually active men who have sex with men: the EXPLORE Study. J Infect Dis. 2004;190:2070-2076.
4. Chin-Hong PV, Berry JM, Cheng SC, et al. Comparison of patient- and clinician-collected anal cytology samples to screen for human papillomavirus-associated anal intraepithelial neoplasia in men who have sex with men. Ann Intern Med. 2008;149:300-306.
5. Nathan M, Singh N, Garrett N, et al. Performance of anal cytology in a clinical setting when measured against histology and high-resolution anoscopy findings. AIDS. 2010;24:373-379.
6. Scott H, Khoury J, Moore BA, et al. Routine anal cytology screening for anal squamous intraepithelial lesions in an urban HIV clinic. Sex Transm Dis. 2008;35:197-202.
7. Palefsky JM, Holly EA, Hogeboom CJ, et al. Anal cytology as a screening tool for anal squamous intraepithelial lesions. J Acquir Immune Defic Syndr Hum Retrovirol. 1997;14:415-422.
8. Mathews C, Caperna J, Cachay ER, et al. Early impact and performance characteristics of an established anal dysplasia screening program: program evaluation considerations. Open AIDS J. 2007;1:11-20.
9. Gao L, Zhou F, Li X, et al. Anal HPV infection in HIV-positive men who have sex with men from China. PLoS ONE. 2010;5:e15256.-
10. Silverberg MJ, Chao C, Leyden WA, et al. HIV infection and the risk of cancers with and without a known infectious cause. AIDS. 2009;23:2337-2345.
IT’S UNCLEAR whether anal cancer screening benefits men who have sex with men because high-quality studies on this subject are lacking. In the absence of high-quality data, anal pap smears aren’t recommended for routine screening of men who have sex with men (strength of recommendation: C, expert opinion).
Evidence summary
The National Cancer Institute reports an annual anal cancer incidence of 1.6 per 100,000 and as of November of last year, expected that 5820 men and women would receive the diagnosis in 2011.1 The 5-year survival rate is 64.9%. For men who have sex with men, the incidence ranges from 35 to 100 per 100,000, with a higher incidence in HIV-positive men.2
Men who have sex with men also have a higher prevalence of human papillomavirus (HPV) than the general population.3,4 HPV is the most common cause of anal squamous intraepithelial lesions. In theory, screening for anal cancer may reduce morbidity and mortality by identifying and treating anal cancer precursors, much as screening has done for cervical cancer.
Small studies suggest that screening may be effective
One study has demonstrated that anal pap smears are potentially effective as a screening tool for detecting anal intraepithelial neoplasia.5 The study was limited by small sample size and failure to address patient-centered outcomes, however. It included only 395 subjects, most of whom (54%) were HIV positive. Additional studies evaluated 265 HIV-positive men and 658 men, of whom 407 were HIV positive, with similar findings.6,7
But a larger study shows no impact
The largest study to date, which included 5083 HIV-positive patients (contributing 13,411 patient-years), didn’t demonstrate a decrease in invasive anal carcinoma during the screening period.8 The difference in HPV prevalence between HIV-positive and HIV-negative men who have sex with men (96% vs 58.9%; P<.001) limits the ability to generalize the conclusions of this study to all men who have sex with men.9
Recommendations
No consensus guidelines exist on screening for anal cancer in men who have sex with men, regardless of HIV status.
The New York State Department of Health recommends baseline cytology and annual anal cancer screening for all HIV-positive men who have sex with men.
Based on the high prevalence of HPV in the HIV-positive population, some experts suggest anal cancer screening for HIV-positive men who have sex with men.10
IT’S UNCLEAR whether anal cancer screening benefits men who have sex with men because high-quality studies on this subject are lacking. In the absence of high-quality data, anal pap smears aren’t recommended for routine screening of men who have sex with men (strength of recommendation: C, expert opinion).
Evidence summary
The National Cancer Institute reports an annual anal cancer incidence of 1.6 per 100,000 and as of November of last year, expected that 5820 men and women would receive the diagnosis in 2011.1 The 5-year survival rate is 64.9%. For men who have sex with men, the incidence ranges from 35 to 100 per 100,000, with a higher incidence in HIV-positive men.2
Men who have sex with men also have a higher prevalence of human papillomavirus (HPV) than the general population.3,4 HPV is the most common cause of anal squamous intraepithelial lesions. In theory, screening for anal cancer may reduce morbidity and mortality by identifying and treating anal cancer precursors, much as screening has done for cervical cancer.
Small studies suggest that screening may be effective
One study has demonstrated that anal pap smears are potentially effective as a screening tool for detecting anal intraepithelial neoplasia.5 The study was limited by small sample size and failure to address patient-centered outcomes, however. It included only 395 subjects, most of whom (54%) were HIV positive. Additional studies evaluated 265 HIV-positive men and 658 men, of whom 407 were HIV positive, with similar findings.6,7
But a larger study shows no impact
The largest study to date, which included 5083 HIV-positive patients (contributing 13,411 patient-years), didn’t demonstrate a decrease in invasive anal carcinoma during the screening period.8 The difference in HPV prevalence between HIV-positive and HIV-negative men who have sex with men (96% vs 58.9%; P<.001) limits the ability to generalize the conclusions of this study to all men who have sex with men.9
Recommendations
No consensus guidelines exist on screening for anal cancer in men who have sex with men, regardless of HIV status.
The New York State Department of Health recommends baseline cytology and annual anal cancer screening for all HIV-positive men who have sex with men.
Based on the high prevalence of HPV in the HIV-positive population, some experts suggest anal cancer screening for HIV-positive men who have sex with men.10
1. National Cancer Institute. SEER stat fact sheets: anal cancer. November 10, 2011. Available at: http://seer.cancer.gov/statfacts/html/anus.html. Accessed February 24, 2012.
2. Altekruse SF, Kosary CL, Krapcho M, et al. eds. SEER cancer statistics review, 1975-2007. Available at: http://seer.cancer.gov/csr/1975_2007. Accessed February 24, 2012.
3. Chin-Hong PV, Vittinghoff E, Cranston RD, et al. Age-specific prevalence of anal human papillomavirus infection in HIV-negative sexually active men who have sex with men: the EXPLORE Study. J Infect Dis. 2004;190:2070-2076.
4. Chin-Hong PV, Berry JM, Cheng SC, et al. Comparison of patient- and clinician-collected anal cytology samples to screen for human papillomavirus-associated anal intraepithelial neoplasia in men who have sex with men. Ann Intern Med. 2008;149:300-306.
5. Nathan M, Singh N, Garrett N, et al. Performance of anal cytology in a clinical setting when measured against histology and high-resolution anoscopy findings. AIDS. 2010;24:373-379.
6. Scott H, Khoury J, Moore BA, et al. Routine anal cytology screening for anal squamous intraepithelial lesions in an urban HIV clinic. Sex Transm Dis. 2008;35:197-202.
7. Palefsky JM, Holly EA, Hogeboom CJ, et al. Anal cytology as a screening tool for anal squamous intraepithelial lesions. J Acquir Immune Defic Syndr Hum Retrovirol. 1997;14:415-422.
8. Mathews C, Caperna J, Cachay ER, et al. Early impact and performance characteristics of an established anal dysplasia screening program: program evaluation considerations. Open AIDS J. 2007;1:11-20.
9. Gao L, Zhou F, Li X, et al. Anal HPV infection in HIV-positive men who have sex with men from China. PLoS ONE. 2010;5:e15256.-
10. Silverberg MJ, Chao C, Leyden WA, et al. HIV infection and the risk of cancers with and without a known infectious cause. AIDS. 2009;23:2337-2345.
1. National Cancer Institute. SEER stat fact sheets: anal cancer. November 10, 2011. Available at: http://seer.cancer.gov/statfacts/html/anus.html. Accessed February 24, 2012.
2. Altekruse SF, Kosary CL, Krapcho M, et al. eds. SEER cancer statistics review, 1975-2007. Available at: http://seer.cancer.gov/csr/1975_2007. Accessed February 24, 2012.
3. Chin-Hong PV, Vittinghoff E, Cranston RD, et al. Age-specific prevalence of anal human papillomavirus infection in HIV-negative sexually active men who have sex with men: the EXPLORE Study. J Infect Dis. 2004;190:2070-2076.
4. Chin-Hong PV, Berry JM, Cheng SC, et al. Comparison of patient- and clinician-collected anal cytology samples to screen for human papillomavirus-associated anal intraepithelial neoplasia in men who have sex with men. Ann Intern Med. 2008;149:300-306.
5. Nathan M, Singh N, Garrett N, et al. Performance of anal cytology in a clinical setting when measured against histology and high-resolution anoscopy findings. AIDS. 2010;24:373-379.
6. Scott H, Khoury J, Moore BA, et al. Routine anal cytology screening for anal squamous intraepithelial lesions in an urban HIV clinic. Sex Transm Dis. 2008;35:197-202.
7. Palefsky JM, Holly EA, Hogeboom CJ, et al. Anal cytology as a screening tool for anal squamous intraepithelial lesions. J Acquir Immune Defic Syndr Hum Retrovirol. 1997;14:415-422.
8. Mathews C, Caperna J, Cachay ER, et al. Early impact and performance characteristics of an established anal dysplasia screening program: program evaluation considerations. Open AIDS J. 2007;1:11-20.
9. Gao L, Zhou F, Li X, et al. Anal HPV infection in HIV-positive men who have sex with men from China. PLoS ONE. 2010;5:e15256.-
10. Silverberg MJ, Chao C, Leyden WA, et al. HIV infection and the risk of cancers with and without a known infectious cause. AIDS. 2009;23:2337-2345.
Evidence-based answers from the Family Physicians Inquiries Network
The effect of insurance-driven medication changes on patient care
Purpose Insurance plans periodically change their formularies to enhance medical efficacy and cost savings. Patients face challenges when formulary changes affect their treatment. This study assessed the impact of insurance-driven medication changes on primary care patients and examined implications for patient care.
Methods We mailed questionnaires to a cross-sectional random sample of 1200 adult patients who had visited one of 3 family medicine practices within the past 6 months, asking them to describe problems they had encountered in filling medication prescriptions. We performed descriptive analyses of the frequency and distribution of demographic variables and conditions being treated. Using logistic regression analysis, we identified demographic and health-related variables independently associated with patient-reported problems caused by formulary changes.
Results Three variables—a greater number of prescription medications taken, younger patient age, and reliance on government insurance—were independently associated with an increased likelihood of encountering a problem filling a medication. Patients who reported an insurance-related issue filling a new or existing prescription over the past year (23%) encountered an average of 3 distinct problems. Patients experienced adverse medical outcomes (41%), decreased satisfaction with the health care system (68%), and problems that burdened the physician practice (83%). Formulary changes involving cardiac/hypertension/lipid and neurologic/psychiatric medications caused the most problems.
Conclusions Insurance-driven medication changes adversely affect patient care and access to treatment, particularly for patients with government insurance. A better understanding of the negative impact of formulary changes on patient care and indirect health care expenditures should inform formulary change practices in order to minimize cost-shifting and maximize continuity of care.
To maintain the cost-effectiveness of health insurance, many organizations, including government agencies, routinely evaluate and choose to adopt alternative treatment modalities. But how do such changes affect patient outcomes? And do near-term cost savings from formulary changes lead to long-term cost benefits?
Chronic disease management and associated complex medication regimens account for most health insurance expenditures.1,2 Changes to prescription formularies are common,3 with medications being added or removed to reduce costs or to respond to revised practice guidelines.4,5
Researchers have examined the clinical risks and merits of changing from one drug to another, as well as the impact of implementing formulary changes on administrative and other costs, overall effectiveness of disease management, and the operational adeptness of health systems.6-10 Routine formulary changes may yield immediate cost savings, but net costs may increase downstream due to disruptions in patient care.11,12 Insurance-driven medication changes have also been shown to negatively affect patient adherence to medical treatment and also disease outcomes.13,14
Patient-level data related to formulary restrictions are limited,15 and analyses of patients’ experiences of medication changes are rare. A better understanding of patients’ experiences in this context could guide interventions to minimize treatment delays and improve outcomes. Our study assessed the effect of insurance-driven medication changes on primary care patients; specifically, the prevalence of difficulty in filling a prescription, resultant problems, and patient characteristics associated with reporting a problem.
Methods
Data collection
We mailed questionnaires to a random sample of 1200 adult patients (≥40 years) who had been seen within the previous 6 months at one of 3 family practices in northeastern Ohio. We asked respondents to quantify and describe any insurance-driven problems they encountered while attempting to fill or refill a prescription over the past year. We recorded each respondent’s insurance status, the name of the medication at issue and other medications they were taking, and demographic data. Comparative data for age and sex were collected for nonrespondents. The University Hospitals Case Medical Center Institutional Review Board approved all data collection procedures and methods for this cross-sectional study.
Data analysis
We tabulated and analyzed data from the surveys using Statistical Package for the Social Sciences (SPSS). We compared age and sex data (using t-test and chi-square test, respectively) between respondents and nonrespondents. We calculated descriptive statistics for all demographic, control, and outcome variables, and computed measures of association between demographic and health-related variables and insurance-driven problems encountered while filling a prescription. Using logistic regression analysis, we identified demographic and health-related variables independently associated with a problematic prescription.
We calculated the frequencies of problems encountered while trying to fill a prescription, and grouped the problems into 3 mutually exclusive categories: adverse medical outcomes, decreased patient satisfaction, and burden on physician practice. Adverse medical outcomes included missed doses of medication, inability to obtain medication, worsened medical condition, new medication adverse effects, and having to go to the emergency department (ED) because of a medication issue. We sorted medications into categories, and calculated the frequency of problems associated with each category.
We based our decision to mail 1200 surveys on a power calculation assuming a 40% response rate and approximately 25% of patients reporting a problem. A sample size of 480 or more provides 80% power to detect moderate differences in characteristics between those reporting a problem and those not reporting a problem.
Results
Four-hundred thirty-four patients returned the survey (36% response rate). We excluded 6 participants from analysis due to incomplete data for the primary outcome variable (problem with a prescription). Respondents and nonrespondents were similar in sex ratio, but respondents on average were 3 years older (P<.001). The average number of prescriptions taken was 3.4, and most patients (85%) had some form of private insurance ( TABLE 1 ). Most patients were female, in good health, and well educated.
Of the 428 study participants, 100 (23%) reported at least one problem obtaining a prescribed medication due to insurance. Generally, those who experienced a problem were younger, more likely to be female, and reported poorer health status than those reporting no problem ( TABLE 1 ). Additionally, those who encountered a problem were more than twice as likely to rely solely on Medicaid or Medicare, and were also taking more prescription medications. Problems filling a prescription were also reported more often in an urban setting than in suburban or semirural areas.
TABLE 1
Demographic variables for patients who did and didn’t report problems filling prescriptions
| Variable | Total (n=428) | Problem (n=100) | No problem (n=328) | P |
|---|---|---|---|---|
| Number of prescriptions, mean (SD) | 3.4 (3.2) | 4.8 (3.2) | 3.0 (3.1) | .001 |
| Age, mean (SD) | 60.0 (12) | 57.8 (13) | 60.6 (12) | .04 |
| Sex, n (%) | .02 | |||
| Male | 139 (32) | 23 (23) | 116 (35) | |
| Female | 289 (68) | 77 (77) | 212 (65) | |
| Health status, n (%) | .002 | |||
| Excellent/very good | 342 (81) | 69 (70)* | 273 (84)* | |
| Fair/poor | 80 (19) | 29 (30)* | 51 (16)* | |
| Education, n (%) | .46 | |||
| High school or less | 112 (27) | 30 (31)* | 82 (25)* | |
| Some college/trade | 140 (33) | 31 (33)* | 109 (34)* | |
| College graduate | 166 (40) | 34 (36)* | 132 (41) | |
| Insurance, n (%) | .001 | |||
| Government (Medicaid or Medicare) | 65 (15) | 27 (27) | 38 (12)* | |
| Nongovernment | 356 (85) | 72 (73)* | 284 (88)* | |
| Practice, n (%) | .005 | |||
| Semirural | 191 (45) | 35 (35) | 156 (48) | |
| Suburban | 116 (27) | 24 (24) | 92 (28) | |
| Urban | 121 (28) | 41 (41) | 80 (24) | |
| SD, standard deviation. *Some data are missing (<2.5%) from columns 2 and 3. | ||||
Using logistic regression, we analyzed a model that included all significant variables (age, total number of prescription drugs taken, sex, health status, insurance type, and practice location). The final logistic regression model showed statistical significance for only 3 variables: type of insurance, total number of prescription drugs taken, and age. (When we included type of insurance in the analysis, practice location was not associated with a problem filling a prescription.)
Specifically, the independent predictors of an insurance-related problem in filling a prescription were reliance solely on government-provided insurance, as opposed to private insurance or government insurance supplemented with private insurance (odds ratio [OR]=1.90; 95% confidence interval [CI], 1.02-3.61); taking more prescription medications (OR=1.19; 95% CI, 1.10-1.29); and being younger (OR=0.96; 95% CI, 0.94-0.99).
Respondents reporting at least one insurance-driven impediment to filling a prescription encountered an average of 2.9 different types of resultant problems ( TABLE 2 ). Insurance-related problems with medications were not limited to new prescriptions. Of the 100 patients reporting a problem with a medication, 21% had a problem with a new prescription, 42% with a medication they were already taking, and 37% with both a new and a previously prescribed medication.
TABLE 2
Resultant problems when patients had at least one insurance-related issue filling a prescription in the previous year
| Problem encountered | Percent of patients reporting problem* (n=100) |
|---|---|
| Adverse medical outcomes | |
| Missed doses of medication | 23 |
| Couldn’t get any medication | 19 |
| Medical condition got worse | 8 |
| New medication adverse effects | 6 |
| Had to go to emergency department | 5 |
| Overall any adverse outcome | 41 |
| Decreased patient satisfaction | |
| Got upset with insurance company | 44 |
| Got upset with pharmacist | 15 |
| Got upset with doctor | 12 |
| Overall any decreased satisfaction | 68 |
| Increased practice burden | |
| Had to wait for pharmacist authorization | 69 |
| Made extra phone calls to practice | 36 |
| Had to get a different medication | 36 |
| Had extra doctor visits | 13 |
| Overall any increased burden | 83 |
| *Patients reported, on average, 2.9 problems; therefore, categories exceed 100%. | |
Forty-one percent of patients reporting a problem experienced adverse medical outcomes. The most serious adverse medical outcomes were reported least often, but occurred nonetheless: worsening of medical condition (8%), new medication adverse effects (6%), and requiring a visit to the ED (5%). More commonly reported was decreased satisfaction with the health care system (68%). Patients were less likely to report being upset with their physician than their insurance company or pharmacist. Problems that burdened the physician practice were reported most frequently (83%).
TABLE 3 shows the medication categories that were affected when respondents reported at least one problem. Formulary changes or restrictions involving cardiac/hypertension/lipid and neurologic/psychiatric medications were linked to the most problems.
TABLE 3
Which medication categories were most affected when patients had a problem filling a prescription?
| Medication category | Frequency of occurrence |
|---|---|
| Cardiac/HTN/lipids | 23 |
| Neurologic/psychiatric | 23 |
| Metabolic/endocrine | 16 |
| Gastrointestinal | 15 |
| Pain | 13 |
| Respiratory | 11 |
| Other | 9 |
| Dermatologic | 7 |
| Total | 117* |
| HTN, hypertension. *Total exceeds 100 because some of the 100 patients had problems with medications in more than one category. | |
Discussion
Nearly one quarter of patients in our sample (23%) experienced problems caused by insurance constraints while they attempted to follow the treatment regimens prescribed by their physicians. Although the most commonly reported insurance-related problems (waiting for pharmacist authorization, making extra phone calls to the physician’s office) could be perceived as minor inconveniences, serious consequences were also common. Our study showed that patients who rely solely on Medicaid or Medicare bore the greatest burden of insurance-related obstacles when filling prescriptions, although others were also affected.
Consistent with prior research in Medicare and Medicaid populations, our study found that medication access restrictions can negatively affect patient adherence.13,16,17 Our study showed that 41% of patients who encountered a problem experienced a medically meaningful adverse outcome; 19% reported they received no medication for their condition. Similarly, a study of Medicare beneficiaries who had failed to fill or refill a prescription found that 20% cited lack of insurance coverage for the medication as a reason for not filling the prescription.17
In our study, 23% of patients reported missing doses of their medication due to insurance-related difficulties, and 8% reported a worsening of their medical condition. The increased costs associated with poor chronic care management are well documented.18 Less well described is the potential net savings produced when insurance formularies are adjusted to expand coverage and lower patient costs for prescription treatments for chronic conditions. In an analysis of cost data from the Pitney-Bowes Corporation, Mahoney12 revealed a significant net savings in health care costs and lost productivity when treatments for chronic conditions were moved to the lowest tier of the formulary, thereby making them available to health plan participants at the lowest cost.
We could not link patient reports of treatment disruptions empirically to medical outcomes or increased costs, due to the constraints of our research question and study design. However, it is reasonable to suggest that longer-term insurance costs for these patients could, in fact, negate any short-term cost savings generated from formulary restrictions. In particular, the 5% of our patient sample who reported using the ED as a consequence of an insurance-related disruption of their prescribed treatment likely added significant unnecessary cost to their treatment. This effect has been seen in other studies.19,20 In our study, cardiac/hypertension/lipid medications and medications for neurologic or psychiatric conditions were the most likely to be problematic. In these categories, competition of branded products may contribute to more frequent formulary changes. Furthermore, increases in morbidity and mortality associated with inadequate treatment of the conditions represented in these 2 categories of medications represent a significant burden to the US health system, including insurers, employers, and individuals.21-23
Although patients were less likely to report being upset with their physician than their insurance company or pharmacist, physicians bore a considerable burden for resolving a number of prevalent patient issues. Most of these problems required extra phone calls to the practice, additional medication authorization, or extra office visits. Physicians and their support staff may serve as buffers between patients and the insurance formulary rules, but at significant cost in their time and effort.
Electronic prescribing systems with real-time pharmacy benefit verification may provide additional efficiencies and help physicians and patients avoid some of the problems cited by our respondents. Providers with such systems receive immediate notification of formulary status, including tier and co-pay levels, which can aid in shared decision-making at the point of prescribing. Physicians without access to e-prescribing may want to use newer formulary search engines that can check formulary status of medications across multiple insurance plans. However, these electronic tools often fail to account for variations in formularies within the same insurance plan for different employers based on their benefit structure. Still, when a medication is not on formulary or a co-payment is required, the physician may be forced to play the role of apologist for the constraints imposed by the insurance formulary.
In cases where formularies restrict the patient’s potential access to a preferred treatment plan, the burden of prior authorizations continues to be borne by physicians. Coverage limitations lead to financial and medical consequences that must be managed in partnership with the patient. A system should be put in place by insurance companies that facilitates out-of-formulary authorizations to prevent lapses in patient care or deleterious changes in medical management.
Study limitations
The findings reported here should be interpreted in light of some limitations of this study. The response rate to our mailed patient survey was modest (36%), although typical for this method. The sex mix of respondents was similar to that of nonrespondents, but nonrespondents were slightly younger. Given that younger age is associated with a greater likelihood of experiencing a problem filling a medication, our findings may underestimate the frequency of this dilemma. In addition, our survey asked patients to recall events that occurred over the past year, introducing a potential for recall bias.
While the overall sample size was relatively small (n=428), it is close to the number calculated for sufficient power to conduct the analyses (n=480). Furthermore, data were collected from 3 distinct patient populations: urban, suburban and semirural. Although the scope of our study included only one geographic region, variability in practice setting lends some tentative support to the generalizability of the findings.
Looking forward
As a standard method to control costs and update treatment guidelines, insurance-mediated medication changes will continue to present unique challenges for patients and health care providers. Formulary changes burden the downstream delivery of medical care with expensive administrative responsibilities and disrupt effective disease management and prevention. Until insurance companies and pharmacy benefit managers start paying heed to total costs of care when contemplating formulary changes, physicians should try to identify formulary conflicts as early as possible in the prescribing process so as to save time for all parties later and improve compliance.
As practices proceed toward adoption of electronic health records, e-prescribing, and the Centers for Medicare & Medicaid Services’ “meaningful use” criteria, physicians may use systems that provide real-time formulary information, which can flag issues before the patient leaves the exam room. Future research should explore the ways formulary changes might be implemented to provide the strongest continuity of patient care with the least amount of cost shifting.
CORRESPONDENCE
Susan A. Flocke, PhD, CWRU Department of Family Medicine & Community Health, 11000 Cedar Avenue, Suite 402, Cleveland, OH 44106; [email protected]
1. Centers for Disease Control and Prevention National Center for Chronic Disease Prevention and Health Promotion. Chronic diseases: the power to prevent, the call to control: at a glance 2009. Available at: http://www.cdc.gov/chronicdisease/resources/publications/aag/chronic.htm. Page last updated December 17, 2009. Accessed June 21, 2012.
2. Mueller C, Schur C, O’Connell J. Prescription drug spending: the impact of age and chronic disease status. Am J Public Health. 1997;87:1626-1629.
3. Kaiser Family Foundation. Prescription drug trends. Available at: http://www.kff.org/rxdrugs/upload/3057_07.pdf. Published September 2008. Accessed December 1, 2009.
4. Neumann PJ. Evidence-based and value-based formulary guidelines. Health Aff (Millwood). 2004;23:124-134.
5. Simon GE, Psaty BM, Hrachovec JB, et al. Principles for evidence-based drug formulary policy. J Gen Intern Med. 2005;20:964-968.
6. Huskamp HA, Deverka PA, Epstein AM, et al. The effect of incentive-based formularies on prescription-drug utilization and spending. N Engl J Med. 2003;349:2224-2232.
7. Meissner B, Dickson M, Shinogle J, et al. Drug and medical cost effects of a drug formulary change with therapeutic interchange for statin drugs in a multistate managed Medicaid organization. J Manag Care Pharm. 2006;12:331-340.
8. Ovsag K, Hydery S, Mousa SA. Preferred drug lists: potential impact on healthcare economics. Vasc Health Risk Manag. 2008;4:403-413.
9. Raisch DW, Klaurens LM, Hayden C, et al. Impact of a formulary change in proton pump inhibitors on health care costs and patients’ symptoms. Dig Dis Sci. 2001;46:1533-1539.
10. Soumerai SB. Benefits and risks of increasing restrictions on access to costly drugs in Medicaid. Health Aff (Millwood). 2004;23:135-146.
11. Johnson TJ, Stahl-Moncada S. Medicaid prescription formulary restrictions and arthritis treatment costs. Am J Public Health. 2008;98:1300-1305.
12. Mahoney JJ. Reducing patient drug acquisition costs can lower diabetes health claims. Am J Manag Care. 2005;11(5 suppl):S170-S176.
13. Ridley DB, Axelsen KJ. Impact of Medicaid preferred drug lists on therapeutic adherence. Pharmacoeconomics. 2006;24 (suppl 3):65-78.
14. Steinman MA, Sands LP, Covinsky KE. Self-restriction of medications due to cost in seniors without prescription coverage. J Gen Intern Med. 2001;16:793-799.
15. Jean CD, Triplett JW. Investigating patient experiences after a formulary change. Am J Health Syst Pharm. 2000;57:1052-1054.
16. Wilson J, Axelsen K, Tang S. Medicaid prescription drug access restrictions: exploring the effect on patient persistence with hypertension medications. Am J Manag Care. 2005;11 (spec no):SP27-SP34.
17. Kennedy J, Tuleu I, Mackay K. Unfilled prescriptions of Medicare beneficiaries: prevalence, reasons, and types of medicines prescribed. J Manag Care Pharm. 2008;14:553-560.
18. Mensah GA, Brown DW. An overview of cardiovascular disease burden in the United States. Health Aff (Millwood). 2007;26:38-48.
19. Sokol MC, McGuigan KA, Verbrugge RR, et al. Impact of medication adherence on hospitalization risk and healthcare cost. Med Care. 2005;43:521-530.
20. Tamblyn R, Laprise R, Hanley JA, et al. Adverse events associated with prescription drug cost-sharing among poor and elderly persons. JAMA. 2001;285:421-429.
21. Flack JM, Casciano R, Casciano J, et al. Cardiovascular disease costs associated with uncontrolled hypertension. Manag Care Interface. 2002;15:28-36.
22. Hall RC, Wise MG. The clinical and financial burden of mood disorders. Cost and outcome. Psychosomatics. 1995;36:S11-S18.
23. McCombs JS, Nichol MB, Newman CM, et al. The costs of interrupting antihypertensive drug therapy in a Medicaid population. Med Care. 1994;32:214-226.
Purpose Insurance plans periodically change their formularies to enhance medical efficacy and cost savings. Patients face challenges when formulary changes affect their treatment. This study assessed the impact of insurance-driven medication changes on primary care patients and examined implications for patient care.
Methods We mailed questionnaires to a cross-sectional random sample of 1200 adult patients who had visited one of 3 family medicine practices within the past 6 months, asking them to describe problems they had encountered in filling medication prescriptions. We performed descriptive analyses of the frequency and distribution of demographic variables and conditions being treated. Using logistic regression analysis, we identified demographic and health-related variables independently associated with patient-reported problems caused by formulary changes.
Results Three variables—a greater number of prescription medications taken, younger patient age, and reliance on government insurance—were independently associated with an increased likelihood of encountering a problem filling a medication. Patients who reported an insurance-related issue filling a new or existing prescription over the past year (23%) encountered an average of 3 distinct problems. Patients experienced adverse medical outcomes (41%), decreased satisfaction with the health care system (68%), and problems that burdened the physician practice (83%). Formulary changes involving cardiac/hypertension/lipid and neurologic/psychiatric medications caused the most problems.
Conclusions Insurance-driven medication changes adversely affect patient care and access to treatment, particularly for patients with government insurance. A better understanding of the negative impact of formulary changes on patient care and indirect health care expenditures should inform formulary change practices in order to minimize cost-shifting and maximize continuity of care.
To maintain the cost-effectiveness of health insurance, many organizations, including government agencies, routinely evaluate and choose to adopt alternative treatment modalities. But how do such changes affect patient outcomes? And do near-term cost savings from formulary changes lead to long-term cost benefits?
Chronic disease management and associated complex medication regimens account for most health insurance expenditures.1,2 Changes to prescription formularies are common,3 with medications being added or removed to reduce costs or to respond to revised practice guidelines.4,5
Researchers have examined the clinical risks and merits of changing from one drug to another, as well as the impact of implementing formulary changes on administrative and other costs, overall effectiveness of disease management, and the operational adeptness of health systems.6-10 Routine formulary changes may yield immediate cost savings, but net costs may increase downstream due to disruptions in patient care.11,12 Insurance-driven medication changes have also been shown to negatively affect patient adherence to medical treatment and also disease outcomes.13,14
Patient-level data related to formulary restrictions are limited,15 and analyses of patients’ experiences of medication changes are rare. A better understanding of patients’ experiences in this context could guide interventions to minimize treatment delays and improve outcomes. Our study assessed the effect of insurance-driven medication changes on primary care patients; specifically, the prevalence of difficulty in filling a prescription, resultant problems, and patient characteristics associated with reporting a problem.
Methods
Data collection
We mailed questionnaires to a random sample of 1200 adult patients (≥40 years) who had been seen within the previous 6 months at one of 3 family practices in northeastern Ohio. We asked respondents to quantify and describe any insurance-driven problems they encountered while attempting to fill or refill a prescription over the past year. We recorded each respondent’s insurance status, the name of the medication at issue and other medications they were taking, and demographic data. Comparative data for age and sex were collected for nonrespondents. The University Hospitals Case Medical Center Institutional Review Board approved all data collection procedures and methods for this cross-sectional study.
Data analysis
We tabulated and analyzed data from the surveys using Statistical Package for the Social Sciences (SPSS). We compared age and sex data (using t-test and chi-square test, respectively) between respondents and nonrespondents. We calculated descriptive statistics for all demographic, control, and outcome variables, and computed measures of association between demographic and health-related variables and insurance-driven problems encountered while filling a prescription. Using logistic regression analysis, we identified demographic and health-related variables independently associated with a problematic prescription.
We calculated the frequencies of problems encountered while trying to fill a prescription, and grouped the problems into 3 mutually exclusive categories: adverse medical outcomes, decreased patient satisfaction, and burden on physician practice. Adverse medical outcomes included missed doses of medication, inability to obtain medication, worsened medical condition, new medication adverse effects, and having to go to the emergency department (ED) because of a medication issue. We sorted medications into categories, and calculated the frequency of problems associated with each category.
We based our decision to mail 1200 surveys on a power calculation assuming a 40% response rate and approximately 25% of patients reporting a problem. A sample size of 480 or more provides 80% power to detect moderate differences in characteristics between those reporting a problem and those not reporting a problem.
Results
Four-hundred thirty-four patients returned the survey (36% response rate). We excluded 6 participants from analysis due to incomplete data for the primary outcome variable (problem with a prescription). Respondents and nonrespondents were similar in sex ratio, but respondents on average were 3 years older (P<.001). The average number of prescriptions taken was 3.4, and most patients (85%) had some form of private insurance ( TABLE 1 ). Most patients were female, in good health, and well educated.
Of the 428 study participants, 100 (23%) reported at least one problem obtaining a prescribed medication due to insurance. Generally, those who experienced a problem were younger, more likely to be female, and reported poorer health status than those reporting no problem ( TABLE 1 ). Additionally, those who encountered a problem were more than twice as likely to rely solely on Medicaid or Medicare, and were also taking more prescription medications. Problems filling a prescription were also reported more often in an urban setting than in suburban or semirural areas.
TABLE 1
Demographic variables for patients who did and didn’t report problems filling prescriptions
| Variable | Total (n=428) | Problem (n=100) | No problem (n=328) | P |
|---|---|---|---|---|
| Number of prescriptions, mean (SD) | 3.4 (3.2) | 4.8 (3.2) | 3.0 (3.1) | .001 |
| Age, mean (SD) | 60.0 (12) | 57.8 (13) | 60.6 (12) | .04 |
| Sex, n (%) | .02 | |||
| Male | 139 (32) | 23 (23) | 116 (35) | |
| Female | 289 (68) | 77 (77) | 212 (65) | |
| Health status, n (%) | .002 | |||
| Excellent/very good | 342 (81) | 69 (70)* | 273 (84)* | |
| Fair/poor | 80 (19) | 29 (30)* | 51 (16)* | |
| Education, n (%) | .46 | |||
| High school or less | 112 (27) | 30 (31)* | 82 (25)* | |
| Some college/trade | 140 (33) | 31 (33)* | 109 (34)* | |
| College graduate | 166 (40) | 34 (36)* | 132 (41) | |
| Insurance, n (%) | .001 | |||
| Government (Medicaid or Medicare) | 65 (15) | 27 (27) | 38 (12)* | |
| Nongovernment | 356 (85) | 72 (73)* | 284 (88)* | |
| Practice, n (%) | .005 | |||
| Semirural | 191 (45) | 35 (35) | 156 (48) | |
| Suburban | 116 (27) | 24 (24) | 92 (28) | |
| Urban | 121 (28) | 41 (41) | 80 (24) | |
| SD, standard deviation. *Some data are missing (<2.5%) from columns 2 and 3. | ||||
Using logistic regression, we analyzed a model that included all significant variables (age, total number of prescription drugs taken, sex, health status, insurance type, and practice location). The final logistic regression model showed statistical significance for only 3 variables: type of insurance, total number of prescription drugs taken, and age. (When we included type of insurance in the analysis, practice location was not associated with a problem filling a prescription.)
Specifically, the independent predictors of an insurance-related problem in filling a prescription were reliance solely on government-provided insurance, as opposed to private insurance or government insurance supplemented with private insurance (odds ratio [OR]=1.90; 95% confidence interval [CI], 1.02-3.61); taking more prescription medications (OR=1.19; 95% CI, 1.10-1.29); and being younger (OR=0.96; 95% CI, 0.94-0.99).
Respondents reporting at least one insurance-driven impediment to filling a prescription encountered an average of 2.9 different types of resultant problems ( TABLE 2 ). Insurance-related problems with medications were not limited to new prescriptions. Of the 100 patients reporting a problem with a medication, 21% had a problem with a new prescription, 42% with a medication they were already taking, and 37% with both a new and a previously prescribed medication.
TABLE 2
Resultant problems when patients had at least one insurance-related issue filling a prescription in the previous year
| Problem encountered | Percent of patients reporting problem* (n=100) |
|---|---|
| Adverse medical outcomes | |
| Missed doses of medication | 23 |
| Couldn’t get any medication | 19 |
| Medical condition got worse | 8 |
| New medication adverse effects | 6 |
| Had to go to emergency department | 5 |
| Overall any adverse outcome | 41 |
| Decreased patient satisfaction | |
| Got upset with insurance company | 44 |
| Got upset with pharmacist | 15 |
| Got upset with doctor | 12 |
| Overall any decreased satisfaction | 68 |
| Increased practice burden | |
| Had to wait for pharmacist authorization | 69 |
| Made extra phone calls to practice | 36 |
| Had to get a different medication | 36 |
| Had extra doctor visits | 13 |
| Overall any increased burden | 83 |
| *Patients reported, on average, 2.9 problems; therefore, categories exceed 100%. | |
Forty-one percent of patients reporting a problem experienced adverse medical outcomes. The most serious adverse medical outcomes were reported least often, but occurred nonetheless: worsening of medical condition (8%), new medication adverse effects (6%), and requiring a visit to the ED (5%). More commonly reported was decreased satisfaction with the health care system (68%). Patients were less likely to report being upset with their physician than their insurance company or pharmacist. Problems that burdened the physician practice were reported most frequently (83%).
TABLE 3 shows the medication categories that were affected when respondents reported at least one problem. Formulary changes or restrictions involving cardiac/hypertension/lipid and neurologic/psychiatric medications were linked to the most problems.
TABLE 3
Which medication categories were most affected when patients had a problem filling a prescription?
| Medication category | Frequency of occurrence |
|---|---|
| Cardiac/HTN/lipids | 23 |
| Neurologic/psychiatric | 23 |
| Metabolic/endocrine | 16 |
| Gastrointestinal | 15 |
| Pain | 13 |
| Respiratory | 11 |
| Other | 9 |
| Dermatologic | 7 |
| Total | 117* |
| HTN, hypertension. *Total exceeds 100 because some of the 100 patients had problems with medications in more than one category. | |
Discussion
Nearly one quarter of patients in our sample (23%) experienced problems caused by insurance constraints while they attempted to follow the treatment regimens prescribed by their physicians. Although the most commonly reported insurance-related problems (waiting for pharmacist authorization, making extra phone calls to the physician’s office) could be perceived as minor inconveniences, serious consequences were also common. Our study showed that patients who rely solely on Medicaid or Medicare bore the greatest burden of insurance-related obstacles when filling prescriptions, although others were also affected.
Consistent with prior research in Medicare and Medicaid populations, our study found that medication access restrictions can negatively affect patient adherence.13,16,17 Our study showed that 41% of patients who encountered a problem experienced a medically meaningful adverse outcome; 19% reported they received no medication for their condition. Similarly, a study of Medicare beneficiaries who had failed to fill or refill a prescription found that 20% cited lack of insurance coverage for the medication as a reason for not filling the prescription.17
In our study, 23% of patients reported missing doses of their medication due to insurance-related difficulties, and 8% reported a worsening of their medical condition. The increased costs associated with poor chronic care management are well documented.18 Less well described is the potential net savings produced when insurance formularies are adjusted to expand coverage and lower patient costs for prescription treatments for chronic conditions. In an analysis of cost data from the Pitney-Bowes Corporation, Mahoney12 revealed a significant net savings in health care costs and lost productivity when treatments for chronic conditions were moved to the lowest tier of the formulary, thereby making them available to health plan participants at the lowest cost.
We could not link patient reports of treatment disruptions empirically to medical outcomes or increased costs, due to the constraints of our research question and study design. However, it is reasonable to suggest that longer-term insurance costs for these patients could, in fact, negate any short-term cost savings generated from formulary restrictions. In particular, the 5% of our patient sample who reported using the ED as a consequence of an insurance-related disruption of their prescribed treatment likely added significant unnecessary cost to their treatment. This effect has been seen in other studies.19,20 In our study, cardiac/hypertension/lipid medications and medications for neurologic or psychiatric conditions were the most likely to be problematic. In these categories, competition of branded products may contribute to more frequent formulary changes. Furthermore, increases in morbidity and mortality associated with inadequate treatment of the conditions represented in these 2 categories of medications represent a significant burden to the US health system, including insurers, employers, and individuals.21-23
Although patients were less likely to report being upset with their physician than their insurance company or pharmacist, physicians bore a considerable burden for resolving a number of prevalent patient issues. Most of these problems required extra phone calls to the practice, additional medication authorization, or extra office visits. Physicians and their support staff may serve as buffers between patients and the insurance formulary rules, but at significant cost in their time and effort.
Electronic prescribing systems with real-time pharmacy benefit verification may provide additional efficiencies and help physicians and patients avoid some of the problems cited by our respondents. Providers with such systems receive immediate notification of formulary status, including tier and co-pay levels, which can aid in shared decision-making at the point of prescribing. Physicians without access to e-prescribing may want to use newer formulary search engines that can check formulary status of medications across multiple insurance plans. However, these electronic tools often fail to account for variations in formularies within the same insurance plan for different employers based on their benefit structure. Still, when a medication is not on formulary or a co-payment is required, the physician may be forced to play the role of apologist for the constraints imposed by the insurance formulary.
In cases where formularies restrict the patient’s potential access to a preferred treatment plan, the burden of prior authorizations continues to be borne by physicians. Coverage limitations lead to financial and medical consequences that must be managed in partnership with the patient. A system should be put in place by insurance companies that facilitates out-of-formulary authorizations to prevent lapses in patient care or deleterious changes in medical management.
Study limitations
The findings reported here should be interpreted in light of some limitations of this study. The response rate to our mailed patient survey was modest (36%), although typical for this method. The sex mix of respondents was similar to that of nonrespondents, but nonrespondents were slightly younger. Given that younger age is associated with a greater likelihood of experiencing a problem filling a medication, our findings may underestimate the frequency of this dilemma. In addition, our survey asked patients to recall events that occurred over the past year, introducing a potential for recall bias.
While the overall sample size was relatively small (n=428), it is close to the number calculated for sufficient power to conduct the analyses (n=480). Furthermore, data were collected from 3 distinct patient populations: urban, suburban and semirural. Although the scope of our study included only one geographic region, variability in practice setting lends some tentative support to the generalizability of the findings.
Looking forward
As a standard method to control costs and update treatment guidelines, insurance-mediated medication changes will continue to present unique challenges for patients and health care providers. Formulary changes burden the downstream delivery of medical care with expensive administrative responsibilities and disrupt effective disease management and prevention. Until insurance companies and pharmacy benefit managers start paying heed to total costs of care when contemplating formulary changes, physicians should try to identify formulary conflicts as early as possible in the prescribing process so as to save time for all parties later and improve compliance.
As practices proceed toward adoption of electronic health records, e-prescribing, and the Centers for Medicare & Medicaid Services’ “meaningful use” criteria, physicians may use systems that provide real-time formulary information, which can flag issues before the patient leaves the exam room. Future research should explore the ways formulary changes might be implemented to provide the strongest continuity of patient care with the least amount of cost shifting.
CORRESPONDENCE
Susan A. Flocke, PhD, CWRU Department of Family Medicine & Community Health, 11000 Cedar Avenue, Suite 402, Cleveland, OH 44106; [email protected]
Purpose Insurance plans periodically change their formularies to enhance medical efficacy and cost savings. Patients face challenges when formulary changes affect their treatment. This study assessed the impact of insurance-driven medication changes on primary care patients and examined implications for patient care.
Methods We mailed questionnaires to a cross-sectional random sample of 1200 adult patients who had visited one of 3 family medicine practices within the past 6 months, asking them to describe problems they had encountered in filling medication prescriptions. We performed descriptive analyses of the frequency and distribution of demographic variables and conditions being treated. Using logistic regression analysis, we identified demographic and health-related variables independently associated with patient-reported problems caused by formulary changes.
Results Three variables—a greater number of prescription medications taken, younger patient age, and reliance on government insurance—were independently associated with an increased likelihood of encountering a problem filling a medication. Patients who reported an insurance-related issue filling a new or existing prescription over the past year (23%) encountered an average of 3 distinct problems. Patients experienced adverse medical outcomes (41%), decreased satisfaction with the health care system (68%), and problems that burdened the physician practice (83%). Formulary changes involving cardiac/hypertension/lipid and neurologic/psychiatric medications caused the most problems.
Conclusions Insurance-driven medication changes adversely affect patient care and access to treatment, particularly for patients with government insurance. A better understanding of the negative impact of formulary changes on patient care and indirect health care expenditures should inform formulary change practices in order to minimize cost-shifting and maximize continuity of care.
To maintain the cost-effectiveness of health insurance, many organizations, including government agencies, routinely evaluate and choose to adopt alternative treatment modalities. But how do such changes affect patient outcomes? And do near-term cost savings from formulary changes lead to long-term cost benefits?
Chronic disease management and associated complex medication regimens account for most health insurance expenditures.1,2 Changes to prescription formularies are common,3 with medications being added or removed to reduce costs or to respond to revised practice guidelines.4,5
Researchers have examined the clinical risks and merits of changing from one drug to another, as well as the impact of implementing formulary changes on administrative and other costs, overall effectiveness of disease management, and the operational adeptness of health systems.6-10 Routine formulary changes may yield immediate cost savings, but net costs may increase downstream due to disruptions in patient care.11,12 Insurance-driven medication changes have also been shown to negatively affect patient adherence to medical treatment and also disease outcomes.13,14
Patient-level data related to formulary restrictions are limited,15 and analyses of patients’ experiences of medication changes are rare. A better understanding of patients’ experiences in this context could guide interventions to minimize treatment delays and improve outcomes. Our study assessed the effect of insurance-driven medication changes on primary care patients; specifically, the prevalence of difficulty in filling a prescription, resultant problems, and patient characteristics associated with reporting a problem.
Methods
Data collection
We mailed questionnaires to a random sample of 1200 adult patients (≥40 years) who had been seen within the previous 6 months at one of 3 family practices in northeastern Ohio. We asked respondents to quantify and describe any insurance-driven problems they encountered while attempting to fill or refill a prescription over the past year. We recorded each respondent’s insurance status, the name of the medication at issue and other medications they were taking, and demographic data. Comparative data for age and sex were collected for nonrespondents. The University Hospitals Case Medical Center Institutional Review Board approved all data collection procedures and methods for this cross-sectional study.
Data analysis
We tabulated and analyzed data from the surveys using Statistical Package for the Social Sciences (SPSS). We compared age and sex data (using t-test and chi-square test, respectively) between respondents and nonrespondents. We calculated descriptive statistics for all demographic, control, and outcome variables, and computed measures of association between demographic and health-related variables and insurance-driven problems encountered while filling a prescription. Using logistic regression analysis, we identified demographic and health-related variables independently associated with a problematic prescription.
We calculated the frequencies of problems encountered while trying to fill a prescription, and grouped the problems into 3 mutually exclusive categories: adverse medical outcomes, decreased patient satisfaction, and burden on physician practice. Adverse medical outcomes included missed doses of medication, inability to obtain medication, worsened medical condition, new medication adverse effects, and having to go to the emergency department (ED) because of a medication issue. We sorted medications into categories, and calculated the frequency of problems associated with each category.
We based our decision to mail 1200 surveys on a power calculation assuming a 40% response rate and approximately 25% of patients reporting a problem. A sample size of 480 or more provides 80% power to detect moderate differences in characteristics between those reporting a problem and those not reporting a problem.
Results
Four-hundred thirty-four patients returned the survey (36% response rate). We excluded 6 participants from analysis due to incomplete data for the primary outcome variable (problem with a prescription). Respondents and nonrespondents were similar in sex ratio, but respondents on average were 3 years older (P<.001). The average number of prescriptions taken was 3.4, and most patients (85%) had some form of private insurance ( TABLE 1 ). Most patients were female, in good health, and well educated.
Of the 428 study participants, 100 (23%) reported at least one problem obtaining a prescribed medication due to insurance. Generally, those who experienced a problem were younger, more likely to be female, and reported poorer health status than those reporting no problem ( TABLE 1 ). Additionally, those who encountered a problem were more than twice as likely to rely solely on Medicaid or Medicare, and were also taking more prescription medications. Problems filling a prescription were also reported more often in an urban setting than in suburban or semirural areas.
TABLE 1
Demographic variables for patients who did and didn’t report problems filling prescriptions
| Variable | Total (n=428) | Problem (n=100) | No problem (n=328) | P |
|---|---|---|---|---|
| Number of prescriptions, mean (SD) | 3.4 (3.2) | 4.8 (3.2) | 3.0 (3.1) | .001 |
| Age, mean (SD) | 60.0 (12) | 57.8 (13) | 60.6 (12) | .04 |
| Sex, n (%) | .02 | |||
| Male | 139 (32) | 23 (23) | 116 (35) | |
| Female | 289 (68) | 77 (77) | 212 (65) | |
| Health status, n (%) | .002 | |||
| Excellent/very good | 342 (81) | 69 (70)* | 273 (84)* | |
| Fair/poor | 80 (19) | 29 (30)* | 51 (16)* | |
| Education, n (%) | .46 | |||
| High school or less | 112 (27) | 30 (31)* | 82 (25)* | |
| Some college/trade | 140 (33) | 31 (33)* | 109 (34)* | |
| College graduate | 166 (40) | 34 (36)* | 132 (41) | |
| Insurance, n (%) | .001 | |||
| Government (Medicaid or Medicare) | 65 (15) | 27 (27) | 38 (12)* | |
| Nongovernment | 356 (85) | 72 (73)* | 284 (88)* | |
| Practice, n (%) | .005 | |||
| Semirural | 191 (45) | 35 (35) | 156 (48) | |
| Suburban | 116 (27) | 24 (24) | 92 (28) | |
| Urban | 121 (28) | 41 (41) | 80 (24) | |
| SD, standard deviation. *Some data are missing (<2.5%) from columns 2 and 3. | ||||
Using logistic regression, we analyzed a model that included all significant variables (age, total number of prescription drugs taken, sex, health status, insurance type, and practice location). The final logistic regression model showed statistical significance for only 3 variables: type of insurance, total number of prescription drugs taken, and age. (When we included type of insurance in the analysis, practice location was not associated with a problem filling a prescription.)
Specifically, the independent predictors of an insurance-related problem in filling a prescription were reliance solely on government-provided insurance, as opposed to private insurance or government insurance supplemented with private insurance (odds ratio [OR]=1.90; 95% confidence interval [CI], 1.02-3.61); taking more prescription medications (OR=1.19; 95% CI, 1.10-1.29); and being younger (OR=0.96; 95% CI, 0.94-0.99).
Respondents reporting at least one insurance-driven impediment to filling a prescription encountered an average of 2.9 different types of resultant problems ( TABLE 2 ). Insurance-related problems with medications were not limited to new prescriptions. Of the 100 patients reporting a problem with a medication, 21% had a problem with a new prescription, 42% with a medication they were already taking, and 37% with both a new and a previously prescribed medication.
TABLE 2
Resultant problems when patients had at least one insurance-related issue filling a prescription in the previous year
| Problem encountered | Percent of patients reporting problem* (n=100) |
|---|---|
| Adverse medical outcomes | |
| Missed doses of medication | 23 |
| Couldn’t get any medication | 19 |
| Medical condition got worse | 8 |
| New medication adverse effects | 6 |
| Had to go to emergency department | 5 |
| Overall any adverse outcome | 41 |
| Decreased patient satisfaction | |
| Got upset with insurance company | 44 |
| Got upset with pharmacist | 15 |
| Got upset with doctor | 12 |
| Overall any decreased satisfaction | 68 |
| Increased practice burden | |
| Had to wait for pharmacist authorization | 69 |
| Made extra phone calls to practice | 36 |
| Had to get a different medication | 36 |
| Had extra doctor visits | 13 |
| Overall any increased burden | 83 |
| *Patients reported, on average, 2.9 problems; therefore, categories exceed 100%. | |
Forty-one percent of patients reporting a problem experienced adverse medical outcomes. The most serious adverse medical outcomes were reported least often, but occurred nonetheless: worsening of medical condition (8%), new medication adverse effects (6%), and requiring a visit to the ED (5%). More commonly reported was decreased satisfaction with the health care system (68%). Patients were less likely to report being upset with their physician than their insurance company or pharmacist. Problems that burdened the physician practice were reported most frequently (83%).
TABLE 3 shows the medication categories that were affected when respondents reported at least one problem. Formulary changes or restrictions involving cardiac/hypertension/lipid and neurologic/psychiatric medications were linked to the most problems.
TABLE 3
Which medication categories were most affected when patients had a problem filling a prescription?
| Medication category | Frequency of occurrence |
|---|---|
| Cardiac/HTN/lipids | 23 |
| Neurologic/psychiatric | 23 |
| Metabolic/endocrine | 16 |
| Gastrointestinal | 15 |
| Pain | 13 |
| Respiratory | 11 |
| Other | 9 |
| Dermatologic | 7 |
| Total | 117* |
| HTN, hypertension. *Total exceeds 100 because some of the 100 patients had problems with medications in more than one category. | |
Discussion
Nearly one quarter of patients in our sample (23%) experienced problems caused by insurance constraints while they attempted to follow the treatment regimens prescribed by their physicians. Although the most commonly reported insurance-related problems (waiting for pharmacist authorization, making extra phone calls to the physician’s office) could be perceived as minor inconveniences, serious consequences were also common. Our study showed that patients who rely solely on Medicaid or Medicare bore the greatest burden of insurance-related obstacles when filling prescriptions, although others were also affected.
Consistent with prior research in Medicare and Medicaid populations, our study found that medication access restrictions can negatively affect patient adherence.13,16,17 Our study showed that 41% of patients who encountered a problem experienced a medically meaningful adverse outcome; 19% reported they received no medication for their condition. Similarly, a study of Medicare beneficiaries who had failed to fill or refill a prescription found that 20% cited lack of insurance coverage for the medication as a reason for not filling the prescription.17
In our study, 23% of patients reported missing doses of their medication due to insurance-related difficulties, and 8% reported a worsening of their medical condition. The increased costs associated with poor chronic care management are well documented.18 Less well described is the potential net savings produced when insurance formularies are adjusted to expand coverage and lower patient costs for prescription treatments for chronic conditions. In an analysis of cost data from the Pitney-Bowes Corporation, Mahoney12 revealed a significant net savings in health care costs and lost productivity when treatments for chronic conditions were moved to the lowest tier of the formulary, thereby making them available to health plan participants at the lowest cost.
We could not link patient reports of treatment disruptions empirically to medical outcomes or increased costs, due to the constraints of our research question and study design. However, it is reasonable to suggest that longer-term insurance costs for these patients could, in fact, negate any short-term cost savings generated from formulary restrictions. In particular, the 5% of our patient sample who reported using the ED as a consequence of an insurance-related disruption of their prescribed treatment likely added significant unnecessary cost to their treatment. This effect has been seen in other studies.19,20 In our study, cardiac/hypertension/lipid medications and medications for neurologic or psychiatric conditions were the most likely to be problematic. In these categories, competition of branded products may contribute to more frequent formulary changes. Furthermore, increases in morbidity and mortality associated with inadequate treatment of the conditions represented in these 2 categories of medications represent a significant burden to the US health system, including insurers, employers, and individuals.21-23
Although patients were less likely to report being upset with their physician than their insurance company or pharmacist, physicians bore a considerable burden for resolving a number of prevalent patient issues. Most of these problems required extra phone calls to the practice, additional medication authorization, or extra office visits. Physicians and their support staff may serve as buffers between patients and the insurance formulary rules, but at significant cost in their time and effort.
Electronic prescribing systems with real-time pharmacy benefit verification may provide additional efficiencies and help physicians and patients avoid some of the problems cited by our respondents. Providers with such systems receive immediate notification of formulary status, including tier and co-pay levels, which can aid in shared decision-making at the point of prescribing. Physicians without access to e-prescribing may want to use newer formulary search engines that can check formulary status of medications across multiple insurance plans. However, these electronic tools often fail to account for variations in formularies within the same insurance plan for different employers based on their benefit structure. Still, when a medication is not on formulary or a co-payment is required, the physician may be forced to play the role of apologist for the constraints imposed by the insurance formulary.
In cases where formularies restrict the patient’s potential access to a preferred treatment plan, the burden of prior authorizations continues to be borne by physicians. Coverage limitations lead to financial and medical consequences that must be managed in partnership with the patient. A system should be put in place by insurance companies that facilitates out-of-formulary authorizations to prevent lapses in patient care or deleterious changes in medical management.
Study limitations
The findings reported here should be interpreted in light of some limitations of this study. The response rate to our mailed patient survey was modest (36%), although typical for this method. The sex mix of respondents was similar to that of nonrespondents, but nonrespondents were slightly younger. Given that younger age is associated with a greater likelihood of experiencing a problem filling a medication, our findings may underestimate the frequency of this dilemma. In addition, our survey asked patients to recall events that occurred over the past year, introducing a potential for recall bias.
While the overall sample size was relatively small (n=428), it is close to the number calculated for sufficient power to conduct the analyses (n=480). Furthermore, data were collected from 3 distinct patient populations: urban, suburban and semirural. Although the scope of our study included only one geographic region, variability in practice setting lends some tentative support to the generalizability of the findings.
Looking forward
As a standard method to control costs and update treatment guidelines, insurance-mediated medication changes will continue to present unique challenges for patients and health care providers. Formulary changes burden the downstream delivery of medical care with expensive administrative responsibilities and disrupt effective disease management and prevention. Until insurance companies and pharmacy benefit managers start paying heed to total costs of care when contemplating formulary changes, physicians should try to identify formulary conflicts as early as possible in the prescribing process so as to save time for all parties later and improve compliance.
As practices proceed toward adoption of electronic health records, e-prescribing, and the Centers for Medicare & Medicaid Services’ “meaningful use” criteria, physicians may use systems that provide real-time formulary information, which can flag issues before the patient leaves the exam room. Future research should explore the ways formulary changes might be implemented to provide the strongest continuity of patient care with the least amount of cost shifting.
CORRESPONDENCE
Susan A. Flocke, PhD, CWRU Department of Family Medicine & Community Health, 11000 Cedar Avenue, Suite 402, Cleveland, OH 44106; [email protected]
1. Centers for Disease Control and Prevention National Center for Chronic Disease Prevention and Health Promotion. Chronic diseases: the power to prevent, the call to control: at a glance 2009. Available at: http://www.cdc.gov/chronicdisease/resources/publications/aag/chronic.htm. Page last updated December 17, 2009. Accessed June 21, 2012.
2. Mueller C, Schur C, O’Connell J. Prescription drug spending: the impact of age and chronic disease status. Am J Public Health. 1997;87:1626-1629.
3. Kaiser Family Foundation. Prescription drug trends. Available at: http://www.kff.org/rxdrugs/upload/3057_07.pdf. Published September 2008. Accessed December 1, 2009.
4. Neumann PJ. Evidence-based and value-based formulary guidelines. Health Aff (Millwood). 2004;23:124-134.
5. Simon GE, Psaty BM, Hrachovec JB, et al. Principles for evidence-based drug formulary policy. J Gen Intern Med. 2005;20:964-968.
6. Huskamp HA, Deverka PA, Epstein AM, et al. The effect of incentive-based formularies on prescription-drug utilization and spending. N Engl J Med. 2003;349:2224-2232.
7. Meissner B, Dickson M, Shinogle J, et al. Drug and medical cost effects of a drug formulary change with therapeutic interchange for statin drugs in a multistate managed Medicaid organization. J Manag Care Pharm. 2006;12:331-340.
8. Ovsag K, Hydery S, Mousa SA. Preferred drug lists: potential impact on healthcare economics. Vasc Health Risk Manag. 2008;4:403-413.
9. Raisch DW, Klaurens LM, Hayden C, et al. Impact of a formulary change in proton pump inhibitors on health care costs and patients’ symptoms. Dig Dis Sci. 2001;46:1533-1539.
10. Soumerai SB. Benefits and risks of increasing restrictions on access to costly drugs in Medicaid. Health Aff (Millwood). 2004;23:135-146.
11. Johnson TJ, Stahl-Moncada S. Medicaid prescription formulary restrictions and arthritis treatment costs. Am J Public Health. 2008;98:1300-1305.
12. Mahoney JJ. Reducing patient drug acquisition costs can lower diabetes health claims. Am J Manag Care. 2005;11(5 suppl):S170-S176.
13. Ridley DB, Axelsen KJ. Impact of Medicaid preferred drug lists on therapeutic adherence. Pharmacoeconomics. 2006;24 (suppl 3):65-78.
14. Steinman MA, Sands LP, Covinsky KE. Self-restriction of medications due to cost in seniors without prescription coverage. J Gen Intern Med. 2001;16:793-799.
15. Jean CD, Triplett JW. Investigating patient experiences after a formulary change. Am J Health Syst Pharm. 2000;57:1052-1054.
16. Wilson J, Axelsen K, Tang S. Medicaid prescription drug access restrictions: exploring the effect on patient persistence with hypertension medications. Am J Manag Care. 2005;11 (spec no):SP27-SP34.
17. Kennedy J, Tuleu I, Mackay K. Unfilled prescriptions of Medicare beneficiaries: prevalence, reasons, and types of medicines prescribed. J Manag Care Pharm. 2008;14:553-560.
18. Mensah GA, Brown DW. An overview of cardiovascular disease burden in the United States. Health Aff (Millwood). 2007;26:38-48.
19. Sokol MC, McGuigan KA, Verbrugge RR, et al. Impact of medication adherence on hospitalization risk and healthcare cost. Med Care. 2005;43:521-530.
20. Tamblyn R, Laprise R, Hanley JA, et al. Adverse events associated with prescription drug cost-sharing among poor and elderly persons. JAMA. 2001;285:421-429.
21. Flack JM, Casciano R, Casciano J, et al. Cardiovascular disease costs associated with uncontrolled hypertension. Manag Care Interface. 2002;15:28-36.
22. Hall RC, Wise MG. The clinical and financial burden of mood disorders. Cost and outcome. Psychosomatics. 1995;36:S11-S18.
23. McCombs JS, Nichol MB, Newman CM, et al. The costs of interrupting antihypertensive drug therapy in a Medicaid population. Med Care. 1994;32:214-226.
1. Centers for Disease Control and Prevention National Center for Chronic Disease Prevention and Health Promotion. Chronic diseases: the power to prevent, the call to control: at a glance 2009. Available at: http://www.cdc.gov/chronicdisease/resources/publications/aag/chronic.htm. Page last updated December 17, 2009. Accessed June 21, 2012.
2. Mueller C, Schur C, O’Connell J. Prescription drug spending: the impact of age and chronic disease status. Am J Public Health. 1997;87:1626-1629.
3. Kaiser Family Foundation. Prescription drug trends. Available at: http://www.kff.org/rxdrugs/upload/3057_07.pdf. Published September 2008. Accessed December 1, 2009.
4. Neumann PJ. Evidence-based and value-based formulary guidelines. Health Aff (Millwood). 2004;23:124-134.
5. Simon GE, Psaty BM, Hrachovec JB, et al. Principles for evidence-based drug formulary policy. J Gen Intern Med. 2005;20:964-968.
6. Huskamp HA, Deverka PA, Epstein AM, et al. The effect of incentive-based formularies on prescription-drug utilization and spending. N Engl J Med. 2003;349:2224-2232.
7. Meissner B, Dickson M, Shinogle J, et al. Drug and medical cost effects of a drug formulary change with therapeutic interchange for statin drugs in a multistate managed Medicaid organization. J Manag Care Pharm. 2006;12:331-340.
8. Ovsag K, Hydery S, Mousa SA. Preferred drug lists: potential impact on healthcare economics. Vasc Health Risk Manag. 2008;4:403-413.
9. Raisch DW, Klaurens LM, Hayden C, et al. Impact of a formulary change in proton pump inhibitors on health care costs and patients’ symptoms. Dig Dis Sci. 2001;46:1533-1539.
10. Soumerai SB. Benefits and risks of increasing restrictions on access to costly drugs in Medicaid. Health Aff (Millwood). 2004;23:135-146.
11. Johnson TJ, Stahl-Moncada S. Medicaid prescription formulary restrictions and arthritis treatment costs. Am J Public Health. 2008;98:1300-1305.
12. Mahoney JJ. Reducing patient drug acquisition costs can lower diabetes health claims. Am J Manag Care. 2005;11(5 suppl):S170-S176.
13. Ridley DB, Axelsen KJ. Impact of Medicaid preferred drug lists on therapeutic adherence. Pharmacoeconomics. 2006;24 (suppl 3):65-78.
14. Steinman MA, Sands LP, Covinsky KE. Self-restriction of medications due to cost in seniors without prescription coverage. J Gen Intern Med. 2001;16:793-799.
15. Jean CD, Triplett JW. Investigating patient experiences after a formulary change. Am J Health Syst Pharm. 2000;57:1052-1054.
16. Wilson J, Axelsen K, Tang S. Medicaid prescription drug access restrictions: exploring the effect on patient persistence with hypertension medications. Am J Manag Care. 2005;11 (spec no):SP27-SP34.
17. Kennedy J, Tuleu I, Mackay K. Unfilled prescriptions of Medicare beneficiaries: prevalence, reasons, and types of medicines prescribed. J Manag Care Pharm. 2008;14:553-560.
18. Mensah GA, Brown DW. An overview of cardiovascular disease burden in the United States. Health Aff (Millwood). 2007;26:38-48.
19. Sokol MC, McGuigan KA, Verbrugge RR, et al. Impact of medication adherence on hospitalization risk and healthcare cost. Med Care. 2005;43:521-530.
20. Tamblyn R, Laprise R, Hanley JA, et al. Adverse events associated with prescription drug cost-sharing among poor and elderly persons. JAMA. 2001;285:421-429.
21. Flack JM, Casciano R, Casciano J, et al. Cardiovascular disease costs associated with uncontrolled hypertension. Manag Care Interface. 2002;15:28-36.
22. Hall RC, Wise MG. The clinical and financial burden of mood disorders. Cost and outcome. Psychosomatics. 1995;36:S11-S18.
23. McCombs JS, Nichol MB, Newman CM, et al. The costs of interrupting antihypertensive drug therapy in a Medicaid population. Med Care. 1994;32:214-226.