User login
Marijuana smoking is an independent risk factor for lung disease in HIV+
Long-term marijuana smoking was associated with lung disease in HIV-infected (HIV+) but not HIV uninfected (HIV–) men who have sex with men (MSM), according to the results of a large, prospective cohort study.

“There were no significant interactions between marijuana and tobacco smoking in any multivariable model tested for HIV+ participants, indicating independent effects of these factors,” wrote David R. Lorenz, PhD, of the Dana-Farber Cancer Institute, Boston, and his colleagues.
These findings are especially important given that the proportion of HIV+ individuals who frequently smoke marijuana is higher than in the general population in the United States, and has increased in recent years, according to the report, published online in EClinicalMedicine.
The study examined 2,704 MSM who met eligibility criteria (1,352 HIV+ and 1,352 HIV− individuals), with a median age of 44 years at baseline and a median follow-up of 10.5 years. A total of 27% of HIV+ participants reported daily or weekly marijuana smoking for 1 year or more during follow-up, compared with 18% of the HIV− participants.
HIV+ participants who smoked marijuana were more likely to report one or more pulmonary diagnoses, versus nonsmoking HIV+ individuals during follow-up (41.0% vs. 30.0% infectious, and 24.8% vs. 19.0% noninfectious), according to the authors. In contrast, there was no association between marijuana smoking and either an infectious or noninfectious pulmonary diagnosis among HIV− participants (24.2% vs. 20.9%, and 14.8% vs. 17.7%, respectively).
For HIV+ individuals, each 10 days/month increase in marijuana smoking in the prior 2-year period was found to be associated with a 6% increased risk of infectious pulmonary diagnosis (hazard risk 1.06 [95% confidence interval 1.00-1.11]; P = .041). Overall, they found that from the 53,000 person-visits in the study, marijuana smoking was associated with increased risk of both infectious and noninfectious pulmonary diagnoses among the 1,352 HIV-infected participants independent of CD4 count, antiretroviral therapy (ART) adherence, and demographic factors as well.
In particular, viral suppression did not seem to interfere with this association between marijuana smoking and infectious pulmonary diagnoses, as it remained significant in models restricted to those person-visits with suppressed HIV viral load (HR 1.41 [1.03-1.91], P = .029).
The authors suggested that HIV-specific factors such as lung immune cell depletion and dysfunction, persistent immune cell activation, systemic inflammation, respiratory microbiome alterations, and oxidative stress, or a combination of these effects, may interact with the alveolar macrophage dysfunction seen in both humans and mouse models exposed to marijuana smoke. Thus, “a potential additive risk of marijuana smoking and HIV disease may explain the increased prevalence of infectious pulmonary diagnoses in our adjusted analyses,” Dr. Lorenz and his colleagues stated.
“These findings suggest that marijuana smoking is a modifiable risk factor that healthcare providers should consider when seeking to prevent or treat lung disease in people infected with HIV, particularly those with other known risk factors including heavy tobacco smoking, and low CD4 T cell count or advanced HIV disease,” they concluded.
The National Institutes of Health funded the study. The authors reported that they had no relevant disclosures.
SOURCE: Lorenz DR et al. EClinicalMedicine. 2019 Jan 24. doi: 10.1016/j.eclinm.2019.01.003.
Long-term marijuana smoking was associated with lung disease in HIV-infected (HIV+) but not HIV uninfected (HIV–) men who have sex with men (MSM), according to the results of a large, prospective cohort study.
“There were no significant interactions between marijuana and tobacco smoking in any multivariable model tested for HIV+ participants, indicating independent effects of these factors,” wrote David R. Lorenz, PhD, of the Dana-Farber Cancer Institute, Boston, and his colleagues.
These findings are especially important given that the proportion of HIV+ individuals who frequently smoke marijuana is higher than in the general population in the United States, and has increased in recent years, according to the report, published online in EClinicalMedicine.
The study examined 2,704 MSM who met eligibility criteria (1,352 HIV+ and 1,352 HIV− individuals), with a median age of 44 years at baseline and a median follow-up of 10.5 years. A total of 27% of HIV+ participants reported daily or weekly marijuana smoking for 1 year or more during follow-up, compared with 18% of the HIV− participants.
HIV+ participants who smoked marijuana were more likely to report one or more pulmonary diagnoses, versus nonsmoking HIV+ individuals during follow-up (41.0% vs. 30.0% infectious, and 24.8% vs. 19.0% noninfectious), according to the authors. In contrast, there was no association between marijuana smoking and either an infectious or noninfectious pulmonary diagnosis among HIV− participants (24.2% vs. 20.9%, and 14.8% vs. 17.7%, respectively).
For HIV+ individuals, each 10 days/month increase in marijuana smoking in the prior 2-year period was found to be associated with a 6% increased risk of infectious pulmonary diagnosis (hazard risk 1.06 [95% confidence interval 1.00-1.11]; P = .041). Overall, they found that from the 53,000 person-visits in the study, marijuana smoking was associated with increased risk of both infectious and noninfectious pulmonary diagnoses among the 1,352 HIV-infected participants independent of CD4 count, antiretroviral therapy (ART) adherence, and demographic factors as well.
In particular, viral suppression did not seem to interfere with this association between marijuana smoking and infectious pulmonary diagnoses, as it remained significant in models restricted to those person-visits with suppressed HIV viral load (HR 1.41 [1.03-1.91], P = .029).
The authors suggested that HIV-specific factors such as lung immune cell depletion and dysfunction, persistent immune cell activation, systemic inflammation, respiratory microbiome alterations, and oxidative stress, or a combination of these effects, may interact with the alveolar macrophage dysfunction seen in both humans and mouse models exposed to marijuana smoke. Thus, “a potential additive risk of marijuana smoking and HIV disease may explain the increased prevalence of infectious pulmonary diagnoses in our adjusted analyses,” Dr. Lorenz and his colleagues stated.
“These findings suggest that marijuana smoking is a modifiable risk factor that healthcare providers should consider when seeking to prevent or treat lung disease in people infected with HIV, particularly those with other known risk factors including heavy tobacco smoking, and low CD4 T cell count or advanced HIV disease,” they concluded.
The National Institutes of Health funded the study. The authors reported that they had no relevant disclosures.
SOURCE: Lorenz DR et al. EClinicalMedicine. 2019 Jan 24. doi: 10.1016/j.eclinm.2019.01.003.
Long-term marijuana smoking was associated with lung disease in HIV-infected (HIV+) but not HIV uninfected (HIV–) men who have sex with men (MSM), according to the results of a large, prospective cohort study.
“There were no significant interactions between marijuana and tobacco smoking in any multivariable model tested for HIV+ participants, indicating independent effects of these factors,” wrote David R. Lorenz, PhD, of the Dana-Farber Cancer Institute, Boston, and his colleagues.
These findings are especially important given that the proportion of HIV+ individuals who frequently smoke marijuana is higher than in the general population in the United States, and has increased in recent years, according to the report, published online in EClinicalMedicine.
The study examined 2,704 MSM who met eligibility criteria (1,352 HIV+ and 1,352 HIV− individuals), with a median age of 44 years at baseline and a median follow-up of 10.5 years. A total of 27% of HIV+ participants reported daily or weekly marijuana smoking for 1 year or more during follow-up, compared with 18% of the HIV− participants.
HIV+ participants who smoked marijuana were more likely to report one or more pulmonary diagnoses, versus nonsmoking HIV+ individuals during follow-up (41.0% vs. 30.0% infectious, and 24.8% vs. 19.0% noninfectious), according to the authors. In contrast, there was no association between marijuana smoking and either an infectious or noninfectious pulmonary diagnosis among HIV− participants (24.2% vs. 20.9%, and 14.8% vs. 17.7%, respectively).
For HIV+ individuals, each 10 days/month increase in marijuana smoking in the prior 2-year period was found to be associated with a 6% increased risk of infectious pulmonary diagnosis (hazard risk 1.06 [95% confidence interval 1.00-1.11]; P = .041). Overall, they found that from the 53,000 person-visits in the study, marijuana smoking was associated with increased risk of both infectious and noninfectious pulmonary diagnoses among the 1,352 HIV-infected participants independent of CD4 count, antiretroviral therapy (ART) adherence, and demographic factors as well.
In particular, viral suppression did not seem to interfere with this association between marijuana smoking and infectious pulmonary diagnoses, as it remained significant in models restricted to those person-visits with suppressed HIV viral load (HR 1.41 [1.03-1.91], P = .029).
The authors suggested that HIV-specific factors such as lung immune cell depletion and dysfunction, persistent immune cell activation, systemic inflammation, respiratory microbiome alterations, and oxidative stress, or a combination of these effects, may interact with the alveolar macrophage dysfunction seen in both humans and mouse models exposed to marijuana smoke. Thus, “a potential additive risk of marijuana smoking and HIV disease may explain the increased prevalence of infectious pulmonary diagnoses in our adjusted analyses,” Dr. Lorenz and his colleagues stated.
“These findings suggest that marijuana smoking is a modifiable risk factor that healthcare providers should consider when seeking to prevent or treat lung disease in people infected with HIV, particularly those with other known risk factors including heavy tobacco smoking, and low CD4 T cell count or advanced HIV disease,” they concluded.
The National Institutes of Health funded the study. The authors reported that they had no relevant disclosures.
SOURCE: Lorenz DR et al. EClinicalMedicine. 2019 Jan 24. doi: 10.1016/j.eclinm.2019.01.003.
FROM ECLINICALMEDICINE
Key clinical point: HIV+ but not HIV– marijuana smokers had an increased rate of pulmonary diagnoses.
Major finding: HIV+ marijuana smokers were more likely to report one or more infectious or noninfectious pulmonary diagnoses, compared with nonsmoking HIV+ individuals (41.0% vs. 30.0%, and 24.8% vs. 19.0%, respectively).
Study details: A prospective cohort study of 1,352 HIV+ vs. 1,352 HIV– men who have sex with men.
Disclosures: The National Institutes of Health funded the study. The authors reported that they had no relevant disclosures.
Source: Lorenz DR et al. EClinicalMedicine. 2019 Jan 24. doi: 10.1016/j.eclinm.2019.01.003.
Another look at overdiagnosis/remission of asthma
I appreciated the PURL, “Should you reassess your patient’s asthma diagnosis?” (J Fam Pract. 2018;67:704-707) that reminded clinicians to taper asthma controller medications in asymptomatic patients. The articles cited1,2 by Drs. Stevermer and Hayes documented that one-third of the adults enrolled in the respective study with physician-diagnosed asthma did not have objective evidence for asthma and were either over-diagnosed or had remitted. These articles also contained evidence that: 1) over-diagnosis was likely much more common than remission,1 and 2) there was a significant temporal trend towards increasing over-diagnosis/remission during the last several decades. The authors of the cited article1 suggested that the temporal trend could be explained by increased public awareness of respiratory symptoms, more aggressive marketing of asthma medications, and a lack of objective measurement of reversible airway obstruction in primary care. These assertions deserve careful consideration as we strive to diagnose asthma appropriately.
Over-diagnosis/remission is almost certainly not as prevalent (33%) as the authors of the cited articles1,2 reported. The reason is simple selection bias: 1) the cited study2 excluded asthma patients who smoked >10 pack-years (it enrolled 701 asthma patients and excluded 812 asthma patients with a >10 pack-year smoking history), and 2) this study likely did not include asthma patients with the asthma-COPD overlap syndrome, which is treated as asthma and comprises an additional 30% of our patients with chronic airflow limitation (the asthma-COPD spectrum).3 Asthma patients who smoke and/or have the overlap syndrome are prone to severe asthma that is refractory to inhaled corticosteroids.3,4
In addition to making the correct diagnosis, it is equally important to be aware of efficacious therapies for severe refractory asthma that primary care clinicians can easily use. There is now good evidence that azithromycin is efficacious for severe refractory asthma5 and should be considered prior to referral for immunomodulatory asthma therapies.6
David L. Hahn, MD, MS
Madison, Wis
1. Aaron SD, Vandemheen KL, Boulet LP, et al; Canadian Respiratory Clinical Research Consortium. Overdiagnosis of asthma in obese and nonobese adults. CMAJ. 2008;179:1121-1131.
2. Aaron SD, Vandemheen KL, FitzGerald JM, et al; Canadian Respiratory Research Network. Reevaluation of diagnosis in adults with physician-diagnosed asthma. JAMA. 2017;317:269-279.
3. Gibson PG, Simpson JL. The overlap syndrome of asthma and COPD: what are its features and how important is it? Thorax. 2009;64:728-735.
4. Stapleton M, Howard-Thompson A, George C, et al. Smoking and asthma. J Am Board Fam Med. 2011;24;313-322.
5. Gibson PG, Yang IA, Upham JW, et al. Effect of azithromycin on asthma exacerbations and quality of life in adults with persistent uncontrolled asthma (AMAZES): a randomised, double-blind, placebo-controlled trial. Lancet. 2017:390659-668.
6. Hahn DL, Grasmick M, Hetzel S, et al; AZMATICS (AZithroMycin-Asthma Trial In Community Settings) Study Group. Azithromycin for bronchial asthma in adults: an effectiveness trial. J Am Board Fam Med. 2012;25:442-459.
Continue to: Authors' response...
Authors’ response:
We appreciate Dr. Hahn’s observations about the PURL1 on overdiagnosis of asthma. This article focused on the results of a prospective, multicenter cohort study2 that evaluated the feasibility of tapering, and in many patients, stopping asthma medications. We agree that if the study had included people diagnosed with asthma who also had smoked at least 10 pack-years or who also had COPD, the proportion of those who would eventually no longer meet diagnostic criteria for asthma would be lower than in this study. We are uncertain of the relative proportion of cases that were overdiagnosis, when compared with true remission of disease, as only 43% of those no longer meeting the diagnostic criteria for asthma had evidence of prior lung function testing, whether by formal spirometry, serial peak function testing, or bronchial challenge testing.
We agree that using efficacious therapies for severe refractory asthma is essential, but the selection of those therapies was outside the scope of this PURL.
James J. Stevermer, MD, MSPH; Alisa Hayes, MD
Columbia, Mo
1. Stevermer JJ, Hayes A. Should you reassess your patient’s asthma diagnosis? J Fam Pract. 2018;67:704-707.
2. Aaron SD, Vandemheen KL, FitzGerald JM, et al; Canadian Respiratory Research Network. Reevaluation of diagnosis in adults with physician-diagnosed asthma. JAMA. 2017;317:269-279.
I appreciated the PURL, “Should you reassess your patient’s asthma diagnosis?” (J Fam Pract. 2018;67:704-707) that reminded clinicians to taper asthma controller medications in asymptomatic patients. The articles cited1,2 by Drs. Stevermer and Hayes documented that one-third of the adults enrolled in the respective study with physician-diagnosed asthma did not have objective evidence for asthma and were either over-diagnosed or had remitted. These articles also contained evidence that: 1) over-diagnosis was likely much more common than remission,1 and 2) there was a significant temporal trend towards increasing over-diagnosis/remission during the last several decades. The authors of the cited article1 suggested that the temporal trend could be explained by increased public awareness of respiratory symptoms, more aggressive marketing of asthma medications, and a lack of objective measurement of reversible airway obstruction in primary care. These assertions deserve careful consideration as we strive to diagnose asthma appropriately.
Over-diagnosis/remission is almost certainly not as prevalent (33%) as the authors of the cited articles1,2 reported. The reason is simple selection bias: 1) the cited study2 excluded asthma patients who smoked >10 pack-years (it enrolled 701 asthma patients and excluded 812 asthma patients with a >10 pack-year smoking history), and 2) this study likely did not include asthma patients with the asthma-COPD overlap syndrome, which is treated as asthma and comprises an additional 30% of our patients with chronic airflow limitation (the asthma-COPD spectrum).3 Asthma patients who smoke and/or have the overlap syndrome are prone to severe asthma that is refractory to inhaled corticosteroids.3,4
In addition to making the correct diagnosis, it is equally important to be aware of efficacious therapies for severe refractory asthma that primary care clinicians can easily use. There is now good evidence that azithromycin is efficacious for severe refractory asthma5 and should be considered prior to referral for immunomodulatory asthma therapies.6
David L. Hahn, MD, MS
Madison, Wis
1. Aaron SD, Vandemheen KL, Boulet LP, et al; Canadian Respiratory Clinical Research Consortium. Overdiagnosis of asthma in obese and nonobese adults. CMAJ. 2008;179:1121-1131.
2. Aaron SD, Vandemheen KL, FitzGerald JM, et al; Canadian Respiratory Research Network. Reevaluation of diagnosis in adults with physician-diagnosed asthma. JAMA. 2017;317:269-279.
3. Gibson PG, Simpson JL. The overlap syndrome of asthma and COPD: what are its features and how important is it? Thorax. 2009;64:728-735.
4. Stapleton M, Howard-Thompson A, George C, et al. Smoking and asthma. J Am Board Fam Med. 2011;24;313-322.
5. Gibson PG, Yang IA, Upham JW, et al. Effect of azithromycin on asthma exacerbations and quality of life in adults with persistent uncontrolled asthma (AMAZES): a randomised, double-blind, placebo-controlled trial. Lancet. 2017:390659-668.
6. Hahn DL, Grasmick M, Hetzel S, et al; AZMATICS (AZithroMycin-Asthma Trial In Community Settings) Study Group. Azithromycin for bronchial asthma in adults: an effectiveness trial. J Am Board Fam Med. 2012;25:442-459.
Continue to: Authors' response...
Authors’ response:
We appreciate Dr. Hahn’s observations about the PURL1 on overdiagnosis of asthma. This article focused on the results of a prospective, multicenter cohort study2 that evaluated the feasibility of tapering, and in many patients, stopping asthma medications. We agree that if the study had included people diagnosed with asthma who also had smoked at least 10 pack-years or who also had COPD, the proportion of those who would eventually no longer meet diagnostic criteria for asthma would be lower than in this study. We are uncertain of the relative proportion of cases that were overdiagnosis, when compared with true remission of disease, as only 43% of those no longer meeting the diagnostic criteria for asthma had evidence of prior lung function testing, whether by formal spirometry, serial peak function testing, or bronchial challenge testing.
We agree that using efficacious therapies for severe refractory asthma is essential, but the selection of those therapies was outside the scope of this PURL.
James J. Stevermer, MD, MSPH; Alisa Hayes, MD
Columbia, Mo
1. Stevermer JJ, Hayes A. Should you reassess your patient’s asthma diagnosis? J Fam Pract. 2018;67:704-707.
2. Aaron SD, Vandemheen KL, FitzGerald JM, et al; Canadian Respiratory Research Network. Reevaluation of diagnosis in adults with physician-diagnosed asthma. JAMA. 2017;317:269-279.
I appreciated the PURL, “Should you reassess your patient’s asthma diagnosis?” (J Fam Pract. 2018;67:704-707) that reminded clinicians to taper asthma controller medications in asymptomatic patients. The articles cited1,2 by Drs. Stevermer and Hayes documented that one-third of the adults enrolled in the respective study with physician-diagnosed asthma did not have objective evidence for asthma and were either over-diagnosed or had remitted. These articles also contained evidence that: 1) over-diagnosis was likely much more common than remission,1 and 2) there was a significant temporal trend towards increasing over-diagnosis/remission during the last several decades. The authors of the cited article1 suggested that the temporal trend could be explained by increased public awareness of respiratory symptoms, more aggressive marketing of asthma medications, and a lack of objective measurement of reversible airway obstruction in primary care. These assertions deserve careful consideration as we strive to diagnose asthma appropriately.
Over-diagnosis/remission is almost certainly not as prevalent (33%) as the authors of the cited articles1,2 reported. The reason is simple selection bias: 1) the cited study2 excluded asthma patients who smoked >10 pack-years (it enrolled 701 asthma patients and excluded 812 asthma patients with a >10 pack-year smoking history), and 2) this study likely did not include asthma patients with the asthma-COPD overlap syndrome, which is treated as asthma and comprises an additional 30% of our patients with chronic airflow limitation (the asthma-COPD spectrum).3 Asthma patients who smoke and/or have the overlap syndrome are prone to severe asthma that is refractory to inhaled corticosteroids.3,4
In addition to making the correct diagnosis, it is equally important to be aware of efficacious therapies for severe refractory asthma that primary care clinicians can easily use. There is now good evidence that azithromycin is efficacious for severe refractory asthma5 and should be considered prior to referral for immunomodulatory asthma therapies.6
David L. Hahn, MD, MS
Madison, Wis
1. Aaron SD, Vandemheen KL, Boulet LP, et al; Canadian Respiratory Clinical Research Consortium. Overdiagnosis of asthma in obese and nonobese adults. CMAJ. 2008;179:1121-1131.
2. Aaron SD, Vandemheen KL, FitzGerald JM, et al; Canadian Respiratory Research Network. Reevaluation of diagnosis in adults with physician-diagnosed asthma. JAMA. 2017;317:269-279.
3. Gibson PG, Simpson JL. The overlap syndrome of asthma and COPD: what are its features and how important is it? Thorax. 2009;64:728-735.
4. Stapleton M, Howard-Thompson A, George C, et al. Smoking and asthma. J Am Board Fam Med. 2011;24;313-322.
5. Gibson PG, Yang IA, Upham JW, et al. Effect of azithromycin on asthma exacerbations and quality of life in adults with persistent uncontrolled asthma (AMAZES): a randomised, double-blind, placebo-controlled trial. Lancet. 2017:390659-668.
6. Hahn DL, Grasmick M, Hetzel S, et al; AZMATICS (AZithroMycin-Asthma Trial In Community Settings) Study Group. Azithromycin for bronchial asthma in adults: an effectiveness trial. J Am Board Fam Med. 2012;25:442-459.
Continue to: Authors' response...
Authors’ response:
We appreciate Dr. Hahn’s observations about the PURL1 on overdiagnosis of asthma. This article focused on the results of a prospective, multicenter cohort study2 that evaluated the feasibility of tapering, and in many patients, stopping asthma medications. We agree that if the study had included people diagnosed with asthma who also had smoked at least 10 pack-years or who also had COPD, the proportion of those who would eventually no longer meet diagnostic criteria for asthma would be lower than in this study. We are uncertain of the relative proportion of cases that were overdiagnosis, when compared with true remission of disease, as only 43% of those no longer meeting the diagnostic criteria for asthma had evidence of prior lung function testing, whether by formal spirometry, serial peak function testing, or bronchial challenge testing.
We agree that using efficacious therapies for severe refractory asthma is essential, but the selection of those therapies was outside the scope of this PURL.
James J. Stevermer, MD, MSPH; Alisa Hayes, MD
Columbia, Mo
1. Stevermer JJ, Hayes A. Should you reassess your patient’s asthma diagnosis? J Fam Pract. 2018;67:704-707.
2. Aaron SD, Vandemheen KL, FitzGerald JM, et al; Canadian Respiratory Research Network. Reevaluation of diagnosis in adults with physician-diagnosed asthma. JAMA. 2017;317:269-279.
Does left atrial appendage closure reduce stroke rates as well as oral anticoagulants and antiplatelet meds in A-fib patients?
EVIDENCE SUMMARY
A 2017 network meta-analysis included 19 RCTs and 87,831 patients receiving anticoagulation, antiplatelet therapy, or LAAC for NVAF.1 LAAC was superior to antiplatelet therapy (hazard ratio [HR]=0.44; 95% confidence interval [CI], 0.23-0.86; P<.05) and similar to NOACs (HR=1.01; 95% CI, 0.53-1.92; P=.969) for reducing risk of stroke.
LAAC and NOACs found “most effective”
A network meta-analysis of 21 RCTs, which included data from 96,017 patients, examined the effectiveness of 7 interventions to prevent stroke in patients with NVAF: 4 NOACs, VKA, aspirin, and LAAC; the analysis compared VKA with the other interventions.2 The 2 trials that investigated LAAC accounted for only 1114 patients.
When the 7 interventions were ranked simultaneously on 2 efficacy outcomes (stroke/systemic embolism and all-cause mortality), all 4 NOACs and LAAC clustered together as “the most effective and lifesaving.”
Fewer hemorrhagic strokes with LAAC than VKA
A 2016 meta-analysis of 6 RCTs compared risk of stroke for adults with NVAF who received LAAC, VKA, or NOACs.3 No significant differences were found between NOACs and VKA or LAAC and VKA. The LAAC group had a significantly smaller number of patients.
A 2015 meta-analysis of 2406 patients with NVAF found that patients who received LAAC had significantly fewer hemorrhagic strokes (HR=0.22; P<.05) than patients who received VKA.4 No differences in all-cause stroke were found between the 2 groups during an average follow-up of 2.69 years.
LAAC found superior to warfarin for stroke prevention in one trial
A 2014 multicenter, randomized study (PROTECT-AF) of 707 patients with NVAF plus 1 additional stroke risk factor compared LAAC with VKA (warfarin).5 LAAC met criteria at 3.8 years for both noninferiority and superiority in preventing stroke, based on 2.3 events per 100 patient-years compared with 3.8 events per 100 patient-years for VKA. The number needed to treat with LAAC was 67 to result in 1 less event per patient-year.
A 2014 RCT (PREVAIL) evaluated patients with NVAF plus 1 additional stroke risk factor. LAAC was noninferior to warfarin for ischemic stroke prevention.6
Continue to: RECOMMENDATIONS
RECOMMENDATIONS
The American College of Cardiology (ACC) recommends LAAC for patients with NVAF who are not candidates for long-term anticoagulation.7 Similarly, the 2016 European Society of Cardiology guidelines issued a Class IIb recommendation for LAAC for stroke prevention in those with contraindications for long-term anticoagulation.8 Lastly, in a 2014 guideline, the American Heart Association, ACC, and the Heart Rhythm Society issued a Class IIb recommendation for surgical excision of the left atrial appendage in patients with atrial fibrillation undergoing cardiac surgery, but did not provide recommendations regarding LAAC.9
1. Sahay S, Nombela-Franco L, Rodes-Cabau J, et al. Efficacy and safety of left atrial appendage closure versus medical treatment in atrial fibrillation: a network meta-analysis from randomised trials. Heart. 2017;103:139-147.
2. Tereshchenko LG, Henrikson CA, Cigarroa, J, et al. Comparative effectiveness of interventions for stroke prevention in atrial fibrillation: a network meta-analysis. J Am Heart Assoc. 2016; 5:e003206.
3. Bajaj NS, Kalra R, Patel N, et al. Comparison of approaches for stroke prophylaxis in patients with non-valvular atrial fibrillation: network meta-analyses of randomized clinical trials. PLoS One. 2016;11:e0163608.
4. Holmes DR Jr, Doshi SK, Kar S, et al. Left atrial appendage closure as an alternative to warfarin for stroke prevention in atrial fibrillation: a patient-level meta-analysis. J Am Coll Cardiol. 2015;65:2614-2623.
5. Reddy VY, Sievert H, Halperin J, et al. Percutaneous left atrial appendage closure vs warfarin for atrial fibrillation: a randomized clinical trial. JAMA. 2014;312:1988-1998.
6. Holmes DR Jr, Kar S, Price MJ, et al. Prospective randomized evaluation of the Watchman Left Atrial Appendage Closure device in patients with atrial fibrillation versus long-term warfarin therapy: the PREVAIL trial. J Am Coll Cardiol. 2014;64:1-12.
7. Panaich S, Holmes DR. Left atrial appendage occlusion: Expert analysis. http://www.acc.org/latest-in-cardiology/articles/2017/ 01/31/13/08/left-atrial-appendage-occlusion. Accessed April 5, 2018.
8. Kirchof P, Benussi S, Kotecha D, et al. 2016 ESC guidelines for management of atrial fibrillation developed in collaboration with EACTS. Europace. 2016;18:1609-1678.
9. January CT, Wann LS, Alpert LS, et al. 2014 AHA/ACC/HRS guideline for the management of patients with atrial fibrillation: executive summary. JACC. 2014;64:2246-2280.
EVIDENCE SUMMARY
A 2017 network meta-analysis included 19 RCTs and 87,831 patients receiving anticoagulation, antiplatelet therapy, or LAAC for NVAF.1 LAAC was superior to antiplatelet therapy (hazard ratio [HR]=0.44; 95% confidence interval [CI], 0.23-0.86; P<.05) and similar to NOACs (HR=1.01; 95% CI, 0.53-1.92; P=.969) for reducing risk of stroke.
LAAC and NOACs found “most effective”
A network meta-analysis of 21 RCTs, which included data from 96,017 patients, examined the effectiveness of 7 interventions to prevent stroke in patients with NVAF: 4 NOACs, VKA, aspirin, and LAAC; the analysis compared VKA with the other interventions.2 The 2 trials that investigated LAAC accounted for only 1114 patients.
When the 7 interventions were ranked simultaneously on 2 efficacy outcomes (stroke/systemic embolism and all-cause mortality), all 4 NOACs and LAAC clustered together as “the most effective and lifesaving.”
Fewer hemorrhagic strokes with LAAC than VKA
A 2016 meta-analysis of 6 RCTs compared risk of stroke for adults with NVAF who received LAAC, VKA, or NOACs.3 No significant differences were found between NOACs and VKA or LAAC and VKA. The LAAC group had a significantly smaller number of patients.
A 2015 meta-analysis of 2406 patients with NVAF found that patients who received LAAC had significantly fewer hemorrhagic strokes (HR=0.22; P<.05) than patients who received VKA.4 No differences in all-cause stroke were found between the 2 groups during an average follow-up of 2.69 years.
LAAC found superior to warfarin for stroke prevention in one trial
A 2014 multicenter, randomized study (PROTECT-AF) of 707 patients with NVAF plus 1 additional stroke risk factor compared LAAC with VKA (warfarin).5 LAAC met criteria at 3.8 years for both noninferiority and superiority in preventing stroke, based on 2.3 events per 100 patient-years compared with 3.8 events per 100 patient-years for VKA. The number needed to treat with LAAC was 67 to result in 1 less event per patient-year.
A 2014 RCT (PREVAIL) evaluated patients with NVAF plus 1 additional stroke risk factor. LAAC was noninferior to warfarin for ischemic stroke prevention.6
Continue to: RECOMMENDATIONS
RECOMMENDATIONS
The American College of Cardiology (ACC) recommends LAAC for patients with NVAF who are not candidates for long-term anticoagulation.7 Similarly, the 2016 European Society of Cardiology guidelines issued a Class IIb recommendation for LAAC for stroke prevention in those with contraindications for long-term anticoagulation.8 Lastly, in a 2014 guideline, the American Heart Association, ACC, and the Heart Rhythm Society issued a Class IIb recommendation for surgical excision of the left atrial appendage in patients with atrial fibrillation undergoing cardiac surgery, but did not provide recommendations regarding LAAC.9
EVIDENCE SUMMARY
A 2017 network meta-analysis included 19 RCTs and 87,831 patients receiving anticoagulation, antiplatelet therapy, or LAAC for NVAF.1 LAAC was superior to antiplatelet therapy (hazard ratio [HR]=0.44; 95% confidence interval [CI], 0.23-0.86; P<.05) and similar to NOACs (HR=1.01; 95% CI, 0.53-1.92; P=.969) for reducing risk of stroke.
LAAC and NOACs found “most effective”
A network meta-analysis of 21 RCTs, which included data from 96,017 patients, examined the effectiveness of 7 interventions to prevent stroke in patients with NVAF: 4 NOACs, VKA, aspirin, and LAAC; the analysis compared VKA with the other interventions.2 The 2 trials that investigated LAAC accounted for only 1114 patients.
When the 7 interventions were ranked simultaneously on 2 efficacy outcomes (stroke/systemic embolism and all-cause mortality), all 4 NOACs and LAAC clustered together as “the most effective and lifesaving.”
Fewer hemorrhagic strokes with LAAC than VKA
A 2016 meta-analysis of 6 RCTs compared risk of stroke for adults with NVAF who received LAAC, VKA, or NOACs.3 No significant differences were found between NOACs and VKA or LAAC and VKA. The LAAC group had a significantly smaller number of patients.
A 2015 meta-analysis of 2406 patients with NVAF found that patients who received LAAC had significantly fewer hemorrhagic strokes (HR=0.22; P<.05) than patients who received VKA.4 No differences in all-cause stroke were found between the 2 groups during an average follow-up of 2.69 years.
LAAC found superior to warfarin for stroke prevention in one trial
A 2014 multicenter, randomized study (PROTECT-AF) of 707 patients with NVAF plus 1 additional stroke risk factor compared LAAC with VKA (warfarin).5 LAAC met criteria at 3.8 years for both noninferiority and superiority in preventing stroke, based on 2.3 events per 100 patient-years compared with 3.8 events per 100 patient-years for VKA. The number needed to treat with LAAC was 67 to result in 1 less event per patient-year.
A 2014 RCT (PREVAIL) evaluated patients with NVAF plus 1 additional stroke risk factor. LAAC was noninferior to warfarin for ischemic stroke prevention.6
Continue to: RECOMMENDATIONS
RECOMMENDATIONS
The American College of Cardiology (ACC) recommends LAAC for patients with NVAF who are not candidates for long-term anticoagulation.7 Similarly, the 2016 European Society of Cardiology guidelines issued a Class IIb recommendation for LAAC for stroke prevention in those with contraindications for long-term anticoagulation.8 Lastly, in a 2014 guideline, the American Heart Association, ACC, and the Heart Rhythm Society issued a Class IIb recommendation for surgical excision of the left atrial appendage in patients with atrial fibrillation undergoing cardiac surgery, but did not provide recommendations regarding LAAC.9
1. Sahay S, Nombela-Franco L, Rodes-Cabau J, et al. Efficacy and safety of left atrial appendage closure versus medical treatment in atrial fibrillation: a network meta-analysis from randomised trials. Heart. 2017;103:139-147.
2. Tereshchenko LG, Henrikson CA, Cigarroa, J, et al. Comparative effectiveness of interventions for stroke prevention in atrial fibrillation: a network meta-analysis. J Am Heart Assoc. 2016; 5:e003206.
3. Bajaj NS, Kalra R, Patel N, et al. Comparison of approaches for stroke prophylaxis in patients with non-valvular atrial fibrillation: network meta-analyses of randomized clinical trials. PLoS One. 2016;11:e0163608.
4. Holmes DR Jr, Doshi SK, Kar S, et al. Left atrial appendage closure as an alternative to warfarin for stroke prevention in atrial fibrillation: a patient-level meta-analysis. J Am Coll Cardiol. 2015;65:2614-2623.
5. Reddy VY, Sievert H, Halperin J, et al. Percutaneous left atrial appendage closure vs warfarin for atrial fibrillation: a randomized clinical trial. JAMA. 2014;312:1988-1998.
6. Holmes DR Jr, Kar S, Price MJ, et al. Prospective randomized evaluation of the Watchman Left Atrial Appendage Closure device in patients with atrial fibrillation versus long-term warfarin therapy: the PREVAIL trial. J Am Coll Cardiol. 2014;64:1-12.
7. Panaich S, Holmes DR. Left atrial appendage occlusion: Expert analysis. http://www.acc.org/latest-in-cardiology/articles/2017/ 01/31/13/08/left-atrial-appendage-occlusion. Accessed April 5, 2018.
8. Kirchof P, Benussi S, Kotecha D, et al. 2016 ESC guidelines for management of atrial fibrillation developed in collaboration with EACTS. Europace. 2016;18:1609-1678.
9. January CT, Wann LS, Alpert LS, et al. 2014 AHA/ACC/HRS guideline for the management of patients with atrial fibrillation: executive summary. JACC. 2014;64:2246-2280.
1. Sahay S, Nombela-Franco L, Rodes-Cabau J, et al. Efficacy and safety of left atrial appendage closure versus medical treatment in atrial fibrillation: a network meta-analysis from randomised trials. Heart. 2017;103:139-147.
2. Tereshchenko LG, Henrikson CA, Cigarroa, J, et al. Comparative effectiveness of interventions for stroke prevention in atrial fibrillation: a network meta-analysis. J Am Heart Assoc. 2016; 5:e003206.
3. Bajaj NS, Kalra R, Patel N, et al. Comparison of approaches for stroke prophylaxis in patients with non-valvular atrial fibrillation: network meta-analyses of randomized clinical trials. PLoS One. 2016;11:e0163608.
4. Holmes DR Jr, Doshi SK, Kar S, et al. Left atrial appendage closure as an alternative to warfarin for stroke prevention in atrial fibrillation: a patient-level meta-analysis. J Am Coll Cardiol. 2015;65:2614-2623.
5. Reddy VY, Sievert H, Halperin J, et al. Percutaneous left atrial appendage closure vs warfarin for atrial fibrillation: a randomized clinical trial. JAMA. 2014;312:1988-1998.
6. Holmes DR Jr, Kar S, Price MJ, et al. Prospective randomized evaluation of the Watchman Left Atrial Appendage Closure device in patients with atrial fibrillation versus long-term warfarin therapy: the PREVAIL trial. J Am Coll Cardiol. 2014;64:1-12.
7. Panaich S, Holmes DR. Left atrial appendage occlusion: Expert analysis. http://www.acc.org/latest-in-cardiology/articles/2017/ 01/31/13/08/left-atrial-appendage-occlusion. Accessed April 5, 2018.
8. Kirchof P, Benussi S, Kotecha D, et al. 2016 ESC guidelines for management of atrial fibrillation developed in collaboration with EACTS. Europace. 2016;18:1609-1678.
9. January CT, Wann LS, Alpert LS, et al. 2014 AHA/ACC/HRS guideline for the management of patients with atrial fibrillation: executive summary. JACC. 2014;64:2246-2280.
EVIDENCE-BASED ANSWER:
Yes. Left atrial appendage closure (LAAC) with the Watchman device is noninferior to vitamin K antagonists (VKAs) and non-VKA oral anticoagulants (NOACs) for adults with nonvalvular atrial fibrillation (NVAF) and 1 additional stroke risk factor (strength of recommendation [SOR]: A, multiple meta-analyses).
LAAC has consistently been shown to be superior to antiplatelet therapy (SOR: A, single meta-analysis). One randomized controlled trial (RCT) demonstrated superiority of LAAC to VKA (SOR: B, single RCT).

Did this COPD Clinical Inquiry miss the mark—or not?
In the Clinical Inquiry, “Does prophylactic azithromycin reduce the number of COPD exacerbations or hospitalizations?” (J Fam Pract. 2018;67:384-385), Lyon et al state that azithromycin “doesn’t benefit patients ≤65 years, patients with GOLD [Global Initiative for Obstructive Lung Disease] stage IV COPD [chronic obstructive pulmonary disease], current smokers, or patients not using oxygen (strength of recommendation [SOR]: B, randomized controlled trials [RCTs]).” These categorical statements are misleading, and clinicians should ignore most of them when considering azithromycin for their patients with severe COPD.
The authors cited groups that were identified in a posthoc analysis1 of the only large trial involving azithromycin for the treatment of COPD to date.2P values for the interaction of azithromycin with GOLD stage (P=.04), smoking (P=.03), and age (P=.02) were significant, but the mean effects (hazard ratios [HRs]) for GOLD stage IV, smoking, and age ≤65 were .84, .99, and .84, respectively. It would be more accurate to say that there may be a diminished efficacy of azithromycin for patients with GOLD IV COPD and age ≤65 years. Only smokers appear to show no response, although the lower end of the 95% confidence interval was 0.71. The P value for the interaction of azithromycin with no long-term oxygen use (P=.23) was not significant, and it is incorrect to infer that oxygen use or nonuse predicts response.
The authors correctly state that the “significance of the results is limited because the study was not originally powered for this level of subgroup analysis,” but this statement is buried later in the article.
David L. Hahn, MD, MS
Madison, Wis
1. Han MK, Tayob N, Murray S, et al. Predictors of chronic obstructive pulmonary disease exacerbation reduction in response to daily azithromycin therapy. Am J Respir Crit Care Med. 2014;189:1503-1508.
2. Albert RK, Connett J, Bailey WC, et al. Azithromycin for prevention of exacerbations of COPD. N Engl J Med. 2011;365:689-698.
Continue to: Author's response...
Author’s response:
Your statement that the evidence-based answer regarding the lack of benefit of azithromycin in patients ≤65 years of age, with stage IV COPD, current smokers, and patients not using oxygen is “misleading” is a bit of an overstatement.
It is fair to say, however, that our statement regarding lack of efficacy among these subgroups of patients should be softened a bit since the data are from subgroup analyses, which should never be the source of definitive conclusions. And you point out that the 95% confidence intervals [CIs] of the HRs for these subgroups of patients do not include a potentially significant effect (0.68, 0.71, 0.61, and 0.65, respectively), so it is possible there is a Type II error, which would lead one to conclude there is no effect for these subgroups when there is one.
Regarding oxygen therapy, in this Clinical Inquiry, we presented data from the direct subgroup analysis, which revealed no difference in COPD exacerbations between the azithromycin and placebo groups for patients not receiving long-term supplemental oxygen (HR=0.80; 95% CI, 0.62-1.03); however, you are correct to point out that the oxygen use subgroup interaction (patients on oxygen vs patients not on oxygen), which we did not include in this Clinical Inquiry, did not reach significance (P=.23), casting some doubt on the authors’ conclusion of no effect for patients not on oxygen.
On the whole, I feel this Clinical Inquiry accurately summarized the existing evidence and that additional research is needed to better define the utility of azithromycin in these subgroups of patients.
Corey Lyon, DO
Denver, Colo
In the Clinical Inquiry, “Does prophylactic azithromycin reduce the number of COPD exacerbations or hospitalizations?” (J Fam Pract. 2018;67:384-385), Lyon et al state that azithromycin “doesn’t benefit patients ≤65 years, patients with GOLD [Global Initiative for Obstructive Lung Disease] stage IV COPD [chronic obstructive pulmonary disease], current smokers, or patients not using oxygen (strength of recommendation [SOR]: B, randomized controlled trials [RCTs]).” These categorical statements are misleading, and clinicians should ignore most of them when considering azithromycin for their patients with severe COPD.
The authors cited groups that were identified in a posthoc analysis1 of the only large trial involving azithromycin for the treatment of COPD to date.2P values for the interaction of azithromycin with GOLD stage (P=.04), smoking (P=.03), and age (P=.02) were significant, but the mean effects (hazard ratios [HRs]) for GOLD stage IV, smoking, and age ≤65 were .84, .99, and .84, respectively. It would be more accurate to say that there may be a diminished efficacy of azithromycin for patients with GOLD IV COPD and age ≤65 years. Only smokers appear to show no response, although the lower end of the 95% confidence interval was 0.71. The P value for the interaction of azithromycin with no long-term oxygen use (P=.23) was not significant, and it is incorrect to infer that oxygen use or nonuse predicts response.
The authors correctly state that the “significance of the results is limited because the study was not originally powered for this level of subgroup analysis,” but this statement is buried later in the article.
David L. Hahn, MD, MS
Madison, Wis
1. Han MK, Tayob N, Murray S, et al. Predictors of chronic obstructive pulmonary disease exacerbation reduction in response to daily azithromycin therapy. Am J Respir Crit Care Med. 2014;189:1503-1508.
2. Albert RK, Connett J, Bailey WC, et al. Azithromycin for prevention of exacerbations of COPD. N Engl J Med. 2011;365:689-698.
Continue to: Author's response...
Author’s response:
Your statement that the evidence-based answer regarding the lack of benefit of azithromycin in patients ≤65 years of age, with stage IV COPD, current smokers, and patients not using oxygen is “misleading” is a bit of an overstatement.
It is fair to say, however, that our statement regarding lack of efficacy among these subgroups of patients should be softened a bit since the data are from subgroup analyses, which should never be the source of definitive conclusions. And you point out that the 95% confidence intervals [CIs] of the HRs for these subgroups of patients do not include a potentially significant effect (0.68, 0.71, 0.61, and 0.65, respectively), so it is possible there is a Type II error, which would lead one to conclude there is no effect for these subgroups when there is one.
Regarding oxygen therapy, in this Clinical Inquiry, we presented data from the direct subgroup analysis, which revealed no difference in COPD exacerbations between the azithromycin and placebo groups for patients not receiving long-term supplemental oxygen (HR=0.80; 95% CI, 0.62-1.03); however, you are correct to point out that the oxygen use subgroup interaction (patients on oxygen vs patients not on oxygen), which we did not include in this Clinical Inquiry, did not reach significance (P=.23), casting some doubt on the authors’ conclusion of no effect for patients not on oxygen.
On the whole, I feel this Clinical Inquiry accurately summarized the existing evidence and that additional research is needed to better define the utility of azithromycin in these subgroups of patients.
Corey Lyon, DO
Denver, Colo
In the Clinical Inquiry, “Does prophylactic azithromycin reduce the number of COPD exacerbations or hospitalizations?” (J Fam Pract. 2018;67:384-385), Lyon et al state that azithromycin “doesn’t benefit patients ≤65 years, patients with GOLD [Global Initiative for Obstructive Lung Disease] stage IV COPD [chronic obstructive pulmonary disease], current smokers, or patients not using oxygen (strength of recommendation [SOR]: B, randomized controlled trials [RCTs]).” These categorical statements are misleading, and clinicians should ignore most of them when considering azithromycin for their patients with severe COPD.
The authors cited groups that were identified in a posthoc analysis1 of the only large trial involving azithromycin for the treatment of COPD to date.2P values for the interaction of azithromycin with GOLD stage (P=.04), smoking (P=.03), and age (P=.02) were significant, but the mean effects (hazard ratios [HRs]) for GOLD stage IV, smoking, and age ≤65 were .84, .99, and .84, respectively. It would be more accurate to say that there may be a diminished efficacy of azithromycin for patients with GOLD IV COPD and age ≤65 years. Only smokers appear to show no response, although the lower end of the 95% confidence interval was 0.71. The P value for the interaction of azithromycin with no long-term oxygen use (P=.23) was not significant, and it is incorrect to infer that oxygen use or nonuse predicts response.
The authors correctly state that the “significance of the results is limited because the study was not originally powered for this level of subgroup analysis,” but this statement is buried later in the article.
David L. Hahn, MD, MS
Madison, Wis
1. Han MK, Tayob N, Murray S, et al. Predictors of chronic obstructive pulmonary disease exacerbation reduction in response to daily azithromycin therapy. Am J Respir Crit Care Med. 2014;189:1503-1508.
2. Albert RK, Connett J, Bailey WC, et al. Azithromycin for prevention of exacerbations of COPD. N Engl J Med. 2011;365:689-698.
Continue to: Author's response...
Author’s response:
Your statement that the evidence-based answer regarding the lack of benefit of azithromycin in patients ≤65 years of age, with stage IV COPD, current smokers, and patients not using oxygen is “misleading” is a bit of an overstatement.
It is fair to say, however, that our statement regarding lack of efficacy among these subgroups of patients should be softened a bit since the data are from subgroup analyses, which should never be the source of definitive conclusions. And you point out that the 95% confidence intervals [CIs] of the HRs for these subgroups of patients do not include a potentially significant effect (0.68, 0.71, 0.61, and 0.65, respectively), so it is possible there is a Type II error, which would lead one to conclude there is no effect for these subgroups when there is one.
Regarding oxygen therapy, in this Clinical Inquiry, we presented data from the direct subgroup analysis, which revealed no difference in COPD exacerbations between the azithromycin and placebo groups for patients not receiving long-term supplemental oxygen (HR=0.80; 95% CI, 0.62-1.03); however, you are correct to point out that the oxygen use subgroup interaction (patients on oxygen vs patients not on oxygen), which we did not include in this Clinical Inquiry, did not reach significance (P=.23), casting some doubt on the authors’ conclusion of no effect for patients not on oxygen.
On the whole, I feel this Clinical Inquiry accurately summarized the existing evidence and that additional research is needed to better define the utility of azithromycin in these subgroups of patients.
Corey Lyon, DO
Denver, Colo
Asthma: Guideline-Informed Practice

What are the benefits/risks of giving betamethasone to women at risk of late preterm labor?
EVIDENCE SUMMARY
A 2016 systematic review and meta-analysis of 3 RCTs that included 3200 women with late preterm labor (between 34 weeks 0 days and 36 weeks 6 days) found that women who were given betamethasone had a significantly lower incidence of transient tachypnea of the newborn (number needed to treat [NNT]=37; relative risk [RR]=0.72; 95% confidence interval [CI], 0.56-0.92), severe respiratory distress syndrome (NNT=114; RR=0.60; 95% CI, 0.33-0.94), and use of surfactant (NNT=92; RR=0.61; 95% CI, 0.38-0.99).1
A composite outcome measure also favors betamethasone
In addition to these 3 outcomes, the largest RCT in the meta-analysis evaluated a composite outcome and found that betamethasone improved it by 20%. The RCT, comparing 1427 women in the experimental arm with 1400 controls, found benefit to administering 12 mg betamethasone intramuscularly every 24 hours for 2 days for women at high risk of late preterm delivery.2 Enrollment criteria included women with 3 cm dilation or 75% effacement, preterm premature rupture of membranes, or a planned delivery scheduled in the late preterm period.
The primary outcome was a composite score based on one or more of the following within 72 hours after birth: continuous positive airway pressure or high-flow nasal cannula for at least 2 continuous hours, supplemental oxygen with a fraction of inspired oxygen of 0.30 or more for at least 4 continuous hours, mechanical ventilation, stillbirth or neonatal death, or the need for extracorporeal circulation membrane oxygenation. The betamethasone group had 165 women (11.6%) with the primary outcome compared with 202 (14.4%) in the control arm (NNT=34; RR=0.80; 95% CI, 0.66–0.97; P<.02).
Neonatal hypoglycemia may increase, but not dangerously
The same RCT explored the risks of late preterm betamethasone. There was no increase in chorioamnionitis nor neonatal sepsis in the betamethasone group.2 Although neonatal hypoglycemia increased (24% vs 15%; number needed to harm=11.1; RR=1.60; 95% CI, 1.37-1.87; P<.001), no increase was seen in intermediate care nursery or ICU stays (41.8% vs 44.9%; RR=0.93; 95% CI, 0.85-1.01; P=.09) nor length of hospital stay (7 vs 8 days; P=.20).
Three letters to the editor questioned whether hypoglycemia from late-term corticosteroids may lead to long-term neurocognitive delays.3 The authors responded that meta-analyses of RCTs haven’t found an association between antenatal steroid use and neurocognitive delay and that studies that have found an association between hypoglycemia and neurocognitive delay looked at profound and prolonged hypoglycemia, which wasn’t seen in the late preterm betamethasone study.
Continue to: RECOMMENDATIONS
RECOMMENDATIONS
Both the American College of Obstetricians and Gynecologists and the Society for Maternal-Fetal Medicine have published recommendations supporting corticosteroids for threatened late preterm delivery with certain caveats.4-6 Because of a lack of evidence, maternity care providers shouldn’t give corticosteroids for threatened late preterm delivery to women with multiple gestation, diabetes, previous exposure to steroids during pregnancy, or pregnancies with major nonlethal fetal malformations.4-6 Evidence doesn’t support tocolysis when steroids are given in the late preterm period.4,5
1. Saccone G, Berghella V. Antenatal corticosteroids for maturity of term or near term fetuses: systematic review and meta-analysis of randomized controlled trials. BMJ. 2016;355:5044.
2. Gyamfi-Bannerman C, Thom E, Blackwell S, et al. Antenatal betamethasone for women at risk for late preterm delivery. N Engl J Med. 2016;374:1311-1320.
3. Gyamfi-Bannerman C, Thom E. Antenatal betamethasone for women at risk for late preterm delivery. N Engl J Med. 2016;375:486-487.
4. American College of Obstetricians and Gynecologists. Committee Opinion No. 677: Antenatal Corticosteroid Therapy for Fetal Maturation. Obstet Gynecol. 2016;128:e187-e194.
5. Society for Maternal-Fetal Medicine (SMFM) Publications Committee. Implementation of the use of antenatal corticosteroids in the late preterm birth period in women at risk for preterm delivery. Am J Obstet Gynecol. 2016;215:B13-B15.
6. American College of Obstetricians and Gynecologists. Practice Bulletin No. 159: Management of Preterm Labor. Obstet Gynecol. 2016;127:e29-e38.
EVIDENCE SUMMARY
A 2016 systematic review and meta-analysis of 3 RCTs that included 3200 women with late preterm labor (between 34 weeks 0 days and 36 weeks 6 days) found that women who were given betamethasone had a significantly lower incidence of transient tachypnea of the newborn (number needed to treat [NNT]=37; relative risk [RR]=0.72; 95% confidence interval [CI], 0.56-0.92), severe respiratory distress syndrome (NNT=114; RR=0.60; 95% CI, 0.33-0.94), and use of surfactant (NNT=92; RR=0.61; 95% CI, 0.38-0.99).1
A composite outcome measure also favors betamethasone
In addition to these 3 outcomes, the largest RCT in the meta-analysis evaluated a composite outcome and found that betamethasone improved it by 20%. The RCT, comparing 1427 women in the experimental arm with 1400 controls, found benefit to administering 12 mg betamethasone intramuscularly every 24 hours for 2 days for women at high risk of late preterm delivery.2 Enrollment criteria included women with 3 cm dilation or 75% effacement, preterm premature rupture of membranes, or a planned delivery scheduled in the late preterm period.
The primary outcome was a composite score based on one or more of the following within 72 hours after birth: continuous positive airway pressure or high-flow nasal cannula for at least 2 continuous hours, supplemental oxygen with a fraction of inspired oxygen of 0.30 or more for at least 4 continuous hours, mechanical ventilation, stillbirth or neonatal death, or the need for extracorporeal circulation membrane oxygenation. The betamethasone group had 165 women (11.6%) with the primary outcome compared with 202 (14.4%) in the control arm (NNT=34; RR=0.80; 95% CI, 0.66–0.97; P<.02).
Neonatal hypoglycemia may increase, but not dangerously
The same RCT explored the risks of late preterm betamethasone. There was no increase in chorioamnionitis nor neonatal sepsis in the betamethasone group.2 Although neonatal hypoglycemia increased (24% vs 15%; number needed to harm=11.1; RR=1.60; 95% CI, 1.37-1.87; P<.001), no increase was seen in intermediate care nursery or ICU stays (41.8% vs 44.9%; RR=0.93; 95% CI, 0.85-1.01; P=.09) nor length of hospital stay (7 vs 8 days; P=.20).
Three letters to the editor questioned whether hypoglycemia from late-term corticosteroids may lead to long-term neurocognitive delays.3 The authors responded that meta-analyses of RCTs haven’t found an association between antenatal steroid use and neurocognitive delay and that studies that have found an association between hypoglycemia and neurocognitive delay looked at profound and prolonged hypoglycemia, which wasn’t seen in the late preterm betamethasone study.
Continue to: RECOMMENDATIONS
RECOMMENDATIONS
Both the American College of Obstetricians and Gynecologists and the Society for Maternal-Fetal Medicine have published recommendations supporting corticosteroids for threatened late preterm delivery with certain caveats.4-6 Because of a lack of evidence, maternity care providers shouldn’t give corticosteroids for threatened late preterm delivery to women with multiple gestation, diabetes, previous exposure to steroids during pregnancy, or pregnancies with major nonlethal fetal malformations.4-6 Evidence doesn’t support tocolysis when steroids are given in the late preterm period.4,5
EVIDENCE SUMMARY
A 2016 systematic review and meta-analysis of 3 RCTs that included 3200 women with late preterm labor (between 34 weeks 0 days and 36 weeks 6 days) found that women who were given betamethasone had a significantly lower incidence of transient tachypnea of the newborn (number needed to treat [NNT]=37; relative risk [RR]=0.72; 95% confidence interval [CI], 0.56-0.92), severe respiratory distress syndrome (NNT=114; RR=0.60; 95% CI, 0.33-0.94), and use of surfactant (NNT=92; RR=0.61; 95% CI, 0.38-0.99).1
A composite outcome measure also favors betamethasone
In addition to these 3 outcomes, the largest RCT in the meta-analysis evaluated a composite outcome and found that betamethasone improved it by 20%. The RCT, comparing 1427 women in the experimental arm with 1400 controls, found benefit to administering 12 mg betamethasone intramuscularly every 24 hours for 2 days for women at high risk of late preterm delivery.2 Enrollment criteria included women with 3 cm dilation or 75% effacement, preterm premature rupture of membranes, or a planned delivery scheduled in the late preterm period.
The primary outcome was a composite score based on one or more of the following within 72 hours after birth: continuous positive airway pressure or high-flow nasal cannula for at least 2 continuous hours, supplemental oxygen with a fraction of inspired oxygen of 0.30 or more for at least 4 continuous hours, mechanical ventilation, stillbirth or neonatal death, or the need for extracorporeal circulation membrane oxygenation. The betamethasone group had 165 women (11.6%) with the primary outcome compared with 202 (14.4%) in the control arm (NNT=34; RR=0.80; 95% CI, 0.66–0.97; P<.02).
Neonatal hypoglycemia may increase, but not dangerously
The same RCT explored the risks of late preterm betamethasone. There was no increase in chorioamnionitis nor neonatal sepsis in the betamethasone group.2 Although neonatal hypoglycemia increased (24% vs 15%; number needed to harm=11.1; RR=1.60; 95% CI, 1.37-1.87; P<.001), no increase was seen in intermediate care nursery or ICU stays (41.8% vs 44.9%; RR=0.93; 95% CI, 0.85-1.01; P=.09) nor length of hospital stay (7 vs 8 days; P=.20).
Three letters to the editor questioned whether hypoglycemia from late-term corticosteroids may lead to long-term neurocognitive delays.3 The authors responded that meta-analyses of RCTs haven’t found an association between antenatal steroid use and neurocognitive delay and that studies that have found an association between hypoglycemia and neurocognitive delay looked at profound and prolonged hypoglycemia, which wasn’t seen in the late preterm betamethasone study.
Continue to: RECOMMENDATIONS
RECOMMENDATIONS
Both the American College of Obstetricians and Gynecologists and the Society for Maternal-Fetal Medicine have published recommendations supporting corticosteroids for threatened late preterm delivery with certain caveats.4-6 Because of a lack of evidence, maternity care providers shouldn’t give corticosteroids for threatened late preterm delivery to women with multiple gestation, diabetes, previous exposure to steroids during pregnancy, or pregnancies with major nonlethal fetal malformations.4-6 Evidence doesn’t support tocolysis when steroids are given in the late preterm period.4,5
1. Saccone G, Berghella V. Antenatal corticosteroids for maturity of term or near term fetuses: systematic review and meta-analysis of randomized controlled trials. BMJ. 2016;355:5044.
2. Gyamfi-Bannerman C, Thom E, Blackwell S, et al. Antenatal betamethasone for women at risk for late preterm delivery. N Engl J Med. 2016;374:1311-1320.
3. Gyamfi-Bannerman C, Thom E. Antenatal betamethasone for women at risk for late preterm delivery. N Engl J Med. 2016;375:486-487.
4. American College of Obstetricians and Gynecologists. Committee Opinion No. 677: Antenatal Corticosteroid Therapy for Fetal Maturation. Obstet Gynecol. 2016;128:e187-e194.
5. Society for Maternal-Fetal Medicine (SMFM) Publications Committee. Implementation of the use of antenatal corticosteroids in the late preterm birth period in women at risk for preterm delivery. Am J Obstet Gynecol. 2016;215:B13-B15.
6. American College of Obstetricians and Gynecologists. Practice Bulletin No. 159: Management of Preterm Labor. Obstet Gynecol. 2016;127:e29-e38.
1. Saccone G, Berghella V. Antenatal corticosteroids for maturity of term or near term fetuses: systematic review and meta-analysis of randomized controlled trials. BMJ. 2016;355:5044.
2. Gyamfi-Bannerman C, Thom E, Blackwell S, et al. Antenatal betamethasone for women at risk for late preterm delivery. N Engl J Med. 2016;374:1311-1320.
3. Gyamfi-Bannerman C, Thom E. Antenatal betamethasone for women at risk for late preterm delivery. N Engl J Med. 2016;375:486-487.
4. American College of Obstetricians and Gynecologists. Committee Opinion No. 677: Antenatal Corticosteroid Therapy for Fetal Maturation. Obstet Gynecol. 2016;128:e187-e194.
5. Society for Maternal-Fetal Medicine (SMFM) Publications Committee. Implementation of the use of antenatal corticosteroids in the late preterm birth period in women at risk for preterm delivery. Am J Obstet Gynecol. 2016;215:B13-B15.
6. American College of Obstetricians and Gynecologists. Practice Bulletin No. 159: Management of Preterm Labor. Obstet Gynecol. 2016;127:e29-e38.
EVIDENCE-BASED ANSWER:
Giving betamethasone to women at risk for delivery between 34 weeks 0 days and 36 weeks 6 days can lower by almost 40% the incidence of transient tachypnea of the newborn, severe respiratory distress syndrome, and the use of surfactant (strength of recommendation [SOR]: A, systematic review of randomized controlled trials [RCTs]).
Betamethasone may increase neonatal hypoglycemia, but the hypoglycemia isn’t associated with a prolonged hospital stay or other negative outcomes.

Don’t overlook these uses of point-of-care ultrasound
In the article, “Point-of-care ultrasound: Coming soon to primary care?” (J Fam Pract. 2018;67:70-79), Bornemann et al outline potential uses for point-of-care ultrasound (POCUS), describing in detail its role in cardiovascular and pulmonary exams, screening for abdominal aortic aneurysms, and diagnosing deep vein thrombosis. The American Academy of Family Physicians, in the Recommended Curriculum Guidelines for Family Medicine Residents (available at: https://www.aafp.org/medical-school-residency/program-directors/curriculum.html), also discusses obstetric and gynecologic uses for POCUS, such as determining fetal presentation and distinguishing viable pregnancy from miscarriage.
In my practice, I most often use POCUS for gynecologic and pregnancy-related issues, such as to ensure proper placement of an intrauterine device (IUD) when the strings are not visible, to determine gestational age in patients with uncertain last menstrual periods, and to confirm pregnancy location when patients have risk factors for, or symptoms suggestive of, ectopic pregnancy.
The breadth of care provided in family medicine is what makes it special. We must make sure that as we expand our care with new technologies, we do not trade tried and true uses of those technologies for newer ones.
Zoey Thill, MD, MPP
Bronx, NY
In the article, “Point-of-care ultrasound: Coming soon to primary care?” (J Fam Pract. 2018;67:70-79), Bornemann et al outline potential uses for point-of-care ultrasound (POCUS), describing in detail its role in cardiovascular and pulmonary exams, screening for abdominal aortic aneurysms, and diagnosing deep vein thrombosis. The American Academy of Family Physicians, in the Recommended Curriculum Guidelines for Family Medicine Residents (available at: https://www.aafp.org/medical-school-residency/program-directors/curriculum.html), also discusses obstetric and gynecologic uses for POCUS, such as determining fetal presentation and distinguishing viable pregnancy from miscarriage.
In my practice, I most often use POCUS for gynecologic and pregnancy-related issues, such as to ensure proper placement of an intrauterine device (IUD) when the strings are not visible, to determine gestational age in patients with uncertain last menstrual periods, and to confirm pregnancy location when patients have risk factors for, or symptoms suggestive of, ectopic pregnancy.
The breadth of care provided in family medicine is what makes it special. We must make sure that as we expand our care with new technologies, we do not trade tried and true uses of those technologies for newer ones.
Zoey Thill, MD, MPP
Bronx, NY
In the article, “Point-of-care ultrasound: Coming soon to primary care?” (J Fam Pract. 2018;67:70-79), Bornemann et al outline potential uses for point-of-care ultrasound (POCUS), describing in detail its role in cardiovascular and pulmonary exams, screening for abdominal aortic aneurysms, and diagnosing deep vein thrombosis. The American Academy of Family Physicians, in the Recommended Curriculum Guidelines for Family Medicine Residents (available at: https://www.aafp.org/medical-school-residency/program-directors/curriculum.html), also discusses obstetric and gynecologic uses for POCUS, such as determining fetal presentation and distinguishing viable pregnancy from miscarriage.
In my practice, I most often use POCUS for gynecologic and pregnancy-related issues, such as to ensure proper placement of an intrauterine device (IUD) when the strings are not visible, to determine gestational age in patients with uncertain last menstrual periods, and to confirm pregnancy location when patients have risk factors for, or symptoms suggestive of, ectopic pregnancy.
The breadth of care provided in family medicine is what makes it special. We must make sure that as we expand our care with new technologies, we do not trade tried and true uses of those technologies for newer ones.
Zoey Thill, MD, MPP
Bronx, NY
Does prophylactic azithromycin reduce the number of COPD exacerbations or hospitalizations?
EVIDENCE SUMMARY
A randomized, placebo-controlled trial including 1142 patients with COPD (forced expiratory volume in one second [FEV1] <70%, postbronchodilator FEV1 <80%) found that daily azithromycin 250 mg reduced acute exacerbations more than placebo over one year.1 Researchers recruited patients who were using supplemental oxygen, had required glucocorticoids, or had been hospitalized for an acute exacerbation in the last year. Patients with asthma, resting heart rate >100 beats/min, prolonged QTc interval (or on prolonging medications), or hearing impairment were excluded.
Azithromycin increased the median time to first exacerbation (defined as increase or new onset of cough, sputum, wheeze, and chest tightness for 3 days requiring antibiotics or systemic steroids) compared with the placebo group (266 days vs 174 days; P<.001) and reduced the risk of an acute exacerbation per patient year (hazard ratio [HR]=0.73; 95% confidence [CI], 0.63-0.84). It also reduced the rate of acute exacerbations per patient year (1.83 vs 1.43; P=.01; rate ratio=0.83; 95% CI, 0.72-0.95). The number needed to treat to prevent one exacerbation was 2.86.
No differences in death from any cause (3% vs 4%; P=.87), death from respiratory cause (2% vs 1%; P=.48), or death from cardiovascular cause (0.2% vs 0.2%; P=1.0) were found between azithromycin and placebo. Nor did rates of hospitalizations for acute exacerbations differ.
The groups also showed no significant difference in serious adverse events leading to discontinuation of medication. Notably, more patients in the azithromycin group had audiogram-confirmed hearing loss (25% vs 20%; P=.04), although the authors state that their criteria for hearing loss may have been too stringent because hearing improved on repeat testing whether or not the study drug was discontinued. In addition, more patients in the placebo group developed nasopharyngeal colonization with methicillin-resistant Staphylococcus aureus (31% vs 12%; P<.001).
Older ex-smokers on long-term O2 benefit most from the antibiotic
A retrospective subgroup analysis of the RCT identified patients who benefited most from daily azithromycin therapy.2 Compared with placebo, azithromycin decreased the time to first exacerbation in patients >65 years (542 patients; HR=0.59; 95% CI, 0.47-0.74), but not patients ≤65 years (571 patients; HR=0.84; 95% CI, 0.68-1.04).
The azithromycin group also demonstrated decreased time to first exacerbation in ex-smokers (867 patients; HR=0.65; 95% CI, 0.55-0.77) and patients on long-term oxygen (659 patients; HR=0.66; 95% CI, 0.55-0.80) but not current smokers (246 patients; HR=0.99; 95% CI, 0.71-1.38) or patients not using long-term oxygen (454 patients; HR=0.80; 95% CI, 0.62-1.03).
Azithromycin administration decreased exacerbations in patients with GOLD stages II (292 patients; HR=0.55; 95% CI, 0.40-0.75) and III (451 patients; HR=0.71; 95% CI, 0.56-0.90), but not stage IV (370 patients; HR=0.84; 95% CI, 0.65-1.08). The significance of the results is limited because the study was not originally powered for this level of subgroup analysis.
Continue to: Smaller study shows similar results
Smaller study shows similar results
A smaller RCT of 92 patients that evaluated exacerbation rates with azithromycin and placebo recruited patients with at least 3 acute COPD exacerbations in the previous year.3
Compared with placebo, oral azithromycin 500 mg 3 times a week (Monday, Wednesday, and Friday) increased the time between exacerbations over a 12-month period (59 days vs 130 days; P=.001). It also reduced the exacerbation rate per person per year (1.94 vs 3.22; risk ratio=0.60; 95% CI, 0.43-0.84) but didn’t change the hospitalization rate (odds ratio=1.34; 95% CI, 0.67-2.7).
No difference in serious adverse events was found between the azithromycin and placebo groups (3 patients vs 5 patients; P=NS), but an increase in diarrhea (9 patients vs 1 patient; P=.015) was noted.
RECOMMENDATIONS
An evidence-based guideline by the American College of Chest Physicians and Canadian Thoracic Society recommends long-term macrolide therapy to prevent acute exacerbations in patients >40 years with moderate or severe COPD and a history of ≥1 moderate or severe exacerbation in the previous year despite maximized inhaler therapy (Grade 2A, weak recommendation, high-quality evidence).4 The guideline also states that the duration and optimal dosages are unknown.
1. Albert RK, Connett J, Bailey WC, et al. Azithromycin for prevention of exacerbations of COPD. N Engl J Med. 2011;365:689-698.
2. Han M, Tayob N, Murray S, et al. Predictors of chronic obstructive pulmonary disease exacerbation reduction in response to daily azithromycin therapy. Am J Resp Crit Care. 2014;189:1503-1508.
3. Pomares X, Montón C, Espasa M, et al. Long-term azithromycin therapy in patients with severe COPD and repeated exacerbations. Int J Chron Obstruct Pulmon Dis. 2011;6:449-456.
4. Criner GJ, Bourbeau J, Diekemper RL, et al. Prevention of acute exacerbations of COPD: American College of Chest Physicians and Canadian Thoracic Society Guideline. Chest. 2015;147:894-942.
EVIDENCE SUMMARY
A randomized, placebo-controlled trial including 1142 patients with COPD (forced expiratory volume in one second [FEV1] <70%, postbronchodilator FEV1 <80%) found that daily azithromycin 250 mg reduced acute exacerbations more than placebo over one year.1 Researchers recruited patients who were using supplemental oxygen, had required glucocorticoids, or had been hospitalized for an acute exacerbation in the last year. Patients with asthma, resting heart rate >100 beats/min, prolonged QTc interval (or on prolonging medications), or hearing impairment were excluded.
Azithromycin increased the median time to first exacerbation (defined as increase or new onset of cough, sputum, wheeze, and chest tightness for 3 days requiring antibiotics or systemic steroids) compared with the placebo group (266 days vs 174 days; P<.001) and reduced the risk of an acute exacerbation per patient year (hazard ratio [HR]=0.73; 95% confidence [CI], 0.63-0.84). It also reduced the rate of acute exacerbations per patient year (1.83 vs 1.43; P=.01; rate ratio=0.83; 95% CI, 0.72-0.95). The number needed to treat to prevent one exacerbation was 2.86.
No differences in death from any cause (3% vs 4%; P=.87), death from respiratory cause (2% vs 1%; P=.48), or death from cardiovascular cause (0.2% vs 0.2%; P=1.0) were found between azithromycin and placebo. Nor did rates of hospitalizations for acute exacerbations differ.
The groups also showed no significant difference in serious adverse events leading to discontinuation of medication. Notably, more patients in the azithromycin group had audiogram-confirmed hearing loss (25% vs 20%; P=.04), although the authors state that their criteria for hearing loss may have been too stringent because hearing improved on repeat testing whether or not the study drug was discontinued. In addition, more patients in the placebo group developed nasopharyngeal colonization with methicillin-resistant Staphylococcus aureus (31% vs 12%; P<.001).
Older ex-smokers on long-term O2 benefit most from the antibiotic
A retrospective subgroup analysis of the RCT identified patients who benefited most from daily azithromycin therapy.2 Compared with placebo, azithromycin decreased the time to first exacerbation in patients >65 years (542 patients; HR=0.59; 95% CI, 0.47-0.74), but not patients ≤65 years (571 patients; HR=0.84; 95% CI, 0.68-1.04).
The azithromycin group also demonstrated decreased time to first exacerbation in ex-smokers (867 patients; HR=0.65; 95% CI, 0.55-0.77) and patients on long-term oxygen (659 patients; HR=0.66; 95% CI, 0.55-0.80) but not current smokers (246 patients; HR=0.99; 95% CI, 0.71-1.38) or patients not using long-term oxygen (454 patients; HR=0.80; 95% CI, 0.62-1.03).
Azithromycin administration decreased exacerbations in patients with GOLD stages II (292 patients; HR=0.55; 95% CI, 0.40-0.75) and III (451 patients; HR=0.71; 95% CI, 0.56-0.90), but not stage IV (370 patients; HR=0.84; 95% CI, 0.65-1.08). The significance of the results is limited because the study was not originally powered for this level of subgroup analysis.
Continue to: Smaller study shows similar results
Smaller study shows similar results
A smaller RCT of 92 patients that evaluated exacerbation rates with azithromycin and placebo recruited patients with at least 3 acute COPD exacerbations in the previous year.3
Compared with placebo, oral azithromycin 500 mg 3 times a week (Monday, Wednesday, and Friday) increased the time between exacerbations over a 12-month period (59 days vs 130 days; P=.001). It also reduced the exacerbation rate per person per year (1.94 vs 3.22; risk ratio=0.60; 95% CI, 0.43-0.84) but didn’t change the hospitalization rate (odds ratio=1.34; 95% CI, 0.67-2.7).
No difference in serious adverse events was found between the azithromycin and placebo groups (3 patients vs 5 patients; P=NS), but an increase in diarrhea (9 patients vs 1 patient; P=.015) was noted.
RECOMMENDATIONS
An evidence-based guideline by the American College of Chest Physicians and Canadian Thoracic Society recommends long-term macrolide therapy to prevent acute exacerbations in patients >40 years with moderate or severe COPD and a history of ≥1 moderate or severe exacerbation in the previous year despite maximized inhaler therapy (Grade 2A, weak recommendation, high-quality evidence).4 The guideline also states that the duration and optimal dosages are unknown.
EVIDENCE SUMMARY
A randomized, placebo-controlled trial including 1142 patients with COPD (forced expiratory volume in one second [FEV1] <70%, postbronchodilator FEV1 <80%) found that daily azithromycin 250 mg reduced acute exacerbations more than placebo over one year.1 Researchers recruited patients who were using supplemental oxygen, had required glucocorticoids, or had been hospitalized for an acute exacerbation in the last year. Patients with asthma, resting heart rate >100 beats/min, prolonged QTc interval (or on prolonging medications), or hearing impairment were excluded.
Azithromycin increased the median time to first exacerbation (defined as increase or new onset of cough, sputum, wheeze, and chest tightness for 3 days requiring antibiotics or systemic steroids) compared with the placebo group (266 days vs 174 days; P<.001) and reduced the risk of an acute exacerbation per patient year (hazard ratio [HR]=0.73; 95% confidence [CI], 0.63-0.84). It also reduced the rate of acute exacerbations per patient year (1.83 vs 1.43; P=.01; rate ratio=0.83; 95% CI, 0.72-0.95). The number needed to treat to prevent one exacerbation was 2.86.
No differences in death from any cause (3% vs 4%; P=.87), death from respiratory cause (2% vs 1%; P=.48), or death from cardiovascular cause (0.2% vs 0.2%; P=1.0) were found between azithromycin and placebo. Nor did rates of hospitalizations for acute exacerbations differ.
The groups also showed no significant difference in serious adverse events leading to discontinuation of medication. Notably, more patients in the azithromycin group had audiogram-confirmed hearing loss (25% vs 20%; P=.04), although the authors state that their criteria for hearing loss may have been too stringent because hearing improved on repeat testing whether or not the study drug was discontinued. In addition, more patients in the placebo group developed nasopharyngeal colonization with methicillin-resistant Staphylococcus aureus (31% vs 12%; P<.001).
Older ex-smokers on long-term O2 benefit most from the antibiotic
A retrospective subgroup analysis of the RCT identified patients who benefited most from daily azithromycin therapy.2 Compared with placebo, azithromycin decreased the time to first exacerbation in patients >65 years (542 patients; HR=0.59; 95% CI, 0.47-0.74), but not patients ≤65 years (571 patients; HR=0.84; 95% CI, 0.68-1.04).
The azithromycin group also demonstrated decreased time to first exacerbation in ex-smokers (867 patients; HR=0.65; 95% CI, 0.55-0.77) and patients on long-term oxygen (659 patients; HR=0.66; 95% CI, 0.55-0.80) but not current smokers (246 patients; HR=0.99; 95% CI, 0.71-1.38) or patients not using long-term oxygen (454 patients; HR=0.80; 95% CI, 0.62-1.03).
Azithromycin administration decreased exacerbations in patients with GOLD stages II (292 patients; HR=0.55; 95% CI, 0.40-0.75) and III (451 patients; HR=0.71; 95% CI, 0.56-0.90), but not stage IV (370 patients; HR=0.84; 95% CI, 0.65-1.08). The significance of the results is limited because the study was not originally powered for this level of subgroup analysis.
Continue to: Smaller study shows similar results
Smaller study shows similar results
A smaller RCT of 92 patients that evaluated exacerbation rates with azithromycin and placebo recruited patients with at least 3 acute COPD exacerbations in the previous year.3
Compared with placebo, oral azithromycin 500 mg 3 times a week (Monday, Wednesday, and Friday) increased the time between exacerbations over a 12-month period (59 days vs 130 days; P=.001). It also reduced the exacerbation rate per person per year (1.94 vs 3.22; risk ratio=0.60; 95% CI, 0.43-0.84) but didn’t change the hospitalization rate (odds ratio=1.34; 95% CI, 0.67-2.7).
No difference in serious adverse events was found between the azithromycin and placebo groups (3 patients vs 5 patients; P=NS), but an increase in diarrhea (9 patients vs 1 patient; P=.015) was noted.
RECOMMENDATIONS
An evidence-based guideline by the American College of Chest Physicians and Canadian Thoracic Society recommends long-term macrolide therapy to prevent acute exacerbations in patients >40 years with moderate or severe COPD and a history of ≥1 moderate or severe exacerbation in the previous year despite maximized inhaler therapy (Grade 2A, weak recommendation, high-quality evidence).4 The guideline also states that the duration and optimal dosages are unknown.
1. Albert RK, Connett J, Bailey WC, et al. Azithromycin for prevention of exacerbations of COPD. N Engl J Med. 2011;365:689-698.
2. Han M, Tayob N, Murray S, et al. Predictors of chronic obstructive pulmonary disease exacerbation reduction in response to daily azithromycin therapy. Am J Resp Crit Care. 2014;189:1503-1508.
3. Pomares X, Montón C, Espasa M, et al. Long-term azithromycin therapy in patients with severe COPD and repeated exacerbations. Int J Chron Obstruct Pulmon Dis. 2011;6:449-456.
4. Criner GJ, Bourbeau J, Diekemper RL, et al. Prevention of acute exacerbations of COPD: American College of Chest Physicians and Canadian Thoracic Society Guideline. Chest. 2015;147:894-942.
1. Albert RK, Connett J, Bailey WC, et al. Azithromycin for prevention of exacerbations of COPD. N Engl J Med. 2011;365:689-698.
2. Han M, Tayob N, Murray S, et al. Predictors of chronic obstructive pulmonary disease exacerbation reduction in response to daily azithromycin therapy. Am J Resp Crit Care. 2014;189:1503-1508.
3. Pomares X, Montón C, Espasa M, et al. Long-term azithromycin therapy in patients with severe COPD and repeated exacerbations. Int J Chron Obstruct Pulmon Dis. 2011;6:449-456.
4. Criner GJ, Bourbeau J, Diekemper RL, et al. Prevention of acute exacerbations of COPD: American College of Chest Physicians and Canadian Thoracic Society Guideline. Chest. 2015;147:894-942.
EVIDENCE-BASED ANSWER:
Yes for exacerbations, no for hospitalizations. Prophylactic azithromycin reduces the number of exacerbations by about 25%. It also extends the time between exacerbations by approximately 90 days for patients with moderate-to-severe chronic obstructive pulmonary disease (COPD). Azithromycin benefits patients who are >65 years, patients with Global Initiative for Obstructive Lung Disease (GOLD) stage II or III COPD, former smokers, and patients using long-term oxygen; it doesn’t benefit patients ≤65 years, patients with GOLD stage IV COPD, current smokers, or patients not using oxygen (strength of recommendation [SOR]: B, randomized controlled trials [RCTs]).
Prophylactic azithromycin doesn’t reduce hospitalizations overall (SOR: B, single small RCT).

When the correct Dx is elusive
In this issue of JFP, Dr. Mendoza reminds us that “Parkinson’s disease can be a tough diagnosis to navigate.”1 Classically, Parkinson’s disease (PD) is associated with a resting tremor, but bradykinesia is actually the hallmark of the disease. PD can also present with subtle movement disorders, as well as depression and early dementia. It is, indeed, a difficult clinical diagnosis, and consultation with an expert to confirm or deny its presence can be quite helpful.
Other conundrums. PD, however, is not the only illness whose signs and symptoms can present a challenge. Chronic and intermittent shortness of breath, for example, can be very difficult to sort out. Is the shortness of breath due to congestive heart failure, chronic obstructive pulmonary disease, asthma, or a neurologic condition such as myasthenia gravis? Or is it the result of several causes?
When asthma isn’t asthma. Because it is a common illness, physicians often diagnose asthma in patients with shortness of breath or wheezing. But a recent study suggests that as many as 30% of primary care patients with a current diagnosis of asthma do not have asthma at all.2
In the study, Canadian researchers recruited 701 adults with physician-diagnosed asthma, all of whom were taking asthma medications regularly. The researchers did baseline pulmonary function testing (including methacholine challenge testing, if needed) and monitored symptoms frequently. Then they gradually withdrew asthma medications from those who did not appear to have a definitive diagnosis of asthma. They followed these patients for one year. One-third (203 of 613) of the patients with complete follow-up data were no longer taking asthma medications one year later and had no symptoms of asthma. Twelve patients had serious alternative diagnoses such as coronary artery disease and bronchiectasis.
Closer to home. In my practice, I found 2 patients with long-standing diagnoses of asthma who didn’t, in fact, have the condition at all. In both cases, my suspicion was raised by lung examination. In one case, fine bibasilar rales suggested pulmonary fibrosis, which was the correct diagnosis, and the patient is now on the lung transplant list. In the other case, a loud venous hum suggested an arteriovenous malformation. Surgery corrected the patient’s “asthma.”
I urge you to reevaluate your asthma patients to be sure they have the correct diagnosis and to keep PD in your differential for patients who present with atypical symptoms. Primary care clinicians must be expert diagnosticians, willing to question prior diagnoses.
1. Young J, Mendoza M. Parkinson’s disease: a treatment guide. J Fam Pract. 2018;67:276-286.
2. Aaron SD, Vandemheen KL, FitzGerald JM, et al for the Canadian Respiratory Research Network. Reevaluation of diagnosis in adults with physician-diagnosed asthma. JAMA. 2017:317:269-279.
Editor-in-Chief
Editor-in-Chief
Editor-in-Chief
In this issue of JFP, Dr. Mendoza reminds us that “Parkinson’s disease can be a tough diagnosis to navigate.”1 Classically, Parkinson’s disease (PD) is associated with a resting tremor, but bradykinesia is actually the hallmark of the disease. PD can also present with subtle movement disorders, as well as depression and early dementia. It is, indeed, a difficult clinical diagnosis, and consultation with an expert to confirm or deny its presence can be quite helpful.
Other conundrums. PD, however, is not the only illness whose signs and symptoms can present a challenge. Chronic and intermittent shortness of breath, for example, can be very difficult to sort out. Is the shortness of breath due to congestive heart failure, chronic obstructive pulmonary disease, asthma, or a neurologic condition such as myasthenia gravis? Or is it the result of several causes?
When asthma isn’t asthma. Because it is a common illness, physicians often diagnose asthma in patients with shortness of breath or wheezing. But a recent study suggests that as many as 30% of primary care patients with a current diagnosis of asthma do not have asthma at all.2
In the study, Canadian researchers recruited 701 adults with physician-diagnosed asthma, all of whom were taking asthma medications regularly. The researchers did baseline pulmonary function testing (including methacholine challenge testing, if needed) and monitored symptoms frequently. Then they gradually withdrew asthma medications from those who did not appear to have a definitive diagnosis of asthma. They followed these patients for one year. One-third (203 of 613) of the patients with complete follow-up data were no longer taking asthma medications one year later and had no symptoms of asthma. Twelve patients had serious alternative diagnoses such as coronary artery disease and bronchiectasis.
Closer to home. In my practice, I found 2 patients with long-standing diagnoses of asthma who didn’t, in fact, have the condition at all. In both cases, my suspicion was raised by lung examination. In one case, fine bibasilar rales suggested pulmonary fibrosis, which was the correct diagnosis, and the patient is now on the lung transplant list. In the other case, a loud venous hum suggested an arteriovenous malformation. Surgery corrected the patient’s “asthma.”
I urge you to reevaluate your asthma patients to be sure they have the correct diagnosis and to keep PD in your differential for patients who present with atypical symptoms. Primary care clinicians must be expert diagnosticians, willing to question prior diagnoses.
In this issue of JFP, Dr. Mendoza reminds us that “Parkinson’s disease can be a tough diagnosis to navigate.”1 Classically, Parkinson’s disease (PD) is associated with a resting tremor, but bradykinesia is actually the hallmark of the disease. PD can also present with subtle movement disorders, as well as depression and early dementia. It is, indeed, a difficult clinical diagnosis, and consultation with an expert to confirm or deny its presence can be quite helpful.
Other conundrums. PD, however, is not the only illness whose signs and symptoms can present a challenge. Chronic and intermittent shortness of breath, for example, can be very difficult to sort out. Is the shortness of breath due to congestive heart failure, chronic obstructive pulmonary disease, asthma, or a neurologic condition such as myasthenia gravis? Or is it the result of several causes?
When asthma isn’t asthma. Because it is a common illness, physicians often diagnose asthma in patients with shortness of breath or wheezing. But a recent study suggests that as many as 30% of primary care patients with a current diagnosis of asthma do not have asthma at all.2
In the study, Canadian researchers recruited 701 adults with physician-diagnosed asthma, all of whom were taking asthma medications regularly. The researchers did baseline pulmonary function testing (including methacholine challenge testing, if needed) and monitored symptoms frequently. Then they gradually withdrew asthma medications from those who did not appear to have a definitive diagnosis of asthma. They followed these patients for one year. One-third (203 of 613) of the patients with complete follow-up data were no longer taking asthma medications one year later and had no symptoms of asthma. Twelve patients had serious alternative diagnoses such as coronary artery disease and bronchiectasis.
Closer to home. In my practice, I found 2 patients with long-standing diagnoses of asthma who didn’t, in fact, have the condition at all. In both cases, my suspicion was raised by lung examination. In one case, fine bibasilar rales suggested pulmonary fibrosis, which was the correct diagnosis, and the patient is now on the lung transplant list. In the other case, a loud venous hum suggested an arteriovenous malformation. Surgery corrected the patient’s “asthma.”
I urge you to reevaluate your asthma patients to be sure they have the correct diagnosis and to keep PD in your differential for patients who present with atypical symptoms. Primary care clinicians must be expert diagnosticians, willing to question prior diagnoses.
1. Young J, Mendoza M. Parkinson’s disease: a treatment guide. J Fam Pract. 2018;67:276-286.
2. Aaron SD, Vandemheen KL, FitzGerald JM, et al for the Canadian Respiratory Research Network. Reevaluation of diagnosis in adults with physician-diagnosed asthma. JAMA. 2017:317:269-279.
1. Young J, Mendoza M. Parkinson’s disease: a treatment guide. J Fam Pract. 2018;67:276-286.
2. Aaron SD, Vandemheen KL, FitzGerald JM, et al for the Canadian Respiratory Research Network. Reevaluation of diagnosis in adults with physician-diagnosed asthma. JAMA. 2017:317:269-279.
Pharmacologic Treatments for Idiopathic Pulmonary Fibrosis
IN THIS ARTICLE
- Confirming the diagnosis
- Pirfenidone treatment
- Nintedanib treatment
A 64-year-old man has a one-year history of dyspnea on exertion and a nonproductive cough. His symptoms are gradually worsening and increasingly bothersome to him.
His medical history includes mild seasonal allergies and GERD, which is well-controlled by oral antihistamines and proton pump inhibitors. He has spent the past 30 years working a desk job as an accountant. He denies a history of smoking, exposure to secondhand smoke, and initiation of new medication.
He admits to increased fatigue, but denies fever, chills, lymphadenopathy, weight change, chest pain, wheezing, abdominal pain, diarrhea, vomiting, claudication, and swelling in the extremities. The rest of the review of systems is negative.
Lab results—complete blood count, comprehensive metabolic panel, TSH, antinuclear antibodies, erythrocyte sedimentation rate, and C-reactive protein—are within normal limits. Spirometry shows very mild restriction. A chest x-ray is abnormal but nonspecific, showing peripheral opacities. An ECG shows normal sinus rhythm.
The patient is given a trial of an inhaled steroid, which yields no improvement. Six months later, the patient is seen by a pulmonologist. Idiopathic pulmonary fibrosis (IPF) is diagnosed based on high-resolution CT (HRCT) and lung biopsy results.
IPF is a chronic, progressive, fibrosing interstitial disease that is limited to lung tissue. It most commonly manifests in older adults with vague symptoms of dyspnea on exertion and nonproductive cough, but symptoms can also include fatigue, muscle and joint aches, clubbing of the fingernails, and weight loss.1 The average life expectancy following diagnosis of IPF is two to five years, and the mortality rate is estimated at 64.3 per million men and 58.4 per million women per year.2,3
Continue to: DIAGNOSIS
DIAGNOSIS
IPF belongs in the general class of idiopathic interstitial pneumonias (IIPs), which are characterized by varying degrees of inflammation and fibrosis of lung interstitium.4 All subtypes of IIPs cause dyspnea and diffuse abnormalities on HRCT, and all vary from each other histologically. Table 1 outlines the key features of each.5-8
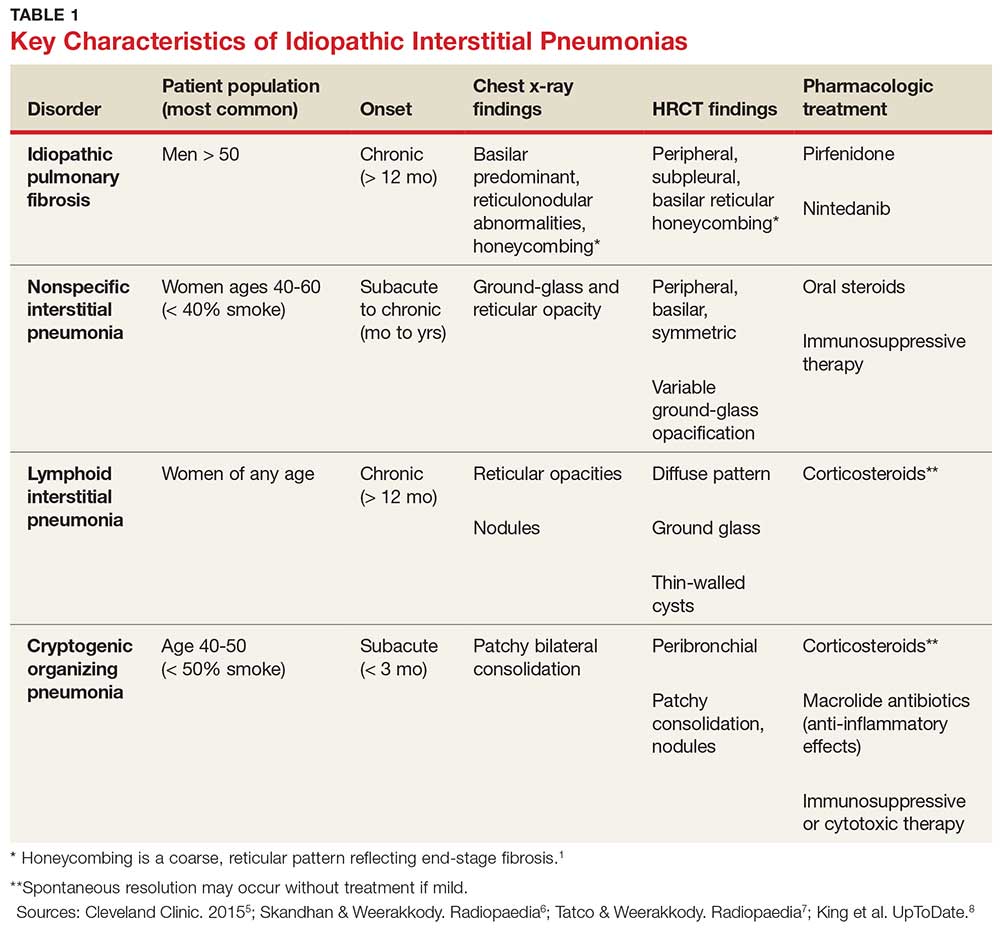
Because of its vague symptomology and the extensive workup needed to rule out other diseases, patients with IPF often have symptoms for one to two years before a diagnosis is made.1 Physical exam may reveal fine inspiratory rales in both lung bases and digital clubbing; eventual signs of pulmonary hypertension and right-sided heart failure may be appreciated.1,9
There are no specific diagnostic laboratory tests to confirm IPF; however, baseline labwork (as outlined in the case presentation) is typically ordered to rule out infection, thyroid disease, or connective tissue disease.10 Many patients are referred to a cardiologist before being seen by a pulmonologist; cardiac stress testing may be done, and an echocardiogram may be performed to rule out heart failure.
Diagnostic testing may include pulmonary function testing, HRCT of the chest, and lung biopsy.10 Tissue samples from patients with IPF reveal different stages of disease, including dense fibrosis with honeycombing, subpleural or paraseptal distribution, fibroblast foci, and normal tissue.11 Pulmonary function test results will show a restrictive pattern. Both forced expiratory volume in one second (FEV1) and forced vital capacity (FVC) will be reduced, and the FEV1/FVC ratio preserved. Due to decreased functional lung volume, diffusing capacity of the lung for carbon monoxide (DLCO) will also be reduced.4,12
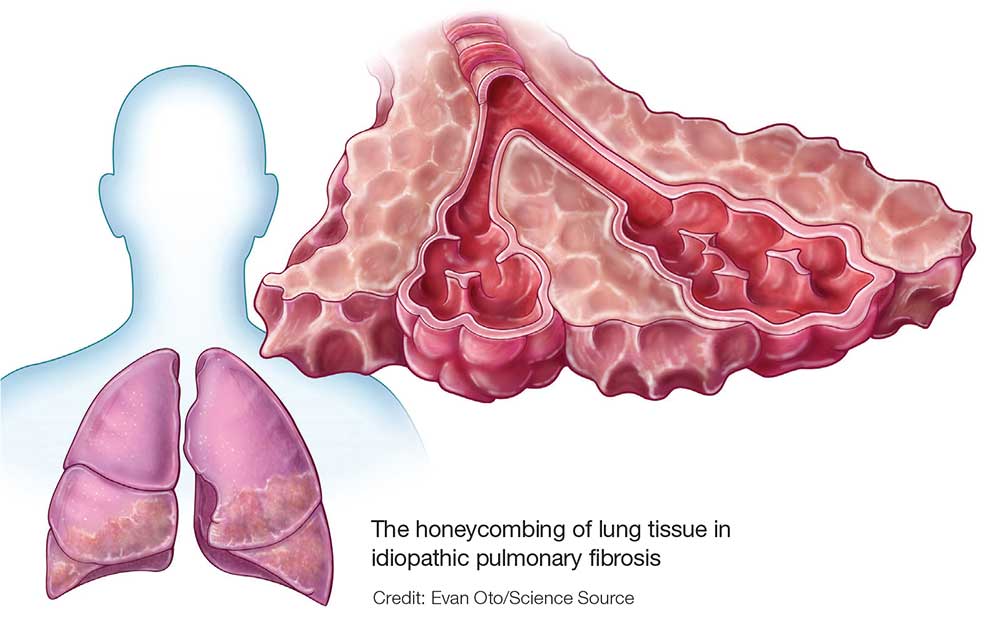
The differential is broad and includes allergic asthma, bronchitis, COPD, lung cancer, hypersensitivity pneumonitis, asbestosis, or pulmonary embolism.
Continue to: TREATMENT HISTORY
TREATMENT HISTORY
IPF has a long history of tried and failed treatment options. The American Thoracic Society (ATS), in concert with other professional organizations, has published comprehensive guidelines and recommendations pertaining to the use of pharmacologic medications to control disease progression. Warfarin and other anticoagulants have been studied, based on the observation that a procoagulant state promotes fibrotic changes in the lung tissue.13 However, anticoagulant use is not recommended in patients with IPF due to lack of efficacy and high potential for harm.13
Immunosuppressants have also been in the spotlight as possible treatment for IPF, but a clinical study investigating the efficacy of a three-drug regimen including prednisone, azathioprine, and N-acetylcysteine was stopped early due to increased risk for harm. Endothelin antagonists and potent tyrosine kinase inhibitors are also not recommended in the most recent edition of IPF guidelines, as they lack benefit.13
In fact, prior to the 2015 edition of the guidelines, no single medication was routinely recommended for patients with IPF. But this is now changing, following the 2014 FDA approval of two new drugs, nintedanib and pirfenidone, designed specifically to treat IPF.14 These drugs have shown promise in clinical trials (results of which are summarized in Table 2).
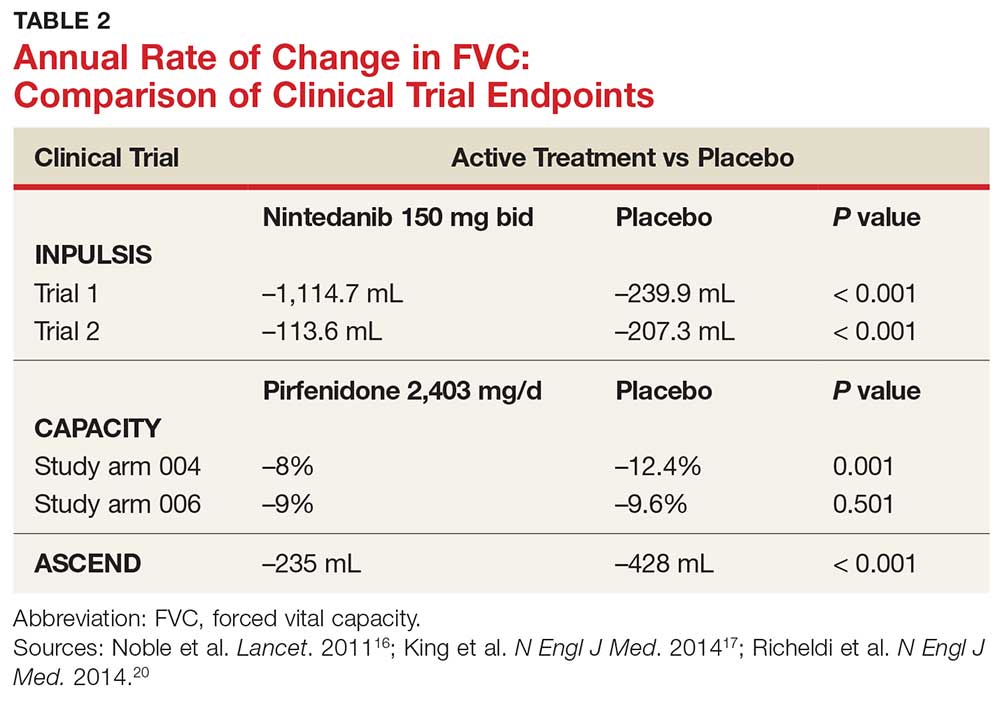
Continue to: NEW PHARMACOLOGIC OPTIONS
NEW PHARMACOLOGIC OPTIONS
Pirfenidone
In 2008, a study was conducted in Japan to determine the mechanism of action of pirfenidone.15 Through in vitro studies of healthy adult lung fibroblasts with added pro-fibrotic factor and transforming growth factor (TGF-ß 1), the researchers found that pirfenidone was effective at decreasing the production of a collagen-binding protein called HSP47. This protein is ubiquitous in fibrotic tissue. The study also showed that pirfenidone decreased the production of collagen type 1, which, when uninhibited, increases fibrosis.15
CAPACITY trials. In the CAPACITY trials, two phase 3 multinational studies conducted from 2006 to 2008, patients were given either pirfenidone or placebo.16 In the first study arm, patients were assigned to pirfenidone 2,403 mg/d (n = 174), pirfenidone 1,197 mg/d (n = 87), or placebo (n = 174). In the second study arm, 171 patients received pirfenidone 2,403 mg/d and 173 patients received placebo. Endpoints were measured at baseline and up to week 72.
The first study arm found that the mean rate of decline of FVC—the primary endpoint—was 4.4% less in the treatment group than in the placebo group (p = 0.001), and there was a 36% decrease in risk for death or disease progression in the treatment group (HR, 0.64; p, 0.023). (Endpoints were defined as: time to confirmed > 10% decline in percentage predicted FVC, > 15% decline in percentage predicted DLCO, or death.) The researchers found no clinically significant change in the six-minute walk test—a secondary endpoint of the study.16
The second study arm, however, found no statistically significant change in FVC between the treatment and placebo groups (with a 0.6% smaller decrease in FVC in the pirfenidone group), nor did they see a difference in progression-free survival. However, there was a significant change in the six-minute walk test between the treatment and placebo groups (p = 0.0009). Throughout the study, the most common adverse effects included nausea (36%), rash (32%), and dyspepsia (19%).16
ASCEND trial. The 2014 Assessment of Pirfenidone to Confirm Efficacy and Safety in Idiopathic Pulmonary Fibrosis (ASCEND) trial was a phase 3, multinational, randomized, double-blind, placebo-controlled study of the use of pirfenidone 2,403 mg/d.17 The study was conducted from 2012 to 2013. Of the total number of patients (N = 522), half received pirfenidone and half received placebo. After 52 weeks of treatment (the end of the study), the researchers found a smaller decline in FVC—the primary endpoint—in the treatment group compared to placebo (mean decline, 235 mL vs 428 mL, respectively [p < 0.001]). Regarding the six-minute walk test, the investigators found that 25.9% of the treatment group exhibited a decrease of ≥ 50 meters, compared to 35.7% of the placebo group (p = 0.04). (Progression-free survival was defined as a confirmed ≥ 10% decrease in predicted FVC, a confirmed decrease of 50 meters in the six-minute walk test, or death.)
The pirfenidone group in the ASCEND trial showed a 43% reduced risk for death or disease progression (HR, 0.57; p, < 0.001).16,17 All-cause mortality was lower in the pirfenidone group (4%) than in the placebo group (7.2%), but this was not statistically significant. Deaths from IPF in the pirfenidone group totaled three patients (1.1%) versus seven patients (2.5%) in the placebo group; this was also not statistically significant. The most common adverse effects seen during the study were nausea (36%), rash (28.1%), and headache (25.9%).17
Recommendations for use. Liver function testing should be performed at baseline, monthly for six months, and every three months afterward, as elevations in liver enzymes have been observed.18 Pirfenidone is a CYP1A2 substrate; moderate-to-strong CYP1A2 inhibitors should therefore be discontinued prior to initiation, as they are likely to decrease exposure and efficacy of pirfenidone. There are currently no black box warnings.18
Continue to: Nintedanib
Nintedanib
Hostettler et al studied lung samples from patients with IPF to determine the mechanism of action of nintedanib.19 Evaluation of fibroblasts derived from IPF samples revealed that they contained higher levels of platelet-derived growth factor (PDGF) than did nonfibrotic control cells. They also found that nintedanib, a tyrosine kinase inhibitor, significantly inhibited the phosphorylation of fibrotic-inducing growth factors—PDGF as well as vascular endothelial growth factor (VEGF).
INPULSIS trials. A phase 3 replicate of randomized, double-blind, multinational studies, the INPULSIS trials were performed between 2011 and 2012.20 Two study arms were used to evaluate a total of 638 patients who received nintedanib 150 mg bid for 52 weeks. The primary endpoint was annual rate of decline of FVC.

The researchers also evaluated efficacy through two other endpoints: patient-reported quality of life and symptoms via the St. George’s Respiratory Questionnaire (SGRQ) and evaluation of time to acute exacerbation. The latter was defined as worsening or new dyspnea, new diffuse pulmonary infiltrates visualized on chest radiography and/or HRCT, or the development of parenchymal abnormalities with no pneumothorax or pleural effusion since the preceding visit; and exclusion of any known causes of acute worsening, including infection, heart failure, pulmonary embolism, and any identifiable cause of acute lung injury.20
INPULSIS 1 (first arm) included 309 patients in the treatment group. Results showed an adjusted annual rate of decline in FVC of 114.7 mL/year, versus 239.9 mL/year in the placebo group (p < 0.001). In the treatment group, 52.8% exhibited ≤ 5% decline in FVC, compared to 38.2% in the placebo group (p = 0.001). No significant between-group differences were found in SGRQ score or time to acute exacerbation.20
INPULSIS 2 had 329 patients receiving nintedanib. An annual rate of decline in FVC of 113.6 mL/year from baseline was observed in the treatment group, compared to 207.3 mL/year in the placebo group (p < 0.001). In the treatment group, 53.2% showed ≤ 5% decline in FVC, versus 39.3% in the placebo group (p = 0.001). There was also a significantly smaller increase in total SGRQ score (meaning, less deterioration in quality of life) in the nintedanib group versus the placebo group (p = 0.02). A statistically significant increase in time to first acute exacerbation was observed in the nintedanib group (p = 0.005).20
There was no significant difference between groups in death from any cause, death from respiratory causation, or death that occurred between randomization and 28 days post treatment. The most common adverse effects seen throughout the two trials included diarrhea (trial 1, 61.5%; trial 2, 63.2%), nausea (trial 1, 22.7%; trial 2, 26.1%), and nasopharyngitis (trial 1, 12.6%; trial 2, 14.6%).20
Recommendations for use. Liver function testing should be performed at baseline, at regular intervals during the first three months, then periodically thereafter; patients in the treatment group of both INPULSIS trials had elevated liver enzymes, and cases of drug-induced liver injury have been observed with use of nintedanib.21 This medication may increase risk for bleeding due to its mechanism of action (VEGFR inhibition). Coadministration with CYP3A4 inhibitors may increase concentration of nintedanib; therefore, close monitoring is recommended. Avoid coadministration with CYP3A4 inducers, as this may decrease concentration of nintedanib by 50%. There are currently no black box warnings.21
Continue to: Patient monitoring
Patient monitoring
The ATS recommends measuring FVC and DLCO every three to six months, or sooner if clinically indicated.13 Pulse oximetry should be measured at rest and on exertion in all patients, regardless of symptoms, to assure proper saturation and identify the need for supplemental oxygen; this should also be done every three to six months.
The ATS recommends prompt detection and treatment of comorbidities such as pulmonary hypertension, emphysema, airflow obstruction, GERD, sleep apnea, and coronary artery disease.13 These recommendations are based on the organization’s 2015 guidelines.
OUTCOME FOR THE CASE PATIENT
The patient was started on pirfenidone (2,403 mg/d). He is continuing treatment and showing improvements in quality of life and slowed deterioration of lung function.
CONCLUSION
IPF causes progressive fibrosis of lung interstitium. The etiology is unknown, the symptoms and signs are vague, and mean life expectancy following diagnosis is two to five years. The most recent IPF guidelines recommend avoiding use of anticoagulants and immunosuppressants (eg, steroids, azathioprine, and N-acetylcysteine), due to their proven ineffectiveness and harm to patients with IPF.
Since the FDA’s approval of pirfenidone and nintedanib, the ATS has made recommendations for their use in patients with IPF. Despite mixed results in clinical trials, both drugs have demonstrated the ability to slow the decline in FVC over time, with relatively benign adverse effects. It is difficult to compare pirfenidone and nintedanib, or to recommend use of one drug over the other. However, it is promising that patients with this routinely fatal disease now have treatment options that can potentially modulate their disease progression.
1. Kim DS, Collard HR, King TE Jr. Classification and natural history of the idiopathic interstitial pneumonias. Proc Am Thorac Soc. 2006;3(4):285-292.
2. Frankel SK, Schwarz MI. Update in idiopathic pulmonary fibrosis. Curr Opin Pulm Med. 2009;15(5):463-469.
3. Olson AL, Swigris JJ, Lezotte DC, et al. Mortality from pulmonary fibrosis increased in the United States from 1992 to 2003. Am J Respir Crit Care Med. 2007;176(3):277-284.
4. Chapman JT. Interstitial lung disease. Cleveland Clinic. August 2010. www.clevelandclinicmeded.com/medical pubs/diseasemanagement/pulmonary/interstitial-lung-disease. Accessed March 12, 2018.
5. Cleveland Clinic. Nonspecific interstitial pneumonia. January 16, 2015. https://my.clevelandclinic.org/health/articles/nonspecific-interstitial-pneumonia. Accessed March 12, 2018.
6. Skandhan AKP, Weerakkody Y. Non-specific interstitial pneumonia. Radiopaedia. https://radiopaedia.org/articles/non-specific-interstitial-pneumonia-1. Accessed March 12, 2018.
7. Tatco V, Weerakkody Y. Lymphocytic interstitial pneumonitis. Radiopaedia. https://radiopaedia.org/articles/lymphocytic-interstitial-pneumonitis-1. Accessed March 12, 2018.
8. King TE Jr, Flaherty KR, Hollingsworth H. Cryptogenic organizing pneumonia. UpToDate. www.uptodate.com/contents/cryptogenic-organizing-pneumonia#H12. Accessed March 12, 2018.
9. Patel NM, Lederer DJ, Borczuk AC, Kawut SM. Pulmonary hypertension in idiopathic pulmonary fibrosis. Chest. 2007; 132(3):998-1006.
10. Lee J. Overview of idiopathic interstitial pneumonias. April 2016. www.merckmanuals.com/professional/pulmonary-disorders/interstitial-lung-diseases/overview-of-idiopathic-interstitial-pneumonias. Accessed March 12, 2018.
11. Lynch DA, Sverzellati N, Travis WD, et al. Diagnostic criteria for idiopathic pulmonary fibrosis: a Fleischner Society White Paper. Lancet Respir Med. 2018;6(2):138-153.
12. Martinez FJ, Flaherty K. Pulmonary function testing in idiopathic interstitial pneumonias. Proc Am Thorac Soc. 2006; 3(4):315-321.
13. Raghu G, Rochwerg B, Zhang Y, et al; American Thoracic Society; European Respiratory Society; Japanese Respiratory Society; Latin American Thoracic Association. An official ATS/ERS/JRS/ALAT clinical practice guideline: treatment of idiopathic pulmonary fibrosis. An update of the 2011 Clinical Practice Guideline. Am J Respir Crit Care Med. 2015; 192(2):e3-e19.
14. Chowdhury BA; FDA. Two FDA drug approvals for idiopathic pulmonary fibrosis (IPF). October 15, 2014. https://blogs.fda.gov/fdavoice/index.php/2014/10/two-fda-drug-approvals-for-idiopathic-pulmonary-fibrosis-ipf/. Accessed March 12, 2018.
15. Nakayama S, Mukae H, Sakamoto N, et al. Pirfenidone inhibits the expression of HSP47 in TGF-beta1-stimulated human lung fibroblasts. Life Sci. 2008; 82(3-4):210-217.
16. Noble PW, Albera C, Bradford WZ, et al; CAPACITY Study Group. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomized trials. Lancet. 2011;377: 1760-1769.
17. King TE Jr, Bradford WZ, Castro-Bernardini S, et al; ASCEND Study Group. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22): 2083-2092.
18. Esbriet [package insert]. South San Francisco, CA: Genentech, Inc; 2016.
19. Hostettler KE, Zhong J, Papakonstantinou E, et al. Anti-fibrotic effects of nintedanib in lung fibroblasts derived from patients with idiopathic pulmonary fibrosis. Respir Res. 2014;15(1):157.
20. Richeldi L, du Bois RM, Raghu G, et al; INPULSIS Trial Investigators. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2071-2082.
21. OFEV [package insert]. Ridgefield, CT: Boehringer Ingelheim Pharmaceuticals, Inc; 2018.
IN THIS ARTICLE
- Confirming the diagnosis
- Pirfenidone treatment
- Nintedanib treatment
A 64-year-old man has a one-year history of dyspnea on exertion and a nonproductive cough. His symptoms are gradually worsening and increasingly bothersome to him.
His medical history includes mild seasonal allergies and GERD, which is well-controlled by oral antihistamines and proton pump inhibitors. He has spent the past 30 years working a desk job as an accountant. He denies a history of smoking, exposure to secondhand smoke, and initiation of new medication.
He admits to increased fatigue, but denies fever, chills, lymphadenopathy, weight change, chest pain, wheezing, abdominal pain, diarrhea, vomiting, claudication, and swelling in the extremities. The rest of the review of systems is negative.
Lab results—complete blood count, comprehensive metabolic panel, TSH, antinuclear antibodies, erythrocyte sedimentation rate, and C-reactive protein—are within normal limits. Spirometry shows very mild restriction. A chest x-ray is abnormal but nonspecific, showing peripheral opacities. An ECG shows normal sinus rhythm.
The patient is given a trial of an inhaled steroid, which yields no improvement. Six months later, the patient is seen by a pulmonologist. Idiopathic pulmonary fibrosis (IPF) is diagnosed based on high-resolution CT (HRCT) and lung biopsy results.
IPF is a chronic, progressive, fibrosing interstitial disease that is limited to lung tissue. It most commonly manifests in older adults with vague symptoms of dyspnea on exertion and nonproductive cough, but symptoms can also include fatigue, muscle and joint aches, clubbing of the fingernails, and weight loss.1 The average life expectancy following diagnosis of IPF is two to five years, and the mortality rate is estimated at 64.3 per million men and 58.4 per million women per year.2,3
Continue to: DIAGNOSIS
DIAGNOSIS
IPF belongs in the general class of idiopathic interstitial pneumonias (IIPs), which are characterized by varying degrees of inflammation and fibrosis of lung interstitium.4 All subtypes of IIPs cause dyspnea and diffuse abnormalities on HRCT, and all vary from each other histologically. Table 1 outlines the key features of each.5-8
Because of its vague symptomology and the extensive workup needed to rule out other diseases, patients with IPF often have symptoms for one to two years before a diagnosis is made.1 Physical exam may reveal fine inspiratory rales in both lung bases and digital clubbing; eventual signs of pulmonary hypertension and right-sided heart failure may be appreciated.1,9
There are no specific diagnostic laboratory tests to confirm IPF; however, baseline labwork (as outlined in the case presentation) is typically ordered to rule out infection, thyroid disease, or connective tissue disease.10 Many patients are referred to a cardiologist before being seen by a pulmonologist; cardiac stress testing may be done, and an echocardiogram may be performed to rule out heart failure.
Diagnostic testing may include pulmonary function testing, HRCT of the chest, and lung biopsy.10 Tissue samples from patients with IPF reveal different stages of disease, including dense fibrosis with honeycombing, subpleural or paraseptal distribution, fibroblast foci, and normal tissue.11 Pulmonary function test results will show a restrictive pattern. Both forced expiratory volume in one second (FEV1) and forced vital capacity (FVC) will be reduced, and the FEV1/FVC ratio preserved. Due to decreased functional lung volume, diffusing capacity of the lung for carbon monoxide (DLCO) will also be reduced.4,12
The differential is broad and includes allergic asthma, bronchitis, COPD, lung cancer, hypersensitivity pneumonitis, asbestosis, or pulmonary embolism.
Continue to: TREATMENT HISTORY
TREATMENT HISTORY
IPF has a long history of tried and failed treatment options. The American Thoracic Society (ATS), in concert with other professional organizations, has published comprehensive guidelines and recommendations pertaining to the use of pharmacologic medications to control disease progression. Warfarin and other anticoagulants have been studied, based on the observation that a procoagulant state promotes fibrotic changes in the lung tissue.13 However, anticoagulant use is not recommended in patients with IPF due to lack of efficacy and high potential for harm.13
Immunosuppressants have also been in the spotlight as possible treatment for IPF, but a clinical study investigating the efficacy of a three-drug regimen including prednisone, azathioprine, and N-acetylcysteine was stopped early due to increased risk for harm. Endothelin antagonists and potent tyrosine kinase inhibitors are also not recommended in the most recent edition of IPF guidelines, as they lack benefit.13
In fact, prior to the 2015 edition of the guidelines, no single medication was routinely recommended for patients with IPF. But this is now changing, following the 2014 FDA approval of two new drugs, nintedanib and pirfenidone, designed specifically to treat IPF.14 These drugs have shown promise in clinical trials (results of which are summarized in Table 2).
Continue to: NEW PHARMACOLOGIC OPTIONS
NEW PHARMACOLOGIC OPTIONS
Pirfenidone
In 2008, a study was conducted in Japan to determine the mechanism of action of pirfenidone.15 Through in vitro studies of healthy adult lung fibroblasts with added pro-fibrotic factor and transforming growth factor (TGF-ß 1), the researchers found that pirfenidone was effective at decreasing the production of a collagen-binding protein called HSP47. This protein is ubiquitous in fibrotic tissue. The study also showed that pirfenidone decreased the production of collagen type 1, which, when uninhibited, increases fibrosis.15
CAPACITY trials. In the CAPACITY trials, two phase 3 multinational studies conducted from 2006 to 2008, patients were given either pirfenidone or placebo.16 In the first study arm, patients were assigned to pirfenidone 2,403 mg/d (n = 174), pirfenidone 1,197 mg/d (n = 87), or placebo (n = 174). In the second study arm, 171 patients received pirfenidone 2,403 mg/d and 173 patients received placebo. Endpoints were measured at baseline and up to week 72.
The first study arm found that the mean rate of decline of FVC—the primary endpoint—was 4.4% less in the treatment group than in the placebo group (p = 0.001), and there was a 36% decrease in risk for death or disease progression in the treatment group (HR, 0.64; p, 0.023). (Endpoints were defined as: time to confirmed > 10% decline in percentage predicted FVC, > 15% decline in percentage predicted DLCO, or death.) The researchers found no clinically significant change in the six-minute walk test—a secondary endpoint of the study.16
The second study arm, however, found no statistically significant change in FVC between the treatment and placebo groups (with a 0.6% smaller decrease in FVC in the pirfenidone group), nor did they see a difference in progression-free survival. However, there was a significant change in the six-minute walk test between the treatment and placebo groups (p = 0.0009). Throughout the study, the most common adverse effects included nausea (36%), rash (32%), and dyspepsia (19%).16
ASCEND trial. The 2014 Assessment of Pirfenidone to Confirm Efficacy and Safety in Idiopathic Pulmonary Fibrosis (ASCEND) trial was a phase 3, multinational, randomized, double-blind, placebo-controlled study of the use of pirfenidone 2,403 mg/d.17 The study was conducted from 2012 to 2013. Of the total number of patients (N = 522), half received pirfenidone and half received placebo. After 52 weeks of treatment (the end of the study), the researchers found a smaller decline in FVC—the primary endpoint—in the treatment group compared to placebo (mean decline, 235 mL vs 428 mL, respectively [p < 0.001]). Regarding the six-minute walk test, the investigators found that 25.9% of the treatment group exhibited a decrease of ≥ 50 meters, compared to 35.7% of the placebo group (p = 0.04). (Progression-free survival was defined as a confirmed ≥ 10% decrease in predicted FVC, a confirmed decrease of 50 meters in the six-minute walk test, or death.)
The pirfenidone group in the ASCEND trial showed a 43% reduced risk for death or disease progression (HR, 0.57; p, < 0.001).16,17 All-cause mortality was lower in the pirfenidone group (4%) than in the placebo group (7.2%), but this was not statistically significant. Deaths from IPF in the pirfenidone group totaled three patients (1.1%) versus seven patients (2.5%) in the placebo group; this was also not statistically significant. The most common adverse effects seen during the study were nausea (36%), rash (28.1%), and headache (25.9%).17
Recommendations for use. Liver function testing should be performed at baseline, monthly for six months, and every three months afterward, as elevations in liver enzymes have been observed.18 Pirfenidone is a CYP1A2 substrate; moderate-to-strong CYP1A2 inhibitors should therefore be discontinued prior to initiation, as they are likely to decrease exposure and efficacy of pirfenidone. There are currently no black box warnings.18
Continue to: Nintedanib
Nintedanib
Hostettler et al studied lung samples from patients with IPF to determine the mechanism of action of nintedanib.19 Evaluation of fibroblasts derived from IPF samples revealed that they contained higher levels of platelet-derived growth factor (PDGF) than did nonfibrotic control cells. They also found that nintedanib, a tyrosine kinase inhibitor, significantly inhibited the phosphorylation of fibrotic-inducing growth factors—PDGF as well as vascular endothelial growth factor (VEGF).
INPULSIS trials. A phase 3 replicate of randomized, double-blind, multinational studies, the INPULSIS trials were performed between 2011 and 2012.20 Two study arms were used to evaluate a total of 638 patients who received nintedanib 150 mg bid for 52 weeks. The primary endpoint was annual rate of decline of FVC.
The researchers also evaluated efficacy through two other endpoints: patient-reported quality of life and symptoms via the St. George’s Respiratory Questionnaire (SGRQ) and evaluation of time to acute exacerbation. The latter was defined as worsening or new dyspnea, new diffuse pulmonary infiltrates visualized on chest radiography and/or HRCT, or the development of parenchymal abnormalities with no pneumothorax or pleural effusion since the preceding visit; and exclusion of any known causes of acute worsening, including infection, heart failure, pulmonary embolism, and any identifiable cause of acute lung injury.20
INPULSIS 1 (first arm) included 309 patients in the treatment group. Results showed an adjusted annual rate of decline in FVC of 114.7 mL/year, versus 239.9 mL/year in the placebo group (p < 0.001). In the treatment group, 52.8% exhibited ≤ 5% decline in FVC, compared to 38.2% in the placebo group (p = 0.001). No significant between-group differences were found in SGRQ score or time to acute exacerbation.20
INPULSIS 2 had 329 patients receiving nintedanib. An annual rate of decline in FVC of 113.6 mL/year from baseline was observed in the treatment group, compared to 207.3 mL/year in the placebo group (p < 0.001). In the treatment group, 53.2% showed ≤ 5% decline in FVC, versus 39.3% in the placebo group (p = 0.001). There was also a significantly smaller increase in total SGRQ score (meaning, less deterioration in quality of life) in the nintedanib group versus the placebo group (p = 0.02). A statistically significant increase in time to first acute exacerbation was observed in the nintedanib group (p = 0.005).20
There was no significant difference between groups in death from any cause, death from respiratory causation, or death that occurred between randomization and 28 days post treatment. The most common adverse effects seen throughout the two trials included diarrhea (trial 1, 61.5%; trial 2, 63.2%), nausea (trial 1, 22.7%; trial 2, 26.1%), and nasopharyngitis (trial 1, 12.6%; trial 2, 14.6%).20
Recommendations for use. Liver function testing should be performed at baseline, at regular intervals during the first three months, then periodically thereafter; patients in the treatment group of both INPULSIS trials had elevated liver enzymes, and cases of drug-induced liver injury have been observed with use of nintedanib.21 This medication may increase risk for bleeding due to its mechanism of action (VEGFR inhibition). Coadministration with CYP3A4 inhibitors may increase concentration of nintedanib; therefore, close monitoring is recommended. Avoid coadministration with CYP3A4 inducers, as this may decrease concentration of nintedanib by 50%. There are currently no black box warnings.21
Continue to: Patient monitoring
Patient monitoring
The ATS recommends measuring FVC and DLCO every three to six months, or sooner if clinically indicated.13 Pulse oximetry should be measured at rest and on exertion in all patients, regardless of symptoms, to assure proper saturation and identify the need for supplemental oxygen; this should also be done every three to six months.
The ATS recommends prompt detection and treatment of comorbidities such as pulmonary hypertension, emphysema, airflow obstruction, GERD, sleep apnea, and coronary artery disease.13 These recommendations are based on the organization’s 2015 guidelines.
OUTCOME FOR THE CASE PATIENT
The patient was started on pirfenidone (2,403 mg/d). He is continuing treatment and showing improvements in quality of life and slowed deterioration of lung function.
CONCLUSION
IPF causes progressive fibrosis of lung interstitium. The etiology is unknown, the symptoms and signs are vague, and mean life expectancy following diagnosis is two to five years. The most recent IPF guidelines recommend avoiding use of anticoagulants and immunosuppressants (eg, steroids, azathioprine, and N-acetylcysteine), due to their proven ineffectiveness and harm to patients with IPF.
Since the FDA’s approval of pirfenidone and nintedanib, the ATS has made recommendations for their use in patients with IPF. Despite mixed results in clinical trials, both drugs have demonstrated the ability to slow the decline in FVC over time, with relatively benign adverse effects. It is difficult to compare pirfenidone and nintedanib, or to recommend use of one drug over the other. However, it is promising that patients with this routinely fatal disease now have treatment options that can potentially modulate their disease progression.
IN THIS ARTICLE
- Confirming the diagnosis
- Pirfenidone treatment
- Nintedanib treatment
A 64-year-old man has a one-year history of dyspnea on exertion and a nonproductive cough. His symptoms are gradually worsening and increasingly bothersome to him.
His medical history includes mild seasonal allergies and GERD, which is well-controlled by oral antihistamines and proton pump inhibitors. He has spent the past 30 years working a desk job as an accountant. He denies a history of smoking, exposure to secondhand smoke, and initiation of new medication.
He admits to increased fatigue, but denies fever, chills, lymphadenopathy, weight change, chest pain, wheezing, abdominal pain, diarrhea, vomiting, claudication, and swelling in the extremities. The rest of the review of systems is negative.
Lab results—complete blood count, comprehensive metabolic panel, TSH, antinuclear antibodies, erythrocyte sedimentation rate, and C-reactive protein—are within normal limits. Spirometry shows very mild restriction. A chest x-ray is abnormal but nonspecific, showing peripheral opacities. An ECG shows normal sinus rhythm.
The patient is given a trial of an inhaled steroid, which yields no improvement. Six months later, the patient is seen by a pulmonologist. Idiopathic pulmonary fibrosis (IPF) is diagnosed based on high-resolution CT (HRCT) and lung biopsy results.
IPF is a chronic, progressive, fibrosing interstitial disease that is limited to lung tissue. It most commonly manifests in older adults with vague symptoms of dyspnea on exertion and nonproductive cough, but symptoms can also include fatigue, muscle and joint aches, clubbing of the fingernails, and weight loss.1 The average life expectancy following diagnosis of IPF is two to five years, and the mortality rate is estimated at 64.3 per million men and 58.4 per million women per year.2,3
Continue to: DIAGNOSIS
DIAGNOSIS
IPF belongs in the general class of idiopathic interstitial pneumonias (IIPs), which are characterized by varying degrees of inflammation and fibrosis of lung interstitium.4 All subtypes of IIPs cause dyspnea and diffuse abnormalities on HRCT, and all vary from each other histologically. Table 1 outlines the key features of each.5-8
Because of its vague symptomology and the extensive workup needed to rule out other diseases, patients with IPF often have symptoms for one to two years before a diagnosis is made.1 Physical exam may reveal fine inspiratory rales in both lung bases and digital clubbing; eventual signs of pulmonary hypertension and right-sided heart failure may be appreciated.1,9
There are no specific diagnostic laboratory tests to confirm IPF; however, baseline labwork (as outlined in the case presentation) is typically ordered to rule out infection, thyroid disease, or connective tissue disease.10 Many patients are referred to a cardiologist before being seen by a pulmonologist; cardiac stress testing may be done, and an echocardiogram may be performed to rule out heart failure.
Diagnostic testing may include pulmonary function testing, HRCT of the chest, and lung biopsy.10 Tissue samples from patients with IPF reveal different stages of disease, including dense fibrosis with honeycombing, subpleural or paraseptal distribution, fibroblast foci, and normal tissue.11 Pulmonary function test results will show a restrictive pattern. Both forced expiratory volume in one second (FEV1) and forced vital capacity (FVC) will be reduced, and the FEV1/FVC ratio preserved. Due to decreased functional lung volume, diffusing capacity of the lung for carbon monoxide (DLCO) will also be reduced.4,12
The differential is broad and includes allergic asthma, bronchitis, COPD, lung cancer, hypersensitivity pneumonitis, asbestosis, or pulmonary embolism.
Continue to: TREATMENT HISTORY
TREATMENT HISTORY
IPF has a long history of tried and failed treatment options. The American Thoracic Society (ATS), in concert with other professional organizations, has published comprehensive guidelines and recommendations pertaining to the use of pharmacologic medications to control disease progression. Warfarin and other anticoagulants have been studied, based on the observation that a procoagulant state promotes fibrotic changes in the lung tissue.13 However, anticoagulant use is not recommended in patients with IPF due to lack of efficacy and high potential for harm.13
Immunosuppressants have also been in the spotlight as possible treatment for IPF, but a clinical study investigating the efficacy of a three-drug regimen including prednisone, azathioprine, and N-acetylcysteine was stopped early due to increased risk for harm. Endothelin antagonists and potent tyrosine kinase inhibitors are also not recommended in the most recent edition of IPF guidelines, as they lack benefit.13
In fact, prior to the 2015 edition of the guidelines, no single medication was routinely recommended for patients with IPF. But this is now changing, following the 2014 FDA approval of two new drugs, nintedanib and pirfenidone, designed specifically to treat IPF.14 These drugs have shown promise in clinical trials (results of which are summarized in Table 2).
Continue to: NEW PHARMACOLOGIC OPTIONS
NEW PHARMACOLOGIC OPTIONS
Pirfenidone
In 2008, a study was conducted in Japan to determine the mechanism of action of pirfenidone.15 Through in vitro studies of healthy adult lung fibroblasts with added pro-fibrotic factor and transforming growth factor (TGF-ß 1), the researchers found that pirfenidone was effective at decreasing the production of a collagen-binding protein called HSP47. This protein is ubiquitous in fibrotic tissue. The study also showed that pirfenidone decreased the production of collagen type 1, which, when uninhibited, increases fibrosis.15
CAPACITY trials. In the CAPACITY trials, two phase 3 multinational studies conducted from 2006 to 2008, patients were given either pirfenidone or placebo.16 In the first study arm, patients were assigned to pirfenidone 2,403 mg/d (n = 174), pirfenidone 1,197 mg/d (n = 87), or placebo (n = 174). In the second study arm, 171 patients received pirfenidone 2,403 mg/d and 173 patients received placebo. Endpoints were measured at baseline and up to week 72.
The first study arm found that the mean rate of decline of FVC—the primary endpoint—was 4.4% less in the treatment group than in the placebo group (p = 0.001), and there was a 36% decrease in risk for death or disease progression in the treatment group (HR, 0.64; p, 0.023). (Endpoints were defined as: time to confirmed > 10% decline in percentage predicted FVC, > 15% decline in percentage predicted DLCO, or death.) The researchers found no clinically significant change in the six-minute walk test—a secondary endpoint of the study.16
The second study arm, however, found no statistically significant change in FVC between the treatment and placebo groups (with a 0.6% smaller decrease in FVC in the pirfenidone group), nor did they see a difference in progression-free survival. However, there was a significant change in the six-minute walk test between the treatment and placebo groups (p = 0.0009). Throughout the study, the most common adverse effects included nausea (36%), rash (32%), and dyspepsia (19%).16
ASCEND trial. The 2014 Assessment of Pirfenidone to Confirm Efficacy and Safety in Idiopathic Pulmonary Fibrosis (ASCEND) trial was a phase 3, multinational, randomized, double-blind, placebo-controlled study of the use of pirfenidone 2,403 mg/d.17 The study was conducted from 2012 to 2013. Of the total number of patients (N = 522), half received pirfenidone and half received placebo. After 52 weeks of treatment (the end of the study), the researchers found a smaller decline in FVC—the primary endpoint—in the treatment group compared to placebo (mean decline, 235 mL vs 428 mL, respectively [p < 0.001]). Regarding the six-minute walk test, the investigators found that 25.9% of the treatment group exhibited a decrease of ≥ 50 meters, compared to 35.7% of the placebo group (p = 0.04). (Progression-free survival was defined as a confirmed ≥ 10% decrease in predicted FVC, a confirmed decrease of 50 meters in the six-minute walk test, or death.)
The pirfenidone group in the ASCEND trial showed a 43% reduced risk for death or disease progression (HR, 0.57; p, < 0.001).16,17 All-cause mortality was lower in the pirfenidone group (4%) than in the placebo group (7.2%), but this was not statistically significant. Deaths from IPF in the pirfenidone group totaled three patients (1.1%) versus seven patients (2.5%) in the placebo group; this was also not statistically significant. The most common adverse effects seen during the study were nausea (36%), rash (28.1%), and headache (25.9%).17
Recommendations for use. Liver function testing should be performed at baseline, monthly for six months, and every three months afterward, as elevations in liver enzymes have been observed.18 Pirfenidone is a CYP1A2 substrate; moderate-to-strong CYP1A2 inhibitors should therefore be discontinued prior to initiation, as they are likely to decrease exposure and efficacy of pirfenidone. There are currently no black box warnings.18
Continue to: Nintedanib
Nintedanib
Hostettler et al studied lung samples from patients with IPF to determine the mechanism of action of nintedanib.19 Evaluation of fibroblasts derived from IPF samples revealed that they contained higher levels of platelet-derived growth factor (PDGF) than did nonfibrotic control cells. They also found that nintedanib, a tyrosine kinase inhibitor, significantly inhibited the phosphorylation of fibrotic-inducing growth factors—PDGF as well as vascular endothelial growth factor (VEGF).
INPULSIS trials. A phase 3 replicate of randomized, double-blind, multinational studies, the INPULSIS trials were performed between 2011 and 2012.20 Two study arms were used to evaluate a total of 638 patients who received nintedanib 150 mg bid for 52 weeks. The primary endpoint was annual rate of decline of FVC.
The researchers also evaluated efficacy through two other endpoints: patient-reported quality of life and symptoms via the St. George’s Respiratory Questionnaire (SGRQ) and evaluation of time to acute exacerbation. The latter was defined as worsening or new dyspnea, new diffuse pulmonary infiltrates visualized on chest radiography and/or HRCT, or the development of parenchymal abnormalities with no pneumothorax or pleural effusion since the preceding visit; and exclusion of any known causes of acute worsening, including infection, heart failure, pulmonary embolism, and any identifiable cause of acute lung injury.20
INPULSIS 1 (first arm) included 309 patients in the treatment group. Results showed an adjusted annual rate of decline in FVC of 114.7 mL/year, versus 239.9 mL/year in the placebo group (p < 0.001). In the treatment group, 52.8% exhibited ≤ 5% decline in FVC, compared to 38.2% in the placebo group (p = 0.001). No significant between-group differences were found in SGRQ score or time to acute exacerbation.20
INPULSIS 2 had 329 patients receiving nintedanib. An annual rate of decline in FVC of 113.6 mL/year from baseline was observed in the treatment group, compared to 207.3 mL/year in the placebo group (p < 0.001). In the treatment group, 53.2% showed ≤ 5% decline in FVC, versus 39.3% in the placebo group (p = 0.001). There was also a significantly smaller increase in total SGRQ score (meaning, less deterioration in quality of life) in the nintedanib group versus the placebo group (p = 0.02). A statistically significant increase in time to first acute exacerbation was observed in the nintedanib group (p = 0.005).20
There was no significant difference between groups in death from any cause, death from respiratory causation, or death that occurred between randomization and 28 days post treatment. The most common adverse effects seen throughout the two trials included diarrhea (trial 1, 61.5%; trial 2, 63.2%), nausea (trial 1, 22.7%; trial 2, 26.1%), and nasopharyngitis (trial 1, 12.6%; trial 2, 14.6%).20
Recommendations for use. Liver function testing should be performed at baseline, at regular intervals during the first three months, then periodically thereafter; patients in the treatment group of both INPULSIS trials had elevated liver enzymes, and cases of drug-induced liver injury have been observed with use of nintedanib.21 This medication may increase risk for bleeding due to its mechanism of action (VEGFR inhibition). Coadministration with CYP3A4 inhibitors may increase concentration of nintedanib; therefore, close monitoring is recommended. Avoid coadministration with CYP3A4 inducers, as this may decrease concentration of nintedanib by 50%. There are currently no black box warnings.21
Continue to: Patient monitoring
Patient monitoring
The ATS recommends measuring FVC and DLCO every three to six months, or sooner if clinically indicated.13 Pulse oximetry should be measured at rest and on exertion in all patients, regardless of symptoms, to assure proper saturation and identify the need for supplemental oxygen; this should also be done every three to six months.
The ATS recommends prompt detection and treatment of comorbidities such as pulmonary hypertension, emphysema, airflow obstruction, GERD, sleep apnea, and coronary artery disease.13 These recommendations are based on the organization’s 2015 guidelines.
OUTCOME FOR THE CASE PATIENT
The patient was started on pirfenidone (2,403 mg/d). He is continuing treatment and showing improvements in quality of life and slowed deterioration of lung function.
CONCLUSION
IPF causes progressive fibrosis of lung interstitium. The etiology is unknown, the symptoms and signs are vague, and mean life expectancy following diagnosis is two to five years. The most recent IPF guidelines recommend avoiding use of anticoagulants and immunosuppressants (eg, steroids, azathioprine, and N-acetylcysteine), due to their proven ineffectiveness and harm to patients with IPF.
Since the FDA’s approval of pirfenidone and nintedanib, the ATS has made recommendations for their use in patients with IPF. Despite mixed results in clinical trials, both drugs have demonstrated the ability to slow the decline in FVC over time, with relatively benign adverse effects. It is difficult to compare pirfenidone and nintedanib, or to recommend use of one drug over the other. However, it is promising that patients with this routinely fatal disease now have treatment options that can potentially modulate their disease progression.
1. Kim DS, Collard HR, King TE Jr. Classification and natural history of the idiopathic interstitial pneumonias. Proc Am Thorac Soc. 2006;3(4):285-292.
2. Frankel SK, Schwarz MI. Update in idiopathic pulmonary fibrosis. Curr Opin Pulm Med. 2009;15(5):463-469.
3. Olson AL, Swigris JJ, Lezotte DC, et al. Mortality from pulmonary fibrosis increased in the United States from 1992 to 2003. Am J Respir Crit Care Med. 2007;176(3):277-284.
4. Chapman JT. Interstitial lung disease. Cleveland Clinic. August 2010. www.clevelandclinicmeded.com/medical pubs/diseasemanagement/pulmonary/interstitial-lung-disease. Accessed March 12, 2018.
5. Cleveland Clinic. Nonspecific interstitial pneumonia. January 16, 2015. https://my.clevelandclinic.org/health/articles/nonspecific-interstitial-pneumonia. Accessed March 12, 2018.
6. Skandhan AKP, Weerakkody Y. Non-specific interstitial pneumonia. Radiopaedia. https://radiopaedia.org/articles/non-specific-interstitial-pneumonia-1. Accessed March 12, 2018.
7. Tatco V, Weerakkody Y. Lymphocytic interstitial pneumonitis. Radiopaedia. https://radiopaedia.org/articles/lymphocytic-interstitial-pneumonitis-1. Accessed March 12, 2018.
8. King TE Jr, Flaherty KR, Hollingsworth H. Cryptogenic organizing pneumonia. UpToDate. www.uptodate.com/contents/cryptogenic-organizing-pneumonia#H12. Accessed March 12, 2018.
9. Patel NM, Lederer DJ, Borczuk AC, Kawut SM. Pulmonary hypertension in idiopathic pulmonary fibrosis. Chest. 2007; 132(3):998-1006.
10. Lee J. Overview of idiopathic interstitial pneumonias. April 2016. www.merckmanuals.com/professional/pulmonary-disorders/interstitial-lung-diseases/overview-of-idiopathic-interstitial-pneumonias. Accessed March 12, 2018.
11. Lynch DA, Sverzellati N, Travis WD, et al. Diagnostic criteria for idiopathic pulmonary fibrosis: a Fleischner Society White Paper. Lancet Respir Med. 2018;6(2):138-153.
12. Martinez FJ, Flaherty K. Pulmonary function testing in idiopathic interstitial pneumonias. Proc Am Thorac Soc. 2006; 3(4):315-321.
13. Raghu G, Rochwerg B, Zhang Y, et al; American Thoracic Society; European Respiratory Society; Japanese Respiratory Society; Latin American Thoracic Association. An official ATS/ERS/JRS/ALAT clinical practice guideline: treatment of idiopathic pulmonary fibrosis. An update of the 2011 Clinical Practice Guideline. Am J Respir Crit Care Med. 2015; 192(2):e3-e19.
14. Chowdhury BA; FDA. Two FDA drug approvals for idiopathic pulmonary fibrosis (IPF). October 15, 2014. https://blogs.fda.gov/fdavoice/index.php/2014/10/two-fda-drug-approvals-for-idiopathic-pulmonary-fibrosis-ipf/. Accessed March 12, 2018.
15. Nakayama S, Mukae H, Sakamoto N, et al. Pirfenidone inhibits the expression of HSP47 in TGF-beta1-stimulated human lung fibroblasts. Life Sci. 2008; 82(3-4):210-217.
16. Noble PW, Albera C, Bradford WZ, et al; CAPACITY Study Group. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomized trials. Lancet. 2011;377: 1760-1769.
17. King TE Jr, Bradford WZ, Castro-Bernardini S, et al; ASCEND Study Group. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22): 2083-2092.
18. Esbriet [package insert]. South San Francisco, CA: Genentech, Inc; 2016.
19. Hostettler KE, Zhong J, Papakonstantinou E, et al. Anti-fibrotic effects of nintedanib in lung fibroblasts derived from patients with idiopathic pulmonary fibrosis. Respir Res. 2014;15(1):157.
20. Richeldi L, du Bois RM, Raghu G, et al; INPULSIS Trial Investigators. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2071-2082.
21. OFEV [package insert]. Ridgefield, CT: Boehringer Ingelheim Pharmaceuticals, Inc; 2018.
1. Kim DS, Collard HR, King TE Jr. Classification and natural history of the idiopathic interstitial pneumonias. Proc Am Thorac Soc. 2006;3(4):285-292.
2. Frankel SK, Schwarz MI. Update in idiopathic pulmonary fibrosis. Curr Opin Pulm Med. 2009;15(5):463-469.
3. Olson AL, Swigris JJ, Lezotte DC, et al. Mortality from pulmonary fibrosis increased in the United States from 1992 to 2003. Am J Respir Crit Care Med. 2007;176(3):277-284.
4. Chapman JT. Interstitial lung disease. Cleveland Clinic. August 2010. www.clevelandclinicmeded.com/medical pubs/diseasemanagement/pulmonary/interstitial-lung-disease. Accessed March 12, 2018.
5. Cleveland Clinic. Nonspecific interstitial pneumonia. January 16, 2015. https://my.clevelandclinic.org/health/articles/nonspecific-interstitial-pneumonia. Accessed March 12, 2018.
6. Skandhan AKP, Weerakkody Y. Non-specific interstitial pneumonia. Radiopaedia. https://radiopaedia.org/articles/non-specific-interstitial-pneumonia-1. Accessed March 12, 2018.
7. Tatco V, Weerakkody Y. Lymphocytic interstitial pneumonitis. Radiopaedia. https://radiopaedia.org/articles/lymphocytic-interstitial-pneumonitis-1. Accessed March 12, 2018.
8. King TE Jr, Flaherty KR, Hollingsworth H. Cryptogenic organizing pneumonia. UpToDate. www.uptodate.com/contents/cryptogenic-organizing-pneumonia#H12. Accessed March 12, 2018.
9. Patel NM, Lederer DJ, Borczuk AC, Kawut SM. Pulmonary hypertension in idiopathic pulmonary fibrosis. Chest. 2007; 132(3):998-1006.
10. Lee J. Overview of idiopathic interstitial pneumonias. April 2016. www.merckmanuals.com/professional/pulmonary-disorders/interstitial-lung-diseases/overview-of-idiopathic-interstitial-pneumonias. Accessed March 12, 2018.
11. Lynch DA, Sverzellati N, Travis WD, et al. Diagnostic criteria for idiopathic pulmonary fibrosis: a Fleischner Society White Paper. Lancet Respir Med. 2018;6(2):138-153.
12. Martinez FJ, Flaherty K. Pulmonary function testing in idiopathic interstitial pneumonias. Proc Am Thorac Soc. 2006; 3(4):315-321.
13. Raghu G, Rochwerg B, Zhang Y, et al; American Thoracic Society; European Respiratory Society; Japanese Respiratory Society; Latin American Thoracic Association. An official ATS/ERS/JRS/ALAT clinical practice guideline: treatment of idiopathic pulmonary fibrosis. An update of the 2011 Clinical Practice Guideline. Am J Respir Crit Care Med. 2015; 192(2):e3-e19.
14. Chowdhury BA; FDA. Two FDA drug approvals for idiopathic pulmonary fibrosis (IPF). October 15, 2014. https://blogs.fda.gov/fdavoice/index.php/2014/10/two-fda-drug-approvals-for-idiopathic-pulmonary-fibrosis-ipf/. Accessed March 12, 2018.
15. Nakayama S, Mukae H, Sakamoto N, et al. Pirfenidone inhibits the expression of HSP47 in TGF-beta1-stimulated human lung fibroblasts. Life Sci. 2008; 82(3-4):210-217.
16. Noble PW, Albera C, Bradford WZ, et al; CAPACITY Study Group. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomized trials. Lancet. 2011;377: 1760-1769.
17. King TE Jr, Bradford WZ, Castro-Bernardini S, et al; ASCEND Study Group. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22): 2083-2092.
18. Esbriet [package insert]. South San Francisco, CA: Genentech, Inc; 2016.
19. Hostettler KE, Zhong J, Papakonstantinou E, et al. Anti-fibrotic effects of nintedanib in lung fibroblasts derived from patients with idiopathic pulmonary fibrosis. Respir Res. 2014;15(1):157.
20. Richeldi L, du Bois RM, Raghu G, et al; INPULSIS Trial Investigators. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2071-2082.
21. OFEV [package insert]. Ridgefield, CT: Boehringer Ingelheim Pharmaceuticals, Inc; 2018.