User login
‘Scared’ and short of breath
Discuss this article at www.facebook.com/CurrentPsychiatry
CASE: Paranoid and scared
Police bring Mr. C, age 42, to a local crisis center after he is found masturbating in public the same day he was released from jail after serving time for the same behavior. Previously, Mr. C was diagnosed with schizophrenia, paranoid type, and alcohol dependence. He is single, unemployed, and lives with his parents. He has had 3 previous admissions to a psychiatric hospital, but no preexisting medical illness. A judge involuntarily commits Mr. C to our psychiatric facility.
Mr. C looks older than his age and has poor hygiene. He appears bizarre, makes poor eye contact, and speaks slowly but with normal volume. His speech is not coherent, relevant, or goal-directed. He is not able to answer questions properly, chanting “it’s eternity, eternity, eternity.” He shows no tremors, repetitive motor behavior, or muscle rigidity. His affect is flat and he has no suicidal or homicidal ideations. Based on Mr. C’s history, we diagnose him with schizophrenia, paranoid type and alcohol dependence.
Over the next 9 days, Mr. C receives trials of haloperidol, lorazepam, diphenhydramine, ziprasidone, olanzapine, hydroxyzine, trazodone, and benztropine to treat his schizophrenia. From days 1 to 3, all medications are given on an as-needed basis. On day 1, Mr. C receives haloperidol, 20 mg, lorazepam, 9 mg, diphenhydramine, 150 mg, and ziprasidone, 20 mg. On day 2, he receives haloperidol, 15 mg, lorazepam, 10 mg, olanzapine, 20 mg, hydroxyzine, 100 mg, and trazodone, 50 mg. On day 3, he receives haloperidol, 20 mg, lorazepam, 6 mg, and trazodone, 100 mg. On days 4 to 8, in addition to scheduled haloperidol, 30 mg/d, benztropine, 1 mg/d, and trazodone, 100 mg/d, he receives haloperidol, 5 mg, and lorazepam, 2 mg, as needed. On day 9, he receives the scheduled haloperidol, 30 mg/d, benztropine, 1 mg/d, and trazodone, 100 mg/d.
During his stay, Mr. C is incoherent and disorganized. On day 9, he eats all of his lunch, none of his dinner, but sips milk and juice and eats snacks. He drinks 2 small cups of water with medication and 2 small cups of water during oral care. His mucosa and tongue are dry. At 11:30 pm, while lying in bed mumbling “scared, scared,” he experiences shortness of breath. His temperature is 99.6°F, blood pressure is 151/93 mm Hg, pulse is 125 beats per minute, respiratory rate is 40 breaths per minute, and oxygen saturation is 91% on ambient air. Twenty minutes later, his blood pressure increases to 180/120 mm Hg. On physical examination, he has “lead pipe” rigidity of both arms. He is awake, confused, and not able to communicate, still mumbling “scared, scared.” Changes in his blood pressure, pulse, and temperature during his stay in the psychiatric hospital are depicted in Figures 1 and 2, respectively.
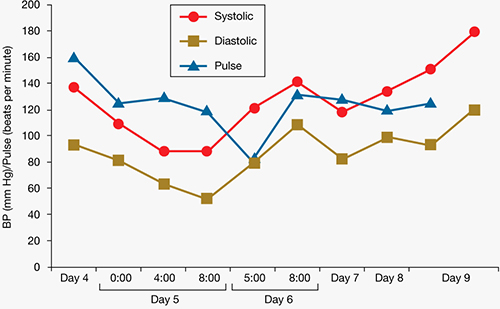
Figure 1: Mr. C’s blood pressure and pulse changes from day 4 to day 9 in the psychiatric hospital
BP: blood pressure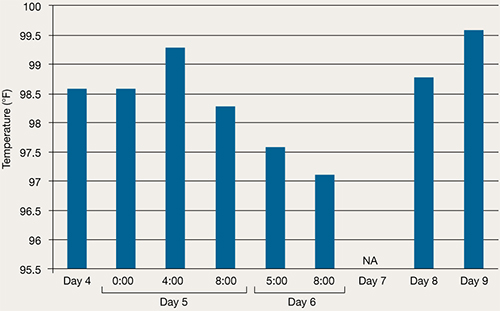
Figure 2: Mr. C’s temperature changes from day 4 to day 9 in the psychiatric hospital
The authors’ observations
NMS is a life-threatening, iatrogenic neurologic emergency associated with antipsychotic use. Early incidence rate estimates ran as high as 3% of patients treated with antipsychotics; however, more recent data suggest an incidence of 0.01% to 0.02%.1 This decrease in frequency likely reflects increased awareness of the disorder, more conservative prescribing patterns, and a shift to using atypical antipsychotics.2 In the mid 1980s and early 1990s the mortality rate was 25% to 30% if NMS was not promptly recognized and treated3; however, progression to more fulminant, lethal NMS episodes now occurs less often and the mortality rate ranges from 10% to 20%.4
If NMS is suspected, immediate transfer to an emergency department (ED) is necessary. Even with early diagnosis, however, complications of NMS are still likely, including:
- rhabdomyolysis
- renal failure
- seizures
- respiratory failure
- aspiration pneumonia
- disseminated intravascular coagulation
- venous thromboembolism.5-9
Caroff et al reported observing a residual catatonic state after acute NMS symptoms subsided.10
Although the pathophysiology of NMS is complex—involving a cascade of dysregulation in multiple neurochemical and neuroendocrine systems—dopamine blockade likely plays a pivotal role in triggering the condition.2 In addition, evidence supports the hypothesis that dysregulated sympathetic nervous system hyperactivity is responsible for most NMS features.11
TREATMENT: Arrival in the ED
Based on his elevated blood pressure (151/93 mm Hg), “lead pipe” rigidity, and increased body temperature associated with Mr. C’s history of haloperidol use for 9 days, the treatment team suspects NMS. Labile blood pressure, which changed from 151/93 to 180/120 mm Hg in 20 minutes, reinforces the NMS diagnosis. Approximately 30 minutes after Mr. C shows signs of NMS, he is transferred to a local ED. He is awake, alert, and communicative after he arrives in the ED, but becomes confused and noncommunicative the next morning. When he arrives in the ED, he is found to have tachycardia (114 beats per minute), tachypnea (26 breaths per minute), blood pressure of 132/84 mm Hg, and temperature of 102°F. In the ED, he is given IV normal saline, diphenhydramine, 25 mg, and IV lorazepam, 1 mg. His rigidity slightly improves.
Early the next morning, his blood pressure is 182/89 mm Hg, respirations are 30 to 40 breaths per minute, and heart rate is 120 beats per minute. He then receives IV lorazepam, 2 mg, after which his tachypnea, tachycardia, and elevated blood pressure improve.
The authors’ observations
A case-control study by Keck et al12 comparing 18 patients with NMS and 36 matched neuroleptic-treated patients with no history of the syndrome identified greater psychomotor agitation, significantly higher doses of neuroleptics, greater rates of dosage increase, and a greater number of IM injections as potential risk factors. Other potential risk factors include use of restraints, pre-existing CNS dopamine activity or receptor function abnormalities, and iron deficiency.2 Agitation, dehydration, and exhaustion were found to be the most consistent systemic factors predisposing patients taking antipsychotics to NMS in small case-control studies.13,14 Well-supported risk factors also include use of high-potency antipsychotics, prior episodes of NMS, age <40, male sex, malnutrition, organic brain syndromes, and lithium use.3,5,15
There is no way to predict the risk of NMS for an individual patient. Usually, symptoms develop within 4 weeks of starting an antipsychotic, but can occur after taking the same dose for many months. The onset may be within hours, but on average it is 4 to 14 days after initiating therapy. Among patients who develop NMS, 90% do so within 10 days.3,5
Mr. C’s risk factors include high-potency antipsychotic use, male sex, relatively high dose (haloperidol, 30 to 35 mg/d), agitation, dehydration, and exhaustion.
Managing NMS
The standard approaches for managing patients with NMS include discontinuing suspected triggering drugs and providing supportive care. Beyond supportive care, oral or IV benzodiazepines may relieve symptoms and speed recovery.2 Dopaminergic drugs, such as bromocriptine or amantadine, used alone or with other treatments, can reduce parkinsonism and disease duration and mortality.2 Dantrolene may be useful only for NMS patients who exhibit extreme temperature elevations, rigidity, and true hypermetabolism.16 Electroconvulsive therapy may be effective for NMS patients whose symptoms do not respond to supportive care and drug therapy or those with residual catatonic or parkinsonian symptoms.2
OUTCOME: Improvement, discharge
Mr. C is admitted to the hospital with the diagnosis of NMS and transferred to the intensive care unit (ICU) for treatment. After Mr. C is admitted to the ICU, apart from continuing the medication given in the ED, he also receives dantrolene, 2 mg/kg, then 1 mg/kg, 4 times a day, as well as IV lorazepam, 1 mg every 6 hours. His other medications include IV pantoprazole, 40 mg/d, for prophylaxis of stress ulcer. Diphenhydramine administration is changed to as needed. On the second day in the ICU, he has only mild upper extremity rigidity but no lower extremity rigidity. However, he suffers 1 seizure, which is treated with IV fosphenytoin at the loading dose, 18 mg/kg, then a maintaining dose of 5 mg phenytoin equivalent/kg/d.
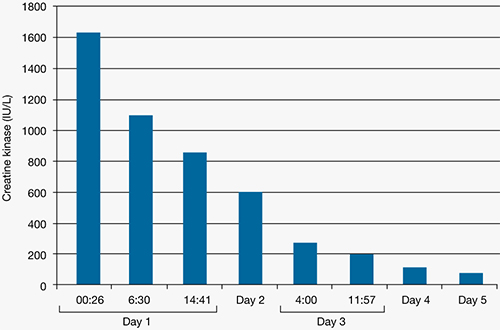
Figure 3: Mr. C’s creatine kinase level (IU/L) during the first 5 days in the intensive care unit
Figure 4: Mr. C’s blood pressure before and after admission
Figure 5: Mr. C’s temperature before and after admissionMr. C remains in the ICU for 7 days. There he receives valproic acid, titrated to 500 mg in the morning and 1,000 mg at bedtime, for agitation. He also receives olanzapine, 5 mg/d, for psychotic symptoms. He develops deep vein thrombosis in the right cephalic vein, which is treated with subcutaneous enoxaparin, 1 mg/kg, and warfarin, 5 mg/d.
He is discharged from the hospital after 2 weeks and returns to the psychiatric facility. He continues to be treated for paranoid schizophrenia with olanzapine, 5 mg/d.
The authors’ observations
High-potency, typical antipsychotics can cause NMS, as shown in Mr. C’s case. It also can be caused by typical low-potency antipsychotics,3 atypical antipsychotics,17 antiemetic drugs,18 and lithium,19,20 and can occur after the withdrawal of levodopa and similar dopaminergic agents during Parkinson’s disease treatment.21 Atypical antipsychotics reported to be associated with NMS include clozapine, risperidone, olanzapine, quetiapine, aripiprazole, ziprasidone, and paliperidone.22-27 Atypical antipsychotic-induced NMS also has been reported in children and adolescents.22,28-30
With the broad application of atypical antipsychotics, physicians should be aware of atypical NMS presentation. Although NMS diagnosis commonly requires core symptoms of hyperthermia and muscle rigidity (Table 1 and 2),31 atypical presentations may not demonstrate temperature changes and/or muscle rigidity or may progress slowly over several days, leading to a delay in diagnosis and treatment.28,30,32,33 Therefore, clinicians should evaluate any patient taking antipsychotics for features of NMS and not prematurely exclude a NMS diagnosis in cases where severe rigidity or hyperthermia is not initially apparent.33
Table 1
DSM-IV-TR criteria for neuroleptic malignant syndrome
| A. The development of severe muscle rigidity and elevated temperature associated with the use of neuroleptic medication |
| B. 2 (or more) of the following: |
|
| Source: Reference 31 |
Table 2
Diagnostic features of neuroleptic malignant syndrome
| Essential features: severe muscle rigidity and elevated temperature in an individual using neuroleptic medication |
| Elevated temperature: from mild (eg, 99º to 100ºF) to markedly hyperthermic states (eg, 106ºF) |
| Creatine kinase: typically elevated, ranging from minor elevations to extremely high levels (exceeding 16,000 IU) |
| Other features: mental status changes, unstable blood pressure, diaphoresis, other signs of autonomic dysfunction |
| Source: Reference 31 |
Related Resource
- Neuroleptic Malignant Syndrome Information Service. www.nmsis.org.
Drug Brand Names
- Amantadine • Symmetrel
- Aripiprazole • Abilify
- Benztropine • Cogentin
- Bromocriptine • Parlodel
- Clozapine • Clozaril
- Dantrolene • Dantrium
- Diphenhydramine • Benadryl
- Enoxaparin • Lovenox
- Fosphenytoin • Cerebyx
- Haloperidol • Haldol
- Hydroxyzine • Vistaril
- Levodopa • Sinemet
- Lithium • Eskalith, Lithobid, others
- Lorazepam • Ativan
- Olanzapine • Zyprexa
- Paliperidone • Invega
- Pantoprazole • Protonix
- Phenytoin • Dilantin
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Trazodone • Desyrel, Oleptro
- Valproic acid • Depakote
- Warfarin • Coumadin
- Ziprasidone • Geodon
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Acknowledgements
The authors are very grateful for the critical reviews by James R. Allen, MD, MPH, professor of Child and Adolescent Psychiatry Fellowship Program at the University of Oklahoma and Lori Hake, DO, director of Psychiatry Residency Training Program at Griffin Memorial Hospital in Norman, OK.
1. Stubner S, Rustenbeck E, Grohmann R, et al. Severe and uncommon involuntary movement disorders due to psychotropic drugs. Pharmacopsychiatry. 2004;37:S54-S64.
2. Strawn JR, Keck PE, Jr, Caroff SN. Neuroleptic malignant syndrome. Am J Psychiatry. 2007;164:870-876.
3. Ropper AH, Brown RH. Adams and Victor’s principles of neurology. 8th ed. New York, NY: McGraw Hill; 2005;1025-1026.
4. Sheil AT, Collins KA, Schandl CA, et al. Fetal neurotoxic response to neuroleptic medications: case report and review of the literature. Am J Forensic Med Pathol. 2007;28:116-120.
5. Balzan MV. The neuroleptic malignant syndrome: a logical approach to the patient with temperature and rigidity. Postgrad Med J. 1998;74:72-76.
6. Caroff SN, Mann SC. Neuroleptic malignant syndrome. Med Clin North Am. 1993;77:185-202.
7. Caroff SN, Rosenberg H, Mann SC, et al. Neuroleptic malignant syndrome in the critical care unit. Crit Care Med. 2002;30:2609-2610.
8. Caroff SN, Campbell EC, Sullivan KA. Neuroleptic malignant syndrome in elderly patients. Expert Rev Neurother. 2007;7:423-431.
9. Gurrera RJ, Simpson JC, Tsuang MT. Meta-analytic evidence of systematic bias in estimates of neuroleptic malignant syndrome incidence. Compr Psychiatry. 2007;48:205-211.
10. Caroff SN, Mann SC, Keck PE, Jr, et al. Residual catatonic state following neuroleptic malignant syndrome. J Clin Psychopharmacol. 2001;21:121-122.
11. Gurrera RJ. Sympathoadrenal hyperactivity and the etiology of neuroleptic malignant syndrome. Am J Psychiatry. 1999;156:169-180.
12. Keck PE, Jr, Pope HG, Jr, Cohen BM, et al. Risk factors for neuroleptic malignant syndrome. A case-control study. Arch Gen Psychiatry. 1989;46:914-918.
13. Berardi D, Amore M, Keck PE, Jr, et al. Clinical and pharmacologic risk factors for neuroleptic malignant syndrome: a case-control study. Biol Psychiatry. 1998;44:748-754.
14. Rosebush PI, Stewart TD. A prospective analysis of 24 episodes of neuroleptic malignant syndrome. Am J Psychiatry. 1989;146:717-725.
15. Martinez M, Marangell LB, Martinez JM. Psychopharmacology. In: Hales RE, Yudofsky SC, Gabbard GO, eds. American Psychiatric Publishing textbook of psychiatry. Arlington, VA: American Psychiatric Publishing, Inc.; 2008:1059-1132.
16. Caroff SN. Neuroleptic malignant syndrome. In: Mann SC, Caroff SN, Keck PE Jr, et al, eds. Neuroleptic malignant syndrome and related conditions. Washington, DC: American Psychiatric Publishing; 2003:1-44.
17. Hammerman S, Lam C, Caroff SN. Neuroleptic malignant syndrome and aripiprazole. J Am Acad Child Adolesc Psychiatry. 2006;45:639-641.
18. Stein MH, Sorscher M, Caroff SN. Neuroleptic malignant syndrome induced by metoclopramide in an infant with Freeman-Sheldon syndrome. Anesth Analg. 2006;103:786-787.
19. Borovicka MC, Bond LC, Gaughan KM. Ziprasidone- and lithium-induced neuroleptic malignant syndrome. Ann Pharmacother. 2006;40:139-142.
20. Gill J, Singh H, Nugent K. Acute lithium intoxication and neuroleptic malignant syndrome. Pharmacotherapy. 2003;23:811-815.
21. Ward C. Neuroleptic malignant syndrome in a patient with Parkinson’s disease: a case study. J Neurosci Nurs. 2005;37:160-162.
22. Leibold J, Patel V, Hasan RA. Neuroleptic malignant syndrome associated with ziprasidone in an adolescent. Clin Ther. 2004;26:1105-1108.
23. Corallo CE, Ernest D. Atypical neuroleptic malignant syndrome with long-term clozapine. Crit Care Resusc. 2007;9:338-340.
24. Molina D, Tingle LE, Lu X. Aripiprazole as the causative agent of neuroleptic malignant syndrome: a case report. Prim Care Companion J Clin Psychiatry. 2007;9:148-150.
25. Trollor JN, Chen X, Sachdev PS. Neuroleptic malignant syndrome associated with atypical antipsychotic drugs. CNS Drugs. 2009;23:477-492.
26. Gortney JS, Fagan A, Kissack JC. Neuroleptic malignant syndrome secondary to quetiapine. Ann Pharmacother. 2009;43:785-791.
27. Han C, Lee SJ, Pae CU. Paliperidone-associated atypical neuroleptic malignant syndrome: a case report. Prog Neuropsychopharmacol Biol Psychiatry. 2011;35:650-651.
28. Hanft A, Eggleston CF, Bourgeois JA. Neuroleptic malignant syndrome in an adolescent after brief exposure to olanzapine. J Child Adolesc Psychopharmacol. 2004;14:481-487.
29. Abu-Kishk I, Toledano M, Reis A, et al. Neuroleptic malignant syndrome in a child treated with an atypical antipsychotic. J Toxicol Clin Toxicol. 2004;42:921-925.
30. Neuhut R, Lindenmayer JP, Silva R. Neuroleptic malignant syndrome in children and adolescents on atypical antipsychotic medication: a review. J Child Adolesc Psychopharmacol. 2009;19:415-422.
31. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association: 2000.
32. Carroll BT, Surber SA. The problem of atypical neuroleptic malignant syndrome: a case report. Psychiatry (Edgmont). 2009;6:45-47.
33. Picard LS, Lindsay S, Strawn JR, et al. Atypical neuroleptic malignant syndrome: diagnostic controversies and considerations. Pharmacotherapy. 2008;28:530-535.
Discuss this article at www.facebook.com/CurrentPsychiatry
CASE: Paranoid and scared
Police bring Mr. C, age 42, to a local crisis center after he is found masturbating in public the same day he was released from jail after serving time for the same behavior. Previously, Mr. C was diagnosed with schizophrenia, paranoid type, and alcohol dependence. He is single, unemployed, and lives with his parents. He has had 3 previous admissions to a psychiatric hospital, but no preexisting medical illness. A judge involuntarily commits Mr. C to our psychiatric facility.
Mr. C looks older than his age and has poor hygiene. He appears bizarre, makes poor eye contact, and speaks slowly but with normal volume. His speech is not coherent, relevant, or goal-directed. He is not able to answer questions properly, chanting “it’s eternity, eternity, eternity.” He shows no tremors, repetitive motor behavior, or muscle rigidity. His affect is flat and he has no suicidal or homicidal ideations. Based on Mr. C’s history, we diagnose him with schizophrenia, paranoid type and alcohol dependence.
Over the next 9 days, Mr. C receives trials of haloperidol, lorazepam, diphenhydramine, ziprasidone, olanzapine, hydroxyzine, trazodone, and benztropine to treat his schizophrenia. From days 1 to 3, all medications are given on an as-needed basis. On day 1, Mr. C receives haloperidol, 20 mg, lorazepam, 9 mg, diphenhydramine, 150 mg, and ziprasidone, 20 mg. On day 2, he receives haloperidol, 15 mg, lorazepam, 10 mg, olanzapine, 20 mg, hydroxyzine, 100 mg, and trazodone, 50 mg. On day 3, he receives haloperidol, 20 mg, lorazepam, 6 mg, and trazodone, 100 mg. On days 4 to 8, in addition to scheduled haloperidol, 30 mg/d, benztropine, 1 mg/d, and trazodone, 100 mg/d, he receives haloperidol, 5 mg, and lorazepam, 2 mg, as needed. On day 9, he receives the scheduled haloperidol, 30 mg/d, benztropine, 1 mg/d, and trazodone, 100 mg/d.
During his stay, Mr. C is incoherent and disorganized. On day 9, he eats all of his lunch, none of his dinner, but sips milk and juice and eats snacks. He drinks 2 small cups of water with medication and 2 small cups of water during oral care. His mucosa and tongue are dry. At 11:30 pm, while lying in bed mumbling “scared, scared,” he experiences shortness of breath. His temperature is 99.6°F, blood pressure is 151/93 mm Hg, pulse is 125 beats per minute, respiratory rate is 40 breaths per minute, and oxygen saturation is 91% on ambient air. Twenty minutes later, his blood pressure increases to 180/120 mm Hg. On physical examination, he has “lead pipe” rigidity of both arms. He is awake, confused, and not able to communicate, still mumbling “scared, scared.” Changes in his blood pressure, pulse, and temperature during his stay in the psychiatric hospital are depicted in Figures 1 and 2, respectively.
Figure 1: Mr. C’s blood pressure and pulse changes from day 4 to day 9 in the psychiatric hospital
BP: blood pressure
Figure 2: Mr. C’s temperature changes from day 4 to day 9 in the psychiatric hospital
The authors’ observations
NMS is a life-threatening, iatrogenic neurologic emergency associated with antipsychotic use. Early incidence rate estimates ran as high as 3% of patients treated with antipsychotics; however, more recent data suggest an incidence of 0.01% to 0.02%.1 This decrease in frequency likely reflects increased awareness of the disorder, more conservative prescribing patterns, and a shift to using atypical antipsychotics.2 In the mid 1980s and early 1990s the mortality rate was 25% to 30% if NMS was not promptly recognized and treated3; however, progression to more fulminant, lethal NMS episodes now occurs less often and the mortality rate ranges from 10% to 20%.4
If NMS is suspected, immediate transfer to an emergency department (ED) is necessary. Even with early diagnosis, however, complications of NMS are still likely, including:
- rhabdomyolysis
- renal failure
- seizures
- respiratory failure
- aspiration pneumonia
- disseminated intravascular coagulation
- venous thromboembolism.5-9
Caroff et al reported observing a residual catatonic state after acute NMS symptoms subsided.10
Although the pathophysiology of NMS is complex—involving a cascade of dysregulation in multiple neurochemical and neuroendocrine systems—dopamine blockade likely plays a pivotal role in triggering the condition.2 In addition, evidence supports the hypothesis that dysregulated sympathetic nervous system hyperactivity is responsible for most NMS features.11
TREATMENT: Arrival in the ED
Based on his elevated blood pressure (151/93 mm Hg), “lead pipe” rigidity, and increased body temperature associated with Mr. C’s history of haloperidol use for 9 days, the treatment team suspects NMS. Labile blood pressure, which changed from 151/93 to 180/120 mm Hg in 20 minutes, reinforces the NMS diagnosis. Approximately 30 minutes after Mr. C shows signs of NMS, he is transferred to a local ED. He is awake, alert, and communicative after he arrives in the ED, but becomes confused and noncommunicative the next morning. When he arrives in the ED, he is found to have tachycardia (114 beats per minute), tachypnea (26 breaths per minute), blood pressure of 132/84 mm Hg, and temperature of 102°F. In the ED, he is given IV normal saline, diphenhydramine, 25 mg, and IV lorazepam, 1 mg. His rigidity slightly improves.
Early the next morning, his blood pressure is 182/89 mm Hg, respirations are 30 to 40 breaths per minute, and heart rate is 120 beats per minute. He then receives IV lorazepam, 2 mg, after which his tachypnea, tachycardia, and elevated blood pressure improve.
The authors’ observations
A case-control study by Keck et al12 comparing 18 patients with NMS and 36 matched neuroleptic-treated patients with no history of the syndrome identified greater psychomotor agitation, significantly higher doses of neuroleptics, greater rates of dosage increase, and a greater number of IM injections as potential risk factors. Other potential risk factors include use of restraints, pre-existing CNS dopamine activity or receptor function abnormalities, and iron deficiency.2 Agitation, dehydration, and exhaustion were found to be the most consistent systemic factors predisposing patients taking antipsychotics to NMS in small case-control studies.13,14 Well-supported risk factors also include use of high-potency antipsychotics, prior episodes of NMS, age <40, male sex, malnutrition, organic brain syndromes, and lithium use.3,5,15
There is no way to predict the risk of NMS for an individual patient. Usually, symptoms develop within 4 weeks of starting an antipsychotic, but can occur after taking the same dose for many months. The onset may be within hours, but on average it is 4 to 14 days after initiating therapy. Among patients who develop NMS, 90% do so within 10 days.3,5
Mr. C’s risk factors include high-potency antipsychotic use, male sex, relatively high dose (haloperidol, 30 to 35 mg/d), agitation, dehydration, and exhaustion.
Managing NMS
The standard approaches for managing patients with NMS include discontinuing suspected triggering drugs and providing supportive care. Beyond supportive care, oral or IV benzodiazepines may relieve symptoms and speed recovery.2 Dopaminergic drugs, such as bromocriptine or amantadine, used alone or with other treatments, can reduce parkinsonism and disease duration and mortality.2 Dantrolene may be useful only for NMS patients who exhibit extreme temperature elevations, rigidity, and true hypermetabolism.16 Electroconvulsive therapy may be effective for NMS patients whose symptoms do not respond to supportive care and drug therapy or those with residual catatonic or parkinsonian symptoms.2
OUTCOME: Improvement, discharge
Mr. C is admitted to the hospital with the diagnosis of NMS and transferred to the intensive care unit (ICU) for treatment. After Mr. C is admitted to the ICU, apart from continuing the medication given in the ED, he also receives dantrolene, 2 mg/kg, then 1 mg/kg, 4 times a day, as well as IV lorazepam, 1 mg every 6 hours. His other medications include IV pantoprazole, 40 mg/d, for prophylaxis of stress ulcer. Diphenhydramine administration is changed to as needed. On the second day in the ICU, he has only mild upper extremity rigidity but no lower extremity rigidity. However, he suffers 1 seizure, which is treated with IV fosphenytoin at the loading dose, 18 mg/kg, then a maintaining dose of 5 mg phenytoin equivalent/kg/d.
Figure 3: Mr. C’s creatine kinase level (IU/L) during the first 5 days in the intensive care unit
Figure 4: Mr. C’s blood pressure before and after admission
Figure 5: Mr. C’s temperature before and after admissionMr. C remains in the ICU for 7 days. There he receives valproic acid, titrated to 500 mg in the morning and 1,000 mg at bedtime, for agitation. He also receives olanzapine, 5 mg/d, for psychotic symptoms. He develops deep vein thrombosis in the right cephalic vein, which is treated with subcutaneous enoxaparin, 1 mg/kg, and warfarin, 5 mg/d.
He is discharged from the hospital after 2 weeks and returns to the psychiatric facility. He continues to be treated for paranoid schizophrenia with olanzapine, 5 mg/d.
The authors’ observations
High-potency, typical antipsychotics can cause NMS, as shown in Mr. C’s case. It also can be caused by typical low-potency antipsychotics,3 atypical antipsychotics,17 antiemetic drugs,18 and lithium,19,20 and can occur after the withdrawal of levodopa and similar dopaminergic agents during Parkinson’s disease treatment.21 Atypical antipsychotics reported to be associated with NMS include clozapine, risperidone, olanzapine, quetiapine, aripiprazole, ziprasidone, and paliperidone.22-27 Atypical antipsychotic-induced NMS also has been reported in children and adolescents.22,28-30
With the broad application of atypical antipsychotics, physicians should be aware of atypical NMS presentation. Although NMS diagnosis commonly requires core symptoms of hyperthermia and muscle rigidity (Table 1 and 2),31 atypical presentations may not demonstrate temperature changes and/or muscle rigidity or may progress slowly over several days, leading to a delay in diagnosis and treatment.28,30,32,33 Therefore, clinicians should evaluate any patient taking antipsychotics for features of NMS and not prematurely exclude a NMS diagnosis in cases where severe rigidity or hyperthermia is not initially apparent.33
Table 1
DSM-IV-TR criteria for neuroleptic malignant syndrome
| A. The development of severe muscle rigidity and elevated temperature associated with the use of neuroleptic medication |
| B. 2 (or more) of the following: |
|
| Source: Reference 31 |
Table 2
Diagnostic features of neuroleptic malignant syndrome
| Essential features: severe muscle rigidity and elevated temperature in an individual using neuroleptic medication |
| Elevated temperature: from mild (eg, 99º to 100ºF) to markedly hyperthermic states (eg, 106ºF) |
| Creatine kinase: typically elevated, ranging from minor elevations to extremely high levels (exceeding 16,000 IU) |
| Other features: mental status changes, unstable blood pressure, diaphoresis, other signs of autonomic dysfunction |
| Source: Reference 31 |
Related Resource
- Neuroleptic Malignant Syndrome Information Service. www.nmsis.org.
Drug Brand Names
- Amantadine • Symmetrel
- Aripiprazole • Abilify
- Benztropine • Cogentin
- Bromocriptine • Parlodel
- Clozapine • Clozaril
- Dantrolene • Dantrium
- Diphenhydramine • Benadryl
- Enoxaparin • Lovenox
- Fosphenytoin • Cerebyx
- Haloperidol • Haldol
- Hydroxyzine • Vistaril
- Levodopa • Sinemet
- Lithium • Eskalith, Lithobid, others
- Lorazepam • Ativan
- Olanzapine • Zyprexa
- Paliperidone • Invega
- Pantoprazole • Protonix
- Phenytoin • Dilantin
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Trazodone • Desyrel, Oleptro
- Valproic acid • Depakote
- Warfarin • Coumadin
- Ziprasidone • Geodon
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Acknowledgements
The authors are very grateful for the critical reviews by James R. Allen, MD, MPH, professor of Child and Adolescent Psychiatry Fellowship Program at the University of Oklahoma and Lori Hake, DO, director of Psychiatry Residency Training Program at Griffin Memorial Hospital in Norman, OK.
Discuss this article at www.facebook.com/CurrentPsychiatry
CASE: Paranoid and scared
Police bring Mr. C, age 42, to a local crisis center after he is found masturbating in public the same day he was released from jail after serving time for the same behavior. Previously, Mr. C was diagnosed with schizophrenia, paranoid type, and alcohol dependence. He is single, unemployed, and lives with his parents. He has had 3 previous admissions to a psychiatric hospital, but no preexisting medical illness. A judge involuntarily commits Mr. C to our psychiatric facility.
Mr. C looks older than his age and has poor hygiene. He appears bizarre, makes poor eye contact, and speaks slowly but with normal volume. His speech is not coherent, relevant, or goal-directed. He is not able to answer questions properly, chanting “it’s eternity, eternity, eternity.” He shows no tremors, repetitive motor behavior, or muscle rigidity. His affect is flat and he has no suicidal or homicidal ideations. Based on Mr. C’s history, we diagnose him with schizophrenia, paranoid type and alcohol dependence.
Over the next 9 days, Mr. C receives trials of haloperidol, lorazepam, diphenhydramine, ziprasidone, olanzapine, hydroxyzine, trazodone, and benztropine to treat his schizophrenia. From days 1 to 3, all medications are given on an as-needed basis. On day 1, Mr. C receives haloperidol, 20 mg, lorazepam, 9 mg, diphenhydramine, 150 mg, and ziprasidone, 20 mg. On day 2, he receives haloperidol, 15 mg, lorazepam, 10 mg, olanzapine, 20 mg, hydroxyzine, 100 mg, and trazodone, 50 mg. On day 3, he receives haloperidol, 20 mg, lorazepam, 6 mg, and trazodone, 100 mg. On days 4 to 8, in addition to scheduled haloperidol, 30 mg/d, benztropine, 1 mg/d, and trazodone, 100 mg/d, he receives haloperidol, 5 mg, and lorazepam, 2 mg, as needed. On day 9, he receives the scheduled haloperidol, 30 mg/d, benztropine, 1 mg/d, and trazodone, 100 mg/d.
During his stay, Mr. C is incoherent and disorganized. On day 9, he eats all of his lunch, none of his dinner, but sips milk and juice and eats snacks. He drinks 2 small cups of water with medication and 2 small cups of water during oral care. His mucosa and tongue are dry. At 11:30 pm, while lying in bed mumbling “scared, scared,” he experiences shortness of breath. His temperature is 99.6°F, blood pressure is 151/93 mm Hg, pulse is 125 beats per minute, respiratory rate is 40 breaths per minute, and oxygen saturation is 91% on ambient air. Twenty minutes later, his blood pressure increases to 180/120 mm Hg. On physical examination, he has “lead pipe” rigidity of both arms. He is awake, confused, and not able to communicate, still mumbling “scared, scared.” Changes in his blood pressure, pulse, and temperature during his stay in the psychiatric hospital are depicted in Figures 1 and 2, respectively.
Figure 1: Mr. C’s blood pressure and pulse changes from day 4 to day 9 in the psychiatric hospital
BP: blood pressure
Figure 2: Mr. C’s temperature changes from day 4 to day 9 in the psychiatric hospital
The authors’ observations
NMS is a life-threatening, iatrogenic neurologic emergency associated with antipsychotic use. Early incidence rate estimates ran as high as 3% of patients treated with antipsychotics; however, more recent data suggest an incidence of 0.01% to 0.02%.1 This decrease in frequency likely reflects increased awareness of the disorder, more conservative prescribing patterns, and a shift to using atypical antipsychotics.2 In the mid 1980s and early 1990s the mortality rate was 25% to 30% if NMS was not promptly recognized and treated3; however, progression to more fulminant, lethal NMS episodes now occurs less often and the mortality rate ranges from 10% to 20%.4
If NMS is suspected, immediate transfer to an emergency department (ED) is necessary. Even with early diagnosis, however, complications of NMS are still likely, including:
- rhabdomyolysis
- renal failure
- seizures
- respiratory failure
- aspiration pneumonia
- disseminated intravascular coagulation
- venous thromboembolism.5-9
Caroff et al reported observing a residual catatonic state after acute NMS symptoms subsided.10
Although the pathophysiology of NMS is complex—involving a cascade of dysregulation in multiple neurochemical and neuroendocrine systems—dopamine blockade likely plays a pivotal role in triggering the condition.2 In addition, evidence supports the hypothesis that dysregulated sympathetic nervous system hyperactivity is responsible for most NMS features.11
TREATMENT: Arrival in the ED
Based on his elevated blood pressure (151/93 mm Hg), “lead pipe” rigidity, and increased body temperature associated with Mr. C’s history of haloperidol use for 9 days, the treatment team suspects NMS. Labile blood pressure, which changed from 151/93 to 180/120 mm Hg in 20 minutes, reinforces the NMS diagnosis. Approximately 30 minutes after Mr. C shows signs of NMS, he is transferred to a local ED. He is awake, alert, and communicative after he arrives in the ED, but becomes confused and noncommunicative the next morning. When he arrives in the ED, he is found to have tachycardia (114 beats per minute), tachypnea (26 breaths per minute), blood pressure of 132/84 mm Hg, and temperature of 102°F. In the ED, he is given IV normal saline, diphenhydramine, 25 mg, and IV lorazepam, 1 mg. His rigidity slightly improves.
Early the next morning, his blood pressure is 182/89 mm Hg, respirations are 30 to 40 breaths per minute, and heart rate is 120 beats per minute. He then receives IV lorazepam, 2 mg, after which his tachypnea, tachycardia, and elevated blood pressure improve.
The authors’ observations
A case-control study by Keck et al12 comparing 18 patients with NMS and 36 matched neuroleptic-treated patients with no history of the syndrome identified greater psychomotor agitation, significantly higher doses of neuroleptics, greater rates of dosage increase, and a greater number of IM injections as potential risk factors. Other potential risk factors include use of restraints, pre-existing CNS dopamine activity or receptor function abnormalities, and iron deficiency.2 Agitation, dehydration, and exhaustion were found to be the most consistent systemic factors predisposing patients taking antipsychotics to NMS in small case-control studies.13,14 Well-supported risk factors also include use of high-potency antipsychotics, prior episodes of NMS, age <40, male sex, malnutrition, organic brain syndromes, and lithium use.3,5,15
There is no way to predict the risk of NMS for an individual patient. Usually, symptoms develop within 4 weeks of starting an antipsychotic, but can occur after taking the same dose for many months. The onset may be within hours, but on average it is 4 to 14 days after initiating therapy. Among patients who develop NMS, 90% do so within 10 days.3,5
Mr. C’s risk factors include high-potency antipsychotic use, male sex, relatively high dose (haloperidol, 30 to 35 mg/d), agitation, dehydration, and exhaustion.
Managing NMS
The standard approaches for managing patients with NMS include discontinuing suspected triggering drugs and providing supportive care. Beyond supportive care, oral or IV benzodiazepines may relieve symptoms and speed recovery.2 Dopaminergic drugs, such as bromocriptine or amantadine, used alone or with other treatments, can reduce parkinsonism and disease duration and mortality.2 Dantrolene may be useful only for NMS patients who exhibit extreme temperature elevations, rigidity, and true hypermetabolism.16 Electroconvulsive therapy may be effective for NMS patients whose symptoms do not respond to supportive care and drug therapy or those with residual catatonic or parkinsonian symptoms.2
OUTCOME: Improvement, discharge
Mr. C is admitted to the hospital with the diagnosis of NMS and transferred to the intensive care unit (ICU) for treatment. After Mr. C is admitted to the ICU, apart from continuing the medication given in the ED, he also receives dantrolene, 2 mg/kg, then 1 mg/kg, 4 times a day, as well as IV lorazepam, 1 mg every 6 hours. His other medications include IV pantoprazole, 40 mg/d, for prophylaxis of stress ulcer. Diphenhydramine administration is changed to as needed. On the second day in the ICU, he has only mild upper extremity rigidity but no lower extremity rigidity. However, he suffers 1 seizure, which is treated with IV fosphenytoin at the loading dose, 18 mg/kg, then a maintaining dose of 5 mg phenytoin equivalent/kg/d.
Figure 3: Mr. C’s creatine kinase level (IU/L) during the first 5 days in the intensive care unit
Figure 4: Mr. C’s blood pressure before and after admission
Figure 5: Mr. C’s temperature before and after admissionMr. C remains in the ICU for 7 days. There he receives valproic acid, titrated to 500 mg in the morning and 1,000 mg at bedtime, for agitation. He also receives olanzapine, 5 mg/d, for psychotic symptoms. He develops deep vein thrombosis in the right cephalic vein, which is treated with subcutaneous enoxaparin, 1 mg/kg, and warfarin, 5 mg/d.
He is discharged from the hospital after 2 weeks and returns to the psychiatric facility. He continues to be treated for paranoid schizophrenia with olanzapine, 5 mg/d.
The authors’ observations
High-potency, typical antipsychotics can cause NMS, as shown in Mr. C’s case. It also can be caused by typical low-potency antipsychotics,3 atypical antipsychotics,17 antiemetic drugs,18 and lithium,19,20 and can occur after the withdrawal of levodopa and similar dopaminergic agents during Parkinson’s disease treatment.21 Atypical antipsychotics reported to be associated with NMS include clozapine, risperidone, olanzapine, quetiapine, aripiprazole, ziprasidone, and paliperidone.22-27 Atypical antipsychotic-induced NMS also has been reported in children and adolescents.22,28-30
With the broad application of atypical antipsychotics, physicians should be aware of atypical NMS presentation. Although NMS diagnosis commonly requires core symptoms of hyperthermia and muscle rigidity (Table 1 and 2),31 atypical presentations may not demonstrate temperature changes and/or muscle rigidity or may progress slowly over several days, leading to a delay in diagnosis and treatment.28,30,32,33 Therefore, clinicians should evaluate any patient taking antipsychotics for features of NMS and not prematurely exclude a NMS diagnosis in cases where severe rigidity or hyperthermia is not initially apparent.33
Table 1
DSM-IV-TR criteria for neuroleptic malignant syndrome
| A. The development of severe muscle rigidity and elevated temperature associated with the use of neuroleptic medication |
| B. 2 (or more) of the following: |
|
| Source: Reference 31 |
Table 2
Diagnostic features of neuroleptic malignant syndrome
| Essential features: severe muscle rigidity and elevated temperature in an individual using neuroleptic medication |
| Elevated temperature: from mild (eg, 99º to 100ºF) to markedly hyperthermic states (eg, 106ºF) |
| Creatine kinase: typically elevated, ranging from minor elevations to extremely high levels (exceeding 16,000 IU) |
| Other features: mental status changes, unstable blood pressure, diaphoresis, other signs of autonomic dysfunction |
| Source: Reference 31 |
Related Resource
- Neuroleptic Malignant Syndrome Information Service. www.nmsis.org.
Drug Brand Names
- Amantadine • Symmetrel
- Aripiprazole • Abilify
- Benztropine • Cogentin
- Bromocriptine • Parlodel
- Clozapine • Clozaril
- Dantrolene • Dantrium
- Diphenhydramine • Benadryl
- Enoxaparin • Lovenox
- Fosphenytoin • Cerebyx
- Haloperidol • Haldol
- Hydroxyzine • Vistaril
- Levodopa • Sinemet
- Lithium • Eskalith, Lithobid, others
- Lorazepam • Ativan
- Olanzapine • Zyprexa
- Paliperidone • Invega
- Pantoprazole • Protonix
- Phenytoin • Dilantin
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Trazodone • Desyrel, Oleptro
- Valproic acid • Depakote
- Warfarin • Coumadin
- Ziprasidone • Geodon
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Acknowledgements
The authors are very grateful for the critical reviews by James R. Allen, MD, MPH, professor of Child and Adolescent Psychiatry Fellowship Program at the University of Oklahoma and Lori Hake, DO, director of Psychiatry Residency Training Program at Griffin Memorial Hospital in Norman, OK.
1. Stubner S, Rustenbeck E, Grohmann R, et al. Severe and uncommon involuntary movement disorders due to psychotropic drugs. Pharmacopsychiatry. 2004;37:S54-S64.
2. Strawn JR, Keck PE, Jr, Caroff SN. Neuroleptic malignant syndrome. Am J Psychiatry. 2007;164:870-876.
3. Ropper AH, Brown RH. Adams and Victor’s principles of neurology. 8th ed. New York, NY: McGraw Hill; 2005;1025-1026.
4. Sheil AT, Collins KA, Schandl CA, et al. Fetal neurotoxic response to neuroleptic medications: case report and review of the literature. Am J Forensic Med Pathol. 2007;28:116-120.
5. Balzan MV. The neuroleptic malignant syndrome: a logical approach to the patient with temperature and rigidity. Postgrad Med J. 1998;74:72-76.
6. Caroff SN, Mann SC. Neuroleptic malignant syndrome. Med Clin North Am. 1993;77:185-202.
7. Caroff SN, Rosenberg H, Mann SC, et al. Neuroleptic malignant syndrome in the critical care unit. Crit Care Med. 2002;30:2609-2610.
8. Caroff SN, Campbell EC, Sullivan KA. Neuroleptic malignant syndrome in elderly patients. Expert Rev Neurother. 2007;7:423-431.
9. Gurrera RJ, Simpson JC, Tsuang MT. Meta-analytic evidence of systematic bias in estimates of neuroleptic malignant syndrome incidence. Compr Psychiatry. 2007;48:205-211.
10. Caroff SN, Mann SC, Keck PE, Jr, et al. Residual catatonic state following neuroleptic malignant syndrome. J Clin Psychopharmacol. 2001;21:121-122.
11. Gurrera RJ. Sympathoadrenal hyperactivity and the etiology of neuroleptic malignant syndrome. Am J Psychiatry. 1999;156:169-180.
12. Keck PE, Jr, Pope HG, Jr, Cohen BM, et al. Risk factors for neuroleptic malignant syndrome. A case-control study. Arch Gen Psychiatry. 1989;46:914-918.
13. Berardi D, Amore M, Keck PE, Jr, et al. Clinical and pharmacologic risk factors for neuroleptic malignant syndrome: a case-control study. Biol Psychiatry. 1998;44:748-754.
14. Rosebush PI, Stewart TD. A prospective analysis of 24 episodes of neuroleptic malignant syndrome. Am J Psychiatry. 1989;146:717-725.
15. Martinez M, Marangell LB, Martinez JM. Psychopharmacology. In: Hales RE, Yudofsky SC, Gabbard GO, eds. American Psychiatric Publishing textbook of psychiatry. Arlington, VA: American Psychiatric Publishing, Inc.; 2008:1059-1132.
16. Caroff SN. Neuroleptic malignant syndrome. In: Mann SC, Caroff SN, Keck PE Jr, et al, eds. Neuroleptic malignant syndrome and related conditions. Washington, DC: American Psychiatric Publishing; 2003:1-44.
17. Hammerman S, Lam C, Caroff SN. Neuroleptic malignant syndrome and aripiprazole. J Am Acad Child Adolesc Psychiatry. 2006;45:639-641.
18. Stein MH, Sorscher M, Caroff SN. Neuroleptic malignant syndrome induced by metoclopramide in an infant with Freeman-Sheldon syndrome. Anesth Analg. 2006;103:786-787.
19. Borovicka MC, Bond LC, Gaughan KM. Ziprasidone- and lithium-induced neuroleptic malignant syndrome. Ann Pharmacother. 2006;40:139-142.
20. Gill J, Singh H, Nugent K. Acute lithium intoxication and neuroleptic malignant syndrome. Pharmacotherapy. 2003;23:811-815.
21. Ward C. Neuroleptic malignant syndrome in a patient with Parkinson’s disease: a case study. J Neurosci Nurs. 2005;37:160-162.
22. Leibold J, Patel V, Hasan RA. Neuroleptic malignant syndrome associated with ziprasidone in an adolescent. Clin Ther. 2004;26:1105-1108.
23. Corallo CE, Ernest D. Atypical neuroleptic malignant syndrome with long-term clozapine. Crit Care Resusc. 2007;9:338-340.
24. Molina D, Tingle LE, Lu X. Aripiprazole as the causative agent of neuroleptic malignant syndrome: a case report. Prim Care Companion J Clin Psychiatry. 2007;9:148-150.
25. Trollor JN, Chen X, Sachdev PS. Neuroleptic malignant syndrome associated with atypical antipsychotic drugs. CNS Drugs. 2009;23:477-492.
26. Gortney JS, Fagan A, Kissack JC. Neuroleptic malignant syndrome secondary to quetiapine. Ann Pharmacother. 2009;43:785-791.
27. Han C, Lee SJ, Pae CU. Paliperidone-associated atypical neuroleptic malignant syndrome: a case report. Prog Neuropsychopharmacol Biol Psychiatry. 2011;35:650-651.
28. Hanft A, Eggleston CF, Bourgeois JA. Neuroleptic malignant syndrome in an adolescent after brief exposure to olanzapine. J Child Adolesc Psychopharmacol. 2004;14:481-487.
29. Abu-Kishk I, Toledano M, Reis A, et al. Neuroleptic malignant syndrome in a child treated with an atypical antipsychotic. J Toxicol Clin Toxicol. 2004;42:921-925.
30. Neuhut R, Lindenmayer JP, Silva R. Neuroleptic malignant syndrome in children and adolescents on atypical antipsychotic medication: a review. J Child Adolesc Psychopharmacol. 2009;19:415-422.
31. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association: 2000.
32. Carroll BT, Surber SA. The problem of atypical neuroleptic malignant syndrome: a case report. Psychiatry (Edgmont). 2009;6:45-47.
33. Picard LS, Lindsay S, Strawn JR, et al. Atypical neuroleptic malignant syndrome: diagnostic controversies and considerations. Pharmacotherapy. 2008;28:530-535.
1. Stubner S, Rustenbeck E, Grohmann R, et al. Severe and uncommon involuntary movement disorders due to psychotropic drugs. Pharmacopsychiatry. 2004;37:S54-S64.
2. Strawn JR, Keck PE, Jr, Caroff SN. Neuroleptic malignant syndrome. Am J Psychiatry. 2007;164:870-876.
3. Ropper AH, Brown RH. Adams and Victor’s principles of neurology. 8th ed. New York, NY: McGraw Hill; 2005;1025-1026.
4. Sheil AT, Collins KA, Schandl CA, et al. Fetal neurotoxic response to neuroleptic medications: case report and review of the literature. Am J Forensic Med Pathol. 2007;28:116-120.
5. Balzan MV. The neuroleptic malignant syndrome: a logical approach to the patient with temperature and rigidity. Postgrad Med J. 1998;74:72-76.
6. Caroff SN, Mann SC. Neuroleptic malignant syndrome. Med Clin North Am. 1993;77:185-202.
7. Caroff SN, Rosenberg H, Mann SC, et al. Neuroleptic malignant syndrome in the critical care unit. Crit Care Med. 2002;30:2609-2610.
8. Caroff SN, Campbell EC, Sullivan KA. Neuroleptic malignant syndrome in elderly patients. Expert Rev Neurother. 2007;7:423-431.
9. Gurrera RJ, Simpson JC, Tsuang MT. Meta-analytic evidence of systematic bias in estimates of neuroleptic malignant syndrome incidence. Compr Psychiatry. 2007;48:205-211.
10. Caroff SN, Mann SC, Keck PE, Jr, et al. Residual catatonic state following neuroleptic malignant syndrome. J Clin Psychopharmacol. 2001;21:121-122.
11. Gurrera RJ. Sympathoadrenal hyperactivity and the etiology of neuroleptic malignant syndrome. Am J Psychiatry. 1999;156:169-180.
12. Keck PE, Jr, Pope HG, Jr, Cohen BM, et al. Risk factors for neuroleptic malignant syndrome. A case-control study. Arch Gen Psychiatry. 1989;46:914-918.
13. Berardi D, Amore M, Keck PE, Jr, et al. Clinical and pharmacologic risk factors for neuroleptic malignant syndrome: a case-control study. Biol Psychiatry. 1998;44:748-754.
14. Rosebush PI, Stewart TD. A prospective analysis of 24 episodes of neuroleptic malignant syndrome. Am J Psychiatry. 1989;146:717-725.
15. Martinez M, Marangell LB, Martinez JM. Psychopharmacology. In: Hales RE, Yudofsky SC, Gabbard GO, eds. American Psychiatric Publishing textbook of psychiatry. Arlington, VA: American Psychiatric Publishing, Inc.; 2008:1059-1132.
16. Caroff SN. Neuroleptic malignant syndrome. In: Mann SC, Caroff SN, Keck PE Jr, et al, eds. Neuroleptic malignant syndrome and related conditions. Washington, DC: American Psychiatric Publishing; 2003:1-44.
17. Hammerman S, Lam C, Caroff SN. Neuroleptic malignant syndrome and aripiprazole. J Am Acad Child Adolesc Psychiatry. 2006;45:639-641.
18. Stein MH, Sorscher M, Caroff SN. Neuroleptic malignant syndrome induced by metoclopramide in an infant with Freeman-Sheldon syndrome. Anesth Analg. 2006;103:786-787.
19. Borovicka MC, Bond LC, Gaughan KM. Ziprasidone- and lithium-induced neuroleptic malignant syndrome. Ann Pharmacother. 2006;40:139-142.
20. Gill J, Singh H, Nugent K. Acute lithium intoxication and neuroleptic malignant syndrome. Pharmacotherapy. 2003;23:811-815.
21. Ward C. Neuroleptic malignant syndrome in a patient with Parkinson’s disease: a case study. J Neurosci Nurs. 2005;37:160-162.
22. Leibold J, Patel V, Hasan RA. Neuroleptic malignant syndrome associated with ziprasidone in an adolescent. Clin Ther. 2004;26:1105-1108.
23. Corallo CE, Ernest D. Atypical neuroleptic malignant syndrome with long-term clozapine. Crit Care Resusc. 2007;9:338-340.
24. Molina D, Tingle LE, Lu X. Aripiprazole as the causative agent of neuroleptic malignant syndrome: a case report. Prim Care Companion J Clin Psychiatry. 2007;9:148-150.
25. Trollor JN, Chen X, Sachdev PS. Neuroleptic malignant syndrome associated with atypical antipsychotic drugs. CNS Drugs. 2009;23:477-492.
26. Gortney JS, Fagan A, Kissack JC. Neuroleptic malignant syndrome secondary to quetiapine. Ann Pharmacother. 2009;43:785-791.
27. Han C, Lee SJ, Pae CU. Paliperidone-associated atypical neuroleptic malignant syndrome: a case report. Prog Neuropsychopharmacol Biol Psychiatry. 2011;35:650-651.
28. Hanft A, Eggleston CF, Bourgeois JA. Neuroleptic malignant syndrome in an adolescent after brief exposure to olanzapine. J Child Adolesc Psychopharmacol. 2004;14:481-487.
29. Abu-Kishk I, Toledano M, Reis A, et al. Neuroleptic malignant syndrome in a child treated with an atypical antipsychotic. J Toxicol Clin Toxicol. 2004;42:921-925.
30. Neuhut R, Lindenmayer JP, Silva R. Neuroleptic malignant syndrome in children and adolescents on atypical antipsychotic medication: a review. J Child Adolesc Psychopharmacol. 2009;19:415-422.
31. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association: 2000.
32. Carroll BT, Surber SA. The problem of atypical neuroleptic malignant syndrome: a case report. Psychiatry (Edgmont). 2009;6:45-47.
33. Picard LS, Lindsay S, Strawn JR, et al. Atypical neuroleptic malignant syndrome: diagnostic controversies and considerations. Pharmacotherapy. 2008;28:530-535.
Unexpected improvement
Discuss this article at www.facebook.com/CurrentPsychiatry
CASE: Relapsing psychosis
Ms. U, age 53, was diagnosed with paranoid schizophrenia at age 21 and has a continuous pattern of frequent relapses and inpatient admissions. She has received therapeutic doses of trifluoperazine, sertindole, haloperidol, loxapine, thioridazine, olanzapine, risperidone, clozapine, and several other antipsychotics not available in the United States. Clozapine had been prescribed at 600 mg/d (average blood level was 350 ng/mL), at times in combination with other antipsychotics or lithium.
Despite treatment, Ms. U has never achieved clinical stability. She has fluctuating yet persistent auditory hallucinations (eg, voices threatening to “announce disasters” or songs of a religious nature), associated disorganized behavior (eg, covering her ears or asking third parties “to turn off the radio”), severe hyponatremia secondary to potomania, paranoid ideation (eg, being followed by a “hidden camera”), and a strong tendency toward negativism, mutism, and emotional lability secondary to her psychotic symptoms. Her affect is predominantly poor and flattened, with very poor insight. Her symptoms are associated with progressive social isolation and poor grooming. Because of her worsening status, Ms. U was admitted to a residential facility 3 years ago.
Ms. U is single and the eldest of 2 siblings. Her parents are deceased; one parent may have committed suicide. She reports a family history of psychosis in her first cousins, but no history of hereditary neurologic disorders. Ms. U is a heavy smoker, did not complete college, and has a job in a family business.
The authors’ observations
Historically, the prevailing theory to explain the pathophysiology of schizophrenia has been the dopamine hypothesis, which links a hyperdopaminergic state in the mesolimbic system with acute psychosis. This theory could explain positive symptoms of schizophrenia but not other core domains, such as negative symptoms and cognitive dysfunction.1-3 The glutamate hypothesis postulates a hypoglutamatergic state can be the cause, at least in part, of various symptoms of psychosis, similar to those induced by phencyclidine and ketamine. Antagonists at the glycine modulatory site of the N-methyl-d-aspartate (NMDA) receptor are being studied as a way to influence this pathway,1 which is believed to be influenced by genetic factors.4
Glutamate, an amino acid, is the primary excitatory neurotransmitter in the brain. Its action is exerted in 2 types of receptors on the postsynaptic neuron: ionotropic and metabotropic.
The activation of NMDA receptors generated by glutamate and glycine coagonist can stimulate an uncontrolled release of calcium and subsequent cell death known as excitotoxicity. This phenomenon has been described in amyotrophic lateral sclerosis (ALS), Alzheimer’s disease, and Huntington’s disease. Although overstimulation of NMDA receptors induces neurodegeneration, NMDA hypoactivity has been observed in psychotic states.5
EVALUATION: Neurologic symptoms
A few months after arriving at the residential facility, Ms. U develops dysarthria and drooling, which the treatment team initially interprets as secondary to high doses of clozapine. In the absence of clinical response after clozapine dose reduction and with the subsequent appearance of dysphagia with solid foods and liquids, Ms. U is evaluated by a ear, nose, and throat physician, and later by a neurologist. Both clinicians describe frontal release signs, anarthria, facial hypomimia, bilateral mild central paresis, absence of soft palate elevation with symmetrical phonation, decreased gag reflex and palatal atrophy, fasciculations, and bilateral lingual mandibular reflex and diagnose Ms. U with progressive bulbar palsy, a variant of ALS.
The authors’ observations
ALS is a progressive, degenerative neuromuscular condition of unknown etiology affecting the corticospinal tracts and the anterior horn of the spinal cord, leading to dysfunction of the upper and lower motor neurons.6 It is more common in men, persons with diets rich in glutamate, and smokers.7,8
Riluzole is the only FDA-approved medication for ALS.9 It interferes with the responses mediated by the NMDA receptor, stabilizes inactive sodium voltage-dependent channels, inhibits glutamate release from synaptic endings, and activates extracellular reuptake of glutamate, all of which are thought to confer a neuroprotective effect.10
TREATMENT: Psychosis improves
As suggested by the neurology team, we begin riluzole, 50 mg every 12 hours. At this time Ms. U also is taking clozapine, 600 mg/d; lithium, 1200 mg/d; and haloperidol, 6 mg/d; her psychiatric symptoms have not changed since the initial evaluation at the residential facility.
Seven months after initiating riluzole Ms. U is more receptive, less querulant, and no longer experiences delusions or hallucinations. At the same time, she develops an interest in her clinical status regarding her ALS diagnosis, which reflects improved insight. One year after starting riluzole, she is more cooperative and adherent with treatment. Ms. U is able to reestablish relationships with her family. Clozapine and haloperidol are tapered and discontinued. Ms. U’s medication regimen includes risperidone, 1 mg/d; methotrimeprazine, 10 mg/d; venlafaxine, 75 mg/d; trazodone, 100 mg/d; and lithium, 600 mg/d, in addition to riluzole, 50 mg every 12 hours.
An assessment 18 months after starting riluzole describes a Positive and Negative Syndrome Scale (PANSS) score of 9 for positive symptoms, 11 for negative, 35 for the general psychopathology, and -2 for the composite (Table 1). Laboratory tests are normal except for a mild normocytic, normochromic anemia. MRI shows no detectable lesions or changes in comparison with previous images.
Table 1
Ms. U’s clinical course
| PANSS score | Treatment | Mental status |
|---|---|---|
| Before starting riluzole | ||
| No PANSS reported | Clozapine, 600 mg/d; lithium, 1200 mg/d; haloperidol, 6 mg/d | Persistent auditory hallucinations. Persistent hallucinatory behavior. Paranoid delirious ideas. Negativism, mutism, and liability reactive to her psychosis state. Poor and flattened affect. Lack of disease awareness. Progressive social isolation. Loss of self care |
| After starting riluzole | ||
| Positive subscale: 9 (below 5th percentile) Negative subscale: 11 (between 5th-25th percentile) General psychopathology subscale: 35 (between 5th-25th percentile) Composite score: -2 (between 25th-50th percentiles) | Riluzole, 50 mg every 12 hours; risperidone, 1 mg/d; methotrimeprazine, 10 mg/d; venlafaxine, 75 mg/d; trazodone, 100 mg/d; lithium, 600 mg/d | Re-establishes relationships with family because she no longer experiences paranoid delusions. Behavioral improvement. Allows physical proximity to nursing and medical personnel. Attention to physical appearance. Participates in social and recreational activities outside the hospital. Absence of auditory hallucinations. Affective improvement with appropriate responses. Realistic anxiety and fear about ALS diagnosis |
| ALS: amyotrophic lateral sclerosis; PANSS: Positive and Negative Syndrome Scale | ||
The authors’ observations
We present a patient with schizophrenia and a continuous pattern of relapses, functional and social impairment, and partial remission of her psychosis despite the use of multiple typical and atypical antipsychotics at therapeutic doses. Ms. U received treatment with clozapine at therapeutic doses for >6 months without sustained improvement. After beginning riluzole, a glutamate pathway antagonist, and with no other changes to her medication regimen, Ms. U experienced substantial improvement in her mental status. This was evidenced by a significant decline in her paranoid delusions, disappearance of auditory hallucinations, and substantial improvement on her social performance.
This fact is consistent with previous observations where modulation of the glutamate pathway has been associated with improvement in depression and anxiety levels in different populations. This case report provides further evidence to the possibility that blocking this receptor is a promising approach to psychotic disorders.
Riluzole for psychiatric illness
Currently, there are 11 clinical trials investigating riluzole for psychiatric disorders, including OCD, depression, bipolar disorder, schizophrenia, and Tourette’s syndrome.11 Consistent with the altered glutamatergic neurotransmission implicated in mood and anxiety disorders, preliminary evidence suggests riluzole can effectively treat OCD, bipolar depression, unipolar depression, and comorbid OCD and depression (Table 2). Some investigators consider the glutamatergic pathway an essential target for future antidepressants and mood-stabilizing agents.12
Other drugs such as memantine, acamprosate, and lamotrigine act on this same pathway and therefore have a role in treating psychiatric and neurologic conditions. In the case of lamotrigine, the drug inhibits glutamate release through inhibition of voltage-dependent sodium and calcium channels13 and postsynaptic AMPA receptors14 and has been shown to effectively treat generalized epilepsies,15 bipolar depression,13,16 and depression and mood swings associated with Huntington’s disease.17
Acamprosate’s attenuation of hyperglutamatergic states through NMDA antagonism and metabotropic glutamate receptors and reduction of intracellular calcium release—therefore balancing the glutamatergic and GABAergic systems and conferring neuroprotective properties—has been effective in patients with alcohol use disorders.18,19
Memantine and amantadine act through NMDA antagonism and by modulating dopaminergic transmission and may have clinical roles beyond dementia treatment.
Table 2
Evidence of efficacy of riluzole for OCD and depression
| Study | Disorder | Findings |
|---|---|---|
| Pittenger et al, 2006a | OCD | Brain imaging reveals elevated glutamate levels in OCD patients; agents that reduce glutamate hyperactivity may be effective |
| Coric et al, 2005b | OCD | Among 13 patients with OCD who received riluzole, 54% demonstrated >35% reduction in Y-BOCS scores and 39% were considered treatment responders |
| Zarate et al, 2005c | Bipolar depression | In an 8-week add-on study of riluzole in combination with lithium of 14 patients with bipolar depression, riluzole showed efficacy as measured by MADRS score and was well tolerated |
| Singh et al, 2004d | Bipolar depression | Case report of a patient with bipolar II disorder and depression who had a good response to riluzole when lamotrigine was discontinued because of a maculopapular erythematic rash |
| Zarate et al, 2004e | Unipolar depression | In a 6-week, open-label trial, 19 treatment-resistant depressed patients received riluzole; significant improvement measured by MADRS, CGI-S, and HAM-A were noted at weeks 3 through 6 |
| Coric et al, 2003f | Comorbid OCD and major depressive disorder | Case report of a patient with symptomatic OCD and depression who did not respond to appropriate pharmacotherapy, including augmentation strategies; adding riluzole significantly attenuated both obsessions and depressive symptoms |
| CGI-S: Clinical Global Impressions-Severity; HAM-A: Hamilton Anxiety Rating Scale; MADRS: Montgomery-Åsberg Depression Rating Scale; OCD: obsessive-compulsive disorder; Y-BOCS: Yale-Brown Obsessive Compulsive Scale Source: a. Pittenger C, Krystal JH, Coric V. Glutamate-modulating drugs as novel pharmacotherapeutic agents in the treatment of obsessive-compulsive disorder. Neurotherapeutics. 2006;3(1):69-81. b. Coric V, Taskiran S, Pittenger C, et al. Riluzole augmentation in treatment-resistant obsessive-compulsive disorder: an open-label trial. Biol Psychiatry. 2005;58(5):424-428. c. Zarate CA Jr, Quiroz JA, Singh JB, et al. An open-label trial of the glutamate-modulating agent riluzole in combination with lithium for the treatment of bipolar depression. Biol Psychiatry. 2005;57(4):430-432. d. Singh J, Zarate CA, Krystal AD. Case report: successful riluzole augmentation therapy in treatment-resistant bipolar depression following the development of rash with lamotrigine. Psychopharmacology. 2004;173(1-2):227-228. e. Zarate CA Jr, Payne JL, Quiroz J, et al. An open-label trial of riluzole in patients with treatment-resistant major depression. Am J Psychiatry. 2004;161(1):171-174. f. Coric V, Milanovic S, Wasylink S, et al. Beneficial effects of the antiglutamatergic agent riluzole in a patient diagnosed with obsessive-compulsive disorder and major depressive disorder. Psychopharmacology. 2003;167(2):219-220. | ||
Schizophrenia-ALS comorbidity
Some investigators have suggested20 the relative rarity of ALS in patients with schizophrenia is attributable to the neuroprotective effects of antipsychotics and antidepressants.21 If this is true, it is possible resistance to antipsychotics among some schizophrenia patients may be underpinned by the degree of cell injury and therefore of neurodegeneration, which may be the case with Ms. U.
Controlled, randomized, double-blind studies are needed to confirm our team’s assumptions. Our observation is limited by the lack of standardized scale measurements to assess all schizophrenia domains before starting riluzole and Ms. U’s clinical improvement could be associated with other factors such as passage of time or schizophrenia “burning out.” However, clinical observation and description from family members and hospital staff are important to consider in this case.
The improvement in schizophrenia symptoms observed from a drug with no action on dopamine blockade—a quality observed in all antipsychotics22—reinforces the possibility that targeting different pathways involved in the genesis of schizophrenia is a reasonable topic for future research. The possible use of riluzole and other glutamate-modulating drugs might influence positive, negative, and cognitive symptoms of schizophrenia.
Related Resources
- Kantrowitz JT, Javitt DC. Glutamate: new hope for schizophrenia treatment. Current Psychiatry. 2011;10(4):68-74.
- Vinson PN, Conn PJ. Metabotropic glutamate receptors as therapeutic targets for schizophrenia. Neuropharmacology. 2011. Epub ahead of print.
Drug Brand Names
- Acamprosate • Campral
- Amantadine • Symmetrel
- Clozapine • Clozaril
- Haloperidol • Haldol
- Ketamine • Ketalar
- Lamotrigine • Lamictal
- Lithium • Eskalith, Lithobid
- Loxapine • Loxitane
- Methotrimeprazine • Nozinan
- Memantine • Namenda
- Olanzapine • Zyprexa
- Riluzole • Rilutek
- Risperidone • Risperdal
- Sertindole • Serdolect
- Thioridazine • Mellaril
- Trazodone • Desyrel, Oleptro
- Trifluoperazine • Stelazine
- Venlafaxine • Effexor
Disclosures
Dr. Millán-González is a consultant to AstraZeneca CAMCAR. Drs. Loizaga-Arniaz and Zúñiga-Montes report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Freudenreich O, Weiss AP, Goff DC. Psychosis and schizophrenia. In: Stern T Rosenbaum, JF, Fava M, et al, eds. Massachusetts general hospital comprehensive clinical psychiatry. Philadelphia, PA: Mosby, an Imprint of Elsevier; 2008:371–389.
2. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington DC: American Psychiatric Association; 2000.
3. Bowie CR, Harvey PD. Cognition in schizophrenia: impairments determinants, and functional importance. Psychiatr Clin North Am. 2005;28(3):613-633.
4. Waddington JL, Corvin AP, Donohoe G, et al. Functional genomics and schizophrenia: endophenotypes and mutant models. Psychiatr Clin North Am. 2007;30(3):365-399.
5. Morrow EM, Roffman JL, Wolf DH, et al. Psychiatric neuroscience: incorporating pathophysiology into clinical case formulation. In: Stern T, Rosenbaum, JF, Fava M, et al, eds. Massachusetts General Hospital comprehensive clinical psychiatry. Philadelphia, PA: Mosby, an Imprint of Elsevier; 2008:543–564.
6. Harrison T. Amyotrophic lateral sclerosis. In: Ferri’s clinical advisor 2010. Philadelphia PA. Mosby, an Imprint of Elsevier; 2011:57.
7. Ringel SP, Murphy JR, Alderson MK, et al. The natural history of amyotrophic lateral sclerosis. Neurology. 1993;43(7):1316-1322.
8. Chancellor AM, Warlow CP. Adult onset motor neuron disease: worldwide mortality incidence and distribution since 1950. J Neurol Neurosurg Psychiatry. 1992;55(12):1106-1115.
9. Practice advisory on the treatment of amyotrophic lateral sclerosis with riluzole: report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 1997;49(3):657-659.
10. Distad BJ, Meekins GD, Liou LL, et al. Drug therapy in amyotrophic lateral sclerosis. Phys Med Rehabil Clin N Am. 2008;19(3):633-651.
11. ClinicalTrials.gov. U.S. National Institutes of Health. Available at: http://clinicaltrials.gov/ct2/results?intr=%22Riluzole%22. Accessed June 27, 2011.
12. Krystal JH, Sanacora G, Blumberg H, et al. Glutamate and GABA systems as targets for novel antidepressant and mood-stabilizing treatments. Mol Psychiatry. 2002;7(suppl 1):S71-80.
13. Calabrese JR, Bowden CL, Sachs GS, et al. A double-blind placebo-controlled study of lamotrigine monotherapy in outpatients with bipolar I depression. Lamictal 602 Study Group. J Clin Psychiatry. 1999;60(2):79-88.
14. Lee CY, Fu WM, Chen CC, et al. Lamotrigine inhibits postsynaptic AMPA receptor and glutamate release in the dentate gyrus. Epilepsia. 2008;49(5):888-897.
15. Patsalos PN. Properties of antiepileptic drugs in the treatment of idiopathic generalized epilepsies. Epilepsia. 2005;46(suppl 9):140-148.
16. Yatham LN, Kennedy SH, Schaffer A, et al. Canadian Network for Mood and Anxiety Treatments (CANMAT) and International Society for Bipolar Disorders (ISBD) collaborative update of CANMAT guidelines for the management of patients with bipolar disorder: update 2009. Bipolar Disord. 2009;11(3):225-255.
17. Shen YC. Lamotrigine in motor and mood symptoms of Huntington’s disease. World J Biol Psychiatry. 2008;9(2):147-149.
18. Scott LJ, Figgitt DP, Keam SJ, et al. Acamprosate: a review of its use in the maintenance of abstinence in patients with alcohol dependence. CNS Drugs. 2005;19(5):445-464.
19. De Witte P, Littleton J, Parot P, et al. Neuroprotective and abstinence-promoting effects of acamprosate: elucidating the mechanism of action. CNS Drugs. 2005;19(6):517-537.
20. Stommel EW, Graber D, Montanye J, et al. Does treating schizophrenia reduce the chances of developing amyotrophic lateral sclerosis? Med Hypotheses. 2007;69(5):1021-1028.
21. Howland RH. Schizophrenia and amyotrophic lateral sclerosis. Compr Psychiatry. 1990;31(4):327-336.
22. Seeman P. Atypical antipsychotics: mechanism of action. Can J Psychiatry. 2002;47(1):27-38.
Discuss this article at www.facebook.com/CurrentPsychiatry
CASE: Relapsing psychosis
Ms. U, age 53, was diagnosed with paranoid schizophrenia at age 21 and has a continuous pattern of frequent relapses and inpatient admissions. She has received therapeutic doses of trifluoperazine, sertindole, haloperidol, loxapine, thioridazine, olanzapine, risperidone, clozapine, and several other antipsychotics not available in the United States. Clozapine had been prescribed at 600 mg/d (average blood level was 350 ng/mL), at times in combination with other antipsychotics or lithium.
Despite treatment, Ms. U has never achieved clinical stability. She has fluctuating yet persistent auditory hallucinations (eg, voices threatening to “announce disasters” or songs of a religious nature), associated disorganized behavior (eg, covering her ears or asking third parties “to turn off the radio”), severe hyponatremia secondary to potomania, paranoid ideation (eg, being followed by a “hidden camera”), and a strong tendency toward negativism, mutism, and emotional lability secondary to her psychotic symptoms. Her affect is predominantly poor and flattened, with very poor insight. Her symptoms are associated with progressive social isolation and poor grooming. Because of her worsening status, Ms. U was admitted to a residential facility 3 years ago.
Ms. U is single and the eldest of 2 siblings. Her parents are deceased; one parent may have committed suicide. She reports a family history of psychosis in her first cousins, but no history of hereditary neurologic disorders. Ms. U is a heavy smoker, did not complete college, and has a job in a family business.
The authors’ observations
Historically, the prevailing theory to explain the pathophysiology of schizophrenia has been the dopamine hypothesis, which links a hyperdopaminergic state in the mesolimbic system with acute psychosis. This theory could explain positive symptoms of schizophrenia but not other core domains, such as negative symptoms and cognitive dysfunction.1-3 The glutamate hypothesis postulates a hypoglutamatergic state can be the cause, at least in part, of various symptoms of psychosis, similar to those induced by phencyclidine and ketamine. Antagonists at the glycine modulatory site of the N-methyl-d-aspartate (NMDA) receptor are being studied as a way to influence this pathway,1 which is believed to be influenced by genetic factors.4
Glutamate, an amino acid, is the primary excitatory neurotransmitter in the brain. Its action is exerted in 2 types of receptors on the postsynaptic neuron: ionotropic and metabotropic.
The activation of NMDA receptors generated by glutamate and glycine coagonist can stimulate an uncontrolled release of calcium and subsequent cell death known as excitotoxicity. This phenomenon has been described in amyotrophic lateral sclerosis (ALS), Alzheimer’s disease, and Huntington’s disease. Although overstimulation of NMDA receptors induces neurodegeneration, NMDA hypoactivity has been observed in psychotic states.5
EVALUATION: Neurologic symptoms
A few months after arriving at the residential facility, Ms. U develops dysarthria and drooling, which the treatment team initially interprets as secondary to high doses of clozapine. In the absence of clinical response after clozapine dose reduction and with the subsequent appearance of dysphagia with solid foods and liquids, Ms. U is evaluated by a ear, nose, and throat physician, and later by a neurologist. Both clinicians describe frontal release signs, anarthria, facial hypomimia, bilateral mild central paresis, absence of soft palate elevation with symmetrical phonation, decreased gag reflex and palatal atrophy, fasciculations, and bilateral lingual mandibular reflex and diagnose Ms. U with progressive bulbar palsy, a variant of ALS.
The authors’ observations
ALS is a progressive, degenerative neuromuscular condition of unknown etiology affecting the corticospinal tracts and the anterior horn of the spinal cord, leading to dysfunction of the upper and lower motor neurons.6 It is more common in men, persons with diets rich in glutamate, and smokers.7,8
Riluzole is the only FDA-approved medication for ALS.9 It interferes with the responses mediated by the NMDA receptor, stabilizes inactive sodium voltage-dependent channels, inhibits glutamate release from synaptic endings, and activates extracellular reuptake of glutamate, all of which are thought to confer a neuroprotective effect.10
TREATMENT: Psychosis improves
As suggested by the neurology team, we begin riluzole, 50 mg every 12 hours. At this time Ms. U also is taking clozapine, 600 mg/d; lithium, 1200 mg/d; and haloperidol, 6 mg/d; her psychiatric symptoms have not changed since the initial evaluation at the residential facility.
Seven months after initiating riluzole Ms. U is more receptive, less querulant, and no longer experiences delusions or hallucinations. At the same time, she develops an interest in her clinical status regarding her ALS diagnosis, which reflects improved insight. One year after starting riluzole, she is more cooperative and adherent with treatment. Ms. U is able to reestablish relationships with her family. Clozapine and haloperidol are tapered and discontinued. Ms. U’s medication regimen includes risperidone, 1 mg/d; methotrimeprazine, 10 mg/d; venlafaxine, 75 mg/d; trazodone, 100 mg/d; and lithium, 600 mg/d, in addition to riluzole, 50 mg every 12 hours.
An assessment 18 months after starting riluzole describes a Positive and Negative Syndrome Scale (PANSS) score of 9 for positive symptoms, 11 for negative, 35 for the general psychopathology, and -2 for the composite (Table 1). Laboratory tests are normal except for a mild normocytic, normochromic anemia. MRI shows no detectable lesions or changes in comparison with previous images.
Table 1
Ms. U’s clinical course
| PANSS score | Treatment | Mental status |
|---|---|---|
| Before starting riluzole | ||
| No PANSS reported | Clozapine, 600 mg/d; lithium, 1200 mg/d; haloperidol, 6 mg/d | Persistent auditory hallucinations. Persistent hallucinatory behavior. Paranoid delirious ideas. Negativism, mutism, and liability reactive to her psychosis state. Poor and flattened affect. Lack of disease awareness. Progressive social isolation. Loss of self care |
| After starting riluzole | ||
| Positive subscale: 9 (below 5th percentile) Negative subscale: 11 (between 5th-25th percentile) General psychopathology subscale: 35 (between 5th-25th percentile) Composite score: -2 (between 25th-50th percentiles) | Riluzole, 50 mg every 12 hours; risperidone, 1 mg/d; methotrimeprazine, 10 mg/d; venlafaxine, 75 mg/d; trazodone, 100 mg/d; lithium, 600 mg/d | Re-establishes relationships with family because she no longer experiences paranoid delusions. Behavioral improvement. Allows physical proximity to nursing and medical personnel. Attention to physical appearance. Participates in social and recreational activities outside the hospital. Absence of auditory hallucinations. Affective improvement with appropriate responses. Realistic anxiety and fear about ALS diagnosis |
| ALS: amyotrophic lateral sclerosis; PANSS: Positive and Negative Syndrome Scale | ||
The authors’ observations
We present a patient with schizophrenia and a continuous pattern of relapses, functional and social impairment, and partial remission of her psychosis despite the use of multiple typical and atypical antipsychotics at therapeutic doses. Ms. U received treatment with clozapine at therapeutic doses for >6 months without sustained improvement. After beginning riluzole, a glutamate pathway antagonist, and with no other changes to her medication regimen, Ms. U experienced substantial improvement in her mental status. This was evidenced by a significant decline in her paranoid delusions, disappearance of auditory hallucinations, and substantial improvement on her social performance.
This fact is consistent with previous observations where modulation of the glutamate pathway has been associated with improvement in depression and anxiety levels in different populations. This case report provides further evidence to the possibility that blocking this receptor is a promising approach to psychotic disorders.
Riluzole for psychiatric illness
Currently, there are 11 clinical trials investigating riluzole for psychiatric disorders, including OCD, depression, bipolar disorder, schizophrenia, and Tourette’s syndrome.11 Consistent with the altered glutamatergic neurotransmission implicated in mood and anxiety disorders, preliminary evidence suggests riluzole can effectively treat OCD, bipolar depression, unipolar depression, and comorbid OCD and depression (Table 2). Some investigators consider the glutamatergic pathway an essential target for future antidepressants and mood-stabilizing agents.12
Other drugs such as memantine, acamprosate, and lamotrigine act on this same pathway and therefore have a role in treating psychiatric and neurologic conditions. In the case of lamotrigine, the drug inhibits glutamate release through inhibition of voltage-dependent sodium and calcium channels13 and postsynaptic AMPA receptors14 and has been shown to effectively treat generalized epilepsies,15 bipolar depression,13,16 and depression and mood swings associated with Huntington’s disease.17
Acamprosate’s attenuation of hyperglutamatergic states through NMDA antagonism and metabotropic glutamate receptors and reduction of intracellular calcium release—therefore balancing the glutamatergic and GABAergic systems and conferring neuroprotective properties—has been effective in patients with alcohol use disorders.18,19
Memantine and amantadine act through NMDA antagonism and by modulating dopaminergic transmission and may have clinical roles beyond dementia treatment.
Table 2
Evidence of efficacy of riluzole for OCD and depression
| Study | Disorder | Findings |
|---|---|---|
| Pittenger et al, 2006a | OCD | Brain imaging reveals elevated glutamate levels in OCD patients; agents that reduce glutamate hyperactivity may be effective |
| Coric et al, 2005b | OCD | Among 13 patients with OCD who received riluzole, 54% demonstrated >35% reduction in Y-BOCS scores and 39% were considered treatment responders |
| Zarate et al, 2005c | Bipolar depression | In an 8-week add-on study of riluzole in combination with lithium of 14 patients with bipolar depression, riluzole showed efficacy as measured by MADRS score and was well tolerated |
| Singh et al, 2004d | Bipolar depression | Case report of a patient with bipolar II disorder and depression who had a good response to riluzole when lamotrigine was discontinued because of a maculopapular erythematic rash |
| Zarate et al, 2004e | Unipolar depression | In a 6-week, open-label trial, 19 treatment-resistant depressed patients received riluzole; significant improvement measured by MADRS, CGI-S, and HAM-A were noted at weeks 3 through 6 |
| Coric et al, 2003f | Comorbid OCD and major depressive disorder | Case report of a patient with symptomatic OCD and depression who did not respond to appropriate pharmacotherapy, including augmentation strategies; adding riluzole significantly attenuated both obsessions and depressive symptoms |
| CGI-S: Clinical Global Impressions-Severity; HAM-A: Hamilton Anxiety Rating Scale; MADRS: Montgomery-Åsberg Depression Rating Scale; OCD: obsessive-compulsive disorder; Y-BOCS: Yale-Brown Obsessive Compulsive Scale Source: a. Pittenger C, Krystal JH, Coric V. Glutamate-modulating drugs as novel pharmacotherapeutic agents in the treatment of obsessive-compulsive disorder. Neurotherapeutics. 2006;3(1):69-81. b. Coric V, Taskiran S, Pittenger C, et al. Riluzole augmentation in treatment-resistant obsessive-compulsive disorder: an open-label trial. Biol Psychiatry. 2005;58(5):424-428. c. Zarate CA Jr, Quiroz JA, Singh JB, et al. An open-label trial of the glutamate-modulating agent riluzole in combination with lithium for the treatment of bipolar depression. Biol Psychiatry. 2005;57(4):430-432. d. Singh J, Zarate CA, Krystal AD. Case report: successful riluzole augmentation therapy in treatment-resistant bipolar depression following the development of rash with lamotrigine. Psychopharmacology. 2004;173(1-2):227-228. e. Zarate CA Jr, Payne JL, Quiroz J, et al. An open-label trial of riluzole in patients with treatment-resistant major depression. Am J Psychiatry. 2004;161(1):171-174. f. Coric V, Milanovic S, Wasylink S, et al. Beneficial effects of the antiglutamatergic agent riluzole in a patient diagnosed with obsessive-compulsive disorder and major depressive disorder. Psychopharmacology. 2003;167(2):219-220. | ||
Schizophrenia-ALS comorbidity
Some investigators have suggested20 the relative rarity of ALS in patients with schizophrenia is attributable to the neuroprotective effects of antipsychotics and antidepressants.21 If this is true, it is possible resistance to antipsychotics among some schizophrenia patients may be underpinned by the degree of cell injury and therefore of neurodegeneration, which may be the case with Ms. U.
Controlled, randomized, double-blind studies are needed to confirm our team’s assumptions. Our observation is limited by the lack of standardized scale measurements to assess all schizophrenia domains before starting riluzole and Ms. U’s clinical improvement could be associated with other factors such as passage of time or schizophrenia “burning out.” However, clinical observation and description from family members and hospital staff are important to consider in this case.
The improvement in schizophrenia symptoms observed from a drug with no action on dopamine blockade—a quality observed in all antipsychotics22—reinforces the possibility that targeting different pathways involved in the genesis of schizophrenia is a reasonable topic for future research. The possible use of riluzole and other glutamate-modulating drugs might influence positive, negative, and cognitive symptoms of schizophrenia.
Related Resources
- Kantrowitz JT, Javitt DC. Glutamate: new hope for schizophrenia treatment. Current Psychiatry. 2011;10(4):68-74.
- Vinson PN, Conn PJ. Metabotropic glutamate receptors as therapeutic targets for schizophrenia. Neuropharmacology. 2011. Epub ahead of print.
Drug Brand Names
- Acamprosate • Campral
- Amantadine • Symmetrel
- Clozapine • Clozaril
- Haloperidol • Haldol
- Ketamine • Ketalar
- Lamotrigine • Lamictal
- Lithium • Eskalith, Lithobid
- Loxapine • Loxitane
- Methotrimeprazine • Nozinan
- Memantine • Namenda
- Olanzapine • Zyprexa
- Riluzole • Rilutek
- Risperidone • Risperdal
- Sertindole • Serdolect
- Thioridazine • Mellaril
- Trazodone • Desyrel, Oleptro
- Trifluoperazine • Stelazine
- Venlafaxine • Effexor
Disclosures
Dr. Millán-González is a consultant to AstraZeneca CAMCAR. Drs. Loizaga-Arniaz and Zúñiga-Montes report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Discuss this article at www.facebook.com/CurrentPsychiatry
CASE: Relapsing psychosis
Ms. U, age 53, was diagnosed with paranoid schizophrenia at age 21 and has a continuous pattern of frequent relapses and inpatient admissions. She has received therapeutic doses of trifluoperazine, sertindole, haloperidol, loxapine, thioridazine, olanzapine, risperidone, clozapine, and several other antipsychotics not available in the United States. Clozapine had been prescribed at 600 mg/d (average blood level was 350 ng/mL), at times in combination with other antipsychotics or lithium.
Despite treatment, Ms. U has never achieved clinical stability. She has fluctuating yet persistent auditory hallucinations (eg, voices threatening to “announce disasters” or songs of a religious nature), associated disorganized behavior (eg, covering her ears or asking third parties “to turn off the radio”), severe hyponatremia secondary to potomania, paranoid ideation (eg, being followed by a “hidden camera”), and a strong tendency toward negativism, mutism, and emotional lability secondary to her psychotic symptoms. Her affect is predominantly poor and flattened, with very poor insight. Her symptoms are associated with progressive social isolation and poor grooming. Because of her worsening status, Ms. U was admitted to a residential facility 3 years ago.
Ms. U is single and the eldest of 2 siblings. Her parents are deceased; one parent may have committed suicide. She reports a family history of psychosis in her first cousins, but no history of hereditary neurologic disorders. Ms. U is a heavy smoker, did not complete college, and has a job in a family business.
The authors’ observations
Historically, the prevailing theory to explain the pathophysiology of schizophrenia has been the dopamine hypothesis, which links a hyperdopaminergic state in the mesolimbic system with acute psychosis. This theory could explain positive symptoms of schizophrenia but not other core domains, such as negative symptoms and cognitive dysfunction.1-3 The glutamate hypothesis postulates a hypoglutamatergic state can be the cause, at least in part, of various symptoms of psychosis, similar to those induced by phencyclidine and ketamine. Antagonists at the glycine modulatory site of the N-methyl-d-aspartate (NMDA) receptor are being studied as a way to influence this pathway,1 which is believed to be influenced by genetic factors.4
Glutamate, an amino acid, is the primary excitatory neurotransmitter in the brain. Its action is exerted in 2 types of receptors on the postsynaptic neuron: ionotropic and metabotropic.
The activation of NMDA receptors generated by glutamate and glycine coagonist can stimulate an uncontrolled release of calcium and subsequent cell death known as excitotoxicity. This phenomenon has been described in amyotrophic lateral sclerosis (ALS), Alzheimer’s disease, and Huntington’s disease. Although overstimulation of NMDA receptors induces neurodegeneration, NMDA hypoactivity has been observed in psychotic states.5
EVALUATION: Neurologic symptoms
A few months after arriving at the residential facility, Ms. U develops dysarthria and drooling, which the treatment team initially interprets as secondary to high doses of clozapine. In the absence of clinical response after clozapine dose reduction and with the subsequent appearance of dysphagia with solid foods and liquids, Ms. U is evaluated by a ear, nose, and throat physician, and later by a neurologist. Both clinicians describe frontal release signs, anarthria, facial hypomimia, bilateral mild central paresis, absence of soft palate elevation with symmetrical phonation, decreased gag reflex and palatal atrophy, fasciculations, and bilateral lingual mandibular reflex and diagnose Ms. U with progressive bulbar palsy, a variant of ALS.
The authors’ observations
ALS is a progressive, degenerative neuromuscular condition of unknown etiology affecting the corticospinal tracts and the anterior horn of the spinal cord, leading to dysfunction of the upper and lower motor neurons.6 It is more common in men, persons with diets rich in glutamate, and smokers.7,8
Riluzole is the only FDA-approved medication for ALS.9 It interferes with the responses mediated by the NMDA receptor, stabilizes inactive sodium voltage-dependent channels, inhibits glutamate release from synaptic endings, and activates extracellular reuptake of glutamate, all of which are thought to confer a neuroprotective effect.10
TREATMENT: Psychosis improves
As suggested by the neurology team, we begin riluzole, 50 mg every 12 hours. At this time Ms. U also is taking clozapine, 600 mg/d; lithium, 1200 mg/d; and haloperidol, 6 mg/d; her psychiatric symptoms have not changed since the initial evaluation at the residential facility.
Seven months after initiating riluzole Ms. U is more receptive, less querulant, and no longer experiences delusions or hallucinations. At the same time, she develops an interest in her clinical status regarding her ALS diagnosis, which reflects improved insight. One year after starting riluzole, she is more cooperative and adherent with treatment. Ms. U is able to reestablish relationships with her family. Clozapine and haloperidol are tapered and discontinued. Ms. U’s medication regimen includes risperidone, 1 mg/d; methotrimeprazine, 10 mg/d; venlafaxine, 75 mg/d; trazodone, 100 mg/d; and lithium, 600 mg/d, in addition to riluzole, 50 mg every 12 hours.
An assessment 18 months after starting riluzole describes a Positive and Negative Syndrome Scale (PANSS) score of 9 for positive symptoms, 11 for negative, 35 for the general psychopathology, and -2 for the composite (Table 1). Laboratory tests are normal except for a mild normocytic, normochromic anemia. MRI shows no detectable lesions or changes in comparison with previous images.
Table 1
Ms. U’s clinical course
| PANSS score | Treatment | Mental status |
|---|---|---|
| Before starting riluzole | ||
| No PANSS reported | Clozapine, 600 mg/d; lithium, 1200 mg/d; haloperidol, 6 mg/d | Persistent auditory hallucinations. Persistent hallucinatory behavior. Paranoid delirious ideas. Negativism, mutism, and liability reactive to her psychosis state. Poor and flattened affect. Lack of disease awareness. Progressive social isolation. Loss of self care |
| After starting riluzole | ||
| Positive subscale: 9 (below 5th percentile) Negative subscale: 11 (between 5th-25th percentile) General psychopathology subscale: 35 (between 5th-25th percentile) Composite score: -2 (between 25th-50th percentiles) | Riluzole, 50 mg every 12 hours; risperidone, 1 mg/d; methotrimeprazine, 10 mg/d; venlafaxine, 75 mg/d; trazodone, 100 mg/d; lithium, 600 mg/d | Re-establishes relationships with family because she no longer experiences paranoid delusions. Behavioral improvement. Allows physical proximity to nursing and medical personnel. Attention to physical appearance. Participates in social and recreational activities outside the hospital. Absence of auditory hallucinations. Affective improvement with appropriate responses. Realistic anxiety and fear about ALS diagnosis |
| ALS: amyotrophic lateral sclerosis; PANSS: Positive and Negative Syndrome Scale | ||
The authors’ observations
We present a patient with schizophrenia and a continuous pattern of relapses, functional and social impairment, and partial remission of her psychosis despite the use of multiple typical and atypical antipsychotics at therapeutic doses. Ms. U received treatment with clozapine at therapeutic doses for >6 months without sustained improvement. After beginning riluzole, a glutamate pathway antagonist, and with no other changes to her medication regimen, Ms. U experienced substantial improvement in her mental status. This was evidenced by a significant decline in her paranoid delusions, disappearance of auditory hallucinations, and substantial improvement on her social performance.
This fact is consistent with previous observations where modulation of the glutamate pathway has been associated with improvement in depression and anxiety levels in different populations. This case report provides further evidence to the possibility that blocking this receptor is a promising approach to psychotic disorders.
Riluzole for psychiatric illness
Currently, there are 11 clinical trials investigating riluzole for psychiatric disorders, including OCD, depression, bipolar disorder, schizophrenia, and Tourette’s syndrome.11 Consistent with the altered glutamatergic neurotransmission implicated in mood and anxiety disorders, preliminary evidence suggests riluzole can effectively treat OCD, bipolar depression, unipolar depression, and comorbid OCD and depression (Table 2). Some investigators consider the glutamatergic pathway an essential target for future antidepressants and mood-stabilizing agents.12
Other drugs such as memantine, acamprosate, and lamotrigine act on this same pathway and therefore have a role in treating psychiatric and neurologic conditions. In the case of lamotrigine, the drug inhibits glutamate release through inhibition of voltage-dependent sodium and calcium channels13 and postsynaptic AMPA receptors14 and has been shown to effectively treat generalized epilepsies,15 bipolar depression,13,16 and depression and mood swings associated with Huntington’s disease.17
Acamprosate’s attenuation of hyperglutamatergic states through NMDA antagonism and metabotropic glutamate receptors and reduction of intracellular calcium release—therefore balancing the glutamatergic and GABAergic systems and conferring neuroprotective properties—has been effective in patients with alcohol use disorders.18,19
Memantine and amantadine act through NMDA antagonism and by modulating dopaminergic transmission and may have clinical roles beyond dementia treatment.
Table 2
Evidence of efficacy of riluzole for OCD and depression
| Study | Disorder | Findings |
|---|---|---|
| Pittenger et al, 2006a | OCD | Brain imaging reveals elevated glutamate levels in OCD patients; agents that reduce glutamate hyperactivity may be effective |
| Coric et al, 2005b | OCD | Among 13 patients with OCD who received riluzole, 54% demonstrated >35% reduction in Y-BOCS scores and 39% were considered treatment responders |
| Zarate et al, 2005c | Bipolar depression | In an 8-week add-on study of riluzole in combination with lithium of 14 patients with bipolar depression, riluzole showed efficacy as measured by MADRS score and was well tolerated |
| Singh et al, 2004d | Bipolar depression | Case report of a patient with bipolar II disorder and depression who had a good response to riluzole when lamotrigine was discontinued because of a maculopapular erythematic rash |
| Zarate et al, 2004e | Unipolar depression | In a 6-week, open-label trial, 19 treatment-resistant depressed patients received riluzole; significant improvement measured by MADRS, CGI-S, and HAM-A were noted at weeks 3 through 6 |
| Coric et al, 2003f | Comorbid OCD and major depressive disorder | Case report of a patient with symptomatic OCD and depression who did not respond to appropriate pharmacotherapy, including augmentation strategies; adding riluzole significantly attenuated both obsessions and depressive symptoms |
| CGI-S: Clinical Global Impressions-Severity; HAM-A: Hamilton Anxiety Rating Scale; MADRS: Montgomery-Åsberg Depression Rating Scale; OCD: obsessive-compulsive disorder; Y-BOCS: Yale-Brown Obsessive Compulsive Scale Source: a. Pittenger C, Krystal JH, Coric V. Glutamate-modulating drugs as novel pharmacotherapeutic agents in the treatment of obsessive-compulsive disorder. Neurotherapeutics. 2006;3(1):69-81. b. Coric V, Taskiran S, Pittenger C, et al. Riluzole augmentation in treatment-resistant obsessive-compulsive disorder: an open-label trial. Biol Psychiatry. 2005;58(5):424-428. c. Zarate CA Jr, Quiroz JA, Singh JB, et al. An open-label trial of the glutamate-modulating agent riluzole in combination with lithium for the treatment of bipolar depression. Biol Psychiatry. 2005;57(4):430-432. d. Singh J, Zarate CA, Krystal AD. Case report: successful riluzole augmentation therapy in treatment-resistant bipolar depression following the development of rash with lamotrigine. Psychopharmacology. 2004;173(1-2):227-228. e. Zarate CA Jr, Payne JL, Quiroz J, et al. An open-label trial of riluzole in patients with treatment-resistant major depression. Am J Psychiatry. 2004;161(1):171-174. f. Coric V, Milanovic S, Wasylink S, et al. Beneficial effects of the antiglutamatergic agent riluzole in a patient diagnosed with obsessive-compulsive disorder and major depressive disorder. Psychopharmacology. 2003;167(2):219-220. | ||
Schizophrenia-ALS comorbidity
Some investigators have suggested20 the relative rarity of ALS in patients with schizophrenia is attributable to the neuroprotective effects of antipsychotics and antidepressants.21 If this is true, it is possible resistance to antipsychotics among some schizophrenia patients may be underpinned by the degree of cell injury and therefore of neurodegeneration, which may be the case with Ms. U.
Controlled, randomized, double-blind studies are needed to confirm our team’s assumptions. Our observation is limited by the lack of standardized scale measurements to assess all schizophrenia domains before starting riluzole and Ms. U’s clinical improvement could be associated with other factors such as passage of time or schizophrenia “burning out.” However, clinical observation and description from family members and hospital staff are important to consider in this case.
The improvement in schizophrenia symptoms observed from a drug with no action on dopamine blockade—a quality observed in all antipsychotics22—reinforces the possibility that targeting different pathways involved in the genesis of schizophrenia is a reasonable topic for future research. The possible use of riluzole and other glutamate-modulating drugs might influence positive, negative, and cognitive symptoms of schizophrenia.
Related Resources
- Kantrowitz JT, Javitt DC. Glutamate: new hope for schizophrenia treatment. Current Psychiatry. 2011;10(4):68-74.
- Vinson PN, Conn PJ. Metabotropic glutamate receptors as therapeutic targets for schizophrenia. Neuropharmacology. 2011. Epub ahead of print.
Drug Brand Names
- Acamprosate • Campral
- Amantadine • Symmetrel
- Clozapine • Clozaril
- Haloperidol • Haldol
- Ketamine • Ketalar
- Lamotrigine • Lamictal
- Lithium • Eskalith, Lithobid
- Loxapine • Loxitane
- Methotrimeprazine • Nozinan
- Memantine • Namenda
- Olanzapine • Zyprexa
- Riluzole • Rilutek
- Risperidone • Risperdal
- Sertindole • Serdolect
- Thioridazine • Mellaril
- Trazodone • Desyrel, Oleptro
- Trifluoperazine • Stelazine
- Venlafaxine • Effexor
Disclosures
Dr. Millán-González is a consultant to AstraZeneca CAMCAR. Drs. Loizaga-Arniaz and Zúñiga-Montes report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Freudenreich O, Weiss AP, Goff DC. Psychosis and schizophrenia. In: Stern T Rosenbaum, JF, Fava M, et al, eds. Massachusetts general hospital comprehensive clinical psychiatry. Philadelphia, PA: Mosby, an Imprint of Elsevier; 2008:371–389.
2. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington DC: American Psychiatric Association; 2000.
3. Bowie CR, Harvey PD. Cognition in schizophrenia: impairments determinants, and functional importance. Psychiatr Clin North Am. 2005;28(3):613-633.
4. Waddington JL, Corvin AP, Donohoe G, et al. Functional genomics and schizophrenia: endophenotypes and mutant models. Psychiatr Clin North Am. 2007;30(3):365-399.
5. Morrow EM, Roffman JL, Wolf DH, et al. Psychiatric neuroscience: incorporating pathophysiology into clinical case formulation. In: Stern T, Rosenbaum, JF, Fava M, et al, eds. Massachusetts General Hospital comprehensive clinical psychiatry. Philadelphia, PA: Mosby, an Imprint of Elsevier; 2008:543–564.
6. Harrison T. Amyotrophic lateral sclerosis. In: Ferri’s clinical advisor 2010. Philadelphia PA. Mosby, an Imprint of Elsevier; 2011:57.
7. Ringel SP, Murphy JR, Alderson MK, et al. The natural history of amyotrophic lateral sclerosis. Neurology. 1993;43(7):1316-1322.
8. Chancellor AM, Warlow CP. Adult onset motor neuron disease: worldwide mortality incidence and distribution since 1950. J Neurol Neurosurg Psychiatry. 1992;55(12):1106-1115.
9. Practice advisory on the treatment of amyotrophic lateral sclerosis with riluzole: report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 1997;49(3):657-659.
10. Distad BJ, Meekins GD, Liou LL, et al. Drug therapy in amyotrophic lateral sclerosis. Phys Med Rehabil Clin N Am. 2008;19(3):633-651.
11. ClinicalTrials.gov. U.S. National Institutes of Health. Available at: http://clinicaltrials.gov/ct2/results?intr=%22Riluzole%22. Accessed June 27, 2011.
12. Krystal JH, Sanacora G, Blumberg H, et al. Glutamate and GABA systems as targets for novel antidepressant and mood-stabilizing treatments. Mol Psychiatry. 2002;7(suppl 1):S71-80.
13. Calabrese JR, Bowden CL, Sachs GS, et al. A double-blind placebo-controlled study of lamotrigine monotherapy in outpatients with bipolar I depression. Lamictal 602 Study Group. J Clin Psychiatry. 1999;60(2):79-88.
14. Lee CY, Fu WM, Chen CC, et al. Lamotrigine inhibits postsynaptic AMPA receptor and glutamate release in the dentate gyrus. Epilepsia. 2008;49(5):888-897.
15. Patsalos PN. Properties of antiepileptic drugs in the treatment of idiopathic generalized epilepsies. Epilepsia. 2005;46(suppl 9):140-148.
16. Yatham LN, Kennedy SH, Schaffer A, et al. Canadian Network for Mood and Anxiety Treatments (CANMAT) and International Society for Bipolar Disorders (ISBD) collaborative update of CANMAT guidelines for the management of patients with bipolar disorder: update 2009. Bipolar Disord. 2009;11(3):225-255.
17. Shen YC. Lamotrigine in motor and mood symptoms of Huntington’s disease. World J Biol Psychiatry. 2008;9(2):147-149.
18. Scott LJ, Figgitt DP, Keam SJ, et al. Acamprosate: a review of its use in the maintenance of abstinence in patients with alcohol dependence. CNS Drugs. 2005;19(5):445-464.
19. De Witte P, Littleton J, Parot P, et al. Neuroprotective and abstinence-promoting effects of acamprosate: elucidating the mechanism of action. CNS Drugs. 2005;19(6):517-537.
20. Stommel EW, Graber D, Montanye J, et al. Does treating schizophrenia reduce the chances of developing amyotrophic lateral sclerosis? Med Hypotheses. 2007;69(5):1021-1028.
21. Howland RH. Schizophrenia and amyotrophic lateral sclerosis. Compr Psychiatry. 1990;31(4):327-336.
22. Seeman P. Atypical antipsychotics: mechanism of action. Can J Psychiatry. 2002;47(1):27-38.
1. Freudenreich O, Weiss AP, Goff DC. Psychosis and schizophrenia. In: Stern T Rosenbaum, JF, Fava M, et al, eds. Massachusetts general hospital comprehensive clinical psychiatry. Philadelphia, PA: Mosby, an Imprint of Elsevier; 2008:371–389.
2. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington DC: American Psychiatric Association; 2000.
3. Bowie CR, Harvey PD. Cognition in schizophrenia: impairments determinants, and functional importance. Psychiatr Clin North Am. 2005;28(3):613-633.
4. Waddington JL, Corvin AP, Donohoe G, et al. Functional genomics and schizophrenia: endophenotypes and mutant models. Psychiatr Clin North Am. 2007;30(3):365-399.
5. Morrow EM, Roffman JL, Wolf DH, et al. Psychiatric neuroscience: incorporating pathophysiology into clinical case formulation. In: Stern T, Rosenbaum, JF, Fava M, et al, eds. Massachusetts General Hospital comprehensive clinical psychiatry. Philadelphia, PA: Mosby, an Imprint of Elsevier; 2008:543–564.
6. Harrison T. Amyotrophic lateral sclerosis. In: Ferri’s clinical advisor 2010. Philadelphia PA. Mosby, an Imprint of Elsevier; 2011:57.
7. Ringel SP, Murphy JR, Alderson MK, et al. The natural history of amyotrophic lateral sclerosis. Neurology. 1993;43(7):1316-1322.
8. Chancellor AM, Warlow CP. Adult onset motor neuron disease: worldwide mortality incidence and distribution since 1950. J Neurol Neurosurg Psychiatry. 1992;55(12):1106-1115.
9. Practice advisory on the treatment of amyotrophic lateral sclerosis with riluzole: report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 1997;49(3):657-659.
10. Distad BJ, Meekins GD, Liou LL, et al. Drug therapy in amyotrophic lateral sclerosis. Phys Med Rehabil Clin N Am. 2008;19(3):633-651.
11. ClinicalTrials.gov. U.S. National Institutes of Health. Available at: http://clinicaltrials.gov/ct2/results?intr=%22Riluzole%22. Accessed June 27, 2011.
12. Krystal JH, Sanacora G, Blumberg H, et al. Glutamate and GABA systems as targets for novel antidepressant and mood-stabilizing treatments. Mol Psychiatry. 2002;7(suppl 1):S71-80.
13. Calabrese JR, Bowden CL, Sachs GS, et al. A double-blind placebo-controlled study of lamotrigine monotherapy in outpatients with bipolar I depression. Lamictal 602 Study Group. J Clin Psychiatry. 1999;60(2):79-88.
14. Lee CY, Fu WM, Chen CC, et al. Lamotrigine inhibits postsynaptic AMPA receptor and glutamate release in the dentate gyrus. Epilepsia. 2008;49(5):888-897.
15. Patsalos PN. Properties of antiepileptic drugs in the treatment of idiopathic generalized epilepsies. Epilepsia. 2005;46(suppl 9):140-148.
16. Yatham LN, Kennedy SH, Schaffer A, et al. Canadian Network for Mood and Anxiety Treatments (CANMAT) and International Society for Bipolar Disorders (ISBD) collaborative update of CANMAT guidelines for the management of patients with bipolar disorder: update 2009. Bipolar Disord. 2009;11(3):225-255.
17. Shen YC. Lamotrigine in motor and mood symptoms of Huntington’s disease. World J Biol Psychiatry. 2008;9(2):147-149.
18. Scott LJ, Figgitt DP, Keam SJ, et al. Acamprosate: a review of its use in the maintenance of abstinence in patients with alcohol dependence. CNS Drugs. 2005;19(5):445-464.
19. De Witte P, Littleton J, Parot P, et al. Neuroprotective and abstinence-promoting effects of acamprosate: elucidating the mechanism of action. CNS Drugs. 2005;19(6):517-537.
20. Stommel EW, Graber D, Montanye J, et al. Does treating schizophrenia reduce the chances of developing amyotrophic lateral sclerosis? Med Hypotheses. 2007;69(5):1021-1028.
21. Howland RH. Schizophrenia and amyotrophic lateral sclerosis. Compr Psychiatry. 1990;31(4):327-336.
22. Seeman P. Atypical antipsychotics: mechanism of action. Can J Psychiatry. 2002;47(1):27-38.
A dangerous GI complication
Discuss this article at www.facebook.com/CurrentPsychiatry
CASE: Gl surgery
Ms. X, age 61, presents to the emergency department (ED) complaining of nausea, vomiting, and abdominal pain and distension. CT scan of her abdomen reveals segmental ischemia in her colon with abscess formation, which leads to immediate surgery, including ileocecostomy with primary anastomosis. After surgery, Ms. X suffers from gastrointestinal (GI) dysmotility. The gastroenterology team recommends daily enemas along with a soft diet after she is discharged.
Ms. X has chronic paranoid schizophrenia, which has been treated successfully for 18 years with clozapine, 500 mg/d. During acute psychotic episodes, she experienced paranoid delusions and command auditory hallucinations telling her to kill herself. She had previous trials of several antipsychotics, including quetiapine, thiothixene, thioridazine, trifluoperazine, chlorpromazine, and haloperidol, all of which were ineffective and poorly tolerated because of serious side effects.
Within 1 month of discharge, Ms. X returns to the ED with nausea, vomiting, and abdominal distension. Abdominal CT scan suggests partial small bowel obstruction and significantly dilated loops of small bowel with decompressed rectum and sigmoid colon. Considering her recent GI surgery and absence of abdominal pain, she is managed with conservative measures, including nasogastric tube decompression and total parenteral nutrition. CT enterography demonstrates no areas of stricture formation with interval decompression.
The psychiatric service is consulted to evaluate the possibility of clozapine-induced paralytic ileus. During initial assessment, Ms. X denies any psychotic symptoms, including paranoid ideations, delusions, and auditory or visual hallucinations, and firmly believes that clozapine helps keep her stable. She also denies mood symptoms that could indicate mania or depression. She shows no signs or symptoms that suggest anticholinergic delirium.
The authors’ observations
Clozapine has proven efficacy in managing treatment-resistant schizophrenia,1-3 but the drug has been associated with life-threatening side effects, including agranulocytosis/neutropenia, myocarditis/cardiomyopathy, arrhythmia, seizures, diabetic ketoacidosis, fulminant hepatic failure, pulmonary embolism, and GI complications.4
Clozapine-induced GI side effects include anorexia, nausea, vomiting, heartburn, abdominal discomfort, diarrhea, and constipation. Clozapine-induced gastrointestinal hypomotility (CIGH) can lead to fecalith formation, which may result in intestinal obstruction/pseudo-obstruction, intestinal distension, necrosis, perforation, sepsis, aspiration from inhalation of feculent vomitus, or dysphagia.5 Constipation has been reported in 14% to 60% of patients who take clozapine,6 although other psychiatric medications also can cause constipation (Table 1). Severe constipation can lead to potentially fatal GI complications such as intestinal obstruction, necrosis, perforation, and sepsis, which is associated with significant morbidity due to bowel resection and a 27.5% mortality rate.5
The underlying mechanism of clozapine-induced constipation has been well established. The gut is innervated mainly by cholinergic and serotonergic receptors (5-HT3) and these receptors are responsible for peristalsis. Clozapine has a potent anticholinergic effect and acts as a strong antagonist of serotonin receptors (5-HT2, 5-HT3, 5-HT6, 5-HT7), which can lead to gut hypomotility.7 Risk factors associated with CIGH include:
- high dose of clozapine (mean dosage >428 mg/d)
- high serum clozapine levels (>500 ng/mL)
- coadministration of anticholinergic medications
- concomitant use of cytochrome P450 (CYP) enzyme inhibitors (medications inhibiting CYP1A2 enzyme)
- comorbid medical illnesses
- fever
- history of surgical bowel resection, GI pathology, and constipation.5
Table 1
Psychotropics associated with constipation
| Class | Medications |
|---|---|
| Atypical antipsychotics | Clozapine, risperidone |
| Typical antipsychotics | Chlorpromazine, haloperidol, pimozide, thioridazine, thiothixene, trifluoperazine |
| Anticholinergics | Benztropine, trihexyphenidyl |
| Antidepressants | Amitriptyline, clomipramine, doxepin, imipramine, nortriptyline, trimipramine |
HISTORY: Medical comorbidities
Ms. X’s medical history is significant for chronic constipation, hypertension, obstructive pulmonary disease, and hyperthyroidism. Her medications include trazodone, 25 mg/d; fluoxetine, 40 mg/d, for negative symptoms and insomnia; docusate sodium, 200 mg/d; polyethylene glycol, 17 g/d; and bisacodyl suppository, 10 mg as needed for constipation. On admission, her laboratory test results—including complete blood count, liver function tests, kidney function tests, thyroid function profile, and serum calcium levels—all were within normal range.
The authors’ observations
Because the prevalence and severity of clozapine-induced constipation seem to be dose-dependent,8 minimizing the dosage is a logical management strategy.9 The life-threatening nature of clozapine-induced GI complications may require rapid dose reduction, which could compromise a patient’s stability. There is a little evidence regarding systematic management of clozapine-induced GI complications (Table 2).
Table 2
Clinical pearls for treating clozapine-induced constipation
| Serum clozapine levels >500 to 700 ng/mL have been associated with increased incidence of severe GI complications |
| Serum clozapine levels can guide reduction of clozapine dosage because of its linear kinetics (ie, halving the clozapine dose will halve the serum clozapine level) |
| Clozapine dosages should be reduced by no more than 25 mg/d to a maximum of 100 mg/week |
TREATMENT: Clozapine reduction
We obtain a serum clozapine level, which is elevated at 553 ng/mL. We recommend gradual reducing Ms. X’s clozapine dosage by 50 mg every 3 to 4 days to reach a target dose of 300 to 350 mg/d, to attain serum clozapine levels 350 to 400 ng/mL. Because of trazodone’s potential anticholinergic action, which could be worsening Ms. X’s constipation, we stop the drug and begin zolpidem, 5 to 10 mg/d, to manage her insomnia. During these medication changes, we closely monitor Ms. X for reemerging psychotic symptoms.
The authors’ observations
In addition to risk factors such as chronic constipation and recent GI surgery, Ms. X’s supra-therapeutic serum clozapine level (553 ng/mL) significantly increased her risk of clozapine-induced paralytic ileus. Antidepressants such as selective serotonin reuptake inhibitors (SSRIs) are known to increase tissue concentrations of clozapine and its major metabolite, norclozapine, by primarily inhibiting CYP1A2 and perhaps CYP2D6.10 As a potent inhibitor of CYPA12, fluvoxamine can inhibit clozapine metabolism, resulting in higher plasma concentrations.11 In Ms. X’s case, fluoxetine could have increased serum clozapine levels because of its ability to inhibit clozapine metabolism via CYP2D6-mediated mechanisms.12
Although clozapine serum levels are not routinely measured, such testing may be indicated in patients who do not respond to or are unable to tolerate clozapine. Clozapine levels should be obtained 12 hours after the bedtime dose (trough levels), several days after clozapine initiation. Serum clozapine levels <350 ng/mL are associated with lack of clinical response.13 Higher serum levels (500 to 700 ng/mL) have been associated with greater incidences of serious GI complications. Serum clozapine levels also help guide clozapine dosage reduction because of its linear kinetics—halving the dose will halve the serum clozapine level.14
OUTCOME: GI symptoms improve
Ms. X shows improved GI motility within few days of the first decrease in her clozapine dosage. Nausea, vomiting, and abdominal distension gradually resolve over 2 weeks with concomitant reduction in clozapine dosage to 300 mg/d (50 mg in the morning and 250 mg at bedtime) without reemergence of psychotic symptoms. She is able to tolerate a soft diet, and conservative GI measures are no longer required. She is discharged home with outpatient surgical and psychiatric follow-up.
The authors’ observations
Successful reversal of severe clozapine-induced constipation—occurring at serum clozapine level of 490 ng/mL—has been reported in a 45-year-old man with treatment-resistant schizophrenia. This was accomplished by cautious reduction of clozapine dosage (400 mg/d to 250 mg/d) over 1 week.15 Slower clozapine titration—reducing the dose by no more than 25 mg/d to a maximum of 100 mg/week—has been recommended.16 It also has been suggested to replace part of the clozapine dose with a less antimuscarinic antipsychotic, such as quetiapine or haloperidol, thereby using the second antipsychotic as a clozapine-sparing agent.9 For example, the clozapine dose could be reduced by 25% by substituting 2 mg of quetiapine for every 1 mg of clozapine.
Prevention
Psychiatrists who prescribe clozapine should take a careful history of risk factors that might predispose patients to clozapine-induced GI side effects. Caution patients to whom you prescribe clozapine about possible development of constipation and the risk of serious GI complications. Enlist family members and caseworkers to keep a close eye on GI side effects in patients receiving clozapine. Advise patients to prevent constipation by eating a high fiber diet, drinking adequate fluids, and getting regular exercise. Patients should be treated aggressively with laxatives to relieve constipation and educated about the warning signs of intestinal obstruction, such as worsening constipation, abdominal pain, vomiting, and inability to pass flatus.17
Rapidly fatal bowel ischemia caused by clozapine has been reported.18 Therefore, urgently refer patients for medical evaluation if you have any concerns about worsening constipation or observe signs of intestinal obstruction. Vigilant consideration of clozapine as a likely culprit in severe GI complications in inpatient settings can prevent morbidity and mortality.
In our case, cautious reduction of clozapine dosage, guided by serum clozapine levels, had obviated the need for surgery and prevented reemergence of psychotic symptoms.
Related Resources
- Drew L, Herdson P. Clozapine and constipation: a serious issue. Aust N Z J Psychiatry. 1997;31(1):149-150.
- Winstead NS, Winstead DK. 5-step plan to treat constipation in psychiatric patients. Current Psychiatry. 2008;7(5):29-39.
Drug Brand Names
- Amitriptyline • Elavil
- Benztropine • Cogentin
- Bisacodyl suppository • Dulcolax, others
- Chlorpromazine • Thorazine
- Clomipramine • Anafranil
- Clozapine • Clozaril
- Docusate sodium • Colace, others
- Doxepin • Adapin, Sinequan
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Haloperidol • Haldol
- Imipramine • Tofranil
- Nortriptyline • Aventyl, Pamelor
- Pimozide • Orap
- Polyethylene glycol • MiraLax
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Thioridazine • Mellaril
- Thiothixene • Navane
- Trazodone • Desyrel, Oleptro
- Trifluoperazine • Stelazine
- Trihexyphenidyl • Artane, Trihexane
- Trimipramine • Surmontil
- Zolpidem • Ambien
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. American Psychiatric Association. Treatment of patients with schizophrenia, second edition. Recommendations for patients with schizophrenia. Arlington, VA: American Psychiatric Publishing, Inc; 2004.
2. Stahl S. Essential psychopharmacology. Cambridge, United Kingdom: Cambridge University Press; 1999.
3. Hardman J, Limbird L, Molinoff P. Goodman & Gilman’s pharmacological basis of therapeutics. New York, NY: McGraw-Hill; 1996.
4. Flanagan RJ, Ball RY. Gastrointestinal hypomotility: an under-recognized life-threatening adverse effect of clozapine. Forensic Sci Int. 2011;206(1-3):e31-36.
5. Palmer SE, McLean RM, Ellis PM, et al. Life-threatening clozapine-induced gastrointestinal hypomotility: an analysis of 102 cases. J Clin Psychiatry. 2008;69(5):759-768.
6. Claghorn J, Honigfeld G, Abuzzahab F, Sr, et al. The risks and benefits of clozapine versus chlorpromazine. J Clin Psychopharmacol. 1987;7:337-384.
7. Perrott J. Serious gastrointestinal adverse effects of clozapine. Psychopharmacology Newsletter. 2009;1-5.
8. Pare J, Riffand P, Baurdeix I. The clozapine in France. Information Psychiatric. 1993;4:389-397.
9. Levin TT, Barrett J, Mendelowitz A. Death from clozapine-induced constipation: case report and literature review. Psychosomatics. 2002;43:71-73.
10. Centorrino F, Baldessarini RJ, Frankenburg FR, et al. Serum levels of clozapine and norclozapine in patients treated with selective serotonin reuptake inhibitors. Am J Psychiatry. 1996;153(6):820-822.
11. Sproule BA, Naranjo CA, Brenmer KE, et al. Selective serotonin reuptake inhibitors and CNS drug interactions. A critical review of the evidence. Clin Pharmacokinet. 1997;33(6):454-471.
12. Urichuk L, Prior TI, Dursun S, et al. Metabolism of atypical antipsychotics: involvement of cytochrome p450 enzymes and relevance for drug-drug interactions. Curr Drug Metab. 2008;9(5):410-418.
13. Perry P, Miller DD, Arndt SV, et al. Clozapine and norclozapine plasma concentrations and clinical responses of treatment-refractory schizophrenic patients. Am J Psychiatry. 1991;148:231-235.
14. Freudenreich O. Clozapine drug levels guide dosing. Current Psychiatry. 2009;8(3):78.-
15. Pelizza L, De Luca P, La Pesa M, et al. Clozapine-induced intestinal occlusion: a serious side effect. Acta Biomed. 2007;78:144-148.
16. Hayes G, Gibler B. Clozapine-induced constipation. Am J Psychiatry. 1995;152:298.-
17. American College of Gastroenterology Chronic Constipation Task Force. An evidence-based approach to the management of chronic constipation in North America. Am J Gastroenterol. 2005;100(suppl 1):S1-4.
18. Townsend G, Curtis D. Case report: rapidly fatal bowel ischaemia on clozapine treatment. BMC Psychiatry. 2006;6:43.-
Discuss this article at www.facebook.com/CurrentPsychiatry
CASE: Gl surgery
Ms. X, age 61, presents to the emergency department (ED) complaining of nausea, vomiting, and abdominal pain and distension. CT scan of her abdomen reveals segmental ischemia in her colon with abscess formation, which leads to immediate surgery, including ileocecostomy with primary anastomosis. After surgery, Ms. X suffers from gastrointestinal (GI) dysmotility. The gastroenterology team recommends daily enemas along with a soft diet after she is discharged.
Ms. X has chronic paranoid schizophrenia, which has been treated successfully for 18 years with clozapine, 500 mg/d. During acute psychotic episodes, she experienced paranoid delusions and command auditory hallucinations telling her to kill herself. She had previous trials of several antipsychotics, including quetiapine, thiothixene, thioridazine, trifluoperazine, chlorpromazine, and haloperidol, all of which were ineffective and poorly tolerated because of serious side effects.
Within 1 month of discharge, Ms. X returns to the ED with nausea, vomiting, and abdominal distension. Abdominal CT scan suggests partial small bowel obstruction and significantly dilated loops of small bowel with decompressed rectum and sigmoid colon. Considering her recent GI surgery and absence of abdominal pain, she is managed with conservative measures, including nasogastric tube decompression and total parenteral nutrition. CT enterography demonstrates no areas of stricture formation with interval decompression.
The psychiatric service is consulted to evaluate the possibility of clozapine-induced paralytic ileus. During initial assessment, Ms. X denies any psychotic symptoms, including paranoid ideations, delusions, and auditory or visual hallucinations, and firmly believes that clozapine helps keep her stable. She also denies mood symptoms that could indicate mania or depression. She shows no signs or symptoms that suggest anticholinergic delirium.
The authors’ observations
Clozapine has proven efficacy in managing treatment-resistant schizophrenia,1-3 but the drug has been associated with life-threatening side effects, including agranulocytosis/neutropenia, myocarditis/cardiomyopathy, arrhythmia, seizures, diabetic ketoacidosis, fulminant hepatic failure, pulmonary embolism, and GI complications.4
Clozapine-induced GI side effects include anorexia, nausea, vomiting, heartburn, abdominal discomfort, diarrhea, and constipation. Clozapine-induced gastrointestinal hypomotility (CIGH) can lead to fecalith formation, which may result in intestinal obstruction/pseudo-obstruction, intestinal distension, necrosis, perforation, sepsis, aspiration from inhalation of feculent vomitus, or dysphagia.5 Constipation has been reported in 14% to 60% of patients who take clozapine,6 although other psychiatric medications also can cause constipation (Table 1). Severe constipation can lead to potentially fatal GI complications such as intestinal obstruction, necrosis, perforation, and sepsis, which is associated with significant morbidity due to bowel resection and a 27.5% mortality rate.5
The underlying mechanism of clozapine-induced constipation has been well established. The gut is innervated mainly by cholinergic and serotonergic receptors (5-HT3) and these receptors are responsible for peristalsis. Clozapine has a potent anticholinergic effect and acts as a strong antagonist of serotonin receptors (5-HT2, 5-HT3, 5-HT6, 5-HT7), which can lead to gut hypomotility.7 Risk factors associated with CIGH include:
- high dose of clozapine (mean dosage >428 mg/d)
- high serum clozapine levels (>500 ng/mL)
- coadministration of anticholinergic medications
- concomitant use of cytochrome P450 (CYP) enzyme inhibitors (medications inhibiting CYP1A2 enzyme)
- comorbid medical illnesses
- fever
- history of surgical bowel resection, GI pathology, and constipation.5
Table 1
Psychotropics associated with constipation
| Class | Medications |
|---|---|
| Atypical antipsychotics | Clozapine, risperidone |
| Typical antipsychotics | Chlorpromazine, haloperidol, pimozide, thioridazine, thiothixene, trifluoperazine |
| Anticholinergics | Benztropine, trihexyphenidyl |
| Antidepressants | Amitriptyline, clomipramine, doxepin, imipramine, nortriptyline, trimipramine |
HISTORY: Medical comorbidities
Ms. X’s medical history is significant for chronic constipation, hypertension, obstructive pulmonary disease, and hyperthyroidism. Her medications include trazodone, 25 mg/d; fluoxetine, 40 mg/d, for negative symptoms and insomnia; docusate sodium, 200 mg/d; polyethylene glycol, 17 g/d; and bisacodyl suppository, 10 mg as needed for constipation. On admission, her laboratory test results—including complete blood count, liver function tests, kidney function tests, thyroid function profile, and serum calcium levels—all were within normal range.
The authors’ observations
Because the prevalence and severity of clozapine-induced constipation seem to be dose-dependent,8 minimizing the dosage is a logical management strategy.9 The life-threatening nature of clozapine-induced GI complications may require rapid dose reduction, which could compromise a patient’s stability. There is a little evidence regarding systematic management of clozapine-induced GI complications (Table 2).
Table 2
Clinical pearls for treating clozapine-induced constipation
| Serum clozapine levels >500 to 700 ng/mL have been associated with increased incidence of severe GI complications |
| Serum clozapine levels can guide reduction of clozapine dosage because of its linear kinetics (ie, halving the clozapine dose will halve the serum clozapine level) |
| Clozapine dosages should be reduced by no more than 25 mg/d to a maximum of 100 mg/week |
TREATMENT: Clozapine reduction
We obtain a serum clozapine level, which is elevated at 553 ng/mL. We recommend gradual reducing Ms. X’s clozapine dosage by 50 mg every 3 to 4 days to reach a target dose of 300 to 350 mg/d, to attain serum clozapine levels 350 to 400 ng/mL. Because of trazodone’s potential anticholinergic action, which could be worsening Ms. X’s constipation, we stop the drug and begin zolpidem, 5 to 10 mg/d, to manage her insomnia. During these medication changes, we closely monitor Ms. X for reemerging psychotic symptoms.
The authors’ observations
In addition to risk factors such as chronic constipation and recent GI surgery, Ms. X’s supra-therapeutic serum clozapine level (553 ng/mL) significantly increased her risk of clozapine-induced paralytic ileus. Antidepressants such as selective serotonin reuptake inhibitors (SSRIs) are known to increase tissue concentrations of clozapine and its major metabolite, norclozapine, by primarily inhibiting CYP1A2 and perhaps CYP2D6.10 As a potent inhibitor of CYPA12, fluvoxamine can inhibit clozapine metabolism, resulting in higher plasma concentrations.11 In Ms. X’s case, fluoxetine could have increased serum clozapine levels because of its ability to inhibit clozapine metabolism via CYP2D6-mediated mechanisms.12
Although clozapine serum levels are not routinely measured, such testing may be indicated in patients who do not respond to or are unable to tolerate clozapine. Clozapine levels should be obtained 12 hours after the bedtime dose (trough levels), several days after clozapine initiation. Serum clozapine levels <350 ng/mL are associated with lack of clinical response.13 Higher serum levels (500 to 700 ng/mL) have been associated with greater incidences of serious GI complications. Serum clozapine levels also help guide clozapine dosage reduction because of its linear kinetics—halving the dose will halve the serum clozapine level.14
OUTCOME: GI symptoms improve
Ms. X shows improved GI motility within few days of the first decrease in her clozapine dosage. Nausea, vomiting, and abdominal distension gradually resolve over 2 weeks with concomitant reduction in clozapine dosage to 300 mg/d (50 mg in the morning and 250 mg at bedtime) without reemergence of psychotic symptoms. She is able to tolerate a soft diet, and conservative GI measures are no longer required. She is discharged home with outpatient surgical and psychiatric follow-up.
The authors’ observations
Successful reversal of severe clozapine-induced constipation—occurring at serum clozapine level of 490 ng/mL—has been reported in a 45-year-old man with treatment-resistant schizophrenia. This was accomplished by cautious reduction of clozapine dosage (400 mg/d to 250 mg/d) over 1 week.15 Slower clozapine titration—reducing the dose by no more than 25 mg/d to a maximum of 100 mg/week—has been recommended.16 It also has been suggested to replace part of the clozapine dose with a less antimuscarinic antipsychotic, such as quetiapine or haloperidol, thereby using the second antipsychotic as a clozapine-sparing agent.9 For example, the clozapine dose could be reduced by 25% by substituting 2 mg of quetiapine for every 1 mg of clozapine.
Prevention
Psychiatrists who prescribe clozapine should take a careful history of risk factors that might predispose patients to clozapine-induced GI side effects. Caution patients to whom you prescribe clozapine about possible development of constipation and the risk of serious GI complications. Enlist family members and caseworkers to keep a close eye on GI side effects in patients receiving clozapine. Advise patients to prevent constipation by eating a high fiber diet, drinking adequate fluids, and getting regular exercise. Patients should be treated aggressively with laxatives to relieve constipation and educated about the warning signs of intestinal obstruction, such as worsening constipation, abdominal pain, vomiting, and inability to pass flatus.17
Rapidly fatal bowel ischemia caused by clozapine has been reported.18 Therefore, urgently refer patients for medical evaluation if you have any concerns about worsening constipation or observe signs of intestinal obstruction. Vigilant consideration of clozapine as a likely culprit in severe GI complications in inpatient settings can prevent morbidity and mortality.
In our case, cautious reduction of clozapine dosage, guided by serum clozapine levels, had obviated the need for surgery and prevented reemergence of psychotic symptoms.
Related Resources
- Drew L, Herdson P. Clozapine and constipation: a serious issue. Aust N Z J Psychiatry. 1997;31(1):149-150.
- Winstead NS, Winstead DK. 5-step plan to treat constipation in psychiatric patients. Current Psychiatry. 2008;7(5):29-39.
Drug Brand Names
- Amitriptyline • Elavil
- Benztropine • Cogentin
- Bisacodyl suppository • Dulcolax, others
- Chlorpromazine • Thorazine
- Clomipramine • Anafranil
- Clozapine • Clozaril
- Docusate sodium • Colace, others
- Doxepin • Adapin, Sinequan
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Haloperidol • Haldol
- Imipramine • Tofranil
- Nortriptyline • Aventyl, Pamelor
- Pimozide • Orap
- Polyethylene glycol • MiraLax
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Thioridazine • Mellaril
- Thiothixene • Navane
- Trazodone • Desyrel, Oleptro
- Trifluoperazine • Stelazine
- Trihexyphenidyl • Artane, Trihexane
- Trimipramine • Surmontil
- Zolpidem • Ambien
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Discuss this article at www.facebook.com/CurrentPsychiatry
CASE: Gl surgery
Ms. X, age 61, presents to the emergency department (ED) complaining of nausea, vomiting, and abdominal pain and distension. CT scan of her abdomen reveals segmental ischemia in her colon with abscess formation, which leads to immediate surgery, including ileocecostomy with primary anastomosis. After surgery, Ms. X suffers from gastrointestinal (GI) dysmotility. The gastroenterology team recommends daily enemas along with a soft diet after she is discharged.
Ms. X has chronic paranoid schizophrenia, which has been treated successfully for 18 years with clozapine, 500 mg/d. During acute psychotic episodes, she experienced paranoid delusions and command auditory hallucinations telling her to kill herself. She had previous trials of several antipsychotics, including quetiapine, thiothixene, thioridazine, trifluoperazine, chlorpromazine, and haloperidol, all of which were ineffective and poorly tolerated because of serious side effects.
Within 1 month of discharge, Ms. X returns to the ED with nausea, vomiting, and abdominal distension. Abdominal CT scan suggests partial small bowel obstruction and significantly dilated loops of small bowel with decompressed rectum and sigmoid colon. Considering her recent GI surgery and absence of abdominal pain, she is managed with conservative measures, including nasogastric tube decompression and total parenteral nutrition. CT enterography demonstrates no areas of stricture formation with interval decompression.
The psychiatric service is consulted to evaluate the possibility of clozapine-induced paralytic ileus. During initial assessment, Ms. X denies any psychotic symptoms, including paranoid ideations, delusions, and auditory or visual hallucinations, and firmly believes that clozapine helps keep her stable. She also denies mood symptoms that could indicate mania or depression. She shows no signs or symptoms that suggest anticholinergic delirium.
The authors’ observations
Clozapine has proven efficacy in managing treatment-resistant schizophrenia,1-3 but the drug has been associated with life-threatening side effects, including agranulocytosis/neutropenia, myocarditis/cardiomyopathy, arrhythmia, seizures, diabetic ketoacidosis, fulminant hepatic failure, pulmonary embolism, and GI complications.4
Clozapine-induced GI side effects include anorexia, nausea, vomiting, heartburn, abdominal discomfort, diarrhea, and constipation. Clozapine-induced gastrointestinal hypomotility (CIGH) can lead to fecalith formation, which may result in intestinal obstruction/pseudo-obstruction, intestinal distension, necrosis, perforation, sepsis, aspiration from inhalation of feculent vomitus, or dysphagia.5 Constipation has been reported in 14% to 60% of patients who take clozapine,6 although other psychiatric medications also can cause constipation (Table 1). Severe constipation can lead to potentially fatal GI complications such as intestinal obstruction, necrosis, perforation, and sepsis, which is associated with significant morbidity due to bowel resection and a 27.5% mortality rate.5
The underlying mechanism of clozapine-induced constipation has been well established. The gut is innervated mainly by cholinergic and serotonergic receptors (5-HT3) and these receptors are responsible for peristalsis. Clozapine has a potent anticholinergic effect and acts as a strong antagonist of serotonin receptors (5-HT2, 5-HT3, 5-HT6, 5-HT7), which can lead to gut hypomotility.7 Risk factors associated with CIGH include:
- high dose of clozapine (mean dosage >428 mg/d)
- high serum clozapine levels (>500 ng/mL)
- coadministration of anticholinergic medications
- concomitant use of cytochrome P450 (CYP) enzyme inhibitors (medications inhibiting CYP1A2 enzyme)
- comorbid medical illnesses
- fever
- history of surgical bowel resection, GI pathology, and constipation.5
Table 1
Psychotropics associated with constipation
| Class | Medications |
|---|---|
| Atypical antipsychotics | Clozapine, risperidone |
| Typical antipsychotics | Chlorpromazine, haloperidol, pimozide, thioridazine, thiothixene, trifluoperazine |
| Anticholinergics | Benztropine, trihexyphenidyl |
| Antidepressants | Amitriptyline, clomipramine, doxepin, imipramine, nortriptyline, trimipramine |
HISTORY: Medical comorbidities
Ms. X’s medical history is significant for chronic constipation, hypertension, obstructive pulmonary disease, and hyperthyroidism. Her medications include trazodone, 25 mg/d; fluoxetine, 40 mg/d, for negative symptoms and insomnia; docusate sodium, 200 mg/d; polyethylene glycol, 17 g/d; and bisacodyl suppository, 10 mg as needed for constipation. On admission, her laboratory test results—including complete blood count, liver function tests, kidney function tests, thyroid function profile, and serum calcium levels—all were within normal range.
The authors’ observations
Because the prevalence and severity of clozapine-induced constipation seem to be dose-dependent,8 minimizing the dosage is a logical management strategy.9 The life-threatening nature of clozapine-induced GI complications may require rapid dose reduction, which could compromise a patient’s stability. There is a little evidence regarding systematic management of clozapine-induced GI complications (Table 2).
Table 2
Clinical pearls for treating clozapine-induced constipation
| Serum clozapine levels >500 to 700 ng/mL have been associated with increased incidence of severe GI complications |
| Serum clozapine levels can guide reduction of clozapine dosage because of its linear kinetics (ie, halving the clozapine dose will halve the serum clozapine level) |
| Clozapine dosages should be reduced by no more than 25 mg/d to a maximum of 100 mg/week |
TREATMENT: Clozapine reduction
We obtain a serum clozapine level, which is elevated at 553 ng/mL. We recommend gradual reducing Ms. X’s clozapine dosage by 50 mg every 3 to 4 days to reach a target dose of 300 to 350 mg/d, to attain serum clozapine levels 350 to 400 ng/mL. Because of trazodone’s potential anticholinergic action, which could be worsening Ms. X’s constipation, we stop the drug and begin zolpidem, 5 to 10 mg/d, to manage her insomnia. During these medication changes, we closely monitor Ms. X for reemerging psychotic symptoms.
The authors’ observations
In addition to risk factors such as chronic constipation and recent GI surgery, Ms. X’s supra-therapeutic serum clozapine level (553 ng/mL) significantly increased her risk of clozapine-induced paralytic ileus. Antidepressants such as selective serotonin reuptake inhibitors (SSRIs) are known to increase tissue concentrations of clozapine and its major metabolite, norclozapine, by primarily inhibiting CYP1A2 and perhaps CYP2D6.10 As a potent inhibitor of CYPA12, fluvoxamine can inhibit clozapine metabolism, resulting in higher plasma concentrations.11 In Ms. X’s case, fluoxetine could have increased serum clozapine levels because of its ability to inhibit clozapine metabolism via CYP2D6-mediated mechanisms.12
Although clozapine serum levels are not routinely measured, such testing may be indicated in patients who do not respond to or are unable to tolerate clozapine. Clozapine levels should be obtained 12 hours after the bedtime dose (trough levels), several days after clozapine initiation. Serum clozapine levels <350 ng/mL are associated with lack of clinical response.13 Higher serum levels (500 to 700 ng/mL) have been associated with greater incidences of serious GI complications. Serum clozapine levels also help guide clozapine dosage reduction because of its linear kinetics—halving the dose will halve the serum clozapine level.14
OUTCOME: GI symptoms improve
Ms. X shows improved GI motility within few days of the first decrease in her clozapine dosage. Nausea, vomiting, and abdominal distension gradually resolve over 2 weeks with concomitant reduction in clozapine dosage to 300 mg/d (50 mg in the morning and 250 mg at bedtime) without reemergence of psychotic symptoms. She is able to tolerate a soft diet, and conservative GI measures are no longer required. She is discharged home with outpatient surgical and psychiatric follow-up.
The authors’ observations
Successful reversal of severe clozapine-induced constipation—occurring at serum clozapine level of 490 ng/mL—has been reported in a 45-year-old man with treatment-resistant schizophrenia. This was accomplished by cautious reduction of clozapine dosage (400 mg/d to 250 mg/d) over 1 week.15 Slower clozapine titration—reducing the dose by no more than 25 mg/d to a maximum of 100 mg/week—has been recommended.16 It also has been suggested to replace part of the clozapine dose with a less antimuscarinic antipsychotic, such as quetiapine or haloperidol, thereby using the second antipsychotic as a clozapine-sparing agent.9 For example, the clozapine dose could be reduced by 25% by substituting 2 mg of quetiapine for every 1 mg of clozapine.
Prevention
Psychiatrists who prescribe clozapine should take a careful history of risk factors that might predispose patients to clozapine-induced GI side effects. Caution patients to whom you prescribe clozapine about possible development of constipation and the risk of serious GI complications. Enlist family members and caseworkers to keep a close eye on GI side effects in patients receiving clozapine. Advise patients to prevent constipation by eating a high fiber diet, drinking adequate fluids, and getting regular exercise. Patients should be treated aggressively with laxatives to relieve constipation and educated about the warning signs of intestinal obstruction, such as worsening constipation, abdominal pain, vomiting, and inability to pass flatus.17
Rapidly fatal bowel ischemia caused by clozapine has been reported.18 Therefore, urgently refer patients for medical evaluation if you have any concerns about worsening constipation or observe signs of intestinal obstruction. Vigilant consideration of clozapine as a likely culprit in severe GI complications in inpatient settings can prevent morbidity and mortality.
In our case, cautious reduction of clozapine dosage, guided by serum clozapine levels, had obviated the need for surgery and prevented reemergence of psychotic symptoms.
Related Resources
- Drew L, Herdson P. Clozapine and constipation: a serious issue. Aust N Z J Psychiatry. 1997;31(1):149-150.
- Winstead NS, Winstead DK. 5-step plan to treat constipation in psychiatric patients. Current Psychiatry. 2008;7(5):29-39.
Drug Brand Names
- Amitriptyline • Elavil
- Benztropine • Cogentin
- Bisacodyl suppository • Dulcolax, others
- Chlorpromazine • Thorazine
- Clomipramine • Anafranil
- Clozapine • Clozaril
- Docusate sodium • Colace, others
- Doxepin • Adapin, Sinequan
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Haloperidol • Haldol
- Imipramine • Tofranil
- Nortriptyline • Aventyl, Pamelor
- Pimozide • Orap
- Polyethylene glycol • MiraLax
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Thioridazine • Mellaril
- Thiothixene • Navane
- Trazodone • Desyrel, Oleptro
- Trifluoperazine • Stelazine
- Trihexyphenidyl • Artane, Trihexane
- Trimipramine • Surmontil
- Zolpidem • Ambien
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. American Psychiatric Association. Treatment of patients with schizophrenia, second edition. Recommendations for patients with schizophrenia. Arlington, VA: American Psychiatric Publishing, Inc; 2004.
2. Stahl S. Essential psychopharmacology. Cambridge, United Kingdom: Cambridge University Press; 1999.
3. Hardman J, Limbird L, Molinoff P. Goodman & Gilman’s pharmacological basis of therapeutics. New York, NY: McGraw-Hill; 1996.
4. Flanagan RJ, Ball RY. Gastrointestinal hypomotility: an under-recognized life-threatening adverse effect of clozapine. Forensic Sci Int. 2011;206(1-3):e31-36.
5. Palmer SE, McLean RM, Ellis PM, et al. Life-threatening clozapine-induced gastrointestinal hypomotility: an analysis of 102 cases. J Clin Psychiatry. 2008;69(5):759-768.
6. Claghorn J, Honigfeld G, Abuzzahab F, Sr, et al. The risks and benefits of clozapine versus chlorpromazine. J Clin Psychopharmacol. 1987;7:337-384.
7. Perrott J. Serious gastrointestinal adverse effects of clozapine. Psychopharmacology Newsletter. 2009;1-5.
8. Pare J, Riffand P, Baurdeix I. The clozapine in France. Information Psychiatric. 1993;4:389-397.
9. Levin TT, Barrett J, Mendelowitz A. Death from clozapine-induced constipation: case report and literature review. Psychosomatics. 2002;43:71-73.
10. Centorrino F, Baldessarini RJ, Frankenburg FR, et al. Serum levels of clozapine and norclozapine in patients treated with selective serotonin reuptake inhibitors. Am J Psychiatry. 1996;153(6):820-822.
11. Sproule BA, Naranjo CA, Brenmer KE, et al. Selective serotonin reuptake inhibitors and CNS drug interactions. A critical review of the evidence. Clin Pharmacokinet. 1997;33(6):454-471.
12. Urichuk L, Prior TI, Dursun S, et al. Metabolism of atypical antipsychotics: involvement of cytochrome p450 enzymes and relevance for drug-drug interactions. Curr Drug Metab. 2008;9(5):410-418.
13. Perry P, Miller DD, Arndt SV, et al. Clozapine and norclozapine plasma concentrations and clinical responses of treatment-refractory schizophrenic patients. Am J Psychiatry. 1991;148:231-235.
14. Freudenreich O. Clozapine drug levels guide dosing. Current Psychiatry. 2009;8(3):78.-
15. Pelizza L, De Luca P, La Pesa M, et al. Clozapine-induced intestinal occlusion: a serious side effect. Acta Biomed. 2007;78:144-148.
16. Hayes G, Gibler B. Clozapine-induced constipation. Am J Psychiatry. 1995;152:298.-
17. American College of Gastroenterology Chronic Constipation Task Force. An evidence-based approach to the management of chronic constipation in North America. Am J Gastroenterol. 2005;100(suppl 1):S1-4.
18. Townsend G, Curtis D. Case report: rapidly fatal bowel ischaemia on clozapine treatment. BMC Psychiatry. 2006;6:43.-
1. American Psychiatric Association. Treatment of patients with schizophrenia, second edition. Recommendations for patients with schizophrenia. Arlington, VA: American Psychiatric Publishing, Inc; 2004.
2. Stahl S. Essential psychopharmacology. Cambridge, United Kingdom: Cambridge University Press; 1999.
3. Hardman J, Limbird L, Molinoff P. Goodman & Gilman’s pharmacological basis of therapeutics. New York, NY: McGraw-Hill; 1996.
4. Flanagan RJ, Ball RY. Gastrointestinal hypomotility: an under-recognized life-threatening adverse effect of clozapine. Forensic Sci Int. 2011;206(1-3):e31-36.
5. Palmer SE, McLean RM, Ellis PM, et al. Life-threatening clozapine-induced gastrointestinal hypomotility: an analysis of 102 cases. J Clin Psychiatry. 2008;69(5):759-768.
6. Claghorn J, Honigfeld G, Abuzzahab F, Sr, et al. The risks and benefits of clozapine versus chlorpromazine. J Clin Psychopharmacol. 1987;7:337-384.
7. Perrott J. Serious gastrointestinal adverse effects of clozapine. Psychopharmacology Newsletter. 2009;1-5.
8. Pare J, Riffand P, Baurdeix I. The clozapine in France. Information Psychiatric. 1993;4:389-397.
9. Levin TT, Barrett J, Mendelowitz A. Death from clozapine-induced constipation: case report and literature review. Psychosomatics. 2002;43:71-73.
10. Centorrino F, Baldessarini RJ, Frankenburg FR, et al. Serum levels of clozapine and norclozapine in patients treated with selective serotonin reuptake inhibitors. Am J Psychiatry. 1996;153(6):820-822.
11. Sproule BA, Naranjo CA, Brenmer KE, et al. Selective serotonin reuptake inhibitors and CNS drug interactions. A critical review of the evidence. Clin Pharmacokinet. 1997;33(6):454-471.
12. Urichuk L, Prior TI, Dursun S, et al. Metabolism of atypical antipsychotics: involvement of cytochrome p450 enzymes and relevance for drug-drug interactions. Curr Drug Metab. 2008;9(5):410-418.
13. Perry P, Miller DD, Arndt SV, et al. Clozapine and norclozapine plasma concentrations and clinical responses of treatment-refractory schizophrenic patients. Am J Psychiatry. 1991;148:231-235.
14. Freudenreich O. Clozapine drug levels guide dosing. Current Psychiatry. 2009;8(3):78.-
15. Pelizza L, De Luca P, La Pesa M, et al. Clozapine-induced intestinal occlusion: a serious side effect. Acta Biomed. 2007;78:144-148.
16. Hayes G, Gibler B. Clozapine-induced constipation. Am J Psychiatry. 1995;152:298.-
17. American College of Gastroenterology Chronic Constipation Task Force. An evidence-based approach to the management of chronic constipation in North America. Am J Gastroenterol. 2005;100(suppl 1):S1-4.
18. Townsend G, Curtis D. Case report: rapidly fatal bowel ischaemia on clozapine treatment. BMC Psychiatry. 2006;6:43.-
Pregnant and moving involuntarily
CASE: Abnormal movements
Pregnant and unsure of her due date, Ms. A, age 35, presents to the emergency room complaining of hourly uterine contractions for the last 3 days and new onset vaginal bleeding. Ms. A is admitted to the obstetrics (OB) service for preterm labor at 34 and 3/7 weeks as dated by a triage ultrasound.
During initial examination by the OB service, Ms. A’s blood pressure is 155/112 mm Hg with a pulse of 126. Her cervix is dilated to 4 centimeters. Her physical exam is notable for rapid, repetitive, involuntary movements in her upper extremities and to a lesser degree in lower extremities. Ms. A is started on IV fluids and hydralazine, 10 mg/d, for elevated blood pressure. Later that day, she delivers a preterm female weighing 2,360 grams in a spontaneous vaginal delivery without any complications.
After delivery, the OB service requests a psychiatric consultation to evaluate Ms. A’s “blunted affect,” history of heavy alcohol use, and abnormal movements. During examination, Ms. A is alert and oriented to her surroundings. She states that this was her eleventh pregnancy; however, she is unable to recall details of most previous pregnancies. She also cannot remember any significant medical, surgical, or mental health history. Ms. A appears distracted, has difficulty participating in the interview, and gives contradictory histories to different team members. She is well groomed but shows repetitive circular movements of her hands, feet, and jaw that are nearly continuous. In addition, Ms. A has intermittent lip biting and smacking. Her speech is delayed, with increased latency of her responses to basic questions.
Her mood is neutral, her affect is blunted, and she denies any current suicidal or homicidal ideations, delusions, and auditory or visual hallucinations. Although her chart indicates a history of alcohol abuse, she denies this history and current drug or alcohol use. Her Mini-Mental State Exam score is a 22/30, missing points in her ability to copy shapes and write a sentence, complicated by her chorea-like upper body movements. She also demonstrates marked inattentiveness and is unwilling to cooperate with spelling “world.” On physical exam, her gait is wide-based but steady.
The authors’ observations
Determining the cause of Ms. A’s abnormal movements, delayed speech, and neutral mood initially proves difficult because she is minimally cooperative with the interview and we find discrepancies between information she provides and her medical records from previous OB admissions. It is unclear whether these inconsistencies are because of her faltering memory—which she admits has worsened in the last year—or unwillingness to provide a complete medical history.
We consider possible substance intoxication given her documented history of substance use. However, an extended drug screen is negative and her laboratory values do not suggest heavy alcohol use.
HISTORY: Depression and confusion
The next day, Ms. A is more cooperative with the interview. She says that she began feeling depressed 8 years ago, around the time her brother was killed in a violent crime. She denies previous psychiatric hospitalizations, but says she attempted suicide 4 years ago by stabbing herself in the throat with a fork. After that attempt, she was referred to an outpatient psychiatrist whom she continues to see intermittently. She says that her abnormal movements started 2 years before she first saw her outpatient psychiatrist.
She says she has been prescribed several medications, but remembers only taking quetiapine for depressive symptoms and insomnia. After a discussion with her psychiatrist about the possible effects of quetiapine on the fetus, she discontinued the drug approximately 8 weeks into her pregnancy. Quetiapine decreased her movement symptoms slightly, and she feels her movements have become uncontrollable since discontinuing it.
She reports increased feelings of sadness, worthlessness, guilt, decreased energy, irritability, and difficulty sleeping during her pregnancy. She denies current or past psychotic symptoms or mania. Ms. A says she has noticed problems with her memory as well as increased confusion over recent months. She often gets lost and cannot remember where she lives after leaving her home.
Based on hospital records, we learn that an MRI of the brain without contrast was completed 1 year ago to “evaluate choreiform movements.” The scan showed mild atrophy and abnormal signal within the caudate and putamen, as well as volume loss. We consult with the neurology service to evaluate Ms. A’s abnormal movements and her previous abnormal brain imaging. The neurologic exam notes that Ms. A has orofacial dyskinesias and near-continuous choreiform movements in her arms and hands. Her gait remains wide-based and she is unable to tandem walk. Because Ms. A shows no new neurologic symptoms, the neurology service does not feel that additional neuroimaging is indicated.
The authors’ observations
In consultation with neurology, the leading differential diagnoses include tardive dyskinesia, chorea gravidarum, and Huntington’s disease. See the Table1,2 for the differential diagnosis of chorea.
Ms. A reports taking quetiapine for 3 years, which suggests possible tardive dyskinesia. Although second-generation antipsychotics have a lower incidence of movement disorders than first-generation antipsychotics, the risk still exists. Withdrawal dyskinesias can occur after suddenly stopping or tapering antipsychotics and appear as extrapyramidal symptoms, including choreoathetosis similar to what Ms. A experienced.3,4 This type of dyskinesia is thought to be secondary to chronic dopamine antagonism leading to increased postsynaptic receptors and dopamine hypersensitivity.5 Because Ms. A discontinued quetiapine early in her pregnancy, withdrawal dyskinesias are less likely.
Because Ms. A presented with a movement disorder while pregnant, the neurology service considers chorea gravidarum, the term given to chorea occurring during pregnancy. This syndrome is thought to be caused by the effects of pregnancy on the basal ganglia.6 Historically, chorea gravidarum was associated with rheumatic fever (RF); however, with the decline in prevalence of RF, most choreiform movements that appear during pregnancy typically are caused by other diseases, such as systemic lupus erythematosus or Huntington’s disease. Approximately one-half of chorea gravidarum cases are idiopathic, with RF and antiphospholipid syndrome accounting for the remainder.7 Huntington’s disease during pregnancy is rare because it tends to present in women beyond childbearing age.
Based on Ms. A’s symptoms and previous MRI findings, we ask her if she has a known family history of Huntington’s disease. She denies this, but says she has not seen her father since she was very young and is uncertain of his medical history.
Table
Differential diagnosis for chorea
| Genetic | Huntington’s disease, benign hereditary chorea, neuroacanthocytosis, dentatorubral-pallidoluysian atrophy, Wilson’s disease, spinocerebellar ataxia, Friedreich’s ataxia |
| Rheumatic disorders | Sydenham’s chorea, chorea gravidarum |
| Drug-induced/toxicity | Neuroleptic drugs, steroids, anticonvulsants, antiparkinson agents, stimulants (amphetamines, cocaine), lithium, dopamine agonists |
| Systemic disorders | Systemic lupus erythematosus, thyrotoxicosis, polycythemia vera, hyperglycemia, AIDS, paraneoplastic syndrome |
| Vascular/trauma | Cerebral hemorrhage, vasculitis, stroke, antiphospholipid antibody syndrome |
| AIDS: acquired immune deficiency syndrome Source: References 1,2 | |
TREATMENT: Restart medication
Ms. A’s laboratory results show a slightly low hemoglobin of 10.5 g/dL and hematocrit of 32.8%. Her mean corpuscular volume is slightly decreased at 77 fL. Her urinalysis is negative, and blood glucose and thyroid-stimulating hormone are within normal limits. Rapid plasma regain, anti-nuclear antibody, and human immunodeficiency virus (HIV) are negative. Based on hospital records, we learn that during the previous admission a year ago a serum ceruloplasmin and serum copper were drawn and were normal.
We contact Ms. A’s outpatient psychiatrist for collateral information. The psychiatrist says he first evaluated Ms. A 3 years ago after a friend brought her in because of strange behavior, including talking to herself, making odd facial gestures, and laughing inappropriately. Although Ms. A denies past psychiatric hospitalizations, her psychiatrist states that she was hospitalized for 1 week after the suicide attempt 4 years ago and prescribed lorazepam and sertraline during that admission. He speculates that the suicide attempt may have been related to 5 of her children being taken from her by the Department of Family and Child Services after police raided her home to search for drugs. Custody was awarded to their respective fathers, causing Ms. A to “snap,” according to her friend.
Since then, neither Ms. A nor her psychiatrist have reported any further psychotic symptoms. Her psychiatrist confirms that Ms. A’s abnormal movements were present before her first appointment with him. He says that he referred Ms. A to a local hospital for a neurology work-up, but she did not schedule an appointment.
When we follow up with Ms. A 2 days after delivery, she continues to deny depressive symptoms, although her affect remains blunted. She says she is looking forward to going home with the baby, whom she plans to bottle feed. Her choreiform movements appear unchanged. She also continues to experience lip smacking. Although Ms. A recognizes that she has some movements, she minimizes them and says they do not bother her. She continues to demonstrate latency in her verbal responses to questions. Based on the collateral history and positive response with quetiapine, we recommend that Ms. A be restarted on quetiapine, 200 mg/d.
The authors’ observations
Ms. A’s choreiform movements started before her psychotic symptoms and subsequent usage of neuroleptic medication, which makes tardive dyskinesia less likely. Laboratory studies rule out systemic lupus erythematosus, HIV, and Wilson’s disease as the cause of her abnormal movements.
Ms. A’s history is highly suggestive of Huntington’s disease. She exhibits classic motor signs, including involuntary choreiform movements in her extremities. She also has psychiatric symptoms that are commonly associated with Huntington’s disease, including depression—which preceded her motor symptoms—cognitive decline, apathy, and psychotic symptoms. In addition, her MRI findings of volume changes in the caudate nucleus and the putamen and inability to rule out a family history make Huntington’s disease more likely (Box).1,8-11
Huntington’s disease is an autosomal dominant disorder characterized by progressive motor, cognitive, and psychiatric disturbances and is the most common genetic cause of chorea. The underlying genetic mutation is a CAG repeat expansion in the Huntington’s disease gene. A Huntington’s disease diagnosis generally is considered in the presence of the characteristic choreiform movements and slowly progressive cognitive decline.8 Physical symptoms can present at any age, although they usually begin between age 35 and 44. In early stages of the disease, patients may experience subtle changes in personality, cognition, and physical skills. Although most Huntington’s disease patients eventually exhibit similar physical symptoms, the onset, progression, and extent of cognitive and psychiatric symptoms vary among individuals. However, psychiatric symptoms frequently are present during the early stages of the disease, often before motor symptoms begin and can include personality changes, irritability, agitation, apathy, and depression. In addition, up to 23% of patients with Huntington’s disease develop psychotic symptoms.1,9 There is no cure for Huntington’s disease, and mean disease duration is 17 to 20 years. The most common cause of death among Huntington’s disease patients is pneumonia, followed by suicide.1
A Huntington’s disease diagnosis is based on clinical symptoms and signs in an individual who has a parent with proven Huntington’s disease and is confirmed by DNA tests.1 Typical neuroanatomic findings include initial neuronal loss in the striatum followed by a diffuse involvement of cortical and subcortical areas.10 Volume changes in the caudate nucleus and the putamen may be a reliable measure of Huntington’s disease and potentially serve as a biomarker.11
Psychiatric symptoms
Psychiatric symptoms frequently are evident in the early stages of Huntington’s disease, often before onset of motor symptoms.1 Depression is the most common sign, and can be difficult to diagnose because weight loss, apathy, and inactivity also occur in Huntington’s disease. Feelings of low self-esteem, guilt, and anxiety can help distinguish depression from symptoms of Huntington’s disease. Cognitive decline also may present before the first motor symptoms occur. Cognitive changes typically are related to executive functions and affected individuals may develop impairments in organization and planning. Psychotic symptoms may be present, but are more common in later stages of the disease.1
Ms. A reported that quetiapine seemed to lessen her choreiform movements, and dopamine receptor blocking agents (ie, antipsychotics) often are considered for managing chorea and psychosis in Huntington’s disease. However, there are few double-blind, placebo-controlled studies evaluating the efficacy of these agents.12 Small, uncontrolled, nonrandomized trials found quetiapine has some efficacy for both motor and psychiatric symptoms in Huntington’s disease.12-15
OUTCOME: Lost to follow-up
Ms. A is discharged from the hospital 3 days after she delivers her daughter and is given an appointment in 6 weeks at an affiliated movement disorders clinic. Before discharge, she is tested for the Huntington’s disease gene mutation with a plan to receive her results during her follow-up visit. During the informed consent process for the genetic testing, Ms. A states that she was tested previously and was quite sure that the test was positive for Huntington’s disease, although she could not recall where or when this testing was completed.
Ms. A also is scheduled to follow up with her obstetrician for a 6-week postpartum check-up and tubal ligation. We encourage Ms. A to make an appointment with her psychiatrist soon after discharge. We also make a referral to the Department of Family and Children Services to provide adequate support and resources to her and her children because of her physical and psychiatric issues.
Ms. A does not show up for her follow-up appointment at the movement disorders clinic. The genetic test is not completed during this admission because of a clerical error, and the serum sample subsequently expires.
The authors’ observations
Although Huntington’s disease is the most likely cause of Ms. A’s presentation, we were unable to confirm the diagnosis with genetic testing. If Ms. A returns to the neurology service and the genetic test is negative for Huntington’s disease, other causes of chorea must be investigated.
Related Resources
- De Marchi N, Mennella R. Huntington’s disease and its association with psychopathology. Harv Rev Psychiatry. 2000; 7(5):278-289.
- Revilla FJ, Grutzendler J, Larsh TR. Huntington disease. Medscape. http://emedicine.medscape.com/article/1150165-overview.
Drug Brand Names
- Hydralazine • Apresoline
- Lithium • Eskalith, Lithobid, others
- Lorazepam • Ativan
- Quetiapine • Seroquel
- Sertraline • Zoloft
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Roos RA. Huntington’s disease: a clinical review. Orphanet J Rare Dis. 2010;5(1):40.-
2. Wild EJ, Tabrizi SJ. The differential diagnosis of chorea. Pract Neurol. 2007;7:360-373.
3. Urbano M, Spiegel D, Rai A. Atypical antipsychotic withdrawal dyskinesia in 4 patients with mood disorders. J Clin Psychopharmacol. 2007;27(6):705-707.
4. Kafantaris V, Hirsch J, Saito E, et al. Treatment of withdrawal dyskinesia. J Am Acad Child Adolesc Psychiatry. 2005;44(11):1102-1103.
5. Creese I, Burt DR, Snyder SH. Dopamine receptor binding enhancement accompanies lesion-induced behavioral supersensitivity. Science. 1977;197(4303):596-598.
6. Kranick SM, Mowry EM, Colcher A, et al. Movement disorders and pregnancy: a review of the literature. Mov Disord. 2010;25(6):665-671.
7. Ramachandran TS. Chorea gravidarum. Medscape. Available at: http://emedicine.medscape.com/article/1149725-overview. Accessed May 4 2011.
8. Panegyres PK, Goh JG. The neurology and natural history of patients with indeterminate CAG repeat length mutations of the Huntington disease gene. J Neurol Sci. 2011;301(1-2):14-20.
9. Shiwach R. Psychopathology in Huntington’s disease patients. Acta Psychiatr Scand. 1994;90:241-246.
10. De Marchi N, Mennella R. Huntington’s disease and its association with psychopathology. Harv Rev Psychiatry. 2000;7:278-289.
11. van den Bogaard SJ, Dumas EM, Acharya TP, et al. and the TRACK-HD Investigator Group. Early atrophy of pallidum and accumbens nucleus in Huntington’s disease. J Neurol. 2011;258(3):412-420.
12. Frank S, Jankovic J. Advances in the pharmacological management of Huntington’s disease. Drugs. 2010;70(5):561-571.
13. Alpay M, Koroshetz WJ. Quetiapine in the treatment of behavioral disturbances in patients with Huntington’s disease. Psychosomatics. 2006;47(1):70-72.
14. Seitz DP, Millson RC. Quetiapine in the management of psychosis secondary to Huntington’s disease: a case report. Can J Psychiatry. 2004;49(6):413.-
15. Bonelli RM, Niederwieser G. Quetiapine in Huntington’s disease: a first case report. J Neurol. 2002;249(8):1114-1115.
CASE: Abnormal movements
Pregnant and unsure of her due date, Ms. A, age 35, presents to the emergency room complaining of hourly uterine contractions for the last 3 days and new onset vaginal bleeding. Ms. A is admitted to the obstetrics (OB) service for preterm labor at 34 and 3/7 weeks as dated by a triage ultrasound.
During initial examination by the OB service, Ms. A’s blood pressure is 155/112 mm Hg with a pulse of 126. Her cervix is dilated to 4 centimeters. Her physical exam is notable for rapid, repetitive, involuntary movements in her upper extremities and to a lesser degree in lower extremities. Ms. A is started on IV fluids and hydralazine, 10 mg/d, for elevated blood pressure. Later that day, she delivers a preterm female weighing 2,360 grams in a spontaneous vaginal delivery without any complications.
After delivery, the OB service requests a psychiatric consultation to evaluate Ms. A’s “blunted affect,” history of heavy alcohol use, and abnormal movements. During examination, Ms. A is alert and oriented to her surroundings. She states that this was her eleventh pregnancy; however, she is unable to recall details of most previous pregnancies. She also cannot remember any significant medical, surgical, or mental health history. Ms. A appears distracted, has difficulty participating in the interview, and gives contradictory histories to different team members. She is well groomed but shows repetitive circular movements of her hands, feet, and jaw that are nearly continuous. In addition, Ms. A has intermittent lip biting and smacking. Her speech is delayed, with increased latency of her responses to basic questions.
Her mood is neutral, her affect is blunted, and she denies any current suicidal or homicidal ideations, delusions, and auditory or visual hallucinations. Although her chart indicates a history of alcohol abuse, she denies this history and current drug or alcohol use. Her Mini-Mental State Exam score is a 22/30, missing points in her ability to copy shapes and write a sentence, complicated by her chorea-like upper body movements. She also demonstrates marked inattentiveness and is unwilling to cooperate with spelling “world.” On physical exam, her gait is wide-based but steady.
The authors’ observations
Determining the cause of Ms. A’s abnormal movements, delayed speech, and neutral mood initially proves difficult because she is minimally cooperative with the interview and we find discrepancies between information she provides and her medical records from previous OB admissions. It is unclear whether these inconsistencies are because of her faltering memory—which she admits has worsened in the last year—or unwillingness to provide a complete medical history.
We consider possible substance intoxication given her documented history of substance use. However, an extended drug screen is negative and her laboratory values do not suggest heavy alcohol use.
HISTORY: Depression and confusion
The next day, Ms. A is more cooperative with the interview. She says that she began feeling depressed 8 years ago, around the time her brother was killed in a violent crime. She denies previous psychiatric hospitalizations, but says she attempted suicide 4 years ago by stabbing herself in the throat with a fork. After that attempt, she was referred to an outpatient psychiatrist whom she continues to see intermittently. She says that her abnormal movements started 2 years before she first saw her outpatient psychiatrist.
She says she has been prescribed several medications, but remembers only taking quetiapine for depressive symptoms and insomnia. After a discussion with her psychiatrist about the possible effects of quetiapine on the fetus, she discontinued the drug approximately 8 weeks into her pregnancy. Quetiapine decreased her movement symptoms slightly, and she feels her movements have become uncontrollable since discontinuing it.
She reports increased feelings of sadness, worthlessness, guilt, decreased energy, irritability, and difficulty sleeping during her pregnancy. She denies current or past psychotic symptoms or mania. Ms. A says she has noticed problems with her memory as well as increased confusion over recent months. She often gets lost and cannot remember where she lives after leaving her home.
Based on hospital records, we learn that an MRI of the brain without contrast was completed 1 year ago to “evaluate choreiform movements.” The scan showed mild atrophy and abnormal signal within the caudate and putamen, as well as volume loss. We consult with the neurology service to evaluate Ms. A’s abnormal movements and her previous abnormal brain imaging. The neurologic exam notes that Ms. A has orofacial dyskinesias and near-continuous choreiform movements in her arms and hands. Her gait remains wide-based and she is unable to tandem walk. Because Ms. A shows no new neurologic symptoms, the neurology service does not feel that additional neuroimaging is indicated.
The authors’ observations
In consultation with neurology, the leading differential diagnoses include tardive dyskinesia, chorea gravidarum, and Huntington’s disease. See the Table1,2 for the differential diagnosis of chorea.
Ms. A reports taking quetiapine for 3 years, which suggests possible tardive dyskinesia. Although second-generation antipsychotics have a lower incidence of movement disorders than first-generation antipsychotics, the risk still exists. Withdrawal dyskinesias can occur after suddenly stopping or tapering antipsychotics and appear as extrapyramidal symptoms, including choreoathetosis similar to what Ms. A experienced.3,4 This type of dyskinesia is thought to be secondary to chronic dopamine antagonism leading to increased postsynaptic receptors and dopamine hypersensitivity.5 Because Ms. A discontinued quetiapine early in her pregnancy, withdrawal dyskinesias are less likely.
Because Ms. A presented with a movement disorder while pregnant, the neurology service considers chorea gravidarum, the term given to chorea occurring during pregnancy. This syndrome is thought to be caused by the effects of pregnancy on the basal ganglia.6 Historically, chorea gravidarum was associated with rheumatic fever (RF); however, with the decline in prevalence of RF, most choreiform movements that appear during pregnancy typically are caused by other diseases, such as systemic lupus erythematosus or Huntington’s disease. Approximately one-half of chorea gravidarum cases are idiopathic, with RF and antiphospholipid syndrome accounting for the remainder.7 Huntington’s disease during pregnancy is rare because it tends to present in women beyond childbearing age.
Based on Ms. A’s symptoms and previous MRI findings, we ask her if she has a known family history of Huntington’s disease. She denies this, but says she has not seen her father since she was very young and is uncertain of his medical history.
Table
Differential diagnosis for chorea
| Genetic | Huntington’s disease, benign hereditary chorea, neuroacanthocytosis, dentatorubral-pallidoluysian atrophy, Wilson’s disease, spinocerebellar ataxia, Friedreich’s ataxia |
| Rheumatic disorders | Sydenham’s chorea, chorea gravidarum |
| Drug-induced/toxicity | Neuroleptic drugs, steroids, anticonvulsants, antiparkinson agents, stimulants (amphetamines, cocaine), lithium, dopamine agonists |
| Systemic disorders | Systemic lupus erythematosus, thyrotoxicosis, polycythemia vera, hyperglycemia, AIDS, paraneoplastic syndrome |
| Vascular/trauma | Cerebral hemorrhage, vasculitis, stroke, antiphospholipid antibody syndrome |
| AIDS: acquired immune deficiency syndrome Source: References 1,2 | |
TREATMENT: Restart medication
Ms. A’s laboratory results show a slightly low hemoglobin of 10.5 g/dL and hematocrit of 32.8%. Her mean corpuscular volume is slightly decreased at 77 fL. Her urinalysis is negative, and blood glucose and thyroid-stimulating hormone are within normal limits. Rapid plasma regain, anti-nuclear antibody, and human immunodeficiency virus (HIV) are negative. Based on hospital records, we learn that during the previous admission a year ago a serum ceruloplasmin and serum copper were drawn and were normal.
We contact Ms. A’s outpatient psychiatrist for collateral information. The psychiatrist says he first evaluated Ms. A 3 years ago after a friend brought her in because of strange behavior, including talking to herself, making odd facial gestures, and laughing inappropriately. Although Ms. A denies past psychiatric hospitalizations, her psychiatrist states that she was hospitalized for 1 week after the suicide attempt 4 years ago and prescribed lorazepam and sertraline during that admission. He speculates that the suicide attempt may have been related to 5 of her children being taken from her by the Department of Family and Child Services after police raided her home to search for drugs. Custody was awarded to their respective fathers, causing Ms. A to “snap,” according to her friend.
Since then, neither Ms. A nor her psychiatrist have reported any further psychotic symptoms. Her psychiatrist confirms that Ms. A’s abnormal movements were present before her first appointment with him. He says that he referred Ms. A to a local hospital for a neurology work-up, but she did not schedule an appointment.
When we follow up with Ms. A 2 days after delivery, she continues to deny depressive symptoms, although her affect remains blunted. She says she is looking forward to going home with the baby, whom she plans to bottle feed. Her choreiform movements appear unchanged. She also continues to experience lip smacking. Although Ms. A recognizes that she has some movements, she minimizes them and says they do not bother her. She continues to demonstrate latency in her verbal responses to questions. Based on the collateral history and positive response with quetiapine, we recommend that Ms. A be restarted on quetiapine, 200 mg/d.
The authors’ observations
Ms. A’s choreiform movements started before her psychotic symptoms and subsequent usage of neuroleptic medication, which makes tardive dyskinesia less likely. Laboratory studies rule out systemic lupus erythematosus, HIV, and Wilson’s disease as the cause of her abnormal movements.
Ms. A’s history is highly suggestive of Huntington’s disease. She exhibits classic motor signs, including involuntary choreiform movements in her extremities. She also has psychiatric symptoms that are commonly associated with Huntington’s disease, including depression—which preceded her motor symptoms—cognitive decline, apathy, and psychotic symptoms. In addition, her MRI findings of volume changes in the caudate nucleus and the putamen and inability to rule out a family history make Huntington’s disease more likely (Box).1,8-11
Huntington’s disease is an autosomal dominant disorder characterized by progressive motor, cognitive, and psychiatric disturbances and is the most common genetic cause of chorea. The underlying genetic mutation is a CAG repeat expansion in the Huntington’s disease gene. A Huntington’s disease diagnosis generally is considered in the presence of the characteristic choreiform movements and slowly progressive cognitive decline.8 Physical symptoms can present at any age, although they usually begin between age 35 and 44. In early stages of the disease, patients may experience subtle changes in personality, cognition, and physical skills. Although most Huntington’s disease patients eventually exhibit similar physical symptoms, the onset, progression, and extent of cognitive and psychiatric symptoms vary among individuals. However, psychiatric symptoms frequently are present during the early stages of the disease, often before motor symptoms begin and can include personality changes, irritability, agitation, apathy, and depression. In addition, up to 23% of patients with Huntington’s disease develop psychotic symptoms.1,9 There is no cure for Huntington’s disease, and mean disease duration is 17 to 20 years. The most common cause of death among Huntington’s disease patients is pneumonia, followed by suicide.1
A Huntington’s disease diagnosis is based on clinical symptoms and signs in an individual who has a parent with proven Huntington’s disease and is confirmed by DNA tests.1 Typical neuroanatomic findings include initial neuronal loss in the striatum followed by a diffuse involvement of cortical and subcortical areas.10 Volume changes in the caudate nucleus and the putamen may be a reliable measure of Huntington’s disease and potentially serve as a biomarker.11
Psychiatric symptoms
Psychiatric symptoms frequently are evident in the early stages of Huntington’s disease, often before onset of motor symptoms.1 Depression is the most common sign, and can be difficult to diagnose because weight loss, apathy, and inactivity also occur in Huntington’s disease. Feelings of low self-esteem, guilt, and anxiety can help distinguish depression from symptoms of Huntington’s disease. Cognitive decline also may present before the first motor symptoms occur. Cognitive changes typically are related to executive functions and affected individuals may develop impairments in organization and planning. Psychotic symptoms may be present, but are more common in later stages of the disease.1
Ms. A reported that quetiapine seemed to lessen her choreiform movements, and dopamine receptor blocking agents (ie, antipsychotics) often are considered for managing chorea and psychosis in Huntington’s disease. However, there are few double-blind, placebo-controlled studies evaluating the efficacy of these agents.12 Small, uncontrolled, nonrandomized trials found quetiapine has some efficacy for both motor and psychiatric symptoms in Huntington’s disease.12-15
OUTCOME: Lost to follow-up
Ms. A is discharged from the hospital 3 days after she delivers her daughter and is given an appointment in 6 weeks at an affiliated movement disorders clinic. Before discharge, she is tested for the Huntington’s disease gene mutation with a plan to receive her results during her follow-up visit. During the informed consent process for the genetic testing, Ms. A states that she was tested previously and was quite sure that the test was positive for Huntington’s disease, although she could not recall where or when this testing was completed.
Ms. A also is scheduled to follow up with her obstetrician for a 6-week postpartum check-up and tubal ligation. We encourage Ms. A to make an appointment with her psychiatrist soon after discharge. We also make a referral to the Department of Family and Children Services to provide adequate support and resources to her and her children because of her physical and psychiatric issues.
Ms. A does not show up for her follow-up appointment at the movement disorders clinic. The genetic test is not completed during this admission because of a clerical error, and the serum sample subsequently expires.
The authors’ observations
Although Huntington’s disease is the most likely cause of Ms. A’s presentation, we were unable to confirm the diagnosis with genetic testing. If Ms. A returns to the neurology service and the genetic test is negative for Huntington’s disease, other causes of chorea must be investigated.
Related Resources
- De Marchi N, Mennella R. Huntington’s disease and its association with psychopathology. Harv Rev Psychiatry. 2000; 7(5):278-289.
- Revilla FJ, Grutzendler J, Larsh TR. Huntington disease. Medscape. http://emedicine.medscape.com/article/1150165-overview.
Drug Brand Names
- Hydralazine • Apresoline
- Lithium • Eskalith, Lithobid, others
- Lorazepam • Ativan
- Quetiapine • Seroquel
- Sertraline • Zoloft
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
CASE: Abnormal movements
Pregnant and unsure of her due date, Ms. A, age 35, presents to the emergency room complaining of hourly uterine contractions for the last 3 days and new onset vaginal bleeding. Ms. A is admitted to the obstetrics (OB) service for preterm labor at 34 and 3/7 weeks as dated by a triage ultrasound.
During initial examination by the OB service, Ms. A’s blood pressure is 155/112 mm Hg with a pulse of 126. Her cervix is dilated to 4 centimeters. Her physical exam is notable for rapid, repetitive, involuntary movements in her upper extremities and to a lesser degree in lower extremities. Ms. A is started on IV fluids and hydralazine, 10 mg/d, for elevated blood pressure. Later that day, she delivers a preterm female weighing 2,360 grams in a spontaneous vaginal delivery without any complications.
After delivery, the OB service requests a psychiatric consultation to evaluate Ms. A’s “blunted affect,” history of heavy alcohol use, and abnormal movements. During examination, Ms. A is alert and oriented to her surroundings. She states that this was her eleventh pregnancy; however, she is unable to recall details of most previous pregnancies. She also cannot remember any significant medical, surgical, or mental health history. Ms. A appears distracted, has difficulty participating in the interview, and gives contradictory histories to different team members. She is well groomed but shows repetitive circular movements of her hands, feet, and jaw that are nearly continuous. In addition, Ms. A has intermittent lip biting and smacking. Her speech is delayed, with increased latency of her responses to basic questions.
Her mood is neutral, her affect is blunted, and she denies any current suicidal or homicidal ideations, delusions, and auditory or visual hallucinations. Although her chart indicates a history of alcohol abuse, she denies this history and current drug or alcohol use. Her Mini-Mental State Exam score is a 22/30, missing points in her ability to copy shapes and write a sentence, complicated by her chorea-like upper body movements. She also demonstrates marked inattentiveness and is unwilling to cooperate with spelling “world.” On physical exam, her gait is wide-based but steady.
The authors’ observations
Determining the cause of Ms. A’s abnormal movements, delayed speech, and neutral mood initially proves difficult because she is minimally cooperative with the interview and we find discrepancies between information she provides and her medical records from previous OB admissions. It is unclear whether these inconsistencies are because of her faltering memory—which she admits has worsened in the last year—or unwillingness to provide a complete medical history.
We consider possible substance intoxication given her documented history of substance use. However, an extended drug screen is negative and her laboratory values do not suggest heavy alcohol use.
HISTORY: Depression and confusion
The next day, Ms. A is more cooperative with the interview. She says that she began feeling depressed 8 years ago, around the time her brother was killed in a violent crime. She denies previous psychiatric hospitalizations, but says she attempted suicide 4 years ago by stabbing herself in the throat with a fork. After that attempt, she was referred to an outpatient psychiatrist whom she continues to see intermittently. She says that her abnormal movements started 2 years before she first saw her outpatient psychiatrist.
She says she has been prescribed several medications, but remembers only taking quetiapine for depressive symptoms and insomnia. After a discussion with her psychiatrist about the possible effects of quetiapine on the fetus, she discontinued the drug approximately 8 weeks into her pregnancy. Quetiapine decreased her movement symptoms slightly, and she feels her movements have become uncontrollable since discontinuing it.
She reports increased feelings of sadness, worthlessness, guilt, decreased energy, irritability, and difficulty sleeping during her pregnancy. She denies current or past psychotic symptoms or mania. Ms. A says she has noticed problems with her memory as well as increased confusion over recent months. She often gets lost and cannot remember where she lives after leaving her home.
Based on hospital records, we learn that an MRI of the brain without contrast was completed 1 year ago to “evaluate choreiform movements.” The scan showed mild atrophy and abnormal signal within the caudate and putamen, as well as volume loss. We consult with the neurology service to evaluate Ms. A’s abnormal movements and her previous abnormal brain imaging. The neurologic exam notes that Ms. A has orofacial dyskinesias and near-continuous choreiform movements in her arms and hands. Her gait remains wide-based and she is unable to tandem walk. Because Ms. A shows no new neurologic symptoms, the neurology service does not feel that additional neuroimaging is indicated.
The authors’ observations
In consultation with neurology, the leading differential diagnoses include tardive dyskinesia, chorea gravidarum, and Huntington’s disease. See the Table1,2 for the differential diagnosis of chorea.
Ms. A reports taking quetiapine for 3 years, which suggests possible tardive dyskinesia. Although second-generation antipsychotics have a lower incidence of movement disorders than first-generation antipsychotics, the risk still exists. Withdrawal dyskinesias can occur after suddenly stopping or tapering antipsychotics and appear as extrapyramidal symptoms, including choreoathetosis similar to what Ms. A experienced.3,4 This type of dyskinesia is thought to be secondary to chronic dopamine antagonism leading to increased postsynaptic receptors and dopamine hypersensitivity.5 Because Ms. A discontinued quetiapine early in her pregnancy, withdrawal dyskinesias are less likely.
Because Ms. A presented with a movement disorder while pregnant, the neurology service considers chorea gravidarum, the term given to chorea occurring during pregnancy. This syndrome is thought to be caused by the effects of pregnancy on the basal ganglia.6 Historically, chorea gravidarum was associated with rheumatic fever (RF); however, with the decline in prevalence of RF, most choreiform movements that appear during pregnancy typically are caused by other diseases, such as systemic lupus erythematosus or Huntington’s disease. Approximately one-half of chorea gravidarum cases are idiopathic, with RF and antiphospholipid syndrome accounting for the remainder.7 Huntington’s disease during pregnancy is rare because it tends to present in women beyond childbearing age.
Based on Ms. A’s symptoms and previous MRI findings, we ask her if she has a known family history of Huntington’s disease. She denies this, but says she has not seen her father since she was very young and is uncertain of his medical history.
Table
Differential diagnosis for chorea
| Genetic | Huntington’s disease, benign hereditary chorea, neuroacanthocytosis, dentatorubral-pallidoluysian atrophy, Wilson’s disease, spinocerebellar ataxia, Friedreich’s ataxia |
| Rheumatic disorders | Sydenham’s chorea, chorea gravidarum |
| Drug-induced/toxicity | Neuroleptic drugs, steroids, anticonvulsants, antiparkinson agents, stimulants (amphetamines, cocaine), lithium, dopamine agonists |
| Systemic disorders | Systemic lupus erythematosus, thyrotoxicosis, polycythemia vera, hyperglycemia, AIDS, paraneoplastic syndrome |
| Vascular/trauma | Cerebral hemorrhage, vasculitis, stroke, antiphospholipid antibody syndrome |
| AIDS: acquired immune deficiency syndrome Source: References 1,2 | |
TREATMENT: Restart medication
Ms. A’s laboratory results show a slightly low hemoglobin of 10.5 g/dL and hematocrit of 32.8%. Her mean corpuscular volume is slightly decreased at 77 fL. Her urinalysis is negative, and blood glucose and thyroid-stimulating hormone are within normal limits. Rapid plasma regain, anti-nuclear antibody, and human immunodeficiency virus (HIV) are negative. Based on hospital records, we learn that during the previous admission a year ago a serum ceruloplasmin and serum copper were drawn and were normal.
We contact Ms. A’s outpatient psychiatrist for collateral information. The psychiatrist says he first evaluated Ms. A 3 years ago after a friend brought her in because of strange behavior, including talking to herself, making odd facial gestures, and laughing inappropriately. Although Ms. A denies past psychiatric hospitalizations, her psychiatrist states that she was hospitalized for 1 week after the suicide attempt 4 years ago and prescribed lorazepam and sertraline during that admission. He speculates that the suicide attempt may have been related to 5 of her children being taken from her by the Department of Family and Child Services after police raided her home to search for drugs. Custody was awarded to their respective fathers, causing Ms. A to “snap,” according to her friend.
Since then, neither Ms. A nor her psychiatrist have reported any further psychotic symptoms. Her psychiatrist confirms that Ms. A’s abnormal movements were present before her first appointment with him. He says that he referred Ms. A to a local hospital for a neurology work-up, but she did not schedule an appointment.
When we follow up with Ms. A 2 days after delivery, she continues to deny depressive symptoms, although her affect remains blunted. She says she is looking forward to going home with the baby, whom she plans to bottle feed. Her choreiform movements appear unchanged. She also continues to experience lip smacking. Although Ms. A recognizes that she has some movements, she minimizes them and says they do not bother her. She continues to demonstrate latency in her verbal responses to questions. Based on the collateral history and positive response with quetiapine, we recommend that Ms. A be restarted on quetiapine, 200 mg/d.
The authors’ observations
Ms. A’s choreiform movements started before her psychotic symptoms and subsequent usage of neuroleptic medication, which makes tardive dyskinesia less likely. Laboratory studies rule out systemic lupus erythematosus, HIV, and Wilson’s disease as the cause of her abnormal movements.
Ms. A’s history is highly suggestive of Huntington’s disease. She exhibits classic motor signs, including involuntary choreiform movements in her extremities. She also has psychiatric symptoms that are commonly associated with Huntington’s disease, including depression—which preceded her motor symptoms—cognitive decline, apathy, and psychotic symptoms. In addition, her MRI findings of volume changes in the caudate nucleus and the putamen and inability to rule out a family history make Huntington’s disease more likely (Box).1,8-11
Huntington’s disease is an autosomal dominant disorder characterized by progressive motor, cognitive, and psychiatric disturbances and is the most common genetic cause of chorea. The underlying genetic mutation is a CAG repeat expansion in the Huntington’s disease gene. A Huntington’s disease diagnosis generally is considered in the presence of the characteristic choreiform movements and slowly progressive cognitive decline.8 Physical symptoms can present at any age, although they usually begin between age 35 and 44. In early stages of the disease, patients may experience subtle changes in personality, cognition, and physical skills. Although most Huntington’s disease patients eventually exhibit similar physical symptoms, the onset, progression, and extent of cognitive and psychiatric symptoms vary among individuals. However, psychiatric symptoms frequently are present during the early stages of the disease, often before motor symptoms begin and can include personality changes, irritability, agitation, apathy, and depression. In addition, up to 23% of patients with Huntington’s disease develop psychotic symptoms.1,9 There is no cure for Huntington’s disease, and mean disease duration is 17 to 20 years. The most common cause of death among Huntington’s disease patients is pneumonia, followed by suicide.1
A Huntington’s disease diagnosis is based on clinical symptoms and signs in an individual who has a parent with proven Huntington’s disease and is confirmed by DNA tests.1 Typical neuroanatomic findings include initial neuronal loss in the striatum followed by a diffuse involvement of cortical and subcortical areas.10 Volume changes in the caudate nucleus and the putamen may be a reliable measure of Huntington’s disease and potentially serve as a biomarker.11
Psychiatric symptoms
Psychiatric symptoms frequently are evident in the early stages of Huntington’s disease, often before onset of motor symptoms.1 Depression is the most common sign, and can be difficult to diagnose because weight loss, apathy, and inactivity also occur in Huntington’s disease. Feelings of low self-esteem, guilt, and anxiety can help distinguish depression from symptoms of Huntington’s disease. Cognitive decline also may present before the first motor symptoms occur. Cognitive changes typically are related to executive functions and affected individuals may develop impairments in organization and planning. Psychotic symptoms may be present, but are more common in later stages of the disease.1
Ms. A reported that quetiapine seemed to lessen her choreiform movements, and dopamine receptor blocking agents (ie, antipsychotics) often are considered for managing chorea and psychosis in Huntington’s disease. However, there are few double-blind, placebo-controlled studies evaluating the efficacy of these agents.12 Small, uncontrolled, nonrandomized trials found quetiapine has some efficacy for both motor and psychiatric symptoms in Huntington’s disease.12-15
OUTCOME: Lost to follow-up
Ms. A is discharged from the hospital 3 days after she delivers her daughter and is given an appointment in 6 weeks at an affiliated movement disorders clinic. Before discharge, she is tested for the Huntington’s disease gene mutation with a plan to receive her results during her follow-up visit. During the informed consent process for the genetic testing, Ms. A states that she was tested previously and was quite sure that the test was positive for Huntington’s disease, although she could not recall where or when this testing was completed.
Ms. A also is scheduled to follow up with her obstetrician for a 6-week postpartum check-up and tubal ligation. We encourage Ms. A to make an appointment with her psychiatrist soon after discharge. We also make a referral to the Department of Family and Children Services to provide adequate support and resources to her and her children because of her physical and psychiatric issues.
Ms. A does not show up for her follow-up appointment at the movement disorders clinic. The genetic test is not completed during this admission because of a clerical error, and the serum sample subsequently expires.
The authors’ observations
Although Huntington’s disease is the most likely cause of Ms. A’s presentation, we were unable to confirm the diagnosis with genetic testing. If Ms. A returns to the neurology service and the genetic test is negative for Huntington’s disease, other causes of chorea must be investigated.
Related Resources
- De Marchi N, Mennella R. Huntington’s disease and its association with psychopathology. Harv Rev Psychiatry. 2000; 7(5):278-289.
- Revilla FJ, Grutzendler J, Larsh TR. Huntington disease. Medscape. http://emedicine.medscape.com/article/1150165-overview.
Drug Brand Names
- Hydralazine • Apresoline
- Lithium • Eskalith, Lithobid, others
- Lorazepam • Ativan
- Quetiapine • Seroquel
- Sertraline • Zoloft
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Roos RA. Huntington’s disease: a clinical review. Orphanet J Rare Dis. 2010;5(1):40.-
2. Wild EJ, Tabrizi SJ. The differential diagnosis of chorea. Pract Neurol. 2007;7:360-373.
3. Urbano M, Spiegel D, Rai A. Atypical antipsychotic withdrawal dyskinesia in 4 patients with mood disorders. J Clin Psychopharmacol. 2007;27(6):705-707.
4. Kafantaris V, Hirsch J, Saito E, et al. Treatment of withdrawal dyskinesia. J Am Acad Child Adolesc Psychiatry. 2005;44(11):1102-1103.
5. Creese I, Burt DR, Snyder SH. Dopamine receptor binding enhancement accompanies lesion-induced behavioral supersensitivity. Science. 1977;197(4303):596-598.
6. Kranick SM, Mowry EM, Colcher A, et al. Movement disorders and pregnancy: a review of the literature. Mov Disord. 2010;25(6):665-671.
7. Ramachandran TS. Chorea gravidarum. Medscape. Available at: http://emedicine.medscape.com/article/1149725-overview. Accessed May 4 2011.
8. Panegyres PK, Goh JG. The neurology and natural history of patients with indeterminate CAG repeat length mutations of the Huntington disease gene. J Neurol Sci. 2011;301(1-2):14-20.
9. Shiwach R. Psychopathology in Huntington’s disease patients. Acta Psychiatr Scand. 1994;90:241-246.
10. De Marchi N, Mennella R. Huntington’s disease and its association with psychopathology. Harv Rev Psychiatry. 2000;7:278-289.
11. van den Bogaard SJ, Dumas EM, Acharya TP, et al. and the TRACK-HD Investigator Group. Early atrophy of pallidum and accumbens nucleus in Huntington’s disease. J Neurol. 2011;258(3):412-420.
12. Frank S, Jankovic J. Advances in the pharmacological management of Huntington’s disease. Drugs. 2010;70(5):561-571.
13. Alpay M, Koroshetz WJ. Quetiapine in the treatment of behavioral disturbances in patients with Huntington’s disease. Psychosomatics. 2006;47(1):70-72.
14. Seitz DP, Millson RC. Quetiapine in the management of psychosis secondary to Huntington’s disease: a case report. Can J Psychiatry. 2004;49(6):413.-
15. Bonelli RM, Niederwieser G. Quetiapine in Huntington’s disease: a first case report. J Neurol. 2002;249(8):1114-1115.
1. Roos RA. Huntington’s disease: a clinical review. Orphanet J Rare Dis. 2010;5(1):40.-
2. Wild EJ, Tabrizi SJ. The differential diagnosis of chorea. Pract Neurol. 2007;7:360-373.
3. Urbano M, Spiegel D, Rai A. Atypical antipsychotic withdrawal dyskinesia in 4 patients with mood disorders. J Clin Psychopharmacol. 2007;27(6):705-707.
4. Kafantaris V, Hirsch J, Saito E, et al. Treatment of withdrawal dyskinesia. J Am Acad Child Adolesc Psychiatry. 2005;44(11):1102-1103.
5. Creese I, Burt DR, Snyder SH. Dopamine receptor binding enhancement accompanies lesion-induced behavioral supersensitivity. Science. 1977;197(4303):596-598.
6. Kranick SM, Mowry EM, Colcher A, et al. Movement disorders and pregnancy: a review of the literature. Mov Disord. 2010;25(6):665-671.
7. Ramachandran TS. Chorea gravidarum. Medscape. Available at: http://emedicine.medscape.com/article/1149725-overview. Accessed May 4 2011.
8. Panegyres PK, Goh JG. The neurology and natural history of patients with indeterminate CAG repeat length mutations of the Huntington disease gene. J Neurol Sci. 2011;301(1-2):14-20.
9. Shiwach R. Psychopathology in Huntington’s disease patients. Acta Psychiatr Scand. 1994;90:241-246.
10. De Marchi N, Mennella R. Huntington’s disease and its association with psychopathology. Harv Rev Psychiatry. 2000;7:278-289.
11. van den Bogaard SJ, Dumas EM, Acharya TP, et al. and the TRACK-HD Investigator Group. Early atrophy of pallidum and accumbens nucleus in Huntington’s disease. J Neurol. 2011;258(3):412-420.
12. Frank S, Jankovic J. Advances in the pharmacological management of Huntington’s disease. Drugs. 2010;70(5):561-571.
13. Alpay M, Koroshetz WJ. Quetiapine in the treatment of behavioral disturbances in patients with Huntington’s disease. Psychosomatics. 2006;47(1):70-72.
14. Seitz DP, Millson RC. Quetiapine in the management of psychosis secondary to Huntington’s disease: a case report. Can J Psychiatry. 2004;49(6):413.-
15. Bonelli RM, Niederwieser G. Quetiapine in Huntington’s disease: a first case report. J Neurol. 2002;249(8):1114-1115.
Opiates and psychotropics: Pharmacokinetics for practitioners
• When choosing pharmacologic therapy, make sure that all medications your patient takes are documented, consider drug-drug interactions, and instruct the patient to notify you of any new medications.
• In addition to toxicity, loss of efficacy of some opiate drugs may occur as a result of metabolic inhibition or induction by psychotropic medications.
• Collaborate with the physician who is prescribing the opioid if psychotropic choices are limited. The patient’s pain may be treated adequately with another analgesic that does not interact with the psychotropic that has been chosen.
As prescribed by his internist, Mr. G, age 44, takes 10 mg of methadone every 4 hours for chronic back pain secondary to a work-related injury 3 years ago. He experiences minimal sedation. Mr. G presents for psychiatric evaluation with complaints of increasing irritability, poor focus, low energy, and lack of interest in usual activities. The psychiatrist diagnoses him with depressive disorder not otherwise specified, and prescribes fluoxetine, 20 mg/d. Three weeks later, Mr. G’s wife contacts the psychiatrist reporting that her husband seems “overmedicated” and describes excess drowsiness and slowed thought processing.
After discussion with Mr. G’s internist and pharmacist, the psychiatrist decides that this oversedation may represent a drug-drug interaction between methadone and fluoxetine resulting in higher-than-expected methadone serum levels. Mr. G is instructed to stop fluoxetine with no taper, and his methadone dose is lowered with good results. Over the next 2 weeks Mr. G is titrated back to his original methadone dose and is re-evaluated by the psychiatrist to discuss medication options to address his depression.
Psychiatrists commonly encounter patients who receive opiate medications for chronic pain. Being aware of potential drug-drug interactions between opiate medications and psychotropics can help avoid adverse effects and combinations that may affect the efficacy of either drug. Pharmacokinetic interactions may affect your choice of psychiatric medication and should be taken into account when addressing adverse effects in any patient who takes opiates and psychotropics.
Metabolic pathways
The primary metabolic pathways for opiate metabolism are the cytochrome P450 (CYP) 2D6 and 3A4 isoenzymes. Depending on the agent used, prescribers may need to consider interactions for both pathways (Table 11,2 and Table 21). For example, oxycodone is metabolized via 2D6 and 3A4 isoenzymes and is a potent analgesic with oxymorphone and noroxycodone as its active metabolites. These metabolites, however, make a negligible contribution to oxycodone’s analgesic effect.3,4 Metabolism by the 3A4 isoenzyme is the principal oxidative pathway and the 2D6 site accounts for approximately 10% of oxycodone metabolism. A randomized, placebo-controlled, crossover study showed that 2D6 inhibition by paroxetine had no significant effect on oxycodone levels; however, a combination of paroxetine and itraconazole, a potent 3A4 inhibitor, resulted in substantial increases in oxycodone plasma levels.5 Remain vigilant for possible opiate toxicity when administering oxycodone with 3A4 inhibitors.
Methadone and meperidine also involve dual pathways. Methadone is metabolized primarily by 3A4 and 2B6, with 2D6 playing a smaller role.6 CYP2D6 seems to play an important part in metabolizing the R-enantiomer of methadone, which is largely responsible for the drug’s opiate effects, such as analgesia and respiratory depression.7,8 Induction of the 3A4 isoenzyme may result in methadone withdrawal, and inhibition may cause methadone toxicity.9 Inducers of 3A4, such as carbamazepine, and inhibitors, such as fluoxetine and fluvoxamine, should be avoided or used very cautiously in patients taking methadone. The 2B6 and 2D6 isoenzymes also may increase or decrease methadone levels and should be treated similarly. In Mr. G’s case, fluoxetine inhibited all 3 isoenzymes that are primarily responsible for methadone metabolism. A better antidepressant choice for Mr. G may have been venlafaxine, which is known to only mildly inhibit 2D6, or mirtazapine, which does not seem to inhibit the major CYP isoforms to an appreciable degree.10
Although the full scope of meperidine metabolism has not been identified,9 an in vitro test demonstrated that 2B6 and 3A4 play important roles in metabolizing meperidine to normeperidine, its major metabolite.11 Normeperidine does not provide analgesia and is associated with neurotoxicity, including anxiety, tremor, muscle twitching, and seizure.12 Agents that induce 3A4—such as carbamazepine or St. John’s wort—may contribute to neurotoxicity.9 Inhibition of these isoenzymes may increase meperidine levels and lead to anticholinergic toxicity or respiratory and central nervous system depression.13,14
Opiates metabolized by the 2D6 isoenzyme include codeine, hydrocodone, and tramadol. The analgesic effect of codeine seems dependent on 2D6 metabolism. Via this pathway, codeine is converted into morphine, which has a 300-times stronger affinity for the μ opioid receptor compared with codeine. 2D6 poor metabolizers have shown codeine intolerance and toxicity.3 Psychotropics known to strongly inhibit 2D6 isoenzyme processes—such as paroxetine, fluoxetine, and bupropion—should be avoided in patients taking codeine to prevent adverse effects and potential loss of efficacy. Better antidepressant choices include citalopram or venlafaxine, which inhibit 2D6 to a lesser degree.
Hydrocodone may be a viable option for patients taking 2D6 inhibitors. Hydrocodone is metabolized by 2D6 into hydromorphone, which is 7 to 33 times more potent than hydrocodone.15 Unlike codeine, 2D6 inhibition may have little effect on hydrocodone’s analgesic properties. Animal studies have shown that inhibition of the CYP analog to 2D6 does not affect analgesic response. In humans, 2D6 inhibition does not seem to affect hydrocodone’s abuse liability.16 Two case reports describe known 2D6 poor metabolizers who showed at least a partial response to hydrocodone.15,16
Tramadol’s analgesic properties may be related to serotonin and norepinephrine reuptake inhibition. It is less potent than codeine but is metabolized via the 2D6 isoenzyme into 0-desmethyltramadol, which is up to 200 times stronger than its parent compound.17 Clinicians should be aware that tramadol’s efficacy may be decreased when coadministered with 2D6 inhibitors. In a randomized, placebo-controlled trial, paroxetine, a potent 2D6 inhibitor, was shown to lessen the analgesic effect of tramadol.18
The 3A4 site is the primary pathway for fentanyl metabolism. Agents that inhibit 3A4 could increase fentanyl plasma concentration, leading to respiratory depression.19 Examples of 3A4 inhibitors include fluoxetine and fluvoxamine.
Psychotropics may inhibit or induce P450 isoenzymes to varying degrees. For example, paroxetine and citalopram are known to inhibit 2D6 but paroxetine is a stronger inhibitor; therefore, a significant drug-drug interaction is more likely with paroxetine and a 2D6 substrate than the same substrate administered with citalopram.
Table 1
Cytochrome P450 isoenzymes inhibited and induced by psychotropics
| Isoenzyme | Potency | Psychotropic(s) |
|---|---|---|
| 2B6 inducer | Moderate | Carbamazepine |
| 2B6 inhibitors | Mild to moderate | Fluoxetine, fluvoxamine |
| Moderate | Sertraline | |
| Potent | Paroxetine | |
| 2D6 inhibitors | Mild | Venlafaxine |
| Mild to moderate | Citalopram, escitalopram, fluvoxamine, risperidone | |
| Moderate | Duloxetine | |
| Moderate to potent | Bupropion | |
| Potent | Fluoxetine, haloperidol, paroxetine | |
| Dose-dependent | Sertraline | |
| 3A4 inducer | Potent | Carbamazepine |
| 3A4 inhibitors | Mild | Sertraline |
| Mild to moderate | Fluoxetine, fluvoxamine | |
| Source: References 1,2 | ||
Table 2
Cytochrome P450 isoenzymes inhibiting and inducing opiate metabolism
| Isoenzyme | Opiates |
|---|---|
| 2B6 inducer | Methadone |
| 2B6 inhibitors | Meperidine, methadone |
| 2D6 inhibitors | Codeine (may involve loss of efficacy as well as toxicity), methadone, tramadol (may involve loss of efficacy) |
| 3A4 inducer | Meperidine, methadone |
| 3A4 inhibitors | Fentanyl, oxycodone, meperidine, methadone |
| Source: Reference 1 | |
Other considerations
In addition to pharmacokinetic interactions, it is important to consider synergistic effects of some opiates and psychotropics. Examples include:
- additive effect on respiratory depression by benzodiazepines and opiates
- increased risk of serotonin syndrome and seizure when using tramadol with selective serotonin reuptake inhibitors or tricyclic antidepressants
- additive prolongation of the QTc interval by methadone when used with psychotropics known to prolong the QTc, such as ziprasidone.9,17,20
Careful attention to these interactions and collaboration among providers can ensure the best outcome for our patients. In Mr. G’s case, collaboration with his internist would be in order, particularly if antidepressant choices are limited. In consultation with the psychiatrist, the internist might choose another opiate to treat Mr. G’s pain that would not interact with fluoxetine. If Mr. G and his physician have struggled to manage his pain and if he is stable on the current regimen, selecting a different antidepressant may be warranted.
Related Resources
- Indiana University School of Medicine drug interactions: cytochrome P450 drug interaction table. http://medicine.iupui.edu/clinpharm/ddis/table.asp.
- Ferrando SJ, Levenson JL, Owen JA, eds. Clinical manual of psychopharmacology in the medically ill. Arlington, VA: American Psychiatric Publishing, Inc; 2010.
Drug Brand Names
- Bupropion • Wellbutrin, Zyban
- Carbamazepine • Tegretol
- Citalopram • Celexa
- Duloxetine • Cymbalta
- Escitalopram • Lexapro
- Fentanyl • Duragesic, Actiq
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Haloperidol • Haldol
- Hydrocodone • Lortab, Vicodin, others
- Itraconazole • Sporanox
- Meperidine • Demerol
- Methadone • Dolophine, Methadose
- Mirtazapine • Remeron
- Morphine • Avinza, Duramorph, others
- Oxycodone • OxyContin, Roxicodone
- Paroxetine • Paxil
- Risperidone • Risperdal
- Sertraline • Zoloft
- Tramadol • Ultram
- Venlafaxine • Effexor
- Ziprasidone • Geodon
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Sandson NB, Armstrong SC, Cozza KL. An overview of psychotropic drug-drug interactions. Psychosomatics. 2005;46:464-494.
2. Faucette SR, Wang H, Hamilton GA. Regulation of CYP2B6 in primary human hepatocytes by prototypical inducers. Drug Metab Dispos. 2004;32(3):348-358.
3. Smith HS. Opioid metabolism. Mayo Clin Proc. 2009;84:613-624.
4. Armstrong SC, Cozza KL. Pharmacokinetic drug interactions of morphine codeine, and their derivatives: theory and clinical reality, part II. Psychosomatics. 2003;44:515-520.
5. Grönlund J, Saari TI, Hagelberg NM, et al. Exposure to oral oxycodone is increased by concomitant inhibition of CYP2D6 and 3A4 pathways, but not by inhibition of CYP2D6 alone. Br J Clin Pharmacol. 2010;70:78-87.
6. Leavitt SB. Methadone-drug* interactions. (*medications illicit drugs, and other substances). 3rd ed. Mundelein, IL: Addiction Treatment Forum; 2005.
7. Pérez de los Cobos J, Siñol N, Trujols J, et al. Association of CYP2D6 ultrarapid metabolizer genotype with deficient patient satisfaction regarding methadone maintenance treatment. Drug Alcohol Depend. 2007;89:190-194.
8. Kristensen K, Christensen CB, Christrup LL. The mu1 mu2, delta, kappa opioid receptor binding profiles of methadone stereoisomers and morphine. Life Sci. 1995;56:PL45-50.
9. Armstrong SC, Wynn GH, Sandson NB. Pharmacokinetic drug interactions of synthetic opiate analgesics. Psychosomatics. 2009;50:169-176.
10. Spina E, Santoro V, D’Arrigo C. Clinically relevant pharmacokinetic drug interactions with second-generation antidepressants: an update. Clin Ther. 2008;30:1206-1227.
11. Ramírez J, Innocenti F, Schuetz EG, et al. CYP2B6, CYP3A4, and CYP2C19 are responsible for the in vitro N-demethylation of meperidine in human liver microsomes. Drug Metab Dispos. 2004;32:930-936.
12. Kaiko RF, Foley KM, Grabinski PY, et al. Central nervous system excitatory effects of meperidine in cancer patients. Ann Neurol. 1983;13:180-185.
13. Chalverus C. Clinically important meperidine toxicities. Journal of Pharmaceutical Care in Pain and Symptom Control. 2001;9:37-55.
14. Beckwith MC, Fox ER, Chandramouli J. Removing meperidine from the health-system formulary—frequently asked questions. J Pain Palliat Care Pharmacother. 2002;16:45-59.
15. Foster A, Mobley E, Wang Z. Complicated pain management in a CYP450 2D6 poor metabolizer. Pain Pract. 2007;7:352-356.
16. Susce MT, Murray-Carmichael E, de Leon J. Response to hydrocodone codeine and oxycodone in a CYP2D6 poor metabolizer. Prog Neuropsychopharmacol Biol Psychiatry. 2006;30:1356-1358.
17. Sansone RA, Sansone LA. Tramadol: seizures serotonin syndrome, and coadministered antidepressants. Psychiatry (Edgmont). 2009;6:17-21.
18. Laugesen S, Enggaard TP, Pedersen RS, et al. Paroxetine, a cytochrome P450 2D6 inhibitor, diminishes the stereoselective O-demethylation and reduces the hypoalgesic effect of tramadol. Clin Pharmacol Ther. 2005;77:312-323.
19. Duragesic [package insert]. Raritan NJ: Ortho-McNeil-Janssen Pharmaceuticals, Inc; 2009.
20. Caplehorn JR, Drummer OH. Fatal methadone toxicity: signs and circumstances and the role of benzodiazepines. Aust N Z J Public Health. 2002;26:358-362;discussion 362–363.
Aaron M. Pierce, DO
Dr. Pierce is Medical Director, Psychiatry Clinic, Assistant Professor, Department of Psychiatry, University of Oklahoma School of Community Medicine, Tulsa, OK.
Nancy C. Brahm, PharmD, MS, BCPP
Dr. Brahm is Clinical Associate Professor, Department of Pharmacy Practice: Clinical and Administrative Sciences-Tulsa, University of Oklahoma College of Pharmacy, Tulsa, OK.
Vicki L. Ellingrod, PharmD, BCPP, FCCP
Series Editor
Aaron M. Pierce, DO
Dr. Pierce is Medical Director, Psychiatry Clinic, Assistant Professor, Department of Psychiatry, University of Oklahoma School of Community Medicine, Tulsa, OK.
Nancy C. Brahm, PharmD, MS, BCPP
Dr. Brahm is Clinical Associate Professor, Department of Pharmacy Practice: Clinical and Administrative Sciences-Tulsa, University of Oklahoma College of Pharmacy, Tulsa, OK.
Vicki L. Ellingrod, PharmD, BCPP, FCCP
Series Editor
Aaron M. Pierce, DO
Dr. Pierce is Medical Director, Psychiatry Clinic, Assistant Professor, Department of Psychiatry, University of Oklahoma School of Community Medicine, Tulsa, OK.
Nancy C. Brahm, PharmD, MS, BCPP
Dr. Brahm is Clinical Associate Professor, Department of Pharmacy Practice: Clinical and Administrative Sciences-Tulsa, University of Oklahoma College of Pharmacy, Tulsa, OK.
Vicki L. Ellingrod, PharmD, BCPP, FCCP
Series Editor
• When choosing pharmacologic therapy, make sure that all medications your patient takes are documented, consider drug-drug interactions, and instruct the patient to notify you of any new medications.
• In addition to toxicity, loss of efficacy of some opiate drugs may occur as a result of metabolic inhibition or induction by psychotropic medications.
• Collaborate with the physician who is prescribing the opioid if psychotropic choices are limited. The patient’s pain may be treated adequately with another analgesic that does not interact with the psychotropic that has been chosen.
As prescribed by his internist, Mr. G, age 44, takes 10 mg of methadone every 4 hours for chronic back pain secondary to a work-related injury 3 years ago. He experiences minimal sedation. Mr. G presents for psychiatric evaluation with complaints of increasing irritability, poor focus, low energy, and lack of interest in usual activities. The psychiatrist diagnoses him with depressive disorder not otherwise specified, and prescribes fluoxetine, 20 mg/d. Three weeks later, Mr. G’s wife contacts the psychiatrist reporting that her husband seems “overmedicated” and describes excess drowsiness and slowed thought processing.
After discussion with Mr. G’s internist and pharmacist, the psychiatrist decides that this oversedation may represent a drug-drug interaction between methadone and fluoxetine resulting in higher-than-expected methadone serum levels. Mr. G is instructed to stop fluoxetine with no taper, and his methadone dose is lowered with good results. Over the next 2 weeks Mr. G is titrated back to his original methadone dose and is re-evaluated by the psychiatrist to discuss medication options to address his depression.
Psychiatrists commonly encounter patients who receive opiate medications for chronic pain. Being aware of potential drug-drug interactions between opiate medications and psychotropics can help avoid adverse effects and combinations that may affect the efficacy of either drug. Pharmacokinetic interactions may affect your choice of psychiatric medication and should be taken into account when addressing adverse effects in any patient who takes opiates and psychotropics.
Metabolic pathways
The primary metabolic pathways for opiate metabolism are the cytochrome P450 (CYP) 2D6 and 3A4 isoenzymes. Depending on the agent used, prescribers may need to consider interactions for both pathways (Table 11,2 and Table 21). For example, oxycodone is metabolized via 2D6 and 3A4 isoenzymes and is a potent analgesic with oxymorphone and noroxycodone as its active metabolites. These metabolites, however, make a negligible contribution to oxycodone’s analgesic effect.3,4 Metabolism by the 3A4 isoenzyme is the principal oxidative pathway and the 2D6 site accounts for approximately 10% of oxycodone metabolism. A randomized, placebo-controlled, crossover study showed that 2D6 inhibition by paroxetine had no significant effect on oxycodone levels; however, a combination of paroxetine and itraconazole, a potent 3A4 inhibitor, resulted in substantial increases in oxycodone plasma levels.5 Remain vigilant for possible opiate toxicity when administering oxycodone with 3A4 inhibitors.
Methadone and meperidine also involve dual pathways. Methadone is metabolized primarily by 3A4 and 2B6, with 2D6 playing a smaller role.6 CYP2D6 seems to play an important part in metabolizing the R-enantiomer of methadone, which is largely responsible for the drug’s opiate effects, such as analgesia and respiratory depression.7,8 Induction of the 3A4 isoenzyme may result in methadone withdrawal, and inhibition may cause methadone toxicity.9 Inducers of 3A4, such as carbamazepine, and inhibitors, such as fluoxetine and fluvoxamine, should be avoided or used very cautiously in patients taking methadone. The 2B6 and 2D6 isoenzymes also may increase or decrease methadone levels and should be treated similarly. In Mr. G’s case, fluoxetine inhibited all 3 isoenzymes that are primarily responsible for methadone metabolism. A better antidepressant choice for Mr. G may have been venlafaxine, which is known to only mildly inhibit 2D6, or mirtazapine, which does not seem to inhibit the major CYP isoforms to an appreciable degree.10
Although the full scope of meperidine metabolism has not been identified,9 an in vitro test demonstrated that 2B6 and 3A4 play important roles in metabolizing meperidine to normeperidine, its major metabolite.11 Normeperidine does not provide analgesia and is associated with neurotoxicity, including anxiety, tremor, muscle twitching, and seizure.12 Agents that induce 3A4—such as carbamazepine or St. John’s wort—may contribute to neurotoxicity.9 Inhibition of these isoenzymes may increase meperidine levels and lead to anticholinergic toxicity or respiratory and central nervous system depression.13,14
Opiates metabolized by the 2D6 isoenzyme include codeine, hydrocodone, and tramadol. The analgesic effect of codeine seems dependent on 2D6 metabolism. Via this pathway, codeine is converted into morphine, which has a 300-times stronger affinity for the μ opioid receptor compared with codeine. 2D6 poor metabolizers have shown codeine intolerance and toxicity.3 Psychotropics known to strongly inhibit 2D6 isoenzyme processes—such as paroxetine, fluoxetine, and bupropion—should be avoided in patients taking codeine to prevent adverse effects and potential loss of efficacy. Better antidepressant choices include citalopram or venlafaxine, which inhibit 2D6 to a lesser degree.
Hydrocodone may be a viable option for patients taking 2D6 inhibitors. Hydrocodone is metabolized by 2D6 into hydromorphone, which is 7 to 33 times more potent than hydrocodone.15 Unlike codeine, 2D6 inhibition may have little effect on hydrocodone’s analgesic properties. Animal studies have shown that inhibition of the CYP analog to 2D6 does not affect analgesic response. In humans, 2D6 inhibition does not seem to affect hydrocodone’s abuse liability.16 Two case reports describe known 2D6 poor metabolizers who showed at least a partial response to hydrocodone.15,16
Tramadol’s analgesic properties may be related to serotonin and norepinephrine reuptake inhibition. It is less potent than codeine but is metabolized via the 2D6 isoenzyme into 0-desmethyltramadol, which is up to 200 times stronger than its parent compound.17 Clinicians should be aware that tramadol’s efficacy may be decreased when coadministered with 2D6 inhibitors. In a randomized, placebo-controlled trial, paroxetine, a potent 2D6 inhibitor, was shown to lessen the analgesic effect of tramadol.18
The 3A4 site is the primary pathway for fentanyl metabolism. Agents that inhibit 3A4 could increase fentanyl plasma concentration, leading to respiratory depression.19 Examples of 3A4 inhibitors include fluoxetine and fluvoxamine.
Psychotropics may inhibit or induce P450 isoenzymes to varying degrees. For example, paroxetine and citalopram are known to inhibit 2D6 but paroxetine is a stronger inhibitor; therefore, a significant drug-drug interaction is more likely with paroxetine and a 2D6 substrate than the same substrate administered with citalopram.
Table 1
Cytochrome P450 isoenzymes inhibited and induced by psychotropics
| Isoenzyme | Potency | Psychotropic(s) |
|---|---|---|
| 2B6 inducer | Moderate | Carbamazepine |
| 2B6 inhibitors | Mild to moderate | Fluoxetine, fluvoxamine |
| Moderate | Sertraline | |
| Potent | Paroxetine | |
| 2D6 inhibitors | Mild | Venlafaxine |
| Mild to moderate | Citalopram, escitalopram, fluvoxamine, risperidone | |
| Moderate | Duloxetine | |
| Moderate to potent | Bupropion | |
| Potent | Fluoxetine, haloperidol, paroxetine | |
| Dose-dependent | Sertraline | |
| 3A4 inducer | Potent | Carbamazepine |
| 3A4 inhibitors | Mild | Sertraline |
| Mild to moderate | Fluoxetine, fluvoxamine | |
| Source: References 1,2 | ||
Table 2
Cytochrome P450 isoenzymes inhibiting and inducing opiate metabolism
| Isoenzyme | Opiates |
|---|---|
| 2B6 inducer | Methadone |
| 2B6 inhibitors | Meperidine, methadone |
| 2D6 inhibitors | Codeine (may involve loss of efficacy as well as toxicity), methadone, tramadol (may involve loss of efficacy) |
| 3A4 inducer | Meperidine, methadone |
| 3A4 inhibitors | Fentanyl, oxycodone, meperidine, methadone |
| Source: Reference 1 | |
Other considerations
In addition to pharmacokinetic interactions, it is important to consider synergistic effects of some opiates and psychotropics. Examples include:
- additive effect on respiratory depression by benzodiazepines and opiates
- increased risk of serotonin syndrome and seizure when using tramadol with selective serotonin reuptake inhibitors or tricyclic antidepressants
- additive prolongation of the QTc interval by methadone when used with psychotropics known to prolong the QTc, such as ziprasidone.9,17,20
Careful attention to these interactions and collaboration among providers can ensure the best outcome for our patients. In Mr. G’s case, collaboration with his internist would be in order, particularly if antidepressant choices are limited. In consultation with the psychiatrist, the internist might choose another opiate to treat Mr. G’s pain that would not interact with fluoxetine. If Mr. G and his physician have struggled to manage his pain and if he is stable on the current regimen, selecting a different antidepressant may be warranted.
Related Resources
- Indiana University School of Medicine drug interactions: cytochrome P450 drug interaction table. http://medicine.iupui.edu/clinpharm/ddis/table.asp.
- Ferrando SJ, Levenson JL, Owen JA, eds. Clinical manual of psychopharmacology in the medically ill. Arlington, VA: American Psychiatric Publishing, Inc; 2010.
Drug Brand Names
- Bupropion • Wellbutrin, Zyban
- Carbamazepine • Tegretol
- Citalopram • Celexa
- Duloxetine • Cymbalta
- Escitalopram • Lexapro
- Fentanyl • Duragesic, Actiq
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Haloperidol • Haldol
- Hydrocodone • Lortab, Vicodin, others
- Itraconazole • Sporanox
- Meperidine • Demerol
- Methadone • Dolophine, Methadose
- Mirtazapine • Remeron
- Morphine • Avinza, Duramorph, others
- Oxycodone • OxyContin, Roxicodone
- Paroxetine • Paxil
- Risperidone • Risperdal
- Sertraline • Zoloft
- Tramadol • Ultram
- Venlafaxine • Effexor
- Ziprasidone • Geodon
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
• When choosing pharmacologic therapy, make sure that all medications your patient takes are documented, consider drug-drug interactions, and instruct the patient to notify you of any new medications.
• In addition to toxicity, loss of efficacy of some opiate drugs may occur as a result of metabolic inhibition or induction by psychotropic medications.
• Collaborate with the physician who is prescribing the opioid if psychotropic choices are limited. The patient’s pain may be treated adequately with another analgesic that does not interact with the psychotropic that has been chosen.
As prescribed by his internist, Mr. G, age 44, takes 10 mg of methadone every 4 hours for chronic back pain secondary to a work-related injury 3 years ago. He experiences minimal sedation. Mr. G presents for psychiatric evaluation with complaints of increasing irritability, poor focus, low energy, and lack of interest in usual activities. The psychiatrist diagnoses him with depressive disorder not otherwise specified, and prescribes fluoxetine, 20 mg/d. Three weeks later, Mr. G’s wife contacts the psychiatrist reporting that her husband seems “overmedicated” and describes excess drowsiness and slowed thought processing.
After discussion with Mr. G’s internist and pharmacist, the psychiatrist decides that this oversedation may represent a drug-drug interaction between methadone and fluoxetine resulting in higher-than-expected methadone serum levels. Mr. G is instructed to stop fluoxetine with no taper, and his methadone dose is lowered with good results. Over the next 2 weeks Mr. G is titrated back to his original methadone dose and is re-evaluated by the psychiatrist to discuss medication options to address his depression.
Psychiatrists commonly encounter patients who receive opiate medications for chronic pain. Being aware of potential drug-drug interactions between opiate medications and psychotropics can help avoid adverse effects and combinations that may affect the efficacy of either drug. Pharmacokinetic interactions may affect your choice of psychiatric medication and should be taken into account when addressing adverse effects in any patient who takes opiates and psychotropics.
Metabolic pathways
The primary metabolic pathways for opiate metabolism are the cytochrome P450 (CYP) 2D6 and 3A4 isoenzymes. Depending on the agent used, prescribers may need to consider interactions for both pathways (Table 11,2 and Table 21). For example, oxycodone is metabolized via 2D6 and 3A4 isoenzymes and is a potent analgesic with oxymorphone and noroxycodone as its active metabolites. These metabolites, however, make a negligible contribution to oxycodone’s analgesic effect.3,4 Metabolism by the 3A4 isoenzyme is the principal oxidative pathway and the 2D6 site accounts for approximately 10% of oxycodone metabolism. A randomized, placebo-controlled, crossover study showed that 2D6 inhibition by paroxetine had no significant effect on oxycodone levels; however, a combination of paroxetine and itraconazole, a potent 3A4 inhibitor, resulted in substantial increases in oxycodone plasma levels.5 Remain vigilant for possible opiate toxicity when administering oxycodone with 3A4 inhibitors.
Methadone and meperidine also involve dual pathways. Methadone is metabolized primarily by 3A4 and 2B6, with 2D6 playing a smaller role.6 CYP2D6 seems to play an important part in metabolizing the R-enantiomer of methadone, which is largely responsible for the drug’s opiate effects, such as analgesia and respiratory depression.7,8 Induction of the 3A4 isoenzyme may result in methadone withdrawal, and inhibition may cause methadone toxicity.9 Inducers of 3A4, such as carbamazepine, and inhibitors, such as fluoxetine and fluvoxamine, should be avoided or used very cautiously in patients taking methadone. The 2B6 and 2D6 isoenzymes also may increase or decrease methadone levels and should be treated similarly. In Mr. G’s case, fluoxetine inhibited all 3 isoenzymes that are primarily responsible for methadone metabolism. A better antidepressant choice for Mr. G may have been venlafaxine, which is known to only mildly inhibit 2D6, or mirtazapine, which does not seem to inhibit the major CYP isoforms to an appreciable degree.10
Although the full scope of meperidine metabolism has not been identified,9 an in vitro test demonstrated that 2B6 and 3A4 play important roles in metabolizing meperidine to normeperidine, its major metabolite.11 Normeperidine does not provide analgesia and is associated with neurotoxicity, including anxiety, tremor, muscle twitching, and seizure.12 Agents that induce 3A4—such as carbamazepine or St. John’s wort—may contribute to neurotoxicity.9 Inhibition of these isoenzymes may increase meperidine levels and lead to anticholinergic toxicity or respiratory and central nervous system depression.13,14
Opiates metabolized by the 2D6 isoenzyme include codeine, hydrocodone, and tramadol. The analgesic effect of codeine seems dependent on 2D6 metabolism. Via this pathway, codeine is converted into morphine, which has a 300-times stronger affinity for the μ opioid receptor compared with codeine. 2D6 poor metabolizers have shown codeine intolerance and toxicity.3 Psychotropics known to strongly inhibit 2D6 isoenzyme processes—such as paroxetine, fluoxetine, and bupropion—should be avoided in patients taking codeine to prevent adverse effects and potential loss of efficacy. Better antidepressant choices include citalopram or venlafaxine, which inhibit 2D6 to a lesser degree.
Hydrocodone may be a viable option for patients taking 2D6 inhibitors. Hydrocodone is metabolized by 2D6 into hydromorphone, which is 7 to 33 times more potent than hydrocodone.15 Unlike codeine, 2D6 inhibition may have little effect on hydrocodone’s analgesic properties. Animal studies have shown that inhibition of the CYP analog to 2D6 does not affect analgesic response. In humans, 2D6 inhibition does not seem to affect hydrocodone’s abuse liability.16 Two case reports describe known 2D6 poor metabolizers who showed at least a partial response to hydrocodone.15,16
Tramadol’s analgesic properties may be related to serotonin and norepinephrine reuptake inhibition. It is less potent than codeine but is metabolized via the 2D6 isoenzyme into 0-desmethyltramadol, which is up to 200 times stronger than its parent compound.17 Clinicians should be aware that tramadol’s efficacy may be decreased when coadministered with 2D6 inhibitors. In a randomized, placebo-controlled trial, paroxetine, a potent 2D6 inhibitor, was shown to lessen the analgesic effect of tramadol.18
The 3A4 site is the primary pathway for fentanyl metabolism. Agents that inhibit 3A4 could increase fentanyl plasma concentration, leading to respiratory depression.19 Examples of 3A4 inhibitors include fluoxetine and fluvoxamine.
Psychotropics may inhibit or induce P450 isoenzymes to varying degrees. For example, paroxetine and citalopram are known to inhibit 2D6 but paroxetine is a stronger inhibitor; therefore, a significant drug-drug interaction is more likely with paroxetine and a 2D6 substrate than the same substrate administered with citalopram.
Table 1
Cytochrome P450 isoenzymes inhibited and induced by psychotropics
| Isoenzyme | Potency | Psychotropic(s) |
|---|---|---|
| 2B6 inducer | Moderate | Carbamazepine |
| 2B6 inhibitors | Mild to moderate | Fluoxetine, fluvoxamine |
| Moderate | Sertraline | |
| Potent | Paroxetine | |
| 2D6 inhibitors | Mild | Venlafaxine |
| Mild to moderate | Citalopram, escitalopram, fluvoxamine, risperidone | |
| Moderate | Duloxetine | |
| Moderate to potent | Bupropion | |
| Potent | Fluoxetine, haloperidol, paroxetine | |
| Dose-dependent | Sertraline | |
| 3A4 inducer | Potent | Carbamazepine |
| 3A4 inhibitors | Mild | Sertraline |
| Mild to moderate | Fluoxetine, fluvoxamine | |
| Source: References 1,2 | ||
Table 2
Cytochrome P450 isoenzymes inhibiting and inducing opiate metabolism
| Isoenzyme | Opiates |
|---|---|
| 2B6 inducer | Methadone |
| 2B6 inhibitors | Meperidine, methadone |
| 2D6 inhibitors | Codeine (may involve loss of efficacy as well as toxicity), methadone, tramadol (may involve loss of efficacy) |
| 3A4 inducer | Meperidine, methadone |
| 3A4 inhibitors | Fentanyl, oxycodone, meperidine, methadone |
| Source: Reference 1 | |
Other considerations
In addition to pharmacokinetic interactions, it is important to consider synergistic effects of some opiates and psychotropics. Examples include:
- additive effect on respiratory depression by benzodiazepines and opiates
- increased risk of serotonin syndrome and seizure when using tramadol with selective serotonin reuptake inhibitors or tricyclic antidepressants
- additive prolongation of the QTc interval by methadone when used with psychotropics known to prolong the QTc, such as ziprasidone.9,17,20
Careful attention to these interactions and collaboration among providers can ensure the best outcome for our patients. In Mr. G’s case, collaboration with his internist would be in order, particularly if antidepressant choices are limited. In consultation with the psychiatrist, the internist might choose another opiate to treat Mr. G’s pain that would not interact with fluoxetine. If Mr. G and his physician have struggled to manage his pain and if he is stable on the current regimen, selecting a different antidepressant may be warranted.
Related Resources
- Indiana University School of Medicine drug interactions: cytochrome P450 drug interaction table. http://medicine.iupui.edu/clinpharm/ddis/table.asp.
- Ferrando SJ, Levenson JL, Owen JA, eds. Clinical manual of psychopharmacology in the medically ill. Arlington, VA: American Psychiatric Publishing, Inc; 2010.
Drug Brand Names
- Bupropion • Wellbutrin, Zyban
- Carbamazepine • Tegretol
- Citalopram • Celexa
- Duloxetine • Cymbalta
- Escitalopram • Lexapro
- Fentanyl • Duragesic, Actiq
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Haloperidol • Haldol
- Hydrocodone • Lortab, Vicodin, others
- Itraconazole • Sporanox
- Meperidine • Demerol
- Methadone • Dolophine, Methadose
- Mirtazapine • Remeron
- Morphine • Avinza, Duramorph, others
- Oxycodone • OxyContin, Roxicodone
- Paroxetine • Paxil
- Risperidone • Risperdal
- Sertraline • Zoloft
- Tramadol • Ultram
- Venlafaxine • Effexor
- Ziprasidone • Geodon
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Sandson NB, Armstrong SC, Cozza KL. An overview of psychotropic drug-drug interactions. Psychosomatics. 2005;46:464-494.
2. Faucette SR, Wang H, Hamilton GA. Regulation of CYP2B6 in primary human hepatocytes by prototypical inducers. Drug Metab Dispos. 2004;32(3):348-358.
3. Smith HS. Opioid metabolism. Mayo Clin Proc. 2009;84:613-624.
4. Armstrong SC, Cozza KL. Pharmacokinetic drug interactions of morphine codeine, and their derivatives: theory and clinical reality, part II. Psychosomatics. 2003;44:515-520.
5. Grönlund J, Saari TI, Hagelberg NM, et al. Exposure to oral oxycodone is increased by concomitant inhibition of CYP2D6 and 3A4 pathways, but not by inhibition of CYP2D6 alone. Br J Clin Pharmacol. 2010;70:78-87.
6. Leavitt SB. Methadone-drug* interactions. (*medications illicit drugs, and other substances). 3rd ed. Mundelein, IL: Addiction Treatment Forum; 2005.
7. Pérez de los Cobos J, Siñol N, Trujols J, et al. Association of CYP2D6 ultrarapid metabolizer genotype with deficient patient satisfaction regarding methadone maintenance treatment. Drug Alcohol Depend. 2007;89:190-194.
8. Kristensen K, Christensen CB, Christrup LL. The mu1 mu2, delta, kappa opioid receptor binding profiles of methadone stereoisomers and morphine. Life Sci. 1995;56:PL45-50.
9. Armstrong SC, Wynn GH, Sandson NB. Pharmacokinetic drug interactions of synthetic opiate analgesics. Psychosomatics. 2009;50:169-176.
10. Spina E, Santoro V, D’Arrigo C. Clinically relevant pharmacokinetic drug interactions with second-generation antidepressants: an update. Clin Ther. 2008;30:1206-1227.
11. Ramírez J, Innocenti F, Schuetz EG, et al. CYP2B6, CYP3A4, and CYP2C19 are responsible for the in vitro N-demethylation of meperidine in human liver microsomes. Drug Metab Dispos. 2004;32:930-936.
12. Kaiko RF, Foley KM, Grabinski PY, et al. Central nervous system excitatory effects of meperidine in cancer patients. Ann Neurol. 1983;13:180-185.
13. Chalverus C. Clinically important meperidine toxicities. Journal of Pharmaceutical Care in Pain and Symptom Control. 2001;9:37-55.
14. Beckwith MC, Fox ER, Chandramouli J. Removing meperidine from the health-system formulary—frequently asked questions. J Pain Palliat Care Pharmacother. 2002;16:45-59.
15. Foster A, Mobley E, Wang Z. Complicated pain management in a CYP450 2D6 poor metabolizer. Pain Pract. 2007;7:352-356.
16. Susce MT, Murray-Carmichael E, de Leon J. Response to hydrocodone codeine and oxycodone in a CYP2D6 poor metabolizer. Prog Neuropsychopharmacol Biol Psychiatry. 2006;30:1356-1358.
17. Sansone RA, Sansone LA. Tramadol: seizures serotonin syndrome, and coadministered antidepressants. Psychiatry (Edgmont). 2009;6:17-21.
18. Laugesen S, Enggaard TP, Pedersen RS, et al. Paroxetine, a cytochrome P450 2D6 inhibitor, diminishes the stereoselective O-demethylation and reduces the hypoalgesic effect of tramadol. Clin Pharmacol Ther. 2005;77:312-323.
19. Duragesic [package insert]. Raritan NJ: Ortho-McNeil-Janssen Pharmaceuticals, Inc; 2009.
20. Caplehorn JR, Drummer OH. Fatal methadone toxicity: signs and circumstances and the role of benzodiazepines. Aust N Z J Public Health. 2002;26:358-362;discussion 362–363.
1. Sandson NB, Armstrong SC, Cozza KL. An overview of psychotropic drug-drug interactions. Psychosomatics. 2005;46:464-494.
2. Faucette SR, Wang H, Hamilton GA. Regulation of CYP2B6 in primary human hepatocytes by prototypical inducers. Drug Metab Dispos. 2004;32(3):348-358.
3. Smith HS. Opioid metabolism. Mayo Clin Proc. 2009;84:613-624.
4. Armstrong SC, Cozza KL. Pharmacokinetic drug interactions of morphine codeine, and their derivatives: theory and clinical reality, part II. Psychosomatics. 2003;44:515-520.
5. Grönlund J, Saari TI, Hagelberg NM, et al. Exposure to oral oxycodone is increased by concomitant inhibition of CYP2D6 and 3A4 pathways, but not by inhibition of CYP2D6 alone. Br J Clin Pharmacol. 2010;70:78-87.
6. Leavitt SB. Methadone-drug* interactions. (*medications illicit drugs, and other substances). 3rd ed. Mundelein, IL: Addiction Treatment Forum; 2005.
7. Pérez de los Cobos J, Siñol N, Trujols J, et al. Association of CYP2D6 ultrarapid metabolizer genotype with deficient patient satisfaction regarding methadone maintenance treatment. Drug Alcohol Depend. 2007;89:190-194.
8. Kristensen K, Christensen CB, Christrup LL. The mu1 mu2, delta, kappa opioid receptor binding profiles of methadone stereoisomers and morphine. Life Sci. 1995;56:PL45-50.
9. Armstrong SC, Wynn GH, Sandson NB. Pharmacokinetic drug interactions of synthetic opiate analgesics. Psychosomatics. 2009;50:169-176.
10. Spina E, Santoro V, D’Arrigo C. Clinically relevant pharmacokinetic drug interactions with second-generation antidepressants: an update. Clin Ther. 2008;30:1206-1227.
11. Ramírez J, Innocenti F, Schuetz EG, et al. CYP2B6, CYP3A4, and CYP2C19 are responsible for the in vitro N-demethylation of meperidine in human liver microsomes. Drug Metab Dispos. 2004;32:930-936.
12. Kaiko RF, Foley KM, Grabinski PY, et al. Central nervous system excitatory effects of meperidine in cancer patients. Ann Neurol. 1983;13:180-185.
13. Chalverus C. Clinically important meperidine toxicities. Journal of Pharmaceutical Care in Pain and Symptom Control. 2001;9:37-55.
14. Beckwith MC, Fox ER, Chandramouli J. Removing meperidine from the health-system formulary—frequently asked questions. J Pain Palliat Care Pharmacother. 2002;16:45-59.
15. Foster A, Mobley E, Wang Z. Complicated pain management in a CYP450 2D6 poor metabolizer. Pain Pract. 2007;7:352-356.
16. Susce MT, Murray-Carmichael E, de Leon J. Response to hydrocodone codeine and oxycodone in a CYP2D6 poor metabolizer. Prog Neuropsychopharmacol Biol Psychiatry. 2006;30:1356-1358.
17. Sansone RA, Sansone LA. Tramadol: seizures serotonin syndrome, and coadministered antidepressants. Psychiatry (Edgmont). 2009;6:17-21.
18. Laugesen S, Enggaard TP, Pedersen RS, et al. Paroxetine, a cytochrome P450 2D6 inhibitor, diminishes the stereoselective O-demethylation and reduces the hypoalgesic effect of tramadol. Clin Pharmacol Ther. 2005;77:312-323.
19. Duragesic [package insert]. Raritan NJ: Ortho-McNeil-Janssen Pharmaceuticals, Inc; 2009.
20. Caplehorn JR, Drummer OH. Fatal methadone toxicity: signs and circumstances and the role of benzodiazepines. Aust N Z J Public Health. 2002;26:358-362;discussion 362–363.
Depression or delirium?
CASE: Agitated and paranoid
Police bring Mr. L, age 85, to the emergency department (ED) because he threatened his wife, claiming she is having an affair. Two days earlier, he was discharged from a different hospital, where he was treated for progressive and fluctuating irritability, depressed mood, confusion, disorientation, inattention, and delusional thinking that had started 4 to 5 months earlier. He has no other psychiatric history.
Mr. L has a history of atrial fibrillation, hypertension, benign prostatic hypertrophy, and noninsulin-dependent diabetes mellitus. Several months ago, he had hip surgery, which was complicated by a surgical wound infection. Medications include digoxin, 0.125 mg/d; atenolol, 100 mg/d; warfarin, 1 mg/d on Monday, Wednesday, Friday, Saturday, and Sunday and 0.5 mg/d Tuesday and Thursday; lisinopril, 40 mg/d; tamsulosin, 0.4 mg/d; and glyburide, 1.25 mg/d. During the previous hospitalization, physicians discovered he had myasthenia gravis, which they treated with prednisone and pyridostigmine. Mr. L also was diagnosed with hyperaldosteronism. An adrenal mass was found in an abdominal CT. At that time, he also was diagnosed with major depressive disorder (MDD) with psychotic features and started on aripiprazole, 10 mg/d, mirtazapine, 30 mg/d, and trazodone, 50 mg/d for sleep.
The authors’ observations
When evaluating mental status changes in older patients, consider the time course and characteristics of the changes, especially if the patient’s cognitive function changes. Acute mental status changes that occur over hours to days often represent delirium caused by a medical condition such as a coronary event or infection. Changes that develop over weeks to months often signal a primary psychiatric disorder such as depression, mania, or dementia. Mr. L’s mood and psychotic symptoms developed over 4 to 5 months and were thought to be a result of MDD with psychotic features. However, his fluctuating cognitive symptoms, confusion, and lack of psychiatric history suggest that the differential diagnosis should include a cognitive disorder such as delirium or dementia. The hypoactive form of delirium often is unrecognized or misdiagnosed as sedation or depression, particularly in older patients.1
Multiple medical conditions and polypharmacy are important factors to consider when evaluating mental status changes in geriatric patients. In Mr. L’s case, atrial fibrillation, hypertension, and diabetes increase his risk of an acute cardiovascular or cerebrovascular event and chronic cerebrovascular disease. Hyperaldosteronism can lead to electrolyte abnormalities that may produce mental status changes. Treatment with an oral hypoglycemic raises the possibility that hypoglycemia is contributing to his mental status changes. Prednisone can cause psychosis, anxiety, and mania. Digoxin toxicity is associated with psychosis and irritability. Pyridostigmine also has been reported to cause psychosis. Use of an antidepressant, such as mirtazapine, could have exacerbated an underlying undiagnosed bipolar disorder. Antipsychotics, such as aripiprazole, may cause akathisia or activation. Substance intoxication or withdrawal should not be excluded solely because a patient is older. In older patients, medications with anti-cholinergic effects are common culprits for cognitive impairment (Table 1).2,3
Table 1
Medications that could contribute to mental status changes
| Anticholinergics (atropine, benztropine, oxybutynin, some OTC medications) |
| Hypnotics/sedatives (benzodiazepines) |
| Opiate analgesics (meperidine) |
| Neuroleptics (clozapine, thioridazine, olanzapine) |
| Antiparkinsonian medications (levodopa, selegiline, pergolide, amantadine) |
| Antidepressants (amitriptyline) |
| Anticonvulsants (phenytoin) |
| Histamine H2 receptor antagonists (ranitidine, cimetidine, omeprazole) |
| Cardiac drugs (digoxin) |
| Nonsteroidal anti-inflammatory drugs (aspirin) |
| Corticosteroids (prednisolone) |
| Antibiotics (penicillins, cephalosporins, quinolones) |
| OTC: over the counter Source: References 2,3 |
ASSESSMENT: More problems
At admission to the medical unit, Mr. L’s temperature is 36.7°C (98°F), with a heart rate of 77 beats per minute, respiratory rate of 24 breaths per minute, and blood pressure of 164/84 mm Hg with oxygen saturation of 96% at room air. Physical exam is notable for 2+ pitting edema in the lower extremities. Mr. L is oriented to person, place, and time and is psychomotorically activated. Neurologic examination is within normal limits.
Laboratory data reveal a potassium level of 2.5 mEq/L. Other results, including complete blood count, comprehensive metabolic panel, thyroid-stimulating hormone, urinalysis, urine toxicology screen, B12, folate, venereal disease research laboratory, and ammonia are unremarkable. Chest radiography reveals an enlarged cardiomediastinum. A CT scan of the brain without contrast shows cortical volume loss and periventricular white matter disease without evidence of acute intracranial abnormality. ECG shows atrial fibrillation with a rate of 67 beats per minute.
Mr. L’s hypokalemia is corrected with potassium chloride and his hyperaldosteronism is treated with spironolactone, 25 mg/d. Physicians on the medical unit discontinue digoxin because Mr. L’s heart rate is controlled with atenolol and he is anticoagulated with warfarin.
Mr. L continues to be depressed and irritable with delusional jealousy. Mirtazapine is continued at 30 mg/d at bedtime. Aripiprazole and trazodone are discontinued and Mr. L is started on olanzapine, 10 mg/d, and haloperidol, 1 mg 4 times a day as needed for agitation. He requires multiple “as needed” haloperidol doses because of intermittent episodes of agitation. Mr. L is then transferred to the inpatient psychiatric unit for continued evaluation and treatment.
The authors’ observations
The fact that Mr. L is alert and oriented is encouraging; however, it does not rule out delirium because this condition is characterized by fluctuating levels of consciousness. Therefore, it is important to reassess him over time and perform a more thorough evaluation of cognitive function, especially attention and concentration, in addition to alertness and orientation. Psychomotor activation could suggest agitated depression, anxiety, mania, psychosis, substance intoxication, akathisia from antipsychotics, or delirium (Table 2).4 Initial evaluation— especially in older patients—should include a thorough history (including collateral sources) and be guided by the clinical presentation and physical examination, taking into consideration life-threatening conditions and common causes of mental status change such as infections, hypoxia, substance or medication effects, acute coronary syndromes, acute neurologic events, and metabolic conditions.
Table 2
DSM-IV-TR criteria for delirium caused by a medical condition
| A. Disturbance of consciousness (ie, reduced clarity of awareness of the environment) with reduced ability to focus, sustain, or shift attention |
| B. A change in cognition (such as memory deficit, disorientation, language disturbance) or the development of a perceptual disturbance that is not better accounted for by a preexisting, established, or evolving dementia |
| C. The disturbance develops over a short period of time (usually hours to days) and tends to fluctuate during the course of the day |
| D. There is evidence from the history, physical examination, or laboratory findings that the disturbance is caused by the direct physiological consequences of a general medical condition |
| Source: Reference 4 |
Reconsider the diagnosis
Even after being treated for hyperaldosteronism and discontinuing unnecessary medications, Mr. L continued to be treated for MDD with psychotic features despite intermittent confusion and agitation. At this point, it might have been useful to reconsider whether MDD with psychotic features was the most appropriate diagnosis to explain his mental status changes.
Mental status changes caused by medical disorders or medications do not immediately clear after the medical disorder is corrected or the medication is discontinued; it could take days or weeks for a patient to return to baseline. In Mr. L’s case it may be useful to simplify his medication regimen because polypharmacy contributes to delirium. Finally, olanzapine could worsen his condition because of its anticholinergic effects.5
EVALUATION: Poor cognitive status
Mental status examination upon admission to the psychiatric unit reveals a poorly cooperative patient with irritable mood and affect with slowed psychomotor activity. Mr. L’s thought process is organized with normal associations and thought content does not reveal suicidality or homicidality. However, he verbalizes delusions about his wife having an affair with a neighbor. He is partially oriented to time but believes he is in Germany. His insight is limited and he demonstrates impaired attention and concentration. We cannot complete a Mini-Mental State Exam (MMSE) because Mr. L does not cooperate.
After admission, Mr. L is intermittently confused, agitated, and disoriented. Between these episodes he is pleasant, cooperative, and oriented. Jealous delusions regarding his wife continue. Olanzapine and mirtazapine are tapered and discontinued. Haloperidol dose is changed to 1 mg 3 times a day, then to 1.5 mg in the morning and 3 mg in the evening. Prednisone is tapered and discontinued.
The authors’ observations
Cognitive testing is essential for the diagnosis and treatment of patients with mental status changes and for evaluating their response to treatment. Although the MMSE is widely used, other scales—including the Confusion Assessment Method, the Organic Brain Syndrome Scale, the Memorial Delirium Assessment Scale, and the delirium severity index6—may be more sensitive for detecting delirium. All of these scales can be difficult to complete when evaluating confused and combative patients. Quick screening instruments for inattention, such as the digit span test and listing days of the week backwards, could be used as well.
HISTORY: Surgical complications
Further questioning of Mr. L’s family reveals that his behavior started to change 7 months ago; this was 1 month after undergoing hip replacement surgery, which was complicated by a surgical wound infection and worsened his medical illnesses. Within a month, Mr. L became withdrawn and appeared depressed. He was confused and intermittently disoriented to place and time. He became irritable and started reporting concerns about his wife having an affair. During this time different medications were introduced, including steroids and several antibiotics.
The authors’ observations
A thorough history from the patient and caregivers, including the time course of mental status changes, new medication use, and history of medical and psychiatric disorders—especially depression and dementia—are important to obtain, especially early in the evaluation.
Although Mr. L’s irritability, delusions, and psychomotor slowing could be signs of psychotic depression, his fluctuating mental status, disorientation, poor attention, and impaired concentration suggest delirium (Table 3).4,7 This diagnosis is supported by the fact that Mr. L’s symptoms emerged after orthopedic surgery. Delirium after orthopedic surgery is common among older patients.8 Contributing and perpetuating factors in Mr. L’s case may have included postoperative complications, hypokalemia (hyperaldosteronism), medications (prednisone, digoxin, and olanzapine), and environmental unfamiliarity during hospitalization. A delirium diagnosis should be based on a high index of suspicion and a careful clinical assessment rather than diagnostic tests.
Table 3
Deconstructing delirium
| Defining characteristics |
| Confusional state of fluctuating course |
| Acute or subacute onset |
| Inattention |
| Disorganized thinking |
| Alteration and fluctuation of level of consciousness |
| Other characteristics |
| Cognitive: Memory impairment, perseveration |
| Motor: Hyperactive, hypoactive, mixed |
| Psychiatric: Thought disorganization, mood changes, delusions, hallucinations |
| Etiologies* |
| Predisposing factors: Age, functional status (ie, immobility), nutritional status (ie, dehydration), sensory impairment, medical conditions, psychiatric conditions (ie, dementia, TBI), medications, illicit drugs |
| Precipitating factors: Acute neurologic conditions (ie, stroke), intercurrent illnesses (ie, infections, hypoxia, anemia), surgery, environmental factors (ie, ICU, restraints, pain), illicit drugs (alcohol withdrawal), medications (ie, polypharmacy, anticholinergics), sleep depravation |
| *Usually >1 etiology ICU: intensive care unit; TBI: traumatic brain injury Source: References 4,7 |
OUTCOME: Return home
Mr. L’s confusion and delusional jealousy decrease over time, as do his disorientation and inattention, as evidenced by improvement on MMSE scores. His last MMSE score is 27/30, failing mostly in attention and recall.
After sustained improvement in cognition and behavior, Mr. L is discharged home on haloperidol and the remainder of his nonpsychiatric medications with outpatient medical and psychiatric follow-up. Over several months, he continues to show improvement and haloperidol is discontinued.
The authors’ observations
Delirium treatment should focus on prompt identification and management of precipitating and contributing factors.7 Antipsychotics are considered first-line treatment for patients with delirium, agitation, or psychosis who pose a risk to themselves or others. Benzodiazepines should be avoided in older patients unless symptoms are secondary to CNS-depressant withdrawal (ie, alcohol, benzodiazepines).9
Although there are no-FDA approved medications for delirium, haloperidol has been widely studied and used for treatment of agitation and psychosis in delirium. There is no evidence that low-dose haloperidol is any less effective than olanzapine or risperidone, or is more likely to cause adverse drug effects such as extrapyramidal syndrome.10 Antipsychotic use in a confused or agitated dementia patient increases risk of mortality compared with dementia patients who do not receive antipsychotics.11 The use of typical or atypical antipsychotics for delirium should be guided by the patient’s characteristics, such as cardiovascular status and presence or absence of underlying dementia. Atypical antipsychotics should be used carefully because—as in Mr. L’s case—anticholinergic side effects of medications such as olanzapine could worsen delirium.5 Once delirium has resolved, antipsychotics should be tapered and discontinued.
Other components of delirium treatment and prevention include:
- reorientation (verbally, with clocks, calendars, etc.)
- safe ambulation
- adequate sleep, food, and fluid intake
- adaptive equipment for vision and hearing impairment
- adequate management of pain and other comorbidities.12
Related Resources
- Khan RA, Kahn D, Bourgeois JA. Delirium: sifting through the confusion. Curr Psychiatry Rep. 2009;11(3):226-234.
- Maldonado JR. Delirium in the acute care setting: characteristics, diagnosis, and treatment. Crit Care Clin. 2008;24:657-722.
- Young J, Inouye SK. Delirium in older people. BMJ. 2007;334(7598):842-846.
Drug Brand Names
- Amantadine • Symmetrel
- Amitriptyline • Elavil
- Aripiprazole • Abilify
- Atenolol • Tenormin
- Atropine • AtroPen
- Benztropine • Cogentin
- Cimetidine • Tagamet
- Clozapine • Clozaril
- Digoxin • Lanoxicaps, Lanoxin
- Glyburide • DiaBeta, Micronase
- Haloperidol • Haldol
- Levodopa/carbidopa • Parcopa, Sinemet
- Lisinopril • Prinivil, Zestril
- Lorazepam • Ativan
- Meperidine • Demerol
- Mirtazapine • Remeron
- Olanzapine • Zyprexa
- Omeprazole • Prilosec
- Oxybutynin • Ditropan
- Pergolide • Permax
- Phenytoin • Dilantin, Phenytek
- Prednisolone • Orapred, Prelone, others
- Prednisone • Deltasone, Meticorten
- Pyridostigmine • Mestinon
- Ranitidine • Zantac
- Risperidone • Risperdal
- Selegiline • Eldepryl, Zelapar
- Spironolactone • Aldactone
- Tamsulosin • Flomax
- Thioridazine • Mellaril
- Trazodone • Desyrel
- Warfarin • Coumadin
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. McAvay GJ, Van Ness PH, Bogardus ST, Jr, et al. Depressive symptoms and the risk of incident delirium in older hospitalized adults. J Am Geriatr Soc. 2007;55:684-691.
2. Mintzer J, Burns A. Anticholinergic side-effects of drugs in elderly people. J R Soc Med. 2000;93(9):457-462.
3. Moore AR, O’Keeffe ST. Drug-induced cognitive impairment in the elderly. Drugs Aging. 1999;15(1):15-28.
4. Diagnostic and statistical manual of mental disorders. 4th ed text rev. Washington, DC: American Psychiatric Association; 2000.
5. Lim CJ, Trevino C, Tampi RR. Can olanzapine cause delirium in the elderly? Ann Pharmacother. 2006;40(1):135-138.
6. Woodford HJ, George J. Cognitive assessment in the elderly: a review of clinical methods. QJM. 2007;100:469-484.
7. Young J, Inouye SK. Delirium in older people. BMJ. 2007;334(7598):842-846.
8. Bruce AJ, Ritchie CW, Blizard R, et al. The incidence of delirium following orthopedic surgery: a meta-analytic review. Int Psychogeriatr. 2007;19(2):197-214.
9. Attard A, Ranjith G, Taylor D. Delirium and its treatment. CNS Drugs. 2008;22:631-644.
10. Lonergan E, Britton AM, Luxenberg J, et al. Antipsychotics for delirium. Cochrane Database Syst Rev. 2007;(2):CD005594.-
11. Schneider LS, Dagerman KS, Insel P. Risk of death with atypical antipsychotic drug treatment for dementia: meta-analysis of randomized placebo-controlled trials. JAMA. 2005;294(15):1934-1943.
12. Tabet N, Howard R. Non-pharmacological interventions in the prevention of delirium. Age Ageing. 2009;38(4):374-379.
CASE: Agitated and paranoid
Police bring Mr. L, age 85, to the emergency department (ED) because he threatened his wife, claiming she is having an affair. Two days earlier, he was discharged from a different hospital, where he was treated for progressive and fluctuating irritability, depressed mood, confusion, disorientation, inattention, and delusional thinking that had started 4 to 5 months earlier. He has no other psychiatric history.
Mr. L has a history of atrial fibrillation, hypertension, benign prostatic hypertrophy, and noninsulin-dependent diabetes mellitus. Several months ago, he had hip surgery, which was complicated by a surgical wound infection. Medications include digoxin, 0.125 mg/d; atenolol, 100 mg/d; warfarin, 1 mg/d on Monday, Wednesday, Friday, Saturday, and Sunday and 0.5 mg/d Tuesday and Thursday; lisinopril, 40 mg/d; tamsulosin, 0.4 mg/d; and glyburide, 1.25 mg/d. During the previous hospitalization, physicians discovered he had myasthenia gravis, which they treated with prednisone and pyridostigmine. Mr. L also was diagnosed with hyperaldosteronism. An adrenal mass was found in an abdominal CT. At that time, he also was diagnosed with major depressive disorder (MDD) with psychotic features and started on aripiprazole, 10 mg/d, mirtazapine, 30 mg/d, and trazodone, 50 mg/d for sleep.
The authors’ observations
When evaluating mental status changes in older patients, consider the time course and characteristics of the changes, especially if the patient’s cognitive function changes. Acute mental status changes that occur over hours to days often represent delirium caused by a medical condition such as a coronary event or infection. Changes that develop over weeks to months often signal a primary psychiatric disorder such as depression, mania, or dementia. Mr. L’s mood and psychotic symptoms developed over 4 to 5 months and were thought to be a result of MDD with psychotic features. However, his fluctuating cognitive symptoms, confusion, and lack of psychiatric history suggest that the differential diagnosis should include a cognitive disorder such as delirium or dementia. The hypoactive form of delirium often is unrecognized or misdiagnosed as sedation or depression, particularly in older patients.1
Multiple medical conditions and polypharmacy are important factors to consider when evaluating mental status changes in geriatric patients. In Mr. L’s case, atrial fibrillation, hypertension, and diabetes increase his risk of an acute cardiovascular or cerebrovascular event and chronic cerebrovascular disease. Hyperaldosteronism can lead to electrolyte abnormalities that may produce mental status changes. Treatment with an oral hypoglycemic raises the possibility that hypoglycemia is contributing to his mental status changes. Prednisone can cause psychosis, anxiety, and mania. Digoxin toxicity is associated with psychosis and irritability. Pyridostigmine also has been reported to cause psychosis. Use of an antidepressant, such as mirtazapine, could have exacerbated an underlying undiagnosed bipolar disorder. Antipsychotics, such as aripiprazole, may cause akathisia or activation. Substance intoxication or withdrawal should not be excluded solely because a patient is older. In older patients, medications with anti-cholinergic effects are common culprits for cognitive impairment (Table 1).2,3
Table 1
Medications that could contribute to mental status changes
| Anticholinergics (atropine, benztropine, oxybutynin, some OTC medications) |
| Hypnotics/sedatives (benzodiazepines) |
| Opiate analgesics (meperidine) |
| Neuroleptics (clozapine, thioridazine, olanzapine) |
| Antiparkinsonian medications (levodopa, selegiline, pergolide, amantadine) |
| Antidepressants (amitriptyline) |
| Anticonvulsants (phenytoin) |
| Histamine H2 receptor antagonists (ranitidine, cimetidine, omeprazole) |
| Cardiac drugs (digoxin) |
| Nonsteroidal anti-inflammatory drugs (aspirin) |
| Corticosteroids (prednisolone) |
| Antibiotics (penicillins, cephalosporins, quinolones) |
| OTC: over the counter Source: References 2,3 |
ASSESSMENT: More problems
At admission to the medical unit, Mr. L’s temperature is 36.7°C (98°F), with a heart rate of 77 beats per minute, respiratory rate of 24 breaths per minute, and blood pressure of 164/84 mm Hg with oxygen saturation of 96% at room air. Physical exam is notable for 2+ pitting edema in the lower extremities. Mr. L is oriented to person, place, and time and is psychomotorically activated. Neurologic examination is within normal limits.
Laboratory data reveal a potassium level of 2.5 mEq/L. Other results, including complete blood count, comprehensive metabolic panel, thyroid-stimulating hormone, urinalysis, urine toxicology screen, B12, folate, venereal disease research laboratory, and ammonia are unremarkable. Chest radiography reveals an enlarged cardiomediastinum. A CT scan of the brain without contrast shows cortical volume loss and periventricular white matter disease without evidence of acute intracranial abnormality. ECG shows atrial fibrillation with a rate of 67 beats per minute.
Mr. L’s hypokalemia is corrected with potassium chloride and his hyperaldosteronism is treated with spironolactone, 25 mg/d. Physicians on the medical unit discontinue digoxin because Mr. L’s heart rate is controlled with atenolol and he is anticoagulated with warfarin.
Mr. L continues to be depressed and irritable with delusional jealousy. Mirtazapine is continued at 30 mg/d at bedtime. Aripiprazole and trazodone are discontinued and Mr. L is started on olanzapine, 10 mg/d, and haloperidol, 1 mg 4 times a day as needed for agitation. He requires multiple “as needed” haloperidol doses because of intermittent episodes of agitation. Mr. L is then transferred to the inpatient psychiatric unit for continued evaluation and treatment.
The authors’ observations
The fact that Mr. L is alert and oriented is encouraging; however, it does not rule out delirium because this condition is characterized by fluctuating levels of consciousness. Therefore, it is important to reassess him over time and perform a more thorough evaluation of cognitive function, especially attention and concentration, in addition to alertness and orientation. Psychomotor activation could suggest agitated depression, anxiety, mania, psychosis, substance intoxication, akathisia from antipsychotics, or delirium (Table 2).4 Initial evaluation— especially in older patients—should include a thorough history (including collateral sources) and be guided by the clinical presentation and physical examination, taking into consideration life-threatening conditions and common causes of mental status change such as infections, hypoxia, substance or medication effects, acute coronary syndromes, acute neurologic events, and metabolic conditions.
Table 2
DSM-IV-TR criteria for delirium caused by a medical condition
| A. Disturbance of consciousness (ie, reduced clarity of awareness of the environment) with reduced ability to focus, sustain, or shift attention |
| B. A change in cognition (such as memory deficit, disorientation, language disturbance) or the development of a perceptual disturbance that is not better accounted for by a preexisting, established, or evolving dementia |
| C. The disturbance develops over a short period of time (usually hours to days) and tends to fluctuate during the course of the day |
| D. There is evidence from the history, physical examination, or laboratory findings that the disturbance is caused by the direct physiological consequences of a general medical condition |
| Source: Reference 4 |
Reconsider the diagnosis
Even after being treated for hyperaldosteronism and discontinuing unnecessary medications, Mr. L continued to be treated for MDD with psychotic features despite intermittent confusion and agitation. At this point, it might have been useful to reconsider whether MDD with psychotic features was the most appropriate diagnosis to explain his mental status changes.
Mental status changes caused by medical disorders or medications do not immediately clear after the medical disorder is corrected or the medication is discontinued; it could take days or weeks for a patient to return to baseline. In Mr. L’s case it may be useful to simplify his medication regimen because polypharmacy contributes to delirium. Finally, olanzapine could worsen his condition because of its anticholinergic effects.5
EVALUATION: Poor cognitive status
Mental status examination upon admission to the psychiatric unit reveals a poorly cooperative patient with irritable mood and affect with slowed psychomotor activity. Mr. L’s thought process is organized with normal associations and thought content does not reveal suicidality or homicidality. However, he verbalizes delusions about his wife having an affair with a neighbor. He is partially oriented to time but believes he is in Germany. His insight is limited and he demonstrates impaired attention and concentration. We cannot complete a Mini-Mental State Exam (MMSE) because Mr. L does not cooperate.
After admission, Mr. L is intermittently confused, agitated, and disoriented. Between these episodes he is pleasant, cooperative, and oriented. Jealous delusions regarding his wife continue. Olanzapine and mirtazapine are tapered and discontinued. Haloperidol dose is changed to 1 mg 3 times a day, then to 1.5 mg in the morning and 3 mg in the evening. Prednisone is tapered and discontinued.
The authors’ observations
Cognitive testing is essential for the diagnosis and treatment of patients with mental status changes and for evaluating their response to treatment. Although the MMSE is widely used, other scales—including the Confusion Assessment Method, the Organic Brain Syndrome Scale, the Memorial Delirium Assessment Scale, and the delirium severity index6—may be more sensitive for detecting delirium. All of these scales can be difficult to complete when evaluating confused and combative patients. Quick screening instruments for inattention, such as the digit span test and listing days of the week backwards, could be used as well.
HISTORY: Surgical complications
Further questioning of Mr. L’s family reveals that his behavior started to change 7 months ago; this was 1 month after undergoing hip replacement surgery, which was complicated by a surgical wound infection and worsened his medical illnesses. Within a month, Mr. L became withdrawn and appeared depressed. He was confused and intermittently disoriented to place and time. He became irritable and started reporting concerns about his wife having an affair. During this time different medications were introduced, including steroids and several antibiotics.
The authors’ observations
A thorough history from the patient and caregivers, including the time course of mental status changes, new medication use, and history of medical and psychiatric disorders—especially depression and dementia—are important to obtain, especially early in the evaluation.
Although Mr. L’s irritability, delusions, and psychomotor slowing could be signs of psychotic depression, his fluctuating mental status, disorientation, poor attention, and impaired concentration suggest delirium (Table 3).4,7 This diagnosis is supported by the fact that Mr. L’s symptoms emerged after orthopedic surgery. Delirium after orthopedic surgery is common among older patients.8 Contributing and perpetuating factors in Mr. L’s case may have included postoperative complications, hypokalemia (hyperaldosteronism), medications (prednisone, digoxin, and olanzapine), and environmental unfamiliarity during hospitalization. A delirium diagnosis should be based on a high index of suspicion and a careful clinical assessment rather than diagnostic tests.
Table 3
Deconstructing delirium
| Defining characteristics |
| Confusional state of fluctuating course |
| Acute or subacute onset |
| Inattention |
| Disorganized thinking |
| Alteration and fluctuation of level of consciousness |
| Other characteristics |
| Cognitive: Memory impairment, perseveration |
| Motor: Hyperactive, hypoactive, mixed |
| Psychiatric: Thought disorganization, mood changes, delusions, hallucinations |
| Etiologies* |
| Predisposing factors: Age, functional status (ie, immobility), nutritional status (ie, dehydration), sensory impairment, medical conditions, psychiatric conditions (ie, dementia, TBI), medications, illicit drugs |
| Precipitating factors: Acute neurologic conditions (ie, stroke), intercurrent illnesses (ie, infections, hypoxia, anemia), surgery, environmental factors (ie, ICU, restraints, pain), illicit drugs (alcohol withdrawal), medications (ie, polypharmacy, anticholinergics), sleep depravation |
| *Usually >1 etiology ICU: intensive care unit; TBI: traumatic brain injury Source: References 4,7 |
OUTCOME: Return home
Mr. L’s confusion and delusional jealousy decrease over time, as do his disorientation and inattention, as evidenced by improvement on MMSE scores. His last MMSE score is 27/30, failing mostly in attention and recall.
After sustained improvement in cognition and behavior, Mr. L is discharged home on haloperidol and the remainder of his nonpsychiatric medications with outpatient medical and psychiatric follow-up. Over several months, he continues to show improvement and haloperidol is discontinued.
The authors’ observations
Delirium treatment should focus on prompt identification and management of precipitating and contributing factors.7 Antipsychotics are considered first-line treatment for patients with delirium, agitation, or psychosis who pose a risk to themselves or others. Benzodiazepines should be avoided in older patients unless symptoms are secondary to CNS-depressant withdrawal (ie, alcohol, benzodiazepines).9
Although there are no-FDA approved medications for delirium, haloperidol has been widely studied and used for treatment of agitation and psychosis in delirium. There is no evidence that low-dose haloperidol is any less effective than olanzapine or risperidone, or is more likely to cause adverse drug effects such as extrapyramidal syndrome.10 Antipsychotic use in a confused or agitated dementia patient increases risk of mortality compared with dementia patients who do not receive antipsychotics.11 The use of typical or atypical antipsychotics for delirium should be guided by the patient’s characteristics, such as cardiovascular status and presence or absence of underlying dementia. Atypical antipsychotics should be used carefully because—as in Mr. L’s case—anticholinergic side effects of medications such as olanzapine could worsen delirium.5 Once delirium has resolved, antipsychotics should be tapered and discontinued.
Other components of delirium treatment and prevention include:
- reorientation (verbally, with clocks, calendars, etc.)
- safe ambulation
- adequate sleep, food, and fluid intake
- adaptive equipment for vision and hearing impairment
- adequate management of pain and other comorbidities.12
Related Resources
- Khan RA, Kahn D, Bourgeois JA. Delirium: sifting through the confusion. Curr Psychiatry Rep. 2009;11(3):226-234.
- Maldonado JR. Delirium in the acute care setting: characteristics, diagnosis, and treatment. Crit Care Clin. 2008;24:657-722.
- Young J, Inouye SK. Delirium in older people. BMJ. 2007;334(7598):842-846.
Drug Brand Names
- Amantadine • Symmetrel
- Amitriptyline • Elavil
- Aripiprazole • Abilify
- Atenolol • Tenormin
- Atropine • AtroPen
- Benztropine • Cogentin
- Cimetidine • Tagamet
- Clozapine • Clozaril
- Digoxin • Lanoxicaps, Lanoxin
- Glyburide • DiaBeta, Micronase
- Haloperidol • Haldol
- Levodopa/carbidopa • Parcopa, Sinemet
- Lisinopril • Prinivil, Zestril
- Lorazepam • Ativan
- Meperidine • Demerol
- Mirtazapine • Remeron
- Olanzapine • Zyprexa
- Omeprazole • Prilosec
- Oxybutynin • Ditropan
- Pergolide • Permax
- Phenytoin • Dilantin, Phenytek
- Prednisolone • Orapred, Prelone, others
- Prednisone • Deltasone, Meticorten
- Pyridostigmine • Mestinon
- Ranitidine • Zantac
- Risperidone • Risperdal
- Selegiline • Eldepryl, Zelapar
- Spironolactone • Aldactone
- Tamsulosin • Flomax
- Thioridazine • Mellaril
- Trazodone • Desyrel
- Warfarin • Coumadin
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
CASE: Agitated and paranoid
Police bring Mr. L, age 85, to the emergency department (ED) because he threatened his wife, claiming she is having an affair. Two days earlier, he was discharged from a different hospital, where he was treated for progressive and fluctuating irritability, depressed mood, confusion, disorientation, inattention, and delusional thinking that had started 4 to 5 months earlier. He has no other psychiatric history.
Mr. L has a history of atrial fibrillation, hypertension, benign prostatic hypertrophy, and noninsulin-dependent diabetes mellitus. Several months ago, he had hip surgery, which was complicated by a surgical wound infection. Medications include digoxin, 0.125 mg/d; atenolol, 100 mg/d; warfarin, 1 mg/d on Monday, Wednesday, Friday, Saturday, and Sunday and 0.5 mg/d Tuesday and Thursday; lisinopril, 40 mg/d; tamsulosin, 0.4 mg/d; and glyburide, 1.25 mg/d. During the previous hospitalization, physicians discovered he had myasthenia gravis, which they treated with prednisone and pyridostigmine. Mr. L also was diagnosed with hyperaldosteronism. An adrenal mass was found in an abdominal CT. At that time, he also was diagnosed with major depressive disorder (MDD) with psychotic features and started on aripiprazole, 10 mg/d, mirtazapine, 30 mg/d, and trazodone, 50 mg/d for sleep.
The authors’ observations
When evaluating mental status changes in older patients, consider the time course and characteristics of the changes, especially if the patient’s cognitive function changes. Acute mental status changes that occur over hours to days often represent delirium caused by a medical condition such as a coronary event or infection. Changes that develop over weeks to months often signal a primary psychiatric disorder such as depression, mania, or dementia. Mr. L’s mood and psychotic symptoms developed over 4 to 5 months and were thought to be a result of MDD with psychotic features. However, his fluctuating cognitive symptoms, confusion, and lack of psychiatric history suggest that the differential diagnosis should include a cognitive disorder such as delirium or dementia. The hypoactive form of delirium often is unrecognized or misdiagnosed as sedation or depression, particularly in older patients.1
Multiple medical conditions and polypharmacy are important factors to consider when evaluating mental status changes in geriatric patients. In Mr. L’s case, atrial fibrillation, hypertension, and diabetes increase his risk of an acute cardiovascular or cerebrovascular event and chronic cerebrovascular disease. Hyperaldosteronism can lead to electrolyte abnormalities that may produce mental status changes. Treatment with an oral hypoglycemic raises the possibility that hypoglycemia is contributing to his mental status changes. Prednisone can cause psychosis, anxiety, and mania. Digoxin toxicity is associated with psychosis and irritability. Pyridostigmine also has been reported to cause psychosis. Use of an antidepressant, such as mirtazapine, could have exacerbated an underlying undiagnosed bipolar disorder. Antipsychotics, such as aripiprazole, may cause akathisia or activation. Substance intoxication or withdrawal should not be excluded solely because a patient is older. In older patients, medications with anti-cholinergic effects are common culprits for cognitive impairment (Table 1).2,3
Table 1
Medications that could contribute to mental status changes
| Anticholinergics (atropine, benztropine, oxybutynin, some OTC medications) |
| Hypnotics/sedatives (benzodiazepines) |
| Opiate analgesics (meperidine) |
| Neuroleptics (clozapine, thioridazine, olanzapine) |
| Antiparkinsonian medications (levodopa, selegiline, pergolide, amantadine) |
| Antidepressants (amitriptyline) |
| Anticonvulsants (phenytoin) |
| Histamine H2 receptor antagonists (ranitidine, cimetidine, omeprazole) |
| Cardiac drugs (digoxin) |
| Nonsteroidal anti-inflammatory drugs (aspirin) |
| Corticosteroids (prednisolone) |
| Antibiotics (penicillins, cephalosporins, quinolones) |
| OTC: over the counter Source: References 2,3 |
ASSESSMENT: More problems
At admission to the medical unit, Mr. L’s temperature is 36.7°C (98°F), with a heart rate of 77 beats per minute, respiratory rate of 24 breaths per minute, and blood pressure of 164/84 mm Hg with oxygen saturation of 96% at room air. Physical exam is notable for 2+ pitting edema in the lower extremities. Mr. L is oriented to person, place, and time and is psychomotorically activated. Neurologic examination is within normal limits.
Laboratory data reveal a potassium level of 2.5 mEq/L. Other results, including complete blood count, comprehensive metabolic panel, thyroid-stimulating hormone, urinalysis, urine toxicology screen, B12, folate, venereal disease research laboratory, and ammonia are unremarkable. Chest radiography reveals an enlarged cardiomediastinum. A CT scan of the brain without contrast shows cortical volume loss and periventricular white matter disease without evidence of acute intracranial abnormality. ECG shows atrial fibrillation with a rate of 67 beats per minute.
Mr. L’s hypokalemia is corrected with potassium chloride and his hyperaldosteronism is treated with spironolactone, 25 mg/d. Physicians on the medical unit discontinue digoxin because Mr. L’s heart rate is controlled with atenolol and he is anticoagulated with warfarin.
Mr. L continues to be depressed and irritable with delusional jealousy. Mirtazapine is continued at 30 mg/d at bedtime. Aripiprazole and trazodone are discontinued and Mr. L is started on olanzapine, 10 mg/d, and haloperidol, 1 mg 4 times a day as needed for agitation. He requires multiple “as needed” haloperidol doses because of intermittent episodes of agitation. Mr. L is then transferred to the inpatient psychiatric unit for continued evaluation and treatment.
The authors’ observations
The fact that Mr. L is alert and oriented is encouraging; however, it does not rule out delirium because this condition is characterized by fluctuating levels of consciousness. Therefore, it is important to reassess him over time and perform a more thorough evaluation of cognitive function, especially attention and concentration, in addition to alertness and orientation. Psychomotor activation could suggest agitated depression, anxiety, mania, psychosis, substance intoxication, akathisia from antipsychotics, or delirium (Table 2).4 Initial evaluation— especially in older patients—should include a thorough history (including collateral sources) and be guided by the clinical presentation and physical examination, taking into consideration life-threatening conditions and common causes of mental status change such as infections, hypoxia, substance or medication effects, acute coronary syndromes, acute neurologic events, and metabolic conditions.
Table 2
DSM-IV-TR criteria for delirium caused by a medical condition
| A. Disturbance of consciousness (ie, reduced clarity of awareness of the environment) with reduced ability to focus, sustain, or shift attention |
| B. A change in cognition (such as memory deficit, disorientation, language disturbance) or the development of a perceptual disturbance that is not better accounted for by a preexisting, established, or evolving dementia |
| C. The disturbance develops over a short period of time (usually hours to days) and tends to fluctuate during the course of the day |
| D. There is evidence from the history, physical examination, or laboratory findings that the disturbance is caused by the direct physiological consequences of a general medical condition |
| Source: Reference 4 |
Reconsider the diagnosis
Even after being treated for hyperaldosteronism and discontinuing unnecessary medications, Mr. L continued to be treated for MDD with psychotic features despite intermittent confusion and agitation. At this point, it might have been useful to reconsider whether MDD with psychotic features was the most appropriate diagnosis to explain his mental status changes.
Mental status changes caused by medical disorders or medications do not immediately clear after the medical disorder is corrected or the medication is discontinued; it could take days or weeks for a patient to return to baseline. In Mr. L’s case it may be useful to simplify his medication regimen because polypharmacy contributes to delirium. Finally, olanzapine could worsen his condition because of its anticholinergic effects.5
EVALUATION: Poor cognitive status
Mental status examination upon admission to the psychiatric unit reveals a poorly cooperative patient with irritable mood and affect with slowed psychomotor activity. Mr. L’s thought process is organized with normal associations and thought content does not reveal suicidality or homicidality. However, he verbalizes delusions about his wife having an affair with a neighbor. He is partially oriented to time but believes he is in Germany. His insight is limited and he demonstrates impaired attention and concentration. We cannot complete a Mini-Mental State Exam (MMSE) because Mr. L does not cooperate.
After admission, Mr. L is intermittently confused, agitated, and disoriented. Between these episodes he is pleasant, cooperative, and oriented. Jealous delusions regarding his wife continue. Olanzapine and mirtazapine are tapered and discontinued. Haloperidol dose is changed to 1 mg 3 times a day, then to 1.5 mg in the morning and 3 mg in the evening. Prednisone is tapered and discontinued.
The authors’ observations
Cognitive testing is essential for the diagnosis and treatment of patients with mental status changes and for evaluating their response to treatment. Although the MMSE is widely used, other scales—including the Confusion Assessment Method, the Organic Brain Syndrome Scale, the Memorial Delirium Assessment Scale, and the delirium severity index6—may be more sensitive for detecting delirium. All of these scales can be difficult to complete when evaluating confused and combative patients. Quick screening instruments for inattention, such as the digit span test and listing days of the week backwards, could be used as well.
HISTORY: Surgical complications
Further questioning of Mr. L’s family reveals that his behavior started to change 7 months ago; this was 1 month after undergoing hip replacement surgery, which was complicated by a surgical wound infection and worsened his medical illnesses. Within a month, Mr. L became withdrawn and appeared depressed. He was confused and intermittently disoriented to place and time. He became irritable and started reporting concerns about his wife having an affair. During this time different medications were introduced, including steroids and several antibiotics.
The authors’ observations
A thorough history from the patient and caregivers, including the time course of mental status changes, new medication use, and history of medical and psychiatric disorders—especially depression and dementia—are important to obtain, especially early in the evaluation.
Although Mr. L’s irritability, delusions, and psychomotor slowing could be signs of psychotic depression, his fluctuating mental status, disorientation, poor attention, and impaired concentration suggest delirium (Table 3).4,7 This diagnosis is supported by the fact that Mr. L’s symptoms emerged after orthopedic surgery. Delirium after orthopedic surgery is common among older patients.8 Contributing and perpetuating factors in Mr. L’s case may have included postoperative complications, hypokalemia (hyperaldosteronism), medications (prednisone, digoxin, and olanzapine), and environmental unfamiliarity during hospitalization. A delirium diagnosis should be based on a high index of suspicion and a careful clinical assessment rather than diagnostic tests.
Table 3
Deconstructing delirium
| Defining characteristics |
| Confusional state of fluctuating course |
| Acute or subacute onset |
| Inattention |
| Disorganized thinking |
| Alteration and fluctuation of level of consciousness |
| Other characteristics |
| Cognitive: Memory impairment, perseveration |
| Motor: Hyperactive, hypoactive, mixed |
| Psychiatric: Thought disorganization, mood changes, delusions, hallucinations |
| Etiologies* |
| Predisposing factors: Age, functional status (ie, immobility), nutritional status (ie, dehydration), sensory impairment, medical conditions, psychiatric conditions (ie, dementia, TBI), medications, illicit drugs |
| Precipitating factors: Acute neurologic conditions (ie, stroke), intercurrent illnesses (ie, infections, hypoxia, anemia), surgery, environmental factors (ie, ICU, restraints, pain), illicit drugs (alcohol withdrawal), medications (ie, polypharmacy, anticholinergics), sleep depravation |
| *Usually >1 etiology ICU: intensive care unit; TBI: traumatic brain injury Source: References 4,7 |
OUTCOME: Return home
Mr. L’s confusion and delusional jealousy decrease over time, as do his disorientation and inattention, as evidenced by improvement on MMSE scores. His last MMSE score is 27/30, failing mostly in attention and recall.
After sustained improvement in cognition and behavior, Mr. L is discharged home on haloperidol and the remainder of his nonpsychiatric medications with outpatient medical and psychiatric follow-up. Over several months, he continues to show improvement and haloperidol is discontinued.
The authors’ observations
Delirium treatment should focus on prompt identification and management of precipitating and contributing factors.7 Antipsychotics are considered first-line treatment for patients with delirium, agitation, or psychosis who pose a risk to themselves or others. Benzodiazepines should be avoided in older patients unless symptoms are secondary to CNS-depressant withdrawal (ie, alcohol, benzodiazepines).9
Although there are no-FDA approved medications for delirium, haloperidol has been widely studied and used for treatment of agitation and psychosis in delirium. There is no evidence that low-dose haloperidol is any less effective than olanzapine or risperidone, or is more likely to cause adverse drug effects such as extrapyramidal syndrome.10 Antipsychotic use in a confused or agitated dementia patient increases risk of mortality compared with dementia patients who do not receive antipsychotics.11 The use of typical or atypical antipsychotics for delirium should be guided by the patient’s characteristics, such as cardiovascular status and presence or absence of underlying dementia. Atypical antipsychotics should be used carefully because—as in Mr. L’s case—anticholinergic side effects of medications such as olanzapine could worsen delirium.5 Once delirium has resolved, antipsychotics should be tapered and discontinued.
Other components of delirium treatment and prevention include:
- reorientation (verbally, with clocks, calendars, etc.)
- safe ambulation
- adequate sleep, food, and fluid intake
- adaptive equipment for vision and hearing impairment
- adequate management of pain and other comorbidities.12
Related Resources
- Khan RA, Kahn D, Bourgeois JA. Delirium: sifting through the confusion. Curr Psychiatry Rep. 2009;11(3):226-234.
- Maldonado JR. Delirium in the acute care setting: characteristics, diagnosis, and treatment. Crit Care Clin. 2008;24:657-722.
- Young J, Inouye SK. Delirium in older people. BMJ. 2007;334(7598):842-846.
Drug Brand Names
- Amantadine • Symmetrel
- Amitriptyline • Elavil
- Aripiprazole • Abilify
- Atenolol • Tenormin
- Atropine • AtroPen
- Benztropine • Cogentin
- Cimetidine • Tagamet
- Clozapine • Clozaril
- Digoxin • Lanoxicaps, Lanoxin
- Glyburide • DiaBeta, Micronase
- Haloperidol • Haldol
- Levodopa/carbidopa • Parcopa, Sinemet
- Lisinopril • Prinivil, Zestril
- Lorazepam • Ativan
- Meperidine • Demerol
- Mirtazapine • Remeron
- Olanzapine • Zyprexa
- Omeprazole • Prilosec
- Oxybutynin • Ditropan
- Pergolide • Permax
- Phenytoin • Dilantin, Phenytek
- Prednisolone • Orapred, Prelone, others
- Prednisone • Deltasone, Meticorten
- Pyridostigmine • Mestinon
- Ranitidine • Zantac
- Risperidone • Risperdal
- Selegiline • Eldepryl, Zelapar
- Spironolactone • Aldactone
- Tamsulosin • Flomax
- Thioridazine • Mellaril
- Trazodone • Desyrel
- Warfarin • Coumadin
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. McAvay GJ, Van Ness PH, Bogardus ST, Jr, et al. Depressive symptoms and the risk of incident delirium in older hospitalized adults. J Am Geriatr Soc. 2007;55:684-691.
2. Mintzer J, Burns A. Anticholinergic side-effects of drugs in elderly people. J R Soc Med. 2000;93(9):457-462.
3. Moore AR, O’Keeffe ST. Drug-induced cognitive impairment in the elderly. Drugs Aging. 1999;15(1):15-28.
4. Diagnostic and statistical manual of mental disorders. 4th ed text rev. Washington, DC: American Psychiatric Association; 2000.
5. Lim CJ, Trevino C, Tampi RR. Can olanzapine cause delirium in the elderly? Ann Pharmacother. 2006;40(1):135-138.
6. Woodford HJ, George J. Cognitive assessment in the elderly: a review of clinical methods. QJM. 2007;100:469-484.
7. Young J, Inouye SK. Delirium in older people. BMJ. 2007;334(7598):842-846.
8. Bruce AJ, Ritchie CW, Blizard R, et al. The incidence of delirium following orthopedic surgery: a meta-analytic review. Int Psychogeriatr. 2007;19(2):197-214.
9. Attard A, Ranjith G, Taylor D. Delirium and its treatment. CNS Drugs. 2008;22:631-644.
10. Lonergan E, Britton AM, Luxenberg J, et al. Antipsychotics for delirium. Cochrane Database Syst Rev. 2007;(2):CD005594.-
11. Schneider LS, Dagerman KS, Insel P. Risk of death with atypical antipsychotic drug treatment for dementia: meta-analysis of randomized placebo-controlled trials. JAMA. 2005;294(15):1934-1943.
12. Tabet N, Howard R. Non-pharmacological interventions in the prevention of delirium. Age Ageing. 2009;38(4):374-379.
1. McAvay GJ, Van Ness PH, Bogardus ST, Jr, et al. Depressive symptoms and the risk of incident delirium in older hospitalized adults. J Am Geriatr Soc. 2007;55:684-691.
2. Mintzer J, Burns A. Anticholinergic side-effects of drugs in elderly people. J R Soc Med. 2000;93(9):457-462.
3. Moore AR, O’Keeffe ST. Drug-induced cognitive impairment in the elderly. Drugs Aging. 1999;15(1):15-28.
4. Diagnostic and statistical manual of mental disorders. 4th ed text rev. Washington, DC: American Psychiatric Association; 2000.
5. Lim CJ, Trevino C, Tampi RR. Can olanzapine cause delirium in the elderly? Ann Pharmacother. 2006;40(1):135-138.
6. Woodford HJ, George J. Cognitive assessment in the elderly: a review of clinical methods. QJM. 2007;100:469-484.
7. Young J, Inouye SK. Delirium in older people. BMJ. 2007;334(7598):842-846.
8. Bruce AJ, Ritchie CW, Blizard R, et al. The incidence of delirium following orthopedic surgery: a meta-analytic review. Int Psychogeriatr. 2007;19(2):197-214.
9. Attard A, Ranjith G, Taylor D. Delirium and its treatment. CNS Drugs. 2008;22:631-644.
10. Lonergan E, Britton AM, Luxenberg J, et al. Antipsychotics for delirium. Cochrane Database Syst Rev. 2007;(2):CD005594.-
11. Schneider LS, Dagerman KS, Insel P. Risk of death with atypical antipsychotic drug treatment for dementia: meta-analysis of randomized placebo-controlled trials. JAMA. 2005;294(15):1934-1943.
12. Tabet N, Howard R. Non-pharmacological interventions in the prevention of delirium. Age Ageing. 2009;38(4):374-379.
Abuse of second-generation antipsychotics: What prescribers need to know
• Antipsychotics have been abused and misused by inpatients and outpatients.
• Most published case reports of antipsychotic abuse involve quetiapine, although some describe misuse of other agents, including olanzapine.
• Serotonin, histamine, and α-adrenergic neurotransmitter systems may play a role in second-generation antipsychotics’ abuse potential.
• Although individuals have misused quetiapine and olanzapine, evidence indicates that these drugs may be effective for treating substance use disorders.
Mr. Z, age 27, seeks treatment for substance abuse at a mental health clinic. He has a 7-year substance use history and his last urine drug screen 1 month ago was positive for marijuana, opiates, and benzodiazepines. Mr. Z reveals that he purchases prescription drugs on the street, including hydrocodone, diazepam, and quetiapine. He states that when he takes a 100-mg dose of quetiapine, he feels happy, relaxed, and “drunk without the mind-numbing effects that you get with alcohol.” Mr. Z often takes quetiapine while smoking marijuana. He sleeps well with this and does not experience a hangover effect.
Although clinicians always are vigilant about patients’ misuse of psychoactive substances, recent case reports have described abuse of antipsychotics, particularly second-generation antipsychotics (SGAs). A PubMed and PsycINFO literature search revealed several case reports of quetiapine abuse (Table)1-6 and 2 case reports of olanzapine misuse.
Table
Case reports of quetiapine abuse
| Reference | Patient | Setting | Description of abuse |
|---|---|---|---|
| Hussain et al, 20051 | Woman, age 34, with history of polysubstance abuse, depression, and borderline personality traits | Prison | Crushed tablets dissolved in water and injected intravenously |
| Morin, 20072 | Woman, age 28, with history of schizoaffective disorder, polysubstance abuse, and personality disorder not otherwise specified | Hospital | Tablets crushed with aspirin and inhaled intranasally |
| Waters et al, 20073 | Man, age 33, with history of polysubstance abuse | Outpatient | Crushed tablets dissolved in water and injected intravenously |
| Reeves et al, 20074 | Man, age 49, with history of alcohol dependence and benzodiazepine abuse | Outpatient | Misuse without psychiatric symptoms or a diagnosed psychiatric disorder |
| Man, age 23, with history of benzodiazepine dependence | Outpatient | Misuse without psychiatric symptoms or a diagnosed psychiatric disorder | |
| Man, age 39, with history of bipolar disorder | Outpatient | Oral use in doses more than the prescribed amount | |
| Murphy et al, 20085 | Man, age 29, with unclear history of schizophrenia | Psychiatric walk-in clinic | Malingering psychiatric symptoms to obtain an oral dose and overnight stay |
| Fischer et al, 20096 | Man, age 53, with history of depressive symptoms | Court-mandated outpatient clinic | Malingering psychiatric symptoms to obtain higher oral doses |
Quetiapine
Methods of quetiapine misuse include ingesting pills, inhaling crushed tablets, and injecting a solution of dissolved tablets.1-7 In case studies, patients report abusing quetiapine for its sedative, anxiolytic, and calming effects.1,2,4-7 One patient reported snorting crushed quetiapine tablets combined with cocaine for “hallucinogenic” effects.3 Street names for quetiapine include “quell,” “Susie-Q,” and “baby heroin,” and “Q-ball” refers to a combination of cocaine and quetiapine.8 Quetiapine tablets have a street value of $3 to $8 for doses ranging from 25 mg to 100 mg.9 Although outpatient misuse of quetiapine is common, abuse in correctional settings also is becoming more frequent.10 Residents of jails and prisons misuse quetiapine for reasons similar to those cited by outpatients: sedation, relief of anxiety, and hallucinogenic effects or “getting high.”1,2,10 Clinicians must differentiate inmates who have legitimate psychiatric symptoms that require antipsychotic treatment from those who are malingering to obtain the drug. Efforts to treat inmates for substance use disorders may be thwarted by the easy availability of drugs in correctional settings.10
Other SGAs
The incidence of misuse of olanzapine and other SGAs is more difficult to ascertain. Only 2 case reports describe olanzapine abuse, both in outpatient settings. One describes a patient treated for depression with psychosis who was using increasingly higher doses of olanzapine to obtain euphoric effects.11 Switching to aripiprazole effectively treated her illness and addressed her olanzapine misuse.
In the other case, a patient with bipolar disorder was able to obtain olanzapine, 40 mg/d, by complaining of worsened manic symptoms.12 He described the experience of misusing olanzapine as getting a “buzz,” feeling “very relaxed,” and blunting the negative jitteriness he felt when he used cocaine.12 This patient stated that he had observed others abusing olanzapine, both orally and intravenously.
Although the literature lacks reports on the risks of antipsychotic abuse, numerous Web sites purport to sell these drugs without a prescription and some describe the experience of illicit use of drugs such as haloperidol, risperidone, quetiapine, and olanzapine and ways to “enhance” the experience by combining drugs.13 Reported experiences with risperidone tend to be negative, citing extrapyramidal side effects and feeling “numb,” whereas olanzapine and quetiapine users describe feeling “drunk without the bad effects of alcohol” and “really happy, calm.” These sites also describe hallucinogenic effects of these agents.13
Mechanism of action
The neuropharmacologic reasons for antipsychotics’ abuse potential are difficult to quantify. Quetiapine and olanzapine have been used to treat cocaine and alcohol abuse, and work perhaps by decreasing the dopamine reward system response to substance use.14,15 Quetiapine’s rapid dissociation from the dopamine receptor has been theorized to contribute to the drug’s abuse potential, possibly through relatively lower potency and decreased residence time at the dopamine receptor.14-16 This mechanism also contributes to quetiapine’s lower risk of extrapyramidal side effects, which make the drug easier to tolerate.
Although dopamine is a factor in substance abuse and treatment of psychotic disorders, other neuropharmacologic mechanisms must be considered. SGAs are theorized to cause dopamine release in the frontal cortex through effects as 5-HT1A agonists and 5-HT2A antagonists.16 Antagonism of α-adrenergic and histaminic receptors may account for these agents’ anxiolytic and sedative properties.8
Misuse of anticholinergic agents has been reported for >50 years.17 Psychiatric patients have been reported to increase use of anticholinergics for their movement side effects as well as hallucinogenic effects.18
Treatment
Regardless of the substance that patients abuse, the treatment goals are the same: to reduce use and achieve recovery. If a patient with psychosis is abusing an SGA, consider switching to an antipsychotic with less abuse potential. Another option is to limit the supply of the abused drug by prescribing smaller quantities or increase the frequency of follow-up visits to ensure compliant use.
Related Resources
- Substance Abuse and Mental Health Services Administration. www.samhsa.gov.
- Galanter M, Kelber HD. The American Psychiatric Publishing textbook of substance abuse treatment. Arlington, VA: American Psychiatric Publishing, Inc; 2008.
Drug Brand Names
- Aripiprazole • Abilify
- Diazepam • Valium
- Haloperidol • Haldol
- Hydrocodone/acetaminophen • Vicodin
- Olanzapine • Zyprexa
- Quetiapine • Seroquel
- Risperidone • Risperdal
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Hussain MZ, Waheed W, Hussain S. Intravenous quetiapine abuse. Am J Psychiatry. 2005;162:1755-1756.
2. Morin AK. Possible intranasal quetiapine misuse. Am J Health Syst Pharm. 2007;64:723-725.
3. Waters BM, Joshi KG. Intravenous quetiapine-cocaine use (“Q-ball”). Am J Psychiatry. 2007;164:1.-
4. Reeves RR, Brister JC. Additional evidence of the abuse potential of quetiapine. S Med J. 2007;100:834-836.
5. Murphy D, Bailey K, Stone M, et al. Addictive potential of quetiapine. Am J Psychiatry. 2008;165:7.-
6. Fischer BA, Boggs DL. The role of antihistaminic effects in the misuse of quetiapine: a case report and review of the literature. Neurosci Biobehav Rev. 2009;34:555-558.
7. Pierre JM, Shnayder I, Wirshing DA, et al. Intranasal quetiapine abuse. Am J Psychiatry. 2004;161(9):1718.-
8. Sansone RA, Sansone LA. Is seroquel developing an illicit reputation for misuse/abuse? Psychiatry (Edgemont). 2010;7(1):13-16.
9. Tarasoff G, Osti K. Black-market value of antipsychotics antidepressants, and hypnotics in Las Vegas, Nevada. Am J Psychiatry. 2007;164:350.-
10. Keltner NL, Vance DE. Biological perspectives: incarcerated care and quetiapine abuse. Perspect Psychiatr Care. 2008;44(3):202-206.
11. Lai CH. Olanzapine abuse was relieved after switching to aripiprazole in a patient with psychotic depression. Prog Neuropsychopharmacol Biol Psychiatry. 2010;34(7):1363-1364.
12. Reeves RR. Abuse of olanzapine by substance abusers. J Psychoactive Drugs. 2007;39(3):297-299.
13. The Vaults of Erowid. Available at: . Accessed April 1 2011.
14. Hanley NJ, Kenna GA. Quetiapine: treatment for substance abuse and drug of abuse. Am J Health Syst Pharm. 2008;65:611-618.
15. Tcheremissine OV. Is quetiapine a drug of abuse? Reexamining the issue of addiction. Expert Opin Drug Saf. 2008;7:739-748.
16. Kuroki T, Nagao N, Nakahara T. Neuropharmacology of second-generation antipsychotic drugs: a validity of the serotonin-dopamine hypothesis. Prog Brain Res. 2008;172:199-212.
17. Smith JM. Abuse of the antiparkinson drugs: a review of the literature. J Clin Psychiatry. 1980;41(10):351-354.
18. Land W, Pinsky D, Salzman C. Abuse and misuse of anticholinergic medications. Hosp Community Psychiatry. 1991;42:580-581.
Gregory T. Bogart, PharmD
Dr. Bogart is a Second-Year Psychiatric Pharmacy Resident
Carol A. Ott, PharmD, BCPP
Dr. Ott is a Clinical Assistant Professor of Pharmacy Practice, Purdue University College of Pharmacy, Indianapolis, IN.
Vicki L. Ellingrod, PharmD, BCPP, FCCP
Series Editor
Gregory T. Bogart, PharmD
Dr. Bogart is a Second-Year Psychiatric Pharmacy Resident
Carol A. Ott, PharmD, BCPP
Dr. Ott is a Clinical Assistant Professor of Pharmacy Practice, Purdue University College of Pharmacy, Indianapolis, IN.
Vicki L. Ellingrod, PharmD, BCPP, FCCP
Series Editor
Gregory T. Bogart, PharmD
Dr. Bogart is a Second-Year Psychiatric Pharmacy Resident
Carol A. Ott, PharmD, BCPP
Dr. Ott is a Clinical Assistant Professor of Pharmacy Practice, Purdue University College of Pharmacy, Indianapolis, IN.
Vicki L. Ellingrod, PharmD, BCPP, FCCP
Series Editor
• Antipsychotics have been abused and misused by inpatients and outpatients.
• Most published case reports of antipsychotic abuse involve quetiapine, although some describe misuse of other agents, including olanzapine.
• Serotonin, histamine, and α-adrenergic neurotransmitter systems may play a role in second-generation antipsychotics’ abuse potential.
• Although individuals have misused quetiapine and olanzapine, evidence indicates that these drugs may be effective for treating substance use disorders.
Mr. Z, age 27, seeks treatment for substance abuse at a mental health clinic. He has a 7-year substance use history and his last urine drug screen 1 month ago was positive for marijuana, opiates, and benzodiazepines. Mr. Z reveals that he purchases prescription drugs on the street, including hydrocodone, diazepam, and quetiapine. He states that when he takes a 100-mg dose of quetiapine, he feels happy, relaxed, and “drunk without the mind-numbing effects that you get with alcohol.” Mr. Z often takes quetiapine while smoking marijuana. He sleeps well with this and does not experience a hangover effect.
Although clinicians always are vigilant about patients’ misuse of psychoactive substances, recent case reports have described abuse of antipsychotics, particularly second-generation antipsychotics (SGAs). A PubMed and PsycINFO literature search revealed several case reports of quetiapine abuse (Table)1-6 and 2 case reports of olanzapine misuse.
Table
Case reports of quetiapine abuse
| Reference | Patient | Setting | Description of abuse |
|---|---|---|---|
| Hussain et al, 20051 | Woman, age 34, with history of polysubstance abuse, depression, and borderline personality traits | Prison | Crushed tablets dissolved in water and injected intravenously |
| Morin, 20072 | Woman, age 28, with history of schizoaffective disorder, polysubstance abuse, and personality disorder not otherwise specified | Hospital | Tablets crushed with aspirin and inhaled intranasally |
| Waters et al, 20073 | Man, age 33, with history of polysubstance abuse | Outpatient | Crushed tablets dissolved in water and injected intravenously |
| Reeves et al, 20074 | Man, age 49, with history of alcohol dependence and benzodiazepine abuse | Outpatient | Misuse without psychiatric symptoms or a diagnosed psychiatric disorder |
| Man, age 23, with history of benzodiazepine dependence | Outpatient | Misuse without psychiatric symptoms or a diagnosed psychiatric disorder | |
| Man, age 39, with history of bipolar disorder | Outpatient | Oral use in doses more than the prescribed amount | |
| Murphy et al, 20085 | Man, age 29, with unclear history of schizophrenia | Psychiatric walk-in clinic | Malingering psychiatric symptoms to obtain an oral dose and overnight stay |
| Fischer et al, 20096 | Man, age 53, with history of depressive symptoms | Court-mandated outpatient clinic | Malingering psychiatric symptoms to obtain higher oral doses |
Quetiapine
Methods of quetiapine misuse include ingesting pills, inhaling crushed tablets, and injecting a solution of dissolved tablets.1-7 In case studies, patients report abusing quetiapine for its sedative, anxiolytic, and calming effects.1,2,4-7 One patient reported snorting crushed quetiapine tablets combined with cocaine for “hallucinogenic” effects.3 Street names for quetiapine include “quell,” “Susie-Q,” and “baby heroin,” and “Q-ball” refers to a combination of cocaine and quetiapine.8 Quetiapine tablets have a street value of $3 to $8 for doses ranging from 25 mg to 100 mg.9 Although outpatient misuse of quetiapine is common, abuse in correctional settings also is becoming more frequent.10 Residents of jails and prisons misuse quetiapine for reasons similar to those cited by outpatients: sedation, relief of anxiety, and hallucinogenic effects or “getting high.”1,2,10 Clinicians must differentiate inmates who have legitimate psychiatric symptoms that require antipsychotic treatment from those who are malingering to obtain the drug. Efforts to treat inmates for substance use disorders may be thwarted by the easy availability of drugs in correctional settings.10
Other SGAs
The incidence of misuse of olanzapine and other SGAs is more difficult to ascertain. Only 2 case reports describe olanzapine abuse, both in outpatient settings. One describes a patient treated for depression with psychosis who was using increasingly higher doses of olanzapine to obtain euphoric effects.11 Switching to aripiprazole effectively treated her illness and addressed her olanzapine misuse.
In the other case, a patient with bipolar disorder was able to obtain olanzapine, 40 mg/d, by complaining of worsened manic symptoms.12 He described the experience of misusing olanzapine as getting a “buzz,” feeling “very relaxed,” and blunting the negative jitteriness he felt when he used cocaine.12 This patient stated that he had observed others abusing olanzapine, both orally and intravenously.
Although the literature lacks reports on the risks of antipsychotic abuse, numerous Web sites purport to sell these drugs without a prescription and some describe the experience of illicit use of drugs such as haloperidol, risperidone, quetiapine, and olanzapine and ways to “enhance” the experience by combining drugs.13 Reported experiences with risperidone tend to be negative, citing extrapyramidal side effects and feeling “numb,” whereas olanzapine and quetiapine users describe feeling “drunk without the bad effects of alcohol” and “really happy, calm.” These sites also describe hallucinogenic effects of these agents.13
Mechanism of action
The neuropharmacologic reasons for antipsychotics’ abuse potential are difficult to quantify. Quetiapine and olanzapine have been used to treat cocaine and alcohol abuse, and work perhaps by decreasing the dopamine reward system response to substance use.14,15 Quetiapine’s rapid dissociation from the dopamine receptor has been theorized to contribute to the drug’s abuse potential, possibly through relatively lower potency and decreased residence time at the dopamine receptor.14-16 This mechanism also contributes to quetiapine’s lower risk of extrapyramidal side effects, which make the drug easier to tolerate.
Although dopamine is a factor in substance abuse and treatment of psychotic disorders, other neuropharmacologic mechanisms must be considered. SGAs are theorized to cause dopamine release in the frontal cortex through effects as 5-HT1A agonists and 5-HT2A antagonists.16 Antagonism of α-adrenergic and histaminic receptors may account for these agents’ anxiolytic and sedative properties.8
Misuse of anticholinergic agents has been reported for >50 years.17 Psychiatric patients have been reported to increase use of anticholinergics for their movement side effects as well as hallucinogenic effects.18
Treatment
Regardless of the substance that patients abuse, the treatment goals are the same: to reduce use and achieve recovery. If a patient with psychosis is abusing an SGA, consider switching to an antipsychotic with less abuse potential. Another option is to limit the supply of the abused drug by prescribing smaller quantities or increase the frequency of follow-up visits to ensure compliant use.
Related Resources
- Substance Abuse and Mental Health Services Administration. www.samhsa.gov.
- Galanter M, Kelber HD. The American Psychiatric Publishing textbook of substance abuse treatment. Arlington, VA: American Psychiatric Publishing, Inc; 2008.
Drug Brand Names
- Aripiprazole • Abilify
- Diazepam • Valium
- Haloperidol • Haldol
- Hydrocodone/acetaminophen • Vicodin
- Olanzapine • Zyprexa
- Quetiapine • Seroquel
- Risperidone • Risperdal
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
• Antipsychotics have been abused and misused by inpatients and outpatients.
• Most published case reports of antipsychotic abuse involve quetiapine, although some describe misuse of other agents, including olanzapine.
• Serotonin, histamine, and α-adrenergic neurotransmitter systems may play a role in second-generation antipsychotics’ abuse potential.
• Although individuals have misused quetiapine and olanzapine, evidence indicates that these drugs may be effective for treating substance use disorders.
Mr. Z, age 27, seeks treatment for substance abuse at a mental health clinic. He has a 7-year substance use history and his last urine drug screen 1 month ago was positive for marijuana, opiates, and benzodiazepines. Mr. Z reveals that he purchases prescription drugs on the street, including hydrocodone, diazepam, and quetiapine. He states that when he takes a 100-mg dose of quetiapine, he feels happy, relaxed, and “drunk without the mind-numbing effects that you get with alcohol.” Mr. Z often takes quetiapine while smoking marijuana. He sleeps well with this and does not experience a hangover effect.
Although clinicians always are vigilant about patients’ misuse of psychoactive substances, recent case reports have described abuse of antipsychotics, particularly second-generation antipsychotics (SGAs). A PubMed and PsycINFO literature search revealed several case reports of quetiapine abuse (Table)1-6 and 2 case reports of olanzapine misuse.
Table
Case reports of quetiapine abuse
| Reference | Patient | Setting | Description of abuse |
|---|---|---|---|
| Hussain et al, 20051 | Woman, age 34, with history of polysubstance abuse, depression, and borderline personality traits | Prison | Crushed tablets dissolved in water and injected intravenously |
| Morin, 20072 | Woman, age 28, with history of schizoaffective disorder, polysubstance abuse, and personality disorder not otherwise specified | Hospital | Tablets crushed with aspirin and inhaled intranasally |
| Waters et al, 20073 | Man, age 33, with history of polysubstance abuse | Outpatient | Crushed tablets dissolved in water and injected intravenously |
| Reeves et al, 20074 | Man, age 49, with history of alcohol dependence and benzodiazepine abuse | Outpatient | Misuse without psychiatric symptoms or a diagnosed psychiatric disorder |
| Man, age 23, with history of benzodiazepine dependence | Outpatient | Misuse without psychiatric symptoms or a diagnosed psychiatric disorder | |
| Man, age 39, with history of bipolar disorder | Outpatient | Oral use in doses more than the prescribed amount | |
| Murphy et al, 20085 | Man, age 29, with unclear history of schizophrenia | Psychiatric walk-in clinic | Malingering psychiatric symptoms to obtain an oral dose and overnight stay |
| Fischer et al, 20096 | Man, age 53, with history of depressive symptoms | Court-mandated outpatient clinic | Malingering psychiatric symptoms to obtain higher oral doses |
Quetiapine
Methods of quetiapine misuse include ingesting pills, inhaling crushed tablets, and injecting a solution of dissolved tablets.1-7 In case studies, patients report abusing quetiapine for its sedative, anxiolytic, and calming effects.1,2,4-7 One patient reported snorting crushed quetiapine tablets combined with cocaine for “hallucinogenic” effects.3 Street names for quetiapine include “quell,” “Susie-Q,” and “baby heroin,” and “Q-ball” refers to a combination of cocaine and quetiapine.8 Quetiapine tablets have a street value of $3 to $8 for doses ranging from 25 mg to 100 mg.9 Although outpatient misuse of quetiapine is common, abuse in correctional settings also is becoming more frequent.10 Residents of jails and prisons misuse quetiapine for reasons similar to those cited by outpatients: sedation, relief of anxiety, and hallucinogenic effects or “getting high.”1,2,10 Clinicians must differentiate inmates who have legitimate psychiatric symptoms that require antipsychotic treatment from those who are malingering to obtain the drug. Efforts to treat inmates for substance use disorders may be thwarted by the easy availability of drugs in correctional settings.10
Other SGAs
The incidence of misuse of olanzapine and other SGAs is more difficult to ascertain. Only 2 case reports describe olanzapine abuse, both in outpatient settings. One describes a patient treated for depression with psychosis who was using increasingly higher doses of olanzapine to obtain euphoric effects.11 Switching to aripiprazole effectively treated her illness and addressed her olanzapine misuse.
In the other case, a patient with bipolar disorder was able to obtain olanzapine, 40 mg/d, by complaining of worsened manic symptoms.12 He described the experience of misusing olanzapine as getting a “buzz,” feeling “very relaxed,” and blunting the negative jitteriness he felt when he used cocaine.12 This patient stated that he had observed others abusing olanzapine, both orally and intravenously.
Although the literature lacks reports on the risks of antipsychotic abuse, numerous Web sites purport to sell these drugs without a prescription and some describe the experience of illicit use of drugs such as haloperidol, risperidone, quetiapine, and olanzapine and ways to “enhance” the experience by combining drugs.13 Reported experiences with risperidone tend to be negative, citing extrapyramidal side effects and feeling “numb,” whereas olanzapine and quetiapine users describe feeling “drunk without the bad effects of alcohol” and “really happy, calm.” These sites also describe hallucinogenic effects of these agents.13
Mechanism of action
The neuropharmacologic reasons for antipsychotics’ abuse potential are difficult to quantify. Quetiapine and olanzapine have been used to treat cocaine and alcohol abuse, and work perhaps by decreasing the dopamine reward system response to substance use.14,15 Quetiapine’s rapid dissociation from the dopamine receptor has been theorized to contribute to the drug’s abuse potential, possibly through relatively lower potency and decreased residence time at the dopamine receptor.14-16 This mechanism also contributes to quetiapine’s lower risk of extrapyramidal side effects, which make the drug easier to tolerate.
Although dopamine is a factor in substance abuse and treatment of psychotic disorders, other neuropharmacologic mechanisms must be considered. SGAs are theorized to cause dopamine release in the frontal cortex through effects as 5-HT1A agonists and 5-HT2A antagonists.16 Antagonism of α-adrenergic and histaminic receptors may account for these agents’ anxiolytic and sedative properties.8
Misuse of anticholinergic agents has been reported for >50 years.17 Psychiatric patients have been reported to increase use of anticholinergics for their movement side effects as well as hallucinogenic effects.18
Treatment
Regardless of the substance that patients abuse, the treatment goals are the same: to reduce use and achieve recovery. If a patient with psychosis is abusing an SGA, consider switching to an antipsychotic with less abuse potential. Another option is to limit the supply of the abused drug by prescribing smaller quantities or increase the frequency of follow-up visits to ensure compliant use.
Related Resources
- Substance Abuse and Mental Health Services Administration. www.samhsa.gov.
- Galanter M, Kelber HD. The American Psychiatric Publishing textbook of substance abuse treatment. Arlington, VA: American Psychiatric Publishing, Inc; 2008.
Drug Brand Names
- Aripiprazole • Abilify
- Diazepam • Valium
- Haloperidol • Haldol
- Hydrocodone/acetaminophen • Vicodin
- Olanzapine • Zyprexa
- Quetiapine • Seroquel
- Risperidone • Risperdal
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Hussain MZ, Waheed W, Hussain S. Intravenous quetiapine abuse. Am J Psychiatry. 2005;162:1755-1756.
2. Morin AK. Possible intranasal quetiapine misuse. Am J Health Syst Pharm. 2007;64:723-725.
3. Waters BM, Joshi KG. Intravenous quetiapine-cocaine use (“Q-ball”). Am J Psychiatry. 2007;164:1.-
4. Reeves RR, Brister JC. Additional evidence of the abuse potential of quetiapine. S Med J. 2007;100:834-836.
5. Murphy D, Bailey K, Stone M, et al. Addictive potential of quetiapine. Am J Psychiatry. 2008;165:7.-
6. Fischer BA, Boggs DL. The role of antihistaminic effects in the misuse of quetiapine: a case report and review of the literature. Neurosci Biobehav Rev. 2009;34:555-558.
7. Pierre JM, Shnayder I, Wirshing DA, et al. Intranasal quetiapine abuse. Am J Psychiatry. 2004;161(9):1718.-
8. Sansone RA, Sansone LA. Is seroquel developing an illicit reputation for misuse/abuse? Psychiatry (Edgemont). 2010;7(1):13-16.
9. Tarasoff G, Osti K. Black-market value of antipsychotics antidepressants, and hypnotics in Las Vegas, Nevada. Am J Psychiatry. 2007;164:350.-
10. Keltner NL, Vance DE. Biological perspectives: incarcerated care and quetiapine abuse. Perspect Psychiatr Care. 2008;44(3):202-206.
11. Lai CH. Olanzapine abuse was relieved after switching to aripiprazole in a patient with psychotic depression. Prog Neuropsychopharmacol Biol Psychiatry. 2010;34(7):1363-1364.
12. Reeves RR. Abuse of olanzapine by substance abusers. J Psychoactive Drugs. 2007;39(3):297-299.
13. The Vaults of Erowid. Available at: . Accessed April 1 2011.
14. Hanley NJ, Kenna GA. Quetiapine: treatment for substance abuse and drug of abuse. Am J Health Syst Pharm. 2008;65:611-618.
15. Tcheremissine OV. Is quetiapine a drug of abuse? Reexamining the issue of addiction. Expert Opin Drug Saf. 2008;7:739-748.
16. Kuroki T, Nagao N, Nakahara T. Neuropharmacology of second-generation antipsychotic drugs: a validity of the serotonin-dopamine hypothesis. Prog Brain Res. 2008;172:199-212.
17. Smith JM. Abuse of the antiparkinson drugs: a review of the literature. J Clin Psychiatry. 1980;41(10):351-354.
18. Land W, Pinsky D, Salzman C. Abuse and misuse of anticholinergic medications. Hosp Community Psychiatry. 1991;42:580-581.
1. Hussain MZ, Waheed W, Hussain S. Intravenous quetiapine abuse. Am J Psychiatry. 2005;162:1755-1756.
2. Morin AK. Possible intranasal quetiapine misuse. Am J Health Syst Pharm. 2007;64:723-725.
3. Waters BM, Joshi KG. Intravenous quetiapine-cocaine use (“Q-ball”). Am J Psychiatry. 2007;164:1.-
4. Reeves RR, Brister JC. Additional evidence of the abuse potential of quetiapine. S Med J. 2007;100:834-836.
5. Murphy D, Bailey K, Stone M, et al. Addictive potential of quetiapine. Am J Psychiatry. 2008;165:7.-
6. Fischer BA, Boggs DL. The role of antihistaminic effects in the misuse of quetiapine: a case report and review of the literature. Neurosci Biobehav Rev. 2009;34:555-558.
7. Pierre JM, Shnayder I, Wirshing DA, et al. Intranasal quetiapine abuse. Am J Psychiatry. 2004;161(9):1718.-
8. Sansone RA, Sansone LA. Is seroquel developing an illicit reputation for misuse/abuse? Psychiatry (Edgemont). 2010;7(1):13-16.
9. Tarasoff G, Osti K. Black-market value of antipsychotics antidepressants, and hypnotics in Las Vegas, Nevada. Am J Psychiatry. 2007;164:350.-
10. Keltner NL, Vance DE. Biological perspectives: incarcerated care and quetiapine abuse. Perspect Psychiatr Care. 2008;44(3):202-206.
11. Lai CH. Olanzapine abuse was relieved after switching to aripiprazole in a patient with psychotic depression. Prog Neuropsychopharmacol Biol Psychiatry. 2010;34(7):1363-1364.
12. Reeves RR. Abuse of olanzapine by substance abusers. J Psychoactive Drugs. 2007;39(3):297-299.
13. The Vaults of Erowid. Available at: . Accessed April 1 2011.
14. Hanley NJ, Kenna GA. Quetiapine: treatment for substance abuse and drug of abuse. Am J Health Syst Pharm. 2008;65:611-618.
15. Tcheremissine OV. Is quetiapine a drug of abuse? Reexamining the issue of addiction. Expert Opin Drug Saf. 2008;7:739-748.
16. Kuroki T, Nagao N, Nakahara T. Neuropharmacology of second-generation antipsychotic drugs: a validity of the serotonin-dopamine hypothesis. Prog Brain Res. 2008;172:199-212.
17. Smith JM. Abuse of the antiparkinson drugs: a review of the literature. J Clin Psychiatry. 1980;41(10):351-354.
18. Land W, Pinsky D, Salzman C. Abuse and misuse of anticholinergic medications. Hosp Community Psychiatry. 1991;42:580-581.
Diabetic and depressed
CASE: Worsening depression
Mr. N, age 64, is a disabled factory worker with a complicated medical history. He has poorly controlled type II diabetes mellitus; obesity (body mass index 40 kg/m2); complicated cryptogenic cirrhosis with prior esophageal varices, portal gastropathy, splenomegaly, and no encephalopathy; surgically treated colon adenocarcinoma; and bilateral thalamic and right occipital infarcts with residual left homonymous hemianopsia and vertical gaze paresis. Mr. N sustained a perioperative stroke 18 months ago while undergoing a colectomy procedure for colon adenocarcinoma; an MRI done at that time showed the bilateral thalamic and right occipital infarcts. While in the internal medicine consultation clinic, Mr. N expresses suicidal and homicidal thoughts, which prompted the internal medicine team to refer him to the emergency department (ED). The team deems Mr. N’s medical problems stable except for diabetic dyscontrol.
In the ED, Mr. N says he feels sad, worthless, and “tired” of his complex family issues and multiple medical conditions. He says he’s had these feeling for at least a year, but his depression has worsened in the last few days. Mr. N is tearful while explaining his discouragement with following a diet for diabetes; earlier that day he ate an entire chocolate cake. He says all 3 of his children have ongoing substance abuse and relationship problems, but he is particularly focused on his youngest daughter, who is involved with a man who is addicted to drugs and physically abuses her and her children. Mr. N describes a detailed plan to shoot him and then commit suicide. This disclosure prompts the ED physician to admit Mr. N to ensure his safety and stabilize his mood.
Mr. N’s temperature is 36. 4°C (97. 5°F), blood pressure is 123/60 mm Hg, pulse is 81 beats per minute, respiratory rate is 24 breaths per minute, and oxygen saturation is 96% on ambient air. His physical exam is notable only for dysphoria and mild gynecomastia. He shows no evidence of acute cardiopulmonary, gastrointestinal, or other neurologic changes. His serum glucose is 650 mg/dL, and his recent hemoglobin A1c (HbA1c) is 10. 9%. His other laboratory tests include a hemoglobin of 11. 7 g/dL; white cell count, 3500/mm3; platelet count, 41, 000/mm3; sodium, 129 mEq/L; potassium, 5. 0 mEq/L; alkaline phosphatase, 90 U/L; aspartate aminotransferase, 23 U/L; alanine aminotransferase, 21 U/L; total bilirubin, 1. 8 mg/dL; creatinine, 1. 2 mg/dL; prothrombin time, 10. 4 sec; and arterial ammonia, <50 ?g/dL. Arterial blood gases are normal.
A year ago, his primary care physician prescribed fluoxetine, 20 mg/d, for fatigue and chronic back pain and neuropathic pain related to diabetes. We continue Mr. N’s outpatient prescription of fluoxetine, 20 mg/d, and low-dose acetaminophen as needed for pain. Furosemide, 40 mg/d, spironolactone, 100 mg/d, and propranolol sustained release, 60 mg/d, are maintained for complications of cirrhosis. Insulin aspart, 22 units with breakfast, 24 units with lunch, and 24 units with supper, also are administered routinely.
We consult with the internal medicine, ophthalmology, neurology, endocrinology, and diabetes services to assist in evaluating and managing Mr. N’s multiple medical conditions.
The authors’ observations
Depression and other forms of psychopathology may be underrecognized in geriatric patients because older adults may not report psychiatric symptoms that are secondary to physical conditions. Cognitive impairment in some older adults also may lead to underreporting of symptoms. Mr. N denies a history of depression, which we confirmed with his wife, daughter, and primary care physician. The late onset of his initial presentation prompted close investigation for a potential medical etiology (Table 1).1,2
We considered post-stroke depression because shortly after Mr. N’s stroke, his neurologist described emotional lability and frustration related to his poor vision. Depression occurs in one-third of chronic stroke survivors and is prevalent among patients referred for neurologic rehabilitation.1 Premorbid neuroticism3 and a history of mental illness are predictors of post-stroke depression. Stroke laterality is not related to risk of post-stroke depressive symptoms,3 but women have a higher risk of developing post-stroke depression.3
Table 1
When to consider medical causes of depressive symptoms
| Late onset of initial depressive presentation |
| Known underlying medical condition, such as cancer, diabetes, or stroke |
| Atypical symptoms and signs of depression, such as hypersomnia, hyperphagia, or agitation |
| Absence of personal or family history of psychiatric illnesses |
| Illicit substance use |
| Medication use (eg, opioids, reserpine, methyldopa, chemotherapy agents, steroids, and oral contraceptives) |
| Treatment resistance or unusual response to treatment |
| Sudden onset of mental symptoms (eg, sudden episode of uncontrollable crying) |
| Source: References 1,2 |
Diabetes and depression
Up to 30% of patients with type 2 diabetes mellitus report a lifetime history of major depression.2 Depression increases the risk of hyperglycemia and accompanying long-term metabolic complications.4,5 Few studies have explored the effects of poor glycemic control on depressive symptoms among diabetic patients.6-9 A literature review revealed no large-scale study investigating worsened depressive symptoms in patients with poor glycemic control.10,11
The cross-sectional difference between a single episode of major depression and adjustment disorder can be subtle. DSM-IV-TR describes the latter as a maladaptive reaction to an identifiable psychosocial stressor, or stressors, that occurs within 3 months of onset of that stressor (Table 2).12 Because we did not deem Mr. N’s depressive symptoms, which were evident only when he was hyperglycemic, to be grossly disproportionate to his stressors, we diagnose him with major depression rather than adjustment disorder.
Table 2
DSM-IV-TR diagnostic criteria for adjustment disorder
| A. The development of emotional or behavioral symptoms in response to an identifiable stressor(s) that occurs within 3 months of the onset of the stressor(s) |
B. These symptoms or behaviors are clinically significant, as evidenced by either of the following:
|
| C. The stress-related disturbance does not meet criteria for another specific axis I disorder and is not merely an exacerbation of a pre-existing axis I or axis II disorder |
| D. The symptoms do not represent bereavement |
| E. Once the stressor (or its consequences) has terminated, the symptoms do not persist for more than an additional 6 months |
Specify whether the condition is acute or chronic, as follows:
|
| Source: Reference 12 |
EVALUATION: No psychiatric history
On admission, Mr. N is overwhelmed, tearful, and dysphoric when describing his situation. He displays no evidence of psychosis, but his judgment and insight are impaired. He shows no change in consciousness or ability to stay awake. Mr. N acknowledges hypersomnolence, anhedonia, anergia, and decreased concentration and continues to express suicidal and homicidal thoughts. He repeatedly denies any personal or family psychiatric history or personal substance abuse, including alcohol and nicotine.
TREATMENT: Glycemic control
Mr. N receives 1 L of saline in the ED and is encouraged to drink more water during hospitalization. With appropriate insulin dosing, Mr. N’s serum glucose levels improve from 650 to 426 mg/dL by the next morning. On his third hospital day, Mr. N’s glucose level is 155 mg/dL in the morning. With tighter glycemic control, his dysphoria improves. He is future-oriented, markedly less dysphoric, and denies homicidal or suicidal ideation. Mr. N is interested in participating in group therapy, and his insight and judgment regarding his homicidal and suicidal thoughts improve. He still doesn’t fully understand the importance of diabetic control, and he struggles with his diet.
On the fourth hospital day, Mr. N’s glucose level rises to 325 mg/dL in the early evening. Subsequently, his mood deteriorates; he becomes increasingly withdrawn and somnolent. With appropriate attention to his dietary intake and supplemental insulin, his serum glucose improves to the 100 to 200 mg/dL range overnight, and his mood improves. When the glucose is controlled, he attends group therapy, tends to his hygiene, denies feeling hopeless, and offers several ideas about how to manage his family situation. After his glucose rises, Mr. N becomes isolative, hopeless, and unable to cope with stressors. With considerable education about the importance of glycemic control, Mr. N is hopeful and future-oriented when he is discharged after 9 days of hospitalization. At outpatient evaluation, he continues to report euthymia with adequate glycemic control.
The authors’ observations
Patients with hyperglycemia may experience symptoms secondary to volume depletion and hyperosmolality. The severity of these symptoms generally is proportional to the extent and duration of the hyperosmolar state. Initially, most patients complain of polyuria and polydipsia, but in more severe cases, mental status changes may evolve and include lethargy, twitching, cloudiness, motor or sensory defects, seizures, and coma. Some evidence shows that hyperglycemic patients with hyperosmolality are symptomatic only if hypernatremia is present.13 Mr. N was hyponatremic, which resolved with aggressive hydration and insulin administration.
Traditionally, depression has been observed to worsen glycemic control in diabetic patients; discussion of increased glucose levels leading to worsened depression rarely has been reported. In a meta-analysis, Lustman et al7 revealed that depression is significantly associated with hyperglycemia. A review by Li et al14 demonstrated that depression is much more common in patients with diabetes compared with general population and 45% of diabetes patients with depression were undiagnosed. Calhoun et al15 showed that for every 1-unit increase in HbA1c the odds of depressive symptoms increase by 22%. Researchers also found a positive relationship between depression and glycemic control in American Indians.15
Mr. N’s case is further evidence that the relationship between diabetes and depression is bidirectional and diagnosis and treatment of each illness impacts the other. Although this case does not confirm causality, it highlights the importance of aggressive approaches to screening and treatment of depression in patients with diabetes, and vice versa.
Related Resources
- Katon W, Russo J, Lin EH, et al. Depression and diabetes: factors associated with major depression at five-year follow-up. Psychosomatics. 2009; 50(6): 570-579.
- Biessels GJ, Luchsinger JA. Diabetes and the brain. New York, NY: Humana Press; 2009.
Drug Brand Names
- Fluoxetine • Prozac
- Furosemide • Lasix
- Insulin aspart • NovoLog
- Insulin glargine • Lantus
- Methyldopa • Aldomet
- Propranolol • Inderal
- Reserpine • Serpasil
- Spironolactone • Aldactone
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Srivastava A, Taly AB, Gupta A, et al. Post-stroke depression: prevalence and relationship with disability in chronic stroke survivors. Ann Indian Acad Neurol. 2010;13(2):123-127.
2. Marcus MD, Wing RR, Guare J, et al. Lifetime prevalence of major depression and its effect on treatment outcome in obese type II diabetic patients. Diabetes Care. 1992;15(2):253-255.
3. Storor DL, Byrne GJ. Pre-morbid personality and depression following stroke. Int Psychogeriatr. 2006;18(3):457-469.
4. Songar A, Kocabasoglu N, Balcioglu I, et al. The relationship between diabetics’ metabolic control levels and psychiatric symptomatology. Integrative Psychiatry. 1993;9:34-40.
5. Von Dras DD, Lichty W. Correlates of depression in diabetic adults. Behav Health Aging. 1990;1:79-84.
6. Lustman PJ, Clouse RE. Depression in diabetic patients: the relationship between mood and glycemic control. J Diabetes Complications. 2005;19(2):113-122.
7. Lustman PJ, Anderson RJ, Freedland KE, et al. Depression and poor glycemic control: a meta-analytic review of the literature. Diabetes Care. 2000;23(7):934-942.
8. Lustman PJ, Griffith LS, Clouse RE. Depression in adults with diabetes: results of a 5-yr follow-up study. Diabetes Care. 1988;11:605-612.
9. Van der Does FE, De Neeling JN, Snoek FJ, et al. Symptoms and well-being in relation to glycemic control in type II diabetes. Diabetes Care. 1996;19:204-210.
10. Genuth S. A case for blood glucose control. Adv Intern Med. 1995;40:573-623.
11. Wrigley M, Mayou R. Psychological factors and admission for poor glycaemic control: a study of psychological and social factors in poorly controlled insulin dependent diabetic patients. J Psychosom Res. 1991;35:335-343.
12. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington DC: American Psychiatric Association; 2000.
13. Magee MF, Bhatt BA. Management of decompensated diabetes. Diabetic ketoacidosis and hyperglycemic hyperosmolar syndrome. Crit Care Clin. 2001;17(1):75-106.
14. Li C, Ford ES, Zhao G, et al. Prevalence and correlates of undiagnosed depression among U.S. adults with diabetes: the Behavioral Risk Factor Surveillance System, 2006. Diabetes Res Clin Pract. 2009;83(2):268-279.
15. Calhoun D, Beals J, Carter EA, et al. Relationship between glycemic control and depression among American Indians in the Strong Heart Study. J Diabetes Complications. 2010;24:217-222.
CASE: Worsening depression
Mr. N, age 64, is a disabled factory worker with a complicated medical history. He has poorly controlled type II diabetes mellitus; obesity (body mass index 40 kg/m2); complicated cryptogenic cirrhosis with prior esophageal varices, portal gastropathy, splenomegaly, and no encephalopathy; surgically treated colon adenocarcinoma; and bilateral thalamic and right occipital infarcts with residual left homonymous hemianopsia and vertical gaze paresis. Mr. N sustained a perioperative stroke 18 months ago while undergoing a colectomy procedure for colon adenocarcinoma; an MRI done at that time showed the bilateral thalamic and right occipital infarcts. While in the internal medicine consultation clinic, Mr. N expresses suicidal and homicidal thoughts, which prompted the internal medicine team to refer him to the emergency department (ED). The team deems Mr. N’s medical problems stable except for diabetic dyscontrol.
In the ED, Mr. N says he feels sad, worthless, and “tired” of his complex family issues and multiple medical conditions. He says he’s had these feeling for at least a year, but his depression has worsened in the last few days. Mr. N is tearful while explaining his discouragement with following a diet for diabetes; earlier that day he ate an entire chocolate cake. He says all 3 of his children have ongoing substance abuse and relationship problems, but he is particularly focused on his youngest daughter, who is involved with a man who is addicted to drugs and physically abuses her and her children. Mr. N describes a detailed plan to shoot him and then commit suicide. This disclosure prompts the ED physician to admit Mr. N to ensure his safety and stabilize his mood.
Mr. N’s temperature is 36. 4°C (97. 5°F), blood pressure is 123/60 mm Hg, pulse is 81 beats per minute, respiratory rate is 24 breaths per minute, and oxygen saturation is 96% on ambient air. His physical exam is notable only for dysphoria and mild gynecomastia. He shows no evidence of acute cardiopulmonary, gastrointestinal, or other neurologic changes. His serum glucose is 650 mg/dL, and his recent hemoglobin A1c (HbA1c) is 10. 9%. His other laboratory tests include a hemoglobin of 11. 7 g/dL; white cell count, 3500/mm3; platelet count, 41, 000/mm3; sodium, 129 mEq/L; potassium, 5. 0 mEq/L; alkaline phosphatase, 90 U/L; aspartate aminotransferase, 23 U/L; alanine aminotransferase, 21 U/L; total bilirubin, 1. 8 mg/dL; creatinine, 1. 2 mg/dL; prothrombin time, 10. 4 sec; and arterial ammonia, <50 ?g/dL. Arterial blood gases are normal.
A year ago, his primary care physician prescribed fluoxetine, 20 mg/d, for fatigue and chronic back pain and neuropathic pain related to diabetes. We continue Mr. N’s outpatient prescription of fluoxetine, 20 mg/d, and low-dose acetaminophen as needed for pain. Furosemide, 40 mg/d, spironolactone, 100 mg/d, and propranolol sustained release, 60 mg/d, are maintained for complications of cirrhosis. Insulin aspart, 22 units with breakfast, 24 units with lunch, and 24 units with supper, also are administered routinely.
We consult with the internal medicine, ophthalmology, neurology, endocrinology, and diabetes services to assist in evaluating and managing Mr. N’s multiple medical conditions.
The authors’ observations
Depression and other forms of psychopathology may be underrecognized in geriatric patients because older adults may not report psychiatric symptoms that are secondary to physical conditions. Cognitive impairment in some older adults also may lead to underreporting of symptoms. Mr. N denies a history of depression, which we confirmed with his wife, daughter, and primary care physician. The late onset of his initial presentation prompted close investigation for a potential medical etiology (Table 1).1,2
We considered post-stroke depression because shortly after Mr. N’s stroke, his neurologist described emotional lability and frustration related to his poor vision. Depression occurs in one-third of chronic stroke survivors and is prevalent among patients referred for neurologic rehabilitation.1 Premorbid neuroticism3 and a history of mental illness are predictors of post-stroke depression. Stroke laterality is not related to risk of post-stroke depressive symptoms,3 but women have a higher risk of developing post-stroke depression.3
Table 1
When to consider medical causes of depressive symptoms
| Late onset of initial depressive presentation |
| Known underlying medical condition, such as cancer, diabetes, or stroke |
| Atypical symptoms and signs of depression, such as hypersomnia, hyperphagia, or agitation |
| Absence of personal or family history of psychiatric illnesses |
| Illicit substance use |
| Medication use (eg, opioids, reserpine, methyldopa, chemotherapy agents, steroids, and oral contraceptives) |
| Treatment resistance or unusual response to treatment |
| Sudden onset of mental symptoms (eg, sudden episode of uncontrollable crying) |
| Source: References 1,2 |
Diabetes and depression
Up to 30% of patients with type 2 diabetes mellitus report a lifetime history of major depression.2 Depression increases the risk of hyperglycemia and accompanying long-term metabolic complications.4,5 Few studies have explored the effects of poor glycemic control on depressive symptoms among diabetic patients.6-9 A literature review revealed no large-scale study investigating worsened depressive symptoms in patients with poor glycemic control.10,11
The cross-sectional difference between a single episode of major depression and adjustment disorder can be subtle. DSM-IV-TR describes the latter as a maladaptive reaction to an identifiable psychosocial stressor, or stressors, that occurs within 3 months of onset of that stressor (Table 2).12 Because we did not deem Mr. N’s depressive symptoms, which were evident only when he was hyperglycemic, to be grossly disproportionate to his stressors, we diagnose him with major depression rather than adjustment disorder.
Table 2
DSM-IV-TR diagnostic criteria for adjustment disorder
| A. The development of emotional or behavioral symptoms in response to an identifiable stressor(s) that occurs within 3 months of the onset of the stressor(s) |
B. These symptoms or behaviors are clinically significant, as evidenced by either of the following:
|
| C. The stress-related disturbance does not meet criteria for another specific axis I disorder and is not merely an exacerbation of a pre-existing axis I or axis II disorder |
| D. The symptoms do not represent bereavement |
| E. Once the stressor (or its consequences) has terminated, the symptoms do not persist for more than an additional 6 months |
Specify whether the condition is acute or chronic, as follows:
|
| Source: Reference 12 |
EVALUATION: No psychiatric history
On admission, Mr. N is overwhelmed, tearful, and dysphoric when describing his situation. He displays no evidence of psychosis, but his judgment and insight are impaired. He shows no change in consciousness or ability to stay awake. Mr. N acknowledges hypersomnolence, anhedonia, anergia, and decreased concentration and continues to express suicidal and homicidal thoughts. He repeatedly denies any personal or family psychiatric history or personal substance abuse, including alcohol and nicotine.
TREATMENT: Glycemic control
Mr. N receives 1 L of saline in the ED and is encouraged to drink more water during hospitalization. With appropriate insulin dosing, Mr. N’s serum glucose levels improve from 650 to 426 mg/dL by the next morning. On his third hospital day, Mr. N’s glucose level is 155 mg/dL in the morning. With tighter glycemic control, his dysphoria improves. He is future-oriented, markedly less dysphoric, and denies homicidal or suicidal ideation. Mr. N is interested in participating in group therapy, and his insight and judgment regarding his homicidal and suicidal thoughts improve. He still doesn’t fully understand the importance of diabetic control, and he struggles with his diet.
On the fourth hospital day, Mr. N’s glucose level rises to 325 mg/dL in the early evening. Subsequently, his mood deteriorates; he becomes increasingly withdrawn and somnolent. With appropriate attention to his dietary intake and supplemental insulin, his serum glucose improves to the 100 to 200 mg/dL range overnight, and his mood improves. When the glucose is controlled, he attends group therapy, tends to his hygiene, denies feeling hopeless, and offers several ideas about how to manage his family situation. After his glucose rises, Mr. N becomes isolative, hopeless, and unable to cope with stressors. With considerable education about the importance of glycemic control, Mr. N is hopeful and future-oriented when he is discharged after 9 days of hospitalization. At outpatient evaluation, he continues to report euthymia with adequate glycemic control.
The authors’ observations
Patients with hyperglycemia may experience symptoms secondary to volume depletion and hyperosmolality. The severity of these symptoms generally is proportional to the extent and duration of the hyperosmolar state. Initially, most patients complain of polyuria and polydipsia, but in more severe cases, mental status changes may evolve and include lethargy, twitching, cloudiness, motor or sensory defects, seizures, and coma. Some evidence shows that hyperglycemic patients with hyperosmolality are symptomatic only if hypernatremia is present.13 Mr. N was hyponatremic, which resolved with aggressive hydration and insulin administration.
Traditionally, depression has been observed to worsen glycemic control in diabetic patients; discussion of increased glucose levels leading to worsened depression rarely has been reported. In a meta-analysis, Lustman et al7 revealed that depression is significantly associated with hyperglycemia. A review by Li et al14 demonstrated that depression is much more common in patients with diabetes compared with general population and 45% of diabetes patients with depression were undiagnosed. Calhoun et al15 showed that for every 1-unit increase in HbA1c the odds of depressive symptoms increase by 22%. Researchers also found a positive relationship between depression and glycemic control in American Indians.15
Mr. N’s case is further evidence that the relationship between diabetes and depression is bidirectional and diagnosis and treatment of each illness impacts the other. Although this case does not confirm causality, it highlights the importance of aggressive approaches to screening and treatment of depression in patients with diabetes, and vice versa.
Related Resources
- Katon W, Russo J, Lin EH, et al. Depression and diabetes: factors associated with major depression at five-year follow-up. Psychosomatics. 2009; 50(6): 570-579.
- Biessels GJ, Luchsinger JA. Diabetes and the brain. New York, NY: Humana Press; 2009.
Drug Brand Names
- Fluoxetine • Prozac
- Furosemide • Lasix
- Insulin aspart • NovoLog
- Insulin glargine • Lantus
- Methyldopa • Aldomet
- Propranolol • Inderal
- Reserpine • Serpasil
- Spironolactone • Aldactone
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
CASE: Worsening depression
Mr. N, age 64, is a disabled factory worker with a complicated medical history. He has poorly controlled type II diabetes mellitus; obesity (body mass index 40 kg/m2); complicated cryptogenic cirrhosis with prior esophageal varices, portal gastropathy, splenomegaly, and no encephalopathy; surgically treated colon adenocarcinoma; and bilateral thalamic and right occipital infarcts with residual left homonymous hemianopsia and vertical gaze paresis. Mr. N sustained a perioperative stroke 18 months ago while undergoing a colectomy procedure for colon adenocarcinoma; an MRI done at that time showed the bilateral thalamic and right occipital infarcts. While in the internal medicine consultation clinic, Mr. N expresses suicidal and homicidal thoughts, which prompted the internal medicine team to refer him to the emergency department (ED). The team deems Mr. N’s medical problems stable except for diabetic dyscontrol.
In the ED, Mr. N says he feels sad, worthless, and “tired” of his complex family issues and multiple medical conditions. He says he’s had these feeling for at least a year, but his depression has worsened in the last few days. Mr. N is tearful while explaining his discouragement with following a diet for diabetes; earlier that day he ate an entire chocolate cake. He says all 3 of his children have ongoing substance abuse and relationship problems, but he is particularly focused on his youngest daughter, who is involved with a man who is addicted to drugs and physically abuses her and her children. Mr. N describes a detailed plan to shoot him and then commit suicide. This disclosure prompts the ED physician to admit Mr. N to ensure his safety and stabilize his mood.
Mr. N’s temperature is 36. 4°C (97. 5°F), blood pressure is 123/60 mm Hg, pulse is 81 beats per minute, respiratory rate is 24 breaths per minute, and oxygen saturation is 96% on ambient air. His physical exam is notable only for dysphoria and mild gynecomastia. He shows no evidence of acute cardiopulmonary, gastrointestinal, or other neurologic changes. His serum glucose is 650 mg/dL, and his recent hemoglobin A1c (HbA1c) is 10. 9%. His other laboratory tests include a hemoglobin of 11. 7 g/dL; white cell count, 3500/mm3; platelet count, 41, 000/mm3; sodium, 129 mEq/L; potassium, 5. 0 mEq/L; alkaline phosphatase, 90 U/L; aspartate aminotransferase, 23 U/L; alanine aminotransferase, 21 U/L; total bilirubin, 1. 8 mg/dL; creatinine, 1. 2 mg/dL; prothrombin time, 10. 4 sec; and arterial ammonia, <50 ?g/dL. Arterial blood gases are normal.
A year ago, his primary care physician prescribed fluoxetine, 20 mg/d, for fatigue and chronic back pain and neuropathic pain related to diabetes. We continue Mr. N’s outpatient prescription of fluoxetine, 20 mg/d, and low-dose acetaminophen as needed for pain. Furosemide, 40 mg/d, spironolactone, 100 mg/d, and propranolol sustained release, 60 mg/d, are maintained for complications of cirrhosis. Insulin aspart, 22 units with breakfast, 24 units with lunch, and 24 units with supper, also are administered routinely.
We consult with the internal medicine, ophthalmology, neurology, endocrinology, and diabetes services to assist in evaluating and managing Mr. N’s multiple medical conditions.
The authors’ observations
Depression and other forms of psychopathology may be underrecognized in geriatric patients because older adults may not report psychiatric symptoms that are secondary to physical conditions. Cognitive impairment in some older adults also may lead to underreporting of symptoms. Mr. N denies a history of depression, which we confirmed with his wife, daughter, and primary care physician. The late onset of his initial presentation prompted close investigation for a potential medical etiology (Table 1).1,2
We considered post-stroke depression because shortly after Mr. N’s stroke, his neurologist described emotional lability and frustration related to his poor vision. Depression occurs in one-third of chronic stroke survivors and is prevalent among patients referred for neurologic rehabilitation.1 Premorbid neuroticism3 and a history of mental illness are predictors of post-stroke depression. Stroke laterality is not related to risk of post-stroke depressive symptoms,3 but women have a higher risk of developing post-stroke depression.3
Table 1
When to consider medical causes of depressive symptoms
| Late onset of initial depressive presentation |
| Known underlying medical condition, such as cancer, diabetes, or stroke |
| Atypical symptoms and signs of depression, such as hypersomnia, hyperphagia, or agitation |
| Absence of personal or family history of psychiatric illnesses |
| Illicit substance use |
| Medication use (eg, opioids, reserpine, methyldopa, chemotherapy agents, steroids, and oral contraceptives) |
| Treatment resistance or unusual response to treatment |
| Sudden onset of mental symptoms (eg, sudden episode of uncontrollable crying) |
| Source: References 1,2 |
Diabetes and depression
Up to 30% of patients with type 2 diabetes mellitus report a lifetime history of major depression.2 Depression increases the risk of hyperglycemia and accompanying long-term metabolic complications.4,5 Few studies have explored the effects of poor glycemic control on depressive symptoms among diabetic patients.6-9 A literature review revealed no large-scale study investigating worsened depressive symptoms in patients with poor glycemic control.10,11
The cross-sectional difference between a single episode of major depression and adjustment disorder can be subtle. DSM-IV-TR describes the latter as a maladaptive reaction to an identifiable psychosocial stressor, or stressors, that occurs within 3 months of onset of that stressor (Table 2).12 Because we did not deem Mr. N’s depressive symptoms, which were evident only when he was hyperglycemic, to be grossly disproportionate to his stressors, we diagnose him with major depression rather than adjustment disorder.
Table 2
DSM-IV-TR diagnostic criteria for adjustment disorder
| A. The development of emotional or behavioral symptoms in response to an identifiable stressor(s) that occurs within 3 months of the onset of the stressor(s) |
B. These symptoms or behaviors are clinically significant, as evidenced by either of the following:
|
| C. The stress-related disturbance does not meet criteria for another specific axis I disorder and is not merely an exacerbation of a pre-existing axis I or axis II disorder |
| D. The symptoms do not represent bereavement |
| E. Once the stressor (or its consequences) has terminated, the symptoms do not persist for more than an additional 6 months |
Specify whether the condition is acute or chronic, as follows:
|
| Source: Reference 12 |
EVALUATION: No psychiatric history
On admission, Mr. N is overwhelmed, tearful, and dysphoric when describing his situation. He displays no evidence of psychosis, but his judgment and insight are impaired. He shows no change in consciousness or ability to stay awake. Mr. N acknowledges hypersomnolence, anhedonia, anergia, and decreased concentration and continues to express suicidal and homicidal thoughts. He repeatedly denies any personal or family psychiatric history or personal substance abuse, including alcohol and nicotine.
TREATMENT: Glycemic control
Mr. N receives 1 L of saline in the ED and is encouraged to drink more water during hospitalization. With appropriate insulin dosing, Mr. N’s serum glucose levels improve from 650 to 426 mg/dL by the next morning. On his third hospital day, Mr. N’s glucose level is 155 mg/dL in the morning. With tighter glycemic control, his dysphoria improves. He is future-oriented, markedly less dysphoric, and denies homicidal or suicidal ideation. Mr. N is interested in participating in group therapy, and his insight and judgment regarding his homicidal and suicidal thoughts improve. He still doesn’t fully understand the importance of diabetic control, and he struggles with his diet.
On the fourth hospital day, Mr. N’s glucose level rises to 325 mg/dL in the early evening. Subsequently, his mood deteriorates; he becomes increasingly withdrawn and somnolent. With appropriate attention to his dietary intake and supplemental insulin, his serum glucose improves to the 100 to 200 mg/dL range overnight, and his mood improves. When the glucose is controlled, he attends group therapy, tends to his hygiene, denies feeling hopeless, and offers several ideas about how to manage his family situation. After his glucose rises, Mr. N becomes isolative, hopeless, and unable to cope with stressors. With considerable education about the importance of glycemic control, Mr. N is hopeful and future-oriented when he is discharged after 9 days of hospitalization. At outpatient evaluation, he continues to report euthymia with adequate glycemic control.
The authors’ observations
Patients with hyperglycemia may experience symptoms secondary to volume depletion and hyperosmolality. The severity of these symptoms generally is proportional to the extent and duration of the hyperosmolar state. Initially, most patients complain of polyuria and polydipsia, but in more severe cases, mental status changes may evolve and include lethargy, twitching, cloudiness, motor or sensory defects, seizures, and coma. Some evidence shows that hyperglycemic patients with hyperosmolality are symptomatic only if hypernatremia is present.13 Mr. N was hyponatremic, which resolved with aggressive hydration and insulin administration.
Traditionally, depression has been observed to worsen glycemic control in diabetic patients; discussion of increased glucose levels leading to worsened depression rarely has been reported. In a meta-analysis, Lustman et al7 revealed that depression is significantly associated with hyperglycemia. A review by Li et al14 demonstrated that depression is much more common in patients with diabetes compared with general population and 45% of diabetes patients with depression were undiagnosed. Calhoun et al15 showed that for every 1-unit increase in HbA1c the odds of depressive symptoms increase by 22%. Researchers also found a positive relationship between depression and glycemic control in American Indians.15
Mr. N’s case is further evidence that the relationship between diabetes and depression is bidirectional and diagnosis and treatment of each illness impacts the other. Although this case does not confirm causality, it highlights the importance of aggressive approaches to screening and treatment of depression in patients with diabetes, and vice versa.
Related Resources
- Katon W, Russo J, Lin EH, et al. Depression and diabetes: factors associated with major depression at five-year follow-up. Psychosomatics. 2009; 50(6): 570-579.
- Biessels GJ, Luchsinger JA. Diabetes and the brain. New York, NY: Humana Press; 2009.
Drug Brand Names
- Fluoxetine • Prozac
- Furosemide • Lasix
- Insulin aspart • NovoLog
- Insulin glargine • Lantus
- Methyldopa • Aldomet
- Propranolol • Inderal
- Reserpine • Serpasil
- Spironolactone • Aldactone
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Srivastava A, Taly AB, Gupta A, et al. Post-stroke depression: prevalence and relationship with disability in chronic stroke survivors. Ann Indian Acad Neurol. 2010;13(2):123-127.
2. Marcus MD, Wing RR, Guare J, et al. Lifetime prevalence of major depression and its effect on treatment outcome in obese type II diabetic patients. Diabetes Care. 1992;15(2):253-255.
3. Storor DL, Byrne GJ. Pre-morbid personality and depression following stroke. Int Psychogeriatr. 2006;18(3):457-469.
4. Songar A, Kocabasoglu N, Balcioglu I, et al. The relationship between diabetics’ metabolic control levels and psychiatric symptomatology. Integrative Psychiatry. 1993;9:34-40.
5. Von Dras DD, Lichty W. Correlates of depression in diabetic adults. Behav Health Aging. 1990;1:79-84.
6. Lustman PJ, Clouse RE. Depression in diabetic patients: the relationship between mood and glycemic control. J Diabetes Complications. 2005;19(2):113-122.
7. Lustman PJ, Anderson RJ, Freedland KE, et al. Depression and poor glycemic control: a meta-analytic review of the literature. Diabetes Care. 2000;23(7):934-942.
8. Lustman PJ, Griffith LS, Clouse RE. Depression in adults with diabetes: results of a 5-yr follow-up study. Diabetes Care. 1988;11:605-612.
9. Van der Does FE, De Neeling JN, Snoek FJ, et al. Symptoms and well-being in relation to glycemic control in type II diabetes. Diabetes Care. 1996;19:204-210.
10. Genuth S. A case for blood glucose control. Adv Intern Med. 1995;40:573-623.
11. Wrigley M, Mayou R. Psychological factors and admission for poor glycaemic control: a study of psychological and social factors in poorly controlled insulin dependent diabetic patients. J Psychosom Res. 1991;35:335-343.
12. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington DC: American Psychiatric Association; 2000.
13. Magee MF, Bhatt BA. Management of decompensated diabetes. Diabetic ketoacidosis and hyperglycemic hyperosmolar syndrome. Crit Care Clin. 2001;17(1):75-106.
14. Li C, Ford ES, Zhao G, et al. Prevalence and correlates of undiagnosed depression among U.S. adults with diabetes: the Behavioral Risk Factor Surveillance System, 2006. Diabetes Res Clin Pract. 2009;83(2):268-279.
15. Calhoun D, Beals J, Carter EA, et al. Relationship between glycemic control and depression among American Indians in the Strong Heart Study. J Diabetes Complications. 2010;24:217-222.
1. Srivastava A, Taly AB, Gupta A, et al. Post-stroke depression: prevalence and relationship with disability in chronic stroke survivors. Ann Indian Acad Neurol. 2010;13(2):123-127.
2. Marcus MD, Wing RR, Guare J, et al. Lifetime prevalence of major depression and its effect on treatment outcome in obese type II diabetic patients. Diabetes Care. 1992;15(2):253-255.
3. Storor DL, Byrne GJ. Pre-morbid personality and depression following stroke. Int Psychogeriatr. 2006;18(3):457-469.
4. Songar A, Kocabasoglu N, Balcioglu I, et al. The relationship between diabetics’ metabolic control levels and psychiatric symptomatology. Integrative Psychiatry. 1993;9:34-40.
5. Von Dras DD, Lichty W. Correlates of depression in diabetic adults. Behav Health Aging. 1990;1:79-84.
6. Lustman PJ, Clouse RE. Depression in diabetic patients: the relationship between mood and glycemic control. J Diabetes Complications. 2005;19(2):113-122.
7. Lustman PJ, Anderson RJ, Freedland KE, et al. Depression and poor glycemic control: a meta-analytic review of the literature. Diabetes Care. 2000;23(7):934-942.
8. Lustman PJ, Griffith LS, Clouse RE. Depression in adults with diabetes: results of a 5-yr follow-up study. Diabetes Care. 1988;11:605-612.
9. Van der Does FE, De Neeling JN, Snoek FJ, et al. Symptoms and well-being in relation to glycemic control in type II diabetes. Diabetes Care. 1996;19:204-210.
10. Genuth S. A case for blood glucose control. Adv Intern Med. 1995;40:573-623.
11. Wrigley M, Mayou R. Psychological factors and admission for poor glycaemic control: a study of psychological and social factors in poorly controlled insulin dependent diabetic patients. J Psychosom Res. 1991;35:335-343.
12. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington DC: American Psychiatric Association; 2000.
13. Magee MF, Bhatt BA. Management of decompensated diabetes. Diabetic ketoacidosis and hyperglycemic hyperosmolar syndrome. Crit Care Clin. 2001;17(1):75-106.
14. Li C, Ford ES, Zhao G, et al. Prevalence and correlates of undiagnosed depression among U.S. adults with diabetes: the Behavioral Risk Factor Surveillance System, 2006. Diabetes Res Clin Pract. 2009;83(2):268-279.
15. Calhoun D, Beals J, Carter EA, et al. Relationship between glycemic control and depression among American Indians in the Strong Heart Study. J Diabetes Complications. 2010;24:217-222.
Dermatologic Manifestations of Musicians: A Case Report and Review of Skin Conditions in Musicians
The mysterious foreign accent
CASE: Disruptive and withdrawn
Police bring Ms. D, age 33, to our psychiatric facility because of violent behavior at her group home. When confronted for allegedly stealing, she became upset, fought with a housemate, and spat. Six months before coming to our facility she was admitted to a private hospital for psychotic disorder, not otherwise specified (NOS) where she was mute, refused all food and medications, lay in her room, and covered her face with a sheet when someone tried to talk to her.
Ms. D denies having depressive symptoms, sleep disturbance, racing thoughts, thoughts of hurting herself or others, or auditory or visual hallucinations. She complains of poor appetite. Ms. D denies a history of mental illness and says she is not taking any medication. She is upset about being hospitalized and says she will not cooperate with treatment. We cannot obtain her complete psychiatric history but available records indicate that she has 1 previous psychiatric hospitalization for psychotic disorder NOS, and has received trials of haloperidol, lorazepam, diphenhydramine, escitalopram, ziprasidone, and benztropine. Her records do not indicate the dosages of these medications or how she responded to pharmacotherapy.
During her mental status exam, Ms. D is well dressed, covers her hair with a scarf, has no unusual body movements, and responds to questions appropriately. She describes her mood as “okay” but appears upset and anxious about being in the hospital. She exhibits no overt psychotic symptoms and does not appear to be responding to auditory hallucinations or having delusional thoughts. Her cognitive function is intact and her intelligence is judged to be average with impaired insight and judgment. However, she speaks with a distinct accent that sounds Jamaican; otherwise, her speech is articulate with normal rate and tone. When we ask about her accent, Ms. D, who is African American, does not disclose her ethnicity and seems to be unaware of her accent. We did not question the authenticity of her accent until after we obtained collateral information from her family.
The authors’ observations
Based on the available information, we make a provisional diagnosis of psychotic disorder NOS and Ms. D is admitted involuntarily because of concerns about her safety. She is reluctant to accept any treatment and receives an involuntary probate commitment for 90 days. At admission, Ms. D is evasive, guarded, secretive, and at times hostile. Her physical examination reveals no signs or symptoms of focal neurologic deficits. Laboratory testing, including urine toxicology, is unremarkable. She refuses an MRI. Later testing reveals a critical ammonia level of 143 μg/dL, warranting an axis III diagnosis of asymptomatic hyperammonemia.
HISTORY: Paranoia and delusions
Ms. D says she was born and raised in a southern state. She reports that she was born to an Egyptian mother who died during childbirth; her father, who is white, was an ambassador stationed abroad. Ms. D attended school until the 11thgrade and was married at age 19 to a Secret Service agent. She says she has a son who was kidnapped by her husband’s enemies, rescued by paying ransom, and currently lives with his grandfather. Ms. D is paranoid and fears that her life is in danger. She also believes that she has gluten sensitivity that could discolor and damage her hair, which is why she always keeps a scarf on her head for protection.
Through an Internet search, we find articles about Ms. D’s son’s kidnapping. The 7-year-old had been missing for weeks when police found him with his mother in safe condition in another state, after Ms. D called her mother to ask for money and a place to stay. The child was taken from Ms. D’s custody because of concerns for his safety. We also find Ms. D’s mother. Although Ms. D insists her mother is deceased, after some persuasion, she signs a release allowing us to talk to her.
Ms. D’s mother reports that her daughter’s psychiatric problems began when she was pregnant. At the time Ms. D did not have a foreign accent. She had started to “talk funny” when her psychiatric symptoms emerged after she married and became pregnant.
Foreign accent syndrome
A foreign accent can be acquired by normal phenomena, such as being immersed in a foreign language, or a pathological process,1 which can include psychiatric (functional) or neurologic illness (organic causes). Foreign accent syndrome (FAS) is a rare speech disorder characterized by the appearance of a new accent, different from the speaker’s native language, that is perceived as foreign by the listener and in most cases also by the speaker.2 Usually an FAS patient has had no exposure to the accent, although in some cases an old accent has re-emerged.3,4
FAS can result from lesions in brain areas involved in speech production, including precentral gyrus, premotor mid-frontal gyrus, left subcortical prerolandic gyrus, postrolandic gyri, and left parietal area.4 Most FAS cases are secondary to a structural lesion in the brain caused by stroke, traumatic brain injury, cerebral hemorrhage, or multiple sclerosis.2 There are a few cases in the literature of acquired foreign accent with psychogenic etiology in patients with schizophrenia and bipolar disorder with psychotic features.5
TREATMENT: Combination therapy
Based on Ms. D’s unstable mood, irritability, delusional beliefs, and paranoid ideas, we start divalproex, 500 mg/d titrated to 1, 750 mg/d, and risperidone, 3 mg in the morning and 4 mg at bedtime.
The unit psychologist evaluates Ms. D and provides individual psychotherapy, which is mainly supportive and psychoeducational. Ms. D gradually becomes cooperative and friendly. She is not willing to talk about her accent or its origin; however, as her psychiatric symptoms improve, her accent gradually diminishes. The accent never completely resolves, but reduces until it is barely noticeable.
The authors’ observations
Ms. D’s foreign accent was more prominent when she displayed positive psychotic symptoms, such as delusions and disorganized thinking, and gradually disappeared as her psychotic symptoms improved. Ms. D’s case was peculiar because her accent was 1 of the first symptoms before her psychosis fully manifested.
How are FAS and psychosis linked?
Language dysfunction in schizophrenia is common and characterized by derailment and disorganization. Severity of language dysfunction in schizophrenia is directly proportional to overall disease severity.6,7 Various hypotheses have suggested the origin of FAS. In patients with FAS secondary to a neurologic disorder, a lesion usually is found in the dominant brain hemisphere, but the cause is not clear in patients with psychosis who have normal MRI findings. One hypothesis by Reeves et al links development of FAS to the functional disconnection between the left dorsolateral prefrontal cortex (DLPFC) and the superior temporal gyrus (STG) during active psychosis.5 In normal speech production, electric impulses originate in the DLPFC and are transmitted to STG in Wernicke’s area. From there, information goes to Broca’s area, which activates the primary motor cortex to pronounce words. In healthy individuals, word generation activates the DLPFC and causes deactivation of the bilateral STG.8 In schizophrenia, the left STG fails to deactivate in the presence of activation of the left DLPFC.9 Interestingly, STG dysfunction is seen only during active phase of psychosis. Its absence in asymptomatic patients with schizophrenia and bipolar disorder10,11 suggest that a foreign accent-like syndrome may be linked to the functional disconnection between the left DLPFC and left STG dysfunction in patients with active psychosis.5
Performing functional neuroimaging, including positron-emission tomography, functional MRI, and single-photon emission computed tomography, of patients with FAS could shed more light on the possible link between FAS and psychosis. In a case report of a patient with bipolar disorder who developed FAS, MRI initially showed no structural lesion but a later functional imaging scan revealed a cerebral infarct in the left insular and anterior temporal cortex.2
One of the limitations in Ms. D’s case is the lack of neuroimaging studies. For the first few weeks of her hospitalization, it was difficult to communicate with Ms. D. She did not acknowledge her illness and would not cooperate with treatment. She was withdrawn and seemed to experience hysterical mutism, which she perceived as caused by extreme food allergies. Later, as her symptoms continued to improve with pharmacologic and psychotherapeutic interventions, neuroimaging was no longer clinically necessary.
OUTCOME: Accent disappears
As Ms. D improves, psychotherapy evolves to gently and carefully challenging her delusions and providing insight-oriented interventions and trauma therapy. As her delusions gradually start to loosen, Ms. D reveals she had been physically and emotionally abused by her husband.
At discharge after 90 days in the hospital, Ms. D’s symptoms are well managed and she no longer shows signs of a thought disorder. Her thinking is clear, rational, and logical. She demonstrates incredible insight and appreciation that she needs to stay in treatment and continue to take divalproex and risperidone. Her delusions appear to be completely resolved and she is focused on reuniting with her son. Many of her previous delusions appear to be related to trauma and partly dissociative.
Ms. D contacts the psychologist several months later to report she is doing well in the community, staying in treatment, and working on legal means to reunite with her son. No trace of any foreign accent is detectable in her voice.
Related Resources
- Miller N, Lowit A, O’Sullivan H. What makes acquired foreign accent syndrome foreign? Journal of Neurolinguistics. 2006; 19: 385-409.
- Tsuruga K, Kobayashi T, Hirai N, et al. Foreign accent syndrome in a case of dissociative (conversion) disorder. Seishin Shinkeigaku Zasshi. 2008; 110(2): 79-87.
Drug Brand Names
- Benztropine • Cogentin
- Diphenhydramine • Benadryl
- Divalproex • Depakote
- Escitalopram • Lexapro
- Haloperidol • Haldol
- Lorazepam • Ativan
- Risperidone • Risperdal
- Ziprasidone • Geodon
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Miller N, Lowit A, O’Sullivan H. What makes acquired foreign accent syndrome foreign? J Neurolinguistics. 2006;19(5):385-409.
2. Poulin S, Macoir J, Paquet N, et al. Psychogenic or neurogenic origin of agrammatism and foreign accent syndrome in a bipolar patient: a case report. Ann Gen Psychiatry. 2007;6:1.-
3. Takayama Y, Sugishita M, Kido T, et al. A case of foreign accent syndrome without aphasia caused by a lesion of the left precentral gyrus. Neurology. 1993;43:1361-1363.
4. Roth EJ, Fink K, Cherney LR, et al. Reversion to a previously learned foreign accent after stroke. Arch Phys Med Rehabil. 1997;78:550-552.
5. Reeves RR, Burke RS, Parker JD. Characteristics of psychotic patients with foreign accent syndrome. J Neuropsychiatry Clin Neurosci. 2007;19:70-76.
6. Ceccherini-Nelli A, Crow TJ. Disintegration of the components of language as the path to a revision of Bleuler’s and Schneider’s concepts of schizophrenia: linguistic disturbances compared with first-rank symptoms in acute psychosis. Br J Psychiatry. 2003;182:233-240.
7. Harrow M, O’Connell EM, Herbener ES, et al. Disordered verbalizations in schizophrenia: a speech disturbance or thought disorder? Compr Psychiatry. 2003;44:353-359.
8. Friston KJ, Frith CD, Liddle PF, et al. Investigating a network of word generation with positron emission tomography. Proc R Soc Lond B Biol Sci. 1991;244:101-106.
9. Frith CD, Friston K, Herold S, et al. Regional brain activity in chronic schizophrenic patients during the performance of a verbal fluency task. Br J Psychiatry. 1995;167:343-349.
10. Spence SA, Liddle PF, Stefan MD, et al. Functional anatomy of verbal fluency in people with schizophrenia and those at genetic risk. Focal dysfunction and distributed disconnectivity reappraised. Br J Psychiatry. 2011;176:52-60.
11. Dye SM, Spence SA, Bench CJ, et al. No evidence for left superior temporal dysfunction in asymptomatic schizophrenia and bipolar disorder. PET study of verbal fluency. Br J Psychiatry. 1999;175:367-374.
CASE: Disruptive and withdrawn
Police bring Ms. D, age 33, to our psychiatric facility because of violent behavior at her group home. When confronted for allegedly stealing, she became upset, fought with a housemate, and spat. Six months before coming to our facility she was admitted to a private hospital for psychotic disorder, not otherwise specified (NOS) where she was mute, refused all food and medications, lay in her room, and covered her face with a sheet when someone tried to talk to her.
Ms. D denies having depressive symptoms, sleep disturbance, racing thoughts, thoughts of hurting herself or others, or auditory or visual hallucinations. She complains of poor appetite. Ms. D denies a history of mental illness and says she is not taking any medication. She is upset about being hospitalized and says she will not cooperate with treatment. We cannot obtain her complete psychiatric history but available records indicate that she has 1 previous psychiatric hospitalization for psychotic disorder NOS, and has received trials of haloperidol, lorazepam, diphenhydramine, escitalopram, ziprasidone, and benztropine. Her records do not indicate the dosages of these medications or how she responded to pharmacotherapy.
During her mental status exam, Ms. D is well dressed, covers her hair with a scarf, has no unusual body movements, and responds to questions appropriately. She describes her mood as “okay” but appears upset and anxious about being in the hospital. She exhibits no overt psychotic symptoms and does not appear to be responding to auditory hallucinations or having delusional thoughts. Her cognitive function is intact and her intelligence is judged to be average with impaired insight and judgment. However, she speaks with a distinct accent that sounds Jamaican; otherwise, her speech is articulate with normal rate and tone. When we ask about her accent, Ms. D, who is African American, does not disclose her ethnicity and seems to be unaware of her accent. We did not question the authenticity of her accent until after we obtained collateral information from her family.
The authors’ observations
Based on the available information, we make a provisional diagnosis of psychotic disorder NOS and Ms. D is admitted involuntarily because of concerns about her safety. She is reluctant to accept any treatment and receives an involuntary probate commitment for 90 days. At admission, Ms. D is evasive, guarded, secretive, and at times hostile. Her physical examination reveals no signs or symptoms of focal neurologic deficits. Laboratory testing, including urine toxicology, is unremarkable. She refuses an MRI. Later testing reveals a critical ammonia level of 143 μg/dL, warranting an axis III diagnosis of asymptomatic hyperammonemia.
HISTORY: Paranoia and delusions
Ms. D says she was born and raised in a southern state. She reports that she was born to an Egyptian mother who died during childbirth; her father, who is white, was an ambassador stationed abroad. Ms. D attended school until the 11thgrade and was married at age 19 to a Secret Service agent. She says she has a son who was kidnapped by her husband’s enemies, rescued by paying ransom, and currently lives with his grandfather. Ms. D is paranoid and fears that her life is in danger. She also believes that she has gluten sensitivity that could discolor and damage her hair, which is why she always keeps a scarf on her head for protection.
Through an Internet search, we find articles about Ms. D’s son’s kidnapping. The 7-year-old had been missing for weeks when police found him with his mother in safe condition in another state, after Ms. D called her mother to ask for money and a place to stay. The child was taken from Ms. D’s custody because of concerns for his safety. We also find Ms. D’s mother. Although Ms. D insists her mother is deceased, after some persuasion, she signs a release allowing us to talk to her.
Ms. D’s mother reports that her daughter’s psychiatric problems began when she was pregnant. At the time Ms. D did not have a foreign accent. She had started to “talk funny” when her psychiatric symptoms emerged after she married and became pregnant.
Foreign accent syndrome
A foreign accent can be acquired by normal phenomena, such as being immersed in a foreign language, or a pathological process,1 which can include psychiatric (functional) or neurologic illness (organic causes). Foreign accent syndrome (FAS) is a rare speech disorder characterized by the appearance of a new accent, different from the speaker’s native language, that is perceived as foreign by the listener and in most cases also by the speaker.2 Usually an FAS patient has had no exposure to the accent, although in some cases an old accent has re-emerged.3,4
FAS can result from lesions in brain areas involved in speech production, including precentral gyrus, premotor mid-frontal gyrus, left subcortical prerolandic gyrus, postrolandic gyri, and left parietal area.4 Most FAS cases are secondary to a structural lesion in the brain caused by stroke, traumatic brain injury, cerebral hemorrhage, or multiple sclerosis.2 There are a few cases in the literature of acquired foreign accent with psychogenic etiology in patients with schizophrenia and bipolar disorder with psychotic features.5
TREATMENT: Combination therapy
Based on Ms. D’s unstable mood, irritability, delusional beliefs, and paranoid ideas, we start divalproex, 500 mg/d titrated to 1, 750 mg/d, and risperidone, 3 mg in the morning and 4 mg at bedtime.
The unit psychologist evaluates Ms. D and provides individual psychotherapy, which is mainly supportive and psychoeducational. Ms. D gradually becomes cooperative and friendly. She is not willing to talk about her accent or its origin; however, as her psychiatric symptoms improve, her accent gradually diminishes. The accent never completely resolves, but reduces until it is barely noticeable.
The authors’ observations
Ms. D’s foreign accent was more prominent when she displayed positive psychotic symptoms, such as delusions and disorganized thinking, and gradually disappeared as her psychotic symptoms improved. Ms. D’s case was peculiar because her accent was 1 of the first symptoms before her psychosis fully manifested.
How are FAS and psychosis linked?
Language dysfunction in schizophrenia is common and characterized by derailment and disorganization. Severity of language dysfunction in schizophrenia is directly proportional to overall disease severity.6,7 Various hypotheses have suggested the origin of FAS. In patients with FAS secondary to a neurologic disorder, a lesion usually is found in the dominant brain hemisphere, but the cause is not clear in patients with psychosis who have normal MRI findings. One hypothesis by Reeves et al links development of FAS to the functional disconnection between the left dorsolateral prefrontal cortex (DLPFC) and the superior temporal gyrus (STG) during active psychosis.5 In normal speech production, electric impulses originate in the DLPFC and are transmitted to STG in Wernicke’s area. From there, information goes to Broca’s area, which activates the primary motor cortex to pronounce words. In healthy individuals, word generation activates the DLPFC and causes deactivation of the bilateral STG.8 In schizophrenia, the left STG fails to deactivate in the presence of activation of the left DLPFC.9 Interestingly, STG dysfunction is seen only during active phase of psychosis. Its absence in asymptomatic patients with schizophrenia and bipolar disorder10,11 suggest that a foreign accent-like syndrome may be linked to the functional disconnection between the left DLPFC and left STG dysfunction in patients with active psychosis.5
Performing functional neuroimaging, including positron-emission tomography, functional MRI, and single-photon emission computed tomography, of patients with FAS could shed more light on the possible link between FAS and psychosis. In a case report of a patient with bipolar disorder who developed FAS, MRI initially showed no structural lesion but a later functional imaging scan revealed a cerebral infarct in the left insular and anterior temporal cortex.2
One of the limitations in Ms. D’s case is the lack of neuroimaging studies. For the first few weeks of her hospitalization, it was difficult to communicate with Ms. D. She did not acknowledge her illness and would not cooperate with treatment. She was withdrawn and seemed to experience hysterical mutism, which she perceived as caused by extreme food allergies. Later, as her symptoms continued to improve with pharmacologic and psychotherapeutic interventions, neuroimaging was no longer clinically necessary.
OUTCOME: Accent disappears
As Ms. D improves, psychotherapy evolves to gently and carefully challenging her delusions and providing insight-oriented interventions and trauma therapy. As her delusions gradually start to loosen, Ms. D reveals she had been physically and emotionally abused by her husband.
At discharge after 90 days in the hospital, Ms. D’s symptoms are well managed and she no longer shows signs of a thought disorder. Her thinking is clear, rational, and logical. She demonstrates incredible insight and appreciation that she needs to stay in treatment and continue to take divalproex and risperidone. Her delusions appear to be completely resolved and she is focused on reuniting with her son. Many of her previous delusions appear to be related to trauma and partly dissociative.
Ms. D contacts the psychologist several months later to report she is doing well in the community, staying in treatment, and working on legal means to reunite with her son. No trace of any foreign accent is detectable in her voice.
Related Resources
- Miller N, Lowit A, O’Sullivan H. What makes acquired foreign accent syndrome foreign? Journal of Neurolinguistics. 2006; 19: 385-409.
- Tsuruga K, Kobayashi T, Hirai N, et al. Foreign accent syndrome in a case of dissociative (conversion) disorder. Seishin Shinkeigaku Zasshi. 2008; 110(2): 79-87.
Drug Brand Names
- Benztropine • Cogentin
- Diphenhydramine • Benadryl
- Divalproex • Depakote
- Escitalopram • Lexapro
- Haloperidol • Haldol
- Lorazepam • Ativan
- Risperidone • Risperdal
- Ziprasidone • Geodon
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
CASE: Disruptive and withdrawn
Police bring Ms. D, age 33, to our psychiatric facility because of violent behavior at her group home. When confronted for allegedly stealing, she became upset, fought with a housemate, and spat. Six months before coming to our facility she was admitted to a private hospital for psychotic disorder, not otherwise specified (NOS) where she was mute, refused all food and medications, lay in her room, and covered her face with a sheet when someone tried to talk to her.
Ms. D denies having depressive symptoms, sleep disturbance, racing thoughts, thoughts of hurting herself or others, or auditory or visual hallucinations. She complains of poor appetite. Ms. D denies a history of mental illness and says she is not taking any medication. She is upset about being hospitalized and says she will not cooperate with treatment. We cannot obtain her complete psychiatric history but available records indicate that she has 1 previous psychiatric hospitalization for psychotic disorder NOS, and has received trials of haloperidol, lorazepam, diphenhydramine, escitalopram, ziprasidone, and benztropine. Her records do not indicate the dosages of these medications or how she responded to pharmacotherapy.
During her mental status exam, Ms. D is well dressed, covers her hair with a scarf, has no unusual body movements, and responds to questions appropriately. She describes her mood as “okay” but appears upset and anxious about being in the hospital. She exhibits no overt psychotic symptoms and does not appear to be responding to auditory hallucinations or having delusional thoughts. Her cognitive function is intact and her intelligence is judged to be average with impaired insight and judgment. However, she speaks with a distinct accent that sounds Jamaican; otherwise, her speech is articulate with normal rate and tone. When we ask about her accent, Ms. D, who is African American, does not disclose her ethnicity and seems to be unaware of her accent. We did not question the authenticity of her accent until after we obtained collateral information from her family.
The authors’ observations
Based on the available information, we make a provisional diagnosis of psychotic disorder NOS and Ms. D is admitted involuntarily because of concerns about her safety. She is reluctant to accept any treatment and receives an involuntary probate commitment for 90 days. At admission, Ms. D is evasive, guarded, secretive, and at times hostile. Her physical examination reveals no signs or symptoms of focal neurologic deficits. Laboratory testing, including urine toxicology, is unremarkable. She refuses an MRI. Later testing reveals a critical ammonia level of 143 μg/dL, warranting an axis III diagnosis of asymptomatic hyperammonemia.
HISTORY: Paranoia and delusions
Ms. D says she was born and raised in a southern state. She reports that she was born to an Egyptian mother who died during childbirth; her father, who is white, was an ambassador stationed abroad. Ms. D attended school until the 11thgrade and was married at age 19 to a Secret Service agent. She says she has a son who was kidnapped by her husband’s enemies, rescued by paying ransom, and currently lives with his grandfather. Ms. D is paranoid and fears that her life is in danger. She also believes that she has gluten sensitivity that could discolor and damage her hair, which is why she always keeps a scarf on her head for protection.
Through an Internet search, we find articles about Ms. D’s son’s kidnapping. The 7-year-old had been missing for weeks when police found him with his mother in safe condition in another state, after Ms. D called her mother to ask for money and a place to stay. The child was taken from Ms. D’s custody because of concerns for his safety. We also find Ms. D’s mother. Although Ms. D insists her mother is deceased, after some persuasion, she signs a release allowing us to talk to her.
Ms. D’s mother reports that her daughter’s psychiatric problems began when she was pregnant. At the time Ms. D did not have a foreign accent. She had started to “talk funny” when her psychiatric symptoms emerged after she married and became pregnant.
Foreign accent syndrome
A foreign accent can be acquired by normal phenomena, such as being immersed in a foreign language, or a pathological process,1 which can include psychiatric (functional) or neurologic illness (organic causes). Foreign accent syndrome (FAS) is a rare speech disorder characterized by the appearance of a new accent, different from the speaker’s native language, that is perceived as foreign by the listener and in most cases also by the speaker.2 Usually an FAS patient has had no exposure to the accent, although in some cases an old accent has re-emerged.3,4
FAS can result from lesions in brain areas involved in speech production, including precentral gyrus, premotor mid-frontal gyrus, left subcortical prerolandic gyrus, postrolandic gyri, and left parietal area.4 Most FAS cases are secondary to a structural lesion in the brain caused by stroke, traumatic brain injury, cerebral hemorrhage, or multiple sclerosis.2 There are a few cases in the literature of acquired foreign accent with psychogenic etiology in patients with schizophrenia and bipolar disorder with psychotic features.5
TREATMENT: Combination therapy
Based on Ms. D’s unstable mood, irritability, delusional beliefs, and paranoid ideas, we start divalproex, 500 mg/d titrated to 1, 750 mg/d, and risperidone, 3 mg in the morning and 4 mg at bedtime.
The unit psychologist evaluates Ms. D and provides individual psychotherapy, which is mainly supportive and psychoeducational. Ms. D gradually becomes cooperative and friendly. She is not willing to talk about her accent or its origin; however, as her psychiatric symptoms improve, her accent gradually diminishes. The accent never completely resolves, but reduces until it is barely noticeable.
The authors’ observations
Ms. D’s foreign accent was more prominent when she displayed positive psychotic symptoms, such as delusions and disorganized thinking, and gradually disappeared as her psychotic symptoms improved. Ms. D’s case was peculiar because her accent was 1 of the first symptoms before her psychosis fully manifested.
How are FAS and psychosis linked?
Language dysfunction in schizophrenia is common and characterized by derailment and disorganization. Severity of language dysfunction in schizophrenia is directly proportional to overall disease severity.6,7 Various hypotheses have suggested the origin of FAS. In patients with FAS secondary to a neurologic disorder, a lesion usually is found in the dominant brain hemisphere, but the cause is not clear in patients with psychosis who have normal MRI findings. One hypothesis by Reeves et al links development of FAS to the functional disconnection between the left dorsolateral prefrontal cortex (DLPFC) and the superior temporal gyrus (STG) during active psychosis.5 In normal speech production, electric impulses originate in the DLPFC and are transmitted to STG in Wernicke’s area. From there, information goes to Broca’s area, which activates the primary motor cortex to pronounce words. In healthy individuals, word generation activates the DLPFC and causes deactivation of the bilateral STG.8 In schizophrenia, the left STG fails to deactivate in the presence of activation of the left DLPFC.9 Interestingly, STG dysfunction is seen only during active phase of psychosis. Its absence in asymptomatic patients with schizophrenia and bipolar disorder10,11 suggest that a foreign accent-like syndrome may be linked to the functional disconnection between the left DLPFC and left STG dysfunction in patients with active psychosis.5
Performing functional neuroimaging, including positron-emission tomography, functional MRI, and single-photon emission computed tomography, of patients with FAS could shed more light on the possible link between FAS and psychosis. In a case report of a patient with bipolar disorder who developed FAS, MRI initially showed no structural lesion but a later functional imaging scan revealed a cerebral infarct in the left insular and anterior temporal cortex.2
One of the limitations in Ms. D’s case is the lack of neuroimaging studies. For the first few weeks of her hospitalization, it was difficult to communicate with Ms. D. She did not acknowledge her illness and would not cooperate with treatment. She was withdrawn and seemed to experience hysterical mutism, which she perceived as caused by extreme food allergies. Later, as her symptoms continued to improve with pharmacologic and psychotherapeutic interventions, neuroimaging was no longer clinically necessary.
OUTCOME: Accent disappears
As Ms. D improves, psychotherapy evolves to gently and carefully challenging her delusions and providing insight-oriented interventions and trauma therapy. As her delusions gradually start to loosen, Ms. D reveals she had been physically and emotionally abused by her husband.
At discharge after 90 days in the hospital, Ms. D’s symptoms are well managed and she no longer shows signs of a thought disorder. Her thinking is clear, rational, and logical. She demonstrates incredible insight and appreciation that she needs to stay in treatment and continue to take divalproex and risperidone. Her delusions appear to be completely resolved and she is focused on reuniting with her son. Many of her previous delusions appear to be related to trauma and partly dissociative.
Ms. D contacts the psychologist several months later to report she is doing well in the community, staying in treatment, and working on legal means to reunite with her son. No trace of any foreign accent is detectable in her voice.
Related Resources
- Miller N, Lowit A, O’Sullivan H. What makes acquired foreign accent syndrome foreign? Journal of Neurolinguistics. 2006; 19: 385-409.
- Tsuruga K, Kobayashi T, Hirai N, et al. Foreign accent syndrome in a case of dissociative (conversion) disorder. Seishin Shinkeigaku Zasshi. 2008; 110(2): 79-87.
Drug Brand Names
- Benztropine • Cogentin
- Diphenhydramine • Benadryl
- Divalproex • Depakote
- Escitalopram • Lexapro
- Haloperidol • Haldol
- Lorazepam • Ativan
- Risperidone • Risperdal
- Ziprasidone • Geodon
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Miller N, Lowit A, O’Sullivan H. What makes acquired foreign accent syndrome foreign? J Neurolinguistics. 2006;19(5):385-409.
2. Poulin S, Macoir J, Paquet N, et al. Psychogenic or neurogenic origin of agrammatism and foreign accent syndrome in a bipolar patient: a case report. Ann Gen Psychiatry. 2007;6:1.-
3. Takayama Y, Sugishita M, Kido T, et al. A case of foreign accent syndrome without aphasia caused by a lesion of the left precentral gyrus. Neurology. 1993;43:1361-1363.
4. Roth EJ, Fink K, Cherney LR, et al. Reversion to a previously learned foreign accent after stroke. Arch Phys Med Rehabil. 1997;78:550-552.
5. Reeves RR, Burke RS, Parker JD. Characteristics of psychotic patients with foreign accent syndrome. J Neuropsychiatry Clin Neurosci. 2007;19:70-76.
6. Ceccherini-Nelli A, Crow TJ. Disintegration of the components of language as the path to a revision of Bleuler’s and Schneider’s concepts of schizophrenia: linguistic disturbances compared with first-rank symptoms in acute psychosis. Br J Psychiatry. 2003;182:233-240.
7. Harrow M, O’Connell EM, Herbener ES, et al. Disordered verbalizations in schizophrenia: a speech disturbance or thought disorder? Compr Psychiatry. 2003;44:353-359.
8. Friston KJ, Frith CD, Liddle PF, et al. Investigating a network of word generation with positron emission tomography. Proc R Soc Lond B Biol Sci. 1991;244:101-106.
9. Frith CD, Friston K, Herold S, et al. Regional brain activity in chronic schizophrenic patients during the performance of a verbal fluency task. Br J Psychiatry. 1995;167:343-349.
10. Spence SA, Liddle PF, Stefan MD, et al. Functional anatomy of verbal fluency in people with schizophrenia and those at genetic risk. Focal dysfunction and distributed disconnectivity reappraised. Br J Psychiatry. 2011;176:52-60.
11. Dye SM, Spence SA, Bench CJ, et al. No evidence for left superior temporal dysfunction in asymptomatic schizophrenia and bipolar disorder. PET study of verbal fluency. Br J Psychiatry. 1999;175:367-374.
1. Miller N, Lowit A, O’Sullivan H. What makes acquired foreign accent syndrome foreign? J Neurolinguistics. 2006;19(5):385-409.
2. Poulin S, Macoir J, Paquet N, et al. Psychogenic or neurogenic origin of agrammatism and foreign accent syndrome in a bipolar patient: a case report. Ann Gen Psychiatry. 2007;6:1.-
3. Takayama Y, Sugishita M, Kido T, et al. A case of foreign accent syndrome without aphasia caused by a lesion of the left precentral gyrus. Neurology. 1993;43:1361-1363.
4. Roth EJ, Fink K, Cherney LR, et al. Reversion to a previously learned foreign accent after stroke. Arch Phys Med Rehabil. 1997;78:550-552.
5. Reeves RR, Burke RS, Parker JD. Characteristics of psychotic patients with foreign accent syndrome. J Neuropsychiatry Clin Neurosci. 2007;19:70-76.
6. Ceccherini-Nelli A, Crow TJ. Disintegration of the components of language as the path to a revision of Bleuler’s and Schneider’s concepts of schizophrenia: linguistic disturbances compared with first-rank symptoms in acute psychosis. Br J Psychiatry. 2003;182:233-240.
7. Harrow M, O’Connell EM, Herbener ES, et al. Disordered verbalizations in schizophrenia: a speech disturbance or thought disorder? Compr Psychiatry. 2003;44:353-359.
8. Friston KJ, Frith CD, Liddle PF, et al. Investigating a network of word generation with positron emission tomography. Proc R Soc Lond B Biol Sci. 1991;244:101-106.
9. Frith CD, Friston K, Herold S, et al. Regional brain activity in chronic schizophrenic patients during the performance of a verbal fluency task. Br J Psychiatry. 1995;167:343-349.
10. Spence SA, Liddle PF, Stefan MD, et al. Functional anatomy of verbal fluency in people with schizophrenia and those at genetic risk. Focal dysfunction and distributed disconnectivity reappraised. Br J Psychiatry. 2011;176:52-60.
11. Dye SM, Spence SA, Bench CJ, et al. No evidence for left superior temporal dysfunction in asymptomatic schizophrenia and bipolar disorder. PET study of verbal fluency. Br J Psychiatry. 1999;175:367-374.