User login
Cutaneous Pemphigus Vegetans Co-occurring With Oral Pemphigus Vulgaris
To the Editor:
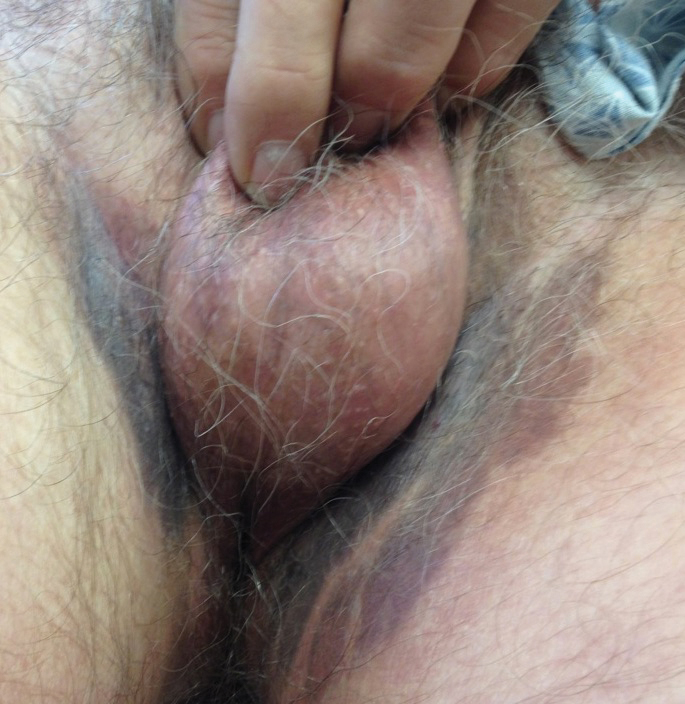
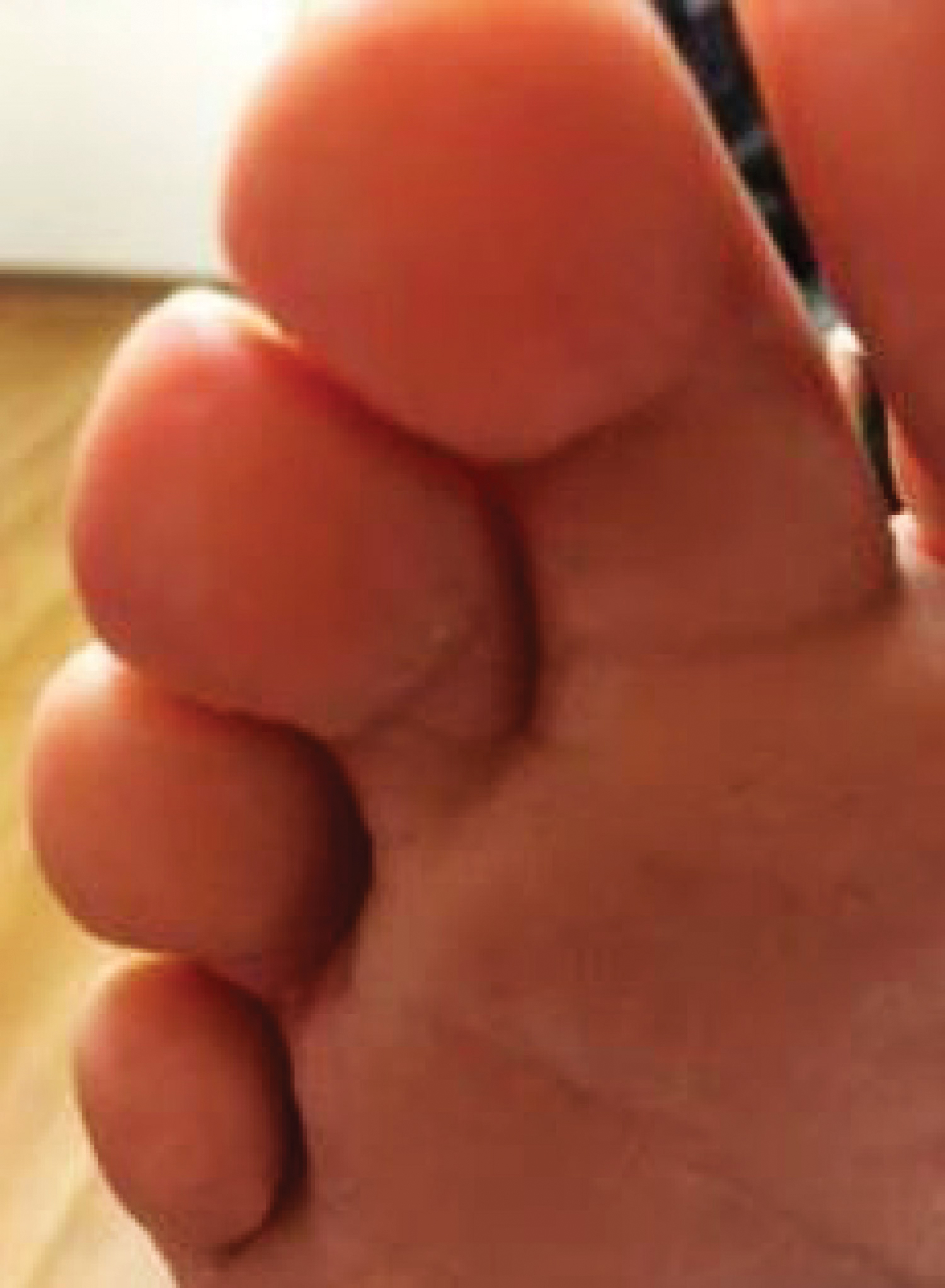
A 74-year-old man with a history of colon cancer and no history of sexually transmitted diseases presented with tender, moist, vegetating, and verrucous plaques localized to the inguinal creases and behind the scrotum of 3 weeks’ duration (Figure 1). The patient recently had taken lisinopril prescribed by his primary care physician for a couple of years for hypertension before switching to losartan prior to the current presentation. He later noticed the groin eruptions. He also noticed white tongue plaques temporally associated with the groin plaques and a long history of recurrent oral ulcerations. Prior to being seen in our clinic, outside physicians cultured methicillin-sensitive Staphylococcus aureus from the groin plaques and treated him with oral clindamycin, cephalexin, and topical mupirocin without a clinical response.
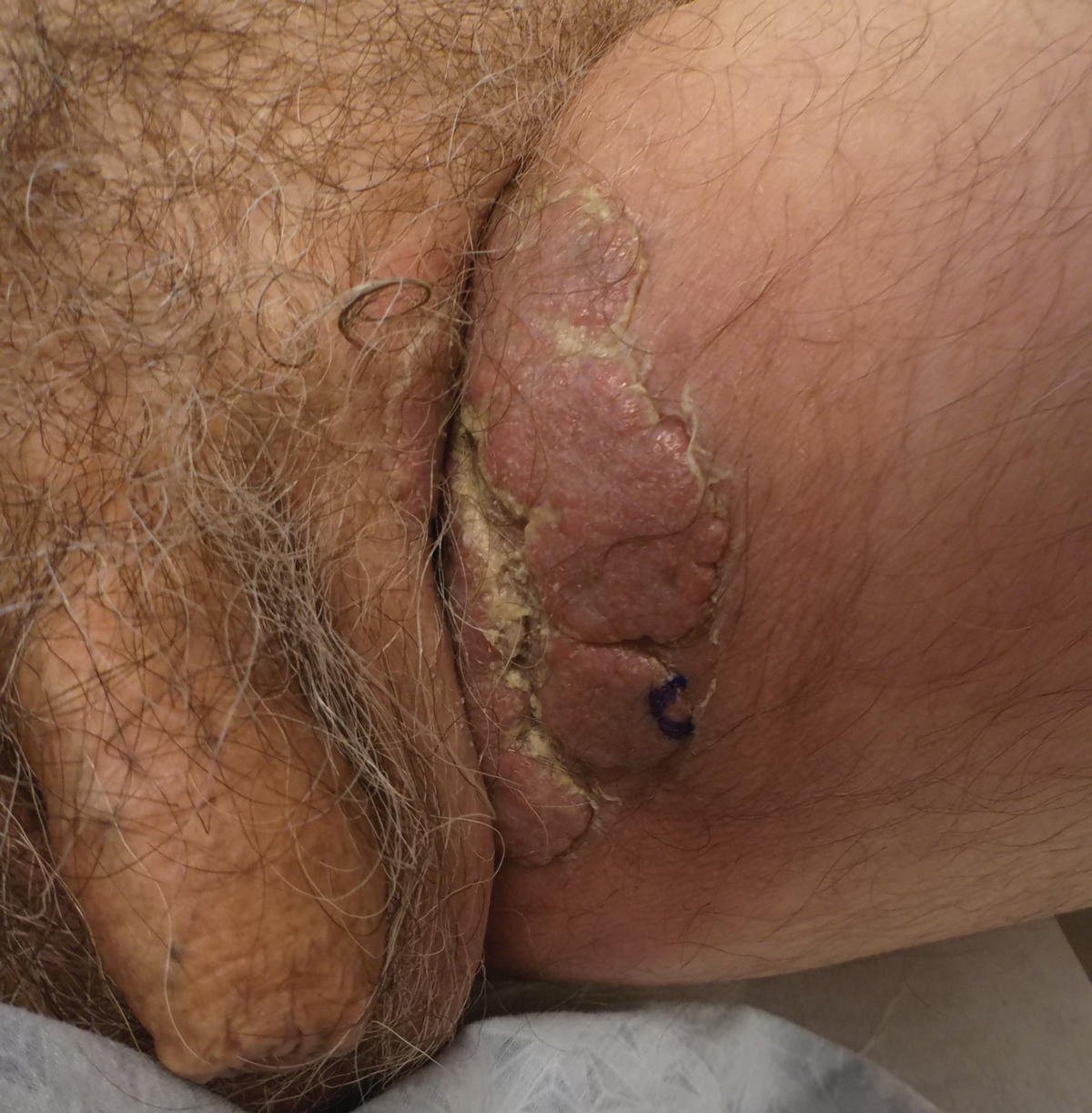
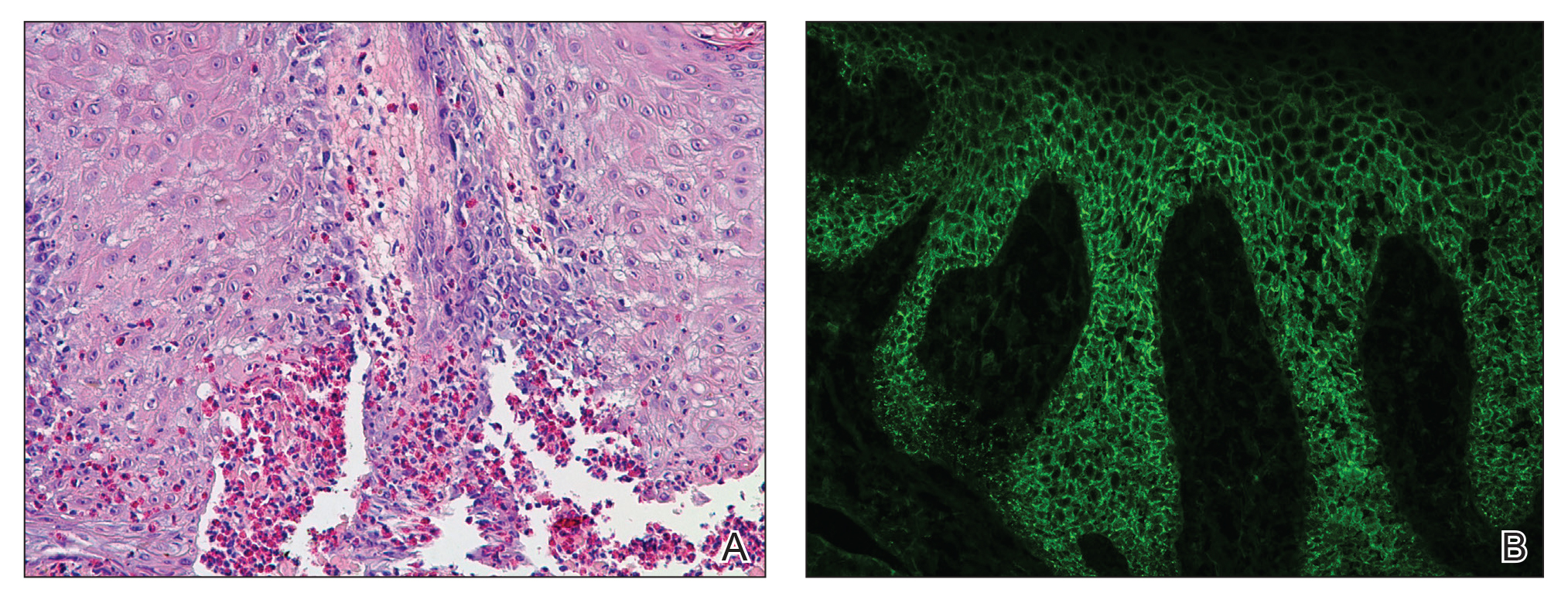
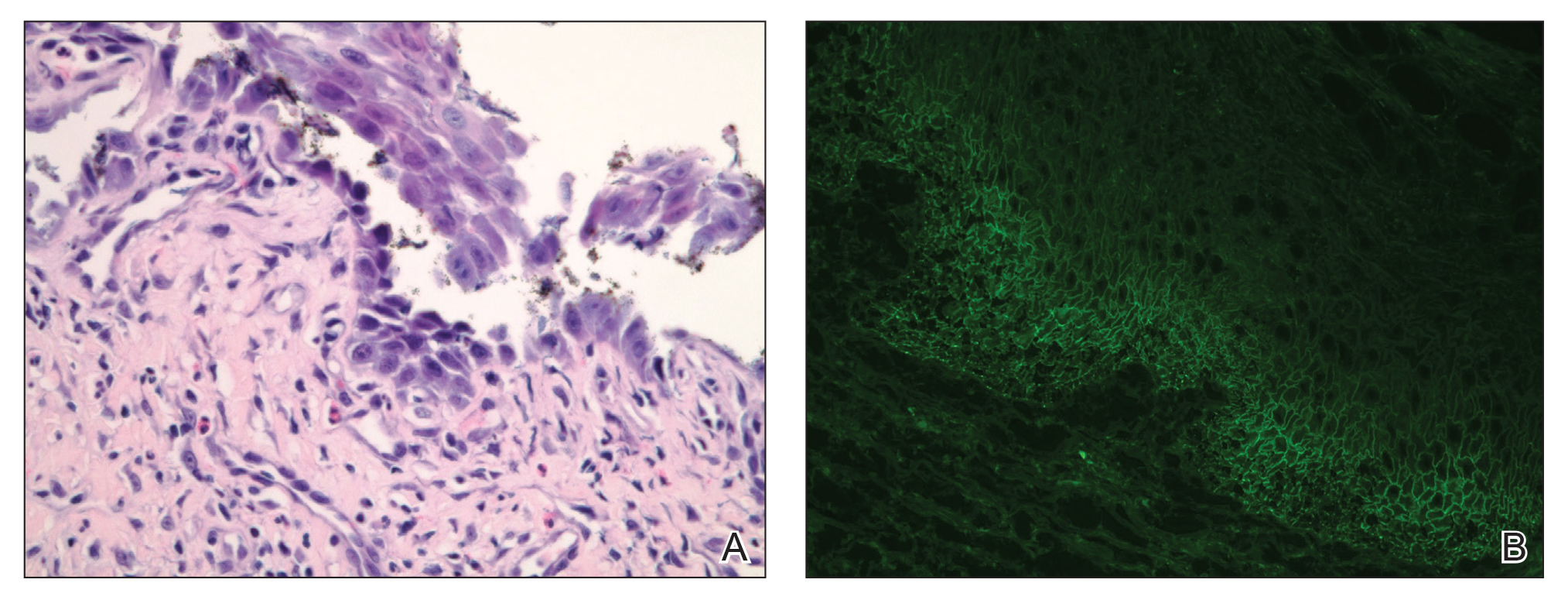
Our differential diagnosis included condyloma acuminata, condyloma lata, and cutaneous pemphigus vegetans. Laboratory testing revealed a nonreactive rapid plasma reagin test and peripheral eosinophilia of 14.9% (reference range, 0%–6%). Biopsy of a left groin plaque revealed epidermal hyperplasia with spongiosis and an eosinophilic-rich infiltrate on hematoxylin and eosin staining (Figure 2A), and direct immunofluorescence revealed diffuse epidermal intercellular IgG deposits (Figure 2B). The patient’s clinical and histologic presentation was consistent with cutaneous pemphigus vegetans. Biopsy of an oral ulcer revealed denuded acantholytic mucositis with eosinophilic-rich submucosal infiltrate and fibrosis (Figure 3A). Direct immunofluorescence was positive for lacelike intercellular staining for IgG and C3 within the squamous epithelium (Figure 3B). Together the clinical and histologic findings were consistent with oral pemphigus vulgaris.
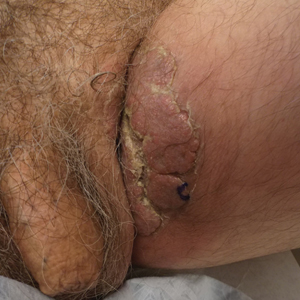
The patient initially was started on oral minocycline 100 mg twice daily and mometasone furoate cream 0.1% twice daily to affected groin areas. With these interventions, the groin plaques almost completely resolved after several months, leaving only residual hyperpigmentation (Figure 4). The oral pemphigus vulgaris initially was treated with dexamethasone 0.5 mg/5 mL solution 2 to 3 times daily, but the lesions were refractory to this approach and also did not improve after the losartan was discontinued for several months. As such, mycophenolate mofetil was started. He was titrated to the lowest effective dose and showed near-complete resolution with 500 mg 3 times daily.
Cutaneous pemphigus vegetans, a rare variant of pemphigus vulgaris, is characterized by vegetating plaques commonly localized to the skin folds, scalp, face, and mucous membranes.1 Involvement of the oral mucosa occurs in a majority of cases. Although our patient had oral ulcerations, he did not have characteristic cerebriform changes of the dorsal tongue or associated verrucous hyperkeratotic lesions involving the buccal mucosa, hard and soft palate, or vermilion border of the lips that typically are seen in pemphigus vegetans.2-5 Subsequent biopsy of the oral mucosa confirmed oral pemphigus vulgaris in our patient.
This case presentation of co-occurring cutaneous pemphigus vegetans and oral pemphigus vulgaris is uncommonly reported in the literature. Although the etiology of this co-occurrence is not clear, it could represent a form of epitope spread, with the mechanism similar to that proposed for the progression of pemphigus vulgaris from the mucosal to the mucocutaneous stage by Chan et al,6 who suggested that an autoimmune reaction against specific desmoglein 3 epitopes (an important protein component for desmosomes and the autoantigen in pemphigus vulgaris) on mucosal membranes could induce local damage. These injuries could then expose the autoreactive immune cells to a secondary desmoglein 3 epitope present in the skin, leading to the development of cutaneous lesions.6 Salato et al7 also supported this idea of intramolecular epitope spread in pemphigus vulgaris, explaining that at various stages of the disease (mucosal and mucocutaneous), the antibodies have “different tissue-binding patterns and pathogenic activities, suggesting that they may recognize distinct epitopes.” This concept of epitope spread from the oral mucosal form to the cutaneous form of pemphigus vulgaris also could help explain our patient’s presentation, as he had a long history of recurrent oral ulcerations prior to developing the vegetating cutaneous plaques of cutaneous pemphigus vegetans.
We also appreciate that either the cutaneous pemphigus vegetans or oral pemphigus vulgaris could have been drug induced in our case. Captopril has been reported to cause pemphigus vulgaris,8 so it is conceivable that the related medication lisinopril was the culprit in our case. A prior case report described an elderly man who developed lisinopril-induced pemphigus foliaceus; however, there was no oral involvement in this case and no further blister formation within 48 hours of discontinuing lisinopril.9 An additional case report implicated lisinopril in the development of a bullous eruption on the oral mucosa in a female patient, though direct and indirect immunofluorescence did not reveal the autoantibodies that typically are seen in pemphigus vulgaris.10 Our patient’s blood eosinophilia also could support an adverse drug reaction. Our patient’s losartan was discontinued for several months without respite of the oral ulcerations and thus was restarted. The cutaneous pemphigus vegetans continues to be in remission and was unaffected by restarting the losartan, making it a less likely culprit for his presentation.
We identified another case in the literature in which an individual with a history of colon cancer was diagnosed with cutaneous pemphigus vegetans.11 As such, we considered a possible link between the 2 diagnoses; however, the temporal disconnect between both conditions in our patient makes this less likely, unlike the other reported case in which the internal neoplasm and pemphigus vegetans appeared nearly simultaneously.11
Finally, our case supports a combination of topical steroids and minocycline for treatment of cutaneous pemphigus vegetans.
Our case demonstrates the importance of considering cutaneous pemphigus vegetans in the differential diagnosis, despite its rarity, when patients present with vegetating plaques. In addition, although oral involvement is common with this condition, if the patient’s oral lesions do not fit the characteristic oral findings seen in pemphigus vegetans, alternative diagnoses should be considered.
- de Almeida HL Jr, Neugebauer MG, Guarenti IM, et al. Pemphigus vegetans associated with verrucous lesions: expanding a phenotype. Clinics (Sao Paulo). 2006;61:279-282.
- Danopoulou I, Stavropoulos P, Stratigos A, et al. Pemphigus vegetans confined to the scalp. Int J Dermatol. 2006;45:1008-1009.
- Markopoulos AK, Antoniades DZ, Zaraboukas T. Pemphigus vegetans of the oral cavity. Int J Dermatol. 2006;45:425-428.
- Woo TY, Solomon AR, Fairley JA. Pemphigus vegetans limited to the lips and oral mucosa. Arch Dermatol. 1985;121:271-272.
- Yuen KL, Yau KC. An old gentleman with vegetative plaques and erosions: a case of pemphigus vegetans. Hong Kong J Dermatol Venereol. 2012;20:179-182.
- Chan LS, Vanderlugt CJ, Hashimoto T, et al. Epitope spreading: lessons from autoimmune skin diseases. J Invest Dermatol. 1998;110:103-109.
- Salato VK, Hacker-Foegen MK, Lazarova Z, et al. Role of intramolecular epitope spreading in pemphigus vulgaris. Clin Immunol. 2005;116:54-64.
- Dashore A, Choudhary SD. Captopril induced pemphigus vulgaris. Indian J Dermatol Venereol Leprol. 1987;53:293-294.
- Patterson CR, Davies MG. Pemphigus foliaceus: an adverse reaction to lisinopril. J Dermatolog Treat. 2004;15:60-62.
- Baričević M, Mravak Stipeti´c M, Situm M, et al. Oral bullous eruption after taking lisinopril—case report and literature review. Wien Klin Wochenschr. 2013;125:408-411.
- Torres T, Ferreira M, Sanches M, et al. Pemphigus vegetans in a patient with colonic cancer. Indian J Dermatol Venereol Leprol. 2009;75:603-605.
To the Editor:
A 74-year-old man with a history of colon cancer and no history of sexually transmitted diseases presented with tender, moist, vegetating, and verrucous plaques localized to the inguinal creases and behind the scrotum of 3 weeks’ duration (Figure 1). The patient recently had taken lisinopril prescribed by his primary care physician for a couple of years for hypertension before switching to losartan prior to the current presentation. He later noticed the groin eruptions. He also noticed white tongue plaques temporally associated with the groin plaques and a long history of recurrent oral ulcerations. Prior to being seen in our clinic, outside physicians cultured methicillin-sensitive Staphylococcus aureus from the groin plaques and treated him with oral clindamycin, cephalexin, and topical mupirocin without a clinical response.
Our differential diagnosis included condyloma acuminata, condyloma lata, and cutaneous pemphigus vegetans. Laboratory testing revealed a nonreactive rapid plasma reagin test and peripheral eosinophilia of 14.9% (reference range, 0%–6%). Biopsy of a left groin plaque revealed epidermal hyperplasia with spongiosis and an eosinophilic-rich infiltrate on hematoxylin and eosin staining (Figure 2A), and direct immunofluorescence revealed diffuse epidermal intercellular IgG deposits (Figure 2B). The patient’s clinical and histologic presentation was consistent with cutaneous pemphigus vegetans. Biopsy of an oral ulcer revealed denuded acantholytic mucositis with eosinophilic-rich submucosal infiltrate and fibrosis (Figure 3A). Direct immunofluorescence was positive for lacelike intercellular staining for IgG and C3 within the squamous epithelium (Figure 3B). Together the clinical and histologic findings were consistent with oral pemphigus vulgaris.
The patient initially was started on oral minocycline 100 mg twice daily and mometasone furoate cream 0.1% twice daily to affected groin areas. With these interventions, the groin plaques almost completely resolved after several months, leaving only residual hyperpigmentation (Figure 4). The oral pemphigus vulgaris initially was treated with dexamethasone 0.5 mg/5 mL solution 2 to 3 times daily, but the lesions were refractory to this approach and also did not improve after the losartan was discontinued for several months. As such, mycophenolate mofetil was started. He was titrated to the lowest effective dose and showed near-complete resolution with 500 mg 3 times daily.
Cutaneous pemphigus vegetans, a rare variant of pemphigus vulgaris, is characterized by vegetating plaques commonly localized to the skin folds, scalp, face, and mucous membranes.1 Involvement of the oral mucosa occurs in a majority of cases. Although our patient had oral ulcerations, he did not have characteristic cerebriform changes of the dorsal tongue or associated verrucous hyperkeratotic lesions involving the buccal mucosa, hard and soft palate, or vermilion border of the lips that typically are seen in pemphigus vegetans.2-5 Subsequent biopsy of the oral mucosa confirmed oral pemphigus vulgaris in our patient.
This case presentation of co-occurring cutaneous pemphigus vegetans and oral pemphigus vulgaris is uncommonly reported in the literature. Although the etiology of this co-occurrence is not clear, it could represent a form of epitope spread, with the mechanism similar to that proposed for the progression of pemphigus vulgaris from the mucosal to the mucocutaneous stage by Chan et al,6 who suggested that an autoimmune reaction against specific desmoglein 3 epitopes (an important protein component for desmosomes and the autoantigen in pemphigus vulgaris) on mucosal membranes could induce local damage. These injuries could then expose the autoreactive immune cells to a secondary desmoglein 3 epitope present in the skin, leading to the development of cutaneous lesions.6 Salato et al7 also supported this idea of intramolecular epitope spread in pemphigus vulgaris, explaining that at various stages of the disease (mucosal and mucocutaneous), the antibodies have “different tissue-binding patterns and pathogenic activities, suggesting that they may recognize distinct epitopes.” This concept of epitope spread from the oral mucosal form to the cutaneous form of pemphigus vulgaris also could help explain our patient’s presentation, as he had a long history of recurrent oral ulcerations prior to developing the vegetating cutaneous plaques of cutaneous pemphigus vegetans.
We also appreciate that either the cutaneous pemphigus vegetans or oral pemphigus vulgaris could have been drug induced in our case. Captopril has been reported to cause pemphigus vulgaris,8 so it is conceivable that the related medication lisinopril was the culprit in our case. A prior case report described an elderly man who developed lisinopril-induced pemphigus foliaceus; however, there was no oral involvement in this case and no further blister formation within 48 hours of discontinuing lisinopril.9 An additional case report implicated lisinopril in the development of a bullous eruption on the oral mucosa in a female patient, though direct and indirect immunofluorescence did not reveal the autoantibodies that typically are seen in pemphigus vulgaris.10 Our patient’s blood eosinophilia also could support an adverse drug reaction. Our patient’s losartan was discontinued for several months without respite of the oral ulcerations and thus was restarted. The cutaneous pemphigus vegetans continues to be in remission and was unaffected by restarting the losartan, making it a less likely culprit for his presentation.
We identified another case in the literature in which an individual with a history of colon cancer was diagnosed with cutaneous pemphigus vegetans.11 As such, we considered a possible link between the 2 diagnoses; however, the temporal disconnect between both conditions in our patient makes this less likely, unlike the other reported case in which the internal neoplasm and pemphigus vegetans appeared nearly simultaneously.11
Finally, our case supports a combination of topical steroids and minocycline for treatment of cutaneous pemphigus vegetans.
Our case demonstrates the importance of considering cutaneous pemphigus vegetans in the differential diagnosis, despite its rarity, when patients present with vegetating plaques. In addition, although oral involvement is common with this condition, if the patient’s oral lesions do not fit the characteristic oral findings seen in pemphigus vegetans, alternative diagnoses should be considered.
To the Editor:
A 74-year-old man with a history of colon cancer and no history of sexually transmitted diseases presented with tender, moist, vegetating, and verrucous plaques localized to the inguinal creases and behind the scrotum of 3 weeks’ duration (Figure 1). The patient recently had taken lisinopril prescribed by his primary care physician for a couple of years for hypertension before switching to losartan prior to the current presentation. He later noticed the groin eruptions. He also noticed white tongue plaques temporally associated with the groin plaques and a long history of recurrent oral ulcerations. Prior to being seen in our clinic, outside physicians cultured methicillin-sensitive Staphylococcus aureus from the groin plaques and treated him with oral clindamycin, cephalexin, and topical mupirocin without a clinical response.
Our differential diagnosis included condyloma acuminata, condyloma lata, and cutaneous pemphigus vegetans. Laboratory testing revealed a nonreactive rapid plasma reagin test and peripheral eosinophilia of 14.9% (reference range, 0%–6%). Biopsy of a left groin plaque revealed epidermal hyperplasia with spongiosis and an eosinophilic-rich infiltrate on hematoxylin and eosin staining (Figure 2A), and direct immunofluorescence revealed diffuse epidermal intercellular IgG deposits (Figure 2B). The patient’s clinical and histologic presentation was consistent with cutaneous pemphigus vegetans. Biopsy of an oral ulcer revealed denuded acantholytic mucositis with eosinophilic-rich submucosal infiltrate and fibrosis (Figure 3A). Direct immunofluorescence was positive for lacelike intercellular staining for IgG and C3 within the squamous epithelium (Figure 3B). Together the clinical and histologic findings were consistent with oral pemphigus vulgaris.
The patient initially was started on oral minocycline 100 mg twice daily and mometasone furoate cream 0.1% twice daily to affected groin areas. With these interventions, the groin plaques almost completely resolved after several months, leaving only residual hyperpigmentation (Figure 4). The oral pemphigus vulgaris initially was treated with dexamethasone 0.5 mg/5 mL solution 2 to 3 times daily, but the lesions were refractory to this approach and also did not improve after the losartan was discontinued for several months. As such, mycophenolate mofetil was started. He was titrated to the lowest effective dose and showed near-complete resolution with 500 mg 3 times daily.
Cutaneous pemphigus vegetans, a rare variant of pemphigus vulgaris, is characterized by vegetating plaques commonly localized to the skin folds, scalp, face, and mucous membranes.1 Involvement of the oral mucosa occurs in a majority of cases. Although our patient had oral ulcerations, he did not have characteristic cerebriform changes of the dorsal tongue or associated verrucous hyperkeratotic lesions involving the buccal mucosa, hard and soft palate, or vermilion border of the lips that typically are seen in pemphigus vegetans.2-5 Subsequent biopsy of the oral mucosa confirmed oral pemphigus vulgaris in our patient.
This case presentation of co-occurring cutaneous pemphigus vegetans and oral pemphigus vulgaris is uncommonly reported in the literature. Although the etiology of this co-occurrence is not clear, it could represent a form of epitope spread, with the mechanism similar to that proposed for the progression of pemphigus vulgaris from the mucosal to the mucocutaneous stage by Chan et al,6 who suggested that an autoimmune reaction against specific desmoglein 3 epitopes (an important protein component for desmosomes and the autoantigen in pemphigus vulgaris) on mucosal membranes could induce local damage. These injuries could then expose the autoreactive immune cells to a secondary desmoglein 3 epitope present in the skin, leading to the development of cutaneous lesions.6 Salato et al7 also supported this idea of intramolecular epitope spread in pemphigus vulgaris, explaining that at various stages of the disease (mucosal and mucocutaneous), the antibodies have “different tissue-binding patterns and pathogenic activities, suggesting that they may recognize distinct epitopes.” This concept of epitope spread from the oral mucosal form to the cutaneous form of pemphigus vulgaris also could help explain our patient’s presentation, as he had a long history of recurrent oral ulcerations prior to developing the vegetating cutaneous plaques of cutaneous pemphigus vegetans.
We also appreciate that either the cutaneous pemphigus vegetans or oral pemphigus vulgaris could have been drug induced in our case. Captopril has been reported to cause pemphigus vulgaris,8 so it is conceivable that the related medication lisinopril was the culprit in our case. A prior case report described an elderly man who developed lisinopril-induced pemphigus foliaceus; however, there was no oral involvement in this case and no further blister formation within 48 hours of discontinuing lisinopril.9 An additional case report implicated lisinopril in the development of a bullous eruption on the oral mucosa in a female patient, though direct and indirect immunofluorescence did not reveal the autoantibodies that typically are seen in pemphigus vulgaris.10 Our patient’s blood eosinophilia also could support an adverse drug reaction. Our patient’s losartan was discontinued for several months without respite of the oral ulcerations and thus was restarted. The cutaneous pemphigus vegetans continues to be in remission and was unaffected by restarting the losartan, making it a less likely culprit for his presentation.
We identified another case in the literature in which an individual with a history of colon cancer was diagnosed with cutaneous pemphigus vegetans.11 As such, we considered a possible link between the 2 diagnoses; however, the temporal disconnect between both conditions in our patient makes this less likely, unlike the other reported case in which the internal neoplasm and pemphigus vegetans appeared nearly simultaneously.11
Finally, our case supports a combination of topical steroids and minocycline for treatment of cutaneous pemphigus vegetans.
Our case demonstrates the importance of considering cutaneous pemphigus vegetans in the differential diagnosis, despite its rarity, when patients present with vegetating plaques. In addition, although oral involvement is common with this condition, if the patient’s oral lesions do not fit the characteristic oral findings seen in pemphigus vegetans, alternative diagnoses should be considered.
- de Almeida HL Jr, Neugebauer MG, Guarenti IM, et al. Pemphigus vegetans associated with verrucous lesions: expanding a phenotype. Clinics (Sao Paulo). 2006;61:279-282.
- Danopoulou I, Stavropoulos P, Stratigos A, et al. Pemphigus vegetans confined to the scalp. Int J Dermatol. 2006;45:1008-1009.
- Markopoulos AK, Antoniades DZ, Zaraboukas T. Pemphigus vegetans of the oral cavity. Int J Dermatol. 2006;45:425-428.
- Woo TY, Solomon AR, Fairley JA. Pemphigus vegetans limited to the lips and oral mucosa. Arch Dermatol. 1985;121:271-272.
- Yuen KL, Yau KC. An old gentleman with vegetative plaques and erosions: a case of pemphigus vegetans. Hong Kong J Dermatol Venereol. 2012;20:179-182.
- Chan LS, Vanderlugt CJ, Hashimoto T, et al. Epitope spreading: lessons from autoimmune skin diseases. J Invest Dermatol. 1998;110:103-109.
- Salato VK, Hacker-Foegen MK, Lazarova Z, et al. Role of intramolecular epitope spreading in pemphigus vulgaris. Clin Immunol. 2005;116:54-64.
- Dashore A, Choudhary SD. Captopril induced pemphigus vulgaris. Indian J Dermatol Venereol Leprol. 1987;53:293-294.
- Patterson CR, Davies MG. Pemphigus foliaceus: an adverse reaction to lisinopril. J Dermatolog Treat. 2004;15:60-62.
- Baričević M, Mravak Stipeti´c M, Situm M, et al. Oral bullous eruption after taking lisinopril—case report and literature review. Wien Klin Wochenschr. 2013;125:408-411.
- Torres T, Ferreira M, Sanches M, et al. Pemphigus vegetans in a patient with colonic cancer. Indian J Dermatol Venereol Leprol. 2009;75:603-605.
- de Almeida HL Jr, Neugebauer MG, Guarenti IM, et al. Pemphigus vegetans associated with verrucous lesions: expanding a phenotype. Clinics (Sao Paulo). 2006;61:279-282.
- Danopoulou I, Stavropoulos P, Stratigos A, et al. Pemphigus vegetans confined to the scalp. Int J Dermatol. 2006;45:1008-1009.
- Markopoulos AK, Antoniades DZ, Zaraboukas T. Pemphigus vegetans of the oral cavity. Int J Dermatol. 2006;45:425-428.
- Woo TY, Solomon AR, Fairley JA. Pemphigus vegetans limited to the lips and oral mucosa. Arch Dermatol. 1985;121:271-272.
- Yuen KL, Yau KC. An old gentleman with vegetative plaques and erosions: a case of pemphigus vegetans. Hong Kong J Dermatol Venereol. 2012;20:179-182.
- Chan LS, Vanderlugt CJ, Hashimoto T, et al. Epitope spreading: lessons from autoimmune skin diseases. J Invest Dermatol. 1998;110:103-109.
- Salato VK, Hacker-Foegen MK, Lazarova Z, et al. Role of intramolecular epitope spreading in pemphigus vulgaris. Clin Immunol. 2005;116:54-64.
- Dashore A, Choudhary SD. Captopril induced pemphigus vulgaris. Indian J Dermatol Venereol Leprol. 1987;53:293-294.
- Patterson CR, Davies MG. Pemphigus foliaceus: an adverse reaction to lisinopril. J Dermatolog Treat. 2004;15:60-62.
- Baričević M, Mravak Stipeti´c M, Situm M, et al. Oral bullous eruption after taking lisinopril—case report and literature review. Wien Klin Wochenschr. 2013;125:408-411.
- Torres T, Ferreira M, Sanches M, et al. Pemphigus vegetans in a patient with colonic cancer. Indian J Dermatol Venereol Leprol. 2009;75:603-605.
Practice Points
- Recognize the clinical and histologic features of pemphigus vegetans, a rare variant of pemphigus vulgaris.
- Consider mechanisms of co-occurring cutaneous pemphigus vegetans and oral pemphigus vulgaris.
Verrucous Psoriasis Treated With Methotrexate and Acitretin Combination Therapy
To the Editor:
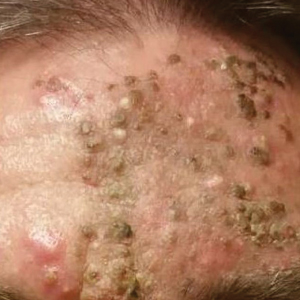
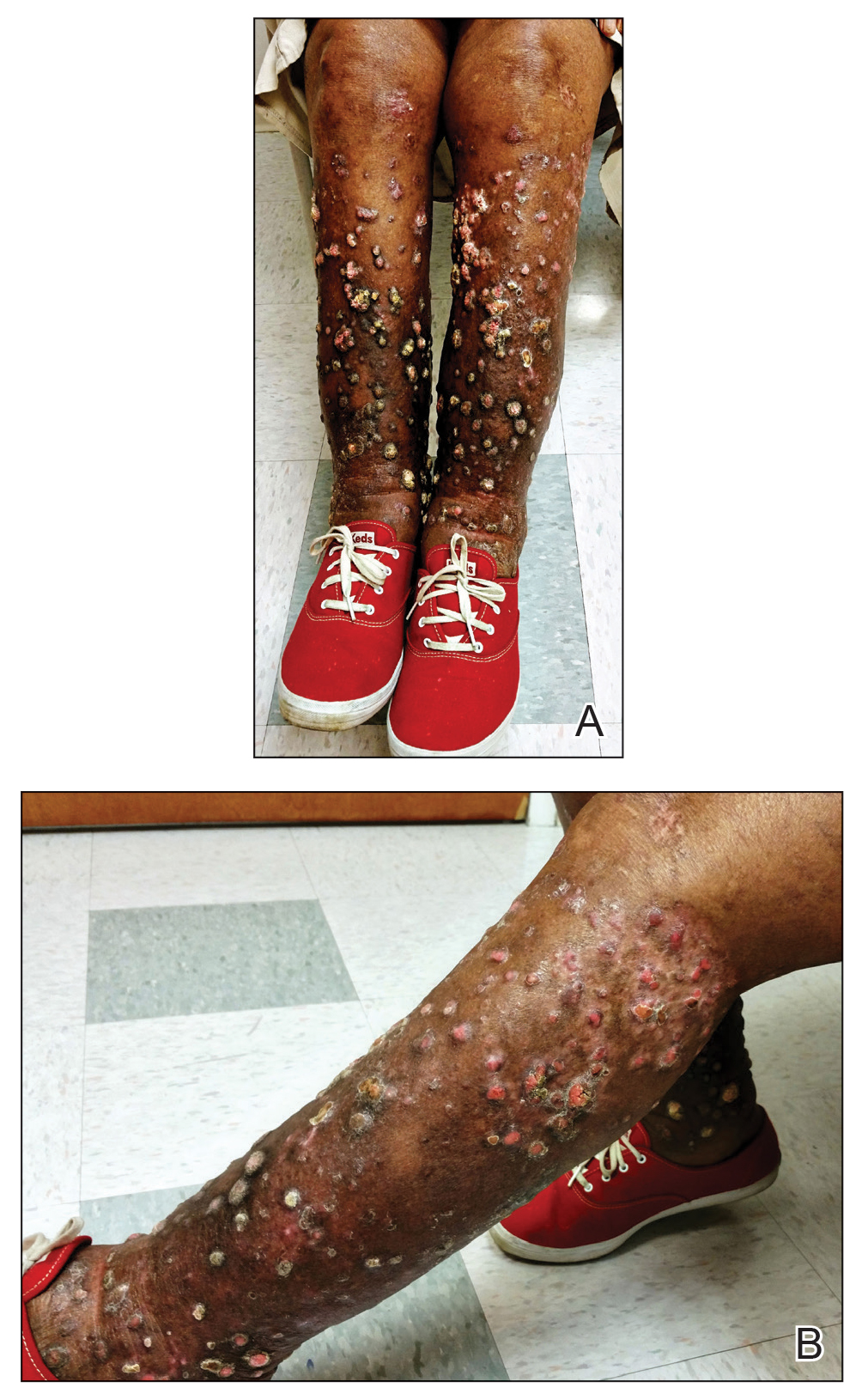
A 76-year-old woman with venous insufficiency presented with numerous thick, hyperkeratotic, confluent papules and plaques involving both legs and thighs as well as the lower back. She initially developed lesions on the distal legs, which progressed to involve the thighs and lower back, slowly enlarging over 7 years (Figure 1). The eruption was associated with pruritus and was profoundly malodorous. The patient had been unsuccessfully treated with triamcinolone ointment, bleach baths, and several courses of oral antibiotics. Her history was remarkable for marked venous insufficiency and mild anemia, with a hemoglobin level of 11.9 g/dL (reference range, 14.0–17.5 g/dL). She had no other abnormalities on a comprehensive blood test, basic metabolic panel, or liver function test.
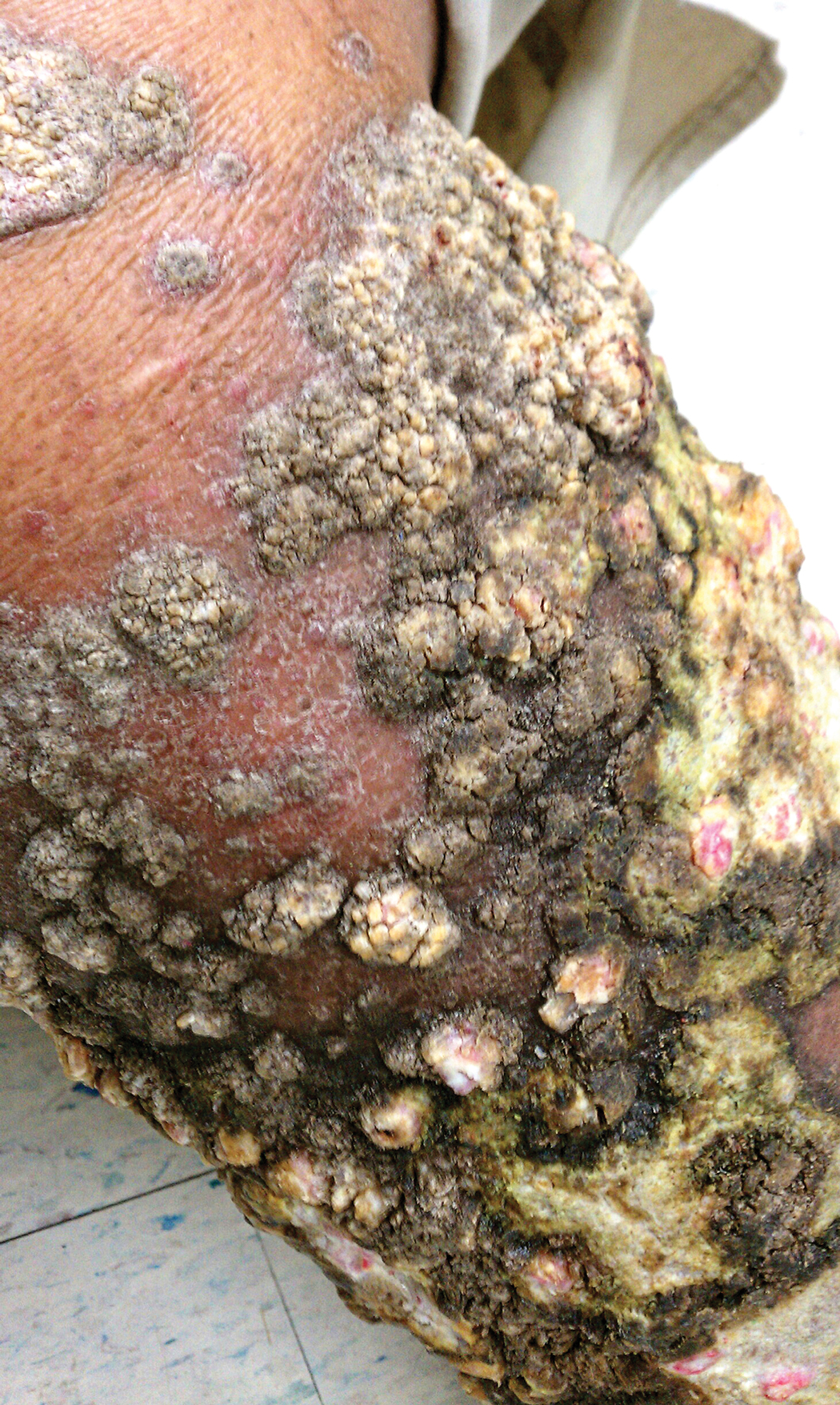
A punch biopsy specimen from the left lower back was obtained and demonstrated papillomatous psoriasiform epidermal hyperplasia with broad parakeratosis, few intracorneal neutrophils, hypogranulosis, and suprapapillary thinning (Figure 2). She was initially treated with oral methotrexate (20 mg weekly), resulting in partial improvement of plaques and complete resolution of pruritus and malodor. After 15 months of treatment with methotrexate, low-dose methotrexate (10 mg weekly) in combination with acitretin 25 mg daily was started, resulting in further improvement of hyperkeratosis (Figure 3). The patient also was given a compounded corticosteroid ointment containing liquor carbonis detergens, salicylic acid, and fluocinonide ointment, achieving minor additional benefit. Comprehensive metabolic panel, lipid panel, and liver function tests were obtained quarterly. Hemoglobin levels remained low, similar to baseline (11.3–12.5 g/dL), while all other values were within reference range. The patient tolerated treatment well, reporting mild dryness of lips on review of systems, which was attributed to acitretin and was treated with emollients.
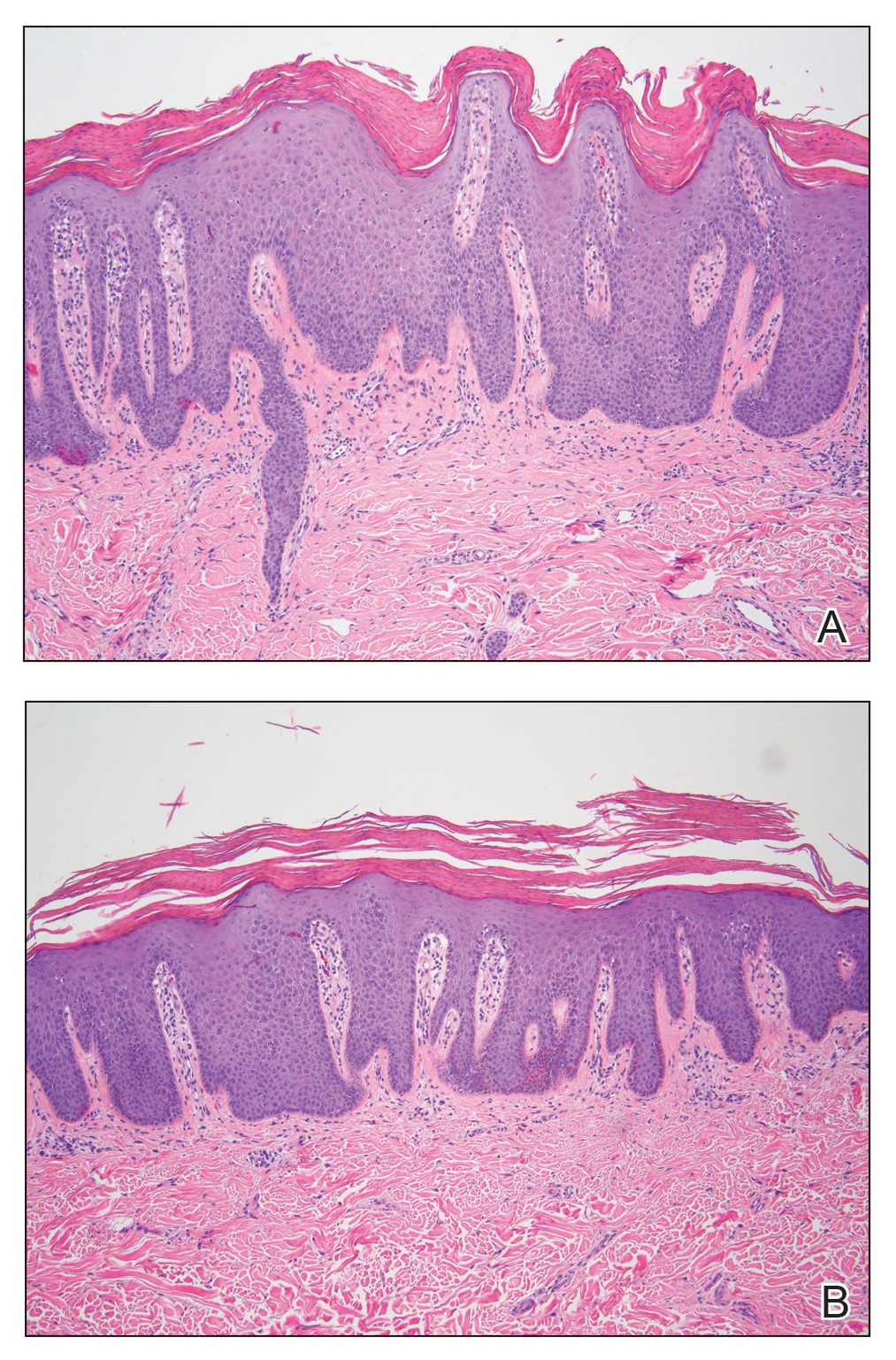
Verrucous psoriasis is an uncommon variant of psoriasis that presents as localized annular, erythrodermic, or drug-induced disease, as reported in a patient with preexisting psoriasis after interferon treatment of hepatitis C.1,2 It is characterized by symmetric hypertrophic verrucous plaques that may have an erythematous base and involve the legs, arms, trunk, and dorsal aspect of the hands3; malodor is frequent.1 Histopathologically, overlapping features of verruca vulgaris and psoriasis have been described. Specifically, lesions display typical psoriasiform changes, including parakeratosis, epidermal acanthosis with elongation of rete ridges, suprapapillary thinning, epidermal hypogranulosis, dilated or tortuous capillaries, and neutrophil collections in the stratum corneum (Munro microabscesses) or stratum spinosum (spongiform pustules of Kogoj).3 Additional findings of papillomatosis and epithelial buttressing are highly suggestive of verrucous psoriasis,3 though epithelial buttressing is not universally present.4-6 Similarly, although eosinophils and plasma cells have been described in some patients with verrucous psoriasis, this finding has not been consistently reported.4-6 Our biopsy specimen (Figure 2) lacks the epithelial buttressing but does exhibit subtle papillomatous hyperplasia consistent with the diagnosis of psoriasis.
The etiology of this entity is unknown. An association with diabetes mellitus, pulmonary disease, lymphatic circulation disorders, and immunosuppression has been proposed. Others have reported repeated trauma as contributing to the pathogenesis.1 For our patient, trauma secondary to scratching, long-standing venous insufficiency, and neglect likely contributed to the development of verrucous plaques.
The diagnosis of verrucous psoriasis can be challenging because of its similarity to several other entities, including verruca vulgaris; epidermal nevus; and squamous cell carcinoma, particularly verrucous carcinoma.4,6,7 The diagnosis has been less challenging in areas where prior typical psoriatic lesions evolved into a verrucous morphology. Our patient presented a diagnostic challenge and draws attention to this unique variant of psoriasis that could easily be misdiagnosed and lead to inappropriate treatment.
Verrucous psoriasis can be recalcitrant to therapy. Although studies addressing treatment modalities are lacking, several recommendations can be derived from case reports and our patient. The use of topical therapies, including topical corticosteroids (eg, fluocinonide, clobetasol, halobetasol), keratolytic agents (eg, urea, salicylic acid), and calcipotriene, provide only minimal improvement when used as monotherapy.1 Better success has been reported with systemic therapies, mainly methotrexate and acitretin, with anecdotal reports favoring the use of oral retinoids.1,6 Conversely, biologic medications such as etanercept, ustekinumab, adalimumab, and infliximab have only provided a partial response.1 Combination therapies including intralesional triamcinolone plus methotrexate4 or methotrexate plus acitretin, as in our patient, seem to provide additional benefit. Methotrexate and acitretin combination therapy has traditionally been avoided because of the risk for hepatotoxicity. However, a case series has demonstrated a moderate safety profile with concurrent use of these drugs in treatment-resistant psoriasis.8 In our case, clinical response was most pronounced with combination therapy of methotrexate 10 mg weekly and acitretin 25 mg daily. Thus, strong consideration should be given for combination methotrexate-acitretin therapy in patients with recalcitrant verrucous psoriasis who lack comorbid conditions.
We present a case of verrucous psoriasis, a variant of psoriasis characterized by hypertrophic plaques. We propose that venous insufficiency and long-standing untreated disease was instrumental to the development of these lesions. Furthermore, retinoids, particularly in combination with methotrexate, provided the most benefit for our patient.
Acknowledgment
We thank Stephen Somach, MD (Cleveland, Ohio), for his help interpreting the microscopic findings in our biopsy specimen. He received no compensation.
- Curtis AR, Yosipovitch G. Erythrodermic verrucous psoriasis. J Dermatolog Treat. 2012;23:215-218.
- Scavo S, Gurrera A, Mazzaglia C, et al. Verrucous psoriasis in a patient with chronic C hepatitis treated with interferon. Clin Drug Investig. 2004;24:427-429.
- Khalil FK, Keehn CA, Saeed S, et al. Verrucous psoriasis: a distinctive clinicopathologic variant of psoriasis. Am J Dermatopathol. 2005;27:204-207.
- Hall L, Marks V, Tyler W. Verrucous psoriasis: a clinical and histopathologic mimicker of verruca vulgaris [abstract]. J Am Acad Dermatol. 2013;68(suppl 1):AB218.
- Monroe HR, Hillman JD, Chiu MW. A case of verrucous psoriasis. Dermatol Online J. 2011;17:10.
- Larsen F, Susa JS, Cockerell CJ, et al. Case of multiple verrucous carcinomas responding to treatment with acetretin more likely to have been a case of verrucous psoriasis. J Am Acad Dermatol. 2007;57:534-535.
- Kuan YZ, Hsu HC, Kuo TT, et al. Multiple verrucous carcinomas treated with acitretin. J Am Acad Dermatol. 2007;56(2 suppl):S29-S32.
- Lowenthal KE, Horn PJ, Kalb RE. Concurrent use of methotrexate and acitretin revisited. J Dermatolog Treat. 2008;19:22-26.
To the Editor:
A 76-year-old woman with venous insufficiency presented with numerous thick, hyperkeratotic, confluent papules and plaques involving both legs and thighs as well as the lower back. She initially developed lesions on the distal legs, which progressed to involve the thighs and lower back, slowly enlarging over 7 years (Figure 1). The eruption was associated with pruritus and was profoundly malodorous. The patient had been unsuccessfully treated with triamcinolone ointment, bleach baths, and several courses of oral antibiotics. Her history was remarkable for marked venous insufficiency and mild anemia, with a hemoglobin level of 11.9 g/dL (reference range, 14.0–17.5 g/dL). She had no other abnormalities on a comprehensive blood test, basic metabolic panel, or liver function test.
A punch biopsy specimen from the left lower back was obtained and demonstrated papillomatous psoriasiform epidermal hyperplasia with broad parakeratosis, few intracorneal neutrophils, hypogranulosis, and suprapapillary thinning (Figure 2). She was initially treated with oral methotrexate (20 mg weekly), resulting in partial improvement of plaques and complete resolution of pruritus and malodor. After 15 months of treatment with methotrexate, low-dose methotrexate (10 mg weekly) in combination with acitretin 25 mg daily was started, resulting in further improvement of hyperkeratosis (Figure 3). The patient also was given a compounded corticosteroid ointment containing liquor carbonis detergens, salicylic acid, and fluocinonide ointment, achieving minor additional benefit. Comprehensive metabolic panel, lipid panel, and liver function tests were obtained quarterly. Hemoglobin levels remained low, similar to baseline (11.3–12.5 g/dL), while all other values were within reference range. The patient tolerated treatment well, reporting mild dryness of lips on review of systems, which was attributed to acitretin and was treated with emollients.
Verrucous psoriasis is an uncommon variant of psoriasis that presents as localized annular, erythrodermic, or drug-induced disease, as reported in a patient with preexisting psoriasis after interferon treatment of hepatitis C.1,2 It is characterized by symmetric hypertrophic verrucous plaques that may have an erythematous base and involve the legs, arms, trunk, and dorsal aspect of the hands3; malodor is frequent.1 Histopathologically, overlapping features of verruca vulgaris and psoriasis have been described. Specifically, lesions display typical psoriasiform changes, including parakeratosis, epidermal acanthosis with elongation of rete ridges, suprapapillary thinning, epidermal hypogranulosis, dilated or tortuous capillaries, and neutrophil collections in the stratum corneum (Munro microabscesses) or stratum spinosum (spongiform pustules of Kogoj).3 Additional findings of papillomatosis and epithelial buttressing are highly suggestive of verrucous psoriasis,3 though epithelial buttressing is not universally present.4-6 Similarly, although eosinophils and plasma cells have been described in some patients with verrucous psoriasis, this finding has not been consistently reported.4-6 Our biopsy specimen (Figure 2) lacks the epithelial buttressing but does exhibit subtle papillomatous hyperplasia consistent with the diagnosis of psoriasis.
The etiology of this entity is unknown. An association with diabetes mellitus, pulmonary disease, lymphatic circulation disorders, and immunosuppression has been proposed. Others have reported repeated trauma as contributing to the pathogenesis.1 For our patient, trauma secondary to scratching, long-standing venous insufficiency, and neglect likely contributed to the development of verrucous plaques.
The diagnosis of verrucous psoriasis can be challenging because of its similarity to several other entities, including verruca vulgaris; epidermal nevus; and squamous cell carcinoma, particularly verrucous carcinoma.4,6,7 The diagnosis has been less challenging in areas where prior typical psoriatic lesions evolved into a verrucous morphology. Our patient presented a diagnostic challenge and draws attention to this unique variant of psoriasis that could easily be misdiagnosed and lead to inappropriate treatment.
Verrucous psoriasis can be recalcitrant to therapy. Although studies addressing treatment modalities are lacking, several recommendations can be derived from case reports and our patient. The use of topical therapies, including topical corticosteroids (eg, fluocinonide, clobetasol, halobetasol), keratolytic agents (eg, urea, salicylic acid), and calcipotriene, provide only minimal improvement when used as monotherapy.1 Better success has been reported with systemic therapies, mainly methotrexate and acitretin, with anecdotal reports favoring the use of oral retinoids.1,6 Conversely, biologic medications such as etanercept, ustekinumab, adalimumab, and infliximab have only provided a partial response.1 Combination therapies including intralesional triamcinolone plus methotrexate4 or methotrexate plus acitretin, as in our patient, seem to provide additional benefit. Methotrexate and acitretin combination therapy has traditionally been avoided because of the risk for hepatotoxicity. However, a case series has demonstrated a moderate safety profile with concurrent use of these drugs in treatment-resistant psoriasis.8 In our case, clinical response was most pronounced with combination therapy of methotrexate 10 mg weekly and acitretin 25 mg daily. Thus, strong consideration should be given for combination methotrexate-acitretin therapy in patients with recalcitrant verrucous psoriasis who lack comorbid conditions.
We present a case of verrucous psoriasis, a variant of psoriasis characterized by hypertrophic plaques. We propose that venous insufficiency and long-standing untreated disease was instrumental to the development of these lesions. Furthermore, retinoids, particularly in combination with methotrexate, provided the most benefit for our patient.
Acknowledgment
We thank Stephen Somach, MD (Cleveland, Ohio), for his help interpreting the microscopic findings in our biopsy specimen. He received no compensation.
To the Editor:
A 76-year-old woman with venous insufficiency presented with numerous thick, hyperkeratotic, confluent papules and plaques involving both legs and thighs as well as the lower back. She initially developed lesions on the distal legs, which progressed to involve the thighs and lower back, slowly enlarging over 7 years (Figure 1). The eruption was associated with pruritus and was profoundly malodorous. The patient had been unsuccessfully treated with triamcinolone ointment, bleach baths, and several courses of oral antibiotics. Her history was remarkable for marked venous insufficiency and mild anemia, with a hemoglobin level of 11.9 g/dL (reference range, 14.0–17.5 g/dL). She had no other abnormalities on a comprehensive blood test, basic metabolic panel, or liver function test.
A punch biopsy specimen from the left lower back was obtained and demonstrated papillomatous psoriasiform epidermal hyperplasia with broad parakeratosis, few intracorneal neutrophils, hypogranulosis, and suprapapillary thinning (Figure 2). She was initially treated with oral methotrexate (20 mg weekly), resulting in partial improvement of plaques and complete resolution of pruritus and malodor. After 15 months of treatment with methotrexate, low-dose methotrexate (10 mg weekly) in combination with acitretin 25 mg daily was started, resulting in further improvement of hyperkeratosis (Figure 3). The patient also was given a compounded corticosteroid ointment containing liquor carbonis detergens, salicylic acid, and fluocinonide ointment, achieving minor additional benefit. Comprehensive metabolic panel, lipid panel, and liver function tests were obtained quarterly. Hemoglobin levels remained low, similar to baseline (11.3–12.5 g/dL), while all other values were within reference range. The patient tolerated treatment well, reporting mild dryness of lips on review of systems, which was attributed to acitretin and was treated with emollients.
Verrucous psoriasis is an uncommon variant of psoriasis that presents as localized annular, erythrodermic, or drug-induced disease, as reported in a patient with preexisting psoriasis after interferon treatment of hepatitis C.1,2 It is characterized by symmetric hypertrophic verrucous plaques that may have an erythematous base and involve the legs, arms, trunk, and dorsal aspect of the hands3; malodor is frequent.1 Histopathologically, overlapping features of verruca vulgaris and psoriasis have been described. Specifically, lesions display typical psoriasiform changes, including parakeratosis, epidermal acanthosis with elongation of rete ridges, suprapapillary thinning, epidermal hypogranulosis, dilated or tortuous capillaries, and neutrophil collections in the stratum corneum (Munro microabscesses) or stratum spinosum (spongiform pustules of Kogoj).3 Additional findings of papillomatosis and epithelial buttressing are highly suggestive of verrucous psoriasis,3 though epithelial buttressing is not universally present.4-6 Similarly, although eosinophils and plasma cells have been described in some patients with verrucous psoriasis, this finding has not been consistently reported.4-6 Our biopsy specimen (Figure 2) lacks the epithelial buttressing but does exhibit subtle papillomatous hyperplasia consistent with the diagnosis of psoriasis.
The etiology of this entity is unknown. An association with diabetes mellitus, pulmonary disease, lymphatic circulation disorders, and immunosuppression has been proposed. Others have reported repeated trauma as contributing to the pathogenesis.1 For our patient, trauma secondary to scratching, long-standing venous insufficiency, and neglect likely contributed to the development of verrucous plaques.
The diagnosis of verrucous psoriasis can be challenging because of its similarity to several other entities, including verruca vulgaris; epidermal nevus; and squamous cell carcinoma, particularly verrucous carcinoma.4,6,7 The diagnosis has been less challenging in areas where prior typical psoriatic lesions evolved into a verrucous morphology. Our patient presented a diagnostic challenge and draws attention to this unique variant of psoriasis that could easily be misdiagnosed and lead to inappropriate treatment.
Verrucous psoriasis can be recalcitrant to therapy. Although studies addressing treatment modalities are lacking, several recommendations can be derived from case reports and our patient. The use of topical therapies, including topical corticosteroids (eg, fluocinonide, clobetasol, halobetasol), keratolytic agents (eg, urea, salicylic acid), and calcipotriene, provide only minimal improvement when used as monotherapy.1 Better success has been reported with systemic therapies, mainly methotrexate and acitretin, with anecdotal reports favoring the use of oral retinoids.1,6 Conversely, biologic medications such as etanercept, ustekinumab, adalimumab, and infliximab have only provided a partial response.1 Combination therapies including intralesional triamcinolone plus methotrexate4 or methotrexate plus acitretin, as in our patient, seem to provide additional benefit. Methotrexate and acitretin combination therapy has traditionally been avoided because of the risk for hepatotoxicity. However, a case series has demonstrated a moderate safety profile with concurrent use of these drugs in treatment-resistant psoriasis.8 In our case, clinical response was most pronounced with combination therapy of methotrexate 10 mg weekly and acitretin 25 mg daily. Thus, strong consideration should be given for combination methotrexate-acitretin therapy in patients with recalcitrant verrucous psoriasis who lack comorbid conditions.
We present a case of verrucous psoriasis, a variant of psoriasis characterized by hypertrophic plaques. We propose that venous insufficiency and long-standing untreated disease was instrumental to the development of these lesions. Furthermore, retinoids, particularly in combination with methotrexate, provided the most benefit for our patient.
Acknowledgment
We thank Stephen Somach, MD (Cleveland, Ohio), for his help interpreting the microscopic findings in our biopsy specimen. He received no compensation.
- Curtis AR, Yosipovitch G. Erythrodermic verrucous psoriasis. J Dermatolog Treat. 2012;23:215-218.
- Scavo S, Gurrera A, Mazzaglia C, et al. Verrucous psoriasis in a patient with chronic C hepatitis treated with interferon. Clin Drug Investig. 2004;24:427-429.
- Khalil FK, Keehn CA, Saeed S, et al. Verrucous psoriasis: a distinctive clinicopathologic variant of psoriasis. Am J Dermatopathol. 2005;27:204-207.
- Hall L, Marks V, Tyler W. Verrucous psoriasis: a clinical and histopathologic mimicker of verruca vulgaris [abstract]. J Am Acad Dermatol. 2013;68(suppl 1):AB218.
- Monroe HR, Hillman JD, Chiu MW. A case of verrucous psoriasis. Dermatol Online J. 2011;17:10.
- Larsen F, Susa JS, Cockerell CJ, et al. Case of multiple verrucous carcinomas responding to treatment with acetretin more likely to have been a case of verrucous psoriasis. J Am Acad Dermatol. 2007;57:534-535.
- Kuan YZ, Hsu HC, Kuo TT, et al. Multiple verrucous carcinomas treated with acitretin. J Am Acad Dermatol. 2007;56(2 suppl):S29-S32.
- Lowenthal KE, Horn PJ, Kalb RE. Concurrent use of methotrexate and acitretin revisited. J Dermatolog Treat. 2008;19:22-26.
- Curtis AR, Yosipovitch G. Erythrodermic verrucous psoriasis. J Dermatolog Treat. 2012;23:215-218.
- Scavo S, Gurrera A, Mazzaglia C, et al. Verrucous psoriasis in a patient with chronic C hepatitis treated with interferon. Clin Drug Investig. 2004;24:427-429.
- Khalil FK, Keehn CA, Saeed S, et al. Verrucous psoriasis: a distinctive clinicopathologic variant of psoriasis. Am J Dermatopathol. 2005;27:204-207.
- Hall L, Marks V, Tyler W. Verrucous psoriasis: a clinical and histopathologic mimicker of verruca vulgaris [abstract]. J Am Acad Dermatol. 2013;68(suppl 1):AB218.
- Monroe HR, Hillman JD, Chiu MW. A case of verrucous psoriasis. Dermatol Online J. 2011;17:10.
- Larsen F, Susa JS, Cockerell CJ, et al. Case of multiple verrucous carcinomas responding to treatment with acetretin more likely to have been a case of verrucous psoriasis. J Am Acad Dermatol. 2007;57:534-535.
- Kuan YZ, Hsu HC, Kuo TT, et al. Multiple verrucous carcinomas treated with acitretin. J Am Acad Dermatol. 2007;56(2 suppl):S29-S32.
- Lowenthal KE, Horn PJ, Kalb RE. Concurrent use of methotrexate and acitretin revisited. J Dermatolog Treat. 2008;19:22-26.
Practice Points
- Verrucous psoriasis in an uncommon but recalcitrant-to-treatment variant of psoriasis that is characterized by hypertrophic plaques.
- The diagnosis of verrucous psoriasis is challenging, as it can mimic other entities such as verruca vulgaris and squamous cell carcinoma.
- Although the etiology of this entity is unknown, an association with diabetes mellitus, pulmonary disease, lymphatic circulation disorders, and immunosuppression has been described.
- The combination of methotrexate and acitretin is a safe and effective option for these patients in the absence of comorbid conditions.
Melanoma In Situ Within a Port-Wine Stain
To the Editor:
Port-wine stains (PWSs) are the most common type of vascular malformations. Patients rarely develop cancers in the overlying skin. However, we describe a case of melanoma in situ occurring within a long-standing facial PWS.
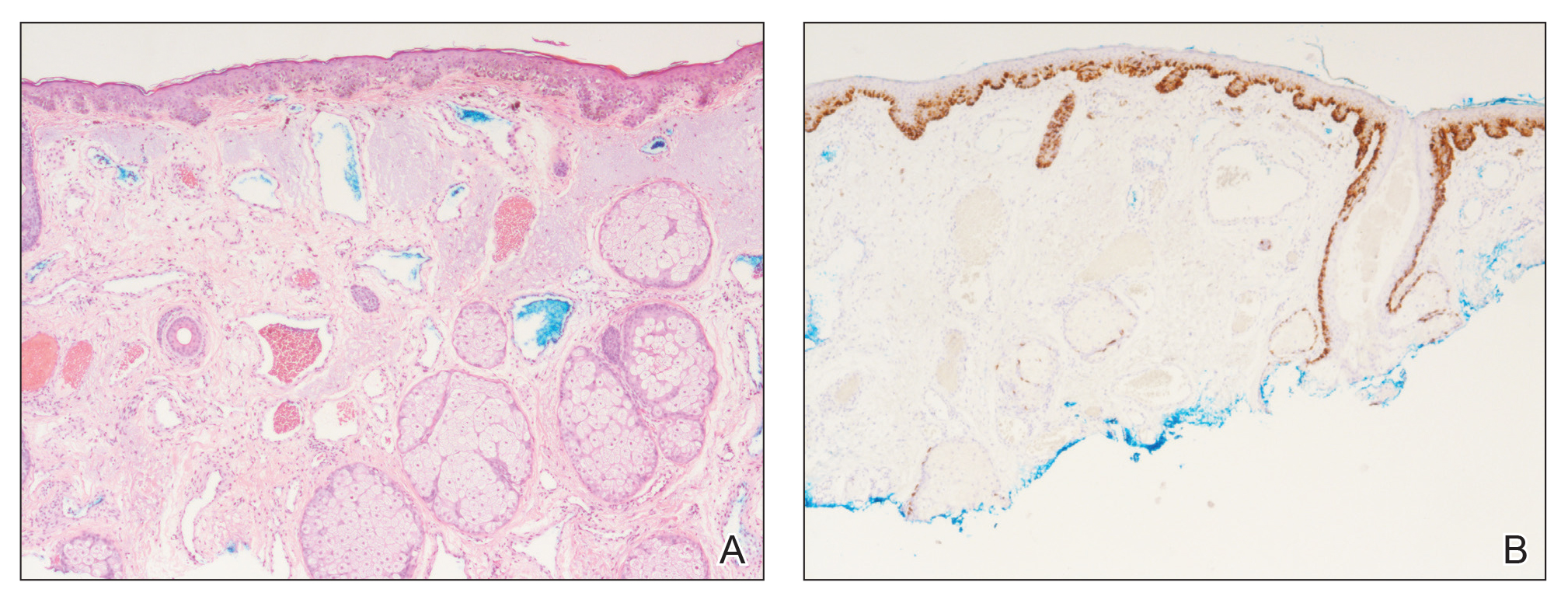
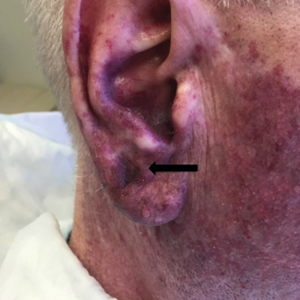
A 60-year-old white man with a history of a large unilateral facial PWS covering the right ear, lateral cheek, jaw, and neck presented to clinic with a new dark lesion on the right ear that had been growing for a few weeks or more. His PWS had been previously treated intermittently with a pulsed dye laser (PDL) for decades with variable improvement. He had not undergone any laser procedures in the last 8 months but wanted to restart treatment with the PDL. Upon further discussion, he reported a new darker area on the right earlobe that was growing. He had no personal or family history of skin cancer and was otherwise healthy. Physical examination revealed a large red vascular patch encompassing the ear, cheek, chin, and lateral neck. Within the PWS there was a black and dark brown patch with irregular borders on the right earlobe (Figure 1A). A shave biopsy was performed for histopathologic examination. The biopsy showed a confluent proliferation of atypical melanocytes along the dermoepidermal junction extending down adnexal structures (Figure 2A) that stained positive for MART-1/Melan-A (Figure 2B). In the dermis, solar elastosis and prominent dilated and thin-walled vessels were present. These findings were consistent with a melanoma in situ, lentigo maligna type, overlying a capillary malformation.
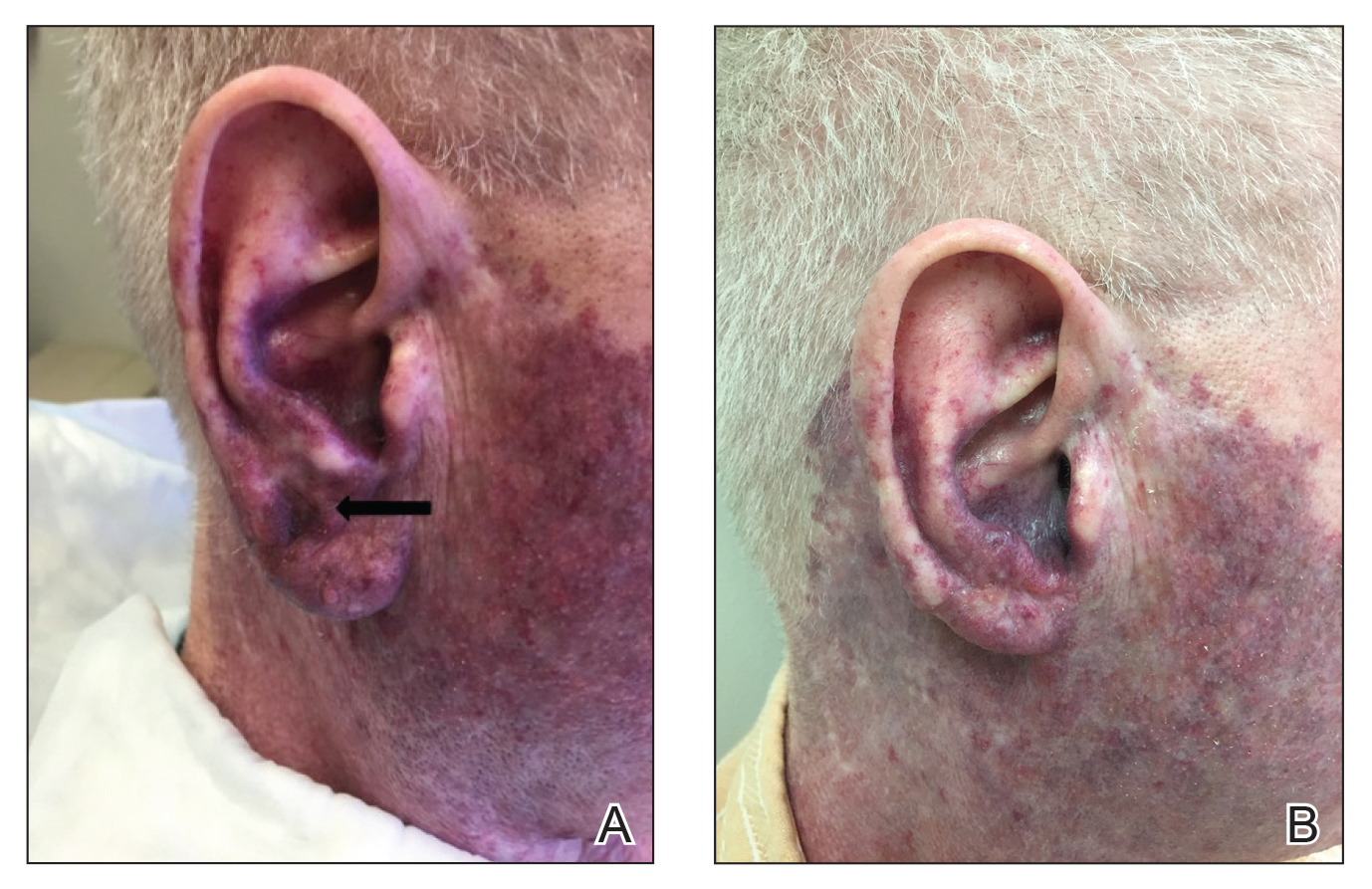
The patient underwent a wedge excision of the lesion with 5-mm margins, resulting in a final postoperative size of 2.5×3.5 cm. There was no excessive bleeding with surgery. A delayed repair was done after clear margins were confirmed by pathology (Figure 1B).
Port-wine stains are congenital vascular malformations that affect approximately 0.3% of individuals.1 Most are located on the head and neck along the distribution of the trigeminal nerve. Cases are thought to occur sporadically, with recent evidence for somatic GNAQ mutations in both nonsyndromic cases and in Sturge-Weber syndrome.2 These lesions become progressively larger with time due to dilation of the capillary proliferation.3 Melanoma in situ, lentigo maligna type, usually affects white men in the sixth and seventh decades of life. It commonly arises on skin with chronic sun damage, particularly on the head and neck.4
Although uncommon, skin cancers have been known to arise in PWSs. Reports of basal cell carcinomas (BCCs) and squamous cell carcinomas (SCCs) have been published, but to date, there are no reports of melanoma or melanoma in situ arising in a PWS. According to a PubMed search of articles indexed for MEDLINE using the terms melanoma and port wine stain, squamous cell carcinoma and port wine stain, and basal cell carcinoma and port wine stain, fewer than 30 cases of BCCs in a PWS and only 4 cases of SCCs in a PWS have been documented, with 1 patient developing multiple BCCs and SCCs.1,5 Most BCCs (approximately 75%) and SCCs have been associated with historical treatments used to treat PWS before the development of laser therapy, such as grenz rays, topical thorium X, and other radiotherapy techniques.5,6 Interestingly, our patient’s PWS had only been treated with a PDL. Other risk factors for skin cancer in a PWS include sun exposure and smoking.5 There is no evidence that a PDL contributes to the development of skin cancer, but radiotherapy is a major factor.7
Treatment of these skin cancers is no different, with both Mohs micrographic surgery and standard excision used when appropriate. Despite the vascular nature of the lesion, there is only a minimal increase in bleeding risk.3 Most reports indicate no increase in perioperative bleeding.5,7 One case documented a hematoma developing postoperatively.6
This case of melanoma in situ arising in a PWS expands the range of skin cancer types known to arise in these malformations. Because of the potential for skin cancer to develop in a PWS, it is important to routinely examine these vascular proliferations.
- Hackett CB, Langtry JA. Basal cell carcinoma of the ala nasi arising in a port wine stain treated using Mohs micrographic surgery and local flap reconstruction. Dermatol Surg. 2014;40:590-592.
- Shirley MD, Tang H, Gallione CJ, et al. Sturge-Weber syndrome and port-wine stains caused by somatic mutation in GNAQ. N Engl J Med. 2013;368:1971-1979.
- Cerrati EW, O TM, Binetter D, et al. Surgical treatment of head and neck port-wine stains by means of a staged zonal approach. Plast Reconstr Surg. 2014;134:1003-1012.
- Kallini JR, Jain SK, Khachemoune A. Lentigo maligna: review of salient characteristics and management. Am J Clin Dermatol. 2013;14:473-480.
- Rajan N, Ryan J, Langtry JA. Squamous cell carcinoma arising within a facial port-wine stain treated by Mohs micrographic surgical excision. Dermatol Surg. 2006;32:864-866.
- Silapunt S, Goldberg LH, Thurber M, et al. Basal cell carcinoma arising in a port-wine stain. Dermatol Surg. 2004;30:1241-1245.
- Jasim ZF, Woo WK, Walsh MY, et al. Multifocal basal cell carcinoma developing in a facial port wine stain treated with argon and pulsed dye laser: a possible role for previous radiotherapy. Dermatol Surg. 2004;30:1155-1157.
To the Editor:
Port-wine stains (PWSs) are the most common type of vascular malformations. Patients rarely develop cancers in the overlying skin. However, we describe a case of melanoma in situ occurring within a long-standing facial PWS.
A 60-year-old white man with a history of a large unilateral facial PWS covering the right ear, lateral cheek, jaw, and neck presented to clinic with a new dark lesion on the right ear that had been growing for a few weeks or more. His PWS had been previously treated intermittently with a pulsed dye laser (PDL) for decades with variable improvement. He had not undergone any laser procedures in the last 8 months but wanted to restart treatment with the PDL. Upon further discussion, he reported a new darker area on the right earlobe that was growing. He had no personal or family history of skin cancer and was otherwise healthy. Physical examination revealed a large red vascular patch encompassing the ear, cheek, chin, and lateral neck. Within the PWS there was a black and dark brown patch with irregular borders on the right earlobe (Figure 1A). A shave biopsy was performed for histopathologic examination. The biopsy showed a confluent proliferation of atypical melanocytes along the dermoepidermal junction extending down adnexal structures (Figure 2A) that stained positive for MART-1/Melan-A (Figure 2B). In the dermis, solar elastosis and prominent dilated and thin-walled vessels were present. These findings were consistent with a melanoma in situ, lentigo maligna type, overlying a capillary malformation.
The patient underwent a wedge excision of the lesion with 5-mm margins, resulting in a final postoperative size of 2.5×3.5 cm. There was no excessive bleeding with surgery. A delayed repair was done after clear margins were confirmed by pathology (Figure 1B).
Port-wine stains are congenital vascular malformations that affect approximately 0.3% of individuals.1 Most are located on the head and neck along the distribution of the trigeminal nerve. Cases are thought to occur sporadically, with recent evidence for somatic GNAQ mutations in both nonsyndromic cases and in Sturge-Weber syndrome.2 These lesions become progressively larger with time due to dilation of the capillary proliferation.3 Melanoma in situ, lentigo maligna type, usually affects white men in the sixth and seventh decades of life. It commonly arises on skin with chronic sun damage, particularly on the head and neck.4
Although uncommon, skin cancers have been known to arise in PWSs. Reports of basal cell carcinomas (BCCs) and squamous cell carcinomas (SCCs) have been published, but to date, there are no reports of melanoma or melanoma in situ arising in a PWS. According to a PubMed search of articles indexed for MEDLINE using the terms melanoma and port wine stain, squamous cell carcinoma and port wine stain, and basal cell carcinoma and port wine stain, fewer than 30 cases of BCCs in a PWS and only 4 cases of SCCs in a PWS have been documented, with 1 patient developing multiple BCCs and SCCs.1,5 Most BCCs (approximately 75%) and SCCs have been associated with historical treatments used to treat PWS before the development of laser therapy, such as grenz rays, topical thorium X, and other radiotherapy techniques.5,6 Interestingly, our patient’s PWS had only been treated with a PDL. Other risk factors for skin cancer in a PWS include sun exposure and smoking.5 There is no evidence that a PDL contributes to the development of skin cancer, but radiotherapy is a major factor.7
Treatment of these skin cancers is no different, with both Mohs micrographic surgery and standard excision used when appropriate. Despite the vascular nature of the lesion, there is only a minimal increase in bleeding risk.3 Most reports indicate no increase in perioperative bleeding.5,7 One case documented a hematoma developing postoperatively.6
This case of melanoma in situ arising in a PWS expands the range of skin cancer types known to arise in these malformations. Because of the potential for skin cancer to develop in a PWS, it is important to routinely examine these vascular proliferations.
To the Editor:
Port-wine stains (PWSs) are the most common type of vascular malformations. Patients rarely develop cancers in the overlying skin. However, we describe a case of melanoma in situ occurring within a long-standing facial PWS.
A 60-year-old white man with a history of a large unilateral facial PWS covering the right ear, lateral cheek, jaw, and neck presented to clinic with a new dark lesion on the right ear that had been growing for a few weeks or more. His PWS had been previously treated intermittently with a pulsed dye laser (PDL) for decades with variable improvement. He had not undergone any laser procedures in the last 8 months but wanted to restart treatment with the PDL. Upon further discussion, he reported a new darker area on the right earlobe that was growing. He had no personal or family history of skin cancer and was otherwise healthy. Physical examination revealed a large red vascular patch encompassing the ear, cheek, chin, and lateral neck. Within the PWS there was a black and dark brown patch with irregular borders on the right earlobe (Figure 1A). A shave biopsy was performed for histopathologic examination. The biopsy showed a confluent proliferation of atypical melanocytes along the dermoepidermal junction extending down adnexal structures (Figure 2A) that stained positive for MART-1/Melan-A (Figure 2B). In the dermis, solar elastosis and prominent dilated and thin-walled vessels were present. These findings were consistent with a melanoma in situ, lentigo maligna type, overlying a capillary malformation.
The patient underwent a wedge excision of the lesion with 5-mm margins, resulting in a final postoperative size of 2.5×3.5 cm. There was no excessive bleeding with surgery. A delayed repair was done after clear margins were confirmed by pathology (Figure 1B).
Port-wine stains are congenital vascular malformations that affect approximately 0.3% of individuals.1 Most are located on the head and neck along the distribution of the trigeminal nerve. Cases are thought to occur sporadically, with recent evidence for somatic GNAQ mutations in both nonsyndromic cases and in Sturge-Weber syndrome.2 These lesions become progressively larger with time due to dilation of the capillary proliferation.3 Melanoma in situ, lentigo maligna type, usually affects white men in the sixth and seventh decades of life. It commonly arises on skin with chronic sun damage, particularly on the head and neck.4
Although uncommon, skin cancers have been known to arise in PWSs. Reports of basal cell carcinomas (BCCs) and squamous cell carcinomas (SCCs) have been published, but to date, there are no reports of melanoma or melanoma in situ arising in a PWS. According to a PubMed search of articles indexed for MEDLINE using the terms melanoma and port wine stain, squamous cell carcinoma and port wine stain, and basal cell carcinoma and port wine stain, fewer than 30 cases of BCCs in a PWS and only 4 cases of SCCs in a PWS have been documented, with 1 patient developing multiple BCCs and SCCs.1,5 Most BCCs (approximately 75%) and SCCs have been associated with historical treatments used to treat PWS before the development of laser therapy, such as grenz rays, topical thorium X, and other radiotherapy techniques.5,6 Interestingly, our patient’s PWS had only been treated with a PDL. Other risk factors for skin cancer in a PWS include sun exposure and smoking.5 There is no evidence that a PDL contributes to the development of skin cancer, but radiotherapy is a major factor.7
Treatment of these skin cancers is no different, with both Mohs micrographic surgery and standard excision used when appropriate. Despite the vascular nature of the lesion, there is only a minimal increase in bleeding risk.3 Most reports indicate no increase in perioperative bleeding.5,7 One case documented a hematoma developing postoperatively.6
This case of melanoma in situ arising in a PWS expands the range of skin cancer types known to arise in these malformations. Because of the potential for skin cancer to develop in a PWS, it is important to routinely examine these vascular proliferations.
- Hackett CB, Langtry JA. Basal cell carcinoma of the ala nasi arising in a port wine stain treated using Mohs micrographic surgery and local flap reconstruction. Dermatol Surg. 2014;40:590-592.
- Shirley MD, Tang H, Gallione CJ, et al. Sturge-Weber syndrome and port-wine stains caused by somatic mutation in GNAQ. N Engl J Med. 2013;368:1971-1979.
- Cerrati EW, O TM, Binetter D, et al. Surgical treatment of head and neck port-wine stains by means of a staged zonal approach. Plast Reconstr Surg. 2014;134:1003-1012.
- Kallini JR, Jain SK, Khachemoune A. Lentigo maligna: review of salient characteristics and management. Am J Clin Dermatol. 2013;14:473-480.
- Rajan N, Ryan J, Langtry JA. Squamous cell carcinoma arising within a facial port-wine stain treated by Mohs micrographic surgical excision. Dermatol Surg. 2006;32:864-866.
- Silapunt S, Goldberg LH, Thurber M, et al. Basal cell carcinoma arising in a port-wine stain. Dermatol Surg. 2004;30:1241-1245.
- Jasim ZF, Woo WK, Walsh MY, et al. Multifocal basal cell carcinoma developing in a facial port wine stain treated with argon and pulsed dye laser: a possible role for previous radiotherapy. Dermatol Surg. 2004;30:1155-1157.
- Hackett CB, Langtry JA. Basal cell carcinoma of the ala nasi arising in a port wine stain treated using Mohs micrographic surgery and local flap reconstruction. Dermatol Surg. 2014;40:590-592.
- Shirley MD, Tang H, Gallione CJ, et al. Sturge-Weber syndrome and port-wine stains caused by somatic mutation in GNAQ. N Engl J Med. 2013;368:1971-1979.
- Cerrati EW, O TM, Binetter D, et al. Surgical treatment of head and neck port-wine stains by means of a staged zonal approach. Plast Reconstr Surg. 2014;134:1003-1012.
- Kallini JR, Jain SK, Khachemoune A. Lentigo maligna: review of salient characteristics and management. Am J Clin Dermatol. 2013;14:473-480.
- Rajan N, Ryan J, Langtry JA. Squamous cell carcinoma arising within a facial port-wine stain treated by Mohs micrographic surgical excision. Dermatol Surg. 2006;32:864-866.
- Silapunt S, Goldberg LH, Thurber M, et al. Basal cell carcinoma arising in a port-wine stain. Dermatol Surg. 2004;30:1241-1245.
- Jasim ZF, Woo WK, Walsh MY, et al. Multifocal basal cell carcinoma developing in a facial port wine stain treated with argon and pulsed dye laser: a possible role for previous radiotherapy. Dermatol Surg. 2004;30:1155-1157.
Practice Points
- Nonmelanoma skin cancer is known to develop in port-wine stains, most commonly basal cell carcinoma.
- The range of skin cancer types known to arise in these malformations can be expanded to include melanoma in situ.
- It is important to routinely examine these vascular proliferations for new lesions.
Kaposi Sarcoma in a Patient With Postpolio Syndrome
Kaposi sarcoma (KS) is a low-grade vascular tumor that is rare among the general US population, with an incidence rate of less than 1 per 100,000.1 The tumor is more common among certain groups of individuals due to geographic differences in the prevalence of KS-associated herpesvirus (also referred to as human herpesvirus 8) as well as host immune factors.2 Kaposi sarcoma often is defined by the patient's predisposing characteristics yielding the following distinct epidemiologic subtypes: (1) classic KS is a rare disease affecting older men of Mediterranean descent; (2) African KS is an endemic cancer with male predominance in sub-Saharan Africa; (3) AIDS-associated KS is an often aggressive AIDS-defining illness; and (4) iatrogenic KS occurs in patients on immunosuppressive therapy.3 When evaluating a patient without any of these risk factors, the clinical suspicion for KS may be low. We report a patient with postpolio syndrome (PPS) who presented with KS of the right leg, ankle, and foot.
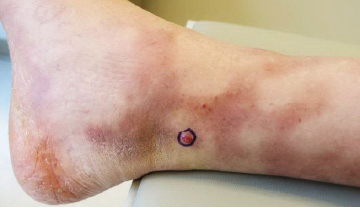
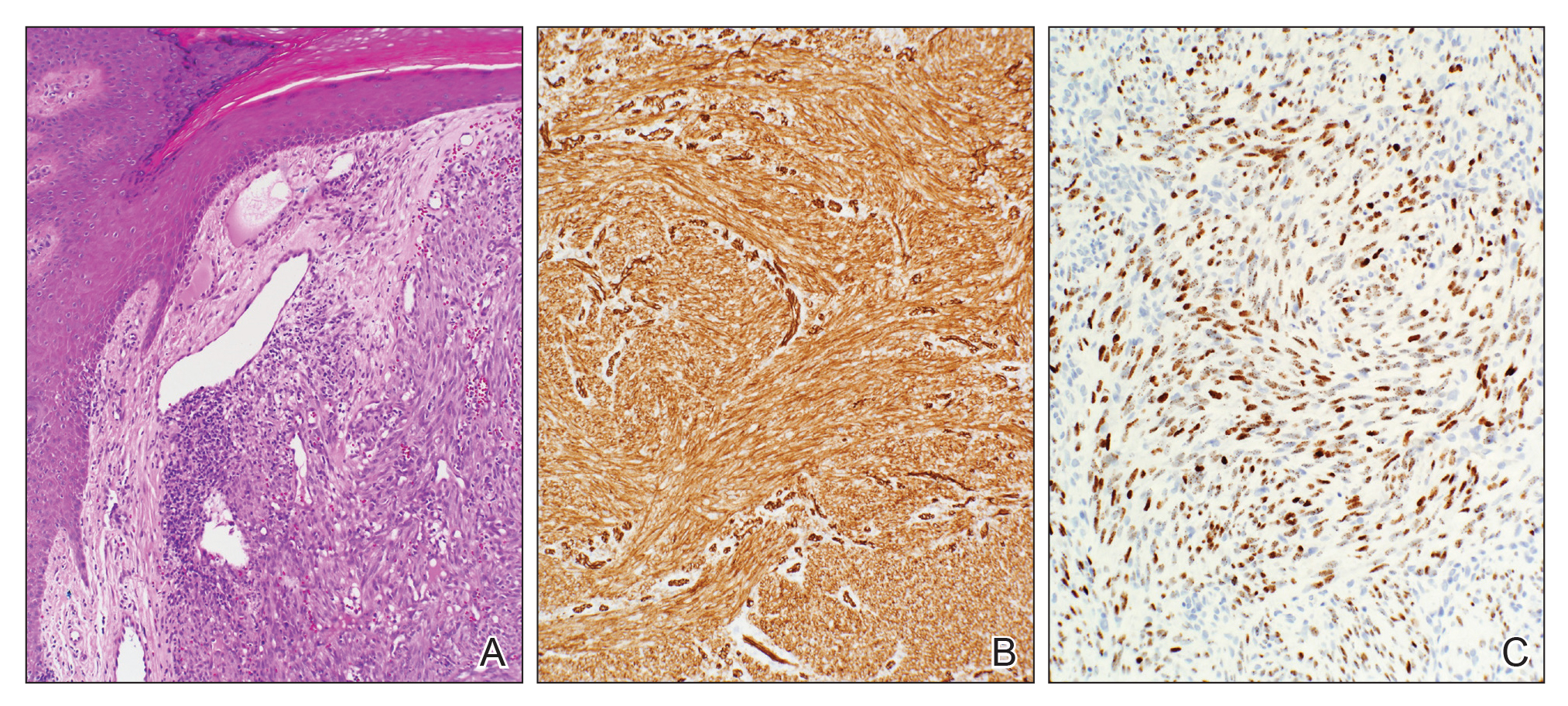
A 77-year-old man with a distant history of paralytic poliomyelitis presented for an annual skin examination with concern for a new lesion on the right ankle. The patient had a history of PPS primarily affecting the right leg. Physical examination revealed residual weakness in an atrophic right lower extremity with a mottled appearance and mild pitting edema to the knee. Two red, dome-shaped, vascular papules were appreciated on the medial aspect of the right ankle (Figure 1), and a shave biopsy of the larger papule was performed. Microscopic examination of the biopsy specimen was consistent with KS (Figure 2). This patient had no history of human immunodeficiency virus or immunosuppressive therapy and was not of Mediterranean descent.
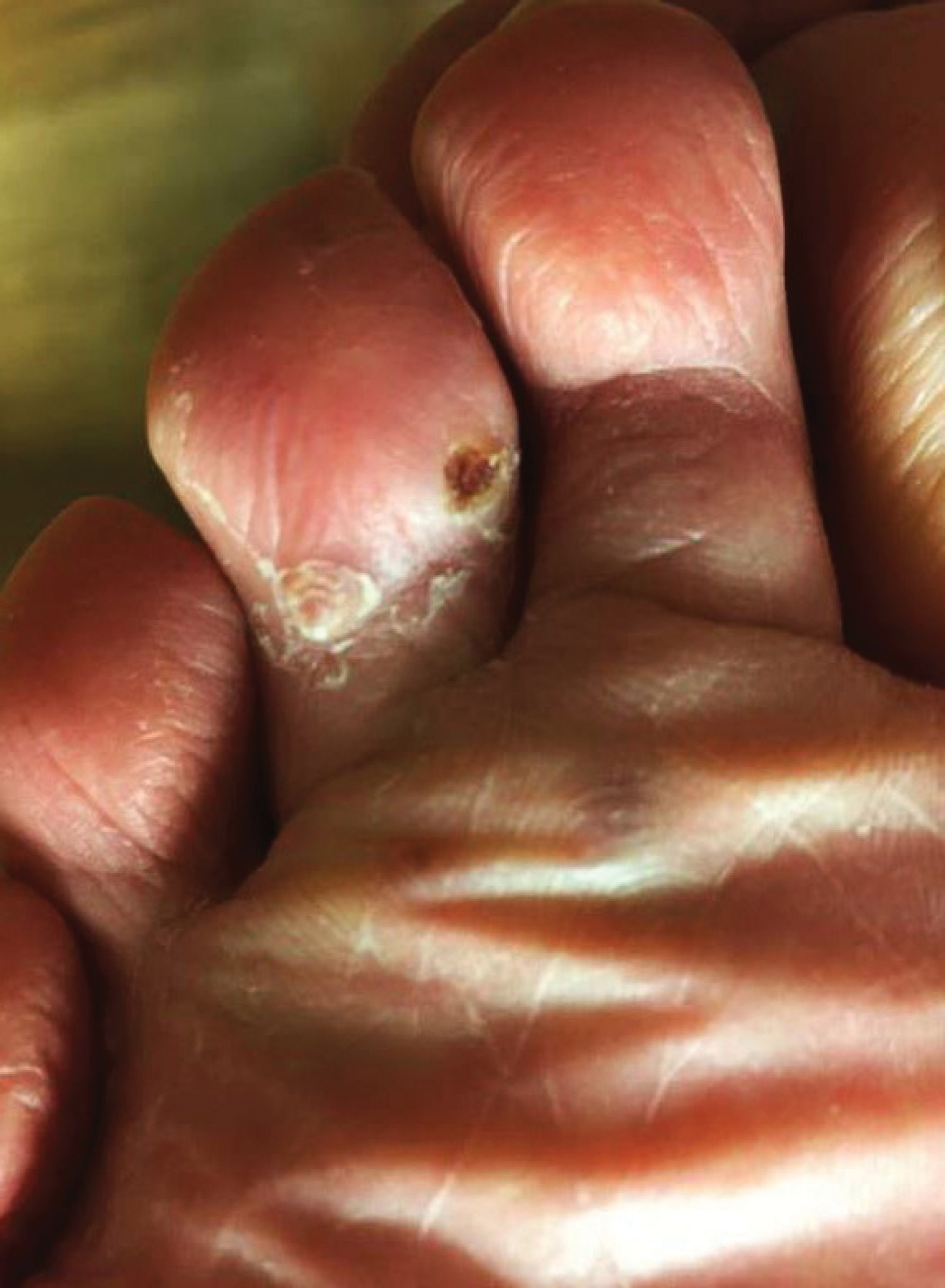
Because KS is a radiosensitive vascular neoplasm and radiation therapy (RT) alone can achieve local control,4 the patient was treated with 6 megaelectron-volt electron-beam RT. He received 30 Gy in 10 fractions to the affected area of the medial ankle. The patient tolerated RT well. Three weeks after completing treatment, he was found to have mild lichenification on the right medial ankle with no clinical evidence of disease. Four months later, he presented with multiple additional vascular papules on the right third toe and in the interdigital web space (Figure 3). Shave biopsy of one of these lesions was consistent with KS. Contrast computed tomography of the chest, abdomen, and pelvis was performed, revealing no evidence of metastatic disease. The patient was treated with 30 Gy in 15 fractions using opposed lateral 6 megaelectron-volt photon fields to the entire right lower extremity below the knee to treat all of the skin affected by the PPS. His posttreatment course was complicated by edema in the affected leg that resolved after daily pneumatic compression. He had no evidence of residual or recurrent disease 6 months after completing RT (Figure 4).
Cutaneous KS is a human herpesvirus 8-positive tumor of endothelial origin typically seen in older men of Mediterranean or African descent and among immunosuppressed patients.4 Our patient did not have any classic risk factors for KS, but his disease did arise in the setting of a right lower extremity that was notably affected by PPS. Postpolio syndrome is characterized by muscle atrophy due to denervation of the motor unit.5 Bruno et al6 found that such deficits in motor innervation could lead to impairments in venous outflow causing cutaneous venous congestion. Acroangiodermatitis clinically resembles KS but is a benign reactive vasoproliferative disorder and is well known to occur in the lower extremities as a sequela of chronic venous insufficiency.7 A case of bilateral lower extremity pseudo-KS was reported in a patient with notable PPS.8 A report of 2 patients describes KS arising in the setting of chronic venous insufficiency without any classic risk factors.9 Therefore, patients with PPS characterized by venous insufficiency may represent a population at increased risk for KS.
- Surveillance, Epidemiology, and End Results (SEER) Program. US Population Data--1969-2017. https://seer.cancer.gov/popdata/. Published January 2019. Accessed November 25, 2019.
- Uldrick TS, Whitby D. Update on KSHV epidemiology, kaposi sarcoma pathogenesis, and treatment of saposi sarcoma. Cancer Lett. 2011;305:150-162.
- Schwartz RA, Micali G, Nasca MR, et al. Kaposi sarcoma: a continuing conundrum. J Am Acad Dermatol. 2008;59:179-206.
- Arnold HL, Odom RB, James WD, et al. Andrews' Diseases of the Skin: Clinical Dermatology. Philadelphia, PA: Saunders; 1990.
- Boyer FV, Tiffreau V, Rapin A, et al. Post-polio syndrome: pathophysiological hypotheses, diagnosis criteria, drug therapy. Ann Phys Rehabil Med. 2010;53:34-41.
- Bruno RL, Johnson JC, Berman WS. Vasomotor abnormalities as post-polio sequelae: functional and clinical implications. Orthopedics. 1985;8:865-869.
- Palmer B, Xia Y, Cho S, Lewis FS. Acroangiodermatitis secondary to chronic venous insufficiency. Cutis. 2010;86:239-240.
- Rotbart G. Kaposi's disease and venous insufficiency. Phlebologie. 1978;31:439-443.
- Que SK, DeFelice T, Abdulla FR, et al. Non-HIV-related kaposi sarcoma in 2 Hispanic patients arising in the setting of chronic venous insufficiency. Cutis. 2015;95:E30-E33.
Kaposi sarcoma (KS) is a low-grade vascular tumor that is rare among the general US population, with an incidence rate of less than 1 per 100,000.1 The tumor is more common among certain groups of individuals due to geographic differences in the prevalence of KS-associated herpesvirus (also referred to as human herpesvirus 8) as well as host immune factors.2 Kaposi sarcoma often is defined by the patient's predisposing characteristics yielding the following distinct epidemiologic subtypes: (1) classic KS is a rare disease affecting older men of Mediterranean descent; (2) African KS is an endemic cancer with male predominance in sub-Saharan Africa; (3) AIDS-associated KS is an often aggressive AIDS-defining illness; and (4) iatrogenic KS occurs in patients on immunosuppressive therapy.3 When evaluating a patient without any of these risk factors, the clinical suspicion for KS may be low. We report a patient with postpolio syndrome (PPS) who presented with KS of the right leg, ankle, and foot.
A 77-year-old man with a distant history of paralytic poliomyelitis presented for an annual skin examination with concern for a new lesion on the right ankle. The patient had a history of PPS primarily affecting the right leg. Physical examination revealed residual weakness in an atrophic right lower extremity with a mottled appearance and mild pitting edema to the knee. Two red, dome-shaped, vascular papules were appreciated on the medial aspect of the right ankle (Figure 1), and a shave biopsy of the larger papule was performed. Microscopic examination of the biopsy specimen was consistent with KS (Figure 2). This patient had no history of human immunodeficiency virus or immunosuppressive therapy and was not of Mediterranean descent.
Because KS is a radiosensitive vascular neoplasm and radiation therapy (RT) alone can achieve local control,4 the patient was treated with 6 megaelectron-volt electron-beam RT. He received 30 Gy in 10 fractions to the affected area of the medial ankle. The patient tolerated RT well. Three weeks after completing treatment, he was found to have mild lichenification on the right medial ankle with no clinical evidence of disease. Four months later, he presented with multiple additional vascular papules on the right third toe and in the interdigital web space (Figure 3). Shave biopsy of one of these lesions was consistent with KS. Contrast computed tomography of the chest, abdomen, and pelvis was performed, revealing no evidence of metastatic disease. The patient was treated with 30 Gy in 15 fractions using opposed lateral 6 megaelectron-volt photon fields to the entire right lower extremity below the knee to treat all of the skin affected by the PPS. His posttreatment course was complicated by edema in the affected leg that resolved after daily pneumatic compression. He had no evidence of residual or recurrent disease 6 months after completing RT (Figure 4).
Cutaneous KS is a human herpesvirus 8-positive tumor of endothelial origin typically seen in older men of Mediterranean or African descent and among immunosuppressed patients.4 Our patient did not have any classic risk factors for KS, but his disease did arise in the setting of a right lower extremity that was notably affected by PPS. Postpolio syndrome is characterized by muscle atrophy due to denervation of the motor unit.5 Bruno et al6 found that such deficits in motor innervation could lead to impairments in venous outflow causing cutaneous venous congestion. Acroangiodermatitis clinically resembles KS but is a benign reactive vasoproliferative disorder and is well known to occur in the lower extremities as a sequela of chronic venous insufficiency.7 A case of bilateral lower extremity pseudo-KS was reported in a patient with notable PPS.8 A report of 2 patients describes KS arising in the setting of chronic venous insufficiency without any classic risk factors.9 Therefore, patients with PPS characterized by venous insufficiency may represent a population at increased risk for KS.
Kaposi sarcoma (KS) is a low-grade vascular tumor that is rare among the general US population, with an incidence rate of less than 1 per 100,000.1 The tumor is more common among certain groups of individuals due to geographic differences in the prevalence of KS-associated herpesvirus (also referred to as human herpesvirus 8) as well as host immune factors.2 Kaposi sarcoma often is defined by the patient's predisposing characteristics yielding the following distinct epidemiologic subtypes: (1) classic KS is a rare disease affecting older men of Mediterranean descent; (2) African KS is an endemic cancer with male predominance in sub-Saharan Africa; (3) AIDS-associated KS is an often aggressive AIDS-defining illness; and (4) iatrogenic KS occurs in patients on immunosuppressive therapy.3 When evaluating a patient without any of these risk factors, the clinical suspicion for KS may be low. We report a patient with postpolio syndrome (PPS) who presented with KS of the right leg, ankle, and foot.
A 77-year-old man with a distant history of paralytic poliomyelitis presented for an annual skin examination with concern for a new lesion on the right ankle. The patient had a history of PPS primarily affecting the right leg. Physical examination revealed residual weakness in an atrophic right lower extremity with a mottled appearance and mild pitting edema to the knee. Two red, dome-shaped, vascular papules were appreciated on the medial aspect of the right ankle (Figure 1), and a shave biopsy of the larger papule was performed. Microscopic examination of the biopsy specimen was consistent with KS (Figure 2). This patient had no history of human immunodeficiency virus or immunosuppressive therapy and was not of Mediterranean descent.
Because KS is a radiosensitive vascular neoplasm and radiation therapy (RT) alone can achieve local control,4 the patient was treated with 6 megaelectron-volt electron-beam RT. He received 30 Gy in 10 fractions to the affected area of the medial ankle. The patient tolerated RT well. Three weeks after completing treatment, he was found to have mild lichenification on the right medial ankle with no clinical evidence of disease. Four months later, he presented with multiple additional vascular papules on the right third toe and in the interdigital web space (Figure 3). Shave biopsy of one of these lesions was consistent with KS. Contrast computed tomography of the chest, abdomen, and pelvis was performed, revealing no evidence of metastatic disease. The patient was treated with 30 Gy in 15 fractions using opposed lateral 6 megaelectron-volt photon fields to the entire right lower extremity below the knee to treat all of the skin affected by the PPS. His posttreatment course was complicated by edema in the affected leg that resolved after daily pneumatic compression. He had no evidence of residual or recurrent disease 6 months after completing RT (Figure 4).
Cutaneous KS is a human herpesvirus 8-positive tumor of endothelial origin typically seen in older men of Mediterranean or African descent and among immunosuppressed patients.4 Our patient did not have any classic risk factors for KS, but his disease did arise in the setting of a right lower extremity that was notably affected by PPS. Postpolio syndrome is characterized by muscle atrophy due to denervation of the motor unit.5 Bruno et al6 found that such deficits in motor innervation could lead to impairments in venous outflow causing cutaneous venous congestion. Acroangiodermatitis clinically resembles KS but is a benign reactive vasoproliferative disorder and is well known to occur in the lower extremities as a sequela of chronic venous insufficiency.7 A case of bilateral lower extremity pseudo-KS was reported in a patient with notable PPS.8 A report of 2 patients describes KS arising in the setting of chronic venous insufficiency without any classic risk factors.9 Therefore, patients with PPS characterized by venous insufficiency may represent a population at increased risk for KS.
- Surveillance, Epidemiology, and End Results (SEER) Program. US Population Data--1969-2017. https://seer.cancer.gov/popdata/. Published January 2019. Accessed November 25, 2019.
- Uldrick TS, Whitby D. Update on KSHV epidemiology, kaposi sarcoma pathogenesis, and treatment of saposi sarcoma. Cancer Lett. 2011;305:150-162.
- Schwartz RA, Micali G, Nasca MR, et al. Kaposi sarcoma: a continuing conundrum. J Am Acad Dermatol. 2008;59:179-206.
- Arnold HL, Odom RB, James WD, et al. Andrews' Diseases of the Skin: Clinical Dermatology. Philadelphia, PA: Saunders; 1990.
- Boyer FV, Tiffreau V, Rapin A, et al. Post-polio syndrome: pathophysiological hypotheses, diagnosis criteria, drug therapy. Ann Phys Rehabil Med. 2010;53:34-41.
- Bruno RL, Johnson JC, Berman WS. Vasomotor abnormalities as post-polio sequelae: functional and clinical implications. Orthopedics. 1985;8:865-869.
- Palmer B, Xia Y, Cho S, Lewis FS. Acroangiodermatitis secondary to chronic venous insufficiency. Cutis. 2010;86:239-240.
- Rotbart G. Kaposi's disease and venous insufficiency. Phlebologie. 1978;31:439-443.
- Que SK, DeFelice T, Abdulla FR, et al. Non-HIV-related kaposi sarcoma in 2 Hispanic patients arising in the setting of chronic venous insufficiency. Cutis. 2015;95:E30-E33.
- Surveillance, Epidemiology, and End Results (SEER) Program. US Population Data--1969-2017. https://seer.cancer.gov/popdata/. Published January 2019. Accessed November 25, 2019.
- Uldrick TS, Whitby D. Update on KSHV epidemiology, kaposi sarcoma pathogenesis, and treatment of saposi sarcoma. Cancer Lett. 2011;305:150-162.
- Schwartz RA, Micali G, Nasca MR, et al. Kaposi sarcoma: a continuing conundrum. J Am Acad Dermatol. 2008;59:179-206.
- Arnold HL, Odom RB, James WD, et al. Andrews' Diseases of the Skin: Clinical Dermatology. Philadelphia, PA: Saunders; 1990.
- Boyer FV, Tiffreau V, Rapin A, et al. Post-polio syndrome: pathophysiological hypotheses, diagnosis criteria, drug therapy. Ann Phys Rehabil Med. 2010;53:34-41.
- Bruno RL, Johnson JC, Berman WS. Vasomotor abnormalities as post-polio sequelae: functional and clinical implications. Orthopedics. 1985;8:865-869.
- Palmer B, Xia Y, Cho S, Lewis FS. Acroangiodermatitis secondary to chronic venous insufficiency. Cutis. 2010;86:239-240.
- Rotbart G. Kaposi's disease and venous insufficiency. Phlebologie. 1978;31:439-443.
- Que SK, DeFelice T, Abdulla FR, et al. Non-HIV-related kaposi sarcoma in 2 Hispanic patients arising in the setting of chronic venous insufficiency. Cutis. 2015;95:E30-E33.
Practice Points
- Cutaneous Kaposi sarcoma (KS) is a human herpesvirus 8–positive tumor of endothelial origin typically seen in older men of Mediterranean or African descent and among immunosuppressed patients.
- In addition, patients with postpolio syndrome characterized by venous insufficiency may represent a population at increased risk for KS.
- Kaposi sarcoma is a radiosensitive vascular neoplasm, and radiation therapy can achieve local control.
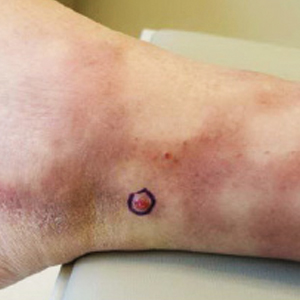
Papulonecrotic Tuberculid Secondary to Mycobacterium avium Complex
To the Editor:
Papulonecrotic tuberculid (PNT) is a cutaneous hypersensitivity reaction to antigenic components of Mycobacterium species, most commonly Mycobacterium tuberculosis. According to a PubMed search of articles indexed for MEDLINE using the terms papulonecrotic tuberculid, Mycobacterium avium complex, and Mycobacterium, only 1 case of PNT secondary to infection with Mycobacterium avium complex (MAC) has been reported.1,2 Papulonecrotic tuberculid classically presents with symmetrical, dusky red papules with necrosis on the extremities.3 Patients may or may not have associated symptoms of fever and weight loss. It is diagnosed through skin biopsy as well as identification of a distant source of mycobacterial infection. Papulonecrotic tuberculid is considered a reactive process to a distant site of mycobacterial infection, and skin lesions contain few, if any, mycobacteria.4
A 65-year-old man was admitted to the hospital for expedited workup of chronic fevers, 20-lb weight loss, and night sweats of 8 months’ duration. He had a medical history of myelodysplastic syndrome and autoimmune hemolytic anemia. During hospitalization, positron emission tomography revealed multilevel vertebral lytic and sclerotic lesions. Subsequent T10 vertebral biopsy showed necrotizing granulomatous inflammation with extensive necrosis and acid-fast bacilli–positive organisms. The patient was empirically started on rifampicin, isoniazid, pyrazinamide, ethambutol, and pyridoxine for presumed M tuberculosis and placed on respiratory isolation.
Dermatology was consulted for a recurrent tender rash on the bilateral upper and lower extremities of 5 years’ duration. Physical examination revealed numerous erythematous papulonecrotic lesions in various states of healing on the bilateral upper and lower extremities (Figure 1). Three years prior to the current presentation, 2 lesions were biopsied and demonstrated leukocytoclastic vasculitis with neutrophilic panniculitis and vasculopathy. A presumptive diagnosis of Sweet syndrome was made given the history of myelodysplastic syndrome, though an infectious etiology could not be ruled out at that time. Concurrently, the patient was diagnosed with autoimmune hemolytic anemia and was started on prednisone. Initially, the skin lesions improved with prednisone but never fully resolved; however, as the dosage of oral steroids decreased, the skin lesions worsened and presented in larger numbers with more frequency. The patient was titrated down to prednisone 5 mg daily with no additional treatment of the skin lesions at that time.
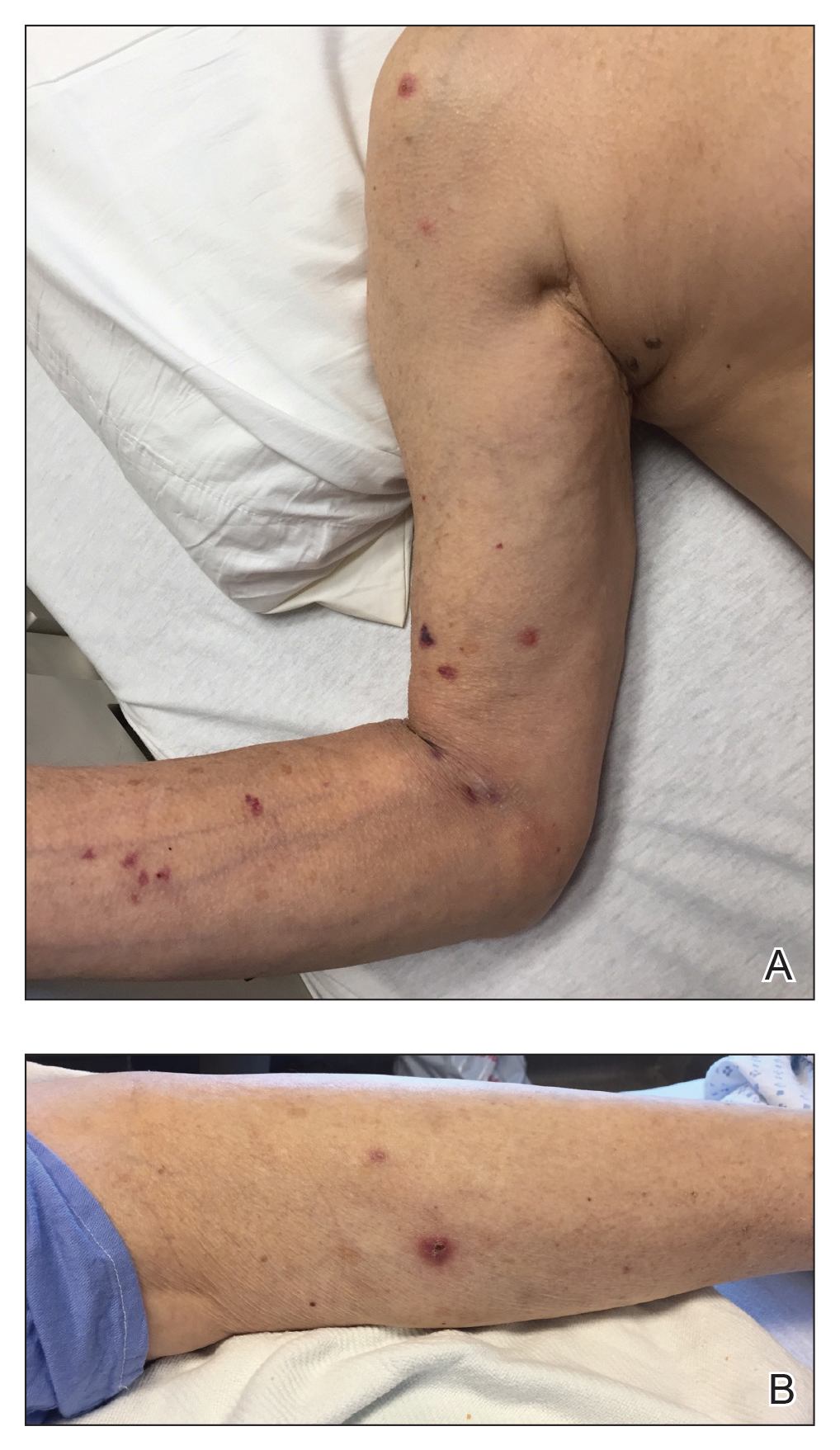
During the current hospitalization, 2 additional biopsies were taken from the arm for routine histopathology and tissue culture. Dermatopathology revealed robust neutrophilic and granulomatous inflammation as well as remarkable necrosis with a few mycobacteria identified on acid-fast and Fite stains (Figure 2). Tissue culture was negative. Additionally, the patient’s spinal biopsy was sent for polymerase chain reaction analysis for Mycobacterium typing, which confirmed MAC. The patient was diagnosed with Pott disease, a mycobacterial infection of the spine, as well as cutaneous papulonecrotic tuberculid secondary to MAC.
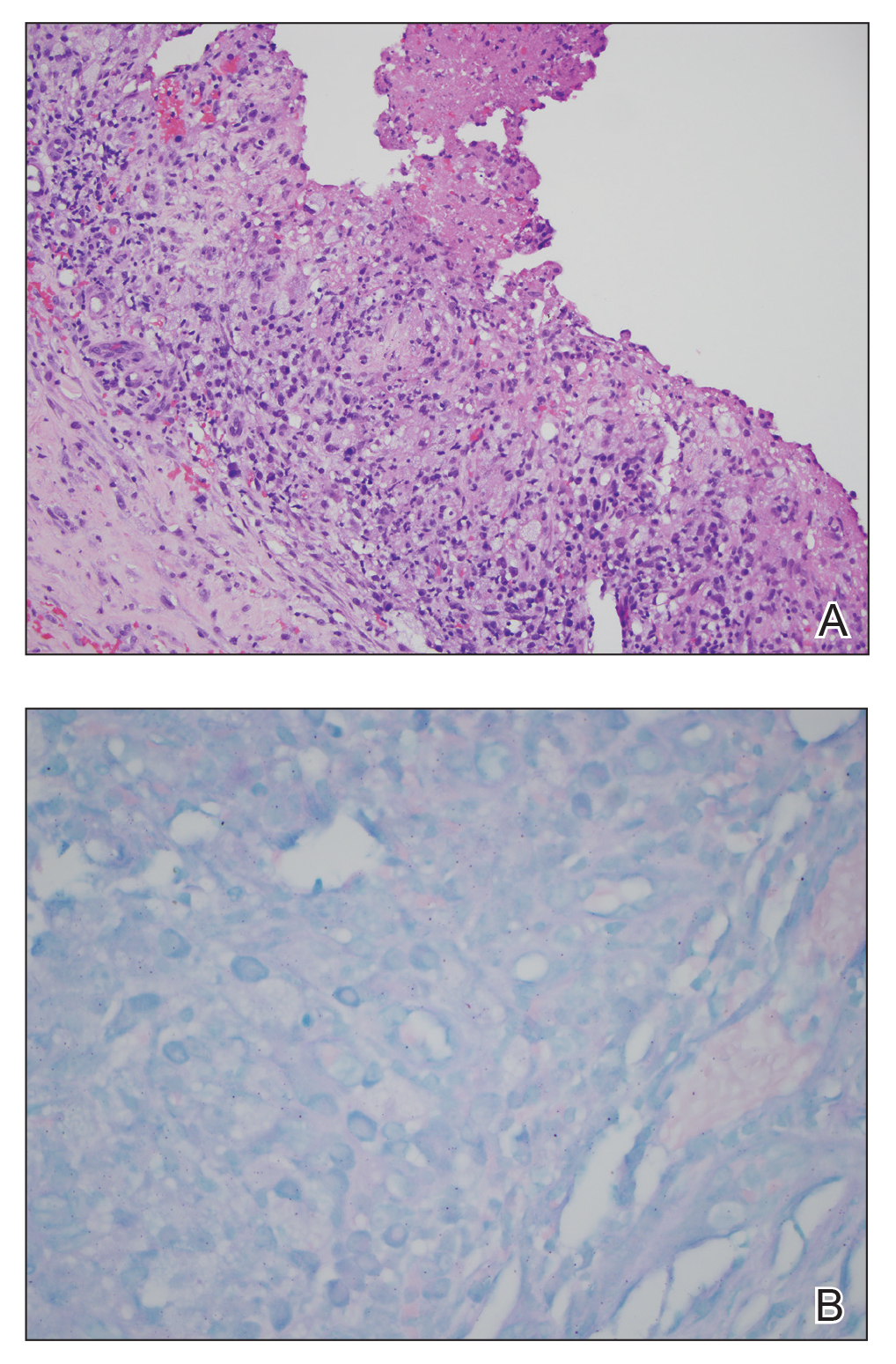
Papulonecrotic tuberculid is the rarest form of cutaneous tuberculosis infection and rarely has been reported in connection to MAC.1 This condition is considered a hypersensitivity reaction that occurs in response to antigenic components of mycobacteria.4 Patients with PNT typically present with recurrent crops of painful papulonecrotic lesions distributed on the extremities. Histopathology in PNT classically reveals necrosis, notable inflammatory infiltrate, and lack of observed organisms.5 Diagnosis often is made through skin biopsy, though histopathology varies based on lesion maturity.4 Early lesions often reveal leukocytoclastic vasculitis, whereas late lesions usually demonstrate granulomatous inflammation.4 Mycobacterium avium complex is difficult to culture, as it is a slow-growing, fastidious bacterium and therefore polymerase chain reaction genotyping is useful for bacterial classification.6
Disseminated MAC infection also was on the differential for our patient; however, we felt it was less likely than PNT for several reasons. First, disseminated infection rarely presents with cutaneous involvement and is associated with pulmonary involvement in 90% of cases.7-9 Second, the granuloma formation noted on our patient’s skin biopsy was not typical for disseminated MAC but is well described in cases of PNT.4,8,9 Finally, in the rare cases in which cutaneous involvement has occurred with disseminated mycobacterial infections, skin biopsies typically revealed numerous Mycobacterium organisms.8,10 In contrast, skin lesions associated with PNT usually reveal few, if any, organisms, as was seen with our patient.2
The patient’s initial biopsies also supported a diagnosis of PNT, as early lesions of PNT typically show leukocytoclastic vasculitis. His response to low and high doses of prednisone also fit well with a PNT diagnosis. In fact, a case of PNT secondary to Mycobacterium bovis similarly showed an improvement in the rash with high-dose steroids but progression with lower doses.11 It is possible that our patient’s response to steroids complicated the diagnosis of his rash.
The treatment of PNT is clearance of the underlying infection. Macrolide antibiotics, such as clarithromycin and azithromycin, have the best efficacy against MAC, in combination with ethambutol and/or rifabutin.6,12 Treatment duration should be 1 year. Amikacin or streptomycin may be added to this regimen during early treatment.6 Mycobacterium avium complex is resistant to many antibiotics, including typical antituberculosis drugs, and sensitivities should be identified at the onset of treatment.11,12
Albeit rare, clinicians should be aware of PNT secondary to MAC or other mycobacterial infections. Because this condition is difficult to diagnose with varying histologic findings and often negative tissue cultures, a high index of suspicion is necessary when a patient presents with recurrent papulonecrotic lesions, especially in immunocompromised hosts and patients with exposure to mycobacteria.
- Williams JT, Pulitzer DR, DeVillez RL. Papulonecrotic tuberculid secondary to disseminated Mycobacterium avium complex. Int J Dermatol. 1994;33:109-112.
- Jordaan HF, Schneider JW. Papulonecrotic tuberculid. Int J Dermatol. 1995;34:217-219.
- Scollard DM, Dacso MM, Abad-Venida ML. Tuberculosis and leprosy: classical granulomatous diseases in the twenty-first century. Dermatol Clin. 2015;33:541-562.
- Kim GW, Park HJ, Kim HS, et al. Simultaneous occurrence of papulonecrotic tuberculid and erythema induratum in a patient with pulmonary tuberculosis. Pediatr Dermatol. 2013;30:256-259.
- Spelta K, Diniz LM. Cutaneous tuberculosis: a 26-year retrospective study in an endemic area. Rev Inst Med Trop Sao Paulo. 2016;58:49.
- Griffith DE, Aksamit T, Brown-Elliott BA, et al. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med. 2007;175:367-416.
- Dyer J, Weiss J, Steiner WS, et al. Primary cutaneous Mycobacterium avium complex infection following squamous cell carcinoma excision. Cutis. 2016;98:E8-E11.
- Kollipara R, Richards K, Tschen J, et al. Disseminated Mycobacterium avium complex with cutaneous lesions. J Cutan Med Surg. 2016;20:272-274.
- Endly DC, Ackerman LS. Disseminated cutaneous Mycobacterium avium complex in a person with AIDS. Dermatol Online J. 2014;20:22616.
- Li JJ, Beresford R, Fyfe J, et al. Clinical and histopathological features of cutaneous nontuberculous mycobacterial infection: a review of 13 cases. J Cutan Pathol. 2017;44:433-443.
- Iden DL, Rogers RS 3rd, Schroeter AL. Papulonecrotic tuberculid secondary to Mycobacterium bovis. Arch Dermatol. 1978;114:564-566.
- Wong NM, Sun LK, Lau PY. Spinal infection caused by Mycobacterium avium complex in a patient with no acquired immune deficiency syndrome: a case report. J Orthop Surg (Hong Kong). 2008;16:359-363.
To the Editor:
Papulonecrotic tuberculid (PNT) is a cutaneous hypersensitivity reaction to antigenic components of Mycobacterium species, most commonly Mycobacterium tuberculosis. According to a PubMed search of articles indexed for MEDLINE using the terms papulonecrotic tuberculid, Mycobacterium avium complex, and Mycobacterium, only 1 case of PNT secondary to infection with Mycobacterium avium complex (MAC) has been reported.1,2 Papulonecrotic tuberculid classically presents with symmetrical, dusky red papules with necrosis on the extremities.3 Patients may or may not have associated symptoms of fever and weight loss. It is diagnosed through skin biopsy as well as identification of a distant source of mycobacterial infection. Papulonecrotic tuberculid is considered a reactive process to a distant site of mycobacterial infection, and skin lesions contain few, if any, mycobacteria.4
A 65-year-old man was admitted to the hospital for expedited workup of chronic fevers, 20-lb weight loss, and night sweats of 8 months’ duration. He had a medical history of myelodysplastic syndrome and autoimmune hemolytic anemia. During hospitalization, positron emission tomography revealed multilevel vertebral lytic and sclerotic lesions. Subsequent T10 vertebral biopsy showed necrotizing granulomatous inflammation with extensive necrosis and acid-fast bacilli–positive organisms. The patient was empirically started on rifampicin, isoniazid, pyrazinamide, ethambutol, and pyridoxine for presumed M tuberculosis and placed on respiratory isolation.
Dermatology was consulted for a recurrent tender rash on the bilateral upper and lower extremities of 5 years’ duration. Physical examination revealed numerous erythematous papulonecrotic lesions in various states of healing on the bilateral upper and lower extremities (Figure 1). Three years prior to the current presentation, 2 lesions were biopsied and demonstrated leukocytoclastic vasculitis with neutrophilic panniculitis and vasculopathy. A presumptive diagnosis of Sweet syndrome was made given the history of myelodysplastic syndrome, though an infectious etiology could not be ruled out at that time. Concurrently, the patient was diagnosed with autoimmune hemolytic anemia and was started on prednisone. Initially, the skin lesions improved with prednisone but never fully resolved; however, as the dosage of oral steroids decreased, the skin lesions worsened and presented in larger numbers with more frequency. The patient was titrated down to prednisone 5 mg daily with no additional treatment of the skin lesions at that time.
During the current hospitalization, 2 additional biopsies were taken from the arm for routine histopathology and tissue culture. Dermatopathology revealed robust neutrophilic and granulomatous inflammation as well as remarkable necrosis with a few mycobacteria identified on acid-fast and Fite stains (Figure 2). Tissue culture was negative. Additionally, the patient’s spinal biopsy was sent for polymerase chain reaction analysis for Mycobacterium typing, which confirmed MAC. The patient was diagnosed with Pott disease, a mycobacterial infection of the spine, as well as cutaneous papulonecrotic tuberculid secondary to MAC.
Papulonecrotic tuberculid is the rarest form of cutaneous tuberculosis infection and rarely has been reported in connection to MAC.1 This condition is considered a hypersensitivity reaction that occurs in response to antigenic components of mycobacteria.4 Patients with PNT typically present with recurrent crops of painful papulonecrotic lesions distributed on the extremities. Histopathology in PNT classically reveals necrosis, notable inflammatory infiltrate, and lack of observed organisms.5 Diagnosis often is made through skin biopsy, though histopathology varies based on lesion maturity.4 Early lesions often reveal leukocytoclastic vasculitis, whereas late lesions usually demonstrate granulomatous inflammation.4 Mycobacterium avium complex is difficult to culture, as it is a slow-growing, fastidious bacterium and therefore polymerase chain reaction genotyping is useful for bacterial classification.6
Disseminated MAC infection also was on the differential for our patient; however, we felt it was less likely than PNT for several reasons. First, disseminated infection rarely presents with cutaneous involvement and is associated with pulmonary involvement in 90% of cases.7-9 Second, the granuloma formation noted on our patient’s skin biopsy was not typical for disseminated MAC but is well described in cases of PNT.4,8,9 Finally, in the rare cases in which cutaneous involvement has occurred with disseminated mycobacterial infections, skin biopsies typically revealed numerous Mycobacterium organisms.8,10 In contrast, skin lesions associated with PNT usually reveal few, if any, organisms, as was seen with our patient.2
The patient’s initial biopsies also supported a diagnosis of PNT, as early lesions of PNT typically show leukocytoclastic vasculitis. His response to low and high doses of prednisone also fit well with a PNT diagnosis. In fact, a case of PNT secondary to Mycobacterium bovis similarly showed an improvement in the rash with high-dose steroids but progression with lower doses.11 It is possible that our patient’s response to steroids complicated the diagnosis of his rash.
The treatment of PNT is clearance of the underlying infection. Macrolide antibiotics, such as clarithromycin and azithromycin, have the best efficacy against MAC, in combination with ethambutol and/or rifabutin.6,12 Treatment duration should be 1 year. Amikacin or streptomycin may be added to this regimen during early treatment.6 Mycobacterium avium complex is resistant to many antibiotics, including typical antituberculosis drugs, and sensitivities should be identified at the onset of treatment.11,12
Albeit rare, clinicians should be aware of PNT secondary to MAC or other mycobacterial infections. Because this condition is difficult to diagnose with varying histologic findings and often negative tissue cultures, a high index of suspicion is necessary when a patient presents with recurrent papulonecrotic lesions, especially in immunocompromised hosts and patients with exposure to mycobacteria.
To the Editor:
Papulonecrotic tuberculid (PNT) is a cutaneous hypersensitivity reaction to antigenic components of Mycobacterium species, most commonly Mycobacterium tuberculosis. According to a PubMed search of articles indexed for MEDLINE using the terms papulonecrotic tuberculid, Mycobacterium avium complex, and Mycobacterium, only 1 case of PNT secondary to infection with Mycobacterium avium complex (MAC) has been reported.1,2 Papulonecrotic tuberculid classically presents with symmetrical, dusky red papules with necrosis on the extremities.3 Patients may or may not have associated symptoms of fever and weight loss. It is diagnosed through skin biopsy as well as identification of a distant source of mycobacterial infection. Papulonecrotic tuberculid is considered a reactive process to a distant site of mycobacterial infection, and skin lesions contain few, if any, mycobacteria.4
A 65-year-old man was admitted to the hospital for expedited workup of chronic fevers, 20-lb weight loss, and night sweats of 8 months’ duration. He had a medical history of myelodysplastic syndrome and autoimmune hemolytic anemia. During hospitalization, positron emission tomography revealed multilevel vertebral lytic and sclerotic lesions. Subsequent T10 vertebral biopsy showed necrotizing granulomatous inflammation with extensive necrosis and acid-fast bacilli–positive organisms. The patient was empirically started on rifampicin, isoniazid, pyrazinamide, ethambutol, and pyridoxine for presumed M tuberculosis and placed on respiratory isolation.
Dermatology was consulted for a recurrent tender rash on the bilateral upper and lower extremities of 5 years’ duration. Physical examination revealed numerous erythematous papulonecrotic lesions in various states of healing on the bilateral upper and lower extremities (Figure 1). Three years prior to the current presentation, 2 lesions were biopsied and demonstrated leukocytoclastic vasculitis with neutrophilic panniculitis and vasculopathy. A presumptive diagnosis of Sweet syndrome was made given the history of myelodysplastic syndrome, though an infectious etiology could not be ruled out at that time. Concurrently, the patient was diagnosed with autoimmune hemolytic anemia and was started on prednisone. Initially, the skin lesions improved with prednisone but never fully resolved; however, as the dosage of oral steroids decreased, the skin lesions worsened and presented in larger numbers with more frequency. The patient was titrated down to prednisone 5 mg daily with no additional treatment of the skin lesions at that time.
During the current hospitalization, 2 additional biopsies were taken from the arm for routine histopathology and tissue culture. Dermatopathology revealed robust neutrophilic and granulomatous inflammation as well as remarkable necrosis with a few mycobacteria identified on acid-fast and Fite stains (Figure 2). Tissue culture was negative. Additionally, the patient’s spinal biopsy was sent for polymerase chain reaction analysis for Mycobacterium typing, which confirmed MAC. The patient was diagnosed with Pott disease, a mycobacterial infection of the spine, as well as cutaneous papulonecrotic tuberculid secondary to MAC.
Papulonecrotic tuberculid is the rarest form of cutaneous tuberculosis infection and rarely has been reported in connection to MAC.1 This condition is considered a hypersensitivity reaction that occurs in response to antigenic components of mycobacteria.4 Patients with PNT typically present with recurrent crops of painful papulonecrotic lesions distributed on the extremities. Histopathology in PNT classically reveals necrosis, notable inflammatory infiltrate, and lack of observed organisms.5 Diagnosis often is made through skin biopsy, though histopathology varies based on lesion maturity.4 Early lesions often reveal leukocytoclastic vasculitis, whereas late lesions usually demonstrate granulomatous inflammation.4 Mycobacterium avium complex is difficult to culture, as it is a slow-growing, fastidious bacterium and therefore polymerase chain reaction genotyping is useful for bacterial classification.6
Disseminated MAC infection also was on the differential for our patient; however, we felt it was less likely than PNT for several reasons. First, disseminated infection rarely presents with cutaneous involvement and is associated with pulmonary involvement in 90% of cases.7-9 Second, the granuloma formation noted on our patient’s skin biopsy was not typical for disseminated MAC but is well described in cases of PNT.4,8,9 Finally, in the rare cases in which cutaneous involvement has occurred with disseminated mycobacterial infections, skin biopsies typically revealed numerous Mycobacterium organisms.8,10 In contrast, skin lesions associated with PNT usually reveal few, if any, organisms, as was seen with our patient.2
The patient’s initial biopsies also supported a diagnosis of PNT, as early lesions of PNT typically show leukocytoclastic vasculitis. His response to low and high doses of prednisone also fit well with a PNT diagnosis. In fact, a case of PNT secondary to Mycobacterium bovis similarly showed an improvement in the rash with high-dose steroids but progression with lower doses.11 It is possible that our patient’s response to steroids complicated the diagnosis of his rash.
The treatment of PNT is clearance of the underlying infection. Macrolide antibiotics, such as clarithromycin and azithromycin, have the best efficacy against MAC, in combination with ethambutol and/or rifabutin.6,12 Treatment duration should be 1 year. Amikacin or streptomycin may be added to this regimen during early treatment.6 Mycobacterium avium complex is resistant to many antibiotics, including typical antituberculosis drugs, and sensitivities should be identified at the onset of treatment.11,12
Albeit rare, clinicians should be aware of PNT secondary to MAC or other mycobacterial infections. Because this condition is difficult to diagnose with varying histologic findings and often negative tissue cultures, a high index of suspicion is necessary when a patient presents with recurrent papulonecrotic lesions, especially in immunocompromised hosts and patients with exposure to mycobacteria.
- Williams JT, Pulitzer DR, DeVillez RL. Papulonecrotic tuberculid secondary to disseminated Mycobacterium avium complex. Int J Dermatol. 1994;33:109-112.
- Jordaan HF, Schneider JW. Papulonecrotic tuberculid. Int J Dermatol. 1995;34:217-219.
- Scollard DM, Dacso MM, Abad-Venida ML. Tuberculosis and leprosy: classical granulomatous diseases in the twenty-first century. Dermatol Clin. 2015;33:541-562.
- Kim GW, Park HJ, Kim HS, et al. Simultaneous occurrence of papulonecrotic tuberculid and erythema induratum in a patient with pulmonary tuberculosis. Pediatr Dermatol. 2013;30:256-259.
- Spelta K, Diniz LM. Cutaneous tuberculosis: a 26-year retrospective study in an endemic area. Rev Inst Med Trop Sao Paulo. 2016;58:49.
- Griffith DE, Aksamit T, Brown-Elliott BA, et al. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med. 2007;175:367-416.
- Dyer J, Weiss J, Steiner WS, et al. Primary cutaneous Mycobacterium avium complex infection following squamous cell carcinoma excision. Cutis. 2016;98:E8-E11.
- Kollipara R, Richards K, Tschen J, et al. Disseminated Mycobacterium avium complex with cutaneous lesions. J Cutan Med Surg. 2016;20:272-274.
- Endly DC, Ackerman LS. Disseminated cutaneous Mycobacterium avium complex in a person with AIDS. Dermatol Online J. 2014;20:22616.
- Li JJ, Beresford R, Fyfe J, et al. Clinical and histopathological features of cutaneous nontuberculous mycobacterial infection: a review of 13 cases. J Cutan Pathol. 2017;44:433-443.
- Iden DL, Rogers RS 3rd, Schroeter AL. Papulonecrotic tuberculid secondary to Mycobacterium bovis. Arch Dermatol. 1978;114:564-566.
- Wong NM, Sun LK, Lau PY. Spinal infection caused by Mycobacterium avium complex in a patient with no acquired immune deficiency syndrome: a case report. J Orthop Surg (Hong Kong). 2008;16:359-363.
- Williams JT, Pulitzer DR, DeVillez RL. Papulonecrotic tuberculid secondary to disseminated Mycobacterium avium complex. Int J Dermatol. 1994;33:109-112.
- Jordaan HF, Schneider JW. Papulonecrotic tuberculid. Int J Dermatol. 1995;34:217-219.
- Scollard DM, Dacso MM, Abad-Venida ML. Tuberculosis and leprosy: classical granulomatous diseases in the twenty-first century. Dermatol Clin. 2015;33:541-562.
- Kim GW, Park HJ, Kim HS, et al. Simultaneous occurrence of papulonecrotic tuberculid and erythema induratum in a patient with pulmonary tuberculosis. Pediatr Dermatol. 2013;30:256-259.
- Spelta K, Diniz LM. Cutaneous tuberculosis: a 26-year retrospective study in an endemic area. Rev Inst Med Trop Sao Paulo. 2016;58:49.
- Griffith DE, Aksamit T, Brown-Elliott BA, et al. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med. 2007;175:367-416.
- Dyer J, Weiss J, Steiner WS, et al. Primary cutaneous Mycobacterium avium complex infection following squamous cell carcinoma excision. Cutis. 2016;98:E8-E11.
- Kollipara R, Richards K, Tschen J, et al. Disseminated Mycobacterium avium complex with cutaneous lesions. J Cutan Med Surg. 2016;20:272-274.
- Endly DC, Ackerman LS. Disseminated cutaneous Mycobacterium avium complex in a person with AIDS. Dermatol Online J. 2014;20:22616.
- Li JJ, Beresford R, Fyfe J, et al. Clinical and histopathological features of cutaneous nontuberculous mycobacterial infection: a review of 13 cases. J Cutan Pathol. 2017;44:433-443.
- Iden DL, Rogers RS 3rd, Schroeter AL. Papulonecrotic tuberculid secondary to Mycobacterium bovis. Arch Dermatol. 1978;114:564-566.
- Wong NM, Sun LK, Lau PY. Spinal infection caused by Mycobacterium avium complex in a patient with no acquired immune deficiency syndrome: a case report. J Orthop Surg (Hong Kong). 2008;16:359-363.
Practice Points
- Papulonecrotic tuberculid (PNT) is a hypersensitivity reaction that presents with reddish papules with central necrosis on the extremities.
- Early PNT histopathology shows leukocytoclastic vasculitis. Later lesions demonstrate granulomatous inflammation on histopathology.
- Mycobacterium avium is difficult to culture; therefore, if you suspect it, we recommend polymerase chain reaction genotyping for bacterial classification.
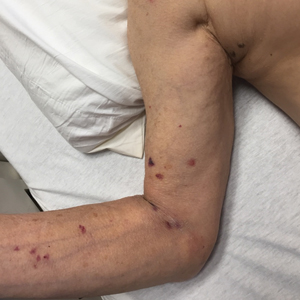
Antecedent Chronic Lymphocytic Leukemia May Be Associated With More Aggressive Mycosis Fungoides
To the Editor:
Mycosis fungoides (MF) is the most common form of primary cutaneous T-cell lymphoma. It has been associated with increased risk for other visceral and hematologic malignancies.1 Chronic lymphocytic leukemia (CLL) is one of the most common hematologic malignancies. In the United States, a patient’s lifetime risk for CLL is 0.6%. Chronic lymphocytic leukemia often is diagnosed as an incidental finding and typically is not detrimental to a patient’s health. Six cases of MF with antecedent or concomitant CLL were identified in a cohort of patients treated at the University of Minnesota (Minneapolis, Minnesota) from 2005 to 2017 (Table).
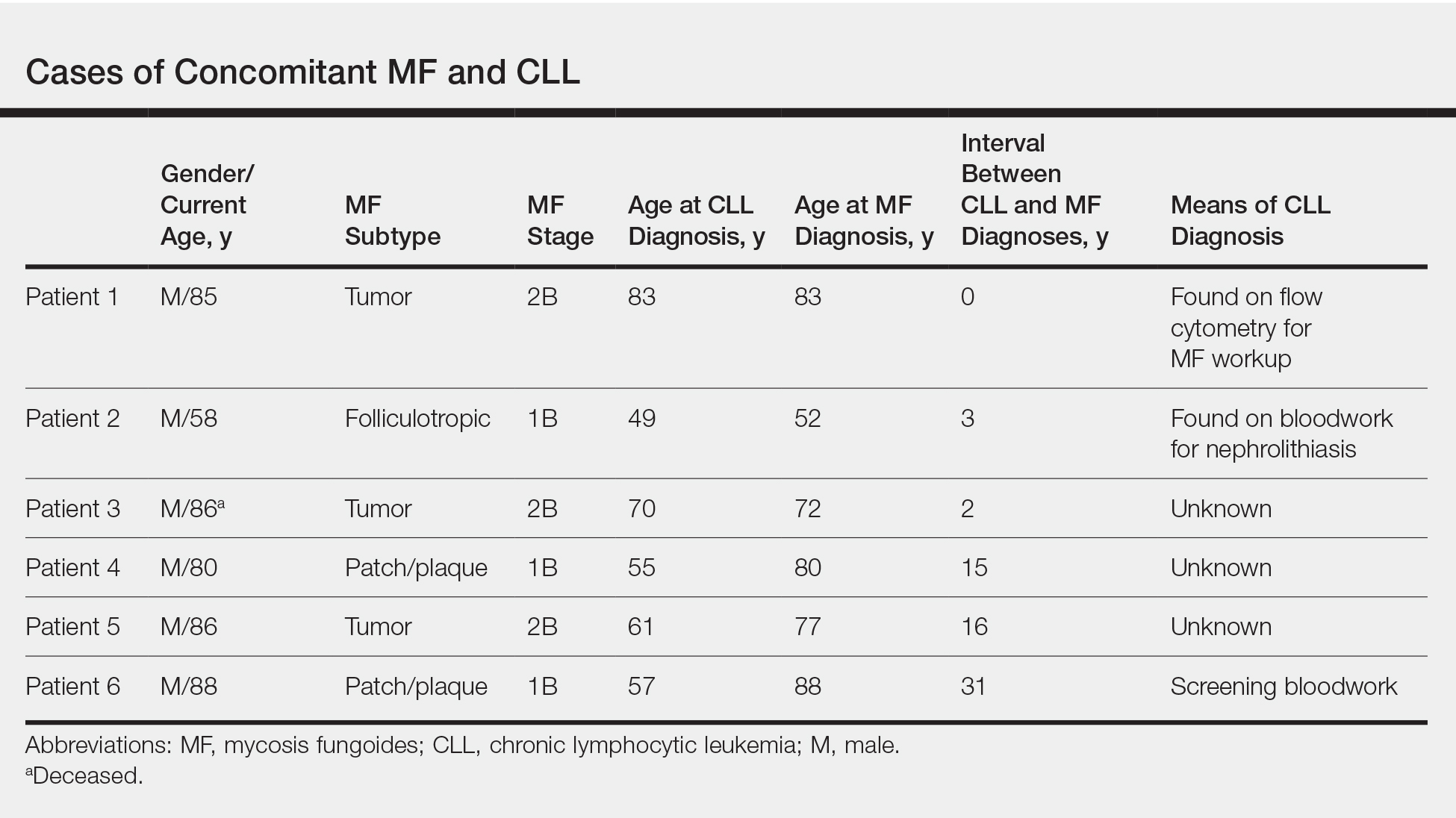
All 6 patients were male, with a mean age of 80.5 years. The mean age at CLL diagnosis was 62.5 years, while the mean age at MF diagnosis was 75.3 years. Three patients were younger than 60 years when their CLL was diagnosed: 49, 55, and 57 years. Notably, 4 patients had more aggressive types of MF: 3 with tumor-stage disease, and 1 with folliculotropic MF. Five patients were diagnosed with CLL before their MF was diagnosed (mean, 13.4 years prior; range, 3–31 years), and 1 was diagnosed as part of the initial MF workup.
Given the frequency of both MF and CLL, the co-occurrence of these diseases is not surprising, as other case reports and a larger case series have described the relationship between MF and malignancy.2 It is possible that CLL patients are more likely to be diagnosed with MF because of their regular hematology/oncology follow-up; however, none of our patients were referred from hematology/oncology to dermatology. Alternatively, patients with MF may be more likely to be diagnosed with CLL because of repeated bloodwork performed for diagnosis and screening, which occurred in only 1 of 6 cases. Most of the other patients were diagnosed with MF more than a decade after being diagnosed with CLL.
Does having CLL make patients more likely to develop MF? It is known that patients with CLL may experience immunodeficiency secondary to immune dysregulation, making them more susceptible to infection and secondary malignancies.3 Of our 6 cases, 4 had aggressive or advanced forms of MF, which is similar to the findings of Chang et al.2 In their report, of 8 patients with MF, 2 had tumor-stage disease and 2 had erythrodermic MF. They determined that these patients had worse overall survival.2 Our data corroborate the finding that patients with CLL may develop more severe MF, which leads to the conclusion that patients diagnosed with CLL before, concomitantly, or after their diagnosis of MF should be closely monitored. It is notable that patients with more advanced disease tend to be older at the time of diagnosis and that patients who are diagnosed at 57 years or older have been found to have worse disease-specific survival.4,5
This report is limited by the small sample size (6 cases), but it serves to draw attention to the phenomenon of co-occurrence of MF and CLL, and the concern that patients with CLL may develop more aggressive MF.
- Huang KP, Weinstock MA, Clarke CA, et al. Second lymphomas and other malignant neoplasms in patients with mycosis fungoides and Sézary syndrome. Arch Dermatol. 2007;143:45-50.
- Chang MB, Weaver AL, Brewer JD. Cutaneous T-cell lymphoma in patients with chronic lymphocytic leukemia: clinical characteristics, temporal relationships, and survival data in a series of 14 patients at Mayo Clinic. Int J Dermatol. 2014;53:966-970.
- Hamblin AD, Hamblin TJ. The immunodeficiency of chronic lymphocytic leukaemia. Br Med Bull. 2008;87:49-62.
- Kim YH, Liu HL, Mraz-Gernhard S, et al. Long-term outcome of 525 patients with mycosis fungoides and Sezary syndrome: clinical prognostic factors and risk for disease progression. Arch Dermatol. 2003;139:857-866.
- Agar NS, Wedgeworth E, Crichton S, et al. Survival outcomes and prognostic factors in mycosis fungoides/Sézary syndrome: validation of the revised International Society for Cutaneous Lymphomas/European Organisation for Research and Treatment of Cancer staging proposal. J Clin Oncol. 2010;28:4730-4739.
To the Editor:
Mycosis fungoides (MF) is the most common form of primary cutaneous T-cell lymphoma. It has been associated with increased risk for other visceral and hematologic malignancies.1 Chronic lymphocytic leukemia (CLL) is one of the most common hematologic malignancies. In the United States, a patient’s lifetime risk for CLL is 0.6%. Chronic lymphocytic leukemia often is diagnosed as an incidental finding and typically is not detrimental to a patient’s health. Six cases of MF with antecedent or concomitant CLL were identified in a cohort of patients treated at the University of Minnesota (Minneapolis, Minnesota) from 2005 to 2017 (Table).
All 6 patients were male, with a mean age of 80.5 years. The mean age at CLL diagnosis was 62.5 years, while the mean age at MF diagnosis was 75.3 years. Three patients were younger than 60 years when their CLL was diagnosed: 49, 55, and 57 years. Notably, 4 patients had more aggressive types of MF: 3 with tumor-stage disease, and 1 with folliculotropic MF. Five patients were diagnosed with CLL before their MF was diagnosed (mean, 13.4 years prior; range, 3–31 years), and 1 was diagnosed as part of the initial MF workup.
Given the frequency of both MF and CLL, the co-occurrence of these diseases is not surprising, as other case reports and a larger case series have described the relationship between MF and malignancy.2 It is possible that CLL patients are more likely to be diagnosed with MF because of their regular hematology/oncology follow-up; however, none of our patients were referred from hematology/oncology to dermatology. Alternatively, patients with MF may be more likely to be diagnosed with CLL because of repeated bloodwork performed for diagnosis and screening, which occurred in only 1 of 6 cases. Most of the other patients were diagnosed with MF more than a decade after being diagnosed with CLL.
Does having CLL make patients more likely to develop MF? It is known that patients with CLL may experience immunodeficiency secondary to immune dysregulation, making them more susceptible to infection and secondary malignancies.3 Of our 6 cases, 4 had aggressive or advanced forms of MF, which is similar to the findings of Chang et al.2 In their report, of 8 patients with MF, 2 had tumor-stage disease and 2 had erythrodermic MF. They determined that these patients had worse overall survival.2 Our data corroborate the finding that patients with CLL may develop more severe MF, which leads to the conclusion that patients diagnosed with CLL before, concomitantly, or after their diagnosis of MF should be closely monitored. It is notable that patients with more advanced disease tend to be older at the time of diagnosis and that patients who are diagnosed at 57 years or older have been found to have worse disease-specific survival.4,5
This report is limited by the small sample size (6 cases), but it serves to draw attention to the phenomenon of co-occurrence of MF and CLL, and the concern that patients with CLL may develop more aggressive MF.
To the Editor:
Mycosis fungoides (MF) is the most common form of primary cutaneous T-cell lymphoma. It has been associated with increased risk for other visceral and hematologic malignancies.1 Chronic lymphocytic leukemia (CLL) is one of the most common hematologic malignancies. In the United States, a patient’s lifetime risk for CLL is 0.6%. Chronic lymphocytic leukemia often is diagnosed as an incidental finding and typically is not detrimental to a patient’s health. Six cases of MF with antecedent or concomitant CLL were identified in a cohort of patients treated at the University of Minnesota (Minneapolis, Minnesota) from 2005 to 2017 (Table).
All 6 patients were male, with a mean age of 80.5 years. The mean age at CLL diagnosis was 62.5 years, while the mean age at MF diagnosis was 75.3 years. Three patients were younger than 60 years when their CLL was diagnosed: 49, 55, and 57 years. Notably, 4 patients had more aggressive types of MF: 3 with tumor-stage disease, and 1 with folliculotropic MF. Five patients were diagnosed with CLL before their MF was diagnosed (mean, 13.4 years prior; range, 3–31 years), and 1 was diagnosed as part of the initial MF workup.
Given the frequency of both MF and CLL, the co-occurrence of these diseases is not surprising, as other case reports and a larger case series have described the relationship between MF and malignancy.2 It is possible that CLL patients are more likely to be diagnosed with MF because of their regular hematology/oncology follow-up; however, none of our patients were referred from hematology/oncology to dermatology. Alternatively, patients with MF may be more likely to be diagnosed with CLL because of repeated bloodwork performed for diagnosis and screening, which occurred in only 1 of 6 cases. Most of the other patients were diagnosed with MF more than a decade after being diagnosed with CLL.
Does having CLL make patients more likely to develop MF? It is known that patients with CLL may experience immunodeficiency secondary to immune dysregulation, making them more susceptible to infection and secondary malignancies.3 Of our 6 cases, 4 had aggressive or advanced forms of MF, which is similar to the findings of Chang et al.2 In their report, of 8 patients with MF, 2 had tumor-stage disease and 2 had erythrodermic MF. They determined that these patients had worse overall survival.2 Our data corroborate the finding that patients with CLL may develop more severe MF, which leads to the conclusion that patients diagnosed with CLL before, concomitantly, or after their diagnosis of MF should be closely monitored. It is notable that patients with more advanced disease tend to be older at the time of diagnosis and that patients who are diagnosed at 57 years or older have been found to have worse disease-specific survival.4,5
This report is limited by the small sample size (6 cases), but it serves to draw attention to the phenomenon of co-occurrence of MF and CLL, and the concern that patients with CLL may develop more aggressive MF.
- Huang KP, Weinstock MA, Clarke CA, et al. Second lymphomas and other malignant neoplasms in patients with mycosis fungoides and Sézary syndrome. Arch Dermatol. 2007;143:45-50.
- Chang MB, Weaver AL, Brewer JD. Cutaneous T-cell lymphoma in patients with chronic lymphocytic leukemia: clinical characteristics, temporal relationships, and survival data in a series of 14 patients at Mayo Clinic. Int J Dermatol. 2014;53:966-970.
- Hamblin AD, Hamblin TJ. The immunodeficiency of chronic lymphocytic leukaemia. Br Med Bull. 2008;87:49-62.
- Kim YH, Liu HL, Mraz-Gernhard S, et al. Long-term outcome of 525 patients with mycosis fungoides and Sezary syndrome: clinical prognostic factors and risk for disease progression. Arch Dermatol. 2003;139:857-866.
- Agar NS, Wedgeworth E, Crichton S, et al. Survival outcomes and prognostic factors in mycosis fungoides/Sézary syndrome: validation of the revised International Society for Cutaneous Lymphomas/European Organisation for Research and Treatment of Cancer staging proposal. J Clin Oncol. 2010;28:4730-4739.
- Huang KP, Weinstock MA, Clarke CA, et al. Second lymphomas and other malignant neoplasms in patients with mycosis fungoides and Sézary syndrome. Arch Dermatol. 2007;143:45-50.
- Chang MB, Weaver AL, Brewer JD. Cutaneous T-cell lymphoma in patients with chronic lymphocytic leukemia: clinical characteristics, temporal relationships, and survival data in a series of 14 patients at Mayo Clinic. Int J Dermatol. 2014;53:966-970.
- Hamblin AD, Hamblin TJ. The immunodeficiency of chronic lymphocytic leukaemia. Br Med Bull. 2008;87:49-62.
- Kim YH, Liu HL, Mraz-Gernhard S, et al. Long-term outcome of 525 patients with mycosis fungoides and Sezary syndrome: clinical prognostic factors and risk for disease progression. Arch Dermatol. 2003;139:857-866.
- Agar NS, Wedgeworth E, Crichton S, et al. Survival outcomes and prognostic factors in mycosis fungoides/Sézary syndrome: validation of the revised International Society for Cutaneous Lymphomas/European Organisation for Research and Treatment of Cancer staging proposal. J Clin Oncol. 2010;28:4730-4739.
Practice Points
- Patients with mycosis fungoides (MF) are at increased risk for second hematologic malignancies, including chronic lymphocytic leukemia (CLL).
- Anecdotal information suggests that patients with CLL prior to developing MF may have more severe phenotypes of MF.
Basal Cell Carcinoma Arising in Nevus Sebaceous During Pregnancy
To the Editor:
Nevus sebaceous of Jadassohn (or nevus sebaceous [NS]) is a congenital hamartomatous disorder initially described by Jadassohn1 in 1895. Nevus sebaceous occurs in 0.3% of newborns2 and is most commonly identified on the face and scalp.3,4 Mehregan and Pinkus5 characterized NS as an organoid tumor containing multiple skin components with 3 life stages. The first stage—occurring during infancy—consists of immature hair follicles and sebaceous glands. The second stage—beginning at puberty—shows development of sebaceous glands, epidermal hyperplasia, and maturation of apocrine glands. The final stage involves formation of secondary benign and malignant neoplasms.
Historically, basal cell carcinoma (BCC) was thought to be the most common neoplasm arising in NS.5-8 In 1993, Ackerman et al9 introduced a new definition of trichoblastoma (TB), expanding the definition to encompass previously excluded benign follicular neoplasms. Large studies conducted after this new definition was proposed suggested that syringocystadenoma papilliferum and TB develop more frequently than does BCC.3,4,10-15 Furthermore, Cribier et al4 and Merrot et al15 reviewed prior cases of NS using the new definition and asserted that the majority of previously diagnosed cases of BCC were considered to be TB under the new criteria. With the advent of modern diagnostic testing, the rate of secondary benign neoplasm growth is now thought to be between 7% and 19%, with syringocystadenoma papilliferum arising in 2% to 13% of cases and TB in 1.5% to 7%.3,4,10-14 Malignant neoplasms are observed much less frequently, with BCC arising in 0% to 1% of NS cases.
Nevus sebaceous lesions typically enlarge during puberty, while malignant neoplasms occur almost exclusively in adulthood,4,10-12 suggesting that hormones contribute to NS stage progression. We present the case of a woman who developed BCC in a previously asymptomatic NS during pregnancy.
A 32-year-old woman who was otherwise healthy presented to our dermatology clinic with a pink-yellow verrucous plaque on the right temporal hairline extending to the preauricular area of the face. The patient had no personal or family history of skin cancer and no history of tanning bed use. She reported that the lesion had been present since birth. A diagnosis of NS was made.
Two years later, she presented with a new bleeding growth atop the previously diagnosed NS that had been present for approximately 4 months (Figure). At this visit she was pregnant (30 weeks’ gestation). Physical examination revealed a 4-mm, brown, pearly papule at the inferior margin of the previously noted pink verrucous plaque on the right temporal hairline. A biopsy was performed and histopathology displayed aggregates of basaloid cells with a high nuclear to cytoplasmic ratio, peripheral palisading, and abundant melanin, consistent with pigmented BCC. The patient was referred for Mohs micrographic surgery; the lesion was removed with clear margins. The patient had no recurrence of BCC at 36-month follow-up.

Few studies have looked at the signal transduction pathways leading to malignant neoplasm formation in NS. Nevus sebaceous lesions are theorized to result from postzygotic genetic mutations in HRAS and KRAS oncogenes,16,17 which also are altered in squamous cell carcinoma and BCC.18 Similarly, Xin et al19 detected loss of heterozygosity of the human patched gene, PTCH, a tumor suppressor in the hedgehog pathway that has been implicated in sporadic BCC formation, suggesting that this loss of heterozygosity may predispose to secondary BCC formation.20,21 However, loss of PTCH heterozygosity could not be replicated by Takata et al22 and Levinsohn et al.16
Increased numbers of androgen receptors have been demonstrated in NS basal keratinocytes and sebaceous glands.23 Nevus sebaceous lesions enlarge during puberty,5 and malignant neoplasms arise almost exclusively in adulthood.3,4,10-13 The androgen surge during puberty and increased androgen levels in adulthood may promote sebaceous gland development and epidermal hyperplasia that result in progression of NS lesions from the first stage to the second stage. Basal cell carcinomas also express androgen receptors and have abnormal androgen hormone metabolism,24,25 though they do not display a notable number of estrogen or progesterone receptors.26 Therefore, increased androgen levels in adulthood also may contribute to progression to secondary neoplasm formation in the third stage.
Similarly, cases of rapid growth of NS lesions during pregnancy, a state of increased testosterone production,27 have
- Jadassohn J. Bemerkugen zur Histologie der systematisirten Naevi und uber “Talgdru˝sen-Naevi”. Arch Dermatol Syph. 1895;33:355-372.
- Alper J, Holmes LB, Mihm MC Jr. Birthmarks with serious medical significance: nevocullular nevi, sebaceous nevi, and multiple café au lait spots. J Pediatr. 1979;95:696-700.
- Muñoz-Pérez MA, García-Hernandez MJ, Ríos JJ, et al. Sebaceus naevi: a clinicopathologic study. J Eur Acad Dermatol Venereol. 2002;16:319-324.
- Cribier B, Scrivener Y, Grosshans E. Tumors arising in nevus sebaceus: a study of 596 cases. J Am Acad Dermatol. 2000;42:263-268.
- Mehregan AH, Pinkus H. Life history of organoid nevi. Special reference to nevus sebaceus of Jadassohn. Arch Dermatol. 1965;91:574-588.
- Jones EW, Heyl T. Naevus sebaceus. a report of 140 cases with special regard to the development of secondary malignant tumours. Br J Dermatol. 1970;82:99-117.
- Serpas de López RM, Hernández-Pérez E. Jadassohn’s sebaceous nevus. J Dermatol Surg Oncol. 1985;11:68-72.
- Smolin T, Hundeiker M. Squamous epithelial and basal cell carcinomas in naevus sebaceus (Jadassohn). Z Hautkr. 1986;61:267-282.
- Ackerman B, Reddy VB, Soyer HP. Neoplasms with Follicular Differentiation. New York, NY: Ardor Scribendi; 1993.
- Kaddu S, Schäppi H, Kerl H, et al. Trichoblastoma and sebaceoma in nevus sebaceus. Am J Dermatopathol. 1999;21:552-556.
- Idriss MH, Elston DM. Secondary neoplasms associated with nevus sebaceus of Jadassohn: a study of 707 cases. J Am Acad Dermatol. 2014;70:332-337.
- Hsu MC, Liau JY, Hong JL, et al. Secondary neoplasms arising from nevus sebaceus: a retrospective study of 450 cases in Taiwan. J Dermatol. 2016;43:175-180.
- Santibanez-Gallerani A, Marshall D, Duarte AM, et al. Should nevus sebaceus of Jadassohn in children be excised? a study of 757 cases, and literature review. J Craniofac Surg. 2003;14:658-660.
- Jaqueti G, Requena L, Sánchez Yus E. Trichoblastoma is the most common neoplasm developed in nevus sebaceus of Jadassohn: a clinicopathologic study of a series of 155 cases. Am J Dermatopathol. 2000;22:108-118.
- Merrot O, Cotten H, Patenotre P, et al. Sebaceous hamartoma of Jadassohn: trichoblastoma mimicking basal cell carcinoma? Ann Chir Plast Esthet. 2002;47:210-213.
- Levinsohn JL, Tian LC, Boyden LM, et al. Whole-exome sequencing reveals somatic mutations in HRAS and KRAS, which cause nevus sebaceus. J Invest Dermatol. 2013;133:827-830.
- Groesser L, Herschberger E, Ruetten A, et al. Postzygotic HRAS and KRAS mutations cause nevus sebaceous and Schimmelpenning syndrome. Nat Genet. 2012;44:783-787.
- Pierceall WE, Goldberg LH, Tainsky MA, et al. Ras gene mutation and amplification in human nonmelanoma skin cancers. Mol Carcinog. 1991;4:196-202.
- Xin H, Matt D, Qin JZ, et al. The sebaceous nevus: a nevus with deletions of the PTCH gene. Cancer Res. 1999;59:1834-1836.
- Johnson RL, Rothman AL, Xie J, et al. Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science. 1996;272:1668-1671.
- Gailani MR, Ståhle-Bäckdahl M, Leffell DJ, et al. The role of the human homologue of Drosophila patched in sporadic basal cell carcinomas. Nat Genet. 1996;14:78-81.
- Takata M, Tojo M, Hatta N, et al. No evidence of deregulated patched-hedgehog signaling pathway in trichoblastomas and other tumors arising within nevus sebaceous. J Invest Dermatol. 2001;117:1666-1670.
- Hamilton KS, Johnson S, Smoller BR. The role of androgen receptors in the clinical course of nevus sebaceus of Jadassohn. Mod Pathol. 2001;14:539-542.
- Moretti G, Cardo P, Rampini E, et al. Testosterone metabolism in basal cell epitheliomas. J Invest Dermatol. 1978;71:361-362.
- Bayer-Garner IB, Givens V, Smoller B. Immunohistochemical staining for androgen receptors: a sensitive marker of sebaceous differentiation. Am J Dermatopathol. 1999;21:426-431.
- Rogers GS, Flowers JL, Pollack SV, et al. Determination of sex steroid receptor in human basal cell carcinoma. J Am Acad Dermatol. 1988;18:1039-1043.
- Bammann BL, Coulam CB, Jiang NS. Total and free testosterone during pregnancy. Am J Obstet Gynecol. 1980;137:293-298.
- Terenzi V, Indrizzi E, Buonaccorsi S, et al. Nevus sebaceus of Jadassohn. J Craniofac Surg. 2006;17:1234-1239.
- Moody MN, Landau JM, Goldberg LH. Nevus sebaceous revisited. Pediatr Dermatol. 2012;29:15-23.
- Lillis PJ, Ceilley RI. Multiple tumors arising in nevus sebaceus. Cutis. 1979;23:310-314.
- Chun K, Vázquez M, Sánchez JL. Nevus sebaceus: clinical outcomeand considerations for prophylactic excision. Int J Dermatol. 1995;34:538-541.
To the Editor:
Nevus sebaceous of Jadassohn (or nevus sebaceous [NS]) is a congenital hamartomatous disorder initially described by Jadassohn1 in 1895. Nevus sebaceous occurs in 0.3% of newborns2 and is most commonly identified on the face and scalp.3,4 Mehregan and Pinkus5 characterized NS as an organoid tumor containing multiple skin components with 3 life stages. The first stage—occurring during infancy—consists of immature hair follicles and sebaceous glands. The second stage—beginning at puberty—shows development of sebaceous glands, epidermal hyperplasia, and maturation of apocrine glands. The final stage involves formation of secondary benign and malignant neoplasms.
Historically, basal cell carcinoma (BCC) was thought to be the most common neoplasm arising in NS.5-8 In 1993, Ackerman et al9 introduced a new definition of trichoblastoma (TB), expanding the definition to encompass previously excluded benign follicular neoplasms. Large studies conducted after this new definition was proposed suggested that syringocystadenoma papilliferum and TB develop more frequently than does BCC.3,4,10-15 Furthermore, Cribier et al4 and Merrot et al15 reviewed prior cases of NS using the new definition and asserted that the majority of previously diagnosed cases of BCC were considered to be TB under the new criteria. With the advent of modern diagnostic testing, the rate of secondary benign neoplasm growth is now thought to be between 7% and 19%, with syringocystadenoma papilliferum arising in 2% to 13% of cases and TB in 1.5% to 7%.3,4,10-14 Malignant neoplasms are observed much less frequently, with BCC arising in 0% to 1% of NS cases.
Nevus sebaceous lesions typically enlarge during puberty, while malignant neoplasms occur almost exclusively in adulthood,4,10-12 suggesting that hormones contribute to NS stage progression. We present the case of a woman who developed BCC in a previously asymptomatic NS during pregnancy.
A 32-year-old woman who was otherwise healthy presented to our dermatology clinic with a pink-yellow verrucous plaque on the right temporal hairline extending to the preauricular area of the face. The patient had no personal or family history of skin cancer and no history of tanning bed use. She reported that the lesion had been present since birth. A diagnosis of NS was made.
Two years later, she presented with a new bleeding growth atop the previously diagnosed NS that had been present for approximately 4 months (Figure). At this visit she was pregnant (30 weeks’ gestation). Physical examination revealed a 4-mm, brown, pearly papule at the inferior margin of the previously noted pink verrucous plaque on the right temporal hairline. A biopsy was performed and histopathology displayed aggregates of basaloid cells with a high nuclear to cytoplasmic ratio, peripheral palisading, and abundant melanin, consistent with pigmented BCC. The patient was referred for Mohs micrographic surgery; the lesion was removed with clear margins. The patient had no recurrence of BCC at 36-month follow-up.
Few studies have looked at the signal transduction pathways leading to malignant neoplasm formation in NS. Nevus sebaceous lesions are theorized to result from postzygotic genetic mutations in HRAS and KRAS oncogenes,16,17 which also are altered in squamous cell carcinoma and BCC.18 Similarly, Xin et al19 detected loss of heterozygosity of the human patched gene, PTCH, a tumor suppressor in the hedgehog pathway that has been implicated in sporadic BCC formation, suggesting that this loss of heterozygosity may predispose to secondary BCC formation.20,21 However, loss of PTCH heterozygosity could not be replicated by Takata et al22 and Levinsohn et al.16
Increased numbers of androgen receptors have been demonstrated in NS basal keratinocytes and sebaceous glands.23 Nevus sebaceous lesions enlarge during puberty,5 and malignant neoplasms arise almost exclusively in adulthood.3,4,10-13 The androgen surge during puberty and increased androgen levels in adulthood may promote sebaceous gland development and epidermal hyperplasia that result in progression of NS lesions from the first stage to the second stage. Basal cell carcinomas also express androgen receptors and have abnormal androgen hormone metabolism,24,25 though they do not display a notable number of estrogen or progesterone receptors.26 Therefore, increased androgen levels in adulthood also may contribute to progression to secondary neoplasm formation in the third stage.
Similarly, cases of rapid growth of NS lesions during pregnancy, a state of increased testosterone production,27 have
To the Editor:
Nevus sebaceous of Jadassohn (or nevus sebaceous [NS]) is a congenital hamartomatous disorder initially described by Jadassohn1 in 1895. Nevus sebaceous occurs in 0.3% of newborns2 and is most commonly identified on the face and scalp.3,4 Mehregan and Pinkus5 characterized NS as an organoid tumor containing multiple skin components with 3 life stages. The first stage—occurring during infancy—consists of immature hair follicles and sebaceous glands. The second stage—beginning at puberty—shows development of sebaceous glands, epidermal hyperplasia, and maturation of apocrine glands. The final stage involves formation of secondary benign and malignant neoplasms.
Historically, basal cell carcinoma (BCC) was thought to be the most common neoplasm arising in NS.5-8 In 1993, Ackerman et al9 introduced a new definition of trichoblastoma (TB), expanding the definition to encompass previously excluded benign follicular neoplasms. Large studies conducted after this new definition was proposed suggested that syringocystadenoma papilliferum and TB develop more frequently than does BCC.3,4,10-15 Furthermore, Cribier et al4 and Merrot et al15 reviewed prior cases of NS using the new definition and asserted that the majority of previously diagnosed cases of BCC were considered to be TB under the new criteria. With the advent of modern diagnostic testing, the rate of secondary benign neoplasm growth is now thought to be between 7% and 19%, with syringocystadenoma papilliferum arising in 2% to 13% of cases and TB in 1.5% to 7%.3,4,10-14 Malignant neoplasms are observed much less frequently, with BCC arising in 0% to 1% of NS cases.
Nevus sebaceous lesions typically enlarge during puberty, while malignant neoplasms occur almost exclusively in adulthood,4,10-12 suggesting that hormones contribute to NS stage progression. We present the case of a woman who developed BCC in a previously asymptomatic NS during pregnancy.
A 32-year-old woman who was otherwise healthy presented to our dermatology clinic with a pink-yellow verrucous plaque on the right temporal hairline extending to the preauricular area of the face. The patient had no personal or family history of skin cancer and no history of tanning bed use. She reported that the lesion had been present since birth. A diagnosis of NS was made.
Two years later, she presented with a new bleeding growth atop the previously diagnosed NS that had been present for approximately 4 months (Figure). At this visit she was pregnant (30 weeks’ gestation). Physical examination revealed a 4-mm, brown, pearly papule at the inferior margin of the previously noted pink verrucous plaque on the right temporal hairline. A biopsy was performed and histopathology displayed aggregates of basaloid cells with a high nuclear to cytoplasmic ratio, peripheral palisading, and abundant melanin, consistent with pigmented BCC. The patient was referred for Mohs micrographic surgery; the lesion was removed with clear margins. The patient had no recurrence of BCC at 36-month follow-up.
Few studies have looked at the signal transduction pathways leading to malignant neoplasm formation in NS. Nevus sebaceous lesions are theorized to result from postzygotic genetic mutations in HRAS and KRAS oncogenes,16,17 which also are altered in squamous cell carcinoma and BCC.18 Similarly, Xin et al19 detected loss of heterozygosity of the human patched gene, PTCH, a tumor suppressor in the hedgehog pathway that has been implicated in sporadic BCC formation, suggesting that this loss of heterozygosity may predispose to secondary BCC formation.20,21 However, loss of PTCH heterozygosity could not be replicated by Takata et al22 and Levinsohn et al.16
Increased numbers of androgen receptors have been demonstrated in NS basal keratinocytes and sebaceous glands.23 Nevus sebaceous lesions enlarge during puberty,5 and malignant neoplasms arise almost exclusively in adulthood.3,4,10-13 The androgen surge during puberty and increased androgen levels in adulthood may promote sebaceous gland development and epidermal hyperplasia that result in progression of NS lesions from the first stage to the second stage. Basal cell carcinomas also express androgen receptors and have abnormal androgen hormone metabolism,24,25 though they do not display a notable number of estrogen or progesterone receptors.26 Therefore, increased androgen levels in adulthood also may contribute to progression to secondary neoplasm formation in the third stage.
Similarly, cases of rapid growth of NS lesions during pregnancy, a state of increased testosterone production,27 have
- Jadassohn J. Bemerkugen zur Histologie der systematisirten Naevi und uber “Talgdru˝sen-Naevi”. Arch Dermatol Syph. 1895;33:355-372.
- Alper J, Holmes LB, Mihm MC Jr. Birthmarks with serious medical significance: nevocullular nevi, sebaceous nevi, and multiple café au lait spots. J Pediatr. 1979;95:696-700.
- Muñoz-Pérez MA, García-Hernandez MJ, Ríos JJ, et al. Sebaceus naevi: a clinicopathologic study. J Eur Acad Dermatol Venereol. 2002;16:319-324.
- Cribier B, Scrivener Y, Grosshans E. Tumors arising in nevus sebaceus: a study of 596 cases. J Am Acad Dermatol. 2000;42:263-268.
- Mehregan AH, Pinkus H. Life history of organoid nevi. Special reference to nevus sebaceus of Jadassohn. Arch Dermatol. 1965;91:574-588.
- Jones EW, Heyl T. Naevus sebaceus. a report of 140 cases with special regard to the development of secondary malignant tumours. Br J Dermatol. 1970;82:99-117.
- Serpas de López RM, Hernández-Pérez E. Jadassohn’s sebaceous nevus. J Dermatol Surg Oncol. 1985;11:68-72.
- Smolin T, Hundeiker M. Squamous epithelial and basal cell carcinomas in naevus sebaceus (Jadassohn). Z Hautkr. 1986;61:267-282.
- Ackerman B, Reddy VB, Soyer HP. Neoplasms with Follicular Differentiation. New York, NY: Ardor Scribendi; 1993.
- Kaddu S, Schäppi H, Kerl H, et al. Trichoblastoma and sebaceoma in nevus sebaceus. Am J Dermatopathol. 1999;21:552-556.
- Idriss MH, Elston DM. Secondary neoplasms associated with nevus sebaceus of Jadassohn: a study of 707 cases. J Am Acad Dermatol. 2014;70:332-337.
- Hsu MC, Liau JY, Hong JL, et al. Secondary neoplasms arising from nevus sebaceus: a retrospective study of 450 cases in Taiwan. J Dermatol. 2016;43:175-180.
- Santibanez-Gallerani A, Marshall D, Duarte AM, et al. Should nevus sebaceus of Jadassohn in children be excised? a study of 757 cases, and literature review. J Craniofac Surg. 2003;14:658-660.
- Jaqueti G, Requena L, Sánchez Yus E. Trichoblastoma is the most common neoplasm developed in nevus sebaceus of Jadassohn: a clinicopathologic study of a series of 155 cases. Am J Dermatopathol. 2000;22:108-118.
- Merrot O, Cotten H, Patenotre P, et al. Sebaceous hamartoma of Jadassohn: trichoblastoma mimicking basal cell carcinoma? Ann Chir Plast Esthet. 2002;47:210-213.
- Levinsohn JL, Tian LC, Boyden LM, et al. Whole-exome sequencing reveals somatic mutations in HRAS and KRAS, which cause nevus sebaceus. J Invest Dermatol. 2013;133:827-830.
- Groesser L, Herschberger E, Ruetten A, et al. Postzygotic HRAS and KRAS mutations cause nevus sebaceous and Schimmelpenning syndrome. Nat Genet. 2012;44:783-787.
- Pierceall WE, Goldberg LH, Tainsky MA, et al. Ras gene mutation and amplification in human nonmelanoma skin cancers. Mol Carcinog. 1991;4:196-202.
- Xin H, Matt D, Qin JZ, et al. The sebaceous nevus: a nevus with deletions of the PTCH gene. Cancer Res. 1999;59:1834-1836.
- Johnson RL, Rothman AL, Xie J, et al. Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science. 1996;272:1668-1671.
- Gailani MR, Ståhle-Bäckdahl M, Leffell DJ, et al. The role of the human homologue of Drosophila patched in sporadic basal cell carcinomas. Nat Genet. 1996;14:78-81.
- Takata M, Tojo M, Hatta N, et al. No evidence of deregulated patched-hedgehog signaling pathway in trichoblastomas and other tumors arising within nevus sebaceous. J Invest Dermatol. 2001;117:1666-1670.
- Hamilton KS, Johnson S, Smoller BR. The role of androgen receptors in the clinical course of nevus sebaceus of Jadassohn. Mod Pathol. 2001;14:539-542.
- Moretti G, Cardo P, Rampini E, et al. Testosterone metabolism in basal cell epitheliomas. J Invest Dermatol. 1978;71:361-362.
- Bayer-Garner IB, Givens V, Smoller B. Immunohistochemical staining for androgen receptors: a sensitive marker of sebaceous differentiation. Am J Dermatopathol. 1999;21:426-431.
- Rogers GS, Flowers JL, Pollack SV, et al. Determination of sex steroid receptor in human basal cell carcinoma. J Am Acad Dermatol. 1988;18:1039-1043.
- Bammann BL, Coulam CB, Jiang NS. Total and free testosterone during pregnancy. Am J Obstet Gynecol. 1980;137:293-298.
- Terenzi V, Indrizzi E, Buonaccorsi S, et al. Nevus sebaceus of Jadassohn. J Craniofac Surg. 2006;17:1234-1239.
- Moody MN, Landau JM, Goldberg LH. Nevus sebaceous revisited. Pediatr Dermatol. 2012;29:15-23.
- Lillis PJ, Ceilley RI. Multiple tumors arising in nevus sebaceus. Cutis. 1979;23:310-314.
- Chun K, Vázquez M, Sánchez JL. Nevus sebaceus: clinical outcomeand considerations for prophylactic excision. Int J Dermatol. 1995;34:538-541.
- Jadassohn J. Bemerkugen zur Histologie der systematisirten Naevi und uber “Talgdru˝sen-Naevi”. Arch Dermatol Syph. 1895;33:355-372.
- Alper J, Holmes LB, Mihm MC Jr. Birthmarks with serious medical significance: nevocullular nevi, sebaceous nevi, and multiple café au lait spots. J Pediatr. 1979;95:696-700.
- Muñoz-Pérez MA, García-Hernandez MJ, Ríos JJ, et al. Sebaceus naevi: a clinicopathologic study. J Eur Acad Dermatol Venereol. 2002;16:319-324.
- Cribier B, Scrivener Y, Grosshans E. Tumors arising in nevus sebaceus: a study of 596 cases. J Am Acad Dermatol. 2000;42:263-268.
- Mehregan AH, Pinkus H. Life history of organoid nevi. Special reference to nevus sebaceus of Jadassohn. Arch Dermatol. 1965;91:574-588.
- Jones EW, Heyl T. Naevus sebaceus. a report of 140 cases with special regard to the development of secondary malignant tumours. Br J Dermatol. 1970;82:99-117.
- Serpas de López RM, Hernández-Pérez E. Jadassohn’s sebaceous nevus. J Dermatol Surg Oncol. 1985;11:68-72.
- Smolin T, Hundeiker M. Squamous epithelial and basal cell carcinomas in naevus sebaceus (Jadassohn). Z Hautkr. 1986;61:267-282.
- Ackerman B, Reddy VB, Soyer HP. Neoplasms with Follicular Differentiation. New York, NY: Ardor Scribendi; 1993.
- Kaddu S, Schäppi H, Kerl H, et al. Trichoblastoma and sebaceoma in nevus sebaceus. Am J Dermatopathol. 1999;21:552-556.
- Idriss MH, Elston DM. Secondary neoplasms associated with nevus sebaceus of Jadassohn: a study of 707 cases. J Am Acad Dermatol. 2014;70:332-337.
- Hsu MC, Liau JY, Hong JL, et al. Secondary neoplasms arising from nevus sebaceus: a retrospective study of 450 cases in Taiwan. J Dermatol. 2016;43:175-180.
- Santibanez-Gallerani A, Marshall D, Duarte AM, et al. Should nevus sebaceus of Jadassohn in children be excised? a study of 757 cases, and literature review. J Craniofac Surg. 2003;14:658-660.
- Jaqueti G, Requena L, Sánchez Yus E. Trichoblastoma is the most common neoplasm developed in nevus sebaceus of Jadassohn: a clinicopathologic study of a series of 155 cases. Am J Dermatopathol. 2000;22:108-118.
- Merrot O, Cotten H, Patenotre P, et al. Sebaceous hamartoma of Jadassohn: trichoblastoma mimicking basal cell carcinoma? Ann Chir Plast Esthet. 2002;47:210-213.
- Levinsohn JL, Tian LC, Boyden LM, et al. Whole-exome sequencing reveals somatic mutations in HRAS and KRAS, which cause nevus sebaceus. J Invest Dermatol. 2013;133:827-830.
- Groesser L, Herschberger E, Ruetten A, et al. Postzygotic HRAS and KRAS mutations cause nevus sebaceous and Schimmelpenning syndrome. Nat Genet. 2012;44:783-787.
- Pierceall WE, Goldberg LH, Tainsky MA, et al. Ras gene mutation and amplification in human nonmelanoma skin cancers. Mol Carcinog. 1991;4:196-202.
- Xin H, Matt D, Qin JZ, et al. The sebaceous nevus: a nevus with deletions of the PTCH gene. Cancer Res. 1999;59:1834-1836.
- Johnson RL, Rothman AL, Xie J, et al. Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science. 1996;272:1668-1671.
- Gailani MR, Ståhle-Bäckdahl M, Leffell DJ, et al. The role of the human homologue of Drosophila patched in sporadic basal cell carcinomas. Nat Genet. 1996;14:78-81.
- Takata M, Tojo M, Hatta N, et al. No evidence of deregulated patched-hedgehog signaling pathway in trichoblastomas and other tumors arising within nevus sebaceous. J Invest Dermatol. 2001;117:1666-1670.
- Hamilton KS, Johnson S, Smoller BR. The role of androgen receptors in the clinical course of nevus sebaceus of Jadassohn. Mod Pathol. 2001;14:539-542.
- Moretti G, Cardo P, Rampini E, et al. Testosterone metabolism in basal cell epitheliomas. J Invest Dermatol. 1978;71:361-362.
- Bayer-Garner IB, Givens V, Smoller B. Immunohistochemical staining for androgen receptors: a sensitive marker of sebaceous differentiation. Am J Dermatopathol. 1999;21:426-431.
- Rogers GS, Flowers JL, Pollack SV, et al. Determination of sex steroid receptor in human basal cell carcinoma. J Am Acad Dermatol. 1988;18:1039-1043.
- Bammann BL, Coulam CB, Jiang NS. Total and free testosterone during pregnancy. Am J Obstet Gynecol. 1980;137:293-298.
- Terenzi V, Indrizzi E, Buonaccorsi S, et al. Nevus sebaceus of Jadassohn. J Craniofac Surg. 2006;17:1234-1239.
- Moody MN, Landau JM, Goldberg LH. Nevus sebaceous revisited. Pediatr Dermatol. 2012;29:15-23.
- Lillis PJ, Ceilley RI. Multiple tumors arising in nevus sebaceus. Cutis. 1979;23:310-314.
- Chun K, Vázquez M, Sánchez JL. Nevus sebaceus: clinical outcomeand considerations for prophylactic excision. Int J Dermatol. 1995;34:538-541.
Practice Points
- Benign neoplasms arise more frequently in nevus sebaceous (NS) lesions than do malignant neoplasms.
- The hormonal changes that occur during pregnancy and puberty appear to play a role in the development of neoplasms in NS lesions.
- Monitoring NS lesions more closely during periods of hormonal change may help diagnose malignant transformations in these patients.
Multiple Keratoacanthomas Arising Within Red Tattoo Pigment
To the Editor:
Keratoacanthoma (KA)–type squamous cell carcinomas (SCCs) are rapidly evolving neoplasms of the epithelium that often spontaneously regress but rarely metastasize.1,2 Keratoacanthomas are thought to ascend from the hair follicle,1 and they clinically present as an enlarging solitary crateriform nodule with a keratin-filled center. Multiple KAs are rare2; histologically, KAs can be difficult to distinguish from conventional SCCs and are frequently treated by standard surgical excision.1 Reactive KAs are a subtype of KA that are induced by trauma including UV exposure, electromagnetic radiation, surgical procedures, chemical peels, laser treatments, and rarely tattoos.3-5
A 56-year-old man presented to the clinic with 3 asymptomatic enlarging papulonodules within a multicolored tattoo along the right forearm and elbow of 5 months’ duration (Figure 1). The lesions developed 1 month after the tattoo was placed and were localized to the areas of red pigment. The patient had several other tattoos. Histologic examination of the lesions revealed a well-differentiated squamous neoplasm with a crateriform invagination consistent with the superficial portion of a KA (Figures 2A–C). The specimen also revealed exogenous red pigment that was consistent with the background tattoo (Figure 2D). The patient underwent excisions of all 3 KAs, and free surgical margins were obtained.

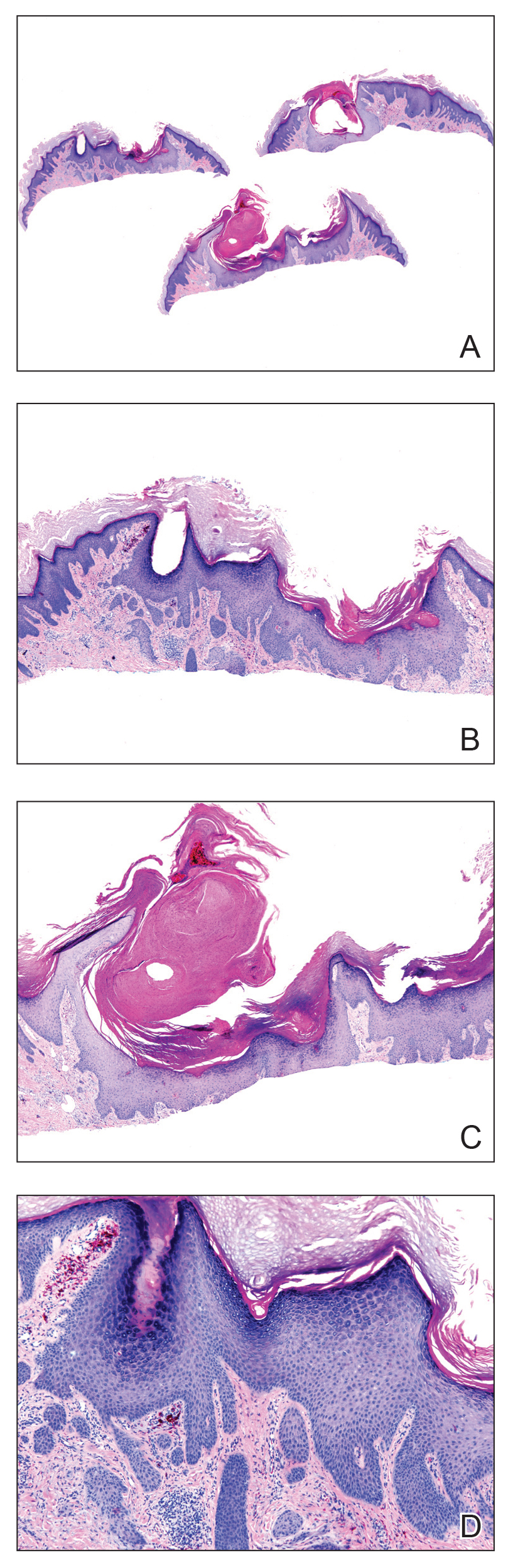
Tattooing is a popular practice dating back to 3000
Cipollaro10 reported the first case of a KA in a tattoo in 1973. Although there have been reports of melanoma and basal cell carcinoma occurring within tattoos, KAs and conventional SCCs are the most common cutaneous neoplasms arising in tattoos.
The pathogenesis underlying the development of malignancies in tattoos is unclear. It has been hypothesized that trauma from tattooing may play a role given the temporal relationship between tattoo placement and malignancy development.11 Another theory is that tattoo pigment causes a chronic inflammatory foreign body reaction that triggers carcinogenesis.12 Lastly, it has been postulated that tattoo pigment may alter UV light absorption in the skin that could potentially impact mutagenesis.11
The most common treatment of KAs is standard surgical excision.4 Mohs micrographic surgery is an option if the KA is located in a cosmetically sensitive area. Although there are no reports of recurrence after excision of tattoo-related KAs, new KAs forming adjacent to a previously excised KA have been reported.13
Currently, tattoos are not regulated by the US Food and Drug Administration before going to market. Although many states regulate the practice of tattooing, few regulate the contents of tattoo ink, and ink is only investigated when safety issues arise.14 This case provides further evidence of an association between KAs, tattooing, and potentially carcinogenic pigments, especially in red dye, supporting the need for further research on the safety of pigment components and more regulation of tattoo ink.
- Schwartz RA. Keratoacanthoma: a clinico-pathologic enigma. Dermatol Surg. 2004;30:326-333.
- Kwiek B, Schwartz RA. Keratoacanthoma (KA): an update and review. J Am Acad Dermatol. 2016;74:1220-1233.
- McGrouther DA, Downie PA, Thompson WD. Reactions to red tattoos. Br J Plas Surg. 1977;30:84-85.
- Sowden JM, Byrne JP, Smith AG, et al. Red tattoo reactions: x-ray microanalysis and patch-test studies. Br J Dermatol. 1991;124:576-580.
- Wiener DA, Scher RK. Basal cell carcinoma arising in a tattoo. Cutis. 1987;39:125-126.
- Pesapane F, Nazzaro G, Gianotti R, et al. A short history of tattoo. JAMA Dermatol. 2014;150:145.
- Junqueira AL, Wanat, KA, Farah RS. Squamous neoplasms arising within tattoos: clinical presentation, histopathology and management. Clin Exp Dermatol. 2017;42:601-606.
- Tammaro A, Toniolo C, Giulianelli V, et al. Chemical research on red pigments after adverse reactions to tattoo. Eur Ann Allergy Clin Immunol. 2016;48:46-48.
- Forbat E, Al-Niaimi F. Patterns of reactions to red pigment tattoo and treatment methods. Dermatol Therapy (Heidelb). 2016;6:13-23.
- Cipollaro VA. Keratoacanthoma developing in a tattoo. Cutis. 1973;11:809.
- Kluger N, Koljonen V. Tattoos, inks, and cancer. Lancet Oncol. 2012;13:E161-E168.
- Müller KM, Schmitz I, Hupe-Nörenberg L. Reaction patterns to cutaneous particulate and ornamental tattoos. Pathologe. 2002;23:46-53.
- Maxim E, Higgins H, D’Souza L. A case of multiple squamous cell carcinomas arising from red tattoo pigment. Int J Womens Dermatol. 2017;3:228-230.
- MacDonald J. Why doesn’t the FDA regulate tattoo ink? JSTOR Daily. September 21, 2017. https://daily.jstor.org/why-doesnt-the-fda-regulate-tattoo-ink/. Accessed October 15, 2019.
To the Editor:
Keratoacanthoma (KA)–type squamous cell carcinomas (SCCs) are rapidly evolving neoplasms of the epithelium that often spontaneously regress but rarely metastasize.1,2 Keratoacanthomas are thought to ascend from the hair follicle,1 and they clinically present as an enlarging solitary crateriform nodule with a keratin-filled center. Multiple KAs are rare2; histologically, KAs can be difficult to distinguish from conventional SCCs and are frequently treated by standard surgical excision.1 Reactive KAs are a subtype of KA that are induced by trauma including UV exposure, electromagnetic radiation, surgical procedures, chemical peels, laser treatments, and rarely tattoos.3-5
A 56-year-old man presented to the clinic with 3 asymptomatic enlarging papulonodules within a multicolored tattoo along the right forearm and elbow of 5 months’ duration (Figure 1). The lesions developed 1 month after the tattoo was placed and were localized to the areas of red pigment. The patient had several other tattoos. Histologic examination of the lesions revealed a well-differentiated squamous neoplasm with a crateriform invagination consistent with the superficial portion of a KA (Figures 2A–C). The specimen also revealed exogenous red pigment that was consistent with the background tattoo (Figure 2D). The patient underwent excisions of all 3 KAs, and free surgical margins were obtained.
Tattooing is a popular practice dating back to 3000
Cipollaro10 reported the first case of a KA in a tattoo in 1973. Although there have been reports of melanoma and basal cell carcinoma occurring within tattoos, KAs and conventional SCCs are the most common cutaneous neoplasms arising in tattoos.
The pathogenesis underlying the development of malignancies in tattoos is unclear. It has been hypothesized that trauma from tattooing may play a role given the temporal relationship between tattoo placement and malignancy development.11 Another theory is that tattoo pigment causes a chronic inflammatory foreign body reaction that triggers carcinogenesis.12 Lastly, it has been postulated that tattoo pigment may alter UV light absorption in the skin that could potentially impact mutagenesis.11
The most common treatment of KAs is standard surgical excision.4 Mohs micrographic surgery is an option if the KA is located in a cosmetically sensitive area. Although there are no reports of recurrence after excision of tattoo-related KAs, new KAs forming adjacent to a previously excised KA have been reported.13
Currently, tattoos are not regulated by the US Food and Drug Administration before going to market. Although many states regulate the practice of tattooing, few regulate the contents of tattoo ink, and ink is only investigated when safety issues arise.14 This case provides further evidence of an association between KAs, tattooing, and potentially carcinogenic pigments, especially in red dye, supporting the need for further research on the safety of pigment components and more regulation of tattoo ink.
To the Editor:
Keratoacanthoma (KA)–type squamous cell carcinomas (SCCs) are rapidly evolving neoplasms of the epithelium that often spontaneously regress but rarely metastasize.1,2 Keratoacanthomas are thought to ascend from the hair follicle,1 and they clinically present as an enlarging solitary crateriform nodule with a keratin-filled center. Multiple KAs are rare2; histologically, KAs can be difficult to distinguish from conventional SCCs and are frequently treated by standard surgical excision.1 Reactive KAs are a subtype of KA that are induced by trauma including UV exposure, electromagnetic radiation, surgical procedures, chemical peels, laser treatments, and rarely tattoos.3-5
A 56-year-old man presented to the clinic with 3 asymptomatic enlarging papulonodules within a multicolored tattoo along the right forearm and elbow of 5 months’ duration (Figure 1). The lesions developed 1 month after the tattoo was placed and were localized to the areas of red pigment. The patient had several other tattoos. Histologic examination of the lesions revealed a well-differentiated squamous neoplasm with a crateriform invagination consistent with the superficial portion of a KA (Figures 2A–C). The specimen also revealed exogenous red pigment that was consistent with the background tattoo (Figure 2D). The patient underwent excisions of all 3 KAs, and free surgical margins were obtained.
Tattooing is a popular practice dating back to 3000
Cipollaro10 reported the first case of a KA in a tattoo in 1973. Although there have been reports of melanoma and basal cell carcinoma occurring within tattoos, KAs and conventional SCCs are the most common cutaneous neoplasms arising in tattoos.
The pathogenesis underlying the development of malignancies in tattoos is unclear. It has been hypothesized that trauma from tattooing may play a role given the temporal relationship between tattoo placement and malignancy development.11 Another theory is that tattoo pigment causes a chronic inflammatory foreign body reaction that triggers carcinogenesis.12 Lastly, it has been postulated that tattoo pigment may alter UV light absorption in the skin that could potentially impact mutagenesis.11
The most common treatment of KAs is standard surgical excision.4 Mohs micrographic surgery is an option if the KA is located in a cosmetically sensitive area. Although there are no reports of recurrence after excision of tattoo-related KAs, new KAs forming adjacent to a previously excised KA have been reported.13
Currently, tattoos are not regulated by the US Food and Drug Administration before going to market. Although many states regulate the practice of tattooing, few regulate the contents of tattoo ink, and ink is only investigated when safety issues arise.14 This case provides further evidence of an association between KAs, tattooing, and potentially carcinogenic pigments, especially in red dye, supporting the need for further research on the safety of pigment components and more regulation of tattoo ink.
- Schwartz RA. Keratoacanthoma: a clinico-pathologic enigma. Dermatol Surg. 2004;30:326-333.
- Kwiek B, Schwartz RA. Keratoacanthoma (KA): an update and review. J Am Acad Dermatol. 2016;74:1220-1233.
- McGrouther DA, Downie PA, Thompson WD. Reactions to red tattoos. Br J Plas Surg. 1977;30:84-85.
- Sowden JM, Byrne JP, Smith AG, et al. Red tattoo reactions: x-ray microanalysis and patch-test studies. Br J Dermatol. 1991;124:576-580.
- Wiener DA, Scher RK. Basal cell carcinoma arising in a tattoo. Cutis. 1987;39:125-126.
- Pesapane F, Nazzaro G, Gianotti R, et al. A short history of tattoo. JAMA Dermatol. 2014;150:145.
- Junqueira AL, Wanat, KA, Farah RS. Squamous neoplasms arising within tattoos: clinical presentation, histopathology and management. Clin Exp Dermatol. 2017;42:601-606.
- Tammaro A, Toniolo C, Giulianelli V, et al. Chemical research on red pigments after adverse reactions to tattoo. Eur Ann Allergy Clin Immunol. 2016;48:46-48.
- Forbat E, Al-Niaimi F. Patterns of reactions to red pigment tattoo and treatment methods. Dermatol Therapy (Heidelb). 2016;6:13-23.
- Cipollaro VA. Keratoacanthoma developing in a tattoo. Cutis. 1973;11:809.
- Kluger N, Koljonen V. Tattoos, inks, and cancer. Lancet Oncol. 2012;13:E161-E168.
- Müller KM, Schmitz I, Hupe-Nörenberg L. Reaction patterns to cutaneous particulate and ornamental tattoos. Pathologe. 2002;23:46-53.
- Maxim E, Higgins H, D’Souza L. A case of multiple squamous cell carcinomas arising from red tattoo pigment. Int J Womens Dermatol. 2017;3:228-230.
- MacDonald J. Why doesn’t the FDA regulate tattoo ink? JSTOR Daily. September 21, 2017. https://daily.jstor.org/why-doesnt-the-fda-regulate-tattoo-ink/. Accessed October 15, 2019.
- Schwartz RA. Keratoacanthoma: a clinico-pathologic enigma. Dermatol Surg. 2004;30:326-333.
- Kwiek B, Schwartz RA. Keratoacanthoma (KA): an update and review. J Am Acad Dermatol. 2016;74:1220-1233.
- McGrouther DA, Downie PA, Thompson WD. Reactions to red tattoos. Br J Plas Surg. 1977;30:84-85.
- Sowden JM, Byrne JP, Smith AG, et al. Red tattoo reactions: x-ray microanalysis and patch-test studies. Br J Dermatol. 1991;124:576-580.
- Wiener DA, Scher RK. Basal cell carcinoma arising in a tattoo. Cutis. 1987;39:125-126.
- Pesapane F, Nazzaro G, Gianotti R, et al. A short history of tattoo. JAMA Dermatol. 2014;150:145.
- Junqueira AL, Wanat, KA, Farah RS. Squamous neoplasms arising within tattoos: clinical presentation, histopathology and management. Clin Exp Dermatol. 2017;42:601-606.
- Tammaro A, Toniolo C, Giulianelli V, et al. Chemical research on red pigments after adverse reactions to tattoo. Eur Ann Allergy Clin Immunol. 2016;48:46-48.
- Forbat E, Al-Niaimi F. Patterns of reactions to red pigment tattoo and treatment methods. Dermatol Therapy (Heidelb). 2016;6:13-23.
- Cipollaro VA. Keratoacanthoma developing in a tattoo. Cutis. 1973;11:809.
- Kluger N, Koljonen V. Tattoos, inks, and cancer. Lancet Oncol. 2012;13:E161-E168.
- Müller KM, Schmitz I, Hupe-Nörenberg L. Reaction patterns to cutaneous particulate and ornamental tattoos. Pathologe. 2002;23:46-53.
- Maxim E, Higgins H, D’Souza L. A case of multiple squamous cell carcinomas arising from red tattoo pigment. Int J Womens Dermatol. 2017;3:228-230.
- MacDonald J. Why doesn’t the FDA regulate tattoo ink? JSTOR Daily. September 21, 2017. https://daily.jstor.org/why-doesnt-the-fda-regulate-tattoo-ink/. Accessed October 15, 2019.
Practice Points
- Tattoo reactions range from infectious and inflammatory dermatoses to the development of malignant neoplasms.
- Red pigment is the most common cause of adverse tattoo reactions.
- The management of tattoo-associated keratoacanthoma (KA)–type squamous cell carcinomas (SCCs) has not been widely published, but they can be approached similarly to nontattoo-associated KA-SCCs.

Pulmonary Hemorrhage as the Initial Presentation of AIDS-Related Kaposi Sarcoma
To the Editor:
Kaposi sarcoma (KS) is an angioproliferative tumor of endothelial origin associated with human herpesvirus 8 infection. It is one of the most prevalent opportunistic infections associated with AIDS and is considered an AIDS-defining illness. In the general population, the incidence of KS is 1 in 100,000 worldwide.1 At the onset of the human immunodeficiency virus (HIV) epidemic in the early 1980s, 25% of individuals with AIDS were found to have KS at the time of AIDS diagnosis. Beginning in the mid-1980s and early 1990s with the introduction of highly active antiretroviral therapy (HAART), the incidence of KS declined to 2% to 4%,2 likely secondary to restoration of immune response.3
The clinical course of KS ranges from benign to severe, involving both cutaneous and visceral forms of disease. Cutaneous KS is the most common form of disease and typically characterizes the initial presentation. It is classically described as violaceous patches, papules, or plaques that can become confluent, forming larger tumors over time. Biopsy of cutaneous lesions may vary based on the clinical morphology. The patch stage typically is characterized by abnormal proliferating vessels surrounding larger ectatic vessels.4 Vascular spaces are more jagged and lined by thin endothelial cells extending into the dermis, forming the classic promontory sign.5 In the plaque stage, the vascular infiltrate becomes more diffuse, involving the dermis and subcutis, and there is proliferation of spindle cells.4 In the nodular stage, spindle-shaped tumor cells form fascicles and vascular spaces become more dilated.4,5 Advanced lesions are further associated with hyaline globules staining positive with periodic acid–Schiff.4 Lymphocytes, plasma cells, and hemosiderin-laden macrophages are admixed within this pathologic architecture.4,5
Visceral KS most commonly occurs in the oropharynx, respiratory tract, and gastrointestinal tract, and rarely is the initial presentation of disease. Classically, visceral KS is an aggressive, potentially life-threatening form of disease and has been found to have a much worse prognosis than cutaneous KS alone. Pulmonary involvement is the second most common site of extracutaneous KS and is known as the most severely life-threatening form of disease.1 Interestingly, since the advent of HAART, the incidence of KS with involvement of the visceral organs has declined at a more dramatic rate than cutaneous KS alone.3 Therefore, although more aggressive in nature, KS with visceral features has become increasingly rare and should be largely preventable given advances in AIDS therapy. We present a case of advanced AIDS-related KS with pulmonary involvement that is rarely seen after the advent of HAART.
A 39-year-old man with HIV diagnosed 8 years prior presented with fever, chest pain, progressive dyspnea, and hemoptysis of 5 months’ duration. At the time, he was nonadherent to medications and had poor follow-up with primary care physicians. At presentation he was tachycardic (149 beats per minute), tachypneic (26 breaths per minute), and his oxygen saturation was 80% on room air. Physical examination of the skin revealed asymptomatic violaceous penile lesions that the patient reported had been present for the last 8 months (Figure 1). Pertinent laboratory values included an HIV-1 viral load of 480,135 copies/mL (reference range, <20 copies/mL) and CD4 count of 14 cells/mm3 (reference range, 480–1700 cells/mm3). A chest radiograph was obtained and revealed bibasilar opacities compatible with a pleural and/or parenchymal process. Bronchoscopy was then performed and revealed bloody secretions throughout the tracheobronchial tree.

Histologic examination of biopsies of the penile lesions revealed spindle cell proliferation with hemorrhage (Figure 2A) that stained positively for HHV-8 (Figure 2B), consistent with KS. Biopsies taken during bronchoscopy similarly revealed spindle cells with hemorrhage (Figure 3). The patient was diagnosed with AIDS-related KS with visceral involvement of the lung parenchyma and tracheobronchial tree. The patient was then admitted to the medical intensive care unit and intubated. Therapy with HAART and paclitaxel was initiated. After 7 days of poor response to therapy, the family opted for terminal extubation and comfort care measures. The patient died hours later.
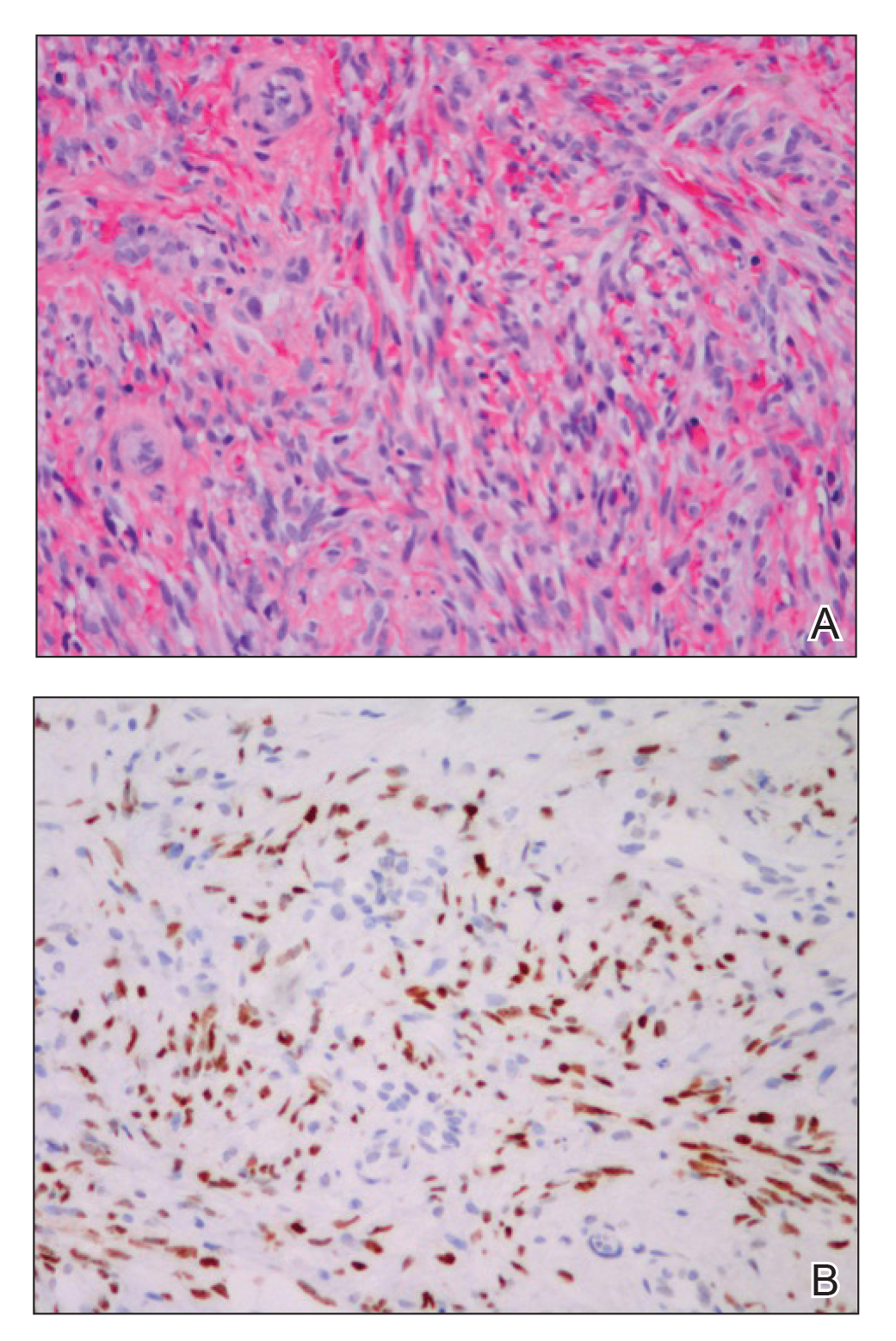
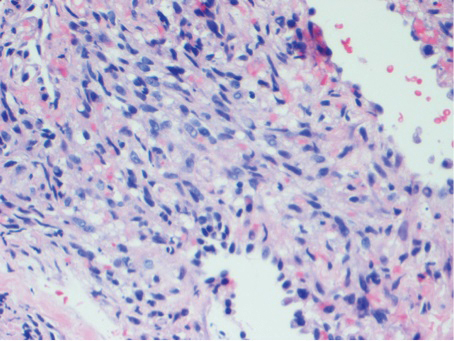
This case report describes the classic phenomenon of AIDS-related KS in a patient with a long-standing history of immunocompromise. Even in the era of HAART, this patient developed a severe form of visceral KS with involvement of the respiratory tract and lung parenchyma.
Since the advent of HAART for the treatment of HIV/AIDS, the incidence of KS, both visceral and cutaneous forms, has dramatically declined; the risk for visceral KS declined by more than 50% but less than 30% for cutaneous KS, supporting the observation that although visceral involvement has classically been noted as the more aggressive and life-threatening form of disease, HAART appears to have a stronger effect on visceral disease than cutaneous disease.3 Although the overall impact of AIDS-defining illnesses has substantially improved over the years, those with AIDS infection remain at risk for opportunistic illness.2
It has been shown that HAART therapy leads to response in more than 50% of cases of KS.5 The administration of HAART in KS patients is associated with improved survival and an 80% reduced risk of death, even when started after KS is diagnosed.6 In a comparison of the differences in clinical manifestations of KS between patients who were already receiving HAART at the time of KS diagnosis to those who were not on HAART, it was shown that patients already on therapy presented with less aggressive clinical features. A smaller percentage of patients who were already on HAART at KS diagnosis presented with visceral disease compared to those who were not on therapy.7
It is evident that treatment of AIDS patients with HAART is not only first-line therapy for the disease but also the best preventative measure against development of KS. Management of KS also centers around the initiation of HAART if the patient is not already maintained on the proper therapy.8 In addition to HAART, treatment options for visceral KS include a variety of chemotherapeutic agents, including but not limited to the use of single-agent adriamycin, vinblastine, paclitaxel, and thalidomide, or combination therapies.
Although notable advances have been made in the management of AIDS patients, this case highlights the need for clinicians to be aware of the risk for KS in the context of immunocompromise. Specifically, patients with advanced AIDS who are not adherent to HAART or who have a poor response to therapy have an amplified risk for developing KS in general as well as an increased risk for developing more severe visceral KS. Maintenance of patients with HAART is shown to greatly reduce the risk for both cutaneous and visceral KS; therefore, patient adherence with therapy is of utmost importance in preventing the occurrence of this deadly disease and its complications. Appropriate follow-up should be made, ensuring that these patients at high risk are adherent to therapy and have proper access to medical care to allow for prevention and early identification of potential complications.
- La Ferla L, Pinzone MR, Nunnari G, et al. Kaposi’s sarcoma in HIV-positive patients: the state of art in the HARRT-era. Eur Rev Med Pharmacol Sci. 2013;17:2354-2365.
- Engels EA, Pfeiffer RM, Goedert JJ, et al; HIV/AIDS Cancer Match Study. Trends in cancer risk among people with AIDS in the United States 1980-2002. AIDS. 2006;20:1645-1654.
- Grabar S, Abraham B, Mahamat A, et al. Differential impact of combination antiretroviral therapy in preventing Kaposi’s sarcoma with and without visceral involvement. JCO. 2006;24:3408-3414.
- Grayson W, Pantanowitz L. Histological variants of cutaneous Kaposi sarcoma [published online July 25, 2008]. Diagn Pathol. 2008;3:31.
- Radu O, Pantanowitz L. Kaposi sarcoma. Arch Pathol Lab Med. 2013;137:289-294.
- Tam HK, Zhang ZF, Jacobson LP, et al. Effect of highly active antiretroviral therapy on survival among HIV-infected men with Kaposi sarcoma or non-Hodgkin lymphoma. Int J Cancer. 2002;98:916-922.
- Nasti G, Martellotta F, Berretta M, et al. Impact of highly active antiretroviral therapy on the presenting features and outcome of patients with acquired immunodeficiency syndrome-related Kaposi sarcoma. Cancer. 2003;98:2440-2446.
- Dupont C, Vasseur E, Beauchet A, et al. Long-term efficacy on Kaposi’s sarcoma of highly active antriretroviral therapy in a cohort of HIV-positive patients. AIDS. 2000;14:987-993.
To the Editor:
Kaposi sarcoma (KS) is an angioproliferative tumor of endothelial origin associated with human herpesvirus 8 infection. It is one of the most prevalent opportunistic infections associated with AIDS and is considered an AIDS-defining illness. In the general population, the incidence of KS is 1 in 100,000 worldwide.1 At the onset of the human immunodeficiency virus (HIV) epidemic in the early 1980s, 25% of individuals with AIDS were found to have KS at the time of AIDS diagnosis. Beginning in the mid-1980s and early 1990s with the introduction of highly active antiretroviral therapy (HAART), the incidence of KS declined to 2% to 4%,2 likely secondary to restoration of immune response.3
The clinical course of KS ranges from benign to severe, involving both cutaneous and visceral forms of disease. Cutaneous KS is the most common form of disease and typically characterizes the initial presentation. It is classically described as violaceous patches, papules, or plaques that can become confluent, forming larger tumors over time. Biopsy of cutaneous lesions may vary based on the clinical morphology. The patch stage typically is characterized by abnormal proliferating vessels surrounding larger ectatic vessels.4 Vascular spaces are more jagged and lined by thin endothelial cells extending into the dermis, forming the classic promontory sign.5 In the plaque stage, the vascular infiltrate becomes more diffuse, involving the dermis and subcutis, and there is proliferation of spindle cells.4 In the nodular stage, spindle-shaped tumor cells form fascicles and vascular spaces become more dilated.4,5 Advanced lesions are further associated with hyaline globules staining positive with periodic acid–Schiff.4 Lymphocytes, plasma cells, and hemosiderin-laden macrophages are admixed within this pathologic architecture.4,5
Visceral KS most commonly occurs in the oropharynx, respiratory tract, and gastrointestinal tract, and rarely is the initial presentation of disease. Classically, visceral KS is an aggressive, potentially life-threatening form of disease and has been found to have a much worse prognosis than cutaneous KS alone. Pulmonary involvement is the second most common site of extracutaneous KS and is known as the most severely life-threatening form of disease.1 Interestingly, since the advent of HAART, the incidence of KS with involvement of the visceral organs has declined at a more dramatic rate than cutaneous KS alone.3 Therefore, although more aggressive in nature, KS with visceral features has become increasingly rare and should be largely preventable given advances in AIDS therapy. We present a case of advanced AIDS-related KS with pulmonary involvement that is rarely seen after the advent of HAART.
A 39-year-old man with HIV diagnosed 8 years prior presented with fever, chest pain, progressive dyspnea, and hemoptysis of 5 months’ duration. At the time, he was nonadherent to medications and had poor follow-up with primary care physicians. At presentation he was tachycardic (149 beats per minute), tachypneic (26 breaths per minute), and his oxygen saturation was 80% on room air. Physical examination of the skin revealed asymptomatic violaceous penile lesions that the patient reported had been present for the last 8 months (Figure 1). Pertinent laboratory values included an HIV-1 viral load of 480,135 copies/mL (reference range, <20 copies/mL) and CD4 count of 14 cells/mm3 (reference range, 480–1700 cells/mm3). A chest radiograph was obtained and revealed bibasilar opacities compatible with a pleural and/or parenchymal process. Bronchoscopy was then performed and revealed bloody secretions throughout the tracheobronchial tree.
Histologic examination of biopsies of the penile lesions revealed spindle cell proliferation with hemorrhage (Figure 2A) that stained positively for HHV-8 (Figure 2B), consistent with KS. Biopsies taken during bronchoscopy similarly revealed spindle cells with hemorrhage (Figure 3). The patient was diagnosed with AIDS-related KS with visceral involvement of the lung parenchyma and tracheobronchial tree. The patient was then admitted to the medical intensive care unit and intubated. Therapy with HAART and paclitaxel was initiated. After 7 days of poor response to therapy, the family opted for terminal extubation and comfort care measures. The patient died hours later.
This case report describes the classic phenomenon of AIDS-related KS in a patient with a long-standing history of immunocompromise. Even in the era of HAART, this patient developed a severe form of visceral KS with involvement of the respiratory tract and lung parenchyma.
Since the advent of HAART for the treatment of HIV/AIDS, the incidence of KS, both visceral and cutaneous forms, has dramatically declined; the risk for visceral KS declined by more than 50% but less than 30% for cutaneous KS, supporting the observation that although visceral involvement has classically been noted as the more aggressive and life-threatening form of disease, HAART appears to have a stronger effect on visceral disease than cutaneous disease.3 Although the overall impact of AIDS-defining illnesses has substantially improved over the years, those with AIDS infection remain at risk for opportunistic illness.2
It has been shown that HAART therapy leads to response in more than 50% of cases of KS.5 The administration of HAART in KS patients is associated with improved survival and an 80% reduced risk of death, even when started after KS is diagnosed.6 In a comparison of the differences in clinical manifestations of KS between patients who were already receiving HAART at the time of KS diagnosis to those who were not on HAART, it was shown that patients already on therapy presented with less aggressive clinical features. A smaller percentage of patients who were already on HAART at KS diagnosis presented with visceral disease compared to those who were not on therapy.7
It is evident that treatment of AIDS patients with HAART is not only first-line therapy for the disease but also the best preventative measure against development of KS. Management of KS also centers around the initiation of HAART if the patient is not already maintained on the proper therapy.8 In addition to HAART, treatment options for visceral KS include a variety of chemotherapeutic agents, including but not limited to the use of single-agent adriamycin, vinblastine, paclitaxel, and thalidomide, or combination therapies.
Although notable advances have been made in the management of AIDS patients, this case highlights the need for clinicians to be aware of the risk for KS in the context of immunocompromise. Specifically, patients with advanced AIDS who are not adherent to HAART or who have a poor response to therapy have an amplified risk for developing KS in general as well as an increased risk for developing more severe visceral KS. Maintenance of patients with HAART is shown to greatly reduce the risk for both cutaneous and visceral KS; therefore, patient adherence with therapy is of utmost importance in preventing the occurrence of this deadly disease and its complications. Appropriate follow-up should be made, ensuring that these patients at high risk are adherent to therapy and have proper access to medical care to allow for prevention and early identification of potential complications.
To the Editor:
Kaposi sarcoma (KS) is an angioproliferative tumor of endothelial origin associated with human herpesvirus 8 infection. It is one of the most prevalent opportunistic infections associated with AIDS and is considered an AIDS-defining illness. In the general population, the incidence of KS is 1 in 100,000 worldwide.1 At the onset of the human immunodeficiency virus (HIV) epidemic in the early 1980s, 25% of individuals with AIDS were found to have KS at the time of AIDS diagnosis. Beginning in the mid-1980s and early 1990s with the introduction of highly active antiretroviral therapy (HAART), the incidence of KS declined to 2% to 4%,2 likely secondary to restoration of immune response.3
The clinical course of KS ranges from benign to severe, involving both cutaneous and visceral forms of disease. Cutaneous KS is the most common form of disease and typically characterizes the initial presentation. It is classically described as violaceous patches, papules, or plaques that can become confluent, forming larger tumors over time. Biopsy of cutaneous lesions may vary based on the clinical morphology. The patch stage typically is characterized by abnormal proliferating vessels surrounding larger ectatic vessels.4 Vascular spaces are more jagged and lined by thin endothelial cells extending into the dermis, forming the classic promontory sign.5 In the plaque stage, the vascular infiltrate becomes more diffuse, involving the dermis and subcutis, and there is proliferation of spindle cells.4 In the nodular stage, spindle-shaped tumor cells form fascicles and vascular spaces become more dilated.4,5 Advanced lesions are further associated with hyaline globules staining positive with periodic acid–Schiff.4 Lymphocytes, plasma cells, and hemosiderin-laden macrophages are admixed within this pathologic architecture.4,5
Visceral KS most commonly occurs in the oropharynx, respiratory tract, and gastrointestinal tract, and rarely is the initial presentation of disease. Classically, visceral KS is an aggressive, potentially life-threatening form of disease and has been found to have a much worse prognosis than cutaneous KS alone. Pulmonary involvement is the second most common site of extracutaneous KS and is known as the most severely life-threatening form of disease.1 Interestingly, since the advent of HAART, the incidence of KS with involvement of the visceral organs has declined at a more dramatic rate than cutaneous KS alone.3 Therefore, although more aggressive in nature, KS with visceral features has become increasingly rare and should be largely preventable given advances in AIDS therapy. We present a case of advanced AIDS-related KS with pulmonary involvement that is rarely seen after the advent of HAART.
A 39-year-old man with HIV diagnosed 8 years prior presented with fever, chest pain, progressive dyspnea, and hemoptysis of 5 months’ duration. At the time, he was nonadherent to medications and had poor follow-up with primary care physicians. At presentation he was tachycardic (149 beats per minute), tachypneic (26 breaths per minute), and his oxygen saturation was 80% on room air. Physical examination of the skin revealed asymptomatic violaceous penile lesions that the patient reported had been present for the last 8 months (Figure 1). Pertinent laboratory values included an HIV-1 viral load of 480,135 copies/mL (reference range, <20 copies/mL) and CD4 count of 14 cells/mm3 (reference range, 480–1700 cells/mm3). A chest radiograph was obtained and revealed bibasilar opacities compatible with a pleural and/or parenchymal process. Bronchoscopy was then performed and revealed bloody secretions throughout the tracheobronchial tree.
Histologic examination of biopsies of the penile lesions revealed spindle cell proliferation with hemorrhage (Figure 2A) that stained positively for HHV-8 (Figure 2B), consistent with KS. Biopsies taken during bronchoscopy similarly revealed spindle cells with hemorrhage (Figure 3). The patient was diagnosed with AIDS-related KS with visceral involvement of the lung parenchyma and tracheobronchial tree. The patient was then admitted to the medical intensive care unit and intubated. Therapy with HAART and paclitaxel was initiated. After 7 days of poor response to therapy, the family opted for terminal extubation and comfort care measures. The patient died hours later.
This case report describes the classic phenomenon of AIDS-related KS in a patient with a long-standing history of immunocompromise. Even in the era of HAART, this patient developed a severe form of visceral KS with involvement of the respiratory tract and lung parenchyma.
Since the advent of HAART for the treatment of HIV/AIDS, the incidence of KS, both visceral and cutaneous forms, has dramatically declined; the risk for visceral KS declined by more than 50% but less than 30% for cutaneous KS, supporting the observation that although visceral involvement has classically been noted as the more aggressive and life-threatening form of disease, HAART appears to have a stronger effect on visceral disease than cutaneous disease.3 Although the overall impact of AIDS-defining illnesses has substantially improved over the years, those with AIDS infection remain at risk for opportunistic illness.2
It has been shown that HAART therapy leads to response in more than 50% of cases of KS.5 The administration of HAART in KS patients is associated with improved survival and an 80% reduced risk of death, even when started after KS is diagnosed.6 In a comparison of the differences in clinical manifestations of KS between patients who were already receiving HAART at the time of KS diagnosis to those who were not on HAART, it was shown that patients already on therapy presented with less aggressive clinical features. A smaller percentage of patients who were already on HAART at KS diagnosis presented with visceral disease compared to those who were not on therapy.7
It is evident that treatment of AIDS patients with HAART is not only first-line therapy for the disease but also the best preventative measure against development of KS. Management of KS also centers around the initiation of HAART if the patient is not already maintained on the proper therapy.8 In addition to HAART, treatment options for visceral KS include a variety of chemotherapeutic agents, including but not limited to the use of single-agent adriamycin, vinblastine, paclitaxel, and thalidomide, or combination therapies.
Although notable advances have been made in the management of AIDS patients, this case highlights the need for clinicians to be aware of the risk for KS in the context of immunocompromise. Specifically, patients with advanced AIDS who are not adherent to HAART or who have a poor response to therapy have an amplified risk for developing KS in general as well as an increased risk for developing more severe visceral KS. Maintenance of patients with HAART is shown to greatly reduce the risk for both cutaneous and visceral KS; therefore, patient adherence with therapy is of utmost importance in preventing the occurrence of this deadly disease and its complications. Appropriate follow-up should be made, ensuring that these patients at high risk are adherent to therapy and have proper access to medical care to allow for prevention and early identification of potential complications.
- La Ferla L, Pinzone MR, Nunnari G, et al. Kaposi’s sarcoma in HIV-positive patients: the state of art in the HARRT-era. Eur Rev Med Pharmacol Sci. 2013;17:2354-2365.
- Engels EA, Pfeiffer RM, Goedert JJ, et al; HIV/AIDS Cancer Match Study. Trends in cancer risk among people with AIDS in the United States 1980-2002. AIDS. 2006;20:1645-1654.
- Grabar S, Abraham B, Mahamat A, et al. Differential impact of combination antiretroviral therapy in preventing Kaposi’s sarcoma with and without visceral involvement. JCO. 2006;24:3408-3414.
- Grayson W, Pantanowitz L. Histological variants of cutaneous Kaposi sarcoma [published online July 25, 2008]. Diagn Pathol. 2008;3:31.
- Radu O, Pantanowitz L. Kaposi sarcoma. Arch Pathol Lab Med. 2013;137:289-294.
- Tam HK, Zhang ZF, Jacobson LP, et al. Effect of highly active antiretroviral therapy on survival among HIV-infected men with Kaposi sarcoma or non-Hodgkin lymphoma. Int J Cancer. 2002;98:916-922.
- Nasti G, Martellotta F, Berretta M, et al. Impact of highly active antiretroviral therapy on the presenting features and outcome of patients with acquired immunodeficiency syndrome-related Kaposi sarcoma. Cancer. 2003;98:2440-2446.
- Dupont C, Vasseur E, Beauchet A, et al. Long-term efficacy on Kaposi’s sarcoma of highly active antriretroviral therapy in a cohort of HIV-positive patients. AIDS. 2000;14:987-993.
- La Ferla L, Pinzone MR, Nunnari G, et al. Kaposi’s sarcoma in HIV-positive patients: the state of art in the HARRT-era. Eur Rev Med Pharmacol Sci. 2013;17:2354-2365.
- Engels EA, Pfeiffer RM, Goedert JJ, et al; HIV/AIDS Cancer Match Study. Trends in cancer risk among people with AIDS in the United States 1980-2002. AIDS. 2006;20:1645-1654.
- Grabar S, Abraham B, Mahamat A, et al. Differential impact of combination antiretroviral therapy in preventing Kaposi’s sarcoma with and without visceral involvement. JCO. 2006;24:3408-3414.
- Grayson W, Pantanowitz L. Histological variants of cutaneous Kaposi sarcoma [published online July 25, 2008]. Diagn Pathol. 2008;3:31.
- Radu O, Pantanowitz L. Kaposi sarcoma. Arch Pathol Lab Med. 2013;137:289-294.
- Tam HK, Zhang ZF, Jacobson LP, et al. Effect of highly active antiretroviral therapy on survival among HIV-infected men with Kaposi sarcoma or non-Hodgkin lymphoma. Int J Cancer. 2002;98:916-922.
- Nasti G, Martellotta F, Berretta M, et al. Impact of highly active antiretroviral therapy on the presenting features and outcome of patients with acquired immunodeficiency syndrome-related Kaposi sarcoma. Cancer. 2003;98:2440-2446.
- Dupont C, Vasseur E, Beauchet A, et al. Long-term efficacy on Kaposi’s sarcoma of highly active antriretroviral therapy in a cohort of HIV-positive patients. AIDS. 2000;14:987-993.
Practice Points
- Visceral Kaposi sarcoma (KS) should be considered in patients with unexplained systemic symptoms in the setting of poorly controlled human immunodeficiency virus (HIV).
- If cutaneous KS is diagnosed in an HIV patient, a detailed history and physical examination should be undertaken to evaluate for signs of systemic disease.
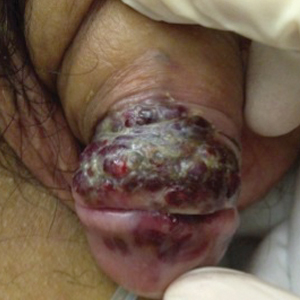
Crusted Demodicosis in an Immunocompetent Patient
To the Editor:
Demodicosis is an infection of humans caused by species of the genus of saprophytic mites Demodex (most commonly Demodex brevis and Demodex folliculorum) that feed on the pilosebaceous unit.1Demodex mites are believed to be a commensal species in humans; an increase in mite concentration or mite penetration of the dermis, however, can cause a shift from a commensal to a pathologic form.2 Demodicosis manifests in a variety of forms, including pityriasis folliculorum, rosacealike demodicosis, and demodicosis gravis. The likelihood of colonization increases with age; the mite rarely is observed in children but is found at a rate approaching 100% in the elderly population.3 It is hypothesized that manifestation of disease might be due to a decrease in immune function or an inherited HLA antigen that causes local immunosuppression.4
A 51-year-old man who was otherwise healthy presented to our clinic with a crusting rash on the face of 9 weeks’ duration. The rash began a few days after he demolished a rotting wooden shed in his backyard. Lesions began as pustules on the left cheek, which then developed notable crusting over the next 5 to 7 days and spread to involve the forehead, nose, and right cheek (Figure 1A).
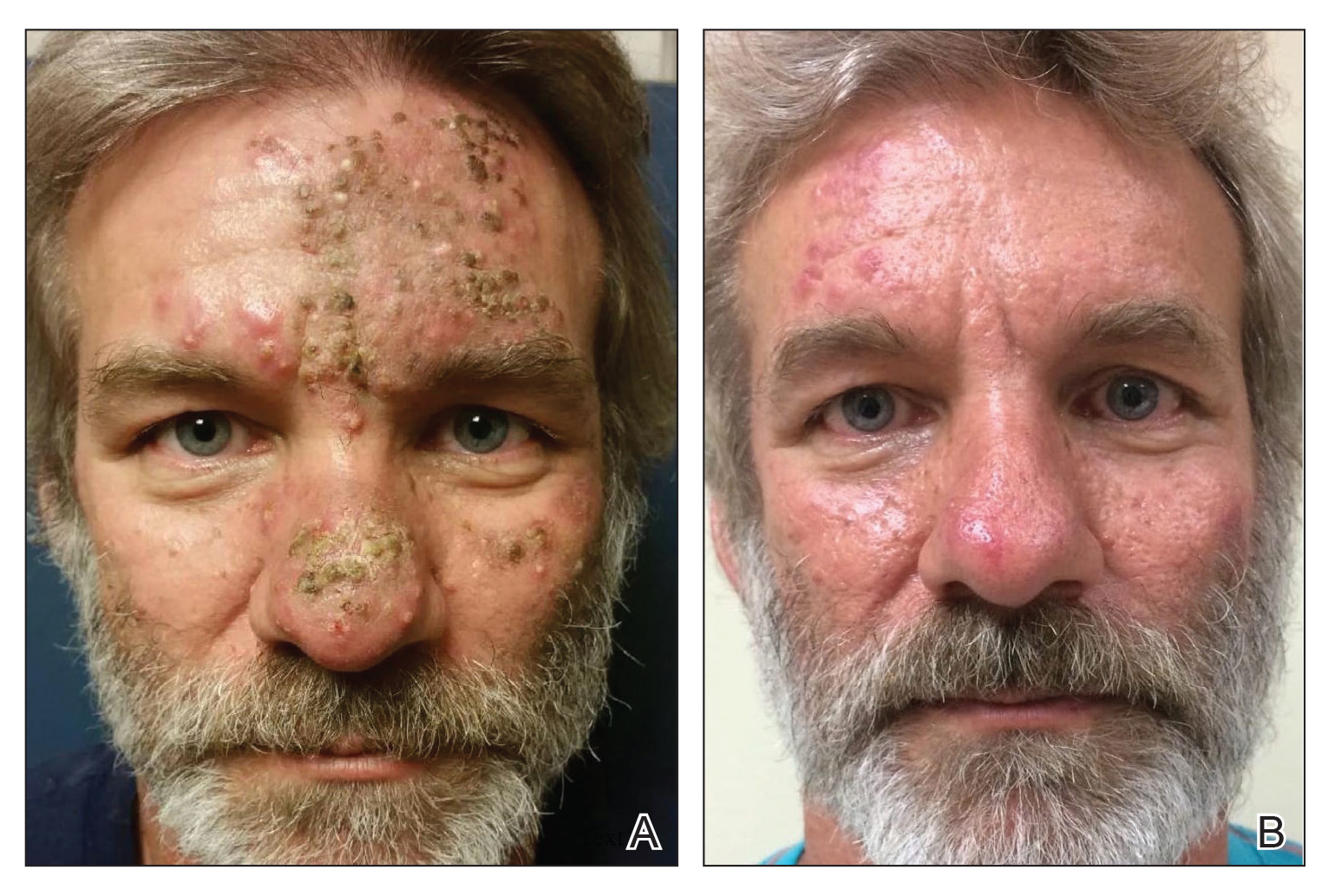
The patient had no underlying immunosuppressive disease; a human immunodeficiency virus screen, complete blood cell count, and tests of hepatic function were all unremarkable. He denied a history of frequent or recurrent sinopulmonary infections, skin infections, or infectious diarrheal illnesses. He had been seen by his primary care physician who had treated him for herpes zoster without improvement.
At our initial evaluation, biopsy was performed; specimens were sent for histopathologic analysis and culture. Findings included a dermal neutrophilic inflammation, a dense perivascular and perifollicular lymphoplasmacytic infiltrate with foci of neutrophilic pustules within the follicles (Figure 2), numerous intrafollicular Demodex mites (Figure 3), perifollicular vague noncaseating granuloma, and mild sebaceous hyperplasia. Grocott methenamine-silver stain and acid-fast bacilli stain were negative.
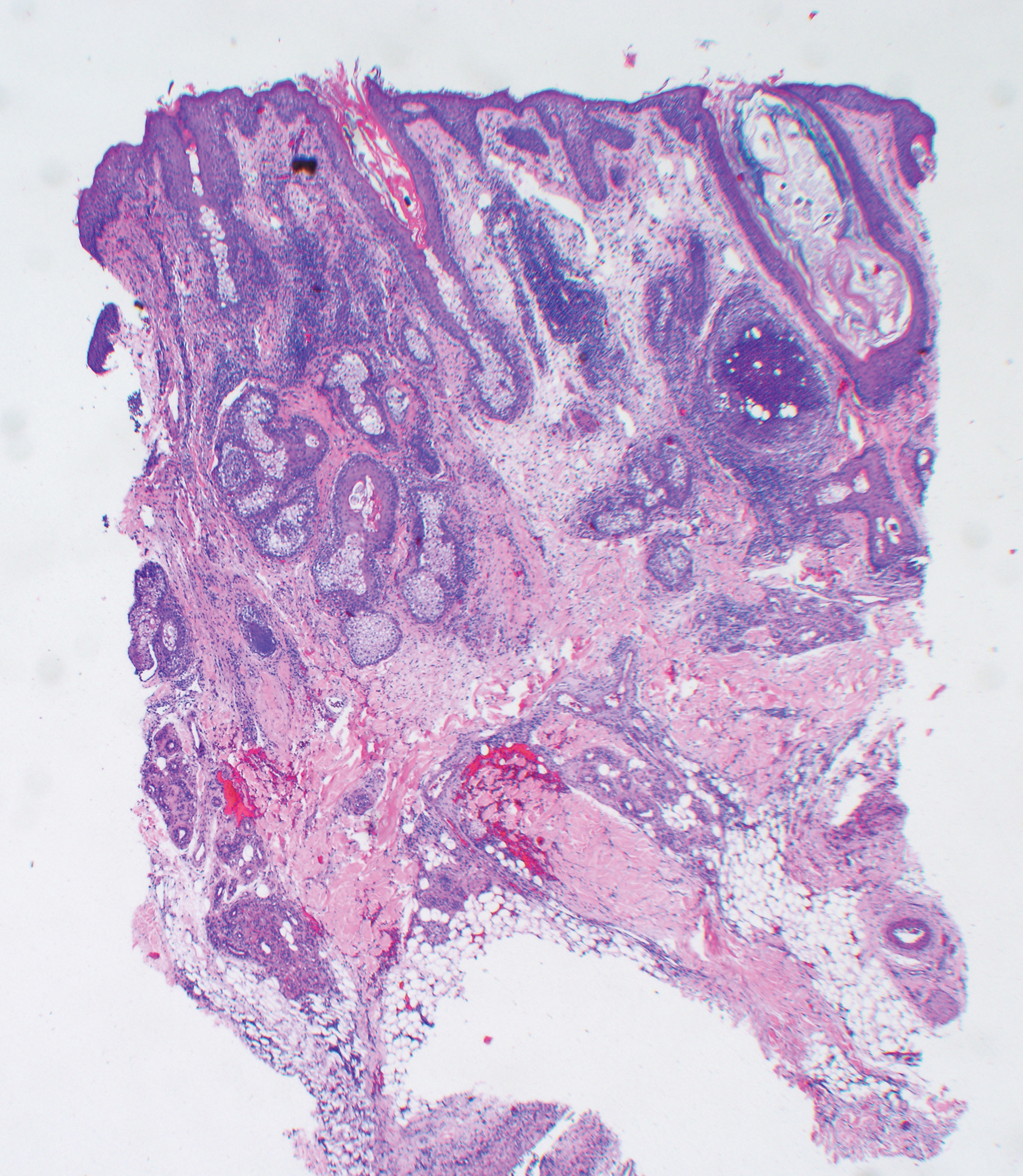
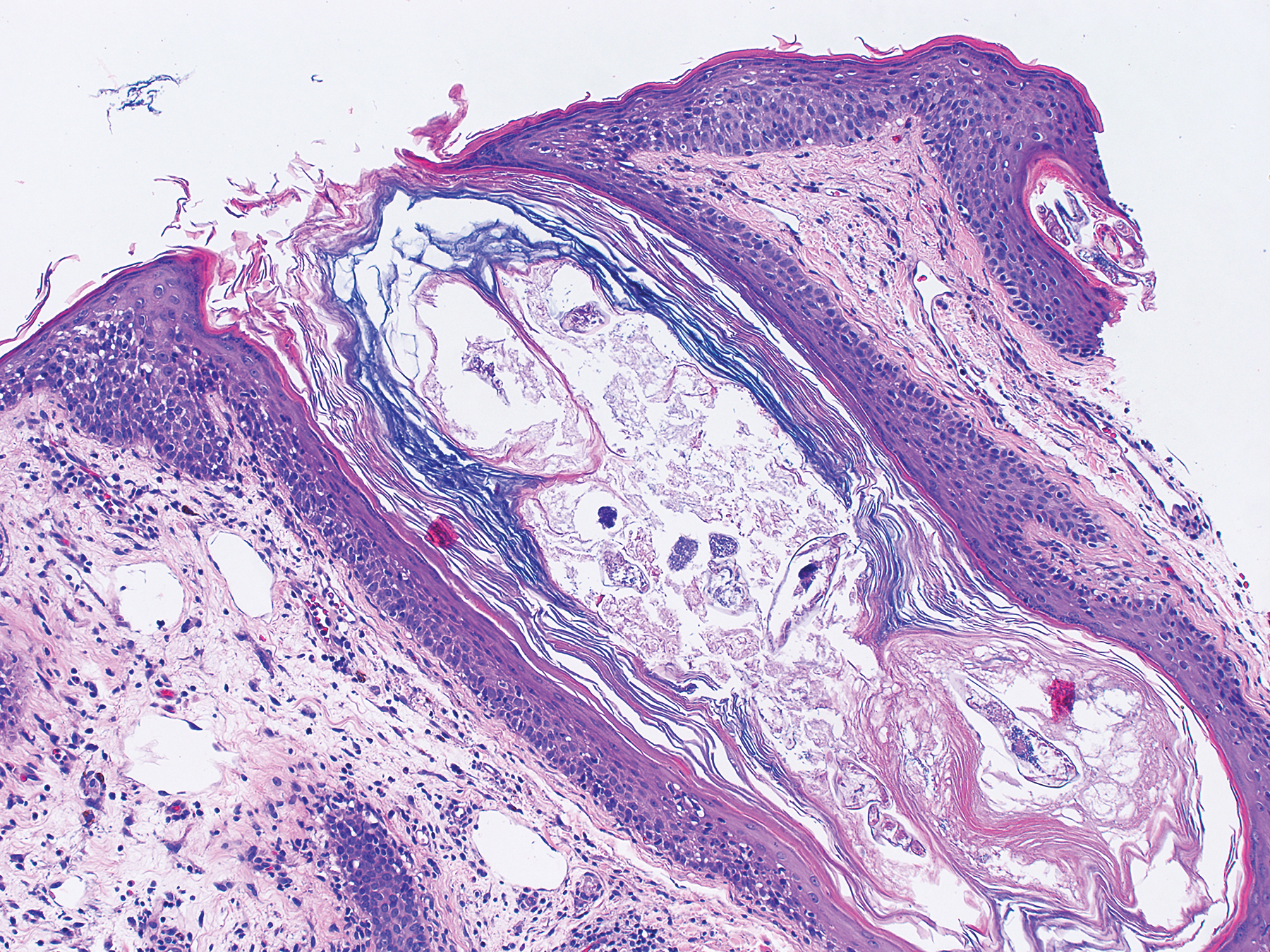
Review of clinical and pathological data yielded a final diagnosis of crusted demodicosis with a background of rosacea. The patient was ultimately treated with a single dose of oral ivermectin 15 mg with a second dose 7 days later in addition to daily application of ivermectin cream 1% to affected areas of his rash. He had notable improvement with this regimen, with complete resolution within 6 weeks (Figure 1B). The patient noted mild recurrence 14 to 21 days after discontinuing topical ivermectin.
The 2 species of Demodex that cause disease in humans each behave distinctively: D folliculorum, with a cigar-shaped body, favors superficial hair follicles; D brevis, a smaller form, burrows deeper into skin where it feeds on the pilosebaceous unit.1 Colonization occurs through direct skin-skin contact that begins as early as infancy and becomes more common with age due to development of sebaceous glands, the main source of nourishment for the mites.2
Demodicosis is classified as primary and secondary. In a prospective study of patients with clinical findings of demodicosis, Akilov et al1 discovered that the 2 forms can be differentiated by skin distribution, seasonality, mite species, and preexisting dermatoses. Primary demodicosis is categorized by sudden onset of symptoms on healthy skin, usually the face. Secondary demodicosis develops progressively in patients with preexisting skin disease, such as rosacea, and can have a broader distribution, involving the face and trunk.2 Clinical manifestations of demodicosis are broad and include pruritic papulopustular, nodulocystic, crusted, and abscesslike lesions.5
Most cases of demodicosis reported in the literature are associated with either local or systemic immunosuppression.6-8 In a case report, an otherwise immunocompetent child developed facial demodicosis after local immunosuppression from chronic use of 2 topical steroid agents.9
Demodex infestation can be diagnosed using a variety of methods, including standardized skin surface biopsy, punch biopsy, and potassium hydroxide analysis. Standardized skin surface biopsy is the preferred method to diagnose demodicosis because it is noninvasive and samples the superficial follicle where Demodex mites typically reside. Diagnosis is made by identifying 5 or more Demodex mites in a low-power field or more than 5 mites per square centimeter in standardized skin surface biopsy.2 Other potential diagnostic tools reported in the literature include dermoscopy and confocal laser scanning microscopy.10,11
There is no standard therapeutic regimen for demodicosis because evidence-based trials regarding the efficacy of treatments are lacking. Oral ivermectin 200 µg/kg in a single dose is considered the preferred treatment; it can be combined with oral erythromycin, topical permethrin, or topical metronidazole.5-7,9
Our case is unique, as crusted demodicosis developed in an immunocompetent adult. Demodicosis usually causes severe eruptions in immunocompromised persons, with only 1 case report detailing a papulopustular rash in an immunocompetent adult.12,13
The pathogenesis of demodicosis remains unclear. Many mechanisms have been hypothesized to play a role in its pathogenesis, including mechanical obstruction of hair follicles, hypersensitivity reaction to Demodex mites, immune dysregulation, and a foreign-body granulomatous reaction to the skeleton of the mite.2,3 Our patient’s particular infestation could have been caused by an exuberant reaction to Demodex; however, it is likely that many factors played a role in his disease process to cause an increase in mite density and subsequent manifestations of disease.
- Akilov OE, Butov YS, Mumcuoglu KY. A clinico-pathological approach to the classification of human demodicosis. J Dtsch Dermatol Ges. 2005;3:607-614.
- Karincaoglu Y, Bayram N, Aycan O, et al. The clinical importance of Demodex folliculorum presenting with nonspecific facial signs and symptoms. J Dermatol. 2004;31:618-626.
- Baima B, Sticherling M. Demodicidosis revisited. Acta Derm Venereol. 2002;82:3-6.
- Noy ML, Hughes S, Bunker CB. Another face of demodicosis. Clin Exp Dermatol. 2016;41:958-959.
- Chen W, Plewig G. Human demodicosis: revisit and a proposed classification. Br J Dermatol. 2014;170:1219-1225.
- Morrás PG, Santos SP, Imedio IL, et al. Rosacea-like demodicidosis in an immunocompromised child. Pediatr Dermatol. 2003;20:28-30.
- Damian D, Rogers M. Demodex infestation in a child with leukaemia: treatment with ivermectin and permethrin. Int J Dermatol. 2003;42:724-726.
- Clyti E, Nacher M, Sainte-Marie D, et al. Ivermectin treatment of three cases of demodecidosis during human immunodeficiency virus infection. Int J Dermatol. 2006;45:1066-1068.
- Guerrero-González GA, Herz-Ruelas ME, Gómez-Flores M, et al. Crusted demodicosis in an immunocompetent pediatric patient. Case Rep Dermatol Med. 2014;2014:458046.
- Friedman P, Sabban EC, Cabo H. Usefulness of dermoscopy in the diagnosis and monitoring treatment of demodicidosis. Dermatol Pract Concept. 2017;7:35-38.
- Harmelin Y, Delaunay P, Erfan N, et al. Interest of confocal laser scanning microscopy for the diagnosis and treatment monitoring of demodicosis. J Eur Acad Dermatol Venereol. 2014;28:255-257.
- Elston CA, Elston DM. Demodex mites. Clin Dermatol. 2014;32:739-743.
- Kaur T, Jindal N, Bansal R, et al. Facial demodicidosis: a diagnostic challenge. Indian J Dermatol. 2012;57:72-73.
To the Editor:
Demodicosis is an infection of humans caused by species of the genus of saprophytic mites Demodex (most commonly Demodex brevis and Demodex folliculorum) that feed on the pilosebaceous unit.1Demodex mites are believed to be a commensal species in humans; an increase in mite concentration or mite penetration of the dermis, however, can cause a shift from a commensal to a pathologic form.2 Demodicosis manifests in a variety of forms, including pityriasis folliculorum, rosacealike demodicosis, and demodicosis gravis. The likelihood of colonization increases with age; the mite rarely is observed in children but is found at a rate approaching 100% in the elderly population.3 It is hypothesized that manifestation of disease might be due to a decrease in immune function or an inherited HLA antigen that causes local immunosuppression.4
A 51-year-old man who was otherwise healthy presented to our clinic with a crusting rash on the face of 9 weeks’ duration. The rash began a few days after he demolished a rotting wooden shed in his backyard. Lesions began as pustules on the left cheek, which then developed notable crusting over the next 5 to 7 days and spread to involve the forehead, nose, and right cheek (Figure 1A).
The patient had no underlying immunosuppressive disease; a human immunodeficiency virus screen, complete blood cell count, and tests of hepatic function were all unremarkable. He denied a history of frequent or recurrent sinopulmonary infections, skin infections, or infectious diarrheal illnesses. He had been seen by his primary care physician who had treated him for herpes zoster without improvement.
At our initial evaluation, biopsy was performed; specimens were sent for histopathologic analysis and culture. Findings included a dermal neutrophilic inflammation, a dense perivascular and perifollicular lymphoplasmacytic infiltrate with foci of neutrophilic pustules within the follicles (Figure 2), numerous intrafollicular Demodex mites (Figure 3), perifollicular vague noncaseating granuloma, and mild sebaceous hyperplasia. Grocott methenamine-silver stain and acid-fast bacilli stain were negative.
Review of clinical and pathological data yielded a final diagnosis of crusted demodicosis with a background of rosacea. The patient was ultimately treated with a single dose of oral ivermectin 15 mg with a second dose 7 days later in addition to daily application of ivermectin cream 1% to affected areas of his rash. He had notable improvement with this regimen, with complete resolution within 6 weeks (Figure 1B). The patient noted mild recurrence 14 to 21 days after discontinuing topical ivermectin.
The 2 species of Demodex that cause disease in humans each behave distinctively: D folliculorum, with a cigar-shaped body, favors superficial hair follicles; D brevis, a smaller form, burrows deeper into skin where it feeds on the pilosebaceous unit.1 Colonization occurs through direct skin-skin contact that begins as early as infancy and becomes more common with age due to development of sebaceous glands, the main source of nourishment for the mites.2
Demodicosis is classified as primary and secondary. In a prospective study of patients with clinical findings of demodicosis, Akilov et al1 discovered that the 2 forms can be differentiated by skin distribution, seasonality, mite species, and preexisting dermatoses. Primary demodicosis is categorized by sudden onset of symptoms on healthy skin, usually the face. Secondary demodicosis develops progressively in patients with preexisting skin disease, such as rosacea, and can have a broader distribution, involving the face and trunk.2 Clinical manifestations of demodicosis are broad and include pruritic papulopustular, nodulocystic, crusted, and abscesslike lesions.5
Most cases of demodicosis reported in the literature are associated with either local or systemic immunosuppression.6-8 In a case report, an otherwise immunocompetent child developed facial demodicosis after local immunosuppression from chronic use of 2 topical steroid agents.9
Demodex infestation can be diagnosed using a variety of methods, including standardized skin surface biopsy, punch biopsy, and potassium hydroxide analysis. Standardized skin surface biopsy is the preferred method to diagnose demodicosis because it is noninvasive and samples the superficial follicle where Demodex mites typically reside. Diagnosis is made by identifying 5 or more Demodex mites in a low-power field or more than 5 mites per square centimeter in standardized skin surface biopsy.2 Other potential diagnostic tools reported in the literature include dermoscopy and confocal laser scanning microscopy.10,11
There is no standard therapeutic regimen for demodicosis because evidence-based trials regarding the efficacy of treatments are lacking. Oral ivermectin 200 µg/kg in a single dose is considered the preferred treatment; it can be combined with oral erythromycin, topical permethrin, or topical metronidazole.5-7,9
Our case is unique, as crusted demodicosis developed in an immunocompetent adult. Demodicosis usually causes severe eruptions in immunocompromised persons, with only 1 case report detailing a papulopustular rash in an immunocompetent adult.12,13
The pathogenesis of demodicosis remains unclear. Many mechanisms have been hypothesized to play a role in its pathogenesis, including mechanical obstruction of hair follicles, hypersensitivity reaction to Demodex mites, immune dysregulation, and a foreign-body granulomatous reaction to the skeleton of the mite.2,3 Our patient’s particular infestation could have been caused by an exuberant reaction to Demodex; however, it is likely that many factors played a role in his disease process to cause an increase in mite density and subsequent manifestations of disease.
To the Editor:
Demodicosis is an infection of humans caused by species of the genus of saprophytic mites Demodex (most commonly Demodex brevis and Demodex folliculorum) that feed on the pilosebaceous unit.1Demodex mites are believed to be a commensal species in humans; an increase in mite concentration or mite penetration of the dermis, however, can cause a shift from a commensal to a pathologic form.2 Demodicosis manifests in a variety of forms, including pityriasis folliculorum, rosacealike demodicosis, and demodicosis gravis. The likelihood of colonization increases with age; the mite rarely is observed in children but is found at a rate approaching 100% in the elderly population.3 It is hypothesized that manifestation of disease might be due to a decrease in immune function or an inherited HLA antigen that causes local immunosuppression.4
A 51-year-old man who was otherwise healthy presented to our clinic with a crusting rash on the face of 9 weeks’ duration. The rash began a few days after he demolished a rotting wooden shed in his backyard. Lesions began as pustules on the left cheek, which then developed notable crusting over the next 5 to 7 days and spread to involve the forehead, nose, and right cheek (Figure 1A).
The patient had no underlying immunosuppressive disease; a human immunodeficiency virus screen, complete blood cell count, and tests of hepatic function were all unremarkable. He denied a history of frequent or recurrent sinopulmonary infections, skin infections, or infectious diarrheal illnesses. He had been seen by his primary care physician who had treated him for herpes zoster without improvement.
At our initial evaluation, biopsy was performed; specimens were sent for histopathologic analysis and culture. Findings included a dermal neutrophilic inflammation, a dense perivascular and perifollicular lymphoplasmacytic infiltrate with foci of neutrophilic pustules within the follicles (Figure 2), numerous intrafollicular Demodex mites (Figure 3), perifollicular vague noncaseating granuloma, and mild sebaceous hyperplasia. Grocott methenamine-silver stain and acid-fast bacilli stain were negative.
Review of clinical and pathological data yielded a final diagnosis of crusted demodicosis with a background of rosacea. The patient was ultimately treated with a single dose of oral ivermectin 15 mg with a second dose 7 days later in addition to daily application of ivermectin cream 1% to affected areas of his rash. He had notable improvement with this regimen, with complete resolution within 6 weeks (Figure 1B). The patient noted mild recurrence 14 to 21 days after discontinuing topical ivermectin.
The 2 species of Demodex that cause disease in humans each behave distinctively: D folliculorum, with a cigar-shaped body, favors superficial hair follicles; D brevis, a smaller form, burrows deeper into skin where it feeds on the pilosebaceous unit.1 Colonization occurs through direct skin-skin contact that begins as early as infancy and becomes more common with age due to development of sebaceous glands, the main source of nourishment for the mites.2
Demodicosis is classified as primary and secondary. In a prospective study of patients with clinical findings of demodicosis, Akilov et al1 discovered that the 2 forms can be differentiated by skin distribution, seasonality, mite species, and preexisting dermatoses. Primary demodicosis is categorized by sudden onset of symptoms on healthy skin, usually the face. Secondary demodicosis develops progressively in patients with preexisting skin disease, such as rosacea, and can have a broader distribution, involving the face and trunk.2 Clinical manifestations of demodicosis are broad and include pruritic papulopustular, nodulocystic, crusted, and abscesslike lesions.5
Most cases of demodicosis reported in the literature are associated with either local or systemic immunosuppression.6-8 In a case report, an otherwise immunocompetent child developed facial demodicosis after local immunosuppression from chronic use of 2 topical steroid agents.9
Demodex infestation can be diagnosed using a variety of methods, including standardized skin surface biopsy, punch biopsy, and potassium hydroxide analysis. Standardized skin surface biopsy is the preferred method to diagnose demodicosis because it is noninvasive and samples the superficial follicle where Demodex mites typically reside. Diagnosis is made by identifying 5 or more Demodex mites in a low-power field or more than 5 mites per square centimeter in standardized skin surface biopsy.2 Other potential diagnostic tools reported in the literature include dermoscopy and confocal laser scanning microscopy.10,11
There is no standard therapeutic regimen for demodicosis because evidence-based trials regarding the efficacy of treatments are lacking. Oral ivermectin 200 µg/kg in a single dose is considered the preferred treatment; it can be combined with oral erythromycin, topical permethrin, or topical metronidazole.5-7,9
Our case is unique, as crusted demodicosis developed in an immunocompetent adult. Demodicosis usually causes severe eruptions in immunocompromised persons, with only 1 case report detailing a papulopustular rash in an immunocompetent adult.12,13
The pathogenesis of demodicosis remains unclear. Many mechanisms have been hypothesized to play a role in its pathogenesis, including mechanical obstruction of hair follicles, hypersensitivity reaction to Demodex mites, immune dysregulation, and a foreign-body granulomatous reaction to the skeleton of the mite.2,3 Our patient’s particular infestation could have been caused by an exuberant reaction to Demodex; however, it is likely that many factors played a role in his disease process to cause an increase in mite density and subsequent manifestations of disease.
- Akilov OE, Butov YS, Mumcuoglu KY. A clinico-pathological approach to the classification of human demodicosis. J Dtsch Dermatol Ges. 2005;3:607-614.
- Karincaoglu Y, Bayram N, Aycan O, et al. The clinical importance of Demodex folliculorum presenting with nonspecific facial signs and symptoms. J Dermatol. 2004;31:618-626.
- Baima B, Sticherling M. Demodicidosis revisited. Acta Derm Venereol. 2002;82:3-6.
- Noy ML, Hughes S, Bunker CB. Another face of demodicosis. Clin Exp Dermatol. 2016;41:958-959.
- Chen W, Plewig G. Human demodicosis: revisit and a proposed classification. Br J Dermatol. 2014;170:1219-1225.
- Morrás PG, Santos SP, Imedio IL, et al. Rosacea-like demodicidosis in an immunocompromised child. Pediatr Dermatol. 2003;20:28-30.
- Damian D, Rogers M. Demodex infestation in a child with leukaemia: treatment with ivermectin and permethrin. Int J Dermatol. 2003;42:724-726.
- Clyti E, Nacher M, Sainte-Marie D, et al. Ivermectin treatment of three cases of demodecidosis during human immunodeficiency virus infection. Int J Dermatol. 2006;45:1066-1068.
- Guerrero-González GA, Herz-Ruelas ME, Gómez-Flores M, et al. Crusted demodicosis in an immunocompetent pediatric patient. Case Rep Dermatol Med. 2014;2014:458046.
- Friedman P, Sabban EC, Cabo H. Usefulness of dermoscopy in the diagnosis and monitoring treatment of demodicidosis. Dermatol Pract Concept. 2017;7:35-38.
- Harmelin Y, Delaunay P, Erfan N, et al. Interest of confocal laser scanning microscopy for the diagnosis and treatment monitoring of demodicosis. J Eur Acad Dermatol Venereol. 2014;28:255-257.
- Elston CA, Elston DM. Demodex mites. Clin Dermatol. 2014;32:739-743.
- Kaur T, Jindal N, Bansal R, et al. Facial demodicidosis: a diagnostic challenge. Indian J Dermatol. 2012;57:72-73.
- Akilov OE, Butov YS, Mumcuoglu KY. A clinico-pathological approach to the classification of human demodicosis. J Dtsch Dermatol Ges. 2005;3:607-614.
- Karincaoglu Y, Bayram N, Aycan O, et al. The clinical importance of Demodex folliculorum presenting with nonspecific facial signs and symptoms. J Dermatol. 2004;31:618-626.
- Baima B, Sticherling M. Demodicidosis revisited. Acta Derm Venereol. 2002;82:3-6.
- Noy ML, Hughes S, Bunker CB. Another face of demodicosis. Clin Exp Dermatol. 2016;41:958-959.
- Chen W, Plewig G. Human demodicosis: revisit and a proposed classification. Br J Dermatol. 2014;170:1219-1225.
- Morrás PG, Santos SP, Imedio IL, et al. Rosacea-like demodicidosis in an immunocompromised child. Pediatr Dermatol. 2003;20:28-30.
- Damian D, Rogers M. Demodex infestation in a child with leukaemia: treatment with ivermectin and permethrin. Int J Dermatol. 2003;42:724-726.
- Clyti E, Nacher M, Sainte-Marie D, et al. Ivermectin treatment of three cases of demodecidosis during human immunodeficiency virus infection. Int J Dermatol. 2006;45:1066-1068.
- Guerrero-González GA, Herz-Ruelas ME, Gómez-Flores M, et al. Crusted demodicosis in an immunocompetent pediatric patient. Case Rep Dermatol Med. 2014;2014:458046.
- Friedman P, Sabban EC, Cabo H. Usefulness of dermoscopy in the diagnosis and monitoring treatment of demodicidosis. Dermatol Pract Concept. 2017;7:35-38.
- Harmelin Y, Delaunay P, Erfan N, et al. Interest of confocal laser scanning microscopy for the diagnosis and treatment monitoring of demodicosis. J Eur Acad Dermatol Venereol. 2014;28:255-257.
- Elston CA, Elston DM. Demodex mites. Clin Dermatol. 2014;32:739-743.
- Kaur T, Jindal N, Bansal R, et al. Facial demodicidosis: a diagnostic challenge. Indian J Dermatol. 2012;57:72-73.
Practice Points
- The Demodex mite, believed to be a commensal species in humans, has the ability to shift to a pathologic form in immunocompromised patients.
- Demodicosis can manifest in a variety of forms including pityriasis folliculorum, rosacealike demodicosis, and demodicosis gravis.