User login
Rapid Development of Perifolliculitis Following Mesotherapy
To the Editor:
Mesotherapy, also known as intradermotherapy, is a cosmetic procedure in which multiple intradermal or subcutaneous injections of homeopathic substances, vitamins, chemicals, and plant extracts are administered.1 First conceived in Europe, mesotherapy is not approved by the US Food and Drug Administration but is gaining popularity in the United States as an alternative cosmetic procedure for various purposes, including lipolysis, body contouring, stretch marks, acne scars, actinic damage, and skin rejuvenation.1,2 We report a case of a healthy woman who developed perifolliculitis, transaminitis, and neutropenia 2 weeks after mesotherapy administration to the face, neck, and chest. We also review other potential side effects of this procedure.
A 36-year-old woman with no notable medical history presented to the emergency department with a worsening pruritic and painful rash on the face, chest, and neck of 2 weeks’ duration. The rash had developed 3 days after the patient received mesotherapy with an unknown substance for cosmetic rejuvenation; the rash was localized only to the injection sites. She did not note any fever, chills, nausea, vomiting, diarrhea, headache, arthralgia, or upper respiratory tract symptoms. She further denied starting any new medications, herbal products, or topical therapies apart from the procedure she had received 2 weeks prior.
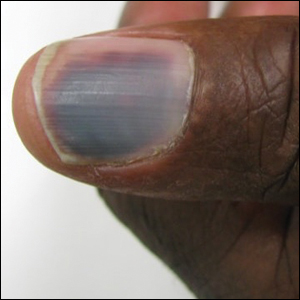
The patient was found to be in no acute distress and vital signs were stable. Laboratory testing was remarkable for elevations in alanine aminotransferase (62 U/L [reference range, 10–40 U/L]) and aspartate aminotransferase (72 U/L [reference range 10–30 U/L]). Moreover, she had an absolute neutrophil count of 0.5×103 cells/µL (reference range 1.8–8.0×103 cells/µL). An electrolyte panel, creatinine level, and urinalysis were normal. Physical examination revealed numerous 4- to 5-mm erythematous papules in a gridlike distribution across the face, neck, and chest (Figure 1). No pustules or nodules were present. There was no discharge, crust, excoriations, or secondary lesions. Additionally, there was no lymphadenopathy and no mucous membrane or ocular involvement.

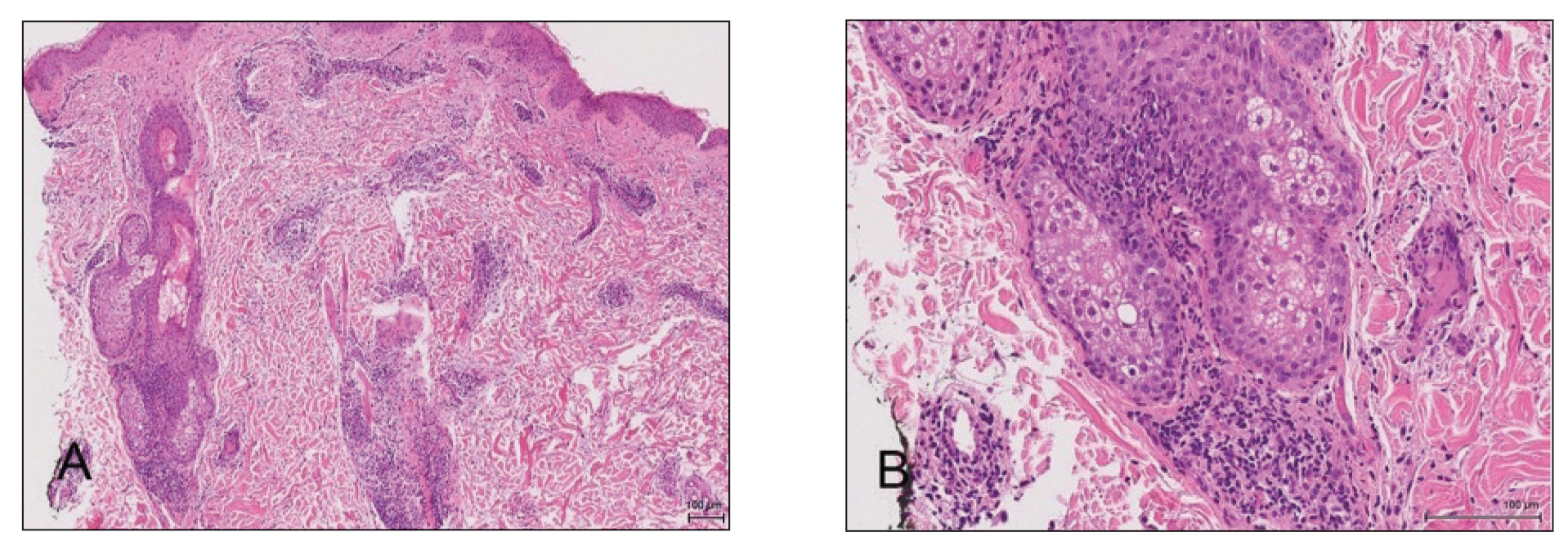
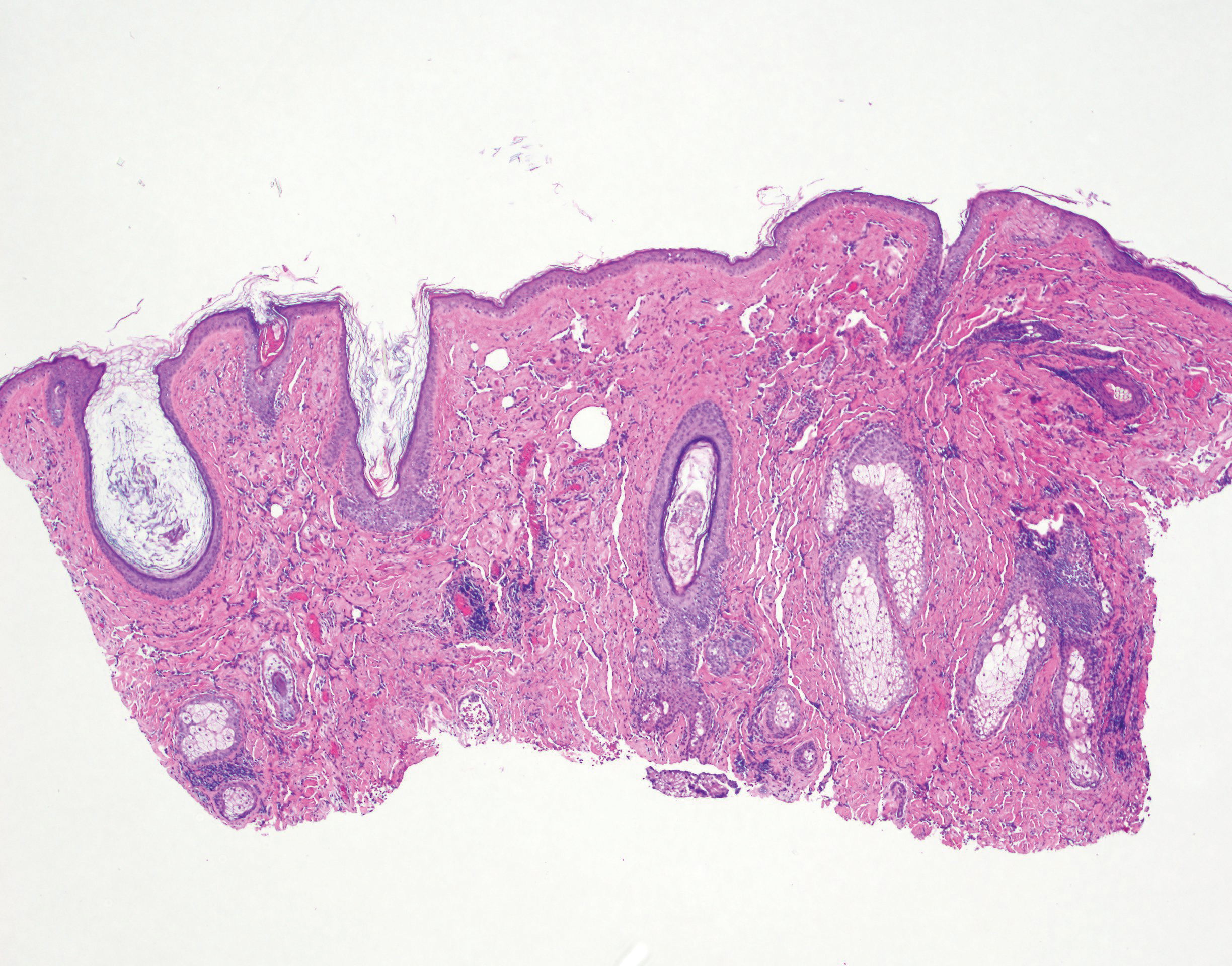
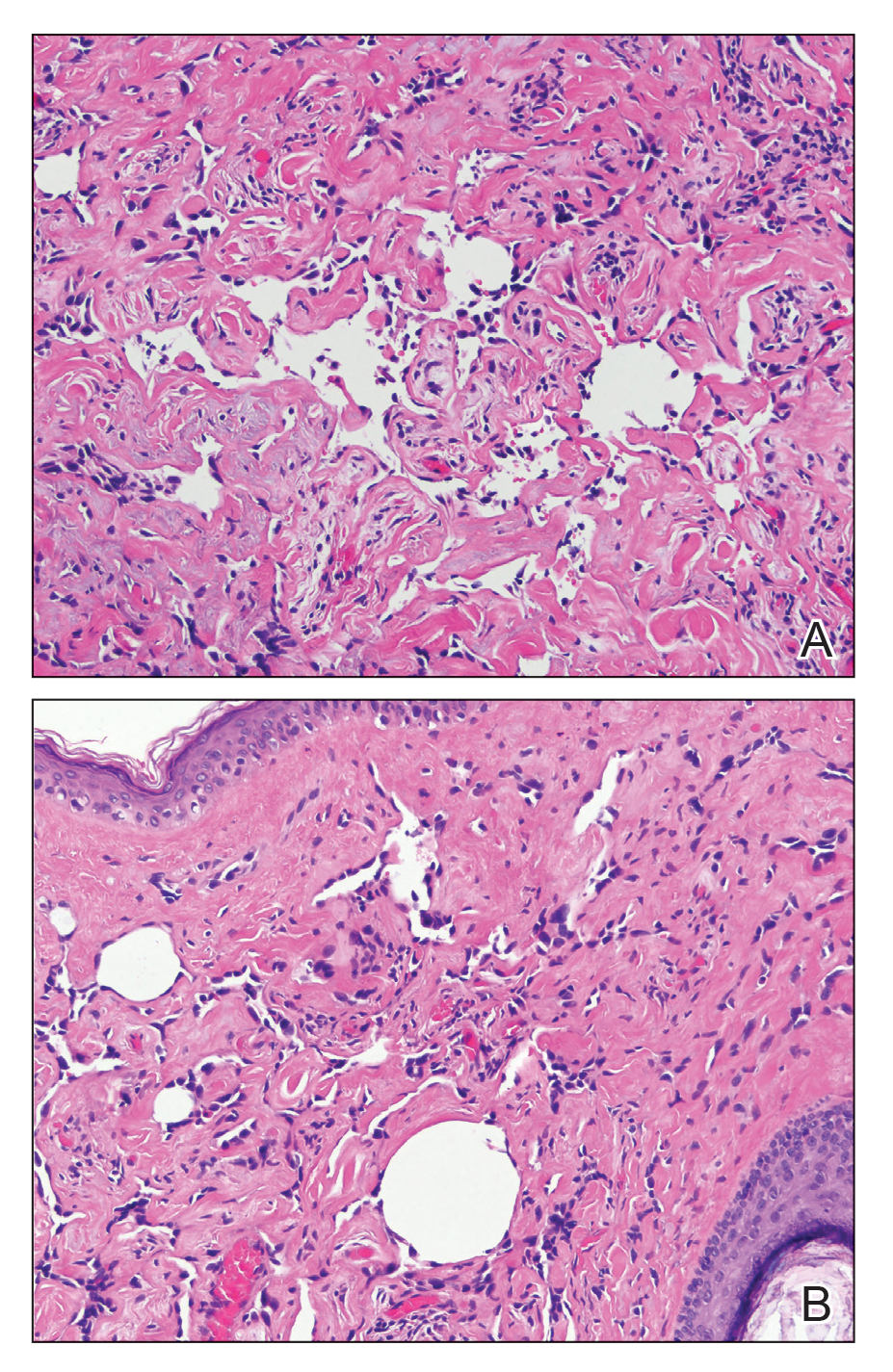
A 4-mm punch biopsy from a representative papule on the right lateral aspect of the neck demonstrated a perifollicular and perivascular lymphohistiocytic infiltrate with some focal granulomatous changes. No polarizable foreign body material was found (Figure 2). Bacterial, fungal, mycobacterial, and skin cultures were obtained, and results were all negative after several weeks.
A diagnosis of perifolliculitis from the mesotherapy procedure was on the top of the differential vs a fast-growing mycobacterial or granulomatous reaction. The patient was started on a prednisone taper at 40 mg once daily tapered down completely over 3 weeks in addition to triamcinolone cream 0.1% applied 2 to 4 times daily as needed. Although she did not return to our outpatient clinic for follow-up, she informed us that her rash had improved 1 month after starting the prednisone taper. She was later lost to follow-up. It is unclear if the transaminitis and neutropenia were related to the materials injected during the mesotherapy procedure or from long-standing health issues.
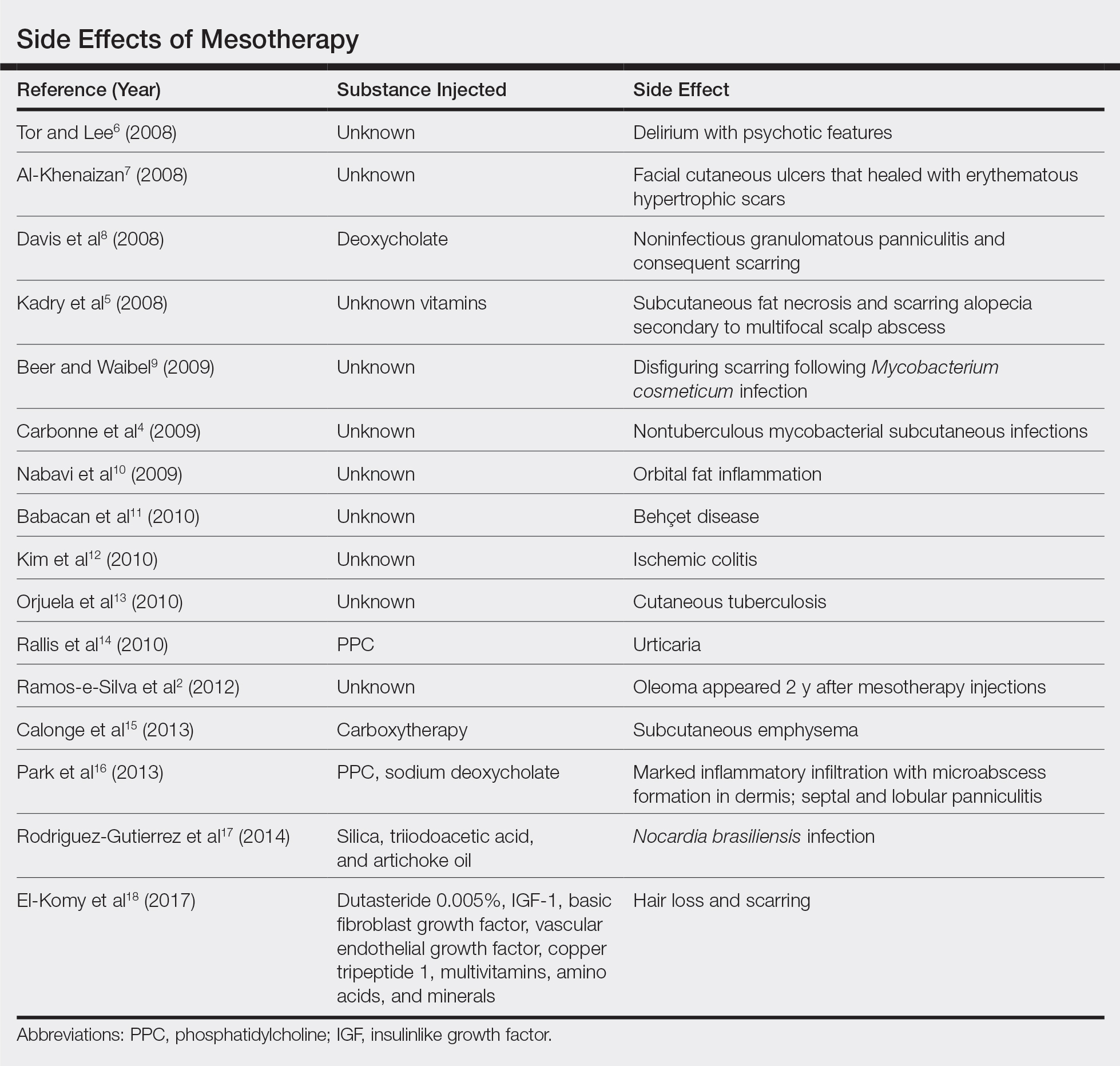
Mesotherapy promises aesthetic benefits through a minimally invasive procedure and therefore is rapidly gaining popularity in aesthetic spas and treatment centers. Due to the lack of regulation in treatment protocols and substances used, there have been numerous reported cases of adverse side effects following mesotherapy, such as pain, allergic reactions, urticaria, panniculitis, ulceration, hair loss, necrosis, paraffinoma, cutaneous tuberculosis, and rapidly growing nontuberculous mycobacterial infections.1-5 More serious side effects also have been reported, such as permanent scarring, deformities, delirium, and massive subcutaneous emphysema (Table).2,4-18
Given the potential complications of mesotherapy documented in the literature, we believe clinical investigations and trials must be performed to appropriately assess the safety and efficacy of this potentially hazardous procedure. Because there currently is insufficient research showing why certain patients are developing these adverse side effects, aesthetic spas and treatment centers should inform patients of all potential side effects associated with mesotherapy for the patient to make an informed decision about the procedure. Mesotherapy should be a point of focus for both the US Food and Drug Administration and researchers to determine its efficacy, safety, and standardization of the procedure.
- Bishara AS, Ibrahim AE, Dibo SA. Cosmetic mesotherapy: between scientific evidence, science fiction, and lucrative business. Aesth Plast Surg. 2008;32:842-849.
- Ramos-e-Silva M, Pereira AL, Ramos-e-Silva S, et al. Oleoma: a rare complication of mesotherapy for cellulite. Int J Dermatol. 2012;51:162-167.
- Rotunda AM, Kolodney MS. Mesotherapy and phosphatidylcholine injections: historical clarification and review. Dermatol Surg. 2006;32:465-480.
- Carbonne A, Brossier F, Arnaud I, et al. Outbreak of nontuberculous mycobacterial subcutaneous infections related to multiple mesotherapy injections. J Clin Microbiol. 2009;47:1961-1964.
- Kadry R, Hamadah I, Al-Issa A, et al. Multifocal scalp abscess with subcutaneous fat necrosis and scarring alopecia as a complication of scalp mesotherapy. J Drugs Dermatol. 2008;7:72-73.
- Tor PC, Lee TS. Delirium with psychotic features possibly associated with mesotherapy. Psychosomatics. 2008;49:273-274.
- Al-Khenaizan S. Facial cutaneous ulcers following mesotherapy. Dermatol Surg. 2008;34:832-834.
- Davis MD, Wright TI, Shehan JM. A complication of mesotherapy: noninfectious granulomatous panniculitis. Arch Dermatol. 2008;144:808-809.
- Beer K, Waibel J. Disfiguring scarring following mesotherapy-associated Mycobacterium cosmeticum infection. J Drugs Dermatol. 2009;8:391-393.
- Nabavi CB, Minckler DS, Tao JP. Histologic features of mesotherapy-induced orbital fat inflammation. Opthalmic Plast Reconstr Surg. 2009;25:69-70.
- Babacan T, Onat AM, Pehlivan Y, et al. A case of Behçet’s disease diagnosed by the panniculitis after mesotherapy. Rheumatol Int. 2010;30:1657-1659.
- Kim JB, Moon W, Park SJ, et al. Ischemic colitis after mesotherapy combined with anti-obesity medications. World J Gastroenterol. 2010;16:1537-1540.
- Orjuela D, Puerto G, Mejia G, et al. Cutaneous tuberculosis after mesotherapy: report of six cases. Biomedica. 2010;30:321-326.
- Rallis E, Kintzoglou S, Moussatou V, et al. Mesotherapy-induced urticaria. Dermatol Surg. 2010;36:1355-1356.
- Calonge WM, Lesbros-Pantoflickova D, Hodina M, et al. Massive subcutaneous emphysema after carbon dioxide mesotherapy. Aesthetic Plast Surg. 2013;37:194-197.
- Park EJ, Kim HS, Kim M, et al. Histological changes after treatment for localized fat deposits with phosphatidylcholine and sodium deoxycholate. J Cosmet Dermatol. 2013;3:240-243.
- Rodriguez-Gutierrez G, Toussaint S, Hernandez-Castro R, et al. Norcardia brasiliensis infection: an emergent suppurative granuloma after mesotherapy. Int J Dermatol. 2014;53:888-890.
- El-Komy M, Hassan A, Tawdy A, et al. Hair loss at injection sites of mesotherapy for alopecia [published online February 3, 2017]. J Cosmet Dermatol. 2017;16:E28-E30.
To the Editor:
Mesotherapy, also known as intradermotherapy, is a cosmetic procedure in which multiple intradermal or subcutaneous injections of homeopathic substances, vitamins, chemicals, and plant extracts are administered.1 First conceived in Europe, mesotherapy is not approved by the US Food and Drug Administration but is gaining popularity in the United States as an alternative cosmetic procedure for various purposes, including lipolysis, body contouring, stretch marks, acne scars, actinic damage, and skin rejuvenation.1,2 We report a case of a healthy woman who developed perifolliculitis, transaminitis, and neutropenia 2 weeks after mesotherapy administration to the face, neck, and chest. We also review other potential side effects of this procedure.
A 36-year-old woman with no notable medical history presented to the emergency department with a worsening pruritic and painful rash on the face, chest, and neck of 2 weeks’ duration. The rash had developed 3 days after the patient received mesotherapy with an unknown substance for cosmetic rejuvenation; the rash was localized only to the injection sites. She did not note any fever, chills, nausea, vomiting, diarrhea, headache, arthralgia, or upper respiratory tract symptoms. She further denied starting any new medications, herbal products, or topical therapies apart from the procedure she had received 2 weeks prior.
The patient was found to be in no acute distress and vital signs were stable. Laboratory testing was remarkable for elevations in alanine aminotransferase (62 U/L [reference range, 10–40 U/L]) and aspartate aminotransferase (72 U/L [reference range 10–30 U/L]). Moreover, she had an absolute neutrophil count of 0.5×103 cells/µL (reference range 1.8–8.0×103 cells/µL). An electrolyte panel, creatinine level, and urinalysis were normal. Physical examination revealed numerous 4- to 5-mm erythematous papules in a gridlike distribution across the face, neck, and chest (Figure 1). No pustules or nodules were present. There was no discharge, crust, excoriations, or secondary lesions. Additionally, there was no lymphadenopathy and no mucous membrane or ocular involvement.
A 4-mm punch biopsy from a representative papule on the right lateral aspect of the neck demonstrated a perifollicular and perivascular lymphohistiocytic infiltrate with some focal granulomatous changes. No polarizable foreign body material was found (Figure 2). Bacterial, fungal, mycobacterial, and skin cultures were obtained, and results were all negative after several weeks.
A diagnosis of perifolliculitis from the mesotherapy procedure was on the top of the differential vs a fast-growing mycobacterial or granulomatous reaction. The patient was started on a prednisone taper at 40 mg once daily tapered down completely over 3 weeks in addition to triamcinolone cream 0.1% applied 2 to 4 times daily as needed. Although she did not return to our outpatient clinic for follow-up, she informed us that her rash had improved 1 month after starting the prednisone taper. She was later lost to follow-up. It is unclear if the transaminitis and neutropenia were related to the materials injected during the mesotherapy procedure or from long-standing health issues.
Mesotherapy promises aesthetic benefits through a minimally invasive procedure and therefore is rapidly gaining popularity in aesthetic spas and treatment centers. Due to the lack of regulation in treatment protocols and substances used, there have been numerous reported cases of adverse side effects following mesotherapy, such as pain, allergic reactions, urticaria, panniculitis, ulceration, hair loss, necrosis, paraffinoma, cutaneous tuberculosis, and rapidly growing nontuberculous mycobacterial infections.1-5 More serious side effects also have been reported, such as permanent scarring, deformities, delirium, and massive subcutaneous emphysema (Table).2,4-18
Given the potential complications of mesotherapy documented in the literature, we believe clinical investigations and trials must be performed to appropriately assess the safety and efficacy of this potentially hazardous procedure. Because there currently is insufficient research showing why certain patients are developing these adverse side effects, aesthetic spas and treatment centers should inform patients of all potential side effects associated with mesotherapy for the patient to make an informed decision about the procedure. Mesotherapy should be a point of focus for both the US Food and Drug Administration and researchers to determine its efficacy, safety, and standardization of the procedure.
To the Editor:
Mesotherapy, also known as intradermotherapy, is a cosmetic procedure in which multiple intradermal or subcutaneous injections of homeopathic substances, vitamins, chemicals, and plant extracts are administered.1 First conceived in Europe, mesotherapy is not approved by the US Food and Drug Administration but is gaining popularity in the United States as an alternative cosmetic procedure for various purposes, including lipolysis, body contouring, stretch marks, acne scars, actinic damage, and skin rejuvenation.1,2 We report a case of a healthy woman who developed perifolliculitis, transaminitis, and neutropenia 2 weeks after mesotherapy administration to the face, neck, and chest. We also review other potential side effects of this procedure.
A 36-year-old woman with no notable medical history presented to the emergency department with a worsening pruritic and painful rash on the face, chest, and neck of 2 weeks’ duration. The rash had developed 3 days after the patient received mesotherapy with an unknown substance for cosmetic rejuvenation; the rash was localized only to the injection sites. She did not note any fever, chills, nausea, vomiting, diarrhea, headache, arthralgia, or upper respiratory tract symptoms. She further denied starting any new medications, herbal products, or topical therapies apart from the procedure she had received 2 weeks prior.
The patient was found to be in no acute distress and vital signs were stable. Laboratory testing was remarkable for elevations in alanine aminotransferase (62 U/L [reference range, 10–40 U/L]) and aspartate aminotransferase (72 U/L [reference range 10–30 U/L]). Moreover, she had an absolute neutrophil count of 0.5×103 cells/µL (reference range 1.8–8.0×103 cells/µL). An electrolyte panel, creatinine level, and urinalysis were normal. Physical examination revealed numerous 4- to 5-mm erythematous papules in a gridlike distribution across the face, neck, and chest (Figure 1). No pustules or nodules were present. There was no discharge, crust, excoriations, or secondary lesions. Additionally, there was no lymphadenopathy and no mucous membrane or ocular involvement.
A 4-mm punch biopsy from a representative papule on the right lateral aspect of the neck demonstrated a perifollicular and perivascular lymphohistiocytic infiltrate with some focal granulomatous changes. No polarizable foreign body material was found (Figure 2). Bacterial, fungal, mycobacterial, and skin cultures were obtained, and results were all negative after several weeks.
A diagnosis of perifolliculitis from the mesotherapy procedure was on the top of the differential vs a fast-growing mycobacterial or granulomatous reaction. The patient was started on a prednisone taper at 40 mg once daily tapered down completely over 3 weeks in addition to triamcinolone cream 0.1% applied 2 to 4 times daily as needed. Although she did not return to our outpatient clinic for follow-up, she informed us that her rash had improved 1 month after starting the prednisone taper. She was later lost to follow-up. It is unclear if the transaminitis and neutropenia were related to the materials injected during the mesotherapy procedure or from long-standing health issues.
Mesotherapy promises aesthetic benefits through a minimally invasive procedure and therefore is rapidly gaining popularity in aesthetic spas and treatment centers. Due to the lack of regulation in treatment protocols and substances used, there have been numerous reported cases of adverse side effects following mesotherapy, such as pain, allergic reactions, urticaria, panniculitis, ulceration, hair loss, necrosis, paraffinoma, cutaneous tuberculosis, and rapidly growing nontuberculous mycobacterial infections.1-5 More serious side effects also have been reported, such as permanent scarring, deformities, delirium, and massive subcutaneous emphysema (Table).2,4-18
Given the potential complications of mesotherapy documented in the literature, we believe clinical investigations and trials must be performed to appropriately assess the safety and efficacy of this potentially hazardous procedure. Because there currently is insufficient research showing why certain patients are developing these adverse side effects, aesthetic spas and treatment centers should inform patients of all potential side effects associated with mesotherapy for the patient to make an informed decision about the procedure. Mesotherapy should be a point of focus for both the US Food and Drug Administration and researchers to determine its efficacy, safety, and standardization of the procedure.
- Bishara AS, Ibrahim AE, Dibo SA. Cosmetic mesotherapy: between scientific evidence, science fiction, and lucrative business. Aesth Plast Surg. 2008;32:842-849.
- Ramos-e-Silva M, Pereira AL, Ramos-e-Silva S, et al. Oleoma: a rare complication of mesotherapy for cellulite. Int J Dermatol. 2012;51:162-167.
- Rotunda AM, Kolodney MS. Mesotherapy and phosphatidylcholine injections: historical clarification and review. Dermatol Surg. 2006;32:465-480.
- Carbonne A, Brossier F, Arnaud I, et al. Outbreak of nontuberculous mycobacterial subcutaneous infections related to multiple mesotherapy injections. J Clin Microbiol. 2009;47:1961-1964.
- Kadry R, Hamadah I, Al-Issa A, et al. Multifocal scalp abscess with subcutaneous fat necrosis and scarring alopecia as a complication of scalp mesotherapy. J Drugs Dermatol. 2008;7:72-73.
- Tor PC, Lee TS. Delirium with psychotic features possibly associated with mesotherapy. Psychosomatics. 2008;49:273-274.
- Al-Khenaizan S. Facial cutaneous ulcers following mesotherapy. Dermatol Surg. 2008;34:832-834.
- Davis MD, Wright TI, Shehan JM. A complication of mesotherapy: noninfectious granulomatous panniculitis. Arch Dermatol. 2008;144:808-809.
- Beer K, Waibel J. Disfiguring scarring following mesotherapy-associated Mycobacterium cosmeticum infection. J Drugs Dermatol. 2009;8:391-393.
- Nabavi CB, Minckler DS, Tao JP. Histologic features of mesotherapy-induced orbital fat inflammation. Opthalmic Plast Reconstr Surg. 2009;25:69-70.
- Babacan T, Onat AM, Pehlivan Y, et al. A case of Behçet’s disease diagnosed by the panniculitis after mesotherapy. Rheumatol Int. 2010;30:1657-1659.
- Kim JB, Moon W, Park SJ, et al. Ischemic colitis after mesotherapy combined with anti-obesity medications. World J Gastroenterol. 2010;16:1537-1540.
- Orjuela D, Puerto G, Mejia G, et al. Cutaneous tuberculosis after mesotherapy: report of six cases. Biomedica. 2010;30:321-326.
- Rallis E, Kintzoglou S, Moussatou V, et al. Mesotherapy-induced urticaria. Dermatol Surg. 2010;36:1355-1356.
- Calonge WM, Lesbros-Pantoflickova D, Hodina M, et al. Massive subcutaneous emphysema after carbon dioxide mesotherapy. Aesthetic Plast Surg. 2013;37:194-197.
- Park EJ, Kim HS, Kim M, et al. Histological changes after treatment for localized fat deposits with phosphatidylcholine and sodium deoxycholate. J Cosmet Dermatol. 2013;3:240-243.
- Rodriguez-Gutierrez G, Toussaint S, Hernandez-Castro R, et al. Norcardia brasiliensis infection: an emergent suppurative granuloma after mesotherapy. Int J Dermatol. 2014;53:888-890.
- El-Komy M, Hassan A, Tawdy A, et al. Hair loss at injection sites of mesotherapy for alopecia [published online February 3, 2017]. J Cosmet Dermatol. 2017;16:E28-E30.
- Bishara AS, Ibrahim AE, Dibo SA. Cosmetic mesotherapy: between scientific evidence, science fiction, and lucrative business. Aesth Plast Surg. 2008;32:842-849.
- Ramos-e-Silva M, Pereira AL, Ramos-e-Silva S, et al. Oleoma: a rare complication of mesotherapy for cellulite. Int J Dermatol. 2012;51:162-167.
- Rotunda AM, Kolodney MS. Mesotherapy and phosphatidylcholine injections: historical clarification and review. Dermatol Surg. 2006;32:465-480.
- Carbonne A, Brossier F, Arnaud I, et al. Outbreak of nontuberculous mycobacterial subcutaneous infections related to multiple mesotherapy injections. J Clin Microbiol. 2009;47:1961-1964.
- Kadry R, Hamadah I, Al-Issa A, et al. Multifocal scalp abscess with subcutaneous fat necrosis and scarring alopecia as a complication of scalp mesotherapy. J Drugs Dermatol. 2008;7:72-73.
- Tor PC, Lee TS. Delirium with psychotic features possibly associated with mesotherapy. Psychosomatics. 2008;49:273-274.
- Al-Khenaizan S. Facial cutaneous ulcers following mesotherapy. Dermatol Surg. 2008;34:832-834.
- Davis MD, Wright TI, Shehan JM. A complication of mesotherapy: noninfectious granulomatous panniculitis. Arch Dermatol. 2008;144:808-809.
- Beer K, Waibel J. Disfiguring scarring following mesotherapy-associated Mycobacterium cosmeticum infection. J Drugs Dermatol. 2009;8:391-393.
- Nabavi CB, Minckler DS, Tao JP. Histologic features of mesotherapy-induced orbital fat inflammation. Opthalmic Plast Reconstr Surg. 2009;25:69-70.
- Babacan T, Onat AM, Pehlivan Y, et al. A case of Behçet’s disease diagnosed by the panniculitis after mesotherapy. Rheumatol Int. 2010;30:1657-1659.
- Kim JB, Moon W, Park SJ, et al. Ischemic colitis after mesotherapy combined with anti-obesity medications. World J Gastroenterol. 2010;16:1537-1540.
- Orjuela D, Puerto G, Mejia G, et al. Cutaneous tuberculosis after mesotherapy: report of six cases. Biomedica. 2010;30:321-326.
- Rallis E, Kintzoglou S, Moussatou V, et al. Mesotherapy-induced urticaria. Dermatol Surg. 2010;36:1355-1356.
- Calonge WM, Lesbros-Pantoflickova D, Hodina M, et al. Massive subcutaneous emphysema after carbon dioxide mesotherapy. Aesthetic Plast Surg. 2013;37:194-197.
- Park EJ, Kim HS, Kim M, et al. Histological changes after treatment for localized fat deposits with phosphatidylcholine and sodium deoxycholate. J Cosmet Dermatol. 2013;3:240-243.
- Rodriguez-Gutierrez G, Toussaint S, Hernandez-Castro R, et al. Norcardia brasiliensis infection: an emergent suppurative granuloma after mesotherapy. Int J Dermatol. 2014;53:888-890.
- El-Komy M, Hassan A, Tawdy A, et al. Hair loss at injection sites of mesotherapy for alopecia [published online February 3, 2017]. J Cosmet Dermatol. 2017;16:E28-E30.
Practice Points
- Mesotherapy—the delivery of vitamins, chemicals, and plant extracts directly into the dermis via injections—is a common procedure performed in both medical and nonmedical settings for cosmetic rejuvenation.
- Complications can occur from mesotherapy treatment.
- Patients should be advised to seek medical care with US Food and Drug Administration–approved cosmetic techniques and substances only

Human Immunodeficiency Virus Infection in a Hepatitis B Virus–Positive Psoriasis Patient Treated With Ustekinumab
To the Editor:
The incidence of psoriasis in human immunodeficiency virus (HIV)–infected patients is similar to the general population, but it usually becomes more severe as immunosuppression increases. Additionally, it tends to be more resistant to conventional therapies, and the incidence and severity of psoriatic arthropathy is increased. Psoriasis often worsens at the time of HIV primary infection.1 We describe a case of a patient with hepatitis B virus (HBV) whose severe plaque psoriasis was controlled on ustekinumab; he was later diagnosed with HIV infection.
A 42-year-old man with HBV treated with entecavir (HBV DNA viral load, <20 copies/mL [inactive carrier, <2000 copies/mL]) presented to our dermatology unit with severe plaque psoriasis (psoriasis area and severity index 23) that caused notable psychologic difficulties such as anxiety and depression. Treatment was attempted with cyclosporine; acitretin; psoralen plus UVA; infliximab; adalimumab; and eventually ustekinumab (45 mg every 3 months), which controlled the condition well (psoriasis area and severity index 0) in an almost completely sustained manner.
Serologic tests requested at one of his analytical control appointments 2 years after initiating treatment with ustekinumab revealed he was HIV positive. The patient reported unprotected sexual intercourse 4 months prior. He was referred to the infectious disease unit and was classified in subtype A1 of HIV infection (CD4 count, 583 cells/µL [reference range, 500-1200 cells/µL]; viral load, 159,268 copies/mL [rapid progression to AIDS, >100,000 copies/mL]). Treatment was initiated with raltegravir, ritonavir, darunavir, and abacavir; tolerance was suitable. Because of the patient’s history of severe psoriasis, treatment with ustekinumab was maintained. Normally, treatment with this drug would be contraindicated in patients with HIV, as it can lead to viral reactivation. Four years after his HIV diagnosis, neither the patient’s cutaneous nor HIV-associated condition had worsened.
For patients with HIV and mild or moderate psoriasis, topical therapies (eg, corticosteroids, vitamin D analogues, tazarotene) are recommended, similar to patients who are HIV negative. Human immunodeficiency virus–positive patients with severe psoriasis who do not respond to topical treatment should receive phototherapy (UVB or psoralen plus UVA) or acitretin along with their antiretroviral drugs.2 In refractory cases, immunosuppressants, including cyclosporine, methotrexate, or tumor necrosis factor α inhibitors, might be used, though experience with them is limited.3,4 Maintaining antiretroviral therapy and prophylaxis against opportunist disease is important in patients who receive such immunosuppressants, as is close monitoring of the viral load.
Ustekinumab is an IL-12/IL-23 monoclonal antibody indicated for the treatment of moderate to severe plaque psoriasis, active psoriatic arthritis, and inflammatory bowel disease. It is contraindicated in patients with clinically important active infections, such as HBV and hepatitis C virus infections.5 However, it was shown to be safe in a group of 18 patients with HBV who had received antiviral prophylaxis6; a degree of reactivation was observed in similar patients who received no such prophylaxis and in others with hepatitis C virus infection.7 The simultaneous use of methotrexate with ustekinumab in the treatment of psoriatic arthritis does not appear to affect the safety of the latter drug.8 Paparizos et al9 described a patient with HIV controlled with antiretroviral drugs who was treated with ustekinumab for psoriasis; no adverse effects were observed.
We report the case of a patient with HBV and psoriasis who was treated with ustekinumab and later became infected with HIV. His ustekinumab treatment was maintained without subsequent cutaneous or systemic complications.
- Menon K, Van Voorhees V, Bebo B, et al. Psoriasis in patients with HIV infection: from the Medical Board of the National Psoriasis Foundation. J Am Acad Dermatol. 2010;62:291-299.
- Chiricozzi A, Saraceno R, Cannizzaro MV. Complete resolution of erythrodermic psoriasis in an HIV and HCV patient unresponsive to antipsoriatic treatments after highly active antiretroviral therapy. Dermatology. 2012;225:333-337.
- Barco D, Puig L, Alomar A. Treatment of moderate-severe psoriasis with etanercept in patients with chronic human immunodeficiency virus infection. Actas Dermosifiliogr. 2010;101(suppl 1):77-81.
- Lindsey SF, Weiss J, Lee ES, et al. Treatment of severe psoriasis and psoriatic arthritis with adalimumab in an HIV positive patient. J Drugs Dermatol. 2014;13:869-871.
- Rustin MH. Long-term safety of biologics in the treatment of moderate to severe plaque psoriasis: review of the current data. Br J Dermatol. 2012;167(suppl 3):3-11.
- Navarro R, Vilarrasa E, Herranz P, et al. Safety and effectiveness of ustekinumab and antitumour necrosis factor therapy in patients with psoriasis and chronic viral hepatitis B or C: a retrospective, multicentre study in a clinical setting. Br J Dermatol. 2013;168:609-616.
- Chiu HY, Chen CH, Wu MS, et al. The safety profile of ustekinumab in the treatment of patients with psoriasis and concurrent hepatitis B or C. Br J Dermatol. 2013;169:1295-1303.
- Weitz JE, Ritchlin CT. Ustekinumab: targeting the IL-17 pathway to improve outcomes in psoriatic arthritis. Expert Opin Biol Ther. 2014;14:515-526.
- Paparizos V, Rallis E, Kirsten L, et al. Ustekinumab for the treatment of HIV psoriasis. J Dermatol Treat. 2012;23:398-399.
To the Editor:
The incidence of psoriasis in human immunodeficiency virus (HIV)–infected patients is similar to the general population, but it usually becomes more severe as immunosuppression increases. Additionally, it tends to be more resistant to conventional therapies, and the incidence and severity of psoriatic arthropathy is increased. Psoriasis often worsens at the time of HIV primary infection.1 We describe a case of a patient with hepatitis B virus (HBV) whose severe plaque psoriasis was controlled on ustekinumab; he was later diagnosed with HIV infection.
A 42-year-old man with HBV treated with entecavir (HBV DNA viral load, <20 copies/mL [inactive carrier, <2000 copies/mL]) presented to our dermatology unit with severe plaque psoriasis (psoriasis area and severity index 23) that caused notable psychologic difficulties such as anxiety and depression. Treatment was attempted with cyclosporine; acitretin; psoralen plus UVA; infliximab; adalimumab; and eventually ustekinumab (45 mg every 3 months), which controlled the condition well (psoriasis area and severity index 0) in an almost completely sustained manner.
Serologic tests requested at one of his analytical control appointments 2 years after initiating treatment with ustekinumab revealed he was HIV positive. The patient reported unprotected sexual intercourse 4 months prior. He was referred to the infectious disease unit and was classified in subtype A1 of HIV infection (CD4 count, 583 cells/µL [reference range, 500-1200 cells/µL]; viral load, 159,268 copies/mL [rapid progression to AIDS, >100,000 copies/mL]). Treatment was initiated with raltegravir, ritonavir, darunavir, and abacavir; tolerance was suitable. Because of the patient’s history of severe psoriasis, treatment with ustekinumab was maintained. Normally, treatment with this drug would be contraindicated in patients with HIV, as it can lead to viral reactivation. Four years after his HIV diagnosis, neither the patient’s cutaneous nor HIV-associated condition had worsened.
For patients with HIV and mild or moderate psoriasis, topical therapies (eg, corticosteroids, vitamin D analogues, tazarotene) are recommended, similar to patients who are HIV negative. Human immunodeficiency virus–positive patients with severe psoriasis who do not respond to topical treatment should receive phototherapy (UVB or psoralen plus UVA) or acitretin along with their antiretroviral drugs.2 In refractory cases, immunosuppressants, including cyclosporine, methotrexate, or tumor necrosis factor α inhibitors, might be used, though experience with them is limited.3,4 Maintaining antiretroviral therapy and prophylaxis against opportunist disease is important in patients who receive such immunosuppressants, as is close monitoring of the viral load.
Ustekinumab is an IL-12/IL-23 monoclonal antibody indicated for the treatment of moderate to severe plaque psoriasis, active psoriatic arthritis, and inflammatory bowel disease. It is contraindicated in patients with clinically important active infections, such as HBV and hepatitis C virus infections.5 However, it was shown to be safe in a group of 18 patients with HBV who had received antiviral prophylaxis6; a degree of reactivation was observed in similar patients who received no such prophylaxis and in others with hepatitis C virus infection.7 The simultaneous use of methotrexate with ustekinumab in the treatment of psoriatic arthritis does not appear to affect the safety of the latter drug.8 Paparizos et al9 described a patient with HIV controlled with antiretroviral drugs who was treated with ustekinumab for psoriasis; no adverse effects were observed.
We report the case of a patient with HBV and psoriasis who was treated with ustekinumab and later became infected with HIV. His ustekinumab treatment was maintained without subsequent cutaneous or systemic complications.
To the Editor:
The incidence of psoriasis in human immunodeficiency virus (HIV)–infected patients is similar to the general population, but it usually becomes more severe as immunosuppression increases. Additionally, it tends to be more resistant to conventional therapies, and the incidence and severity of psoriatic arthropathy is increased. Psoriasis often worsens at the time of HIV primary infection.1 We describe a case of a patient with hepatitis B virus (HBV) whose severe plaque psoriasis was controlled on ustekinumab; he was later diagnosed with HIV infection.
A 42-year-old man with HBV treated with entecavir (HBV DNA viral load, <20 copies/mL [inactive carrier, <2000 copies/mL]) presented to our dermatology unit with severe plaque psoriasis (psoriasis area and severity index 23) that caused notable psychologic difficulties such as anxiety and depression. Treatment was attempted with cyclosporine; acitretin; psoralen plus UVA; infliximab; adalimumab; and eventually ustekinumab (45 mg every 3 months), which controlled the condition well (psoriasis area and severity index 0) in an almost completely sustained manner.
Serologic tests requested at one of his analytical control appointments 2 years after initiating treatment with ustekinumab revealed he was HIV positive. The patient reported unprotected sexual intercourse 4 months prior. He was referred to the infectious disease unit and was classified in subtype A1 of HIV infection (CD4 count, 583 cells/µL [reference range, 500-1200 cells/µL]; viral load, 159,268 copies/mL [rapid progression to AIDS, >100,000 copies/mL]). Treatment was initiated with raltegravir, ritonavir, darunavir, and abacavir; tolerance was suitable. Because of the patient’s history of severe psoriasis, treatment with ustekinumab was maintained. Normally, treatment with this drug would be contraindicated in patients with HIV, as it can lead to viral reactivation. Four years after his HIV diagnosis, neither the patient’s cutaneous nor HIV-associated condition had worsened.
For patients with HIV and mild or moderate psoriasis, topical therapies (eg, corticosteroids, vitamin D analogues, tazarotene) are recommended, similar to patients who are HIV negative. Human immunodeficiency virus–positive patients with severe psoriasis who do not respond to topical treatment should receive phototherapy (UVB or psoralen plus UVA) or acitretin along with their antiretroviral drugs.2 In refractory cases, immunosuppressants, including cyclosporine, methotrexate, or tumor necrosis factor α inhibitors, might be used, though experience with them is limited.3,4 Maintaining antiretroviral therapy and prophylaxis against opportunist disease is important in patients who receive such immunosuppressants, as is close monitoring of the viral load.
Ustekinumab is an IL-12/IL-23 monoclonal antibody indicated for the treatment of moderate to severe plaque psoriasis, active psoriatic arthritis, and inflammatory bowel disease. It is contraindicated in patients with clinically important active infections, such as HBV and hepatitis C virus infections.5 However, it was shown to be safe in a group of 18 patients with HBV who had received antiviral prophylaxis6; a degree of reactivation was observed in similar patients who received no such prophylaxis and in others with hepatitis C virus infection.7 The simultaneous use of methotrexate with ustekinumab in the treatment of psoriatic arthritis does not appear to affect the safety of the latter drug.8 Paparizos et al9 described a patient with HIV controlled with antiretroviral drugs who was treated with ustekinumab for psoriasis; no adverse effects were observed.
We report the case of a patient with HBV and psoriasis who was treated with ustekinumab and later became infected with HIV. His ustekinumab treatment was maintained without subsequent cutaneous or systemic complications.
- Menon K, Van Voorhees V, Bebo B, et al. Psoriasis in patients with HIV infection: from the Medical Board of the National Psoriasis Foundation. J Am Acad Dermatol. 2010;62:291-299.
- Chiricozzi A, Saraceno R, Cannizzaro MV. Complete resolution of erythrodermic psoriasis in an HIV and HCV patient unresponsive to antipsoriatic treatments after highly active antiretroviral therapy. Dermatology. 2012;225:333-337.
- Barco D, Puig L, Alomar A. Treatment of moderate-severe psoriasis with etanercept in patients with chronic human immunodeficiency virus infection. Actas Dermosifiliogr. 2010;101(suppl 1):77-81.
- Lindsey SF, Weiss J, Lee ES, et al. Treatment of severe psoriasis and psoriatic arthritis with adalimumab in an HIV positive patient. J Drugs Dermatol. 2014;13:869-871.
- Rustin MH. Long-term safety of biologics in the treatment of moderate to severe plaque psoriasis: review of the current data. Br J Dermatol. 2012;167(suppl 3):3-11.
- Navarro R, Vilarrasa E, Herranz P, et al. Safety and effectiveness of ustekinumab and antitumour necrosis factor therapy in patients with psoriasis and chronic viral hepatitis B or C: a retrospective, multicentre study in a clinical setting. Br J Dermatol. 2013;168:609-616.
- Chiu HY, Chen CH, Wu MS, et al. The safety profile of ustekinumab in the treatment of patients with psoriasis and concurrent hepatitis B or C. Br J Dermatol. 2013;169:1295-1303.
- Weitz JE, Ritchlin CT. Ustekinumab: targeting the IL-17 pathway to improve outcomes in psoriatic arthritis. Expert Opin Biol Ther. 2014;14:515-526.
- Paparizos V, Rallis E, Kirsten L, et al. Ustekinumab for the treatment of HIV psoriasis. J Dermatol Treat. 2012;23:398-399.
- Menon K, Van Voorhees V, Bebo B, et al. Psoriasis in patients with HIV infection: from the Medical Board of the National Psoriasis Foundation. J Am Acad Dermatol. 2010;62:291-299.
- Chiricozzi A, Saraceno R, Cannizzaro MV. Complete resolution of erythrodermic psoriasis in an HIV and HCV patient unresponsive to antipsoriatic treatments after highly active antiretroviral therapy. Dermatology. 2012;225:333-337.
- Barco D, Puig L, Alomar A. Treatment of moderate-severe psoriasis with etanercept in patients with chronic human immunodeficiency virus infection. Actas Dermosifiliogr. 2010;101(suppl 1):77-81.
- Lindsey SF, Weiss J, Lee ES, et al. Treatment of severe psoriasis and psoriatic arthritis with adalimumab in an HIV positive patient. J Drugs Dermatol. 2014;13:869-871.
- Rustin MH. Long-term safety of biologics in the treatment of moderate to severe plaque psoriasis: review of the current data. Br J Dermatol. 2012;167(suppl 3):3-11.
- Navarro R, Vilarrasa E, Herranz P, et al. Safety and effectiveness of ustekinumab and antitumour necrosis factor therapy in patients with psoriasis and chronic viral hepatitis B or C: a retrospective, multicentre study in a clinical setting. Br J Dermatol. 2013;168:609-616.
- Chiu HY, Chen CH, Wu MS, et al. The safety profile of ustekinumab in the treatment of patients with psoriasis and concurrent hepatitis B or C. Br J Dermatol. 2013;169:1295-1303.
- Weitz JE, Ritchlin CT. Ustekinumab: targeting the IL-17 pathway to improve outcomes in psoriatic arthritis. Expert Opin Biol Ther. 2014;14:515-526.
- Paparizos V, Rallis E, Kirsten L, et al. Ustekinumab for the treatment of HIV psoriasis. J Dermatol Treat. 2012;23:398-399.
Practice Points
- Psoriasis in patients with human immunodeficiency virus (HIV) tends to be more resistant to conventional therapies.
- Experience is limited in the use of immunosuppressants and biologics to treat psoriasis in HIV patients.
- Maintaining antiretroviral therapy and prophylaxis against opportunist disease is important in HIV patients who receive biologics, as is close monitoring of the viral load.
Angiosarcoma Imitating a Morpheaform Basal Cell Carcinoma
To the Editor:
Basal cell carcinoma (BCC) is the most common of the nonmelanoma skin cancers and is a highly curable skin growth.1,2 Conversely, angiosarcomas are aggressive vascular tumors of endothelial origin that classically appear as reddish purple patches or plaques that exhibit rapid growth and invasion.3 Sporadic cutaneous angiosarcomas are the most common type of this soft tissue tumor, occurring most often in the head and neck regions in men older than 70 years.4,5 Other types of angiosarcomas include those associated with radiation therapy and chronic lymphedema. Postradiation angiosarcomas have been most frequently reported after treatment of breast cancer and appear as infiltrative plaques over the irradiated area.4,5 Patients with chronic lymphedema, which most commonly is related to axillary lymph node dissection for breast cancer (90% of cases), may develop angiosarcoma presenting as a violaceous indurated plaque.5 Although angiosarcomas most often are seen with these distinct clinical characteristics, especially their violaceous color, they have been shown to mimic a few other skin disorders such as eczema and keratoacanthoma, but a limited number of cases of angiosarcoma mimicking BCC have been reported.1,6,7 We present a case of an elderly man with a unique presentation of a lesion that clinically appeared as a morpheaform BCC but was confirmed to be an angiosarcoma on histopathology.
A 75-year-old man was referred to our dermatology clinic for evaluation of a flesh-colored plaque on the face that initially had developed 2 years prior on the right central malar cheek. Computed tomography of the head and neck 1 year prior, which the patient reported was for workup of the lesion, was found to be negative; however, these medical records were not obtained for confirmation. The lesion had been stable in size and remained flesh colored until 6 months prior to the current presentation when it exhibited a rapid increase in size. An initial biopsy was performed 1 month prior to presentation by an outside dermatology office and had been read as an angiosarcoma.
Physical examination revealed a 6-cm, flesh-colored, indurated, ill-defined plaque distributed on the right malar cheek below the eye and extending to the nasal bridge (Figure 1). There was no cervical or facial lymphadenopathy. The clinical features resembled a morpheaform BCC, and the lesion did not exhibit any reddish or purple color indicating it was of vascular origin. However, due to the prior histopathology report and recent rapid enlargement, a repeat sampling with a larger punch biopsy was performed, which confirmed the diagnosis of angiosarcoma. Histopathology demonstrated multiple atypical vascular channels lined by hyperchromatic cells extending from the upper dermis to the base of the biopsy site (Figure 2). Large, oval, atypical nuclei were present in multiple endothelial cells in the vascular channels, with some forming irregularly contoured and slitlike formations (Figure 3). Immunochemical staining was intensely and uniformly positive for CD31 and CD34, both endothelial markers. Diffuse positive staining with CD31 is considered to have high sensitivity and specificity for the diagnosis of angiosarcoma.4 Other pertinent staining demonstrated 2+ positivity for factor VIII and 1+ positivity for D2-40; CD45, AE1/AE3, S-100, and human herpesvirus 8 were negative, consistent with angiosarcoma. The patient was referred to radiation oncology and otolaryngology at our Multidisciplinary Head and Neck Oncology Center for further investigation of the extent of the disease and discussion of treatment. Computed tomography of the head and neck region at this time showed extensive disease extending into the medial canthal area without metastasis. Due to the extent of disease and facial location, he was not deemed a candidate for surgery. He was treated with 6 weeks of targeted radiation therapy with concurrent chemotherapy. He tolerated this treatment with minimal side effects and was found to be free from clinical disease 1 year after diagnosis. He was followed for 20 months by our Multidisciplinary Oncology Clinic without recurrence of his disease but was then lost to follow-up.
This case illustrates a rare presentation of an angiosarcoma clinically mimicking a BCC, which has been described in a small number of case reports and retrospective reviews. One study of 656 patients diagnosed with BCC based on clinical features revealed that 48 of these lesions were proven to be a BCC-mimicking lesion and only 1 was an angiosarcoma.1 Cutaneous lesions that appear on physical examination to be a highly curable BCC may not induce the same urgency for treatment as an angiosarcoma. Although the clinical presentation may mimic a morpheaform BCC, our case demonstrates that it is imperative to include angiosarcoma in the differential diagnosis and underscores the utility of tissue sampling. Angiosarcoma has a poor overall 5-year survival rate, and patients often are found to have multiple metastatic lesions at diagnosis. However, diagnosis prior to metastasis may improve prognosis.8
Our patient’s angiosarcoma did not exhibit metastasis at the time of diagnosis, and he was able to achieve a favorable outcome. However, the 5-year survival rate is only 40%, and close clinical monitoring after diagnosis is required.8 Including angiosarcoma in the differential diagnosis for our patient, particularly upon lesion appearance 2 years prior, may have resulted in diagnosis antecedent to local invasion, possibly providing more treatment options. Employing a higher index of clinical suspicion for angiosarcoma may lead to decreased mortality in other patients due to increased detection.
- Kim HS, Kim TW, Mun JH, et al. Basal cell carcinoma–mimicking lesions in Korean clinical settings. Ann Dermatol. 2014;26:431-436.
- Christenson LJ, Borrowman TA, Vachon CM, et al. Incidence of basal cell and squamous cell carcinomas in a population younger than 40 years. JAMA. 2005;294:681-690.
- Goldsmith LA, Katz S, Gilchrest BA. Fitzpatrick’s Dermatology in General Medicine. New York, NY: McGraw Hill; 2012.
- Dosset LA, Harrington M, Cruse CW, et al. Cutaneous angiosarcoma. Curr Probl Cancer. 2015;39:258-263.
- North PE, Kincannon J. Vascular neoplasms and neoplastic-like proliferations. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Saunders; 2012:1915-1942.
- Kong YL, Chandran NS, Goh SG, et al. Cutaneous angiosarcoma of the scalp mimicking a keratoacanthoma. Dermatol Online J. 2013;19:18566.
- Trinh NQ, Rashed I, Hutchens KA, et al. Unusual clinical presentation of cutaneous angiosarcoma masquerading as eczema: a case report and review of the literature. Case Rep Dermatol Med. 2013;2013:906426.
- Buehler D, Rice SR, Moody JS, et al. Angiosarcoma outcomes and prognostic factors. a 25-year single institution experience. Am J Clin Oncol. 2014;37:473-479.
To the Editor:
Basal cell carcinoma (BCC) is the most common of the nonmelanoma skin cancers and is a highly curable skin growth.1,2 Conversely, angiosarcomas are aggressive vascular tumors of endothelial origin that classically appear as reddish purple patches or plaques that exhibit rapid growth and invasion.3 Sporadic cutaneous angiosarcomas are the most common type of this soft tissue tumor, occurring most often in the head and neck regions in men older than 70 years.4,5 Other types of angiosarcomas include those associated with radiation therapy and chronic lymphedema. Postradiation angiosarcomas have been most frequently reported after treatment of breast cancer and appear as infiltrative plaques over the irradiated area.4,5 Patients with chronic lymphedema, which most commonly is related to axillary lymph node dissection for breast cancer (90% of cases), may develop angiosarcoma presenting as a violaceous indurated plaque.5 Although angiosarcomas most often are seen with these distinct clinical characteristics, especially their violaceous color, they have been shown to mimic a few other skin disorders such as eczema and keratoacanthoma, but a limited number of cases of angiosarcoma mimicking BCC have been reported.1,6,7 We present a case of an elderly man with a unique presentation of a lesion that clinically appeared as a morpheaform BCC but was confirmed to be an angiosarcoma on histopathology.
A 75-year-old man was referred to our dermatology clinic for evaluation of a flesh-colored plaque on the face that initially had developed 2 years prior on the right central malar cheek. Computed tomography of the head and neck 1 year prior, which the patient reported was for workup of the lesion, was found to be negative; however, these medical records were not obtained for confirmation. The lesion had been stable in size and remained flesh colored until 6 months prior to the current presentation when it exhibited a rapid increase in size. An initial biopsy was performed 1 month prior to presentation by an outside dermatology office and had been read as an angiosarcoma.
Physical examination revealed a 6-cm, flesh-colored, indurated, ill-defined plaque distributed on the right malar cheek below the eye and extending to the nasal bridge (Figure 1). There was no cervical or facial lymphadenopathy. The clinical features resembled a morpheaform BCC, and the lesion did not exhibit any reddish or purple color indicating it was of vascular origin. However, due to the prior histopathology report and recent rapid enlargement, a repeat sampling with a larger punch biopsy was performed, which confirmed the diagnosis of angiosarcoma. Histopathology demonstrated multiple atypical vascular channels lined by hyperchromatic cells extending from the upper dermis to the base of the biopsy site (Figure 2). Large, oval, atypical nuclei were present in multiple endothelial cells in the vascular channels, with some forming irregularly contoured and slitlike formations (Figure 3). Immunochemical staining was intensely and uniformly positive for CD31 and CD34, both endothelial markers. Diffuse positive staining with CD31 is considered to have high sensitivity and specificity for the diagnosis of angiosarcoma.4 Other pertinent staining demonstrated 2+ positivity for factor VIII and 1+ positivity for D2-40; CD45, AE1/AE3, S-100, and human herpesvirus 8 were negative, consistent with angiosarcoma. The patient was referred to radiation oncology and otolaryngology at our Multidisciplinary Head and Neck Oncology Center for further investigation of the extent of the disease and discussion of treatment. Computed tomography of the head and neck region at this time showed extensive disease extending into the medial canthal area without metastasis. Due to the extent of disease and facial location, he was not deemed a candidate for surgery. He was treated with 6 weeks of targeted radiation therapy with concurrent chemotherapy. He tolerated this treatment with minimal side effects and was found to be free from clinical disease 1 year after diagnosis. He was followed for 20 months by our Multidisciplinary Oncology Clinic without recurrence of his disease but was then lost to follow-up.
This case illustrates a rare presentation of an angiosarcoma clinically mimicking a BCC, which has been described in a small number of case reports and retrospective reviews. One study of 656 patients diagnosed with BCC based on clinical features revealed that 48 of these lesions were proven to be a BCC-mimicking lesion and only 1 was an angiosarcoma.1 Cutaneous lesions that appear on physical examination to be a highly curable BCC may not induce the same urgency for treatment as an angiosarcoma. Although the clinical presentation may mimic a morpheaform BCC, our case demonstrates that it is imperative to include angiosarcoma in the differential diagnosis and underscores the utility of tissue sampling. Angiosarcoma has a poor overall 5-year survival rate, and patients often are found to have multiple metastatic lesions at diagnosis. However, diagnosis prior to metastasis may improve prognosis.8
Our patient’s angiosarcoma did not exhibit metastasis at the time of diagnosis, and he was able to achieve a favorable outcome. However, the 5-year survival rate is only 40%, and close clinical monitoring after diagnosis is required.8 Including angiosarcoma in the differential diagnosis for our patient, particularly upon lesion appearance 2 years prior, may have resulted in diagnosis antecedent to local invasion, possibly providing more treatment options. Employing a higher index of clinical suspicion for angiosarcoma may lead to decreased mortality in other patients due to increased detection.
To the Editor:
Basal cell carcinoma (BCC) is the most common of the nonmelanoma skin cancers and is a highly curable skin growth.1,2 Conversely, angiosarcomas are aggressive vascular tumors of endothelial origin that classically appear as reddish purple patches or plaques that exhibit rapid growth and invasion.3 Sporadic cutaneous angiosarcomas are the most common type of this soft tissue tumor, occurring most often in the head and neck regions in men older than 70 years.4,5 Other types of angiosarcomas include those associated with radiation therapy and chronic lymphedema. Postradiation angiosarcomas have been most frequently reported after treatment of breast cancer and appear as infiltrative plaques over the irradiated area.4,5 Patients with chronic lymphedema, which most commonly is related to axillary lymph node dissection for breast cancer (90% of cases), may develop angiosarcoma presenting as a violaceous indurated plaque.5 Although angiosarcomas most often are seen with these distinct clinical characteristics, especially their violaceous color, they have been shown to mimic a few other skin disorders such as eczema and keratoacanthoma, but a limited number of cases of angiosarcoma mimicking BCC have been reported.1,6,7 We present a case of an elderly man with a unique presentation of a lesion that clinically appeared as a morpheaform BCC but was confirmed to be an angiosarcoma on histopathology.
A 75-year-old man was referred to our dermatology clinic for evaluation of a flesh-colored plaque on the face that initially had developed 2 years prior on the right central malar cheek. Computed tomography of the head and neck 1 year prior, which the patient reported was for workup of the lesion, was found to be negative; however, these medical records were not obtained for confirmation. The lesion had been stable in size and remained flesh colored until 6 months prior to the current presentation when it exhibited a rapid increase in size. An initial biopsy was performed 1 month prior to presentation by an outside dermatology office and had been read as an angiosarcoma.
Physical examination revealed a 6-cm, flesh-colored, indurated, ill-defined plaque distributed on the right malar cheek below the eye and extending to the nasal bridge (Figure 1). There was no cervical or facial lymphadenopathy. The clinical features resembled a morpheaform BCC, and the lesion did not exhibit any reddish or purple color indicating it was of vascular origin. However, due to the prior histopathology report and recent rapid enlargement, a repeat sampling with a larger punch biopsy was performed, which confirmed the diagnosis of angiosarcoma. Histopathology demonstrated multiple atypical vascular channels lined by hyperchromatic cells extending from the upper dermis to the base of the biopsy site (Figure 2). Large, oval, atypical nuclei were present in multiple endothelial cells in the vascular channels, with some forming irregularly contoured and slitlike formations (Figure 3). Immunochemical staining was intensely and uniformly positive for CD31 and CD34, both endothelial markers. Diffuse positive staining with CD31 is considered to have high sensitivity and specificity for the diagnosis of angiosarcoma.4 Other pertinent staining demonstrated 2+ positivity for factor VIII and 1+ positivity for D2-40; CD45, AE1/AE3, S-100, and human herpesvirus 8 were negative, consistent with angiosarcoma. The patient was referred to radiation oncology and otolaryngology at our Multidisciplinary Head and Neck Oncology Center for further investigation of the extent of the disease and discussion of treatment. Computed tomography of the head and neck region at this time showed extensive disease extending into the medial canthal area without metastasis. Due to the extent of disease and facial location, he was not deemed a candidate for surgery. He was treated with 6 weeks of targeted radiation therapy with concurrent chemotherapy. He tolerated this treatment with minimal side effects and was found to be free from clinical disease 1 year after diagnosis. He was followed for 20 months by our Multidisciplinary Oncology Clinic without recurrence of his disease but was then lost to follow-up.
This case illustrates a rare presentation of an angiosarcoma clinically mimicking a BCC, which has been described in a small number of case reports and retrospective reviews. One study of 656 patients diagnosed with BCC based on clinical features revealed that 48 of these lesions were proven to be a BCC-mimicking lesion and only 1 was an angiosarcoma.1 Cutaneous lesions that appear on physical examination to be a highly curable BCC may not induce the same urgency for treatment as an angiosarcoma. Although the clinical presentation may mimic a morpheaform BCC, our case demonstrates that it is imperative to include angiosarcoma in the differential diagnosis and underscores the utility of tissue sampling. Angiosarcoma has a poor overall 5-year survival rate, and patients often are found to have multiple metastatic lesions at diagnosis. However, diagnosis prior to metastasis may improve prognosis.8
Our patient’s angiosarcoma did not exhibit metastasis at the time of diagnosis, and he was able to achieve a favorable outcome. However, the 5-year survival rate is only 40%, and close clinical monitoring after diagnosis is required.8 Including angiosarcoma in the differential diagnosis for our patient, particularly upon lesion appearance 2 years prior, may have resulted in diagnosis antecedent to local invasion, possibly providing more treatment options. Employing a higher index of clinical suspicion for angiosarcoma may lead to decreased mortality in other patients due to increased detection.
- Kim HS, Kim TW, Mun JH, et al. Basal cell carcinoma–mimicking lesions in Korean clinical settings. Ann Dermatol. 2014;26:431-436.
- Christenson LJ, Borrowman TA, Vachon CM, et al. Incidence of basal cell and squamous cell carcinomas in a population younger than 40 years. JAMA. 2005;294:681-690.
- Goldsmith LA, Katz S, Gilchrest BA. Fitzpatrick’s Dermatology in General Medicine. New York, NY: McGraw Hill; 2012.
- Dosset LA, Harrington M, Cruse CW, et al. Cutaneous angiosarcoma. Curr Probl Cancer. 2015;39:258-263.
- North PE, Kincannon J. Vascular neoplasms and neoplastic-like proliferations. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Saunders; 2012:1915-1942.
- Kong YL, Chandran NS, Goh SG, et al. Cutaneous angiosarcoma of the scalp mimicking a keratoacanthoma. Dermatol Online J. 2013;19:18566.
- Trinh NQ, Rashed I, Hutchens KA, et al. Unusual clinical presentation of cutaneous angiosarcoma masquerading as eczema: a case report and review of the literature. Case Rep Dermatol Med. 2013;2013:906426.
- Buehler D, Rice SR, Moody JS, et al. Angiosarcoma outcomes and prognostic factors. a 25-year single institution experience. Am J Clin Oncol. 2014;37:473-479.
- Kim HS, Kim TW, Mun JH, et al. Basal cell carcinoma–mimicking lesions in Korean clinical settings. Ann Dermatol. 2014;26:431-436.
- Christenson LJ, Borrowman TA, Vachon CM, et al. Incidence of basal cell and squamous cell carcinomas in a population younger than 40 years. JAMA. 2005;294:681-690.
- Goldsmith LA, Katz S, Gilchrest BA. Fitzpatrick’s Dermatology in General Medicine. New York, NY: McGraw Hill; 2012.
- Dosset LA, Harrington M, Cruse CW, et al. Cutaneous angiosarcoma. Curr Probl Cancer. 2015;39:258-263.
- North PE, Kincannon J. Vascular neoplasms and neoplastic-like proliferations. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Saunders; 2012:1915-1942.
- Kong YL, Chandran NS, Goh SG, et al. Cutaneous angiosarcoma of the scalp mimicking a keratoacanthoma. Dermatol Online J. 2013;19:18566.
- Trinh NQ, Rashed I, Hutchens KA, et al. Unusual clinical presentation of cutaneous angiosarcoma masquerading as eczema: a case report and review of the literature. Case Rep Dermatol Med. 2013;2013:906426.
- Buehler D, Rice SR, Moody JS, et al. Angiosarcoma outcomes and prognostic factors. a 25-year single institution experience. Am J Clin Oncol. 2014;37:473-479.
Practice Points
- Angiosarcoma is an aggressive vascular tumor with a poor prognosis.
- Angiosarcomas can arise in the setting of chronic lymphedema or prior radiation therapy or can arise spontaneously.
- Classically, angiosarcoma presents as a violaceous patch or plaque but occasionally can exhibit atypical clinical features. Angiosarcomas should be considered on the differential for any changing plaque on the head or neck.
Cutaneous Collagenous Vasculopathy
To the Editor:
Cutaneous collagenous vasculopathy (CCV) is a rare idiopathic microangiopathy characterized by diffuse blanchable telangiectases that usually develop in late adulthood. It appears morphologically identical to generalized essential telangiectasia (GET), but skin biopsy characteristically shows dilated superficial blood vessels in the papillary dermis that are surrounded by a thickened layer of type IV collagen.1 We report a case of CCV occurring in an elderly white man.
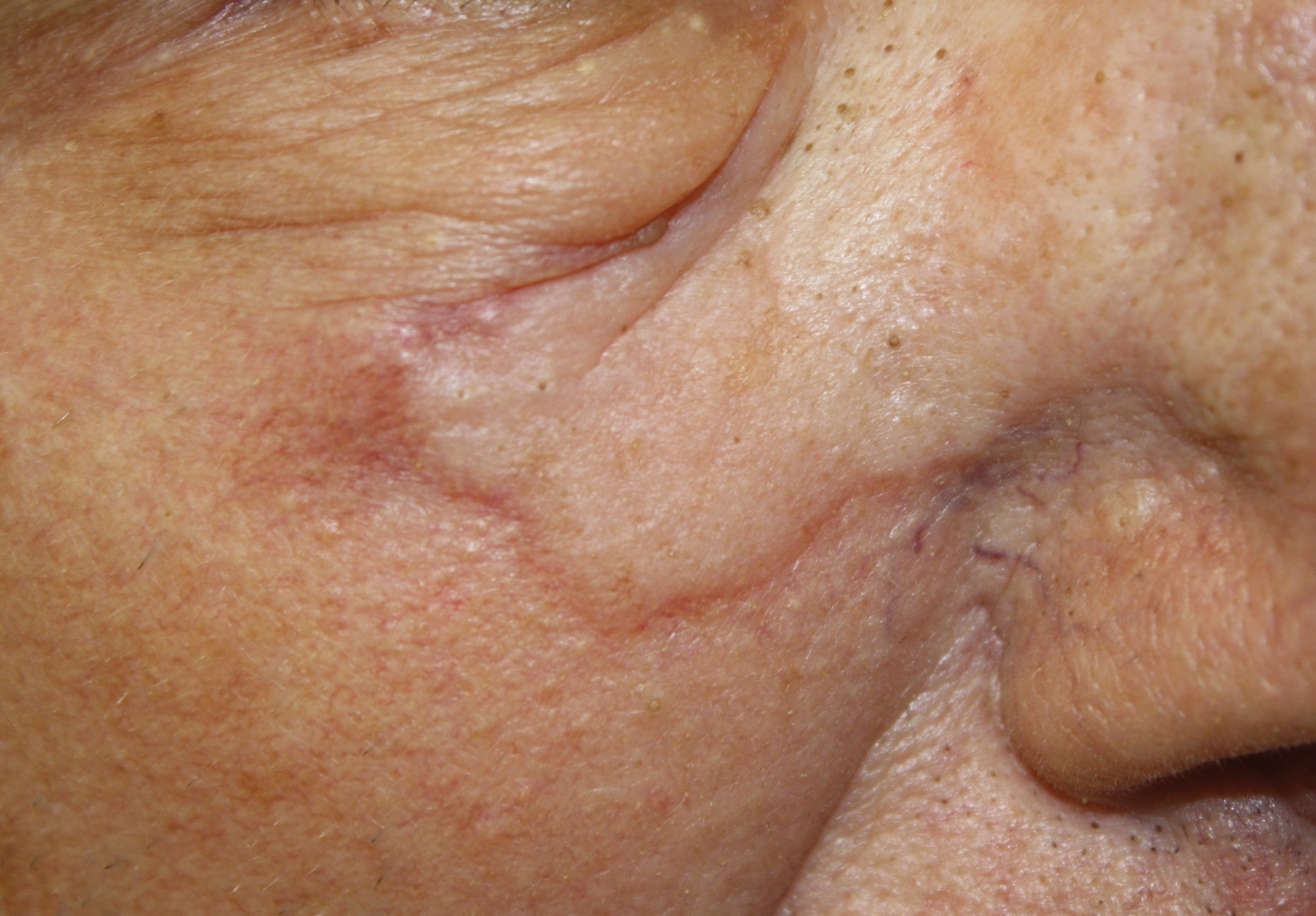
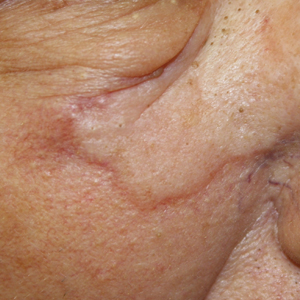
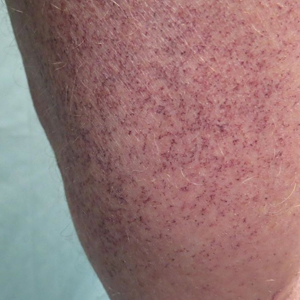
A 72-year-old man presented with an asymptomatic rash on the arms, legs, and abdomen of 3 years’ duration. His medical history was remarkable for hypothyroidism, hypertension, reflex sympathetic dystrophy syndrome, coronary artery disease, and nonmelanoma skin cancer. He denied any changes in medications or illnesses prior to onset of the rash. Physical examination revealed diffuse, erythematous, blanchable telangiectases on the arms, legs, and trunk (Figure 1). No petechiae, atrophy, or epidermal changes were appreciated. Darier sign was negative.

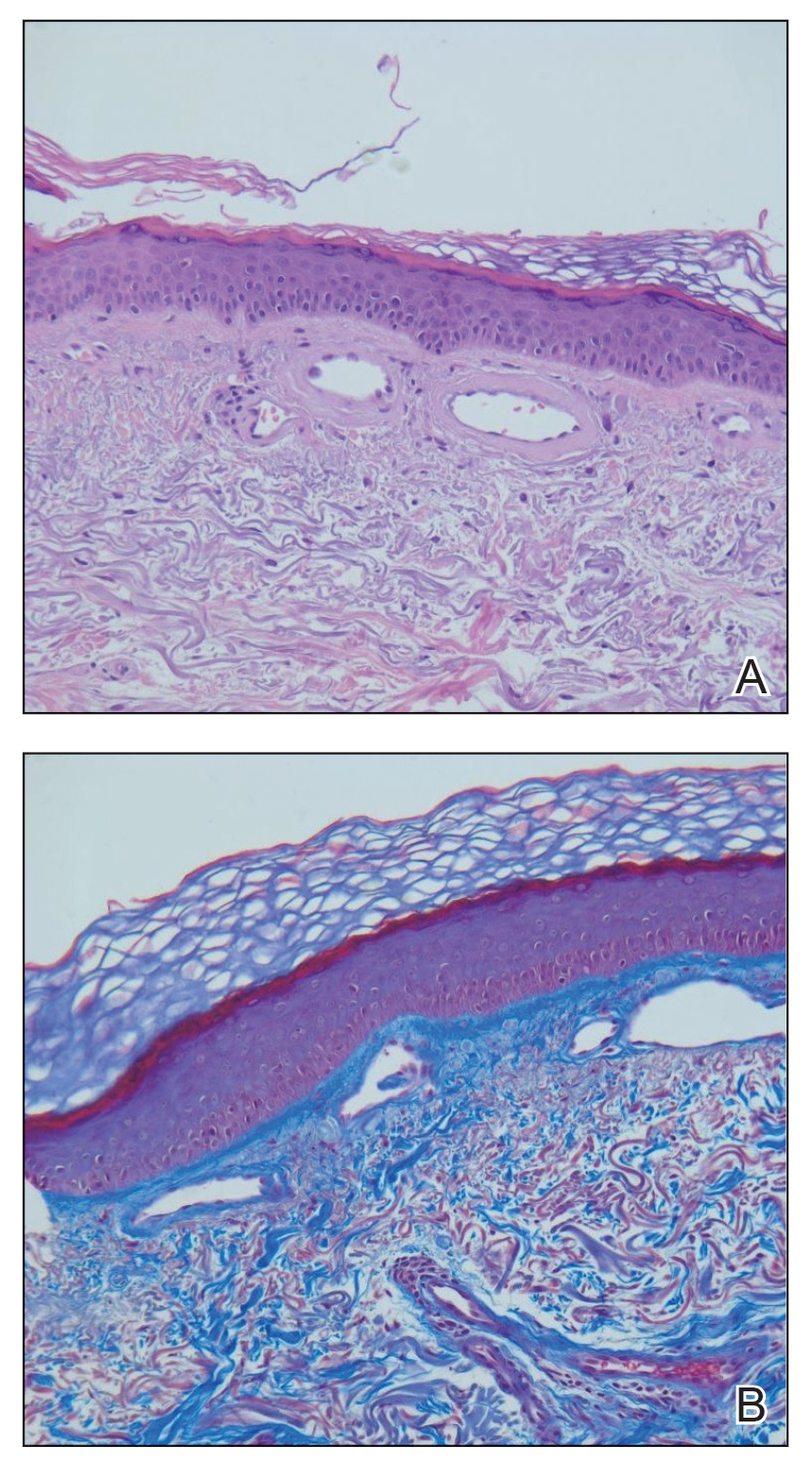
Hematoxylin and eosin–stained sections of skin from the abdomen showed an unremarkable epidermis overlying a superficial dermis with dilated blood vessels with thickened walls that contained eosinophilic amorphous hyaline material (Figure 2A). This material stained positive with Masson trichrome (Figure 2B), a finding that was consistent with increased collagen fiber deposition within the vessel walls. Phosphotungstic acid–hematoxylin and Congo red stains were negative. No histologic features of a vaso-occlusive disorder or vasculitis were identified. These histologic findings were consistent with the rare diagnosis of CCV.
Cutaneous collagenous vasculopathy is a rare idiopathic microangiopathy that was first reported by Salama and Rosenthal1 in 2000. They reported the case of a 54-year-old man with spreading, asymptomatic, generalized cutaneous telangiectases of 5 years’ duration. Similar to our patient, skin biopsy showed dilated superficial dermal vasculature with deposition of eosinophilic hyaline material, which stained positive with periodic acid–Schiff with diastase and exhibited immunoreactivity to type IV collagen.1
A PubMed search of articles indexed for MEDLINE using the search term cutaneous collagenous vasculopathy yielded 19 additional patients with biopsy-proven CCV.2-6 The condition has shown no gender prevalence but generally is seen in middle-aged or elderly white individuals, with the exception of a white pediatric patient.4 Cutaneous collagenous vasculopathy usually presents as telangiectases on the legs that progress to involve the trunk and arms while sparing the head and neck, nail beds, and mucous membranes.5 However, it also has been described as first presenting on the bilateral breasts2 as well as a nonprogressive localization on the thigh.6
Skin biopsy is essential to differentiate CCV from GET, which appears morphologically identical. Cutaneous collagenous vasculopathy may be underreported as a result of clinician choice not to biopsy due to a presumptive diagnosis of GET.3 Successful treatment with a pulsed dye laser has been reported,7 though the extent of disease may make complete destruction of the lesions difficult to accomplish. Although it is theorized that CCV may be a marker for underlying systemic disease or even a genetic defect causing abnormal collagen deposition, its cause has yet to be ascertained.5 Previously reported patients have had a variety of comorbidities, including several cases of type 2 diabetes mellitus.6 Another patient was reported to have recently started treatment with an angiotensin receptor blocker prior to onset of CCV.5
Our case contributes to the small series of reported patients with this rare diagnosis and further suggests that it may be underreported at this time. Similar to previously reported cases, our patient was an elderly white individual. Although our patient had long-standing iatrogenic hypothyroidism, no recent medication changes or underlying comorbidities could be tied to the development of CCV. Further studies are needed to determine if this disease process is associated with any underlying systemic illnesses, medications, or family history.
- Salama S, Rosenthal D. Cutaneous collagenous vasculopathy with generalized telangiectasia: an immunohistochemical and ultrastructural study. J Cutan Pathol. 2000;27:40-48.
- Borroni RG, Derlino F, Agozzino M, et al. Hypothermic cutaneous collagenous vasculopathy with centrifugal spreading [published online March 31, 2014]. J Eur Acad Dermatol Venereol. 2015;29:1444-1446.
- Moulonguet I, Hershkovitch D, Fraitag S. Widespread cutaneous telangiectasias: challenge. Am J Dermatopathol. 2013;35:661-662, 688-669.
- Lloyd BM, Pruden SJ 2nd, Lind AC, et al. Cutaneous collagenous vasculopathy: report of the first pediatric case. Pediatr Dermatol. 2011;28:598-599.
- Kanitakis J, Faisant M, Wagschal D, et al. Cutaneous collagenous vasculopathy: ultrastructural and immunohistochemical study of a new case. Am J Clin Dermatol. 2010;11:63-66.
- Davis TL, Mandal RV, Bevona C, et al. Collagenous vasculopathy: a report of three cases. J Cutan Pathol. 2008;35:967-970.
- Echeverría B, Sanmartín O, Botella-Estrada R, et al. Cutaneous collagenous vasculopathy successfully treated with pulsed dye laser. Int J Dermatol. 2012;51:1359-1362.
To the Editor:
Cutaneous collagenous vasculopathy (CCV) is a rare idiopathic microangiopathy characterized by diffuse blanchable telangiectases that usually develop in late adulthood. It appears morphologically identical to generalized essential telangiectasia (GET), but skin biopsy characteristically shows dilated superficial blood vessels in the papillary dermis that are surrounded by a thickened layer of type IV collagen.1 We report a case of CCV occurring in an elderly white man.
A 72-year-old man presented with an asymptomatic rash on the arms, legs, and abdomen of 3 years’ duration. His medical history was remarkable for hypothyroidism, hypertension, reflex sympathetic dystrophy syndrome, coronary artery disease, and nonmelanoma skin cancer. He denied any changes in medications or illnesses prior to onset of the rash. Physical examination revealed diffuse, erythematous, blanchable telangiectases on the arms, legs, and trunk (Figure 1). No petechiae, atrophy, or epidermal changes were appreciated. Darier sign was negative.
Hematoxylin and eosin–stained sections of skin from the abdomen showed an unremarkable epidermis overlying a superficial dermis with dilated blood vessels with thickened walls that contained eosinophilic amorphous hyaline material (Figure 2A). This material stained positive with Masson trichrome (Figure 2B), a finding that was consistent with increased collagen fiber deposition within the vessel walls. Phosphotungstic acid–hematoxylin and Congo red stains were negative. No histologic features of a vaso-occlusive disorder or vasculitis were identified. These histologic findings were consistent with the rare diagnosis of CCV.
Cutaneous collagenous vasculopathy is a rare idiopathic microangiopathy that was first reported by Salama and Rosenthal1 in 2000. They reported the case of a 54-year-old man with spreading, asymptomatic, generalized cutaneous telangiectases of 5 years’ duration. Similar to our patient, skin biopsy showed dilated superficial dermal vasculature with deposition of eosinophilic hyaline material, which stained positive with periodic acid–Schiff with diastase and exhibited immunoreactivity to type IV collagen.1
A PubMed search of articles indexed for MEDLINE using the search term cutaneous collagenous vasculopathy yielded 19 additional patients with biopsy-proven CCV.2-6 The condition has shown no gender prevalence but generally is seen in middle-aged or elderly white individuals, with the exception of a white pediatric patient.4 Cutaneous collagenous vasculopathy usually presents as telangiectases on the legs that progress to involve the trunk and arms while sparing the head and neck, nail beds, and mucous membranes.5 However, it also has been described as first presenting on the bilateral breasts2 as well as a nonprogressive localization on the thigh.6
Skin biopsy is essential to differentiate CCV from GET, which appears morphologically identical. Cutaneous collagenous vasculopathy may be underreported as a result of clinician choice not to biopsy due to a presumptive diagnosis of GET.3 Successful treatment with a pulsed dye laser has been reported,7 though the extent of disease may make complete destruction of the lesions difficult to accomplish. Although it is theorized that CCV may be a marker for underlying systemic disease or even a genetic defect causing abnormal collagen deposition, its cause has yet to be ascertained.5 Previously reported patients have had a variety of comorbidities, including several cases of type 2 diabetes mellitus.6 Another patient was reported to have recently started treatment with an angiotensin receptor blocker prior to onset of CCV.5
Our case contributes to the small series of reported patients with this rare diagnosis and further suggests that it may be underreported at this time. Similar to previously reported cases, our patient was an elderly white individual. Although our patient had long-standing iatrogenic hypothyroidism, no recent medication changes or underlying comorbidities could be tied to the development of CCV. Further studies are needed to determine if this disease process is associated with any underlying systemic illnesses, medications, or family history.
To the Editor:
Cutaneous collagenous vasculopathy (CCV) is a rare idiopathic microangiopathy characterized by diffuse blanchable telangiectases that usually develop in late adulthood. It appears morphologically identical to generalized essential telangiectasia (GET), but skin biopsy characteristically shows dilated superficial blood vessels in the papillary dermis that are surrounded by a thickened layer of type IV collagen.1 We report a case of CCV occurring in an elderly white man.
A 72-year-old man presented with an asymptomatic rash on the arms, legs, and abdomen of 3 years’ duration. His medical history was remarkable for hypothyroidism, hypertension, reflex sympathetic dystrophy syndrome, coronary artery disease, and nonmelanoma skin cancer. He denied any changes in medications or illnesses prior to onset of the rash. Physical examination revealed diffuse, erythematous, blanchable telangiectases on the arms, legs, and trunk (Figure 1). No petechiae, atrophy, or epidermal changes were appreciated. Darier sign was negative.
Hematoxylin and eosin–stained sections of skin from the abdomen showed an unremarkable epidermis overlying a superficial dermis with dilated blood vessels with thickened walls that contained eosinophilic amorphous hyaline material (Figure 2A). This material stained positive with Masson trichrome (Figure 2B), a finding that was consistent with increased collagen fiber deposition within the vessel walls. Phosphotungstic acid–hematoxylin and Congo red stains were negative. No histologic features of a vaso-occlusive disorder or vasculitis were identified. These histologic findings were consistent with the rare diagnosis of CCV.
Cutaneous collagenous vasculopathy is a rare idiopathic microangiopathy that was first reported by Salama and Rosenthal1 in 2000. They reported the case of a 54-year-old man with spreading, asymptomatic, generalized cutaneous telangiectases of 5 years’ duration. Similar to our patient, skin biopsy showed dilated superficial dermal vasculature with deposition of eosinophilic hyaline material, which stained positive with periodic acid–Schiff with diastase and exhibited immunoreactivity to type IV collagen.1
A PubMed search of articles indexed for MEDLINE using the search term cutaneous collagenous vasculopathy yielded 19 additional patients with biopsy-proven CCV.2-6 The condition has shown no gender prevalence but generally is seen in middle-aged or elderly white individuals, with the exception of a white pediatric patient.4 Cutaneous collagenous vasculopathy usually presents as telangiectases on the legs that progress to involve the trunk and arms while sparing the head and neck, nail beds, and mucous membranes.5 However, it also has been described as first presenting on the bilateral breasts2 as well as a nonprogressive localization on the thigh.6
Skin biopsy is essential to differentiate CCV from GET, which appears morphologically identical. Cutaneous collagenous vasculopathy may be underreported as a result of clinician choice not to biopsy due to a presumptive diagnosis of GET.3 Successful treatment with a pulsed dye laser has been reported,7 though the extent of disease may make complete destruction of the lesions difficult to accomplish. Although it is theorized that CCV may be a marker for underlying systemic disease or even a genetic defect causing abnormal collagen deposition, its cause has yet to be ascertained.5 Previously reported patients have had a variety of comorbidities, including several cases of type 2 diabetes mellitus.6 Another patient was reported to have recently started treatment with an angiotensin receptor blocker prior to onset of CCV.5
Our case contributes to the small series of reported patients with this rare diagnosis and further suggests that it may be underreported at this time. Similar to previously reported cases, our patient was an elderly white individual. Although our patient had long-standing iatrogenic hypothyroidism, no recent medication changes or underlying comorbidities could be tied to the development of CCV. Further studies are needed to determine if this disease process is associated with any underlying systemic illnesses, medications, or family history.
- Salama S, Rosenthal D. Cutaneous collagenous vasculopathy with generalized telangiectasia: an immunohistochemical and ultrastructural study. J Cutan Pathol. 2000;27:40-48.
- Borroni RG, Derlino F, Agozzino M, et al. Hypothermic cutaneous collagenous vasculopathy with centrifugal spreading [published online March 31, 2014]. J Eur Acad Dermatol Venereol. 2015;29:1444-1446.
- Moulonguet I, Hershkovitch D, Fraitag S. Widespread cutaneous telangiectasias: challenge. Am J Dermatopathol. 2013;35:661-662, 688-669.
- Lloyd BM, Pruden SJ 2nd, Lind AC, et al. Cutaneous collagenous vasculopathy: report of the first pediatric case. Pediatr Dermatol. 2011;28:598-599.
- Kanitakis J, Faisant M, Wagschal D, et al. Cutaneous collagenous vasculopathy: ultrastructural and immunohistochemical study of a new case. Am J Clin Dermatol. 2010;11:63-66.
- Davis TL, Mandal RV, Bevona C, et al. Collagenous vasculopathy: a report of three cases. J Cutan Pathol. 2008;35:967-970.
- Echeverría B, Sanmartín O, Botella-Estrada R, et al. Cutaneous collagenous vasculopathy successfully treated with pulsed dye laser. Int J Dermatol. 2012;51:1359-1362.
- Salama S, Rosenthal D. Cutaneous collagenous vasculopathy with generalized telangiectasia: an immunohistochemical and ultrastructural study. J Cutan Pathol. 2000;27:40-48.
- Borroni RG, Derlino F, Agozzino M, et al. Hypothermic cutaneous collagenous vasculopathy with centrifugal spreading [published online March 31, 2014]. J Eur Acad Dermatol Venereol. 2015;29:1444-1446.
- Moulonguet I, Hershkovitch D, Fraitag S. Widespread cutaneous telangiectasias: challenge. Am J Dermatopathol. 2013;35:661-662, 688-669.
- Lloyd BM, Pruden SJ 2nd, Lind AC, et al. Cutaneous collagenous vasculopathy: report of the first pediatric case. Pediatr Dermatol. 2011;28:598-599.
- Kanitakis J, Faisant M, Wagschal D, et al. Cutaneous collagenous vasculopathy: ultrastructural and immunohistochemical study of a new case. Am J Clin Dermatol. 2010;11:63-66.
- Davis TL, Mandal RV, Bevona C, et al. Collagenous vasculopathy: a report of three cases. J Cutan Pathol. 2008;35:967-970.
- Echeverría B, Sanmartín O, Botella-Estrada R, et al. Cutaneous collagenous vasculopathy successfully treated with pulsed dye laser. Int J Dermatol. 2012;51:1359-1362.
Practice Points
- Cutaneous collagenous vasculopathy (CCV) should be in the differential diagnosis of widespread telangiectases.
- Biopsy is needed to differentiate between CCV and generalized essential telangiectasia because of their similar clinical features.
- There may be underlying comorbidities associated with CCV, but the exact cause of the condition has yet to be found.
Persistent Chlorotrichosis With Chronic Sun Exposure
To the Editor:
Chlorotrichosis, or green hair discoloration, is a dermatologic condition secondary to copper deposition on the hair. It most often is seen among swimmers who have prolonged exposure to chlorinated pools. The classic patient has predisposing chemical, heat, or mechanical damage to the hair shaft and usually lighter-colored hair.1-3 We present a case of chlorotrichosis in a young brunette patient who did not have predisposing factors except for chronic sun exposure.
A 13-year-old healthy adolescent girl with brown hair presented with persistent green hair for 2 years (Figure 1A). She had first noted hair discoloration after swimming in a neighbor’s chlorinated outdoor pool during summertime but experienced year-round persistence even without swimming. She denied any history of typical risk factors for hair damage, including exposure to hair dye or bleach, styling products, heat, or mechanical damage from excessive brushing. Her sister had blonde hair with a history of similar activities and exposures, and although she did style her hair with heat, she did not develop hair discoloration. The patient lived in a newer home, and prior tap water testing did not show elevated levels of copper. She admitted to strictly wearing her hair down at all times, including during strenuous activity and swimming. Excessive teasing at school prompted her mother to seek advice from hair salons. Bleaching test strips of hair reportedly caused paradoxical intensification of green, and the patient declined recommendations for red hair dye. The patient also tried Internet-based suggestions such as topically applying crushed aspirin, lemon juice, tea tree oil, and clarifying shampoos, which all failed to result in notable improvement.
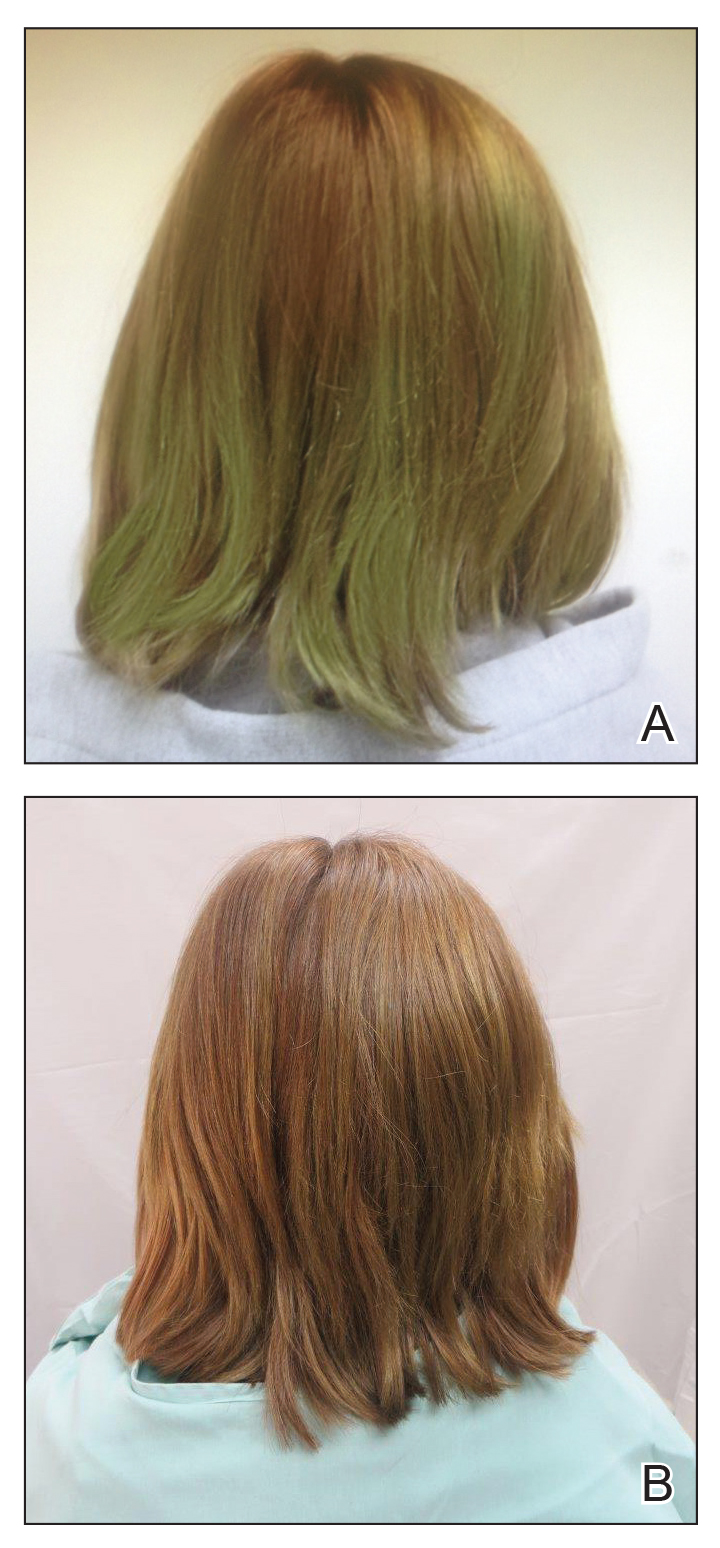
Physical examination revealed a sun-exposed distribution of ashy green hair that was worse at the distal hair ends and completely spared the roots. Trichoscopy of discolored hair (Figure 2A) revealed diffuse cuticle thinning, whereas unaffected hair appeared normal (Figure 2B). Because the patient reported slight improvement with tea tree oil, treatment was initiated with twice-weekly hot vegetable oil treatments applied for 20 minutes, which ultimately proved unsuccessful. Penicillamine shampoo (250-mg capsule of penicillamine into 5-mL purified water and 5-mL pH-balanced clear shampoo) was then recommended. At 3-month follow-up, the patient exhibited notable improvement of the hair discoloration, with only mild persistence at the distal ends of sun-damaged hair, visible only under fluorescent lighting (Figure 1B). Our recommendations thereafter were focused on prevention (Table).

The source of exogenous copper in chlorotrichosis commonly is tap water flowing through copper pipes or swimming pools rich in chlorine and copper-containing algaecides.2,4,8 The acidity of tap water is thought to cause the release of copper from the pipes.2,5 Such acidity could result from the effects of acid rain on water reservoirs or from water additives such as fluoride2 or those used in decalcification systems.5 Additionally, the attachment of electrical grounds to copper piping can cause copper to solubilize through an electric current, increasing water levels of copper.3 Although low pH facilitates copper solubility, high pH within the hair facilitates copper precipitation, which is quickly followed by adhesion to anionic molecules within hair shafts. Therefore, it is postulated that chlorotrichosis may persist in insufficiently rinsed hair with residual alkaline shampoo.6
Beyond pH flux in the induction of chlorotrichosis, other environmental agents have been suspected to play a role. A case report of green hair in a black patient following use of selenium sulfide 2.5% shampoo identified hair damage from tinea capitis infection as predisposing to chlorotrichosis.9 Other reports have cited tar shampoo and industrial exposure to cobalt, nickel, brass, mercury, or chromium as causative factors.2,3,6,7 Interestingly, green hair discoloration also has been observed in the metabolic disorder phenylketonuria.1
Few individuals exposed to elevated levels of copper will develop chlorotrichosis, which emphasizes the critical role of predisposing hair damage in its pathogenesis. With violation of the hair cuticle, chlorine can crystallize and copper can adhere to the hair shaft.10 Bleaching and waving of the hair also appear to alter the composition of keratin by increasing the number of cysteic acid and similar anionic sulfonate groups, which can bind copper.8
Although not harmful, chlorotrichosis may be aesthetically undesirable and lead to considerable social ostracism. Without intrinsic hair defects or obvious differences in predisposing factors, the question was raised as to why our patient, as a brunette, experienced dramatic hair discoloration while her blonde sister was entirely unaffected. We postulated that our patient’s persistent green hair may have been due to her unique predisposition to extensive sun-induced and mechanical hair damage because of her unwavering tendency to wear her hair down at all times. A variety of treatments of variable reported efficacy have been proposed (Table); fortunately, if treatments fail, the discoloration resolves with hair growth.
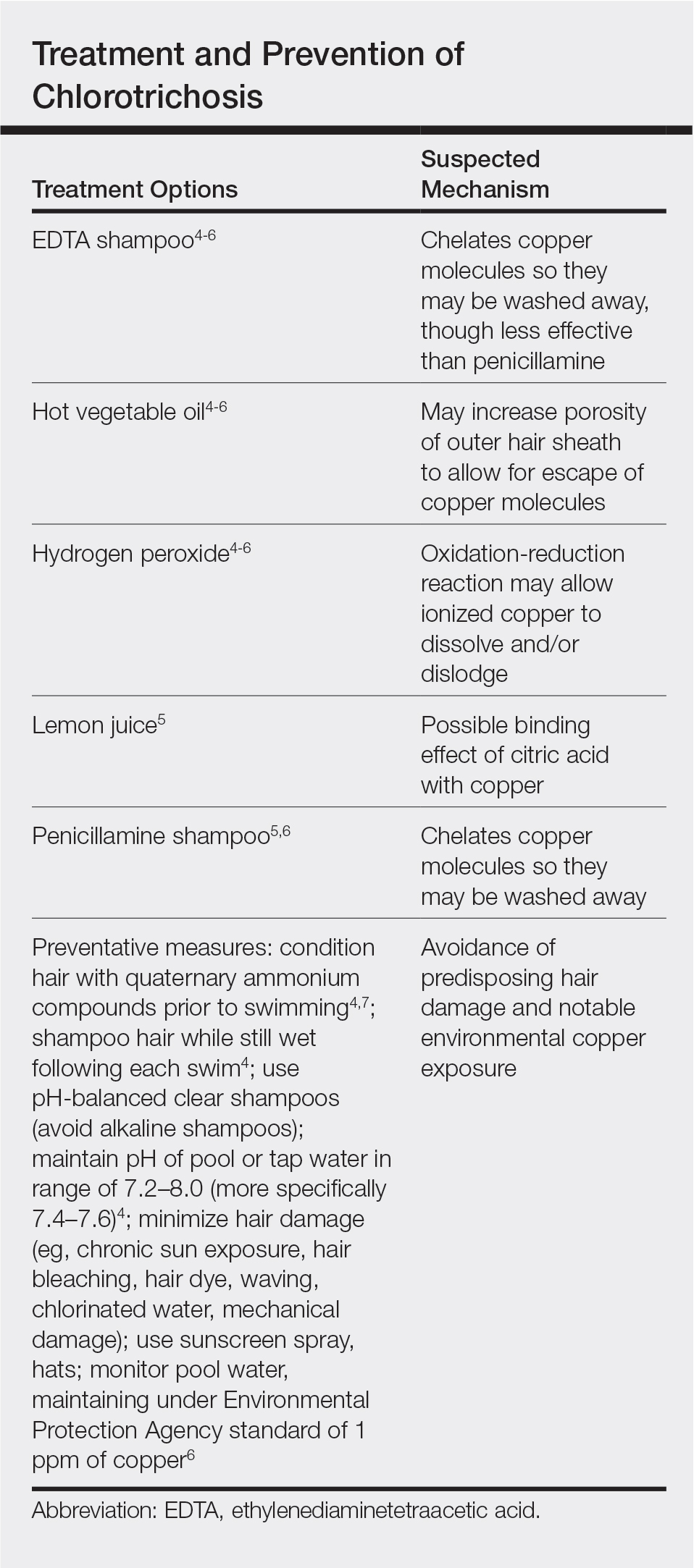
This case is unique in that it presented in a brunette patient with seemingly minimal hair damage with an unaffected blonde-haired sibling and with persistence over years. Furthermore, it lends credence to the use of penicillamine shampoo in treating chlorotrichosis, even in particularly difficult cases in which other treatments have failed.
- Holmes LB, Goldsmith LA. The man with green hair [letter]. N Engl J Med. 1974;291:1037.
Lampe RM, Henderson AL, Hansen GH. Green hair. JAMA. 1977;237:2092. - Nordlund JJ, Hartley C, Fister J. On the cause of green hair. Arch Dermatol. 1977;113:1700.
- Goldschmidt H. Green hair. Arch Dermatol. 1979;115:1288.
- Hinz T, Klingmuller K, Bieber T, et al. The mystery of green hair. Eur J Dermatol. 2009;19:409-410.
- Mascaro JM Jr, Ferrando J, Fontarnau R, et al. Green hair. Cutis. 1995;56:37-40.
- Bhat GR, Lukenbach ER, Kennedy RR, et al. The green hair problem: a preliminary investigation. J Soc Cosmet Chem. 1979;30:1-8.
- Blanc D, Zultak M, Rochefort A, et al. Green hair: clinical, chemical and epidemiologic study. apropos of a case. Ann Dermatol Venereol. 1988;115:807-812.
- Fitzgerald EA, Purcell SM, Goldman HM. Green hair discoloration due to selenium sulfide. Int J Dermatol. 1997;36:238-239.
- Fair NB, Gupta BS. The chlorine-hair interaction. II. effect of chlorination at varied pH levels on hair properties. J Soc Cosmet Chem. 1987;38:371-384.
To the Editor:
Chlorotrichosis, or green hair discoloration, is a dermatologic condition secondary to copper deposition on the hair. It most often is seen among swimmers who have prolonged exposure to chlorinated pools. The classic patient has predisposing chemical, heat, or mechanical damage to the hair shaft and usually lighter-colored hair.1-3 We present a case of chlorotrichosis in a young brunette patient who did not have predisposing factors except for chronic sun exposure.
A 13-year-old healthy adolescent girl with brown hair presented with persistent green hair for 2 years (Figure 1A). She had first noted hair discoloration after swimming in a neighbor’s chlorinated outdoor pool during summertime but experienced year-round persistence even without swimming. She denied any history of typical risk factors for hair damage, including exposure to hair dye or bleach, styling products, heat, or mechanical damage from excessive brushing. Her sister had blonde hair with a history of similar activities and exposures, and although she did style her hair with heat, she did not develop hair discoloration. The patient lived in a newer home, and prior tap water testing did not show elevated levels of copper. She admitted to strictly wearing her hair down at all times, including during strenuous activity and swimming. Excessive teasing at school prompted her mother to seek advice from hair salons. Bleaching test strips of hair reportedly caused paradoxical intensification of green, and the patient declined recommendations for red hair dye. The patient also tried Internet-based suggestions such as topically applying crushed aspirin, lemon juice, tea tree oil, and clarifying shampoos, which all failed to result in notable improvement.
Physical examination revealed a sun-exposed distribution of ashy green hair that was worse at the distal hair ends and completely spared the roots. Trichoscopy of discolored hair (Figure 2A) revealed diffuse cuticle thinning, whereas unaffected hair appeared normal (Figure 2B). Because the patient reported slight improvement with tea tree oil, treatment was initiated with twice-weekly hot vegetable oil treatments applied for 20 minutes, which ultimately proved unsuccessful. Penicillamine shampoo (250-mg capsule of penicillamine into 5-mL purified water and 5-mL pH-balanced clear shampoo) was then recommended. At 3-month follow-up, the patient exhibited notable improvement of the hair discoloration, with only mild persistence at the distal ends of sun-damaged hair, visible only under fluorescent lighting (Figure 1B). Our recommendations thereafter were focused on prevention (Table).
The source of exogenous copper in chlorotrichosis commonly is tap water flowing through copper pipes or swimming pools rich in chlorine and copper-containing algaecides.2,4,8 The acidity of tap water is thought to cause the release of copper from the pipes.2,5 Such acidity could result from the effects of acid rain on water reservoirs or from water additives such as fluoride2 or those used in decalcification systems.5 Additionally, the attachment of electrical grounds to copper piping can cause copper to solubilize through an electric current, increasing water levels of copper.3 Although low pH facilitates copper solubility, high pH within the hair facilitates copper precipitation, which is quickly followed by adhesion to anionic molecules within hair shafts. Therefore, it is postulated that chlorotrichosis may persist in insufficiently rinsed hair with residual alkaline shampoo.6
Beyond pH flux in the induction of chlorotrichosis, other environmental agents have been suspected to play a role. A case report of green hair in a black patient following use of selenium sulfide 2.5% shampoo identified hair damage from tinea capitis infection as predisposing to chlorotrichosis.9 Other reports have cited tar shampoo and industrial exposure to cobalt, nickel, brass, mercury, or chromium as causative factors.2,3,6,7 Interestingly, green hair discoloration also has been observed in the metabolic disorder phenylketonuria.1
Few individuals exposed to elevated levels of copper will develop chlorotrichosis, which emphasizes the critical role of predisposing hair damage in its pathogenesis. With violation of the hair cuticle, chlorine can crystallize and copper can adhere to the hair shaft.10 Bleaching and waving of the hair also appear to alter the composition of keratin by increasing the number of cysteic acid and similar anionic sulfonate groups, which can bind copper.8
Although not harmful, chlorotrichosis may be aesthetically undesirable and lead to considerable social ostracism. Without intrinsic hair defects or obvious differences in predisposing factors, the question was raised as to why our patient, as a brunette, experienced dramatic hair discoloration while her blonde sister was entirely unaffected. We postulated that our patient’s persistent green hair may have been due to her unique predisposition to extensive sun-induced and mechanical hair damage because of her unwavering tendency to wear her hair down at all times. A variety of treatments of variable reported efficacy have been proposed (Table); fortunately, if treatments fail, the discoloration resolves with hair growth.
This case is unique in that it presented in a brunette patient with seemingly minimal hair damage with an unaffected blonde-haired sibling and with persistence over years. Furthermore, it lends credence to the use of penicillamine shampoo in treating chlorotrichosis, even in particularly difficult cases in which other treatments have failed.
To the Editor:
Chlorotrichosis, or green hair discoloration, is a dermatologic condition secondary to copper deposition on the hair. It most often is seen among swimmers who have prolonged exposure to chlorinated pools. The classic patient has predisposing chemical, heat, or mechanical damage to the hair shaft and usually lighter-colored hair.1-3 We present a case of chlorotrichosis in a young brunette patient who did not have predisposing factors except for chronic sun exposure.
A 13-year-old healthy adolescent girl with brown hair presented with persistent green hair for 2 years (Figure 1A). She had first noted hair discoloration after swimming in a neighbor’s chlorinated outdoor pool during summertime but experienced year-round persistence even without swimming. She denied any history of typical risk factors for hair damage, including exposure to hair dye or bleach, styling products, heat, or mechanical damage from excessive brushing. Her sister had blonde hair with a history of similar activities and exposures, and although she did style her hair with heat, she did not develop hair discoloration. The patient lived in a newer home, and prior tap water testing did not show elevated levels of copper. She admitted to strictly wearing her hair down at all times, including during strenuous activity and swimming. Excessive teasing at school prompted her mother to seek advice from hair salons. Bleaching test strips of hair reportedly caused paradoxical intensification of green, and the patient declined recommendations for red hair dye. The patient also tried Internet-based suggestions such as topically applying crushed aspirin, lemon juice, tea tree oil, and clarifying shampoos, which all failed to result in notable improvement.
Physical examination revealed a sun-exposed distribution of ashy green hair that was worse at the distal hair ends and completely spared the roots. Trichoscopy of discolored hair (Figure 2A) revealed diffuse cuticle thinning, whereas unaffected hair appeared normal (Figure 2B). Because the patient reported slight improvement with tea tree oil, treatment was initiated with twice-weekly hot vegetable oil treatments applied for 20 minutes, which ultimately proved unsuccessful. Penicillamine shampoo (250-mg capsule of penicillamine into 5-mL purified water and 5-mL pH-balanced clear shampoo) was then recommended. At 3-month follow-up, the patient exhibited notable improvement of the hair discoloration, with only mild persistence at the distal ends of sun-damaged hair, visible only under fluorescent lighting (Figure 1B). Our recommendations thereafter were focused on prevention (Table).
The source of exogenous copper in chlorotrichosis commonly is tap water flowing through copper pipes or swimming pools rich in chlorine and copper-containing algaecides.2,4,8 The acidity of tap water is thought to cause the release of copper from the pipes.2,5 Such acidity could result from the effects of acid rain on water reservoirs or from water additives such as fluoride2 or those used in decalcification systems.5 Additionally, the attachment of electrical grounds to copper piping can cause copper to solubilize through an electric current, increasing water levels of copper.3 Although low pH facilitates copper solubility, high pH within the hair facilitates copper precipitation, which is quickly followed by adhesion to anionic molecules within hair shafts. Therefore, it is postulated that chlorotrichosis may persist in insufficiently rinsed hair with residual alkaline shampoo.6
Beyond pH flux in the induction of chlorotrichosis, other environmental agents have been suspected to play a role. A case report of green hair in a black patient following use of selenium sulfide 2.5% shampoo identified hair damage from tinea capitis infection as predisposing to chlorotrichosis.9 Other reports have cited tar shampoo and industrial exposure to cobalt, nickel, brass, mercury, or chromium as causative factors.2,3,6,7 Interestingly, green hair discoloration also has been observed in the metabolic disorder phenylketonuria.1
Few individuals exposed to elevated levels of copper will develop chlorotrichosis, which emphasizes the critical role of predisposing hair damage in its pathogenesis. With violation of the hair cuticle, chlorine can crystallize and copper can adhere to the hair shaft.10 Bleaching and waving of the hair also appear to alter the composition of keratin by increasing the number of cysteic acid and similar anionic sulfonate groups, which can bind copper.8
Although not harmful, chlorotrichosis may be aesthetically undesirable and lead to considerable social ostracism. Without intrinsic hair defects or obvious differences in predisposing factors, the question was raised as to why our patient, as a brunette, experienced dramatic hair discoloration while her blonde sister was entirely unaffected. We postulated that our patient’s persistent green hair may have been due to her unique predisposition to extensive sun-induced and mechanical hair damage because of her unwavering tendency to wear her hair down at all times. A variety of treatments of variable reported efficacy have been proposed (Table); fortunately, if treatments fail, the discoloration resolves with hair growth.
This case is unique in that it presented in a brunette patient with seemingly minimal hair damage with an unaffected blonde-haired sibling and with persistence over years. Furthermore, it lends credence to the use of penicillamine shampoo in treating chlorotrichosis, even in particularly difficult cases in which other treatments have failed.
- Holmes LB, Goldsmith LA. The man with green hair [letter]. N Engl J Med. 1974;291:1037.
Lampe RM, Henderson AL, Hansen GH. Green hair. JAMA. 1977;237:2092. - Nordlund JJ, Hartley C, Fister J. On the cause of green hair. Arch Dermatol. 1977;113:1700.
- Goldschmidt H. Green hair. Arch Dermatol. 1979;115:1288.
- Hinz T, Klingmuller K, Bieber T, et al. The mystery of green hair. Eur J Dermatol. 2009;19:409-410.
- Mascaro JM Jr, Ferrando J, Fontarnau R, et al. Green hair. Cutis. 1995;56:37-40.
- Bhat GR, Lukenbach ER, Kennedy RR, et al. The green hair problem: a preliminary investigation. J Soc Cosmet Chem. 1979;30:1-8.
- Blanc D, Zultak M, Rochefort A, et al. Green hair: clinical, chemical and epidemiologic study. apropos of a case. Ann Dermatol Venereol. 1988;115:807-812.
- Fitzgerald EA, Purcell SM, Goldman HM. Green hair discoloration due to selenium sulfide. Int J Dermatol. 1997;36:238-239.
- Fair NB, Gupta BS. The chlorine-hair interaction. II. effect of chlorination at varied pH levels on hair properties. J Soc Cosmet Chem. 1987;38:371-384.
- Holmes LB, Goldsmith LA. The man with green hair [letter]. N Engl J Med. 1974;291:1037.
Lampe RM, Henderson AL, Hansen GH. Green hair. JAMA. 1977;237:2092. - Nordlund JJ, Hartley C, Fister J. On the cause of green hair. Arch Dermatol. 1977;113:1700.
- Goldschmidt H. Green hair. Arch Dermatol. 1979;115:1288.
- Hinz T, Klingmuller K, Bieber T, et al. The mystery of green hair. Eur J Dermatol. 2009;19:409-410.
- Mascaro JM Jr, Ferrando J, Fontarnau R, et al. Green hair. Cutis. 1995;56:37-40.
- Bhat GR, Lukenbach ER, Kennedy RR, et al. The green hair problem: a preliminary investigation. J Soc Cosmet Chem. 1979;30:1-8.
- Blanc D, Zultak M, Rochefort A, et al. Green hair: clinical, chemical and epidemiologic study. apropos of a case. Ann Dermatol Venereol. 1988;115:807-812.
- Fitzgerald EA, Purcell SM, Goldman HM. Green hair discoloration due to selenium sulfide. Int J Dermatol. 1997;36:238-239.
- Fair NB, Gupta BS. The chlorine-hair interaction. II. effect of chlorination at varied pH levels on hair properties. J Soc Cosmet Chem. 1987;38:371-384.
Practice Points
- Chlorotrichosis is the deposition of copper onto hair, which causes a green discoloration and most commonly occurs in blonde patients with excessive exposure to chlorinated water.
- Hair cuticle damage from hair care practices, such as use of heat or chemicals, can predispose patients to the development of chlorotrichosis.
- Although a number of treatments have been proposed, the use of penicillamine shampoo seems to be particularly effective and works via chelation of the adherent copper molecules.

Localized Acanthosis Nigricans at the Site of Repetitive Insulin Injections
To the Editor:
Acanthosis nigricans (AN) is characterized by asymptomatic, hyperpigmented, velvety plaques that can occur anywhere on the body but most often present on the skin of the neck, axillae, groin, and other body folds.1-12 Although there are 5 subtypes, benign AN is the most common and is related to insulin resistance.1-4 Insulin can bind to insulinlike growth factor 1 (IGF-1) on keratinocytes, stimulating their proliferation. In type 2 diabetes mellitus, endogenous insulin levels are high enough to bind IGF-1 and activate keratinocytes, leading to the development of AN. Insulin injections have been associated with cutaneous side effects including lipoatrophy, lipohypertrophy, and postinflammatory hyperpigmentation.3 Acanthosis nigricans at insulin injection sites is a rare clinical condition.
A 64-year-old man presented for evaluation of a growing asymptomatic lesion on the abdomen of 6 years’ duration. He had a 17-year history of type 2 diabetes mellitus treated with insulin injections for 14 years after failing oral hypoglycemic agents. The patient reported injecting at the same site on the abdomen for the last 14 years. Physical examination revealed a lichenified, hyperpigmented, verrucous plaque on the lower abdomen under the umbilicus (Figure 1). No similar skin lesions were observed elsewhere on the body. A punch biopsy of the plaque showed hyperkeratosis, papillomatosis, acanthosis, and hyperpigmentation, which was consistent with AN (Figure 2). The patient was instructed to rotate injection sites and avoid the affected area. Over-the-counter ammonium lactate cream applied twice daily to the affected site also was recommended. After 2 months of treatment with this regimen, the patient reported softening and lightening of the lesion on the abdomen.
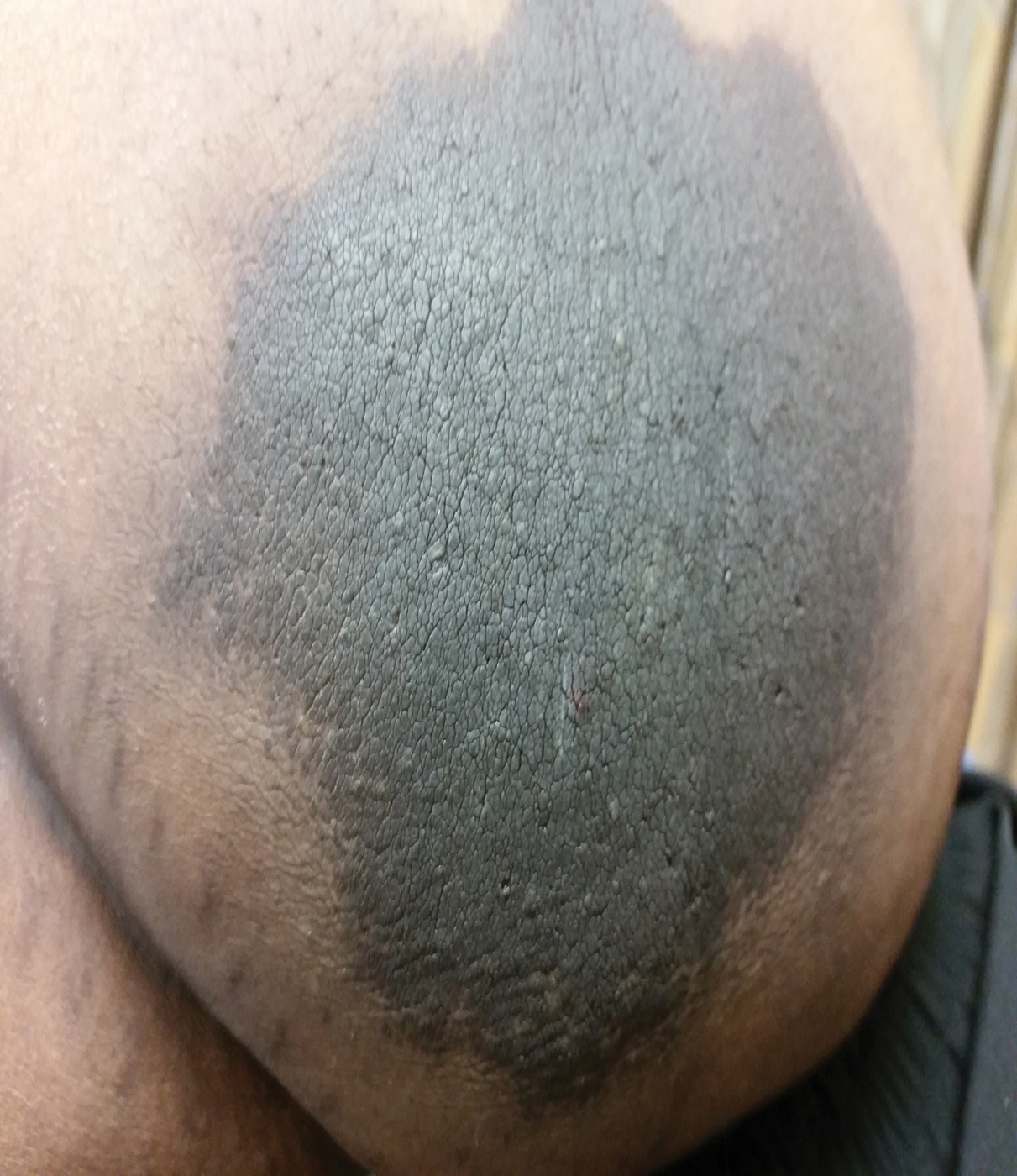

A PubMed search of articles indexed for MEDLINE for all English-language studies with human participants using the terms acanthosis nigricans and insulin injections yielded 20 results. Of them, 13 discussed localized AN at insulin injection sites: 12 case reports (Table)1-12 and 1 observational study in a group of diabetic patients.13
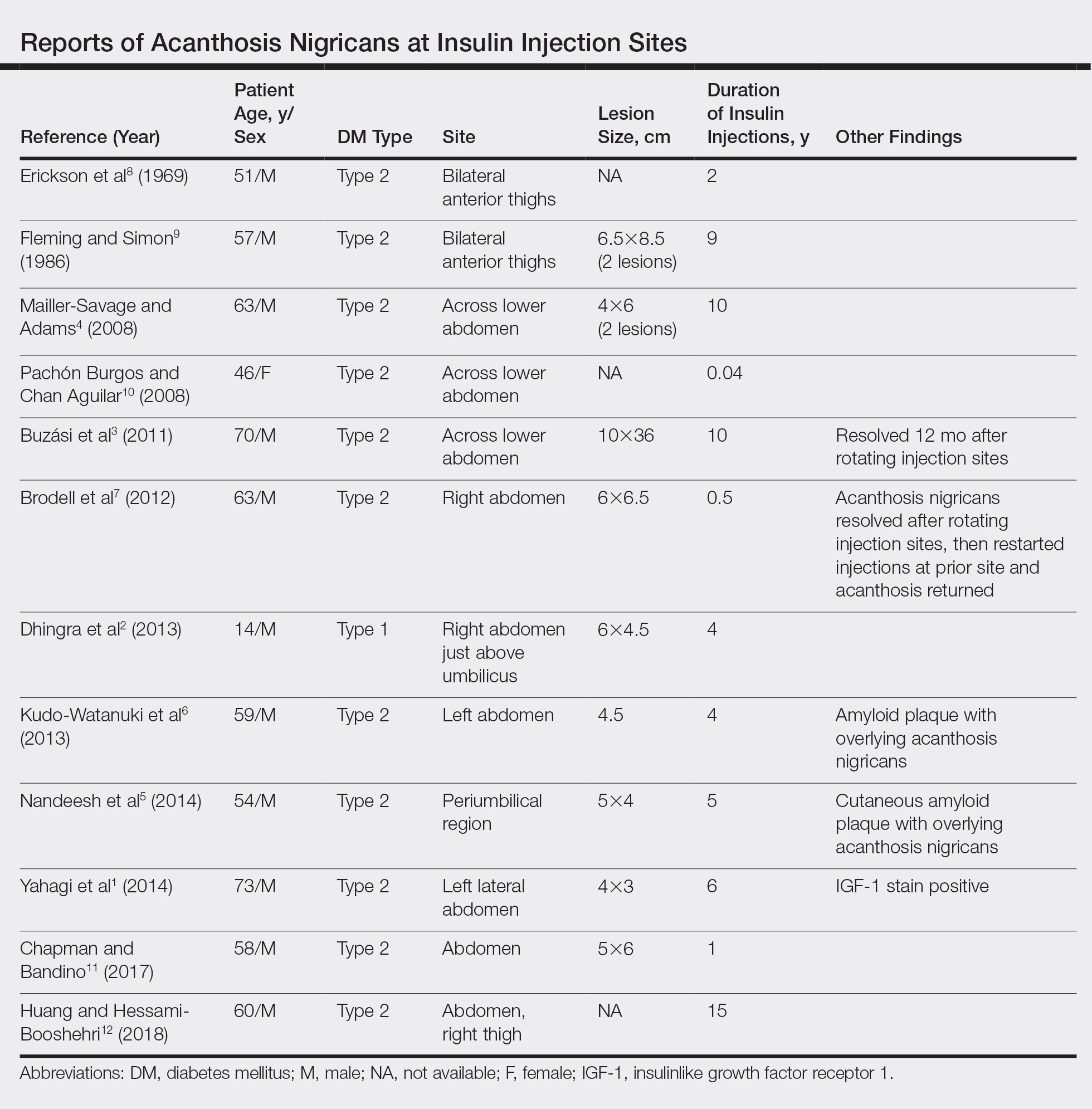
In the observational study, 500 diabetic patients were examined for insulin injection-site dermatoses and only 2 had localized injection-site AN. No other information was provided for these 2 patients.13 In the 12 published case reports,1-12 all patients did not rotate sites for their insulin injections and repeatedly injected into the affected area. The abdomen was the most commonly affected site, seen in 83% (10/12) of cases, while 25% (3/12) involved the thighs. All but 1 patient had type 2 diabetes mellitus. In 2 patients, “amyloid” was noted on the biopsy report in addition to changes consistent with AN. At least 2 patients had clearance after rotating injection sites.3,12
It has been suggested that localized AN at insulin injection sites develops through similar mechanisms as benign AN. Contributing factors to the development of benign AN may be IGF-1, fibroblast growth factor, and epidermal growth factor.1-3 Insulin is similar in structure to IGF-1 and can bind to IGF-1 receptors at high enough concentrations. Insulinlike growth factor 1 receptors are present on keratinocytes and fibroblasts, which proliferate upon activation, leading to the development of AN.1-3 Localized AN is thought to occur when repetitive insulin at the injection site saturates IGF-1 receptors on local keratinocytes.
Based on our patient and prior reports in the literature, localized AN is an uncommon cutaneous complication of insulin injections. Physicians should ask about repetitive insulin injections in the same site when localized AN occurs in patients with diabetes mellitus on insulin therapy. They also should discuss the importance of rotating sites for insulin adminstration to prevent the development of cutaneous complications including AN.
- Yahagi E, Mabuchi T, Nuruki H, et al. Case of exogenous insulin-derived acanthosis nigricans caused by insulin injections. Tokai J Exp Clin Med. 2014;39:5-9.
- Dhingra M, Garg G, Gupta M, et al. Exogenous insulin-derived acanthosis nigricans: could it be a cause of increased insulin requirement? Dermatol Online J. 2013;19:9.
- Buzási K, Sápi Z, Jermendy G. Acanthosis nigricans as a local cutaneous side effect of repeated human insulin injections. Diabetes Res Clin Pract. 2011;94:E34-E36.
- Mailler-Savage EA, Adams BB. Exogenous insulin-derived acanthosis nigricans. Arch Dermatol. 2008;144:126-127.
- Nandeesh BN, Rajalakshmi T, Shubha B. Cutaneous amyloidosis and insulin with coexistence of acanthosis nigricans. Indian J Pathol Microbiol. 2014;57:127-129.
- Kudo-Watanuki S, Kurihara E, Yamamoto K, et al. Coexistence of insulin-derived amyloidosis and an overlying acanthosis nigricans-like lesion at the site of insulin injection. Clin Exp Dermatol. 2013;38:25-29.
- Brodell JD Jr, Cannella JD, Helms SE. Case report: acanthosis nigricans resulting from repetitive same-site insulin injections. J Drugs Dermatol. 2012;11:E85-E87.
- Erickson L, Lipschutz DE, Wrigley W, et al. A peculiar cutaneous reaction to repeated injections of insulin. JAMA. 1969;209:934-935.
- Fleming MG, Simon SI. Cutaneous insulin reaction resembling acanthosis nigricans. Arch Dermatol. 1986;122:1054-1056.
- Pachon Burgos A, Chan Aguilar MP. Visual vignette. hyperpigmented hyperkeratotic cutaneous insulin reaction that resembles acanthosis nigricans with lipohypertrophy. Endocr Pract. 2008;14:514.
- Chapman SE, Bandino JP. A verrucous plaque on the abdomen: challenge. Am J Dermatopathol. 2017;39:E163.
- Huang Y, Hessami-Booshehri M. Acanthosis nigricans at sites of insulin injection in a man with diabetes. CMAJ. 2018;190:E1390.
- Sawatkar GU, Kanwar AJ, Dogra S, et al. Spectrum of cutaneous manifestations of type 1 diabetes mellitus in 500 South Asian patients. Br J Dermatol. 2014;171:1402-1406.
To the Editor:
Acanthosis nigricans (AN) is characterized by asymptomatic, hyperpigmented, velvety plaques that can occur anywhere on the body but most often present on the skin of the neck, axillae, groin, and other body folds.1-12 Although there are 5 subtypes, benign AN is the most common and is related to insulin resistance.1-4 Insulin can bind to insulinlike growth factor 1 (IGF-1) on keratinocytes, stimulating their proliferation. In type 2 diabetes mellitus, endogenous insulin levels are high enough to bind IGF-1 and activate keratinocytes, leading to the development of AN. Insulin injections have been associated with cutaneous side effects including lipoatrophy, lipohypertrophy, and postinflammatory hyperpigmentation.3 Acanthosis nigricans at insulin injection sites is a rare clinical condition.
A 64-year-old man presented for evaluation of a growing asymptomatic lesion on the abdomen of 6 years’ duration. He had a 17-year history of type 2 diabetes mellitus treated with insulin injections for 14 years after failing oral hypoglycemic agents. The patient reported injecting at the same site on the abdomen for the last 14 years. Physical examination revealed a lichenified, hyperpigmented, verrucous plaque on the lower abdomen under the umbilicus (Figure 1). No similar skin lesions were observed elsewhere on the body. A punch biopsy of the plaque showed hyperkeratosis, papillomatosis, acanthosis, and hyperpigmentation, which was consistent with AN (Figure 2). The patient was instructed to rotate injection sites and avoid the affected area. Over-the-counter ammonium lactate cream applied twice daily to the affected site also was recommended. After 2 months of treatment with this regimen, the patient reported softening and lightening of the lesion on the abdomen.
A PubMed search of articles indexed for MEDLINE for all English-language studies with human participants using the terms acanthosis nigricans and insulin injections yielded 20 results. Of them, 13 discussed localized AN at insulin injection sites: 12 case reports (Table)1-12 and 1 observational study in a group of diabetic patients.13
In the observational study, 500 diabetic patients were examined for insulin injection-site dermatoses and only 2 had localized injection-site AN. No other information was provided for these 2 patients.13 In the 12 published case reports,1-12 all patients did not rotate sites for their insulin injections and repeatedly injected into the affected area. The abdomen was the most commonly affected site, seen in 83% (10/12) of cases, while 25% (3/12) involved the thighs. All but 1 patient had type 2 diabetes mellitus. In 2 patients, “amyloid” was noted on the biopsy report in addition to changes consistent with AN. At least 2 patients had clearance after rotating injection sites.3,12
It has been suggested that localized AN at insulin injection sites develops through similar mechanisms as benign AN. Contributing factors to the development of benign AN may be IGF-1, fibroblast growth factor, and epidermal growth factor.1-3 Insulin is similar in structure to IGF-1 and can bind to IGF-1 receptors at high enough concentrations. Insulinlike growth factor 1 receptors are present on keratinocytes and fibroblasts, which proliferate upon activation, leading to the development of AN.1-3 Localized AN is thought to occur when repetitive insulin at the injection site saturates IGF-1 receptors on local keratinocytes.
Based on our patient and prior reports in the literature, localized AN is an uncommon cutaneous complication of insulin injections. Physicians should ask about repetitive insulin injections in the same site when localized AN occurs in patients with diabetes mellitus on insulin therapy. They also should discuss the importance of rotating sites for insulin adminstration to prevent the development of cutaneous complications including AN.
To the Editor:
Acanthosis nigricans (AN) is characterized by asymptomatic, hyperpigmented, velvety plaques that can occur anywhere on the body but most often present on the skin of the neck, axillae, groin, and other body folds.1-12 Although there are 5 subtypes, benign AN is the most common and is related to insulin resistance.1-4 Insulin can bind to insulinlike growth factor 1 (IGF-1) on keratinocytes, stimulating their proliferation. In type 2 diabetes mellitus, endogenous insulin levels are high enough to bind IGF-1 and activate keratinocytes, leading to the development of AN. Insulin injections have been associated with cutaneous side effects including lipoatrophy, lipohypertrophy, and postinflammatory hyperpigmentation.3 Acanthosis nigricans at insulin injection sites is a rare clinical condition.
A 64-year-old man presented for evaluation of a growing asymptomatic lesion on the abdomen of 6 years’ duration. He had a 17-year history of type 2 diabetes mellitus treated with insulin injections for 14 years after failing oral hypoglycemic agents. The patient reported injecting at the same site on the abdomen for the last 14 years. Physical examination revealed a lichenified, hyperpigmented, verrucous plaque on the lower abdomen under the umbilicus (Figure 1). No similar skin lesions were observed elsewhere on the body. A punch biopsy of the plaque showed hyperkeratosis, papillomatosis, acanthosis, and hyperpigmentation, which was consistent with AN (Figure 2). The patient was instructed to rotate injection sites and avoid the affected area. Over-the-counter ammonium lactate cream applied twice daily to the affected site also was recommended. After 2 months of treatment with this regimen, the patient reported softening and lightening of the lesion on the abdomen.
A PubMed search of articles indexed for MEDLINE for all English-language studies with human participants using the terms acanthosis nigricans and insulin injections yielded 20 results. Of them, 13 discussed localized AN at insulin injection sites: 12 case reports (Table)1-12 and 1 observational study in a group of diabetic patients.13
In the observational study, 500 diabetic patients were examined for insulin injection-site dermatoses and only 2 had localized injection-site AN. No other information was provided for these 2 patients.13 In the 12 published case reports,1-12 all patients did not rotate sites for their insulin injections and repeatedly injected into the affected area. The abdomen was the most commonly affected site, seen in 83% (10/12) of cases, while 25% (3/12) involved the thighs. All but 1 patient had type 2 diabetes mellitus. In 2 patients, “amyloid” was noted on the biopsy report in addition to changes consistent with AN. At least 2 patients had clearance after rotating injection sites.3,12
It has been suggested that localized AN at insulin injection sites develops through similar mechanisms as benign AN. Contributing factors to the development of benign AN may be IGF-1, fibroblast growth factor, and epidermal growth factor.1-3 Insulin is similar in structure to IGF-1 and can bind to IGF-1 receptors at high enough concentrations. Insulinlike growth factor 1 receptors are present on keratinocytes and fibroblasts, which proliferate upon activation, leading to the development of AN.1-3 Localized AN is thought to occur when repetitive insulin at the injection site saturates IGF-1 receptors on local keratinocytes.
Based on our patient and prior reports in the literature, localized AN is an uncommon cutaneous complication of insulin injections. Physicians should ask about repetitive insulin injections in the same site when localized AN occurs in patients with diabetes mellitus on insulin therapy. They also should discuss the importance of rotating sites for insulin adminstration to prevent the development of cutaneous complications including AN.
- Yahagi E, Mabuchi T, Nuruki H, et al. Case of exogenous insulin-derived acanthosis nigricans caused by insulin injections. Tokai J Exp Clin Med. 2014;39:5-9.
- Dhingra M, Garg G, Gupta M, et al. Exogenous insulin-derived acanthosis nigricans: could it be a cause of increased insulin requirement? Dermatol Online J. 2013;19:9.
- Buzási K, Sápi Z, Jermendy G. Acanthosis nigricans as a local cutaneous side effect of repeated human insulin injections. Diabetes Res Clin Pract. 2011;94:E34-E36.
- Mailler-Savage EA, Adams BB. Exogenous insulin-derived acanthosis nigricans. Arch Dermatol. 2008;144:126-127.
- Nandeesh BN, Rajalakshmi T, Shubha B. Cutaneous amyloidosis and insulin with coexistence of acanthosis nigricans. Indian J Pathol Microbiol. 2014;57:127-129.
- Kudo-Watanuki S, Kurihara E, Yamamoto K, et al. Coexistence of insulin-derived amyloidosis and an overlying acanthosis nigricans-like lesion at the site of insulin injection. Clin Exp Dermatol. 2013;38:25-29.
- Brodell JD Jr, Cannella JD, Helms SE. Case report: acanthosis nigricans resulting from repetitive same-site insulin injections. J Drugs Dermatol. 2012;11:E85-E87.
- Erickson L, Lipschutz DE, Wrigley W, et al. A peculiar cutaneous reaction to repeated injections of insulin. JAMA. 1969;209:934-935.
- Fleming MG, Simon SI. Cutaneous insulin reaction resembling acanthosis nigricans. Arch Dermatol. 1986;122:1054-1056.
- Pachon Burgos A, Chan Aguilar MP. Visual vignette. hyperpigmented hyperkeratotic cutaneous insulin reaction that resembles acanthosis nigricans with lipohypertrophy. Endocr Pract. 2008;14:514.
- Chapman SE, Bandino JP. A verrucous plaque on the abdomen: challenge. Am J Dermatopathol. 2017;39:E163.
- Huang Y, Hessami-Booshehri M. Acanthosis nigricans at sites of insulin injection in a man with diabetes. CMAJ. 2018;190:E1390.
- Sawatkar GU, Kanwar AJ, Dogra S, et al. Spectrum of cutaneous manifestations of type 1 diabetes mellitus in 500 South Asian patients. Br J Dermatol. 2014;171:1402-1406.
- Yahagi E, Mabuchi T, Nuruki H, et al. Case of exogenous insulin-derived acanthosis nigricans caused by insulin injections. Tokai J Exp Clin Med. 2014;39:5-9.
- Dhingra M, Garg G, Gupta M, et al. Exogenous insulin-derived acanthosis nigricans: could it be a cause of increased insulin requirement? Dermatol Online J. 2013;19:9.
- Buzási K, Sápi Z, Jermendy G. Acanthosis nigricans as a local cutaneous side effect of repeated human insulin injections. Diabetes Res Clin Pract. 2011;94:E34-E36.
- Mailler-Savage EA, Adams BB. Exogenous insulin-derived acanthosis nigricans. Arch Dermatol. 2008;144:126-127.
- Nandeesh BN, Rajalakshmi T, Shubha B. Cutaneous amyloidosis and insulin with coexistence of acanthosis nigricans. Indian J Pathol Microbiol. 2014;57:127-129.
- Kudo-Watanuki S, Kurihara E, Yamamoto K, et al. Coexistence of insulin-derived amyloidosis and an overlying acanthosis nigricans-like lesion at the site of insulin injection. Clin Exp Dermatol. 2013;38:25-29.
- Brodell JD Jr, Cannella JD, Helms SE. Case report: acanthosis nigricans resulting from repetitive same-site insulin injections. J Drugs Dermatol. 2012;11:E85-E87.
- Erickson L, Lipschutz DE, Wrigley W, et al. A peculiar cutaneous reaction to repeated injections of insulin. JAMA. 1969;209:934-935.
- Fleming MG, Simon SI. Cutaneous insulin reaction resembling acanthosis nigricans. Arch Dermatol. 1986;122:1054-1056.
- Pachon Burgos A, Chan Aguilar MP. Visual vignette. hyperpigmented hyperkeratotic cutaneous insulin reaction that resembles acanthosis nigricans with lipohypertrophy. Endocr Pract. 2008;14:514.
- Chapman SE, Bandino JP. A verrucous plaque on the abdomen: challenge. Am J Dermatopathol. 2017;39:E163.
- Huang Y, Hessami-Booshehri M. Acanthosis nigricans at sites of insulin injection in a man with diabetes. CMAJ. 2018;190:E1390.
- Sawatkar GU, Kanwar AJ, Dogra S, et al. Spectrum of cutaneous manifestations of type 1 diabetes mellitus in 500 South Asian patients. Br J Dermatol. 2014;171:1402-1406.
Practice Points
- Benign acanthosis nigricans (AN) is most often related to insulin resistance and presents as asymptomatic, hyperpigmented, velvety plaques on the neck, axillae, groin, and other body folds.
- Benign AN related to insulin resistance occurs when insulin binds to insulinlike growth factor 1 on keratinocytes and stimulates proliferations.
- Although insulin injections have been associated with several cutaneous side effects, including lipoatrophy, lipohypertrophy, and postinflammatory hyperpigmentation, localized AN is an uncommonly reported cutaneous adverse effect.
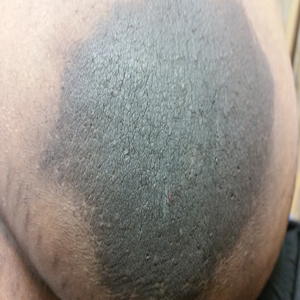
Not So Classic, Classic Kaposi Sarcoma
To the Editor:
Kaposi sarcoma (KS) is a lymphatic endothelial cell neoplasm that frequently presents as multiple vascular cutaneous and mucosal nodules.1 The classic KS variant typically is described as the presentation of KS in otherwise healthy elderly men of Jewish or Mediterranean descent.2 We present 2 cases of classic KS presenting in Mexican women living in Los Angeles County, California, with atypical clinical features.
A 65-year-old woman of Mexican descent presented to the dermatology clinic for evaluation of an asymptomatic growth on the right ventral forearm of 4 months’ duration. Biopsy of the lesion demonstrated a spindle cell proliferation suspicious for KS. Physical examination several months following the initial biopsy revealed a mildly indurated scar on the right ventral forearm with an adjacent faintly erythematous papule (Figure 1A). Repeat biopsy revealed a dermal spindle cell proliferation suggestive of KS (Figures 1B and 1C). S-100 and cytokeratin stains were negative, but a latent nuclear antigen 1 stain for human herpesvirus 8 was positive. Human immunodeficiency virus screening also was negative. Given the isolated findings, the oncology service determined that observation alone was the most appropriate management. Over time, the patient developed several similar scattered erythematous papules, and treatment with imiquimod cream 5% was initiated for 1 month without improvement. She was subsequently given a trial of alitretinoin ointment 0.1% twice daily. The lesions improved, and she continues to be well controlled on this topical therapy alone.
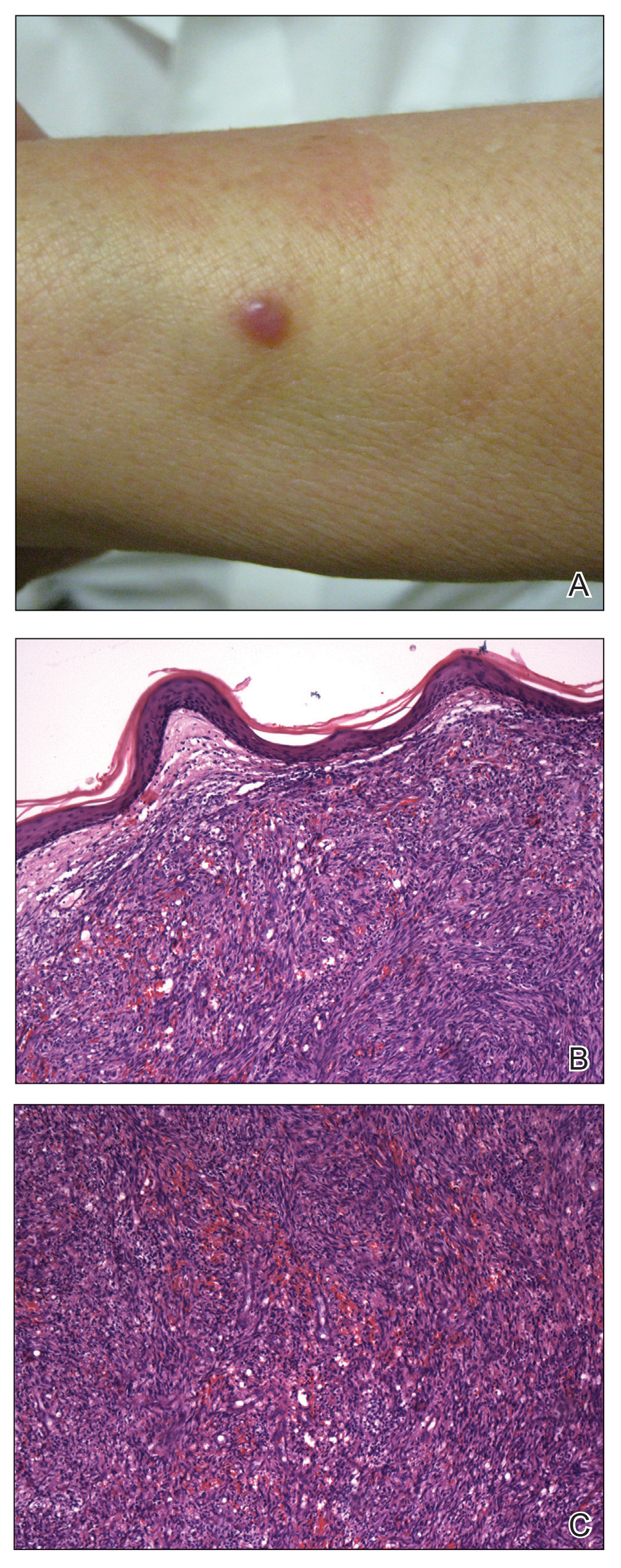
A 62-year-old woman of Mexican descent with end-stage cryptogenic cirrhosis was admitted to the hospital for evaluation of transplant candidacy. Dermatology was consulted to assess a 5×3-cm, asymptomatic, solitary, violaceous plaque on the right plantar foot (Figure 2A) with no palpable lymph nodes. Biopsy revealed a dermal proliferation of slit-like vascular channels infiltrating through the collagen and surrounding preexisting vascular spaces and adnexal structures (Figure 2B). Extravasation of erythrocytes and plasma cells also was appreciated. Latent nuclear antigen 1 staining showed strong nuclear positivity consistent with KS (Figure 2C), and a human immunodeficiency virus test and workup for underlying immunosuppression were negative. The patient had no history of treatment with immunosuppressive medications. Further workup revealed involvement of the lymphatic system. The patient was removed from the transplant list and was not a candidate for chemotherapy due to liver failure.
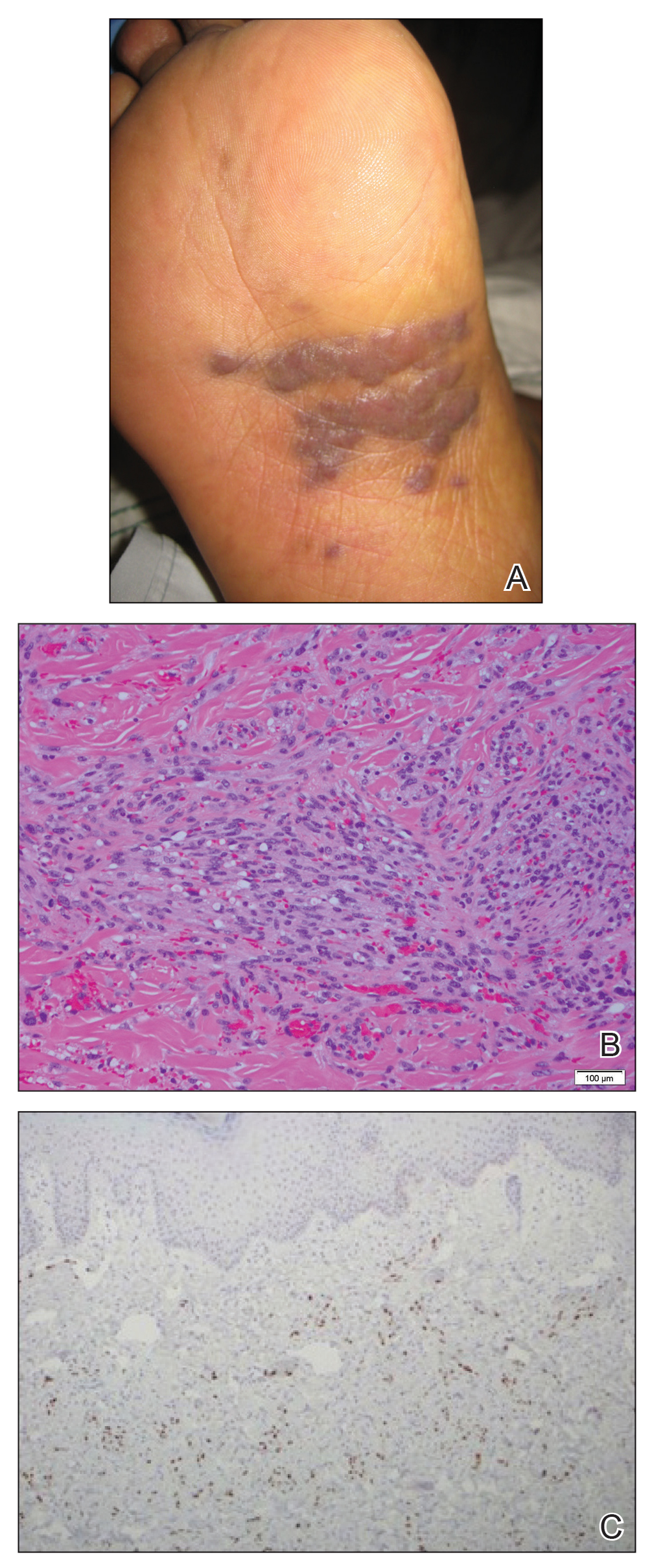
Kaposi sarcoma is a vascular neoplasm associated with human herpesvirus 8. It typically presents as erythematous to violaceous papules and plaques on the extremities.1 At least 10 morphologic variants of KS have been identified.3 The indolent classic variant of KS most commonly is found in immunocompetent individuals and has been reported to primarily affect elderly men of Jewish or Mediterranean descent.
Epidemiologic analyses of this disease in the South American population are rare. More than 250 cases have been published from South American countries with the largest series published from patients in Argentina, Peru, and Colombia.4 The incidence of classic KS in Peru is 2.54 per 10,000 individuals,5 and the disease has been diagnosed in 1 of 1000 malignant neoplasms at the National Cancer Institute of Colombia.6
A search of the Armed Forces Institute of Pathology Soft Tissue Pathology registry for classic KS patients (1980-2000) showed that 18% of 438 cases of classic KS were diagnosed in South American Hispanics,7 a percentage that nearly approximates the proportion of patients with KS of Mediterranean descent. Interestingly, in an analysis conducted within Los Angeles County, classic KS was most frequently diagnosed in Jewish, Eastern European–born men who were 55 years or older, followed by European-born women. In this study, white, Spanish, and black populations were diagnosed with classic KS with equal frequency.8
Our 2 cases of classic KS from Los Angeles County had several notable features. Both patients were women from Mexico, a demographic not previously associated with classic KS,8 and they did not have risk factors commonly associated with classic KS. We emphasize that classic KS likely is underreported and understudied in the Mexican and South American populations with need for further epidemiological and clinical analyses.
- Kaposi M. Idiopathisches multiples Pigmentsarkom der Haut. Arch Dermatol Syphilol. 1872;4:265-272.
- Akasbi Y, Awada A, Arifi S, et al. Non-HIV Kaposi’s sarcoma: a review and therapeutic perspectives. Bull Cancer. 2012;99:92-99.
- Schwartz RA. Kaposi’s sarcoma: an update. J Surg Oncol. 2004;87:146-151.
- Mohanna S, Maco V, Bravo F, et al. Epidemiology and clinical characteristics of classic Kaposi’s sarcoma, seroprevalence, and variants of human herpesvirus 8 in South America: a critical review of an old disease. Int J Infect Dis. 2005;9:2392-2350.
- Mohanna S, Ferrufino JC, Sanchez J, et al. Epidemiological and clinical characteristics of classic Kaposi’s sarcoma in Peru. J Am Acad Dermatol. 2005;53:435-441.
- García A, Olivella F, Valderrama S, et al. Kaposi’s sarcoma in Colombia. Cancer. 1989;64:2393-2398.
- Hiatt KM, Nelson AM, Lichy JH, et al. Classic Kaposi Sarcoma in the United States over the last two decades: a clinicopathologic and molecular study of 438 non-HIV related Kaposi Sarcoma patients with comparison to HIV-related Kaposi Sarcoma. Mod Pathol. 2008;21:572-582.
- Ross RK, Casagrande JT, Dworsky RL, et al. Kaposi’s sarcoma in Los Angeles, California. J Natl Cancer Inst. 1985;75:1011-1015.
To the Editor:
Kaposi sarcoma (KS) is a lymphatic endothelial cell neoplasm that frequently presents as multiple vascular cutaneous and mucosal nodules.1 The classic KS variant typically is described as the presentation of KS in otherwise healthy elderly men of Jewish or Mediterranean descent.2 We present 2 cases of classic KS presenting in Mexican women living in Los Angeles County, California, with atypical clinical features.
A 65-year-old woman of Mexican descent presented to the dermatology clinic for evaluation of an asymptomatic growth on the right ventral forearm of 4 months’ duration. Biopsy of the lesion demonstrated a spindle cell proliferation suspicious for KS. Physical examination several months following the initial biopsy revealed a mildly indurated scar on the right ventral forearm with an adjacent faintly erythematous papule (Figure 1A). Repeat biopsy revealed a dermal spindle cell proliferation suggestive of KS (Figures 1B and 1C). S-100 and cytokeratin stains were negative, but a latent nuclear antigen 1 stain for human herpesvirus 8 was positive. Human immunodeficiency virus screening also was negative. Given the isolated findings, the oncology service determined that observation alone was the most appropriate management. Over time, the patient developed several similar scattered erythematous papules, and treatment with imiquimod cream 5% was initiated for 1 month without improvement. She was subsequently given a trial of alitretinoin ointment 0.1% twice daily. The lesions improved, and she continues to be well controlled on this topical therapy alone.
A 62-year-old woman of Mexican descent with end-stage cryptogenic cirrhosis was admitted to the hospital for evaluation of transplant candidacy. Dermatology was consulted to assess a 5×3-cm, asymptomatic, solitary, violaceous plaque on the right plantar foot (Figure 2A) with no palpable lymph nodes. Biopsy revealed a dermal proliferation of slit-like vascular channels infiltrating through the collagen and surrounding preexisting vascular spaces and adnexal structures (Figure 2B). Extravasation of erythrocytes and plasma cells also was appreciated. Latent nuclear antigen 1 staining showed strong nuclear positivity consistent with KS (Figure 2C), and a human immunodeficiency virus test and workup for underlying immunosuppression were negative. The patient had no history of treatment with immunosuppressive medications. Further workup revealed involvement of the lymphatic system. The patient was removed from the transplant list and was not a candidate for chemotherapy due to liver failure.
Kaposi sarcoma is a vascular neoplasm associated with human herpesvirus 8. It typically presents as erythematous to violaceous papules and plaques on the extremities.1 At least 10 morphologic variants of KS have been identified.3 The indolent classic variant of KS most commonly is found in immunocompetent individuals and has been reported to primarily affect elderly men of Jewish or Mediterranean descent.
Epidemiologic analyses of this disease in the South American population are rare. More than 250 cases have been published from South American countries with the largest series published from patients in Argentina, Peru, and Colombia.4 The incidence of classic KS in Peru is 2.54 per 10,000 individuals,5 and the disease has been diagnosed in 1 of 1000 malignant neoplasms at the National Cancer Institute of Colombia.6
A search of the Armed Forces Institute of Pathology Soft Tissue Pathology registry for classic KS patients (1980-2000) showed that 18% of 438 cases of classic KS were diagnosed in South American Hispanics,7 a percentage that nearly approximates the proportion of patients with KS of Mediterranean descent. Interestingly, in an analysis conducted within Los Angeles County, classic KS was most frequently diagnosed in Jewish, Eastern European–born men who were 55 years or older, followed by European-born women. In this study, white, Spanish, and black populations were diagnosed with classic KS with equal frequency.8
Our 2 cases of classic KS from Los Angeles County had several notable features. Both patients were women from Mexico, a demographic not previously associated with classic KS,8 and they did not have risk factors commonly associated with classic KS. We emphasize that classic KS likely is underreported and understudied in the Mexican and South American populations with need for further epidemiological and clinical analyses.
To the Editor:
Kaposi sarcoma (KS) is a lymphatic endothelial cell neoplasm that frequently presents as multiple vascular cutaneous and mucosal nodules.1 The classic KS variant typically is described as the presentation of KS in otherwise healthy elderly men of Jewish or Mediterranean descent.2 We present 2 cases of classic KS presenting in Mexican women living in Los Angeles County, California, with atypical clinical features.
A 65-year-old woman of Mexican descent presented to the dermatology clinic for evaluation of an asymptomatic growth on the right ventral forearm of 4 months’ duration. Biopsy of the lesion demonstrated a spindle cell proliferation suspicious for KS. Physical examination several months following the initial biopsy revealed a mildly indurated scar on the right ventral forearm with an adjacent faintly erythematous papule (Figure 1A). Repeat biopsy revealed a dermal spindle cell proliferation suggestive of KS (Figures 1B and 1C). S-100 and cytokeratin stains were negative, but a latent nuclear antigen 1 stain for human herpesvirus 8 was positive. Human immunodeficiency virus screening also was negative. Given the isolated findings, the oncology service determined that observation alone was the most appropriate management. Over time, the patient developed several similar scattered erythematous papules, and treatment with imiquimod cream 5% was initiated for 1 month without improvement. She was subsequently given a trial of alitretinoin ointment 0.1% twice daily. The lesions improved, and she continues to be well controlled on this topical therapy alone.
A 62-year-old woman of Mexican descent with end-stage cryptogenic cirrhosis was admitted to the hospital for evaluation of transplant candidacy. Dermatology was consulted to assess a 5×3-cm, asymptomatic, solitary, violaceous plaque on the right plantar foot (Figure 2A) with no palpable lymph nodes. Biopsy revealed a dermal proliferation of slit-like vascular channels infiltrating through the collagen and surrounding preexisting vascular spaces and adnexal structures (Figure 2B). Extravasation of erythrocytes and plasma cells also was appreciated. Latent nuclear antigen 1 staining showed strong nuclear positivity consistent with KS (Figure 2C), and a human immunodeficiency virus test and workup for underlying immunosuppression were negative. The patient had no history of treatment with immunosuppressive medications. Further workup revealed involvement of the lymphatic system. The patient was removed from the transplant list and was not a candidate for chemotherapy due to liver failure.
Kaposi sarcoma is a vascular neoplasm associated with human herpesvirus 8. It typically presents as erythematous to violaceous papules and plaques on the extremities.1 At least 10 morphologic variants of KS have been identified.3 The indolent classic variant of KS most commonly is found in immunocompetent individuals and has been reported to primarily affect elderly men of Jewish or Mediterranean descent.
Epidemiologic analyses of this disease in the South American population are rare. More than 250 cases have been published from South American countries with the largest series published from patients in Argentina, Peru, and Colombia.4 The incidence of classic KS in Peru is 2.54 per 10,000 individuals,5 and the disease has been diagnosed in 1 of 1000 malignant neoplasms at the National Cancer Institute of Colombia.6
A search of the Armed Forces Institute of Pathology Soft Tissue Pathology registry for classic KS patients (1980-2000) showed that 18% of 438 cases of classic KS were diagnosed in South American Hispanics,7 a percentage that nearly approximates the proportion of patients with KS of Mediterranean descent. Interestingly, in an analysis conducted within Los Angeles County, classic KS was most frequently diagnosed in Jewish, Eastern European–born men who were 55 years or older, followed by European-born women. In this study, white, Spanish, and black populations were diagnosed with classic KS with equal frequency.8
Our 2 cases of classic KS from Los Angeles County had several notable features. Both patients were women from Mexico, a demographic not previously associated with classic KS,8 and they did not have risk factors commonly associated with classic KS. We emphasize that classic KS likely is underreported and understudied in the Mexican and South American populations with need for further epidemiological and clinical analyses.
- Kaposi M. Idiopathisches multiples Pigmentsarkom der Haut. Arch Dermatol Syphilol. 1872;4:265-272.
- Akasbi Y, Awada A, Arifi S, et al. Non-HIV Kaposi’s sarcoma: a review and therapeutic perspectives. Bull Cancer. 2012;99:92-99.
- Schwartz RA. Kaposi’s sarcoma: an update. J Surg Oncol. 2004;87:146-151.
- Mohanna S, Maco V, Bravo F, et al. Epidemiology and clinical characteristics of classic Kaposi’s sarcoma, seroprevalence, and variants of human herpesvirus 8 in South America: a critical review of an old disease. Int J Infect Dis. 2005;9:2392-2350.
- Mohanna S, Ferrufino JC, Sanchez J, et al. Epidemiological and clinical characteristics of classic Kaposi’s sarcoma in Peru. J Am Acad Dermatol. 2005;53:435-441.
- García A, Olivella F, Valderrama S, et al. Kaposi’s sarcoma in Colombia. Cancer. 1989;64:2393-2398.
- Hiatt KM, Nelson AM, Lichy JH, et al. Classic Kaposi Sarcoma in the United States over the last two decades: a clinicopathologic and molecular study of 438 non-HIV related Kaposi Sarcoma patients with comparison to HIV-related Kaposi Sarcoma. Mod Pathol. 2008;21:572-582.
- Ross RK, Casagrande JT, Dworsky RL, et al. Kaposi’s sarcoma in Los Angeles, California. J Natl Cancer Inst. 1985;75:1011-1015.
- Kaposi M. Idiopathisches multiples Pigmentsarkom der Haut. Arch Dermatol Syphilol. 1872;4:265-272.
- Akasbi Y, Awada A, Arifi S, et al. Non-HIV Kaposi’s sarcoma: a review and therapeutic perspectives. Bull Cancer. 2012;99:92-99.
- Schwartz RA. Kaposi’s sarcoma: an update. J Surg Oncol. 2004;87:146-151.
- Mohanna S, Maco V, Bravo F, et al. Epidemiology and clinical characteristics of classic Kaposi’s sarcoma, seroprevalence, and variants of human herpesvirus 8 in South America: a critical review of an old disease. Int J Infect Dis. 2005;9:2392-2350.
- Mohanna S, Ferrufino JC, Sanchez J, et al. Epidemiological and clinical characteristics of classic Kaposi’s sarcoma in Peru. J Am Acad Dermatol. 2005;53:435-441.
- García A, Olivella F, Valderrama S, et al. Kaposi’s sarcoma in Colombia. Cancer. 1989;64:2393-2398.
- Hiatt KM, Nelson AM, Lichy JH, et al. Classic Kaposi Sarcoma in the United States over the last two decades: a clinicopathologic and molecular study of 438 non-HIV related Kaposi Sarcoma patients with comparison to HIV-related Kaposi Sarcoma. Mod Pathol. 2008;21:572-582.
- Ross RK, Casagrande JT, Dworsky RL, et al. Kaposi’s sarcoma in Los Angeles, California. J Natl Cancer Inst. 1985;75:1011-1015.
Practice Points
- Classic Kaposi sarcoma is an indolent neoplasm that can be diagnosed in immunocompetent females of Mexican descent.
- Common diagnoses appearing in skin of color may appear morphologically disparate, and a high index of suspicion is needed for correct diagnosis.
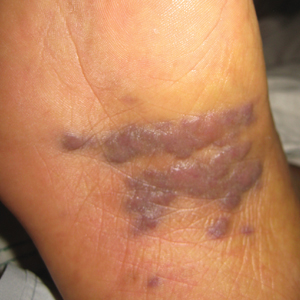
Antineutrophil Cytoplasmic Antibody Vasculitis Induced by Hydralazine
To the Editor:
Hydralazine-induced antineutrophil cytoplasmic antibody vasculitis (HIAV) is a rare side effect that may develop in patients treated with hydralazine. Without early recognition and hydralazine cessation, patients often develop acute renal failure and pulmonary hemorrhage that may result in death. We present a case of HIAV.
A 67-year-old woman presented with progressive, tense, hemorrhagic, and necrotic bullae on both sides of the face and neck as well as the extremities of 2 weeks’ duration. She had a history of hypertension and a thyroid nodule after unilateral thyroid lobectomy. A review of symptoms was positive for worsening dyspnea and progressive generalized weakness. Noteworthy medications included amlodipine, metoprolol, levothyroxine, and oral hydralazine 75 mg 3 times daily for 13 months.
Bullae first appeared on the patient’s scalp and quickly progressed with a cephalocaudal pattern with a propensity for the eyes, nostrils, and labial mucosa (Figure 1). The tongue was covered by an eschar, and she had diffuse periorbital edema. Additionally, concentric purpuric patches were noted on the thighs and lower legs (Figure 2).
Pertinent laboratory findings included a positive antinuclear antibody titer of 1:320 and perinuclear antineutrophil cytoplasmic antibody (ANCA) titer of 1:160, along with an elevated serum creatinine level (2.31 mg/dL [reference range, 0.6–1.2 mg/dL]). Bilateral perihilar infiltrates with bilateral pleural effusions were noted on a chest radiograph.
While hospitalized, she developed pulmonary hemorrhages and a progressive decline in respiratory status. She subsequently was admitted to the medical intensive care unit. Aggressive support was administered, and several skin biopsy specimens were obtained along with an endobronchial biopsy of the right middle lobe.
Skin histopathology revealed a necrotic vasculitis (Figure 3). Direct immunofluorescence was not performed. Lung histopathology showed fragments of bronchial tissue with acute and chronic inflammation, focal necrosis, granulation tissue formation, edema, and squamous metaplasia. Together with the clinical history, these findings were consistent with HIAV.

Hydralazine was immediately discontinued, and the patient was started on 65 mg daily of intravenous methylprednisolone; methylprednisolone was later changed to oral prednisone 30 mg daily. Due to multiple organ involvement—lung and kidney—intravenous rituximab 375 mg/m2 every week for 4 weeks, per lymphoma protocol, was started. Within 2 weeks of beginning therapy, her renal function and respiratory status improved, and by week 4, the skin lesions had completely resolved. Although initially she did well on immunosuppressive therapy with resolution of all symptoms, the patient contracted Clostridium difficile–induced systemic inflammatory response syndrome after 5 weeks of therapy and died.
Hydralazine was first introduced in 1951 for adjunctive hypertension therapy due to its vasodilation effects.1-3 Since its introduction, it has been implicated in 2 important disease processes: HIAV and hydralazine-induced lupus.
Hydralazine-induced ANCA vasculitis was first documented in 1980; by 2011, multiple cases had been reported.1-7 Hydralazine-induced ANCA vasculitis has occurred in patients aged 11 to 79 years taking 50 to 300 mg daily. Symptom onset varies from 6 months to 14 years, with a mean exposure duration of 4.7 years and mean daily dose of 142 mg.1-7
Clinical manifestations range from less specific, such as fever, malaise, arthralgia, myalgia, and weight loss, to single tissue or organ involvement that may be fatal. The most frequent clinical features include kidney involvement (81%), cutaneous vasculitis (25%), arthralgia (24%), and pleuropulmonary involvement (19%). Cutaneous manifestations include but are not limited to palpable lower extremity purpura; morbilliform eruptions; and hemorrhagic blisters on the lower legs, arms, trunk, nasal septum, and uvula.1-4,8
The most commonly affected organ is the kidney, which commonly presents as hematuria, proteinuria, and elevated serum creatinine level. Histopathologically, patients most likely will have necrotizing and crescentic glomerulonephritis that is pauci-immune by immunofluorescence.7,9 The lungs are the next most commonly affected organ, with a classic presentation of cough, dyspnea, and hemoptysis in the setting of intra-alveolar hemorrhage.6,8 When both the kidneys and lungs are involved, the patient is said to have pulmonary-renal syndrome that is characterized by lung infiltrates or nodules with or without hemorrhage, hemoptysis, and pleuritis in the setting of glomerulonephritis.1,6
Clear data on incidence and prevalence of HIAV does not exist due to the rarity of the disease and the lack of prospective studies. To identify a clear incidence and prevalence, prospective longitudinal studies with larger cohorts along with better recognition and diagnosis are needed.2,8,10 A few predisposing risk factors have been identified, including older age, a cumulative dose of 100 g at the time of presentation, female sex, a history of thyroid disease, HLA-DR4 genotypes, slow hepatic acetylation, and the null gene for C4.1,3,5,9-11 Our patient was an older woman with a history of thyroid disease who had been taking oral hydralazine 75 mg 3 times daily for 13 months. During this 13-month duration, she had no dose adjustments.
Currently, the pathomechanism for HIAV is unclear and may be multifactorial. There are 4 main theories2,8-10,12,13:
1. Hydralazine and its metabolites accumulate inside neutrophils, then subsequently bind and alter the configuration of myeloperoxidase (MPO). This alteration leads to spreading of the autoimmune response to other autoantigens, making neutrophil proteins (eg, elastase, lactoferrin, nuclear antigens) immunogenic.
2. Hydralazine binds MPO in neutrophils, creating cytotoxic products that induce neutrophil apoptosis. Neutrophil apoptosis without priming then results in ANCA antigen presence on the neutrophil cell membrane and the formation of MPO-ANCA. Myeloperoxidase-ANCA then binds to these membrane-bound antigens that cause self-perpetuating, constitutive activation through cross-linking with proteinase 3 or MPO and Fcγ receptors.
3. Activated neutrophils in the presence of hydrogen peroxidase release MPO that converts hydralazine into a cytotoxic product that is immunogenic for T cells that activate ANCA-producing B cells.
4. Histone H3 trimethyl Lys27 (H3K27me3) levels are perturbed in HIAV, which leads to aberrant gene silencing of proteinase 3 and MPO.In contrast, the demethylase Jumonji domain-containing protein 3 for the H3K27me3 histone is increased in patients without HIAV. Based on this data and the data showing a role for hydralazine in reversing epigenetic silencing of tumor suppressor genes in cancer cells,13 it has been proposed that hydralazine may reverse epigenetic silencing of proteinase 3 and MPO.
Diagnosing HIAV is still difficult because physicians do not recognize the drug as the etiologic agent, there is extensive variability in duration between starting the drug and onset of symptoms, and there often is a failure to order the appropriate laboratory and invasive tests needed for evaluation and diagnosis.3,5,8,10,12 Despite these difficulties, a set of criteria and practices for diagnosis are delineated in Table 1, with the key diagnostic feature being resolution with hydralazine cessation.1,5,7,8,12
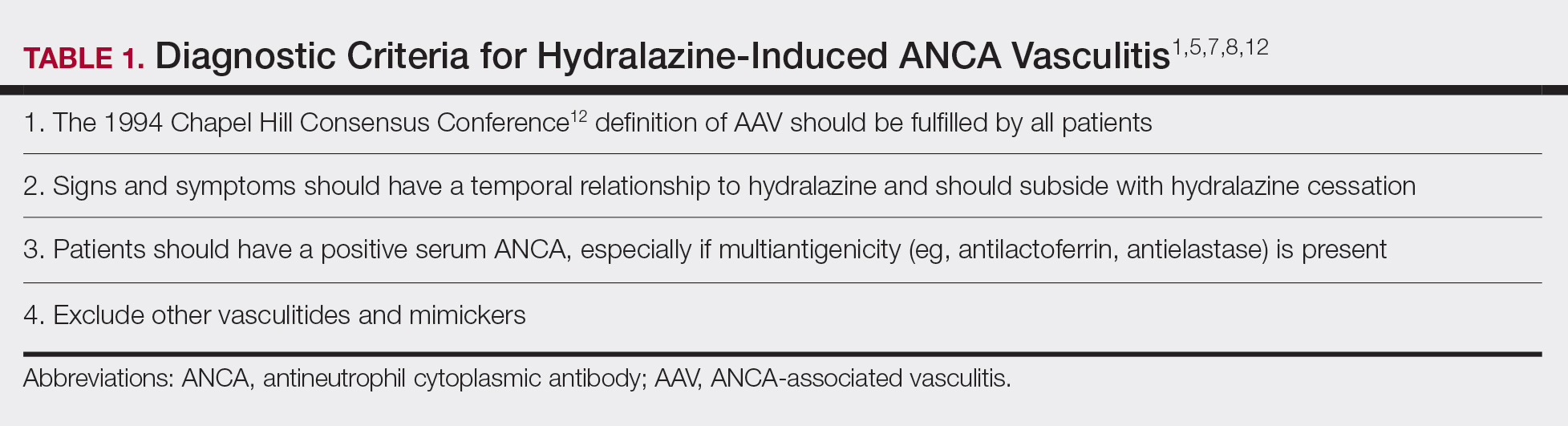
A comprehensive drug history from at least 6 months prior to presentation is essential. Biopsies also are strongly encouraged to confirm the presence of vasculitis and to determine its severity.8,12 If renal biopsies are performed, they typically show scant IgG, IgM, and C3 deposition that is characteristic of ANCA-positive pauci-immune glomerulonephritis. Compared to hydralazine-induced lupus, renal involvement in the setting of HIAV has a relative lack of immunoglobulin and complement deposition with histopathology and immunostaining.14
Laboratory test results including serum MPO-ANCA (perinuclear ANCA) with coexisting elastase and/or lactoferrin autoantibodies is characteristic of HIAV. Antinuclear antibody, antihistone, anti–double-stranded DNA, and antiphospholipid antibodies along with low complement levels also may be present.2,4,9,10,13,15 It is recommended that ANCA assays combine indirect immunofluorescence with antigen-specific enzyme-linked immunosorbent assay.8 With respect to its idiopathic counterpart, patients may only present with MPO-ANCA, while other aforementioned antibodies (eg, antihistone, anti–double-stranded DNA) are rarely found or are entirely absent.2,9 Patients with HIAV often have higher titers of MPO-ANCA.9,15 In hydralazine-induced lupus, patients rarely have MPO-ANCA.
When a diagnosis of HIAV is made, it cannot be confirmed until hydralazine is discontinued and the patient’s symptoms resolve. Therefore, it is both diagnostic and therapeutic to discontinue hydralazine when HIAV is suspected. If recognized when the patient is only presenting with nonspecific symptoms, simple hydralazine cessation may be all that is needed; however, because recognition and diagnosis of HIAV is difficult, most patients present when the disease is severe and has progressed to organ involvement.8-10
Treatment recommendations are highlighted in Table 2.8,9,12 Glucocorticoid therapy is believed to work by preventing T-cell and B-cell maturation needed to produce MPO-ANCA. Rituximab, on the other hand, is suspected to act by clearing the peripheral blood of MPO-ANCA B cells.12,16 Of note, patients with HIAV are different from their idiopathic counterparts because they usually need shorter courses of immunosuppressive therapy, long-term maintenance usually is unnecessary, and their prognosis generally is good if the offending agent is withdrawn.7-9,12 Once the appropriate therapy is instituted, vasculitic manifestations are expected to resolve 10 days to 8 months after hydralazine cessation; however, a response often is seen within 1 to 4 weeks after initiation of systemic treatment.4,8 Serum ANCA should be monitored, and there should be surveillance for the emergence of a chronic underlying vasculitis.8,12
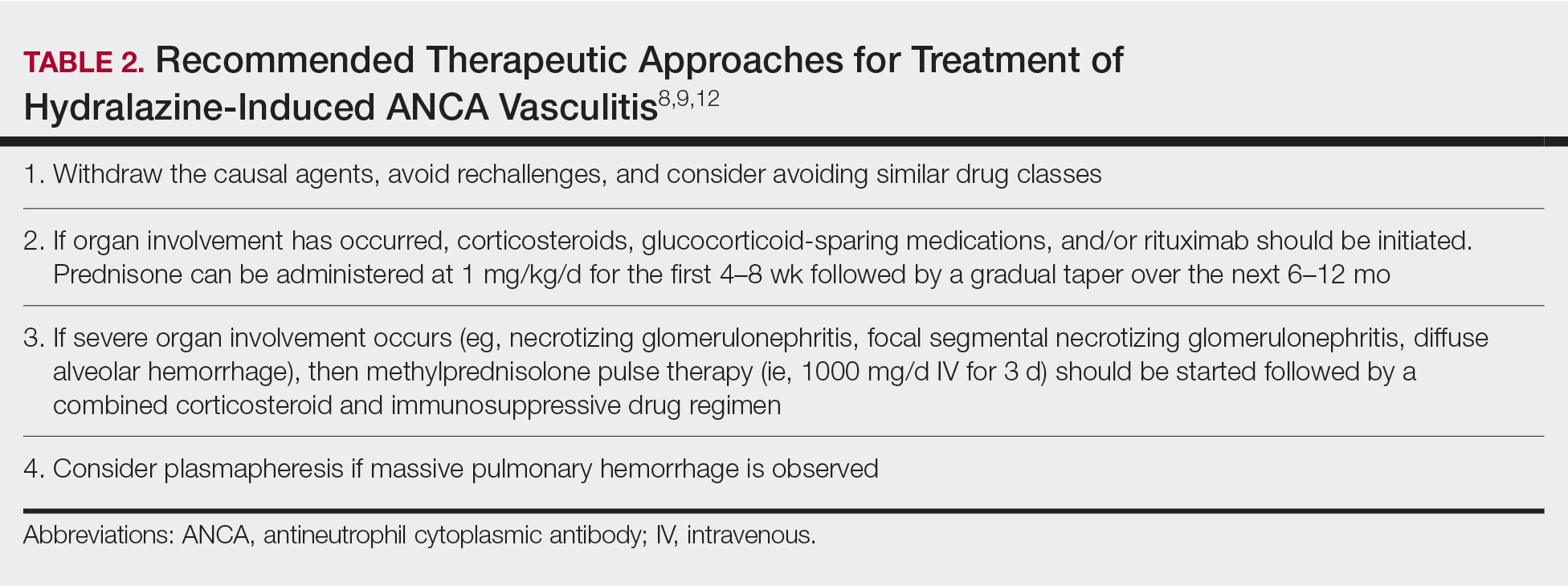
Our patient highlights the importance of identifying individuals at risk for HIAV. We seek to increase recognition of this entity, as it is not commonly seen in a dermatologic setting and is associated with high morbidity and mortality, as seen in our patient.
- Yokogawa N, Vivino FB. Hydralazine-induced autoimmune disease: comparison to idiopathic lupus and ANCA-positive vasculitis. Mod Rheumatol. 2009;19:338-347.
- Agarwal G, Sultan G, Werner SL, et al. Hydralazine induces myeloperoxidase and proteinase 3 anti-neutrophil cytoplasmic antibody vasculitis and leads to pulmonary renal syndrome. Case Rep Nephrol. 2014;2014:868590.
- Keasberry J, Frazier J, Isbel NM, et al. Hydralazine-induced anti-neutrophilic cytoplasmic antibody-positive renal vasculitis presenting with a vasculitic syndrome, acute nephritis and a puzzling skin rash: a case report. J Med Case Rep. 2013;7:20.
- ten Holder SM, Joy MS, Falk RJ. Cutaneous and systemic manifestations of drug-induced vasculitis. Ann Pharmacother. 2002;36:130-147.
- Namas R, Rubin B, Adwar W, et al. A challenging twist in pulmonary renal syndrome. Case Rep Rheumatol. 2014;2014:516362.
- Dobre M, Wish J, Negrea L. Hydralazine-induced ANCA-positive pauci-immune glomerulonephritis. Ren Fail. 2009;31:745-748.
- Hogan JJ, Markowitz GS, Radhakrishnan J. Drug-induced glomerular disease: immune-mediated injury. Clin J Am Soc Nephrol. 2015;10:1300-1310.
- Radic M, Martinovic Kaliterna D, Radic J. Drug-induced vasculitis: a clinical and pathological review. Neth J Med. 2012;70:12-17.
- Babar F, Posner JN, Obah EA. Hydralazine-induced pauci-immune glomerulonephritis: intriguing case series misleading diagnoses. J Community Hosp Intern Med Perspect. 2016;6:30632.
- Marina VP, Malhotra D, Kaw D. Hydralazine-induced ANCA vasculitis with pulmonary renal syndrome: a rare clinical presentation. Int Urol Nephrol. 2012;44:1907-1909.
- Magro CM. Associated ANCA positive vasculitis. The Dermatologist. 2015;23(7). http://www.the-dermatologist.com/content/associated-anca-positive-vasculitis. Accessed January 30, 2020.
- Gao Y, Zhao MH. Review article: Drug-induced anti-neutrophil cytoplasmic antibody-associated vasculitis. Nephrology (Carlton). 2009;14:33-41.
- Grau RG. Drug-induced vasculitis: new insights and a changing lineup of suspects. Curr Rheumatol Rep. 2015;17:71.
- Sangala N, Lee RW, Horsfield C, et al. Combined ANCA-associated vasculitis and lupus syndrome following prolonged use of hydralazine: a timely reminder of an old foe. Int Urol Nephrol. 2010;42:503-506.
- Choi HK, Merkel PA, Walker AM, et al. Drug-associated antineutrophil cytoplasmic antibody-positive vasculitis: prevalence among patients with high titers of antimyeloperoxidase antibodies. Arthritis Rheum. 2000;43:405-413.
- Coutinho AE, Chapman KE. The anti-inflammatory and immunosuppressive effects of glucocorticoids, recent developments and mechanistic insights. Mol Cell Endocrinol. 2011;335:2-13.
To the Editor:
Hydralazine-induced antineutrophil cytoplasmic antibody vasculitis (HIAV) is a rare side effect that may develop in patients treated with hydralazine. Without early recognition and hydralazine cessation, patients often develop acute renal failure and pulmonary hemorrhage that may result in death. We present a case of HIAV.
A 67-year-old woman presented with progressive, tense, hemorrhagic, and necrotic bullae on both sides of the face and neck as well as the extremities of 2 weeks’ duration. She had a history of hypertension and a thyroid nodule after unilateral thyroid lobectomy. A review of symptoms was positive for worsening dyspnea and progressive generalized weakness. Noteworthy medications included amlodipine, metoprolol, levothyroxine, and oral hydralazine 75 mg 3 times daily for 13 months.
Bullae first appeared on the patient’s scalp and quickly progressed with a cephalocaudal pattern with a propensity for the eyes, nostrils, and labial mucosa (Figure 1). The tongue was covered by an eschar, and she had diffuse periorbital edema. Additionally, concentric purpuric patches were noted on the thighs and lower legs (Figure 2).
Pertinent laboratory findings included a positive antinuclear antibody titer of 1:320 and perinuclear antineutrophil cytoplasmic antibody (ANCA) titer of 1:160, along with an elevated serum creatinine level (2.31 mg/dL [reference range, 0.6–1.2 mg/dL]). Bilateral perihilar infiltrates with bilateral pleural effusions were noted on a chest radiograph.
While hospitalized, she developed pulmonary hemorrhages and a progressive decline in respiratory status. She subsequently was admitted to the medical intensive care unit. Aggressive support was administered, and several skin biopsy specimens were obtained along with an endobronchial biopsy of the right middle lobe.
Skin histopathology revealed a necrotic vasculitis (Figure 3). Direct immunofluorescence was not performed. Lung histopathology showed fragments of bronchial tissue with acute and chronic inflammation, focal necrosis, granulation tissue formation, edema, and squamous metaplasia. Together with the clinical history, these findings were consistent with HIAV.
Hydralazine was immediately discontinued, and the patient was started on 65 mg daily of intravenous methylprednisolone; methylprednisolone was later changed to oral prednisone 30 mg daily. Due to multiple organ involvement—lung and kidney—intravenous rituximab 375 mg/m2 every week for 4 weeks, per lymphoma protocol, was started. Within 2 weeks of beginning therapy, her renal function and respiratory status improved, and by week 4, the skin lesions had completely resolved. Although initially she did well on immunosuppressive therapy with resolution of all symptoms, the patient contracted Clostridium difficile–induced systemic inflammatory response syndrome after 5 weeks of therapy and died.
Hydralazine was first introduced in 1951 for adjunctive hypertension therapy due to its vasodilation effects.1-3 Since its introduction, it has been implicated in 2 important disease processes: HIAV and hydralazine-induced lupus.
Hydralazine-induced ANCA vasculitis was first documented in 1980; by 2011, multiple cases had been reported.1-7 Hydralazine-induced ANCA vasculitis has occurred in patients aged 11 to 79 years taking 50 to 300 mg daily. Symptom onset varies from 6 months to 14 years, with a mean exposure duration of 4.7 years and mean daily dose of 142 mg.1-7
Clinical manifestations range from less specific, such as fever, malaise, arthralgia, myalgia, and weight loss, to single tissue or organ involvement that may be fatal. The most frequent clinical features include kidney involvement (81%), cutaneous vasculitis (25%), arthralgia (24%), and pleuropulmonary involvement (19%). Cutaneous manifestations include but are not limited to palpable lower extremity purpura; morbilliform eruptions; and hemorrhagic blisters on the lower legs, arms, trunk, nasal septum, and uvula.1-4,8
The most commonly affected organ is the kidney, which commonly presents as hematuria, proteinuria, and elevated serum creatinine level. Histopathologically, patients most likely will have necrotizing and crescentic glomerulonephritis that is pauci-immune by immunofluorescence.7,9 The lungs are the next most commonly affected organ, with a classic presentation of cough, dyspnea, and hemoptysis in the setting of intra-alveolar hemorrhage.6,8 When both the kidneys and lungs are involved, the patient is said to have pulmonary-renal syndrome that is characterized by lung infiltrates or nodules with or without hemorrhage, hemoptysis, and pleuritis in the setting of glomerulonephritis.1,6
Clear data on incidence and prevalence of HIAV does not exist due to the rarity of the disease and the lack of prospective studies. To identify a clear incidence and prevalence, prospective longitudinal studies with larger cohorts along with better recognition and diagnosis are needed.2,8,10 A few predisposing risk factors have been identified, including older age, a cumulative dose of 100 g at the time of presentation, female sex, a history of thyroid disease, HLA-DR4 genotypes, slow hepatic acetylation, and the null gene for C4.1,3,5,9-11 Our patient was an older woman with a history of thyroid disease who had been taking oral hydralazine 75 mg 3 times daily for 13 months. During this 13-month duration, she had no dose adjustments.
Currently, the pathomechanism for HIAV is unclear and may be multifactorial. There are 4 main theories2,8-10,12,13:
1. Hydralazine and its metabolites accumulate inside neutrophils, then subsequently bind and alter the configuration of myeloperoxidase (MPO). This alteration leads to spreading of the autoimmune response to other autoantigens, making neutrophil proteins (eg, elastase, lactoferrin, nuclear antigens) immunogenic.
2. Hydralazine binds MPO in neutrophils, creating cytotoxic products that induce neutrophil apoptosis. Neutrophil apoptosis without priming then results in ANCA antigen presence on the neutrophil cell membrane and the formation of MPO-ANCA. Myeloperoxidase-ANCA then binds to these membrane-bound antigens that cause self-perpetuating, constitutive activation through cross-linking with proteinase 3 or MPO and Fcγ receptors.
3. Activated neutrophils in the presence of hydrogen peroxidase release MPO that converts hydralazine into a cytotoxic product that is immunogenic for T cells that activate ANCA-producing B cells.
4. Histone H3 trimethyl Lys27 (H3K27me3) levels are perturbed in HIAV, which leads to aberrant gene silencing of proteinase 3 and MPO.In contrast, the demethylase Jumonji domain-containing protein 3 for the H3K27me3 histone is increased in patients without HIAV. Based on this data and the data showing a role for hydralazine in reversing epigenetic silencing of tumor suppressor genes in cancer cells,13 it has been proposed that hydralazine may reverse epigenetic silencing of proteinase 3 and MPO.
Diagnosing HIAV is still difficult because physicians do not recognize the drug as the etiologic agent, there is extensive variability in duration between starting the drug and onset of symptoms, and there often is a failure to order the appropriate laboratory and invasive tests needed for evaluation and diagnosis.3,5,8,10,12 Despite these difficulties, a set of criteria and practices for diagnosis are delineated in Table 1, with the key diagnostic feature being resolution with hydralazine cessation.1,5,7,8,12
A comprehensive drug history from at least 6 months prior to presentation is essential. Biopsies also are strongly encouraged to confirm the presence of vasculitis and to determine its severity.8,12 If renal biopsies are performed, they typically show scant IgG, IgM, and C3 deposition that is characteristic of ANCA-positive pauci-immune glomerulonephritis. Compared to hydralazine-induced lupus, renal involvement in the setting of HIAV has a relative lack of immunoglobulin and complement deposition with histopathology and immunostaining.14
Laboratory test results including serum MPO-ANCA (perinuclear ANCA) with coexisting elastase and/or lactoferrin autoantibodies is characteristic of HIAV. Antinuclear antibody, antihistone, anti–double-stranded DNA, and antiphospholipid antibodies along with low complement levels also may be present.2,4,9,10,13,15 It is recommended that ANCA assays combine indirect immunofluorescence with antigen-specific enzyme-linked immunosorbent assay.8 With respect to its idiopathic counterpart, patients may only present with MPO-ANCA, while other aforementioned antibodies (eg, antihistone, anti–double-stranded DNA) are rarely found or are entirely absent.2,9 Patients with HIAV often have higher titers of MPO-ANCA.9,15 In hydralazine-induced lupus, patients rarely have MPO-ANCA.
When a diagnosis of HIAV is made, it cannot be confirmed until hydralazine is discontinued and the patient’s symptoms resolve. Therefore, it is both diagnostic and therapeutic to discontinue hydralazine when HIAV is suspected. If recognized when the patient is only presenting with nonspecific symptoms, simple hydralazine cessation may be all that is needed; however, because recognition and diagnosis of HIAV is difficult, most patients present when the disease is severe and has progressed to organ involvement.8-10
Treatment recommendations are highlighted in Table 2.8,9,12 Glucocorticoid therapy is believed to work by preventing T-cell and B-cell maturation needed to produce MPO-ANCA. Rituximab, on the other hand, is suspected to act by clearing the peripheral blood of MPO-ANCA B cells.12,16 Of note, patients with HIAV are different from their idiopathic counterparts because they usually need shorter courses of immunosuppressive therapy, long-term maintenance usually is unnecessary, and their prognosis generally is good if the offending agent is withdrawn.7-9,12 Once the appropriate therapy is instituted, vasculitic manifestations are expected to resolve 10 days to 8 months after hydralazine cessation; however, a response often is seen within 1 to 4 weeks after initiation of systemic treatment.4,8 Serum ANCA should be monitored, and there should be surveillance for the emergence of a chronic underlying vasculitis.8,12
Our patient highlights the importance of identifying individuals at risk for HIAV. We seek to increase recognition of this entity, as it is not commonly seen in a dermatologic setting and is associated with high morbidity and mortality, as seen in our patient.
To the Editor:
Hydralazine-induced antineutrophil cytoplasmic antibody vasculitis (HIAV) is a rare side effect that may develop in patients treated with hydralazine. Without early recognition and hydralazine cessation, patients often develop acute renal failure and pulmonary hemorrhage that may result in death. We present a case of HIAV.
A 67-year-old woman presented with progressive, tense, hemorrhagic, and necrotic bullae on both sides of the face and neck as well as the extremities of 2 weeks’ duration. She had a history of hypertension and a thyroid nodule after unilateral thyroid lobectomy. A review of symptoms was positive for worsening dyspnea and progressive generalized weakness. Noteworthy medications included amlodipine, metoprolol, levothyroxine, and oral hydralazine 75 mg 3 times daily for 13 months.
Bullae first appeared on the patient’s scalp and quickly progressed with a cephalocaudal pattern with a propensity for the eyes, nostrils, and labial mucosa (Figure 1). The tongue was covered by an eschar, and she had diffuse periorbital edema. Additionally, concentric purpuric patches were noted on the thighs and lower legs (Figure 2).
Pertinent laboratory findings included a positive antinuclear antibody titer of 1:320 and perinuclear antineutrophil cytoplasmic antibody (ANCA) titer of 1:160, along with an elevated serum creatinine level (2.31 mg/dL [reference range, 0.6–1.2 mg/dL]). Bilateral perihilar infiltrates with bilateral pleural effusions were noted on a chest radiograph.
While hospitalized, she developed pulmonary hemorrhages and a progressive decline in respiratory status. She subsequently was admitted to the medical intensive care unit. Aggressive support was administered, and several skin biopsy specimens were obtained along with an endobronchial biopsy of the right middle lobe.
Skin histopathology revealed a necrotic vasculitis (Figure 3). Direct immunofluorescence was not performed. Lung histopathology showed fragments of bronchial tissue with acute and chronic inflammation, focal necrosis, granulation tissue formation, edema, and squamous metaplasia. Together with the clinical history, these findings were consistent with HIAV.
Hydralazine was immediately discontinued, and the patient was started on 65 mg daily of intravenous methylprednisolone; methylprednisolone was later changed to oral prednisone 30 mg daily. Due to multiple organ involvement—lung and kidney—intravenous rituximab 375 mg/m2 every week for 4 weeks, per lymphoma protocol, was started. Within 2 weeks of beginning therapy, her renal function and respiratory status improved, and by week 4, the skin lesions had completely resolved. Although initially she did well on immunosuppressive therapy with resolution of all symptoms, the patient contracted Clostridium difficile–induced systemic inflammatory response syndrome after 5 weeks of therapy and died.
Hydralazine was first introduced in 1951 for adjunctive hypertension therapy due to its vasodilation effects.1-3 Since its introduction, it has been implicated in 2 important disease processes: HIAV and hydralazine-induced lupus.
Hydralazine-induced ANCA vasculitis was first documented in 1980; by 2011, multiple cases had been reported.1-7 Hydralazine-induced ANCA vasculitis has occurred in patients aged 11 to 79 years taking 50 to 300 mg daily. Symptom onset varies from 6 months to 14 years, with a mean exposure duration of 4.7 years and mean daily dose of 142 mg.1-7
Clinical manifestations range from less specific, such as fever, malaise, arthralgia, myalgia, and weight loss, to single tissue or organ involvement that may be fatal. The most frequent clinical features include kidney involvement (81%), cutaneous vasculitis (25%), arthralgia (24%), and pleuropulmonary involvement (19%). Cutaneous manifestations include but are not limited to palpable lower extremity purpura; morbilliform eruptions; and hemorrhagic blisters on the lower legs, arms, trunk, nasal septum, and uvula.1-4,8
The most commonly affected organ is the kidney, which commonly presents as hematuria, proteinuria, and elevated serum creatinine level. Histopathologically, patients most likely will have necrotizing and crescentic glomerulonephritis that is pauci-immune by immunofluorescence.7,9 The lungs are the next most commonly affected organ, with a classic presentation of cough, dyspnea, and hemoptysis in the setting of intra-alveolar hemorrhage.6,8 When both the kidneys and lungs are involved, the patient is said to have pulmonary-renal syndrome that is characterized by lung infiltrates or nodules with or without hemorrhage, hemoptysis, and pleuritis in the setting of glomerulonephritis.1,6
Clear data on incidence and prevalence of HIAV does not exist due to the rarity of the disease and the lack of prospective studies. To identify a clear incidence and prevalence, prospective longitudinal studies with larger cohorts along with better recognition and diagnosis are needed.2,8,10 A few predisposing risk factors have been identified, including older age, a cumulative dose of 100 g at the time of presentation, female sex, a history of thyroid disease, HLA-DR4 genotypes, slow hepatic acetylation, and the null gene for C4.1,3,5,9-11 Our patient was an older woman with a history of thyroid disease who had been taking oral hydralazine 75 mg 3 times daily for 13 months. During this 13-month duration, she had no dose adjustments.
Currently, the pathomechanism for HIAV is unclear and may be multifactorial. There are 4 main theories2,8-10,12,13:
1. Hydralazine and its metabolites accumulate inside neutrophils, then subsequently bind and alter the configuration of myeloperoxidase (MPO). This alteration leads to spreading of the autoimmune response to other autoantigens, making neutrophil proteins (eg, elastase, lactoferrin, nuclear antigens) immunogenic.
2. Hydralazine binds MPO in neutrophils, creating cytotoxic products that induce neutrophil apoptosis. Neutrophil apoptosis without priming then results in ANCA antigen presence on the neutrophil cell membrane and the formation of MPO-ANCA. Myeloperoxidase-ANCA then binds to these membrane-bound antigens that cause self-perpetuating, constitutive activation through cross-linking with proteinase 3 or MPO and Fcγ receptors.
3. Activated neutrophils in the presence of hydrogen peroxidase release MPO that converts hydralazine into a cytotoxic product that is immunogenic for T cells that activate ANCA-producing B cells.
4. Histone H3 trimethyl Lys27 (H3K27me3) levels are perturbed in HIAV, which leads to aberrant gene silencing of proteinase 3 and MPO.In contrast, the demethylase Jumonji domain-containing protein 3 for the H3K27me3 histone is increased in patients without HIAV. Based on this data and the data showing a role for hydralazine in reversing epigenetic silencing of tumor suppressor genes in cancer cells,13 it has been proposed that hydralazine may reverse epigenetic silencing of proteinase 3 and MPO.
Diagnosing HIAV is still difficult because physicians do not recognize the drug as the etiologic agent, there is extensive variability in duration between starting the drug and onset of symptoms, and there often is a failure to order the appropriate laboratory and invasive tests needed for evaluation and diagnosis.3,5,8,10,12 Despite these difficulties, a set of criteria and practices for diagnosis are delineated in Table 1, with the key diagnostic feature being resolution with hydralazine cessation.1,5,7,8,12
A comprehensive drug history from at least 6 months prior to presentation is essential. Biopsies also are strongly encouraged to confirm the presence of vasculitis and to determine its severity.8,12 If renal biopsies are performed, they typically show scant IgG, IgM, and C3 deposition that is characteristic of ANCA-positive pauci-immune glomerulonephritis. Compared to hydralazine-induced lupus, renal involvement in the setting of HIAV has a relative lack of immunoglobulin and complement deposition with histopathology and immunostaining.14
Laboratory test results including serum MPO-ANCA (perinuclear ANCA) with coexisting elastase and/or lactoferrin autoantibodies is characteristic of HIAV. Antinuclear antibody, antihistone, anti–double-stranded DNA, and antiphospholipid antibodies along with low complement levels also may be present.2,4,9,10,13,15 It is recommended that ANCA assays combine indirect immunofluorescence with antigen-specific enzyme-linked immunosorbent assay.8 With respect to its idiopathic counterpart, patients may only present with MPO-ANCA, while other aforementioned antibodies (eg, antihistone, anti–double-stranded DNA) are rarely found or are entirely absent.2,9 Patients with HIAV often have higher titers of MPO-ANCA.9,15 In hydralazine-induced lupus, patients rarely have MPO-ANCA.
When a diagnosis of HIAV is made, it cannot be confirmed until hydralazine is discontinued and the patient’s symptoms resolve. Therefore, it is both diagnostic and therapeutic to discontinue hydralazine when HIAV is suspected. If recognized when the patient is only presenting with nonspecific symptoms, simple hydralazine cessation may be all that is needed; however, because recognition and diagnosis of HIAV is difficult, most patients present when the disease is severe and has progressed to organ involvement.8-10
Treatment recommendations are highlighted in Table 2.8,9,12 Glucocorticoid therapy is believed to work by preventing T-cell and B-cell maturation needed to produce MPO-ANCA. Rituximab, on the other hand, is suspected to act by clearing the peripheral blood of MPO-ANCA B cells.12,16 Of note, patients with HIAV are different from their idiopathic counterparts because they usually need shorter courses of immunosuppressive therapy, long-term maintenance usually is unnecessary, and their prognosis generally is good if the offending agent is withdrawn.7-9,12 Once the appropriate therapy is instituted, vasculitic manifestations are expected to resolve 10 days to 8 months after hydralazine cessation; however, a response often is seen within 1 to 4 weeks after initiation of systemic treatment.4,8 Serum ANCA should be monitored, and there should be surveillance for the emergence of a chronic underlying vasculitis.8,12
Our patient highlights the importance of identifying individuals at risk for HIAV. We seek to increase recognition of this entity, as it is not commonly seen in a dermatologic setting and is associated with high morbidity and mortality, as seen in our patient.
- Yokogawa N, Vivino FB. Hydralazine-induced autoimmune disease: comparison to idiopathic lupus and ANCA-positive vasculitis. Mod Rheumatol. 2009;19:338-347.
- Agarwal G, Sultan G, Werner SL, et al. Hydralazine induces myeloperoxidase and proteinase 3 anti-neutrophil cytoplasmic antibody vasculitis and leads to pulmonary renal syndrome. Case Rep Nephrol. 2014;2014:868590.
- Keasberry J, Frazier J, Isbel NM, et al. Hydralazine-induced anti-neutrophilic cytoplasmic antibody-positive renal vasculitis presenting with a vasculitic syndrome, acute nephritis and a puzzling skin rash: a case report. J Med Case Rep. 2013;7:20.
- ten Holder SM, Joy MS, Falk RJ. Cutaneous and systemic manifestations of drug-induced vasculitis. Ann Pharmacother. 2002;36:130-147.
- Namas R, Rubin B, Adwar W, et al. A challenging twist in pulmonary renal syndrome. Case Rep Rheumatol. 2014;2014:516362.
- Dobre M, Wish J, Negrea L. Hydralazine-induced ANCA-positive pauci-immune glomerulonephritis. Ren Fail. 2009;31:745-748.
- Hogan JJ, Markowitz GS, Radhakrishnan J. Drug-induced glomerular disease: immune-mediated injury. Clin J Am Soc Nephrol. 2015;10:1300-1310.
- Radic M, Martinovic Kaliterna D, Radic J. Drug-induced vasculitis: a clinical and pathological review. Neth J Med. 2012;70:12-17.
- Babar F, Posner JN, Obah EA. Hydralazine-induced pauci-immune glomerulonephritis: intriguing case series misleading diagnoses. J Community Hosp Intern Med Perspect. 2016;6:30632.
- Marina VP, Malhotra D, Kaw D. Hydralazine-induced ANCA vasculitis with pulmonary renal syndrome: a rare clinical presentation. Int Urol Nephrol. 2012;44:1907-1909.
- Magro CM. Associated ANCA positive vasculitis. The Dermatologist. 2015;23(7). http://www.the-dermatologist.com/content/associated-anca-positive-vasculitis. Accessed January 30, 2020.
- Gao Y, Zhao MH. Review article: Drug-induced anti-neutrophil cytoplasmic antibody-associated vasculitis. Nephrology (Carlton). 2009;14:33-41.
- Grau RG. Drug-induced vasculitis: new insights and a changing lineup of suspects. Curr Rheumatol Rep. 2015;17:71.
- Sangala N, Lee RW, Horsfield C, et al. Combined ANCA-associated vasculitis and lupus syndrome following prolonged use of hydralazine: a timely reminder of an old foe. Int Urol Nephrol. 2010;42:503-506.
- Choi HK, Merkel PA, Walker AM, et al. Drug-associated antineutrophil cytoplasmic antibody-positive vasculitis: prevalence among patients with high titers of antimyeloperoxidase antibodies. Arthritis Rheum. 2000;43:405-413.
- Coutinho AE, Chapman KE. The anti-inflammatory and immunosuppressive effects of glucocorticoids, recent developments and mechanistic insights. Mol Cell Endocrinol. 2011;335:2-13.
- Yokogawa N, Vivino FB. Hydralazine-induced autoimmune disease: comparison to idiopathic lupus and ANCA-positive vasculitis. Mod Rheumatol. 2009;19:338-347.
- Agarwal G, Sultan G, Werner SL, et al. Hydralazine induces myeloperoxidase and proteinase 3 anti-neutrophil cytoplasmic antibody vasculitis and leads to pulmonary renal syndrome. Case Rep Nephrol. 2014;2014:868590.
- Keasberry J, Frazier J, Isbel NM, et al. Hydralazine-induced anti-neutrophilic cytoplasmic antibody-positive renal vasculitis presenting with a vasculitic syndrome, acute nephritis and a puzzling skin rash: a case report. J Med Case Rep. 2013;7:20.
- ten Holder SM, Joy MS, Falk RJ. Cutaneous and systemic manifestations of drug-induced vasculitis. Ann Pharmacother. 2002;36:130-147.
- Namas R, Rubin B, Adwar W, et al. A challenging twist in pulmonary renal syndrome. Case Rep Rheumatol. 2014;2014:516362.
- Dobre M, Wish J, Negrea L. Hydralazine-induced ANCA-positive pauci-immune glomerulonephritis. Ren Fail. 2009;31:745-748.
- Hogan JJ, Markowitz GS, Radhakrishnan J. Drug-induced glomerular disease: immune-mediated injury. Clin J Am Soc Nephrol. 2015;10:1300-1310.
- Radic M, Martinovic Kaliterna D, Radic J. Drug-induced vasculitis: a clinical and pathological review. Neth J Med. 2012;70:12-17.
- Babar F, Posner JN, Obah EA. Hydralazine-induced pauci-immune glomerulonephritis: intriguing case series misleading diagnoses. J Community Hosp Intern Med Perspect. 2016;6:30632.
- Marina VP, Malhotra D, Kaw D. Hydralazine-induced ANCA vasculitis with pulmonary renal syndrome: a rare clinical presentation. Int Urol Nephrol. 2012;44:1907-1909.
- Magro CM. Associated ANCA positive vasculitis. The Dermatologist. 2015;23(7). http://www.the-dermatologist.com/content/associated-anca-positive-vasculitis. Accessed January 30, 2020.
- Gao Y, Zhao MH. Review article: Drug-induced anti-neutrophil cytoplasmic antibody-associated vasculitis. Nephrology (Carlton). 2009;14:33-41.
- Grau RG. Drug-induced vasculitis: new insights and a changing lineup of suspects. Curr Rheumatol Rep. 2015;17:71.
- Sangala N, Lee RW, Horsfield C, et al. Combined ANCA-associated vasculitis and lupus syndrome following prolonged use of hydralazine: a timely reminder of an old foe. Int Urol Nephrol. 2010;42:503-506.
- Choi HK, Merkel PA, Walker AM, et al. Drug-associated antineutrophil cytoplasmic antibody-positive vasculitis: prevalence among patients with high titers of antimyeloperoxidase antibodies. Arthritis Rheum. 2000;43:405-413.
- Coutinho AE, Chapman KE. The anti-inflammatory and immunosuppressive effects of glucocorticoids, recent developments and mechanistic insights. Mol Cell Endocrinol. 2011;335:2-13.
Practice Points
- Hydralazine-induced antineutrophil cytoplasmic antibody vasculitis (HIAV) is a rare side effect of hydralazine treatment and can have notable morbidity and mortality.
- Incidence and prevalence of HIAV is unclear due to its rarity, but risk factors that have been identified are older age, a cumulative dose of 100 g of hydralazine at the time of presentation, female sex, thyroid disease, HLA-DR4 genotypes, slow hepatic acetylation, and the null gene for C4.
- Symptoms of HIAV can include fever, malaise, arthralgia, weight loss, or even involvement of organs such as the kidneys and lungs.
- If recognized early, cessation of hydralazine and supportive therapy generally are sufficient; however, severe cases may need management with high-dose corticosteroids, rituximab, and even plasmapheresis.
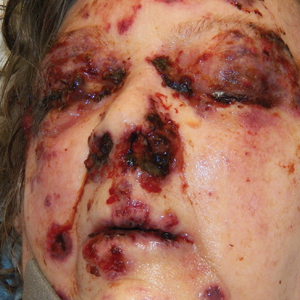
Pembrolizumab-Induced Lobular Panniculitis in the Setting of Metastatic Melanoma
To the Editor:
Pembrolizumab is an anti–programmed death receptor 1 humanized monoclonal antibody used for treating advanced or metastatic melanoma.1 It is associated with several immune-related adverse events because it blocks a T-cell receptor checkpoint.2 The most common dermatologic immune-related adverse event seen with anti–programmed death receptor 1 medications is a nonspecific morbilliform rash, usually seen after the second treatment cycle; however, pruritus, vitiligo, bullous disorders, and lichenoid reactions also have been reported.3 We report a case of pembrolizumab-induced, self-limited lobular panniculitis in a patient with metastatic melanoma.
A 37-year-old woman with malignant melanoma presented with tender, erythematous, subcutaneous nodules on the hips and legs of 2 weeks’ duration (Figure 1). Twelve years prior to the current presentation, she was diagnosed with metastases to the cecum, lung, and brain. A review of systems was otherwise negative. She had been receiving pembrolizumab infusions (2 mg/kg every 3 weeks) for the last 2.7 years as second-line therapy after previously undergoing chemotherapy, radiation, and resection. She was not taking oral contraceptives or other hormone-based medications and did not report any new medications.
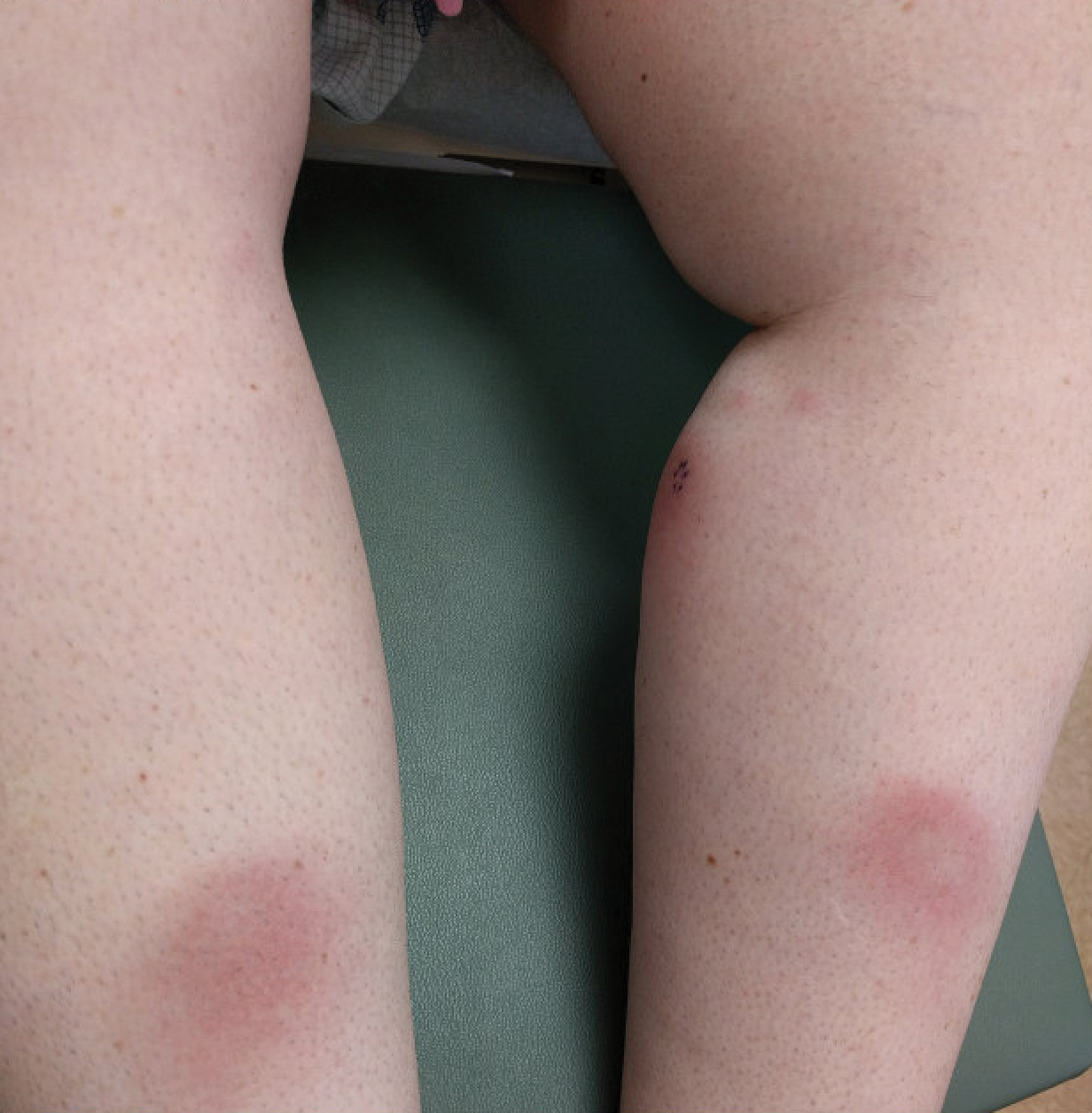
Laboratory testing was negative for infectious processes including Lyme disease, tuberculosis, and Streptococcus due to recent upper respiratory infection. Punch biopsy of a left shin lesion revealed a lobular panniculitis with lymphohistiocytic inflammation, a focal lymphocytic vasculitis, and small granulomas (Figure 2). Periodic acid–Schiff, Gram, and acid-fast bacilli stains were negative. After ruling out alternative causes, the etiology of the panniculitis was deemed to be a pembrolizumab side effect. The patient was treated conservatively with ibuprofen; pembrolizumab was not discontinued. Two weeks later, the panniculitis had resolved without additional treatment. She remains on pembrolizumab and is doing well.
Panniculitis is known to be associated with certain BRAF inhibitors used for the treatment of melanoma positive for the BRAF V600E mutation, including vemurafenib and dabrafenib.4,5 Reports of panniculitis in the setting of pembrolizumab are limited and are seen within the larger context of sarcoidosis. One patient on pembrolizumab for metastatic melanoma developed granulomatous lobular panniculitis with oligoarthritis, high fever, and hilar/mediastinal adenopathy, consistent with pembrolizumab-induced sarcoidosis. It developed after her second pembrolizumab infusion and resolved with prednisone and temporary pembrolizumab cessation.6 In another case, pembrolizumab triggered a flare of sarcoidosis with similar granulomatous subcutaneous nodules in a patient with stage IV lymphoma who was previously diagnosed with sarcoidosis but lacked cutaneous manifestations. The lesions resolved with prednisone therapy.7
Chest computed tomography was normal in our patient, and she reported no systemic symptoms. Additional laboratory studies to evaluate for sarcoidosis were not obtained. Furthermore, the lesions quickly resolved despite continued use of pembrolizumab. We report this case to highlight that pembrolizumab may induce an isolated, self-limited lobular panniculitis years after medication initiation.
- Poole RM. Pembrolizumab: first global approval. Drugs. 2014;74:1973-1981.
- Michot JM, Bigenwald C, Champiat S, et al. Immune-related adverse events with immune checkpoint blockade: a comprehensive review. Eur J Cancer. 2016;54:139-148.
- Naidoo J, Page DB, Li BT, et al. Toxicities of the anti-PD-1 and anti-PD-L1 immune checkpoint antibodies. Ann Oncol. 2016;27:1362.
- Boussemart L, Routier E, Mateus C, et al. Prospective study of cutaneous side-effects associated with the BRAF inhibitor vemurafenib: a study of 42 patients. Ann Oncol. 2013;24:1691-1697.
- Ramani NS, Curry JL, Kapil J, et al. Panniculitis with necrotizing granulomata in a patient on BRAF inhibitor (dabrafenib) therapy for metastatic melanoma. Am J Dermatopathol. 2015;37:E96-E99.
- Burillo-Martinez S, Morales-Raya C, Prieto-Barrios M, et al. Pembrolizumab-induced extensive panniculitis and nevus regression: two novel cutaneous manifestations of the post-immunotherapy granulomatous reactions spectrum. JAMA Dermatol. 2017;153:721-722.
- Cotliar J, Querfeld C, Boswell WJ, et al. Pembrolizumab-associated sarcoidosis. JAAD Case Rep. 2016;2:290-293.
To the Editor:
Pembrolizumab is an anti–programmed death receptor 1 humanized monoclonal antibody used for treating advanced or metastatic melanoma.1 It is associated with several immune-related adverse events because it blocks a T-cell receptor checkpoint.2 The most common dermatologic immune-related adverse event seen with anti–programmed death receptor 1 medications is a nonspecific morbilliform rash, usually seen after the second treatment cycle; however, pruritus, vitiligo, bullous disorders, and lichenoid reactions also have been reported.3 We report a case of pembrolizumab-induced, self-limited lobular panniculitis in a patient with metastatic melanoma.
A 37-year-old woman with malignant melanoma presented with tender, erythematous, subcutaneous nodules on the hips and legs of 2 weeks’ duration (Figure 1). Twelve years prior to the current presentation, she was diagnosed with metastases to the cecum, lung, and brain. A review of systems was otherwise negative. She had been receiving pembrolizumab infusions (2 mg/kg every 3 weeks) for the last 2.7 years as second-line therapy after previously undergoing chemotherapy, radiation, and resection. She was not taking oral contraceptives or other hormone-based medications and did not report any new medications.
Laboratory testing was negative for infectious processes including Lyme disease, tuberculosis, and Streptococcus due to recent upper respiratory infection. Punch biopsy of a left shin lesion revealed a lobular panniculitis with lymphohistiocytic inflammation, a focal lymphocytic vasculitis, and small granulomas (Figure 2). Periodic acid–Schiff, Gram, and acid-fast bacilli stains were negative. After ruling out alternative causes, the etiology of the panniculitis was deemed to be a pembrolizumab side effect. The patient was treated conservatively with ibuprofen; pembrolizumab was not discontinued. Two weeks later, the panniculitis had resolved without additional treatment. She remains on pembrolizumab and is doing well.
Panniculitis is known to be associated with certain BRAF inhibitors used for the treatment of melanoma positive for the BRAF V600E mutation, including vemurafenib and dabrafenib.4,5 Reports of panniculitis in the setting of pembrolizumab are limited and are seen within the larger context of sarcoidosis. One patient on pembrolizumab for metastatic melanoma developed granulomatous lobular panniculitis with oligoarthritis, high fever, and hilar/mediastinal adenopathy, consistent with pembrolizumab-induced sarcoidosis. It developed after her second pembrolizumab infusion and resolved with prednisone and temporary pembrolizumab cessation.6 In another case, pembrolizumab triggered a flare of sarcoidosis with similar granulomatous subcutaneous nodules in a patient with stage IV lymphoma who was previously diagnosed with sarcoidosis but lacked cutaneous manifestations. The lesions resolved with prednisone therapy.7
Chest computed tomography was normal in our patient, and she reported no systemic symptoms. Additional laboratory studies to evaluate for sarcoidosis were not obtained. Furthermore, the lesions quickly resolved despite continued use of pembrolizumab. We report this case to highlight that pembrolizumab may induce an isolated, self-limited lobular panniculitis years after medication initiation.
To the Editor:
Pembrolizumab is an anti–programmed death receptor 1 humanized monoclonal antibody used for treating advanced or metastatic melanoma.1 It is associated with several immune-related adverse events because it blocks a T-cell receptor checkpoint.2 The most common dermatologic immune-related adverse event seen with anti–programmed death receptor 1 medications is a nonspecific morbilliform rash, usually seen after the second treatment cycle; however, pruritus, vitiligo, bullous disorders, and lichenoid reactions also have been reported.3 We report a case of pembrolizumab-induced, self-limited lobular panniculitis in a patient with metastatic melanoma.
A 37-year-old woman with malignant melanoma presented with tender, erythematous, subcutaneous nodules on the hips and legs of 2 weeks’ duration (Figure 1). Twelve years prior to the current presentation, she was diagnosed with metastases to the cecum, lung, and brain. A review of systems was otherwise negative. She had been receiving pembrolizumab infusions (2 mg/kg every 3 weeks) for the last 2.7 years as second-line therapy after previously undergoing chemotherapy, radiation, and resection. She was not taking oral contraceptives or other hormone-based medications and did not report any new medications.
Laboratory testing was negative for infectious processes including Lyme disease, tuberculosis, and Streptococcus due to recent upper respiratory infection. Punch biopsy of a left shin lesion revealed a lobular panniculitis with lymphohistiocytic inflammation, a focal lymphocytic vasculitis, and small granulomas (Figure 2). Periodic acid–Schiff, Gram, and acid-fast bacilli stains were negative. After ruling out alternative causes, the etiology of the panniculitis was deemed to be a pembrolizumab side effect. The patient was treated conservatively with ibuprofen; pembrolizumab was not discontinued. Two weeks later, the panniculitis had resolved without additional treatment. She remains on pembrolizumab and is doing well.
Panniculitis is known to be associated with certain BRAF inhibitors used for the treatment of melanoma positive for the BRAF V600E mutation, including vemurafenib and dabrafenib.4,5 Reports of panniculitis in the setting of pembrolizumab are limited and are seen within the larger context of sarcoidosis. One patient on pembrolizumab for metastatic melanoma developed granulomatous lobular panniculitis with oligoarthritis, high fever, and hilar/mediastinal adenopathy, consistent with pembrolizumab-induced sarcoidosis. It developed after her second pembrolizumab infusion and resolved with prednisone and temporary pembrolizumab cessation.6 In another case, pembrolizumab triggered a flare of sarcoidosis with similar granulomatous subcutaneous nodules in a patient with stage IV lymphoma who was previously diagnosed with sarcoidosis but lacked cutaneous manifestations. The lesions resolved with prednisone therapy.7
Chest computed tomography was normal in our patient, and she reported no systemic symptoms. Additional laboratory studies to evaluate for sarcoidosis were not obtained. Furthermore, the lesions quickly resolved despite continued use of pembrolizumab. We report this case to highlight that pembrolizumab may induce an isolated, self-limited lobular panniculitis years after medication initiation.
- Poole RM. Pembrolizumab: first global approval. Drugs. 2014;74:1973-1981.
- Michot JM, Bigenwald C, Champiat S, et al. Immune-related adverse events with immune checkpoint blockade: a comprehensive review. Eur J Cancer. 2016;54:139-148.
- Naidoo J, Page DB, Li BT, et al. Toxicities of the anti-PD-1 and anti-PD-L1 immune checkpoint antibodies. Ann Oncol. 2016;27:1362.
- Boussemart L, Routier E, Mateus C, et al. Prospective study of cutaneous side-effects associated with the BRAF inhibitor vemurafenib: a study of 42 patients. Ann Oncol. 2013;24:1691-1697.
- Ramani NS, Curry JL, Kapil J, et al. Panniculitis with necrotizing granulomata in a patient on BRAF inhibitor (dabrafenib) therapy for metastatic melanoma. Am J Dermatopathol. 2015;37:E96-E99.
- Burillo-Martinez S, Morales-Raya C, Prieto-Barrios M, et al. Pembrolizumab-induced extensive panniculitis and nevus regression: two novel cutaneous manifestations of the post-immunotherapy granulomatous reactions spectrum. JAMA Dermatol. 2017;153:721-722.
- Cotliar J, Querfeld C, Boswell WJ, et al. Pembrolizumab-associated sarcoidosis. JAAD Case Rep. 2016;2:290-293.
- Poole RM. Pembrolizumab: first global approval. Drugs. 2014;74:1973-1981.
- Michot JM, Bigenwald C, Champiat S, et al. Immune-related adverse events with immune checkpoint blockade: a comprehensive review. Eur J Cancer. 2016;54:139-148.
- Naidoo J, Page DB, Li BT, et al. Toxicities of the anti-PD-1 and anti-PD-L1 immune checkpoint antibodies. Ann Oncol. 2016;27:1362.
- Boussemart L, Routier E, Mateus C, et al. Prospective study of cutaneous side-effects associated with the BRAF inhibitor vemurafenib: a study of 42 patients. Ann Oncol. 2013;24:1691-1697.
- Ramani NS, Curry JL, Kapil J, et al. Panniculitis with necrotizing granulomata in a patient on BRAF inhibitor (dabrafenib) therapy for metastatic melanoma. Am J Dermatopathol. 2015;37:E96-E99.
- Burillo-Martinez S, Morales-Raya C, Prieto-Barrios M, et al. Pembrolizumab-induced extensive panniculitis and nevus regression: two novel cutaneous manifestations of the post-immunotherapy granulomatous reactions spectrum. JAMA Dermatol. 2017;153:721-722.
- Cotliar J, Querfeld C, Boswell WJ, et al. Pembrolizumab-associated sarcoidosis. JAAD Case Rep. 2016;2:290-293.
Practice Points
- Pembrolizumab may cause lobular panniculitis years after treatment initiation.
- Pembrolizumab-induced lobular panniculitis may self-resolve without discontinuing the medication.
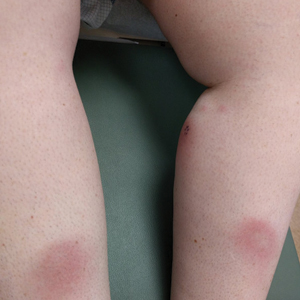
Subungual Hemorrhage From an Epidermal Growth Factor Receptor Inhibitor
To the Editor:
The epidermal growth factor receptor (EGFR) signaling pathway plays a role in the differentiation, proliferation, and survival of several cell types.1 Erlotinib is an EGFR inhibitor that targets aberrant cells that overexpress this receptor and has been used in the treatment of various solid malignant tumors.2,3 Common dermatologic side effects associated with EGFR inhibitors include papulopustular rash, xeroderma, and paronychia.2,3 We present a unique finding of subungual hemorrhage of the thumbnails in a patient taking erlotinib.
A 50-year-old man presented with acute-onset tenderness and discoloration of the thumbnails of 1 week’s duration. There was no preceding trauma or history of similar symptoms. His medical history was notable for recurrent lung adenocarcinoma with EGFR L858R mutation. Erlotinib therapy was initiated 5 weeks prior to symptom onset. He developed notable xeroderma of the palms and soles that preceded nail changes by a few days. He completed treatment with carboplatin and pemetrexed 16 months prior to relapse after paclitaxel failed due to a severe allergic reaction. There were no nail symptoms during that time. The patient did not have a documented coagulation disorder and was not on any known medications that would predispose him to bleeding. Physical examination demonstrated subungual hemorrhage of the thumbnails with tenderness on palpation (Figure). There was no evidence of periungual changes or nail plate abnormality. All other nails appeared normal. Laboratory test results showed normal platelets. Supportive therapeutic measures were recommended, and the patient was advised to avoid trauma to the nails.
Nail toxicities reported with EGFR inhibitors include paronychia, periungual pyogenic granulomas, and ingrown nails.1-3 Inflammation of the nail bed also can lead to secondary nail changes, such as onychodystrophy or onycholysis.2 Subungual hemorrhage has been reported as a side effect of taxanes, anticoagulants, anthracyclines, anti-inflammatory agents, and retinoids.4,5
The pathogenesis of nail toxicity secondary to EGFR inhibitors is not entirely clear. Symptoms commonly occur several weeks to months after therapy initiation.6 Epidermal growth factor receptor inhibitors disrupt proliferation and promote apoptosis of keratinocytes that is thought to enhance fragility of the periungual skin and nail plate.1,3 Under the influence of EGFR inhibition, a proinflammatory microenvironment in the skin is created through a type I interferon response leading to tissue damage.7 These changes may predispose patients to develop subungual hemorrhage in response to repeated nail microtrauma. Subungual asymptomatic splinter hemorrhage is a nail finding described in patients treated with the multikinase inhibitors sorafenib and sunitinib. Splinter hemorrhages of the nails are thought to be secondary to capillary microinjuries of the digits that cannot be repaired due to inhibition of vascular EGFRs.4
The time course of erlotinib administration and the simultaneous onset of xeroderma, a known side effect of the drug, in our patient are consistent with other cases.6 Subungual hemorrhage, which the patient reported observing only days after the onset of xeroderma, provides increased support that the anti-EGFR medication was likely responsible for both side effects concurrently. Bilateral involvement of the thumbs makes trauma as an inciting event unlikely.
Incidence of nail changes secondary to anti-EGFR drugs are likely underestimated and underreported.3 Subungual hemorrhage should be considered as an additional, less common nail side effect of EGFR inhibitors that clinicians and patients may encounter. Improved awareness and understanding of nail toxicities associated with EGFR inhibitors may offer better insight into the pathogenesis of these side effects and management options.
- Piraccini BM, Alessandrini A. Drug-related nail disease. Clin Dermatol. 2013;31:618-626.
- Kiyohara Y, Yamazaki N, Kishi A. Erlotinib-related skin toxicities: treatment strategies in patients with metastatic non-small cell lung cancer. J Am Acad Dermatol. 2013;69:463-472.
- Minisini AM, Tosti A, Sobrero AF, et al. Taxane-induced nail changes: incidence, clinical presentation and outcome. Ann Oncol. 2003;333-337.
- Garden BC, Wu S, Lacouture ME. The risk of nail changes with epidermal growth factor receptor inhibitors: a systematic review of the literature and meta-analysis. J Am Acad Dermatol. 2012;67:400-408.
- Fox LP. Nail toxicity associated with epidermal growth factor receptor inhibitor therapy. J Am Acad Dermatol. 2007;56:460-465.
- Chen KL, Lin CC, Cho YT, et al. Comparison of skin toxic effects associated with gefitinib, erlotinib or afatinib treatment for non-small cell lung cancer. JAMA Dermatol. 2016;152:340-342.
- Lulli D, Carbone ML, Pastore S. Epidermal growth factor receptor inhibitors trigger a type I interferon response in human skin. Oncotarget. 2016;7:47777-47793.
To the Editor:
The epidermal growth factor receptor (EGFR) signaling pathway plays a role in the differentiation, proliferation, and survival of several cell types.1 Erlotinib is an EGFR inhibitor that targets aberrant cells that overexpress this receptor and has been used in the treatment of various solid malignant tumors.2,3 Common dermatologic side effects associated with EGFR inhibitors include papulopustular rash, xeroderma, and paronychia.2,3 We present a unique finding of subungual hemorrhage of the thumbnails in a patient taking erlotinib.
A 50-year-old man presented with acute-onset tenderness and discoloration of the thumbnails of 1 week’s duration. There was no preceding trauma or history of similar symptoms. His medical history was notable for recurrent lung adenocarcinoma with EGFR L858R mutation. Erlotinib therapy was initiated 5 weeks prior to symptom onset. He developed notable xeroderma of the palms and soles that preceded nail changes by a few days. He completed treatment with carboplatin and pemetrexed 16 months prior to relapse after paclitaxel failed due to a severe allergic reaction. There were no nail symptoms during that time. The patient did not have a documented coagulation disorder and was not on any known medications that would predispose him to bleeding. Physical examination demonstrated subungual hemorrhage of the thumbnails with tenderness on palpation (Figure). There was no evidence of periungual changes or nail plate abnormality. All other nails appeared normal. Laboratory test results showed normal platelets. Supportive therapeutic measures were recommended, and the patient was advised to avoid trauma to the nails.
Nail toxicities reported with EGFR inhibitors include paronychia, periungual pyogenic granulomas, and ingrown nails.1-3 Inflammation of the nail bed also can lead to secondary nail changes, such as onychodystrophy or onycholysis.2 Subungual hemorrhage has been reported as a side effect of taxanes, anticoagulants, anthracyclines, anti-inflammatory agents, and retinoids.4,5
The pathogenesis of nail toxicity secondary to EGFR inhibitors is not entirely clear. Symptoms commonly occur several weeks to months after therapy initiation.6 Epidermal growth factor receptor inhibitors disrupt proliferation and promote apoptosis of keratinocytes that is thought to enhance fragility of the periungual skin and nail plate.1,3 Under the influence of EGFR inhibition, a proinflammatory microenvironment in the skin is created through a type I interferon response leading to tissue damage.7 These changes may predispose patients to develop subungual hemorrhage in response to repeated nail microtrauma. Subungual asymptomatic splinter hemorrhage is a nail finding described in patients treated with the multikinase inhibitors sorafenib and sunitinib. Splinter hemorrhages of the nails are thought to be secondary to capillary microinjuries of the digits that cannot be repaired due to inhibition of vascular EGFRs.4
The time course of erlotinib administration and the simultaneous onset of xeroderma, a known side effect of the drug, in our patient are consistent with other cases.6 Subungual hemorrhage, which the patient reported observing only days after the onset of xeroderma, provides increased support that the anti-EGFR medication was likely responsible for both side effects concurrently. Bilateral involvement of the thumbs makes trauma as an inciting event unlikely.
Incidence of nail changes secondary to anti-EGFR drugs are likely underestimated and underreported.3 Subungual hemorrhage should be considered as an additional, less common nail side effect of EGFR inhibitors that clinicians and patients may encounter. Improved awareness and understanding of nail toxicities associated with EGFR inhibitors may offer better insight into the pathogenesis of these side effects and management options.
To the Editor:
The epidermal growth factor receptor (EGFR) signaling pathway plays a role in the differentiation, proliferation, and survival of several cell types.1 Erlotinib is an EGFR inhibitor that targets aberrant cells that overexpress this receptor and has been used in the treatment of various solid malignant tumors.2,3 Common dermatologic side effects associated with EGFR inhibitors include papulopustular rash, xeroderma, and paronychia.2,3 We present a unique finding of subungual hemorrhage of the thumbnails in a patient taking erlotinib.
A 50-year-old man presented with acute-onset tenderness and discoloration of the thumbnails of 1 week’s duration. There was no preceding trauma or history of similar symptoms. His medical history was notable for recurrent lung adenocarcinoma with EGFR L858R mutation. Erlotinib therapy was initiated 5 weeks prior to symptom onset. He developed notable xeroderma of the palms and soles that preceded nail changes by a few days. He completed treatment with carboplatin and pemetrexed 16 months prior to relapse after paclitaxel failed due to a severe allergic reaction. There were no nail symptoms during that time. The patient did not have a documented coagulation disorder and was not on any known medications that would predispose him to bleeding. Physical examination demonstrated subungual hemorrhage of the thumbnails with tenderness on palpation (Figure). There was no evidence of periungual changes or nail plate abnormality. All other nails appeared normal. Laboratory test results showed normal platelets. Supportive therapeutic measures were recommended, and the patient was advised to avoid trauma to the nails.
Nail toxicities reported with EGFR inhibitors include paronychia, periungual pyogenic granulomas, and ingrown nails.1-3 Inflammation of the nail bed also can lead to secondary nail changes, such as onychodystrophy or onycholysis.2 Subungual hemorrhage has been reported as a side effect of taxanes, anticoagulants, anthracyclines, anti-inflammatory agents, and retinoids.4,5
The pathogenesis of nail toxicity secondary to EGFR inhibitors is not entirely clear. Symptoms commonly occur several weeks to months after therapy initiation.6 Epidermal growth factor receptor inhibitors disrupt proliferation and promote apoptosis of keratinocytes that is thought to enhance fragility of the periungual skin and nail plate.1,3 Under the influence of EGFR inhibition, a proinflammatory microenvironment in the skin is created through a type I interferon response leading to tissue damage.7 These changes may predispose patients to develop subungual hemorrhage in response to repeated nail microtrauma. Subungual asymptomatic splinter hemorrhage is a nail finding described in patients treated with the multikinase inhibitors sorafenib and sunitinib. Splinter hemorrhages of the nails are thought to be secondary to capillary microinjuries of the digits that cannot be repaired due to inhibition of vascular EGFRs.4
The time course of erlotinib administration and the simultaneous onset of xeroderma, a known side effect of the drug, in our patient are consistent with other cases.6 Subungual hemorrhage, which the patient reported observing only days after the onset of xeroderma, provides increased support that the anti-EGFR medication was likely responsible for both side effects concurrently. Bilateral involvement of the thumbs makes trauma as an inciting event unlikely.
Incidence of nail changes secondary to anti-EGFR drugs are likely underestimated and underreported.3 Subungual hemorrhage should be considered as an additional, less common nail side effect of EGFR inhibitors that clinicians and patients may encounter. Improved awareness and understanding of nail toxicities associated with EGFR inhibitors may offer better insight into the pathogenesis of these side effects and management options.
- Piraccini BM, Alessandrini A. Drug-related nail disease. Clin Dermatol. 2013;31:618-626.
- Kiyohara Y, Yamazaki N, Kishi A. Erlotinib-related skin toxicities: treatment strategies in patients with metastatic non-small cell lung cancer. J Am Acad Dermatol. 2013;69:463-472.
- Minisini AM, Tosti A, Sobrero AF, et al. Taxane-induced nail changes: incidence, clinical presentation and outcome. Ann Oncol. 2003;333-337.
- Garden BC, Wu S, Lacouture ME. The risk of nail changes with epidermal growth factor receptor inhibitors: a systematic review of the literature and meta-analysis. J Am Acad Dermatol. 2012;67:400-408.
- Fox LP. Nail toxicity associated with epidermal growth factor receptor inhibitor therapy. J Am Acad Dermatol. 2007;56:460-465.
- Chen KL, Lin CC, Cho YT, et al. Comparison of skin toxic effects associated with gefitinib, erlotinib or afatinib treatment for non-small cell lung cancer. JAMA Dermatol. 2016;152:340-342.
- Lulli D, Carbone ML, Pastore S. Epidermal growth factor receptor inhibitors trigger a type I interferon response in human skin. Oncotarget. 2016;7:47777-47793.
- Piraccini BM, Alessandrini A. Drug-related nail disease. Clin Dermatol. 2013;31:618-626.
- Kiyohara Y, Yamazaki N, Kishi A. Erlotinib-related skin toxicities: treatment strategies in patients with metastatic non-small cell lung cancer. J Am Acad Dermatol. 2013;69:463-472.
- Minisini AM, Tosti A, Sobrero AF, et al. Taxane-induced nail changes: incidence, clinical presentation and outcome. Ann Oncol. 2003;333-337.
- Garden BC, Wu S, Lacouture ME. The risk of nail changes with epidermal growth factor receptor inhibitors: a systematic review of the literature and meta-analysis. J Am Acad Dermatol. 2012;67:400-408.
- Fox LP. Nail toxicity associated with epidermal growth factor receptor inhibitor therapy. J Am Acad Dermatol. 2007;56:460-465.
- Chen KL, Lin CC, Cho YT, et al. Comparison of skin toxic effects associated with gefitinib, erlotinib or afatinib treatment for non-small cell lung cancer. JAMA Dermatol. 2016;152:340-342.
- Lulli D, Carbone ML, Pastore S. Epidermal growth factor receptor inhibitors trigger a type I interferon response in human skin. Oncotarget. 2016;7:47777-47793.
Practice Points
- Subungual hemorrhage is a potential adverse side effect of epidermal growth factor receptor inhibitors.
- Epidermal growth factor receptor inhibition may lead to enhanced fragility of the periungual skin and nail plate as well as a proinflammatory microenvironment in the skin, predisposing patients to nail toxicity.