User login
Cutaneous Collagenous Vasculopathy
To the Editor:
Cutaneous microangiopathy describes pathology of the small blood vessels within the dermis.
We report a case of CCV in a 41-year-old woman who presented for evaluation of a rash on the bilateral lower extremities of 7 to 8 months’ duration. The eruption had started on the left ankle and spread over several weeks to the bilateral dorsal feet followed by the ankles and shins. The patient noted associated swelling and a pressure like dysesthesia of the lower legs. She was otherwise in good health, though she had started an oral contraceptive 1 year prior for heavy menstrual bleeding. A review of systems was negative for deep vein thrombosis, pulmonary embolus, and other thromboembolic phenomena, and the patient had no history of hepatic or renal dysfunction, cancer, or heart disease. Her family history was negative for clotting disorders or bleeding diatheses.
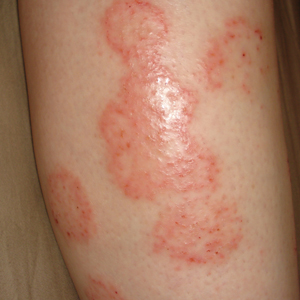
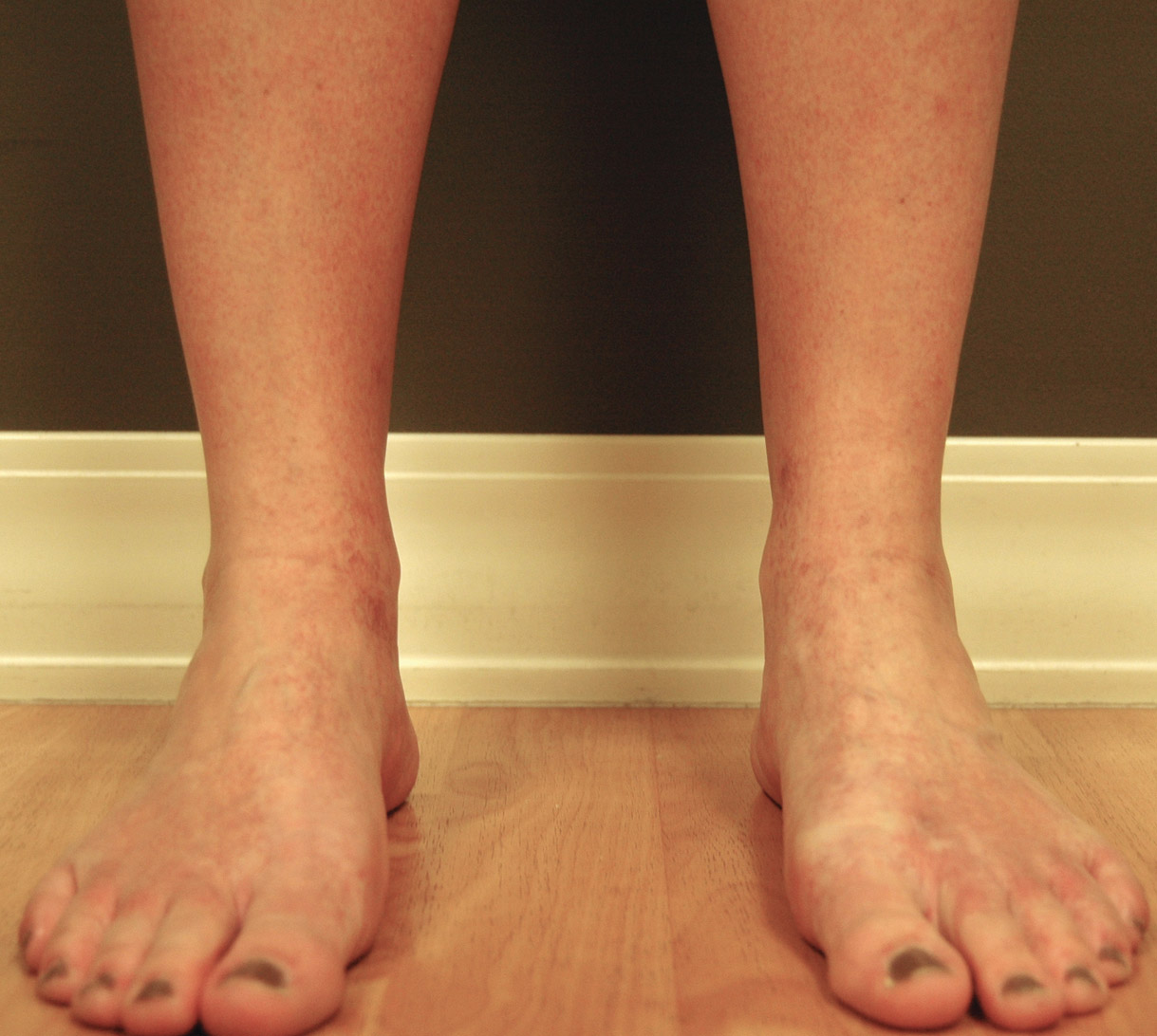
On physical examination, telangiectatic matting was present on the bilateral ankles and dorsal feet with an associated blanchable erythema (Figure 1). The matting extended into a fine, mottled, pretibial telangiectasia associated with Schamberg purpura. She had no pitting edema, and both dorsalis pedis and posterior popliteal pulses were intact and symmetric bilaterally. No popliteal lymphadenopathy or palpable cords were present.
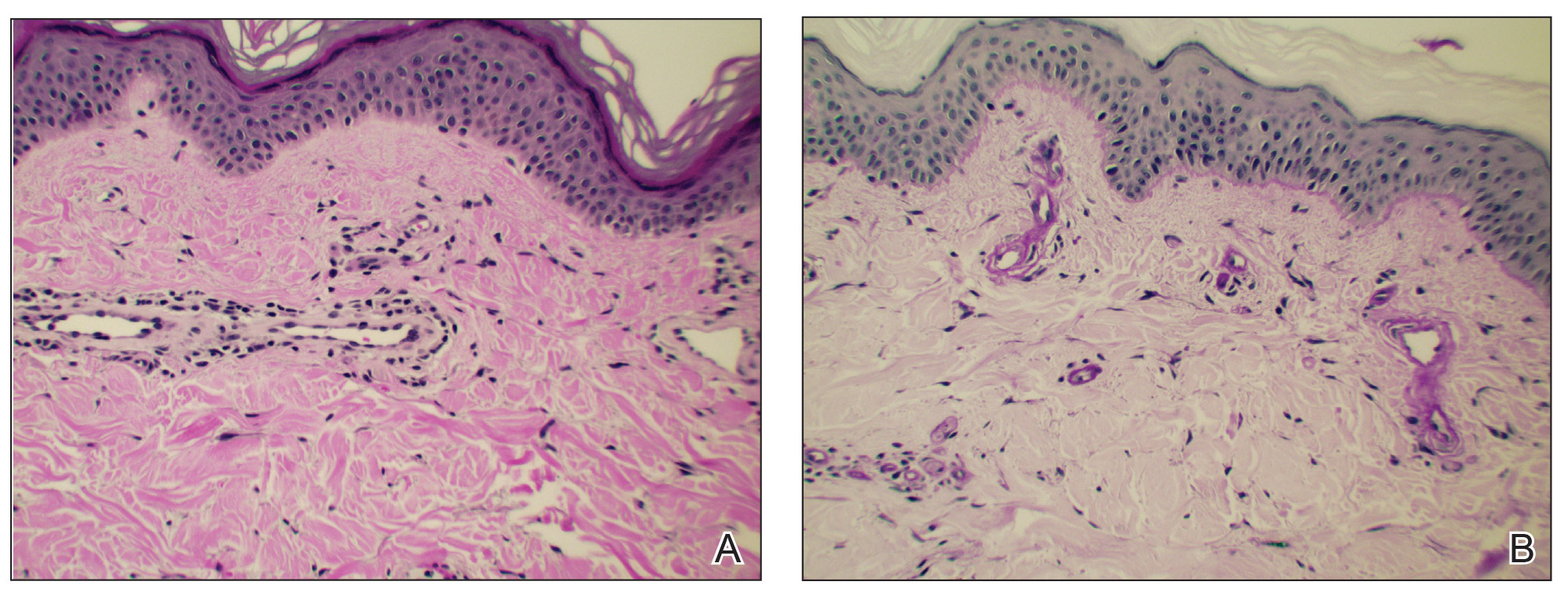
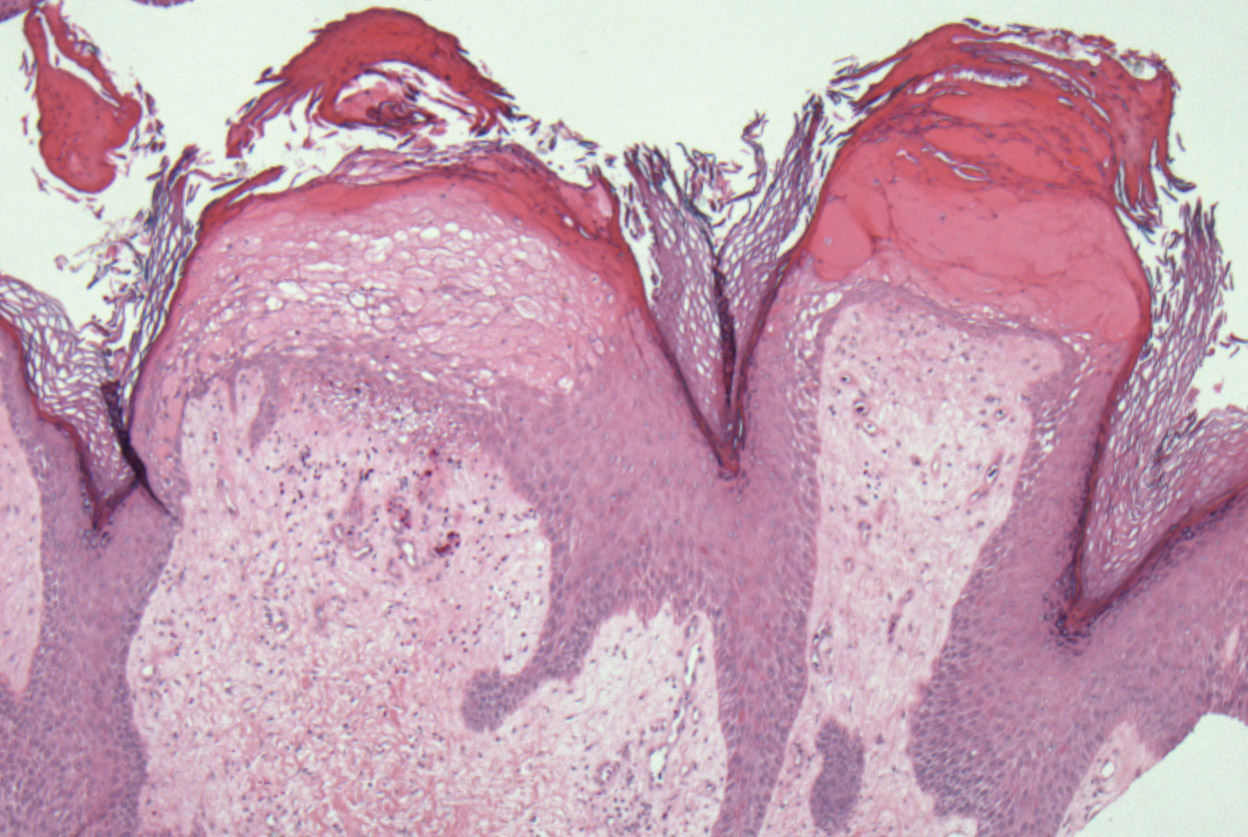
Two punch biopsies taken from the erythematous telangiectatic area on the left foot and metatarsal region demonstrated an unremarkable epidermis without interface change, thickening of the epidermal basement membrane, or single-cell dyskeratosis. There was mild dilatation of blood vessels within the superficial dermis with mild perivascular lymphocytic inflammation and rare extravasated erythrocytes. Leukocytoclastic debris, fibrinoid necrosis of vessel walls, and endothelial cell necrosis were not seen. As is classic in CCV, the vessel walls appeared thickened by eosinophilic hyaline material, which was periodic acid–Schiff positive and diastase resistant (Figure 2). Sclerotic thickening of collagen bundles or absence of periadnexal adipose tissue was not seen. CD34 immunohistochemical staining demonstrated normal retained CD34 interstitial dermal positivity, which excluded morphea. Additionally, direct immunofluorescence testing was negative for IgG, IgA, IgM, C3, fibrin, and C1q. Nodular reduplication of vessels or other changes of stasis were not seen. Fibrin thrombi or neoplastic cells were not identified. The clinical and histopathologic findings were suggestive of CCV.
Prior case reports of CCV have described a similar clinical manifestation with blanching macules that occur symmetrically on the lower extremities and spread cephalically.1-6 A distinction from hereditary hemorrhagic telangiectasia is the noninvolvement of mucous membranes and nails. The etiology of this rare microangiopathy has not been elucidated, though disease concurrence with local trauma, stressful events such as childbirth, and diabetes mellitus has been documented.6 As the body of literature continues to grow, more research regarding the etiology, mechanism, prognosis, and treatment options will enhance our understanding of CCV.
- Bondier L, Tardieu M, Leveque P, et al. Cutaneous collagenous vasculopathy: report of two cases presenting as disseminated telangiectasias and review of the literature. Am J Dermatopathol. 2017;39:682-688.
- Salama S, Rosenthal D. Cutaneous collagenous vasculopathy with generalized telangiectasia: an immunohistochemical and ultrastructural study. J Cutan Pathol. 2000;27:40-48.
- Lloyd BM, Pruden SJ, Lind AC, et al. Cutaneous collagenous vasculopathy: report of the first pediatric case. Pediatr Dermatol. 2011;28:598-599.
- Salama S, Chorneyko K, Belovic B. Cutaneous collagenous vasculopathy associated with intravascular occlusive fibrin thrombi. J Cutan Pathol. 2014;41:386-393.
- Perez A, Wain ME, Robson A, et al. Cutaneous collagenous vasculopathy with generalized telangiectasia in two female patients. J Am Acad Dermatol. 2010;63:882-885.
- Burdick LM, Losher S, Somach SC, et al. Cutaneous collagenous vasculopathy: a rare cutaneous microangiopathy. J Cutan Pathol. 2012;39:741-746.
To the Editor:
Cutaneous microangiopathy describes pathology of the small blood vessels within the dermis.
We report a case of CCV in a 41-year-old woman who presented for evaluation of a rash on the bilateral lower extremities of 7 to 8 months’ duration. The eruption had started on the left ankle and spread over several weeks to the bilateral dorsal feet followed by the ankles and shins. The patient noted associated swelling and a pressure like dysesthesia of the lower legs. She was otherwise in good health, though she had started an oral contraceptive 1 year prior for heavy menstrual bleeding. A review of systems was negative for deep vein thrombosis, pulmonary embolus, and other thromboembolic phenomena, and the patient had no history of hepatic or renal dysfunction, cancer, or heart disease. Her family history was negative for clotting disorders or bleeding diatheses.
On physical examination, telangiectatic matting was present on the bilateral ankles and dorsal feet with an associated blanchable erythema (Figure 1). The matting extended into a fine, mottled, pretibial telangiectasia associated with Schamberg purpura. She had no pitting edema, and both dorsalis pedis and posterior popliteal pulses were intact and symmetric bilaterally. No popliteal lymphadenopathy or palpable cords were present.
Two punch biopsies taken from the erythematous telangiectatic area on the left foot and metatarsal region demonstrated an unremarkable epidermis without interface change, thickening of the epidermal basement membrane, or single-cell dyskeratosis. There was mild dilatation of blood vessels within the superficial dermis with mild perivascular lymphocytic inflammation and rare extravasated erythrocytes. Leukocytoclastic debris, fibrinoid necrosis of vessel walls, and endothelial cell necrosis were not seen. As is classic in CCV, the vessel walls appeared thickened by eosinophilic hyaline material, which was periodic acid–Schiff positive and diastase resistant (Figure 2). Sclerotic thickening of collagen bundles or absence of periadnexal adipose tissue was not seen. CD34 immunohistochemical staining demonstrated normal retained CD34 interstitial dermal positivity, which excluded morphea. Additionally, direct immunofluorescence testing was negative for IgG, IgA, IgM, C3, fibrin, and C1q. Nodular reduplication of vessels or other changes of stasis were not seen. Fibrin thrombi or neoplastic cells were not identified. The clinical and histopathologic findings were suggestive of CCV.
Prior case reports of CCV have described a similar clinical manifestation with blanching macules that occur symmetrically on the lower extremities and spread cephalically.1-6 A distinction from hereditary hemorrhagic telangiectasia is the noninvolvement of mucous membranes and nails. The etiology of this rare microangiopathy has not been elucidated, though disease concurrence with local trauma, stressful events such as childbirth, and diabetes mellitus has been documented.6 As the body of literature continues to grow, more research regarding the etiology, mechanism, prognosis, and treatment options will enhance our understanding of CCV.
To the Editor:
Cutaneous microangiopathy describes pathology of the small blood vessels within the dermis.
We report a case of CCV in a 41-year-old woman who presented for evaluation of a rash on the bilateral lower extremities of 7 to 8 months’ duration. The eruption had started on the left ankle and spread over several weeks to the bilateral dorsal feet followed by the ankles and shins. The patient noted associated swelling and a pressure like dysesthesia of the lower legs. She was otherwise in good health, though she had started an oral contraceptive 1 year prior for heavy menstrual bleeding. A review of systems was negative for deep vein thrombosis, pulmonary embolus, and other thromboembolic phenomena, and the patient had no history of hepatic or renal dysfunction, cancer, or heart disease. Her family history was negative for clotting disorders or bleeding diatheses.
On physical examination, telangiectatic matting was present on the bilateral ankles and dorsal feet with an associated blanchable erythema (Figure 1). The matting extended into a fine, mottled, pretibial telangiectasia associated with Schamberg purpura. She had no pitting edema, and both dorsalis pedis and posterior popliteal pulses were intact and symmetric bilaterally. No popliteal lymphadenopathy or palpable cords were present.
Two punch biopsies taken from the erythematous telangiectatic area on the left foot and metatarsal region demonstrated an unremarkable epidermis without interface change, thickening of the epidermal basement membrane, or single-cell dyskeratosis. There was mild dilatation of blood vessels within the superficial dermis with mild perivascular lymphocytic inflammation and rare extravasated erythrocytes. Leukocytoclastic debris, fibrinoid necrosis of vessel walls, and endothelial cell necrosis were not seen. As is classic in CCV, the vessel walls appeared thickened by eosinophilic hyaline material, which was periodic acid–Schiff positive and diastase resistant (Figure 2). Sclerotic thickening of collagen bundles or absence of periadnexal adipose tissue was not seen. CD34 immunohistochemical staining demonstrated normal retained CD34 interstitial dermal positivity, which excluded morphea. Additionally, direct immunofluorescence testing was negative for IgG, IgA, IgM, C3, fibrin, and C1q. Nodular reduplication of vessels or other changes of stasis were not seen. Fibrin thrombi or neoplastic cells were not identified. The clinical and histopathologic findings were suggestive of CCV.
Prior case reports of CCV have described a similar clinical manifestation with blanching macules that occur symmetrically on the lower extremities and spread cephalically.1-6 A distinction from hereditary hemorrhagic telangiectasia is the noninvolvement of mucous membranes and nails. The etiology of this rare microangiopathy has not been elucidated, though disease concurrence with local trauma, stressful events such as childbirth, and diabetes mellitus has been documented.6 As the body of literature continues to grow, more research regarding the etiology, mechanism, prognosis, and treatment options will enhance our understanding of CCV.
- Bondier L, Tardieu M, Leveque P, et al. Cutaneous collagenous vasculopathy: report of two cases presenting as disseminated telangiectasias and review of the literature. Am J Dermatopathol. 2017;39:682-688.
- Salama S, Rosenthal D. Cutaneous collagenous vasculopathy with generalized telangiectasia: an immunohistochemical and ultrastructural study. J Cutan Pathol. 2000;27:40-48.
- Lloyd BM, Pruden SJ, Lind AC, et al. Cutaneous collagenous vasculopathy: report of the first pediatric case. Pediatr Dermatol. 2011;28:598-599.
- Salama S, Chorneyko K, Belovic B. Cutaneous collagenous vasculopathy associated with intravascular occlusive fibrin thrombi. J Cutan Pathol. 2014;41:386-393.
- Perez A, Wain ME, Robson A, et al. Cutaneous collagenous vasculopathy with generalized telangiectasia in two female patients. J Am Acad Dermatol. 2010;63:882-885.
- Burdick LM, Losher S, Somach SC, et al. Cutaneous collagenous vasculopathy: a rare cutaneous microangiopathy. J Cutan Pathol. 2012;39:741-746.
- Bondier L, Tardieu M, Leveque P, et al. Cutaneous collagenous vasculopathy: report of two cases presenting as disseminated telangiectasias and review of the literature. Am J Dermatopathol. 2017;39:682-688.
- Salama S, Rosenthal D. Cutaneous collagenous vasculopathy with generalized telangiectasia: an immunohistochemical and ultrastructural study. J Cutan Pathol. 2000;27:40-48.
- Lloyd BM, Pruden SJ, Lind AC, et al. Cutaneous collagenous vasculopathy: report of the first pediatric case. Pediatr Dermatol. 2011;28:598-599.
- Salama S, Chorneyko K, Belovic B. Cutaneous collagenous vasculopathy associated with intravascular occlusive fibrin thrombi. J Cutan Pathol. 2014;41:386-393.
- Perez A, Wain ME, Robson A, et al. Cutaneous collagenous vasculopathy with generalized telangiectasia in two female patients. J Am Acad Dermatol. 2010;63:882-885.
- Burdick LM, Losher S, Somach SC, et al. Cutaneous collagenous vasculopathy: a rare cutaneous microangiopathy. J Cutan Pathol. 2012;39:741-746.
Practice Points
- In cutaneous collagenous vasculopathy (CCV), skin biopsy may demonstrate eosinophilic hyaline thickening of superficial dermal blood vessels with mild perivascular lymphocytic inflammation and rare extravasated erythrocytes.
- Lack of mucous membrane and nail involvement differentiates CCV from hereditary hemorrhagic telangiectasia.
Elephantiasis Nostras Verrucosa Secondary to Scleroderma
To the Editor:
Elephantiasis nostras verrucosa (ENV) is a skin disorder caused by marked underlying lymphedema that leads to hyperkeratosis, papillomatosis, and verrucous growths on the epidermis.1 The pathophysiology of ENV relates to noninfectious lymphatic obstruction and lymphatic fibrosis secondary to venous stasis, malignancy, radiation therapy, or trauma.2 We present an unusual case of lymphedema and subsequent ENV limited to the arms and hands in a patient with scleroderma, an autoimmune fibrosing disorder.
A 54-year-old woman with a 5-year history of scleroderma presented to our dermatology clinic for treatment of progressive skin changes including pruritus, tightness, finger ulcerations, and pus exuding from papules on the dorsal arms and hands. She had been experiencing several systemic symptoms including dysphagia and lung involvement, necessitating oxygen therapy and a continuous positive airway pressure device for pulmonary arterial hypertension. A computed tomography scan of the lungs demonstrated an increase in ground-glass infiltrates in the right lower lobe and an air-fluid level in the esophagus. At the time of presentation, she was being treated with bosentan and sildenafil for pulmonary arterial hypertension, in addition to prednisone, venlafaxine, lansoprazole, metoclopramide, levothyroxine, temazepam, aspirin, and oxycodone. In the 2 years prior to presentation, she had been treated with intravenous cyclophosphamide once monthly for 6 months, adalimumab for 1 year, and 1 session of photodynamic therapy to the arms, all without benefit.
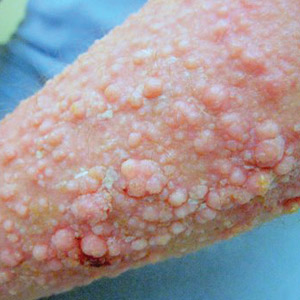
Physical examination showed cutaneous signs of scleroderma including marked sclerosis of the skin on the face, hands, V of the neck, proximal arms, and mid and proximal thighs. Excoriated papules with overlying crusting and pustulation were superimposed on the sclerotic skin of the arms (Figure 1).

A superinfection was diagnosed and treated with cephalexin 500 mg 4 times daily for 2 weeks; thereafter, mupirocin cream twice daily was used as needed. She was prescribed fexofenadine 180 mg twice daily and doxepin 20 mg at bedtime for pruritus.
At 3-week follow-up, a trial of narrowband UVB therapy was recommended for control of pruritus. Two weeks later, a modified wet-wrap regimen using clobetasol ointment 0.5% twice daily covered with wet gauze followed by a self-adherent dressing was initiated only on the right arm for comparison purposes. This treatment was not successful. A biopsy taken from the left arm showed lymphedema with perivascular fibroplasia and epidermal hyperplasia consistent with ENV (Figure 2).
Two months after her initial presentation, we instituted treatment with tazarotene gel 0.1% twice daily to the arms as well as a water-based topical emulsion to the finger ulcerations and a healing ointment to the hands. A month later, the patient reported no benefit with tazarotene. She desired more flexibility in her arms and hands; therefore, after a discussion with her rheumatologist, biweekly psoralen plus UVA (PUVA) therapy was initiated. Five months after presentation, methotrexate (MTX) 15 mg once weekly with folic acid 1 mg once daily was added. The PUVA therapy and MTX were stopped 3 months later due to lack of treatment benefit.
The patient was referred to vascular medicine for possible compression therapy. It was determined that her vasculature was intact, but compression therapy was contraindicated due to underlying systemic sclerosis. She was subsequently prescribed mycophenolate mofetil 1000 mg twice daily by her rheumatologist. The options of serial excisions or laser resurfacing were presented, but she declined.
Elephantiasis nostras verrucosa is differentiated from elephantiasis tropica, which is caused by a filarial infection of the lymphatic system. The chronic obstructive lymphedema characteristic of ENV can present as a result of various primary or secondary etiologies including trauma, malignancy, venous stasis, inflammation, or infection.3 In systemic sclerosis, extravascular fibrosis theoretically can lead to lymphatic obstruction and subsequent lymphatic stasis. In turn, the pathophysiology of dermal and subcutaneous fibrosis likely reflects autoantibodies (eg, anticardiolipin antibodies) that can damage lymphatic and nonlymphatic vessels.4,5
As the underlying mechanism of ENV, fibrosis of lymphatic vessels in systemic sclerosis is not well documented. Characteristic features of systemic sclerosis include extensive fibrosis, fibroproliferative vasculopathy, and inflammation, which are all possible mechanisms for the internal lymphatic obstruction resulting in the skin changes observed in ENV.6 It seemed unusual that the fibrotic changes of lymphatic vessels in our patient were extensive enough to cause ENV of the upper extremities; lower extremity involvement is the more common presentation because of the greater likelihood of lymphedema manifesting in the legs and feet. Lower extremity ENV has been reported in association with scleroderma.7,8
Regarding therapeutic options, Boyd et al9 reported a good response in a patient with ENV of the abdomen who was treated with topical tazarotene. Additionally, PUVA and MTX have been reported to be beneficial for the progressive skin changes of systemic sclerosis.10 Mycophenolate mofetil has been used in patients who fail MTX therapy because of its antifibrotic properties without the side-effect profiles of other immunosuppressives, such as imatinib.10,11 In our patient, skin lesions persisted following these varied approaches, and compression therapy was not advised due to the underlying sclerosis.
Because options for medical treatment of severe ENV are limited, surgical debridement of the affected limb often remains the only viable option in advanced cases.12 A PubMed search of articles indexed for MEDLINE using the terms elephantiasis (MeSH terms) or elephantiasis (all fields) and scleroderma, systemic (MeSH terms) or scleroderma (all fields) and systemic (all fields) or systemic scleroderma (all fields) or scleroderma (all fields) or scleroderma, localized (MeSH terms) or scleroderma (all fields) and localized (all fields) or localized scleroderma (all fields) yielded only 1 other case report of lower extremity ENV in a patient with systemic sclerosis who ultimately required bilateral leg amputation.8 When possible, avoiding lymphostasis through compression and control of any underlying infections is important in the treatment and prevention of ENV.3
- Sisto K, Khachemoune A. Elephantiasis nostras verrucosa: a review. Am J Clin Dermatol. 2008;9:141-146.
- Schissel DJ, Hivnor C, Elston DM. Elephantiasis nostras verrucosa. Cutis. 1998;62:77-80.
- Duckworth A, Husain J, DeHeer P. Elephantiasis nostras verrucosa or ‘mossy foot lesions’ in lymphedema praecox. J Am Podiatr Med Assoc. 2008;98:66-69.
- Assous N, Allanore Y, Batteaux F, et al. Prevalence of antiphospholipid antibodies in systemic sclerosis and association with primitive pulmonary arterial hypertension and endothelial injury. Clin Exp Rheumatol. 2005;23:199-204.
- Derrett-Smith EC, Dooley A, Gilbane AJ, et al. Endothelial injury in a transforming growth-factor-dependent mouse model of scleroderma induces pulmonary arterial hypertension. Arthritis Rheum. 2013;65:2928-2939.
- Pattanaik M, Brown M, Postlethwaite A. Vascular involvement in systemic sclerosis (scleroderma). J Inflamm Res. 2011;4:105-125.
- Kerchner K, Fleischer A, Yosipovitch G. Lower extremity lymphedema update: pathophysiology, diagnosis and treatment guidelines. J Am Acad Dermatol. 2008;59:324-331.
- Chatterjee S, Karai L. Elephantiasis nostras verrucosa in a patient with systemic sclerosis. Clin Exp Dermatol. 2009;34:e696-e698.
- Boyd J, Sloan S, Meffert J. Elephantiasis nostrum verrucosa of the abdomen: clinical results with tazarotene. J Drugs Dermatol. 2004;3:446-448.
- Fett, N. Scleroderma: nomenclature, etiology, pathogenesis, prognosis, and treatments: facts and controversies. Clin Dermatol. 2013;31:432-437.
- Moinzadeh P, Krieg T, Hunzelmann N. Imatinib treatment of generalized localized scleroderma (morphea). J Am Acad Dermatol. 2010;63:e102-e104.
- Iwao F, Sato-Matsumura KC, Sawamura D, et al. Elephantiasis nostras verrucosa successfully treated by surgical debridement. Dermatol Surg. 2004;30:939-941.
To the Editor:
Elephantiasis nostras verrucosa (ENV) is a skin disorder caused by marked underlying lymphedema that leads to hyperkeratosis, papillomatosis, and verrucous growths on the epidermis.1 The pathophysiology of ENV relates to noninfectious lymphatic obstruction and lymphatic fibrosis secondary to venous stasis, malignancy, radiation therapy, or trauma.2 We present an unusual case of lymphedema and subsequent ENV limited to the arms and hands in a patient with scleroderma, an autoimmune fibrosing disorder.
A 54-year-old woman with a 5-year history of scleroderma presented to our dermatology clinic for treatment of progressive skin changes including pruritus, tightness, finger ulcerations, and pus exuding from papules on the dorsal arms and hands. She had been experiencing several systemic symptoms including dysphagia and lung involvement, necessitating oxygen therapy and a continuous positive airway pressure device for pulmonary arterial hypertension. A computed tomography scan of the lungs demonstrated an increase in ground-glass infiltrates in the right lower lobe and an air-fluid level in the esophagus. At the time of presentation, she was being treated with bosentan and sildenafil for pulmonary arterial hypertension, in addition to prednisone, venlafaxine, lansoprazole, metoclopramide, levothyroxine, temazepam, aspirin, and oxycodone. In the 2 years prior to presentation, she had been treated with intravenous cyclophosphamide once monthly for 6 months, adalimumab for 1 year, and 1 session of photodynamic therapy to the arms, all without benefit.
Physical examination showed cutaneous signs of scleroderma including marked sclerosis of the skin on the face, hands, V of the neck, proximal arms, and mid and proximal thighs. Excoriated papules with overlying crusting and pustulation were superimposed on the sclerotic skin of the arms (Figure 1).
A superinfection was diagnosed and treated with cephalexin 500 mg 4 times daily for 2 weeks; thereafter, mupirocin cream twice daily was used as needed. She was prescribed fexofenadine 180 mg twice daily and doxepin 20 mg at bedtime for pruritus.
At 3-week follow-up, a trial of narrowband UVB therapy was recommended for control of pruritus. Two weeks later, a modified wet-wrap regimen using clobetasol ointment 0.5% twice daily covered with wet gauze followed by a self-adherent dressing was initiated only on the right arm for comparison purposes. This treatment was not successful. A biopsy taken from the left arm showed lymphedema with perivascular fibroplasia and epidermal hyperplasia consistent with ENV (Figure 2).
Two months after her initial presentation, we instituted treatment with tazarotene gel 0.1% twice daily to the arms as well as a water-based topical emulsion to the finger ulcerations and a healing ointment to the hands. A month later, the patient reported no benefit with tazarotene. She desired more flexibility in her arms and hands; therefore, after a discussion with her rheumatologist, biweekly psoralen plus UVA (PUVA) therapy was initiated. Five months after presentation, methotrexate (MTX) 15 mg once weekly with folic acid 1 mg once daily was added. The PUVA therapy and MTX were stopped 3 months later due to lack of treatment benefit.
The patient was referred to vascular medicine for possible compression therapy. It was determined that her vasculature was intact, but compression therapy was contraindicated due to underlying systemic sclerosis. She was subsequently prescribed mycophenolate mofetil 1000 mg twice daily by her rheumatologist. The options of serial excisions or laser resurfacing were presented, but she declined.
Elephantiasis nostras verrucosa is differentiated from elephantiasis tropica, which is caused by a filarial infection of the lymphatic system. The chronic obstructive lymphedema characteristic of ENV can present as a result of various primary or secondary etiologies including trauma, malignancy, venous stasis, inflammation, or infection.3 In systemic sclerosis, extravascular fibrosis theoretically can lead to lymphatic obstruction and subsequent lymphatic stasis. In turn, the pathophysiology of dermal and subcutaneous fibrosis likely reflects autoantibodies (eg, anticardiolipin antibodies) that can damage lymphatic and nonlymphatic vessels.4,5
As the underlying mechanism of ENV, fibrosis of lymphatic vessels in systemic sclerosis is not well documented. Characteristic features of systemic sclerosis include extensive fibrosis, fibroproliferative vasculopathy, and inflammation, which are all possible mechanisms for the internal lymphatic obstruction resulting in the skin changes observed in ENV.6 It seemed unusual that the fibrotic changes of lymphatic vessels in our patient were extensive enough to cause ENV of the upper extremities; lower extremity involvement is the more common presentation because of the greater likelihood of lymphedema manifesting in the legs and feet. Lower extremity ENV has been reported in association with scleroderma.7,8
Regarding therapeutic options, Boyd et al9 reported a good response in a patient with ENV of the abdomen who was treated with topical tazarotene. Additionally, PUVA and MTX have been reported to be beneficial for the progressive skin changes of systemic sclerosis.10 Mycophenolate mofetil has been used in patients who fail MTX therapy because of its antifibrotic properties without the side-effect profiles of other immunosuppressives, such as imatinib.10,11 In our patient, skin lesions persisted following these varied approaches, and compression therapy was not advised due to the underlying sclerosis.
Because options for medical treatment of severe ENV are limited, surgical debridement of the affected limb often remains the only viable option in advanced cases.12 A PubMed search of articles indexed for MEDLINE using the terms elephantiasis (MeSH terms) or elephantiasis (all fields) and scleroderma, systemic (MeSH terms) or scleroderma (all fields) and systemic (all fields) or systemic scleroderma (all fields) or scleroderma (all fields) or scleroderma, localized (MeSH terms) or scleroderma (all fields) and localized (all fields) or localized scleroderma (all fields) yielded only 1 other case report of lower extremity ENV in a patient with systemic sclerosis who ultimately required bilateral leg amputation.8 When possible, avoiding lymphostasis through compression and control of any underlying infections is important in the treatment and prevention of ENV.3
To the Editor:
Elephantiasis nostras verrucosa (ENV) is a skin disorder caused by marked underlying lymphedema that leads to hyperkeratosis, papillomatosis, and verrucous growths on the epidermis.1 The pathophysiology of ENV relates to noninfectious lymphatic obstruction and lymphatic fibrosis secondary to venous stasis, malignancy, radiation therapy, or trauma.2 We present an unusual case of lymphedema and subsequent ENV limited to the arms and hands in a patient with scleroderma, an autoimmune fibrosing disorder.
A 54-year-old woman with a 5-year history of scleroderma presented to our dermatology clinic for treatment of progressive skin changes including pruritus, tightness, finger ulcerations, and pus exuding from papules on the dorsal arms and hands. She had been experiencing several systemic symptoms including dysphagia and lung involvement, necessitating oxygen therapy and a continuous positive airway pressure device for pulmonary arterial hypertension. A computed tomography scan of the lungs demonstrated an increase in ground-glass infiltrates in the right lower lobe and an air-fluid level in the esophagus. At the time of presentation, she was being treated with bosentan and sildenafil for pulmonary arterial hypertension, in addition to prednisone, venlafaxine, lansoprazole, metoclopramide, levothyroxine, temazepam, aspirin, and oxycodone. In the 2 years prior to presentation, she had been treated with intravenous cyclophosphamide once monthly for 6 months, adalimumab for 1 year, and 1 session of photodynamic therapy to the arms, all without benefit.
Physical examination showed cutaneous signs of scleroderma including marked sclerosis of the skin on the face, hands, V of the neck, proximal arms, and mid and proximal thighs. Excoriated papules with overlying crusting and pustulation were superimposed on the sclerotic skin of the arms (Figure 1).
A superinfection was diagnosed and treated with cephalexin 500 mg 4 times daily for 2 weeks; thereafter, mupirocin cream twice daily was used as needed. She was prescribed fexofenadine 180 mg twice daily and doxepin 20 mg at bedtime for pruritus.
At 3-week follow-up, a trial of narrowband UVB therapy was recommended for control of pruritus. Two weeks later, a modified wet-wrap regimen using clobetasol ointment 0.5% twice daily covered with wet gauze followed by a self-adherent dressing was initiated only on the right arm for comparison purposes. This treatment was not successful. A biopsy taken from the left arm showed lymphedema with perivascular fibroplasia and epidermal hyperplasia consistent with ENV (Figure 2).
Two months after her initial presentation, we instituted treatment with tazarotene gel 0.1% twice daily to the arms as well as a water-based topical emulsion to the finger ulcerations and a healing ointment to the hands. A month later, the patient reported no benefit with tazarotene. She desired more flexibility in her arms and hands; therefore, after a discussion with her rheumatologist, biweekly psoralen plus UVA (PUVA) therapy was initiated. Five months after presentation, methotrexate (MTX) 15 mg once weekly with folic acid 1 mg once daily was added. The PUVA therapy and MTX were stopped 3 months later due to lack of treatment benefit.
The patient was referred to vascular medicine for possible compression therapy. It was determined that her vasculature was intact, but compression therapy was contraindicated due to underlying systemic sclerosis. She was subsequently prescribed mycophenolate mofetil 1000 mg twice daily by her rheumatologist. The options of serial excisions or laser resurfacing were presented, but she declined.
Elephantiasis nostras verrucosa is differentiated from elephantiasis tropica, which is caused by a filarial infection of the lymphatic system. The chronic obstructive lymphedema characteristic of ENV can present as a result of various primary or secondary etiologies including trauma, malignancy, venous stasis, inflammation, or infection.3 In systemic sclerosis, extravascular fibrosis theoretically can lead to lymphatic obstruction and subsequent lymphatic stasis. In turn, the pathophysiology of dermal and subcutaneous fibrosis likely reflects autoantibodies (eg, anticardiolipin antibodies) that can damage lymphatic and nonlymphatic vessels.4,5
As the underlying mechanism of ENV, fibrosis of lymphatic vessels in systemic sclerosis is not well documented. Characteristic features of systemic sclerosis include extensive fibrosis, fibroproliferative vasculopathy, and inflammation, which are all possible mechanisms for the internal lymphatic obstruction resulting in the skin changes observed in ENV.6 It seemed unusual that the fibrotic changes of lymphatic vessels in our patient were extensive enough to cause ENV of the upper extremities; lower extremity involvement is the more common presentation because of the greater likelihood of lymphedema manifesting in the legs and feet. Lower extremity ENV has been reported in association with scleroderma.7,8
Regarding therapeutic options, Boyd et al9 reported a good response in a patient with ENV of the abdomen who was treated with topical tazarotene. Additionally, PUVA and MTX have been reported to be beneficial for the progressive skin changes of systemic sclerosis.10 Mycophenolate mofetil has been used in patients who fail MTX therapy because of its antifibrotic properties without the side-effect profiles of other immunosuppressives, such as imatinib.10,11 In our patient, skin lesions persisted following these varied approaches, and compression therapy was not advised due to the underlying sclerosis.
Because options for medical treatment of severe ENV are limited, surgical debridement of the affected limb often remains the only viable option in advanced cases.12 A PubMed search of articles indexed for MEDLINE using the terms elephantiasis (MeSH terms) or elephantiasis (all fields) and scleroderma, systemic (MeSH terms) or scleroderma (all fields) and systemic (all fields) or systemic scleroderma (all fields) or scleroderma (all fields) or scleroderma, localized (MeSH terms) or scleroderma (all fields) and localized (all fields) or localized scleroderma (all fields) yielded only 1 other case report of lower extremity ENV in a patient with systemic sclerosis who ultimately required bilateral leg amputation.8 When possible, avoiding lymphostasis through compression and control of any underlying infections is important in the treatment and prevention of ENV.3
- Sisto K, Khachemoune A. Elephantiasis nostras verrucosa: a review. Am J Clin Dermatol. 2008;9:141-146.
- Schissel DJ, Hivnor C, Elston DM. Elephantiasis nostras verrucosa. Cutis. 1998;62:77-80.
- Duckworth A, Husain J, DeHeer P. Elephantiasis nostras verrucosa or ‘mossy foot lesions’ in lymphedema praecox. J Am Podiatr Med Assoc. 2008;98:66-69.
- Assous N, Allanore Y, Batteaux F, et al. Prevalence of antiphospholipid antibodies in systemic sclerosis and association with primitive pulmonary arterial hypertension and endothelial injury. Clin Exp Rheumatol. 2005;23:199-204.
- Derrett-Smith EC, Dooley A, Gilbane AJ, et al. Endothelial injury in a transforming growth-factor-dependent mouse model of scleroderma induces pulmonary arterial hypertension. Arthritis Rheum. 2013;65:2928-2939.
- Pattanaik M, Brown M, Postlethwaite A. Vascular involvement in systemic sclerosis (scleroderma). J Inflamm Res. 2011;4:105-125.
- Kerchner K, Fleischer A, Yosipovitch G. Lower extremity lymphedema update: pathophysiology, diagnosis and treatment guidelines. J Am Acad Dermatol. 2008;59:324-331.
- Chatterjee S, Karai L. Elephantiasis nostras verrucosa in a patient with systemic sclerosis. Clin Exp Dermatol. 2009;34:e696-e698.
- Boyd J, Sloan S, Meffert J. Elephantiasis nostrum verrucosa of the abdomen: clinical results with tazarotene. J Drugs Dermatol. 2004;3:446-448.
- Fett, N. Scleroderma: nomenclature, etiology, pathogenesis, prognosis, and treatments: facts and controversies. Clin Dermatol. 2013;31:432-437.
- Moinzadeh P, Krieg T, Hunzelmann N. Imatinib treatment of generalized localized scleroderma (morphea). J Am Acad Dermatol. 2010;63:e102-e104.
- Iwao F, Sato-Matsumura KC, Sawamura D, et al. Elephantiasis nostras verrucosa successfully treated by surgical debridement. Dermatol Surg. 2004;30:939-941.
- Sisto K, Khachemoune A. Elephantiasis nostras verrucosa: a review. Am J Clin Dermatol. 2008;9:141-146.
- Schissel DJ, Hivnor C, Elston DM. Elephantiasis nostras verrucosa. Cutis. 1998;62:77-80.
- Duckworth A, Husain J, DeHeer P. Elephantiasis nostras verrucosa or ‘mossy foot lesions’ in lymphedema praecox. J Am Podiatr Med Assoc. 2008;98:66-69.
- Assous N, Allanore Y, Batteaux F, et al. Prevalence of antiphospholipid antibodies in systemic sclerosis and association with primitive pulmonary arterial hypertension and endothelial injury. Clin Exp Rheumatol. 2005;23:199-204.
- Derrett-Smith EC, Dooley A, Gilbane AJ, et al. Endothelial injury in a transforming growth-factor-dependent mouse model of scleroderma induces pulmonary arterial hypertension. Arthritis Rheum. 2013;65:2928-2939.
- Pattanaik M, Brown M, Postlethwaite A. Vascular involvement in systemic sclerosis (scleroderma). J Inflamm Res. 2011;4:105-125.
- Kerchner K, Fleischer A, Yosipovitch G. Lower extremity lymphedema update: pathophysiology, diagnosis and treatment guidelines. J Am Acad Dermatol. 2008;59:324-331.
- Chatterjee S, Karai L. Elephantiasis nostras verrucosa in a patient with systemic sclerosis. Clin Exp Dermatol. 2009;34:e696-e698.
- Boyd J, Sloan S, Meffert J. Elephantiasis nostrum verrucosa of the abdomen: clinical results with tazarotene. J Drugs Dermatol. 2004;3:446-448.
- Fett, N. Scleroderma: nomenclature, etiology, pathogenesis, prognosis, and treatments: facts and controversies. Clin Dermatol. 2013;31:432-437.
- Moinzadeh P, Krieg T, Hunzelmann N. Imatinib treatment of generalized localized scleroderma (morphea). J Am Acad Dermatol. 2010;63:e102-e104.
- Iwao F, Sato-Matsumura KC, Sawamura D, et al. Elephantiasis nostras verrucosa successfully treated by surgical debridement. Dermatol Surg. 2004;30:939-941.
Practice Points
- Scleroderma rarely may lead to elephantiasis nostras verrucosa (ENV) of the upper extremities.
- Avoiding lymphostasis through compression and control of concomitant skin and soft tissue infections is important in the treatment and prevention of ENV.
Neurofibromatosis Type 1 in the Setting of Systemic Lupus Erythematosus
To the Editor:
Patients with concurrent neurofibromatosis type 1 (NF-1) and systemic lupus erythematosus (SLE) rarely have been reported in the literature. Neurofibromatosis type 1 is one of the most common genetic disorders, with a worldwide birth incidence of 1 in 2500 individuals and prevalence of 1 in 4000 individuals.1 The incidence and prevalence of SLE varies widely depending on race and geographic location. Estimated incidence rates for SLE range from 1 to 25 per 100,000 individuals annually in North America, South America, Europe, and Asia.2,3 The reported worldwide prevalence is 20 to 150 cases per 100,000 individuals annually.2,4,5
Given the high prevalence of both conditions, the association between SLE and NF-1 likely is underrecognized; therefore, identifying more patients with concurrent SLE and NF-1 and describing the interplay between the 2 conditions may have important therapeutic implications. We present the case of a middle-aged woman with a history of SLE who had cutaneous lesions characteristic of NF-1 to further the understanding of these concurrent conditions.
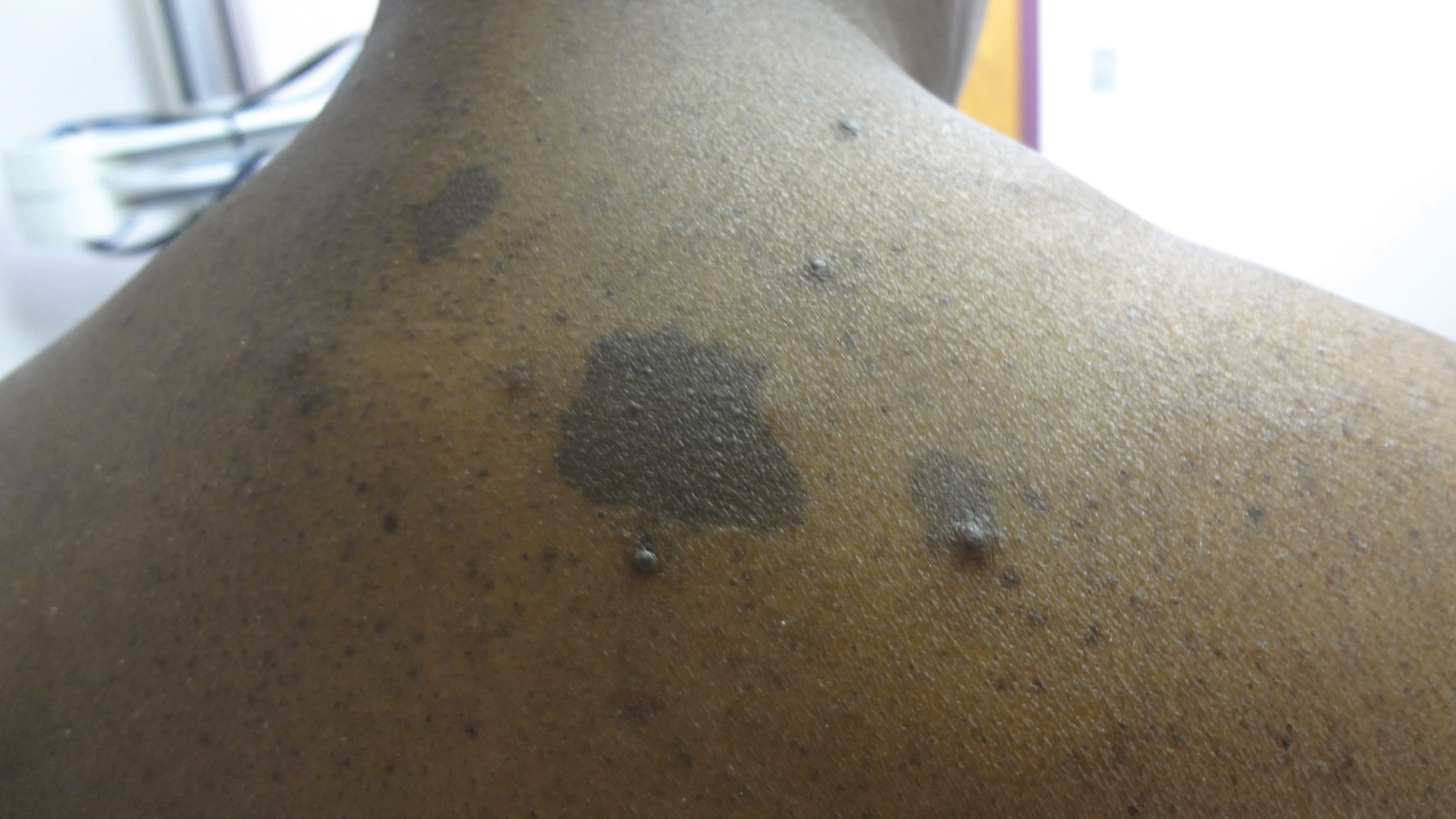
A middle-aged woman presented to our academic dermatology clinic for evaluation and removal of dark spots that had been present diffusely on the trunk and extremities since birth. She reported a history of SLE with lupus nephritis, hypertension, and a nodular goiter following a partial thyroidectomy. She noted that she did not seek treatment for the skin findings sooner because she was more concerned about her other medical conditions; however, because she felt these conditions were now stable, she decided to seek treatment for the “rash.” Physical examination revealed hundreds of café au lait macules and numerous neurofibromas diffusely distributed on the trunk and extremities (Figure 1) as well as bilateral axillary freckling. A clinical diagnosis of NF-1 was made.
When questioned, the patient reported that she may have been diagnosed with NF-1 in the past by another physician, but she did not recall it specifically. The patient was advised that there were no treatments for the café au lait macules. We notified her other physicians of the NF-1 diagnosis so she could be monitored for systemic conditions related to NF-1, including optic gliomas, pheochromocytoma, renal artery stenosis, and internal neurofibromas. We also referred the patient for genetic counseling; of note, the patient reported she had 4 children without any evidence of similar skin lesions or chronic health problems.
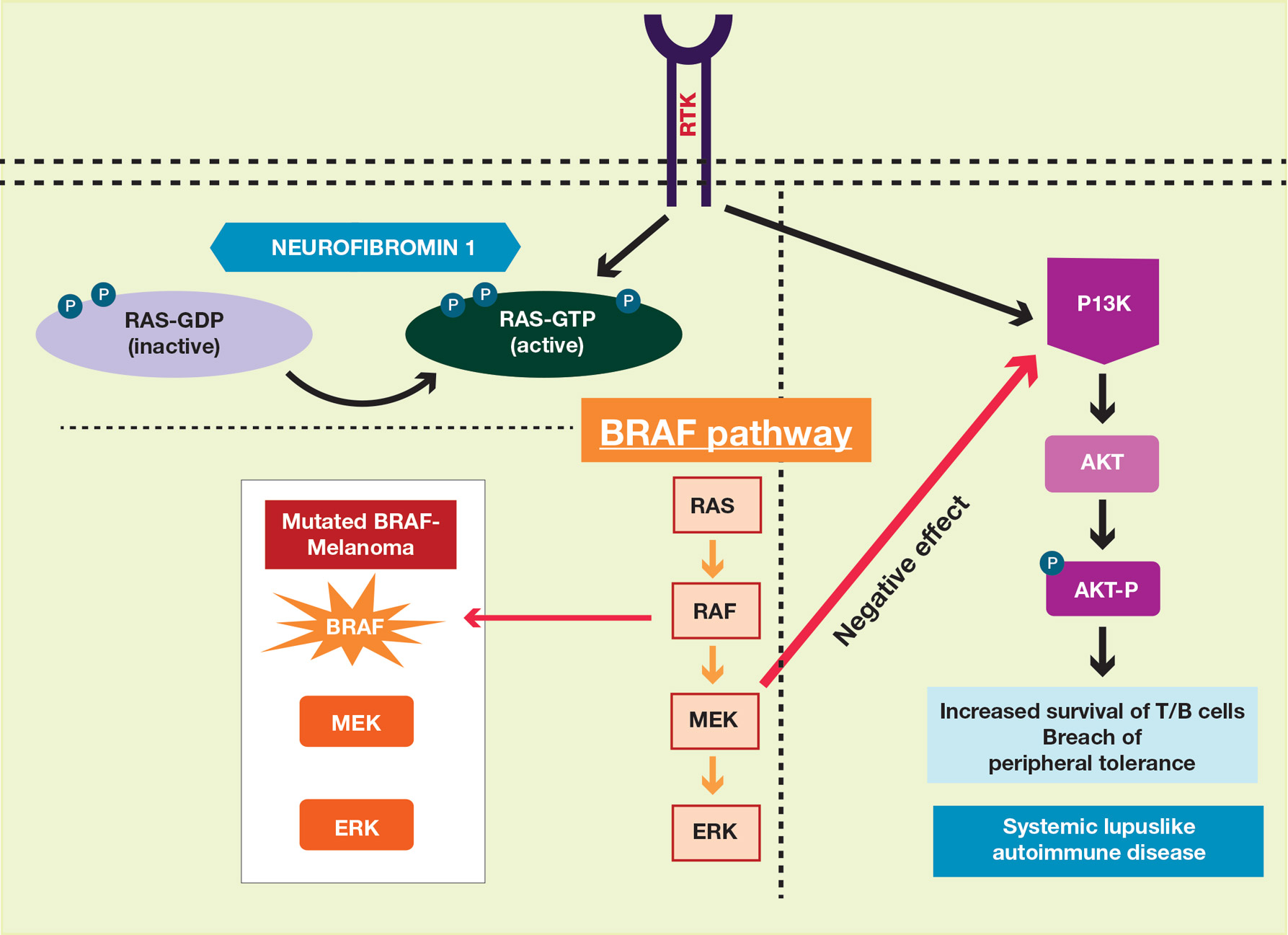
A PubMed search of articles indexed for MEDLINE using the terms systemic lupus and neurofibromatosis yielded 8 cases of patients having both SLE and NF-1 (including our case).6-11 Our patient reported having multiple lesions since birth, decades before the onset and diagnosis of SLE. In 3 other cases, patients were diagnosed with SLE and then presented with neurofibromas, leading to NF-1 diagnosis.In the discussion of those 3 cases, it was proposed that immune system alterations caused by SLE leading to viral illness may have predisposed the patients to the development of tumors and other collagen diseases, or it could be coincidental.6,7 In another case, a patient with NF-1 developed SLE, which was thought to be coincidental.8 Akyuz et al9 described the case of a pediatric patient with NF-1 who subsequently was diagnosed with SLE. The authors suggested that the lack of neurofibromin contributed to the development of SLE, an autoimmune condition. Under normal circumstances, neurofibromin acts as a guanosine triphosphatase–activating protein for RAS in T cells.10 CD8+ T-cell function also is impaired in patients with SLE.9 Additionally, it has been reported that anti–double-stranded DNA antibodies and immune complexes were present in NF-1 patients, even though there were low titers.12 Thus, the authors proposed that the lack of neurofibromin led to dysregulation of the RAS pathway and impairment of T cells, creating an immune milieu that predisposed the patient to development of SLE. Our case gives additional credence to this theory, as our patient had a similar clinical course: the café au lait macules were present since birth and the symptoms of SLE surfaced much later in her late 20s and 30s. Another case by Makino and Tampo10 described a patient with a history of SLE who was later diagnosed with NF-1 based on choroidal findings highly specific for NF-1 but did not have other classic findings of NF-1. The authors mentioned that there might be a potential relationship between these two disorders but did not speculate any theory in particular for their case.10
The interplay between an autoimmune condition such as SLE and NF-1, a condition traditionally thought to be due to a genetic mutation, may have greater clinical and therapeutic implications beyond just these two disorders. Although it is well established that RAS pathway disruption causes NF-1, it has been uncovered that dysfunction in the RAS pathway also can contribute to melanoma oncogenesis.13,14 These insights have led to the development of and approval of targeted drugs designed to inhibit the RAS pathway (eg, vemurafenib, dabrafenib, trametinib).14-17 Melanoma also is considered a “model” tumor for studying the relationship between the immune system and cancer.18
Our case also is instructive in another point: our patient had never sought treatment for her skin lesions, as she said she had other more serious health conditions. Closer evaluation of her skin condition may have led to earlier diagnosis of NF-1, which has important health implications. The average lifespan of patients with NF-1 is 10 to 15 years lower than the general population, with cancer being the leading cause of death.20 Malignant peripheral nerve sheath tumors are the most common malignant tumors observed in such patients.21-23 Other cancers that are associated with NF-1 include rhabdomyosarcomas, gastrointestinal stromal tumors, neuroectodermal tumors, pheochromocytomas, and breast carcinomas.23
To make a clinical diagnosis of NF-1, a patient must have 2 of 7 cardinal clinical features as defined by the National Institutes of Health (Table).24 In our patient with hundreds of café au lait macules and dozens of neurofibromas, the diagnosis was clear; however, in other patients, the skin findings of NF-1 may not be as prominent. A patient could meet criteria for NF-1 diagnosis with the inconspicuous presentation of 6 café au lait macules and either 1 plexiform neurofibroma or 2 neurofibromas (of any type) on the entire body.

We recommend that patients with SLE undergo skin examinations to look for more subtle presentations of NF-1. Earlier diagnosis will help to initiate close monitoring of the disorder’s associated systemic health risks. In addition, the identification of more patients with both NF-1 and SLE may help shed light on the etiology of both conditions.
- Carey JC, Baty BJ, Johnson JP, et al. The genetic aspects of neurofibromatosis. Ann N Y Acad Sci. 1986;486:45-56.
- Pons-Estel GJ, Alarcón GS, Scofield L, et al. Understanding the epidemiology and progression of systemic lupus erythematosus. Semin Arthritis Rheum. 2010;39:257-268.
- Danchenko N, Satia JA, Anthony MS. Epidemiology of systemic lupus erythematosus: a comparison of worldwide disease burden. Lupus. 2006;15:308-318.
- Lawrence RC, Helmick CG, Arnett FC, et al. Estimates of the prevalence of arthritis and selected musculoskeletal disorders in the United States. Arthritis Rheum. 1998;41:778-799.
- Chakravarty EF, Bush TM, Manzi S, et al. Prevalence of adult systemic lupus erythematosus in California and Pennsylvania in 2000: estimates obtained using hospitalization data. Arthritis Rheum. 2007;56:2092-2094.
- Bitnun S, Bassan H. Letter: neurofibromatosis and SLE. N Engl J Med. 1975;292:429-430.
- Riccardi VM. Neurofibromatosis in a patient with systemic lupus erythematosus. Arthritis Rheum. 1983;26:574.
- Corominas H, Guardiola JM, Matas L, et al. Neurofibromatosis and systemic lupus erythematosus. a matter of coincidence? Clin Rhematol. 2003;22:496-497.
- Akyuz SG, Caltik A, Bulbul M, et al. An unusual pediatric case with neurofibromatosis and systemic lupus erythematosus. Rheumatol Int. 2012;32:2345-47.
- Makino S, Tampo H. Rare and unusual choroidal abnormalities in a patient with systemic lupus erythematosus. Case Rep Ophthalmol. 2013;4:81-86.
- Galvan JM, Hofkamp MP. Usefulness of intrapartum magnetic resonance imaging for a parturient with neurofibromatosis type I during induction of labor for preeclampsia. Proc (Bayl Univ Med Cent). 2018;31:92-93.
- Gerosa PL, Vai C, Bizzozer L, et al. Immunological and clinical surveillance in Recklinghausen’s neurofibromatosis (NF1). Panminerva Med. 1993;35:80-85.
- Busca R, Abbe P, Mantoux F, et al. RAS mediates the cAMP-dependent activation of extracellular signal-regulated kinases (ERKs) in melanocytes. EMBO J. 2000;19:2900-2910.
- Sullivan RJ, Flaherty K. MAP kinase signaling and inhibition in melanoma. Oncogene. 2013;32:2373-2379.
- Hennessy BT, Smith DL, Ram PT, et al. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov. 2005;12:988-1004.
- Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507-2516.
- Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380:358-365.
- Maio M. Melanoma as a model tumour for immuno-oncology. Ann Oncol. 2012;23:(suppl 8):viii10-4.
- Zmajkovicova K, Jesenberger V, Catalanotti F, et al. MEK1 is required for PTEN membrane recruitment, AKT regulation, and the maintenance of peripheral tolerance. Mol Cell. 2013;50:43-55.
- Patil S, Chamberlain RS. Neoplasms associated with germline and somatic NF1 gene mutations. Oncologist. 2012;17:101-116.
- Carroll SL, Ratner N. How does the Schwann cell lineage form tumors in NF1? Glia. 2008;56:1590-1605.
- Rasmussen SA, Friedman JM. NF1 gene and neurofibromatosis 1. Am J Epidemiol. 2000;151:33-40.
- Yohay K. Neurofibromatosis type 1 and associated malignancies. Curr Neurol Neurosci Rep. 2009;9:247-253.
- Neurofibromatosis. conference statement. National Institutes of Health Consensus Development Conference. Arch Neurol. 1988;45:575-78.
To the Editor:
Patients with concurrent neurofibromatosis type 1 (NF-1) and systemic lupus erythematosus (SLE) rarely have been reported in the literature. Neurofibromatosis type 1 is one of the most common genetic disorders, with a worldwide birth incidence of 1 in 2500 individuals and prevalence of 1 in 4000 individuals.1 The incidence and prevalence of SLE varies widely depending on race and geographic location. Estimated incidence rates for SLE range from 1 to 25 per 100,000 individuals annually in North America, South America, Europe, and Asia.2,3 The reported worldwide prevalence is 20 to 150 cases per 100,000 individuals annually.2,4,5
Given the high prevalence of both conditions, the association between SLE and NF-1 likely is underrecognized; therefore, identifying more patients with concurrent SLE and NF-1 and describing the interplay between the 2 conditions may have important therapeutic implications. We present the case of a middle-aged woman with a history of SLE who had cutaneous lesions characteristic of NF-1 to further the understanding of these concurrent conditions.
A middle-aged woman presented to our academic dermatology clinic for evaluation and removal of dark spots that had been present diffusely on the trunk and extremities since birth. She reported a history of SLE with lupus nephritis, hypertension, and a nodular goiter following a partial thyroidectomy. She noted that she did not seek treatment for the skin findings sooner because she was more concerned about her other medical conditions; however, because she felt these conditions were now stable, she decided to seek treatment for the “rash.” Physical examination revealed hundreds of café au lait macules and numerous neurofibromas diffusely distributed on the trunk and extremities (Figure 1) as well as bilateral axillary freckling. A clinical diagnosis of NF-1 was made.
When questioned, the patient reported that she may have been diagnosed with NF-1 in the past by another physician, but she did not recall it specifically. The patient was advised that there were no treatments for the café au lait macules. We notified her other physicians of the NF-1 diagnosis so she could be monitored for systemic conditions related to NF-1, including optic gliomas, pheochromocytoma, renal artery stenosis, and internal neurofibromas. We also referred the patient for genetic counseling; of note, the patient reported she had 4 children without any evidence of similar skin lesions or chronic health problems.
A PubMed search of articles indexed for MEDLINE using the terms systemic lupus and neurofibromatosis yielded 8 cases of patients having both SLE and NF-1 (including our case).6-11 Our patient reported having multiple lesions since birth, decades before the onset and diagnosis of SLE. In 3 other cases, patients were diagnosed with SLE and then presented with neurofibromas, leading to NF-1 diagnosis.In the discussion of those 3 cases, it was proposed that immune system alterations caused by SLE leading to viral illness may have predisposed the patients to the development of tumors and other collagen diseases, or it could be coincidental.6,7 In another case, a patient with NF-1 developed SLE, which was thought to be coincidental.8 Akyuz et al9 described the case of a pediatric patient with NF-1 who subsequently was diagnosed with SLE. The authors suggested that the lack of neurofibromin contributed to the development of SLE, an autoimmune condition. Under normal circumstances, neurofibromin acts as a guanosine triphosphatase–activating protein for RAS in T cells.10 CD8+ T-cell function also is impaired in patients with SLE.9 Additionally, it has been reported that anti–double-stranded DNA antibodies and immune complexes were present in NF-1 patients, even though there were low titers.12 Thus, the authors proposed that the lack of neurofibromin led to dysregulation of the RAS pathway and impairment of T cells, creating an immune milieu that predisposed the patient to development of SLE. Our case gives additional credence to this theory, as our patient had a similar clinical course: the café au lait macules were present since birth and the symptoms of SLE surfaced much later in her late 20s and 30s. Another case by Makino and Tampo10 described a patient with a history of SLE who was later diagnosed with NF-1 based on choroidal findings highly specific for NF-1 but did not have other classic findings of NF-1. The authors mentioned that there might be a potential relationship between these two disorders but did not speculate any theory in particular for their case.10
The interplay between an autoimmune condition such as SLE and NF-1, a condition traditionally thought to be due to a genetic mutation, may have greater clinical and therapeutic implications beyond just these two disorders. Although it is well established that RAS pathway disruption causes NF-1, it has been uncovered that dysfunction in the RAS pathway also can contribute to melanoma oncogenesis.13,14 These insights have led to the development of and approval of targeted drugs designed to inhibit the RAS pathway (eg, vemurafenib, dabrafenib, trametinib).14-17 Melanoma also is considered a “model” tumor for studying the relationship between the immune system and cancer.18
Our case also is instructive in another point: our patient had never sought treatment for her skin lesions, as she said she had other more serious health conditions. Closer evaluation of her skin condition may have led to earlier diagnosis of NF-1, which has important health implications. The average lifespan of patients with NF-1 is 10 to 15 years lower than the general population, with cancer being the leading cause of death.20 Malignant peripheral nerve sheath tumors are the most common malignant tumors observed in such patients.21-23 Other cancers that are associated with NF-1 include rhabdomyosarcomas, gastrointestinal stromal tumors, neuroectodermal tumors, pheochromocytomas, and breast carcinomas.23
To make a clinical diagnosis of NF-1, a patient must have 2 of 7 cardinal clinical features as defined by the National Institutes of Health (Table).24 In our patient with hundreds of café au lait macules and dozens of neurofibromas, the diagnosis was clear; however, in other patients, the skin findings of NF-1 may not be as prominent. A patient could meet criteria for NF-1 diagnosis with the inconspicuous presentation of 6 café au lait macules and either 1 plexiform neurofibroma or 2 neurofibromas (of any type) on the entire body.
We recommend that patients with SLE undergo skin examinations to look for more subtle presentations of NF-1. Earlier diagnosis will help to initiate close monitoring of the disorder’s associated systemic health risks. In addition, the identification of more patients with both NF-1 and SLE may help shed light on the etiology of both conditions.
To the Editor:
Patients with concurrent neurofibromatosis type 1 (NF-1) and systemic lupus erythematosus (SLE) rarely have been reported in the literature. Neurofibromatosis type 1 is one of the most common genetic disorders, with a worldwide birth incidence of 1 in 2500 individuals and prevalence of 1 in 4000 individuals.1 The incidence and prevalence of SLE varies widely depending on race and geographic location. Estimated incidence rates for SLE range from 1 to 25 per 100,000 individuals annually in North America, South America, Europe, and Asia.2,3 The reported worldwide prevalence is 20 to 150 cases per 100,000 individuals annually.2,4,5
Given the high prevalence of both conditions, the association between SLE and NF-1 likely is underrecognized; therefore, identifying more patients with concurrent SLE and NF-1 and describing the interplay between the 2 conditions may have important therapeutic implications. We present the case of a middle-aged woman with a history of SLE who had cutaneous lesions characteristic of NF-1 to further the understanding of these concurrent conditions.
A middle-aged woman presented to our academic dermatology clinic for evaluation and removal of dark spots that had been present diffusely on the trunk and extremities since birth. She reported a history of SLE with lupus nephritis, hypertension, and a nodular goiter following a partial thyroidectomy. She noted that she did not seek treatment for the skin findings sooner because she was more concerned about her other medical conditions; however, because she felt these conditions were now stable, she decided to seek treatment for the “rash.” Physical examination revealed hundreds of café au lait macules and numerous neurofibromas diffusely distributed on the trunk and extremities (Figure 1) as well as bilateral axillary freckling. A clinical diagnosis of NF-1 was made.
When questioned, the patient reported that she may have been diagnosed with NF-1 in the past by another physician, but she did not recall it specifically. The patient was advised that there were no treatments for the café au lait macules. We notified her other physicians of the NF-1 diagnosis so she could be monitored for systemic conditions related to NF-1, including optic gliomas, pheochromocytoma, renal artery stenosis, and internal neurofibromas. We also referred the patient for genetic counseling; of note, the patient reported she had 4 children without any evidence of similar skin lesions or chronic health problems.
A PubMed search of articles indexed for MEDLINE using the terms systemic lupus and neurofibromatosis yielded 8 cases of patients having both SLE and NF-1 (including our case).6-11 Our patient reported having multiple lesions since birth, decades before the onset and diagnosis of SLE. In 3 other cases, patients were diagnosed with SLE and then presented with neurofibromas, leading to NF-1 diagnosis.In the discussion of those 3 cases, it was proposed that immune system alterations caused by SLE leading to viral illness may have predisposed the patients to the development of tumors and other collagen diseases, or it could be coincidental.6,7 In another case, a patient with NF-1 developed SLE, which was thought to be coincidental.8 Akyuz et al9 described the case of a pediatric patient with NF-1 who subsequently was diagnosed with SLE. The authors suggested that the lack of neurofibromin contributed to the development of SLE, an autoimmune condition. Under normal circumstances, neurofibromin acts as a guanosine triphosphatase–activating protein for RAS in T cells.10 CD8+ T-cell function also is impaired in patients with SLE.9 Additionally, it has been reported that anti–double-stranded DNA antibodies and immune complexes were present in NF-1 patients, even though there were low titers.12 Thus, the authors proposed that the lack of neurofibromin led to dysregulation of the RAS pathway and impairment of T cells, creating an immune milieu that predisposed the patient to development of SLE. Our case gives additional credence to this theory, as our patient had a similar clinical course: the café au lait macules were present since birth and the symptoms of SLE surfaced much later in her late 20s and 30s. Another case by Makino and Tampo10 described a patient with a history of SLE who was later diagnosed with NF-1 based on choroidal findings highly specific for NF-1 but did not have other classic findings of NF-1. The authors mentioned that there might be a potential relationship between these two disorders but did not speculate any theory in particular for their case.10
The interplay between an autoimmune condition such as SLE and NF-1, a condition traditionally thought to be due to a genetic mutation, may have greater clinical and therapeutic implications beyond just these two disorders. Although it is well established that RAS pathway disruption causes NF-1, it has been uncovered that dysfunction in the RAS pathway also can contribute to melanoma oncogenesis.13,14 These insights have led to the development of and approval of targeted drugs designed to inhibit the RAS pathway (eg, vemurafenib, dabrafenib, trametinib).14-17 Melanoma also is considered a “model” tumor for studying the relationship between the immune system and cancer.18
Our case also is instructive in another point: our patient had never sought treatment for her skin lesions, as she said she had other more serious health conditions. Closer evaluation of her skin condition may have led to earlier diagnosis of NF-1, which has important health implications. The average lifespan of patients with NF-1 is 10 to 15 years lower than the general population, with cancer being the leading cause of death.20 Malignant peripheral nerve sheath tumors are the most common malignant tumors observed in such patients.21-23 Other cancers that are associated with NF-1 include rhabdomyosarcomas, gastrointestinal stromal tumors, neuroectodermal tumors, pheochromocytomas, and breast carcinomas.23
To make a clinical diagnosis of NF-1, a patient must have 2 of 7 cardinal clinical features as defined by the National Institutes of Health (Table).24 In our patient with hundreds of café au lait macules and dozens of neurofibromas, the diagnosis was clear; however, in other patients, the skin findings of NF-1 may not be as prominent. A patient could meet criteria for NF-1 diagnosis with the inconspicuous presentation of 6 café au lait macules and either 1 plexiform neurofibroma or 2 neurofibromas (of any type) on the entire body.
We recommend that patients with SLE undergo skin examinations to look for more subtle presentations of NF-1. Earlier diagnosis will help to initiate close monitoring of the disorder’s associated systemic health risks. In addition, the identification of more patients with both NF-1 and SLE may help shed light on the etiology of both conditions.
- Carey JC, Baty BJ, Johnson JP, et al. The genetic aspects of neurofibromatosis. Ann N Y Acad Sci. 1986;486:45-56.
- Pons-Estel GJ, Alarcón GS, Scofield L, et al. Understanding the epidemiology and progression of systemic lupus erythematosus. Semin Arthritis Rheum. 2010;39:257-268.
- Danchenko N, Satia JA, Anthony MS. Epidemiology of systemic lupus erythematosus: a comparison of worldwide disease burden. Lupus. 2006;15:308-318.
- Lawrence RC, Helmick CG, Arnett FC, et al. Estimates of the prevalence of arthritis and selected musculoskeletal disorders in the United States. Arthritis Rheum. 1998;41:778-799.
- Chakravarty EF, Bush TM, Manzi S, et al. Prevalence of adult systemic lupus erythematosus in California and Pennsylvania in 2000: estimates obtained using hospitalization data. Arthritis Rheum. 2007;56:2092-2094.
- Bitnun S, Bassan H. Letter: neurofibromatosis and SLE. N Engl J Med. 1975;292:429-430.
- Riccardi VM. Neurofibromatosis in a patient with systemic lupus erythematosus. Arthritis Rheum. 1983;26:574.
- Corominas H, Guardiola JM, Matas L, et al. Neurofibromatosis and systemic lupus erythematosus. a matter of coincidence? Clin Rhematol. 2003;22:496-497.
- Akyuz SG, Caltik A, Bulbul M, et al. An unusual pediatric case with neurofibromatosis and systemic lupus erythematosus. Rheumatol Int. 2012;32:2345-47.
- Makino S, Tampo H. Rare and unusual choroidal abnormalities in a patient with systemic lupus erythematosus. Case Rep Ophthalmol. 2013;4:81-86.
- Galvan JM, Hofkamp MP. Usefulness of intrapartum magnetic resonance imaging for a parturient with neurofibromatosis type I during induction of labor for preeclampsia. Proc (Bayl Univ Med Cent). 2018;31:92-93.
- Gerosa PL, Vai C, Bizzozer L, et al. Immunological and clinical surveillance in Recklinghausen’s neurofibromatosis (NF1). Panminerva Med. 1993;35:80-85.
- Busca R, Abbe P, Mantoux F, et al. RAS mediates the cAMP-dependent activation of extracellular signal-regulated kinases (ERKs) in melanocytes. EMBO J. 2000;19:2900-2910.
- Sullivan RJ, Flaherty K. MAP kinase signaling and inhibition in melanoma. Oncogene. 2013;32:2373-2379.
- Hennessy BT, Smith DL, Ram PT, et al. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov. 2005;12:988-1004.
- Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507-2516.
- Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380:358-365.
- Maio M. Melanoma as a model tumour for immuno-oncology. Ann Oncol. 2012;23:(suppl 8):viii10-4.
- Zmajkovicova K, Jesenberger V, Catalanotti F, et al. MEK1 is required for PTEN membrane recruitment, AKT regulation, and the maintenance of peripheral tolerance. Mol Cell. 2013;50:43-55.
- Patil S, Chamberlain RS. Neoplasms associated with germline and somatic NF1 gene mutations. Oncologist. 2012;17:101-116.
- Carroll SL, Ratner N. How does the Schwann cell lineage form tumors in NF1? Glia. 2008;56:1590-1605.
- Rasmussen SA, Friedman JM. NF1 gene and neurofibromatosis 1. Am J Epidemiol. 2000;151:33-40.
- Yohay K. Neurofibromatosis type 1 and associated malignancies. Curr Neurol Neurosci Rep. 2009;9:247-253.
- Neurofibromatosis. conference statement. National Institutes of Health Consensus Development Conference. Arch Neurol. 1988;45:575-78.
- Carey JC, Baty BJ, Johnson JP, et al. The genetic aspects of neurofibromatosis. Ann N Y Acad Sci. 1986;486:45-56.
- Pons-Estel GJ, Alarcón GS, Scofield L, et al. Understanding the epidemiology and progression of systemic lupus erythematosus. Semin Arthritis Rheum. 2010;39:257-268.
- Danchenko N, Satia JA, Anthony MS. Epidemiology of systemic lupus erythematosus: a comparison of worldwide disease burden. Lupus. 2006;15:308-318.
- Lawrence RC, Helmick CG, Arnett FC, et al. Estimates of the prevalence of arthritis and selected musculoskeletal disorders in the United States. Arthritis Rheum. 1998;41:778-799.
- Chakravarty EF, Bush TM, Manzi S, et al. Prevalence of adult systemic lupus erythematosus in California and Pennsylvania in 2000: estimates obtained using hospitalization data. Arthritis Rheum. 2007;56:2092-2094.
- Bitnun S, Bassan H. Letter: neurofibromatosis and SLE. N Engl J Med. 1975;292:429-430.
- Riccardi VM. Neurofibromatosis in a patient with systemic lupus erythematosus. Arthritis Rheum. 1983;26:574.
- Corominas H, Guardiola JM, Matas L, et al. Neurofibromatosis and systemic lupus erythematosus. a matter of coincidence? Clin Rhematol. 2003;22:496-497.
- Akyuz SG, Caltik A, Bulbul M, et al. An unusual pediatric case with neurofibromatosis and systemic lupus erythematosus. Rheumatol Int. 2012;32:2345-47.
- Makino S, Tampo H. Rare and unusual choroidal abnormalities in a patient with systemic lupus erythematosus. Case Rep Ophthalmol. 2013;4:81-86.
- Galvan JM, Hofkamp MP. Usefulness of intrapartum magnetic resonance imaging for a parturient with neurofibromatosis type I during induction of labor for preeclampsia. Proc (Bayl Univ Med Cent). 2018;31:92-93.
- Gerosa PL, Vai C, Bizzozer L, et al. Immunological and clinical surveillance in Recklinghausen’s neurofibromatosis (NF1). Panminerva Med. 1993;35:80-85.
- Busca R, Abbe P, Mantoux F, et al. RAS mediates the cAMP-dependent activation of extracellular signal-regulated kinases (ERKs) in melanocytes. EMBO J. 2000;19:2900-2910.
- Sullivan RJ, Flaherty K. MAP kinase signaling and inhibition in melanoma. Oncogene. 2013;32:2373-2379.
- Hennessy BT, Smith DL, Ram PT, et al. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov. 2005;12:988-1004.
- Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507-2516.
- Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380:358-365.
- Maio M. Melanoma as a model tumour for immuno-oncology. Ann Oncol. 2012;23:(suppl 8):viii10-4.
- Zmajkovicova K, Jesenberger V, Catalanotti F, et al. MEK1 is required for PTEN membrane recruitment, AKT regulation, and the maintenance of peripheral tolerance. Mol Cell. 2013;50:43-55.
- Patil S, Chamberlain RS. Neoplasms associated with germline and somatic NF1 gene mutations. Oncologist. 2012;17:101-116.
- Carroll SL, Ratner N. How does the Schwann cell lineage form tumors in NF1? Glia. 2008;56:1590-1605.
- Rasmussen SA, Friedman JM. NF1 gene and neurofibromatosis 1. Am J Epidemiol. 2000;151:33-40.
- Yohay K. Neurofibromatosis type 1 and associated malignancies. Curr Neurol Neurosci Rep. 2009;9:247-253.
- Neurofibromatosis. conference statement. National Institutes of Health Consensus Development Conference. Arch Neurol. 1988;45:575-78.
Practice Points
- Patients with neurofibromatosis type 1 (NF-1) benefit from early diagnosis and long-term follow-up.
- Patients with systemic lupus erythematosus (SLE) may develop different malignancies given the immune dysregulation. We recommend that patients with SLE undergo detailed skin examinations to check for subtle clues for NF-1.
- Similarly, patients with NF-1 can develop SLE later in life.
Locally Destructive Metastatic Basal Cell Carcinoma
To the Editor:
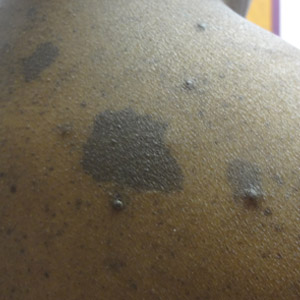
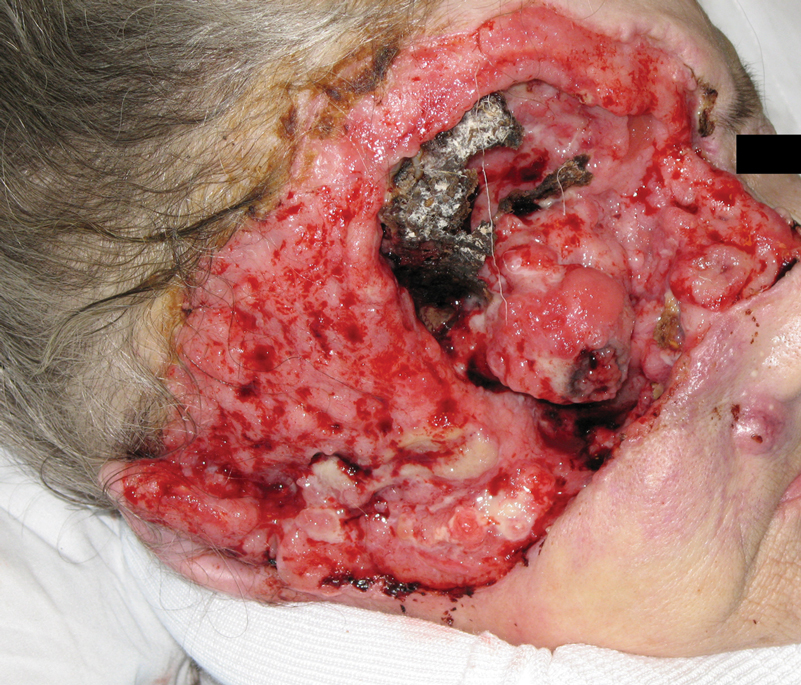
A 60-year-old woman with a history of lymphoma presented to the emergency department for evaluation of intermittent diarrhea and vomiting of 2 weeks’ duration. On presentation, a rather large dressing covering the entire right half of the face was noted. Removal of the bandage revealed a necrotic, extensively destructive, right-sided facial lesion with a fully exposed ocular globe (Figure 1). The patient lived alone and was accompanied by a neighbor, who disclosed that the lesion had been neglected and enlarged over the last 15 years. Moreover, the neighbor reported that the patient had recently experienced several episodes of vertigo and frequent falls.
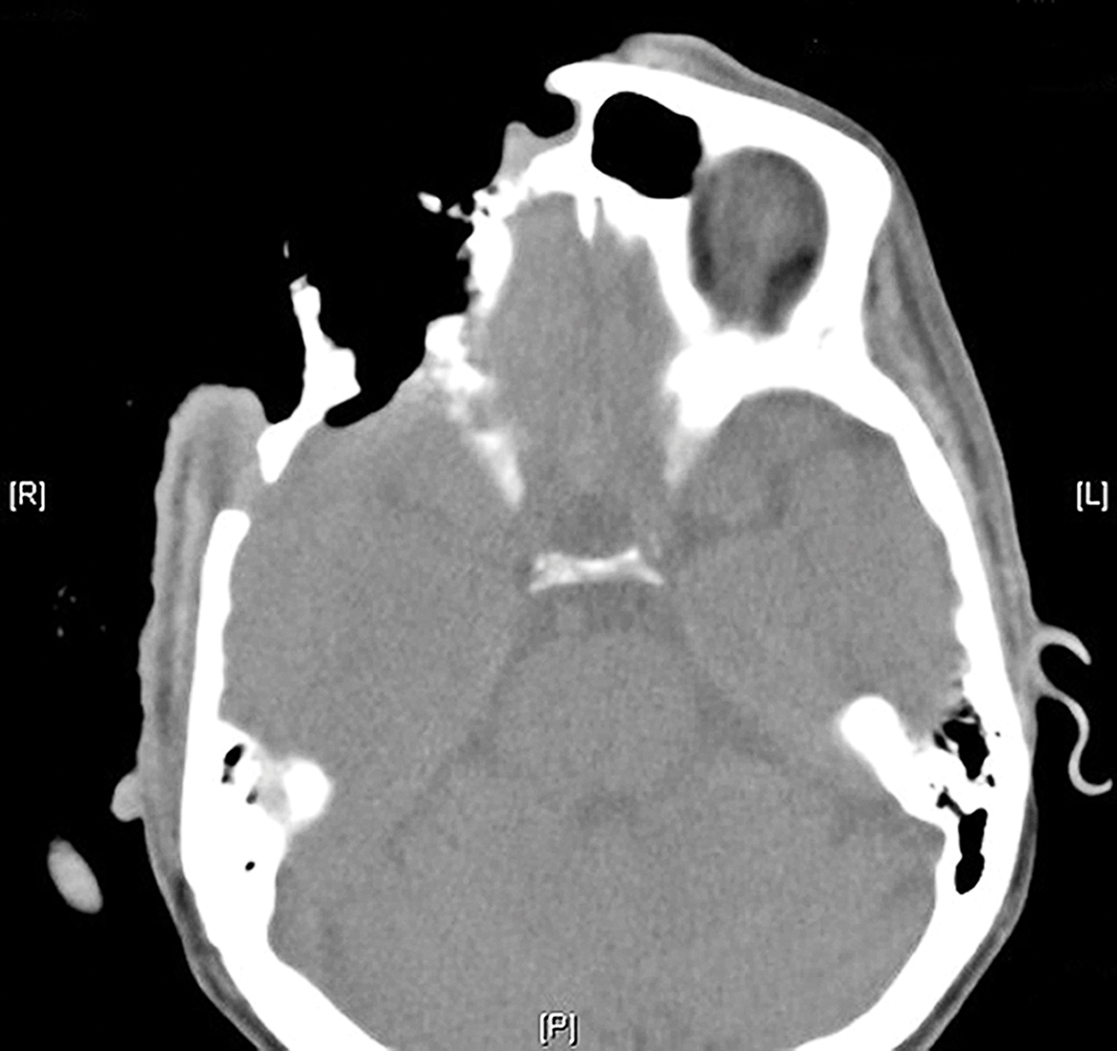
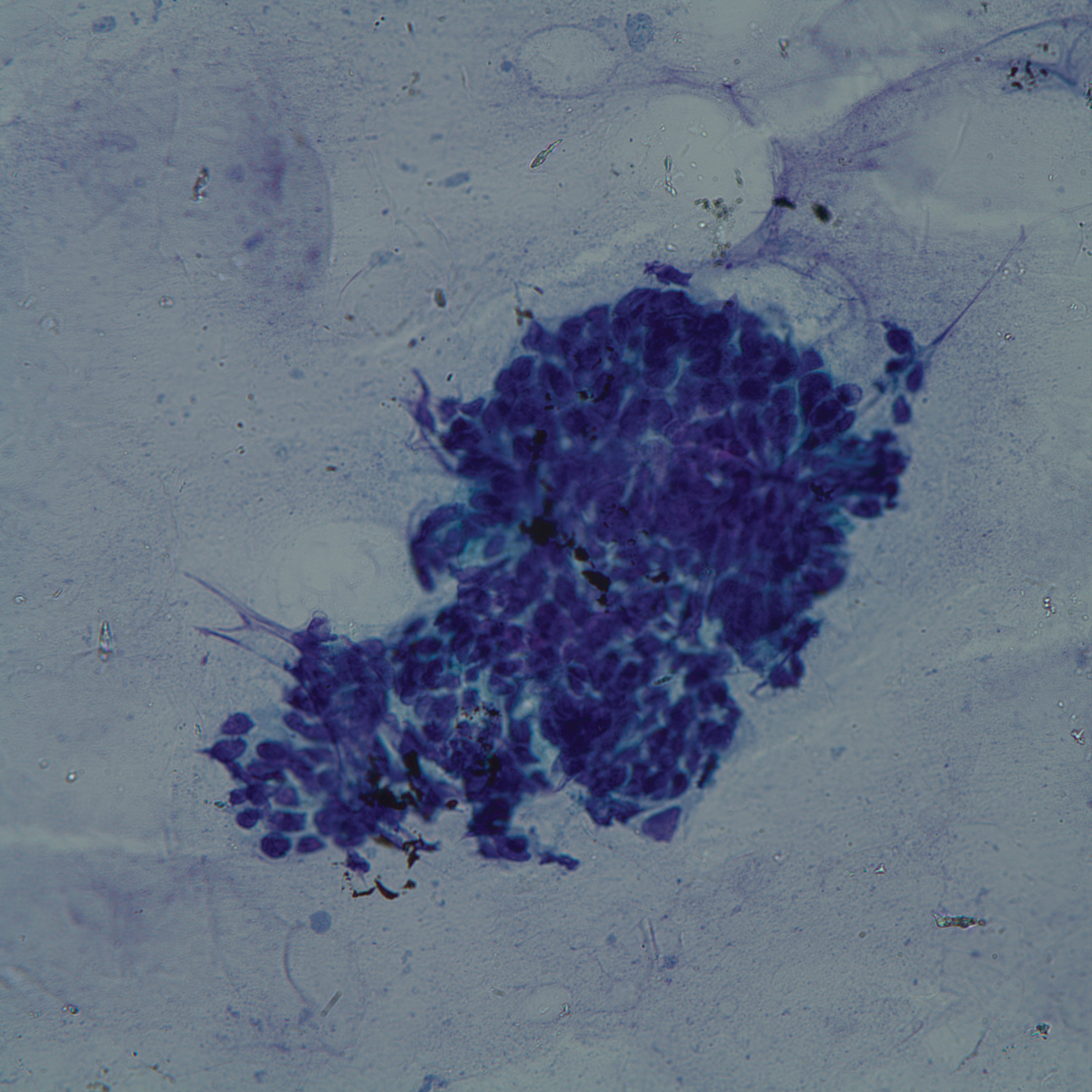
On admission to the hospital, dermatology was consulted and initial workup included computed tomography (CT) scan of the head and maxillofacial region, which showed a destructive process involving the right frontotemporal bone, maxillofacial region, sphenoid, and skull base with exposure of intracranial contents (Figure 2). An aggressive wound care regimen was instituted. Biopsy of the wound margin revealed nodular and focally infiltrative basal cell carcinoma (BCC) (Figure 3).
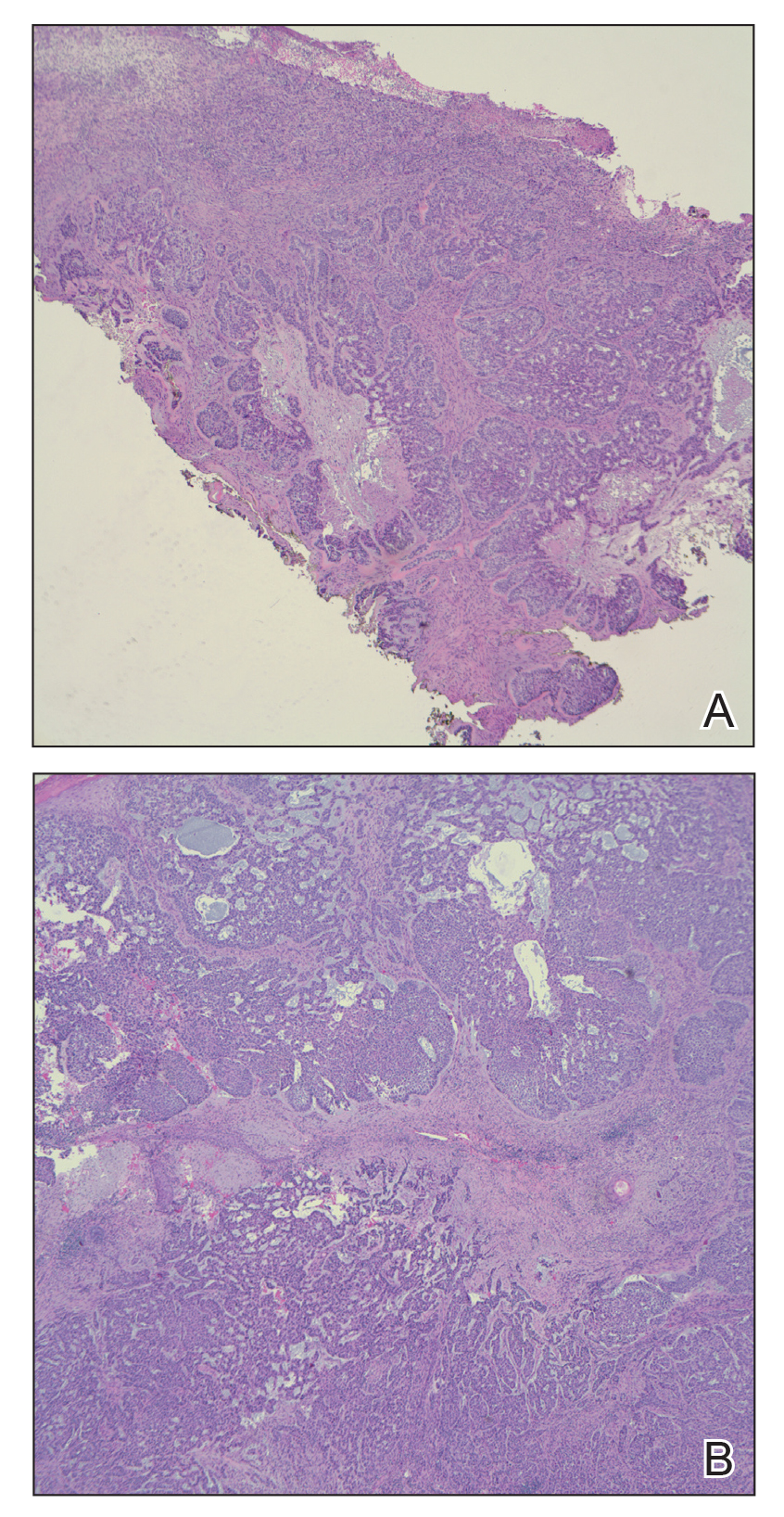
and focally infiltrative basal cell carcinoma (H&E, original magnifications×4 and ×10).
Several days into her hospitalization, the patient developed radicular pain in both arms and weakness in all 4 extremities. A CT scan of the neck revealed a pathologic fracture of the C7 vertebrae. Several medical and surgical services as well as psychiatry were consulted. Given the extensive nature of the disease involvement with limited treatment options, the patient sought to forego further interventions and was discharged to hospice care.
Basal cell carcinomas rarely metastasize, with a reported incidence of 0.0028% to 0.5%.1 The likelihood of metastasis is most closely related to tumor size and depth of invasion. Tumors greater than 3 cm in diameter have a 2% incidence of metastatic spread and/or death. The incidence of metastatic spread and/or death is estimated to be 25% for tumors with a diameter of 5 cm and 50% for tumors with a diameter of 10 cm or greater.2 Other risk factors for metastatic spread include long duration of disease, failure to respond to conventional treatment, and prior radiation treatment in the affected area.1 In one review, the median interval between onset of BCC and metastasis was 9 years.3 In our case, 15 years of neglect most likely led to the aggressiveness of the tumor. Although the workup in our patient was limited per her request, there was no evidence that her lymphoma had recurred or that she was in any other way immunocompromised. Unfortunately, in this patient’s case, the local destructiveness of the carcinoma with subsequent bony invasion and necrosis was complicated with secondary Rhizopus infection. A PubMed search of articles indexed for MEDLINE using the terms basal cell carcinoma and mucormycosis revealed no other reported cases of BCC associated with mucormycosis; therefore, our case represents a rare presentation of this association. Rhinocerebral mucormycosis is the most common manifestation of mucormycosis and more commonly occurs in diabetics with ketoacidosis and in severely debilitated or immunosuppressed individuals.4 The extensive bony destruction, especially of the nasal region, of our patient’s tumor likely led to secondary infection with Rhizopus.
Approximately 85% of all metastatic BCCs originate in the head and neck region, with lymph nodes being the first site of metastasis and involved in approximately half of all cases.1,4 Metastases to the lungs, bone, liver, and other viscera can occur with advanced disease. Metastasis generally portends a poor prognosis, with survival rarely exceeding 1.5 years. Until recently, therapeutic options for metastatic disease were limited, with marginal response to chemotherapy with methotrexate, fluorouracil, bleomycin, and cisplatin.4 Vismodegib, a novel smoothened receptor inhibitor that blocks the sonic hedgehog pathway implicated in BCC carcinogenesis, offers a new promising treatment for management and control of advanced disease.5
- Junior W, Ribeiro SC, Vieira SC, et al. Metastatic basal cell carcinoma: a case report. Dermatol Online J. 2003;9:18.
- Snow SN, Sahl WJ, Lo J, et al. Metastatic basal cell carcinoma: report of 5 cases. Cancer. 1994;73:328-335.
- Von Domarus H, Stevens PJ. Metastatic basal cell carcinoma: report of five cases and review of 170 cases in the literature. J Am Acad Dermatol. 1984;10:1043-1060.
- Kolekar JS. Rhinocerebral mucormycosis: a retrospective study. Indian J Otolaryngol Head Neck Surg. 2015;67:93-96.
- Koehlblinger P, Lang R. New developments in the treatment of basal cell carcinoma: update on current and emerging treatment options with a focus on vismodegib. Onc Targets Ther. 2018;11:8327-8340.
To the Editor:
A 60-year-old woman with a history of lymphoma presented to the emergency department for evaluation of intermittent diarrhea and vomiting of 2 weeks’ duration. On presentation, a rather large dressing covering the entire right half of the face was noted. Removal of the bandage revealed a necrotic, extensively destructive, right-sided facial lesion with a fully exposed ocular globe (Figure 1). The patient lived alone and was accompanied by a neighbor, who disclosed that the lesion had been neglected and enlarged over the last 15 years. Moreover, the neighbor reported that the patient had recently experienced several episodes of vertigo and frequent falls.
On admission to the hospital, dermatology was consulted and initial workup included computed tomography (CT) scan of the head and maxillofacial region, which showed a destructive process involving the right frontotemporal bone, maxillofacial region, sphenoid, and skull base with exposure of intracranial contents (Figure 2). An aggressive wound care regimen was instituted. Biopsy of the wound margin revealed nodular and focally infiltrative basal cell carcinoma (BCC) (Figure 3).
and focally infiltrative basal cell carcinoma (H&E, original magnifications×4 and ×10).
Several days into her hospitalization, the patient developed radicular pain in both arms and weakness in all 4 extremities. A CT scan of the neck revealed a pathologic fracture of the C7 vertebrae. Several medical and surgical services as well as psychiatry were consulted. Given the extensive nature of the disease involvement with limited treatment options, the patient sought to forego further interventions and was discharged to hospice care.
Basal cell carcinomas rarely metastasize, with a reported incidence of 0.0028% to 0.5%.1 The likelihood of metastasis is most closely related to tumor size and depth of invasion. Tumors greater than 3 cm in diameter have a 2% incidence of metastatic spread and/or death. The incidence of metastatic spread and/or death is estimated to be 25% for tumors with a diameter of 5 cm and 50% for tumors with a diameter of 10 cm or greater.2 Other risk factors for metastatic spread include long duration of disease, failure to respond to conventional treatment, and prior radiation treatment in the affected area.1 In one review, the median interval between onset of BCC and metastasis was 9 years.3 In our case, 15 years of neglect most likely led to the aggressiveness of the tumor. Although the workup in our patient was limited per her request, there was no evidence that her lymphoma had recurred or that she was in any other way immunocompromised. Unfortunately, in this patient’s case, the local destructiveness of the carcinoma with subsequent bony invasion and necrosis was complicated with secondary Rhizopus infection. A PubMed search of articles indexed for MEDLINE using the terms basal cell carcinoma and mucormycosis revealed no other reported cases of BCC associated with mucormycosis; therefore, our case represents a rare presentation of this association. Rhinocerebral mucormycosis is the most common manifestation of mucormycosis and more commonly occurs in diabetics with ketoacidosis and in severely debilitated or immunosuppressed individuals.4 The extensive bony destruction, especially of the nasal region, of our patient’s tumor likely led to secondary infection with Rhizopus.
Approximately 85% of all metastatic BCCs originate in the head and neck region, with lymph nodes being the first site of metastasis and involved in approximately half of all cases.1,4 Metastases to the lungs, bone, liver, and other viscera can occur with advanced disease. Metastasis generally portends a poor prognosis, with survival rarely exceeding 1.5 years. Until recently, therapeutic options for metastatic disease were limited, with marginal response to chemotherapy with methotrexate, fluorouracil, bleomycin, and cisplatin.4 Vismodegib, a novel smoothened receptor inhibitor that blocks the sonic hedgehog pathway implicated in BCC carcinogenesis, offers a new promising treatment for management and control of advanced disease.5
To the Editor:
A 60-year-old woman with a history of lymphoma presented to the emergency department for evaluation of intermittent diarrhea and vomiting of 2 weeks’ duration. On presentation, a rather large dressing covering the entire right half of the face was noted. Removal of the bandage revealed a necrotic, extensively destructive, right-sided facial lesion with a fully exposed ocular globe (Figure 1). The patient lived alone and was accompanied by a neighbor, who disclosed that the lesion had been neglected and enlarged over the last 15 years. Moreover, the neighbor reported that the patient had recently experienced several episodes of vertigo and frequent falls.
On admission to the hospital, dermatology was consulted and initial workup included computed tomography (CT) scan of the head and maxillofacial region, which showed a destructive process involving the right frontotemporal bone, maxillofacial region, sphenoid, and skull base with exposure of intracranial contents (Figure 2). An aggressive wound care regimen was instituted. Biopsy of the wound margin revealed nodular and focally infiltrative basal cell carcinoma (BCC) (Figure 3).
and focally infiltrative basal cell carcinoma (H&E, original magnifications×4 and ×10).
Several days into her hospitalization, the patient developed radicular pain in both arms and weakness in all 4 extremities. A CT scan of the neck revealed a pathologic fracture of the C7 vertebrae. Several medical and surgical services as well as psychiatry were consulted. Given the extensive nature of the disease involvement with limited treatment options, the patient sought to forego further interventions and was discharged to hospice care.
Basal cell carcinomas rarely metastasize, with a reported incidence of 0.0028% to 0.5%.1 The likelihood of metastasis is most closely related to tumor size and depth of invasion. Tumors greater than 3 cm in diameter have a 2% incidence of metastatic spread and/or death. The incidence of metastatic spread and/or death is estimated to be 25% for tumors with a diameter of 5 cm and 50% for tumors with a diameter of 10 cm or greater.2 Other risk factors for metastatic spread include long duration of disease, failure to respond to conventional treatment, and prior radiation treatment in the affected area.1 In one review, the median interval between onset of BCC and metastasis was 9 years.3 In our case, 15 years of neglect most likely led to the aggressiveness of the tumor. Although the workup in our patient was limited per her request, there was no evidence that her lymphoma had recurred or that she was in any other way immunocompromised. Unfortunately, in this patient’s case, the local destructiveness of the carcinoma with subsequent bony invasion and necrosis was complicated with secondary Rhizopus infection. A PubMed search of articles indexed for MEDLINE using the terms basal cell carcinoma and mucormycosis revealed no other reported cases of BCC associated with mucormycosis; therefore, our case represents a rare presentation of this association. Rhinocerebral mucormycosis is the most common manifestation of mucormycosis and more commonly occurs in diabetics with ketoacidosis and in severely debilitated or immunosuppressed individuals.4 The extensive bony destruction, especially of the nasal region, of our patient’s tumor likely led to secondary infection with Rhizopus.
Approximately 85% of all metastatic BCCs originate in the head and neck region, with lymph nodes being the first site of metastasis and involved in approximately half of all cases.1,4 Metastases to the lungs, bone, liver, and other viscera can occur with advanced disease. Metastasis generally portends a poor prognosis, with survival rarely exceeding 1.5 years. Until recently, therapeutic options for metastatic disease were limited, with marginal response to chemotherapy with methotrexate, fluorouracil, bleomycin, and cisplatin.4 Vismodegib, a novel smoothened receptor inhibitor that blocks the sonic hedgehog pathway implicated in BCC carcinogenesis, offers a new promising treatment for management and control of advanced disease.5
- Junior W, Ribeiro SC, Vieira SC, et al. Metastatic basal cell carcinoma: a case report. Dermatol Online J. 2003;9:18.
- Snow SN, Sahl WJ, Lo J, et al. Metastatic basal cell carcinoma: report of 5 cases. Cancer. 1994;73:328-335.
- Von Domarus H, Stevens PJ. Metastatic basal cell carcinoma: report of five cases and review of 170 cases in the literature. J Am Acad Dermatol. 1984;10:1043-1060.
- Kolekar JS. Rhinocerebral mucormycosis: a retrospective study. Indian J Otolaryngol Head Neck Surg. 2015;67:93-96.
- Koehlblinger P, Lang R. New developments in the treatment of basal cell carcinoma: update on current and emerging treatment options with a focus on vismodegib. Onc Targets Ther. 2018;11:8327-8340.
- Junior W, Ribeiro SC, Vieira SC, et al. Metastatic basal cell carcinoma: a case report. Dermatol Online J. 2003;9:18.
- Snow SN, Sahl WJ, Lo J, et al. Metastatic basal cell carcinoma: report of 5 cases. Cancer. 1994;73:328-335.
- Von Domarus H, Stevens PJ. Metastatic basal cell carcinoma: report of five cases and review of 170 cases in the literature. J Am Acad Dermatol. 1984;10:1043-1060.
- Kolekar JS. Rhinocerebral mucormycosis: a retrospective study. Indian J Otolaryngol Head Neck Surg. 2015;67:93-96.
- Koehlblinger P, Lang R. New developments in the treatment of basal cell carcinoma: update on current and emerging treatment options with a focus on vismodegib. Onc Targets Ther. 2018;11:8327-8340.
Practice Points
- Risk factors associated with metastatic spread of basal cell carcinoma (BCC) include larger tumor size, greater depth of invasion, long duration of disease, failure to respond to conventional treatment, and prior radiation treatment in the affected area.
- The median interval between onset of BCC and metastasis has been shown to be approximately 9 years.
- Vismodegib can be an effective oral therapy for patients with metastatic BCC, locally advanced BCC, recurrence following surgery, or those who are not candidates for surgery or radiation.
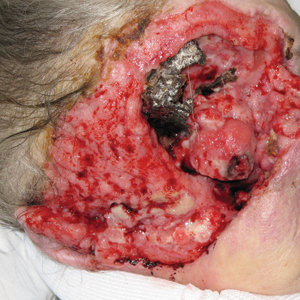
Multicentric Reticulohistiocytosis With Arthralgia and Red-Orange Papulonodules
To the Editor:
A 50-year-old woman presented with an asymptomatic eruption on the dorsal aspect of the hands, abdomen, and face of 6 months’ duration. The eruption was associated with generalized arthralgia and fatigue. Within several weeks of onset of the cutaneous eruption, the patient developed swelling in the hands as well as worsening arthralgia. She was treated for presumed Lyme borreliosis but reported no improvement in the symptoms. She was then referred to dermatology for further management.
Physical examination revealed red-orange, edematous, monomorphic papulonodules scattered on the nasolabial folds, upper lip, and along the dorsal aspect of the hands and fingers (Figure 1). A brown rippled plaque was present on the left lower abdomen. The oral mucosa and nails were unremarkable. Laboratory studies showed elevated total cholesterol (244 mg/dL [reference range, <200 mg/dL]), low-density lipoproteins (130 mg/dL [reference range, 10–30 mg/dL]), aspartate aminotransferase (140 U/L [reference range, 10–30 U/L]), alanine aminotransferase (110 U/L [reference range, 10–40 U/L]), and total bilirubin (1.5 mg/dL [reference range, 0.3–1.2 mg/dL]). White blood cell count and C-reactive protein levels were within reference range. An antinuclear antibody titer of 1:80 with a homogenous pattern was found, and aldolase levels were elevated. Laboratory investigations for rheumatoid factor, Lyme disease, tuberculosis, hepatitis, and human immunodeficiency virus were negative. A chest radiograph was normal.
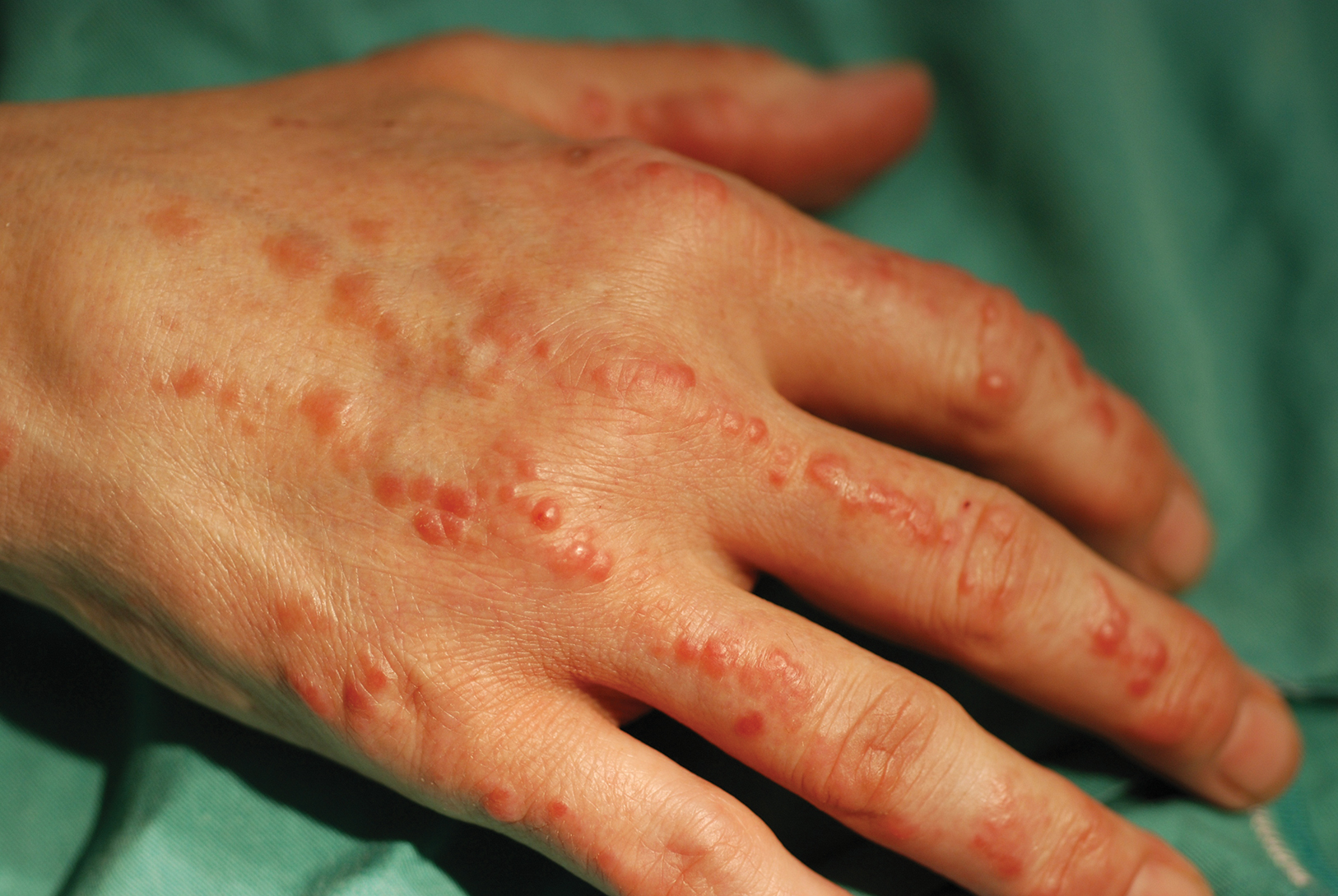
A punch biopsy from the right dorsal hand revealed a dermal proliferation of mononucleated and multinucleated epithelioid histiocytes with ample amounts of eosinophilic ground-glass cytoplasm (Figure 2). Immunohistochemistry revealed epithelioid histiocytes reactive for CD68, CD163, and factor XIIIA, and negative for S-100 and CD1a.
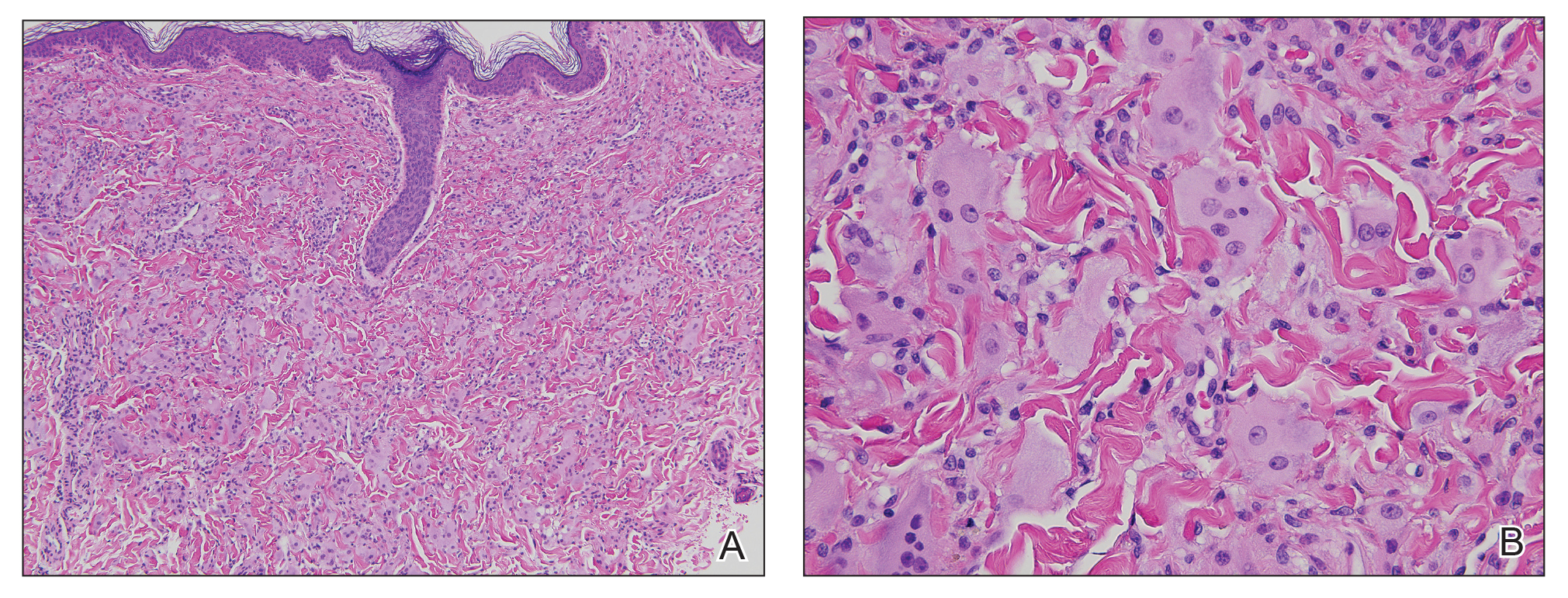
The patient was diagnosed with multicentric reticulohistiocytosis (MRH) and was initially treated with prednisone. Treatment was later augmented with etanercept and methotrexate with improvement in both the skin and joint symptoms.
Multicentric reticulohistiocytosis is a rare, non–Langerhans cell histiocytosis with both cutaneous and systemic features. Although case reports date back to the late 1800s, the term multicentric reticulohistiocytosis was first used in 1954.1 Multicentric reticulohistiocytosis is extremely uncommon and precludes thorough investigation of its etiology and management. The condition typically presents in the fifth to sixth decades of life and occurs more frequently in women with a female to male ratio estimated at 3 to 1.2,3 Pediatric cases have been reported but are exceedingly rare.4
Multicentric reticulohistiocytosis typically presents with a severe erosive arthropathy known as arthritis mutilans. Patients display a symmetric polyarthritis that commonly involves the elbows, wrists, and proximal and distal aspects of the interphalangeal joints. Onset and progression can be rapid, and the erosive nature leads to deformities in up to 45% of patients.2,5,6 Cutaneous findings arise an average of 3 years after the development of arthritis, though one-fifth of patients will initially present with cutaneous findings followed by the development of arthritis at any time.3,6 Clinical features include flesh-colored to reddish brown or yellow papulonodules that range in size from several millimeters to 2 cm. The lesions most commonly occur on the face (eg, ears, nose, paranasal cheeks), scalp, dorsal and lateral aspects of the hands and fingers, and overlying articular regions of the extremities. Characteristic periungual lesions classically are referred to as coral beads.4,6 Patients commonly report pruritus that may precede the development of the papules and nodules. Other cutaneous manifestations include xanthelasma, nail changes, and a photodistributed erythematous maculopapular eruption that may mimic dermatomyositis.6
Cutaneous findings of MRH can mimic rheumatoid nodules, gout, Gottron papules of dermatomyositis, lipoid proteinosis, sarcoidosis, lepromatous leprosy, granuloma annulare, xanthoma, xanthogranuloma, and fibroxanthoma.6,7 Histopathologic features may distinguish MRH from such entities. Findings include fairly well-circumscribed aggregates of large multinucleated giant cells with characteristic eosinophilic ground-glass cytoplasm. Histiocytes stain positively for CD68, HAM56, CD11b, and CD14, and variably for factor XIIIa. CD68, which is expressed by monocytes/macrophages, has been universally reported to be the most reliable marker of MRH. Negative staining for S-100 and CD1a supports a non-Langerhans origin for the involved histiocytes. If arthritic symptoms predominate, MRH must be distinguished from rheumatoid and psoriatic arthritis.6,7
Mucosal involvement occurs in approximately 50% of patients and includes the presence of nodules in the oral, nasal, and pharyngeal mucosae, as well as eye structures.2,3 Histiocytic infiltration has been documented in the heart, lungs, thyroid, liver, stomach, kidneys, muscle, bone marrow, and urogenital tract. Histiocytes also can invade the cartilage of the ears and nose causing disfigurement and characteristic leonine facies. Pathologic fractures may occur with bone involvement.5
Systemic features associated with MRH include hyperlipidemia, diabetes mellitus, thyroid disease, hypergammaglobulinemia, and various autoimmune diseases. Patients less frequently report fever and weight loss.2,5,6,8 Additionally, a positive tuberculin test occurs in 12% to 50% of patients.6 Various autoimmune diseases occur in 6% to 17% of cases including systemic lupus erythematosus, systemic sclerosis, rheumatoid arthritis, dermatomyositis, Sjögren syndrome, and primary biliary cirrhosis.2,5,6,8 The most clinically salient feature of MRH is its association with malignant conditions, which occur in up to 31% of patients. A variety of cancers have been reported in association with MRH, including breast, cervical, ovarian, stomach, penile, lymphoma, mesothelioma, and melanoma.7
The etiology of MRH is unclear. Although onset may precede the development of a malignant condition and regress with treatment, it cannot be considered a true paraneoplastic disorder, as it has no association with a specific cancer and does not typically parallel the disease course.6,9 Reports of increased levels of inflammatory mediators released from macrophages and endothelial cells, specifically IL-12, IL-1β, IL-6, and tumor necrosis factor α (TNF-α), have been thought to drive the destruction of bone and cartilage.6 In particular, TNF-α acts to indirectly induce destruction by stimulating proteolytic activity in macrophages, similar to the pathogenesis of joint damage in rheumatoid arthritis.8 Osteoclastic activity may play a role in the pathogenesis of MRH, as multinucleated giant cells in MRH can mature into osteoclasts by receptor activated nuclear factor–κB ligand signaling. In addition, patients treated with bisphosphonates have had decreased lacunar resorption.2,8
Initial management of MRH should include screening for hyperlipidemia, hypergammaglobulinemia, hyperglycemia, thyroid dysfunction, and autoimmune diseases, as well as age-appropriate cancer screening. Imaging studies should evaluate for the presence of erosive arthritis. There are no well-defined treatment algorithms for MRH due to the rarity of the disease, and recommendations largely rely on case reports. Although spontaneous remission typically occurs within 5 to 10 years, the risk for joint destruction argues for early pharmacologic intervention. Current management includes the use of nonsteroidal anti-inflammatory drugs and various immunosuppressants including oral glucocorticoids, cyclophosphamide, chlorambucil, methotrexate, or azathioprine.2 A combination of methotrexate with cyclophosphamide or glucocorticoids also has shown efficacy.10 Anti–TNF-α agents, such as etanercept, adalimumab, and infliximab, have been used with some success.2 Tumor necrosis factor α inhibitors used in combination with oral glucocorticoids and methotrexate may have an increased benefit.2,9,11 Evidence suggesting that TNF-α plays a role in the destruction of bone and cartilage led to the successful use of infliximab in combination with oral glucocorticoids and methotrexate, which prevented possible development of antibodies to infliximab and increased its efficacy.12 Bisphosphonate use in combination with glucocorticoids and methotrexate may prevent joint destruction without the serious adverse events associated with anti–TNF-α agents.2,9,13,14
- Goltz RW, Laymon CW. Multicentric reticulohistiocytosis of the skin and synovia; reticulohistiocytoma or ganglioneuroma. AMA Arch Derma Syphilol. 1954;69:717-731.
- Islam AD, Naguwa SM, Cheema GS, et al. Multicentric reticulohistiocytosis: a rare yet challenging disease. Clin Rev Allergy Immunol. 2013;45:281-289.
- West KL, Sporn T, Puri PK. Multicentric reticulohistiocytosis: a unique case with pulmonary fibrosis. Arch Dermatol. 2012;148:228-232.
- Outland JD, Keiran SJ, Schikler KN, et al. Multicentric reticulohistiocytosis in a 14-year-old girl. Pediatr Dermatol. 2002;19:527-531.
- Gorman JD, Danning C, Schumacher HR, et al. Multicentric reticulohistiocytosis: case report with immunohistochemical analysis and literature review. Arthritis Rheum. 2000;43:930-938.
- Tajirian AL, Malik MK, Robinson-Bostom L, et al. Multicentric reticulohistiocytosis. Clin Dermatol. 2006;24:486-492.
- Luz FB, Gaspar TAP, Kalil-Gaspar N, et al. Multicentric reticulohistiocytosis. J Eur Acad Dermatol Venereol. 2001;15:524-531.
- Trotta F, Castellino G, Lo Monaco A. Multicentric reticulohistiocytosis. Best Pract Res Clin Rheumatol. 2004;18:759-772.
- Kalajian AH, Callen JP. Multicentric reticulohistiocytosis successfully treated with infliximab: an illustrative case and evaluation of cytokine expression supporting anti-tumor necrosis factor therapy. Arch Derm. 2008;144:1360-1366.
- Liang GC, Granston AS. Complete remission of multicentric reticulohistiocytosis with combination therapy of steroid, cyclophosphamide, and low-dose pulse methotrexate. case report, review of the literature, and proposal for treatment. Arthritis Rheum. 1996;39:171-174.
- Lovelace K, Loyd A, Adelson D, et al. Etanercept and the treatment of multicentric reticulohistiocytosis. Arch Dermatol. 2005;141:1167-1168.
- Lee MW, Lee EY, Jeong YI, et al. Successful treatment of multicentric reticulohistiocytosis with a combination of infliximab, prednisolone and methotrexate. Acta Derm Venereol. 2004;84:478-479.
- Adamopoulos IE, Wordsworth PB, Edwards JR, et al. Osteoclast differentiation and bone resorption in multicentric reticulohistiocytosis. Hum Pathol. 2006;37:1176-1185.
- Satoh M, Oyama N, Yamada H, et al. Treatment trial of multicentric reticulohistiocytosis with a combination of predonisolone, methotrexate and alendronate. J Dermatol. 2008;35:168-171.
To the Editor:
A 50-year-old woman presented with an asymptomatic eruption on the dorsal aspect of the hands, abdomen, and face of 6 months’ duration. The eruption was associated with generalized arthralgia and fatigue. Within several weeks of onset of the cutaneous eruption, the patient developed swelling in the hands as well as worsening arthralgia. She was treated for presumed Lyme borreliosis but reported no improvement in the symptoms. She was then referred to dermatology for further management.
Physical examination revealed red-orange, edematous, monomorphic papulonodules scattered on the nasolabial folds, upper lip, and along the dorsal aspect of the hands and fingers (Figure 1). A brown rippled plaque was present on the left lower abdomen. The oral mucosa and nails were unremarkable. Laboratory studies showed elevated total cholesterol (244 mg/dL [reference range, <200 mg/dL]), low-density lipoproteins (130 mg/dL [reference range, 10–30 mg/dL]), aspartate aminotransferase (140 U/L [reference range, 10–30 U/L]), alanine aminotransferase (110 U/L [reference range, 10–40 U/L]), and total bilirubin (1.5 mg/dL [reference range, 0.3–1.2 mg/dL]). White blood cell count and C-reactive protein levels were within reference range. An antinuclear antibody titer of 1:80 with a homogenous pattern was found, and aldolase levels were elevated. Laboratory investigations for rheumatoid factor, Lyme disease, tuberculosis, hepatitis, and human immunodeficiency virus were negative. A chest radiograph was normal.
A punch biopsy from the right dorsal hand revealed a dermal proliferation of mononucleated and multinucleated epithelioid histiocytes with ample amounts of eosinophilic ground-glass cytoplasm (Figure 2). Immunohistochemistry revealed epithelioid histiocytes reactive for CD68, CD163, and factor XIIIA, and negative for S-100 and CD1a.
The patient was diagnosed with multicentric reticulohistiocytosis (MRH) and was initially treated with prednisone. Treatment was later augmented with etanercept and methotrexate with improvement in both the skin and joint symptoms.
Multicentric reticulohistiocytosis is a rare, non–Langerhans cell histiocytosis with both cutaneous and systemic features. Although case reports date back to the late 1800s, the term multicentric reticulohistiocytosis was first used in 1954.1 Multicentric reticulohistiocytosis is extremely uncommon and precludes thorough investigation of its etiology and management. The condition typically presents in the fifth to sixth decades of life and occurs more frequently in women with a female to male ratio estimated at 3 to 1.2,3 Pediatric cases have been reported but are exceedingly rare.4
Multicentric reticulohistiocytosis typically presents with a severe erosive arthropathy known as arthritis mutilans. Patients display a symmetric polyarthritis that commonly involves the elbows, wrists, and proximal and distal aspects of the interphalangeal joints. Onset and progression can be rapid, and the erosive nature leads to deformities in up to 45% of patients.2,5,6 Cutaneous findings arise an average of 3 years after the development of arthritis, though one-fifth of patients will initially present with cutaneous findings followed by the development of arthritis at any time.3,6 Clinical features include flesh-colored to reddish brown or yellow papulonodules that range in size from several millimeters to 2 cm. The lesions most commonly occur on the face (eg, ears, nose, paranasal cheeks), scalp, dorsal and lateral aspects of the hands and fingers, and overlying articular regions of the extremities. Characteristic periungual lesions classically are referred to as coral beads.4,6 Patients commonly report pruritus that may precede the development of the papules and nodules. Other cutaneous manifestations include xanthelasma, nail changes, and a photodistributed erythematous maculopapular eruption that may mimic dermatomyositis.6
Cutaneous findings of MRH can mimic rheumatoid nodules, gout, Gottron papules of dermatomyositis, lipoid proteinosis, sarcoidosis, lepromatous leprosy, granuloma annulare, xanthoma, xanthogranuloma, and fibroxanthoma.6,7 Histopathologic features may distinguish MRH from such entities. Findings include fairly well-circumscribed aggregates of large multinucleated giant cells with characteristic eosinophilic ground-glass cytoplasm. Histiocytes stain positively for CD68, HAM56, CD11b, and CD14, and variably for factor XIIIa. CD68, which is expressed by monocytes/macrophages, has been universally reported to be the most reliable marker of MRH. Negative staining for S-100 and CD1a supports a non-Langerhans origin for the involved histiocytes. If arthritic symptoms predominate, MRH must be distinguished from rheumatoid and psoriatic arthritis.6,7
Mucosal involvement occurs in approximately 50% of patients and includes the presence of nodules in the oral, nasal, and pharyngeal mucosae, as well as eye structures.2,3 Histiocytic infiltration has been documented in the heart, lungs, thyroid, liver, stomach, kidneys, muscle, bone marrow, and urogenital tract. Histiocytes also can invade the cartilage of the ears and nose causing disfigurement and characteristic leonine facies. Pathologic fractures may occur with bone involvement.5
Systemic features associated with MRH include hyperlipidemia, diabetes mellitus, thyroid disease, hypergammaglobulinemia, and various autoimmune diseases. Patients less frequently report fever and weight loss.2,5,6,8 Additionally, a positive tuberculin test occurs in 12% to 50% of patients.6 Various autoimmune diseases occur in 6% to 17% of cases including systemic lupus erythematosus, systemic sclerosis, rheumatoid arthritis, dermatomyositis, Sjögren syndrome, and primary biliary cirrhosis.2,5,6,8 The most clinically salient feature of MRH is its association with malignant conditions, which occur in up to 31% of patients. A variety of cancers have been reported in association with MRH, including breast, cervical, ovarian, stomach, penile, lymphoma, mesothelioma, and melanoma.7
The etiology of MRH is unclear. Although onset may precede the development of a malignant condition and regress with treatment, it cannot be considered a true paraneoplastic disorder, as it has no association with a specific cancer and does not typically parallel the disease course.6,9 Reports of increased levels of inflammatory mediators released from macrophages and endothelial cells, specifically IL-12, IL-1β, IL-6, and tumor necrosis factor α (TNF-α), have been thought to drive the destruction of bone and cartilage.6 In particular, TNF-α acts to indirectly induce destruction by stimulating proteolytic activity in macrophages, similar to the pathogenesis of joint damage in rheumatoid arthritis.8 Osteoclastic activity may play a role in the pathogenesis of MRH, as multinucleated giant cells in MRH can mature into osteoclasts by receptor activated nuclear factor–κB ligand signaling. In addition, patients treated with bisphosphonates have had decreased lacunar resorption.2,8
Initial management of MRH should include screening for hyperlipidemia, hypergammaglobulinemia, hyperglycemia, thyroid dysfunction, and autoimmune diseases, as well as age-appropriate cancer screening. Imaging studies should evaluate for the presence of erosive arthritis. There are no well-defined treatment algorithms for MRH due to the rarity of the disease, and recommendations largely rely on case reports. Although spontaneous remission typically occurs within 5 to 10 years, the risk for joint destruction argues for early pharmacologic intervention. Current management includes the use of nonsteroidal anti-inflammatory drugs and various immunosuppressants including oral glucocorticoids, cyclophosphamide, chlorambucil, methotrexate, or azathioprine.2 A combination of methotrexate with cyclophosphamide or glucocorticoids also has shown efficacy.10 Anti–TNF-α agents, such as etanercept, adalimumab, and infliximab, have been used with some success.2 Tumor necrosis factor α inhibitors used in combination with oral glucocorticoids and methotrexate may have an increased benefit.2,9,11 Evidence suggesting that TNF-α plays a role in the destruction of bone and cartilage led to the successful use of infliximab in combination with oral glucocorticoids and methotrexate, which prevented possible development of antibodies to infliximab and increased its efficacy.12 Bisphosphonate use in combination with glucocorticoids and methotrexate may prevent joint destruction without the serious adverse events associated with anti–TNF-α agents.2,9,13,14
To the Editor:
A 50-year-old woman presented with an asymptomatic eruption on the dorsal aspect of the hands, abdomen, and face of 6 months’ duration. The eruption was associated with generalized arthralgia and fatigue. Within several weeks of onset of the cutaneous eruption, the patient developed swelling in the hands as well as worsening arthralgia. She was treated for presumed Lyme borreliosis but reported no improvement in the symptoms. She was then referred to dermatology for further management.
Physical examination revealed red-orange, edematous, monomorphic papulonodules scattered on the nasolabial folds, upper lip, and along the dorsal aspect of the hands and fingers (Figure 1). A brown rippled plaque was present on the left lower abdomen. The oral mucosa and nails were unremarkable. Laboratory studies showed elevated total cholesterol (244 mg/dL [reference range, <200 mg/dL]), low-density lipoproteins (130 mg/dL [reference range, 10–30 mg/dL]), aspartate aminotransferase (140 U/L [reference range, 10–30 U/L]), alanine aminotransferase (110 U/L [reference range, 10–40 U/L]), and total bilirubin (1.5 mg/dL [reference range, 0.3–1.2 mg/dL]). White blood cell count and C-reactive protein levels were within reference range. An antinuclear antibody titer of 1:80 with a homogenous pattern was found, and aldolase levels were elevated. Laboratory investigations for rheumatoid factor, Lyme disease, tuberculosis, hepatitis, and human immunodeficiency virus were negative. A chest radiograph was normal.
A punch biopsy from the right dorsal hand revealed a dermal proliferation of mononucleated and multinucleated epithelioid histiocytes with ample amounts of eosinophilic ground-glass cytoplasm (Figure 2). Immunohistochemistry revealed epithelioid histiocytes reactive for CD68, CD163, and factor XIIIA, and negative for S-100 and CD1a.
The patient was diagnosed with multicentric reticulohistiocytosis (MRH) and was initially treated with prednisone. Treatment was later augmented with etanercept and methotrexate with improvement in both the skin and joint symptoms.
Multicentric reticulohistiocytosis is a rare, non–Langerhans cell histiocytosis with both cutaneous and systemic features. Although case reports date back to the late 1800s, the term multicentric reticulohistiocytosis was first used in 1954.1 Multicentric reticulohistiocytosis is extremely uncommon and precludes thorough investigation of its etiology and management. The condition typically presents in the fifth to sixth decades of life and occurs more frequently in women with a female to male ratio estimated at 3 to 1.2,3 Pediatric cases have been reported but are exceedingly rare.4
Multicentric reticulohistiocytosis typically presents with a severe erosive arthropathy known as arthritis mutilans. Patients display a symmetric polyarthritis that commonly involves the elbows, wrists, and proximal and distal aspects of the interphalangeal joints. Onset and progression can be rapid, and the erosive nature leads to deformities in up to 45% of patients.2,5,6 Cutaneous findings arise an average of 3 years after the development of arthritis, though one-fifth of patients will initially present with cutaneous findings followed by the development of arthritis at any time.3,6 Clinical features include flesh-colored to reddish brown or yellow papulonodules that range in size from several millimeters to 2 cm. The lesions most commonly occur on the face (eg, ears, nose, paranasal cheeks), scalp, dorsal and lateral aspects of the hands and fingers, and overlying articular regions of the extremities. Characteristic periungual lesions classically are referred to as coral beads.4,6 Patients commonly report pruritus that may precede the development of the papules and nodules. Other cutaneous manifestations include xanthelasma, nail changes, and a photodistributed erythematous maculopapular eruption that may mimic dermatomyositis.6
Cutaneous findings of MRH can mimic rheumatoid nodules, gout, Gottron papules of dermatomyositis, lipoid proteinosis, sarcoidosis, lepromatous leprosy, granuloma annulare, xanthoma, xanthogranuloma, and fibroxanthoma.6,7 Histopathologic features may distinguish MRH from such entities. Findings include fairly well-circumscribed aggregates of large multinucleated giant cells with characteristic eosinophilic ground-glass cytoplasm. Histiocytes stain positively for CD68, HAM56, CD11b, and CD14, and variably for factor XIIIa. CD68, which is expressed by monocytes/macrophages, has been universally reported to be the most reliable marker of MRH. Negative staining for S-100 and CD1a supports a non-Langerhans origin for the involved histiocytes. If arthritic symptoms predominate, MRH must be distinguished from rheumatoid and psoriatic arthritis.6,7
Mucosal involvement occurs in approximately 50% of patients and includes the presence of nodules in the oral, nasal, and pharyngeal mucosae, as well as eye structures.2,3 Histiocytic infiltration has been documented in the heart, lungs, thyroid, liver, stomach, kidneys, muscle, bone marrow, and urogenital tract. Histiocytes also can invade the cartilage of the ears and nose causing disfigurement and characteristic leonine facies. Pathologic fractures may occur with bone involvement.5
Systemic features associated with MRH include hyperlipidemia, diabetes mellitus, thyroid disease, hypergammaglobulinemia, and various autoimmune diseases. Patients less frequently report fever and weight loss.2,5,6,8 Additionally, a positive tuberculin test occurs in 12% to 50% of patients.6 Various autoimmune diseases occur in 6% to 17% of cases including systemic lupus erythematosus, systemic sclerosis, rheumatoid arthritis, dermatomyositis, Sjögren syndrome, and primary biliary cirrhosis.2,5,6,8 The most clinically salient feature of MRH is its association with malignant conditions, which occur in up to 31% of patients. A variety of cancers have been reported in association with MRH, including breast, cervical, ovarian, stomach, penile, lymphoma, mesothelioma, and melanoma.7
The etiology of MRH is unclear. Although onset may precede the development of a malignant condition and regress with treatment, it cannot be considered a true paraneoplastic disorder, as it has no association with a specific cancer and does not typically parallel the disease course.6,9 Reports of increased levels of inflammatory mediators released from macrophages and endothelial cells, specifically IL-12, IL-1β, IL-6, and tumor necrosis factor α (TNF-α), have been thought to drive the destruction of bone and cartilage.6 In particular, TNF-α acts to indirectly induce destruction by stimulating proteolytic activity in macrophages, similar to the pathogenesis of joint damage in rheumatoid arthritis.8 Osteoclastic activity may play a role in the pathogenesis of MRH, as multinucleated giant cells in MRH can mature into osteoclasts by receptor activated nuclear factor–κB ligand signaling. In addition, patients treated with bisphosphonates have had decreased lacunar resorption.2,8
Initial management of MRH should include screening for hyperlipidemia, hypergammaglobulinemia, hyperglycemia, thyroid dysfunction, and autoimmune diseases, as well as age-appropriate cancer screening. Imaging studies should evaluate for the presence of erosive arthritis. There are no well-defined treatment algorithms for MRH due to the rarity of the disease, and recommendations largely rely on case reports. Although spontaneous remission typically occurs within 5 to 10 years, the risk for joint destruction argues for early pharmacologic intervention. Current management includes the use of nonsteroidal anti-inflammatory drugs and various immunosuppressants including oral glucocorticoids, cyclophosphamide, chlorambucil, methotrexate, or azathioprine.2 A combination of methotrexate with cyclophosphamide or glucocorticoids also has shown efficacy.10 Anti–TNF-α agents, such as etanercept, adalimumab, and infliximab, have been used with some success.2 Tumor necrosis factor α inhibitors used in combination with oral glucocorticoids and methotrexate may have an increased benefit.2,9,11 Evidence suggesting that TNF-α plays a role in the destruction of bone and cartilage led to the successful use of infliximab in combination with oral glucocorticoids and methotrexate, which prevented possible development of antibodies to infliximab and increased its efficacy.12 Bisphosphonate use in combination with glucocorticoids and methotrexate may prevent joint destruction without the serious adverse events associated with anti–TNF-α agents.2,9,13,14
- Goltz RW, Laymon CW. Multicentric reticulohistiocytosis of the skin and synovia; reticulohistiocytoma or ganglioneuroma. AMA Arch Derma Syphilol. 1954;69:717-731.
- Islam AD, Naguwa SM, Cheema GS, et al. Multicentric reticulohistiocytosis: a rare yet challenging disease. Clin Rev Allergy Immunol. 2013;45:281-289.
- West KL, Sporn T, Puri PK. Multicentric reticulohistiocytosis: a unique case with pulmonary fibrosis. Arch Dermatol. 2012;148:228-232.
- Outland JD, Keiran SJ, Schikler KN, et al. Multicentric reticulohistiocytosis in a 14-year-old girl. Pediatr Dermatol. 2002;19:527-531.
- Gorman JD, Danning C, Schumacher HR, et al. Multicentric reticulohistiocytosis: case report with immunohistochemical analysis and literature review. Arthritis Rheum. 2000;43:930-938.
- Tajirian AL, Malik MK, Robinson-Bostom L, et al. Multicentric reticulohistiocytosis. Clin Dermatol. 2006;24:486-492.
- Luz FB, Gaspar TAP, Kalil-Gaspar N, et al. Multicentric reticulohistiocytosis. J Eur Acad Dermatol Venereol. 2001;15:524-531.
- Trotta F, Castellino G, Lo Monaco A. Multicentric reticulohistiocytosis. Best Pract Res Clin Rheumatol. 2004;18:759-772.
- Kalajian AH, Callen JP. Multicentric reticulohistiocytosis successfully treated with infliximab: an illustrative case and evaluation of cytokine expression supporting anti-tumor necrosis factor therapy. Arch Derm. 2008;144:1360-1366.
- Liang GC, Granston AS. Complete remission of multicentric reticulohistiocytosis with combination therapy of steroid, cyclophosphamide, and low-dose pulse methotrexate. case report, review of the literature, and proposal for treatment. Arthritis Rheum. 1996;39:171-174.
- Lovelace K, Loyd A, Adelson D, et al. Etanercept and the treatment of multicentric reticulohistiocytosis. Arch Dermatol. 2005;141:1167-1168.
- Lee MW, Lee EY, Jeong YI, et al. Successful treatment of multicentric reticulohistiocytosis with a combination of infliximab, prednisolone and methotrexate. Acta Derm Venereol. 2004;84:478-479.
- Adamopoulos IE, Wordsworth PB, Edwards JR, et al. Osteoclast differentiation and bone resorption in multicentric reticulohistiocytosis. Hum Pathol. 2006;37:1176-1185.
- Satoh M, Oyama N, Yamada H, et al. Treatment trial of multicentric reticulohistiocytosis with a combination of predonisolone, methotrexate and alendronate. J Dermatol. 2008;35:168-171.
- Goltz RW, Laymon CW. Multicentric reticulohistiocytosis of the skin and synovia; reticulohistiocytoma or ganglioneuroma. AMA Arch Derma Syphilol. 1954;69:717-731.
- Islam AD, Naguwa SM, Cheema GS, et al. Multicentric reticulohistiocytosis: a rare yet challenging disease. Clin Rev Allergy Immunol. 2013;45:281-289.
- West KL, Sporn T, Puri PK. Multicentric reticulohistiocytosis: a unique case with pulmonary fibrosis. Arch Dermatol. 2012;148:228-232.
- Outland JD, Keiran SJ, Schikler KN, et al. Multicentric reticulohistiocytosis in a 14-year-old girl. Pediatr Dermatol. 2002;19:527-531.
- Gorman JD, Danning C, Schumacher HR, et al. Multicentric reticulohistiocytosis: case report with immunohistochemical analysis and literature review. Arthritis Rheum. 2000;43:930-938.
- Tajirian AL, Malik MK, Robinson-Bostom L, et al. Multicentric reticulohistiocytosis. Clin Dermatol. 2006;24:486-492.
- Luz FB, Gaspar TAP, Kalil-Gaspar N, et al. Multicentric reticulohistiocytosis. J Eur Acad Dermatol Venereol. 2001;15:524-531.
- Trotta F, Castellino G, Lo Monaco A. Multicentric reticulohistiocytosis. Best Pract Res Clin Rheumatol. 2004;18:759-772.
- Kalajian AH, Callen JP. Multicentric reticulohistiocytosis successfully treated with infliximab: an illustrative case and evaluation of cytokine expression supporting anti-tumor necrosis factor therapy. Arch Derm. 2008;144:1360-1366.
- Liang GC, Granston AS. Complete remission of multicentric reticulohistiocytosis with combination therapy of steroid, cyclophosphamide, and low-dose pulse methotrexate. case report, review of the literature, and proposal for treatment. Arthritis Rheum. 1996;39:171-174.
- Lovelace K, Loyd A, Adelson D, et al. Etanercept and the treatment of multicentric reticulohistiocytosis. Arch Dermatol. 2005;141:1167-1168.
- Lee MW, Lee EY, Jeong YI, et al. Successful treatment of multicentric reticulohistiocytosis with a combination of infliximab, prednisolone and methotrexate. Acta Derm Venereol. 2004;84:478-479.
- Adamopoulos IE, Wordsworth PB, Edwards JR, et al. Osteoclast differentiation and bone resorption in multicentric reticulohistiocytosis. Hum Pathol. 2006;37:1176-1185.
- Satoh M, Oyama N, Yamada H, et al. Treatment trial of multicentric reticulohistiocytosis with a combination of predonisolone, methotrexate and alendronate. J Dermatol. 2008;35:168-171.
Practice Points
- Multicentric reticulohistiocytosis (MRH) is an important entity to recognize given its association with underlying malignancy and irreversible destructive arthritis.
- Diagnosis of MRH warrants extensive review of systems, age-appropriate cancer screening, and relevant systemic workup.
- Early pharmacologic intervention should be initiated with nonsteroidal anti-inflammatory agents or immunosuppressant agents.
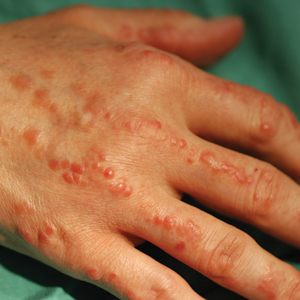
Paraneoplastic Dermatomyositis Presenting With Interesting Cutaneous Findings
To the Editor:
We report an interesting clinical case of dermatomyositis (DM) that presented with an associated malignancy (small cell lung cancer). This patient also had an unusual clinical finding of predominantly unilateral, confluent, erythematous papules on the knee, a cutaneous sign that is seldom described in the DM literature. This case serves to reinforce the classic findings and associations of DM, in addition to the uncommon manifestation of predominantly unilateral papules on the knee.
A 68-year-old woman presented with several cutaneous manifestations including the classic findings of photo distributed erythema on the arms and face, a heliotrope rash, Gottron papules, and confluent pink papules on the left knee (Figure 1). The patient also had one of the more rare manifestations of DM, flagellate erythema on the back (Figure 2). She had a history of breast cancer and was found to have metastatic small cell lung cancer at the time of the DM diagnosis.
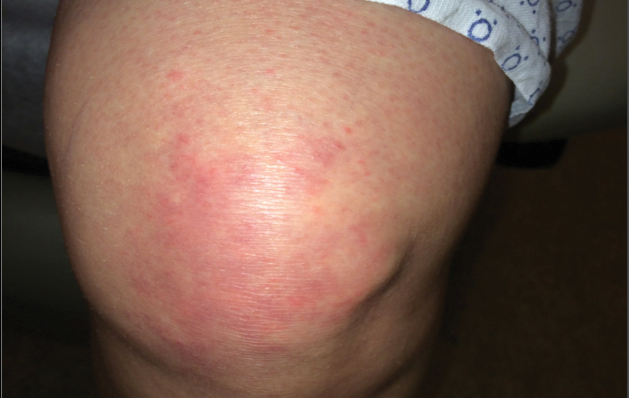
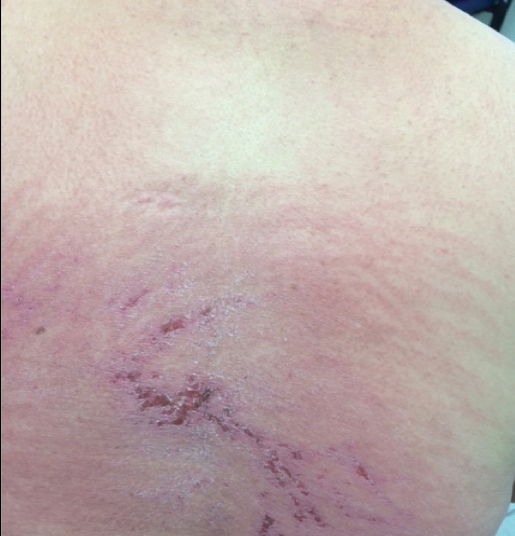
A punch biopsy from an area of flagellate erythema on the back revealed an interface dermatitis with a superficial, perivascular, lymphocyte-predominant inflammatory infiltrate (Figure 3). Alcian blue and colloidal iron stains revealed a marked increase in papillary dermal mucin. With the characteristic changes on skin biopsy and the classic skin findings present in our patient, we felt confident diagnosing her with DM. At the time of diagnosis, the patient also was found to have metastatic small cell lung cancer, suggesting a true paraneoplastic relationship.
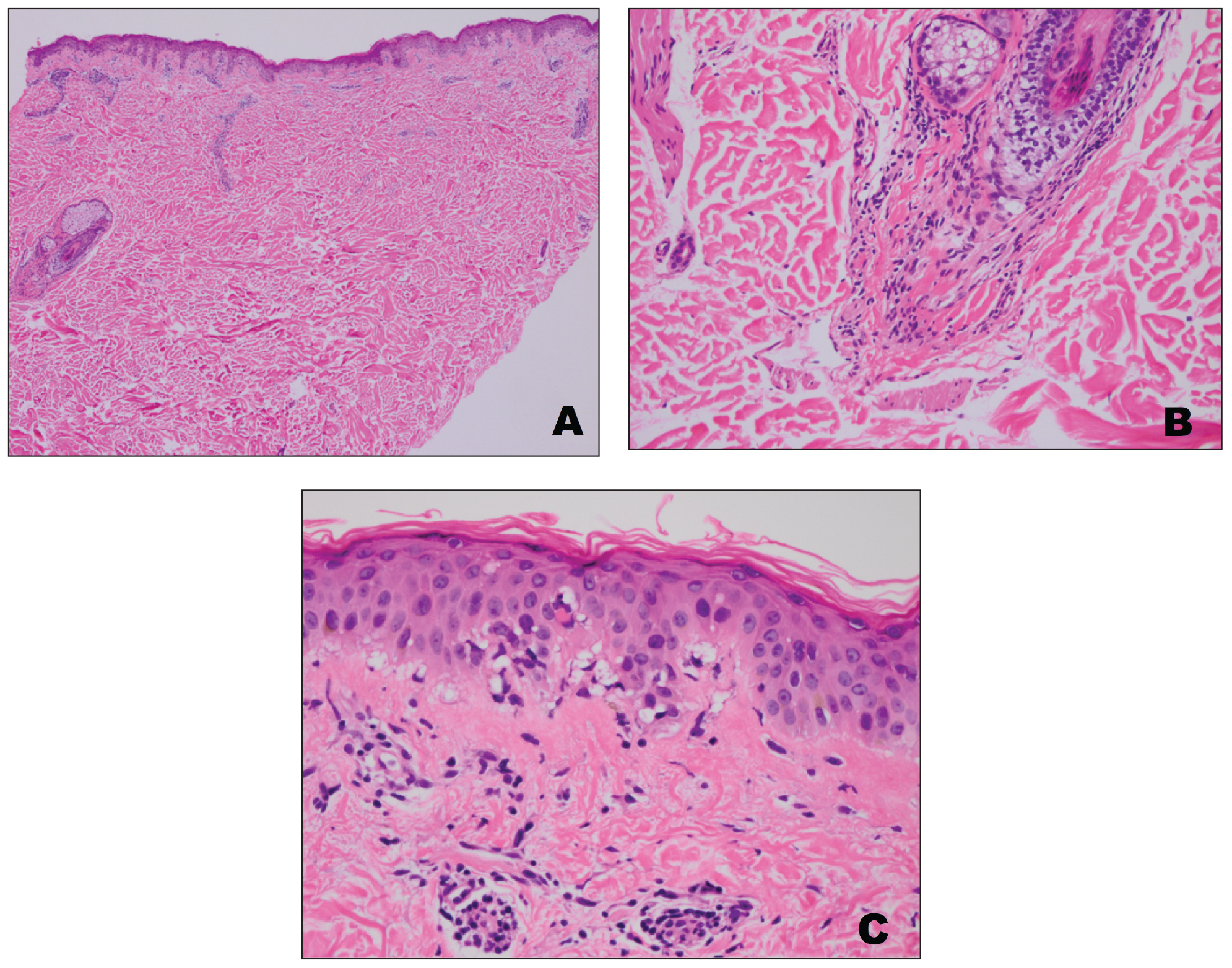

The association of DM and amyopathic DM with internal malignancy is well known. Bohan and Peter1 noted an overall figure ranging from 15% to 34% with an increased frequency in patients with skin and muscle involvement.1 Hill et al5 examined this link in a population-based study that identified corresponding malignancies. Specifically, they noted cancers to arise most frequently in the airway (eg, lung, trachea, bronchus), ovaries, breasts, colorectal region, and stomach.5 There also has been work performed to identify if certain dermatologic findings may be associated with a higher risk of malignancy.6,7 A meta-analysis by Wang et al6 showed that Gottron sign did not have an association with cancer, but findings of cutaneous necrosis did have an association. It is unknown if the specific cutaneous findings in our patient, including the predominantly unilateral papules on the knee, may have been a clue to the underlying malignancy.
In summary, we believe that our patient presented with the classic manifestations of DM in addition to the curious cutaneous sign of predominantly unilateral, confluent, erythematous papules on the knee, a clinical finding that may aid in the diagnosis of DM and also may alert the clinician to a possible underlying malignancy.
- Bohan A, Peter JB. Polymyositis and dermatomyositis (first of two parts). N Engl J Med. 1975;292:344-347.
- Santmyire-Rosenberger B, Dugan EM. Skin involvement in dermatomyositis. Curr Opin Rheumatol. 2003;15:714-722.
- Callen JP. Dermatomyositis. Lancet. 2000;355:53-57.
- Lister RK, Cooper ES, Paige DG. Papules and pustules of the elbows and knees: an uncommon clinical sign of dermatomyositis in oriental children. Pediatr Dermatol. 2000;17:37-40.
- Hill CL, Zhang Y, Sigurgeirsson B, et al. Frequency of specific cancer types in dermatomyositis and polymyositis: a population-based study. Lancet. 2001;357:96-100.
- Wang J, Guo G, Chen G, et al. Meta‐analysis of the association of dermatomyositis and polymyositis with cancer. Br J Dermatol. 2013;169:838-847.
- Chen YJ, Wu CY, Shen JL. Predicting factors of malignancy in dermatomyositis and polymyositis: a case–control study. Br J Dermatol. 2001;144:825-831.
To the Editor:
We report an interesting clinical case of dermatomyositis (DM) that presented with an associated malignancy (small cell lung cancer). This patient also had an unusual clinical finding of predominantly unilateral, confluent, erythematous papules on the knee, a cutaneous sign that is seldom described in the DM literature. This case serves to reinforce the classic findings and associations of DM, in addition to the uncommon manifestation of predominantly unilateral papules on the knee.
A 68-year-old woman presented with several cutaneous manifestations including the classic findings of photo distributed erythema on the arms and face, a heliotrope rash, Gottron papules, and confluent pink papules on the left knee (Figure 1). The patient also had one of the more rare manifestations of DM, flagellate erythema on the back (Figure 2). She had a history of breast cancer and was found to have metastatic small cell lung cancer at the time of the DM diagnosis.
A punch biopsy from an area of flagellate erythema on the back revealed an interface dermatitis with a superficial, perivascular, lymphocyte-predominant inflammatory infiltrate (Figure 3). Alcian blue and colloidal iron stains revealed a marked increase in papillary dermal mucin. With the characteristic changes on skin biopsy and the classic skin findings present in our patient, we felt confident diagnosing her with DM. At the time of diagnosis, the patient also was found to have metastatic small cell lung cancer, suggesting a true paraneoplastic relationship.
The association of DM and amyopathic DM with internal malignancy is well known. Bohan and Peter1 noted an overall figure ranging from 15% to 34% with an increased frequency in patients with skin and muscle involvement.1 Hill et al5 examined this link in a population-based study that identified corresponding malignancies. Specifically, they noted cancers to arise most frequently in the airway (eg, lung, trachea, bronchus), ovaries, breasts, colorectal region, and stomach.5 There also has been work performed to identify if certain dermatologic findings may be associated with a higher risk of malignancy.6,7 A meta-analysis by Wang et al6 showed that Gottron sign did not have an association with cancer, but findings of cutaneous necrosis did have an association. It is unknown if the specific cutaneous findings in our patient, including the predominantly unilateral papules on the knee, may have been a clue to the underlying malignancy.
In summary, we believe that our patient presented with the classic manifestations of DM in addition to the curious cutaneous sign of predominantly unilateral, confluent, erythematous papules on the knee, a clinical finding that may aid in the diagnosis of DM and also may alert the clinician to a possible underlying malignancy.
To the Editor:
We report an interesting clinical case of dermatomyositis (DM) that presented with an associated malignancy (small cell lung cancer). This patient also had an unusual clinical finding of predominantly unilateral, confluent, erythematous papules on the knee, a cutaneous sign that is seldom described in the DM literature. This case serves to reinforce the classic findings and associations of DM, in addition to the uncommon manifestation of predominantly unilateral papules on the knee.
A 68-year-old woman presented with several cutaneous manifestations including the classic findings of photo distributed erythema on the arms and face, a heliotrope rash, Gottron papules, and confluent pink papules on the left knee (Figure 1). The patient also had one of the more rare manifestations of DM, flagellate erythema on the back (Figure 2). She had a history of breast cancer and was found to have metastatic small cell lung cancer at the time of the DM diagnosis.
A punch biopsy from an area of flagellate erythema on the back revealed an interface dermatitis with a superficial, perivascular, lymphocyte-predominant inflammatory infiltrate (Figure 3). Alcian blue and colloidal iron stains revealed a marked increase in papillary dermal mucin. With the characteristic changes on skin biopsy and the classic skin findings present in our patient, we felt confident diagnosing her with DM. At the time of diagnosis, the patient also was found to have metastatic small cell lung cancer, suggesting a true paraneoplastic relationship.
The association of DM and amyopathic DM with internal malignancy is well known. Bohan and Peter1 noted an overall figure ranging from 15% to 34% with an increased frequency in patients with skin and muscle involvement.1 Hill et al5 examined this link in a population-based study that identified corresponding malignancies. Specifically, they noted cancers to arise most frequently in the airway (eg, lung, trachea, bronchus), ovaries, breasts, colorectal region, and stomach.5 There also has been work performed to identify if certain dermatologic findings may be associated with a higher risk of malignancy.6,7 A meta-analysis by Wang et al6 showed that Gottron sign did not have an association with cancer, but findings of cutaneous necrosis did have an association. It is unknown if the specific cutaneous findings in our patient, including the predominantly unilateral papules on the knee, may have been a clue to the underlying malignancy.
In summary, we believe that our patient presented with the classic manifestations of DM in addition to the curious cutaneous sign of predominantly unilateral, confluent, erythematous papules on the knee, a clinical finding that may aid in the diagnosis of DM and also may alert the clinician to a possible underlying malignancy.
- Bohan A, Peter JB. Polymyositis and dermatomyositis (first of two parts). N Engl J Med. 1975;292:344-347.
- Santmyire-Rosenberger B, Dugan EM. Skin involvement in dermatomyositis. Curr Opin Rheumatol. 2003;15:714-722.
- Callen JP. Dermatomyositis. Lancet. 2000;355:53-57.
- Lister RK, Cooper ES, Paige DG. Papules and pustules of the elbows and knees: an uncommon clinical sign of dermatomyositis in oriental children. Pediatr Dermatol. 2000;17:37-40.
- Hill CL, Zhang Y, Sigurgeirsson B, et al. Frequency of specific cancer types in dermatomyositis and polymyositis: a population-based study. Lancet. 2001;357:96-100.
- Wang J, Guo G, Chen G, et al. Meta‐analysis of the association of dermatomyositis and polymyositis with cancer. Br J Dermatol. 2013;169:838-847.
- Chen YJ, Wu CY, Shen JL. Predicting factors of malignancy in dermatomyositis and polymyositis: a case–control study. Br J Dermatol. 2001;144:825-831.
- Bohan A, Peter JB. Polymyositis and dermatomyositis (first of two parts). N Engl J Med. 1975;292:344-347.
- Santmyire-Rosenberger B, Dugan EM. Skin involvement in dermatomyositis. Curr Opin Rheumatol. 2003;15:714-722.
- Callen JP. Dermatomyositis. Lancet. 2000;355:53-57.
- Lister RK, Cooper ES, Paige DG. Papules and pustules of the elbows and knees: an uncommon clinical sign of dermatomyositis in oriental children. Pediatr Dermatol. 2000;17:37-40.
- Hill CL, Zhang Y, Sigurgeirsson B, et al. Frequency of specific cancer types in dermatomyositis and polymyositis: a population-based study. Lancet. 2001;357:96-100.
- Wang J, Guo G, Chen G, et al. Meta‐analysis of the association of dermatomyositis and polymyositis with cancer. Br J Dermatol. 2013;169:838-847.
- Chen YJ, Wu CY, Shen JL. Predicting factors of malignancy in dermatomyositis and polymyositis: a case–control study. Br J Dermatol. 2001;144:825-831.
Practice Points
- Dermatomyositis has myriad cutaneous features including the shawl sign, the heliotrope sign, and Gottron papules.
- Less commonly, patients can present with the Holster sign (poikiloderma of the lateral thighs).
- Even less commonly, as in this report, patients can present with a psoriasiform papular eruption on the knees or with flagellate erythema on the back.
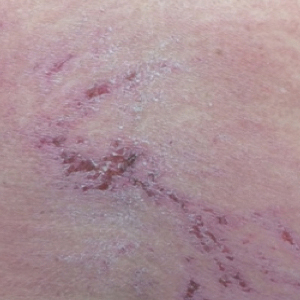
Acute Kwashiorkor in the Setting of Cerebral Palsy and Pancreatic Insufficiency
To the Editor:
Kwashiorkor, or protein-calorie malnutrition, is a common issue in developing countries subject to starvation. In economically advanced nations, however, kwashiorkor is extremely rare and may appear in children placed on restrictive diets instituted by well-meaning guardians. Kwashiorkor also may occur because of gastrointestinal malabsorption. We present a unique case of kwashiorkor that revealed an underlying diagnosis of pancreatic insufficiency.
A 12-year-old girl presented to the hospital with 4 days of watery nonbloody diarrhea occurring with every feeding as well as new onset of presumed diaper dermatitis that had not responded to nystatin cream. Facial swelling also was noted the day prior to admission. Her medical history was notable for cerebral palsy secondary to nonaccidental trauma, leaving the patient nonverbal and quadriplegic. She had numerous prior admissions for sepsis with marked hypotension and more recently was diagnosed with insulin-dependent type 2 diabetes mellitus. She had never lived outside the United States and resided at home with her adoptive parents.
Physical examination revealed a nonverbal underweight girl (weight, 25 kg). Large areas of denudation with surrounding desquamated skin resembling flaking enamel paint covered the buttocks and posterior legs bilaterally (Figure). She had linear hyperpigmented patches on the dorsal hands with one superficial erosion on the left wrist. Marked periorbital edema as well as nonpitting edema of the face, arms, and legs were present.
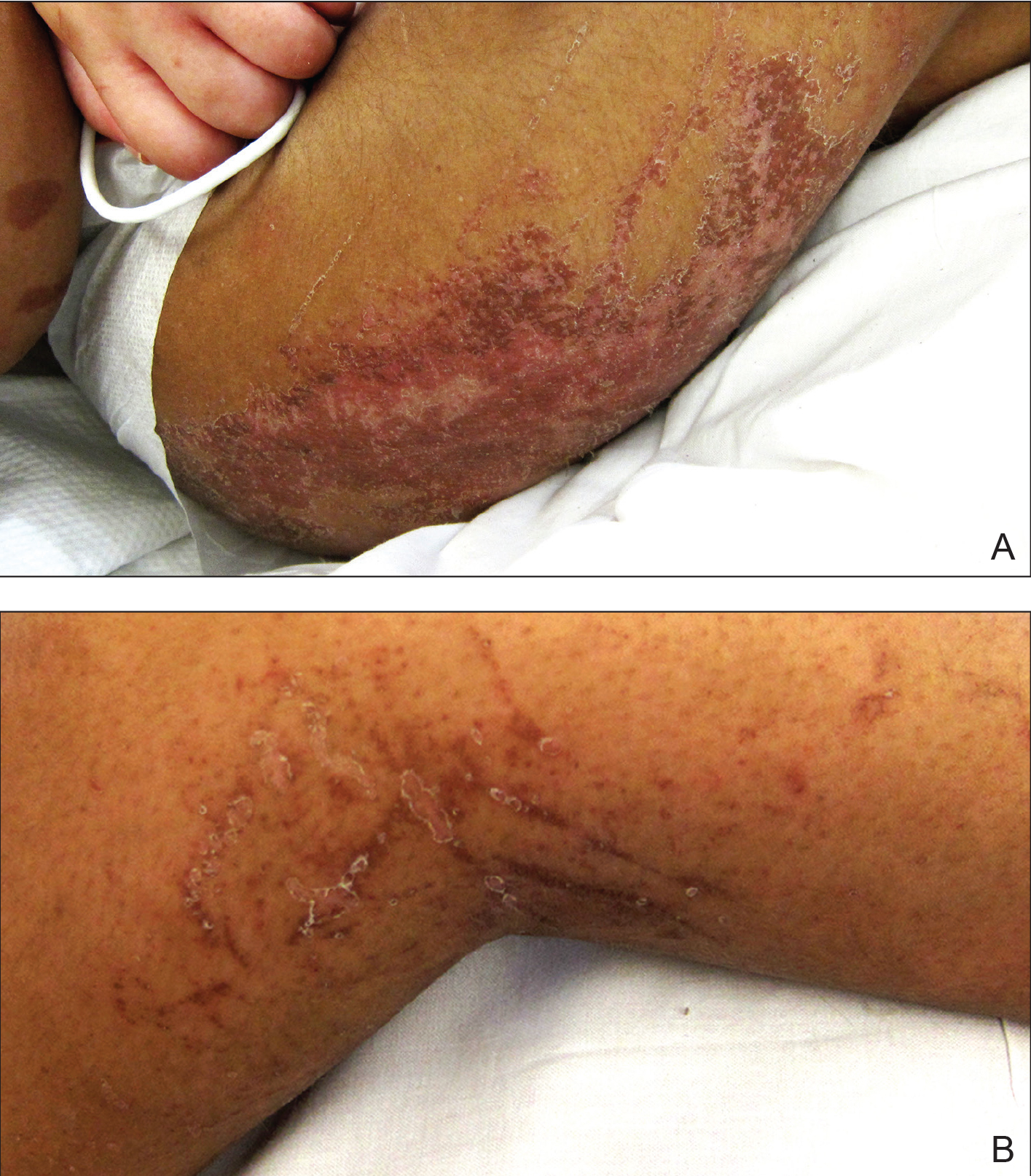
Upon additional questioning, the patient’s adoptive parent reported a diet of formula containing 1.0 cal/mL with 200-mL feedings 3 times daily through a Geiger-Müller tube, providing a daily protein intake of approximately 17.7 g per day (0.7 g/kg per day). On the day of admission, abnormal laboratory findings included low protein and albumin levels at 4.6 g/dL (reference range, 5.7–8.2 g/dL) and 2.1 g/dL (reference range, 3.2–4.8 g/dL), respectively; an elevated aspartate aminotransferase level of 73 U/L (reference range, 10–34 U/L); and an elevated alanine aminotransferase level of 80 U/L (reference range, 10–40 U/L). Based on the patient’s characteristic clinical findings and abnormal laboratory values, a diagnosis of acute kwashiorkor was made. Although the zinc level was low at 0.29 µg/mL (reference range, 0.66–1.10 µg/mL), the patient did not have any periorificial involvement to support a diagnosis of acrodermatitis enteropathica.
Upon further workup, stool elastase was measured at less than 50 µg per gram of stool (reference range, >200 µg pancreatic elastase per gram of stool), confirming a diagnosis of severe pancreatic insufficiency. Pancreatic enzyme supplementation was initiated along with an increase in protein intake to 1.5 g/kg per day. The patient’s hospital course was complicated by respiratory distress and sepsis, leading to a prolonged hospital stay. A component of refeeding syndrome may have contributed to the patient’s respiratory distress.
Kwashiorkor, a form of protein malnutrition, is caused by inadequate protein intake and usually is seen in developing countries when children are weaned from breastmilk to a diet high in starch and low in protein. It is characterized by edema, growth retardation, a characteristic dermatosis, depigmentation of hair, lethargy, and irritability.1 If left untreated, kwashiorkor can be fatal. Skin changes associated with kwashiorkor first occur in areas of friction or pressure. The skin develops patches of hyperpigmentation that subsequently desquamate in a pattern likened to flaky paint. In the current case of a nonmobile child with diarrhea, prominent involvement of the buttocks and thighs would be expected. This dermatosis does not appear in marasmus and is pathognomonic for kwashiorkor when seen in a child with edema.2
Children in the United States developing kwashiorkor secondary to severely restrictive diets has been reported.3 However, kwashiorkor also may occur due to underlying chronic malabsorptive disease. There have been rare reports of children with cystic fibrosis presenting with kwashiorkor,4 as well as a case of kwashiorkor secondary to underlying infantile Crohn disease.5
Cerebral palsy is associated with multiple different risk factors for malnutrition. Musculoskeletal deformities, oral-motor difficulties, medication side effects, limited communication skills, compromised pulmonary status, and poor muscle tone can all contribute to energy and nutrient deprivation.6 A 2018 study including 728 children registered into the Bangladesh Cerebral Palsy Register between January 2015 and December 2016 demonstrated that more than two-thirds were underweight (70.0%) and stunted (73.1%) and that children with tri/quadriplegic cerebral palsy presented with the highest proportion of severe malnutrition.7 In another report (N=142), up to 85% of children with spastic quadriplegia had severe feeding problems,8 making this population particularly high risk for poor nutritional status.
Pancreatic exocrine insufficiency is characterized by reduced secretion of amylase, lipase, and protease, and it may result in diarrhea, weight loss, malabsorption of essential nutrients, and malnutrition. Pancreatic exocrine insufficiency may occur in the setting of chronic pancreatitis, pancreatic surgery, and cystic fibrosis. Our patient had numerous hospitalizations for sepsis marked by hypotension, and in the absence of more typical causes, we postulate that both endocrine and exocrine pancreatic damage resulted from prolonged hypotension. A sweat chloride test was not performed, as the patient had not experienced frequent pulmonary infections or other signs of cystic fibrosis.
According to a report from the Food and Agriculture Organization of the United Nations/World Health Organization/United Nations University (FAO/WHO/UNU), protein should provide at least 10% of the total caloric intake in a child.9 Although the adoptive parent approximated that our patient received 12% of her daily calories in the form of protein, the amount that she absorbed in the context of pancreatic insufficiency was undoubtedly much lower.
In this case, the diagnosis of kwashiorkor led to the discovery of underlying pancreatic exocrine insufficiency. Low stool elastase confirmed the diagnosis. Because kwashiorkor is rare in developed countries, the classic signs and symptoms may go unrecognized, which can lead to delayed diagnosis and notable morbidity and mortality. New-onset edema and desquamative rash in a child, especially a child with cerebral palsy, should alert physicians to the possibility of acute kwashiorkor and prompt investigation into underlying medical issues that may have contributed to its development.
1. Trowell HC, Davies JN, Dean RF. Kwashiorkor. II. clinical picture, pathology, and differential diagnosis. Br Med J. 1952;2:798-801.
2. Latham MC. The dermatosis of kwashiorkor in young children. Semin Dermatol. 1991;10:270-272.
3. Liu T, Howard RM, Mancini AJ, et al. Kwashiorkor in the United States: fad diets, perceived and true milk allergy, and nutritional ignorance. Arch Dermatol. 2001;137:630-636.
4. Phillips RJ, Crock CM, Dillon MJ, et al. Cystic fibrosis presenting as kwashiorkor with florid skin rash. Arch Dis Childhood. 1993;69:446-448.
5. Al-Mubarak L, Al-Khenaizan S, Al Goufi T. Cutaneous presentation of kwashiorkor due to infantile Crohn’s disease. Eur J Pediatr. 2010;169:117-119.
6. Wittenbrook W. Nutritional assessment and intervention in cerebral palsy. Practical Gastroenterol. Feb 2011;92:16-32. http://www.practicalgastro.com/pdf/February11/WittenbrookArticle.pdf.
7. Jahan I, Muhit M, Karim T, et al. What makes children with cerebral palsy vulnerable to malnutrition? findings from the Bangladesh cerebral palsy register (BCPR)[published online April 16, 2018]. Disabil Rehabil. doi:10.1080/09638288.2018.1461260.
8. Stallings VA, Charney EB, Davies JC, et al. Nutrition-related growth failure of children with quadriplegic cerebral palsy. Dev Med Child Neurol. 1993;35:126-138.
9. World Health Organization. Energy and Protein Requirements: Report of a Joint FAO/WHO/UNU Expert Consultation. Geneva, Switzerland: World Health Organization; 1985. Technical Report Series 724.
To the Editor:
Kwashiorkor, or protein-calorie malnutrition, is a common issue in developing countries subject to starvation. In economically advanced nations, however, kwashiorkor is extremely rare and may appear in children placed on restrictive diets instituted by well-meaning guardians. Kwashiorkor also may occur because of gastrointestinal malabsorption. We present a unique case of kwashiorkor that revealed an underlying diagnosis of pancreatic insufficiency.
A 12-year-old girl presented to the hospital with 4 days of watery nonbloody diarrhea occurring with every feeding as well as new onset of presumed diaper dermatitis that had not responded to nystatin cream. Facial swelling also was noted the day prior to admission. Her medical history was notable for cerebral palsy secondary to nonaccidental trauma, leaving the patient nonverbal and quadriplegic. She had numerous prior admissions for sepsis with marked hypotension and more recently was diagnosed with insulin-dependent type 2 diabetes mellitus. She had never lived outside the United States and resided at home with her adoptive parents.
Physical examination revealed a nonverbal underweight girl (weight, 25 kg). Large areas of denudation with surrounding desquamated skin resembling flaking enamel paint covered the buttocks and posterior legs bilaterally (Figure). She had linear hyperpigmented patches on the dorsal hands with one superficial erosion on the left wrist. Marked periorbital edema as well as nonpitting edema of the face, arms, and legs were present.
Upon additional questioning, the patient’s adoptive parent reported a diet of formula containing 1.0 cal/mL with 200-mL feedings 3 times daily through a Geiger-Müller tube, providing a daily protein intake of approximately 17.7 g per day (0.7 g/kg per day). On the day of admission, abnormal laboratory findings included low protein and albumin levels at 4.6 g/dL (reference range, 5.7–8.2 g/dL) and 2.1 g/dL (reference range, 3.2–4.8 g/dL), respectively; an elevated aspartate aminotransferase level of 73 U/L (reference range, 10–34 U/L); and an elevated alanine aminotransferase level of 80 U/L (reference range, 10–40 U/L). Based on the patient’s characteristic clinical findings and abnormal laboratory values, a diagnosis of acute kwashiorkor was made. Although the zinc level was low at 0.29 µg/mL (reference range, 0.66–1.10 µg/mL), the patient did not have any periorificial involvement to support a diagnosis of acrodermatitis enteropathica.
Upon further workup, stool elastase was measured at less than 50 µg per gram of stool (reference range, >200 µg pancreatic elastase per gram of stool), confirming a diagnosis of severe pancreatic insufficiency. Pancreatic enzyme supplementation was initiated along with an increase in protein intake to 1.5 g/kg per day. The patient’s hospital course was complicated by respiratory distress and sepsis, leading to a prolonged hospital stay. A component of refeeding syndrome may have contributed to the patient’s respiratory distress.
Kwashiorkor, a form of protein malnutrition, is caused by inadequate protein intake and usually is seen in developing countries when children are weaned from breastmilk to a diet high in starch and low in protein. It is characterized by edema, growth retardation, a characteristic dermatosis, depigmentation of hair, lethargy, and irritability.1 If left untreated, kwashiorkor can be fatal. Skin changes associated with kwashiorkor first occur in areas of friction or pressure. The skin develops patches of hyperpigmentation that subsequently desquamate in a pattern likened to flaky paint. In the current case of a nonmobile child with diarrhea, prominent involvement of the buttocks and thighs would be expected. This dermatosis does not appear in marasmus and is pathognomonic for kwashiorkor when seen in a child with edema.2
Children in the United States developing kwashiorkor secondary to severely restrictive diets has been reported.3 However, kwashiorkor also may occur due to underlying chronic malabsorptive disease. There have been rare reports of children with cystic fibrosis presenting with kwashiorkor,4 as well as a case of kwashiorkor secondary to underlying infantile Crohn disease.5
Cerebral palsy is associated with multiple different risk factors for malnutrition. Musculoskeletal deformities, oral-motor difficulties, medication side effects, limited communication skills, compromised pulmonary status, and poor muscle tone can all contribute to energy and nutrient deprivation.6 A 2018 study including 728 children registered into the Bangladesh Cerebral Palsy Register between January 2015 and December 2016 demonstrated that more than two-thirds were underweight (70.0%) and stunted (73.1%) and that children with tri/quadriplegic cerebral palsy presented with the highest proportion of severe malnutrition.7 In another report (N=142), up to 85% of children with spastic quadriplegia had severe feeding problems,8 making this population particularly high risk for poor nutritional status.
Pancreatic exocrine insufficiency is characterized by reduced secretion of amylase, lipase, and protease, and it may result in diarrhea, weight loss, malabsorption of essential nutrients, and malnutrition. Pancreatic exocrine insufficiency may occur in the setting of chronic pancreatitis, pancreatic surgery, and cystic fibrosis. Our patient had numerous hospitalizations for sepsis marked by hypotension, and in the absence of more typical causes, we postulate that both endocrine and exocrine pancreatic damage resulted from prolonged hypotension. A sweat chloride test was not performed, as the patient had not experienced frequent pulmonary infections or other signs of cystic fibrosis.
According to a report from the Food and Agriculture Organization of the United Nations/World Health Organization/United Nations University (FAO/WHO/UNU), protein should provide at least 10% of the total caloric intake in a child.9 Although the adoptive parent approximated that our patient received 12% of her daily calories in the form of protein, the amount that she absorbed in the context of pancreatic insufficiency was undoubtedly much lower.
In this case, the diagnosis of kwashiorkor led to the discovery of underlying pancreatic exocrine insufficiency. Low stool elastase confirmed the diagnosis. Because kwashiorkor is rare in developed countries, the classic signs and symptoms may go unrecognized, which can lead to delayed diagnosis and notable morbidity and mortality. New-onset edema and desquamative rash in a child, especially a child with cerebral palsy, should alert physicians to the possibility of acute kwashiorkor and prompt investigation into underlying medical issues that may have contributed to its development.
To the Editor:
Kwashiorkor, or protein-calorie malnutrition, is a common issue in developing countries subject to starvation. In economically advanced nations, however, kwashiorkor is extremely rare and may appear in children placed on restrictive diets instituted by well-meaning guardians. Kwashiorkor also may occur because of gastrointestinal malabsorption. We present a unique case of kwashiorkor that revealed an underlying diagnosis of pancreatic insufficiency.
A 12-year-old girl presented to the hospital with 4 days of watery nonbloody diarrhea occurring with every feeding as well as new onset of presumed diaper dermatitis that had not responded to nystatin cream. Facial swelling also was noted the day prior to admission. Her medical history was notable for cerebral palsy secondary to nonaccidental trauma, leaving the patient nonverbal and quadriplegic. She had numerous prior admissions for sepsis with marked hypotension and more recently was diagnosed with insulin-dependent type 2 diabetes mellitus. She had never lived outside the United States and resided at home with her adoptive parents.
Physical examination revealed a nonverbal underweight girl (weight, 25 kg). Large areas of denudation with surrounding desquamated skin resembling flaking enamel paint covered the buttocks and posterior legs bilaterally (Figure). She had linear hyperpigmented patches on the dorsal hands with one superficial erosion on the left wrist. Marked periorbital edema as well as nonpitting edema of the face, arms, and legs were present.
Upon additional questioning, the patient’s adoptive parent reported a diet of formula containing 1.0 cal/mL with 200-mL feedings 3 times daily through a Geiger-Müller tube, providing a daily protein intake of approximately 17.7 g per day (0.7 g/kg per day). On the day of admission, abnormal laboratory findings included low protein and albumin levels at 4.6 g/dL (reference range, 5.7–8.2 g/dL) and 2.1 g/dL (reference range, 3.2–4.8 g/dL), respectively; an elevated aspartate aminotransferase level of 73 U/L (reference range, 10–34 U/L); and an elevated alanine aminotransferase level of 80 U/L (reference range, 10–40 U/L). Based on the patient’s characteristic clinical findings and abnormal laboratory values, a diagnosis of acute kwashiorkor was made. Although the zinc level was low at 0.29 µg/mL (reference range, 0.66–1.10 µg/mL), the patient did not have any periorificial involvement to support a diagnosis of acrodermatitis enteropathica.
Upon further workup, stool elastase was measured at less than 50 µg per gram of stool (reference range, >200 µg pancreatic elastase per gram of stool), confirming a diagnosis of severe pancreatic insufficiency. Pancreatic enzyme supplementation was initiated along with an increase in protein intake to 1.5 g/kg per day. The patient’s hospital course was complicated by respiratory distress and sepsis, leading to a prolonged hospital stay. A component of refeeding syndrome may have contributed to the patient’s respiratory distress.
Kwashiorkor, a form of protein malnutrition, is caused by inadequate protein intake and usually is seen in developing countries when children are weaned from breastmilk to a diet high in starch and low in protein. It is characterized by edema, growth retardation, a characteristic dermatosis, depigmentation of hair, lethargy, and irritability.1 If left untreated, kwashiorkor can be fatal. Skin changes associated with kwashiorkor first occur in areas of friction or pressure. The skin develops patches of hyperpigmentation that subsequently desquamate in a pattern likened to flaky paint. In the current case of a nonmobile child with diarrhea, prominent involvement of the buttocks and thighs would be expected. This dermatosis does not appear in marasmus and is pathognomonic for kwashiorkor when seen in a child with edema.2
Children in the United States developing kwashiorkor secondary to severely restrictive diets has been reported.3 However, kwashiorkor also may occur due to underlying chronic malabsorptive disease. There have been rare reports of children with cystic fibrosis presenting with kwashiorkor,4 as well as a case of kwashiorkor secondary to underlying infantile Crohn disease.5
Cerebral palsy is associated with multiple different risk factors for malnutrition. Musculoskeletal deformities, oral-motor difficulties, medication side effects, limited communication skills, compromised pulmonary status, and poor muscle tone can all contribute to energy and nutrient deprivation.6 A 2018 study including 728 children registered into the Bangladesh Cerebral Palsy Register between January 2015 and December 2016 demonstrated that more than two-thirds were underweight (70.0%) and stunted (73.1%) and that children with tri/quadriplegic cerebral palsy presented with the highest proportion of severe malnutrition.7 In another report (N=142), up to 85% of children with spastic quadriplegia had severe feeding problems,8 making this population particularly high risk for poor nutritional status.
Pancreatic exocrine insufficiency is characterized by reduced secretion of amylase, lipase, and protease, and it may result in diarrhea, weight loss, malabsorption of essential nutrients, and malnutrition. Pancreatic exocrine insufficiency may occur in the setting of chronic pancreatitis, pancreatic surgery, and cystic fibrosis. Our patient had numerous hospitalizations for sepsis marked by hypotension, and in the absence of more typical causes, we postulate that both endocrine and exocrine pancreatic damage resulted from prolonged hypotension. A sweat chloride test was not performed, as the patient had not experienced frequent pulmonary infections or other signs of cystic fibrosis.
According to a report from the Food and Agriculture Organization of the United Nations/World Health Organization/United Nations University (FAO/WHO/UNU), protein should provide at least 10% of the total caloric intake in a child.9 Although the adoptive parent approximated that our patient received 12% of her daily calories in the form of protein, the amount that she absorbed in the context of pancreatic insufficiency was undoubtedly much lower.
In this case, the diagnosis of kwashiorkor led to the discovery of underlying pancreatic exocrine insufficiency. Low stool elastase confirmed the diagnosis. Because kwashiorkor is rare in developed countries, the classic signs and symptoms may go unrecognized, which can lead to delayed diagnosis and notable morbidity and mortality. New-onset edema and desquamative rash in a child, especially a child with cerebral palsy, should alert physicians to the possibility of acute kwashiorkor and prompt investigation into underlying medical issues that may have contributed to its development.
1. Trowell HC, Davies JN, Dean RF. Kwashiorkor. II. clinical picture, pathology, and differential diagnosis. Br Med J. 1952;2:798-801.
2. Latham MC. The dermatosis of kwashiorkor in young children. Semin Dermatol. 1991;10:270-272.
3. Liu T, Howard RM, Mancini AJ, et al. Kwashiorkor in the United States: fad diets, perceived and true milk allergy, and nutritional ignorance. Arch Dermatol. 2001;137:630-636.
4. Phillips RJ, Crock CM, Dillon MJ, et al. Cystic fibrosis presenting as kwashiorkor with florid skin rash. Arch Dis Childhood. 1993;69:446-448.
5. Al-Mubarak L, Al-Khenaizan S, Al Goufi T. Cutaneous presentation of kwashiorkor due to infantile Crohn’s disease. Eur J Pediatr. 2010;169:117-119.
6. Wittenbrook W. Nutritional assessment and intervention in cerebral palsy. Practical Gastroenterol. Feb 2011;92:16-32. http://www.practicalgastro.com/pdf/February11/WittenbrookArticle.pdf.
7. Jahan I, Muhit M, Karim T, et al. What makes children with cerebral palsy vulnerable to malnutrition? findings from the Bangladesh cerebral palsy register (BCPR)[published online April 16, 2018]. Disabil Rehabil. doi:10.1080/09638288.2018.1461260.
8. Stallings VA, Charney EB, Davies JC, et al. Nutrition-related growth failure of children with quadriplegic cerebral palsy. Dev Med Child Neurol. 1993;35:126-138.
9. World Health Organization. Energy and Protein Requirements: Report of a Joint FAO/WHO/UNU Expert Consultation. Geneva, Switzerland: World Health Organization; 1985. Technical Report Series 724.
1. Trowell HC, Davies JN, Dean RF. Kwashiorkor. II. clinical picture, pathology, and differential diagnosis. Br Med J. 1952;2:798-801.
2. Latham MC. The dermatosis of kwashiorkor in young children. Semin Dermatol. 1991;10:270-272.
3. Liu T, Howard RM, Mancini AJ, et al. Kwashiorkor in the United States: fad diets, perceived and true milk allergy, and nutritional ignorance. Arch Dermatol. 2001;137:630-636.
4. Phillips RJ, Crock CM, Dillon MJ, et al. Cystic fibrosis presenting as kwashiorkor with florid skin rash. Arch Dis Childhood. 1993;69:446-448.
5. Al-Mubarak L, Al-Khenaizan S, Al Goufi T. Cutaneous presentation of kwashiorkor due to infantile Crohn’s disease. Eur J Pediatr. 2010;169:117-119.
6. Wittenbrook W. Nutritional assessment and intervention in cerebral palsy. Practical Gastroenterol. Feb 2011;92:16-32. http://www.practicalgastro.com/pdf/February11/WittenbrookArticle.pdf.
7. Jahan I, Muhit M, Karim T, et al. What makes children with cerebral palsy vulnerable to malnutrition? findings from the Bangladesh cerebral palsy register (BCPR)[published online April 16, 2018]. Disabil Rehabil. doi:10.1080/09638288.2018.1461260.
8. Stallings VA, Charney EB, Davies JC, et al. Nutrition-related growth failure of children with quadriplegic cerebral palsy. Dev Med Child Neurol. 1993;35:126-138.
9. World Health Organization. Energy and Protein Requirements: Report of a Joint FAO/WHO/UNU Expert Consultation. Geneva, Switzerland: World Health Organization; 1985. Technical Report Series 724.
Practice Points
• Pancreatic exocrine deficiency, confirmed by low stool elastase, can lead to kwashiorkor and requires a high index of suspicion for diagnosis.
• Kwashiorkor is not only seen in developing countries but also in certain at-risk populations in economically advantaged countries.
• For multiple reasons, patients with cerebral palsy are at particular risk for nutritional deficiencies including kwashiorkor.
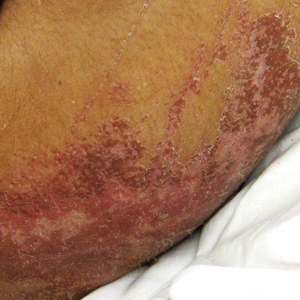
Annular Elastolytic Giant Cell Granuloma: Mysterious Enlarging Scarring Lesions
To the Editor:
A 52-year-old woman with a medical history of migraines and cervicalgia presented with lesions on the right arm, back, and right calf. The patient stated that the lesions began as small papules that had grown over 13 months, with the largest papule on the right forearm. She reported no itching, bleeding, pain, discharge, or other symptoms associated with the lesions. She had a multiple-year history of similar lesions that did not respond to treatment with antifungals, moderate-potency steroids, and other over-the-counter creams. The lesions would resolve spontaneously with scarring and subsequently recur. Prior skin biopsies were inconclusive. The patient did not report any systemic symptoms or a personal or family history of connective tissue diseases.
Physical examination revealed a 4-cm asymmetric, annular, erythematous plaque with central clearing on the right dorsal forearm with defined margins except over the distal aspect (Figure 1). She also had several 1- to 2-cm erythematous, nummular, asymmetric plaques on the right upper arm with well-defined margins. She had several lesions over the central and left sides of the upper back that were similar to the lesions on the upper arm.
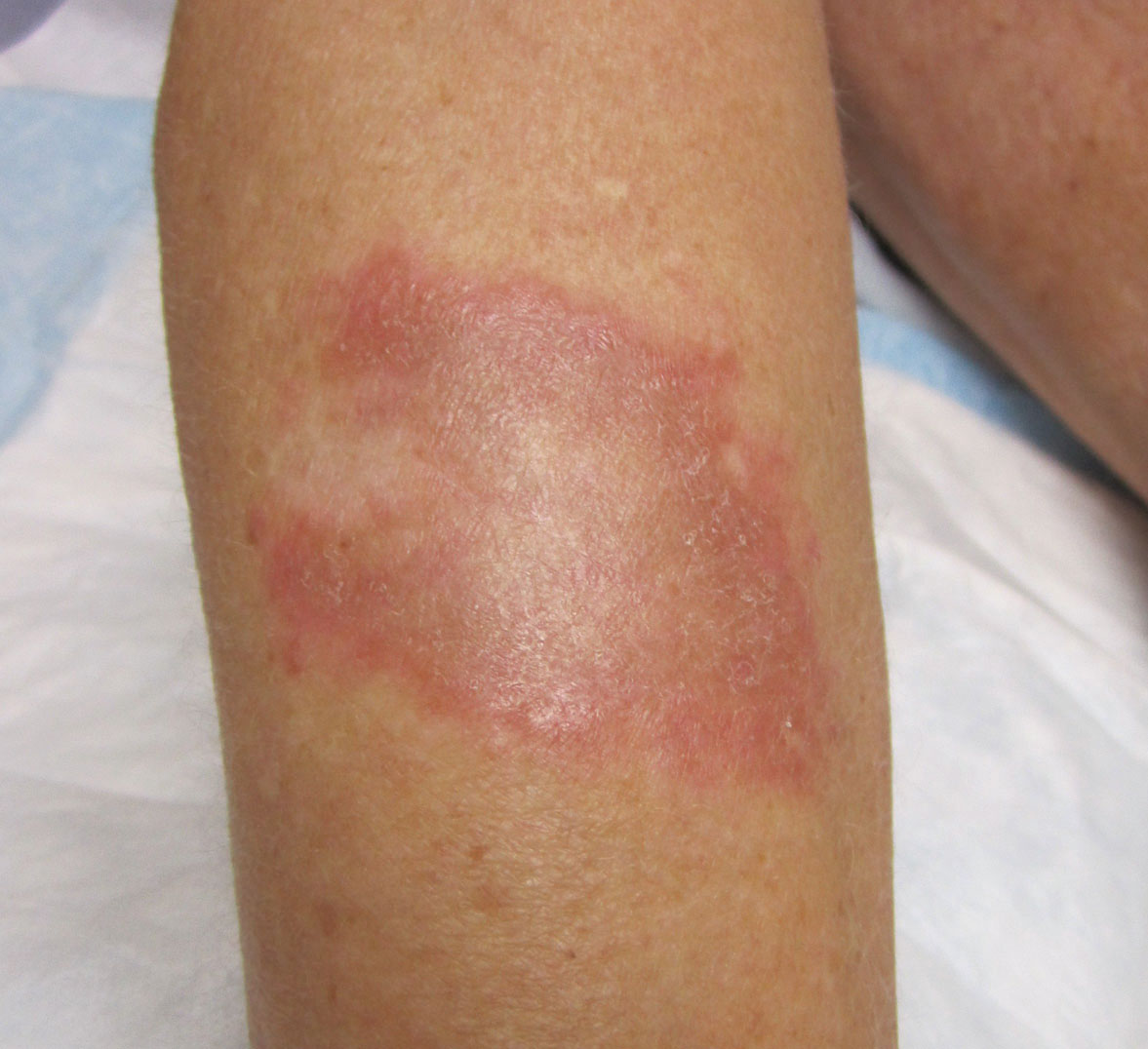
Two 4-mm punch biopsies of the right dorsal forearm and left side of the upper back revealed similar histologic features with a predominantly unremarkable epidermis. The dermis revealed a lymphohistiocytic infiltrate with prominent multinucleated giant cells organized into foreign body–type granulomas that extended into the deep dermis and subcutaneous tissue (Figure 2). In the granulomatous areas, there was a near-complete loss of elastic fibers with focal elastophagocytosis highlighted with Verhoeff-van Gieson (elastin) stain (Figure 3). Grocott-Gomori methenamine-silver and Fite stains for microorganisms were negative, and there was an absence of necrobiosis, lipids, and mucin.
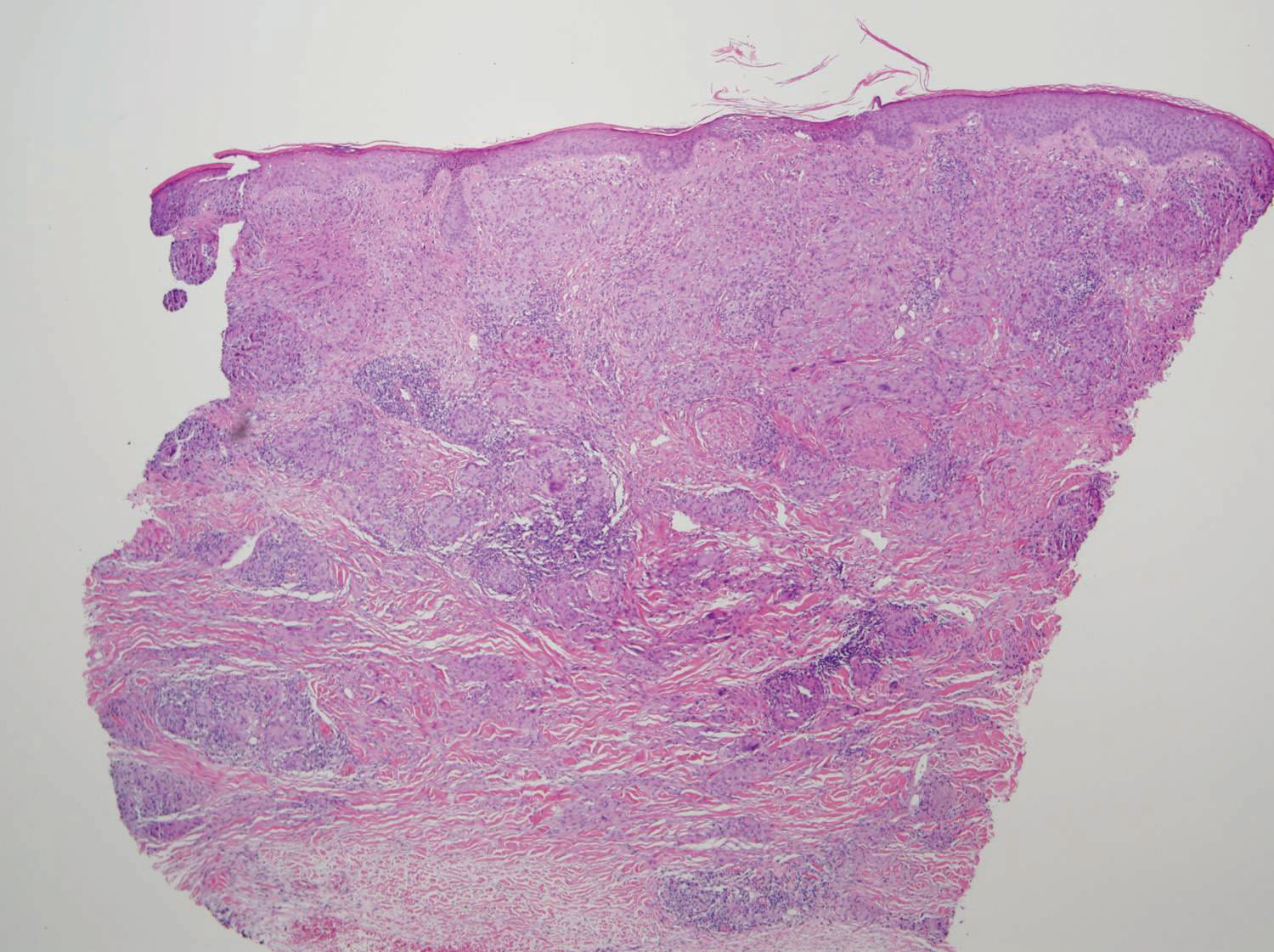
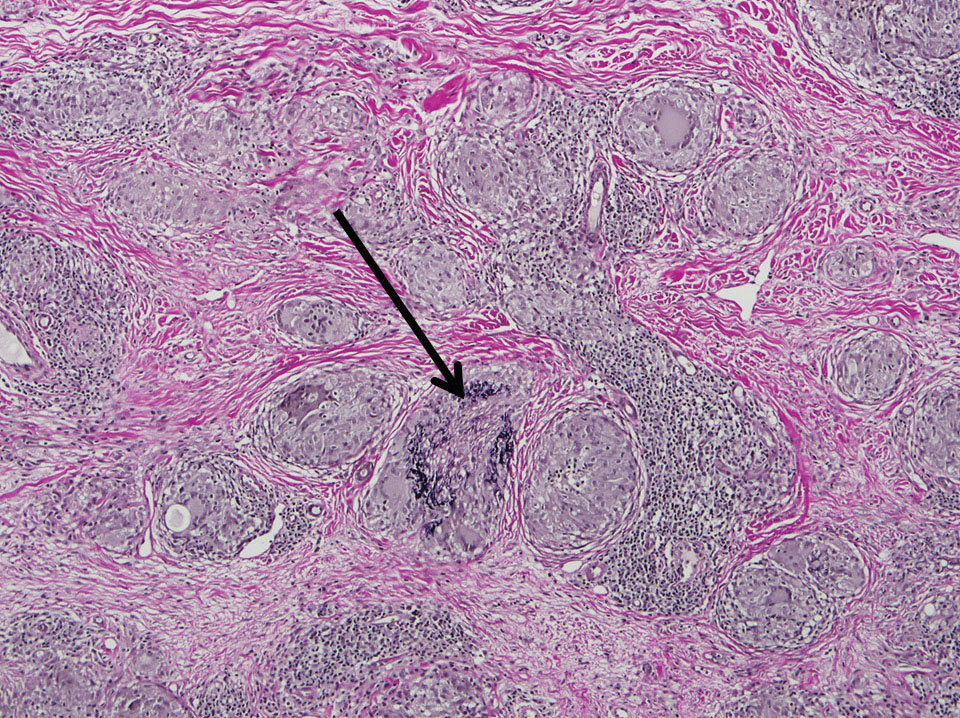
The histologic findings of a granulomatous dermatitis with loss of elastic fibers and elastophagocytosis in addition to the patient’s clinical presentation and history were consistent with the diagnosis of annular elastolytic giant cell granuloma (AEGCG). Infectious and other granulomatous diseases including sarcoidosis were ruled out via clinical history, unremarkable laboratory analysis (ie, complete blood cell count, chemistry panel, antinuclear antibody, urinalysis), and a normal chest radiograph. The histologic findings via the various stains were instrumental to the diagnosis. The patient was treated with fluocinonide and subsequently lost to follow-up.
Annular elastolytic giant cell granuloma is an uncommon cutaneous disease that presents with recurring annular plaques with raised erythematous borders and subsequent residual scarring.1 O’Brien2 originally described this condition in 1975 as an actinic granuloma due to similar histologic findings in areas of the patient’s sun-exposed skin. Ragaz and Ackerman3 disputed O’Brien’s2 description, claiming granulomatous inflammation was a primary pathologic process and not a consequence to damaged elastotic material. In 1979, Hanke et al4 termed the lesions as AEGCG because he did not find a correlation to the sun-exposed areas of the patients and did not see solar elastosis.
Although AEGCG has an unclear pathogenesis, cellular immunologic reactions induced by modified function of elastic fibers’ antigenicity contribute to AEGCG formation.5 Therefore, environmental and host factors may play a role in its etiopathogenesis. In one study, 37% of 38 Japanese patients with AEGCG were found to have definitive or latent diabetes mellitus, raising the possible role of diabetes in the structural damage of the elastic fibers.6
Patients typically are middle-aged women who present clinically with red or atrophic plaques that have slightly elevated borders. They have centripetal spread with a resulting atrophic center.7 Clinically, the differential diagnosis of this condition includes actinic granuloma, granuloma annulare, and granuloma multiforme.8
Histologically, AEGCG has a granulomatous component with multinucleated giant cells in the upper and mid dermis. This component typically is distributed peripherally to a central zone that lacks elastic tissue. Elastophagocytosis, a classic finding in AEGCG, is the phagocytosis of elastic fibers that can microscopically be seen in the cytoplasm of histiocytes and multinucleated giant cells. There also is an absence of necrobiosis, lipids, mucin, and a palisading arrangement of the granulomas. These findings distinguish AEGCG from granuloma annulare and necrobiosis lipoidica, the primary histologic differential diagnoses.9 In addition, consideration of entities consistently exhibiting elastophagocytosis such as mid-dermal elastolysis, papillary dermal elastolysis, actinic granuloma, and granulomatous slack skin should be considered.5,10,11
Therapy for AEGCG is broad and includes topical, intralesional, and systemic corticosteroids. Hydroxychloroquine, isotretinoin, clofazimine, dapsone, photochemotherapy, and cyclosporine also have been utilized with varying results. Other reports show improvement with surgical excision, cryotherapy, or cauterization of small lesions.12-15
1. Tock CL, Cohen PR. Annular elastolytic giant cell granuloma. Cutis. 1998;62:181-187.
2. O’Brien JP. Actinic granuloma: an annular connective tissue disorder affecting sun- and heat-damaged (elastotic) skin. Arch Dermatol. 1975;111:460-466.
3. Ragaz A, Ackerman AB. Is actinic granuloma a specific condition? Am J Dermatopathol. 1979;1:43-50.
4. Hanke CW, Bailin PL, Roenigk HH Jr. Annular elastolytic giant cell granuloma. a clinicopathologic study of five cases and a review of similar entities. J Am Acad Dermatol. 1979;1:413-421.
5. El-Khoury J, Kurban M, Abbas O. Elastophagocytosis: underlying mechanisms and associated cutaneous entities. J Am Acad Dermatol. 2014;70:934-44.
6. Aso Y, Izaki Y, Teraki Y. Annular elastolytic giant cell granuloma associated with diabetes mellitus: a case report and review of the Japanese literature. Clin Exp Dermatol. 2011;36:917-919.
7. Pestoni C, Pereiro M Jr, Toribio J. Annular elastolytic giant cell granuloma produced on an old burn scar and spreading after a mechanical trauma. Acta Derm Venereol. 2003;83:312-313.
8. Oka M, Kunisada M, Nishigori C. Generalized annular elastolytic giant cell granuloma with sparing of striae distensae. J Dermatol. 2013;40:220-222.
9. Limas C. The spectrum of primary cutaneous elastolytic granulomas and their distinction from granuloma annulare: a clinicopathological analysis. Histopathology. 2004;44:277-282.
10. McGrae JD Jr. Actinic granuloma: a clinical, histopathologic, and immunocytochemical study. Arch Dermatol. 1986;122:43-47.
11. Shah A, Safaya A. Granulomatous slack skin disease: a review, in comparison with mycosis fungoides. J Eur Acad Dermatol Venereol. 2012;26:1472-1478.
12. Chou WT, Tsai TF, Hung CM, et al. Multiple annular erythematous plaques on the back. Annular elastolytic giant cell granuloma (AEGCG). Indian J Dermatol Venereol Leprol. 2011;77:727-728.
13. Pérez-Pérez L, Garcia-Gavin J, Alleque F, et al. Successful treatment of generalized elastolytic giant cell granuloma with psoralen-ultraviolet A. Photodermatol Photoimmunol Photomed. 2012;28:264-266.
14. Babuna G, Buyukbabani N, Yazganoglu KD, et al. Effective treatment with hydroxychloroquine in a case of annular elastolytic giant cell granuloma. Indian J Dermatol Venereol Leprol. 2011;77:110-111.
15. Can B, Kavala M, Türkoglu Z, et al. Successful treatment of annular elastolytic giant cell granuloma with hydroxylchloroquine. Int J Dermatol. 2013;52:509-511.
To the Editor:
A 52-year-old woman with a medical history of migraines and cervicalgia presented with lesions on the right arm, back, and right calf. The patient stated that the lesions began as small papules that had grown over 13 months, with the largest papule on the right forearm. She reported no itching, bleeding, pain, discharge, or other symptoms associated with the lesions. She had a multiple-year history of similar lesions that did not respond to treatment with antifungals, moderate-potency steroids, and other over-the-counter creams. The lesions would resolve spontaneously with scarring and subsequently recur. Prior skin biopsies were inconclusive. The patient did not report any systemic symptoms or a personal or family history of connective tissue diseases.
Physical examination revealed a 4-cm asymmetric, annular, erythematous plaque with central clearing on the right dorsal forearm with defined margins except over the distal aspect (Figure 1). She also had several 1- to 2-cm erythematous, nummular, asymmetric plaques on the right upper arm with well-defined margins. She had several lesions over the central and left sides of the upper back that were similar to the lesions on the upper arm.
Two 4-mm punch biopsies of the right dorsal forearm and left side of the upper back revealed similar histologic features with a predominantly unremarkable epidermis. The dermis revealed a lymphohistiocytic infiltrate with prominent multinucleated giant cells organized into foreign body–type granulomas that extended into the deep dermis and subcutaneous tissue (Figure 2). In the granulomatous areas, there was a near-complete loss of elastic fibers with focal elastophagocytosis highlighted with Verhoeff-van Gieson (elastin) stain (Figure 3). Grocott-Gomori methenamine-silver and Fite stains for microorganisms were negative, and there was an absence of necrobiosis, lipids, and mucin.
The histologic findings of a granulomatous dermatitis with loss of elastic fibers and elastophagocytosis in addition to the patient’s clinical presentation and history were consistent with the diagnosis of annular elastolytic giant cell granuloma (AEGCG). Infectious and other granulomatous diseases including sarcoidosis were ruled out via clinical history, unremarkable laboratory analysis (ie, complete blood cell count, chemistry panel, antinuclear antibody, urinalysis), and a normal chest radiograph. The histologic findings via the various stains were instrumental to the diagnosis. The patient was treated with fluocinonide and subsequently lost to follow-up.
Annular elastolytic giant cell granuloma is an uncommon cutaneous disease that presents with recurring annular plaques with raised erythematous borders and subsequent residual scarring.1 O’Brien2 originally described this condition in 1975 as an actinic granuloma due to similar histologic findings in areas of the patient’s sun-exposed skin. Ragaz and Ackerman3 disputed O’Brien’s2 description, claiming granulomatous inflammation was a primary pathologic process and not a consequence to damaged elastotic material. In 1979, Hanke et al4 termed the lesions as AEGCG because he did not find a correlation to the sun-exposed areas of the patients and did not see solar elastosis.
Although AEGCG has an unclear pathogenesis, cellular immunologic reactions induced by modified function of elastic fibers’ antigenicity contribute to AEGCG formation.5 Therefore, environmental and host factors may play a role in its etiopathogenesis. In one study, 37% of 38 Japanese patients with AEGCG were found to have definitive or latent diabetes mellitus, raising the possible role of diabetes in the structural damage of the elastic fibers.6
Patients typically are middle-aged women who present clinically with red or atrophic plaques that have slightly elevated borders. They have centripetal spread with a resulting atrophic center.7 Clinically, the differential diagnosis of this condition includes actinic granuloma, granuloma annulare, and granuloma multiforme.8
Histologically, AEGCG has a granulomatous component with multinucleated giant cells in the upper and mid dermis. This component typically is distributed peripherally to a central zone that lacks elastic tissue. Elastophagocytosis, a classic finding in AEGCG, is the phagocytosis of elastic fibers that can microscopically be seen in the cytoplasm of histiocytes and multinucleated giant cells. There also is an absence of necrobiosis, lipids, mucin, and a palisading arrangement of the granulomas. These findings distinguish AEGCG from granuloma annulare and necrobiosis lipoidica, the primary histologic differential diagnoses.9 In addition, consideration of entities consistently exhibiting elastophagocytosis such as mid-dermal elastolysis, papillary dermal elastolysis, actinic granuloma, and granulomatous slack skin should be considered.5,10,11
Therapy for AEGCG is broad and includes topical, intralesional, and systemic corticosteroids. Hydroxychloroquine, isotretinoin, clofazimine, dapsone, photochemotherapy, and cyclosporine also have been utilized with varying results. Other reports show improvement with surgical excision, cryotherapy, or cauterization of small lesions.12-15
To the Editor:
A 52-year-old woman with a medical history of migraines and cervicalgia presented with lesions on the right arm, back, and right calf. The patient stated that the lesions began as small papules that had grown over 13 months, with the largest papule on the right forearm. She reported no itching, bleeding, pain, discharge, or other symptoms associated with the lesions. She had a multiple-year history of similar lesions that did not respond to treatment with antifungals, moderate-potency steroids, and other over-the-counter creams. The lesions would resolve spontaneously with scarring and subsequently recur. Prior skin biopsies were inconclusive. The patient did not report any systemic symptoms or a personal or family history of connective tissue diseases.
Physical examination revealed a 4-cm asymmetric, annular, erythematous plaque with central clearing on the right dorsal forearm with defined margins except over the distal aspect (Figure 1). She also had several 1- to 2-cm erythematous, nummular, asymmetric plaques on the right upper arm with well-defined margins. She had several lesions over the central and left sides of the upper back that were similar to the lesions on the upper arm.
Two 4-mm punch biopsies of the right dorsal forearm and left side of the upper back revealed similar histologic features with a predominantly unremarkable epidermis. The dermis revealed a lymphohistiocytic infiltrate with prominent multinucleated giant cells organized into foreign body–type granulomas that extended into the deep dermis and subcutaneous tissue (Figure 2). In the granulomatous areas, there was a near-complete loss of elastic fibers with focal elastophagocytosis highlighted with Verhoeff-van Gieson (elastin) stain (Figure 3). Grocott-Gomori methenamine-silver and Fite stains for microorganisms were negative, and there was an absence of necrobiosis, lipids, and mucin.
The histologic findings of a granulomatous dermatitis with loss of elastic fibers and elastophagocytosis in addition to the patient’s clinical presentation and history were consistent with the diagnosis of annular elastolytic giant cell granuloma (AEGCG). Infectious and other granulomatous diseases including sarcoidosis were ruled out via clinical history, unremarkable laboratory analysis (ie, complete blood cell count, chemistry panel, antinuclear antibody, urinalysis), and a normal chest radiograph. The histologic findings via the various stains were instrumental to the diagnosis. The patient was treated with fluocinonide and subsequently lost to follow-up.
Annular elastolytic giant cell granuloma is an uncommon cutaneous disease that presents with recurring annular plaques with raised erythematous borders and subsequent residual scarring.1 O’Brien2 originally described this condition in 1975 as an actinic granuloma due to similar histologic findings in areas of the patient’s sun-exposed skin. Ragaz and Ackerman3 disputed O’Brien’s2 description, claiming granulomatous inflammation was a primary pathologic process and not a consequence to damaged elastotic material. In 1979, Hanke et al4 termed the lesions as AEGCG because he did not find a correlation to the sun-exposed areas of the patients and did not see solar elastosis.
Although AEGCG has an unclear pathogenesis, cellular immunologic reactions induced by modified function of elastic fibers’ antigenicity contribute to AEGCG formation.5 Therefore, environmental and host factors may play a role in its etiopathogenesis. In one study, 37% of 38 Japanese patients with AEGCG were found to have definitive or latent diabetes mellitus, raising the possible role of diabetes in the structural damage of the elastic fibers.6
Patients typically are middle-aged women who present clinically with red or atrophic plaques that have slightly elevated borders. They have centripetal spread with a resulting atrophic center.7 Clinically, the differential diagnosis of this condition includes actinic granuloma, granuloma annulare, and granuloma multiforme.8
Histologically, AEGCG has a granulomatous component with multinucleated giant cells in the upper and mid dermis. This component typically is distributed peripherally to a central zone that lacks elastic tissue. Elastophagocytosis, a classic finding in AEGCG, is the phagocytosis of elastic fibers that can microscopically be seen in the cytoplasm of histiocytes and multinucleated giant cells. There also is an absence of necrobiosis, lipids, mucin, and a palisading arrangement of the granulomas. These findings distinguish AEGCG from granuloma annulare and necrobiosis lipoidica, the primary histologic differential diagnoses.9 In addition, consideration of entities consistently exhibiting elastophagocytosis such as mid-dermal elastolysis, papillary dermal elastolysis, actinic granuloma, and granulomatous slack skin should be considered.5,10,11
Therapy for AEGCG is broad and includes topical, intralesional, and systemic corticosteroids. Hydroxychloroquine, isotretinoin, clofazimine, dapsone, photochemotherapy, and cyclosporine also have been utilized with varying results. Other reports show improvement with surgical excision, cryotherapy, or cauterization of small lesions.12-15
1. Tock CL, Cohen PR. Annular elastolytic giant cell granuloma. Cutis. 1998;62:181-187.
2. O’Brien JP. Actinic granuloma: an annular connective tissue disorder affecting sun- and heat-damaged (elastotic) skin. Arch Dermatol. 1975;111:460-466.
3. Ragaz A, Ackerman AB. Is actinic granuloma a specific condition? Am J Dermatopathol. 1979;1:43-50.
4. Hanke CW, Bailin PL, Roenigk HH Jr. Annular elastolytic giant cell granuloma. a clinicopathologic study of five cases and a review of similar entities. J Am Acad Dermatol. 1979;1:413-421.
5. El-Khoury J, Kurban M, Abbas O. Elastophagocytosis: underlying mechanisms and associated cutaneous entities. J Am Acad Dermatol. 2014;70:934-44.
6. Aso Y, Izaki Y, Teraki Y. Annular elastolytic giant cell granuloma associated with diabetes mellitus: a case report and review of the Japanese literature. Clin Exp Dermatol. 2011;36:917-919.
7. Pestoni C, Pereiro M Jr, Toribio J. Annular elastolytic giant cell granuloma produced on an old burn scar and spreading after a mechanical trauma. Acta Derm Venereol. 2003;83:312-313.
8. Oka M, Kunisada M, Nishigori C. Generalized annular elastolytic giant cell granuloma with sparing of striae distensae. J Dermatol. 2013;40:220-222.
9. Limas C. The spectrum of primary cutaneous elastolytic granulomas and their distinction from granuloma annulare: a clinicopathological analysis. Histopathology. 2004;44:277-282.
10. McGrae JD Jr. Actinic granuloma: a clinical, histopathologic, and immunocytochemical study. Arch Dermatol. 1986;122:43-47.
11. Shah A, Safaya A. Granulomatous slack skin disease: a review, in comparison with mycosis fungoides. J Eur Acad Dermatol Venereol. 2012;26:1472-1478.
12. Chou WT, Tsai TF, Hung CM, et al. Multiple annular erythematous plaques on the back. Annular elastolytic giant cell granuloma (AEGCG). Indian J Dermatol Venereol Leprol. 2011;77:727-728.
13. Pérez-Pérez L, Garcia-Gavin J, Alleque F, et al. Successful treatment of generalized elastolytic giant cell granuloma with psoralen-ultraviolet A. Photodermatol Photoimmunol Photomed. 2012;28:264-266.
14. Babuna G, Buyukbabani N, Yazganoglu KD, et al. Effective treatment with hydroxychloroquine in a case of annular elastolytic giant cell granuloma. Indian J Dermatol Venereol Leprol. 2011;77:110-111.
15. Can B, Kavala M, Türkoglu Z, et al. Successful treatment of annular elastolytic giant cell granuloma with hydroxylchloroquine. Int J Dermatol. 2013;52:509-511.
1. Tock CL, Cohen PR. Annular elastolytic giant cell granuloma. Cutis. 1998;62:181-187.
2. O’Brien JP. Actinic granuloma: an annular connective tissue disorder affecting sun- and heat-damaged (elastotic) skin. Arch Dermatol. 1975;111:460-466.
3. Ragaz A, Ackerman AB. Is actinic granuloma a specific condition? Am J Dermatopathol. 1979;1:43-50.
4. Hanke CW, Bailin PL, Roenigk HH Jr. Annular elastolytic giant cell granuloma. a clinicopathologic study of five cases and a review of similar entities. J Am Acad Dermatol. 1979;1:413-421.
5. El-Khoury J, Kurban M, Abbas O. Elastophagocytosis: underlying mechanisms and associated cutaneous entities. J Am Acad Dermatol. 2014;70:934-44.
6. Aso Y, Izaki Y, Teraki Y. Annular elastolytic giant cell granuloma associated with diabetes mellitus: a case report and review of the Japanese literature. Clin Exp Dermatol. 2011;36:917-919.
7. Pestoni C, Pereiro M Jr, Toribio J. Annular elastolytic giant cell granuloma produced on an old burn scar and spreading after a mechanical trauma. Acta Derm Venereol. 2003;83:312-313.
8. Oka M, Kunisada M, Nishigori C. Generalized annular elastolytic giant cell granuloma with sparing of striae distensae. J Dermatol. 2013;40:220-222.
9. Limas C. The spectrum of primary cutaneous elastolytic granulomas and their distinction from granuloma annulare: a clinicopathological analysis. Histopathology. 2004;44:277-282.
10. McGrae JD Jr. Actinic granuloma: a clinical, histopathologic, and immunocytochemical study. Arch Dermatol. 1986;122:43-47.
11. Shah A, Safaya A. Granulomatous slack skin disease: a review, in comparison with mycosis fungoides. J Eur Acad Dermatol Venereol. 2012;26:1472-1478.
12. Chou WT, Tsai TF, Hung CM, et al. Multiple annular erythematous plaques on the back. Annular elastolytic giant cell granuloma (AEGCG). Indian J Dermatol Venereol Leprol. 2011;77:727-728.
13. Pérez-Pérez L, Garcia-Gavin J, Alleque F, et al. Successful treatment of generalized elastolytic giant cell granuloma with psoralen-ultraviolet A. Photodermatol Photoimmunol Photomed. 2012;28:264-266.
14. Babuna G, Buyukbabani N, Yazganoglu KD, et al. Effective treatment with hydroxychloroquine in a case of annular elastolytic giant cell granuloma. Indian J Dermatol Venereol Leprol. 2011;77:110-111.
15. Can B, Kavala M, Türkoglu Z, et al. Successful treatment of annular elastolytic giant cell granuloma with hydroxylchloroquine. Int J Dermatol. 2013;52:509-511.
Practice Points
- Annular elastolytic giant cell granuloma (AEGCG) should be kept in the differential diagnosis when assessing a middle-aged woman with recurring annular plaques with a raised border and an atrophic center on both sun-exposed and sun-protected areas of the body.
- Histologically, AEGCG classically has a granulomatous component in the dermis that lacks elastic tissue and has no necrobiosis, lipids, or mucin. Staining with elastin may be necessary to highlight these areas as well as demonstrate elastophagocytosis.
Lipoblastoma of the Scalp in a Child
To the Editor:
A 2-year-old boy was referred to our pediatric dermatology clinic by his pediatrician for an enlarging mass on the mid frontal scalp. The lesion had been present since birth and slowly enlarged. His parents thought the lesion was mostly asymptomatic; however, if it was irritated, the child would cry. He was otherwise healthy and had no history of skin conditions. There was no family history of skin conditions, birthmarks, or vascular malformations. On physical examination, we observed an isolated, approximately 3-cm, well-circumscribed, mobile, flesh-colored and violaceous nodule on the mid frontal scalp (Figure 1). At that time our differential diagnosis included a complex hemangioma or other vascular proliferation, nevus lipomatosis, or even a soft-tissue malignancy such as a sarcoma. Prior to biopsy, we ordered magnetic resonance imaging (MRI) to evaluate for intracranial extension of the lesion. The MRI revealed a 3.2-cm frontal midline scalp mass with complex imaging characteristics, and the radiologist gave a differential diagnosis of hemangioma, teratoma, or less likely liposarcoma (Figure 2). Fortunately, there was no evidence of central nervous system or intracranial invasion. We then proceeded with excisional biopsy, which grossly revealed a nodular, well-circumscribed, yellow mass (Figure 3). The wound was closed with primary closure. Histologically, there was a lobulated tumor with thin, well-vascularized connective tissue septa within a myxoid stroma (Figure 4A). The tumor was composed of lipocytes in varying stages of maturity without obvious nuclear atypia (Figures 4B and 4C), leading to a diagnosis of lipoblastoma.

Lipoblastoma (also known as an embryonic lipoma) is a rare variant of lipoma. It is a benign neoplasm of immature white fat cells primarily seen in children younger than 3 years. It is reportedly twice as common in boys versus girls. Lipoblastomas present as enlarging, soft, mobile, painless nodules, usually 3 to 5 cm in diameter. The extremities are the favored location, but they also have been described on the head, neck, and trunk. Additionally, mediastinal and retroperitoneal lipoblastomas have been documented.1 The tumors may be symptomatic, particularly when involving the neck or mediastinum. In rare instances, they may present with respiratory distress.2 Multiple cases of head and neck lipoblastomas have been published in the English-language literature.3 Growing evidence supports that a chromosomal breakpoint abnormality at 8q11-q13 may be implicated in the pathogenesis.4

mass without intracranial extension.
Most lipoblastomas are well circumscribed, encapsulated, and limited to the subcutis. However, lipoblastomatosis is the diffuse counterpart to lipoblastoma, affecting deeper soft tissue and often infiltrating adjacent skeletal muscle.1

Diagnosis is made by histologic evaluation. Lipoblastoma appears as immature fat cells in varying stages of maturity with septa separating them into lobules. There should not be nuclear atypia.5 The histologic differential diagnosis includes other adipose tumors, most chiefly myxoid liposarcoma. These tumors have a lobular pattern without prominent septae and contain nuclear atypia with atypical mitotic figures. Myxoid liposarcoma has an infiltrating pattern similar to lipoblastomatosis and has a metastatic rate up to 60%.6 Imaging studies such as MRI are helpful in diagnosis, particularly in head and neck or visceral cases.3 The treatment of choice of lipoblastoma is wide excision. With complete removal, tumors rarely recur. Recurrences are more common in lipoblastomatosis or with incompletely excised primary lesions.3 A 14% to 24% recurrence rate has been recognized. Cytogenic analysis of lipomatous tumors has begun to reveal translocations in chromosome 8q11-13 region breakpoint abnormalities and translocations, specifically involving the pleomorphic adenoma gene 1, PLAG1, as the oncogenic target in lipoblastoma.6 Identification of these molecular mutations may provide aid in differentiating histologically similar-appearing tumors in the future.
 (H&E, original magnification ×2). Higher-power view showed mature fat cells intermingled with spindle-shaped mesenchymal cells and lipobl")
This case illustrates a rare benign childhood tumor that can be difficult to diagnose prior to histologic examination. Our patient did not fit the typical description of a lipoblastoma, as his tumor was axially located as opposed to the more common peripheral presentation. We aim to raise awareness of this diagnosis as more cases are being recognized.
- Kaddu S, Kohler S. Muscle, adipose, and cartilage neoplasms. In: Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. St. Louis, MO: Mosby; 2003:1988-1989.
- Benato C, Falezza G, Lonardoni A, et al. Acute respiratory distress caused by a giant mediastinal lipoblastoma in a 16-month-old boy. Ann Thorac Surg. 2011;92:119-120.
- Pham NS, Poirier B, Fuller SC, et al. Pediatric lipoblastoma in the head and neck: a systematic review of 48 reported cases. Int J Pediatr Otorhinolaryngol. 2010;74:723-728.
- Chen Z, Coffin CM, Scott S, et al. Evidence by spectral karyotyping that 8q11.2 is nonrandomly involved in lipoblastoma. J Mol Diagn. 2000;2:73-77.
- Weedon, D. Weedon’s Skin Pathology. 3rd ed. China: Churchill Livingstone; 2010.
- Hicks J, Dilley A, Patal D. Lipoblastoma and lipoblastomatosis in infancy and childhood: histopathologic, ultrastructural, and cytogenetic features. Ultrastruct Pathol. 2001;25:321-333.
To the Editor:
A 2-year-old boy was referred to our pediatric dermatology clinic by his pediatrician for an enlarging mass on the mid frontal scalp. The lesion had been present since birth and slowly enlarged. His parents thought the lesion was mostly asymptomatic; however, if it was irritated, the child would cry. He was otherwise healthy and had no history of skin conditions. There was no family history of skin conditions, birthmarks, or vascular malformations. On physical examination, we observed an isolated, approximately 3-cm, well-circumscribed, mobile, flesh-colored and violaceous nodule on the mid frontal scalp (Figure 1). At that time our differential diagnosis included a complex hemangioma or other vascular proliferation, nevus lipomatosis, or even a soft-tissue malignancy such as a sarcoma. Prior to biopsy, we ordered magnetic resonance imaging (MRI) to evaluate for intracranial extension of the lesion. The MRI revealed a 3.2-cm frontal midline scalp mass with complex imaging characteristics, and the radiologist gave a differential diagnosis of hemangioma, teratoma, or less likely liposarcoma (Figure 2). Fortunately, there was no evidence of central nervous system or intracranial invasion. We then proceeded with excisional biopsy, which grossly revealed a nodular, well-circumscribed, yellow mass (Figure 3). The wound was closed with primary closure. Histologically, there was a lobulated tumor with thin, well-vascularized connective tissue septa within a myxoid stroma (Figure 4A). The tumor was composed of lipocytes in varying stages of maturity without obvious nuclear atypia (Figures 4B and 4C), leading to a diagnosis of lipoblastoma.
Lipoblastoma (also known as an embryonic lipoma) is a rare variant of lipoma. It is a benign neoplasm of immature white fat cells primarily seen in children younger than 3 years. It is reportedly twice as common in boys versus girls. Lipoblastomas present as enlarging, soft, mobile, painless nodules, usually 3 to 5 cm in diameter. The extremities are the favored location, but they also have been described on the head, neck, and trunk. Additionally, mediastinal and retroperitoneal lipoblastomas have been documented.1 The tumors may be symptomatic, particularly when involving the neck or mediastinum. In rare instances, they may present with respiratory distress.2 Multiple cases of head and neck lipoblastomas have been published in the English-language literature.3 Growing evidence supports that a chromosomal breakpoint abnormality at 8q11-q13 may be implicated in the pathogenesis.4
mass without intracranial extension.
Most lipoblastomas are well circumscribed, encapsulated, and limited to the subcutis. However, lipoblastomatosis is the diffuse counterpart to lipoblastoma, affecting deeper soft tissue and often infiltrating adjacent skeletal muscle.1
Diagnosis is made by histologic evaluation. Lipoblastoma appears as immature fat cells in varying stages of maturity with septa separating them into lobules. There should not be nuclear atypia.5 The histologic differential diagnosis includes other adipose tumors, most chiefly myxoid liposarcoma. These tumors have a lobular pattern without prominent septae and contain nuclear atypia with atypical mitotic figures. Myxoid liposarcoma has an infiltrating pattern similar to lipoblastomatosis and has a metastatic rate up to 60%.6 Imaging studies such as MRI are helpful in diagnosis, particularly in head and neck or visceral cases.3 The treatment of choice of lipoblastoma is wide excision. With complete removal, tumors rarely recur. Recurrences are more common in lipoblastomatosis or with incompletely excised primary lesions.3 A 14% to 24% recurrence rate has been recognized. Cytogenic analysis of lipomatous tumors has begun to reveal translocations in chromosome 8q11-13 region breakpoint abnormalities and translocations, specifically involving the pleomorphic adenoma gene 1, PLAG1, as the oncogenic target in lipoblastoma.6 Identification of these molecular mutations may provide aid in differentiating histologically similar-appearing tumors in the future.
This case illustrates a rare benign childhood tumor that can be difficult to diagnose prior to histologic examination. Our patient did not fit the typical description of a lipoblastoma, as his tumor was axially located as opposed to the more common peripheral presentation. We aim to raise awareness of this diagnosis as more cases are being recognized.
To the Editor:
A 2-year-old boy was referred to our pediatric dermatology clinic by his pediatrician for an enlarging mass on the mid frontal scalp. The lesion had been present since birth and slowly enlarged. His parents thought the lesion was mostly asymptomatic; however, if it was irritated, the child would cry. He was otherwise healthy and had no history of skin conditions. There was no family history of skin conditions, birthmarks, or vascular malformations. On physical examination, we observed an isolated, approximately 3-cm, well-circumscribed, mobile, flesh-colored and violaceous nodule on the mid frontal scalp (Figure 1). At that time our differential diagnosis included a complex hemangioma or other vascular proliferation, nevus lipomatosis, or even a soft-tissue malignancy such as a sarcoma. Prior to biopsy, we ordered magnetic resonance imaging (MRI) to evaluate for intracranial extension of the lesion. The MRI revealed a 3.2-cm frontal midline scalp mass with complex imaging characteristics, and the radiologist gave a differential diagnosis of hemangioma, teratoma, or less likely liposarcoma (Figure 2). Fortunately, there was no evidence of central nervous system or intracranial invasion. We then proceeded with excisional biopsy, which grossly revealed a nodular, well-circumscribed, yellow mass (Figure 3). The wound was closed with primary closure. Histologically, there was a lobulated tumor with thin, well-vascularized connective tissue septa within a myxoid stroma (Figure 4A). The tumor was composed of lipocytes in varying stages of maturity without obvious nuclear atypia (Figures 4B and 4C), leading to a diagnosis of lipoblastoma.
Lipoblastoma (also known as an embryonic lipoma) is a rare variant of lipoma. It is a benign neoplasm of immature white fat cells primarily seen in children younger than 3 years. It is reportedly twice as common in boys versus girls. Lipoblastomas present as enlarging, soft, mobile, painless nodules, usually 3 to 5 cm in diameter. The extremities are the favored location, but they also have been described on the head, neck, and trunk. Additionally, mediastinal and retroperitoneal lipoblastomas have been documented.1 The tumors may be symptomatic, particularly when involving the neck or mediastinum. In rare instances, they may present with respiratory distress.2 Multiple cases of head and neck lipoblastomas have been published in the English-language literature.3 Growing evidence supports that a chromosomal breakpoint abnormality at 8q11-q13 may be implicated in the pathogenesis.4
mass without intracranial extension.
Most lipoblastomas are well circumscribed, encapsulated, and limited to the subcutis. However, lipoblastomatosis is the diffuse counterpart to lipoblastoma, affecting deeper soft tissue and often infiltrating adjacent skeletal muscle.1
Diagnosis is made by histologic evaluation. Lipoblastoma appears as immature fat cells in varying stages of maturity with septa separating them into lobules. There should not be nuclear atypia.5 The histologic differential diagnosis includes other adipose tumors, most chiefly myxoid liposarcoma. These tumors have a lobular pattern without prominent septae and contain nuclear atypia with atypical mitotic figures. Myxoid liposarcoma has an infiltrating pattern similar to lipoblastomatosis and has a metastatic rate up to 60%.6 Imaging studies such as MRI are helpful in diagnosis, particularly in head and neck or visceral cases.3 The treatment of choice of lipoblastoma is wide excision. With complete removal, tumors rarely recur. Recurrences are more common in lipoblastomatosis or with incompletely excised primary lesions.3 A 14% to 24% recurrence rate has been recognized. Cytogenic analysis of lipomatous tumors has begun to reveal translocations in chromosome 8q11-13 region breakpoint abnormalities and translocations, specifically involving the pleomorphic adenoma gene 1, PLAG1, as the oncogenic target in lipoblastoma.6 Identification of these molecular mutations may provide aid in differentiating histologically similar-appearing tumors in the future.
This case illustrates a rare benign childhood tumor that can be difficult to diagnose prior to histologic examination. Our patient did not fit the typical description of a lipoblastoma, as his tumor was axially located as opposed to the more common peripheral presentation. We aim to raise awareness of this diagnosis as more cases are being recognized.
- Kaddu S, Kohler S. Muscle, adipose, and cartilage neoplasms. In: Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. St. Louis, MO: Mosby; 2003:1988-1989.
- Benato C, Falezza G, Lonardoni A, et al. Acute respiratory distress caused by a giant mediastinal lipoblastoma in a 16-month-old boy. Ann Thorac Surg. 2011;92:119-120.
- Pham NS, Poirier B, Fuller SC, et al. Pediatric lipoblastoma in the head and neck: a systematic review of 48 reported cases. Int J Pediatr Otorhinolaryngol. 2010;74:723-728.
- Chen Z, Coffin CM, Scott S, et al. Evidence by spectral karyotyping that 8q11.2 is nonrandomly involved in lipoblastoma. J Mol Diagn. 2000;2:73-77.
- Weedon, D. Weedon’s Skin Pathology. 3rd ed. China: Churchill Livingstone; 2010.
- Hicks J, Dilley A, Patal D. Lipoblastoma and lipoblastomatosis in infancy and childhood: histopathologic, ultrastructural, and cytogenetic features. Ultrastruct Pathol. 2001;25:321-333.
- Kaddu S, Kohler S. Muscle, adipose, and cartilage neoplasms. In: Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. St. Louis, MO: Mosby; 2003:1988-1989.
- Benato C, Falezza G, Lonardoni A, et al. Acute respiratory distress caused by a giant mediastinal lipoblastoma in a 16-month-old boy. Ann Thorac Surg. 2011;92:119-120.
- Pham NS, Poirier B, Fuller SC, et al. Pediatric lipoblastoma in the head and neck: a systematic review of 48 reported cases. Int J Pediatr Otorhinolaryngol. 2010;74:723-728.
- Chen Z, Coffin CM, Scott S, et al. Evidence by spectral karyotyping that 8q11.2 is nonrandomly involved in lipoblastoma. J Mol Diagn. 2000;2:73-77.
- Weedon, D. Weedon’s Skin Pathology. 3rd ed. China: Churchill Livingstone; 2010.
- Hicks J, Dilley A, Patal D. Lipoblastoma and lipoblastomatosis in infancy and childhood: histopathologic, ultrastructural, and cytogenetic features. Ultrastruct Pathol. 2001;25:321-333.
Practice Points
- Lipoblastoma is a benign neoplasm of immature white fat cells primarily seen in children younger than 3 years.
- Lipoblastomas often present as painless nodules located on the extremities.
- Histologically, lipoblastoma reveals immature adipose cells in varying stages of maturity arranged into lobules separated by septae.
- Consider magnetic resonance imaging if visceral extension is a concern; otherwise, surgical excision is curative in most cases.

Primary Cutaneous Cryptococcosis in an Immunocompetent Iraq War Veteran
To the Editor:
Disseminated cryptococcosis is a well-known opportunistic infection in patients with advanced human immunodeficiency virus (HIV) infection, but it is not frequently seen as a primary infection of the skin in immunocompetent hosts. We report a case of primary cutaneous cryptococcosis (PCC) of the lower legs in an immunocompetent Iraq War veteran.
A 28-year-old female service member presented to the dermatology clinic with progressively enlarging plaquelike lesions on the shins of 6 months’ duration. The patient had resided and worked as a deployed soldier in the lower level of a bullet hole–laden, pigeon-infested observation tower in southern Iraq 9 months prior to the current presentation. During her 7-month deployment, she reported daily exposure to pigeon excreta on equipment and frequently sustained superficial abrasions and lacerations to the legs due to the cramped and hazardous working environment. The patient noticed intensely pruritic, bugbitelike papular lesions on the shins and calves 1 month after residing in the observation tower. She sought medical treatment and was given hydrocortisone cream 1% and calamine lotion for a presumed irritant dermatitis. Over the ensuing 3 months, the pruritus worsened, and the primary lesions coalesced into annular erythematous plaques (Figure).
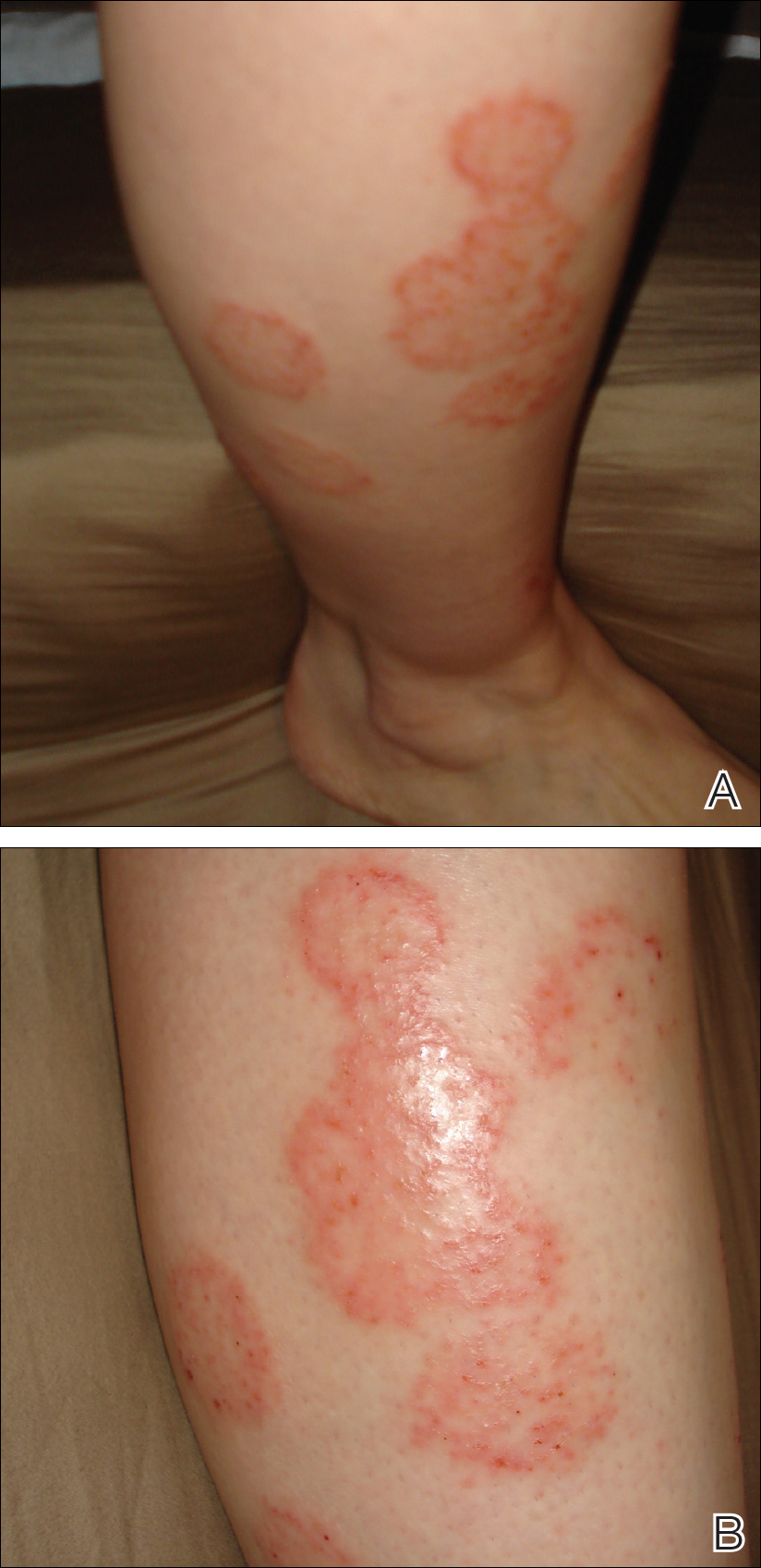
After returning to the United States, the patient presented again for medical care and was given ketoconazole cream 1% for presumed tinea corporis, which resulted in no improvement. A dermatologic consultation and evaluation ensued with subsequent microbial workup showing no bacterial growth on wound culture and no fungal elements on a potassium hydroxide preparation. Hematoxylin and eosin, periodic acid–Schiff, and Grocott-Gomori methenamine-silver staining did not demonstrate any organisms. Tissue cultures for bacteria and acid-fast bacilli showed no growth. A fungal tissue culture ultimately confirmed the presence of Cryptococcus neoformans. A lumbar puncture showed no evidence of Cryptococcus on DNA probe testing. Serologic testing for HIV was negative, and brain magnetic resonance imaging showed no lesions. Sputum culture and staining showed no fungal elements, and a chest radiograph was normal. A diagnosis of PCC was made and therapy with oral fluconazole 200 mg twice daily was initiated, with the intention of completing a 6-month course. During the treatment, the pruritus resolved within 3 weeks and the lesions involuted over 3 months. From the time of onset of the lesions throughout treatment, the patient showed no pulmonary, neurologic, or other systemic symptoms. She currently is healthy with no evidence of recurrence.
Primary cutaneous cryptococcosis mainly affects individuals with underlying immunosuppression, most commonly due to advanced HIV, prolonged treatment with immunosuppressive medications, or organ transplantation.1 The most common route of inoculation is by inhalation of Cryptococcus spores with subsequent hematogenous dissemination.2 Primary cutaneous cryptococcosis with skin lesions and no concomitant systemic involvement has rarely been reported, and
Due to the worldwide deployment of US military service members, exotic cutaneous infectious diseases such as PCC may be encountered in dermatology practice. Prompt clinical and histologic diagnosis is imperative to assess for systemic disease and avoid cutaneous spread and morbidity in US service members and travelers returning home from the Middle East.
- Antony SA, Antony SJ. Primary cutaneous Cryptococcus in nonimmunocompromised patients. Cutis. 1995;56:96-98.
- Mirza SA, Phelan M, Rimland D, et al. The changing epidemiology of cryptococcosis: an update from population-based active surveillance in 2 large metropolitan areas, 1992-2000. Clin Infect Dis. 2003;36:789-94.
- Kielstein P, Hotzel H, Schmalreck A, et al. Occurrence of Cryptococcus spp. in excreta of pigeons and pet birds. Mycoses. 2000;43:7-15.
- Leão CA, Ferreira-Paim K, Andrade-Silva L, et al. Primary cutaneous cryptococcosis caused by Cryptococcus gattii in an immunocompetent host [published online October 28, 2010]. Med Mycol. 2011;49:352-355.
- Zorman JV, Zupanc TL, Parac Z, et al. Primary cutaneous cryptococcosis in a renal transplant recipient: case report. Mycoses. 2010;53:535-537.
To the Editor:
Disseminated cryptococcosis is a well-known opportunistic infection in patients with advanced human immunodeficiency virus (HIV) infection, but it is not frequently seen as a primary infection of the skin in immunocompetent hosts. We report a case of primary cutaneous cryptococcosis (PCC) of the lower legs in an immunocompetent Iraq War veteran.
A 28-year-old female service member presented to the dermatology clinic with progressively enlarging plaquelike lesions on the shins of 6 months’ duration. The patient had resided and worked as a deployed soldier in the lower level of a bullet hole–laden, pigeon-infested observation tower in southern Iraq 9 months prior to the current presentation. During her 7-month deployment, she reported daily exposure to pigeon excreta on equipment and frequently sustained superficial abrasions and lacerations to the legs due to the cramped and hazardous working environment. The patient noticed intensely pruritic, bugbitelike papular lesions on the shins and calves 1 month after residing in the observation tower. She sought medical treatment and was given hydrocortisone cream 1% and calamine lotion for a presumed irritant dermatitis. Over the ensuing 3 months, the pruritus worsened, and the primary lesions coalesced into annular erythematous plaques (Figure).
After returning to the United States, the patient presented again for medical care and was given ketoconazole cream 1% for presumed tinea corporis, which resulted in no improvement. A dermatologic consultation and evaluation ensued with subsequent microbial workup showing no bacterial growth on wound culture and no fungal elements on a potassium hydroxide preparation. Hematoxylin and eosin, periodic acid–Schiff, and Grocott-Gomori methenamine-silver staining did not demonstrate any organisms. Tissue cultures for bacteria and acid-fast bacilli showed no growth. A fungal tissue culture ultimately confirmed the presence of Cryptococcus neoformans. A lumbar puncture showed no evidence of Cryptococcus on DNA probe testing. Serologic testing for HIV was negative, and brain magnetic resonance imaging showed no lesions. Sputum culture and staining showed no fungal elements, and a chest radiograph was normal. A diagnosis of PCC was made and therapy with oral fluconazole 200 mg twice daily was initiated, with the intention of completing a 6-month course. During the treatment, the pruritus resolved within 3 weeks and the lesions involuted over 3 months. From the time of onset of the lesions throughout treatment, the patient showed no pulmonary, neurologic, or other systemic symptoms. She currently is healthy with no evidence of recurrence.
Primary cutaneous cryptococcosis mainly affects individuals with underlying immunosuppression, most commonly due to advanced HIV, prolonged treatment with immunosuppressive medications, or organ transplantation.1 The most common route of inoculation is by inhalation of Cryptococcus spores with subsequent hematogenous dissemination.2 Primary cutaneous cryptococcosis with skin lesions and no concomitant systemic involvement has rarely been reported, and
Due to the worldwide deployment of US military service members, exotic cutaneous infectious diseases such as PCC may be encountered in dermatology practice. Prompt clinical and histologic diagnosis is imperative to assess for systemic disease and avoid cutaneous spread and morbidity in US service members and travelers returning home from the Middle East.
To the Editor:
Disseminated cryptococcosis is a well-known opportunistic infection in patients with advanced human immunodeficiency virus (HIV) infection, but it is not frequently seen as a primary infection of the skin in immunocompetent hosts. We report a case of primary cutaneous cryptococcosis (PCC) of the lower legs in an immunocompetent Iraq War veteran.
A 28-year-old female service member presented to the dermatology clinic with progressively enlarging plaquelike lesions on the shins of 6 months’ duration. The patient had resided and worked as a deployed soldier in the lower level of a bullet hole–laden, pigeon-infested observation tower in southern Iraq 9 months prior to the current presentation. During her 7-month deployment, she reported daily exposure to pigeon excreta on equipment and frequently sustained superficial abrasions and lacerations to the legs due to the cramped and hazardous working environment. The patient noticed intensely pruritic, bugbitelike papular lesions on the shins and calves 1 month after residing in the observation tower. She sought medical treatment and was given hydrocortisone cream 1% and calamine lotion for a presumed irritant dermatitis. Over the ensuing 3 months, the pruritus worsened, and the primary lesions coalesced into annular erythematous plaques (Figure).
After returning to the United States, the patient presented again for medical care and was given ketoconazole cream 1% for presumed tinea corporis, which resulted in no improvement. A dermatologic consultation and evaluation ensued with subsequent microbial workup showing no bacterial growth on wound culture and no fungal elements on a potassium hydroxide preparation. Hematoxylin and eosin, periodic acid–Schiff, and Grocott-Gomori methenamine-silver staining did not demonstrate any organisms. Tissue cultures for bacteria and acid-fast bacilli showed no growth. A fungal tissue culture ultimately confirmed the presence of Cryptococcus neoformans. A lumbar puncture showed no evidence of Cryptococcus on DNA probe testing. Serologic testing for HIV was negative, and brain magnetic resonance imaging showed no lesions. Sputum culture and staining showed no fungal elements, and a chest radiograph was normal. A diagnosis of PCC was made and therapy with oral fluconazole 200 mg twice daily was initiated, with the intention of completing a 6-month course. During the treatment, the pruritus resolved within 3 weeks and the lesions involuted over 3 months. From the time of onset of the lesions throughout treatment, the patient showed no pulmonary, neurologic, or other systemic symptoms. She currently is healthy with no evidence of recurrence.
Primary cutaneous cryptococcosis mainly affects individuals with underlying immunosuppression, most commonly due to advanced HIV, prolonged treatment with immunosuppressive medications, or organ transplantation.1 The most common route of inoculation is by inhalation of Cryptococcus spores with subsequent hematogenous dissemination.2 Primary cutaneous cryptococcosis with skin lesions and no concomitant systemic involvement has rarely been reported, and
Due to the worldwide deployment of US military service members, exotic cutaneous infectious diseases such as PCC may be encountered in dermatology practice. Prompt clinical and histologic diagnosis is imperative to assess for systemic disease and avoid cutaneous spread and morbidity in US service members and travelers returning home from the Middle East.
- Antony SA, Antony SJ. Primary cutaneous Cryptococcus in nonimmunocompromised patients. Cutis. 1995;56:96-98.
- Mirza SA, Phelan M, Rimland D, et al. The changing epidemiology of cryptococcosis: an update from population-based active surveillance in 2 large metropolitan areas, 1992-2000. Clin Infect Dis. 2003;36:789-94.
- Kielstein P, Hotzel H, Schmalreck A, et al. Occurrence of Cryptococcus spp. in excreta of pigeons and pet birds. Mycoses. 2000;43:7-15.
- Leão CA, Ferreira-Paim K, Andrade-Silva L, et al. Primary cutaneous cryptococcosis caused by Cryptococcus gattii in an immunocompetent host [published online October 28, 2010]. Med Mycol. 2011;49:352-355.
- Zorman JV, Zupanc TL, Parac Z, et al. Primary cutaneous cryptococcosis in a renal transplant recipient: case report. Mycoses. 2010;53:535-537.
- Antony SA, Antony SJ. Primary cutaneous Cryptococcus in nonimmunocompromised patients. Cutis. 1995;56:96-98.
- Mirza SA, Phelan M, Rimland D, et al. The changing epidemiology of cryptococcosis: an update from population-based active surveillance in 2 large metropolitan areas, 1992-2000. Clin Infect Dis. 2003;36:789-94.
- Kielstein P, Hotzel H, Schmalreck A, et al. Occurrence of Cryptococcus spp. in excreta of pigeons and pet birds. Mycoses. 2000;43:7-15.
- Leão CA, Ferreira-Paim K, Andrade-Silva L, et al. Primary cutaneous cryptococcosis caused by Cryptococcus gattii in an immunocompetent host [published online October 28, 2010]. Med Mycol. 2011;49:352-355.
- Zorman JV, Zupanc TL, Parac Z, et al. Primary cutaneous cryptococcosis in a renal transplant recipient: case report. Mycoses. 2010;53:535-537.
Practice Points
- Disseminated cryptococcosis is not commonly seen as a primary cutaneous infection in immunocompetent hosts.
- When encountered, primary cutaneous cryptococcosis (PCC) usually is associated with environments that predispose patients to skin wounds with simultaneous exposure to soil or vegetative debris contaminated with bird excreta.
- The variable presentation of PCC can cause clinical confusion and diagnostic delay; therefore, a high index of suspicion is required for timely diagnosis, particularly in US service members and travelers returning home from endemic areas.