User login
Bilateral wrist pain • limited range of motion • tenderness to palpation • Dx?
THE CASE
A 12-year-old girl presented to my office (JH) with bilateral wrist pain. She had fallen on both wrists palmar-flexed and then, while trying to get up, landed on both wrists dorsi-flexed. The patient did not hear any “pops,” but felt immediate pain when her wrists hyperextended. Hand, wrist, and forearm x-rays were negative bilaterally for fractures. She was placed in bilateral thumb spica splints.
At follow-up one week later, the patient reported 6/10 pain in her left wrist and 7/10 pain in her right wrist. The pain increased to 10/10 bilaterally with movement and was not relieved by icing or nonsteroidal anti-inflammatory drugs. On physical exam, there was bilateral swelling of the wrists without ecchymosis or erythema. The patient had limited passive and active range of motion, especially during wrist extension. She also had tenderness to palpation over the anatomical snuff box, extending proximally to the distal radius bilaterally. She had no tenderness over the ulna or metacarpals, no loss of sensation in any area nerves, and she was neurovascularly intact bilaterally.
Based on the mechanism of injury, undetected fracture or full thickness ligament tear were both possible. Because of this, and because magnetic resonance imaging (MRI) entails no radiation exposure, MRI was chosen for additional imaging of both wrists.
THE DIAGNOSIS

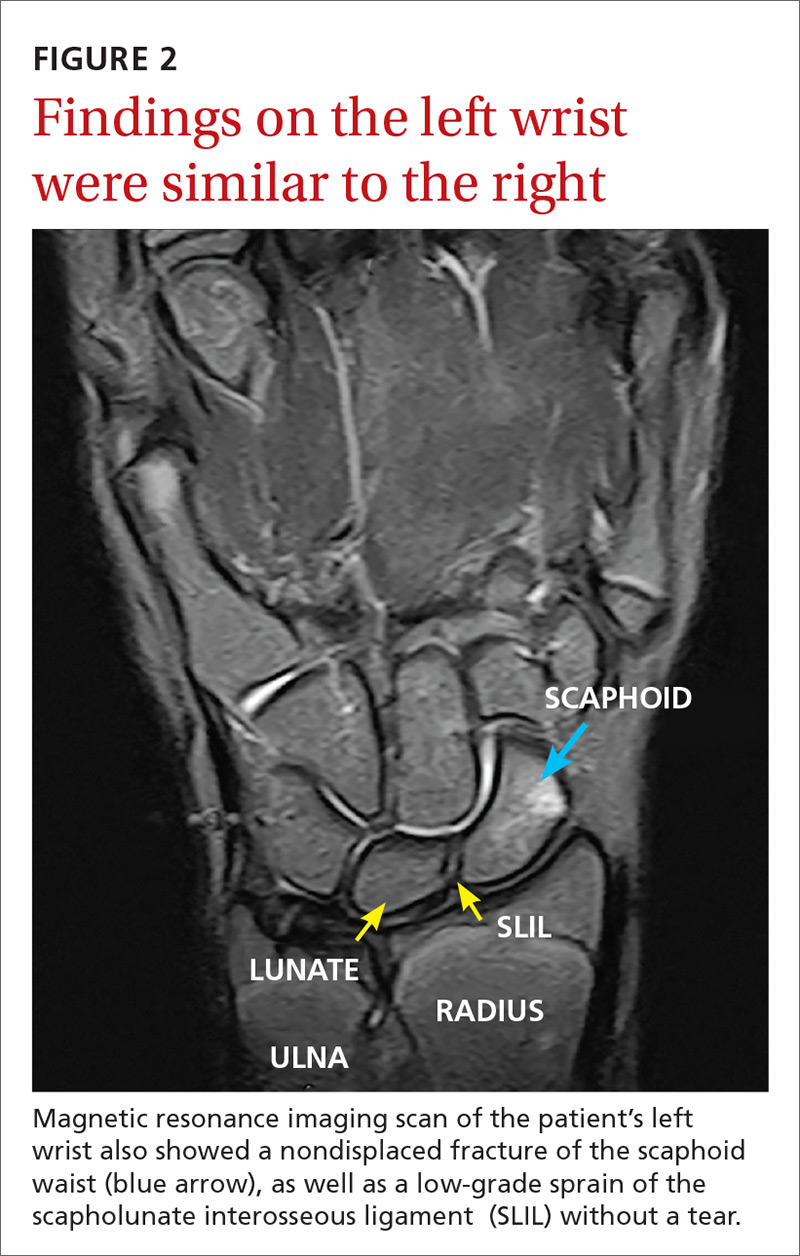
The MRI revealed bilateral, nondisplaced, extra-articular fractures extending through the scaphoid waist, with surrounding bone marrow edema. In the right wrist, the patient also had a low-grade partial tear of the membranous portion of the scapholunate interosseous ligament (SLIL) at the scaphoid attachment (FIGURE 1). In the left wrist, she also had a low-grade sprain of the SLIL without tear (FIGURE 2).
DISCUSSION
Carpal fractures account for 6% of all fractures.1 Scaphoid fractures are the most common carpal bone fracture among all age groups, but account for only 0.4% of all pediatric fractures.1-3 They’re commonly missed on x-rays because they are usually nondisplaced and hidden by other structures superimposed on the image.1,2,4 Undetected, scaphoid fractures can cause prolonged interruption to the bone’s architecture, leading to avascular necrosis of the proximal portion of the scaphoid bone.5,6
Bilateral scaphoid fractures are extremely rare and account for less than 1% of all scaphoid fractures.7 Very few of these cases have been published in the literature, and those that have been published have talked about the fractures being secondary to chronic stress fractures and as being treated with internal fixation (regardless of whether the fractures were nondisplaced or if the ligaments were intact).6-9
Our patient was placed in bilateral fiberglass short-arm thumb spica casts. We tried conservative treatment measures first because she had help with her activities of daily living (ADLs). At a follow-up visit 2 weeks later, we switched the casts to long-arm thumb spica casts because of the patient’s ability to pronate and supinate her wrists in the short-arm versions. After one month of wearing the long-arm casts, we placed her back in bilateral short-arm casts for 2 weeks. Eight weeks after the fall, we removed the short-arm casts for reevaluation.
We obtained x-rays to assess for any new changes to the wrist and specifically the scaphoid bones. The x-rays showed almost completely healed scaphoid bones with good alignment, but the patient still had 5/10 pain in the left wrist and 8/10 pain in the right wrist with movement. We placed her in adjustable thermoformable polymer braces, which were removed when she bathed.
Due to the uniqueness of her injuries, our patient had weekly visits with her primary care provider (PCP) for the first 2 months of treatment, followed by bimonthly visits for the remainder. At 10 weeks after the fall, her pain with movement was almost gone and she began physical therapy. She also began removing the braces during sedentary activity in order to practice range-of-motion exercises to prevent excessive stiffness in her wrists. Our patient regained full strength and range of motion one month later.
One other published case report describes the successful union of bilateral scaphoid fractures using bilateral long-arm casts followed by short-arm casts.7 Similar to our patient’s case, full union of the scaphoid bones was achieved within 12 weeks.7 Together, these cases suggest that conservative treatment methods are a viable alternative to surgery.
TAKEAWAY
For patients presenting with wrist pain after trauma to the wrists, assess anatomical snuffbox tenderness and obtain x-rays. Do not be falsely reassured by negative x-rays in the presence of a positive physical exam, however, as scaphoid fractures are often hidden on x-rays. If tenderness at the anatomical snuffbox is present and doesn’t subside within a few days, apply a short-arm thumb splint and obtain subsequent imaging.
If bilateral, nondisplaced, stable scaphoid fractures are diagnosed, conservative treatment with long-arm and short-arm casts is a viable alternative to surgery. This treatment decision should be made on an individual basis, however, as it requires the patient to have frequent PCP visits, assistance with ADLs, and complete adherence to the treatment plan.
1. Pillai A, Jain M. Management of clinical fractures of the scaphoid: results of an audit and literature review. Eur J Emerg Med. 2005;12:47-51.
2. Evenski AJ, Adamczyk MJ, Steiner RP, et al. Clinically suspected scaphoid fractures in children. J Pediatr Orthop. 2009;29:352-355.
3. Wulff R, Schmidt T. Carpal fractures in children. J Pediatr Orthop. 1998;18:462-465.
4. Nellans KW, Chung KC. Pediatric hand fractures. Hand Clin. 2013;29:569-578.
5. Jernigan EW, Smetana BS, Patterson JM. Pediatric scaphoid proximal pole nonunion with avascular necrosis. J Hand Surgery. 2017;42:299.e1-299.e4.
6. Pidemunt G, Torres-Claramunt R, Ginés A, et al. Bilateral stress fracture of the carpal scaphoid: report in a child and review of the literature. Clin J Sport Med. 2012;22:511-513.
7. Saglam F, Gulabi D, Baysal Ö, et al. Chronic wrist pain in a goalkeeper; bilateral scaphoid stress fracture: a case report. Int J Surg Case Rep. 2015;7:20-22.
8. Muzaffar N, Wani I, Ehsan M, et al. Simultaneous bilateral scaphoid fractures in a soldier managed conservatively by scaphoid casts. Arch Clin Exp Surg. 2016;5:63-64.
9. Mohamed Haflah NH, Mat Nor NF, Abdullah S, et al. Bilateral scaphoid stress fracture in a platform diver presenting with unilateral symptoms. Singapore Med J. 2014;55:e159-e161.
THE CASE
A 12-year-old girl presented to my office (JH) with bilateral wrist pain. She had fallen on both wrists palmar-flexed and then, while trying to get up, landed on both wrists dorsi-flexed. The patient did not hear any “pops,” but felt immediate pain when her wrists hyperextended. Hand, wrist, and forearm x-rays were negative bilaterally for fractures. She was placed in bilateral thumb spica splints.
At follow-up one week later, the patient reported 6/10 pain in her left wrist and 7/10 pain in her right wrist. The pain increased to 10/10 bilaterally with movement and was not relieved by icing or nonsteroidal anti-inflammatory drugs. On physical exam, there was bilateral swelling of the wrists without ecchymosis or erythema. The patient had limited passive and active range of motion, especially during wrist extension. She also had tenderness to palpation over the anatomical snuff box, extending proximally to the distal radius bilaterally. She had no tenderness over the ulna or metacarpals, no loss of sensation in any area nerves, and she was neurovascularly intact bilaterally.
Based on the mechanism of injury, undetected fracture or full thickness ligament tear were both possible. Because of this, and because magnetic resonance imaging (MRI) entails no radiation exposure, MRI was chosen for additional imaging of both wrists.
THE DIAGNOSIS
The MRI revealed bilateral, nondisplaced, extra-articular fractures extending through the scaphoid waist, with surrounding bone marrow edema. In the right wrist, the patient also had a low-grade partial tear of the membranous portion of the scapholunate interosseous ligament (SLIL) at the scaphoid attachment (FIGURE 1). In the left wrist, she also had a low-grade sprain of the SLIL without tear (FIGURE 2).
DISCUSSION
Carpal fractures account for 6% of all fractures.1 Scaphoid fractures are the most common carpal bone fracture among all age groups, but account for only 0.4% of all pediatric fractures.1-3 They’re commonly missed on x-rays because they are usually nondisplaced and hidden by other structures superimposed on the image.1,2,4 Undetected, scaphoid fractures can cause prolonged interruption to the bone’s architecture, leading to avascular necrosis of the proximal portion of the scaphoid bone.5,6
Bilateral scaphoid fractures are extremely rare and account for less than 1% of all scaphoid fractures.7 Very few of these cases have been published in the literature, and those that have been published have talked about the fractures being secondary to chronic stress fractures and as being treated with internal fixation (regardless of whether the fractures were nondisplaced or if the ligaments were intact).6-9
Our patient was placed in bilateral fiberglass short-arm thumb spica casts. We tried conservative treatment measures first because she had help with her activities of daily living (ADLs). At a follow-up visit 2 weeks later, we switched the casts to long-arm thumb spica casts because of the patient’s ability to pronate and supinate her wrists in the short-arm versions. After one month of wearing the long-arm casts, we placed her back in bilateral short-arm casts for 2 weeks. Eight weeks after the fall, we removed the short-arm casts for reevaluation.
We obtained x-rays to assess for any new changes to the wrist and specifically the scaphoid bones. The x-rays showed almost completely healed scaphoid bones with good alignment, but the patient still had 5/10 pain in the left wrist and 8/10 pain in the right wrist with movement. We placed her in adjustable thermoformable polymer braces, which were removed when she bathed.
Due to the uniqueness of her injuries, our patient had weekly visits with her primary care provider (PCP) for the first 2 months of treatment, followed by bimonthly visits for the remainder. At 10 weeks after the fall, her pain with movement was almost gone and she began physical therapy. She also began removing the braces during sedentary activity in order to practice range-of-motion exercises to prevent excessive stiffness in her wrists. Our patient regained full strength and range of motion one month later.
One other published case report describes the successful union of bilateral scaphoid fractures using bilateral long-arm casts followed by short-arm casts.7 Similar to our patient’s case, full union of the scaphoid bones was achieved within 12 weeks.7 Together, these cases suggest that conservative treatment methods are a viable alternative to surgery.
TAKEAWAY
For patients presenting with wrist pain after trauma to the wrists, assess anatomical snuffbox tenderness and obtain x-rays. Do not be falsely reassured by negative x-rays in the presence of a positive physical exam, however, as scaphoid fractures are often hidden on x-rays. If tenderness at the anatomical snuffbox is present and doesn’t subside within a few days, apply a short-arm thumb splint and obtain subsequent imaging.
If bilateral, nondisplaced, stable scaphoid fractures are diagnosed, conservative treatment with long-arm and short-arm casts is a viable alternative to surgery. This treatment decision should be made on an individual basis, however, as it requires the patient to have frequent PCP visits, assistance with ADLs, and complete adherence to the treatment plan.
THE CASE
A 12-year-old girl presented to my office (JH) with bilateral wrist pain. She had fallen on both wrists palmar-flexed and then, while trying to get up, landed on both wrists dorsi-flexed. The patient did not hear any “pops,” but felt immediate pain when her wrists hyperextended. Hand, wrist, and forearm x-rays were negative bilaterally for fractures. She was placed in bilateral thumb spica splints.
At follow-up one week later, the patient reported 6/10 pain in her left wrist and 7/10 pain in her right wrist. The pain increased to 10/10 bilaterally with movement and was not relieved by icing or nonsteroidal anti-inflammatory drugs. On physical exam, there was bilateral swelling of the wrists without ecchymosis or erythema. The patient had limited passive and active range of motion, especially during wrist extension. She also had tenderness to palpation over the anatomical snuff box, extending proximally to the distal radius bilaterally. She had no tenderness over the ulna or metacarpals, no loss of sensation in any area nerves, and she was neurovascularly intact bilaterally.
Based on the mechanism of injury, undetected fracture or full thickness ligament tear were both possible. Because of this, and because magnetic resonance imaging (MRI) entails no radiation exposure, MRI was chosen for additional imaging of both wrists.
THE DIAGNOSIS
The MRI revealed bilateral, nondisplaced, extra-articular fractures extending through the scaphoid waist, with surrounding bone marrow edema. In the right wrist, the patient also had a low-grade partial tear of the membranous portion of the scapholunate interosseous ligament (SLIL) at the scaphoid attachment (FIGURE 1). In the left wrist, she also had a low-grade sprain of the SLIL without tear (FIGURE 2).
DISCUSSION
Carpal fractures account for 6% of all fractures.1 Scaphoid fractures are the most common carpal bone fracture among all age groups, but account for only 0.4% of all pediatric fractures.1-3 They’re commonly missed on x-rays because they are usually nondisplaced and hidden by other structures superimposed on the image.1,2,4 Undetected, scaphoid fractures can cause prolonged interruption to the bone’s architecture, leading to avascular necrosis of the proximal portion of the scaphoid bone.5,6
Bilateral scaphoid fractures are extremely rare and account for less than 1% of all scaphoid fractures.7 Very few of these cases have been published in the literature, and those that have been published have talked about the fractures being secondary to chronic stress fractures and as being treated with internal fixation (regardless of whether the fractures were nondisplaced or if the ligaments were intact).6-9
Our patient was placed in bilateral fiberglass short-arm thumb spica casts. We tried conservative treatment measures first because she had help with her activities of daily living (ADLs). At a follow-up visit 2 weeks later, we switched the casts to long-arm thumb spica casts because of the patient’s ability to pronate and supinate her wrists in the short-arm versions. After one month of wearing the long-arm casts, we placed her back in bilateral short-arm casts for 2 weeks. Eight weeks after the fall, we removed the short-arm casts for reevaluation.
We obtained x-rays to assess for any new changes to the wrist and specifically the scaphoid bones. The x-rays showed almost completely healed scaphoid bones with good alignment, but the patient still had 5/10 pain in the left wrist and 8/10 pain in the right wrist with movement. We placed her in adjustable thermoformable polymer braces, which were removed when she bathed.
Due to the uniqueness of her injuries, our patient had weekly visits with her primary care provider (PCP) for the first 2 months of treatment, followed by bimonthly visits for the remainder. At 10 weeks after the fall, her pain with movement was almost gone and she began physical therapy. She also began removing the braces during sedentary activity in order to practice range-of-motion exercises to prevent excessive stiffness in her wrists. Our patient regained full strength and range of motion one month later.
One other published case report describes the successful union of bilateral scaphoid fractures using bilateral long-arm casts followed by short-arm casts.7 Similar to our patient’s case, full union of the scaphoid bones was achieved within 12 weeks.7 Together, these cases suggest that conservative treatment methods are a viable alternative to surgery.
TAKEAWAY
For patients presenting with wrist pain after trauma to the wrists, assess anatomical snuffbox tenderness and obtain x-rays. Do not be falsely reassured by negative x-rays in the presence of a positive physical exam, however, as scaphoid fractures are often hidden on x-rays. If tenderness at the anatomical snuffbox is present and doesn’t subside within a few days, apply a short-arm thumb splint and obtain subsequent imaging.
If bilateral, nondisplaced, stable scaphoid fractures are diagnosed, conservative treatment with long-arm and short-arm casts is a viable alternative to surgery. This treatment decision should be made on an individual basis, however, as it requires the patient to have frequent PCP visits, assistance with ADLs, and complete adherence to the treatment plan.
1. Pillai A, Jain M. Management of clinical fractures of the scaphoid: results of an audit and literature review. Eur J Emerg Med. 2005;12:47-51.
2. Evenski AJ, Adamczyk MJ, Steiner RP, et al. Clinically suspected scaphoid fractures in children. J Pediatr Orthop. 2009;29:352-355.
3. Wulff R, Schmidt T. Carpal fractures in children. J Pediatr Orthop. 1998;18:462-465.
4. Nellans KW, Chung KC. Pediatric hand fractures. Hand Clin. 2013;29:569-578.
5. Jernigan EW, Smetana BS, Patterson JM. Pediatric scaphoid proximal pole nonunion with avascular necrosis. J Hand Surgery. 2017;42:299.e1-299.e4.
6. Pidemunt G, Torres-Claramunt R, Ginés A, et al. Bilateral stress fracture of the carpal scaphoid: report in a child and review of the literature. Clin J Sport Med. 2012;22:511-513.
7. Saglam F, Gulabi D, Baysal Ö, et al. Chronic wrist pain in a goalkeeper; bilateral scaphoid stress fracture: a case report. Int J Surg Case Rep. 2015;7:20-22.
8. Muzaffar N, Wani I, Ehsan M, et al. Simultaneous bilateral scaphoid fractures in a soldier managed conservatively by scaphoid casts. Arch Clin Exp Surg. 2016;5:63-64.
9. Mohamed Haflah NH, Mat Nor NF, Abdullah S, et al. Bilateral scaphoid stress fracture in a platform diver presenting with unilateral symptoms. Singapore Med J. 2014;55:e159-e161.
1. Pillai A, Jain M. Management of clinical fractures of the scaphoid: results of an audit and literature review. Eur J Emerg Med. 2005;12:47-51.
2. Evenski AJ, Adamczyk MJ, Steiner RP, et al. Clinically suspected scaphoid fractures in children. J Pediatr Orthop. 2009;29:352-355.
3. Wulff R, Schmidt T. Carpal fractures in children. J Pediatr Orthop. 1998;18:462-465.
4. Nellans KW, Chung KC. Pediatric hand fractures. Hand Clin. 2013;29:569-578.
5. Jernigan EW, Smetana BS, Patterson JM. Pediatric scaphoid proximal pole nonunion with avascular necrosis. J Hand Surgery. 2017;42:299.e1-299.e4.
6. Pidemunt G, Torres-Claramunt R, Ginés A, et al. Bilateral stress fracture of the carpal scaphoid: report in a child and review of the literature. Clin J Sport Med. 2012;22:511-513.
7. Saglam F, Gulabi D, Baysal Ö, et al. Chronic wrist pain in a goalkeeper; bilateral scaphoid stress fracture: a case report. Int J Surg Case Rep. 2015;7:20-22.
8. Muzaffar N, Wani I, Ehsan M, et al. Simultaneous bilateral scaphoid fractures in a soldier managed conservatively by scaphoid casts. Arch Clin Exp Surg. 2016;5:63-64.
9. Mohamed Haflah NH, Mat Nor NF, Abdullah S, et al. Bilateral scaphoid stress fracture in a platform diver presenting with unilateral symptoms. Singapore Med J. 2014;55:e159-e161.
Concurrent ipilimumab and CMV colitis refractory to oral steroids
Immune checkpoint inhibitors, including anti-cytotoxic T-lymphocyte antigen 4 (anti-CTLA4) and anti-programmed cell death protein-1 (anti-PD-1) antibodies, have demonstrated clinical and survival benefits in a variety of malignancies, which has led to an expansion in their role in oncology. In melanoma, the anti-CTLA-4 antibody, ipilimumab, has demonstrated a survival benefit in patients with advanced metastatic melanoma and in patients with resectable disease with lymph node involvement.1,2
Ipilimumab exerts its effect by binding CTLA-4 on conventional and regulatory T cells, thus blocking inhibitory signals on T cells, which leads to an antitumor response.3 The increased immune response counteracts the immune-evading mechanisms of the tumor. With increased use of these agents, immune-related adverse events (irAEs) have become more prevalent. The most common irAEs secondary to ipilimumab are skin rash, colitis/diarrhea, hepatitis, pneumonitis, and various endocrinopathies.4 In a phase 3 trial of adjuvant ipilimumab in patients with resected stage III melanoma, grade 3 or 4 adverse events occurred in 54.1% of participants in the ipilimumab arm, the most common being diarrhea and colitis (9.8% and 6.8%, respectively).2Recognition and management of irAEs has led to the implementation of treatment guidelines.4,5 Management of irAEs includes checkpoint inhibitor discontinuation and reversal of the immune response by institution of immunosuppression with corticosteroids.
Case presentation and summary
A 40-year-old white woman with stage IIIB BRAF V600E-positive melanoma presented with diarrhea refractory to high-dose prednisone (1 mg/kg BID). She had recently undergone wide local excision and sentinel node biopsy and received her inaugural dose of ipilimumab (10 mg/kg).
The patient first presented with loose, watery stools that had begun 8 days after she had received her first dose of adjuvant ipilimumab. She was admitted to the hospital, and intravenous methylprednisolone was initiated along with empiric ciprofloxacin (400 mg, IVPB Q12h) and metronidazole (500 mg, IVPB Q8h) as infectious causes were concurrently ruled out. During this initial admission, the patient’s stool was negative for Clostridium difficile toxin, ova, and parasites, as well as enteric pathogens by culture. After infectious causes were excluded, she was diagnosed with ipilimumab-induced colitis. Antibiotics were discontinued, and the patient ultimately noted improvement in her symptoms. On hospital day 7, she was experiencing only 2 bowel movements a day and was discharged on 80 mg of prednisone twice daily.
After discharge the patient noted persistence of her symptoms. At her follow-up, 9 days after discharge, the patient noted continued symptoms of low-grade diarrhea. She failed a trial of steroid tapering due to exacerbation of her abdominal pain and frequency of diarrhea. Further investigation was negative for C. diff toxin and a computed-tomography scan was consistent with continuing colitis. The patient’s symptoms continued to worsen, with recurrence of grade 3 diarrhea, and she was ultimately readmitted 17 days after her earlier discharge (36 days after her first ipilimumab dosing).
On re-admission, the patient was again given intravenous methylprednisolone and experienced interval improvement in the frequency of diarrhea. A gastroenterology expert was consulted, and the patient underwent a flexible sigmoidoscopy that demonstrated findings of diffuse and severe inflammation and biopsies were obtained (Figure 1). After several days of continued symptoms, the patient received infliximab 5 mg/kg for treatment of her adverse autoimmune reaction. After administration, the patient noted improvement in the frequency and volume of diarrhea, however, her symptoms still persisted.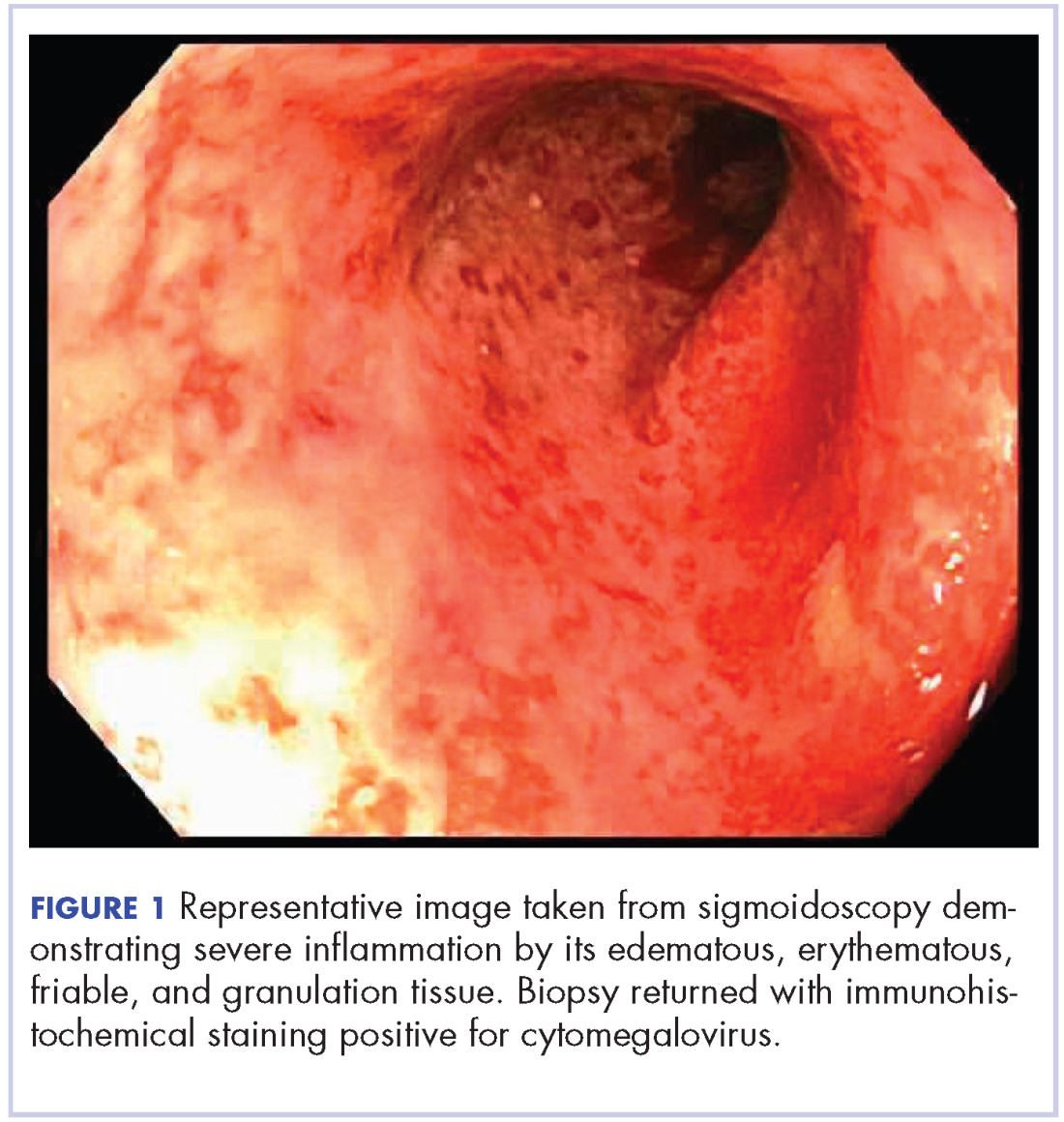
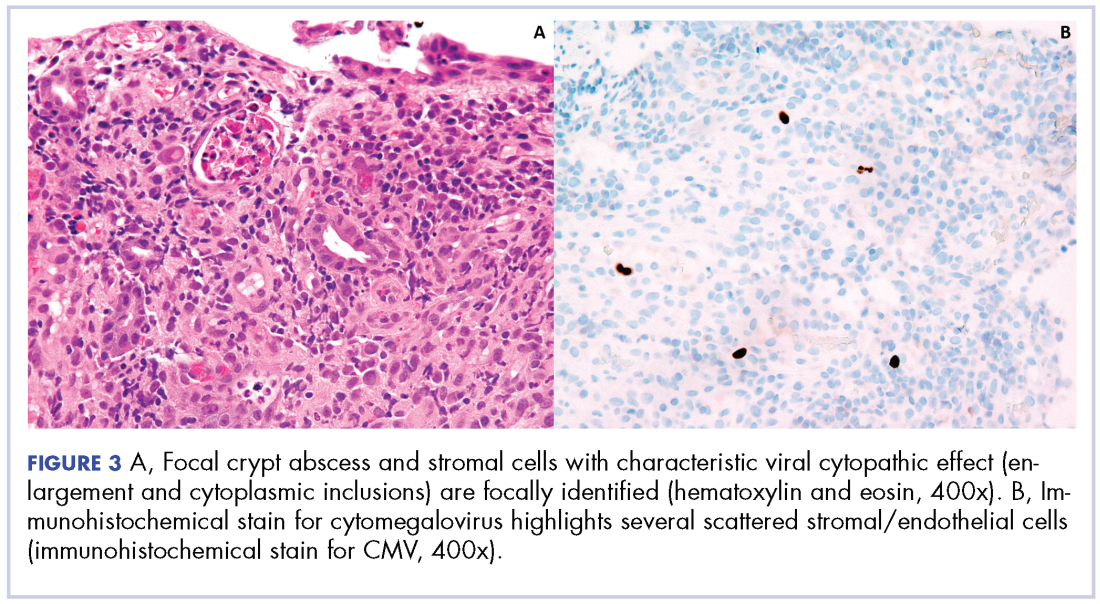
Biopsy results subsequently revealed findings compatible with ipilimumab-induced colitis, and immunohistochemical staining demonstrated positivity for cytomegalovirus (CMV). Specifically, histologic examination showed lymphoplasmacytic expansion of the lamina propria, some architectural distortion, and increased crypt apoptosis. Scattered cryptitis and crypt abscesses were also noted, as were rare stromal and endothelial cells with characteristic CMV inclusions (Figure 2 and Figure 3).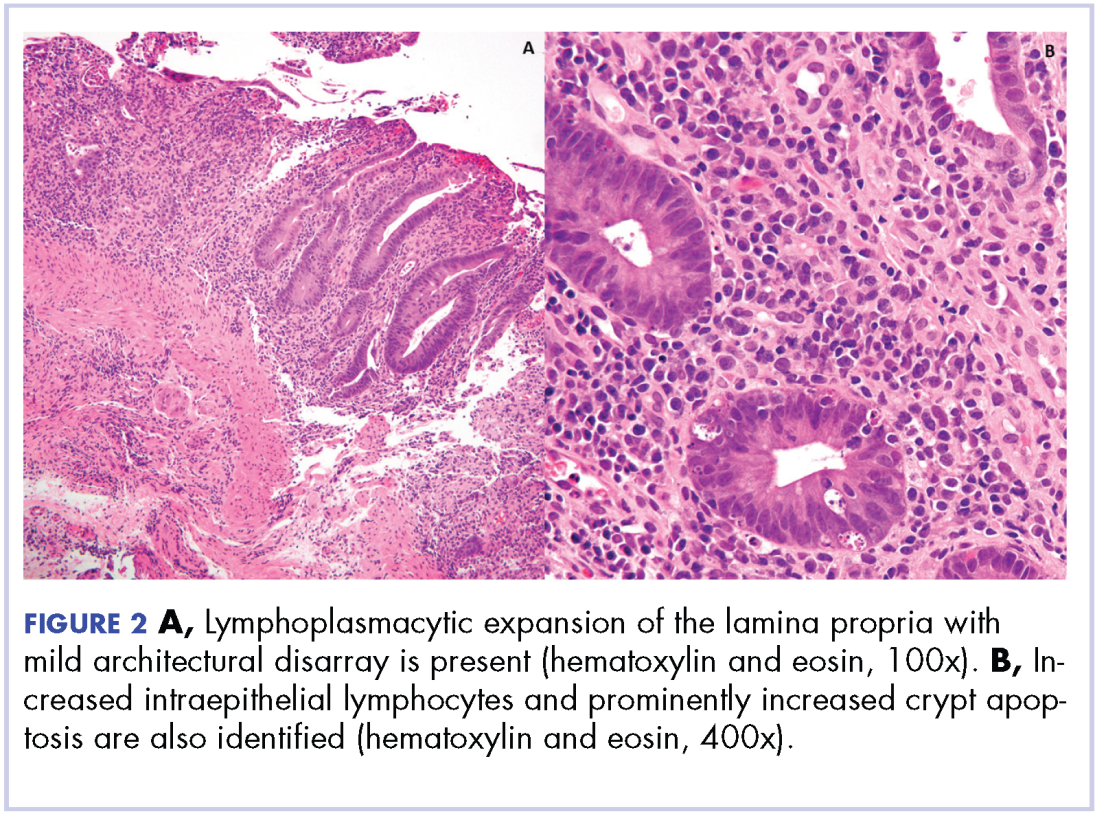
Serum CMV polymerase chain reaction (PCR) was also positive at 652,000 IU/mL (lower limit of detection 100 IU/mL). Induction dosing of ganciclovir (5 mg/kg IV Q12h) was initiated. The combined treatment with intravenous methylprednisone and ganciclovir led to an improvement in diarrhea frequency and resolution of blood in the stool. She was transitioned to oral prednisone, but it resulted in redevelopment of grade 3 diarrhea. The patient was therefore resumed on and discharged on daily intravenous methylprednisolone.
After discharge, the patient was started on budesonide 9 mg daily. Her serum CMV PCR level reduced and she was transitioned to oral valgancyclovir (900 mg daily) for maintenance. Another unsuccessful attempt was made to switch her to oral prednisone.
About 14 weeks after the initial ipilimumab dosing, the patient underwent another flexible sigmoidoscopy that again demonstrated severe colitis from the rectum to sigmoid colon. Biopsies were negative for CMV. Patient was readmitted for recurrence of diarrhea the following week. Treatment with IV methylprednisone (1mg/kg BID) and infliximab (5 mg/kg) again led to an improvement of symptoms. She was again discharged on IV methylprednisone (1 mg/kg BID) with a taper.
In the 15th week after her initial ipilimumab dose, the patient presented with a perforated bowel, requiring a subtotal colectomy and end ileostomy. She continued on a slow taper of oral prednisone (50 mg daily and decrease by 10 mg every 5 days).
At her last documented follow-up, 8 months after her first ipilimumab dose, she was having normal output from her ileostomy. She developed secondary adrenal insufficiency because of the long-term steroids and continued to take prednisone 5 mg daily.
Discussion
Diarrhea and colitis are common irAEs attributable to checkpoint-inhibitor therapy used for the treatment of melanoma. This case of ipilimumab-induced colitis refractory to high-dose oral steroids demonstrates the risks associated with management of anti-CTLA-4 induced colitis. In particular, the high-dose corticosteroids required to treat the autoimmune component of this patient’s colitis increased her susceptibility to CMV reactivation.
The diagnosis of colitis secondary to ipilimumab is made primarily in the appropriate clinical setting, and typically onsets during the induction period (within 12 weeks of initial dosing) and most resolve within 6-8 weeks.6 Histopathologically, there is lymphoplasmacytic expansion of lamina propria, increased intraepithelial lymphocytes, and increased epithelial apoptosis of crypts. One can also see acute cryptitis and crypt abscesses. Reactive epithelial changes with mucin depletion are also often seen in epithelial cells.
Findings from immunohistochemical studies have shown the increased intraepithelial lymphocytes to be predominantly CD8-positive T cells, while the lamina propria contains an increase in the mixture of CD4- and CD8-positive T cells. In addition, small intestinal samples show villous blunting. There is an absence of significant architectural distortion and well-developed basal lymphoplasmacytic infiltrates characteristic of chronic mucosal injury, such as idiopathic inflammatory bowel disease.7 Granulomas are also absent in most series, though they have been reported in some cases.8 The features are similar to those seen in autoimmune enteropathy, but goblet and endocrine cells remain preserved. Graft-versus-host disease has similar histologic features, however, the clinical setting usually makes the distinction between these obvious.
Current treatment algorithms for ipilimumab-related diarrhea, begin with immediate treatment with intravenous methylprednisolone (125 mg once). This is followed with oral prednisone at a dose of 1-2 mg/kg tapered over 4 to 8 weeks.4 In patients with persistent symptoms despite adequate doses of corticosteroids, infliximab (5 mg/kg every 2 weeks) is recommended until the resolution of symptoms, and a longer taper of prednisone is often necessary.
Institution of high-dose corticosteroids to treat grade 3 or 4 irAEs can increase the risk for infection, including opportunistic infections. One retrospective review of patients administered checkpoint inhibitors at a single institution revealed that 7.3% of 740 patients developed a severe infection that lead to hospitalization or treatment with intravenous antibiotics.9 In that patient cohort, only 0.6% had a serious infection secondary to a viral etiology, and 1 patient developed CMV enterocolitis. Most patients who developed an infection in this cohort had received corticosteroids (46/54 patients, 85%) and/or infliximab (13/54 patients, 24%).9
CMV is a member of the Herpesviridae family. After a primary infection, which can often go unrecognized in an immunocompetent host, CMV can persist in a latent state.10 In a study by Bate and colleagues, the age-adjusted seropositivity of CMV was found to be 50.4%.11 Based on those results, immunosuppression in a patient who has previously been infected with CMV can lead to a risk of reactivation or even reinfection. In the era of checkpoint-inhibitor therapy, reactivation of CMV has been described previously in a case of CMV hepatitis and a report of CMV colitis.12,13 Immunosuppression, such as that caused by corticosteroids, is a risk factor for CMV infection.14 Colitis caused by CMV usually presents with abdominal pain, diarrhea, and bloody diarrhea.15 In suspected cases of CMV colitis, endoscopy should be pursued with biopsy for tissue examination. A tissue diagnosis is required for CMV colitis because serum PCR can be negative in isolated cases of gastrointestinal CMV infection.15
Conclusion
Despite appropriate treatment with ganciclovir and the noted response in the patient’s serum CMV PCR, symptom exacerbation was observed with the transition to oral prednisone. The requirement for intravenous corticosteroids in the present case demonstrates the prolonged effects exerted by irAEs secondary to checkpoint-inhibitor therapy. Those effects are attributable to the design of the antibody – ipilimumab is a fully humanized monoclonal antibody and has a plasma half-life of about 15 days.1,4
By the identification of CMV histopathologically, this case, along with the case presented by Lankes and colleagues,13 illustrates the importance of considering CMV colitis in patients who are being treated with ipilimumab and who develop persistent or worsening diarrhea after initial treatment with high-dose steroids.
Early recognition of possible coexistent CMV colitis in patients with a history of treatment with ipilimumab can have important clinical consequences. It can lead to quicker implementation of proper antiviral therapy and minimization of immune suppression to levels required to maintain control of the patient’s symptoms.
1. Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711-723.
2. Eggermont AM, Chiarion-Sileni V, Grob JJ, et al. Prolonged survival in stage III melanoma with ipilimumab adjuvant therapy. N Engl J Med. 2016;375(19):1845-1855.
3. Glassman PM, Balthasar JP. Mechanistic considerations for the use of monoclonal antibodies for cancer therapy. Cancer Biol Med. 2014;11(1):20-33.
4. Weber JS, Kahler KC, Hauschild A. Management of immune-related adverse events and kinetics of response with ipilimumab. J Clin Oncol. 2012;30(21):2691-2697.
5. Fecher LA, Agarwala SS, Hodi FS, Weber JS. Ipilimumab and its toxicities: a multidisciplinary approach. Oncologist. 2013;18(6):733-743.
6. Weber JS, Dummer R, de Pril V, Lebbe C, Hodi FS, Investigators MDX. Patterns of onset and resolution of immune-related adverse events of special interest with ipilimumab: detailed safety analysis from a phase 3 trial in patients with advanced melanoma. Cancer. 2013;119(9):1675-1682.
7. Oble DA, Mino-Kenudson M, Goldsmith J, et al. Alpha-CTLA-4 mAb-associated panenteritis: a histologic and immunohistochemical analysis. Am J Surg Pathol. 2008;32(8):1130-1137.
8. Beck KE, Blansfield JA, Tran KQ, et al. Enterocolitis in patients with cancer after antibody blockade of cytotoxic T-lymphocyte-associated antigen 4. J Clin Oncol. 2006;24(15):2283-2289.
9. Del Castillo M, Romero FA, Arguello E, Kyi C, Postow MA, Redelman-Sidi G. The spectrum of serious infections among patients receiving immune checkpoint blockade for the treatment of melanoma. Clin Infect Dis. 2016;63(11):1490-1493.
10. Pillet S, Pozzetto B, Roblin X. Cytomegalovirus and ulcerative colitis: place of antiviral therapy. World J Gastroenterol. 2016;22(6):2030-2045.
11. Bate SL, Dollard SC, Cannon MJ. Cytomegalovirus seroprevalence in the United States: the national health and nutrition examination surveys, 1988-2004. Clin Infect Dis. 2010;50(11):1439-1447.
12. Uslu U, Agaimy A, Hundorfean G, Harrer T, Schuler G, Heinzerling L. autoimmune colitis and subsequent CMV-induced hepatitis after treatment with ipilimumab. J Immunother. 2015;38(5):212-215.
13. Lankes K, Hundorfean G, Harrer T, et al. Anti-TNF-refractory colitis after checkpoint inhibitor therapy: possible role of CMV-mediated immunopathogenesis. Oncoimmunology. 2016;5(6):e1128611.
14. Ko JH, Peck KR, Lee WJ, et al. Clinical presentation and risk factors for cytomegalovirus colitis in immunocompetent adult patients. Clin Infect Dis. 2015;60(6):e20-26.
15. You DM, Johnson MD. Cytomegalovirus infection and the gastrointestinal tract. Curr Gastroenterol Rep. 2012;14(4):334-342.
Immune checkpoint inhibitors, including anti-cytotoxic T-lymphocyte antigen 4 (anti-CTLA4) and anti-programmed cell death protein-1 (anti-PD-1) antibodies, have demonstrated clinical and survival benefits in a variety of malignancies, which has led to an expansion in their role in oncology. In melanoma, the anti-CTLA-4 antibody, ipilimumab, has demonstrated a survival benefit in patients with advanced metastatic melanoma and in patients with resectable disease with lymph node involvement.1,2
Ipilimumab exerts its effect by binding CTLA-4 on conventional and regulatory T cells, thus blocking inhibitory signals on T cells, which leads to an antitumor response.3 The increased immune response counteracts the immune-evading mechanisms of the tumor. With increased use of these agents, immune-related adverse events (irAEs) have become more prevalent. The most common irAEs secondary to ipilimumab are skin rash, colitis/diarrhea, hepatitis, pneumonitis, and various endocrinopathies.4 In a phase 3 trial of adjuvant ipilimumab in patients with resected stage III melanoma, grade 3 or 4 adverse events occurred in 54.1% of participants in the ipilimumab arm, the most common being diarrhea and colitis (9.8% and 6.8%, respectively).2Recognition and management of irAEs has led to the implementation of treatment guidelines.4,5 Management of irAEs includes checkpoint inhibitor discontinuation and reversal of the immune response by institution of immunosuppression with corticosteroids.
Case presentation and summary
A 40-year-old white woman with stage IIIB BRAF V600E-positive melanoma presented with diarrhea refractory to high-dose prednisone (1 mg/kg BID). She had recently undergone wide local excision and sentinel node biopsy and received her inaugural dose of ipilimumab (10 mg/kg).
The patient first presented with loose, watery stools that had begun 8 days after she had received her first dose of adjuvant ipilimumab. She was admitted to the hospital, and intravenous methylprednisolone was initiated along with empiric ciprofloxacin (400 mg, IVPB Q12h) and metronidazole (500 mg, IVPB Q8h) as infectious causes were concurrently ruled out. During this initial admission, the patient’s stool was negative for Clostridium difficile toxin, ova, and parasites, as well as enteric pathogens by culture. After infectious causes were excluded, she was diagnosed with ipilimumab-induced colitis. Antibiotics were discontinued, and the patient ultimately noted improvement in her symptoms. On hospital day 7, she was experiencing only 2 bowel movements a day and was discharged on 80 mg of prednisone twice daily.
After discharge the patient noted persistence of her symptoms. At her follow-up, 9 days after discharge, the patient noted continued symptoms of low-grade diarrhea. She failed a trial of steroid tapering due to exacerbation of her abdominal pain and frequency of diarrhea. Further investigation was negative for C. diff toxin and a computed-tomography scan was consistent with continuing colitis. The patient’s symptoms continued to worsen, with recurrence of grade 3 diarrhea, and she was ultimately readmitted 17 days after her earlier discharge (36 days after her first ipilimumab dosing).
On re-admission, the patient was again given intravenous methylprednisolone and experienced interval improvement in the frequency of diarrhea. A gastroenterology expert was consulted, and the patient underwent a flexible sigmoidoscopy that demonstrated findings of diffuse and severe inflammation and biopsies were obtained (Figure 1). After several days of continued symptoms, the patient received infliximab 5 mg/kg for treatment of her adverse autoimmune reaction. After administration, the patient noted improvement in the frequency and volume of diarrhea, however, her symptoms still persisted.
Biopsy results subsequently revealed findings compatible with ipilimumab-induced colitis, and immunohistochemical staining demonstrated positivity for cytomegalovirus (CMV). Specifically, histologic examination showed lymphoplasmacytic expansion of the lamina propria, some architectural distortion, and increased crypt apoptosis. Scattered cryptitis and crypt abscesses were also noted, as were rare stromal and endothelial cells with characteristic CMV inclusions (Figure 2 and Figure 3).
Serum CMV polymerase chain reaction (PCR) was also positive at 652,000 IU/mL (lower limit of detection 100 IU/mL). Induction dosing of ganciclovir (5 mg/kg IV Q12h) was initiated. The combined treatment with intravenous methylprednisone and ganciclovir led to an improvement in diarrhea frequency and resolution of blood in the stool. She was transitioned to oral prednisone, but it resulted in redevelopment of grade 3 diarrhea. The patient was therefore resumed on and discharged on daily intravenous methylprednisolone.
After discharge, the patient was started on budesonide 9 mg daily. Her serum CMV PCR level reduced and she was transitioned to oral valgancyclovir (900 mg daily) for maintenance. Another unsuccessful attempt was made to switch her to oral prednisone.
About 14 weeks after the initial ipilimumab dosing, the patient underwent another flexible sigmoidoscopy that again demonstrated severe colitis from the rectum to sigmoid colon. Biopsies were negative for CMV. Patient was readmitted for recurrence of diarrhea the following week. Treatment with IV methylprednisone (1mg/kg BID) and infliximab (5 mg/kg) again led to an improvement of symptoms. She was again discharged on IV methylprednisone (1 mg/kg BID) with a taper.
In the 15th week after her initial ipilimumab dose, the patient presented with a perforated bowel, requiring a subtotal colectomy and end ileostomy. She continued on a slow taper of oral prednisone (50 mg daily and decrease by 10 mg every 5 days).
At her last documented follow-up, 8 months after her first ipilimumab dose, she was having normal output from her ileostomy. She developed secondary adrenal insufficiency because of the long-term steroids and continued to take prednisone 5 mg daily.
Discussion
Diarrhea and colitis are common irAEs attributable to checkpoint-inhibitor therapy used for the treatment of melanoma. This case of ipilimumab-induced colitis refractory to high-dose oral steroids demonstrates the risks associated with management of anti-CTLA-4 induced colitis. In particular, the high-dose corticosteroids required to treat the autoimmune component of this patient’s colitis increased her susceptibility to CMV reactivation.
The diagnosis of colitis secondary to ipilimumab is made primarily in the appropriate clinical setting, and typically onsets during the induction period (within 12 weeks of initial dosing) and most resolve within 6-8 weeks.6 Histopathologically, there is lymphoplasmacytic expansion of lamina propria, increased intraepithelial lymphocytes, and increased epithelial apoptosis of crypts. One can also see acute cryptitis and crypt abscesses. Reactive epithelial changes with mucin depletion are also often seen in epithelial cells.
Findings from immunohistochemical studies have shown the increased intraepithelial lymphocytes to be predominantly CD8-positive T cells, while the lamina propria contains an increase in the mixture of CD4- and CD8-positive T cells. In addition, small intestinal samples show villous blunting. There is an absence of significant architectural distortion and well-developed basal lymphoplasmacytic infiltrates characteristic of chronic mucosal injury, such as idiopathic inflammatory bowel disease.7 Granulomas are also absent in most series, though they have been reported in some cases.8 The features are similar to those seen in autoimmune enteropathy, but goblet and endocrine cells remain preserved. Graft-versus-host disease has similar histologic features, however, the clinical setting usually makes the distinction between these obvious.
Current treatment algorithms for ipilimumab-related diarrhea, begin with immediate treatment with intravenous methylprednisolone (125 mg once). This is followed with oral prednisone at a dose of 1-2 mg/kg tapered over 4 to 8 weeks.4 In patients with persistent symptoms despite adequate doses of corticosteroids, infliximab (5 mg/kg every 2 weeks) is recommended until the resolution of symptoms, and a longer taper of prednisone is often necessary.
Institution of high-dose corticosteroids to treat grade 3 or 4 irAEs can increase the risk for infection, including opportunistic infections. One retrospective review of patients administered checkpoint inhibitors at a single institution revealed that 7.3% of 740 patients developed a severe infection that lead to hospitalization or treatment with intravenous antibiotics.9 In that patient cohort, only 0.6% had a serious infection secondary to a viral etiology, and 1 patient developed CMV enterocolitis. Most patients who developed an infection in this cohort had received corticosteroids (46/54 patients, 85%) and/or infliximab (13/54 patients, 24%).9
CMV is a member of the Herpesviridae family. After a primary infection, which can often go unrecognized in an immunocompetent host, CMV can persist in a latent state.10 In a study by Bate and colleagues, the age-adjusted seropositivity of CMV was found to be 50.4%.11 Based on those results, immunosuppression in a patient who has previously been infected with CMV can lead to a risk of reactivation or even reinfection. In the era of checkpoint-inhibitor therapy, reactivation of CMV has been described previously in a case of CMV hepatitis and a report of CMV colitis.12,13 Immunosuppression, such as that caused by corticosteroids, is a risk factor for CMV infection.14 Colitis caused by CMV usually presents with abdominal pain, diarrhea, and bloody diarrhea.15 In suspected cases of CMV colitis, endoscopy should be pursued with biopsy for tissue examination. A tissue diagnosis is required for CMV colitis because serum PCR can be negative in isolated cases of gastrointestinal CMV infection.15
Conclusion
Despite appropriate treatment with ganciclovir and the noted response in the patient’s serum CMV PCR, symptom exacerbation was observed with the transition to oral prednisone. The requirement for intravenous corticosteroids in the present case demonstrates the prolonged effects exerted by irAEs secondary to checkpoint-inhibitor therapy. Those effects are attributable to the design of the antibody – ipilimumab is a fully humanized monoclonal antibody and has a plasma half-life of about 15 days.1,4
By the identification of CMV histopathologically, this case, along with the case presented by Lankes and colleagues,13 illustrates the importance of considering CMV colitis in patients who are being treated with ipilimumab and who develop persistent or worsening diarrhea after initial treatment with high-dose steroids.
Early recognition of possible coexistent CMV colitis in patients with a history of treatment with ipilimumab can have important clinical consequences. It can lead to quicker implementation of proper antiviral therapy and minimization of immune suppression to levels required to maintain control of the patient’s symptoms.
Immune checkpoint inhibitors, including anti-cytotoxic T-lymphocyte antigen 4 (anti-CTLA4) and anti-programmed cell death protein-1 (anti-PD-1) antibodies, have demonstrated clinical and survival benefits in a variety of malignancies, which has led to an expansion in their role in oncology. In melanoma, the anti-CTLA-4 antibody, ipilimumab, has demonstrated a survival benefit in patients with advanced metastatic melanoma and in patients with resectable disease with lymph node involvement.1,2
Ipilimumab exerts its effect by binding CTLA-4 on conventional and regulatory T cells, thus blocking inhibitory signals on T cells, which leads to an antitumor response.3 The increased immune response counteracts the immune-evading mechanisms of the tumor. With increased use of these agents, immune-related adverse events (irAEs) have become more prevalent. The most common irAEs secondary to ipilimumab are skin rash, colitis/diarrhea, hepatitis, pneumonitis, and various endocrinopathies.4 In a phase 3 trial of adjuvant ipilimumab in patients with resected stage III melanoma, grade 3 or 4 adverse events occurred in 54.1% of participants in the ipilimumab arm, the most common being diarrhea and colitis (9.8% and 6.8%, respectively).2Recognition and management of irAEs has led to the implementation of treatment guidelines.4,5 Management of irAEs includes checkpoint inhibitor discontinuation and reversal of the immune response by institution of immunosuppression with corticosteroids.
Case presentation and summary
A 40-year-old white woman with stage IIIB BRAF V600E-positive melanoma presented with diarrhea refractory to high-dose prednisone (1 mg/kg BID). She had recently undergone wide local excision and sentinel node biopsy and received her inaugural dose of ipilimumab (10 mg/kg).
The patient first presented with loose, watery stools that had begun 8 days after she had received her first dose of adjuvant ipilimumab. She was admitted to the hospital, and intravenous methylprednisolone was initiated along with empiric ciprofloxacin (400 mg, IVPB Q12h) and metronidazole (500 mg, IVPB Q8h) as infectious causes were concurrently ruled out. During this initial admission, the patient’s stool was negative for Clostridium difficile toxin, ova, and parasites, as well as enteric pathogens by culture. After infectious causes were excluded, she was diagnosed with ipilimumab-induced colitis. Antibiotics were discontinued, and the patient ultimately noted improvement in her symptoms. On hospital day 7, she was experiencing only 2 bowel movements a day and was discharged on 80 mg of prednisone twice daily.
After discharge the patient noted persistence of her symptoms. At her follow-up, 9 days after discharge, the patient noted continued symptoms of low-grade diarrhea. She failed a trial of steroid tapering due to exacerbation of her abdominal pain and frequency of diarrhea. Further investigation was negative for C. diff toxin and a computed-tomography scan was consistent with continuing colitis. The patient’s symptoms continued to worsen, with recurrence of grade 3 diarrhea, and she was ultimately readmitted 17 days after her earlier discharge (36 days after her first ipilimumab dosing).
On re-admission, the patient was again given intravenous methylprednisolone and experienced interval improvement in the frequency of diarrhea. A gastroenterology expert was consulted, and the patient underwent a flexible sigmoidoscopy that demonstrated findings of diffuse and severe inflammation and biopsies were obtained (Figure 1). After several days of continued symptoms, the patient received infliximab 5 mg/kg for treatment of her adverse autoimmune reaction. After administration, the patient noted improvement in the frequency and volume of diarrhea, however, her symptoms still persisted.
Biopsy results subsequently revealed findings compatible with ipilimumab-induced colitis, and immunohistochemical staining demonstrated positivity for cytomegalovirus (CMV). Specifically, histologic examination showed lymphoplasmacytic expansion of the lamina propria, some architectural distortion, and increased crypt apoptosis. Scattered cryptitis and crypt abscesses were also noted, as were rare stromal and endothelial cells with characteristic CMV inclusions (Figure 2 and Figure 3).
Serum CMV polymerase chain reaction (PCR) was also positive at 652,000 IU/mL (lower limit of detection 100 IU/mL). Induction dosing of ganciclovir (5 mg/kg IV Q12h) was initiated. The combined treatment with intravenous methylprednisone and ganciclovir led to an improvement in diarrhea frequency and resolution of blood in the stool. She was transitioned to oral prednisone, but it resulted in redevelopment of grade 3 diarrhea. The patient was therefore resumed on and discharged on daily intravenous methylprednisolone.
After discharge, the patient was started on budesonide 9 mg daily. Her serum CMV PCR level reduced and she was transitioned to oral valgancyclovir (900 mg daily) for maintenance. Another unsuccessful attempt was made to switch her to oral prednisone.
About 14 weeks after the initial ipilimumab dosing, the patient underwent another flexible sigmoidoscopy that again demonstrated severe colitis from the rectum to sigmoid colon. Biopsies were negative for CMV. Patient was readmitted for recurrence of diarrhea the following week. Treatment with IV methylprednisone (1mg/kg BID) and infliximab (5 mg/kg) again led to an improvement of symptoms. She was again discharged on IV methylprednisone (1 mg/kg BID) with a taper.
In the 15th week after her initial ipilimumab dose, the patient presented with a perforated bowel, requiring a subtotal colectomy and end ileostomy. She continued on a slow taper of oral prednisone (50 mg daily and decrease by 10 mg every 5 days).
At her last documented follow-up, 8 months after her first ipilimumab dose, she was having normal output from her ileostomy. She developed secondary adrenal insufficiency because of the long-term steroids and continued to take prednisone 5 mg daily.
Discussion
Diarrhea and colitis are common irAEs attributable to checkpoint-inhibitor therapy used for the treatment of melanoma. This case of ipilimumab-induced colitis refractory to high-dose oral steroids demonstrates the risks associated with management of anti-CTLA-4 induced colitis. In particular, the high-dose corticosteroids required to treat the autoimmune component of this patient’s colitis increased her susceptibility to CMV reactivation.
The diagnosis of colitis secondary to ipilimumab is made primarily in the appropriate clinical setting, and typically onsets during the induction period (within 12 weeks of initial dosing) and most resolve within 6-8 weeks.6 Histopathologically, there is lymphoplasmacytic expansion of lamina propria, increased intraepithelial lymphocytes, and increased epithelial apoptosis of crypts. One can also see acute cryptitis and crypt abscesses. Reactive epithelial changes with mucin depletion are also often seen in epithelial cells.
Findings from immunohistochemical studies have shown the increased intraepithelial lymphocytes to be predominantly CD8-positive T cells, while the lamina propria contains an increase in the mixture of CD4- and CD8-positive T cells. In addition, small intestinal samples show villous blunting. There is an absence of significant architectural distortion and well-developed basal lymphoplasmacytic infiltrates characteristic of chronic mucosal injury, such as idiopathic inflammatory bowel disease.7 Granulomas are also absent in most series, though they have been reported in some cases.8 The features are similar to those seen in autoimmune enteropathy, but goblet and endocrine cells remain preserved. Graft-versus-host disease has similar histologic features, however, the clinical setting usually makes the distinction between these obvious.
Current treatment algorithms for ipilimumab-related diarrhea, begin with immediate treatment with intravenous methylprednisolone (125 mg once). This is followed with oral prednisone at a dose of 1-2 mg/kg tapered over 4 to 8 weeks.4 In patients with persistent symptoms despite adequate doses of corticosteroids, infliximab (5 mg/kg every 2 weeks) is recommended until the resolution of symptoms, and a longer taper of prednisone is often necessary.
Institution of high-dose corticosteroids to treat grade 3 or 4 irAEs can increase the risk for infection, including opportunistic infections. One retrospective review of patients administered checkpoint inhibitors at a single institution revealed that 7.3% of 740 patients developed a severe infection that lead to hospitalization or treatment with intravenous antibiotics.9 In that patient cohort, only 0.6% had a serious infection secondary to a viral etiology, and 1 patient developed CMV enterocolitis. Most patients who developed an infection in this cohort had received corticosteroids (46/54 patients, 85%) and/or infliximab (13/54 patients, 24%).9
CMV is a member of the Herpesviridae family. After a primary infection, which can often go unrecognized in an immunocompetent host, CMV can persist in a latent state.10 In a study by Bate and colleagues, the age-adjusted seropositivity of CMV was found to be 50.4%.11 Based on those results, immunosuppression in a patient who has previously been infected with CMV can lead to a risk of reactivation or even reinfection. In the era of checkpoint-inhibitor therapy, reactivation of CMV has been described previously in a case of CMV hepatitis and a report of CMV colitis.12,13 Immunosuppression, such as that caused by corticosteroids, is a risk factor for CMV infection.14 Colitis caused by CMV usually presents with abdominal pain, diarrhea, and bloody diarrhea.15 In suspected cases of CMV colitis, endoscopy should be pursued with biopsy for tissue examination. A tissue diagnosis is required for CMV colitis because serum PCR can be negative in isolated cases of gastrointestinal CMV infection.15
Conclusion
Despite appropriate treatment with ganciclovir and the noted response in the patient’s serum CMV PCR, symptom exacerbation was observed with the transition to oral prednisone. The requirement for intravenous corticosteroids in the present case demonstrates the prolonged effects exerted by irAEs secondary to checkpoint-inhibitor therapy. Those effects are attributable to the design of the antibody – ipilimumab is a fully humanized monoclonal antibody and has a plasma half-life of about 15 days.1,4
By the identification of CMV histopathologically, this case, along with the case presented by Lankes and colleagues,13 illustrates the importance of considering CMV colitis in patients who are being treated with ipilimumab and who develop persistent or worsening diarrhea after initial treatment with high-dose steroids.
Early recognition of possible coexistent CMV colitis in patients with a history of treatment with ipilimumab can have important clinical consequences. It can lead to quicker implementation of proper antiviral therapy and minimization of immune suppression to levels required to maintain control of the patient’s symptoms.
1. Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711-723.
2. Eggermont AM, Chiarion-Sileni V, Grob JJ, et al. Prolonged survival in stage III melanoma with ipilimumab adjuvant therapy. N Engl J Med. 2016;375(19):1845-1855.
3. Glassman PM, Balthasar JP. Mechanistic considerations for the use of monoclonal antibodies for cancer therapy. Cancer Biol Med. 2014;11(1):20-33.
4. Weber JS, Kahler KC, Hauschild A. Management of immune-related adverse events and kinetics of response with ipilimumab. J Clin Oncol. 2012;30(21):2691-2697.
5. Fecher LA, Agarwala SS, Hodi FS, Weber JS. Ipilimumab and its toxicities: a multidisciplinary approach. Oncologist. 2013;18(6):733-743.
6. Weber JS, Dummer R, de Pril V, Lebbe C, Hodi FS, Investigators MDX. Patterns of onset and resolution of immune-related adverse events of special interest with ipilimumab: detailed safety analysis from a phase 3 trial in patients with advanced melanoma. Cancer. 2013;119(9):1675-1682.
7. Oble DA, Mino-Kenudson M, Goldsmith J, et al. Alpha-CTLA-4 mAb-associated panenteritis: a histologic and immunohistochemical analysis. Am J Surg Pathol. 2008;32(8):1130-1137.
8. Beck KE, Blansfield JA, Tran KQ, et al. Enterocolitis in patients with cancer after antibody blockade of cytotoxic T-lymphocyte-associated antigen 4. J Clin Oncol. 2006;24(15):2283-2289.
9. Del Castillo M, Romero FA, Arguello E, Kyi C, Postow MA, Redelman-Sidi G. The spectrum of serious infections among patients receiving immune checkpoint blockade for the treatment of melanoma. Clin Infect Dis. 2016;63(11):1490-1493.
10. Pillet S, Pozzetto B, Roblin X. Cytomegalovirus and ulcerative colitis: place of antiviral therapy. World J Gastroenterol. 2016;22(6):2030-2045.
11. Bate SL, Dollard SC, Cannon MJ. Cytomegalovirus seroprevalence in the United States: the national health and nutrition examination surveys, 1988-2004. Clin Infect Dis. 2010;50(11):1439-1447.
12. Uslu U, Agaimy A, Hundorfean G, Harrer T, Schuler G, Heinzerling L. autoimmune colitis and subsequent CMV-induced hepatitis after treatment with ipilimumab. J Immunother. 2015;38(5):212-215.
13. Lankes K, Hundorfean G, Harrer T, et al. Anti-TNF-refractory colitis after checkpoint inhibitor therapy: possible role of CMV-mediated immunopathogenesis. Oncoimmunology. 2016;5(6):e1128611.
14. Ko JH, Peck KR, Lee WJ, et al. Clinical presentation and risk factors for cytomegalovirus colitis in immunocompetent adult patients. Clin Infect Dis. 2015;60(6):e20-26.
15. You DM, Johnson MD. Cytomegalovirus infection and the gastrointestinal tract. Curr Gastroenterol Rep. 2012;14(4):334-342.
1. Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711-723.
2. Eggermont AM, Chiarion-Sileni V, Grob JJ, et al. Prolonged survival in stage III melanoma with ipilimumab adjuvant therapy. N Engl J Med. 2016;375(19):1845-1855.
3. Glassman PM, Balthasar JP. Mechanistic considerations for the use of monoclonal antibodies for cancer therapy. Cancer Biol Med. 2014;11(1):20-33.
4. Weber JS, Kahler KC, Hauschild A. Management of immune-related adverse events and kinetics of response with ipilimumab. J Clin Oncol. 2012;30(21):2691-2697.
5. Fecher LA, Agarwala SS, Hodi FS, Weber JS. Ipilimumab and its toxicities: a multidisciplinary approach. Oncologist. 2013;18(6):733-743.
6. Weber JS, Dummer R, de Pril V, Lebbe C, Hodi FS, Investigators MDX. Patterns of onset and resolution of immune-related adverse events of special interest with ipilimumab: detailed safety analysis from a phase 3 trial in patients with advanced melanoma. Cancer. 2013;119(9):1675-1682.
7. Oble DA, Mino-Kenudson M, Goldsmith J, et al. Alpha-CTLA-4 mAb-associated panenteritis: a histologic and immunohistochemical analysis. Am J Surg Pathol. 2008;32(8):1130-1137.
8. Beck KE, Blansfield JA, Tran KQ, et al. Enterocolitis in patients with cancer after antibody blockade of cytotoxic T-lymphocyte-associated antigen 4. J Clin Oncol. 2006;24(15):2283-2289.
9. Del Castillo M, Romero FA, Arguello E, Kyi C, Postow MA, Redelman-Sidi G. The spectrum of serious infections among patients receiving immune checkpoint blockade for the treatment of melanoma. Clin Infect Dis. 2016;63(11):1490-1493.
10. Pillet S, Pozzetto B, Roblin X. Cytomegalovirus and ulcerative colitis: place of antiviral therapy. World J Gastroenterol. 2016;22(6):2030-2045.
11. Bate SL, Dollard SC, Cannon MJ. Cytomegalovirus seroprevalence in the United States: the national health and nutrition examination surveys, 1988-2004. Clin Infect Dis. 2010;50(11):1439-1447.
12. Uslu U, Agaimy A, Hundorfean G, Harrer T, Schuler G, Heinzerling L. autoimmune colitis and subsequent CMV-induced hepatitis after treatment with ipilimumab. J Immunother. 2015;38(5):212-215.
13. Lankes K, Hundorfean G, Harrer T, et al. Anti-TNF-refractory colitis after checkpoint inhibitor therapy: possible role of CMV-mediated immunopathogenesis. Oncoimmunology. 2016;5(6):e1128611.
14. Ko JH, Peck KR, Lee WJ, et al. Clinical presentation and risk factors for cytomegalovirus colitis in immunocompetent adult patients. Clin Infect Dis. 2015;60(6):e20-26.
15. You DM, Johnson MD. Cytomegalovirus infection and the gastrointestinal tract. Curr Gastroenterol Rep. 2012;14(4):334-342.
Recurrent head and neck cancer presenting as a large retroperitoneal mass
Worldwide, head and neck cancers account for more than half a million cases annually and nearly 400,000 deaths.1 Although the exact incidence of metastatic disease of these primarily squamous cell tumors is difficult to determine, the incidence is thought to be much lower than that of other solid tumors.2 When the different sites of metastatic disease of these tumors have been studied previously, the most common have been (in descending order of frequency) the lungs, bones, liver, skin, mediastinum, and bone marrow.2,3 It is extremely rare area for head and neck squamous cell cancers to metastasize to the retroperitoneum. To our knowledge, only 2 other such cases have been reported in the literature.4,5 In those two cases, the metastatic recurrence occurred at 6 and 13 months after definitive treatment of the primary cancer.
Case presentation and summary
The patient in this case is a 60-year-old man with a history of stage IV moderately differentiated invasive squamous cell carcinoma (p16 negative, Bcl-2 negative, EGFR positive) of the hypopharynx that had been initially diagnosed in 2012. At that time, he underwent a total laryngectomy, partial pharyngectomy, and total thyroidectomy. A 2-centimeter mediastinal mass was also identified on a computed-tomography scan of the thorax and resected during the initial curative surgery. Final surgical pathology on the primary hypopharygeal tumor revealed a 4.1-cm moderately differentiated squamous cell carcinoma with negative margins, but positive lymphovascular invasion (Figure 1). The 2-cm mediastinal mass also revealed the same squamous cell carcinoma as the hypopharyngeal primary. Final surgical margins were negative.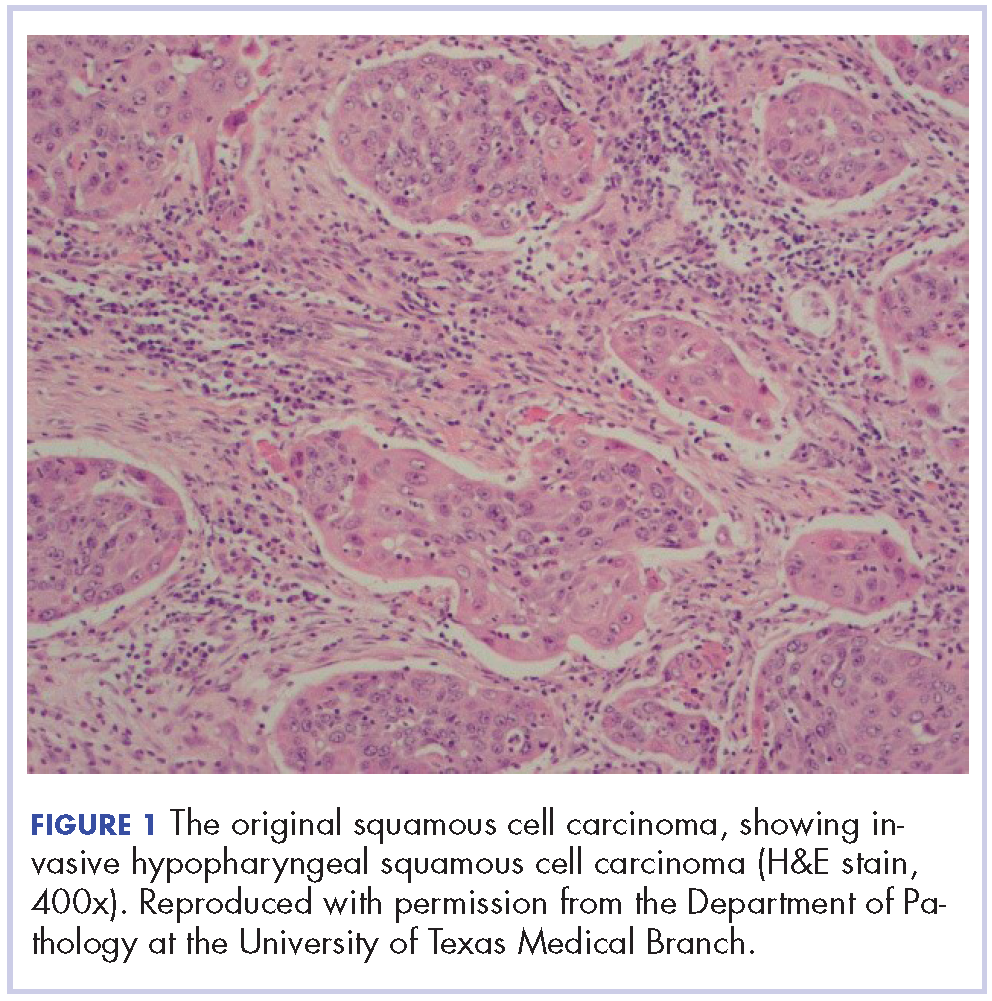
The patient went on to receive adjuvant treatment in the form of concurrent chemoradiation with cisplatin (100 mg/m2 every 21 days for 3 doses, with 70 Gy of radiation]. After his initial treatment, he was followed closely by a multidisciplinary team, including otolaryngology, radiation oncology, and medical oncology specialists. He underwent a positron-emission tomography–CT scan 1 year after the conclusion of adjuvant therapy that showed no evidence of local or distant disease. The patient underwent 12 fiberoptic pharyngoscopy procedures over the course of 4 years without any evidence of local disease recurrence. He underwent a CT scan of the neck in October of 2016 without any evidence of local disease recurrence.
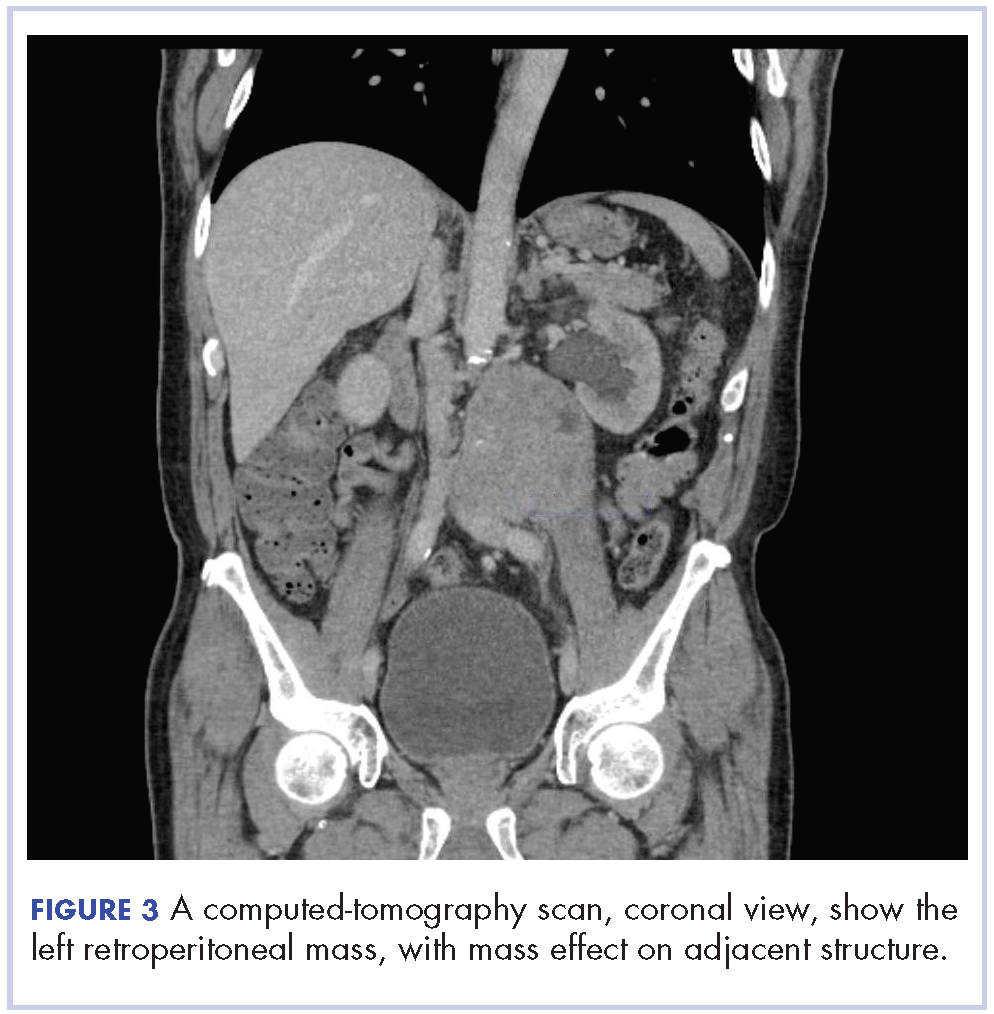
In early 2017, the patient presented with fatigue, abdominal pain, and back pain during the previous month. CT imaging revealed a left retroperitoneal mass of 8.8 x 4.0 x 6.6 cm, with bony destruction of L3-L4 causing left hydronephrosis (Figure 2 and Figure 3). Other staging work-up and imaging did not reveal any other distant disease or locoregional disease recurrence in the head and neck. Lab work was significant for an acute kidney injury that was likely secondary to mass effect from the tumor.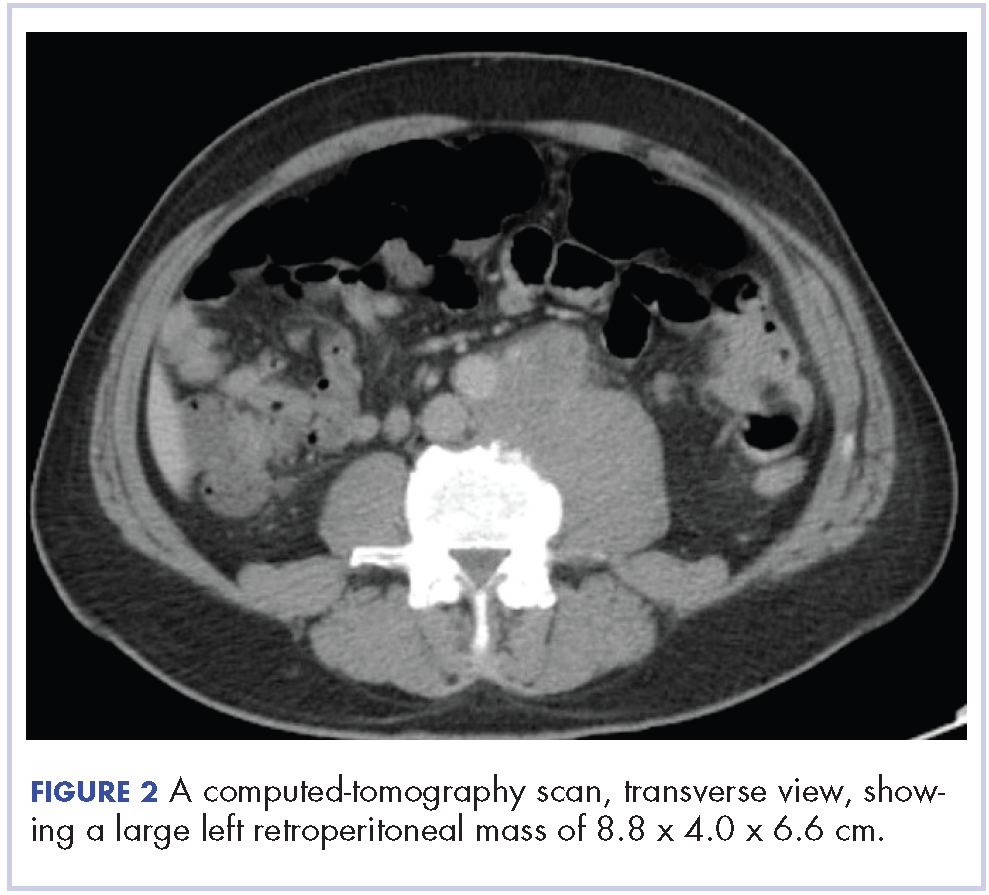
The mass was biopsied, with pathology revealing squamous cell carcinoma consistent with metastatic, recurrent disease from the previously known head and neck primary, and it was also p16 negative, Bcl-2 negative, and EGFR positive (Figure 4).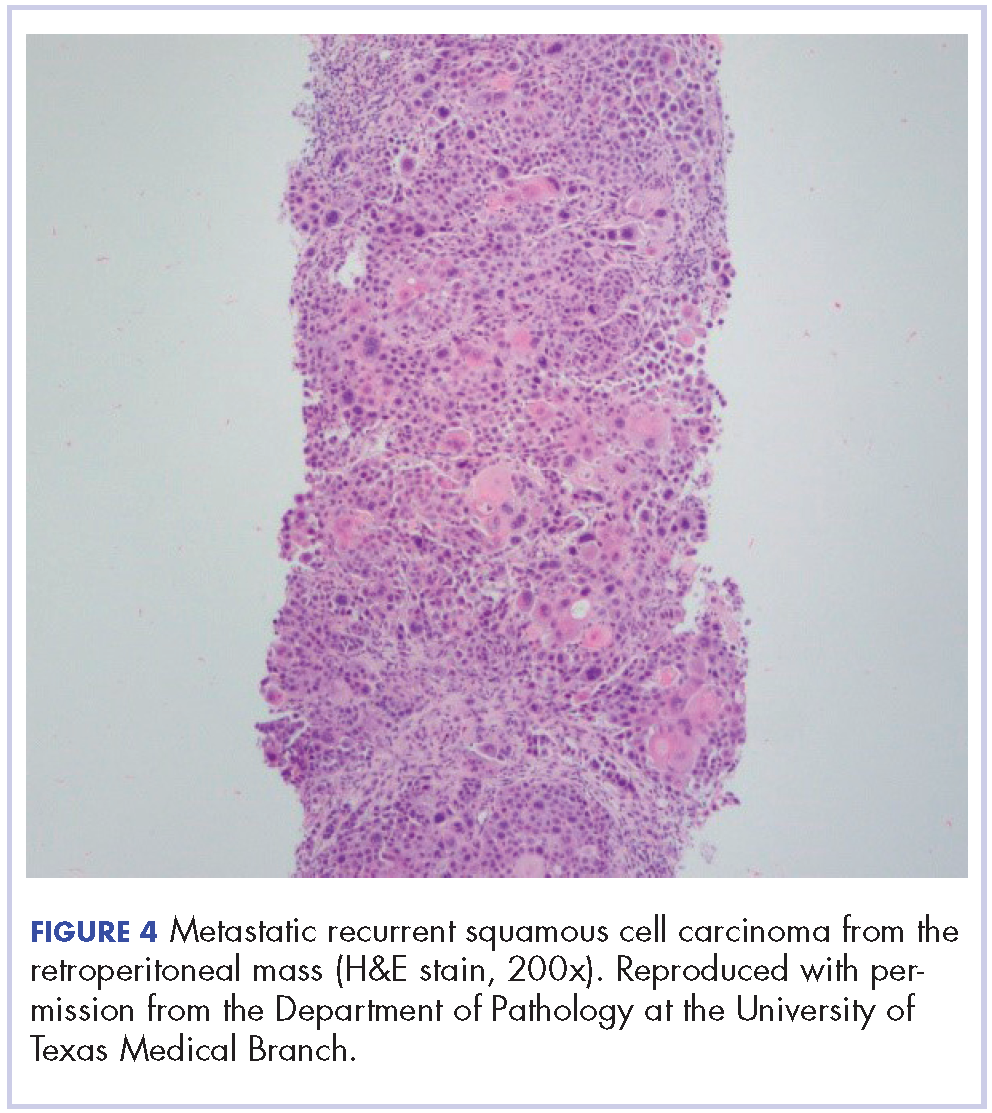
After a multidisciplinary discussion it was determined that the best front-line treatment option would be to treat with definitive concurrent chemoradiation. However, due to the size and location of the mass, it was not possible to deliver an effective therapeutic dose of radiation without unacceptable toxicity to the adjacent structures. Therefore, palliative systemic therapy was the only option. These treatment options, including systemic chemotherapy and immunotherapy, were discussed with the patient. However, he did not want to pursue any further cancer treatment and wanted instead to focus on palliation (pain control, antiemetics and nephrostomy to relieve obstruction) and hospice. He passed away 3 months later.
Discussion
Masses of the retroperitoneum have a wide differential diagnosis.6 Primary malignancies including lymphomas, sarcomas, neurogenic tumors, and germ cell tumors may all present primarily as retroperitoneal masses.6,7 Nonmalignant processes such as retroperitoneal fibrosis may also present in this manner.7 Certain tumors are known to metastasize to the retroperitoneum, namely carcinomas of the gastrointestinal tract and ovary as well as lung cancer or melanoma.5,8 Some primary retroperitoneal masses in women have been described in the literature as being HPV-associated squamous cell cancers of unknown primaries.9
When head and neck cancers metastasize they typically metastasize to the lungs, bone, liver, mediastinum, skin, and bone marrow. Most metastasis is pulmonary in origin, with the literature indicating it accounts for 52%-66% of head and neck cancer metastases, with bone metastases next in frequency at 12%-22%.2,3,10 In general, the incidence of distant metastatic disease in head and neck cancers is not as common as its other solid tumor counterparts, and even metastasis to other lymph node groups other than locoregional cervical nodes is rare.11 Furthermore, late metastasis occurring more than 2 years after definitive treatment is also an infrequent occurrence.12
When discussing distant metastatic disease in head and neck cancer, previous literature has described an increasing likelihood of distant metastases when there is locoregional disease recurrence.13 Moreover, the retroperitoneum is an exceedingly rare site of distant metastatic disease for head and neck cancer. There have been only 2 previous cases that have described this phenomenon, and in both cases the metastases occurred within or close to 1 year of definitive locoregional treatment.4,5
Conclusion
We present our case to present an exceedingly rare case of distant metastatic, recurrent disease from head and neck cancer to the retroperitoneum (without locoregional recurrence) that occurred 4 years after definitive treatment. We believe this to be the first case of its kind to be described when taking into consideration the site of metastases, when the metastatic recurrence occurred and that it happened without loco-regional disease recurrence. This case highlights the importance of keeping a wide differential diagnosis when encountering a retroperitoneal mass in a patient with even a remote history of head and neck cancer.
Acknowledgments
The authors thank the following members of the Department of Pathology at the University of Texas Medical Branch: Asad Ahmad, MD; Eduardo Eyzaguirre, MD; Timothy C Allen, MD, JD, FACP; and Suimmin Qiu, MD, PHD.
1. Fitzmaurice C, Allen C, Barber RM, et al. Global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life-years for 32 cancer groups, 1990 to 2015: a systematic analysis for the Global Burden of Disease Study. JAMA Oncol. 2017;3:524-548.
2. Ferlito A, Shaha AR, Silver CE, Rinaldo A, Mondin V. Incidence and sites of distant metastases from head and neck cancer. ORL J Otorhinolaryngol Relat Spec. 2001;63:202-207.
3. Wiegand S, Zimmermann A, Wilhelm T, Werner JA. Survival after distant metastasis in head and neck cancer. Anticancer Res. 2015;35:5499-5502.
4. Hofmann U, O’Connor JP, Biyani CS, Harnden P, Selby P, Weston PM. Retroperitoneal metastatic squamous cell carcinoma of the tonsil (with elevated beta human chorionic gonadotrophin): a misdiagnosis as extra-gonadal germ cell tumour. J Laryngol Otol. 2006;120:885-887.
5. Purkayastha A, Sharma N, Suhag V. Extremely rare and unusual case of retroperitoneal and pelvic metastasis from squamous cell carcinoma of vallecula. Int J Cancer Ther Oncol. 2016;4(2):1-4.
6. Rajiah P, Sinha R, Cuevas C, Dubinsky TJ, Bush WH, Kolokythas O. Imaging of uncommon retroperitoneal masses. Radiographics 2011;31:949-976.
7. Scali EP, Chandler TM, Heffernan EJ, Coyle J, Harris AC, Chang SD. Primary retroperitoneal masses: what is the differential diagnosis? Abdom Imaging. 2015;40:1887-1903.
8. Levy AD, Shaw JC, Sobin LH. Secondary tumors and tumorlike lesions of the peritoneal cavity: imaging features with pathologic correlation. Radiographics. 2009;29:347-373.
9. Isbell A, Fields EC. Three cases of women with HPV-related squamous cell carcinoma of unknown primary in the pelvis and retroperitoneum: a case series. Gynecol Oncol Rep. 2016;16:5-8.
10. León X, Quer M, Orús C, del Prado Venegas M, López M. Distant metastases in head and neck cancer patients who achieved loco-regional control. Head Neck. 2000;22:680-686.
11. Alavi S, Namazie A, Sercarz JA, Wang MB, Blackwell KE. Distant lymphatic metastasis from head and neck cancer. Ann Otol Rhinol Laryngol. 1999;108:860-863.
12. Krishnatry R, Gupta T, Murthy V, et al. Factors predicting ‘time to distant metastasis’ in radically treated head and neck cancer. Indian J Cancer. 2014;51:231-235.
13. Goodwin WJ. Distant metastases from oropharyngeal cancer. ORL J Otorhinolaryngol Relat Spec. 2001;63:222-223.
Worldwide, head and neck cancers account for more than half a million cases annually and nearly 400,000 deaths.1 Although the exact incidence of metastatic disease of these primarily squamous cell tumors is difficult to determine, the incidence is thought to be much lower than that of other solid tumors.2 When the different sites of metastatic disease of these tumors have been studied previously, the most common have been (in descending order of frequency) the lungs, bones, liver, skin, mediastinum, and bone marrow.2,3 It is extremely rare area for head and neck squamous cell cancers to metastasize to the retroperitoneum. To our knowledge, only 2 other such cases have been reported in the literature.4,5 In those two cases, the metastatic recurrence occurred at 6 and 13 months after definitive treatment of the primary cancer.
Case presentation and summary
The patient in this case is a 60-year-old man with a history of stage IV moderately differentiated invasive squamous cell carcinoma (p16 negative, Bcl-2 negative, EGFR positive) of the hypopharynx that had been initially diagnosed in 2012. At that time, he underwent a total laryngectomy, partial pharyngectomy, and total thyroidectomy. A 2-centimeter mediastinal mass was also identified on a computed-tomography scan of the thorax and resected during the initial curative surgery. Final surgical pathology on the primary hypopharygeal tumor revealed a 4.1-cm moderately differentiated squamous cell carcinoma with negative margins, but positive lymphovascular invasion (Figure 1). The 2-cm mediastinal mass also revealed the same squamous cell carcinoma as the hypopharyngeal primary. Final surgical margins were negative.
The patient went on to receive adjuvant treatment in the form of concurrent chemoradiation with cisplatin (100 mg/m2 every 21 days for 3 doses, with 70 Gy of radiation]. After his initial treatment, he was followed closely by a multidisciplinary team, including otolaryngology, radiation oncology, and medical oncology specialists. He underwent a positron-emission tomography–CT scan 1 year after the conclusion of adjuvant therapy that showed no evidence of local or distant disease. The patient underwent 12 fiberoptic pharyngoscopy procedures over the course of 4 years without any evidence of local disease recurrence. He underwent a CT scan of the neck in October of 2016 without any evidence of local disease recurrence.
In early 2017, the patient presented with fatigue, abdominal pain, and back pain during the previous month. CT imaging revealed a left retroperitoneal mass of 8.8 x 4.0 x 6.6 cm, with bony destruction of L3-L4 causing left hydronephrosis (Figure 2 and Figure 3). Other staging work-up and imaging did not reveal any other distant disease or locoregional disease recurrence in the head and neck. Lab work was significant for an acute kidney injury that was likely secondary to mass effect from the tumor.
The mass was biopsied, with pathology revealing squamous cell carcinoma consistent with metastatic, recurrent disease from the previously known head and neck primary, and it was also p16 negative, Bcl-2 negative, and EGFR positive (Figure 4).
After a multidisciplinary discussion it was determined that the best front-line treatment option would be to treat with definitive concurrent chemoradiation. However, due to the size and location of the mass, it was not possible to deliver an effective therapeutic dose of radiation without unacceptable toxicity to the adjacent structures. Therefore, palliative systemic therapy was the only option. These treatment options, including systemic chemotherapy and immunotherapy, were discussed with the patient. However, he did not want to pursue any further cancer treatment and wanted instead to focus on palliation (pain control, antiemetics and nephrostomy to relieve obstruction) and hospice. He passed away 3 months later.
Discussion
Masses of the retroperitoneum have a wide differential diagnosis.6 Primary malignancies including lymphomas, sarcomas, neurogenic tumors, and germ cell tumors may all present primarily as retroperitoneal masses.6,7 Nonmalignant processes such as retroperitoneal fibrosis may also present in this manner.7 Certain tumors are known to metastasize to the retroperitoneum, namely carcinomas of the gastrointestinal tract and ovary as well as lung cancer or melanoma.5,8 Some primary retroperitoneal masses in women have been described in the literature as being HPV-associated squamous cell cancers of unknown primaries.9
When head and neck cancers metastasize they typically metastasize to the lungs, bone, liver, mediastinum, skin, and bone marrow. Most metastasis is pulmonary in origin, with the literature indicating it accounts for 52%-66% of head and neck cancer metastases, with bone metastases next in frequency at 12%-22%.2,3,10 In general, the incidence of distant metastatic disease in head and neck cancers is not as common as its other solid tumor counterparts, and even metastasis to other lymph node groups other than locoregional cervical nodes is rare.11 Furthermore, late metastasis occurring more than 2 years after definitive treatment is also an infrequent occurrence.12
When discussing distant metastatic disease in head and neck cancer, previous literature has described an increasing likelihood of distant metastases when there is locoregional disease recurrence.13 Moreover, the retroperitoneum is an exceedingly rare site of distant metastatic disease for head and neck cancer. There have been only 2 previous cases that have described this phenomenon, and in both cases the metastases occurred within or close to 1 year of definitive locoregional treatment.4,5
Conclusion
We present our case to present an exceedingly rare case of distant metastatic, recurrent disease from head and neck cancer to the retroperitoneum (without locoregional recurrence) that occurred 4 years after definitive treatment. We believe this to be the first case of its kind to be described when taking into consideration the site of metastases, when the metastatic recurrence occurred and that it happened without loco-regional disease recurrence. This case highlights the importance of keeping a wide differential diagnosis when encountering a retroperitoneal mass in a patient with even a remote history of head and neck cancer.
Acknowledgments
The authors thank the following members of the Department of Pathology at the University of Texas Medical Branch: Asad Ahmad, MD; Eduardo Eyzaguirre, MD; Timothy C Allen, MD, JD, FACP; and Suimmin Qiu, MD, PHD.
Worldwide, head and neck cancers account for more than half a million cases annually and nearly 400,000 deaths.1 Although the exact incidence of metastatic disease of these primarily squamous cell tumors is difficult to determine, the incidence is thought to be much lower than that of other solid tumors.2 When the different sites of metastatic disease of these tumors have been studied previously, the most common have been (in descending order of frequency) the lungs, bones, liver, skin, mediastinum, and bone marrow.2,3 It is extremely rare area for head and neck squamous cell cancers to metastasize to the retroperitoneum. To our knowledge, only 2 other such cases have been reported in the literature.4,5 In those two cases, the metastatic recurrence occurred at 6 and 13 months after definitive treatment of the primary cancer.
Case presentation and summary
The patient in this case is a 60-year-old man with a history of stage IV moderately differentiated invasive squamous cell carcinoma (p16 negative, Bcl-2 negative, EGFR positive) of the hypopharynx that had been initially diagnosed in 2012. At that time, he underwent a total laryngectomy, partial pharyngectomy, and total thyroidectomy. A 2-centimeter mediastinal mass was also identified on a computed-tomography scan of the thorax and resected during the initial curative surgery. Final surgical pathology on the primary hypopharygeal tumor revealed a 4.1-cm moderately differentiated squamous cell carcinoma with negative margins, but positive lymphovascular invasion (Figure 1). The 2-cm mediastinal mass also revealed the same squamous cell carcinoma as the hypopharyngeal primary. Final surgical margins were negative.
The patient went on to receive adjuvant treatment in the form of concurrent chemoradiation with cisplatin (100 mg/m2 every 21 days for 3 doses, with 70 Gy of radiation]. After his initial treatment, he was followed closely by a multidisciplinary team, including otolaryngology, radiation oncology, and medical oncology specialists. He underwent a positron-emission tomography–CT scan 1 year after the conclusion of adjuvant therapy that showed no evidence of local or distant disease. The patient underwent 12 fiberoptic pharyngoscopy procedures over the course of 4 years without any evidence of local disease recurrence. He underwent a CT scan of the neck in October of 2016 without any evidence of local disease recurrence.
In early 2017, the patient presented with fatigue, abdominal pain, and back pain during the previous month. CT imaging revealed a left retroperitoneal mass of 8.8 x 4.0 x 6.6 cm, with bony destruction of L3-L4 causing left hydronephrosis (Figure 2 and Figure 3). Other staging work-up and imaging did not reveal any other distant disease or locoregional disease recurrence in the head and neck. Lab work was significant for an acute kidney injury that was likely secondary to mass effect from the tumor.
The mass was biopsied, with pathology revealing squamous cell carcinoma consistent with metastatic, recurrent disease from the previously known head and neck primary, and it was also p16 negative, Bcl-2 negative, and EGFR positive (Figure 4).
After a multidisciplinary discussion it was determined that the best front-line treatment option would be to treat with definitive concurrent chemoradiation. However, due to the size and location of the mass, it was not possible to deliver an effective therapeutic dose of radiation without unacceptable toxicity to the adjacent structures. Therefore, palliative systemic therapy was the only option. These treatment options, including systemic chemotherapy and immunotherapy, were discussed with the patient. However, he did not want to pursue any further cancer treatment and wanted instead to focus on palliation (pain control, antiemetics and nephrostomy to relieve obstruction) and hospice. He passed away 3 months later.
Discussion
Masses of the retroperitoneum have a wide differential diagnosis.6 Primary malignancies including lymphomas, sarcomas, neurogenic tumors, and germ cell tumors may all present primarily as retroperitoneal masses.6,7 Nonmalignant processes such as retroperitoneal fibrosis may also present in this manner.7 Certain tumors are known to metastasize to the retroperitoneum, namely carcinomas of the gastrointestinal tract and ovary as well as lung cancer or melanoma.5,8 Some primary retroperitoneal masses in women have been described in the literature as being HPV-associated squamous cell cancers of unknown primaries.9
When head and neck cancers metastasize they typically metastasize to the lungs, bone, liver, mediastinum, skin, and bone marrow. Most metastasis is pulmonary in origin, with the literature indicating it accounts for 52%-66% of head and neck cancer metastases, with bone metastases next in frequency at 12%-22%.2,3,10 In general, the incidence of distant metastatic disease in head and neck cancers is not as common as its other solid tumor counterparts, and even metastasis to other lymph node groups other than locoregional cervical nodes is rare.11 Furthermore, late metastasis occurring more than 2 years after definitive treatment is also an infrequent occurrence.12
When discussing distant metastatic disease in head and neck cancer, previous literature has described an increasing likelihood of distant metastases when there is locoregional disease recurrence.13 Moreover, the retroperitoneum is an exceedingly rare site of distant metastatic disease for head and neck cancer. There have been only 2 previous cases that have described this phenomenon, and in both cases the metastases occurred within or close to 1 year of definitive locoregional treatment.4,5
Conclusion
We present our case to present an exceedingly rare case of distant metastatic, recurrent disease from head and neck cancer to the retroperitoneum (without locoregional recurrence) that occurred 4 years after definitive treatment. We believe this to be the first case of its kind to be described when taking into consideration the site of metastases, when the metastatic recurrence occurred and that it happened without loco-regional disease recurrence. This case highlights the importance of keeping a wide differential diagnosis when encountering a retroperitoneal mass in a patient with even a remote history of head and neck cancer.
Acknowledgments
The authors thank the following members of the Department of Pathology at the University of Texas Medical Branch: Asad Ahmad, MD; Eduardo Eyzaguirre, MD; Timothy C Allen, MD, JD, FACP; and Suimmin Qiu, MD, PHD.
1. Fitzmaurice C, Allen C, Barber RM, et al. Global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life-years for 32 cancer groups, 1990 to 2015: a systematic analysis for the Global Burden of Disease Study. JAMA Oncol. 2017;3:524-548.
2. Ferlito A, Shaha AR, Silver CE, Rinaldo A, Mondin V. Incidence and sites of distant metastases from head and neck cancer. ORL J Otorhinolaryngol Relat Spec. 2001;63:202-207.
3. Wiegand S, Zimmermann A, Wilhelm T, Werner JA. Survival after distant metastasis in head and neck cancer. Anticancer Res. 2015;35:5499-5502.
4. Hofmann U, O’Connor JP, Biyani CS, Harnden P, Selby P, Weston PM. Retroperitoneal metastatic squamous cell carcinoma of the tonsil (with elevated beta human chorionic gonadotrophin): a misdiagnosis as extra-gonadal germ cell tumour. J Laryngol Otol. 2006;120:885-887.
5. Purkayastha A, Sharma N, Suhag V. Extremely rare and unusual case of retroperitoneal and pelvic metastasis from squamous cell carcinoma of vallecula. Int J Cancer Ther Oncol. 2016;4(2):1-4.
6. Rajiah P, Sinha R, Cuevas C, Dubinsky TJ, Bush WH, Kolokythas O. Imaging of uncommon retroperitoneal masses. Radiographics 2011;31:949-976.
7. Scali EP, Chandler TM, Heffernan EJ, Coyle J, Harris AC, Chang SD. Primary retroperitoneal masses: what is the differential diagnosis? Abdom Imaging. 2015;40:1887-1903.
8. Levy AD, Shaw JC, Sobin LH. Secondary tumors and tumorlike lesions of the peritoneal cavity: imaging features with pathologic correlation. Radiographics. 2009;29:347-373.
9. Isbell A, Fields EC. Three cases of women with HPV-related squamous cell carcinoma of unknown primary in the pelvis and retroperitoneum: a case series. Gynecol Oncol Rep. 2016;16:5-8.
10. León X, Quer M, Orús C, del Prado Venegas M, López M. Distant metastases in head and neck cancer patients who achieved loco-regional control. Head Neck. 2000;22:680-686.
11. Alavi S, Namazie A, Sercarz JA, Wang MB, Blackwell KE. Distant lymphatic metastasis from head and neck cancer. Ann Otol Rhinol Laryngol. 1999;108:860-863.
12. Krishnatry R, Gupta T, Murthy V, et al. Factors predicting ‘time to distant metastasis’ in radically treated head and neck cancer. Indian J Cancer. 2014;51:231-235.
13. Goodwin WJ. Distant metastases from oropharyngeal cancer. ORL J Otorhinolaryngol Relat Spec. 2001;63:222-223.
1. Fitzmaurice C, Allen C, Barber RM, et al. Global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life-years for 32 cancer groups, 1990 to 2015: a systematic analysis for the Global Burden of Disease Study. JAMA Oncol. 2017;3:524-548.
2. Ferlito A, Shaha AR, Silver CE, Rinaldo A, Mondin V. Incidence and sites of distant metastases from head and neck cancer. ORL J Otorhinolaryngol Relat Spec. 2001;63:202-207.
3. Wiegand S, Zimmermann A, Wilhelm T, Werner JA. Survival after distant metastasis in head and neck cancer. Anticancer Res. 2015;35:5499-5502.
4. Hofmann U, O’Connor JP, Biyani CS, Harnden P, Selby P, Weston PM. Retroperitoneal metastatic squamous cell carcinoma of the tonsil (with elevated beta human chorionic gonadotrophin): a misdiagnosis as extra-gonadal germ cell tumour. J Laryngol Otol. 2006;120:885-887.
5. Purkayastha A, Sharma N, Suhag V. Extremely rare and unusual case of retroperitoneal and pelvic metastasis from squamous cell carcinoma of vallecula. Int J Cancer Ther Oncol. 2016;4(2):1-4.
6. Rajiah P, Sinha R, Cuevas C, Dubinsky TJ, Bush WH, Kolokythas O. Imaging of uncommon retroperitoneal masses. Radiographics 2011;31:949-976.
7. Scali EP, Chandler TM, Heffernan EJ, Coyle J, Harris AC, Chang SD. Primary retroperitoneal masses: what is the differential diagnosis? Abdom Imaging. 2015;40:1887-1903.
8. Levy AD, Shaw JC, Sobin LH. Secondary tumors and tumorlike lesions of the peritoneal cavity: imaging features with pathologic correlation. Radiographics. 2009;29:347-373.
9. Isbell A, Fields EC. Three cases of women with HPV-related squamous cell carcinoma of unknown primary in the pelvis and retroperitoneum: a case series. Gynecol Oncol Rep. 2016;16:5-8.
10. León X, Quer M, Orús C, del Prado Venegas M, López M. Distant metastases in head and neck cancer patients who achieved loco-regional control. Head Neck. 2000;22:680-686.
11. Alavi S, Namazie A, Sercarz JA, Wang MB, Blackwell KE. Distant lymphatic metastasis from head and neck cancer. Ann Otol Rhinol Laryngol. 1999;108:860-863.
12. Krishnatry R, Gupta T, Murthy V, et al. Factors predicting ‘time to distant metastasis’ in radically treated head and neck cancer. Indian J Cancer. 2014;51:231-235.
13. Goodwin WJ. Distant metastases from oropharyngeal cancer. ORL J Otorhinolaryngol Relat Spec. 2001;63:222-223.
Massive liver metastasis from colon adenocarcinoma causing cardiac tamponade
Colorectal cancer is the third most commonly diagnosed cancer in the United States.1 About 5% of Americans will be diagnosed with colorectal cancer in their lifetime, of which 20% will present with distant metastasis.2 The most common sites of metastasis are regional lymph nodes, liver, lung and peritoneum, and patients may present with signs or symptoms related to disease burden at any of these organs.
Case presentation and summary
A 55-year-old man had presented to an outside hospital in August of 2014 with 6 months of hematochezia and a 40-lb weight loss. He was found to be severely anemic on admission (hemoglobin, 4.9 g/dL [normal, 13-17 g/dL], hematocrit, 16% [normal, 35%-45%]). A computed-tomography (CT) scan of the abdomen and pelvis with contrast revealed a mass of 6.9 x 4.7 x 6.3 cm in the rectosigmoid colon and a mass of 10.0 x 12.0 x 10.7 cm in the right hepatic lobe consistent with metastatic disease. The patient was taken to the operating room where the rectosigmoid mass was resected completely. The liver mass was deemed unresectable because of its large size, and surgically directed therapy could not be performed. Pathology was consistent with a T3N1 moderately differentiated adenocarcinoma 11 cm from the anal verge. Further molecular tumor studies revealed wild type KRAS and NRAS, as well as a BRAF mutation.
About 4 weeks after the surgery, the patient was seen at our institution for an initial consultation and was noted to have significant anasarca, including 4+ pitting lower extremity edema and scrotal edema. He complained of dyspnea on exertion, which he attributed to deconditioning. His resting heart rate was found to be 123 beats per minute (normal, 60-100 bpm). Jugular venous distention was present. The patient was sent for an urgent echocardiogram, which showed external compression of the right atrium and ventricle by his liver metastasis resulting in tamponade physiology without the presence of any pericardial effusion (Figure 1).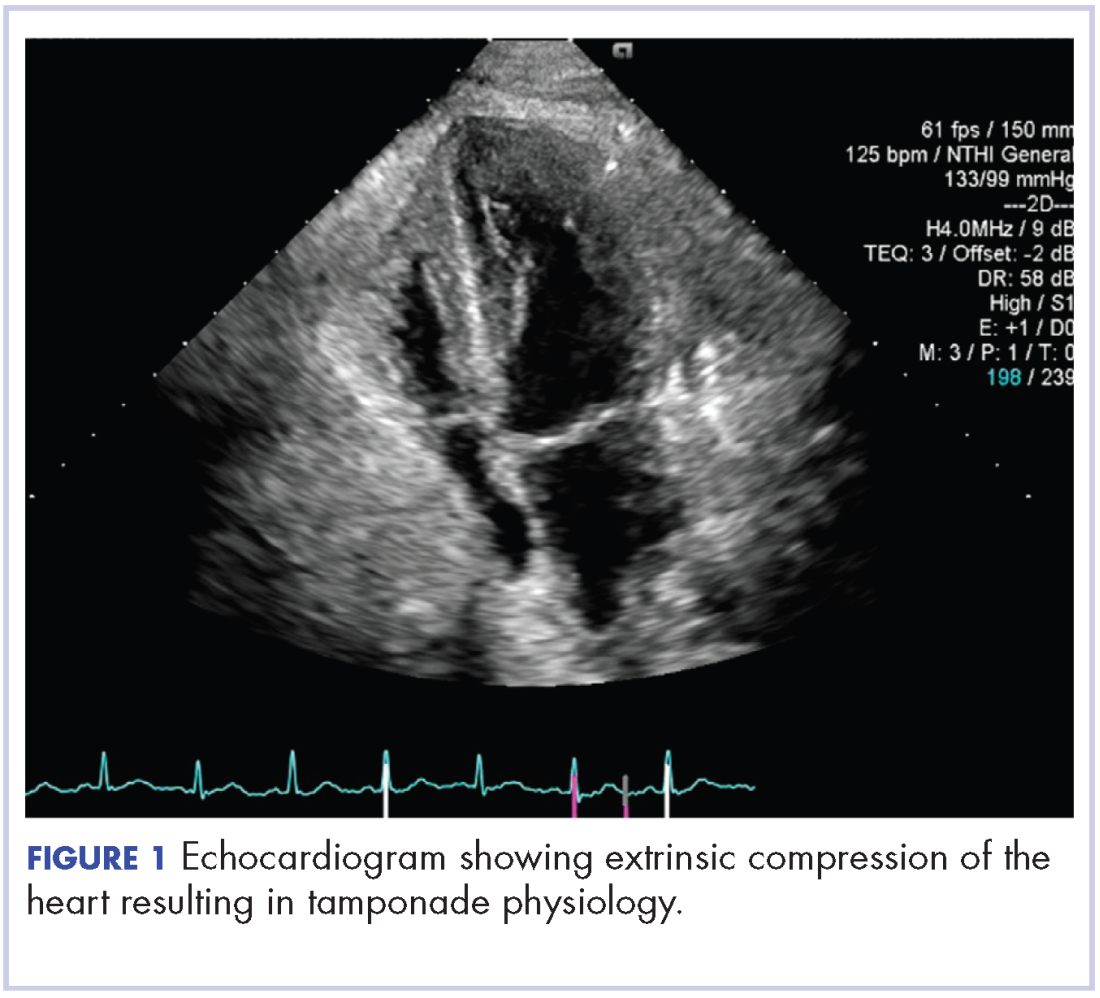
A CT of the abdomen and pelvis at that time showed that the liver mass had increased to 17.6 x 12.1 x 16.1 cm, exerting pressure on the heart and causing atelectasis of the right middle and lower lung lobes (Figure 2).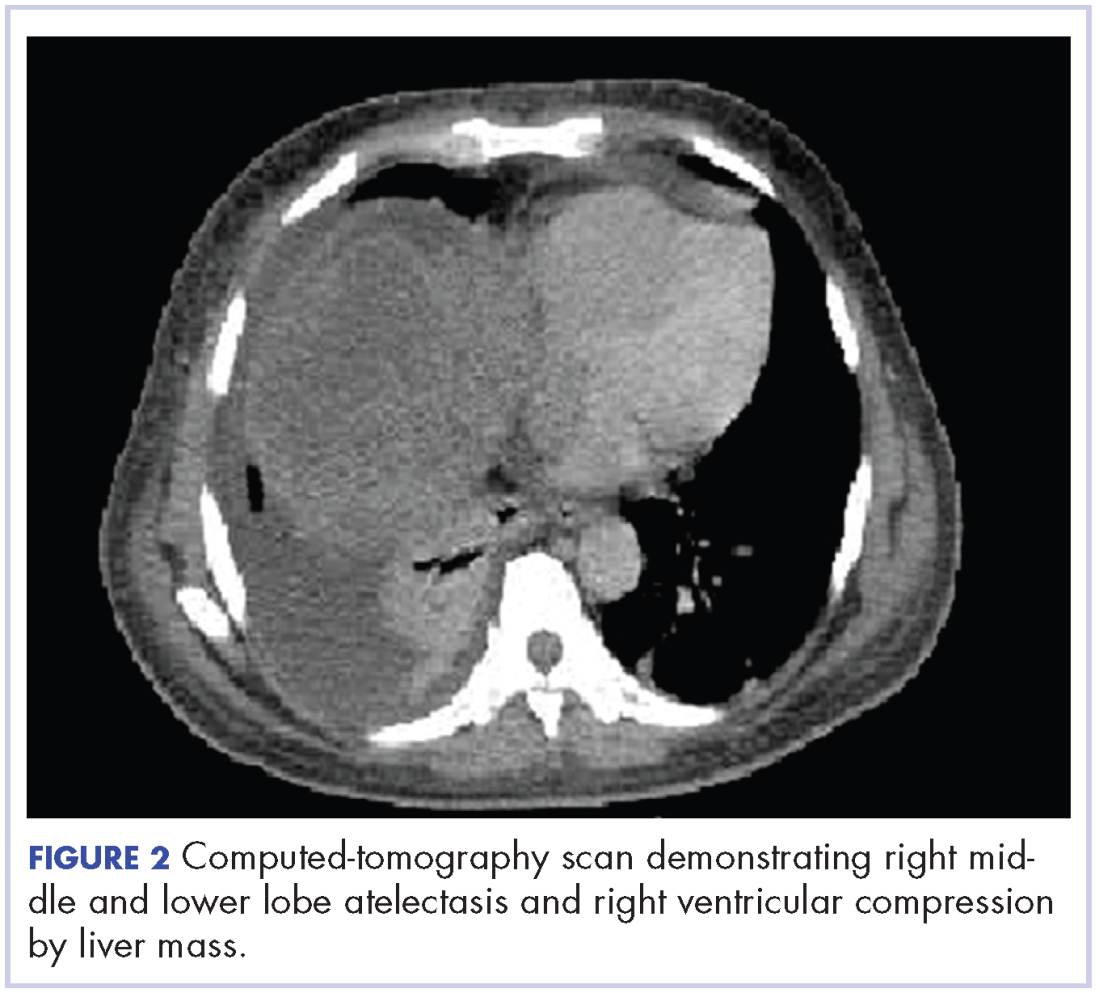
Treatment plan
The patient was evaluated by surgical oncology for resection, but his cardiovascular status placed him at high risk for perioperative complications, so such surgery was not pursued. Radioembolization was considered but not pursued because the process needed to evaluate, plan, and treat was not considered sufficiently timely. We consulted with our radiation oncology colleagues about external beam radiotherapy (EBRT) for rapid palliation. They evaluated the patient and recommended the EBRT, and the patient signed consent for treatment. We performed a CT-based simulation and generated an external beam, linear-accelerator–based treatment plan. The plan consisted of three 15-megavoltage photon fields delivering 3,000 cGy in 10 fractions to the whole liver, with appropriate multileaf collimation blocking to minimize dose to adjacent heart, right lung, and bilateral kidneys (Figure 3).
Before initiation of the EBRT, the patient received systemic chemotherapy with a dose-adjusted FOLFOX regimen (5-FU bolus 200 mg/m2, leucovorin 200 mg/m2, oxaliplatin 85 mg/m2, with infusional 5-FU 2,400 mg/m2 over 46 hours). After completing 1 dose of modified FOLFOX, he completed 10 fractions of whole liver radiotherapy with the aforementioned plan. He tolerated the initial treatment well and his subjective symptoms improved. The patient then proceeded to further systemic therapy. After recent data demonstrated improved median progression-free survival and response rates with FOLFOXIRI plus bevacizumab (infusional 5-FU 3200 mg/m2, leucovorin 200 mg/m2, irinotecan 165 mg/m2, and oxaliplatin 85 mg/m2, bevacizumab 5 mg/kg) versus FOLFIRI plus bevacizumab,3 we decided to modify his systemic therapy to FOLFOXIRI with bevacizumab to induce a better response.
Treatment response
After 2 doses of chemotherapy and completion of radiotherapy, the edema and shortness of breath improved. A follow-up echocardiogram performed a month after completion of EBRT, 1 dose of FOLFOX, and 1 dose of FOLFOXIRI showed resolution of the cardiac compression (Figure 4).
A CT scan of the abdomen and pelvis obtained after 3 cycles of FOLFOXIRI showed marked decrease in the size of the right lobe hepatic mass from 17.6 x 12.1 cm to 12.0 x 8.0 cm. Given the survival benefit of VEGF inhibition in colon cancer, bevacizumab (5 mg/kg) was added to the FOLFOXIRI regimen with cycle 4. Unfortunately, after the 5th cycle, a CT scan of the abdomen showed an increase in size of the hepatic lesions. At this time, FOLFOXIRI and bevacizumab were stopped, and given the tumor’s KRAS/NRAS wild type status, systemic therapy was changed to panitumumab (6 mg/kg). The patient initially tolerated treatment well, but after 9 cycles, the total bilirubin started to increase. CT abdomen at this point was consistent with progression of disease. The patient was not eligible for a clinical trial targeting BRAF mutation given the elevated bilirubin. Regorafanib (80 mg daily for 3 weeks on and 1 week off) was started. After the first cycle, the total bilirubin increased further and the regorafanib was dose reduced to 40 mg daily. Unfortunately, a repeat CT scan of the abdomen demonstrated progression of disease, and given that he developed a progressive transaminitis and hyperbilirubinemia, hospice care was recommended. The patient died shortly thereafter, about 15 months after his initial diagnosis.
Discussion
Massive liver metastasis in the setting of disseminated cancer is not an uncommon manifestation of advanced cancer that can have life-threatening consequences. In te present case, a bulky liver metastasis caused extrinsic compression of the right atrium, resulting in obvious clinical and echocardiogram-proven cardiac tamponade physiology. To our knowledge, this is the first reported case of the treatment of a bulky hepatic metastasis causing cardiac tamponade. In this patient’s case, both radiotherapy and chemotherapy were given safely in rapid sequence resulting in quick resolution of the patient’s symptoms and echocardiogram findings. The presence of a BRAF mutation conferred a poor prognosis and poor response to systemic chemotherapy. Nevertheless, the patient showed good response to a FOLFOXIRI regimen, chosen in this emergent situation given its significantly higher response rates compared with the standard FOLFIRI regimen, which was tolerated well with minimal adverse effects.
Findings from randomized controlled trials examining the role of palliative radiotherapy for metastatic liver disease have suggested that dose escalation above 30 Gy to the whole liver may lead to unacceptably high rates of radiation-induced liver disease, which typically leads to mortality.4-8 Two prospective trials comparing twice daily with daily fractionation have shown no benefit to hyperfractionation, with possibly increased rates of acute toxicity in the setting of hepatocellular carcinoma.9,10 There is emerging evidence that partial liver irradiation, in the appropriate setting in the form of boost after whole-liver RT or stereotactic body radiotherapy, may allow for further dose escalation while avoiding clinical hepatitis.11 Although there is no clear consensus about optimal RT dose and fractionation, the aforementioned studies show that dose and fractionation schemes ranging between 21 Gy and 30 Gy in 1.5 Gy to 3 Gy daily fractions likely provide the best therapeutic ratio for whole-liver irradiation.
In conclusion, this case demonstrates the resolution of cardiac tamponade from a massive liver colorectal metastasis after chemoradiation and illustrates the potential utility of adding radiotherapy to chemotherapy in an urgent scenario where the former might not typically be considered.
1. American Cancer Society. Cancer Facts & Figures 2015. https://www.cancer.org/research/cancer-facts-statistics/all-cancer-facts-figures/cancer-facts-figures-2015.html. Published 2015. Accessed October 10, 2017.
2. Siegel R, Desantis C, Jemal A. Colorectal cancer statistics, 2014. CA Cancer J Clin. 2014;64(2):104-117.
3. Loupakis F, Cremolini C, Masi G, et al. Initial therapy with FOLFOXIRI and bevacizumab for metastatic colorectal cancer. N Engl J Med. 2014;371:1609-1618.
4. Russell AH, Clyde C, Wasserman TH, Turner SS, Rotman M. Accelerated hyperfractionated hepatic irradiation in the management of patients with liver metastases: results of the RTOG dose escalating protocol. Int J Radiat Oncol Biol Phys. 1993;27(1):117-123.
5. Turek-Maischeider M, Kazem I. Palliative irradiation for liver metastases. JAMA. 1975;232(6):625-628.
6. Sherman DM, Weichselbaum R, Order SE, Cloud L, Trey C, Piro AJ. Palliation of hepatic metastasis. Cancer. 1978;41(5):2013-2017.
7. Prasad B, Lee MS, Hendrickson FR. Irradiation of hepatic metastases. Int J Radiat Oncol Biol Phys. 1977;2:129-132.
8. Borgelt BB, Gelber R, Brady LW, Griffin T, Hendrickson FR. The palliation of hepatic metastases: results of the Radiation Therapy Oncology Group pilot study. Int J Radiat Oncol Biol Phys. 1981;7(5):587-591.
9. Raju PI, Maruyama Y, DeSimone P, MacDonald J. Treatment of liver metastases with a combination of chemotherapy and hyperfractionated external radiation therapy. Am J Clin Oncol. 1987;10(1):41-43.
10. Stillwagon GB, Order SE, Guse C, et al. 194 hepatocellular cancers treated by radiation and chemotherapy combinations: toxicity and response: a Radiation Therapy Oncology Group Study. Int J Radiat Oncol Biol Phys. 1989;17(6):1223-1229.
11. Mohiuddin M, Chen E, Ahmad N. Combined liver radiation and chemotherapy for palliation of hepatic metastases from colorectal cancer. J Clin Oncol. 1996;14(3):722-728.
Colorectal cancer is the third most commonly diagnosed cancer in the United States.1 About 5% of Americans will be diagnosed with colorectal cancer in their lifetime, of which 20% will present with distant metastasis.2 The most common sites of metastasis are regional lymph nodes, liver, lung and peritoneum, and patients may present with signs or symptoms related to disease burden at any of these organs.
Case presentation and summary
A 55-year-old man had presented to an outside hospital in August of 2014 with 6 months of hematochezia and a 40-lb weight loss. He was found to be severely anemic on admission (hemoglobin, 4.9 g/dL [normal, 13-17 g/dL], hematocrit, 16% [normal, 35%-45%]). A computed-tomography (CT) scan of the abdomen and pelvis with contrast revealed a mass of 6.9 x 4.7 x 6.3 cm in the rectosigmoid colon and a mass of 10.0 x 12.0 x 10.7 cm in the right hepatic lobe consistent with metastatic disease. The patient was taken to the operating room where the rectosigmoid mass was resected completely. The liver mass was deemed unresectable because of its large size, and surgically directed therapy could not be performed. Pathology was consistent with a T3N1 moderately differentiated adenocarcinoma 11 cm from the anal verge. Further molecular tumor studies revealed wild type KRAS and NRAS, as well as a BRAF mutation.
About 4 weeks after the surgery, the patient was seen at our institution for an initial consultation and was noted to have significant anasarca, including 4+ pitting lower extremity edema and scrotal edema. He complained of dyspnea on exertion, which he attributed to deconditioning. His resting heart rate was found to be 123 beats per minute (normal, 60-100 bpm). Jugular venous distention was present. The patient was sent for an urgent echocardiogram, which showed external compression of the right atrium and ventricle by his liver metastasis resulting in tamponade physiology without the presence of any pericardial effusion (Figure 1).
A CT of the abdomen and pelvis at that time showed that the liver mass had increased to 17.6 x 12.1 x 16.1 cm, exerting pressure on the heart and causing atelectasis of the right middle and lower lung lobes (Figure 2).
Treatment plan
The patient was evaluated by surgical oncology for resection, but his cardiovascular status placed him at high risk for perioperative complications, so such surgery was not pursued. Radioembolization was considered but not pursued because the process needed to evaluate, plan, and treat was not considered sufficiently timely. We consulted with our radiation oncology colleagues about external beam radiotherapy (EBRT) for rapid palliation. They evaluated the patient and recommended the EBRT, and the patient signed consent for treatment. We performed a CT-based simulation and generated an external beam, linear-accelerator–based treatment plan. The plan consisted of three 15-megavoltage photon fields delivering 3,000 cGy in 10 fractions to the whole liver, with appropriate multileaf collimation blocking to minimize dose to adjacent heart, right lung, and bilateral kidneys (Figure 3).
Before initiation of the EBRT, the patient received systemic chemotherapy with a dose-adjusted FOLFOX regimen (5-FU bolus 200 mg/m2, leucovorin 200 mg/m2, oxaliplatin 85 mg/m2, with infusional 5-FU 2,400 mg/m2 over 46 hours). After completing 1 dose of modified FOLFOX, he completed 10 fractions of whole liver radiotherapy with the aforementioned plan. He tolerated the initial treatment well and his subjective symptoms improved. The patient then proceeded to further systemic therapy. After recent data demonstrated improved median progression-free survival and response rates with FOLFOXIRI plus bevacizumab (infusional 5-FU 3200 mg/m2, leucovorin 200 mg/m2, irinotecan 165 mg/m2, and oxaliplatin 85 mg/m2, bevacizumab 5 mg/kg) versus FOLFIRI plus bevacizumab,3 we decided to modify his systemic therapy to FOLFOXIRI with bevacizumab to induce a better response.
Treatment response
After 2 doses of chemotherapy and completion of radiotherapy, the edema and shortness of breath improved. A follow-up echocardiogram performed a month after completion of EBRT, 1 dose of FOLFOX, and 1 dose of FOLFOXIRI showed resolution of the cardiac compression (Figure 4).
A CT scan of the abdomen and pelvis obtained after 3 cycles of FOLFOXIRI showed marked decrease in the size of the right lobe hepatic mass from 17.6 x 12.1 cm to 12.0 x 8.0 cm. Given the survival benefit of VEGF inhibition in colon cancer, bevacizumab (5 mg/kg) was added to the FOLFOXIRI regimen with cycle 4. Unfortunately, after the 5th cycle, a CT scan of the abdomen showed an increase in size of the hepatic lesions. At this time, FOLFOXIRI and bevacizumab were stopped, and given the tumor’s KRAS/NRAS wild type status, systemic therapy was changed to panitumumab (6 mg/kg). The patient initially tolerated treatment well, but after 9 cycles, the total bilirubin started to increase. CT abdomen at this point was consistent with progression of disease. The patient was not eligible for a clinical trial targeting BRAF mutation given the elevated bilirubin. Regorafanib (80 mg daily for 3 weeks on and 1 week off) was started. After the first cycle, the total bilirubin increased further and the regorafanib was dose reduced to 40 mg daily. Unfortunately, a repeat CT scan of the abdomen demonstrated progression of disease, and given that he developed a progressive transaminitis and hyperbilirubinemia, hospice care was recommended. The patient died shortly thereafter, about 15 months after his initial diagnosis.
Discussion
Massive liver metastasis in the setting of disseminated cancer is not an uncommon manifestation of advanced cancer that can have life-threatening consequences. In te present case, a bulky liver metastasis caused extrinsic compression of the right atrium, resulting in obvious clinical and echocardiogram-proven cardiac tamponade physiology. To our knowledge, this is the first reported case of the treatment of a bulky hepatic metastasis causing cardiac tamponade. In this patient’s case, both radiotherapy and chemotherapy were given safely in rapid sequence resulting in quick resolution of the patient’s symptoms and echocardiogram findings. The presence of a BRAF mutation conferred a poor prognosis and poor response to systemic chemotherapy. Nevertheless, the patient showed good response to a FOLFOXIRI regimen, chosen in this emergent situation given its significantly higher response rates compared with the standard FOLFIRI regimen, which was tolerated well with minimal adverse effects.
Findings from randomized controlled trials examining the role of palliative radiotherapy for metastatic liver disease have suggested that dose escalation above 30 Gy to the whole liver may lead to unacceptably high rates of radiation-induced liver disease, which typically leads to mortality.4-8 Two prospective trials comparing twice daily with daily fractionation have shown no benefit to hyperfractionation, with possibly increased rates of acute toxicity in the setting of hepatocellular carcinoma.9,10 There is emerging evidence that partial liver irradiation, in the appropriate setting in the form of boost after whole-liver RT or stereotactic body radiotherapy, may allow for further dose escalation while avoiding clinical hepatitis.11 Although there is no clear consensus about optimal RT dose and fractionation, the aforementioned studies show that dose and fractionation schemes ranging between 21 Gy and 30 Gy in 1.5 Gy to 3 Gy daily fractions likely provide the best therapeutic ratio for whole-liver irradiation.
In conclusion, this case demonstrates the resolution of cardiac tamponade from a massive liver colorectal metastasis after chemoradiation and illustrates the potential utility of adding radiotherapy to chemotherapy in an urgent scenario where the former might not typically be considered.
Colorectal cancer is the third most commonly diagnosed cancer in the United States.1 About 5% of Americans will be diagnosed with colorectal cancer in their lifetime, of which 20% will present with distant metastasis.2 The most common sites of metastasis are regional lymph nodes, liver, lung and peritoneum, and patients may present with signs or symptoms related to disease burden at any of these organs.
Case presentation and summary
A 55-year-old man had presented to an outside hospital in August of 2014 with 6 months of hematochezia and a 40-lb weight loss. He was found to be severely anemic on admission (hemoglobin, 4.9 g/dL [normal, 13-17 g/dL], hematocrit, 16% [normal, 35%-45%]). A computed-tomography (CT) scan of the abdomen and pelvis with contrast revealed a mass of 6.9 x 4.7 x 6.3 cm in the rectosigmoid colon and a mass of 10.0 x 12.0 x 10.7 cm in the right hepatic lobe consistent with metastatic disease. The patient was taken to the operating room where the rectosigmoid mass was resected completely. The liver mass was deemed unresectable because of its large size, and surgically directed therapy could not be performed. Pathology was consistent with a T3N1 moderately differentiated adenocarcinoma 11 cm from the anal verge. Further molecular tumor studies revealed wild type KRAS and NRAS, as well as a BRAF mutation.
About 4 weeks after the surgery, the patient was seen at our institution for an initial consultation and was noted to have significant anasarca, including 4+ pitting lower extremity edema and scrotal edema. He complained of dyspnea on exertion, which he attributed to deconditioning. His resting heart rate was found to be 123 beats per minute (normal, 60-100 bpm). Jugular venous distention was present. The patient was sent for an urgent echocardiogram, which showed external compression of the right atrium and ventricle by his liver metastasis resulting in tamponade physiology without the presence of any pericardial effusion (Figure 1).
A CT of the abdomen and pelvis at that time showed that the liver mass had increased to 17.6 x 12.1 x 16.1 cm, exerting pressure on the heart and causing atelectasis of the right middle and lower lung lobes (Figure 2).
Treatment plan
The patient was evaluated by surgical oncology for resection, but his cardiovascular status placed him at high risk for perioperative complications, so such surgery was not pursued. Radioembolization was considered but not pursued because the process needed to evaluate, plan, and treat was not considered sufficiently timely. We consulted with our radiation oncology colleagues about external beam radiotherapy (EBRT) for rapid palliation. They evaluated the patient and recommended the EBRT, and the patient signed consent for treatment. We performed a CT-based simulation and generated an external beam, linear-accelerator–based treatment plan. The plan consisted of three 15-megavoltage photon fields delivering 3,000 cGy in 10 fractions to the whole liver, with appropriate multileaf collimation blocking to minimize dose to adjacent heart, right lung, and bilateral kidneys (Figure 3).
Before initiation of the EBRT, the patient received systemic chemotherapy with a dose-adjusted FOLFOX regimen (5-FU bolus 200 mg/m2, leucovorin 200 mg/m2, oxaliplatin 85 mg/m2, with infusional 5-FU 2,400 mg/m2 over 46 hours). After completing 1 dose of modified FOLFOX, he completed 10 fractions of whole liver radiotherapy with the aforementioned plan. He tolerated the initial treatment well and his subjective symptoms improved. The patient then proceeded to further systemic therapy. After recent data demonstrated improved median progression-free survival and response rates with FOLFOXIRI plus bevacizumab (infusional 5-FU 3200 mg/m2, leucovorin 200 mg/m2, irinotecan 165 mg/m2, and oxaliplatin 85 mg/m2, bevacizumab 5 mg/kg) versus FOLFIRI plus bevacizumab,3 we decided to modify his systemic therapy to FOLFOXIRI with bevacizumab to induce a better response.
Treatment response
After 2 doses of chemotherapy and completion of radiotherapy, the edema and shortness of breath improved. A follow-up echocardiogram performed a month after completion of EBRT, 1 dose of FOLFOX, and 1 dose of FOLFOXIRI showed resolution of the cardiac compression (Figure 4).
A CT scan of the abdomen and pelvis obtained after 3 cycles of FOLFOXIRI showed marked decrease in the size of the right lobe hepatic mass from 17.6 x 12.1 cm to 12.0 x 8.0 cm. Given the survival benefit of VEGF inhibition in colon cancer, bevacizumab (5 mg/kg) was added to the FOLFOXIRI regimen with cycle 4. Unfortunately, after the 5th cycle, a CT scan of the abdomen showed an increase in size of the hepatic lesions. At this time, FOLFOXIRI and bevacizumab were stopped, and given the tumor’s KRAS/NRAS wild type status, systemic therapy was changed to panitumumab (6 mg/kg). The patient initially tolerated treatment well, but after 9 cycles, the total bilirubin started to increase. CT abdomen at this point was consistent with progression of disease. The patient was not eligible for a clinical trial targeting BRAF mutation given the elevated bilirubin. Regorafanib (80 mg daily for 3 weeks on and 1 week off) was started. After the first cycle, the total bilirubin increased further and the regorafanib was dose reduced to 40 mg daily. Unfortunately, a repeat CT scan of the abdomen demonstrated progression of disease, and given that he developed a progressive transaminitis and hyperbilirubinemia, hospice care was recommended. The patient died shortly thereafter, about 15 months after his initial diagnosis.
Discussion
Massive liver metastasis in the setting of disseminated cancer is not an uncommon manifestation of advanced cancer that can have life-threatening consequences. In te present case, a bulky liver metastasis caused extrinsic compression of the right atrium, resulting in obvious clinical and echocardiogram-proven cardiac tamponade physiology. To our knowledge, this is the first reported case of the treatment of a bulky hepatic metastasis causing cardiac tamponade. In this patient’s case, both radiotherapy and chemotherapy were given safely in rapid sequence resulting in quick resolution of the patient’s symptoms and echocardiogram findings. The presence of a BRAF mutation conferred a poor prognosis and poor response to systemic chemotherapy. Nevertheless, the patient showed good response to a FOLFOXIRI regimen, chosen in this emergent situation given its significantly higher response rates compared with the standard FOLFIRI regimen, which was tolerated well with minimal adverse effects.
Findings from randomized controlled trials examining the role of palliative radiotherapy for metastatic liver disease have suggested that dose escalation above 30 Gy to the whole liver may lead to unacceptably high rates of radiation-induced liver disease, which typically leads to mortality.4-8 Two prospective trials comparing twice daily with daily fractionation have shown no benefit to hyperfractionation, with possibly increased rates of acute toxicity in the setting of hepatocellular carcinoma.9,10 There is emerging evidence that partial liver irradiation, in the appropriate setting in the form of boost after whole-liver RT or stereotactic body radiotherapy, may allow for further dose escalation while avoiding clinical hepatitis.11 Although there is no clear consensus about optimal RT dose and fractionation, the aforementioned studies show that dose and fractionation schemes ranging between 21 Gy and 30 Gy in 1.5 Gy to 3 Gy daily fractions likely provide the best therapeutic ratio for whole-liver irradiation.
In conclusion, this case demonstrates the resolution of cardiac tamponade from a massive liver colorectal metastasis after chemoradiation and illustrates the potential utility of adding radiotherapy to chemotherapy in an urgent scenario where the former might not typically be considered.
1. American Cancer Society. Cancer Facts & Figures 2015. https://www.cancer.org/research/cancer-facts-statistics/all-cancer-facts-figures/cancer-facts-figures-2015.html. Published 2015. Accessed October 10, 2017.
2. Siegel R, Desantis C, Jemal A. Colorectal cancer statistics, 2014. CA Cancer J Clin. 2014;64(2):104-117.
3. Loupakis F, Cremolini C, Masi G, et al. Initial therapy with FOLFOXIRI and bevacizumab for metastatic colorectal cancer. N Engl J Med. 2014;371:1609-1618.
4. Russell AH, Clyde C, Wasserman TH, Turner SS, Rotman M. Accelerated hyperfractionated hepatic irradiation in the management of patients with liver metastases: results of the RTOG dose escalating protocol. Int J Radiat Oncol Biol Phys. 1993;27(1):117-123.
5. Turek-Maischeider M, Kazem I. Palliative irradiation for liver metastases. JAMA. 1975;232(6):625-628.
6. Sherman DM, Weichselbaum R, Order SE, Cloud L, Trey C, Piro AJ. Palliation of hepatic metastasis. Cancer. 1978;41(5):2013-2017.
7. Prasad B, Lee MS, Hendrickson FR. Irradiation of hepatic metastases. Int J Radiat Oncol Biol Phys. 1977;2:129-132.
8. Borgelt BB, Gelber R, Brady LW, Griffin T, Hendrickson FR. The palliation of hepatic metastases: results of the Radiation Therapy Oncology Group pilot study. Int J Radiat Oncol Biol Phys. 1981;7(5):587-591.
9. Raju PI, Maruyama Y, DeSimone P, MacDonald J. Treatment of liver metastases with a combination of chemotherapy and hyperfractionated external radiation therapy. Am J Clin Oncol. 1987;10(1):41-43.
10. Stillwagon GB, Order SE, Guse C, et al. 194 hepatocellular cancers treated by radiation and chemotherapy combinations: toxicity and response: a Radiation Therapy Oncology Group Study. Int J Radiat Oncol Biol Phys. 1989;17(6):1223-1229.
11. Mohiuddin M, Chen E, Ahmad N. Combined liver radiation and chemotherapy for palliation of hepatic metastases from colorectal cancer. J Clin Oncol. 1996;14(3):722-728.
1. American Cancer Society. Cancer Facts & Figures 2015. https://www.cancer.org/research/cancer-facts-statistics/all-cancer-facts-figures/cancer-facts-figures-2015.html. Published 2015. Accessed October 10, 2017.
2. Siegel R, Desantis C, Jemal A. Colorectal cancer statistics, 2014. CA Cancer J Clin. 2014;64(2):104-117.
3. Loupakis F, Cremolini C, Masi G, et al. Initial therapy with FOLFOXIRI and bevacizumab for metastatic colorectal cancer. N Engl J Med. 2014;371:1609-1618.
4. Russell AH, Clyde C, Wasserman TH, Turner SS, Rotman M. Accelerated hyperfractionated hepatic irradiation in the management of patients with liver metastases: results of the RTOG dose escalating protocol. Int J Radiat Oncol Biol Phys. 1993;27(1):117-123.
5. Turek-Maischeider M, Kazem I. Palliative irradiation for liver metastases. JAMA. 1975;232(6):625-628.
6. Sherman DM, Weichselbaum R, Order SE, Cloud L, Trey C, Piro AJ. Palliation of hepatic metastasis. Cancer. 1978;41(5):2013-2017.
7. Prasad B, Lee MS, Hendrickson FR. Irradiation of hepatic metastases. Int J Radiat Oncol Biol Phys. 1977;2:129-132.
8. Borgelt BB, Gelber R, Brady LW, Griffin T, Hendrickson FR. The palliation of hepatic metastases: results of the Radiation Therapy Oncology Group pilot study. Int J Radiat Oncol Biol Phys. 1981;7(5):587-591.
9. Raju PI, Maruyama Y, DeSimone P, MacDonald J. Treatment of liver metastases with a combination of chemotherapy and hyperfractionated external radiation therapy. Am J Clin Oncol. 1987;10(1):41-43.
10. Stillwagon GB, Order SE, Guse C, et al. 194 hepatocellular cancers treated by radiation and chemotherapy combinations: toxicity and response: a Radiation Therapy Oncology Group Study. Int J Radiat Oncol Biol Phys. 1989;17(6):1223-1229.
11. Mohiuddin M, Chen E, Ahmad N. Combined liver radiation and chemotherapy for palliation of hepatic metastases from colorectal cancer. J Clin Oncol. 1996;14(3):722-728.
Cardiac pleomorphic sarcoma after placement of Dacron graft
Primary cardiac tumors, either benign or malignant, are very rare. The combined incidence is 0.002% on pooled autopsy series.1 The benign tumors account for 63% of primary cardiac tumors and include myxoma, the most common, and followed by papillary fibroelastoma, fibroma, and hemangioma. The remaining 37% are malignant tumors, essentially predominated by sarcomas.1
Although myxoma is the most common tumor arising in the left atrium, we present a case that shows that sarcoma can also arise from the same chamber. In fact, sarcomas could mimic cardiac myxoma.2 The cardiac sarcomas can have similar clinical presentation and more importantly can share similar histopathological features. Sarcomas may have myxoid features.2 Cases diagnosed as cardiac myxomas should be diligently worked up to rule out the presence of sarcomas with myxoid features. In addition, foreign bodies have been found to induce sarcomas in experimental animals.3,4 In particular, 2 case reports have described sarcomas arising in association with Dacron vascular prostheses in humans.5,6 We present here the case of a patient who was diagnosed with cardiac pleomorphic sarcoma 8 years after the placement of a Dacron graft.
Case presentation and summary
A 56-year-old woman with history of left atrial myxoma status after resection in 2005 and placement of a Dacron graft, morbid obesity, hypertension, and asthma presented to the emergency department with progressively worsening shortness of breath and blurry vision over period of 2 months. Acute coronary syndrome was ruled out by electrocardiogram and serial biomarkers. A computed-tomography angiogram was pursued because of her history of left atrial myxoma, and the results suggested the presence of a left atrial tumor. She underwent a transesophageal echocardiogram, which confirmed the presence of a large left atrial mass that likely was attached to the interatrial septum prolapsing across the mitral valve and was suggestive for recurrent left atrial myxoma (Figure 1). The results of a cardiac catheterization showed normal coronaries.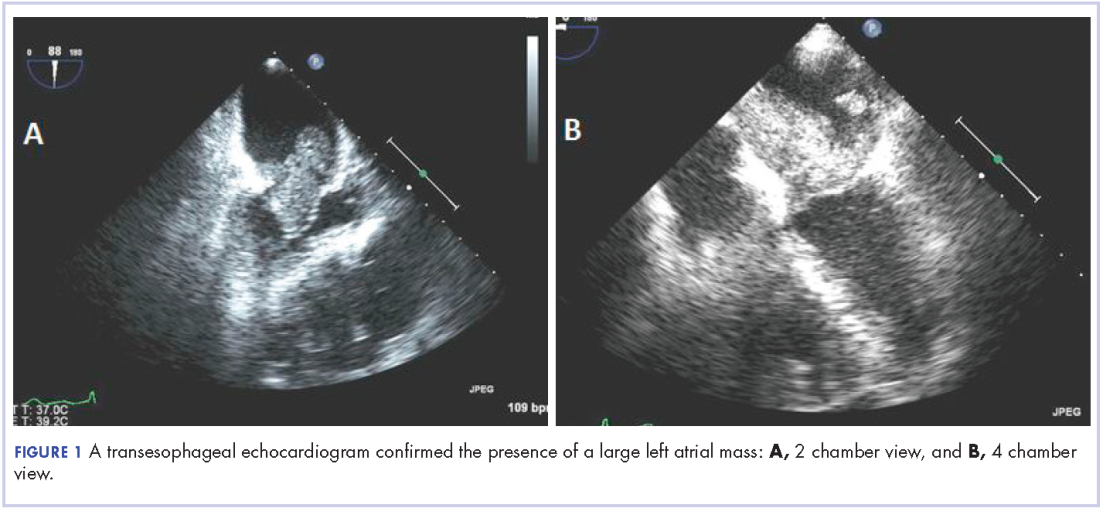
The patient subsequently underwent an excision of the left atrial tumor with profound internal and external myocardial cooling using antegrade blood cardioplegia under mildly hypothermic cardiopulmonary bypass. Frozen sections showed high-grade malignancy in favor of sarcoma. The hematoxylin and eosin stained permanent sections showed sheets of malignant pleomorphic spindle cells focally arranged in a storiform pattern. There were areas of necrosis and abundant mitotic activity. By immunohistochemical (IHC) stains, the tumor cells were diffusely positive for vimentin, and negative for pan-cytokeratin antibody (AE1/AE3), S-100 protein, Melan-A antibody, HMB45, CD34, CD31, myogenin, and MYOD1. IHC stains for CK-OSCAR, desmin, and smooth muscle actin were focally positive, and a ki-67 stain showed a proliferation index of about 80%. The histologic and IHC findings were consistent with a final diagnosis of high-grade undifferentiated pleomorphic sarcoma (Figure 2).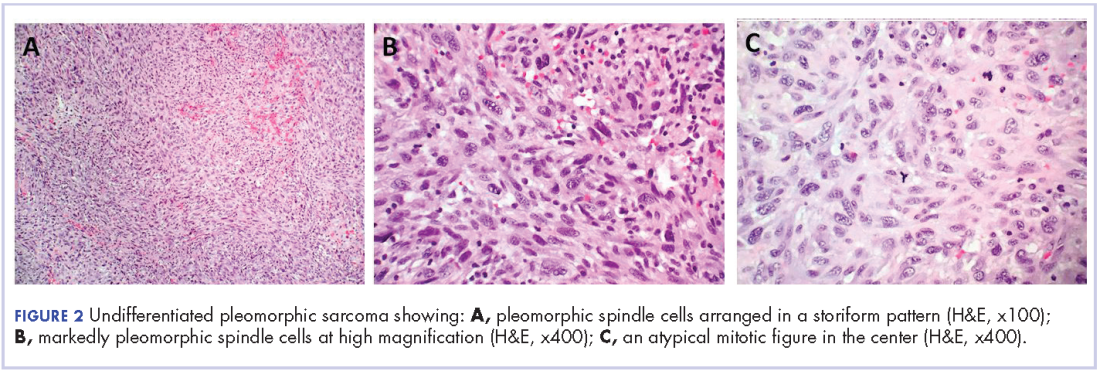
A positron emission tomography scan performed November 2013 did not show any other activity. The patient was scheduled for chemotherapy with adriamycin and ifosfamide with a plan for total of 6 cycles. Before her admission for the chemotherapy, the patient was admitted to the hospital for atrial fibrillation with rapid ventricular response and had multiple complications requiring prolonged hospitalization and rehabilitation. Repeat imaging 2 months later showed diffuse metastatic disease. However, her performance status had declined and she was not eligible for chemotherapy. She was placed under hospice care.
Discussion
This case demonstrates development of a cardiac pleomorphic sarcoma, a rare tumor, after placement of a Dacron graft. Given that foreign bodies have been found to induce sarcomas in experimental animals,3,4 and a few case reports have described sarcomas arising in association with Dacron vascular prostheses, 5-10 it seems that an exuberant host response around the foreign body might represent an important intermediate step in the development of the sarcoma.
There is no clearly defined pathogenesis that explains the link between a Dacron graft and sarcomas. In 1950s, Oppenheimer and colleagues described the formation of malignant tumors by various types of plastics, including Dacron, that were embedded in rats. 3,4 Most of the tumors were some form of sarcomas. It was inferred that physical properties of the plastics may have some role in tumor development. Plastics in sheet form or film that remained in situ for more than 6 months induced significant number of tumors compared with other forms such as sponges, films with holes, or powders.3,4 The 3-dimensional polymeric structure of the Dacron graft seems to play a role in induction of sarcoma as well. A pore diameter of less than 0.4 mm may increase tumorigenicity.11 The removal of the material before the 6-months mark does not lead to malignant tumors, which further supports the link between Dacron graft and formation of tumor. A pocket is formed around the foreign material after a certain period, as has been shown in histologic studies as the site of tumor origin.9,10
At the molecular level, the MDM-2/p53 pathway has been cited as possible mechanism for pathogenesis of intimal sarcoma.12,13 It has been suggested that endothelial dysplasia occurs as a precursor lesion in these sarcomas.14 The Dacron graft may cause a dysplastic effect on the endothelium leading to this precursor lesion and in certain cases transforming into sarcoma. Further definitive studies are required.
The primary treatment for cardiac sarcoma is surgical removal, although it is not always feasible. Findings in a Mayo clinic study showed that the median survival was 17 months for patients who underwent complete surgical excision, compared with 6 months for those who complete resection was not possible.15 In addition, a 10% survival rate at 1 year has been reported in primary cardiac sarcomas that are treated without any type of surgery.16
There is no clear-cut evidence supporting or refuting adjuvant chemotherapy for cardiac sarcoma. Some have inferred a potential benefit of adjuvant chemotherapy although definitive conclusions cannot be drawn. The median survival was 16.5 months in a case series of patients who received adjuvant chemotherapy, compared with 9 months and 11 months in 2 other case series.17,18,19 Multiple chemotherapy regimens have been used in the past for treatment. A retrospective s
Radiation showed some benefit in progression-free survival in a French retrospective study.21 Radiation therapies have been tried in other cases, as well in addition to chemotherapy. However, there is not enough data to support or refute it at this time.15,17,20 Several sporadic cases reported show benefit of cardiac transplantation.21,22
Conclusion
In consideration of the placement of the Dacron graft 8 years before the tumor occurrence, the anatomic proximity of the tumor to the Dacron graft, and the association between sarcoma with Dacron in medical literature, it seems logical to infer that this unusual malignancy in our patient is associated with the Dacron prosthesis.
1. Patil HR, Singh D, Hajdu M. Cardiac sarcoma presenting as heart failure and diagnosed as recurrent myxoma by echocardiogram. Eur J Echocardiogr. 2010;11(4):E12.
2. Awamleh P, Alberca MT, Gamallo C, Enrech S, Sarraj A. Left atrium myxosarcoma: an exceptional cardiac malignant primary tumor. Clin Cardiol. 2007;30(6):306-308.
3. Oppenheimer BS, Oppenheimer ET, Stout AP, Danishefsky I. Malignant tumors resulting from embedding plastics in rodents. Science. 1953;118:305-306.
4. Oppenheimer BS, Oppenheimer ET, Stout AP, Willhite M, Danishefski, I. The latent period in carcinogenesis by plastics in rats and its relation to the presarcomatous stage. Cancer. 1958;11(1):204-213.
5. Almeida NJ, Hoang P, Biddle P, Arouni A, Esterbrooks D. Primary cardiac angiosarcoma: in a patient with a Dacron aortic prosthesis. Tex Heart Inst J. 2011;38(1):61-65; discussion 65.
6. Stewart B, Manglik N, Zhao B, et al. Aortic intimal sarcoma: report of two cases with immunohistochemical analysis for pathogenesis. Cardiovasc Pathol. 2013;22(5):351-356.
7. Umscheid TW, Rouhani G, Morlang T, et al. Hemangiosarcoma after endovascular aortic aneurysm repair. J Endovasc Ther. 2007;14(1):101-105.
8. Ben-Izhak O, Vlodavsky E, Ofer A, Engel A, Nitecky S, Hoffman A. Epithelioid angiosarcoma associated with a Dacron vascular graft. Am J Surg Pathol. 1999;23(11):1418-1422.
9. Fyfe BS, Quintana CS, Kaneko M, Griepp RB. Aortic sarcoma four years after Dacron graft insertion. Ann Thorac Surg. 1994;58(6):1752-1754.
10. O’Connell TX, Fee HJ, Golding A. Sarcoma associated with Dacron prosthetic material: case report and review of the literature. J Thorac Cardiovasc Surg. 1976;72(1):94-96.
11. Karp RD, Johnson KH, Buoen LC, et al. Tumorogenesis by millipore filters in mice: histology and ultastructure of tissue reactions, as related to pore size. J Natl Cancer Inst. 1973;51:1275-1285.
12. Bode-Lesniewska B, Zhao J, Speel EJ, et al. Gains of 12q13-14 and overexpression of mdm2 are frequent findings in intimal sarcomas of the pulmonary artery. Virchows Arch. 2001;438:57-65.
13. Zeitz C, Rossle M, Haas C, et al. MDM-2 oncoprotein overexpression, p53 gene mutation, and VEGF up-regulation in angiosarcomas. Am J Surg Pathol. 1998;153:1425-1433.
14. Haber LM, Truong L. Immunohistochemical demonstration of the endothelialnature of aortic intimal sarcoma. Am J Surg Pathol. 1988 Oct;12(10):798-802. PubMed PMID: 3138923.
15. Simpson L, Kumar SK, Okuno SH, et al. Malignant primary cardiac tumors: review of a single institution experience. Cancer. 2008;112(11):2440-2446.
16. Leja MJ, Shah DJ, Reardon MJ. Primary cardiac tumors. Tex Heart Inst J. 2011;38(3):261-262.
17. Donsbeck AV, Ranchere D, Coindre JM, Le Gall F, Cordier JF, Loire R. Primary cardiac sarcomas: an immunohistochemical and grading study with long-term follow-up of 24 cases. Histopathology. 1999;34(4):295-304.
18. Putnam JB, Sweeney MS, Colon R, Lanza LA, Frazier OH, Cooley DC. Primary cardiac sarcomas. Ann Thorac Surg. 1990; 51; 906-910.
19. Murphy WR, Sweeney MS, Putnam JB et al. Surgical treatment of cardiac tumors: a 25-year experience. Ann Thorac Surg. 1990;49;612-618.
20. Llombart-Cussac A, Pivot X, Contesso G, et al. Adjuvant chemotherapy for primary cardiac sarcomas: the IGR experience. Br J Cancer. 1998;78(12):1624-1628.
21. Isambert N, Ray-Coquard I, Italiano A, et al. Primary cardiac sarcomas: a retrospective study of the French Sarcoma Group. Eur J Cancer. 2014;50(1):128-136.
22. Agaimy A, Rösch J, Weyand M, Strecker T. Primary and metastatic cardiac sarcomas: a 12-year experience at a German heart center. Int J Clin Exp Pathol. 2012;5(9):928-938.
Primary cardiac tumors, either benign or malignant, are very rare. The combined incidence is 0.002% on pooled autopsy series.1 The benign tumors account for 63% of primary cardiac tumors and include myxoma, the most common, and followed by papillary fibroelastoma, fibroma, and hemangioma. The remaining 37% are malignant tumors, essentially predominated by sarcomas.1
Although myxoma is the most common tumor arising in the left atrium, we present a case that shows that sarcoma can also arise from the same chamber. In fact, sarcomas could mimic cardiac myxoma.2 The cardiac sarcomas can have similar clinical presentation and more importantly can share similar histopathological features. Sarcomas may have myxoid features.2 Cases diagnosed as cardiac myxomas should be diligently worked up to rule out the presence of sarcomas with myxoid features. In addition, foreign bodies have been found to induce sarcomas in experimental animals.3,4 In particular, 2 case reports have described sarcomas arising in association with Dacron vascular prostheses in humans.5,6 We present here the case of a patient who was diagnosed with cardiac pleomorphic sarcoma 8 years after the placement of a Dacron graft.
Case presentation and summary
A 56-year-old woman with history of left atrial myxoma status after resection in 2005 and placement of a Dacron graft, morbid obesity, hypertension, and asthma presented to the emergency department with progressively worsening shortness of breath and blurry vision over period of 2 months. Acute coronary syndrome was ruled out by electrocardiogram and serial biomarkers. A computed-tomography angiogram was pursued because of her history of left atrial myxoma, and the results suggested the presence of a left atrial tumor. She underwent a transesophageal echocardiogram, which confirmed the presence of a large left atrial mass that likely was attached to the interatrial septum prolapsing across the mitral valve and was suggestive for recurrent left atrial myxoma (Figure 1). The results of a cardiac catheterization showed normal coronaries.
The patient subsequently underwent an excision of the left atrial tumor with profound internal and external myocardial cooling using antegrade blood cardioplegia under mildly hypothermic cardiopulmonary bypass. Frozen sections showed high-grade malignancy in favor of sarcoma. The hematoxylin and eosin stained permanent sections showed sheets of malignant pleomorphic spindle cells focally arranged in a storiform pattern. There were areas of necrosis and abundant mitotic activity. By immunohistochemical (IHC) stains, the tumor cells were diffusely positive for vimentin, and negative for pan-cytokeratin antibody (AE1/AE3), S-100 protein, Melan-A antibody, HMB45, CD34, CD31, myogenin, and MYOD1. IHC stains for CK-OSCAR, desmin, and smooth muscle actin were focally positive, and a ki-67 stain showed a proliferation index of about 80%. The histologic and IHC findings were consistent with a final diagnosis of high-grade undifferentiated pleomorphic sarcoma (Figure 2).
A positron emission tomography scan performed November 2013 did not show any other activity. The patient was scheduled for chemotherapy with adriamycin and ifosfamide with a plan for total of 6 cycles. Before her admission for the chemotherapy, the patient was admitted to the hospital for atrial fibrillation with rapid ventricular response and had multiple complications requiring prolonged hospitalization and rehabilitation. Repeat imaging 2 months later showed diffuse metastatic disease. However, her performance status had declined and she was not eligible for chemotherapy. She was placed under hospice care.
Discussion
This case demonstrates development of a cardiac pleomorphic sarcoma, a rare tumor, after placement of a Dacron graft. Given that foreign bodies have been found to induce sarcomas in experimental animals,3,4 and a few case reports have described sarcomas arising in association with Dacron vascular prostheses, 5-10 it seems that an exuberant host response around the foreign body might represent an important intermediate step in the development of the sarcoma.
There is no clearly defined pathogenesis that explains the link between a Dacron graft and sarcomas. In 1950s, Oppenheimer and colleagues described the formation of malignant tumors by various types of plastics, including Dacron, that were embedded in rats. 3,4 Most of the tumors were some form of sarcomas. It was inferred that physical properties of the plastics may have some role in tumor development. Plastics in sheet form or film that remained in situ for more than 6 months induced significant number of tumors compared with other forms such as sponges, films with holes, or powders.3,4 The 3-dimensional polymeric structure of the Dacron graft seems to play a role in induction of sarcoma as well. A pore diameter of less than 0.4 mm may increase tumorigenicity.11 The removal of the material before the 6-months mark does not lead to malignant tumors, which further supports the link between Dacron graft and formation of tumor. A pocket is formed around the foreign material after a certain period, as has been shown in histologic studies as the site of tumor origin.9,10
At the molecular level, the MDM-2/p53 pathway has been cited as possible mechanism for pathogenesis of intimal sarcoma.12,13 It has been suggested that endothelial dysplasia occurs as a precursor lesion in these sarcomas.14 The Dacron graft may cause a dysplastic effect on the endothelium leading to this precursor lesion and in certain cases transforming into sarcoma. Further definitive studies are required.
The primary treatment for cardiac sarcoma is surgical removal, although it is not always feasible. Findings in a Mayo clinic study showed that the median survival was 17 months for patients who underwent complete surgical excision, compared with 6 months for those who complete resection was not possible.15 In addition, a 10% survival rate at 1 year has been reported in primary cardiac sarcomas that are treated without any type of surgery.16
There is no clear-cut evidence supporting or refuting adjuvant chemotherapy for cardiac sarcoma. Some have inferred a potential benefit of adjuvant chemotherapy although definitive conclusions cannot be drawn. The median survival was 16.5 months in a case series of patients who received adjuvant chemotherapy, compared with 9 months and 11 months in 2 other case series.17,18,19 Multiple chemotherapy regimens have been used in the past for treatment. A retrospective s
Radiation showed some benefit in progression-free survival in a French retrospective study.21 Radiation therapies have been tried in other cases, as well in addition to chemotherapy. However, there is not enough data to support or refute it at this time.15,17,20 Several sporadic cases reported show benefit of cardiac transplantation.21,22
Conclusion
In consideration of the placement of the Dacron graft 8 years before the tumor occurrence, the anatomic proximity of the tumor to the Dacron graft, and the association between sarcoma with Dacron in medical literature, it seems logical to infer that this unusual malignancy in our patient is associated with the Dacron prosthesis.
Primary cardiac tumors, either benign or malignant, are very rare. The combined incidence is 0.002% on pooled autopsy series.1 The benign tumors account for 63% of primary cardiac tumors and include myxoma, the most common, and followed by papillary fibroelastoma, fibroma, and hemangioma. The remaining 37% are malignant tumors, essentially predominated by sarcomas.1
Although myxoma is the most common tumor arising in the left atrium, we present a case that shows that sarcoma can also arise from the same chamber. In fact, sarcomas could mimic cardiac myxoma.2 The cardiac sarcomas can have similar clinical presentation and more importantly can share similar histopathological features. Sarcomas may have myxoid features.2 Cases diagnosed as cardiac myxomas should be diligently worked up to rule out the presence of sarcomas with myxoid features. In addition, foreign bodies have been found to induce sarcomas in experimental animals.3,4 In particular, 2 case reports have described sarcomas arising in association with Dacron vascular prostheses in humans.5,6 We present here the case of a patient who was diagnosed with cardiac pleomorphic sarcoma 8 years after the placement of a Dacron graft.
Case presentation and summary
A 56-year-old woman with history of left atrial myxoma status after resection in 2005 and placement of a Dacron graft, morbid obesity, hypertension, and asthma presented to the emergency department with progressively worsening shortness of breath and blurry vision over period of 2 months. Acute coronary syndrome was ruled out by electrocardiogram and serial biomarkers. A computed-tomography angiogram was pursued because of her history of left atrial myxoma, and the results suggested the presence of a left atrial tumor. She underwent a transesophageal echocardiogram, which confirmed the presence of a large left atrial mass that likely was attached to the interatrial septum prolapsing across the mitral valve and was suggestive for recurrent left atrial myxoma (Figure 1). The results of a cardiac catheterization showed normal coronaries.
The patient subsequently underwent an excision of the left atrial tumor with profound internal and external myocardial cooling using antegrade blood cardioplegia under mildly hypothermic cardiopulmonary bypass. Frozen sections showed high-grade malignancy in favor of sarcoma. The hematoxylin and eosin stained permanent sections showed sheets of malignant pleomorphic spindle cells focally arranged in a storiform pattern. There were areas of necrosis and abundant mitotic activity. By immunohistochemical (IHC) stains, the tumor cells were diffusely positive for vimentin, and negative for pan-cytokeratin antibody (AE1/AE3), S-100 protein, Melan-A antibody, HMB45, CD34, CD31, myogenin, and MYOD1. IHC stains for CK-OSCAR, desmin, and smooth muscle actin were focally positive, and a ki-67 stain showed a proliferation index of about 80%. The histologic and IHC findings were consistent with a final diagnosis of high-grade undifferentiated pleomorphic sarcoma (Figure 2).
A positron emission tomography scan performed November 2013 did not show any other activity. The patient was scheduled for chemotherapy with adriamycin and ifosfamide with a plan for total of 6 cycles. Before her admission for the chemotherapy, the patient was admitted to the hospital for atrial fibrillation with rapid ventricular response and had multiple complications requiring prolonged hospitalization and rehabilitation. Repeat imaging 2 months later showed diffuse metastatic disease. However, her performance status had declined and she was not eligible for chemotherapy. She was placed under hospice care.
Discussion
This case demonstrates development of a cardiac pleomorphic sarcoma, a rare tumor, after placement of a Dacron graft. Given that foreign bodies have been found to induce sarcomas in experimental animals,3,4 and a few case reports have described sarcomas arising in association with Dacron vascular prostheses, 5-10 it seems that an exuberant host response around the foreign body might represent an important intermediate step in the development of the sarcoma.
There is no clearly defined pathogenesis that explains the link between a Dacron graft and sarcomas. In 1950s, Oppenheimer and colleagues described the formation of malignant tumors by various types of plastics, including Dacron, that were embedded in rats. 3,4 Most of the tumors were some form of sarcomas. It was inferred that physical properties of the plastics may have some role in tumor development. Plastics in sheet form or film that remained in situ for more than 6 months induced significant number of tumors compared with other forms such as sponges, films with holes, or powders.3,4 The 3-dimensional polymeric structure of the Dacron graft seems to play a role in induction of sarcoma as well. A pore diameter of less than 0.4 mm may increase tumorigenicity.11 The removal of the material before the 6-months mark does not lead to malignant tumors, which further supports the link between Dacron graft and formation of tumor. A pocket is formed around the foreign material after a certain period, as has been shown in histologic studies as the site of tumor origin.9,10
At the molecular level, the MDM-2/p53 pathway has been cited as possible mechanism for pathogenesis of intimal sarcoma.12,13 It has been suggested that endothelial dysplasia occurs as a precursor lesion in these sarcomas.14 The Dacron graft may cause a dysplastic effect on the endothelium leading to this precursor lesion and in certain cases transforming into sarcoma. Further definitive studies are required.
The primary treatment for cardiac sarcoma is surgical removal, although it is not always feasible. Findings in a Mayo clinic study showed that the median survival was 17 months for patients who underwent complete surgical excision, compared with 6 months for those who complete resection was not possible.15 In addition, a 10% survival rate at 1 year has been reported in primary cardiac sarcomas that are treated without any type of surgery.16
There is no clear-cut evidence supporting or refuting adjuvant chemotherapy for cardiac sarcoma. Some have inferred a potential benefit of adjuvant chemotherapy although definitive conclusions cannot be drawn. The median survival was 16.5 months in a case series of patients who received adjuvant chemotherapy, compared with 9 months and 11 months in 2 other case series.17,18,19 Multiple chemotherapy regimens have been used in the past for treatment. A retrospective s
Radiation showed some benefit in progression-free survival in a French retrospective study.21 Radiation therapies have been tried in other cases, as well in addition to chemotherapy. However, there is not enough data to support or refute it at this time.15,17,20 Several sporadic cases reported show benefit of cardiac transplantation.21,22
Conclusion
In consideration of the placement of the Dacron graft 8 years before the tumor occurrence, the anatomic proximity of the tumor to the Dacron graft, and the association between sarcoma with Dacron in medical literature, it seems logical to infer that this unusual malignancy in our patient is associated with the Dacron prosthesis.
1. Patil HR, Singh D, Hajdu M. Cardiac sarcoma presenting as heart failure and diagnosed as recurrent myxoma by echocardiogram. Eur J Echocardiogr. 2010;11(4):E12.
2. Awamleh P, Alberca MT, Gamallo C, Enrech S, Sarraj A. Left atrium myxosarcoma: an exceptional cardiac malignant primary tumor. Clin Cardiol. 2007;30(6):306-308.
3. Oppenheimer BS, Oppenheimer ET, Stout AP, Danishefsky I. Malignant tumors resulting from embedding plastics in rodents. Science. 1953;118:305-306.
4. Oppenheimer BS, Oppenheimer ET, Stout AP, Willhite M, Danishefski, I. The latent period in carcinogenesis by plastics in rats and its relation to the presarcomatous stage. Cancer. 1958;11(1):204-213.
5. Almeida NJ, Hoang P, Biddle P, Arouni A, Esterbrooks D. Primary cardiac angiosarcoma: in a patient with a Dacron aortic prosthesis. Tex Heart Inst J. 2011;38(1):61-65; discussion 65.
6. Stewart B, Manglik N, Zhao B, et al. Aortic intimal sarcoma: report of two cases with immunohistochemical analysis for pathogenesis. Cardiovasc Pathol. 2013;22(5):351-356.
7. Umscheid TW, Rouhani G, Morlang T, et al. Hemangiosarcoma after endovascular aortic aneurysm repair. J Endovasc Ther. 2007;14(1):101-105.
8. Ben-Izhak O, Vlodavsky E, Ofer A, Engel A, Nitecky S, Hoffman A. Epithelioid angiosarcoma associated with a Dacron vascular graft. Am J Surg Pathol. 1999;23(11):1418-1422.
9. Fyfe BS, Quintana CS, Kaneko M, Griepp RB. Aortic sarcoma four years after Dacron graft insertion. Ann Thorac Surg. 1994;58(6):1752-1754.
10. O’Connell TX, Fee HJ, Golding A. Sarcoma associated with Dacron prosthetic material: case report and review of the literature. J Thorac Cardiovasc Surg. 1976;72(1):94-96.
11. Karp RD, Johnson KH, Buoen LC, et al. Tumorogenesis by millipore filters in mice: histology and ultastructure of tissue reactions, as related to pore size. J Natl Cancer Inst. 1973;51:1275-1285.
12. Bode-Lesniewska B, Zhao J, Speel EJ, et al. Gains of 12q13-14 and overexpression of mdm2 are frequent findings in intimal sarcomas of the pulmonary artery. Virchows Arch. 2001;438:57-65.
13. Zeitz C, Rossle M, Haas C, et al. MDM-2 oncoprotein overexpression, p53 gene mutation, and VEGF up-regulation in angiosarcomas. Am J Surg Pathol. 1998;153:1425-1433.
14. Haber LM, Truong L. Immunohistochemical demonstration of the endothelialnature of aortic intimal sarcoma. Am J Surg Pathol. 1988 Oct;12(10):798-802. PubMed PMID: 3138923.
15. Simpson L, Kumar SK, Okuno SH, et al. Malignant primary cardiac tumors: review of a single institution experience. Cancer. 2008;112(11):2440-2446.
16. Leja MJ, Shah DJ, Reardon MJ. Primary cardiac tumors. Tex Heart Inst J. 2011;38(3):261-262.
17. Donsbeck AV, Ranchere D, Coindre JM, Le Gall F, Cordier JF, Loire R. Primary cardiac sarcomas: an immunohistochemical and grading study with long-term follow-up of 24 cases. Histopathology. 1999;34(4):295-304.
18. Putnam JB, Sweeney MS, Colon R, Lanza LA, Frazier OH, Cooley DC. Primary cardiac sarcomas. Ann Thorac Surg. 1990; 51; 906-910.
19. Murphy WR, Sweeney MS, Putnam JB et al. Surgical treatment of cardiac tumors: a 25-year experience. Ann Thorac Surg. 1990;49;612-618.
20. Llombart-Cussac A, Pivot X, Contesso G, et al. Adjuvant chemotherapy for primary cardiac sarcomas: the IGR experience. Br J Cancer. 1998;78(12):1624-1628.
21. Isambert N, Ray-Coquard I, Italiano A, et al. Primary cardiac sarcomas: a retrospective study of the French Sarcoma Group. Eur J Cancer. 2014;50(1):128-136.
22. Agaimy A, Rösch J, Weyand M, Strecker T. Primary and metastatic cardiac sarcomas: a 12-year experience at a German heart center. Int J Clin Exp Pathol. 2012;5(9):928-938.
1. Patil HR, Singh D, Hajdu M. Cardiac sarcoma presenting as heart failure and diagnosed as recurrent myxoma by echocardiogram. Eur J Echocardiogr. 2010;11(4):E12.
2. Awamleh P, Alberca MT, Gamallo C, Enrech S, Sarraj A. Left atrium myxosarcoma: an exceptional cardiac malignant primary tumor. Clin Cardiol. 2007;30(6):306-308.
3. Oppenheimer BS, Oppenheimer ET, Stout AP, Danishefsky I. Malignant tumors resulting from embedding plastics in rodents. Science. 1953;118:305-306.
4. Oppenheimer BS, Oppenheimer ET, Stout AP, Willhite M, Danishefski, I. The latent period in carcinogenesis by plastics in rats and its relation to the presarcomatous stage. Cancer. 1958;11(1):204-213.
5. Almeida NJ, Hoang P, Biddle P, Arouni A, Esterbrooks D. Primary cardiac angiosarcoma: in a patient with a Dacron aortic prosthesis. Tex Heart Inst J. 2011;38(1):61-65; discussion 65.
6. Stewart B, Manglik N, Zhao B, et al. Aortic intimal sarcoma: report of two cases with immunohistochemical analysis for pathogenesis. Cardiovasc Pathol. 2013;22(5):351-356.
7. Umscheid TW, Rouhani G, Morlang T, et al. Hemangiosarcoma after endovascular aortic aneurysm repair. J Endovasc Ther. 2007;14(1):101-105.
8. Ben-Izhak O, Vlodavsky E, Ofer A, Engel A, Nitecky S, Hoffman A. Epithelioid angiosarcoma associated with a Dacron vascular graft. Am J Surg Pathol. 1999;23(11):1418-1422.
9. Fyfe BS, Quintana CS, Kaneko M, Griepp RB. Aortic sarcoma four years after Dacron graft insertion. Ann Thorac Surg. 1994;58(6):1752-1754.
10. O’Connell TX, Fee HJ, Golding A. Sarcoma associated with Dacron prosthetic material: case report and review of the literature. J Thorac Cardiovasc Surg. 1976;72(1):94-96.
11. Karp RD, Johnson KH, Buoen LC, et al. Tumorogenesis by millipore filters in mice: histology and ultastructure of tissue reactions, as related to pore size. J Natl Cancer Inst. 1973;51:1275-1285.
12. Bode-Lesniewska B, Zhao J, Speel EJ, et al. Gains of 12q13-14 and overexpression of mdm2 are frequent findings in intimal sarcomas of the pulmonary artery. Virchows Arch. 2001;438:57-65.
13. Zeitz C, Rossle M, Haas C, et al. MDM-2 oncoprotein overexpression, p53 gene mutation, and VEGF up-regulation in angiosarcomas. Am J Surg Pathol. 1998;153:1425-1433.
14. Haber LM, Truong L. Immunohistochemical demonstration of the endothelialnature of aortic intimal sarcoma. Am J Surg Pathol. 1988 Oct;12(10):798-802. PubMed PMID: 3138923.
15. Simpson L, Kumar SK, Okuno SH, et al. Malignant primary cardiac tumors: review of a single institution experience. Cancer. 2008;112(11):2440-2446.
16. Leja MJ, Shah DJ, Reardon MJ. Primary cardiac tumors. Tex Heart Inst J. 2011;38(3):261-262.
17. Donsbeck AV, Ranchere D, Coindre JM, Le Gall F, Cordier JF, Loire R. Primary cardiac sarcomas: an immunohistochemical and grading study with long-term follow-up of 24 cases. Histopathology. 1999;34(4):295-304.
18. Putnam JB, Sweeney MS, Colon R, Lanza LA, Frazier OH, Cooley DC. Primary cardiac sarcomas. Ann Thorac Surg. 1990; 51; 906-910.
19. Murphy WR, Sweeney MS, Putnam JB et al. Surgical treatment of cardiac tumors: a 25-year experience. Ann Thorac Surg. 1990;49;612-618.
20. Llombart-Cussac A, Pivot X, Contesso G, et al. Adjuvant chemotherapy for primary cardiac sarcomas: the IGR experience. Br J Cancer. 1998;78(12):1624-1628.
21. Isambert N, Ray-Coquard I, Italiano A, et al. Primary cardiac sarcomas: a retrospective study of the French Sarcoma Group. Eur J Cancer. 2014;50(1):128-136.
22. Agaimy A, Rösch J, Weyand M, Strecker T. Primary and metastatic cardiac sarcomas: a 12-year experience at a German heart center. Int J Clin Exp Pathol. 2012;5(9):928-938.
Misleading Diagnosis of Idiopathic Pulmonary Fibrosis: A Clinical Concern
Sjogren syndrome (SS) is a chronic inflammatory autoimmune disorder characterized by lymphocytic infiltration of lacrimal and salivary glands causing sicca syndrome.¹ The disease can extend beyond the exocrine glands, and systemic manifestations, including vasculitis, lung, renal or neurologic involvement, can occur.² Lung disease associated with SS is more commonly seen in women aged ≥ 60 years. The most common symptoms include dry cough, chest pain, and dyspnea on exertion. Sjogren syndrome also may produce several respiratory complications, including bronchial hyperresponsiveness, bronchiolitis, bronchiectasis, pulmonary infections, pulmonary amyloidosis, pulmonary embolism, pulmonary hypertension, lymphomas, and interstitial lung diseases (ILD).² Although ILD typically occurs 5 to 10 years after the onset of SS, lung disease can precede SS.
Pulmonary involvement is associated with systemic manifestations, hypergammaglobulinemia, and anti-SSA and anti-SSB antibodies.² Laboratory tests that confirm a diagnosis of SS include antinuclear antibody (ANA), anti-Ro/SSA, and anti-La/SSB antibodies.² Pulmonary function test (PFT) results appear to reflect impairment of either the lung (restrictive syndrome) or airways (obstructive syndrome).² Imaging abnormalities may include ground-glass attenuation, subpleural small nodules, nonseptal linear opacities, interlobular septal thickening, bronchiectasis, and cysts.³ Therefore, many ILD cases show similar imaging and pathologic findings; nevertheless, they have identifiable etiology that are not idiopathic.
Case Presentation
A 67-year-old man with a medical history of hypertension, peripheral vascular disease, and keratoconjunctivitis sicca (treated with eye drops) developed progressive shortness of breath, dyspnea on exertion, and weight loss (40 pounds) over the course of 6 months. A pulmonary function test showed a restrictive abnormality with decreased diffusing capacity of the lungs for carbon monoxide (eFigure available online at www.fedprac.com). A chest computed tomography (CT) scan showed the presence of significant thickening of the interlobular septi that was more pronounced in the subpleural regions of the lungs and lower lobes, which was consistent with usual interstitial pneumonia. A chest X-ray conducted 4 months prior showed no significant acute cardiopulmonary abnormalities (Figure 1). An open lung wedge biopsy revealed chronic organizing pneumonia with mild interstitial chronic inflammation, smooth muscle hypertrophy, and honeycomb changes consistent with usual interstitial pneumonia. 
The patient had been diagnosed with idiopathic pulmonary fibrosis (IPF) by a private physician and started pirfenidone and oxygen therapy. Three months later the patient presented to the VA Caribbean Healthcare System in San Juan Puerto Rico when he developed an exacerbation of IPF. The patient reported having fever, chills, dry cough, night sweats, and marked shortness of breath. He was found hypoxemic (partial pressure of O2 was 50 mm Hg) and required a venturi mask set to 50% fractioned of inspired O2 to maintain a peripheral oxygen saturation around 90%. A chest X-ray showed decreased lung volume with bilateral interstitial and alveolar disease (Figure 2). Leukocytosis was present at 17×10-3/µl. The chest CT scan showed interval worsening of diffuse ground-glass airspace opacities and worsening of interstitial opacities; there 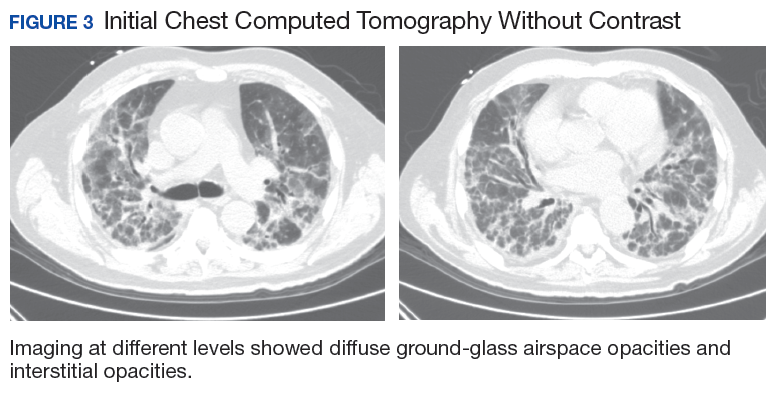
After careful clinical assessment (+ dry eyes) and radiographic pattern evaluation (diffuse bilateral interstitial and ground-glass opacities), the clinical diagnosis of IPF was queried after the patient’s rheumatologic workup came back positive for ANA and anti-Ro/SSA tests. Since the etiology of ILD was secondary to SS, pirfenidone was discontinued, and the patient was started on steroid therapy with subsequent marked clinical improvement. Parotid biopsy revealed the presence of inflammatory cells supporting the diagnosis of ILD associated to SS. The patient was discharged home on a tapering dose of steroids. Four months after therapy with steroids, a follow-up chest CT scan without contrast showed a chronic ILD with improved ground-glass opacities (Figure 4). The patient currently is in good health without oxygen supplementation.
Discussion
Diagnosis of SS is challenging, since it may mimic other conditions such as IPF. The most common type of SS-associated ILD is nonspecific interstitial pneumonia (NSIP), although usual interstitial pneumonia (UIP) can be visualized, as in this case study. Usual interstitial pneumonia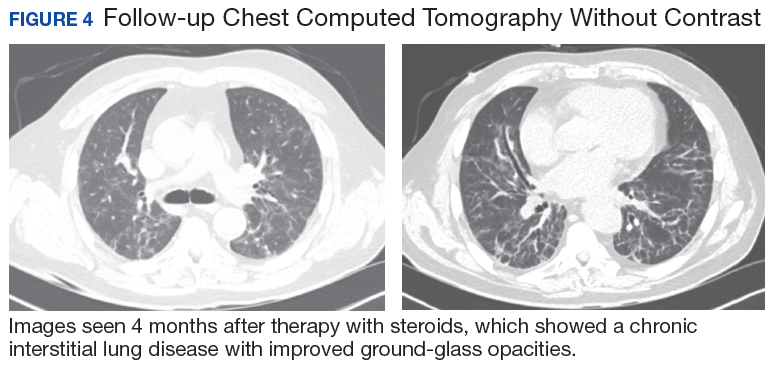
Diagnosing IPF cannot be solely based on a lung biopsy consistent with UIP. Appropriate diagnosis should consider the clinical presentation; PFT, laboratory findings (including rheumatologic workup), imaging (especially radiographic patterns), and biopsies. Moreover, the pathologic characteristic of IPF, which is UIP, can be found with other diseases, such as SS. Thus, it is important to make an accurate diagnosis to provide the appropriate treatment available. Patients with ILD associated with SS who have worsening symptoms, PFT, and radiographic abnormalities may be treated with oral prednisone (daily dose: 1 mg/kg).
Conclusion
This case highlights the importance of making an adequate diagnosis of ILD considering that available treatments differ for all possible etiologies other than IPF. This is a true clinical concern taking into account that many patients might be receiving inappropriate therapy for IPF diagnosis, as illustrated in the case study.
1. Ito I, Nagai S, Kitaichi M, et al. Pulmonary manifestations of primary Sjogren’s syndrome: a clinical, radiologic, and pathologic study. Am J Respir Crit Care Med. 2005;171(6):632-638.
2. Flament T, Bigot A, Chaigne B, Henique H, Diot E, Marchand-Adam S. Pulmonary manifestations of Sjögren’s syndrome. Eur Respir Rev. 2016;25(140):110-123.
3. Koyama M, Johkoh T, Honda O, et al. Pulmonary involvement in primary Sjögren’s syndrome: spectrum of pulmonary abnormalities and computed tomography findings in 60 patients. J Thorac Imaging. 2001;16(4):290-296.
4. Wuyts WA, Cavazza A, Rossi G, Bonella F, Sverzellati N, Spagnolo P. Differential diagnosis of usual interstitial pneumonia: when is it truly idiopathic? Eur Respir Rev. 2014;23(133):308-319.
Sjogren syndrome (SS) is a chronic inflammatory autoimmune disorder characterized by lymphocytic infiltration of lacrimal and salivary glands causing sicca syndrome.¹ The disease can extend beyond the exocrine glands, and systemic manifestations, including vasculitis, lung, renal or neurologic involvement, can occur.² Lung disease associated with SS is more commonly seen in women aged ≥ 60 years. The most common symptoms include dry cough, chest pain, and dyspnea on exertion. Sjogren syndrome also may produce several respiratory complications, including bronchial hyperresponsiveness, bronchiolitis, bronchiectasis, pulmonary infections, pulmonary amyloidosis, pulmonary embolism, pulmonary hypertension, lymphomas, and interstitial lung diseases (ILD).² Although ILD typically occurs 5 to 10 years after the onset of SS, lung disease can precede SS.
Pulmonary involvement is associated with systemic manifestations, hypergammaglobulinemia, and anti-SSA and anti-SSB antibodies.² Laboratory tests that confirm a diagnosis of SS include antinuclear antibody (ANA), anti-Ro/SSA, and anti-La/SSB antibodies.² Pulmonary function test (PFT) results appear to reflect impairment of either the lung (restrictive syndrome) or airways (obstructive syndrome).² Imaging abnormalities may include ground-glass attenuation, subpleural small nodules, nonseptal linear opacities, interlobular septal thickening, bronchiectasis, and cysts.³ Therefore, many ILD cases show similar imaging and pathologic findings; nevertheless, they have identifiable etiology that are not idiopathic.
Case Presentation
A 67-year-old man with a medical history of hypertension, peripheral vascular disease, and keratoconjunctivitis sicca (treated with eye drops) developed progressive shortness of breath, dyspnea on exertion, and weight loss (40 pounds) over the course of 6 months. A pulmonary function test showed a restrictive abnormality with decreased diffusing capacity of the lungs for carbon monoxide (eFigure available online at www.fedprac.com). A chest computed tomography (CT) scan showed the presence of significant thickening of the interlobular septi that was more pronounced in the subpleural regions of the lungs and lower lobes, which was consistent with usual interstitial pneumonia. A chest X-ray conducted 4 months prior showed no significant acute cardiopulmonary abnormalities (Figure 1). An open lung wedge biopsy revealed chronic organizing pneumonia with mild interstitial chronic inflammation, smooth muscle hypertrophy, and honeycomb changes consistent with usual interstitial pneumonia.
The patient had been diagnosed with idiopathic pulmonary fibrosis (IPF) by a private physician and started pirfenidone and oxygen therapy. Three months later the patient presented to the VA Caribbean Healthcare System in San Juan Puerto Rico when he developed an exacerbation of IPF. The patient reported having fever, chills, dry cough, night sweats, and marked shortness of breath. He was found hypoxemic (partial pressure of O2 was 50 mm Hg) and required a venturi mask set to 50% fractioned of inspired O2 to maintain a peripheral oxygen saturation around 90%. A chest X-ray showed decreased lung volume with bilateral interstitial and alveolar disease (Figure 2). Leukocytosis was present at 17×10-3/µl. The chest CT scan showed interval worsening of diffuse ground-glass airspace opacities and worsening of interstitial opacities; there
After careful clinical assessment (+ dry eyes) and radiographic pattern evaluation (diffuse bilateral interstitial and ground-glass opacities), the clinical diagnosis of IPF was queried after the patient’s rheumatologic workup came back positive for ANA and anti-Ro/SSA tests. Since the etiology of ILD was secondary to SS, pirfenidone was discontinued, and the patient was started on steroid therapy with subsequent marked clinical improvement. Parotid biopsy revealed the presence of inflammatory cells supporting the diagnosis of ILD associated to SS. The patient was discharged home on a tapering dose of steroids. Four months after therapy with steroids, a follow-up chest CT scan without contrast showed a chronic ILD with improved ground-glass opacities (Figure 4). The patient currently is in good health without oxygen supplementation.
Discussion
Diagnosis of SS is challenging, since it may mimic other conditions such as IPF. The most common type of SS-associated ILD is nonspecific interstitial pneumonia (NSIP), although usual interstitial pneumonia (UIP) can be visualized, as in this case study. Usual interstitial pneumonia
Diagnosing IPF cannot be solely based on a lung biopsy consistent with UIP. Appropriate diagnosis should consider the clinical presentation; PFT, laboratory findings (including rheumatologic workup), imaging (especially radiographic patterns), and biopsies. Moreover, the pathologic characteristic of IPF, which is UIP, can be found with other diseases, such as SS. Thus, it is important to make an accurate diagnosis to provide the appropriate treatment available. Patients with ILD associated with SS who have worsening symptoms, PFT, and radiographic abnormalities may be treated with oral prednisone (daily dose: 1 mg/kg).
Conclusion
This case highlights the importance of making an adequate diagnosis of ILD considering that available treatments differ for all possible etiologies other than IPF. This is a true clinical concern taking into account that many patients might be receiving inappropriate therapy for IPF diagnosis, as illustrated in the case study.
Sjogren syndrome (SS) is a chronic inflammatory autoimmune disorder characterized by lymphocytic infiltration of lacrimal and salivary glands causing sicca syndrome.¹ The disease can extend beyond the exocrine glands, and systemic manifestations, including vasculitis, lung, renal or neurologic involvement, can occur.² Lung disease associated with SS is more commonly seen in women aged ≥ 60 years. The most common symptoms include dry cough, chest pain, and dyspnea on exertion. Sjogren syndrome also may produce several respiratory complications, including bronchial hyperresponsiveness, bronchiolitis, bronchiectasis, pulmonary infections, pulmonary amyloidosis, pulmonary embolism, pulmonary hypertension, lymphomas, and interstitial lung diseases (ILD).² Although ILD typically occurs 5 to 10 years after the onset of SS, lung disease can precede SS.
Pulmonary involvement is associated with systemic manifestations, hypergammaglobulinemia, and anti-SSA and anti-SSB antibodies.² Laboratory tests that confirm a diagnosis of SS include antinuclear antibody (ANA), anti-Ro/SSA, and anti-La/SSB antibodies.² Pulmonary function test (PFT) results appear to reflect impairment of either the lung (restrictive syndrome) or airways (obstructive syndrome).² Imaging abnormalities may include ground-glass attenuation, subpleural small nodules, nonseptal linear opacities, interlobular septal thickening, bronchiectasis, and cysts.³ Therefore, many ILD cases show similar imaging and pathologic findings; nevertheless, they have identifiable etiology that are not idiopathic.
Case Presentation
A 67-year-old man with a medical history of hypertension, peripheral vascular disease, and keratoconjunctivitis sicca (treated with eye drops) developed progressive shortness of breath, dyspnea on exertion, and weight loss (40 pounds) over the course of 6 months. A pulmonary function test showed a restrictive abnormality with decreased diffusing capacity of the lungs for carbon monoxide (eFigure available online at www.fedprac.com). A chest computed tomography (CT) scan showed the presence of significant thickening of the interlobular septi that was more pronounced in the subpleural regions of the lungs and lower lobes, which was consistent with usual interstitial pneumonia. A chest X-ray conducted 4 months prior showed no significant acute cardiopulmonary abnormalities (Figure 1). An open lung wedge biopsy revealed chronic organizing pneumonia with mild interstitial chronic inflammation, smooth muscle hypertrophy, and honeycomb changes consistent with usual interstitial pneumonia.
The patient had been diagnosed with idiopathic pulmonary fibrosis (IPF) by a private physician and started pirfenidone and oxygen therapy. Three months later the patient presented to the VA Caribbean Healthcare System in San Juan Puerto Rico when he developed an exacerbation of IPF. The patient reported having fever, chills, dry cough, night sweats, and marked shortness of breath. He was found hypoxemic (partial pressure of O2 was 50 mm Hg) and required a venturi mask set to 50% fractioned of inspired O2 to maintain a peripheral oxygen saturation around 90%. A chest X-ray showed decreased lung volume with bilateral interstitial and alveolar disease (Figure 2). Leukocytosis was present at 17×10-3/µl. The chest CT scan showed interval worsening of diffuse ground-glass airspace opacities and worsening of interstitial opacities; there
After careful clinical assessment (+ dry eyes) and radiographic pattern evaluation (diffuse bilateral interstitial and ground-glass opacities), the clinical diagnosis of IPF was queried after the patient’s rheumatologic workup came back positive for ANA and anti-Ro/SSA tests. Since the etiology of ILD was secondary to SS, pirfenidone was discontinued, and the patient was started on steroid therapy with subsequent marked clinical improvement. Parotid biopsy revealed the presence of inflammatory cells supporting the diagnosis of ILD associated to SS. The patient was discharged home on a tapering dose of steroids. Four months after therapy with steroids, a follow-up chest CT scan without contrast showed a chronic ILD with improved ground-glass opacities (Figure 4). The patient currently is in good health without oxygen supplementation.
Discussion
Diagnosis of SS is challenging, since it may mimic other conditions such as IPF. The most common type of SS-associated ILD is nonspecific interstitial pneumonia (NSIP), although usual interstitial pneumonia (UIP) can be visualized, as in this case study. Usual interstitial pneumonia
Diagnosing IPF cannot be solely based on a lung biopsy consistent with UIP. Appropriate diagnosis should consider the clinical presentation; PFT, laboratory findings (including rheumatologic workup), imaging (especially radiographic patterns), and biopsies. Moreover, the pathologic characteristic of IPF, which is UIP, can be found with other diseases, such as SS. Thus, it is important to make an accurate diagnosis to provide the appropriate treatment available. Patients with ILD associated with SS who have worsening symptoms, PFT, and radiographic abnormalities may be treated with oral prednisone (daily dose: 1 mg/kg).
Conclusion
This case highlights the importance of making an adequate diagnosis of ILD considering that available treatments differ for all possible etiologies other than IPF. This is a true clinical concern taking into account that many patients might be receiving inappropriate therapy for IPF diagnosis, as illustrated in the case study.
1. Ito I, Nagai S, Kitaichi M, et al. Pulmonary manifestations of primary Sjogren’s syndrome: a clinical, radiologic, and pathologic study. Am J Respir Crit Care Med. 2005;171(6):632-638.
2. Flament T, Bigot A, Chaigne B, Henique H, Diot E, Marchand-Adam S. Pulmonary manifestations of Sjögren’s syndrome. Eur Respir Rev. 2016;25(140):110-123.
3. Koyama M, Johkoh T, Honda O, et al. Pulmonary involvement in primary Sjögren’s syndrome: spectrum of pulmonary abnormalities and computed tomography findings in 60 patients. J Thorac Imaging. 2001;16(4):290-296.
4. Wuyts WA, Cavazza A, Rossi G, Bonella F, Sverzellati N, Spagnolo P. Differential diagnosis of usual interstitial pneumonia: when is it truly idiopathic? Eur Respir Rev. 2014;23(133):308-319.
1. Ito I, Nagai S, Kitaichi M, et al. Pulmonary manifestations of primary Sjogren’s syndrome: a clinical, radiologic, and pathologic study. Am J Respir Crit Care Med. 2005;171(6):632-638.
2. Flament T, Bigot A, Chaigne B, Henique H, Diot E, Marchand-Adam S. Pulmonary manifestations of Sjögren’s syndrome. Eur Respir Rev. 2016;25(140):110-123.
3. Koyama M, Johkoh T, Honda O, et al. Pulmonary involvement in primary Sjögren’s syndrome: spectrum of pulmonary abnormalities and computed tomography findings in 60 patients. J Thorac Imaging. 2001;16(4):290-296.
4. Wuyts WA, Cavazza A, Rossi G, Bonella F, Sverzellati N, Spagnolo P. Differential diagnosis of usual interstitial pneumonia: when is it truly idiopathic? Eur Respir Rev. 2014;23(133):308-319.
Panniculitis, Pancreatitis, and Polyarthritis: A Rare Clinical Syndrome
Pancreatic panniculitis is a rare disease contributing to widespread fat necrosis in patients with underlying pancreatic disorders. This entity was first described in 1883,1 but it was not until 1947 that it was reported in the English-language literature.2 Patients with pancreatitis infrequently develop extrapancreatic manifestations. It has been estimated that only 2% to 3% of patients worldwide with an underlying pancreatic disease develop cutaneous lesions.3 Patients who develop pancreatic panniculitis typically present with tender, edematous, erythematous to brown, subcutaneous nodules on the lower legs with the tendency for spontaneous ulceration. Lesions tend to exude a viscous, yellow-brown, oily substance that represents liquefactive necrosis of enzymatic fat in subcutaneous tissue. Cutaneous lesions may precede, occur simultaneously, or follow the development of an underlying pancreatic disorder. Rarely, patients may develop inflammatory arthritis secondary to intraosseous fat necrosis, completing the triad of findings diagnostic for panniculitis, pancreatitis, and polyarthritis (PPP) syndrome. Although the underlying pancreatic pathology may vary, roughly 80% of cases worldwide have acute/chronic pancreatitis or pancreatic carcinoma, most commonly acinar cell carcinoma.4-6 Less common pancreatic disorders include pancreatic pseudocyst, pancreatic divisum, and vascular pancreatic fistulas.7 Narváez et al8 found that of the 25 cases of PPP syndrome reported in the literature, 68% (17/25) were men, 32% (8/25) were women, 56% (14/25) were younger than 50 years, and 64% (16/25) had a history of prior or current alcohol abuse.
Case Report
A 68-year-old man with a history of hypertension, gastroesophageal reflux disease, chronic pancreatitis of unknown etiology, and arthritis presented to our clinic for evaluation of painful skin nodules on the lower legs of 8 months’ duration, in addition to joint pain and swelling of the metacarpophalangeal (MCP), metatarsophalangeal, and ankle joints. He had a history of numerous hospital admissions over the last 2 years for pancreatitis and was being managed by the rheumatology department for arthritic symptoms.
Physical examination revealed multiple 1- to 4-cm, ill-defined, erythematous to brown, subcutaneous nodules on the bilateral lower legs (Figure 1) and right inferomedial thigh that were tender to palpation. Marked erythema and edema of the MCP and metatarsophalangeal joints (Figure 2) and bilateral ankles were observed. Diffuse 2+ pitting edema was present in the bilateral lower extremities, along with areas of hyperpigmentation overlying resolving lesions.

Laboratory data revealed an elevated lipase level (>16,000 U/L [reference range, 31–186 U/L]), amylase level (>4700 U/L [reference range, 27–131 U/L]), erythrocyte sedimentation rate (94 mm/h [reference range, 0–20 mm/h]), and C-reactive protein level (93.5 mg/L [0.08–3.1 mg/L]). The patient had more than 6 episodes of recurrent idiopathic pancreatitis over the last 2 years, though symptoms of abdominal pain were minimal to nonexistent. Liver function tests and alcohol, calcium, and triglyceride levels all were within reference range. Rheumatoid factor and antinuclear antibodies were negative.
Ultrasonography showed no evidence of cholelithiasis. Computed tomography of the abdomen and pelvis demonstrated a 1.8×1.4-cm hypodense lesion within the pancreatic head with calcifications and mild proximal pancreatic ductal dilatation (Figure 3). However, multiple magnetic resonance cholangiopancreatography examinations and endoscopic ultrasounds with fine-needle aspiration specimens were performed, all negative for malignancy. Computed tomography of the left ankle demonstrated evidence of bony cortical destruction in the lateral aspect of the posterior calcaneus. Bone biopsy specimens demonstrated mild chronic inflammation with no evidence of osteomyelitis. A serum uric acid level was found to be 4.4 mg/dL (reference range, 4.0–8.0 mg/dL) and a joint aspirate demonstrated turbid fluid with lipoid material and no evidence of crystals or organisms on culture. Furthermore, a 4-mm punch biopsy of a nodule on the right leg revealed extensive lobular and septal liquefactive adipocyte necrosis with scattered neutrophils and lymphocytes (Figure 4). Aggregates of fine granular basophilic material were observed with prominent adipocyte degeneration and calcification.

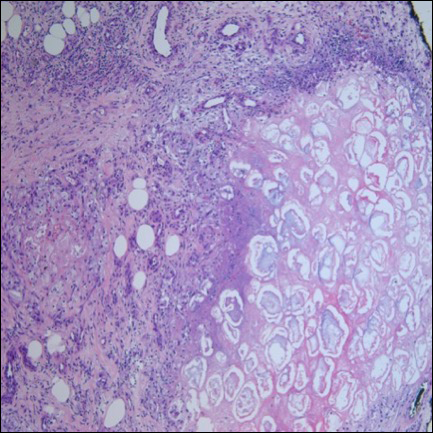
Symptomatic treatment with nonsteroidal anti-inflammatory drugs (NSAIDs) along with intralesional, topical, and oral corticosteroids had proven ineffective in the management of this patient. He was subsequently referred to the surgery department for a pancreaticoduodenectomy (Whipple procedure) with notable improvement in pancreatic enzyme levels, lower leg subcutaneous nodules, and arthritis weeks after surgery.
Comment
A triad of pancreatic panniculitis, pancreatitis, and polyarthritis characterizes a rare entity known as PPP syndrome. Pancreatic panniculitis is a rare form of subcutaneous lobular fat necrosis associated with various underlying pancreatic disorders. Approximately 0.3% to 3.0% of patients with an underlying pancreatic disorder are affected with pancreatic panniculitis.9 Pancreatic panniculitis has been found in roughly 2% to 3% of patients with acute or chronic pancreatitis and pancreatic carcinoma, most commonly the acinar cell type.10 Narváez et al8 reported that nearly two-thirds of patients diagnosed with PPP syndrome have minimal to absent abdominal symptoms that often lead to misdiagnosis and affect the overall prognosis of patients with pancreatic disease. Any delay in the diagnosis of PPP syndrome leads to a worse prognosis, with a mortality rate reported to be approximately 24%.8 Potts et al5 provided a review of 27 patients with pancreatic panniculitis in which all 8 patients with pancreatic carcinoma and 42% (8/19) of patients with pancreatitis died.
Pancreatic panniculitis in the setting of PPP syndrome commonly presents with erythematous to brown, exquisitely tender, edematous, subcutaneous nodules on the lower legs. Lesions can range in size from several millimeters to 5 cm. The subcutaneous nodules may spontaneously ulcerate and exude oily viscous material from the liquefactive necrosis of adipocytes. In approximately 40% of patients, skin lesions are the presenting feature.11 Lesions typically resolve only after the pancreatic inflammation regresses, leaving behind atrophic hyperpigmented scars.3 Other presenting symptoms may include joint pain, pitting edema, and subcutaneous nodules, which can precede the diagnosis by up to 9 months.
The exact pathogenesis of PPP syndrome remains unclear. The most widely recognized hypothesis suggests that pancreatic enzymes (eg, trypsin, amylase, lipase, phospholipase A) released from the damaged pancreas are transported through the bloodstream to distant visceral and soft tissue sites, leading to lipolysis and inflammation to the surrounding subcutis and bone marrow.3 Ferrari et al12 reported this effect as a product of the accumulation of high levels of free fatty acids within the joint space by the action of lipolytic pancreatic enzymes on adipose cell membranes, resulting in acute arthritis.
Histopathologic findings of pancreatic panniculitis vary based on the acuity of the disease. Acute lesions typically demonstrate lobular and septal panniculitis. Szymanski and Bluefarb13 described the pathognomonic histologic findings of focal liquefactive necrosis and anucleate necrotic adipocytes surrounded by a shadowy and thickened cell membrane signifying the characteristic ghost cells. Fine basophilic material also may be seen intermixed with the necrotic adipocytes, representing saponified calcium. A brisk inflammatory infiltrate involving lymphocytes, macrophages, and neutrophils tends to surround the areas of necrotic adipocytes. Chronic lesions often demonstrate a paucity of fat necrosis and ghost cells and more granulomatous infiltrate. Langerhans giant cells, macrophages, and lymphocytes predominate in the subcutaneous fat.
Laboratory findings associated with pancreatic panniculitis may include elevated serum amylase, lipase, and/or trypsin levels. Not all the enzymes have to be elevated simultaneously. On occasion, one enzyme may be within reference range while the others are elevated. Rarely, patients may have an elevated lipase level with no signs of underlying pancreatic disease, which demonstrates that panniculitis does not correlate with the enzyme levels. In all cases of suspected pancreatic panniculitis, a complete laboratory workup is recommended including lipase, amylase, and trypsin serum levels. Eosinophilia may be a prominent finding in patients with pancreatic panniculitis and tends to occur in association with an underlying pancreatic carcinoma. Patients with pancreatic panniculitis associated with pancreatic carcinoma tend to have more severe, diffuse, and persistent subcutaneous nodules that often are refractory to treatment with frequent recurrence. A rare constellation of findings known as Schmid triad is comprised of panniculitis, polyarthritis, and eosinophilia and typically portends a poor prognosis secondary to an underlying pancreatic tumor.14 Cutaneous nodules may predate the diagnosis of pancreatic carcinoma by several months, thus signifying the need for a high index of suspicion in patients with lower leg subcutaneous nodules.
Joint disease most commonly involves the ankles, knees, wrists, and MCP joints.5,6,11 It has been suggested that arthritic symptoms are from periarticular fat necrosis or a direct extension from the necrotic subcutaneous tissue to the adjacent joint space.15 Dahl et al3 reported the composition of joint effusion fluid in 3 patients with PPP syndrome. The aspirate in all 3 patients contained viscous yellow material similar to the necrotic adipose tissue seen draining from subcutaneous nodules. Joint aspirate analysis demonstrated increased concentration of free fatty acids in the joint fluid consistent with severe lipolysis.3
The PPP syndrome acronym may be misleading to physicians, as arthritis is not always polyarticular. Dahl et al3 reported that monoarticular or oligoarticular arthritic symptoms were present in 56% of patients studied. In rare cases, the arthritic symptoms antedated the diagnosis of clinically asymptomatic pancreatic disease. Arthritis can be either symmetric or asymmetric and infrequently follows a chronic course, leading to radiographic lytic lesions and symptoms that often are unresponsive to conventional therapy.16
Treatment of PPP syndrome is largely supportive, with a focus on correcting the underlying pancreatic disease. It is imperative to identify any complicating factors contributing to high levels of circulating pancreatic enzymes. Pseudocysts must be addressed if discovered in these patients, as they often perpetuate the substantial release of pancreatic enzymes into the serum, leading to characteristic subcutaneous fat necrosis and arthritis. Sepsis also is a concern, likely secondary to bacterial colonization of the ulcerated subcutaneous nodules and compromised skin barrier. Nonsteroidal anti-inflammatory drugs and corticosteroids have been used for symptomatic relief but usually are ineffective and have not been shown to reduce the duration of the disease.12,16 Octreotide has been utilized and may potentially reduce pancreatic enzyme secretion leading to improvement in cutaneous and musculoskeletal lesions.17 Plasmapheresis has been used as an adjuvant treatment in patients with persistent hyperamylasemia and hyperlipasemia, but reports are anecdotal. Often reserved for severe disease, cholecystectomy, pancreatic duct removal, and pancreaticoduodenectomy have demonstrated success in the management of chronic pancreatitis and panniculitis. Dahl et al3 reported 2 cases in which cholecystectomy was performed with complete resolution of the skin and pancreatic disease. Our patient was initially treated symptomatically with NSAIDs and corticosteroids but there was no clinical response. The patient eventually underwent a pancreaticoduodenectomy 9 months after the onset of symptoms with complete resolution of joint pain and swelling, greater than 50% resolution of his lower leg subcutaneous nodules, and remarkable reduction in amylase and lipase levels on 1-month follow-up.
Conclusion
Panniculitis, pancreatitis, and polyarthritis syndrome is a rare diagnosis characterized by a triad of pancreatic panniculitis, pancreatitis, and polyarthritis. Adjuvant therapies for PPP syndrome, such as NSAIDs, corticosteroids, plasmapheresis, and octreotide, have been used with equivocal results, but definitive treatment requires correction of the primary pancreatic disorder. More importantly, many pancreatic diseases can cause pancreatic panniculitis, but extensive, refractory, or ulcerated cases could be an early indicator of an occult pancreatic malignancy and should prompt early evaluation with a multidisciplinary approach. This approach should incorporate management from dermatology, internal medicine, rheumatology, gastroenterology, surgery, and primary care.
- Chiari H. Uber die Sogenannte Fettnekrose. Prag Med Wochenschr. 1883;8:285-286, 299-301.
- Blauvelt H. Case of acute pancreatitis with subcutaneous fat necrosis. Br J Surg. 1946;34:207-208.
- Dahl PR, Su D, Cullimore KC, et al. Pancreatic panniculitis. J Am Acad Dermatol. 1995;33:413-417.
- Mullen GT, Caperton EM Jr, Crespin SR, et al. Arthritis and skin lesions resembling erythema nodosum in pancreatic disease. Ann Intern Med. 1968;68:75-87.
- Potts DE, Mass MF, Iseman MD. Syndrome and pancreatic disease, subcutaneous fat necrosis and polyserositis: case report and review of literature. Am J Med. 1975;58:417-423.
- Sorensen EV. Subcutaneous fat necrosis in pancreatic disease: a review and two new case reports. J Clin Gastroenterol. 1988;10:71-75.
- García-Romero D, Vanaclocha F. Pancreatic panniculitis. Dermatol Clin. 2008;26:465-470.
- Narváez J, Bianchi M, Santo P, et al. Pancreatitis, panniculitis, and polyarthritis. Semin Arthritis Rheum. 2010;39:417-423.
- Rongioetti F, Caputo V. Pancreatic panniculitis. G Ital Dermatol Venereol. 2013;148:419-425.
- Poelman SM, Nguyen K. Pancreatic panniculitis associated with acinar cell pancreatic carcinoma. J Cutan Med Surg. 2008;12:38-42.
- Hughes SH, Apisarnthanarax P, Mullins F. Subcutaneous fat necrosis associated with pancreatic disease. Arch Dermatol. 1975:111:506-510.
- Ferrari R, Wendelboe M, Ford PM, et al. Pancreatitis arthritis with periarticular fat necrosis. J Rheumatol. 1993;20:1436-1437.
- Szymanski FJ, Bluefarb SM. Nodular fat necrosis and pancreatic diseases. Arch Dermatol. 1961;83:224-229.
- Beltraminelly HS, Buechner SA, Hausermann P. Pancreatic panniculitis in a patient with an acinar cell cystadenocarcinoma of the pancreas. Dermatology. 2004;208:265-267.
- Burns WA, Matthews MJ, Hamosh M, et al. Lipase-secreting acinar cell carcinoma of the pancreas with polyarthropathy: a light and electron microscopic, histochemical, and biochemical study. Cancer. 1974;33:1002-1009.
- Baron M, Paltiel H, Lander P. Aseptic necrosis of the talus and calcaneal insufficiency fractures in a patient with pancreatitis, subcutaneous fat necrosis, and arthritis. Arthritis Rheum. 1984;27:1309-1313.
- Zundler S, Erber R, Agaimy A, et al. Pancreatic panniculitis in a patient with pancreatic-type acinar cell carcinoma of the liver—case report and review of literature. BMC Cancer. 2016;16:130.
Pancreatic panniculitis is a rare disease contributing to widespread fat necrosis in patients with underlying pancreatic disorders. This entity was first described in 1883,1 but it was not until 1947 that it was reported in the English-language literature.2 Patients with pancreatitis infrequently develop extrapancreatic manifestations. It has been estimated that only 2% to 3% of patients worldwide with an underlying pancreatic disease develop cutaneous lesions.3 Patients who develop pancreatic panniculitis typically present with tender, edematous, erythematous to brown, subcutaneous nodules on the lower legs with the tendency for spontaneous ulceration. Lesions tend to exude a viscous, yellow-brown, oily substance that represents liquefactive necrosis of enzymatic fat in subcutaneous tissue. Cutaneous lesions may precede, occur simultaneously, or follow the development of an underlying pancreatic disorder. Rarely, patients may develop inflammatory arthritis secondary to intraosseous fat necrosis, completing the triad of findings diagnostic for panniculitis, pancreatitis, and polyarthritis (PPP) syndrome. Although the underlying pancreatic pathology may vary, roughly 80% of cases worldwide have acute/chronic pancreatitis or pancreatic carcinoma, most commonly acinar cell carcinoma.4-6 Less common pancreatic disorders include pancreatic pseudocyst, pancreatic divisum, and vascular pancreatic fistulas.7 Narváez et al8 found that of the 25 cases of PPP syndrome reported in the literature, 68% (17/25) were men, 32% (8/25) were women, 56% (14/25) were younger than 50 years, and 64% (16/25) had a history of prior or current alcohol abuse.
Case Report
A 68-year-old man with a history of hypertension, gastroesophageal reflux disease, chronic pancreatitis of unknown etiology, and arthritis presented to our clinic for evaluation of painful skin nodules on the lower legs of 8 months’ duration, in addition to joint pain and swelling of the metacarpophalangeal (MCP), metatarsophalangeal, and ankle joints. He had a history of numerous hospital admissions over the last 2 years for pancreatitis and was being managed by the rheumatology department for arthritic symptoms.
Physical examination revealed multiple 1- to 4-cm, ill-defined, erythematous to brown, subcutaneous nodules on the bilateral lower legs (Figure 1) and right inferomedial thigh that were tender to palpation. Marked erythema and edema of the MCP and metatarsophalangeal joints (Figure 2) and bilateral ankles were observed. Diffuse 2+ pitting edema was present in the bilateral lower extremities, along with areas of hyperpigmentation overlying resolving lesions.
Laboratory data revealed an elevated lipase level (>16,000 U/L [reference range, 31–186 U/L]), amylase level (>4700 U/L [reference range, 27–131 U/L]), erythrocyte sedimentation rate (94 mm/h [reference range, 0–20 mm/h]), and C-reactive protein level (93.5 mg/L [0.08–3.1 mg/L]). The patient had more than 6 episodes of recurrent idiopathic pancreatitis over the last 2 years, though symptoms of abdominal pain were minimal to nonexistent. Liver function tests and alcohol, calcium, and triglyceride levels all were within reference range. Rheumatoid factor and antinuclear antibodies were negative.
Ultrasonography showed no evidence of cholelithiasis. Computed tomography of the abdomen and pelvis demonstrated a 1.8×1.4-cm hypodense lesion within the pancreatic head with calcifications and mild proximal pancreatic ductal dilatation (Figure 3). However, multiple magnetic resonance cholangiopancreatography examinations and endoscopic ultrasounds with fine-needle aspiration specimens were performed, all negative for malignancy. Computed tomography of the left ankle demonstrated evidence of bony cortical destruction in the lateral aspect of the posterior calcaneus. Bone biopsy specimens demonstrated mild chronic inflammation with no evidence of osteomyelitis. A serum uric acid level was found to be 4.4 mg/dL (reference range, 4.0–8.0 mg/dL) and a joint aspirate demonstrated turbid fluid with lipoid material and no evidence of crystals or organisms on culture. Furthermore, a 4-mm punch biopsy of a nodule on the right leg revealed extensive lobular and septal liquefactive adipocyte necrosis with scattered neutrophils and lymphocytes (Figure 4). Aggregates of fine granular basophilic material were observed with prominent adipocyte degeneration and calcification.
Symptomatic treatment with nonsteroidal anti-inflammatory drugs (NSAIDs) along with intralesional, topical, and oral corticosteroids had proven ineffective in the management of this patient. He was subsequently referred to the surgery department for a pancreaticoduodenectomy (Whipple procedure) with notable improvement in pancreatic enzyme levels, lower leg subcutaneous nodules, and arthritis weeks after surgery.
Comment
A triad of pancreatic panniculitis, pancreatitis, and polyarthritis characterizes a rare entity known as PPP syndrome. Pancreatic panniculitis is a rare form of subcutaneous lobular fat necrosis associated with various underlying pancreatic disorders. Approximately 0.3% to 3.0% of patients with an underlying pancreatic disorder are affected with pancreatic panniculitis.9 Pancreatic panniculitis has been found in roughly 2% to 3% of patients with acute or chronic pancreatitis and pancreatic carcinoma, most commonly the acinar cell type.10 Narváez et al8 reported that nearly two-thirds of patients diagnosed with PPP syndrome have minimal to absent abdominal symptoms that often lead to misdiagnosis and affect the overall prognosis of patients with pancreatic disease. Any delay in the diagnosis of PPP syndrome leads to a worse prognosis, with a mortality rate reported to be approximately 24%.8 Potts et al5 provided a review of 27 patients with pancreatic panniculitis in which all 8 patients with pancreatic carcinoma and 42% (8/19) of patients with pancreatitis died.
Pancreatic panniculitis in the setting of PPP syndrome commonly presents with erythematous to brown, exquisitely tender, edematous, subcutaneous nodules on the lower legs. Lesions can range in size from several millimeters to 5 cm. The subcutaneous nodules may spontaneously ulcerate and exude oily viscous material from the liquefactive necrosis of adipocytes. In approximately 40% of patients, skin lesions are the presenting feature.11 Lesions typically resolve only after the pancreatic inflammation regresses, leaving behind atrophic hyperpigmented scars.3 Other presenting symptoms may include joint pain, pitting edema, and subcutaneous nodules, which can precede the diagnosis by up to 9 months.
The exact pathogenesis of PPP syndrome remains unclear. The most widely recognized hypothesis suggests that pancreatic enzymes (eg, trypsin, amylase, lipase, phospholipase A) released from the damaged pancreas are transported through the bloodstream to distant visceral and soft tissue sites, leading to lipolysis and inflammation to the surrounding subcutis and bone marrow.3 Ferrari et al12 reported this effect as a product of the accumulation of high levels of free fatty acids within the joint space by the action of lipolytic pancreatic enzymes on adipose cell membranes, resulting in acute arthritis.
Histopathologic findings of pancreatic panniculitis vary based on the acuity of the disease. Acute lesions typically demonstrate lobular and septal panniculitis. Szymanski and Bluefarb13 described the pathognomonic histologic findings of focal liquefactive necrosis and anucleate necrotic adipocytes surrounded by a shadowy and thickened cell membrane signifying the characteristic ghost cells. Fine basophilic material also may be seen intermixed with the necrotic adipocytes, representing saponified calcium. A brisk inflammatory infiltrate involving lymphocytes, macrophages, and neutrophils tends to surround the areas of necrotic adipocytes. Chronic lesions often demonstrate a paucity of fat necrosis and ghost cells and more granulomatous infiltrate. Langerhans giant cells, macrophages, and lymphocytes predominate in the subcutaneous fat.
Laboratory findings associated with pancreatic panniculitis may include elevated serum amylase, lipase, and/or trypsin levels. Not all the enzymes have to be elevated simultaneously. On occasion, one enzyme may be within reference range while the others are elevated. Rarely, patients may have an elevated lipase level with no signs of underlying pancreatic disease, which demonstrates that panniculitis does not correlate with the enzyme levels. In all cases of suspected pancreatic panniculitis, a complete laboratory workup is recommended including lipase, amylase, and trypsin serum levels. Eosinophilia may be a prominent finding in patients with pancreatic panniculitis and tends to occur in association with an underlying pancreatic carcinoma. Patients with pancreatic panniculitis associated with pancreatic carcinoma tend to have more severe, diffuse, and persistent subcutaneous nodules that often are refractory to treatment with frequent recurrence. A rare constellation of findings known as Schmid triad is comprised of panniculitis, polyarthritis, and eosinophilia and typically portends a poor prognosis secondary to an underlying pancreatic tumor.14 Cutaneous nodules may predate the diagnosis of pancreatic carcinoma by several months, thus signifying the need for a high index of suspicion in patients with lower leg subcutaneous nodules.
Joint disease most commonly involves the ankles, knees, wrists, and MCP joints.5,6,11 It has been suggested that arthritic symptoms are from periarticular fat necrosis or a direct extension from the necrotic subcutaneous tissue to the adjacent joint space.15 Dahl et al3 reported the composition of joint effusion fluid in 3 patients with PPP syndrome. The aspirate in all 3 patients contained viscous yellow material similar to the necrotic adipose tissue seen draining from subcutaneous nodules. Joint aspirate analysis demonstrated increased concentration of free fatty acids in the joint fluid consistent with severe lipolysis.3
The PPP syndrome acronym may be misleading to physicians, as arthritis is not always polyarticular. Dahl et al3 reported that monoarticular or oligoarticular arthritic symptoms were present in 56% of patients studied. In rare cases, the arthritic symptoms antedated the diagnosis of clinically asymptomatic pancreatic disease. Arthritis can be either symmetric or asymmetric and infrequently follows a chronic course, leading to radiographic lytic lesions and symptoms that often are unresponsive to conventional therapy.16
Treatment of PPP syndrome is largely supportive, with a focus on correcting the underlying pancreatic disease. It is imperative to identify any complicating factors contributing to high levels of circulating pancreatic enzymes. Pseudocysts must be addressed if discovered in these patients, as they often perpetuate the substantial release of pancreatic enzymes into the serum, leading to characteristic subcutaneous fat necrosis and arthritis. Sepsis also is a concern, likely secondary to bacterial colonization of the ulcerated subcutaneous nodules and compromised skin barrier. Nonsteroidal anti-inflammatory drugs and corticosteroids have been used for symptomatic relief but usually are ineffective and have not been shown to reduce the duration of the disease.12,16 Octreotide has been utilized and may potentially reduce pancreatic enzyme secretion leading to improvement in cutaneous and musculoskeletal lesions.17 Plasmapheresis has been used as an adjuvant treatment in patients with persistent hyperamylasemia and hyperlipasemia, but reports are anecdotal. Often reserved for severe disease, cholecystectomy, pancreatic duct removal, and pancreaticoduodenectomy have demonstrated success in the management of chronic pancreatitis and panniculitis. Dahl et al3 reported 2 cases in which cholecystectomy was performed with complete resolution of the skin and pancreatic disease. Our patient was initially treated symptomatically with NSAIDs and corticosteroids but there was no clinical response. The patient eventually underwent a pancreaticoduodenectomy 9 months after the onset of symptoms with complete resolution of joint pain and swelling, greater than 50% resolution of his lower leg subcutaneous nodules, and remarkable reduction in amylase and lipase levels on 1-month follow-up.
Conclusion
Panniculitis, pancreatitis, and polyarthritis syndrome is a rare diagnosis characterized by a triad of pancreatic panniculitis, pancreatitis, and polyarthritis. Adjuvant therapies for PPP syndrome, such as NSAIDs, corticosteroids, plasmapheresis, and octreotide, have been used with equivocal results, but definitive treatment requires correction of the primary pancreatic disorder. More importantly, many pancreatic diseases can cause pancreatic panniculitis, but extensive, refractory, or ulcerated cases could be an early indicator of an occult pancreatic malignancy and should prompt early evaluation with a multidisciplinary approach. This approach should incorporate management from dermatology, internal medicine, rheumatology, gastroenterology, surgery, and primary care.
Pancreatic panniculitis is a rare disease contributing to widespread fat necrosis in patients with underlying pancreatic disorders. This entity was first described in 1883,1 but it was not until 1947 that it was reported in the English-language literature.2 Patients with pancreatitis infrequently develop extrapancreatic manifestations. It has been estimated that only 2% to 3% of patients worldwide with an underlying pancreatic disease develop cutaneous lesions.3 Patients who develop pancreatic panniculitis typically present with tender, edematous, erythematous to brown, subcutaneous nodules on the lower legs with the tendency for spontaneous ulceration. Lesions tend to exude a viscous, yellow-brown, oily substance that represents liquefactive necrosis of enzymatic fat in subcutaneous tissue. Cutaneous lesions may precede, occur simultaneously, or follow the development of an underlying pancreatic disorder. Rarely, patients may develop inflammatory arthritis secondary to intraosseous fat necrosis, completing the triad of findings diagnostic for panniculitis, pancreatitis, and polyarthritis (PPP) syndrome. Although the underlying pancreatic pathology may vary, roughly 80% of cases worldwide have acute/chronic pancreatitis or pancreatic carcinoma, most commonly acinar cell carcinoma.4-6 Less common pancreatic disorders include pancreatic pseudocyst, pancreatic divisum, and vascular pancreatic fistulas.7 Narváez et al8 found that of the 25 cases of PPP syndrome reported in the literature, 68% (17/25) were men, 32% (8/25) were women, 56% (14/25) were younger than 50 years, and 64% (16/25) had a history of prior or current alcohol abuse.
Case Report
A 68-year-old man with a history of hypertension, gastroesophageal reflux disease, chronic pancreatitis of unknown etiology, and arthritis presented to our clinic for evaluation of painful skin nodules on the lower legs of 8 months’ duration, in addition to joint pain and swelling of the metacarpophalangeal (MCP), metatarsophalangeal, and ankle joints. He had a history of numerous hospital admissions over the last 2 years for pancreatitis and was being managed by the rheumatology department for arthritic symptoms.
Physical examination revealed multiple 1- to 4-cm, ill-defined, erythematous to brown, subcutaneous nodules on the bilateral lower legs (Figure 1) and right inferomedial thigh that were tender to palpation. Marked erythema and edema of the MCP and metatarsophalangeal joints (Figure 2) and bilateral ankles were observed. Diffuse 2+ pitting edema was present in the bilateral lower extremities, along with areas of hyperpigmentation overlying resolving lesions.
Laboratory data revealed an elevated lipase level (>16,000 U/L [reference range, 31–186 U/L]), amylase level (>4700 U/L [reference range, 27–131 U/L]), erythrocyte sedimentation rate (94 mm/h [reference range, 0–20 mm/h]), and C-reactive protein level (93.5 mg/L [0.08–3.1 mg/L]). The patient had more than 6 episodes of recurrent idiopathic pancreatitis over the last 2 years, though symptoms of abdominal pain were minimal to nonexistent. Liver function tests and alcohol, calcium, and triglyceride levels all were within reference range. Rheumatoid factor and antinuclear antibodies were negative.
Ultrasonography showed no evidence of cholelithiasis. Computed tomography of the abdomen and pelvis demonstrated a 1.8×1.4-cm hypodense lesion within the pancreatic head with calcifications and mild proximal pancreatic ductal dilatation (Figure 3). However, multiple magnetic resonance cholangiopancreatography examinations and endoscopic ultrasounds with fine-needle aspiration specimens were performed, all negative for malignancy. Computed tomography of the left ankle demonstrated evidence of bony cortical destruction in the lateral aspect of the posterior calcaneus. Bone biopsy specimens demonstrated mild chronic inflammation with no evidence of osteomyelitis. A serum uric acid level was found to be 4.4 mg/dL (reference range, 4.0–8.0 mg/dL) and a joint aspirate demonstrated turbid fluid with lipoid material and no evidence of crystals or organisms on culture. Furthermore, a 4-mm punch biopsy of a nodule on the right leg revealed extensive lobular and septal liquefactive adipocyte necrosis with scattered neutrophils and lymphocytes (Figure 4). Aggregates of fine granular basophilic material were observed with prominent adipocyte degeneration and calcification.
Symptomatic treatment with nonsteroidal anti-inflammatory drugs (NSAIDs) along with intralesional, topical, and oral corticosteroids had proven ineffective in the management of this patient. He was subsequently referred to the surgery department for a pancreaticoduodenectomy (Whipple procedure) with notable improvement in pancreatic enzyme levels, lower leg subcutaneous nodules, and arthritis weeks after surgery.
Comment
A triad of pancreatic panniculitis, pancreatitis, and polyarthritis characterizes a rare entity known as PPP syndrome. Pancreatic panniculitis is a rare form of subcutaneous lobular fat necrosis associated with various underlying pancreatic disorders. Approximately 0.3% to 3.0% of patients with an underlying pancreatic disorder are affected with pancreatic panniculitis.9 Pancreatic panniculitis has been found in roughly 2% to 3% of patients with acute or chronic pancreatitis and pancreatic carcinoma, most commonly the acinar cell type.10 Narváez et al8 reported that nearly two-thirds of patients diagnosed with PPP syndrome have minimal to absent abdominal symptoms that often lead to misdiagnosis and affect the overall prognosis of patients with pancreatic disease. Any delay in the diagnosis of PPP syndrome leads to a worse prognosis, with a mortality rate reported to be approximately 24%.8 Potts et al5 provided a review of 27 patients with pancreatic panniculitis in which all 8 patients with pancreatic carcinoma and 42% (8/19) of patients with pancreatitis died.
Pancreatic panniculitis in the setting of PPP syndrome commonly presents with erythematous to brown, exquisitely tender, edematous, subcutaneous nodules on the lower legs. Lesions can range in size from several millimeters to 5 cm. The subcutaneous nodules may spontaneously ulcerate and exude oily viscous material from the liquefactive necrosis of adipocytes. In approximately 40% of patients, skin lesions are the presenting feature.11 Lesions typically resolve only after the pancreatic inflammation regresses, leaving behind atrophic hyperpigmented scars.3 Other presenting symptoms may include joint pain, pitting edema, and subcutaneous nodules, which can precede the diagnosis by up to 9 months.
The exact pathogenesis of PPP syndrome remains unclear. The most widely recognized hypothesis suggests that pancreatic enzymes (eg, trypsin, amylase, lipase, phospholipase A) released from the damaged pancreas are transported through the bloodstream to distant visceral and soft tissue sites, leading to lipolysis and inflammation to the surrounding subcutis and bone marrow.3 Ferrari et al12 reported this effect as a product of the accumulation of high levels of free fatty acids within the joint space by the action of lipolytic pancreatic enzymes on adipose cell membranes, resulting in acute arthritis.
Histopathologic findings of pancreatic panniculitis vary based on the acuity of the disease. Acute lesions typically demonstrate lobular and septal panniculitis. Szymanski and Bluefarb13 described the pathognomonic histologic findings of focal liquefactive necrosis and anucleate necrotic adipocytes surrounded by a shadowy and thickened cell membrane signifying the characteristic ghost cells. Fine basophilic material also may be seen intermixed with the necrotic adipocytes, representing saponified calcium. A brisk inflammatory infiltrate involving lymphocytes, macrophages, and neutrophils tends to surround the areas of necrotic adipocytes. Chronic lesions often demonstrate a paucity of fat necrosis and ghost cells and more granulomatous infiltrate. Langerhans giant cells, macrophages, and lymphocytes predominate in the subcutaneous fat.
Laboratory findings associated with pancreatic panniculitis may include elevated serum amylase, lipase, and/or trypsin levels. Not all the enzymes have to be elevated simultaneously. On occasion, one enzyme may be within reference range while the others are elevated. Rarely, patients may have an elevated lipase level with no signs of underlying pancreatic disease, which demonstrates that panniculitis does not correlate with the enzyme levels. In all cases of suspected pancreatic panniculitis, a complete laboratory workup is recommended including lipase, amylase, and trypsin serum levels. Eosinophilia may be a prominent finding in patients with pancreatic panniculitis and tends to occur in association with an underlying pancreatic carcinoma. Patients with pancreatic panniculitis associated with pancreatic carcinoma tend to have more severe, diffuse, and persistent subcutaneous nodules that often are refractory to treatment with frequent recurrence. A rare constellation of findings known as Schmid triad is comprised of panniculitis, polyarthritis, and eosinophilia and typically portends a poor prognosis secondary to an underlying pancreatic tumor.14 Cutaneous nodules may predate the diagnosis of pancreatic carcinoma by several months, thus signifying the need for a high index of suspicion in patients with lower leg subcutaneous nodules.
Joint disease most commonly involves the ankles, knees, wrists, and MCP joints.5,6,11 It has been suggested that arthritic symptoms are from periarticular fat necrosis or a direct extension from the necrotic subcutaneous tissue to the adjacent joint space.15 Dahl et al3 reported the composition of joint effusion fluid in 3 patients with PPP syndrome. The aspirate in all 3 patients contained viscous yellow material similar to the necrotic adipose tissue seen draining from subcutaneous nodules. Joint aspirate analysis demonstrated increased concentration of free fatty acids in the joint fluid consistent with severe lipolysis.3
The PPP syndrome acronym may be misleading to physicians, as arthritis is not always polyarticular. Dahl et al3 reported that monoarticular or oligoarticular arthritic symptoms were present in 56% of patients studied. In rare cases, the arthritic symptoms antedated the diagnosis of clinically asymptomatic pancreatic disease. Arthritis can be either symmetric or asymmetric and infrequently follows a chronic course, leading to radiographic lytic lesions and symptoms that often are unresponsive to conventional therapy.16
Treatment of PPP syndrome is largely supportive, with a focus on correcting the underlying pancreatic disease. It is imperative to identify any complicating factors contributing to high levels of circulating pancreatic enzymes. Pseudocysts must be addressed if discovered in these patients, as they often perpetuate the substantial release of pancreatic enzymes into the serum, leading to characteristic subcutaneous fat necrosis and arthritis. Sepsis also is a concern, likely secondary to bacterial colonization of the ulcerated subcutaneous nodules and compromised skin barrier. Nonsteroidal anti-inflammatory drugs and corticosteroids have been used for symptomatic relief but usually are ineffective and have not been shown to reduce the duration of the disease.12,16 Octreotide has been utilized and may potentially reduce pancreatic enzyme secretion leading to improvement in cutaneous and musculoskeletal lesions.17 Plasmapheresis has been used as an adjuvant treatment in patients with persistent hyperamylasemia and hyperlipasemia, but reports are anecdotal. Often reserved for severe disease, cholecystectomy, pancreatic duct removal, and pancreaticoduodenectomy have demonstrated success in the management of chronic pancreatitis and panniculitis. Dahl et al3 reported 2 cases in which cholecystectomy was performed with complete resolution of the skin and pancreatic disease. Our patient was initially treated symptomatically with NSAIDs and corticosteroids but there was no clinical response. The patient eventually underwent a pancreaticoduodenectomy 9 months after the onset of symptoms with complete resolution of joint pain and swelling, greater than 50% resolution of his lower leg subcutaneous nodules, and remarkable reduction in amylase and lipase levels on 1-month follow-up.
Conclusion
Panniculitis, pancreatitis, and polyarthritis syndrome is a rare diagnosis characterized by a triad of pancreatic panniculitis, pancreatitis, and polyarthritis. Adjuvant therapies for PPP syndrome, such as NSAIDs, corticosteroids, plasmapheresis, and octreotide, have been used with equivocal results, but definitive treatment requires correction of the primary pancreatic disorder. More importantly, many pancreatic diseases can cause pancreatic panniculitis, but extensive, refractory, or ulcerated cases could be an early indicator of an occult pancreatic malignancy and should prompt early evaluation with a multidisciplinary approach. This approach should incorporate management from dermatology, internal medicine, rheumatology, gastroenterology, surgery, and primary care.
- Chiari H. Uber die Sogenannte Fettnekrose. Prag Med Wochenschr. 1883;8:285-286, 299-301.
- Blauvelt H. Case of acute pancreatitis with subcutaneous fat necrosis. Br J Surg. 1946;34:207-208.
- Dahl PR, Su D, Cullimore KC, et al. Pancreatic panniculitis. J Am Acad Dermatol. 1995;33:413-417.
- Mullen GT, Caperton EM Jr, Crespin SR, et al. Arthritis and skin lesions resembling erythema nodosum in pancreatic disease. Ann Intern Med. 1968;68:75-87.
- Potts DE, Mass MF, Iseman MD. Syndrome and pancreatic disease, subcutaneous fat necrosis and polyserositis: case report and review of literature. Am J Med. 1975;58:417-423.
- Sorensen EV. Subcutaneous fat necrosis in pancreatic disease: a review and two new case reports. J Clin Gastroenterol. 1988;10:71-75.
- García-Romero D, Vanaclocha F. Pancreatic panniculitis. Dermatol Clin. 2008;26:465-470.
- Narváez J, Bianchi M, Santo P, et al. Pancreatitis, panniculitis, and polyarthritis. Semin Arthritis Rheum. 2010;39:417-423.
- Rongioetti F, Caputo V. Pancreatic panniculitis. G Ital Dermatol Venereol. 2013;148:419-425.
- Poelman SM, Nguyen K. Pancreatic panniculitis associated with acinar cell pancreatic carcinoma. J Cutan Med Surg. 2008;12:38-42.
- Hughes SH, Apisarnthanarax P, Mullins F. Subcutaneous fat necrosis associated with pancreatic disease. Arch Dermatol. 1975:111:506-510.
- Ferrari R, Wendelboe M, Ford PM, et al. Pancreatitis arthritis with periarticular fat necrosis. J Rheumatol. 1993;20:1436-1437.
- Szymanski FJ, Bluefarb SM. Nodular fat necrosis and pancreatic diseases. Arch Dermatol. 1961;83:224-229.
- Beltraminelly HS, Buechner SA, Hausermann P. Pancreatic panniculitis in a patient with an acinar cell cystadenocarcinoma of the pancreas. Dermatology. 2004;208:265-267.
- Burns WA, Matthews MJ, Hamosh M, et al. Lipase-secreting acinar cell carcinoma of the pancreas with polyarthropathy: a light and electron microscopic, histochemical, and biochemical study. Cancer. 1974;33:1002-1009.
- Baron M, Paltiel H, Lander P. Aseptic necrosis of the talus and calcaneal insufficiency fractures in a patient with pancreatitis, subcutaneous fat necrosis, and arthritis. Arthritis Rheum. 1984;27:1309-1313.
- Zundler S, Erber R, Agaimy A, et al. Pancreatic panniculitis in a patient with pancreatic-type acinar cell carcinoma of the liver—case report and review of literature. BMC Cancer. 2016;16:130.
- Chiari H. Uber die Sogenannte Fettnekrose. Prag Med Wochenschr. 1883;8:285-286, 299-301.
- Blauvelt H. Case of acute pancreatitis with subcutaneous fat necrosis. Br J Surg. 1946;34:207-208.
- Dahl PR, Su D, Cullimore KC, et al. Pancreatic panniculitis. J Am Acad Dermatol. 1995;33:413-417.
- Mullen GT, Caperton EM Jr, Crespin SR, et al. Arthritis and skin lesions resembling erythema nodosum in pancreatic disease. Ann Intern Med. 1968;68:75-87.
- Potts DE, Mass MF, Iseman MD. Syndrome and pancreatic disease, subcutaneous fat necrosis and polyserositis: case report and review of literature. Am J Med. 1975;58:417-423.
- Sorensen EV. Subcutaneous fat necrosis in pancreatic disease: a review and two new case reports. J Clin Gastroenterol. 1988;10:71-75.
- García-Romero D, Vanaclocha F. Pancreatic panniculitis. Dermatol Clin. 2008;26:465-470.
- Narváez J, Bianchi M, Santo P, et al. Pancreatitis, panniculitis, and polyarthritis. Semin Arthritis Rheum. 2010;39:417-423.
- Rongioetti F, Caputo V. Pancreatic panniculitis. G Ital Dermatol Venereol. 2013;148:419-425.
- Poelman SM, Nguyen K. Pancreatic panniculitis associated with acinar cell pancreatic carcinoma. J Cutan Med Surg. 2008;12:38-42.
- Hughes SH, Apisarnthanarax P, Mullins F. Subcutaneous fat necrosis associated with pancreatic disease. Arch Dermatol. 1975:111:506-510.
- Ferrari R, Wendelboe M, Ford PM, et al. Pancreatitis arthritis with periarticular fat necrosis. J Rheumatol. 1993;20:1436-1437.
- Szymanski FJ, Bluefarb SM. Nodular fat necrosis and pancreatic diseases. Arch Dermatol. 1961;83:224-229.
- Beltraminelly HS, Buechner SA, Hausermann P. Pancreatic panniculitis in a patient with an acinar cell cystadenocarcinoma of the pancreas. Dermatology. 2004;208:265-267.
- Burns WA, Matthews MJ, Hamosh M, et al. Lipase-secreting acinar cell carcinoma of the pancreas with polyarthropathy: a light and electron microscopic, histochemical, and biochemical study. Cancer. 1974;33:1002-1009.
- Baron M, Paltiel H, Lander P. Aseptic necrosis of the talus and calcaneal insufficiency fractures in a patient with pancreatitis, subcutaneous fat necrosis, and arthritis. Arthritis Rheum. 1984;27:1309-1313.
- Zundler S, Erber R, Agaimy A, et al. Pancreatic panniculitis in a patient with pancreatic-type acinar cell carcinoma of the liver—case report and review of literature. BMC Cancer. 2016;16:130.
Practice Points
- Recognition of skin lesions in a patient with a history of pancreatitis may represent a rare entity known as pancreatic panniculitis.
- Panniculitis, pancreatitis, and polyarthritis (PPP) syndrome is a rare diagnosis characterized by a triad of pancreatic panniculitis, pancreatitis, and polyarthritis.
- A rare constellation of findings known as Schmid triad is comprised of panniculitis, polyarthritis, and eosinophilia and typically portends a poor prognosis secondary to an underlying pancreatic tumor.
- These findings should prompt early evaluation with a multidisciplinary approach.
A Veteran With Alcohol Use Disorder and Acute Pancreatitis
Case Presentation. A 23-year-old male U.S. Army veteran with a history of alcohol use disorder and posttraumatic stress disorder (PTSD) presented to the VA Boston Healthcare System (VABHS) West Roxbury campus emergency department (ED) with epigastric abdominal pain in the setting of consuming alcohol. The patient had served in the infantry in Afghanistan during Operation Enduring Freedom. He consumed up to 12 alcoholic drinks per day (both beer and hard liquor) for the past 3 years and had been hospitalized 3 times previously; twice for alcohol detoxification and once for PTSD. He is a former tobacco smoker with fewer than 5 pack-years, he uses marijuana often and does not use IV drugs. In the ED, his physical examination was notable for a heart rate of 130 beats per minute and blood pressure of 161/111 mm Hg. He was alert and oriented and had a mild tremor. The patient was diaphoretic with dry mucous membranes, tenderness to palpation in the epigastrium, and abdominal guarding. A computed tomography (CT) scan of the abdomen revealed acute pancreatitis without necrosis. The patient received 1 L of normal saline and was admitted to the medical ward for presumed alcoholic pancreatitis.
► Rahul Ganatra, MD, MPH, Chief Medical Resident, VABHS and Beth Israel Deaconess Medical Center. Dr. Weber, we care for many young people who drink more than they should and almost none of them end up with alcoholic pancreatitis. What are the relevant risk factors that make individuals like this patient more susceptible to alcoholic pancreatitis?
►Horst Christian Weber, MD, Gastroenterology Service, VABHS, and Assistant Professor of Medicine, Boston University School of Medicine. While we don’t have a good understanding of the precise mechanism of alcoholic pancreatitis, we do know that in the U.S., alcohol consumption is responsible for about one-third of all cases.1 Acute pancreatitis in general may present with a wide range of disease severity. It is the most common cause of gastrointestinal-related hospitalization,2 and the mortality of hospital inpatients with pancreatitis is about 5%.3,4 Therefore, acute pancreatitis represents a prevalent condition with a critical impact on morbidity and mortality. Alcoholic pancreatitis typically occurs after many years of heavy alcohol use, not after a single drinking 
► Dr. Ganatra. At this point, the chemistry laboratory paged the admitting resident with the notification that the patient’s blood was grossly lipemic. Ultracentrifugation was performed to separate the lipid layer and his laboratory values result (Table). Notable abnormalities included polycythemia with a hemoglobin of 17.4 g/dL, hyponatremia with a sodium of 129 mmol/L, normal renal function, elevated aspartate aminotransferase (AST) and alanine aminotransferase (ALT) (AST 258 IU/L and ALT 153 IU/L, respectively), hyperbilirubinemia with a total bilirubin of 2.7 mg/dL, and a serum alcohol level of 147 mg/dL. Due to anticipated requirement for a higher level of care, the patient was transferred to the Medical Intensive Care Unit (MICU).
Dr. Breu, can you help us interpret this patient’s numerous laboratory abnormalities? Without yet having the triglyceride level available, how does the fact that the patient’s blood was lipemic affect our interpretation of his labs? What further workup is warranted?
► Anthony Breu, MD, Medical Service, VABHS, Assistant Professor of Medicine, Harvard Medical School. First, the positive alcohol level confirms a recent ingestion. Second, he has elevated transaminases with the AST greater than the ALT, which is consistent with alcoholic liver disease. While the initial assumption is that this patient has alcohol-induced pancreatitis, the elevations in bilirubin and alkaline phosphatase may suggest gallstone pancreatitis, and the lipemic appearing serum could suggest triglyceride-mediated pancreatitis. If the patient does have elevated triglyceride levels, the sodium level may indicate pseudohyponatremia, a laboratory artifact seen if a dilution step is used. To further evaluate the patient, I would obtain a triglyceride level and a right upper quadrant ultrasound. Direct ion-selective electrode analysis of the sodium level can be done with a device used to measure blood gases to exclude pseudohyponatremia.
► Dr. Ganatra. A right upper quadrant ultrasound was obtained in the MICU, which showed hepatic steatosis and hepatomegaly to 19 cm, but no evidence of biliary obstruction by stones or sludge. The common bile duct measured 3.2 mm in diameter. A triglyceride level returned above assay at > 3,392 mg/dL. A review of the medical record revealed a triglyceride level of 105 mg/dL 16 months prior. The Gastroenterology Department was consulted.
Dr. Weber, we now have 2 etiologies for pancreatitis in this patient: alcohol and hypertriglyceridemia. How do each cause pancreatitis? Is it possible to determine in this case which one is the more likely driver?
► Dr. Weber. The mechanism for alcohol-induced pancreatitis is not fully known, but there are several hypotheses. One is that alcohol may increase the synthesis or activation of pancreatic digestive enzymes.6 Another is that metabolites of alcohol are directly toxic to the pancreas.6 Based on the epidemiologic observation that alcoholic pancreatitis usually happens in long-standing users, all we can say is that it is not very likely to be the effect of an acute insult. For hypertriglyceridemic pancreatitis, we believe the injury is due to the toxic effect of free fatty acids in the pancreas liberated by lipolysis of triglycerides by pancreatic lipases. Higher triglycerides are associated with higher risk, suggesting a dose-response relationship: This risk is not greatly increased until triglycerides exceed 500 mg/dL; above 1,000 mg/dL, the risk is about 5%, and above 2,000 mg/dL, the risk is between 10% and 20%.7 In summary, we cannot really determine whether the alcohol or the triglycerides are the main cause of his pancreatitis, but given his markedly elevated triglycerides, he should be treated for hypertriglyceridemic pancreatitis.
►Dr. Ganatra. Dr. Breu, regardless of the underlying etiology, this patient requires treatment. What does the literature suggest as the best course of action regarding crystalloid administration in patients with acute pancreatitis?
►Dr. Breu. There are 2 issues to discuss regarding IV fluids in acute pancreatitis: choice of crystalloid and rate of administration. For the choice of IV fluid, lactated Ringer solution (LR) may be preferred over normal saline (NS). There are both pathophysiologic and evidence-based rationales for this choice. As Dr. Weber alluded to, trypsinogen activation is an important step in the pathogenesis of acute pancreatitis and requires a low pH compartment. As most clinicians have experienced, NS may cause a metabolic acidosis; however, the use of LR may mitigate this. A 2011 randomized clinical trial showed that patients who received LR had less systemic inflammatory response syndrome (SIRS) and lower C-reactive protein (CRP) levels at 24 hours compared with patients who received NS.8 While these are surrogate outcomes, they, along with the theoretical basis, suggest LR is preferred.
Regarding rate, the key is fast and early.9 In my experience, internists often underdose IV rehydration within the first 12 to 24 hours, fail to change the rate based on clinical response, and leave patients on high rates too long. In a patient like this, a rate of 350 cc/h is a reasonable place to start. But, one must reassess response (ie, ensure there is a decrease in hematocrit and/or blood urea nitrogen) every 6 hours and increase the rate as needed. After the first 24 to 48 hours have passed, the rate should be lowered.
►Dr. Ganatra. The patient received 2 mg of IV hydromorphone and a 2 L bolus of LR. This was followed by a continuous infusion of LR at 200 cc/h. Dr. Weber, apart from the standard therapies for pancreatitis, what are our treatment options in hypertriglyceridemic pancreatitis?
►Dr. Weber. In the acute setting, IV insulin with or without dextrose is the most extensively studied therapy. Insulin rapidly decreases triglyceride levels by activating lipoprotein lipase and inhibiting hormone- sensitive lipase. The net effect is reduction in serum triglycerides available to be hydrolyzed to free fatty acids in the pancreas.7 For severe cases (ie, where acute pancreatitis is accompanied by hypocalcemia, lactic acidosis or a markedly elevated lipase), apheresis with therapeutic plasma exchange to more rapidly reduce triglyceride concentration is the preferred therapy. The goal is to reduce triglycerides to levels 
►Dr. Ganatra. Due to the possibility that the patient would require apheresis, which was not available at the VABHS West Roxbury campus, the patient was transferred to an affiliate hospital. The patient was started on 10% dextrose at 300 cc/h and an IV insulin infusion. His triglycerides fell to < 500 mg/dL over the subsequent 48 hours, and ultimately, apheresis was not required. Enteral nutrition by nasogastric (NG) tube was initiated on hospital day 6. The patient’s hospital course was notable for acute respiratory distress syndrome that required intubation for 7 days, hyperbilirubinemia (with a peak bilirubin of 10.5 mg/dL), acute kidney injury (with a peak creatinine 4.7 mg/dL), fever without an identified infectious source, alcohol withdrawal syndrome that required phenobarbital, and delirium. Nine days later, he was transferred back to the VABHS West Roxbury campus. His condition stabilized, and he was transferred to the medical floor. On hospital day 14, the patient’s mental status improved, and he began tolerating oral nutrition.
Dr. Breu, over the years, the standard of care regarding when to start enteral nutrition in pancreatitis has changed considerably. This patient received enteral nutrition via NG tube but also had periods of being NPO (nothing by mouth) for up to 6 days. What is the current best practice for timing of initiating enteral nutrition in acute pancreatitis?
►Dr. Breu. It is true that the standard of care has changed and continues to evolve. Many decades ago, patients with acute pancreatitis would routinely undergo NG tube suction to reduce delivery of gastric contents to the duodenum, thereby decreasing pancreas activation, allowing it to rest.11 The NG tube also allowed for decompression of any ileus that had formed. Beginning in the 1970s, several clinical trials were performed, showing that NG tube suction was no better than simply making the patient NPO.12,13 More recently, we have begun to move toward earlier feeding. Again, there is a pathophysiologic rationale (bowel rest is associated with intestinal atrophy, predisposing to bacterial translocation and resulting infectious complications) and increasing evidence supporting this practice.9 Even in severe pancreatitis, hunger may be used to initiate oral intake.14
►Dr. Ganatra. On hospital day 16, the patient developed sudden-onset right-sided back and flank pain, and his hemoglobin dropped to 6.1 mg/dL, which required transfusion of packed red blood cells. He remained afebrile and hemodynamically stable. Dr. Weber, what are the major complications of acute pancreatitis, and when should we suspect them? Should we be worried about complications of pancreatitis in this patient?
►Dr. Weber. Organ failure in the acute setting can occur due to activation of cytokine cascades and the systemic inflammatory response syndrome and is described by clinical and radiologic criteria called the Atlanta Classification.15 Apart from organ failure, the most serious complications of acute pancreatitis are necrosis of pancreatic tissue leading to walled-off pancreatic necrosis and the formation of peripancreatic fluid collections and pseudocysts, which occur in about 15% of patients with acute pancreatitis. These complications are serious because they can become infected, which portends a higher mortality and in some cases require surgical resection.
Other complications of acute pancreatitis include pseudoaneurysm formation, which is when a vessel bleeds into a pancreatic pseudocyst, and thromboses of the splenic, portal, or mesenteric veins. Thrombotic complications may occur in up to half of patients with pancreatic necrosis but are uncommon without some degree of necrosis.16 No necrosis was noted on this patient’s initial CT scan, so the probability of thrombosis is low. Also, as it takes several weeks for pseudocyst formation to occur, a bleeding pseudoaneurysm is unlikely at this early stage. Therefore, a complication of pancreatitis is unlikely in this patient, and evaluation for other causes of abdominal pain should be considered.
►Dr. Ganatra. A noncontrast CT of the abdomen and pelvis was obtained and revealed no evidence of complications or other acute pathology. His pain was managed conservatively, and hemoglobin remained stable. Over the next 5 days, the patient’s symptoms gradually resolved, his oral intake improved, and he was discharged home on gemfibrozil 600 mg twice daily 19 days after admission. He declined psychiatry follow-up for his PTSD, and after discharge he did not keep his scheduled gastroenterology (GI) follow-up appointment. Four months later, the patient presented again with epigastric abdominal pain similar to his initial presentation. The patient had resumed drinking, stating that “alcohol is the only thing that helps [with the PTSD].” He had not been taking the gemfibrozil. He was admitted with a recurrent episode of pancreatitis; however, his triglycerides on admission were 119 mg/dL.
Dr. Weber, this patient’s triglycerides declined rapidly over a period of just 4 months with questionable adherence to gemfibrozil. However, he was admitted again with another episode of pancreatitis, this time in the setting of alcohol use alone without markedly elevated triglycerides. What do we know about recurrence risk for pancreatitis? Are some etiologies of pancreatitis more likely to present with recurrent attacks than are others?
►Dr. Weber. The rate of recurrence following an episode of acute pancreatitis varies according to the cause, but in general, about 20% to 30% of patients will experience a recurrence, and 5% to 10% will go on to develop chronic pancreatitis.17 Alcoholic pancreatitis does carry a higher risk of recurrence than pancreatitis due to other causes; the risk is as high as 50%. Not surprisingly, recurrence of acute pancreatitis increases risk for development of chronic pancreatitis. As this patient is a smoker, it is worth noting that smoking potentiates pancreatic damage from alcohol and increases the risk for both recurrent and chronic pancreatitis.5
►Dr. Ganatra. The patient was treated with IV hydromorphone and IV LR at 350 cc/h. Oral nutrition was begun immediately. He manifested no organ dysfunction, and his symptoms improved over the course of 48 hours. He was discharged home with psychiatry and GI follow-up scheduled. Dr. Breu and Dr. Weber, how should we counsel this patient to reduce his risk of recurrent attacks of pancreatitis in the future, and what options do we have for pharmacotherapy to decrease his risk?
►Dr. Breu. I’ll let Dr. Weber comment on mitigating the risk of hypertriglyceride-induced pancreatitis and reserve my comments to pharmacotherapy in alcohol use disorder. This patient may be a candidate for naltrexone therapy, either in oral or intramuscular formulations. Both have been showed to reduce the risk of returning to heavy drinking and may be particularly beneficial in those with a family history.18,19 Acamprosate is also an option.
►Dr. Weber. Data on recurrence risk in hypertriglyceridemic pancreatitis are limited, but there are case reports suggesting that a fatty diet and alcohol use are implicated in recurrence.20 I would counsel the patient on lifestyle modifications that are known to reduce this risk. I agree with Dr. Breu that devoting our efforts to helping him reduce or eliminate his alcohol consumption is the single most important thing we can do to reduce his risk for recurrent attacks. Since the patient reports that he drinks alcohol in order to cope with his PTSD, establishing care with a mental health provider to address this is of the utmost importance. In addition, smoking cessation and promoting medication adherence with gemfibrozil will also reduce risk for future episodes, but continued alcohol use is his strongest risk factor.
►Dr. Ganatra. After discharge, the patient engaged with outpatient psychiatry and GI. He still reports feeling that alcohol is the only thing that alleviates his PTSD and anxiety symptoms. He is not currently interested in pharmacotherapy for cessation of alcohol use.
Acknowledgments
The authors thank Ivana Jankovic, MD, Matthew Lewis Chase, MD
1. Dufour MC, Adamson MD. The epidemiology of alcohol-induced pancreatitis. Pancreas. 2003;27(4):286-290.
2. Peery AF, Dellon ES, Lund J, et al. Burden of gastrointestinal disease in the United States: 2012 update. Gastroenterology. 2012;143(5):1179-1187.e1-e3.
3. Cavallini G, Frulloni L, Bassi C, et al; ProInf-AISP Study Group. Prospective multicentre survey on acute pancreatitis in Italy (ProInf-AISP): results on 1005 patients. Dig Liver Dis. 2004;36(3):205-211.
4. Banks PA, Freeman ML; Practice Parameters Committee of the American College of Gastroenterology. Practice guidelines in acute pancreatitis. Am J Gastroenterol. 2006;101(10):2379-2400.
5. Hartwig W, Werner J, Ryschich E, et al. Cigarette smoke enhances ethanol-induced pancreatic injury. Pancreas. 2000;21(3):272-278.
6. Chowdhury P, Gupta P. Pathophysiology of alcoholic pancreatitis: an overview. World J Gastroenterol. 2006;12(46):7421-7427.
7. Scherer J, Singh VP, Pitchumoni CS, Yadav D. Issues in hypertriglyceridemic pancreatitis: an update. J Clin Gastroenterol. 2014;48(3):195-203.
8. Wu BU, Hwang JQ, Gardner TH, et al. Lactated Ringer’s solution reduces systemic inflammation compared with saline in patients with acute pancreatitis. Clin Gastroenterol Hepatol. 2011;9(8):710-717.e1.
9. Tenner S, Baillie J, DeWitt J, Vege SS; American College of Gastroenterology. American College of Gastroenterology guideline: management of acute pancreatitis. Am J Gastroenterol. 2013;108(9):1400-1415; 1416.
10. Preiss D, Tikkanen MJ, Welsh P, et al. Lipid-modifying therapies and risk of pancreatitis: a meta-analysis. JAMA. 2012;308(8):804-811.
11. Nardi GL. Pancreatitis. N Engl J Med. 1963;268(19):1065-1067.
12. Naeije R, Salingret E, Clumeck N, De Troyer A, Devis G. Is nasogastric suction necessary in acute pancreatitis? Br Med J. 1978;2(6138):659-660.
13. Levant JA, Secrist DM, Resin H, Sturdevant RA, Guth PH. Nasogastric suction in the treatment of alcoholic pancreatitis: a controlled study. JAMA. 1974;229(1):51-52.
14. Zhao XL, Zhu SF, Xue GJ, et al. Early oral refeeding based on hunger in moderate and severe acute pancreatitis: a prospective controlled, randomized clinical trial. Nutrition. 2015;31(1):171-175.
15. Banks PA, Bollen TL, Dervenis C, et al; Acute Pancreatitis Classification Working Group. Classification of acute pancreatitis—2012: revision of the Atlanta classification and definitions by international consensus. Gut. 2013;62(1):102-111.
16. Easler J, Muddana V, Furlan A, et al. Portosplenomesenteric venous thrombosis in patients with acute pancreatitis is associated with pancreatic necrosis and usually has a benign course. Clin Gastroenterol Hepatol. 2014;12(5):854-862.
17. Yadav D, O’Connell M, Papachristou GI. Natural history following the first attack of acute pancreatitis. Am J Gastroenterol. 2012;107(7):1096-1103.
18. Jonas DE, Amick HR, Feltner C, et al. Pharmacotherapy for adults with alcohol use disorders in outpatient settings: a systematic review and meta-analysis. JAMA. 2014;311(18):1889-1900.
19. Garbutt JC, Kranzler HR, O’Malley SS, et al. Efficacy and tolerability of long-acting injectable naltrexone for alcohol dependence: a randomized controlled trial. JAMA. 2005;293(13):1617-1625.
20. Piolot A, Nadler F, Cavallero E, Coquard JL, Jacotot B. Prevention of recurrent acute pancreatitis in patients with severe hypertriglyceridemia: value of regular plasmapheresis. Pancreas. 1996;13(1):96-99.
Case Presentation. A 23-year-old male U.S. Army veteran with a history of alcohol use disorder and posttraumatic stress disorder (PTSD) presented to the VA Boston Healthcare System (VABHS) West Roxbury campus emergency department (ED) with epigastric abdominal pain in the setting of consuming alcohol. The patient had served in the infantry in Afghanistan during Operation Enduring Freedom. He consumed up to 12 alcoholic drinks per day (both beer and hard liquor) for the past 3 years and had been hospitalized 3 times previously; twice for alcohol detoxification and once for PTSD. He is a former tobacco smoker with fewer than 5 pack-years, he uses marijuana often and does not use IV drugs. In the ED, his physical examination was notable for a heart rate of 130 beats per minute and blood pressure of 161/111 mm Hg. He was alert and oriented and had a mild tremor. The patient was diaphoretic with dry mucous membranes, tenderness to palpation in the epigastrium, and abdominal guarding. A computed tomography (CT) scan of the abdomen revealed acute pancreatitis without necrosis. The patient received 1 L of normal saline and was admitted to the medical ward for presumed alcoholic pancreatitis.
► Rahul Ganatra, MD, MPH, Chief Medical Resident, VABHS and Beth Israel Deaconess Medical Center. Dr. Weber, we care for many young people who drink more than they should and almost none of them end up with alcoholic pancreatitis. What are the relevant risk factors that make individuals like this patient more susceptible to alcoholic pancreatitis?
►Horst Christian Weber, MD, Gastroenterology Service, VABHS, and Assistant Professor of Medicine, Boston University School of Medicine. While we don’t have a good understanding of the precise mechanism of alcoholic pancreatitis, we do know that in the U.S., alcohol consumption is responsible for about one-third of all cases.1 Acute pancreatitis in general may present with a wide range of disease severity. It is the most common cause of gastrointestinal-related hospitalization,2 and the mortality of hospital inpatients with pancreatitis is about 5%.3,4 Therefore, acute pancreatitis represents a prevalent condition with a critical impact on morbidity and mortality. Alcoholic pancreatitis typically occurs after many years of heavy alcohol use, not after a single drinking
► Dr. Ganatra. At this point, the chemistry laboratory paged the admitting resident with the notification that the patient’s blood was grossly lipemic. Ultracentrifugation was performed to separate the lipid layer and his laboratory values result (Table). Notable abnormalities included polycythemia with a hemoglobin of 17.4 g/dL, hyponatremia with a sodium of 129 mmol/L, normal renal function, elevated aspartate aminotransferase (AST) and alanine aminotransferase (ALT) (AST 258 IU/L and ALT 153 IU/L, respectively), hyperbilirubinemia with a total bilirubin of 2.7 mg/dL, and a serum alcohol level of 147 mg/dL. Due to anticipated requirement for a higher level of care, the patient was transferred to the Medical Intensive Care Unit (MICU).
Dr. Breu, can you help us interpret this patient’s numerous laboratory abnormalities? Without yet having the triglyceride level available, how does the fact that the patient’s blood was lipemic affect our interpretation of his labs? What further workup is warranted?
► Anthony Breu, MD, Medical Service, VABHS, Assistant Professor of Medicine, Harvard Medical School. First, the positive alcohol level confirms a recent ingestion. Second, he has elevated transaminases with the AST greater than the ALT, which is consistent with alcoholic liver disease. While the initial assumption is that this patient has alcohol-induced pancreatitis, the elevations in bilirubin and alkaline phosphatase may suggest gallstone pancreatitis, and the lipemic appearing serum could suggest triglyceride-mediated pancreatitis. If the patient does have elevated triglyceride levels, the sodium level may indicate pseudohyponatremia, a laboratory artifact seen if a dilution step is used. To further evaluate the patient, I would obtain a triglyceride level and a right upper quadrant ultrasound. Direct ion-selective electrode analysis of the sodium level can be done with a device used to measure blood gases to exclude pseudohyponatremia.
► Dr. Ganatra. A right upper quadrant ultrasound was obtained in the MICU, which showed hepatic steatosis and hepatomegaly to 19 cm, but no evidence of biliary obstruction by stones or sludge. The common bile duct measured 3.2 mm in diameter. A triglyceride level returned above assay at > 3,392 mg/dL. A review of the medical record revealed a triglyceride level of 105 mg/dL 16 months prior. The Gastroenterology Department was consulted.
Dr. Weber, we now have 2 etiologies for pancreatitis in this patient: alcohol and hypertriglyceridemia. How do each cause pancreatitis? Is it possible to determine in this case which one is the more likely driver?
► Dr. Weber. The mechanism for alcohol-induced pancreatitis is not fully known, but there are several hypotheses. One is that alcohol may increase the synthesis or activation of pancreatic digestive enzymes.6 Another is that metabolites of alcohol are directly toxic to the pancreas.6 Based on the epidemiologic observation that alcoholic pancreatitis usually happens in long-standing users, all we can say is that it is not very likely to be the effect of an acute insult. For hypertriglyceridemic pancreatitis, we believe the injury is due to the toxic effect of free fatty acids in the pancreas liberated by lipolysis of triglycerides by pancreatic lipases. Higher triglycerides are associated with higher risk, suggesting a dose-response relationship: This risk is not greatly increased until triglycerides exceed 500 mg/dL; above 1,000 mg/dL, the risk is about 5%, and above 2,000 mg/dL, the risk is between 10% and 20%.7 In summary, we cannot really determine whether the alcohol or the triglycerides are the main cause of his pancreatitis, but given his markedly elevated triglycerides, he should be treated for hypertriglyceridemic pancreatitis.
►Dr. Ganatra. Dr. Breu, regardless of the underlying etiology, this patient requires treatment. What does the literature suggest as the best course of action regarding crystalloid administration in patients with acute pancreatitis?
►Dr. Breu. There are 2 issues to discuss regarding IV fluids in acute pancreatitis: choice of crystalloid and rate of administration. For the choice of IV fluid, lactated Ringer solution (LR) may be preferred over normal saline (NS). There are both pathophysiologic and evidence-based rationales for this choice. As Dr. Weber alluded to, trypsinogen activation is an important step in the pathogenesis of acute pancreatitis and requires a low pH compartment. As most clinicians have experienced, NS may cause a metabolic acidosis; however, the use of LR may mitigate this. A 2011 randomized clinical trial showed that patients who received LR had less systemic inflammatory response syndrome (SIRS) and lower C-reactive protein (CRP) levels at 24 hours compared with patients who received NS.8 While these are surrogate outcomes, they, along with the theoretical basis, suggest LR is preferred.
Regarding rate, the key is fast and early.9 In my experience, internists often underdose IV rehydration within the first 12 to 24 hours, fail to change the rate based on clinical response, and leave patients on high rates too long. In a patient like this, a rate of 350 cc/h is a reasonable place to start. But, one must reassess response (ie, ensure there is a decrease in hematocrit and/or blood urea nitrogen) every 6 hours and increase the rate as needed. After the first 24 to 48 hours have passed, the rate should be lowered.
►Dr. Ganatra. The patient received 2 mg of IV hydromorphone and a 2 L bolus of LR. This was followed by a continuous infusion of LR at 200 cc/h. Dr. Weber, apart from the standard therapies for pancreatitis, what are our treatment options in hypertriglyceridemic pancreatitis?
►Dr. Weber. In the acute setting, IV insulin with or without dextrose is the most extensively studied therapy. Insulin rapidly decreases triglyceride levels by activating lipoprotein lipase and inhibiting hormone- sensitive lipase. The net effect is reduction in serum triglycerides available to be hydrolyzed to free fatty acids in the pancreas.7 For severe cases (ie, where acute pancreatitis is accompanied by hypocalcemia, lactic acidosis or a markedly elevated lipase), apheresis with therapeutic plasma exchange to more rapidly reduce triglyceride concentration is the preferred therapy. The goal is to reduce triglycerides to levels
►Dr. Ganatra. Due to the possibility that the patient would require apheresis, which was not available at the VABHS West Roxbury campus, the patient was transferred to an affiliate hospital. The patient was started on 10% dextrose at 300 cc/h and an IV insulin infusion. His triglycerides fell to < 500 mg/dL over the subsequent 48 hours, and ultimately, apheresis was not required. Enteral nutrition by nasogastric (NG) tube was initiated on hospital day 6. The patient’s hospital course was notable for acute respiratory distress syndrome that required intubation for 7 days, hyperbilirubinemia (with a peak bilirubin of 10.5 mg/dL), acute kidney injury (with a peak creatinine 4.7 mg/dL), fever without an identified infectious source, alcohol withdrawal syndrome that required phenobarbital, and delirium. Nine days later, he was transferred back to the VABHS West Roxbury campus. His condition stabilized, and he was transferred to the medical floor. On hospital day 14, the patient’s mental status improved, and he began tolerating oral nutrition.
Dr. Breu, over the years, the standard of care regarding when to start enteral nutrition in pancreatitis has changed considerably. This patient received enteral nutrition via NG tube but also had periods of being NPO (nothing by mouth) for up to 6 days. What is the current best practice for timing of initiating enteral nutrition in acute pancreatitis?
►Dr. Breu. It is true that the standard of care has changed and continues to evolve. Many decades ago, patients with acute pancreatitis would routinely undergo NG tube suction to reduce delivery of gastric contents to the duodenum, thereby decreasing pancreas activation, allowing it to rest.11 The NG tube also allowed for decompression of any ileus that had formed. Beginning in the 1970s, several clinical trials were performed, showing that NG tube suction was no better than simply making the patient NPO.12,13 More recently, we have begun to move toward earlier feeding. Again, there is a pathophysiologic rationale (bowel rest is associated with intestinal atrophy, predisposing to bacterial translocation and resulting infectious complications) and increasing evidence supporting this practice.9 Even in severe pancreatitis, hunger may be used to initiate oral intake.14
►Dr. Ganatra. On hospital day 16, the patient developed sudden-onset right-sided back and flank pain, and his hemoglobin dropped to 6.1 mg/dL, which required transfusion of packed red blood cells. He remained afebrile and hemodynamically stable. Dr. Weber, what are the major complications of acute pancreatitis, and when should we suspect them? Should we be worried about complications of pancreatitis in this patient?
►Dr. Weber. Organ failure in the acute setting can occur due to activation of cytokine cascades and the systemic inflammatory response syndrome and is described by clinical and radiologic criteria called the Atlanta Classification.15 Apart from organ failure, the most serious complications of acute pancreatitis are necrosis of pancreatic tissue leading to walled-off pancreatic necrosis and the formation of peripancreatic fluid collections and pseudocysts, which occur in about 15% of patients with acute pancreatitis. These complications are serious because they can become infected, which portends a higher mortality and in some cases require surgical resection.
Other complications of acute pancreatitis include pseudoaneurysm formation, which is when a vessel bleeds into a pancreatic pseudocyst, and thromboses of the splenic, portal, or mesenteric veins. Thrombotic complications may occur in up to half of patients with pancreatic necrosis but are uncommon without some degree of necrosis.16 No necrosis was noted on this patient’s initial CT scan, so the probability of thrombosis is low. Also, as it takes several weeks for pseudocyst formation to occur, a bleeding pseudoaneurysm is unlikely at this early stage. Therefore, a complication of pancreatitis is unlikely in this patient, and evaluation for other causes of abdominal pain should be considered.
►Dr. Ganatra. A noncontrast CT of the abdomen and pelvis was obtained and revealed no evidence of complications or other acute pathology. His pain was managed conservatively, and hemoglobin remained stable. Over the next 5 days, the patient’s symptoms gradually resolved, his oral intake improved, and he was discharged home on gemfibrozil 600 mg twice daily 19 days after admission. He declined psychiatry follow-up for his PTSD, and after discharge he did not keep his scheduled gastroenterology (GI) follow-up appointment. Four months later, the patient presented again with epigastric abdominal pain similar to his initial presentation. The patient had resumed drinking, stating that “alcohol is the only thing that helps [with the PTSD].” He had not been taking the gemfibrozil. He was admitted with a recurrent episode of pancreatitis; however, his triglycerides on admission were 119 mg/dL.
Dr. Weber, this patient’s triglycerides declined rapidly over a period of just 4 months with questionable adherence to gemfibrozil. However, he was admitted again with another episode of pancreatitis, this time in the setting of alcohol use alone without markedly elevated triglycerides. What do we know about recurrence risk for pancreatitis? Are some etiologies of pancreatitis more likely to present with recurrent attacks than are others?
►Dr. Weber. The rate of recurrence following an episode of acute pancreatitis varies according to the cause, but in general, about 20% to 30% of patients will experience a recurrence, and 5% to 10% will go on to develop chronic pancreatitis.17 Alcoholic pancreatitis does carry a higher risk of recurrence than pancreatitis due to other causes; the risk is as high as 50%. Not surprisingly, recurrence of acute pancreatitis increases risk for development of chronic pancreatitis. As this patient is a smoker, it is worth noting that smoking potentiates pancreatic damage from alcohol and increases the risk for both recurrent and chronic pancreatitis.5
►Dr. Ganatra. The patient was treated with IV hydromorphone and IV LR at 350 cc/h. Oral nutrition was begun immediately. He manifested no organ dysfunction, and his symptoms improved over the course of 48 hours. He was discharged home with psychiatry and GI follow-up scheduled. Dr. Breu and Dr. Weber, how should we counsel this patient to reduce his risk of recurrent attacks of pancreatitis in the future, and what options do we have for pharmacotherapy to decrease his risk?
►Dr. Breu. I’ll let Dr. Weber comment on mitigating the risk of hypertriglyceride-induced pancreatitis and reserve my comments to pharmacotherapy in alcohol use disorder. This patient may be a candidate for naltrexone therapy, either in oral or intramuscular formulations. Both have been showed to reduce the risk of returning to heavy drinking and may be particularly beneficial in those with a family history.18,19 Acamprosate is also an option.
►Dr. Weber. Data on recurrence risk in hypertriglyceridemic pancreatitis are limited, but there are case reports suggesting that a fatty diet and alcohol use are implicated in recurrence.20 I would counsel the patient on lifestyle modifications that are known to reduce this risk. I agree with Dr. Breu that devoting our efforts to helping him reduce or eliminate his alcohol consumption is the single most important thing we can do to reduce his risk for recurrent attacks. Since the patient reports that he drinks alcohol in order to cope with his PTSD, establishing care with a mental health provider to address this is of the utmost importance. In addition, smoking cessation and promoting medication adherence with gemfibrozil will also reduce risk for future episodes, but continued alcohol use is his strongest risk factor.
►Dr. Ganatra. After discharge, the patient engaged with outpatient psychiatry and GI. He still reports feeling that alcohol is the only thing that alleviates his PTSD and anxiety symptoms. He is not currently interested in pharmacotherapy for cessation of alcohol use.
Acknowledgments
The authors thank Ivana Jankovic, MD, Matthew Lewis Chase, MD
Case Presentation. A 23-year-old male U.S. Army veteran with a history of alcohol use disorder and posttraumatic stress disorder (PTSD) presented to the VA Boston Healthcare System (VABHS) West Roxbury campus emergency department (ED) with epigastric abdominal pain in the setting of consuming alcohol. The patient had served in the infantry in Afghanistan during Operation Enduring Freedom. He consumed up to 12 alcoholic drinks per day (both beer and hard liquor) for the past 3 years and had been hospitalized 3 times previously; twice for alcohol detoxification and once for PTSD. He is a former tobacco smoker with fewer than 5 pack-years, he uses marijuana often and does not use IV drugs. In the ED, his physical examination was notable for a heart rate of 130 beats per minute and blood pressure of 161/111 mm Hg. He was alert and oriented and had a mild tremor. The patient was diaphoretic with dry mucous membranes, tenderness to palpation in the epigastrium, and abdominal guarding. A computed tomography (CT) scan of the abdomen revealed acute pancreatitis without necrosis. The patient received 1 L of normal saline and was admitted to the medical ward for presumed alcoholic pancreatitis.
► Rahul Ganatra, MD, MPH, Chief Medical Resident, VABHS and Beth Israel Deaconess Medical Center. Dr. Weber, we care for many young people who drink more than they should and almost none of them end up with alcoholic pancreatitis. What are the relevant risk factors that make individuals like this patient more susceptible to alcoholic pancreatitis?
►Horst Christian Weber, MD, Gastroenterology Service, VABHS, and Assistant Professor of Medicine, Boston University School of Medicine. While we don’t have a good understanding of the precise mechanism of alcoholic pancreatitis, we do know that in the U.S., alcohol consumption is responsible for about one-third of all cases.1 Acute pancreatitis in general may present with a wide range of disease severity. It is the most common cause of gastrointestinal-related hospitalization,2 and the mortality of hospital inpatients with pancreatitis is about 5%.3,4 Therefore, acute pancreatitis represents a prevalent condition with a critical impact on morbidity and mortality. Alcoholic pancreatitis typically occurs after many years of heavy alcohol use, not after a single drinking
► Dr. Ganatra. At this point, the chemistry laboratory paged the admitting resident with the notification that the patient’s blood was grossly lipemic. Ultracentrifugation was performed to separate the lipid layer and his laboratory values result (Table). Notable abnormalities included polycythemia with a hemoglobin of 17.4 g/dL, hyponatremia with a sodium of 129 mmol/L, normal renal function, elevated aspartate aminotransferase (AST) and alanine aminotransferase (ALT) (AST 258 IU/L and ALT 153 IU/L, respectively), hyperbilirubinemia with a total bilirubin of 2.7 mg/dL, and a serum alcohol level of 147 mg/dL. Due to anticipated requirement for a higher level of care, the patient was transferred to the Medical Intensive Care Unit (MICU).
Dr. Breu, can you help us interpret this patient’s numerous laboratory abnormalities? Without yet having the triglyceride level available, how does the fact that the patient’s blood was lipemic affect our interpretation of his labs? What further workup is warranted?
► Anthony Breu, MD, Medical Service, VABHS, Assistant Professor of Medicine, Harvard Medical School. First, the positive alcohol level confirms a recent ingestion. Second, he has elevated transaminases with the AST greater than the ALT, which is consistent with alcoholic liver disease. While the initial assumption is that this patient has alcohol-induced pancreatitis, the elevations in bilirubin and alkaline phosphatase may suggest gallstone pancreatitis, and the lipemic appearing serum could suggest triglyceride-mediated pancreatitis. If the patient does have elevated triglyceride levels, the sodium level may indicate pseudohyponatremia, a laboratory artifact seen if a dilution step is used. To further evaluate the patient, I would obtain a triglyceride level and a right upper quadrant ultrasound. Direct ion-selective electrode analysis of the sodium level can be done with a device used to measure blood gases to exclude pseudohyponatremia.
► Dr. Ganatra. A right upper quadrant ultrasound was obtained in the MICU, which showed hepatic steatosis and hepatomegaly to 19 cm, but no evidence of biliary obstruction by stones or sludge. The common bile duct measured 3.2 mm in diameter. A triglyceride level returned above assay at > 3,392 mg/dL. A review of the medical record revealed a triglyceride level of 105 mg/dL 16 months prior. The Gastroenterology Department was consulted.
Dr. Weber, we now have 2 etiologies for pancreatitis in this patient: alcohol and hypertriglyceridemia. How do each cause pancreatitis? Is it possible to determine in this case which one is the more likely driver?
► Dr. Weber. The mechanism for alcohol-induced pancreatitis is not fully known, but there are several hypotheses. One is that alcohol may increase the synthesis or activation of pancreatic digestive enzymes.6 Another is that metabolites of alcohol are directly toxic to the pancreas.6 Based on the epidemiologic observation that alcoholic pancreatitis usually happens in long-standing users, all we can say is that it is not very likely to be the effect of an acute insult. For hypertriglyceridemic pancreatitis, we believe the injury is due to the toxic effect of free fatty acids in the pancreas liberated by lipolysis of triglycerides by pancreatic lipases. Higher triglycerides are associated with higher risk, suggesting a dose-response relationship: This risk is not greatly increased until triglycerides exceed 500 mg/dL; above 1,000 mg/dL, the risk is about 5%, and above 2,000 mg/dL, the risk is between 10% and 20%.7 In summary, we cannot really determine whether the alcohol or the triglycerides are the main cause of his pancreatitis, but given his markedly elevated triglycerides, he should be treated for hypertriglyceridemic pancreatitis.
►Dr. Ganatra. Dr. Breu, regardless of the underlying etiology, this patient requires treatment. What does the literature suggest as the best course of action regarding crystalloid administration in patients with acute pancreatitis?
►Dr. Breu. There are 2 issues to discuss regarding IV fluids in acute pancreatitis: choice of crystalloid and rate of administration. For the choice of IV fluid, lactated Ringer solution (LR) may be preferred over normal saline (NS). There are both pathophysiologic and evidence-based rationales for this choice. As Dr. Weber alluded to, trypsinogen activation is an important step in the pathogenesis of acute pancreatitis and requires a low pH compartment. As most clinicians have experienced, NS may cause a metabolic acidosis; however, the use of LR may mitigate this. A 2011 randomized clinical trial showed that patients who received LR had less systemic inflammatory response syndrome (SIRS) and lower C-reactive protein (CRP) levels at 24 hours compared with patients who received NS.8 While these are surrogate outcomes, they, along with the theoretical basis, suggest LR is preferred.
Regarding rate, the key is fast and early.9 In my experience, internists often underdose IV rehydration within the first 12 to 24 hours, fail to change the rate based on clinical response, and leave patients on high rates too long. In a patient like this, a rate of 350 cc/h is a reasonable place to start. But, one must reassess response (ie, ensure there is a decrease in hematocrit and/or blood urea nitrogen) every 6 hours and increase the rate as needed. After the first 24 to 48 hours have passed, the rate should be lowered.
►Dr. Ganatra. The patient received 2 mg of IV hydromorphone and a 2 L bolus of LR. This was followed by a continuous infusion of LR at 200 cc/h. Dr. Weber, apart from the standard therapies for pancreatitis, what are our treatment options in hypertriglyceridemic pancreatitis?
►Dr. Weber. In the acute setting, IV insulin with or without dextrose is the most extensively studied therapy. Insulin rapidly decreases triglyceride levels by activating lipoprotein lipase and inhibiting hormone- sensitive lipase. The net effect is reduction in serum triglycerides available to be hydrolyzed to free fatty acids in the pancreas.7 For severe cases (ie, where acute pancreatitis is accompanied by hypocalcemia, lactic acidosis or a markedly elevated lipase), apheresis with therapeutic plasma exchange to more rapidly reduce triglyceride concentration is the preferred therapy. The goal is to reduce triglycerides to levels
►Dr. Ganatra. Due to the possibility that the patient would require apheresis, which was not available at the VABHS West Roxbury campus, the patient was transferred to an affiliate hospital. The patient was started on 10% dextrose at 300 cc/h and an IV insulin infusion. His triglycerides fell to < 500 mg/dL over the subsequent 48 hours, and ultimately, apheresis was not required. Enteral nutrition by nasogastric (NG) tube was initiated on hospital day 6. The patient’s hospital course was notable for acute respiratory distress syndrome that required intubation for 7 days, hyperbilirubinemia (with a peak bilirubin of 10.5 mg/dL), acute kidney injury (with a peak creatinine 4.7 mg/dL), fever without an identified infectious source, alcohol withdrawal syndrome that required phenobarbital, and delirium. Nine days later, he was transferred back to the VABHS West Roxbury campus. His condition stabilized, and he was transferred to the medical floor. On hospital day 14, the patient’s mental status improved, and he began tolerating oral nutrition.
Dr. Breu, over the years, the standard of care regarding when to start enteral nutrition in pancreatitis has changed considerably. This patient received enteral nutrition via NG tube but also had periods of being NPO (nothing by mouth) for up to 6 days. What is the current best practice for timing of initiating enteral nutrition in acute pancreatitis?
►Dr. Breu. It is true that the standard of care has changed and continues to evolve. Many decades ago, patients with acute pancreatitis would routinely undergo NG tube suction to reduce delivery of gastric contents to the duodenum, thereby decreasing pancreas activation, allowing it to rest.11 The NG tube also allowed for decompression of any ileus that had formed. Beginning in the 1970s, several clinical trials were performed, showing that NG tube suction was no better than simply making the patient NPO.12,13 More recently, we have begun to move toward earlier feeding. Again, there is a pathophysiologic rationale (bowel rest is associated with intestinal atrophy, predisposing to bacterial translocation and resulting infectious complications) and increasing evidence supporting this practice.9 Even in severe pancreatitis, hunger may be used to initiate oral intake.14
►Dr. Ganatra. On hospital day 16, the patient developed sudden-onset right-sided back and flank pain, and his hemoglobin dropped to 6.1 mg/dL, which required transfusion of packed red blood cells. He remained afebrile and hemodynamically stable. Dr. Weber, what are the major complications of acute pancreatitis, and when should we suspect them? Should we be worried about complications of pancreatitis in this patient?
►Dr. Weber. Organ failure in the acute setting can occur due to activation of cytokine cascades and the systemic inflammatory response syndrome and is described by clinical and radiologic criteria called the Atlanta Classification.15 Apart from organ failure, the most serious complications of acute pancreatitis are necrosis of pancreatic tissue leading to walled-off pancreatic necrosis and the formation of peripancreatic fluid collections and pseudocysts, which occur in about 15% of patients with acute pancreatitis. These complications are serious because they can become infected, which portends a higher mortality and in some cases require surgical resection.
Other complications of acute pancreatitis include pseudoaneurysm formation, which is when a vessel bleeds into a pancreatic pseudocyst, and thromboses of the splenic, portal, or mesenteric veins. Thrombotic complications may occur in up to half of patients with pancreatic necrosis but are uncommon without some degree of necrosis.16 No necrosis was noted on this patient’s initial CT scan, so the probability of thrombosis is low. Also, as it takes several weeks for pseudocyst formation to occur, a bleeding pseudoaneurysm is unlikely at this early stage. Therefore, a complication of pancreatitis is unlikely in this patient, and evaluation for other causes of abdominal pain should be considered.
►Dr. Ganatra. A noncontrast CT of the abdomen and pelvis was obtained and revealed no evidence of complications or other acute pathology. His pain was managed conservatively, and hemoglobin remained stable. Over the next 5 days, the patient’s symptoms gradually resolved, his oral intake improved, and he was discharged home on gemfibrozil 600 mg twice daily 19 days after admission. He declined psychiatry follow-up for his PTSD, and after discharge he did not keep his scheduled gastroenterology (GI) follow-up appointment. Four months later, the patient presented again with epigastric abdominal pain similar to his initial presentation. The patient had resumed drinking, stating that “alcohol is the only thing that helps [with the PTSD].” He had not been taking the gemfibrozil. He was admitted with a recurrent episode of pancreatitis; however, his triglycerides on admission were 119 mg/dL.
Dr. Weber, this patient’s triglycerides declined rapidly over a period of just 4 months with questionable adherence to gemfibrozil. However, he was admitted again with another episode of pancreatitis, this time in the setting of alcohol use alone without markedly elevated triglycerides. What do we know about recurrence risk for pancreatitis? Are some etiologies of pancreatitis more likely to present with recurrent attacks than are others?
►Dr. Weber. The rate of recurrence following an episode of acute pancreatitis varies according to the cause, but in general, about 20% to 30% of patients will experience a recurrence, and 5% to 10% will go on to develop chronic pancreatitis.17 Alcoholic pancreatitis does carry a higher risk of recurrence than pancreatitis due to other causes; the risk is as high as 50%. Not surprisingly, recurrence of acute pancreatitis increases risk for development of chronic pancreatitis. As this patient is a smoker, it is worth noting that smoking potentiates pancreatic damage from alcohol and increases the risk for both recurrent and chronic pancreatitis.5
►Dr. Ganatra. The patient was treated with IV hydromorphone and IV LR at 350 cc/h. Oral nutrition was begun immediately. He manifested no organ dysfunction, and his symptoms improved over the course of 48 hours. He was discharged home with psychiatry and GI follow-up scheduled. Dr. Breu and Dr. Weber, how should we counsel this patient to reduce his risk of recurrent attacks of pancreatitis in the future, and what options do we have for pharmacotherapy to decrease his risk?
►Dr. Breu. I’ll let Dr. Weber comment on mitigating the risk of hypertriglyceride-induced pancreatitis and reserve my comments to pharmacotherapy in alcohol use disorder. This patient may be a candidate for naltrexone therapy, either in oral or intramuscular formulations. Both have been showed to reduce the risk of returning to heavy drinking and may be particularly beneficial in those with a family history.18,19 Acamprosate is also an option.
►Dr. Weber. Data on recurrence risk in hypertriglyceridemic pancreatitis are limited, but there are case reports suggesting that a fatty diet and alcohol use are implicated in recurrence.20 I would counsel the patient on lifestyle modifications that are known to reduce this risk. I agree with Dr. Breu that devoting our efforts to helping him reduce or eliminate his alcohol consumption is the single most important thing we can do to reduce his risk for recurrent attacks. Since the patient reports that he drinks alcohol in order to cope with his PTSD, establishing care with a mental health provider to address this is of the utmost importance. In addition, smoking cessation and promoting medication adherence with gemfibrozil will also reduce risk for future episodes, but continued alcohol use is his strongest risk factor.
►Dr. Ganatra. After discharge, the patient engaged with outpatient psychiatry and GI. He still reports feeling that alcohol is the only thing that alleviates his PTSD and anxiety symptoms. He is not currently interested in pharmacotherapy for cessation of alcohol use.
Acknowledgments
The authors thank Ivana Jankovic, MD, Matthew Lewis Chase, MD
1. Dufour MC, Adamson MD. The epidemiology of alcohol-induced pancreatitis. Pancreas. 2003;27(4):286-290.
2. Peery AF, Dellon ES, Lund J, et al. Burden of gastrointestinal disease in the United States: 2012 update. Gastroenterology. 2012;143(5):1179-1187.e1-e3.
3. Cavallini G, Frulloni L, Bassi C, et al; ProInf-AISP Study Group. Prospective multicentre survey on acute pancreatitis in Italy (ProInf-AISP): results on 1005 patients. Dig Liver Dis. 2004;36(3):205-211.
4. Banks PA, Freeman ML; Practice Parameters Committee of the American College of Gastroenterology. Practice guidelines in acute pancreatitis. Am J Gastroenterol. 2006;101(10):2379-2400.
5. Hartwig W, Werner J, Ryschich E, et al. Cigarette smoke enhances ethanol-induced pancreatic injury. Pancreas. 2000;21(3):272-278.
6. Chowdhury P, Gupta P. Pathophysiology of alcoholic pancreatitis: an overview. World J Gastroenterol. 2006;12(46):7421-7427.
7. Scherer J, Singh VP, Pitchumoni CS, Yadav D. Issues in hypertriglyceridemic pancreatitis: an update. J Clin Gastroenterol. 2014;48(3):195-203.
8. Wu BU, Hwang JQ, Gardner TH, et al. Lactated Ringer’s solution reduces systemic inflammation compared with saline in patients with acute pancreatitis. Clin Gastroenterol Hepatol. 2011;9(8):710-717.e1.
9. Tenner S, Baillie J, DeWitt J, Vege SS; American College of Gastroenterology. American College of Gastroenterology guideline: management of acute pancreatitis. Am J Gastroenterol. 2013;108(9):1400-1415; 1416.
10. Preiss D, Tikkanen MJ, Welsh P, et al. Lipid-modifying therapies and risk of pancreatitis: a meta-analysis. JAMA. 2012;308(8):804-811.
11. Nardi GL. Pancreatitis. N Engl J Med. 1963;268(19):1065-1067.
12. Naeije R, Salingret E, Clumeck N, De Troyer A, Devis G. Is nasogastric suction necessary in acute pancreatitis? Br Med J. 1978;2(6138):659-660.
13. Levant JA, Secrist DM, Resin H, Sturdevant RA, Guth PH. Nasogastric suction in the treatment of alcoholic pancreatitis: a controlled study. JAMA. 1974;229(1):51-52.
14. Zhao XL, Zhu SF, Xue GJ, et al. Early oral refeeding based on hunger in moderate and severe acute pancreatitis: a prospective controlled, randomized clinical trial. Nutrition. 2015;31(1):171-175.
15. Banks PA, Bollen TL, Dervenis C, et al; Acute Pancreatitis Classification Working Group. Classification of acute pancreatitis—2012: revision of the Atlanta classification and definitions by international consensus. Gut. 2013;62(1):102-111.
16. Easler J, Muddana V, Furlan A, et al. Portosplenomesenteric venous thrombosis in patients with acute pancreatitis is associated with pancreatic necrosis and usually has a benign course. Clin Gastroenterol Hepatol. 2014;12(5):854-862.
17. Yadav D, O’Connell M, Papachristou GI. Natural history following the first attack of acute pancreatitis. Am J Gastroenterol. 2012;107(7):1096-1103.
18. Jonas DE, Amick HR, Feltner C, et al. Pharmacotherapy for adults with alcohol use disorders in outpatient settings: a systematic review and meta-analysis. JAMA. 2014;311(18):1889-1900.
19. Garbutt JC, Kranzler HR, O’Malley SS, et al. Efficacy and tolerability of long-acting injectable naltrexone for alcohol dependence: a randomized controlled trial. JAMA. 2005;293(13):1617-1625.
20. Piolot A, Nadler F, Cavallero E, Coquard JL, Jacotot B. Prevention of recurrent acute pancreatitis in patients with severe hypertriglyceridemia: value of regular plasmapheresis. Pancreas. 1996;13(1):96-99.
1. Dufour MC, Adamson MD. The epidemiology of alcohol-induced pancreatitis. Pancreas. 2003;27(4):286-290.
2. Peery AF, Dellon ES, Lund J, et al. Burden of gastrointestinal disease in the United States: 2012 update. Gastroenterology. 2012;143(5):1179-1187.e1-e3.
3. Cavallini G, Frulloni L, Bassi C, et al; ProInf-AISP Study Group. Prospective multicentre survey on acute pancreatitis in Italy (ProInf-AISP): results on 1005 patients. Dig Liver Dis. 2004;36(3):205-211.
4. Banks PA, Freeman ML; Practice Parameters Committee of the American College of Gastroenterology. Practice guidelines in acute pancreatitis. Am J Gastroenterol. 2006;101(10):2379-2400.
5. Hartwig W, Werner J, Ryschich E, et al. Cigarette smoke enhances ethanol-induced pancreatic injury. Pancreas. 2000;21(3):272-278.
6. Chowdhury P, Gupta P. Pathophysiology of alcoholic pancreatitis: an overview. World J Gastroenterol. 2006;12(46):7421-7427.
7. Scherer J, Singh VP, Pitchumoni CS, Yadav D. Issues in hypertriglyceridemic pancreatitis: an update. J Clin Gastroenterol. 2014;48(3):195-203.
8. Wu BU, Hwang JQ, Gardner TH, et al. Lactated Ringer’s solution reduces systemic inflammation compared with saline in patients with acute pancreatitis. Clin Gastroenterol Hepatol. 2011;9(8):710-717.e1.
9. Tenner S, Baillie J, DeWitt J, Vege SS; American College of Gastroenterology. American College of Gastroenterology guideline: management of acute pancreatitis. Am J Gastroenterol. 2013;108(9):1400-1415; 1416.
10. Preiss D, Tikkanen MJ, Welsh P, et al. Lipid-modifying therapies and risk of pancreatitis: a meta-analysis. JAMA. 2012;308(8):804-811.
11. Nardi GL. Pancreatitis. N Engl J Med. 1963;268(19):1065-1067.
12. Naeije R, Salingret E, Clumeck N, De Troyer A, Devis G. Is nasogastric suction necessary in acute pancreatitis? Br Med J. 1978;2(6138):659-660.
13. Levant JA, Secrist DM, Resin H, Sturdevant RA, Guth PH. Nasogastric suction in the treatment of alcoholic pancreatitis: a controlled study. JAMA. 1974;229(1):51-52.
14. Zhao XL, Zhu SF, Xue GJ, et al. Early oral refeeding based on hunger in moderate and severe acute pancreatitis: a prospective controlled, randomized clinical trial. Nutrition. 2015;31(1):171-175.
15. Banks PA, Bollen TL, Dervenis C, et al; Acute Pancreatitis Classification Working Group. Classification of acute pancreatitis—2012: revision of the Atlanta classification and definitions by international consensus. Gut. 2013;62(1):102-111.
16. Easler J, Muddana V, Furlan A, et al. Portosplenomesenteric venous thrombosis in patients with acute pancreatitis is associated with pancreatic necrosis and usually has a benign course. Clin Gastroenterol Hepatol. 2014;12(5):854-862.
17. Yadav D, O’Connell M, Papachristou GI. Natural history following the first attack of acute pancreatitis. Am J Gastroenterol. 2012;107(7):1096-1103.
18. Jonas DE, Amick HR, Feltner C, et al. Pharmacotherapy for adults with alcohol use disorders in outpatient settings: a systematic review and meta-analysis. JAMA. 2014;311(18):1889-1900.
19. Garbutt JC, Kranzler HR, O’Malley SS, et al. Efficacy and tolerability of long-acting injectable naltrexone for alcohol dependence: a randomized controlled trial. JAMA. 2005;293(13):1617-1625.
20. Piolot A, Nadler F, Cavallero E, Coquard JL, Jacotot B. Prevention of recurrent acute pancreatitis in patients with severe hypertriglyceridemia: value of regular plasmapheresis. Pancreas. 1996;13(1):96-99.