User login
Molluscum Contagiosum in Immunocompromised Patients: AIDS Presenting as Molluscum Contagiosum in a Patient With Psoriasis on Biologic Therapy
Molluscum contagiosum (MC) is a double-stranded DNA virus of the Poxviridae family, which commonly infects human keratinocytes resulting in small, umbilicated, flesh-colored papules. The greatest incidence of MC is seen in the pediatric population and sexually active young adults, and it is considered a self-limited disease in immunocompetent individuals.1 With the emergence of the human immunodeficiency virus (HIV) and subsequent AIDS epidemic in the 1980s, a new population of immunocompromised individuals has been observed to be increasingly susceptible to MC with an atypical clinical presentation and a recalcitrant disease course.2 Although the increased prevalence of MC in the HIV population has been well-documented, it has been observed in other disease states or iatrogenically induced immunosuppression due to a deficiency in function or absolute number of T lymphocytes.
We present a case of a patient with long-standing psoriasis on biologic therapy who presented with MC with a subsequent workup that revealed AIDS. This case reiterates the importance of MC as a potential indicator of underlying immunosuppression. We review the literature to evaluate the occurrence of MC in immunosuppressed patients.
Case Report
A 33-year-old man initially presented for evaluation of severe plaque-type psoriasis associated with pain, erythema, and swelling of the joints of the hands of 10 years’ duration. He was started on methotrexate 5 mg weekly and topical corticosteroids but was unable to tolerate methotrexate due to headaches. He also had difficulty affording topical medications and adjunctive phototherapy. The patient was sporadically seen in follow-up with persistence of psoriatic plaques involving up to 60% body surface area (BSA) with the only treatment consisting of occasional topical steroids. Five years later, the patient was restarted on methotrexate 5 to 7.5 mg weekly, which resulted in moderate improvement. However, because of persistent elevation of liver enzymes, this treatment was stopped. Several months later he was evaluated for treatment with a biologic agent, and after a negative tuberculin skin test, he began treatment with etanercept 50 mg subcutaneous injection twice weekly, which provided notable improvement and allowed for reduction of dose frequency to once weekly.
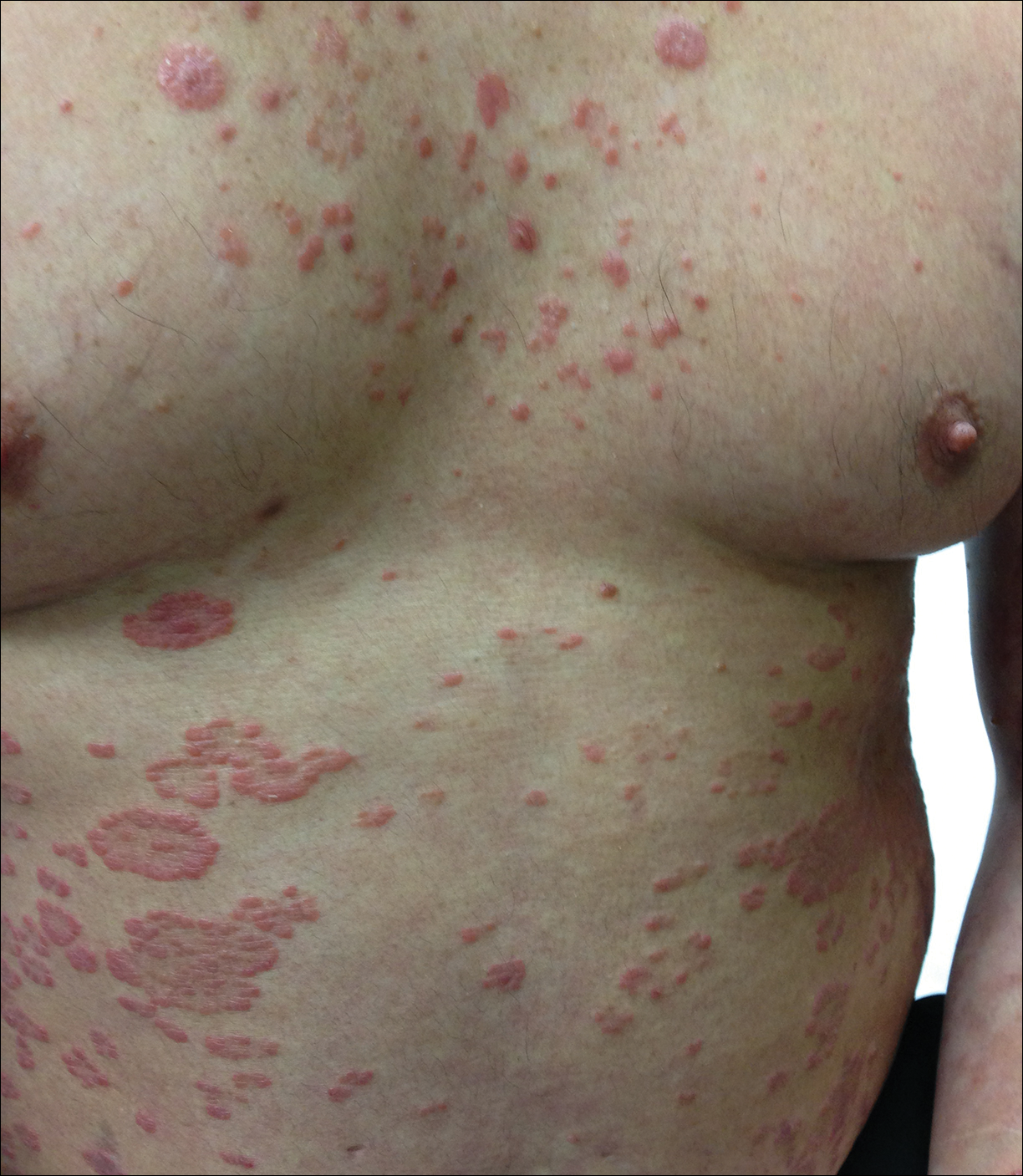
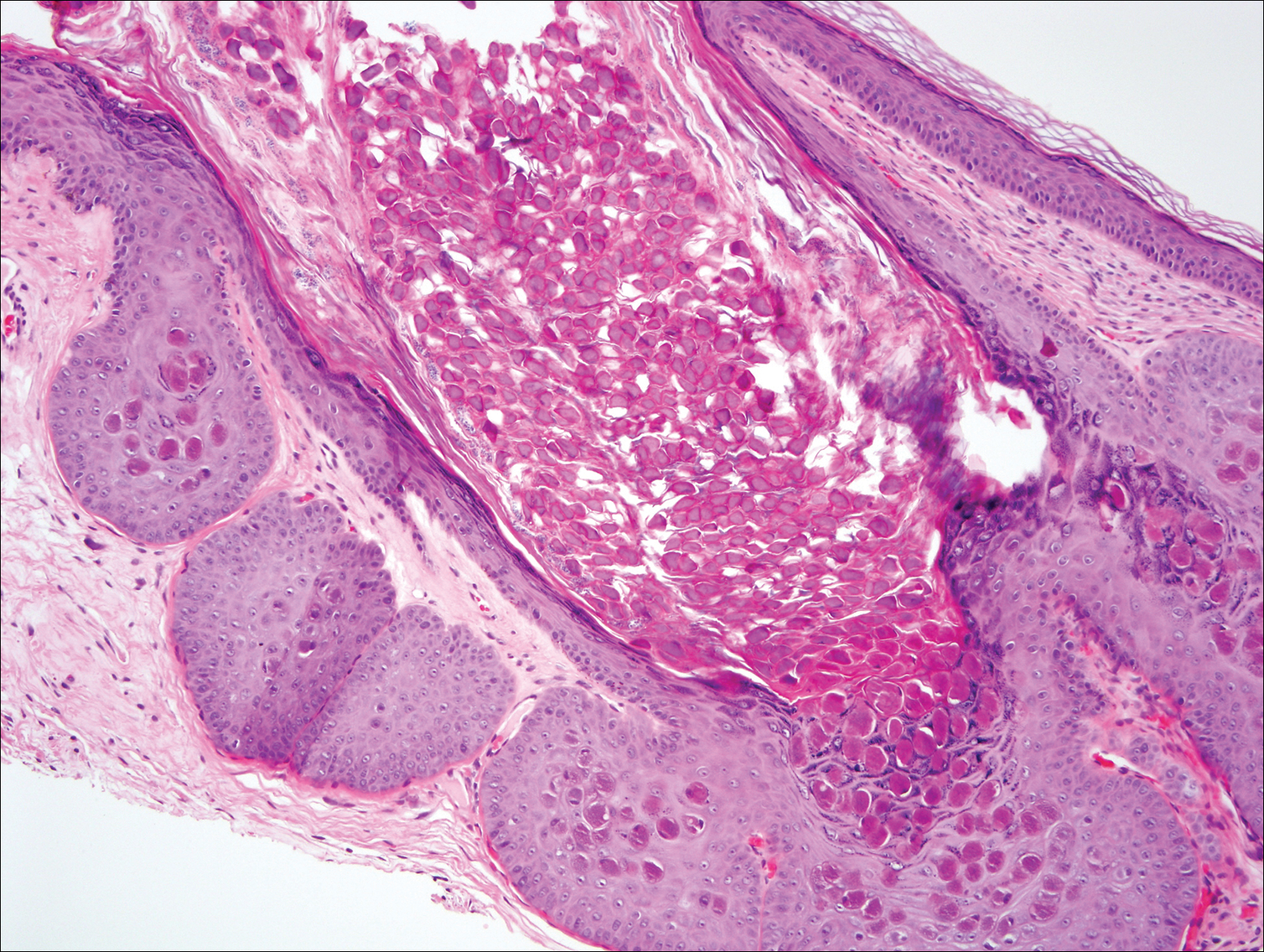
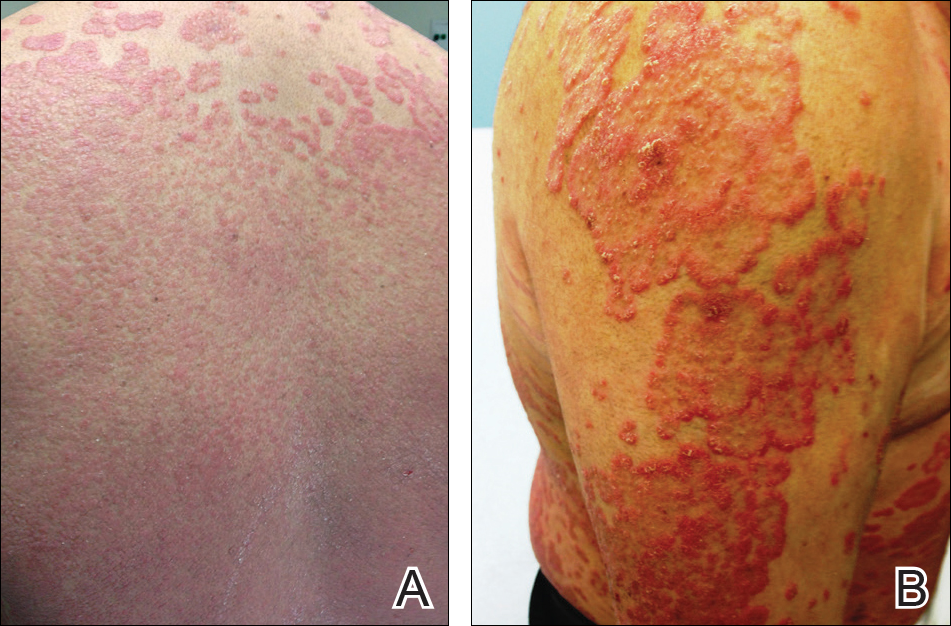
At follow-up 1 year later, the patient had continued improvement of psoriasis with approximately 30% BSA on a treatment regimen of etanercept 50 mg weekly injection and topical corticosteroids. However, on physical examination, there were multiple small semitranslucent papules with telangiectases on the chest and upper back (Figure 1). Biopsy of a representative papule on the chest revealed MC (Figure 2). The patient was subsequently advised to stop etanercept and to return immediately to the clinic for HIV testing. He returned for follow-up 3 months later with pronounced worsening of disease and a new onset of blurred vision of the right eye. Cutaneous examination revealed numerous large erythematous plaques with superficial scale and cerebriform surface on the chest, back, abdomen, and upper and lower extremities involving 80% BSA (Figure 3). Biopsy of a plaque demonstrated psoriasiform dermatitis with neutrophils and parakeratosis consistent with psoriasis. Extensive blood work was notable for reactive HIV antibody and lymphopenia, CD4 lymphocyte count of 60 cells/mm3, and an HIV viral load of 247,000 copies/mL, meeting diagnostic criteria for AIDS. Additionally, ophthalmologic evaluation revealed toxoplasma retinitis. Upon initiation of highly active antiretroviral therapy (HAART) and continued use of topical corticosteroids, the patient experienced notable improvement of disease severity with approximately 20% BSA.
Comment
Molluscum contagiosum is a common skin infection. Among patients with HIV and other types of impaired cellular immunity, the prevalence of MC is estimated to be as high as 20%.3 The MC poxvirus survives and proliferates within the epidermis by interfering with tumor necrosis factor–induced apoptosis of virally infected cells; therefore, intact cell-mediated immunity is an important component of prevention and clearance of poxvirus infections. In immunocompromised patients, the presentation of MC varies widely, and the disease is often difficult to eradicate. This review will highlight the prevalence, presentation, and treatment of MC in the context of immunosuppressed states.
HIV/AIDS
Molluscum contagiosum in HIV-positive patients was first recognized in 1983,2 and its prevalence is estimated to range from 5% to 18% in AIDS patients.3 Molluscum contagiosum is a clinical sign of HIV progression, and its incidence appears to increase with reduced immune function (ie, a CD4 cell count <200/mm3).3 In a study of 456 patients with HIV-associated skin disorders, the majority of patients with MC had notable immunosuppression with a median survival time of 12 months. Thus, MC was not an independent prognostic marker but a clinical indicator of markedly reduced immune status.4
Molluscum contagiosum is transmitted in both sexual and nonsexual patterns in HIV-positive individuals, with the distribution of the latter involving primarily the face and neck. Although it may present with typical umbilicated papules, MC has a wide range of atypical clinical presentations in patients with AIDS that can make it difficult to diagnose. Complicated cases of eyelid MC have been reported in advanced HIV in both adults and children, resulting in obstruction of vision due to large lesions (up to 2 cm) or hundreds of confluent lesions.5 Giant MC, which appears as large exophytic nodules, is another presentation that has been frequently described in patients with advanced HIV. In these patients, the lesions often are too voluminous for conservative therapy and require excision.6 Atypical MC lesions also can resemble other dermatologic conditions, including condyloma acuminatum,7 nevus sebaceous of Jadassohn, ecthyma,8 and cutaneous horns,9,10 as well as other bacterial and fungal infections in HIV-positive patients, such as cutaneous Cryptococcus neoformans,11,12 disseminated histoplasmosis,13 and infections caused by Penicillium marneffei14 and Bartonella henselae.15 In most cases of MC in HIV-positive patients, diagnosis is dependent on the examination of biopsy specimens, which maintain the same histopathologic features regardless of immune status.
The management of MC in patients with HIV/AIDS is difficult. Molluscum contagiosum has shown no evidence of spontaneous resolution in patients with HIV, and treatment with one modality is often insufficient. Treatment is most successful when a combination approach is utilized with destructive procedures (eg, curettage, cryosurgery) and adjunctive agents (eg, retinoids, cantharidin, trichloroacetic acid). Imiquimod and cidofovir have been used off label for MC in AIDS patients.16 Imiquimod, which is used to treat genital warts, another cutaneous viral infection seen in patients with HIV, has demonstrated efficacy in treating MC.16 In a randomized controlled trial comparing imiquimod cream 5% to cryotherapy for MC in healthy children, imiquimod was slow acting but better suited than cryotherapy for patients with eruptions of many small lesions.17 For HIV patients, numerous reports have described successful treatment of disseminated or recalcitrant MC with topical imiquimod.18-20 Cidofovir, an antiviral used to treat cytomegalovirus retinitis in patients with AIDS, is a promising antiviral agent against the poxvirus family. In a study of viral DNA polymerase genes of MC virus, cidofovir inhibited MC virus DNA polymerase activity.21 It has been used in both topical (1% to 3%) and intravenous form to successfully treat recalcitrant and exuberant giant MC.6,22 However, the use of cidofovir is limited by its high costs, especially when compounded into a topical formulation.23
From a systemic standpoint, numerous reports have shown that treating the underlying HIV by optimizing HAART is the most important first step in clearing MC.24-27 However, a special concern regarding the initiation of HAART in patients with MC as well as a markedly impaired immune function is the development of an inflammatory reaction called immune reconstitution inflammatory syndrome (IRIS). This reaction is thought to be a result of immune recovery in severely immunosuppressed patients. During the initial phase of reconstitution when CD4 lymphocyte counts rise and viral load decreases, IRIS occurs due to an inflammatory reaction to microbial and autoimmune antigens, leading to temporary clinical deterioration.28 The incidence has been reported in up to 25% of patients starting HAART, and 52% to 78% of IRIS cases involve dermatologic manifestations such as varicella-zoster virus, cytomegalovirus infections, genital warts, and MC.29,30 In a cohort study of 199 patients, 2% of patients developed MC within 6 months of initiating HAART.31 In a case of exuberant MC lesions after beginning HAART, the lesions spontaneously resolved with the progression of immune reconstitution.28
Malignancies
Patients with hematologic malignancies such as lymphoma and leukemia comprise another subset of patients at risk for atypical presentations of MC. Molluscum contagiosum has been described in patients with hematologic malignancies such as adult T-cell leukemia/lymphoma, multiple myeloma, chronic myeloid leukemia, acute lymphoblastic leukemia, lymphomatoid papulosis, and non-Hodgkin lymphoma. In a review of MC in children with cancer, 0.5% were diagnosed with MC.32,33 Reports also have documented eruptive MC in the presence of solid organ cancers, including lung cancer.34
In patients with malignancies, the differential diagnosis should include other common dermatologic conditions such as varicella, herpes simplex, papillomas, pyoderma, and cutaneous cryptococcosis, as well as MC. Similar to HIV-positive patients, the lesions of MC described in patients with malignancies do not tend to spontaneously resolve. In a report of a pediatric patient with acute lymphoblastic leukemia, MC presented as an ulcerated lesion without any classic features, requiring biopsy for definitive diagnosis. Only partial resolution was achieved with cryotherapy and crusting of the lesion in an attempt to slow the progression.35 In a series of 5 children with hematologic malignancies and MC, little improvement was noted after treatment with surgical scraping, liquid nitrogen, and salicylic acid ointment 5%. Similar to patients with HIV, improvement of immune status and function help clear the disease, and patients who reach remission and discontinue chemotherapeutic agents have a higher rate of spontaneous resolution of previously recalcitrant MC lesions.36
Transplant Patients
Molluscum contagiosum in transplant patients has features similar to patients with HIV/AIDS. In organ transplant recipients, there is an increased risk for cutaneous disease from iatrogenic immunosuppression or immunosuppression through infectious or neoplastic processes.37 As in other immunocompromised populations, MC often has an atypical presentation in transplant patients with more extensive involvement and recalcitrant, rapidly recurring lesions.
In a review of 145 pediatric organ transplant recipients, MC was the fourth most common skin infection after verruca vulgaris, tinea versicolor, and herpes simplex/zoster. Affecting 7% of patients, the majority of patients demonstrated clinically typical lesions; however, the disease was difficult to eradicate if multiple lesions were present.37 In other reports in adults, fulminant and giant MC have been described after renal and other solid organ transplants.38,39 Molluscum contagiosum also has been reported to mimic other skin diseases in transplant patients including tinea barbae40 and nodular basal cell carcinomas.41
The standard treatments are identical to those used in patients with HIV, including ablative methods via liquid nitrogen, electrocautery, cantharidin, trichloroacetic acid, and topical retinoids. Similar to MC in other immunocompromised states, treatment can be difficult and usually requires multiple modalities. For children, imiquimod cream 5% has been recommended due to high clearance rates (up to 92%) and the painless nature of the treatment.42,43
Other Iatrogenic Immunosuppressive States
Immunosuppression through the use of steroids, chemotherapeutic agents, and biologic drugs often is the result of treatment of various diseases. In patients with psoriasis treated with systemic immunosuppressive agents, there are numerous reports that describe the appearance of eruptive MC in association with methotrexate, cyclosporine, and biologics. Methotrexate acts as an immunosuppressive agent by binding to dihydrofolate reductase, which inhibits DNA synthesis in immunologically competent cells.44 It also may block host defense mechanisms against MC by suppressing the expression of serum inflammatory cytokines such as tumor necrosis factor α (TNF-α) and IFN-γ and suppressing the activity of TNF-α inducing apoptosis of virus-infected cells. Cyclosporine used in conjunction with methotrexate may exacerbate the insult to the immune system by inhibiting the production of IFN-γ.45 Biologics are an emerging class of drugs that have demonstrated efficacy in moderate to severe psoriasis by inhibiting TNF-α or other inflammatory molecules. Several published reports have described eruptive or atypical MC in patients on biologic medications. In one case, within 2 weeks after initiation of infliximab, a monoclonal antibody against TNF-α, a patient developed an eruption of MC involving the entire body.46 In another report, an anti–TNF-α agent for rheumatoid arthritis was associated with atypical MC with eyelid lesions.47
There are other skin disorders treated with immunosuppressive agents that also have been associated with MC. In a patient with pemphigus vulgaris treated with prednisolone, pimecrolimus, and azathioprine, MC lesions were observed on the face and within healed pemphigus vulgaris sites.48 Pimecrolimus and tacrolimus, corticosteroid-sparing agents, suppress cell-mediated immunity and inhibit inflammatory cytokines such as IL-2. The infection resolved with a gradual tapering of immunosuppressive therapy and 10 sessions of cryotherapy.48 In a case of topical pimecrolimus for pityriasis alba, the patient developed biopsy-proven MC within 2 weeks of initiating treatment in the areas that were treated with tacrolimus.49
In nontransplant patients with iatrogenic immunosuppression, MC treatment has not been documented to be as challenging as in patients with inherent immunosuppression. Most patients respond to either withdrawal of the drug alone or to simple ablative treatments such as cryotherapy.45,46,48 This important difference is most likely due to the presence of an otherwise intact immune system.
Conclusion
This case describes the appearance of MC in a patient with psoriasis treated with a TNF-α inhibitor who was ultimately diagnosed with AIDS. Although atypical MC infections have been documented in patients with psoriasis undergoing treatment with biologics, it is thought to be more common for MC to occur in more remarkably immunocompromised states such as AIDS. Thus, the persistence and progression of MC in our patient despite discontinuation of etanercept suggested a separate underlying process. Subsequent workup led to the diagnosis of AIDS along with the opportunistic ocular infection of toxoplasmosis retinitis. This clinical sequence consisting of psoriasis treated with a biologic agent, development of MC, and subsequent diagnosis of AIDS is unique and clinically significant to dermatologists. The presentation of psoriasis in patients with HIV can be diverse with different levels of severity and atypical clinical features. In many cases, HIV is known to exacerbate the classic clinical presentation of psoriasis. However, there are other particular presentations of psoriasis in HIV patients that have been observed, which include a predilection for scalp lesions, palmoplantar keratoderma, flexural involvement, and higher levels of immunodeficiency.50 Although tuberculin skin tests are required prior to initiating biologic therapy due to the potential for disease reactivation, there are no requirements for HIV antibody testing. In cases of severe recalcitrant psoriasis, an HIV test should be ordered during the workup to establish an early diagnosis so that an HIV-positive patient can avoid poor outcomes from either the disease processes, the use of certain therapeutic agents, or both. Furthermore, the benefit of avoiding possible harm to the patient and potential legal action outweighs the cost of performing surveillance HIV testing in this subset of patients. Thus, due to the potential additive immunosuppressive effect of HIV with biologic therapy, providers should always assess for risk factors and consider testing for HIV in all patients before initiating treatment with immunosuppressive agents such as biologics.
- Dohil MA, Lin P, Lee J, et al. The epidemiology of molluscum contagiosum in children. J Am Acad Dermatol. 2006;54:47-54.
- Reichert CM, O’Leary TJ, Levens DL, et al. Autopsy pathology in the acquired immune deficiency syndrome. Am J Pathol. 1983;112:357-382.
- Czelusta A, Yen-Moore A, Van der Straten M, et al. An overview of sexually transmitted diseases. Part III. Sexually transmitted diseases in HIV-infected patients. J Am Acad Dermatol. 2000;43:409-432.
- Husak R, Garbe C, Orfanos CE. Mollusca contagiosa in HIV infection. Clinical manifestation, relation to immune status and prognostic value in 39 patients [in German]. Hautarzt. 1997;48:103-109.
- Averbuch D, Jaouni T, Pe’er J, et al. Confluent molluscum contagiosum covering the eyelids of an HIV-positive child. Clin Exp Ophthalmol. 2009;37:525-527.
- Erickson C, Driscoll M, Gaspari A. Efficacy of intravenous cidofovir in the treatment of giant molluscum contagiosum in a patient with human immunodeficiency virus. Arch Dermatol. 2011;147:652-654.
- Mastrolorenzo A, Urbano FG, Salimbeni L, et al. Atypical molluscum contagiosum infection in an HIV-infected patient. Int J Dermatol. 1998;37:378-380.
- Itin PH, Gilli L. Molluscum contagiosum mimicking sebaceous nevus of Jadassohn, ecthyma and giant condylomata acuminata in HIV-infected patients. Dermatology. 1994;189:396-398.
- Sim JH, Lee ES. Molluscum contagiosum presenting as a cutaneous horn. Ann Dermatol. 2011;23:262-263.
- Manchanda Y, Sethuraman G, Paderwani PP, et al. Molluscum contagiosum presenting as penile horn in an HIV positive patient. Sex Transm Infect. 2005;81:183-184.
- Miller SJ. Cutaneous cryptococcus resembling molluscum contagiosum in a patient with acquired immunodeficiency syndrome. Cutis. 1988;41:411-412.
- Sornum A. A mistaken diagnosis of molluscum contagiosum in a HIV-positive patient in rural South Africa. BMJ Case Rep. 2012;14.
- Corti M, Villafañe MF, Palmieri O, et al. Rupioid histoplasmosis: first case reported in an AIDS patient in Argentina. Rev Inst Med Trop Sao Paulo. 2010;52:279-280.
- Saikia L, Nath R, Hazarika D, et al. Atypical cutaneous lesions of Penicillium marneffei infection as a manifestation of the immune reconstitution inflammatory syndrome after highly active antiretroviral therapy. Indian J Dermatol Venereol Leprol. 2010;76:45-48.
- de Souza JA. Molluscum or a mimic? Am J Med. 2006;119:927-929.
- Conant MA. Immunomodulatory therapy in the management of viral infections in patients with HIV infection. J Am Acad Dermatol. 2000;43:S27-S30.
- Gamble RG, Echols KF, Dellavalle RP. Imiquimod vs cryotherapy for molluscum contagiosum: a randomized controlled trial. Arch Dermatol. 2012;148:109-112.
- Brown CW Jr, O’Donoghue M, Moore J, et al. Recalcitrant molluscum contagiosum in an HIV-afflicted male treated successfully with topical imiquimod. Cutis. 2000;65:363-366.
- Strauss RM, Doyle EL, Mohsen AH, et al. Successful treatment of molluscum contagiosum with topical imiquimod in a severely immunocompromised HIV-positive patient. Int J STD AIDS. 2001;12:264-266.
- Theiler M, Kempf W, Kerl K, et al. Disseminated molluscum contagiosum in a HIV-positive child. improvement after therapy with 5% imiquimod. J Dermatol Case Rep. 2011;5:19-23.
- Watanabe T, Tamaki K. Cidofovir diphosphate inhibits molluscum contagiosum virus DNA polymerase activity. J Invest Dermatol. 2008;128:1327-1329.
- Calista D. Topical cidofovir for severe cutaneous human papillomavirus and molluscum contagiosum infections in patients with HIV/AIDS. a pilot study. J Eur Acad Dermatol Venereol. 2000;14:484-488.
- Toro JR, Sanchez S, Turiansky G, et al. Topical cidofovir for the treatment of dermatologic conditions: verruca, condyloma, intraepithelial neoplasia, herpes simplex and its potential use in smallpox. Dermatol Clin. 2003;21:301-309.
- Calista D, Boschini A, Landi G. Resolution of disseminated molluscum contagiosum with highly active anti-retroviral therapy (HAART) in patients with AIDS. Eur J Dermatol. 1999;9:211-213.
- Cattelan AM, Sasset L, Corti L, et al. A complete remission of recalcitrant molluscum contagiosum in an AIDS patient following highly active antiretroviral therapy (HAART). J Infect. 1999;38:58-60.
- Sen S, Bhaumik P. Resolution of giant molluscum contagiosum with antiretroviral therapy. Indian J Dermatol Venereol Leprol. 2008;74:267-268.
- Sen S, Goswami BK, Karjyi N, et al. Disfiguring molluscum contagiosum in a HIV-positive patient responding to antiretroviral therapy. Indian J Dermatol. 2009;54:180-182.
- Pereira B, Fernandes C, Nachiambo E, et al. Exuberant molluscum contagiosum as a manifestation of the immune reconstitution inflammatory syndrome. Dermatol Online J. 2007;13:6.
- Osei-Sekyere B, Karstaedt AS. Immune reconstitution inflammatory syndrome involving the skin. Clin Exp Dermatol. 2010;35:477-481.
- Sung KU, Lee HE, Choi WR, et al. Molluscum contagiosum as a skin manifestation of immune reconstitution inflammatory syndrome in an AIDS patient who is receiving HAART. Korean J Fam Med. 2012;33:182-185.
- Ratnam I, Chiu C, Kandala NB, et al. Incidence and risk factors for immune reconstitution inflammatory syndrome in an ethnically diverse HIV type 1-infected cohort. Clin Infect Dis. 2006;42:418-427.
- Chen KW, Yang CF, Huang CT, et al. Molluscum contagiosum in a patient with adult T-cell leukaemia/lymphoma. Br J Haematol. 2011;155:286.
- Fernandez KH, Bream M, Ali MA, et al. Investigation of molluscum contagiosum virus, orf and other parapoxviruses in lymphomatoid papulosis. J Am Acad Dermatol. 2013;68:1046-1047.
- Nakamura-Wakatsuki T, Kato Y, Miura T, et al. Eruptive molluscum contagiosums in a patient with rheumatoid arthritis and lung cancer. Rheumatol Int. 2011;31:1117-1118.
- Ozyürek E, Sentürk N, Kefeli M, et al. Ulcerating molluscum contagiosum in a boy with relapsed acute lymphoblastic leukemia. J Pediatr Hematol Oncol. 2011;33:E114-E116.
- Hughes WT, Parham DM. Molluscum contagiosum in children with cancer or acquired immunodeficiency syndrome. Pediatr Infect Dis J. 1991;10:152-156.
- Euvrard S, Kanitakis J, Cochat P, et al. Skin diseases in children with organ transplants. J Am Acad Dermatol. 2001;44:932-939.
- Gardner LS, Ormond PJ. Treatment of multiple giant molluscum contagiosum in a renal transplant patient with imiquimod 5% cream. Clin Exp Dermatol. 2006;31:452-453.
- Mansur AT, Göktay F, Gündüz S, et al. Multiple giant molluscum contagiosum in a renal transplant recipient. Transpl Infect Dis. 2004;6:120-123.
- Feldmeyer L, Kamarashev J, Boehler A, et al. Molluscum contagiosum folliculitis mimicking tinea barbae in a lung transplant recipient. J Am Acad Dermatol. 2010;63:169-171.
- Tas¸kapan O, Yenicesu M, Aksu A. A giant solitary molluscum contagiosum, resembling nodular basal cell carcinoma, in a renal transplant recipient. Acta Derm Venereol. 1996;76:247-248.
- Tan HH, Goh CL. Viral infections affecting the skin in organ transplant recipients: epidemiology and current management strategies. Am J Clin Dermatol. 2006;7:13-29.
- Al-Mutairi N, Al-Doukhi A, Al-Farag S, et al. Comparative study on the efficacy, safety, and acceptability of imiquimod 5% cream versus cryotherapy for molluscum contagiosum in children. Pediatr Dermatol. 2010;27:388-394.
- Lim KS, Foo CC. Disseminated molluscum contagiosum in a patient with chronic plaque psoriasis taking methotrexate. Clin Exp Dermatol. 2007;32:591-593.
- Fotiadou C, Lazaridou E, Lekkas D, et al. Disseminated, eruptive molluscum contagiosum lesions in a psoriasis patient under treatment with methotrexate and cyclosporine. Eur J Dermatol. 2012;22:147-148.
- Antoniou C, Kosmadaki MG, Stratigos AJ, et al. Genital HPV lesions and molluscum contagiosum occurring in patients receiving anti-TNF-alpha therapy. Dermatology. 2008;216:364-365.
- Cursiefen C, Grunke M, Dechant C, et al. Multiple bilateral eyelid molluscum contagiosum lesions associated with TNFalpha-antibody and methotrexate therapy. Am J Ophthalmol. 2002;134:270-271.
- Heng YK, Lee JS, Neoh CY. Verrucous plaques in a pemphigus vulgaris patient on immunosuppressive therapy. Int J Dermatol. 2012;51:1044-1046.
- Goksugur N, Ozbostanci B, Goksugur SB. Molluscum contagiosum infection associated with pimecrolimus use in pityriasis alba. Pediatr Dermatol. 2007;24:E63-E65.
- Fernandes S, Pinto GM, Cardoso J. Particular clinical presentations of psoriasis in HIV patients. Int J STD AIDS. 2011;22:653-654.
Molluscum contagiosum (MC) is a double-stranded DNA virus of the Poxviridae family, which commonly infects human keratinocytes resulting in small, umbilicated, flesh-colored papules. The greatest incidence of MC is seen in the pediatric population and sexually active young adults, and it is considered a self-limited disease in immunocompetent individuals.1 With the emergence of the human immunodeficiency virus (HIV) and subsequent AIDS epidemic in the 1980s, a new population of immunocompromised individuals has been observed to be increasingly susceptible to MC with an atypical clinical presentation and a recalcitrant disease course.2 Although the increased prevalence of MC in the HIV population has been well-documented, it has been observed in other disease states or iatrogenically induced immunosuppression due to a deficiency in function or absolute number of T lymphocytes.
We present a case of a patient with long-standing psoriasis on biologic therapy who presented with MC with a subsequent workup that revealed AIDS. This case reiterates the importance of MC as a potential indicator of underlying immunosuppression. We review the literature to evaluate the occurrence of MC in immunosuppressed patients.
Case Report
A 33-year-old man initially presented for evaluation of severe plaque-type psoriasis associated with pain, erythema, and swelling of the joints of the hands of 10 years’ duration. He was started on methotrexate 5 mg weekly and topical corticosteroids but was unable to tolerate methotrexate due to headaches. He also had difficulty affording topical medications and adjunctive phototherapy. The patient was sporadically seen in follow-up with persistence of psoriatic plaques involving up to 60% body surface area (BSA) with the only treatment consisting of occasional topical steroids. Five years later, the patient was restarted on methotrexate 5 to 7.5 mg weekly, which resulted in moderate improvement. However, because of persistent elevation of liver enzymes, this treatment was stopped. Several months later he was evaluated for treatment with a biologic agent, and after a negative tuberculin skin test, he began treatment with etanercept 50 mg subcutaneous injection twice weekly, which provided notable improvement and allowed for reduction of dose frequency to once weekly.
At follow-up 1 year later, the patient had continued improvement of psoriasis with approximately 30% BSA on a treatment regimen of etanercept 50 mg weekly injection and topical corticosteroids. However, on physical examination, there were multiple small semitranslucent papules with telangiectases on the chest and upper back (Figure 1). Biopsy of a representative papule on the chest revealed MC (Figure 2). The patient was subsequently advised to stop etanercept and to return immediately to the clinic for HIV testing. He returned for follow-up 3 months later with pronounced worsening of disease and a new onset of blurred vision of the right eye. Cutaneous examination revealed numerous large erythematous plaques with superficial scale and cerebriform surface on the chest, back, abdomen, and upper and lower extremities involving 80% BSA (Figure 3). Biopsy of a plaque demonstrated psoriasiform dermatitis with neutrophils and parakeratosis consistent with psoriasis. Extensive blood work was notable for reactive HIV antibody and lymphopenia, CD4 lymphocyte count of 60 cells/mm3, and an HIV viral load of 247,000 copies/mL, meeting diagnostic criteria for AIDS. Additionally, ophthalmologic evaluation revealed toxoplasma retinitis. Upon initiation of highly active antiretroviral therapy (HAART) and continued use of topical corticosteroids, the patient experienced notable improvement of disease severity with approximately 20% BSA.
Comment
Molluscum contagiosum is a common skin infection. Among patients with HIV and other types of impaired cellular immunity, the prevalence of MC is estimated to be as high as 20%.3 The MC poxvirus survives and proliferates within the epidermis by interfering with tumor necrosis factor–induced apoptosis of virally infected cells; therefore, intact cell-mediated immunity is an important component of prevention and clearance of poxvirus infections. In immunocompromised patients, the presentation of MC varies widely, and the disease is often difficult to eradicate. This review will highlight the prevalence, presentation, and treatment of MC in the context of immunosuppressed states.
HIV/AIDS
Molluscum contagiosum in HIV-positive patients was first recognized in 1983,2 and its prevalence is estimated to range from 5% to 18% in AIDS patients.3 Molluscum contagiosum is a clinical sign of HIV progression, and its incidence appears to increase with reduced immune function (ie, a CD4 cell count <200/mm3).3 In a study of 456 patients with HIV-associated skin disorders, the majority of patients with MC had notable immunosuppression with a median survival time of 12 months. Thus, MC was not an independent prognostic marker but a clinical indicator of markedly reduced immune status.4
Molluscum contagiosum is transmitted in both sexual and nonsexual patterns in HIV-positive individuals, with the distribution of the latter involving primarily the face and neck. Although it may present with typical umbilicated papules, MC has a wide range of atypical clinical presentations in patients with AIDS that can make it difficult to diagnose. Complicated cases of eyelid MC have been reported in advanced HIV in both adults and children, resulting in obstruction of vision due to large lesions (up to 2 cm) or hundreds of confluent lesions.5 Giant MC, which appears as large exophytic nodules, is another presentation that has been frequently described in patients with advanced HIV. In these patients, the lesions often are too voluminous for conservative therapy and require excision.6 Atypical MC lesions also can resemble other dermatologic conditions, including condyloma acuminatum,7 nevus sebaceous of Jadassohn, ecthyma,8 and cutaneous horns,9,10 as well as other bacterial and fungal infections in HIV-positive patients, such as cutaneous Cryptococcus neoformans,11,12 disseminated histoplasmosis,13 and infections caused by Penicillium marneffei14 and Bartonella henselae.15 In most cases of MC in HIV-positive patients, diagnosis is dependent on the examination of biopsy specimens, which maintain the same histopathologic features regardless of immune status.
The management of MC in patients with HIV/AIDS is difficult. Molluscum contagiosum has shown no evidence of spontaneous resolution in patients with HIV, and treatment with one modality is often insufficient. Treatment is most successful when a combination approach is utilized with destructive procedures (eg, curettage, cryosurgery) and adjunctive agents (eg, retinoids, cantharidin, trichloroacetic acid). Imiquimod and cidofovir have been used off label for MC in AIDS patients.16 Imiquimod, which is used to treat genital warts, another cutaneous viral infection seen in patients with HIV, has demonstrated efficacy in treating MC.16 In a randomized controlled trial comparing imiquimod cream 5% to cryotherapy for MC in healthy children, imiquimod was slow acting but better suited than cryotherapy for patients with eruptions of many small lesions.17 For HIV patients, numerous reports have described successful treatment of disseminated or recalcitrant MC with topical imiquimod.18-20 Cidofovir, an antiviral used to treat cytomegalovirus retinitis in patients with AIDS, is a promising antiviral agent against the poxvirus family. In a study of viral DNA polymerase genes of MC virus, cidofovir inhibited MC virus DNA polymerase activity.21 It has been used in both topical (1% to 3%) and intravenous form to successfully treat recalcitrant and exuberant giant MC.6,22 However, the use of cidofovir is limited by its high costs, especially when compounded into a topical formulation.23
From a systemic standpoint, numerous reports have shown that treating the underlying HIV by optimizing HAART is the most important first step in clearing MC.24-27 However, a special concern regarding the initiation of HAART in patients with MC as well as a markedly impaired immune function is the development of an inflammatory reaction called immune reconstitution inflammatory syndrome (IRIS). This reaction is thought to be a result of immune recovery in severely immunosuppressed patients. During the initial phase of reconstitution when CD4 lymphocyte counts rise and viral load decreases, IRIS occurs due to an inflammatory reaction to microbial and autoimmune antigens, leading to temporary clinical deterioration.28 The incidence has been reported in up to 25% of patients starting HAART, and 52% to 78% of IRIS cases involve dermatologic manifestations such as varicella-zoster virus, cytomegalovirus infections, genital warts, and MC.29,30 In a cohort study of 199 patients, 2% of patients developed MC within 6 months of initiating HAART.31 In a case of exuberant MC lesions after beginning HAART, the lesions spontaneously resolved with the progression of immune reconstitution.28
Malignancies
Patients with hematologic malignancies such as lymphoma and leukemia comprise another subset of patients at risk for atypical presentations of MC. Molluscum contagiosum has been described in patients with hematologic malignancies such as adult T-cell leukemia/lymphoma, multiple myeloma, chronic myeloid leukemia, acute lymphoblastic leukemia, lymphomatoid papulosis, and non-Hodgkin lymphoma. In a review of MC in children with cancer, 0.5% were diagnosed with MC.32,33 Reports also have documented eruptive MC in the presence of solid organ cancers, including lung cancer.34
In patients with malignancies, the differential diagnosis should include other common dermatologic conditions such as varicella, herpes simplex, papillomas, pyoderma, and cutaneous cryptococcosis, as well as MC. Similar to HIV-positive patients, the lesions of MC described in patients with malignancies do not tend to spontaneously resolve. In a report of a pediatric patient with acute lymphoblastic leukemia, MC presented as an ulcerated lesion without any classic features, requiring biopsy for definitive diagnosis. Only partial resolution was achieved with cryotherapy and crusting of the lesion in an attempt to slow the progression.35 In a series of 5 children with hematologic malignancies and MC, little improvement was noted after treatment with surgical scraping, liquid nitrogen, and salicylic acid ointment 5%. Similar to patients with HIV, improvement of immune status and function help clear the disease, and patients who reach remission and discontinue chemotherapeutic agents have a higher rate of spontaneous resolution of previously recalcitrant MC lesions.36
Transplant Patients
Molluscum contagiosum in transplant patients has features similar to patients with HIV/AIDS. In organ transplant recipients, there is an increased risk for cutaneous disease from iatrogenic immunosuppression or immunosuppression through infectious or neoplastic processes.37 As in other immunocompromised populations, MC often has an atypical presentation in transplant patients with more extensive involvement and recalcitrant, rapidly recurring lesions.
In a review of 145 pediatric organ transplant recipients, MC was the fourth most common skin infection after verruca vulgaris, tinea versicolor, and herpes simplex/zoster. Affecting 7% of patients, the majority of patients demonstrated clinically typical lesions; however, the disease was difficult to eradicate if multiple lesions were present.37 In other reports in adults, fulminant and giant MC have been described after renal and other solid organ transplants.38,39 Molluscum contagiosum also has been reported to mimic other skin diseases in transplant patients including tinea barbae40 and nodular basal cell carcinomas.41
The standard treatments are identical to those used in patients with HIV, including ablative methods via liquid nitrogen, electrocautery, cantharidin, trichloroacetic acid, and topical retinoids. Similar to MC in other immunocompromised states, treatment can be difficult and usually requires multiple modalities. For children, imiquimod cream 5% has been recommended due to high clearance rates (up to 92%) and the painless nature of the treatment.42,43
Other Iatrogenic Immunosuppressive States
Immunosuppression through the use of steroids, chemotherapeutic agents, and biologic drugs often is the result of treatment of various diseases. In patients with psoriasis treated with systemic immunosuppressive agents, there are numerous reports that describe the appearance of eruptive MC in association with methotrexate, cyclosporine, and biologics. Methotrexate acts as an immunosuppressive agent by binding to dihydrofolate reductase, which inhibits DNA synthesis in immunologically competent cells.44 It also may block host defense mechanisms against MC by suppressing the expression of serum inflammatory cytokines such as tumor necrosis factor α (TNF-α) and IFN-γ and suppressing the activity of TNF-α inducing apoptosis of virus-infected cells. Cyclosporine used in conjunction with methotrexate may exacerbate the insult to the immune system by inhibiting the production of IFN-γ.45 Biologics are an emerging class of drugs that have demonstrated efficacy in moderate to severe psoriasis by inhibiting TNF-α or other inflammatory molecules. Several published reports have described eruptive or atypical MC in patients on biologic medications. In one case, within 2 weeks after initiation of infliximab, a monoclonal antibody against TNF-α, a patient developed an eruption of MC involving the entire body.46 In another report, an anti–TNF-α agent for rheumatoid arthritis was associated with atypical MC with eyelid lesions.47
There are other skin disorders treated with immunosuppressive agents that also have been associated with MC. In a patient with pemphigus vulgaris treated with prednisolone, pimecrolimus, and azathioprine, MC lesions were observed on the face and within healed pemphigus vulgaris sites.48 Pimecrolimus and tacrolimus, corticosteroid-sparing agents, suppress cell-mediated immunity and inhibit inflammatory cytokines such as IL-2. The infection resolved with a gradual tapering of immunosuppressive therapy and 10 sessions of cryotherapy.48 In a case of topical pimecrolimus for pityriasis alba, the patient developed biopsy-proven MC within 2 weeks of initiating treatment in the areas that were treated with tacrolimus.49
In nontransplant patients with iatrogenic immunosuppression, MC treatment has not been documented to be as challenging as in patients with inherent immunosuppression. Most patients respond to either withdrawal of the drug alone or to simple ablative treatments such as cryotherapy.45,46,48 This important difference is most likely due to the presence of an otherwise intact immune system.
Conclusion
This case describes the appearance of MC in a patient with psoriasis treated with a TNF-α inhibitor who was ultimately diagnosed with AIDS. Although atypical MC infections have been documented in patients with psoriasis undergoing treatment with biologics, it is thought to be more common for MC to occur in more remarkably immunocompromised states such as AIDS. Thus, the persistence and progression of MC in our patient despite discontinuation of etanercept suggested a separate underlying process. Subsequent workup led to the diagnosis of AIDS along with the opportunistic ocular infection of toxoplasmosis retinitis. This clinical sequence consisting of psoriasis treated with a biologic agent, development of MC, and subsequent diagnosis of AIDS is unique and clinically significant to dermatologists. The presentation of psoriasis in patients with HIV can be diverse with different levels of severity and atypical clinical features. In many cases, HIV is known to exacerbate the classic clinical presentation of psoriasis. However, there are other particular presentations of psoriasis in HIV patients that have been observed, which include a predilection for scalp lesions, palmoplantar keratoderma, flexural involvement, and higher levels of immunodeficiency.50 Although tuberculin skin tests are required prior to initiating biologic therapy due to the potential for disease reactivation, there are no requirements for HIV antibody testing. In cases of severe recalcitrant psoriasis, an HIV test should be ordered during the workup to establish an early diagnosis so that an HIV-positive patient can avoid poor outcomes from either the disease processes, the use of certain therapeutic agents, or both. Furthermore, the benefit of avoiding possible harm to the patient and potential legal action outweighs the cost of performing surveillance HIV testing in this subset of patients. Thus, due to the potential additive immunosuppressive effect of HIV with biologic therapy, providers should always assess for risk factors and consider testing for HIV in all patients before initiating treatment with immunosuppressive agents such as biologics.
Molluscum contagiosum (MC) is a double-stranded DNA virus of the Poxviridae family, which commonly infects human keratinocytes resulting in small, umbilicated, flesh-colored papules. The greatest incidence of MC is seen in the pediatric population and sexually active young adults, and it is considered a self-limited disease in immunocompetent individuals.1 With the emergence of the human immunodeficiency virus (HIV) and subsequent AIDS epidemic in the 1980s, a new population of immunocompromised individuals has been observed to be increasingly susceptible to MC with an atypical clinical presentation and a recalcitrant disease course.2 Although the increased prevalence of MC in the HIV population has been well-documented, it has been observed in other disease states or iatrogenically induced immunosuppression due to a deficiency in function or absolute number of T lymphocytes.
We present a case of a patient with long-standing psoriasis on biologic therapy who presented with MC with a subsequent workup that revealed AIDS. This case reiterates the importance of MC as a potential indicator of underlying immunosuppression. We review the literature to evaluate the occurrence of MC in immunosuppressed patients.
Case Report
A 33-year-old man initially presented for evaluation of severe plaque-type psoriasis associated with pain, erythema, and swelling of the joints of the hands of 10 years’ duration. He was started on methotrexate 5 mg weekly and topical corticosteroids but was unable to tolerate methotrexate due to headaches. He also had difficulty affording topical medications and adjunctive phototherapy. The patient was sporadically seen in follow-up with persistence of psoriatic plaques involving up to 60% body surface area (BSA) with the only treatment consisting of occasional topical steroids. Five years later, the patient was restarted on methotrexate 5 to 7.5 mg weekly, which resulted in moderate improvement. However, because of persistent elevation of liver enzymes, this treatment was stopped. Several months later he was evaluated for treatment with a biologic agent, and after a negative tuberculin skin test, he began treatment with etanercept 50 mg subcutaneous injection twice weekly, which provided notable improvement and allowed for reduction of dose frequency to once weekly.
At follow-up 1 year later, the patient had continued improvement of psoriasis with approximately 30% BSA on a treatment regimen of etanercept 50 mg weekly injection and topical corticosteroids. However, on physical examination, there were multiple small semitranslucent papules with telangiectases on the chest and upper back (Figure 1). Biopsy of a representative papule on the chest revealed MC (Figure 2). The patient was subsequently advised to stop etanercept and to return immediately to the clinic for HIV testing. He returned for follow-up 3 months later with pronounced worsening of disease and a new onset of blurred vision of the right eye. Cutaneous examination revealed numerous large erythematous plaques with superficial scale and cerebriform surface on the chest, back, abdomen, and upper and lower extremities involving 80% BSA (Figure 3). Biopsy of a plaque demonstrated psoriasiform dermatitis with neutrophils and parakeratosis consistent with psoriasis. Extensive blood work was notable for reactive HIV antibody and lymphopenia, CD4 lymphocyte count of 60 cells/mm3, and an HIV viral load of 247,000 copies/mL, meeting diagnostic criteria for AIDS. Additionally, ophthalmologic evaluation revealed toxoplasma retinitis. Upon initiation of highly active antiretroviral therapy (HAART) and continued use of topical corticosteroids, the patient experienced notable improvement of disease severity with approximately 20% BSA.
Comment
Molluscum contagiosum is a common skin infection. Among patients with HIV and other types of impaired cellular immunity, the prevalence of MC is estimated to be as high as 20%.3 The MC poxvirus survives and proliferates within the epidermis by interfering with tumor necrosis factor–induced apoptosis of virally infected cells; therefore, intact cell-mediated immunity is an important component of prevention and clearance of poxvirus infections. In immunocompromised patients, the presentation of MC varies widely, and the disease is often difficult to eradicate. This review will highlight the prevalence, presentation, and treatment of MC in the context of immunosuppressed states.
HIV/AIDS
Molluscum contagiosum in HIV-positive patients was first recognized in 1983,2 and its prevalence is estimated to range from 5% to 18% in AIDS patients.3 Molluscum contagiosum is a clinical sign of HIV progression, and its incidence appears to increase with reduced immune function (ie, a CD4 cell count <200/mm3).3 In a study of 456 patients with HIV-associated skin disorders, the majority of patients with MC had notable immunosuppression with a median survival time of 12 months. Thus, MC was not an independent prognostic marker but a clinical indicator of markedly reduced immune status.4
Molluscum contagiosum is transmitted in both sexual and nonsexual patterns in HIV-positive individuals, with the distribution of the latter involving primarily the face and neck. Although it may present with typical umbilicated papules, MC has a wide range of atypical clinical presentations in patients with AIDS that can make it difficult to diagnose. Complicated cases of eyelid MC have been reported in advanced HIV in both adults and children, resulting in obstruction of vision due to large lesions (up to 2 cm) or hundreds of confluent lesions.5 Giant MC, which appears as large exophytic nodules, is another presentation that has been frequently described in patients with advanced HIV. In these patients, the lesions often are too voluminous for conservative therapy and require excision.6 Atypical MC lesions also can resemble other dermatologic conditions, including condyloma acuminatum,7 nevus sebaceous of Jadassohn, ecthyma,8 and cutaneous horns,9,10 as well as other bacterial and fungal infections in HIV-positive patients, such as cutaneous Cryptococcus neoformans,11,12 disseminated histoplasmosis,13 and infections caused by Penicillium marneffei14 and Bartonella henselae.15 In most cases of MC in HIV-positive patients, diagnosis is dependent on the examination of biopsy specimens, which maintain the same histopathologic features regardless of immune status.
The management of MC in patients with HIV/AIDS is difficult. Molluscum contagiosum has shown no evidence of spontaneous resolution in patients with HIV, and treatment with one modality is often insufficient. Treatment is most successful when a combination approach is utilized with destructive procedures (eg, curettage, cryosurgery) and adjunctive agents (eg, retinoids, cantharidin, trichloroacetic acid). Imiquimod and cidofovir have been used off label for MC in AIDS patients.16 Imiquimod, which is used to treat genital warts, another cutaneous viral infection seen in patients with HIV, has demonstrated efficacy in treating MC.16 In a randomized controlled trial comparing imiquimod cream 5% to cryotherapy for MC in healthy children, imiquimod was slow acting but better suited than cryotherapy for patients with eruptions of many small lesions.17 For HIV patients, numerous reports have described successful treatment of disseminated or recalcitrant MC with topical imiquimod.18-20 Cidofovir, an antiviral used to treat cytomegalovirus retinitis in patients with AIDS, is a promising antiviral agent against the poxvirus family. In a study of viral DNA polymerase genes of MC virus, cidofovir inhibited MC virus DNA polymerase activity.21 It has been used in both topical (1% to 3%) and intravenous form to successfully treat recalcitrant and exuberant giant MC.6,22 However, the use of cidofovir is limited by its high costs, especially when compounded into a topical formulation.23
From a systemic standpoint, numerous reports have shown that treating the underlying HIV by optimizing HAART is the most important first step in clearing MC.24-27 However, a special concern regarding the initiation of HAART in patients with MC as well as a markedly impaired immune function is the development of an inflammatory reaction called immune reconstitution inflammatory syndrome (IRIS). This reaction is thought to be a result of immune recovery in severely immunosuppressed patients. During the initial phase of reconstitution when CD4 lymphocyte counts rise and viral load decreases, IRIS occurs due to an inflammatory reaction to microbial and autoimmune antigens, leading to temporary clinical deterioration.28 The incidence has been reported in up to 25% of patients starting HAART, and 52% to 78% of IRIS cases involve dermatologic manifestations such as varicella-zoster virus, cytomegalovirus infections, genital warts, and MC.29,30 In a cohort study of 199 patients, 2% of patients developed MC within 6 months of initiating HAART.31 In a case of exuberant MC lesions after beginning HAART, the lesions spontaneously resolved with the progression of immune reconstitution.28
Malignancies
Patients with hematologic malignancies such as lymphoma and leukemia comprise another subset of patients at risk for atypical presentations of MC. Molluscum contagiosum has been described in patients with hematologic malignancies such as adult T-cell leukemia/lymphoma, multiple myeloma, chronic myeloid leukemia, acute lymphoblastic leukemia, lymphomatoid papulosis, and non-Hodgkin lymphoma. In a review of MC in children with cancer, 0.5% were diagnosed with MC.32,33 Reports also have documented eruptive MC in the presence of solid organ cancers, including lung cancer.34
In patients with malignancies, the differential diagnosis should include other common dermatologic conditions such as varicella, herpes simplex, papillomas, pyoderma, and cutaneous cryptococcosis, as well as MC. Similar to HIV-positive patients, the lesions of MC described in patients with malignancies do not tend to spontaneously resolve. In a report of a pediatric patient with acute lymphoblastic leukemia, MC presented as an ulcerated lesion without any classic features, requiring biopsy for definitive diagnosis. Only partial resolution was achieved with cryotherapy and crusting of the lesion in an attempt to slow the progression.35 In a series of 5 children with hematologic malignancies and MC, little improvement was noted after treatment with surgical scraping, liquid nitrogen, and salicylic acid ointment 5%. Similar to patients with HIV, improvement of immune status and function help clear the disease, and patients who reach remission and discontinue chemotherapeutic agents have a higher rate of spontaneous resolution of previously recalcitrant MC lesions.36
Transplant Patients
Molluscum contagiosum in transplant patients has features similar to patients with HIV/AIDS. In organ transplant recipients, there is an increased risk for cutaneous disease from iatrogenic immunosuppression or immunosuppression through infectious or neoplastic processes.37 As in other immunocompromised populations, MC often has an atypical presentation in transplant patients with more extensive involvement and recalcitrant, rapidly recurring lesions.
In a review of 145 pediatric organ transplant recipients, MC was the fourth most common skin infection after verruca vulgaris, tinea versicolor, and herpes simplex/zoster. Affecting 7% of patients, the majority of patients demonstrated clinically typical lesions; however, the disease was difficult to eradicate if multiple lesions were present.37 In other reports in adults, fulminant and giant MC have been described after renal and other solid organ transplants.38,39 Molluscum contagiosum also has been reported to mimic other skin diseases in transplant patients including tinea barbae40 and nodular basal cell carcinomas.41
The standard treatments are identical to those used in patients with HIV, including ablative methods via liquid nitrogen, electrocautery, cantharidin, trichloroacetic acid, and topical retinoids. Similar to MC in other immunocompromised states, treatment can be difficult and usually requires multiple modalities. For children, imiquimod cream 5% has been recommended due to high clearance rates (up to 92%) and the painless nature of the treatment.42,43
Other Iatrogenic Immunosuppressive States
Immunosuppression through the use of steroids, chemotherapeutic agents, and biologic drugs often is the result of treatment of various diseases. In patients with psoriasis treated with systemic immunosuppressive agents, there are numerous reports that describe the appearance of eruptive MC in association with methotrexate, cyclosporine, and biologics. Methotrexate acts as an immunosuppressive agent by binding to dihydrofolate reductase, which inhibits DNA synthesis in immunologically competent cells.44 It also may block host defense mechanisms against MC by suppressing the expression of serum inflammatory cytokines such as tumor necrosis factor α (TNF-α) and IFN-γ and suppressing the activity of TNF-α inducing apoptosis of virus-infected cells. Cyclosporine used in conjunction with methotrexate may exacerbate the insult to the immune system by inhibiting the production of IFN-γ.45 Biologics are an emerging class of drugs that have demonstrated efficacy in moderate to severe psoriasis by inhibiting TNF-α or other inflammatory molecules. Several published reports have described eruptive or atypical MC in patients on biologic medications. In one case, within 2 weeks after initiation of infliximab, a monoclonal antibody against TNF-α, a patient developed an eruption of MC involving the entire body.46 In another report, an anti–TNF-α agent for rheumatoid arthritis was associated with atypical MC with eyelid lesions.47
There are other skin disorders treated with immunosuppressive agents that also have been associated with MC. In a patient with pemphigus vulgaris treated with prednisolone, pimecrolimus, and azathioprine, MC lesions were observed on the face and within healed pemphigus vulgaris sites.48 Pimecrolimus and tacrolimus, corticosteroid-sparing agents, suppress cell-mediated immunity and inhibit inflammatory cytokines such as IL-2. The infection resolved with a gradual tapering of immunosuppressive therapy and 10 sessions of cryotherapy.48 In a case of topical pimecrolimus for pityriasis alba, the patient developed biopsy-proven MC within 2 weeks of initiating treatment in the areas that were treated with tacrolimus.49
In nontransplant patients with iatrogenic immunosuppression, MC treatment has not been documented to be as challenging as in patients with inherent immunosuppression. Most patients respond to either withdrawal of the drug alone or to simple ablative treatments such as cryotherapy.45,46,48 This important difference is most likely due to the presence of an otherwise intact immune system.
Conclusion
This case describes the appearance of MC in a patient with psoriasis treated with a TNF-α inhibitor who was ultimately diagnosed with AIDS. Although atypical MC infections have been documented in patients with psoriasis undergoing treatment with biologics, it is thought to be more common for MC to occur in more remarkably immunocompromised states such as AIDS. Thus, the persistence and progression of MC in our patient despite discontinuation of etanercept suggested a separate underlying process. Subsequent workup led to the diagnosis of AIDS along with the opportunistic ocular infection of toxoplasmosis retinitis. This clinical sequence consisting of psoriasis treated with a biologic agent, development of MC, and subsequent diagnosis of AIDS is unique and clinically significant to dermatologists. The presentation of psoriasis in patients with HIV can be diverse with different levels of severity and atypical clinical features. In many cases, HIV is known to exacerbate the classic clinical presentation of psoriasis. However, there are other particular presentations of psoriasis in HIV patients that have been observed, which include a predilection for scalp lesions, palmoplantar keratoderma, flexural involvement, and higher levels of immunodeficiency.50 Although tuberculin skin tests are required prior to initiating biologic therapy due to the potential for disease reactivation, there are no requirements for HIV antibody testing. In cases of severe recalcitrant psoriasis, an HIV test should be ordered during the workup to establish an early diagnosis so that an HIV-positive patient can avoid poor outcomes from either the disease processes, the use of certain therapeutic agents, or both. Furthermore, the benefit of avoiding possible harm to the patient and potential legal action outweighs the cost of performing surveillance HIV testing in this subset of patients. Thus, due to the potential additive immunosuppressive effect of HIV with biologic therapy, providers should always assess for risk factors and consider testing for HIV in all patients before initiating treatment with immunosuppressive agents such as biologics.
- Dohil MA, Lin P, Lee J, et al. The epidemiology of molluscum contagiosum in children. J Am Acad Dermatol. 2006;54:47-54.
- Reichert CM, O’Leary TJ, Levens DL, et al. Autopsy pathology in the acquired immune deficiency syndrome. Am J Pathol. 1983;112:357-382.
- Czelusta A, Yen-Moore A, Van der Straten M, et al. An overview of sexually transmitted diseases. Part III. Sexually transmitted diseases in HIV-infected patients. J Am Acad Dermatol. 2000;43:409-432.
- Husak R, Garbe C, Orfanos CE. Mollusca contagiosa in HIV infection. Clinical manifestation, relation to immune status and prognostic value in 39 patients [in German]. Hautarzt. 1997;48:103-109.
- Averbuch D, Jaouni T, Pe’er J, et al. Confluent molluscum contagiosum covering the eyelids of an HIV-positive child. Clin Exp Ophthalmol. 2009;37:525-527.
- Erickson C, Driscoll M, Gaspari A. Efficacy of intravenous cidofovir in the treatment of giant molluscum contagiosum in a patient with human immunodeficiency virus. Arch Dermatol. 2011;147:652-654.
- Mastrolorenzo A, Urbano FG, Salimbeni L, et al. Atypical molluscum contagiosum infection in an HIV-infected patient. Int J Dermatol. 1998;37:378-380.
- Itin PH, Gilli L. Molluscum contagiosum mimicking sebaceous nevus of Jadassohn, ecthyma and giant condylomata acuminata in HIV-infected patients. Dermatology. 1994;189:396-398.
- Sim JH, Lee ES. Molluscum contagiosum presenting as a cutaneous horn. Ann Dermatol. 2011;23:262-263.
- Manchanda Y, Sethuraman G, Paderwani PP, et al. Molluscum contagiosum presenting as penile horn in an HIV positive patient. Sex Transm Infect. 2005;81:183-184.
- Miller SJ. Cutaneous cryptococcus resembling molluscum contagiosum in a patient with acquired immunodeficiency syndrome. Cutis. 1988;41:411-412.
- Sornum A. A mistaken diagnosis of molluscum contagiosum in a HIV-positive patient in rural South Africa. BMJ Case Rep. 2012;14.
- Corti M, Villafañe MF, Palmieri O, et al. Rupioid histoplasmosis: first case reported in an AIDS patient in Argentina. Rev Inst Med Trop Sao Paulo. 2010;52:279-280.
- Saikia L, Nath R, Hazarika D, et al. Atypical cutaneous lesions of Penicillium marneffei infection as a manifestation of the immune reconstitution inflammatory syndrome after highly active antiretroviral therapy. Indian J Dermatol Venereol Leprol. 2010;76:45-48.
- de Souza JA. Molluscum or a mimic? Am J Med. 2006;119:927-929.
- Conant MA. Immunomodulatory therapy in the management of viral infections in patients with HIV infection. J Am Acad Dermatol. 2000;43:S27-S30.
- Gamble RG, Echols KF, Dellavalle RP. Imiquimod vs cryotherapy for molluscum contagiosum: a randomized controlled trial. Arch Dermatol. 2012;148:109-112.
- Brown CW Jr, O’Donoghue M, Moore J, et al. Recalcitrant molluscum contagiosum in an HIV-afflicted male treated successfully with topical imiquimod. Cutis. 2000;65:363-366.
- Strauss RM, Doyle EL, Mohsen AH, et al. Successful treatment of molluscum contagiosum with topical imiquimod in a severely immunocompromised HIV-positive patient. Int J STD AIDS. 2001;12:264-266.
- Theiler M, Kempf W, Kerl K, et al. Disseminated molluscum contagiosum in a HIV-positive child. improvement after therapy with 5% imiquimod. J Dermatol Case Rep. 2011;5:19-23.
- Watanabe T, Tamaki K. Cidofovir diphosphate inhibits molluscum contagiosum virus DNA polymerase activity. J Invest Dermatol. 2008;128:1327-1329.
- Calista D. Topical cidofovir for severe cutaneous human papillomavirus and molluscum contagiosum infections in patients with HIV/AIDS. a pilot study. J Eur Acad Dermatol Venereol. 2000;14:484-488.
- Toro JR, Sanchez S, Turiansky G, et al. Topical cidofovir for the treatment of dermatologic conditions: verruca, condyloma, intraepithelial neoplasia, herpes simplex and its potential use in smallpox. Dermatol Clin. 2003;21:301-309.
- Calista D, Boschini A, Landi G. Resolution of disseminated molluscum contagiosum with highly active anti-retroviral therapy (HAART) in patients with AIDS. Eur J Dermatol. 1999;9:211-213.
- Cattelan AM, Sasset L, Corti L, et al. A complete remission of recalcitrant molluscum contagiosum in an AIDS patient following highly active antiretroviral therapy (HAART). J Infect. 1999;38:58-60.
- Sen S, Bhaumik P. Resolution of giant molluscum contagiosum with antiretroviral therapy. Indian J Dermatol Venereol Leprol. 2008;74:267-268.
- Sen S, Goswami BK, Karjyi N, et al. Disfiguring molluscum contagiosum in a HIV-positive patient responding to antiretroviral therapy. Indian J Dermatol. 2009;54:180-182.
- Pereira B, Fernandes C, Nachiambo E, et al. Exuberant molluscum contagiosum as a manifestation of the immune reconstitution inflammatory syndrome. Dermatol Online J. 2007;13:6.
- Osei-Sekyere B, Karstaedt AS. Immune reconstitution inflammatory syndrome involving the skin. Clin Exp Dermatol. 2010;35:477-481.
- Sung KU, Lee HE, Choi WR, et al. Molluscum contagiosum as a skin manifestation of immune reconstitution inflammatory syndrome in an AIDS patient who is receiving HAART. Korean J Fam Med. 2012;33:182-185.
- Ratnam I, Chiu C, Kandala NB, et al. Incidence and risk factors for immune reconstitution inflammatory syndrome in an ethnically diverse HIV type 1-infected cohort. Clin Infect Dis. 2006;42:418-427.
- Chen KW, Yang CF, Huang CT, et al. Molluscum contagiosum in a patient with adult T-cell leukaemia/lymphoma. Br J Haematol. 2011;155:286.
- Fernandez KH, Bream M, Ali MA, et al. Investigation of molluscum contagiosum virus, orf and other parapoxviruses in lymphomatoid papulosis. J Am Acad Dermatol. 2013;68:1046-1047.
- Nakamura-Wakatsuki T, Kato Y, Miura T, et al. Eruptive molluscum contagiosums in a patient with rheumatoid arthritis and lung cancer. Rheumatol Int. 2011;31:1117-1118.
- Ozyürek E, Sentürk N, Kefeli M, et al. Ulcerating molluscum contagiosum in a boy with relapsed acute lymphoblastic leukemia. J Pediatr Hematol Oncol. 2011;33:E114-E116.
- Hughes WT, Parham DM. Molluscum contagiosum in children with cancer or acquired immunodeficiency syndrome. Pediatr Infect Dis J. 1991;10:152-156.
- Euvrard S, Kanitakis J, Cochat P, et al. Skin diseases in children with organ transplants. J Am Acad Dermatol. 2001;44:932-939.
- Gardner LS, Ormond PJ. Treatment of multiple giant molluscum contagiosum in a renal transplant patient with imiquimod 5% cream. Clin Exp Dermatol. 2006;31:452-453.
- Mansur AT, Göktay F, Gündüz S, et al. Multiple giant molluscum contagiosum in a renal transplant recipient. Transpl Infect Dis. 2004;6:120-123.
- Feldmeyer L, Kamarashev J, Boehler A, et al. Molluscum contagiosum folliculitis mimicking tinea barbae in a lung transplant recipient. J Am Acad Dermatol. 2010;63:169-171.
- Tas¸kapan O, Yenicesu M, Aksu A. A giant solitary molluscum contagiosum, resembling nodular basal cell carcinoma, in a renal transplant recipient. Acta Derm Venereol. 1996;76:247-248.
- Tan HH, Goh CL. Viral infections affecting the skin in organ transplant recipients: epidemiology and current management strategies. Am J Clin Dermatol. 2006;7:13-29.
- Al-Mutairi N, Al-Doukhi A, Al-Farag S, et al. Comparative study on the efficacy, safety, and acceptability of imiquimod 5% cream versus cryotherapy for molluscum contagiosum in children. Pediatr Dermatol. 2010;27:388-394.
- Lim KS, Foo CC. Disseminated molluscum contagiosum in a patient with chronic plaque psoriasis taking methotrexate. Clin Exp Dermatol. 2007;32:591-593.
- Fotiadou C, Lazaridou E, Lekkas D, et al. Disseminated, eruptive molluscum contagiosum lesions in a psoriasis patient under treatment with methotrexate and cyclosporine. Eur J Dermatol. 2012;22:147-148.
- Antoniou C, Kosmadaki MG, Stratigos AJ, et al. Genital HPV lesions and molluscum contagiosum occurring in patients receiving anti-TNF-alpha therapy. Dermatology. 2008;216:364-365.
- Cursiefen C, Grunke M, Dechant C, et al. Multiple bilateral eyelid molluscum contagiosum lesions associated with TNFalpha-antibody and methotrexate therapy. Am J Ophthalmol. 2002;134:270-271.
- Heng YK, Lee JS, Neoh CY. Verrucous plaques in a pemphigus vulgaris patient on immunosuppressive therapy. Int J Dermatol. 2012;51:1044-1046.
- Goksugur N, Ozbostanci B, Goksugur SB. Molluscum contagiosum infection associated with pimecrolimus use in pityriasis alba. Pediatr Dermatol. 2007;24:E63-E65.
- Fernandes S, Pinto GM, Cardoso J. Particular clinical presentations of psoriasis in HIV patients. Int J STD AIDS. 2011;22:653-654.
- Dohil MA, Lin P, Lee J, et al. The epidemiology of molluscum contagiosum in children. J Am Acad Dermatol. 2006;54:47-54.
- Reichert CM, O’Leary TJ, Levens DL, et al. Autopsy pathology in the acquired immune deficiency syndrome. Am J Pathol. 1983;112:357-382.
- Czelusta A, Yen-Moore A, Van der Straten M, et al. An overview of sexually transmitted diseases. Part III. Sexually transmitted diseases in HIV-infected patients. J Am Acad Dermatol. 2000;43:409-432.
- Husak R, Garbe C, Orfanos CE. Mollusca contagiosa in HIV infection. Clinical manifestation, relation to immune status and prognostic value in 39 patients [in German]. Hautarzt. 1997;48:103-109.
- Averbuch D, Jaouni T, Pe’er J, et al. Confluent molluscum contagiosum covering the eyelids of an HIV-positive child. Clin Exp Ophthalmol. 2009;37:525-527.
- Erickson C, Driscoll M, Gaspari A. Efficacy of intravenous cidofovir in the treatment of giant molluscum contagiosum in a patient with human immunodeficiency virus. Arch Dermatol. 2011;147:652-654.
- Mastrolorenzo A, Urbano FG, Salimbeni L, et al. Atypical molluscum contagiosum infection in an HIV-infected patient. Int J Dermatol. 1998;37:378-380.
- Itin PH, Gilli L. Molluscum contagiosum mimicking sebaceous nevus of Jadassohn, ecthyma and giant condylomata acuminata in HIV-infected patients. Dermatology. 1994;189:396-398.
- Sim JH, Lee ES. Molluscum contagiosum presenting as a cutaneous horn. Ann Dermatol. 2011;23:262-263.
- Manchanda Y, Sethuraman G, Paderwani PP, et al. Molluscum contagiosum presenting as penile horn in an HIV positive patient. Sex Transm Infect. 2005;81:183-184.
- Miller SJ. Cutaneous cryptococcus resembling molluscum contagiosum in a patient with acquired immunodeficiency syndrome. Cutis. 1988;41:411-412.
- Sornum A. A mistaken diagnosis of molluscum contagiosum in a HIV-positive patient in rural South Africa. BMJ Case Rep. 2012;14.
- Corti M, Villafañe MF, Palmieri O, et al. Rupioid histoplasmosis: first case reported in an AIDS patient in Argentina. Rev Inst Med Trop Sao Paulo. 2010;52:279-280.
- Saikia L, Nath R, Hazarika D, et al. Atypical cutaneous lesions of Penicillium marneffei infection as a manifestation of the immune reconstitution inflammatory syndrome after highly active antiretroviral therapy. Indian J Dermatol Venereol Leprol. 2010;76:45-48.
- de Souza JA. Molluscum or a mimic? Am J Med. 2006;119:927-929.
- Conant MA. Immunomodulatory therapy in the management of viral infections in patients with HIV infection. J Am Acad Dermatol. 2000;43:S27-S30.
- Gamble RG, Echols KF, Dellavalle RP. Imiquimod vs cryotherapy for molluscum contagiosum: a randomized controlled trial. Arch Dermatol. 2012;148:109-112.
- Brown CW Jr, O’Donoghue M, Moore J, et al. Recalcitrant molluscum contagiosum in an HIV-afflicted male treated successfully with topical imiquimod. Cutis. 2000;65:363-366.
- Strauss RM, Doyle EL, Mohsen AH, et al. Successful treatment of molluscum contagiosum with topical imiquimod in a severely immunocompromised HIV-positive patient. Int J STD AIDS. 2001;12:264-266.
- Theiler M, Kempf W, Kerl K, et al. Disseminated molluscum contagiosum in a HIV-positive child. improvement after therapy with 5% imiquimod. J Dermatol Case Rep. 2011;5:19-23.
- Watanabe T, Tamaki K. Cidofovir diphosphate inhibits molluscum contagiosum virus DNA polymerase activity. J Invest Dermatol. 2008;128:1327-1329.
- Calista D. Topical cidofovir for severe cutaneous human papillomavirus and molluscum contagiosum infections in patients with HIV/AIDS. a pilot study. J Eur Acad Dermatol Venereol. 2000;14:484-488.
- Toro JR, Sanchez S, Turiansky G, et al. Topical cidofovir for the treatment of dermatologic conditions: verruca, condyloma, intraepithelial neoplasia, herpes simplex and its potential use in smallpox. Dermatol Clin. 2003;21:301-309.
- Calista D, Boschini A, Landi G. Resolution of disseminated molluscum contagiosum with highly active anti-retroviral therapy (HAART) in patients with AIDS. Eur J Dermatol. 1999;9:211-213.
- Cattelan AM, Sasset L, Corti L, et al. A complete remission of recalcitrant molluscum contagiosum in an AIDS patient following highly active antiretroviral therapy (HAART). J Infect. 1999;38:58-60.
- Sen S, Bhaumik P. Resolution of giant molluscum contagiosum with antiretroviral therapy. Indian J Dermatol Venereol Leprol. 2008;74:267-268.
- Sen S, Goswami BK, Karjyi N, et al. Disfiguring molluscum contagiosum in a HIV-positive patient responding to antiretroviral therapy. Indian J Dermatol. 2009;54:180-182.
- Pereira B, Fernandes C, Nachiambo E, et al. Exuberant molluscum contagiosum as a manifestation of the immune reconstitution inflammatory syndrome. Dermatol Online J. 2007;13:6.
- Osei-Sekyere B, Karstaedt AS. Immune reconstitution inflammatory syndrome involving the skin. Clin Exp Dermatol. 2010;35:477-481.
- Sung KU, Lee HE, Choi WR, et al. Molluscum contagiosum as a skin manifestation of immune reconstitution inflammatory syndrome in an AIDS patient who is receiving HAART. Korean J Fam Med. 2012;33:182-185.
- Ratnam I, Chiu C, Kandala NB, et al. Incidence and risk factors for immune reconstitution inflammatory syndrome in an ethnically diverse HIV type 1-infected cohort. Clin Infect Dis. 2006;42:418-427.
- Chen KW, Yang CF, Huang CT, et al. Molluscum contagiosum in a patient with adult T-cell leukaemia/lymphoma. Br J Haematol. 2011;155:286.
- Fernandez KH, Bream M, Ali MA, et al. Investigation of molluscum contagiosum virus, orf and other parapoxviruses in lymphomatoid papulosis. J Am Acad Dermatol. 2013;68:1046-1047.
- Nakamura-Wakatsuki T, Kato Y, Miura T, et al. Eruptive molluscum contagiosums in a patient with rheumatoid arthritis and lung cancer. Rheumatol Int. 2011;31:1117-1118.
- Ozyürek E, Sentürk N, Kefeli M, et al. Ulcerating molluscum contagiosum in a boy with relapsed acute lymphoblastic leukemia. J Pediatr Hematol Oncol. 2011;33:E114-E116.
- Hughes WT, Parham DM. Molluscum contagiosum in children with cancer or acquired immunodeficiency syndrome. Pediatr Infect Dis J. 1991;10:152-156.
- Euvrard S, Kanitakis J, Cochat P, et al. Skin diseases in children with organ transplants. J Am Acad Dermatol. 2001;44:932-939.
- Gardner LS, Ormond PJ. Treatment of multiple giant molluscum contagiosum in a renal transplant patient with imiquimod 5% cream. Clin Exp Dermatol. 2006;31:452-453.
- Mansur AT, Göktay F, Gündüz S, et al. Multiple giant molluscum contagiosum in a renal transplant recipient. Transpl Infect Dis. 2004;6:120-123.
- Feldmeyer L, Kamarashev J, Boehler A, et al. Molluscum contagiosum folliculitis mimicking tinea barbae in a lung transplant recipient. J Am Acad Dermatol. 2010;63:169-171.
- Tas¸kapan O, Yenicesu M, Aksu A. A giant solitary molluscum contagiosum, resembling nodular basal cell carcinoma, in a renal transplant recipient. Acta Derm Venereol. 1996;76:247-248.
- Tan HH, Goh CL. Viral infections affecting the skin in organ transplant recipients: epidemiology and current management strategies. Am J Clin Dermatol. 2006;7:13-29.
- Al-Mutairi N, Al-Doukhi A, Al-Farag S, et al. Comparative study on the efficacy, safety, and acceptability of imiquimod 5% cream versus cryotherapy for molluscum contagiosum in children. Pediatr Dermatol. 2010;27:388-394.
- Lim KS, Foo CC. Disseminated molluscum contagiosum in a patient with chronic plaque psoriasis taking methotrexate. Clin Exp Dermatol. 2007;32:591-593.
- Fotiadou C, Lazaridou E, Lekkas D, et al. Disseminated, eruptive molluscum contagiosum lesions in a psoriasis patient under treatment with methotrexate and cyclosporine. Eur J Dermatol. 2012;22:147-148.
- Antoniou C, Kosmadaki MG, Stratigos AJ, et al. Genital HPV lesions and molluscum contagiosum occurring in patients receiving anti-TNF-alpha therapy. Dermatology. 2008;216:364-365.
- Cursiefen C, Grunke M, Dechant C, et al. Multiple bilateral eyelid molluscum contagiosum lesions associated with TNFalpha-antibody and methotrexate therapy. Am J Ophthalmol. 2002;134:270-271.
- Heng YK, Lee JS, Neoh CY. Verrucous plaques in a pemphigus vulgaris patient on immunosuppressive therapy. Int J Dermatol. 2012;51:1044-1046.
- Goksugur N, Ozbostanci B, Goksugur SB. Molluscum contagiosum infection associated with pimecrolimus use in pityriasis alba. Pediatr Dermatol. 2007;24:E63-E65.
- Fernandes S, Pinto GM, Cardoso J. Particular clinical presentations of psoriasis in HIV patients. Int J STD AIDS. 2011;22:653-654.
Practice Points
- Molluscum contagiosum (MC) is highly prevalent and can have a wide range of atypical clinical presentations in patients with impaired cellular immunity (eg, human immunodeficiency virus [HIV]).
- Treatment of MC should include destructive procedures, if possible, as well as adjunctive agents such as topical retinoids, cantharidin, trichloroacetic acid, imiquimod, or cidofovir.
- Clinicians should consider screening patients with severe recalcitrant psoriasis for HIV to avoid poor outcomes from therapeutic agents.
Nail-Patella Syndrome: Clinical Clues for Making the Diagnosis
Nail-patella syndrome (NPS), also known as hereditary osteo-onychodysplasia syndrome, is a rare autosomal-dominant disorder with an estimated incidence of 1 per 50,000 individuals in the United States. Nail-patella syndrome presents due to a heterozygous loss-of-function mutation in the LIM homeobox transcription factor 1 beta gene, LMX1B, on chromosome 9q34.1 LMX1B gene mutations are fully penetrant, but there is variable expressivity, even within families.2
Case Report
A 69-year-old man presented to the dermatology clinic for a routine skin cancer screening. The patient’s history was remarkable for dystrophic fingernails and toenails since birth. In his 20s he developed progressively worsening instability of the left knee and chronic back pain due to scoliosis, lumbar lordosis, and spinal disc herniation. Since then, he underwent knee surgery and 7 back surgeries for rheumatologic disease. His medical history also was remarkable for osteoporosis, hypertension, and glaucoma. Family history was notable for similar findings in the patient’s sister; mother; and maternal aunt, uncle, and grandmother, all with varying disease severity.
Physical examination was remarkable for bilateral fingernail hypoplasia that was most prominent on the thumb, with improvement in each nail on progression toward the fifth digit (Figure 1A). Triangular fingernail lunulae, longitudinal ridging, and nail splitting were present (Figure 1A and 1B). Hypoplastic crumbly toenails also were appreciated (Figure 1C). Skin creases over the distal interphalangeal joints of the fingers and toes were conspicuously absent. Limited range of motion was noted in multiple joints, with profound limitation of bilateral elbow extension. Review of prior imaging reports revealed bilateral iliac horns as well as left patellar absence and right patellar hypoplasia (Figure 2). Urinalysis was remarkable for proteinuria and microscopic hematuria. Given the constellation of examination findings and positive family history, a diagnosis of NPS was made.
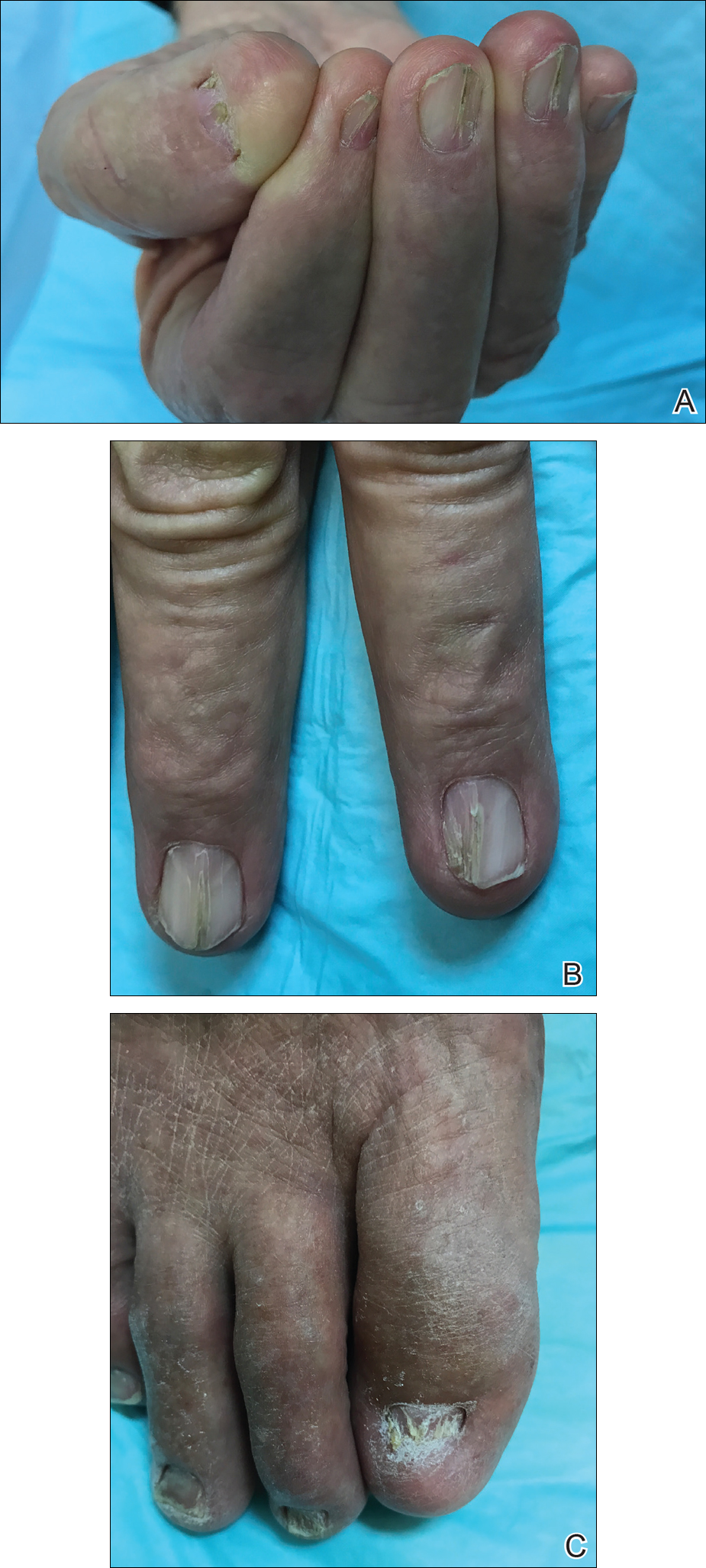
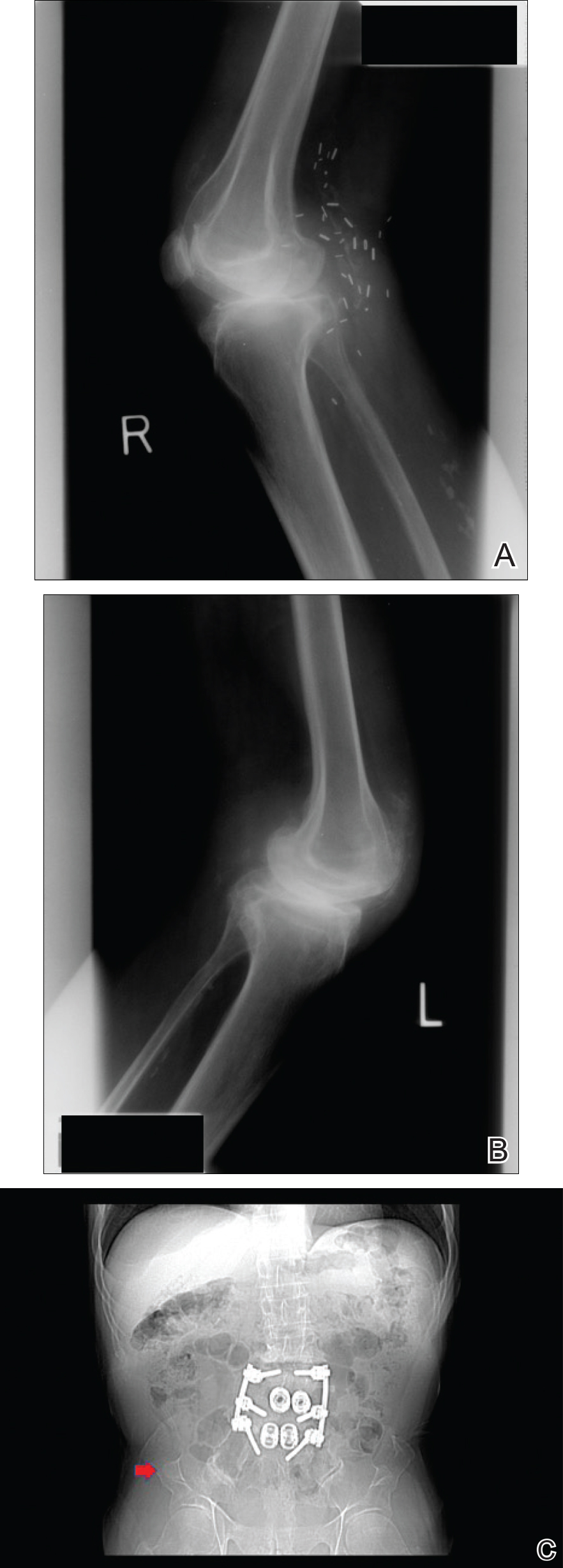
Comment
Nail-patella syndrome is characterized by variable dermatologic, neurologic, nephrogenic, ophthalmologic, and orthopedic clinical manifestations.3 Almost all patients with NPS have bilateral and symmetric nail changes, including absent or hypoplastic nails with ridging, splitting, or discoloration and triangular-shaped lunulae.1,4 Nail findings are the most consistent findings of NPS, as they are present in more than 98% of patients.5 The thumb often is the most severely affected nail, with improvement appreciated on progression toward the fifth digit, as seen in our patient (Figure 1A).5 Each individual nail usually is more severely affected on its ulnar side. When toenails are involved, the abnormalities tend to be less severe, and the little toenail is most commonly affected. Distal digital changes also are observed in almost all patients. Loss of dorsal creases in the skin overlying the distal interphalangeal joints can be considered as a diagnostic clue.3,4
There are a variety of orthopedic manifestations of NPS. Hypoplastic or absent patellae leading to recurrent subluxations or dislocations is a common finding.4 Bilateral symmetric bone formations (horns) arising from the iliac crest are pathognomonic but only found on radiography 70% of the time.6 Occasionally these protuberances can be palpated on physical examination,5 though this finding was not appreciated in our patient. Dysplasia of the elbows may result in limited elbow extension and limited pronation and supination. Early degenerative arthritis, lumbar lordosis, and scoliosis also are not uncommon. In addition, skeletal integrity is compromised, leading to early osteoporosis and increased risk for fractures.5
Nephropathy develops in approximately 30% to 40% of patients and is a major determinant of mortality in these patients.2 Mutations in the LMX1B gene lead to abnormal development of podocytes and reduction in collagen in the glomerular basement membrane. The first sign of renal involvement usually is proteinuria, with or without microscopic hematuria. As in our patient, many patients develop hypertension. Patients may progress to develop nephrotic syndrome and end-stage renal failure (5%–10%).7 Death from NPS-related nephropathy has occurred, even in childhood.4,5
Primary open-angle glaucoma has been recognized as a feature of NPS.8 It is the most frequent ocular abnormality observed, followed by ocular hypertension and Lester sign of the iris.3,5 These conditions also are more common in younger patients with NPS than in the general population.5 Important neurologic findings include epilepsy, peripheral neuropathy, attention deficit disorder, major depressive disorder, and vasomotor problems.9
Our case highlights the importance of recognizing this rare condition to provide a multidisciplinary approach to care that addresses all aspects of LMX1B-associated disease in affected individuals. Nail findings may be the first clue to the need for additional screenings in these patients. Nail-patella syndrome patients should undergo thorough ophthalmologic examinations every 2 years, including measurement of intraocular pressure, examination of the optic disc, and assessment of visual fields. Given the variability in severity of joint problems and the unpredictable anatomy of the joints, magnetic resonance imaging of the joints is recommended prior to orthopedic intervention. Most importantly, physicians should recognize this genodermatosis to implement periodic screenings for renal disease, as up to 40% of NPS patients develop kidney failure. Annual blood pressure measurements, urinalysis, and measurement of the protein to creatinine ratio in the urine are recommended. For patients with end-stage renal failure, renal transplantation results in cure of nephropathy and may even result in nail regrowth.10 Further, this case is notable in that it describes a patient with NPS who is older than most other individuals presenting with the condition, thereby revealing novel information about NPS in its more advanced stages.
- Harita Y, Kitanaka S, Isojima T, et al. Spectrum of LMX1B mutations: from nail-patella syndrome to isolated nephropathy [published online July 23, 2016]. Pediatr Nephrol. doi:10.1007/s00467-016-3462-x.
- Ghoumid J, Petit F, Holder-Espinasse M, et al. Nail-patella syndrome: clinical and molecular data in 55 families raising the hypothesis of a genetic heterogeneity [published online April 22, 2015]. Eur J Hum Genet. 2016;24:44-50.
- Tong SY, Luk HM, Tong TM, et al. The nail points to the diagnosis. Fong disease or hereditary osteo-onychodysplasia. Hong Kong Med J. 2015;21:573.e3-573.e5.
- Figueroa-Silva O, Vicente A, Agudo A, et al. Nail-patella syndrome: report of 11 pediatric cases. J Eur Acad Dermatol Venereol. 2016;30:1614-1617.
- Sweeney E, Fryer A, Mountford R, et al. Nail patella syndrome: a review of the phenotype aided by developmental biology. J Med Genet. 2003;40:153-162.
- Tigchelaar S, Lenting A, Bongers EM, et al. Nail patella syndrome: knee symptoms and surgical outcomes. a questionnaire-based survey [published online November 17, 2015]. Orthop Traumatol Surg Res. 2015;101:959-962.
- Lemley KV. Kidney disease in nail-patella syndrome [published online June 6, 2008]. Pediatr Nephrol. 2009;24:2345-2354.
- Sweeney E, Hoover-Fong JE, McIntosh I. Nail-patella syndrome. In: Adam MP, Ardinger HH, Pagon RA, et al, eds. GeneReviews. Seattle, WA: University of Washington, Seattle; 2003. https://www.ncbi.nlm.nih.gov/books/NBK1132/. Updated November 13, 2014. Accessed January 30, 2018.
- Lopez-Arvizu C, Sparrow EP, Strube MJ, et al. Increased symptoms of attention deficit hyperactivity disorder and major depressive disorder symptoms in nail-patella syndrome: potential association with LMX1B loss-of-function [published online November 2, 2010]. Am J Med Genet B Neuropsychiatr Genet. 2011;156B:59-66.
- Chan PC, Chan KW, Cheng IK, et al. Living-related renal transplantation in a patient with nail-patella syndrome. Nephron. 1988;50:164-166.
Nail-patella syndrome (NPS), also known as hereditary osteo-onychodysplasia syndrome, is a rare autosomal-dominant disorder with an estimated incidence of 1 per 50,000 individuals in the United States. Nail-patella syndrome presents due to a heterozygous loss-of-function mutation in the LIM homeobox transcription factor 1 beta gene, LMX1B, on chromosome 9q34.1 LMX1B gene mutations are fully penetrant, but there is variable expressivity, even within families.2
Case Report
A 69-year-old man presented to the dermatology clinic for a routine skin cancer screening. The patient’s history was remarkable for dystrophic fingernails and toenails since birth. In his 20s he developed progressively worsening instability of the left knee and chronic back pain due to scoliosis, lumbar lordosis, and spinal disc herniation. Since then, he underwent knee surgery and 7 back surgeries for rheumatologic disease. His medical history also was remarkable for osteoporosis, hypertension, and glaucoma. Family history was notable for similar findings in the patient’s sister; mother; and maternal aunt, uncle, and grandmother, all with varying disease severity.
Physical examination was remarkable for bilateral fingernail hypoplasia that was most prominent on the thumb, with improvement in each nail on progression toward the fifth digit (Figure 1A). Triangular fingernail lunulae, longitudinal ridging, and nail splitting were present (Figure 1A and 1B). Hypoplastic crumbly toenails also were appreciated (Figure 1C). Skin creases over the distal interphalangeal joints of the fingers and toes were conspicuously absent. Limited range of motion was noted in multiple joints, with profound limitation of bilateral elbow extension. Review of prior imaging reports revealed bilateral iliac horns as well as left patellar absence and right patellar hypoplasia (Figure 2). Urinalysis was remarkable for proteinuria and microscopic hematuria. Given the constellation of examination findings and positive family history, a diagnosis of NPS was made.
Comment
Nail-patella syndrome is characterized by variable dermatologic, neurologic, nephrogenic, ophthalmologic, and orthopedic clinical manifestations.3 Almost all patients with NPS have bilateral and symmetric nail changes, including absent or hypoplastic nails with ridging, splitting, or discoloration and triangular-shaped lunulae.1,4 Nail findings are the most consistent findings of NPS, as they are present in more than 98% of patients.5 The thumb often is the most severely affected nail, with improvement appreciated on progression toward the fifth digit, as seen in our patient (Figure 1A).5 Each individual nail usually is more severely affected on its ulnar side. When toenails are involved, the abnormalities tend to be less severe, and the little toenail is most commonly affected. Distal digital changes also are observed in almost all patients. Loss of dorsal creases in the skin overlying the distal interphalangeal joints can be considered as a diagnostic clue.3,4
There are a variety of orthopedic manifestations of NPS. Hypoplastic or absent patellae leading to recurrent subluxations or dislocations is a common finding.4 Bilateral symmetric bone formations (horns) arising from the iliac crest are pathognomonic but only found on radiography 70% of the time.6 Occasionally these protuberances can be palpated on physical examination,5 though this finding was not appreciated in our patient. Dysplasia of the elbows may result in limited elbow extension and limited pronation and supination. Early degenerative arthritis, lumbar lordosis, and scoliosis also are not uncommon. In addition, skeletal integrity is compromised, leading to early osteoporosis and increased risk for fractures.5
Nephropathy develops in approximately 30% to 40% of patients and is a major determinant of mortality in these patients.2 Mutations in the LMX1B gene lead to abnormal development of podocytes and reduction in collagen in the glomerular basement membrane. The first sign of renal involvement usually is proteinuria, with or without microscopic hematuria. As in our patient, many patients develop hypertension. Patients may progress to develop nephrotic syndrome and end-stage renal failure (5%–10%).7 Death from NPS-related nephropathy has occurred, even in childhood.4,5
Primary open-angle glaucoma has been recognized as a feature of NPS.8 It is the most frequent ocular abnormality observed, followed by ocular hypertension and Lester sign of the iris.3,5 These conditions also are more common in younger patients with NPS than in the general population.5 Important neurologic findings include epilepsy, peripheral neuropathy, attention deficit disorder, major depressive disorder, and vasomotor problems.9
Our case highlights the importance of recognizing this rare condition to provide a multidisciplinary approach to care that addresses all aspects of LMX1B-associated disease in affected individuals. Nail findings may be the first clue to the need for additional screenings in these patients. Nail-patella syndrome patients should undergo thorough ophthalmologic examinations every 2 years, including measurement of intraocular pressure, examination of the optic disc, and assessment of visual fields. Given the variability in severity of joint problems and the unpredictable anatomy of the joints, magnetic resonance imaging of the joints is recommended prior to orthopedic intervention. Most importantly, physicians should recognize this genodermatosis to implement periodic screenings for renal disease, as up to 40% of NPS patients develop kidney failure. Annual blood pressure measurements, urinalysis, and measurement of the protein to creatinine ratio in the urine are recommended. For patients with end-stage renal failure, renal transplantation results in cure of nephropathy and may even result in nail regrowth.10 Further, this case is notable in that it describes a patient with NPS who is older than most other individuals presenting with the condition, thereby revealing novel information about NPS in its more advanced stages.
Nail-patella syndrome (NPS), also known as hereditary osteo-onychodysplasia syndrome, is a rare autosomal-dominant disorder with an estimated incidence of 1 per 50,000 individuals in the United States. Nail-patella syndrome presents due to a heterozygous loss-of-function mutation in the LIM homeobox transcription factor 1 beta gene, LMX1B, on chromosome 9q34.1 LMX1B gene mutations are fully penetrant, but there is variable expressivity, even within families.2
Case Report
A 69-year-old man presented to the dermatology clinic for a routine skin cancer screening. The patient’s history was remarkable for dystrophic fingernails and toenails since birth. In his 20s he developed progressively worsening instability of the left knee and chronic back pain due to scoliosis, lumbar lordosis, and spinal disc herniation. Since then, he underwent knee surgery and 7 back surgeries for rheumatologic disease. His medical history also was remarkable for osteoporosis, hypertension, and glaucoma. Family history was notable for similar findings in the patient’s sister; mother; and maternal aunt, uncle, and grandmother, all with varying disease severity.
Physical examination was remarkable for bilateral fingernail hypoplasia that was most prominent on the thumb, with improvement in each nail on progression toward the fifth digit (Figure 1A). Triangular fingernail lunulae, longitudinal ridging, and nail splitting were present (Figure 1A and 1B). Hypoplastic crumbly toenails also were appreciated (Figure 1C). Skin creases over the distal interphalangeal joints of the fingers and toes were conspicuously absent. Limited range of motion was noted in multiple joints, with profound limitation of bilateral elbow extension. Review of prior imaging reports revealed bilateral iliac horns as well as left patellar absence and right patellar hypoplasia (Figure 2). Urinalysis was remarkable for proteinuria and microscopic hematuria. Given the constellation of examination findings and positive family history, a diagnosis of NPS was made.
Comment
Nail-patella syndrome is characterized by variable dermatologic, neurologic, nephrogenic, ophthalmologic, and orthopedic clinical manifestations.3 Almost all patients with NPS have bilateral and symmetric nail changes, including absent or hypoplastic nails with ridging, splitting, or discoloration and triangular-shaped lunulae.1,4 Nail findings are the most consistent findings of NPS, as they are present in more than 98% of patients.5 The thumb often is the most severely affected nail, with improvement appreciated on progression toward the fifth digit, as seen in our patient (Figure 1A).5 Each individual nail usually is more severely affected on its ulnar side. When toenails are involved, the abnormalities tend to be less severe, and the little toenail is most commonly affected. Distal digital changes also are observed in almost all patients. Loss of dorsal creases in the skin overlying the distal interphalangeal joints can be considered as a diagnostic clue.3,4
There are a variety of orthopedic manifestations of NPS. Hypoplastic or absent patellae leading to recurrent subluxations or dislocations is a common finding.4 Bilateral symmetric bone formations (horns) arising from the iliac crest are pathognomonic but only found on radiography 70% of the time.6 Occasionally these protuberances can be palpated on physical examination,5 though this finding was not appreciated in our patient. Dysplasia of the elbows may result in limited elbow extension and limited pronation and supination. Early degenerative arthritis, lumbar lordosis, and scoliosis also are not uncommon. In addition, skeletal integrity is compromised, leading to early osteoporosis and increased risk for fractures.5
Nephropathy develops in approximately 30% to 40% of patients and is a major determinant of mortality in these patients.2 Mutations in the LMX1B gene lead to abnormal development of podocytes and reduction in collagen in the glomerular basement membrane. The first sign of renal involvement usually is proteinuria, with or without microscopic hematuria. As in our patient, many patients develop hypertension. Patients may progress to develop nephrotic syndrome and end-stage renal failure (5%–10%).7 Death from NPS-related nephropathy has occurred, even in childhood.4,5
Primary open-angle glaucoma has been recognized as a feature of NPS.8 It is the most frequent ocular abnormality observed, followed by ocular hypertension and Lester sign of the iris.3,5 These conditions also are more common in younger patients with NPS than in the general population.5 Important neurologic findings include epilepsy, peripheral neuropathy, attention deficit disorder, major depressive disorder, and vasomotor problems.9
Our case highlights the importance of recognizing this rare condition to provide a multidisciplinary approach to care that addresses all aspects of LMX1B-associated disease in affected individuals. Nail findings may be the first clue to the need for additional screenings in these patients. Nail-patella syndrome patients should undergo thorough ophthalmologic examinations every 2 years, including measurement of intraocular pressure, examination of the optic disc, and assessment of visual fields. Given the variability in severity of joint problems and the unpredictable anatomy of the joints, magnetic resonance imaging of the joints is recommended prior to orthopedic intervention. Most importantly, physicians should recognize this genodermatosis to implement periodic screenings for renal disease, as up to 40% of NPS patients develop kidney failure. Annual blood pressure measurements, urinalysis, and measurement of the protein to creatinine ratio in the urine are recommended. For patients with end-stage renal failure, renal transplantation results in cure of nephropathy and may even result in nail regrowth.10 Further, this case is notable in that it describes a patient with NPS who is older than most other individuals presenting with the condition, thereby revealing novel information about NPS in its more advanced stages.
- Harita Y, Kitanaka S, Isojima T, et al. Spectrum of LMX1B mutations: from nail-patella syndrome to isolated nephropathy [published online July 23, 2016]. Pediatr Nephrol. doi:10.1007/s00467-016-3462-x.
- Ghoumid J, Petit F, Holder-Espinasse M, et al. Nail-patella syndrome: clinical and molecular data in 55 families raising the hypothesis of a genetic heterogeneity [published online April 22, 2015]. Eur J Hum Genet. 2016;24:44-50.
- Tong SY, Luk HM, Tong TM, et al. The nail points to the diagnosis. Fong disease or hereditary osteo-onychodysplasia. Hong Kong Med J. 2015;21:573.e3-573.e5.
- Figueroa-Silva O, Vicente A, Agudo A, et al. Nail-patella syndrome: report of 11 pediatric cases. J Eur Acad Dermatol Venereol. 2016;30:1614-1617.
- Sweeney E, Fryer A, Mountford R, et al. Nail patella syndrome: a review of the phenotype aided by developmental biology. J Med Genet. 2003;40:153-162.
- Tigchelaar S, Lenting A, Bongers EM, et al. Nail patella syndrome: knee symptoms and surgical outcomes. a questionnaire-based survey [published online November 17, 2015]. Orthop Traumatol Surg Res. 2015;101:959-962.
- Lemley KV. Kidney disease in nail-patella syndrome [published online June 6, 2008]. Pediatr Nephrol. 2009;24:2345-2354.
- Sweeney E, Hoover-Fong JE, McIntosh I. Nail-patella syndrome. In: Adam MP, Ardinger HH, Pagon RA, et al, eds. GeneReviews. Seattle, WA: University of Washington, Seattle; 2003. https://www.ncbi.nlm.nih.gov/books/NBK1132/. Updated November 13, 2014. Accessed January 30, 2018.
- Lopez-Arvizu C, Sparrow EP, Strube MJ, et al. Increased symptoms of attention deficit hyperactivity disorder and major depressive disorder symptoms in nail-patella syndrome: potential association with LMX1B loss-of-function [published online November 2, 2010]. Am J Med Genet B Neuropsychiatr Genet. 2011;156B:59-66.
- Chan PC, Chan KW, Cheng IK, et al. Living-related renal transplantation in a patient with nail-patella syndrome. Nephron. 1988;50:164-166.
- Harita Y, Kitanaka S, Isojima T, et al. Spectrum of LMX1B mutations: from nail-patella syndrome to isolated nephropathy [published online July 23, 2016]. Pediatr Nephrol. doi:10.1007/s00467-016-3462-x.
- Ghoumid J, Petit F, Holder-Espinasse M, et al. Nail-patella syndrome: clinical and molecular data in 55 families raising the hypothesis of a genetic heterogeneity [published online April 22, 2015]. Eur J Hum Genet. 2016;24:44-50.
- Tong SY, Luk HM, Tong TM, et al. The nail points to the diagnosis. Fong disease or hereditary osteo-onychodysplasia. Hong Kong Med J. 2015;21:573.e3-573.e5.
- Figueroa-Silva O, Vicente A, Agudo A, et al. Nail-patella syndrome: report of 11 pediatric cases. J Eur Acad Dermatol Venereol. 2016;30:1614-1617.
- Sweeney E, Fryer A, Mountford R, et al. Nail patella syndrome: a review of the phenotype aided by developmental biology. J Med Genet. 2003;40:153-162.
- Tigchelaar S, Lenting A, Bongers EM, et al. Nail patella syndrome: knee symptoms and surgical outcomes. a questionnaire-based survey [published online November 17, 2015]. Orthop Traumatol Surg Res. 2015;101:959-962.
- Lemley KV. Kidney disease in nail-patella syndrome [published online June 6, 2008]. Pediatr Nephrol. 2009;24:2345-2354.
- Sweeney E, Hoover-Fong JE, McIntosh I. Nail-patella syndrome. In: Adam MP, Ardinger HH, Pagon RA, et al, eds. GeneReviews. Seattle, WA: University of Washington, Seattle; 2003. https://www.ncbi.nlm.nih.gov/books/NBK1132/. Updated November 13, 2014. Accessed January 30, 2018.
- Lopez-Arvizu C, Sparrow EP, Strube MJ, et al. Increased symptoms of attention deficit hyperactivity disorder and major depressive disorder symptoms in nail-patella syndrome: potential association with LMX1B loss-of-function [published online November 2, 2010]. Am J Med Genet B Neuropsychiatr Genet. 2011;156B:59-66.
- Chan PC, Chan KW, Cheng IK, et al. Living-related renal transplantation in a patient with nail-patella syndrome. Nephron. 1988;50:164-166.
Practice Points
- Nail-patella syndrome (NPS) is a multisystem disease.
- Nail findings (eg, triangular lunulae) may be the first clue to NPS and should prompt investigation of associated renal, ocular, neurologic, skeletal, and orthopedic abnormalities.
- Early intervention and a multidisciplinary approach to care can improve morbidity and mortality in patients with NPS.
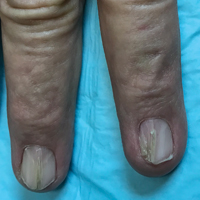
Melkersson-Rosenthal Syndrome Successfully Treated With Adalimumab
Melkersson-Rosenthal syndrome (MRS) is a rare condition comprised of unilateral peripheral facial nerve palsy, episodic or progressive facial edema, and lingua plicata (also known as fissured tongue). Melkersson-Rosenthal syndrome is a subtype of orofacial granulomatosis and often is mistaken for angioedema or pseudoangioedema due to the swelling of the lips and eyelids. We present a case of MRS that cleared in response to adalimumab therapy.
Case Report
A 69-year-old woman presented to our dermatology clinic with facial edema and a fissured tongue of 4 years’ duration. These symptoms had failed to improve with doxycycline, tacrolimus ointment 0.1%, and cortisone injections of the upper lip, as well as a balsam-free diet, fragrance-free skin products, and flavor-free toothpaste prescribed by multiple physicians over 4 years. Two weeks prior to the current presentation the patient developed left facial nerve palsy that was diagnosed by an outside physician as Bell palsy, and the patient completed a 7-day course of prednisone 1 day prior to presentation. The patient’s medical history was remarkable for type 2 diabetes mellitus controlled with metformin, hyperlipidemia controlled with ezetimibe-simvastatin, and psoriasis. She reported no family history of autoimmune or dermatologic disorders and denied any fever, unintentional weight loss, nausea, vomiting, or diarrhea.
On physical examination, the patient had considerable perioral edema and erythema without warmth or tenderness (Figure 1A). The tongue was fissured with notable scalloping at the lateral margins (Figure 1B). There were no aphthous ulcers or lymphadenopathy, and the remainder of the neurologic examination was normal. The patient had erythematous plaques with scaling on the bilateral elbows. Cardiopulmonary, musculoskeletal, and abdominal examinations were otherwise normal.
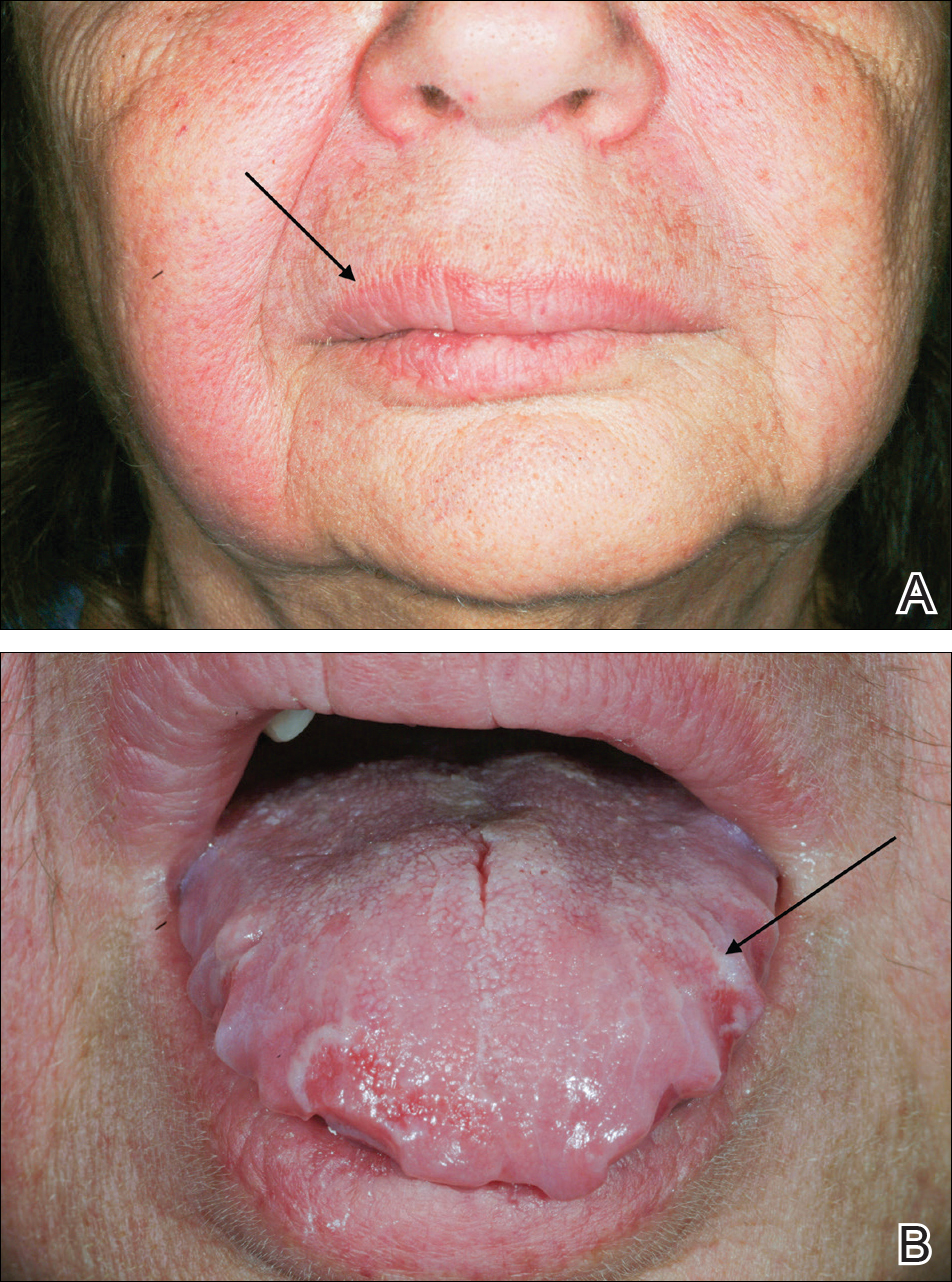
Laboratory data revealed an elevated white blood cell count of 17,500/µL (reference range, 4500–11,000/µL), an elevated absolute neutrophil count of 14,018/µL (reference range, 0–700/µL), and an absolute eosinophil count of 0/µL (reference range, 0–450/µL), with the rest of the complete blood cell count within reference range. A basic metabolic panel showed an elevated glucose level of 326 mg/dL (reference range, 70–110 mg/dL), consistent with diabetes and most likely exacerbated by the recent steroid course. A lipid panel was consistent with diagnosed hyperlipidemia (total cholesterol, 236 mg/dL [reference range, <200 mg/dL]; low-density lipoprotein, 134 mg/dL [reference range, 10–30 mg/dL]; triglycerides, 188 mg/dL [reference range, <160 mg/dL]). Hepatitis B and C tests were negative. A punch biopsy of the buccal and labial mucosa was taken, revealing a parakeratinized stratified squamous epithelium with an unusual pattern of surface keratinization with foci of intracellular and extracellular edema in the spinous layer. The underlying fibrous connective tissue was edematous with infiltrates of lymphocytes, mast cells, macrophages, and a few plasma cells. The pathology report listed the diagnosis as nonspecific “chronic mucositis,” with a list of differential diagnoses that included angioedema, hypersensitivity reaction, or other possible autoimmune disorders.
On consideration of these differential diagnoses, it was felt most likely to be MRS, which remains a primarily clinical diagnosis characterized by the triad of symptoms seen in this patient. Treatment of this condition emphasizes inflammation, and steroid therapy often is utilized, as it was in our patient. After the diagnosis of MRS was made, the patient received adalimumab 80 mg subcutaneously on day 1 and 40 mg on day 8 as a loading dose; she subsequently began a course of subcutaneous injections of adalimumab 40 mg once every other week for treatment of psoriasis with the goal of simultaneously treating the MRS. The symptoms did not completely resolve at this dose, so it was increased to 40 mg once weekly. The patient reported that the facial edema, lingua plicata, and facial nerve palsy resolved concomitantly over approximately 3 months with greater improvement at 5 months (Figure 2). The patient has had no relapses as of the last follow-up at 11 months.
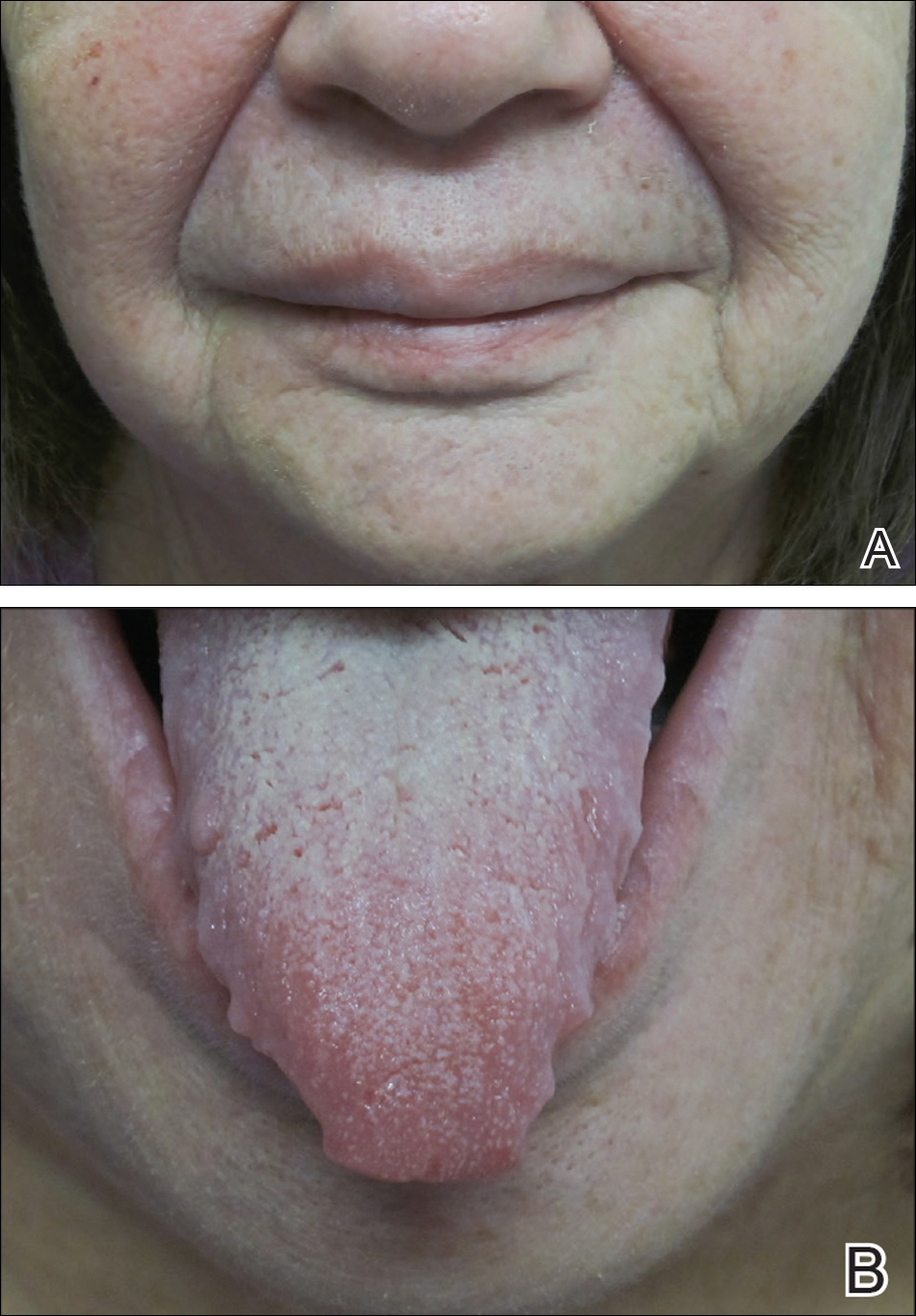
Comment
Melkersson-Rosenthal syndrome usually presents sporadically, though there are reports of familial association,1-3 and only 8% to 25% of patients worldwide present with the complete triad of symptoms.4 The pathogenesis of the syndrome is controversial. Granulomatous changes have been found in patients experiencing chronic edema. However, according to Zimmer et al5 in a study of 42 MRS patients, only 46% (19/42) had granulomatous changes; 36% (15/42) had nonspecific inflammation, 11% (5/42) had incidental findings, and 7% (3/42) showed no histopathologic abnormalities. Granulomatous cheilitis is a subtype of orofacial granulomatosis, an idiopathic process that causes swelling of the face and lips as well as intraoral swelling and ulceration. Orofacial granulomatosis is referred to as granulomatous cheilitis when the lip is involved. Melkersson-Rosenthal syndrome is another subtype of orofacial granulomatosis that includes facial palsy and fissured tongue.6,7
In a clinical study of 7 patients with MRS, Liu and Yu1 found 3 (42%) patients to have dysarthria, dysphagia, and tongue muscle atrophy; 1 patient to have migrainelike headaches; 1 patient to have decreased vision and an ocular movement disorder; 1 patient to have ipsilateral hearing loss; and 1 patient to lack any other symptoms. Halevy et al8 suggested a possible association of MRS with psoriasis. In their review of 12 patients, 1 (8%) had psoriatic arthritis, 2 (17%) had skin biopsy–proven psoriasis, and 3 (25%) had a family history of psoriasis.8 Because the disease is quite rare, it is difficult to determine other symptoms that may be associated with the disease.
Tumor necrosis factor α (TNF-α) is needed for granuloma formation, and TNF-α antagonists have been used to treat a number of granulomatous conditions including Crohn disease and sarcoidosis.9-11 Two case reports indicate that infliximab, a mouse/human chimeric monoclonal antibody to TNF-α, has been used successfully to clear MRS.12,13 One report cited the use of adalimumab for maintenance therapy of MRS,12 and more recently, adalimumab has been reported for refractory MRS.14 However, there currently are no known reports regarding the efficacy of adalimumab as a first-line treatment of MRS.
Adalimumab is a fully human monoclonal antibody to TNF-α, which is administered via subcutaneous injections. Infliximab must be administered at an infusion center, making treatment logistically more difficult for patients, and can be associated with the development of infusion reactions, though the exact data on infusion reactions are difficult to estimate due to variations in reporting.15,16
In 2014, Stein et al
Conclusion
We present a case of a 69-year-old woman who presented with facial nerve palsy, facial edema, and a fissured tongue, which is the classic triad of MRS, and all 3 symptoms improved with adalimumab.
- Liu R, Yu S. Melk
ersson-Rosenthal syndrome: a review of seven patients [published online May 7, 2013]. J Clin Neurosci. 2013;20:993-995. - Sun B, Zhou C, Han Z. Facial palsy in Melkersson-Rosenthal syndrome and Bell’s palsy: familial history and recurrence tendency [published online August 13, 2014]. Ann Otol Rhinol Laryngol. 2015;124:107-109.
- Meisel-Stosiek M, Hornstein OP, Stosiek N. Family study on Melkersson-Rosenthal syndrome. some hereditary aspects of the disease and review of literature. Acta Derm Venereol. 1990;70:221-226.
- Sciubba JJ, Said-Al-Naief N. Orofacial granulomatosis: presentation, pathology and management of 13 cases. J Oral Pathol Med. 2003;32:576-585.
- Zimmer WM, Rogers
RS 3rd, Reeve CM, et al. Orofacial manifestations of Melkersson-Rosenthal syndrome. a study of 42 patients and review of 220 cases from the literature. Oral Surg Oral Med Oral Pathol. 1992;74:610-619. - Critchlow WA, Cha
ng D. Cheilitis granulomatosa: a review [published online September 22, 2013]. Head Neck Pathol. 2014;8:209-213. - Allen CM, Camisa
C. Oral disease. In: Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. Philadelphia, PA: Elsevier; 2012:1157-1160. - Halevy S, Shalom
G, Trattner A, et al. Melkersson-Rosenthal syndrome: a possible association with psoriasis. J Am Acad Dermatol. 2012;67:795-796. - Algood HM, Lin P
L, Flynn JL. Tumor necrosis factor and chemokine interactions in the formation and maintenance of granulomas in tuberculosis. Clin Infect Dis. 2005;41(suppl 3):S189-S193. - Yee AM, Pochapin
MB. Treatment of complicated sarcoidosis with infliximab anti-tumor necrosis factor-alpha therapy. Ann Intern Med. 2001;135:27-31. - Targan SR, Hanau
er SB, van Deventer SJ, et al. A short-term study of chimeric monoclonal antibody cA2 to tumor necrosis factor alpha for Crohn’s disease. Crohn’s Disease cA2 Study Group. N Engl J Med. 1997;337:1029-1035. - Kakimoto C, Spar
ks C, White AA. Melkersson-Rosenthal syndrome: a form of pseudoangioedema. Ann Allergy Asthma Immunol. 2007;99:185-189. - Wickramasinghe N
, Gunasekara CN, Fernando WS, et al. Vulvitis granulomatosa, Melkersson-Rosenthal syndrome, and Crohn’s disease: dramatic response to infliximab therapy. Int J Dermatol. 2012;51:966-968. - Stein J, Paulke
A, Schacher B, et al. An extraordinary form of the Melkersson-Rosenthal syndrome successfully treated with the tumour necrosis factor-α blocker adalimumab [published online May 14, 2014]. BMJ Case Rep. doi:10.1136/bcr-2014-204674. - Cheifetz A, Smedley M, Martin S, et al. The incidence and management of infusion reactions to infliximab: a large center experience. Am J Gastroenterol. 2003;98:1315-1324.
- Choquette D, Faraawi R, Chow A, et al. Incidence and management of infusion reactions to infliximab in a prospective real-world community registry. J Rheumatol. 2015;42:1105-1111.
- Ruiz Villaverde R, Sánchez Cano D. Successful treatment of granulomatous cheilitis with adalimumab. Int J Dermatol. 2012;51:118-120.
Melkersson-Rosenthal syndrome (MRS) is a rare condition comprised of unilateral peripheral facial nerve palsy, episodic or progressive facial edema, and lingua plicata (also known as fissured tongue). Melkersson-Rosenthal syndrome is a subtype of orofacial granulomatosis and often is mistaken for angioedema or pseudoangioedema due to the swelling of the lips and eyelids. We present a case of MRS that cleared in response to adalimumab therapy.
Case Report
A 69-year-old woman presented to our dermatology clinic with facial edema and a fissured tongue of 4 years’ duration. These symptoms had failed to improve with doxycycline, tacrolimus ointment 0.1%, and cortisone injections of the upper lip, as well as a balsam-free diet, fragrance-free skin products, and flavor-free toothpaste prescribed by multiple physicians over 4 years. Two weeks prior to the current presentation the patient developed left facial nerve palsy that was diagnosed by an outside physician as Bell palsy, and the patient completed a 7-day course of prednisone 1 day prior to presentation. The patient’s medical history was remarkable for type 2 diabetes mellitus controlled with metformin, hyperlipidemia controlled with ezetimibe-simvastatin, and psoriasis. She reported no family history of autoimmune or dermatologic disorders and denied any fever, unintentional weight loss, nausea, vomiting, or diarrhea.
On physical examination, the patient had considerable perioral edema and erythema without warmth or tenderness (Figure 1A). The tongue was fissured with notable scalloping at the lateral margins (Figure 1B). There were no aphthous ulcers or lymphadenopathy, and the remainder of the neurologic examination was normal. The patient had erythematous plaques with scaling on the bilateral elbows. Cardiopulmonary, musculoskeletal, and abdominal examinations were otherwise normal.
Laboratory data revealed an elevated white blood cell count of 17,500/µL (reference range, 4500–11,000/µL), an elevated absolute neutrophil count of 14,018/µL (reference range, 0–700/µL), and an absolute eosinophil count of 0/µL (reference range, 0–450/µL), with the rest of the complete blood cell count within reference range. A basic metabolic panel showed an elevated glucose level of 326 mg/dL (reference range, 70–110 mg/dL), consistent with diabetes and most likely exacerbated by the recent steroid course. A lipid panel was consistent with diagnosed hyperlipidemia (total cholesterol, 236 mg/dL [reference range, <200 mg/dL]; low-density lipoprotein, 134 mg/dL [reference range, 10–30 mg/dL]; triglycerides, 188 mg/dL [reference range, <160 mg/dL]). Hepatitis B and C tests were negative. A punch biopsy of the buccal and labial mucosa was taken, revealing a parakeratinized stratified squamous epithelium with an unusual pattern of surface keratinization with foci of intracellular and extracellular edema in the spinous layer. The underlying fibrous connective tissue was edematous with infiltrates of lymphocytes, mast cells, macrophages, and a few plasma cells. The pathology report listed the diagnosis as nonspecific “chronic mucositis,” with a list of differential diagnoses that included angioedema, hypersensitivity reaction, or other possible autoimmune disorders.
On consideration of these differential diagnoses, it was felt most likely to be MRS, which remains a primarily clinical diagnosis characterized by the triad of symptoms seen in this patient. Treatment of this condition emphasizes inflammation, and steroid therapy often is utilized, as it was in our patient. After the diagnosis of MRS was made, the patient received adalimumab 80 mg subcutaneously on day 1 and 40 mg on day 8 as a loading dose; she subsequently began a course of subcutaneous injections of adalimumab 40 mg once every other week for treatment of psoriasis with the goal of simultaneously treating the MRS. The symptoms did not completely resolve at this dose, so it was increased to 40 mg once weekly. The patient reported that the facial edema, lingua plicata, and facial nerve palsy resolved concomitantly over approximately 3 months with greater improvement at 5 months (Figure 2). The patient has had no relapses as of the last follow-up at 11 months.
Comment
Melkersson-Rosenthal syndrome usually presents sporadically, though there are reports of familial association,1-3 and only 8% to 25% of patients worldwide present with the complete triad of symptoms.4 The pathogenesis of the syndrome is controversial. Granulomatous changes have been found in patients experiencing chronic edema. However, according to Zimmer et al5 in a study of 42 MRS patients, only 46% (19/42) had granulomatous changes; 36% (15/42) had nonspecific inflammation, 11% (5/42) had incidental findings, and 7% (3/42) showed no histopathologic abnormalities. Granulomatous cheilitis is a subtype of orofacial granulomatosis, an idiopathic process that causes swelling of the face and lips as well as intraoral swelling and ulceration. Orofacial granulomatosis is referred to as granulomatous cheilitis when the lip is involved. Melkersson-Rosenthal syndrome is another subtype of orofacial granulomatosis that includes facial palsy and fissured tongue.6,7
In a clinical study of 7 patients with MRS, Liu and Yu1 found 3 (42%) patients to have dysarthria, dysphagia, and tongue muscle atrophy; 1 patient to have migrainelike headaches; 1 patient to have decreased vision and an ocular movement disorder; 1 patient to have ipsilateral hearing loss; and 1 patient to lack any other symptoms. Halevy et al8 suggested a possible association of MRS with psoriasis. In their review of 12 patients, 1 (8%) had psoriatic arthritis, 2 (17%) had skin biopsy–proven psoriasis, and 3 (25%) had a family history of psoriasis.8 Because the disease is quite rare, it is difficult to determine other symptoms that may be associated with the disease.
Tumor necrosis factor α (TNF-α) is needed for granuloma formation, and TNF-α antagonists have been used to treat a number of granulomatous conditions including Crohn disease and sarcoidosis.9-11 Two case reports indicate that infliximab, a mouse/human chimeric monoclonal antibody to TNF-α, has been used successfully to clear MRS.12,13 One report cited the use of adalimumab for maintenance therapy of MRS,12 and more recently, adalimumab has been reported for refractory MRS.14 However, there currently are no known reports regarding the efficacy of adalimumab as a first-line treatment of MRS.
Adalimumab is a fully human monoclonal antibody to TNF-α, which is administered via subcutaneous injections. Infliximab must be administered at an infusion center, making treatment logistically more difficult for patients, and can be associated with the development of infusion reactions, though the exact data on infusion reactions are difficult to estimate due to variations in reporting.15,16
In 2014, Stein et al
Conclusion
We present a case of a 69-year-old woman who presented with facial nerve palsy, facial edema, and a fissured tongue, which is the classic triad of MRS, and all 3 symptoms improved with adalimumab.
Melkersson-Rosenthal syndrome (MRS) is a rare condition comprised of unilateral peripheral facial nerve palsy, episodic or progressive facial edema, and lingua plicata (also known as fissured tongue). Melkersson-Rosenthal syndrome is a subtype of orofacial granulomatosis and often is mistaken for angioedema or pseudoangioedema due to the swelling of the lips and eyelids. We present a case of MRS that cleared in response to adalimumab therapy.
Case Report
A 69-year-old woman presented to our dermatology clinic with facial edema and a fissured tongue of 4 years’ duration. These symptoms had failed to improve with doxycycline, tacrolimus ointment 0.1%, and cortisone injections of the upper lip, as well as a balsam-free diet, fragrance-free skin products, and flavor-free toothpaste prescribed by multiple physicians over 4 years. Two weeks prior to the current presentation the patient developed left facial nerve palsy that was diagnosed by an outside physician as Bell palsy, and the patient completed a 7-day course of prednisone 1 day prior to presentation. The patient’s medical history was remarkable for type 2 diabetes mellitus controlled with metformin, hyperlipidemia controlled with ezetimibe-simvastatin, and psoriasis. She reported no family history of autoimmune or dermatologic disorders and denied any fever, unintentional weight loss, nausea, vomiting, or diarrhea.
On physical examination, the patient had considerable perioral edema and erythema without warmth or tenderness (Figure 1A). The tongue was fissured with notable scalloping at the lateral margins (Figure 1B). There were no aphthous ulcers or lymphadenopathy, and the remainder of the neurologic examination was normal. The patient had erythematous plaques with scaling on the bilateral elbows. Cardiopulmonary, musculoskeletal, and abdominal examinations were otherwise normal.
Laboratory data revealed an elevated white blood cell count of 17,500/µL (reference range, 4500–11,000/µL), an elevated absolute neutrophil count of 14,018/µL (reference range, 0–700/µL), and an absolute eosinophil count of 0/µL (reference range, 0–450/µL), with the rest of the complete blood cell count within reference range. A basic metabolic panel showed an elevated glucose level of 326 mg/dL (reference range, 70–110 mg/dL), consistent with diabetes and most likely exacerbated by the recent steroid course. A lipid panel was consistent with diagnosed hyperlipidemia (total cholesterol, 236 mg/dL [reference range, <200 mg/dL]; low-density lipoprotein, 134 mg/dL [reference range, 10–30 mg/dL]; triglycerides, 188 mg/dL [reference range, <160 mg/dL]). Hepatitis B and C tests were negative. A punch biopsy of the buccal and labial mucosa was taken, revealing a parakeratinized stratified squamous epithelium with an unusual pattern of surface keratinization with foci of intracellular and extracellular edema in the spinous layer. The underlying fibrous connective tissue was edematous with infiltrates of lymphocytes, mast cells, macrophages, and a few plasma cells. The pathology report listed the diagnosis as nonspecific “chronic mucositis,” with a list of differential diagnoses that included angioedema, hypersensitivity reaction, or other possible autoimmune disorders.
On consideration of these differential diagnoses, it was felt most likely to be MRS, which remains a primarily clinical diagnosis characterized by the triad of symptoms seen in this patient. Treatment of this condition emphasizes inflammation, and steroid therapy often is utilized, as it was in our patient. After the diagnosis of MRS was made, the patient received adalimumab 80 mg subcutaneously on day 1 and 40 mg on day 8 as a loading dose; she subsequently began a course of subcutaneous injections of adalimumab 40 mg once every other week for treatment of psoriasis with the goal of simultaneously treating the MRS. The symptoms did not completely resolve at this dose, so it was increased to 40 mg once weekly. The patient reported that the facial edema, lingua plicata, and facial nerve palsy resolved concomitantly over approximately 3 months with greater improvement at 5 months (Figure 2). The patient has had no relapses as of the last follow-up at 11 months.
Comment
Melkersson-Rosenthal syndrome usually presents sporadically, though there are reports of familial association,1-3 and only 8% to 25% of patients worldwide present with the complete triad of symptoms.4 The pathogenesis of the syndrome is controversial. Granulomatous changes have been found in patients experiencing chronic edema. However, according to Zimmer et al5 in a study of 42 MRS patients, only 46% (19/42) had granulomatous changes; 36% (15/42) had nonspecific inflammation, 11% (5/42) had incidental findings, and 7% (3/42) showed no histopathologic abnormalities. Granulomatous cheilitis is a subtype of orofacial granulomatosis, an idiopathic process that causes swelling of the face and lips as well as intraoral swelling and ulceration. Orofacial granulomatosis is referred to as granulomatous cheilitis when the lip is involved. Melkersson-Rosenthal syndrome is another subtype of orofacial granulomatosis that includes facial palsy and fissured tongue.6,7
In a clinical study of 7 patients with MRS, Liu and Yu1 found 3 (42%) patients to have dysarthria, dysphagia, and tongue muscle atrophy; 1 patient to have migrainelike headaches; 1 patient to have decreased vision and an ocular movement disorder; 1 patient to have ipsilateral hearing loss; and 1 patient to lack any other symptoms. Halevy et al8 suggested a possible association of MRS with psoriasis. In their review of 12 patients, 1 (8%) had psoriatic arthritis, 2 (17%) had skin biopsy–proven psoriasis, and 3 (25%) had a family history of psoriasis.8 Because the disease is quite rare, it is difficult to determine other symptoms that may be associated with the disease.
Tumor necrosis factor α (TNF-α) is needed for granuloma formation, and TNF-α antagonists have been used to treat a number of granulomatous conditions including Crohn disease and sarcoidosis.9-11 Two case reports indicate that infliximab, a mouse/human chimeric monoclonal antibody to TNF-α, has been used successfully to clear MRS.12,13 One report cited the use of adalimumab for maintenance therapy of MRS,12 and more recently, adalimumab has been reported for refractory MRS.14 However, there currently are no known reports regarding the efficacy of adalimumab as a first-line treatment of MRS.
Adalimumab is a fully human monoclonal antibody to TNF-α, which is administered via subcutaneous injections. Infliximab must be administered at an infusion center, making treatment logistically more difficult for patients, and can be associated with the development of infusion reactions, though the exact data on infusion reactions are difficult to estimate due to variations in reporting.15,16
In 2014, Stein et al
Conclusion
We present a case of a 69-year-old woman who presented with facial nerve palsy, facial edema, and a fissured tongue, which is the classic triad of MRS, and all 3 symptoms improved with adalimumab.
- Liu R, Yu S. Melk
ersson-Rosenthal syndrome: a review of seven patients [published online May 7, 2013]. J Clin Neurosci. 2013;20:993-995. - Sun B, Zhou C, Han Z. Facial palsy in Melkersson-Rosenthal syndrome and Bell’s palsy: familial history and recurrence tendency [published online August 13, 2014]. Ann Otol Rhinol Laryngol. 2015;124:107-109.
- Meisel-Stosiek M, Hornstein OP, Stosiek N. Family study on Melkersson-Rosenthal syndrome. some hereditary aspects of the disease and review of literature. Acta Derm Venereol. 1990;70:221-226.
- Sciubba JJ, Said-Al-Naief N. Orofacial granulomatosis: presentation, pathology and management of 13 cases. J Oral Pathol Med. 2003;32:576-585.
- Zimmer WM, Rogers
RS 3rd, Reeve CM, et al. Orofacial manifestations of Melkersson-Rosenthal syndrome. a study of 42 patients and review of 220 cases from the literature. Oral Surg Oral Med Oral Pathol. 1992;74:610-619. - Critchlow WA, Cha
ng D. Cheilitis granulomatosa: a review [published online September 22, 2013]. Head Neck Pathol. 2014;8:209-213. - Allen CM, Camisa
C. Oral disease. In: Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. Philadelphia, PA: Elsevier; 2012:1157-1160. - Halevy S, Shalom
G, Trattner A, et al. Melkersson-Rosenthal syndrome: a possible association with psoriasis. J Am Acad Dermatol. 2012;67:795-796. - Algood HM, Lin P
L, Flynn JL. Tumor necrosis factor and chemokine interactions in the formation and maintenance of granulomas in tuberculosis. Clin Infect Dis. 2005;41(suppl 3):S189-S193. - Yee AM, Pochapin
MB. Treatment of complicated sarcoidosis with infliximab anti-tumor necrosis factor-alpha therapy. Ann Intern Med. 2001;135:27-31. - Targan SR, Hanau
er SB, van Deventer SJ, et al. A short-term study of chimeric monoclonal antibody cA2 to tumor necrosis factor alpha for Crohn’s disease. Crohn’s Disease cA2 Study Group. N Engl J Med. 1997;337:1029-1035. - Kakimoto C, Spar
ks C, White AA. Melkersson-Rosenthal syndrome: a form of pseudoangioedema. Ann Allergy Asthma Immunol. 2007;99:185-189. - Wickramasinghe N
, Gunasekara CN, Fernando WS, et al. Vulvitis granulomatosa, Melkersson-Rosenthal syndrome, and Crohn’s disease: dramatic response to infliximab therapy. Int J Dermatol. 2012;51:966-968. - Stein J, Paulke
A, Schacher B, et al. An extraordinary form of the Melkersson-Rosenthal syndrome successfully treated with the tumour necrosis factor-α blocker adalimumab [published online May 14, 2014]. BMJ Case Rep. doi:10.1136/bcr-2014-204674. - Cheifetz A, Smedley M, Martin S, et al. The incidence and management of infusion reactions to infliximab: a large center experience. Am J Gastroenterol. 2003;98:1315-1324.
- Choquette D, Faraawi R, Chow A, et al. Incidence and management of infusion reactions to infliximab in a prospective real-world community registry. J Rheumatol. 2015;42:1105-1111.
- Ruiz Villaverde R, Sánchez Cano D. Successful treatment of granulomatous cheilitis with adalimumab. Int J Dermatol. 2012;51:118-120.
- Liu R, Yu S. Melk
ersson-Rosenthal syndrome: a review of seven patients [published online May 7, 2013]. J Clin Neurosci. 2013;20:993-995. - Sun B, Zhou C, Han Z. Facial palsy in Melkersson-Rosenthal syndrome and Bell’s palsy: familial history and recurrence tendency [published online August 13, 2014]. Ann Otol Rhinol Laryngol. 2015;124:107-109.
- Meisel-Stosiek M, Hornstein OP, Stosiek N. Family study on Melkersson-Rosenthal syndrome. some hereditary aspects of the disease and review of literature. Acta Derm Venereol. 1990;70:221-226.
- Sciubba JJ, Said-Al-Naief N. Orofacial granulomatosis: presentation, pathology and management of 13 cases. J Oral Pathol Med. 2003;32:576-585.
- Zimmer WM, Rogers
RS 3rd, Reeve CM, et al. Orofacial manifestations of Melkersson-Rosenthal syndrome. a study of 42 patients and review of 220 cases from the literature. Oral Surg Oral Med Oral Pathol. 1992;74:610-619. - Critchlow WA, Cha
ng D. Cheilitis granulomatosa: a review [published online September 22, 2013]. Head Neck Pathol. 2014;8:209-213. - Allen CM, Camisa
C. Oral disease. In: Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. Philadelphia, PA: Elsevier; 2012:1157-1160. - Halevy S, Shalom
G, Trattner A, et al. Melkersson-Rosenthal syndrome: a possible association with psoriasis. J Am Acad Dermatol. 2012;67:795-796. - Algood HM, Lin P
L, Flynn JL. Tumor necrosis factor and chemokine interactions in the formation and maintenance of granulomas in tuberculosis. Clin Infect Dis. 2005;41(suppl 3):S189-S193. - Yee AM, Pochapin
MB. Treatment of complicated sarcoidosis with infliximab anti-tumor necrosis factor-alpha therapy. Ann Intern Med. 2001;135:27-31. - Targan SR, Hanau
er SB, van Deventer SJ, et al. A short-term study of chimeric monoclonal antibody cA2 to tumor necrosis factor alpha for Crohn’s disease. Crohn’s Disease cA2 Study Group. N Engl J Med. 1997;337:1029-1035. - Kakimoto C, Spar
ks C, White AA. Melkersson-Rosenthal syndrome: a form of pseudoangioedema. Ann Allergy Asthma Immunol. 2007;99:185-189. - Wickramasinghe N
, Gunasekara CN, Fernando WS, et al. Vulvitis granulomatosa, Melkersson-Rosenthal syndrome, and Crohn’s disease: dramatic response to infliximab therapy. Int J Dermatol. 2012;51:966-968. - Stein J, Paulke
A, Schacher B, et al. An extraordinary form of the Melkersson-Rosenthal syndrome successfully treated with the tumour necrosis factor-α blocker adalimumab [published online May 14, 2014]. BMJ Case Rep. doi:10.1136/bcr-2014-204674. - Cheifetz A, Smedley M, Martin S, et al. The incidence and management of infusion reactions to infliximab: a large center experience. Am J Gastroenterol. 2003;98:1315-1324.
- Choquette D, Faraawi R, Chow A, et al. Incidence and management of infusion reactions to infliximab in a prospective real-world community registry. J Rheumatol. 2015;42:1105-1111.
- Ruiz Villaverde R, Sánchez Cano D. Successful treatment of granulomatous cheilitis with adalimumab. Int J Dermatol. 2012;51:118-120.
Practice Points
- The classical triad of Melkersson-Rosenthal syndrome (MRS), which includes facial nerve palsy, facial edema, and lingua plicata, can present gradually over time and should therefore be kept in the differential of cheilitis.
- Tumor necrosis factor α therapy may play a crucial role in rare granulomatous diseases, including MRS.
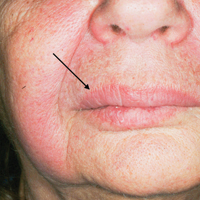
Postpartum Psychosis in a Young VA Patient: Diagnosis, Implications, and Treatment Recommendations
Postpartum psychosis is a psychiatric emergency that can endanger the life of the mother and the newborn child if untreated. About 1 to 2 mothers in 1,000 experiences postpartum psychosis after delivery.1 This rate is much higher among women with an established diagnosis of bipolar disorder before pregnancy.1
Expedient recognition, diagnosis, and referral to a high-level psychiatric facility (usually a locked inpatient unit) are critical for ensuring the safety of mother and infant. A diligent medical workup followed by thorough education for the patient and family are important steps in caring for patients with postpartum psychosis. Close mental health follow-up, pharmacologic interventions, informed decision making regarding breastfeeding, and preserving the sleep-wake cycle are critical for stabilization.2
The authors present the case of a patient admitted to VA Central California Health Care System (VACCHCS) with postpartum psychosis and a discussion on existing research on the prevalence of postpartum psychosis, relevant risk factors, the association with bipolar disorder, and treatment strategies.
Case Presentation
A 31-year-old active-duty female with no history of mental illness was admitted to the psychiatric unit because new-onset disorganized behavior was preventing her from functioning at her workplace. Two weeks after giving birth to her second child, the patient began exhibiting an uncharacteristic, debilitating labile mood and disorganized behavior. Her supervisors required her to present for medical attention about 3 months after the birth of her child. She was transferred to VACCHCS for higher level medical care on military orders. The patient’s husband initially attributed these psychiatric symptoms to vocational stress and taking care of 2 young children. He observed the patient exhibiting tearfulness about her job, which quickly alternated with euphoric episodes of singing and dancing at inappropriate times, such as when the children had quieted down and were being prepared to go to bed.
At the initial psychiatric evaluation after transfer to VACCHCS, the patient appeared well-kept and slightly overweight. In general her appearance was unremarkable. Throughout the examination she sang both subtly and loudly and at times was confrontational and irritable.
She related oddly and often was guarded and difficult to engage; she sang and played with her blanket in a childlike way. She smiled and laughed inappropriately, mumbled incoherently to herself, scanned the room suspiciously, and often made intense eye contact. Her affect was labile, both tearful and euphoric at several points in the examination. Her thought process was tangential, illogical, grandiose, difficult to redirect, and with loose associations. Her thought content consisted of delusions (“I’ve got the devil on my back”) and grandiosity (“I am everyone, I am you…the president, the mayor”), and she often stated that she planned to become a singer or performer.
The patient claimed she was neither suicidal nor had thoughts of infanticide. She reported having no visual and auditory hallucinations but often seemed to be responding to internal stimuli: She mumbled to herself and looked intensely at parts of the room. Cognitively, the patient was fully intact to recent and remote events but displayed a poor attention span. She did not exhibit any motor abnormalities, such as tremor, rigidity, weakness, sensory loss, or abnormal gait.
The patient’s workup included full chemistry, complete blood count, thyroid-stimulating hormone, antithyroid antibodies, calcium, rapid plasma reagin to rule out syphilis, toxicology, folate, vitamin B12, and vitamin D. All laboratory results were negative or within normal limits, although the urine drug screen was positive for cannabis. The patient’s husband noted that his wife never used cannabis except the weekend before her admission, when she impulsively went dancing, which was out of character for her. Her psychotic symptoms had been present weeks before the cannabis use; therefore, the her symptoms could not be attributed to a substance-induced psychotic disorder. A test for synthetic cannabis derivatives was negative. Newer synthetic compounds can cause more severe substance-induced psychotic symptoms than those of cannabis.3
The patient was diagnosed with postpartum psychosis and was started on the oral antipsychotic olanzapine 10 mg at bedtime. Additional doses were administered to control ongoing symptoms, which included a disorganized thought process; loose associations; euphoria; grandiosity; delusional content, such as “You are just a tool in place to help me;” reports of feeling as though she were in “outer space, outside in the galaxy;” decreased need for sleep; and irritability. The patient spent an entire interview with her eyes closed, stating that she could “hear” better because she was overstimulated if her eyes were open. She also described olfactory hallucinations of “strong perfume,” which the 2 providers present could not detect.
Olanzapine was not well tolerated because of sedation and was discontinued in favor of risperidone, 2 mg twice daily. Risperidone was more effective and better tolerated. Lithium was initiated the next day with target dosing at 300 mg in the morning and 600 mg at night. The patient became capable of linear, organized discussion and planning but remained euphoric with high energy; she exhibited grandiosity with frequent singing and dancing throughout her hospital stay. She often described her mood as “good, excellent, exuberant, exciting,” perseverating on the way words sounded and giggling in a childlike manner. She continued to have intrusive dreams of “hell and the devil” and that she was killed by gunshot.
The patient was continued on lithium and risperidone and transferred to a larger military hospital for further inpatient management, respecting military orders. Before discharge, a family conference was held with the patient and her husband to educate them on the importance of continued treatment, close follow-up, regular sleep patterns, and not breastfeeding while taking the prescribed medications. Although she was not back to her baseline at the time of transfer, the patient had stabilized significantly and gained sufficient insight into her condition.
Discussion
Postpartum psychosis can present with a prodromal phase consisting of fatigue, insomnia, restlessness, tearfulness, and emotional lability, making early identification difficult. Later, florid psychotic symptoms can include suspiciousness, confusion, incoherence, irrational statements, obsessive concern about the infant’s health, and delusions, including a belief that the baby is dead or defective. Some women might deny that the birth occurred or feel that they are unmarried, virginal, or persecuted.1 More concerning symptoms include auditory hallucinations commanding the mother to harm or kill the infant and/or herself. Symptoms often begin within days to weeks of birth, usually 2 to 3 weeks after delivery but can occur as long as 8 weeks postpartum.1 Several cases of infanticide and suicide have been documented.1 The risk of experiencing another psychotic episode in subsequent pregnancies can be as high as 50%.4-6 Regardless of symptom severity at onset, postpartum psychosis is a psychiatric emergency and must be treated as such.
Bipolar Disorder and Postpartum Psychosis
A close relationship exists between postpartum psychosis and development of bipolar disorder. A postpartum psychotic episode often is the harbinger of bipolar illness.7 About two-thirds of women who have an episode of postpartum psychosis will experience an underlying affective disorder within a year of childbirth.1,8 It is unclear what triggers the psychotic episode, but it has been theorized that major systemic shifts in hormone levelsor trauma of delivery could instigate development of symptoms.1,9
Risk factors include obstetric complications; perinatal infant mortality; previous episodes of bipolar disorder, psychosis, or postpartum psychosis; family history of bipolar disorder or postpartum psychosis; sleep deprivation; increased environmental stress; and lack of partner support.10 The strongest risk factor for developing postpartum psychosis is a personal or family history of bipolar disorder or a related psychotic disorder.11 This risk factor is identified in about 40% to 50% cases of postpartum psychosis.11
Treatment
Standard treatment for postpartum psychosis includes an antipsychotic and often lithium and benzodiazepines.1,7,10,11 This treatment approach differs slightly from treating a patient with a nonpostpartum psychotic illness, who generally would not receive mood stabilizers, such as lithium. Including a mood stabilizer for postpartum psychosis is warranted because of the association between postpartum psychosis and bipolar disorder, which is treated with a mood stabilizer.
Prevalence
Postpartum psychosis is identified in 1 to 2 per 1,000 childbirths. In women who have had an earlier episode of postpartum psychosis or have a diagnosis of bipolar disorder, the rate is up to 100 times higher.1 Kendell and colleagues found that psychiatric admissions occurred at a rate 7 times higher in the 30 days after birth than in the prepregnancy period, suggesting that metabolic factors might be involved in triggering postpartum psychotic symptoms.12 An abrupt hormonal loss occurs at childbirth; hormones peak 200-fold during gestation and decline rapidly within a day after birth.9 Despite the severity of symptoms in postpartum psychosis, these patients tend to have a better prognosis than that of women with psychotic episodes not related to pregnancy.4
Brockington and colleagues found that patients with postpartum psychosis had more mood lability, distractibility, and confusion than those with psychosis unrelated to pregnancy.15 Patients with postpartum psychosis were more likely to have impaired sensorium, bizarre quality of delusions, and memory loss. Psychosis with onset after childbirth included high levels of thought disorganization, delusions of reference, delusions of persecution, and greater levels of homicidal ideation and behavior.16 This study also reported symptoms such as visual, tactile, and olfactory hallucinations and a presentation similar to that of delirium.
Chandra and colleagues found that 53% of women with postpartum psychosis had delusions about the infant, including beliefs that someone would harm or kill the baby or that the baby would be harmed by their breast milk.17 Compared with women with bipolar disorder, Oostheuizen and colleagues found that women with postpartum psychosis had delusions of control, such as feeling under the influence of an overpowering force that controlled their actions.18 Infanticidal thoughts are common among patients with postpartum psychosis, and about 4% of women committed infanticide.1
Rapid stabilization and treatment are important because postpartum psychosis is considered a psychiatric emergency.7 Potential consequences of delayed diagnosis and treatment include harm or death of the infant by infanticide and death of the mother by suicide. A thorough physical examination is important to rule out metabolic or neuroendocrine causes of psychosis other than postpartum hormonal shifts. These could include causes of altered mental status: stroke, pulmonary embolism, amniotic fluid emboli, Sheehan syndrome, thyroid disorders, electrolyte abnormalities, acute hemorrhage, sepsis, and substance toxicity or withdrawal.10 A complete blood count, full chemistry, thyroid function tests, antithyroid antibody tests, calcium, vitamin B12, and folate should be measured.7,10
Initial treatment should include antipsychotics and often mood stabilizers such as lithium. Managing insomnia aggressively is also necessary for initial stabilization and to prevent a repeat manic episode if the patient develops bipolar disorder.2 Many experts argue that sleep loss in combination with other risk factors might be the final common pathway for development of postpartum psychosis in women predisposed to this disorder.19,20
Treating insomnia in an outpatient setting includes teaching sleep hygiene practices and relaxation techniques. Although these methods to regulate sleep could be encouraged during the emergent inpatient stabilization of a patient with postpartum psychosis, pharmacologic approaches are necessary for acute mania and psychosis. Concern about possible dependence on benzodiazepines and other sedating sleep aids are valid; however, the benefit of acute stabilization of psychotic symptoms outweighs the potential risk of dependence.
Typically, first-line treatment is an antipsychotic, and second-generation antipsychotics generally are preferred over first-generation antipsychotics because of their more benign adverse effect profile.21,22 There are no controlled trials that compare antipsychotics with placebo or other interventions for postpartum psychosis. Therefore, use of atypical antipsychotics is based on randomized trials demonstrating efficacy in reducing psychosis in bipolar disorder, depression with psychotic features, and schizoaffective disorder.23,24 Once the patient is treated with an antipsychotic, further use of psychotropic medications, such as lithium or other mood stabilizers, should be based on the patient’s clinical presentation. For example, the patient in this case study primarily had manic symptoms consistent with bipolar disorder, making lithium or another mood stabilizer an appropriate choice.
Bergink and colleagues demonstrated positive outcomes with a treatment algorithm involving sequential use of benzodiazepines to improve sleep, an antipsychotic to decrease acute manic symptoms, lithium to stabilize mood based on symptoms, and electroconvulsive therapy if other treatments were not successful.25 Case studies document that administering estrogen led to recovery from postpartum psychosis, although patients often relapsed when estrogen was stopped.26 Electroconvulsive therapy has shown promising results, especially in patients who do not respond to antipsychotic medications or lithium.27,28
Antipsychotic and Other Psychotropic Medications
Choice of an antipsychotic and other psychotropic medications to treat postpartum psychosis is based on the patient’s breastfeeding status. The benefits of treatment should be weighed against the risks of a breastfeeding infant’s exposure to the medication. Because postpartum psychosis is a psychiatric emergency, the benefits of the medication are considered to outweigh any potential adverse effect to the breastfeeding infant exposed to the medication. Risks of untreated postpartum psychosis to the infant include rejection of the infant, poor parental relationships, suicide, infanticide, long-term failure to bond with the child, delayed infant development, and failure to thrive.29 Also, many mothers—including the patient in this presentation—decide that the benefits of treatment outweigh those of breastfeeding and choose to feed their infant with formula. Even if the patient chooses to bottle-feed her infant, consider administering medications that are considered safer for breastfeeding because the patient may need to continue the psychotropic during later pregnancies to prevent future psychotic episodes.30 All psychotropic medications pass into breast milk.29 Studies on the long-term effect of these medications on the infant are limited, but experts tend to recommend olanzapine, quetiapine, and risperidone over aripiprazole and ziprasidone.21,31-33
Lithium often is used to treat postpartum psychosis. Studies examining risk to the infant after long-term exposure to lithium through breast milk have not been conducted, but the American Academy of Pediatrics discourages its use during breastfeeding because of concerns about toxicity in the infant.34-36
Sleep regulation is important to treat bipolar disorder and to prevent future episodes.2,20,21 To ensure safety of the infant and mother before discharge, family education is imperative to establish close follow-up, adequate sleep, and reduction of stressors.7,10 Separation from the infant might be necessary after discharge, and someone should monitor the infant at all times until the outpatient mental health provider confirms that all psychotic symptoms have resolved.7,10 Successful treatment of postpartum psychosis requires close communication among the mental health provider, the pediatrician, and the obstetrician or women’s health provider.10 Because a close-knit team approach after discharge from the acute psychiatric unit is necessary, the care of such a patient and her child provides an educational opportunity for individuals working in integrated care clinics.
Conclusion
Postpartum psychosis is a psychiatric emergency requiring immediate treatment to prevent dire outcomes such as suicide or infanticide. Treatment considerations include the cost-benefit analysis of breastfeeding and the toxicity of psychotropic medications when ingested by the infant via breast milk. A close relationship has been demonstrated between postpartum psychosis and bipolar disorder.
Preferred treatment regimens include lithium and an antipsychotic. Educate the family as a unit about the diagnosis and treatment, the importance of adequate sleep for treatment and prophylaxis, and the decision on whether to discontinue breastfeeding despite its well-known benefits for mother and infant. Stabilization is a multifaceted process and needs to be reinforced with a solid plan for support and follow-up appointments. Because of the higher risk of relapse, educate patients about prophylactic treatment during subsequent pregnancies and monitor for development of bipolar disorder in the future.
1. Sadock B, Sadock V, Ruiz P. Kaplan & Sadock’s Synopsis of Psychiatry. 11th ed. Philadelphia, PA: Wolters Kluwer; 2015.
2. Sharma V. Pharmacotherapy of postpartum psychosis. Expert Opin Pharmacother. 2003;4(10):1651-1658.
3. Bassir Nia A, Medrano B, Perkel C, Galynker I, Hurd YL. Psychiatric comorbidity associated with synthetic cannabinoid use compared to cannabis. J Psychopharmacol. 2016;30(12):1321-1330.
4. Rhohde A, Marneros A. Postpartum psychoses: onset and long-term course. Psychopathology. 1993;26(3-4):203-209.
5. Videbech P, Gouliaev G. First admission with puerperal psychosis: 7-14 years of follow-up. Acta Psychiatr Scand. 1995;91(3):167-173.
6. Terp IM, Engholm G, Møller H, Mortensen PB. A follow-up study of postpartum psychoses: prognosis and risk factors for readmission. Acta Psychiatr Scand. 1999;100(1):40-46.
7. Spinelli MG. Postpartum psychosis: detection of risk and management. Am J Psychiatry. 2009;166(4):405-408.
8. Blackmore ER, Rubinow DR, O’Connor TG, et al. Reproductive outcomes and risk of subsequent illness in women diagnosed with postpartum psychosis. Bipolar Disord. 2013;15(4):394-404.
9. Bloch M, Schmidt PJ, Danaceau M, Murphy J, Nieman L, Rubinow DR. Effects of gonadal steroids in women with a history of postpartum depression. Am J Psychiatry. 2000;157(6):924-930.
10. Monzon C, Lanza di Scalea T, Pearlstein T. Postpartum psychosis: updates and clinical issues. Psychiatric Times. 2014. http://www.psychiatrictimes.com/special-reports/postpartum -psychosis-updates-and-clinical-issues. Published January 15, 2014. Accessed December 14, 2017.
11. Davies W. Understanding the pathophysiology of postpartum psychosis: challenges and new approaches. World J Psychiatry. 2017;7(2):77-88.
12. Kendell RE, Chalmers JC, Platz C. Epidemiology of puerperal psychoses. Br J Psychiatry. 1987;150:662-673.
13. Leibenluft E. Women with bipolar illness: clinical and research issues. Am J Psychiatry. 1996;153(2):163-173.
14. Chaudron LH, Pies R. The relationship between postpartum psychosis and bipolar disorder: a review. J Clin Psychiatry. 2003;64(11):1284-1292.
15. Brockington IF, Cernik KF, Schofield EM, Downing AR, Francis AF, Keelan C. Puerperal psychosis: phenomena and diagnosis. Arch Gen Psychiatry. 1981;38(7):829-833.
16. Wisner KL, Peindl K, Hanusa BH. Symptomatology of affective and psychotic illnesses related to childbearing. J Affect Disord. 1994;30(2):77-87.
17. Chandra PS, Bhargavaraman RP, Raghunandan VN, Shaligram D. Delusions related to infant and their association with mother-infant interactions in postpartum psychotic disorders. Arch Womens Ment Health. 2006;9(5):285-288.
18. Oosthuizen P, Russouw H, Roberts M. Is puerperal psychosis bipolar mood disorder? A phenomenological comparison. Compr Psychiatry. 1995;36(1):77-81.
19. Sharma V, Mazmanian D. Sleep loss and postpartum psychosis. Bipolar Disord. 2003;5(2):98-105.
20 Bilszta JL, Meyer D, Buist AE. Bipolar affective disorder in the postnatal period: investigating the role of sleep. Bipolar Disord. 2010;12(5):568-578.
21. Doucet S, Jones I, Letourneau N, Dennis CL, Blackmore ER. Interventions for the prevention and treatment of postpartum psychosis: a systematic review. Arch Womens Ment Health. 2011;14(2):89-98.
22 Perlis RH, Welge JA, Vornik LA, Hirschfeld RM, Keck PE Jr. Atypical antipsychotics in the treatment of mania: a meta-analysis of randomized, placebo-controlled trials. J Clin Psychiatry. 2006;67(4):509-516.
23 Wijkstra J, Lijmer J, Balk FJ, Geddes JR, Nolen WA. Pharmacological treatment for unipolar psychotic depression: systematic review and meta-analysis. Br J Psychiatry. 2006;188:410-415.
24. Smith LA, Cornelius V, Warnock A, Tacchi MJ, Taylor D. Pharmacological interventions for acute bipolar mania: a systematic review of randomized placebo-controlled trials. Bipolar Disord. 2007;9(6):551-560.
25. Bergink V, Burgerhout KM, Koorengevel KM, et al. Treatment of psychosis and mania in the postpartum period. Am J Psychiatry. 2015;172(2):115-123.
26. Ahokas A, Aito M, Rimón R. Positive treatment effect of estradiol in postpartum psychosis: a pilot study. J Clin Psychiatry. 2000;61(3):166-169.
27. Reed P, Sermin N, Appleby L, Faragher B. A comparison of clinical response to electroconvulsive therapy in puerperal and non-puerperal psychoses. J Affect Disord. 1999;54(3):255-260.
28. Forray A, Ostroff RB. The use of electroconvulsive therapy in postpartum affective disorders. J ECT. 2007;23(3):188-193.
29. Robinson GE. Psychopharmacology in pregnancy and postpartum. Focus. 2012;10(1):3-14.
30. Wesseloo R, Kamperman AM, Munk-Olsen T, Pop VJ, Kushner SA, Bergink V. Risk of postpartum relapse in bipolar disorder and postpartum psychosis: a systematic review and meta-analysis. Am J Psychiatry. 2016;173(2):117-127.
31. Sharma V, Smith A, Mazmanian D. Olanzapine in the prevention of postpartum psychosis and mood episodes in bipolar disorder. Bipolar Disord. 2006;8(4):400-404.
32. Gobbi G. Quetiapine in postpartum psychosis. J Clin Psychopharmacol. 2014;34(6):744-745.
33. Uguz F. Second-generation antipsychotics during the lactation period: a comparative systematic review on infant safety. J Clin Psychopharmacol. 2016;36(3):244-252.
34. Sachs HC; Committee on Drugs. The transfer of drugs and therapeutics into human breast milk: an update on selected topics. Pediatrics. 2013;132(3):e796-e809.
35. Lithium [package insert]. Columbus, OH: Roxane Laboratories Inc; 2011.
36. Grandjean EM, Aubry JM. Lithium: updated human knowledge using an evidence-based approach: part III: clinical safety. CNS Drugs. 2009;23(5):397-418.
Postpartum psychosis is a psychiatric emergency that can endanger the life of the mother and the newborn child if untreated. About 1 to 2 mothers in 1,000 experiences postpartum psychosis after delivery.1 This rate is much higher among women with an established diagnosis of bipolar disorder before pregnancy.1
Expedient recognition, diagnosis, and referral to a high-level psychiatric facility (usually a locked inpatient unit) are critical for ensuring the safety of mother and infant. A diligent medical workup followed by thorough education for the patient and family are important steps in caring for patients with postpartum psychosis. Close mental health follow-up, pharmacologic interventions, informed decision making regarding breastfeeding, and preserving the sleep-wake cycle are critical for stabilization.2
The authors present the case of a patient admitted to VA Central California Health Care System (VACCHCS) with postpartum psychosis and a discussion on existing research on the prevalence of postpartum psychosis, relevant risk factors, the association with bipolar disorder, and treatment strategies.
Case Presentation
A 31-year-old active-duty female with no history of mental illness was admitted to the psychiatric unit because new-onset disorganized behavior was preventing her from functioning at her workplace. Two weeks after giving birth to her second child, the patient began exhibiting an uncharacteristic, debilitating labile mood and disorganized behavior. Her supervisors required her to present for medical attention about 3 months after the birth of her child. She was transferred to VACCHCS for higher level medical care on military orders. The patient’s husband initially attributed these psychiatric symptoms to vocational stress and taking care of 2 young children. He observed the patient exhibiting tearfulness about her job, which quickly alternated with euphoric episodes of singing and dancing at inappropriate times, such as when the children had quieted down and were being prepared to go to bed.
At the initial psychiatric evaluation after transfer to VACCHCS, the patient appeared well-kept and slightly overweight. In general her appearance was unremarkable. Throughout the examination she sang both subtly and loudly and at times was confrontational and irritable.
She related oddly and often was guarded and difficult to engage; she sang and played with her blanket in a childlike way. She smiled and laughed inappropriately, mumbled incoherently to herself, scanned the room suspiciously, and often made intense eye contact. Her affect was labile, both tearful and euphoric at several points in the examination. Her thought process was tangential, illogical, grandiose, difficult to redirect, and with loose associations. Her thought content consisted of delusions (“I’ve got the devil on my back”) and grandiosity (“I am everyone, I am you…the president, the mayor”), and she often stated that she planned to become a singer or performer.
The patient claimed she was neither suicidal nor had thoughts of infanticide. She reported having no visual and auditory hallucinations but often seemed to be responding to internal stimuli: She mumbled to herself and looked intensely at parts of the room. Cognitively, the patient was fully intact to recent and remote events but displayed a poor attention span. She did not exhibit any motor abnormalities, such as tremor, rigidity, weakness, sensory loss, or abnormal gait.
The patient’s workup included full chemistry, complete blood count, thyroid-stimulating hormone, antithyroid antibodies, calcium, rapid plasma reagin to rule out syphilis, toxicology, folate, vitamin B12, and vitamin D. All laboratory results were negative or within normal limits, although the urine drug screen was positive for cannabis. The patient’s husband noted that his wife never used cannabis except the weekend before her admission, when she impulsively went dancing, which was out of character for her. Her psychotic symptoms had been present weeks before the cannabis use; therefore, the her symptoms could not be attributed to a substance-induced psychotic disorder. A test for synthetic cannabis derivatives was negative. Newer synthetic compounds can cause more severe substance-induced psychotic symptoms than those of cannabis.3
The patient was diagnosed with postpartum psychosis and was started on the oral antipsychotic olanzapine 10 mg at bedtime. Additional doses were administered to control ongoing symptoms, which included a disorganized thought process; loose associations; euphoria; grandiosity; delusional content, such as “You are just a tool in place to help me;” reports of feeling as though she were in “outer space, outside in the galaxy;” decreased need for sleep; and irritability. The patient spent an entire interview with her eyes closed, stating that she could “hear” better because she was overstimulated if her eyes were open. She also described olfactory hallucinations of “strong perfume,” which the 2 providers present could not detect.
Olanzapine was not well tolerated because of sedation and was discontinued in favor of risperidone, 2 mg twice daily. Risperidone was more effective and better tolerated. Lithium was initiated the next day with target dosing at 300 mg in the morning and 600 mg at night. The patient became capable of linear, organized discussion and planning but remained euphoric with high energy; she exhibited grandiosity with frequent singing and dancing throughout her hospital stay. She often described her mood as “good, excellent, exuberant, exciting,” perseverating on the way words sounded and giggling in a childlike manner. She continued to have intrusive dreams of “hell and the devil” and that she was killed by gunshot.
The patient was continued on lithium and risperidone and transferred to a larger military hospital for further inpatient management, respecting military orders. Before discharge, a family conference was held with the patient and her husband to educate them on the importance of continued treatment, close follow-up, regular sleep patterns, and not breastfeeding while taking the prescribed medications. Although she was not back to her baseline at the time of transfer, the patient had stabilized significantly and gained sufficient insight into her condition.
Discussion
Postpartum psychosis can present with a prodromal phase consisting of fatigue, insomnia, restlessness, tearfulness, and emotional lability, making early identification difficult. Later, florid psychotic symptoms can include suspiciousness, confusion, incoherence, irrational statements, obsessive concern about the infant’s health, and delusions, including a belief that the baby is dead or defective. Some women might deny that the birth occurred or feel that they are unmarried, virginal, or persecuted.1 More concerning symptoms include auditory hallucinations commanding the mother to harm or kill the infant and/or herself. Symptoms often begin within days to weeks of birth, usually 2 to 3 weeks after delivery but can occur as long as 8 weeks postpartum.1 Several cases of infanticide and suicide have been documented.1 The risk of experiencing another psychotic episode in subsequent pregnancies can be as high as 50%.4-6 Regardless of symptom severity at onset, postpartum psychosis is a psychiatric emergency and must be treated as such.
Bipolar Disorder and Postpartum Psychosis
A close relationship exists between postpartum psychosis and development of bipolar disorder. A postpartum psychotic episode often is the harbinger of bipolar illness.7 About two-thirds of women who have an episode of postpartum psychosis will experience an underlying affective disorder within a year of childbirth.1,8 It is unclear what triggers the psychotic episode, but it has been theorized that major systemic shifts in hormone levelsor trauma of delivery could instigate development of symptoms.1,9
Risk factors include obstetric complications; perinatal infant mortality; previous episodes of bipolar disorder, psychosis, or postpartum psychosis; family history of bipolar disorder or postpartum psychosis; sleep deprivation; increased environmental stress; and lack of partner support.10 The strongest risk factor for developing postpartum psychosis is a personal or family history of bipolar disorder or a related psychotic disorder.11 This risk factor is identified in about 40% to 50% cases of postpartum psychosis.11
Treatment
Standard treatment for postpartum psychosis includes an antipsychotic and often lithium and benzodiazepines.1,7,10,11 This treatment approach differs slightly from treating a patient with a nonpostpartum psychotic illness, who generally would not receive mood stabilizers, such as lithium. Including a mood stabilizer for postpartum psychosis is warranted because of the association between postpartum psychosis and bipolar disorder, which is treated with a mood stabilizer.
Prevalence
Postpartum psychosis is identified in 1 to 2 per 1,000 childbirths. In women who have had an earlier episode of postpartum psychosis or have a diagnosis of bipolar disorder, the rate is up to 100 times higher.1 Kendell and colleagues found that psychiatric admissions occurred at a rate 7 times higher in the 30 days after birth than in the prepregnancy period, suggesting that metabolic factors might be involved in triggering postpartum psychotic symptoms.12 An abrupt hormonal loss occurs at childbirth; hormones peak 200-fold during gestation and decline rapidly within a day after birth.9 Despite the severity of symptoms in postpartum psychosis, these patients tend to have a better prognosis than that of women with psychotic episodes not related to pregnancy.4
Brockington and colleagues found that patients with postpartum psychosis had more mood lability, distractibility, and confusion than those with psychosis unrelated to pregnancy.15 Patients with postpartum psychosis were more likely to have impaired sensorium, bizarre quality of delusions, and memory loss. Psychosis with onset after childbirth included high levels of thought disorganization, delusions of reference, delusions of persecution, and greater levels of homicidal ideation and behavior.16 This study also reported symptoms such as visual, tactile, and olfactory hallucinations and a presentation similar to that of delirium.
Chandra and colleagues found that 53% of women with postpartum psychosis had delusions about the infant, including beliefs that someone would harm or kill the baby or that the baby would be harmed by their breast milk.17 Compared with women with bipolar disorder, Oostheuizen and colleagues found that women with postpartum psychosis had delusions of control, such as feeling under the influence of an overpowering force that controlled their actions.18 Infanticidal thoughts are common among patients with postpartum psychosis, and about 4% of women committed infanticide.1
Rapid stabilization and treatment are important because postpartum psychosis is considered a psychiatric emergency.7 Potential consequences of delayed diagnosis and treatment include harm or death of the infant by infanticide and death of the mother by suicide. A thorough physical examination is important to rule out metabolic or neuroendocrine causes of psychosis other than postpartum hormonal shifts. These could include causes of altered mental status: stroke, pulmonary embolism, amniotic fluid emboli, Sheehan syndrome, thyroid disorders, electrolyte abnormalities, acute hemorrhage, sepsis, and substance toxicity or withdrawal.10 A complete blood count, full chemistry, thyroid function tests, antithyroid antibody tests, calcium, vitamin B12, and folate should be measured.7,10
Initial treatment should include antipsychotics and often mood stabilizers such as lithium. Managing insomnia aggressively is also necessary for initial stabilization and to prevent a repeat manic episode if the patient develops bipolar disorder.2 Many experts argue that sleep loss in combination with other risk factors might be the final common pathway for development of postpartum psychosis in women predisposed to this disorder.19,20
Treating insomnia in an outpatient setting includes teaching sleep hygiene practices and relaxation techniques. Although these methods to regulate sleep could be encouraged during the emergent inpatient stabilization of a patient with postpartum psychosis, pharmacologic approaches are necessary for acute mania and psychosis. Concern about possible dependence on benzodiazepines and other sedating sleep aids are valid; however, the benefit of acute stabilization of psychotic symptoms outweighs the potential risk of dependence.
Typically, first-line treatment is an antipsychotic, and second-generation antipsychotics generally are preferred over first-generation antipsychotics because of their more benign adverse effect profile.21,22 There are no controlled trials that compare antipsychotics with placebo or other interventions for postpartum psychosis. Therefore, use of atypical antipsychotics is based on randomized trials demonstrating efficacy in reducing psychosis in bipolar disorder, depression with psychotic features, and schizoaffective disorder.23,24 Once the patient is treated with an antipsychotic, further use of psychotropic medications, such as lithium or other mood stabilizers, should be based on the patient’s clinical presentation. For example, the patient in this case study primarily had manic symptoms consistent with bipolar disorder, making lithium or another mood stabilizer an appropriate choice.
Bergink and colleagues demonstrated positive outcomes with a treatment algorithm involving sequential use of benzodiazepines to improve sleep, an antipsychotic to decrease acute manic symptoms, lithium to stabilize mood based on symptoms, and electroconvulsive therapy if other treatments were not successful.25 Case studies document that administering estrogen led to recovery from postpartum psychosis, although patients often relapsed when estrogen was stopped.26 Electroconvulsive therapy has shown promising results, especially in patients who do not respond to antipsychotic medications or lithium.27,28
Antipsychotic and Other Psychotropic Medications
Choice of an antipsychotic and other psychotropic medications to treat postpartum psychosis is based on the patient’s breastfeeding status. The benefits of treatment should be weighed against the risks of a breastfeeding infant’s exposure to the medication. Because postpartum psychosis is a psychiatric emergency, the benefits of the medication are considered to outweigh any potential adverse effect to the breastfeeding infant exposed to the medication. Risks of untreated postpartum psychosis to the infant include rejection of the infant, poor parental relationships, suicide, infanticide, long-term failure to bond with the child, delayed infant development, and failure to thrive.29 Also, many mothers—including the patient in this presentation—decide that the benefits of treatment outweigh those of breastfeeding and choose to feed their infant with formula. Even if the patient chooses to bottle-feed her infant, consider administering medications that are considered safer for breastfeeding because the patient may need to continue the psychotropic during later pregnancies to prevent future psychotic episodes.30 All psychotropic medications pass into breast milk.29 Studies on the long-term effect of these medications on the infant are limited, but experts tend to recommend olanzapine, quetiapine, and risperidone over aripiprazole and ziprasidone.21,31-33
Lithium often is used to treat postpartum psychosis. Studies examining risk to the infant after long-term exposure to lithium through breast milk have not been conducted, but the American Academy of Pediatrics discourages its use during breastfeeding because of concerns about toxicity in the infant.34-36
Sleep regulation is important to treat bipolar disorder and to prevent future episodes.2,20,21 To ensure safety of the infant and mother before discharge, family education is imperative to establish close follow-up, adequate sleep, and reduction of stressors.7,10 Separation from the infant might be necessary after discharge, and someone should monitor the infant at all times until the outpatient mental health provider confirms that all psychotic symptoms have resolved.7,10 Successful treatment of postpartum psychosis requires close communication among the mental health provider, the pediatrician, and the obstetrician or women’s health provider.10 Because a close-knit team approach after discharge from the acute psychiatric unit is necessary, the care of such a patient and her child provides an educational opportunity for individuals working in integrated care clinics.
Conclusion
Postpartum psychosis is a psychiatric emergency requiring immediate treatment to prevent dire outcomes such as suicide or infanticide. Treatment considerations include the cost-benefit analysis of breastfeeding and the toxicity of psychotropic medications when ingested by the infant via breast milk. A close relationship has been demonstrated between postpartum psychosis and bipolar disorder.
Preferred treatment regimens include lithium and an antipsychotic. Educate the family as a unit about the diagnosis and treatment, the importance of adequate sleep for treatment and prophylaxis, and the decision on whether to discontinue breastfeeding despite its well-known benefits for mother and infant. Stabilization is a multifaceted process and needs to be reinforced with a solid plan for support and follow-up appointments. Because of the higher risk of relapse, educate patients about prophylactic treatment during subsequent pregnancies and monitor for development of bipolar disorder in the future.
Postpartum psychosis is a psychiatric emergency that can endanger the life of the mother and the newborn child if untreated. About 1 to 2 mothers in 1,000 experiences postpartum psychosis after delivery.1 This rate is much higher among women with an established diagnosis of bipolar disorder before pregnancy.1
Expedient recognition, diagnosis, and referral to a high-level psychiatric facility (usually a locked inpatient unit) are critical for ensuring the safety of mother and infant. A diligent medical workup followed by thorough education for the patient and family are important steps in caring for patients with postpartum psychosis. Close mental health follow-up, pharmacologic interventions, informed decision making regarding breastfeeding, and preserving the sleep-wake cycle are critical for stabilization.2
The authors present the case of a patient admitted to VA Central California Health Care System (VACCHCS) with postpartum psychosis and a discussion on existing research on the prevalence of postpartum psychosis, relevant risk factors, the association with bipolar disorder, and treatment strategies.
Case Presentation
A 31-year-old active-duty female with no history of mental illness was admitted to the psychiatric unit because new-onset disorganized behavior was preventing her from functioning at her workplace. Two weeks after giving birth to her second child, the patient began exhibiting an uncharacteristic, debilitating labile mood and disorganized behavior. Her supervisors required her to present for medical attention about 3 months after the birth of her child. She was transferred to VACCHCS for higher level medical care on military orders. The patient’s husband initially attributed these psychiatric symptoms to vocational stress and taking care of 2 young children. He observed the patient exhibiting tearfulness about her job, which quickly alternated with euphoric episodes of singing and dancing at inappropriate times, such as when the children had quieted down and were being prepared to go to bed.
At the initial psychiatric evaluation after transfer to VACCHCS, the patient appeared well-kept and slightly overweight. In general her appearance was unremarkable. Throughout the examination she sang both subtly and loudly and at times was confrontational and irritable.
She related oddly and often was guarded and difficult to engage; she sang and played with her blanket in a childlike way. She smiled and laughed inappropriately, mumbled incoherently to herself, scanned the room suspiciously, and often made intense eye contact. Her affect was labile, both tearful and euphoric at several points in the examination. Her thought process was tangential, illogical, grandiose, difficult to redirect, and with loose associations. Her thought content consisted of delusions (“I’ve got the devil on my back”) and grandiosity (“I am everyone, I am you…the president, the mayor”), and she often stated that she planned to become a singer or performer.
The patient claimed she was neither suicidal nor had thoughts of infanticide. She reported having no visual and auditory hallucinations but often seemed to be responding to internal stimuli: She mumbled to herself and looked intensely at parts of the room. Cognitively, the patient was fully intact to recent and remote events but displayed a poor attention span. She did not exhibit any motor abnormalities, such as tremor, rigidity, weakness, sensory loss, or abnormal gait.
The patient’s workup included full chemistry, complete blood count, thyroid-stimulating hormone, antithyroid antibodies, calcium, rapid plasma reagin to rule out syphilis, toxicology, folate, vitamin B12, and vitamin D. All laboratory results were negative or within normal limits, although the urine drug screen was positive for cannabis. The patient’s husband noted that his wife never used cannabis except the weekend before her admission, when she impulsively went dancing, which was out of character for her. Her psychotic symptoms had been present weeks before the cannabis use; therefore, the her symptoms could not be attributed to a substance-induced psychotic disorder. A test for synthetic cannabis derivatives was negative. Newer synthetic compounds can cause more severe substance-induced psychotic symptoms than those of cannabis.3
The patient was diagnosed with postpartum psychosis and was started on the oral antipsychotic olanzapine 10 mg at bedtime. Additional doses were administered to control ongoing symptoms, which included a disorganized thought process; loose associations; euphoria; grandiosity; delusional content, such as “You are just a tool in place to help me;” reports of feeling as though she were in “outer space, outside in the galaxy;” decreased need for sleep; and irritability. The patient spent an entire interview with her eyes closed, stating that she could “hear” better because she was overstimulated if her eyes were open. She also described olfactory hallucinations of “strong perfume,” which the 2 providers present could not detect.
Olanzapine was not well tolerated because of sedation and was discontinued in favor of risperidone, 2 mg twice daily. Risperidone was more effective and better tolerated. Lithium was initiated the next day with target dosing at 300 mg in the morning and 600 mg at night. The patient became capable of linear, organized discussion and planning but remained euphoric with high energy; she exhibited grandiosity with frequent singing and dancing throughout her hospital stay. She often described her mood as “good, excellent, exuberant, exciting,” perseverating on the way words sounded and giggling in a childlike manner. She continued to have intrusive dreams of “hell and the devil” and that she was killed by gunshot.
The patient was continued on lithium and risperidone and transferred to a larger military hospital for further inpatient management, respecting military orders. Before discharge, a family conference was held with the patient and her husband to educate them on the importance of continued treatment, close follow-up, regular sleep patterns, and not breastfeeding while taking the prescribed medications. Although she was not back to her baseline at the time of transfer, the patient had stabilized significantly and gained sufficient insight into her condition.
Discussion
Postpartum psychosis can present with a prodromal phase consisting of fatigue, insomnia, restlessness, tearfulness, and emotional lability, making early identification difficult. Later, florid psychotic symptoms can include suspiciousness, confusion, incoherence, irrational statements, obsessive concern about the infant’s health, and delusions, including a belief that the baby is dead or defective. Some women might deny that the birth occurred or feel that they are unmarried, virginal, or persecuted.1 More concerning symptoms include auditory hallucinations commanding the mother to harm or kill the infant and/or herself. Symptoms often begin within days to weeks of birth, usually 2 to 3 weeks after delivery but can occur as long as 8 weeks postpartum.1 Several cases of infanticide and suicide have been documented.1 The risk of experiencing another psychotic episode in subsequent pregnancies can be as high as 50%.4-6 Regardless of symptom severity at onset, postpartum psychosis is a psychiatric emergency and must be treated as such.
Bipolar Disorder and Postpartum Psychosis
A close relationship exists between postpartum psychosis and development of bipolar disorder. A postpartum psychotic episode often is the harbinger of bipolar illness.7 About two-thirds of women who have an episode of postpartum psychosis will experience an underlying affective disorder within a year of childbirth.1,8 It is unclear what triggers the psychotic episode, but it has been theorized that major systemic shifts in hormone levelsor trauma of delivery could instigate development of symptoms.1,9
Risk factors include obstetric complications; perinatal infant mortality; previous episodes of bipolar disorder, psychosis, or postpartum psychosis; family history of bipolar disorder or postpartum psychosis; sleep deprivation; increased environmental stress; and lack of partner support.10 The strongest risk factor for developing postpartum psychosis is a personal or family history of bipolar disorder or a related psychotic disorder.11 This risk factor is identified in about 40% to 50% cases of postpartum psychosis.11
Treatment
Standard treatment for postpartum psychosis includes an antipsychotic and often lithium and benzodiazepines.1,7,10,11 This treatment approach differs slightly from treating a patient with a nonpostpartum psychotic illness, who generally would not receive mood stabilizers, such as lithium. Including a mood stabilizer for postpartum psychosis is warranted because of the association between postpartum psychosis and bipolar disorder, which is treated with a mood stabilizer.
Prevalence
Postpartum psychosis is identified in 1 to 2 per 1,000 childbirths. In women who have had an earlier episode of postpartum psychosis or have a diagnosis of bipolar disorder, the rate is up to 100 times higher.1 Kendell and colleagues found that psychiatric admissions occurred at a rate 7 times higher in the 30 days after birth than in the prepregnancy period, suggesting that metabolic factors might be involved in triggering postpartum psychotic symptoms.12 An abrupt hormonal loss occurs at childbirth; hormones peak 200-fold during gestation and decline rapidly within a day after birth.9 Despite the severity of symptoms in postpartum psychosis, these patients tend to have a better prognosis than that of women with psychotic episodes not related to pregnancy.4
Brockington and colleagues found that patients with postpartum psychosis had more mood lability, distractibility, and confusion than those with psychosis unrelated to pregnancy.15 Patients with postpartum psychosis were more likely to have impaired sensorium, bizarre quality of delusions, and memory loss. Psychosis with onset after childbirth included high levels of thought disorganization, delusions of reference, delusions of persecution, and greater levels of homicidal ideation and behavior.16 This study also reported symptoms such as visual, tactile, and olfactory hallucinations and a presentation similar to that of delirium.
Chandra and colleagues found that 53% of women with postpartum psychosis had delusions about the infant, including beliefs that someone would harm or kill the baby or that the baby would be harmed by their breast milk.17 Compared with women with bipolar disorder, Oostheuizen and colleagues found that women with postpartum psychosis had delusions of control, such as feeling under the influence of an overpowering force that controlled their actions.18 Infanticidal thoughts are common among patients with postpartum psychosis, and about 4% of women committed infanticide.1
Rapid stabilization and treatment are important because postpartum psychosis is considered a psychiatric emergency.7 Potential consequences of delayed diagnosis and treatment include harm or death of the infant by infanticide and death of the mother by suicide. A thorough physical examination is important to rule out metabolic or neuroendocrine causes of psychosis other than postpartum hormonal shifts. These could include causes of altered mental status: stroke, pulmonary embolism, amniotic fluid emboli, Sheehan syndrome, thyroid disorders, electrolyte abnormalities, acute hemorrhage, sepsis, and substance toxicity or withdrawal.10 A complete blood count, full chemistry, thyroid function tests, antithyroid antibody tests, calcium, vitamin B12, and folate should be measured.7,10
Initial treatment should include antipsychotics and often mood stabilizers such as lithium. Managing insomnia aggressively is also necessary for initial stabilization and to prevent a repeat manic episode if the patient develops bipolar disorder.2 Many experts argue that sleep loss in combination with other risk factors might be the final common pathway for development of postpartum psychosis in women predisposed to this disorder.19,20
Treating insomnia in an outpatient setting includes teaching sleep hygiene practices and relaxation techniques. Although these methods to regulate sleep could be encouraged during the emergent inpatient stabilization of a patient with postpartum psychosis, pharmacologic approaches are necessary for acute mania and psychosis. Concern about possible dependence on benzodiazepines and other sedating sleep aids are valid; however, the benefit of acute stabilization of psychotic symptoms outweighs the potential risk of dependence.
Typically, first-line treatment is an antipsychotic, and second-generation antipsychotics generally are preferred over first-generation antipsychotics because of their more benign adverse effect profile.21,22 There are no controlled trials that compare antipsychotics with placebo or other interventions for postpartum psychosis. Therefore, use of atypical antipsychotics is based on randomized trials demonstrating efficacy in reducing psychosis in bipolar disorder, depression with psychotic features, and schizoaffective disorder.23,24 Once the patient is treated with an antipsychotic, further use of psychotropic medications, such as lithium or other mood stabilizers, should be based on the patient’s clinical presentation. For example, the patient in this case study primarily had manic symptoms consistent with bipolar disorder, making lithium or another mood stabilizer an appropriate choice.
Bergink and colleagues demonstrated positive outcomes with a treatment algorithm involving sequential use of benzodiazepines to improve sleep, an antipsychotic to decrease acute manic symptoms, lithium to stabilize mood based on symptoms, and electroconvulsive therapy if other treatments were not successful.25 Case studies document that administering estrogen led to recovery from postpartum psychosis, although patients often relapsed when estrogen was stopped.26 Electroconvulsive therapy has shown promising results, especially in patients who do not respond to antipsychotic medications or lithium.27,28
Antipsychotic and Other Psychotropic Medications
Choice of an antipsychotic and other psychotropic medications to treat postpartum psychosis is based on the patient’s breastfeeding status. The benefits of treatment should be weighed against the risks of a breastfeeding infant’s exposure to the medication. Because postpartum psychosis is a psychiatric emergency, the benefits of the medication are considered to outweigh any potential adverse effect to the breastfeeding infant exposed to the medication. Risks of untreated postpartum psychosis to the infant include rejection of the infant, poor parental relationships, suicide, infanticide, long-term failure to bond with the child, delayed infant development, and failure to thrive.29 Also, many mothers—including the patient in this presentation—decide that the benefits of treatment outweigh those of breastfeeding and choose to feed their infant with formula. Even if the patient chooses to bottle-feed her infant, consider administering medications that are considered safer for breastfeeding because the patient may need to continue the psychotropic during later pregnancies to prevent future psychotic episodes.30 All psychotropic medications pass into breast milk.29 Studies on the long-term effect of these medications on the infant are limited, but experts tend to recommend olanzapine, quetiapine, and risperidone over aripiprazole and ziprasidone.21,31-33
Lithium often is used to treat postpartum psychosis. Studies examining risk to the infant after long-term exposure to lithium through breast milk have not been conducted, but the American Academy of Pediatrics discourages its use during breastfeeding because of concerns about toxicity in the infant.34-36
Sleep regulation is important to treat bipolar disorder and to prevent future episodes.2,20,21 To ensure safety of the infant and mother before discharge, family education is imperative to establish close follow-up, adequate sleep, and reduction of stressors.7,10 Separation from the infant might be necessary after discharge, and someone should monitor the infant at all times until the outpatient mental health provider confirms that all psychotic symptoms have resolved.7,10 Successful treatment of postpartum psychosis requires close communication among the mental health provider, the pediatrician, and the obstetrician or women’s health provider.10 Because a close-knit team approach after discharge from the acute psychiatric unit is necessary, the care of such a patient and her child provides an educational opportunity for individuals working in integrated care clinics.
Conclusion
Postpartum psychosis is a psychiatric emergency requiring immediate treatment to prevent dire outcomes such as suicide or infanticide. Treatment considerations include the cost-benefit analysis of breastfeeding and the toxicity of psychotropic medications when ingested by the infant via breast milk. A close relationship has been demonstrated between postpartum psychosis and bipolar disorder.
Preferred treatment regimens include lithium and an antipsychotic. Educate the family as a unit about the diagnosis and treatment, the importance of adequate sleep for treatment and prophylaxis, and the decision on whether to discontinue breastfeeding despite its well-known benefits for mother and infant. Stabilization is a multifaceted process and needs to be reinforced with a solid plan for support and follow-up appointments. Because of the higher risk of relapse, educate patients about prophylactic treatment during subsequent pregnancies and monitor for development of bipolar disorder in the future.
1. Sadock B, Sadock V, Ruiz P. Kaplan & Sadock’s Synopsis of Psychiatry. 11th ed. Philadelphia, PA: Wolters Kluwer; 2015.
2. Sharma V. Pharmacotherapy of postpartum psychosis. Expert Opin Pharmacother. 2003;4(10):1651-1658.
3. Bassir Nia A, Medrano B, Perkel C, Galynker I, Hurd YL. Psychiatric comorbidity associated with synthetic cannabinoid use compared to cannabis. J Psychopharmacol. 2016;30(12):1321-1330.
4. Rhohde A, Marneros A. Postpartum psychoses: onset and long-term course. Psychopathology. 1993;26(3-4):203-209.
5. Videbech P, Gouliaev G. First admission with puerperal psychosis: 7-14 years of follow-up. Acta Psychiatr Scand. 1995;91(3):167-173.
6. Terp IM, Engholm G, Møller H, Mortensen PB. A follow-up study of postpartum psychoses: prognosis and risk factors for readmission. Acta Psychiatr Scand. 1999;100(1):40-46.
7. Spinelli MG. Postpartum psychosis: detection of risk and management. Am J Psychiatry. 2009;166(4):405-408.
8. Blackmore ER, Rubinow DR, O’Connor TG, et al. Reproductive outcomes and risk of subsequent illness in women diagnosed with postpartum psychosis. Bipolar Disord. 2013;15(4):394-404.
9. Bloch M, Schmidt PJ, Danaceau M, Murphy J, Nieman L, Rubinow DR. Effects of gonadal steroids in women with a history of postpartum depression. Am J Psychiatry. 2000;157(6):924-930.
10. Monzon C, Lanza di Scalea T, Pearlstein T. Postpartum psychosis: updates and clinical issues. Psychiatric Times. 2014. http://www.psychiatrictimes.com/special-reports/postpartum -psychosis-updates-and-clinical-issues. Published January 15, 2014. Accessed December 14, 2017.
11. Davies W. Understanding the pathophysiology of postpartum psychosis: challenges and new approaches. World J Psychiatry. 2017;7(2):77-88.
12. Kendell RE, Chalmers JC, Platz C. Epidemiology of puerperal psychoses. Br J Psychiatry. 1987;150:662-673.
13. Leibenluft E. Women with bipolar illness: clinical and research issues. Am J Psychiatry. 1996;153(2):163-173.
14. Chaudron LH, Pies R. The relationship between postpartum psychosis and bipolar disorder: a review. J Clin Psychiatry. 2003;64(11):1284-1292.
15. Brockington IF, Cernik KF, Schofield EM, Downing AR, Francis AF, Keelan C. Puerperal psychosis: phenomena and diagnosis. Arch Gen Psychiatry. 1981;38(7):829-833.
16. Wisner KL, Peindl K, Hanusa BH. Symptomatology of affective and psychotic illnesses related to childbearing. J Affect Disord. 1994;30(2):77-87.
17. Chandra PS, Bhargavaraman RP, Raghunandan VN, Shaligram D. Delusions related to infant and their association with mother-infant interactions in postpartum psychotic disorders. Arch Womens Ment Health. 2006;9(5):285-288.
18. Oosthuizen P, Russouw H, Roberts M. Is puerperal psychosis bipolar mood disorder? A phenomenological comparison. Compr Psychiatry. 1995;36(1):77-81.
19. Sharma V, Mazmanian D. Sleep loss and postpartum psychosis. Bipolar Disord. 2003;5(2):98-105.
20 Bilszta JL, Meyer D, Buist AE. Bipolar affective disorder in the postnatal period: investigating the role of sleep. Bipolar Disord. 2010;12(5):568-578.
21. Doucet S, Jones I, Letourneau N, Dennis CL, Blackmore ER. Interventions for the prevention and treatment of postpartum psychosis: a systematic review. Arch Womens Ment Health. 2011;14(2):89-98.
22 Perlis RH, Welge JA, Vornik LA, Hirschfeld RM, Keck PE Jr. Atypical antipsychotics in the treatment of mania: a meta-analysis of randomized, placebo-controlled trials. J Clin Psychiatry. 2006;67(4):509-516.
23 Wijkstra J, Lijmer J, Balk FJ, Geddes JR, Nolen WA. Pharmacological treatment for unipolar psychotic depression: systematic review and meta-analysis. Br J Psychiatry. 2006;188:410-415.
24. Smith LA, Cornelius V, Warnock A, Tacchi MJ, Taylor D. Pharmacological interventions for acute bipolar mania: a systematic review of randomized placebo-controlled trials. Bipolar Disord. 2007;9(6):551-560.
25. Bergink V, Burgerhout KM, Koorengevel KM, et al. Treatment of psychosis and mania in the postpartum period. Am J Psychiatry. 2015;172(2):115-123.
26. Ahokas A, Aito M, Rimón R. Positive treatment effect of estradiol in postpartum psychosis: a pilot study. J Clin Psychiatry. 2000;61(3):166-169.
27. Reed P, Sermin N, Appleby L, Faragher B. A comparison of clinical response to electroconvulsive therapy in puerperal and non-puerperal psychoses. J Affect Disord. 1999;54(3):255-260.
28. Forray A, Ostroff RB. The use of electroconvulsive therapy in postpartum affective disorders. J ECT. 2007;23(3):188-193.
29. Robinson GE. Psychopharmacology in pregnancy and postpartum. Focus. 2012;10(1):3-14.
30. Wesseloo R, Kamperman AM, Munk-Olsen T, Pop VJ, Kushner SA, Bergink V. Risk of postpartum relapse in bipolar disorder and postpartum psychosis: a systematic review and meta-analysis. Am J Psychiatry. 2016;173(2):117-127.
31. Sharma V, Smith A, Mazmanian D. Olanzapine in the prevention of postpartum psychosis and mood episodes in bipolar disorder. Bipolar Disord. 2006;8(4):400-404.
32. Gobbi G. Quetiapine in postpartum psychosis. J Clin Psychopharmacol. 2014;34(6):744-745.
33. Uguz F. Second-generation antipsychotics during the lactation period: a comparative systematic review on infant safety. J Clin Psychopharmacol. 2016;36(3):244-252.
34. Sachs HC; Committee on Drugs. The transfer of drugs and therapeutics into human breast milk: an update on selected topics. Pediatrics. 2013;132(3):e796-e809.
35. Lithium [package insert]. Columbus, OH: Roxane Laboratories Inc; 2011.
36. Grandjean EM, Aubry JM. Lithium: updated human knowledge using an evidence-based approach: part III: clinical safety. CNS Drugs. 2009;23(5):397-418.
1. Sadock B, Sadock V, Ruiz P. Kaplan & Sadock’s Synopsis of Psychiatry. 11th ed. Philadelphia, PA: Wolters Kluwer; 2015.
2. Sharma V. Pharmacotherapy of postpartum psychosis. Expert Opin Pharmacother. 2003;4(10):1651-1658.
3. Bassir Nia A, Medrano B, Perkel C, Galynker I, Hurd YL. Psychiatric comorbidity associated with synthetic cannabinoid use compared to cannabis. J Psychopharmacol. 2016;30(12):1321-1330.
4. Rhohde A, Marneros A. Postpartum psychoses: onset and long-term course. Psychopathology. 1993;26(3-4):203-209.
5. Videbech P, Gouliaev G. First admission with puerperal psychosis: 7-14 years of follow-up. Acta Psychiatr Scand. 1995;91(3):167-173.
6. Terp IM, Engholm G, Møller H, Mortensen PB. A follow-up study of postpartum psychoses: prognosis and risk factors for readmission. Acta Psychiatr Scand. 1999;100(1):40-46.
7. Spinelli MG. Postpartum psychosis: detection of risk and management. Am J Psychiatry. 2009;166(4):405-408.
8. Blackmore ER, Rubinow DR, O’Connor TG, et al. Reproductive outcomes and risk of subsequent illness in women diagnosed with postpartum psychosis. Bipolar Disord. 2013;15(4):394-404.
9. Bloch M, Schmidt PJ, Danaceau M, Murphy J, Nieman L, Rubinow DR. Effects of gonadal steroids in women with a history of postpartum depression. Am J Psychiatry. 2000;157(6):924-930.
10. Monzon C, Lanza di Scalea T, Pearlstein T. Postpartum psychosis: updates and clinical issues. Psychiatric Times. 2014. http://www.psychiatrictimes.com/special-reports/postpartum -psychosis-updates-and-clinical-issues. Published January 15, 2014. Accessed December 14, 2017.
11. Davies W. Understanding the pathophysiology of postpartum psychosis: challenges and new approaches. World J Psychiatry. 2017;7(2):77-88.
12. Kendell RE, Chalmers JC, Platz C. Epidemiology of puerperal psychoses. Br J Psychiatry. 1987;150:662-673.
13. Leibenluft E. Women with bipolar illness: clinical and research issues. Am J Psychiatry. 1996;153(2):163-173.
14. Chaudron LH, Pies R. The relationship between postpartum psychosis and bipolar disorder: a review. J Clin Psychiatry. 2003;64(11):1284-1292.
15. Brockington IF, Cernik KF, Schofield EM, Downing AR, Francis AF, Keelan C. Puerperal psychosis: phenomena and diagnosis. Arch Gen Psychiatry. 1981;38(7):829-833.
16. Wisner KL, Peindl K, Hanusa BH. Symptomatology of affective and psychotic illnesses related to childbearing. J Affect Disord. 1994;30(2):77-87.
17. Chandra PS, Bhargavaraman RP, Raghunandan VN, Shaligram D. Delusions related to infant and their association with mother-infant interactions in postpartum psychotic disorders. Arch Womens Ment Health. 2006;9(5):285-288.
18. Oosthuizen P, Russouw H, Roberts M. Is puerperal psychosis bipolar mood disorder? A phenomenological comparison. Compr Psychiatry. 1995;36(1):77-81.
19. Sharma V, Mazmanian D. Sleep loss and postpartum psychosis. Bipolar Disord. 2003;5(2):98-105.
20 Bilszta JL, Meyer D, Buist AE. Bipolar affective disorder in the postnatal period: investigating the role of sleep. Bipolar Disord. 2010;12(5):568-578.
21. Doucet S, Jones I, Letourneau N, Dennis CL, Blackmore ER. Interventions for the prevention and treatment of postpartum psychosis: a systematic review. Arch Womens Ment Health. 2011;14(2):89-98.
22 Perlis RH, Welge JA, Vornik LA, Hirschfeld RM, Keck PE Jr. Atypical antipsychotics in the treatment of mania: a meta-analysis of randomized, placebo-controlled trials. J Clin Psychiatry. 2006;67(4):509-516.
23 Wijkstra J, Lijmer J, Balk FJ, Geddes JR, Nolen WA. Pharmacological treatment for unipolar psychotic depression: systematic review and meta-analysis. Br J Psychiatry. 2006;188:410-415.
24. Smith LA, Cornelius V, Warnock A, Tacchi MJ, Taylor D. Pharmacological interventions for acute bipolar mania: a systematic review of randomized placebo-controlled trials. Bipolar Disord. 2007;9(6):551-560.
25. Bergink V, Burgerhout KM, Koorengevel KM, et al. Treatment of psychosis and mania in the postpartum period. Am J Psychiatry. 2015;172(2):115-123.
26. Ahokas A, Aito M, Rimón R. Positive treatment effect of estradiol in postpartum psychosis: a pilot study. J Clin Psychiatry. 2000;61(3):166-169.
27. Reed P, Sermin N, Appleby L, Faragher B. A comparison of clinical response to electroconvulsive therapy in puerperal and non-puerperal psychoses. J Affect Disord. 1999;54(3):255-260.
28. Forray A, Ostroff RB. The use of electroconvulsive therapy in postpartum affective disorders. J ECT. 2007;23(3):188-193.
29. Robinson GE. Psychopharmacology in pregnancy and postpartum. Focus. 2012;10(1):3-14.
30. Wesseloo R, Kamperman AM, Munk-Olsen T, Pop VJ, Kushner SA, Bergink V. Risk of postpartum relapse in bipolar disorder and postpartum psychosis: a systematic review and meta-analysis. Am J Psychiatry. 2016;173(2):117-127.
31. Sharma V, Smith A, Mazmanian D. Olanzapine in the prevention of postpartum psychosis and mood episodes in bipolar disorder. Bipolar Disord. 2006;8(4):400-404.
32. Gobbi G. Quetiapine in postpartum psychosis. J Clin Psychopharmacol. 2014;34(6):744-745.
33. Uguz F. Second-generation antipsychotics during the lactation period: a comparative systematic review on infant safety. J Clin Psychopharmacol. 2016;36(3):244-252.
34. Sachs HC; Committee on Drugs. The transfer of drugs and therapeutics into human breast milk: an update on selected topics. Pediatrics. 2013;132(3):e796-e809.
35. Lithium [package insert]. Columbus, OH: Roxane Laboratories Inc; 2011.
36. Grandjean EM, Aubry JM. Lithium: updated human knowledge using an evidence-based approach: part III: clinical safety. CNS Drugs. 2009;23(5):397-418.
Mild cough • wheezing • loud heart sounds • Dx?
THE CASE
A 25-year-old man, who was an active duty US Navy sailor, went to his ship’s medical department complaining of a mild cough that he’d had for 2 days. He denied having any fevers, chills, night sweats, angina, or dyspnea. He said he hadn’t experienced any exertional fatigue or difficulty completing the rigorous physical tasks of his occupation as an engineman on the ship. The patient had no medical or surgical history of significance, and he wasn’t taking any medications or supplements.
On exam, he was not in acute distress and his vital signs were within normal limits. Auscultation revealed mild wheezing throughout the upper lung fields and loud heart sounds throughout his chest that were audible even with gentle contact of the stethoscope diaphragm. He had no discernible murmurs, rubs, or gallops.
In light of the unusually loud heart sounds heard on exam, we performed an electrocardiogram. The EKG revealed a normal sinus rhythm, slight right axis deviation indicated by tall R-waves in V1 (also suggestive of right ventricular hypertrophy), an incomplete right bundle branch block, and a crochetage sign (a notch in the R-waves of the inferior leads).1 A chest x-ray (FIGURE 1) revealed a normal-sized heart and dilated pulmonary vasculature suggestive of pulmonary hypertension.

THE DIAGNOSIS
To further evaluate the cardiopulmonary findings, ultrasound studies (transthoracic and transesophageal echocardiography) were performed. These demonstrated a very large secundum-type atrial septal defect (ASD), measuring at its largest point about 30 × 48 mm (FIGURE 2 and FIGURE 3C). Doppler flow analysis and a bubble study (VIDEOS 1 and 2) demonstrated significant shunting across the ASD. Gated cardiac computed tomography (CT) was also used to characterize the ASD (FIGURE 3). It revealed that the superior and posterior rims of the ASD were essentially absent and that the right atrium and ventricle were severely enlarged, while the left chambers were normal in size and function with an ejection fraction >55%. The notching of the R-waves of the inferior leads, seen in our patient’s EKG, is typically seen with large ASDs.1,2
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
Transthoracic echocardiography with color Doppler flow (red) demonstrated significant shunting across a large atrial septal defect (white box). The largest white dot is positioned near the center of the defect.
LA, left atrium; LV, left ventricle; RA, right atrium; RV, right ventricle.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
Transthoracic echocardiography with a bubble study showed injected air bubbles traversing the atrial septal defect.
LA, left atrium; LV, left ventricle; RA, right atrium; RV, right ventricle.
DISCUSSION
ASDs are typically uncovered on exam via auscultation of heart sounds, which might reveal a split of the second heart sound (S2) and diastolic murmurs. ASDs are typically classified by size, and their management depends on this factor, along with the patient’s age and symptoms. In children with small defects (<6 mm), treatment usually consists of conservative observation, as more than half of these ASDs will spontaneously close.3 But, as children age, they are more likely to engage in exertional activity (work, recreational sports) and an unrepaired ASD may yield symptoms (angina, dyspnea, fatigue, other cardiopulmonary strain). With such symptoms and when closure is not spontaneously achieved by adolescence or adulthood, an invasive approach is often necessary to correct the defect.
ASD repair. Traditionally, repair has involved some form of open thoracotomy. More recently, several minimally invasive techniques have been developed. Catheter-based device closure, in which a catheter is percutaneously guided to the defect and a patch is deployed to seal the ASD, is a technique that has been shown to successfully correct large ASDs of up to 40 mm in size.4 Robotic procedures have also been developed to correct ASDs through much smaller incisions.5 Both of these techniques require a significant rim of residual septal tissue around the defect.
Individualized approach. Since our patient had a rather large ASD that did not have sufficient residual septal rim tissue, percutaneous and robotic approaches were not feasible. Instead, he required more invasive cardiothoracic surgery. In cases such as this, the exact technique and type of incision (sternotomy vs access through the lateral chest wall) depend on age, gender, and the presence of other comorbidities.6
Our patient. Because there was concern that any approach other than a median one might not afford enough space to fix an ASD of such considerable size, our patient underwent a median sternotomy by a pediatric cardiothoracic surgeon who specialized in these repairs (in children as well as young adults). During the procedure, the ASD was accessed and confirmed to be as large as predicted by diagnostic imaging. A surgical patch was sutured in place to correct the defect. There were no intra-operative or postop complications.
Four weeks later, the patient had a mild pericardial effusion that was managed medically with daily furosemide and aspirin. At his 8-week postop appointment, the fluid accumulation had resolved, and he was completely asymptomatic. The patient returned to full-time active duty in the US Navy.
Adults with rather large ASDs can present in a relatively asymptomatic manner and report none of the classic complaints (angina, dyspnea, fatigue). They may even engage in heavy exertional activity with no difficulty. The underlying defect may be discovered incidentally on exam by noting a split of the S2 on auscultation. If pulmonary hypertension exists, the clinician may also note a loud S2. An exam that raises suspicion for an ASD can then be followed by tests that solidify the diagnosis. Surgery is usually necessary to correct an ASD in an adult who is symptomatic or exhibits significant cardiopulmonary strain.
1. Heller J, Hagège AA, Besse B, et al. “Crochetage” (notch) on R wave in inferior limb leads: a new independent electrocardiographic sign of atrial septal defect. J Am Coll Cardiol. 1996;27:877-882.
2. Kuijpers JM, Mulder BJM, Bouma BJ. Secundum atrial septal defect in adults: a practical review and recent developments. Neth Heart J. 2015;23:205-211.
3. McMahon CJ, Feltes TF, Fraley JK, et al. Natural history of growth of secundum atrial septal defects and implications for transcatheter closure. Heart. 2002;87:256-259.
4. Lopez K, Dalvi BV, Balzer D, et al. Transcatheter closure of large secundum atrial septal defects using the 40 mm amplatzer septal occluder: results of an international registry. Catheter Cardiovasc Interv. 2005;66:580-584.
5. Argenziano M, Oz MC, Kohmoto T, et al. Totally endoscopic atrial septal defect repair with robotic assistance. Circulation. 2003;108 Suppl 1:II191-II194.
6. Hopkins RA, Bert AA, Buchholz B, et al. Surgical patch closure of atrial septal defects. Ann Thorac Surg. 2004;77:2144-2149.
THE CASE
A 25-year-old man, who was an active duty US Navy sailor, went to his ship’s medical department complaining of a mild cough that he’d had for 2 days. He denied having any fevers, chills, night sweats, angina, or dyspnea. He said he hadn’t experienced any exertional fatigue or difficulty completing the rigorous physical tasks of his occupation as an engineman on the ship. The patient had no medical or surgical history of significance, and he wasn’t taking any medications or supplements.
On exam, he was not in acute distress and his vital signs were within normal limits. Auscultation revealed mild wheezing throughout the upper lung fields and loud heart sounds throughout his chest that were audible even with gentle contact of the stethoscope diaphragm. He had no discernible murmurs, rubs, or gallops.
In light of the unusually loud heart sounds heard on exam, we performed an electrocardiogram. The EKG revealed a normal sinus rhythm, slight right axis deviation indicated by tall R-waves in V1 (also suggestive of right ventricular hypertrophy), an incomplete right bundle branch block, and a crochetage sign (a notch in the R-waves of the inferior leads).1 A chest x-ray (FIGURE 1) revealed a normal-sized heart and dilated pulmonary vasculature suggestive of pulmonary hypertension.
THE DIAGNOSIS
To further evaluate the cardiopulmonary findings, ultrasound studies (transthoracic and transesophageal echocardiography) were performed. These demonstrated a very large secundum-type atrial septal defect (ASD), measuring at its largest point about 30 × 48 mm (FIGURE 2 and FIGURE 3C). Doppler flow analysis and a bubble study (VIDEOS 1 and 2) demonstrated significant shunting across the ASD. Gated cardiac computed tomography (CT) was also used to characterize the ASD (FIGURE 3). It revealed that the superior and posterior rims of the ASD were essentially absent and that the right atrium and ventricle were severely enlarged, while the left chambers were normal in size and function with an ejection fraction >55%. The notching of the R-waves of the inferior leads, seen in our patient’s EKG, is typically seen with large ASDs.1,2
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
Transthoracic echocardiography with color Doppler flow (red) demonstrated significant shunting across a large atrial septal defect (white box). The largest white dot is positioned near the center of the defect.
LA, left atrium; LV, left ventricle; RA, right atrium; RV, right ventricle.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
Transthoracic echocardiography with a bubble study showed injected air bubbles traversing the atrial septal defect.
LA, left atrium; LV, left ventricle; RA, right atrium; RV, right ventricle.
DISCUSSION
ASDs are typically uncovered on exam via auscultation of heart sounds, which might reveal a split of the second heart sound (S2) and diastolic murmurs. ASDs are typically classified by size, and their management depends on this factor, along with the patient’s age and symptoms. In children with small defects (<6 mm), treatment usually consists of conservative observation, as more than half of these ASDs will spontaneously close.3 But, as children age, they are more likely to engage in exertional activity (work, recreational sports) and an unrepaired ASD may yield symptoms (angina, dyspnea, fatigue, other cardiopulmonary strain). With such symptoms and when closure is not spontaneously achieved by adolescence or adulthood, an invasive approach is often necessary to correct the defect.
ASD repair. Traditionally, repair has involved some form of open thoracotomy. More recently, several minimally invasive techniques have been developed. Catheter-based device closure, in which a catheter is percutaneously guided to the defect and a patch is deployed to seal the ASD, is a technique that has been shown to successfully correct large ASDs of up to 40 mm in size.4 Robotic procedures have also been developed to correct ASDs through much smaller incisions.5 Both of these techniques require a significant rim of residual septal tissue around the defect.
Individualized approach. Since our patient had a rather large ASD that did not have sufficient residual septal rim tissue, percutaneous and robotic approaches were not feasible. Instead, he required more invasive cardiothoracic surgery. In cases such as this, the exact technique and type of incision (sternotomy vs access through the lateral chest wall) depend on age, gender, and the presence of other comorbidities.6
Our patient. Because there was concern that any approach other than a median one might not afford enough space to fix an ASD of such considerable size, our patient underwent a median sternotomy by a pediatric cardiothoracic surgeon who specialized in these repairs (in children as well as young adults). During the procedure, the ASD was accessed and confirmed to be as large as predicted by diagnostic imaging. A surgical patch was sutured in place to correct the defect. There were no intra-operative or postop complications.
Four weeks later, the patient had a mild pericardial effusion that was managed medically with daily furosemide and aspirin. At his 8-week postop appointment, the fluid accumulation had resolved, and he was completely asymptomatic. The patient returned to full-time active duty in the US Navy.
Adults with rather large ASDs can present in a relatively asymptomatic manner and report none of the classic complaints (angina, dyspnea, fatigue). They may even engage in heavy exertional activity with no difficulty. The underlying defect may be discovered incidentally on exam by noting a split of the S2 on auscultation. If pulmonary hypertension exists, the clinician may also note a loud S2. An exam that raises suspicion for an ASD can then be followed by tests that solidify the diagnosis. Surgery is usually necessary to correct an ASD in an adult who is symptomatic or exhibits significant cardiopulmonary strain.
THE CASE
A 25-year-old man, who was an active duty US Navy sailor, went to his ship’s medical department complaining of a mild cough that he’d had for 2 days. He denied having any fevers, chills, night sweats, angina, or dyspnea. He said he hadn’t experienced any exertional fatigue or difficulty completing the rigorous physical tasks of his occupation as an engineman on the ship. The patient had no medical or surgical history of significance, and he wasn’t taking any medications or supplements.
On exam, he was not in acute distress and his vital signs were within normal limits. Auscultation revealed mild wheezing throughout the upper lung fields and loud heart sounds throughout his chest that were audible even with gentle contact of the stethoscope diaphragm. He had no discernible murmurs, rubs, or gallops.
In light of the unusually loud heart sounds heard on exam, we performed an electrocardiogram. The EKG revealed a normal sinus rhythm, slight right axis deviation indicated by tall R-waves in V1 (also suggestive of right ventricular hypertrophy), an incomplete right bundle branch block, and a crochetage sign (a notch in the R-waves of the inferior leads).1 A chest x-ray (FIGURE 1) revealed a normal-sized heart and dilated pulmonary vasculature suggestive of pulmonary hypertension.
THE DIAGNOSIS
To further evaluate the cardiopulmonary findings, ultrasound studies (transthoracic and transesophageal echocardiography) were performed. These demonstrated a very large secundum-type atrial septal defect (ASD), measuring at its largest point about 30 × 48 mm (FIGURE 2 and FIGURE 3C). Doppler flow analysis and a bubble study (VIDEOS 1 and 2) demonstrated significant shunting across the ASD. Gated cardiac computed tomography (CT) was also used to characterize the ASD (FIGURE 3). It revealed that the superior and posterior rims of the ASD were essentially absent and that the right atrium and ventricle were severely enlarged, while the left chambers were normal in size and function with an ejection fraction >55%. The notching of the R-waves of the inferior leads, seen in our patient’s EKG, is typically seen with large ASDs.1,2
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
Transthoracic echocardiography with color Doppler flow (red) demonstrated significant shunting across a large atrial septal defect (white box). The largest white dot is positioned near the center of the defect.
LA, left atrium; LV, left ventricle; RA, right atrium; RV, right ventricle.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
Transthoracic echocardiography with a bubble study showed injected air bubbles traversing the atrial septal defect.
LA, left atrium; LV, left ventricle; RA, right atrium; RV, right ventricle.
DISCUSSION
ASDs are typically uncovered on exam via auscultation of heart sounds, which might reveal a split of the second heart sound (S2) and diastolic murmurs. ASDs are typically classified by size, and their management depends on this factor, along with the patient’s age and symptoms. In children with small defects (<6 mm), treatment usually consists of conservative observation, as more than half of these ASDs will spontaneously close.3 But, as children age, they are more likely to engage in exertional activity (work, recreational sports) and an unrepaired ASD may yield symptoms (angina, dyspnea, fatigue, other cardiopulmonary strain). With such symptoms and when closure is not spontaneously achieved by adolescence or adulthood, an invasive approach is often necessary to correct the defect.
ASD repair. Traditionally, repair has involved some form of open thoracotomy. More recently, several minimally invasive techniques have been developed. Catheter-based device closure, in which a catheter is percutaneously guided to the defect and a patch is deployed to seal the ASD, is a technique that has been shown to successfully correct large ASDs of up to 40 mm in size.4 Robotic procedures have also been developed to correct ASDs through much smaller incisions.5 Both of these techniques require a significant rim of residual septal tissue around the defect.
Individualized approach. Since our patient had a rather large ASD that did not have sufficient residual septal rim tissue, percutaneous and robotic approaches were not feasible. Instead, he required more invasive cardiothoracic surgery. In cases such as this, the exact technique and type of incision (sternotomy vs access through the lateral chest wall) depend on age, gender, and the presence of other comorbidities.6
Our patient. Because there was concern that any approach other than a median one might not afford enough space to fix an ASD of such considerable size, our patient underwent a median sternotomy by a pediatric cardiothoracic surgeon who specialized in these repairs (in children as well as young adults). During the procedure, the ASD was accessed and confirmed to be as large as predicted by diagnostic imaging. A surgical patch was sutured in place to correct the defect. There were no intra-operative or postop complications.
Four weeks later, the patient had a mild pericardial effusion that was managed medically with daily furosemide and aspirin. At his 8-week postop appointment, the fluid accumulation had resolved, and he was completely asymptomatic. The patient returned to full-time active duty in the US Navy.
Adults with rather large ASDs can present in a relatively asymptomatic manner and report none of the classic complaints (angina, dyspnea, fatigue). They may even engage in heavy exertional activity with no difficulty. The underlying defect may be discovered incidentally on exam by noting a split of the S2 on auscultation. If pulmonary hypertension exists, the clinician may also note a loud S2. An exam that raises suspicion for an ASD can then be followed by tests that solidify the diagnosis. Surgery is usually necessary to correct an ASD in an adult who is symptomatic or exhibits significant cardiopulmonary strain.
1. Heller J, Hagège AA, Besse B, et al. “Crochetage” (notch) on R wave in inferior limb leads: a new independent electrocardiographic sign of atrial septal defect. J Am Coll Cardiol. 1996;27:877-882.
2. Kuijpers JM, Mulder BJM, Bouma BJ. Secundum atrial septal defect in adults: a practical review and recent developments. Neth Heart J. 2015;23:205-211.
3. McMahon CJ, Feltes TF, Fraley JK, et al. Natural history of growth of secundum atrial septal defects and implications for transcatheter closure. Heart. 2002;87:256-259.
4. Lopez K, Dalvi BV, Balzer D, et al. Transcatheter closure of large secundum atrial septal defects using the 40 mm amplatzer septal occluder: results of an international registry. Catheter Cardiovasc Interv. 2005;66:580-584.
5. Argenziano M, Oz MC, Kohmoto T, et al. Totally endoscopic atrial septal defect repair with robotic assistance. Circulation. 2003;108 Suppl 1:II191-II194.
6. Hopkins RA, Bert AA, Buchholz B, et al. Surgical patch closure of atrial septal defects. Ann Thorac Surg. 2004;77:2144-2149.
1. Heller J, Hagège AA, Besse B, et al. “Crochetage” (notch) on R wave in inferior limb leads: a new independent electrocardiographic sign of atrial septal defect. J Am Coll Cardiol. 1996;27:877-882.
2. Kuijpers JM, Mulder BJM, Bouma BJ. Secundum atrial septal defect in adults: a practical review and recent developments. Neth Heart J. 2015;23:205-211.
3. McMahon CJ, Feltes TF, Fraley JK, et al. Natural history of growth of secundum atrial septal defects and implications for transcatheter closure. Heart. 2002;87:256-259.
4. Lopez K, Dalvi BV, Balzer D, et al. Transcatheter closure of large secundum atrial septal defects using the 40 mm amplatzer septal occluder: results of an international registry. Catheter Cardiovasc Interv. 2005;66:580-584.
5. Argenziano M, Oz MC, Kohmoto T, et al. Totally endoscopic atrial septal defect repair with robotic assistance. Circulation. 2003;108 Suppl 1:II191-II194.
6. Hopkins RA, Bert AA, Buchholz B, et al. Surgical patch closure of atrial septal defects. Ann Thorac Surg. 2004;77:2144-2149.

Elevated serum alkaline phosphatase • generalized pruritus • Dx?
THE CASE
A 34-year-old woman was referred to the hepatology clinic for evaluation of an increased serum alkaline phosphatase (ALP) level. She was gravida 5 and in her 38th week of gestation. Her obstetric history was significant for 2 uncomplicated spontaneous term vaginal deliveries resulting in live births and 2 spontaneous abortions. The patient reported generalized pruritus for 2 months prior to the visit. She had no comorbidities and denied any other symptoms. She reported no family history of liver disease or complications during pregnancy in relatives. The patient did not smoke or drink, and had come to our hospital for her prenatal care visits.
The physical exam revealed normal vital signs, no jaundice, a gravid uterus, and acanthosis nigricans on the neck and axilla with scattered excoriations on the arms, legs, and abdomen. Her serum ALP level was 1093 U/L (normal: 50-136 U/L). Immediately before this pregnancy, her serum ALP had been normal at 95 U/L, but it had since been increasing with a peak value of 1134 U/L by 37 weeks’ gestation. Serum transaminase activities and albumin and bilirubin concentrations were normal, as was her prothrombin time. The rest of her lab tests were also normal, including her fasting serum bile acid concentration, which was 9 mcmol/L (normal: 4.5-19.2 mcmol/L).
THE DIAGNOSIS
Although cholestasis of pregnancy was considered, the patient’s markedly elevated serum ALP level suggested the presence of another cholestatic liver disease. Additional tests revealed an antimitochondrial antibody (AMA) titer of 1:320 (normal: <1:20) and immunoglobulin A, G, and M levels within normal limits. Accordingly, we diagnosed primary biliary cholangitis (PBC).
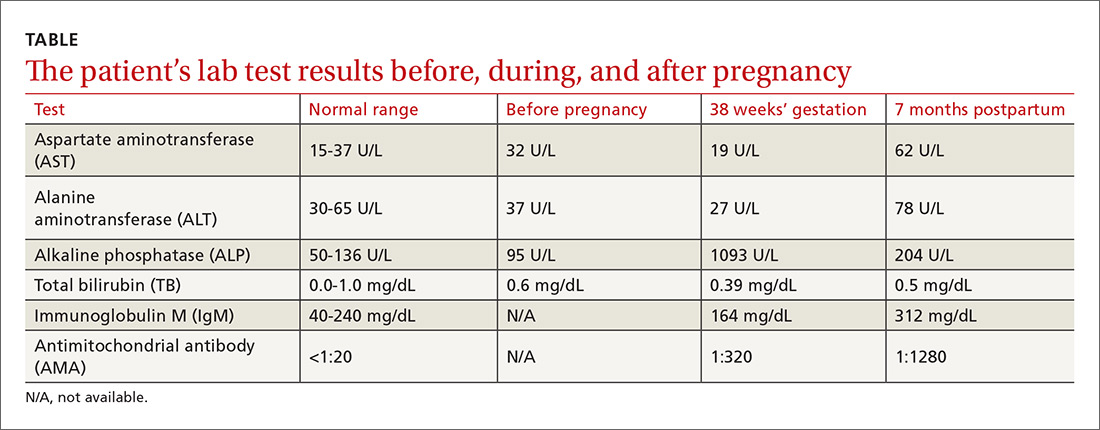
The patient delivered vaginally at another institution uneventfully and returned to the hepatology clinic 7 months postpartum. Repeat laboratory tests (TABLE) revealed increased AMA titer and immunoglobulin M levels from baseline (38 weeks’ gestation). The physical exam was notable for the absence of both jaundice and stigmata of chronic liver disease. A liver ultrasound was normal. The patient still reported pruritus, as well as a new symptom—fatigue. A liver biopsy was performed, and findings were consistent with PBC, stage 1 (FIGURE).
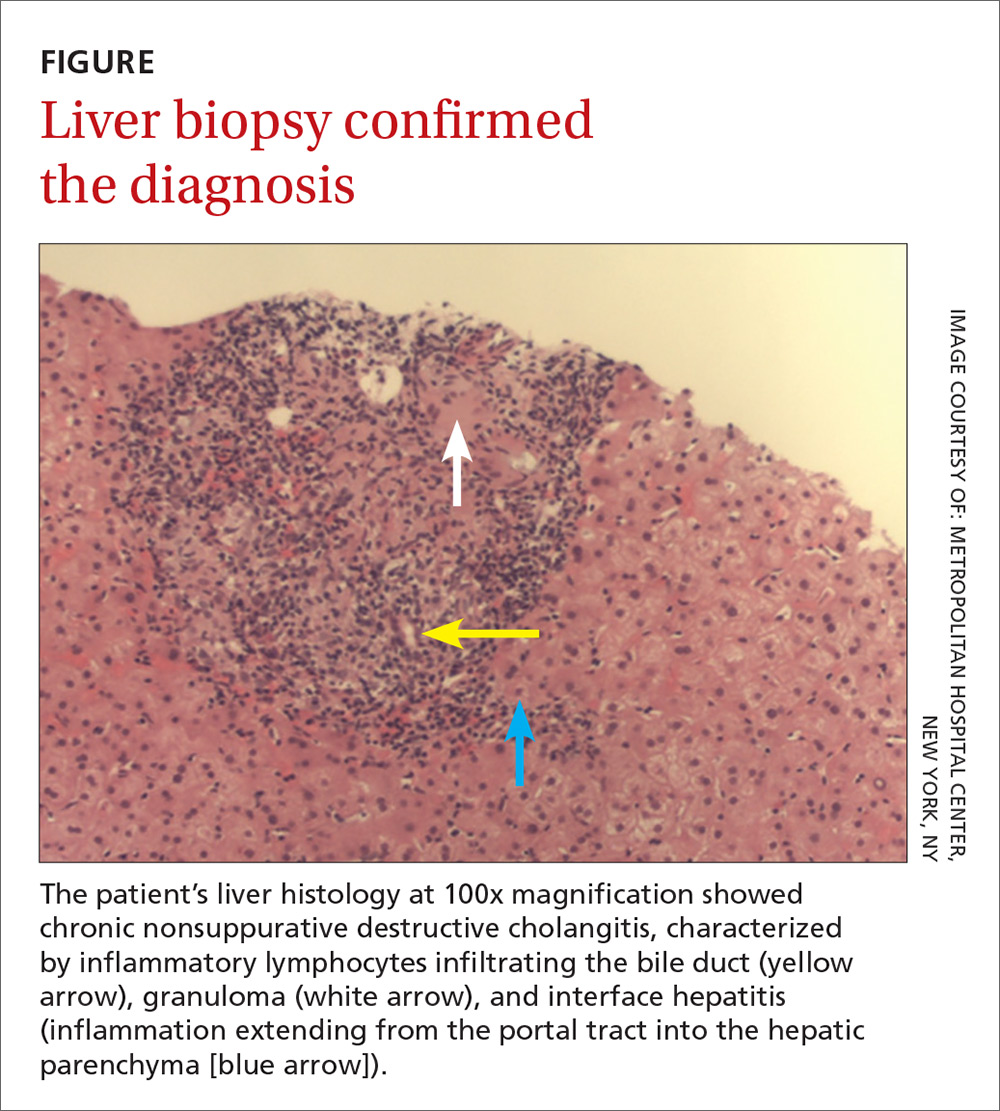
DISCUSSION
PBC, historically known as primary biliary cirrhosis, is a chronic, likely immune-mediated, cholestatic liver disease characterized by the progressive inflammatory destruction of intrahepatic bile ducts. The disease has a female to male predominance of 10:1, with age of diagnosis most often between 40 and 50 years, although about a quarter of female patients present during their reproductive years.1,2
PBC in pregnant women
During pregnancy, the profound physiologic changes and adaptations in the endocrine, metabolic, and immune systems that are necessary for normal fetal development can affect the maternal hepatobiliary system. In patients with prior autoimmune liver disease, the liver is known to adapt itself to these physiologic changes by entering a state of immune tolerance. This is induced by relative hypercortisolism, a shift from predominantly cell-mediated immunity to humoral immunity, and inhibition of T-cell activation. These changes can result in remission of autoimmune disease activity during pregnancy and postpartum flaring when these protective mechanisms are lost (although neither remission nor postpartum flaring occurred in this patient’s case).1-3
While a well-compensated state is associated with better fetal and maternal outcomes than a decompensated condition, cirrhosis is not a contraindication to pregnancy. Vaginal delivery is generally safe for patients with PBC, and studies have reported no childbirth complications or adverse maternal outcomes.1,3,4
The approved treatment for PBC, ursodeoxycholic acid (UDCA), was classified as a category B agent according to the Food and Drug Administration’s now defunct classification system for drugs used during pregnancy and lactation. It’s considered to be the treatment of choice for intrahepatic cholestasis of pregnancy, but there are no recommendations for its use in pregnant patients with PBC. Several studies have observed no significant teratogenic effect in babies whose mothers were treated with UDCA for PBC during pregnancy.1-4 Postpartum, 60% to 70% of PBC patients have been reported to exhibit biochemical disease activity,1,3 and in one case, a liver transplant was required due to liver failure.5
Look for AMA, elevated ALP
The diagnosis of the disease in this case was made by the detection of AMA, which has a specificity of 98% for PBC. However, isolated instances of the presence of AMA are not uncommon; they have been documented in up to 64% of healthy individuals.6 In addition, while one would expect to see a 2- to 4-fold rise in ALP levels during pregnancy (due to placental isoenzyme production),2,7 our patient’s serum ALP level was much higher, suggesting probable cholestatic liver disease such as PBC. The diagnosis in this case was confirmed by liver biopsy.
Our patient was started on UDCA 13 to 15 mg/kg/d. She remained clinically stable at subsequent follow-ups.
THE TAKEAWAY
Typically seen in middle-aged women, PBC can be detected by the presence of AMA and elevated ALP levels. Pregnant patients with chronic liver disease, including PBC, should be followed by a hepatologist and a high-risk obstetrician. They should be carefully monitored and frequently reassessed throughout the pregnancy, delivery, and postpartum period, even though studies have documented favorable outcomes for both mother and baby.1,3,4
1. Trivedi PJ, Kumagi T, Al-Harthy N, et al. Good maternal and fetal outcomes for pregnant women with primary biliary cirrhosis. Clin Gastroenterol Hepatol. 2014;12:1179-1185.
2. Marchioni Beery RM, Vaziri H, Forouhar F. Primary biliary cirrhosis and primary sclerosing cholangitis: a review featuring a women’s health perspective. J Clin Transl Hepatol. 2014;2:266-284.
3. Efe C, Kahramanoğlu-Aksoy E, Yilmaz B, et al. Pregnancy in women with primary biliary cirrhosis. Autoimmun Rev. 2014;13:931-935.
4. Floreani A, Infantolino C, Franceschet I, et al. Pregnancy and primary biliary cirrhosis: a case control study. Clin Rev Allergy Immunol. 2015;48:236-242.
5. Rabinovitz M, Appasamy R, Finkelstein S. Primary biliary cirrhosis diagnosed during pregnancy. Does it have a different outcome? Dig Dis Sci. 1995;40:571-574.
6. Carey EJ, Ali AH, Lindor KD. Primary biliary cirrhosis. Lancet. 2015;386:1565-1575.
7. The Johns Hopkins School of Medicine Department of Gynecology. Hurt KJ, Guile MW, Bienstock JL, et al, eds. The Johns Hopkins Manual of Gynecology and Obstetrics. 4th edition. Philadelphia, PA: Lippincott Williams and Wilkins; 2011.
THE CASE
A 34-year-old woman was referred to the hepatology clinic for evaluation of an increased serum alkaline phosphatase (ALP) level. She was gravida 5 and in her 38th week of gestation. Her obstetric history was significant for 2 uncomplicated spontaneous term vaginal deliveries resulting in live births and 2 spontaneous abortions. The patient reported generalized pruritus for 2 months prior to the visit. She had no comorbidities and denied any other symptoms. She reported no family history of liver disease or complications during pregnancy in relatives. The patient did not smoke or drink, and had come to our hospital for her prenatal care visits.
The physical exam revealed normal vital signs, no jaundice, a gravid uterus, and acanthosis nigricans on the neck and axilla with scattered excoriations on the arms, legs, and abdomen. Her serum ALP level was 1093 U/L (normal: 50-136 U/L). Immediately before this pregnancy, her serum ALP had been normal at 95 U/L, but it had since been increasing with a peak value of 1134 U/L by 37 weeks’ gestation. Serum transaminase activities and albumin and bilirubin concentrations were normal, as was her prothrombin time. The rest of her lab tests were also normal, including her fasting serum bile acid concentration, which was 9 mcmol/L (normal: 4.5-19.2 mcmol/L).
THE DIAGNOSIS
Although cholestasis of pregnancy was considered, the patient’s markedly elevated serum ALP level suggested the presence of another cholestatic liver disease. Additional tests revealed an antimitochondrial antibody (AMA) titer of 1:320 (normal: <1:20) and immunoglobulin A, G, and M levels within normal limits. Accordingly, we diagnosed primary biliary cholangitis (PBC).
The patient delivered vaginally at another institution uneventfully and returned to the hepatology clinic 7 months postpartum. Repeat laboratory tests (TABLE) revealed increased AMA titer and immunoglobulin M levels from baseline (38 weeks’ gestation). The physical exam was notable for the absence of both jaundice and stigmata of chronic liver disease. A liver ultrasound was normal. The patient still reported pruritus, as well as a new symptom—fatigue. A liver biopsy was performed, and findings were consistent with PBC, stage 1 (FIGURE).
DISCUSSION
PBC, historically known as primary biliary cirrhosis, is a chronic, likely immune-mediated, cholestatic liver disease characterized by the progressive inflammatory destruction of intrahepatic bile ducts. The disease has a female to male predominance of 10:1, with age of diagnosis most often between 40 and 50 years, although about a quarter of female patients present during their reproductive years.1,2
PBC in pregnant women
During pregnancy, the profound physiologic changes and adaptations in the endocrine, metabolic, and immune systems that are necessary for normal fetal development can affect the maternal hepatobiliary system. In patients with prior autoimmune liver disease, the liver is known to adapt itself to these physiologic changes by entering a state of immune tolerance. This is induced by relative hypercortisolism, a shift from predominantly cell-mediated immunity to humoral immunity, and inhibition of T-cell activation. These changes can result in remission of autoimmune disease activity during pregnancy and postpartum flaring when these protective mechanisms are lost (although neither remission nor postpartum flaring occurred in this patient’s case).1-3
While a well-compensated state is associated with better fetal and maternal outcomes than a decompensated condition, cirrhosis is not a contraindication to pregnancy. Vaginal delivery is generally safe for patients with PBC, and studies have reported no childbirth complications or adverse maternal outcomes.1,3,4
The approved treatment for PBC, ursodeoxycholic acid (UDCA), was classified as a category B agent according to the Food and Drug Administration’s now defunct classification system for drugs used during pregnancy and lactation. It’s considered to be the treatment of choice for intrahepatic cholestasis of pregnancy, but there are no recommendations for its use in pregnant patients with PBC. Several studies have observed no significant teratogenic effect in babies whose mothers were treated with UDCA for PBC during pregnancy.1-4 Postpartum, 60% to 70% of PBC patients have been reported to exhibit biochemical disease activity,1,3 and in one case, a liver transplant was required due to liver failure.5
Look for AMA, elevated ALP
The diagnosis of the disease in this case was made by the detection of AMA, which has a specificity of 98% for PBC. However, isolated instances of the presence of AMA are not uncommon; they have been documented in up to 64% of healthy individuals.6 In addition, while one would expect to see a 2- to 4-fold rise in ALP levels during pregnancy (due to placental isoenzyme production),2,7 our patient’s serum ALP level was much higher, suggesting probable cholestatic liver disease such as PBC. The diagnosis in this case was confirmed by liver biopsy.
Our patient was started on UDCA 13 to 15 mg/kg/d. She remained clinically stable at subsequent follow-ups.
THE TAKEAWAY
Typically seen in middle-aged women, PBC can be detected by the presence of AMA and elevated ALP levels. Pregnant patients with chronic liver disease, including PBC, should be followed by a hepatologist and a high-risk obstetrician. They should be carefully monitored and frequently reassessed throughout the pregnancy, delivery, and postpartum period, even though studies have documented favorable outcomes for both mother and baby.1,3,4
THE CASE
A 34-year-old woman was referred to the hepatology clinic for evaluation of an increased serum alkaline phosphatase (ALP) level. She was gravida 5 and in her 38th week of gestation. Her obstetric history was significant for 2 uncomplicated spontaneous term vaginal deliveries resulting in live births and 2 spontaneous abortions. The patient reported generalized pruritus for 2 months prior to the visit. She had no comorbidities and denied any other symptoms. She reported no family history of liver disease or complications during pregnancy in relatives. The patient did not smoke or drink, and had come to our hospital for her prenatal care visits.
The physical exam revealed normal vital signs, no jaundice, a gravid uterus, and acanthosis nigricans on the neck and axilla with scattered excoriations on the arms, legs, and abdomen. Her serum ALP level was 1093 U/L (normal: 50-136 U/L). Immediately before this pregnancy, her serum ALP had been normal at 95 U/L, but it had since been increasing with a peak value of 1134 U/L by 37 weeks’ gestation. Serum transaminase activities and albumin and bilirubin concentrations were normal, as was her prothrombin time. The rest of her lab tests were also normal, including her fasting serum bile acid concentration, which was 9 mcmol/L (normal: 4.5-19.2 mcmol/L).
THE DIAGNOSIS
Although cholestasis of pregnancy was considered, the patient’s markedly elevated serum ALP level suggested the presence of another cholestatic liver disease. Additional tests revealed an antimitochondrial antibody (AMA) titer of 1:320 (normal: <1:20) and immunoglobulin A, G, and M levels within normal limits. Accordingly, we diagnosed primary biliary cholangitis (PBC).
The patient delivered vaginally at another institution uneventfully and returned to the hepatology clinic 7 months postpartum. Repeat laboratory tests (TABLE) revealed increased AMA titer and immunoglobulin M levels from baseline (38 weeks’ gestation). The physical exam was notable for the absence of both jaundice and stigmata of chronic liver disease. A liver ultrasound was normal. The patient still reported pruritus, as well as a new symptom—fatigue. A liver biopsy was performed, and findings were consistent with PBC, stage 1 (FIGURE).
DISCUSSION
PBC, historically known as primary biliary cirrhosis, is a chronic, likely immune-mediated, cholestatic liver disease characterized by the progressive inflammatory destruction of intrahepatic bile ducts. The disease has a female to male predominance of 10:1, with age of diagnosis most often between 40 and 50 years, although about a quarter of female patients present during their reproductive years.1,2
PBC in pregnant women
During pregnancy, the profound physiologic changes and adaptations in the endocrine, metabolic, and immune systems that are necessary for normal fetal development can affect the maternal hepatobiliary system. In patients with prior autoimmune liver disease, the liver is known to adapt itself to these physiologic changes by entering a state of immune tolerance. This is induced by relative hypercortisolism, a shift from predominantly cell-mediated immunity to humoral immunity, and inhibition of T-cell activation. These changes can result in remission of autoimmune disease activity during pregnancy and postpartum flaring when these protective mechanisms are lost (although neither remission nor postpartum flaring occurred in this patient’s case).1-3
While a well-compensated state is associated with better fetal and maternal outcomes than a decompensated condition, cirrhosis is not a contraindication to pregnancy. Vaginal delivery is generally safe for patients with PBC, and studies have reported no childbirth complications or adverse maternal outcomes.1,3,4
The approved treatment for PBC, ursodeoxycholic acid (UDCA), was classified as a category B agent according to the Food and Drug Administration’s now defunct classification system for drugs used during pregnancy and lactation. It’s considered to be the treatment of choice for intrahepatic cholestasis of pregnancy, but there are no recommendations for its use in pregnant patients with PBC. Several studies have observed no significant teratogenic effect in babies whose mothers were treated with UDCA for PBC during pregnancy.1-4 Postpartum, 60% to 70% of PBC patients have been reported to exhibit biochemical disease activity,1,3 and in one case, a liver transplant was required due to liver failure.5
Look for AMA, elevated ALP
The diagnosis of the disease in this case was made by the detection of AMA, which has a specificity of 98% for PBC. However, isolated instances of the presence of AMA are not uncommon; they have been documented in up to 64% of healthy individuals.6 In addition, while one would expect to see a 2- to 4-fold rise in ALP levels during pregnancy (due to placental isoenzyme production),2,7 our patient’s serum ALP level was much higher, suggesting probable cholestatic liver disease such as PBC. The diagnosis in this case was confirmed by liver biopsy.
Our patient was started on UDCA 13 to 15 mg/kg/d. She remained clinically stable at subsequent follow-ups.
THE TAKEAWAY
Typically seen in middle-aged women, PBC can be detected by the presence of AMA and elevated ALP levels. Pregnant patients with chronic liver disease, including PBC, should be followed by a hepatologist and a high-risk obstetrician. They should be carefully monitored and frequently reassessed throughout the pregnancy, delivery, and postpartum period, even though studies have documented favorable outcomes for both mother and baby.1,3,4
1. Trivedi PJ, Kumagi T, Al-Harthy N, et al. Good maternal and fetal outcomes for pregnant women with primary biliary cirrhosis. Clin Gastroenterol Hepatol. 2014;12:1179-1185.
2. Marchioni Beery RM, Vaziri H, Forouhar F. Primary biliary cirrhosis and primary sclerosing cholangitis: a review featuring a women’s health perspective. J Clin Transl Hepatol. 2014;2:266-284.
3. Efe C, Kahramanoğlu-Aksoy E, Yilmaz B, et al. Pregnancy in women with primary biliary cirrhosis. Autoimmun Rev. 2014;13:931-935.
4. Floreani A, Infantolino C, Franceschet I, et al. Pregnancy and primary biliary cirrhosis: a case control study. Clin Rev Allergy Immunol. 2015;48:236-242.
5. Rabinovitz M, Appasamy R, Finkelstein S. Primary biliary cirrhosis diagnosed during pregnancy. Does it have a different outcome? Dig Dis Sci. 1995;40:571-574.
6. Carey EJ, Ali AH, Lindor KD. Primary biliary cirrhosis. Lancet. 2015;386:1565-1575.
7. The Johns Hopkins School of Medicine Department of Gynecology. Hurt KJ, Guile MW, Bienstock JL, et al, eds. The Johns Hopkins Manual of Gynecology and Obstetrics. 4th edition. Philadelphia, PA: Lippincott Williams and Wilkins; 2011.
1. Trivedi PJ, Kumagi T, Al-Harthy N, et al. Good maternal and fetal outcomes for pregnant women with primary biliary cirrhosis. Clin Gastroenterol Hepatol. 2014;12:1179-1185.
2. Marchioni Beery RM, Vaziri H, Forouhar F. Primary biliary cirrhosis and primary sclerosing cholangitis: a review featuring a women’s health perspective. J Clin Transl Hepatol. 2014;2:266-284.
3. Efe C, Kahramanoğlu-Aksoy E, Yilmaz B, et al. Pregnancy in women with primary biliary cirrhosis. Autoimmun Rev. 2014;13:931-935.
4. Floreani A, Infantolino C, Franceschet I, et al. Pregnancy and primary biliary cirrhosis: a case control study. Clin Rev Allergy Immunol. 2015;48:236-242.
5. Rabinovitz M, Appasamy R, Finkelstein S. Primary biliary cirrhosis diagnosed during pregnancy. Does it have a different outcome? Dig Dis Sci. 1995;40:571-574.
6. Carey EJ, Ali AH, Lindor KD. Primary biliary cirrhosis. Lancet. 2015;386:1565-1575.
7. The Johns Hopkins School of Medicine Department of Gynecology. Hurt KJ, Guile MW, Bienstock JL, et al, eds. The Johns Hopkins Manual of Gynecology and Obstetrics. 4th edition. Philadelphia, PA: Lippincott Williams and Wilkins; 2011.
Primary Cutaneous Follicle Center Lymphoma Mimicking Folliculitis
The 2008 World Health Organization and European Organization for Treatment of Cancer joint classification has distinguished 3 categories of primary cutaneous B-cell lymphomas: primary cutaneous follicle center lymphoma (PCFCL), primary cutaneous diffuse large B-cell lymphoma, and primary cutaneous marginal zone lymphoma.1-3 Primary cutaneous follicle center lymphoma is the most common type of cutaneous B-cell lymphoma, accounting for approximately 60% of cases worldwide.4 The median age at diagnosis is 60 years, and most lesions are located on the scalp, forehead, neck, and trunk.5 Histologically, PCFCL is characterized by dermal proliferation of centrocytes and centroblasts derived from germinal center B cells that are arranged in either a follicular, diffuse, or mixed growth pattern.1 The cutaneous manifestations of PCFCL include solitary erythematous or violaceous plaques, nodules, or tumors of varying sizes.4 Grouped lesions also may be observed, but multifocal disease is rare.1 We report a rare presentation of PCFCL mimicking folliculitis with multiple multifocal papules on the back.
Case Report
A 54-year-old woman presented with fever and leukocytosis of 4 days’ duration and was admitted to the hospital for presumed sepsis. She had a history of mastectomy for treatment of ductal carcinoma in situ of the right breast 5 years prior to the current presentation and endocrine therapy with tamoxifen. Her symptoms were thought to be a complication from a surgery for implantation of a tissue expander in the right breast 5 years prior to presentation.
During her hospital admission, she developed a papular and cystic eruption on the back that was clinically suggestive of folliculitis, transient acantholytic dermatosis (Grover disease), or miliaria rubra (Figure 1). This papular and cystic eruption initially was managed conservatively with observation as she recovered from an occult infection. Due to the persistent nature of the eruption on the back, an excisional biopsy of the cystic component was performed 2 months after her discharge from the hospital. Histologic studies showed a dense infiltrate of lymphocytes, which expanded into the deep dermis in a nodular and diffuse growth pattern that was accentuated in the periadnexal areas. The B lymphocytes were small and hyperchromatic with few scattered centroblasts (Figure 2). Further immunohistochemical studies demonstrated that the neoplastic cells were positive for CD20, CD79a, BCL-2, and BCL-6; CD3, CD5, and cyclin D1 were negative. Staining for antigen Ki-67 revealed a proliferation index of 15% to 20% among the neoplastic cells (Figure 3). These findings were consistent with either PCFCL or secondary cutaneous follicle center lymphoma.
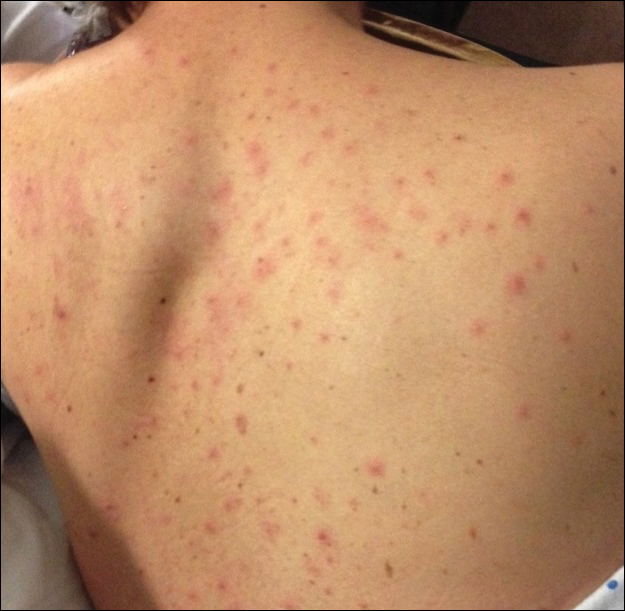
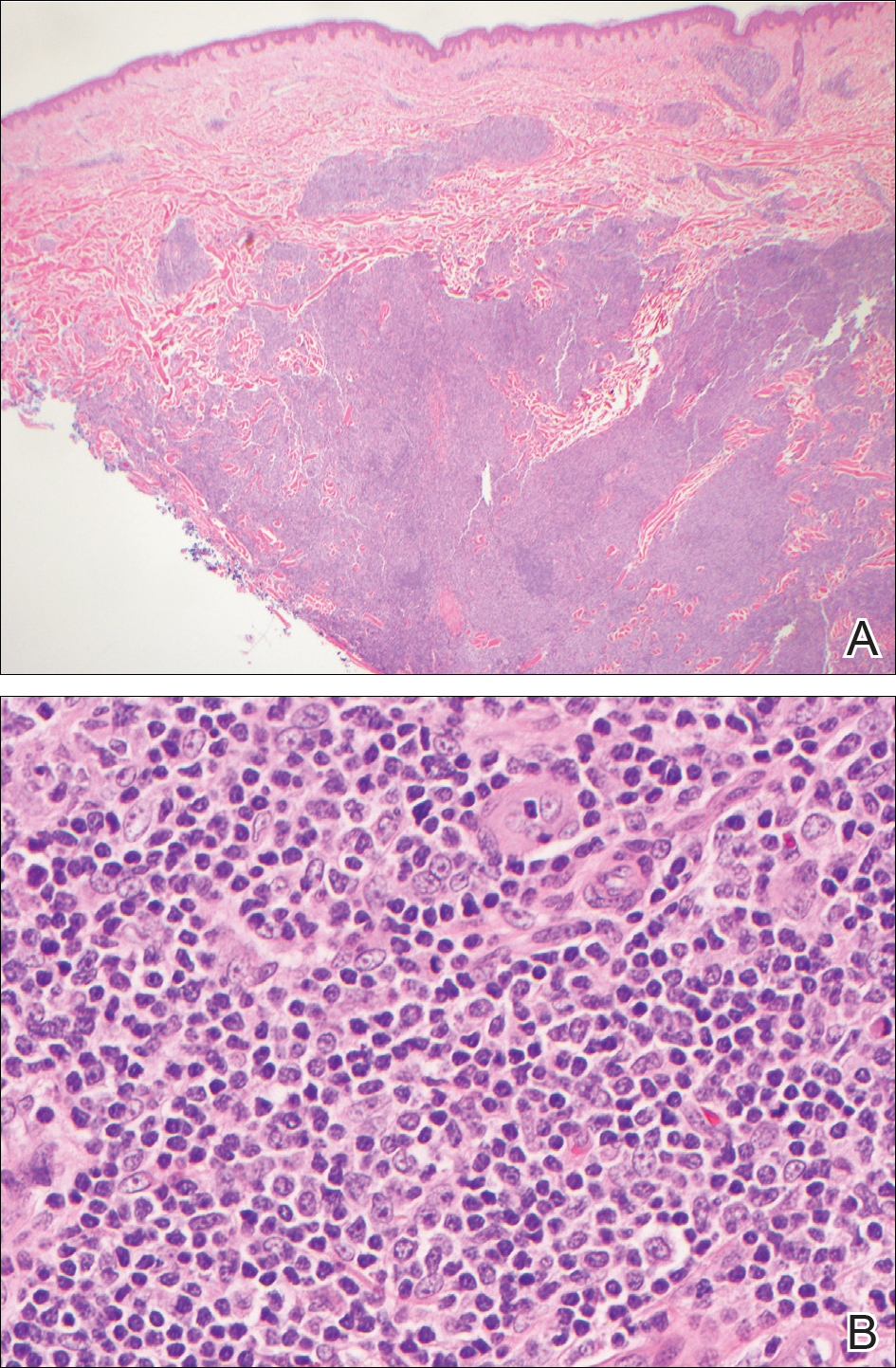
Further evaluation for systemic disease was unremarkable. Positron emission tomography–computed tomography revealed no evidence of nodal lymphoma, and a bone marrow biopsy was negative. Other laboratory studies including lactate dehydrogenase were within reference range, which conferred a diagnosis of PCFCL. The patient was treated with localized electron beam radiation therapy to the skin of the mid back for a total dose of 24 Gy in 12 fractions at 2 Gy per fraction once daily over a 12-day period. She tolerated the treatment well and has remained clinically and radiographically without evidence of disease for more than 3 years.
Comment
Because the incidence of cutaneous B-cell lymphomas has been increasing, especially among males, non-Hispanic whites, and adults older than 50 years,1 it is important for clinicians to have a high index of suspicion for this entity. In our patient, the clinical findings of a papular, largely asymptomatic eruption on the back with acute onset were initially thought to be consistent with folliculitis; the differential diagnosis included transient acantholytic dermatosis and miliaria rubra. Lymphoma was not in the initial clinical differential, and we only arrived at this diagnosis based on histopathologic evaluation.
The neoplastic cells typically are positive for CD20, CD79a, and BCL-6, and negative for BCL-2.4 Most cases of PCFCL do not express the t(14;18) translocation involving the BCL-2 locus, in contrast to systemic follicular lymphoma.1 Systemic imaging and evaluation is needed to definitively differentiate PCFCL from systemic lymphoma with cutaneous involvement. Our patient was unusual in that BCL-2 was strongly staining in the setting of a negative systemic workup.
With regard to treatment of PCFCL, electron beam radiation therapy is highly effective and safe in patients with solitary lesions, as the remission rate is close to 100%.1 For patients with multiple lesions confined to one area, electron beam radiation therapy also can be helpful, as in our patient. In patients with more extensive skin involvement, rituximab therapy may be preferable. Relapse following treatment with either radiation or rituximab occurs in approximately one-third of patients, but these relapses generally are limited to the skin.1 The International Extranodal Lymphoma Study Group has noted that elevated lactate dehydrogenase, presence of more than 2 skin lesions, and presence of nodular lesions are negative prognostic factors in patients with PCFCL6; however, PCFCL has an excellent prognosis overall with a 5-year survival rate of 95%.1
Other rare heterogeneous presentations of PCFCL have been reported in the literature. A large multinodular mass on the scalp with multifocal facial lesions has been described in a patient with essential thrombocytopenia.7 Another report identified a variant of PCFCL characterized by multiple erythematous firm papules that were distributed in a miliary pattern, predominantly on the forehead and cheeks.8 Barzilai et al9 described 4 patients with PCFCL who developed lesions that were clinically similar to rosacea or rhinophyma, including papulonodular eruptions on the cheeks; infiltrated erythematous nasal plaques; and small flesh-colored to erythematous papules on the cheeks, nose, helices, and upper back. Hodak et al10 identified 2 cases of PCFCL that manifested as anetoderma, a condition characterized by the focal loss of elastic tissue. In the setting of chronic lymphocytic leukemia, PCFCL has been observed as a red or violaceous nodule with a centrally depressed scar on the legs.11 In one case, PCFCL manifested as recurrent episodes of extraorbital swelling and a multifocal red-blue macular lesion that extended from the inferior orbital rim to the nasojugal fold.12 An interesting presentation of PCFCL was noted as a small, recurring, blood-filled blister on the cheek with perineural spread of the tumor along cranial nerves V2, V3, VII, and VIII.13 In the pediatric literature, PCFCL has been reported to present as an erythematous nodule with a smooth surface and a hard elastic consistency that appeared on the nose and nasolabial fold and spread to the ipsilateral cheek, maxillary sinus, and soft palate.14 In many of these unusual cases, the diagnosis of PCFCL was made after treatment with topical or systemic anti-inflammatory therapies failed.
Increased recognition of anomalous presentations of PCFCL among dermatologists can lead to more timely diagnoses and treatment. Based on our experience with this patient, we recommend considering biopsy for histopathologic evaluation when treating patients with presumed folliculitis or transient acantholytic dermatosis that does not improve with routine treatment or is accompanied by systemic symptoms.
- Wilcox RA. Cutaneous B-cell lymphomas: 2015 update on diagnosis, risk-stratification, and management. Am J Hematol. 2015;90:73-76.
- Kim YH, Willemze R, Pimpinelli N, et al. TNM classification system for primary cutaneous lymphomas other than mycosis fungoides and Sézary syndrome: a proposal of the International Society for Cutaneous Lymphomas (ISCL) and the Cutaneous Lymphoma Task Force of the European Organization of Research and Treatment of Cancer (EORTC). Blood. 2007;110:479-484.
- World Health Organization. WHO Classification of Tumors of Haematopoietic and Lymphoid Tissues. Lyon, France: World Health Organization; 2008: 227.
- Dilly M, Ben-Rejeb H, Vergier B, et al. Primary cutaneous follicle center lymphoma with Hodgkin and Reed-Sternberg-like cells: a new histopathologic variant. J Cutan Pathol. 2014;41:797-801.
- Suárez AL, Pulitzer M, Horwitz S, et al. Primary cutaneous B-cell lymphomas: part I. clinical features, diagnosis, and classification. J Am Acad Dermatol. 2013;69:329.e1-13; quiz 341-342.
- Mian M, Marcheselli L, Luminari S, et al. CLIPI: a new prognostic index for indolent cutaneous B cell lymphoma proposed by the International Extranodal Lymphoma Study Group (IELSG 11) [published online September 25, 2010]. Ann Hematol. 2011;90:401-408.
- Tirefort Y, Pham XC, Ibrahim YL, et al. A rare case of primary cutaneous follicle centre lymphoma presenting as a giant tumour of the scalp and combined with JAK2V617F positive essential thrombocythaemia. Biomark Res. 2014;2:7.
- Massone C, Fink-Puches R, Laimer M, et al. Miliary and agminated-type primary cutaneous follicle center lymphoma: report of 18 cases.J Am Acad Dermatol. 2011;65:749-755.
- Barzilai A, Feuerman H, Quaglino P, et al. Cutaneous B-cell neoplasms mimicking granulomatous rosacea or rhinophyma. Arch Dermatol. 2012;148:824-831.
- Hodak E, Feuerman H, Barzilai A, et al. Anetodermic primary cutaneous B-cell lymphoma: a unique clinicopathological presentation of lymphoma possibly associated with antiphospholipid antibodies. Arch Dermatol. 2010;146:175-182.
- Konda S, Beckford A, Demierre MF, et al. Primary cutaneous follicle center lymphoma in the setting of chronic lymphocytic leukemia. Indian J Dermatol Venereol Leprol. 2011;77:314-317.
- Pandya VB, Conway RM, Taylor SF. Primary cutaneous B cell lymphoma presenting as recurrent eyelid swelling. Clin Exp Ophthalmol. 2008;36:672-674.
- Buda-Okreglak EM, Walden MJ, Brissette MD. Perineural CNS invasion in primary cutaneous follicular center lymphoma. J Clin Oncol. 2007;25:4684-4686.
- Ghislanzoni M, Gambini D, Perrone T, et al. Primary cutaneous follicular center cell lymphoma of the nose with maxillary sinus involvement in a pediatric patient. J Am Acad Dermatol. 2005;52(5 suppl 1):S73-S75.
The 2008 World Health Organization and European Organization for Treatment of Cancer joint classification has distinguished 3 categories of primary cutaneous B-cell lymphomas: primary cutaneous follicle center lymphoma (PCFCL), primary cutaneous diffuse large B-cell lymphoma, and primary cutaneous marginal zone lymphoma.1-3 Primary cutaneous follicle center lymphoma is the most common type of cutaneous B-cell lymphoma, accounting for approximately 60% of cases worldwide.4 The median age at diagnosis is 60 years, and most lesions are located on the scalp, forehead, neck, and trunk.5 Histologically, PCFCL is characterized by dermal proliferation of centrocytes and centroblasts derived from germinal center B cells that are arranged in either a follicular, diffuse, or mixed growth pattern.1 The cutaneous manifestations of PCFCL include solitary erythematous or violaceous plaques, nodules, or tumors of varying sizes.4 Grouped lesions also may be observed, but multifocal disease is rare.1 We report a rare presentation of PCFCL mimicking folliculitis with multiple multifocal papules on the back.
Case Report
A 54-year-old woman presented with fever and leukocytosis of 4 days’ duration and was admitted to the hospital for presumed sepsis. She had a history of mastectomy for treatment of ductal carcinoma in situ of the right breast 5 years prior to the current presentation and endocrine therapy with tamoxifen. Her symptoms were thought to be a complication from a surgery for implantation of a tissue expander in the right breast 5 years prior to presentation.
During her hospital admission, she developed a papular and cystic eruption on the back that was clinically suggestive of folliculitis, transient acantholytic dermatosis (Grover disease), or miliaria rubra (Figure 1). This papular and cystic eruption initially was managed conservatively with observation as she recovered from an occult infection. Due to the persistent nature of the eruption on the back, an excisional biopsy of the cystic component was performed 2 months after her discharge from the hospital. Histologic studies showed a dense infiltrate of lymphocytes, which expanded into the deep dermis in a nodular and diffuse growth pattern that was accentuated in the periadnexal areas. The B lymphocytes were small and hyperchromatic with few scattered centroblasts (Figure 2). Further immunohistochemical studies demonstrated that the neoplastic cells were positive for CD20, CD79a, BCL-2, and BCL-6; CD3, CD5, and cyclin D1 were negative. Staining for antigen Ki-67 revealed a proliferation index of 15% to 20% among the neoplastic cells (Figure 3). These findings were consistent with either PCFCL or secondary cutaneous follicle center lymphoma.
Further evaluation for systemic disease was unremarkable. Positron emission tomography–computed tomography revealed no evidence of nodal lymphoma, and a bone marrow biopsy was negative. Other laboratory studies including lactate dehydrogenase were within reference range, which conferred a diagnosis of PCFCL. The patient was treated with localized electron beam radiation therapy to the skin of the mid back for a total dose of 24 Gy in 12 fractions at 2 Gy per fraction once daily over a 12-day period. She tolerated the treatment well and has remained clinically and radiographically without evidence of disease for more than 3 years.
Comment
Because the incidence of cutaneous B-cell lymphomas has been increasing, especially among males, non-Hispanic whites, and adults older than 50 years,1 it is important for clinicians to have a high index of suspicion for this entity. In our patient, the clinical findings of a papular, largely asymptomatic eruption on the back with acute onset were initially thought to be consistent with folliculitis; the differential diagnosis included transient acantholytic dermatosis and miliaria rubra. Lymphoma was not in the initial clinical differential, and we only arrived at this diagnosis based on histopathologic evaluation.
The neoplastic cells typically are positive for CD20, CD79a, and BCL-6, and negative for BCL-2.4 Most cases of PCFCL do not express the t(14;18) translocation involving the BCL-2 locus, in contrast to systemic follicular lymphoma.1 Systemic imaging and evaluation is needed to definitively differentiate PCFCL from systemic lymphoma with cutaneous involvement. Our patient was unusual in that BCL-2 was strongly staining in the setting of a negative systemic workup.
With regard to treatment of PCFCL, electron beam radiation therapy is highly effective and safe in patients with solitary lesions, as the remission rate is close to 100%.1 For patients with multiple lesions confined to one area, electron beam radiation therapy also can be helpful, as in our patient. In patients with more extensive skin involvement, rituximab therapy may be preferable. Relapse following treatment with either radiation or rituximab occurs in approximately one-third of patients, but these relapses generally are limited to the skin.1 The International Extranodal Lymphoma Study Group has noted that elevated lactate dehydrogenase, presence of more than 2 skin lesions, and presence of nodular lesions are negative prognostic factors in patients with PCFCL6; however, PCFCL has an excellent prognosis overall with a 5-year survival rate of 95%.1
Other rare heterogeneous presentations of PCFCL have been reported in the literature. A large multinodular mass on the scalp with multifocal facial lesions has been described in a patient with essential thrombocytopenia.7 Another report identified a variant of PCFCL characterized by multiple erythematous firm papules that were distributed in a miliary pattern, predominantly on the forehead and cheeks.8 Barzilai et al9 described 4 patients with PCFCL who developed lesions that were clinically similar to rosacea or rhinophyma, including papulonodular eruptions on the cheeks; infiltrated erythematous nasal plaques; and small flesh-colored to erythematous papules on the cheeks, nose, helices, and upper back. Hodak et al10 identified 2 cases of PCFCL that manifested as anetoderma, a condition characterized by the focal loss of elastic tissue. In the setting of chronic lymphocytic leukemia, PCFCL has been observed as a red or violaceous nodule with a centrally depressed scar on the legs.11 In one case, PCFCL manifested as recurrent episodes of extraorbital swelling and a multifocal red-blue macular lesion that extended from the inferior orbital rim to the nasojugal fold.12 An interesting presentation of PCFCL was noted as a small, recurring, blood-filled blister on the cheek with perineural spread of the tumor along cranial nerves V2, V3, VII, and VIII.13 In the pediatric literature, PCFCL has been reported to present as an erythematous nodule with a smooth surface and a hard elastic consistency that appeared on the nose and nasolabial fold and spread to the ipsilateral cheek, maxillary sinus, and soft palate.14 In many of these unusual cases, the diagnosis of PCFCL was made after treatment with topical or systemic anti-inflammatory therapies failed.
Increased recognition of anomalous presentations of PCFCL among dermatologists can lead to more timely diagnoses and treatment. Based on our experience with this patient, we recommend considering biopsy for histopathologic evaluation when treating patients with presumed folliculitis or transient acantholytic dermatosis that does not improve with routine treatment or is accompanied by systemic symptoms.
The 2008 World Health Organization and European Organization for Treatment of Cancer joint classification has distinguished 3 categories of primary cutaneous B-cell lymphomas: primary cutaneous follicle center lymphoma (PCFCL), primary cutaneous diffuse large B-cell lymphoma, and primary cutaneous marginal zone lymphoma.1-3 Primary cutaneous follicle center lymphoma is the most common type of cutaneous B-cell lymphoma, accounting for approximately 60% of cases worldwide.4 The median age at diagnosis is 60 years, and most lesions are located on the scalp, forehead, neck, and trunk.5 Histologically, PCFCL is characterized by dermal proliferation of centrocytes and centroblasts derived from germinal center B cells that are arranged in either a follicular, diffuse, or mixed growth pattern.1 The cutaneous manifestations of PCFCL include solitary erythematous or violaceous plaques, nodules, or tumors of varying sizes.4 Grouped lesions also may be observed, but multifocal disease is rare.1 We report a rare presentation of PCFCL mimicking folliculitis with multiple multifocal papules on the back.
Case Report
A 54-year-old woman presented with fever and leukocytosis of 4 days’ duration and was admitted to the hospital for presumed sepsis. She had a history of mastectomy for treatment of ductal carcinoma in situ of the right breast 5 years prior to the current presentation and endocrine therapy with tamoxifen. Her symptoms were thought to be a complication from a surgery for implantation of a tissue expander in the right breast 5 years prior to presentation.
During her hospital admission, she developed a papular and cystic eruption on the back that was clinically suggestive of folliculitis, transient acantholytic dermatosis (Grover disease), or miliaria rubra (Figure 1). This papular and cystic eruption initially was managed conservatively with observation as she recovered from an occult infection. Due to the persistent nature of the eruption on the back, an excisional biopsy of the cystic component was performed 2 months after her discharge from the hospital. Histologic studies showed a dense infiltrate of lymphocytes, which expanded into the deep dermis in a nodular and diffuse growth pattern that was accentuated in the periadnexal areas. The B lymphocytes were small and hyperchromatic with few scattered centroblasts (Figure 2). Further immunohistochemical studies demonstrated that the neoplastic cells were positive for CD20, CD79a, BCL-2, and BCL-6; CD3, CD5, and cyclin D1 were negative. Staining for antigen Ki-67 revealed a proliferation index of 15% to 20% among the neoplastic cells (Figure 3). These findings were consistent with either PCFCL or secondary cutaneous follicle center lymphoma.
Further evaluation for systemic disease was unremarkable. Positron emission tomography–computed tomography revealed no evidence of nodal lymphoma, and a bone marrow biopsy was negative. Other laboratory studies including lactate dehydrogenase were within reference range, which conferred a diagnosis of PCFCL. The patient was treated with localized electron beam radiation therapy to the skin of the mid back for a total dose of 24 Gy in 12 fractions at 2 Gy per fraction once daily over a 12-day period. She tolerated the treatment well and has remained clinically and radiographically without evidence of disease for more than 3 years.
Comment
Because the incidence of cutaneous B-cell lymphomas has been increasing, especially among males, non-Hispanic whites, and adults older than 50 years,1 it is important for clinicians to have a high index of suspicion for this entity. In our patient, the clinical findings of a papular, largely asymptomatic eruption on the back with acute onset were initially thought to be consistent with folliculitis; the differential diagnosis included transient acantholytic dermatosis and miliaria rubra. Lymphoma was not in the initial clinical differential, and we only arrived at this diagnosis based on histopathologic evaluation.
The neoplastic cells typically are positive for CD20, CD79a, and BCL-6, and negative for BCL-2.4 Most cases of PCFCL do not express the t(14;18) translocation involving the BCL-2 locus, in contrast to systemic follicular lymphoma.1 Systemic imaging and evaluation is needed to definitively differentiate PCFCL from systemic lymphoma with cutaneous involvement. Our patient was unusual in that BCL-2 was strongly staining in the setting of a negative systemic workup.
With regard to treatment of PCFCL, electron beam radiation therapy is highly effective and safe in patients with solitary lesions, as the remission rate is close to 100%.1 For patients with multiple lesions confined to one area, electron beam radiation therapy also can be helpful, as in our patient. In patients with more extensive skin involvement, rituximab therapy may be preferable. Relapse following treatment with either radiation or rituximab occurs in approximately one-third of patients, but these relapses generally are limited to the skin.1 The International Extranodal Lymphoma Study Group has noted that elevated lactate dehydrogenase, presence of more than 2 skin lesions, and presence of nodular lesions are negative prognostic factors in patients with PCFCL6; however, PCFCL has an excellent prognosis overall with a 5-year survival rate of 95%.1
Other rare heterogeneous presentations of PCFCL have been reported in the literature. A large multinodular mass on the scalp with multifocal facial lesions has been described in a patient with essential thrombocytopenia.7 Another report identified a variant of PCFCL characterized by multiple erythematous firm papules that were distributed in a miliary pattern, predominantly on the forehead and cheeks.8 Barzilai et al9 described 4 patients with PCFCL who developed lesions that were clinically similar to rosacea or rhinophyma, including papulonodular eruptions on the cheeks; infiltrated erythematous nasal plaques; and small flesh-colored to erythematous papules on the cheeks, nose, helices, and upper back. Hodak et al10 identified 2 cases of PCFCL that manifested as anetoderma, a condition characterized by the focal loss of elastic tissue. In the setting of chronic lymphocytic leukemia, PCFCL has been observed as a red or violaceous nodule with a centrally depressed scar on the legs.11 In one case, PCFCL manifested as recurrent episodes of extraorbital swelling and a multifocal red-blue macular lesion that extended from the inferior orbital rim to the nasojugal fold.12 An interesting presentation of PCFCL was noted as a small, recurring, blood-filled blister on the cheek with perineural spread of the tumor along cranial nerves V2, V3, VII, and VIII.13 In the pediatric literature, PCFCL has been reported to present as an erythematous nodule with a smooth surface and a hard elastic consistency that appeared on the nose and nasolabial fold and spread to the ipsilateral cheek, maxillary sinus, and soft palate.14 In many of these unusual cases, the diagnosis of PCFCL was made after treatment with topical or systemic anti-inflammatory therapies failed.
Increased recognition of anomalous presentations of PCFCL among dermatologists can lead to more timely diagnoses and treatment. Based on our experience with this patient, we recommend considering biopsy for histopathologic evaluation when treating patients with presumed folliculitis or transient acantholytic dermatosis that does not improve with routine treatment or is accompanied by systemic symptoms.
- Wilcox RA. Cutaneous B-cell lymphomas: 2015 update on diagnosis, risk-stratification, and management. Am J Hematol. 2015;90:73-76.
- Kim YH, Willemze R, Pimpinelli N, et al. TNM classification system for primary cutaneous lymphomas other than mycosis fungoides and Sézary syndrome: a proposal of the International Society for Cutaneous Lymphomas (ISCL) and the Cutaneous Lymphoma Task Force of the European Organization of Research and Treatment of Cancer (EORTC). Blood. 2007;110:479-484.
- World Health Organization. WHO Classification of Tumors of Haematopoietic and Lymphoid Tissues. Lyon, France: World Health Organization; 2008: 227.
- Dilly M, Ben-Rejeb H, Vergier B, et al. Primary cutaneous follicle center lymphoma with Hodgkin and Reed-Sternberg-like cells: a new histopathologic variant. J Cutan Pathol. 2014;41:797-801.
- Suárez AL, Pulitzer M, Horwitz S, et al. Primary cutaneous B-cell lymphomas: part I. clinical features, diagnosis, and classification. J Am Acad Dermatol. 2013;69:329.e1-13; quiz 341-342.
- Mian M, Marcheselli L, Luminari S, et al. CLIPI: a new prognostic index for indolent cutaneous B cell lymphoma proposed by the International Extranodal Lymphoma Study Group (IELSG 11) [published online September 25, 2010]. Ann Hematol. 2011;90:401-408.
- Tirefort Y, Pham XC, Ibrahim YL, et al. A rare case of primary cutaneous follicle centre lymphoma presenting as a giant tumour of the scalp and combined with JAK2V617F positive essential thrombocythaemia. Biomark Res. 2014;2:7.
- Massone C, Fink-Puches R, Laimer M, et al. Miliary and agminated-type primary cutaneous follicle center lymphoma: report of 18 cases.J Am Acad Dermatol. 2011;65:749-755.
- Barzilai A, Feuerman H, Quaglino P, et al. Cutaneous B-cell neoplasms mimicking granulomatous rosacea or rhinophyma. Arch Dermatol. 2012;148:824-831.
- Hodak E, Feuerman H, Barzilai A, et al. Anetodermic primary cutaneous B-cell lymphoma: a unique clinicopathological presentation of lymphoma possibly associated with antiphospholipid antibodies. Arch Dermatol. 2010;146:175-182.
- Konda S, Beckford A, Demierre MF, et al. Primary cutaneous follicle center lymphoma in the setting of chronic lymphocytic leukemia. Indian J Dermatol Venereol Leprol. 2011;77:314-317.
- Pandya VB, Conway RM, Taylor SF. Primary cutaneous B cell lymphoma presenting as recurrent eyelid swelling. Clin Exp Ophthalmol. 2008;36:672-674.
- Buda-Okreglak EM, Walden MJ, Brissette MD. Perineural CNS invasion in primary cutaneous follicular center lymphoma. J Clin Oncol. 2007;25:4684-4686.
- Ghislanzoni M, Gambini D, Perrone T, et al. Primary cutaneous follicular center cell lymphoma of the nose with maxillary sinus involvement in a pediatric patient. J Am Acad Dermatol. 2005;52(5 suppl 1):S73-S75.
- Wilcox RA. Cutaneous B-cell lymphomas: 2015 update on diagnosis, risk-stratification, and management. Am J Hematol. 2015;90:73-76.
- Kim YH, Willemze R, Pimpinelli N, et al. TNM classification system for primary cutaneous lymphomas other than mycosis fungoides and Sézary syndrome: a proposal of the International Society for Cutaneous Lymphomas (ISCL) and the Cutaneous Lymphoma Task Force of the European Organization of Research and Treatment of Cancer (EORTC). Blood. 2007;110:479-484.
- World Health Organization. WHO Classification of Tumors of Haematopoietic and Lymphoid Tissues. Lyon, France: World Health Organization; 2008: 227.
- Dilly M, Ben-Rejeb H, Vergier B, et al. Primary cutaneous follicle center lymphoma with Hodgkin and Reed-Sternberg-like cells: a new histopathologic variant. J Cutan Pathol. 2014;41:797-801.
- Suárez AL, Pulitzer M, Horwitz S, et al. Primary cutaneous B-cell lymphomas: part I. clinical features, diagnosis, and classification. J Am Acad Dermatol. 2013;69:329.e1-13; quiz 341-342.
- Mian M, Marcheselli L, Luminari S, et al. CLIPI: a new prognostic index for indolent cutaneous B cell lymphoma proposed by the International Extranodal Lymphoma Study Group (IELSG 11) [published online September 25, 2010]. Ann Hematol. 2011;90:401-408.
- Tirefort Y, Pham XC, Ibrahim YL, et al. A rare case of primary cutaneous follicle centre lymphoma presenting as a giant tumour of the scalp and combined with JAK2V617F positive essential thrombocythaemia. Biomark Res. 2014;2:7.
- Massone C, Fink-Puches R, Laimer M, et al. Miliary and agminated-type primary cutaneous follicle center lymphoma: report of 18 cases.J Am Acad Dermatol. 2011;65:749-755.
- Barzilai A, Feuerman H, Quaglino P, et al. Cutaneous B-cell neoplasms mimicking granulomatous rosacea or rhinophyma. Arch Dermatol. 2012;148:824-831.
- Hodak E, Feuerman H, Barzilai A, et al. Anetodermic primary cutaneous B-cell lymphoma: a unique clinicopathological presentation of lymphoma possibly associated with antiphospholipid antibodies. Arch Dermatol. 2010;146:175-182.
- Konda S, Beckford A, Demierre MF, et al. Primary cutaneous follicle center lymphoma in the setting of chronic lymphocytic leukemia. Indian J Dermatol Venereol Leprol. 2011;77:314-317.
- Pandya VB, Conway RM, Taylor SF. Primary cutaneous B cell lymphoma presenting as recurrent eyelid swelling. Clin Exp Ophthalmol. 2008;36:672-674.
- Buda-Okreglak EM, Walden MJ, Brissette MD. Perineural CNS invasion in primary cutaneous follicular center lymphoma. J Clin Oncol. 2007;25:4684-4686.
- Ghislanzoni M, Gambini D, Perrone T, et al. Primary cutaneous follicular center cell lymphoma of the nose with maxillary sinus involvement in a pediatric patient. J Am Acad Dermatol. 2005;52(5 suppl 1):S73-S75.
Practice Points
- Atypical or unresponsive folliculitis should be biopsied.
- Primary cutaneous follicle center lymphoma can mimic folliculitis or Grover disease.

Ethanol Intoxication From Hand Sanitizer Ingestion
Case
A 29-year-old man presented to the ED several hours after ingesting what he described as a “hefty” bottle of hand sanitizer. The patient stated that he ingested such a considerable quantity of liquid hand sanitizer because he was unable to obtain beer or liquor. He further admitted to drinking two 40-ounce beers daily for the past several years, noting that he last consumed drinking alcohol the preceding day.
The patient denied any other coingestants. He also denied nausea, vomiting, abdominal pain, or other somatic complaints. The patient’s medical history was significant for hypertension and hepatitis C, and his social history was significant for daily alcohol consumption, tobacco abuse, and former benzodiazepine, marijuana, and intravenous heroin abuse. His psychiatric history was significant for borderline personality disorder, major depression, and bulimia. The patient’s home medications included a daily multivitamin, folate, thiamine, sertraline, mirtazapine, and prazosin.
Initial vital signs at presentation were: blood pressure, 124/77 mm Hg; heart rate, 86 beats/min; respiratory rate, 15 breaths/min; and temperature, 98.0°F. On physical examination, he was noted to have slurred speech and nystagmus. His pupils were equal and reactive, without scleral icterus. The abdomen was nontender and nondistended, with regular bowel sounds, and without ascites or varicosities visualized. The rest of the examination was unremarkable. The patient did express thoughts of suicidality, but denied any homicidal ideation.
Laboratory studies revealed a serum ethanol concentration of 446 mg/dL. The patient’s basic metabolic panel was unremarkable, and liver function test results showed mildly elevated enzymes. The coagulation panel was within normal limits.
Is alcohol-based hand sanitizer consumption an emerging public health concern?
Excessive alcohol consumption is a recognized public health problem in the United States and is associated with an average of 88,000 deaths per year.1 In a select population of patients, an untoward effect has developed from another public health target—that of hand hygiene.
Alcohol-based liquid hand sanitizers have become ubiquitous as a weapon in the antimicrobial arsenal with recommendations for its use as an alternative to soap and water in certain clinical settings. Liquid hand sanitizers are ideal for hospital or community use as they are faster, more effective, and less irritating to the skin than traditional hand-washing techniques.2
The downside to the widespread availability of hand sanitizers is that they offer easy access to individuals in search of clandestine sources of alcohol. Prior case reports have discussed the practice of consuming alcohol-based hand sanitizers for the purpose of intoxication in institutionalized persons, such as prisoners or patients in psychiatric facilities who are restricted to conventional sources of alcohol.
Children and confused elderly patients are also at risk for unintentional ingestions.3,4 An article reviewed exposures reported to the American Association of Poison Control Center’s National Poison Data System over a 5-year period from 2005 to 2009.3 Of the 68,712 reported cases in this cohort, 80.5% were in children younger than 6 years of age. The investigators also noted an increased incidence of exposure over this period with an average of 1,894 additional cases per year.3There were 17,154 children aged 12 years and younger reported in 2017 to poison centers with exposures to hand sanitizers. Young children may be enticed by the bright colorful packaging and similarity to food and candy smells.5
What are the clinical manifestations of alcohol-based hand sanitizer ingestion?
Significant hazards exist from ingesting liquid hand sanitizer, including the high alcohol content, which varies from 40% to 85%.2 Because isopropanol is commonly one of the components (if not the sole component) of many hand-sanitizer preparations, isopropanol toxicity may occur when ingested. The effects of isopropanol are similar to those of ethanol, with clinical effects reported after ingestion of as little as 100 mL of 70% isopropanol solution.4
Hand sanitizer formulations vary by manufacturer and contain different concentrations of ethanol and/or isopropanol, as well as additional potential inactive ingredients such as acetone, 1-propanol, 2-propanol, benzyl alcohol, hydrogen peroxide, glycerin, water, and different perfumes.3,4
Persons who consume hand sanitizers recreationally are often unaware of the large alcohol content by volume that they are consuming. Recreational ingestion of hand sanitizer is believed to be the cause of at least one case of lethal ethanol intoxication. An articlereported a case of a male patient who suffered respiratory arrest after consuming an ethanol-based hand sanitizer.6 This patient was noted to have a serum ethanol of 536 mg/dL after consuming an unknown quantity of a 354 mL container of a 62% ethanol by volume hand sanitizer.6
Institutionalized individuals seeking alcohol through this source have discovered novel ways to yield a stronger product. Through the use of table salt and a cotton sock, it is possible to extract a liquid from a gel hand sanitizer preparation, yielding an alcohol context 30% higher by volume than the parent mixture.7
Alcohol intoxication poses a host of health effects. In nonhabituated individuals, a lethal load of alcohol can be achieved by consuming a volume of as little as 400 mL of an 80% alcohol-based solution.4 Symptoms from ingestion of an alcohol-based liquid hand sanitizer typically appear 1 to 2 hours after ingestion and mirror that of the alcohol toxidrome. Most commonly, this includes nausea, vomiting, epigastric pain, and varying degrees of central nervous system (CNS) depression.4 The life-threatening clinical manifestation of alcohol intoxication includes severe CNS and respiratory depression resulting in respiratory arrest, hypothermia, cardiac dysrhythmias with possible cardiac arrest, hypoglycemia, ketoacidosis, and hypotension.3
How is alcohol-based hand sanitizer ingestion managed?
The management of patients with alcohol-based hand sanitizer ingestion is the same as the management of alcohol ingestion from more socially acceptable sources and is mainly supportive.3,4 These measures are directed at managing the patient’s airway with intubation and mechanical ventilation when appropriate, as well as supportive measures to address any underlying metabolic derangement or hypotension.2 While hemodialysis has been used in some patients who had severe organ dysfunction and did not respond to supportive measures, it is usually not necessary.1,3
Case Conclusion
The patient in this case was subsequently admitted to an intermediate level of care. He did not require intubation or further hemodynamic support during his initial acute intoxication. Later in the patient’s hospital course, he was noted to be in alcohol withdrawal, and proper management was initiated. He also required therapeutic one-to-one supervision after members of the nursing staff observed the patient consuming the hand sanitizer gel present in patient-care areas. He was later seen by psychiatry services. The psychiatrist recommended transfer to an inpatient psychiatric facility upon medical clearance for treatment of his psychiatric illness as well as alcohol dependence.
1. Esser MB, Hedden SL, Kanny D, Brewer RD, Gfroerer JC, Naimi TS. Prevalence of alcohol dependence among US adult drinkers, 2009-2011. Prev Chronic Dis. 2014;11:E206. doi:10.5888/pcd11.140329.
2. Pittet D, Boyce JM. Revolutionizing hand hygiene in health-care settings: guidelines revisted. Lancet Infect Dis. 2003;3(5):269-270.
3. Gormley NJ, Bronstein AC, Rasimas JJ, et al. The rising incidence of intentional ingestion of ethanol-containing hand sanitizers. Crit Care Med. 2012:40(1):290-294. doi:10.1097/CCM.0b013e31822f09c0.
4. Archer JR, Wood DM, Tizzard Z, Jones AL, Dargan PI. Alcohol hand rubs: hygiene and hazard. BMJ. 2007;335(7630):1154-1155.
5. Hand sanitizer. American Association of Poison Control Centers Web site. http://www.aapcc.org/alerts/hand-sanitizer/. Accessed December 27, 2017.
6. Schneir AB, Clark RF. Death caused by ingestion of an ethanol-based hand sanitizer. J Emerg Med. 2013;45(3):358-360. doi:10.1016/j.jemermed.2013.03.018.
7. Darracq MA, Ghafouri N, Pesce A, Cantrell FL. Hand sanitizer intoxication following a crude extraction method. Am J Drug Alcohol Abuse. 2013;39(3):217-218. doi:10.3109/00952990.2013.773335.
Case
A 29-year-old man presented to the ED several hours after ingesting what he described as a “hefty” bottle of hand sanitizer. The patient stated that he ingested such a considerable quantity of liquid hand sanitizer because he was unable to obtain beer or liquor. He further admitted to drinking two 40-ounce beers daily for the past several years, noting that he last consumed drinking alcohol the preceding day.
The patient denied any other coingestants. He also denied nausea, vomiting, abdominal pain, or other somatic complaints. The patient’s medical history was significant for hypertension and hepatitis C, and his social history was significant for daily alcohol consumption, tobacco abuse, and former benzodiazepine, marijuana, and intravenous heroin abuse. His psychiatric history was significant for borderline personality disorder, major depression, and bulimia. The patient’s home medications included a daily multivitamin, folate, thiamine, sertraline, mirtazapine, and prazosin.
Initial vital signs at presentation were: blood pressure, 124/77 mm Hg; heart rate, 86 beats/min; respiratory rate, 15 breaths/min; and temperature, 98.0°F. On physical examination, he was noted to have slurred speech and nystagmus. His pupils were equal and reactive, without scleral icterus. The abdomen was nontender and nondistended, with regular bowel sounds, and without ascites or varicosities visualized. The rest of the examination was unremarkable. The patient did express thoughts of suicidality, but denied any homicidal ideation.
Laboratory studies revealed a serum ethanol concentration of 446 mg/dL. The patient’s basic metabolic panel was unremarkable, and liver function test results showed mildly elevated enzymes. The coagulation panel was within normal limits.
Is alcohol-based hand sanitizer consumption an emerging public health concern?
Excessive alcohol consumption is a recognized public health problem in the United States and is associated with an average of 88,000 deaths per year.1 In a select population of patients, an untoward effect has developed from another public health target—that of hand hygiene.
Alcohol-based liquid hand sanitizers have become ubiquitous as a weapon in the antimicrobial arsenal with recommendations for its use as an alternative to soap and water in certain clinical settings. Liquid hand sanitizers are ideal for hospital or community use as they are faster, more effective, and less irritating to the skin than traditional hand-washing techniques.2
The downside to the widespread availability of hand sanitizers is that they offer easy access to individuals in search of clandestine sources of alcohol. Prior case reports have discussed the practice of consuming alcohol-based hand sanitizers for the purpose of intoxication in institutionalized persons, such as prisoners or patients in psychiatric facilities who are restricted to conventional sources of alcohol.
Children and confused elderly patients are also at risk for unintentional ingestions.3,4 An article reviewed exposures reported to the American Association of Poison Control Center’s National Poison Data System over a 5-year period from 2005 to 2009.3 Of the 68,712 reported cases in this cohort, 80.5% were in children younger than 6 years of age. The investigators also noted an increased incidence of exposure over this period with an average of 1,894 additional cases per year.3There were 17,154 children aged 12 years and younger reported in 2017 to poison centers with exposures to hand sanitizers. Young children may be enticed by the bright colorful packaging and similarity to food and candy smells.5
What are the clinical manifestations of alcohol-based hand sanitizer ingestion?
Significant hazards exist from ingesting liquid hand sanitizer, including the high alcohol content, which varies from 40% to 85%.2 Because isopropanol is commonly one of the components (if not the sole component) of many hand-sanitizer preparations, isopropanol toxicity may occur when ingested. The effects of isopropanol are similar to those of ethanol, with clinical effects reported after ingestion of as little as 100 mL of 70% isopropanol solution.4
Hand sanitizer formulations vary by manufacturer and contain different concentrations of ethanol and/or isopropanol, as well as additional potential inactive ingredients such as acetone, 1-propanol, 2-propanol, benzyl alcohol, hydrogen peroxide, glycerin, water, and different perfumes.3,4
Persons who consume hand sanitizers recreationally are often unaware of the large alcohol content by volume that they are consuming. Recreational ingestion of hand sanitizer is believed to be the cause of at least one case of lethal ethanol intoxication. An articlereported a case of a male patient who suffered respiratory arrest after consuming an ethanol-based hand sanitizer.6 This patient was noted to have a serum ethanol of 536 mg/dL after consuming an unknown quantity of a 354 mL container of a 62% ethanol by volume hand sanitizer.6
Institutionalized individuals seeking alcohol through this source have discovered novel ways to yield a stronger product. Through the use of table salt and a cotton sock, it is possible to extract a liquid from a gel hand sanitizer preparation, yielding an alcohol context 30% higher by volume than the parent mixture.7
Alcohol intoxication poses a host of health effects. In nonhabituated individuals, a lethal load of alcohol can be achieved by consuming a volume of as little as 400 mL of an 80% alcohol-based solution.4 Symptoms from ingestion of an alcohol-based liquid hand sanitizer typically appear 1 to 2 hours after ingestion and mirror that of the alcohol toxidrome. Most commonly, this includes nausea, vomiting, epigastric pain, and varying degrees of central nervous system (CNS) depression.4 The life-threatening clinical manifestation of alcohol intoxication includes severe CNS and respiratory depression resulting in respiratory arrest, hypothermia, cardiac dysrhythmias with possible cardiac arrest, hypoglycemia, ketoacidosis, and hypotension.3
How is alcohol-based hand sanitizer ingestion managed?
The management of patients with alcohol-based hand sanitizer ingestion is the same as the management of alcohol ingestion from more socially acceptable sources and is mainly supportive.3,4 These measures are directed at managing the patient’s airway with intubation and mechanical ventilation when appropriate, as well as supportive measures to address any underlying metabolic derangement or hypotension.2 While hemodialysis has been used in some patients who had severe organ dysfunction and did not respond to supportive measures, it is usually not necessary.1,3
Case Conclusion
The patient in this case was subsequently admitted to an intermediate level of care. He did not require intubation or further hemodynamic support during his initial acute intoxication. Later in the patient’s hospital course, he was noted to be in alcohol withdrawal, and proper management was initiated. He also required therapeutic one-to-one supervision after members of the nursing staff observed the patient consuming the hand sanitizer gel present in patient-care areas. He was later seen by psychiatry services. The psychiatrist recommended transfer to an inpatient psychiatric facility upon medical clearance for treatment of his psychiatric illness as well as alcohol dependence.
Case
A 29-year-old man presented to the ED several hours after ingesting what he described as a “hefty” bottle of hand sanitizer. The patient stated that he ingested such a considerable quantity of liquid hand sanitizer because he was unable to obtain beer or liquor. He further admitted to drinking two 40-ounce beers daily for the past several years, noting that he last consumed drinking alcohol the preceding day.
The patient denied any other coingestants. He also denied nausea, vomiting, abdominal pain, or other somatic complaints. The patient’s medical history was significant for hypertension and hepatitis C, and his social history was significant for daily alcohol consumption, tobacco abuse, and former benzodiazepine, marijuana, and intravenous heroin abuse. His psychiatric history was significant for borderline personality disorder, major depression, and bulimia. The patient’s home medications included a daily multivitamin, folate, thiamine, sertraline, mirtazapine, and prazosin.
Initial vital signs at presentation were: blood pressure, 124/77 mm Hg; heart rate, 86 beats/min; respiratory rate, 15 breaths/min; and temperature, 98.0°F. On physical examination, he was noted to have slurred speech and nystagmus. His pupils were equal and reactive, without scleral icterus. The abdomen was nontender and nondistended, with regular bowel sounds, and without ascites or varicosities visualized. The rest of the examination was unremarkable. The patient did express thoughts of suicidality, but denied any homicidal ideation.
Laboratory studies revealed a serum ethanol concentration of 446 mg/dL. The patient’s basic metabolic panel was unremarkable, and liver function test results showed mildly elevated enzymes. The coagulation panel was within normal limits.
Is alcohol-based hand sanitizer consumption an emerging public health concern?
Excessive alcohol consumption is a recognized public health problem in the United States and is associated with an average of 88,000 deaths per year.1 In a select population of patients, an untoward effect has developed from another public health target—that of hand hygiene.
Alcohol-based liquid hand sanitizers have become ubiquitous as a weapon in the antimicrobial arsenal with recommendations for its use as an alternative to soap and water in certain clinical settings. Liquid hand sanitizers are ideal for hospital or community use as they are faster, more effective, and less irritating to the skin than traditional hand-washing techniques.2
The downside to the widespread availability of hand sanitizers is that they offer easy access to individuals in search of clandestine sources of alcohol. Prior case reports have discussed the practice of consuming alcohol-based hand sanitizers for the purpose of intoxication in institutionalized persons, such as prisoners or patients in psychiatric facilities who are restricted to conventional sources of alcohol.
Children and confused elderly patients are also at risk for unintentional ingestions.3,4 An article reviewed exposures reported to the American Association of Poison Control Center’s National Poison Data System over a 5-year period from 2005 to 2009.3 Of the 68,712 reported cases in this cohort, 80.5% were in children younger than 6 years of age. The investigators also noted an increased incidence of exposure over this period with an average of 1,894 additional cases per year.3There were 17,154 children aged 12 years and younger reported in 2017 to poison centers with exposures to hand sanitizers. Young children may be enticed by the bright colorful packaging and similarity to food and candy smells.5
What are the clinical manifestations of alcohol-based hand sanitizer ingestion?
Significant hazards exist from ingesting liquid hand sanitizer, including the high alcohol content, which varies from 40% to 85%.2 Because isopropanol is commonly one of the components (if not the sole component) of many hand-sanitizer preparations, isopropanol toxicity may occur when ingested. The effects of isopropanol are similar to those of ethanol, with clinical effects reported after ingestion of as little as 100 mL of 70% isopropanol solution.4
Hand sanitizer formulations vary by manufacturer and contain different concentrations of ethanol and/or isopropanol, as well as additional potential inactive ingredients such as acetone, 1-propanol, 2-propanol, benzyl alcohol, hydrogen peroxide, glycerin, water, and different perfumes.3,4
Persons who consume hand sanitizers recreationally are often unaware of the large alcohol content by volume that they are consuming. Recreational ingestion of hand sanitizer is believed to be the cause of at least one case of lethal ethanol intoxication. An articlereported a case of a male patient who suffered respiratory arrest after consuming an ethanol-based hand sanitizer.6 This patient was noted to have a serum ethanol of 536 mg/dL after consuming an unknown quantity of a 354 mL container of a 62% ethanol by volume hand sanitizer.6
Institutionalized individuals seeking alcohol through this source have discovered novel ways to yield a stronger product. Through the use of table salt and a cotton sock, it is possible to extract a liquid from a gel hand sanitizer preparation, yielding an alcohol context 30% higher by volume than the parent mixture.7
Alcohol intoxication poses a host of health effects. In nonhabituated individuals, a lethal load of alcohol can be achieved by consuming a volume of as little as 400 mL of an 80% alcohol-based solution.4 Symptoms from ingestion of an alcohol-based liquid hand sanitizer typically appear 1 to 2 hours after ingestion and mirror that of the alcohol toxidrome. Most commonly, this includes nausea, vomiting, epigastric pain, and varying degrees of central nervous system (CNS) depression.4 The life-threatening clinical manifestation of alcohol intoxication includes severe CNS and respiratory depression resulting in respiratory arrest, hypothermia, cardiac dysrhythmias with possible cardiac arrest, hypoglycemia, ketoacidosis, and hypotension.3
How is alcohol-based hand sanitizer ingestion managed?
The management of patients with alcohol-based hand sanitizer ingestion is the same as the management of alcohol ingestion from more socially acceptable sources and is mainly supportive.3,4 These measures are directed at managing the patient’s airway with intubation and mechanical ventilation when appropriate, as well as supportive measures to address any underlying metabolic derangement or hypotension.2 While hemodialysis has been used in some patients who had severe organ dysfunction and did not respond to supportive measures, it is usually not necessary.1,3
Case Conclusion
The patient in this case was subsequently admitted to an intermediate level of care. He did not require intubation or further hemodynamic support during his initial acute intoxication. Later in the patient’s hospital course, he was noted to be in alcohol withdrawal, and proper management was initiated. He also required therapeutic one-to-one supervision after members of the nursing staff observed the patient consuming the hand sanitizer gel present in patient-care areas. He was later seen by psychiatry services. The psychiatrist recommended transfer to an inpatient psychiatric facility upon medical clearance for treatment of his psychiatric illness as well as alcohol dependence.
1. Esser MB, Hedden SL, Kanny D, Brewer RD, Gfroerer JC, Naimi TS. Prevalence of alcohol dependence among US adult drinkers, 2009-2011. Prev Chronic Dis. 2014;11:E206. doi:10.5888/pcd11.140329.
2. Pittet D, Boyce JM. Revolutionizing hand hygiene in health-care settings: guidelines revisted. Lancet Infect Dis. 2003;3(5):269-270.
3. Gormley NJ, Bronstein AC, Rasimas JJ, et al. The rising incidence of intentional ingestion of ethanol-containing hand sanitizers. Crit Care Med. 2012:40(1):290-294. doi:10.1097/CCM.0b013e31822f09c0.
4. Archer JR, Wood DM, Tizzard Z, Jones AL, Dargan PI. Alcohol hand rubs: hygiene and hazard. BMJ. 2007;335(7630):1154-1155.
5. Hand sanitizer. American Association of Poison Control Centers Web site. http://www.aapcc.org/alerts/hand-sanitizer/. Accessed December 27, 2017.
6. Schneir AB, Clark RF. Death caused by ingestion of an ethanol-based hand sanitizer. J Emerg Med. 2013;45(3):358-360. doi:10.1016/j.jemermed.2013.03.018.
7. Darracq MA, Ghafouri N, Pesce A, Cantrell FL. Hand sanitizer intoxication following a crude extraction method. Am J Drug Alcohol Abuse. 2013;39(3):217-218. doi:10.3109/00952990.2013.773335.
1. Esser MB, Hedden SL, Kanny D, Brewer RD, Gfroerer JC, Naimi TS. Prevalence of alcohol dependence among US adult drinkers, 2009-2011. Prev Chronic Dis. 2014;11:E206. doi:10.5888/pcd11.140329.
2. Pittet D, Boyce JM. Revolutionizing hand hygiene in health-care settings: guidelines revisted. Lancet Infect Dis. 2003;3(5):269-270.
3. Gormley NJ, Bronstein AC, Rasimas JJ, et al. The rising incidence of intentional ingestion of ethanol-containing hand sanitizers. Crit Care Med. 2012:40(1):290-294. doi:10.1097/CCM.0b013e31822f09c0.
4. Archer JR, Wood DM, Tizzard Z, Jones AL, Dargan PI. Alcohol hand rubs: hygiene and hazard. BMJ. 2007;335(7630):1154-1155.
5. Hand sanitizer. American Association of Poison Control Centers Web site. http://www.aapcc.org/alerts/hand-sanitizer/. Accessed December 27, 2017.
6. Schneir AB, Clark RF. Death caused by ingestion of an ethanol-based hand sanitizer. J Emerg Med. 2013;45(3):358-360. doi:10.1016/j.jemermed.2013.03.018.
7. Darracq MA, Ghafouri N, Pesce A, Cantrell FL. Hand sanitizer intoxication following a crude extraction method. Am J Drug Alcohol Abuse. 2013;39(3):217-218. doi:10.3109/00952990.2013.773335.

Pulmonary Mucormycosis in a Patient With Uncontrolled Diabetes
Mucorales fungi are ubiquitous organisms commonly inhabiting soil and can cause opportunistic infections. The majority of infections are caused by 3 genera: Rhizopus, Mucor, and Rhizomucor.1 Infection occurs by inhalation or by direct contact with damaged skin. Mucorales infections can have cutaneous, rhinocerebral, pulmonary, gastrointestinal, and central nervous system manifestations. Pulmonary mucormycosis is often rapidly progressive with angioinvasion and fulminant necrosis causing acute dyspnea, hemoptysis, and chest pain. More indolent pulmonary Mucorales infections can mimic a pulmonary mass with occasional cavitation found on imaging studies similar to other fungal infections (eg, Aspergillus).2 Risk factors include severe uncontrolled diabetes mellitus (DM), recurrent diabetic ketoacidosis (DKA), immunosuppression due to congenital or acquired causes, hematologic malignancies, and chronic renal failure.3 The authors present a case of a patient with recurrent DKA and pulmonary mucormycosis.
Case Presentation
A 62-year-old male with DM and a more than 30-pack-year smoking history presented to the emergency department with abdominal pain and chest pain ongoing for about 1 week. The patient had a history of frequent admissions with DKA and medication nonadherence.
On admission, the patient was hemodynamically stable. His vital signs were: temperature 97.4° F, heart rate 89 bpm, respirations 24 breathes per minute, blood pressure 146/86 mm Hg, and oxygen saturation 94% on ambient air. The patient appeared ill but the physical examination was otherwise unremarkable. Laboratory results revealed a white blood cell count of 24,400 with neutrophilic predominance, blood glucose 658 mg/dL, creatinine clearance 2.16 mL/min/1.73 m2, sodium level 124 mEq/L, bicarbonate 6 mEq/L, anion gap 27 mEq/L, 6.8 pH, partial pressure of CO2 11 mm Hg, and lactic acid 2.3 mmol/L.
The patient admitted for DKA management and placed on an insulin drip. Although he did not have a fever or cough productive of sputum or hemoptysis, there was concern that pneumonia might have precipitated DKA. A chest X-ray revealed a patchy, right suprahilar opacity (Figure 1). 
The patient was placed on vancomycin 1,000 mg every 12 hours and cefepime 2,000 mg every 12 hours for possible hospital-acquired pneumonia because of his history of recent DKA hospitalization. Once the patient’s anion gap was closed and metabolic acidosis was resolved, the insulin drip was discontinued, and the patient was transferred to the general medical ward for further management. There, he continued to report having chest pain. A computed tomography (CT) scan without contrast of the chest (contrast was held due to recent acute kidney injury) revealed right hilar soft tissue density obstructing the bronchus intermidius, which had resulted in a right-lung collapse and right-sided pleural effusion (Figure 2). The left lung was clear, and there was no evidence of nodularity. 
Given the patient’s extensive smoking history, the initial concern was for pulmonary malignancy. The decision was made to proceed with bronchoscopy with endobronchial ultrasound-guided transbronchial needle biopsy. Endobronchial brushings and biopsies of R11, 7, right bronchus intermedius, and right upper lobe were obtained. Gross inspection of the airway revealed markedly abnormal-appearing mucosa involving the take off to the right upper lobe and the entire bronchus intermedius with friable, cobblestoned, and edematous mucosa. Biopsies and immunostaining for occult carcinoma markers, including CD-56, TTF-1, Synaptophysin A, chromogranin, AE1/AE3, and CK-5/6, were negative for malignancy. Final microbiologic analysis was positive for Mucor. There was no evidence of bacterial or mycobacterial growth.
Due to continued suspicion for malignancy and lack of histologic yield, the patient underwent a repeat endobronchial ultrasound-guided needle biopsy. On this occasion, gross inspection revealed significant mucosal necrosis and extensive, extrinsic bronchial compression starting from the right bronchial division and notable throughout the right middle and lower lobes (Figure 3). 
Bronchial washings revealed necrotic material with rare fungal hyphae present. Biopsies yielded necrotic material or lung tissue containing nonseptate hyphae with rare, right-angle branching consistent with Mucor (Figures 4 and 5). Malignancy was not present in the specimens obtained. 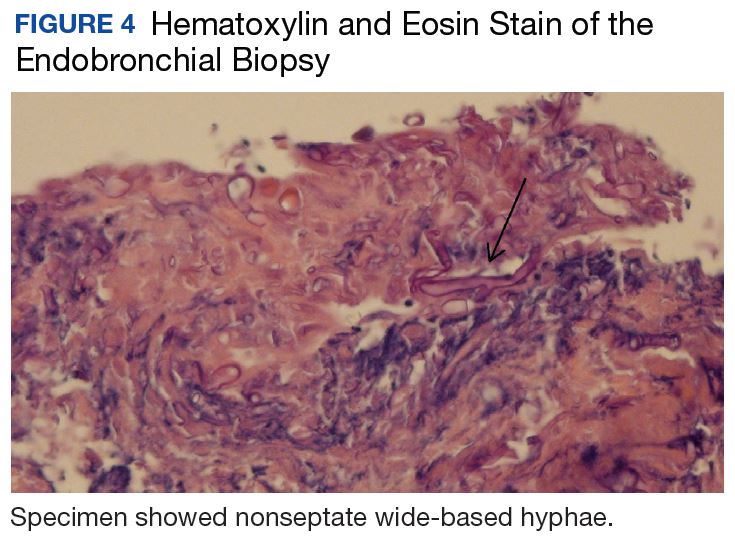
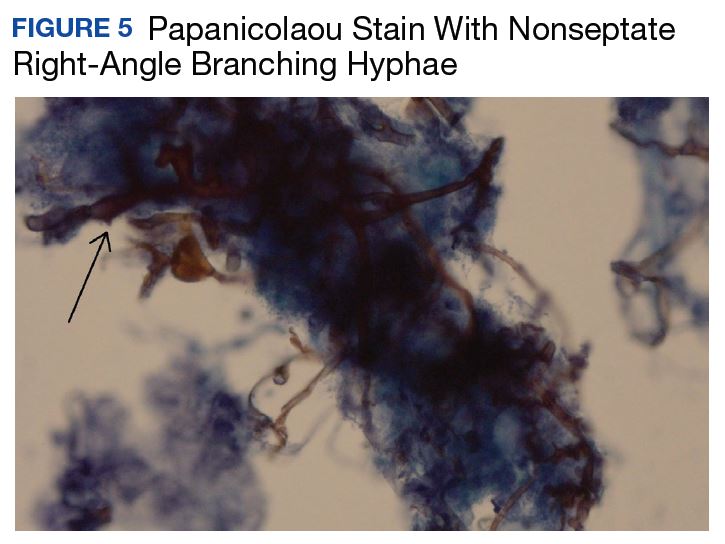
Based on the bronchoscopy results, thoracic surgery and infectious disease specialists were consulted. Surgical intervention was not recommended because of concerns for potential postoperative complications. The infectious disease specialists recommended initiation of liposomal amphotericin B at 10 mg/kg/d. Magnetic resonance imaging of the head showed parietal lobe enhancement with restricted diffusion most consistent with prior infarct. Paranasal sinus disease also was demonstrated. The latter findings prompted further evaluation. The patient underwent right and left endoscopic resection of concha bullosa as well as left maxillary endoscopic antrostomy. Gross examination showed thick mucosa in left concha bullosa, polypoid changes anterior to bulla ethmoidalis, and clear left maxillary sinus. The procedure had to be aborted when the patient experienced cardiac arrest secondary to ventricular fibrillation; he was successfully resuscitated.
Samples from the contents of right and left sinuses as well as left concha bullosa were submitted to pathology, showing benign respiratory mucosa with chronic inflammation and foci of bone without fungal elements. There was no other evidence of disseminated mucormycosis. The patient had a prolonged hospital course complicated by progressive hypoxemia, acute kidney injury, and toxic metabolic encephalopathy. Three months after his original diagnosis, he sustained another cardiac arrest in the hospital. Shortly after achieving return of spontaneous circulation and initiation of invasive mechanical ventilation, the family elected to withdraw care. The family declined an autopsy.
Discussion
This article describes a case of subacute pulmonary mucormycosis in a patient with recurrent DKA. Although patients with poorly controlled DM commonly present with the rhinocerebral form of mucormycosis, pulmonary involvement with a subacute course has been described. Determining the final diagnosis for the current patient was challenging due to the subtlety of his respiratory symptoms and the inconsistent initial findings on chest radiography. A pulmonary disease was finally suspected when a mass was found on the CT scan. However, the middle mediastinal mass was more suspicious for malignancy, particularly given the patient’s smoking history and persistent hyponatremia. In fact, the lack of any neoplastic findings on the initial endobronchial biopsy prompted the health care team to pursue a second biopsy that was consistent with mucormycosis.
This case demonstrates the challenges of prompt diagnosis and treatment of this potentially fatal infection. Furthermore, the extent of the disease at diagnosis precluded this patient from having a surgical intervention, which has been associated with better outcomes than those of medical management alone. Finally, it remains unknown whether the patient had an underlying malignancy, which could have increased the likelihood of pulmonary mucormycosis; the biopsy yield may have been confounded by repeated sampling of necrotic material caused by mucormycosis. Further investigation of any potential pulmonary neoplasm was limited by the patient’s clinical condition and the poor prognosis due to the extent of infection.
Mucorales is an order of fungi comprised of 6 main families that have potential to cause a variety of infections. The genera Mucor, Rhizopus, and Rhizomucor cause the majority of infections.1 Mucormycosis (infection with Mucorales) is generally a rare fungal infection with an incidence of about 500 cases per year in the U.S. However, the incidence is increasing with an aging population, higher prevalence of DM and chronic kidney disease, and a growing population of immunocompromised patients due to advances in cancer therapy and transplantation. Risk factors for pulmonary mucormycosis include conditions associated with congenital and acquired immunodeficiency: hematologic malignancies, uncontrolled DM, solid tumors, and organ transplantation.2
Presentation
Notably, there seems to be an association between specific organ system involvement and predisposing conditions. Pulmonary mucormycosis occurs much less frequently than does the rhinocerebral form in patients with DKA but occurs more commonly in patients with neutropenia that is due to chemotherapy or hematopoietic stem cell transplantation (HSCT) for the treatment of hematologic malignancies.2
The mechanisms for preferential site infection are not well understood with current knowledge of mucormycosis pathogenesis. Current research demonstrates monocytes and neutrophils may play a vital role in the body’s defense against Mucor by both phagocytosis and oxidative damage. Chemotaxis and oxidative cell lysis seem to be compromised in states of hyperglycemia and acidosis. Iron metabolism repeatedly has been shown to play a role in the pathogenesis of mucormycosis. Specifically, patients receiving deferoxamine seem to have a predisposition to Mucorales infections, presumably due to the increased iron supply to the fungus.4 Notably, systemic acidosis also facilitates higher concentrations of available serum iron.
One of the main characteristics of mucormycosis is its ability to aggressively invade blood vessels, causing thrombosis and necrosis and subsequently disseminate hematogenously or through the lymphatic system. This property, at least in large part, depends on endothelial cell damage following phagocytosis of fungus by these cells.
Of note, some of the azole class of drugs (eg, voriconazole), which may be used for antifungal prophylaxis in patients with hematologic malignancies accompanied by neutropenia, have been implicated in predisposition to mucormycosis.2 It also is commonly seen in patients undergoing HSCT. Patients with DM and DKA also can present with pulmonary mucormycosis but generally have a more indolent course unless they develop pulmonary hemorrhage.3 Infection usually occurs by inhalation.
Patients may report dyspnea, cough, and chest pain, which is sometimes accompanied by a fever. Presentation is generally indistinguishable from other causes of pneumonia, and the routinely obtained sputum cultures are usually not diagnostically significant.
Radiographic findings are variable and may include pulmonary nodules, consolidations, masses, and cavitary lesions.1 Due to tissue invasion, a CT scan of the chest might demonstrate a mass crossing mediastinal tissue planes. Definitive diagnosis requires a biopsy with a demonstration of characteristic broad-based nonseptate hyphae with tissue invasion as well as a positive culture (Figures 4 and 5).5 Due to nonspecific symptoms as well as laboratory and imaging findings, a biopsy and, therefore, definitive diagnosis are often delayed. However, postponing medical and surgical therapy for mucormycosis has been associated with worse outcomes.6 With the absence of easily available serologic tests and unspecific symptoms in early disease, many mucormycosis cases are diagnosed postmortem.
Treatments
Recently described therapy advancements have indicated improved outcomes.7 Nevertheless, prognosis remains universally poor with 65% to 70% mortality for patients with cases of isolated pulmonary mucormycosis.8 Many of these patients succumb to sepsis, respiratory failure, and hemoptysis. Patients with pulmonary mucormycosis usually die of dissemination rather than of the sequelae of the pulmonary disease. In fact, pulmonary infection seems to have the highest incidence of dissemination in patients with neutropenia. Surgical therapy seems to have more favorable outcomes than treatment with antifungals alone, especially when considering infection primarily affecting 1 lung.8
Amphotericin B remains the first-line agent for treatment of pulmonary mucormycosis. Retrospective studies show that this agent remains one of the few with activity against Mucor with reported successful outcomes. Specifically, the liposomal formulation seems to have greater efficacy.9 Strong prospective data are lacking. An increasing body of evidence supports a potential benefit from adding echinocandins.10 Although these agents have minimal activity against mucormycosis in vitro, adjunctive therapy to amphotericin resulted in better survival. Alternative regimens include the combination of amphotericin with posaconazole or itraconazole. Both these agents seem to have in vitro activity against mucormycosis pathogens, although poor absorption of these agents puts the potential benefit of such combinations in question.
In patients unable to tolerate polyenes due to adverse effects (AEs), the use of posaconazole as monotherapy has been reported with positive results. One retrospective study reported treatment success in up to 60% and stable disease in 21% of patients at 12 weeks. This study included 24 out of 36 patients with pulmonary mucormycosis.11 Significantly fewer AEs and oral administration makes posaconazole an attractive alternative treatment for mucormycosis and needs further prospective evaluation.
Novel therapies have been attempted, though without success thus far. One randomized clinical trial conducted on patients with mucormycosis attempted to determine whether capitalizing on iron metabolism by Mucor by providing adjunctive deferasirox, an iron chelator, would lead to an initial improvement in mortality. However, outcomes did not improve and resulted in higher mortality rates at 90 days in the intervention group.12
Reversal of underlying conditions remains the cornerstone of successful therapy. If possible, it is important to cease immunosuppression by avoiding corticosteroids, correcting acidosis and hyperglycemia, and discontinuing aluminum and iron chelators.13 This approach becomes problematic in patients with DM with poor glucose control due to nonadherence or lack of resources and in situations where the underlying condition is difficult to treat or the treatment puts patients at risk for mucormycosis (eg, malignancies). Surgery in addition to antifungal therapy should be pursued wherever possible for definitive therapy.
1. Ribes JA, Vanover-Sams CL, Baker DJ. Zygomycetes in human disease. Clin Microbiol Rev. 2000;13(2):236-301.
2. Smith JA, Kauffman CA. Pulmonary fungal infections. Respirology. 2012;17(6):913-926.
3. Spellberg B, Edwards J Jr, Ibrahim A. Novel perspectives on mucormycosis: pathophysiology, presentation, and management. Clin Microbiol Rev. 2005;18(3):556-569.
4. Prokopowicz GP, Bradley SF, Kauffman CA. Indolent zygomycosis associated with deferoxamine chelation therapy. Mycoses. 1994;37(11-12):427-431.
5. Hamilos G, Samonis G, Kontoyiannis DP. Pulmonary mucormycosis. Semin Respir Crit Care Med. 2011;32(6):693-702.
6. Chamilos G, Lewis RE, Kontoyiannis DP. Delaying amphotericin B-based frontline therapy significantly increases mortality among patients with hematologic malignancy who have zygomycosis. Clin Infect Dis. 2008;47(4):503-509.
7. Parfrey NA. Improved diagnosis and prognosis of mucormycosis. A clinicopathologic study of 33 cases. Medicine (Baltimore). 1986;65(2):113-1
8. Tedder M, Spratt JA, Anstadt MP, Hegde SS, Tedder SD, Lowe JE. Pulmonary mucormycosis: results of medical and surgical therapy. Ann Thorac Surg. 1994;57(4):1044-1050.
9.
10. Reed C, Bryant R, Ibrahim AS, et al. Combination polyene-caspofungin treatment of rhino-orbital-cerebral mucormycosis. Clin Infect Dis. 2008;47(3):364-371.
11. van Burik JA, Hare RS, Solomon HF, Corrado ML, Kontoyiannis DP. Posaconazole is effective as salvage therapy in zygomycosis: a retrospective summary of 91 cases. Clin Infect Dis. 2006;42(7):e61-e65.
12. Spellberg B, Ibrahim AS, Chin-Hong PV, et al. The Deferasirox-AmBisome Therapy for Mucormycosis (DEFEAT Mucor) study: a randomized, double-blinded, placebo-controlled trial. J Antimicrob Chemother. 2012;67(3):715-722.
13. de Locht M, Boelaert JR, Schneider YJ. Iron uptake from ferrioxamine and from ferrirhizoferrin by germinating spores of Rhizopus microsporus. Biochem Pharmacol. 1994; 47(10):1843-1850.
Mucorales fungi are ubiquitous organisms commonly inhabiting soil and can cause opportunistic infections. The majority of infections are caused by 3 genera: Rhizopus, Mucor, and Rhizomucor.1 Infection occurs by inhalation or by direct contact with damaged skin. Mucorales infections can have cutaneous, rhinocerebral, pulmonary, gastrointestinal, and central nervous system manifestations. Pulmonary mucormycosis is often rapidly progressive with angioinvasion and fulminant necrosis causing acute dyspnea, hemoptysis, and chest pain. More indolent pulmonary Mucorales infections can mimic a pulmonary mass with occasional cavitation found on imaging studies similar to other fungal infections (eg, Aspergillus).2 Risk factors include severe uncontrolled diabetes mellitus (DM), recurrent diabetic ketoacidosis (DKA), immunosuppression due to congenital or acquired causes, hematologic malignancies, and chronic renal failure.3 The authors present a case of a patient with recurrent DKA and pulmonary mucormycosis.
Case Presentation
A 62-year-old male with DM and a more than 30-pack-year smoking history presented to the emergency department with abdominal pain and chest pain ongoing for about 1 week. The patient had a history of frequent admissions with DKA and medication nonadherence.
On admission, the patient was hemodynamically stable. His vital signs were: temperature 97.4° F, heart rate 89 bpm, respirations 24 breathes per minute, blood pressure 146/86 mm Hg, and oxygen saturation 94% on ambient air. The patient appeared ill but the physical examination was otherwise unremarkable. Laboratory results revealed a white blood cell count of 24,400 with neutrophilic predominance, blood glucose 658 mg/dL, creatinine clearance 2.16 mL/min/1.73 m2, sodium level 124 mEq/L, bicarbonate 6 mEq/L, anion gap 27 mEq/L, 6.8 pH, partial pressure of CO2 11 mm Hg, and lactic acid 2.3 mmol/L.
The patient admitted for DKA management and placed on an insulin drip. Although he did not have a fever or cough productive of sputum or hemoptysis, there was concern that pneumonia might have precipitated DKA. A chest X-ray revealed a patchy, right suprahilar opacity (Figure 1).
The patient was placed on vancomycin 1,000 mg every 12 hours and cefepime 2,000 mg every 12 hours for possible hospital-acquired pneumonia because of his history of recent DKA hospitalization. Once the patient’s anion gap was closed and metabolic acidosis was resolved, the insulin drip was discontinued, and the patient was transferred to the general medical ward for further management. There, he continued to report having chest pain. A computed tomography (CT) scan without contrast of the chest (contrast was held due to recent acute kidney injury) revealed right hilar soft tissue density obstructing the bronchus intermidius, which had resulted in a right-lung collapse and right-sided pleural effusion (Figure 2). The left lung was clear, and there was no evidence of nodularity.
Given the patient’s extensive smoking history, the initial concern was for pulmonary malignancy. The decision was made to proceed with bronchoscopy with endobronchial ultrasound-guided transbronchial needle biopsy. Endobronchial brushings and biopsies of R11, 7, right bronchus intermedius, and right upper lobe were obtained. Gross inspection of the airway revealed markedly abnormal-appearing mucosa involving the take off to the right upper lobe and the entire bronchus intermedius with friable, cobblestoned, and edematous mucosa. Biopsies and immunostaining for occult carcinoma markers, including CD-56, TTF-1, Synaptophysin A, chromogranin, AE1/AE3, and CK-5/6, were negative for malignancy. Final microbiologic analysis was positive for Mucor. There was no evidence of bacterial or mycobacterial growth.
Due to continued suspicion for malignancy and lack of histologic yield, the patient underwent a repeat endobronchial ultrasound-guided needle biopsy. On this occasion, gross inspection revealed significant mucosal necrosis and extensive, extrinsic bronchial compression starting from the right bronchial division and notable throughout the right middle and lower lobes (Figure 3).
Bronchial washings revealed necrotic material with rare fungal hyphae present. Biopsies yielded necrotic material or lung tissue containing nonseptate hyphae with rare, right-angle branching consistent with Mucor (Figures 4 and 5). Malignancy was not present in the specimens obtained.
Based on the bronchoscopy results, thoracic surgery and infectious disease specialists were consulted. Surgical intervention was not recommended because of concerns for potential postoperative complications. The infectious disease specialists recommended initiation of liposomal amphotericin B at 10 mg/kg/d. Magnetic resonance imaging of the head showed parietal lobe enhancement with restricted diffusion most consistent with prior infarct. Paranasal sinus disease also was demonstrated. The latter findings prompted further evaluation. The patient underwent right and left endoscopic resection of concha bullosa as well as left maxillary endoscopic antrostomy. Gross examination showed thick mucosa in left concha bullosa, polypoid changes anterior to bulla ethmoidalis, and clear left maxillary sinus. The procedure had to be aborted when the patient experienced cardiac arrest secondary to ventricular fibrillation; he was successfully resuscitated.
Samples from the contents of right and left sinuses as well as left concha bullosa were submitted to pathology, showing benign respiratory mucosa with chronic inflammation and foci of bone without fungal elements. There was no other evidence of disseminated mucormycosis. The patient had a prolonged hospital course complicated by progressive hypoxemia, acute kidney injury, and toxic metabolic encephalopathy. Three months after his original diagnosis, he sustained another cardiac arrest in the hospital. Shortly after achieving return of spontaneous circulation and initiation of invasive mechanical ventilation, the family elected to withdraw care. The family declined an autopsy.
Discussion
This article describes a case of subacute pulmonary mucormycosis in a patient with recurrent DKA. Although patients with poorly controlled DM commonly present with the rhinocerebral form of mucormycosis, pulmonary involvement with a subacute course has been described. Determining the final diagnosis for the current patient was challenging due to the subtlety of his respiratory symptoms and the inconsistent initial findings on chest radiography. A pulmonary disease was finally suspected when a mass was found on the CT scan. However, the middle mediastinal mass was more suspicious for malignancy, particularly given the patient’s smoking history and persistent hyponatremia. In fact, the lack of any neoplastic findings on the initial endobronchial biopsy prompted the health care team to pursue a second biopsy that was consistent with mucormycosis.
This case demonstrates the challenges of prompt diagnosis and treatment of this potentially fatal infection. Furthermore, the extent of the disease at diagnosis precluded this patient from having a surgical intervention, which has been associated with better outcomes than those of medical management alone. Finally, it remains unknown whether the patient had an underlying malignancy, which could have increased the likelihood of pulmonary mucormycosis; the biopsy yield may have been confounded by repeated sampling of necrotic material caused by mucormycosis. Further investigation of any potential pulmonary neoplasm was limited by the patient’s clinical condition and the poor prognosis due to the extent of infection.
Mucorales is an order of fungi comprised of 6 main families that have potential to cause a variety of infections. The genera Mucor, Rhizopus, and Rhizomucor cause the majority of infections.1 Mucormycosis (infection with Mucorales) is generally a rare fungal infection with an incidence of about 500 cases per year in the U.S. However, the incidence is increasing with an aging population, higher prevalence of DM and chronic kidney disease, and a growing population of immunocompromised patients due to advances in cancer therapy and transplantation. Risk factors for pulmonary mucormycosis include conditions associated with congenital and acquired immunodeficiency: hematologic malignancies, uncontrolled DM, solid tumors, and organ transplantation.2
Presentation
Notably, there seems to be an association between specific organ system involvement and predisposing conditions. Pulmonary mucormycosis occurs much less frequently than does the rhinocerebral form in patients with DKA but occurs more commonly in patients with neutropenia that is due to chemotherapy or hematopoietic stem cell transplantation (HSCT) for the treatment of hematologic malignancies.2
The mechanisms for preferential site infection are not well understood with current knowledge of mucormycosis pathogenesis. Current research demonstrates monocytes and neutrophils may play a vital role in the body’s defense against Mucor by both phagocytosis and oxidative damage. Chemotaxis and oxidative cell lysis seem to be compromised in states of hyperglycemia and acidosis. Iron metabolism repeatedly has been shown to play a role in the pathogenesis of mucormycosis. Specifically, patients receiving deferoxamine seem to have a predisposition to Mucorales infections, presumably due to the increased iron supply to the fungus.4 Notably, systemic acidosis also facilitates higher concentrations of available serum iron.
One of the main characteristics of mucormycosis is its ability to aggressively invade blood vessels, causing thrombosis and necrosis and subsequently disseminate hematogenously or through the lymphatic system. This property, at least in large part, depends on endothelial cell damage following phagocytosis of fungus by these cells.
Of note, some of the azole class of drugs (eg, voriconazole), which may be used for antifungal prophylaxis in patients with hematologic malignancies accompanied by neutropenia, have been implicated in predisposition to mucormycosis.2 It also is commonly seen in patients undergoing HSCT. Patients with DM and DKA also can present with pulmonary mucormycosis but generally have a more indolent course unless they develop pulmonary hemorrhage.3 Infection usually occurs by inhalation.
Patients may report dyspnea, cough, and chest pain, which is sometimes accompanied by a fever. Presentation is generally indistinguishable from other causes of pneumonia, and the routinely obtained sputum cultures are usually not diagnostically significant.
Radiographic findings are variable and may include pulmonary nodules, consolidations, masses, and cavitary lesions.1 Due to tissue invasion, a CT scan of the chest might demonstrate a mass crossing mediastinal tissue planes. Definitive diagnosis requires a biopsy with a demonstration of characteristic broad-based nonseptate hyphae with tissue invasion as well as a positive culture (Figures 4 and 5).5 Due to nonspecific symptoms as well as laboratory and imaging findings, a biopsy and, therefore, definitive diagnosis are often delayed. However, postponing medical and surgical therapy for mucormycosis has been associated with worse outcomes.6 With the absence of easily available serologic tests and unspecific symptoms in early disease, many mucormycosis cases are diagnosed postmortem.
Treatments
Recently described therapy advancements have indicated improved outcomes.7 Nevertheless, prognosis remains universally poor with 65% to 70% mortality for patients with cases of isolated pulmonary mucormycosis.8 Many of these patients succumb to sepsis, respiratory failure, and hemoptysis. Patients with pulmonary mucormycosis usually die of dissemination rather than of the sequelae of the pulmonary disease. In fact, pulmonary infection seems to have the highest incidence of dissemination in patients with neutropenia. Surgical therapy seems to have more favorable outcomes than treatment with antifungals alone, especially when considering infection primarily affecting 1 lung.8
Amphotericin B remains the first-line agent for treatment of pulmonary mucormycosis. Retrospective studies show that this agent remains one of the few with activity against Mucor with reported successful outcomes. Specifically, the liposomal formulation seems to have greater efficacy.9 Strong prospective data are lacking. An increasing body of evidence supports a potential benefit from adding echinocandins.10 Although these agents have minimal activity against mucormycosis in vitro, adjunctive therapy to amphotericin resulted in better survival. Alternative regimens include the combination of amphotericin with posaconazole or itraconazole. Both these agents seem to have in vitro activity against mucormycosis pathogens, although poor absorption of these agents puts the potential benefit of such combinations in question.
In patients unable to tolerate polyenes due to adverse effects (AEs), the use of posaconazole as monotherapy has been reported with positive results. One retrospective study reported treatment success in up to 60% and stable disease in 21% of patients at 12 weeks. This study included 24 out of 36 patients with pulmonary mucormycosis.11 Significantly fewer AEs and oral administration makes posaconazole an attractive alternative treatment for mucormycosis and needs further prospective evaluation.
Novel therapies have been attempted, though without success thus far. One randomized clinical trial conducted on patients with mucormycosis attempted to determine whether capitalizing on iron metabolism by Mucor by providing adjunctive deferasirox, an iron chelator, would lead to an initial improvement in mortality. However, outcomes did not improve and resulted in higher mortality rates at 90 days in the intervention group.12
Reversal of underlying conditions remains the cornerstone of successful therapy. If possible, it is important to cease immunosuppression by avoiding corticosteroids, correcting acidosis and hyperglycemia, and discontinuing aluminum and iron chelators.13 This approach becomes problematic in patients with DM with poor glucose control due to nonadherence or lack of resources and in situations where the underlying condition is difficult to treat or the treatment puts patients at risk for mucormycosis (eg, malignancies). Surgery in addition to antifungal therapy should be pursued wherever possible for definitive therapy.
Mucorales fungi are ubiquitous organisms commonly inhabiting soil and can cause opportunistic infections. The majority of infections are caused by 3 genera: Rhizopus, Mucor, and Rhizomucor.1 Infection occurs by inhalation or by direct contact with damaged skin. Mucorales infections can have cutaneous, rhinocerebral, pulmonary, gastrointestinal, and central nervous system manifestations. Pulmonary mucormycosis is often rapidly progressive with angioinvasion and fulminant necrosis causing acute dyspnea, hemoptysis, and chest pain. More indolent pulmonary Mucorales infections can mimic a pulmonary mass with occasional cavitation found on imaging studies similar to other fungal infections (eg, Aspergillus).2 Risk factors include severe uncontrolled diabetes mellitus (DM), recurrent diabetic ketoacidosis (DKA), immunosuppression due to congenital or acquired causes, hematologic malignancies, and chronic renal failure.3 The authors present a case of a patient with recurrent DKA and pulmonary mucormycosis.
Case Presentation
A 62-year-old male with DM and a more than 30-pack-year smoking history presented to the emergency department with abdominal pain and chest pain ongoing for about 1 week. The patient had a history of frequent admissions with DKA and medication nonadherence.
On admission, the patient was hemodynamically stable. His vital signs were: temperature 97.4° F, heart rate 89 bpm, respirations 24 breathes per minute, blood pressure 146/86 mm Hg, and oxygen saturation 94% on ambient air. The patient appeared ill but the physical examination was otherwise unremarkable. Laboratory results revealed a white blood cell count of 24,400 with neutrophilic predominance, blood glucose 658 mg/dL, creatinine clearance 2.16 mL/min/1.73 m2, sodium level 124 mEq/L, bicarbonate 6 mEq/L, anion gap 27 mEq/L, 6.8 pH, partial pressure of CO2 11 mm Hg, and lactic acid 2.3 mmol/L.
The patient admitted for DKA management and placed on an insulin drip. Although he did not have a fever or cough productive of sputum or hemoptysis, there was concern that pneumonia might have precipitated DKA. A chest X-ray revealed a patchy, right suprahilar opacity (Figure 1).
The patient was placed on vancomycin 1,000 mg every 12 hours and cefepime 2,000 mg every 12 hours for possible hospital-acquired pneumonia because of his history of recent DKA hospitalization. Once the patient’s anion gap was closed and metabolic acidosis was resolved, the insulin drip was discontinued, and the patient was transferred to the general medical ward for further management. There, he continued to report having chest pain. A computed tomography (CT) scan without contrast of the chest (contrast was held due to recent acute kidney injury) revealed right hilar soft tissue density obstructing the bronchus intermidius, which had resulted in a right-lung collapse and right-sided pleural effusion (Figure 2). The left lung was clear, and there was no evidence of nodularity.
Given the patient’s extensive smoking history, the initial concern was for pulmonary malignancy. The decision was made to proceed with bronchoscopy with endobronchial ultrasound-guided transbronchial needle biopsy. Endobronchial brushings and biopsies of R11, 7, right bronchus intermedius, and right upper lobe were obtained. Gross inspection of the airway revealed markedly abnormal-appearing mucosa involving the take off to the right upper lobe and the entire bronchus intermedius with friable, cobblestoned, and edematous mucosa. Biopsies and immunostaining for occult carcinoma markers, including CD-56, TTF-1, Synaptophysin A, chromogranin, AE1/AE3, and CK-5/6, were negative for malignancy. Final microbiologic analysis was positive for Mucor. There was no evidence of bacterial or mycobacterial growth.
Due to continued suspicion for malignancy and lack of histologic yield, the patient underwent a repeat endobronchial ultrasound-guided needle biopsy. On this occasion, gross inspection revealed significant mucosal necrosis and extensive, extrinsic bronchial compression starting from the right bronchial division and notable throughout the right middle and lower lobes (Figure 3).
Bronchial washings revealed necrotic material with rare fungal hyphae present. Biopsies yielded necrotic material or lung tissue containing nonseptate hyphae with rare, right-angle branching consistent with Mucor (Figures 4 and 5). Malignancy was not present in the specimens obtained.
Based on the bronchoscopy results, thoracic surgery and infectious disease specialists were consulted. Surgical intervention was not recommended because of concerns for potential postoperative complications. The infectious disease specialists recommended initiation of liposomal amphotericin B at 10 mg/kg/d. Magnetic resonance imaging of the head showed parietal lobe enhancement with restricted diffusion most consistent with prior infarct. Paranasal sinus disease also was demonstrated. The latter findings prompted further evaluation. The patient underwent right and left endoscopic resection of concha bullosa as well as left maxillary endoscopic antrostomy. Gross examination showed thick mucosa in left concha bullosa, polypoid changes anterior to bulla ethmoidalis, and clear left maxillary sinus. The procedure had to be aborted when the patient experienced cardiac arrest secondary to ventricular fibrillation; he was successfully resuscitated.
Samples from the contents of right and left sinuses as well as left concha bullosa were submitted to pathology, showing benign respiratory mucosa with chronic inflammation and foci of bone without fungal elements. There was no other evidence of disseminated mucormycosis. The patient had a prolonged hospital course complicated by progressive hypoxemia, acute kidney injury, and toxic metabolic encephalopathy. Three months after his original diagnosis, he sustained another cardiac arrest in the hospital. Shortly after achieving return of spontaneous circulation and initiation of invasive mechanical ventilation, the family elected to withdraw care. The family declined an autopsy.
Discussion
This article describes a case of subacute pulmonary mucormycosis in a patient with recurrent DKA. Although patients with poorly controlled DM commonly present with the rhinocerebral form of mucormycosis, pulmonary involvement with a subacute course has been described. Determining the final diagnosis for the current patient was challenging due to the subtlety of his respiratory symptoms and the inconsistent initial findings on chest radiography. A pulmonary disease was finally suspected when a mass was found on the CT scan. However, the middle mediastinal mass was more suspicious for malignancy, particularly given the patient’s smoking history and persistent hyponatremia. In fact, the lack of any neoplastic findings on the initial endobronchial biopsy prompted the health care team to pursue a second biopsy that was consistent with mucormycosis.
This case demonstrates the challenges of prompt diagnosis and treatment of this potentially fatal infection. Furthermore, the extent of the disease at diagnosis precluded this patient from having a surgical intervention, which has been associated with better outcomes than those of medical management alone. Finally, it remains unknown whether the patient had an underlying malignancy, which could have increased the likelihood of pulmonary mucormycosis; the biopsy yield may have been confounded by repeated sampling of necrotic material caused by mucormycosis. Further investigation of any potential pulmonary neoplasm was limited by the patient’s clinical condition and the poor prognosis due to the extent of infection.
Mucorales is an order of fungi comprised of 6 main families that have potential to cause a variety of infections. The genera Mucor, Rhizopus, and Rhizomucor cause the majority of infections.1 Mucormycosis (infection with Mucorales) is generally a rare fungal infection with an incidence of about 500 cases per year in the U.S. However, the incidence is increasing with an aging population, higher prevalence of DM and chronic kidney disease, and a growing population of immunocompromised patients due to advances in cancer therapy and transplantation. Risk factors for pulmonary mucormycosis include conditions associated with congenital and acquired immunodeficiency: hematologic malignancies, uncontrolled DM, solid tumors, and organ transplantation.2
Presentation
Notably, there seems to be an association between specific organ system involvement and predisposing conditions. Pulmonary mucormycosis occurs much less frequently than does the rhinocerebral form in patients with DKA but occurs more commonly in patients with neutropenia that is due to chemotherapy or hematopoietic stem cell transplantation (HSCT) for the treatment of hematologic malignancies.2
The mechanisms for preferential site infection are not well understood with current knowledge of mucormycosis pathogenesis. Current research demonstrates monocytes and neutrophils may play a vital role in the body’s defense against Mucor by both phagocytosis and oxidative damage. Chemotaxis and oxidative cell lysis seem to be compromised in states of hyperglycemia and acidosis. Iron metabolism repeatedly has been shown to play a role in the pathogenesis of mucormycosis. Specifically, patients receiving deferoxamine seem to have a predisposition to Mucorales infections, presumably due to the increased iron supply to the fungus.4 Notably, systemic acidosis also facilitates higher concentrations of available serum iron.
One of the main characteristics of mucormycosis is its ability to aggressively invade blood vessels, causing thrombosis and necrosis and subsequently disseminate hematogenously or through the lymphatic system. This property, at least in large part, depends on endothelial cell damage following phagocytosis of fungus by these cells.
Of note, some of the azole class of drugs (eg, voriconazole), which may be used for antifungal prophylaxis in patients with hematologic malignancies accompanied by neutropenia, have been implicated in predisposition to mucormycosis.2 It also is commonly seen in patients undergoing HSCT. Patients with DM and DKA also can present with pulmonary mucormycosis but generally have a more indolent course unless they develop pulmonary hemorrhage.3 Infection usually occurs by inhalation.
Patients may report dyspnea, cough, and chest pain, which is sometimes accompanied by a fever. Presentation is generally indistinguishable from other causes of pneumonia, and the routinely obtained sputum cultures are usually not diagnostically significant.
Radiographic findings are variable and may include pulmonary nodules, consolidations, masses, and cavitary lesions.1 Due to tissue invasion, a CT scan of the chest might demonstrate a mass crossing mediastinal tissue planes. Definitive diagnosis requires a biopsy with a demonstration of characteristic broad-based nonseptate hyphae with tissue invasion as well as a positive culture (Figures 4 and 5).5 Due to nonspecific symptoms as well as laboratory and imaging findings, a biopsy and, therefore, definitive diagnosis are often delayed. However, postponing medical and surgical therapy for mucormycosis has been associated with worse outcomes.6 With the absence of easily available serologic tests and unspecific symptoms in early disease, many mucormycosis cases are diagnosed postmortem.
Treatments
Recently described therapy advancements have indicated improved outcomes.7 Nevertheless, prognosis remains universally poor with 65% to 70% mortality for patients with cases of isolated pulmonary mucormycosis.8 Many of these patients succumb to sepsis, respiratory failure, and hemoptysis. Patients with pulmonary mucormycosis usually die of dissemination rather than of the sequelae of the pulmonary disease. In fact, pulmonary infection seems to have the highest incidence of dissemination in patients with neutropenia. Surgical therapy seems to have more favorable outcomes than treatment with antifungals alone, especially when considering infection primarily affecting 1 lung.8
Amphotericin B remains the first-line agent for treatment of pulmonary mucormycosis. Retrospective studies show that this agent remains one of the few with activity against Mucor with reported successful outcomes. Specifically, the liposomal formulation seems to have greater efficacy.9 Strong prospective data are lacking. An increasing body of evidence supports a potential benefit from adding echinocandins.10 Although these agents have minimal activity against mucormycosis in vitro, adjunctive therapy to amphotericin resulted in better survival. Alternative regimens include the combination of amphotericin with posaconazole or itraconazole. Both these agents seem to have in vitro activity against mucormycosis pathogens, although poor absorption of these agents puts the potential benefit of such combinations in question.
In patients unable to tolerate polyenes due to adverse effects (AEs), the use of posaconazole as monotherapy has been reported with positive results. One retrospective study reported treatment success in up to 60% and stable disease in 21% of patients at 12 weeks. This study included 24 out of 36 patients with pulmonary mucormycosis.11 Significantly fewer AEs and oral administration makes posaconazole an attractive alternative treatment for mucormycosis and needs further prospective evaluation.
Novel therapies have been attempted, though without success thus far. One randomized clinical trial conducted on patients with mucormycosis attempted to determine whether capitalizing on iron metabolism by Mucor by providing adjunctive deferasirox, an iron chelator, would lead to an initial improvement in mortality. However, outcomes did not improve and resulted in higher mortality rates at 90 days in the intervention group.12
Reversal of underlying conditions remains the cornerstone of successful therapy. If possible, it is important to cease immunosuppression by avoiding corticosteroids, correcting acidosis and hyperglycemia, and discontinuing aluminum and iron chelators.13 This approach becomes problematic in patients with DM with poor glucose control due to nonadherence or lack of resources and in situations where the underlying condition is difficult to treat or the treatment puts patients at risk for mucormycosis (eg, malignancies). Surgery in addition to antifungal therapy should be pursued wherever possible for definitive therapy.
1. Ribes JA, Vanover-Sams CL, Baker DJ. Zygomycetes in human disease. Clin Microbiol Rev. 2000;13(2):236-301.
2. Smith JA, Kauffman CA. Pulmonary fungal infections. Respirology. 2012;17(6):913-926.
3. Spellberg B, Edwards J Jr, Ibrahim A. Novel perspectives on mucormycosis: pathophysiology, presentation, and management. Clin Microbiol Rev. 2005;18(3):556-569.
4. Prokopowicz GP, Bradley SF, Kauffman CA. Indolent zygomycosis associated with deferoxamine chelation therapy. Mycoses. 1994;37(11-12):427-431.
5. Hamilos G, Samonis G, Kontoyiannis DP. Pulmonary mucormycosis. Semin Respir Crit Care Med. 2011;32(6):693-702.
6. Chamilos G, Lewis RE, Kontoyiannis DP. Delaying amphotericin B-based frontline therapy significantly increases mortality among patients with hematologic malignancy who have zygomycosis. Clin Infect Dis. 2008;47(4):503-509.
7. Parfrey NA. Improved diagnosis and prognosis of mucormycosis. A clinicopathologic study of 33 cases. Medicine (Baltimore). 1986;65(2):113-1
8. Tedder M, Spratt JA, Anstadt MP, Hegde SS, Tedder SD, Lowe JE. Pulmonary mucormycosis: results of medical and surgical therapy. Ann Thorac Surg. 1994;57(4):1044-1050.
9.
10. Reed C, Bryant R, Ibrahim AS, et al. Combination polyene-caspofungin treatment of rhino-orbital-cerebral mucormycosis. Clin Infect Dis. 2008;47(3):364-371.
11. van Burik JA, Hare RS, Solomon HF, Corrado ML, Kontoyiannis DP. Posaconazole is effective as salvage therapy in zygomycosis: a retrospective summary of 91 cases. Clin Infect Dis. 2006;42(7):e61-e65.
12. Spellberg B, Ibrahim AS, Chin-Hong PV, et al. The Deferasirox-AmBisome Therapy for Mucormycosis (DEFEAT Mucor) study: a randomized, double-blinded, placebo-controlled trial. J Antimicrob Chemother. 2012;67(3):715-722.
13. de Locht M, Boelaert JR, Schneider YJ. Iron uptake from ferrioxamine and from ferrirhizoferrin by germinating spores of Rhizopus microsporus. Biochem Pharmacol. 1994; 47(10):1843-1850.
1. Ribes JA, Vanover-Sams CL, Baker DJ. Zygomycetes in human disease. Clin Microbiol Rev. 2000;13(2):236-301.
2. Smith JA, Kauffman CA. Pulmonary fungal infections. Respirology. 2012;17(6):913-926.
3. Spellberg B, Edwards J Jr, Ibrahim A. Novel perspectives on mucormycosis: pathophysiology, presentation, and management. Clin Microbiol Rev. 2005;18(3):556-569.
4. Prokopowicz GP, Bradley SF, Kauffman CA. Indolent zygomycosis associated with deferoxamine chelation therapy. Mycoses. 1994;37(11-12):427-431.
5. Hamilos G, Samonis G, Kontoyiannis DP. Pulmonary mucormycosis. Semin Respir Crit Care Med. 2011;32(6):693-702.
6. Chamilos G, Lewis RE, Kontoyiannis DP. Delaying amphotericin B-based frontline therapy significantly increases mortality among patients with hematologic malignancy who have zygomycosis. Clin Infect Dis. 2008;47(4):503-509.
7. Parfrey NA. Improved diagnosis and prognosis of mucormycosis. A clinicopathologic study of 33 cases. Medicine (Baltimore). 1986;65(2):113-1
8. Tedder M, Spratt JA, Anstadt MP, Hegde SS, Tedder SD, Lowe JE. Pulmonary mucormycosis: results of medical and surgical therapy. Ann Thorac Surg. 1994;57(4):1044-1050.
9.
10. Reed C, Bryant R, Ibrahim AS, et al. Combination polyene-caspofungin treatment of rhino-orbital-cerebral mucormycosis. Clin Infect Dis. 2008;47(3):364-371.
11. van Burik JA, Hare RS, Solomon HF, Corrado ML, Kontoyiannis DP. Posaconazole is effective as salvage therapy in zygomycosis: a retrospective summary of 91 cases. Clin Infect Dis. 2006;42(7):e61-e65.
12. Spellberg B, Ibrahim AS, Chin-Hong PV, et al. The Deferasirox-AmBisome Therapy for Mucormycosis (DEFEAT Mucor) study: a randomized, double-blinded, placebo-controlled trial. J Antimicrob Chemother. 2012;67(3):715-722.
13. de Locht M, Boelaert JR, Schneider YJ. Iron uptake from ferrioxamine and from ferrirhizoferrin by germinating spores of Rhizopus microsporus. Biochem Pharmacol. 1994; 47(10):1843-1850.