User login
Growing Pink Nodule on the Ankle
Growing Pink Nodule on the Ankle
THE DIAGNOSIS: Epithelioid Fibrous Histiocytoma
In our patient, immunohistochemical stains for Factor XIIIa, CD68, and anaplastic lymphoma kinase (ALK) 1 confirmed the diagnosis of epithelioid fibrous histiocytoma (EFH). The location and relatively large size of the lesion led to a joint decision by the patient and physician to perform a complete excision, which was done with no complications.
Once considered a rare variant of dermatofibroma, EFH most commonly manifests as a solitary, vascular-appearing or flesh-colored papule or nodule on the legs. It often develops in the fifth decade of life with greater prevalence in men.1-5 Our patient is one of the few known cases of EFH in children that have been reported in the literature.3,6 Although EFH is benign, complete excision typically is performed due to the rarity of the lesion.3
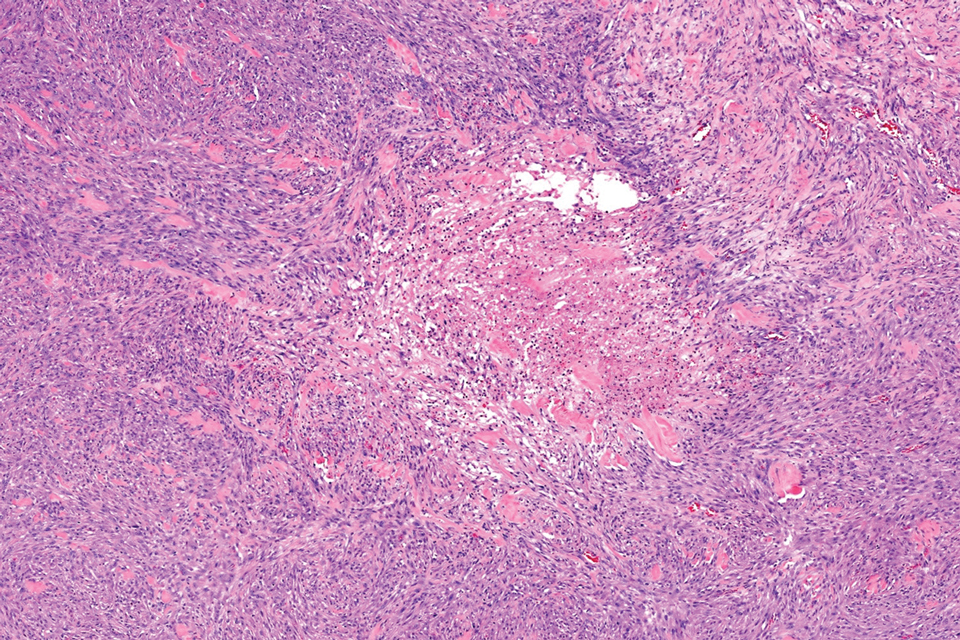
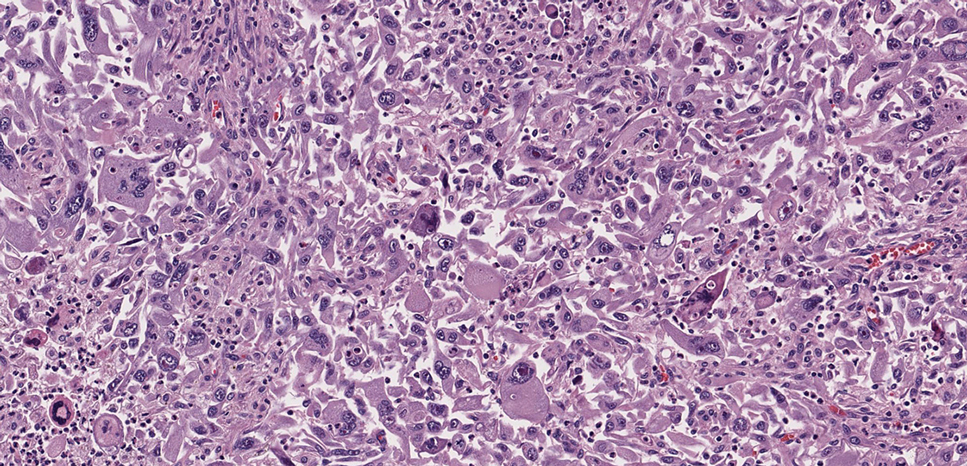
The overexpression of ALK distinguishes EFH from other fibrohistiocytic lesions (Figure 1).5 The most common fusion partners are sequestosome 1 and vinculin (VCL), which account for more than 70% of cases.3,5,7 Interestingly, VCL-ALK fusions have been reported to occur in a subset of pediatric renal cell carcinomas and recently in an ovoid spindle cell neoplasm considered to be a low-grade sarcoma.3,7-9 Further studies have identified less common fusion partners, including the dynactin subunit 1, ETS variant transcription factor 6, protein-tyrosine phosphatase, receptor-type, F polypeptide-interacting protein-binding protein 1, sperm antigen with calponin homology and coiled-coil domains 1, tropomyosin 3, protein kinase cAMP-dependent type II regulatory subunit alpha, melanophilin, and Echinoderm microtubule-associated protein-like 4 genes.3,8
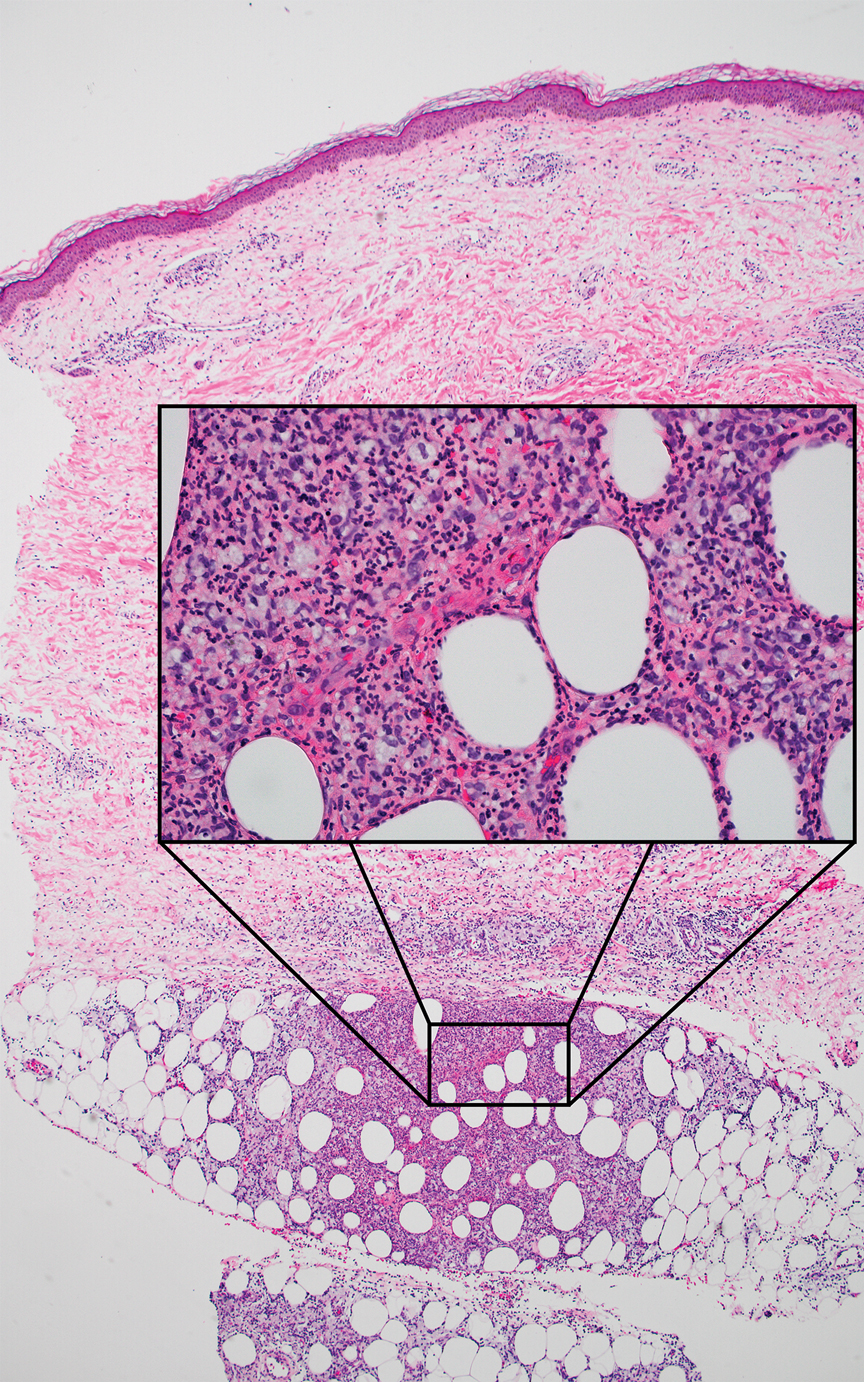
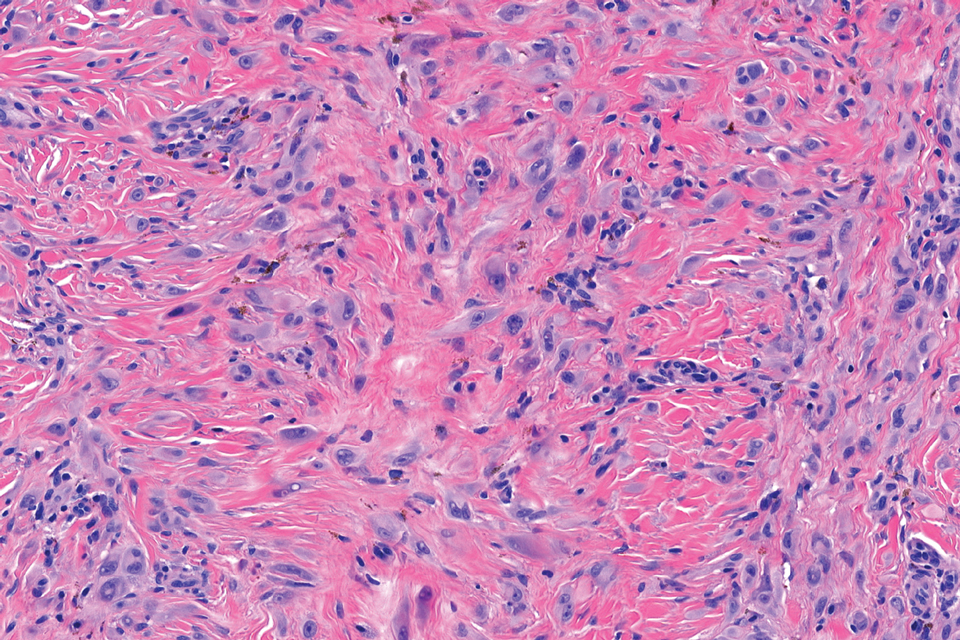
In contrast to benign fibrous histiocytomas, EFHs primarily consist of epithelioid cells, have well-defined borders, exhibit prominent vascularity, usually are situated close to the epidermis, and lack multinucleated cells or histiocytes laden with lipids or hemosiderin.2 The characteristic histopathologic finding is rounded or angulated epithelioid cells, with eosinophilic cytoplasm accounting for more than 50% of the tumor cell population.1-3,5 The nuclei of the epithelioid cells are rounded and vesicular with small eosinophilic nucleoli and low mitotic activity. Common clinical features include an exophytic nodule with a classic epidermal collarette and an epidermis that exhibits variable degrees of hyperplasia.1-3,5 Epithelioid fibrous histiocytomas often are confined to the superficial dermis and rarely extend to the subcutaneous layer. The stroma is collagenous with prominent vascularity, although older lesions can become more hyalinized and sclerotic.3 Histopathologically, these tumors can be a diagnostic challenge, as they often are mistaken for other fibrohistiocytic or melanocytic lesions.
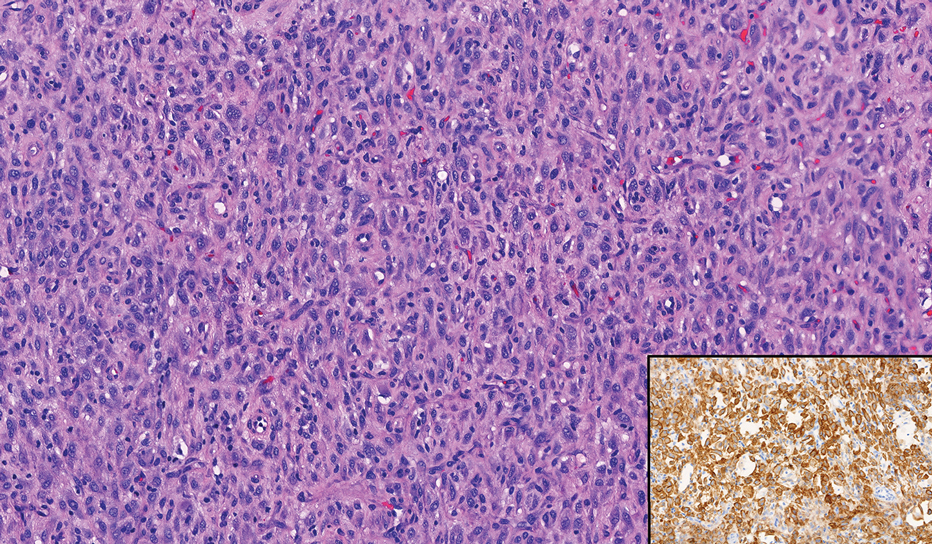
Atypical fibroxanthoma (AFX) manifests as a dome-shaped exophytic nodule that can rapidly grow to 1 to 2 cm. Historically, it was thought to be a pseudomalignancy, but most investigators consider it within the spectrum of pleomorphic dermal sarcoma and undifferentiated pleomorphic sarcoma. Atypical fibroxanthoma usually occurs on the head and neck in elderly patients with sun-damaged skin. Histopathologically, the neoplastic cells of AFX range from atypical spindle cells and pleomorphic round to polygonal epithelioid cells to large, irregularly shaped multinucleated cells, some with foamy cytoplasm (Figure 2). The atypical spindle cells stain diffusely positive for CD10 and vimentin, while small subpopulations stain positively for CD68 or CD163 and procollagen 1. Smooth muscle actin inconsistently stains the tumor, and when it does, the staining typically is faint and patchy. Atypical fibroxanthomas usually do not stain positively for melanocytic, skeletal muscle, or keratinocytic markers.
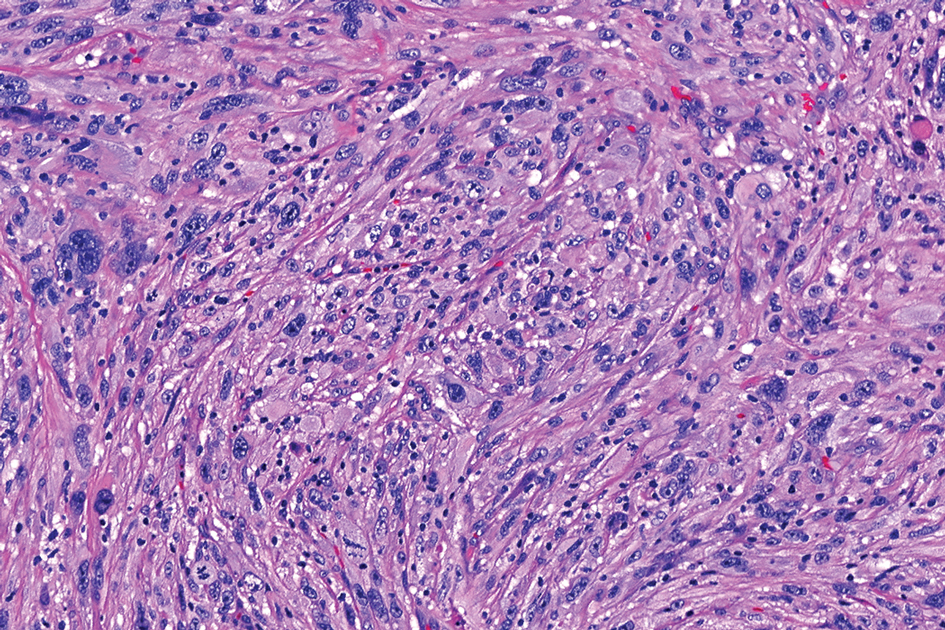
Cellular dermatofibroma typically manifests as small, dome-shaped papules on the arms and legs that normally range from a few millimeters to 1 cm but occasionally measure up to 2 cm. Histopathologically, there are interweaving fascicles of spindle cells with hyperchromatic nuclei and peripheral splaying of the plump spindle cells that wrap around collagen bundles, known as collagen trapping (Figure 3). Unlike EFH, multinucleated cells and histiocytes with abundant lipids and hemosiderin often accompany the spindle cells in cellular dermatofibromas, which stain strongly positive for CD10 and vimentin, similar to AFX and EFH. The smooth muscle actin–staining pattern usually is faint and patchy, and in some cases, cellular dermatofibroma may not stain at all. Factor XIIIa and CD68 highlight the 2 populations of cells—fibroblasts and histiocytes—that make up the lesion.4
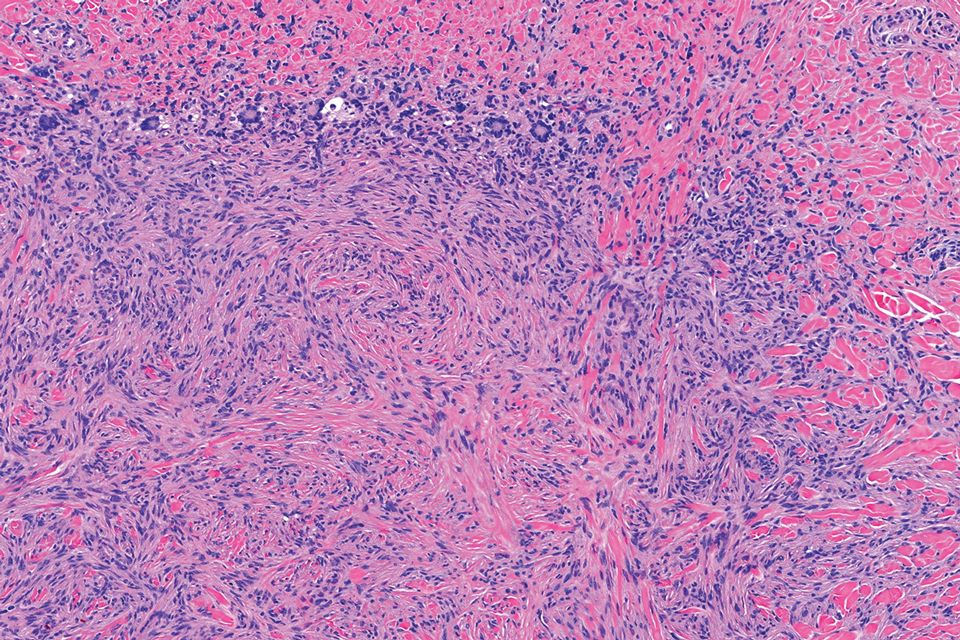
Epithelioid sarcoma comprises 2 types: distal (or conventional) type occurring on the distal arms and legs, particularly the hands and fingers of young adults, and proximal type occurring on the trunk and proximal extremities, including the upper arms and thighs.10 Epithelioid sarcoma is a rare aggressive malignancy that usually manifests as a firm nodule, sometimes with ulceration depending on the size. Histopathologically, diffuse dermal proliferation of ovoid to polygonal epithelioid cells arranged in short fascicles and nodular aggregations is observed (Figure 4). Spindle cells may be observed at the periphery of the lesion. Areas of necrosis are a frequent finding and a helpful diagnostic clue. Nearly all cases stain positively for pancytokeratin, CAM5.2, epithelial membrane antigen, and vimentin, and approximately half stain positively for CD34; there are variable expressions of ERG and smooth muscle actin.10 In most cases, epithelioid sarcoma does not stain positively for S100 or CD68. The majority (90%) of cases harbor a mutation in the SWI/SNF-related matrix-associated actin-dependent regulator of chromatin subfamily B member 1 gene, resulting in the loss of INI1 protein expression, which can be demonstrated by immunohistochemistry. 10 As the cytologic atypia usually is minimal, epithelioid sarcoma may be misdiagnosed as a necrotizing granuloma and benign fibrous lesions, particularly when superficial or small partial biopsies are performed.
Intradermal Spitz nevi can measure from a few millimeters to more than 2 cm and can range from pink to brown to black. The most common locations are the lower extremities as well as the head and neck. Histopathologically, intradermal Spitz nevi have nests of large epithelioid melanocytes with large nuclei and abundant cytoplasm (eFigure). Nuclear pseudo-inclusions, which are cytoplasmic invaginations into the nucleus, are frequent. Unlike the other conditions in the differential, these entities stain positively for melanocytic markers—S100, SOX10, and Melan-A—but not CD68 or CD163.11 A variety of kinase fusions are observed in Spitz nevi, including the ALK gene, neurotrophic tyrosine receptor kinase, ROS proto-oncogene 1, megakaryocyte-erythroid progenitor, and v-raf murine sarcoma viral oncogene homolog B1 genes.12
- Jones EW, Cerio R, Smith NP. Epithelioid cell histiocytoma: a new entity. Br J Dermatol. 1989;120:185-195.
- Glusac EJ, McNiff JM. Epithelioid cell histiocytoma: a simulant of vascular and melanocytic neoplasms. Am J Dermatopathol. 1999;21:1-7.
- Felty CC, Linos K. Epithelioid fibrous histiocytoma: a concise review [published correction appears in Am J Dermatopathol. 2020 Aug;42(8):628]. Am J Dermatopathol. 2019;41:879-883.
- Luzar B, Calonje E. Cutaneous fibrohistiocytic tumours—an update. Histopathology. 2010;56:148-165. doi:10.1111/j.1365-2559.2009.03447.x
- Doyle LA, Mariño-Enriquez A, Fletcher CD, et al. ALK rearrangement and overexpression in epithelioid fibrous histiocytoma. Mod Pathol. 2015;28:904-912.
- Singh Gomez C, Calonje E, Fletcher CD. Epithelioid benign fibrous histiocytoma of skin: clinico-pathological analysis of 20 cases of a poorly known variant. Histopathology. 1994;24:123-129.
- Jedrych J, Nikiforova M, Kennedy TF, et al. Epithelioid cell histiocytoma of the skin with clonal ALK gene rearrangement resulting in VCL- and SQSTM1-ALK gene fusions. Br J Dermatol. 2015;172: 1427-1429.
- Dickson BC, Swanson D, Charames GS, et al. Epithelioid fibrous histiocytoma: molecular characterization of ALK fusion partners in 23 cases. Mod Pathol. 2018;31:753-762.
- Helm M, Chang A, Fanburg-Smith JC, et al. Cutaneous VCL::ALK fusion ovoid-spindle cell neoplasm. J Cutan Pathol. 2023;50:405-409.
- Thway K, Jones RL, Noujaim J, et al. Epithelioid sarcoma: diagnostic features and genetics. Adv Anat Pathol. 2016;23:41-49.
- Bolognia JL, Jorizzo JJ, Schaffer JV et al. Dermatology, 4th ed. Philadelphia: Elsevier; 2018.
- Wiesner T, He J, Yelensky R, et al. Kinase fusions are frequent in Spitz tumours and spitzoid melanomas. Nat Commun. 2014;5:3116.
THE DIAGNOSIS: Epithelioid Fibrous Histiocytoma
In our patient, immunohistochemical stains for Factor XIIIa, CD68, and anaplastic lymphoma kinase (ALK) 1 confirmed the diagnosis of epithelioid fibrous histiocytoma (EFH). The location and relatively large size of the lesion led to a joint decision by the patient and physician to perform a complete excision, which was done with no complications.
Once considered a rare variant of dermatofibroma, EFH most commonly manifests as a solitary, vascular-appearing or flesh-colored papule or nodule on the legs. It often develops in the fifth decade of life with greater prevalence in men.1-5 Our patient is one of the few known cases of EFH in children that have been reported in the literature.3,6 Although EFH is benign, complete excision typically is performed due to the rarity of the lesion.3
The overexpression of ALK distinguishes EFH from other fibrohistiocytic lesions (Figure 1).5 The most common fusion partners are sequestosome 1 and vinculin (VCL), which account for more than 70% of cases.3,5,7 Interestingly, VCL-ALK fusions have been reported to occur in a subset of pediatric renal cell carcinomas and recently in an ovoid spindle cell neoplasm considered to be a low-grade sarcoma.3,7-9 Further studies have identified less common fusion partners, including the dynactin subunit 1, ETS variant transcription factor 6, protein-tyrosine phosphatase, receptor-type, F polypeptide-interacting protein-binding protein 1, sperm antigen with calponin homology and coiled-coil domains 1, tropomyosin 3, protein kinase cAMP-dependent type II regulatory subunit alpha, melanophilin, and Echinoderm microtubule-associated protein-like 4 genes.3,8
In contrast to benign fibrous histiocytomas, EFHs primarily consist of epithelioid cells, have well-defined borders, exhibit prominent vascularity, usually are situated close to the epidermis, and lack multinucleated cells or histiocytes laden with lipids or hemosiderin.2 The characteristic histopathologic finding is rounded or angulated epithelioid cells, with eosinophilic cytoplasm accounting for more than 50% of the tumor cell population.1-3,5 The nuclei of the epithelioid cells are rounded and vesicular with small eosinophilic nucleoli and low mitotic activity. Common clinical features include an exophytic nodule with a classic epidermal collarette and an epidermis that exhibits variable degrees of hyperplasia.1-3,5 Epithelioid fibrous histiocytomas often are confined to the superficial dermis and rarely extend to the subcutaneous layer. The stroma is collagenous with prominent vascularity, although older lesions can become more hyalinized and sclerotic.3 Histopathologically, these tumors can be a diagnostic challenge, as they often are mistaken for other fibrohistiocytic or melanocytic lesions.
Atypical fibroxanthoma (AFX) manifests as a dome-shaped exophytic nodule that can rapidly grow to 1 to 2 cm. Historically, it was thought to be a pseudomalignancy, but most investigators consider it within the spectrum of pleomorphic dermal sarcoma and undifferentiated pleomorphic sarcoma. Atypical fibroxanthoma usually occurs on the head and neck in elderly patients with sun-damaged skin. Histopathologically, the neoplastic cells of AFX range from atypical spindle cells and pleomorphic round to polygonal epithelioid cells to large, irregularly shaped multinucleated cells, some with foamy cytoplasm (Figure 2). The atypical spindle cells stain diffusely positive for CD10 and vimentin, while small subpopulations stain positively for CD68 or CD163 and procollagen 1. Smooth muscle actin inconsistently stains the tumor, and when it does, the staining typically is faint and patchy. Atypical fibroxanthomas usually do not stain positively for melanocytic, skeletal muscle, or keratinocytic markers.
Cellular dermatofibroma typically manifests as small, dome-shaped papules on the arms and legs that normally range from a few millimeters to 1 cm but occasionally measure up to 2 cm. Histopathologically, there are interweaving fascicles of spindle cells with hyperchromatic nuclei and peripheral splaying of the plump spindle cells that wrap around collagen bundles, known as collagen trapping (Figure 3). Unlike EFH, multinucleated cells and histiocytes with abundant lipids and hemosiderin often accompany the spindle cells in cellular dermatofibromas, which stain strongly positive for CD10 and vimentin, similar to AFX and EFH. The smooth muscle actin–staining pattern usually is faint and patchy, and in some cases, cellular dermatofibroma may not stain at all. Factor XIIIa and CD68 highlight the 2 populations of cells—fibroblasts and histiocytes—that make up the lesion.4
Epithelioid sarcoma comprises 2 types: distal (or conventional) type occurring on the distal arms and legs, particularly the hands and fingers of young adults, and proximal type occurring on the trunk and proximal extremities, including the upper arms and thighs.10 Epithelioid sarcoma is a rare aggressive malignancy that usually manifests as a firm nodule, sometimes with ulceration depending on the size. Histopathologically, diffuse dermal proliferation of ovoid to polygonal epithelioid cells arranged in short fascicles and nodular aggregations is observed (Figure 4). Spindle cells may be observed at the periphery of the lesion. Areas of necrosis are a frequent finding and a helpful diagnostic clue. Nearly all cases stain positively for pancytokeratin, CAM5.2, epithelial membrane antigen, and vimentin, and approximately half stain positively for CD34; there are variable expressions of ERG and smooth muscle actin.10 In most cases, epithelioid sarcoma does not stain positively for S100 or CD68. The majority (90%) of cases harbor a mutation in the SWI/SNF-related matrix-associated actin-dependent regulator of chromatin subfamily B member 1 gene, resulting in the loss of INI1 protein expression, which can be demonstrated by immunohistochemistry. 10 As the cytologic atypia usually is minimal, epithelioid sarcoma may be misdiagnosed as a necrotizing granuloma and benign fibrous lesions, particularly when superficial or small partial biopsies are performed.
Intradermal Spitz nevi can measure from a few millimeters to more than 2 cm and can range from pink to brown to black. The most common locations are the lower extremities as well as the head and neck. Histopathologically, intradermal Spitz nevi have nests of large epithelioid melanocytes with large nuclei and abundant cytoplasm (eFigure). Nuclear pseudo-inclusions, which are cytoplasmic invaginations into the nucleus, are frequent. Unlike the other conditions in the differential, these entities stain positively for melanocytic markers—S100, SOX10, and Melan-A—but not CD68 or CD163.11 A variety of kinase fusions are observed in Spitz nevi, including the ALK gene, neurotrophic tyrosine receptor kinase, ROS proto-oncogene 1, megakaryocyte-erythroid progenitor, and v-raf murine sarcoma viral oncogene homolog B1 genes.12
THE DIAGNOSIS: Epithelioid Fibrous Histiocytoma
In our patient, immunohistochemical stains for Factor XIIIa, CD68, and anaplastic lymphoma kinase (ALK) 1 confirmed the diagnosis of epithelioid fibrous histiocytoma (EFH). The location and relatively large size of the lesion led to a joint decision by the patient and physician to perform a complete excision, which was done with no complications.
Once considered a rare variant of dermatofibroma, EFH most commonly manifests as a solitary, vascular-appearing or flesh-colored papule or nodule on the legs. It often develops in the fifth decade of life with greater prevalence in men.1-5 Our patient is one of the few known cases of EFH in children that have been reported in the literature.3,6 Although EFH is benign, complete excision typically is performed due to the rarity of the lesion.3
The overexpression of ALK distinguishes EFH from other fibrohistiocytic lesions (Figure 1).5 The most common fusion partners are sequestosome 1 and vinculin (VCL), which account for more than 70% of cases.3,5,7 Interestingly, VCL-ALK fusions have been reported to occur in a subset of pediatric renal cell carcinomas and recently in an ovoid spindle cell neoplasm considered to be a low-grade sarcoma.3,7-9 Further studies have identified less common fusion partners, including the dynactin subunit 1, ETS variant transcription factor 6, protein-tyrosine phosphatase, receptor-type, F polypeptide-interacting protein-binding protein 1, sperm antigen with calponin homology and coiled-coil domains 1, tropomyosin 3, protein kinase cAMP-dependent type II regulatory subunit alpha, melanophilin, and Echinoderm microtubule-associated protein-like 4 genes.3,8
In contrast to benign fibrous histiocytomas, EFHs primarily consist of epithelioid cells, have well-defined borders, exhibit prominent vascularity, usually are situated close to the epidermis, and lack multinucleated cells or histiocytes laden with lipids or hemosiderin.2 The characteristic histopathologic finding is rounded or angulated epithelioid cells, with eosinophilic cytoplasm accounting for more than 50% of the tumor cell population.1-3,5 The nuclei of the epithelioid cells are rounded and vesicular with small eosinophilic nucleoli and low mitotic activity. Common clinical features include an exophytic nodule with a classic epidermal collarette and an epidermis that exhibits variable degrees of hyperplasia.1-3,5 Epithelioid fibrous histiocytomas often are confined to the superficial dermis and rarely extend to the subcutaneous layer. The stroma is collagenous with prominent vascularity, although older lesions can become more hyalinized and sclerotic.3 Histopathologically, these tumors can be a diagnostic challenge, as they often are mistaken for other fibrohistiocytic or melanocytic lesions.
Atypical fibroxanthoma (AFX) manifests as a dome-shaped exophytic nodule that can rapidly grow to 1 to 2 cm. Historically, it was thought to be a pseudomalignancy, but most investigators consider it within the spectrum of pleomorphic dermal sarcoma and undifferentiated pleomorphic sarcoma. Atypical fibroxanthoma usually occurs on the head and neck in elderly patients with sun-damaged skin. Histopathologically, the neoplastic cells of AFX range from atypical spindle cells and pleomorphic round to polygonal epithelioid cells to large, irregularly shaped multinucleated cells, some with foamy cytoplasm (Figure 2). The atypical spindle cells stain diffusely positive for CD10 and vimentin, while small subpopulations stain positively for CD68 or CD163 and procollagen 1. Smooth muscle actin inconsistently stains the tumor, and when it does, the staining typically is faint and patchy. Atypical fibroxanthomas usually do not stain positively for melanocytic, skeletal muscle, or keratinocytic markers.
Cellular dermatofibroma typically manifests as small, dome-shaped papules on the arms and legs that normally range from a few millimeters to 1 cm but occasionally measure up to 2 cm. Histopathologically, there are interweaving fascicles of spindle cells with hyperchromatic nuclei and peripheral splaying of the plump spindle cells that wrap around collagen bundles, known as collagen trapping (Figure 3). Unlike EFH, multinucleated cells and histiocytes with abundant lipids and hemosiderin often accompany the spindle cells in cellular dermatofibromas, which stain strongly positive for CD10 and vimentin, similar to AFX and EFH. The smooth muscle actin–staining pattern usually is faint and patchy, and in some cases, cellular dermatofibroma may not stain at all. Factor XIIIa and CD68 highlight the 2 populations of cells—fibroblasts and histiocytes—that make up the lesion.4
Epithelioid sarcoma comprises 2 types: distal (or conventional) type occurring on the distal arms and legs, particularly the hands and fingers of young adults, and proximal type occurring on the trunk and proximal extremities, including the upper arms and thighs.10 Epithelioid sarcoma is a rare aggressive malignancy that usually manifests as a firm nodule, sometimes with ulceration depending on the size. Histopathologically, diffuse dermal proliferation of ovoid to polygonal epithelioid cells arranged in short fascicles and nodular aggregations is observed (Figure 4). Spindle cells may be observed at the periphery of the lesion. Areas of necrosis are a frequent finding and a helpful diagnostic clue. Nearly all cases stain positively for pancytokeratin, CAM5.2, epithelial membrane antigen, and vimentin, and approximately half stain positively for CD34; there are variable expressions of ERG and smooth muscle actin.10 In most cases, epithelioid sarcoma does not stain positively for S100 or CD68. The majority (90%) of cases harbor a mutation in the SWI/SNF-related matrix-associated actin-dependent regulator of chromatin subfamily B member 1 gene, resulting in the loss of INI1 protein expression, which can be demonstrated by immunohistochemistry. 10 As the cytologic atypia usually is minimal, epithelioid sarcoma may be misdiagnosed as a necrotizing granuloma and benign fibrous lesions, particularly when superficial or small partial biopsies are performed.
Intradermal Spitz nevi can measure from a few millimeters to more than 2 cm and can range from pink to brown to black. The most common locations are the lower extremities as well as the head and neck. Histopathologically, intradermal Spitz nevi have nests of large epithelioid melanocytes with large nuclei and abundant cytoplasm (eFigure). Nuclear pseudo-inclusions, which are cytoplasmic invaginations into the nucleus, are frequent. Unlike the other conditions in the differential, these entities stain positively for melanocytic markers—S100, SOX10, and Melan-A—but not CD68 or CD163.11 A variety of kinase fusions are observed in Spitz nevi, including the ALK gene, neurotrophic tyrosine receptor kinase, ROS proto-oncogene 1, megakaryocyte-erythroid progenitor, and v-raf murine sarcoma viral oncogene homolog B1 genes.12
- Jones EW, Cerio R, Smith NP. Epithelioid cell histiocytoma: a new entity. Br J Dermatol. 1989;120:185-195.
- Glusac EJ, McNiff JM. Epithelioid cell histiocytoma: a simulant of vascular and melanocytic neoplasms. Am J Dermatopathol. 1999;21:1-7.
- Felty CC, Linos K. Epithelioid fibrous histiocytoma: a concise review [published correction appears in Am J Dermatopathol. 2020 Aug;42(8):628]. Am J Dermatopathol. 2019;41:879-883.
- Luzar B, Calonje E. Cutaneous fibrohistiocytic tumours—an update. Histopathology. 2010;56:148-165. doi:10.1111/j.1365-2559.2009.03447.x
- Doyle LA, Mariño-Enriquez A, Fletcher CD, et al. ALK rearrangement and overexpression in epithelioid fibrous histiocytoma. Mod Pathol. 2015;28:904-912.
- Singh Gomez C, Calonje E, Fletcher CD. Epithelioid benign fibrous histiocytoma of skin: clinico-pathological analysis of 20 cases of a poorly known variant. Histopathology. 1994;24:123-129.
- Jedrych J, Nikiforova M, Kennedy TF, et al. Epithelioid cell histiocytoma of the skin with clonal ALK gene rearrangement resulting in VCL- and SQSTM1-ALK gene fusions. Br J Dermatol. 2015;172: 1427-1429.
- Dickson BC, Swanson D, Charames GS, et al. Epithelioid fibrous histiocytoma: molecular characterization of ALK fusion partners in 23 cases. Mod Pathol. 2018;31:753-762.
- Helm M, Chang A, Fanburg-Smith JC, et al. Cutaneous VCL::ALK fusion ovoid-spindle cell neoplasm. J Cutan Pathol. 2023;50:405-409.
- Thway K, Jones RL, Noujaim J, et al. Epithelioid sarcoma: diagnostic features and genetics. Adv Anat Pathol. 2016;23:41-49.
- Bolognia JL, Jorizzo JJ, Schaffer JV et al. Dermatology, 4th ed. Philadelphia: Elsevier; 2018.
- Wiesner T, He J, Yelensky R, et al. Kinase fusions are frequent in Spitz tumours and spitzoid melanomas. Nat Commun. 2014;5:3116.
- Jones EW, Cerio R, Smith NP. Epithelioid cell histiocytoma: a new entity. Br J Dermatol. 1989;120:185-195.
- Glusac EJ, McNiff JM. Epithelioid cell histiocytoma: a simulant of vascular and melanocytic neoplasms. Am J Dermatopathol. 1999;21:1-7.
- Felty CC, Linos K. Epithelioid fibrous histiocytoma: a concise review [published correction appears in Am J Dermatopathol. 2020 Aug;42(8):628]. Am J Dermatopathol. 2019;41:879-883.
- Luzar B, Calonje E. Cutaneous fibrohistiocytic tumours—an update. Histopathology. 2010;56:148-165. doi:10.1111/j.1365-2559.2009.03447.x
- Doyle LA, Mariño-Enriquez A, Fletcher CD, et al. ALK rearrangement and overexpression in epithelioid fibrous histiocytoma. Mod Pathol. 2015;28:904-912.
- Singh Gomez C, Calonje E, Fletcher CD. Epithelioid benign fibrous histiocytoma of skin: clinico-pathological analysis of 20 cases of a poorly known variant. Histopathology. 1994;24:123-129.
- Jedrych J, Nikiforova M, Kennedy TF, et al. Epithelioid cell histiocytoma of the skin with clonal ALK gene rearrangement resulting in VCL- and SQSTM1-ALK gene fusions. Br J Dermatol. 2015;172: 1427-1429.
- Dickson BC, Swanson D, Charames GS, et al. Epithelioid fibrous histiocytoma: molecular characterization of ALK fusion partners in 23 cases. Mod Pathol. 2018;31:753-762.
- Helm M, Chang A, Fanburg-Smith JC, et al. Cutaneous VCL::ALK fusion ovoid-spindle cell neoplasm. J Cutan Pathol. 2023;50:405-409.
- Thway K, Jones RL, Noujaim J, et al. Epithelioid sarcoma: diagnostic features and genetics. Adv Anat Pathol. 2016;23:41-49.
- Bolognia JL, Jorizzo JJ, Schaffer JV et al. Dermatology, 4th ed. Philadelphia: Elsevier; 2018.
- Wiesner T, He J, Yelensky R, et al. Kinase fusions are frequent in Spitz tumours and spitzoid melanomas. Nat Commun. 2014;5:3116.
Growing Pink Nodule on the Ankle
Growing Pink Nodule on the Ankle
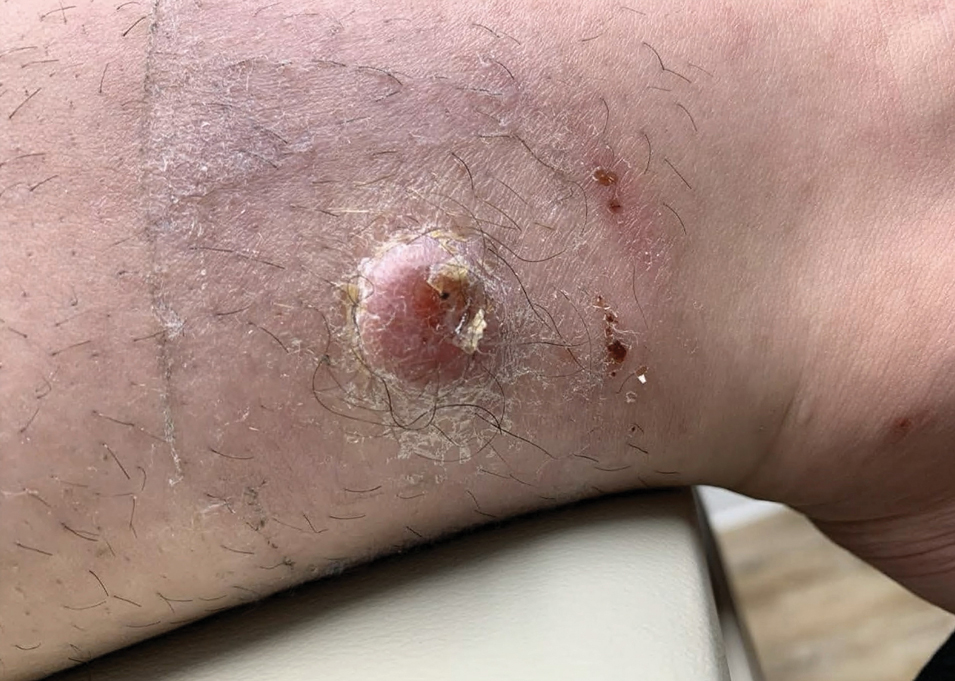
A 17-year-old girl presented to the dermatology department with a slow-growing lesion on the right lower leg that progressed in size over 1 year. The patient reported that the lesion occasionally bled but denied any other associated symptoms or a personal or family history of skin cancer. Physical examination revealed a solitary, well-circumscribed, circular, pink nodule on the right lateral upper ankle. Dermoscopy showed an amorphous mixture of pale and pink areas. A shave biopsy revealed a proliferation of epithelioid cells that diffusely stained positive for Factor XIIIa and anaplastic lymphoma kinase 1 and stained negatively for pancytokeratin, Melan A, CD34, ERG, CD31, SOX10, smooth muscle actin, desmin, and CD30. Next-generation sequencing revealed a vinculin and anaplastic lymphoma kinase gene fusion.

Painful Flesh-Colored Nodule on the Shoulder
Painful Flesh-Colored Nodule on the Shoulder
THE DIAGNOSIS: Dermatofibrosarcoma Protuberans
The histologic findings showed fascicular proliferation of relatively monomorphic spindle cells with extensive entrapment of collagen and adipocytes. Immunohistochemical staining showed that the lesional cells were diffusely positive for CD34 and negative for SOX10, S100, desmin, and factor XIIIa. The decision was made to perform cytogenetic testing with fluorescence in situ hybridization to evaluate for the presence of platelet-derived growth factor receptor beta (PDGFB) polypeptide rearrangement, a key biomarker known to be positive in most patients with dermatofibrosarcoma protuberans (DFSP).1 This rearrangement results in overproduction of PDGFB, continuous activation of platelet-derived growth factor receptor beta, cellular proliferation, and tumor formation.2 In our patient, results were positive for the PDGFB polypeptide rearrangement, which confirmed suspected diagnosis of DFSP with fibrous histiocytoma like morphology. The patient was referred for Mohs micrographic surgery for proper management.
Dermatofibrosarcoma protuberans is a rare soft-tissue tumor that involves the dermis, subcutaneous fat, and sometimes muscle and fascia.2 Dermatofibrosarcoma protuberans primarily affects young to middle-aged adults, with a slight predilection for individuals in the third to fifth decades of life.3 Lesions preferentially involve the trunk, particularly the shoulder and chest regions, and manifest as poorly circumscribed, locally aggressive mesenchymal neoplasms with a high local recurrence rate but low metastatic potential.4,5 Clinically, the lesions appear as flesh-colored, rubbery plaques or nodules. A diagnosis of DFSP requires a high index of clinical suspicion, and histologic, immunohistochemical, and molecular testing usually are required for confirmation.
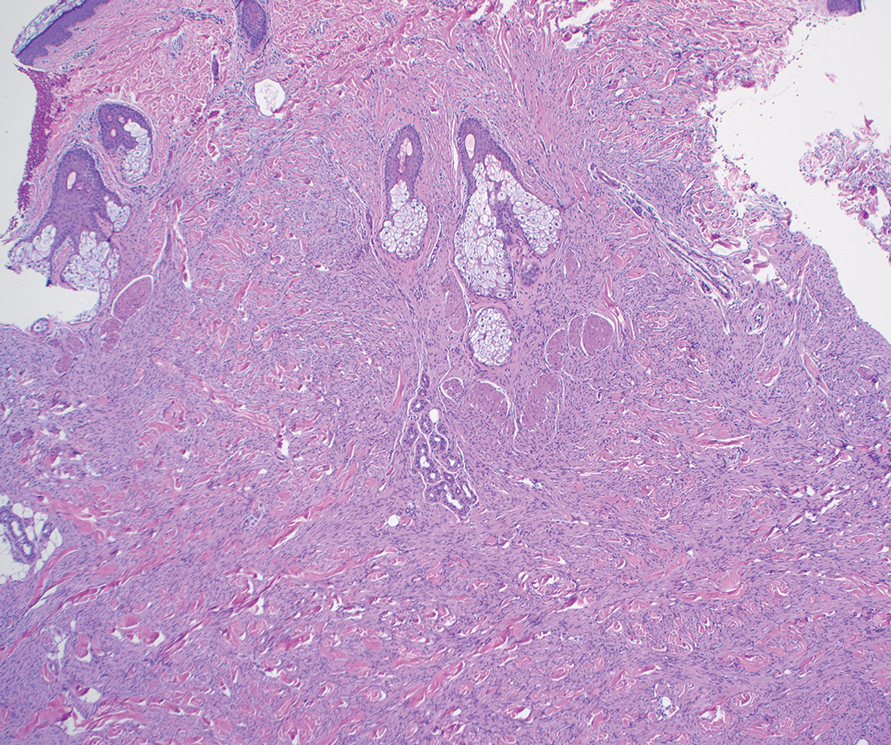
On histopathologic examination, DFSP classically demonstrates uniform, spindle-shaped cells that traditionally are arranged in an intersecting pattern and primarily are based in the dermis (Figure 1).5 Infiltration into the underlying tissue is a common feature, with neoplastic extensions causing a classic honeycomb pattern6 that also can be seen in diffuse neurofibroma and may cause diagnostic challenges; however, the immunohistology staining of neurofibroma differs from DFSP in that it stains positive for CD34, SOX-100, and S100, while DFSP has strong and diffuse CD34 immunoreactivity with negative immunostaining for SOX10, S100, desmin, and factor XIIIa.2,6
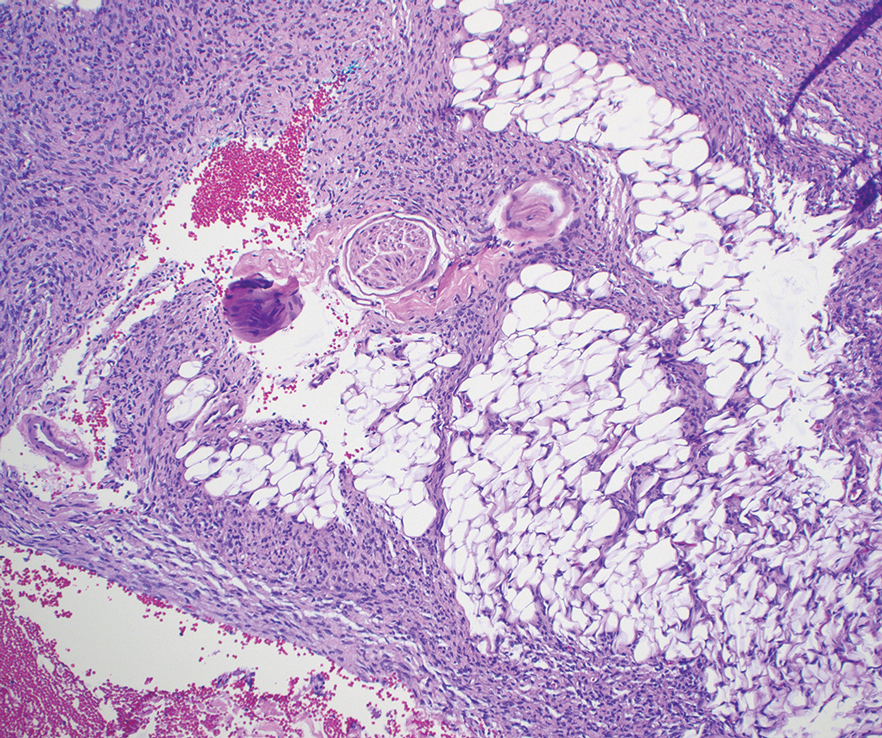
Dermatofibrosarcoma protuberans can cause considerable fat infiltration compared to other soft-tissue neoplasms, making this finding suspicious for—if not characteristic of—DFSP. Collagen trapping also can be observed; however, this is more pathognomonic in cellular fibrous histiocytoma, which is a distinct clinical variant of dermatofibromas. Due to its similarity to other lesions, histopathologic examination along with immunostaining can assist in differentiating and accurately diagnosing DFSP.6
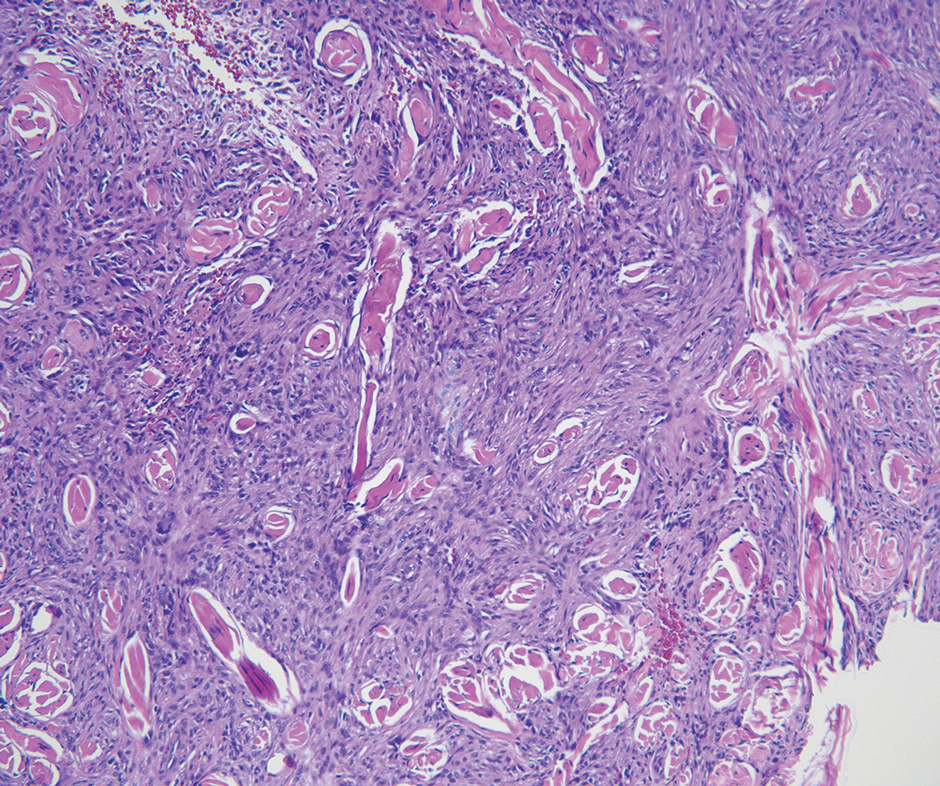
Cellular fibrous histiocytoma (CFH), a distinct clinical variant of dermatofibromas, is a benign tumor of mesenchymal origin that occurs more commonly on the trunk, arms, and legs. On histologic examination, CFH is composed of spindle-shaped cells with variable amounts of eosinophilic cytoplasm and small, oval-shaped eosinophilic nuclei and collagen trapping (Figure 2).7,8 Most CFHs occupy the superficial dermis but can extend into the deep reticular dermis, thus mimicking the honeycomb pattern seen in DFSP. This neoplasm can show a similar architecture to DFSP, which is why further investigation including cytogenetics and immunohistochemical staining can help differentiate the two conditions. Cellular fibrous histiocytoma typically stains negative for CD34 and positive for factor XIIIa.9 However, CD34 can be positive in a subset of CFHs, with a considerable subset showing peripheral CD34 positivity and a smaller subset showing central CD34 the positivity.10 This suggests that CD34 cannot be the only factor differentiating these 2 lesions in making a proper dermatopathologic diagnosis.
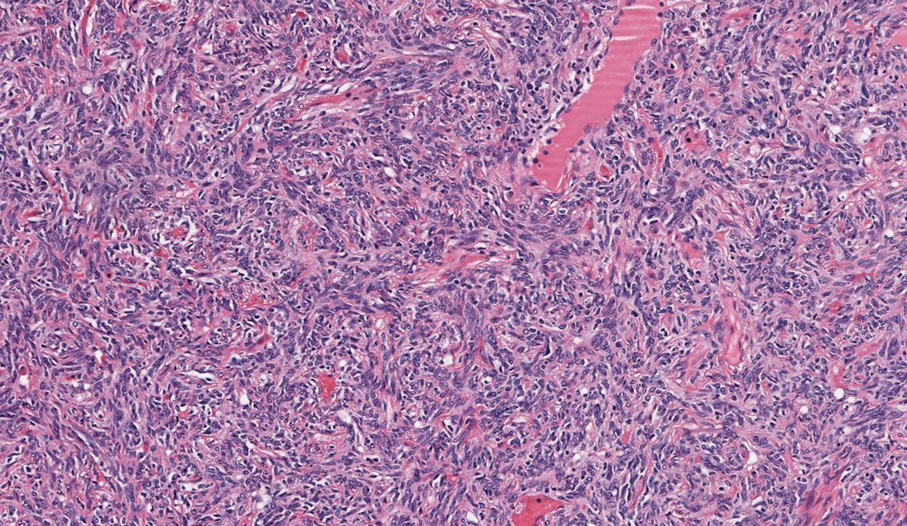
Solitary fibrous tumor (SFT) is a rare mesenchymal tumor that can occur anywhere on the body and typically manifests as a deep, painless, enlarging mass in adults aged 50 to 60 years.11 On histologic examination, SFT consists of randomly arranged cells with a spindle or ovoid shape within a collagenous stroma intermixed with blood vessels with a characteristic staghorn shape (Figure 3).11 Low-grade SFT shows a patternless arrangement with spindle cells, a low number of mitotic figures, and vessels with a staghorn appearance compared to high-grade SFT, which shows hypercellularity with nuclear pleomorphism and a high number of mitotic figures.11 Solitary fibrous tumors are positive for CD34 and STAT-6 and negative for CD31 and typically demonstrate NGFI-A binding protein 2 (NAB2)—signal transducer and activator of transcription 6 (STAT 6) gene fusion.11
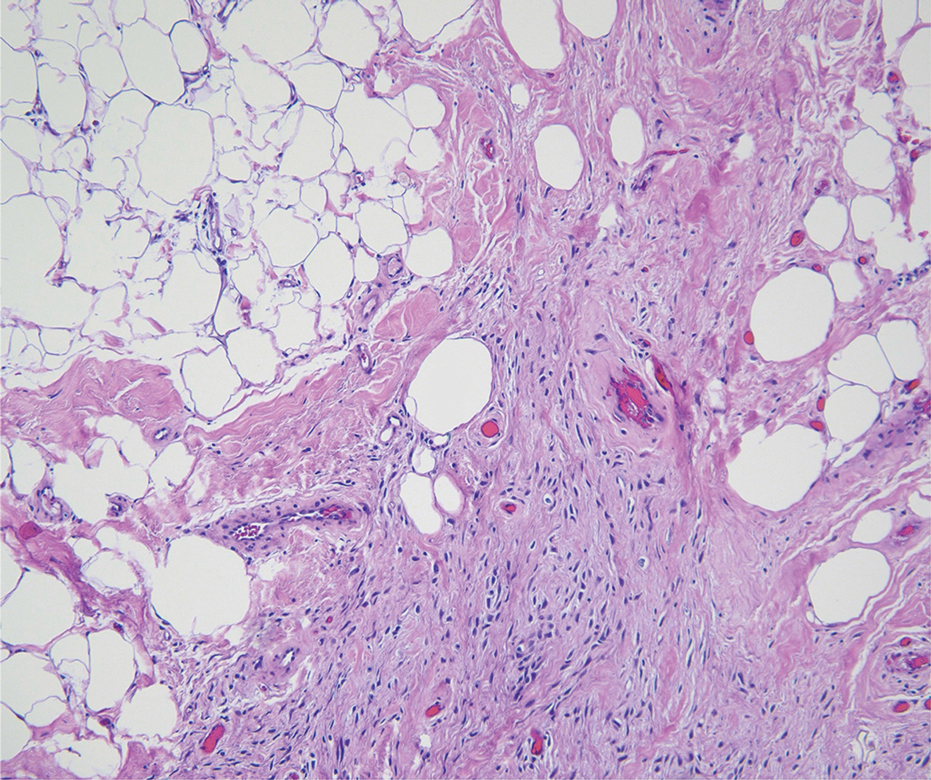
Spindle-cell lipomas are rare, benign, slow-growing, lipomatous tumors that typically manifest in men aged 40 to 70 years.12 These lesions originate most frequently in the subcutaneous tissue of the upper back, posterior neck, and shoulders. The histologic growth pattern of spindle-cell lipomas can mimic other spindle-cell and myxoid tumors, which is why cytogenetic analysis is crucial for differentiating these lesions. On histologic examination, spindle-cell lipomas exhibit a mixture of mature adipocytes, uniform spindle cells, and collagen bundles (eFigure). Spindle-cell lipoma stains positive for CD34 but negative for S100.13 In addition, spindle-cell lipomas tend to show structural rearrangements (mainly deletions) of the long arm of chromosome 13 or even losses of whole chromosome 13, which contains the retinoblastoma (RB1) gene.13
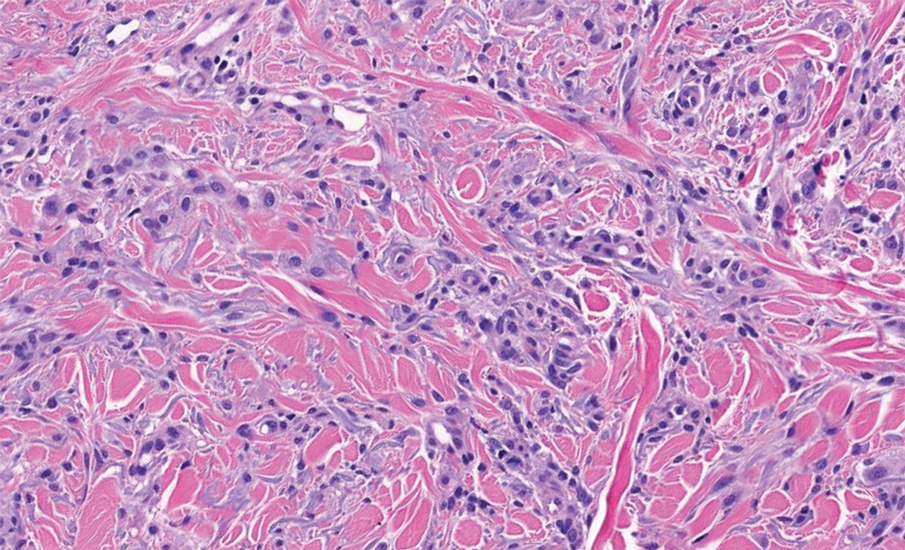
Pleomorphic dermal sarcoma is a rare mesenchymal tumor that can appear clinically and histologically similar to atypical fibroxanthoma.14 This lesion often manifests in elderly patients and is strongly associated with chronic sun exposure.15 Pleomorphic dermal sarcoma is a locally aggressive tumor with metastatic potential to the skin or lymph nodes. On histologic examination, these tumors exhibit pleomorphic atypical epithelioid or spindle cells as well as multinucleated tumor giant cells with possible tumor necrosis, lymphovascular invasion, or perineural infiltration (Figure 4). Pleomorphic dermal sarcoma, typically a diagnosis of exclusion, requires immunohistochemistry to aid in proper identification.16 These lesions stain positive for CD10 and negative for cytokeratins, desmin, HMB45, CD34, p63, p40, SOX10, and S100.15,16
- Ugurel S, Kortmann R, Mohr P, et al. S1 guidelines for dermatofibrosarcoma protuberans (DFSP)—update 2018. J Dtsch Dermatol Ges. 2019;17:663-668. doi:10.1111/ddg.13849
- Brooks J, Ramsey ML. Dermatofibrosarcoma protuberans. StatPearls Publishing; 2024. Updated April 18, 2024. Accessed April 30, 2025.
- Bowne WB, Antonescu CR, Leung DH, et al. Dermatofibrosarcoma protuberans: a clinicopathologic analysis of patients treated and followed at a single institution. Cancer. 2000;88:2711-2720.
- Lim SX, Ramaiya A, Levell NJ, et al. Review of dermatofibrosarcoma protuberans. Clin Exp Dermatol. 2022;48:297-302. doi:10.1093/ced/llac111
- Trinidad CM, Wangsiricharoen S, Prieto VG, et al. Rare variants of dermatofibrosarcoma protuberans: clinical, histologic, and molecular features and diagnostic pitfalls. Dermatopathology. 2023;10:54-62. doi:10.3390/dermatopathology10010008
- Hao X, Billings SD, Wu F, et al. Dermatofibrosarcoma protuberans: update on the diagnosis and treatment. J Clin Med. 2020;9:1752. doi:10.3390/jcm9061752
- Tsunoda K, Oikawa H, Maeda F, et al. A case of cellular fibrous histiocytoma on the right elbow with repeated relapse within a short period. Case Rep Dermatol. 2015;7:10–16. https://doi.org/10.1159/000371790
- Calonje E, Mentzel T, Fletcher CD. Cellular benign fibrous histiocytoma. Clinicopathologic analysis of 74 cases of a distinctive variant of cutaneous fibrous histiocytoma with frequent recurrence. Am J Surg Pathol. 1994;18:668-676.
- Goldblum JR, Tuthill RJ. CD34 and factor-XIIIa immunoreactivity in dermatofibrosarcoma protuberans and dermatofibroma. Am J Dermatopathology. 1997;19:147-153. doi:10.1097/00000372-199704000-00008
- Volpicelli ER, Fletcher CD. Desmin and CD34 positivity in cellular fibrous histiocytoma: an immunohistochemical analysis of 100 cases. J Cutan Pathol. 2012;39:747-752. doi:10.1111/j.1600-0560.2012.01944.x
- Martin-Broto J, Mondaza-Hernandez JL, Moura DS, et al. A comprehensive review on solitary fibrous tumor: new insights for new horizons. Cancers (Basel). 2021;13:2913. doi:10.3390/cancers13122913
- Machol JA, Cusic JG, O’Connor EA, et al. Spindle cell lipoma of the neck: review of the literature and case report. Plast Reconstr Surg Glob Open. 2015;3:E550. doi:10.1097/GOX.0000000000000405
- Domanski HA, Carlén B, Jonsson K, et al. Distinct cytologic features of spindle cell lipoma. a cytologic-histologic study with clinical, radiologic, electron microscopic, and cytogenetic correlations. Cancer. 2001;93:381-389. doi:10.1002/cncr.10142
- Devine RL, Cameron A, Holden AM, et al. The pleomorphic dermal sarcoma: its management, follow-up and the need for more guidance. Adv Oral Maxillofac Surg. 2021;2:100046. doi:10.1016 /j.adoms.2021.100046
- Seretis K, Klaroudas A, Galani V, et al. Pleomorphic dermal sarcoma: it might be rare but it exists [published online August 4, 2023]. J Surg Case Rep. doi:10.1093/jscr/rjad374
- Miller K, Goodlad JR, Brenn T. Pleomorphic dermal sarcoma. Am J Surg Pathol. 2012;36:1317-1326. doi:10.1097/pas.0b013e31825359e1
THE DIAGNOSIS: Dermatofibrosarcoma Protuberans
The histologic findings showed fascicular proliferation of relatively monomorphic spindle cells with extensive entrapment of collagen and adipocytes. Immunohistochemical staining showed that the lesional cells were diffusely positive for CD34 and negative for SOX10, S100, desmin, and factor XIIIa. The decision was made to perform cytogenetic testing with fluorescence in situ hybridization to evaluate for the presence of platelet-derived growth factor receptor beta (PDGFB) polypeptide rearrangement, a key biomarker known to be positive in most patients with dermatofibrosarcoma protuberans (DFSP).1 This rearrangement results in overproduction of PDGFB, continuous activation of platelet-derived growth factor receptor beta, cellular proliferation, and tumor formation.2 In our patient, results were positive for the PDGFB polypeptide rearrangement, which confirmed suspected diagnosis of DFSP with fibrous histiocytoma like morphology. The patient was referred for Mohs micrographic surgery for proper management.
Dermatofibrosarcoma protuberans is a rare soft-tissue tumor that involves the dermis, subcutaneous fat, and sometimes muscle and fascia.2 Dermatofibrosarcoma protuberans primarily affects young to middle-aged adults, with a slight predilection for individuals in the third to fifth decades of life.3 Lesions preferentially involve the trunk, particularly the shoulder and chest regions, and manifest as poorly circumscribed, locally aggressive mesenchymal neoplasms with a high local recurrence rate but low metastatic potential.4,5 Clinically, the lesions appear as flesh-colored, rubbery plaques or nodules. A diagnosis of DFSP requires a high index of clinical suspicion, and histologic, immunohistochemical, and molecular testing usually are required for confirmation.
On histopathologic examination, DFSP classically demonstrates uniform, spindle-shaped cells that traditionally are arranged in an intersecting pattern and primarily are based in the dermis (Figure 1).5 Infiltration into the underlying tissue is a common feature, with neoplastic extensions causing a classic honeycomb pattern6 that also can be seen in diffuse neurofibroma and may cause diagnostic challenges; however, the immunohistology staining of neurofibroma differs from DFSP in that it stains positive for CD34, SOX-100, and S100, while DFSP has strong and diffuse CD34 immunoreactivity with negative immunostaining for SOX10, S100, desmin, and factor XIIIa.2,6
Dermatofibrosarcoma protuberans can cause considerable fat infiltration compared to other soft-tissue neoplasms, making this finding suspicious for—if not characteristic of—DFSP. Collagen trapping also can be observed; however, this is more pathognomonic in cellular fibrous histiocytoma, which is a distinct clinical variant of dermatofibromas. Due to its similarity to other lesions, histopathologic examination along with immunostaining can assist in differentiating and accurately diagnosing DFSP.6
Cellular fibrous histiocytoma (CFH), a distinct clinical variant of dermatofibromas, is a benign tumor of mesenchymal origin that occurs more commonly on the trunk, arms, and legs. On histologic examination, CFH is composed of spindle-shaped cells with variable amounts of eosinophilic cytoplasm and small, oval-shaped eosinophilic nuclei and collagen trapping (Figure 2).7,8 Most CFHs occupy the superficial dermis but can extend into the deep reticular dermis, thus mimicking the honeycomb pattern seen in DFSP. This neoplasm can show a similar architecture to DFSP, which is why further investigation including cytogenetics and immunohistochemical staining can help differentiate the two conditions. Cellular fibrous histiocytoma typically stains negative for CD34 and positive for factor XIIIa.9 However, CD34 can be positive in a subset of CFHs, with a considerable subset showing peripheral CD34 positivity and a smaller subset showing central CD34 the positivity.10 This suggests that CD34 cannot be the only factor differentiating these 2 lesions in making a proper dermatopathologic diagnosis.
Solitary fibrous tumor (SFT) is a rare mesenchymal tumor that can occur anywhere on the body and typically manifests as a deep, painless, enlarging mass in adults aged 50 to 60 years.11 On histologic examination, SFT consists of randomly arranged cells with a spindle or ovoid shape within a collagenous stroma intermixed with blood vessels with a characteristic staghorn shape (Figure 3).11 Low-grade SFT shows a patternless arrangement with spindle cells, a low number of mitotic figures, and vessels with a staghorn appearance compared to high-grade SFT, which shows hypercellularity with nuclear pleomorphism and a high number of mitotic figures.11 Solitary fibrous tumors are positive for CD34 and STAT-6 and negative for CD31 and typically demonstrate NGFI-A binding protein 2 (NAB2)—signal transducer and activator of transcription 6 (STAT 6) gene fusion.11
Spindle-cell lipomas are rare, benign, slow-growing, lipomatous tumors that typically manifest in men aged 40 to 70 years.12 These lesions originate most frequently in the subcutaneous tissue of the upper back, posterior neck, and shoulders. The histologic growth pattern of spindle-cell lipomas can mimic other spindle-cell and myxoid tumors, which is why cytogenetic analysis is crucial for differentiating these lesions. On histologic examination, spindle-cell lipomas exhibit a mixture of mature adipocytes, uniform spindle cells, and collagen bundles (eFigure). Spindle-cell lipoma stains positive for CD34 but negative for S100.13 In addition, spindle-cell lipomas tend to show structural rearrangements (mainly deletions) of the long arm of chromosome 13 or even losses of whole chromosome 13, which contains the retinoblastoma (RB1) gene.13
Pleomorphic dermal sarcoma is a rare mesenchymal tumor that can appear clinically and histologically similar to atypical fibroxanthoma.14 This lesion often manifests in elderly patients and is strongly associated with chronic sun exposure.15 Pleomorphic dermal sarcoma is a locally aggressive tumor with metastatic potential to the skin or lymph nodes. On histologic examination, these tumors exhibit pleomorphic atypical epithelioid or spindle cells as well as multinucleated tumor giant cells with possible tumor necrosis, lymphovascular invasion, or perineural infiltration (Figure 4). Pleomorphic dermal sarcoma, typically a diagnosis of exclusion, requires immunohistochemistry to aid in proper identification.16 These lesions stain positive for CD10 and negative for cytokeratins, desmin, HMB45, CD34, p63, p40, SOX10, and S100.15,16
THE DIAGNOSIS: Dermatofibrosarcoma Protuberans
The histologic findings showed fascicular proliferation of relatively monomorphic spindle cells with extensive entrapment of collagen and adipocytes. Immunohistochemical staining showed that the lesional cells were diffusely positive for CD34 and negative for SOX10, S100, desmin, and factor XIIIa. The decision was made to perform cytogenetic testing with fluorescence in situ hybridization to evaluate for the presence of platelet-derived growth factor receptor beta (PDGFB) polypeptide rearrangement, a key biomarker known to be positive in most patients with dermatofibrosarcoma protuberans (DFSP).1 This rearrangement results in overproduction of PDGFB, continuous activation of platelet-derived growth factor receptor beta, cellular proliferation, and tumor formation.2 In our patient, results were positive for the PDGFB polypeptide rearrangement, which confirmed suspected diagnosis of DFSP with fibrous histiocytoma like morphology. The patient was referred for Mohs micrographic surgery for proper management.
Dermatofibrosarcoma protuberans is a rare soft-tissue tumor that involves the dermis, subcutaneous fat, and sometimes muscle and fascia.2 Dermatofibrosarcoma protuberans primarily affects young to middle-aged adults, with a slight predilection for individuals in the third to fifth decades of life.3 Lesions preferentially involve the trunk, particularly the shoulder and chest regions, and manifest as poorly circumscribed, locally aggressive mesenchymal neoplasms with a high local recurrence rate but low metastatic potential.4,5 Clinically, the lesions appear as flesh-colored, rubbery plaques or nodules. A diagnosis of DFSP requires a high index of clinical suspicion, and histologic, immunohistochemical, and molecular testing usually are required for confirmation.
On histopathologic examination, DFSP classically demonstrates uniform, spindle-shaped cells that traditionally are arranged in an intersecting pattern and primarily are based in the dermis (Figure 1).5 Infiltration into the underlying tissue is a common feature, with neoplastic extensions causing a classic honeycomb pattern6 that also can be seen in diffuse neurofibroma and may cause diagnostic challenges; however, the immunohistology staining of neurofibroma differs from DFSP in that it stains positive for CD34, SOX-100, and S100, while DFSP has strong and diffuse CD34 immunoreactivity with negative immunostaining for SOX10, S100, desmin, and factor XIIIa.2,6
Dermatofibrosarcoma protuberans can cause considerable fat infiltration compared to other soft-tissue neoplasms, making this finding suspicious for—if not characteristic of—DFSP. Collagen trapping also can be observed; however, this is more pathognomonic in cellular fibrous histiocytoma, which is a distinct clinical variant of dermatofibromas. Due to its similarity to other lesions, histopathologic examination along with immunostaining can assist in differentiating and accurately diagnosing DFSP.6
Cellular fibrous histiocytoma (CFH), a distinct clinical variant of dermatofibromas, is a benign tumor of mesenchymal origin that occurs more commonly on the trunk, arms, and legs. On histologic examination, CFH is composed of spindle-shaped cells with variable amounts of eosinophilic cytoplasm and small, oval-shaped eosinophilic nuclei and collagen trapping (Figure 2).7,8 Most CFHs occupy the superficial dermis but can extend into the deep reticular dermis, thus mimicking the honeycomb pattern seen in DFSP. This neoplasm can show a similar architecture to DFSP, which is why further investigation including cytogenetics and immunohistochemical staining can help differentiate the two conditions. Cellular fibrous histiocytoma typically stains negative for CD34 and positive for factor XIIIa.9 However, CD34 can be positive in a subset of CFHs, with a considerable subset showing peripheral CD34 positivity and a smaller subset showing central CD34 the positivity.10 This suggests that CD34 cannot be the only factor differentiating these 2 lesions in making a proper dermatopathologic diagnosis.
Solitary fibrous tumor (SFT) is a rare mesenchymal tumor that can occur anywhere on the body and typically manifests as a deep, painless, enlarging mass in adults aged 50 to 60 years.11 On histologic examination, SFT consists of randomly arranged cells with a spindle or ovoid shape within a collagenous stroma intermixed with blood vessels with a characteristic staghorn shape (Figure 3).11 Low-grade SFT shows a patternless arrangement with spindle cells, a low number of mitotic figures, and vessels with a staghorn appearance compared to high-grade SFT, which shows hypercellularity with nuclear pleomorphism and a high number of mitotic figures.11 Solitary fibrous tumors are positive for CD34 and STAT-6 and negative for CD31 and typically demonstrate NGFI-A binding protein 2 (NAB2)—signal transducer and activator of transcription 6 (STAT 6) gene fusion.11
Spindle-cell lipomas are rare, benign, slow-growing, lipomatous tumors that typically manifest in men aged 40 to 70 years.12 These lesions originate most frequently in the subcutaneous tissue of the upper back, posterior neck, and shoulders. The histologic growth pattern of spindle-cell lipomas can mimic other spindle-cell and myxoid tumors, which is why cytogenetic analysis is crucial for differentiating these lesions. On histologic examination, spindle-cell lipomas exhibit a mixture of mature adipocytes, uniform spindle cells, and collagen bundles (eFigure). Spindle-cell lipoma stains positive for CD34 but negative for S100.13 In addition, spindle-cell lipomas tend to show structural rearrangements (mainly deletions) of the long arm of chromosome 13 or even losses of whole chromosome 13, which contains the retinoblastoma (RB1) gene.13
Pleomorphic dermal sarcoma is a rare mesenchymal tumor that can appear clinically and histologically similar to atypical fibroxanthoma.14 This lesion often manifests in elderly patients and is strongly associated with chronic sun exposure.15 Pleomorphic dermal sarcoma is a locally aggressive tumor with metastatic potential to the skin or lymph nodes. On histologic examination, these tumors exhibit pleomorphic atypical epithelioid or spindle cells as well as multinucleated tumor giant cells with possible tumor necrosis, lymphovascular invasion, or perineural infiltration (Figure 4). Pleomorphic dermal sarcoma, typically a diagnosis of exclusion, requires immunohistochemistry to aid in proper identification.16 These lesions stain positive for CD10 and negative for cytokeratins, desmin, HMB45, CD34, p63, p40, SOX10, and S100.15,16
- Ugurel S, Kortmann R, Mohr P, et al. S1 guidelines for dermatofibrosarcoma protuberans (DFSP)—update 2018. J Dtsch Dermatol Ges. 2019;17:663-668. doi:10.1111/ddg.13849
- Brooks J, Ramsey ML. Dermatofibrosarcoma protuberans. StatPearls Publishing; 2024. Updated April 18, 2024. Accessed April 30, 2025.
- Bowne WB, Antonescu CR, Leung DH, et al. Dermatofibrosarcoma protuberans: a clinicopathologic analysis of patients treated and followed at a single institution. Cancer. 2000;88:2711-2720.
- Lim SX, Ramaiya A, Levell NJ, et al. Review of dermatofibrosarcoma protuberans. Clin Exp Dermatol. 2022;48:297-302. doi:10.1093/ced/llac111
- Trinidad CM, Wangsiricharoen S, Prieto VG, et al. Rare variants of dermatofibrosarcoma protuberans: clinical, histologic, and molecular features and diagnostic pitfalls. Dermatopathology. 2023;10:54-62. doi:10.3390/dermatopathology10010008
- Hao X, Billings SD, Wu F, et al. Dermatofibrosarcoma protuberans: update on the diagnosis and treatment. J Clin Med. 2020;9:1752. doi:10.3390/jcm9061752
- Tsunoda K, Oikawa H, Maeda F, et al. A case of cellular fibrous histiocytoma on the right elbow with repeated relapse within a short period. Case Rep Dermatol. 2015;7:10–16. https://doi.org/10.1159/000371790
- Calonje E, Mentzel T, Fletcher CD. Cellular benign fibrous histiocytoma. Clinicopathologic analysis of 74 cases of a distinctive variant of cutaneous fibrous histiocytoma with frequent recurrence. Am J Surg Pathol. 1994;18:668-676.
- Goldblum JR, Tuthill RJ. CD34 and factor-XIIIa immunoreactivity in dermatofibrosarcoma protuberans and dermatofibroma. Am J Dermatopathology. 1997;19:147-153. doi:10.1097/00000372-199704000-00008
- Volpicelli ER, Fletcher CD. Desmin and CD34 positivity in cellular fibrous histiocytoma: an immunohistochemical analysis of 100 cases. J Cutan Pathol. 2012;39:747-752. doi:10.1111/j.1600-0560.2012.01944.x
- Martin-Broto J, Mondaza-Hernandez JL, Moura DS, et al. A comprehensive review on solitary fibrous tumor: new insights for new horizons. Cancers (Basel). 2021;13:2913. doi:10.3390/cancers13122913
- Machol JA, Cusic JG, O’Connor EA, et al. Spindle cell lipoma of the neck: review of the literature and case report. Plast Reconstr Surg Glob Open. 2015;3:E550. doi:10.1097/GOX.0000000000000405
- Domanski HA, Carlén B, Jonsson K, et al. Distinct cytologic features of spindle cell lipoma. a cytologic-histologic study with clinical, radiologic, electron microscopic, and cytogenetic correlations. Cancer. 2001;93:381-389. doi:10.1002/cncr.10142
- Devine RL, Cameron A, Holden AM, et al. The pleomorphic dermal sarcoma: its management, follow-up and the need for more guidance. Adv Oral Maxillofac Surg. 2021;2:100046. doi:10.1016 /j.adoms.2021.100046
- Seretis K, Klaroudas A, Galani V, et al. Pleomorphic dermal sarcoma: it might be rare but it exists [published online August 4, 2023]. J Surg Case Rep. doi:10.1093/jscr/rjad374
- Miller K, Goodlad JR, Brenn T. Pleomorphic dermal sarcoma. Am J Surg Pathol. 2012;36:1317-1326. doi:10.1097/pas.0b013e31825359e1
- Ugurel S, Kortmann R, Mohr P, et al. S1 guidelines for dermatofibrosarcoma protuberans (DFSP)—update 2018. J Dtsch Dermatol Ges. 2019;17:663-668. doi:10.1111/ddg.13849
- Brooks J, Ramsey ML. Dermatofibrosarcoma protuberans. StatPearls Publishing; 2024. Updated April 18, 2024. Accessed April 30, 2025.
- Bowne WB, Antonescu CR, Leung DH, et al. Dermatofibrosarcoma protuberans: a clinicopathologic analysis of patients treated and followed at a single institution. Cancer. 2000;88:2711-2720.
- Lim SX, Ramaiya A, Levell NJ, et al. Review of dermatofibrosarcoma protuberans. Clin Exp Dermatol. 2022;48:297-302. doi:10.1093/ced/llac111
- Trinidad CM, Wangsiricharoen S, Prieto VG, et al. Rare variants of dermatofibrosarcoma protuberans: clinical, histologic, and molecular features and diagnostic pitfalls. Dermatopathology. 2023;10:54-62. doi:10.3390/dermatopathology10010008
- Hao X, Billings SD, Wu F, et al. Dermatofibrosarcoma protuberans: update on the diagnosis and treatment. J Clin Med. 2020;9:1752. doi:10.3390/jcm9061752
- Tsunoda K, Oikawa H, Maeda F, et al. A case of cellular fibrous histiocytoma on the right elbow with repeated relapse within a short period. Case Rep Dermatol. 2015;7:10–16. https://doi.org/10.1159/000371790
- Calonje E, Mentzel T, Fletcher CD. Cellular benign fibrous histiocytoma. Clinicopathologic analysis of 74 cases of a distinctive variant of cutaneous fibrous histiocytoma with frequent recurrence. Am J Surg Pathol. 1994;18:668-676.
- Goldblum JR, Tuthill RJ. CD34 and factor-XIIIa immunoreactivity in dermatofibrosarcoma protuberans and dermatofibroma. Am J Dermatopathology. 1997;19:147-153. doi:10.1097/00000372-199704000-00008
- Volpicelli ER, Fletcher CD. Desmin and CD34 positivity in cellular fibrous histiocytoma: an immunohistochemical analysis of 100 cases. J Cutan Pathol. 2012;39:747-752. doi:10.1111/j.1600-0560.2012.01944.x
- Martin-Broto J, Mondaza-Hernandez JL, Moura DS, et al. A comprehensive review on solitary fibrous tumor: new insights for new horizons. Cancers (Basel). 2021;13:2913. doi:10.3390/cancers13122913
- Machol JA, Cusic JG, O’Connor EA, et al. Spindle cell lipoma of the neck: review of the literature and case report. Plast Reconstr Surg Glob Open. 2015;3:E550. doi:10.1097/GOX.0000000000000405
- Domanski HA, Carlén B, Jonsson K, et al. Distinct cytologic features of spindle cell lipoma. a cytologic-histologic study with clinical, radiologic, electron microscopic, and cytogenetic correlations. Cancer. 2001;93:381-389. doi:10.1002/cncr.10142
- Devine RL, Cameron A, Holden AM, et al. The pleomorphic dermal sarcoma: its management, follow-up and the need for more guidance. Adv Oral Maxillofac Surg. 2021;2:100046. doi:10.1016 /j.adoms.2021.100046
- Seretis K, Klaroudas A, Galani V, et al. Pleomorphic dermal sarcoma: it might be rare but it exists [published online August 4, 2023]. J Surg Case Rep. doi:10.1093/jscr/rjad374
- Miller K, Goodlad JR, Brenn T. Pleomorphic dermal sarcoma. Am J Surg Pathol. 2012;36:1317-1326. doi:10.1097/pas.0b013e31825359e1
Painful Flesh-Colored Nodule on the Shoulder
Painful Flesh-Colored Nodule on the Shoulder
A 26-year-old man with no notable medical history presented to the dermatology clinic with an inconspicuous, painful, raised lesion on the right posterior shoulder of 6 months’ duration. The patient reported that the lesion was tender to light palpation and bothersome in his daily activities. Physical examination revealed a firm, flesh-colored, 1.8-cm nodule with no erythema or pigmentation on the right shoulder. An elliptical excisional biopsy was performed and submitted for histologic evaluation.

Multiple Firm Papules on the Wrists and Forearms
Multiple Firm Papules on the Wrists and Forearms
THE DIAGNOSIS: Acral Persistent Papular Mucinosis
Histopathologic analysis revealed conspicuous interstitial mucin deposition throughout the upper to mid reticular dermis in the absence of a cellular infiltrate or fibroplasia. Colloidal iron staining confirmed the presence of mucin. In correlation with the clinical presentation, a diagnosis of acral persistent papular mucinosis (APPM) was made. The patient was counseled on the benign disease course and lack of associated comorbidities, and additional treatment was not pursued.
Acral persistent papular mucinosis is a rare distinct subtype of cutaneous mucinosis that initially was described by Rongioletti et al1 in 1986. As a localized form of lichen myxedematosus, APPM is characterized by mucin deposition in the dermis with no systemic involvement. The precise pathogenesis remains unclear, although some investigators have suggested that cytokine-mediated stimulation of glycosaminoglycan production may contribute to increased mucin accumulation in the dermis.2 Acral persistent papular mucinosis predominantly affects middle-aged women with a 5:1 female-to-male predominance.3 Clinically, patients present with discrete, nonfollicular, waxy papules that typically measure 2 to 5 mm and are distributed symmetrically on the extensor surfaces of the wrists and forearms. While the lesions generally are asymptomatic, some patients may report mild pruritus. The condition is chronic, with lesions seldom resolving and often increasing in number over time.3
Histologically, APPM is characterized by focal deposits of mucin in the upper reticular dermis with no evidence of increased fibroblast proliferation or fibrosis.4 This feature is pivotal in differentiating APPM from other subtypes of localized lichen myxedematosus and similar dermatoses. Diagnosis of APPM requires exclusion of systemic involvement, including thyroid abnormalities and monoclonal gammopathy, aligning with its classification as a purely cutaneous condition.5 Management of APPM is unclear due to its rarity. Reassurance for patients of its benign nature as well as clinical observation are recommended, though some reports cite benefits of treatment with topical corticosteroids or calcineurin inhibitors.6,7 The long-term prognosis for patients with APPM is favorable, although the persistence of and potential increase in lesions over time can be a cosmetic concern.
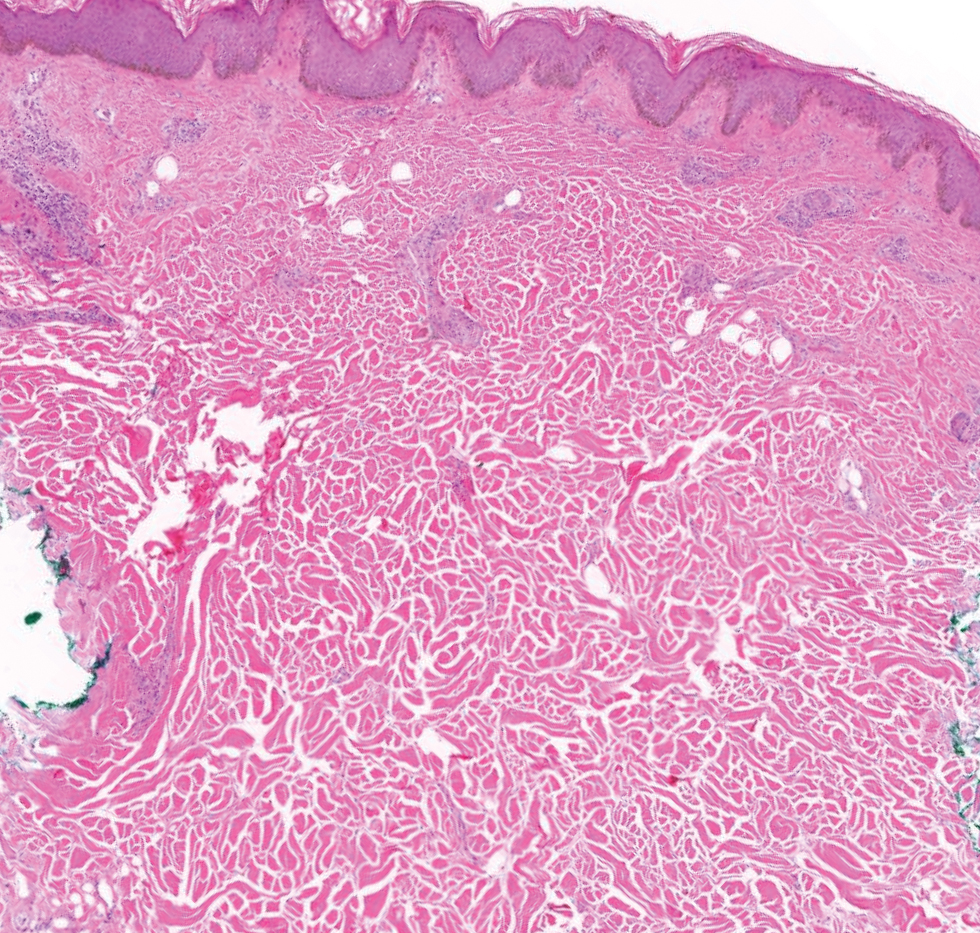
The differential diagnoses for APPM include scleromyxedema, scleredema, and other cutaneous eruptions that manifest as smooth flesh-colored papules, such as granuloma annulare and lichen nitidus.3 Scleromyxedema is a systemic cutaneous mucinosis that is part of the same disease spectrum as lichen myxedematosus. The papular eruption of scleromyxedema is much more widespread, and coalescing of the lesions may lead to characteristic skin thickening, creating leonine facies and deep furrowing over the trunk.8 Extracutaneous manifestations are frequent in scleromyxedema, and up to 90% of patients exhibit evidence of an underlying plasma cell dyscrasia.2 Histopathologically, scleromyxedema shows extensive fibroblast proliferation and fibrosis, in contrast to the findings of APPM (Figure 1).
The histopathology of APPM is most similar to scleredema, a rare fibromucinous disorder of the skin associated with diabetes, infection (especially poststreptococcal), or monoclonal gammopathy.9 Biopsy evaluation of scleredema reveals a normal epidermis with mucin deposition between collagen bundles predominantly in the deep reticular dermis as well as absent fibroblast proliferation (Figure 2). Unlike APPM, scleredema manifests with diffuse woody induration with erythema and hyperpigmentation on the posterior neck and upper back.9 On physical examination, the distinct clinical features of scleredema distinguish this condition from APPM and scleromyxedema.
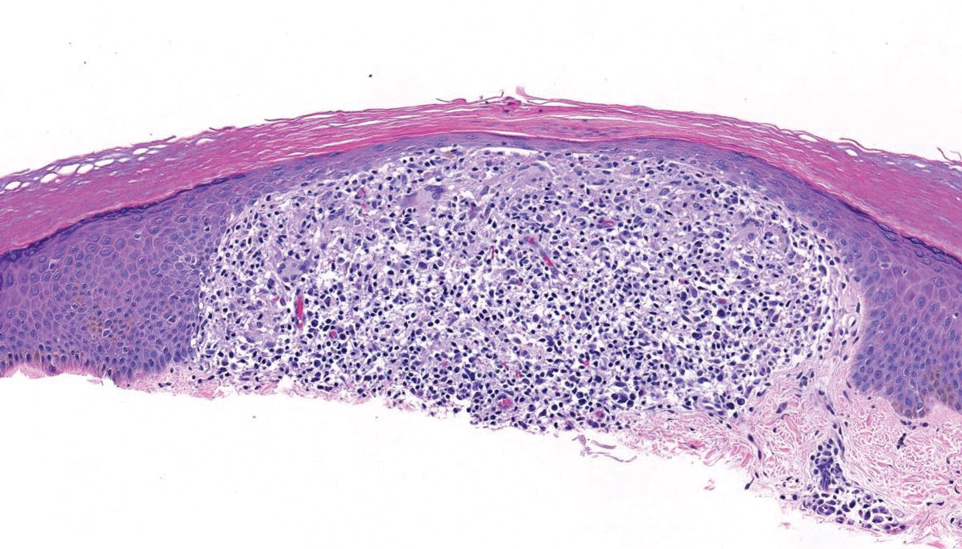
Papular granuloma annulare also was considered in our patient due to the presence of small flesh-colored papules. Histologically, granuloma annulare is characterized by palisading granulomas and mucin deposition in the dermis.10 However, the pattern of mucin deposition differs from that seen in APPM. In granuloma annulare, mucin is observed around foci of degenerated collagen (Figure 3), which was not observed in our patient.10 Additionally, the absence of an inflammatory infiltrate in our patient further ruled out this diagnosis.
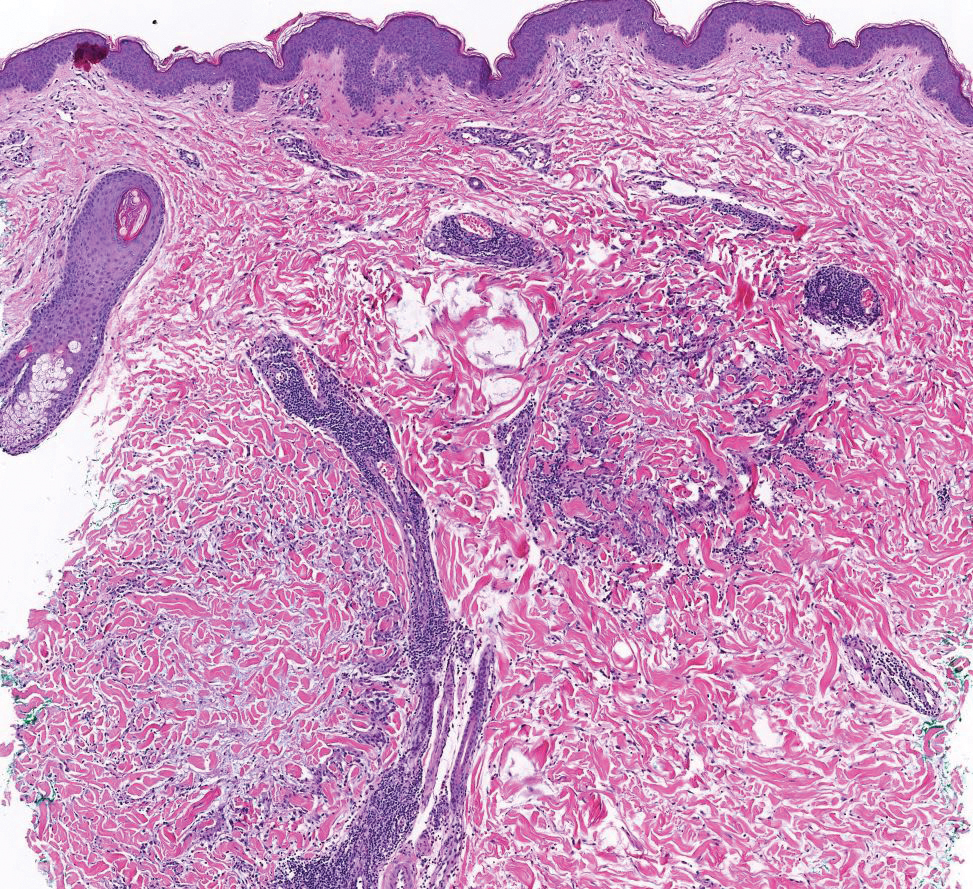
Lichen nitidus also could be considered in the differential diagnosis for ACCM. It typically manifests with minute, clustered, monomorphous papules with a predilection for the chest, abdomen, flexural forearms, and genitalia. The histology of lichen nitidus is distinct, showing a well-circumscribed lymphohistiocytic infiltrate in the papillary dermis bordered by epidermal ridges, resembling a ball and clutch appearance (Figure 4).11
Although the clinical differential diagnosis in our patient was broad, histopathologic evaluation played a crucial role in confirming the diagnosis of APPM. This benign condition could be overlooked by patients and physicians; thorough clinical evaluation is necessary to rule out systemic mucinoses, which are associated with higher risks of morbidity and mortality.
- Rongioletti F, Rebora A. Acral persistent papular mucinosis: a new entity. Arch Dermatol. 1986;122:1237-1239. doi:10.1001 /archderm.1986.01660230027002
- Christman MP, Sukhdeo K, Kim RH, et al. Papular mucinosis, or localized lichen myxedematosus (LM)(discrete papular type). Dermatol Online J. 2017;23:13030/qt3xp109qd.
- Rongioletti F, Ferreli C, Atzori L. Acral persistent papular mucinosis. Clin Dermatol. 2021;39:211-214. doi:10.1016/j.clindermatol.2020.10.001
- Rongioletti F, Rebora A. Cutaneous mucinoses: microscopic criteria for diagnosis. Am J Dermatopathol. 2001;23:257-267. doi:10.1097/00000372- 200106000-00022
- Rongioletti F. Lichen myxedematosus (papular mucinosis): new concepts and perspectives for an old disease. Semin Cutan Med Surg. 2006;25:100-104. doi:10.1016/j.sder.2006.04.001
- Jun JY, Oh SH, Shim JH, et al. Acral persistent papular mucinosis with partial response to tacrolimus ointment. Ann Dermatol. 2016;28:517-519. doi:10.5021/ad.2016.28.4.517
- Rongioletti F, Zaccaria E, Cozzani E, et al. Treatment of localized lichen myxedematosus of discrete type with tacrolimus ointment. J Am Acad Dermatol. 2008;58:530-532. doi:10.1016/j.jaad.2006.10.021
- Rongioletti F, Merlo G, Cinotti E, et al. Scleromyxedema: a multicenter study of characteristics, comorbidities, course, and therapy in 30 patients. J Am Acad Dermatol. 2013;69:66-72. doi:10.1016 /j.jaad.2013.01.007
- Rongioletti F, Kaiser F, Cinotti E, et al. Scleredema. a multicentre study of characteristics, comorbidities, course and therapy in 44 patients. J Eur Acad Dermatol Venereol. 2015;29:2399-2404. doi:10.1111/jdv.13272
- Piette EW, Rosenbach M. Granuloma annulare: clinical and histologic variants, epidemiology, and genetics. J Am Acad Dermatol. 2016;75:457-465. doi:10.1016/j.jaad.2015.03.054
- Al-Mutairi N, Hassanein A, Nour-Eldin O, et al. Generalized lichen nitidus. Pediatr Dermatol. 2005;22:158-160. doi:10.1111 /j.1525-1470.2005.22215.x
THE DIAGNOSIS: Acral Persistent Papular Mucinosis
Histopathologic analysis revealed conspicuous interstitial mucin deposition throughout the upper to mid reticular dermis in the absence of a cellular infiltrate or fibroplasia. Colloidal iron staining confirmed the presence of mucin. In correlation with the clinical presentation, a diagnosis of acral persistent papular mucinosis (APPM) was made. The patient was counseled on the benign disease course and lack of associated comorbidities, and additional treatment was not pursued.
Acral persistent papular mucinosis is a rare distinct subtype of cutaneous mucinosis that initially was described by Rongioletti et al1 in 1986. As a localized form of lichen myxedematosus, APPM is characterized by mucin deposition in the dermis with no systemic involvement. The precise pathogenesis remains unclear, although some investigators have suggested that cytokine-mediated stimulation of glycosaminoglycan production may contribute to increased mucin accumulation in the dermis.2 Acral persistent papular mucinosis predominantly affects middle-aged women with a 5:1 female-to-male predominance.3 Clinically, patients present with discrete, nonfollicular, waxy papules that typically measure 2 to 5 mm and are distributed symmetrically on the extensor surfaces of the wrists and forearms. While the lesions generally are asymptomatic, some patients may report mild pruritus. The condition is chronic, with lesions seldom resolving and often increasing in number over time.3
Histologically, APPM is characterized by focal deposits of mucin in the upper reticular dermis with no evidence of increased fibroblast proliferation or fibrosis.4 This feature is pivotal in differentiating APPM from other subtypes of localized lichen myxedematosus and similar dermatoses. Diagnosis of APPM requires exclusion of systemic involvement, including thyroid abnormalities and monoclonal gammopathy, aligning with its classification as a purely cutaneous condition.5 Management of APPM is unclear due to its rarity. Reassurance for patients of its benign nature as well as clinical observation are recommended, though some reports cite benefits of treatment with topical corticosteroids or calcineurin inhibitors.6,7 The long-term prognosis for patients with APPM is favorable, although the persistence of and potential increase in lesions over time can be a cosmetic concern.
The differential diagnoses for APPM include scleromyxedema, scleredema, and other cutaneous eruptions that manifest as smooth flesh-colored papules, such as granuloma annulare and lichen nitidus.3 Scleromyxedema is a systemic cutaneous mucinosis that is part of the same disease spectrum as lichen myxedematosus. The papular eruption of scleromyxedema is much more widespread, and coalescing of the lesions may lead to characteristic skin thickening, creating leonine facies and deep furrowing over the trunk.8 Extracutaneous manifestations are frequent in scleromyxedema, and up to 90% of patients exhibit evidence of an underlying plasma cell dyscrasia.2 Histopathologically, scleromyxedema shows extensive fibroblast proliferation and fibrosis, in contrast to the findings of APPM (Figure 1).
The histopathology of APPM is most similar to scleredema, a rare fibromucinous disorder of the skin associated with diabetes, infection (especially poststreptococcal), or monoclonal gammopathy.9 Biopsy evaluation of scleredema reveals a normal epidermis with mucin deposition between collagen bundles predominantly in the deep reticular dermis as well as absent fibroblast proliferation (Figure 2). Unlike APPM, scleredema manifests with diffuse woody induration with erythema and hyperpigmentation on the posterior neck and upper back.9 On physical examination, the distinct clinical features of scleredema distinguish this condition from APPM and scleromyxedema.
Papular granuloma annulare also was considered in our patient due to the presence of small flesh-colored papules. Histologically, granuloma annulare is characterized by palisading granulomas and mucin deposition in the dermis.10 However, the pattern of mucin deposition differs from that seen in APPM. In granuloma annulare, mucin is observed around foci of degenerated collagen (Figure 3), which was not observed in our patient.10 Additionally, the absence of an inflammatory infiltrate in our patient further ruled out this diagnosis.
Lichen nitidus also could be considered in the differential diagnosis for ACCM. It typically manifests with minute, clustered, monomorphous papules with a predilection for the chest, abdomen, flexural forearms, and genitalia. The histology of lichen nitidus is distinct, showing a well-circumscribed lymphohistiocytic infiltrate in the papillary dermis bordered by epidermal ridges, resembling a ball and clutch appearance (Figure 4).11
Although the clinical differential diagnosis in our patient was broad, histopathologic evaluation played a crucial role in confirming the diagnosis of APPM. This benign condition could be overlooked by patients and physicians; thorough clinical evaluation is necessary to rule out systemic mucinoses, which are associated with higher risks of morbidity and mortality.
THE DIAGNOSIS: Acral Persistent Papular Mucinosis
Histopathologic analysis revealed conspicuous interstitial mucin deposition throughout the upper to mid reticular dermis in the absence of a cellular infiltrate or fibroplasia. Colloidal iron staining confirmed the presence of mucin. In correlation with the clinical presentation, a diagnosis of acral persistent papular mucinosis (APPM) was made. The patient was counseled on the benign disease course and lack of associated comorbidities, and additional treatment was not pursued.
Acral persistent papular mucinosis is a rare distinct subtype of cutaneous mucinosis that initially was described by Rongioletti et al1 in 1986. As a localized form of lichen myxedematosus, APPM is characterized by mucin deposition in the dermis with no systemic involvement. The precise pathogenesis remains unclear, although some investigators have suggested that cytokine-mediated stimulation of glycosaminoglycan production may contribute to increased mucin accumulation in the dermis.2 Acral persistent papular mucinosis predominantly affects middle-aged women with a 5:1 female-to-male predominance.3 Clinically, patients present with discrete, nonfollicular, waxy papules that typically measure 2 to 5 mm and are distributed symmetrically on the extensor surfaces of the wrists and forearms. While the lesions generally are asymptomatic, some patients may report mild pruritus. The condition is chronic, with lesions seldom resolving and often increasing in number over time.3
Histologically, APPM is characterized by focal deposits of mucin in the upper reticular dermis with no evidence of increased fibroblast proliferation or fibrosis.4 This feature is pivotal in differentiating APPM from other subtypes of localized lichen myxedematosus and similar dermatoses. Diagnosis of APPM requires exclusion of systemic involvement, including thyroid abnormalities and monoclonal gammopathy, aligning with its classification as a purely cutaneous condition.5 Management of APPM is unclear due to its rarity. Reassurance for patients of its benign nature as well as clinical observation are recommended, though some reports cite benefits of treatment with topical corticosteroids or calcineurin inhibitors.6,7 The long-term prognosis for patients with APPM is favorable, although the persistence of and potential increase in lesions over time can be a cosmetic concern.
The differential diagnoses for APPM include scleromyxedema, scleredema, and other cutaneous eruptions that manifest as smooth flesh-colored papules, such as granuloma annulare and lichen nitidus.3 Scleromyxedema is a systemic cutaneous mucinosis that is part of the same disease spectrum as lichen myxedematosus. The papular eruption of scleromyxedema is much more widespread, and coalescing of the lesions may lead to characteristic skin thickening, creating leonine facies and deep furrowing over the trunk.8 Extracutaneous manifestations are frequent in scleromyxedema, and up to 90% of patients exhibit evidence of an underlying plasma cell dyscrasia.2 Histopathologically, scleromyxedema shows extensive fibroblast proliferation and fibrosis, in contrast to the findings of APPM (Figure 1).
The histopathology of APPM is most similar to scleredema, a rare fibromucinous disorder of the skin associated with diabetes, infection (especially poststreptococcal), or monoclonal gammopathy.9 Biopsy evaluation of scleredema reveals a normal epidermis with mucin deposition between collagen bundles predominantly in the deep reticular dermis as well as absent fibroblast proliferation (Figure 2). Unlike APPM, scleredema manifests with diffuse woody induration with erythema and hyperpigmentation on the posterior neck and upper back.9 On physical examination, the distinct clinical features of scleredema distinguish this condition from APPM and scleromyxedema.
Papular granuloma annulare also was considered in our patient due to the presence of small flesh-colored papules. Histologically, granuloma annulare is characterized by palisading granulomas and mucin deposition in the dermis.10 However, the pattern of mucin deposition differs from that seen in APPM. In granuloma annulare, mucin is observed around foci of degenerated collagen (Figure 3), which was not observed in our patient.10 Additionally, the absence of an inflammatory infiltrate in our patient further ruled out this diagnosis.
Lichen nitidus also could be considered in the differential diagnosis for ACCM. It typically manifests with minute, clustered, monomorphous papules with a predilection for the chest, abdomen, flexural forearms, and genitalia. The histology of lichen nitidus is distinct, showing a well-circumscribed lymphohistiocytic infiltrate in the papillary dermis bordered by epidermal ridges, resembling a ball and clutch appearance (Figure 4).11
Although the clinical differential diagnosis in our patient was broad, histopathologic evaluation played a crucial role in confirming the diagnosis of APPM. This benign condition could be overlooked by patients and physicians; thorough clinical evaluation is necessary to rule out systemic mucinoses, which are associated with higher risks of morbidity and mortality.
- Rongioletti F, Rebora A. Acral persistent papular mucinosis: a new entity. Arch Dermatol. 1986;122:1237-1239. doi:10.1001 /archderm.1986.01660230027002
- Christman MP, Sukhdeo K, Kim RH, et al. Papular mucinosis, or localized lichen myxedematosus (LM)(discrete papular type). Dermatol Online J. 2017;23:13030/qt3xp109qd.
- Rongioletti F, Ferreli C, Atzori L. Acral persistent papular mucinosis. Clin Dermatol. 2021;39:211-214. doi:10.1016/j.clindermatol.2020.10.001
- Rongioletti F, Rebora A. Cutaneous mucinoses: microscopic criteria for diagnosis. Am J Dermatopathol. 2001;23:257-267. doi:10.1097/00000372- 200106000-00022
- Rongioletti F. Lichen myxedematosus (papular mucinosis): new concepts and perspectives for an old disease. Semin Cutan Med Surg. 2006;25:100-104. doi:10.1016/j.sder.2006.04.001
- Jun JY, Oh SH, Shim JH, et al. Acral persistent papular mucinosis with partial response to tacrolimus ointment. Ann Dermatol. 2016;28:517-519. doi:10.5021/ad.2016.28.4.517
- Rongioletti F, Zaccaria E, Cozzani E, et al. Treatment of localized lichen myxedematosus of discrete type with tacrolimus ointment. J Am Acad Dermatol. 2008;58:530-532. doi:10.1016/j.jaad.2006.10.021
- Rongioletti F, Merlo G, Cinotti E, et al. Scleromyxedema: a multicenter study of characteristics, comorbidities, course, and therapy in 30 patients. J Am Acad Dermatol. 2013;69:66-72. doi:10.1016 /j.jaad.2013.01.007
- Rongioletti F, Kaiser F, Cinotti E, et al. Scleredema. a multicentre study of characteristics, comorbidities, course and therapy in 44 patients. J Eur Acad Dermatol Venereol. 2015;29:2399-2404. doi:10.1111/jdv.13272
- Piette EW, Rosenbach M. Granuloma annulare: clinical and histologic variants, epidemiology, and genetics. J Am Acad Dermatol. 2016;75:457-465. doi:10.1016/j.jaad.2015.03.054
- Al-Mutairi N, Hassanein A, Nour-Eldin O, et al. Generalized lichen nitidus. Pediatr Dermatol. 2005;22:158-160. doi:10.1111 /j.1525-1470.2005.22215.x
- Rongioletti F, Rebora A. Acral persistent papular mucinosis: a new entity. Arch Dermatol. 1986;122:1237-1239. doi:10.1001 /archderm.1986.01660230027002
- Christman MP, Sukhdeo K, Kim RH, et al. Papular mucinosis, or localized lichen myxedematosus (LM)(discrete papular type). Dermatol Online J. 2017;23:13030/qt3xp109qd.
- Rongioletti F, Ferreli C, Atzori L. Acral persistent papular mucinosis. Clin Dermatol. 2021;39:211-214. doi:10.1016/j.clindermatol.2020.10.001
- Rongioletti F, Rebora A. Cutaneous mucinoses: microscopic criteria for diagnosis. Am J Dermatopathol. 2001;23:257-267. doi:10.1097/00000372- 200106000-00022
- Rongioletti F. Lichen myxedematosus (papular mucinosis): new concepts and perspectives for an old disease. Semin Cutan Med Surg. 2006;25:100-104. doi:10.1016/j.sder.2006.04.001
- Jun JY, Oh SH, Shim JH, et al. Acral persistent papular mucinosis with partial response to tacrolimus ointment. Ann Dermatol. 2016;28:517-519. doi:10.5021/ad.2016.28.4.517
- Rongioletti F, Zaccaria E, Cozzani E, et al. Treatment of localized lichen myxedematosus of discrete type with tacrolimus ointment. J Am Acad Dermatol. 2008;58:530-532. doi:10.1016/j.jaad.2006.10.021
- Rongioletti F, Merlo G, Cinotti E, et al. Scleromyxedema: a multicenter study of characteristics, comorbidities, course, and therapy in 30 patients. J Am Acad Dermatol. 2013;69:66-72. doi:10.1016 /j.jaad.2013.01.007
- Rongioletti F, Kaiser F, Cinotti E, et al. Scleredema. a multicentre study of characteristics, comorbidities, course and therapy in 44 patients. J Eur Acad Dermatol Venereol. 2015;29:2399-2404. doi:10.1111/jdv.13272
- Piette EW, Rosenbach M. Granuloma annulare: clinical and histologic variants, epidemiology, and genetics. J Am Acad Dermatol. 2016;75:457-465. doi:10.1016/j.jaad.2015.03.054
- Al-Mutairi N, Hassanein A, Nour-Eldin O, et al. Generalized lichen nitidus. Pediatr Dermatol. 2005;22:158-160. doi:10.1111 /j.1525-1470.2005.22215.x
Multiple Firm Papules on the Wrists and Forearms
Multiple Firm Papules on the Wrists and Forearms
A 69-year-old woman presented to the dermatology department with persistent asymptomatic skin lesions on the wrists and forearms of several months’ duration. The lesions had slowly grown in number over the past few months with no identifiable triggers. The patient reported no known history of injury or trauma to the affected sites and was not taking any prescription medications other than daily vitamins. She denied any family history of similar lesions and was otherwise healthy. Physical examination revealed multiple waxy, firm, hypopigmented, 3- to 5-mm papules located exclusively on the dorsal wrists and forearms. No extracutaneous involvement was observed. A 4-mm punch biopsy from the forearm was obtained.
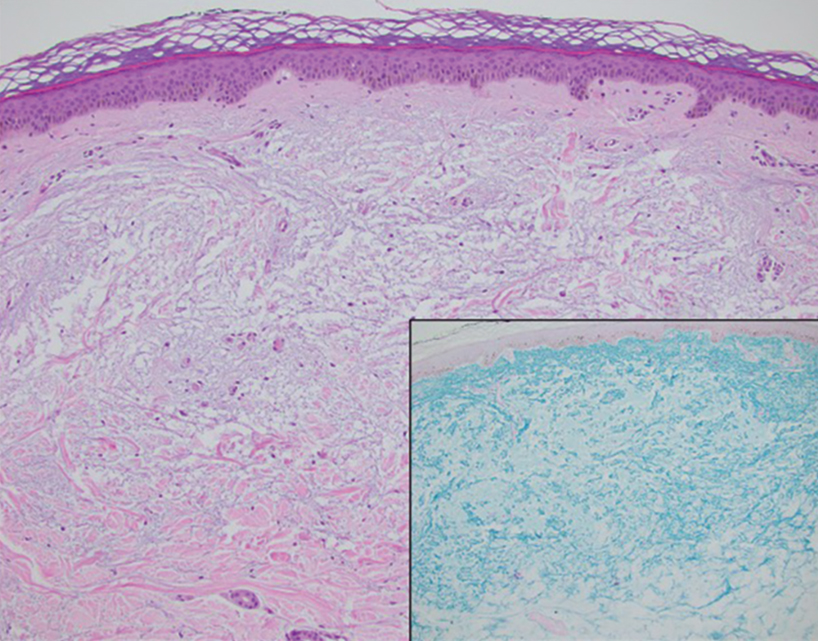
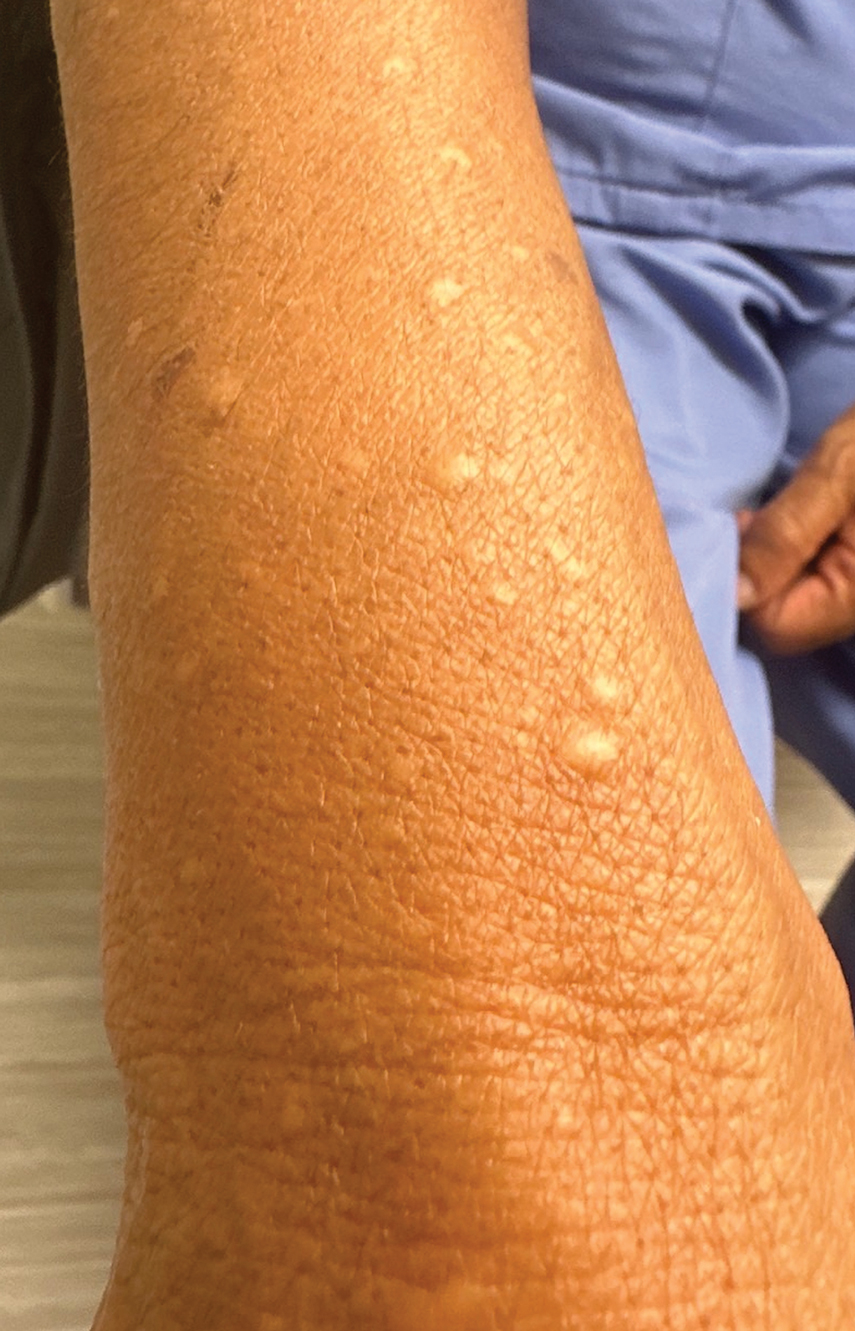

Pink Ulcerated Nodule on the Forearm
Pink Ulcerated Nodule on the Forearm
THE DIAGNOSIS: Cutaneous Cryptococcosis
Biopsy of the ulcerated nodule showed numerous yeastlike organisms within clear mucinous capsules and with some surrounding inflammation. On Grocott methenamine silver staining, the organisms stained black. Workup for disseminated cryptococcus was negative, leading to a diagnosis of primary cutaneous cryptococcosis in the setting of immunosuppression. Notably, cryptococcosis infection has been reported in patients taking fingolimod (a sphingosine-1-phosphate receptor) for multiple sclerosis, which was the case for our patient.1
The genus Cryptococcus comprises more than 30 species of encapsulated basidiomycetous fungi distributed ubiquitously in nature. Currently, only 2 species are known to cause infectious disease in humans: Cryptococcus neoformans, which affects both immunocompromised and immunocompetent patients and frequently is isolated from pigeon droppings, as well as Cryptococcus gatti, which primarily affects immunocompetent patients and is more commonly isolated from soil and decaying wood.2
Primary cutaneous cryptococcosis (PCC), characterized by direct inoculation of C neoformans or C gatti via skin injury, is rare and typically is seen in patients with decreased cell-mediated immunity, such as those on chronic corticosteroid therapy, solid-organ transplant recipients, and those with HIV.3 Primary cutaneous cryptococcosis typically manifests as a solitary or confined lesion on exposed areas of the skin and often is accompanied by regional lymphadenopathy.4,5 The most common cutaneous findings associated with PCC include ulceration, cellulitis, and whitlow.5 In immunocompetent hosts, frequently affected sites include the arms, fingers, and face, while the trunk and lower extremities are more commonly affected in immunocompromised hosts.3 Secondary cutaneous cryptococcosis occurs through hematologic spread in patients with disseminated cryptococcosis after inhalation of Cryptococcosis spores and differs from PCC in that it typically manifests as multiple lesions scattered on both exposed and covered areas of the skin. Patients also may have signs and symptoms of disseminated cryptococcosis such as pneumonia and/or meningitis at presentation.5
Despite the difference between PCC and secondary cutaneous cryptococcosis, almost every type of skin lesion has been observed in cryptococcosis, including pustules, nodules, vesicles, acneform lesions, purpura, ulcers, abscesses, molluscumlike lesions, granulomas, draining sinuses, and cellulitis.6,7
Cutaneous cryptococcosis generally is associated with 2 types of histologic reactions: gelatinous and granulomatous. The gelatinous reaction shows numerous yeastlike organisms ranging from 4 μm to 12 μm in diameter with large mucinous polysaccharide capsules and scant inflammation. Organisms may be seen in mucoid sheets.8 The granulomatous type shows a more pronounced reaction with fewer organisms ranging from 2 μm to 4 μm in diameter found within giant cells, histiocytes, and lymphocytes.6,9 Areas of necrosis occasionally can be observed.8
It is important to consider infection with Blastomyces dermatitidis and Histoplasma capsulatum in the differential Both entities can manifest as necrotizing granulomas on histology (Figures 1 and 2).10 Microscopic morphology can help differentiate these pathogenic fungi from Cryptococcus diagnosis of cryptococcosis. species which show pleomorphic, narrow-based budding yeast with wide capsules. In contrast, H capsulatum is characterized by small, intracellular, yeastlike cells with microconidia and macroconidia, while B dermatitidis is distinguished by spherical, thick-walled cells with broad-based budding.11 Capsular material also can help distinguish Cryptococcus from other pathogenic fungi. Special stains highlighting the polysaccharide capsule of Cryptococcus can best identify the yeast. The capsule stains red with periodic acid–Schiff, blue with Alcian blue, and black with Grocott methenamine silver. Mucicarmine is especially useful as it can stain the mucinous capsule pinkish red and typically does not stain other pathogenic fungi.12 Capsule-deficient organisms can lead to considerable difficulties in diagnosis given the organisms can vary in size and may mimic H capsulatum or B dermatitidis. The Fontana-Masson stain is a valuable tool in identifying capsule-deficient organisms, as melanin is found in Cryptococcus cell walls; thus, positive staining excludes H capsulatum and B dermatitidis.13
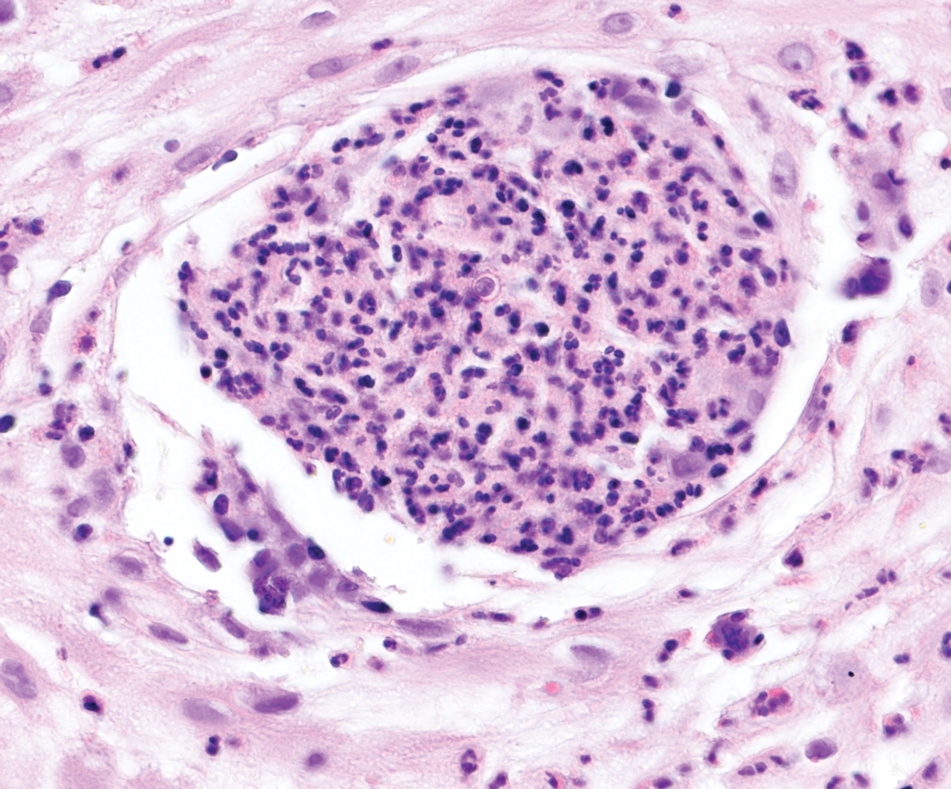
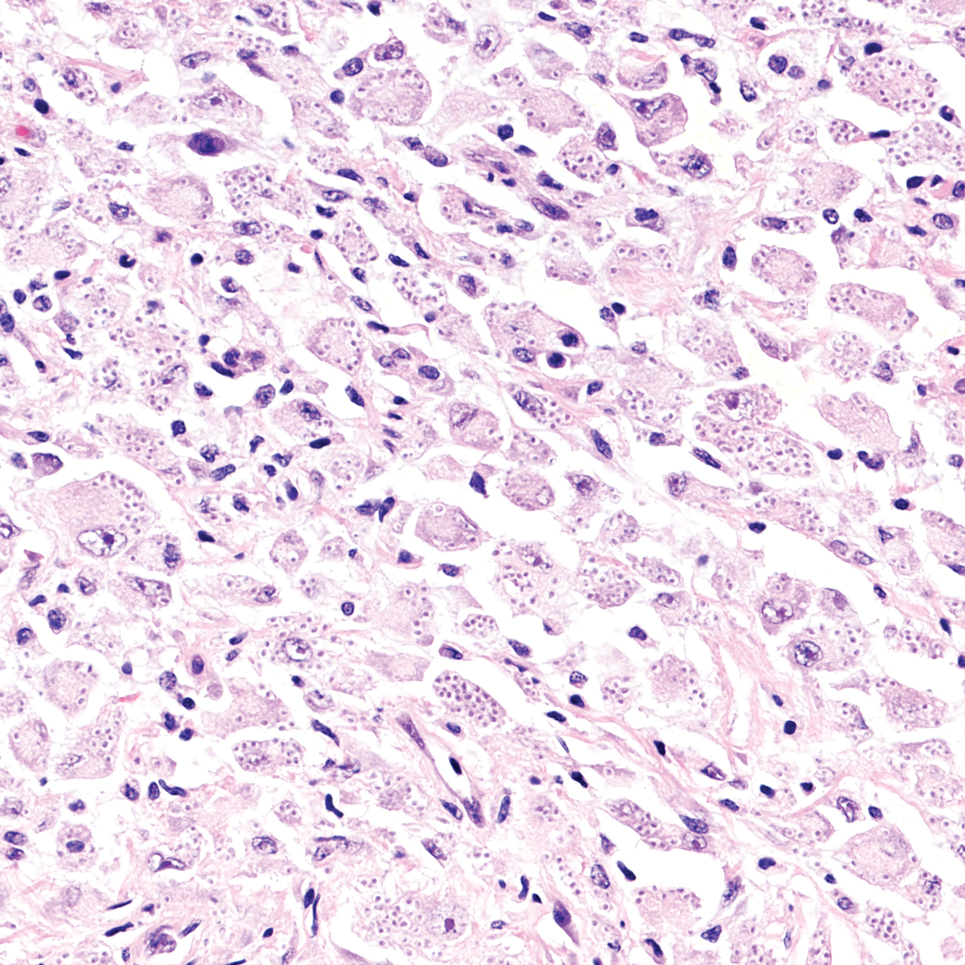
Cutaneous foreign body granuloma, which refers to a granulomatous inflammatory reaction to a foreign body in the skin, is another differential diagnosis that is important to distinguish from cutaneous cryptococcosis. On histology, a collection of histiocytes surround the inert material, forming giant cells without an immune response (Figure 3).10 In contrast, granulomas caused by infectious etiologies (eg, Cryptococcus species) have an associated adaptive immune response and can be further classified as necrotizing or non-necrotizing. Necrotizing granulomas have a distinct central necrosis with a surrounding lymphohistiocytic reaction with peripheral chronic inflammation.10
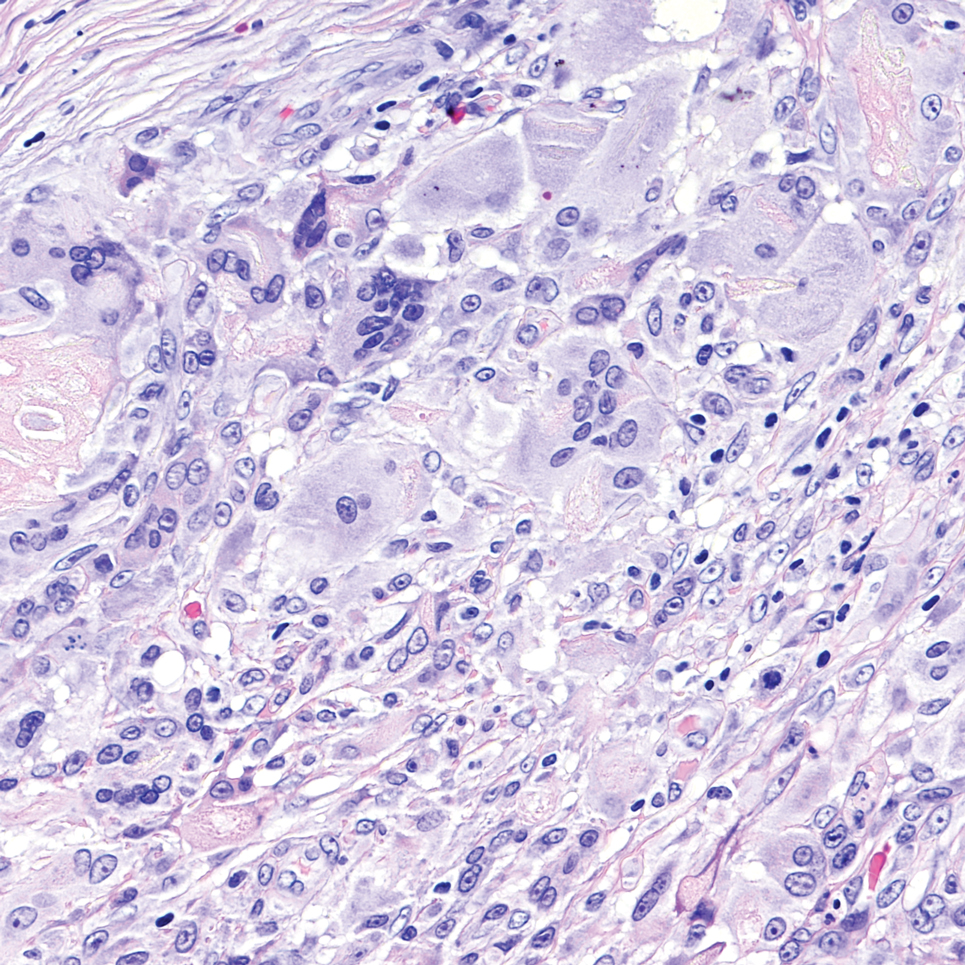
Sweet syndrome is another mimicker of cutaneous cryptococcosis. A histologic variant of Sweet syndrome has been reported that has characteristic cutaneous lesions clinically but shows basophilic bodies with a surrounding halo on pathology that can be mistaken for Cryptococcus yeast. Classic histopathology of Sweet syndrome features papillary dermal edema with neutrophil or histiocytelike inflammatory infiltrate (Figure 4). Identification of Sweet syndrome can be aided by positive myeloperoxidase staining and negative periodic acid–Schiff staining.14,15
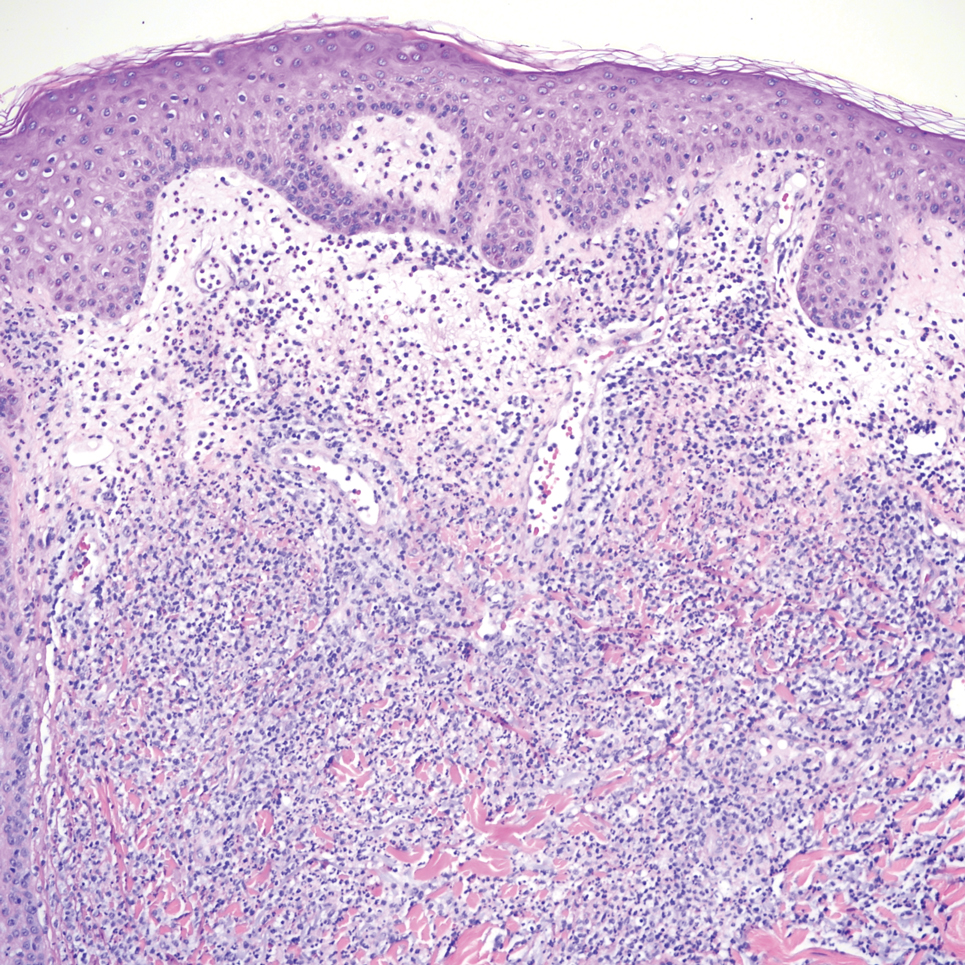
- Lehmann NM, Kammeyer JA. Cerebral venous thrombosis due to Cryptococcus in a multiple sclerosis patient on fingolimod. Case Rep Neurol. 2022; 14:286-290. doi:10.1159/000524359
- Maziarz EK, Perfect JR. Cryptococcosis. Infect Dis Clin North Am. 2016;30:179-206. doi:10.1016/j.idc.2015.10.006.
- Christianson JC, Engber W, Andes D. Primary cutaneous cryptococcosis in immunocompetent and immunocompromised hosts. Med Mycol. 2003;41:177-188. doi:10.1080/1369378031000137224
- Tilak R, Prakash P, Nigam C, et al. Cryptococcal meningitis with an antecedent cutaneous Cryptococcal lesion. Dermatol Online J. 2009;15:12.
- Neuville S, Dromer F, Morin O, et al. Primary cutaneous cryptococcosis: a distinct clinical entity. Clin Infect Dis. 2003;36:337-347. doi:10.1086/345956
- Dimino-Emme L, Gurevitch AW. Cutaneous manifestations of disseminated cryptococcosis. J Am Acad Dermatol. 1995;32:844-850.
- Anderson DJ, Schmidt C, Goodman J, Pomeroy C. Cryptococcal disease presenting as cellulitis. Clin Infect Dis. 1992;14:666-672. doi:10.1093/clinids/14.3.666
- Moore M. Cryptococcosis with cutaneous manifestations: four cases with a review of published reports. J Invest Dermatol. 1957;28(2):159-182. doi: 10.1038/jid.1957.17
- Phan NQ, Tirado M, Moeckel SMC, et al. Cutaneous and pulmonary cryptococcosis in an immunocompetent patient. J Dtsch Dermatol Ges. 2019;17:1283-1286. doi:10.1111/ddg.13997.
- Shah KK, Pritt BS, Alexander MP. Histopathologic review of granulomatous inflammation. J Clin Tuberc Other Mycobact Dis. 2017;7:1-12. doi: 10.1016/j.jctube.2017.02.001
- Fridlington E, Colome-Grimmer M, Kelly E, et al. Tzanck smear as a rapid diagnostic tool for disseminated cryptococcal infection. Arch Dermatol. 2006;142:25-27. doi: 10.1001/archderm.142.1.25
- Hernandez AD. Cutaneous Cryptococcosis. Dermatol Clin. 1989; 7:269-274.
- Ro JY, Lee SS, Ayala AG. Advantage of Fontana-Masson stain in capsule-deficient cryptococcal infection. Arch Pathol Lab Med. 1987;111:53-57.
- Jordan AA, Graciaa DS, Gopalsamy SN, et al. Sweet syndrome imitating cutaneous cryptococcal disease. Open Forum Infect Dis. 2022;9:ofac608. doi: 10.1093/ofid/ofac608
- Ko JS, Fernandez AP, Anderson KA, et al. Morphologic mimickers of Cryptococcus occurring within inflammatory infiltrates in the setting of neutrophilic dermatitis: a series of three cases highlighting clinical dilemmas associated with a novel histopathologic pitfall. J Cutan Pathol. 2013;40:38-45. doi: 10.1111/cup.12019
THE DIAGNOSIS: Cutaneous Cryptococcosis
Biopsy of the ulcerated nodule showed numerous yeastlike organisms within clear mucinous capsules and with some surrounding inflammation. On Grocott methenamine silver staining, the organisms stained black. Workup for disseminated cryptococcus was negative, leading to a diagnosis of primary cutaneous cryptococcosis in the setting of immunosuppression. Notably, cryptococcosis infection has been reported in patients taking fingolimod (a sphingosine-1-phosphate receptor) for multiple sclerosis, which was the case for our patient.1
The genus Cryptococcus comprises more than 30 species of encapsulated basidiomycetous fungi distributed ubiquitously in nature. Currently, only 2 species are known to cause infectious disease in humans: Cryptococcus neoformans, which affects both immunocompromised and immunocompetent patients and frequently is isolated from pigeon droppings, as well as Cryptococcus gatti, which primarily affects immunocompetent patients and is more commonly isolated from soil and decaying wood.2
Primary cutaneous cryptococcosis (PCC), characterized by direct inoculation of C neoformans or C gatti via skin injury, is rare and typically is seen in patients with decreased cell-mediated immunity, such as those on chronic corticosteroid therapy, solid-organ transplant recipients, and those with HIV.3 Primary cutaneous cryptococcosis typically manifests as a solitary or confined lesion on exposed areas of the skin and often is accompanied by regional lymphadenopathy.4,5 The most common cutaneous findings associated with PCC include ulceration, cellulitis, and whitlow.5 In immunocompetent hosts, frequently affected sites include the arms, fingers, and face, while the trunk and lower extremities are more commonly affected in immunocompromised hosts.3 Secondary cutaneous cryptococcosis occurs through hematologic spread in patients with disseminated cryptococcosis after inhalation of Cryptococcosis spores and differs from PCC in that it typically manifests as multiple lesions scattered on both exposed and covered areas of the skin. Patients also may have signs and symptoms of disseminated cryptococcosis such as pneumonia and/or meningitis at presentation.5
Despite the difference between PCC and secondary cutaneous cryptococcosis, almost every type of skin lesion has been observed in cryptococcosis, including pustules, nodules, vesicles, acneform lesions, purpura, ulcers, abscesses, molluscumlike lesions, granulomas, draining sinuses, and cellulitis.6,7
Cutaneous cryptococcosis generally is associated with 2 types of histologic reactions: gelatinous and granulomatous. The gelatinous reaction shows numerous yeastlike organisms ranging from 4 μm to 12 μm in diameter with large mucinous polysaccharide capsules and scant inflammation. Organisms may be seen in mucoid sheets.8 The granulomatous type shows a more pronounced reaction with fewer organisms ranging from 2 μm to 4 μm in diameter found within giant cells, histiocytes, and lymphocytes.6,9 Areas of necrosis occasionally can be observed.8
It is important to consider infection with Blastomyces dermatitidis and Histoplasma capsulatum in the differential Both entities can manifest as necrotizing granulomas on histology (Figures 1 and 2).10 Microscopic morphology can help differentiate these pathogenic fungi from Cryptococcus diagnosis of cryptococcosis. species which show pleomorphic, narrow-based budding yeast with wide capsules. In contrast, H capsulatum is characterized by small, intracellular, yeastlike cells with microconidia and macroconidia, while B dermatitidis is distinguished by spherical, thick-walled cells with broad-based budding.11 Capsular material also can help distinguish Cryptococcus from other pathogenic fungi. Special stains highlighting the polysaccharide capsule of Cryptococcus can best identify the yeast. The capsule stains red with periodic acid–Schiff, blue with Alcian blue, and black with Grocott methenamine silver. Mucicarmine is especially useful as it can stain the mucinous capsule pinkish red and typically does not stain other pathogenic fungi.12 Capsule-deficient organisms can lead to considerable difficulties in diagnosis given the organisms can vary in size and may mimic H capsulatum or B dermatitidis. The Fontana-Masson stain is a valuable tool in identifying capsule-deficient organisms, as melanin is found in Cryptococcus cell walls; thus, positive staining excludes H capsulatum and B dermatitidis.13
Cutaneous foreign body granuloma, which refers to a granulomatous inflammatory reaction to a foreign body in the skin, is another differential diagnosis that is important to distinguish from cutaneous cryptococcosis. On histology, a collection of histiocytes surround the inert material, forming giant cells without an immune response (Figure 3).10 In contrast, granulomas caused by infectious etiologies (eg, Cryptococcus species) have an associated adaptive immune response and can be further classified as necrotizing or non-necrotizing. Necrotizing granulomas have a distinct central necrosis with a surrounding lymphohistiocytic reaction with peripheral chronic inflammation.10
Sweet syndrome is another mimicker of cutaneous cryptococcosis. A histologic variant of Sweet syndrome has been reported that has characteristic cutaneous lesions clinically but shows basophilic bodies with a surrounding halo on pathology that can be mistaken for Cryptococcus yeast. Classic histopathology of Sweet syndrome features papillary dermal edema with neutrophil or histiocytelike inflammatory infiltrate (Figure 4). Identification of Sweet syndrome can be aided by positive myeloperoxidase staining and negative periodic acid–Schiff staining.14,15
THE DIAGNOSIS: Cutaneous Cryptococcosis
Biopsy of the ulcerated nodule showed numerous yeastlike organisms within clear mucinous capsules and with some surrounding inflammation. On Grocott methenamine silver staining, the organisms stained black. Workup for disseminated cryptococcus was negative, leading to a diagnosis of primary cutaneous cryptococcosis in the setting of immunosuppression. Notably, cryptococcosis infection has been reported in patients taking fingolimod (a sphingosine-1-phosphate receptor) for multiple sclerosis, which was the case for our patient.1
The genus Cryptococcus comprises more than 30 species of encapsulated basidiomycetous fungi distributed ubiquitously in nature. Currently, only 2 species are known to cause infectious disease in humans: Cryptococcus neoformans, which affects both immunocompromised and immunocompetent patients and frequently is isolated from pigeon droppings, as well as Cryptococcus gatti, which primarily affects immunocompetent patients and is more commonly isolated from soil and decaying wood.2
Primary cutaneous cryptococcosis (PCC), characterized by direct inoculation of C neoformans or C gatti via skin injury, is rare and typically is seen in patients with decreased cell-mediated immunity, such as those on chronic corticosteroid therapy, solid-organ transplant recipients, and those with HIV.3 Primary cutaneous cryptococcosis typically manifests as a solitary or confined lesion on exposed areas of the skin and often is accompanied by regional lymphadenopathy.4,5 The most common cutaneous findings associated with PCC include ulceration, cellulitis, and whitlow.5 In immunocompetent hosts, frequently affected sites include the arms, fingers, and face, while the trunk and lower extremities are more commonly affected in immunocompromised hosts.3 Secondary cutaneous cryptococcosis occurs through hematologic spread in patients with disseminated cryptococcosis after inhalation of Cryptococcosis spores and differs from PCC in that it typically manifests as multiple lesions scattered on both exposed and covered areas of the skin. Patients also may have signs and symptoms of disseminated cryptococcosis such as pneumonia and/or meningitis at presentation.5
Despite the difference between PCC and secondary cutaneous cryptococcosis, almost every type of skin lesion has been observed in cryptococcosis, including pustules, nodules, vesicles, acneform lesions, purpura, ulcers, abscesses, molluscumlike lesions, granulomas, draining sinuses, and cellulitis.6,7
Cutaneous cryptococcosis generally is associated with 2 types of histologic reactions: gelatinous and granulomatous. The gelatinous reaction shows numerous yeastlike organisms ranging from 4 μm to 12 μm in diameter with large mucinous polysaccharide capsules and scant inflammation. Organisms may be seen in mucoid sheets.8 The granulomatous type shows a more pronounced reaction with fewer organisms ranging from 2 μm to 4 μm in diameter found within giant cells, histiocytes, and lymphocytes.6,9 Areas of necrosis occasionally can be observed.8
It is important to consider infection with Blastomyces dermatitidis and Histoplasma capsulatum in the differential Both entities can manifest as necrotizing granulomas on histology (Figures 1 and 2).10 Microscopic morphology can help differentiate these pathogenic fungi from Cryptococcus diagnosis of cryptococcosis. species which show pleomorphic, narrow-based budding yeast with wide capsules. In contrast, H capsulatum is characterized by small, intracellular, yeastlike cells with microconidia and macroconidia, while B dermatitidis is distinguished by spherical, thick-walled cells with broad-based budding.11 Capsular material also can help distinguish Cryptococcus from other pathogenic fungi. Special stains highlighting the polysaccharide capsule of Cryptococcus can best identify the yeast. The capsule stains red with periodic acid–Schiff, blue with Alcian blue, and black with Grocott methenamine silver. Mucicarmine is especially useful as it can stain the mucinous capsule pinkish red and typically does not stain other pathogenic fungi.12 Capsule-deficient organisms can lead to considerable difficulties in diagnosis given the organisms can vary in size and may mimic H capsulatum or B dermatitidis. The Fontana-Masson stain is a valuable tool in identifying capsule-deficient organisms, as melanin is found in Cryptococcus cell walls; thus, positive staining excludes H capsulatum and B dermatitidis.13
Cutaneous foreign body granuloma, which refers to a granulomatous inflammatory reaction to a foreign body in the skin, is another differential diagnosis that is important to distinguish from cutaneous cryptococcosis. On histology, a collection of histiocytes surround the inert material, forming giant cells without an immune response (Figure 3).10 In contrast, granulomas caused by infectious etiologies (eg, Cryptococcus species) have an associated adaptive immune response and can be further classified as necrotizing or non-necrotizing. Necrotizing granulomas have a distinct central necrosis with a surrounding lymphohistiocytic reaction with peripheral chronic inflammation.10
Sweet syndrome is another mimicker of cutaneous cryptococcosis. A histologic variant of Sweet syndrome has been reported that has characteristic cutaneous lesions clinically but shows basophilic bodies with a surrounding halo on pathology that can be mistaken for Cryptococcus yeast. Classic histopathology of Sweet syndrome features papillary dermal edema with neutrophil or histiocytelike inflammatory infiltrate (Figure 4). Identification of Sweet syndrome can be aided by positive myeloperoxidase staining and negative periodic acid–Schiff staining.14,15
- Lehmann NM, Kammeyer JA. Cerebral venous thrombosis due to Cryptococcus in a multiple sclerosis patient on fingolimod. Case Rep Neurol. 2022; 14:286-290. doi:10.1159/000524359
- Maziarz EK, Perfect JR. Cryptococcosis. Infect Dis Clin North Am. 2016;30:179-206. doi:10.1016/j.idc.2015.10.006.
- Christianson JC, Engber W, Andes D. Primary cutaneous cryptococcosis in immunocompetent and immunocompromised hosts. Med Mycol. 2003;41:177-188. doi:10.1080/1369378031000137224
- Tilak R, Prakash P, Nigam C, et al. Cryptococcal meningitis with an antecedent cutaneous Cryptococcal lesion. Dermatol Online J. 2009;15:12.
- Neuville S, Dromer F, Morin O, et al. Primary cutaneous cryptococcosis: a distinct clinical entity. Clin Infect Dis. 2003;36:337-347. doi:10.1086/345956
- Dimino-Emme L, Gurevitch AW. Cutaneous manifestations of disseminated cryptococcosis. J Am Acad Dermatol. 1995;32:844-850.
- Anderson DJ, Schmidt C, Goodman J, Pomeroy C. Cryptococcal disease presenting as cellulitis. Clin Infect Dis. 1992;14:666-672. doi:10.1093/clinids/14.3.666
- Moore M. Cryptococcosis with cutaneous manifestations: four cases with a review of published reports. J Invest Dermatol. 1957;28(2):159-182. doi: 10.1038/jid.1957.17
- Phan NQ, Tirado M, Moeckel SMC, et al. Cutaneous and pulmonary cryptococcosis in an immunocompetent patient. J Dtsch Dermatol Ges. 2019;17:1283-1286. doi:10.1111/ddg.13997.
- Shah KK, Pritt BS, Alexander MP. Histopathologic review of granulomatous inflammation. J Clin Tuberc Other Mycobact Dis. 2017;7:1-12. doi: 10.1016/j.jctube.2017.02.001
- Fridlington E, Colome-Grimmer M, Kelly E, et al. Tzanck smear as a rapid diagnostic tool for disseminated cryptococcal infection. Arch Dermatol. 2006;142:25-27. doi: 10.1001/archderm.142.1.25
- Hernandez AD. Cutaneous Cryptococcosis. Dermatol Clin. 1989; 7:269-274.
- Ro JY, Lee SS, Ayala AG. Advantage of Fontana-Masson stain in capsule-deficient cryptococcal infection. Arch Pathol Lab Med. 1987;111:53-57.
- Jordan AA, Graciaa DS, Gopalsamy SN, et al. Sweet syndrome imitating cutaneous cryptococcal disease. Open Forum Infect Dis. 2022;9:ofac608. doi: 10.1093/ofid/ofac608
- Ko JS, Fernandez AP, Anderson KA, et al. Morphologic mimickers of Cryptococcus occurring within inflammatory infiltrates in the setting of neutrophilic dermatitis: a series of three cases highlighting clinical dilemmas associated with a novel histopathologic pitfall. J Cutan Pathol. 2013;40:38-45. doi: 10.1111/cup.12019
- Lehmann NM, Kammeyer JA. Cerebral venous thrombosis due to Cryptococcus in a multiple sclerosis patient on fingolimod. Case Rep Neurol. 2022; 14:286-290. doi:10.1159/000524359
- Maziarz EK, Perfect JR. Cryptococcosis. Infect Dis Clin North Am. 2016;30:179-206. doi:10.1016/j.idc.2015.10.006.
- Christianson JC, Engber W, Andes D. Primary cutaneous cryptococcosis in immunocompetent and immunocompromised hosts. Med Mycol. 2003;41:177-188. doi:10.1080/1369378031000137224
- Tilak R, Prakash P, Nigam C, et al. Cryptococcal meningitis with an antecedent cutaneous Cryptococcal lesion. Dermatol Online J. 2009;15:12.
- Neuville S, Dromer F, Morin O, et al. Primary cutaneous cryptococcosis: a distinct clinical entity. Clin Infect Dis. 2003;36:337-347. doi:10.1086/345956
- Dimino-Emme L, Gurevitch AW. Cutaneous manifestations of disseminated cryptococcosis. J Am Acad Dermatol. 1995;32:844-850.
- Anderson DJ, Schmidt C, Goodman J, Pomeroy C. Cryptococcal disease presenting as cellulitis. Clin Infect Dis. 1992;14:666-672. doi:10.1093/clinids/14.3.666
- Moore M. Cryptococcosis with cutaneous manifestations: four cases with a review of published reports. J Invest Dermatol. 1957;28(2):159-182. doi: 10.1038/jid.1957.17
- Phan NQ, Tirado M, Moeckel SMC, et al. Cutaneous and pulmonary cryptococcosis in an immunocompetent patient. J Dtsch Dermatol Ges. 2019;17:1283-1286. doi:10.1111/ddg.13997.
- Shah KK, Pritt BS, Alexander MP. Histopathologic review of granulomatous inflammation. J Clin Tuberc Other Mycobact Dis. 2017;7:1-12. doi: 10.1016/j.jctube.2017.02.001
- Fridlington E, Colome-Grimmer M, Kelly E, et al. Tzanck smear as a rapid diagnostic tool for disseminated cryptococcal infection. Arch Dermatol. 2006;142:25-27. doi: 10.1001/archderm.142.1.25
- Hernandez AD. Cutaneous Cryptococcosis. Dermatol Clin. 1989; 7:269-274.
- Ro JY, Lee SS, Ayala AG. Advantage of Fontana-Masson stain in capsule-deficient cryptococcal infection. Arch Pathol Lab Med. 1987;111:53-57.
- Jordan AA, Graciaa DS, Gopalsamy SN, et al. Sweet syndrome imitating cutaneous cryptococcal disease. Open Forum Infect Dis. 2022;9:ofac608. doi: 10.1093/ofid/ofac608
- Ko JS, Fernandez AP, Anderson KA, et al. Morphologic mimickers of Cryptococcus occurring within inflammatory infiltrates in the setting of neutrophilic dermatitis: a series of three cases highlighting clinical dilemmas associated with a novel histopathologic pitfall. J Cutan Pathol. 2013;40:38-45. doi: 10.1111/cup.12019
Pink Ulcerated Nodule on the Forearm
Pink Ulcerated Nodule on the Forearm
A 51-year-old man with a history of multiple sclerosis treated with fingolimod presented to the dermatology department with an ulcerated lesion on the left forearm of 2 to 3 months’ duration. The patient reported that he recently presented to the emergency department for drainage of the lesion, which was unsuccessful. Shortly after, he traumatized the lesion at his construction job. At the current presentation, physical examination revealed a 1-cm, flesh-colored to faintly pink, ulcerated nodule on the left forearm. A biopsy was performed.
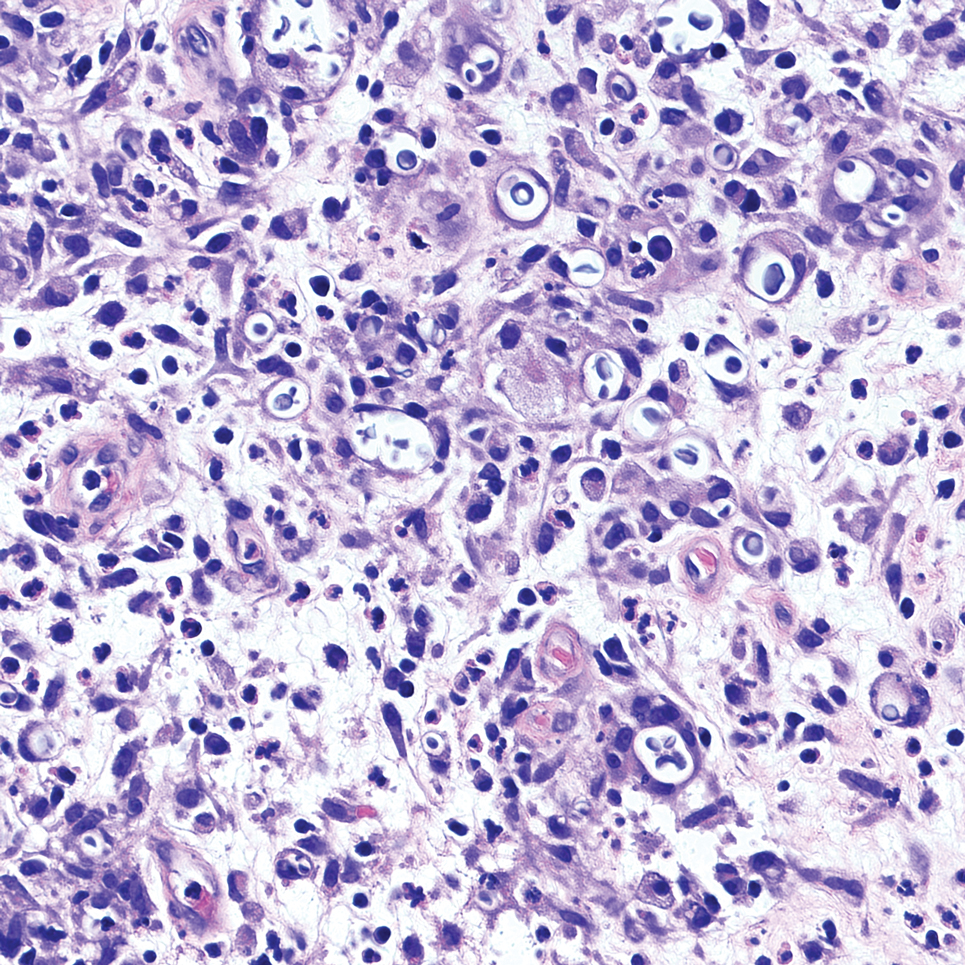
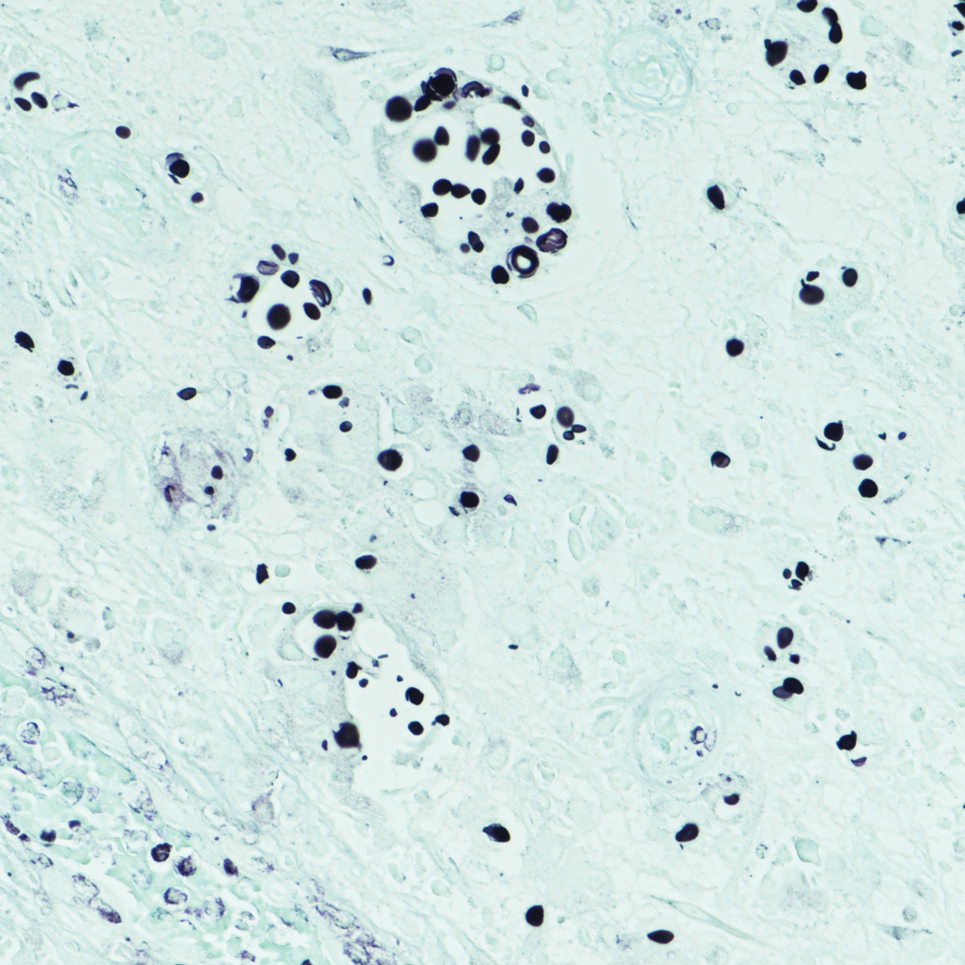

Pink Papule on the Lower Eyelid
Pink Papule on the Lower Eyelid
THE DIAGNOSIS: Poroma
Poromas are benign adnexal neoplasms that often are classified into the broader category of acrospiromas. They most commonly affect areas with a high density of eccrine sweat glands, such as the palms and soles, but also can appear in any area of the body with sweat glands.1 Poromas may have cuboidal eccrine cells with ovoid nuclei and a delicate vascularized stroma on histology or may show apocrinelike features with sebaceous cells.2,3 Immunohistochemically, poromas stain positively for carcinoembryonic antigen, epithelial membrane antigen, and periodic acid–Schiff (PAS) with diastase sensitivity.1,4 Cytokeratin (CK) 1 and CK-10 are expressed in the tumor nests.1
Poromas are the benign counterpart of porocarcinomas, which can recur and may become invasive and metastasize. Porocarcinomas have been shown to undergo malignant transformation from poromas as well as develop de novo.5 Histologic differentiation between the 2 conditions is key in determining excisional margins for treatment and follow-up. Poromas are histologically similar to porocarcinomas, but the latter show invasion into the dermis, nuclear and cytoplasmic pleomorphism, nuclear hyperchromatism, and increased mitotic activity.6 S-100 protein can be positive in porocarcinoma.7 Both poromas and porocarcinomas are associated with Yes-associated protein 1 (YAP1), Mastermind-like protein 2 (MAML2), and NUT midline carcinoma family member 1 (NUTM1) gene fusions.5
Basal cell carcinoma (BCC) is the most common cutaneous malignancy. It rarely metastasizes but can be locally destructive.8 Basal cell carcinomas typically occur on sun-exposed skin in middle-aged and elderly patients and classically manifest as pink or flesh-colored pearly papules with rolled borders and overlying telangiectasia.9 Risk factors for BCC include a chronic sun exposure, lighter skin phenotypes, immunosuppression, and a family history of skin cancer. The 2 most common subtypes of BCC are nodular and superficial, which comprise around 85% of BCCs.10 Histologically, nodular BCCs demonstrate nests of malignant basaloid cells with central disorganization, peripheral palisading, tumor-stroma clefting, and a mucoid stroma with spindle cells (Figure 1). Superficial BCC manifests with small islands of malignant basaloid cells with peripheral palisading that connect with the epidermis, often with a lichenoid inflammatory infiltrate.9 Basal cell carcinomas stain positively for Ber-EP4 and are associated with patched 1 (PTCH1), patched 2 (PTCH2), and tumor protein 53 (TP53) gene mutations.9,11
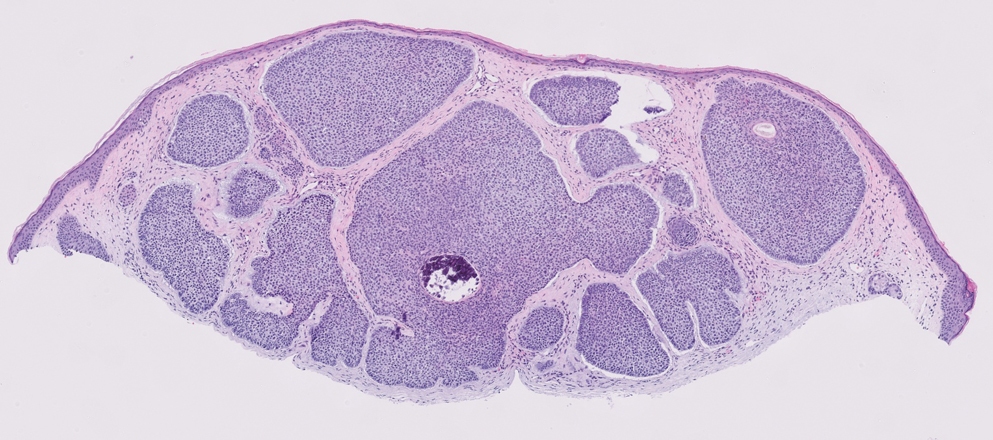
Spiradenomas are benign adnexal tumors manifesting as painful, usually singular, 1- to 3-cm nodules in younger adults.12 Histologically, spiradenomas have large clusters of small irregularly shaped aggregations of small basaloid and large polygonal cells with surrounding hyalinized basement membrane material and intratumoral lymphocytes (Figure 2).4 Spiradenomas stain positive for p63, D2-40, and CK7 and are associated with cylindromatosis lysine 63 deubiquitinase (CYLD) and alpha-protein kinase 1 (ALPK1) gene mutations.5
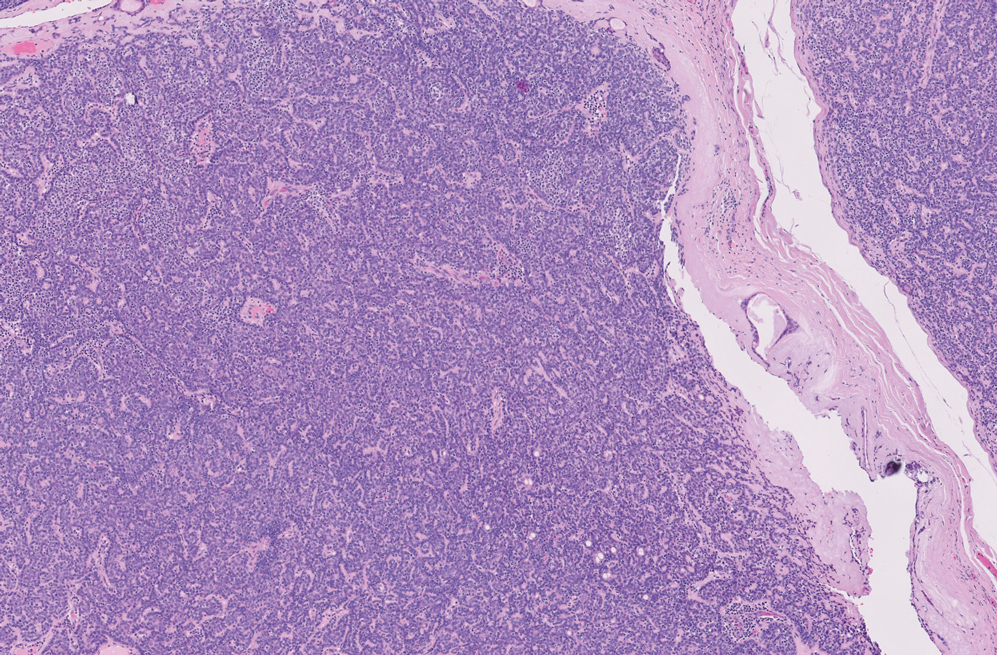
Squamous cell carcinoma (SCC) is the second most common nonmelanoma skin cancer worldwide.13 Lesions typically develop on sun-exposed skin and manifest as red, hyperkeratotic, and sometimes ulcerated plaques or nodules.14 Risk factors for SCC include chronic sun exposure, lighter skin phenotypes, increased age, and immunosuppression. Histologically, there are several variants of SCC: low-risk variants include keratoacanthomas, verrucous carcinomas, and clear cell SCC, and high-risk variants include acantholytic SCC, spindle cell SCC, and adenosquamous carcinoma.14 Generally, low-grade SCC will have well-differentiated or moderately differentiated intercellular bridges or keratin pearls with tumor cells in a solid or sheetlike pattern (Figure 3). High-grade SCC will be poorly differentiated with the presence of infiltrating individual tumor cells.15 Immunohistochemically, SCC stains positive for p63, p40, AE1/AE3, CK5/6, and MNF116 while Ber-Ep4 is negative.14,15 Poorly differentiated SCCs have high rates of mutation, commonly in the tumor protein 53 (TP53), Cyclin-dependent kinase inhibitor 2A (CDKN2A), Ras pathway, and notch receptor 1 (NOTCH-1) genes.13
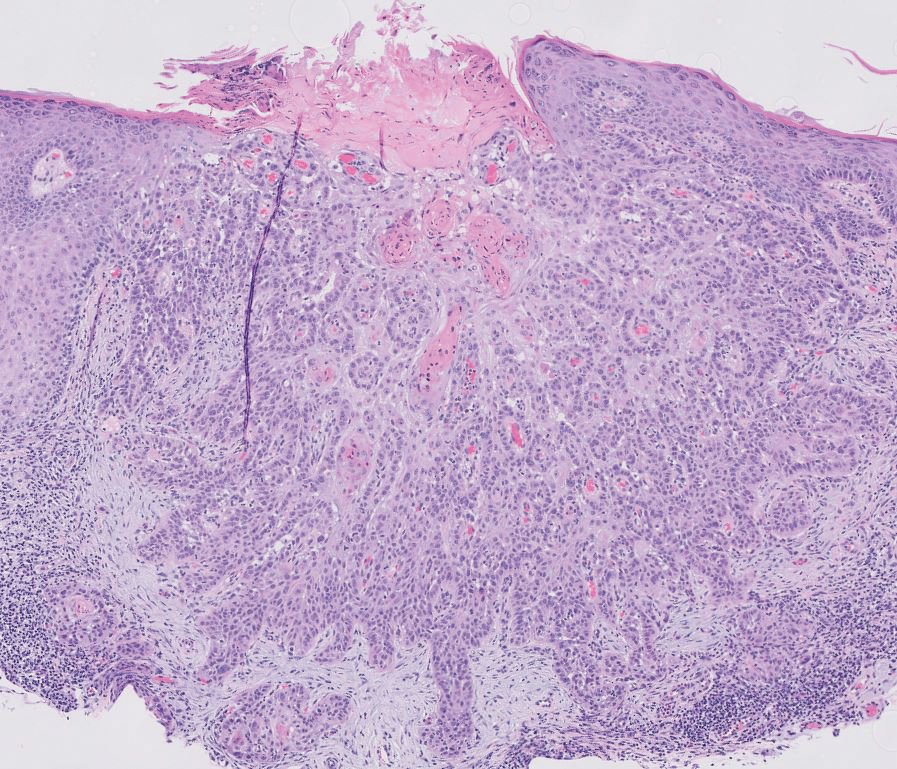
Syringomas are benign adnexal tumors that manifest as multiple soft, yellow to flesh-colored, 1- to 2-mm papules typically located on the lower eyelids, most commonly in women of reproductive age.16 Syringomas are described on histology as small comma-shaped nests with cords of eosinophilic to clear cells with central ducts surrounded by a sclerotic stroma (Figure 4). They stain positively for carcinoembryonic antigen, epithelial membrane antigen, and CK-5 and are associated with genetic mutations in phosphatidylinositol-4, 5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA) and AKT serine/threonine kinase 1 (ATK1).4
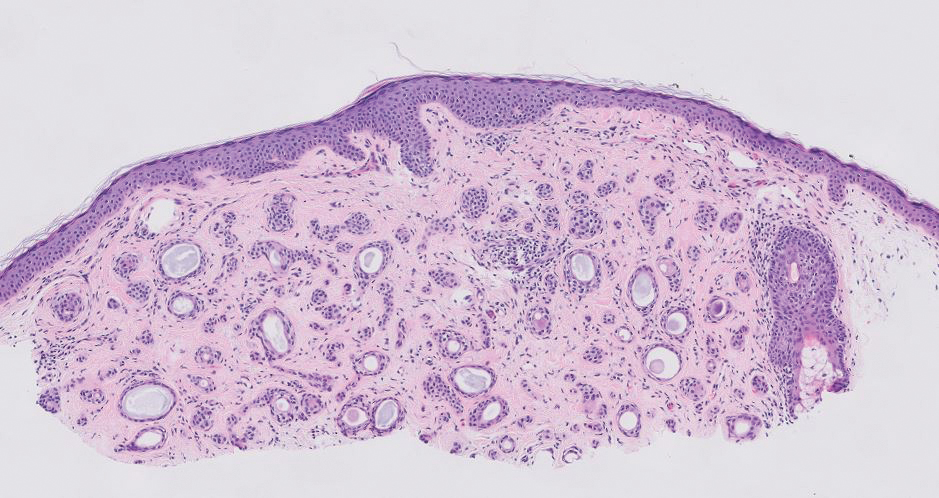
Due to its regular exposure to sunlight, the eyelid accounts for 5% to 10% of all skin malignancies. Common eyelid lesions include squamous papilloma, seborrheic keratosis, epidermal inclusion cyst, hidrocystoma, intradermal nevus, BCC, SCC, and sebaceous carcinoma.17 Aside from syringomas, benign sweat gland tumors like poromas, hidradenomas, and spiradenomas usually do not manifest on the eyelids but should be included in the differential diagnosis of an unidentifiable lesion due to the small risk for malignant transformation. Eyelid poromas manifest polymorphically, most commonly being clinically diagnosed as BCC, making the histologic examination key for proper diagnosis and management.18
- Patterson J. Weedon’s Skin Pathology. 5th ed. Elsevier Limited; 2021.
- Aoki K, Baba S, Nohara T, et al. Eccrine poroma. J Dermatol. 1980; 7:263-269. doi:10.1111/j.1346-8138.1980.tb01967.x
- Harvell JD, Kerschmann RL, LeBoit PE. Eccrine or apocrine poroma? six poromas with divergent adnexal differentiation. Am J Dermatopathol. 1996;18:1-9. doi:10.1097/00000372-199602000-00001
- Miller AC, Adjei S, Temiz LA, et al. Dermal duct tumor: a diagnostic dilemma. Dermatopathology. 2022;9:36-47. doi:10.3390
- Macagno N, Sohier P, Kervarrec T, et al. Recent advances on immunohistochemistry and molecular biology for the diagnosis of adnexal sweat gland tumors. Cancers. 2022;14:476. doi:10.3390/cancers14030476
- Robson A, Greene J, Ansari N, et al. Eccrine porocarcinoma (malignant eccrine poroma): a clinicopathologic study of 69 cases. Am J Surg Pathol. 2001;25:710-720. doi:10.1097/00000478-200106000-00002 /dermatopathology9010007
- Kurisu Y, Tsuji M, Yasuda E, et al. A case of eccrine porocarcinoma: usefulness of immunostain for S-100 protein in the diagnoses of recurrent and metastatic dedifferentiated lesions. Ann Dermatol. 2013;25:348-351. doi:10.5021/ad.2013.25.3.348
- Stanoszek LM, Wang GY, Harms PW. Histologic mimics of basal cell carcinoma. Arch Pathol Lab Med. 2017;141:1490-1502. doi:10.5858 /arpa.2017-0222-RA
- Niculet E, Craescu M, Rebegea L, et al. Basal cell carcinoma: comprehensive clinical and histopathological aspects, novel imaging tools and therapeutic approaches (review). Exp Ther Med. 2022;23:60. doi:10.3892/etm.2021.10982
- Pelucchi C, Di Landro A, Naldi L, et al. Risk factors for histological types and anatomic sites of cutaneous basal-cell carcinoma: an Italian case-control study. J Invest Dermatol. 2007;127:935-944. doi:10.1038/sj.jid.5700598
- Sunjaya AP, Sunjaya AF, Tan ST. The use of BEREP4 immunohistochemistry staining for detection of basal cell carcinoma. J Skin Cancer. 2017;2017:2692604. doi:10.1155/2017/2692604
- Kim J, Yang HJ, Pyo JS. Eccrine spiradenoma of the scalp. Arch Craniofacial Surg. 2017;18:211-213. doi:10.7181/acfs.2017.18.3.211
- Que SKT, Zwald FO, Schmults CD. Cutaneous squamous cell carcinoma: incidence, risk factors, diagnosis, and staging. J Am Acad Dermatol. 2018;78:237-247. doi:10.1016/j.jaad.2017.08.059
- Waldman A, Schmults C. Cutaneous squamous cell carcinoma. Hematol Oncol Clin North Am. 2019;33:1-12. doi:10.1016/j.hoc.2018.08.001
- Yanofsky VR, Mercer SE, Phelps RG. Histopathological variants of cutaneous squamous cell carcinoma: a review. J Skin Cancer. 2011;2011:210813. doi:10.1155/2011/210813
- Lee JH, Chang JY, Lee KH. Syringoma: a clinicopathologic and immunohistologic study and results of treatment. Yonsei Med J. 2007;48:35-40. doi:10.3349/ymj.2007.48.1.35
- Adamski WZ, Maciejewski J, Adamska K, et al. The prevalence of various eyelid skin lesions in a single-centre observation study. Adv Dermatol Allergol Dermatol Alergol. 2021;38:804-807. doi:10.5114 /ada.2020.95652
- Mencía-Gutiérrez E, Navarro-Perea C, Gutiérrez-Díaz E, et al. Eyelid eccrine poroma: a case report and review of literature. Cureus. 202:12:E8906. doi:10.7759/cureus.8906
THE DIAGNOSIS: Poroma
Poromas are benign adnexal neoplasms that often are classified into the broader category of acrospiromas. They most commonly affect areas with a high density of eccrine sweat glands, such as the palms and soles, but also can appear in any area of the body with sweat glands.1 Poromas may have cuboidal eccrine cells with ovoid nuclei and a delicate vascularized stroma on histology or may show apocrinelike features with sebaceous cells.2,3 Immunohistochemically, poromas stain positively for carcinoembryonic antigen, epithelial membrane antigen, and periodic acid–Schiff (PAS) with diastase sensitivity.1,4 Cytokeratin (CK) 1 and CK-10 are expressed in the tumor nests.1
Poromas are the benign counterpart of porocarcinomas, which can recur and may become invasive and metastasize. Porocarcinomas have been shown to undergo malignant transformation from poromas as well as develop de novo.5 Histologic differentiation between the 2 conditions is key in determining excisional margins for treatment and follow-up. Poromas are histologically similar to porocarcinomas, but the latter show invasion into the dermis, nuclear and cytoplasmic pleomorphism, nuclear hyperchromatism, and increased mitotic activity.6 S-100 protein can be positive in porocarcinoma.7 Both poromas and porocarcinomas are associated with Yes-associated protein 1 (YAP1), Mastermind-like protein 2 (MAML2), and NUT midline carcinoma family member 1 (NUTM1) gene fusions.5
Basal cell carcinoma (BCC) is the most common cutaneous malignancy. It rarely metastasizes but can be locally destructive.8 Basal cell carcinomas typically occur on sun-exposed skin in middle-aged and elderly patients and classically manifest as pink or flesh-colored pearly papules with rolled borders and overlying telangiectasia.9 Risk factors for BCC include a chronic sun exposure, lighter skin phenotypes, immunosuppression, and a family history of skin cancer. The 2 most common subtypes of BCC are nodular and superficial, which comprise around 85% of BCCs.10 Histologically, nodular BCCs demonstrate nests of malignant basaloid cells with central disorganization, peripheral palisading, tumor-stroma clefting, and a mucoid stroma with spindle cells (Figure 1). Superficial BCC manifests with small islands of malignant basaloid cells with peripheral palisading that connect with the epidermis, often with a lichenoid inflammatory infiltrate.9 Basal cell carcinomas stain positively for Ber-EP4 and are associated with patched 1 (PTCH1), patched 2 (PTCH2), and tumor protein 53 (TP53) gene mutations.9,11
Spiradenomas are benign adnexal tumors manifesting as painful, usually singular, 1- to 3-cm nodules in younger adults.12 Histologically, spiradenomas have large clusters of small irregularly shaped aggregations of small basaloid and large polygonal cells with surrounding hyalinized basement membrane material and intratumoral lymphocytes (Figure 2).4 Spiradenomas stain positive for p63, D2-40, and CK7 and are associated with cylindromatosis lysine 63 deubiquitinase (CYLD) and alpha-protein kinase 1 (ALPK1) gene mutations.5
Squamous cell carcinoma (SCC) is the second most common nonmelanoma skin cancer worldwide.13 Lesions typically develop on sun-exposed skin and manifest as red, hyperkeratotic, and sometimes ulcerated plaques or nodules.14 Risk factors for SCC include chronic sun exposure, lighter skin phenotypes, increased age, and immunosuppression. Histologically, there are several variants of SCC: low-risk variants include keratoacanthomas, verrucous carcinomas, and clear cell SCC, and high-risk variants include acantholytic SCC, spindle cell SCC, and adenosquamous carcinoma.14 Generally, low-grade SCC will have well-differentiated or moderately differentiated intercellular bridges or keratin pearls with tumor cells in a solid or sheetlike pattern (Figure 3). High-grade SCC will be poorly differentiated with the presence of infiltrating individual tumor cells.15 Immunohistochemically, SCC stains positive for p63, p40, AE1/AE3, CK5/6, and MNF116 while Ber-Ep4 is negative.14,15 Poorly differentiated SCCs have high rates of mutation, commonly in the tumor protein 53 (TP53), Cyclin-dependent kinase inhibitor 2A (CDKN2A), Ras pathway, and notch receptor 1 (NOTCH-1) genes.13
Syringomas are benign adnexal tumors that manifest as multiple soft, yellow to flesh-colored, 1- to 2-mm papules typically located on the lower eyelids, most commonly in women of reproductive age.16 Syringomas are described on histology as small comma-shaped nests with cords of eosinophilic to clear cells with central ducts surrounded by a sclerotic stroma (Figure 4). They stain positively for carcinoembryonic antigen, epithelial membrane antigen, and CK-5 and are associated with genetic mutations in phosphatidylinositol-4, 5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA) and AKT serine/threonine kinase 1 (ATK1).4
Due to its regular exposure to sunlight, the eyelid accounts for 5% to 10% of all skin malignancies. Common eyelid lesions include squamous papilloma, seborrheic keratosis, epidermal inclusion cyst, hidrocystoma, intradermal nevus, BCC, SCC, and sebaceous carcinoma.17 Aside from syringomas, benign sweat gland tumors like poromas, hidradenomas, and spiradenomas usually do not manifest on the eyelids but should be included in the differential diagnosis of an unidentifiable lesion due to the small risk for malignant transformation. Eyelid poromas manifest polymorphically, most commonly being clinically diagnosed as BCC, making the histologic examination key for proper diagnosis and management.18
THE DIAGNOSIS: Poroma
Poromas are benign adnexal neoplasms that often are classified into the broader category of acrospiromas. They most commonly affect areas with a high density of eccrine sweat glands, such as the palms and soles, but also can appear in any area of the body with sweat glands.1 Poromas may have cuboidal eccrine cells with ovoid nuclei and a delicate vascularized stroma on histology or may show apocrinelike features with sebaceous cells.2,3 Immunohistochemically, poromas stain positively for carcinoembryonic antigen, epithelial membrane antigen, and periodic acid–Schiff (PAS) with diastase sensitivity.1,4 Cytokeratin (CK) 1 and CK-10 are expressed in the tumor nests.1
Poromas are the benign counterpart of porocarcinomas, which can recur and may become invasive and metastasize. Porocarcinomas have been shown to undergo malignant transformation from poromas as well as develop de novo.5 Histologic differentiation between the 2 conditions is key in determining excisional margins for treatment and follow-up. Poromas are histologically similar to porocarcinomas, but the latter show invasion into the dermis, nuclear and cytoplasmic pleomorphism, nuclear hyperchromatism, and increased mitotic activity.6 S-100 protein can be positive in porocarcinoma.7 Both poromas and porocarcinomas are associated with Yes-associated protein 1 (YAP1), Mastermind-like protein 2 (MAML2), and NUT midline carcinoma family member 1 (NUTM1) gene fusions.5
Basal cell carcinoma (BCC) is the most common cutaneous malignancy. It rarely metastasizes but can be locally destructive.8 Basal cell carcinomas typically occur on sun-exposed skin in middle-aged and elderly patients and classically manifest as pink or flesh-colored pearly papules with rolled borders and overlying telangiectasia.9 Risk factors for BCC include a chronic sun exposure, lighter skin phenotypes, immunosuppression, and a family history of skin cancer. The 2 most common subtypes of BCC are nodular and superficial, which comprise around 85% of BCCs.10 Histologically, nodular BCCs demonstrate nests of malignant basaloid cells with central disorganization, peripheral palisading, tumor-stroma clefting, and a mucoid stroma with spindle cells (Figure 1). Superficial BCC manifests with small islands of malignant basaloid cells with peripheral palisading that connect with the epidermis, often with a lichenoid inflammatory infiltrate.9 Basal cell carcinomas stain positively for Ber-EP4 and are associated with patched 1 (PTCH1), patched 2 (PTCH2), and tumor protein 53 (TP53) gene mutations.9,11
Spiradenomas are benign adnexal tumors manifesting as painful, usually singular, 1- to 3-cm nodules in younger adults.12 Histologically, spiradenomas have large clusters of small irregularly shaped aggregations of small basaloid and large polygonal cells with surrounding hyalinized basement membrane material and intratumoral lymphocytes (Figure 2).4 Spiradenomas stain positive for p63, D2-40, and CK7 and are associated with cylindromatosis lysine 63 deubiquitinase (CYLD) and alpha-protein kinase 1 (ALPK1) gene mutations.5
Squamous cell carcinoma (SCC) is the second most common nonmelanoma skin cancer worldwide.13 Lesions typically develop on sun-exposed skin and manifest as red, hyperkeratotic, and sometimes ulcerated plaques or nodules.14 Risk factors for SCC include chronic sun exposure, lighter skin phenotypes, increased age, and immunosuppression. Histologically, there are several variants of SCC: low-risk variants include keratoacanthomas, verrucous carcinomas, and clear cell SCC, and high-risk variants include acantholytic SCC, spindle cell SCC, and adenosquamous carcinoma.14 Generally, low-grade SCC will have well-differentiated or moderately differentiated intercellular bridges or keratin pearls with tumor cells in a solid or sheetlike pattern (Figure 3). High-grade SCC will be poorly differentiated with the presence of infiltrating individual tumor cells.15 Immunohistochemically, SCC stains positive for p63, p40, AE1/AE3, CK5/6, and MNF116 while Ber-Ep4 is negative.14,15 Poorly differentiated SCCs have high rates of mutation, commonly in the tumor protein 53 (TP53), Cyclin-dependent kinase inhibitor 2A (CDKN2A), Ras pathway, and notch receptor 1 (NOTCH-1) genes.13
Syringomas are benign adnexal tumors that manifest as multiple soft, yellow to flesh-colored, 1- to 2-mm papules typically located on the lower eyelids, most commonly in women of reproductive age.16 Syringomas are described on histology as small comma-shaped nests with cords of eosinophilic to clear cells with central ducts surrounded by a sclerotic stroma (Figure 4). They stain positively for carcinoembryonic antigen, epithelial membrane antigen, and CK-5 and are associated with genetic mutations in phosphatidylinositol-4, 5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA) and AKT serine/threonine kinase 1 (ATK1).4
Due to its regular exposure to sunlight, the eyelid accounts for 5% to 10% of all skin malignancies. Common eyelid lesions include squamous papilloma, seborrheic keratosis, epidermal inclusion cyst, hidrocystoma, intradermal nevus, BCC, SCC, and sebaceous carcinoma.17 Aside from syringomas, benign sweat gland tumors like poromas, hidradenomas, and spiradenomas usually do not manifest on the eyelids but should be included in the differential diagnosis of an unidentifiable lesion due to the small risk for malignant transformation. Eyelid poromas manifest polymorphically, most commonly being clinically diagnosed as BCC, making the histologic examination key for proper diagnosis and management.18
- Patterson J. Weedon’s Skin Pathology. 5th ed. Elsevier Limited; 2021.
- Aoki K, Baba S, Nohara T, et al. Eccrine poroma. J Dermatol. 1980; 7:263-269. doi:10.1111/j.1346-8138.1980.tb01967.x
- Harvell JD, Kerschmann RL, LeBoit PE. Eccrine or apocrine poroma? six poromas with divergent adnexal differentiation. Am J Dermatopathol. 1996;18:1-9. doi:10.1097/00000372-199602000-00001
- Miller AC, Adjei S, Temiz LA, et al. Dermal duct tumor: a diagnostic dilemma. Dermatopathology. 2022;9:36-47. doi:10.3390
- Macagno N, Sohier P, Kervarrec T, et al. Recent advances on immunohistochemistry and molecular biology for the diagnosis of adnexal sweat gland tumors. Cancers. 2022;14:476. doi:10.3390/cancers14030476
- Robson A, Greene J, Ansari N, et al. Eccrine porocarcinoma (malignant eccrine poroma): a clinicopathologic study of 69 cases. Am J Surg Pathol. 2001;25:710-720. doi:10.1097/00000478-200106000-00002 /dermatopathology9010007
- Kurisu Y, Tsuji M, Yasuda E, et al. A case of eccrine porocarcinoma: usefulness of immunostain for S-100 protein in the diagnoses of recurrent and metastatic dedifferentiated lesions. Ann Dermatol. 2013;25:348-351. doi:10.5021/ad.2013.25.3.348
- Stanoszek LM, Wang GY, Harms PW. Histologic mimics of basal cell carcinoma. Arch Pathol Lab Med. 2017;141:1490-1502. doi:10.5858 /arpa.2017-0222-RA
- Niculet E, Craescu M, Rebegea L, et al. Basal cell carcinoma: comprehensive clinical and histopathological aspects, novel imaging tools and therapeutic approaches (review). Exp Ther Med. 2022;23:60. doi:10.3892/etm.2021.10982
- Pelucchi C, Di Landro A, Naldi L, et al. Risk factors for histological types and anatomic sites of cutaneous basal-cell carcinoma: an Italian case-control study. J Invest Dermatol. 2007;127:935-944. doi:10.1038/sj.jid.5700598
- Sunjaya AP, Sunjaya AF, Tan ST. The use of BEREP4 immunohistochemistry staining for detection of basal cell carcinoma. J Skin Cancer. 2017;2017:2692604. doi:10.1155/2017/2692604
- Kim J, Yang HJ, Pyo JS. Eccrine spiradenoma of the scalp. Arch Craniofacial Surg. 2017;18:211-213. doi:10.7181/acfs.2017.18.3.211
- Que SKT, Zwald FO, Schmults CD. Cutaneous squamous cell carcinoma: incidence, risk factors, diagnosis, and staging. J Am Acad Dermatol. 2018;78:237-247. doi:10.1016/j.jaad.2017.08.059
- Waldman A, Schmults C. Cutaneous squamous cell carcinoma. Hematol Oncol Clin North Am. 2019;33:1-12. doi:10.1016/j.hoc.2018.08.001
- Yanofsky VR, Mercer SE, Phelps RG. Histopathological variants of cutaneous squamous cell carcinoma: a review. J Skin Cancer. 2011;2011:210813. doi:10.1155/2011/210813
- Lee JH, Chang JY, Lee KH. Syringoma: a clinicopathologic and immunohistologic study and results of treatment. Yonsei Med J. 2007;48:35-40. doi:10.3349/ymj.2007.48.1.35
- Adamski WZ, Maciejewski J, Adamska K, et al. The prevalence of various eyelid skin lesions in a single-centre observation study. Adv Dermatol Allergol Dermatol Alergol. 2021;38:804-807. doi:10.5114 /ada.2020.95652
- Mencía-Gutiérrez E, Navarro-Perea C, Gutiérrez-Díaz E, et al. Eyelid eccrine poroma: a case report and review of literature. Cureus. 202:12:E8906. doi:10.7759/cureus.8906
- Patterson J. Weedon’s Skin Pathology. 5th ed. Elsevier Limited; 2021.
- Aoki K, Baba S, Nohara T, et al. Eccrine poroma. J Dermatol. 1980; 7:263-269. doi:10.1111/j.1346-8138.1980.tb01967.x
- Harvell JD, Kerschmann RL, LeBoit PE. Eccrine or apocrine poroma? six poromas with divergent adnexal differentiation. Am J Dermatopathol. 1996;18:1-9. doi:10.1097/00000372-199602000-00001
- Miller AC, Adjei S, Temiz LA, et al. Dermal duct tumor: a diagnostic dilemma. Dermatopathology. 2022;9:36-47. doi:10.3390
- Macagno N, Sohier P, Kervarrec T, et al. Recent advances on immunohistochemistry and molecular biology for the diagnosis of adnexal sweat gland tumors. Cancers. 2022;14:476. doi:10.3390/cancers14030476
- Robson A, Greene J, Ansari N, et al. Eccrine porocarcinoma (malignant eccrine poroma): a clinicopathologic study of 69 cases. Am J Surg Pathol. 2001;25:710-720. doi:10.1097/00000478-200106000-00002 /dermatopathology9010007
- Kurisu Y, Tsuji M, Yasuda E, et al. A case of eccrine porocarcinoma: usefulness of immunostain for S-100 protein in the diagnoses of recurrent and metastatic dedifferentiated lesions. Ann Dermatol. 2013;25:348-351. doi:10.5021/ad.2013.25.3.348
- Stanoszek LM, Wang GY, Harms PW. Histologic mimics of basal cell carcinoma. Arch Pathol Lab Med. 2017;141:1490-1502. doi:10.5858 /arpa.2017-0222-RA
- Niculet E, Craescu M, Rebegea L, et al. Basal cell carcinoma: comprehensive clinical and histopathological aspects, novel imaging tools and therapeutic approaches (review). Exp Ther Med. 2022;23:60. doi:10.3892/etm.2021.10982
- Pelucchi C, Di Landro A, Naldi L, et al. Risk factors for histological types and anatomic sites of cutaneous basal-cell carcinoma: an Italian case-control study. J Invest Dermatol. 2007;127:935-944. doi:10.1038/sj.jid.5700598
- Sunjaya AP, Sunjaya AF, Tan ST. The use of BEREP4 immunohistochemistry staining for detection of basal cell carcinoma. J Skin Cancer. 2017;2017:2692604. doi:10.1155/2017/2692604
- Kim J, Yang HJ, Pyo JS. Eccrine spiradenoma of the scalp. Arch Craniofacial Surg. 2017;18:211-213. doi:10.7181/acfs.2017.18.3.211
- Que SKT, Zwald FO, Schmults CD. Cutaneous squamous cell carcinoma: incidence, risk factors, diagnosis, and staging. J Am Acad Dermatol. 2018;78:237-247. doi:10.1016/j.jaad.2017.08.059
- Waldman A, Schmults C. Cutaneous squamous cell carcinoma. Hematol Oncol Clin North Am. 2019;33:1-12. doi:10.1016/j.hoc.2018.08.001
- Yanofsky VR, Mercer SE, Phelps RG. Histopathological variants of cutaneous squamous cell carcinoma: a review. J Skin Cancer. 2011;2011:210813. doi:10.1155/2011/210813
- Lee JH, Chang JY, Lee KH. Syringoma: a clinicopathologic and immunohistologic study and results of treatment. Yonsei Med J. 2007;48:35-40. doi:10.3349/ymj.2007.48.1.35
- Adamski WZ, Maciejewski J, Adamska K, et al. The prevalence of various eyelid skin lesions in a single-centre observation study. Adv Dermatol Allergol Dermatol Alergol. 2021;38:804-807. doi:10.5114 /ada.2020.95652
- Mencía-Gutiérrez E, Navarro-Perea C, Gutiérrez-Díaz E, et al. Eyelid eccrine poroma: a case report and review of literature. Cureus. 202:12:E8906. doi:10.7759/cureus.8906
Pink Papule on the Lower Eyelid
Pink Papule on the Lower Eyelid
A 57-year-old man with no notable medical history presented to the dermatology clinic for evaluation of an asymptomatic papule on the left lower eyelid. The patient reported that the lesion seemed to wax and wane in size over time. Physical examination revealed a small, pink, verrucous papule on the left lower eyelid. A shave biopsy of the lesion revealed a well-circumscribed collection of small, monomorphic, cuboidal cells with basophilic round nuclei, inconspicuous nucleoli, and compact eosinophilic cytoplasm (top) with focal areas of duct formation (bottom) that was sharply demarcated from normal keratinocytes.
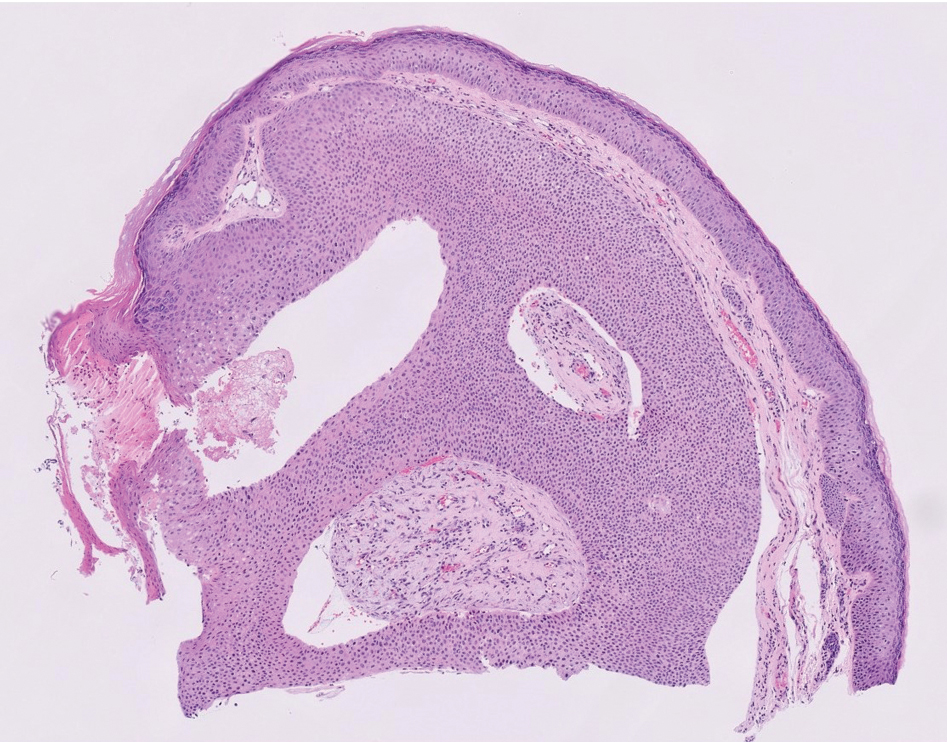
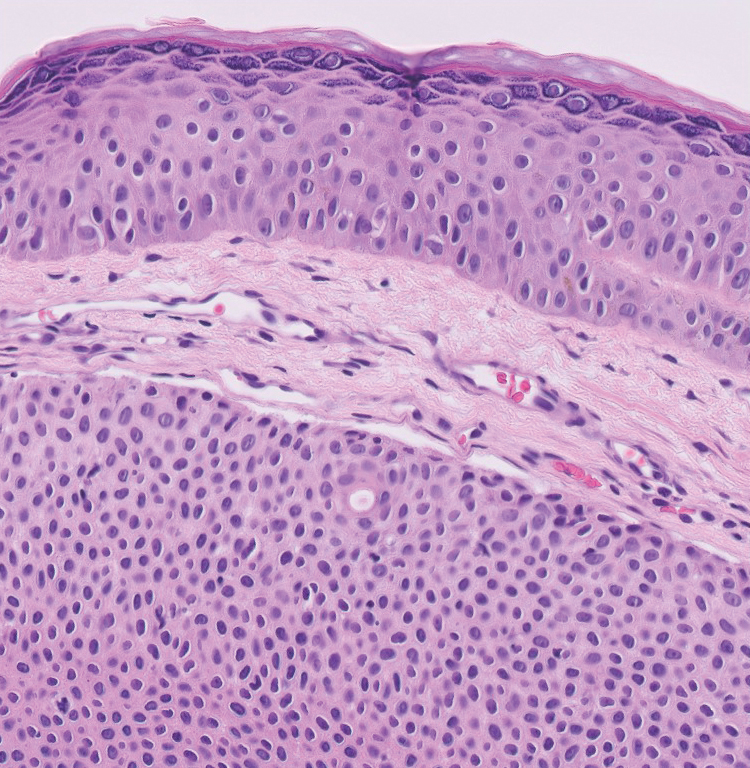

Painful Ulcers on the Elbows, Knees, and Ankles
Painful Ulcers on the Elbows, Knees, and Ankles
THE DIAGNOSIS: Diffuse Dermal Angiomatosis
Diffuse dermal angiomatosis (DDA) is a rare benign condition that manifests as tender, indurated, erythematous or violaceous plaques that can develop ulceration and necrosis. It typically occurs in areas susceptible to chronic hypoxia, such as the arms and legs, as was seen in our patient, as well as on large pendulous breasts in females. This condition is a distinct variant of reactive angioendotheliomatosis associated with smoking, trauma, underlying vaso-occlusion, and hypercoagulability.1,2 Risk factors include a history of smoking as well as conditions associated with chronic hypoxia, such as severe peripheral vascular disease, subclavian artery stenosis, hypercoagulable states, monoclonal gammopathy, steal syndrome from an arteriovenous fistula, end-stage renal failure, calciphylaxis, and obesity.1
Histopathology of DDA reveals a diffuse dermal proliferation of capillaries due to upregulation of vascular endothelial growth factor secondary to chronic ischemia and hypoxia.1,2 Small, well-formed capillaries surrounded by pericytes dissect through dermal collagen into the subcutis (eFigure 1). Spindle-shaped cells with vacuolated cytoplasm and scattered extravasated erythrocytes with hemosiderin may be observed.2 Cellular atypia generally is not seen.2,3 Diffuse dermal angiomatosis is characterized by positive CD31, CD34, and ERG immunostaining1 and HHV-8 and D2-40 negativity.2 In our patient, the areas suggestive of connective tissue calciumlike depositions were concerning for dystrophic calcification related to end-stage renal disease. Although Von Kossa staining failed to highlight vascular calcifications, early calciphylaxis from end-stage renal disease could not be excluded.
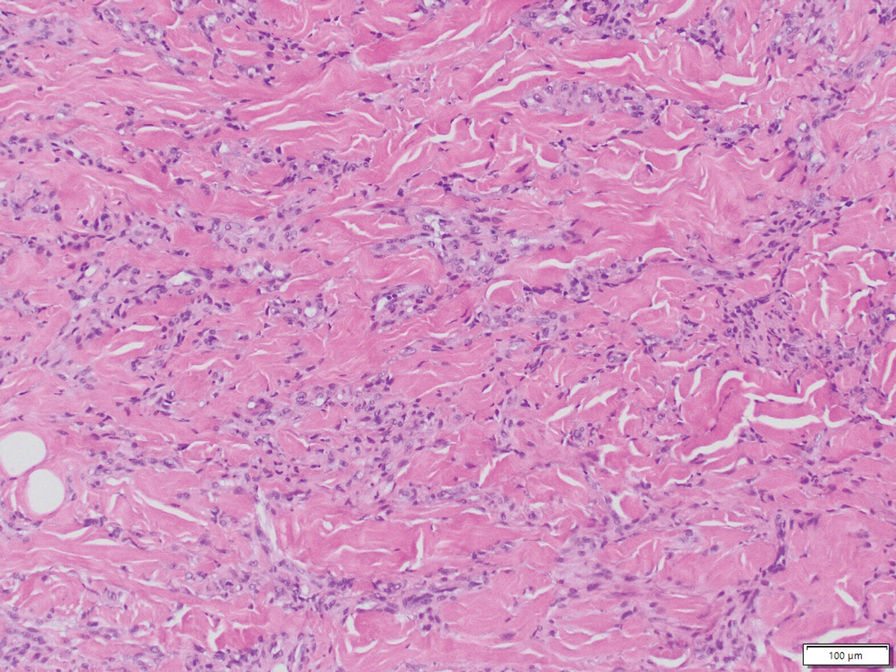
The main goal of DDA treatment is to target tissue hypoxia, and primary preventive measures aim to reduce risk factors associated with atherosclerosis.1 Treatment options for DDA include revascularization, reduction mammoplasty, excision, isotretinoin, oral corticosteroids, smoking cessation, pentoxifylline plus aspirin, and management of underlying calciphylaxis.1,2 Spontaneous resolution of DDA rarely has been reported.1
Acroangiodermatitis, also known as pseudo–Kaposi sarcoma (KS), is a rare angioproliferative disorder that often is associated with vascular anomalies.4,5 It is divided into 2 main variants: Mali type, which is associated with chronic venous insufficiency, and Stewart-Bluefarb type, associated with arteriovenous malformations.4 This condition is characterized by red to violaceous macules, papules, or plaques that may become ulcerated or coalesce to form larger confluent patches, typically arising on the lower extremities.4,6,7 Histopathology of acroangiodermatitis reveals circumscribed lobular proliferation of thick-walled dermal vessels (eFigure 2), in contrast to the diffuse dermal proliferation of endothelial cells between collagen bundles seen in DDA.2,3,6
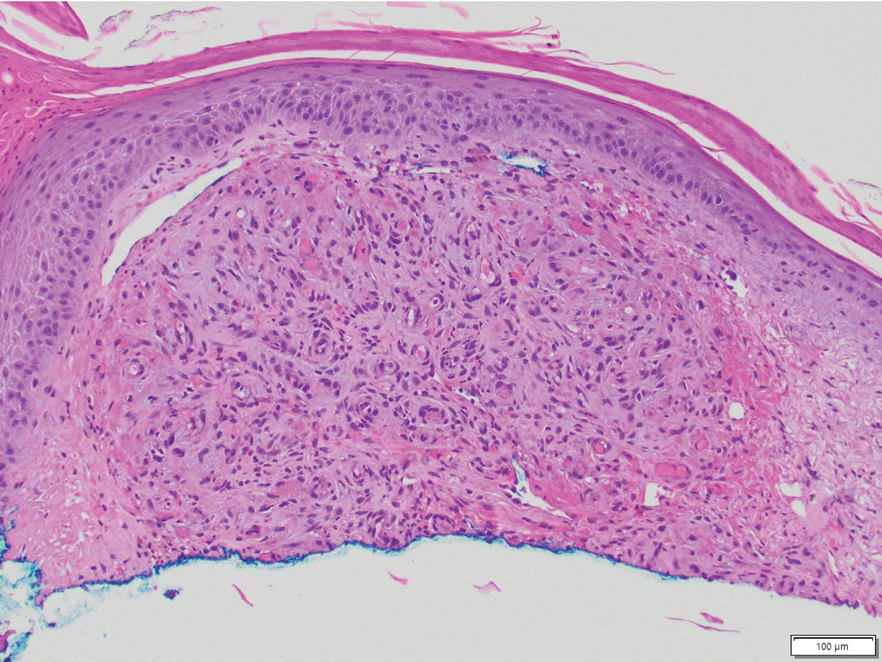
Angiosarcoma is a rare, highly aggressive vascular tumor that originates from vascular or lymphatic endothelial cells. It typically manifests with raised, bruiselike, erythematous to violaceous papules or plaques.8,9 Histopathologically, the hallmark feature of angiosarcoma is abnormal, pleomorphic, malignant endothelial cells with pale, light, eosinophilic cytoplasm and hyperchromatic nuclei (eFigure 3).2,9 In poorly differentiated cases, malignant endothelial cells may exhibit an epithelioid morphology with areas of hemorrhage and necrosis.9 Immunohistochemistry is positive for ERG, CD34, CD31, vascular endothelial growth factor, and D2-40.2,9
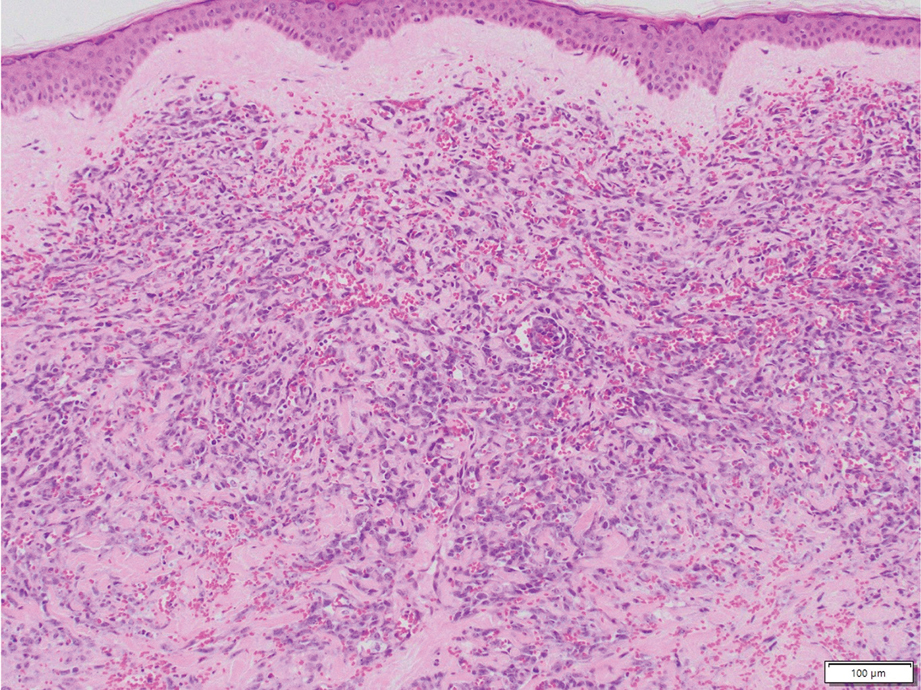
Kaposi sarcoma is a soft tissue malignancy known to occur in immunosuppressed patients such as individuals with AIDS or those undergoing immunosuppressive therapy for organ transplantation.10 There are 4 major forms of KS: classic (appearing on the lower extremities in elderly men of Mediterranean and Eastern European descent), endemic (occurring in children specifically in Africa with generalized lymph node involvement), HIV/ AIDS–related (occurring in patients not taking highly active antiretroviral therapy with diffuse involvement of the skin and internal organs), and iatrogenic (occurring in immunosuppressed patients with diffuse involvement of the skin and internal organs).10,11 Kaposi sarcoma presents as multiple reddish brown, raised or flat, painless, nonblanching mucocutaneous lesions that occasionally can ulcerate and bleed.11 Histopathologic features of KS include vascular proliferation in the dermis with diffuse slitlike lumen formation with the promontory sign, hyaline globules, hemosiderin accumulation, and an inflammatory component that often contains plasma cells (eFigure 4).2,11 Kaposi sarcoma is characterized by positive staining for CD31, CD34, D2-40, and HHV-8; the last 2 are an important distinction from DDA.2
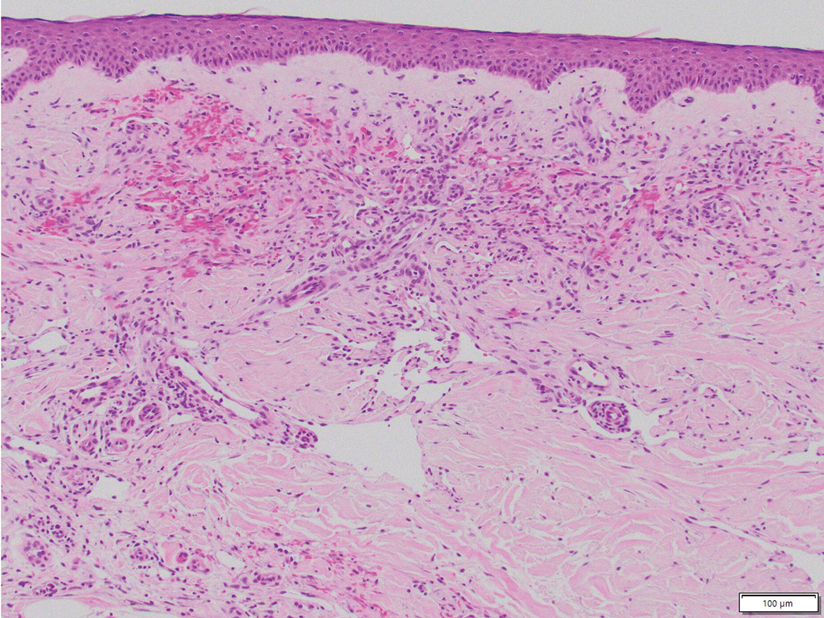
Targetoid hemosiderotic hemangioma, also known as hobnail hemangioma, is a benign vascular lesion that typically manifests as a solitary, brown to violaceous papule or plaque on the trunk or extremities.12 It is sometimes surrounded by a pale area and a peripheral ecchymotic ring, giving the lesion a targetoid appearance.12,13 Histopathologic features include dilated, thin-walled vessels with prominent endothelial hobnailing in the papillary dermis, slit-shaped vascular channels between collagen bundles in the deeper dermis, and an interstitial lymphocytic infiltrate with extravasated erythrocytes and hemosiderin deposits (eFigure 5).12,14 The etiology of targetoid hemosiderotic hemangioma remains unclear. Chronic inflammation, trauma, exposure to ionizing radiation, and vascular obstruction have been suggested as inciting factors, though many cases have been reported without a history of cutaneous injury.12,13 Studies suggest a lymphatic origin instead of its original classification as a hemangioma.13,15 The endothelial cells stain positive with CD31 and may stain with D2-40 and CD34.13,15
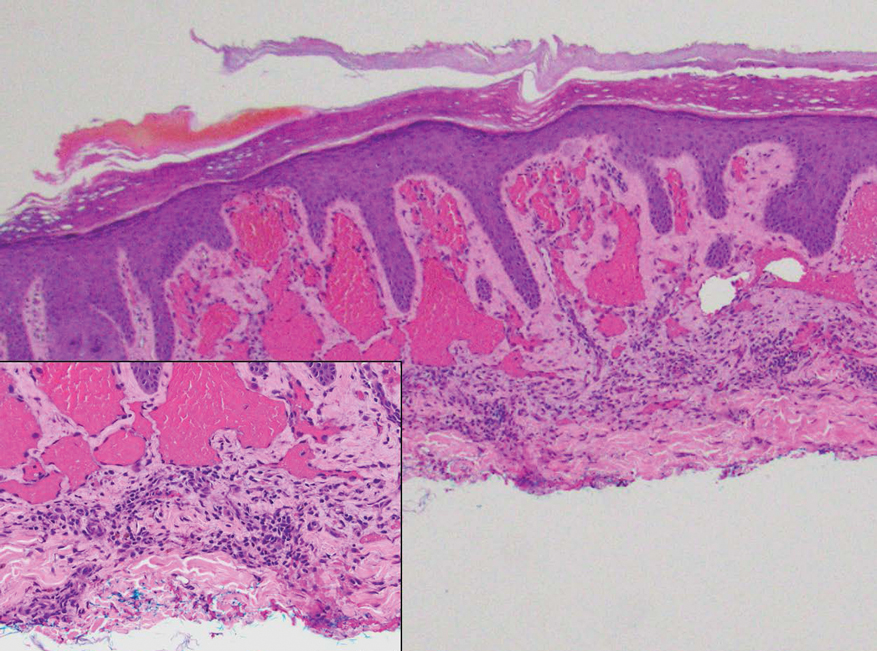
- Nguyen N, Silfvast-Kaiser AS, Frieder J, et al. Diffuse dermal angiomatosis of the breast. Proc Bayl Univ Med Cent. 2020;33:273-275. doi:10.1080/08998280.2020.1722052
- Frikha F, Boudaya S, Abid N, et al. Diffuse dermal angiomatosis of the breast with adjacent fat necrosis: a case report and review of the literature. Dermatol Online J. 2018;24:13030/qt1vq114n7
- Yang H, Ahmed I, Mathew V, et al. Diffuse dermal angiomatosis of the breast. Arch Dermatol. 2006;142:343-347. doi:10.1001 /archderm.142.3.343
- Chhabra G, Verma P, Khullar G, et al. Acroangiodermatitis, Mali and Stewart-Bluefarb type: two additional cases in adolescents. Australas J Dermatol. 2021;62:E156-E157. doi:10.1111/ajd.13386
- Ramírez-Marín HA, Ruben-Castillo C, Barrera-Godínez A, et al. Acroangiodermatitis of the hand secondary to a dysfunctional a rteriovenous fistula. Ann Vasc Surg. 2021;77:350.e13-350.e17. doi:10.1016/j.avsg.2021.05.042
- Sun L, Duarte S, Soares-de-Almeida L. Acroangiodermatitis of Mali—an unusual cause of painful ulcer. Actas Dermo-Sifiliográficas. 2023;114:546. doi:10.1016/j.ad.2022.07.013
- Parsi K, O’Connor A, Bester L. Stewart–Bluefarb syndrome: report of five cases and a review of literature. Phlebology. 2015;30:505-514. doi:10.1177/0268355514548090
- Alharbi A, Kim YC, AlShomer F, et al. Utility of multimodal treatment protocols in the management of scalp cutaneous angiosarcoma. Plast Reconstr Surg Glob Open. 2023;11:E4827. doi:10.1097 /GOX.0000000000004827
- Young RJ, Brown NJ, Reed MW, et al. Angiosarcoma. Lancet Oncol. 2010;11:983-991. doi:10.1016/S1470-2045(10)70023-1
- Bishop BN, Lynch DT. Kaposi sarcoma. StatPearls [Internet]. StatPearls Publishing; 2024. Updated June 5, 2023. Accessed January 7, 2024. http://www.ncbi.nlm.nih.gov/books/NBK534839/
- Cesarman E, Damania B, Krown SE, et al. Kaposi sarcoma. Nat Rev Dis Primer. 2019;5:1-21. doi:10.1038/s41572-019-0060-9
- AbuHilal M, Breslavet M, Ho N, et al. Hobnail hemangioma (superficial hemosiderotic lymphovascular malformation) in children: a series of 6 pediatric cases and review of the literature. J Cutan Med Surg. 2016;20:216-220. doi:10.1177/1203475415612421
- Kakizaki P, Valente NYS, Paiva DLM, et al. Targetoid hemosiderotic hemangioma—case report. An Bras Dermatol. 2014;89:956-959. doi:10.1590/abd1806-4841.20143264
- Trindade F, Kutzner H, Tellechea Ó, et al. Hobnail hemangioma reclassified as superficial lymphatic malformation: a study of 52 cases. J Am Acad Dermatol. 2012;66:112-115. doi:10.1016/j.jaad.2011.05.019
- Hejnold M, Dyduch G, Mojsa I, et al. Hobnail hemangioma: a immunohistochemical study and literature review. Pol J Pathol. 2012;63:189-192. doi:10.5114/pjp.2012.31504
THE DIAGNOSIS: Diffuse Dermal Angiomatosis
Diffuse dermal angiomatosis (DDA) is a rare benign condition that manifests as tender, indurated, erythematous or violaceous plaques that can develop ulceration and necrosis. It typically occurs in areas susceptible to chronic hypoxia, such as the arms and legs, as was seen in our patient, as well as on large pendulous breasts in females. This condition is a distinct variant of reactive angioendotheliomatosis associated with smoking, trauma, underlying vaso-occlusion, and hypercoagulability.1,2 Risk factors include a history of smoking as well as conditions associated with chronic hypoxia, such as severe peripheral vascular disease, subclavian artery stenosis, hypercoagulable states, monoclonal gammopathy, steal syndrome from an arteriovenous fistula, end-stage renal failure, calciphylaxis, and obesity.1
Histopathology of DDA reveals a diffuse dermal proliferation of capillaries due to upregulation of vascular endothelial growth factor secondary to chronic ischemia and hypoxia.1,2 Small, well-formed capillaries surrounded by pericytes dissect through dermal collagen into the subcutis (eFigure 1). Spindle-shaped cells with vacuolated cytoplasm and scattered extravasated erythrocytes with hemosiderin may be observed.2 Cellular atypia generally is not seen.2,3 Diffuse dermal angiomatosis is characterized by positive CD31, CD34, and ERG immunostaining1 and HHV-8 and D2-40 negativity.2 In our patient, the areas suggestive of connective tissue calciumlike depositions were concerning for dystrophic calcification related to end-stage renal disease. Although Von Kossa staining failed to highlight vascular calcifications, early calciphylaxis from end-stage renal disease could not be excluded.
The main goal of DDA treatment is to target tissue hypoxia, and primary preventive measures aim to reduce risk factors associated with atherosclerosis.1 Treatment options for DDA include revascularization, reduction mammoplasty, excision, isotretinoin, oral corticosteroids, smoking cessation, pentoxifylline plus aspirin, and management of underlying calciphylaxis.1,2 Spontaneous resolution of DDA rarely has been reported.1
Acroangiodermatitis, also known as pseudo–Kaposi sarcoma (KS), is a rare angioproliferative disorder that often is associated with vascular anomalies.4,5 It is divided into 2 main variants: Mali type, which is associated with chronic venous insufficiency, and Stewart-Bluefarb type, associated with arteriovenous malformations.4 This condition is characterized by red to violaceous macules, papules, or plaques that may become ulcerated or coalesce to form larger confluent patches, typically arising on the lower extremities.4,6,7 Histopathology of acroangiodermatitis reveals circumscribed lobular proliferation of thick-walled dermal vessels (eFigure 2), in contrast to the diffuse dermal proliferation of endothelial cells between collagen bundles seen in DDA.2,3,6
Angiosarcoma is a rare, highly aggressive vascular tumor that originates from vascular or lymphatic endothelial cells. It typically manifests with raised, bruiselike, erythematous to violaceous papules or plaques.8,9 Histopathologically, the hallmark feature of angiosarcoma is abnormal, pleomorphic, malignant endothelial cells with pale, light, eosinophilic cytoplasm and hyperchromatic nuclei (eFigure 3).2,9 In poorly differentiated cases, malignant endothelial cells may exhibit an epithelioid morphology with areas of hemorrhage and necrosis.9 Immunohistochemistry is positive for ERG, CD34, CD31, vascular endothelial growth factor, and D2-40.2,9
Kaposi sarcoma is a soft tissue malignancy known to occur in immunosuppressed patients such as individuals with AIDS or those undergoing immunosuppressive therapy for organ transplantation.10 There are 4 major forms of KS: classic (appearing on the lower extremities in elderly men of Mediterranean and Eastern European descent), endemic (occurring in children specifically in Africa with generalized lymph node involvement), HIV/ AIDS–related (occurring in patients not taking highly active antiretroviral therapy with diffuse involvement of the skin and internal organs), and iatrogenic (occurring in immunosuppressed patients with diffuse involvement of the skin and internal organs).10,11 Kaposi sarcoma presents as multiple reddish brown, raised or flat, painless, nonblanching mucocutaneous lesions that occasionally can ulcerate and bleed.11 Histopathologic features of KS include vascular proliferation in the dermis with diffuse slitlike lumen formation with the promontory sign, hyaline globules, hemosiderin accumulation, and an inflammatory component that often contains plasma cells (eFigure 4).2,11 Kaposi sarcoma is characterized by positive staining for CD31, CD34, D2-40, and HHV-8; the last 2 are an important distinction from DDA.2
Targetoid hemosiderotic hemangioma, also known as hobnail hemangioma, is a benign vascular lesion that typically manifests as a solitary, brown to violaceous papule or plaque on the trunk or extremities.12 It is sometimes surrounded by a pale area and a peripheral ecchymotic ring, giving the lesion a targetoid appearance.12,13 Histopathologic features include dilated, thin-walled vessels with prominent endothelial hobnailing in the papillary dermis, slit-shaped vascular channels between collagen bundles in the deeper dermis, and an interstitial lymphocytic infiltrate with extravasated erythrocytes and hemosiderin deposits (eFigure 5).12,14 The etiology of targetoid hemosiderotic hemangioma remains unclear. Chronic inflammation, trauma, exposure to ionizing radiation, and vascular obstruction have been suggested as inciting factors, though many cases have been reported without a history of cutaneous injury.12,13 Studies suggest a lymphatic origin instead of its original classification as a hemangioma.13,15 The endothelial cells stain positive with CD31 and may stain with D2-40 and CD34.13,15
THE DIAGNOSIS: Diffuse Dermal Angiomatosis
Diffuse dermal angiomatosis (DDA) is a rare benign condition that manifests as tender, indurated, erythematous or violaceous plaques that can develop ulceration and necrosis. It typically occurs in areas susceptible to chronic hypoxia, such as the arms and legs, as was seen in our patient, as well as on large pendulous breasts in females. This condition is a distinct variant of reactive angioendotheliomatosis associated with smoking, trauma, underlying vaso-occlusion, and hypercoagulability.1,2 Risk factors include a history of smoking as well as conditions associated with chronic hypoxia, such as severe peripheral vascular disease, subclavian artery stenosis, hypercoagulable states, monoclonal gammopathy, steal syndrome from an arteriovenous fistula, end-stage renal failure, calciphylaxis, and obesity.1
Histopathology of DDA reveals a diffuse dermal proliferation of capillaries due to upregulation of vascular endothelial growth factor secondary to chronic ischemia and hypoxia.1,2 Small, well-formed capillaries surrounded by pericytes dissect through dermal collagen into the subcutis (eFigure 1). Spindle-shaped cells with vacuolated cytoplasm and scattered extravasated erythrocytes with hemosiderin may be observed.2 Cellular atypia generally is not seen.2,3 Diffuse dermal angiomatosis is characterized by positive CD31, CD34, and ERG immunostaining1 and HHV-8 and D2-40 negativity.2 In our patient, the areas suggestive of connective tissue calciumlike depositions were concerning for dystrophic calcification related to end-stage renal disease. Although Von Kossa staining failed to highlight vascular calcifications, early calciphylaxis from end-stage renal disease could not be excluded.
The main goal of DDA treatment is to target tissue hypoxia, and primary preventive measures aim to reduce risk factors associated with atherosclerosis.1 Treatment options for DDA include revascularization, reduction mammoplasty, excision, isotretinoin, oral corticosteroids, smoking cessation, pentoxifylline plus aspirin, and management of underlying calciphylaxis.1,2 Spontaneous resolution of DDA rarely has been reported.1
Acroangiodermatitis, also known as pseudo–Kaposi sarcoma (KS), is a rare angioproliferative disorder that often is associated with vascular anomalies.4,5 It is divided into 2 main variants: Mali type, which is associated with chronic venous insufficiency, and Stewart-Bluefarb type, associated with arteriovenous malformations.4 This condition is characterized by red to violaceous macules, papules, or plaques that may become ulcerated or coalesce to form larger confluent patches, typically arising on the lower extremities.4,6,7 Histopathology of acroangiodermatitis reveals circumscribed lobular proliferation of thick-walled dermal vessels (eFigure 2), in contrast to the diffuse dermal proliferation of endothelial cells between collagen bundles seen in DDA.2,3,6
Angiosarcoma is a rare, highly aggressive vascular tumor that originates from vascular or lymphatic endothelial cells. It typically manifests with raised, bruiselike, erythematous to violaceous papules or plaques.8,9 Histopathologically, the hallmark feature of angiosarcoma is abnormal, pleomorphic, malignant endothelial cells with pale, light, eosinophilic cytoplasm and hyperchromatic nuclei (eFigure 3).2,9 In poorly differentiated cases, malignant endothelial cells may exhibit an epithelioid morphology with areas of hemorrhage and necrosis.9 Immunohistochemistry is positive for ERG, CD34, CD31, vascular endothelial growth factor, and D2-40.2,9
Kaposi sarcoma is a soft tissue malignancy known to occur in immunosuppressed patients such as individuals with AIDS or those undergoing immunosuppressive therapy for organ transplantation.10 There are 4 major forms of KS: classic (appearing on the lower extremities in elderly men of Mediterranean and Eastern European descent), endemic (occurring in children specifically in Africa with generalized lymph node involvement), HIV/ AIDS–related (occurring in patients not taking highly active antiretroviral therapy with diffuse involvement of the skin and internal organs), and iatrogenic (occurring in immunosuppressed patients with diffuse involvement of the skin and internal organs).10,11 Kaposi sarcoma presents as multiple reddish brown, raised or flat, painless, nonblanching mucocutaneous lesions that occasionally can ulcerate and bleed.11 Histopathologic features of KS include vascular proliferation in the dermis with diffuse slitlike lumen formation with the promontory sign, hyaline globules, hemosiderin accumulation, and an inflammatory component that often contains plasma cells (eFigure 4).2,11 Kaposi sarcoma is characterized by positive staining for CD31, CD34, D2-40, and HHV-8; the last 2 are an important distinction from DDA.2
Targetoid hemosiderotic hemangioma, also known as hobnail hemangioma, is a benign vascular lesion that typically manifests as a solitary, brown to violaceous papule or plaque on the trunk or extremities.12 It is sometimes surrounded by a pale area and a peripheral ecchymotic ring, giving the lesion a targetoid appearance.12,13 Histopathologic features include dilated, thin-walled vessels with prominent endothelial hobnailing in the papillary dermis, slit-shaped vascular channels between collagen bundles in the deeper dermis, and an interstitial lymphocytic infiltrate with extravasated erythrocytes and hemosiderin deposits (eFigure 5).12,14 The etiology of targetoid hemosiderotic hemangioma remains unclear. Chronic inflammation, trauma, exposure to ionizing radiation, and vascular obstruction have been suggested as inciting factors, though many cases have been reported without a history of cutaneous injury.12,13 Studies suggest a lymphatic origin instead of its original classification as a hemangioma.13,15 The endothelial cells stain positive with CD31 and may stain with D2-40 and CD34.13,15
- Nguyen N, Silfvast-Kaiser AS, Frieder J, et al. Diffuse dermal angiomatosis of the breast. Proc Bayl Univ Med Cent. 2020;33:273-275. doi:10.1080/08998280.2020.1722052
- Frikha F, Boudaya S, Abid N, et al. Diffuse dermal angiomatosis of the breast with adjacent fat necrosis: a case report and review of the literature. Dermatol Online J. 2018;24:13030/qt1vq114n7
- Yang H, Ahmed I, Mathew V, et al. Diffuse dermal angiomatosis of the breast. Arch Dermatol. 2006;142:343-347. doi:10.1001 /archderm.142.3.343
- Chhabra G, Verma P, Khullar G, et al. Acroangiodermatitis, Mali and Stewart-Bluefarb type: two additional cases in adolescents. Australas J Dermatol. 2021;62:E156-E157. doi:10.1111/ajd.13386
- Ramírez-Marín HA, Ruben-Castillo C, Barrera-Godínez A, et al. Acroangiodermatitis of the hand secondary to a dysfunctional a rteriovenous fistula. Ann Vasc Surg. 2021;77:350.e13-350.e17. doi:10.1016/j.avsg.2021.05.042
- Sun L, Duarte S, Soares-de-Almeida L. Acroangiodermatitis of Mali—an unusual cause of painful ulcer. Actas Dermo-Sifiliográficas. 2023;114:546. doi:10.1016/j.ad.2022.07.013
- Parsi K, O’Connor A, Bester L. Stewart–Bluefarb syndrome: report of five cases and a review of literature. Phlebology. 2015;30:505-514. doi:10.1177/0268355514548090
- Alharbi A, Kim YC, AlShomer F, et al. Utility of multimodal treatment protocols in the management of scalp cutaneous angiosarcoma. Plast Reconstr Surg Glob Open. 2023;11:E4827. doi:10.1097 /GOX.0000000000004827
- Young RJ, Brown NJ, Reed MW, et al. Angiosarcoma. Lancet Oncol. 2010;11:983-991. doi:10.1016/S1470-2045(10)70023-1
- Bishop BN, Lynch DT. Kaposi sarcoma. StatPearls [Internet]. StatPearls Publishing; 2024. Updated June 5, 2023. Accessed January 7, 2024. http://www.ncbi.nlm.nih.gov/books/NBK534839/
- Cesarman E, Damania B, Krown SE, et al. Kaposi sarcoma. Nat Rev Dis Primer. 2019;5:1-21. doi:10.1038/s41572-019-0060-9
- AbuHilal M, Breslavet M, Ho N, et al. Hobnail hemangioma (superficial hemosiderotic lymphovascular malformation) in children: a series of 6 pediatric cases and review of the literature. J Cutan Med Surg. 2016;20:216-220. doi:10.1177/1203475415612421
- Kakizaki P, Valente NYS, Paiva DLM, et al. Targetoid hemosiderotic hemangioma—case report. An Bras Dermatol. 2014;89:956-959. doi:10.1590/abd1806-4841.20143264
- Trindade F, Kutzner H, Tellechea Ó, et al. Hobnail hemangioma reclassified as superficial lymphatic malformation: a study of 52 cases. J Am Acad Dermatol. 2012;66:112-115. doi:10.1016/j.jaad.2011.05.019
- Hejnold M, Dyduch G, Mojsa I, et al. Hobnail hemangioma: a immunohistochemical study and literature review. Pol J Pathol. 2012;63:189-192. doi:10.5114/pjp.2012.31504
- Nguyen N, Silfvast-Kaiser AS, Frieder J, et al. Diffuse dermal angiomatosis of the breast. Proc Bayl Univ Med Cent. 2020;33:273-275. doi:10.1080/08998280.2020.1722052
- Frikha F, Boudaya S, Abid N, et al. Diffuse dermal angiomatosis of the breast with adjacent fat necrosis: a case report and review of the literature. Dermatol Online J. 2018;24:13030/qt1vq114n7
- Yang H, Ahmed I, Mathew V, et al. Diffuse dermal angiomatosis of the breast. Arch Dermatol. 2006;142:343-347. doi:10.1001 /archderm.142.3.343
- Chhabra G, Verma P, Khullar G, et al. Acroangiodermatitis, Mali and Stewart-Bluefarb type: two additional cases in adolescents. Australas J Dermatol. 2021;62:E156-E157. doi:10.1111/ajd.13386
- Ramírez-Marín HA, Ruben-Castillo C, Barrera-Godínez A, et al. Acroangiodermatitis of the hand secondary to a dysfunctional a rteriovenous fistula. Ann Vasc Surg. 2021;77:350.e13-350.e17. doi:10.1016/j.avsg.2021.05.042
- Sun L, Duarte S, Soares-de-Almeida L. Acroangiodermatitis of Mali—an unusual cause of painful ulcer. Actas Dermo-Sifiliográficas. 2023;114:546. doi:10.1016/j.ad.2022.07.013
- Parsi K, O’Connor A, Bester L. Stewart–Bluefarb syndrome: report of five cases and a review of literature. Phlebology. 2015;30:505-514. doi:10.1177/0268355514548090
- Alharbi A, Kim YC, AlShomer F, et al. Utility of multimodal treatment protocols in the management of scalp cutaneous angiosarcoma. Plast Reconstr Surg Glob Open. 2023;11:E4827. doi:10.1097 /GOX.0000000000004827
- Young RJ, Brown NJ, Reed MW, et al. Angiosarcoma. Lancet Oncol. 2010;11:983-991. doi:10.1016/S1470-2045(10)70023-1
- Bishop BN, Lynch DT. Kaposi sarcoma. StatPearls [Internet]. StatPearls Publishing; 2024. Updated June 5, 2023. Accessed January 7, 2024. http://www.ncbi.nlm.nih.gov/books/NBK534839/
- Cesarman E, Damania B, Krown SE, et al. Kaposi sarcoma. Nat Rev Dis Primer. 2019;5:1-21. doi:10.1038/s41572-019-0060-9
- AbuHilal M, Breslavet M, Ho N, et al. Hobnail hemangioma (superficial hemosiderotic lymphovascular malformation) in children: a series of 6 pediatric cases and review of the literature. J Cutan Med Surg. 2016;20:216-220. doi:10.1177/1203475415612421
- Kakizaki P, Valente NYS, Paiva DLM, et al. Targetoid hemosiderotic hemangioma—case report. An Bras Dermatol. 2014;89:956-959. doi:10.1590/abd1806-4841.20143264
- Trindade F, Kutzner H, Tellechea Ó, et al. Hobnail hemangioma reclassified as superficial lymphatic malformation: a study of 52 cases. J Am Acad Dermatol. 2012;66:112-115. doi:10.1016/j.jaad.2011.05.019
- Hejnold M, Dyduch G, Mojsa I, et al. Hobnail hemangioma: a immunohistochemical study and literature review. Pol J Pathol. 2012;63:189-192. doi:10.5114/pjp.2012.31504
Painful Ulcers on the Elbows, Knees, and Ankles
Painful Ulcers on the Elbows, Knees, and Ankles
A 46-year-old woman with a history of systemic lupus erythematosus and end-stage renal disease presented to the dermatology department with painful ulcers on the extensor surfaces of the elbows, knees, and ankles of 2 months’ duration. Physical examination revealed angulated ulcers with surrounding pink erythema. A 4-mm punch biopsy and CD31 immunostaining of the left knee revealed dystrophic elastic fibers and purplish calciumlike depositions on connective tissue fibers in the mid to deep dermis.
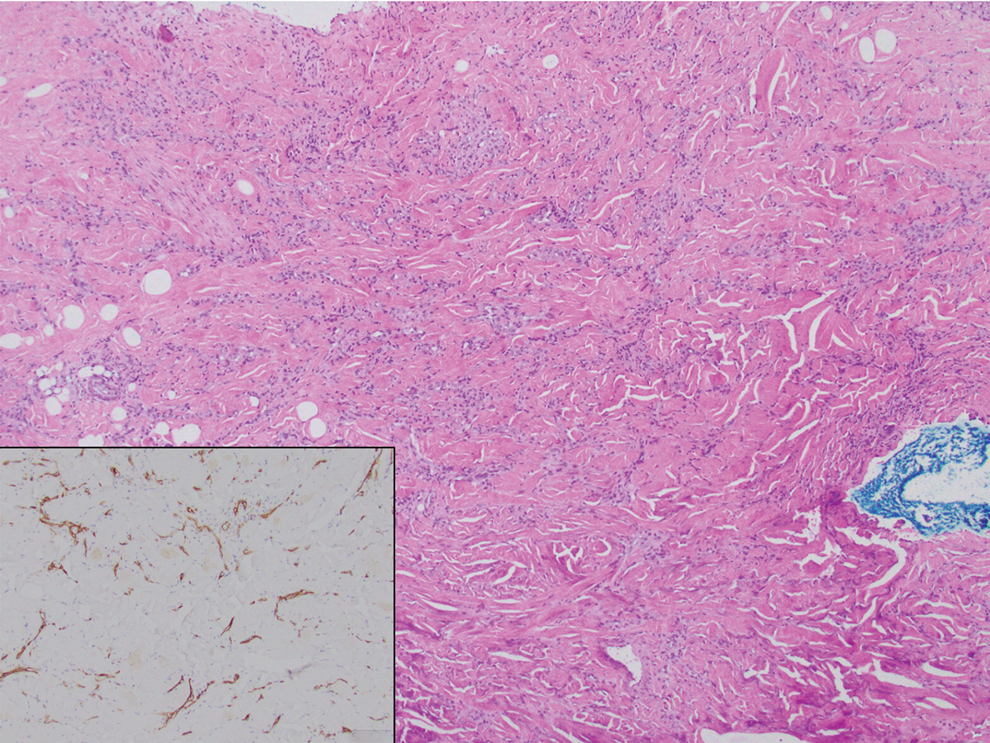

Solitary Lesion on the Umbilicus
Solitary Lesion on the Umbilicus
THE DIAGNOSIS: Cutaneous Endometriosis
Endometriosis is the ectopic presence of endometrial tissue and occurs in approximately 13% of women of childbearing age.1 This non-neoplastic lesion can manifest on the skin in less than 5.5% of endometriosis cases worldwide. Historically, secondary cutaneous endometriosis (CE) most frequently has been associated with prior gynecologic surgery (often cesarean section)2; however, an increased incidence of primary CE in patients without prior surgical history recently has been documented in the literature.3 While secondary CE usually manifests at the site of a surgical scar, primary CE has a predilection for the umbilicus (Villar nodule). In both primary and secondary CE, patients present clinically with a solitary nodule and abdominal pain that may be exacerbated during menstruation. Bleeding without associated pain may be more common in primary CE, while bleeding with pain may be more common in secondary CE. Cutaneous endometriosis often is overlooked given its low incidence, leading to delayed diagnosis. Primary CE often is misdiagnosed clinically as a pyogenic granuloma, Sister Mary Joseph nodule, or keloid, while secondary CE may be mistaken for a fibroma, incisional hernia, or granuloma.2
Primary and secondary CE have identical histopathologic features. Glands of variable size consisting of a single epithelial layer of columnar cells are present in the reticular dermis or subcutis (quiz image).4 The accompanying periglandular stroma often is uniform, consisting of spindle-shaped basophilic cells with abundant vascular structures. The stroma may contain moderate numbers of mitotic figures, a chronic inflammatory infiltrate, and extravasated red blood cells. The ectopic tissue may be inactive or display morphologic changes resembling those of the endometrium in the normal menstrual cycle.4 As the ectopic tissue progresses through the stages of menstruation, the glandular morphology also transforms. The proliferative stage demonstrates increased epithelial mitotic figures, the secretory stage exhibits intraluminal secretion, and during menstruation there are degenerative epithelial cells and evidence of vascular congestion. A mixture of glandular stages may be seen in biopsy results. Robust immunohistochemical expression of CD10 in the endometrial stroma can aid in diagnosis (Figure 1). Estrogen and progesterone receptor immunostaining also shows strong nuclear positivity, except in decidualized tissue.4 Unlike intestinal glands, endometrial glands do not express CDX2 or CK20.5 Complete surgical excision of CE usually is curative; however, recurrence has been documented in 10% (3/30) of cases.2
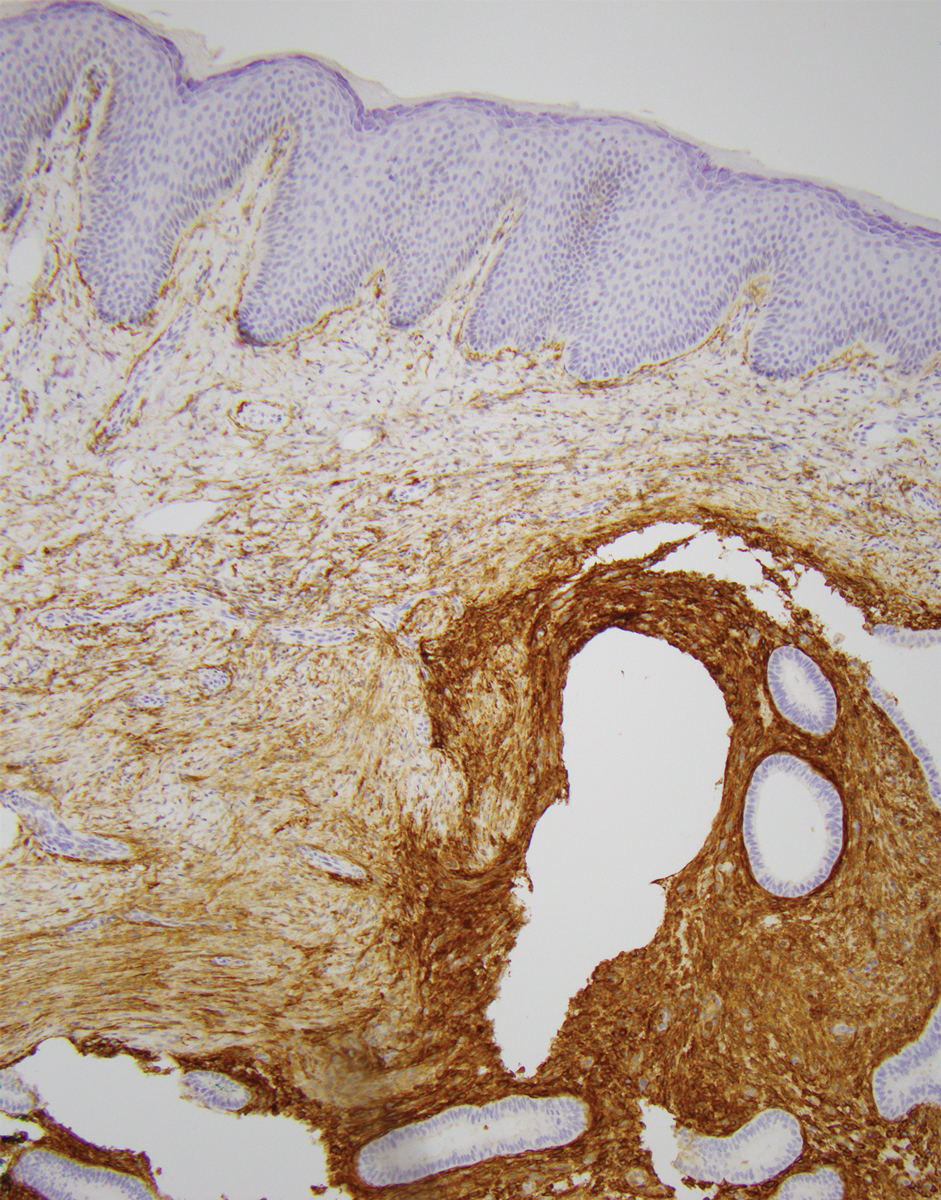
Breast carcinoma is the most common internal malignancy associated with cutaneous metastasis and may develop prior to visceral diagnosis. It is possible that tumor cells travel through the communicating networks of the cutaneous lymphatic ducts and the mammary lymphatic plexus; however, cutaneous manifestation often is located on the ipsilateral breast, and therefore tumor expansion rather than true metastasis cannot always be ruled out. On histopathology, findings of breast adenocarcinoma include tumor cells that tend to show either interstitial, nodular, mixed, or intravascular growth patterns (Figure 2). Tumor cells may invade the stroma in clusters or as individual cells. Sites of distant metastasis may show an increased likelihood of vascular and lymphatic invasion.6
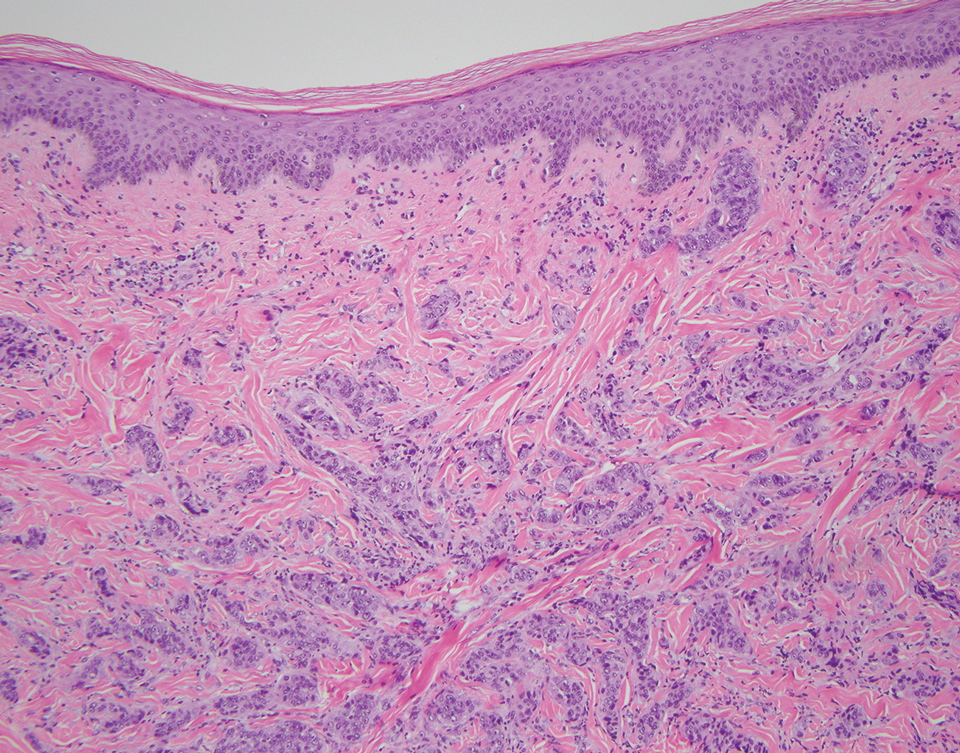
Nodular hidradenoma often manifests as a solitary nodule in the head or neck region, predominantly in women.7 Pathology shows well-demarcated intradermal aggregates of tumor cells within a hyalinized stroma; connection to the epidermis is not a feature of nodular hidradenoma. The epithelial component consists of polygonal cells with eosinophilic to amphophilic cytoplasm as well as large glycogenated cells with pale to clear cytoplasm (leading to the alternative term clear cell hidradenoma)(Figure 3). The cystic portion represents deterioration of tumor cells. Surgical excision usually is curative, although lesions may recur. Malignant transformation is rare.7
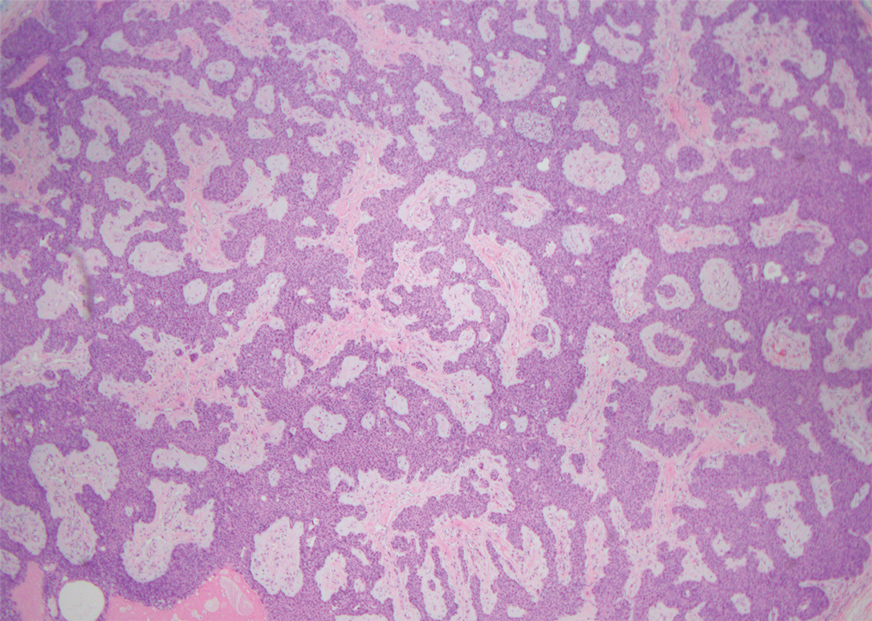
Sister Mary Joseph nodule is a cutaneous involvement of the umbilicus by a metastatic malignancy, often from an intra-abdominal primary malignancy (most commonly ovarian carcinoma in women and colonic carcinoma in men). Clinically, patients present with a solitary firm nodule or plaque within the umbilicus.8,9 Histopathology recapitulates the primary tumor (Figure 4).9 Sister Mary Joseph nodule portends a poor prognosis, with a survival rate of less than 8 months from the time of diagnosis.10
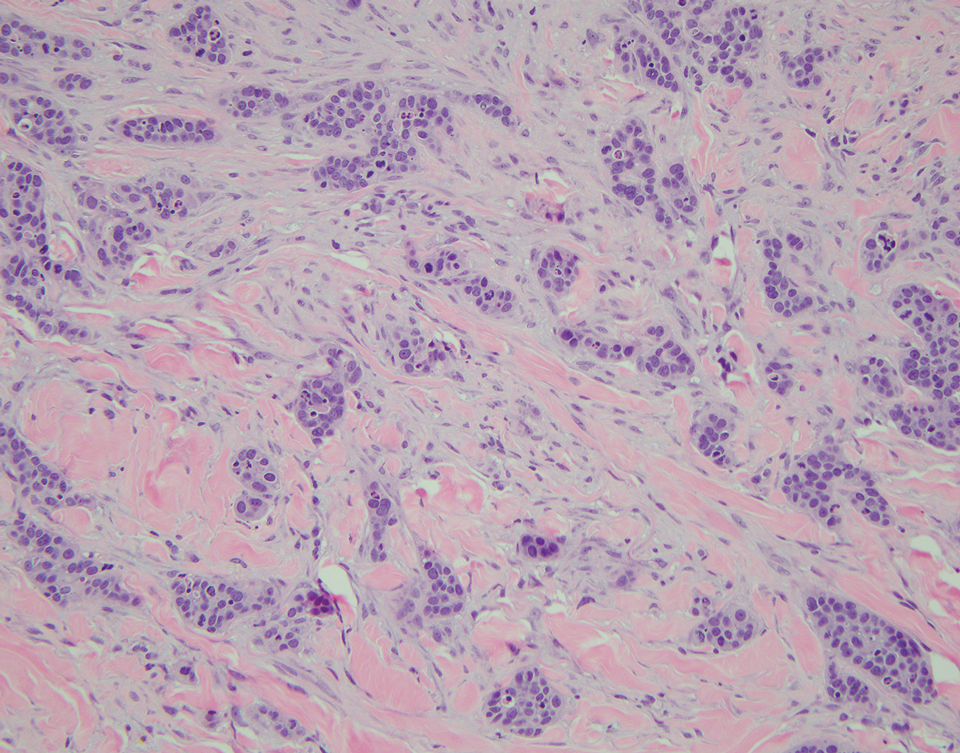
Urachal duct cyst develops from a remnant of the urachus that closed appropriately at the umbilicus and bladder but did not completely regress. It may manifest as an extraperitoneal mass at the umbilicus. Clinically, urachal duct cysts may be asymptomatic until an inciting event (eg, inflammation, deposition of calculus, or malignancy) occurs.11 Histopathology shows cystically dilated structures lined with a transitional epithelium (Figure 5).12 Urachal duct cysts usually are diagnosed in children or young adults and subsequently are excised.11
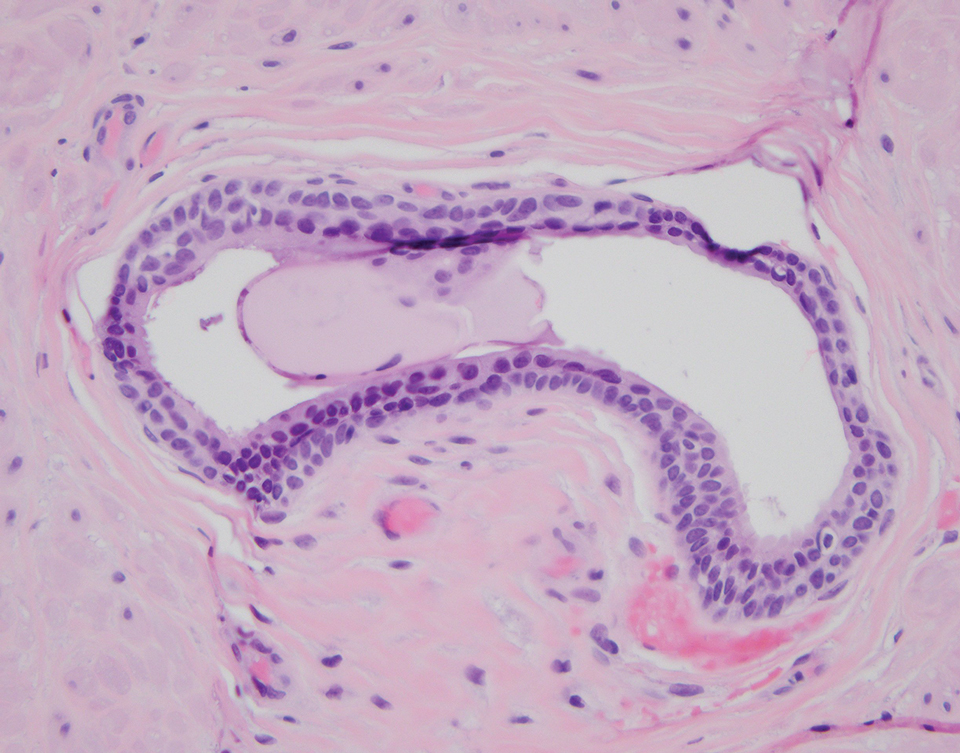
- Harder C, Velho RV, Brandes I, et al. Assessing the true prevalence of endometriosis: a narrative review of literature data. Int J Gynaecol Obstet. 2024;167:883-900. doi:10.1002/ijgo.15756
- Lopez-Soto A, Sanchez-Zapata MI, Martinez-Cendan JP, et al. Cutaneous endometriosis: presentation of 33 cases and literature review. Eur J Obstet Gynecol Reprod Biol. Feb 2018;221:58-63. doi:10.1016 /j.ejogrb.2017.11.024
- Dridi D, Chiaffarino F, Parazzini F, et al. Umbilical endometriosis: a systematic literature review and pathogenic theory proposal. J Clin Med. 2022;11:995. doi:10.3390/jcm11040995
- Farooq U, Laureano AC, Miteva M, Elgart GW. Cutaneous endometriosis: diagnostic immunohistochemistry and clinicopathologic correlation. J Cutan Pathol. 2011;38:525-528. doi:10.1111/j.1600-0560.2011.01681.x
- Gadducci A, Zannoni GF. Endometriosis-associated extraovarian malignancies: a challenging question for the clinician and the pathologist. Anticancer Res. 2020;40:2429-2438. doi:10.21873/anticanres.14212
- Ronen S, Suster D, Chen WS, et al. Histologic patterns of cutaneous metastases of breast carcinoma: a clinicopathologic study of 232 cases. Am J Dermatopathol. 2021;43:401-411. doi:10.1097 /DAD.0000000000001841
- Nandeesh BN, Rajalakshmi T. A study of histopathologic spectrum of nodular hidradenoma. Am J Dermatopathol. 2012;34:461-470. doi:10.1097/DAD.0b013e31821a4d33
- Abu-Hilal M, Newman JS. Sister Mary Joseph and her nodule: historical and clinical perspective. Am J Med Sci. 2009;337:271-273. doi:10.1097/MAJ.0b013e3181954187
- Powell FC, Cooper AJ, Massa MC, et al. Sister Mary Joseph’s nodule: a clinical and histologic study. J Am Acad Dermatol. 1984;10:610-615. doi:10.1016/s0190-9622(84)80265-0
- Hugen N, Kanne H, Simmer F, et al. Umbilical metastases: real-world data shows abysmal outcome. Int J Cancer. 2021;149: 1266-1273. doi:10.1002/ijc.33684
- Al-Salem A. An Illustrated Guide to Pediatric Urology. 1st ed. Springer Cham; 2016.
- Schubert GE, Pavkovic MB, Bethke-Bedürftig BA. Tubular urachal remnants in adult bladders. J Urol. 1982;127:40-42. doi:10.1016/s0022- 5347(17)53595-8
THE DIAGNOSIS: Cutaneous Endometriosis
Endometriosis is the ectopic presence of endometrial tissue and occurs in approximately 13% of women of childbearing age.1 This non-neoplastic lesion can manifest on the skin in less than 5.5% of endometriosis cases worldwide. Historically, secondary cutaneous endometriosis (CE) most frequently has been associated with prior gynecologic surgery (often cesarean section)2; however, an increased incidence of primary CE in patients without prior surgical history recently has been documented in the literature.3 While secondary CE usually manifests at the site of a surgical scar, primary CE has a predilection for the umbilicus (Villar nodule). In both primary and secondary CE, patients present clinically with a solitary nodule and abdominal pain that may be exacerbated during menstruation. Bleeding without associated pain may be more common in primary CE, while bleeding with pain may be more common in secondary CE. Cutaneous endometriosis often is overlooked given its low incidence, leading to delayed diagnosis. Primary CE often is misdiagnosed clinically as a pyogenic granuloma, Sister Mary Joseph nodule, or keloid, while secondary CE may be mistaken for a fibroma, incisional hernia, or granuloma.2
Primary and secondary CE have identical histopathologic features. Glands of variable size consisting of a single epithelial layer of columnar cells are present in the reticular dermis or subcutis (quiz image).4 The accompanying periglandular stroma often is uniform, consisting of spindle-shaped basophilic cells with abundant vascular structures. The stroma may contain moderate numbers of mitotic figures, a chronic inflammatory infiltrate, and extravasated red blood cells. The ectopic tissue may be inactive or display morphologic changes resembling those of the endometrium in the normal menstrual cycle.4 As the ectopic tissue progresses through the stages of menstruation, the glandular morphology also transforms. The proliferative stage demonstrates increased epithelial mitotic figures, the secretory stage exhibits intraluminal secretion, and during menstruation there are degenerative epithelial cells and evidence of vascular congestion. A mixture of glandular stages may be seen in biopsy results. Robust immunohistochemical expression of CD10 in the endometrial stroma can aid in diagnosis (Figure 1). Estrogen and progesterone receptor immunostaining also shows strong nuclear positivity, except in decidualized tissue.4 Unlike intestinal glands, endometrial glands do not express CDX2 or CK20.5 Complete surgical excision of CE usually is curative; however, recurrence has been documented in 10% (3/30) of cases.2
Breast carcinoma is the most common internal malignancy associated with cutaneous metastasis and may develop prior to visceral diagnosis. It is possible that tumor cells travel through the communicating networks of the cutaneous lymphatic ducts and the mammary lymphatic plexus; however, cutaneous manifestation often is located on the ipsilateral breast, and therefore tumor expansion rather than true metastasis cannot always be ruled out. On histopathology, findings of breast adenocarcinoma include tumor cells that tend to show either interstitial, nodular, mixed, or intravascular growth patterns (Figure 2). Tumor cells may invade the stroma in clusters or as individual cells. Sites of distant metastasis may show an increased likelihood of vascular and lymphatic invasion.6
Nodular hidradenoma often manifests as a solitary nodule in the head or neck region, predominantly in women.7 Pathology shows well-demarcated intradermal aggregates of tumor cells within a hyalinized stroma; connection to the epidermis is not a feature of nodular hidradenoma. The epithelial component consists of polygonal cells with eosinophilic to amphophilic cytoplasm as well as large glycogenated cells with pale to clear cytoplasm (leading to the alternative term clear cell hidradenoma)(Figure 3). The cystic portion represents deterioration of tumor cells. Surgical excision usually is curative, although lesions may recur. Malignant transformation is rare.7
Sister Mary Joseph nodule is a cutaneous involvement of the umbilicus by a metastatic malignancy, often from an intra-abdominal primary malignancy (most commonly ovarian carcinoma in women and colonic carcinoma in men). Clinically, patients present with a solitary firm nodule or plaque within the umbilicus.8,9 Histopathology recapitulates the primary tumor (Figure 4).9 Sister Mary Joseph nodule portends a poor prognosis, with a survival rate of less than 8 months from the time of diagnosis.10
Urachal duct cyst develops from a remnant of the urachus that closed appropriately at the umbilicus and bladder but did not completely regress. It may manifest as an extraperitoneal mass at the umbilicus. Clinically, urachal duct cysts may be asymptomatic until an inciting event (eg, inflammation, deposition of calculus, or malignancy) occurs.11 Histopathology shows cystically dilated structures lined with a transitional epithelium (Figure 5).12 Urachal duct cysts usually are diagnosed in children or young adults and subsequently are excised.11
THE DIAGNOSIS: Cutaneous Endometriosis
Endometriosis is the ectopic presence of endometrial tissue and occurs in approximately 13% of women of childbearing age.1 This non-neoplastic lesion can manifest on the skin in less than 5.5% of endometriosis cases worldwide. Historically, secondary cutaneous endometriosis (CE) most frequently has been associated with prior gynecologic surgery (often cesarean section)2; however, an increased incidence of primary CE in patients without prior surgical history recently has been documented in the literature.3 While secondary CE usually manifests at the site of a surgical scar, primary CE has a predilection for the umbilicus (Villar nodule). In both primary and secondary CE, patients present clinically with a solitary nodule and abdominal pain that may be exacerbated during menstruation. Bleeding without associated pain may be more common in primary CE, while bleeding with pain may be more common in secondary CE. Cutaneous endometriosis often is overlooked given its low incidence, leading to delayed diagnosis. Primary CE often is misdiagnosed clinically as a pyogenic granuloma, Sister Mary Joseph nodule, or keloid, while secondary CE may be mistaken for a fibroma, incisional hernia, or granuloma.2
Primary and secondary CE have identical histopathologic features. Glands of variable size consisting of a single epithelial layer of columnar cells are present in the reticular dermis or subcutis (quiz image).4 The accompanying periglandular stroma often is uniform, consisting of spindle-shaped basophilic cells with abundant vascular structures. The stroma may contain moderate numbers of mitotic figures, a chronic inflammatory infiltrate, and extravasated red blood cells. The ectopic tissue may be inactive or display morphologic changes resembling those of the endometrium in the normal menstrual cycle.4 As the ectopic tissue progresses through the stages of menstruation, the glandular morphology also transforms. The proliferative stage demonstrates increased epithelial mitotic figures, the secretory stage exhibits intraluminal secretion, and during menstruation there are degenerative epithelial cells and evidence of vascular congestion. A mixture of glandular stages may be seen in biopsy results. Robust immunohistochemical expression of CD10 in the endometrial stroma can aid in diagnosis (Figure 1). Estrogen and progesterone receptor immunostaining also shows strong nuclear positivity, except in decidualized tissue.4 Unlike intestinal glands, endometrial glands do not express CDX2 or CK20.5 Complete surgical excision of CE usually is curative; however, recurrence has been documented in 10% (3/30) of cases.2
Breast carcinoma is the most common internal malignancy associated with cutaneous metastasis and may develop prior to visceral diagnosis. It is possible that tumor cells travel through the communicating networks of the cutaneous lymphatic ducts and the mammary lymphatic plexus; however, cutaneous manifestation often is located on the ipsilateral breast, and therefore tumor expansion rather than true metastasis cannot always be ruled out. On histopathology, findings of breast adenocarcinoma include tumor cells that tend to show either interstitial, nodular, mixed, or intravascular growth patterns (Figure 2). Tumor cells may invade the stroma in clusters or as individual cells. Sites of distant metastasis may show an increased likelihood of vascular and lymphatic invasion.6
Nodular hidradenoma often manifests as a solitary nodule in the head or neck region, predominantly in women.7 Pathology shows well-demarcated intradermal aggregates of tumor cells within a hyalinized stroma; connection to the epidermis is not a feature of nodular hidradenoma. The epithelial component consists of polygonal cells with eosinophilic to amphophilic cytoplasm as well as large glycogenated cells with pale to clear cytoplasm (leading to the alternative term clear cell hidradenoma)(Figure 3). The cystic portion represents deterioration of tumor cells. Surgical excision usually is curative, although lesions may recur. Malignant transformation is rare.7
Sister Mary Joseph nodule is a cutaneous involvement of the umbilicus by a metastatic malignancy, often from an intra-abdominal primary malignancy (most commonly ovarian carcinoma in women and colonic carcinoma in men). Clinically, patients present with a solitary firm nodule or plaque within the umbilicus.8,9 Histopathology recapitulates the primary tumor (Figure 4).9 Sister Mary Joseph nodule portends a poor prognosis, with a survival rate of less than 8 months from the time of diagnosis.10
Urachal duct cyst develops from a remnant of the urachus that closed appropriately at the umbilicus and bladder but did not completely regress. It may manifest as an extraperitoneal mass at the umbilicus. Clinically, urachal duct cysts may be asymptomatic until an inciting event (eg, inflammation, deposition of calculus, or malignancy) occurs.11 Histopathology shows cystically dilated structures lined with a transitional epithelium (Figure 5).12 Urachal duct cysts usually are diagnosed in children or young adults and subsequently are excised.11
- Harder C, Velho RV, Brandes I, et al. Assessing the true prevalence of endometriosis: a narrative review of literature data. Int J Gynaecol Obstet. 2024;167:883-900. doi:10.1002/ijgo.15756
- Lopez-Soto A, Sanchez-Zapata MI, Martinez-Cendan JP, et al. Cutaneous endometriosis: presentation of 33 cases and literature review. Eur J Obstet Gynecol Reprod Biol. Feb 2018;221:58-63. doi:10.1016 /j.ejogrb.2017.11.024
- Dridi D, Chiaffarino F, Parazzini F, et al. Umbilical endometriosis: a systematic literature review and pathogenic theory proposal. J Clin Med. 2022;11:995. doi:10.3390/jcm11040995
- Farooq U, Laureano AC, Miteva M, Elgart GW. Cutaneous endometriosis: diagnostic immunohistochemistry and clinicopathologic correlation. J Cutan Pathol. 2011;38:525-528. doi:10.1111/j.1600-0560.2011.01681.x
- Gadducci A, Zannoni GF. Endometriosis-associated extraovarian malignancies: a challenging question for the clinician and the pathologist. Anticancer Res. 2020;40:2429-2438. doi:10.21873/anticanres.14212
- Ronen S, Suster D, Chen WS, et al. Histologic patterns of cutaneous metastases of breast carcinoma: a clinicopathologic study of 232 cases. Am J Dermatopathol. 2021;43:401-411. doi:10.1097 /DAD.0000000000001841
- Nandeesh BN, Rajalakshmi T. A study of histopathologic spectrum of nodular hidradenoma. Am J Dermatopathol. 2012;34:461-470. doi:10.1097/DAD.0b013e31821a4d33
- Abu-Hilal M, Newman JS. Sister Mary Joseph and her nodule: historical and clinical perspective. Am J Med Sci. 2009;337:271-273. doi:10.1097/MAJ.0b013e3181954187
- Powell FC, Cooper AJ, Massa MC, et al. Sister Mary Joseph’s nodule: a clinical and histologic study. J Am Acad Dermatol. 1984;10:610-615. doi:10.1016/s0190-9622(84)80265-0
- Hugen N, Kanne H, Simmer F, et al. Umbilical metastases: real-world data shows abysmal outcome. Int J Cancer. 2021;149: 1266-1273. doi:10.1002/ijc.33684
- Al-Salem A. An Illustrated Guide to Pediatric Urology. 1st ed. Springer Cham; 2016.
- Schubert GE, Pavkovic MB, Bethke-Bedürftig BA. Tubular urachal remnants in adult bladders. J Urol. 1982;127:40-42. doi:10.1016/s0022- 5347(17)53595-8
- Harder C, Velho RV, Brandes I, et al. Assessing the true prevalence of endometriosis: a narrative review of literature data. Int J Gynaecol Obstet. 2024;167:883-900. doi:10.1002/ijgo.15756
- Lopez-Soto A, Sanchez-Zapata MI, Martinez-Cendan JP, et al. Cutaneous endometriosis: presentation of 33 cases and literature review. Eur J Obstet Gynecol Reprod Biol. Feb 2018;221:58-63. doi:10.1016 /j.ejogrb.2017.11.024
- Dridi D, Chiaffarino F, Parazzini F, et al. Umbilical endometriosis: a systematic literature review and pathogenic theory proposal. J Clin Med. 2022;11:995. doi:10.3390/jcm11040995
- Farooq U, Laureano AC, Miteva M, Elgart GW. Cutaneous endometriosis: diagnostic immunohistochemistry and clinicopathologic correlation. J Cutan Pathol. 2011;38:525-528. doi:10.1111/j.1600-0560.2011.01681.x
- Gadducci A, Zannoni GF. Endometriosis-associated extraovarian malignancies: a challenging question for the clinician and the pathologist. Anticancer Res. 2020;40:2429-2438. doi:10.21873/anticanres.14212
- Ronen S, Suster D, Chen WS, et al. Histologic patterns of cutaneous metastases of breast carcinoma: a clinicopathologic study of 232 cases. Am J Dermatopathol. 2021;43:401-411. doi:10.1097 /DAD.0000000000001841
- Nandeesh BN, Rajalakshmi T. A study of histopathologic spectrum of nodular hidradenoma. Am J Dermatopathol. 2012;34:461-470. doi:10.1097/DAD.0b013e31821a4d33
- Abu-Hilal M, Newman JS. Sister Mary Joseph and her nodule: historical and clinical perspective. Am J Med Sci. 2009;337:271-273. doi:10.1097/MAJ.0b013e3181954187
- Powell FC, Cooper AJ, Massa MC, et al. Sister Mary Joseph’s nodule: a clinical and histologic study. J Am Acad Dermatol. 1984;10:610-615. doi:10.1016/s0190-9622(84)80265-0
- Hugen N, Kanne H, Simmer F, et al. Umbilical metastases: real-world data shows abysmal outcome. Int J Cancer. 2021;149: 1266-1273. doi:10.1002/ijc.33684
- Al-Salem A. An Illustrated Guide to Pediatric Urology. 1st ed. Springer Cham; 2016.
- Schubert GE, Pavkovic MB, Bethke-Bedürftig BA. Tubular urachal remnants in adult bladders. J Urol. 1982;127:40-42. doi:10.1016/s0022- 5347(17)53595-8
Solitary Lesion on the Umbilicus
Solitary Lesion on the Umbilicus
A 33-year-old woman with no notable medical or surgical history presented to our clinic with a solitary indurated nodule on the umbilicus that had been progressively enlarging for 1 year. The patient reported that she had undergone piercing of the umbilicus more than 5 years prior. She noted that the lesion was uncomfortable and pruritic and occasionally bled spontaneously. Physical examination revealed no other mucosal or cutaneous findings. A shave biopsy of the nodule was performed.
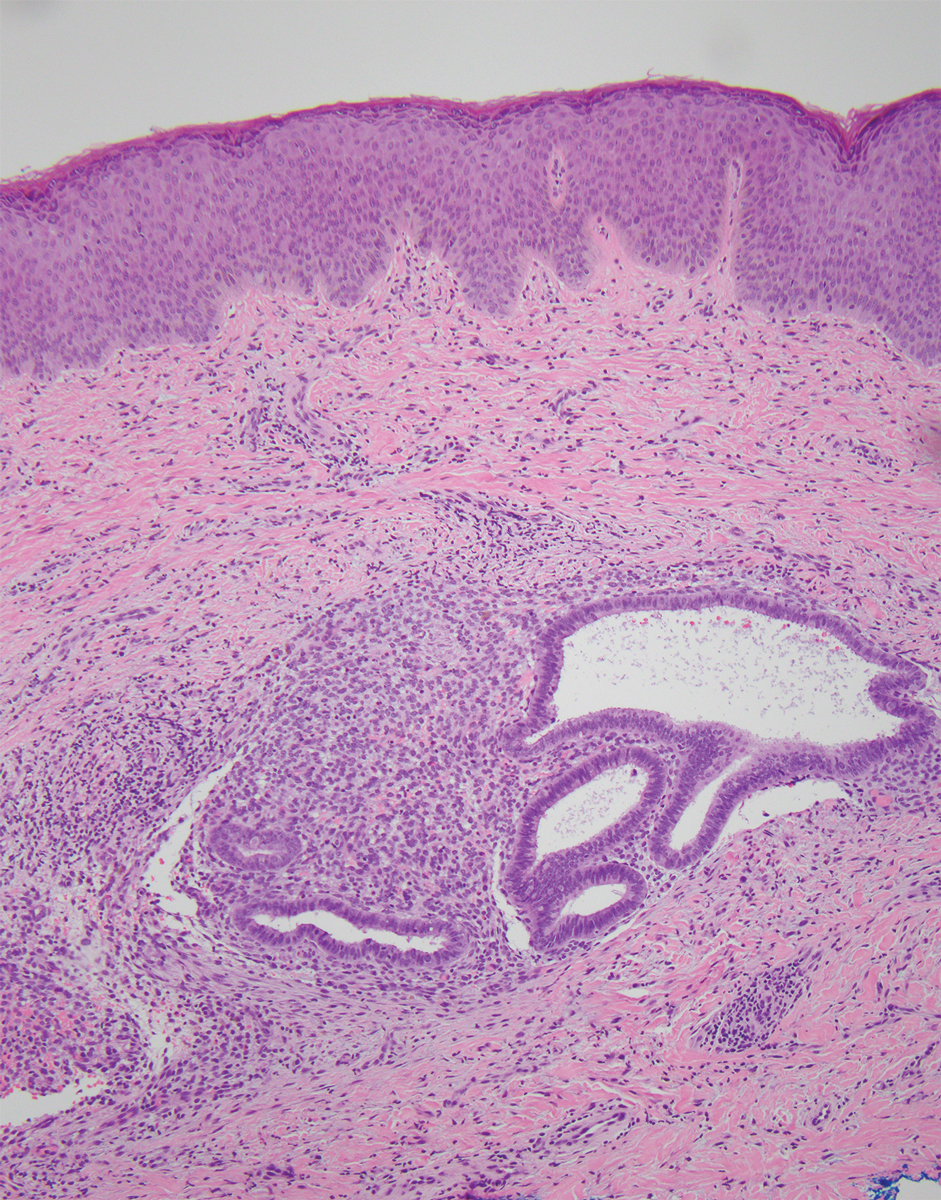

Scalp Nodule With Copious Fluid
Scalp Nodule With Copious Fluid
The Diagnosis: Apocrine Hidrocystoma
Histopathology of the excised nodule revealed a partially collapsed, multiloculated dermal cyst lined with apocrine cells, which was consistent with a diagnosis of apocrine hidrocystoma. Apocrine hidrocystomas are cysts that range from flesh-colored to blue-black and most commonly manifest as solitary lesions on the face, particularly near the eyelids.1,2 Apocrine hidrocystomas typically range from 1 to 10 mm in diameter and contain fluid that can be colorless, yellow-brown, or blue-black.1,2 Apocrine hidrocystomas usually are reported between the ages of 30 and 70 years and have no sex predilection.3
Apocrine hidrocystomas are thought to develop from adenomatous growth of apocrine sweat gland coils.4 The term apocrine hidrocystoma has been used interchangeably with apocrine cystadenoma, though some investigators have recommended using the latter term only for lesions with true papillary projections.5 Definitive diagnosis is obtained through histopathology, which typically shows unilocular or multilocular cystic spaces in the dermis lined by an apocrine secretory epithelium. These secretory cells often demonstrate decapitation secretion and apical snouting. The cyst wall may send pseudopapillary projections into the cystic cavity.1,2 While apocrine and eccrine hidrocystomas previously were recognized as separate entities, it has been suggested that so-called eccrine hidrocystomas are truly apocrine in nature, with a cyst wall that is compressed by the cyst contents.4
Apocrine hidrocystomas are benign and do not require treatment; however, they may be removed for cosmetic purposes, most commonly via surgical excision. Lesions treated with needle puncture as monotherapy frequently recur. Other successful methods for removal include cyst puncture followed by hypertonic glucose sclerotherapy, trichloroacetic acid injection, botulinum toxin A injection, or CO2 laser treatment.3,6
Several clinical and histopathologic findings can distinguish between apocrine hidrocystomas and other diagnoses in the differential. Lipomas are common benign tumors composed of mature fat that typically manifest as solitary, painless, soft nodules with a normal overlying epidermis. They frequently are distributed on the neck, arms, legs, and buttocks. While the differential for our patient initially included lipoma, these lesions do not contain or release fluid, which was present in our patient. On histopathology, lipoma shows a uniform population of mature fat cells with small, uniform, and eccentric nuclei (Figure 1).7
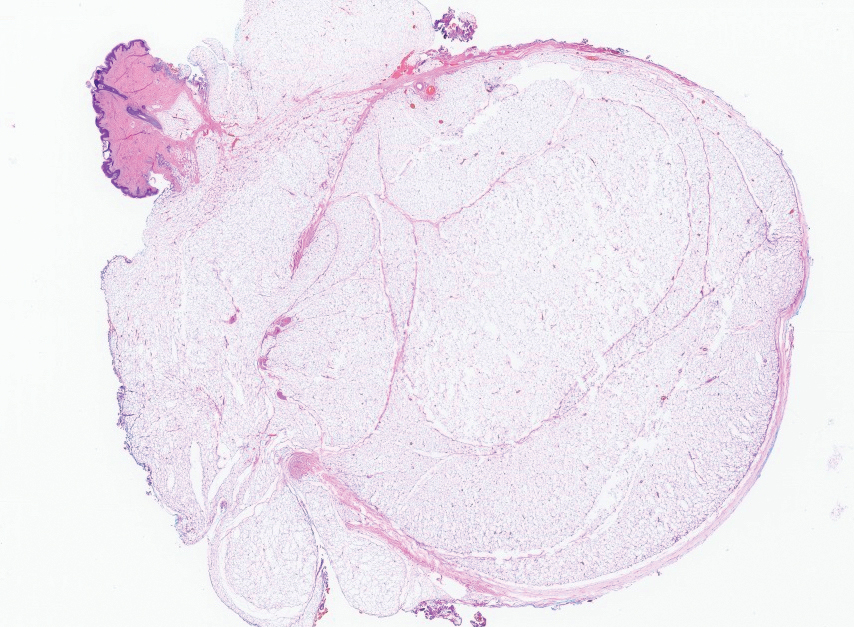
Epidermal inclusion cysts are derived from the follicular infundibulum and commonly are found on the face and upper trunk. They manifest as flesh-colored dermal nodules and may have a visible punctum. As opposed to the cystic cavities lined with apocrine cells seen in apocrine hidrocystomas, epidermal inclusion cysts are lined with a stratified squamous epithelium, are filled with laminated keratin, and have a visible granular layer (Figure 2).8
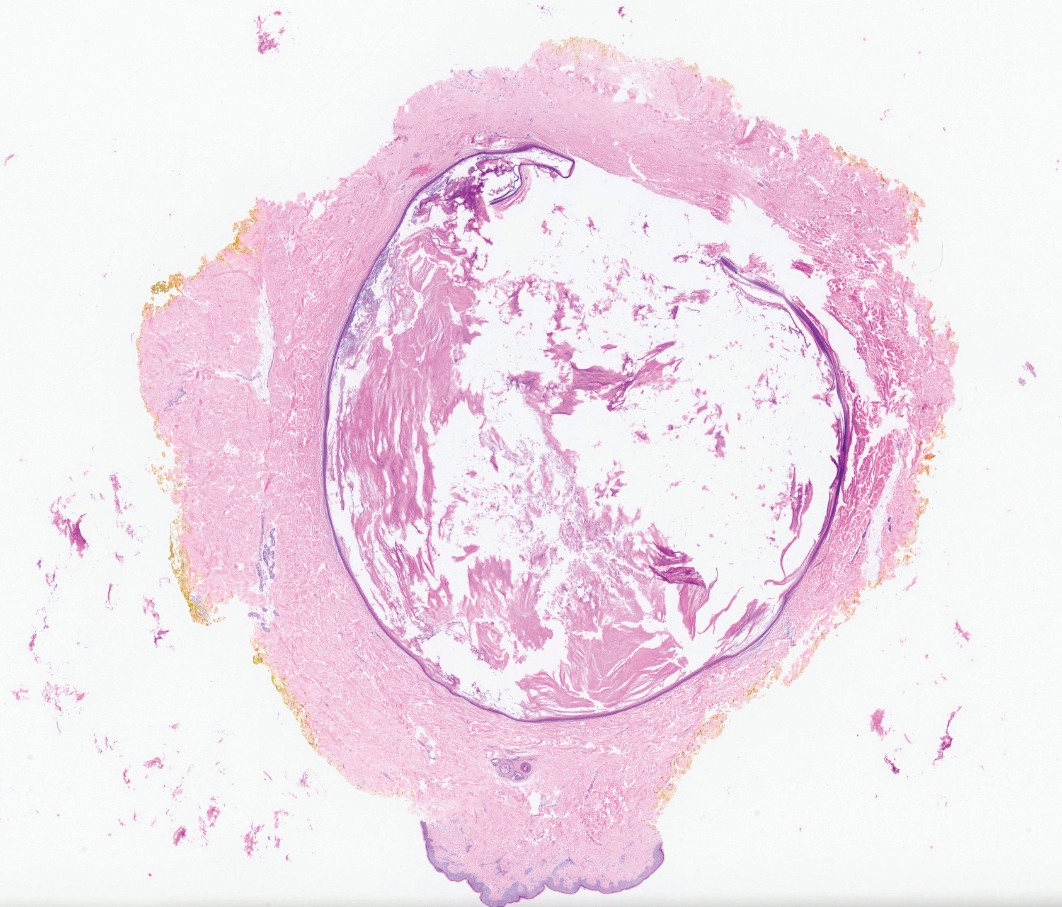
Pilar cysts, also known as trichilemmal cysts, clinically resemble epidermal inclusion cysts but are derived from the outer root sheath of hair follicles, manifesting as flesh-colored dermal nodules almost always found on the scalp. On histopathology, pilar cysts are lined with stratified squamous epithelial cells without a visible granular layer and are filled with compact eosinophilic keratin (Figure 3).8
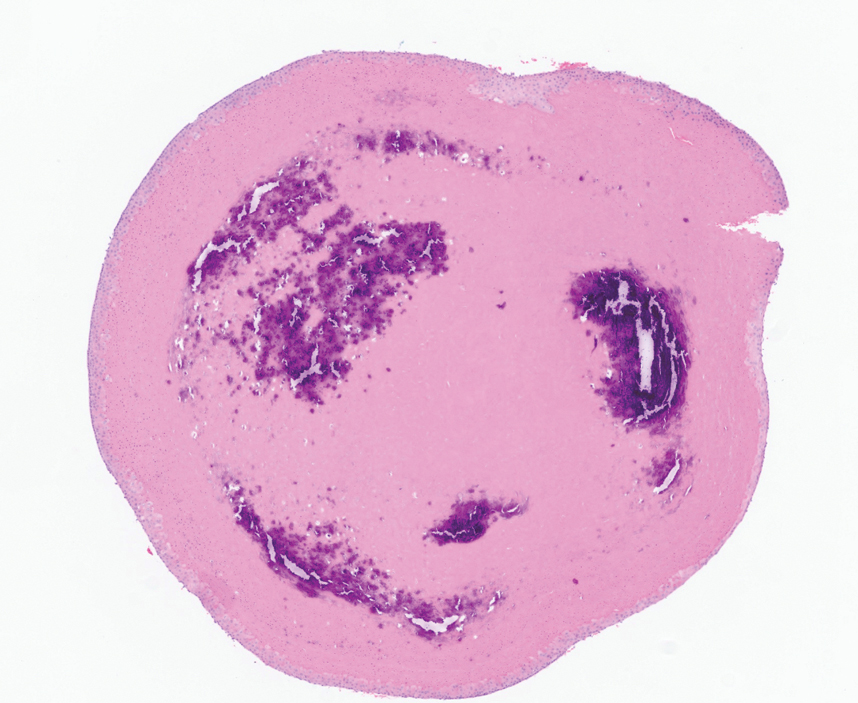
Tubular apocrine adenomas are benign neoplasms of the apocrine glands that manifest as smooth nodules. They are within the same spectrum as papillary eccrine adenomas, appearing more frequently on the legs and less frequently on the face and scalp.9 Histopathology generally demonstrates well-circumscribed lobules of tubular structures in the dermis. Similar to apocrine hidrocystomas, tubular apocrine adenomas will demonstrate an inner layer of columnar apocrine cells with decapitation secretion, but the tubular architecture helps differentiate it from other adnexal tumors (Figure 4).10
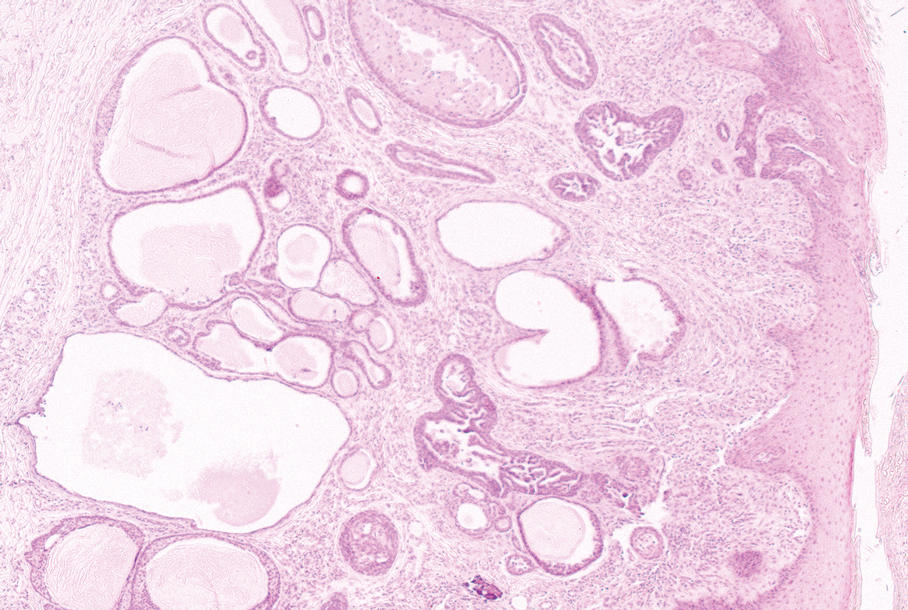
The clinical manifestation of the apocrine hidrocystoma in our patient was unusual due to its size and location. Apocrine hidrocystomas rarely are found on the scalp, with few other cases found in the literature. To our knowledge, this is the largest apocrine hidrocystoma found on the scalp to date, although there is at least 1 other published case of an apocrine hidrocystoma on the scalp measuring at least 3 cm in diameter.11 Our case highlights the importance of recognizing atypical manifestations of apocrine hidrocystomas, as a lesion on the midline scalp that discharges a thin fluid might raise initial concern for an intracranial connection. Awareness of atypical manifestations of common lesions can expand dermatologists’ differential diagnoses and help them to reassure patients.
- Smith JD. Apocrine hidrocystoma (cystadenoma). Arch Dermatol. 1974;109:700. doi:10.1001/archderm.1974.01630050046010
- Mehregan AH. Apocrine cystadenoma: a clinicopathologic study with special reference to the pigmented variety. Arch Dermatol. 1964;90:274. doi:10.1001/archderm.1964.01600030024005
- Hafsi W, Badri T, Shah F. Apocrine hidrocystoma. StatPearls [Internet]. Updated April 13, 2024. Accessed November 6, 2024. http://www.ncbi.nlm.nih.gov/books/NBK448109/
- de Viragh PA, Szeimies RM, Eckert F. Apocrine cystadenoma, apocrine hidrocystoma, and eccrine hidrocystoma: three distinct tumors defined by expression of keratins and human milk fat globulin 1. J Cutan Pathol. 1997;24:249-255. doi:10.1111/j.1600-0560.1997.tb01590.x
- Sugiyama A, Sugiura M, Piris A, et al. Apocrine cystadenoma and apocrine hidrocystoma: examination of 21 cases with emphasis on nomenclature according to proliferative features. J Cutan Pathol. 2007;34:912-917. doi:10.1111/j.1600-0560.2007.00757.x
- Bickley LK, Goldberg DJ, Imaeda S, et al. Treatment of multiple apocrine hidrocystomas with the carbon dioxide (CO2) laser. J Dermatol Surg Oncol. 1989;15:599-602. doi:10.1111/j.1524-4725.1989.tb03597.x
- Kaddu S. Smooth muscle, adipose and cartilage neoplasms. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. Elsevier; 2018:2086-2101.
- Stone MS. Cysts. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. Elsevier; 2018:1057-1074.
- Requena L, Sangüeza O. Tubular adenoma. In: Requena L, Sangüeza O, eds. Cutaneous Adnexal Neoplasms. Springer International Publishing; 2017:127-136. doi:10.1007/978-3-319-45704-8_12
- McCalmont TH, Pincus LB. Adnexal neoplasms. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. Elsevier; 2018:1057-1074.
- Nguyen HP, Barker HS, Bloomquist L, et al. Giant pigmented apocrine hidrocystoma of the scalp. Dermatol Online J. 2020;26:13030/qt7rt3s4pp.
The Diagnosis: Apocrine Hidrocystoma
Histopathology of the excised nodule revealed a partially collapsed, multiloculated dermal cyst lined with apocrine cells, which was consistent with a diagnosis of apocrine hidrocystoma. Apocrine hidrocystomas are cysts that range from flesh-colored to blue-black and most commonly manifest as solitary lesions on the face, particularly near the eyelids.1,2 Apocrine hidrocystomas typically range from 1 to 10 mm in diameter and contain fluid that can be colorless, yellow-brown, or blue-black.1,2 Apocrine hidrocystomas usually are reported between the ages of 30 and 70 years and have no sex predilection.3
Apocrine hidrocystomas are thought to develop from adenomatous growth of apocrine sweat gland coils.4 The term apocrine hidrocystoma has been used interchangeably with apocrine cystadenoma, though some investigators have recommended using the latter term only for lesions with true papillary projections.5 Definitive diagnosis is obtained through histopathology, which typically shows unilocular or multilocular cystic spaces in the dermis lined by an apocrine secretory epithelium. These secretory cells often demonstrate decapitation secretion and apical snouting. The cyst wall may send pseudopapillary projections into the cystic cavity.1,2 While apocrine and eccrine hidrocystomas previously were recognized as separate entities, it has been suggested that so-called eccrine hidrocystomas are truly apocrine in nature, with a cyst wall that is compressed by the cyst contents.4
Apocrine hidrocystomas are benign and do not require treatment; however, they may be removed for cosmetic purposes, most commonly via surgical excision. Lesions treated with needle puncture as monotherapy frequently recur. Other successful methods for removal include cyst puncture followed by hypertonic glucose sclerotherapy, trichloroacetic acid injection, botulinum toxin A injection, or CO2 laser treatment.3,6
Several clinical and histopathologic findings can distinguish between apocrine hidrocystomas and other diagnoses in the differential. Lipomas are common benign tumors composed of mature fat that typically manifest as solitary, painless, soft nodules with a normal overlying epidermis. They frequently are distributed on the neck, arms, legs, and buttocks. While the differential for our patient initially included lipoma, these lesions do not contain or release fluid, which was present in our patient. On histopathology, lipoma shows a uniform population of mature fat cells with small, uniform, and eccentric nuclei (Figure 1).7
Epidermal inclusion cysts are derived from the follicular infundibulum and commonly are found on the face and upper trunk. They manifest as flesh-colored dermal nodules and may have a visible punctum. As opposed to the cystic cavities lined with apocrine cells seen in apocrine hidrocystomas, epidermal inclusion cysts are lined with a stratified squamous epithelium, are filled with laminated keratin, and have a visible granular layer (Figure 2).8
Pilar cysts, also known as trichilemmal cysts, clinically resemble epidermal inclusion cysts but are derived from the outer root sheath of hair follicles, manifesting as flesh-colored dermal nodules almost always found on the scalp. On histopathology, pilar cysts are lined with stratified squamous epithelial cells without a visible granular layer and are filled with compact eosinophilic keratin (Figure 3).8
Tubular apocrine adenomas are benign neoplasms of the apocrine glands that manifest as smooth nodules. They are within the same spectrum as papillary eccrine adenomas, appearing more frequently on the legs and less frequently on the face and scalp.9 Histopathology generally demonstrates well-circumscribed lobules of tubular structures in the dermis. Similar to apocrine hidrocystomas, tubular apocrine adenomas will demonstrate an inner layer of columnar apocrine cells with decapitation secretion, but the tubular architecture helps differentiate it from other adnexal tumors (Figure 4).10
The clinical manifestation of the apocrine hidrocystoma in our patient was unusual due to its size and location. Apocrine hidrocystomas rarely are found on the scalp, with few other cases found in the literature. To our knowledge, this is the largest apocrine hidrocystoma found on the scalp to date, although there is at least 1 other published case of an apocrine hidrocystoma on the scalp measuring at least 3 cm in diameter.11 Our case highlights the importance of recognizing atypical manifestations of apocrine hidrocystomas, as a lesion on the midline scalp that discharges a thin fluid might raise initial concern for an intracranial connection. Awareness of atypical manifestations of common lesions can expand dermatologists’ differential diagnoses and help them to reassure patients.
The Diagnosis: Apocrine Hidrocystoma
Histopathology of the excised nodule revealed a partially collapsed, multiloculated dermal cyst lined with apocrine cells, which was consistent with a diagnosis of apocrine hidrocystoma. Apocrine hidrocystomas are cysts that range from flesh-colored to blue-black and most commonly manifest as solitary lesions on the face, particularly near the eyelids.1,2 Apocrine hidrocystomas typically range from 1 to 10 mm in diameter and contain fluid that can be colorless, yellow-brown, or blue-black.1,2 Apocrine hidrocystomas usually are reported between the ages of 30 and 70 years and have no sex predilection.3
Apocrine hidrocystomas are thought to develop from adenomatous growth of apocrine sweat gland coils.4 The term apocrine hidrocystoma has been used interchangeably with apocrine cystadenoma, though some investigators have recommended using the latter term only for lesions with true papillary projections.5 Definitive diagnosis is obtained through histopathology, which typically shows unilocular or multilocular cystic spaces in the dermis lined by an apocrine secretory epithelium. These secretory cells often demonstrate decapitation secretion and apical snouting. The cyst wall may send pseudopapillary projections into the cystic cavity.1,2 While apocrine and eccrine hidrocystomas previously were recognized as separate entities, it has been suggested that so-called eccrine hidrocystomas are truly apocrine in nature, with a cyst wall that is compressed by the cyst contents.4
Apocrine hidrocystomas are benign and do not require treatment; however, they may be removed for cosmetic purposes, most commonly via surgical excision. Lesions treated with needle puncture as monotherapy frequently recur. Other successful methods for removal include cyst puncture followed by hypertonic glucose sclerotherapy, trichloroacetic acid injection, botulinum toxin A injection, or CO2 laser treatment.3,6
Several clinical and histopathologic findings can distinguish between apocrine hidrocystomas and other diagnoses in the differential. Lipomas are common benign tumors composed of mature fat that typically manifest as solitary, painless, soft nodules with a normal overlying epidermis. They frequently are distributed on the neck, arms, legs, and buttocks. While the differential for our patient initially included lipoma, these lesions do not contain or release fluid, which was present in our patient. On histopathology, lipoma shows a uniform population of mature fat cells with small, uniform, and eccentric nuclei (Figure 1).7
Epidermal inclusion cysts are derived from the follicular infundibulum and commonly are found on the face and upper trunk. They manifest as flesh-colored dermal nodules and may have a visible punctum. As opposed to the cystic cavities lined with apocrine cells seen in apocrine hidrocystomas, epidermal inclusion cysts are lined with a stratified squamous epithelium, are filled with laminated keratin, and have a visible granular layer (Figure 2).8
Pilar cysts, also known as trichilemmal cysts, clinically resemble epidermal inclusion cysts but are derived from the outer root sheath of hair follicles, manifesting as flesh-colored dermal nodules almost always found on the scalp. On histopathology, pilar cysts are lined with stratified squamous epithelial cells without a visible granular layer and are filled with compact eosinophilic keratin (Figure 3).8
Tubular apocrine adenomas are benign neoplasms of the apocrine glands that manifest as smooth nodules. They are within the same spectrum as papillary eccrine adenomas, appearing more frequently on the legs and less frequently on the face and scalp.9 Histopathology generally demonstrates well-circumscribed lobules of tubular structures in the dermis. Similar to apocrine hidrocystomas, tubular apocrine adenomas will demonstrate an inner layer of columnar apocrine cells with decapitation secretion, but the tubular architecture helps differentiate it from other adnexal tumors (Figure 4).10
The clinical manifestation of the apocrine hidrocystoma in our patient was unusual due to its size and location. Apocrine hidrocystomas rarely are found on the scalp, with few other cases found in the literature. To our knowledge, this is the largest apocrine hidrocystoma found on the scalp to date, although there is at least 1 other published case of an apocrine hidrocystoma on the scalp measuring at least 3 cm in diameter.11 Our case highlights the importance of recognizing atypical manifestations of apocrine hidrocystomas, as a lesion on the midline scalp that discharges a thin fluid might raise initial concern for an intracranial connection. Awareness of atypical manifestations of common lesions can expand dermatologists’ differential diagnoses and help them to reassure patients.
- Smith JD. Apocrine hidrocystoma (cystadenoma). Arch Dermatol. 1974;109:700. doi:10.1001/archderm.1974.01630050046010
- Mehregan AH. Apocrine cystadenoma: a clinicopathologic study with special reference to the pigmented variety. Arch Dermatol. 1964;90:274. doi:10.1001/archderm.1964.01600030024005
- Hafsi W, Badri T, Shah F. Apocrine hidrocystoma. StatPearls [Internet]. Updated April 13, 2024. Accessed November 6, 2024. http://www.ncbi.nlm.nih.gov/books/NBK448109/
- de Viragh PA, Szeimies RM, Eckert F. Apocrine cystadenoma, apocrine hidrocystoma, and eccrine hidrocystoma: three distinct tumors defined by expression of keratins and human milk fat globulin 1. J Cutan Pathol. 1997;24:249-255. doi:10.1111/j.1600-0560.1997.tb01590.x
- Sugiyama A, Sugiura M, Piris A, et al. Apocrine cystadenoma and apocrine hidrocystoma: examination of 21 cases with emphasis on nomenclature according to proliferative features. J Cutan Pathol. 2007;34:912-917. doi:10.1111/j.1600-0560.2007.00757.x
- Bickley LK, Goldberg DJ, Imaeda S, et al. Treatment of multiple apocrine hidrocystomas with the carbon dioxide (CO2) laser. J Dermatol Surg Oncol. 1989;15:599-602. doi:10.1111/j.1524-4725.1989.tb03597.x
- Kaddu S. Smooth muscle, adipose and cartilage neoplasms. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. Elsevier; 2018:2086-2101.
- Stone MS. Cysts. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. Elsevier; 2018:1057-1074.
- Requena L, Sangüeza O. Tubular adenoma. In: Requena L, Sangüeza O, eds. Cutaneous Adnexal Neoplasms. Springer International Publishing; 2017:127-136. doi:10.1007/978-3-319-45704-8_12
- McCalmont TH, Pincus LB. Adnexal neoplasms. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. Elsevier; 2018:1057-1074.
- Nguyen HP, Barker HS, Bloomquist L, et al. Giant pigmented apocrine hidrocystoma of the scalp. Dermatol Online J. 2020;26:13030/qt7rt3s4pp.
- Smith JD. Apocrine hidrocystoma (cystadenoma). Arch Dermatol. 1974;109:700. doi:10.1001/archderm.1974.01630050046010
- Mehregan AH. Apocrine cystadenoma: a clinicopathologic study with special reference to the pigmented variety. Arch Dermatol. 1964;90:274. doi:10.1001/archderm.1964.01600030024005
- Hafsi W, Badri T, Shah F. Apocrine hidrocystoma. StatPearls [Internet]. Updated April 13, 2024. Accessed November 6, 2024. http://www.ncbi.nlm.nih.gov/books/NBK448109/
- de Viragh PA, Szeimies RM, Eckert F. Apocrine cystadenoma, apocrine hidrocystoma, and eccrine hidrocystoma: three distinct tumors defined by expression of keratins and human milk fat globulin 1. J Cutan Pathol. 1997;24:249-255. doi:10.1111/j.1600-0560.1997.tb01590.x
- Sugiyama A, Sugiura M, Piris A, et al. Apocrine cystadenoma and apocrine hidrocystoma: examination of 21 cases with emphasis on nomenclature according to proliferative features. J Cutan Pathol. 2007;34:912-917. doi:10.1111/j.1600-0560.2007.00757.x
- Bickley LK, Goldberg DJ, Imaeda S, et al. Treatment of multiple apocrine hidrocystomas with the carbon dioxide (CO2) laser. J Dermatol Surg Oncol. 1989;15:599-602. doi:10.1111/j.1524-4725.1989.tb03597.x
- Kaddu S. Smooth muscle, adipose and cartilage neoplasms. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. Elsevier; 2018:2086-2101.
- Stone MS. Cysts. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. Elsevier; 2018:1057-1074.
- Requena L, Sangüeza O. Tubular adenoma. In: Requena L, Sangüeza O, eds. Cutaneous Adnexal Neoplasms. Springer International Publishing; 2017:127-136. doi:10.1007/978-3-319-45704-8_12
- McCalmont TH, Pincus LB. Adnexal neoplasms. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. Elsevier; 2018:1057-1074.
- Nguyen HP, Barker HS, Bloomquist L, et al. Giant pigmented apocrine hidrocystoma of the scalp. Dermatol Online J. 2020;26:13030/qt7rt3s4pp.
Scalp Nodule With Copious Fluid
Scalp Nodule With Copious Fluid
A 48-year-old woman presented to the dermatology clinic with a suspected cyst on the occipital scalp. The patient noted that the lesion had been present for years and denied any pain, pruritus, or drainage from the site. Physical examination revealed a soft, flesh-colored, subcutaneous nodule measuring 4.2×3.2 cm on the midline occipital scalp. During excision, the lesion drained a copious amount of thin yellowish fluid.
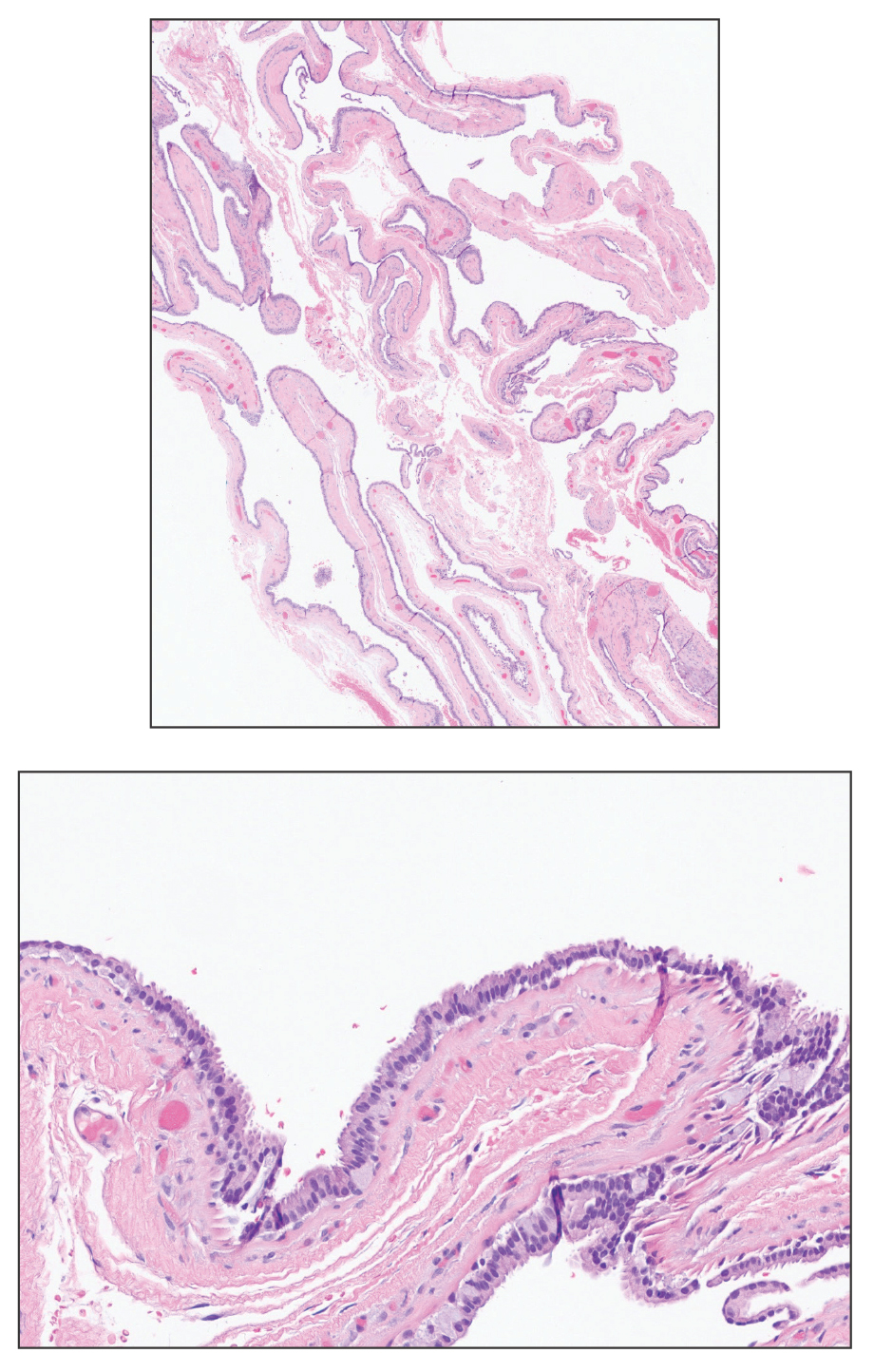
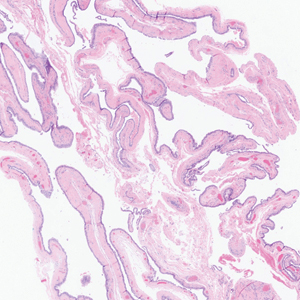
Asymptomatic Papules on the Neck
THE DIAGNOSIS: White Fibrous Papulosis
Given the histopathology findings, location on a sun-exposed site, lack of any additional systemic signs or symptoms, and no family history of similar lesions to suggest an underlying genetic condition, a diagnosis of white fibrous papulosis (WFP) was made. White fibrous papulosis is a relatively rare cutaneous disorder that was first reported by Shimizu et al1 in 1985. It is characterized by numerous grouped, 2- to 3-mm, white to flesh-colored papules that in most cases are confined to the neck in middle-aged to elderly individuals; however, cases involving the upper trunk and axillae also have been reported.1-3 The etiology of this condition is unclear but is thought to be related to aging and chronic exposure to UV light. Although treatment is not required, various modalities including tretinoin, excision, and laser therapy have been trialed with varying success.2,4 Our patient elected not to proceed with treatment.
Histologically, WFP may manifest similarly to connective tissue nevi; the overall architecture is nonspecific with focally thickened collagen and often elastic fibers that may be normal to reduced and/or fragmented, as well as an overall decrease in superficial dermal elastic tissue.3,5 Therefore, the differential diagnosis may include connective tissue nevi and require clinical correlation to make a correct diagnosis.
Pseudoxanthoma elasticum (PXE) is an autosomalrecessive disorder most commonly related to mutations in the ATP binding cassette subfamily C member 6 (ABCC6) gene that tends to manifest clinically on the neck and flexural extremities.6 This disease affects elastic fibers, which may become calcified over time. Pseudoxanthoma elasticum is associated with ocular complications relating to the Bruch membrane of the retina and angioid streaks; choroidal neovascularization involving the damaged Bruch membrane and episodes of acute retinopathy may result in vision loss in later stages of the disease.7 Involvement of the elastic laminae of arteries can be associated with cardiovascular and cerebrovascular complications such as stroke, coronary artery disease, claudication, and aneurysms. Involvement of the gastrointestinal or genitourinary tracts also may occur and most commonly manifests with bleeding. Pathologic alterations in the elastic fibers of the lungs also have been reported in patients with PXE.8 Histologically, PXE exhibits increased abnormally clumped and fragmented elastic fibers in the superficial dermis, often with calcification (Figure 1). Pseudo-PXE related to D-penicillamine use often lacks calcification and has a bramble bush appearance.9

Fibrofolliculomas may manifest alone or in association with an underlying condition such as Birt-Hogg-Dubé syndrome, in which lesions are most frequently seen scattered on the scalp, face, ears, neck, or upper trunk.10 This condition is related to a folliculin (FLCN) gene germline mutation. Birt-Hogg-Dubé syndrome also may be associated with acrochordons, trichodiscomas, renal cancer, and lung cysts with or without spontaneous pneumothorax. Less frequently noted findings include oral papules, epidermal cysts, angiofibromas, lipomas/angiolipomas, parotid gland tumors, and thyroid neoplasms. Connective tissue nevi/collagenomas can appear clinically similar to fibrofolliculomas; true connective tissue nevi are reported less commonly in Birt-Hogg-Dubé syndrome.11 Histologically, a fibrofolliculoma manifests with epidermal strands originating from a hair follicle associated with prominent surrounding connective tissue (Figure 2).
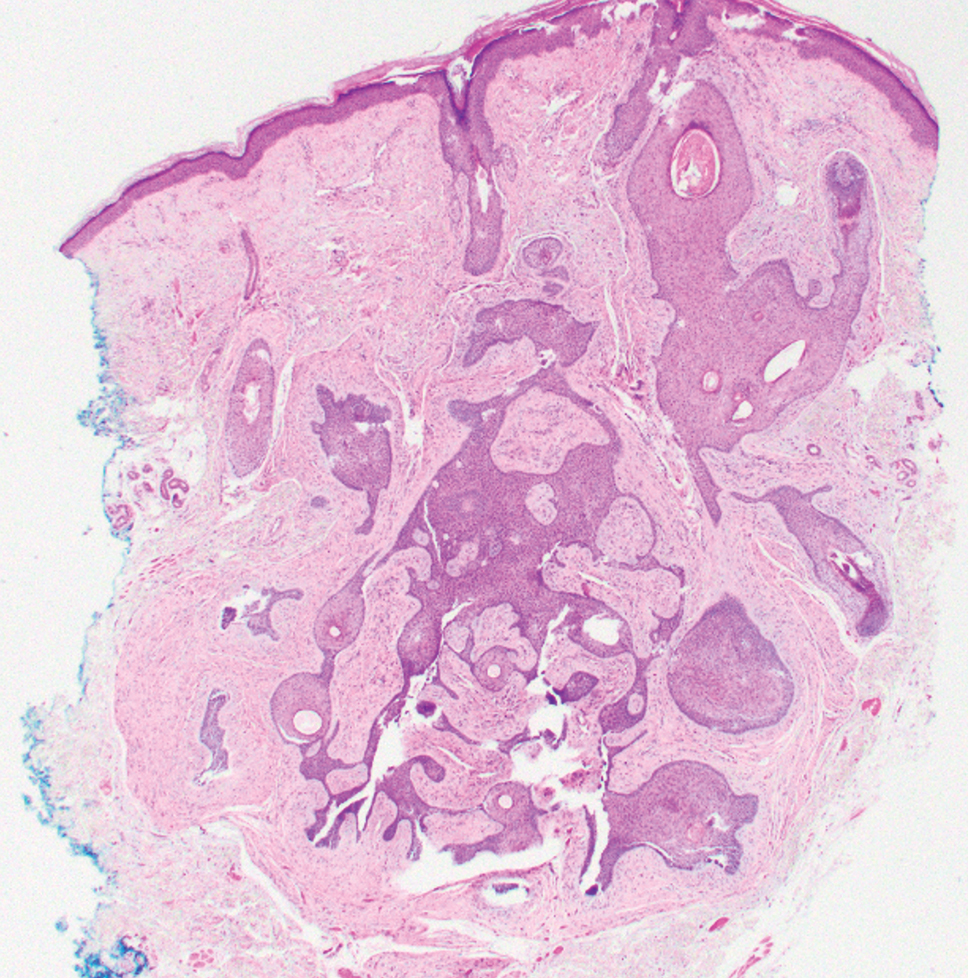
Elastofibroma dorsi is a benign tumor of connective tissue that most commonly manifests clinically as a solitary subcutaneous mass on the back near the inferior angle of the scapula; it typically develops below the rhomboid major and latissimus dorsi muscles.12 The pathogenesis is uncertain, but some patients have reported a family history of the condition or a history of repetitive shoulder movement/trauma prior to onset; the mass may be asymptomatic or associated with pain and/or swelling. Those affected tend to be older than 50 years.13 Histologically, thickened and rounded to beaded elastic fibers are seen admixed with collagen (Figure 3).
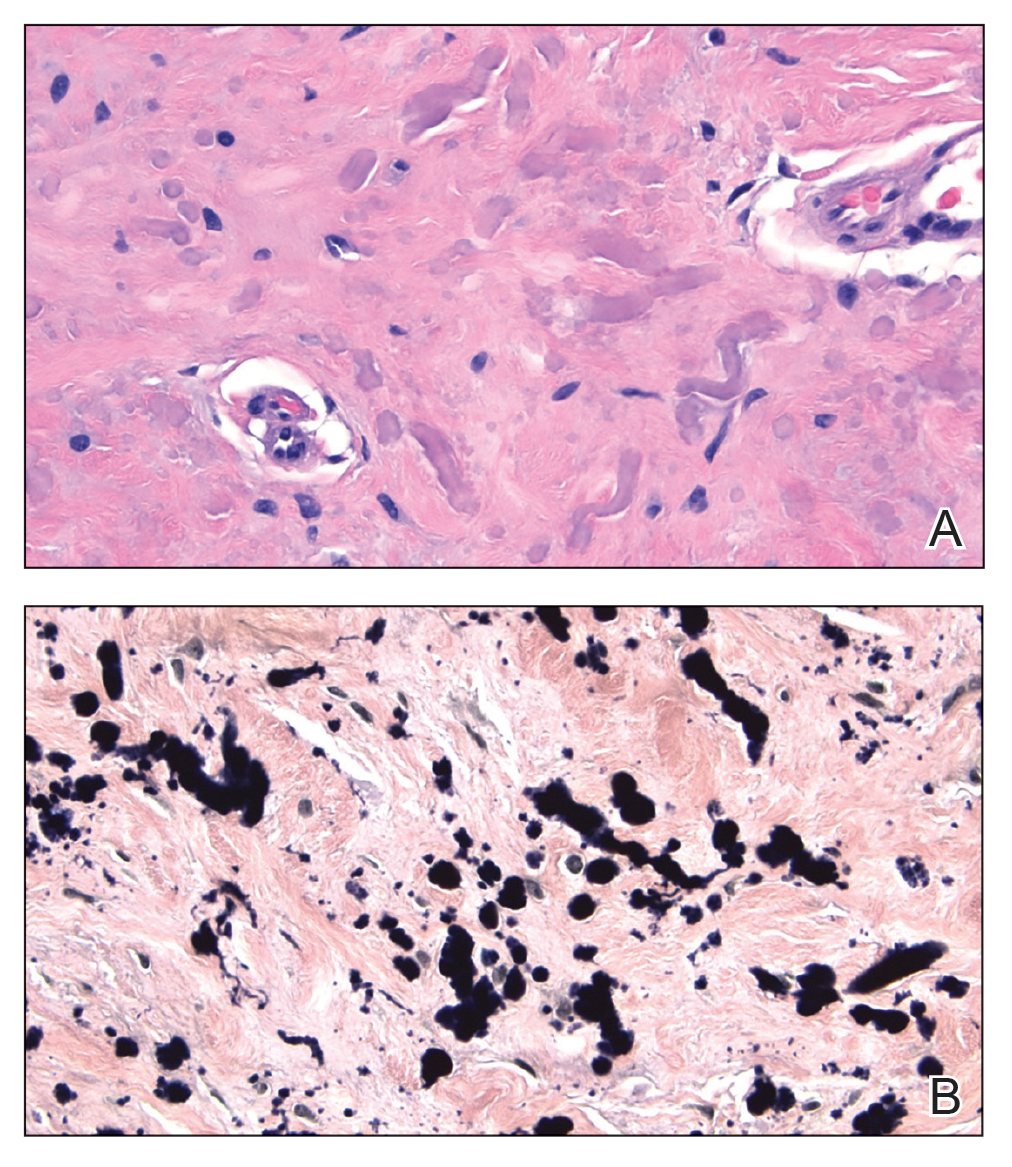
Actinic (solar) elastosis frequently is encountered in many skin biopsies and is caused by chronic photodamage. More hypertrophic variants, such as papular or nodular solar elastosis, may clinically manifest similarly to WFP.14 Histologically, actinic elastosis manifests as a considerable increase in elastic tissue in the papillary and superficial reticular dermis (Figure 4).
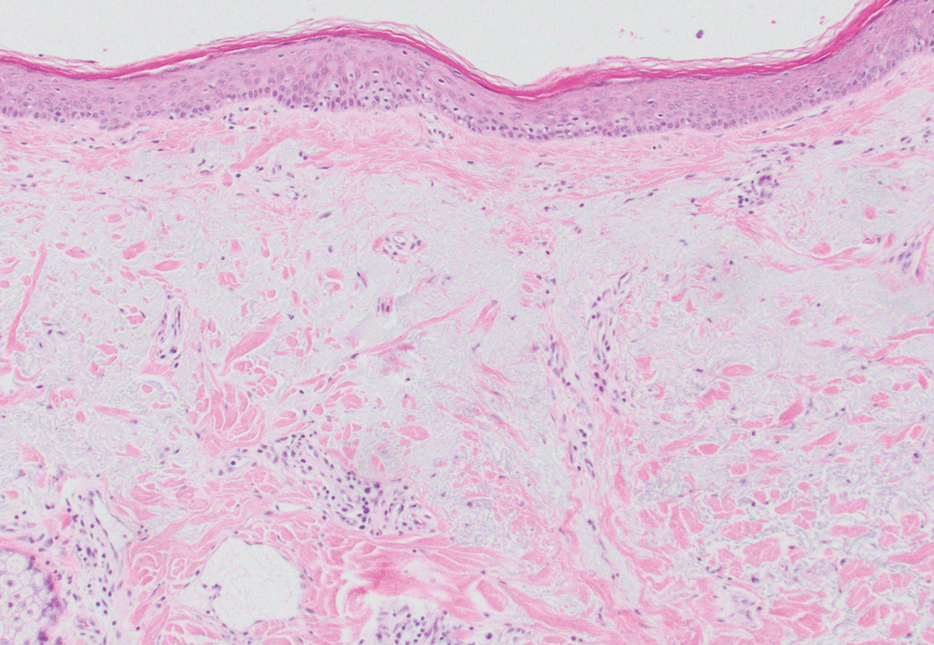
- Shimizu H, Nishikawa T, Kimura S. White fibrous papulosis of the neck: review of our 16 cases. Nihon Hifuka Gakkai Zasshi. 1985;95:1077-1084.
- Teo W, Pang S. White fibrous papulosis of the chest and back. J Am Acad Dermatol. 2012;66:AB33.
- Dokic Y, Tschen J. White fibrous papulosis of the axillae and neck. Cureus. 2020;12:E7635.
- Lueangarun S, Panchaprateep R. White fibrous papulosis of the neck treated with fractionated 1550-nm erbium glass laser: a case report. J Lasers Med Sci. 2016;7:256-258.
- Rios-Gomez M, Ramos-Garibay JA, Perez-Santana ME, et al. White fibrous papulosis of the neck: a case report. Cureus. 2022;14:E25661.
- Váradi A, Szabó Z, Pomozi V, et al. ABCC6 as a target in pseudoxanthoma elasticum. Curr Drug Targets. 2011;12:671-682.
- Gliem M, Birtel J, Müller PL, et al. Acute retinopathy in pseudoxanthoma elasticum. JAMA Ophthalmol. 2019;137:1165-1173.
- Germain DP. Pseudoxanthoma elasticum. Orphanet J Rare Dis. 2017;12:85. doi:10.1186/s13023-017-0639-8
- Chisti MA, Binamer Y, Alfadley A, et al. D-penicillamine-induced pseudo-pseudoxanthoma elasticum and extensive elastosis perforans serpiginosa with excellent response to acitretin. Ann Saudi Med. 2019;39:56-60.
- Criscito MC, Mu EW, Meehan SA, et al. Dermoscopic features of a solitary fibrofolliculoma on the left cheek. J Am Acad Dermatol. 2017;76(2 suppl 1):S8-S9.
- Sattler EC, Steinlein OK. Birt-Hogg-Dubé syndrome. In: Adam MP, Everman DB, Mirzaa GM, et al, eds. GeneReviews® [Internet]. Updated January 30, 2020. Accessed February 23, 2023. https://www.ncbi.nlm.nih.gov/books/NBK1522
- Patnayak R, Jena A, Settipalli S, et al. Elastofibroma: an uncommon tumor revisited. J Cutan Aesthet Surg. 2016;9:34-37. doi:10.4103/0974- 2077.178543
- Chandrasekar CR, Grimer RJ, Carter SR, et al. Elastofibroma dorsi: an uncommon benign pseudotumour. Sarcoma. 2008;2008:756565. doi:10.1155/2008/756565
- Kwittken J. Papular elastosis. Cutis. 2000;66:81-83.
THE DIAGNOSIS: White Fibrous Papulosis
Given the histopathology findings, location on a sun-exposed site, lack of any additional systemic signs or symptoms, and no family history of similar lesions to suggest an underlying genetic condition, a diagnosis of white fibrous papulosis (WFP) was made. White fibrous papulosis is a relatively rare cutaneous disorder that was first reported by Shimizu et al1 in 1985. It is characterized by numerous grouped, 2- to 3-mm, white to flesh-colored papules that in most cases are confined to the neck in middle-aged to elderly individuals; however, cases involving the upper trunk and axillae also have been reported.1-3 The etiology of this condition is unclear but is thought to be related to aging and chronic exposure to UV light. Although treatment is not required, various modalities including tretinoin, excision, and laser therapy have been trialed with varying success.2,4 Our patient elected not to proceed with treatment.
Histologically, WFP may manifest similarly to connective tissue nevi; the overall architecture is nonspecific with focally thickened collagen and often elastic fibers that may be normal to reduced and/or fragmented, as well as an overall decrease in superficial dermal elastic tissue.3,5 Therefore, the differential diagnosis may include connective tissue nevi and require clinical correlation to make a correct diagnosis.
Pseudoxanthoma elasticum (PXE) is an autosomalrecessive disorder most commonly related to mutations in the ATP binding cassette subfamily C member 6 (ABCC6) gene that tends to manifest clinically on the neck and flexural extremities.6 This disease affects elastic fibers, which may become calcified over time. Pseudoxanthoma elasticum is associated with ocular complications relating to the Bruch membrane of the retina and angioid streaks; choroidal neovascularization involving the damaged Bruch membrane and episodes of acute retinopathy may result in vision loss in later stages of the disease.7 Involvement of the elastic laminae of arteries can be associated with cardiovascular and cerebrovascular complications such as stroke, coronary artery disease, claudication, and aneurysms. Involvement of the gastrointestinal or genitourinary tracts also may occur and most commonly manifests with bleeding. Pathologic alterations in the elastic fibers of the lungs also have been reported in patients with PXE.8 Histologically, PXE exhibits increased abnormally clumped and fragmented elastic fibers in the superficial dermis, often with calcification (Figure 1). Pseudo-PXE related to D-penicillamine use often lacks calcification and has a bramble bush appearance.9
Fibrofolliculomas may manifest alone or in association with an underlying condition such as Birt-Hogg-Dubé syndrome, in which lesions are most frequently seen scattered on the scalp, face, ears, neck, or upper trunk.10 This condition is related to a folliculin (FLCN) gene germline mutation. Birt-Hogg-Dubé syndrome also may be associated with acrochordons, trichodiscomas, renal cancer, and lung cysts with or without spontaneous pneumothorax. Less frequently noted findings include oral papules, epidermal cysts, angiofibromas, lipomas/angiolipomas, parotid gland tumors, and thyroid neoplasms. Connective tissue nevi/collagenomas can appear clinically similar to fibrofolliculomas; true connective tissue nevi are reported less commonly in Birt-Hogg-Dubé syndrome.11 Histologically, a fibrofolliculoma manifests with epidermal strands originating from a hair follicle associated with prominent surrounding connective tissue (Figure 2).
Elastofibroma dorsi is a benign tumor of connective tissue that most commonly manifests clinically as a solitary subcutaneous mass on the back near the inferior angle of the scapula; it typically develops below the rhomboid major and latissimus dorsi muscles.12 The pathogenesis is uncertain, but some patients have reported a family history of the condition or a history of repetitive shoulder movement/trauma prior to onset; the mass may be asymptomatic or associated with pain and/or swelling. Those affected tend to be older than 50 years.13 Histologically, thickened and rounded to beaded elastic fibers are seen admixed with collagen (Figure 3).
Actinic (solar) elastosis frequently is encountered in many skin biopsies and is caused by chronic photodamage. More hypertrophic variants, such as papular or nodular solar elastosis, may clinically manifest similarly to WFP.14 Histologically, actinic elastosis manifests as a considerable increase in elastic tissue in the papillary and superficial reticular dermis (Figure 4).
THE DIAGNOSIS: White Fibrous Papulosis
Given the histopathology findings, location on a sun-exposed site, lack of any additional systemic signs or symptoms, and no family history of similar lesions to suggest an underlying genetic condition, a diagnosis of white fibrous papulosis (WFP) was made. White fibrous papulosis is a relatively rare cutaneous disorder that was first reported by Shimizu et al1 in 1985. It is characterized by numerous grouped, 2- to 3-mm, white to flesh-colored papules that in most cases are confined to the neck in middle-aged to elderly individuals; however, cases involving the upper trunk and axillae also have been reported.1-3 The etiology of this condition is unclear but is thought to be related to aging and chronic exposure to UV light. Although treatment is not required, various modalities including tretinoin, excision, and laser therapy have been trialed with varying success.2,4 Our patient elected not to proceed with treatment.
Histologically, WFP may manifest similarly to connective tissue nevi; the overall architecture is nonspecific with focally thickened collagen and often elastic fibers that may be normal to reduced and/or fragmented, as well as an overall decrease in superficial dermal elastic tissue.3,5 Therefore, the differential diagnosis may include connective tissue nevi and require clinical correlation to make a correct diagnosis.
Pseudoxanthoma elasticum (PXE) is an autosomalrecessive disorder most commonly related to mutations in the ATP binding cassette subfamily C member 6 (ABCC6) gene that tends to manifest clinically on the neck and flexural extremities.6 This disease affects elastic fibers, which may become calcified over time. Pseudoxanthoma elasticum is associated with ocular complications relating to the Bruch membrane of the retina and angioid streaks; choroidal neovascularization involving the damaged Bruch membrane and episodes of acute retinopathy may result in vision loss in later stages of the disease.7 Involvement of the elastic laminae of arteries can be associated with cardiovascular and cerebrovascular complications such as stroke, coronary artery disease, claudication, and aneurysms. Involvement of the gastrointestinal or genitourinary tracts also may occur and most commonly manifests with bleeding. Pathologic alterations in the elastic fibers of the lungs also have been reported in patients with PXE.8 Histologically, PXE exhibits increased abnormally clumped and fragmented elastic fibers in the superficial dermis, often with calcification (Figure 1). Pseudo-PXE related to D-penicillamine use often lacks calcification and has a bramble bush appearance.9
Fibrofolliculomas may manifest alone or in association with an underlying condition such as Birt-Hogg-Dubé syndrome, in which lesions are most frequently seen scattered on the scalp, face, ears, neck, or upper trunk.10 This condition is related to a folliculin (FLCN) gene germline mutation. Birt-Hogg-Dubé syndrome also may be associated with acrochordons, trichodiscomas, renal cancer, and lung cysts with or without spontaneous pneumothorax. Less frequently noted findings include oral papules, epidermal cysts, angiofibromas, lipomas/angiolipomas, parotid gland tumors, and thyroid neoplasms. Connective tissue nevi/collagenomas can appear clinically similar to fibrofolliculomas; true connective tissue nevi are reported less commonly in Birt-Hogg-Dubé syndrome.11 Histologically, a fibrofolliculoma manifests with epidermal strands originating from a hair follicle associated with prominent surrounding connective tissue (Figure 2).
Elastofibroma dorsi is a benign tumor of connective tissue that most commonly manifests clinically as a solitary subcutaneous mass on the back near the inferior angle of the scapula; it typically develops below the rhomboid major and latissimus dorsi muscles.12 The pathogenesis is uncertain, but some patients have reported a family history of the condition or a history of repetitive shoulder movement/trauma prior to onset; the mass may be asymptomatic or associated with pain and/or swelling. Those affected tend to be older than 50 years.13 Histologically, thickened and rounded to beaded elastic fibers are seen admixed with collagen (Figure 3).
Actinic (solar) elastosis frequently is encountered in many skin biopsies and is caused by chronic photodamage. More hypertrophic variants, such as papular or nodular solar elastosis, may clinically manifest similarly to WFP.14 Histologically, actinic elastosis manifests as a considerable increase in elastic tissue in the papillary and superficial reticular dermis (Figure 4).
- Shimizu H, Nishikawa T, Kimura S. White fibrous papulosis of the neck: review of our 16 cases. Nihon Hifuka Gakkai Zasshi. 1985;95:1077-1084.
- Teo W, Pang S. White fibrous papulosis of the chest and back. J Am Acad Dermatol. 2012;66:AB33.
- Dokic Y, Tschen J. White fibrous papulosis of the axillae and neck. Cureus. 2020;12:E7635.
- Lueangarun S, Panchaprateep R. White fibrous papulosis of the neck treated with fractionated 1550-nm erbium glass laser: a case report. J Lasers Med Sci. 2016;7:256-258.
- Rios-Gomez M, Ramos-Garibay JA, Perez-Santana ME, et al. White fibrous papulosis of the neck: a case report. Cureus. 2022;14:E25661.
- Váradi A, Szabó Z, Pomozi V, et al. ABCC6 as a target in pseudoxanthoma elasticum. Curr Drug Targets. 2011;12:671-682.
- Gliem M, Birtel J, Müller PL, et al. Acute retinopathy in pseudoxanthoma elasticum. JAMA Ophthalmol. 2019;137:1165-1173.
- Germain DP. Pseudoxanthoma elasticum. Orphanet J Rare Dis. 2017;12:85. doi:10.1186/s13023-017-0639-8
- Chisti MA, Binamer Y, Alfadley A, et al. D-penicillamine-induced pseudo-pseudoxanthoma elasticum and extensive elastosis perforans serpiginosa with excellent response to acitretin. Ann Saudi Med. 2019;39:56-60.
- Criscito MC, Mu EW, Meehan SA, et al. Dermoscopic features of a solitary fibrofolliculoma on the left cheek. J Am Acad Dermatol. 2017;76(2 suppl 1):S8-S9.
- Sattler EC, Steinlein OK. Birt-Hogg-Dubé syndrome. In: Adam MP, Everman DB, Mirzaa GM, et al, eds. GeneReviews® [Internet]. Updated January 30, 2020. Accessed February 23, 2023. https://www.ncbi.nlm.nih.gov/books/NBK1522
- Patnayak R, Jena A, Settipalli S, et al. Elastofibroma: an uncommon tumor revisited. J Cutan Aesthet Surg. 2016;9:34-37. doi:10.4103/0974- 2077.178543
- Chandrasekar CR, Grimer RJ, Carter SR, et al. Elastofibroma dorsi: an uncommon benign pseudotumour. Sarcoma. 2008;2008:756565. doi:10.1155/2008/756565
- Kwittken J. Papular elastosis. Cutis. 2000;66:81-83.
- Shimizu H, Nishikawa T, Kimura S. White fibrous papulosis of the neck: review of our 16 cases. Nihon Hifuka Gakkai Zasshi. 1985;95:1077-1084.
- Teo W, Pang S. White fibrous papulosis of the chest and back. J Am Acad Dermatol. 2012;66:AB33.
- Dokic Y, Tschen J. White fibrous papulosis of the axillae and neck. Cureus. 2020;12:E7635.
- Lueangarun S, Panchaprateep R. White fibrous papulosis of the neck treated with fractionated 1550-nm erbium glass laser: a case report. J Lasers Med Sci. 2016;7:256-258.
- Rios-Gomez M, Ramos-Garibay JA, Perez-Santana ME, et al. White fibrous papulosis of the neck: a case report. Cureus. 2022;14:E25661.
- Váradi A, Szabó Z, Pomozi V, et al. ABCC6 as a target in pseudoxanthoma elasticum. Curr Drug Targets. 2011;12:671-682.
- Gliem M, Birtel J, Müller PL, et al. Acute retinopathy in pseudoxanthoma elasticum. JAMA Ophthalmol. 2019;137:1165-1173.
- Germain DP. Pseudoxanthoma elasticum. Orphanet J Rare Dis. 2017;12:85. doi:10.1186/s13023-017-0639-8
- Chisti MA, Binamer Y, Alfadley A, et al. D-penicillamine-induced pseudo-pseudoxanthoma elasticum and extensive elastosis perforans serpiginosa with excellent response to acitretin. Ann Saudi Med. 2019;39:56-60.
- Criscito MC, Mu EW, Meehan SA, et al. Dermoscopic features of a solitary fibrofolliculoma on the left cheek. J Am Acad Dermatol. 2017;76(2 suppl 1):S8-S9.
- Sattler EC, Steinlein OK. Birt-Hogg-Dubé syndrome. In: Adam MP, Everman DB, Mirzaa GM, et al, eds. GeneReviews® [Internet]. Updated January 30, 2020. Accessed February 23, 2023. https://www.ncbi.nlm.nih.gov/books/NBK1522
- Patnayak R, Jena A, Settipalli S, et al. Elastofibroma: an uncommon tumor revisited. J Cutan Aesthet Surg. 2016;9:34-37. doi:10.4103/0974- 2077.178543
- Chandrasekar CR, Grimer RJ, Carter SR, et al. Elastofibroma dorsi: an uncommon benign pseudotumour. Sarcoma. 2008;2008:756565. doi:10.1155/2008/756565
- Kwittken J. Papular elastosis. Cutis. 2000;66:81-83.
A 70-year-old woman with a history of osteoporosis and breast cancer presented for evaluation of asymptomatic, 2- to 3-mm, white to flesh-colored papules concentrated on the inferior occipital scalp and posterior neck (inset) for at least several months. She had no additional systemic signs or symptoms, and there was no family history of similar skin findings. A punch biopsy was performed.
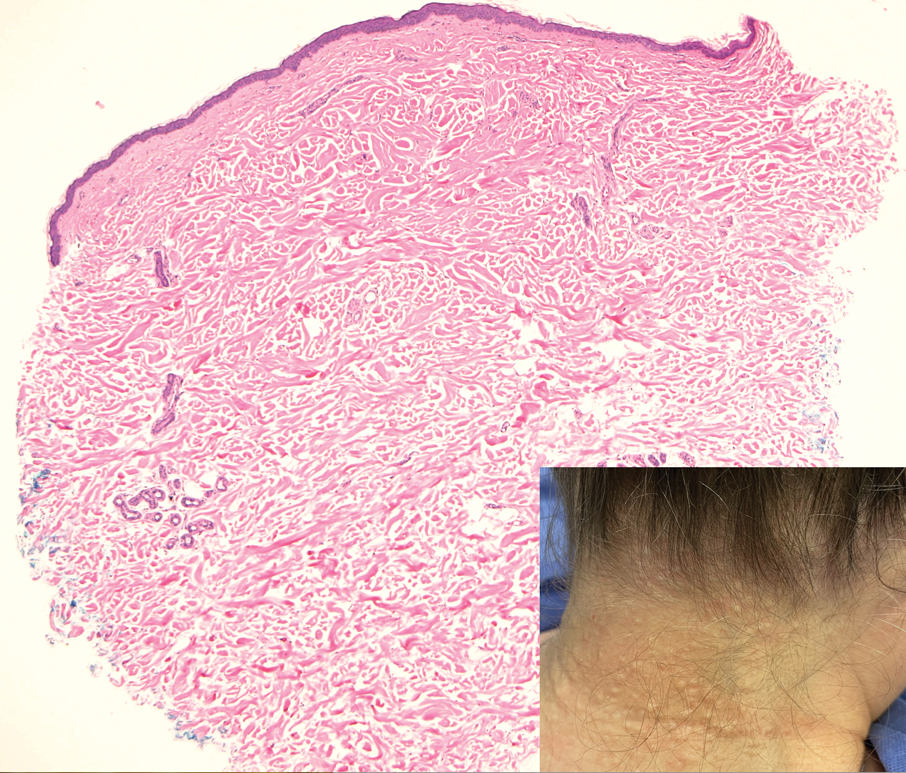
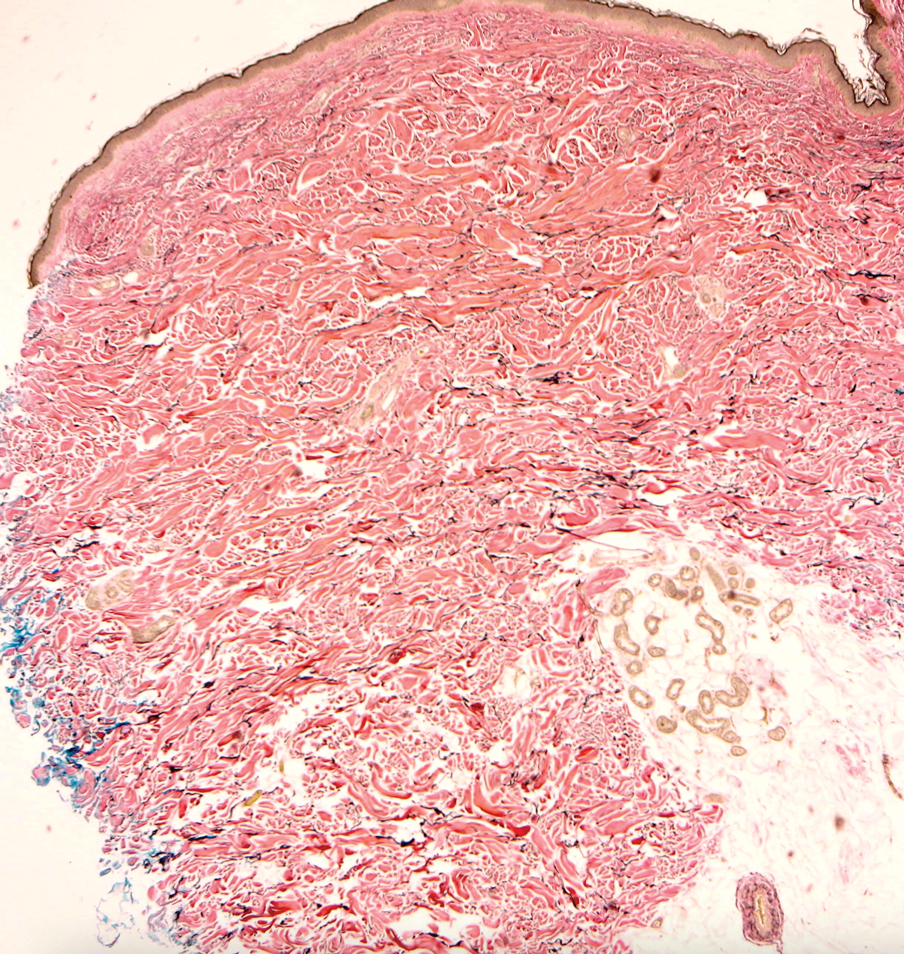
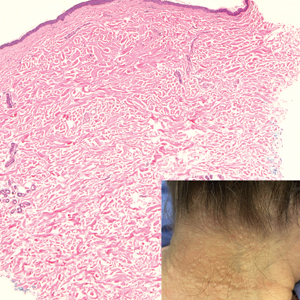
Acute Tender Papules on the Arms and Legs
The Diagnosis: Erythema Nodosum Leprosum
Erythema nodosum leprosum (ENL) is a type 2 reaction sometimes seen in patients infected with Mycobacterium leprae—primarily those with lepromatous or borderline lepromatous subtypes. Clinically, ENL manifests with abrupt onset of tender erythematous papules with associated fevers and general malaise. Studies have demonstrated a complex immune system reaction in ENL, but the detailed pathophysiology is not fully understood.1 Biopsies conducted within 24 hours of lesion formation are most elucidating. Foamy histiocytes admixed with neutrophils are seen in the subcutis, often causing a lobular panniculitis (quiz image).2 Neutrophils rarely are seen in other types of leprosy and thus are a useful diagnostic clue for ENL. Vasculitis of small- to medium-sized vessels can be seen but is not a necessary diagnostic criterion. Fite staining will highlight many acid-fast bacilli within the histiocytes (Figure 1).
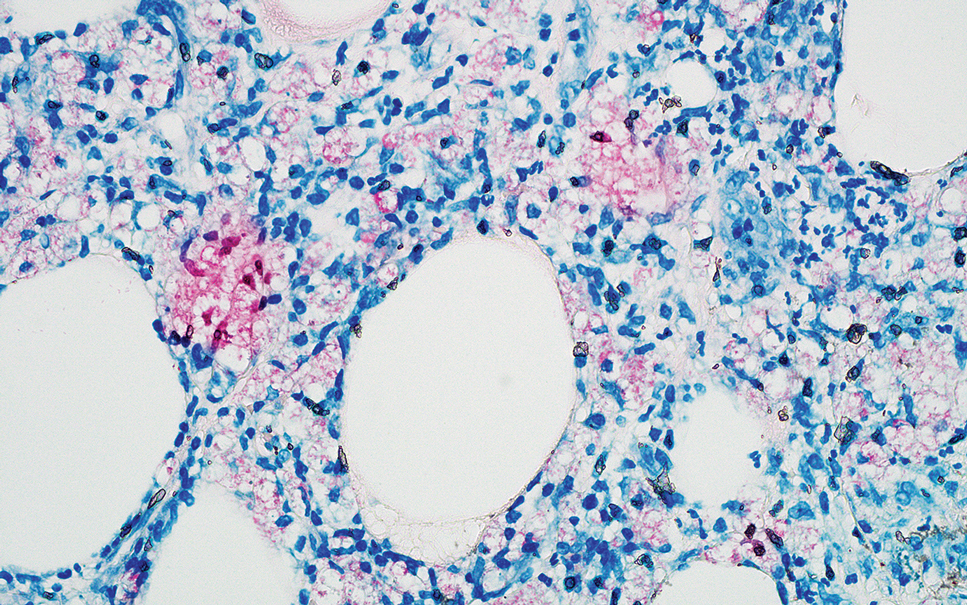
Erythema nodosum leprosum is treated with a combination of immunosuppressants such as prednisone and thalidomide. Our patient was taking triple-antibiotic therapy—dapsone, rifampin, and clofazimine—for lepromatous leprosy when the erythematous papules developed on the arms and legs. After a skin biopsy confirmed the diagnosis of ENL, he was started on prednisone 20 mg daily with plans for close follow-up. Unfortunately, the patient was subsequently lost to follow-up.
Acute febrile neutrophilic dermatosis (also known as Sweet syndrome) is an acute inflammatory disease characterized by abrupt onset of painful erythematous papules, plaques, or nodules on the skin. It often is seen in association with preceding infections (especially those in the upper respiratory or gastrointestinal tracts), hematologic malignancies, inflammatory bowel disease, or exposure to certain classes of medications (eg, granulocyte colony-stimulating factor, tyrosine kinase inhibitors, various antibiotics).3 Histologically, acute febrile neutrophilic dermatosis is characterized by dense neutrophilic infiltrates, often with notable dermal edema (Figure 2).4 Many cases also show leukocytoclastic vasculitis; however, foamy histiocytes are not a notable component of the inflammatory infiltrate, though a histiocytoid form of acute febrile neutrophilic dermatosis has been described.5 Infections must be rigorously ruled out prior to diagnosing a patient with acute febrile neutrophilic dermatosis, making it a diagnosis of exclusion.
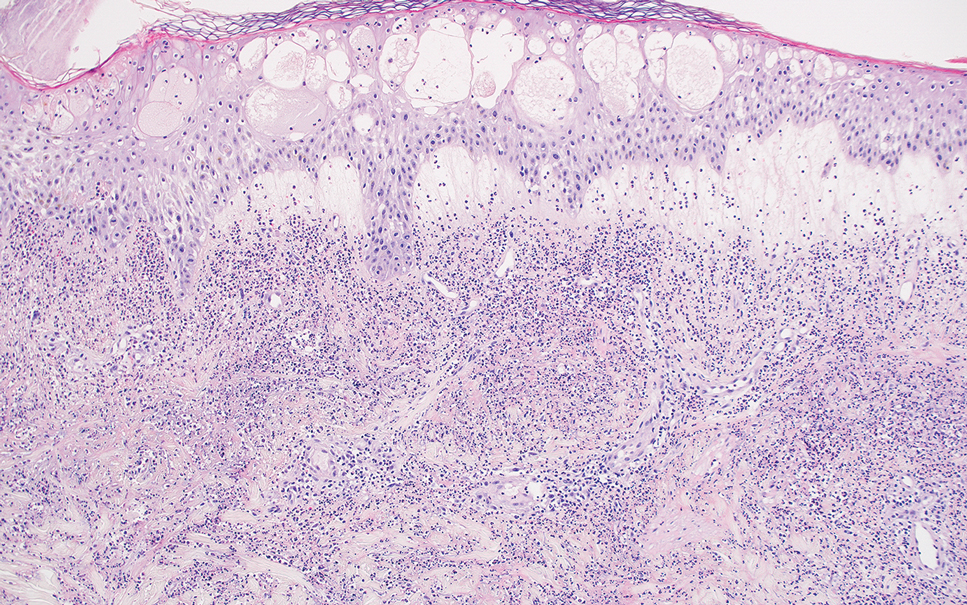
Cutaneous coccidioidomycosis is an infection caused by the dimorphic fungi Coccidioides immitis or Coccidioides posadasii. Cutaneous disease is rare but can occur from direct inoculation or dissemination from pulmonary disease in immunocompetent or immunocompromised patients. Papules, pustules, or plaques are seen clinically. Histologically, cutaneous coccidioidomycosis shows spherules that vary from 10 to 100 μm and are filled with multiple smaller endospores (Figure 3).6 Pseudoepitheliomatous hyperplasia with dense suppurative and granulomatous infiltrates also is seen.
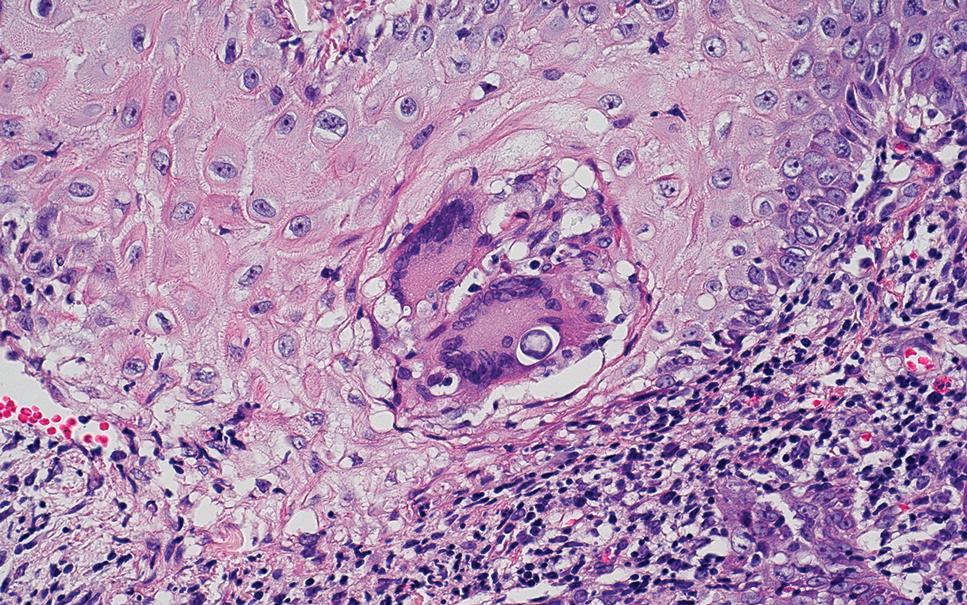
Erythema induratum is characterized by tender nodules on the lower extremities and has a substantial female predominance. Many cases are associated with Mycobacterium tuberculosis infection. The bacteria are not seen directly in the skin but are instead detectable through DNA polymerase chain reaction testing or investigation of other organ systems.7,8 Histologically, lesions show a lobular panniculitis with a mixed infiltrate. Vasculitis is seen in approximately 90% of erythema induratum cases vs approximately 25% of classic ENL cases (Figure 4),2,9 which has led some to use the term nodular vasculitis to describe this disease entity. Nodular vasculitis is considered by others to be a distinct disease entity in which there are clinical and histologic features similar to erythema induratum but no evidence of M tuberculosis infection.9
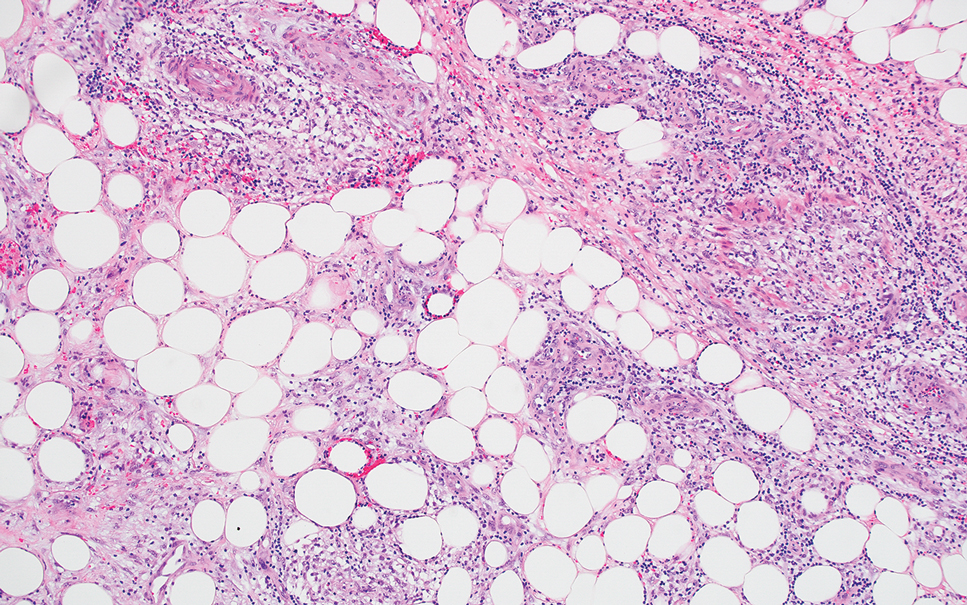
Polyarteritis nodosa is a vasculitis that affects medium- sized vessels of various organ systems. The presenting signs and symptoms vary based on the affected organ systems. Palpable to retiform purpura, livedo racemosa, subcutaneous nodules, or ulcers are seen when the skin is involved. The histologic hallmark is necrotizing vasculitis of medium-sized arterioles (Figure 5), although leukocytoclastic vasculitis of small-caliber vessels also can be seen in biopsies of affected skin.10 The vascular changes are said to be segmental, with uninvolved segments interspersed with involved segments. Antineutrophil cytoplasmic antibody (ANCA)– associated vasculitis also must be considered when one sees leukocytoclastic vasculitis of small-caliber vessels in the skin, as it can be distinguished most readily by detecting circulating antibodies specific for myeloperoxidase (MPO-ANCA) or proteinase 3 (PR3-ANCA).
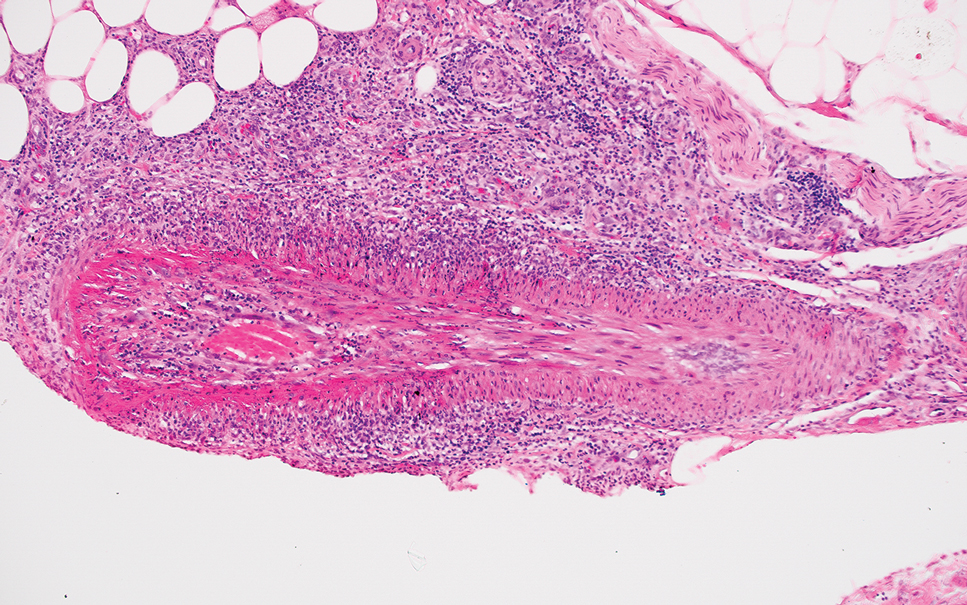
- Polycarpou A, Walker SL, Lockwood DNJ. A systematic review of immunological studies of erythema nodosum leprosum. Front Immunol. 2017;8:233. doi:10.3389/fimmu.2017.00233
- Massone C, Belachew WA, Schettini A. Histopathology of the lepromatous skin biopsy. Clin Dermatol. 2015;33:38-45. doi:10.1016/j.clindermatol.2014.10.003
- Cohen PR. Sweet’s syndrome—a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:1-28. doi:10.1186/1750-1172-2-34
- Ratzinger G, Burgdorf W, Zelger BG, et al. Acute febrile neutrophilic dermatosis: a histopathologic study of 31 cases with review of literature. Am J Dermatopathol. 2007;29:125-133. doi:10.1097/01.dad.0000249887.59810.76
- Wilson TC, Stone MS, Swick BL. Histiocytoid Sweet syndrome with haloed myeloid cells masquerading as a cryptococcal infection. Am J Dermatopathology. 2014;36:264-269. doi:10.1097/DAD.0b013e31828b811b
- Guarner J, Brandt ME. Histopathologic diagnosis of fungal infections in the 21st century. Clin Microbiol Rev. 2011;24:247-280. doi:10.1128/CMR.00053-10
- Schneider JW, Jordaan HF, Geiger DH, et al. Erythema induratum of Bazin: a clinicopathological study of 20 cases of Mycobacterium tuberculosis DNA in skin lesions by polymerase chain reaction. Am J Dermatopathol. 1995;17:350-356. doi:10.1097/00000372-199508000-00008
- Boonchai W, Suthipinittharm P, Mahaisavariya P. Panniculitis in tuberculosis: a clinicopathologic study of nodular panniculitis associated with tuberculosis. Int J Dermatol. 1998;37:361-363. doi:10.1046/j.1365-4362.1998.00299.x
- Segura S, Pujol RM, Trindade F, et al. Vasculitis in erythema induratum of Bazin: a histopathologic study of 101 biopsy specimens from 86 patients. J Am Acad Dermatol. 2008;59:839-851. doi:10.1016/j.jaad.2008.07.030
- Ishiguro N, Kawashima M. Cutaneous polyarteritis nodosa: a report of 16 cases with clinical and histopathological analysis and a review of the published work. J Dermatol. 2010;37:85-93. doi:10.1111/j.1346-8138.2009.00752.x
The Diagnosis: Erythema Nodosum Leprosum
Erythema nodosum leprosum (ENL) is a type 2 reaction sometimes seen in patients infected with Mycobacterium leprae—primarily those with lepromatous or borderline lepromatous subtypes. Clinically, ENL manifests with abrupt onset of tender erythematous papules with associated fevers and general malaise. Studies have demonstrated a complex immune system reaction in ENL, but the detailed pathophysiology is not fully understood.1 Biopsies conducted within 24 hours of lesion formation are most elucidating. Foamy histiocytes admixed with neutrophils are seen in the subcutis, often causing a lobular panniculitis (quiz image).2 Neutrophils rarely are seen in other types of leprosy and thus are a useful diagnostic clue for ENL. Vasculitis of small- to medium-sized vessels can be seen but is not a necessary diagnostic criterion. Fite staining will highlight many acid-fast bacilli within the histiocytes (Figure 1).
Erythema nodosum leprosum is treated with a combination of immunosuppressants such as prednisone and thalidomide. Our patient was taking triple-antibiotic therapy—dapsone, rifampin, and clofazimine—for lepromatous leprosy when the erythematous papules developed on the arms and legs. After a skin biopsy confirmed the diagnosis of ENL, he was started on prednisone 20 mg daily with plans for close follow-up. Unfortunately, the patient was subsequently lost to follow-up.
Acute febrile neutrophilic dermatosis (also known as Sweet syndrome) is an acute inflammatory disease characterized by abrupt onset of painful erythematous papules, plaques, or nodules on the skin. It often is seen in association with preceding infections (especially those in the upper respiratory or gastrointestinal tracts), hematologic malignancies, inflammatory bowel disease, or exposure to certain classes of medications (eg, granulocyte colony-stimulating factor, tyrosine kinase inhibitors, various antibiotics).3 Histologically, acute febrile neutrophilic dermatosis is characterized by dense neutrophilic infiltrates, often with notable dermal edema (Figure 2).4 Many cases also show leukocytoclastic vasculitis; however, foamy histiocytes are not a notable component of the inflammatory infiltrate, though a histiocytoid form of acute febrile neutrophilic dermatosis has been described.5 Infections must be rigorously ruled out prior to diagnosing a patient with acute febrile neutrophilic dermatosis, making it a diagnosis of exclusion.
Cutaneous coccidioidomycosis is an infection caused by the dimorphic fungi Coccidioides immitis or Coccidioides posadasii. Cutaneous disease is rare but can occur from direct inoculation or dissemination from pulmonary disease in immunocompetent or immunocompromised patients. Papules, pustules, or plaques are seen clinically. Histologically, cutaneous coccidioidomycosis shows spherules that vary from 10 to 100 μm and are filled with multiple smaller endospores (Figure 3).6 Pseudoepitheliomatous hyperplasia with dense suppurative and granulomatous infiltrates also is seen.
Erythema induratum is characterized by tender nodules on the lower extremities and has a substantial female predominance. Many cases are associated with Mycobacterium tuberculosis infection. The bacteria are not seen directly in the skin but are instead detectable through DNA polymerase chain reaction testing or investigation of other organ systems.7,8 Histologically, lesions show a lobular panniculitis with a mixed infiltrate. Vasculitis is seen in approximately 90% of erythema induratum cases vs approximately 25% of classic ENL cases (Figure 4),2,9 which has led some to use the term nodular vasculitis to describe this disease entity. Nodular vasculitis is considered by others to be a distinct disease entity in which there are clinical and histologic features similar to erythema induratum but no evidence of M tuberculosis infection.9
Polyarteritis nodosa is a vasculitis that affects medium- sized vessels of various organ systems. The presenting signs and symptoms vary based on the affected organ systems. Palpable to retiform purpura, livedo racemosa, subcutaneous nodules, or ulcers are seen when the skin is involved. The histologic hallmark is necrotizing vasculitis of medium-sized arterioles (Figure 5), although leukocytoclastic vasculitis of small-caliber vessels also can be seen in biopsies of affected skin.10 The vascular changes are said to be segmental, with uninvolved segments interspersed with involved segments. Antineutrophil cytoplasmic antibody (ANCA)– associated vasculitis also must be considered when one sees leukocytoclastic vasculitis of small-caliber vessels in the skin, as it can be distinguished most readily by detecting circulating antibodies specific for myeloperoxidase (MPO-ANCA) or proteinase 3 (PR3-ANCA).
The Diagnosis: Erythema Nodosum Leprosum
Erythema nodosum leprosum (ENL) is a type 2 reaction sometimes seen in patients infected with Mycobacterium leprae—primarily those with lepromatous or borderline lepromatous subtypes. Clinically, ENL manifests with abrupt onset of tender erythematous papules with associated fevers and general malaise. Studies have demonstrated a complex immune system reaction in ENL, but the detailed pathophysiology is not fully understood.1 Biopsies conducted within 24 hours of lesion formation are most elucidating. Foamy histiocytes admixed with neutrophils are seen in the subcutis, often causing a lobular panniculitis (quiz image).2 Neutrophils rarely are seen in other types of leprosy and thus are a useful diagnostic clue for ENL. Vasculitis of small- to medium-sized vessels can be seen but is not a necessary diagnostic criterion. Fite staining will highlight many acid-fast bacilli within the histiocytes (Figure 1).
Erythema nodosum leprosum is treated with a combination of immunosuppressants such as prednisone and thalidomide. Our patient was taking triple-antibiotic therapy—dapsone, rifampin, and clofazimine—for lepromatous leprosy when the erythematous papules developed on the arms and legs. After a skin biopsy confirmed the diagnosis of ENL, he was started on prednisone 20 mg daily with plans for close follow-up. Unfortunately, the patient was subsequently lost to follow-up.
Acute febrile neutrophilic dermatosis (also known as Sweet syndrome) is an acute inflammatory disease characterized by abrupt onset of painful erythematous papules, plaques, or nodules on the skin. It often is seen in association with preceding infections (especially those in the upper respiratory or gastrointestinal tracts), hematologic malignancies, inflammatory bowel disease, or exposure to certain classes of medications (eg, granulocyte colony-stimulating factor, tyrosine kinase inhibitors, various antibiotics).3 Histologically, acute febrile neutrophilic dermatosis is characterized by dense neutrophilic infiltrates, often with notable dermal edema (Figure 2).4 Many cases also show leukocytoclastic vasculitis; however, foamy histiocytes are not a notable component of the inflammatory infiltrate, though a histiocytoid form of acute febrile neutrophilic dermatosis has been described.5 Infections must be rigorously ruled out prior to diagnosing a patient with acute febrile neutrophilic dermatosis, making it a diagnosis of exclusion.
Cutaneous coccidioidomycosis is an infection caused by the dimorphic fungi Coccidioides immitis or Coccidioides posadasii. Cutaneous disease is rare but can occur from direct inoculation or dissemination from pulmonary disease in immunocompetent or immunocompromised patients. Papules, pustules, or plaques are seen clinically. Histologically, cutaneous coccidioidomycosis shows spherules that vary from 10 to 100 μm and are filled with multiple smaller endospores (Figure 3).6 Pseudoepitheliomatous hyperplasia with dense suppurative and granulomatous infiltrates also is seen.
Erythema induratum is characterized by tender nodules on the lower extremities and has a substantial female predominance. Many cases are associated with Mycobacterium tuberculosis infection. The bacteria are not seen directly in the skin but are instead detectable through DNA polymerase chain reaction testing or investigation of other organ systems.7,8 Histologically, lesions show a lobular panniculitis with a mixed infiltrate. Vasculitis is seen in approximately 90% of erythema induratum cases vs approximately 25% of classic ENL cases (Figure 4),2,9 which has led some to use the term nodular vasculitis to describe this disease entity. Nodular vasculitis is considered by others to be a distinct disease entity in which there are clinical and histologic features similar to erythema induratum but no evidence of M tuberculosis infection.9
Polyarteritis nodosa is a vasculitis that affects medium- sized vessels of various organ systems. The presenting signs and symptoms vary based on the affected organ systems. Palpable to retiform purpura, livedo racemosa, subcutaneous nodules, or ulcers are seen when the skin is involved. The histologic hallmark is necrotizing vasculitis of medium-sized arterioles (Figure 5), although leukocytoclastic vasculitis of small-caliber vessels also can be seen in biopsies of affected skin.10 The vascular changes are said to be segmental, with uninvolved segments interspersed with involved segments. Antineutrophil cytoplasmic antibody (ANCA)– associated vasculitis also must be considered when one sees leukocytoclastic vasculitis of small-caliber vessels in the skin, as it can be distinguished most readily by detecting circulating antibodies specific for myeloperoxidase (MPO-ANCA) or proteinase 3 (PR3-ANCA).
- Polycarpou A, Walker SL, Lockwood DNJ. A systematic review of immunological studies of erythema nodosum leprosum. Front Immunol. 2017;8:233. doi:10.3389/fimmu.2017.00233
- Massone C, Belachew WA, Schettini A. Histopathology of the lepromatous skin biopsy. Clin Dermatol. 2015;33:38-45. doi:10.1016/j.clindermatol.2014.10.003
- Cohen PR. Sweet’s syndrome—a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:1-28. doi:10.1186/1750-1172-2-34
- Ratzinger G, Burgdorf W, Zelger BG, et al. Acute febrile neutrophilic dermatosis: a histopathologic study of 31 cases with review of literature. Am J Dermatopathol. 2007;29:125-133. doi:10.1097/01.dad.0000249887.59810.76
- Wilson TC, Stone MS, Swick BL. Histiocytoid Sweet syndrome with haloed myeloid cells masquerading as a cryptococcal infection. Am J Dermatopathology. 2014;36:264-269. doi:10.1097/DAD.0b013e31828b811b
- Guarner J, Brandt ME. Histopathologic diagnosis of fungal infections in the 21st century. Clin Microbiol Rev. 2011;24:247-280. doi:10.1128/CMR.00053-10
- Schneider JW, Jordaan HF, Geiger DH, et al. Erythema induratum of Bazin: a clinicopathological study of 20 cases of Mycobacterium tuberculosis DNA in skin lesions by polymerase chain reaction. Am J Dermatopathol. 1995;17:350-356. doi:10.1097/00000372-199508000-00008
- Boonchai W, Suthipinittharm P, Mahaisavariya P. Panniculitis in tuberculosis: a clinicopathologic study of nodular panniculitis associated with tuberculosis. Int J Dermatol. 1998;37:361-363. doi:10.1046/j.1365-4362.1998.00299.x
- Segura S, Pujol RM, Trindade F, et al. Vasculitis in erythema induratum of Bazin: a histopathologic study of 101 biopsy specimens from 86 patients. J Am Acad Dermatol. 2008;59:839-851. doi:10.1016/j.jaad.2008.07.030
- Ishiguro N, Kawashima M. Cutaneous polyarteritis nodosa: a report of 16 cases with clinical and histopathological analysis and a review of the published work. J Dermatol. 2010;37:85-93. doi:10.1111/j.1346-8138.2009.00752.x
- Polycarpou A, Walker SL, Lockwood DNJ. A systematic review of immunological studies of erythema nodosum leprosum. Front Immunol. 2017;8:233. doi:10.3389/fimmu.2017.00233
- Massone C, Belachew WA, Schettini A. Histopathology of the lepromatous skin biopsy. Clin Dermatol. 2015;33:38-45. doi:10.1016/j.clindermatol.2014.10.003
- Cohen PR. Sweet’s syndrome—a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:1-28. doi:10.1186/1750-1172-2-34
- Ratzinger G, Burgdorf W, Zelger BG, et al. Acute febrile neutrophilic dermatosis: a histopathologic study of 31 cases with review of literature. Am J Dermatopathol. 2007;29:125-133. doi:10.1097/01.dad.0000249887.59810.76
- Wilson TC, Stone MS, Swick BL. Histiocytoid Sweet syndrome with haloed myeloid cells masquerading as a cryptococcal infection. Am J Dermatopathology. 2014;36:264-269. doi:10.1097/DAD.0b013e31828b811b
- Guarner J, Brandt ME. Histopathologic diagnosis of fungal infections in the 21st century. Clin Microbiol Rev. 2011;24:247-280. doi:10.1128/CMR.00053-10
- Schneider JW, Jordaan HF, Geiger DH, et al. Erythema induratum of Bazin: a clinicopathological study of 20 cases of Mycobacterium tuberculosis DNA in skin lesions by polymerase chain reaction. Am J Dermatopathol. 1995;17:350-356. doi:10.1097/00000372-199508000-00008
- Boonchai W, Suthipinittharm P, Mahaisavariya P. Panniculitis in tuberculosis: a clinicopathologic study of nodular panniculitis associated with tuberculosis. Int J Dermatol. 1998;37:361-363. doi:10.1046/j.1365-4362.1998.00299.x
- Segura S, Pujol RM, Trindade F, et al. Vasculitis in erythema induratum of Bazin: a histopathologic study of 101 biopsy specimens from 86 patients. J Am Acad Dermatol. 2008;59:839-851. doi:10.1016/j.jaad.2008.07.030
- Ishiguro N, Kawashima M. Cutaneous polyarteritis nodosa: a report of 16 cases with clinical and histopathological analysis and a review of the published work. J Dermatol. 2010;37:85-93. doi:10.1111/j.1346-8138.2009.00752.x
A 66-year-old man presented with new tender erythematous papules scattered over the arms and legs. A biopsy of a lesion on the left thigh was performed.