User login
RAP device cleared for short-term improvement in appearance of cellulite
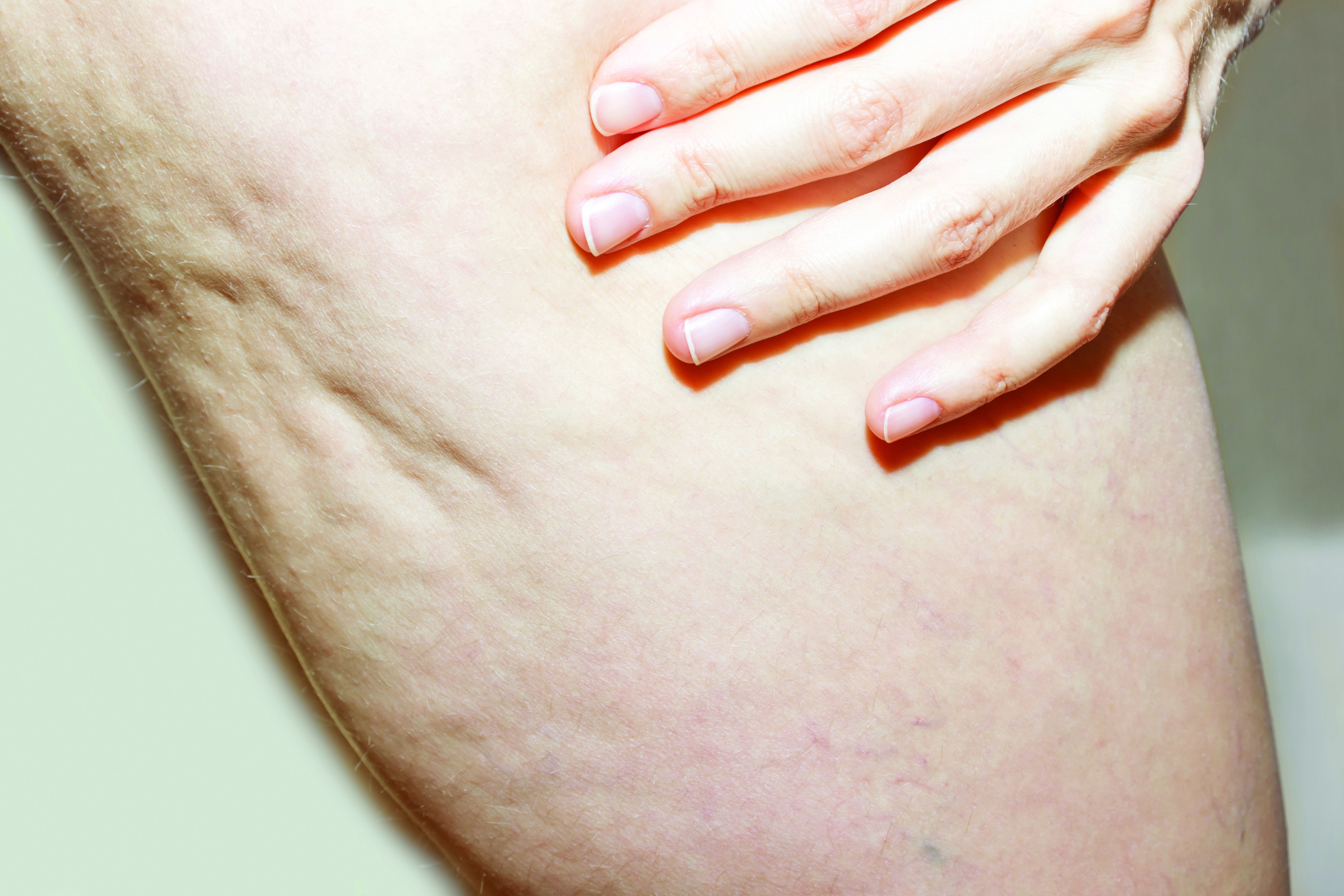
As described in a press release issued by Soliton, the RAP device emits rapid acoustic pulses (shock waves) that are transmitted through the skin at a rate of up to 100 pulses per second to rupture or “shear” the fibrotic septa. This causes the release of septa, which results in a smoothening of skin dimples. The procedure takes 40-60 minutes to perform.
“This is a novel, noninvasive treatment for cellulite that appears to be safe, with little pain and little downtime,” Mathew M. Avram, MD, JD, director of laser, cosmetics, and dermatologic surgery at Massachusetts General Hospital, Boston, said in an interview. “Further study and experience will determine the efficacy of this device and the optimization of its parameters going forward.”
In clinical trials that were part of the FDA’s 510(k) application process, patients underwent a single, noninvasive treatment that required no anesthesia and caused no unexpected or serious adverse events. The procedure also received strong patient satisfaction ratings, and clinical trial participants rated their average pain score as 2.4 out of 10.
Soliton plans to begin selling the device for both tattoo removal and cellulite treatment in the first half of 2021. “While the technology is broadly the same, the replaceable treatment cartridges [for tattoo removal and cellulite treatment] differ in significant ways,” Dr. Avram said.
Dr. Avram disclosed that he has received consulting fees from Allergan, Merz, Sciton, and Soliton. He also reported having ownership and/or shareholder interest in Cytrellis.
As described in a press release issued by Soliton, the RAP device emits rapid acoustic pulses (shock waves) that are transmitted through the skin at a rate of up to 100 pulses per second to rupture or “shear” the fibrotic septa. This causes the release of septa, which results in a smoothening of skin dimples. The procedure takes 40-60 minutes to perform.
“This is a novel, noninvasive treatment for cellulite that appears to be safe, with little pain and little downtime,” Mathew M. Avram, MD, JD, director of laser, cosmetics, and dermatologic surgery at Massachusetts General Hospital, Boston, said in an interview. “Further study and experience will determine the efficacy of this device and the optimization of its parameters going forward.”
In clinical trials that were part of the FDA’s 510(k) application process, patients underwent a single, noninvasive treatment that required no anesthesia and caused no unexpected or serious adverse events. The procedure also received strong patient satisfaction ratings, and clinical trial participants rated their average pain score as 2.4 out of 10.
Soliton plans to begin selling the device for both tattoo removal and cellulite treatment in the first half of 2021. “While the technology is broadly the same, the replaceable treatment cartridges [for tattoo removal and cellulite treatment] differ in significant ways,” Dr. Avram said.
Dr. Avram disclosed that he has received consulting fees from Allergan, Merz, Sciton, and Soliton. He also reported having ownership and/or shareholder interest in Cytrellis.
As described in a press release issued by Soliton, the RAP device emits rapid acoustic pulses (shock waves) that are transmitted through the skin at a rate of up to 100 pulses per second to rupture or “shear” the fibrotic septa. This causes the release of septa, which results in a smoothening of skin dimples. The procedure takes 40-60 minutes to perform.
“This is a novel, noninvasive treatment for cellulite that appears to be safe, with little pain and little downtime,” Mathew M. Avram, MD, JD, director of laser, cosmetics, and dermatologic surgery at Massachusetts General Hospital, Boston, said in an interview. “Further study and experience will determine the efficacy of this device and the optimization of its parameters going forward.”
In clinical trials that were part of the FDA’s 510(k) application process, patients underwent a single, noninvasive treatment that required no anesthesia and caused no unexpected or serious adverse events. The procedure also received strong patient satisfaction ratings, and clinical trial participants rated their average pain score as 2.4 out of 10.
Soliton plans to begin selling the device for both tattoo removal and cellulite treatment in the first half of 2021. “While the technology is broadly the same, the replaceable treatment cartridges [for tattoo removal and cellulite treatment] differ in significant ways,” Dr. Avram said.
Dr. Avram disclosed that he has received consulting fees from Allergan, Merz, Sciton, and Soliton. He also reported having ownership and/or shareholder interest in Cytrellis.
Novel oral testosterone replacement therapy headed to FDA
Marius Pharmaceuticals has submitted a new drug application (NDA) to the Food and Drug Administration for Kyzatrex, an oral testosterone replacement therapy (TRT).
With this NDA, the company is seeking approval for Kyzatrex as a treatment for adult men with primary and secondary hypogonadism, also known as testosterone deficiency. Marius has requested a priority review that, if accepted, would result in an anticipated 6-month review period.
Current treatment options for hypogonadal men consist of therapies with safety concerns, such as cardiovascular and metabolic risks, that make patient adherence to treatment very low.
Kyzatrex is a novel oral formulation of testosterone undecanoate administered twice daily in a soft gelatin capsule.
“TRT remains a therapeutic challenge because there are worrisome and conflicting data related to increased cardiovascular disease risk, which has special relevance to high-risk diabetic populations,” Paul S. Jellinger, MD, professor of clinical medicine at the University of Miami, told this news organization. Furthermore, “injectable depot testosterone may be associated with peak supraphysiological levels and a substantial increase in hemoglobin. Topical testosterone offers more stable levels without a peak and trough, but in some men achieving physiologic levels may be difficult.”
The NDA is supported by results from a 6-month treatment extension of the pivotal phase 3 MRS-TU-2019 study (NCT04467697). Final results from this study have not been presented, but the company wrote in a press release that the results will be published some time in 2021.
They further reported that Kyzatrex was well tolerated by patients, with more than 96% of study participants completing 90 days of treatment in the pivotal phase 3 study. Study patients achieved average testosterone levels in the normal range.
Across the pooled phase 3 trials, the most frequent treatment-related treatment-emergent adverse event (TEAE) was hypertension, and no serious TEAEs were considered treatment related.
“We are extremely proud to have generated compelling efficacy and safety data in our phase 3 trials,” said Om Dhingra, PhD, cofounder and CEO of Marius. “We look forward to continuing to work collaboratively with the FDA on the review of our application, and if approved, Kyzatrex has the potential to become the standard of care for the treatment of primary and secondary hypogonadism globally.”
“An oral [testosterone] preparation with steady state physiologic levels would be a welcome addition to our choices for therapy assuming, of course, the absence of adverse effects,” explained Dr. Jellinger. “However, the greater challenge of testosterone therapy is the appropriate selection of those suited for testosterone replacement therapy.”
The company also plans to submit a marketing authorization application with the European Medicines Agency in the first half of 2022.
Marius Pharmaceuticals has submitted a new drug application (NDA) to the Food and Drug Administration for Kyzatrex, an oral testosterone replacement therapy (TRT).
With this NDA, the company is seeking approval for Kyzatrex as a treatment for adult men with primary and secondary hypogonadism, also known as testosterone deficiency. Marius has requested a priority review that, if accepted, would result in an anticipated 6-month review period.
Current treatment options for hypogonadal men consist of therapies with safety concerns, such as cardiovascular and metabolic risks, that make patient adherence to treatment very low.
Kyzatrex is a novel oral formulation of testosterone undecanoate administered twice daily in a soft gelatin capsule.
“TRT remains a therapeutic challenge because there are worrisome and conflicting data related to increased cardiovascular disease risk, which has special relevance to high-risk diabetic populations,” Paul S. Jellinger, MD, professor of clinical medicine at the University of Miami, told this news organization. Furthermore, “injectable depot testosterone may be associated with peak supraphysiological levels and a substantial increase in hemoglobin. Topical testosterone offers more stable levels without a peak and trough, but in some men achieving physiologic levels may be difficult.”
The NDA is supported by results from a 6-month treatment extension of the pivotal phase 3 MRS-TU-2019 study (NCT04467697). Final results from this study have not been presented, but the company wrote in a press release that the results will be published some time in 2021.
They further reported that Kyzatrex was well tolerated by patients, with more than 96% of study participants completing 90 days of treatment in the pivotal phase 3 study. Study patients achieved average testosterone levels in the normal range.
Across the pooled phase 3 trials, the most frequent treatment-related treatment-emergent adverse event (TEAE) was hypertension, and no serious TEAEs were considered treatment related.
“We are extremely proud to have generated compelling efficacy and safety data in our phase 3 trials,” said Om Dhingra, PhD, cofounder and CEO of Marius. “We look forward to continuing to work collaboratively with the FDA on the review of our application, and if approved, Kyzatrex has the potential to become the standard of care for the treatment of primary and secondary hypogonadism globally.”
“An oral [testosterone] preparation with steady state physiologic levels would be a welcome addition to our choices for therapy assuming, of course, the absence of adverse effects,” explained Dr. Jellinger. “However, the greater challenge of testosterone therapy is the appropriate selection of those suited for testosterone replacement therapy.”
The company also plans to submit a marketing authorization application with the European Medicines Agency in the first half of 2022.
Marius Pharmaceuticals has submitted a new drug application (NDA) to the Food and Drug Administration for Kyzatrex, an oral testosterone replacement therapy (TRT).
With this NDA, the company is seeking approval for Kyzatrex as a treatment for adult men with primary and secondary hypogonadism, also known as testosterone deficiency. Marius has requested a priority review that, if accepted, would result in an anticipated 6-month review period.
Current treatment options for hypogonadal men consist of therapies with safety concerns, such as cardiovascular and metabolic risks, that make patient adherence to treatment very low.
Kyzatrex is a novel oral formulation of testosterone undecanoate administered twice daily in a soft gelatin capsule.
“TRT remains a therapeutic challenge because there are worrisome and conflicting data related to increased cardiovascular disease risk, which has special relevance to high-risk diabetic populations,” Paul S. Jellinger, MD, professor of clinical medicine at the University of Miami, told this news organization. Furthermore, “injectable depot testosterone may be associated with peak supraphysiological levels and a substantial increase in hemoglobin. Topical testosterone offers more stable levels without a peak and trough, but in some men achieving physiologic levels may be difficult.”
The NDA is supported by results from a 6-month treatment extension of the pivotal phase 3 MRS-TU-2019 study (NCT04467697). Final results from this study have not been presented, but the company wrote in a press release that the results will be published some time in 2021.
They further reported that Kyzatrex was well tolerated by patients, with more than 96% of study participants completing 90 days of treatment in the pivotal phase 3 study. Study patients achieved average testosterone levels in the normal range.
Across the pooled phase 3 trials, the most frequent treatment-related treatment-emergent adverse event (TEAE) was hypertension, and no serious TEAEs were considered treatment related.
“We are extremely proud to have generated compelling efficacy and safety data in our phase 3 trials,” said Om Dhingra, PhD, cofounder and CEO of Marius. “We look forward to continuing to work collaboratively with the FDA on the review of our application, and if approved, Kyzatrex has the potential to become the standard of care for the treatment of primary and secondary hypogonadism globally.”
“An oral [testosterone] preparation with steady state physiologic levels would be a welcome addition to our choices for therapy assuming, of course, the absence of adverse effects,” explained Dr. Jellinger. “However, the greater challenge of testosterone therapy is the appropriate selection of those suited for testosterone replacement therapy.”
The company also plans to submit a marketing authorization application with the European Medicines Agency in the first half of 2022.
FDA approves voclosporin for lupus nephritis
The Food and Drug Administration has approved voclosporin (Lupkynis) for the treatment of lupus nephritis, according to a Jan. 22 press release from manufacturer Aurinia Pharmaceuticals.
Lupkynis is a calcineurin-inhibitor immunosuppressant, and is the first oral medication to show effectiveness in lupus nephritis, according to the company. The drug is indicated for the treatment of adult patients with active lupus nephritis in combination with a background immunosuppressive therapy regimen, according to the drug label, which also has a boxed warning describing the increased risk of infections and malignancies, including lymphoma.
The approval of voclosporin was based on data from two studies, the AURORA phase 3 study and the AURA-LV phase 2 study. The studies included 533 adults with lupus nephritis who were randomized to 23.7 mg or placebo of voclosporin twice daily in the form of oral capsules, or placebo capsules, in addition to standard of care (mycophenolate mofetil plus low-dose glucocorticoids).
In the AURORA phase 3 study of 357 patients, close to twice as many patients in the treatment group showed a complete renal response, compared with the placebo group after 1 year (40.8% vs. 22.5%). In addition, patients treated with voclosporin more quickly achieved a significant reduction in urine protein to creatinine ratio, compared with the placebo patients (169 days vs. 372 days).
Severe adverse events were similar between the groups, including the most common complication of infection (10.1% and 11.2% for voclosporin and control groups, respectively). Other adverse reactions reported in at least 3% of the study participants included a decrease in glomerular filtration rate, hypertension, diarrhea, headache, anemia, cough, urinary tract infection, upper abdominal pain, dyspepsia, alopecia, renal impairment, abdominal pain, mouth ulceration, fatigue, tremor, acute kidney injury, and decreased appetite, according to the company press release.
Full clinical trial information for the AURORA study is available here.
The Food and Drug Administration has approved voclosporin (Lupkynis) for the treatment of lupus nephritis, according to a Jan. 22 press release from manufacturer Aurinia Pharmaceuticals.
Lupkynis is a calcineurin-inhibitor immunosuppressant, and is the first oral medication to show effectiveness in lupus nephritis, according to the company. The drug is indicated for the treatment of adult patients with active lupus nephritis in combination with a background immunosuppressive therapy regimen, according to the drug label, which also has a boxed warning describing the increased risk of infections and malignancies, including lymphoma.
The approval of voclosporin was based on data from two studies, the AURORA phase 3 study and the AURA-LV phase 2 study. The studies included 533 adults with lupus nephritis who were randomized to 23.7 mg or placebo of voclosporin twice daily in the form of oral capsules, or placebo capsules, in addition to standard of care (mycophenolate mofetil plus low-dose glucocorticoids).
In the AURORA phase 3 study of 357 patients, close to twice as many patients in the treatment group showed a complete renal response, compared with the placebo group after 1 year (40.8% vs. 22.5%). In addition, patients treated with voclosporin more quickly achieved a significant reduction in urine protein to creatinine ratio, compared with the placebo patients (169 days vs. 372 days).
Severe adverse events were similar between the groups, including the most common complication of infection (10.1% and 11.2% for voclosporin and control groups, respectively). Other adverse reactions reported in at least 3% of the study participants included a decrease in glomerular filtration rate, hypertension, diarrhea, headache, anemia, cough, urinary tract infection, upper abdominal pain, dyspepsia, alopecia, renal impairment, abdominal pain, mouth ulceration, fatigue, tremor, acute kidney injury, and decreased appetite, according to the company press release.
Full clinical trial information for the AURORA study is available here.
The Food and Drug Administration has approved voclosporin (Lupkynis) for the treatment of lupus nephritis, according to a Jan. 22 press release from manufacturer Aurinia Pharmaceuticals.
Lupkynis is a calcineurin-inhibitor immunosuppressant, and is the first oral medication to show effectiveness in lupus nephritis, according to the company. The drug is indicated for the treatment of adult patients with active lupus nephritis in combination with a background immunosuppressive therapy regimen, according to the drug label, which also has a boxed warning describing the increased risk of infections and malignancies, including lymphoma.
The approval of voclosporin was based on data from two studies, the AURORA phase 3 study and the AURA-LV phase 2 study. The studies included 533 adults with lupus nephritis who were randomized to 23.7 mg or placebo of voclosporin twice daily in the form of oral capsules, or placebo capsules, in addition to standard of care (mycophenolate mofetil plus low-dose glucocorticoids).
In the AURORA phase 3 study of 357 patients, close to twice as many patients in the treatment group showed a complete renal response, compared with the placebo group after 1 year (40.8% vs. 22.5%). In addition, patients treated with voclosporin more quickly achieved a significant reduction in urine protein to creatinine ratio, compared with the placebo patients (169 days vs. 372 days).
Severe adverse events were similar between the groups, including the most common complication of infection (10.1% and 11.2% for voclosporin and control groups, respectively). Other adverse reactions reported in at least 3% of the study participants included a decrease in glomerular filtration rate, hypertension, diarrhea, headache, anemia, cough, urinary tract infection, upper abdominal pain, dyspepsia, alopecia, renal impairment, abdominal pain, mouth ulceration, fatigue, tremor, acute kidney injury, and decreased appetite, according to the company press release.
Full clinical trial information for the AURORA study is available here.
First monthly injectable HIV treatment approved by FDA
Cabenuva (cabotegravir and rilpivirine, a once-per-month injectable formulation) was approved by the Food and Drug Administration as a complete regimen for treatment of HIV-1 infection in adults. It is intended to replace current antiretroviral regimens in those patients who are virologically suppressed with no history of treatment failure and with no known or suspected resistance to either of the two component drugs.
Cabenuva is the first FDA-approved monthly injectable, complete regimen for HIV-infected adults, according to the agency’s announcement.
In addition, the FDA-approved Vocabria (cabotegravir, tablet formulation), a preparatory treatment intended to be taken in combination with oral rilpivirine (Edurant) for 1 month prior to starting treatment with Cabenuva to ensure the medications are well tolerated before switching to the extended-release injectable formulation. The FDA granted the approval of Cabenuva and Vocabria to ViiV Healthcare.
Cabotegravir is as an integrase strand transfer inhibitor that blocks HIV integrase by attaching to the active integrase site and inhibiting retroviral DNA integration, which is necessary in order for HIV to replicate. In contrast, rilpivirine acts as a diarylpyrimidine nonnucleoside reverse transcriptase inhibitor of HIV-1.
Approval of Cabenuva was based upon two randomized, open-label, controlled clinical trials in 1,182 HIV-infected adults who were virologically suppressed (HIV-1 RNA less than 50 copies/mL) before initiation of treatment with Cabenuva. The two pivotal phase three clinical studies were: Antiretroviral Therapy as Long-Acting Suppression (ATLAS; NCT02951052) and First Long-Acting Injectable Regimen (FLAIR; NCT02938520). Patients in both trials continued to show virologic suppression at the conclusion of each study, and no clinically relevant change from baseline in CD4+ cell counts was observed, according to the FDA announcement.
Adverse reactions with Cabenuva included injection-site reactions, fever, fatigue, headache, musculoskeletal pain, nausea, sleep disorders, dizziness, and rash. The FDA warned that Cabenuva should not be used if there is a known previous hypersensitivity reaction to cabotegravir or rilpivirine, or in patients who are not virally suppressed (HIV-1 RNA greater than 50 copies/mL).
Cabenuva and Vocabria were granted Fast Track and Priority Review designation by the FDA. Prescribing information for Cabenuva is available on the ViiV Healthcare website.
Cabenuva (cabotegravir and rilpivirine, a once-per-month injectable formulation) was approved by the Food and Drug Administration as a complete regimen for treatment of HIV-1 infection in adults. It is intended to replace current antiretroviral regimens in those patients who are virologically suppressed with no history of treatment failure and with no known or suspected resistance to either of the two component drugs.
Cabenuva is the first FDA-approved monthly injectable, complete regimen for HIV-infected adults, according to the agency’s announcement.
In addition, the FDA-approved Vocabria (cabotegravir, tablet formulation), a preparatory treatment intended to be taken in combination with oral rilpivirine (Edurant) for 1 month prior to starting treatment with Cabenuva to ensure the medications are well tolerated before switching to the extended-release injectable formulation. The FDA granted the approval of Cabenuva and Vocabria to ViiV Healthcare.
Cabotegravir is as an integrase strand transfer inhibitor that blocks HIV integrase by attaching to the active integrase site and inhibiting retroviral DNA integration, which is necessary in order for HIV to replicate. In contrast, rilpivirine acts as a diarylpyrimidine nonnucleoside reverse transcriptase inhibitor of HIV-1.
Approval of Cabenuva was based upon two randomized, open-label, controlled clinical trials in 1,182 HIV-infected adults who were virologically suppressed (HIV-1 RNA less than 50 copies/mL) before initiation of treatment with Cabenuva. The two pivotal phase three clinical studies were: Antiretroviral Therapy as Long-Acting Suppression (ATLAS; NCT02951052) and First Long-Acting Injectable Regimen (FLAIR; NCT02938520). Patients in both trials continued to show virologic suppression at the conclusion of each study, and no clinically relevant change from baseline in CD4+ cell counts was observed, according to the FDA announcement.
Adverse reactions with Cabenuva included injection-site reactions, fever, fatigue, headache, musculoskeletal pain, nausea, sleep disorders, dizziness, and rash. The FDA warned that Cabenuva should not be used if there is a known previous hypersensitivity reaction to cabotegravir or rilpivirine, or in patients who are not virally suppressed (HIV-1 RNA greater than 50 copies/mL).
Cabenuva and Vocabria were granted Fast Track and Priority Review designation by the FDA. Prescribing information for Cabenuva is available on the ViiV Healthcare website.
Cabenuva (cabotegravir and rilpivirine, a once-per-month injectable formulation) was approved by the Food and Drug Administration as a complete regimen for treatment of HIV-1 infection in adults. It is intended to replace current antiretroviral regimens in those patients who are virologically suppressed with no history of treatment failure and with no known or suspected resistance to either of the two component drugs.
Cabenuva is the first FDA-approved monthly injectable, complete regimen for HIV-infected adults, according to the agency’s announcement.
In addition, the FDA-approved Vocabria (cabotegravir, tablet formulation), a preparatory treatment intended to be taken in combination with oral rilpivirine (Edurant) for 1 month prior to starting treatment with Cabenuva to ensure the medications are well tolerated before switching to the extended-release injectable formulation. The FDA granted the approval of Cabenuva and Vocabria to ViiV Healthcare.
Cabotegravir is as an integrase strand transfer inhibitor that blocks HIV integrase by attaching to the active integrase site and inhibiting retroviral DNA integration, which is necessary in order for HIV to replicate. In contrast, rilpivirine acts as a diarylpyrimidine nonnucleoside reverse transcriptase inhibitor of HIV-1.
Approval of Cabenuva was based upon two randomized, open-label, controlled clinical trials in 1,182 HIV-infected adults who were virologically suppressed (HIV-1 RNA less than 50 copies/mL) before initiation of treatment with Cabenuva. The two pivotal phase three clinical studies were: Antiretroviral Therapy as Long-Acting Suppression (ATLAS; NCT02951052) and First Long-Acting Injectable Regimen (FLAIR; NCT02938520). Patients in both trials continued to show virologic suppression at the conclusion of each study, and no clinically relevant change from baseline in CD4+ cell counts was observed, according to the FDA announcement.
Adverse reactions with Cabenuva included injection-site reactions, fever, fatigue, headache, musculoskeletal pain, nausea, sleep disorders, dizziness, and rash. The FDA warned that Cabenuva should not be used if there is a known previous hypersensitivity reaction to cabotegravir or rilpivirine, or in patients who are not virally suppressed (HIV-1 RNA greater than 50 copies/mL).
Cabenuva and Vocabria were granted Fast Track and Priority Review designation by the FDA. Prescribing information for Cabenuva is available on the ViiV Healthcare website.
NEWS FROM THE FDA
Sotagliflozin’s trial data receives FDA welcome for NDA filing
The Food and Drug Administration has determined that data collected on the dual SGLT1/2 inhibitor sotagliflozin (Zynquista) for treating patients with type 2 diabetes in the SOLOIST and SCORED pivotal trials can help support a New Drug Application (NDA) submission, according to a statement released on Jan. 14 by Lexicon Pharmaceuticals, the company developing this drug. Lexicon concurrently said that it hopes to potentially file this NDA later in 2021.
The statement said the FDA’s decision related to an NDA for “an indication to reduce the risk of cardiovascular death, hospitalization for heart failure, and urgent visits for heart failure in adult patients with type 2 diabetes with either worsening heart failure or additional risk factors for heart failure.”
Results from SOLOIST and SCORED, first reported in November 2020 at the American Heart Association scientific sessions, showed statistically significant benefits for their respective primary endpoints.
The findings also demonstrated several novel benefits from the first advanced clinical trials of an SGLT inhibitor that blocks both the SGLT2 protein in kidneys as well as the SGLT1 protein, which resides primarily in the gastrointestinal system and is the main route for glucose out of the gut.
In both SOLOIST and SCORED, patient outcomes on sotagliflozin tracked the benefits and adverse effects previously seen with several SGLT2 inhibitors (canagliflozin, dapagliflozin, empagliflozin, and ertugliflozin), but in addition showed several unprecedented benefits: An ability to lower hemoglobin A1c in patients with severely depressed renal function, safe initiation in patients recently hospitalized for heart failure, the first prospective data to show improvements in patients with heart failure with preserved ejection fraction, and a higher level of protection against MIs and strokes than the SGLT2 inhibitors.
The FDA’s willingness to consider data from both trials in an NDA was not a given, as the primary endpoints for both trials underwent tweaking while they were underway to compensate for an unexpectedly early end to patient enrollment and follow-up caused by changes in drug company sponsorship and challenges introduced by the COVID-19 pandemic.
In 2019, the FDA denied the NDA for sotagliflozin as a treatment for patients with type 1 diabetes, but this indication received approval in Europe.
SOLOIST and SCORED were sponsored initially by Sanofi, and more recently by Lexicon.
The Food and Drug Administration has determined that data collected on the dual SGLT1/2 inhibitor sotagliflozin (Zynquista) for treating patients with type 2 diabetes in the SOLOIST and SCORED pivotal trials can help support a New Drug Application (NDA) submission, according to a statement released on Jan. 14 by Lexicon Pharmaceuticals, the company developing this drug. Lexicon concurrently said that it hopes to potentially file this NDA later in 2021.
The statement said the FDA’s decision related to an NDA for “an indication to reduce the risk of cardiovascular death, hospitalization for heart failure, and urgent visits for heart failure in adult patients with type 2 diabetes with either worsening heart failure or additional risk factors for heart failure.”
Results from SOLOIST and SCORED, first reported in November 2020 at the American Heart Association scientific sessions, showed statistically significant benefits for their respective primary endpoints.
The findings also demonstrated several novel benefits from the first advanced clinical trials of an SGLT inhibitor that blocks both the SGLT2 protein in kidneys as well as the SGLT1 protein, which resides primarily in the gastrointestinal system and is the main route for glucose out of the gut.
In both SOLOIST and SCORED, patient outcomes on sotagliflozin tracked the benefits and adverse effects previously seen with several SGLT2 inhibitors (canagliflozin, dapagliflozin, empagliflozin, and ertugliflozin), but in addition showed several unprecedented benefits: An ability to lower hemoglobin A1c in patients with severely depressed renal function, safe initiation in patients recently hospitalized for heart failure, the first prospective data to show improvements in patients with heart failure with preserved ejection fraction, and a higher level of protection against MIs and strokes than the SGLT2 inhibitors.
The FDA’s willingness to consider data from both trials in an NDA was not a given, as the primary endpoints for both trials underwent tweaking while they were underway to compensate for an unexpectedly early end to patient enrollment and follow-up caused by changes in drug company sponsorship and challenges introduced by the COVID-19 pandemic.
In 2019, the FDA denied the NDA for sotagliflozin as a treatment for patients with type 1 diabetes, but this indication received approval in Europe.
SOLOIST and SCORED were sponsored initially by Sanofi, and more recently by Lexicon.
The Food and Drug Administration has determined that data collected on the dual SGLT1/2 inhibitor sotagliflozin (Zynquista) for treating patients with type 2 diabetes in the SOLOIST and SCORED pivotal trials can help support a New Drug Application (NDA) submission, according to a statement released on Jan. 14 by Lexicon Pharmaceuticals, the company developing this drug. Lexicon concurrently said that it hopes to potentially file this NDA later in 2021.
The statement said the FDA’s decision related to an NDA for “an indication to reduce the risk of cardiovascular death, hospitalization for heart failure, and urgent visits for heart failure in adult patients with type 2 diabetes with either worsening heart failure or additional risk factors for heart failure.”
Results from SOLOIST and SCORED, first reported in November 2020 at the American Heart Association scientific sessions, showed statistically significant benefits for their respective primary endpoints.
The findings also demonstrated several novel benefits from the first advanced clinical trials of an SGLT inhibitor that blocks both the SGLT2 protein in kidneys as well as the SGLT1 protein, which resides primarily in the gastrointestinal system and is the main route for glucose out of the gut.
In both SOLOIST and SCORED, patient outcomes on sotagliflozin tracked the benefits and adverse effects previously seen with several SGLT2 inhibitors (canagliflozin, dapagliflozin, empagliflozin, and ertugliflozin), but in addition showed several unprecedented benefits: An ability to lower hemoglobin A1c in patients with severely depressed renal function, safe initiation in patients recently hospitalized for heart failure, the first prospective data to show improvements in patients with heart failure with preserved ejection fraction, and a higher level of protection against MIs and strokes than the SGLT2 inhibitors.
The FDA’s willingness to consider data from both trials in an NDA was not a given, as the primary endpoints for both trials underwent tweaking while they were underway to compensate for an unexpectedly early end to patient enrollment and follow-up caused by changes in drug company sponsorship and challenges introduced by the COVID-19 pandemic.
In 2019, the FDA denied the NDA for sotagliflozin as a treatment for patients with type 1 diabetes, but this indication received approval in Europe.
SOLOIST and SCORED were sponsored initially by Sanofi, and more recently by Lexicon.
Another lot of extended-release metformin is recalled in the U.S.
Nostrum Laboratories has voluntarily recalled another lot of metformin HCl extended-release tablets 750-mg dosage, expanding their initial announcement in November 2020. According to the new notice, issued by the Food and Drug Administration earlier this week, the recalled tablets are off-white and oblong with a debossed ID “NM7.”

The lot number, NDC, and expiration dates can be found on the FDA website.
Nostrum noted that the tablets were distributed across the United States to wholesalers; these distributors are being notified of the recall and the company is arranging for the drug to be returned.
Metformin is the most prescribed medication worldwide for the treatment of type 2 diabetes.
Nostrum said that anyone in possession of any of the affected lots should consult their physician or pharmacist to obtain a replacement treatment option because it can be dangerous for patients with type 2 diabetes to stop taking metformin.
This new announcement expands further the number of metformin HCl extended-release tablets recalled in the United States because they contain potentially high levels of nitrosamines, also known as N-nitrosodimethylamine (NDMA), which are possible carcinogens.
The risks of nitrosamines are not clear. The FDA said they may increase the risk of cancer in people who are exposed to high levels over a long period of time, “but we do not anticipate that shorter-term exposure at levels above the acceptable intake limit would lead to an increase in the risk of cancer.”
As well as the November recall of 2 lots of metformin by Nostrum, 76 more lots of metformin extended-release tablets were flagged in October 2020 from various manufacturers for possible contamination with NDMA, on top of an earlier recall for the same problem in May 2020.
More than 175 different drug combinations, all extended release with either 500 mg or 750 mg of metformin, have now been recalled since late May 2020, and a list of those recalled to November 2020 is available here.
A version of this article first appeared on Medscape.com.
Nostrum Laboratories has voluntarily recalled another lot of metformin HCl extended-release tablets 750-mg dosage, expanding their initial announcement in November 2020. According to the new notice, issued by the Food and Drug Administration earlier this week, the recalled tablets are off-white and oblong with a debossed ID “NM7.”
The lot number, NDC, and expiration dates can be found on the FDA website.
Nostrum noted that the tablets were distributed across the United States to wholesalers; these distributors are being notified of the recall and the company is arranging for the drug to be returned.
Metformin is the most prescribed medication worldwide for the treatment of type 2 diabetes.
Nostrum said that anyone in possession of any of the affected lots should consult their physician or pharmacist to obtain a replacement treatment option because it can be dangerous for patients with type 2 diabetes to stop taking metformin.
This new announcement expands further the number of metformin HCl extended-release tablets recalled in the United States because they contain potentially high levels of nitrosamines, also known as N-nitrosodimethylamine (NDMA), which are possible carcinogens.
The risks of nitrosamines are not clear. The FDA said they may increase the risk of cancer in people who are exposed to high levels over a long period of time, “but we do not anticipate that shorter-term exposure at levels above the acceptable intake limit would lead to an increase in the risk of cancer.”
As well as the November recall of 2 lots of metformin by Nostrum, 76 more lots of metformin extended-release tablets were flagged in October 2020 from various manufacturers for possible contamination with NDMA, on top of an earlier recall for the same problem in May 2020.
More than 175 different drug combinations, all extended release with either 500 mg or 750 mg of metformin, have now been recalled since late May 2020, and a list of those recalled to November 2020 is available here.
A version of this article first appeared on Medscape.com.
Nostrum Laboratories has voluntarily recalled another lot of metformin HCl extended-release tablets 750-mg dosage, expanding their initial announcement in November 2020. According to the new notice, issued by the Food and Drug Administration earlier this week, the recalled tablets are off-white and oblong with a debossed ID “NM7.”
The lot number, NDC, and expiration dates can be found on the FDA website.
Nostrum noted that the tablets were distributed across the United States to wholesalers; these distributors are being notified of the recall and the company is arranging for the drug to be returned.
Metformin is the most prescribed medication worldwide for the treatment of type 2 diabetes.
Nostrum said that anyone in possession of any of the affected lots should consult their physician or pharmacist to obtain a replacement treatment option because it can be dangerous for patients with type 2 diabetes to stop taking metformin.
This new announcement expands further the number of metformin HCl extended-release tablets recalled in the United States because they contain potentially high levels of nitrosamines, also known as N-nitrosodimethylamine (NDMA), which are possible carcinogens.
The risks of nitrosamines are not clear. The FDA said they may increase the risk of cancer in people who are exposed to high levels over a long period of time, “but we do not anticipate that shorter-term exposure at levels above the acceptable intake limit would lead to an increase in the risk of cancer.”
As well as the November recall of 2 lots of metformin by Nostrum, 76 more lots of metformin extended-release tablets were flagged in October 2020 from various manufacturers for possible contamination with NDMA, on top of an earlier recall for the same problem in May 2020.
More than 175 different drug combinations, all extended release with either 500 mg or 750 mg of metformin, have now been recalled since late May 2020, and a list of those recalled to November 2020 is available here.
A version of this article first appeared on Medscape.com.
Feds to states: Give COVID-19 vaccine to 65+ and those with comorbidities
Federal health officials are urging states to vaccinate all Americans over age 65 and those aged 16-64 who have a documented underlying health condition that makes them more vulnerable to COVID-19.
U.S. Department of Health and Human Services (HHS) Secretary Alex Azar and Centers for Disease Control and Prevention Director Robert Redfield, MD, made the recommendation in a briefing with reporters on Jan. 12, saying that the current vaccine supply was sufficient to meet demand for the next phase of immunization as recommended by the CDC’s Advisory Committee on Immunization Practices.
“We are ready for a transition that we outlined last September in the playbook we sent to states,” Mr. Azar said. Both he and U.S. Army General Gustave F. Perna, chief operations officer for Operation Warp Speed, said that confidence in the distribution system had led to the decision to urge wider access.
The federal government will also increase the number of sites eligible to receive vaccine – including some 13,000 federally qualified community health centers – and will not keep doses in reserve as insurance against issues that might prevent people from receiving a second dose on a timely basis.
“We don’t need to hold back reserve doses,” Mr. Azar said, noting that if there were any “glitches in production” the federal government would move to fulfill obligations for second doses first and delay initial doses.
Azar: Use it or lose it
In a move that is sure to generate pushback, Mr. Azar said that states that don’t quickly administer vaccines will receive fewer doses in the future. That policy will not go into effect until later in February, which leaves open the possibility that it could be reversed by the incoming Biden administration.
“We have too much vaccine sitting in freezers at hospitals with hospitals not using it,” said Mr. Azar, who also blamed the slow administration process on a reporting lag and states being what he called “overly prescriptive” in who has been eligible to receive a shot.
“I would rather have people working to get appointments to get vaccinated than having vaccine going to waste sitting in freezers,” he told reporters.
Mr. Azar had already been pushing for broader vaccination, telling states to do so in an Operation Warp Speed briefing on Jan. 6. At that briefing, he also said that the federal government would be stepping up vaccination through an “early launch” of a federal partnership with 19 pharmacy chains, which will let states allocate vaccines directly to some 40,000 pharmacy sites.
Gen. Perna said during the Jan. 12 briefing that the aim is to further expand that to some 70,000 locations total.
The CDC reported that as of Jan. 11 some 25.4 million doses have been distributed, with 8.9 million administered. An additional 4.2 million doses were distributed to long-term care facilities, and 937,000 residents and staff have received a dose.
“Pace of administration”
Alaska, Connecticut, North Dakota, South Dakota, the District of Columbia, West Virginia, and the Northern Mariana Islands have administered the most vaccines per capita, according to the CDC. But even these locations have immunized only 4%-5% of their populations, the New York Times reports. At the bottom: Alabama, Arizona, Arkansas, Georgia, Mississippi, and South Carolina.
The federal government can encourage but not require states to move on to new phases of vaccination.
“States ultimately determine how they will proceed with vaccination,” said Marcus Plescia, MD, MPH, chief medical officer for the Association of State and Territorial Health Officials. “Most will be cautious about assuring there are doses for those needing a second dose,” he said in an interview.
Dr. Plescia said that ensuring a second dose is available is especially important for health care workers “who need to be confident that they are protected and not inadvertently transmitting the disease themselves.”
He added that “once we reach a steady state of supply and administration, the rate-limiting factor will be supply of vaccine.”
That supply could now be threatened if states don’t comply with a just-announced federal action that will change how doses are allocated.
Beginning in late February, vaccine allocations to states will be based on “the pace of administration reported by states,” and the size of the 65-and-older population, said Mr. Azar, who has previously criticized New York Governor Andrew Cuomo for fining hospitals that didn’t use up vaccine supply within a week.
“This new system gives states a strong incentive to ensure that all vaccinations are being promptly reported, which they currently are not,” he said.
Currently, allocations are based on a state’s or territory’s population.
Prepandemic, states were required to report vaccinations within 30 days. Since COVID-19 vaccines became available, the CDC has required reporting of shots within 72 hours.
Dr. Redfield said the requirement has caused some difficulty, and that the CDC is investigating why some states have reported using only 15% of doses while others have used 80%.
States have been scrambling to ramp up vaccinations.
Just ahead of the federal briefing, Gov. Cuomo tweeted that New York would be opening up vaccinations to anyone older than 65.
The Associated Press is reporting that some states have started mass vaccination sites.
Arizona has begun operating a 24/7 appointment-only vaccination program at State Farm Stadium outside of Phoenix, with the aim of immunizing 6,000 people each day, according to local radio station KJZZ.
California and Florida have also taken steps to use stadiums, while Michigan, New Jersey, New York, and Texas will use convention centers and fairgrounds, Axios has reported.
In Florida, Palm Beach County Health Director Alina Alonso, MD, told county commissioners on Jan. 12 that there isn’t enough vaccine to meet demand, WPTV reported. “We need to realize that there’s a shortage of vaccine. So it’s not the plan, it’s not our ability to do it. It’s simply supply and demand at this point,” Dr. Alonso said, according to the TV station report.
A version of this article first appeared on Medscape.com.
Federal health officials are urging states to vaccinate all Americans over age 65 and those aged 16-64 who have a documented underlying health condition that makes them more vulnerable to COVID-19.
U.S. Department of Health and Human Services (HHS) Secretary Alex Azar and Centers for Disease Control and Prevention Director Robert Redfield, MD, made the recommendation in a briefing with reporters on Jan. 12, saying that the current vaccine supply was sufficient to meet demand for the next phase of immunization as recommended by the CDC’s Advisory Committee on Immunization Practices.
“We are ready for a transition that we outlined last September in the playbook we sent to states,” Mr. Azar said. Both he and U.S. Army General Gustave F. Perna, chief operations officer for Operation Warp Speed, said that confidence in the distribution system had led to the decision to urge wider access.
The federal government will also increase the number of sites eligible to receive vaccine – including some 13,000 federally qualified community health centers – and will not keep doses in reserve as insurance against issues that might prevent people from receiving a second dose on a timely basis.
“We don’t need to hold back reserve doses,” Mr. Azar said, noting that if there were any “glitches in production” the federal government would move to fulfill obligations for second doses first and delay initial doses.
Azar: Use it or lose it
In a move that is sure to generate pushback, Mr. Azar said that states that don’t quickly administer vaccines will receive fewer doses in the future. That policy will not go into effect until later in February, which leaves open the possibility that it could be reversed by the incoming Biden administration.
“We have too much vaccine sitting in freezers at hospitals with hospitals not using it,” said Mr. Azar, who also blamed the slow administration process on a reporting lag and states being what he called “overly prescriptive” in who has been eligible to receive a shot.
“I would rather have people working to get appointments to get vaccinated than having vaccine going to waste sitting in freezers,” he told reporters.
Mr. Azar had already been pushing for broader vaccination, telling states to do so in an Operation Warp Speed briefing on Jan. 6. At that briefing, he also said that the federal government would be stepping up vaccination through an “early launch” of a federal partnership with 19 pharmacy chains, which will let states allocate vaccines directly to some 40,000 pharmacy sites.
Gen. Perna said during the Jan. 12 briefing that the aim is to further expand that to some 70,000 locations total.
The CDC reported that as of Jan. 11 some 25.4 million doses have been distributed, with 8.9 million administered. An additional 4.2 million doses were distributed to long-term care facilities, and 937,000 residents and staff have received a dose.
“Pace of administration”
Alaska, Connecticut, North Dakota, South Dakota, the District of Columbia, West Virginia, and the Northern Mariana Islands have administered the most vaccines per capita, according to the CDC. But even these locations have immunized only 4%-5% of their populations, the New York Times reports. At the bottom: Alabama, Arizona, Arkansas, Georgia, Mississippi, and South Carolina.
The federal government can encourage but not require states to move on to new phases of vaccination.
“States ultimately determine how they will proceed with vaccination,” said Marcus Plescia, MD, MPH, chief medical officer for the Association of State and Territorial Health Officials. “Most will be cautious about assuring there are doses for those needing a second dose,” he said in an interview.
Dr. Plescia said that ensuring a second dose is available is especially important for health care workers “who need to be confident that they are protected and not inadvertently transmitting the disease themselves.”
He added that “once we reach a steady state of supply and administration, the rate-limiting factor will be supply of vaccine.”
That supply could now be threatened if states don’t comply with a just-announced federal action that will change how doses are allocated.
Beginning in late February, vaccine allocations to states will be based on “the pace of administration reported by states,” and the size of the 65-and-older population, said Mr. Azar, who has previously criticized New York Governor Andrew Cuomo for fining hospitals that didn’t use up vaccine supply within a week.
“This new system gives states a strong incentive to ensure that all vaccinations are being promptly reported, which they currently are not,” he said.
Currently, allocations are based on a state’s or territory’s population.
Prepandemic, states were required to report vaccinations within 30 days. Since COVID-19 vaccines became available, the CDC has required reporting of shots within 72 hours.
Dr. Redfield said the requirement has caused some difficulty, and that the CDC is investigating why some states have reported using only 15% of doses while others have used 80%.
States have been scrambling to ramp up vaccinations.
Just ahead of the federal briefing, Gov. Cuomo tweeted that New York would be opening up vaccinations to anyone older than 65.
The Associated Press is reporting that some states have started mass vaccination sites.
Arizona has begun operating a 24/7 appointment-only vaccination program at State Farm Stadium outside of Phoenix, with the aim of immunizing 6,000 people each day, according to local radio station KJZZ.
California and Florida have also taken steps to use stadiums, while Michigan, New Jersey, New York, and Texas will use convention centers and fairgrounds, Axios has reported.
In Florida, Palm Beach County Health Director Alina Alonso, MD, told county commissioners on Jan. 12 that there isn’t enough vaccine to meet demand, WPTV reported. “We need to realize that there’s a shortage of vaccine. So it’s not the plan, it’s not our ability to do it. It’s simply supply and demand at this point,” Dr. Alonso said, according to the TV station report.
A version of this article first appeared on Medscape.com.
Federal health officials are urging states to vaccinate all Americans over age 65 and those aged 16-64 who have a documented underlying health condition that makes them more vulnerable to COVID-19.
U.S. Department of Health and Human Services (HHS) Secretary Alex Azar and Centers for Disease Control and Prevention Director Robert Redfield, MD, made the recommendation in a briefing with reporters on Jan. 12, saying that the current vaccine supply was sufficient to meet demand for the next phase of immunization as recommended by the CDC’s Advisory Committee on Immunization Practices.
“We are ready for a transition that we outlined last September in the playbook we sent to states,” Mr. Azar said. Both he and U.S. Army General Gustave F. Perna, chief operations officer for Operation Warp Speed, said that confidence in the distribution system had led to the decision to urge wider access.
The federal government will also increase the number of sites eligible to receive vaccine – including some 13,000 federally qualified community health centers – and will not keep doses in reserve as insurance against issues that might prevent people from receiving a second dose on a timely basis.
“We don’t need to hold back reserve doses,” Mr. Azar said, noting that if there were any “glitches in production” the federal government would move to fulfill obligations for second doses first and delay initial doses.
Azar: Use it or lose it
In a move that is sure to generate pushback, Mr. Azar said that states that don’t quickly administer vaccines will receive fewer doses in the future. That policy will not go into effect until later in February, which leaves open the possibility that it could be reversed by the incoming Biden administration.
“We have too much vaccine sitting in freezers at hospitals with hospitals not using it,” said Mr. Azar, who also blamed the slow administration process on a reporting lag and states being what he called “overly prescriptive” in who has been eligible to receive a shot.
“I would rather have people working to get appointments to get vaccinated than having vaccine going to waste sitting in freezers,” he told reporters.
Mr. Azar had already been pushing for broader vaccination, telling states to do so in an Operation Warp Speed briefing on Jan. 6. At that briefing, he also said that the federal government would be stepping up vaccination through an “early launch” of a federal partnership with 19 pharmacy chains, which will let states allocate vaccines directly to some 40,000 pharmacy sites.
Gen. Perna said during the Jan. 12 briefing that the aim is to further expand that to some 70,000 locations total.
The CDC reported that as of Jan. 11 some 25.4 million doses have been distributed, with 8.9 million administered. An additional 4.2 million doses were distributed to long-term care facilities, and 937,000 residents and staff have received a dose.
“Pace of administration”
Alaska, Connecticut, North Dakota, South Dakota, the District of Columbia, West Virginia, and the Northern Mariana Islands have administered the most vaccines per capita, according to the CDC. But even these locations have immunized only 4%-5% of their populations, the New York Times reports. At the bottom: Alabama, Arizona, Arkansas, Georgia, Mississippi, and South Carolina.
The federal government can encourage but not require states to move on to new phases of vaccination.
“States ultimately determine how they will proceed with vaccination,” said Marcus Plescia, MD, MPH, chief medical officer for the Association of State and Territorial Health Officials. “Most will be cautious about assuring there are doses for those needing a second dose,” he said in an interview.
Dr. Plescia said that ensuring a second dose is available is especially important for health care workers “who need to be confident that they are protected and not inadvertently transmitting the disease themselves.”
He added that “once we reach a steady state of supply and administration, the rate-limiting factor will be supply of vaccine.”
That supply could now be threatened if states don’t comply with a just-announced federal action that will change how doses are allocated.
Beginning in late February, vaccine allocations to states will be based on “the pace of administration reported by states,” and the size of the 65-and-older population, said Mr. Azar, who has previously criticized New York Governor Andrew Cuomo for fining hospitals that didn’t use up vaccine supply within a week.
“This new system gives states a strong incentive to ensure that all vaccinations are being promptly reported, which they currently are not,” he said.
Currently, allocations are based on a state’s or territory’s population.
Prepandemic, states were required to report vaccinations within 30 days. Since COVID-19 vaccines became available, the CDC has required reporting of shots within 72 hours.
Dr. Redfield said the requirement has caused some difficulty, and that the CDC is investigating why some states have reported using only 15% of doses while others have used 80%.
States have been scrambling to ramp up vaccinations.
Just ahead of the federal briefing, Gov. Cuomo tweeted that New York would be opening up vaccinations to anyone older than 65.
The Associated Press is reporting that some states have started mass vaccination sites.
Arizona has begun operating a 24/7 appointment-only vaccination program at State Farm Stadium outside of Phoenix, with the aim of immunizing 6,000 people each day, according to local radio station KJZZ.
California and Florida have also taken steps to use stadiums, while Michigan, New Jersey, New York, and Texas will use convention centers and fairgrounds, Axios has reported.
In Florida, Palm Beach County Health Director Alina Alonso, MD, told county commissioners on Jan. 12 that there isn’t enough vaccine to meet demand, WPTV reported. “We need to realize that there’s a shortage of vaccine. So it’s not the plan, it’s not our ability to do it. It’s simply supply and demand at this point,” Dr. Alonso said, according to the TV station report.
A version of this article first appeared on Medscape.com.
FDA finalizes guidance for power morcellators in gynecologic surgery
The agency noted that physicians should conduct a thorough preoperative screening and that the devices should only be used for hysterectomies and myomectomies. Clinicians should not use the devices in cases involving uterine malignancy or suspected uterine malignancy.
In addition, clinicians should not use morcellators to remove uterine tissue containing suspected fibroids in women older than 50 years or who are postmenopausal. Nor should the devices be used for women who are “candidates for removal of tissue (en bloc) through the vagina or via a minilaparotomy incision,” the agency said.
The safety communication, which was issued on Dec. 29, 2020, updates previous guidance from February 2020. The updated recommendations are consistent with final labeling guidance for laparoscopic power morcellators, also issued by the FDA on Dec. 29.
Risk of disease spread
Prior evidence suggests that use of uncontained power morcellators in women with malignant uterine tissue can spread disease.
Even among women who do not have malignant uterine tissue, containment is important. The agency noted an association between uncontained power morcellation and the spread of benign uterine tissue, such as parasitic myomas and disseminated peritoneal leiomyomatosis, which could require additional surgeries.
In 2016, the FDA approved the PneumoLiner, a containment system for isolating uterine tissue that is not suspected of containing cancer.
“While unsuspected cancer can occur at any age, the prevalence of unsuspected cancer in women undergoing hysterectomy for fibroids increases with age such that the benefit-risk profile of using [laparoscopic power morcellators] is worse in older women when compared to younger women,” according to the new labeling guidance. “Also, the surgical technique of en bloc tissue removal eliminates the need to perform morcellation, thereby reducing the risk of iatrogenic dissemination and upstaging of an occult sarcoma. A thorough preoperative screening should be conducted; however, it is important to note that no screening procedure that can reliably detect sarcoma preoperatively has been identified.”
“The FDA will continue to review the latest data and scientific literature on laparoscopic power morcellation, including gathering real-world evidence from patients, providers and others, and encouraging innovation to better detect uterine cancer and develop containment systems for gynecologic surgery,” said Jeffrey Shuren, MD, JD, director of the FDA’s Center for Devices and Radiological Health, in a news release. “The FDA seeks to ensure women and their health care providers are fully informed when considering laparoscopic power morcellators for gynecologic surgeries.”
A version of this article first appeared on Medscape.com.
The agency noted that physicians should conduct a thorough preoperative screening and that the devices should only be used for hysterectomies and myomectomies. Clinicians should not use the devices in cases involving uterine malignancy or suspected uterine malignancy.
In addition, clinicians should not use morcellators to remove uterine tissue containing suspected fibroids in women older than 50 years or who are postmenopausal. Nor should the devices be used for women who are “candidates for removal of tissue (en bloc) through the vagina or via a minilaparotomy incision,” the agency said.
The safety communication, which was issued on Dec. 29, 2020, updates previous guidance from February 2020. The updated recommendations are consistent with final labeling guidance for laparoscopic power morcellators, also issued by the FDA on Dec. 29.
Risk of disease spread
Prior evidence suggests that use of uncontained power morcellators in women with malignant uterine tissue can spread disease.
Even among women who do not have malignant uterine tissue, containment is important. The agency noted an association between uncontained power morcellation and the spread of benign uterine tissue, such as parasitic myomas and disseminated peritoneal leiomyomatosis, which could require additional surgeries.
In 2016, the FDA approved the PneumoLiner, a containment system for isolating uterine tissue that is not suspected of containing cancer.
“While unsuspected cancer can occur at any age, the prevalence of unsuspected cancer in women undergoing hysterectomy for fibroids increases with age such that the benefit-risk profile of using [laparoscopic power morcellators] is worse in older women when compared to younger women,” according to the new labeling guidance. “Also, the surgical technique of en bloc tissue removal eliminates the need to perform morcellation, thereby reducing the risk of iatrogenic dissemination and upstaging of an occult sarcoma. A thorough preoperative screening should be conducted; however, it is important to note that no screening procedure that can reliably detect sarcoma preoperatively has been identified.”
“The FDA will continue to review the latest data and scientific literature on laparoscopic power morcellation, including gathering real-world evidence from patients, providers and others, and encouraging innovation to better detect uterine cancer and develop containment systems for gynecologic surgery,” said Jeffrey Shuren, MD, JD, director of the FDA’s Center for Devices and Radiological Health, in a news release. “The FDA seeks to ensure women and their health care providers are fully informed when considering laparoscopic power morcellators for gynecologic surgeries.”
A version of this article first appeared on Medscape.com.
The agency noted that physicians should conduct a thorough preoperative screening and that the devices should only be used for hysterectomies and myomectomies. Clinicians should not use the devices in cases involving uterine malignancy or suspected uterine malignancy.
In addition, clinicians should not use morcellators to remove uterine tissue containing suspected fibroids in women older than 50 years or who are postmenopausal. Nor should the devices be used for women who are “candidates for removal of tissue (en bloc) through the vagina or via a minilaparotomy incision,” the agency said.
The safety communication, which was issued on Dec. 29, 2020, updates previous guidance from February 2020. The updated recommendations are consistent with final labeling guidance for laparoscopic power morcellators, also issued by the FDA on Dec. 29.
Risk of disease spread
Prior evidence suggests that use of uncontained power morcellators in women with malignant uterine tissue can spread disease.
Even among women who do not have malignant uterine tissue, containment is important. The agency noted an association between uncontained power morcellation and the spread of benign uterine tissue, such as parasitic myomas and disseminated peritoneal leiomyomatosis, which could require additional surgeries.
In 2016, the FDA approved the PneumoLiner, a containment system for isolating uterine tissue that is not suspected of containing cancer.
“While unsuspected cancer can occur at any age, the prevalence of unsuspected cancer in women undergoing hysterectomy for fibroids increases with age such that the benefit-risk profile of using [laparoscopic power morcellators] is worse in older women when compared to younger women,” according to the new labeling guidance. “Also, the surgical technique of en bloc tissue removal eliminates the need to perform morcellation, thereby reducing the risk of iatrogenic dissemination and upstaging of an occult sarcoma. A thorough preoperative screening should be conducted; however, it is important to note that no screening procedure that can reliably detect sarcoma preoperatively has been identified.”
“The FDA will continue to review the latest data and scientific literature on laparoscopic power morcellation, including gathering real-world evidence from patients, providers and others, and encouraging innovation to better detect uterine cancer and develop containment systems for gynecologic surgery,” said Jeffrey Shuren, MD, JD, director of the FDA’s Center for Devices and Radiological Health, in a news release. “The FDA seeks to ensure women and their health care providers are fully informed when considering laparoscopic power morcellators for gynecologic surgeries.”
A version of this article first appeared on Medscape.com.
Anaphylaxis cases after COVID-19 vaccine rising but still rare: CDC
Health care providers should be ready to treat rare cases of anaphylaxis following administration of COVID-19 vaccines, federal medical officials have urged. The officials also stressed the importance of continuing vaccinations, despite reports of the rare side effect.
There have been 29 cases of anaphylaxis to date following administration of a COVID-19 vaccine, officials from the Centers for Disease Control and Prevention said in a call with reporters on Jan. 6.
The severe allergic reaction, which appears to be rare, can happen with either the Pfizer-BioNTech vaccine or the rival Moderna product. The Food and Drug Administration granted emergency use authorizations for these two vaccines in December.
Even with the cases seen to date, the COVID-19 vaccines remain a “good value proposition,” Nancy Messonnier, MD, director of the CDC’s National Center for Immunization, said in the call.
There have been about 11.1 cases of anaphylaxis per million doses with the Pfizer-BioNTech COVID-19 vaccine, which is higher than the estimated 1.3 cases per million doses with influenza vaccines, she said. But the low risk of anaphylaxis must be balanced against the threat of COVID-19, which currently claims about 2,000 lives a day in the United States, she said. In addition, many people are reporting long-term complications with COVID-19 even if they recover.
Kept in context, the data on anaphylaxis should not scare people away from getting a COVID-19 vaccine, she added.
“Their risk from COVID and poor outcomes is still more than the risk of a severe outcome from the vaccine,” Dr. Messonnier said. “And fortunately, we know how to treat anaphylaxis.”
Dr. Messonnier urged health care workers administering COVID-19 vaccines to be prepared.
“Anybody administering vaccines needs not just to have the EpiPen available, but frankly, to know how to use it,” Dr. Messonnier said.
MMWR details
The CDC on Jan. 6 also provided an update on anaphylaxis in Morbidity and Mortality Weekly Report (MMWR).
The information included in the report was based on cases reported with the Pfizer-BioNTech vaccine – the first to get emergency use authorization from the FDA. On the call with reporters, CDC officials confirmed there have been additional reports since then and anaphylaxis has been reported with both the Pfizer-BioNTech and Moderna vaccines. CDC officials said they could not give a breakdown of how many cases were linked to each of these products at this time.
Between Dec. 14 and 23, 2020, monitoring by the Vaccine Adverse Event Reporting System detected 21 cases of anaphylaxis after administration of a reported 1,893,360 first doses of the Pfizer-BioNTech COVID-19 vaccine. Most reactions – 71% – occurred within 15 minutes of vaccination.
A version of this article first appeared on Medscape.com.
Health care providers should be ready to treat rare cases of anaphylaxis following administration of COVID-19 vaccines, federal medical officials have urged. The officials also stressed the importance of continuing vaccinations, despite reports of the rare side effect.
There have been 29 cases of anaphylaxis to date following administration of a COVID-19 vaccine, officials from the Centers for Disease Control and Prevention said in a call with reporters on Jan. 6.
The severe allergic reaction, which appears to be rare, can happen with either the Pfizer-BioNTech vaccine or the rival Moderna product. The Food and Drug Administration granted emergency use authorizations for these two vaccines in December.
Even with the cases seen to date, the COVID-19 vaccines remain a “good value proposition,” Nancy Messonnier, MD, director of the CDC’s National Center for Immunization, said in the call.
There have been about 11.1 cases of anaphylaxis per million doses with the Pfizer-BioNTech COVID-19 vaccine, which is higher than the estimated 1.3 cases per million doses with influenza vaccines, she said. But the low risk of anaphylaxis must be balanced against the threat of COVID-19, which currently claims about 2,000 lives a day in the United States, she said. In addition, many people are reporting long-term complications with COVID-19 even if they recover.
Kept in context, the data on anaphylaxis should not scare people away from getting a COVID-19 vaccine, she added.
“Their risk from COVID and poor outcomes is still more than the risk of a severe outcome from the vaccine,” Dr. Messonnier said. “And fortunately, we know how to treat anaphylaxis.”
Dr. Messonnier urged health care workers administering COVID-19 vaccines to be prepared.
“Anybody administering vaccines needs not just to have the EpiPen available, but frankly, to know how to use it,” Dr. Messonnier said.
MMWR details
The CDC on Jan. 6 also provided an update on anaphylaxis in Morbidity and Mortality Weekly Report (MMWR).
The information included in the report was based on cases reported with the Pfizer-BioNTech vaccine – the first to get emergency use authorization from the FDA. On the call with reporters, CDC officials confirmed there have been additional reports since then and anaphylaxis has been reported with both the Pfizer-BioNTech and Moderna vaccines. CDC officials said they could not give a breakdown of how many cases were linked to each of these products at this time.
Between Dec. 14 and 23, 2020, monitoring by the Vaccine Adverse Event Reporting System detected 21 cases of anaphylaxis after administration of a reported 1,893,360 first doses of the Pfizer-BioNTech COVID-19 vaccine. Most reactions – 71% – occurred within 15 minutes of vaccination.
A version of this article first appeared on Medscape.com.
Health care providers should be ready to treat rare cases of anaphylaxis following administration of COVID-19 vaccines, federal medical officials have urged. The officials also stressed the importance of continuing vaccinations, despite reports of the rare side effect.
There have been 29 cases of anaphylaxis to date following administration of a COVID-19 vaccine, officials from the Centers for Disease Control and Prevention said in a call with reporters on Jan. 6.
The severe allergic reaction, which appears to be rare, can happen with either the Pfizer-BioNTech vaccine or the rival Moderna product. The Food and Drug Administration granted emergency use authorizations for these two vaccines in December.
Even with the cases seen to date, the COVID-19 vaccines remain a “good value proposition,” Nancy Messonnier, MD, director of the CDC’s National Center for Immunization, said in the call.
There have been about 11.1 cases of anaphylaxis per million doses with the Pfizer-BioNTech COVID-19 vaccine, which is higher than the estimated 1.3 cases per million doses with influenza vaccines, she said. But the low risk of anaphylaxis must be balanced against the threat of COVID-19, which currently claims about 2,000 lives a day in the United States, she said. In addition, many people are reporting long-term complications with COVID-19 even if they recover.
Kept in context, the data on anaphylaxis should not scare people away from getting a COVID-19 vaccine, she added.
“Their risk from COVID and poor outcomes is still more than the risk of a severe outcome from the vaccine,” Dr. Messonnier said. “And fortunately, we know how to treat anaphylaxis.”
Dr. Messonnier urged health care workers administering COVID-19 vaccines to be prepared.
“Anybody administering vaccines needs not just to have the EpiPen available, but frankly, to know how to use it,” Dr. Messonnier said.
MMWR details
The CDC on Jan. 6 also provided an update on anaphylaxis in Morbidity and Mortality Weekly Report (MMWR).
The information included in the report was based on cases reported with the Pfizer-BioNTech vaccine – the first to get emergency use authorization from the FDA. On the call with reporters, CDC officials confirmed there have been additional reports since then and anaphylaxis has been reported with both the Pfizer-BioNTech and Moderna vaccines. CDC officials said they could not give a breakdown of how many cases were linked to each of these products at this time.
Between Dec. 14 and 23, 2020, monitoring by the Vaccine Adverse Event Reporting System detected 21 cases of anaphylaxis after administration of a reported 1,893,360 first doses of the Pfizer-BioNTech COVID-19 vaccine. Most reactions – 71% – occurred within 15 minutes of vaccination.
A version of this article first appeared on Medscape.com.
FDA okays first generic injected glucagon for hypoglycemia
The FDA determined that Amphastar’s Glucagon for Injection Emergency Kit, 1 mg, a synthetic peptide product, is bioequivalent and therapeutically equivalent to Eli Lilly’s recombinant DNA Glucagon Emergency Kit for Low Blood Sugar.
Both require a multistep mixing process that means they are complicated to use.
In 2019, FDA approved two branded, easier-to-use formulations of glucagon – one nasally administered (Baqsimi, Eli Lilly & Co) and the other a prefilled pen or syringe (Gvoke HypoPen and Gvoke PFS, respectively, Xeris Pharmaceuticals).
The new generic will have the advantage of lower cost, Amphastar spokesman Dan Dischner said in an interview.
“Our generic glucagon will be priced as a generic product so that patients will benefit from a lower price. As we are just at the beginning of the commercialization of the product, we are unable to discuss our specific product price,” he wrote.
As with the branded Lilly injectable glucagon, the new generic is also indicated as a diagnostic aid in gastrointestinal radiologic imaging, as glucagon slows gastric motility.
According to an FDA statement, glucagon is a “complex product” that has been difficult to manufacture generically despite the lifting of patent protection. This approval was the result of the FDA’s efforts to encourage the development and submission of applications for such drugs.
Amphastar specializes in “developing, manufacturing, marketing, and selling technically-challenging generic and proprietary injectable, inhalation, and intranasal products,” the company website says.
Mr. Dischner said, “Glucagon is a complex product that requires R&D and manufacturing capabilities to develop a highly purified synthetic peptide product bioequivalent and therapeutically equivalent to the recombinant DNA origin Glucagon. Given that this product has been through various review cycles, its complexity, and the technological capabilities required to manufacture, it is no surprise that there hasn’t been a generic of glucagon until now.”
Side effects of injected glucagon include nausea, vomiting, transient increase in heart rate, and redness/swelling of the injection site.
Mr. Dischner added, “We are confident that our generic to Lilly’s time-tested glucagon will provide a favorable option, at a reasonable price, to patients who rely on this product.”
A version of this article first appeared on Medscape.com.
The FDA determined that Amphastar’s Glucagon for Injection Emergency Kit, 1 mg, a synthetic peptide product, is bioequivalent and therapeutically equivalent to Eli Lilly’s recombinant DNA Glucagon Emergency Kit for Low Blood Sugar.
Both require a multistep mixing process that means they are complicated to use.
In 2019, FDA approved two branded, easier-to-use formulations of glucagon – one nasally administered (Baqsimi, Eli Lilly & Co) and the other a prefilled pen or syringe (Gvoke HypoPen and Gvoke PFS, respectively, Xeris Pharmaceuticals).
The new generic will have the advantage of lower cost, Amphastar spokesman Dan Dischner said in an interview.
“Our generic glucagon will be priced as a generic product so that patients will benefit from a lower price. As we are just at the beginning of the commercialization of the product, we are unable to discuss our specific product price,” he wrote.
As with the branded Lilly injectable glucagon, the new generic is also indicated as a diagnostic aid in gastrointestinal radiologic imaging, as glucagon slows gastric motility.
According to an FDA statement, glucagon is a “complex product” that has been difficult to manufacture generically despite the lifting of patent protection. This approval was the result of the FDA’s efforts to encourage the development and submission of applications for such drugs.
Amphastar specializes in “developing, manufacturing, marketing, and selling technically-challenging generic and proprietary injectable, inhalation, and intranasal products,” the company website says.
Mr. Dischner said, “Glucagon is a complex product that requires R&D and manufacturing capabilities to develop a highly purified synthetic peptide product bioequivalent and therapeutically equivalent to the recombinant DNA origin Glucagon. Given that this product has been through various review cycles, its complexity, and the technological capabilities required to manufacture, it is no surprise that there hasn’t been a generic of glucagon until now.”
Side effects of injected glucagon include nausea, vomiting, transient increase in heart rate, and redness/swelling of the injection site.
Mr. Dischner added, “We are confident that our generic to Lilly’s time-tested glucagon will provide a favorable option, at a reasonable price, to patients who rely on this product.”
A version of this article first appeared on Medscape.com.
The FDA determined that Amphastar’s Glucagon for Injection Emergency Kit, 1 mg, a synthetic peptide product, is bioequivalent and therapeutically equivalent to Eli Lilly’s recombinant DNA Glucagon Emergency Kit for Low Blood Sugar.
Both require a multistep mixing process that means they are complicated to use.
In 2019, FDA approved two branded, easier-to-use formulations of glucagon – one nasally administered (Baqsimi, Eli Lilly & Co) and the other a prefilled pen or syringe (Gvoke HypoPen and Gvoke PFS, respectively, Xeris Pharmaceuticals).
The new generic will have the advantage of lower cost, Amphastar spokesman Dan Dischner said in an interview.
“Our generic glucagon will be priced as a generic product so that patients will benefit from a lower price. As we are just at the beginning of the commercialization of the product, we are unable to discuss our specific product price,” he wrote.
As with the branded Lilly injectable glucagon, the new generic is also indicated as a diagnostic aid in gastrointestinal radiologic imaging, as glucagon slows gastric motility.
According to an FDA statement, glucagon is a “complex product” that has been difficult to manufacture generically despite the lifting of patent protection. This approval was the result of the FDA’s efforts to encourage the development and submission of applications for such drugs.
Amphastar specializes in “developing, manufacturing, marketing, and selling technically-challenging generic and proprietary injectable, inhalation, and intranasal products,” the company website says.
Mr. Dischner said, “Glucagon is a complex product that requires R&D and manufacturing capabilities to develop a highly purified synthetic peptide product bioequivalent and therapeutically equivalent to the recombinant DNA origin Glucagon. Given that this product has been through various review cycles, its complexity, and the technological capabilities required to manufacture, it is no surprise that there hasn’t been a generic of glucagon until now.”
Side effects of injected glucagon include nausea, vomiting, transient increase in heart rate, and redness/swelling of the injection site.
Mr. Dischner added, “We are confident that our generic to Lilly’s time-tested glucagon will provide a favorable option, at a reasonable price, to patients who rely on this product.”
A version of this article first appeared on Medscape.com.