User login
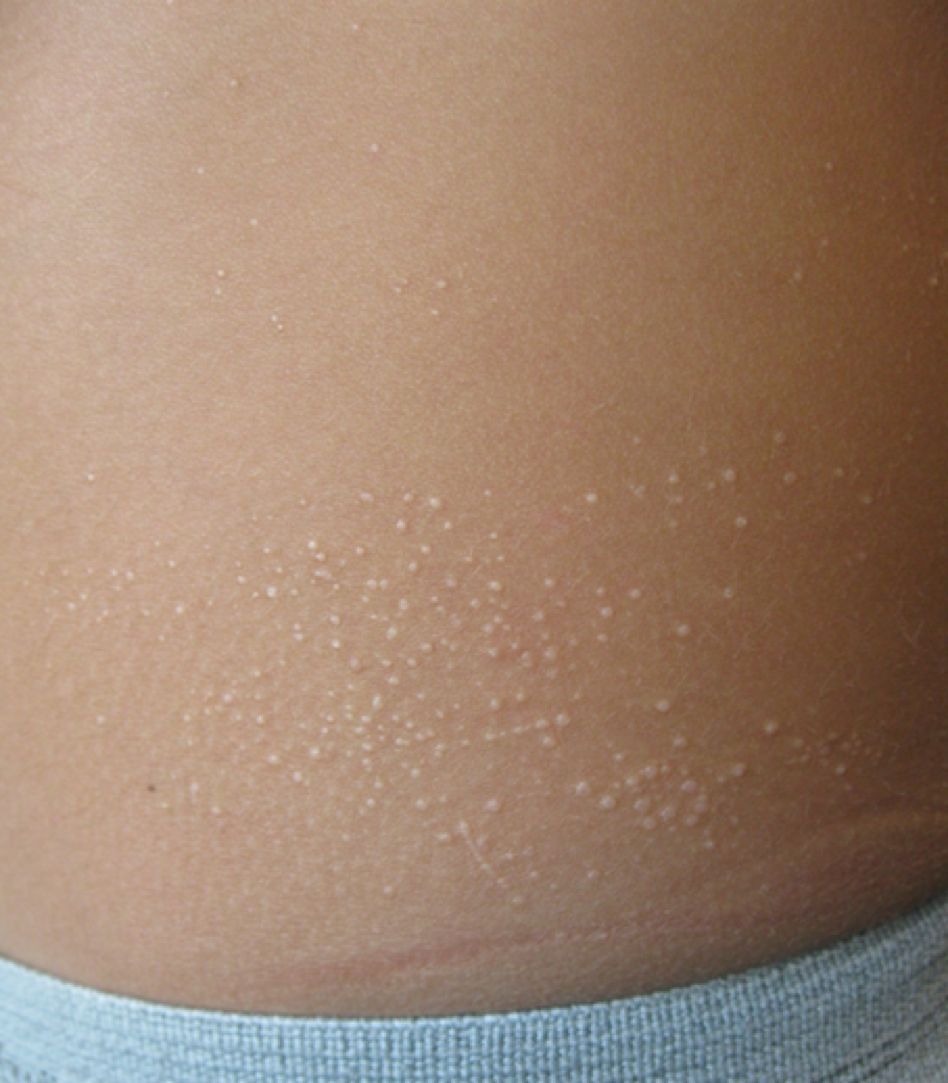
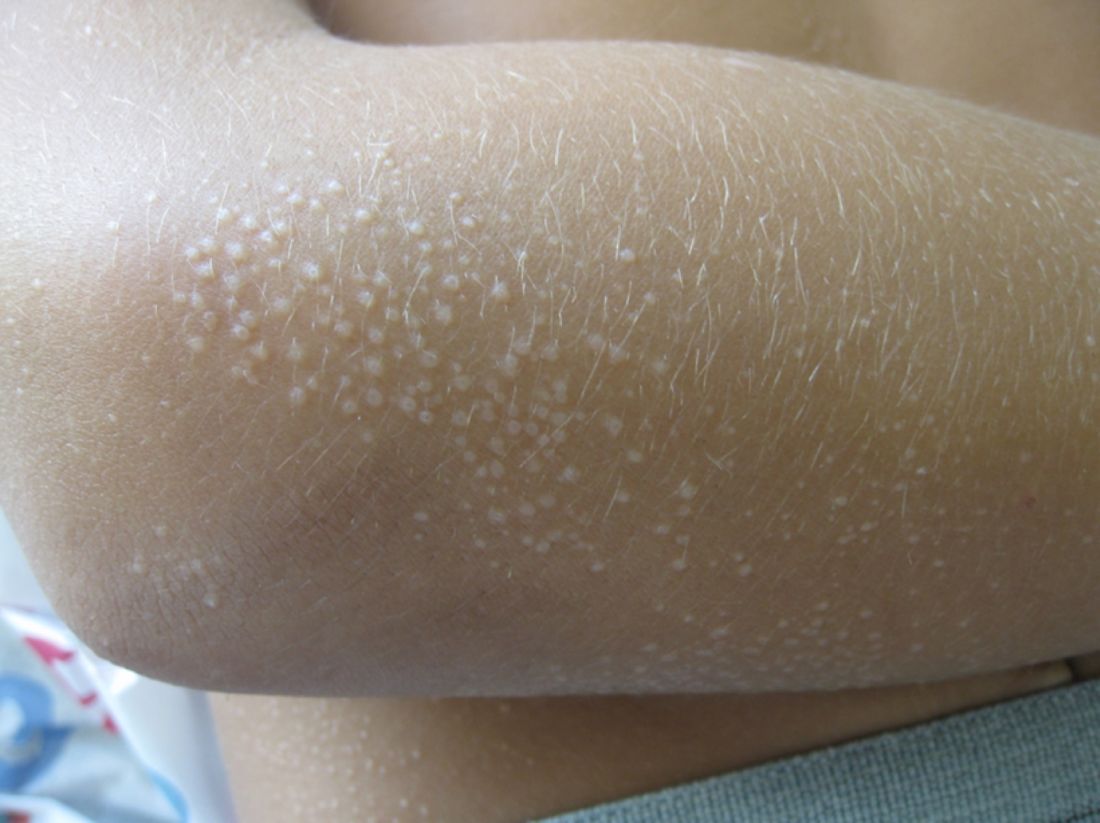
A 13-month-old, healthy black male presented with a 6-month history of dry, scaly skin on the body
Ichthyosis vulgaris
Ichthyoses describe a group of disorders of cornification in which the epidermis differentiates abnormally, leading to generalized scaling of the skin. Ichthyosis is derived from the Greek word for fish, “ichthys.” Ichthyosis vulgaris is the most common of these conditions and often presents in early childhood during the first year of life. It is inherited in an autosomal-dominant pattern. Skin is dry and scaly over the entire body, although the antecubital and popliteal fossa may be uninvolved. The scalp may be involved as well. Atopy and keratosis pilaris may be associated. By adulthood, symptoms tend to abate.
X-linked ichthyosis is an X-linked recessive trait, in which males are affected and mothers are carriers. The condition is caused by a deficiency of steroid sulfatase. This deficiency can result in low levels of estrogen during pregnancy in the mother of an affected fetus, hampering labor progression, and often requiring C-section. Children usually present before 3 months of age. Scales are large and dark. The antecubital and popliteal fossa are usually spared. The neck almost always is involved, coining the term “dirty neck disease.” Corneal opacities are present upon ophthalmologic examination. There is an increased risk of cryptorchidism and testicular cancer. Skin symptoms tend to worsen into adulthood.
Lamellar ichthyosis generally occurs at birth with a striking collodion-type membrane covering the body and underlying erythroderma, which then desquamates. Ectropion is usually present as well. Resulting scales are large and gray-brown. Lamellar ichthyosis is inherited in an autosomal recessive pattern. Mutations in transglutaminase 1 (TGM1), ALOXE3, ALOX12B, and ABCA12 genes have been implicated in this disorder.
Acquired ichthyosis can appear clinically similar to ichthyosis vulgaris. It occurs in patients with systemic diseases such as Hodgkin disease, non-Hodgkin lymphoma, mycosis fungoides, multiple myeloma, hypothyroidism, sarcoidosis, AIDS, and others.
improve hyperkeratosis. Urea-containing products can be helpful. Salicylic acid may be used but merit caution in children because of salicylate toxicity. Oral and topical retinoid can be helpful in lamellar ichthyosis.

This case and photo were submitted by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/edermatologynews.com. To submit a case for possible publication, send an email to [email protected].
Ichthyosis vulgaris
Ichthyoses describe a group of disorders of cornification in which the epidermis differentiates abnormally, leading to generalized scaling of the skin. Ichthyosis is derived from the Greek word for fish, “ichthys.” Ichthyosis vulgaris is the most common of these conditions and often presents in early childhood during the first year of life. It is inherited in an autosomal-dominant pattern. Skin is dry and scaly over the entire body, although the antecubital and popliteal fossa may be uninvolved. The scalp may be involved as well. Atopy and keratosis pilaris may be associated. By adulthood, symptoms tend to abate.
X-linked ichthyosis is an X-linked recessive trait, in which males are affected and mothers are carriers. The condition is caused by a deficiency of steroid sulfatase. This deficiency can result in low levels of estrogen during pregnancy in the mother of an affected fetus, hampering labor progression, and often requiring C-section. Children usually present before 3 months of age. Scales are large and dark. The antecubital and popliteal fossa are usually spared. The neck almost always is involved, coining the term “dirty neck disease.” Corneal opacities are present upon ophthalmologic examination. There is an increased risk of cryptorchidism and testicular cancer. Skin symptoms tend to worsen into adulthood.
Lamellar ichthyosis generally occurs at birth with a striking collodion-type membrane covering the body and underlying erythroderma, which then desquamates. Ectropion is usually present as well. Resulting scales are large and gray-brown. Lamellar ichthyosis is inherited in an autosomal recessive pattern. Mutations in transglutaminase 1 (TGM1), ALOXE3, ALOX12B, and ABCA12 genes have been implicated in this disorder.
Acquired ichthyosis can appear clinically similar to ichthyosis vulgaris. It occurs in patients with systemic diseases such as Hodgkin disease, non-Hodgkin lymphoma, mycosis fungoides, multiple myeloma, hypothyroidism, sarcoidosis, AIDS, and others.
improve hyperkeratosis. Urea-containing products can be helpful. Salicylic acid may be used but merit caution in children because of salicylate toxicity. Oral and topical retinoid can be helpful in lamellar ichthyosis.
This case and photo were submitted by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/edermatologynews.com. To submit a case for possible publication, send an email to [email protected].
Ichthyosis vulgaris
Ichthyoses describe a group of disorders of cornification in which the epidermis differentiates abnormally, leading to generalized scaling of the skin. Ichthyosis is derived from the Greek word for fish, “ichthys.” Ichthyosis vulgaris is the most common of these conditions and often presents in early childhood during the first year of life. It is inherited in an autosomal-dominant pattern. Skin is dry and scaly over the entire body, although the antecubital and popliteal fossa may be uninvolved. The scalp may be involved as well. Atopy and keratosis pilaris may be associated. By adulthood, symptoms tend to abate.
X-linked ichthyosis is an X-linked recessive trait, in which males are affected and mothers are carriers. The condition is caused by a deficiency of steroid sulfatase. This deficiency can result in low levels of estrogen during pregnancy in the mother of an affected fetus, hampering labor progression, and often requiring C-section. Children usually present before 3 months of age. Scales are large and dark. The antecubital and popliteal fossa are usually spared. The neck almost always is involved, coining the term “dirty neck disease.” Corneal opacities are present upon ophthalmologic examination. There is an increased risk of cryptorchidism and testicular cancer. Skin symptoms tend to worsen into adulthood.
Lamellar ichthyosis generally occurs at birth with a striking collodion-type membrane covering the body and underlying erythroderma, which then desquamates. Ectropion is usually present as well. Resulting scales are large and gray-brown. Lamellar ichthyosis is inherited in an autosomal recessive pattern. Mutations in transglutaminase 1 (TGM1), ALOXE3, ALOX12B, and ABCA12 genes have been implicated in this disorder.
Acquired ichthyosis can appear clinically similar to ichthyosis vulgaris. It occurs in patients with systemic diseases such as Hodgkin disease, non-Hodgkin lymphoma, mycosis fungoides, multiple myeloma, hypothyroidism, sarcoidosis, AIDS, and others.
improve hyperkeratosis. Urea-containing products can be helpful. Salicylic acid may be used but merit caution in children because of salicylate toxicity. Oral and topical retinoid can be helpful in lamellar ichthyosis.
This case and photo were submitted by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/edermatologynews.com. To submit a case for possible publication, send an email to [email protected].
A 13-month-old, healthy black male presented with a 6-month history of dry, scaly skin on the body, including scalp and extremities. His neck was unaffected. His mother reports an uneventful pregnancy and natural childbirth. He had been prescribed triamcinolone in the past for eczema.
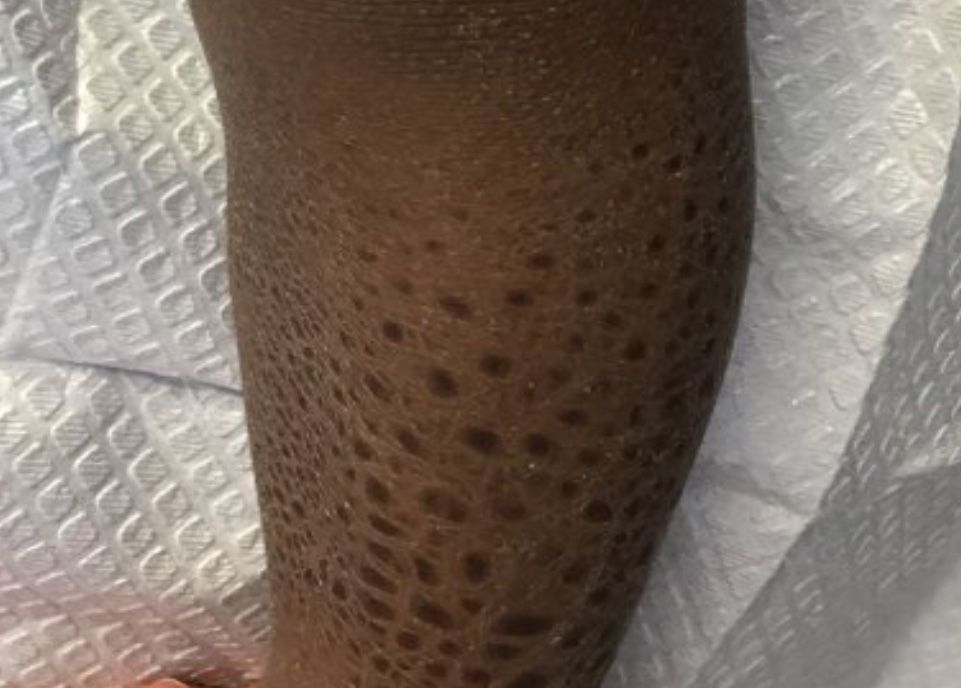
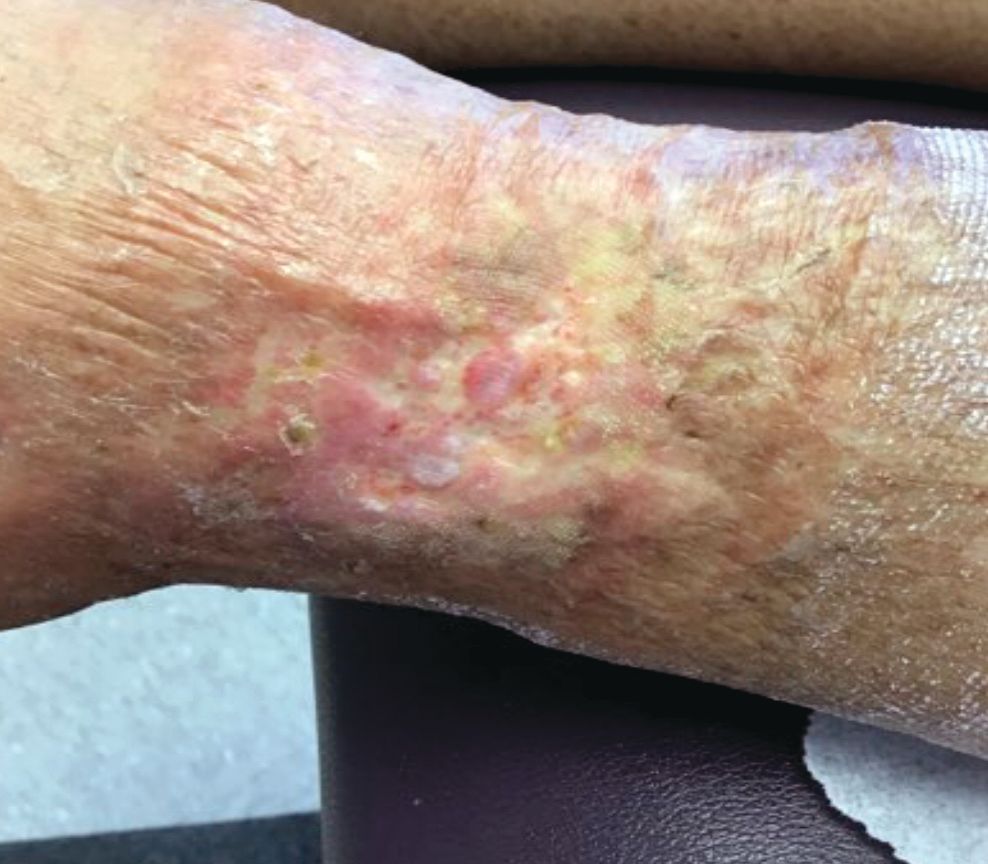
A 60-year-old white woman presented with a 3-month history of a painful, nonhealing ulceration on her left lateral lower leg
It is a vasculopathy rather than a vasculitis as the former is caused by occlusion of blood vessels and the latter results from inflammation of the vessels. Middle-aged women tend to be affected more frequently. Although the exact cause is unclear, systemic diseases, such as hypercoagulable states, may predispose vessels to develop occlusion. Associated disorders include antiphospholipid syndrome, protein C deficiency, factor V mutation, arteriosclerosis, hyperhomocysteinemia, and hepatitis C.
Typically, lesions begin as painful purpura or reticulated macules on the lower extremities that ulcerate and heal very slowly. Ankles, particularly malleoli, are more frequently affected. When they heal, they form painless white stellate scars typical of atrophie blanche. Surrounding erythema, telangiectasias, and sclerosis may be present; livedo reticularis may be seen as well.
Histologically, the epidermis may be atrophic or necrotic. Hyaline thickening of the blood vessel walls is seen. Thrombi may be present. Direct immunofluorescence of perilesional skin may be positive for complement C3 and immunoglobulin (IgM) in dermal blood vessels.
Livedoid vasculopathy can be difficult to treat. Treatment is aimed at reducing clotting and improving blood flow and includes antiplatelet drugs (low-dose aspirin, dipyridamole), anticoagulants, and vasodilating agents (nifedipine). Pentoxifylline two or three times daily may help by altering blood viscosity. A recent literature search reports success in topical dapsone applied to lesions twice daily under occlusion. Leg elevation and compression stockings help healing. Livedoid vasculopathy may have periods of activity and remission.
The case and photo were submitted by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/edermatologynews.com. To submit a case for possible publication, send an email to [email protected]
It is a vasculopathy rather than a vasculitis as the former is caused by occlusion of blood vessels and the latter results from inflammation of the vessels. Middle-aged women tend to be affected more frequently. Although the exact cause is unclear, systemic diseases, such as hypercoagulable states, may predispose vessels to develop occlusion. Associated disorders include antiphospholipid syndrome, protein C deficiency, factor V mutation, arteriosclerosis, hyperhomocysteinemia, and hepatitis C.
Typically, lesions begin as painful purpura or reticulated macules on the lower extremities that ulcerate and heal very slowly. Ankles, particularly malleoli, are more frequently affected. When they heal, they form painless white stellate scars typical of atrophie blanche. Surrounding erythema, telangiectasias, and sclerosis may be present; livedo reticularis may be seen as well.
Histologically, the epidermis may be atrophic or necrotic. Hyaline thickening of the blood vessel walls is seen. Thrombi may be present. Direct immunofluorescence of perilesional skin may be positive for complement C3 and immunoglobulin (IgM) in dermal blood vessels.
Livedoid vasculopathy can be difficult to treat. Treatment is aimed at reducing clotting and improving blood flow and includes antiplatelet drugs (low-dose aspirin, dipyridamole), anticoagulants, and vasodilating agents (nifedipine). Pentoxifylline two or three times daily may help by altering blood viscosity. A recent literature search reports success in topical dapsone applied to lesions twice daily under occlusion. Leg elevation and compression stockings help healing. Livedoid vasculopathy may have periods of activity and remission.
The case and photo were submitted by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/edermatologynews.com. To submit a case for possible publication, send an email to [email protected]
It is a vasculopathy rather than a vasculitis as the former is caused by occlusion of blood vessels and the latter results from inflammation of the vessels. Middle-aged women tend to be affected more frequently. Although the exact cause is unclear, systemic diseases, such as hypercoagulable states, may predispose vessels to develop occlusion. Associated disorders include antiphospholipid syndrome, protein C deficiency, factor V mutation, arteriosclerosis, hyperhomocysteinemia, and hepatitis C.
Typically, lesions begin as painful purpura or reticulated macules on the lower extremities that ulcerate and heal very slowly. Ankles, particularly malleoli, are more frequently affected. When they heal, they form painless white stellate scars typical of atrophie blanche. Surrounding erythema, telangiectasias, and sclerosis may be present; livedo reticularis may be seen as well.
Histologically, the epidermis may be atrophic or necrotic. Hyaline thickening of the blood vessel walls is seen. Thrombi may be present. Direct immunofluorescence of perilesional skin may be positive for complement C3 and immunoglobulin (IgM) in dermal blood vessels.
Livedoid vasculopathy can be difficult to treat. Treatment is aimed at reducing clotting and improving blood flow and includes antiplatelet drugs (low-dose aspirin, dipyridamole), anticoagulants, and vasodilating agents (nifedipine). Pentoxifylline two or three times daily may help by altering blood viscosity. A recent literature search reports success in topical dapsone applied to lesions twice daily under occlusion. Leg elevation and compression stockings help healing. Livedoid vasculopathy may have periods of activity and remission.
The case and photo were submitted by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/edermatologynews.com. To submit a case for possible publication, send an email to [email protected]
What is your diagnosis?
It most commonly affects young girls. The pathogenesis of LAHS is thought to involve a sporadic, autosomal dominant mutation that leads to a defect between the hair cuticle and the inner root sheath.1 This defect results in the hair being poorly anchored to the scalp, and therefore easily and painlessly plucked or lost during normal hair care.

The classic presentation of LAHS is that of hair thinning and hair that may be unruly and/or lackluster; the hair rarely, if ever, requires cutting.2 The key feature is the ability to easily and painlessly pluck hairs from the patient’s scalp. The affected area is limited to the scalp, and loss of eyebrows, eyelashes, and body hair should not be seen.
Diagnosis and consideration of the differential
The diagnosis of LAHS can in some cases be made on history and physical exam alone. Patients with LAHS typically will show hair thinning with or without dullness or unruliness. They lack evidence of scalp inflammation, such as erythema, scale, pruritus, and pain. Areas of hair thinning or aberration are typically not well demarcated, and there are typically not areas of complete hair loss. There is no scarring or atrophy of the scalp itself.

Diagnostic tests include the “hair pull test,” as well as trichogram testing. In the “hair pull test” a provider grasps a set of hair at the proximal shaft near the scalp. The traction applied should result in the painless and easy extraction of more than 10% of grasped hairs in a patient with LAHS. Removal of less than 10% of hair is a normal finding, as patients without LAHS typically have about 10% of their scalp hair in the telogen phase at any given time, which would result in removal during the hair pull test.3 In trichography, plucked hairs are examined under magnification, with or without the use of selective dyes. Cinnamaldehyde is a dye that stains citrulline, which is abundant in the inner root sheath, and can be a tool in identifying its presence and/or aberrations.4 A trichogram of the pulled hairs in a patient with LAHS may classically show ruffled appearance of the cuticle, misshapen anagen hair bulbs, and absence of the inner root sheath.5 Examination under magnification also allows providers to better identify telogen versus anagen hairs, which aids in the diagnosis. By carefully considering the patient history, physical exam, and results of additional hair tests, providers can make the diagnosis of LAHS and avoid unnecessary blood work and invasive procedures like scalp biopsies.
The differential diagnosis of hair loss frequently includes alopecia areata. However, in alopecia areata, patients typically have sharply demarcated areas of hair loss, which may involve the eyebrows, eyelids, and body hairs. In alopecia areata, providers may be able to identify the “exclamation point sign” in which the hair shaft thins proximally, leading to the appearance of more pigmented, thicker hairs floating above the scalp.
Telogen effluvium is a condition in which a medical illness or stress, such as systemic illness, surgery, severe emotional distress, childbirth, dietary changes, or another traumatic event, causes a disruption in the natural cycle of hair growth such that the percentage of hairs in the telogen phase increases from about 10% to up to 70%.6 Unlike in LAHS, in which shed hairs are in the anagen phase, the hair that is shed in telogen effluvium is in the telogen phase and will have a different appearance when magnified.
Anagen effluvium, loss of hairs in their growing phase, is typically associated with chemotherapy. The hairs become broken and fractured at the shaft leading to breakage at different points throughout the scalp. Affected areas can include the eyebrows, eyelashes, and body hair. In the absence of a history of administration of a chemotherapy agent (or other drug known to trigger hair loss), the diagnosis of anagen effluvium should not be made.
Patients with trichotillosis (also known as trichotillomania) present with areas of hair loss caused by intentional or subconscious hair pulling. It is considered a psychological condition that can be associated with obsessive compulsive disorder, although the presence of a secondary psychological diagnosis is not required. Providers may see irregular geometric shapes of hair loss, and on close inspection see broken hair shafts of different lengths. Patients most often pull hair from their scalps (over 70% of patients), but also can pull eyelashes, eyebrow hairs, and pubic hairs.7
Treatment
LAHS is self-limited and does not necessitate treatment. However, if patients or parents feel there is significant disease burden, perhaps with poor effects on quality of life or with psychosocial impairment, treatment with minoxidil 5% solution has been studied with some success reported in the literature.1,8,9
Ms. Natsis is a medical student at the University of California, San Diego. Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital-San Diego. He is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego. Ms. Natsis and Dr. Eichenfield had no relevant financial disclosures. Email them at [email protected].
References
1. Arch Dermatol. 2002;138(4):501-6.
2. Int J Trichology. 2010;2(2):96-100.
3. Pediatric Dermatol. 2016:33(50):507-10.
4. Dermatol Clin. 1986;14:745-51.
5. Arch Dermatol. 2009;145(10):1123-8.
6. J Clin Diagn Res. 2015;9(9):WE01-3.
7. Am J Psychiatry. 2016;173(9):868-74.
8. Australas J Dermatol. 2018;59:e286-e287.
9. Pediatr Dermatol. 2014;31:389-90.
It most commonly affects young girls. The pathogenesis of LAHS is thought to involve a sporadic, autosomal dominant mutation that leads to a defect between the hair cuticle and the inner root sheath.1 This defect results in the hair being poorly anchored to the scalp, and therefore easily and painlessly plucked or lost during normal hair care.
The classic presentation of LAHS is that of hair thinning and hair that may be unruly and/or lackluster; the hair rarely, if ever, requires cutting.2 The key feature is the ability to easily and painlessly pluck hairs from the patient’s scalp. The affected area is limited to the scalp, and loss of eyebrows, eyelashes, and body hair should not be seen.
Diagnosis and consideration of the differential
The diagnosis of LAHS can in some cases be made on history and physical exam alone. Patients with LAHS typically will show hair thinning with or without dullness or unruliness. They lack evidence of scalp inflammation, such as erythema, scale, pruritus, and pain. Areas of hair thinning or aberration are typically not well demarcated, and there are typically not areas of complete hair loss. There is no scarring or atrophy of the scalp itself.
Diagnostic tests include the “hair pull test,” as well as trichogram testing. In the “hair pull test” a provider grasps a set of hair at the proximal shaft near the scalp. The traction applied should result in the painless and easy extraction of more than 10% of grasped hairs in a patient with LAHS. Removal of less than 10% of hair is a normal finding, as patients without LAHS typically have about 10% of their scalp hair in the telogen phase at any given time, which would result in removal during the hair pull test.3 In trichography, plucked hairs are examined under magnification, with or without the use of selective dyes. Cinnamaldehyde is a dye that stains citrulline, which is abundant in the inner root sheath, and can be a tool in identifying its presence and/or aberrations.4 A trichogram of the pulled hairs in a patient with LAHS may classically show ruffled appearance of the cuticle, misshapen anagen hair bulbs, and absence of the inner root sheath.5 Examination under magnification also allows providers to better identify telogen versus anagen hairs, which aids in the diagnosis. By carefully considering the patient history, physical exam, and results of additional hair tests, providers can make the diagnosis of LAHS and avoid unnecessary blood work and invasive procedures like scalp biopsies.
The differential diagnosis of hair loss frequently includes alopecia areata. However, in alopecia areata, patients typically have sharply demarcated areas of hair loss, which may involve the eyebrows, eyelids, and body hairs. In alopecia areata, providers may be able to identify the “exclamation point sign” in which the hair shaft thins proximally, leading to the appearance of more pigmented, thicker hairs floating above the scalp.
Telogen effluvium is a condition in which a medical illness or stress, such as systemic illness, surgery, severe emotional distress, childbirth, dietary changes, or another traumatic event, causes a disruption in the natural cycle of hair growth such that the percentage of hairs in the telogen phase increases from about 10% to up to 70%.6 Unlike in LAHS, in which shed hairs are in the anagen phase, the hair that is shed in telogen effluvium is in the telogen phase and will have a different appearance when magnified.
Anagen effluvium, loss of hairs in their growing phase, is typically associated with chemotherapy. The hairs become broken and fractured at the shaft leading to breakage at different points throughout the scalp. Affected areas can include the eyebrows, eyelashes, and body hair. In the absence of a history of administration of a chemotherapy agent (or other drug known to trigger hair loss), the diagnosis of anagen effluvium should not be made.
Patients with trichotillosis (also known as trichotillomania) present with areas of hair loss caused by intentional or subconscious hair pulling. It is considered a psychological condition that can be associated with obsessive compulsive disorder, although the presence of a secondary psychological diagnosis is not required. Providers may see irregular geometric shapes of hair loss, and on close inspection see broken hair shafts of different lengths. Patients most often pull hair from their scalps (over 70% of patients), but also can pull eyelashes, eyebrow hairs, and pubic hairs.7
Treatment
LAHS is self-limited and does not necessitate treatment. However, if patients or parents feel there is significant disease burden, perhaps with poor effects on quality of life or with psychosocial impairment, treatment with minoxidil 5% solution has been studied with some success reported in the literature.1,8,9
Ms. Natsis is a medical student at the University of California, San Diego. Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital-San Diego. He is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego. Ms. Natsis and Dr. Eichenfield had no relevant financial disclosures. Email them at [email protected].
References
1. Arch Dermatol. 2002;138(4):501-6.
2. Int J Trichology. 2010;2(2):96-100.
3. Pediatric Dermatol. 2016:33(50):507-10.
4. Dermatol Clin. 1986;14:745-51.
5. Arch Dermatol. 2009;145(10):1123-8.
6. J Clin Diagn Res. 2015;9(9):WE01-3.
7. Am J Psychiatry. 2016;173(9):868-74.
8. Australas J Dermatol. 2018;59:e286-e287.
9. Pediatr Dermatol. 2014;31:389-90.
It most commonly affects young girls. The pathogenesis of LAHS is thought to involve a sporadic, autosomal dominant mutation that leads to a defect between the hair cuticle and the inner root sheath.1 This defect results in the hair being poorly anchored to the scalp, and therefore easily and painlessly plucked or lost during normal hair care.
The classic presentation of LAHS is that of hair thinning and hair that may be unruly and/or lackluster; the hair rarely, if ever, requires cutting.2 The key feature is the ability to easily and painlessly pluck hairs from the patient’s scalp. The affected area is limited to the scalp, and loss of eyebrows, eyelashes, and body hair should not be seen.
Diagnosis and consideration of the differential
The diagnosis of LAHS can in some cases be made on history and physical exam alone. Patients with LAHS typically will show hair thinning with or without dullness or unruliness. They lack evidence of scalp inflammation, such as erythema, scale, pruritus, and pain. Areas of hair thinning or aberration are typically not well demarcated, and there are typically not areas of complete hair loss. There is no scarring or atrophy of the scalp itself.
Diagnostic tests include the “hair pull test,” as well as trichogram testing. In the “hair pull test” a provider grasps a set of hair at the proximal shaft near the scalp. The traction applied should result in the painless and easy extraction of more than 10% of grasped hairs in a patient with LAHS. Removal of less than 10% of hair is a normal finding, as patients without LAHS typically have about 10% of their scalp hair in the telogen phase at any given time, which would result in removal during the hair pull test.3 In trichography, plucked hairs are examined under magnification, with or without the use of selective dyes. Cinnamaldehyde is a dye that stains citrulline, which is abundant in the inner root sheath, and can be a tool in identifying its presence and/or aberrations.4 A trichogram of the pulled hairs in a patient with LAHS may classically show ruffled appearance of the cuticle, misshapen anagen hair bulbs, and absence of the inner root sheath.5 Examination under magnification also allows providers to better identify telogen versus anagen hairs, which aids in the diagnosis. By carefully considering the patient history, physical exam, and results of additional hair tests, providers can make the diagnosis of LAHS and avoid unnecessary blood work and invasive procedures like scalp biopsies.
The differential diagnosis of hair loss frequently includes alopecia areata. However, in alopecia areata, patients typically have sharply demarcated areas of hair loss, which may involve the eyebrows, eyelids, and body hairs. In alopecia areata, providers may be able to identify the “exclamation point sign” in which the hair shaft thins proximally, leading to the appearance of more pigmented, thicker hairs floating above the scalp.
Telogen effluvium is a condition in which a medical illness or stress, such as systemic illness, surgery, severe emotional distress, childbirth, dietary changes, or another traumatic event, causes a disruption in the natural cycle of hair growth such that the percentage of hairs in the telogen phase increases from about 10% to up to 70%.6 Unlike in LAHS, in which shed hairs are in the anagen phase, the hair that is shed in telogen effluvium is in the telogen phase and will have a different appearance when magnified.
Anagen effluvium, loss of hairs in their growing phase, is typically associated with chemotherapy. The hairs become broken and fractured at the shaft leading to breakage at different points throughout the scalp. Affected areas can include the eyebrows, eyelashes, and body hair. In the absence of a history of administration of a chemotherapy agent (or other drug known to trigger hair loss), the diagnosis of anagen effluvium should not be made.
Patients with trichotillosis (also known as trichotillomania) present with areas of hair loss caused by intentional or subconscious hair pulling. It is considered a psychological condition that can be associated with obsessive compulsive disorder, although the presence of a secondary psychological diagnosis is not required. Providers may see irregular geometric shapes of hair loss, and on close inspection see broken hair shafts of different lengths. Patients most often pull hair from their scalps (over 70% of patients), but also can pull eyelashes, eyebrow hairs, and pubic hairs.7
Treatment
LAHS is self-limited and does not necessitate treatment. However, if patients or parents feel there is significant disease burden, perhaps with poor effects on quality of life or with psychosocial impairment, treatment with minoxidil 5% solution has been studied with some success reported in the literature.1,8,9
Ms. Natsis is a medical student at the University of California, San Diego. Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital-San Diego. He is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego. Ms. Natsis and Dr. Eichenfield had no relevant financial disclosures. Email them at [email protected].
References
1. Arch Dermatol. 2002;138(4):501-6.
2. Int J Trichology. 2010;2(2):96-100.
3. Pediatric Dermatol. 2016:33(50):507-10.
4. Dermatol Clin. 1986;14:745-51.
5. Arch Dermatol. 2009;145(10):1123-8.
6. J Clin Diagn Res. 2015;9(9):WE01-3.
7. Am J Psychiatry. 2016;173(9):868-74.
8. Australas J Dermatol. 2018;59:e286-e287.
9. Pediatr Dermatol. 2014;31:389-90.
A 5-year-old female is brought to clinic for hair loss. The mother reports that when styling her daughter's hair, she has noticed areas of hair thinning, especially at the temples and at the occiput. The mother denies scale, pruritus, and erythema. The patient reports that her scalp does not hurt. She has never had a haircut because her hair hasn't grown long enough to cut. There is no history of specific bald spots. The patient has no personal history of psoriasis, seborrheic dermatitis, or autoimmune disease. No picking has been noted, and there is no history of compulsive behaviors or anxiety. The patient's mother has a history of Graves disease. The mother reports that the patient's older sister may have had hair thinning when she was younger as well, but no longer has thin hair.
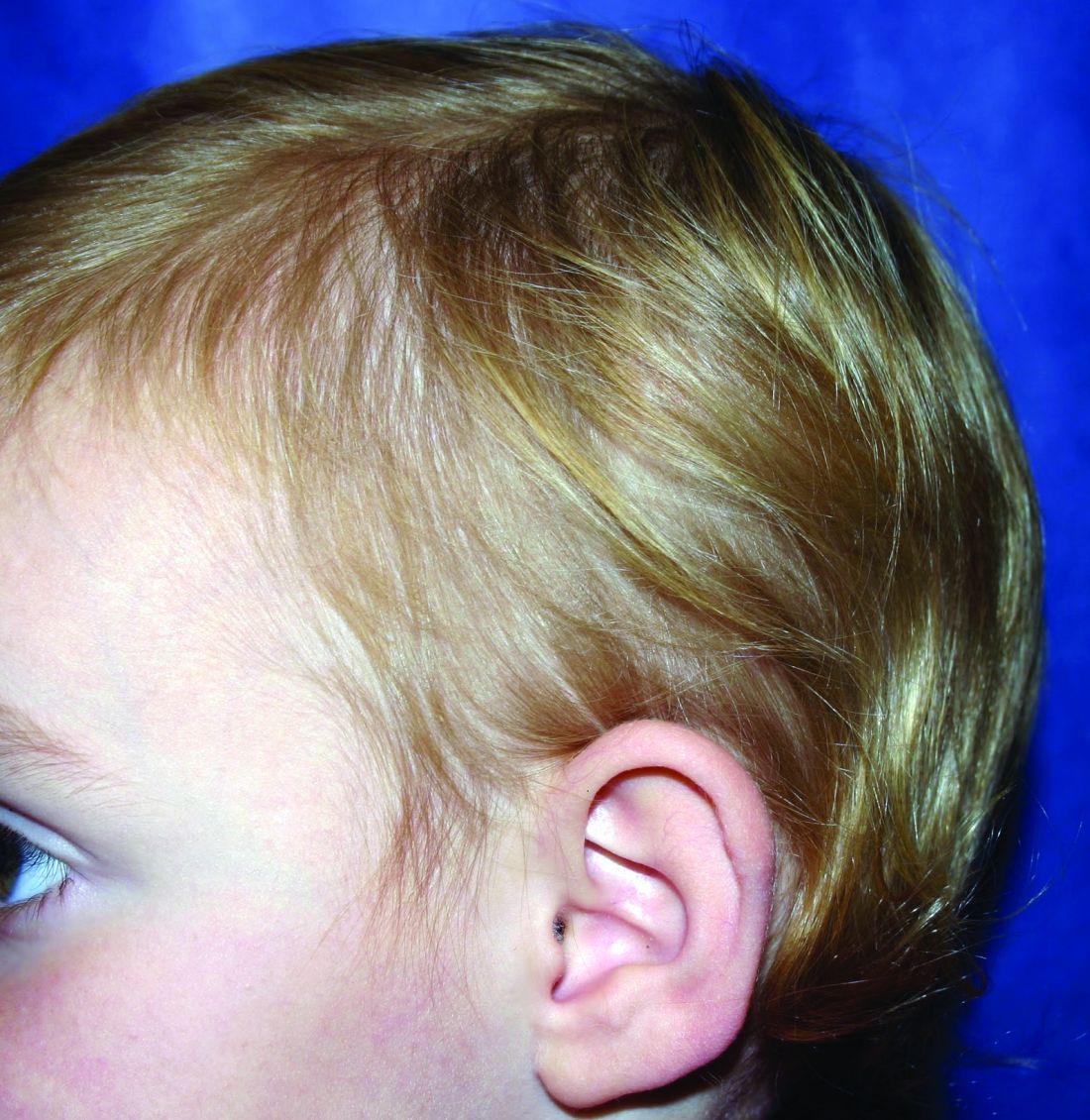
The patient was previously seen by another provider who prescribed hydrocortisone 2.5% ointment, which the mother has been applying nightly without improvement.
The child is otherwise medically well and thriving, with no recent change in activity level and with growth parameters consistently around the 75th percentile for height and weight. On physical exam, the patient has blondish, fine hair with areas of poorly demarcated hair thinning at the left temple and at the occiput. The hair remaining at the occiput is normal in texture. There are no areas of complete hair loss. There is no scale, erythema, or abnormal pigmentation, and no cervical or occipital adenopathy. The patient has intact eyebrows and eyelashes.
What's your diagnosis?
The patient was diagnosed with lichen spinulosus (LS) based on the physical appearance of the lesions (hyperkeratotic spiny papules forming plaques), the lack of pruritus, and negative personal history of atopic dermatitis.
Lichen spinulosus is an underreported entity, first described in 1908 by Adamson as superficial circumscribed chronic dermatitis in children and adolescents. The median age of presentation is age 16 years. There are several reports of possible associations with systemic infections such as HIV, fungi, and syphilis, as well as chronic diseases such as Crohn’s disease, Hodgkin disease, seborrhea, and secondary to certain medications such as omeprazole. There are no prior reports of infliximab being associated with LS, but it has been reported to cause other lichenoid reactions such as lichen planus and lichen planopilaris.
Clinically the lesions are characterized by asymptomatic, small (1 cm), skin color, hyperkeratotic, follicular papules that coalesce into plaques. The lesions usually occur on the extensor surfaces of the arms, neck, torso, and buttocks. Mild pruritus can occur in some patients.
The lesions in keratosis pilaris can be very similar to lichen spinulosus, but usually they don’t coalesce into plaques and are commonly present on the extensor surfaces of the arms, thighs, and cheeks. Histopathology of both conditions is very similar.

Another condition to consider includes papular eczema. The lesions in papular eczema tend to be pruritic and are not as circumscribed as LS lesions. Papular eczema responds well to the use of topical corticosteroids, while LS lesions usually do not. Lichen nitidus (LN) is characterized by monomorphic, skin color, 1-mm, flat-topped papules. Lesions tend to occur in crops rather than circumscribed papules forming plaques. LN most commonly presents on the extensor surface of the arms, trunk, dorsal hands, and genitalia. Koebner phenomenon is usually seen. Although uncommon in children, a more generalized type of follicular mucinosis can present very similar to lichen spinulosus. A recent study found LS-like lesions with associated hypopigmentation and hair loss should be suspicious for folliculotropic mycosis fungoides.
Keratolytics such as lactic acid, urea, and salicylic acid can help improve LS, although they do not cure it. Other reported treatments include the use of topical adapalene, tacalcitol cream, and tretinoin gel with hydroactive adhesive.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. Email her at [email protected].
The patient was diagnosed with lichen spinulosus (LS) based on the physical appearance of the lesions (hyperkeratotic spiny papules forming plaques), the lack of pruritus, and negative personal history of atopic dermatitis.
Lichen spinulosus is an underreported entity, first described in 1908 by Adamson as superficial circumscribed chronic dermatitis in children and adolescents. The median age of presentation is age 16 years. There are several reports of possible associations with systemic infections such as HIV, fungi, and syphilis, as well as chronic diseases such as Crohn’s disease, Hodgkin disease, seborrhea, and secondary to certain medications such as omeprazole. There are no prior reports of infliximab being associated with LS, but it has been reported to cause other lichenoid reactions such as lichen planus and lichen planopilaris.
Clinically the lesions are characterized by asymptomatic, small (1 cm), skin color, hyperkeratotic, follicular papules that coalesce into plaques. The lesions usually occur on the extensor surfaces of the arms, neck, torso, and buttocks. Mild pruritus can occur in some patients.
The lesions in keratosis pilaris can be very similar to lichen spinulosus, but usually they don’t coalesce into plaques and are commonly present on the extensor surfaces of the arms, thighs, and cheeks. Histopathology of both conditions is very similar.
Another condition to consider includes papular eczema. The lesions in papular eczema tend to be pruritic and are not as circumscribed as LS lesions. Papular eczema responds well to the use of topical corticosteroids, while LS lesions usually do not. Lichen nitidus (LN) is characterized by monomorphic, skin color, 1-mm, flat-topped papules. Lesions tend to occur in crops rather than circumscribed papules forming plaques. LN most commonly presents on the extensor surface of the arms, trunk, dorsal hands, and genitalia. Koebner phenomenon is usually seen. Although uncommon in children, a more generalized type of follicular mucinosis can present very similar to lichen spinulosus. A recent study found LS-like lesions with associated hypopigmentation and hair loss should be suspicious for folliculotropic mycosis fungoides.
Keratolytics such as lactic acid, urea, and salicylic acid can help improve LS, although they do not cure it. Other reported treatments include the use of topical adapalene, tacalcitol cream, and tretinoin gel with hydroactive adhesive.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. Email her at [email protected].
The patient was diagnosed with lichen spinulosus (LS) based on the physical appearance of the lesions (hyperkeratotic spiny papules forming plaques), the lack of pruritus, and negative personal history of atopic dermatitis.
Lichen spinulosus is an underreported entity, first described in 1908 by Adamson as superficial circumscribed chronic dermatitis in children and adolescents. The median age of presentation is age 16 years. There are several reports of possible associations with systemic infections such as HIV, fungi, and syphilis, as well as chronic diseases such as Crohn’s disease, Hodgkin disease, seborrhea, and secondary to certain medications such as omeprazole. There are no prior reports of infliximab being associated with LS, but it has been reported to cause other lichenoid reactions such as lichen planus and lichen planopilaris.
Clinically the lesions are characterized by asymptomatic, small (1 cm), skin color, hyperkeratotic, follicular papules that coalesce into plaques. The lesions usually occur on the extensor surfaces of the arms, neck, torso, and buttocks. Mild pruritus can occur in some patients.
The lesions in keratosis pilaris can be very similar to lichen spinulosus, but usually they don’t coalesce into plaques and are commonly present on the extensor surfaces of the arms, thighs, and cheeks. Histopathology of both conditions is very similar.
Another condition to consider includes papular eczema. The lesions in papular eczema tend to be pruritic and are not as circumscribed as LS lesions. Papular eczema responds well to the use of topical corticosteroids, while LS lesions usually do not. Lichen nitidus (LN) is characterized by monomorphic, skin color, 1-mm, flat-topped papules. Lesions tend to occur in crops rather than circumscribed papules forming plaques. LN most commonly presents on the extensor surface of the arms, trunk, dorsal hands, and genitalia. Koebner phenomenon is usually seen. Although uncommon in children, a more generalized type of follicular mucinosis can present very similar to lichen spinulosus. A recent study found LS-like lesions with associated hypopigmentation and hair loss should be suspicious for folliculotropic mycosis fungoides.
Keratolytics such as lactic acid, urea, and salicylic acid can help improve LS, although they do not cure it. Other reported treatments include the use of topical adapalene, tacalcitol cream, and tretinoin gel with hydroactive adhesive.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. Email her at [email protected].
A 7-year-old male with a history of Crohn's disease presents with 6 months of asymptomatic, bumpy lesions on the torso and extremities. He has been using over-the-counter hydrocortisone and moisturizers without it helping. His Crohn's disease has been controlled with infliximab infusions for 2 years. The mother is concerned the rash could be a side effect of the medication.
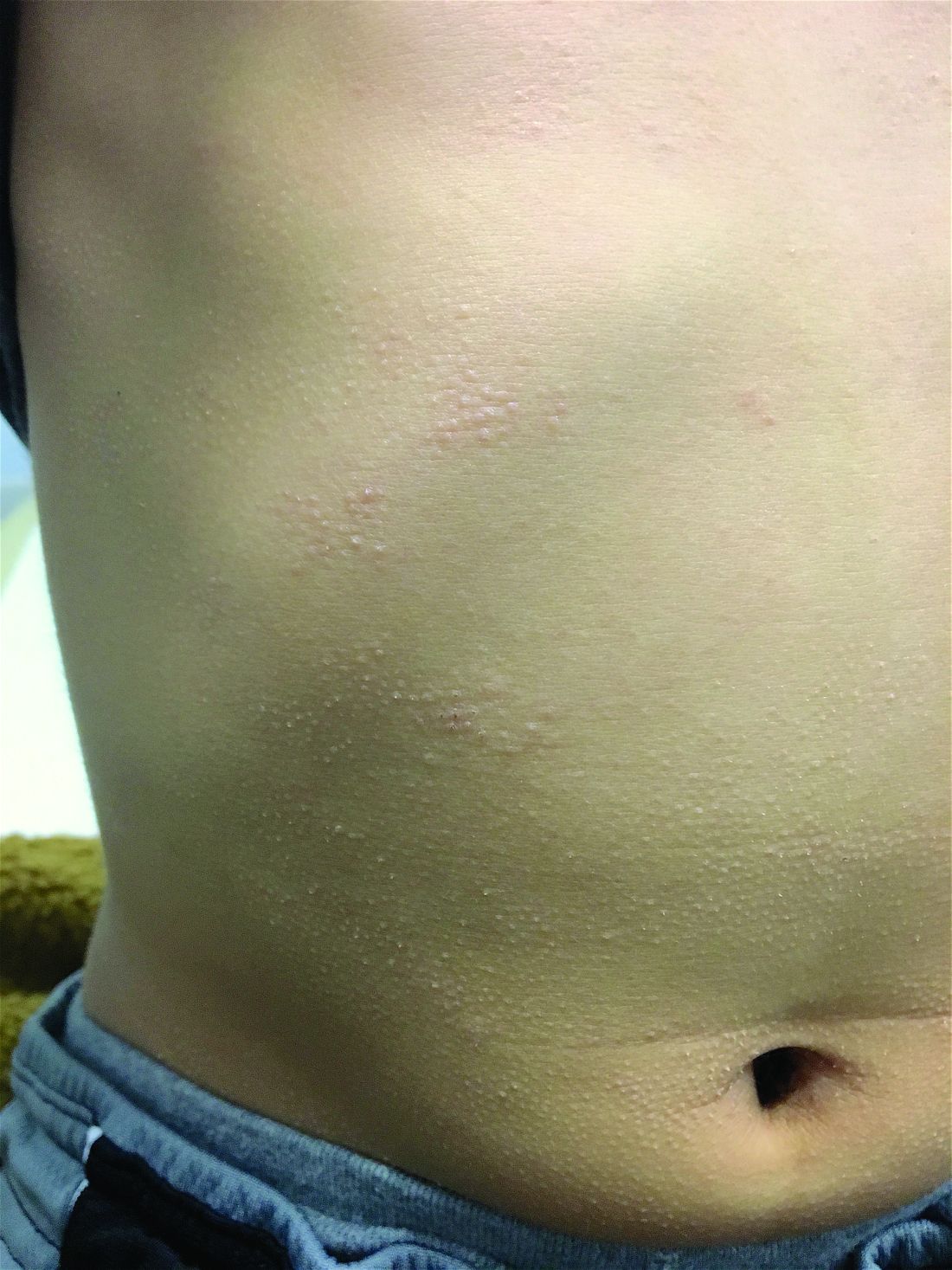
He denies any prior history of atopic dermatitis or psoriasis. The mother had eczema as a child. He has two brothers who have been diagnosed and treated for allergic rhinitis.
On physical examination, he is a thin, pleasant young boy in no distress.
His skin is somewhat dry, and there are several hyperkeratotic follicular papules forming plaques on the torso and extremities. There is no associated hair loss on the affected areas or inflammation noted.
December 2018
Vasculitis is a process in which blood vessels become inflamed and necrotic. Classic small-vessel vasculitis reveals a leukocytoclastic vasculitis and most commonly presents as palpable purpura. In addition to skin, organs such as joints, kidneys, and intestines can be involved.
where immunoglobulin A (IgA) is deposited in the vessel walls. It is the most common form of vasculitis in children (usually aged 4-8 years). The incidence is higher in the winter. Some patients experience a prodrome of fever, colicky abdominal pain, and joint pain prior to the development of cutaneous symptoms. Disease in children tends to be self-limited. Adults may present with HSP as well, and often exhibit more severe disease that may become chronic with relapses and is more difficult to treat. In both children and adults, infectious causes, such as streptococcus pharyngitis, are the most common trigger. In adults, malignancy may be associated with HSP. A literature search revealed medications implicated in HSP such as antibiotics (vancomycin, penicillin, cephalosporins, clarithromycin), ACE inhibitors, and nonsteroidal anti-inflammatories. Many cases of HSP are idiopathic.
Patients present with erythematous macules that progress to purpura on the extremities. Lesions may be vesicular or bullous and may become necrotic and ulcerate. Arthralgias, often of lower-extremity joints, may be present. Abdominal pain and renal disease may occur in both children and adults. Adults are more likely to develop chronic kidney disease and must be followed carefully with serial blood work and urinalysis to evaluate for hematuria and proteinuria. Severe abdominal pain is an emergency as intussusception may occur.
Histologically, leukocytoclastic vasculitis of small vessels is present. On direct immunofluorescence of perilesional skin, IgA, C3, and fibrin deposits can be seen. Serum IgA is unreliable and may be seen in healthy adults as well.
Treatment is generally supportive as the disease is self-limited. The use of corticosteroids is controversial. This may be effective for joint inflammation, abdominal disease, active nephritis, and ulcerated skin lesions, but doesn’t prevent the recurrence of skin lesions. Dapsone or colchicine can be used for resistant cutaneous lesions. In severe cases, intravenous immunoglobulin may be warranted.
This case and photo were submitted by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at edermatologynews.com. To submit a case for possible publication, send an email to [email protected].
Vasculitis is a process in which blood vessels become inflamed and necrotic. Classic small-vessel vasculitis reveals a leukocytoclastic vasculitis and most commonly presents as palpable purpura. In addition to skin, organs such as joints, kidneys, and intestines can be involved.
where immunoglobulin A (IgA) is deposited in the vessel walls. It is the most common form of vasculitis in children (usually aged 4-8 years). The incidence is higher in the winter. Some patients experience a prodrome of fever, colicky abdominal pain, and joint pain prior to the development of cutaneous symptoms. Disease in children tends to be self-limited. Adults may present with HSP as well, and often exhibit more severe disease that may become chronic with relapses and is more difficult to treat. In both children and adults, infectious causes, such as streptococcus pharyngitis, are the most common trigger. In adults, malignancy may be associated with HSP. A literature search revealed medications implicated in HSP such as antibiotics (vancomycin, penicillin, cephalosporins, clarithromycin), ACE inhibitors, and nonsteroidal anti-inflammatories. Many cases of HSP are idiopathic.
Patients present with erythematous macules that progress to purpura on the extremities. Lesions may be vesicular or bullous and may become necrotic and ulcerate. Arthralgias, often of lower-extremity joints, may be present. Abdominal pain and renal disease may occur in both children and adults. Adults are more likely to develop chronic kidney disease and must be followed carefully with serial blood work and urinalysis to evaluate for hematuria and proteinuria. Severe abdominal pain is an emergency as intussusception may occur.
Histologically, leukocytoclastic vasculitis of small vessels is present. On direct immunofluorescence of perilesional skin, IgA, C3, and fibrin deposits can be seen. Serum IgA is unreliable and may be seen in healthy adults as well.
Treatment is generally supportive as the disease is self-limited. The use of corticosteroids is controversial. This may be effective for joint inflammation, abdominal disease, active nephritis, and ulcerated skin lesions, but doesn’t prevent the recurrence of skin lesions. Dapsone or colchicine can be used for resistant cutaneous lesions. In severe cases, intravenous immunoglobulin may be warranted.
This case and photo were submitted by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at edermatologynews.com. To submit a case for possible publication, send an email to [email protected].
Vasculitis is a process in which blood vessels become inflamed and necrotic. Classic small-vessel vasculitis reveals a leukocytoclastic vasculitis and most commonly presents as palpable purpura. In addition to skin, organs such as joints, kidneys, and intestines can be involved.
where immunoglobulin A (IgA) is deposited in the vessel walls. It is the most common form of vasculitis in children (usually aged 4-8 years). The incidence is higher in the winter. Some patients experience a prodrome of fever, colicky abdominal pain, and joint pain prior to the development of cutaneous symptoms. Disease in children tends to be self-limited. Adults may present with HSP as well, and often exhibit more severe disease that may become chronic with relapses and is more difficult to treat. In both children and adults, infectious causes, such as streptococcus pharyngitis, are the most common trigger. In adults, malignancy may be associated with HSP. A literature search revealed medications implicated in HSP such as antibiotics (vancomycin, penicillin, cephalosporins, clarithromycin), ACE inhibitors, and nonsteroidal anti-inflammatories. Many cases of HSP are idiopathic.
Patients present with erythematous macules that progress to purpura on the extremities. Lesions may be vesicular or bullous and may become necrotic and ulcerate. Arthralgias, often of lower-extremity joints, may be present. Abdominal pain and renal disease may occur in both children and adults. Adults are more likely to develop chronic kidney disease and must be followed carefully with serial blood work and urinalysis to evaluate for hematuria and proteinuria. Severe abdominal pain is an emergency as intussusception may occur.
Histologically, leukocytoclastic vasculitis of small vessels is present. On direct immunofluorescence of perilesional skin, IgA, C3, and fibrin deposits can be seen. Serum IgA is unreliable and may be seen in healthy adults as well.
Treatment is generally supportive as the disease is self-limited. The use of corticosteroids is controversial. This may be effective for joint inflammation, abdominal disease, active nephritis, and ulcerated skin lesions, but doesn’t prevent the recurrence of skin lesions. Dapsone or colchicine can be used for resistant cutaneous lesions. In severe cases, intravenous immunoglobulin may be warranted.
This case and photo were submitted by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at edermatologynews.com. To submit a case for possible publication, send an email to [email protected].
A 63-year-old white female presented with a 2-week history of hemorrhagic purpuric lesions and necrotic vesicles on the bilateral lower extremities.
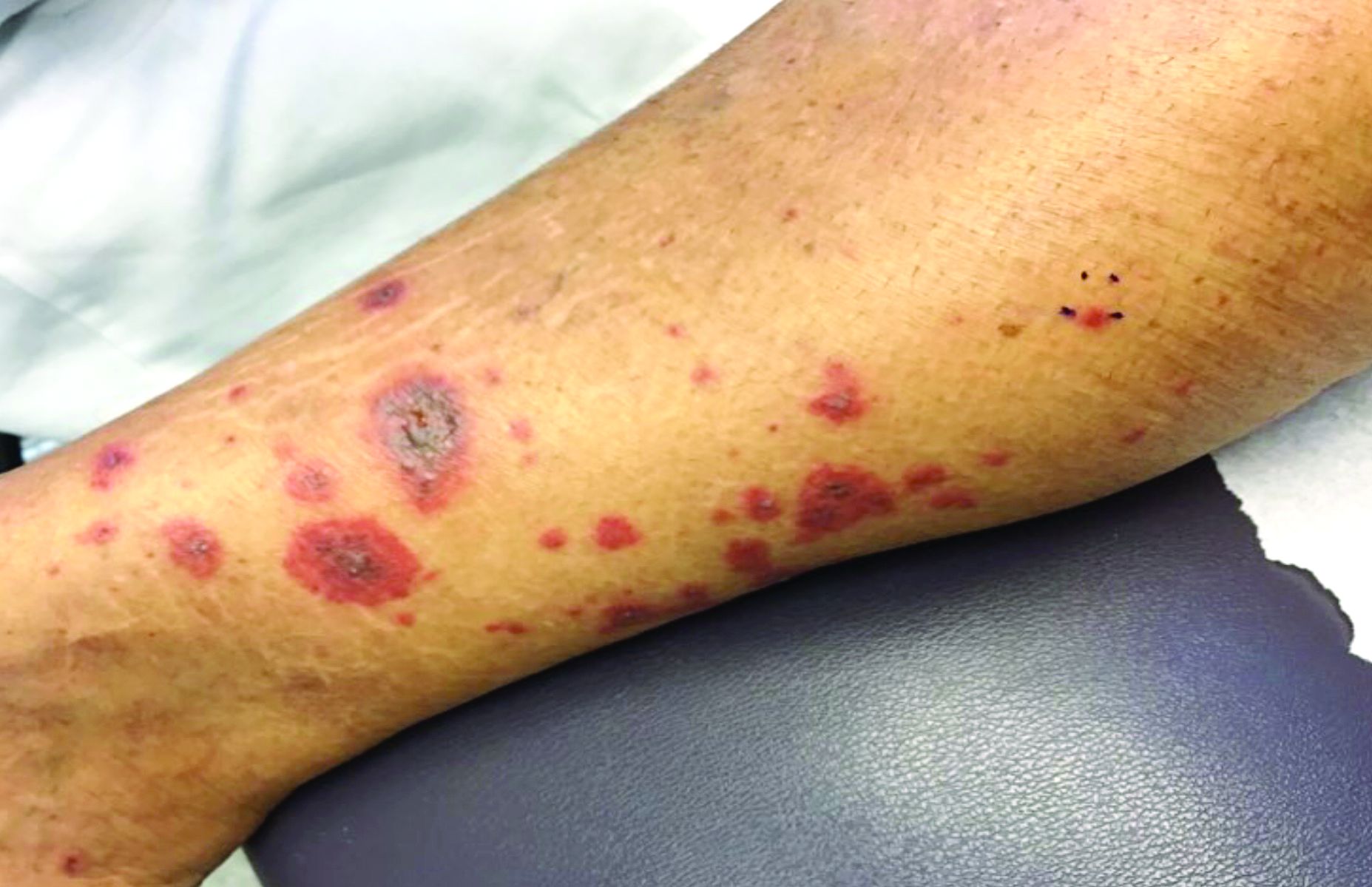
Nearly 1 year prior to presentation, the patient underwent surgical resection for lung cancer. The patient also complained of joint swelling and pain in her ankles. She denied abdominal pain. She denied recent illness, including sore throat and upper respiratory infection. Skin biopsies were performed, including for direct immunofluorescence.
What is your diagnosis?
Epidermal nevi are a subset of cutaneous hamartomas resulting from somatic mutations of epidermal cells, presenting as keratinocyte or epidermal appendage overgrowths. The most common type appear in a linear distribution and are termed linear epidermal nevi or linear verrucous epidermal nevi.

There are variations of epidermal nevi (EN) that can be composed of superficial epidermal keratinocytes, sebaceous glands, apocrine or eccrine glands, hair follicles, or smooth muscle. For example, many consider a nevus sebaceous to be a type of epidermal nevus as well. The incidence of EN is approximately 1 in 1,000 newborns. Postzygotic cell mutations result in a mosaic distribution that follows embryonic migration patterns, appearing in a Blaschkoid distribution.
EN present most frequently as unilateral linear or whorled hyperpigmented coalescing papules. The lesions can be present at birth or during childhood, and after appearing, grow with the patient. Typically the lesions become more raised and verrucous around puberty. The differential diagnosis of linear EN include lichen striatus, warts, and incontinentia pigmenti. Lichen striatus can be differentiated because it presents later in life and self-resolves. Verrucae are the most commonly mistaken diagnosis for EN; warts do not usually persist in the same pattern over time with proportionate growth and typically respond to locally destructive treatments such as liquid nitrogen, unlike EN. Incontinentia pigmenti presents as vesicles initially and shows a quick evolution, differentiating it from EN. Inflammatory linear verrucous epidermal nevus (ILVEN) is a variant of linear EN that has associated chronic and intermittent erythema, scale, and pruritus. Lichen nitidus often has a pruritic presentation; however, it is flat topped and skin colored, helping differentiate it from linear EN.
There has been recent research advancing gene associations for linear EN displaying many lesions associated with mosaic mutations in oncogenes. Multiple genes have been identified with EN including RAS, FGFR3, and PIK3CA1. FGFR3 and PIK3CA mutations are associated with 50% of keratinocytic nevi. Of the RAS family, the HRAS pathway has been most closely associated with nevus sebaceous. While KRAS and NRAS genes have been associated with EN, it is to a lesser degree. However, there are multiple recent case reports demonstrating a potential association of G12D mosaicism of the KRAS gene in EN with rhabdomyosarcoma and bladder cancers2.
The diagnosis of epidermal nevus syndrome should be considered when there is a nevus with associated developmental abnormality of the central nervous system, eyes, or musculoskeletal systems. The most common systemic symptoms include delays in developmental milestones, seizure disorders, coloboma, strabismus, muscle weakness, and hemihypertrophy. To date, there are six specific epidermal nevus syndromes identified: sebaceous nevus syndrome, nevus comedonicus syndrome, Becker nevus syndrome, phakomatosis pigmentokeratotica, Proteus syndrome, congenital hemidysplasia with ichthyosiform nevus and limb defects, and cutaneous-skeletal hypophosphatemia syndrome3. In addition to the syndromes described, there are reports of associations between keratinocytic nevi and ILVEN with hypophosphatemic rickets and precocious puberty.
Linear EN are rarely associated with malignant transformation to basal cell carcinoma or squamous cell carcinoma, depending on the cell type involved. Given the low risk of malignancy, the lesions do not need to be removed routinely. For small lesions, monitoring often is the preferred management. However, lesions with functional significance, or causing strangulation or deformity, can be treated with surgical excision, curettage, or laser destruction
Dr. Kaushik is with the division of pediatric and adolescent dermatology at Rady Children’s Hospital–San Diego, and Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital–San Diego. He is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego. There are no conflicts of interest or financial disclosures for Dr. Kaushik or Dr. Eichenfield. Email them at [email protected].
References
1. Pediatr Dermatol. 2004 Jul-Aug;21(4):432-9.
2. J Med Genet. 2010 Dec;47(12):859-62.
3. Pediatr Dermatol. 2018 Jan;35(1):21-9.
Epidermal nevi are a subset of cutaneous hamartomas resulting from somatic mutations of epidermal cells, presenting as keratinocyte or epidermal appendage overgrowths. The most common type appear in a linear distribution and are termed linear epidermal nevi or linear verrucous epidermal nevi.
There are variations of epidermal nevi (EN) that can be composed of superficial epidermal keratinocytes, sebaceous glands, apocrine or eccrine glands, hair follicles, or smooth muscle. For example, many consider a nevus sebaceous to be a type of epidermal nevus as well. The incidence of EN is approximately 1 in 1,000 newborns. Postzygotic cell mutations result in a mosaic distribution that follows embryonic migration patterns, appearing in a Blaschkoid distribution.
EN present most frequently as unilateral linear or whorled hyperpigmented coalescing papules. The lesions can be present at birth or during childhood, and after appearing, grow with the patient. Typically the lesions become more raised and verrucous around puberty. The differential diagnosis of linear EN include lichen striatus, warts, and incontinentia pigmenti. Lichen striatus can be differentiated because it presents later in life and self-resolves. Verrucae are the most commonly mistaken diagnosis for EN; warts do not usually persist in the same pattern over time with proportionate growth and typically respond to locally destructive treatments such as liquid nitrogen, unlike EN. Incontinentia pigmenti presents as vesicles initially and shows a quick evolution, differentiating it from EN. Inflammatory linear verrucous epidermal nevus (ILVEN) is a variant of linear EN that has associated chronic and intermittent erythema, scale, and pruritus. Lichen nitidus often has a pruritic presentation; however, it is flat topped and skin colored, helping differentiate it from linear EN.
There has been recent research advancing gene associations for linear EN displaying many lesions associated with mosaic mutations in oncogenes. Multiple genes have been identified with EN including RAS, FGFR3, and PIK3CA1. FGFR3 and PIK3CA mutations are associated with 50% of keratinocytic nevi. Of the RAS family, the HRAS pathway has been most closely associated with nevus sebaceous. While KRAS and NRAS genes have been associated with EN, it is to a lesser degree. However, there are multiple recent case reports demonstrating a potential association of G12D mosaicism of the KRAS gene in EN with rhabdomyosarcoma and bladder cancers2.
The diagnosis of epidermal nevus syndrome should be considered when there is a nevus with associated developmental abnormality of the central nervous system, eyes, or musculoskeletal systems. The most common systemic symptoms include delays in developmental milestones, seizure disorders, coloboma, strabismus, muscle weakness, and hemihypertrophy. To date, there are six specific epidermal nevus syndromes identified: sebaceous nevus syndrome, nevus comedonicus syndrome, Becker nevus syndrome, phakomatosis pigmentokeratotica, Proteus syndrome, congenital hemidysplasia with ichthyosiform nevus and limb defects, and cutaneous-skeletal hypophosphatemia syndrome3. In addition to the syndromes described, there are reports of associations between keratinocytic nevi and ILVEN with hypophosphatemic rickets and precocious puberty.
Linear EN are rarely associated with malignant transformation to basal cell carcinoma or squamous cell carcinoma, depending on the cell type involved. Given the low risk of malignancy, the lesions do not need to be removed routinely. For small lesions, monitoring often is the preferred management. However, lesions with functional significance, or causing strangulation or deformity, can be treated with surgical excision, curettage, or laser destruction
Dr. Kaushik is with the division of pediatric and adolescent dermatology at Rady Children’s Hospital–San Diego, and Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital–San Diego. He is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego. There are no conflicts of interest or financial disclosures for Dr. Kaushik or Dr. Eichenfield. Email them at [email protected].
References
1. Pediatr Dermatol. 2004 Jul-Aug;21(4):432-9.
2. J Med Genet. 2010 Dec;47(12):859-62.
3. Pediatr Dermatol. 2018 Jan;35(1):21-9.
Epidermal nevi are a subset of cutaneous hamartomas resulting from somatic mutations of epidermal cells, presenting as keratinocyte or epidermal appendage overgrowths. The most common type appear in a linear distribution and are termed linear epidermal nevi or linear verrucous epidermal nevi.
There are variations of epidermal nevi (EN) that can be composed of superficial epidermal keratinocytes, sebaceous glands, apocrine or eccrine glands, hair follicles, or smooth muscle. For example, many consider a nevus sebaceous to be a type of epidermal nevus as well. The incidence of EN is approximately 1 in 1,000 newborns. Postzygotic cell mutations result in a mosaic distribution that follows embryonic migration patterns, appearing in a Blaschkoid distribution.
EN present most frequently as unilateral linear or whorled hyperpigmented coalescing papules. The lesions can be present at birth or during childhood, and after appearing, grow with the patient. Typically the lesions become more raised and verrucous around puberty. The differential diagnosis of linear EN include lichen striatus, warts, and incontinentia pigmenti. Lichen striatus can be differentiated because it presents later in life and self-resolves. Verrucae are the most commonly mistaken diagnosis for EN; warts do not usually persist in the same pattern over time with proportionate growth and typically respond to locally destructive treatments such as liquid nitrogen, unlike EN. Incontinentia pigmenti presents as vesicles initially and shows a quick evolution, differentiating it from EN. Inflammatory linear verrucous epidermal nevus (ILVEN) is a variant of linear EN that has associated chronic and intermittent erythema, scale, and pruritus. Lichen nitidus often has a pruritic presentation; however, it is flat topped and skin colored, helping differentiate it from linear EN.
There has been recent research advancing gene associations for linear EN displaying many lesions associated with mosaic mutations in oncogenes. Multiple genes have been identified with EN including RAS, FGFR3, and PIK3CA1. FGFR3 and PIK3CA mutations are associated with 50% of keratinocytic nevi. Of the RAS family, the HRAS pathway has been most closely associated with nevus sebaceous. While KRAS and NRAS genes have been associated with EN, it is to a lesser degree. However, there are multiple recent case reports demonstrating a potential association of G12D mosaicism of the KRAS gene in EN with rhabdomyosarcoma and bladder cancers2.
The diagnosis of epidermal nevus syndrome should be considered when there is a nevus with associated developmental abnormality of the central nervous system, eyes, or musculoskeletal systems. The most common systemic symptoms include delays in developmental milestones, seizure disorders, coloboma, strabismus, muscle weakness, and hemihypertrophy. To date, there are six specific epidermal nevus syndromes identified: sebaceous nevus syndrome, nevus comedonicus syndrome, Becker nevus syndrome, phakomatosis pigmentokeratotica, Proteus syndrome, congenital hemidysplasia with ichthyosiform nevus and limb defects, and cutaneous-skeletal hypophosphatemia syndrome3. In addition to the syndromes described, there are reports of associations between keratinocytic nevi and ILVEN with hypophosphatemic rickets and precocious puberty.
Linear EN are rarely associated with malignant transformation to basal cell carcinoma or squamous cell carcinoma, depending on the cell type involved. Given the low risk of malignancy, the lesions do not need to be removed routinely. For small lesions, monitoring often is the preferred management. However, lesions with functional significance, or causing strangulation or deformity, can be treated with surgical excision, curettage, or laser destruction
Dr. Kaushik is with the division of pediatric and adolescent dermatology at Rady Children’s Hospital–San Diego, and Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital–San Diego. He is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego. There are no conflicts of interest or financial disclosures for Dr. Kaushik or Dr. Eichenfield. Email them at [email protected].
References
1. Pediatr Dermatol. 2004 Jul-Aug;21(4):432-9.
2. J Med Genet. 2010 Dec;47(12):859-62.
3. Pediatr Dermatol. 2018 Jan;35(1):21-9.
A 6-year-old, otherwise-healthy male is brought into clinic for evaluation of papules on his neck. The rash has been present since 1 year of age and has been growing in size proportionately. He claims there is occasional itching but no pain or redness. He does not seem to be disturbed by his rash. He has two siblings, aged 2 and 4 years, without lesions.
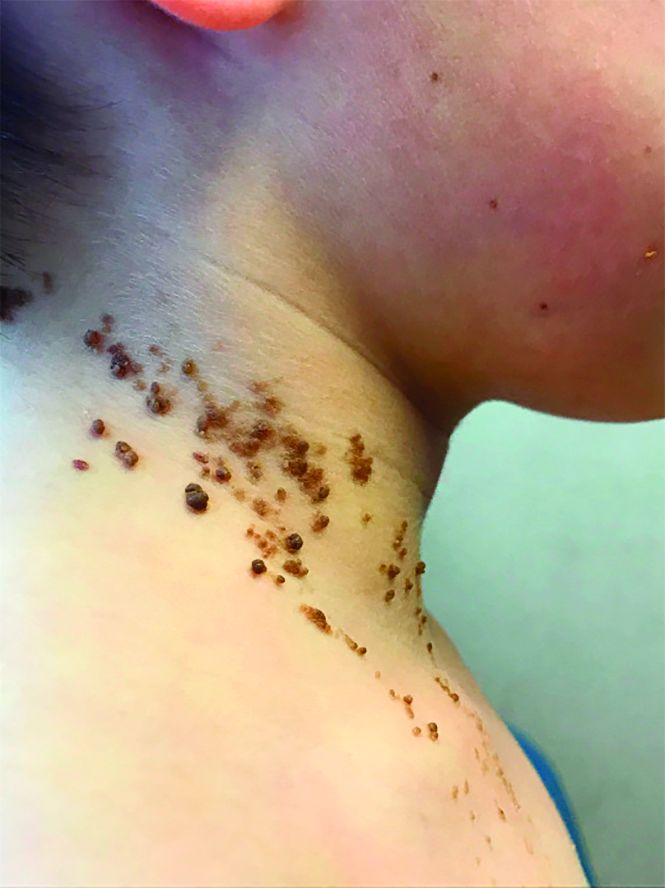
On physical exam, he is noted to have a linear plaque of hyperpigmented verrucous papules on his neck.
What is your diagnosis? - December 2018
A KOH (potassium hydroxide) test done at the visit was negative as well as a fungal culture of each toenail.
The patient was diagnosed with congenital malalignment of the great toenails (CMGTN) based on history and morphologic appearance.
Congenital malalignment of the great toenails is an underrecognized and underreported nail disorder characterized by lateral deviation of the nail plate, which is not parallel to the longitudinal axis of the distal phalanx.1 The cause is unknown. Some reports suggest a genetic cause being transmitted in an autosomal dominant fashion with variable expression.2 There have been reports of CMGTN in monozygotic and dizygotic twins making this theory likely.3 Other authors consider an external cause such as amniotic bands, neonatal asphyxia, vascular malformations, and uterine pressure. This condition also has been reported in patients with Rubinstein-Taybi syndrome.4
The nail changes can occur at birth but in some cases, such as our patient, the nails become dystrophic months to years after birth. Characteristic nail changes include shorter, discolored, hyperkeratotic nails with transverse groove or ridges. In some cases, the dystrophic nails may cause inflammation and tenderness and is the most common cause of ingrown toenails in children.
The differential diagnosis includes onychomycosis, traumatic nails, nail psoriasis, pachyonychia congenital (PC), and onychomadesis. Onychomycosis can present with white or yellow discoloration of the nail that in some cases can be associated with nail breakage, hyperkeratosis, onycholysis, and subungual debris. Either fungal culture or periodic acid shift stain of nail clippings can help confirm or exclude this diagnosis. Psoriatic nails present with nail pits, oils spots, and onycholysis. Traumatic nail changes may occur from using small shoes and trauma from running or playing soccer, and presents with subungual hemorrhage and nail dystrophy of the first or second toenail. PC is a genetic disorder caused by a mutation in certain keratin proteins of the skin (k6a, k6b, K16 and K17). These patients usually have other skin findings including palmoplantar keratoderma, white plaques on the mouth, and skin cysts (steatocystoma multiplex and vellus hair cysts). Nail changes characteristic of PC includes subungual hyperkeratosis that causes a wedge shape thickening of the nail bed (pincer nails).5 Onychomadesis can be seen after viral infections such as hand-foot-mouth disease or in patients taking chemotherapy drugs that affect nail growth.
CMGTN usually resolves with time, but some patients with severe deviation and paronychia may need surgical correction.6
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. Email her at [email protected].
References
1. Dermatol Online J. 2014 Jan 15;20(1):21251.
2. J Dtsch Dermatol Ges. 2012 May;10(5):326-30.
3. J Am Acad Dermatol. 2007 Oct;57(4):711-5.
4. Pediatr Dermatol. 2004 Jan-Feb;21(1):44-7.
5. Curr Opin Pediatr. 2014 Aug;26(4):440-5.
6. Skin Appendage Disord. 2018 Oct;4(4):230-5.
A KOH (potassium hydroxide) test done at the visit was negative as well as a fungal culture of each toenail.
The patient was diagnosed with congenital malalignment of the great toenails (CMGTN) based on history and morphologic appearance.
Congenital malalignment of the great toenails is an underrecognized and underreported nail disorder characterized by lateral deviation of the nail plate, which is not parallel to the longitudinal axis of the distal phalanx.1 The cause is unknown. Some reports suggest a genetic cause being transmitted in an autosomal dominant fashion with variable expression.2 There have been reports of CMGTN in monozygotic and dizygotic twins making this theory likely.3 Other authors consider an external cause such as amniotic bands, neonatal asphyxia, vascular malformations, and uterine pressure. This condition also has been reported in patients with Rubinstein-Taybi syndrome.4
The nail changes can occur at birth but in some cases, such as our patient, the nails become dystrophic months to years after birth. Characteristic nail changes include shorter, discolored, hyperkeratotic nails with transverse groove or ridges. In some cases, the dystrophic nails may cause inflammation and tenderness and is the most common cause of ingrown toenails in children.
The differential diagnosis includes onychomycosis, traumatic nails, nail psoriasis, pachyonychia congenital (PC), and onychomadesis. Onychomycosis can present with white or yellow discoloration of the nail that in some cases can be associated with nail breakage, hyperkeratosis, onycholysis, and subungual debris. Either fungal culture or periodic acid shift stain of nail clippings can help confirm or exclude this diagnosis. Psoriatic nails present with nail pits, oils spots, and onycholysis. Traumatic nail changes may occur from using small shoes and trauma from running or playing soccer, and presents with subungual hemorrhage and nail dystrophy of the first or second toenail. PC is a genetic disorder caused by a mutation in certain keratin proteins of the skin (k6a, k6b, K16 and K17). These patients usually have other skin findings including palmoplantar keratoderma, white plaques on the mouth, and skin cysts (steatocystoma multiplex and vellus hair cysts). Nail changes characteristic of PC includes subungual hyperkeratosis that causes a wedge shape thickening of the nail bed (pincer nails).5 Onychomadesis can be seen after viral infections such as hand-foot-mouth disease or in patients taking chemotherapy drugs that affect nail growth.
CMGTN usually resolves with time, but some patients with severe deviation and paronychia may need surgical correction.6
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. Email her at [email protected].
References
1. Dermatol Online J. 2014 Jan 15;20(1):21251.
2. J Dtsch Dermatol Ges. 2012 May;10(5):326-30.
3. J Am Acad Dermatol. 2007 Oct;57(4):711-5.
4. Pediatr Dermatol. 2004 Jan-Feb;21(1):44-7.
5. Curr Opin Pediatr. 2014 Aug;26(4):440-5.
6. Skin Appendage Disord. 2018 Oct;4(4):230-5.
A KOH (potassium hydroxide) test done at the visit was negative as well as a fungal culture of each toenail.
The patient was diagnosed with congenital malalignment of the great toenails (CMGTN) based on history and morphologic appearance.
Congenital malalignment of the great toenails is an underrecognized and underreported nail disorder characterized by lateral deviation of the nail plate, which is not parallel to the longitudinal axis of the distal phalanx.1 The cause is unknown. Some reports suggest a genetic cause being transmitted in an autosomal dominant fashion with variable expression.2 There have been reports of CMGTN in monozygotic and dizygotic twins making this theory likely.3 Other authors consider an external cause such as amniotic bands, neonatal asphyxia, vascular malformations, and uterine pressure. This condition also has been reported in patients with Rubinstein-Taybi syndrome.4
The nail changes can occur at birth but in some cases, such as our patient, the nails become dystrophic months to years after birth. Characteristic nail changes include shorter, discolored, hyperkeratotic nails with transverse groove or ridges. In some cases, the dystrophic nails may cause inflammation and tenderness and is the most common cause of ingrown toenails in children.
The differential diagnosis includes onychomycosis, traumatic nails, nail psoriasis, pachyonychia congenital (PC), and onychomadesis. Onychomycosis can present with white or yellow discoloration of the nail that in some cases can be associated with nail breakage, hyperkeratosis, onycholysis, and subungual debris. Either fungal culture or periodic acid shift stain of nail clippings can help confirm or exclude this diagnosis. Psoriatic nails present with nail pits, oils spots, and onycholysis. Traumatic nail changes may occur from using small shoes and trauma from running or playing soccer, and presents with subungual hemorrhage and nail dystrophy of the first or second toenail. PC is a genetic disorder caused by a mutation in certain keratin proteins of the skin (k6a, k6b, K16 and K17). These patients usually have other skin findings including palmoplantar keratoderma, white plaques on the mouth, and skin cysts (steatocystoma multiplex and vellus hair cysts). Nail changes characteristic of PC includes subungual hyperkeratosis that causes a wedge shape thickening of the nail bed (pincer nails).5 Onychomadesis can be seen after viral infections such as hand-foot-mouth disease or in patients taking chemotherapy drugs that affect nail growth.
CMGTN usually resolves with time, but some patients with severe deviation and paronychia may need surgical correction.6
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. Email her at [email protected].
References
1. Dermatol Online J. 2014 Jan 15;20(1):21251.
2. J Dtsch Dermatol Ges. 2012 May;10(5):326-30.
3. J Am Acad Dermatol. 2007 Oct;57(4):711-5.
4. Pediatr Dermatol. 2004 Jan-Feb;21(1):44-7.
5. Curr Opin Pediatr. 2014 Aug;26(4):440-5.
6. Skin Appendage Disord. 2018 Oct;4(4):230-5.
A 4-year-old boy is brought to our pediatric dermatology clinic by his mother with the concern of difficult to treat toenail fungus.
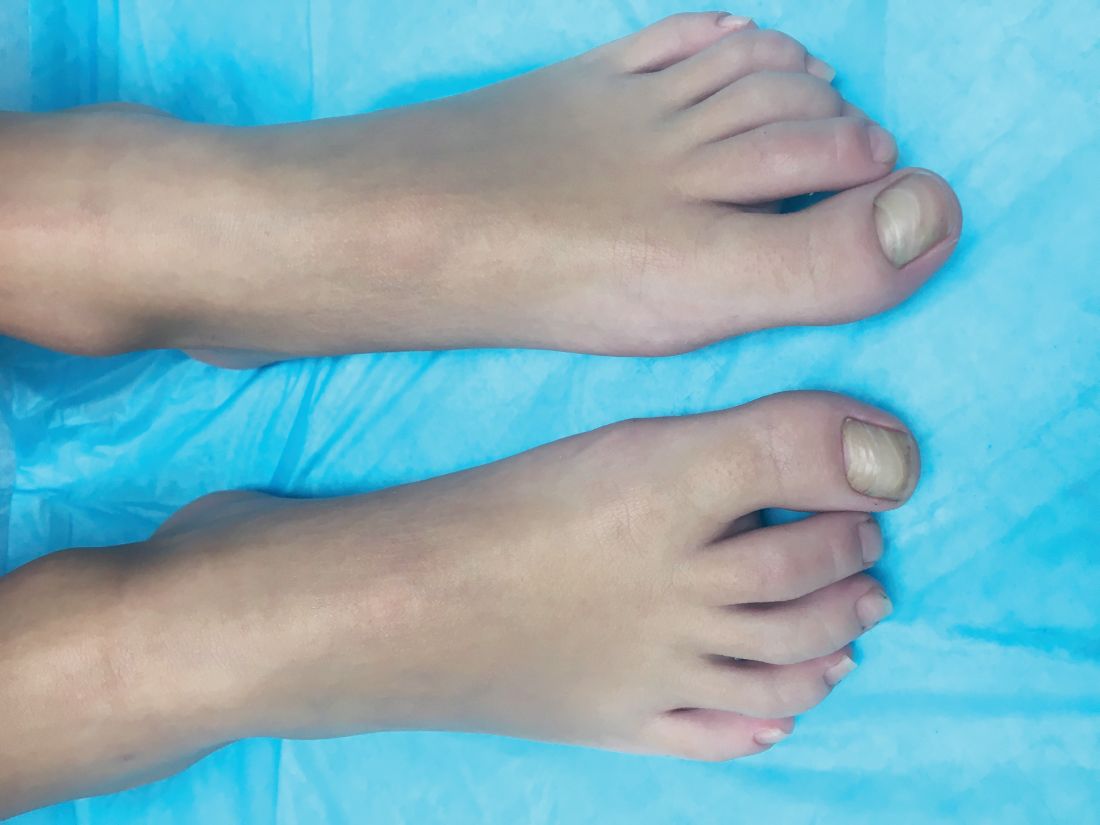
The mother reported that she started noticing the toenail changes at around 8 months of age, and it has been progressively getting worse.
He has been treated with several courses of topical antifungals and 3 months of oral terbinafine without success.
A fungal culture done 1 year prior showed slight growth of Cladosporium Sp., but the nails failed to improve after systemic therapy. He denied any associated pain or inflammation. He likes playing softball and plays soccer sometimes. The mother is very worried because the father also has a history of onychomycosis that he has not been able to clear for years.
On physical exam, he is a very pleasant young boy. His cutaneous exam is normal including hair and teeth except for thickening of the bilateral first toenails associated with transverse ridging and yellow discoloration.
December 2018
. White individuals over aged 50 years are more frequently affected. Both genders are equally affected, and 25% of cases occur on the covered areas (trunk or extremities) of younger patients. Clinically, lesions present as pink to red plaques or nodules that exhibit rapid growth. Ulceration or crusting may be present. Causes of AFX include ultraviolet radiation and ionizing radiation. AFX is considered a superficial variant of malignant fibrous histiocytoma (MFH), the most common soft tissue sarcoma of adults. Clinically, MFH involves deeper tissues than does AFX, often on the thighs or buttocks. MFH is a more aggressive malignancy that regularly metastasizes.
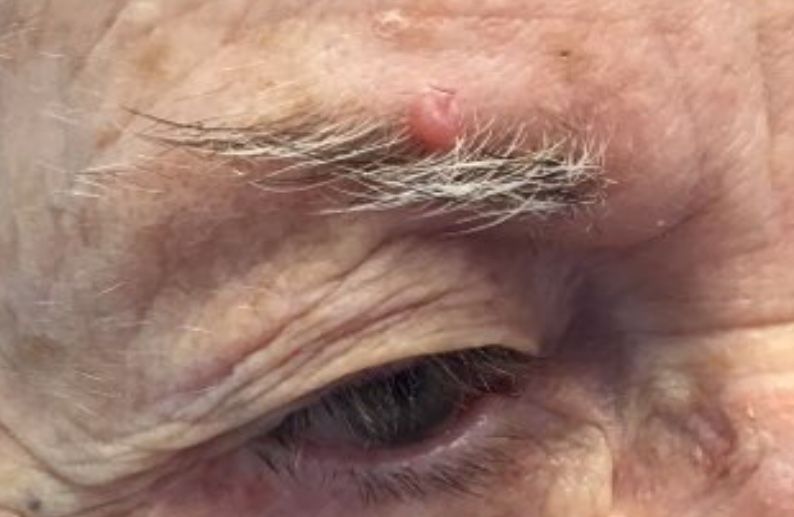
Histologically, the tumor occurs as a dermal proliferation of “bizarre” spindle cells, epithelioid cells, and atypical histiocytes. Vesicular changes may be present in the nucleus or cytoplasm of the spindle cells. Mitotic figures are present. Multinucleated giant cells may be present. Solar elastosis is often seen, as well. Vimentin and histiocyte stains are positive. Unlike melanoma, S-100 staining is minimal. Unlike squamous cell carcinoma, prekeratin staining is negative. CD34 is negative. AFX resembles MFH histologically.
Surgical excision by the Mohs procedure is preferred over wide excision as there is a risk of recurrence. AFX rarely metastasizes. This is more likely if inadequately excised or the patient is immunosuppressed. Sun protective practices, such as applying and reapplying sunscreen regularly, wearing sun protective clothing, and avoiding excessive UV exposure during peak hours is recommended.
This case and photo were submitted by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at edermatologynews.com. To submit a case for possible publication, send an email to [email protected].
. White individuals over aged 50 years are more frequently affected. Both genders are equally affected, and 25% of cases occur on the covered areas (trunk or extremities) of younger patients. Clinically, lesions present as pink to red plaques or nodules that exhibit rapid growth. Ulceration or crusting may be present. Causes of AFX include ultraviolet radiation and ionizing radiation. AFX is considered a superficial variant of malignant fibrous histiocytoma (MFH), the most common soft tissue sarcoma of adults. Clinically, MFH involves deeper tissues than does AFX, often on the thighs or buttocks. MFH is a more aggressive malignancy that regularly metastasizes.
Histologically, the tumor occurs as a dermal proliferation of “bizarre” spindle cells, epithelioid cells, and atypical histiocytes. Vesicular changes may be present in the nucleus or cytoplasm of the spindle cells. Mitotic figures are present. Multinucleated giant cells may be present. Solar elastosis is often seen, as well. Vimentin and histiocyte stains are positive. Unlike melanoma, S-100 staining is minimal. Unlike squamous cell carcinoma, prekeratin staining is negative. CD34 is negative. AFX resembles MFH histologically.
Surgical excision by the Mohs procedure is preferred over wide excision as there is a risk of recurrence. AFX rarely metastasizes. This is more likely if inadequately excised or the patient is immunosuppressed. Sun protective practices, such as applying and reapplying sunscreen regularly, wearing sun protective clothing, and avoiding excessive UV exposure during peak hours is recommended.
This case and photo were submitted by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at edermatologynews.com. To submit a case for possible publication, send an email to [email protected].
. White individuals over aged 50 years are more frequently affected. Both genders are equally affected, and 25% of cases occur on the covered areas (trunk or extremities) of younger patients. Clinically, lesions present as pink to red plaques or nodules that exhibit rapid growth. Ulceration or crusting may be present. Causes of AFX include ultraviolet radiation and ionizing radiation. AFX is considered a superficial variant of malignant fibrous histiocytoma (MFH), the most common soft tissue sarcoma of adults. Clinically, MFH involves deeper tissues than does AFX, often on the thighs or buttocks. MFH is a more aggressive malignancy that regularly metastasizes.
Histologically, the tumor occurs as a dermal proliferation of “bizarre” spindle cells, epithelioid cells, and atypical histiocytes. Vesicular changes may be present in the nucleus or cytoplasm of the spindle cells. Mitotic figures are present. Multinucleated giant cells may be present. Solar elastosis is often seen, as well. Vimentin and histiocyte stains are positive. Unlike melanoma, S-100 staining is minimal. Unlike squamous cell carcinoma, prekeratin staining is negative. CD34 is negative. AFX resembles MFH histologically.
Surgical excision by the Mohs procedure is preferred over wide excision as there is a risk of recurrence. AFX rarely metastasizes. This is more likely if inadequately excised or the patient is immunosuppressed. Sun protective practices, such as applying and reapplying sunscreen regularly, wearing sun protective clothing, and avoiding excessive UV exposure during peak hours is recommended.
This case and photo were submitted by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at edermatologynews.com. To submit a case for possible publication, send an email to [email protected].
November 2018
Desmoplastic trichilemmoma
that presents as a solitary, skin colored lesion on the midface. Lesions may appear smooth or verrucous. Lesions may occur alongside trichoepitheliomas. They may also occur on genital skin and resemble condyloma acuminata.
Histopathology reveals downward lobular growth of the epidermis. Keratinocytes are clear secondary to periodic acid-Schiff (PAS)–positive glycogen in the cells. In desmoplastic trichilemmoma, small clusters of cells are arranged in an infiltrative pattern that resembles invasive carcinoma. Often, the desmoplastic areas are surrounded by benign-appearing trichilemmomas, which helps to make the diagnosis. Desmoplastic trichilemmomas can also occur within nevus sebaceous. As trichilemmoma is a benign growth; no treatment is needed. However, if further removal is desired, electrodesiccation, cryotherapy, shave removal, or excision are treatment options. Rarely seen, the malignant counterpart to trichilemmomas is a trichilemmal carcinoma, which requires surgical excision or Mohs.
The appearance of multiple trichilemmomas is a marker for Cowden syndrome. Cowden syndrome is a rare autosomal dominant disorder in which there is a mutation in a tumor-suppressor gene called PTEN. Patients may have oral mucosal papillomas, sclerotic fibromas, acral keratotic papules, and are at risk for the development of adenocarcinoma of the breast, gastrointestinal tract, and thyroid.
Trichoepithelioma is a benign neoplasm derived from follicular germ cells that presents as a skin-colored papule on the midface, especially the nose. Multiple trichoepitheliomas are a marker for Brooke-Spiegler syndrome. A desmoplastic trichoepithelioma is a variant that has stromal sclerosis on pathology. It is a benign lesion, although may be difficult to differentiate from sclerosing basal cell or microcystic adnexal carcinoma.
Angiofibroma, or fibrous papule, is a commonly seen, benign, skin-colored papule also often occurring on the nose. They can be treated for cosmetic purposes. Multiple lesions are associated with tuberous sclerosis.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at edermatologynews.com. To submit a case for possible publication, send an email to [email protected].
Desmoplastic trichilemmoma
that presents as a solitary, skin colored lesion on the midface. Lesions may appear smooth or verrucous. Lesions may occur alongside trichoepitheliomas. They may also occur on genital skin and resemble condyloma acuminata.
Histopathology reveals downward lobular growth of the epidermis. Keratinocytes are clear secondary to periodic acid-Schiff (PAS)–positive glycogen in the cells. In desmoplastic trichilemmoma, small clusters of cells are arranged in an infiltrative pattern that resembles invasive carcinoma. Often, the desmoplastic areas are surrounded by benign-appearing trichilemmomas, which helps to make the diagnosis. Desmoplastic trichilemmomas can also occur within nevus sebaceous. As trichilemmoma is a benign growth; no treatment is needed. However, if further removal is desired, electrodesiccation, cryotherapy, shave removal, or excision are treatment options. Rarely seen, the malignant counterpart to trichilemmomas is a trichilemmal carcinoma, which requires surgical excision or Mohs.
The appearance of multiple trichilemmomas is a marker for Cowden syndrome. Cowden syndrome is a rare autosomal dominant disorder in which there is a mutation in a tumor-suppressor gene called PTEN. Patients may have oral mucosal papillomas, sclerotic fibromas, acral keratotic papules, and are at risk for the development of adenocarcinoma of the breast, gastrointestinal tract, and thyroid.
Trichoepithelioma is a benign neoplasm derived from follicular germ cells that presents as a skin-colored papule on the midface, especially the nose. Multiple trichoepitheliomas are a marker for Brooke-Spiegler syndrome. A desmoplastic trichoepithelioma is a variant that has stromal sclerosis on pathology. It is a benign lesion, although may be difficult to differentiate from sclerosing basal cell or microcystic adnexal carcinoma.
Angiofibroma, or fibrous papule, is a commonly seen, benign, skin-colored papule also often occurring on the nose. They can be treated for cosmetic purposes. Multiple lesions are associated with tuberous sclerosis.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at edermatologynews.com. To submit a case for possible publication, send an email to [email protected].
Desmoplastic trichilemmoma
that presents as a solitary, skin colored lesion on the midface. Lesions may appear smooth or verrucous. Lesions may occur alongside trichoepitheliomas. They may also occur on genital skin and resemble condyloma acuminata.
Histopathology reveals downward lobular growth of the epidermis. Keratinocytes are clear secondary to periodic acid-Schiff (PAS)–positive glycogen in the cells. In desmoplastic trichilemmoma, small clusters of cells are arranged in an infiltrative pattern that resembles invasive carcinoma. Often, the desmoplastic areas are surrounded by benign-appearing trichilemmomas, which helps to make the diagnosis. Desmoplastic trichilemmomas can also occur within nevus sebaceous. As trichilemmoma is a benign growth; no treatment is needed. However, if further removal is desired, electrodesiccation, cryotherapy, shave removal, or excision are treatment options. Rarely seen, the malignant counterpart to trichilemmomas is a trichilemmal carcinoma, which requires surgical excision or Mohs.
The appearance of multiple trichilemmomas is a marker for Cowden syndrome. Cowden syndrome is a rare autosomal dominant disorder in which there is a mutation in a tumor-suppressor gene called PTEN. Patients may have oral mucosal papillomas, sclerotic fibromas, acral keratotic papules, and are at risk for the development of adenocarcinoma of the breast, gastrointestinal tract, and thyroid.
Trichoepithelioma is a benign neoplasm derived from follicular germ cells that presents as a skin-colored papule on the midface, especially the nose. Multiple trichoepitheliomas are a marker for Brooke-Spiegler syndrome. A desmoplastic trichoepithelioma is a variant that has stromal sclerosis on pathology. It is a benign lesion, although may be difficult to differentiate from sclerosing basal cell or microcystic adnexal carcinoma.
Angiofibroma, or fibrous papule, is a commonly seen, benign, skin-colored papule also often occurring on the nose. They can be treated for cosmetic purposes. Multiple lesions are associated with tuberous sclerosis.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at edermatologynews.com. To submit a case for possible publication, send an email to [email protected].
What is your diagnosis?
Lichen nitidus, which literally means “shiny moss,” is a relatively rare, chronic skin eruption that is characterized clinically by asymptomatic, flat-topped, sharply-demarcated, skin-colored papules, which are sometimes described as being “pinpoint.”
Lichen nitidus mainly affects children and young adults. The most common sites of involvement are the trunk, flexor aspects of upper extremities, dorsal aspects of hands, and genitalia, but lesions can occur anywhere on the skin. The lesions also can develop in sites of trauma (Koebner phenomenon), and this can be a significant clinical clue to aid in the diagnosis of lichen nitidus in favor of other conditions which may present as many small papules.1 Nail changes can occur but are rare, presenting as dystrophy, pitting, riding, or loss of nail(s).2
Lichen nitidus can be a challenging diagnosis to make, especially if a practitioner is not used to seeing it. Many dermatologic conditions present with many fine papules, including the other answer choices in the given quiz question (molluscum contagiosum, keratosis pilaris, verruca vulgaris, papular eczema). What allows for a clearer diagnosis of lichen nitidus is the history provided by the patient as well as the exam. Lichen nitidus lesions typically arise without a known trigger and often persist for months while remaining asymptomatic.
Molluscum contagiosum tends to include papules that are larger and more substantial than lichen nitidus papules and may be accompanied by background hyperpigmentation or erythema, known as the “beginning of the end” sign. Keratosis pilaris is commonly thought of as a skin type more so than a skin condition, and is more commonly seen in fair-skinned individuals along the lateral arms and cheeks. It is commonly paired with a background of erythema and skin than tends to be more xerotic. Verruca are typically larger lesions, sometimes with a rough surface, and are not typically shiny. Verruca are more likely to present as a single lesion or a few lesions at a given location, as opposed to lichen nitidus which has many individual papules at a single location. Papular eczema typically is intensely pruritic and is associated with xerosis and atopy.
The cause of lichen nitidus is unknown, and there are no reported genetic factors that contribute to its presentation.3 It is thought to be a subtype of lichen planus, although this is still debated. There is more work that needs to be done to find answers to these questions and to assess what triggers these fine papules to present and in whom.
The dermatoscopic features of lichen nitidus were reported in a series of eight cases and include absent dermatoglyphics, radial ridges, ill-defined hypopigmentation, diffuse erythema, linear vessels within the lesion, and peripheral scaling.4 These features are distinctive in combination and can in some cases be used to clinically diagnose lichen nitidus without the need for a skin biopsy, which is an invasive procedure that should be avoided when possible, especially given the benign nature of lichen nitidus.
If a biopsy is performed, the histologic features that commonly are seen include well-circumscribed granuloma-like lymphohistiocytic infiltrates in the papillary dermis adjacent to ridges, mimicking a “ball-and-claw” formation.1,5
Lichen nitidus generally is self-limiting, with minimal cosmetic disruption; therefore, treatment usually is not necessary. The lesions typically resolve within 1 year of presentation – and often sooner. Topical steroids can be used for symptomatic relief of pruritus, but generally do not hasten resolution of the papules themselves. Additionally, there have been reports in the literature of the successful resolution of lesions using topical calcineurin inhibitors and UVB therapy.
In a case report of an 8-year-old child with histologically confirmed lichen nitidus that had been present for 2 years, pimecrolimus 1% cream was used twice daily for 2 months with improvement and flattening of the papules.6 This report is compelling because the lesions persisted for twice the expected time of resolution without improvement and then showed relatively quick response to pimecrolimus. In a case of a 32-year-old male with lichen nitidus on his penis, tacrolimus 0.1% was used for 4 weeks with resolution of the papules.7 Although given that lichen nitidus can self-resolve in this same time period, it is unclear in this case whether the tacrolimus was the independent cause of the resolution.
With regard to UVB therapy, there have been reports of lichen nitidus resolution after 17-30 irradiation sessions in patients with lesions present for 3-6 months, although again, it is possible that the resolution observed was simply the natural course of the lichen nitidus for these patients rather than a therapeutic benefit of UVB therapy.8,9
Given that lichen nitidus is benign and typically asymptomatic or with mild pruritus, it is reasonable to monitor the lesions without treating them. If the lesions persist beyond 1 year, it also is reasonable to trial therapies, such as topical calcineurin inhibitors and UVB therapy, although using these treatments earlier in the disease course has only limited supporting data and any improvement seen within 1 year of onset may be attributed to the natural disease course as opposed to an effect of the intervention. Considerations of the cost of therapy, as well as the degree to which the patient is bothered by the lesions and how long the lesions have persisted, should be undertaken when considering whether an intervention should be made.
Ms. Natsis is a medical student at the University of California, San Diego. Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital–San Diego. He is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego. There are no conflicts of interest or financial disclosures for Ms. Natsis or Dr. Eichenfield. Email them at [email protected].
References
1. Cutis. 1999 Aug 1;64(2):135-6.
2. J Am Acad Dermatol. 2004 Oct;51(4):606-24.
3. Papulosquamous diseases, in “Pediatric Dermatology,” 4th ed. (St Louis: Mosby; 2011, Vol. 2.
4. Pediatr Dermatol. 2018. doi: 10.1111/pde.13576.
5. Cutis. 2013 Dec;92(6):288, 297-8.
6. Dermatol Online J. 2011 Jul 15;17(7):11.
7. J Drugs Dermatol. 2004 Nov-Dec;3(6):683-4.
8. Int J Dermatol. 2004 Dec 23;45:615-7.
9. Photodermatol Photoimmunol Photomed. 2013 Aug;29(4):215-7.
Lichen nitidus, which literally means “shiny moss,” is a relatively rare, chronic skin eruption that is characterized clinically by asymptomatic, flat-topped, sharply-demarcated, skin-colored papules, which are sometimes described as being “pinpoint.”
Lichen nitidus mainly affects children and young adults. The most common sites of involvement are the trunk, flexor aspects of upper extremities, dorsal aspects of hands, and genitalia, but lesions can occur anywhere on the skin. The lesions also can develop in sites of trauma (Koebner phenomenon), and this can be a significant clinical clue to aid in the diagnosis of lichen nitidus in favor of other conditions which may present as many small papules.1 Nail changes can occur but are rare, presenting as dystrophy, pitting, riding, or loss of nail(s).2
Lichen nitidus can be a challenging diagnosis to make, especially if a practitioner is not used to seeing it. Many dermatologic conditions present with many fine papules, including the other answer choices in the given quiz question (molluscum contagiosum, keratosis pilaris, verruca vulgaris, papular eczema). What allows for a clearer diagnosis of lichen nitidus is the history provided by the patient as well as the exam. Lichen nitidus lesions typically arise without a known trigger and often persist for months while remaining asymptomatic.
Molluscum contagiosum tends to include papules that are larger and more substantial than lichen nitidus papules and may be accompanied by background hyperpigmentation or erythema, known as the “beginning of the end” sign. Keratosis pilaris is commonly thought of as a skin type more so than a skin condition, and is more commonly seen in fair-skinned individuals along the lateral arms and cheeks. It is commonly paired with a background of erythema and skin than tends to be more xerotic. Verruca are typically larger lesions, sometimes with a rough surface, and are not typically shiny. Verruca are more likely to present as a single lesion or a few lesions at a given location, as opposed to lichen nitidus which has many individual papules at a single location. Papular eczema typically is intensely pruritic and is associated with xerosis and atopy.
The cause of lichen nitidus is unknown, and there are no reported genetic factors that contribute to its presentation.3 It is thought to be a subtype of lichen planus, although this is still debated. There is more work that needs to be done to find answers to these questions and to assess what triggers these fine papules to present and in whom.
The dermatoscopic features of lichen nitidus were reported in a series of eight cases and include absent dermatoglyphics, radial ridges, ill-defined hypopigmentation, diffuse erythema, linear vessels within the lesion, and peripheral scaling.4 These features are distinctive in combination and can in some cases be used to clinically diagnose lichen nitidus without the need for a skin biopsy, which is an invasive procedure that should be avoided when possible, especially given the benign nature of lichen nitidus.
If a biopsy is performed, the histologic features that commonly are seen include well-circumscribed granuloma-like lymphohistiocytic infiltrates in the papillary dermis adjacent to ridges, mimicking a “ball-and-claw” formation.1,5
Lichen nitidus generally is self-limiting, with minimal cosmetic disruption; therefore, treatment usually is not necessary. The lesions typically resolve within 1 year of presentation – and often sooner. Topical steroids can be used for symptomatic relief of pruritus, but generally do not hasten resolution of the papules themselves. Additionally, there have been reports in the literature of the successful resolution of lesions using topical calcineurin inhibitors and UVB therapy.
In a case report of an 8-year-old child with histologically confirmed lichen nitidus that had been present for 2 years, pimecrolimus 1% cream was used twice daily for 2 months with improvement and flattening of the papules.6 This report is compelling because the lesions persisted for twice the expected time of resolution without improvement and then showed relatively quick response to pimecrolimus. In a case of a 32-year-old male with lichen nitidus on his penis, tacrolimus 0.1% was used for 4 weeks with resolution of the papules.7 Although given that lichen nitidus can self-resolve in this same time period, it is unclear in this case whether the tacrolimus was the independent cause of the resolution.
With regard to UVB therapy, there have been reports of lichen nitidus resolution after 17-30 irradiation sessions in patients with lesions present for 3-6 months, although again, it is possible that the resolution observed was simply the natural course of the lichen nitidus for these patients rather than a therapeutic benefit of UVB therapy.8,9
Given that lichen nitidus is benign and typically asymptomatic or with mild pruritus, it is reasonable to monitor the lesions without treating them. If the lesions persist beyond 1 year, it also is reasonable to trial therapies, such as topical calcineurin inhibitors and UVB therapy, although using these treatments earlier in the disease course has only limited supporting data and any improvement seen within 1 year of onset may be attributed to the natural disease course as opposed to an effect of the intervention. Considerations of the cost of therapy, as well as the degree to which the patient is bothered by the lesions and how long the lesions have persisted, should be undertaken when considering whether an intervention should be made.
Ms. Natsis is a medical student at the University of California, San Diego. Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital–San Diego. He is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego. There are no conflicts of interest or financial disclosures for Ms. Natsis or Dr. Eichenfield. Email them at [email protected].
References
1. Cutis. 1999 Aug 1;64(2):135-6.
2. J Am Acad Dermatol. 2004 Oct;51(4):606-24.
3. Papulosquamous diseases, in “Pediatric Dermatology,” 4th ed. (St Louis: Mosby; 2011, Vol. 2.
4. Pediatr Dermatol. 2018. doi: 10.1111/pde.13576.
5. Cutis. 2013 Dec;92(6):288, 297-8.
6. Dermatol Online J. 2011 Jul 15;17(7):11.
7. J Drugs Dermatol. 2004 Nov-Dec;3(6):683-4.
8. Int J Dermatol. 2004 Dec 23;45:615-7.
9. Photodermatol Photoimmunol Photomed. 2013 Aug;29(4):215-7.
Lichen nitidus, which literally means “shiny moss,” is a relatively rare, chronic skin eruption that is characterized clinically by asymptomatic, flat-topped, sharply-demarcated, skin-colored papules, which are sometimes described as being “pinpoint.”
Lichen nitidus mainly affects children and young adults. The most common sites of involvement are the trunk, flexor aspects of upper extremities, dorsal aspects of hands, and genitalia, but lesions can occur anywhere on the skin. The lesions also can develop in sites of trauma (Koebner phenomenon), and this can be a significant clinical clue to aid in the diagnosis of lichen nitidus in favor of other conditions which may present as many small papules.1 Nail changes can occur but are rare, presenting as dystrophy, pitting, riding, or loss of nail(s).2
Lichen nitidus can be a challenging diagnosis to make, especially if a practitioner is not used to seeing it. Many dermatologic conditions present with many fine papules, including the other answer choices in the given quiz question (molluscum contagiosum, keratosis pilaris, verruca vulgaris, papular eczema). What allows for a clearer diagnosis of lichen nitidus is the history provided by the patient as well as the exam. Lichen nitidus lesions typically arise without a known trigger and often persist for months while remaining asymptomatic.
Molluscum contagiosum tends to include papules that are larger and more substantial than lichen nitidus papules and may be accompanied by background hyperpigmentation or erythema, known as the “beginning of the end” sign. Keratosis pilaris is commonly thought of as a skin type more so than a skin condition, and is more commonly seen in fair-skinned individuals along the lateral arms and cheeks. It is commonly paired with a background of erythema and skin than tends to be more xerotic. Verruca are typically larger lesions, sometimes with a rough surface, and are not typically shiny. Verruca are more likely to present as a single lesion or a few lesions at a given location, as opposed to lichen nitidus which has many individual papules at a single location. Papular eczema typically is intensely pruritic and is associated with xerosis and atopy.
The cause of lichen nitidus is unknown, and there are no reported genetic factors that contribute to its presentation.3 It is thought to be a subtype of lichen planus, although this is still debated. There is more work that needs to be done to find answers to these questions and to assess what triggers these fine papules to present and in whom.
The dermatoscopic features of lichen nitidus were reported in a series of eight cases and include absent dermatoglyphics, radial ridges, ill-defined hypopigmentation, diffuse erythema, linear vessels within the lesion, and peripheral scaling.4 These features are distinctive in combination and can in some cases be used to clinically diagnose lichen nitidus without the need for a skin biopsy, which is an invasive procedure that should be avoided when possible, especially given the benign nature of lichen nitidus.
If a biopsy is performed, the histologic features that commonly are seen include well-circumscribed granuloma-like lymphohistiocytic infiltrates in the papillary dermis adjacent to ridges, mimicking a “ball-and-claw” formation.1,5
Lichen nitidus generally is self-limiting, with minimal cosmetic disruption; therefore, treatment usually is not necessary. The lesions typically resolve within 1 year of presentation – and often sooner. Topical steroids can be used for symptomatic relief of pruritus, but generally do not hasten resolution of the papules themselves. Additionally, there have been reports in the literature of the successful resolution of lesions using topical calcineurin inhibitors and UVB therapy.
In a case report of an 8-year-old child with histologically confirmed lichen nitidus that had been present for 2 years, pimecrolimus 1% cream was used twice daily for 2 months with improvement and flattening of the papules.6 This report is compelling because the lesions persisted for twice the expected time of resolution without improvement and then showed relatively quick response to pimecrolimus. In a case of a 32-year-old male with lichen nitidus on his penis, tacrolimus 0.1% was used for 4 weeks with resolution of the papules.7 Although given that lichen nitidus can self-resolve in this same time period, it is unclear in this case whether the tacrolimus was the independent cause of the resolution.
With regard to UVB therapy, there have been reports of lichen nitidus resolution after 17-30 irradiation sessions in patients with lesions present for 3-6 months, although again, it is possible that the resolution observed was simply the natural course of the lichen nitidus for these patients rather than a therapeutic benefit of UVB therapy.8,9
Given that lichen nitidus is benign and typically asymptomatic or with mild pruritus, it is reasonable to monitor the lesions without treating them. If the lesions persist beyond 1 year, it also is reasonable to trial therapies, such as topical calcineurin inhibitors and UVB therapy, although using these treatments earlier in the disease course has only limited supporting data and any improvement seen within 1 year of onset may be attributed to the natural disease course as opposed to an effect of the intervention. Considerations of the cost of therapy, as well as the degree to which the patient is bothered by the lesions and how long the lesions have persisted, should be undertaken when considering whether an intervention should be made.
Ms. Natsis is a medical student at the University of California, San Diego. Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital–San Diego. He is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego. There are no conflicts of interest or financial disclosures for Ms. Natsis or Dr. Eichenfield. Email them at [email protected].
References
1. Cutis. 1999 Aug 1;64(2):135-6.
2. J Am Acad Dermatol. 2004 Oct;51(4):606-24.
3. Papulosquamous diseases, in “Pediatric Dermatology,” 4th ed. (St Louis: Mosby; 2011, Vol. 2.
4. Pediatr Dermatol. 2018. doi: 10.1111/pde.13576.
5. Cutis. 2013 Dec;92(6):288, 297-8.
6. Dermatol Online J. 2011 Jul 15;17(7):11.
7. J Drugs Dermatol. 2004 Nov-Dec;3(6):683-4.
8. Int J Dermatol. 2004 Dec 23;45:615-7.
9. Photodermatol Photoimmunol Photomed. 2013 Aug;29(4):215-7.