User login
Advances in the treatment of dyslipidemia
The 2013 joint guidelines of the American College of Cardiology and American Heart Association (ACC/AHA)1 on the treatment of blood cholesterol to reduce cardiovascular risk recommend high-intensity statin therapy for secondary prevention of cardiovascular events. The question of primary prevention is not so straightforward, and the recommended strategy has come under fire. In addition, the guidelines focus on statins and not on LDL-C levels, and the role of nonstatin lipid-lowering drugs and the value of reducing LDL-C levels to well below levels currently regarded as “normal” remain unclear.
This article comments on the 2013 ACC/AHA guidelines, reviews the data on optimal LDL-C levels, and discusses new nonstatin agents.
ACC/AHA GUIDELINES: A MIXED MESSAGE
The 2013 ACC/AHA cholesterol guidelines1 can be characterized by the title from the famous Western film “The Good, the Bad, and the Ugly.”
The good: A clear message to treat
The guidelines deliver an unambiguous message to treat patients at high risk with high-intensity statin therapy. This mandate is very helpful as it should reduce the undertreatment of patients.
The seemingly bad
Two common misconceptions regarding the guidelines:

They abandon LDL-C targets. Actually, the guidelines do not argue for or against targets; they simply remain silent, citing that randomized trials have not been conducted with LDL-C targets as specific goals. Technically, this statement is true. However, it seems contrived to argue, for example, that the benefit of atorvastatin 80 mg over 10 mg in the Treating to New Targets trial could not be reliably ascribed to the lower LDL-C achieved with the higher dose, but rather to some undefined benefit of high-intensity statin therapy, especially as the guidelines define the intensity of statins by the degree of LDL-C lowering. In fact, by correlating the incidence of coronary heart disease events with the levels of LDL-C achieved in those trials, conclusions can reasonably be drawn from such data (Figure 1).2
The guidelines do not recommend nonstatin drugs. Actually, the guidelines note that clinicians are free to consider other therapies, especially those proven to reduce the risk of cardiovascular events, a central principle of medicine. Since the guidelines were published, data have emerged indicating that the role of nonstatin drugs also needs consideration.
The ugly: Risk calculator untested
The guidelines promote the use of a risk calculator developed by the ACC/AHA to estimate the 10-year risk of an atherosclerotic event for people whose LDL-C levels are between 70 and 189 mg/dL to help decide whether to initiate statin therapy for primary prevention of atherosclerotic cardiovascular disease. Such an approach is reasonable, although the risk score was promulgated without evidence to support its utility.
Media coverage of the risk calculator was fierce. Some physicians found imperfections in the risk score (as is true for all risk scores), resulting in public mistrust of the guidelines and of the medical community as a whole. This needless controversy may have compromised the main message—that LDL-C should be lowered in many people—a message backed by strong evidence.
Alternative strategies proposed
Ridker et al3 have proposed a hybrid strategy to guide statin use for apparently healthy people that combines the ACC/AHA guideline approach with entry criteria for randomized clinical trials that showed statin efficacy for primary prevention.
Genetic analysis may offer another approach. Mega et al4 stratified more than 48,000 people by a genetic risk score based on 27 genetic variants and found a significant association with risk of coronary events. Targeting therapy to people found to be at higher risk on this basis offers greater risk reduction than expected for the general population. Biomarkers and imaging tests are other potentially useful risk determinants.
LDL-C: LOWER IS BETTER
Although no clinical trial has yet targeted specific LDL-C levels, there is plenty of evidence that lower LDL-C levels offer greater benefit (Figure 1).2
In 1994, the Scandinavian Simvastatin Survival Study5 established the benefit of statins in patients with known vascular disease. The mean LDL-C level achieved in the active treatment group was 120 mg/dL. More trials followed supporting the benefits of statins and of reducing LDL-C from average levels in the 120s down to 100 mg/dL.
In 2004, the Pravastatin or Atorvastatin Evaluation and Infection Therapy–Thrombolysis in Myocardial Infarction 22 trial6 observed an even greater risk reduction in patients with known risk by treating with statins; the mean LDL-C level achieved in the group randomized to an intensive regimen of atorvastatin 80 mg per day was 62 mg/dL. The same year, the Adult Treatment Panel III of the National Cholesterol Education Program7 issued updated guidelines including an optional goal of LDL-C less than 70 mg/dL for patients at very high risk.
In 2008, the Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin (JUPITER)8 found a significantly lower incidence of major cardiovascular events at 2 years in apparently healthy men and women with baseline LDL-C levels of less than 130 mg/dL after treatment with rosuvastatin 20 mg daily, with an achieved median LDL-C of 55 mg/dL.
How low should LDL-C go?
Evidence from clinical trials indicates a 20% to 25% reduction in the risk of cardiovascular events for every 39-mg/dL decrease in LDL-C. Extrapolating the data, cardiovascular disease risk would be reduced to zero if LDL-C were brought down below 40 mg/dL.
Brown and Goldstein,9 who won the 1985 Nobel Prize in medicine for their work in cholesterol metabolism, estimated that a plasma level of LDL-C of only 25 mg/dL would be sufficient to nourish cells with cholesterol. Cells can synthesize all the cholesterol they need, underscoring that LDL-C is simply the final end-product that the liver removes from circulation.
Other evidence that lower LDL-C does not have adverse effects comes from non-Western populations as well as from other mammals. Total cholesterol levels range in the low 100s mg/dL in Native American and African tribal populations, with LDL-C estimated to be about 50 to 75 mg/dL. Elephants, baboons, and foxes have even lower levels.10
Clinical trial data also support that LDL-C levels below the current “normal” are better. The Cholesterol Treatment Trialists’ Collaboration11 analyzed data from more than 160,000 patients in 26 trials that evaluated either more- vs less-intensive statin regimens or statin treatment vs control. No baseline level below which lowering LDL-C further was not beneficial was found. Patients who started out with an LDL-C level of less than 77 mg/dL had the same risk reduction of major vascular events when the level was dropped to 50 mg/dL as those who started at higher levels and reduced their LDL-C by the same amount. In the JUPITER trial, even those with a baseline LDL-C of less than 60 mg/dL benefited from statin therapy.12
BEYOND STATINS
Ezetimibe further lowers risk
Ezetimibe is a nonstatin drug that reduces LDL-C by about 15% to 20%. The Improved Reduction of Outcomes: Vytorin Efficacy International Trial13 registered more than 18,000 patients with a baseline LDL-C level of less than 125 mg/dL (or 100 mg/dL if already on lipid-lowering therapy) who had been stabilized shortly after an acute cardiovascular event. They were randomized to receive either simvastatin 40 mg or combined simvastatin 40 mg and ezetimibe 10 mg. The study intended to determine two things: whether ezetimibe could further lower LDL-C when combined with a statin, and whether risk could be reduced further by driving the LDL-C below 70 mg/dL and down to the mid-50s.
After 1 year, the average LDL-C level was 70 mg/dL in the simvastatin group and 53 mg/dL in the combined simvastatin and ezetimibe group. At 7 years, for the primary end point (cardiovascular death, myocardial infarction, unstable angina requiring hospitalization, coronary revascularization, or stroke), there was a 6% reduction of events in the combined drug treatment group, with the number of people needed to treat being 50 to prevent one event. For the narrower end point of cardiovascular death, nonfatal myocardial infarction, and nonfatal stroke, there was a 10% risk reduction in the combined drug treatment arm.14
The amount of risk reduction is exactly what was predicted by the Cholesterol Treatment Trialists’ Collaboration’s plot of reduction in events vs reduction in LDL-C based on the analysis of 26 trials, adding further evidence that it is the LDL-C reduction itself, rather than the means by which LDL-C is reduced, that is critical for benefit.
PCSK9 inhibitors: A new approach
Mutations in the gene for proprotein convertase subtilisin kexin type 9 (PCSK9) have become a new focus of interest for reducing LDL-C and cardiovascular risk.15 PCSK9 binds to the LDL-C receptor on the surface of hepatocytes and escorts it to its destruction in the lysosomes, rather than allowing it to return to the cell surface to take more LDL-C out of circulation.
People with a gain-of-function mutation (conferring too much PCSK9, resulting in fewer LDL-C receptors and more LDL-C in circulation) are a more recently recognized subset of those with autosomal-dominant familial hypercholesterolemia. They have total cholesterol levels in the 90th percentile, tendon xanthomas, and a high risk of myocardial infarction and stroke at a young age.
Conversely, those with a nonsense mutation in PCSK9—leading to loss of function—have a 28% reduction in mean LDL-C and 88% reduction in risk of coronary heart disease compared with those without the mutation.16 Two women (ages 32 and 21, fertile) have been found who have inactivating mutations in both PCSK9 alleles, and both are in apparent good health, with LDL-C levels of 14 mg/dL and 15 mg/dL, respectively.17,18
Dramatic reduction in LDL-C
Monoclonal antibodies have been developed that bind PCSK9 and block its action with the goal of developing new LDL-C–lowering treatments. Phase 2 clinical trials of varying doses of evolocumab (Repatha), a drug in this class, combined with standard therapy (a statin with or without ezetimibe), found a 66% reduction of LDL-C at high doses at 12 weeks compared with standard therapy alone, with concomitant reductions in other atherogenic lipoproteins.19 Patients who could not tolerate statins because of myalgia responded well to evolocumab.20
Patients with heterozygous familial hypercholesterolemia also had a substantial reduction in LDL-C (55% at the highest dosage), even though they have fewer LDL-C receptors for the drug to act upon.21 People with homozygous familial hypercholesterolemia and no LDL-C receptors had a lesser relative reduction in LDL-C that depended on the type of mutations they had. Nonetheless, given how high LDL-C levels are in this population, the absolute decreases in LDL-C level were quite impressive.
Cardiovascular risk reduced
Data at nearly 1 year showed continued reduction of LDL-C by about 60% (absolute reduction: 73 mg/dL), as well as a lower incidence of cardiovascular events starting at just 3 months, much sooner than observed in some statin trials.22 Benefits were found regardless of subgroup (sex, age, statin use, baseline LDL-C level, or known vascular disease). No difference was found in the safety profile between the evolocumab and control arms. Only 2.4% of participants discontinued evolocumab because of adverse events, and the incidence of adverse effects did not correlate with LDL-C level achieved.
Neurocognitive effects occurred in 0.9% of the evolocumab arm vs 0.3% in the control arm. This difference has not been explained: although there is cholesterol in the central nervous system, it is generated locally, and lipoproteins—and evolocumab—are not thought to cross the blood-brain barrier.
Long-term trials of evolocumab are currently under way for patients with cardiovascular disease, as are trials of two other PCSK9 inhibitors, alirocumab and bococizumab, in addition to standard statin therapy.
On July 24, 2015, the US Food and Drug Administration (FDA) approved the first PCSK9 inhibitor, alirocumab (Praluent) for patients with heterozygous familial hypercholesterolemia or those with clinical atherosclerotic cardiovascular disease who require additional lowering of LDL-C. The starting dosage is 75 mg subcutaneously every 2 weeks, which can be increased up to 150 mg every 2 weeks.
Evolocumab was approved by the FDA on August 27, 2015, for the same indications. The dosage is 140 mg subcutaneously every 2 weeks or 420 mg every month.
- Stone NJ, Robinson JG, Lichtenstein AH, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation 2014; 129:S1-S45. Erratum in: Circulation 2014; 129:S46–S48.
- Raymond C, Cho L, Rocco M, Hazen SL. New cholesterol guidelines: worth the wait? Cleve Clin J Med 2014; 81:11–19.
- Ridker PM, Rose L, Cook NR. A proposal to incorporate trial data into a hybrid ACC/AHA algorithm for the allocation of statin therapy in primary prevention. J Am Coll Cardiol 2015; 65:942–948.
- Mega JL, Stitziel NO, Smith JG, et al. Genetic risk, coronary heart disease events, and the clinical benefit of statin therapy: an analysis of primary and secondary prevention trials. Lancet 2015; 385:2264–2271.
- Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S). Lancet 1994; 344:1383–1389.
- Cannon CP, Braunwald E, McCabe CH, et al; Pravastatin or Atorvastatin Evaluation and Infection Therapy-Thrombolysis in Myocardial Infarction 22 Investigators. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med 2004; 350:1495–1504.
- Grundy SM, Cleeman JI, Merz CN, et al; National Heart, Lung, and Blood Institute; American College of Cardiology Foundation; American Heart Association. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines. Circulation 2004; 110:227–239. Erratum in Circulation 2004; 110:763.
- Ridker PM, Danielson E, Fonseca FAH, et al; JUPITER Study Group. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med 2008; 359:2195–2207.
- Brown MS, Goldstein JL. A receptor-mediated pathway for cholesterol homeostasis. Science 1986; 232:34–47.
- Hochholzer W, Giugliano RP. Lipid lowering goals: back to nature? Ther Adv Cardiovasc Dis 2010; 4:185–191.
- Cholesterol Treatment Trialists’ (CTT) Collaboration, Baigent C, Blackwell L, Emberson J, et al. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet 2010; 376:1670–1681.
- Hsia J, MacFadyen JG, Monyak J, Ridker PM. Cardiovascular event reduction and adverse events among subjects attaining low-density lipoprotein cholesterol <50 mg/dl with rosuvastatin. The JUPITER trial (Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin). J Am Coll Cardiol 2011; 57:1666–1675.
- Cannon CP, Giugliano RP, Blazing MA, et al; IMPROVE-IT Investigators. Rationale and design of IMPROVE-IT (IMProved Reduction of Outcomes: Vytorin Efficacy International Trial): comparison of ezetimibe/simvastatin versus simvastatin monotherapy on cardiovascular outcomes in patients with acute coronary syndromes. Am Heart J 2008; 156:826–832.
- Cannon CP, Blazing MA, Giugliano RP, et al for the IMPROVE-IT Investigators. Ezetimibe added to statin therapy after acute coronary syndromes. N Engl J Med 2015; 372:2387–2397.
- Giugliano RP, Sabatine MS. Are PCSK9 Inhibitors the next breakthrough in the cardiovascular field? J Am Coll Cardiol 2015; 65:2638–2651.
- Cohen JC, Boerwinkle E, Mosley TH Jr, Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med 2006; 354:1264–1272.
- Zhao Z, Tuakli-Wosornu Y, Lagace TA, et al. Molecular characterization of loss-of-function mutations in PCSK9 and identification of a compound heterozygote. Am J Hum Genet 2006; 79:514-523.
- Hooper AJ, Marais AD, Tanyanyiwa DM, Burnett JR. The C679X mutation in PCSK9 is present and lowers blood cholesterol in a Southern African population. Atherosclerosis 2007; 193:445–448.
- Giugliano RP, Desai NR, Kohli P, et al; LAPLACE-TIMI 57 Investigators. Efficacy, safety, and tolerability of a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 in combination with a statin in patients with hypercholesterolaemia (LAPLACE-TIMI 57): a randomised, placebo-controlled, dose-ranging, phase 2 study. Lancet 2012; 380:2007–2017.
- Sullivan D, Olsson AG, Scott R, et al. Effect of a monoclonal antibody to PCSK9 on low-density lipoprotein cholesterol levels in statin-intolerant patients: the GAUSS randomized trial. JAMA 2012; 308:2497–2506.
- Raal F, Scott R, Somaratne R, et al. Low-density lipoprotein cholesterol-lowering effects of AMG 145, a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 serine protease in patients with heterozygous familial hypercholesterolemia: the Reduction of LDL-C with PCSK9 Inhibition in Heterozygous Familial Hypercholesterolemia Disorder (RUTHERFORD) randomized trial. Circulation 2012; 126:2408–2417.
- Sabatine MS, Giugliano RP, Wiviott SD, et al; Open-Label Study of Long-Term Evaluation against LDL Cholesterol (OSLER) Investigators. Efficacy and safety of evolocumab in reducing lipids and cardiovascular events. N Engl J Med 2015; 372:1500–1509.
The 2013 joint guidelines of the American College of Cardiology and American Heart Association (ACC/AHA)1 on the treatment of blood cholesterol to reduce cardiovascular risk recommend high-intensity statin therapy for secondary prevention of cardiovascular events. The question of primary prevention is not so straightforward, and the recommended strategy has come under fire. In addition, the guidelines focus on statins and not on LDL-C levels, and the role of nonstatin lipid-lowering drugs and the value of reducing LDL-C levels to well below levels currently regarded as “normal” remain unclear.
This article comments on the 2013 ACC/AHA guidelines, reviews the data on optimal LDL-C levels, and discusses new nonstatin agents.
ACC/AHA GUIDELINES: A MIXED MESSAGE
The 2013 ACC/AHA cholesterol guidelines1 can be characterized by the title from the famous Western film “The Good, the Bad, and the Ugly.”
The good: A clear message to treat
The guidelines deliver an unambiguous message to treat patients at high risk with high-intensity statin therapy. This mandate is very helpful as it should reduce the undertreatment of patients.
The seemingly bad
Two common misconceptions regarding the guidelines:
They abandon LDL-C targets. Actually, the guidelines do not argue for or against targets; they simply remain silent, citing that randomized trials have not been conducted with LDL-C targets as specific goals. Technically, this statement is true. However, it seems contrived to argue, for example, that the benefit of atorvastatin 80 mg over 10 mg in the Treating to New Targets trial could not be reliably ascribed to the lower LDL-C achieved with the higher dose, but rather to some undefined benefit of high-intensity statin therapy, especially as the guidelines define the intensity of statins by the degree of LDL-C lowering. In fact, by correlating the incidence of coronary heart disease events with the levels of LDL-C achieved in those trials, conclusions can reasonably be drawn from such data (Figure 1).2
The guidelines do not recommend nonstatin drugs. Actually, the guidelines note that clinicians are free to consider other therapies, especially those proven to reduce the risk of cardiovascular events, a central principle of medicine. Since the guidelines were published, data have emerged indicating that the role of nonstatin drugs also needs consideration.
The ugly: Risk calculator untested
The guidelines promote the use of a risk calculator developed by the ACC/AHA to estimate the 10-year risk of an atherosclerotic event for people whose LDL-C levels are between 70 and 189 mg/dL to help decide whether to initiate statin therapy for primary prevention of atherosclerotic cardiovascular disease. Such an approach is reasonable, although the risk score was promulgated without evidence to support its utility.
Media coverage of the risk calculator was fierce. Some physicians found imperfections in the risk score (as is true for all risk scores), resulting in public mistrust of the guidelines and of the medical community as a whole. This needless controversy may have compromised the main message—that LDL-C should be lowered in many people—a message backed by strong evidence.
Alternative strategies proposed
Ridker et al3 have proposed a hybrid strategy to guide statin use for apparently healthy people that combines the ACC/AHA guideline approach with entry criteria for randomized clinical trials that showed statin efficacy for primary prevention.
Genetic analysis may offer another approach. Mega et al4 stratified more than 48,000 people by a genetic risk score based on 27 genetic variants and found a significant association with risk of coronary events. Targeting therapy to people found to be at higher risk on this basis offers greater risk reduction than expected for the general population. Biomarkers and imaging tests are other potentially useful risk determinants.
LDL-C: LOWER IS BETTER
Although no clinical trial has yet targeted specific LDL-C levels, there is plenty of evidence that lower LDL-C levels offer greater benefit (Figure 1).2
In 1994, the Scandinavian Simvastatin Survival Study5 established the benefit of statins in patients with known vascular disease. The mean LDL-C level achieved in the active treatment group was 120 mg/dL. More trials followed supporting the benefits of statins and of reducing LDL-C from average levels in the 120s down to 100 mg/dL.
In 2004, the Pravastatin or Atorvastatin Evaluation and Infection Therapy–Thrombolysis in Myocardial Infarction 22 trial6 observed an even greater risk reduction in patients with known risk by treating with statins; the mean LDL-C level achieved in the group randomized to an intensive regimen of atorvastatin 80 mg per day was 62 mg/dL. The same year, the Adult Treatment Panel III of the National Cholesterol Education Program7 issued updated guidelines including an optional goal of LDL-C less than 70 mg/dL for patients at very high risk.
In 2008, the Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin (JUPITER)8 found a significantly lower incidence of major cardiovascular events at 2 years in apparently healthy men and women with baseline LDL-C levels of less than 130 mg/dL after treatment with rosuvastatin 20 mg daily, with an achieved median LDL-C of 55 mg/dL.
How low should LDL-C go?
Evidence from clinical trials indicates a 20% to 25% reduction in the risk of cardiovascular events for every 39-mg/dL decrease in LDL-C. Extrapolating the data, cardiovascular disease risk would be reduced to zero if LDL-C were brought down below 40 mg/dL.
Brown and Goldstein,9 who won the 1985 Nobel Prize in medicine for their work in cholesterol metabolism, estimated that a plasma level of LDL-C of only 25 mg/dL would be sufficient to nourish cells with cholesterol. Cells can synthesize all the cholesterol they need, underscoring that LDL-C is simply the final end-product that the liver removes from circulation.
Other evidence that lower LDL-C does not have adverse effects comes from non-Western populations as well as from other mammals. Total cholesterol levels range in the low 100s mg/dL in Native American and African tribal populations, with LDL-C estimated to be about 50 to 75 mg/dL. Elephants, baboons, and foxes have even lower levels.10
Clinical trial data also support that LDL-C levels below the current “normal” are better. The Cholesterol Treatment Trialists’ Collaboration11 analyzed data from more than 160,000 patients in 26 trials that evaluated either more- vs less-intensive statin regimens or statin treatment vs control. No baseline level below which lowering LDL-C further was not beneficial was found. Patients who started out with an LDL-C level of less than 77 mg/dL had the same risk reduction of major vascular events when the level was dropped to 50 mg/dL as those who started at higher levels and reduced their LDL-C by the same amount. In the JUPITER trial, even those with a baseline LDL-C of less than 60 mg/dL benefited from statin therapy.12
BEYOND STATINS
Ezetimibe further lowers risk
Ezetimibe is a nonstatin drug that reduces LDL-C by about 15% to 20%. The Improved Reduction of Outcomes: Vytorin Efficacy International Trial13 registered more than 18,000 patients with a baseline LDL-C level of less than 125 mg/dL (or 100 mg/dL if already on lipid-lowering therapy) who had been stabilized shortly after an acute cardiovascular event. They were randomized to receive either simvastatin 40 mg or combined simvastatin 40 mg and ezetimibe 10 mg. The study intended to determine two things: whether ezetimibe could further lower LDL-C when combined with a statin, and whether risk could be reduced further by driving the LDL-C below 70 mg/dL and down to the mid-50s.
After 1 year, the average LDL-C level was 70 mg/dL in the simvastatin group and 53 mg/dL in the combined simvastatin and ezetimibe group. At 7 years, for the primary end point (cardiovascular death, myocardial infarction, unstable angina requiring hospitalization, coronary revascularization, or stroke), there was a 6% reduction of events in the combined drug treatment group, with the number of people needed to treat being 50 to prevent one event. For the narrower end point of cardiovascular death, nonfatal myocardial infarction, and nonfatal stroke, there was a 10% risk reduction in the combined drug treatment arm.14
The amount of risk reduction is exactly what was predicted by the Cholesterol Treatment Trialists’ Collaboration’s plot of reduction in events vs reduction in LDL-C based on the analysis of 26 trials, adding further evidence that it is the LDL-C reduction itself, rather than the means by which LDL-C is reduced, that is critical for benefit.
PCSK9 inhibitors: A new approach
Mutations in the gene for proprotein convertase subtilisin kexin type 9 (PCSK9) have become a new focus of interest for reducing LDL-C and cardiovascular risk.15 PCSK9 binds to the LDL-C receptor on the surface of hepatocytes and escorts it to its destruction in the lysosomes, rather than allowing it to return to the cell surface to take more LDL-C out of circulation.
People with a gain-of-function mutation (conferring too much PCSK9, resulting in fewer LDL-C receptors and more LDL-C in circulation) are a more recently recognized subset of those with autosomal-dominant familial hypercholesterolemia. They have total cholesterol levels in the 90th percentile, tendon xanthomas, and a high risk of myocardial infarction and stroke at a young age.
Conversely, those with a nonsense mutation in PCSK9—leading to loss of function—have a 28% reduction in mean LDL-C and 88% reduction in risk of coronary heart disease compared with those without the mutation.16 Two women (ages 32 and 21, fertile) have been found who have inactivating mutations in both PCSK9 alleles, and both are in apparent good health, with LDL-C levels of 14 mg/dL and 15 mg/dL, respectively.17,18
Dramatic reduction in LDL-C
Monoclonal antibodies have been developed that bind PCSK9 and block its action with the goal of developing new LDL-C–lowering treatments. Phase 2 clinical trials of varying doses of evolocumab (Repatha), a drug in this class, combined with standard therapy (a statin with or without ezetimibe), found a 66% reduction of LDL-C at high doses at 12 weeks compared with standard therapy alone, with concomitant reductions in other atherogenic lipoproteins.19 Patients who could not tolerate statins because of myalgia responded well to evolocumab.20
Patients with heterozygous familial hypercholesterolemia also had a substantial reduction in LDL-C (55% at the highest dosage), even though they have fewer LDL-C receptors for the drug to act upon.21 People with homozygous familial hypercholesterolemia and no LDL-C receptors had a lesser relative reduction in LDL-C that depended on the type of mutations they had. Nonetheless, given how high LDL-C levels are in this population, the absolute decreases in LDL-C level were quite impressive.
Cardiovascular risk reduced
Data at nearly 1 year showed continued reduction of LDL-C by about 60% (absolute reduction: 73 mg/dL), as well as a lower incidence of cardiovascular events starting at just 3 months, much sooner than observed in some statin trials.22 Benefits were found regardless of subgroup (sex, age, statin use, baseline LDL-C level, or known vascular disease). No difference was found in the safety profile between the evolocumab and control arms. Only 2.4% of participants discontinued evolocumab because of adverse events, and the incidence of adverse effects did not correlate with LDL-C level achieved.
Neurocognitive effects occurred in 0.9% of the evolocumab arm vs 0.3% in the control arm. This difference has not been explained: although there is cholesterol in the central nervous system, it is generated locally, and lipoproteins—and evolocumab—are not thought to cross the blood-brain barrier.
Long-term trials of evolocumab are currently under way for patients with cardiovascular disease, as are trials of two other PCSK9 inhibitors, alirocumab and bococizumab, in addition to standard statin therapy.
On July 24, 2015, the US Food and Drug Administration (FDA) approved the first PCSK9 inhibitor, alirocumab (Praluent) for patients with heterozygous familial hypercholesterolemia or those with clinical atherosclerotic cardiovascular disease who require additional lowering of LDL-C. The starting dosage is 75 mg subcutaneously every 2 weeks, which can be increased up to 150 mg every 2 weeks.
Evolocumab was approved by the FDA on August 27, 2015, for the same indications. The dosage is 140 mg subcutaneously every 2 weeks or 420 mg every month.
The 2013 joint guidelines of the American College of Cardiology and American Heart Association (ACC/AHA)1 on the treatment of blood cholesterol to reduce cardiovascular risk recommend high-intensity statin therapy for secondary prevention of cardiovascular events. The question of primary prevention is not so straightforward, and the recommended strategy has come under fire. In addition, the guidelines focus on statins and not on LDL-C levels, and the role of nonstatin lipid-lowering drugs and the value of reducing LDL-C levels to well below levels currently regarded as “normal” remain unclear.
This article comments on the 2013 ACC/AHA guidelines, reviews the data on optimal LDL-C levels, and discusses new nonstatin agents.
ACC/AHA GUIDELINES: A MIXED MESSAGE
The 2013 ACC/AHA cholesterol guidelines1 can be characterized by the title from the famous Western film “The Good, the Bad, and the Ugly.”
The good: A clear message to treat
The guidelines deliver an unambiguous message to treat patients at high risk with high-intensity statin therapy. This mandate is very helpful as it should reduce the undertreatment of patients.
The seemingly bad
Two common misconceptions regarding the guidelines:
They abandon LDL-C targets. Actually, the guidelines do not argue for or against targets; they simply remain silent, citing that randomized trials have not been conducted with LDL-C targets as specific goals. Technically, this statement is true. However, it seems contrived to argue, for example, that the benefit of atorvastatin 80 mg over 10 mg in the Treating to New Targets trial could not be reliably ascribed to the lower LDL-C achieved with the higher dose, but rather to some undefined benefit of high-intensity statin therapy, especially as the guidelines define the intensity of statins by the degree of LDL-C lowering. In fact, by correlating the incidence of coronary heart disease events with the levels of LDL-C achieved in those trials, conclusions can reasonably be drawn from such data (Figure 1).2
The guidelines do not recommend nonstatin drugs. Actually, the guidelines note that clinicians are free to consider other therapies, especially those proven to reduce the risk of cardiovascular events, a central principle of medicine. Since the guidelines were published, data have emerged indicating that the role of nonstatin drugs also needs consideration.
The ugly: Risk calculator untested
The guidelines promote the use of a risk calculator developed by the ACC/AHA to estimate the 10-year risk of an atherosclerotic event for people whose LDL-C levels are between 70 and 189 mg/dL to help decide whether to initiate statin therapy for primary prevention of atherosclerotic cardiovascular disease. Such an approach is reasonable, although the risk score was promulgated without evidence to support its utility.
Media coverage of the risk calculator was fierce. Some physicians found imperfections in the risk score (as is true for all risk scores), resulting in public mistrust of the guidelines and of the medical community as a whole. This needless controversy may have compromised the main message—that LDL-C should be lowered in many people—a message backed by strong evidence.
Alternative strategies proposed
Ridker et al3 have proposed a hybrid strategy to guide statin use for apparently healthy people that combines the ACC/AHA guideline approach with entry criteria for randomized clinical trials that showed statin efficacy for primary prevention.
Genetic analysis may offer another approach. Mega et al4 stratified more than 48,000 people by a genetic risk score based on 27 genetic variants and found a significant association with risk of coronary events. Targeting therapy to people found to be at higher risk on this basis offers greater risk reduction than expected for the general population. Biomarkers and imaging tests are other potentially useful risk determinants.
LDL-C: LOWER IS BETTER
Although no clinical trial has yet targeted specific LDL-C levels, there is plenty of evidence that lower LDL-C levels offer greater benefit (Figure 1).2
In 1994, the Scandinavian Simvastatin Survival Study5 established the benefit of statins in patients with known vascular disease. The mean LDL-C level achieved in the active treatment group was 120 mg/dL. More trials followed supporting the benefits of statins and of reducing LDL-C from average levels in the 120s down to 100 mg/dL.
In 2004, the Pravastatin or Atorvastatin Evaluation and Infection Therapy–Thrombolysis in Myocardial Infarction 22 trial6 observed an even greater risk reduction in patients with known risk by treating with statins; the mean LDL-C level achieved in the group randomized to an intensive regimen of atorvastatin 80 mg per day was 62 mg/dL. The same year, the Adult Treatment Panel III of the National Cholesterol Education Program7 issued updated guidelines including an optional goal of LDL-C less than 70 mg/dL for patients at very high risk.
In 2008, the Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin (JUPITER)8 found a significantly lower incidence of major cardiovascular events at 2 years in apparently healthy men and women with baseline LDL-C levels of less than 130 mg/dL after treatment with rosuvastatin 20 mg daily, with an achieved median LDL-C of 55 mg/dL.
How low should LDL-C go?
Evidence from clinical trials indicates a 20% to 25% reduction in the risk of cardiovascular events for every 39-mg/dL decrease in LDL-C. Extrapolating the data, cardiovascular disease risk would be reduced to zero if LDL-C were brought down below 40 mg/dL.
Brown and Goldstein,9 who won the 1985 Nobel Prize in medicine for their work in cholesterol metabolism, estimated that a plasma level of LDL-C of only 25 mg/dL would be sufficient to nourish cells with cholesterol. Cells can synthesize all the cholesterol they need, underscoring that LDL-C is simply the final end-product that the liver removes from circulation.
Other evidence that lower LDL-C does not have adverse effects comes from non-Western populations as well as from other mammals. Total cholesterol levels range in the low 100s mg/dL in Native American and African tribal populations, with LDL-C estimated to be about 50 to 75 mg/dL. Elephants, baboons, and foxes have even lower levels.10
Clinical trial data also support that LDL-C levels below the current “normal” are better. The Cholesterol Treatment Trialists’ Collaboration11 analyzed data from more than 160,000 patients in 26 trials that evaluated either more- vs less-intensive statin regimens or statin treatment vs control. No baseline level below which lowering LDL-C further was not beneficial was found. Patients who started out with an LDL-C level of less than 77 mg/dL had the same risk reduction of major vascular events when the level was dropped to 50 mg/dL as those who started at higher levels and reduced their LDL-C by the same amount. In the JUPITER trial, even those with a baseline LDL-C of less than 60 mg/dL benefited from statin therapy.12
BEYOND STATINS
Ezetimibe further lowers risk
Ezetimibe is a nonstatin drug that reduces LDL-C by about 15% to 20%. The Improved Reduction of Outcomes: Vytorin Efficacy International Trial13 registered more than 18,000 patients with a baseline LDL-C level of less than 125 mg/dL (or 100 mg/dL if already on lipid-lowering therapy) who had been stabilized shortly after an acute cardiovascular event. They were randomized to receive either simvastatin 40 mg or combined simvastatin 40 mg and ezetimibe 10 mg. The study intended to determine two things: whether ezetimibe could further lower LDL-C when combined with a statin, and whether risk could be reduced further by driving the LDL-C below 70 mg/dL and down to the mid-50s.
After 1 year, the average LDL-C level was 70 mg/dL in the simvastatin group and 53 mg/dL in the combined simvastatin and ezetimibe group. At 7 years, for the primary end point (cardiovascular death, myocardial infarction, unstable angina requiring hospitalization, coronary revascularization, or stroke), there was a 6% reduction of events in the combined drug treatment group, with the number of people needed to treat being 50 to prevent one event. For the narrower end point of cardiovascular death, nonfatal myocardial infarction, and nonfatal stroke, there was a 10% risk reduction in the combined drug treatment arm.14
The amount of risk reduction is exactly what was predicted by the Cholesterol Treatment Trialists’ Collaboration’s plot of reduction in events vs reduction in LDL-C based on the analysis of 26 trials, adding further evidence that it is the LDL-C reduction itself, rather than the means by which LDL-C is reduced, that is critical for benefit.
PCSK9 inhibitors: A new approach
Mutations in the gene for proprotein convertase subtilisin kexin type 9 (PCSK9) have become a new focus of interest for reducing LDL-C and cardiovascular risk.15 PCSK9 binds to the LDL-C receptor on the surface of hepatocytes and escorts it to its destruction in the lysosomes, rather than allowing it to return to the cell surface to take more LDL-C out of circulation.
People with a gain-of-function mutation (conferring too much PCSK9, resulting in fewer LDL-C receptors and more LDL-C in circulation) are a more recently recognized subset of those with autosomal-dominant familial hypercholesterolemia. They have total cholesterol levels in the 90th percentile, tendon xanthomas, and a high risk of myocardial infarction and stroke at a young age.
Conversely, those with a nonsense mutation in PCSK9—leading to loss of function—have a 28% reduction in mean LDL-C and 88% reduction in risk of coronary heart disease compared with those without the mutation.16 Two women (ages 32 and 21, fertile) have been found who have inactivating mutations in both PCSK9 alleles, and both are in apparent good health, with LDL-C levels of 14 mg/dL and 15 mg/dL, respectively.17,18
Dramatic reduction in LDL-C
Monoclonal antibodies have been developed that bind PCSK9 and block its action with the goal of developing new LDL-C–lowering treatments. Phase 2 clinical trials of varying doses of evolocumab (Repatha), a drug in this class, combined with standard therapy (a statin with or without ezetimibe), found a 66% reduction of LDL-C at high doses at 12 weeks compared with standard therapy alone, with concomitant reductions in other atherogenic lipoproteins.19 Patients who could not tolerate statins because of myalgia responded well to evolocumab.20
Patients with heterozygous familial hypercholesterolemia also had a substantial reduction in LDL-C (55% at the highest dosage), even though they have fewer LDL-C receptors for the drug to act upon.21 People with homozygous familial hypercholesterolemia and no LDL-C receptors had a lesser relative reduction in LDL-C that depended on the type of mutations they had. Nonetheless, given how high LDL-C levels are in this population, the absolute decreases in LDL-C level were quite impressive.
Cardiovascular risk reduced
Data at nearly 1 year showed continued reduction of LDL-C by about 60% (absolute reduction: 73 mg/dL), as well as a lower incidence of cardiovascular events starting at just 3 months, much sooner than observed in some statin trials.22 Benefits were found regardless of subgroup (sex, age, statin use, baseline LDL-C level, or known vascular disease). No difference was found in the safety profile between the evolocumab and control arms. Only 2.4% of participants discontinued evolocumab because of adverse events, and the incidence of adverse effects did not correlate with LDL-C level achieved.
Neurocognitive effects occurred in 0.9% of the evolocumab arm vs 0.3% in the control arm. This difference has not been explained: although there is cholesterol in the central nervous system, it is generated locally, and lipoproteins—and evolocumab—are not thought to cross the blood-brain barrier.
Long-term trials of evolocumab are currently under way for patients with cardiovascular disease, as are trials of two other PCSK9 inhibitors, alirocumab and bococizumab, in addition to standard statin therapy.
On July 24, 2015, the US Food and Drug Administration (FDA) approved the first PCSK9 inhibitor, alirocumab (Praluent) for patients with heterozygous familial hypercholesterolemia or those with clinical atherosclerotic cardiovascular disease who require additional lowering of LDL-C. The starting dosage is 75 mg subcutaneously every 2 weeks, which can be increased up to 150 mg every 2 weeks.
Evolocumab was approved by the FDA on August 27, 2015, for the same indications. The dosage is 140 mg subcutaneously every 2 weeks or 420 mg every month.
- Stone NJ, Robinson JG, Lichtenstein AH, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation 2014; 129:S1-S45. Erratum in: Circulation 2014; 129:S46–S48.
- Raymond C, Cho L, Rocco M, Hazen SL. New cholesterol guidelines: worth the wait? Cleve Clin J Med 2014; 81:11–19.
- Ridker PM, Rose L, Cook NR. A proposal to incorporate trial data into a hybrid ACC/AHA algorithm for the allocation of statin therapy in primary prevention. J Am Coll Cardiol 2015; 65:942–948.
- Mega JL, Stitziel NO, Smith JG, et al. Genetic risk, coronary heart disease events, and the clinical benefit of statin therapy: an analysis of primary and secondary prevention trials. Lancet 2015; 385:2264–2271.
- Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S). Lancet 1994; 344:1383–1389.
- Cannon CP, Braunwald E, McCabe CH, et al; Pravastatin or Atorvastatin Evaluation and Infection Therapy-Thrombolysis in Myocardial Infarction 22 Investigators. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med 2004; 350:1495–1504.
- Grundy SM, Cleeman JI, Merz CN, et al; National Heart, Lung, and Blood Institute; American College of Cardiology Foundation; American Heart Association. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines. Circulation 2004; 110:227–239. Erratum in Circulation 2004; 110:763.
- Ridker PM, Danielson E, Fonseca FAH, et al; JUPITER Study Group. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med 2008; 359:2195–2207.
- Brown MS, Goldstein JL. A receptor-mediated pathway for cholesterol homeostasis. Science 1986; 232:34–47.
- Hochholzer W, Giugliano RP. Lipid lowering goals: back to nature? Ther Adv Cardiovasc Dis 2010; 4:185–191.
- Cholesterol Treatment Trialists’ (CTT) Collaboration, Baigent C, Blackwell L, Emberson J, et al. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet 2010; 376:1670–1681.
- Hsia J, MacFadyen JG, Monyak J, Ridker PM. Cardiovascular event reduction and adverse events among subjects attaining low-density lipoprotein cholesterol <50 mg/dl with rosuvastatin. The JUPITER trial (Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin). J Am Coll Cardiol 2011; 57:1666–1675.
- Cannon CP, Giugliano RP, Blazing MA, et al; IMPROVE-IT Investigators. Rationale and design of IMPROVE-IT (IMProved Reduction of Outcomes: Vytorin Efficacy International Trial): comparison of ezetimibe/simvastatin versus simvastatin monotherapy on cardiovascular outcomes in patients with acute coronary syndromes. Am Heart J 2008; 156:826–832.
- Cannon CP, Blazing MA, Giugliano RP, et al for the IMPROVE-IT Investigators. Ezetimibe added to statin therapy after acute coronary syndromes. N Engl J Med 2015; 372:2387–2397.
- Giugliano RP, Sabatine MS. Are PCSK9 Inhibitors the next breakthrough in the cardiovascular field? J Am Coll Cardiol 2015; 65:2638–2651.
- Cohen JC, Boerwinkle E, Mosley TH Jr, Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med 2006; 354:1264–1272.
- Zhao Z, Tuakli-Wosornu Y, Lagace TA, et al. Molecular characterization of loss-of-function mutations in PCSK9 and identification of a compound heterozygote. Am J Hum Genet 2006; 79:514-523.
- Hooper AJ, Marais AD, Tanyanyiwa DM, Burnett JR. The C679X mutation in PCSK9 is present and lowers blood cholesterol in a Southern African population. Atherosclerosis 2007; 193:445–448.
- Giugliano RP, Desai NR, Kohli P, et al; LAPLACE-TIMI 57 Investigators. Efficacy, safety, and tolerability of a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 in combination with a statin in patients with hypercholesterolaemia (LAPLACE-TIMI 57): a randomised, placebo-controlled, dose-ranging, phase 2 study. Lancet 2012; 380:2007–2017.
- Sullivan D, Olsson AG, Scott R, et al. Effect of a monoclonal antibody to PCSK9 on low-density lipoprotein cholesterol levels in statin-intolerant patients: the GAUSS randomized trial. JAMA 2012; 308:2497–2506.
- Raal F, Scott R, Somaratne R, et al. Low-density lipoprotein cholesterol-lowering effects of AMG 145, a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 serine protease in patients with heterozygous familial hypercholesterolemia: the Reduction of LDL-C with PCSK9 Inhibition in Heterozygous Familial Hypercholesterolemia Disorder (RUTHERFORD) randomized trial. Circulation 2012; 126:2408–2417.
- Sabatine MS, Giugliano RP, Wiviott SD, et al; Open-Label Study of Long-Term Evaluation against LDL Cholesterol (OSLER) Investigators. Efficacy and safety of evolocumab in reducing lipids and cardiovascular events. N Engl J Med 2015; 372:1500–1509.
- Stone NJ, Robinson JG, Lichtenstein AH, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation 2014; 129:S1-S45. Erratum in: Circulation 2014; 129:S46–S48.
- Raymond C, Cho L, Rocco M, Hazen SL. New cholesterol guidelines: worth the wait? Cleve Clin J Med 2014; 81:11–19.
- Ridker PM, Rose L, Cook NR. A proposal to incorporate trial data into a hybrid ACC/AHA algorithm for the allocation of statin therapy in primary prevention. J Am Coll Cardiol 2015; 65:942–948.
- Mega JL, Stitziel NO, Smith JG, et al. Genetic risk, coronary heart disease events, and the clinical benefit of statin therapy: an analysis of primary and secondary prevention trials. Lancet 2015; 385:2264–2271.
- Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S). Lancet 1994; 344:1383–1389.
- Cannon CP, Braunwald E, McCabe CH, et al; Pravastatin or Atorvastatin Evaluation and Infection Therapy-Thrombolysis in Myocardial Infarction 22 Investigators. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med 2004; 350:1495–1504.
- Grundy SM, Cleeman JI, Merz CN, et al; National Heart, Lung, and Blood Institute; American College of Cardiology Foundation; American Heart Association. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines. Circulation 2004; 110:227–239. Erratum in Circulation 2004; 110:763.
- Ridker PM, Danielson E, Fonseca FAH, et al; JUPITER Study Group. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med 2008; 359:2195–2207.
- Brown MS, Goldstein JL. A receptor-mediated pathway for cholesterol homeostasis. Science 1986; 232:34–47.
- Hochholzer W, Giugliano RP. Lipid lowering goals: back to nature? Ther Adv Cardiovasc Dis 2010; 4:185–191.
- Cholesterol Treatment Trialists’ (CTT) Collaboration, Baigent C, Blackwell L, Emberson J, et al. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet 2010; 376:1670–1681.
- Hsia J, MacFadyen JG, Monyak J, Ridker PM. Cardiovascular event reduction and adverse events among subjects attaining low-density lipoprotein cholesterol <50 mg/dl with rosuvastatin. The JUPITER trial (Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin). J Am Coll Cardiol 2011; 57:1666–1675.
- Cannon CP, Giugliano RP, Blazing MA, et al; IMPROVE-IT Investigators. Rationale and design of IMPROVE-IT (IMProved Reduction of Outcomes: Vytorin Efficacy International Trial): comparison of ezetimibe/simvastatin versus simvastatin monotherapy on cardiovascular outcomes in patients with acute coronary syndromes. Am Heart J 2008; 156:826–832.
- Cannon CP, Blazing MA, Giugliano RP, et al for the IMPROVE-IT Investigators. Ezetimibe added to statin therapy after acute coronary syndromes. N Engl J Med 2015; 372:2387–2397.
- Giugliano RP, Sabatine MS. Are PCSK9 Inhibitors the next breakthrough in the cardiovascular field? J Am Coll Cardiol 2015; 65:2638–2651.
- Cohen JC, Boerwinkle E, Mosley TH Jr, Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med 2006; 354:1264–1272.
- Zhao Z, Tuakli-Wosornu Y, Lagace TA, et al. Molecular characterization of loss-of-function mutations in PCSK9 and identification of a compound heterozygote. Am J Hum Genet 2006; 79:514-523.
- Hooper AJ, Marais AD, Tanyanyiwa DM, Burnett JR. The C679X mutation in PCSK9 is present and lowers blood cholesterol in a Southern African population. Atherosclerosis 2007; 193:445–448.
- Giugliano RP, Desai NR, Kohli P, et al; LAPLACE-TIMI 57 Investigators. Efficacy, safety, and tolerability of a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 in combination with a statin in patients with hypercholesterolaemia (LAPLACE-TIMI 57): a randomised, placebo-controlled, dose-ranging, phase 2 study. Lancet 2012; 380:2007–2017.
- Sullivan D, Olsson AG, Scott R, et al. Effect of a monoclonal antibody to PCSK9 on low-density lipoprotein cholesterol levels in statin-intolerant patients: the GAUSS randomized trial. JAMA 2012; 308:2497–2506.
- Raal F, Scott R, Somaratne R, et al. Low-density lipoprotein cholesterol-lowering effects of AMG 145, a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 serine protease in patients with heterozygous familial hypercholesterolemia: the Reduction of LDL-C with PCSK9 Inhibition in Heterozygous Familial Hypercholesterolemia Disorder (RUTHERFORD) randomized trial. Circulation 2012; 126:2408–2417.
- Sabatine MS, Giugliano RP, Wiviott SD, et al; Open-Label Study of Long-Term Evaluation against LDL Cholesterol (OSLER) Investigators. Efficacy and safety of evolocumab in reducing lipids and cardiovascular events. N Engl J Med 2015; 372:1500–1509.
KEY POINTS
- Patients at high risk of atherosclerotic cardiovascular disease should be treated with high-intensity statin therapy.
- To date, no baseline level has been identified beneath which lowering LDL-C does not provide clinical benefit.
- The benefits of lower LDL-C are seen with a variety of pharmacologic interventions and in people who have naturally low levels due to genetic variants.
- Clinical trial evidence supports that ezetimibe reduces the risk of cardiovascular events.
- Proprotein convertase subtilisin kexin type 9 (PCSK9) inhibitors reduce LDL-C by approximately 60%, and preliminary data show that they reduce the risk of cardiovascular events.

2015 Update on Parkinson disease
This has been a boom year for Parkinson disease, with the US Food and Drug Administration (FDA) approving two new therapies, and with others in the pipeline.
This article details clinical signs of Parkinson disease, discusses functional imaging, provides an update on current thinking on disease pathogenesis, and gives an overview of managing parkinsonian symptoms and dyskinesias.
DIAGNOSIS REMAINS CLINICAL
Although a better understanding of Parkinson disease has been gained in recent years, with the recognition of several premotor features and potential biomarkers, its diagnosis is still primarily based on clinical motor findings. The four cardinal motor features have the mnemonic TRAP:
- Tremor at rest can be subtle, involving just the thumb, best observed when the patient is sitting with the hand resting on the lap; or it can be obvious, involving the entire hand, arm, feet, lips, and chin.
- Rigidity can be felt rather than seen, by slowly passively rotating the patient’s wrist or elbow and feeling resistance. The right and left sides often differ.
- Akinesia or bradykinesia (slowness or lack of movement) can be observed by having the patient walk down a hallway. One may observe reduced arm swing and hesitation in initiating movement.
- Postural instability usually develops later rather than sooner in the disease progression. The patient may need to hold onto someone to maintain balance when getting up or walking.
At least two features must be present to make the diagnosis of parkinsonism. One feature must be tremor or rigidity.
Although the criteria for parkinsonism appear simple, the diagnosis of Parkinson disease is not always clear-cut. For example, shaking can be secondary to a dopamine receptor-blocking medication, to anxiety, or to essential tremor; rigidity and slowness may be due to arthritis; and postural instability can result from a neuropathy. Moreover, other neurodegenerative parkinsonian disorders may respond to levodopa (at least initially) and may present with levodopa-induced dyskinesias. Robust response to levodopa and the occurrence of dyskinesias are two additional features that strongly suggest the diagnosis of Parkinson disease.
Supporting parkinsonian features include stooped posture, masked facies, micrographia (small handwriting), drooling, speech changes (eg, hypophonia or soft speech, stuttering, slurring, monotonic speech), and a shuffling, festinating gait (quick short steps as if falling forward).
PARKINSON MIMICS
Parkinsonism is a broader term than Parkinson disease or idiopathic Parkinson disease. It is characterized by akinetic rigidity and impaired motor activity that leads to reduced function and falls; behavioral changes also may occur.
In the United States, Parkinson disease is the most common cause of parkinsonism. Other nonneurodegenerative causes are drug-induced parkinsonism (due to dopamine receptor antagonists such as antipsychotic or antiemetic drugs), stroke (in the basal ganglia or frontal lobe), and normal-pressure hydrocephalus (causing lower-body parkinsonism). Mimics of parkinsonism include essential tremor and psychogenic parkinsonism.
Parkinsonism can also be caused by Parkinson-plus disorders, ie, neurodegenerative conditions characterized by parkinsonism along with additional signs and symptoms, as listed below. Parkinson-plus disorders include progressive supranuclear palsy, multiple system atrophy, corticobasal degeneration, and Lewy body disease.
Clinical features that suggest a diagnosis other than Parkinson disease include1:
- Poor response to adequate dosages of levodopa
- Early onset of postural instability and falls
- Axial rigidity (eg, stiff neck) more than appendicular rigidity
- Early dementia
- Supranuclear gaze palsy
- Unusual movements besides tremor, eg, limb dystonia, myoclonus, limb levitation or alien limb syndrome
- Profound autonomic dysfunction
- Psychotic symptoms before taking levodopa or dopaminergic medication.
The precise diagnosis of Parkinson-plus disorders is not critical, as the treatment is generally the same for all of them: ie, levodopa (if it shows some efficacy and is well tolerated), with additional symptomatic treatment for features such as depression, cognitive impairment, and autonomic dysfunction, and supportive therapy including physical, occupational, speech, and swallowing therapy.
IMAGING MAY ASSIST IN THE DIAGNOSIS
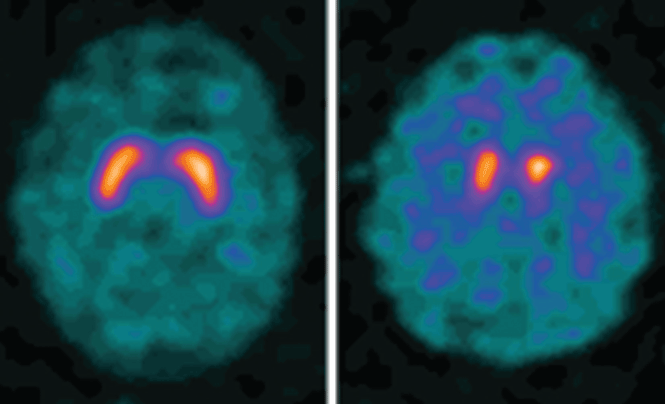
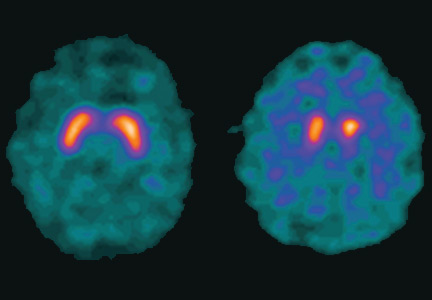
Dopamine transporter single-photon emission computed tomography (SPECT) is a functional imaging technique that supposedly reflects dopamine uptake by surviving presynaptic dopaminergic neurons in the striate bodies of the basal ganglia. Normal uptake shows distinct cashew-shaped enhancement bilaterally. In Parkinson disease, the enhanced areas are smaller and asymmetric, first with diminution of the tail (representing the putamen), then later involving the head (representing the caudate) along with the other striate bodies (Figure 1).

Dopamine transporter SPECT does not distinguish one neurodegenerative parkinsonian disorder from another. Therefore, it should not be used to distinguish Parkinson disease from other Parkinson-plus syndromes. But it does distinguish neurodegenerative parkinsonian disorders from nonneurodegenerative conditions and mimics, which have a normal result on dopamine transporter SPECT (Table 1).
SLOWING DISEASE PROGRESSION
Current treatments for Parkinson disease can significantly improve symptoms but, unfortunately, do not cure the disease or slow its progression. Testing whether agents modify the disease course is particularly difficult with Parkinson disease, because it affects individuals differently, has a wide spectrum of symptoms, has a long time course, and lacks definitive markers to monitor progression. Some agents have shown promise:
Caffeine. People who drink coffee are less likely to develop Parkinson disease, with the risk declining with the number of cups per day.2 For those who have the disease, drinking coffee is associated with reduced symptoms.
Exercise improves Parkinson disease and may prevent it, and some studies suggest that it can delay its progression.3 Exercise has been shown in an animal model to reduce the vulnerability of dopamine neurons to the toxic agent 6-hydroxydopamine.4 Functional magnetic resonance imaging studies have shown blood flow patterns before and after exercise that are similar to those seen in patients with and without Parkinson medication.3
Rasagiline, a monoamine oxidase B (MAO-B) inhibitor used for symptomatic treatment of Parkinson disease, had conflicting results in a neuroprotective clinical trial. Patients who received rasagiline 1 mg daily—but not those who received 2 mg daily—at the beginning of the trial had better Parkinson motor scores compared with patients who received rasagiline 9 months later.5
Inosine is a urate precursor that elevates urate levels in serum and the central nervous system. For unknown reasons, patients with Parkinson disease tend to have a low uric acid level, and higher levels are associated with milder disease. It is hoped that raising the uric acid level to a “pre-gout level” may slow the progression of Parkinson disease.
Isradipine, a calcium channel blocker, was found in an epidemiologic study of elderly patients to be associated with reduced likelihood of developing Parkinson disease.6 The drug is now undergoing clinical trials.
Smoking. Although cigarette smokers have long been recognized as having a very low risk of developing Parkinson disease, smoking is not recommended.
Agents found ineffective. Agents that have been tested and found ineffective in modifying the course of Parkinson disease include vitamin E, coenzyme Q10, riluzole, GPI-1485, pramipexole, cogane, CEP-1347, TCH-346, and creatine.
NOT JUST DOPAMINE—OR TREMORS
Dopamine deficiency is central to the current understanding of the pathogenesis of Parkinson disease and the focus of treatment efforts, but if dopamine deficiency were the only problem, replacing it should completely ameliorate all parkinsonian features. Other neurotransmitters also play roles: norepinephrine is implicated in orthostatic symptoms and apathy, acetylcholine in cognitive behaviors, glutamate in dyskinesias, and serotonin in depression, anxiety, and sleep abnormalities.
The most recognized area of involvement in the brain has traditionally been the substantia nigra in the midbrain. However, current thinking is that the disease starts lower in the caudal area of the brainstem (along with the olfactory tubercle), moves through the pons to the midbrain, then spreads across the cerebrum with extensive neocortical involvement.
Early premotor indicators are now recognized to occur 15 to 20 years before a tremor appears. The first signs are often hyposmia (diminished sense of smell, reflecting involvement of the olfactory tubercle) and constipation (reflecting involvement of the medulla and the vagus nucleus). With pons involvement, the patient can develop rapid eye movement sleep behavior disorder, depression, or anxiety. Only then does the disease spread to the midbrain and cause resting tremor, rigidity, and bradykinesia.7
Identifying the preclinical stages and starting disease-modifying treatments before the onset of motor symptoms may one day prove important, but at this point, the premotor symptoms (anosmia, constipation, depression) are too nonspecific to be useful, and such treatments have not yet been identified.
TREATMENT: LEVODOPA STILL PRIMARY
When to start drug treatment depends primarily on how much the symptoms bother the patient. Regardless of the clinician’s (or patient’s) belief in the benefits of delaying symptomatic treatment, it is universally considered necessary to start medication when gait problems develop because of the danger of a fall and resulting disability.
Carbidopa-levodopa combination therapy remains the most effective treatment; if it is not effective, another diagnosis may need to be considered. Carbidopa-levodopa improves tremor, rigidity, and bradykinesia, particularly in the early stages of Parkinson disease. It is well tolerated, has rapid onset, reduces the risk of death, and is the least expensive of the medications for Parkinson disease.
Immediate-release and continued-release formulations are available, as well as one that dissolves rapidly on the tongue and can be taken without water. An oral extended-release carbidopa-levodopa formulation (Rytary) was approved by the FDA in January 2015. Tablets are filled with drug-containing microbeads that dissolve at different rates to achieve therapeutic levodopa levels as quickly as the immediate-release formulation and maintain them for an extended time.8
The development of dyskinesias is the major psychological drawback of levodopa, occurring in 80% of patients after 5 to 10 years of treatment. Although many patients fear this side effect, most patients who develop it find it preferable to the rigidity and bradykinesia of Parkinson disease. In most cases, bothersome dyskinesias can be controlled by adjusting medications.9,10
Dopamine agonists include pramipexole, ropinirole, and rotigotine. They are available in generic form as three-times-daily dosing; once-daily dosing is also available, but not as a generic formulation. Dopamine agonists have the advantage of potentially improving depression and delaying the onset of dyskinesias.
However, dopamine agonists have a number of disadvantages compared with levodopa: they have a longer titration period, are less effective, and are less well tolerated, especially in the elderly. Side effects occur more frequently than with levodopa and include general and peripheral edema, hallucinations, nausea, lightheadedness, and sleepiness.11,12 These drugs are also associated with “sleep attacks” (sudden falling asleep while active, such as while driving or eating) and with compulsive and impulsive behaviors such as hypersexuality, buying, binge eating, and gambling. Although these behaviors occur in fewer than 10% of patients, they can be devastating, leading to marital, financial, and legal problems. A bothersome clinical state termed dopamine agonist withdrawal syndrome is characterized by anxiety, depression, jitteriness, and palpitations when dopamine agonists are tapered or discontinued because of a side effect.13
MAO-B inhibitors delay the breakdown of dopamine, allowing it to “stay” in the brain for a longer period of time. Rasagiline for early monotherapy has the advantages of once-daily dosing, no titration, and excellent tolerability, even in the elderly. Potential drug interactions should be considered when using this drug. Early warnings about interactions with tyramine-rich foods were lifted after trials showed that this was not a problem.14
Amantadine is an N-methyl-d-aspartate (NMDA) receptor antagonist often used in early Parkinson disease and for treatment of dyskinesias and fatigue. It is the only drug that is intrinsically antidyskinetic and also improves Parkinson symptoms.15 Side effects include leg swelling, livedo reticularis, and neuropsychiatric and anticholinergic effects.
Anticholinergic agents (eg, trihexyphenidyl) improve tremor but are not as useful for bradykinesia or rigidity, and often have anticholinergic effects such as mental dullness, dry mouth, dry eye, and urinary hesitancy, especially in the elderly, so they have a limited role in Parkinson treatment.
MOTOR COMPLICATIONS: FLUCTUATIONS AND DYSKINESIAS
Motor fluctuations are changes between the akinetic and mobile phases of Parkinson disease, or the off-periods and on-periods of drug treatment. A patient who is “off” is generally rigid and feels that the medication is not working. A patient who is “on” feels loose and mobile and that the medication is working. Variants of motor fluctuations include:
- End-of-dose deterioration
- Delayed onset of response (more than half an hour after taking medication)
- Drug-resistant offs—medication has become ineffective
- Random oscillation—on-off phenomenon
- Freezing—unpredictable inability to start or finish a movement.
Dyskinesias are abnormal involuntary movements such as writhing and twisting. They are associated with dopaminergic therapy at peak dose, when the drug starts to turn on or wear off (termed diphasic dyskinesias).16
The storage hypothesis provides a plausible explanation for the development of motor complications as the disease progresses. Although the half-life of levodopa is only 60 to 90 minutes, it is effective in early disease when given three times a day. It is believed that at this stage of the disease, enough dopaminergic neurons survive to “store” dopamine and release it as needed. As the disease progresses and dopaminergic neurons die, storage capacity diminishes, and the clinical effect slowly starts to approximate the pharmacokinetic profile of the drug. Upon taking the medication, the patient gets a surge of drug, causing dyskinesias, followed later by rigidity as the effect wears off since there are fewer surviving dopaminergic cells to store dopamine.
MANAGING DYSKINESIAS
Patients with dyskinesias should first be asked if they are bothered by them; not all patients are troubled by dyskinesias. If the movements only bother others (eg, family members), then education is often the only treatment needed. If the patient is uncomfortable, the following measures can be tried:
- Taking lower, more frequent doses of levodopa (however, risk of wearing off becomes a problem)
- Adding a dopamine agonist or MAO-B inhibitor while lowering the levodopa dose (however, MAO-B inhibitors pose a risk of side effects in elderly patients)
- Adding clozapine (periodic laboratory testing is required to monitor blood levels and liver and kidney function)
- Adding amantadine (however, this poses a risk of cognitive side effects).
Deep-brain-stimulation surgery is appropriate for select patients who are generally physically healthy, cognitively intact, and emotionally stable, with a strong family support system, but who are bothered by symptoms of parkinsonism (such as tremors), motor fluctuations, or dyskinesias.17
Infusion pump. In January 2015, the FDA approved a new system that continuously delivers levodopa-carbidopa in a 4:1 ratio in gel suspension for 16 hours directly into the small intestine, minimizing motor fluctuations. The patient changes the cartridge daily and turns it off at bedtime.
*Dr. Fernandez has received research support from AbbVie, Acadia, Auspex, Biotie Therapies, Civitas, Kyowa/ProStrakan, Michael J. Fox Foundation, Movement Disorders Society, NIH/NINDS, Parkinson Study Group, Rhythm, Synosia, and Teva. He also has received honoraria from Carling Communications, International Parkinson and Movement Disorders Society, The Ohio State University, and PRIME Education, Inc as a speaker in CME events. He has received honoraria from Biogen, GE Health Care, Lundbeck, Merz Pharmaceuticals, and Pfizer as a consultant. He has received royalty payments from Demos Publishing for serving as a book author/editor. Cleveland Clinic has contracts with AbbVie and Merz Pharmaceuticals for Dr. Fernandez’s role as a member of the Global Steering Committee for LCIG studies and as a consultant or speaker, and as Head Principal Investigator for the Xeomin Registry Study. Dr. Fernandez has received a stipend from International Parkinson and Movement Disorders Society for serving as medical editor of the Movement Disorders Society website.
- Wenning GK, Ben-Shlomo Y, Hughes A, Daniel SE, Lees A, Quinn NP. What clinical features are most useful to distinguish definite multiple system atrophy from Parkinson’s disease? J Neurol Neurosurg Psychiatry 2000; 68:434–440.
- Hernán MA, Takkouche B, Caamaño-Isoma F, et al. A meta-analysis of coffee drinking, cigarette smoking, and risk of Parkinson’s disease. Ann Neurol 2002; 52:276–84.
- Ridgel A, Thota A, Vitek JL, Alberts JL. Forced, not voluntary, exercise improves motor function in Parkinson’s disease patients. Neurorehabil Neural Repair 2009; 23:600–608.
- Smith AD, Zigmond MJ. Can the brain be protected through exercise? Lessons from an animal model of parkinsonism. Exp Neurol 2003; 184:31–39.
- Olanow CW, Rascol O, Hauser R, et al, for the ADAGIO Study Investigators. A double-blind, delayed-start trial of rasagiline in Parkinson’s disease. N Engl J Med 2009; 361:1268–1278.
- Pasternak B, Svanström H, Nielsen NM, Fugger L, Melbye M, Hviid A. Use of calcium channel blockers and Parkinson’s disease. Am J Epidemiol 2012; 175:627-635.
- Braak H, Del Tredici K, Rüb U, de Vos RAI, Jansen Steur ENH, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 2003; 24:197–211.
- Hauser RA, Ellenbogen AL, Metman LV, et al. Crossover comparison of IPX066 and a standard levodopa formulation in advanced Parkinson’s disease. Mov Disord 2011; 26:2246–2252.
- Hely MA, Morris JG, Reid WG, Trafficante R. Sydney Multicenter Study of Parkinson’s disease: non-L-dopa-responsive problems dominate at 15 years. Mov Disord 2005; 20:190–199.
- Hung SW, Adeli GM, Arenovich T, Fox SH, Lang AE. Patient perception of dyskinesia in Parkinson’s disease. J Neurol Neurosurg Psychiatry 2010; 81:1112–1115.
- Rascol O, Brooks DJ, Korczyn AD, De Deyn PP, Clarke CE, Lang AE. A five-year study of the incidence of dyskinesia in patients with early Parkinson’s disease who were treated with ropinirole or levodopa. N Engl J Med 2000; 342:1484–1491.
- Parkinson Study Group. Pramipexole vs levodopa as initial treatment for Parkinson disease: a randomized controlled trial. Parkinson Study Group. JAMA 2000; 284:1931–1938.
- Nirenberg MJ. Dopamine agonist withdrawal syndrome: implications for patient care. Drugs Aging 2013; 30:587–592.
- Teva Neuroscience, Inc. Azilect prescribing information. https://www.azilect.com/Content/pdf/azi-40850-azilect-electronic-pi.pdf. Accessed June 29, 2015.
- Snow BJ, Macdonald L, Mcauley D, Wallis W. The effect of amantadine on levodopa-induced dyskinesias in Parkinson’s disease: a double-blind, placebo-controlled study. Clin Neuropharmacol 2000; 23:82–85.
- Adler CH, Ahlskog JE, eds. Parkinson’s Disease and Movement Disorders: Diagnosis and Treatment Guidelines for the Practicing Physician. Totowa, NJ: Humana Press; 2000.
- Machado A, Fernandez HH, Deogaonkar M. Deep brain stimulation: what can patients expect from it? Cleve Clin J Med 2012; 79:113–120.
This has been a boom year for Parkinson disease, with the US Food and Drug Administration (FDA) approving two new therapies, and with others in the pipeline.
This article details clinical signs of Parkinson disease, discusses functional imaging, provides an update on current thinking on disease pathogenesis, and gives an overview of managing parkinsonian symptoms and dyskinesias.
DIAGNOSIS REMAINS CLINICAL
Although a better understanding of Parkinson disease has been gained in recent years, with the recognition of several premotor features and potential biomarkers, its diagnosis is still primarily based on clinical motor findings. The four cardinal motor features have the mnemonic TRAP:
- Tremor at rest can be subtle, involving just the thumb, best observed when the patient is sitting with the hand resting on the lap; or it can be obvious, involving the entire hand, arm, feet, lips, and chin.
- Rigidity can be felt rather than seen, by slowly passively rotating the patient’s wrist or elbow and feeling resistance. The right and left sides often differ.
- Akinesia or bradykinesia (slowness or lack of movement) can be observed by having the patient walk down a hallway. One may observe reduced arm swing and hesitation in initiating movement.
- Postural instability usually develops later rather than sooner in the disease progression. The patient may need to hold onto someone to maintain balance when getting up or walking.
At least two features must be present to make the diagnosis of parkinsonism. One feature must be tremor or rigidity.
Although the criteria for parkinsonism appear simple, the diagnosis of Parkinson disease is not always clear-cut. For example, shaking can be secondary to a dopamine receptor-blocking medication, to anxiety, or to essential tremor; rigidity and slowness may be due to arthritis; and postural instability can result from a neuropathy. Moreover, other neurodegenerative parkinsonian disorders may respond to levodopa (at least initially) and may present with levodopa-induced dyskinesias. Robust response to levodopa and the occurrence of dyskinesias are two additional features that strongly suggest the diagnosis of Parkinson disease.
Supporting parkinsonian features include stooped posture, masked facies, micrographia (small handwriting), drooling, speech changes (eg, hypophonia or soft speech, stuttering, slurring, monotonic speech), and a shuffling, festinating gait (quick short steps as if falling forward).
PARKINSON MIMICS
Parkinsonism is a broader term than Parkinson disease or idiopathic Parkinson disease. It is characterized by akinetic rigidity and impaired motor activity that leads to reduced function and falls; behavioral changes also may occur.
In the United States, Parkinson disease is the most common cause of parkinsonism. Other nonneurodegenerative causes are drug-induced parkinsonism (due to dopamine receptor antagonists such as antipsychotic or antiemetic drugs), stroke (in the basal ganglia or frontal lobe), and normal-pressure hydrocephalus (causing lower-body parkinsonism). Mimics of parkinsonism include essential tremor and psychogenic parkinsonism.
Parkinsonism can also be caused by Parkinson-plus disorders, ie, neurodegenerative conditions characterized by parkinsonism along with additional signs and symptoms, as listed below. Parkinson-plus disorders include progressive supranuclear palsy, multiple system atrophy, corticobasal degeneration, and Lewy body disease.
Clinical features that suggest a diagnosis other than Parkinson disease include1:
- Poor response to adequate dosages of levodopa
- Early onset of postural instability and falls
- Axial rigidity (eg, stiff neck) more than appendicular rigidity
- Early dementia
- Supranuclear gaze palsy
- Unusual movements besides tremor, eg, limb dystonia, myoclonus, limb levitation or alien limb syndrome
- Profound autonomic dysfunction
- Psychotic symptoms before taking levodopa or dopaminergic medication.
The precise diagnosis of Parkinson-plus disorders is not critical, as the treatment is generally the same for all of them: ie, levodopa (if it shows some efficacy and is well tolerated), with additional symptomatic treatment for features such as depression, cognitive impairment, and autonomic dysfunction, and supportive therapy including physical, occupational, speech, and swallowing therapy.
IMAGING MAY ASSIST IN THE DIAGNOSIS
Dopamine transporter single-photon emission computed tomography (SPECT) is a functional imaging technique that supposedly reflects dopamine uptake by surviving presynaptic dopaminergic neurons in the striate bodies of the basal ganglia. Normal uptake shows distinct cashew-shaped enhancement bilaterally. In Parkinson disease, the enhanced areas are smaller and asymmetric, first with diminution of the tail (representing the putamen), then later involving the head (representing the caudate) along with the other striate bodies (Figure 1).
Dopamine transporter SPECT does not distinguish one neurodegenerative parkinsonian disorder from another. Therefore, it should not be used to distinguish Parkinson disease from other Parkinson-plus syndromes. But it does distinguish neurodegenerative parkinsonian disorders from nonneurodegenerative conditions and mimics, which have a normal result on dopamine transporter SPECT (Table 1).
SLOWING DISEASE PROGRESSION
Current treatments for Parkinson disease can significantly improve symptoms but, unfortunately, do not cure the disease or slow its progression. Testing whether agents modify the disease course is particularly difficult with Parkinson disease, because it affects individuals differently, has a wide spectrum of symptoms, has a long time course, and lacks definitive markers to monitor progression. Some agents have shown promise:
Caffeine. People who drink coffee are less likely to develop Parkinson disease, with the risk declining with the number of cups per day.2 For those who have the disease, drinking coffee is associated with reduced symptoms.
Exercise improves Parkinson disease and may prevent it, and some studies suggest that it can delay its progression.3 Exercise has been shown in an animal model to reduce the vulnerability of dopamine neurons to the toxic agent 6-hydroxydopamine.4 Functional magnetic resonance imaging studies have shown blood flow patterns before and after exercise that are similar to those seen in patients with and without Parkinson medication.3
Rasagiline, a monoamine oxidase B (MAO-B) inhibitor used for symptomatic treatment of Parkinson disease, had conflicting results in a neuroprotective clinical trial. Patients who received rasagiline 1 mg daily—but not those who received 2 mg daily—at the beginning of the trial had better Parkinson motor scores compared with patients who received rasagiline 9 months later.5
Inosine is a urate precursor that elevates urate levels in serum and the central nervous system. For unknown reasons, patients with Parkinson disease tend to have a low uric acid level, and higher levels are associated with milder disease. It is hoped that raising the uric acid level to a “pre-gout level” may slow the progression of Parkinson disease.
Isradipine, a calcium channel blocker, was found in an epidemiologic study of elderly patients to be associated with reduced likelihood of developing Parkinson disease.6 The drug is now undergoing clinical trials.
Smoking. Although cigarette smokers have long been recognized as having a very low risk of developing Parkinson disease, smoking is not recommended.
Agents found ineffective. Agents that have been tested and found ineffective in modifying the course of Parkinson disease include vitamin E, coenzyme Q10, riluzole, GPI-1485, pramipexole, cogane, CEP-1347, TCH-346, and creatine.
NOT JUST DOPAMINE—OR TREMORS
Dopamine deficiency is central to the current understanding of the pathogenesis of Parkinson disease and the focus of treatment efforts, but if dopamine deficiency were the only problem, replacing it should completely ameliorate all parkinsonian features. Other neurotransmitters also play roles: norepinephrine is implicated in orthostatic symptoms and apathy, acetylcholine in cognitive behaviors, glutamate in dyskinesias, and serotonin in depression, anxiety, and sleep abnormalities.
The most recognized area of involvement in the brain has traditionally been the substantia nigra in the midbrain. However, current thinking is that the disease starts lower in the caudal area of the brainstem (along with the olfactory tubercle), moves through the pons to the midbrain, then spreads across the cerebrum with extensive neocortical involvement.
Early premotor indicators are now recognized to occur 15 to 20 years before a tremor appears. The first signs are often hyposmia (diminished sense of smell, reflecting involvement of the olfactory tubercle) and constipation (reflecting involvement of the medulla and the vagus nucleus). With pons involvement, the patient can develop rapid eye movement sleep behavior disorder, depression, or anxiety. Only then does the disease spread to the midbrain and cause resting tremor, rigidity, and bradykinesia.7
Identifying the preclinical stages and starting disease-modifying treatments before the onset of motor symptoms may one day prove important, but at this point, the premotor symptoms (anosmia, constipation, depression) are too nonspecific to be useful, and such treatments have not yet been identified.
TREATMENT: LEVODOPA STILL PRIMARY
When to start drug treatment depends primarily on how much the symptoms bother the patient. Regardless of the clinician’s (or patient’s) belief in the benefits of delaying symptomatic treatment, it is universally considered necessary to start medication when gait problems develop because of the danger of a fall and resulting disability.
Carbidopa-levodopa combination therapy remains the most effective treatment; if it is not effective, another diagnosis may need to be considered. Carbidopa-levodopa improves tremor, rigidity, and bradykinesia, particularly in the early stages of Parkinson disease. It is well tolerated, has rapid onset, reduces the risk of death, and is the least expensive of the medications for Parkinson disease.
Immediate-release and continued-release formulations are available, as well as one that dissolves rapidly on the tongue and can be taken without water. An oral extended-release carbidopa-levodopa formulation (Rytary) was approved by the FDA in January 2015. Tablets are filled with drug-containing microbeads that dissolve at different rates to achieve therapeutic levodopa levels as quickly as the immediate-release formulation and maintain them for an extended time.8
The development of dyskinesias is the major psychological drawback of levodopa, occurring in 80% of patients after 5 to 10 years of treatment. Although many patients fear this side effect, most patients who develop it find it preferable to the rigidity and bradykinesia of Parkinson disease. In most cases, bothersome dyskinesias can be controlled by adjusting medications.9,10
Dopamine agonists include pramipexole, ropinirole, and rotigotine. They are available in generic form as three-times-daily dosing; once-daily dosing is also available, but not as a generic formulation. Dopamine agonists have the advantage of potentially improving depression and delaying the onset of dyskinesias.
However, dopamine agonists have a number of disadvantages compared with levodopa: they have a longer titration period, are less effective, and are less well tolerated, especially in the elderly. Side effects occur more frequently than with levodopa and include general and peripheral edema, hallucinations, nausea, lightheadedness, and sleepiness.11,12 These drugs are also associated with “sleep attacks” (sudden falling asleep while active, such as while driving or eating) and with compulsive and impulsive behaviors such as hypersexuality, buying, binge eating, and gambling. Although these behaviors occur in fewer than 10% of patients, they can be devastating, leading to marital, financial, and legal problems. A bothersome clinical state termed dopamine agonist withdrawal syndrome is characterized by anxiety, depression, jitteriness, and palpitations when dopamine agonists are tapered or discontinued because of a side effect.13
MAO-B inhibitors delay the breakdown of dopamine, allowing it to “stay” in the brain for a longer period of time. Rasagiline for early monotherapy has the advantages of once-daily dosing, no titration, and excellent tolerability, even in the elderly. Potential drug interactions should be considered when using this drug. Early warnings about interactions with tyramine-rich foods were lifted after trials showed that this was not a problem.14
Amantadine is an N-methyl-d-aspartate (NMDA) receptor antagonist often used in early Parkinson disease and for treatment of dyskinesias and fatigue. It is the only drug that is intrinsically antidyskinetic and also improves Parkinson symptoms.15 Side effects include leg swelling, livedo reticularis, and neuropsychiatric and anticholinergic effects.
Anticholinergic agents (eg, trihexyphenidyl) improve tremor but are not as useful for bradykinesia or rigidity, and often have anticholinergic effects such as mental dullness, dry mouth, dry eye, and urinary hesitancy, especially in the elderly, so they have a limited role in Parkinson treatment.
MOTOR COMPLICATIONS: FLUCTUATIONS AND DYSKINESIAS
Motor fluctuations are changes between the akinetic and mobile phases of Parkinson disease, or the off-periods and on-periods of drug treatment. A patient who is “off” is generally rigid and feels that the medication is not working. A patient who is “on” feels loose and mobile and that the medication is working. Variants of motor fluctuations include:
- End-of-dose deterioration
- Delayed onset of response (more than half an hour after taking medication)
- Drug-resistant offs—medication has become ineffective
- Random oscillation—on-off phenomenon
- Freezing—unpredictable inability to start or finish a movement.
Dyskinesias are abnormal involuntary movements such as writhing and twisting. They are associated with dopaminergic therapy at peak dose, when the drug starts to turn on or wear off (termed diphasic dyskinesias).16
The storage hypothesis provides a plausible explanation for the development of motor complications as the disease progresses. Although the half-life of levodopa is only 60 to 90 minutes, it is effective in early disease when given three times a day. It is believed that at this stage of the disease, enough dopaminergic neurons survive to “store” dopamine and release it as needed. As the disease progresses and dopaminergic neurons die, storage capacity diminishes, and the clinical effect slowly starts to approximate the pharmacokinetic profile of the drug. Upon taking the medication, the patient gets a surge of drug, causing dyskinesias, followed later by rigidity as the effect wears off since there are fewer surviving dopaminergic cells to store dopamine.
MANAGING DYSKINESIAS
Patients with dyskinesias should first be asked if they are bothered by them; not all patients are troubled by dyskinesias. If the movements only bother others (eg, family members), then education is often the only treatment needed. If the patient is uncomfortable, the following measures can be tried:
- Taking lower, more frequent doses of levodopa (however, risk of wearing off becomes a problem)
- Adding a dopamine agonist or MAO-B inhibitor while lowering the levodopa dose (however, MAO-B inhibitors pose a risk of side effects in elderly patients)
- Adding clozapine (periodic laboratory testing is required to monitor blood levels and liver and kidney function)
- Adding amantadine (however, this poses a risk of cognitive side effects).
Deep-brain-stimulation surgery is appropriate for select patients who are generally physically healthy, cognitively intact, and emotionally stable, with a strong family support system, but who are bothered by symptoms of parkinsonism (such as tremors), motor fluctuations, or dyskinesias.17
Infusion pump. In January 2015, the FDA approved a new system that continuously delivers levodopa-carbidopa in a 4:1 ratio in gel suspension for 16 hours directly into the small intestine, minimizing motor fluctuations. The patient changes the cartridge daily and turns it off at bedtime.
*Dr. Fernandez has received research support from AbbVie, Acadia, Auspex, Biotie Therapies, Civitas, Kyowa/ProStrakan, Michael J. Fox Foundation, Movement Disorders Society, NIH/NINDS, Parkinson Study Group, Rhythm, Synosia, and Teva. He also has received honoraria from Carling Communications, International Parkinson and Movement Disorders Society, The Ohio State University, and PRIME Education, Inc as a speaker in CME events. He has received honoraria from Biogen, GE Health Care, Lundbeck, Merz Pharmaceuticals, and Pfizer as a consultant. He has received royalty payments from Demos Publishing for serving as a book author/editor. Cleveland Clinic has contracts with AbbVie and Merz Pharmaceuticals for Dr. Fernandez’s role as a member of the Global Steering Committee for LCIG studies and as a consultant or speaker, and as Head Principal Investigator for the Xeomin Registry Study. Dr. Fernandez has received a stipend from International Parkinson and Movement Disorders Society for serving as medical editor of the Movement Disorders Society website.
This has been a boom year for Parkinson disease, with the US Food and Drug Administration (FDA) approving two new therapies, and with others in the pipeline.
This article details clinical signs of Parkinson disease, discusses functional imaging, provides an update on current thinking on disease pathogenesis, and gives an overview of managing parkinsonian symptoms and dyskinesias.
DIAGNOSIS REMAINS CLINICAL
Although a better understanding of Parkinson disease has been gained in recent years, with the recognition of several premotor features and potential biomarkers, its diagnosis is still primarily based on clinical motor findings. The four cardinal motor features have the mnemonic TRAP:
- Tremor at rest can be subtle, involving just the thumb, best observed when the patient is sitting with the hand resting on the lap; or it can be obvious, involving the entire hand, arm, feet, lips, and chin.
- Rigidity can be felt rather than seen, by slowly passively rotating the patient’s wrist or elbow and feeling resistance. The right and left sides often differ.
- Akinesia or bradykinesia (slowness or lack of movement) can be observed by having the patient walk down a hallway. One may observe reduced arm swing and hesitation in initiating movement.
- Postural instability usually develops later rather than sooner in the disease progression. The patient may need to hold onto someone to maintain balance when getting up or walking.
At least two features must be present to make the diagnosis of parkinsonism. One feature must be tremor or rigidity.
Although the criteria for parkinsonism appear simple, the diagnosis of Parkinson disease is not always clear-cut. For example, shaking can be secondary to a dopamine receptor-blocking medication, to anxiety, or to essential tremor; rigidity and slowness may be due to arthritis; and postural instability can result from a neuropathy. Moreover, other neurodegenerative parkinsonian disorders may respond to levodopa (at least initially) and may present with levodopa-induced dyskinesias. Robust response to levodopa and the occurrence of dyskinesias are two additional features that strongly suggest the diagnosis of Parkinson disease.
Supporting parkinsonian features include stooped posture, masked facies, micrographia (small handwriting), drooling, speech changes (eg, hypophonia or soft speech, stuttering, slurring, monotonic speech), and a shuffling, festinating gait (quick short steps as if falling forward).
PARKINSON MIMICS
Parkinsonism is a broader term than Parkinson disease or idiopathic Parkinson disease. It is characterized by akinetic rigidity and impaired motor activity that leads to reduced function and falls; behavioral changes also may occur.
In the United States, Parkinson disease is the most common cause of parkinsonism. Other nonneurodegenerative causes are drug-induced parkinsonism (due to dopamine receptor antagonists such as antipsychotic or antiemetic drugs), stroke (in the basal ganglia or frontal lobe), and normal-pressure hydrocephalus (causing lower-body parkinsonism). Mimics of parkinsonism include essential tremor and psychogenic parkinsonism.
Parkinsonism can also be caused by Parkinson-plus disorders, ie, neurodegenerative conditions characterized by parkinsonism along with additional signs and symptoms, as listed below. Parkinson-plus disorders include progressive supranuclear palsy, multiple system atrophy, corticobasal degeneration, and Lewy body disease.
Clinical features that suggest a diagnosis other than Parkinson disease include1:
- Poor response to adequate dosages of levodopa
- Early onset of postural instability and falls
- Axial rigidity (eg, stiff neck) more than appendicular rigidity
- Early dementia
- Supranuclear gaze palsy
- Unusual movements besides tremor, eg, limb dystonia, myoclonus, limb levitation or alien limb syndrome
- Profound autonomic dysfunction
- Psychotic symptoms before taking levodopa or dopaminergic medication.
The precise diagnosis of Parkinson-plus disorders is not critical, as the treatment is generally the same for all of them: ie, levodopa (if it shows some efficacy and is well tolerated), with additional symptomatic treatment for features such as depression, cognitive impairment, and autonomic dysfunction, and supportive therapy including physical, occupational, speech, and swallowing therapy.
IMAGING MAY ASSIST IN THE DIAGNOSIS
Dopamine transporter single-photon emission computed tomography (SPECT) is a functional imaging technique that supposedly reflects dopamine uptake by surviving presynaptic dopaminergic neurons in the striate bodies of the basal ganglia. Normal uptake shows distinct cashew-shaped enhancement bilaterally. In Parkinson disease, the enhanced areas are smaller and asymmetric, first with diminution of the tail (representing the putamen), then later involving the head (representing the caudate) along with the other striate bodies (Figure 1).
Dopamine transporter SPECT does not distinguish one neurodegenerative parkinsonian disorder from another. Therefore, it should not be used to distinguish Parkinson disease from other Parkinson-plus syndromes. But it does distinguish neurodegenerative parkinsonian disorders from nonneurodegenerative conditions and mimics, which have a normal result on dopamine transporter SPECT (Table 1).
SLOWING DISEASE PROGRESSION
Current treatments for Parkinson disease can significantly improve symptoms but, unfortunately, do not cure the disease or slow its progression. Testing whether agents modify the disease course is particularly difficult with Parkinson disease, because it affects individuals differently, has a wide spectrum of symptoms, has a long time course, and lacks definitive markers to monitor progression. Some agents have shown promise:
Caffeine. People who drink coffee are less likely to develop Parkinson disease, with the risk declining with the number of cups per day.2 For those who have the disease, drinking coffee is associated with reduced symptoms.
Exercise improves Parkinson disease and may prevent it, and some studies suggest that it can delay its progression.3 Exercise has been shown in an animal model to reduce the vulnerability of dopamine neurons to the toxic agent 6-hydroxydopamine.4 Functional magnetic resonance imaging studies have shown blood flow patterns before and after exercise that are similar to those seen in patients with and without Parkinson medication.3
Rasagiline, a monoamine oxidase B (MAO-B) inhibitor used for symptomatic treatment of Parkinson disease, had conflicting results in a neuroprotective clinical trial. Patients who received rasagiline 1 mg daily—but not those who received 2 mg daily—at the beginning of the trial had better Parkinson motor scores compared with patients who received rasagiline 9 months later.5
Inosine is a urate precursor that elevates urate levels in serum and the central nervous system. For unknown reasons, patients with Parkinson disease tend to have a low uric acid level, and higher levels are associated with milder disease. It is hoped that raising the uric acid level to a “pre-gout level” may slow the progression of Parkinson disease.
Isradipine, a calcium channel blocker, was found in an epidemiologic study of elderly patients to be associated with reduced likelihood of developing Parkinson disease.6 The drug is now undergoing clinical trials.
Smoking. Although cigarette smokers have long been recognized as having a very low risk of developing Parkinson disease, smoking is not recommended.
Agents found ineffective. Agents that have been tested and found ineffective in modifying the course of Parkinson disease include vitamin E, coenzyme Q10, riluzole, GPI-1485, pramipexole, cogane, CEP-1347, TCH-346, and creatine.
NOT JUST DOPAMINE—OR TREMORS
Dopamine deficiency is central to the current understanding of the pathogenesis of Parkinson disease and the focus of treatment efforts, but if dopamine deficiency were the only problem, replacing it should completely ameliorate all parkinsonian features. Other neurotransmitters also play roles: norepinephrine is implicated in orthostatic symptoms and apathy, acetylcholine in cognitive behaviors, glutamate in dyskinesias, and serotonin in depression, anxiety, and sleep abnormalities.
The most recognized area of involvement in the brain has traditionally been the substantia nigra in the midbrain. However, current thinking is that the disease starts lower in the caudal area of the brainstem (along with the olfactory tubercle), moves through the pons to the midbrain, then spreads across the cerebrum with extensive neocortical involvement.
Early premotor indicators are now recognized to occur 15 to 20 years before a tremor appears. The first signs are often hyposmia (diminished sense of smell, reflecting involvement of the olfactory tubercle) and constipation (reflecting involvement of the medulla and the vagus nucleus). With pons involvement, the patient can develop rapid eye movement sleep behavior disorder, depression, or anxiety. Only then does the disease spread to the midbrain and cause resting tremor, rigidity, and bradykinesia.7
Identifying the preclinical stages and starting disease-modifying treatments before the onset of motor symptoms may one day prove important, but at this point, the premotor symptoms (anosmia, constipation, depression) are too nonspecific to be useful, and such treatments have not yet been identified.
TREATMENT: LEVODOPA STILL PRIMARY
When to start drug treatment depends primarily on how much the symptoms bother the patient. Regardless of the clinician’s (or patient’s) belief in the benefits of delaying symptomatic treatment, it is universally considered necessary to start medication when gait problems develop because of the danger of a fall and resulting disability.
Carbidopa-levodopa combination therapy remains the most effective treatment; if it is not effective, another diagnosis may need to be considered. Carbidopa-levodopa improves tremor, rigidity, and bradykinesia, particularly in the early stages of Parkinson disease. It is well tolerated, has rapid onset, reduces the risk of death, and is the least expensive of the medications for Parkinson disease.
Immediate-release and continued-release formulations are available, as well as one that dissolves rapidly on the tongue and can be taken without water. An oral extended-release carbidopa-levodopa formulation (Rytary) was approved by the FDA in January 2015. Tablets are filled with drug-containing microbeads that dissolve at different rates to achieve therapeutic levodopa levels as quickly as the immediate-release formulation and maintain them for an extended time.8
The development of dyskinesias is the major psychological drawback of levodopa, occurring in 80% of patients after 5 to 10 years of treatment. Although many patients fear this side effect, most patients who develop it find it preferable to the rigidity and bradykinesia of Parkinson disease. In most cases, bothersome dyskinesias can be controlled by adjusting medications.9,10
Dopamine agonists include pramipexole, ropinirole, and rotigotine. They are available in generic form as three-times-daily dosing; once-daily dosing is also available, but not as a generic formulation. Dopamine agonists have the advantage of potentially improving depression and delaying the onset of dyskinesias.
However, dopamine agonists have a number of disadvantages compared with levodopa: they have a longer titration period, are less effective, and are less well tolerated, especially in the elderly. Side effects occur more frequently than with levodopa and include general and peripheral edema, hallucinations, nausea, lightheadedness, and sleepiness.11,12 These drugs are also associated with “sleep attacks” (sudden falling asleep while active, such as while driving or eating) and with compulsive and impulsive behaviors such as hypersexuality, buying, binge eating, and gambling. Although these behaviors occur in fewer than 10% of patients, they can be devastating, leading to marital, financial, and legal problems. A bothersome clinical state termed dopamine agonist withdrawal syndrome is characterized by anxiety, depression, jitteriness, and palpitations when dopamine agonists are tapered or discontinued because of a side effect.13
MAO-B inhibitors delay the breakdown of dopamine, allowing it to “stay” in the brain for a longer period of time. Rasagiline for early monotherapy has the advantages of once-daily dosing, no titration, and excellent tolerability, even in the elderly. Potential drug interactions should be considered when using this drug. Early warnings about interactions with tyramine-rich foods were lifted after trials showed that this was not a problem.14
Amantadine is an N-methyl-d-aspartate (NMDA) receptor antagonist often used in early Parkinson disease and for treatment of dyskinesias and fatigue. It is the only drug that is intrinsically antidyskinetic and also improves Parkinson symptoms.15 Side effects include leg swelling, livedo reticularis, and neuropsychiatric and anticholinergic effects.
Anticholinergic agents (eg, trihexyphenidyl) improve tremor but are not as useful for bradykinesia or rigidity, and often have anticholinergic effects such as mental dullness, dry mouth, dry eye, and urinary hesitancy, especially in the elderly, so they have a limited role in Parkinson treatment.
MOTOR COMPLICATIONS: FLUCTUATIONS AND DYSKINESIAS
Motor fluctuations are changes between the akinetic and mobile phases of Parkinson disease, or the off-periods and on-periods of drug treatment. A patient who is “off” is generally rigid and feels that the medication is not working. A patient who is “on” feels loose and mobile and that the medication is working. Variants of motor fluctuations include:
- End-of-dose deterioration
- Delayed onset of response (more than half an hour after taking medication)
- Drug-resistant offs—medication has become ineffective
- Random oscillation—on-off phenomenon
- Freezing—unpredictable inability to start or finish a movement.
Dyskinesias are abnormal involuntary movements such as writhing and twisting. They are associated with dopaminergic therapy at peak dose, when the drug starts to turn on or wear off (termed diphasic dyskinesias).16
The storage hypothesis provides a plausible explanation for the development of motor complications as the disease progresses. Although the half-life of levodopa is only 60 to 90 minutes, it is effective in early disease when given three times a day. It is believed that at this stage of the disease, enough dopaminergic neurons survive to “store” dopamine and release it as needed. As the disease progresses and dopaminergic neurons die, storage capacity diminishes, and the clinical effect slowly starts to approximate the pharmacokinetic profile of the drug. Upon taking the medication, the patient gets a surge of drug, causing dyskinesias, followed later by rigidity as the effect wears off since there are fewer surviving dopaminergic cells to store dopamine.
MANAGING DYSKINESIAS
Patients with dyskinesias should first be asked if they are bothered by them; not all patients are troubled by dyskinesias. If the movements only bother others (eg, family members), then education is often the only treatment needed. If the patient is uncomfortable, the following measures can be tried:
- Taking lower, more frequent doses of levodopa (however, risk of wearing off becomes a problem)
- Adding a dopamine agonist or MAO-B inhibitor while lowering the levodopa dose (however, MAO-B inhibitors pose a risk of side effects in elderly patients)
- Adding clozapine (periodic laboratory testing is required to monitor blood levels and liver and kidney function)
- Adding amantadine (however, this poses a risk of cognitive side effects).
Deep-brain-stimulation surgery is appropriate for select patients who are generally physically healthy, cognitively intact, and emotionally stable, with a strong family support system, but who are bothered by symptoms of parkinsonism (such as tremors), motor fluctuations, or dyskinesias.17
Infusion pump. In January 2015, the FDA approved a new system that continuously delivers levodopa-carbidopa in a 4:1 ratio in gel suspension for 16 hours directly into the small intestine, minimizing motor fluctuations. The patient changes the cartridge daily and turns it off at bedtime.
*Dr. Fernandez has received research support from AbbVie, Acadia, Auspex, Biotie Therapies, Civitas, Kyowa/ProStrakan, Michael J. Fox Foundation, Movement Disorders Society, NIH/NINDS, Parkinson Study Group, Rhythm, Synosia, and Teva. He also has received honoraria from Carling Communications, International Parkinson and Movement Disorders Society, The Ohio State University, and PRIME Education, Inc as a speaker in CME events. He has received honoraria from Biogen, GE Health Care, Lundbeck, Merz Pharmaceuticals, and Pfizer as a consultant. He has received royalty payments from Demos Publishing for serving as a book author/editor. Cleveland Clinic has contracts with AbbVie and Merz Pharmaceuticals for Dr. Fernandez’s role as a member of the Global Steering Committee for LCIG studies and as a consultant or speaker, and as Head Principal Investigator for the Xeomin Registry Study. Dr. Fernandez has received a stipend from International Parkinson and Movement Disorders Society for serving as medical editor of the Movement Disorders Society website.
- Wenning GK, Ben-Shlomo Y, Hughes A, Daniel SE, Lees A, Quinn NP. What clinical features are most useful to distinguish definite multiple system atrophy from Parkinson’s disease? J Neurol Neurosurg Psychiatry 2000; 68:434–440.
- Hernán MA, Takkouche B, Caamaño-Isoma F, et al. A meta-analysis of coffee drinking, cigarette smoking, and risk of Parkinson’s disease. Ann Neurol 2002; 52:276–84.
- Ridgel A, Thota A, Vitek JL, Alberts JL. Forced, not voluntary, exercise improves motor function in Parkinson’s disease patients. Neurorehabil Neural Repair 2009; 23:600–608.
- Smith AD, Zigmond MJ. Can the brain be protected through exercise? Lessons from an animal model of parkinsonism. Exp Neurol 2003; 184:31–39.
- Olanow CW, Rascol O, Hauser R, et al, for the ADAGIO Study Investigators. A double-blind, delayed-start trial of rasagiline in Parkinson’s disease. N Engl J Med 2009; 361:1268–1278.
- Pasternak B, Svanström H, Nielsen NM, Fugger L, Melbye M, Hviid A. Use of calcium channel blockers and Parkinson’s disease. Am J Epidemiol 2012; 175:627-635.
- Braak H, Del Tredici K, Rüb U, de Vos RAI, Jansen Steur ENH, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 2003; 24:197–211.
- Hauser RA, Ellenbogen AL, Metman LV, et al. Crossover comparison of IPX066 and a standard levodopa formulation in advanced Parkinson’s disease. Mov Disord 2011; 26:2246–2252.
- Hely MA, Morris JG, Reid WG, Trafficante R. Sydney Multicenter Study of Parkinson’s disease: non-L-dopa-responsive problems dominate at 15 years. Mov Disord 2005; 20:190–199.
- Hung SW, Adeli GM, Arenovich T, Fox SH, Lang AE. Patient perception of dyskinesia in Parkinson’s disease. J Neurol Neurosurg Psychiatry 2010; 81:1112–1115.
- Rascol O, Brooks DJ, Korczyn AD, De Deyn PP, Clarke CE, Lang AE. A five-year study of the incidence of dyskinesia in patients with early Parkinson’s disease who were treated with ropinirole or levodopa. N Engl J Med 2000; 342:1484–1491.
- Parkinson Study Group. Pramipexole vs levodopa as initial treatment for Parkinson disease: a randomized controlled trial. Parkinson Study Group. JAMA 2000; 284:1931–1938.
- Nirenberg MJ. Dopamine agonist withdrawal syndrome: implications for patient care. Drugs Aging 2013; 30:587–592.
- Teva Neuroscience, Inc. Azilect prescribing information. https://www.azilect.com/Content/pdf/azi-40850-azilect-electronic-pi.pdf. Accessed June 29, 2015.
- Snow BJ, Macdonald L, Mcauley D, Wallis W. The effect of amantadine on levodopa-induced dyskinesias in Parkinson’s disease: a double-blind, placebo-controlled study. Clin Neuropharmacol 2000; 23:82–85.
- Adler CH, Ahlskog JE, eds. Parkinson’s Disease and Movement Disorders: Diagnosis and Treatment Guidelines for the Practicing Physician. Totowa, NJ: Humana Press; 2000.
- Machado A, Fernandez HH, Deogaonkar M. Deep brain stimulation: what can patients expect from it? Cleve Clin J Med 2012; 79:113–120.
- Wenning GK, Ben-Shlomo Y, Hughes A, Daniel SE, Lees A, Quinn NP. What clinical features are most useful to distinguish definite multiple system atrophy from Parkinson’s disease? J Neurol Neurosurg Psychiatry 2000; 68:434–440.
- Hernán MA, Takkouche B, Caamaño-Isoma F, et al. A meta-analysis of coffee drinking, cigarette smoking, and risk of Parkinson’s disease. Ann Neurol 2002; 52:276–84.
- Ridgel A, Thota A, Vitek JL, Alberts JL. Forced, not voluntary, exercise improves motor function in Parkinson’s disease patients. Neurorehabil Neural Repair 2009; 23:600–608.
- Smith AD, Zigmond MJ. Can the brain be protected through exercise? Lessons from an animal model of parkinsonism. Exp Neurol 2003; 184:31–39.
- Olanow CW, Rascol O, Hauser R, et al, for the ADAGIO Study Investigators. A double-blind, delayed-start trial of rasagiline in Parkinson’s disease. N Engl J Med 2009; 361:1268–1278.
- Pasternak B, Svanström H, Nielsen NM, Fugger L, Melbye M, Hviid A. Use of calcium channel blockers and Parkinson’s disease. Am J Epidemiol 2012; 175:627-635.
- Braak H, Del Tredici K, Rüb U, de Vos RAI, Jansen Steur ENH, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 2003; 24:197–211.
- Hauser RA, Ellenbogen AL, Metman LV, et al. Crossover comparison of IPX066 and a standard levodopa formulation in advanced Parkinson’s disease. Mov Disord 2011; 26:2246–2252.
- Hely MA, Morris JG, Reid WG, Trafficante R. Sydney Multicenter Study of Parkinson’s disease: non-L-dopa-responsive problems dominate at 15 years. Mov Disord 2005; 20:190–199.
- Hung SW, Adeli GM, Arenovich T, Fox SH, Lang AE. Patient perception of dyskinesia in Parkinson’s disease. J Neurol Neurosurg Psychiatry 2010; 81:1112–1115.
- Rascol O, Brooks DJ, Korczyn AD, De Deyn PP, Clarke CE, Lang AE. A five-year study of the incidence of dyskinesia in patients with early Parkinson’s disease who were treated with ropinirole or levodopa. N Engl J Med 2000; 342:1484–1491.
- Parkinson Study Group. Pramipexole vs levodopa as initial treatment for Parkinson disease: a randomized controlled trial. Parkinson Study Group. JAMA 2000; 284:1931–1938.
- Nirenberg MJ. Dopamine agonist withdrawal syndrome: implications for patient care. Drugs Aging 2013; 30:587–592.
- Teva Neuroscience, Inc. Azilect prescribing information. https://www.azilect.com/Content/pdf/azi-40850-azilect-electronic-pi.pdf. Accessed June 29, 2015.
- Snow BJ, Macdonald L, Mcauley D, Wallis W. The effect of amantadine on levodopa-induced dyskinesias in Parkinson’s disease: a double-blind, placebo-controlled study. Clin Neuropharmacol 2000; 23:82–85.
- Adler CH, Ahlskog JE, eds. Parkinson’s Disease and Movement Disorders: Diagnosis and Treatment Guidelines for the Practicing Physician. Totowa, NJ: Humana Press; 2000.
- Machado A, Fernandez HH, Deogaonkar M. Deep brain stimulation: what can patients expect from it? Cleve Clin J Med 2012; 79:113–120.
KEY POINTS
- Parkinson disease is diagnosed by clinical signs with the mnemonic TRAP: Tremor at rest, Rigidity, Akinesia or bradykinesia, and Postural/gait instability.
- A dopamine transporter functional scan can distinguish neurodegenerative parkinsonian disorders from nonneurodegenerative etiologies such as drug-induced parkinsonism and vascular parkinsonism, and from mimics such as psychogenic parkinsonism and essential tremor.
- Coffee consumption and exercise may benefit patients with Parkinson disease.
- Carbidopa-levodopa combination therapy is still the most effective treatment, but most patients develop dyskinesia after 5 to 10 years of treatment.
- Dyskinesias can be managed by adjusting or changing medications, switching to the new levodopa infusion pump system, or with deep-brain-stimulation surgery.