User login
FDA approves first treatment for neuromyelitis optica spectrum disorder

Soliris, a complement inhibitor, is the first FDA-approved treatment for NMOSD, a rare autoimmune disease of the central nervous system that mainly affects the optic nerves and spinal cord, according to a news release.
About 73% of patients with NMOSD test positive for anti-AQP4 antibodies, and complement activation resulting from anti-AQP4 antibodies is an underlying cause of the disease, according to the news release from Alexion, the company that markets the drug. The average age of NMOSD onset is 39 years, and the disease can lead to permanent visual impairment and paralysis. The condition, previously known as Devic’s disease, may affect between 4,000 and 8,000 people in the United States. NMOSD may be confused with other neurologic conditions such as multiple sclerosis.
Investigators studied the drug’s effectiveness in a placebo-controlled clinical trial of 143 patients with NMOSD who had anti-AQP4 antibodies. Compared with placebo, Soliris reduced the number of NMOSD relapses by 94% during the 48-week study. Nearly 98% of patients in the PREVENT trial who received Soliris were relapse-free after 48 weeks, compared with 63% of patients who received placebo.
Soliris also reduced hospitalizations and the need for corticosteroids and plasma exchange to treat acute attacks.
Soliris includes a boxed warning about life-threatening and fatal meningococcal infections that have occurred in patients treated with Soliris. Patients should be monitored and evaluated immediately if infection is suspected, according to the FDA announcement. In addition, health care professionals should use caution when administering Soliris to patients with any other infection. No cases of meningococcal infection were observed in the PREVENT trial.
Soliris is available through a restricted program under a Risk Evaluation and Mitigation Strategy (REMS). Prescribers must counsel patients about the risk of meningococcal infection and ensure that patients have been vaccinated with meningococcal vaccines.
Adverse reactions in the NMOSD clinical trial included upper respiratory infection, nasopharyngitis, diarrhea, back pain, dizziness, influenza, joint pain, sore throat, and confusion.
The drug’s use for NMOSD received Orphan Drug designation, which provides incentives for the development of drugs for rare diseases.
Eculizumab first was approved by the FDA in 2007 and also may be used to treat paroxysmal nocturnal hemoglobinuria, atypical hemolytic uremic syndrome, and myasthenia gravis.
Soliris, a complement inhibitor, is the first FDA-approved treatment for NMOSD, a rare autoimmune disease of the central nervous system that mainly affects the optic nerves and spinal cord, according to a news release.
About 73% of patients with NMOSD test positive for anti-AQP4 antibodies, and complement activation resulting from anti-AQP4 antibodies is an underlying cause of the disease, according to the news release from Alexion, the company that markets the drug. The average age of NMOSD onset is 39 years, and the disease can lead to permanent visual impairment and paralysis. The condition, previously known as Devic’s disease, may affect between 4,000 and 8,000 people in the United States. NMOSD may be confused with other neurologic conditions such as multiple sclerosis.
Investigators studied the drug’s effectiveness in a placebo-controlled clinical trial of 143 patients with NMOSD who had anti-AQP4 antibodies. Compared with placebo, Soliris reduced the number of NMOSD relapses by 94% during the 48-week study. Nearly 98% of patients in the PREVENT trial who received Soliris were relapse-free after 48 weeks, compared with 63% of patients who received placebo.
Soliris also reduced hospitalizations and the need for corticosteroids and plasma exchange to treat acute attacks.
Soliris includes a boxed warning about life-threatening and fatal meningococcal infections that have occurred in patients treated with Soliris. Patients should be monitored and evaluated immediately if infection is suspected, according to the FDA announcement. In addition, health care professionals should use caution when administering Soliris to patients with any other infection. No cases of meningococcal infection were observed in the PREVENT trial.
Soliris is available through a restricted program under a Risk Evaluation and Mitigation Strategy (REMS). Prescribers must counsel patients about the risk of meningococcal infection and ensure that patients have been vaccinated with meningococcal vaccines.
Adverse reactions in the NMOSD clinical trial included upper respiratory infection, nasopharyngitis, diarrhea, back pain, dizziness, influenza, joint pain, sore throat, and confusion.
The drug’s use for NMOSD received Orphan Drug designation, which provides incentives for the development of drugs for rare diseases.
Eculizumab first was approved by the FDA in 2007 and also may be used to treat paroxysmal nocturnal hemoglobinuria, atypical hemolytic uremic syndrome, and myasthenia gravis.
Soliris, a complement inhibitor, is the first FDA-approved treatment for NMOSD, a rare autoimmune disease of the central nervous system that mainly affects the optic nerves and spinal cord, according to a news release.
About 73% of patients with NMOSD test positive for anti-AQP4 antibodies, and complement activation resulting from anti-AQP4 antibodies is an underlying cause of the disease, according to the news release from Alexion, the company that markets the drug. The average age of NMOSD onset is 39 years, and the disease can lead to permanent visual impairment and paralysis. The condition, previously known as Devic’s disease, may affect between 4,000 and 8,000 people in the United States. NMOSD may be confused with other neurologic conditions such as multiple sclerosis.
Investigators studied the drug’s effectiveness in a placebo-controlled clinical trial of 143 patients with NMOSD who had anti-AQP4 antibodies. Compared with placebo, Soliris reduced the number of NMOSD relapses by 94% during the 48-week study. Nearly 98% of patients in the PREVENT trial who received Soliris were relapse-free after 48 weeks, compared with 63% of patients who received placebo.
Soliris also reduced hospitalizations and the need for corticosteroids and plasma exchange to treat acute attacks.
Soliris includes a boxed warning about life-threatening and fatal meningococcal infections that have occurred in patients treated with Soliris. Patients should be monitored and evaluated immediately if infection is suspected, according to the FDA announcement. In addition, health care professionals should use caution when administering Soliris to patients with any other infection. No cases of meningococcal infection were observed in the PREVENT trial.
Soliris is available through a restricted program under a Risk Evaluation and Mitigation Strategy (REMS). Prescribers must counsel patients about the risk of meningococcal infection and ensure that patients have been vaccinated with meningococcal vaccines.
Adverse reactions in the NMOSD clinical trial included upper respiratory infection, nasopharyngitis, diarrhea, back pain, dizziness, influenza, joint pain, sore throat, and confusion.
The drug’s use for NMOSD received Orphan Drug designation, which provides incentives for the development of drugs for rare diseases.
Eculizumab first was approved by the FDA in 2007 and also may be used to treat paroxysmal nocturnal hemoglobinuria, atypical hemolytic uremic syndrome, and myasthenia gravis.
FDA expands Doptelet approval to ITP patients with thrombocytopenia
The Food and Drug Administration has approved a supplemental New Drug Application expanding the indication of avatrombopag (Doptelet) to include treatment of thrombocytopenia in adults with chronic immune thrombocytopenia (ITP) with insufficient response to previous therapy, according to Dova Pharmaceuticals.

FDA approval was based on results of a phase 3 trial in which a majority of patients who received avatrombopag achieved a platelet count of at least 50,000 per mcg after 8 days of therapy. In addition, efficacy was superior to patients in the placebo group in the maintenance of platelet counts during the 6-month treatment period.
Avatrombopag – an oral, thrombopoietin receptor agonist administered with food – was previously indicated for the treatment of chronic liver disease in adult patients who are scheduled to undergo a procedure. The most common adverse reactions in patients with ITP include headache, fatigue, contusion, epistaxis, upper respiratory tract infection, arthralgia, gingival bleeding, petechiae, and nasopharyngitis.
Find the full press release on the Dova Pharmaceuticals website.
The Food and Drug Administration has approved a supplemental New Drug Application expanding the indication of avatrombopag (Doptelet) to include treatment of thrombocytopenia in adults with chronic immune thrombocytopenia (ITP) with insufficient response to previous therapy, according to Dova Pharmaceuticals.
FDA approval was based on results of a phase 3 trial in which a majority of patients who received avatrombopag achieved a platelet count of at least 50,000 per mcg after 8 days of therapy. In addition, efficacy was superior to patients in the placebo group in the maintenance of platelet counts during the 6-month treatment period.
Avatrombopag – an oral, thrombopoietin receptor agonist administered with food – was previously indicated for the treatment of chronic liver disease in adult patients who are scheduled to undergo a procedure. The most common adverse reactions in patients with ITP include headache, fatigue, contusion, epistaxis, upper respiratory tract infection, arthralgia, gingival bleeding, petechiae, and nasopharyngitis.
Find the full press release on the Dova Pharmaceuticals website.
The Food and Drug Administration has approved a supplemental New Drug Application expanding the indication of avatrombopag (Doptelet) to include treatment of thrombocytopenia in adults with chronic immune thrombocytopenia (ITP) with insufficient response to previous therapy, according to Dova Pharmaceuticals.
FDA approval was based on results of a phase 3 trial in which a majority of patients who received avatrombopag achieved a platelet count of at least 50,000 per mcg after 8 days of therapy. In addition, efficacy was superior to patients in the placebo group in the maintenance of platelet counts during the 6-month treatment period.
Avatrombopag – an oral, thrombopoietin receptor agonist administered with food – was previously indicated for the treatment of chronic liver disease in adult patients who are scheduled to undergo a procedure. The most common adverse reactions in patients with ITP include headache, fatigue, contusion, epistaxis, upper respiratory tract infection, arthralgia, gingival bleeding, petechiae, and nasopharyngitis.
Find the full press release on the Dova Pharmaceuticals website.
ACIP adds hexavalent vaccine to VFC program
The pediatric hexavalent vaccine (DTaP-[inactivated poliovirus] IPV-[hepatitis B] HepB-[Haemophilis influenzae type b] Hib) should be included as an option in the Vaccines for Children (VFC) program for the infant series at ages 2, 4, and 6 months, according to unanimous votes at a meeting of the Centers for Disease Control and Prevention’s Advisory Committee on Immunization Practices.
The addition of the vaccine to the VFC program required no motions on the part of the committee, but involved separate votes on each component of the vaccine.
Combination vaccination has been associated with increased coverage and more likely completion of the full infant vaccine series, said Sara Oliver, MD, of the CDC’s National Center for Immunization and Respiratory Diseases.
The new vaccine is being developed jointly by Sanofi and Merck, and has been approved by the Food and Drug Administration for use in children through age 4 years.
Dr. Oliver presented evidence that the safety profile of the combination vaccine is consistent with that of the component vaccines. In addition, “use of combination vaccines can reduce the number of injections patient receive and alleviate concern associated with the number of injections,” she said. However, “considerations should include provider assessment, patient preference, and the potential for adverse events.”
although it will not be available until 2021 in order to ensure sufficient supply, Dr. Oliver noted.
The combination vaccination work group considered whether the new vaccine should be preferentially recommended for American Indian and Alaskan Native populations, but they concluded that post–dose one immunogenicity data are needed before such a preferential recommendation can be made.
The ACIP members had no financial conflicts to disclose.
The pediatric hexavalent vaccine (DTaP-[inactivated poliovirus] IPV-[hepatitis B] HepB-[Haemophilis influenzae type b] Hib) should be included as an option in the Vaccines for Children (VFC) program for the infant series at ages 2, 4, and 6 months, according to unanimous votes at a meeting of the Centers for Disease Control and Prevention’s Advisory Committee on Immunization Practices.
The addition of the vaccine to the VFC program required no motions on the part of the committee, but involved separate votes on each component of the vaccine.
Combination vaccination has been associated with increased coverage and more likely completion of the full infant vaccine series, said Sara Oliver, MD, of the CDC’s National Center for Immunization and Respiratory Diseases.
The new vaccine is being developed jointly by Sanofi and Merck, and has been approved by the Food and Drug Administration for use in children through age 4 years.
Dr. Oliver presented evidence that the safety profile of the combination vaccine is consistent with that of the component vaccines. In addition, “use of combination vaccines can reduce the number of injections patient receive and alleviate concern associated with the number of injections,” she said. However, “considerations should include provider assessment, patient preference, and the potential for adverse events.”
although it will not be available until 2021 in order to ensure sufficient supply, Dr. Oliver noted.
The combination vaccination work group considered whether the new vaccine should be preferentially recommended for American Indian and Alaskan Native populations, but they concluded that post–dose one immunogenicity data are needed before such a preferential recommendation can be made.
The ACIP members had no financial conflicts to disclose.
The pediatric hexavalent vaccine (DTaP-[inactivated poliovirus] IPV-[hepatitis B] HepB-[Haemophilis influenzae type b] Hib) should be included as an option in the Vaccines for Children (VFC) program for the infant series at ages 2, 4, and 6 months, according to unanimous votes at a meeting of the Centers for Disease Control and Prevention’s Advisory Committee on Immunization Practices.
The addition of the vaccine to the VFC program required no motions on the part of the committee, but involved separate votes on each component of the vaccine.
Combination vaccination has been associated with increased coverage and more likely completion of the full infant vaccine series, said Sara Oliver, MD, of the CDC’s National Center for Immunization and Respiratory Diseases.
The new vaccine is being developed jointly by Sanofi and Merck, and has been approved by the Food and Drug Administration for use in children through age 4 years.
Dr. Oliver presented evidence that the safety profile of the combination vaccine is consistent with that of the component vaccines. In addition, “use of combination vaccines can reduce the number of injections patient receive and alleviate concern associated with the number of injections,” she said. However, “considerations should include provider assessment, patient preference, and the potential for adverse events.”
although it will not be available until 2021 in order to ensure sufficient supply, Dr. Oliver noted.
The combination vaccination work group considered whether the new vaccine should be preferentially recommended for American Indian and Alaskan Native populations, but they concluded that post–dose one immunogenicity data are needed before such a preferential recommendation can be made.
The ACIP members had no financial conflicts to disclose.
REPORTING FROM AN ACIP MEETING
ACIP favors shared decision on pneumococcal vaccine for older adults
Pneumococcal vaccination with the 13-valent pneumococcal conjugate vaccine (PCV13) based on shared clinical decision making is recommended for immunocompetent adults aged 65 years and older who have not previously received PCV13, and all adults aged 65 years and older should continue to receive the pneumococcal polysaccharide vaccine (PPSV23), according to a vote at a meeting of the Centers for Disease Control and Prevention’s Advisory Committee on Immunization Practices.

The motion passed with an 11-1 vote after members voted down two other options to either discontinue or continue the current recommendation of PCV13 for all immunocompetent adults aged 65 years and older. The current recommendation for PCV13 for adults aged 65 years and older has been in place since 2014.
The pneumococcal work group assessed indirect effects of the pediatric PCV vaccination on older adults prior to 2014 and since 2014, and what additional benefits might be expected if routine vaccination of older adults continued.
“Indirect effects have been observed in all age groups” said Almea Matanock, MD, of the CDC’s National Center for Immunization and Respiratory Diseases. Although there were no safety concerns, the public health impact of continued vaccination of adults was minimal.
Although PCV13 resulted in a 75% reduction in vaccine-type invasive pneumococcal disease and a 45% reduction in vaccine-type nonbacteremic pneumonia in 2014, the annual number needed to vaccinate to prevent a single case of outpatient pneumonia was 2,600, said Dr. Matanock.
Dr. Matanock presented key issues from the Evidence to Recommendations Framework for and against the recommendation for PCV13 in older adults. Work group comments in favor of continuing the recommendation for PCV13 in older adults included effective disease prevention and the potential negative impact on the importance of adult vaccines if the vaccine was no longer recommended. However, some work group members and committee members expressed concern about resource allocation and steering vaccines away from younger age groups in whom they have been more consistently effective.
Paul Hunter, MD, of the City of Milwaukee Health Department, voted against the shared clinical decision making, and instead favored discontinuing the recommendation for PCV13 for older adults. “I think clinicians need a clear message,” he said, adding that “the public health bang for the buck is with the kids.”
“I think there was a recognition that the population level benefit is minimal,” said work group chair Grace Lee, MD.
Although the work group recognized some benefit for older adults, the burden of disease for PCV-specific disease is low, compared with all-cause pneumonia, said Dr. Lee of Lucile Packard Children’s Hospital at Stanford, Calif. However, the recommendation for shared clinical decision making allows for potential insurance coverage of the vaccine for adults who decide after discussion with their health care provider that they would benefit.
“We are still unpacking this construct” of shared clinical decision making, which in this case applies to adults without immunocompromising conditions, and is more of a provider assessment than a risk assessment, she said.
The ACIP members had no financial conflicts to disclose.
Pneumococcal vaccination with the 13-valent pneumococcal conjugate vaccine (PCV13) based on shared clinical decision making is recommended for immunocompetent adults aged 65 years and older who have not previously received PCV13, and all adults aged 65 years and older should continue to receive the pneumococcal polysaccharide vaccine (PPSV23), according to a vote at a meeting of the Centers for Disease Control and Prevention’s Advisory Committee on Immunization Practices.
The motion passed with an 11-1 vote after members voted down two other options to either discontinue or continue the current recommendation of PCV13 for all immunocompetent adults aged 65 years and older. The current recommendation for PCV13 for adults aged 65 years and older has been in place since 2014.
The pneumococcal work group assessed indirect effects of the pediatric PCV vaccination on older adults prior to 2014 and since 2014, and what additional benefits might be expected if routine vaccination of older adults continued.
“Indirect effects have been observed in all age groups” said Almea Matanock, MD, of the CDC’s National Center for Immunization and Respiratory Diseases. Although there were no safety concerns, the public health impact of continued vaccination of adults was minimal.
Although PCV13 resulted in a 75% reduction in vaccine-type invasive pneumococcal disease and a 45% reduction in vaccine-type nonbacteremic pneumonia in 2014, the annual number needed to vaccinate to prevent a single case of outpatient pneumonia was 2,600, said Dr. Matanock.
Dr. Matanock presented key issues from the Evidence to Recommendations Framework for and against the recommendation for PCV13 in older adults. Work group comments in favor of continuing the recommendation for PCV13 in older adults included effective disease prevention and the potential negative impact on the importance of adult vaccines if the vaccine was no longer recommended. However, some work group members and committee members expressed concern about resource allocation and steering vaccines away from younger age groups in whom they have been more consistently effective.
Paul Hunter, MD, of the City of Milwaukee Health Department, voted against the shared clinical decision making, and instead favored discontinuing the recommendation for PCV13 for older adults. “I think clinicians need a clear message,” he said, adding that “the public health bang for the buck is with the kids.”
“I think there was a recognition that the population level benefit is minimal,” said work group chair Grace Lee, MD.
Although the work group recognized some benefit for older adults, the burden of disease for PCV-specific disease is low, compared with all-cause pneumonia, said Dr. Lee of Lucile Packard Children’s Hospital at Stanford, Calif. However, the recommendation for shared clinical decision making allows for potential insurance coverage of the vaccine for adults who decide after discussion with their health care provider that they would benefit.
“We are still unpacking this construct” of shared clinical decision making, which in this case applies to adults without immunocompromising conditions, and is more of a provider assessment than a risk assessment, she said.
The ACIP members had no financial conflicts to disclose.
Pneumococcal vaccination with the 13-valent pneumococcal conjugate vaccine (PCV13) based on shared clinical decision making is recommended for immunocompetent adults aged 65 years and older who have not previously received PCV13, and all adults aged 65 years and older should continue to receive the pneumococcal polysaccharide vaccine (PPSV23), according to a vote at a meeting of the Centers for Disease Control and Prevention’s Advisory Committee on Immunization Practices.
The motion passed with an 11-1 vote after members voted down two other options to either discontinue or continue the current recommendation of PCV13 for all immunocompetent adults aged 65 years and older. The current recommendation for PCV13 for adults aged 65 years and older has been in place since 2014.
The pneumococcal work group assessed indirect effects of the pediatric PCV vaccination on older adults prior to 2014 and since 2014, and what additional benefits might be expected if routine vaccination of older adults continued.
“Indirect effects have been observed in all age groups” said Almea Matanock, MD, of the CDC’s National Center for Immunization and Respiratory Diseases. Although there were no safety concerns, the public health impact of continued vaccination of adults was minimal.
Although PCV13 resulted in a 75% reduction in vaccine-type invasive pneumococcal disease and a 45% reduction in vaccine-type nonbacteremic pneumonia in 2014, the annual number needed to vaccinate to prevent a single case of outpatient pneumonia was 2,600, said Dr. Matanock.
Dr. Matanock presented key issues from the Evidence to Recommendations Framework for and against the recommendation for PCV13 in older adults. Work group comments in favor of continuing the recommendation for PCV13 in older adults included effective disease prevention and the potential negative impact on the importance of adult vaccines if the vaccine was no longer recommended. However, some work group members and committee members expressed concern about resource allocation and steering vaccines away from younger age groups in whom they have been more consistently effective.
Paul Hunter, MD, of the City of Milwaukee Health Department, voted against the shared clinical decision making, and instead favored discontinuing the recommendation for PCV13 for older adults. “I think clinicians need a clear message,” he said, adding that “the public health bang for the buck is with the kids.”
“I think there was a recognition that the population level benefit is minimal,” said work group chair Grace Lee, MD.
Although the work group recognized some benefit for older adults, the burden of disease for PCV-specific disease is low, compared with all-cause pneumonia, said Dr. Lee of Lucile Packard Children’s Hospital at Stanford, Calif. However, the recommendation for shared clinical decision making allows for potential insurance coverage of the vaccine for adults who decide after discussion with their health care provider that they would benefit.
“We are still unpacking this construct” of shared clinical decision making, which in this case applies to adults without immunocompromising conditions, and is more of a provider assessment than a risk assessment, she said.
The ACIP members had no financial conflicts to disclose.
REPORTING FROM AN ACIP MEETING
ACIP extends HPV vaccine coverage
according to a unanimous vote at a meeting of the Centers for Disease Control and Prevention’s Advisory Committee on Immunization Practices.
This change affects males aged 22 through 26 years; the HPV vaccine is currently recommended for males and females aged 11 or 12 years, with catch-up vaccination through age 21 for males and age 26 for females.
The change was supported in part by increased interest in simplifying and harmonizing the vaccine schedule, said Lauri Markowitz, MD, of the CDC’s National Center for Immunization and Respiratory Diseases (NCIRD), who presented the HPV work group’s considerations.
In addition, the committee voted 10-4 in favor of catch-up HPV vaccination, based on shared clinical decision making, for all adults aged 27 through 45 years.
Although the current program of HPV vaccination for youth has demonstrated effectiveness, data from multiple models suggest that widespread HPV vaccination for adults older than 26 years is much less cost effective, and would yield relatively small additional health benefits, Dr. Markowitz said.
The HPV work group reviewed data from a range of clinical trials, epidemiology, and natural history, as well as results from five different health economic models. They concluded that an assessment of benefits and harms favors expanding the catch-up vaccination to all individuals through 26 years, said Elissa Meites, MD, of the CDC, who presented the official work group opinion. The group’s opinion on the second question was that the additional population level benefit of expanding HPV vaccination to all adults would be minimal and not a reasonable and effective allocation of resources, but that shared clinical decision making would allow flexibility.
The committee expressed strong opinions about the potential for shared clinical decision making as a policy for vaccination for adults older than 26 years. Some felt that this option was a way to include adults at risk for HPV, such as divorced women with new partners, or women getting married for the first time later in life who might not have been exposed to HPV through other relationships. In addition, supporters noted that the shared clinical decision-making option would allow for potential insurance coverage, and would involve discussion between doctors and patients to assess risk.
However, other committee members felt that any recommendation for older adult vaccination would distract clinicians from the importance and value of HPV vaccination for the target age group of 11- and 12-year-olds, and might divert resources from the younger age group in whom it has shown the most benefit.
Resource allocation was a concern voiced by many committee members. Kelly Moore, MD, MPH, of Vanderbilt University, Nashville, Tenn., said she voted no on expanding vaccination to older adults because “we didn’t have details on shared clinical decision making, in the absence of information on what that meant, and in the presence of supply questions, I didn’t feel comfortable expanding vaccination to a huge population,” she said.
Paul Hunter, MD, of the City of Milwaukee Health Department, also voted no, and expressed concern that expanding the HPV vaccination recommendations to older adults would send the message that vaccination for children and teens is not effective or important.
The text of the new recommendations for routine and catch-up vaccination states that the recommendations “also apply to MSM [men who have sex with men], transgender people, and people with immunocompromising conditions.”
The ACIP members had no financial conflicts to disclose.
according to a unanimous vote at a meeting of the Centers for Disease Control and Prevention’s Advisory Committee on Immunization Practices.
This change affects males aged 22 through 26 years; the HPV vaccine is currently recommended for males and females aged 11 or 12 years, with catch-up vaccination through age 21 for males and age 26 for females.
The change was supported in part by increased interest in simplifying and harmonizing the vaccine schedule, said Lauri Markowitz, MD, of the CDC’s National Center for Immunization and Respiratory Diseases (NCIRD), who presented the HPV work group’s considerations.
In addition, the committee voted 10-4 in favor of catch-up HPV vaccination, based on shared clinical decision making, for all adults aged 27 through 45 years.
Although the current program of HPV vaccination for youth has demonstrated effectiveness, data from multiple models suggest that widespread HPV vaccination for adults older than 26 years is much less cost effective, and would yield relatively small additional health benefits, Dr. Markowitz said.
The HPV work group reviewed data from a range of clinical trials, epidemiology, and natural history, as well as results from five different health economic models. They concluded that an assessment of benefits and harms favors expanding the catch-up vaccination to all individuals through 26 years, said Elissa Meites, MD, of the CDC, who presented the official work group opinion. The group’s opinion on the second question was that the additional population level benefit of expanding HPV vaccination to all adults would be minimal and not a reasonable and effective allocation of resources, but that shared clinical decision making would allow flexibility.
The committee expressed strong opinions about the potential for shared clinical decision making as a policy for vaccination for adults older than 26 years. Some felt that this option was a way to include adults at risk for HPV, such as divorced women with new partners, or women getting married for the first time later in life who might not have been exposed to HPV through other relationships. In addition, supporters noted that the shared clinical decision-making option would allow for potential insurance coverage, and would involve discussion between doctors and patients to assess risk.
However, other committee members felt that any recommendation for older adult vaccination would distract clinicians from the importance and value of HPV vaccination for the target age group of 11- and 12-year-olds, and might divert resources from the younger age group in whom it has shown the most benefit.
Resource allocation was a concern voiced by many committee members. Kelly Moore, MD, MPH, of Vanderbilt University, Nashville, Tenn., said she voted no on expanding vaccination to older adults because “we didn’t have details on shared clinical decision making, in the absence of information on what that meant, and in the presence of supply questions, I didn’t feel comfortable expanding vaccination to a huge population,” she said.
Paul Hunter, MD, of the City of Milwaukee Health Department, also voted no, and expressed concern that expanding the HPV vaccination recommendations to older adults would send the message that vaccination for children and teens is not effective or important.
The text of the new recommendations for routine and catch-up vaccination states that the recommendations “also apply to MSM [men who have sex with men], transgender people, and people with immunocompromising conditions.”
The ACIP members had no financial conflicts to disclose.
according to a unanimous vote at a meeting of the Centers for Disease Control and Prevention’s Advisory Committee on Immunization Practices.
This change affects males aged 22 through 26 years; the HPV vaccine is currently recommended for males and females aged 11 or 12 years, with catch-up vaccination through age 21 for males and age 26 for females.
The change was supported in part by increased interest in simplifying and harmonizing the vaccine schedule, said Lauri Markowitz, MD, of the CDC’s National Center for Immunization and Respiratory Diseases (NCIRD), who presented the HPV work group’s considerations.
In addition, the committee voted 10-4 in favor of catch-up HPV vaccination, based on shared clinical decision making, for all adults aged 27 through 45 years.
Although the current program of HPV vaccination for youth has demonstrated effectiveness, data from multiple models suggest that widespread HPV vaccination for adults older than 26 years is much less cost effective, and would yield relatively small additional health benefits, Dr. Markowitz said.
The HPV work group reviewed data from a range of clinical trials, epidemiology, and natural history, as well as results from five different health economic models. They concluded that an assessment of benefits and harms favors expanding the catch-up vaccination to all individuals through 26 years, said Elissa Meites, MD, of the CDC, who presented the official work group opinion. The group’s opinion on the second question was that the additional population level benefit of expanding HPV vaccination to all adults would be minimal and not a reasonable and effective allocation of resources, but that shared clinical decision making would allow flexibility.
The committee expressed strong opinions about the potential for shared clinical decision making as a policy for vaccination for adults older than 26 years. Some felt that this option was a way to include adults at risk for HPV, such as divorced women with new partners, or women getting married for the first time later in life who might not have been exposed to HPV through other relationships. In addition, supporters noted that the shared clinical decision-making option would allow for potential insurance coverage, and would involve discussion between doctors and patients to assess risk.
However, other committee members felt that any recommendation for older adult vaccination would distract clinicians from the importance and value of HPV vaccination for the target age group of 11- and 12-year-olds, and might divert resources from the younger age group in whom it has shown the most benefit.
Resource allocation was a concern voiced by many committee members. Kelly Moore, MD, MPH, of Vanderbilt University, Nashville, Tenn., said she voted no on expanding vaccination to older adults because “we didn’t have details on shared clinical decision making, in the absence of information on what that meant, and in the presence of supply questions, I didn’t feel comfortable expanding vaccination to a huge population,” she said.
Paul Hunter, MD, of the City of Milwaukee Health Department, also voted no, and expressed concern that expanding the HPV vaccination recommendations to older adults would send the message that vaccination for children and teens is not effective or important.
The text of the new recommendations for routine and catch-up vaccination states that the recommendations “also apply to MSM [men who have sex with men], transgender people, and people with immunocompromising conditions.”
The ACIP members had no financial conflicts to disclose.
REPORTING FROM AN ACIP MEETING
FDA approves dupilumab for chronic rhinosinusitis with nasal polyps
The Food and Drug Administration has approved dupilumab (Dupixent) for the treatment of chronic rhinosinusitis accompanied by nasal polyps in adults.
FDA approval is based on results from a pair of studies involving 724 patients aged 18 years or older with chronic rhinosinusitis with nasal polyps who were symptomatic despite undergoing treatment with intranasal corticosteroids and who received either dupilumab or a placebo. Compared with the placebo group, patients receiving dupilumab had statistically significant reductions in nasal polyp size and nasal congestion; they also had an increased ability to smell and required less nasal polyp surgery and oral steroids.
The most commonly reported adverse events were injection-site reactions and eye and eyelid inflammation, which included redness, swelling, and itching. The drug can cause severe allergic reactions and eye problems, such as conjunctivitis or keratitis; patients should also not receive live vaccines while taking dupilumab.
“Nasal polyps can lead to loss of smell, and often patients require surgery to remove the polyps. Dupixent provides an important treatment option for patients whose nasal polyps are not adequately controlled with intranasal steroids. It also reduces the need for nasal polyp surgery and oral steroids,” said Sally Seymour, MD, director of the Division of Pulmonary, Allergy, and Rheumatology Products in the FDA’s Center for Drug Evaluation and Research.
Find the full press release on the FDA website.
The Food and Drug Administration has approved dupilumab (Dupixent) for the treatment of chronic rhinosinusitis accompanied by nasal polyps in adults.
FDA approval is based on results from a pair of studies involving 724 patients aged 18 years or older with chronic rhinosinusitis with nasal polyps who were symptomatic despite undergoing treatment with intranasal corticosteroids and who received either dupilumab or a placebo. Compared with the placebo group, patients receiving dupilumab had statistically significant reductions in nasal polyp size and nasal congestion; they also had an increased ability to smell and required less nasal polyp surgery and oral steroids.
The most commonly reported adverse events were injection-site reactions and eye and eyelid inflammation, which included redness, swelling, and itching. The drug can cause severe allergic reactions and eye problems, such as conjunctivitis or keratitis; patients should also not receive live vaccines while taking dupilumab.
“Nasal polyps can lead to loss of smell, and often patients require surgery to remove the polyps. Dupixent provides an important treatment option for patients whose nasal polyps are not adequately controlled with intranasal steroids. It also reduces the need for nasal polyp surgery and oral steroids,” said Sally Seymour, MD, director of the Division of Pulmonary, Allergy, and Rheumatology Products in the FDA’s Center for Drug Evaluation and Research.
Find the full press release on the FDA website.
The Food and Drug Administration has approved dupilumab (Dupixent) for the treatment of chronic rhinosinusitis accompanied by nasal polyps in adults.
FDA approval is based on results from a pair of studies involving 724 patients aged 18 years or older with chronic rhinosinusitis with nasal polyps who were symptomatic despite undergoing treatment with intranasal corticosteroids and who received either dupilumab or a placebo. Compared with the placebo group, patients receiving dupilumab had statistically significant reductions in nasal polyp size and nasal congestion; they also had an increased ability to smell and required less nasal polyp surgery and oral steroids.
The most commonly reported adverse events were injection-site reactions and eye and eyelid inflammation, which included redness, swelling, and itching. The drug can cause severe allergic reactions and eye problems, such as conjunctivitis or keratitis; patients should also not receive live vaccines while taking dupilumab.
“Nasal polyps can lead to loss of smell, and often patients require surgery to remove the polyps. Dupixent provides an important treatment option for patients whose nasal polyps are not adequately controlled with intranasal steroids. It also reduces the need for nasal polyp surgery and oral steroids,” said Sally Seymour, MD, director of the Division of Pulmonary, Allergy, and Rheumatology Products in the FDA’s Center for Drug Evaluation and Research.
Find the full press release on the FDA website.
Measles incidence has slowed as summer begins
There were 33 new measles cases reported last week, bringing the U.S. total to 1,077 for the year through June 20, according to the Centers for Disease Control and Prevention.
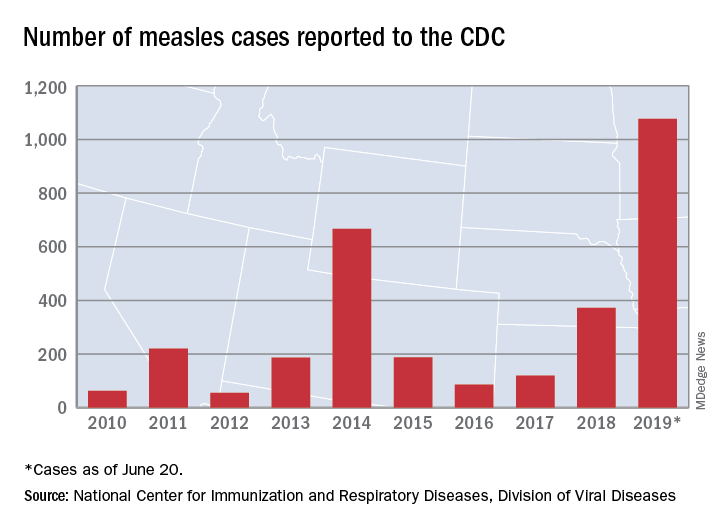
The number of new cases is an increase from the 22 reported the week before, but weekly incidence has been trending downward since hitting a high of 90 in mid-April, CDC data show.
The two continuing outbreaks in New York State made up more than half of the new cases, as Rockland County reported nine cases and New York City reported eight (seven in Brooklyn and one in Queens). Only one new case was reported in California as of the CDC’s June 20 cutoff, but the Los Angeles County Department of Public Health said on June 22 that it was assessing two possible cases, with potential public exposures occurring in a theater and a restaurant.
In a survey conducted in April, a majority of physicians with experience treating measles said that summer travel would lead to increased measles outbreaks and deaths.
There were 33 new measles cases reported last week, bringing the U.S. total to 1,077 for the year through June 20, according to the Centers for Disease Control and Prevention.
The number of new cases is an increase from the 22 reported the week before, but weekly incidence has been trending downward since hitting a high of 90 in mid-April, CDC data show.
The two continuing outbreaks in New York State made up more than half of the new cases, as Rockland County reported nine cases and New York City reported eight (seven in Brooklyn and one in Queens). Only one new case was reported in California as of the CDC’s June 20 cutoff, but the Los Angeles County Department of Public Health said on June 22 that it was assessing two possible cases, with potential public exposures occurring in a theater and a restaurant.
In a survey conducted in April, a majority of physicians with experience treating measles said that summer travel would lead to increased measles outbreaks and deaths.
There were 33 new measles cases reported last week, bringing the U.S. total to 1,077 for the year through June 20, according to the Centers for Disease Control and Prevention.
The number of new cases is an increase from the 22 reported the week before, but weekly incidence has been trending downward since hitting a high of 90 in mid-April, CDC data show.
The two continuing outbreaks in New York State made up more than half of the new cases, as Rockland County reported nine cases and New York City reported eight (seven in Brooklyn and one in Queens). Only one new case was reported in California as of the CDC’s June 20 cutoff, but the Los Angeles County Department of Public Health said on June 22 that it was assessing two possible cases, with potential public exposures occurring in a theater and a restaurant.
In a survey conducted in April, a majority of physicians with experience treating measles said that summer travel would lead to increased measles outbreaks and deaths.
CF drug picks up indication for children as young as 6
to include children as young as 6 years old who have cystic fibrosis.

The drug was approved in 2018 for patients aged 12 years and older who have the most common cause of the disease, two alleles for the F508del mutation in the gene that codes for the cystic fibrosis transmembrane conductance regulator (CFTR) protein, or at least one other CFTR mutation responsive to the combination, as listed in labeling.
The original approval was based on three phase 3, double blind, placebo-controlled trials, which demonstrated improvements in lung function and other key measures of the disease. One trial that found a 6.8% mean improvement in lung function testing over placebo at 8 weeks, and another that found a 4% improvement at 24 weeks, with fewer respiratory exacerbations and improved respiratory-related quality of life. A third trial in patients without the indicated genetic mutations was ended early for futility.
The efficacy in children under 12 years was extrapolated from those trials, plus an open-label study that found similar effects.
Labeling warns of elevated liver enzymes and cataracts in children, and notes that the drug should be taken with food that contains fat. Labeling also recommends against use with strong cytochrome P450 3A4 (CYP3A) inducers – rifampin, phenobarbital, St. John’s wort, among others – because they might reduce efficacy, and against use with CYP3A inhibitors – ketoconazole, clarithromycin, Seville oranges, grapefruit juice, etc. – because of the risk of increased exposure.
The most common side effects are headache, nausea, sinus congestion, and dizziness. The FDA has cleared a CF gene test to check for the required mutations. Symdeko is marketed by Vertex Pharmaceuticals.
to include children as young as 6 years old who have cystic fibrosis.
The drug was approved in 2018 for patients aged 12 years and older who have the most common cause of the disease, two alleles for the F508del mutation in the gene that codes for the cystic fibrosis transmembrane conductance regulator (CFTR) protein, or at least one other CFTR mutation responsive to the combination, as listed in labeling.
The original approval was based on three phase 3, double blind, placebo-controlled trials, which demonstrated improvements in lung function and other key measures of the disease. One trial that found a 6.8% mean improvement in lung function testing over placebo at 8 weeks, and another that found a 4% improvement at 24 weeks, with fewer respiratory exacerbations and improved respiratory-related quality of life. A third trial in patients without the indicated genetic mutations was ended early for futility.
The efficacy in children under 12 years was extrapolated from those trials, plus an open-label study that found similar effects.
Labeling warns of elevated liver enzymes and cataracts in children, and notes that the drug should be taken with food that contains fat. Labeling also recommends against use with strong cytochrome P450 3A4 (CYP3A) inducers – rifampin, phenobarbital, St. John’s wort, among others – because they might reduce efficacy, and against use with CYP3A inhibitors – ketoconazole, clarithromycin, Seville oranges, grapefruit juice, etc. – because of the risk of increased exposure.
The most common side effects are headache, nausea, sinus congestion, and dizziness. The FDA has cleared a CF gene test to check for the required mutations. Symdeko is marketed by Vertex Pharmaceuticals.
to include children as young as 6 years old who have cystic fibrosis.
The drug was approved in 2018 for patients aged 12 years and older who have the most common cause of the disease, two alleles for the F508del mutation in the gene that codes for the cystic fibrosis transmembrane conductance regulator (CFTR) protein, or at least one other CFTR mutation responsive to the combination, as listed in labeling.
The original approval was based on three phase 3, double blind, placebo-controlled trials, which demonstrated improvements in lung function and other key measures of the disease. One trial that found a 6.8% mean improvement in lung function testing over placebo at 8 weeks, and another that found a 4% improvement at 24 weeks, with fewer respiratory exacerbations and improved respiratory-related quality of life. A third trial in patients without the indicated genetic mutations was ended early for futility.
The efficacy in children under 12 years was extrapolated from those trials, plus an open-label study that found similar effects.
Labeling warns of elevated liver enzymes and cataracts in children, and notes that the drug should be taken with food that contains fat. Labeling also recommends against use with strong cytochrome P450 3A4 (CYP3A) inducers – rifampin, phenobarbital, St. John’s wort, among others – because they might reduce efficacy, and against use with CYP3A inhibitors – ketoconazole, clarithromycin, Seville oranges, grapefruit juice, etc. – because of the risk of increased exposure.
The most common side effects are headache, nausea, sinus congestion, and dizziness. The FDA has cleared a CF gene test to check for the required mutations. Symdeko is marketed by Vertex Pharmaceuticals.
FDA panel: Continue using paclitaxel-eluting PAD devices, with caveats
GAITHERSBURG, MD. – There was sufficient evidence of a late mortality signal seen at 2-5 years post procedurally for paclitaxel-eluting stents and coated balloons to warrant a label change for the devices, the Food and Drug Administration’s Circulatory System Devices Panel unanimously agreed after 2 days of deliberation.
That signal was brought to light in a meta-analysis published last December by Konstantinos Katsanos, MD, of Patras University Hospital, Rion, Greece, and colleagues (J Am Heart Assoc. 2018;7:e011245). Although there were concerns about the quality of the industry data used in the study, the caliber of the analysis itself and the subsequent data presented by the FDA to the panel were deemed sufficient to recommend a warning of concern to patients and providers.
Much of the new data from industry and large database registries presented to the panel, which was chaired by Richard A. Lange, MD, indicated a lessening to no evidence of the mortality effect. But this evidence was deemed insufficient to counter the evidence of the randomized controlled trials individually and collectively as presented in the Katsanos meta-analysis and subsequent information presented by the FDA that examined various parameters in a variety of sensitivity analyses that confirmed the late mortality signal. There was also concern that the industry and the registry analyses presented were not peer reviewed.
However, the panel also determined that it would be inappropriate to pull the devices from the market and from general use for several reasons.
One key reason was that, according to the panel, there was no mechanistic cause apparent for the late mortality. In addition, no convincing dose-response data could be teased from the preclinical and clinical trials studied because of their variability of devices, application methods, and lack of appropriate tissue analysis across studies.
Finally, the industry data used to create the meta-analysis were considered to be fundamentally flawed: in blinding, in the relatively small numbers of patients, and in the large percentage of patients lost to follow-up. The latter could have dramatically influenced the perceived results, especially as the studies were not powered or designed to follow mortality over such a period of time, according to the panel.
These limitations to the signal were especially important to the panel because of the obvious benefits with regard to quality of life provided to patients from these devices, which were attested to during the 2-day meeting by numerous presenters from industry, medical organizations – including societies and nonprofits – and providers.
In responding to FDA requests on a variety of concerns, the panel reiterated that there was a credible mortality signal, but that they could not be confident about the magnitude and whether it was caused by the paclitaxel treatment or some factor in the design or conduct of the studies. In addition, the panel members felt that they could neither confirm nor eliminate a class effect, given the fact that the information was based on a meta-analysis and thus none of the included devices could safely be removed from consideration.
They suggested that further safety information should be obtained, potentially by assessing and perhaps altering data collection in 29 ongoing studies over the next 5 years or so in more than 10,000 patients.
In addition, several of the panel members felt that additional animal studies might be performed including the use of older rat models; and using animal models that mimicked the kind of comorbidities present in the treated population, such as diabetes and atherosclerosis. They suggested cross-company industry cooperation with the FDA on these models, including looking at drug interactions and mimicking the dose application of stents/balloons.
Both the FDA representative and the panel were especially concerned with the benefit/risk profile.
The recommendation to still market the devices with a label warning was warranted, according to many members of the panel. They pointed to the clear benefits in quality of life and the lowered need for revascularization despite the evidence of the mortality signal, which, while statistically significant, could not be pinned town with regard to mechanisms or specific causes of death.
Overall, there was a concern that there should be a dialogue between patients and their doctors to discuss clear short-term benefits with unknown long-term risk, and that the label should support this by clearly mentioning the mortality signal that was found, although there was no attempt to develop exact wording.
Panel member Joaquin E. Cigarroa, MD, head of cardiovascular medicine at Oregon Health & Science University, Portland, suggested with regard to labeling that a statement “that ‘there may be’ – not ‘there is’ – a late mortality signal, should be included.”
Panel member John C. Somberg, MD, program director of clinical research and bioinformatics at Rush University in Lake Bluff, Ill., stated: “The label should say something like, ‘when looking at a meta-analysis that combined all studies with stents and balloons that carried paclitaxel, there may be a late mortality, which must be balanced against an early and sustained benefit in terms of pain on walking and potential loss of circulation to your extremity.’ ”
Other panel members thought that the meta-analysis should not be privileged and that somehow the totality of the evidence should somehow be distilled down into the label, including the evidence against the signal.
“We’re meeting because of a signal, of a concern – an honest, well-meaning concern – of increased mortality. And my opinion is that the patients need to be informed of it,” said Dr. Lange, president, Texas Tech University, El Paso.
Some members of the panel felt that it may not be justifiable to use these devices in patients with low intrinsic risk and low recurrence risk, and the whole spectrum of patients may need to be considered in further studies to figure out the subgroups that have more benefits and more risks, and also to consider how to mitigate risks in patients who receive the device, whether through medical therapy or lifestyle modification.
In particular, Frank W. LoGerfo, MD, the William V. McDermott Distinguished Professor of Surgery at Harvard Medical School, Boston, stated: “Interventions for claudication should be extremely rare. It rarely progresses, and the pain should be worked through by exercise with low risk of limb loss.” He added that intervention with these devices “takes away options. Trading life for something that is not limb-threatening is something we should not be considering.”
There was no firm consensus on whether new randomized trials should be done, although they were of course the ideal solution. Kevin E. Kip, PhD, Distinguished USF Health Professor at the University of South Florida, Tampa, and others argued that, whether new trials were necessary or not, to deal with the safety question in a timely fashion, existing trials have to capture as much of the missing data as possible, and carry out follow-up out further.
FDA representative Bram Zuckerman, MD, director of the Office of Cardiovascular Devices at the Center for Devices and Radiological Health, indicated that those things might not be easily be accomplished due to regulatory constraints and the financial costs, and that to do so there would be need for community effort among stakeholders, including collaborative efforts with existing prospective registries such as that run by the Vascular Quality Initiative.
One overall conclusion by both the FDA and panel members was that the quality of these and other such studies going forward must improve, by standardizing definitions and data forms to make studies more uniform across the industry. They reemphasized the need to work with the registries to get common data included, and to incorporate of insurance provider and Social Security death data as much as possible to help alleviate the lost follow-up problem.
SOURCE: Webcasts of the complete 2 days of the FDA panel meeting are available online.
GAITHERSBURG, MD. – There was sufficient evidence of a late mortality signal seen at 2-5 years post procedurally for paclitaxel-eluting stents and coated balloons to warrant a label change for the devices, the Food and Drug Administration’s Circulatory System Devices Panel unanimously agreed after 2 days of deliberation.
That signal was brought to light in a meta-analysis published last December by Konstantinos Katsanos, MD, of Patras University Hospital, Rion, Greece, and colleagues (J Am Heart Assoc. 2018;7:e011245). Although there were concerns about the quality of the industry data used in the study, the caliber of the analysis itself and the subsequent data presented by the FDA to the panel were deemed sufficient to recommend a warning of concern to patients and providers.
Much of the new data from industry and large database registries presented to the panel, which was chaired by Richard A. Lange, MD, indicated a lessening to no evidence of the mortality effect. But this evidence was deemed insufficient to counter the evidence of the randomized controlled trials individually and collectively as presented in the Katsanos meta-analysis and subsequent information presented by the FDA that examined various parameters in a variety of sensitivity analyses that confirmed the late mortality signal. There was also concern that the industry and the registry analyses presented were not peer reviewed.
However, the panel also determined that it would be inappropriate to pull the devices from the market and from general use for several reasons.
One key reason was that, according to the panel, there was no mechanistic cause apparent for the late mortality. In addition, no convincing dose-response data could be teased from the preclinical and clinical trials studied because of their variability of devices, application methods, and lack of appropriate tissue analysis across studies.
Finally, the industry data used to create the meta-analysis were considered to be fundamentally flawed: in blinding, in the relatively small numbers of patients, and in the large percentage of patients lost to follow-up. The latter could have dramatically influenced the perceived results, especially as the studies were not powered or designed to follow mortality over such a period of time, according to the panel.
These limitations to the signal were especially important to the panel because of the obvious benefits with regard to quality of life provided to patients from these devices, which were attested to during the 2-day meeting by numerous presenters from industry, medical organizations – including societies and nonprofits – and providers.
In responding to FDA requests on a variety of concerns, the panel reiterated that there was a credible mortality signal, but that they could not be confident about the magnitude and whether it was caused by the paclitaxel treatment or some factor in the design or conduct of the studies. In addition, the panel members felt that they could neither confirm nor eliminate a class effect, given the fact that the information was based on a meta-analysis and thus none of the included devices could safely be removed from consideration.
They suggested that further safety information should be obtained, potentially by assessing and perhaps altering data collection in 29 ongoing studies over the next 5 years or so in more than 10,000 patients.
In addition, several of the panel members felt that additional animal studies might be performed including the use of older rat models; and using animal models that mimicked the kind of comorbidities present in the treated population, such as diabetes and atherosclerosis. They suggested cross-company industry cooperation with the FDA on these models, including looking at drug interactions and mimicking the dose application of stents/balloons.
Both the FDA representative and the panel were especially concerned with the benefit/risk profile.
The recommendation to still market the devices with a label warning was warranted, according to many members of the panel. They pointed to the clear benefits in quality of life and the lowered need for revascularization despite the evidence of the mortality signal, which, while statistically significant, could not be pinned town with regard to mechanisms or specific causes of death.
Overall, there was a concern that there should be a dialogue between patients and their doctors to discuss clear short-term benefits with unknown long-term risk, and that the label should support this by clearly mentioning the mortality signal that was found, although there was no attempt to develop exact wording.
Panel member Joaquin E. Cigarroa, MD, head of cardiovascular medicine at Oregon Health & Science University, Portland, suggested with regard to labeling that a statement “that ‘there may be’ – not ‘there is’ – a late mortality signal, should be included.”
Panel member John C. Somberg, MD, program director of clinical research and bioinformatics at Rush University in Lake Bluff, Ill., stated: “The label should say something like, ‘when looking at a meta-analysis that combined all studies with stents and balloons that carried paclitaxel, there may be a late mortality, which must be balanced against an early and sustained benefit in terms of pain on walking and potential loss of circulation to your extremity.’ ”
Other panel members thought that the meta-analysis should not be privileged and that somehow the totality of the evidence should somehow be distilled down into the label, including the evidence against the signal.
“We’re meeting because of a signal, of a concern – an honest, well-meaning concern – of increased mortality. And my opinion is that the patients need to be informed of it,” said Dr. Lange, president, Texas Tech University, El Paso.
Some members of the panel felt that it may not be justifiable to use these devices in patients with low intrinsic risk and low recurrence risk, and the whole spectrum of patients may need to be considered in further studies to figure out the subgroups that have more benefits and more risks, and also to consider how to mitigate risks in patients who receive the device, whether through medical therapy or lifestyle modification.
In particular, Frank W. LoGerfo, MD, the William V. McDermott Distinguished Professor of Surgery at Harvard Medical School, Boston, stated: “Interventions for claudication should be extremely rare. It rarely progresses, and the pain should be worked through by exercise with low risk of limb loss.” He added that intervention with these devices “takes away options. Trading life for something that is not limb-threatening is something we should not be considering.”
There was no firm consensus on whether new randomized trials should be done, although they were of course the ideal solution. Kevin E. Kip, PhD, Distinguished USF Health Professor at the University of South Florida, Tampa, and others argued that, whether new trials were necessary or not, to deal with the safety question in a timely fashion, existing trials have to capture as much of the missing data as possible, and carry out follow-up out further.
FDA representative Bram Zuckerman, MD, director of the Office of Cardiovascular Devices at the Center for Devices and Radiological Health, indicated that those things might not be easily be accomplished due to regulatory constraints and the financial costs, and that to do so there would be need for community effort among stakeholders, including collaborative efforts with existing prospective registries such as that run by the Vascular Quality Initiative.
One overall conclusion by both the FDA and panel members was that the quality of these and other such studies going forward must improve, by standardizing definitions and data forms to make studies more uniform across the industry. They reemphasized the need to work with the registries to get common data included, and to incorporate of insurance provider and Social Security death data as much as possible to help alleviate the lost follow-up problem.
SOURCE: Webcasts of the complete 2 days of the FDA panel meeting are available online.
GAITHERSBURG, MD. – There was sufficient evidence of a late mortality signal seen at 2-5 years post procedurally for paclitaxel-eluting stents and coated balloons to warrant a label change for the devices, the Food and Drug Administration’s Circulatory System Devices Panel unanimously agreed after 2 days of deliberation.
That signal was brought to light in a meta-analysis published last December by Konstantinos Katsanos, MD, of Patras University Hospital, Rion, Greece, and colleagues (J Am Heart Assoc. 2018;7:e011245). Although there were concerns about the quality of the industry data used in the study, the caliber of the analysis itself and the subsequent data presented by the FDA to the panel were deemed sufficient to recommend a warning of concern to patients and providers.
Much of the new data from industry and large database registries presented to the panel, which was chaired by Richard A. Lange, MD, indicated a lessening to no evidence of the mortality effect. But this evidence was deemed insufficient to counter the evidence of the randomized controlled trials individually and collectively as presented in the Katsanos meta-analysis and subsequent information presented by the FDA that examined various parameters in a variety of sensitivity analyses that confirmed the late mortality signal. There was also concern that the industry and the registry analyses presented were not peer reviewed.
However, the panel also determined that it would be inappropriate to pull the devices from the market and from general use for several reasons.
One key reason was that, according to the panel, there was no mechanistic cause apparent for the late mortality. In addition, no convincing dose-response data could be teased from the preclinical and clinical trials studied because of their variability of devices, application methods, and lack of appropriate tissue analysis across studies.
Finally, the industry data used to create the meta-analysis were considered to be fundamentally flawed: in blinding, in the relatively small numbers of patients, and in the large percentage of patients lost to follow-up. The latter could have dramatically influenced the perceived results, especially as the studies were not powered or designed to follow mortality over such a period of time, according to the panel.
These limitations to the signal were especially important to the panel because of the obvious benefits with regard to quality of life provided to patients from these devices, which were attested to during the 2-day meeting by numerous presenters from industry, medical organizations – including societies and nonprofits – and providers.
In responding to FDA requests on a variety of concerns, the panel reiterated that there was a credible mortality signal, but that they could not be confident about the magnitude and whether it was caused by the paclitaxel treatment or some factor in the design or conduct of the studies. In addition, the panel members felt that they could neither confirm nor eliminate a class effect, given the fact that the information was based on a meta-analysis and thus none of the included devices could safely be removed from consideration.
They suggested that further safety information should be obtained, potentially by assessing and perhaps altering data collection in 29 ongoing studies over the next 5 years or so in more than 10,000 patients.
In addition, several of the panel members felt that additional animal studies might be performed including the use of older rat models; and using animal models that mimicked the kind of comorbidities present in the treated population, such as diabetes and atherosclerosis. They suggested cross-company industry cooperation with the FDA on these models, including looking at drug interactions and mimicking the dose application of stents/balloons.
Both the FDA representative and the panel were especially concerned with the benefit/risk profile.
The recommendation to still market the devices with a label warning was warranted, according to many members of the panel. They pointed to the clear benefits in quality of life and the lowered need for revascularization despite the evidence of the mortality signal, which, while statistically significant, could not be pinned town with regard to mechanisms or specific causes of death.
Overall, there was a concern that there should be a dialogue between patients and their doctors to discuss clear short-term benefits with unknown long-term risk, and that the label should support this by clearly mentioning the mortality signal that was found, although there was no attempt to develop exact wording.
Panel member Joaquin E. Cigarroa, MD, head of cardiovascular medicine at Oregon Health & Science University, Portland, suggested with regard to labeling that a statement “that ‘there may be’ – not ‘there is’ – a late mortality signal, should be included.”
Panel member John C. Somberg, MD, program director of clinical research and bioinformatics at Rush University in Lake Bluff, Ill., stated: “The label should say something like, ‘when looking at a meta-analysis that combined all studies with stents and balloons that carried paclitaxel, there may be a late mortality, which must be balanced against an early and sustained benefit in terms of pain on walking and potential loss of circulation to your extremity.’ ”
Other panel members thought that the meta-analysis should not be privileged and that somehow the totality of the evidence should somehow be distilled down into the label, including the evidence against the signal.
“We’re meeting because of a signal, of a concern – an honest, well-meaning concern – of increased mortality. And my opinion is that the patients need to be informed of it,” said Dr. Lange, president, Texas Tech University, El Paso.
Some members of the panel felt that it may not be justifiable to use these devices in patients with low intrinsic risk and low recurrence risk, and the whole spectrum of patients may need to be considered in further studies to figure out the subgroups that have more benefits and more risks, and also to consider how to mitigate risks in patients who receive the device, whether through medical therapy or lifestyle modification.
In particular, Frank W. LoGerfo, MD, the William V. McDermott Distinguished Professor of Surgery at Harvard Medical School, Boston, stated: “Interventions for claudication should be extremely rare. It rarely progresses, and the pain should be worked through by exercise with low risk of limb loss.” He added that intervention with these devices “takes away options. Trading life for something that is not limb-threatening is something we should not be considering.”
There was no firm consensus on whether new randomized trials should be done, although they were of course the ideal solution. Kevin E. Kip, PhD, Distinguished USF Health Professor at the University of South Florida, Tampa, and others argued that, whether new trials were necessary or not, to deal with the safety question in a timely fashion, existing trials have to capture as much of the missing data as possible, and carry out follow-up out further.
FDA representative Bram Zuckerman, MD, director of the Office of Cardiovascular Devices at the Center for Devices and Radiological Health, indicated that those things might not be easily be accomplished due to regulatory constraints and the financial costs, and that to do so there would be need for community effort among stakeholders, including collaborative efforts with existing prospective registries such as that run by the Vascular Quality Initiative.
One overall conclusion by both the FDA and panel members was that the quality of these and other such studies going forward must improve, by standardizing definitions and data forms to make studies more uniform across the industry. They reemphasized the need to work with the registries to get common data included, and to incorporate of insurance provider and Social Security death data as much as possible to help alleviate the lost follow-up problem.
SOURCE: Webcasts of the complete 2 days of the FDA panel meeting are available online.
REPORTING FROM AN FDA PANEL MEETING
COPD rates reflect current smoking prevalence
, according to a Centers for Disease Control and Prevention analysis of respondents to a behavioral risk factor survey.
“Population-based strategies for smoking prevention and control have the potential to decrease the prevalence of COPD in the United States,” wrote Anne G. Wheaton, PhD, of the CDC’s National Center for Chronic Disease Prevention and Health Promotion and coauthors. The study was published in the Morbidity and Mortality Weekly Report.
Dr. Wheaton and her fellow researchers analyzed data from 418,378 adult respondents to the 2017 Behavioral Risk Factor Surveillance System survey. Responses came from all 50 states and Washington, D.C.; respondents who had smoked less than 100 lifetime cigarettes were categorized as “never smoked,” while those who had smoked at least 100 cigarettes but no longer smoked were categorized as “former smokers.” Anyone who had smoked at least 100 cigarettes and currently smoked was categorized as a “current smoker.”
The age-adjusted prevalence of COPD among U.S. adults was 6.2% (95% confidence interval, 6.0%-6.3%) in 2017. Current cigarette smokers had a prevalence of 15.2% (95% CI, 14.7%-15.7%); this dipped to 7.6% (95% CI, 7.3%-8.0%) among former smokers and 2.8% (95% CI, 2.7%-2.9%) among adults who had never smoked. Patterns were visible within states: Current smokers had a state-level prevalence of COPD that was strongly correlated with state-level current smoking prevalence (Pearson correlation coefficient, 0.69; P less than .001). State-level COPD prevalence among former smokers (Pearson correlation coefficient, 0.71; P less than .001) and those who never smoked (Pearson correlation coefficient, 0.64; P less than .001) were also strongly correlated with the current smoking prevalence, indicating secondhand smoke as a risk factor for COPD.
The coauthors acknowledged the study’s limitations, including relying on self-reporting for both COPD and smoking status. They also noted that there was no way to measure exposure to secondhand smoke, other indoor or outdoor air pollutants, or respiratory infection history, “all of which might contribute to COPD risk.”
No conflicts of interest were reported.
SOURCE: Wheaton AG et al. MMWR Morb Mortal Wkly Rep. 2019 Jun 21;68(24):533-8.
, according to a Centers for Disease Control and Prevention analysis of respondents to a behavioral risk factor survey.
“Population-based strategies for smoking prevention and control have the potential to decrease the prevalence of COPD in the United States,” wrote Anne G. Wheaton, PhD, of the CDC’s National Center for Chronic Disease Prevention and Health Promotion and coauthors. The study was published in the Morbidity and Mortality Weekly Report.
Dr. Wheaton and her fellow researchers analyzed data from 418,378 adult respondents to the 2017 Behavioral Risk Factor Surveillance System survey. Responses came from all 50 states and Washington, D.C.; respondents who had smoked less than 100 lifetime cigarettes were categorized as “never smoked,” while those who had smoked at least 100 cigarettes but no longer smoked were categorized as “former smokers.” Anyone who had smoked at least 100 cigarettes and currently smoked was categorized as a “current smoker.”
The age-adjusted prevalence of COPD among U.S. adults was 6.2% (95% confidence interval, 6.0%-6.3%) in 2017. Current cigarette smokers had a prevalence of 15.2% (95% CI, 14.7%-15.7%); this dipped to 7.6% (95% CI, 7.3%-8.0%) among former smokers and 2.8% (95% CI, 2.7%-2.9%) among adults who had never smoked. Patterns were visible within states: Current smokers had a state-level prevalence of COPD that was strongly correlated with state-level current smoking prevalence (Pearson correlation coefficient, 0.69; P less than .001). State-level COPD prevalence among former smokers (Pearson correlation coefficient, 0.71; P less than .001) and those who never smoked (Pearson correlation coefficient, 0.64; P less than .001) were also strongly correlated with the current smoking prevalence, indicating secondhand smoke as a risk factor for COPD.
The coauthors acknowledged the study’s limitations, including relying on self-reporting for both COPD and smoking status. They also noted that there was no way to measure exposure to secondhand smoke, other indoor or outdoor air pollutants, or respiratory infection history, “all of which might contribute to COPD risk.”
No conflicts of interest were reported.
SOURCE: Wheaton AG et al. MMWR Morb Mortal Wkly Rep. 2019 Jun 21;68(24):533-8.
, according to a Centers for Disease Control and Prevention analysis of respondents to a behavioral risk factor survey.
“Population-based strategies for smoking prevention and control have the potential to decrease the prevalence of COPD in the United States,” wrote Anne G. Wheaton, PhD, of the CDC’s National Center for Chronic Disease Prevention and Health Promotion and coauthors. The study was published in the Morbidity and Mortality Weekly Report.
Dr. Wheaton and her fellow researchers analyzed data from 418,378 adult respondents to the 2017 Behavioral Risk Factor Surveillance System survey. Responses came from all 50 states and Washington, D.C.; respondents who had smoked less than 100 lifetime cigarettes were categorized as “never smoked,” while those who had smoked at least 100 cigarettes but no longer smoked were categorized as “former smokers.” Anyone who had smoked at least 100 cigarettes and currently smoked was categorized as a “current smoker.”
The age-adjusted prevalence of COPD among U.S. adults was 6.2% (95% confidence interval, 6.0%-6.3%) in 2017. Current cigarette smokers had a prevalence of 15.2% (95% CI, 14.7%-15.7%); this dipped to 7.6% (95% CI, 7.3%-8.0%) among former smokers and 2.8% (95% CI, 2.7%-2.9%) among adults who had never smoked. Patterns were visible within states: Current smokers had a state-level prevalence of COPD that was strongly correlated with state-level current smoking prevalence (Pearson correlation coefficient, 0.69; P less than .001). State-level COPD prevalence among former smokers (Pearson correlation coefficient, 0.71; P less than .001) and those who never smoked (Pearson correlation coefficient, 0.64; P less than .001) were also strongly correlated with the current smoking prevalence, indicating secondhand smoke as a risk factor for COPD.
The coauthors acknowledged the study’s limitations, including relying on self-reporting for both COPD and smoking status. They also noted that there was no way to measure exposure to secondhand smoke, other indoor or outdoor air pollutants, or respiratory infection history, “all of which might contribute to COPD risk.”
No conflicts of interest were reported.
SOURCE: Wheaton AG et al. MMWR Morb Mortal Wkly Rep. 2019 Jun 21;68(24):533-8.
FROM MMWR