User login
FDA approves pembrolizumab for advanced SCLC
The Food and Drug Administration has granted accelerated approval to pembrolizumab for patients with metastatic small cell lung cancer (SCLC) with disease progression on or after platinum-based chemotherapy and at least one other prior line of therapy.

Approval was based on an overall response rate of 19% among 83 patients with SCLC who had disease progression on or after two or more prior lines of therapy enrolled in two nonrandomized trials, according to the FDA.
SCLC cohorts in KEYNOTE-028 and KEYNOTE-158 received either pembrolizumab 200 mg intravenously every 3 weeks (n = 64) or 10 mg/kg intravenously every 2 weeks (n = 19). Treatment continued until documented disease progression, unacceptable toxicity, or for a maximum of 24 months.
The ORR was 19% (95% confidence interval, 11%-29%), while the complete response rate was 2%. Responses were durable for 6 months or longer in 94% of the 16 responding patients.
Common adverse reactions included fatigue, decreased appetite, cough, nausea, and constipation. The most frequent serious adverse reactions were pneumonia and pleural effusion.
The recommended dosage for SCLC treatment is 200 mg, administered as an intravenous infusion over 30 minutes every 3 weeks until disease progression, unacceptable toxicity, or up to 24 months in patients without disease progression, the FDA said.
Pembrolizumab is marketed as Keytruda by Merck.
The Food and Drug Administration has granted accelerated approval to pembrolizumab for patients with metastatic small cell lung cancer (SCLC) with disease progression on or after platinum-based chemotherapy and at least one other prior line of therapy.
Approval was based on an overall response rate of 19% among 83 patients with SCLC who had disease progression on or after two or more prior lines of therapy enrolled in two nonrandomized trials, according to the FDA.
SCLC cohorts in KEYNOTE-028 and KEYNOTE-158 received either pembrolizumab 200 mg intravenously every 3 weeks (n = 64) or 10 mg/kg intravenously every 2 weeks (n = 19). Treatment continued until documented disease progression, unacceptable toxicity, or for a maximum of 24 months.
The ORR was 19% (95% confidence interval, 11%-29%), while the complete response rate was 2%. Responses were durable for 6 months or longer in 94% of the 16 responding patients.
Common adverse reactions included fatigue, decreased appetite, cough, nausea, and constipation. The most frequent serious adverse reactions were pneumonia and pleural effusion.
The recommended dosage for SCLC treatment is 200 mg, administered as an intravenous infusion over 30 minutes every 3 weeks until disease progression, unacceptable toxicity, or up to 24 months in patients without disease progression, the FDA said.
Pembrolizumab is marketed as Keytruda by Merck.
The Food and Drug Administration has granted accelerated approval to pembrolizumab for patients with metastatic small cell lung cancer (SCLC) with disease progression on or after platinum-based chemotherapy and at least one other prior line of therapy.
Approval was based on an overall response rate of 19% among 83 patients with SCLC who had disease progression on or after two or more prior lines of therapy enrolled in two nonrandomized trials, according to the FDA.
SCLC cohorts in KEYNOTE-028 and KEYNOTE-158 received either pembrolizumab 200 mg intravenously every 3 weeks (n = 64) or 10 mg/kg intravenously every 2 weeks (n = 19). Treatment continued until documented disease progression, unacceptable toxicity, or for a maximum of 24 months.
The ORR was 19% (95% confidence interval, 11%-29%), while the complete response rate was 2%. Responses were durable for 6 months or longer in 94% of the 16 responding patients.
Common adverse reactions included fatigue, decreased appetite, cough, nausea, and constipation. The most frequent serious adverse reactions were pneumonia and pleural effusion.
The recommended dosage for SCLC treatment is 200 mg, administered as an intravenous infusion over 30 minutes every 3 weeks until disease progression, unacceptable toxicity, or up to 24 months in patients without disease progression, the FDA said.
Pembrolizumab is marketed as Keytruda by Merck.
Physical activity prevalence shows urban/rural divide
according to the Centers for Disease Control and Prevention.
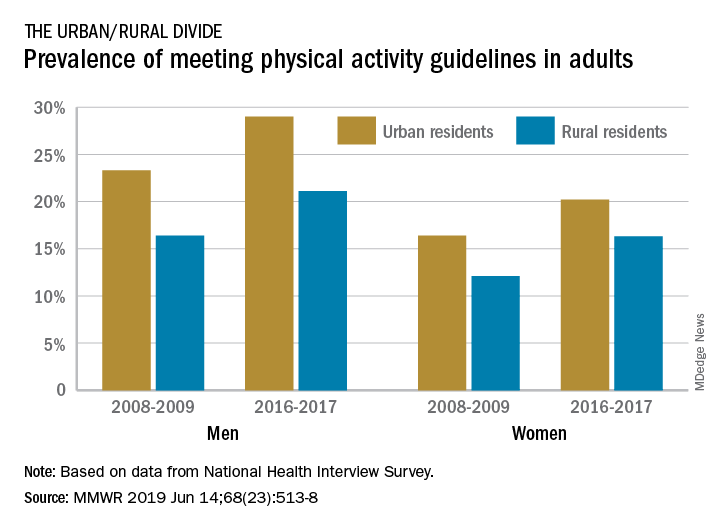
The prevalence of meeting the aerobic and muscle-strengthening recommendations in the 2008 Physical Activity Guidelines for Americans rose from 18.2% of adults in 2008 to 24.3% in 2017, but despite that increase, “insufficient participation in physical activity remains a public health concern,” Geoffrey P. Whitfield, PhD, and his associates said in the Morbidity and Mortality Weekly Report.
There was progress among both urban and rural residents, but those in rural areas were behind at the start of the study period in 2008 and remained behind in 2017. The prevalence of meeting the activity guideline started at 13.3% for rural residents and 19.4% for urbanites and rose to 19.6% and 25.3%, respectively, in 2017 – that’s an annual percentage point change of 0.5% for each population, the investigators reported. Rates among women were well below those of men in both populations.
Rural communities may lack the infrastructure, such as sidewalks, schoolyards, and parks, to support physical activities, or rural residents may get more exercise through occupational and domestic tasks, rather than through the leisure-time activities that are the focus of the National Health Interview Survey, which was the source of the study data, Dr. Whitfield and his associates suggested.
The 2008 federal guidelines recommend that adults get 150-300 minutes of moderate-intensity or 75-150 minutes of vigorous-intensity aerobic physical activity per week, along with muscle-strengthening activities of at least moderate intensity involving all major muscle groups on 2 or more days each week.
SOURCE: Whitfield GP et al. MMWR. 2019 Jun 14;68(23):514-8.
according to the Centers for Disease Control and Prevention.
The prevalence of meeting the aerobic and muscle-strengthening recommendations in the 2008 Physical Activity Guidelines for Americans rose from 18.2% of adults in 2008 to 24.3% in 2017, but despite that increase, “insufficient participation in physical activity remains a public health concern,” Geoffrey P. Whitfield, PhD, and his associates said in the Morbidity and Mortality Weekly Report.
There was progress among both urban and rural residents, but those in rural areas were behind at the start of the study period in 2008 and remained behind in 2017. The prevalence of meeting the activity guideline started at 13.3% for rural residents and 19.4% for urbanites and rose to 19.6% and 25.3%, respectively, in 2017 – that’s an annual percentage point change of 0.5% for each population, the investigators reported. Rates among women were well below those of men in both populations.
Rural communities may lack the infrastructure, such as sidewalks, schoolyards, and parks, to support physical activities, or rural residents may get more exercise through occupational and domestic tasks, rather than through the leisure-time activities that are the focus of the National Health Interview Survey, which was the source of the study data, Dr. Whitfield and his associates suggested.
The 2008 federal guidelines recommend that adults get 150-300 minutes of moderate-intensity or 75-150 minutes of vigorous-intensity aerobic physical activity per week, along with muscle-strengthening activities of at least moderate intensity involving all major muscle groups on 2 or more days each week.
SOURCE: Whitfield GP et al. MMWR. 2019 Jun 14;68(23):514-8.
according to the Centers for Disease Control and Prevention.
The prevalence of meeting the aerobic and muscle-strengthening recommendations in the 2008 Physical Activity Guidelines for Americans rose from 18.2% of adults in 2008 to 24.3% in 2017, but despite that increase, “insufficient participation in physical activity remains a public health concern,” Geoffrey P. Whitfield, PhD, and his associates said in the Morbidity and Mortality Weekly Report.
There was progress among both urban and rural residents, but those in rural areas were behind at the start of the study period in 2008 and remained behind in 2017. The prevalence of meeting the activity guideline started at 13.3% for rural residents and 19.4% for urbanites and rose to 19.6% and 25.3%, respectively, in 2017 – that’s an annual percentage point change of 0.5% for each population, the investigators reported. Rates among women were well below those of men in both populations.
Rural communities may lack the infrastructure, such as sidewalks, schoolyards, and parks, to support physical activities, or rural residents may get more exercise through occupational and domestic tasks, rather than through the leisure-time activities that are the focus of the National Health Interview Survey, which was the source of the study data, Dr. Whitfield and his associates suggested.
The 2008 federal guidelines recommend that adults get 150-300 minutes of moderate-intensity or 75-150 minutes of vigorous-intensity aerobic physical activity per week, along with muscle-strengthening activities of at least moderate intensity involving all major muscle groups on 2 or more days each week.
SOURCE: Whitfield GP et al. MMWR. 2019 Jun 14;68(23):514-8.
FROM MMWR
FDA warns about fecal microbiota for transplantation
Officials at the Food and Drug Administration have issued a safety alert regarding the use of fecal microbiota for transplantation and the risk of serious adverse reactions because of transmission of multidrug-resistant organisms (MDROs).

According to the alert, which was issued on June 13, 2019, the agency became aware of two immunocompromised adult patients who received investigational fecal microbiota for transplantation (FMT) and developed extended-spectrum beta-lactamase (EBSL)–producing Escherichia coli. One of the patients died.
“This is certainly a theoretical risk that we’ve known about,” Lea Ann Chen, MD, a gastroenterologist at New York University, said in an interview. “This announcement is important, because we probably don’t counsel patients specifically about this risk. We say there is a risk for transmission of infectious agents in general, but I think that probably very few counsel patients about a risk for transmission of MDROs.”
The donor stool and FMT used in the two patients were not tested for ESBL-producing gram-negative organisms prior to use.
As a result of these serious adverse reactions, the FDA has determined that the following donor screening and stool testing protections are needed for any investigational use of FMT.
- Donor screening with questions that specifically address risk factors for colonization with MDROs, and exclusion of individuals at higher risk of colonization with MDROs.
- MDRO testing of donor stool and exclusion of stool that tests positive for MDRO. FDA scientists have determined the specific MDRO testing and frequency that should be implemented.
On June 14, the American Gastroenterological Association sent a communication about the FDA alert to its members, which stated that the AGA “is committed to advancing applications of the gut microbiome. Our top priority is ensuring patient safety from microbiome-based therapeutics, such as FMT. Through the AGA FMT National Registry, AGA is working with physicians and patients to track FMT usage, patient outcomes and adverse events. Associated with the registry is a biorepository of donor and patient stool samples, which will allow further investigation of unexpected events such as those described in FDA’s safety alert.”
Dr. Chen, who received the AGA Research Foundation’s 2016 Research Scholar Award for her work on the gut microbiome and inflammatory bowel disease, pointed out that FMT has also been studied as a way to prevent colonization and infection with certain drug resistant organisms, such as vancomycin-resistant Enterococcus.
“Therefore, it’s not that FMT is ‘bad;’ we just have to be more diligent about optimizing the safety of the procedure by screening for of multidrug-resistant organisms,” she said. “We also need to study the use of FMT more, so that we can fully understand the risks associated with the procedure. It’s an important and potentially lifesaving procedure for some, but it’s important that everyone go into the procedure understanding fully what the risks and benefits are.”
Suspected adverse events related to the administration of FMT products can be reported to the FDA at 1-800-332-1088 or via MedWatch.
Officials at the Food and Drug Administration have issued a safety alert regarding the use of fecal microbiota for transplantation and the risk of serious adverse reactions because of transmission of multidrug-resistant organisms (MDROs).
According to the alert, which was issued on June 13, 2019, the agency became aware of two immunocompromised adult patients who received investigational fecal microbiota for transplantation (FMT) and developed extended-spectrum beta-lactamase (EBSL)–producing Escherichia coli. One of the patients died.
“This is certainly a theoretical risk that we’ve known about,” Lea Ann Chen, MD, a gastroenterologist at New York University, said in an interview. “This announcement is important, because we probably don’t counsel patients specifically about this risk. We say there is a risk for transmission of infectious agents in general, but I think that probably very few counsel patients about a risk for transmission of MDROs.”
The donor stool and FMT used in the two patients were not tested for ESBL-producing gram-negative organisms prior to use.
As a result of these serious adverse reactions, the FDA has determined that the following donor screening and stool testing protections are needed for any investigational use of FMT.
- Donor screening with questions that specifically address risk factors for colonization with MDROs, and exclusion of individuals at higher risk of colonization with MDROs.
- MDRO testing of donor stool and exclusion of stool that tests positive for MDRO. FDA scientists have determined the specific MDRO testing and frequency that should be implemented.
On June 14, the American Gastroenterological Association sent a communication about the FDA alert to its members, which stated that the AGA “is committed to advancing applications of the gut microbiome. Our top priority is ensuring patient safety from microbiome-based therapeutics, such as FMT. Through the AGA FMT National Registry, AGA is working with physicians and patients to track FMT usage, patient outcomes and adverse events. Associated with the registry is a biorepository of donor and patient stool samples, which will allow further investigation of unexpected events such as those described in FDA’s safety alert.”
Dr. Chen, who received the AGA Research Foundation’s 2016 Research Scholar Award for her work on the gut microbiome and inflammatory bowel disease, pointed out that FMT has also been studied as a way to prevent colonization and infection with certain drug resistant organisms, such as vancomycin-resistant Enterococcus.
“Therefore, it’s not that FMT is ‘bad;’ we just have to be more diligent about optimizing the safety of the procedure by screening for of multidrug-resistant organisms,” she said. “We also need to study the use of FMT more, so that we can fully understand the risks associated with the procedure. It’s an important and potentially lifesaving procedure for some, but it’s important that everyone go into the procedure understanding fully what the risks and benefits are.”
Suspected adverse events related to the administration of FMT products can be reported to the FDA at 1-800-332-1088 or via MedWatch.
Officials at the Food and Drug Administration have issued a safety alert regarding the use of fecal microbiota for transplantation and the risk of serious adverse reactions because of transmission of multidrug-resistant organisms (MDROs).
According to the alert, which was issued on June 13, 2019, the agency became aware of two immunocompromised adult patients who received investigational fecal microbiota for transplantation (FMT) and developed extended-spectrum beta-lactamase (EBSL)–producing Escherichia coli. One of the patients died.
“This is certainly a theoretical risk that we’ve known about,” Lea Ann Chen, MD, a gastroenterologist at New York University, said in an interview. “This announcement is important, because we probably don’t counsel patients specifically about this risk. We say there is a risk for transmission of infectious agents in general, but I think that probably very few counsel patients about a risk for transmission of MDROs.”
The donor stool and FMT used in the two patients were not tested for ESBL-producing gram-negative organisms prior to use.
As a result of these serious adverse reactions, the FDA has determined that the following donor screening and stool testing protections are needed for any investigational use of FMT.
- Donor screening with questions that specifically address risk factors for colonization with MDROs, and exclusion of individuals at higher risk of colonization with MDROs.
- MDRO testing of donor stool and exclusion of stool that tests positive for MDRO. FDA scientists have determined the specific MDRO testing and frequency that should be implemented.
On June 14, the American Gastroenterological Association sent a communication about the FDA alert to its members, which stated that the AGA “is committed to advancing applications of the gut microbiome. Our top priority is ensuring patient safety from microbiome-based therapeutics, such as FMT. Through the AGA FMT National Registry, AGA is working with physicians and patients to track FMT usage, patient outcomes and adverse events. Associated with the registry is a biorepository of donor and patient stool samples, which will allow further investigation of unexpected events such as those described in FDA’s safety alert.”
Dr. Chen, who received the AGA Research Foundation’s 2016 Research Scholar Award for her work on the gut microbiome and inflammatory bowel disease, pointed out that FMT has also been studied as a way to prevent colonization and infection with certain drug resistant organisms, such as vancomycin-resistant Enterococcus.
“Therefore, it’s not that FMT is ‘bad;’ we just have to be more diligent about optimizing the safety of the procedure by screening for of multidrug-resistant organisms,” she said. “We also need to study the use of FMT more, so that we can fully understand the risks associated with the procedure. It’s an important and potentially lifesaving procedure for some, but it’s important that everyone go into the procedure understanding fully what the risks and benefits are.”
Suspected adverse events related to the administration of FMT products can be reported to the FDA at 1-800-332-1088 or via MedWatch.
FDA approves trastuzumab-anns for HER2-positive breast, gastric cancer
The Food and Drug Administration has approved Amgen’s trastuzumab-anns as a trastuzumab biosimilar for the treatment of HER2-positive breast cancer and gastric cancer.
This biosimilar, to be marketed as Kanjinti, is the fifth trastuzumab biosimilar to be approved by the agency, according to the FDA.
Approval was based in part on the LILAC study, which demonstrated that the biosimilar, previously called ABP-980, had similar efficacy and comparable cardiac safety with trastuzumab.
In the phase 3 study, 725 patients with HER2-positive early breast cancer were randomized to neoadjuvant treatment with trastuzumab-anns or trastuzumab, plus paclitaxel, for four cycles following four cycles of chemotherapy. The primary pathological complete response endpoint was achieved in 48% of those in the biosimilar arm, compared with 40.5% in the trastuzumab arm. Patients then went on to receive adjuvant treatment with ABP 980 or trastuzumab every 3 weeks for up to 1 year following surgery.
Grade 3 or worse adverse events during the neoadjuvant phase occurred in 15% of patients in the ABP 980 group and 14% in the trastuzumab group. The most frequent grade 3 event in both study arms was neutropenia. In the adjuvant phase, grade 3 or worse adverse events occurred in 9% of those continuing ABP 980 and in 6% of those continuing trastuzumab. The most frequent events in both arms were infections, infestations, and neutropenia.
Trastuzumab-anns is indicated for adjuvant treatment of HER2-overexpressing node positive or node negative breast cancer, first-line treatment of HER2-overexpressing metastatic breast cancer, and first-line treatment of patients with HER2-overexpressing metastatic gastric or gastroesophageal junction adenocarcinoma. The FDA indicates patients should be selected based on an FDA-approved companion diagnostic for a trastuzumab product.
The biosimilar includes a boxed warning for cardiomyopathy, infusion reactions, embryo-fetal toxicity, and pulmonary toxicity.
The Food and Drug Administration has approved Amgen’s trastuzumab-anns as a trastuzumab biosimilar for the treatment of HER2-positive breast cancer and gastric cancer.
This biosimilar, to be marketed as Kanjinti, is the fifth trastuzumab biosimilar to be approved by the agency, according to the FDA.
Approval was based in part on the LILAC study, which demonstrated that the biosimilar, previously called ABP-980, had similar efficacy and comparable cardiac safety with trastuzumab.
In the phase 3 study, 725 patients with HER2-positive early breast cancer were randomized to neoadjuvant treatment with trastuzumab-anns or trastuzumab, plus paclitaxel, for four cycles following four cycles of chemotherapy. The primary pathological complete response endpoint was achieved in 48% of those in the biosimilar arm, compared with 40.5% in the trastuzumab arm. Patients then went on to receive adjuvant treatment with ABP 980 or trastuzumab every 3 weeks for up to 1 year following surgery.
Grade 3 or worse adverse events during the neoadjuvant phase occurred in 15% of patients in the ABP 980 group and 14% in the trastuzumab group. The most frequent grade 3 event in both study arms was neutropenia. In the adjuvant phase, grade 3 or worse adverse events occurred in 9% of those continuing ABP 980 and in 6% of those continuing trastuzumab. The most frequent events in both arms were infections, infestations, and neutropenia.
Trastuzumab-anns is indicated for adjuvant treatment of HER2-overexpressing node positive or node negative breast cancer, first-line treatment of HER2-overexpressing metastatic breast cancer, and first-line treatment of patients with HER2-overexpressing metastatic gastric or gastroesophageal junction adenocarcinoma. The FDA indicates patients should be selected based on an FDA-approved companion diagnostic for a trastuzumab product.
The biosimilar includes a boxed warning for cardiomyopathy, infusion reactions, embryo-fetal toxicity, and pulmonary toxicity.
The Food and Drug Administration has approved Amgen’s trastuzumab-anns as a trastuzumab biosimilar for the treatment of HER2-positive breast cancer and gastric cancer.
This biosimilar, to be marketed as Kanjinti, is the fifth trastuzumab biosimilar to be approved by the agency, according to the FDA.
Approval was based in part on the LILAC study, which demonstrated that the biosimilar, previously called ABP-980, had similar efficacy and comparable cardiac safety with trastuzumab.
In the phase 3 study, 725 patients with HER2-positive early breast cancer were randomized to neoadjuvant treatment with trastuzumab-anns or trastuzumab, plus paclitaxel, for four cycles following four cycles of chemotherapy. The primary pathological complete response endpoint was achieved in 48% of those in the biosimilar arm, compared with 40.5% in the trastuzumab arm. Patients then went on to receive adjuvant treatment with ABP 980 or trastuzumab every 3 weeks for up to 1 year following surgery.
Grade 3 or worse adverse events during the neoadjuvant phase occurred in 15% of patients in the ABP 980 group and 14% in the trastuzumab group. The most frequent grade 3 event in both study arms was neutropenia. In the adjuvant phase, grade 3 or worse adverse events occurred in 9% of those continuing ABP 980 and in 6% of those continuing trastuzumab. The most frequent events in both arms were infections, infestations, and neutropenia.
Trastuzumab-anns is indicated for adjuvant treatment of HER2-overexpressing node positive or node negative breast cancer, first-line treatment of HER2-overexpressing metastatic breast cancer, and first-line treatment of patients with HER2-overexpressing metastatic gastric or gastroesophageal junction adenocarcinoma. The FDA indicates patients should be selected based on an FDA-approved companion diagnostic for a trastuzumab product.
The biosimilar includes a boxed warning for cardiomyopathy, infusion reactions, embryo-fetal toxicity, and pulmonary toxicity.
FDA invites sample submission for FDA-ARGOS database
which seeks to support research and regulatory decisions regarding DNA testing for pathogens with quality-controlled and curated genomic sequence data. Such testing and devices could be used as medical countermeasures against biothreats such as Ebola and Zika.

Infectious disease next-generation sequencing could use DNA analysis to help identify pathogens – from viruses to parasites – faster and more efficiently by, in theory, accomplishing with one test what was only possible before with many, according to the FDA. In order to not only further development of such tests and devices but also aid regulatory and scientific review of them, the FDA has collaborated with the Department of Defense, the National Center for Biotechnology Information, and Institute for Genome Sciences at the University of Maryland, Baltimore, to create FDA-ARGOS.
However, the FDA and its collaborators need samples of pathogens to continue developing the database, so they’ve invited health care professionals to submit samples for that purpose. More information, including preferred organism list and submission guidelines, can be found on the FDA-ARGOS website.
which seeks to support research and regulatory decisions regarding DNA testing for pathogens with quality-controlled and curated genomic sequence data. Such testing and devices could be used as medical countermeasures against biothreats such as Ebola and Zika.
Infectious disease next-generation sequencing could use DNA analysis to help identify pathogens – from viruses to parasites – faster and more efficiently by, in theory, accomplishing with one test what was only possible before with many, according to the FDA. In order to not only further development of such tests and devices but also aid regulatory and scientific review of them, the FDA has collaborated with the Department of Defense, the National Center for Biotechnology Information, and Institute for Genome Sciences at the University of Maryland, Baltimore, to create FDA-ARGOS.
However, the FDA and its collaborators need samples of pathogens to continue developing the database, so they’ve invited health care professionals to submit samples for that purpose. More information, including preferred organism list and submission guidelines, can be found on the FDA-ARGOS website.
which seeks to support research and regulatory decisions regarding DNA testing for pathogens with quality-controlled and curated genomic sequence data. Such testing and devices could be used as medical countermeasures against biothreats such as Ebola and Zika.
Infectious disease next-generation sequencing could use DNA analysis to help identify pathogens – from viruses to parasites – faster and more efficiently by, in theory, accomplishing with one test what was only possible before with many, according to the FDA. In order to not only further development of such tests and devices but also aid regulatory and scientific review of them, the FDA has collaborated with the Department of Defense, the National Center for Biotechnology Information, and Institute for Genome Sciences at the University of Maryland, Baltimore, to create FDA-ARGOS.
However, the FDA and its collaborators need samples of pathogens to continue developing the database, so they’ve invited health care professionals to submit samples for that purpose. More information, including preferred organism list and submission guidelines, can be found on the FDA-ARGOS website.
FDA approves Keytruda for metastatic HNSCC
The Food and Drug Administration has approved pembrolizumab (Keytruda) for the first-line treatment of patients with metastatic or unresectable recurrent head and neck squamous cell carcinoma (HNSCC).
FDA approval was based on results of the randomized, multicenter, three-arm, open‑label, active‑controlled KEYNOTE-048 trial. The 882 patients in the trial had metastatic HNSCC, and they received either single-agent pembrolizumab; pembrolizumab, carboplatin or cisplatin, and platinum and fluorouracil; or cetuximab, carboplatin or cisplatin, and platinum and fluorouracil.
Patients who received pembrolizumab plus chemotherapy had a mean overall survival of 13.0 months, compared with 10.7 months in the cetuximab plus chemotherapy group (hazard ratio, 0.77; 95% CI, 0.63-0.93; P = .0067).
In the patient subgroups that received single-agent pembrolizumab, overall survival was 12.3 months in patients with a Combined Positive Score of at least 1, compared with 10.3 months for the cetuximab plus chemotherapy group (HR, 0.78; 95% CI, 0.64-0.96; P = .0171). In patients with a Combined Positive Score of at least 20, overall survival was 14.9 months in the pembrolizumab-only group and 10.7 months in the cetuximab plus chemotherapy group (HR, 0.61; 95% CI, 0.45-0.83; P = .0015).
The most common adverse events in patients who received single-agent pembrolizumab were fatigue, constipation, and rash. In patients who received pembrolizumab plus chemotherapy, the most common adverse events were nausea, fatigue, constipation, vomiting, mucosal inflammation, diarrhea, decreased appetite, stomatitis, and cough.
Pembrolizumab is approved for use in combination with platinum and fluorouracil for all patients and as a single agent for patients whose tumors express programmed death–ligand 1, the FDA said.
The FDA also expanded the intended use for the PD-L1 IHC 22C3 pharmDx kit to include use as a companion diagnostic device for selecting patients with HNSCC for treatment with pembrolizumab as a single agent.
Find the full press release on the FDA website.
The Food and Drug Administration has approved pembrolizumab (Keytruda) for the first-line treatment of patients with metastatic or unresectable recurrent head and neck squamous cell carcinoma (HNSCC).
FDA approval was based on results of the randomized, multicenter, three-arm, open‑label, active‑controlled KEYNOTE-048 trial. The 882 patients in the trial had metastatic HNSCC, and they received either single-agent pembrolizumab; pembrolizumab, carboplatin or cisplatin, and platinum and fluorouracil; or cetuximab, carboplatin or cisplatin, and platinum and fluorouracil.
Patients who received pembrolizumab plus chemotherapy had a mean overall survival of 13.0 months, compared with 10.7 months in the cetuximab plus chemotherapy group (hazard ratio, 0.77; 95% CI, 0.63-0.93; P = .0067).
In the patient subgroups that received single-agent pembrolizumab, overall survival was 12.3 months in patients with a Combined Positive Score of at least 1, compared with 10.3 months for the cetuximab plus chemotherapy group (HR, 0.78; 95% CI, 0.64-0.96; P = .0171). In patients with a Combined Positive Score of at least 20, overall survival was 14.9 months in the pembrolizumab-only group and 10.7 months in the cetuximab plus chemotherapy group (HR, 0.61; 95% CI, 0.45-0.83; P = .0015).
The most common adverse events in patients who received single-agent pembrolizumab were fatigue, constipation, and rash. In patients who received pembrolizumab plus chemotherapy, the most common adverse events were nausea, fatigue, constipation, vomiting, mucosal inflammation, diarrhea, decreased appetite, stomatitis, and cough.
Pembrolizumab is approved for use in combination with platinum and fluorouracil for all patients and as a single agent for patients whose tumors express programmed death–ligand 1, the FDA said.
The FDA also expanded the intended use for the PD-L1 IHC 22C3 pharmDx kit to include use as a companion diagnostic device for selecting patients with HNSCC for treatment with pembrolizumab as a single agent.
Find the full press release on the FDA website.
The Food and Drug Administration has approved pembrolizumab (Keytruda) for the first-line treatment of patients with metastatic or unresectable recurrent head and neck squamous cell carcinoma (HNSCC).
FDA approval was based on results of the randomized, multicenter, three-arm, open‑label, active‑controlled KEYNOTE-048 trial. The 882 patients in the trial had metastatic HNSCC, and they received either single-agent pembrolizumab; pembrolizumab, carboplatin or cisplatin, and platinum and fluorouracil; or cetuximab, carboplatin or cisplatin, and platinum and fluorouracil.
Patients who received pembrolizumab plus chemotherapy had a mean overall survival of 13.0 months, compared with 10.7 months in the cetuximab plus chemotherapy group (hazard ratio, 0.77; 95% CI, 0.63-0.93; P = .0067).
In the patient subgroups that received single-agent pembrolizumab, overall survival was 12.3 months in patients with a Combined Positive Score of at least 1, compared with 10.3 months for the cetuximab plus chemotherapy group (HR, 0.78; 95% CI, 0.64-0.96; P = .0171). In patients with a Combined Positive Score of at least 20, overall survival was 14.9 months in the pembrolizumab-only group and 10.7 months in the cetuximab plus chemotherapy group (HR, 0.61; 95% CI, 0.45-0.83; P = .0015).
The most common adverse events in patients who received single-agent pembrolizumab were fatigue, constipation, and rash. In patients who received pembrolizumab plus chemotherapy, the most common adverse events were nausea, fatigue, constipation, vomiting, mucosal inflammation, diarrhea, decreased appetite, stomatitis, and cough.
Pembrolizumab is approved for use in combination with platinum and fluorouracil for all patients and as a single agent for patients whose tumors express programmed death–ligand 1, the FDA said.
The FDA also expanded the intended use for the PD-L1 IHC 22C3 pharmDx kit to include use as a companion diagnostic device for selecting patients with HNSCC for treatment with pembrolizumab as a single agent.
Find the full press release on the FDA website.
United States now over 1,000 measles cases this year
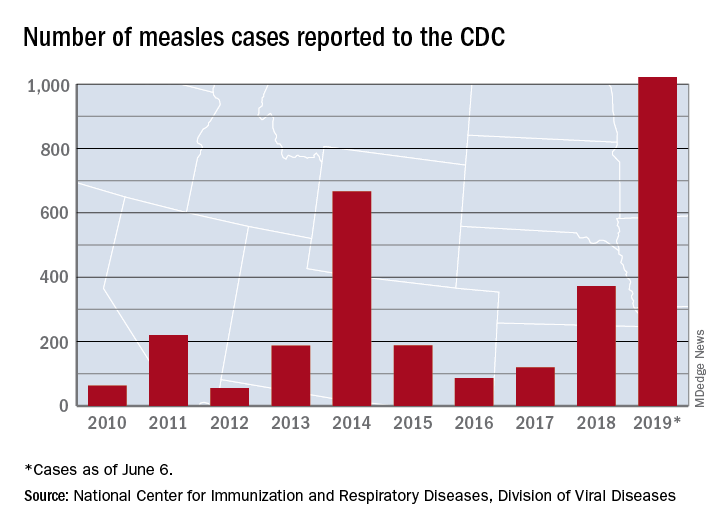
The 41 new cases reported for the week ending June 6 bring the total for the year to 1,022, the CDC reported June 10, and that is more than any year since 1992, when there were 2,237 cases.
Idaho and Virginia reported their first cases of 2019, which makes a total of 28 states with measles cases this year. The Idaho case was reported in Latah County and is the state’s first since 2001. In Virginia, health officials are investigating possible contacts with an infected individual at Dulles International Airport and two other locations on June 2 and 4.
Outbreaks in Georgia, Maryland, and Michigan have ended, while seven others continue in California (Butte, Los Angeles, and Sacramento Counties), New York (Rockland County and New York City), Pennsylvania, and Washington, the CDC said. New York City has the largest outbreak this year with 509 cases through June 3, most of them occurring in Brooklyn.
The 41 new cases reported for the week ending June 6 bring the total for the year to 1,022, the CDC reported June 10, and that is more than any year since 1992, when there were 2,237 cases.
Idaho and Virginia reported their first cases of 2019, which makes a total of 28 states with measles cases this year. The Idaho case was reported in Latah County and is the state’s first since 2001. In Virginia, health officials are investigating possible contacts with an infected individual at Dulles International Airport and two other locations on June 2 and 4.
Outbreaks in Georgia, Maryland, and Michigan have ended, while seven others continue in California (Butte, Los Angeles, and Sacramento Counties), New York (Rockland County and New York City), Pennsylvania, and Washington, the CDC said. New York City has the largest outbreak this year with 509 cases through June 3, most of them occurring in Brooklyn.
The 41 new cases reported for the week ending June 6 bring the total for the year to 1,022, the CDC reported June 10, and that is more than any year since 1992, when there were 2,237 cases.
Idaho and Virginia reported their first cases of 2019, which makes a total of 28 states with measles cases this year. The Idaho case was reported in Latah County and is the state’s first since 2001. In Virginia, health officials are investigating possible contacts with an infected individual at Dulles International Airport and two other locations on June 2 and 4.
Outbreaks in Georgia, Maryland, and Michigan have ended, while seven others continue in California (Butte, Los Angeles, and Sacramento Counties), New York (Rockland County and New York City), Pennsylvania, and Washington, the CDC said. New York City has the largest outbreak this year with 509 cases through June 3, most of them occurring in Brooklyn.
The costs of surviving cancer
Cancer survivors have significantly higher out-of-pocket medical costs than those with no history of cancer, and a quarter of those survivors have some type of material hardship related to their diagnosis, according to the Centers for Disease Control and Prevention.
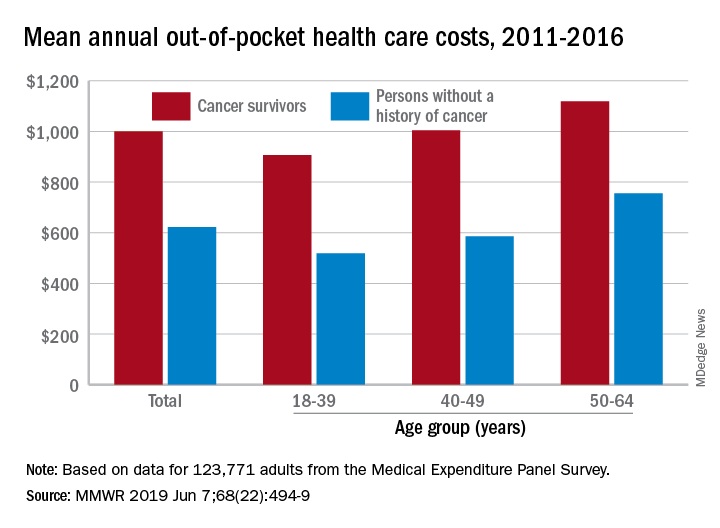
Along with those material financial hardships – the need to borrow money, go into debt, or declare bankruptcy – more than 34% of cancer survivors aged 18-64 years experienced psychological financial hardship, defined as worry about large medical bills, in 2011 and 2016, Donatus U. Ekwueme, PhD, and his associates reported in the Morbidity and Mortality Weekly Report.
Cancer survivors spend 60% more out of pocket than those with no cancer history: $1,000 a year from 2011 to 2016, compared with $622 for adults without a history of cancer. Spending was lowest among younger people (18-39 years) and increased with age, but the prevalence of both material and psychological hardships was highest in the middle age group (40-49 years) and lowest in the oldest group (50-64 years), they said.
Women had higher out-of-pocket costs than men, although the difference was smaller for those with cancer ($1,023 vs. $976) than for those without ($721 vs. $519). Material and psychological hardships were both more common among women, said Dr. Ekwueme of the CDC’s National Center for Chronic Disease Prevention and Health Promotion, Atlanta, and his associates.
Mean out-of-pocket spending was much higher for cancer survivors with private health insurance ($1,114) than for survivors with public insurance ($471), but material hardship was much more prevalent among those with public insurance (33.1% vs. 21.9%). Rates of psychological hardship, however, were much closer: 35.9% for those with public insurance and 32.5% for those with private insurance, the investigators said.
“The number of Americans with a history of cancer is projected to increase in the next decade, and the economic burden associated with living with a cancer diagnosis will likely increase as well,” they wrote, and interventions such as “systematic screening for financial hardship at cancer diagnosis and throughout the cancer care trajectory [are needed] to minimize financial hardship for cancer survivors.”
The analysis was based on data for 123,771 adults aged 18-64 years from the Medical Expenditure Panel Survey. Out-of-pocket costs were calculated using data from 2011 to 2016, with all costs adjusted to 2016 dollars, but the hardship calculations involved data from only 2011 and 2016.
SOURCE: Ekwueme DU et al. MMWR 2019 Jun 7;68(22):494-9.
Cancer survivors have significantly higher out-of-pocket medical costs than those with no history of cancer, and a quarter of those survivors have some type of material hardship related to their diagnosis, according to the Centers for Disease Control and Prevention.
Along with those material financial hardships – the need to borrow money, go into debt, or declare bankruptcy – more than 34% of cancer survivors aged 18-64 years experienced psychological financial hardship, defined as worry about large medical bills, in 2011 and 2016, Donatus U. Ekwueme, PhD, and his associates reported in the Morbidity and Mortality Weekly Report.
Cancer survivors spend 60% more out of pocket than those with no cancer history: $1,000 a year from 2011 to 2016, compared with $622 for adults without a history of cancer. Spending was lowest among younger people (18-39 years) and increased with age, but the prevalence of both material and psychological hardships was highest in the middle age group (40-49 years) and lowest in the oldest group (50-64 years), they said.
Women had higher out-of-pocket costs than men, although the difference was smaller for those with cancer ($1,023 vs. $976) than for those without ($721 vs. $519). Material and psychological hardships were both more common among women, said Dr. Ekwueme of the CDC’s National Center for Chronic Disease Prevention and Health Promotion, Atlanta, and his associates.
Mean out-of-pocket spending was much higher for cancer survivors with private health insurance ($1,114) than for survivors with public insurance ($471), but material hardship was much more prevalent among those with public insurance (33.1% vs. 21.9%). Rates of psychological hardship, however, were much closer: 35.9% for those with public insurance and 32.5% for those with private insurance, the investigators said.
“The number of Americans with a history of cancer is projected to increase in the next decade, and the economic burden associated with living with a cancer diagnosis will likely increase as well,” they wrote, and interventions such as “systematic screening for financial hardship at cancer diagnosis and throughout the cancer care trajectory [are needed] to minimize financial hardship for cancer survivors.”
The analysis was based on data for 123,771 adults aged 18-64 years from the Medical Expenditure Panel Survey. Out-of-pocket costs were calculated using data from 2011 to 2016, with all costs adjusted to 2016 dollars, but the hardship calculations involved data from only 2011 and 2016.
SOURCE: Ekwueme DU et al. MMWR 2019 Jun 7;68(22):494-9.
Cancer survivors have significantly higher out-of-pocket medical costs than those with no history of cancer, and a quarter of those survivors have some type of material hardship related to their diagnosis, according to the Centers for Disease Control and Prevention.
Along with those material financial hardships – the need to borrow money, go into debt, or declare bankruptcy – more than 34% of cancer survivors aged 18-64 years experienced psychological financial hardship, defined as worry about large medical bills, in 2011 and 2016, Donatus U. Ekwueme, PhD, and his associates reported in the Morbidity and Mortality Weekly Report.
Cancer survivors spend 60% more out of pocket than those with no cancer history: $1,000 a year from 2011 to 2016, compared with $622 for adults without a history of cancer. Spending was lowest among younger people (18-39 years) and increased with age, but the prevalence of both material and psychological hardships was highest in the middle age group (40-49 years) and lowest in the oldest group (50-64 years), they said.
Women had higher out-of-pocket costs than men, although the difference was smaller for those with cancer ($1,023 vs. $976) than for those without ($721 vs. $519). Material and psychological hardships were both more common among women, said Dr. Ekwueme of the CDC’s National Center for Chronic Disease Prevention and Health Promotion, Atlanta, and his associates.
Mean out-of-pocket spending was much higher for cancer survivors with private health insurance ($1,114) than for survivors with public insurance ($471), but material hardship was much more prevalent among those with public insurance (33.1% vs. 21.9%). Rates of psychological hardship, however, were much closer: 35.9% for those with public insurance and 32.5% for those with private insurance, the investigators said.
“The number of Americans with a history of cancer is projected to increase in the next decade, and the economic burden associated with living with a cancer diagnosis will likely increase as well,” they wrote, and interventions such as “systematic screening for financial hardship at cancer diagnosis and throughout the cancer care trajectory [are needed] to minimize financial hardship for cancer survivors.”
The analysis was based on data for 123,771 adults aged 18-64 years from the Medical Expenditure Panel Survey. Out-of-pocket costs were calculated using data from 2011 to 2016, with all costs adjusted to 2016 dollars, but the hardship calculations involved data from only 2011 and 2016.
SOURCE: Ekwueme DU et al. MMWR 2019 Jun 7;68(22):494-9.
FROM MMWR
FDA approves Nucala’s new at-home formulations
, according to a press release from the drug’s developer. The biologic will now be available as an autoinjector and as a prefilled safety syringe.
The 100-mg subcutaneous mepolizumab injection is indicated as an add-on treatment for patients 12 years and older with severe eosinophilic asthma, and the three-dose 100-mg subcutaneous injections are indicated for the rare eosinophilic granulomatosis and polyangiitis, with the biologic administered every 4 weeks in either context. The release emphasizes that mepolizumab is not approved for acute bronchospasm or status asthmaticus. Health care professionals should first determine whether self-assisted administration or administration provided by a caregiver is appropriate, and then they should provide patients and/or caregivers with proper training in how to do so.
The approval is based on two open-label, single-arm, phase 3a studies that demonstrated successful administration was possible with these options among patients with severe eosinophilic asthma, at rates of 89%-95% in one study and 100% in the other. These results were followed by those of an open-label, parallel group, single-dose study that confirmed the pharmacokinetic and pharmacodynamic profiles of these new means of administration were comparable with those currently approved.
Mepolizumab is not indicated for those with a history of hypersensitivity to either mepolizumab or to the formulation’s excipients, such as anaphylaxis, angioedema, bronchospasm, hypotension, urticaria, or rash. Any reductions of inhaled corticosteroids after initiation of mepolizumab should be gradual and under the supervision of a health care professional. Some infections by herpes zoster have been observed. The most common adverse reactions (occurring in 3% or more of patients and more often than with placebo) during the first 24 weeks of treatment were headache (19%), injection site reaction (8%), back pain (5%), fatigue (5%), influenza (3%), urinary tract infection (3%), abdominal pain upper (3%), pruritus (3%), eczema (3%), and muscle spasm (3%). Full prescribing information can be found on the FDA website.
, according to a press release from the drug’s developer. The biologic will now be available as an autoinjector and as a prefilled safety syringe.
The 100-mg subcutaneous mepolizumab injection is indicated as an add-on treatment for patients 12 years and older with severe eosinophilic asthma, and the three-dose 100-mg subcutaneous injections are indicated for the rare eosinophilic granulomatosis and polyangiitis, with the biologic administered every 4 weeks in either context. The release emphasizes that mepolizumab is not approved for acute bronchospasm or status asthmaticus. Health care professionals should first determine whether self-assisted administration or administration provided by a caregiver is appropriate, and then they should provide patients and/or caregivers with proper training in how to do so.
The approval is based on two open-label, single-arm, phase 3a studies that demonstrated successful administration was possible with these options among patients with severe eosinophilic asthma, at rates of 89%-95% in one study and 100% in the other. These results were followed by those of an open-label, parallel group, single-dose study that confirmed the pharmacokinetic and pharmacodynamic profiles of these new means of administration were comparable with those currently approved.
Mepolizumab is not indicated for those with a history of hypersensitivity to either mepolizumab or to the formulation’s excipients, such as anaphylaxis, angioedema, bronchospasm, hypotension, urticaria, or rash. Any reductions of inhaled corticosteroids after initiation of mepolizumab should be gradual and under the supervision of a health care professional. Some infections by herpes zoster have been observed. The most common adverse reactions (occurring in 3% or more of patients and more often than with placebo) during the first 24 weeks of treatment were headache (19%), injection site reaction (8%), back pain (5%), fatigue (5%), influenza (3%), urinary tract infection (3%), abdominal pain upper (3%), pruritus (3%), eczema (3%), and muscle spasm (3%). Full prescribing information can be found on the FDA website.
, according to a press release from the drug’s developer. The biologic will now be available as an autoinjector and as a prefilled safety syringe.
The 100-mg subcutaneous mepolizumab injection is indicated as an add-on treatment for patients 12 years and older with severe eosinophilic asthma, and the three-dose 100-mg subcutaneous injections are indicated for the rare eosinophilic granulomatosis and polyangiitis, with the biologic administered every 4 weeks in either context. The release emphasizes that mepolizumab is not approved for acute bronchospasm or status asthmaticus. Health care professionals should first determine whether self-assisted administration or administration provided by a caregiver is appropriate, and then they should provide patients and/or caregivers with proper training in how to do so.
The approval is based on two open-label, single-arm, phase 3a studies that demonstrated successful administration was possible with these options among patients with severe eosinophilic asthma, at rates of 89%-95% in one study and 100% in the other. These results were followed by those of an open-label, parallel group, single-dose study that confirmed the pharmacokinetic and pharmacodynamic profiles of these new means of administration were comparable with those currently approved.
Mepolizumab is not indicated for those with a history of hypersensitivity to either mepolizumab or to the formulation’s excipients, such as anaphylaxis, angioedema, bronchospasm, hypotension, urticaria, or rash. Any reductions of inhaled corticosteroids after initiation of mepolizumab should be gradual and under the supervision of a health care professional. Some infections by herpes zoster have been observed. The most common adverse reactions (occurring in 3% or more of patients and more often than with placebo) during the first 24 weeks of treatment were headache (19%), injection site reaction (8%), back pain (5%), fatigue (5%), influenza (3%), urinary tract infection (3%), abdominal pain upper (3%), pruritus (3%), eczema (3%), and muscle spasm (3%). Full prescribing information can be found on the FDA website.
Stewart Tepper: Emgality approval ‘very exciting’
The drug, a humanized monoclonal antibody that binds to calcitonin gene-related peptide (CGRP), is administered by self-injection in 300-mg doses.
Galcanezumab is the first medication for episodic cluster headache that reduces the frequency of attacks, the agency said in an announcement.
Cluster headache can be more intense than migraine. The pain is unilateral and occurs in the orbital, supraorbital, or temporal regions. It reaches its peak intensity within 5-10 minutes and generally lasts for 30-90 minutes. Symptoms include a burning sensation, conjunctival injection, rhinorrhea, and photosensitivity. Patients often have one to three of these headaches per day, and the headaches appear to be linked to the circadian rhythm. An episodic cluster cycle can last for weeks to months of daily or near daily attacks.
A study presented at the recent meeting of the American Academy of Neurology provided evidence of the drug’s efficacy in cluster headache. In this trial, researchers randomized 106 patients with episodic cluster headache to galcanezumab or placebo. The baseline cluster headache frequency was 17.3 attacks per week, and galcanezumab reduced this frequency to 9.1 attacks per week, compared with 12.1 attacks per week with placebo. The most common side effect reported in this and other clinical trials was injection-site reactions.
Galcanezumab entails a risk of hypersensitivity reactions, according to the FDA. These reactions may occur several days after administration and may be prolonged. “If a serious hypersensitivity reaction occurs, treatment should be discontinued,” the agency said.
“It’s a very exciting day. There had never been a drug approved for prevention of cluster headache,” said Stewart J. Tepper, MD, professor of neurology at the Geisel School of Medicine at Dartmouth and director of the Dartmouth Headache Center, Dartmouth-Hitchcock Medical Center, Lebanon, N.H.

It is difficult to achieve therapeutic concentrations of current preventive medications that do not have FDA approval for this indication, such as verapamil, lithium, or antiepileptic drugs. Galcanezumab, in contrast, works quickly. It is important to note that the approval was for preventive treatment of episodic cluster headache, not for prevention of chronic cluster headache, and not for acute treatment, Dr. Tepper said.
“It’s important to get optimal therapy for cluster headache. It is one of the most disabling, terrible disorders on Earth,” Dr. Tepper said. “The importance [of this approval] cannot be overestimated.”
When asked for comment, Alan M. Rapoport, MD, clinical professor of neurology at the University of California, Los Angeles, said “If this monoclonal antibody to the CGRP ligand works as well in real life as in the trial, it will be an important advance in the treatment of cluster headache.”
Prior to the approval of galcanezumab, noninvasive vagal nerve stimulation was approved in November 2018 for adjunctive use in the preventive treatment of cluster headache in adults.
The FDA granted the application for galcanezumab using a Priority Review and Breakthrough Therapy designation. The agency approved galcanezumab for the preventive treatment of migraine in adults in September 2018. The drug appears to have a similar safety profile in both patient populations. Eli Lilly, which is based in Indianapolis, Indiana, manufactures the drug.
This article was updated June 5, 2019.
The drug, a humanized monoclonal antibody that binds to calcitonin gene-related peptide (CGRP), is administered by self-injection in 300-mg doses.
Galcanezumab is the first medication for episodic cluster headache that reduces the frequency of attacks, the agency said in an announcement.
Cluster headache can be more intense than migraine. The pain is unilateral and occurs in the orbital, supraorbital, or temporal regions. It reaches its peak intensity within 5-10 minutes and generally lasts for 30-90 minutes. Symptoms include a burning sensation, conjunctival injection, rhinorrhea, and photosensitivity. Patients often have one to three of these headaches per day, and the headaches appear to be linked to the circadian rhythm. An episodic cluster cycle can last for weeks to months of daily or near daily attacks.
A study presented at the recent meeting of the American Academy of Neurology provided evidence of the drug’s efficacy in cluster headache. In this trial, researchers randomized 106 patients with episodic cluster headache to galcanezumab or placebo. The baseline cluster headache frequency was 17.3 attacks per week, and galcanezumab reduced this frequency to 9.1 attacks per week, compared with 12.1 attacks per week with placebo. The most common side effect reported in this and other clinical trials was injection-site reactions.
Galcanezumab entails a risk of hypersensitivity reactions, according to the FDA. These reactions may occur several days after administration and may be prolonged. “If a serious hypersensitivity reaction occurs, treatment should be discontinued,” the agency said.
“It’s a very exciting day. There had never been a drug approved for prevention of cluster headache,” said Stewart J. Tepper, MD, professor of neurology at the Geisel School of Medicine at Dartmouth and director of the Dartmouth Headache Center, Dartmouth-Hitchcock Medical Center, Lebanon, N.H.
It is difficult to achieve therapeutic concentrations of current preventive medications that do not have FDA approval for this indication, such as verapamil, lithium, or antiepileptic drugs. Galcanezumab, in contrast, works quickly. It is important to note that the approval was for preventive treatment of episodic cluster headache, not for prevention of chronic cluster headache, and not for acute treatment, Dr. Tepper said.
“It’s important to get optimal therapy for cluster headache. It is one of the most disabling, terrible disorders on Earth,” Dr. Tepper said. “The importance [of this approval] cannot be overestimated.”
When asked for comment, Alan M. Rapoport, MD, clinical professor of neurology at the University of California, Los Angeles, said “If this monoclonal antibody to the CGRP ligand works as well in real life as in the trial, it will be an important advance in the treatment of cluster headache.”
Prior to the approval of galcanezumab, noninvasive vagal nerve stimulation was approved in November 2018 for adjunctive use in the preventive treatment of cluster headache in adults.
The FDA granted the application for galcanezumab using a Priority Review and Breakthrough Therapy designation. The agency approved galcanezumab for the preventive treatment of migraine in adults in September 2018. The drug appears to have a similar safety profile in both patient populations. Eli Lilly, which is based in Indianapolis, Indiana, manufactures the drug.
This article was updated June 5, 2019.
The drug, a humanized monoclonal antibody that binds to calcitonin gene-related peptide (CGRP), is administered by self-injection in 300-mg doses.
Galcanezumab is the first medication for episodic cluster headache that reduces the frequency of attacks, the agency said in an announcement.
Cluster headache can be more intense than migraine. The pain is unilateral and occurs in the orbital, supraorbital, or temporal regions. It reaches its peak intensity within 5-10 minutes and generally lasts for 30-90 minutes. Symptoms include a burning sensation, conjunctival injection, rhinorrhea, and photosensitivity. Patients often have one to three of these headaches per day, and the headaches appear to be linked to the circadian rhythm. An episodic cluster cycle can last for weeks to months of daily or near daily attacks.
A study presented at the recent meeting of the American Academy of Neurology provided evidence of the drug’s efficacy in cluster headache. In this trial, researchers randomized 106 patients with episodic cluster headache to galcanezumab or placebo. The baseline cluster headache frequency was 17.3 attacks per week, and galcanezumab reduced this frequency to 9.1 attacks per week, compared with 12.1 attacks per week with placebo. The most common side effect reported in this and other clinical trials was injection-site reactions.
Galcanezumab entails a risk of hypersensitivity reactions, according to the FDA. These reactions may occur several days after administration and may be prolonged. “If a serious hypersensitivity reaction occurs, treatment should be discontinued,” the agency said.
“It’s a very exciting day. There had never been a drug approved for prevention of cluster headache,” said Stewart J. Tepper, MD, professor of neurology at the Geisel School of Medicine at Dartmouth and director of the Dartmouth Headache Center, Dartmouth-Hitchcock Medical Center, Lebanon, N.H.
It is difficult to achieve therapeutic concentrations of current preventive medications that do not have FDA approval for this indication, such as verapamil, lithium, or antiepileptic drugs. Galcanezumab, in contrast, works quickly. It is important to note that the approval was for preventive treatment of episodic cluster headache, not for prevention of chronic cluster headache, and not for acute treatment, Dr. Tepper said.
“It’s important to get optimal therapy for cluster headache. It is one of the most disabling, terrible disorders on Earth,” Dr. Tepper said. “The importance [of this approval] cannot be overestimated.”
When asked for comment, Alan M. Rapoport, MD, clinical professor of neurology at the University of California, Los Angeles, said “If this monoclonal antibody to the CGRP ligand works as well in real life as in the trial, it will be an important advance in the treatment of cluster headache.”
Prior to the approval of galcanezumab, noninvasive vagal nerve stimulation was approved in November 2018 for adjunctive use in the preventive treatment of cluster headache in adults.
The FDA granted the application for galcanezumab using a Priority Review and Breakthrough Therapy designation. The agency approved galcanezumab for the preventive treatment of migraine in adults in September 2018. The drug appears to have a similar safety profile in both patient populations. Eli Lilly, which is based in Indianapolis, Indiana, manufactures the drug.
This article was updated June 5, 2019.