User login
Vedolizumab looks safer than anti-TNF drugs in older adults with IBD
A large analysis of Medicare data from all 50 states suggests that vedolizumab may be just as effective as anti–tumor necrosis factor (anti-TNF) agents in controlling inflammatory bowel disease (IBD) in patients aged over 65 years, with fewer infectious disease hospitalizations.
The study was prompted by the fact that older adults are greatly underrepresented in clinical trials of approved IBD medications. There is a second peak in IBD diagnosis among people in their 50s and 60s, and IBD patients are living longer with more effective medications. So although a significant number of IBD patients are aged 65 years or older, that group encompasses less than 1% of adults in clinical trials, Bharati Kochar, MD, reported at the annual congress of the Crohn’s & Colitis Foundation and the American Gastroenterological Association.
“Therefore, we don’t know how well these medications work and how safe they are specifically in older adults,” said Dr. Kochar, a gastroenterologist at Massachusetts General Hospital and assistant professor of medicine at Harvard Medical School, both in Boston.
The data largely support what had been known mechanistically about vedolizumab. “It suggests that both drugs work well enough to prevent [IBD-related] hospitalizations, but clearly there was a benefit toward the safer medication, Entyvio [vedolizumab], in the infection-related hospitalizations. That’s not the only readout in infections, but it is an important readout because infections that get hospitalized are the ones that predict mortality and disability,” said Matthew Ciorba, MD, who attended the session. Dr. Ciorba is director of the IBD Center at Washington University in St. Louis and was not involved in the study.
“I think this study is reassuring to clinicians. It provides important clinical data that support what we know about the mechanisms of vedolizumab. The safety data we predicted is borne out in this large and well-done study,” said Dr. Ciorba.

The researchers collected a 20% random sample from a 50-state Medicare claims database, including patients who were aged 65 years or older, who had two or more codes for Crohn’s disease or ulcerative colitis, and had 18 months of continuous enrollment. It excluded Medicare Part C patients; those who used ustekinumab, natalizumab, cyclosporine, or tacrolimus during the look back and study period; and those with two or more codes for rheumatoid arthritis, plaque psoriasis, psoriatic arthritis, or ankylosing spondylitis during the study period.
Among those included, 480 patients were on vedolizumab, while 1,152 were on anti-TNF medications. The two groups were broadly similar in their characteristics: Twenty-nine percent of both groups took budesonide, although the anti-TNF group had a higher frequency use of systemic corticosteroids (68% vs. 57%), 5-ASA drugs (62% vs. 42%), and immunomodulators (32% vs. 28%).
There were no significant differences between the two groups with respect to frequency of IBD-related hospitalizations, IBD-related surgery, steroid prescription rate after induction, or all-cause hospitalization. However, infection-related hospitalizations were less frequent in the vedolizumab group (crude incidence, 0.03 vs. 0.05 per person-year; adjusted hazard ratio, 0.47; 95% confidence interval, 0.25-0.86).
“I think it’s important to use your clinical judgment to treat the patient in front of you, and these data should simply help contextualize risk for older IBD patients newly initiating vedolizumab and anti-TNF agents,” said Dr. Kochar. However, recognizing the limitations of any retrospective study based on administrative data, she called for additional research. “There is a vast need for additional large and robust comparative effectiveness and safety studies in older adults of the rapidly proliferating arsenal of IBD medications,” Dr. Kochar concluded.
Dr. Kochar and Dr. Ciorba have no relevant financial disclosures.
A large analysis of Medicare data from all 50 states suggests that vedolizumab may be just as effective as anti–tumor necrosis factor (anti-TNF) agents in controlling inflammatory bowel disease (IBD) in patients aged over 65 years, with fewer infectious disease hospitalizations.
The study was prompted by the fact that older adults are greatly underrepresented in clinical trials of approved IBD medications. There is a second peak in IBD diagnosis among people in their 50s and 60s, and IBD patients are living longer with more effective medications. So although a significant number of IBD patients are aged 65 years or older, that group encompasses less than 1% of adults in clinical trials, Bharati Kochar, MD, reported at the annual congress of the Crohn’s & Colitis Foundation and the American Gastroenterological Association.
“Therefore, we don’t know how well these medications work and how safe they are specifically in older adults,” said Dr. Kochar, a gastroenterologist at Massachusetts General Hospital and assistant professor of medicine at Harvard Medical School, both in Boston.
The data largely support what had been known mechanistically about vedolizumab. “It suggests that both drugs work well enough to prevent [IBD-related] hospitalizations, but clearly there was a benefit toward the safer medication, Entyvio [vedolizumab], in the infection-related hospitalizations. That’s not the only readout in infections, but it is an important readout because infections that get hospitalized are the ones that predict mortality and disability,” said Matthew Ciorba, MD, who attended the session. Dr. Ciorba is director of the IBD Center at Washington University in St. Louis and was not involved in the study.
“I think this study is reassuring to clinicians. It provides important clinical data that support what we know about the mechanisms of vedolizumab. The safety data we predicted is borne out in this large and well-done study,” said Dr. Ciorba.
The researchers collected a 20% random sample from a 50-state Medicare claims database, including patients who were aged 65 years or older, who had two or more codes for Crohn’s disease or ulcerative colitis, and had 18 months of continuous enrollment. It excluded Medicare Part C patients; those who used ustekinumab, natalizumab, cyclosporine, or tacrolimus during the look back and study period; and those with two or more codes for rheumatoid arthritis, plaque psoriasis, psoriatic arthritis, or ankylosing spondylitis during the study period.
Among those included, 480 patients were on vedolizumab, while 1,152 were on anti-TNF medications. The two groups were broadly similar in their characteristics: Twenty-nine percent of both groups took budesonide, although the anti-TNF group had a higher frequency use of systemic corticosteroids (68% vs. 57%), 5-ASA drugs (62% vs. 42%), and immunomodulators (32% vs. 28%).
There were no significant differences between the two groups with respect to frequency of IBD-related hospitalizations, IBD-related surgery, steroid prescription rate after induction, or all-cause hospitalization. However, infection-related hospitalizations were less frequent in the vedolizumab group (crude incidence, 0.03 vs. 0.05 per person-year; adjusted hazard ratio, 0.47; 95% confidence interval, 0.25-0.86).
“I think it’s important to use your clinical judgment to treat the patient in front of you, and these data should simply help contextualize risk for older IBD patients newly initiating vedolizumab and anti-TNF agents,” said Dr. Kochar. However, recognizing the limitations of any retrospective study based on administrative data, she called for additional research. “There is a vast need for additional large and robust comparative effectiveness and safety studies in older adults of the rapidly proliferating arsenal of IBD medications,” Dr. Kochar concluded.
Dr. Kochar and Dr. Ciorba have no relevant financial disclosures.
A large analysis of Medicare data from all 50 states suggests that vedolizumab may be just as effective as anti–tumor necrosis factor (anti-TNF) agents in controlling inflammatory bowel disease (IBD) in patients aged over 65 years, with fewer infectious disease hospitalizations.
The study was prompted by the fact that older adults are greatly underrepresented in clinical trials of approved IBD medications. There is a second peak in IBD diagnosis among people in their 50s and 60s, and IBD patients are living longer with more effective medications. So although a significant number of IBD patients are aged 65 years or older, that group encompasses less than 1% of adults in clinical trials, Bharati Kochar, MD, reported at the annual congress of the Crohn’s & Colitis Foundation and the American Gastroenterological Association.
“Therefore, we don’t know how well these medications work and how safe they are specifically in older adults,” said Dr. Kochar, a gastroenterologist at Massachusetts General Hospital and assistant professor of medicine at Harvard Medical School, both in Boston.
The data largely support what had been known mechanistically about vedolizumab. “It suggests that both drugs work well enough to prevent [IBD-related] hospitalizations, but clearly there was a benefit toward the safer medication, Entyvio [vedolizumab], in the infection-related hospitalizations. That’s not the only readout in infections, but it is an important readout because infections that get hospitalized are the ones that predict mortality and disability,” said Matthew Ciorba, MD, who attended the session. Dr. Ciorba is director of the IBD Center at Washington University in St. Louis and was not involved in the study.
“I think this study is reassuring to clinicians. It provides important clinical data that support what we know about the mechanisms of vedolizumab. The safety data we predicted is borne out in this large and well-done study,” said Dr. Ciorba.
The researchers collected a 20% random sample from a 50-state Medicare claims database, including patients who were aged 65 years or older, who had two or more codes for Crohn’s disease or ulcerative colitis, and had 18 months of continuous enrollment. It excluded Medicare Part C patients; those who used ustekinumab, natalizumab, cyclosporine, or tacrolimus during the look back and study period; and those with two or more codes for rheumatoid arthritis, plaque psoriasis, psoriatic arthritis, or ankylosing spondylitis during the study period.
Among those included, 480 patients were on vedolizumab, while 1,152 were on anti-TNF medications. The two groups were broadly similar in their characteristics: Twenty-nine percent of both groups took budesonide, although the anti-TNF group had a higher frequency use of systemic corticosteroids (68% vs. 57%), 5-ASA drugs (62% vs. 42%), and immunomodulators (32% vs. 28%).
There were no significant differences between the two groups with respect to frequency of IBD-related hospitalizations, IBD-related surgery, steroid prescription rate after induction, or all-cause hospitalization. However, infection-related hospitalizations were less frequent in the vedolizumab group (crude incidence, 0.03 vs. 0.05 per person-year; adjusted hazard ratio, 0.47; 95% confidence interval, 0.25-0.86).
“I think it’s important to use your clinical judgment to treat the patient in front of you, and these data should simply help contextualize risk for older IBD patients newly initiating vedolizumab and anti-TNF agents,” said Dr. Kochar. However, recognizing the limitations of any retrospective study based on administrative data, she called for additional research. “There is a vast need for additional large and robust comparative effectiveness and safety studies in older adults of the rapidly proliferating arsenal of IBD medications,” Dr. Kochar concluded.
Dr. Kochar and Dr. Ciorba have no relevant financial disclosures.
FROM THE CROHN’S & COLITIS CONGRESS
Prostate drugs tied to lower risk for Parkinson’s disease
new research suggests. Treatment of BPH with terazosin (Hytrin), doxazosin (Cardura), or alfuzosin (Uroxatral), all of which enhance glycolysis, was associated with a lower risk of developing Parkinson’s disease than patients taking a drug used for the same indication, tamsulosin (Flomax), which does not affect glycolysis.
“If giving someone terazosin or similar medications truly reduces their risk of disease, these results could have significant clinical implications for neurologists,” said lead author Jacob E. Simmering, PhD, assistant professor of internal medicine at the University of Iowa, Iowa City.
There are few reliable neuroprotective treatments for Parkinson’s disease, he said. “We can manage some of the symptoms, but we can’t stop it from progressing. If a randomized trial finds the same result, this will provide a new option to slow progression of Parkinson’s disease.”
The pathogenesis of Parkinson’s disease is heterogeneous, however, and not all patients may benefit from glycolysis-enhancing drugs, the investigators noted. Future research will be needed to identify potential candidates for this treatment, and clarify the effects of these drugs, they wrote.
The findings were published online Feb. 1, 2021, in JAMA Neurology.
Time-dependent effects
The major risk factor for Parkinson’s disease is age, which is associated with impaired energy metabolism. Glycolysis is decreased among patients with Parkinson’s disease, yet impaired energy metabolism has not been investigated widely as a pathogenic factor in the disease, the authors wrote.
Studies have indicated that terazosin increases the activity of an enzyme important in glycolysis. Doxazosin and alfuzosin have a similar mechanism of action and enhance energy metabolism. Tamsulosin, a structurally unrelated drug, has the same mechanism of action as the other three drugs, but does not enhance energy metabolism.
In this report, the researchers investigated the hypothesis that patients who received therapy with terazosin, doxazosin, or alfuzosin would have a lower risk of developing Parkinson’s disease than patients receiving tamsulosin. To do that, they used health care utilization data from Denmark and the United States, including the Danish National Prescription Registry, the Danish National Patient Registry, the Danish Civil Registration System, and the Truven Health Analytics MarketScan database.
The investigators searched the records for patients who filled prescriptions for any of the four drugs of interest. They excluded any patients who developed Parkinson’s disease within 1 year of starting medication. Because use of these drugs is rare among women, they included only men in their analysis.
They looked at patient outcomes beginning at 1 year after the initiation of treatment. They also required patients to fill at least two prescriptions before the beginning of follow-up. Patients who switched from tamsulosin to any of the other drugs, or vice versa, were excluded from analysis.
The investigators used propensity-score matching to ensure that patients in the tamsulosin and terazosin/doxazosin/alfuzosin groups were similar in terms of their other potential risk factors. The primary outcome was the development of Parkinson’s disease.
They identified 52,365 propensity score–matched pairs in the Danish registries and 94,883 pairs in the Truven database. The mean age was 67.9 years in the Danish registries and 63.8 years in the Truven database, and follow-up was approximately 5 years and 3 years respectively. Baseline covariates were well balanced between cohorts.
Among Danish patients, those who took terazosin, doxazosin, or alfuzosin had a lower risk of developing Parkinson’s disease versus those who took tamsulosin (hazard ratio, 0.88). Similarly, patients in the Truven database who took terazosin, doxazosin, or alfuzosin had a lower risk of developing Parkinson’s disease than those who took tamsulosin (HR, 0.63).
In both cohorts, the risk for Parkinson’s disease among patients receiving terazosin, doxazosin, or alfuzosin, compared with those receiving tamsulosin, decreased with increasing numbers of prescriptions filled. Long-term treatment with any of the three glycolysis-enhancing drugs was associated with greater risk reduction in the Danish (HR, 0.79) and Truven (HR, 0.46) cohorts versus tamsulosin.
Differences in case definitions, which may reflect how Parkinson’s disease was managed, complicate comparisons between the Danish and Truven cohorts, said Dr. Simmering. Another challenge is the source of the data. “The Truven data set was derived from insurance claims from people with private insurance or Medicare supplemental plans,” he said. “This group is quite large but may not be representative of everyone in the United States. We would also only be able to follow people while they were on one insurance plan. If they switched coverage to a company that doesn’t contribute data, we would lose them.”
The Danish database, however, includes all residents of Denmark. Only people who left the country were lost to follow-up.
The results support the hypothesis that increasing energy in cells slows disease progression, Dr. Simmering added. “There are a few conditions, mostly REM sleep disorders, that are associated with future diagnosis of Parkinson’s disease. Right now, we don’t have anything to offer people at elevated risk of Parkinson’s disease that might prevent the disease. If a controlled trial finds that terazosin slows or prevents Parkinson’s disease, we would have something truly protective to offer these patients.”
Biomarker needed
Commenting on the results, Alberto J. Espay, MD, MSc, professor of neurology at the University of Cincinnati Academic Health Center, was cautious. “These findings are of unclear applicability to any particular patient without a biomarker for a deficit of glycolysis that these drugs are presumed to affect,” Dr. Espay said. “Hence, there is no feasible or warranted change in practice as a result of this study.”
Pathogenic mechanisms are heterogeneous among patients with Parkinson’s disease, Dr. Espay added. “We will need to understand who among the large biological universe of Parkinson’s patients may have impaired energy metabolism as a pathogenic mechanism to be selected for a future clinical trial evaluating terazosin, doxazosin, or alfuzosin as a potential disease-modifying intervention.”
Parkinson’s disease is not one disease, but a group of disorders with unique biological abnormalities, said Dr. Espay. “We know so much about ‘Parkinson’s disease’ and next to nothing about the biology of individuals with Parkinson’s disease.”
This situation has enabled the development of symptomatic treatments, such as dopaminergic therapies, but failed to yield disease-modifying treatments, he said.
The University of Iowa contributed funds for this study. Dr. Simmering has received pilot funding from the University of Iowa Institute for Clinical and Translational Science. He had no conflicts of interest to disclose. Dr. Espay disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
new research suggests. Treatment of BPH with terazosin (Hytrin), doxazosin (Cardura), or alfuzosin (Uroxatral), all of which enhance glycolysis, was associated with a lower risk of developing Parkinson’s disease than patients taking a drug used for the same indication, tamsulosin (Flomax), which does not affect glycolysis.
“If giving someone terazosin or similar medications truly reduces their risk of disease, these results could have significant clinical implications for neurologists,” said lead author Jacob E. Simmering, PhD, assistant professor of internal medicine at the University of Iowa, Iowa City.
There are few reliable neuroprotective treatments for Parkinson’s disease, he said. “We can manage some of the symptoms, but we can’t stop it from progressing. If a randomized trial finds the same result, this will provide a new option to slow progression of Parkinson’s disease.”
The pathogenesis of Parkinson’s disease is heterogeneous, however, and not all patients may benefit from glycolysis-enhancing drugs, the investigators noted. Future research will be needed to identify potential candidates for this treatment, and clarify the effects of these drugs, they wrote.
The findings were published online Feb. 1, 2021, in JAMA Neurology.
Time-dependent effects
The major risk factor for Parkinson’s disease is age, which is associated with impaired energy metabolism. Glycolysis is decreased among patients with Parkinson’s disease, yet impaired energy metabolism has not been investigated widely as a pathogenic factor in the disease, the authors wrote.
Studies have indicated that terazosin increases the activity of an enzyme important in glycolysis. Doxazosin and alfuzosin have a similar mechanism of action and enhance energy metabolism. Tamsulosin, a structurally unrelated drug, has the same mechanism of action as the other three drugs, but does not enhance energy metabolism.
In this report, the researchers investigated the hypothesis that patients who received therapy with terazosin, doxazosin, or alfuzosin would have a lower risk of developing Parkinson’s disease than patients receiving tamsulosin. To do that, they used health care utilization data from Denmark and the United States, including the Danish National Prescription Registry, the Danish National Patient Registry, the Danish Civil Registration System, and the Truven Health Analytics MarketScan database.
The investigators searched the records for patients who filled prescriptions for any of the four drugs of interest. They excluded any patients who developed Parkinson’s disease within 1 year of starting medication. Because use of these drugs is rare among women, they included only men in their analysis.
They looked at patient outcomes beginning at 1 year after the initiation of treatment. They also required patients to fill at least two prescriptions before the beginning of follow-up. Patients who switched from tamsulosin to any of the other drugs, or vice versa, were excluded from analysis.
The investigators used propensity-score matching to ensure that patients in the tamsulosin and terazosin/doxazosin/alfuzosin groups were similar in terms of their other potential risk factors. The primary outcome was the development of Parkinson’s disease.
They identified 52,365 propensity score–matched pairs in the Danish registries and 94,883 pairs in the Truven database. The mean age was 67.9 years in the Danish registries and 63.8 years in the Truven database, and follow-up was approximately 5 years and 3 years respectively. Baseline covariates were well balanced between cohorts.
Among Danish patients, those who took terazosin, doxazosin, or alfuzosin had a lower risk of developing Parkinson’s disease versus those who took tamsulosin (hazard ratio, 0.88). Similarly, patients in the Truven database who took terazosin, doxazosin, or alfuzosin had a lower risk of developing Parkinson’s disease than those who took tamsulosin (HR, 0.63).
In both cohorts, the risk for Parkinson’s disease among patients receiving terazosin, doxazosin, or alfuzosin, compared with those receiving tamsulosin, decreased with increasing numbers of prescriptions filled. Long-term treatment with any of the three glycolysis-enhancing drugs was associated with greater risk reduction in the Danish (HR, 0.79) and Truven (HR, 0.46) cohorts versus tamsulosin.
Differences in case definitions, which may reflect how Parkinson’s disease was managed, complicate comparisons between the Danish and Truven cohorts, said Dr. Simmering. Another challenge is the source of the data. “The Truven data set was derived from insurance claims from people with private insurance or Medicare supplemental plans,” he said. “This group is quite large but may not be representative of everyone in the United States. We would also only be able to follow people while they were on one insurance plan. If they switched coverage to a company that doesn’t contribute data, we would lose them.”
The Danish database, however, includes all residents of Denmark. Only people who left the country were lost to follow-up.
The results support the hypothesis that increasing energy in cells slows disease progression, Dr. Simmering added. “There are a few conditions, mostly REM sleep disorders, that are associated with future diagnosis of Parkinson’s disease. Right now, we don’t have anything to offer people at elevated risk of Parkinson’s disease that might prevent the disease. If a controlled trial finds that terazosin slows or prevents Parkinson’s disease, we would have something truly protective to offer these patients.”
Biomarker needed
Commenting on the results, Alberto J. Espay, MD, MSc, professor of neurology at the University of Cincinnati Academic Health Center, was cautious. “These findings are of unclear applicability to any particular patient without a biomarker for a deficit of glycolysis that these drugs are presumed to affect,” Dr. Espay said. “Hence, there is no feasible or warranted change in practice as a result of this study.”
Pathogenic mechanisms are heterogeneous among patients with Parkinson’s disease, Dr. Espay added. “We will need to understand who among the large biological universe of Parkinson’s patients may have impaired energy metabolism as a pathogenic mechanism to be selected for a future clinical trial evaluating terazosin, doxazosin, or alfuzosin as a potential disease-modifying intervention.”
Parkinson’s disease is not one disease, but a group of disorders with unique biological abnormalities, said Dr. Espay. “We know so much about ‘Parkinson’s disease’ and next to nothing about the biology of individuals with Parkinson’s disease.”
This situation has enabled the development of symptomatic treatments, such as dopaminergic therapies, but failed to yield disease-modifying treatments, he said.
The University of Iowa contributed funds for this study. Dr. Simmering has received pilot funding from the University of Iowa Institute for Clinical and Translational Science. He had no conflicts of interest to disclose. Dr. Espay disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
new research suggests. Treatment of BPH with terazosin (Hytrin), doxazosin (Cardura), or alfuzosin (Uroxatral), all of which enhance glycolysis, was associated with a lower risk of developing Parkinson’s disease than patients taking a drug used for the same indication, tamsulosin (Flomax), which does not affect glycolysis.
“If giving someone terazosin or similar medications truly reduces their risk of disease, these results could have significant clinical implications for neurologists,” said lead author Jacob E. Simmering, PhD, assistant professor of internal medicine at the University of Iowa, Iowa City.
There are few reliable neuroprotective treatments for Parkinson’s disease, he said. “We can manage some of the symptoms, but we can’t stop it from progressing. If a randomized trial finds the same result, this will provide a new option to slow progression of Parkinson’s disease.”
The pathogenesis of Parkinson’s disease is heterogeneous, however, and not all patients may benefit from glycolysis-enhancing drugs, the investigators noted. Future research will be needed to identify potential candidates for this treatment, and clarify the effects of these drugs, they wrote.
The findings were published online Feb. 1, 2021, in JAMA Neurology.
Time-dependent effects
The major risk factor for Parkinson’s disease is age, which is associated with impaired energy metabolism. Glycolysis is decreased among patients with Parkinson’s disease, yet impaired energy metabolism has not been investigated widely as a pathogenic factor in the disease, the authors wrote.
Studies have indicated that terazosin increases the activity of an enzyme important in glycolysis. Doxazosin and alfuzosin have a similar mechanism of action and enhance energy metabolism. Tamsulosin, a structurally unrelated drug, has the same mechanism of action as the other three drugs, but does not enhance energy metabolism.
In this report, the researchers investigated the hypothesis that patients who received therapy with terazosin, doxazosin, or alfuzosin would have a lower risk of developing Parkinson’s disease than patients receiving tamsulosin. To do that, they used health care utilization data from Denmark and the United States, including the Danish National Prescription Registry, the Danish National Patient Registry, the Danish Civil Registration System, and the Truven Health Analytics MarketScan database.
The investigators searched the records for patients who filled prescriptions for any of the four drugs of interest. They excluded any patients who developed Parkinson’s disease within 1 year of starting medication. Because use of these drugs is rare among women, they included only men in their analysis.
They looked at patient outcomes beginning at 1 year after the initiation of treatment. They also required patients to fill at least two prescriptions before the beginning of follow-up. Patients who switched from tamsulosin to any of the other drugs, or vice versa, were excluded from analysis.
The investigators used propensity-score matching to ensure that patients in the tamsulosin and terazosin/doxazosin/alfuzosin groups were similar in terms of their other potential risk factors. The primary outcome was the development of Parkinson’s disease.
They identified 52,365 propensity score–matched pairs in the Danish registries and 94,883 pairs in the Truven database. The mean age was 67.9 years in the Danish registries and 63.8 years in the Truven database, and follow-up was approximately 5 years and 3 years respectively. Baseline covariates were well balanced between cohorts.
Among Danish patients, those who took terazosin, doxazosin, or alfuzosin had a lower risk of developing Parkinson’s disease versus those who took tamsulosin (hazard ratio, 0.88). Similarly, patients in the Truven database who took terazosin, doxazosin, or alfuzosin had a lower risk of developing Parkinson’s disease than those who took tamsulosin (HR, 0.63).
In both cohorts, the risk for Parkinson’s disease among patients receiving terazosin, doxazosin, or alfuzosin, compared with those receiving tamsulosin, decreased with increasing numbers of prescriptions filled. Long-term treatment with any of the three glycolysis-enhancing drugs was associated with greater risk reduction in the Danish (HR, 0.79) and Truven (HR, 0.46) cohorts versus tamsulosin.
Differences in case definitions, which may reflect how Parkinson’s disease was managed, complicate comparisons between the Danish and Truven cohorts, said Dr. Simmering. Another challenge is the source of the data. “The Truven data set was derived from insurance claims from people with private insurance or Medicare supplemental plans,” he said. “This group is quite large but may not be representative of everyone in the United States. We would also only be able to follow people while they were on one insurance plan. If they switched coverage to a company that doesn’t contribute data, we would lose them.”
The Danish database, however, includes all residents of Denmark. Only people who left the country were lost to follow-up.
The results support the hypothesis that increasing energy in cells slows disease progression, Dr. Simmering added. “There are a few conditions, mostly REM sleep disorders, that are associated with future diagnosis of Parkinson’s disease. Right now, we don’t have anything to offer people at elevated risk of Parkinson’s disease that might prevent the disease. If a controlled trial finds that terazosin slows or prevents Parkinson’s disease, we would have something truly protective to offer these patients.”
Biomarker needed
Commenting on the results, Alberto J. Espay, MD, MSc, professor of neurology at the University of Cincinnati Academic Health Center, was cautious. “These findings are of unclear applicability to any particular patient without a biomarker for a deficit of glycolysis that these drugs are presumed to affect,” Dr. Espay said. “Hence, there is no feasible or warranted change in practice as a result of this study.”
Pathogenic mechanisms are heterogeneous among patients with Parkinson’s disease, Dr. Espay added. “We will need to understand who among the large biological universe of Parkinson’s patients may have impaired energy metabolism as a pathogenic mechanism to be selected for a future clinical trial evaluating terazosin, doxazosin, or alfuzosin as a potential disease-modifying intervention.”
Parkinson’s disease is not one disease, but a group of disorders with unique biological abnormalities, said Dr. Espay. “We know so much about ‘Parkinson’s disease’ and next to nothing about the biology of individuals with Parkinson’s disease.”
This situation has enabled the development of symptomatic treatments, such as dopaminergic therapies, but failed to yield disease-modifying treatments, he said.
The University of Iowa contributed funds for this study. Dr. Simmering has received pilot funding from the University of Iowa Institute for Clinical and Translational Science. He had no conflicts of interest to disclose. Dr. Espay disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM NEUROLOGY
ColCORONA: More questions than answers for colchicine in COVID-19
Science by press release and preprint has cooled clinician enthusiasm for the use of colchicine in nonhospitalized patients with COVID-19, despite a pressing need for early treatments.

As previously reported by this news organization, a Jan. 22 press release announced that the massive ColCORONA study missed its primary endpoint of hospitalization or death among 4,488 newly diagnosed patients at increased risk for hospitalization.
But it also touted that use of the anti-inflammatory drug significantly reduced the primary endpoint in 4,159 of those patients with polymerase chain reaction–confirmed COVID and led to reductions of 25%, 50%, and 44%, respectively, for hospitalizations, ventilations, and death.
Lead investigator Jean-Claude Tardif, MD, director of the Montreal Heart Institute Research Centre, deemed the findings a “medical breakthrough.”
When the preprint released a few days later, however, newly revealed confidence intervals showed colchicine did not meaningfully reduce the need for mechanical ventilation (odds ratio, 0.50; 95% confidence interval, 0.23-1.07) or death alone (OR, 0.56; 95% CI, 0.19-1.66).
Further, the significant benefit on the primary outcome came at the cost of a fivefold increase in pulmonary embolism (11 vs. 2; P = .01), which was not mentioned in the press release.
“Whether this represents a real phenomenon or simply the play of chance is not known,” Dr. Tardif and colleagues noted later in the preprint.

“I read the preprint on colchicine and I have so many questions,” Aaron E. Glatt, MD, spokesperson for the Infectious Diseases Society of America and chief of infectious diseases, Mount Sinai South Nassau, Hewlett, N.Y., said in an interview. “I’ve been burned too many times with COVID and prefer to see better data.
“People sometimes say if you wait for perfect data, people are going to die,” he said. “Yeah, but we have no idea if people are going to die from getting this drug more than not getting it. That’s what concerns me. How many pulmonary emboli are going to be fatal versus the slight benefit that the study showed?”
The pushback to the non–peer-reviewed data on social media and via emails was so strong that Dr. Tardif posted a nearly 2,000-word letter responding to the many questions at play.
Chief among them was why the trial, originally planned for 6,000 patients, was stopped early by the investigators without consultation with the data safety monitoring board (DSMB).
The explanation in the letter that logistical issues like running the study call center, budget constraints, and a perceived need to quickly communicate the results left some calling foul that the study wasn’t allowed to finish and come to a more definitive conclusion.
“I can be a little bit sympathetic to their cause but at the same time the DSMB should have said no,” said David Boulware, MD, MPH, who led a recent hydroxychloroquine trial in COVID-19. “The problem is we’re sort of left in limbo, where some people kind of believe it and some say it’s not really a thing. So it’s not really moving the needle, as far as guidelines go.”

Indeed, a Twitter poll by cardiologist James Januzzi Jr., MD, captured the uncertainty, with 28% of respondents saying the trial was “neutral,” 58% saying “maybe but meh,” and 14% saying “colchicine for all.”
Another poll cheekily asked whether ColCORONA was the Gamestop/Reddit equivalent of COVID.
“The press release really didn’t help things because it very much oversold the effect. That, I think, poisoned the well,” said Dr. Boulware, professor of medicine in infectious diseases at the University of Minnesota, Minneapolis.
“The question I’m left with is not whether colchicine works, but who does it work in,” he said. “That’s really the fundamental question because it does seem that there are probably high-risk groups in their trial and others where they benefit, whereas other groups don’t benefit. In the subgroup analysis, there was absolutely no beneficial effect in women.”
According to the authors, the number needed to treat to prevent one death or hospitalization was 71 overall, but 29 for patients with diabetes, 31 for those aged 70 years and older, 53 for patients with respiratory disease, and 25 for those with coronary disease or heart failure.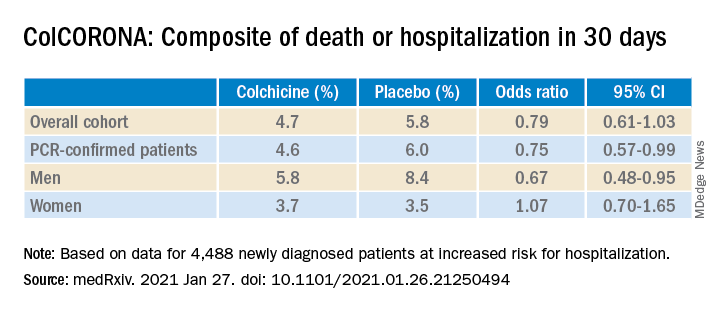
Men are at higher risk overall for poor outcomes. But “the authors didn’t present a multivariable analysis, so it is unclear if another factor, such as a differential prevalence of smoking or cardiovascular risk factors, contributed to the differential benefit,” Rachel Bender Ignacio, MD, MPH, infectious disease specialist, University of Washington, Seattle, said in an interview.
Importantly, in this pragmatic study, duration and severity of symptoms were not reported, observed Dr. Bender Ignacio, who is also a STOP-COVID-2 investigator. “We don’t yet have data as to whether colchicine shortens duration or severity of symptoms or prevents long COVID, so we need more data on that.”
The overall risk for serious adverse events was lower in the colchicine group, but the difference in pulmonary embolism (PE) was striking, she said. This could be caused by a real biologic effect, or it’s possible that persons with shortness of breath and hypoxia, without evident viral pneumonia on chest x-ray after a positive COVID-19 test, were more likely to receive a CT-PE study.
The press release also failed to include information, later noted in the preprint, that the MHI has submitted two patents related to colchicine: “Methods of treating a coronavirus infection using colchicine” and “Early administration of low-dose colchicine after myocardial infarction.”
Reached for clarification, MHI communications adviser Camille Turbide said in an interview that the first patent “simply refers to the novel concept of preventing complications of COVID-19, such as admission to the hospital, with colchicine as tested in the ColCORONA study.”
The second patent, she said, refers to the “novel concept that administering colchicine early after a major adverse cardiovascular event is better than waiting several days,” as supported by the COLCOT study, which Dr. Tardif also led.
The patents are being reviewed by authorities and “Dr. Tardif has waived his rights in these patents and does not stand to benefit financially at all if colchicine becomes used as a treatment for COVID-19,” Ms. Turbide said.
Dr. Tardif did not respond to interview requests for this story. Dr. Glatt said conflicts of interest must be assessed and are “something that is of great concern in any scientific study.”
Cardiologist Steve Nissen, MD, of the Cleveland Clinic said in an interview that, “despite the negative results, the study does suggest that colchicine might have a benefit and should be studied in future trials. These findings are not sufficient evidence to suggest use of the drug in patients infected with COVID-19.”
He noted that adverse effects like diarrhea were expected but that the excess PE was unexpected and needs greater clarification.
“Stopping the trial for administrative reasons is puzzling and undermined the ability of the trial to give a reliable answer,” Dr. Nissen said. “This is a reasonable pilot study that should be viewed as hypothesis generating but inconclusive.”
Several sources said a new trial is unlikely, particularly given the cost and 28 trials already evaluating colchicine. Among these are RECOVERY and COLCOVID, testing whether colchicine can reduce the duration of hospitalization or death in hospitalized patients with COVID-19.
Because there are so many trials ongoing right now, including for antivirals and other immunomodulators, it’s important that, if colchicine comes to routine clinical use, it provides access to treatment for those not able or willing to access clinical trials, rather than impeding clinical trial enrollment, Dr. Bender Ignacio suggested.
“We have already learned the lesson in the pandemic that early adoption of potentially promising therapies can negatively impact our ability to study and develop other promising treatments,” she said.
The trial was coordinated by the Montreal Heart Institute and funded by the government of Quebec; the National Heart, Lung, and Blood Institute of the National Institutes of Health; Montreal philanthropist Sophie Desmarais, and the COVID-19 Therapeutics Accelerator launched by the Bill & Melinda Gates Foundation, Wellcome, and Mastercard. CGI, Dacima, and Pharmascience of Montreal were also collaborators. Dr. Glatt reported no conflicts of interest. Dr. Boulware reported receiving $18 in food and beverages from Gilead Sciences in 2018.
A version of this article first appeared on Medscape.com.
Science by press release and preprint has cooled clinician enthusiasm for the use of colchicine in nonhospitalized patients with COVID-19, despite a pressing need for early treatments.
As previously reported by this news organization, a Jan. 22 press release announced that the massive ColCORONA study missed its primary endpoint of hospitalization or death among 4,488 newly diagnosed patients at increased risk for hospitalization.
But it also touted that use of the anti-inflammatory drug significantly reduced the primary endpoint in 4,159 of those patients with polymerase chain reaction–confirmed COVID and led to reductions of 25%, 50%, and 44%, respectively, for hospitalizations, ventilations, and death.
Lead investigator Jean-Claude Tardif, MD, director of the Montreal Heart Institute Research Centre, deemed the findings a “medical breakthrough.”
When the preprint released a few days later, however, newly revealed confidence intervals showed colchicine did not meaningfully reduce the need for mechanical ventilation (odds ratio, 0.50; 95% confidence interval, 0.23-1.07) or death alone (OR, 0.56; 95% CI, 0.19-1.66).
Further, the significant benefit on the primary outcome came at the cost of a fivefold increase in pulmonary embolism (11 vs. 2; P = .01), which was not mentioned in the press release.
“Whether this represents a real phenomenon or simply the play of chance is not known,” Dr. Tardif and colleagues noted later in the preprint.
“I read the preprint on colchicine and I have so many questions,” Aaron E. Glatt, MD, spokesperson for the Infectious Diseases Society of America and chief of infectious diseases, Mount Sinai South Nassau, Hewlett, N.Y., said in an interview. “I’ve been burned too many times with COVID and prefer to see better data.
“People sometimes say if you wait for perfect data, people are going to die,” he said. “Yeah, but we have no idea if people are going to die from getting this drug more than not getting it. That’s what concerns me. How many pulmonary emboli are going to be fatal versus the slight benefit that the study showed?”
The pushback to the non–peer-reviewed data on social media and via emails was so strong that Dr. Tardif posted a nearly 2,000-word letter responding to the many questions at play.
Chief among them was why the trial, originally planned for 6,000 patients, was stopped early by the investigators without consultation with the data safety monitoring board (DSMB).
The explanation in the letter that logistical issues like running the study call center, budget constraints, and a perceived need to quickly communicate the results left some calling foul that the study wasn’t allowed to finish and come to a more definitive conclusion.
“I can be a little bit sympathetic to their cause but at the same time the DSMB should have said no,” said David Boulware, MD, MPH, who led a recent hydroxychloroquine trial in COVID-19. “The problem is we’re sort of left in limbo, where some people kind of believe it and some say it’s not really a thing. So it’s not really moving the needle, as far as guidelines go.”
Indeed, a Twitter poll by cardiologist James Januzzi Jr., MD, captured the uncertainty, with 28% of respondents saying the trial was “neutral,” 58% saying “maybe but meh,” and 14% saying “colchicine for all.”
Another poll cheekily asked whether ColCORONA was the Gamestop/Reddit equivalent of COVID.
“The press release really didn’t help things because it very much oversold the effect. That, I think, poisoned the well,” said Dr. Boulware, professor of medicine in infectious diseases at the University of Minnesota, Minneapolis.
“The question I’m left with is not whether colchicine works, but who does it work in,” he said. “That’s really the fundamental question because it does seem that there are probably high-risk groups in their trial and others where they benefit, whereas other groups don’t benefit. In the subgroup analysis, there was absolutely no beneficial effect in women.”
According to the authors, the number needed to treat to prevent one death or hospitalization was 71 overall, but 29 for patients with diabetes, 31 for those aged 70 years and older, 53 for patients with respiratory disease, and 25 for those with coronary disease or heart failure.
Men are at higher risk overall for poor outcomes. But “the authors didn’t present a multivariable analysis, so it is unclear if another factor, such as a differential prevalence of smoking or cardiovascular risk factors, contributed to the differential benefit,” Rachel Bender Ignacio, MD, MPH, infectious disease specialist, University of Washington, Seattle, said in an interview.
Importantly, in this pragmatic study, duration and severity of symptoms were not reported, observed Dr. Bender Ignacio, who is also a STOP-COVID-2 investigator. “We don’t yet have data as to whether colchicine shortens duration or severity of symptoms or prevents long COVID, so we need more data on that.”
The overall risk for serious adverse events was lower in the colchicine group, but the difference in pulmonary embolism (PE) was striking, she said. This could be caused by a real biologic effect, or it’s possible that persons with shortness of breath and hypoxia, without evident viral pneumonia on chest x-ray after a positive COVID-19 test, were more likely to receive a CT-PE study.
The press release also failed to include information, later noted in the preprint, that the MHI has submitted two patents related to colchicine: “Methods of treating a coronavirus infection using colchicine” and “Early administration of low-dose colchicine after myocardial infarction.”
Reached for clarification, MHI communications adviser Camille Turbide said in an interview that the first patent “simply refers to the novel concept of preventing complications of COVID-19, such as admission to the hospital, with colchicine as tested in the ColCORONA study.”
The second patent, she said, refers to the “novel concept that administering colchicine early after a major adverse cardiovascular event is better than waiting several days,” as supported by the COLCOT study, which Dr. Tardif also led.
The patents are being reviewed by authorities and “Dr. Tardif has waived his rights in these patents and does not stand to benefit financially at all if colchicine becomes used as a treatment for COVID-19,” Ms. Turbide said.
Dr. Tardif did not respond to interview requests for this story. Dr. Glatt said conflicts of interest must be assessed and are “something that is of great concern in any scientific study.”
Cardiologist Steve Nissen, MD, of the Cleveland Clinic said in an interview that, “despite the negative results, the study does suggest that colchicine might have a benefit and should be studied in future trials. These findings are not sufficient evidence to suggest use of the drug in patients infected with COVID-19.”
He noted that adverse effects like diarrhea were expected but that the excess PE was unexpected and needs greater clarification.
“Stopping the trial for administrative reasons is puzzling and undermined the ability of the trial to give a reliable answer,” Dr. Nissen said. “This is a reasonable pilot study that should be viewed as hypothesis generating but inconclusive.”
Several sources said a new trial is unlikely, particularly given the cost and 28 trials already evaluating colchicine. Among these are RECOVERY and COLCOVID, testing whether colchicine can reduce the duration of hospitalization or death in hospitalized patients with COVID-19.
Because there are so many trials ongoing right now, including for antivirals and other immunomodulators, it’s important that, if colchicine comes to routine clinical use, it provides access to treatment for those not able or willing to access clinical trials, rather than impeding clinical trial enrollment, Dr. Bender Ignacio suggested.
“We have already learned the lesson in the pandemic that early adoption of potentially promising therapies can negatively impact our ability to study and develop other promising treatments,” she said.
The trial was coordinated by the Montreal Heart Institute and funded by the government of Quebec; the National Heart, Lung, and Blood Institute of the National Institutes of Health; Montreal philanthropist Sophie Desmarais, and the COVID-19 Therapeutics Accelerator launched by the Bill & Melinda Gates Foundation, Wellcome, and Mastercard. CGI, Dacima, and Pharmascience of Montreal were also collaborators. Dr. Glatt reported no conflicts of interest. Dr. Boulware reported receiving $18 in food and beverages from Gilead Sciences in 2018.
A version of this article first appeared on Medscape.com.
Science by press release and preprint has cooled clinician enthusiasm for the use of colchicine in nonhospitalized patients with COVID-19, despite a pressing need for early treatments.
As previously reported by this news organization, a Jan. 22 press release announced that the massive ColCORONA study missed its primary endpoint of hospitalization or death among 4,488 newly diagnosed patients at increased risk for hospitalization.
But it also touted that use of the anti-inflammatory drug significantly reduced the primary endpoint in 4,159 of those patients with polymerase chain reaction–confirmed COVID and led to reductions of 25%, 50%, and 44%, respectively, for hospitalizations, ventilations, and death.
Lead investigator Jean-Claude Tardif, MD, director of the Montreal Heart Institute Research Centre, deemed the findings a “medical breakthrough.”
When the preprint released a few days later, however, newly revealed confidence intervals showed colchicine did not meaningfully reduce the need for mechanical ventilation (odds ratio, 0.50; 95% confidence interval, 0.23-1.07) or death alone (OR, 0.56; 95% CI, 0.19-1.66).
Further, the significant benefit on the primary outcome came at the cost of a fivefold increase in pulmonary embolism (11 vs. 2; P = .01), which was not mentioned in the press release.
“Whether this represents a real phenomenon or simply the play of chance is not known,” Dr. Tardif and colleagues noted later in the preprint.
“I read the preprint on colchicine and I have so many questions,” Aaron E. Glatt, MD, spokesperson for the Infectious Diseases Society of America and chief of infectious diseases, Mount Sinai South Nassau, Hewlett, N.Y., said in an interview. “I’ve been burned too many times with COVID and prefer to see better data.
“People sometimes say if you wait for perfect data, people are going to die,” he said. “Yeah, but we have no idea if people are going to die from getting this drug more than not getting it. That’s what concerns me. How many pulmonary emboli are going to be fatal versus the slight benefit that the study showed?”
The pushback to the non–peer-reviewed data on social media and via emails was so strong that Dr. Tardif posted a nearly 2,000-word letter responding to the many questions at play.
Chief among them was why the trial, originally planned for 6,000 patients, was stopped early by the investigators without consultation with the data safety monitoring board (DSMB).
The explanation in the letter that logistical issues like running the study call center, budget constraints, and a perceived need to quickly communicate the results left some calling foul that the study wasn’t allowed to finish and come to a more definitive conclusion.
“I can be a little bit sympathetic to their cause but at the same time the DSMB should have said no,” said David Boulware, MD, MPH, who led a recent hydroxychloroquine trial in COVID-19. “The problem is we’re sort of left in limbo, where some people kind of believe it and some say it’s not really a thing. So it’s not really moving the needle, as far as guidelines go.”
Indeed, a Twitter poll by cardiologist James Januzzi Jr., MD, captured the uncertainty, with 28% of respondents saying the trial was “neutral,” 58% saying “maybe but meh,” and 14% saying “colchicine for all.”
Another poll cheekily asked whether ColCORONA was the Gamestop/Reddit equivalent of COVID.
“The press release really didn’t help things because it very much oversold the effect. That, I think, poisoned the well,” said Dr. Boulware, professor of medicine in infectious diseases at the University of Minnesota, Minneapolis.
“The question I’m left with is not whether colchicine works, but who does it work in,” he said. “That’s really the fundamental question because it does seem that there are probably high-risk groups in their trial and others where they benefit, whereas other groups don’t benefit. In the subgroup analysis, there was absolutely no beneficial effect in women.”
According to the authors, the number needed to treat to prevent one death or hospitalization was 71 overall, but 29 for patients with diabetes, 31 for those aged 70 years and older, 53 for patients with respiratory disease, and 25 for those with coronary disease or heart failure.
Men are at higher risk overall for poor outcomes. But “the authors didn’t present a multivariable analysis, so it is unclear if another factor, such as a differential prevalence of smoking or cardiovascular risk factors, contributed to the differential benefit,” Rachel Bender Ignacio, MD, MPH, infectious disease specialist, University of Washington, Seattle, said in an interview.
Importantly, in this pragmatic study, duration and severity of symptoms were not reported, observed Dr. Bender Ignacio, who is also a STOP-COVID-2 investigator. “We don’t yet have data as to whether colchicine shortens duration or severity of symptoms or prevents long COVID, so we need more data on that.”
The overall risk for serious adverse events was lower in the colchicine group, but the difference in pulmonary embolism (PE) was striking, she said. This could be caused by a real biologic effect, or it’s possible that persons with shortness of breath and hypoxia, without evident viral pneumonia on chest x-ray after a positive COVID-19 test, were more likely to receive a CT-PE study.
The press release also failed to include information, later noted in the preprint, that the MHI has submitted two patents related to colchicine: “Methods of treating a coronavirus infection using colchicine” and “Early administration of low-dose colchicine after myocardial infarction.”
Reached for clarification, MHI communications adviser Camille Turbide said in an interview that the first patent “simply refers to the novel concept of preventing complications of COVID-19, such as admission to the hospital, with colchicine as tested in the ColCORONA study.”
The second patent, she said, refers to the “novel concept that administering colchicine early after a major adverse cardiovascular event is better than waiting several days,” as supported by the COLCOT study, which Dr. Tardif also led.
The patents are being reviewed by authorities and “Dr. Tardif has waived his rights in these patents and does not stand to benefit financially at all if colchicine becomes used as a treatment for COVID-19,” Ms. Turbide said.
Dr. Tardif did not respond to interview requests for this story. Dr. Glatt said conflicts of interest must be assessed and are “something that is of great concern in any scientific study.”
Cardiologist Steve Nissen, MD, of the Cleveland Clinic said in an interview that, “despite the negative results, the study does suggest that colchicine might have a benefit and should be studied in future trials. These findings are not sufficient evidence to suggest use of the drug in patients infected with COVID-19.”
He noted that adverse effects like diarrhea were expected but that the excess PE was unexpected and needs greater clarification.
“Stopping the trial for administrative reasons is puzzling and undermined the ability of the trial to give a reliable answer,” Dr. Nissen said. “This is a reasonable pilot study that should be viewed as hypothesis generating but inconclusive.”
Several sources said a new trial is unlikely, particularly given the cost and 28 trials already evaluating colchicine. Among these are RECOVERY and COLCOVID, testing whether colchicine can reduce the duration of hospitalization or death in hospitalized patients with COVID-19.
Because there are so many trials ongoing right now, including for antivirals and other immunomodulators, it’s important that, if colchicine comes to routine clinical use, it provides access to treatment for those not able or willing to access clinical trials, rather than impeding clinical trial enrollment, Dr. Bender Ignacio suggested.
“We have already learned the lesson in the pandemic that early adoption of potentially promising therapies can negatively impact our ability to study and develop other promising treatments,” she said.
The trial was coordinated by the Montreal Heart Institute and funded by the government of Quebec; the National Heart, Lung, and Blood Institute of the National Institutes of Health; Montreal philanthropist Sophie Desmarais, and the COVID-19 Therapeutics Accelerator launched by the Bill & Melinda Gates Foundation, Wellcome, and Mastercard. CGI, Dacima, and Pharmascience of Montreal were also collaborators. Dr. Glatt reported no conflicts of interest. Dr. Boulware reported receiving $18 in food and beverages from Gilead Sciences in 2018.
A version of this article first appeared on Medscape.com.
Vaccine may blunt effects of deadly synthetic opioids
New experimental vaccines could stop the worst effects of synthetic fentanyl and carfentanil, two drugs that have been major drivers of the opioid epidemic in the United States, according to a new study published in ACS Chemical Biology on Feb. 3, 2021.

During several experiments in mice, the vaccines prevented respiratory depression, which is the main cause of overdose deaths. The vaccines also reduced the amount of drug that was distributed to the brain. Once in the brain, synthetic opioids prompt the body to slow down breathing, and when too much of the drug is consumed, breathing can stop.
“Synthetic opioids are not only extremely deadly but also addictive and easy to manufacture, making them a formidable public health threat, especially when the coronavirus crisis is negatively impacting mental health,” Kim Janda, PhD, a chemist at Scripps Research Institute in La Jolla, Calif., who developed the vaccines, said in a statement.
Fentanyl is up to 100 times stronger than morphine, and carfentanil, which is often used by veterinarians to sedate large animals such as elephants, is up to 10,000 times stronger than morphine. Carfentanil isn’t as well-known as a street drug, but it’s being used more often as an additive in heroin and cocaine.
“We’ve shown it is possible to prevent these unnecessary deaths by eliciting antibodies that stop the drug from reaching the brain,” he said.
The vaccines could be used in emergency situations to treat overdoses and as a therapy for those with substance abuse disorders, Dr. Janda said. In addition, the vaccines could protect military officers who are exposed to opioids as chemical weapons, and they may also help opioid-sniffing police dogs to train for the job.
The vaccines are still in the early stages of testing, but looking at the latest data “brings us hope that this approach will work to treat a number of opioid-related maladies,” Dr. Janda said.
In December, the CDC reported that more than 81,000 drug overdose deaths happened in the United States between May 2019 and May 2020, which was the highest number ever recorded in a 12-month period. Synthetic opioids, particularly illegally created fentanyl, were to blame.
“Unfortunately, currently battling a pandemic,” Dr. Janda said. “We look forward to continuing our vaccine research and translating it to the clinic, where we can begin to make an impact on the opioid crisis.”
A version of this article first appeared on Medscape.com.
New experimental vaccines could stop the worst effects of synthetic fentanyl and carfentanil, two drugs that have been major drivers of the opioid epidemic in the United States, according to a new study published in ACS Chemical Biology on Feb. 3, 2021.
During several experiments in mice, the vaccines prevented respiratory depression, which is the main cause of overdose deaths. The vaccines also reduced the amount of drug that was distributed to the brain. Once in the brain, synthetic opioids prompt the body to slow down breathing, and when too much of the drug is consumed, breathing can stop.
“Synthetic opioids are not only extremely deadly but also addictive and easy to manufacture, making them a formidable public health threat, especially when the coronavirus crisis is negatively impacting mental health,” Kim Janda, PhD, a chemist at Scripps Research Institute in La Jolla, Calif., who developed the vaccines, said in a statement.
Fentanyl is up to 100 times stronger than morphine, and carfentanil, which is often used by veterinarians to sedate large animals such as elephants, is up to 10,000 times stronger than morphine. Carfentanil isn’t as well-known as a street drug, but it’s being used more often as an additive in heroin and cocaine.
“We’ve shown it is possible to prevent these unnecessary deaths by eliciting antibodies that stop the drug from reaching the brain,” he said.
The vaccines could be used in emergency situations to treat overdoses and as a therapy for those with substance abuse disorders, Dr. Janda said. In addition, the vaccines could protect military officers who are exposed to opioids as chemical weapons, and they may also help opioid-sniffing police dogs to train for the job.
The vaccines are still in the early stages of testing, but looking at the latest data “brings us hope that this approach will work to treat a number of opioid-related maladies,” Dr. Janda said.
In December, the CDC reported that more than 81,000 drug overdose deaths happened in the United States between May 2019 and May 2020, which was the highest number ever recorded in a 12-month period. Synthetic opioids, particularly illegally created fentanyl, were to blame.
“Unfortunately, currently battling a pandemic,” Dr. Janda said. “We look forward to continuing our vaccine research and translating it to the clinic, where we can begin to make an impact on the opioid crisis.”
A version of this article first appeared on Medscape.com.
New experimental vaccines could stop the worst effects of synthetic fentanyl and carfentanil, two drugs that have been major drivers of the opioid epidemic in the United States, according to a new study published in ACS Chemical Biology on Feb. 3, 2021.
During several experiments in mice, the vaccines prevented respiratory depression, which is the main cause of overdose deaths. The vaccines also reduced the amount of drug that was distributed to the brain. Once in the brain, synthetic opioids prompt the body to slow down breathing, and when too much of the drug is consumed, breathing can stop.
“Synthetic opioids are not only extremely deadly but also addictive and easy to manufacture, making them a formidable public health threat, especially when the coronavirus crisis is negatively impacting mental health,” Kim Janda, PhD, a chemist at Scripps Research Institute in La Jolla, Calif., who developed the vaccines, said in a statement.
Fentanyl is up to 100 times stronger than morphine, and carfentanil, which is often used by veterinarians to sedate large animals such as elephants, is up to 10,000 times stronger than morphine. Carfentanil isn’t as well-known as a street drug, but it’s being used more often as an additive in heroin and cocaine.
“We’ve shown it is possible to prevent these unnecessary deaths by eliciting antibodies that stop the drug from reaching the brain,” he said.
The vaccines could be used in emergency situations to treat overdoses and as a therapy for those with substance abuse disorders, Dr. Janda said. In addition, the vaccines could protect military officers who are exposed to opioids as chemical weapons, and they may also help opioid-sniffing police dogs to train for the job.
The vaccines are still in the early stages of testing, but looking at the latest data “brings us hope that this approach will work to treat a number of opioid-related maladies,” Dr. Janda said.
In December, the CDC reported that more than 81,000 drug overdose deaths happened in the United States between May 2019 and May 2020, which was the highest number ever recorded in a 12-month period. Synthetic opioids, particularly illegally created fentanyl, were to blame.
“Unfortunately, currently battling a pandemic,” Dr. Janda said. “We look forward to continuing our vaccine research and translating it to the clinic, where we can begin to make an impact on the opioid crisis.”
A version of this article first appeared on Medscape.com.
Neoadjuvant immunotherapy shows promise in stage III melanoma
The next dramatic , John M. Kirkwood, MD, predicted at a virtual forum on cutaneous malignancies jointly presented by the Postgraduate Institute for Medicine and Global Academy for Medical Education.

These agents have already demonstrated profound efficacy, first in stage IV metastatic disease and more recently as adjuvant therapy for resected stage III melanoma. Now, there is a great interest in learning whether by prescribing them preoperatively, patients might reduce their risk of advancing to metastatic disease. And neoadjuvant therapy offers an extremely attractive feature: It yields results in an accelerated fashion.
“The major problem with postoperative adjuvant trials in melanoma since 1984 is the long time to maturity. Many of us don’t want to wait the full 9 or 10 years for a full-bore, phase 3 adjuvant trial in stage III melanoma to mature,” explained Dr. Kirkwood, professor of medicine, dermatology, and translational science and coleader of the melanoma and skin cancer program at the University of Pittsburgh. “The opportunity to treat a patient who presents with a bulky lymph node, has a biopsy, and then can be treated for 3 or 6 weeks or sometimes even longer periods with a therapy that’s promising allows us to ask what’s going on in the tumor tissue, what’s going on in the clinical response at 3 or 6 weeks, and if there’s pathological complete or near-complete response under the microscope.”
Because pathological complete response is a strong predictor of relapse-free survival, this neoadjuvant-forward therapeutic strategy has the potential to provide patients and their physicians with an early forecast of likely clinical outcome only 4-6 weeks into treatment. Also, there is both preclinical and clinical evidence that neoadjuvant therapy may offer a survival advantage over adjuvant therapy, perhaps as a result of early treatment of micrometastatic disease. Another benefit of neoadjuvant therapy for melanoma is the resultant tumor shrinkage, which can permit less extensive surgery.
Dr. Kirkwood highlighted a phase 2 clinical trial conducted at the University of Pittsburgh to illustrate the potential of neoadjuvant therapy in melanoma. The ongoing single-arm study includes 32 patients with stage IIIB or IIIC resectable melanoma along with accessible tumor for biopsy and intratumoral injections of CMP-001, a toll-like receptor 9 agonist. According to the Eighth Edition of the American Joint Committee on Cancer staging manual, stage IIIB melanoma has a 10-year mortality of 23%, and stage IIIC disease has 40%.
CMP-001 triggers type 1 interferon production through activation of plasmacytoid dendritic cells. The resultant inflammatory response draws T cells into the tumor to enhance the response to immunotherapy, which in this study was nivolumab (Opdivo), a human programmed death ligand 1 (PD-L1)–blocking antibody. The neoadjuvant regimen consisted of seven once-weekly intratumoral injections of CMP-001, plus three 240-mg doses of nivolumab given at 2-week intervals. This was followed by resection, then 1 year of adjuvant therapy with nivolumab at 480 mg every 4 weeks and intratumoral CMP-001 every 4 weeks.
In an interim analysis, a major pathologic response occurred in an impressive 15 of 21 patients (71%) after 6 weeks of neoadjuvant therapy. Thirteen of the 15 had a pathologic complete response. Encouragingly, no one with a pathologic complete or near-complete response has relapsed to date.
“A pathologic complete response or near-complete response with neoadjuvant therapy appears to be a biomarker of durable disease control and is associated with excellent outcomes,” Dr. Kirkwood observed, adding that the Pittsburgh experience has been mirrored in reports from the Netherlands, Australia, and University of Texas M.D. Anderson Cancer Center, Houston, involving other neoadjuvant agents.
Other potential early biomarkers of favorable outcome with neoadjuvant therapy include CD8+ T cells in the tumor at baseline, tumor mutational burden, T-cell clonality, and a T-cell–inflamed gene-expression profile.
There were no dose-limiting toxicities or delays in surgery related to the neoadjuvant treatment.
Of note, imaging often inaccurately showed only a partial response in patients who actually had a pathologic complete response, meaning totally devoid of tumor, Dr. Kirkwood said.
Corroboration of these findings is planned in the national multicenter ECOG-ACRIN neoadjuvant trial EA6194.
“Consider referring to this trial any patients who present with bulky nodal disease for whom a treatment assessment at 4-6 weeks is desired in order to predict what the outcome may be,” he suggested.
Dr. Kirkwood reported receiving research grants from Amgen, BMS, Castle Biosciences, Checkmate, Immunocore, Iovance, and Novartis and serving as a consultant to a handful of companies.
Global Academy for Medical Education and this news organization are owned by the same company.
The next dramatic , John M. Kirkwood, MD, predicted at a virtual forum on cutaneous malignancies jointly presented by the Postgraduate Institute for Medicine and Global Academy for Medical Education.
These agents have already demonstrated profound efficacy, first in stage IV metastatic disease and more recently as adjuvant therapy for resected stage III melanoma. Now, there is a great interest in learning whether by prescribing them preoperatively, patients might reduce their risk of advancing to metastatic disease. And neoadjuvant therapy offers an extremely attractive feature: It yields results in an accelerated fashion.
“The major problem with postoperative adjuvant trials in melanoma since 1984 is the long time to maturity. Many of us don’t want to wait the full 9 or 10 years for a full-bore, phase 3 adjuvant trial in stage III melanoma to mature,” explained Dr. Kirkwood, professor of medicine, dermatology, and translational science and coleader of the melanoma and skin cancer program at the University of Pittsburgh. “The opportunity to treat a patient who presents with a bulky lymph node, has a biopsy, and then can be treated for 3 or 6 weeks or sometimes even longer periods with a therapy that’s promising allows us to ask what’s going on in the tumor tissue, what’s going on in the clinical response at 3 or 6 weeks, and if there’s pathological complete or near-complete response under the microscope.”
Because pathological complete response is a strong predictor of relapse-free survival, this neoadjuvant-forward therapeutic strategy has the potential to provide patients and their physicians with an early forecast of likely clinical outcome only 4-6 weeks into treatment. Also, there is both preclinical and clinical evidence that neoadjuvant therapy may offer a survival advantage over adjuvant therapy, perhaps as a result of early treatment of micrometastatic disease. Another benefit of neoadjuvant therapy for melanoma is the resultant tumor shrinkage, which can permit less extensive surgery.
Dr. Kirkwood highlighted a phase 2 clinical trial conducted at the University of Pittsburgh to illustrate the potential of neoadjuvant therapy in melanoma. The ongoing single-arm study includes 32 patients with stage IIIB or IIIC resectable melanoma along with accessible tumor for biopsy and intratumoral injections of CMP-001, a toll-like receptor 9 agonist. According to the Eighth Edition of the American Joint Committee on Cancer staging manual, stage IIIB melanoma has a 10-year mortality of 23%, and stage IIIC disease has 40%.
CMP-001 triggers type 1 interferon production through activation of plasmacytoid dendritic cells. The resultant inflammatory response draws T cells into the tumor to enhance the response to immunotherapy, which in this study was nivolumab (Opdivo), a human programmed death ligand 1 (PD-L1)–blocking antibody. The neoadjuvant regimen consisted of seven once-weekly intratumoral injections of CMP-001, plus three 240-mg doses of nivolumab given at 2-week intervals. This was followed by resection, then 1 year of adjuvant therapy with nivolumab at 480 mg every 4 weeks and intratumoral CMP-001 every 4 weeks.
In an interim analysis, a major pathologic response occurred in an impressive 15 of 21 patients (71%) after 6 weeks of neoadjuvant therapy. Thirteen of the 15 had a pathologic complete response. Encouragingly, no one with a pathologic complete or near-complete response has relapsed to date.
“A pathologic complete response or near-complete response with neoadjuvant therapy appears to be a biomarker of durable disease control and is associated with excellent outcomes,” Dr. Kirkwood observed, adding that the Pittsburgh experience has been mirrored in reports from the Netherlands, Australia, and University of Texas M.D. Anderson Cancer Center, Houston, involving other neoadjuvant agents.
Other potential early biomarkers of favorable outcome with neoadjuvant therapy include CD8+ T cells in the tumor at baseline, tumor mutational burden, T-cell clonality, and a T-cell–inflamed gene-expression profile.
There were no dose-limiting toxicities or delays in surgery related to the neoadjuvant treatment.
Of note, imaging often inaccurately showed only a partial response in patients who actually had a pathologic complete response, meaning totally devoid of tumor, Dr. Kirkwood said.
Corroboration of these findings is planned in the national multicenter ECOG-ACRIN neoadjuvant trial EA6194.
“Consider referring to this trial any patients who present with bulky nodal disease for whom a treatment assessment at 4-6 weeks is desired in order to predict what the outcome may be,” he suggested.
Dr. Kirkwood reported receiving research grants from Amgen, BMS, Castle Biosciences, Checkmate, Immunocore, Iovance, and Novartis and serving as a consultant to a handful of companies.
Global Academy for Medical Education and this news organization are owned by the same company.
The next dramatic , John M. Kirkwood, MD, predicted at a virtual forum on cutaneous malignancies jointly presented by the Postgraduate Institute for Medicine and Global Academy for Medical Education.
These agents have already demonstrated profound efficacy, first in stage IV metastatic disease and more recently as adjuvant therapy for resected stage III melanoma. Now, there is a great interest in learning whether by prescribing them preoperatively, patients might reduce their risk of advancing to metastatic disease. And neoadjuvant therapy offers an extremely attractive feature: It yields results in an accelerated fashion.
“The major problem with postoperative adjuvant trials in melanoma since 1984 is the long time to maturity. Many of us don’t want to wait the full 9 or 10 years for a full-bore, phase 3 adjuvant trial in stage III melanoma to mature,” explained Dr. Kirkwood, professor of medicine, dermatology, and translational science and coleader of the melanoma and skin cancer program at the University of Pittsburgh. “The opportunity to treat a patient who presents with a bulky lymph node, has a biopsy, and then can be treated for 3 or 6 weeks or sometimes even longer periods with a therapy that’s promising allows us to ask what’s going on in the tumor tissue, what’s going on in the clinical response at 3 or 6 weeks, and if there’s pathological complete or near-complete response under the microscope.”
Because pathological complete response is a strong predictor of relapse-free survival, this neoadjuvant-forward therapeutic strategy has the potential to provide patients and their physicians with an early forecast of likely clinical outcome only 4-6 weeks into treatment. Also, there is both preclinical and clinical evidence that neoadjuvant therapy may offer a survival advantage over adjuvant therapy, perhaps as a result of early treatment of micrometastatic disease. Another benefit of neoadjuvant therapy for melanoma is the resultant tumor shrinkage, which can permit less extensive surgery.
Dr. Kirkwood highlighted a phase 2 clinical trial conducted at the University of Pittsburgh to illustrate the potential of neoadjuvant therapy in melanoma. The ongoing single-arm study includes 32 patients with stage IIIB or IIIC resectable melanoma along with accessible tumor for biopsy and intratumoral injections of CMP-001, a toll-like receptor 9 agonist. According to the Eighth Edition of the American Joint Committee on Cancer staging manual, stage IIIB melanoma has a 10-year mortality of 23%, and stage IIIC disease has 40%.
CMP-001 triggers type 1 interferon production through activation of plasmacytoid dendritic cells. The resultant inflammatory response draws T cells into the tumor to enhance the response to immunotherapy, which in this study was nivolumab (Opdivo), a human programmed death ligand 1 (PD-L1)–blocking antibody. The neoadjuvant regimen consisted of seven once-weekly intratumoral injections of CMP-001, plus three 240-mg doses of nivolumab given at 2-week intervals. This was followed by resection, then 1 year of adjuvant therapy with nivolumab at 480 mg every 4 weeks and intratumoral CMP-001 every 4 weeks.
In an interim analysis, a major pathologic response occurred in an impressive 15 of 21 patients (71%) after 6 weeks of neoadjuvant therapy. Thirteen of the 15 had a pathologic complete response. Encouragingly, no one with a pathologic complete or near-complete response has relapsed to date.
“A pathologic complete response or near-complete response with neoadjuvant therapy appears to be a biomarker of durable disease control and is associated with excellent outcomes,” Dr. Kirkwood observed, adding that the Pittsburgh experience has been mirrored in reports from the Netherlands, Australia, and University of Texas M.D. Anderson Cancer Center, Houston, involving other neoadjuvant agents.
Other potential early biomarkers of favorable outcome with neoadjuvant therapy include CD8+ T cells in the tumor at baseline, tumor mutational burden, T-cell clonality, and a T-cell–inflamed gene-expression profile.
There were no dose-limiting toxicities or delays in surgery related to the neoadjuvant treatment.
Of note, imaging often inaccurately showed only a partial response in patients who actually had a pathologic complete response, meaning totally devoid of tumor, Dr. Kirkwood said.
Corroboration of these findings is planned in the national multicenter ECOG-ACRIN neoadjuvant trial EA6194.
“Consider referring to this trial any patients who present with bulky nodal disease for whom a treatment assessment at 4-6 weeks is desired in order to predict what the outcome may be,” he suggested.
Dr. Kirkwood reported receiving research grants from Amgen, BMS, Castle Biosciences, Checkmate, Immunocore, Iovance, and Novartis and serving as a consultant to a handful of companies.
Global Academy for Medical Education and this news organization are owned by the same company.
FROM THE CUTANEOUS MALIGNANCIES FORUM
COVID-19: Peginterferon lambda may prevent clinical deterioration, shorten viral shedding
and shorten the duration of viral shedding, according to results of a double-blind, placebo-controlled trial (NCT04354259).
Reductions in severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) RNA were greater with peginterferon lambda than with placebo from day 3 onward in the phase 2 study led by Jordan J. Feld, MD, of the Toronto Centre for Liver Disease. The findings were reported in The Lancet Respiratory Medicine.
Fewer side effects
To date in randomized clinical trials, efficacy in treatment of COVID-19 has been shown only for remdesivir and dexamethasone in hospitalized patients, and in an interim analysis of accelerated viral clearance for a monoclonal antibody infusion in outpatients.
Activity against respiratory pathogens has been demonstrated for interferon lambda-1, a type III interferon shown to be involved in innate antiviral responses. Interferons, Dr. Feld and coauthors stated, drive induction of genes with antiviral, antiproliferative and immunoregulatory properties, and early treatment with interferons might halt clinical progression and shorten the duration of viral shedding with reduced onward transmission. In addition, interferon lambdas (type III) use a distinct receptor complex with high expression levels limited to epithelial cells in the lung, liver, and intestine, leading to fewer side effects than other interferons, including avoiding risk of promoting cytokine storm syndrome.
The researchers investigated peginterferon lambda safety and efficacy in treatment of patients with laboratory-confirmed, mild to moderate COVID-19. Sixty patients (median age 46 years, about 60% female, about 50% White) were recruited from outpatient testing centers at six institutions in Toronto, and referred to a single ambulatory site. Patients were randomly assigned 1:1 to a single subcutaneous injection of peginterferon lambda 180 mcg or placebo within 7 days of symptom onset or, if asymptomatic, of their first positive swab. Mean time from symptom onset to injection was about 4.5 days, and about 18.5% were asymptomatic. The primary outcome was the proportion of patients negative for SARS-CoV-2 RNA on day 7 after the injection.
Greater benefit with higher baseline load
A higher baseline SARS-CoV-2 RNA concentration found in the peginterferon lambda group was found to be significantly associated with day 7 clearance (odds ratio [OR] 0.69 [95% confidence interval 0.51-0.87]; P = ·001). In the peginterferon lambda group, also, the mean decline in SARS-CoV-2 RNA was significantly larger than in the placebo group across all time points (days 3, 5, 7, and14). While viral load decline was 0.81 log greater in the treatment group (P = .14) by day 3, viral load decline increased to 1.67 log copies per mL by day 5 (P = .013) and 2.42 log copies per mL by day 7 (P = .0041). At day 14, the viral decline was 1.77 log copies per mL larger in the peginterferon lambda group (P = .048). The investigators pointed out that the difference in viral load decline between groups was greater in patients with high baseline viral load (at or above 106 copies per mL). In the peginterferon lambda high baseline viral load group, the reduction was 7.17 log copies per mL, versus 4.92 log copies per mL in the placebo group (P = .004).
More patients SARS-CoV-2 RNA negative
By day 7, 80% of patients in the peginterferon lambda group were negative for SARS-CoV-2 RNA, compared with 63% in the placebo group (P = .15). After baseline load adjustment, however, the peginterferon lambda treatment was significantly associated with day 7 clearance (OR 4·12 [95% CI 1·15-16·73]; P = .029).
Respiratory symptoms improved faster
Most symptoms in both groups were mild to moderate, without difference in frequency or severity. While symptom improvement was generally similar over time for both groups, respiratory symptoms improved faster with peginterferon lambda, with the effect more pronounced in the high baseline viral load group (OR 5·88 (0·81-42·46; P =. 079).
Laboratory adverse events, similar for both groups, were mild.
“Peginterferon lambda has potential to prevent clinical deterioration and shorten duration of viral shedding,” the investigators concluded.
“This clinical trial is important” because it suggests that a single intravenous dose of peginterferon lambda administered to outpatients with a positive SARS-CoV-2 nasopharyngeal swab speeds reduction of SARS-CoV-2 viral load, David L. Bowton, MD, FCCP, professor emeritus, Wake Forest Baptist Health, Winston-Salem, N.C., said in an interview. He observed that the smaller viral load difference observed at day 14 likely reflects host immune responses.

Dr. Bowton also noted that two placebo group baseline characteristics (five placebo group patients with anti-SARS-CoV-2 S protein IgG antibodies; two times more undetectable SARS-CoV-2 RNA at baseline assessment) would tend to reduce differences between the peginterferon lambda and placebo groups. He added that the study findings were concordant with another phase 2 trial of hospitalized COVID-19 patients receiving inhaled interferon beta-1a.
“Thus, interferons may find a place in the treatment of COVID-19 and perhaps other severe viral illnesses,” Dr. Bowton said.
The study was funded by the Toronto COVID-19 Action Initiative, University of Toronto, and the Ontario First COVID-19 Rapid Research Fund, Toronto General & Western Hospital Foundation.
Dr. Bowton had no disclosures. Disclosures for Dr. Feld and coauthors are listed on the Lancet Respiratory Medicine website.
and shorten the duration of viral shedding, according to results of a double-blind, placebo-controlled trial (NCT04354259).
Reductions in severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) RNA were greater with peginterferon lambda than with placebo from day 3 onward in the phase 2 study led by Jordan J. Feld, MD, of the Toronto Centre for Liver Disease. The findings were reported in The Lancet Respiratory Medicine.
Fewer side effects
To date in randomized clinical trials, efficacy in treatment of COVID-19 has been shown only for remdesivir and dexamethasone in hospitalized patients, and in an interim analysis of accelerated viral clearance for a monoclonal antibody infusion in outpatients.
Activity against respiratory pathogens has been demonstrated for interferon lambda-1, a type III interferon shown to be involved in innate antiviral responses. Interferons, Dr. Feld and coauthors stated, drive induction of genes with antiviral, antiproliferative and immunoregulatory properties, and early treatment with interferons might halt clinical progression and shorten the duration of viral shedding with reduced onward transmission. In addition, interferon lambdas (type III) use a distinct receptor complex with high expression levels limited to epithelial cells in the lung, liver, and intestine, leading to fewer side effects than other interferons, including avoiding risk of promoting cytokine storm syndrome.
The researchers investigated peginterferon lambda safety and efficacy in treatment of patients with laboratory-confirmed, mild to moderate COVID-19. Sixty patients (median age 46 years, about 60% female, about 50% White) were recruited from outpatient testing centers at six institutions in Toronto, and referred to a single ambulatory site. Patients were randomly assigned 1:1 to a single subcutaneous injection of peginterferon lambda 180 mcg or placebo within 7 days of symptom onset or, if asymptomatic, of their first positive swab. Mean time from symptom onset to injection was about 4.5 days, and about 18.5% were asymptomatic. The primary outcome was the proportion of patients negative for SARS-CoV-2 RNA on day 7 after the injection.
Greater benefit with higher baseline load
A higher baseline SARS-CoV-2 RNA concentration found in the peginterferon lambda group was found to be significantly associated with day 7 clearance (odds ratio [OR] 0.69 [95% confidence interval 0.51-0.87]; P = ·001). In the peginterferon lambda group, also, the mean decline in SARS-CoV-2 RNA was significantly larger than in the placebo group across all time points (days 3, 5, 7, and14). While viral load decline was 0.81 log greater in the treatment group (P = .14) by day 3, viral load decline increased to 1.67 log copies per mL by day 5 (P = .013) and 2.42 log copies per mL by day 7 (P = .0041). At day 14, the viral decline was 1.77 log copies per mL larger in the peginterferon lambda group (P = .048). The investigators pointed out that the difference in viral load decline between groups was greater in patients with high baseline viral load (at or above 106 copies per mL). In the peginterferon lambda high baseline viral load group, the reduction was 7.17 log copies per mL, versus 4.92 log copies per mL in the placebo group (P = .004).
More patients SARS-CoV-2 RNA negative
By day 7, 80% of patients in the peginterferon lambda group were negative for SARS-CoV-2 RNA, compared with 63% in the placebo group (P = .15). After baseline load adjustment, however, the peginterferon lambda treatment was significantly associated with day 7 clearance (OR 4·12 [95% CI 1·15-16·73]; P = .029).
Respiratory symptoms improved faster
Most symptoms in both groups were mild to moderate, without difference in frequency or severity. While symptom improvement was generally similar over time for both groups, respiratory symptoms improved faster with peginterferon lambda, with the effect more pronounced in the high baseline viral load group (OR 5·88 (0·81-42·46; P =. 079).
Laboratory adverse events, similar for both groups, were mild.
“Peginterferon lambda has potential to prevent clinical deterioration and shorten duration of viral shedding,” the investigators concluded.
“This clinical trial is important” because it suggests that a single intravenous dose of peginterferon lambda administered to outpatients with a positive SARS-CoV-2 nasopharyngeal swab speeds reduction of SARS-CoV-2 viral load, David L. Bowton, MD, FCCP, professor emeritus, Wake Forest Baptist Health, Winston-Salem, N.C., said in an interview. He observed that the smaller viral load difference observed at day 14 likely reflects host immune responses.
Dr. Bowton also noted that two placebo group baseline characteristics (five placebo group patients with anti-SARS-CoV-2 S protein IgG antibodies; two times more undetectable SARS-CoV-2 RNA at baseline assessment) would tend to reduce differences between the peginterferon lambda and placebo groups. He added that the study findings were concordant with another phase 2 trial of hospitalized COVID-19 patients receiving inhaled interferon beta-1a.
“Thus, interferons may find a place in the treatment of COVID-19 and perhaps other severe viral illnesses,” Dr. Bowton said.
The study was funded by the Toronto COVID-19 Action Initiative, University of Toronto, and the Ontario First COVID-19 Rapid Research Fund, Toronto General & Western Hospital Foundation.
Dr. Bowton had no disclosures. Disclosures for Dr. Feld and coauthors are listed on the Lancet Respiratory Medicine website.
and shorten the duration of viral shedding, according to results of a double-blind, placebo-controlled trial (NCT04354259).
Reductions in severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) RNA were greater with peginterferon lambda than with placebo from day 3 onward in the phase 2 study led by Jordan J. Feld, MD, of the Toronto Centre for Liver Disease. The findings were reported in The Lancet Respiratory Medicine.
Fewer side effects
To date in randomized clinical trials, efficacy in treatment of COVID-19 has been shown only for remdesivir and dexamethasone in hospitalized patients, and in an interim analysis of accelerated viral clearance for a monoclonal antibody infusion in outpatients.
Activity against respiratory pathogens has been demonstrated for interferon lambda-1, a type III interferon shown to be involved in innate antiviral responses. Interferons, Dr. Feld and coauthors stated, drive induction of genes with antiviral, antiproliferative and immunoregulatory properties, and early treatment with interferons might halt clinical progression and shorten the duration of viral shedding with reduced onward transmission. In addition, interferon lambdas (type III) use a distinct receptor complex with high expression levels limited to epithelial cells in the lung, liver, and intestine, leading to fewer side effects than other interferons, including avoiding risk of promoting cytokine storm syndrome.
The researchers investigated peginterferon lambda safety and efficacy in treatment of patients with laboratory-confirmed, mild to moderate COVID-19. Sixty patients (median age 46 years, about 60% female, about 50% White) were recruited from outpatient testing centers at six institutions in Toronto, and referred to a single ambulatory site. Patients were randomly assigned 1:1 to a single subcutaneous injection of peginterferon lambda 180 mcg or placebo within 7 days of symptom onset or, if asymptomatic, of their first positive swab. Mean time from symptom onset to injection was about 4.5 days, and about 18.5% were asymptomatic. The primary outcome was the proportion of patients negative for SARS-CoV-2 RNA on day 7 after the injection.
Greater benefit with higher baseline load
A higher baseline SARS-CoV-2 RNA concentration found in the peginterferon lambda group was found to be significantly associated with day 7 clearance (odds ratio [OR] 0.69 [95% confidence interval 0.51-0.87]; P = ·001). In the peginterferon lambda group, also, the mean decline in SARS-CoV-2 RNA was significantly larger than in the placebo group across all time points (days 3, 5, 7, and14). While viral load decline was 0.81 log greater in the treatment group (P = .14) by day 3, viral load decline increased to 1.67 log copies per mL by day 5 (P = .013) and 2.42 log copies per mL by day 7 (P = .0041). At day 14, the viral decline was 1.77 log copies per mL larger in the peginterferon lambda group (P = .048). The investigators pointed out that the difference in viral load decline between groups was greater in patients with high baseline viral load (at or above 106 copies per mL). In the peginterferon lambda high baseline viral load group, the reduction was 7.17 log copies per mL, versus 4.92 log copies per mL in the placebo group (P = .004).
More patients SARS-CoV-2 RNA negative
By day 7, 80% of patients in the peginterferon lambda group were negative for SARS-CoV-2 RNA, compared with 63% in the placebo group (P = .15). After baseline load adjustment, however, the peginterferon lambda treatment was significantly associated with day 7 clearance (OR 4·12 [95% CI 1·15-16·73]; P = .029).
Respiratory symptoms improved faster
Most symptoms in both groups were mild to moderate, without difference in frequency or severity. While symptom improvement was generally similar over time for both groups, respiratory symptoms improved faster with peginterferon lambda, with the effect more pronounced in the high baseline viral load group (OR 5·88 (0·81-42·46; P =. 079).
Laboratory adverse events, similar for both groups, were mild.
“Peginterferon lambda has potential to prevent clinical deterioration and shorten duration of viral shedding,” the investigators concluded.
“This clinical trial is important” because it suggests that a single intravenous dose of peginterferon lambda administered to outpatients with a positive SARS-CoV-2 nasopharyngeal swab speeds reduction of SARS-CoV-2 viral load, David L. Bowton, MD, FCCP, professor emeritus, Wake Forest Baptist Health, Winston-Salem, N.C., said in an interview. He observed that the smaller viral load difference observed at day 14 likely reflects host immune responses.
Dr. Bowton also noted that two placebo group baseline characteristics (five placebo group patients with anti-SARS-CoV-2 S protein IgG antibodies; two times more undetectable SARS-CoV-2 RNA at baseline assessment) would tend to reduce differences between the peginterferon lambda and placebo groups. He added that the study findings were concordant with another phase 2 trial of hospitalized COVID-19 patients receiving inhaled interferon beta-1a.
“Thus, interferons may find a place in the treatment of COVID-19 and perhaps other severe viral illnesses,” Dr. Bowton said.
The study was funded by the Toronto COVID-19 Action Initiative, University of Toronto, and the Ontario First COVID-19 Rapid Research Fund, Toronto General & Western Hospital Foundation.
Dr. Bowton had no disclosures. Disclosures for Dr. Feld and coauthors are listed on the Lancet Respiratory Medicine website.
FROM THE LANCET RESPIRATORY MEDICINE
Inhaled hyaluronan may bring sigh of relief to COPD patients
(COPD), findings of a new study suggest.

HMW-HA was associated with a significantly shorter duration of noninvasive positive-pressure ventilation (NIPPV), lower systemic inflammatory markers, and lower measured peak airway pressure, compared with placebo, reported lead author Flavia Galdi, MD, of Campus Bio-Medico University Hospital, Rome, and colleagues.
“HMW-HA is a naturally occurring sugar that is abundant in the extracellular matrix, including in the lung,” the investigators wrote in Respiratory Research. “[It] has been used routinely, together with hypertonic saline, in cystic fibrosis patients [for several years] with no reported side effects; rather, it improves tolerability and decreases the need for bronchodilators in these patients.”
According to Robert A. Sandhaus, MD, PhD, FCCP, of National Jewish Health, Denver, the role of hyaluronan in lung disease was first recognized decades ago.

“Data stretching back into the 1970s has identified decreases in hyaluronan content in emphysematous lung tissue, protection of lung connective tissue from proteolysis by hyaluronan, and potential therapeutic roles for hyaluronan in a variety of disease, especially of the lungs,” he said in an interview.
For patients with COPD, treatment with HMW-HA may provide benefit by counteracting an imbalance in diseased lung tissue, wrote Dr. Galdi and colleagues.
“Emerging evidence suggests that imbalance between declining HMW-HA levels, and increasing smaller fragments of hyaluronan may contribute to chronic airway disease pathogenesis,” they wrote. “This has led to the hypothesis that exogenous supplementation of HMW-HA may restore hyaluronan homeostasis in favor of undegraded molecules, inhibit inflammation and loss of lung function, and ameliorate COPD progression.”
To test this hypothesis, the investigators screened 44 patients with a history of acute exacerbations of COPD necessitating NIPPV, ultimately excluding 3 patients because of heart failure. Following 1:1 randomization, 20 patients received HMW-HA while 21 received placebo, each twice daily, in conjunction with NIPPV and standard medical therapy. Treatment continued until NIPPV failure or liberation from NIPPV. Most patients received NIPPV in the hospital; however, home/chronic NIPPV was given to four patients in the placebo group and three patients in the HMW-HA group.
The primary outcome was duration of NIPPV. Secondary outcomes included markers of systemic inflammation associated with acute exacerbations of COPD and respiratory physiology parameters. Adverse events were also reported.
Results showed that patients treated with HMW-HA were liberated sooner from NIPPV than were those who received placebo (mean, 5.2 vs 6.4 days; P < .037). Similarly, patients in the HMW-HA group had significantly shorter hospital stay, on average, than those in the placebo group (mean, 7.2 vs 10.2 days; P = .039). Median values followed a similar pattern.
“These data suggest that HMW-HA shortened the duration of acute respiratory failure, need for NIPPV and, consequently, hospital length of stay in these patients,” the investigators wrote.
Secondary outcomes further supported these therapeutic benefits. Compared with placebo, HMW-HA was associated with significantly lower peak pressure and greater improvements in both pCO2/FiO2 ratio and inflammatory markers. No adverse events were reported.
Further analyses involving human bronchial epithelial cell cultures offered some mechanistic insight. Using micro-optical coherence tomography imaging, the investigators found that HMW-HA treatment was associated with “a prominent effect on mucociliary transport” in cell cultures derived from COPD patients and in healthy nonsmoker cell cultures exposed to cigarette smoke extract.
“Our study shows for the first time the therapeutic potential of an extracellular matrix molecule in acute exacerbation of human lung disease,” the investigators concluded, noting a “clinically meaningful salutary effect” on duration of NIPPV.
Dr. Galdi and colleagues went on to predict that benefits in a real-world patient population could be even more meaningful.
“Since the serum samples were collected at the end of NIPPV, HMW-HA–treated patients were on average sampled a day earlier than placebo-treated patients (because they were liberated from NIPPV a day earlier on average),” the investigators wrote. “Thus, HMW-HA treatment effects may have been underestimated in our study.”
According to Dr. Sandhaus, “The current report, while a relatively small single-center study, is well controlled and the results suggest that inhaled hyaluronan decreased time on noninvasive ventilation, decreased hospital stay duration, and decreased some mediators of inflammation.”
He also suggested that HMW-HA may have a role in the prophylactic setting.
“The limitations of this pilot study are appropriately explored by the authors but do not dampen the exciting possibility that this therapeutic approach may hold promise not only in severe exacerbations of COPD but potentially for the prevention of such exacerbations,” Dr. Sandhaus said.

Jerome O. Cantor, MD, FCCP, of St. John’s University, New York, who previously conducted a pilot study for using lower molecular weight hyaluronan in COPD and published a review on the subject, said that more studies are necessary.
“Further clinical trials are needed to better determine the role of hyaluronan as an adjunct to existing therapies for COPD exacerbations,” he said.
The study was supported by the National Institutes of Health. The investigators and Dr. Sandhaus declared no conflicts of interest. Dr. Cantor disclosed a relationship with MatRx Therapeutics.
(COPD), findings of a new study suggest.
HMW-HA was associated with a significantly shorter duration of noninvasive positive-pressure ventilation (NIPPV), lower systemic inflammatory markers, and lower measured peak airway pressure, compared with placebo, reported lead author Flavia Galdi, MD, of Campus Bio-Medico University Hospital, Rome, and colleagues.
“HMW-HA is a naturally occurring sugar that is abundant in the extracellular matrix, including in the lung,” the investigators wrote in Respiratory Research. “[It] has been used routinely, together with hypertonic saline, in cystic fibrosis patients [for several years] with no reported side effects; rather, it improves tolerability and decreases the need for bronchodilators in these patients.”
According to Robert A. Sandhaus, MD, PhD, FCCP, of National Jewish Health, Denver, the role of hyaluronan in lung disease was first recognized decades ago.
“Data stretching back into the 1970s has identified decreases in hyaluronan content in emphysematous lung tissue, protection of lung connective tissue from proteolysis by hyaluronan, and potential therapeutic roles for hyaluronan in a variety of disease, especially of the lungs,” he said in an interview.
For patients with COPD, treatment with HMW-HA may provide benefit by counteracting an imbalance in diseased lung tissue, wrote Dr. Galdi and colleagues.
“Emerging evidence suggests that imbalance between declining HMW-HA levels, and increasing smaller fragments of hyaluronan may contribute to chronic airway disease pathogenesis,” they wrote. “This has led to the hypothesis that exogenous supplementation of HMW-HA may restore hyaluronan homeostasis in favor of undegraded molecules, inhibit inflammation and loss of lung function, and ameliorate COPD progression.”
To test this hypothesis, the investigators screened 44 patients with a history of acute exacerbations of COPD necessitating NIPPV, ultimately excluding 3 patients because of heart failure. Following 1:1 randomization, 20 patients received HMW-HA while 21 received placebo, each twice daily, in conjunction with NIPPV and standard medical therapy. Treatment continued until NIPPV failure or liberation from NIPPV. Most patients received NIPPV in the hospital; however, home/chronic NIPPV was given to four patients in the placebo group and three patients in the HMW-HA group.
The primary outcome was duration of NIPPV. Secondary outcomes included markers of systemic inflammation associated with acute exacerbations of COPD and respiratory physiology parameters. Adverse events were also reported.
Results showed that patients treated with HMW-HA were liberated sooner from NIPPV than were those who received placebo (mean, 5.2 vs 6.4 days; P < .037). Similarly, patients in the HMW-HA group had significantly shorter hospital stay, on average, than those in the placebo group (mean, 7.2 vs 10.2 days; P = .039). Median values followed a similar pattern.
“These data suggest that HMW-HA shortened the duration of acute respiratory failure, need for NIPPV and, consequently, hospital length of stay in these patients,” the investigators wrote.
Secondary outcomes further supported these therapeutic benefits. Compared with placebo, HMW-HA was associated with significantly lower peak pressure and greater improvements in both pCO2/FiO2 ratio and inflammatory markers. No adverse events were reported.
Further analyses involving human bronchial epithelial cell cultures offered some mechanistic insight. Using micro-optical coherence tomography imaging, the investigators found that HMW-HA treatment was associated with “a prominent effect on mucociliary transport” in cell cultures derived from COPD patients and in healthy nonsmoker cell cultures exposed to cigarette smoke extract.
“Our study shows for the first time the therapeutic potential of an extracellular matrix molecule in acute exacerbation of human lung disease,” the investigators concluded, noting a “clinically meaningful salutary effect” on duration of NIPPV.
Dr. Galdi and colleagues went on to predict that benefits in a real-world patient population could be even more meaningful.
“Since the serum samples were collected at the end of NIPPV, HMW-HA–treated patients were on average sampled a day earlier than placebo-treated patients (because they were liberated from NIPPV a day earlier on average),” the investigators wrote. “Thus, HMW-HA treatment effects may have been underestimated in our study.”
According to Dr. Sandhaus, “The current report, while a relatively small single-center study, is well controlled and the results suggest that inhaled hyaluronan decreased time on noninvasive ventilation, decreased hospital stay duration, and decreased some mediators of inflammation.”
He also suggested that HMW-HA may have a role in the prophylactic setting.
“The limitations of this pilot study are appropriately explored by the authors but do not dampen the exciting possibility that this therapeutic approach may hold promise not only in severe exacerbations of COPD but potentially for the prevention of such exacerbations,” Dr. Sandhaus said.
Jerome O. Cantor, MD, FCCP, of St. John’s University, New York, who previously conducted a pilot study for using lower molecular weight hyaluronan in COPD and published a review on the subject, said that more studies are necessary.
“Further clinical trials are needed to better determine the role of hyaluronan as an adjunct to existing therapies for COPD exacerbations,” he said.
The study was supported by the National Institutes of Health. The investigators and Dr. Sandhaus declared no conflicts of interest. Dr. Cantor disclosed a relationship with MatRx Therapeutics.
(COPD), findings of a new study suggest.
HMW-HA was associated with a significantly shorter duration of noninvasive positive-pressure ventilation (NIPPV), lower systemic inflammatory markers, and lower measured peak airway pressure, compared with placebo, reported lead author Flavia Galdi, MD, of Campus Bio-Medico University Hospital, Rome, and colleagues.
“HMW-HA is a naturally occurring sugar that is abundant in the extracellular matrix, including in the lung,” the investigators wrote in Respiratory Research. “[It] has been used routinely, together with hypertonic saline, in cystic fibrosis patients [for several years] with no reported side effects; rather, it improves tolerability and decreases the need for bronchodilators in these patients.”
According to Robert A. Sandhaus, MD, PhD, FCCP, of National Jewish Health, Denver, the role of hyaluronan in lung disease was first recognized decades ago.
“Data stretching back into the 1970s has identified decreases in hyaluronan content in emphysematous lung tissue, protection of lung connective tissue from proteolysis by hyaluronan, and potential therapeutic roles for hyaluronan in a variety of disease, especially of the lungs,” he said in an interview.
For patients with COPD, treatment with HMW-HA may provide benefit by counteracting an imbalance in diseased lung tissue, wrote Dr. Galdi and colleagues.
“Emerging evidence suggests that imbalance between declining HMW-HA levels, and increasing smaller fragments of hyaluronan may contribute to chronic airway disease pathogenesis,” they wrote. “This has led to the hypothesis that exogenous supplementation of HMW-HA may restore hyaluronan homeostasis in favor of undegraded molecules, inhibit inflammation and loss of lung function, and ameliorate COPD progression.”
To test this hypothesis, the investigators screened 44 patients with a history of acute exacerbations of COPD necessitating NIPPV, ultimately excluding 3 patients because of heart failure. Following 1:1 randomization, 20 patients received HMW-HA while 21 received placebo, each twice daily, in conjunction with NIPPV and standard medical therapy. Treatment continued until NIPPV failure or liberation from NIPPV. Most patients received NIPPV in the hospital; however, home/chronic NIPPV was given to four patients in the placebo group and three patients in the HMW-HA group.
The primary outcome was duration of NIPPV. Secondary outcomes included markers of systemic inflammation associated with acute exacerbations of COPD and respiratory physiology parameters. Adverse events were also reported.
Results showed that patients treated with HMW-HA were liberated sooner from NIPPV than were those who received placebo (mean, 5.2 vs 6.4 days; P < .037). Similarly, patients in the HMW-HA group had significantly shorter hospital stay, on average, than those in the placebo group (mean, 7.2 vs 10.2 days; P = .039). Median values followed a similar pattern.
“These data suggest that HMW-HA shortened the duration of acute respiratory failure, need for NIPPV and, consequently, hospital length of stay in these patients,” the investigators wrote.
Secondary outcomes further supported these therapeutic benefits. Compared with placebo, HMW-HA was associated with significantly lower peak pressure and greater improvements in both pCO2/FiO2 ratio and inflammatory markers. No adverse events were reported.
Further analyses involving human bronchial epithelial cell cultures offered some mechanistic insight. Using micro-optical coherence tomography imaging, the investigators found that HMW-HA treatment was associated with “a prominent effect on mucociliary transport” in cell cultures derived from COPD patients and in healthy nonsmoker cell cultures exposed to cigarette smoke extract.
“Our study shows for the first time the therapeutic potential of an extracellular matrix molecule in acute exacerbation of human lung disease,” the investigators concluded, noting a “clinically meaningful salutary effect” on duration of NIPPV.
Dr. Galdi and colleagues went on to predict that benefits in a real-world patient population could be even more meaningful.
“Since the serum samples were collected at the end of NIPPV, HMW-HA–treated patients were on average sampled a day earlier than placebo-treated patients (because they were liberated from NIPPV a day earlier on average),” the investigators wrote. “Thus, HMW-HA treatment effects may have been underestimated in our study.”
According to Dr. Sandhaus, “The current report, while a relatively small single-center study, is well controlled and the results suggest that inhaled hyaluronan decreased time on noninvasive ventilation, decreased hospital stay duration, and decreased some mediators of inflammation.”
He also suggested that HMW-HA may have a role in the prophylactic setting.
“The limitations of this pilot study are appropriately explored by the authors but do not dampen the exciting possibility that this therapeutic approach may hold promise not only in severe exacerbations of COPD but potentially for the prevention of such exacerbations,” Dr. Sandhaus said.
Jerome O. Cantor, MD, FCCP, of St. John’s University, New York, who previously conducted a pilot study for using lower molecular weight hyaluronan in COPD and published a review on the subject, said that more studies are necessary.
“Further clinical trials are needed to better determine the role of hyaluronan as an adjunct to existing therapies for COPD exacerbations,” he said.
The study was supported by the National Institutes of Health. The investigators and Dr. Sandhaus declared no conflicts of interest. Dr. Cantor disclosed a relationship with MatRx Therapeutics.
FROM RESPIRATORY RESEARCH
Women increasingly turn to CBD, with or without doc’s blessing
When 42-year-old Danielle Simone Brand started having hormonal migraines, she first turned to cannabidiol (CBD) oil, eventually adding an occasional pull on a prefilled tetrahydrocannabinol (THC) vape for nighttime use. She was careful to avoid THC during work hours. A parenting and cannabis writer, Ms. Brand had more than a cursory background in cannabinoid medicine and had spent time at her local California dispensary discussing various cannabinoid components that might help alleviate her pain.

A self-professed “do-it-yourselfer,” Ms. Brand continues to use cannabinoids for her monthly headaches, forgoing any other pain medication. “There are times for conventional medicine in partnership with your doctor, but when it comes to health and wellness, women should be empowered to make decisions and self-experiment,” she said in an interview.
Ms. Brand is not alone. Significant numbers of women are replacing or supplementing prescription medications with cannabinoids, often without consulting their primary care physician, ob.gyn., or other specialist. At times, women have tried to have these conversations, only to be met with silence or worse.
Take Linda Fuller, a 58-year-old yoga instructor from Long Island who says that she uses CBD and THC for chronic sacroiliac pain after a car accident and to alleviate stress-triggered eczema flares. “I’ve had doctors turn their backs on me; I’ve had nurse practitioners walk out on me in the middle of a sentence,” she said in an interview.
Ms. Fuller said her conversion to cannabinoid medicine is relatively new; she never used cannabis recreationally before her accident but now considers it a gift. She doesn’t keep aspirin in the house and refused pain medication immediately after she injured her back.
Diana Krach, a 34-year-old writer from Maryland, says she’s encountered roadblocks about her decision to use cannabinoids for endometriosis and for pain from Crohn’s disease. When she tried to discuss her CBD use with a gastroenterologist, he interrupted her: “Whatever pot you’re smoking isn’t going to work, you’re going on biologics.”
Ms. Krach had not been smoking anything but had turned to a CBD tincture for symptom relief after prescription pain medications failed to help.
Ms. Brand, Ms. Fuller, and Ms. Krach are the tip of the iceberg when it comes to women seeking symptom relief outside the medicine cabinet. A recent survey in the Journal of Women’s Health of almost 1,000 women show that 90% (most between the ages of 35 and 44) had used cannabis and would consider using it to treat gynecologic pain. Roughly 80% said they would consider using it for procedure-related pain or other conditions. Additionally, women have reported using cannabinoids for PTSD, sleep disturbances or insomnia, anxiety, and migraine headaches.
Observational survey data have likewise shown that 80% of women with advanced or recurrent gynecologic malignancies who were prescribed cannabis reported that it was equivalent or superior to other medications for relieving pain, neuropathy, nausea, insomnia, decreased appetite, and anxiety.
In another survey, almost half (45%) of women with gynecologic malignancies who used nonprescribed cannabis for the same symptoms reported that they had reduced their use of prescription narcotics after initiating use of cannabis.
The gray zone
There has been a surge in self-reported cannabis use among pregnant women in particular. The National Survey on Drug Use and Health findings for the periods 2002-2003 and 2016-2017 highlight increases in adjusted prevalence rates from 3.4% to 7% in past-month use among pregnant women overall and from 5.7% to 12.1% during the first trimester alone.
“The more that you talk to pregnant women, the more that you realize that a lot are using cannabinoids for something that is basically medicinal, for sleep, for anxiety, or for nausea,” Katrina Mark, MD, an ob.gyn. and associate professor of medicine at the University of Maryland, College Park, said in an interview. “I’m not saying it’s fine to use drugs in pregnancy, but it is a grayer conversation than a lot of colleagues want to believe. Telling women to quit seems foolish since the alternative is to be anxious, don’t sleep, don’t eat, or use a medication that also has risks to it.”
One observational study shows that pregnant women themselves are conflicted. Although the majority believe that cannabis is “natural” and “safe,” compared with prescription drugs, they aren’t entirely in the dark about potential risks. They often express frustration with practitioners’ responses when these topics are broached during office visits. An observational survey among women and practitioners published in 2020 highlights that only half of doctors openly discouraged perinatal cannabis use and that others opted out of the discussion entirely.
This is the experience of many of the women that this news organization spoke with. Ms. Krach pointed out that “there’s a big deficit in listening; the doctor is supposed to be working for our behalf, especially when it comes to reproductive health.”
Dr. Mark believed that a lot of the conversation has been clouded by the illegality of the substance but that cannabinoids deserve as much of a fair chance for discussion and consideration as other medicines, which also carry risks in pregnancy. “There’s literally no evidence that it will work in pregnancy [for these symptoms], but there’s no evidence that it doesn’t, either,” she said in an interview. “When I have this conversation with colleagues who do not share my views, I try to encourage them to look at the actual risks versus the benefits versus the alternatives.”
The ‘entourage effect’
Data supporting cannabinoids have been mostly laboratory based, case based, or observational. However, several well-designed (albeit small) trials have demonstrated efficacy for chronic pain conditions, including neuropathic and headache pain, as well as in Crohn’s disease. Most investigators have concluded that dosage is important and that there is a synergistic interaction between compounds (known as the “entourage effect”) that relates to cannabinoid efficacy or lack thereof, as well as possible adverse effects.
In addition to legality issues, the entourage effect is one of the most important factors related to the medical use of cannabinoids. “There are literally thousands of cultivars of cannabis, each with their own phytocannabinoid and terpenic profiles that may produce distinct therapeutic effects, [so] it is misguided to speak of cannabis in monolithic terms. It is like making broad claims about soup,” wrote coauthor Samoon Ahmad, MD, in Medical Marijuana: A Clinical Handbook.
Additionally, the role that reproductive hormones play is not entirely understood. Reproductive-aged women appear to be more susceptible to a “telescoping” (gender-related progression to dependence) effect in comparison with men. Ziva Cooper, PhD, director of the Cannabis Research Initiative at the University of California, Los Angeles, said in an interview. She explained that research has shown that factors such as the degree of exposure, frequency of use, and menses confound this susceptibility.
It’s the data
Frustration over cannabinoid therapeutics abound, especially when it comes to data, legal issues, and lack of training. “The feedback that I hear from providers is that there isn’t enough information; we just don’t know enough about it,” Dr. Mark said, “but there is information that we do have, and ignoring it is not beneficial.”
Dr. Cooper concurred. Although she readily acknowledges that data from randomized, placebo-controlled trials are mostly lacking, she says, “There are signals in the literature providing evidence for the utility of cannabis and cannabinoids for pain and some other effects.”
Other practitioners said in an interview that some patients admit to using cannabinoids but that they lack the ample information to guide these patients. By and large, many women equate “natural” with “safe,” and some will experiment on their own to see what works.
Those experiments are not without risk, which is why “it’s just as important for physicians to talk to their patients about cannabis use as it is for patients to be forthcoming about that use,” said Dr. Cooper. “It could have implications on their overall health as well as interactions with other drugs that they’re using.”
That balance from a clinical perspective on cannabis is crucial, wrote coauthor Kenneth Hill, MD, in Medical Marijuana: A Clinical Handbook. “Without it,” he wrote, “the window of opportunity for a patient to accept treatment that she needs may not be open very long.”
A version of this article first appeared on Medscape.com.
When 42-year-old Danielle Simone Brand started having hormonal migraines, she first turned to cannabidiol (CBD) oil, eventually adding an occasional pull on a prefilled tetrahydrocannabinol (THC) vape for nighttime use. She was careful to avoid THC during work hours. A parenting and cannabis writer, Ms. Brand had more than a cursory background in cannabinoid medicine and had spent time at her local California dispensary discussing various cannabinoid components that might help alleviate her pain.
A self-professed “do-it-yourselfer,” Ms. Brand continues to use cannabinoids for her monthly headaches, forgoing any other pain medication. “There are times for conventional medicine in partnership with your doctor, but when it comes to health and wellness, women should be empowered to make decisions and self-experiment,” she said in an interview.
Ms. Brand is not alone. Significant numbers of women are replacing or supplementing prescription medications with cannabinoids, often without consulting their primary care physician, ob.gyn., or other specialist. At times, women have tried to have these conversations, only to be met with silence or worse.
Take Linda Fuller, a 58-year-old yoga instructor from Long Island who says that she uses CBD and THC for chronic sacroiliac pain after a car accident and to alleviate stress-triggered eczema flares. “I’ve had doctors turn their backs on me; I’ve had nurse practitioners walk out on me in the middle of a sentence,” she said in an interview.
Ms. Fuller said her conversion to cannabinoid medicine is relatively new; she never used cannabis recreationally before her accident but now considers it a gift. She doesn’t keep aspirin in the house and refused pain medication immediately after she injured her back.
Diana Krach, a 34-year-old writer from Maryland, says she’s encountered roadblocks about her decision to use cannabinoids for endometriosis and for pain from Crohn’s disease. When she tried to discuss her CBD use with a gastroenterologist, he interrupted her: “Whatever pot you’re smoking isn’t going to work, you’re going on biologics.”
Ms. Krach had not been smoking anything but had turned to a CBD tincture for symptom relief after prescription pain medications failed to help.
Ms. Brand, Ms. Fuller, and Ms. Krach are the tip of the iceberg when it comes to women seeking symptom relief outside the medicine cabinet. A recent survey in the Journal of Women’s Health of almost 1,000 women show that 90% (most between the ages of 35 and 44) had used cannabis and would consider using it to treat gynecologic pain. Roughly 80% said they would consider using it for procedure-related pain or other conditions. Additionally, women have reported using cannabinoids for PTSD, sleep disturbances or insomnia, anxiety, and migraine headaches.
Observational survey data have likewise shown that 80% of women with advanced or recurrent gynecologic malignancies who were prescribed cannabis reported that it was equivalent or superior to other medications for relieving pain, neuropathy, nausea, insomnia, decreased appetite, and anxiety.
In another survey, almost half (45%) of women with gynecologic malignancies who used nonprescribed cannabis for the same symptoms reported that they had reduced their use of prescription narcotics after initiating use of cannabis.
The gray zone
There has been a surge in self-reported cannabis use among pregnant women in particular. The National Survey on Drug Use and Health findings for the periods 2002-2003 and 2016-2017 highlight increases in adjusted prevalence rates from 3.4% to 7% in past-month use among pregnant women overall and from 5.7% to 12.1% during the first trimester alone.
“The more that you talk to pregnant women, the more that you realize that a lot are using cannabinoids for something that is basically medicinal, for sleep, for anxiety, or for nausea,” Katrina Mark, MD, an ob.gyn. and associate professor of medicine at the University of Maryland, College Park, said in an interview. “I’m not saying it’s fine to use drugs in pregnancy, but it is a grayer conversation than a lot of colleagues want to believe. Telling women to quit seems foolish since the alternative is to be anxious, don’t sleep, don’t eat, or use a medication that also has risks to it.”
One observational study shows that pregnant women themselves are conflicted. Although the majority believe that cannabis is “natural” and “safe,” compared with prescription drugs, they aren’t entirely in the dark about potential risks. They often express frustration with practitioners’ responses when these topics are broached during office visits. An observational survey among women and practitioners published in 2020 highlights that only half of doctors openly discouraged perinatal cannabis use and that others opted out of the discussion entirely.
This is the experience of many of the women that this news organization spoke with. Ms. Krach pointed out that “there’s a big deficit in listening; the doctor is supposed to be working for our behalf, especially when it comes to reproductive health.”
Dr. Mark believed that a lot of the conversation has been clouded by the illegality of the substance but that cannabinoids deserve as much of a fair chance for discussion and consideration as other medicines, which also carry risks in pregnancy. “There’s literally no evidence that it will work in pregnancy [for these symptoms], but there’s no evidence that it doesn’t, either,” she said in an interview. “When I have this conversation with colleagues who do not share my views, I try to encourage them to look at the actual risks versus the benefits versus the alternatives.”
The ‘entourage effect’
Data supporting cannabinoids have been mostly laboratory based, case based, or observational. However, several well-designed (albeit small) trials have demonstrated efficacy for chronic pain conditions, including neuropathic and headache pain, as well as in Crohn’s disease. Most investigators have concluded that dosage is important and that there is a synergistic interaction between compounds (known as the “entourage effect”) that relates to cannabinoid efficacy or lack thereof, as well as possible adverse effects.
In addition to legality issues, the entourage effect is one of the most important factors related to the medical use of cannabinoids. “There are literally thousands of cultivars of cannabis, each with their own phytocannabinoid and terpenic profiles that may produce distinct therapeutic effects, [so] it is misguided to speak of cannabis in monolithic terms. It is like making broad claims about soup,” wrote coauthor Samoon Ahmad, MD, in Medical Marijuana: A Clinical Handbook.
Additionally, the role that reproductive hormones play is not entirely understood. Reproductive-aged women appear to be more susceptible to a “telescoping” (gender-related progression to dependence) effect in comparison with men. Ziva Cooper, PhD, director of the Cannabis Research Initiative at the University of California, Los Angeles, said in an interview. She explained that research has shown that factors such as the degree of exposure, frequency of use, and menses confound this susceptibility.
It’s the data
Frustration over cannabinoid therapeutics abound, especially when it comes to data, legal issues, and lack of training. “The feedback that I hear from providers is that there isn’t enough information; we just don’t know enough about it,” Dr. Mark said, “but there is information that we do have, and ignoring it is not beneficial.”
Dr. Cooper concurred. Although she readily acknowledges that data from randomized, placebo-controlled trials are mostly lacking, she says, “There are signals in the literature providing evidence for the utility of cannabis and cannabinoids for pain and some other effects.”
Other practitioners said in an interview that some patients admit to using cannabinoids but that they lack the ample information to guide these patients. By and large, many women equate “natural” with “safe,” and some will experiment on their own to see what works.
Those experiments are not without risk, which is why “it’s just as important for physicians to talk to their patients about cannabis use as it is for patients to be forthcoming about that use,” said Dr. Cooper. “It could have implications on their overall health as well as interactions with other drugs that they’re using.”
That balance from a clinical perspective on cannabis is crucial, wrote coauthor Kenneth Hill, MD, in Medical Marijuana: A Clinical Handbook. “Without it,” he wrote, “the window of opportunity for a patient to accept treatment that she needs may not be open very long.”
A version of this article first appeared on Medscape.com.
When 42-year-old Danielle Simone Brand started having hormonal migraines, she first turned to cannabidiol (CBD) oil, eventually adding an occasional pull on a prefilled tetrahydrocannabinol (THC) vape for nighttime use. She was careful to avoid THC during work hours. A parenting and cannabis writer, Ms. Brand had more than a cursory background in cannabinoid medicine and had spent time at her local California dispensary discussing various cannabinoid components that might help alleviate her pain.
A self-professed “do-it-yourselfer,” Ms. Brand continues to use cannabinoids for her monthly headaches, forgoing any other pain medication. “There are times for conventional medicine in partnership with your doctor, but when it comes to health and wellness, women should be empowered to make decisions and self-experiment,” she said in an interview.
Ms. Brand is not alone. Significant numbers of women are replacing or supplementing prescription medications with cannabinoids, often without consulting their primary care physician, ob.gyn., or other specialist. At times, women have tried to have these conversations, only to be met with silence or worse.
Take Linda Fuller, a 58-year-old yoga instructor from Long Island who says that she uses CBD and THC for chronic sacroiliac pain after a car accident and to alleviate stress-triggered eczema flares. “I’ve had doctors turn their backs on me; I’ve had nurse practitioners walk out on me in the middle of a sentence,” she said in an interview.
Ms. Fuller said her conversion to cannabinoid medicine is relatively new; she never used cannabis recreationally before her accident but now considers it a gift. She doesn’t keep aspirin in the house and refused pain medication immediately after she injured her back.
Diana Krach, a 34-year-old writer from Maryland, says she’s encountered roadblocks about her decision to use cannabinoids for endometriosis and for pain from Crohn’s disease. When she tried to discuss her CBD use with a gastroenterologist, he interrupted her: “Whatever pot you’re smoking isn’t going to work, you’re going on biologics.”
Ms. Krach had not been smoking anything but had turned to a CBD tincture for symptom relief after prescription pain medications failed to help.
Ms. Brand, Ms. Fuller, and Ms. Krach are the tip of the iceberg when it comes to women seeking symptom relief outside the medicine cabinet. A recent survey in the Journal of Women’s Health of almost 1,000 women show that 90% (most between the ages of 35 and 44) had used cannabis and would consider using it to treat gynecologic pain. Roughly 80% said they would consider using it for procedure-related pain or other conditions. Additionally, women have reported using cannabinoids for PTSD, sleep disturbances or insomnia, anxiety, and migraine headaches.
Observational survey data have likewise shown that 80% of women with advanced or recurrent gynecologic malignancies who were prescribed cannabis reported that it was equivalent or superior to other medications for relieving pain, neuropathy, nausea, insomnia, decreased appetite, and anxiety.
In another survey, almost half (45%) of women with gynecologic malignancies who used nonprescribed cannabis for the same symptoms reported that they had reduced their use of prescription narcotics after initiating use of cannabis.
The gray zone
There has been a surge in self-reported cannabis use among pregnant women in particular. The National Survey on Drug Use and Health findings for the periods 2002-2003 and 2016-2017 highlight increases in adjusted prevalence rates from 3.4% to 7% in past-month use among pregnant women overall and from 5.7% to 12.1% during the first trimester alone.
“The more that you talk to pregnant women, the more that you realize that a lot are using cannabinoids for something that is basically medicinal, for sleep, for anxiety, or for nausea,” Katrina Mark, MD, an ob.gyn. and associate professor of medicine at the University of Maryland, College Park, said in an interview. “I’m not saying it’s fine to use drugs in pregnancy, but it is a grayer conversation than a lot of colleagues want to believe. Telling women to quit seems foolish since the alternative is to be anxious, don’t sleep, don’t eat, or use a medication that also has risks to it.”
One observational study shows that pregnant women themselves are conflicted. Although the majority believe that cannabis is “natural” and “safe,” compared with prescription drugs, they aren’t entirely in the dark about potential risks. They often express frustration with practitioners’ responses when these topics are broached during office visits. An observational survey among women and practitioners published in 2020 highlights that only half of doctors openly discouraged perinatal cannabis use and that others opted out of the discussion entirely.
This is the experience of many of the women that this news organization spoke with. Ms. Krach pointed out that “there’s a big deficit in listening; the doctor is supposed to be working for our behalf, especially when it comes to reproductive health.”
Dr. Mark believed that a lot of the conversation has been clouded by the illegality of the substance but that cannabinoids deserve as much of a fair chance for discussion and consideration as other medicines, which also carry risks in pregnancy. “There’s literally no evidence that it will work in pregnancy [for these symptoms], but there’s no evidence that it doesn’t, either,” she said in an interview. “When I have this conversation with colleagues who do not share my views, I try to encourage them to look at the actual risks versus the benefits versus the alternatives.”
The ‘entourage effect’
Data supporting cannabinoids have been mostly laboratory based, case based, or observational. However, several well-designed (albeit small) trials have demonstrated efficacy for chronic pain conditions, including neuropathic and headache pain, as well as in Crohn’s disease. Most investigators have concluded that dosage is important and that there is a synergistic interaction between compounds (known as the “entourage effect”) that relates to cannabinoid efficacy or lack thereof, as well as possible adverse effects.
In addition to legality issues, the entourage effect is one of the most important factors related to the medical use of cannabinoids. “There are literally thousands of cultivars of cannabis, each with their own phytocannabinoid and terpenic profiles that may produce distinct therapeutic effects, [so] it is misguided to speak of cannabis in monolithic terms. It is like making broad claims about soup,” wrote coauthor Samoon Ahmad, MD, in Medical Marijuana: A Clinical Handbook.
Additionally, the role that reproductive hormones play is not entirely understood. Reproductive-aged women appear to be more susceptible to a “telescoping” (gender-related progression to dependence) effect in comparison with men. Ziva Cooper, PhD, director of the Cannabis Research Initiative at the University of California, Los Angeles, said in an interview. She explained that research has shown that factors such as the degree of exposure, frequency of use, and menses confound this susceptibility.
It’s the data
Frustration over cannabinoid therapeutics abound, especially when it comes to data, legal issues, and lack of training. “The feedback that I hear from providers is that there isn’t enough information; we just don’t know enough about it,” Dr. Mark said, “but there is information that we do have, and ignoring it is not beneficial.”
Dr. Cooper concurred. Although she readily acknowledges that data from randomized, placebo-controlled trials are mostly lacking, she says, “There are signals in the literature providing evidence for the utility of cannabis and cannabinoids for pain and some other effects.”
Other practitioners said in an interview that some patients admit to using cannabinoids but that they lack the ample information to guide these patients. By and large, many women equate “natural” with “safe,” and some will experiment on their own to see what works.
Those experiments are not without risk, which is why “it’s just as important for physicians to talk to their patients about cannabis use as it is for patients to be forthcoming about that use,” said Dr. Cooper. “It could have implications on their overall health as well as interactions with other drugs that they’re using.”
That balance from a clinical perspective on cannabis is crucial, wrote coauthor Kenneth Hill, MD, in Medical Marijuana: A Clinical Handbook. “Without it,” he wrote, “the window of opportunity for a patient to accept treatment that she needs may not be open very long.”
A version of this article first appeared on Medscape.com.
Customized chemotherapy did not improve survival in early NSCLC
The patients were randomized to receive investigator’s choice of platinum-based chemotherapy or treatment tailored according to messenger RNA (mRNA) expression of two molecular markers – excision repair cross complementation 1 (ERCC1) and thymidylate synthase (TS).
There was no significant difference in overall survival or recurrence-free survival between the treatment approaches. However, toxicity was less common among patients who received customized treatment.
These results, from the phase 3 ITACA trial, were presented at the 2020 World Conference on Lung Cancer (Abstract 1820), which was rescheduled to January 2021.
“There is a clear need to define patients most likely to derive survival benefit from adjuvant therapy and spare patients who do not need adjuvant chemotherapy due to the toxicity of such therapy,” said presenter Silvia Novello, MD, PhD, of the University of Turin in Italy. “mRNA expression of different genes has been correlated with the sensitivity or resistance to specific anticancer agents.”
With this in mind, Dr. Novello and colleagues conducted the ITACA trial. The researchers’ primary goal was to determine whether an adjuvant pharmacogenomic-driven approach was able to improve overall survival in completely resected NSCLC.
Patients and treatment
The researchers randomized 773 NSCLC patients within 5-8 weeks after radical surgery. Genomic analyses were performed soon after surgery, and patients were randomly assigned to investigator’s choice of platinum-based chemotherapy or to tailored treatments defined by mRNA levels of ERCC1 and TS.
Patients with high ERCC1 mRNA expression who were randomized to tailored treatment received single-agent docetaxel if their TS level was high or pemetrexed monotherapy if their TS level was low.
Patients with low ERCC1 mRNA expression who were randomized to tailored treatment received cisplatin-gemcitabine if their TS level was high or cisplatin-pemetrexed if their TS was low.
The most frequent doublets used in control patients were cisplatin-gemcitabine and cisplatin-vinorelbine.
The demographic characteristics of the 384 patients randomized to tailored therapy and the 389 control subjects were well-balanced, Dr. Novello said. Two-thirds of patients had stage II disease, 11% were never smokers, and the vast majority had a lobectomy as the resection method.
Results
At a median follow-up of 28.2 months, the median overall survival was 96.4 months in the tailored therapy arm and 83.5 months in the control arm. The median recurrence-free survival was 64.4 months and 41.5 months, respectively.
“Adjuvant chemotherapy customization based on the primary tumor tissue mRNA expression of ERCC1 and TS did not significantly improve overall survival or recurrence-free survival,” Dr. Novello said. “There was a non–statistically significant trend for overall survival favoring the customized arm.”
Dr. Novello noted that, when the final analysis was performed, the study was underpowered, as only 46% of expected events were collected. Assuming the same hazard ratio point estimate and that the expected 336 events were collected, the hazard ratio estimate would be 0.76 (P = .012).
Grade 3/4 toxicities occurred in 32.6% of patients in the tailored therapy arm and 45.9% of those in the control arm (P < .001).
“It is important to underline that the treatment customization significantly improved the toxicity profile without compromising the efficacy,” Dr. Novello said.
She added that “more comprehensive and high-throughput diagnostic techniques will be needed in order to tailor adjuvant chemotherapy, with or without immunotherapy, in completely resected NSCLC.”
“The ITACA study is the largest adjuvant study tailored to ERCC1/TS status, and the results have been long-awaited,” said Tetsuya Mitsudomi, MD, a professor at Kindai University in Japan and president of the International Association for the Study of Lung Cancer.
“This trial should be praised for the mandated genomic analysis that was accomplished within a reasonably short time frame before random assignment. In addition, this trial confirmed that there is no biomarker strong enough to predict the efficacy of cytotoxic chemotherapy. However, the concept of customizing adjuvant therapy according to the genomic status of patients’ tumors is valid, leading to the recent demonstration in the ADAURA study of the superiority of osimertinib in delaying the postoperative recurrence of disease in patients with EGFR-mutated NSCLC.”
The ITACA study was funded by University of Turin and Eli Lilly. Dr. Novello disclosed relationships with Eli Lilly, Amgen, AstraZeneca, Bohringer Ingelheim, Beigene, Pfizer, Roche, Merck, Bristol-Myers Squibb, Takeda, and Sanofi. Dr. Mitsudomi disclosed relationships with Eli Lilly, AstraZeneca, Boehringer-Ingelheim, Chugai, Pfizer, Merck, Ono Pharmaceutical, Bristol-Myers Squibb, Novartis, ThermoFisher, Guardant, Eisai, Amgen, and Johnson & Johnson.
The patients were randomized to receive investigator’s choice of platinum-based chemotherapy or treatment tailored according to messenger RNA (mRNA) expression of two molecular markers – excision repair cross complementation 1 (ERCC1) and thymidylate synthase (TS).
There was no significant difference in overall survival or recurrence-free survival between the treatment approaches. However, toxicity was less common among patients who received customized treatment.
These results, from the phase 3 ITACA trial, were presented at the 2020 World Conference on Lung Cancer (Abstract 1820), which was rescheduled to January 2021.
“There is a clear need to define patients most likely to derive survival benefit from adjuvant therapy and spare patients who do not need adjuvant chemotherapy due to the toxicity of such therapy,” said presenter Silvia Novello, MD, PhD, of the University of Turin in Italy. “mRNA expression of different genes has been correlated with the sensitivity or resistance to specific anticancer agents.”
With this in mind, Dr. Novello and colleagues conducted the ITACA trial. The researchers’ primary goal was to determine whether an adjuvant pharmacogenomic-driven approach was able to improve overall survival in completely resected NSCLC.
Patients and treatment
The researchers randomized 773 NSCLC patients within 5-8 weeks after radical surgery. Genomic analyses were performed soon after surgery, and patients were randomly assigned to investigator’s choice of platinum-based chemotherapy or to tailored treatments defined by mRNA levels of ERCC1 and TS.
Patients with high ERCC1 mRNA expression who were randomized to tailored treatment received single-agent docetaxel if their TS level was high or pemetrexed monotherapy if their TS level was low.
Patients with low ERCC1 mRNA expression who were randomized to tailored treatment received cisplatin-gemcitabine if their TS level was high or cisplatin-pemetrexed if their TS was low.
The most frequent doublets used in control patients were cisplatin-gemcitabine and cisplatin-vinorelbine.
The demographic characteristics of the 384 patients randomized to tailored therapy and the 389 control subjects were well-balanced, Dr. Novello said. Two-thirds of patients had stage II disease, 11% were never smokers, and the vast majority had a lobectomy as the resection method.
Results
At a median follow-up of 28.2 months, the median overall survival was 96.4 months in the tailored therapy arm and 83.5 months in the control arm. The median recurrence-free survival was 64.4 months and 41.5 months, respectively.
“Adjuvant chemotherapy customization based on the primary tumor tissue mRNA expression of ERCC1 and TS did not significantly improve overall survival or recurrence-free survival,” Dr. Novello said. “There was a non–statistically significant trend for overall survival favoring the customized arm.”
Dr. Novello noted that, when the final analysis was performed, the study was underpowered, as only 46% of expected events were collected. Assuming the same hazard ratio point estimate and that the expected 336 events were collected, the hazard ratio estimate would be 0.76 (P = .012).
Grade 3/4 toxicities occurred in 32.6% of patients in the tailored therapy arm and 45.9% of those in the control arm (P < .001).
“It is important to underline that the treatment customization significantly improved the toxicity profile without compromising the efficacy,” Dr. Novello said.
She added that “more comprehensive and high-throughput diagnostic techniques will be needed in order to tailor adjuvant chemotherapy, with or without immunotherapy, in completely resected NSCLC.”
“The ITACA study is the largest adjuvant study tailored to ERCC1/TS status, and the results have been long-awaited,” said Tetsuya Mitsudomi, MD, a professor at Kindai University in Japan and president of the International Association for the Study of Lung Cancer.
“This trial should be praised for the mandated genomic analysis that was accomplished within a reasonably short time frame before random assignment. In addition, this trial confirmed that there is no biomarker strong enough to predict the efficacy of cytotoxic chemotherapy. However, the concept of customizing adjuvant therapy according to the genomic status of patients’ tumors is valid, leading to the recent demonstration in the ADAURA study of the superiority of osimertinib in delaying the postoperative recurrence of disease in patients with EGFR-mutated NSCLC.”
The ITACA study was funded by University of Turin and Eli Lilly. Dr. Novello disclosed relationships with Eli Lilly, Amgen, AstraZeneca, Bohringer Ingelheim, Beigene, Pfizer, Roche, Merck, Bristol-Myers Squibb, Takeda, and Sanofi. Dr. Mitsudomi disclosed relationships with Eli Lilly, AstraZeneca, Boehringer-Ingelheim, Chugai, Pfizer, Merck, Ono Pharmaceutical, Bristol-Myers Squibb, Novartis, ThermoFisher, Guardant, Eisai, Amgen, and Johnson & Johnson.
The patients were randomized to receive investigator’s choice of platinum-based chemotherapy or treatment tailored according to messenger RNA (mRNA) expression of two molecular markers – excision repair cross complementation 1 (ERCC1) and thymidylate synthase (TS).
There was no significant difference in overall survival or recurrence-free survival between the treatment approaches. However, toxicity was less common among patients who received customized treatment.
These results, from the phase 3 ITACA trial, were presented at the 2020 World Conference on Lung Cancer (Abstract 1820), which was rescheduled to January 2021.
“There is a clear need to define patients most likely to derive survival benefit from adjuvant therapy and spare patients who do not need adjuvant chemotherapy due to the toxicity of such therapy,” said presenter Silvia Novello, MD, PhD, of the University of Turin in Italy. “mRNA expression of different genes has been correlated with the sensitivity or resistance to specific anticancer agents.”
With this in mind, Dr. Novello and colleagues conducted the ITACA trial. The researchers’ primary goal was to determine whether an adjuvant pharmacogenomic-driven approach was able to improve overall survival in completely resected NSCLC.
Patients and treatment
The researchers randomized 773 NSCLC patients within 5-8 weeks after radical surgery. Genomic analyses were performed soon after surgery, and patients were randomly assigned to investigator’s choice of platinum-based chemotherapy or to tailored treatments defined by mRNA levels of ERCC1 and TS.
Patients with high ERCC1 mRNA expression who were randomized to tailored treatment received single-agent docetaxel if their TS level was high or pemetrexed monotherapy if their TS level was low.
Patients with low ERCC1 mRNA expression who were randomized to tailored treatment received cisplatin-gemcitabine if their TS level was high or cisplatin-pemetrexed if their TS was low.
The most frequent doublets used in control patients were cisplatin-gemcitabine and cisplatin-vinorelbine.
The demographic characteristics of the 384 patients randomized to tailored therapy and the 389 control subjects were well-balanced, Dr. Novello said. Two-thirds of patients had stage II disease, 11% were never smokers, and the vast majority had a lobectomy as the resection method.
Results
At a median follow-up of 28.2 months, the median overall survival was 96.4 months in the tailored therapy arm and 83.5 months in the control arm. The median recurrence-free survival was 64.4 months and 41.5 months, respectively.
“Adjuvant chemotherapy customization based on the primary tumor tissue mRNA expression of ERCC1 and TS did not significantly improve overall survival or recurrence-free survival,” Dr. Novello said. “There was a non–statistically significant trend for overall survival favoring the customized arm.”
Dr. Novello noted that, when the final analysis was performed, the study was underpowered, as only 46% of expected events were collected. Assuming the same hazard ratio point estimate and that the expected 336 events were collected, the hazard ratio estimate would be 0.76 (P = .012).
Grade 3/4 toxicities occurred in 32.6% of patients in the tailored therapy arm and 45.9% of those in the control arm (P < .001).
“It is important to underline that the treatment customization significantly improved the toxicity profile without compromising the efficacy,” Dr. Novello said.
She added that “more comprehensive and high-throughput diagnostic techniques will be needed in order to tailor adjuvant chemotherapy, with or without immunotherapy, in completely resected NSCLC.”
“The ITACA study is the largest adjuvant study tailored to ERCC1/TS status, and the results have been long-awaited,” said Tetsuya Mitsudomi, MD, a professor at Kindai University in Japan and president of the International Association for the Study of Lung Cancer.
“This trial should be praised for the mandated genomic analysis that was accomplished within a reasonably short time frame before random assignment. In addition, this trial confirmed that there is no biomarker strong enough to predict the efficacy of cytotoxic chemotherapy. However, the concept of customizing adjuvant therapy according to the genomic status of patients’ tumors is valid, leading to the recent demonstration in the ADAURA study of the superiority of osimertinib in delaying the postoperative recurrence of disease in patients with EGFR-mutated NSCLC.”
The ITACA study was funded by University of Turin and Eli Lilly. Dr. Novello disclosed relationships with Eli Lilly, Amgen, AstraZeneca, Bohringer Ingelheim, Beigene, Pfizer, Roche, Merck, Bristol-Myers Squibb, Takeda, and Sanofi. Dr. Mitsudomi disclosed relationships with Eli Lilly, AstraZeneca, Boehringer-Ingelheim, Chugai, Pfizer, Merck, Ono Pharmaceutical, Bristol-Myers Squibb, Novartis, ThermoFisher, Guardant, Eisai, Amgen, and Johnson & Johnson.
FROM WCLC 2020
Cisplatin tops cetuximab for advanced head and neck cancer
Concurrent cisplatin should remain the standard treatment over cetuximab for patients with locoregionally advanced head and neck squamous cell carcinoma, according to a large comparative phase 3 trial.
Based on the results of an interim analysis of the ARTSCAN III clinical trial, the independent safety data monitoring committee recommended early closure of the study because the results of the study suggest that cetuximab may be inferior to cisplatin.
“This study supports previous retrospective and prospective studies that suggest that concurrent cisplatin with radiation is superior to regimens with concurrent cetuximab in locally advanced head and neck cancers,” commented Sachin Jhawar, MD, MSCI, assistant professor in the department of radiation oncology at Ohio State University Comprehensive Cancer Center, Columbus. “While the previous studies De-ESCALaTE HPV and RTOG 1016 were specific to HPV [human papillomavirus]-positive cancers, this study allowed non–virally mediated tumors, though the majority of cases were HPV related.”
The new study also used a lower-dose weekly regimen of cisplatin than the other two studies, noted Dr. Jhawar. Early trials used a cisplatin dose of 100 mg/m2 every 3 weeks, but “the field is moving toward a dose of 40 mg/m2 weekly, the dose use in the Swedish study. This study had an interesting second randomization of radiation dose-escalation for more advanced primary T stage tumors, but because the study ended early it is difficult to fully interpret those results.”
Swedish researchers, led by Maria Gebre-Medhin, MD, PhD, department of hematology, oncology, and radiation physics, Skåne University Hospital, Lund, Sweden, performed an open-label, randomized, controlled, phase 3 study of patients with locoregionally advanced head and neck squamous cell carcinoma. The patients received IV cetuximab 400 mg/m2 1 week before the start of radiation therapy followed by 250 mg/m2 per week, or weekly IV cisplatin 40 mg/m2 during radiation therapy.
The study results were published in the Journal of Clinical Oncology.
The study was prematurely closed after an unplanned interim analysis when 298 patients had been randomly assigned. The cumulative incidence of locoregional failures at 3 years was more than twice as high in the cetuximab group (23%), compared with the cisplatin group (9%; P = .0036). At 3 years, overall survival was higher in the cisplatin (88%) group than in the cetuximab group (78%), but the difference was not significant (P = .086). The cumulative incidence of distant failures did not differ between the treatment groups, and the toxicity burden was similar.
“Concurrent cisplatin led to improved locoregional control and event-free survival with a trend toward improved overall survival. The types of toxicity were different, as would be expected with the different drug mechanisms, but the rate of toxicity was not,” said Dr. Dr. Jhawar. “Interestingly, the benefit of cisplatin seemed to be limited to patients with p16-positive oropharyngeal cancer. There was clinical equipoise in the p16-negative oropharyngeal cancer group and in the non–oropharyngeal cancer group. The numbers were small, but this is intriguing and suggests that there is more work to be done in this group of patients to tease out if we can escalate or use alternative therapy.”
Cisplatin has been repeatedly proven to be superior in selected populations. The next steps, said Dr. Jhawar, include defining “optimal regimens in cisplatin-ineligible populations based on age, performance status, kidney function, hearing loss, neuropathy, and HIV/AIDS; improvements with new targeted therapies and immunotherapies; and deescalation of systemic and/or radiation regimens in the best outcome groups, such as low-risk HPV-positive patients. And we are going to see more personalized medicine with genetic testing of tumors.”
When patients are ineligible for cisplatin, no optimal regimens have been defined as yet. At Ohio State, Dr. Jhawar and colleagues use carboplatin therapy.
For practicing oncologists, Dr. Jhawar said the bottom line is “patients who are eligible should receive concurrent cisplatin therapy for locoregionally advanced head and neck cancer.”
The ARTSCAN III study was funded by the Swedish Cancer Society and the Mrs. Berta Kamprad Cancer Foundation. One of the study’s coauthors reported a leadership role in ScandiDos. The other authors reported they had no conflicts. Dr. Jhawar reported he had no conflicts of interest.
Concurrent cisplatin should remain the standard treatment over cetuximab for patients with locoregionally advanced head and neck squamous cell carcinoma, according to a large comparative phase 3 trial.
Based on the results of an interim analysis of the ARTSCAN III clinical trial, the independent safety data monitoring committee recommended early closure of the study because the results of the study suggest that cetuximab may be inferior to cisplatin.
“This study supports previous retrospective and prospective studies that suggest that concurrent cisplatin with radiation is superior to regimens with concurrent cetuximab in locally advanced head and neck cancers,” commented Sachin Jhawar, MD, MSCI, assistant professor in the department of radiation oncology at Ohio State University Comprehensive Cancer Center, Columbus. “While the previous studies De-ESCALaTE HPV and RTOG 1016 were specific to HPV [human papillomavirus]-positive cancers, this study allowed non–virally mediated tumors, though the majority of cases were HPV related.”
The new study also used a lower-dose weekly regimen of cisplatin than the other two studies, noted Dr. Jhawar. Early trials used a cisplatin dose of 100 mg/m2 every 3 weeks, but “the field is moving toward a dose of 40 mg/m2 weekly, the dose use in the Swedish study. This study had an interesting second randomization of radiation dose-escalation for more advanced primary T stage tumors, but because the study ended early it is difficult to fully interpret those results.”
Swedish researchers, led by Maria Gebre-Medhin, MD, PhD, department of hematology, oncology, and radiation physics, Skåne University Hospital, Lund, Sweden, performed an open-label, randomized, controlled, phase 3 study of patients with locoregionally advanced head and neck squamous cell carcinoma. The patients received IV cetuximab 400 mg/m2 1 week before the start of radiation therapy followed by 250 mg/m2 per week, or weekly IV cisplatin 40 mg/m2 during radiation therapy.
The study results were published in the Journal of Clinical Oncology.
The study was prematurely closed after an unplanned interim analysis when 298 patients had been randomly assigned. The cumulative incidence of locoregional failures at 3 years was more than twice as high in the cetuximab group (23%), compared with the cisplatin group (9%; P = .0036). At 3 years, overall survival was higher in the cisplatin (88%) group than in the cetuximab group (78%), but the difference was not significant (P = .086). The cumulative incidence of distant failures did not differ between the treatment groups, and the toxicity burden was similar.
“Concurrent cisplatin led to improved locoregional control and event-free survival with a trend toward improved overall survival. The types of toxicity were different, as would be expected with the different drug mechanisms, but the rate of toxicity was not,” said Dr. Dr. Jhawar. “Interestingly, the benefit of cisplatin seemed to be limited to patients with p16-positive oropharyngeal cancer. There was clinical equipoise in the p16-negative oropharyngeal cancer group and in the non–oropharyngeal cancer group. The numbers were small, but this is intriguing and suggests that there is more work to be done in this group of patients to tease out if we can escalate or use alternative therapy.”
Cisplatin has been repeatedly proven to be superior in selected populations. The next steps, said Dr. Jhawar, include defining “optimal regimens in cisplatin-ineligible populations based on age, performance status, kidney function, hearing loss, neuropathy, and HIV/AIDS; improvements with new targeted therapies and immunotherapies; and deescalation of systemic and/or radiation regimens in the best outcome groups, such as low-risk HPV-positive patients. And we are going to see more personalized medicine with genetic testing of tumors.”
When patients are ineligible for cisplatin, no optimal regimens have been defined as yet. At Ohio State, Dr. Jhawar and colleagues use carboplatin therapy.
For practicing oncologists, Dr. Jhawar said the bottom line is “patients who are eligible should receive concurrent cisplatin therapy for locoregionally advanced head and neck cancer.”
The ARTSCAN III study was funded by the Swedish Cancer Society and the Mrs. Berta Kamprad Cancer Foundation. One of the study’s coauthors reported a leadership role in ScandiDos. The other authors reported they had no conflicts. Dr. Jhawar reported he had no conflicts of interest.
Concurrent cisplatin should remain the standard treatment over cetuximab for patients with locoregionally advanced head and neck squamous cell carcinoma, according to a large comparative phase 3 trial.
Based on the results of an interim analysis of the ARTSCAN III clinical trial, the independent safety data monitoring committee recommended early closure of the study because the results of the study suggest that cetuximab may be inferior to cisplatin.
“This study supports previous retrospective and prospective studies that suggest that concurrent cisplatin with radiation is superior to regimens with concurrent cetuximab in locally advanced head and neck cancers,” commented Sachin Jhawar, MD, MSCI, assistant professor in the department of radiation oncology at Ohio State University Comprehensive Cancer Center, Columbus. “While the previous studies De-ESCALaTE HPV and RTOG 1016 were specific to HPV [human papillomavirus]-positive cancers, this study allowed non–virally mediated tumors, though the majority of cases were HPV related.”
The new study also used a lower-dose weekly regimen of cisplatin than the other two studies, noted Dr. Jhawar. Early trials used a cisplatin dose of 100 mg/m2 every 3 weeks, but “the field is moving toward a dose of 40 mg/m2 weekly, the dose use in the Swedish study. This study had an interesting second randomization of radiation dose-escalation for more advanced primary T stage tumors, but because the study ended early it is difficult to fully interpret those results.”
Swedish researchers, led by Maria Gebre-Medhin, MD, PhD, department of hematology, oncology, and radiation physics, Skåne University Hospital, Lund, Sweden, performed an open-label, randomized, controlled, phase 3 study of patients with locoregionally advanced head and neck squamous cell carcinoma. The patients received IV cetuximab 400 mg/m2 1 week before the start of radiation therapy followed by 250 mg/m2 per week, or weekly IV cisplatin 40 mg/m2 during radiation therapy.
The study results were published in the Journal of Clinical Oncology.
The study was prematurely closed after an unplanned interim analysis when 298 patients had been randomly assigned. The cumulative incidence of locoregional failures at 3 years was more than twice as high in the cetuximab group (23%), compared with the cisplatin group (9%; P = .0036). At 3 years, overall survival was higher in the cisplatin (88%) group than in the cetuximab group (78%), but the difference was not significant (P = .086). The cumulative incidence of distant failures did not differ between the treatment groups, and the toxicity burden was similar.
“Concurrent cisplatin led to improved locoregional control and event-free survival with a trend toward improved overall survival. The types of toxicity were different, as would be expected with the different drug mechanisms, but the rate of toxicity was not,” said Dr. Dr. Jhawar. “Interestingly, the benefit of cisplatin seemed to be limited to patients with p16-positive oropharyngeal cancer. There was clinical equipoise in the p16-negative oropharyngeal cancer group and in the non–oropharyngeal cancer group. The numbers were small, but this is intriguing and suggests that there is more work to be done in this group of patients to tease out if we can escalate or use alternative therapy.”
Cisplatin has been repeatedly proven to be superior in selected populations. The next steps, said Dr. Jhawar, include defining “optimal regimens in cisplatin-ineligible populations based on age, performance status, kidney function, hearing loss, neuropathy, and HIV/AIDS; improvements with new targeted therapies and immunotherapies; and deescalation of systemic and/or radiation regimens in the best outcome groups, such as low-risk HPV-positive patients. And we are going to see more personalized medicine with genetic testing of tumors.”
When patients are ineligible for cisplatin, no optimal regimens have been defined as yet. At Ohio State, Dr. Jhawar and colleagues use carboplatin therapy.
For practicing oncologists, Dr. Jhawar said the bottom line is “patients who are eligible should receive concurrent cisplatin therapy for locoregionally advanced head and neck cancer.”
The ARTSCAN III study was funded by the Swedish Cancer Society and the Mrs. Berta Kamprad Cancer Foundation. One of the study’s coauthors reported a leadership role in ScandiDos. The other authors reported they had no conflicts. Dr. Jhawar reported he had no conflicts of interest.
FROM THE JOURNAL OF CLINICAL ONCOLOGY