User login
Vesicles and Bullae on the Leg
The Diagnosis: Cutaneous B-cell Lymphoma
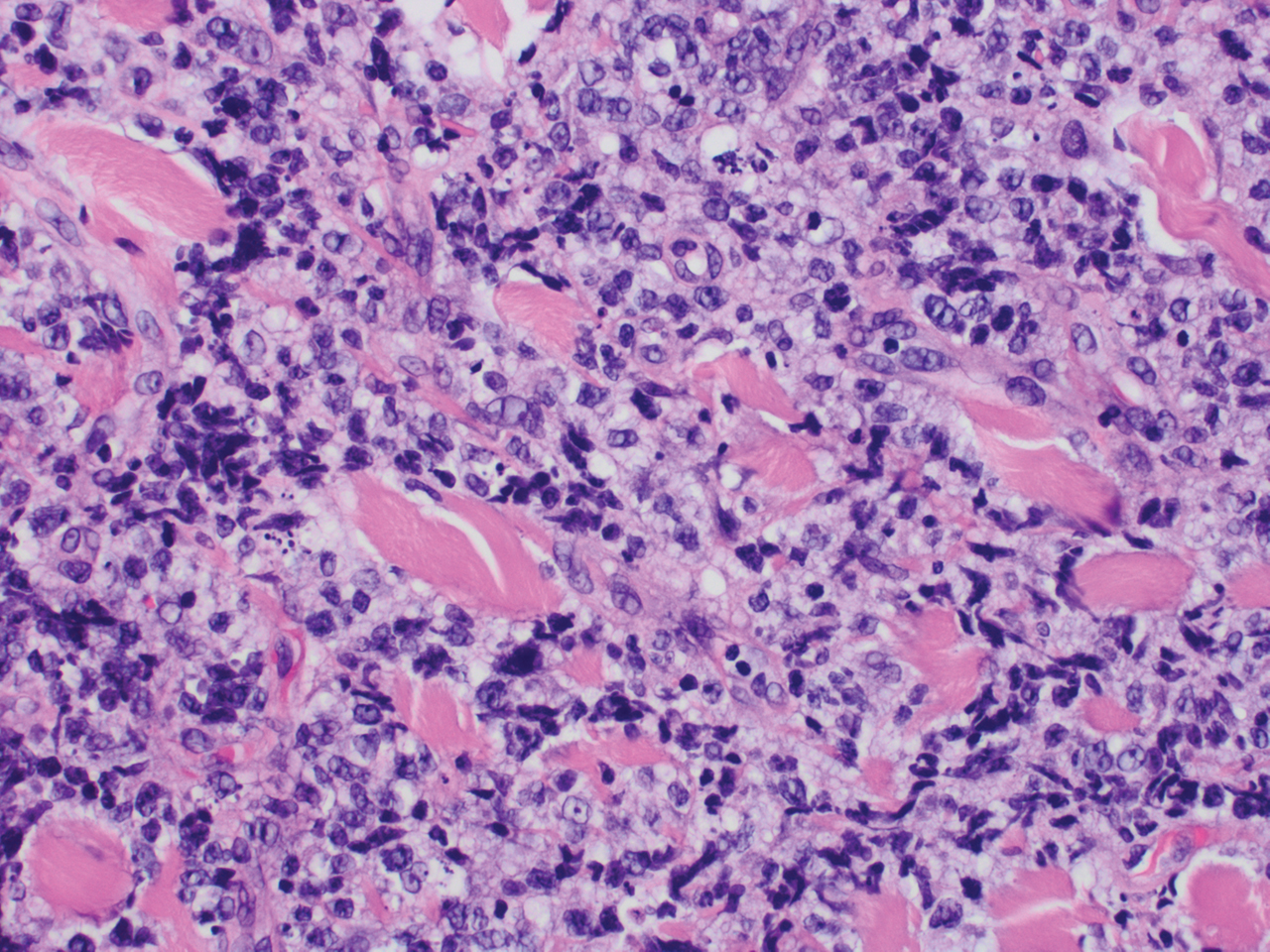
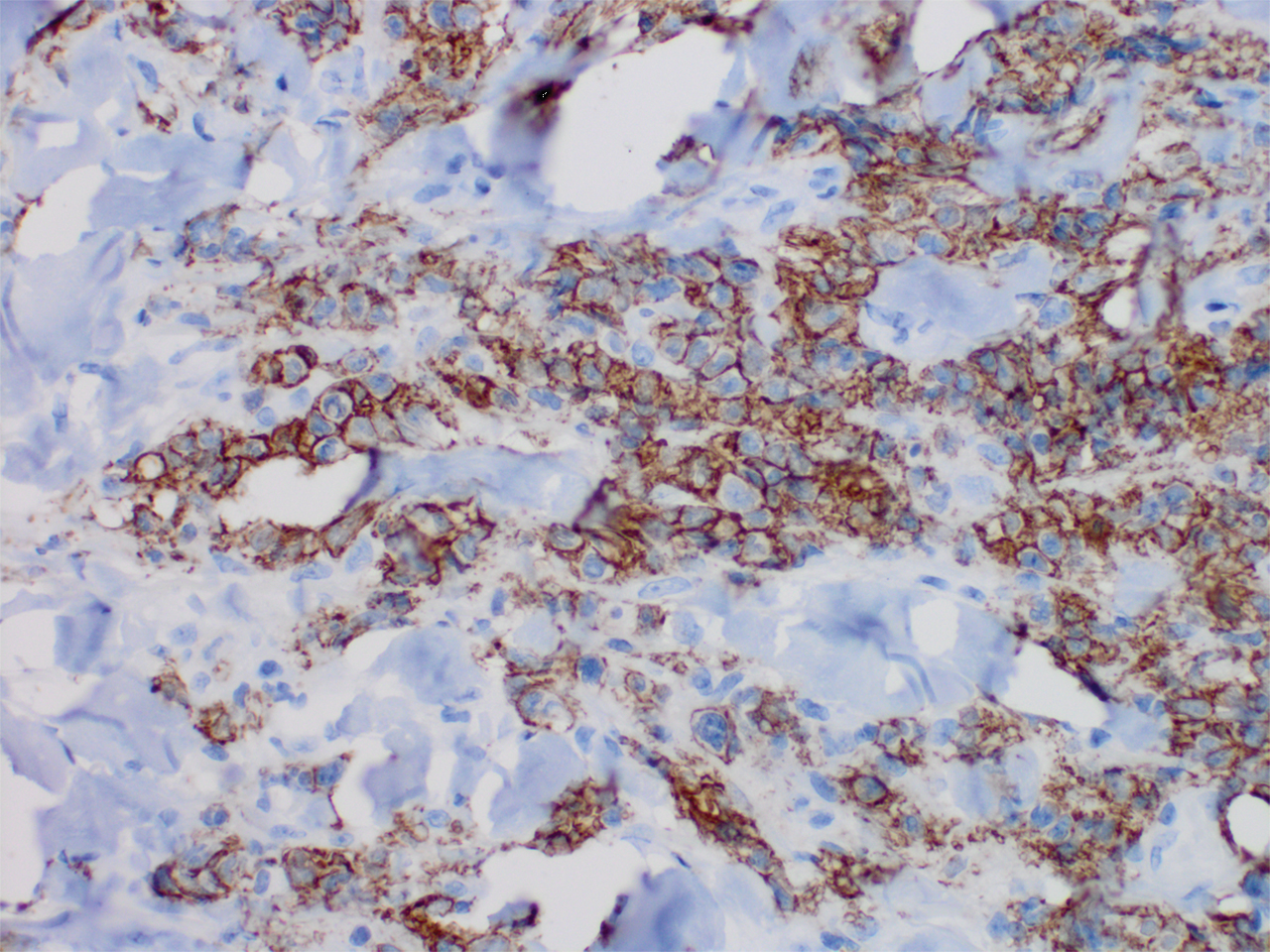
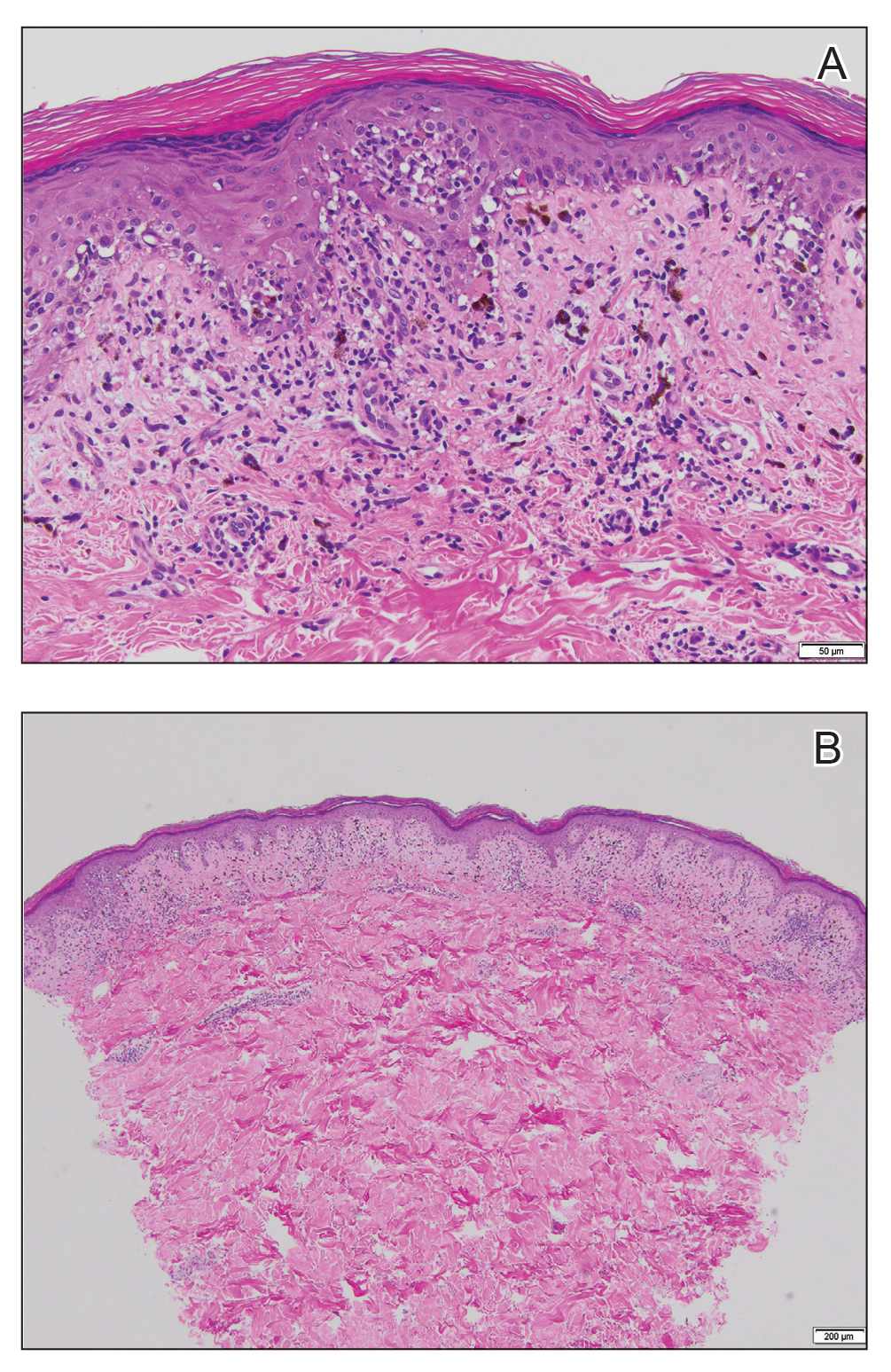
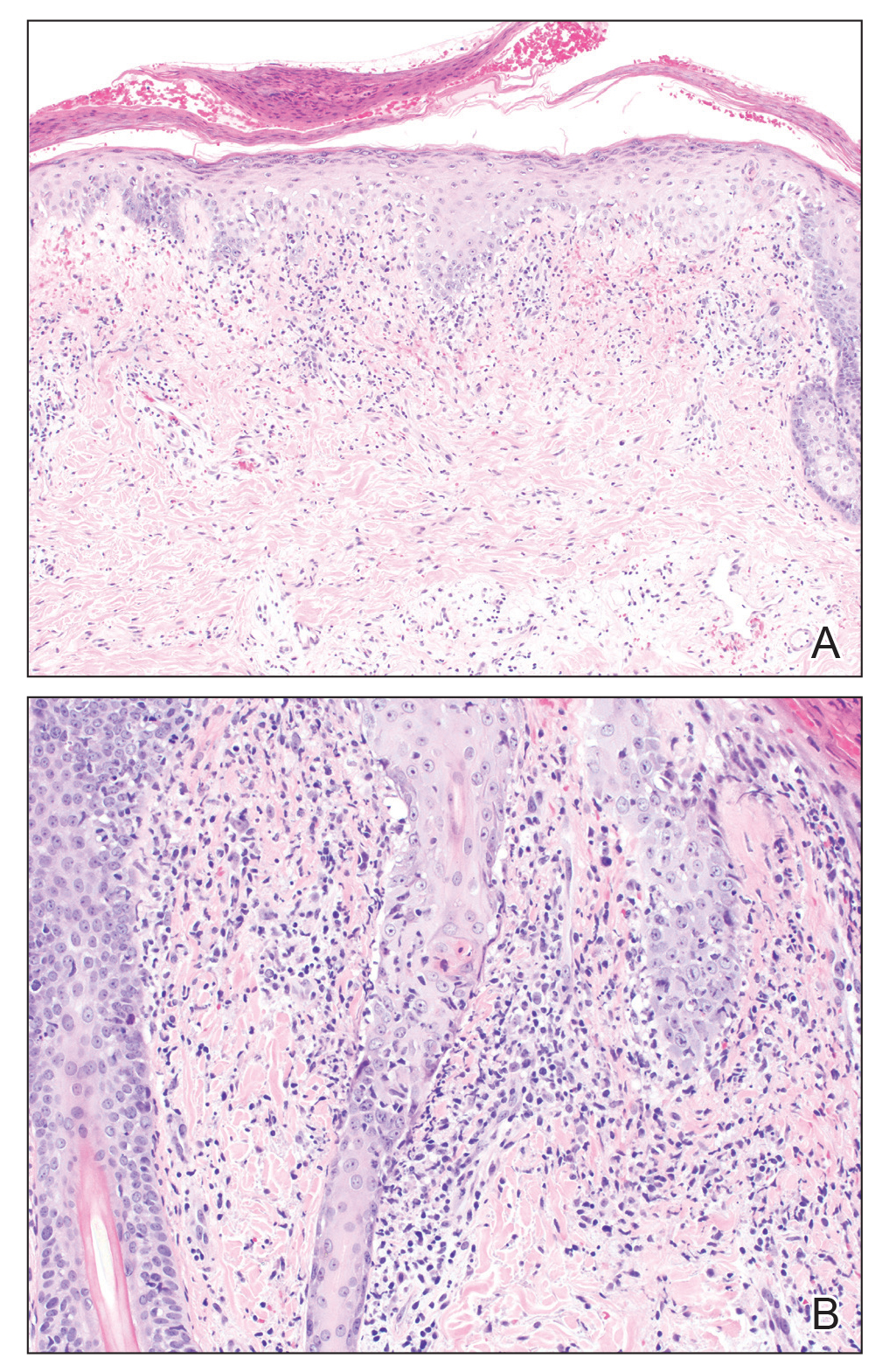
Histopathology revealed a dense and diffuse lymphocytic infiltrate throughout the dermis with occasional individual cell necrosis. On closer inspection, the infiltrate consisted of intermediate-sized lymphocytes, some with a vesiculated nucleus and ample amount of cytoplasm, while others contained hyperchromatic nuclei (Figure 1). These cells stained strongly positive for B-cell marker (CD20), while only a few mature lymphocytes demonstrated T-cell phenotype (CD3)(Figure 2).
Although the patient recounted a 3-month history of lower leg edema, he also reported that the rash began a few weeks after his diagnosis of systemic B-cell follicular lymphoma. Throughout this time, he was seen by various physicians who attributed the edema and skin changes to chronic stasis, peripheral venous insufficiency, and diabetic peripheral neuropathy. His primary care physician prescribed an antifungal lotion, which he discontinued on his own due to lack of improvement. Upon arrival to the emergency department, he was started on intravenous cefazolin and subcutaneous heparin. Doppler ultrasonography of the legs was ordered to rule out a deep venous thrombosis. Dermatology was consulted and proceeded with a punch biopsy to investigate for cutaneous B-cell lymphoma (BCL) with a plan to follow up as an outpatient for results upon discharge. He also was prescribed triamcinolone ointment 0.1% twice daily for symptomatic relief.
The patient's left axillary lymph node was biopsied for pathologic evaluation. Immunohistochemical staining revealed expression of B-cell markers CD20, CD79a, and PAX5, along with the antiapoptotic markers BCL-2 and BCL-6. Fluorescence in situ hybridization displayed gene rearrangements of BCL-2, BCL-6, and t(14;18)/IgH-BCL2 in the majority of cells. CD3 and CD5 immunostains were negative, indicating that T cells were not involved in this process. Flow cytometry identified a monoclonal κ B-cell population in 40% to 50% of the total cells, which co-expressed CD10, CD19, CD22, and CD38; the cells were negative for CD5, CD20, and CD23. Cell size was variably enlarged and CD71 positive, otherwise known as transferrin receptor 1, indicating the mediation of iron transport into cells of erythroid lineage that is necessary for proliferation.1 Bone marrow core biopsy did not identify features of bone marrow involvement by the lymphoma. Based on these results, the patient was diagnosed with systemic B-cell follicular lymphoma grade 3b stage IIIA. Oncology initiated a systemic chemotherapy regimen with obinutuzumab, cyclophosphamide, doxorubicin hydrochloride (hydroxydaunorubicin), vincristine sulfate, and prednisone.
Skin involvement in B-cell follicular lymphoma can be primary or secondary. Although all subtypes of BCL can have secondary cutaneous involvement, it is most common in advanced-stage disease (stages III or IV).2 Cutaneous manifestations of primary cutaneous follicle-center lymphoma (PCFCL) and systemic/nodal follicular lymphoma secondarily involving the skin can be difficult to distinguish clinically and histopathologically; both appear as solitary or grouped plaques and nodules most commonly on the head, neck, or trunk, and rarely on the legs.3 Although the pathologic features of these two diagnoses can seem almost identical, it is important to differentiate them due to their differing prognosis and management. Patients with follicular lymphoma involving the skin are more likely than those with PCFCL to develop lymphadenopathy and B symptoms.3 Primary cutaneous follicle-center lymphoma also generally runs an indolent course and requires local therapy, while secondary involvement of the skin due to systemic/nodal follicular lymphoma has a worse prognosis and requires systemic chemotherapy treatment.4
Immunohistochemical markers are the most helpful tool used to distinguish PCFCL from systemic/nodal follicular lymphoma involving the skin. Tumors of B-cell origin are expected to express associated B-cell markers such as CD20, CD79a, and PAX52; BCL-6, a marker of germinal center cells, also is expected to stain positive.2 CD10 is positive in a majority of cases with a follicular growth pattern, while those with a diffuse pattern of growth may have a negative stain.2 The most valuable histopathologic indicator differentiating primary and secondary skin involvement is the intensity of BCL-2 expression.5 The prognostic significance of the t(14;18)/IgH-BCL2 rearrangement is controversial, with rearrangement identified in more than 75% of systemic/nodal follicular lymphoma cases and less commonly found in PCFCL, with one report arguing an incidence ranging from 1% to 40%.5
A comprehensive history and physical examination are necessary to develop a differential diagnosis. Our patient's lower leg edema and extensive medical history made the diagnosis more complicated. Pitting edema was present on physical examination, making elephantiasis nostras verrucosa less likely, as it would instead present with nonpitting edema and a woody feel.6 Our patient did not have epidemiologic exposure to filariasis through foreign travel and did not present with any classic signs or symptoms of lymphatic filariasis, such as fever, eosinophilia, chyluria, or hydrocele.7 Although a negative history of HIV makes Kaposi sarcoma and bacillary angiomatosis less likely diagnoses, a biopsy would be useful to rule out these conditions. Positive inguinal lymphadenopathy present on physical examination may have contributed to lymphatic flow obstruction leading to the leg lymphedema in our patient.
- Marsee DK, Pinkus GS, Yu H. CD71 (transferrin receptor): an effective marker for erythroid precursors in bone marrow biopsy specimens. Am J Clin Pathol. 2010;134:429-435.
- Jaffe ES. Navigating the cutaneous B-cell lymphomas: avoiding the rocky shoals. Mod Pathol. 2020;33(suppl 1):96-106.
- Skala SL, Hristov B, Hristov AC. Primary cutaneous follicle center lymphoma. Arch Pathol Lab Med. 2018;142:1313-1321.
- Suárez AL, Pulitzer M, Horwitz S, et al. Primary cutaneous B-cell lymphomas: part I. clinical features, diagnosis, and classification. J Am Acad Dermatol. 2013;69:329.e1-13; quiz 341-342.
- Servitje O, Climent F, Colomo L, et al. Primary cutaneous vs secondary cutaneous follicular lymphomas: a comparative study focused on BCL2, CD10, and t(14;18) expression. J Cutan Pathol. 2018;46:182-189.
- Fredman R, Tenenhaus M. Elephantiasis nostras verrucose [published online October 12, 2012]. Eplasty. 2012;12:ic14.
- Lourens GB, Ferrell DK. Lymphatic filariasis. Nurs Clin of North Am. 2019;54:181-192.
The Diagnosis: Cutaneous B-cell Lymphoma
Histopathology revealed a dense and diffuse lymphocytic infiltrate throughout the dermis with occasional individual cell necrosis. On closer inspection, the infiltrate consisted of intermediate-sized lymphocytes, some with a vesiculated nucleus and ample amount of cytoplasm, while others contained hyperchromatic nuclei (Figure 1). These cells stained strongly positive for B-cell marker (CD20), while only a few mature lymphocytes demonstrated T-cell phenotype (CD3)(Figure 2).
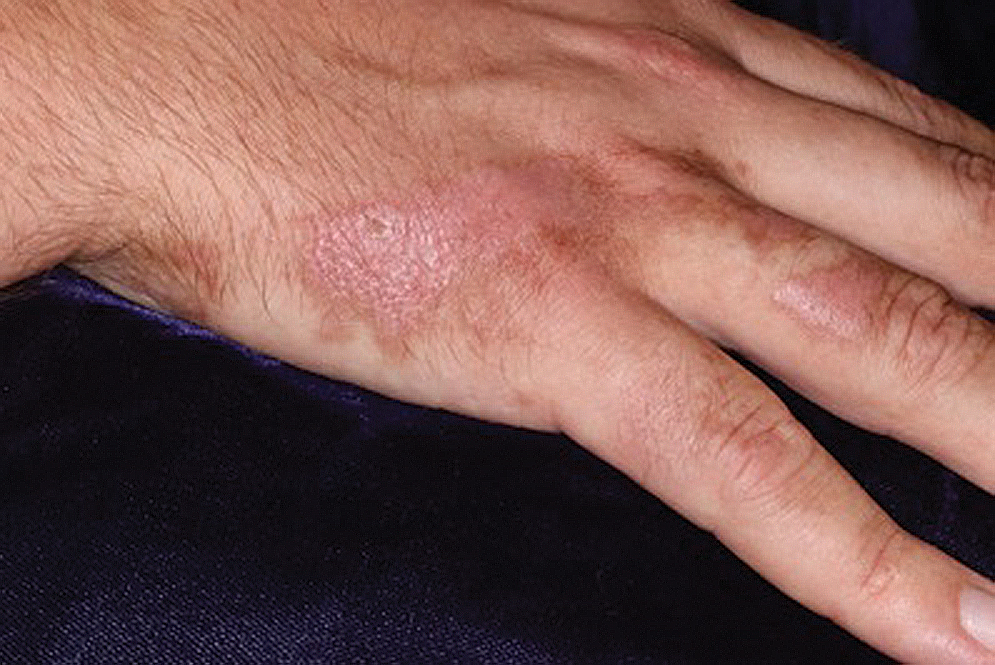
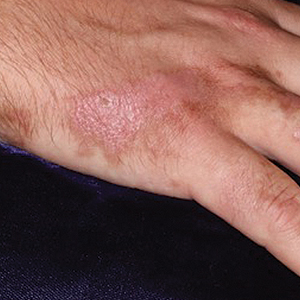
Although the patient recounted a 3-month history of lower leg edema, he also reported that the rash began a few weeks after his diagnosis of systemic B-cell follicular lymphoma. Throughout this time, he was seen by various physicians who attributed the edema and skin changes to chronic stasis, peripheral venous insufficiency, and diabetic peripheral neuropathy. His primary care physician prescribed an antifungal lotion, which he discontinued on his own due to lack of improvement. Upon arrival to the emergency department, he was started on intravenous cefazolin and subcutaneous heparin. Doppler ultrasonography of the legs was ordered to rule out a deep venous thrombosis. Dermatology was consulted and proceeded with a punch biopsy to investigate for cutaneous B-cell lymphoma (BCL) with a plan to follow up as an outpatient for results upon discharge. He also was prescribed triamcinolone ointment 0.1% twice daily for symptomatic relief.
The patient's left axillary lymph node was biopsied for pathologic evaluation. Immunohistochemical staining revealed expression of B-cell markers CD20, CD79a, and PAX5, along with the antiapoptotic markers BCL-2 and BCL-6. Fluorescence in situ hybridization displayed gene rearrangements of BCL-2, BCL-6, and t(14;18)/IgH-BCL2 in the majority of cells. CD3 and CD5 immunostains were negative, indicating that T cells were not involved in this process. Flow cytometry identified a monoclonal κ B-cell population in 40% to 50% of the total cells, which co-expressed CD10, CD19, CD22, and CD38; the cells were negative for CD5, CD20, and CD23. Cell size was variably enlarged and CD71 positive, otherwise known as transferrin receptor 1, indicating the mediation of iron transport into cells of erythroid lineage that is necessary for proliferation.1 Bone marrow core biopsy did not identify features of bone marrow involvement by the lymphoma. Based on these results, the patient was diagnosed with systemic B-cell follicular lymphoma grade 3b stage IIIA. Oncology initiated a systemic chemotherapy regimen with obinutuzumab, cyclophosphamide, doxorubicin hydrochloride (hydroxydaunorubicin), vincristine sulfate, and prednisone.
Skin involvement in B-cell follicular lymphoma can be primary or secondary. Although all subtypes of BCL can have secondary cutaneous involvement, it is most common in advanced-stage disease (stages III or IV).2 Cutaneous manifestations of primary cutaneous follicle-center lymphoma (PCFCL) and systemic/nodal follicular lymphoma secondarily involving the skin can be difficult to distinguish clinically and histopathologically; both appear as solitary or grouped plaques and nodules most commonly on the head, neck, or trunk, and rarely on the legs.3 Although the pathologic features of these two diagnoses can seem almost identical, it is important to differentiate them due to their differing prognosis and management. Patients with follicular lymphoma involving the skin are more likely than those with PCFCL to develop lymphadenopathy and B symptoms.3 Primary cutaneous follicle-center lymphoma also generally runs an indolent course and requires local therapy, while secondary involvement of the skin due to systemic/nodal follicular lymphoma has a worse prognosis and requires systemic chemotherapy treatment.4
Immunohistochemical markers are the most helpful tool used to distinguish PCFCL from systemic/nodal follicular lymphoma involving the skin. Tumors of B-cell origin are expected to express associated B-cell markers such as CD20, CD79a, and PAX52; BCL-6, a marker of germinal center cells, also is expected to stain positive.2 CD10 is positive in a majority of cases with a follicular growth pattern, while those with a diffuse pattern of growth may have a negative stain.2 The most valuable histopathologic indicator differentiating primary and secondary skin involvement is the intensity of BCL-2 expression.5 The prognostic significance of the t(14;18)/IgH-BCL2 rearrangement is controversial, with rearrangement identified in more than 75% of systemic/nodal follicular lymphoma cases and less commonly found in PCFCL, with one report arguing an incidence ranging from 1% to 40%.5
A comprehensive history and physical examination are necessary to develop a differential diagnosis. Our patient's lower leg edema and extensive medical history made the diagnosis more complicated. Pitting edema was present on physical examination, making elephantiasis nostras verrucosa less likely, as it would instead present with nonpitting edema and a woody feel.6 Our patient did not have epidemiologic exposure to filariasis through foreign travel and did not present with any classic signs or symptoms of lymphatic filariasis, such as fever, eosinophilia, chyluria, or hydrocele.7 Although a negative history of HIV makes Kaposi sarcoma and bacillary angiomatosis less likely diagnoses, a biopsy would be useful to rule out these conditions. Positive inguinal lymphadenopathy present on physical examination may have contributed to lymphatic flow obstruction leading to the leg lymphedema in our patient.
The Diagnosis: Cutaneous B-cell Lymphoma
Histopathology revealed a dense and diffuse lymphocytic infiltrate throughout the dermis with occasional individual cell necrosis. On closer inspection, the infiltrate consisted of intermediate-sized lymphocytes, some with a vesiculated nucleus and ample amount of cytoplasm, while others contained hyperchromatic nuclei (Figure 1). These cells stained strongly positive for B-cell marker (CD20), while only a few mature lymphocytes demonstrated T-cell phenotype (CD3)(Figure 2).
Although the patient recounted a 3-month history of lower leg edema, he also reported that the rash began a few weeks after his diagnosis of systemic B-cell follicular lymphoma. Throughout this time, he was seen by various physicians who attributed the edema and skin changes to chronic stasis, peripheral venous insufficiency, and diabetic peripheral neuropathy. His primary care physician prescribed an antifungal lotion, which he discontinued on his own due to lack of improvement. Upon arrival to the emergency department, he was started on intravenous cefazolin and subcutaneous heparin. Doppler ultrasonography of the legs was ordered to rule out a deep venous thrombosis. Dermatology was consulted and proceeded with a punch biopsy to investigate for cutaneous B-cell lymphoma (BCL) with a plan to follow up as an outpatient for results upon discharge. He also was prescribed triamcinolone ointment 0.1% twice daily for symptomatic relief.
The patient's left axillary lymph node was biopsied for pathologic evaluation. Immunohistochemical staining revealed expression of B-cell markers CD20, CD79a, and PAX5, along with the antiapoptotic markers BCL-2 and BCL-6. Fluorescence in situ hybridization displayed gene rearrangements of BCL-2, BCL-6, and t(14;18)/IgH-BCL2 in the majority of cells. CD3 and CD5 immunostains were negative, indicating that T cells were not involved in this process. Flow cytometry identified a monoclonal κ B-cell population in 40% to 50% of the total cells, which co-expressed CD10, CD19, CD22, and CD38; the cells were negative for CD5, CD20, and CD23. Cell size was variably enlarged and CD71 positive, otherwise known as transferrin receptor 1, indicating the mediation of iron transport into cells of erythroid lineage that is necessary for proliferation.1 Bone marrow core biopsy did not identify features of bone marrow involvement by the lymphoma. Based on these results, the patient was diagnosed with systemic B-cell follicular lymphoma grade 3b stage IIIA. Oncology initiated a systemic chemotherapy regimen with obinutuzumab, cyclophosphamide, doxorubicin hydrochloride (hydroxydaunorubicin), vincristine sulfate, and prednisone.
Skin involvement in B-cell follicular lymphoma can be primary or secondary. Although all subtypes of BCL can have secondary cutaneous involvement, it is most common in advanced-stage disease (stages III or IV).2 Cutaneous manifestations of primary cutaneous follicle-center lymphoma (PCFCL) and systemic/nodal follicular lymphoma secondarily involving the skin can be difficult to distinguish clinically and histopathologically; both appear as solitary or grouped plaques and nodules most commonly on the head, neck, or trunk, and rarely on the legs.3 Although the pathologic features of these two diagnoses can seem almost identical, it is important to differentiate them due to their differing prognosis and management. Patients with follicular lymphoma involving the skin are more likely than those with PCFCL to develop lymphadenopathy and B symptoms.3 Primary cutaneous follicle-center lymphoma also generally runs an indolent course and requires local therapy, while secondary involvement of the skin due to systemic/nodal follicular lymphoma has a worse prognosis and requires systemic chemotherapy treatment.4
Immunohistochemical markers are the most helpful tool used to distinguish PCFCL from systemic/nodal follicular lymphoma involving the skin. Tumors of B-cell origin are expected to express associated B-cell markers such as CD20, CD79a, and PAX52; BCL-6, a marker of germinal center cells, also is expected to stain positive.2 CD10 is positive in a majority of cases with a follicular growth pattern, while those with a diffuse pattern of growth may have a negative stain.2 The most valuable histopathologic indicator differentiating primary and secondary skin involvement is the intensity of BCL-2 expression.5 The prognostic significance of the t(14;18)/IgH-BCL2 rearrangement is controversial, with rearrangement identified in more than 75% of systemic/nodal follicular lymphoma cases and less commonly found in PCFCL, with one report arguing an incidence ranging from 1% to 40%.5
A comprehensive history and physical examination are necessary to develop a differential diagnosis. Our patient's lower leg edema and extensive medical history made the diagnosis more complicated. Pitting edema was present on physical examination, making elephantiasis nostras verrucosa less likely, as it would instead present with nonpitting edema and a woody feel.6 Our patient did not have epidemiologic exposure to filariasis through foreign travel and did not present with any classic signs or symptoms of lymphatic filariasis, such as fever, eosinophilia, chyluria, or hydrocele.7 Although a negative history of HIV makes Kaposi sarcoma and bacillary angiomatosis less likely diagnoses, a biopsy would be useful to rule out these conditions. Positive inguinal lymphadenopathy present on physical examination may have contributed to lymphatic flow obstruction leading to the leg lymphedema in our patient.
- Marsee DK, Pinkus GS, Yu H. CD71 (transferrin receptor): an effective marker for erythroid precursors in bone marrow biopsy specimens. Am J Clin Pathol. 2010;134:429-435.
- Jaffe ES. Navigating the cutaneous B-cell lymphomas: avoiding the rocky shoals. Mod Pathol. 2020;33(suppl 1):96-106.
- Skala SL, Hristov B, Hristov AC. Primary cutaneous follicle center lymphoma. Arch Pathol Lab Med. 2018;142:1313-1321.
- Suárez AL, Pulitzer M, Horwitz S, et al. Primary cutaneous B-cell lymphomas: part I. clinical features, diagnosis, and classification. J Am Acad Dermatol. 2013;69:329.e1-13; quiz 341-342.
- Servitje O, Climent F, Colomo L, et al. Primary cutaneous vs secondary cutaneous follicular lymphomas: a comparative study focused on BCL2, CD10, and t(14;18) expression. J Cutan Pathol. 2018;46:182-189.
- Fredman R, Tenenhaus M. Elephantiasis nostras verrucose [published online October 12, 2012]. Eplasty. 2012;12:ic14.
- Lourens GB, Ferrell DK. Lymphatic filariasis. Nurs Clin of North Am. 2019;54:181-192.
- Marsee DK, Pinkus GS, Yu H. CD71 (transferrin receptor): an effective marker for erythroid precursors in bone marrow biopsy specimens. Am J Clin Pathol. 2010;134:429-435.
- Jaffe ES. Navigating the cutaneous B-cell lymphomas: avoiding the rocky shoals. Mod Pathol. 2020;33(suppl 1):96-106.
- Skala SL, Hristov B, Hristov AC. Primary cutaneous follicle center lymphoma. Arch Pathol Lab Med. 2018;142:1313-1321.
- Suárez AL, Pulitzer M, Horwitz S, et al. Primary cutaneous B-cell lymphomas: part I. clinical features, diagnosis, and classification. J Am Acad Dermatol. 2013;69:329.e1-13; quiz 341-342.
- Servitje O, Climent F, Colomo L, et al. Primary cutaneous vs secondary cutaneous follicular lymphomas: a comparative study focused on BCL2, CD10, and t(14;18) expression. J Cutan Pathol. 2018;46:182-189.
- Fredman R, Tenenhaus M. Elephantiasis nostras verrucose [published online October 12, 2012]. Eplasty. 2012;12:ic14.
- Lourens GB, Ferrell DK. Lymphatic filariasis. Nurs Clin of North Am. 2019;54:181-192.
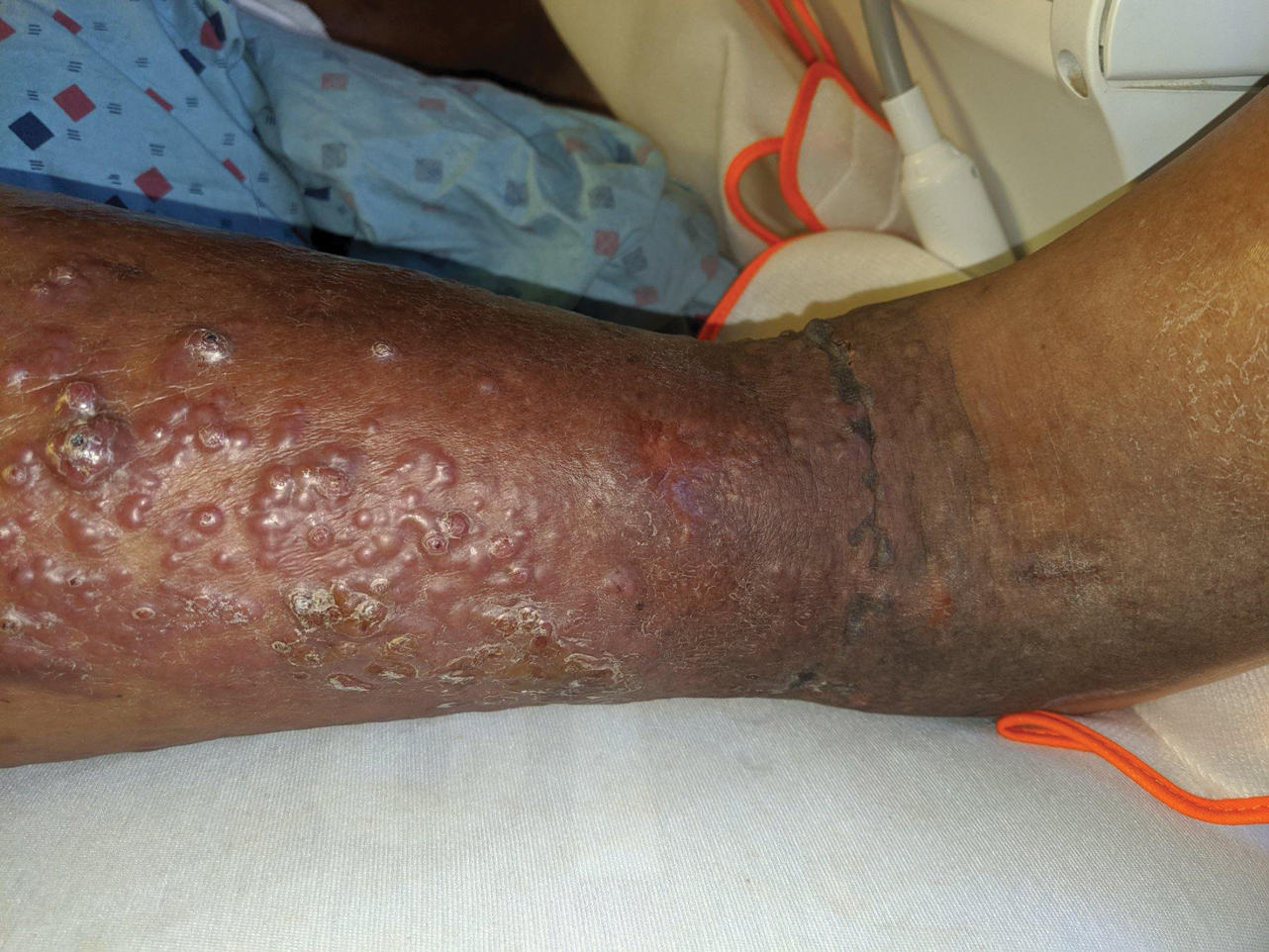
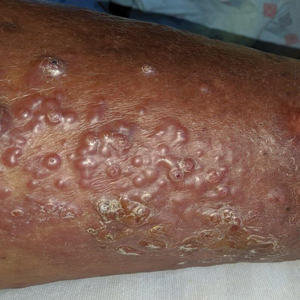
A 60-year-old man presented to the emergency department with slowly progressing edema of the lower legs of 3 months’ duration. In the week prior to presentation to the emergency department, he noticed a sudden eruption of vesicles and bullae on the right leg that drained clear fluid and healed with brown crust. The lesions were associated with mild burning, pruritus, and pain. He denied fever, chills, recent travel, or injury. His medical history was notable for poorly controlled diabetes mellitus, congestive heart failure, hypertension, chronic kidney disease, hyperlipidemia, and chronic anemia. Physical examination revealed multiple scattered erythematous vesicles and bullae on the right leg on a background of hyperpigmentation. Bilateral 2+ pitting edema of the legs also was present. A punch biopsy of a lesion was performed.
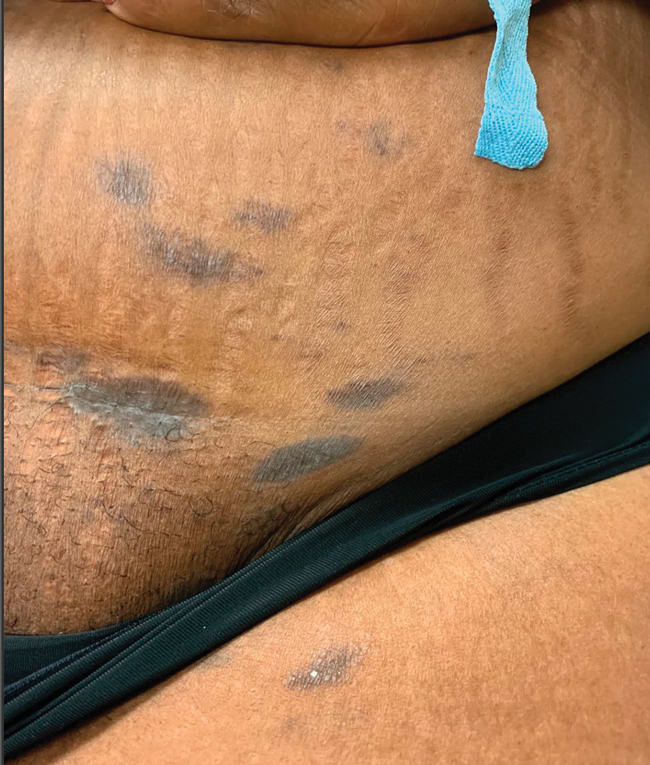
Asymptomatic Discolored Lesions on the Groin
The Diagnosis: Lichen Planus Pigmentosus-Inversus
Histopathologic examination revealed hyperkeratosis with dense, bandlike, lymphocytic inflammation at the dermoepidermal junction with associated melanin-containing macrophages in the papillary dermis (Figure 1). The physical examination and histopathology were consistent with a diagnosis of lichen planus pigmentosus-inversus (LPPI). Treatment was discussed with the patient, with options including phototherapy, tacrolimus, or a high-dose steroid. Given that the lesions were asymptomatic and not bothersome, the patient denied treatment and agreed to routine follow-up.
The first case of LPPI was reported in 20011; since then, approximately 100 cases have been reported in the literature.2 A rare variant of lichen planus, LPPI predominantly occurs in middle-aged women.2,3 Lichen planus pigmentosus-inversus is characterized by well-circumscribed, brown macules confined to non-sun-exposed intertriginous areas such as the axillae and groin.2 Although the rash remains localized, multiple lesions could arise in the same area, such as the groin as seen in our patient (Figure 2). Unlike in lichen planus, the oral mucosa, nails, and scalp are not affected. Furthermore, pruritus typically is absent in most cases of LPPI.2,4 Histopathologic findings include an atrophic epidermis with lichenoid infiltrates of lymphocytes and histocytes as well as substantial pigmentary incontinence with melanin-containing macrophages in the papillary dermis.4,5
Given the gender, age, and clinical features of our patient, this case represents a classic scenario of LPPI. It currently is unknown if ethnicity plays a role in the disorder. Lichen planus pigmentosus-inversus initially was thought to be more prevalent in White patients; however, studies have been reported in individuals with darker skin.1,2
The main differential diagnosis includes erythema dyschromicum perstans, postinflammatory hyperpigmentation, and lichen planus. Although erythema dyschromicum perstans develops in individuals with darker skin, lesions are restricted to the upper torso and limbs.2-4 In both lichen planus and lichen actinicus, skin findings primarily develop in sun-exposed areas, such as the face, neck, and hands.4,6 Given the negative history of trauma, postinflammatory hyperpigmentation was unlikely in our patient. Furthermore, a distinguishing characteristic of LPPI is the deposition of melanin deep within the dermal layer.3
Lesions developing in nonexposed intertriginous skin makes LPPI unique and distinguishes it from other more common conditions. The lesions commonly are hyperpigmented and are not as pruritic as other lichen-associated conditions. Lichen planus pigmentosus-inversus often persists for months, and the rash generally is resistant to treatment.2,5 Topical tacrolimus and high-dose steroids may improve symptoms, though results have varied substantially. In addition, some cases have resolved spontaneously.1,4,6,7 Because LPPI is asymptomatic and benign, spontaneous resolution and routine care is a reasonable treatment strategy. Some cases have supported this strategy as safe and high-value care.2
- Mohamed M, Korbi M, Hammedi F, et al. Lichen planus pigmentosus inversus: a series of 10 Tunisian patients. Int J Dermatol. 2016;55:1088-1091.
- Lichen planus pigmentosus-inversus: a rare variant of lichen planus. J Am Acad Dermatol. 2015;72(suppl 1):AB239. https://doi.org /10.1016/j.jaad.2015.02.959
- Chen S, Sun W, Zhou G, et al. Lichen planus pigmentosus-inversus: report of three Chinese cases and review of the published work. J Dermatol. 2015;42:77-80.
- Tabanlıoǧlu-Onan D, Íncel-Uysal P, Öktem A, et al. Lichen planus pigmentosus-inversus: a peculiar variant of lichen planus. Dermatologica Sinica. 2017;35:210-212.
- Barros HR, Almeida JR, Mattos e Dinato SL, et al. Lichen planus pigmentosus inversus. An Bras Dermatol. 2013;88(6 suppl 1):146-149.
- Bennàssar A, Mas A, Julià M, et al. Annular plaques in the skin folds: 4 cases of lichen planus pigmentosus-inversus [in Spanish]. Actas Dermosifiliogr. 2009;100:602-605.
- Ghorbel HH, Badri T, Ben Brahim E, et al. Lichen planus pigmentosus inversus. Indian J Dermatol Venereol Leprol. 2014;80:580.
The Diagnosis: Lichen Planus Pigmentosus-Inversus
Histopathologic examination revealed hyperkeratosis with dense, bandlike, lymphocytic inflammation at the dermoepidermal junction with associated melanin-containing macrophages in the papillary dermis (Figure 1). The physical examination and histopathology were consistent with a diagnosis of lichen planus pigmentosus-inversus (LPPI). Treatment was discussed with the patient, with options including phototherapy, tacrolimus, or a high-dose steroid. Given that the lesions were asymptomatic and not bothersome, the patient denied treatment and agreed to routine follow-up.
The first case of LPPI was reported in 20011; since then, approximately 100 cases have been reported in the literature.2 A rare variant of lichen planus, LPPI predominantly occurs in middle-aged women.2,3 Lichen planus pigmentosus-inversus is characterized by well-circumscribed, brown macules confined to non-sun-exposed intertriginous areas such as the axillae and groin.2 Although the rash remains localized, multiple lesions could arise in the same area, such as the groin as seen in our patient (Figure 2). Unlike in lichen planus, the oral mucosa, nails, and scalp are not affected. Furthermore, pruritus typically is absent in most cases of LPPI.2,4 Histopathologic findings include an atrophic epidermis with lichenoid infiltrates of lymphocytes and histocytes as well as substantial pigmentary incontinence with melanin-containing macrophages in the papillary dermis.4,5
Given the gender, age, and clinical features of our patient, this case represents a classic scenario of LPPI. It currently is unknown if ethnicity plays a role in the disorder. Lichen planus pigmentosus-inversus initially was thought to be more prevalent in White patients; however, studies have been reported in individuals with darker skin.1,2
The main differential diagnosis includes erythema dyschromicum perstans, postinflammatory hyperpigmentation, and lichen planus. Although erythema dyschromicum perstans develops in individuals with darker skin, lesions are restricted to the upper torso and limbs.2-4 In both lichen planus and lichen actinicus, skin findings primarily develop in sun-exposed areas, such as the face, neck, and hands.4,6 Given the negative history of trauma, postinflammatory hyperpigmentation was unlikely in our patient. Furthermore, a distinguishing characteristic of LPPI is the deposition of melanin deep within the dermal layer.3
Lesions developing in nonexposed intertriginous skin makes LPPI unique and distinguishes it from other more common conditions. The lesions commonly are hyperpigmented and are not as pruritic as other lichen-associated conditions. Lichen planus pigmentosus-inversus often persists for months, and the rash generally is resistant to treatment.2,5 Topical tacrolimus and high-dose steroids may improve symptoms, though results have varied substantially. In addition, some cases have resolved spontaneously.1,4,6,7 Because LPPI is asymptomatic and benign, spontaneous resolution and routine care is a reasonable treatment strategy. Some cases have supported this strategy as safe and high-value care.2
The Diagnosis: Lichen Planus Pigmentosus-Inversus
Histopathologic examination revealed hyperkeratosis with dense, bandlike, lymphocytic inflammation at the dermoepidermal junction with associated melanin-containing macrophages in the papillary dermis (Figure 1). The physical examination and histopathology were consistent with a diagnosis of lichen planus pigmentosus-inversus (LPPI). Treatment was discussed with the patient, with options including phototherapy, tacrolimus, or a high-dose steroid. Given that the lesions were asymptomatic and not bothersome, the patient denied treatment and agreed to routine follow-up.
The first case of LPPI was reported in 20011; since then, approximately 100 cases have been reported in the literature.2 A rare variant of lichen planus, LPPI predominantly occurs in middle-aged women.2,3 Lichen planus pigmentosus-inversus is characterized by well-circumscribed, brown macules confined to non-sun-exposed intertriginous areas such as the axillae and groin.2 Although the rash remains localized, multiple lesions could arise in the same area, such as the groin as seen in our patient (Figure 2). Unlike in lichen planus, the oral mucosa, nails, and scalp are not affected. Furthermore, pruritus typically is absent in most cases of LPPI.2,4 Histopathologic findings include an atrophic epidermis with lichenoid infiltrates of lymphocytes and histocytes as well as substantial pigmentary incontinence with melanin-containing macrophages in the papillary dermis.4,5
Given the gender, age, and clinical features of our patient, this case represents a classic scenario of LPPI. It currently is unknown if ethnicity plays a role in the disorder. Lichen planus pigmentosus-inversus initially was thought to be more prevalent in White patients; however, studies have been reported in individuals with darker skin.1,2
The main differential diagnosis includes erythema dyschromicum perstans, postinflammatory hyperpigmentation, and lichen planus. Although erythema dyschromicum perstans develops in individuals with darker skin, lesions are restricted to the upper torso and limbs.2-4 In both lichen planus and lichen actinicus, skin findings primarily develop in sun-exposed areas, such as the face, neck, and hands.4,6 Given the negative history of trauma, postinflammatory hyperpigmentation was unlikely in our patient. Furthermore, a distinguishing characteristic of LPPI is the deposition of melanin deep within the dermal layer.3
Lesions developing in nonexposed intertriginous skin makes LPPI unique and distinguishes it from other more common conditions. The lesions commonly are hyperpigmented and are not as pruritic as other lichen-associated conditions. Lichen planus pigmentosus-inversus often persists for months, and the rash generally is resistant to treatment.2,5 Topical tacrolimus and high-dose steroids may improve symptoms, though results have varied substantially. In addition, some cases have resolved spontaneously.1,4,6,7 Because LPPI is asymptomatic and benign, spontaneous resolution and routine care is a reasonable treatment strategy. Some cases have supported this strategy as safe and high-value care.2
- Mohamed M, Korbi M, Hammedi F, et al. Lichen planus pigmentosus inversus: a series of 10 Tunisian patients. Int J Dermatol. 2016;55:1088-1091.
- Lichen planus pigmentosus-inversus: a rare variant of lichen planus. J Am Acad Dermatol. 2015;72(suppl 1):AB239. https://doi.org /10.1016/j.jaad.2015.02.959
- Chen S, Sun W, Zhou G, et al. Lichen planus pigmentosus-inversus: report of three Chinese cases and review of the published work. J Dermatol. 2015;42:77-80.
- Tabanlıoǧlu-Onan D, Íncel-Uysal P, Öktem A, et al. Lichen planus pigmentosus-inversus: a peculiar variant of lichen planus. Dermatologica Sinica. 2017;35:210-212.
- Barros HR, Almeida JR, Mattos e Dinato SL, et al. Lichen planus pigmentosus inversus. An Bras Dermatol. 2013;88(6 suppl 1):146-149.
- Bennàssar A, Mas A, Julià M, et al. Annular plaques in the skin folds: 4 cases of lichen planus pigmentosus-inversus [in Spanish]. Actas Dermosifiliogr. 2009;100:602-605.
- Ghorbel HH, Badri T, Ben Brahim E, et al. Lichen planus pigmentosus inversus. Indian J Dermatol Venereol Leprol. 2014;80:580.
- Mohamed M, Korbi M, Hammedi F, et al. Lichen planus pigmentosus inversus: a series of 10 Tunisian patients. Int J Dermatol. 2016;55:1088-1091.
- Lichen planus pigmentosus-inversus: a rare variant of lichen planus. J Am Acad Dermatol. 2015;72(suppl 1):AB239. https://doi.org /10.1016/j.jaad.2015.02.959
- Chen S, Sun W, Zhou G, et al. Lichen planus pigmentosus-inversus: report of three Chinese cases and review of the published work. J Dermatol. 2015;42:77-80.
- Tabanlıoǧlu-Onan D, Íncel-Uysal P, Öktem A, et al. Lichen planus pigmentosus-inversus: a peculiar variant of lichen planus. Dermatologica Sinica. 2017;35:210-212.
- Barros HR, Almeida JR, Mattos e Dinato SL, et al. Lichen planus pigmentosus inversus. An Bras Dermatol. 2013;88(6 suppl 1):146-149.
- Bennàssar A, Mas A, Julià M, et al. Annular plaques in the skin folds: 4 cases of lichen planus pigmentosus-inversus [in Spanish]. Actas Dermosifiliogr. 2009;100:602-605.
- Ghorbel HH, Badri T, Ben Brahim E, et al. Lichen planus pigmentosus inversus. Indian J Dermatol Venereol Leprol. 2014;80:580.
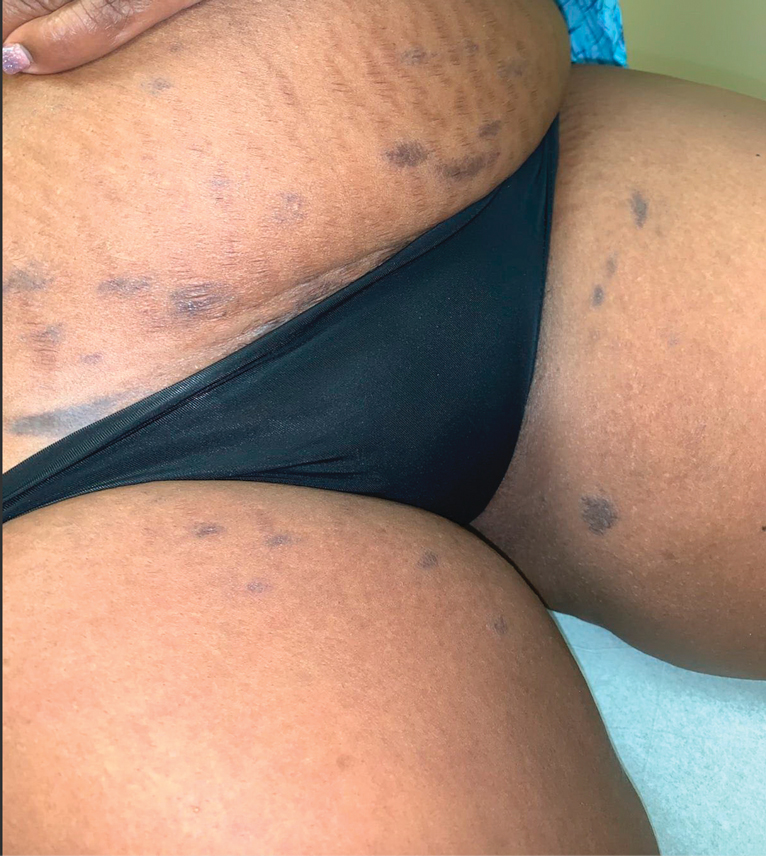
A 45-year-old African American woman presented with an asymptomatic rash that had worsened over the month prior to presentation. It initially began on the upper thighs and then spread to the abdomen, groin, and buttocks. The rash was mildly pruritic and had grown both in size and number of lesions. She had not tried any new over-the-counter medications. Her medical history was notable for late-stage breast cancer diagnosed 4 years prior that was treated with radiation and neoadjuvant NeoPACT—carboplatin, docetaxel, and pembrolizumab. One year prior to presentation, she underwent a lumpectomy that was complicated by gas gangrene of the finger. She has been in remission since the surgery. Physical examination at the current presentation was remarkable for multiple well-circumscribed, hyperpigmented macules on the medial thighs, lower abdomen, and buttocks. Syphilis antibody screening was negative.
Crusted Papules on the Bilateral Helices and Lobules
The Diagnosis: Kikuchi-Fujimoto Disease
A skin biopsy from the left helix was obtained. Histopathologic examination revealed a vacuolar interface reaction with marked papillary dermal edema and a patchy perijunctional lymphocytic infiltrate. The dermis was free of increased mucin (Figure 1). Immunohistochemical staining for CD56 and Epstein-Barr virus (EBV)–encoded small nuclear RNA chromogenic in situ hybridization were negative. Laboratory workup was remarkable for elevated transaminases and inflammatory markers (eg, C-reactive protein, erythrocyte sedimentation rate) but negative for rheumatologic markers (eg, antinuclear antibodies, antineutrophil cytoplasmic antibodies, myeloperoxidase antibodies, serine protease IgG). An extensive infectious workup was unrevealing. Computed tomography highlighted prominent lymphadenopathy throughout the cervical and supraclavicular chains and a large necrotic lymph node in the porta hepatis (Figure 2). Right neck lymph node aspiration revealed necrotizing lymphadenitis in a background of histiocytes and mixed lymphocytes. Coupling the clinical presentation and histomorphology with imaging, a diagnosis of Kikuchi-Fujimoto disease (KD) was rendered.

Kikuchi-Fujimoto disease is a rare illness of unknown etiology characterized by cervical lymphadenopathy and fever. Originally described in Japan, KD affects all racial and ethnic groups1,2 but more commonly is seen in women and patients younger than 40 years.3 It can be associated with systemic lupus erythematosus (SLE) and other autoimmune diseases (eg, relapsing polychondritis, adult-onset Still disease),3 and lymphoma.4 Multiple infections have been implicated in the pathogenesis of KD, including EBV and other human herpesviruses; HIV; human T-cell leukemia virus type 1; dengue virus; parvovirus B19; and Yersinia enterocolitica, Bartonella, Brucella, and Toxoplasma infections.3,5,6
Kikuchi-Fujimoto disease classically presents with fever and cervical lymphadenopathy. In a retrospective review of 244 patients with KD, the 3 most common manifestations included lymphadenopathy, fever, and rash.7 A diagnosis of KD is rendered based on clinical presentation and lymph node histopathologic findings of paracortical necrosis and florid histiocytic infiltrate.1
The cutaneous manifestations of KD are heterogeneous yet mostly transient. Cutaneous involvement is reported in 16.6% to 40% of patients.3,5,6 Common cutaneous manifestations include erythematous macules, papules, patches, and plaques; erosions, nodules, and bullae less commonly can occur.6 A variety of cutaneous manifestations have been reported in KD, including lesions mimicking pigmented purpuric dermatoses, vasculitis, Sweet syndrome, drug eruptions, and viral exanthems.6 Signs and symptoms of KD usually resolve within 1 to 4 months. Although there are no established treatments for this disease, patients with severe or persistent symptoms can be treated with steroids or hydroxychloroquine. Recurrences after treatment have been reported.8
Systemic lupus erythematosus is a multiorgan disease with protean manifestations. Cutaneous manifestations of SLE include malar erythema and discoid, annular, and papulosquamous lesions. Histopathologic patterns frequently observed in cutaneous lesions associated with SLE include interface dermatitis with perivascular infiltrates, dermal mucin, and plasmacytoid dendritic cells (marked by CD123 staining); these findings were notably absent in our case.6
Lupus vulgaris is a form of cutaneous tuberculosis that results from reactivation of Mycobacterium tuberculosis in tubercles formed during preceding hematogenous dissemination. The head and neck region is the most common location, particularly the nose, cheeks, and earlobes. Small, brown-red, soft papules coalesce into gelatinous plaques, demonstrating a characteristic apple jelly appearance on diascopy. Other clinical manifestations include the plaque/plane, hypertrophic/tumorlike, and ulcerative/scarring forms.9 Delayed-type hypersensitivity testing by tuberculin skin test, interferon-gamma release assay, or polymerase chain reaction–based assays can detect Mycobacterium tuberculosis. Histopathology shows well-formed granulomas surrounded by chronic inflammatory cells and central necrosis.
Hydroa vacciniforme–like (HV-like) eruption is a rare photosensitive disorder characterized by vesiculopapules on sun-exposed areas. Hydroa vacciniforme–like eruptions rarely have been reported to progress to EBVassociated malignant lymphoma.10 Unlike typical hydroa vacciniforme, which resolves by early adulthood, HV-like eruptions can become more severe with age and are associated with systemic manifestations, including fevers, lymphadenopathy, and liver damage. Histopathologic examination reveals a dense infiltrate of atypical T lymphocytes or natural killer cells (CD56+), which stain positive for EBV-encoded small nuclear RNA,10 in contrast to the patchy perijunctional lymphocytic infiltrate seen in KD.
This case highlights the protean cutaneous manifestations of a rare rheumatologic entity. It demonstrates the importance of a full systemic workup when considering an enigmatic disease. Our patient was started on prednisone 20 mg and hydroxychloroquine 200 mg daily. Within 24 hours, the fevers and rash both improved.
- Turner RR, Martin J, Dorfman RF. Necrotizing lymphadenitis. a study of 30 cases. Am J Surg Pathol. 1983;7:115-123.
- Dorfman RF, Berry GJ. Kikuchi’s histiocytic necrotizing lymphadenitis: an analysis of 108 cases with emphasis on differential diagnosis. Semin Diagn Pathol. 1988;5:329-345.
- Atwater AR, Longley BJ, Aughenbaugh WD. Kikuchi’s disease: case report and systematic review of cutaneous and histopathologic presentations. J Am Acad Dermatol. 2008;59:130-136.
- Yoshino T, Mannami T, Ichimura K, et al. Two cases of histiocytic necrotizing lymphadenitis (Kikuchi-Fujimoto’s disease) following diffuse large B-cell lymphoma. Hum Pathol. 2000;31:1328-1331.
- Yen A, Fearneyhough P, Raimer SS, et al. EBV-associated Kikuchi’s histiocytic necrotizing lymphadenitis with cutaneous manifestations. J Am Acad Dermatol. 1997;36:342-346.
- Kim JH, Kim YB, In SI, et al. The cutaneous lesions of Kikuchi’s disease: a comprehensive analysis of 16 cases based on the clinicopathologic, immunohistochemical, and immunofluorescence studies with an emphasis on the differential diagnosis. Hum Pathol. 2010;41:1245-1254.
- Kucukardali Y, Solmazgul E, Kunter E, et al. Kikuchi-Fujimoto Disease: analysis of 244 cases. Clin Rheumatol. 2007;26:50-54.
- Smith KG, Becker GJ, Busmanis I. Recurrent Kikuchi’s disease. Lancet. 1992;340:124.
- Macgregor R. Cutaneous tuberculosis. Clin Dermatol. 1995;13:245-255.
- Iwatsuki K, Ohtsuka M, Harada H, et al. Clinicopathologic manifestations of Epstein-Barr virus–associated cutaneous lymphoproliferative disorders. Arch Dermatol. 1997;133:1081-1086.
The Diagnosis: Kikuchi-Fujimoto Disease
A skin biopsy from the left helix was obtained. Histopathologic examination revealed a vacuolar interface reaction with marked papillary dermal edema and a patchy perijunctional lymphocytic infiltrate. The dermis was free of increased mucin (Figure 1). Immunohistochemical staining for CD56 and Epstein-Barr virus (EBV)–encoded small nuclear RNA chromogenic in situ hybridization were negative. Laboratory workup was remarkable for elevated transaminases and inflammatory markers (eg, C-reactive protein, erythrocyte sedimentation rate) but negative for rheumatologic markers (eg, antinuclear antibodies, antineutrophil cytoplasmic antibodies, myeloperoxidase antibodies, serine protease IgG). An extensive infectious workup was unrevealing. Computed tomography highlighted prominent lymphadenopathy throughout the cervical and supraclavicular chains and a large necrotic lymph node in the porta hepatis (Figure 2). Right neck lymph node aspiration revealed necrotizing lymphadenitis in a background of histiocytes and mixed lymphocytes. Coupling the clinical presentation and histomorphology with imaging, a diagnosis of Kikuchi-Fujimoto disease (KD) was rendered.
Kikuchi-Fujimoto disease is a rare illness of unknown etiology characterized by cervical lymphadenopathy and fever. Originally described in Japan, KD affects all racial and ethnic groups1,2 but more commonly is seen in women and patients younger than 40 years.3 It can be associated with systemic lupus erythematosus (SLE) and other autoimmune diseases (eg, relapsing polychondritis, adult-onset Still disease),3 and lymphoma.4 Multiple infections have been implicated in the pathogenesis of KD, including EBV and other human herpesviruses; HIV; human T-cell leukemia virus type 1; dengue virus; parvovirus B19; and Yersinia enterocolitica, Bartonella, Brucella, and Toxoplasma infections.3,5,6
Kikuchi-Fujimoto disease classically presents with fever and cervical lymphadenopathy. In a retrospective review of 244 patients with KD, the 3 most common manifestations included lymphadenopathy, fever, and rash.7 A diagnosis of KD is rendered based on clinical presentation and lymph node histopathologic findings of paracortical necrosis and florid histiocytic infiltrate.1
The cutaneous manifestations of KD are heterogeneous yet mostly transient. Cutaneous involvement is reported in 16.6% to 40% of patients.3,5,6 Common cutaneous manifestations include erythematous macules, papules, patches, and plaques; erosions, nodules, and bullae less commonly can occur.6 A variety of cutaneous manifestations have been reported in KD, including lesions mimicking pigmented purpuric dermatoses, vasculitis, Sweet syndrome, drug eruptions, and viral exanthems.6 Signs and symptoms of KD usually resolve within 1 to 4 months. Although there are no established treatments for this disease, patients with severe or persistent symptoms can be treated with steroids or hydroxychloroquine. Recurrences after treatment have been reported.8
Systemic lupus erythematosus is a multiorgan disease with protean manifestations. Cutaneous manifestations of SLE include malar erythema and discoid, annular, and papulosquamous lesions. Histopathologic patterns frequently observed in cutaneous lesions associated with SLE include interface dermatitis with perivascular infiltrates, dermal mucin, and plasmacytoid dendritic cells (marked by CD123 staining); these findings were notably absent in our case.6
Lupus vulgaris is a form of cutaneous tuberculosis that results from reactivation of Mycobacterium tuberculosis in tubercles formed during preceding hematogenous dissemination. The head and neck region is the most common location, particularly the nose, cheeks, and earlobes. Small, brown-red, soft papules coalesce into gelatinous plaques, demonstrating a characteristic apple jelly appearance on diascopy. Other clinical manifestations include the plaque/plane, hypertrophic/tumorlike, and ulcerative/scarring forms.9 Delayed-type hypersensitivity testing by tuberculin skin test, interferon-gamma release assay, or polymerase chain reaction–based assays can detect Mycobacterium tuberculosis. Histopathology shows well-formed granulomas surrounded by chronic inflammatory cells and central necrosis.
Hydroa vacciniforme–like (HV-like) eruption is a rare photosensitive disorder characterized by vesiculopapules on sun-exposed areas. Hydroa vacciniforme–like eruptions rarely have been reported to progress to EBVassociated malignant lymphoma.10 Unlike typical hydroa vacciniforme, which resolves by early adulthood, HV-like eruptions can become more severe with age and are associated with systemic manifestations, including fevers, lymphadenopathy, and liver damage. Histopathologic examination reveals a dense infiltrate of atypical T lymphocytes or natural killer cells (CD56+), which stain positive for EBV-encoded small nuclear RNA,10 in contrast to the patchy perijunctional lymphocytic infiltrate seen in KD.
This case highlights the protean cutaneous manifestations of a rare rheumatologic entity. It demonstrates the importance of a full systemic workup when considering an enigmatic disease. Our patient was started on prednisone 20 mg and hydroxychloroquine 200 mg daily. Within 24 hours, the fevers and rash both improved.
The Diagnosis: Kikuchi-Fujimoto Disease
A skin biopsy from the left helix was obtained. Histopathologic examination revealed a vacuolar interface reaction with marked papillary dermal edema and a patchy perijunctional lymphocytic infiltrate. The dermis was free of increased mucin (Figure 1). Immunohistochemical staining for CD56 and Epstein-Barr virus (EBV)–encoded small nuclear RNA chromogenic in situ hybridization were negative. Laboratory workup was remarkable for elevated transaminases and inflammatory markers (eg, C-reactive protein, erythrocyte sedimentation rate) but negative for rheumatologic markers (eg, antinuclear antibodies, antineutrophil cytoplasmic antibodies, myeloperoxidase antibodies, serine protease IgG). An extensive infectious workup was unrevealing. Computed tomography highlighted prominent lymphadenopathy throughout the cervical and supraclavicular chains and a large necrotic lymph node in the porta hepatis (Figure 2). Right neck lymph node aspiration revealed necrotizing lymphadenitis in a background of histiocytes and mixed lymphocytes. Coupling the clinical presentation and histomorphology with imaging, a diagnosis of Kikuchi-Fujimoto disease (KD) was rendered.
Kikuchi-Fujimoto disease is a rare illness of unknown etiology characterized by cervical lymphadenopathy and fever. Originally described in Japan, KD affects all racial and ethnic groups1,2 but more commonly is seen in women and patients younger than 40 years.3 It can be associated with systemic lupus erythematosus (SLE) and other autoimmune diseases (eg, relapsing polychondritis, adult-onset Still disease),3 and lymphoma.4 Multiple infections have been implicated in the pathogenesis of KD, including EBV and other human herpesviruses; HIV; human T-cell leukemia virus type 1; dengue virus; parvovirus B19; and Yersinia enterocolitica, Bartonella, Brucella, and Toxoplasma infections.3,5,6
Kikuchi-Fujimoto disease classically presents with fever and cervical lymphadenopathy. In a retrospective review of 244 patients with KD, the 3 most common manifestations included lymphadenopathy, fever, and rash.7 A diagnosis of KD is rendered based on clinical presentation and lymph node histopathologic findings of paracortical necrosis and florid histiocytic infiltrate.1
The cutaneous manifestations of KD are heterogeneous yet mostly transient. Cutaneous involvement is reported in 16.6% to 40% of patients.3,5,6 Common cutaneous manifestations include erythematous macules, papules, patches, and plaques; erosions, nodules, and bullae less commonly can occur.6 A variety of cutaneous manifestations have been reported in KD, including lesions mimicking pigmented purpuric dermatoses, vasculitis, Sweet syndrome, drug eruptions, and viral exanthems.6 Signs and symptoms of KD usually resolve within 1 to 4 months. Although there are no established treatments for this disease, patients with severe or persistent symptoms can be treated with steroids or hydroxychloroquine. Recurrences after treatment have been reported.8
Systemic lupus erythematosus is a multiorgan disease with protean manifestations. Cutaneous manifestations of SLE include malar erythema and discoid, annular, and papulosquamous lesions. Histopathologic patterns frequently observed in cutaneous lesions associated with SLE include interface dermatitis with perivascular infiltrates, dermal mucin, and plasmacytoid dendritic cells (marked by CD123 staining); these findings were notably absent in our case.6
Lupus vulgaris is a form of cutaneous tuberculosis that results from reactivation of Mycobacterium tuberculosis in tubercles formed during preceding hematogenous dissemination. The head and neck region is the most common location, particularly the nose, cheeks, and earlobes. Small, brown-red, soft papules coalesce into gelatinous plaques, demonstrating a characteristic apple jelly appearance on diascopy. Other clinical manifestations include the plaque/plane, hypertrophic/tumorlike, and ulcerative/scarring forms.9 Delayed-type hypersensitivity testing by tuberculin skin test, interferon-gamma release assay, or polymerase chain reaction–based assays can detect Mycobacterium tuberculosis. Histopathology shows well-formed granulomas surrounded by chronic inflammatory cells and central necrosis.
Hydroa vacciniforme–like (HV-like) eruption is a rare photosensitive disorder characterized by vesiculopapules on sun-exposed areas. Hydroa vacciniforme–like eruptions rarely have been reported to progress to EBVassociated malignant lymphoma.10 Unlike typical hydroa vacciniforme, which resolves by early adulthood, HV-like eruptions can become more severe with age and are associated with systemic manifestations, including fevers, lymphadenopathy, and liver damage. Histopathologic examination reveals a dense infiltrate of atypical T lymphocytes or natural killer cells (CD56+), which stain positive for EBV-encoded small nuclear RNA,10 in contrast to the patchy perijunctional lymphocytic infiltrate seen in KD.
This case highlights the protean cutaneous manifestations of a rare rheumatologic entity. It demonstrates the importance of a full systemic workup when considering an enigmatic disease. Our patient was started on prednisone 20 mg and hydroxychloroquine 200 mg daily. Within 24 hours, the fevers and rash both improved.
- Turner RR, Martin J, Dorfman RF. Necrotizing lymphadenitis. a study of 30 cases. Am J Surg Pathol. 1983;7:115-123.
- Dorfman RF, Berry GJ. Kikuchi’s histiocytic necrotizing lymphadenitis: an analysis of 108 cases with emphasis on differential diagnosis. Semin Diagn Pathol. 1988;5:329-345.
- Atwater AR, Longley BJ, Aughenbaugh WD. Kikuchi’s disease: case report and systematic review of cutaneous and histopathologic presentations. J Am Acad Dermatol. 2008;59:130-136.
- Yoshino T, Mannami T, Ichimura K, et al. Two cases of histiocytic necrotizing lymphadenitis (Kikuchi-Fujimoto’s disease) following diffuse large B-cell lymphoma. Hum Pathol. 2000;31:1328-1331.
- Yen A, Fearneyhough P, Raimer SS, et al. EBV-associated Kikuchi’s histiocytic necrotizing lymphadenitis with cutaneous manifestations. J Am Acad Dermatol. 1997;36:342-346.
- Kim JH, Kim YB, In SI, et al. The cutaneous lesions of Kikuchi’s disease: a comprehensive analysis of 16 cases based on the clinicopathologic, immunohistochemical, and immunofluorescence studies with an emphasis on the differential diagnosis. Hum Pathol. 2010;41:1245-1254.
- Kucukardali Y, Solmazgul E, Kunter E, et al. Kikuchi-Fujimoto Disease: analysis of 244 cases. Clin Rheumatol. 2007;26:50-54.
- Smith KG, Becker GJ, Busmanis I. Recurrent Kikuchi’s disease. Lancet. 1992;340:124.
- Macgregor R. Cutaneous tuberculosis. Clin Dermatol. 1995;13:245-255.
- Iwatsuki K, Ohtsuka M, Harada H, et al. Clinicopathologic manifestations of Epstein-Barr virus–associated cutaneous lymphoproliferative disorders. Arch Dermatol. 1997;133:1081-1086.
- Turner RR, Martin J, Dorfman RF. Necrotizing lymphadenitis. a study of 30 cases. Am J Surg Pathol. 1983;7:115-123.
- Dorfman RF, Berry GJ. Kikuchi’s histiocytic necrotizing lymphadenitis: an analysis of 108 cases with emphasis on differential diagnosis. Semin Diagn Pathol. 1988;5:329-345.
- Atwater AR, Longley BJ, Aughenbaugh WD. Kikuchi’s disease: case report and systematic review of cutaneous and histopathologic presentations. J Am Acad Dermatol. 2008;59:130-136.
- Yoshino T, Mannami T, Ichimura K, et al. Two cases of histiocytic necrotizing lymphadenitis (Kikuchi-Fujimoto’s disease) following diffuse large B-cell lymphoma. Hum Pathol. 2000;31:1328-1331.
- Yen A, Fearneyhough P, Raimer SS, et al. EBV-associated Kikuchi’s histiocytic necrotizing lymphadenitis with cutaneous manifestations. J Am Acad Dermatol. 1997;36:342-346.
- Kim JH, Kim YB, In SI, et al. The cutaneous lesions of Kikuchi’s disease: a comprehensive analysis of 16 cases based on the clinicopathologic, immunohistochemical, and immunofluorescence studies with an emphasis on the differential diagnosis. Hum Pathol. 2010;41:1245-1254.
- Kucukardali Y, Solmazgul E, Kunter E, et al. Kikuchi-Fujimoto Disease: analysis of 244 cases. Clin Rheumatol. 2007;26:50-54.
- Smith KG, Becker GJ, Busmanis I. Recurrent Kikuchi’s disease. Lancet. 1992;340:124.
- Macgregor R. Cutaneous tuberculosis. Clin Dermatol. 1995;13:245-255.
- Iwatsuki K, Ohtsuka M, Harada H, et al. Clinicopathologic manifestations of Epstein-Barr virus–associated cutaneous lymphoproliferative disorders. Arch Dermatol. 1997;133:1081-1086.
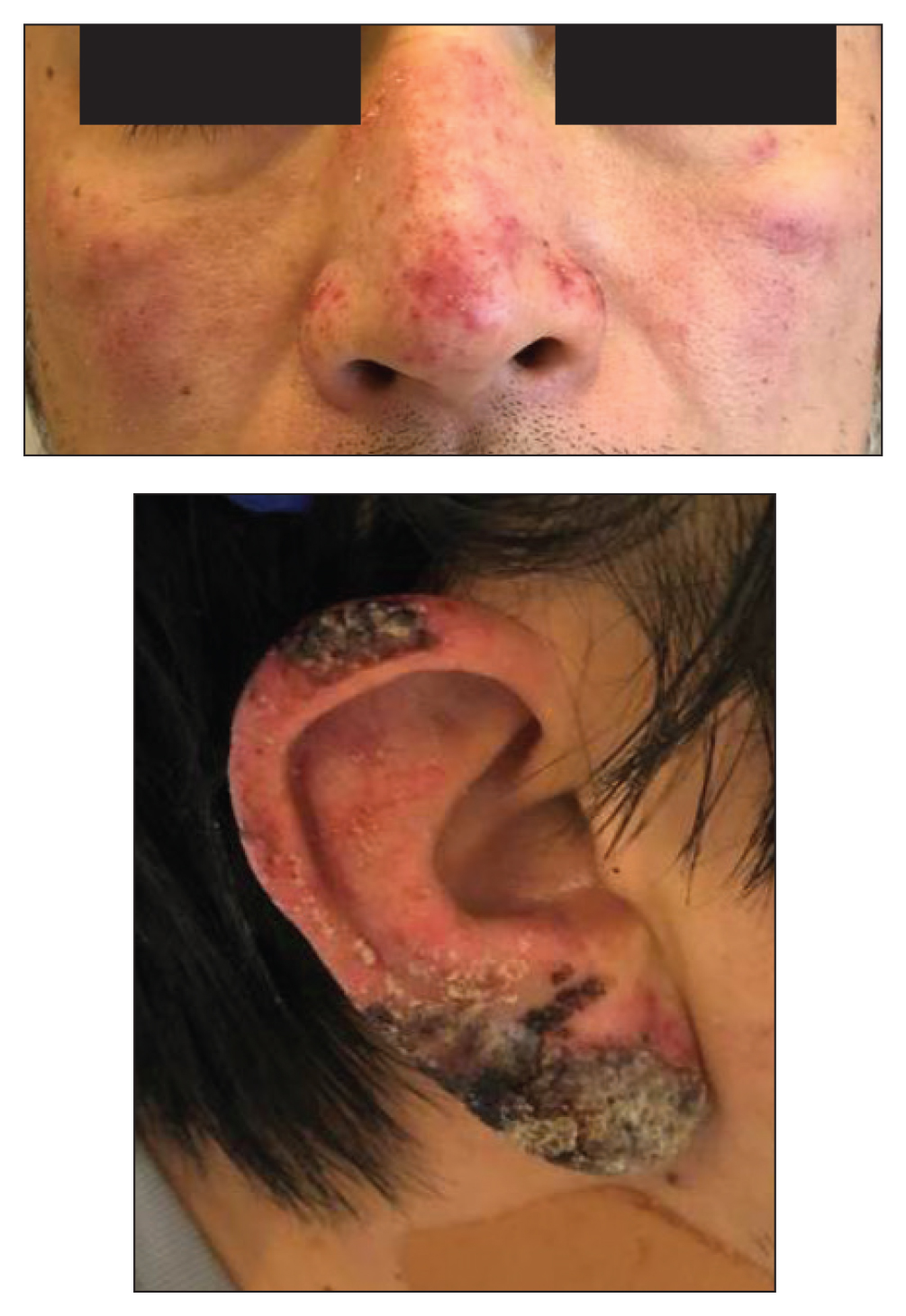
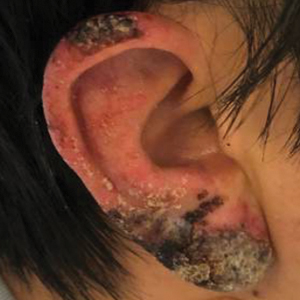
A healthy 42-year-old Japanese man presented with painful lymphadenopathy and fevers of 1 month’s duration as well as a pruritic rash and bilateral ear redness and crusting of 1 week’s duration. He initially was seen at an outside facility and was treated with antibiotics and supportive care for cervical adenitis. During clinical evaluation, he denied joint pain, photosensitivity, and oral lesions. His medical and family history were noncontributory. Although he reported recent travel to multiple countries, he denied exposure to animals, ticks, or sick individuals. Physical examination revealed erythematous blanching papules on the nose and cheeks (top) as well as crusted papules coalescing into plaques on the bilateral helices and lobules (bottom).
Thick Hyperkeratotic Plaques on the Palms and Soles
The Diagnosis: Keratoderma Climactericum
Keratoderma climactericum was first reported in 1934 by Haxthausen1 as nonpruritic circumscribed hyperkeratosis located mainly on the palms and soles. The initial eruption was described as discrete lesions with an oval or round shape that progressed to less-defined, confluent, hyperkeratotic patches with fissures.1 Keratoderma climactericum also may be referred to as Haxthausen disease and is considered an acquired palmoplantar keratoderma.2
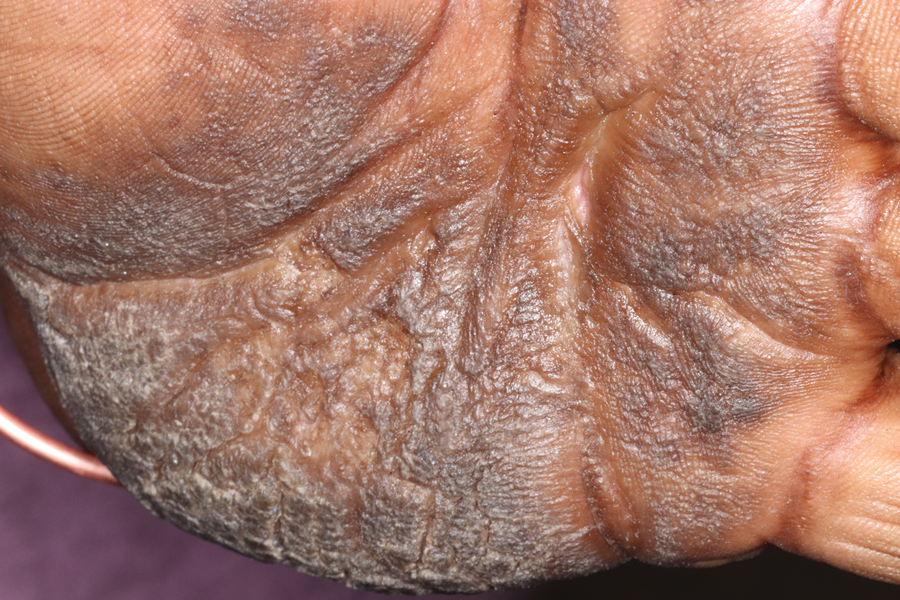
Keratoderma climactericum is a rare dermatologic disorder that presents in women of menopausal age who have no family or personal history of skin disease. Keratoderma climactericum is associated with hypertension and obesity.2 Keratotic lesions usually first occur on the plantar surfaces with eventual development of fissuring and hyperkeratosis that causes painful walking. The keratotic lesions on the plantar surfaces often are nonpruritic and gradually become confluent over time. As the disease progresses, keratotic lesions appear on the central palms, which can lead to confluent hyperkeratosis on the palmar surfaces (Figure 1).2 The exact mechanism of keratoderma climactericum has not been described but is believed to be due to hormonal dysregulation.2
In 1986, Deschamps et al3 presented 10 cases of keratoderma climactericum occurring in menopausal women with an average age of 57 years. The lesions began on the soles at areas of greatest pressure. Histopathology for each patient showed orthokeratotic hyperkeratosis, irregular hyperplasia, interpapillary ridges, and exocytosis of lymphocytes in the epidermis. Seven patients were treated with etretinate, which first led to the removal of palmar lesions, followed by improvement in plantar lesions and pain when walking. There was no association of keratoderma climactericum and sex hormones, as hormone levels were negative or normal for each patient.3
Three cases of keratoderma climactericum following bilateral oophorectomy in young women were reported by Wachtel4 in 1981. Unlike in women of menopausal age, there was no association of keratoderma climactericum with hypertension or obesity. Additionally, the lesions on the palms and soles were more diffusely distributed than in women of menopausal age. Estrogen administration completely reversed each patient's hyperkeratotic palms and soles.4 A definitive pathogenic role of estrogens in the development of keratoderma climactericum has yet to be determined.2
Histopathology is not specific for keratoderma climactericum, making the disease a clinical diagnosis. However, a biopsy may be useful to rule out palmoplantar psoriasis.2 Clinical information such as the age and sex of the patient, distribution of disease, presence of fissuring, and progression of disease from soles to palms should be considered when making a diagnosis of keratoderma climactericum. The differential diagnosis of keratoderma climactericum should include fungal infections, contact dermatitis, irritant dermatitis, psoriasis, atopic dermatitis, underlying malignancy, and pityriasis rubra pilaris.
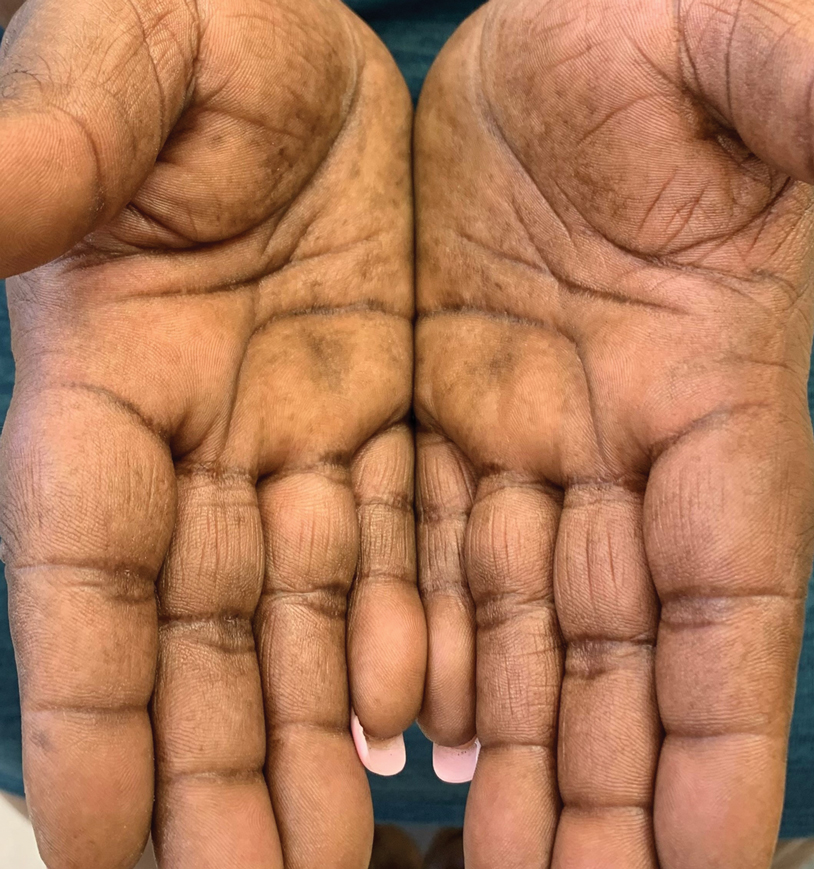
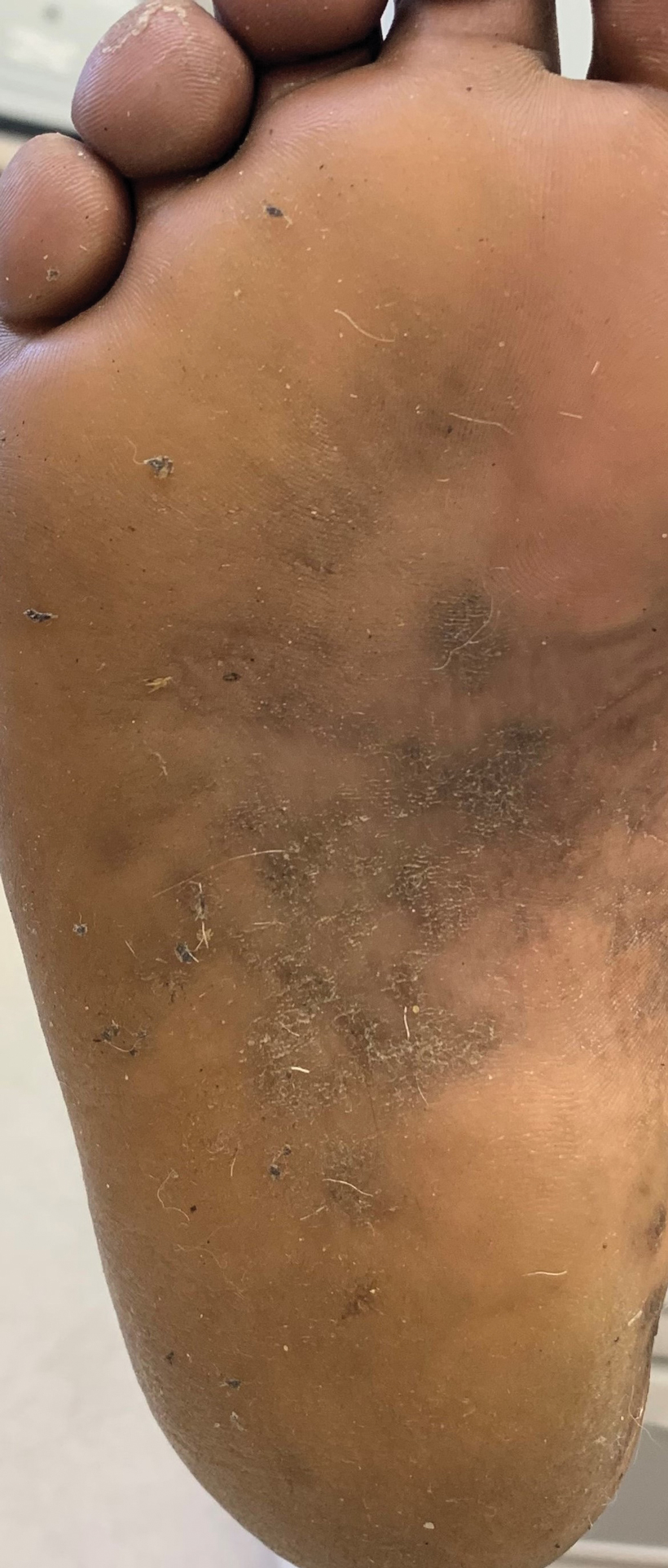
Treatment options for keratoderma climactericum include salicylic acid, emollients, oral retinoids, urea ointments, estriol cream, and topical steroids.5,6 Our patient was prescribed acitretin 25 mg daily and ammonium lactate to apply topically as needed for dry skin. Five months after the initial presentation, fissures and dry skin on the bilateral soles were still present. Ammonium lactate was discontinued, and the patient was prescribed urea cream 40%. Fifteen months after the initial presentation, the patient reported substantial improvement on the hands and feet and noted that she no longer needed the urea cream. Physical examination revealed no presence of hyperkeratosis or fissuring on the palms (Figure 2), and mild hyperkeratosis was present on the plantar surfaces of the feet (Figure 3). The patient continued to use acitretin to prevent disease relapse.
Keratoderma climactericum is an unusual and debilitating condition that occurs in women of menopausal age. It is diagnosed by its specific clinical presentation. More common diagnoses such as tinea and dermatitis should be ruled out before considering keratoderma climactericum.
- Haxthausen H. Keratoderma climactericum. Br J Dermatol. 1934;46:161-167.
- Patel S, Zirwas M, English JC. Acquired palmoplantar keratoderma. Am J Clin Dermatol. 2007;8:1-11.
- Deschamps P, Leroy D, Pedailles S, et al. Keratoderma climactericum (Haxthausen's disease): clinical signs, laboratory findings and etretinate treatment in 10 patients. Dermatologica. 1986;172:258-262.
- Wachtel TJ. Plantar and palmar hyperkeratosis in young castrated women. Int J Dermatol. 1981;20:270-271.
- Bristow I. The management of heel fissures using a steroid impregnated tape (Haelan) in a patient with Keratoderma climactericum. Podiatry Now. 2008;11:22-23.
- Mendes-Bastos P. Plantar keratoderma climactericum: successful improvement with a topical estriol cream. J Cosmet Dermatol. 2018;17:811-813.
The Diagnosis: Keratoderma Climactericum
Keratoderma climactericum was first reported in 1934 by Haxthausen1 as nonpruritic circumscribed hyperkeratosis located mainly on the palms and soles. The initial eruption was described as discrete lesions with an oval or round shape that progressed to less-defined, confluent, hyperkeratotic patches with fissures.1 Keratoderma climactericum also may be referred to as Haxthausen disease and is considered an acquired palmoplantar keratoderma.2
Keratoderma climactericum is a rare dermatologic disorder that presents in women of menopausal age who have no family or personal history of skin disease. Keratoderma climactericum is associated with hypertension and obesity.2 Keratotic lesions usually first occur on the plantar surfaces with eventual development of fissuring and hyperkeratosis that causes painful walking. The keratotic lesions on the plantar surfaces often are nonpruritic and gradually become confluent over time. As the disease progresses, keratotic lesions appear on the central palms, which can lead to confluent hyperkeratosis on the palmar surfaces (Figure 1).2 The exact mechanism of keratoderma climactericum has not been described but is believed to be due to hormonal dysregulation.2
In 1986, Deschamps et al3 presented 10 cases of keratoderma climactericum occurring in menopausal women with an average age of 57 years. The lesions began on the soles at areas of greatest pressure. Histopathology for each patient showed orthokeratotic hyperkeratosis, irregular hyperplasia, interpapillary ridges, and exocytosis of lymphocytes in the epidermis. Seven patients were treated with etretinate, which first led to the removal of palmar lesions, followed by improvement in plantar lesions and pain when walking. There was no association of keratoderma climactericum and sex hormones, as hormone levels were negative or normal for each patient.3
Three cases of keratoderma climactericum following bilateral oophorectomy in young women were reported by Wachtel4 in 1981. Unlike in women of menopausal age, there was no association of keratoderma climactericum with hypertension or obesity. Additionally, the lesions on the palms and soles were more diffusely distributed than in women of menopausal age. Estrogen administration completely reversed each patient's hyperkeratotic palms and soles.4 A definitive pathogenic role of estrogens in the development of keratoderma climactericum has yet to be determined.2
Histopathology is not specific for keratoderma climactericum, making the disease a clinical diagnosis. However, a biopsy may be useful to rule out palmoplantar psoriasis.2 Clinical information such as the age and sex of the patient, distribution of disease, presence of fissuring, and progression of disease from soles to palms should be considered when making a diagnosis of keratoderma climactericum. The differential diagnosis of keratoderma climactericum should include fungal infections, contact dermatitis, irritant dermatitis, psoriasis, atopic dermatitis, underlying malignancy, and pityriasis rubra pilaris.
Treatment options for keratoderma climactericum include salicylic acid, emollients, oral retinoids, urea ointments, estriol cream, and topical steroids.5,6 Our patient was prescribed acitretin 25 mg daily and ammonium lactate to apply topically as needed for dry skin. Five months after the initial presentation, fissures and dry skin on the bilateral soles were still present. Ammonium lactate was discontinued, and the patient was prescribed urea cream 40%. Fifteen months after the initial presentation, the patient reported substantial improvement on the hands and feet and noted that she no longer needed the urea cream. Physical examination revealed no presence of hyperkeratosis or fissuring on the palms (Figure 2), and mild hyperkeratosis was present on the plantar surfaces of the feet (Figure 3). The patient continued to use acitretin to prevent disease relapse.
Keratoderma climactericum is an unusual and debilitating condition that occurs in women of menopausal age. It is diagnosed by its specific clinical presentation. More common diagnoses such as tinea and dermatitis should be ruled out before considering keratoderma climactericum.
The Diagnosis: Keratoderma Climactericum
Keratoderma climactericum was first reported in 1934 by Haxthausen1 as nonpruritic circumscribed hyperkeratosis located mainly on the palms and soles. The initial eruption was described as discrete lesions with an oval or round shape that progressed to less-defined, confluent, hyperkeratotic patches with fissures.1 Keratoderma climactericum also may be referred to as Haxthausen disease and is considered an acquired palmoplantar keratoderma.2
Keratoderma climactericum is a rare dermatologic disorder that presents in women of menopausal age who have no family or personal history of skin disease. Keratoderma climactericum is associated with hypertension and obesity.2 Keratotic lesions usually first occur on the plantar surfaces with eventual development of fissuring and hyperkeratosis that causes painful walking. The keratotic lesions on the plantar surfaces often are nonpruritic and gradually become confluent over time. As the disease progresses, keratotic lesions appear on the central palms, which can lead to confluent hyperkeratosis on the palmar surfaces (Figure 1).2 The exact mechanism of keratoderma climactericum has not been described but is believed to be due to hormonal dysregulation.2
In 1986, Deschamps et al3 presented 10 cases of keratoderma climactericum occurring in menopausal women with an average age of 57 years. The lesions began on the soles at areas of greatest pressure. Histopathology for each patient showed orthokeratotic hyperkeratosis, irregular hyperplasia, interpapillary ridges, and exocytosis of lymphocytes in the epidermis. Seven patients were treated with etretinate, which first led to the removal of palmar lesions, followed by improvement in plantar lesions and pain when walking. There was no association of keratoderma climactericum and sex hormones, as hormone levels were negative or normal for each patient.3
Three cases of keratoderma climactericum following bilateral oophorectomy in young women were reported by Wachtel4 in 1981. Unlike in women of menopausal age, there was no association of keratoderma climactericum with hypertension or obesity. Additionally, the lesions on the palms and soles were more diffusely distributed than in women of menopausal age. Estrogen administration completely reversed each patient's hyperkeratotic palms and soles.4 A definitive pathogenic role of estrogens in the development of keratoderma climactericum has yet to be determined.2
Histopathology is not specific for keratoderma climactericum, making the disease a clinical diagnosis. However, a biopsy may be useful to rule out palmoplantar psoriasis.2 Clinical information such as the age and sex of the patient, distribution of disease, presence of fissuring, and progression of disease from soles to palms should be considered when making a diagnosis of keratoderma climactericum. The differential diagnosis of keratoderma climactericum should include fungal infections, contact dermatitis, irritant dermatitis, psoriasis, atopic dermatitis, underlying malignancy, and pityriasis rubra pilaris.
Treatment options for keratoderma climactericum include salicylic acid, emollients, oral retinoids, urea ointments, estriol cream, and topical steroids.5,6 Our patient was prescribed acitretin 25 mg daily and ammonium lactate to apply topically as needed for dry skin. Five months after the initial presentation, fissures and dry skin on the bilateral soles were still present. Ammonium lactate was discontinued, and the patient was prescribed urea cream 40%. Fifteen months after the initial presentation, the patient reported substantial improvement on the hands and feet and noted that she no longer needed the urea cream. Physical examination revealed no presence of hyperkeratosis or fissuring on the palms (Figure 2), and mild hyperkeratosis was present on the plantar surfaces of the feet (Figure 3). The patient continued to use acitretin to prevent disease relapse.
Keratoderma climactericum is an unusual and debilitating condition that occurs in women of menopausal age. It is diagnosed by its specific clinical presentation. More common diagnoses such as tinea and dermatitis should be ruled out before considering keratoderma climactericum.
- Haxthausen H. Keratoderma climactericum. Br J Dermatol. 1934;46:161-167.
- Patel S, Zirwas M, English JC. Acquired palmoplantar keratoderma. Am J Clin Dermatol. 2007;8:1-11.
- Deschamps P, Leroy D, Pedailles S, et al. Keratoderma climactericum (Haxthausen's disease): clinical signs, laboratory findings and etretinate treatment in 10 patients. Dermatologica. 1986;172:258-262.
- Wachtel TJ. Plantar and palmar hyperkeratosis in young castrated women. Int J Dermatol. 1981;20:270-271.
- Bristow I. The management of heel fissures using a steroid impregnated tape (Haelan) in a patient with Keratoderma climactericum. Podiatry Now. 2008;11:22-23.
- Mendes-Bastos P. Plantar keratoderma climactericum: successful improvement with a topical estriol cream. J Cosmet Dermatol. 2018;17:811-813.
- Haxthausen H. Keratoderma climactericum. Br J Dermatol. 1934;46:161-167.
- Patel S, Zirwas M, English JC. Acquired palmoplantar keratoderma. Am J Clin Dermatol. 2007;8:1-11.
- Deschamps P, Leroy D, Pedailles S, et al. Keratoderma climactericum (Haxthausen's disease): clinical signs, laboratory findings and etretinate treatment in 10 patients. Dermatologica. 1986;172:258-262.
- Wachtel TJ. Plantar and palmar hyperkeratosis in young castrated women. Int J Dermatol. 1981;20:270-271.
- Bristow I. The management of heel fissures using a steroid impregnated tape (Haelan) in a patient with Keratoderma climactericum. Podiatry Now. 2008;11:22-23.
- Mendes-Bastos P. Plantar keratoderma climactericum: successful improvement with a topical estriol cream. J Cosmet Dermatol. 2018;17:811-813.
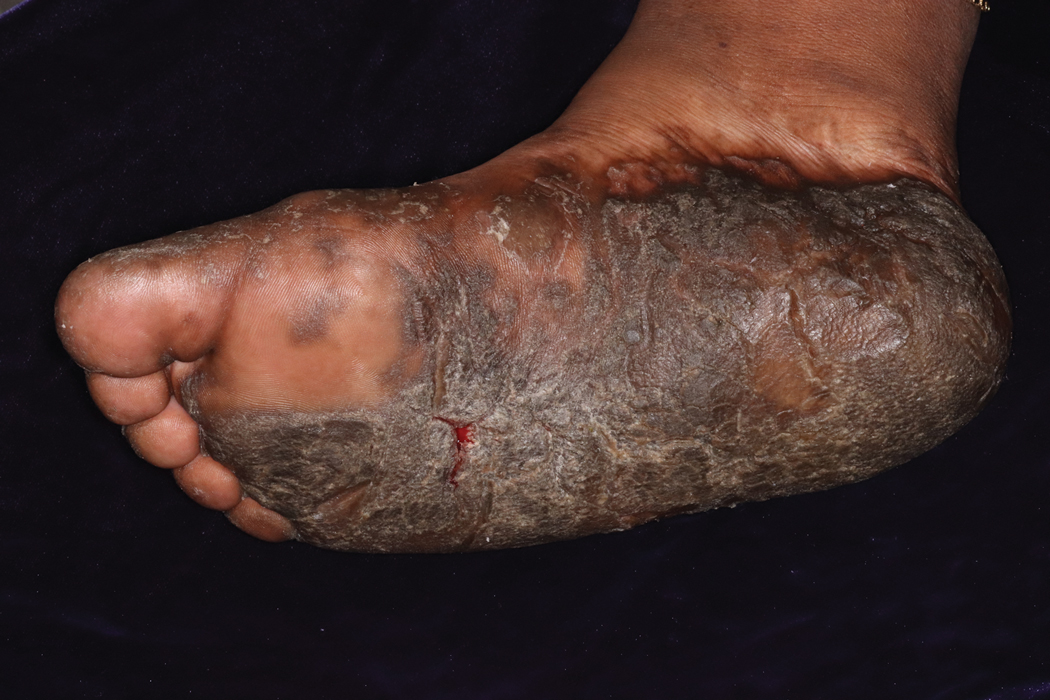
A 52-year-old woman with a history of rheumatoid arthritis presented with a rash on the palms and soles of 7 years' duration that started around the onset of menopause. Physical examination revealed thick hyperkeratotic plaques with multiple deep fissures on the palms and soles. The patient's current medications included methotrexate for rheumatoid arthritis. She previously had been prescribed adalimumab by an outside physician for the rash, which provided no relief, and currently was using urea ointment, which caused a burning sensation on the palms and soles. The patient denied a personal or family history of psoriasis.
Hyperkeratotic Nummular Plaques on the Upper Trunk
The Diagnosis: Extragenital Lichen Sclerosus Et Atrophicus
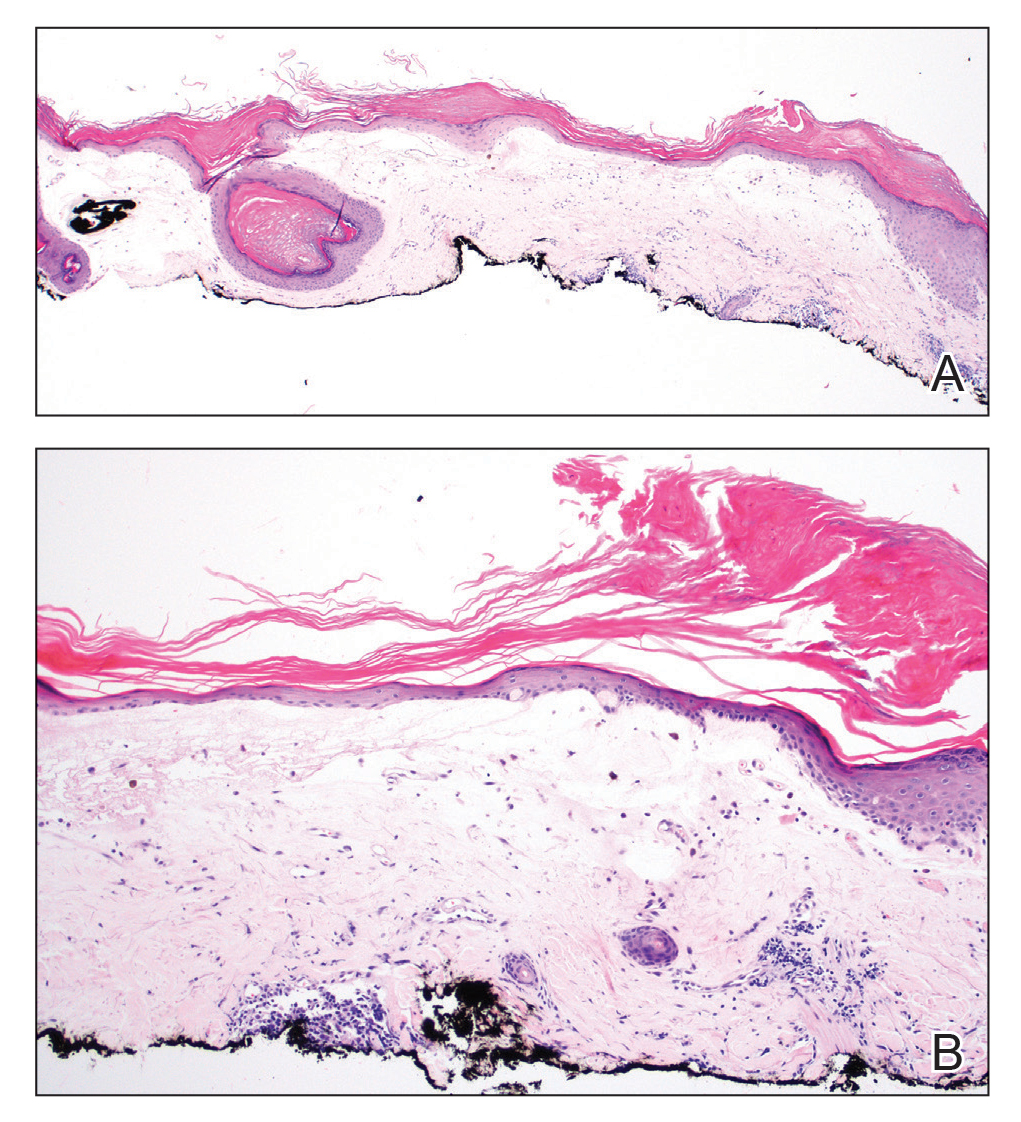
Histopathologic evaluation revealed hyperkeratosis, follicular plugging, epidermal atrophy, and homogenization of papillary dermal collagen with an underlying lymphocytic infiltrate (Figure 1). Direct immunofluorescence of a plaque with a superimposed bulla was negative for deposition of C3, IgG, IgA, IgM, or fibrinogen. Accordingly, clinicopathologic correlation supported a diagnosis of extragenital lichen sclerosus et atrophicus (LSA). Of note, the patient's history of genital irritation was due to genital LSA that preceded the extragenital manifestations.
Lichen sclerosus et atrophicus is an inflammatory dermatosis that typically presents as atrophic white papules of the anogenital area that coalesce into pruritic plaques; the exact etiology remains to be elucidated, yet various circulating autoantibodies have been identified, suggesting a role for autoimmunity.1,2 Lichen sclerosus et atrophicus is more common in women than in men, with a bimodal peak in the age of onset affecting postmenopausal and prepubertal populations.1 In women, affected areas include the labia minora and majora, clitoris, perineum, and perianal skin; LSA spares the mucosal surfaces of the vagina and cervix.2 In men, uncircumscribed genital skin more commonly is affected. Involvement is localized to the foreskin and glans with occasional urethral involvement.2
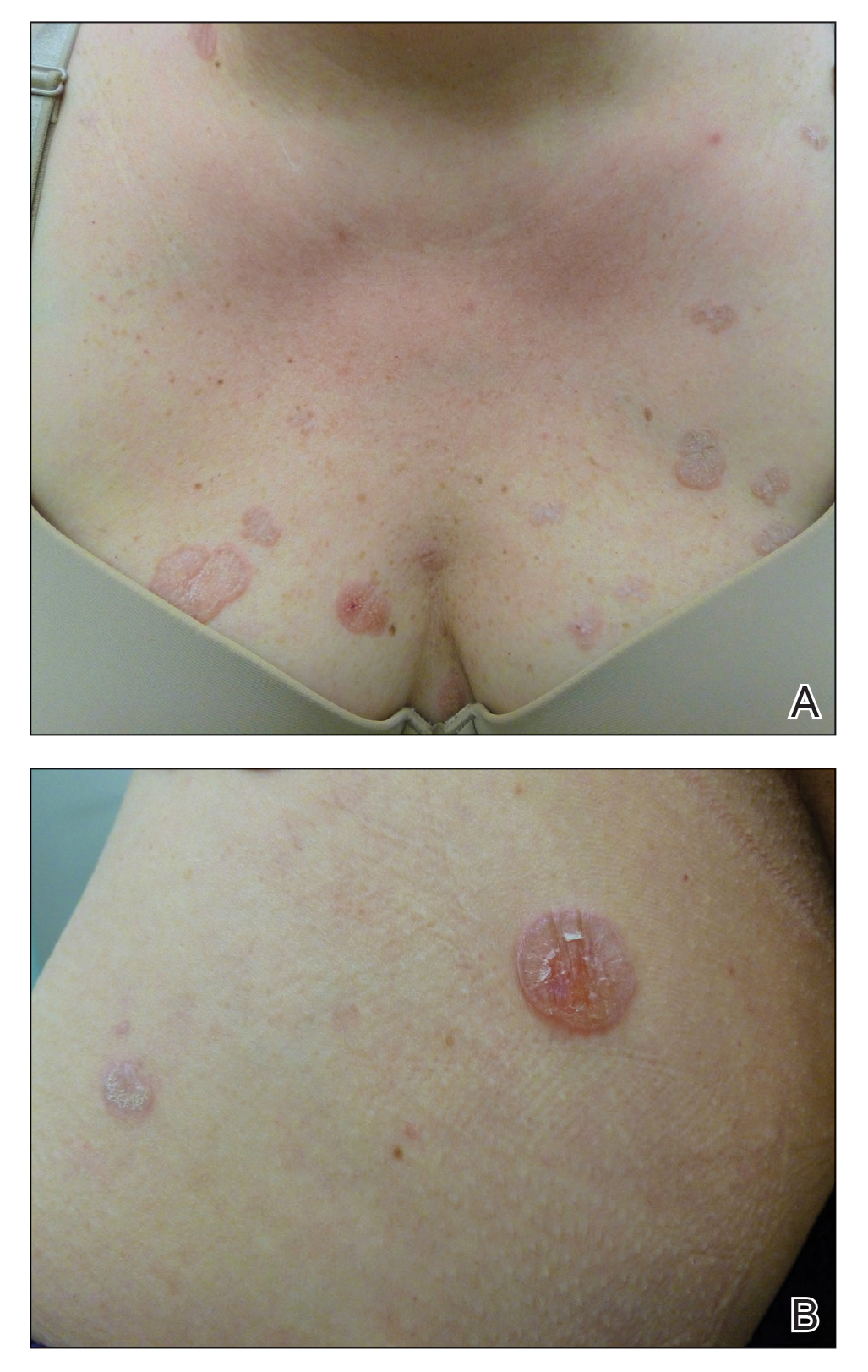
In contrast, extragenital LSA tends to present as asymptomatic papules and plaques that develop atrophy with time, involving the back, shoulders, neck, chest, thighs, axillae, and flexural wrists2,3; an erythematous rim often is present,4 and hyperkeratosis with follicular plugging may be prominent.5 Our patient's case emphasizes the predilection of plaques for the chest and intermammary skin (Figure 2A). Approximately 15% of LSA cases have extragenital involvement, and extragenital-limited disease accounts for roughly 5% of cases.6,7 Unlike genital LSA, extragenital disease has not been associated with an increased risk for squamous cell carcinoma.1 Bullae formation within plaques of genital or extragenital LSA has been reported3,8 and is exemplified in our patient (Figure 2B). Intralesional bullae formation likely is due to a combination of internal and external factors, mainly the inability to withstand shear forces due to an atrophic epidermis with basal vacuolar injury overlying an edematous papillary dermis with altered collagen.8 Dermatoscopic findings may aid in recognizing extragenital LSA9,10; our patient's plaques demonstrated the characteristic findings of comedolike openings, structureless white areas, and pink borders (Figure 3).
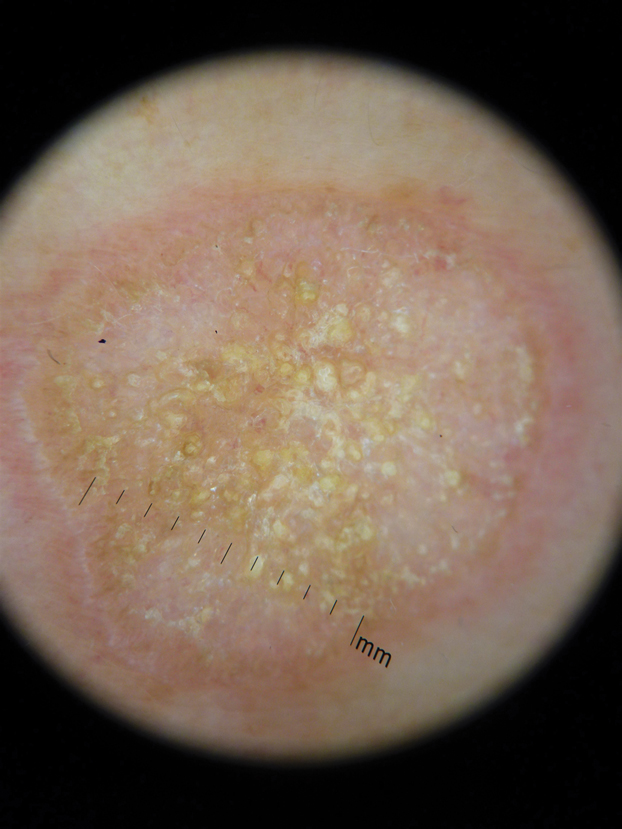
The clinical differential diagnosis for well-demarcated, pink, scaly plaques is broad. Nummular eczema usually presents as coin-shaped eczematous plaques on the dorsal aspects of the hands or lower extremities, and histology shows epidermal spongiosis.11 Nummular eczema may be considered due to the striking round morphology of various plaques, yet our patient's presentation was better served by a consideration of several papulosquamous disorders.
Lichen planus (LP) presents as intensely pruritic, violaceous, polygonal, flat-topped papules with overlying reticular white lines, or Wickham striae, that favor the flexural wrists, lower back, and lower extremities. Lichen planus also may have oral and genital mucosal involvement. Similar to LSA, LP is more common in women and preferentially affects the postmenopausal population.12 Additionally, hypertrophic LP may obscure Wickham striae and mimic extragenital LSA; distinguishing features of hypertrophic LP are intense pruritus and a predilection for the shins. Histology is defined by orthohyperkeratosis, hypergranulosis, sawtooth acanthosis, and vacuolar degeneration of the basal layer with Civatte bodies or dyskeratotic basal keratinocytes overlying a characteristic bandlike infiltrate of lymphocytes.12
Lichen simplex chronicus (LSC) is characterized by intense pruritus and presents as hyperkeratotic plaques with a predilection for accessible regions such as the posterior neck and extremities.13 The striking annular demarcation of this case makes LSC unlikely. Comparable to LSA and LP, LSC also may present with both genital and extragenital findings. Histology of LSC is characterized by irregular acanthosis or thickening of the epidermis with vertical streaking of collagen and vascular bundles of the papillary dermis.13
Subacute cutaneous lupus erythematosus (SCLE) is important to consider for a new papulosquamous eruption with a predilection for the sun-exposed skin of a middle-aged woman. The presence of papules on the volar wrist and history of genital irritation, however, make this entity less likely. Similar to LSA, histologic examination of SCLE reveals epidermal atrophy, basal layer degeneration, and papillary dermal edema with lymphocytic inflammation. However, SCLE lacks the band of inflammation underlying pale homogenized papillary dermal collagen, the most distinguishing feature of LSA; instead, SCLE shows superficial and deep perivascular and periadnexal lymphocytes and mucin in the dermis.14
Lichen sclerosus et atrophicus may be chronic and progressive in nature or cycle through remissions and relapses.2 Treatment is not curative, and management is directed to alleviating symptoms and preventing the progression of disease. First-line management of extragenital LSA is potent topical steroids.1 Adjuvant topical calcineurin inhibitors may be used as steroid-sparing agents.2 Phototherapy is a second-line therapy and even narrowband UVB phototherapy has demonstrated efficacy in managing extragenital LSA.15,16 Our patient was started on mometasone ointment and calcipotriene cream with slight improvement after a 6-month trial. Ongoing management is focused on optimizing application of topical therapies.
- Powell JJ, Wojnarowska F. Lichen sclerosus. Lancet. 1999;353:1777-1783.
- Fistarol SK, Itin PH. Diagnosis and treatment of lichen sclerosus. Am J Clin Dermatol. 2013;14:27-47.
- Meffert JJ, Davis BM, Grimwood RE. Lichen sclerosus. J Am Acad Dermatol. 1995;32:393-416.
- Surkan M, Hull P. A case of lichen sclerosus et atrophicus with distinct erythematous borders. J Cutan Med Surg. 2015;19:600-603.
- Kimura A, Kambe N, Satoh T, et al. Follicular keratosis and bullous formation are typical signs of extragenital lichen sclerosus. J Dermatol. 2011;38:834-836.
- Meyrick Thomas RH, Ridley CM, McGibbon DH, et al. Lichen sclerosus et atrophicus and autoimmunity: a study of 350 women. Br J Dermatol. 1988;118:41-46.
- Wallace HJ. Lichen sclerosus et atrophicus. Trans St Johns Hosp Dermatol Soc. 1971;57:9-30.
- Hallel-Halevy D, Grunwald MH, Yerushalmi J, et al. Bullous lichen sclerosus et atrophicus. J Am Acad Dermatol. 1998;39:500-501.
- Garrido-Ríos AA, Álvarez-Garrido H, Sanz-Muñoz C, et al. Dermoscopy of extragenital lichen sclerosus. Arch Dermatol. 2009;145:1468.
- Larre Borges A, Tiodorovic-Zivkovic D, Lallas A, et al. Clinical, dermoscopic and histopathologic features of genital and extragenital lichen sclerosus. J Eur Acad Dermatol Venereol. 2013;27:1433-1439.
- Rudikoff D. Differential diagnosis of round or discoid lesions. Clin Dermatol. 2011;29:489-497.
- Boyd AS, Neldner KH. Lichen planus. J Am Acad Dermatol. 1991;25:593-619.
- Shaffer B, Beerman H. Lichen simplex chronicus and its variants: a discussion of certain psychodynamic mechanisms and clinical and histopathologic correlations. AMA Arch Derm Syphilol. 1951;64:340-351.
- Walling HW, Sontheimer RD. Cutaneous lupus erythematosus. Am J Clin Dermatol. 2009;10:365-381.
- Sauder MB, Linzon-Smith J, Beecker J. Extragenital bullous lichen sclerosus. J Am Acad Dermatol. 2014;71:981-984.
- Colbert RL, Chiang MP, Carlin CS, et al. Progressive extragenital lichen sclerosus successfully treated with narrowband UV-B phototherapy. Arch Dermatol. 2007;143:19-20.
The Diagnosis: Extragenital Lichen Sclerosus Et Atrophicus
Histopathologic evaluation revealed hyperkeratosis, follicular plugging, epidermal atrophy, and homogenization of papillary dermal collagen with an underlying lymphocytic infiltrate (Figure 1). Direct immunofluorescence of a plaque with a superimposed bulla was negative for deposition of C3, IgG, IgA, IgM, or fibrinogen. Accordingly, clinicopathologic correlation supported a diagnosis of extragenital lichen sclerosus et atrophicus (LSA). Of note, the patient's history of genital irritation was due to genital LSA that preceded the extragenital manifestations.
Lichen sclerosus et atrophicus is an inflammatory dermatosis that typically presents as atrophic white papules of the anogenital area that coalesce into pruritic plaques; the exact etiology remains to be elucidated, yet various circulating autoantibodies have been identified, suggesting a role for autoimmunity.1,2 Lichen sclerosus et atrophicus is more common in women than in men, with a bimodal peak in the age of onset affecting postmenopausal and prepubertal populations.1 In women, affected areas include the labia minora and majora, clitoris, perineum, and perianal skin; LSA spares the mucosal surfaces of the vagina and cervix.2 In men, uncircumscribed genital skin more commonly is affected. Involvement is localized to the foreskin and glans with occasional urethral involvement.2
In contrast, extragenital LSA tends to present as asymptomatic papules and plaques that develop atrophy with time, involving the back, shoulders, neck, chest, thighs, axillae, and flexural wrists2,3; an erythematous rim often is present,4 and hyperkeratosis with follicular plugging may be prominent.5 Our patient's case emphasizes the predilection of plaques for the chest and intermammary skin (Figure 2A). Approximately 15% of LSA cases have extragenital involvement, and extragenital-limited disease accounts for roughly 5% of cases.6,7 Unlike genital LSA, extragenital disease has not been associated with an increased risk for squamous cell carcinoma.1 Bullae formation within plaques of genital or extragenital LSA has been reported3,8 and is exemplified in our patient (Figure 2B). Intralesional bullae formation likely is due to a combination of internal and external factors, mainly the inability to withstand shear forces due to an atrophic epidermis with basal vacuolar injury overlying an edematous papillary dermis with altered collagen.8 Dermatoscopic findings may aid in recognizing extragenital LSA9,10; our patient's plaques demonstrated the characteristic findings of comedolike openings, structureless white areas, and pink borders (Figure 3).
The clinical differential diagnosis for well-demarcated, pink, scaly plaques is broad. Nummular eczema usually presents as coin-shaped eczematous plaques on the dorsal aspects of the hands or lower extremities, and histology shows epidermal spongiosis.11 Nummular eczema may be considered due to the striking round morphology of various plaques, yet our patient's presentation was better served by a consideration of several papulosquamous disorders.
Lichen planus (LP) presents as intensely pruritic, violaceous, polygonal, flat-topped papules with overlying reticular white lines, or Wickham striae, that favor the flexural wrists, lower back, and lower extremities. Lichen planus also may have oral and genital mucosal involvement. Similar to LSA, LP is more common in women and preferentially affects the postmenopausal population.12 Additionally, hypertrophic LP may obscure Wickham striae and mimic extragenital LSA; distinguishing features of hypertrophic LP are intense pruritus and a predilection for the shins. Histology is defined by orthohyperkeratosis, hypergranulosis, sawtooth acanthosis, and vacuolar degeneration of the basal layer with Civatte bodies or dyskeratotic basal keratinocytes overlying a characteristic bandlike infiltrate of lymphocytes.12
Lichen simplex chronicus (LSC) is characterized by intense pruritus and presents as hyperkeratotic plaques with a predilection for accessible regions such as the posterior neck and extremities.13 The striking annular demarcation of this case makes LSC unlikely. Comparable to LSA and LP, LSC also may present with both genital and extragenital findings. Histology of LSC is characterized by irregular acanthosis or thickening of the epidermis with vertical streaking of collagen and vascular bundles of the papillary dermis.13
Subacute cutaneous lupus erythematosus (SCLE) is important to consider for a new papulosquamous eruption with a predilection for the sun-exposed skin of a middle-aged woman. The presence of papules on the volar wrist and history of genital irritation, however, make this entity less likely. Similar to LSA, histologic examination of SCLE reveals epidermal atrophy, basal layer degeneration, and papillary dermal edema with lymphocytic inflammation. However, SCLE lacks the band of inflammation underlying pale homogenized papillary dermal collagen, the most distinguishing feature of LSA; instead, SCLE shows superficial and deep perivascular and periadnexal lymphocytes and mucin in the dermis.14
Lichen sclerosus et atrophicus may be chronic and progressive in nature or cycle through remissions and relapses.2 Treatment is not curative, and management is directed to alleviating symptoms and preventing the progression of disease. First-line management of extragenital LSA is potent topical steroids.1 Adjuvant topical calcineurin inhibitors may be used as steroid-sparing agents.2 Phototherapy is a second-line therapy and even narrowband UVB phototherapy has demonstrated efficacy in managing extragenital LSA.15,16 Our patient was started on mometasone ointment and calcipotriene cream with slight improvement after a 6-month trial. Ongoing management is focused on optimizing application of topical therapies.
The Diagnosis: Extragenital Lichen Sclerosus Et Atrophicus
Histopathologic evaluation revealed hyperkeratosis, follicular plugging, epidermal atrophy, and homogenization of papillary dermal collagen with an underlying lymphocytic infiltrate (Figure 1). Direct immunofluorescence of a plaque with a superimposed bulla was negative for deposition of C3, IgG, IgA, IgM, or fibrinogen. Accordingly, clinicopathologic correlation supported a diagnosis of extragenital lichen sclerosus et atrophicus (LSA). Of note, the patient's history of genital irritation was due to genital LSA that preceded the extragenital manifestations.
Lichen sclerosus et atrophicus is an inflammatory dermatosis that typically presents as atrophic white papules of the anogenital area that coalesce into pruritic plaques; the exact etiology remains to be elucidated, yet various circulating autoantibodies have been identified, suggesting a role for autoimmunity.1,2 Lichen sclerosus et atrophicus is more common in women than in men, with a bimodal peak in the age of onset affecting postmenopausal and prepubertal populations.1 In women, affected areas include the labia minora and majora, clitoris, perineum, and perianal skin; LSA spares the mucosal surfaces of the vagina and cervix.2 In men, uncircumscribed genital skin more commonly is affected. Involvement is localized to the foreskin and glans with occasional urethral involvement.2
In contrast, extragenital LSA tends to present as asymptomatic papules and plaques that develop atrophy with time, involving the back, shoulders, neck, chest, thighs, axillae, and flexural wrists2,3; an erythematous rim often is present,4 and hyperkeratosis with follicular plugging may be prominent.5 Our patient's case emphasizes the predilection of plaques for the chest and intermammary skin (Figure 2A). Approximately 15% of LSA cases have extragenital involvement, and extragenital-limited disease accounts for roughly 5% of cases.6,7 Unlike genital LSA, extragenital disease has not been associated with an increased risk for squamous cell carcinoma.1 Bullae formation within plaques of genital or extragenital LSA has been reported3,8 and is exemplified in our patient (Figure 2B). Intralesional bullae formation likely is due to a combination of internal and external factors, mainly the inability to withstand shear forces due to an atrophic epidermis with basal vacuolar injury overlying an edematous papillary dermis with altered collagen.8 Dermatoscopic findings may aid in recognizing extragenital LSA9,10; our patient's plaques demonstrated the characteristic findings of comedolike openings, structureless white areas, and pink borders (Figure 3).
The clinical differential diagnosis for well-demarcated, pink, scaly plaques is broad. Nummular eczema usually presents as coin-shaped eczematous plaques on the dorsal aspects of the hands or lower extremities, and histology shows epidermal spongiosis.11 Nummular eczema may be considered due to the striking round morphology of various plaques, yet our patient's presentation was better served by a consideration of several papulosquamous disorders.
Lichen planus (LP) presents as intensely pruritic, violaceous, polygonal, flat-topped papules with overlying reticular white lines, or Wickham striae, that favor the flexural wrists, lower back, and lower extremities. Lichen planus also may have oral and genital mucosal involvement. Similar to LSA, LP is more common in women and preferentially affects the postmenopausal population.12 Additionally, hypertrophic LP may obscure Wickham striae and mimic extragenital LSA; distinguishing features of hypertrophic LP are intense pruritus and a predilection for the shins. Histology is defined by orthohyperkeratosis, hypergranulosis, sawtooth acanthosis, and vacuolar degeneration of the basal layer with Civatte bodies or dyskeratotic basal keratinocytes overlying a characteristic bandlike infiltrate of lymphocytes.12
Lichen simplex chronicus (LSC) is characterized by intense pruritus and presents as hyperkeratotic plaques with a predilection for accessible regions such as the posterior neck and extremities.13 The striking annular demarcation of this case makes LSC unlikely. Comparable to LSA and LP, LSC also may present with both genital and extragenital findings. Histology of LSC is characterized by irregular acanthosis or thickening of the epidermis with vertical streaking of collagen and vascular bundles of the papillary dermis.13
Subacute cutaneous lupus erythematosus (SCLE) is important to consider for a new papulosquamous eruption with a predilection for the sun-exposed skin of a middle-aged woman. The presence of papules on the volar wrist and history of genital irritation, however, make this entity less likely. Similar to LSA, histologic examination of SCLE reveals epidermal atrophy, basal layer degeneration, and papillary dermal edema with lymphocytic inflammation. However, SCLE lacks the band of inflammation underlying pale homogenized papillary dermal collagen, the most distinguishing feature of LSA; instead, SCLE shows superficial and deep perivascular and periadnexal lymphocytes and mucin in the dermis.14
Lichen sclerosus et atrophicus may be chronic and progressive in nature or cycle through remissions and relapses.2 Treatment is not curative, and management is directed to alleviating symptoms and preventing the progression of disease. First-line management of extragenital LSA is potent topical steroids.1 Adjuvant topical calcineurin inhibitors may be used as steroid-sparing agents.2 Phototherapy is a second-line therapy and even narrowband UVB phototherapy has demonstrated efficacy in managing extragenital LSA.15,16 Our patient was started on mometasone ointment and calcipotriene cream with slight improvement after a 6-month trial. Ongoing management is focused on optimizing application of topical therapies.
- Powell JJ, Wojnarowska F. Lichen sclerosus. Lancet. 1999;353:1777-1783.
- Fistarol SK, Itin PH. Diagnosis and treatment of lichen sclerosus. Am J Clin Dermatol. 2013;14:27-47.
- Meffert JJ, Davis BM, Grimwood RE. Lichen sclerosus. J Am Acad Dermatol. 1995;32:393-416.
- Surkan M, Hull P. A case of lichen sclerosus et atrophicus with distinct erythematous borders. J Cutan Med Surg. 2015;19:600-603.
- Kimura A, Kambe N, Satoh T, et al. Follicular keratosis and bullous formation are typical signs of extragenital lichen sclerosus. J Dermatol. 2011;38:834-836.
- Meyrick Thomas RH, Ridley CM, McGibbon DH, et al. Lichen sclerosus et atrophicus and autoimmunity: a study of 350 women. Br J Dermatol. 1988;118:41-46.
- Wallace HJ. Lichen sclerosus et atrophicus. Trans St Johns Hosp Dermatol Soc. 1971;57:9-30.
- Hallel-Halevy D, Grunwald MH, Yerushalmi J, et al. Bullous lichen sclerosus et atrophicus. J Am Acad Dermatol. 1998;39:500-501.
- Garrido-Ríos AA, Álvarez-Garrido H, Sanz-Muñoz C, et al. Dermoscopy of extragenital lichen sclerosus. Arch Dermatol. 2009;145:1468.
- Larre Borges A, Tiodorovic-Zivkovic D, Lallas A, et al. Clinical, dermoscopic and histopathologic features of genital and extragenital lichen sclerosus. J Eur Acad Dermatol Venereol. 2013;27:1433-1439.
- Rudikoff D. Differential diagnosis of round or discoid lesions. Clin Dermatol. 2011;29:489-497.
- Boyd AS, Neldner KH. Lichen planus. J Am Acad Dermatol. 1991;25:593-619.
- Shaffer B, Beerman H. Lichen simplex chronicus and its variants: a discussion of certain psychodynamic mechanisms and clinical and histopathologic correlations. AMA Arch Derm Syphilol. 1951;64:340-351.
- Walling HW, Sontheimer RD. Cutaneous lupus erythematosus. Am J Clin Dermatol. 2009;10:365-381.
- Sauder MB, Linzon-Smith J, Beecker J. Extragenital bullous lichen sclerosus. J Am Acad Dermatol. 2014;71:981-984.
- Colbert RL, Chiang MP, Carlin CS, et al. Progressive extragenital lichen sclerosus successfully treated with narrowband UV-B phototherapy. Arch Dermatol. 2007;143:19-20.
- Powell JJ, Wojnarowska F. Lichen sclerosus. Lancet. 1999;353:1777-1783.
- Fistarol SK, Itin PH. Diagnosis and treatment of lichen sclerosus. Am J Clin Dermatol. 2013;14:27-47.
- Meffert JJ, Davis BM, Grimwood RE. Lichen sclerosus. J Am Acad Dermatol. 1995;32:393-416.
- Surkan M, Hull P. A case of lichen sclerosus et atrophicus with distinct erythematous borders. J Cutan Med Surg. 2015;19:600-603.
- Kimura A, Kambe N, Satoh T, et al. Follicular keratosis and bullous formation are typical signs of extragenital lichen sclerosus. J Dermatol. 2011;38:834-836.
- Meyrick Thomas RH, Ridley CM, McGibbon DH, et al. Lichen sclerosus et atrophicus and autoimmunity: a study of 350 women. Br J Dermatol. 1988;118:41-46.
- Wallace HJ. Lichen sclerosus et atrophicus. Trans St Johns Hosp Dermatol Soc. 1971;57:9-30.
- Hallel-Halevy D, Grunwald MH, Yerushalmi J, et al. Bullous lichen sclerosus et atrophicus. J Am Acad Dermatol. 1998;39:500-501.
- Garrido-Ríos AA, Álvarez-Garrido H, Sanz-Muñoz C, et al. Dermoscopy of extragenital lichen sclerosus. Arch Dermatol. 2009;145:1468.
- Larre Borges A, Tiodorovic-Zivkovic D, Lallas A, et al. Clinical, dermoscopic and histopathologic features of genital and extragenital lichen sclerosus. J Eur Acad Dermatol Venereol. 2013;27:1433-1439.
- Rudikoff D. Differential diagnosis of round or discoid lesions. Clin Dermatol. 2011;29:489-497.
- Boyd AS, Neldner KH. Lichen planus. J Am Acad Dermatol. 1991;25:593-619.
- Shaffer B, Beerman H. Lichen simplex chronicus and its variants: a discussion of certain psychodynamic mechanisms and clinical and histopathologic correlations. AMA Arch Derm Syphilol. 1951;64:340-351.
- Walling HW, Sontheimer RD. Cutaneous lupus erythematosus. Am J Clin Dermatol. 2009;10:365-381.
- Sauder MB, Linzon-Smith J, Beecker J. Extragenital bullous lichen sclerosus. J Am Acad Dermatol. 2014;71:981-984.
- Colbert RL, Chiang MP, Carlin CS, et al. Progressive extragenital lichen sclerosus successfully treated with narrowband UV-B phototherapy. Arch Dermatol. 2007;143:19-20.
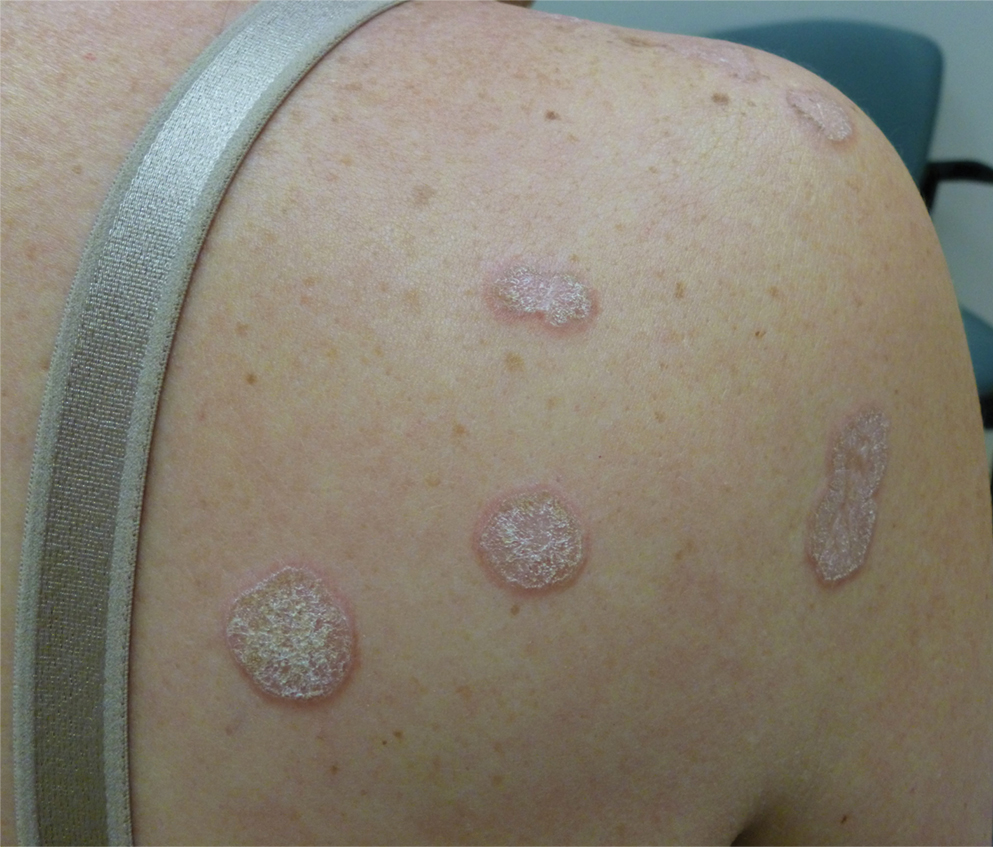
A 48-year-old woman with a history of type 2 diabetes mellitus and nonalcoholic steatohepatitis presented with papules and plaques on the upper trunk, proximal extremities, and volar wrists. Clear fluid–filled bullae occasionally developed within the plaques and subsequently ruptured and healed. Aside from intermittent lesion tenderness and irritation with the formation and rupture of the bullae, the patient’s plaques were asymptomatic, and she specifically denied pruritus. A review of systems revealed a history of genital irritation evaluated by a gynecologist; nystatin–triamcinolone cream 0.1% applied as needed provided relief. The patient denied any recent flares or any new or changing oral mucosa findings or symptoms, preceding medications, or family history of similar lesions. Physical examination revealed well-demarcated, round, pink plaques with keratotic scale scattered across the upper trunk and central chest. The bilateral volar wrists were surfaced by well-circumscribed, thin, pink to violaceous, hyperkeratotic papules.
Painless Mobile Nodule on the Shoulder
The Diagnosis: Cutaneous Metaplastic Synovial Cyst
Gross examination of the excised nodule revealed a 2.5×1.2×1.0-cm, intact, gray-white, thin-walled, smooth-lined nodule filled with clear mucinouslike material. Hematoxylin and eosin-stained sections demonstrated a dermal-based cystlike structure composed of a lining of connective tissue with hyalinized material and fibrin as well as spindle and epithelioid cells with a mild mixed inflammatory infiltrate (Figure). These histopathologic findings led to the diagnosis of cutaneous metaplastic synovial cyst (CMSC).
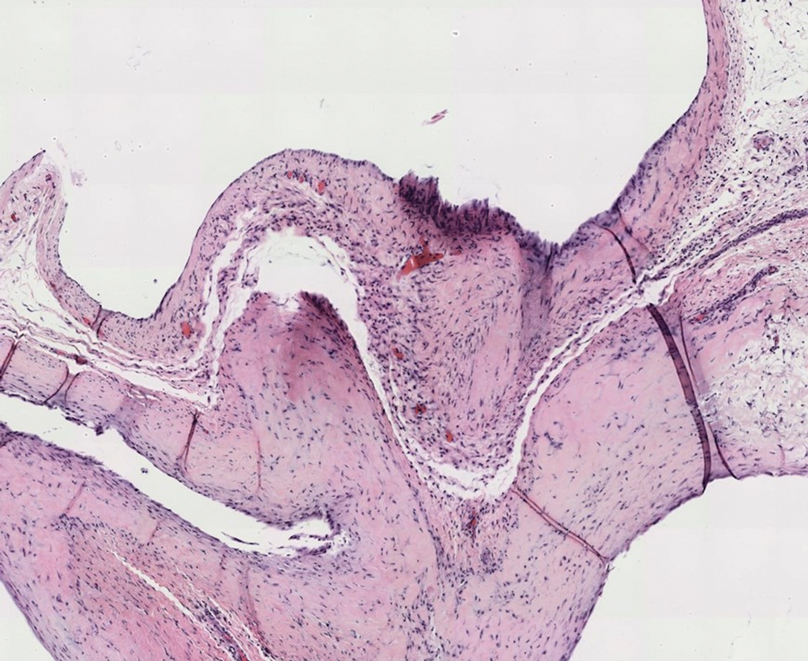
Cutaneous metaplastic synovial cyst, also known as synovial metaplasia of the skin, is an uncommon benign cystic lesion that was first reported by Gonzalez et al1 in 1987. Histologically, CMSC lacks an epithelial lining and therefore is not a true cyst but rather a pseudocyst.2 Clinically, the lesion typically presents as a solitary subcutaneous nodule that may be tender or painless. In a literature review of CMSC cases performed by Fukuyama et al,3 distribution of reported cases according to body site varied; however, limbs were found to be the most commonly involved area. A PubMed search of articles indexed for MEDLINE as well as a Google Scholar search using the term cutaneous metaplastic synovial cyst revealed at least 37 cases reported in the English-language literature,3-9 including our present case. The pathogenesis remains uncertain; however, a majority of previously reported cases of CMSC characteristically have been associated with a pre-existing lesion, with most presentations developing at surgical scar sites secondary to operation or trauma.5 Relative tissue fragility secondary to rheumatoid arthritis10 and Ehlers-Danlos syndrome9,11,12 has been linked to CMSC in some documented reports, while a minority of cases report no antecedent events triggering formation of the lesion.13-15
As evidenced by our patient, CMSC clinically mimics several other benign entities; histopathologic examination is necessary to confirm the diagnosis. Although nodular hidradenoma also may clinically present as a solitary firm intradermal nodule, microscopy reveals a dermal-based lobulated tumor containing cystic spaces and solid areas composed of basophilic polyhedral cells and round glycogen-filled clear cells.16 Epidermoid cysts are differentiated from CMSC by the presence of a cyst wall lining composed of stratified squamous epithelium and associated laminated keratin within the lumen,17 which corresponds to its pearly white appearance on gross examination. Cutaneous ciliated cysts predominantly occur on the lower extremities of young women and are lined by simple cuboidal or columnar ciliated cells that resemble müllerian epithelium.18 Similar to CMSC, ganglion cysts are pseudocysts that lack a true epithelial lining but differ in appearance due to their mucin-filled synovial-lined sac.19 Additionally, ganglion cysts most often occur on the dorsal and volar aspects of the wrist.
Excisional biopsy is indicated as the preferred treatment of CMSC, given the lesion's benign behavior and low recurrence rate.6 Our case highlights this rare entity and reinforces its inclusion in the differential diagnosis of subcutaneous mobile nodules, especially in the setting of prior tissue injury secondary to trauma, surgical procedures, or conditions such as rheumatoid arthritis or Ehlers-Danlos syndrome. Unlike most previously reported cases, our patient reported no preceding tissue injury associated with formation of the lesion, and she was largely asymptomatic on presentation. Considering the limited number of CMSC cases demonstrated in the literature, it is important to continue reporting new cases to better understand characteristics and presentations of this uncommon lesion.
- Gonzalez JG, Ghiselli RW, Santa Cruz DJ. Synovial metaplasia of the skin. Am J Surg Pathol. 1987;11:343-350.
- Calonje E, Brenn T, Lazar A, et al. Cutaneous cysts. In: Calonje E, Brenn T, Lazar A, et al. McKee's Pathology of the Skin. 5th ed. Elsevier Limited; 2020:1680-1697.
- Fukuyama M, Sato Y, Hayakawa J, et al. Cutaneous metaplastic synovial cyst: case report and literature review from the dermatological point of view. Keio J Med. 2016;66:9-13.
- Karaytug K, Kapicioglu M, Can N, et al. Unprecedented recurrence of carpal tunnel syndrome by metaplastic synovial cyst in the carpal tunnel. Acta Orthop Traumatol Turc. 2019;53:230-232.
- Martelli SJ, Silveira FM, Carvalho PH, et al. Asymptomatic subcutaneous swelling of lower face. Oral Surg Oral Med Oral Pathol Oral Radiol. 2019;128:101-105.
- Majdi M, Saffar H, Ghanadan A. Cutaneous metaplastic synovial cyst: a case report. Iran J Pathol. 2016;11:423-426.
- Ramachandra S, Rao L, Al-Kindi M. Cutaneous metaplastic synovial cyst. Sultan Qaboos Univ Med J. 2016;16:E117-E118.
- Heidarian A, Xie Q, Banihashemi A. Cutaneous metaplastic synovial cyst presenting as an axillary mass after modified mastectomy and adjuvant radiotherapy. Am J Clin Pathol. 2016;146:S2.
- Fernandez-Flores A, Barja-Lopez JM. Cutaneous metaplastic synovial cyst in Ehlers-Danlos syndrome. J Cutan Pathol. 2020;47:729-733.
- Choonhakarn C, Tang S. Cutaneous metaplastic synovial cyst. J Dermatol. 2003;30:480-484.
- Guala A, Viglio S, Ottinetti A, et al. Cutaneous metaplastic synovial cyst in Ehlers-Danlos syndrome: report of a second case. Am J Dermatopathol. 2008;30:59-61.
- Nieto S, Buezo GF, Jones-Caballero M, et al. Cutaneous metaplastic synovial cyst in an Ehlers-Danlos patient. Am J Dermatopathol. 1997;19:407-410.
- Goiriz R, Rios-Buceta L, Alonso-Perez A, et al. Cutaneous metaplastic synovial cyst. J Am Acad Dermatol. 2005;53:180-181.
- Kim BC, Choi WJ, Park EJ, et al. Cutaneous metaplastic synovial cyst of the first metatarsal head area. Ann Dermatol. 2011;23(suppl 2):S165-S168.
- Yang HC, Tsai YJ, Hu SL, et al. Cutaneous metaplastic synovial cyst--a case report and review of literature. Dermatol Sinica. 2003;21:275-279.
- Kataria SP, Singh G, Batra A, et al. Nodular hidradenoma: a series of five cases in male subjects and review of literature. Adv Cytol Pathol. 2018;3:46-47.
- Mohamed Haflah N, Mohd Kassim A, Hassan Shukur M. Giant epidermoid cyst of the thigh. Malays Orthop J. 2011;5:17-19.
- Torisu-Itakura H, Itakura E, Horiuchi R, et al. Cutaneous ciliated cyst on the leg of a woman of menopausal age. Acta Derm Venereol. 2009;89:323-324.
- Fullen DR. Cysts and sinuses. In: Busam K, ed. Dermatopathology. Saunders; 2010:300-330.
The Diagnosis: Cutaneous Metaplastic Synovial Cyst
Gross examination of the excised nodule revealed a 2.5×1.2×1.0-cm, intact, gray-white, thin-walled, smooth-lined nodule filled with clear mucinouslike material. Hematoxylin and eosin-stained sections demonstrated a dermal-based cystlike structure composed of a lining of connective tissue with hyalinized material and fibrin as well as spindle and epithelioid cells with a mild mixed inflammatory infiltrate (Figure). These histopathologic findings led to the diagnosis of cutaneous metaplastic synovial cyst (CMSC).
Cutaneous metaplastic synovial cyst, also known as synovial metaplasia of the skin, is an uncommon benign cystic lesion that was first reported by Gonzalez et al1 in 1987. Histologically, CMSC lacks an epithelial lining and therefore is not a true cyst but rather a pseudocyst.2 Clinically, the lesion typically presents as a solitary subcutaneous nodule that may be tender or painless. In a literature review of CMSC cases performed by Fukuyama et al,3 distribution of reported cases according to body site varied; however, limbs were found to be the most commonly involved area. A PubMed search of articles indexed for MEDLINE as well as a Google Scholar search using the term cutaneous metaplastic synovial cyst revealed at least 37 cases reported in the English-language literature,3-9 including our present case. The pathogenesis remains uncertain; however, a majority of previously reported cases of CMSC characteristically have been associated with a pre-existing lesion, with most presentations developing at surgical scar sites secondary to operation or trauma.5 Relative tissue fragility secondary to rheumatoid arthritis10 and Ehlers-Danlos syndrome9,11,12 has been linked to CMSC in some documented reports, while a minority of cases report no antecedent events triggering formation of the lesion.13-15
As evidenced by our patient, CMSC clinically mimics several other benign entities; histopathologic examination is necessary to confirm the diagnosis. Although nodular hidradenoma also may clinically present as a solitary firm intradermal nodule, microscopy reveals a dermal-based lobulated tumor containing cystic spaces and solid areas composed of basophilic polyhedral cells and round glycogen-filled clear cells.16 Epidermoid cysts are differentiated from CMSC by the presence of a cyst wall lining composed of stratified squamous epithelium and associated laminated keratin within the lumen,17 which corresponds to its pearly white appearance on gross examination. Cutaneous ciliated cysts predominantly occur on the lower extremities of young women and are lined by simple cuboidal or columnar ciliated cells that resemble müllerian epithelium.18 Similar to CMSC, ganglion cysts are pseudocysts that lack a true epithelial lining but differ in appearance due to their mucin-filled synovial-lined sac.19 Additionally, ganglion cysts most often occur on the dorsal and volar aspects of the wrist.
Excisional biopsy is indicated as the preferred treatment of CMSC, given the lesion's benign behavior and low recurrence rate.6 Our case highlights this rare entity and reinforces its inclusion in the differential diagnosis of subcutaneous mobile nodules, especially in the setting of prior tissue injury secondary to trauma, surgical procedures, or conditions such as rheumatoid arthritis or Ehlers-Danlos syndrome. Unlike most previously reported cases, our patient reported no preceding tissue injury associated with formation of the lesion, and she was largely asymptomatic on presentation. Considering the limited number of CMSC cases demonstrated in the literature, it is important to continue reporting new cases to better understand characteristics and presentations of this uncommon lesion.
The Diagnosis: Cutaneous Metaplastic Synovial Cyst
Gross examination of the excised nodule revealed a 2.5×1.2×1.0-cm, intact, gray-white, thin-walled, smooth-lined nodule filled with clear mucinouslike material. Hematoxylin and eosin-stained sections demonstrated a dermal-based cystlike structure composed of a lining of connective tissue with hyalinized material and fibrin as well as spindle and epithelioid cells with a mild mixed inflammatory infiltrate (Figure). These histopathologic findings led to the diagnosis of cutaneous metaplastic synovial cyst (CMSC).
Cutaneous metaplastic synovial cyst, also known as synovial metaplasia of the skin, is an uncommon benign cystic lesion that was first reported by Gonzalez et al1 in 1987. Histologically, CMSC lacks an epithelial lining and therefore is not a true cyst but rather a pseudocyst.2 Clinically, the lesion typically presents as a solitary subcutaneous nodule that may be tender or painless. In a literature review of CMSC cases performed by Fukuyama et al,3 distribution of reported cases according to body site varied; however, limbs were found to be the most commonly involved area. A PubMed search of articles indexed for MEDLINE as well as a Google Scholar search using the term cutaneous metaplastic synovial cyst revealed at least 37 cases reported in the English-language literature,3-9 including our present case. The pathogenesis remains uncertain; however, a majority of previously reported cases of CMSC characteristically have been associated with a pre-existing lesion, with most presentations developing at surgical scar sites secondary to operation or trauma.5 Relative tissue fragility secondary to rheumatoid arthritis10 and Ehlers-Danlos syndrome9,11,12 has been linked to CMSC in some documented reports, while a minority of cases report no antecedent events triggering formation of the lesion.13-15
As evidenced by our patient, CMSC clinically mimics several other benign entities; histopathologic examination is necessary to confirm the diagnosis. Although nodular hidradenoma also may clinically present as a solitary firm intradermal nodule, microscopy reveals a dermal-based lobulated tumor containing cystic spaces and solid areas composed of basophilic polyhedral cells and round glycogen-filled clear cells.16 Epidermoid cysts are differentiated from CMSC by the presence of a cyst wall lining composed of stratified squamous epithelium and associated laminated keratin within the lumen,17 which corresponds to its pearly white appearance on gross examination. Cutaneous ciliated cysts predominantly occur on the lower extremities of young women and are lined by simple cuboidal or columnar ciliated cells that resemble müllerian epithelium.18 Similar to CMSC, ganglion cysts are pseudocysts that lack a true epithelial lining but differ in appearance due to their mucin-filled synovial-lined sac.19 Additionally, ganglion cysts most often occur on the dorsal and volar aspects of the wrist.
Excisional biopsy is indicated as the preferred treatment of CMSC, given the lesion's benign behavior and low recurrence rate.6 Our case highlights this rare entity and reinforces its inclusion in the differential diagnosis of subcutaneous mobile nodules, especially in the setting of prior tissue injury secondary to trauma, surgical procedures, or conditions such as rheumatoid arthritis or Ehlers-Danlos syndrome. Unlike most previously reported cases, our patient reported no preceding tissue injury associated with formation of the lesion, and she was largely asymptomatic on presentation. Considering the limited number of CMSC cases demonstrated in the literature, it is important to continue reporting new cases to better understand characteristics and presentations of this uncommon lesion.
- Gonzalez JG, Ghiselli RW, Santa Cruz DJ. Synovial metaplasia of the skin. Am J Surg Pathol. 1987;11:343-350.
- Calonje E, Brenn T, Lazar A, et al. Cutaneous cysts. In: Calonje E, Brenn T, Lazar A, et al. McKee's Pathology of the Skin. 5th ed. Elsevier Limited; 2020:1680-1697.
- Fukuyama M, Sato Y, Hayakawa J, et al. Cutaneous metaplastic synovial cyst: case report and literature review from the dermatological point of view. Keio J Med. 2016;66:9-13.
- Karaytug K, Kapicioglu M, Can N, et al. Unprecedented recurrence of carpal tunnel syndrome by metaplastic synovial cyst in the carpal tunnel. Acta Orthop Traumatol Turc. 2019;53:230-232.
- Martelli SJ, Silveira FM, Carvalho PH, et al. Asymptomatic subcutaneous swelling of lower face. Oral Surg Oral Med Oral Pathol Oral Radiol. 2019;128:101-105.
- Majdi M, Saffar H, Ghanadan A. Cutaneous metaplastic synovial cyst: a case report. Iran J Pathol. 2016;11:423-426.
- Ramachandra S, Rao L, Al-Kindi M. Cutaneous metaplastic synovial cyst. Sultan Qaboos Univ Med J. 2016;16:E117-E118.
- Heidarian A, Xie Q, Banihashemi A. Cutaneous metaplastic synovial cyst presenting as an axillary mass after modified mastectomy and adjuvant radiotherapy. Am J Clin Pathol. 2016;146:S2.
- Fernandez-Flores A, Barja-Lopez JM. Cutaneous metaplastic synovial cyst in Ehlers-Danlos syndrome. J Cutan Pathol. 2020;47:729-733.
- Choonhakarn C, Tang S. Cutaneous metaplastic synovial cyst. J Dermatol. 2003;30:480-484.
- Guala A, Viglio S, Ottinetti A, et al. Cutaneous metaplastic synovial cyst in Ehlers-Danlos syndrome: report of a second case. Am J Dermatopathol. 2008;30:59-61.
- Nieto S, Buezo GF, Jones-Caballero M, et al. Cutaneous metaplastic synovial cyst in an Ehlers-Danlos patient. Am J Dermatopathol. 1997;19:407-410.
- Goiriz R, Rios-Buceta L, Alonso-Perez A, et al. Cutaneous metaplastic synovial cyst. J Am Acad Dermatol. 2005;53:180-181.
- Kim BC, Choi WJ, Park EJ, et al. Cutaneous metaplastic synovial cyst of the first metatarsal head area. Ann Dermatol. 2011;23(suppl 2):S165-S168.
- Yang HC, Tsai YJ, Hu SL, et al. Cutaneous metaplastic synovial cyst--a case report and review of literature. Dermatol Sinica. 2003;21:275-279.
- Kataria SP, Singh G, Batra A, et al. Nodular hidradenoma: a series of five cases in male subjects and review of literature. Adv Cytol Pathol. 2018;3:46-47.
- Mohamed Haflah N, Mohd Kassim A, Hassan Shukur M. Giant epidermoid cyst of the thigh. Malays Orthop J. 2011;5:17-19.
- Torisu-Itakura H, Itakura E, Horiuchi R, et al. Cutaneous ciliated cyst on the leg of a woman of menopausal age. Acta Derm Venereol. 2009;89:323-324.
- Fullen DR. Cysts and sinuses. In: Busam K, ed. Dermatopathology. Saunders; 2010:300-330.
- Gonzalez JG, Ghiselli RW, Santa Cruz DJ. Synovial metaplasia of the skin. Am J Surg Pathol. 1987;11:343-350.
- Calonje E, Brenn T, Lazar A, et al. Cutaneous cysts. In: Calonje E, Brenn T, Lazar A, et al. McKee's Pathology of the Skin. 5th ed. Elsevier Limited; 2020:1680-1697.
- Fukuyama M, Sato Y, Hayakawa J, et al. Cutaneous metaplastic synovial cyst: case report and literature review from the dermatological point of view. Keio J Med. 2016;66:9-13.
- Karaytug K, Kapicioglu M, Can N, et al. Unprecedented recurrence of carpal tunnel syndrome by metaplastic synovial cyst in the carpal tunnel. Acta Orthop Traumatol Turc. 2019;53:230-232.
- Martelli SJ, Silveira FM, Carvalho PH, et al. Asymptomatic subcutaneous swelling of lower face. Oral Surg Oral Med Oral Pathol Oral Radiol. 2019;128:101-105.
- Majdi M, Saffar H, Ghanadan A. Cutaneous metaplastic synovial cyst: a case report. Iran J Pathol. 2016;11:423-426.
- Ramachandra S, Rao L, Al-Kindi M. Cutaneous metaplastic synovial cyst. Sultan Qaboos Univ Med J. 2016;16:E117-E118.
- Heidarian A, Xie Q, Banihashemi A. Cutaneous metaplastic synovial cyst presenting as an axillary mass after modified mastectomy and adjuvant radiotherapy. Am J Clin Pathol. 2016;146:S2.
- Fernandez-Flores A, Barja-Lopez JM. Cutaneous metaplastic synovial cyst in Ehlers-Danlos syndrome. J Cutan Pathol. 2020;47:729-733.
- Choonhakarn C, Tang S. Cutaneous metaplastic synovial cyst. J Dermatol. 2003;30:480-484.
- Guala A, Viglio S, Ottinetti A, et al. Cutaneous metaplastic synovial cyst in Ehlers-Danlos syndrome: report of a second case. Am J Dermatopathol. 2008;30:59-61.
- Nieto S, Buezo GF, Jones-Caballero M, et al. Cutaneous metaplastic synovial cyst in an Ehlers-Danlos patient. Am J Dermatopathol. 1997;19:407-410.
- Goiriz R, Rios-Buceta L, Alonso-Perez A, et al. Cutaneous metaplastic synovial cyst. J Am Acad Dermatol. 2005;53:180-181.
- Kim BC, Choi WJ, Park EJ, et al. Cutaneous metaplastic synovial cyst of the first metatarsal head area. Ann Dermatol. 2011;23(suppl 2):S165-S168.
- Yang HC, Tsai YJ, Hu SL, et al. Cutaneous metaplastic synovial cyst--a case report and review of literature. Dermatol Sinica. 2003;21:275-279.
- Kataria SP, Singh G, Batra A, et al. Nodular hidradenoma: a series of five cases in male subjects and review of literature. Adv Cytol Pathol. 2018;3:46-47.
- Mohamed Haflah N, Mohd Kassim A, Hassan Shukur M. Giant epidermoid cyst of the thigh. Malays Orthop J. 2011;5:17-19.
- Torisu-Itakura H, Itakura E, Horiuchi R, et al. Cutaneous ciliated cyst on the leg of a woman of menopausal age. Acta Derm Venereol. 2009;89:323-324.
- Fullen DR. Cysts and sinuses. In: Busam K, ed. Dermatopathology. Saunders; 2010:300-330.
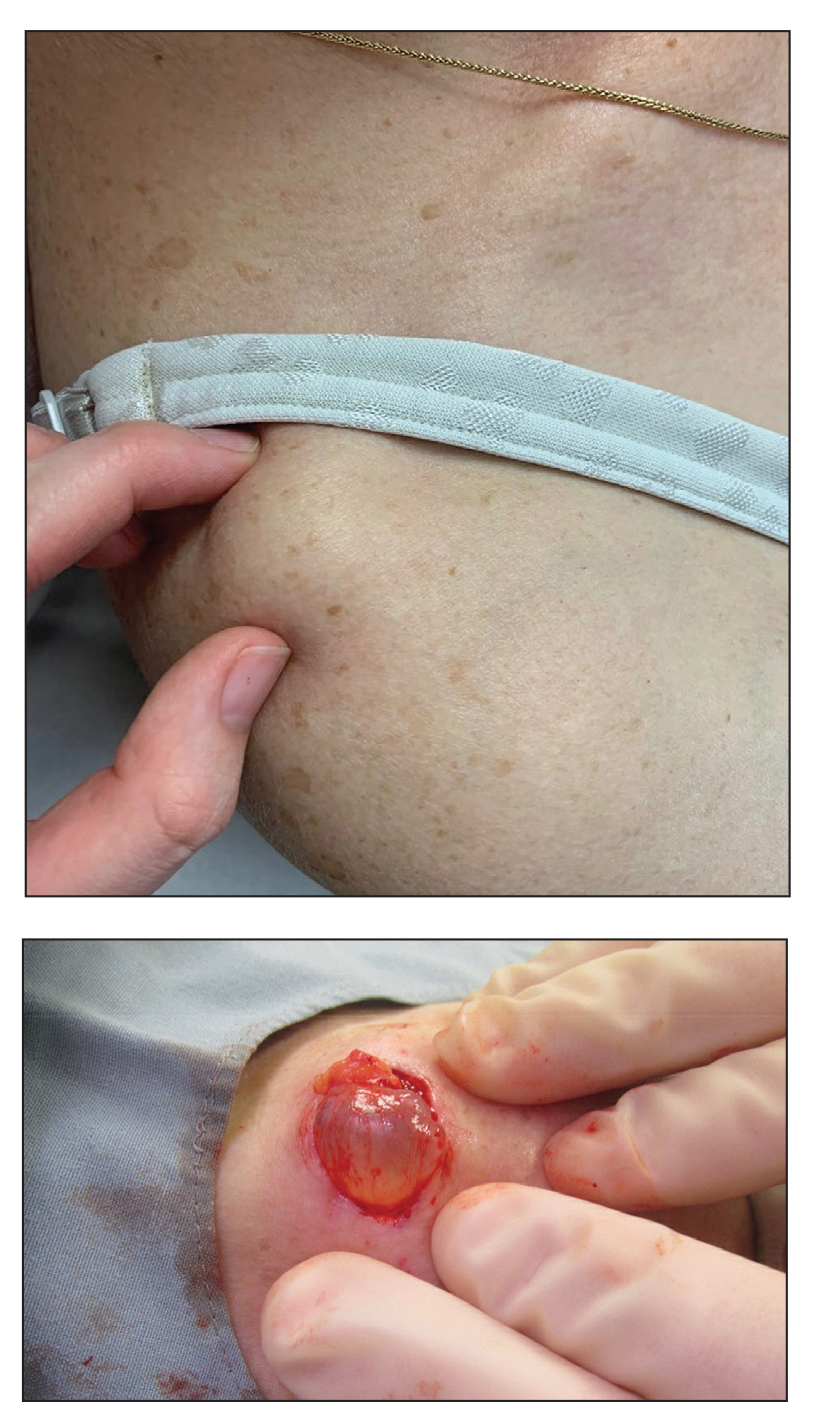
A 70-year-old woman presented to the outpatient dermatology clinic with an acute-onset lesion on the right shoulder. She first noticed a “cyst” developing in the area approximately 3 weeks prior but noted that it may have been present longer. The lesion was bothersome when her undergarments rubbed against it, but she otherwise denied pain, increase in size, or drainage from the site. Her medical history was remarkable for a proliferating trichilemmal tumor on the right parietal scalp treated with Mohs surgery approximately 13 years prior to presentation. She had no personal or family history of skin cancer. Physical examination revealed a 2.5-cm, mobile, nontender, flesh-colored subcutaneous nodule on the right shoulder (top); no ulceration, bleeding, or drainage was present. The surrounding skin demonstrated no clinical changes. The patient was scheduled for outpatient surgical excision of the nodule, which initially was suspected to be a lipoma. During the excision, a translucent cystlike nodule (bottom) was gently dissected and sent for histopathologic examination.
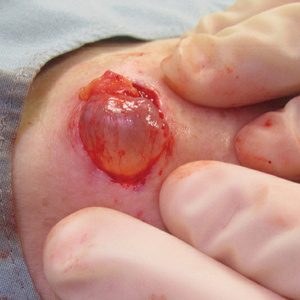
Eruptive Annular Papules on the Trunk of an Organ Transplant Recipient
The Diagnosis: Epidermodysplasia Verruciformis
Histopathologic examination of our patient's biopsy specimen revealed mild acanthosis with prominent hypergranulosis and enlarged keratinocytes with blue-gray cytoplasm (Figure). A diagnosis of acquired epidermodysplasia verruciformis (EV) was rendered. The patient was treated with photodynamic therapy utilizing 5-aminolevulinic acid.
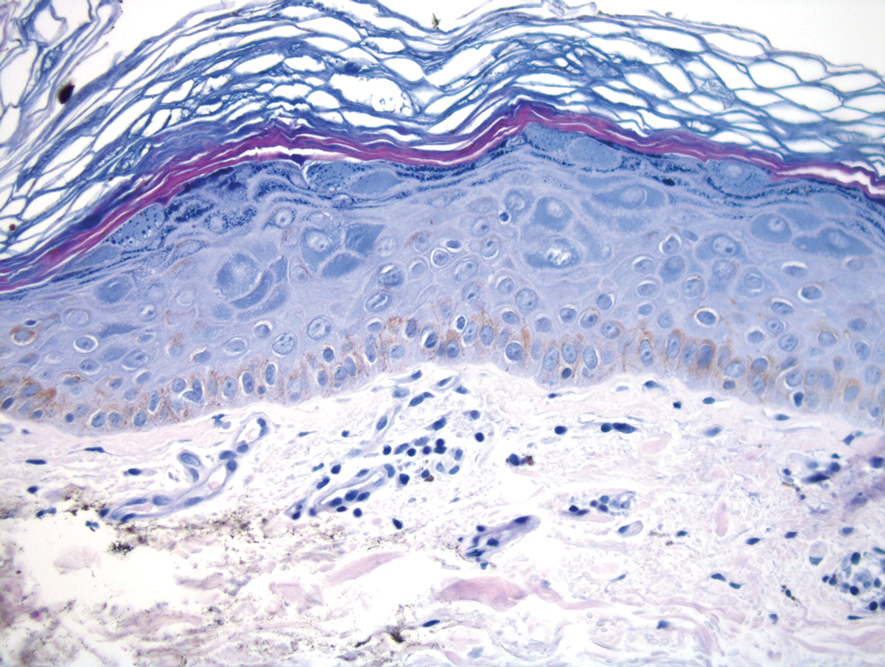
Epidermodysplasia verruciformis is characterized by susceptibility to human papillomavirus (HPV) infections via a defect in cellular immunity. Epidermodysplasia verruciformis was first described as an autosomal-recessive genodermatosis, but it can be acquired in immunosuppressed states with an atypical clinical appearance.1 There are few case reports in skin of color. Acquired EV appears in patients with acquired immunodeficiencies that are susceptible to EV-causing HPVs via a similar mechanism found in inherited EV.2 The most common HPV serotypes involved in EV are HPV-5 and HPV-8. The duration of immunosuppression has been found to be positively correlated with the risk for EV development, with the majority of patients developing lesions after 5 years of immunosuppression.3 There is an approximately 60% risk of malignant transformation of EV lesions into nonmelanoma skin cancer.2 This risk is believed to be lower in patients with darker skin.4
Preventative measures including sun protection and annual surveillance are crucial in EV patients given the high rate of malignant transformation in sun-exposed lesions.5 Treatment options for EV are anecdotal and have variable results, ranging from topicals including 5-fluorouracil and imiquimod to systemic medications including acitretin and interferon.3 Photodynamic therapy can be used for extensive EV. Surgical modalities and other destructive methods also have been tried.6
Epidermodysplasia verruciformis often can be confused with similar dermatoses. Porokeratosis appears as annular pink papules with waferlike peripheral scales. Tinea versicolor is a dermatophyte infection caused by Malassezia furfur and presents as multiple dyspigmented, finely scaling, thin papules and plaques. Subacute cutaneous lupus erythematosus presents as pink, scaly, annular or psoriasiform papules and plaques most commonly on the trunk. Discoid lupus erythematosus presents as pink, hypopigmented or depigmented, atrophic plaques with a peripheral rim of erythema that indicates activity. Secondary syphilis, commonly denoted as the "great mimicker," presents as psoriasiform papules and plaques among other variable morphologies.
- Sa NB, Guerini MB, Barbato MT, et al. Epidermodysplasia verruciformis: clinical presentation with varied forms of lesions. An Bras Dermatol. 2011;86(4 suppl 1):S57-S60.
- Rogers HD, Macgregor JL, Nord KM, et al. Acquired epidermodysplasia verruciformis. J Am Acad Dermatol. 2009;60:315-320.
- Henley JK, Hossler EW. Acquired epidermodysplasia verruciformis occurring in a renal transplant recipient. Cutis. 2017;99:E9-E12.
- Jacyk WK, De Villiers EM. Epidermodysplasia verruciformis in Africans. Int J Dermatol. 1993;32:806-810.
- Fox SH, Elston DM. Epidermodysplasia verruciformis and the risk for malignancy. Cutis. 2016;98:E10-E12.
- Shruti S, Siraj F, Singh A, et al. Epidermodysplasia verruciformis: three case reports and a brief review. Acta Dermatovenerol Alp Pannonica Adriat. 2017;26:59-61.
The Diagnosis: Epidermodysplasia Verruciformis
Histopathologic examination of our patient's biopsy specimen revealed mild acanthosis with prominent hypergranulosis and enlarged keratinocytes with blue-gray cytoplasm (Figure). A diagnosis of acquired epidermodysplasia verruciformis (EV) was rendered. The patient was treated with photodynamic therapy utilizing 5-aminolevulinic acid.
Epidermodysplasia verruciformis is characterized by susceptibility to human papillomavirus (HPV) infections via a defect in cellular immunity. Epidermodysplasia verruciformis was first described as an autosomal-recessive genodermatosis, but it can be acquired in immunosuppressed states with an atypical clinical appearance.1 There are few case reports in skin of color. Acquired EV appears in patients with acquired immunodeficiencies that are susceptible to EV-causing HPVs via a similar mechanism found in inherited EV.2 The most common HPV serotypes involved in EV are HPV-5 and HPV-8. The duration of immunosuppression has been found to be positively correlated with the risk for EV development, with the majority of patients developing lesions after 5 years of immunosuppression.3 There is an approximately 60% risk of malignant transformation of EV lesions into nonmelanoma skin cancer.2 This risk is believed to be lower in patients with darker skin.4
Preventative measures including sun protection and annual surveillance are crucial in EV patients given the high rate of malignant transformation in sun-exposed lesions.5 Treatment options for EV are anecdotal and have variable results, ranging from topicals including 5-fluorouracil and imiquimod to systemic medications including acitretin and interferon.3 Photodynamic therapy can be used for extensive EV. Surgical modalities and other destructive methods also have been tried.6
Epidermodysplasia verruciformis often can be confused with similar dermatoses. Porokeratosis appears as annular pink papules with waferlike peripheral scales. Tinea versicolor is a dermatophyte infection caused by Malassezia furfur and presents as multiple dyspigmented, finely scaling, thin papules and plaques. Subacute cutaneous lupus erythematosus presents as pink, scaly, annular or psoriasiform papules and plaques most commonly on the trunk. Discoid lupus erythematosus presents as pink, hypopigmented or depigmented, atrophic plaques with a peripheral rim of erythema that indicates activity. Secondary syphilis, commonly denoted as the "great mimicker," presents as psoriasiform papules and plaques among other variable morphologies.
The Diagnosis: Epidermodysplasia Verruciformis
Histopathologic examination of our patient's biopsy specimen revealed mild acanthosis with prominent hypergranulosis and enlarged keratinocytes with blue-gray cytoplasm (Figure). A diagnosis of acquired epidermodysplasia verruciformis (EV) was rendered. The patient was treated with photodynamic therapy utilizing 5-aminolevulinic acid.
Epidermodysplasia verruciformis is characterized by susceptibility to human papillomavirus (HPV) infections via a defect in cellular immunity. Epidermodysplasia verruciformis was first described as an autosomal-recessive genodermatosis, but it can be acquired in immunosuppressed states with an atypical clinical appearance.1 There are few case reports in skin of color. Acquired EV appears in patients with acquired immunodeficiencies that are susceptible to EV-causing HPVs via a similar mechanism found in inherited EV.2 The most common HPV serotypes involved in EV are HPV-5 and HPV-8. The duration of immunosuppression has been found to be positively correlated with the risk for EV development, with the majority of patients developing lesions after 5 years of immunosuppression.3 There is an approximately 60% risk of malignant transformation of EV lesions into nonmelanoma skin cancer.2 This risk is believed to be lower in patients with darker skin.4
Preventative measures including sun protection and annual surveillance are crucial in EV patients given the high rate of malignant transformation in sun-exposed lesions.5 Treatment options for EV are anecdotal and have variable results, ranging from topicals including 5-fluorouracil and imiquimod to systemic medications including acitretin and interferon.3 Photodynamic therapy can be used for extensive EV. Surgical modalities and other destructive methods also have been tried.6
Epidermodysplasia verruciformis often can be confused with similar dermatoses. Porokeratosis appears as annular pink papules with waferlike peripheral scales. Tinea versicolor is a dermatophyte infection caused by Malassezia furfur and presents as multiple dyspigmented, finely scaling, thin papules and plaques. Subacute cutaneous lupus erythematosus presents as pink, scaly, annular or psoriasiform papules and plaques most commonly on the trunk. Discoid lupus erythematosus presents as pink, hypopigmented or depigmented, atrophic plaques with a peripheral rim of erythema that indicates activity. Secondary syphilis, commonly denoted as the "great mimicker," presents as psoriasiform papules and plaques among other variable morphologies.
- Sa NB, Guerini MB, Barbato MT, et al. Epidermodysplasia verruciformis: clinical presentation with varied forms of lesions. An Bras Dermatol. 2011;86(4 suppl 1):S57-S60.
- Rogers HD, Macgregor JL, Nord KM, et al. Acquired epidermodysplasia verruciformis. J Am Acad Dermatol. 2009;60:315-320.
- Henley JK, Hossler EW. Acquired epidermodysplasia verruciformis occurring in a renal transplant recipient. Cutis. 2017;99:E9-E12.
- Jacyk WK, De Villiers EM. Epidermodysplasia verruciformis in Africans. Int J Dermatol. 1993;32:806-810.
- Fox SH, Elston DM. Epidermodysplasia verruciformis and the risk for malignancy. Cutis. 2016;98:E10-E12.
- Shruti S, Siraj F, Singh A, et al. Epidermodysplasia verruciformis: three case reports and a brief review. Acta Dermatovenerol Alp Pannonica Adriat. 2017;26:59-61.
- Sa NB, Guerini MB, Barbato MT, et al. Epidermodysplasia verruciformis: clinical presentation with varied forms of lesions. An Bras Dermatol. 2011;86(4 suppl 1):S57-S60.
- Rogers HD, Macgregor JL, Nord KM, et al. Acquired epidermodysplasia verruciformis. J Am Acad Dermatol. 2009;60:315-320.
- Henley JK, Hossler EW. Acquired epidermodysplasia verruciformis occurring in a renal transplant recipient. Cutis. 2017;99:E9-E12.
- Jacyk WK, De Villiers EM. Epidermodysplasia verruciformis in Africans. Int J Dermatol. 1993;32:806-810.
- Fox SH, Elston DM. Epidermodysplasia verruciformis and the risk for malignancy. Cutis. 2016;98:E10-E12.
- Shruti S, Siraj F, Singh A, et al. Epidermodysplasia verruciformis: three case reports and a brief review. Acta Dermatovenerol Alp Pannonica Adriat. 2017;26:59-61.
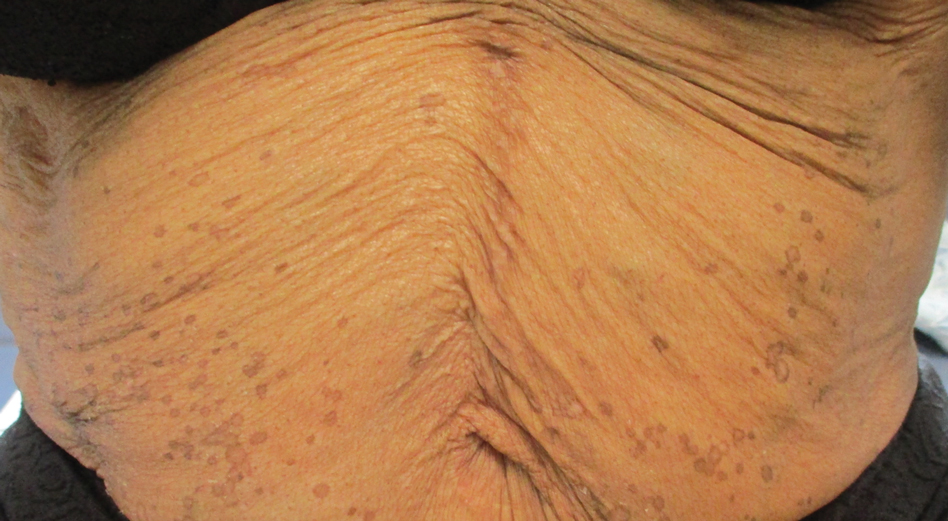
A 50-year-old Black woman with systemic lupus erythematosus and a renal transplant 15 years prior due to lupus nephritis presented with a nonpruritic rash on the abdomen of 1 year’s duration. Her immunosuppressive regimen consisted of tacrolimus, azathioprine, and prednisone. Physical examination revealed numerous monomorphic, annular, hyperpigmented, and thin papules with central clearing present on the abdomen extending to the flanks and groin. The patient denied any family history of similar lesions. A 4-mm punch biopsy of an abdominal lesion was performed.
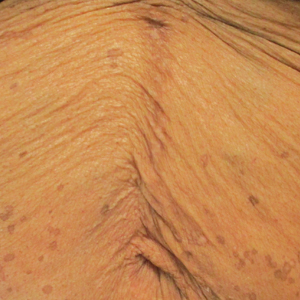
Multifocal Annular Pink Plaques With a Central Violaceous Hue
The Diagnosis: Disseminated Erythema Chronicum Migrans
Empiric treatment with doxycycline 100 mg twice daily for 14 days was initiated for suspected early disseminated Lyme disease manifesting as disseminated multifocal erythema chronicum migrans (Figure). Lyme screening immunoassay and confirmatory IgM Western blot testing subsequently were found to be positive. The clinical history of recent travel to an endemic area and tick bite combined with the recent onset of multifocal erythema migrans lesions, systemic symptoms, elevated erythrocyte sedimentation rate, and positive Lyme serology supported the diagnosis of Lyme disease.
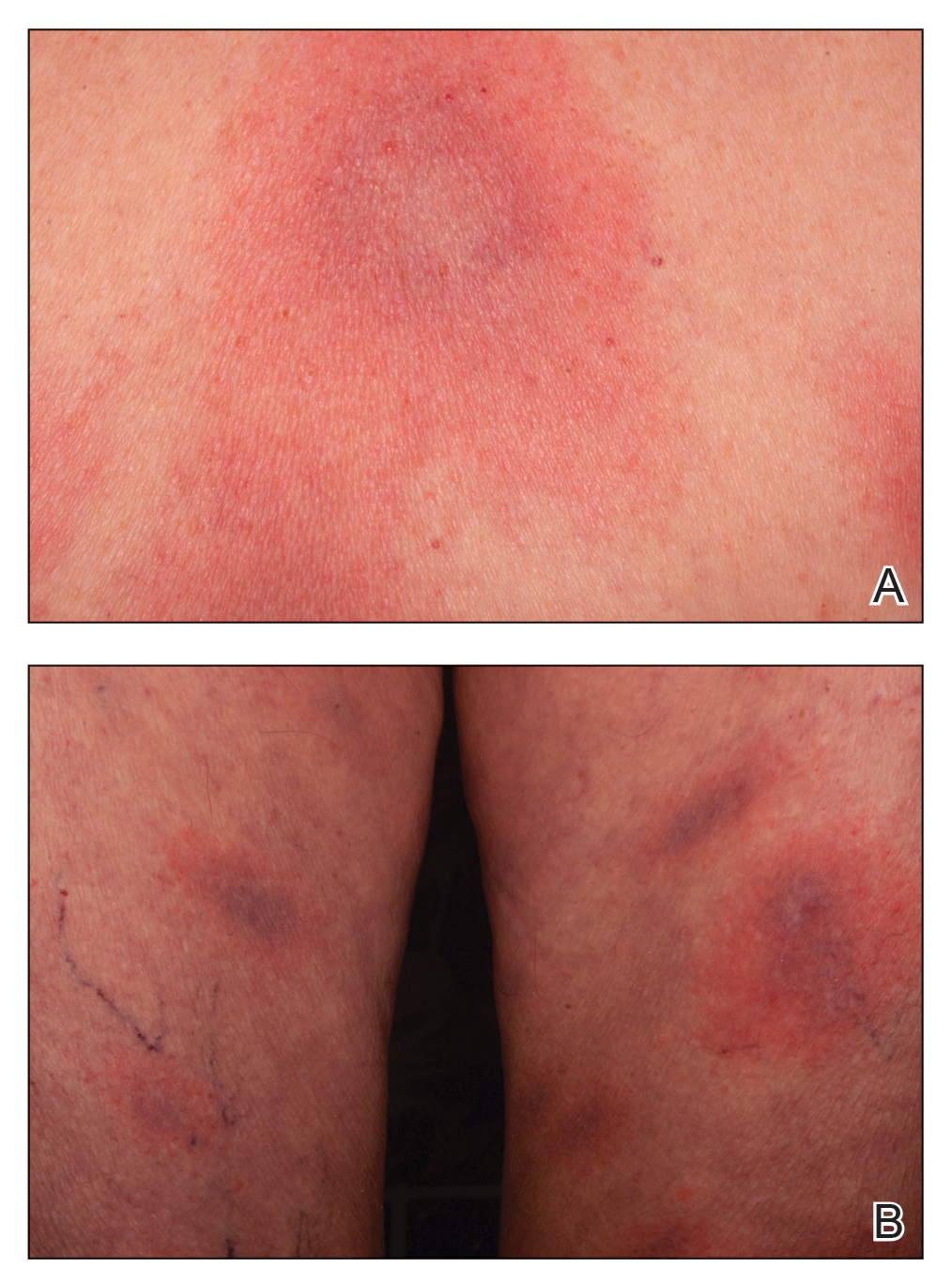
The appropriate clinical context and cutaneous morphology are key when considering the differential diagnosis for multifocal annular lesions. Several entities comprised the differential diagnosis considered in our patient. Sweet syndrome is a neutrophilic dermatosis that can present with fever and varying painful cutaneous lesions. It often is associated with certain medications, underlying illnesses, and infections.1 Our patient’s lesions were not painful, and she had no notable medical history, recent infections, or new medication use, making Sweet syndrome unlikely. A fixed drug eruption was low on the differential, as the patient denied starting any new medications within the 3 months prior to presentation. Erythema multiforme is an acute-onset immunemediated condition of the skin and mucous membranes that typically affects young adults and often is associated with infection (eg, herpes simplex virus, Mycoplasma pneumoniae) or medication use. Cutaneous lesions typically are self-limited, less than 3 cm targets with 3 concentric distinct color zones, often with central bullae or erosions. Although erythema multiforme was higher on the differential, it was less likely, as the patient lacked mucosal lesions and did not have symptoms of underlying herpetic or mycoplasma infection, and the clinical picture was more consistent with Lyme disease. Lastly, the failure for individual skin lesions to resolve within
24 hours excluded the diagnosis of urticaria.
Lyme disease is a tick-borne illness caused by 3 species of the Borrelia spirochete: Borrelia burgdorferi, Borrelia afzelii, and Borrelia garinii.2 In the United States, the disease predominantly is caused by B burgdorferi that is endemic in the upper Midwest and Northeast regions.3 There are 3 stages of Lyme disease: early localized, early disseminated, and late disseminated disease. Early localized disease typically presents with a characteristic single erythema migrans (EM) lesion 3 to 30 days following a bite by the tick Ixodes scapularis.2 The EM lesion gradually can enlarge over a period of several days, reaching up to 12 inches in diameter.2 Early disseminated disease can occur weeks to months following a tick bite and may present with systemic symptoms, multiple widespread
EM lesions, neurologic features such as meningitis or facial nerve palsy, and/or cardiac manifestations such as atrioventricular block or myocarditis. Late disseminated disease can present with chronic arthritis or encephalopathy after months to years if the disease is left untreated.4
Early localized Lyme disease can be diagnosed clinically if the characteristic EM lesion is present in a patient who visited an endemic area. Laboratory testing and Lyme serology are neither required nor recommended in these cases, as the lesion often appears before adequate time has lapsed to develop an adaptive immune response to the organism.5 In contrast, Lyme serology should be ordered in any patient who meets all of the following criteria: (1) patient lives in or has recently traveled to an area endemic for Lyme disease, (2) presence of a risk factor for tick exposure, and (3) symptoms consistent with early disseminated or late Lyme disease. Patients with signs of early or late disseminated disease typically are seropositive, as IgM antibodies can be detected within 2 weeks of onset of the EM lesion and IgG antibodies within 2 to 6 weeks.6 The Centers for Disease Control and Prevention recommends a 2-tiered approach when testing for Lyme disease.7 A screening test with high sensitivity such as an enzyme-linked immunosorbent assay or an immunofluorescence assay initially should be performed.7 If results of the screening test are equivocal or positive, secondary confirmatory testing should be performed via IgM, with or without IgG Western immunoblot assay.7 Biopsy with histologic evaluation can reveal nonspecific findings of vascular endothelial injury and increased mucin deposition. Patients with suspected Lyme disease should immediately be started on empiric treatment with doxycycline 100 mg twice daily for a minimum of 10 days (14–28 days if there is concern for dissemination) to prevent post-Lyme sequelae.5 Our patient’s cutaneous lesions responded to oral doxycycline.
- Sweet’s syndrome. Mayo Clinic. Accessed January 8, 2021. https://www.mayoclinic.org/diseases-conditions/sweets-syndrome /symptoms-causes/syc-20351117
- Steere AC. Lyme disease. N Engl J Med. 2001;345:115-125.
- Lyme disease maps: most recent year. Centers for Disease Control and Prevention. Updated November 22, 2019. Accessed January 8, 2021. https://www.cdc.gov/lyme/datasurveillance /maps-recent.html.
- Steere AC, Sikand VK. The present manifestations of Lyme disease and the outcomes of treatment. N Engl J Med. 2003;348:2472-2474.
- Sanchez E, Vannier E, Wormser GP, et al. Diagnosis, treatment, and prevention of Lyme disease, human granulocytic anaplasmosis, and babesiosis: a review. JAMA. 2016;315:1767-1777.
- Shapiro ED. Borrelia burgdorferi (Lyme disease). Pediatr Rev. 2014; 35:500-509.
- Mead P, Petersen J, Hinckley A. Updated CDC recommendation for serologic diagnosis of Lyme disease. MMWR Morb Mortal Wkly Rep. 2019;68:703
The Diagnosis: Disseminated Erythema Chronicum Migrans
Empiric treatment with doxycycline 100 mg twice daily for 14 days was initiated for suspected early disseminated Lyme disease manifesting as disseminated multifocal erythema chronicum migrans (Figure). Lyme screening immunoassay and confirmatory IgM Western blot testing subsequently were found to be positive. The clinical history of recent travel to an endemic area and tick bite combined with the recent onset of multifocal erythema migrans lesions, systemic symptoms, elevated erythrocyte sedimentation rate, and positive Lyme serology supported the diagnosis of Lyme disease.
The appropriate clinical context and cutaneous morphology are key when considering the differential diagnosis for multifocal annular lesions. Several entities comprised the differential diagnosis considered in our patient. Sweet syndrome is a neutrophilic dermatosis that can present with fever and varying painful cutaneous lesions. It often is associated with certain medications, underlying illnesses, and infections.1 Our patient’s lesions were not painful, and she had no notable medical history, recent infections, or new medication use, making Sweet syndrome unlikely. A fixed drug eruption was low on the differential, as the patient denied starting any new medications within the 3 months prior to presentation. Erythema multiforme is an acute-onset immunemediated condition of the skin and mucous membranes that typically affects young adults and often is associated with infection (eg, herpes simplex virus, Mycoplasma pneumoniae) or medication use. Cutaneous lesions typically are self-limited, less than 3 cm targets with 3 concentric distinct color zones, often with central bullae or erosions. Although erythema multiforme was higher on the differential, it was less likely, as the patient lacked mucosal lesions and did not have symptoms of underlying herpetic or mycoplasma infection, and the clinical picture was more consistent with Lyme disease. Lastly, the failure for individual skin lesions to resolve within
24 hours excluded the diagnosis of urticaria.
Lyme disease is a tick-borne illness caused by 3 species of the Borrelia spirochete: Borrelia burgdorferi, Borrelia afzelii, and Borrelia garinii.2 In the United States, the disease predominantly is caused by B burgdorferi that is endemic in the upper Midwest and Northeast regions.3 There are 3 stages of Lyme disease: early localized, early disseminated, and late disseminated disease. Early localized disease typically presents with a characteristic single erythema migrans (EM) lesion 3 to 30 days following a bite by the tick Ixodes scapularis.2 The EM lesion gradually can enlarge over a period of several days, reaching up to 12 inches in diameter.2 Early disseminated disease can occur weeks to months following a tick bite and may present with systemic symptoms, multiple widespread
EM lesions, neurologic features such as meningitis or facial nerve palsy, and/or cardiac manifestations such as atrioventricular block or myocarditis. Late disseminated disease can present with chronic arthritis or encephalopathy after months to years if the disease is left untreated.4
Early localized Lyme disease can be diagnosed clinically if the characteristic EM lesion is present in a patient who visited an endemic area. Laboratory testing and Lyme serology are neither required nor recommended in these cases, as the lesion often appears before adequate time has lapsed to develop an adaptive immune response to the organism.5 In contrast, Lyme serology should be ordered in any patient who meets all of the following criteria: (1) patient lives in or has recently traveled to an area endemic for Lyme disease, (2) presence of a risk factor for tick exposure, and (3) symptoms consistent with early disseminated or late Lyme disease. Patients with signs of early or late disseminated disease typically are seropositive, as IgM antibodies can be detected within 2 weeks of onset of the EM lesion and IgG antibodies within 2 to 6 weeks.6 The Centers for Disease Control and Prevention recommends a 2-tiered approach when testing for Lyme disease.7 A screening test with high sensitivity such as an enzyme-linked immunosorbent assay or an immunofluorescence assay initially should be performed.7 If results of the screening test are equivocal or positive, secondary confirmatory testing should be performed via IgM, with or without IgG Western immunoblot assay.7 Biopsy with histologic evaluation can reveal nonspecific findings of vascular endothelial injury and increased mucin deposition. Patients with suspected Lyme disease should immediately be started on empiric treatment with doxycycline 100 mg twice daily for a minimum of 10 days (14–28 days if there is concern for dissemination) to prevent post-Lyme sequelae.5 Our patient’s cutaneous lesions responded to oral doxycycline.
The Diagnosis: Disseminated Erythema Chronicum Migrans
Empiric treatment with doxycycline 100 mg twice daily for 14 days was initiated for suspected early disseminated Lyme disease manifesting as disseminated multifocal erythema chronicum migrans (Figure). Lyme screening immunoassay and confirmatory IgM Western blot testing subsequently were found to be positive. The clinical history of recent travel to an endemic area and tick bite combined with the recent onset of multifocal erythema migrans lesions, systemic symptoms, elevated erythrocyte sedimentation rate, and positive Lyme serology supported the diagnosis of Lyme disease.
The appropriate clinical context and cutaneous morphology are key when considering the differential diagnosis for multifocal annular lesions. Several entities comprised the differential diagnosis considered in our patient. Sweet syndrome is a neutrophilic dermatosis that can present with fever and varying painful cutaneous lesions. It often is associated with certain medications, underlying illnesses, and infections.1 Our patient’s lesions were not painful, and she had no notable medical history, recent infections, or new medication use, making Sweet syndrome unlikely. A fixed drug eruption was low on the differential, as the patient denied starting any new medications within the 3 months prior to presentation. Erythema multiforme is an acute-onset immunemediated condition of the skin and mucous membranes that typically affects young adults and often is associated with infection (eg, herpes simplex virus, Mycoplasma pneumoniae) or medication use. Cutaneous lesions typically are self-limited, less than 3 cm targets with 3 concentric distinct color zones, often with central bullae or erosions. Although erythema multiforme was higher on the differential, it was less likely, as the patient lacked mucosal lesions and did not have symptoms of underlying herpetic or mycoplasma infection, and the clinical picture was more consistent with Lyme disease. Lastly, the failure for individual skin lesions to resolve within
24 hours excluded the diagnosis of urticaria.
Lyme disease is a tick-borne illness caused by 3 species of the Borrelia spirochete: Borrelia burgdorferi, Borrelia afzelii, and Borrelia garinii.2 In the United States, the disease predominantly is caused by B burgdorferi that is endemic in the upper Midwest and Northeast regions.3 There are 3 stages of Lyme disease: early localized, early disseminated, and late disseminated disease. Early localized disease typically presents with a characteristic single erythema migrans (EM) lesion 3 to 30 days following a bite by the tick Ixodes scapularis.2 The EM lesion gradually can enlarge over a period of several days, reaching up to 12 inches in diameter.2 Early disseminated disease can occur weeks to months following a tick bite and may present with systemic symptoms, multiple widespread
EM lesions, neurologic features such as meningitis or facial nerve palsy, and/or cardiac manifestations such as atrioventricular block or myocarditis. Late disseminated disease can present with chronic arthritis or encephalopathy after months to years if the disease is left untreated.4
Early localized Lyme disease can be diagnosed clinically if the characteristic EM lesion is present in a patient who visited an endemic area. Laboratory testing and Lyme serology are neither required nor recommended in these cases, as the lesion often appears before adequate time has lapsed to develop an adaptive immune response to the organism.5 In contrast, Lyme serology should be ordered in any patient who meets all of the following criteria: (1) patient lives in or has recently traveled to an area endemic for Lyme disease, (2) presence of a risk factor for tick exposure, and (3) symptoms consistent with early disseminated or late Lyme disease. Patients with signs of early or late disseminated disease typically are seropositive, as IgM antibodies can be detected within 2 weeks of onset of the EM lesion and IgG antibodies within 2 to 6 weeks.6 The Centers for Disease Control and Prevention recommends a 2-tiered approach when testing for Lyme disease.7 A screening test with high sensitivity such as an enzyme-linked immunosorbent assay or an immunofluorescence assay initially should be performed.7 If results of the screening test are equivocal or positive, secondary confirmatory testing should be performed via IgM, with or without IgG Western immunoblot assay.7 Biopsy with histologic evaluation can reveal nonspecific findings of vascular endothelial injury and increased mucin deposition. Patients with suspected Lyme disease should immediately be started on empiric treatment with doxycycline 100 mg twice daily for a minimum of 10 days (14–28 days if there is concern for dissemination) to prevent post-Lyme sequelae.5 Our patient’s cutaneous lesions responded to oral doxycycline.
- Sweet’s syndrome. Mayo Clinic. Accessed January 8, 2021. https://www.mayoclinic.org/diseases-conditions/sweets-syndrome /symptoms-causes/syc-20351117
- Steere AC. Lyme disease. N Engl J Med. 2001;345:115-125.
- Lyme disease maps: most recent year. Centers for Disease Control and Prevention. Updated November 22, 2019. Accessed January 8, 2021. https://www.cdc.gov/lyme/datasurveillance /maps-recent.html.
- Steere AC, Sikand VK. The present manifestations of Lyme disease and the outcomes of treatment. N Engl J Med. 2003;348:2472-2474.
- Sanchez E, Vannier E, Wormser GP, et al. Diagnosis, treatment, and prevention of Lyme disease, human granulocytic anaplasmosis, and babesiosis: a review. JAMA. 2016;315:1767-1777.
- Shapiro ED. Borrelia burgdorferi (Lyme disease). Pediatr Rev. 2014; 35:500-509.
- Mead P, Petersen J, Hinckley A. Updated CDC recommendation for serologic diagnosis of Lyme disease. MMWR Morb Mortal Wkly Rep. 2019;68:703
- Sweet’s syndrome. Mayo Clinic. Accessed January 8, 2021. https://www.mayoclinic.org/diseases-conditions/sweets-syndrome /symptoms-causes/syc-20351117
- Steere AC. Lyme disease. N Engl J Med. 2001;345:115-125.
- Lyme disease maps: most recent year. Centers for Disease Control and Prevention. Updated November 22, 2019. Accessed January 8, 2021. https://www.cdc.gov/lyme/datasurveillance /maps-recent.html.
- Steere AC, Sikand VK. The present manifestations of Lyme disease and the outcomes of treatment. N Engl J Med. 2003;348:2472-2474.
- Sanchez E, Vannier E, Wormser GP, et al. Diagnosis, treatment, and prevention of Lyme disease, human granulocytic anaplasmosis, and babesiosis: a review. JAMA. 2016;315:1767-1777.
- Shapiro ED. Borrelia burgdorferi (Lyme disease). Pediatr Rev. 2014; 35:500-509.
- Mead P, Petersen J, Hinckley A. Updated CDC recommendation for serologic diagnosis of Lyme disease. MMWR Morb Mortal Wkly Rep. 2019;68:703
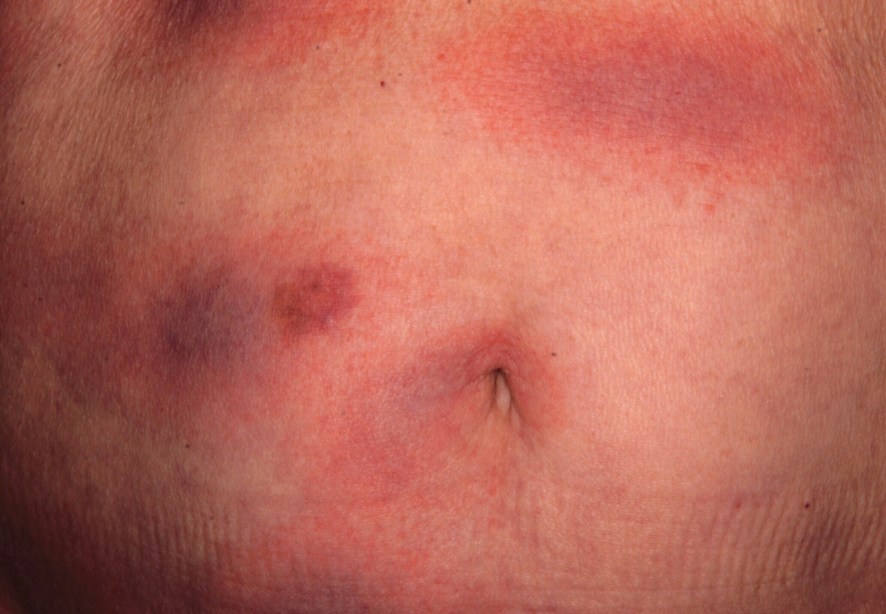
An otherwise healthy 78-year-old woman presented with a diffuse, mildly itchy rash of 5 days’ duration with associated fatigue, chills, decreased appetite, and nausea. She reported waking up with her arms “feeling like they weigh a ton.” She denied any pain, bleeding, or oozing and was unsure if new spots were continuing to develop. The patient reported having allergies to numerous medications but denied any new medications or recent illnesses. She had recently spent time on a farm in Minnesota, and upon further questioning she recalled a tick bite 2 months prior to presentation. She stated that she removed the nonengorged tick and that it could not have been attached for more than 24 hours. Her medical and family history were unremarkable. Physical examination showed multiple annular pink plaques with a central violaceous hue in a generalized distribution involving the face, trunk, arms, and legs with mild erythema of the palms. The plantar surfaces were clear, and there was no evidence of lymphadenopathy. The remainder of the physical examination and review of systems was negative. Laboratory screening was notable for an elevated erythrocyte sedimentation rate and C-reactive protein level with negative antinuclear antibodies.
Pink Patches With a Hyperpigmented Rim
The Diagnosis: Phytophotodermatitis
A more detailed patient history revealed that there was beer with limes on the boat, but the partygoers neglected to bring a knife. The patient volunteered to tear the limes apart with his bare hands. Because he was clad only in swim trunks, lime juice splattered over various regions of his body.
Phytophotodermatitis is a phototoxic blistering rash that follows topical exposure to plant-derived furocoumarins and sunlight. (Figure) Furocoumarins are photosensitizing substances produced by certain plants, possibly as a defense mechanism against predators.1 They cause a nonimmunologic phototoxic reaction when deposited on the skin and exposed to UVA radiation. Exposure to limes is the most common precipitant of phytophotodermatitis, but other potential culprits include lemons, grapefruit, figs, carrots, parsnips, celery, and dill.2
Lesions associated with phytophotodermatitis classically present as painful erythematous patches and bullae in regions of furocoumarin exposure. Affected areas are well demarcated and irregularly shaped and heal with a characteristic hyperpigmented rim. They often have a downward streak pattern from the dripping juice.3 If the furocoumarins are transferred by touch, lesions can appear in the shape of handprints, which may raise alarms for physical abuse in children.4
Photochemical reactions caused by activated furocoumarins cross-link nuclear DNA and damage cell membranes. These changes lead to cellular death resulting in edema and destruction of the epidermis. Other effects include an increase in keratin and thickening of the stratum corneum. The hyperpigmentation is a result of increased concentration of melanosomes and stimulation of melanocytes by activated furocoumarins.5
Management of phytophotodermatitis depends on the severity of skin injury. Mild cases may not require any treatment, whereas the most severe ones require admission to a burn unit for wound care. Anti-inflammatory medications are the mainstay of therapy. Our patient was prescribed desonide cream 0.05% for application to the affected areas. Sunscreen should be applied to prevent worsening of hyperpigmentation, which may take months to years to fade naturally. If hyperpigmentation is cosmetically troubling to the patient, bleaching agents such as hydroquinone and retinoids or Nd:YAG laser can be used to accelerate the resolution of pigment.5
Phototoxicity differs from less common photoallergic reactions caused by preformed antibodies or a delayed cell-mediated response to a trigger. The classic presentation of photoallergy is apruritic, inflammatory, bullous eruption in a sensitized individual.6 Allergic contact dermatitis more commonly is associated with pruritus than pain, and it presents as a papulovesicular eruption that evolves into lichenified plaques.7 Porphyria cutanea tarda would likely be accompanied by other cutaneous features such as hypertrichosis and sclerodermoid plaques with dystrophic calcification, in addition to wine-colored urine-containing porphyrins.8 Bullous fixed drug eruptions develop within 48 hours of exposure to a causative agent. The patient typically would experience pruritus and burning at the site of clearly demarcated erythematous lesions that healed with hyperpigmentation.9 Lesions of bullous lupus erythematosus may appear in areas without sun exposure, and they would be more likely to leave behind hypopigmentation rather than hyperpigmentation.10
- Pathak MA. Phytophotodermatitis. Clin Dermatol. 1986;4:102-121.
- Egan CL, Sterling G. Phytophotodermatitis: a visit to Margaritaville. Cutis. 1993;51:41-42.
- Hankinson A, Lloyd B, Alweis R. Lime-induced phytophotodermatitis [published online ahead of print September 29, 2014]. J Community Hosp Intern Med Perspect. doi:10.3402/jchimp.v4.25090
- Fitzpatrick JK, Kohlwes J. Lime-induced phytophotodermatitis. J Gen Intern Med. 2018;33:975.
- Weber IC, Davis CP, Greeson DM. Phytophotodermatitis: the other "lime" disease. J Emerg Med. 1999;17:235-237.
- Monteiro AF, Rato M, Martins C. Drug-induced photosensitivity: photoallergic and phototoxic reactions. Clin Dermatol. 2016;34:571-581.
- Tan CH, Rasool S, Johnston GA. Contact dermatitis: allergic and irritant. Clin Dermatol. 2014;32:116-124.
- Dawe R. An overview of the cutaneous porphyrias. F1000Res. 2017;6:1906.
- Bandino JP, Wohltmann WE, Bray DW, et al. Naproxen-induced generalized bullous fixed drug eruption. Dermatol Online J. 2009;15:4.
- Contestable JJ, Edhegard KD, Meyerle JH. Bullous systemic lupus erythematosus: a review and update to diagnosis and treatment. Am J Clin Dermatol. 2014;15:517-524.
The Diagnosis: Phytophotodermatitis
A more detailed patient history revealed that there was beer with limes on the boat, but the partygoers neglected to bring a knife. The patient volunteered to tear the limes apart with his bare hands. Because he was clad only in swim trunks, lime juice splattered over various regions of his body.
Phytophotodermatitis is a phototoxic blistering rash that follows topical exposure to plant-derived furocoumarins and sunlight. (Figure) Furocoumarins are photosensitizing substances produced by certain plants, possibly as a defense mechanism against predators.1 They cause a nonimmunologic phototoxic reaction when deposited on the skin and exposed to UVA radiation. Exposure to limes is the most common precipitant of phytophotodermatitis, but other potential culprits include lemons, grapefruit, figs, carrots, parsnips, celery, and dill.2
Lesions associated with phytophotodermatitis classically present as painful erythematous patches and bullae in regions of furocoumarin exposure. Affected areas are well demarcated and irregularly shaped and heal with a characteristic hyperpigmented rim. They often have a downward streak pattern from the dripping juice.3 If the furocoumarins are transferred by touch, lesions can appear in the shape of handprints, which may raise alarms for physical abuse in children.4
Photochemical reactions caused by activated furocoumarins cross-link nuclear DNA and damage cell membranes. These changes lead to cellular death resulting in edema and destruction of the epidermis. Other effects include an increase in keratin and thickening of the stratum corneum. The hyperpigmentation is a result of increased concentration of melanosomes and stimulation of melanocytes by activated furocoumarins.5
Management of phytophotodermatitis depends on the severity of skin injury. Mild cases may not require any treatment, whereas the most severe ones require admission to a burn unit for wound care. Anti-inflammatory medications are the mainstay of therapy. Our patient was prescribed desonide cream 0.05% for application to the affected areas. Sunscreen should be applied to prevent worsening of hyperpigmentation, which may take months to years to fade naturally. If hyperpigmentation is cosmetically troubling to the patient, bleaching agents such as hydroquinone and retinoids or Nd:YAG laser can be used to accelerate the resolution of pigment.5
Phototoxicity differs from less common photoallergic reactions caused by preformed antibodies or a delayed cell-mediated response to a trigger. The classic presentation of photoallergy is apruritic, inflammatory, bullous eruption in a sensitized individual.6 Allergic contact dermatitis more commonly is associated with pruritus than pain, and it presents as a papulovesicular eruption that evolves into lichenified plaques.7 Porphyria cutanea tarda would likely be accompanied by other cutaneous features such as hypertrichosis and sclerodermoid plaques with dystrophic calcification, in addition to wine-colored urine-containing porphyrins.8 Bullous fixed drug eruptions develop within 48 hours of exposure to a causative agent. The patient typically would experience pruritus and burning at the site of clearly demarcated erythematous lesions that healed with hyperpigmentation.9 Lesions of bullous lupus erythematosus may appear in areas without sun exposure, and they would be more likely to leave behind hypopigmentation rather than hyperpigmentation.10
The Diagnosis: Phytophotodermatitis
A more detailed patient history revealed that there was beer with limes on the boat, but the partygoers neglected to bring a knife. The patient volunteered to tear the limes apart with his bare hands. Because he was clad only in swim trunks, lime juice splattered over various regions of his body.
Phytophotodermatitis is a phototoxic blistering rash that follows topical exposure to plant-derived furocoumarins and sunlight. (Figure) Furocoumarins are photosensitizing substances produced by certain plants, possibly as a defense mechanism against predators.1 They cause a nonimmunologic phototoxic reaction when deposited on the skin and exposed to UVA radiation. Exposure to limes is the most common precipitant of phytophotodermatitis, but other potential culprits include lemons, grapefruit, figs, carrots, parsnips, celery, and dill.2
Lesions associated with phytophotodermatitis classically present as painful erythematous patches and bullae in regions of furocoumarin exposure. Affected areas are well demarcated and irregularly shaped and heal with a characteristic hyperpigmented rim. They often have a downward streak pattern from the dripping juice.3 If the furocoumarins are transferred by touch, lesions can appear in the shape of handprints, which may raise alarms for physical abuse in children.4
Photochemical reactions caused by activated furocoumarins cross-link nuclear DNA and damage cell membranes. These changes lead to cellular death resulting in edema and destruction of the epidermis. Other effects include an increase in keratin and thickening of the stratum corneum. The hyperpigmentation is a result of increased concentration of melanosomes and stimulation of melanocytes by activated furocoumarins.5
Management of phytophotodermatitis depends on the severity of skin injury. Mild cases may not require any treatment, whereas the most severe ones require admission to a burn unit for wound care. Anti-inflammatory medications are the mainstay of therapy. Our patient was prescribed desonide cream 0.05% for application to the affected areas. Sunscreen should be applied to prevent worsening of hyperpigmentation, which may take months to years to fade naturally. If hyperpigmentation is cosmetically troubling to the patient, bleaching agents such as hydroquinone and retinoids or Nd:YAG laser can be used to accelerate the resolution of pigment.5
Phototoxicity differs from less common photoallergic reactions caused by preformed antibodies or a delayed cell-mediated response to a trigger. The classic presentation of photoallergy is apruritic, inflammatory, bullous eruption in a sensitized individual.6 Allergic contact dermatitis more commonly is associated with pruritus than pain, and it presents as a papulovesicular eruption that evolves into lichenified plaques.7 Porphyria cutanea tarda would likely be accompanied by other cutaneous features such as hypertrichosis and sclerodermoid plaques with dystrophic calcification, in addition to wine-colored urine-containing porphyrins.8 Bullous fixed drug eruptions develop within 48 hours of exposure to a causative agent. The patient typically would experience pruritus and burning at the site of clearly demarcated erythematous lesions that healed with hyperpigmentation.9 Lesions of bullous lupus erythematosus may appear in areas without sun exposure, and they would be more likely to leave behind hypopigmentation rather than hyperpigmentation.10
- Pathak MA. Phytophotodermatitis. Clin Dermatol. 1986;4:102-121.
- Egan CL, Sterling G. Phytophotodermatitis: a visit to Margaritaville. Cutis. 1993;51:41-42.
- Hankinson A, Lloyd B, Alweis R. Lime-induced phytophotodermatitis [published online ahead of print September 29, 2014]. J Community Hosp Intern Med Perspect. doi:10.3402/jchimp.v4.25090
- Fitzpatrick JK, Kohlwes J. Lime-induced phytophotodermatitis. J Gen Intern Med. 2018;33:975.
- Weber IC, Davis CP, Greeson DM. Phytophotodermatitis: the other "lime" disease. J Emerg Med. 1999;17:235-237.
- Monteiro AF, Rato M, Martins C. Drug-induced photosensitivity: photoallergic and phototoxic reactions. Clin Dermatol. 2016;34:571-581.
- Tan CH, Rasool S, Johnston GA. Contact dermatitis: allergic and irritant. Clin Dermatol. 2014;32:116-124.
- Dawe R. An overview of the cutaneous porphyrias. F1000Res. 2017;6:1906.
- Bandino JP, Wohltmann WE, Bray DW, et al. Naproxen-induced generalized bullous fixed drug eruption. Dermatol Online J. 2009;15:4.
- Contestable JJ, Edhegard KD, Meyerle JH. Bullous systemic lupus erythematosus: a review and update to diagnosis and treatment. Am J Clin Dermatol. 2014;15:517-524.
- Pathak MA. Phytophotodermatitis. Clin Dermatol. 1986;4:102-121.
- Egan CL, Sterling G. Phytophotodermatitis: a visit to Margaritaville. Cutis. 1993;51:41-42.
- Hankinson A, Lloyd B, Alweis R. Lime-induced phytophotodermatitis [published online ahead of print September 29, 2014]. J Community Hosp Intern Med Perspect. doi:10.3402/jchimp.v4.25090
- Fitzpatrick JK, Kohlwes J. Lime-induced phytophotodermatitis. J Gen Intern Med. 2018;33:975.
- Weber IC, Davis CP, Greeson DM. Phytophotodermatitis: the other "lime" disease. J Emerg Med. 1999;17:235-237.
- Monteiro AF, Rato M, Martins C. Drug-induced photosensitivity: photoallergic and phototoxic reactions. Clin Dermatol. 2016;34:571-581.
- Tan CH, Rasool S, Johnston GA. Contact dermatitis: allergic and irritant. Clin Dermatol. 2014;32:116-124.
- Dawe R. An overview of the cutaneous porphyrias. F1000Res. 2017;6:1906.
- Bandino JP, Wohltmann WE, Bray DW, et al. Naproxen-induced generalized bullous fixed drug eruption. Dermatol Online J. 2009;15:4.
- Contestable JJ, Edhegard KD, Meyerle JH. Bullous systemic lupus erythematosus: a review and update to diagnosis and treatment. Am J Clin Dermatol. 2014;15:517-524.
A 25-year-old man presented with a rash on the right hand, chest, abdomen, right thigh, and ankles of 2 weeks’ duration. He reported that the eruption began with bullous lesions following a boat trip. The bullae ruptured over the next several days, and the lesions evolved to the current appearance. Although the patient had experienced pain at the site of active blisters, he denied any current pain, itching, or bleeding from the lesions. No other medical comorbidities were present.