User login
Pitted Depressions on the Hands and Elbows
The Diagnosis: Bazex‐Dupré‐Christol Syndrome
Bazex‐Dupré‐Christol syndrome (BDCS) is a rare X-linked dominant genodermatosis characterized by a triad of hypotrichosis, follicular atrophoderma, and multiple basal cell carcinomas (BCCs). Since first being described in 1964,1 there have been fewer than 200 reported cases of BDCS.2 Although a causative gene has not yet been identified, the mutation has been mapped to an 11.4-Mb interval in the Xq25-27.1 region of the X chromosome.3
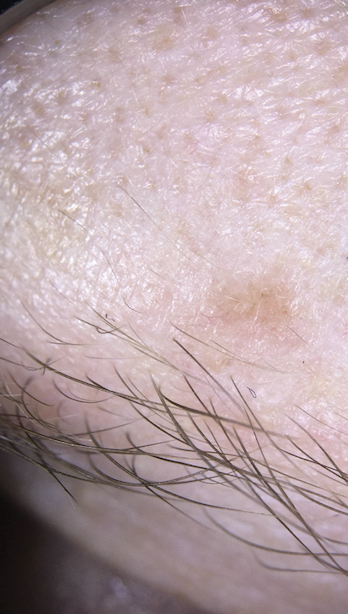
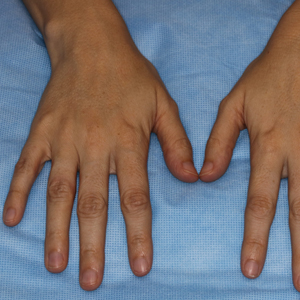
Classically, congenital hypotrichosis is the first observed symptom and can present shortly after birth.4 It typically is widespread, though sometimes it may be confined to the eyebrows, eyelashes, and scalp. Follicular atrophoderma, which occurs due to a laxation and deepening of the follicular ostia, is seen in 80% of cases and typically presents in early childhood as depressions lacking hair.2 It commonly is found on the face, extensor surfaces of the elbows and knees, and dorsal aspects of the hands and feet. Physical examination of our patient revealed follicular atrophoderma on both the dorsal surfaces of the hands and the extensor surfaces of the elbows. Hair shaft anomalies including pili torti, pili bifurcati, and trichorrhexis nodosa are infrequently observed symptoms of BDCS.2
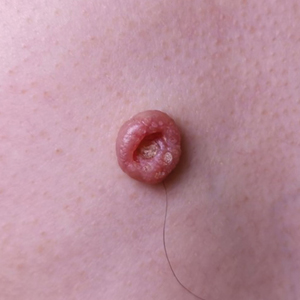
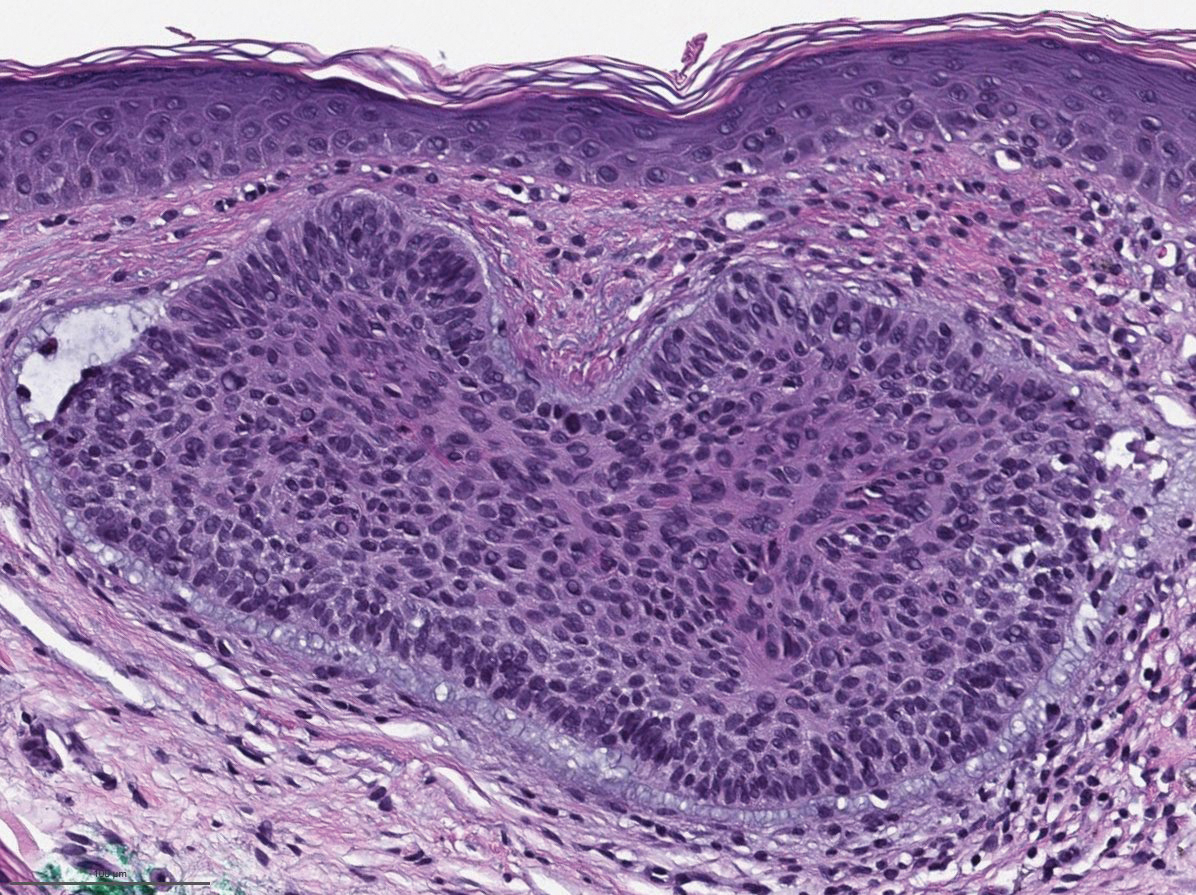
Basal cell carcinoma often manifests in the second or third decades of life, though there are reports of BCC developing in BDCS patients as young as 3 years. Basal cell carcinoma typically arises on sun-exposed areas, especially the face, neck, and chest. These lesions can be pigmented or nonpigmented and range from 2 to 20 mm in diameter.4 Our patient presented with a BCC on the forehead (Figure 1). Histopathologic evaluation showed a proliferation of basaloid cells with peripheral palisading (Figure 2), confirming the diagnosis of BCC.
Milia, which are not considered part of the classic BDCS triad, are seen in 70% of cases.2 They commonly are found on the face and often diminish with age. Milia may precede the formation of follicular atrophoderma and BCC. Hypohidrosis most commonly occurs on the forehead but can be widespread.2 Other less commonly observed features include epidermal cysts, hyperpigmentation of the face, and trichoepitheliomas.4 The management of BDCS involves frequent clinical examinations, BCC treatment, genetic counseling, and photoprotection.2,4
Nevoid BCC syndrome (NBCCS), also known as Gorlin-Goltz syndrome, is an autosomal-dominant disease characterized by multiple nevoid BCCs, macrocephaly with a large forehead, cleft lip or palate, jaw keratocysts, palmar and plantar pits, and calcification of the falx cerebri.5 Nevoid BCC syndrome is caused by a mutation in the PTCH1 gene in the hedgehog signaling pathway.6 The absence of common symptoms of NBCCS including macrocephaly, palmar or plantar pits, and cleft lip or palate, as well as negative genetic testing, suggested that our patient did not have NBCCS.
Rombo syndrome shares features with BDCS. Similar to BDCS, symptoms of Rombo syndrome include follicular atrophy, milialike papules, and BCC. Patients with Rombo syndrome typically present with atrophoderma vermiculatum on the cheeks and forehead in childhood.7 This atrophoderma presents with a pitted atrophic appearance in a reticular pattern on sun-exposed areas. Other distinguishing features from BDCS include cyanotic redness of sun-exposed skin and telangiectatic vessels.8
Multiple hereditary infundibulocystic BCC is another rare genodermatosis that is characterized by the presence of multiple infundibulocystic BCCs on the face and genitals. Infundibulocystic BCC is a well-differentiated subtype of BCC characterized by buds and cords of basaloid cells with scant stroma. Multiple hereditary infundibulocystic BCC is inherited in an autosomal-dominant fashion and has been linked to SUFU mutation in the sonic hedgehog pathway.9
Rothmund-Thomson syndrome is an autosomalrecessive disorder characterized by sparse hair, skeletal and dental abnormalities, and a high risk for developing keratinocyte carcinomas. It is differentiated from BDCS clinically by the presence of erythema, edema, and blistering, resulting in poikiloderma, plantar hyperkeratotic lesions, and bone defects.10
- Bazex A. Génodermatose complexe de type indéterminé associant une hypotrichose, un état atrophodermique généralisé et des dégénérescences cutanées multiples (épitheliomas baso-cellulaires). Bull Soc Fr Derm Syphiligr. 1964;71:206.
- Al Sabbagh MM, Baqi MA. Bazex-Dupre-Christol syndrome: review of clinical and molecular aspects. Int J Dermatol. 2018;57:1102-1106.
- Parren LJ, Abuzahra F, Wagenvoort T, et al. Linkage refinement of Bazex-Dupre-Christol syndrome to an 11.4-Mb interval on chromosome Xq25-27.1. Br J Dermatol. 2011;165:201-203.
- Abuzahra F, Parren LJ, Frank J. Multiple familial and pigmented basal cell carcinomas in early childhood--Bazex-Dupre-Christol syndrome. J Eur Acad Dermatol Venereol. 2012;26:117-121.
- Shevchenko A, Durkin JR, Moon AT. Generalized basaloid follicular hamartoma syndrome versus Gorlin syndrome: a diagnostic challenge. Pediatr Dermatol. 2018;35:E396-E397.
- Fujii K, Miyashita T. Gorlin syndrome (nevoid basal cell carcinoma syndrome): update and literature review. Pediatr Int. 2014;56:667-674.
- van Steensel MA, Jaspers NG, Steijlen PM. A case of Rombo syndrome. Br J Dermatol. 2001;144:1215-1218.
- Lee YC, Son SJ, Han TY, et al. A case of atrophoderma vermiculatum showing a good response to topical tretinoin. Ann Dermatol. 2018;30:116-118.
- Schulman JM, Oh DH, Sanborn JZ, et al. Multiple hereditary infundibulocystic basal cell carcinoma syndrome associated with a germline SUFU mutation. JAMA Dermatol. 2016;152:323-327.
- Larizza L, Roversi G, Volpi L. Rothmund-Thomson syndrome. Orphanet J Rare Dis. 2010;5:2.
The Diagnosis: Bazex‐Dupré‐Christol Syndrome
Bazex‐Dupré‐Christol syndrome (BDCS) is a rare X-linked dominant genodermatosis characterized by a triad of hypotrichosis, follicular atrophoderma, and multiple basal cell carcinomas (BCCs). Since first being described in 1964,1 there have been fewer than 200 reported cases of BDCS.2 Although a causative gene has not yet been identified, the mutation has been mapped to an 11.4-Mb interval in the Xq25-27.1 region of the X chromosome.3
Classically, congenital hypotrichosis is the first observed symptom and can present shortly after birth.4 It typically is widespread, though sometimes it may be confined to the eyebrows, eyelashes, and scalp. Follicular atrophoderma, which occurs due to a laxation and deepening of the follicular ostia, is seen in 80% of cases and typically presents in early childhood as depressions lacking hair.2 It commonly is found on the face, extensor surfaces of the elbows and knees, and dorsal aspects of the hands and feet. Physical examination of our patient revealed follicular atrophoderma on both the dorsal surfaces of the hands and the extensor surfaces of the elbows. Hair shaft anomalies including pili torti, pili bifurcati, and trichorrhexis nodosa are infrequently observed symptoms of BDCS.2
Basal cell carcinoma often manifests in the second or third decades of life, though there are reports of BCC developing in BDCS patients as young as 3 years. Basal cell carcinoma typically arises on sun-exposed areas, especially the face, neck, and chest. These lesions can be pigmented or nonpigmented and range from 2 to 20 mm in diameter.4 Our patient presented with a BCC on the forehead (Figure 1). Histopathologic evaluation showed a proliferation of basaloid cells with peripheral palisading (Figure 2), confirming the diagnosis of BCC.
Milia, which are not considered part of the classic BDCS triad, are seen in 70% of cases.2 They commonly are found on the face and often diminish with age. Milia may precede the formation of follicular atrophoderma and BCC. Hypohidrosis most commonly occurs on the forehead but can be widespread.2 Other less commonly observed features include epidermal cysts, hyperpigmentation of the face, and trichoepitheliomas.4 The management of BDCS involves frequent clinical examinations, BCC treatment, genetic counseling, and photoprotection.2,4
Nevoid BCC syndrome (NBCCS), also known as Gorlin-Goltz syndrome, is an autosomal-dominant disease characterized by multiple nevoid BCCs, macrocephaly with a large forehead, cleft lip or palate, jaw keratocysts, palmar and plantar pits, and calcification of the falx cerebri.5 Nevoid BCC syndrome is caused by a mutation in the PTCH1 gene in the hedgehog signaling pathway.6 The absence of common symptoms of NBCCS including macrocephaly, palmar or plantar pits, and cleft lip or palate, as well as negative genetic testing, suggested that our patient did not have NBCCS.
Rombo syndrome shares features with BDCS. Similar to BDCS, symptoms of Rombo syndrome include follicular atrophy, milialike papules, and BCC. Patients with Rombo syndrome typically present with atrophoderma vermiculatum on the cheeks and forehead in childhood.7 This atrophoderma presents with a pitted atrophic appearance in a reticular pattern on sun-exposed areas. Other distinguishing features from BDCS include cyanotic redness of sun-exposed skin and telangiectatic vessels.8
Multiple hereditary infundibulocystic BCC is another rare genodermatosis that is characterized by the presence of multiple infundibulocystic BCCs on the face and genitals. Infundibulocystic BCC is a well-differentiated subtype of BCC characterized by buds and cords of basaloid cells with scant stroma. Multiple hereditary infundibulocystic BCC is inherited in an autosomal-dominant fashion and has been linked to SUFU mutation in the sonic hedgehog pathway.9
Rothmund-Thomson syndrome is an autosomalrecessive disorder characterized by sparse hair, skeletal and dental abnormalities, and a high risk for developing keratinocyte carcinomas. It is differentiated from BDCS clinically by the presence of erythema, edema, and blistering, resulting in poikiloderma, plantar hyperkeratotic lesions, and bone defects.10
The Diagnosis: Bazex‐Dupré‐Christol Syndrome
Bazex‐Dupré‐Christol syndrome (BDCS) is a rare X-linked dominant genodermatosis characterized by a triad of hypotrichosis, follicular atrophoderma, and multiple basal cell carcinomas (BCCs). Since first being described in 1964,1 there have been fewer than 200 reported cases of BDCS.2 Although a causative gene has not yet been identified, the mutation has been mapped to an 11.4-Mb interval in the Xq25-27.1 region of the X chromosome.3
Classically, congenital hypotrichosis is the first observed symptom and can present shortly after birth.4 It typically is widespread, though sometimes it may be confined to the eyebrows, eyelashes, and scalp. Follicular atrophoderma, which occurs due to a laxation and deepening of the follicular ostia, is seen in 80% of cases and typically presents in early childhood as depressions lacking hair.2 It commonly is found on the face, extensor surfaces of the elbows and knees, and dorsal aspects of the hands and feet. Physical examination of our patient revealed follicular atrophoderma on both the dorsal surfaces of the hands and the extensor surfaces of the elbows. Hair shaft anomalies including pili torti, pili bifurcati, and trichorrhexis nodosa are infrequently observed symptoms of BDCS.2
Basal cell carcinoma often manifests in the second or third decades of life, though there are reports of BCC developing in BDCS patients as young as 3 years. Basal cell carcinoma typically arises on sun-exposed areas, especially the face, neck, and chest. These lesions can be pigmented or nonpigmented and range from 2 to 20 mm in diameter.4 Our patient presented with a BCC on the forehead (Figure 1). Histopathologic evaluation showed a proliferation of basaloid cells with peripheral palisading (Figure 2), confirming the diagnosis of BCC.
Milia, which are not considered part of the classic BDCS triad, are seen in 70% of cases.2 They commonly are found on the face and often diminish with age. Milia may precede the formation of follicular atrophoderma and BCC. Hypohidrosis most commonly occurs on the forehead but can be widespread.2 Other less commonly observed features include epidermal cysts, hyperpigmentation of the face, and trichoepitheliomas.4 The management of BDCS involves frequent clinical examinations, BCC treatment, genetic counseling, and photoprotection.2,4
Nevoid BCC syndrome (NBCCS), also known as Gorlin-Goltz syndrome, is an autosomal-dominant disease characterized by multiple nevoid BCCs, macrocephaly with a large forehead, cleft lip or palate, jaw keratocysts, palmar and plantar pits, and calcification of the falx cerebri.5 Nevoid BCC syndrome is caused by a mutation in the PTCH1 gene in the hedgehog signaling pathway.6 The absence of common symptoms of NBCCS including macrocephaly, palmar or plantar pits, and cleft lip or palate, as well as negative genetic testing, suggested that our patient did not have NBCCS.
Rombo syndrome shares features with BDCS. Similar to BDCS, symptoms of Rombo syndrome include follicular atrophy, milialike papules, and BCC. Patients with Rombo syndrome typically present with atrophoderma vermiculatum on the cheeks and forehead in childhood.7 This atrophoderma presents with a pitted atrophic appearance in a reticular pattern on sun-exposed areas. Other distinguishing features from BDCS include cyanotic redness of sun-exposed skin and telangiectatic vessels.8
Multiple hereditary infundibulocystic BCC is another rare genodermatosis that is characterized by the presence of multiple infundibulocystic BCCs on the face and genitals. Infundibulocystic BCC is a well-differentiated subtype of BCC characterized by buds and cords of basaloid cells with scant stroma. Multiple hereditary infundibulocystic BCC is inherited in an autosomal-dominant fashion and has been linked to SUFU mutation in the sonic hedgehog pathway.9
Rothmund-Thomson syndrome is an autosomalrecessive disorder characterized by sparse hair, skeletal and dental abnormalities, and a high risk for developing keratinocyte carcinomas. It is differentiated from BDCS clinically by the presence of erythema, edema, and blistering, resulting in poikiloderma, plantar hyperkeratotic lesions, and bone defects.10
- Bazex A. Génodermatose complexe de type indéterminé associant une hypotrichose, un état atrophodermique généralisé et des dégénérescences cutanées multiples (épitheliomas baso-cellulaires). Bull Soc Fr Derm Syphiligr. 1964;71:206.
- Al Sabbagh MM, Baqi MA. Bazex-Dupre-Christol syndrome: review of clinical and molecular aspects. Int J Dermatol. 2018;57:1102-1106.
- Parren LJ, Abuzahra F, Wagenvoort T, et al. Linkage refinement of Bazex-Dupre-Christol syndrome to an 11.4-Mb interval on chromosome Xq25-27.1. Br J Dermatol. 2011;165:201-203.
- Abuzahra F, Parren LJ, Frank J. Multiple familial and pigmented basal cell carcinomas in early childhood--Bazex-Dupre-Christol syndrome. J Eur Acad Dermatol Venereol. 2012;26:117-121.
- Shevchenko A, Durkin JR, Moon AT. Generalized basaloid follicular hamartoma syndrome versus Gorlin syndrome: a diagnostic challenge. Pediatr Dermatol. 2018;35:E396-E397.
- Fujii K, Miyashita T. Gorlin syndrome (nevoid basal cell carcinoma syndrome): update and literature review. Pediatr Int. 2014;56:667-674.
- van Steensel MA, Jaspers NG, Steijlen PM. A case of Rombo syndrome. Br J Dermatol. 2001;144:1215-1218.
- Lee YC, Son SJ, Han TY, et al. A case of atrophoderma vermiculatum showing a good response to topical tretinoin. Ann Dermatol. 2018;30:116-118.
- Schulman JM, Oh DH, Sanborn JZ, et al. Multiple hereditary infundibulocystic basal cell carcinoma syndrome associated with a germline SUFU mutation. JAMA Dermatol. 2016;152:323-327.
- Larizza L, Roversi G, Volpi L. Rothmund-Thomson syndrome. Orphanet J Rare Dis. 2010;5:2.
- Bazex A. Génodermatose complexe de type indéterminé associant une hypotrichose, un état atrophodermique généralisé et des dégénérescences cutanées multiples (épitheliomas baso-cellulaires). Bull Soc Fr Derm Syphiligr. 1964;71:206.
- Al Sabbagh MM, Baqi MA. Bazex-Dupre-Christol syndrome: review of clinical and molecular aspects. Int J Dermatol. 2018;57:1102-1106.
- Parren LJ, Abuzahra F, Wagenvoort T, et al. Linkage refinement of Bazex-Dupre-Christol syndrome to an 11.4-Mb interval on chromosome Xq25-27.1. Br J Dermatol. 2011;165:201-203.
- Abuzahra F, Parren LJ, Frank J. Multiple familial and pigmented basal cell carcinomas in early childhood--Bazex-Dupre-Christol syndrome. J Eur Acad Dermatol Venereol. 2012;26:117-121.
- Shevchenko A, Durkin JR, Moon AT. Generalized basaloid follicular hamartoma syndrome versus Gorlin syndrome: a diagnostic challenge. Pediatr Dermatol. 2018;35:E396-E397.
- Fujii K, Miyashita T. Gorlin syndrome (nevoid basal cell carcinoma syndrome): update and literature review. Pediatr Int. 2014;56:667-674.
- van Steensel MA, Jaspers NG, Steijlen PM. A case of Rombo syndrome. Br J Dermatol. 2001;144:1215-1218.
- Lee YC, Son SJ, Han TY, et al. A case of atrophoderma vermiculatum showing a good response to topical tretinoin. Ann Dermatol. 2018;30:116-118.
- Schulman JM, Oh DH, Sanborn JZ, et al. Multiple hereditary infundibulocystic basal cell carcinoma syndrome associated with a germline SUFU mutation. JAMA Dermatol. 2016;152:323-327.
- Larizza L, Roversi G, Volpi L. Rothmund-Thomson syndrome. Orphanet J Rare Dis. 2010;5:2.
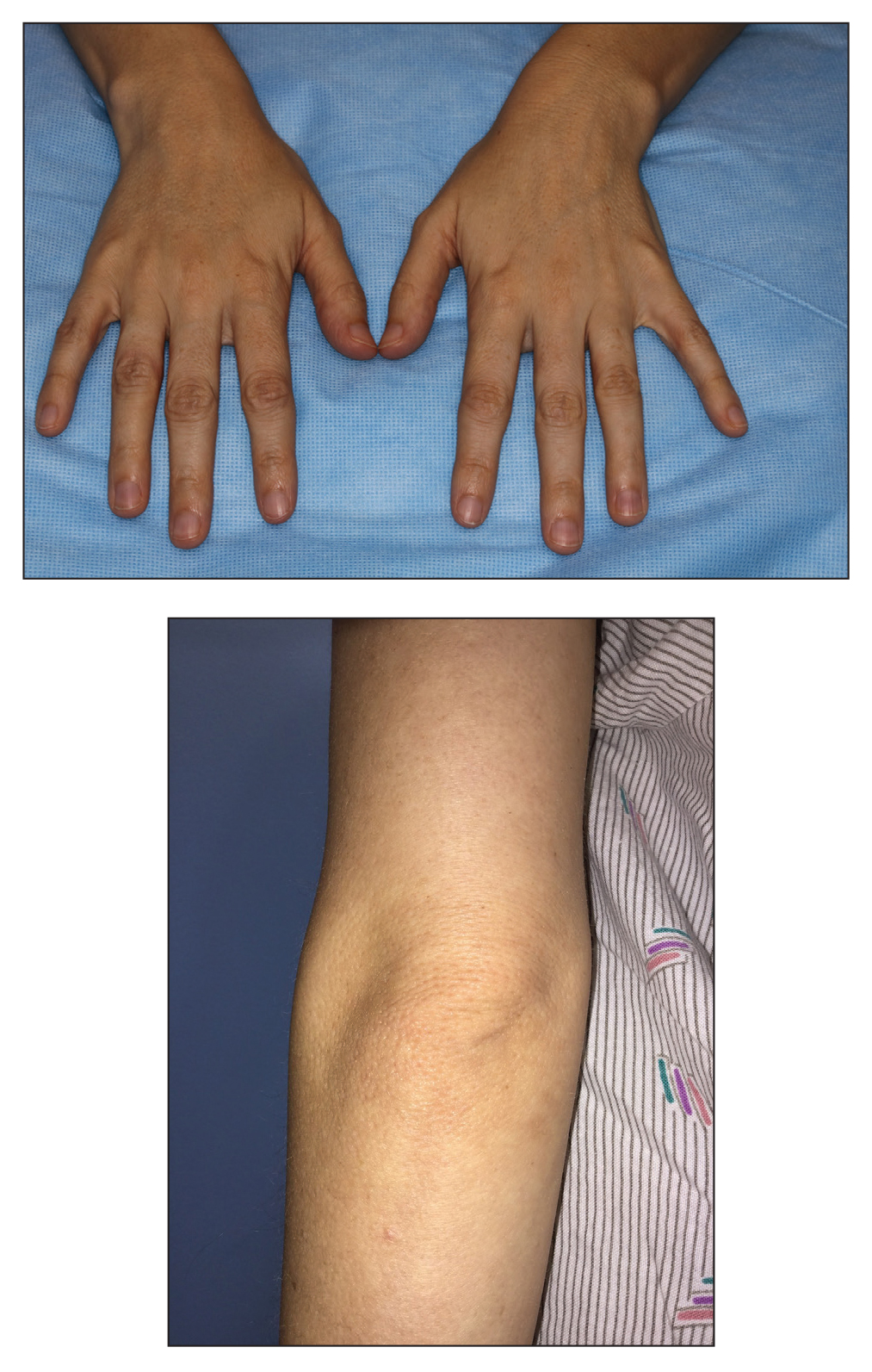
A 28-year-old woman presented for evaluation of a pearly papule on the forehead of several months’ duration that was concerning for basal cell carcinoma (BCC). She had a history of numerous BCCs starting at the age of 17 years. She denied radiation or other carcinogenic exposures and had no other notable medical history. The patient’s mother and grandmother also had numerous BCCs. Physical examination revealed hypotrichosis; numerous 3- to 5-mm white cystic papules on the face, chest, and upper arms; and 1- to 5-mm pitted depressions on the dorsal aspects of the hands (top) and extensor surfaces of the elbows (bottom). A proliferation of basaloid cells with peripheral palisading was seen on histopathologic evaluation. Genetic testing revealed no protein patched homolog 1, PTCH1, or suppressor of fused homolog, SUFU, gene mutations.
Erythema, Blisters, and Scars on the Elbows, Knees, and Legs
The Diagnosis: Epidermolysis Bullosa Acquisita
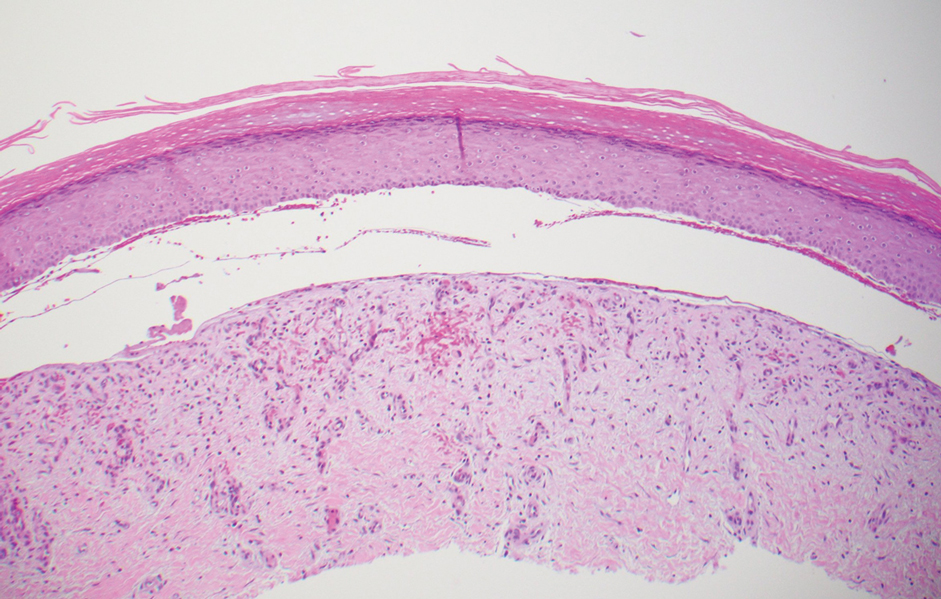
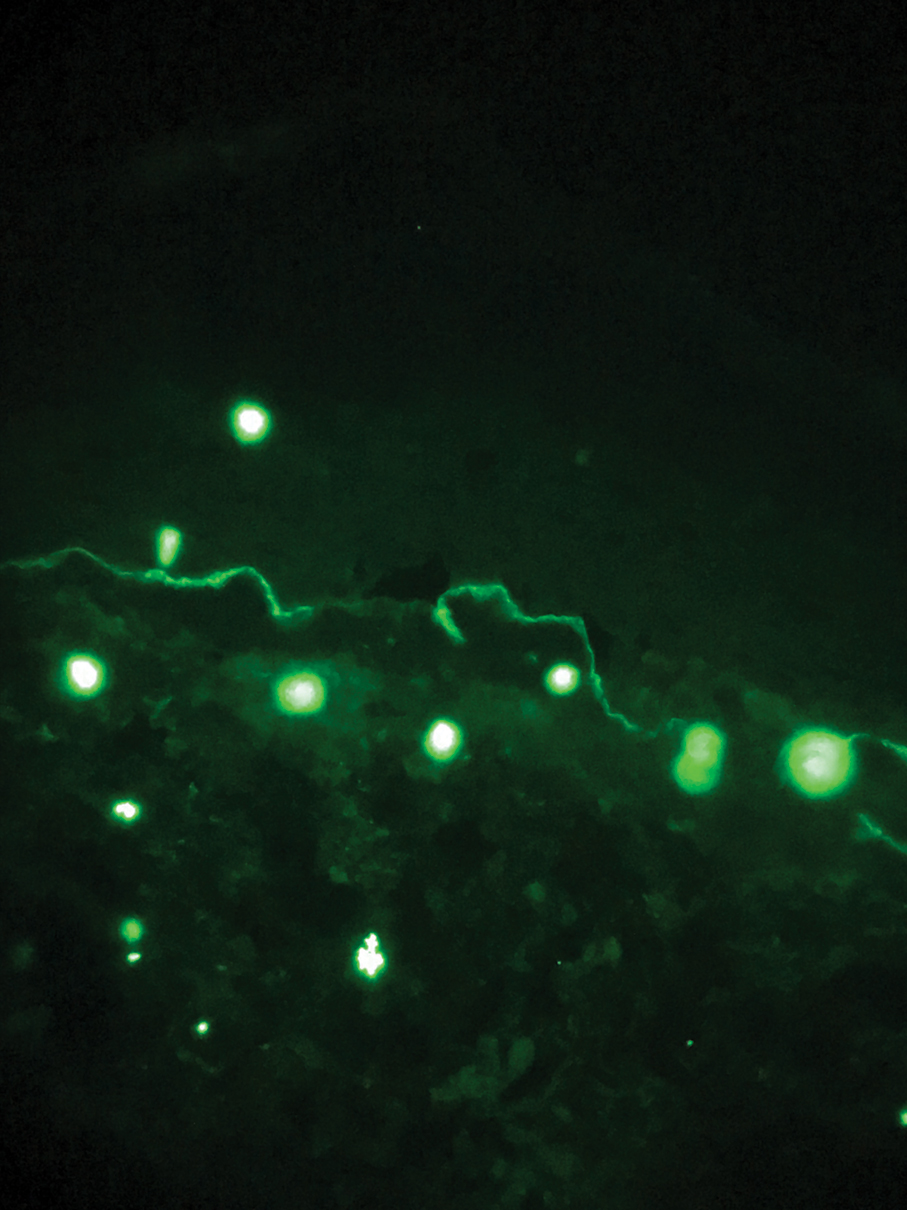
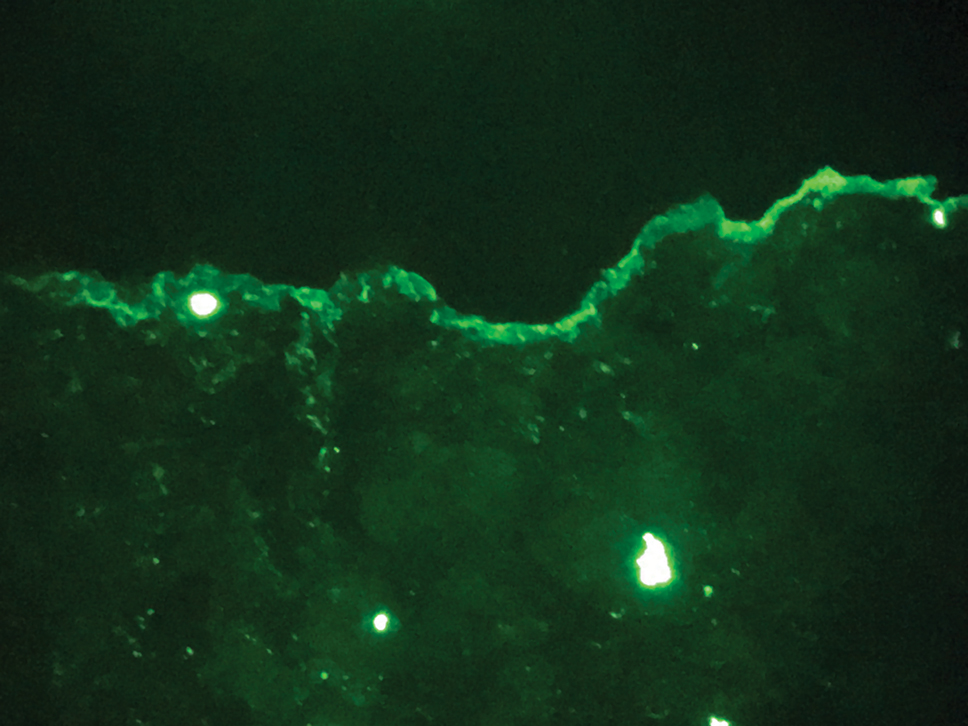
The diagnosis of epidermolysis bullosa acquisita (EBA) was made based on the clinical and pathologic findings. A blistering disorder that resolves with milia is characteristic of EBA. Hematoxylin and eosin staining demonstrated a pauci-inflammatory separation between the epidermis and dermis (Figure 1). Direct immunofluorescence studies showed linear IgG deposition along the basement membrane zone while C3 was negative (Figure 2). Salt-split skin was essential, as it revealed IgG deposition to the floor of the split (Figure 3), a pattern seen in EBA and not bullous pemphigoid (BP).1
Epidermolysis bullosa acquisita is an acquired autoimmune bullous disorder that results from antibodies to type VII collagen, an anchoring fibril that attaches the lamina densa to the dermis. The epidemiology and etiology of the trigger that leads to antibody production are not well known, but an association between EBA and inflammatory bowel disease has been described.2 Although this disease may present in childhood, EBA most commonly is a disorder seen in adults and the elderly. A classic noninflammatory mechanobullous form as well as an inflammatory BP-like form are the most commonly encountered presentations. Light microscopy demonstrates subepidermal cleavage without acantholysis. In the inflammatory BP-like subtype, an inflammatory infiltrate may be present. Direct immunofluorescence is remarkable for a linear band of IgG deposits along the basement membrane zone, with or without C3 deposition in a similar pattern.1
Bullous pemphigoid is within the differential of EBA. It can be difficult to differentiate clinically, especially when a patient has the BP-like variant of EBA because, as the name implies, it mimics BP. Patients with BP often will report a pruritic patch that will then develop into an urticarial plaque. Scarring and milia rarely are seen in BP but can be observed in the multiple presentations of EBA. Hematoxylin and eosin staining and direct immunofluorescence may be almost identical, and differentiating between the 2 disorders can be a challenge. Immunodeposition in EBA occurs in a U-shaped, serrated pattern, while the pattern in BP is N-shaped and serrated.3 Although the U-shaped, serrated pattern is relatively specific, it is not always easy to interpret and requires a high-quality biopsy specimen, which can be difficult to discern with certainty in suboptimal preparations. Another way to differentiate between the 2 entities is to utilize the salt-split skin technique, as performed in our patient. With salt-split skin, the biopsy is placed into a solution of 1 mol/L sodium chloride and incubated at 4 °C (39 °F) for 18 to 24 hours. A blister is then produced at the level of the lamina lucida, which allows for the staining of immunoreactants to occur either above or below that split (commonly referred to as staining on the roof or floor of the blister cavity). With EBA, there is immunoreactant deposition on the floor of the blister, while the opposite occurs in BP.4
Epidermolysis bullosa simplex is the most common type of epidermolysis bullosa, with keratin genes KRT5 and KRT14 as frequent mutations. Patients develop blisters, vesicles, bullae, and milia on traumatized areas of the body such as the hands, elbows, knees, and feet. This disease presents early in childhood. Histology exhibits a cell-poor subepidermal blister.5 With porphyria cutanea tarda, reduced activity of uroporphyrinogen decarboxylase, a major enzyme in the heme synthesis pathway, leads to blisters with erosions and milia on sun-exposed areas of the body. Histologic evaluation reveals a subepidermal pauci-inflammatory vesicle with festooning of the dermal papillae and amphophilic basement membrane within the epidermis. Direct immunofluorescence of porphyria cutanea tarda demonstrates IgM and C3 in the vessels.6 Sweet syndrome is a neutrophilic dermatosis that presents as erythematous, edematous, hot, and tender plaques along with fever and leukocytosis. It is associated with myeloproliferative disorders. Biopsy demonstrates papillary dermal edema along with diffuse neutrophilic infiltrate.7
Numerous medications have been recommended for the treatment of EBA, ranging from steroids to steroid-sparing drugs such as colchicine and dapsone.8,9 Our patient was educated on physical precautions and was started on dapsone alone due to comorbid diabetes mellitus and renal disease. Within a few weeks of initiating dapsone, he observed a reduction in erythema, and within months he experienced a decrease in blister eruption frequency.
- Vorobyev A, Ludwig RJ, Schmidt E. Clinical features and diagnosis of epidermolysis bullosa acquisita. Expert Rev Clin Immunol. 2017;13:157-169.
- Reddy H, Shipman AR, Wojnarowska F. Epidermolysis bullosa acquisita and inflammatory bowel disease: a review of the literature. Clin Exp Dermatol. 2013;38:225-230.
- Vodegel RM, Jonkman MF, Pas HH, et al. U-serrated immunodeposition pattern differentiates type VII collagen targeting bullous diseases from other subepidermal bullous autoimmune diseases. Br J Dermatol. 2004;151:112-118.
- Gardner KM, Crawford RI. Distinguishing epidermolysis bullosa acquisita from bullous pemphigoid without direct immunofluorescence. J Cutan Med Surg. 2018;22:22-24.
- Sprecher E. Epidermolysis bullosa simplex. Dermatol Clin. 2010;28:23-32.
- Maynard B, Peters MS. Histologic and immunofluorescence study of cutaneous porphyrias. J Cutan Pathol. 1992;19:40-47.
- Nelson CA, Stephen S, Ashchyan HJ, et al. Neutrophilic dermatoses: pathogenesis, Sweet syndrome, neutrophilic eccrine hidradenitis, and Behçet disease. J Am Acad Dermatol. 2018:79:987-1006.
- Kirtschig G, Murrell D, Wojnarowska F, et al. Interventions for mucous membrane pemphigoid and epidermolysis bullosa acquisita. Cochrane Database Syst Rev. 2003;1:CD004056
- Gürcan HM, Ahmed AR. Current concepts in the treatment of epidermolysis bullosa acquisita. Expert Opin Pharmacother. 2011;12:1259-1268.
The Diagnosis: Epidermolysis Bullosa Acquisita
The diagnosis of epidermolysis bullosa acquisita (EBA) was made based on the clinical and pathologic findings. A blistering disorder that resolves with milia is characteristic of EBA. Hematoxylin and eosin staining demonstrated a pauci-inflammatory separation between the epidermis and dermis (Figure 1). Direct immunofluorescence studies showed linear IgG deposition along the basement membrane zone while C3 was negative (Figure 2). Salt-split skin was essential, as it revealed IgG deposition to the floor of the split (Figure 3), a pattern seen in EBA and not bullous pemphigoid (BP).1
Epidermolysis bullosa acquisita is an acquired autoimmune bullous disorder that results from antibodies to type VII collagen, an anchoring fibril that attaches the lamina densa to the dermis. The epidemiology and etiology of the trigger that leads to antibody production are not well known, but an association between EBA and inflammatory bowel disease has been described.2 Although this disease may present in childhood, EBA most commonly is a disorder seen in adults and the elderly. A classic noninflammatory mechanobullous form as well as an inflammatory BP-like form are the most commonly encountered presentations. Light microscopy demonstrates subepidermal cleavage without acantholysis. In the inflammatory BP-like subtype, an inflammatory infiltrate may be present. Direct immunofluorescence is remarkable for a linear band of IgG deposits along the basement membrane zone, with or without C3 deposition in a similar pattern.1
Bullous pemphigoid is within the differential of EBA. It can be difficult to differentiate clinically, especially when a patient has the BP-like variant of EBA because, as the name implies, it mimics BP. Patients with BP often will report a pruritic patch that will then develop into an urticarial plaque. Scarring and milia rarely are seen in BP but can be observed in the multiple presentations of EBA. Hematoxylin and eosin staining and direct immunofluorescence may be almost identical, and differentiating between the 2 disorders can be a challenge. Immunodeposition in EBA occurs in a U-shaped, serrated pattern, while the pattern in BP is N-shaped and serrated.3 Although the U-shaped, serrated pattern is relatively specific, it is not always easy to interpret and requires a high-quality biopsy specimen, which can be difficult to discern with certainty in suboptimal preparations. Another way to differentiate between the 2 entities is to utilize the salt-split skin technique, as performed in our patient. With salt-split skin, the biopsy is placed into a solution of 1 mol/L sodium chloride and incubated at 4 °C (39 °F) for 18 to 24 hours. A blister is then produced at the level of the lamina lucida, which allows for the staining of immunoreactants to occur either above or below that split (commonly referred to as staining on the roof or floor of the blister cavity). With EBA, there is immunoreactant deposition on the floor of the blister, while the opposite occurs in BP.4
Epidermolysis bullosa simplex is the most common type of epidermolysis bullosa, with keratin genes KRT5 and KRT14 as frequent mutations. Patients develop blisters, vesicles, bullae, and milia on traumatized areas of the body such as the hands, elbows, knees, and feet. This disease presents early in childhood. Histology exhibits a cell-poor subepidermal blister.5 With porphyria cutanea tarda, reduced activity of uroporphyrinogen decarboxylase, a major enzyme in the heme synthesis pathway, leads to blisters with erosions and milia on sun-exposed areas of the body. Histologic evaluation reveals a subepidermal pauci-inflammatory vesicle with festooning of the dermal papillae and amphophilic basement membrane within the epidermis. Direct immunofluorescence of porphyria cutanea tarda demonstrates IgM and C3 in the vessels.6 Sweet syndrome is a neutrophilic dermatosis that presents as erythematous, edematous, hot, and tender plaques along with fever and leukocytosis. It is associated with myeloproliferative disorders. Biopsy demonstrates papillary dermal edema along with diffuse neutrophilic infiltrate.7
Numerous medications have been recommended for the treatment of EBA, ranging from steroids to steroid-sparing drugs such as colchicine and dapsone.8,9 Our patient was educated on physical precautions and was started on dapsone alone due to comorbid diabetes mellitus and renal disease. Within a few weeks of initiating dapsone, he observed a reduction in erythema, and within months he experienced a decrease in blister eruption frequency.
The Diagnosis: Epidermolysis Bullosa Acquisita
The diagnosis of epidermolysis bullosa acquisita (EBA) was made based on the clinical and pathologic findings. A blistering disorder that resolves with milia is characteristic of EBA. Hematoxylin and eosin staining demonstrated a pauci-inflammatory separation between the epidermis and dermis (Figure 1). Direct immunofluorescence studies showed linear IgG deposition along the basement membrane zone while C3 was negative (Figure 2). Salt-split skin was essential, as it revealed IgG deposition to the floor of the split (Figure 3), a pattern seen in EBA and not bullous pemphigoid (BP).1
Epidermolysis bullosa acquisita is an acquired autoimmune bullous disorder that results from antibodies to type VII collagen, an anchoring fibril that attaches the lamina densa to the dermis. The epidemiology and etiology of the trigger that leads to antibody production are not well known, but an association between EBA and inflammatory bowel disease has been described.2 Although this disease may present in childhood, EBA most commonly is a disorder seen in adults and the elderly. A classic noninflammatory mechanobullous form as well as an inflammatory BP-like form are the most commonly encountered presentations. Light microscopy demonstrates subepidermal cleavage without acantholysis. In the inflammatory BP-like subtype, an inflammatory infiltrate may be present. Direct immunofluorescence is remarkable for a linear band of IgG deposits along the basement membrane zone, with or without C3 deposition in a similar pattern.1
Bullous pemphigoid is within the differential of EBA. It can be difficult to differentiate clinically, especially when a patient has the BP-like variant of EBA because, as the name implies, it mimics BP. Patients with BP often will report a pruritic patch that will then develop into an urticarial plaque. Scarring and milia rarely are seen in BP but can be observed in the multiple presentations of EBA. Hematoxylin and eosin staining and direct immunofluorescence may be almost identical, and differentiating between the 2 disorders can be a challenge. Immunodeposition in EBA occurs in a U-shaped, serrated pattern, while the pattern in BP is N-shaped and serrated.3 Although the U-shaped, serrated pattern is relatively specific, it is not always easy to interpret and requires a high-quality biopsy specimen, which can be difficult to discern with certainty in suboptimal preparations. Another way to differentiate between the 2 entities is to utilize the salt-split skin technique, as performed in our patient. With salt-split skin, the biopsy is placed into a solution of 1 mol/L sodium chloride and incubated at 4 °C (39 °F) for 18 to 24 hours. A blister is then produced at the level of the lamina lucida, which allows for the staining of immunoreactants to occur either above or below that split (commonly referred to as staining on the roof or floor of the blister cavity). With EBA, there is immunoreactant deposition on the floor of the blister, while the opposite occurs in BP.4
Epidermolysis bullosa simplex is the most common type of epidermolysis bullosa, with keratin genes KRT5 and KRT14 as frequent mutations. Patients develop blisters, vesicles, bullae, and milia on traumatized areas of the body such as the hands, elbows, knees, and feet. This disease presents early in childhood. Histology exhibits a cell-poor subepidermal blister.5 With porphyria cutanea tarda, reduced activity of uroporphyrinogen decarboxylase, a major enzyme in the heme synthesis pathway, leads to blisters with erosions and milia on sun-exposed areas of the body. Histologic evaluation reveals a subepidermal pauci-inflammatory vesicle with festooning of the dermal papillae and amphophilic basement membrane within the epidermis. Direct immunofluorescence of porphyria cutanea tarda demonstrates IgM and C3 in the vessels.6 Sweet syndrome is a neutrophilic dermatosis that presents as erythematous, edematous, hot, and tender plaques along with fever and leukocytosis. It is associated with myeloproliferative disorders. Biopsy demonstrates papillary dermal edema along with diffuse neutrophilic infiltrate.7
Numerous medications have been recommended for the treatment of EBA, ranging from steroids to steroid-sparing drugs such as colchicine and dapsone.8,9 Our patient was educated on physical precautions and was started on dapsone alone due to comorbid diabetes mellitus and renal disease. Within a few weeks of initiating dapsone, he observed a reduction in erythema, and within months he experienced a decrease in blister eruption frequency.
- Vorobyev A, Ludwig RJ, Schmidt E. Clinical features and diagnosis of epidermolysis bullosa acquisita. Expert Rev Clin Immunol. 2017;13:157-169.
- Reddy H, Shipman AR, Wojnarowska F. Epidermolysis bullosa acquisita and inflammatory bowel disease: a review of the literature. Clin Exp Dermatol. 2013;38:225-230.
- Vodegel RM, Jonkman MF, Pas HH, et al. U-serrated immunodeposition pattern differentiates type VII collagen targeting bullous diseases from other subepidermal bullous autoimmune diseases. Br J Dermatol. 2004;151:112-118.
- Gardner KM, Crawford RI. Distinguishing epidermolysis bullosa acquisita from bullous pemphigoid without direct immunofluorescence. J Cutan Med Surg. 2018;22:22-24.
- Sprecher E. Epidermolysis bullosa simplex. Dermatol Clin. 2010;28:23-32.
- Maynard B, Peters MS. Histologic and immunofluorescence study of cutaneous porphyrias. J Cutan Pathol. 1992;19:40-47.
- Nelson CA, Stephen S, Ashchyan HJ, et al. Neutrophilic dermatoses: pathogenesis, Sweet syndrome, neutrophilic eccrine hidradenitis, and Behçet disease. J Am Acad Dermatol. 2018:79:987-1006.
- Kirtschig G, Murrell D, Wojnarowska F, et al. Interventions for mucous membrane pemphigoid and epidermolysis bullosa acquisita. Cochrane Database Syst Rev. 2003;1:CD004056
- Gürcan HM, Ahmed AR. Current concepts in the treatment of epidermolysis bullosa acquisita. Expert Opin Pharmacother. 2011;12:1259-1268.
- Vorobyev A, Ludwig RJ, Schmidt E. Clinical features and diagnosis of epidermolysis bullosa acquisita. Expert Rev Clin Immunol. 2017;13:157-169.
- Reddy H, Shipman AR, Wojnarowska F. Epidermolysis bullosa acquisita and inflammatory bowel disease: a review of the literature. Clin Exp Dermatol. 2013;38:225-230.
- Vodegel RM, Jonkman MF, Pas HH, et al. U-serrated immunodeposition pattern differentiates type VII collagen targeting bullous diseases from other subepidermal bullous autoimmune diseases. Br J Dermatol. 2004;151:112-118.
- Gardner KM, Crawford RI. Distinguishing epidermolysis bullosa acquisita from bullous pemphigoid without direct immunofluorescence. J Cutan Med Surg. 2018;22:22-24.
- Sprecher E. Epidermolysis bullosa simplex. Dermatol Clin. 2010;28:23-32.
- Maynard B, Peters MS. Histologic and immunofluorescence study of cutaneous porphyrias. J Cutan Pathol. 1992;19:40-47.
- Nelson CA, Stephen S, Ashchyan HJ, et al. Neutrophilic dermatoses: pathogenesis, Sweet syndrome, neutrophilic eccrine hidradenitis, and Behçet disease. J Am Acad Dermatol. 2018:79:987-1006.
- Kirtschig G, Murrell D, Wojnarowska F, et al. Interventions for mucous membrane pemphigoid and epidermolysis bullosa acquisita. Cochrane Database Syst Rev. 2003;1:CD004056
- Gürcan HM, Ahmed AR. Current concepts in the treatment of epidermolysis bullosa acquisita. Expert Opin Pharmacother. 2011;12:1259-1268.
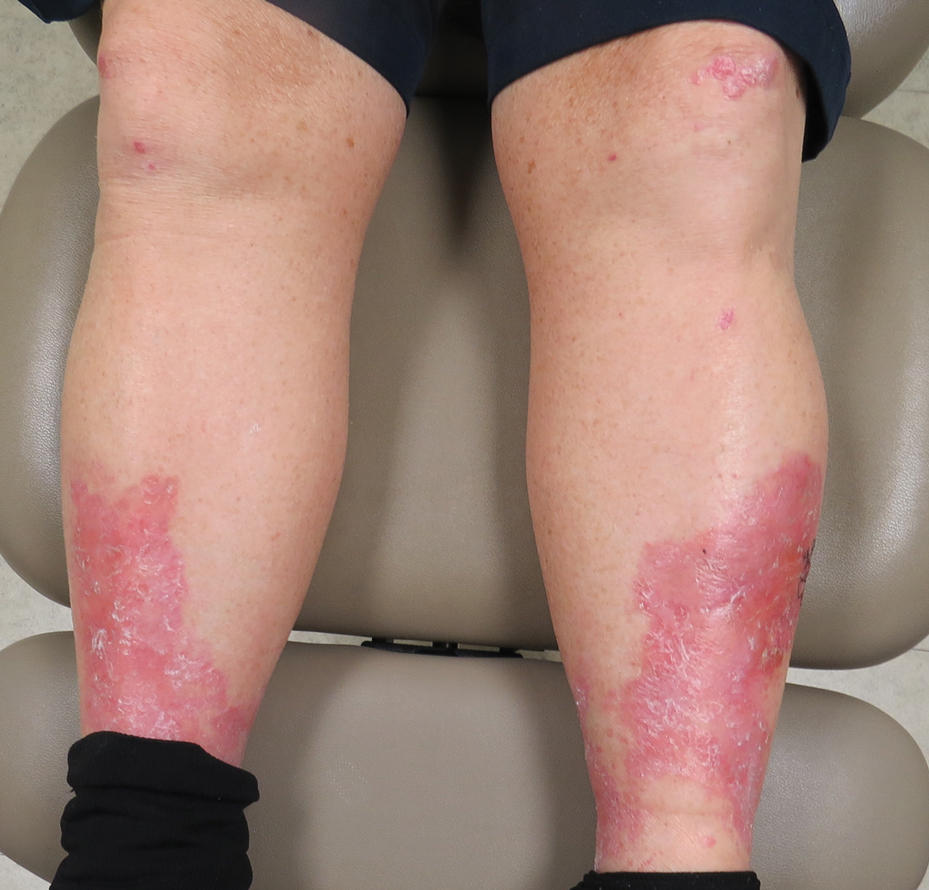
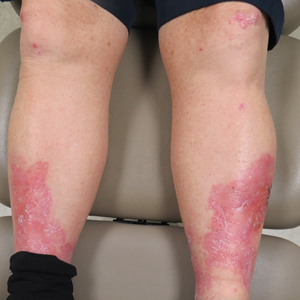
A 69-year-old man presented with an asymptomatic rash on the extensor surfaces of 2 years' duration. He reported recurrent blisters that would then scar over. The lesions did not occur in relation to any known trauma. The patient's medical history revealed dialysis-dependent end-stage renal disease secondary to type 2 diabetes mellitus. His medications were noncontributory, and there was no family history of blistering disorders. He had tried triamcinolone cream without any improvement. Physical examination was remarkable for erythematous blisters and bullae with scales and milia on the elbows, knees, and lower legs. The oral mucosa was unremarkable. Shave biopsies of the skin for direct immunofluorescence and salt-split skin studies were obtained.
Atrophic Lesion on the Abdomen
The Diagnosis: Anetoderma of Prematurity
Anetoderma is a rare benign cutaneous disorder characterized by atrophic patches of skin due to dermal thinning. The term anetoderma is derived from the Greek words anetos (relaxed) and derma (skin).1 The physical appearance of the skin is associated with a reduction or loss of elastic tissue in the dermal layer, as seen on histolopathology.2
Two forms of anetoderma have been described. Primary anetoderma is an idiopathic form with no preceding inflammatory lesions. Secondary anetoderma is a reactive process linked to a known preceding inflammatory, infectious, autoimmune, or drug-induced condition.3 On histopathology, both primary and secondary anetoderma are characterized by a loss of elastic tissue or elastin fibers in the superficial to mid dermis.2
Anetoderma of prematurity was first described in 1996 by Prizant et al4 in 9 extremely premature (24-29 weeks' gestation) infants in neonatal intensive care units (NICUs). Although the exact mechanism behind anetoderma of prematurity is still unknown, Prizant et al4 and other investigators5 postulated that application of adhesive monitoring leads in the NICU played a role in the development of the lesions.
Iatrogenic anetoderma of prematurity is clinically characterized by circumscribed areas of either wrinkled macular depression or pouchlike herniations, ranging from flesh-colored to violaceous hues. Lesion size varies from a few millimeters to several centimeters in diameter, and they often are oval or round in shape.2 Although not common, it is possible for the atrophic patches to be preceded by an area of ecchymosis without necrosis or atrophy and, if present, they usually evolve within a few days to the characteristic appearance of anetoderma.3 They are found at discrete sites where monitoring leads or other medical devices are commonly placed, such as the forehead, abdomen, chest, and proximal limbs.
Lesions of anetoderma of prematurity are not present at birth, which distinguishes them from congenital anetoderma.6 It is unclear if the lesions are associated with the degree of prematurity, extremely low birth weight, or other associated factors of preterm birth. Although often clinically diagnosed, the diagnosis can be confirmed by a loss of elastic fibers on histopathology when stained with Verhoeff-van Gieson stain.1 Over time, the atrophic patches have the potential to evolve into herniated forms of anetoderma. Self-healing or improvement of the lesions often does not occur. Although the lesion is benign, it often requires surgical correction later in life for cosmesis.
Infants in the NICU are at risk for iatrogenic cutaneous injuries, which rarely may include anetoderma. Anetoderma of prematurity has been linked to the use of monitoring leads, adhesive tape, and other medical devices placed on the skin. Prizant et al4 postulated that the cause of anetoderma in these infants was irritants such as skin cleansers, urine, or sweat that may be trapped under the electrodes. Other hypotheses include local hypoxemia due to prolonged pressure from the electrodes on immature skin or excessive traction used when removing adhesive tape from the skin.7,8 Premature infants may be more susceptible to these lesions because of the reduced epidermal thickness of premature skin; immaturity of skin structure; or functional immaturity of elastin deposition regulators, such as elastase, lysyl oxidase, the complement system, and decay-accelerating factor.3 The diagnosis should be differentiated from congenital anetoderma, which also has been described in premature neonates but is characterized by lesions that are present at birth. Its origins are still unclear, despite having histopathologic features similar to iatrogenic anetoderma.9
Focal dermal hypoplasia (FDH) is the hallmark cutaneous finding in Goltz syndrome, a rare set of congenital abnormalities of the skin, oral structures, musculoskeletal system, and central nervous system. Similar to congenital anetoderma, FDH also is characterized by atrophic cutaneous lesions; however, the cutaneous lesions in FDH appear as linear, streaky atrophic lesions often with telangiectasias that follow Blaschko lines.10 The cutaneous lesions in FDH often are associated with other noncutaneous signs such as polydactyly or asymmetric limbs.10 Cutis laxa is caused by an abnormality in the elastic tissue resulting in a loose sagging appearance of the skin and frequently results in an aged facial appearance. There are both acquired and inherited forms that can be either solely cutaneous or present with extracutaneous features, such as cardiac abnormalities or emphysema.11
In contrast to the atrophic appearance of anetodermas, connective tissue nevi and nevus lipomatosus superficialis present as hamartomas that either can be present at birth or arise in infancy. Connective tissue nevi are hamartomas of dermal connective tissue that consist of excessive production of collagen, elastin, or glycosaminoglycans and appear as slightly elevated, flesh-colored to yellow nodules or plaques.12 Connective tissue nevi often are described in association with other diseases, most commonly tuberous sclerosis (shagreen patches) or familial cutaneous collagenoma. Nevus lipomatosus superficialis is an asymptomatic connective tissue hamartoma composed of mature adipocytes in the dermis. The lesions consist of clusters of flesh-colored to yellow, soft, rubbery papules or nodules with a smooth or verrucoid surface that do not cross the midline and may follow Blaschko lines.11
With advances in neonatal infant medical care, survival of extremely premature infants is increasing, and it is possible that this rare cutaneous disorder may become more prevalent. Care should be taken to avoid unnecessary pressure on surfaces where electrodes are placed and tightly applied adhesive tape. When electrodes are placed on the ventral side, the child should be placed supine; similarly, place electrodes on the dorsal side when the child is lying prone.5 A diagnosis of anetoderma of prematurity later in childhood may be difficult, so knowledge and awareness can help guide pediatricians and dermatologists to a correct diagnosis and prevent unnecessary evaluations and/or concerns.
- Misch KJ, Rhodes EL, Allen J, et al. Anetoderma of Jadassohn. J R Soc Med.1988;81:734-736.
- Venencie PY, Winkelmann RK. Histopathologic findings in anetoderma. Arch Dermatol. 1984;120:1040-1044.
- Maffeis L, Pugni L, Pietrasanta C, et al. Case report iatrogenic anetoderma of prematurity: a case report and review of the literature. 2014;2014:781493.
- Prizant TL, Lucky AW, Frieden IJ, et al. Spontaneous atrophic patches in extremely premature infants: anetoderma of prematurity. Arch Dermatol. 1996;132:671-674.
- Goujon E, Beer F, Gay S, et al. Anetoderma of prematurity: an iatrogenic consequence of neonatal intensive care anetoderma of prematurity from NICU. Arch Dermatol. 2010;146:565-567.
- Wain EM, Mellerio JE, Robson A, et al. Congenital anetoderma in a preterm infant. Pediatr Dermatol. 2008;25:626-629.
- Colditz PB, Dunster KR, Joy GJ, et al. Anetoderma of prematurity in association with electrocardiographic electrodes. J Am Acad Dermatol. 1999;41:479-481.
- Goujan E, Beer F, Gay S, et al. Study supervision. Arch Dermatol. 2010;146:565-567.
- Aberer E, Weissenbacher G. Congenital anetoderma induced by intrauterine infection? Arch Dermatol. 1997;133:526-527.
- Mallory SB, Krafchik BR, Moore DJ, et al. Goltz syndrome. Pediatr Dermatol. 1989;6:251-253.
- Bolognia J, Schaffer J, Cerroni L. Dermatology. Elsevier Saunders; 2017.
- Uitto J, Santa Cruz DJ, Eisen AZ. Connective tissue nevi of the skin. clinical, genetic, and histopathologic classification of hamartomas of the collagen, elastin, and proteoglycan type. J Am Acad Dermatol. 1980;3:441-461.
The Diagnosis: Anetoderma of Prematurity
Anetoderma is a rare benign cutaneous disorder characterized by atrophic patches of skin due to dermal thinning. The term anetoderma is derived from the Greek words anetos (relaxed) and derma (skin).1 The physical appearance of the skin is associated with a reduction or loss of elastic tissue in the dermal layer, as seen on histolopathology.2
Two forms of anetoderma have been described. Primary anetoderma is an idiopathic form with no preceding inflammatory lesions. Secondary anetoderma is a reactive process linked to a known preceding inflammatory, infectious, autoimmune, or drug-induced condition.3 On histopathology, both primary and secondary anetoderma are characterized by a loss of elastic tissue or elastin fibers in the superficial to mid dermis.2
Anetoderma of prematurity was first described in 1996 by Prizant et al4 in 9 extremely premature (24-29 weeks' gestation) infants in neonatal intensive care units (NICUs). Although the exact mechanism behind anetoderma of prematurity is still unknown, Prizant et al4 and other investigators5 postulated that application of adhesive monitoring leads in the NICU played a role in the development of the lesions.
Iatrogenic anetoderma of prematurity is clinically characterized by circumscribed areas of either wrinkled macular depression or pouchlike herniations, ranging from flesh-colored to violaceous hues. Lesion size varies from a few millimeters to several centimeters in diameter, and they often are oval or round in shape.2 Although not common, it is possible for the atrophic patches to be preceded by an area of ecchymosis without necrosis or atrophy and, if present, they usually evolve within a few days to the characteristic appearance of anetoderma.3 They are found at discrete sites where monitoring leads or other medical devices are commonly placed, such as the forehead, abdomen, chest, and proximal limbs.
Lesions of anetoderma of prematurity are not present at birth, which distinguishes them from congenital anetoderma.6 It is unclear if the lesions are associated with the degree of prematurity, extremely low birth weight, or other associated factors of preterm birth. Although often clinically diagnosed, the diagnosis can be confirmed by a loss of elastic fibers on histopathology when stained with Verhoeff-van Gieson stain.1 Over time, the atrophic patches have the potential to evolve into herniated forms of anetoderma. Self-healing or improvement of the lesions often does not occur. Although the lesion is benign, it often requires surgical correction later in life for cosmesis.
Infants in the NICU are at risk for iatrogenic cutaneous injuries, which rarely may include anetoderma. Anetoderma of prematurity has been linked to the use of monitoring leads, adhesive tape, and other medical devices placed on the skin. Prizant et al4 postulated that the cause of anetoderma in these infants was irritants such as skin cleansers, urine, or sweat that may be trapped under the electrodes. Other hypotheses include local hypoxemia due to prolonged pressure from the electrodes on immature skin or excessive traction used when removing adhesive tape from the skin.7,8 Premature infants may be more susceptible to these lesions because of the reduced epidermal thickness of premature skin; immaturity of skin structure; or functional immaturity of elastin deposition regulators, such as elastase, lysyl oxidase, the complement system, and decay-accelerating factor.3 The diagnosis should be differentiated from congenital anetoderma, which also has been described in premature neonates but is characterized by lesions that are present at birth. Its origins are still unclear, despite having histopathologic features similar to iatrogenic anetoderma.9
Focal dermal hypoplasia (FDH) is the hallmark cutaneous finding in Goltz syndrome, a rare set of congenital abnormalities of the skin, oral structures, musculoskeletal system, and central nervous system. Similar to congenital anetoderma, FDH also is characterized by atrophic cutaneous lesions; however, the cutaneous lesions in FDH appear as linear, streaky atrophic lesions often with telangiectasias that follow Blaschko lines.10 The cutaneous lesions in FDH often are associated with other noncutaneous signs such as polydactyly or asymmetric limbs.10 Cutis laxa is caused by an abnormality in the elastic tissue resulting in a loose sagging appearance of the skin and frequently results in an aged facial appearance. There are both acquired and inherited forms that can be either solely cutaneous or present with extracutaneous features, such as cardiac abnormalities or emphysema.11
In contrast to the atrophic appearance of anetodermas, connective tissue nevi and nevus lipomatosus superficialis present as hamartomas that either can be present at birth or arise in infancy. Connective tissue nevi are hamartomas of dermal connective tissue that consist of excessive production of collagen, elastin, or glycosaminoglycans and appear as slightly elevated, flesh-colored to yellow nodules or plaques.12 Connective tissue nevi often are described in association with other diseases, most commonly tuberous sclerosis (shagreen patches) or familial cutaneous collagenoma. Nevus lipomatosus superficialis is an asymptomatic connective tissue hamartoma composed of mature adipocytes in the dermis. The lesions consist of clusters of flesh-colored to yellow, soft, rubbery papules or nodules with a smooth or verrucoid surface that do not cross the midline and may follow Blaschko lines.11
With advances in neonatal infant medical care, survival of extremely premature infants is increasing, and it is possible that this rare cutaneous disorder may become more prevalent. Care should be taken to avoid unnecessary pressure on surfaces where electrodes are placed and tightly applied adhesive tape. When electrodes are placed on the ventral side, the child should be placed supine; similarly, place electrodes on the dorsal side when the child is lying prone.5 A diagnosis of anetoderma of prematurity later in childhood may be difficult, so knowledge and awareness can help guide pediatricians and dermatologists to a correct diagnosis and prevent unnecessary evaluations and/or concerns.
The Diagnosis: Anetoderma of Prematurity
Anetoderma is a rare benign cutaneous disorder characterized by atrophic patches of skin due to dermal thinning. The term anetoderma is derived from the Greek words anetos (relaxed) and derma (skin).1 The physical appearance of the skin is associated with a reduction or loss of elastic tissue in the dermal layer, as seen on histolopathology.2
Two forms of anetoderma have been described. Primary anetoderma is an idiopathic form with no preceding inflammatory lesions. Secondary anetoderma is a reactive process linked to a known preceding inflammatory, infectious, autoimmune, or drug-induced condition.3 On histopathology, both primary and secondary anetoderma are characterized by a loss of elastic tissue or elastin fibers in the superficial to mid dermis.2
Anetoderma of prematurity was first described in 1996 by Prizant et al4 in 9 extremely premature (24-29 weeks' gestation) infants in neonatal intensive care units (NICUs). Although the exact mechanism behind anetoderma of prematurity is still unknown, Prizant et al4 and other investigators5 postulated that application of adhesive monitoring leads in the NICU played a role in the development of the lesions.
Iatrogenic anetoderma of prematurity is clinically characterized by circumscribed areas of either wrinkled macular depression or pouchlike herniations, ranging from flesh-colored to violaceous hues. Lesion size varies from a few millimeters to several centimeters in diameter, and they often are oval or round in shape.2 Although not common, it is possible for the atrophic patches to be preceded by an area of ecchymosis without necrosis or atrophy and, if present, they usually evolve within a few days to the characteristic appearance of anetoderma.3 They are found at discrete sites where monitoring leads or other medical devices are commonly placed, such as the forehead, abdomen, chest, and proximal limbs.
Lesions of anetoderma of prematurity are not present at birth, which distinguishes them from congenital anetoderma.6 It is unclear if the lesions are associated with the degree of prematurity, extremely low birth weight, or other associated factors of preterm birth. Although often clinically diagnosed, the diagnosis can be confirmed by a loss of elastic fibers on histopathology when stained with Verhoeff-van Gieson stain.1 Over time, the atrophic patches have the potential to evolve into herniated forms of anetoderma. Self-healing or improvement of the lesions often does not occur. Although the lesion is benign, it often requires surgical correction later in life for cosmesis.
Infants in the NICU are at risk for iatrogenic cutaneous injuries, which rarely may include anetoderma. Anetoderma of prematurity has been linked to the use of monitoring leads, adhesive tape, and other medical devices placed on the skin. Prizant et al4 postulated that the cause of anetoderma in these infants was irritants such as skin cleansers, urine, or sweat that may be trapped under the electrodes. Other hypotheses include local hypoxemia due to prolonged pressure from the electrodes on immature skin or excessive traction used when removing adhesive tape from the skin.7,8 Premature infants may be more susceptible to these lesions because of the reduced epidermal thickness of premature skin; immaturity of skin structure; or functional immaturity of elastin deposition regulators, such as elastase, lysyl oxidase, the complement system, and decay-accelerating factor.3 The diagnosis should be differentiated from congenital anetoderma, which also has been described in premature neonates but is characterized by lesions that are present at birth. Its origins are still unclear, despite having histopathologic features similar to iatrogenic anetoderma.9
Focal dermal hypoplasia (FDH) is the hallmark cutaneous finding in Goltz syndrome, a rare set of congenital abnormalities of the skin, oral structures, musculoskeletal system, and central nervous system. Similar to congenital anetoderma, FDH also is characterized by atrophic cutaneous lesions; however, the cutaneous lesions in FDH appear as linear, streaky atrophic lesions often with telangiectasias that follow Blaschko lines.10 The cutaneous lesions in FDH often are associated with other noncutaneous signs such as polydactyly or asymmetric limbs.10 Cutis laxa is caused by an abnormality in the elastic tissue resulting in a loose sagging appearance of the skin and frequently results in an aged facial appearance. There are both acquired and inherited forms that can be either solely cutaneous or present with extracutaneous features, such as cardiac abnormalities or emphysema.11
In contrast to the atrophic appearance of anetodermas, connective tissue nevi and nevus lipomatosus superficialis present as hamartomas that either can be present at birth or arise in infancy. Connective tissue nevi are hamartomas of dermal connective tissue that consist of excessive production of collagen, elastin, or glycosaminoglycans and appear as slightly elevated, flesh-colored to yellow nodules or plaques.12 Connective tissue nevi often are described in association with other diseases, most commonly tuberous sclerosis (shagreen patches) or familial cutaneous collagenoma. Nevus lipomatosus superficialis is an asymptomatic connective tissue hamartoma composed of mature adipocytes in the dermis. The lesions consist of clusters of flesh-colored to yellow, soft, rubbery papules or nodules with a smooth or verrucoid surface that do not cross the midline and may follow Blaschko lines.11
With advances in neonatal infant medical care, survival of extremely premature infants is increasing, and it is possible that this rare cutaneous disorder may become more prevalent. Care should be taken to avoid unnecessary pressure on surfaces where electrodes are placed and tightly applied adhesive tape. When electrodes are placed on the ventral side, the child should be placed supine; similarly, place electrodes on the dorsal side when the child is lying prone.5 A diagnosis of anetoderma of prematurity later in childhood may be difficult, so knowledge and awareness can help guide pediatricians and dermatologists to a correct diagnosis and prevent unnecessary evaluations and/or concerns.
- Misch KJ, Rhodes EL, Allen J, et al. Anetoderma of Jadassohn. J R Soc Med.1988;81:734-736.
- Venencie PY, Winkelmann RK. Histopathologic findings in anetoderma. Arch Dermatol. 1984;120:1040-1044.
- Maffeis L, Pugni L, Pietrasanta C, et al. Case report iatrogenic anetoderma of prematurity: a case report and review of the literature. 2014;2014:781493.
- Prizant TL, Lucky AW, Frieden IJ, et al. Spontaneous atrophic patches in extremely premature infants: anetoderma of prematurity. Arch Dermatol. 1996;132:671-674.
- Goujon E, Beer F, Gay S, et al. Anetoderma of prematurity: an iatrogenic consequence of neonatal intensive care anetoderma of prematurity from NICU. Arch Dermatol. 2010;146:565-567.
- Wain EM, Mellerio JE, Robson A, et al. Congenital anetoderma in a preterm infant. Pediatr Dermatol. 2008;25:626-629.
- Colditz PB, Dunster KR, Joy GJ, et al. Anetoderma of prematurity in association with electrocardiographic electrodes. J Am Acad Dermatol. 1999;41:479-481.
- Goujan E, Beer F, Gay S, et al. Study supervision. Arch Dermatol. 2010;146:565-567.
- Aberer E, Weissenbacher G. Congenital anetoderma induced by intrauterine infection? Arch Dermatol. 1997;133:526-527.
- Mallory SB, Krafchik BR, Moore DJ, et al. Goltz syndrome. Pediatr Dermatol. 1989;6:251-253.
- Bolognia J, Schaffer J, Cerroni L. Dermatology. Elsevier Saunders; 2017.
- Uitto J, Santa Cruz DJ, Eisen AZ. Connective tissue nevi of the skin. clinical, genetic, and histopathologic classification of hamartomas of the collagen, elastin, and proteoglycan type. J Am Acad Dermatol. 1980;3:441-461.
- Misch KJ, Rhodes EL, Allen J, et al. Anetoderma of Jadassohn. J R Soc Med.1988;81:734-736.
- Venencie PY, Winkelmann RK. Histopathologic findings in anetoderma. Arch Dermatol. 1984;120:1040-1044.
- Maffeis L, Pugni L, Pietrasanta C, et al. Case report iatrogenic anetoderma of prematurity: a case report and review of the literature. 2014;2014:781493.
- Prizant TL, Lucky AW, Frieden IJ, et al. Spontaneous atrophic patches in extremely premature infants: anetoderma of prematurity. Arch Dermatol. 1996;132:671-674.
- Goujon E, Beer F, Gay S, et al. Anetoderma of prematurity: an iatrogenic consequence of neonatal intensive care anetoderma of prematurity from NICU. Arch Dermatol. 2010;146:565-567.
- Wain EM, Mellerio JE, Robson A, et al. Congenital anetoderma in a preterm infant. Pediatr Dermatol. 2008;25:626-629.
- Colditz PB, Dunster KR, Joy GJ, et al. Anetoderma of prematurity in association with electrocardiographic electrodes. J Am Acad Dermatol. 1999;41:479-481.
- Goujan E, Beer F, Gay S, et al. Study supervision. Arch Dermatol. 2010;146:565-567.
- Aberer E, Weissenbacher G. Congenital anetoderma induced by intrauterine infection? Arch Dermatol. 1997;133:526-527.
- Mallory SB, Krafchik BR, Moore DJ, et al. Goltz syndrome. Pediatr Dermatol. 1989;6:251-253.
- Bolognia J, Schaffer J, Cerroni L. Dermatology. Elsevier Saunders; 2017.
- Uitto J, Santa Cruz DJ, Eisen AZ. Connective tissue nevi of the skin. clinical, genetic, and histopathologic classification of hamartomas of the collagen, elastin, and proteoglycan type. J Am Acad Dermatol. 1980;3:441-461.

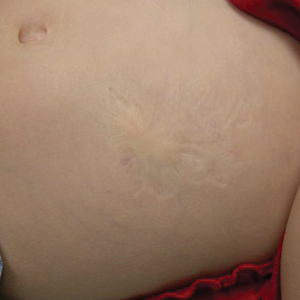
An 18-month-old child presented with a 4-cm, atrophic, flesh-colored plaque on the left lateral aspect of the abdomen with overlying wrinkling of the skin. There was no outpouching of the skin or pain associated with the lesion. No other skin abnormalities were noted. The child was born premature at 30 weeks’ gestation (birth weight, 1400 g). The postnatal course was complicated by respiratory distress syndrome requiring prolonged ventilator support. The infant was in the neonatal intensive care unit for 5 months. The atrophic lesion first developed at 5 months of life and remained stable. Although the lesion was not present at birth, the parents noted that it was preceded by an ecchymotic lesion without necrosis that was first noticed at 2 months of life while the patient was in the neonatal intensive care unit.
Expanding Verrucous Plaque on the Face
The Diagnosis: Blastomycosis
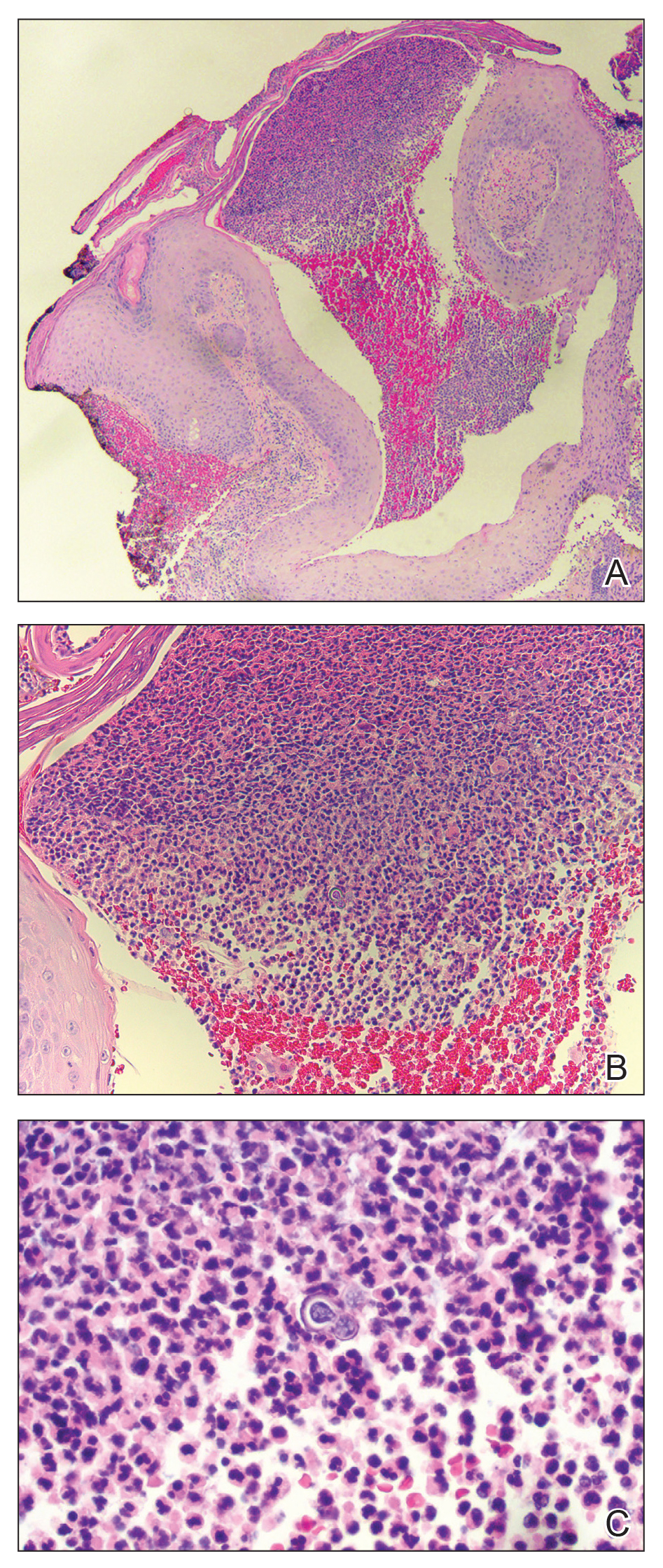
Histopathologic examination of 3 punch biopsies from the left side of the upper lip showed pseudoepitheliomatous hyperplasia with intraepidermal microabscesses and dermal suppurative granulomatous inflammation (Figure 1A). Stains were negative for periodic acid-Schiff, herpes simplex virus, and varicella-zoster virus. Direct and indirect immunofluorescence for skin autoantibodies were negative. Two separate tissue culture specimens showed no bacterial, fungal, or mycobacterial growth. Leishmania polymerase chain reaction and DNA sequencing were negative. An additional punch biopsy revealed yeast forms with broad-based budding and refractile walls (Figures 1B and 1C) that were highlighted with Grocott-Gomori methenamine-silver stain of the tissue (Figure 2). Chest radiography demonstrated no pulmonary involvement. In collaboration with an infectious disease specialist, the patient was started on itraconazole 200 mg twice daily for a total of 6 months.
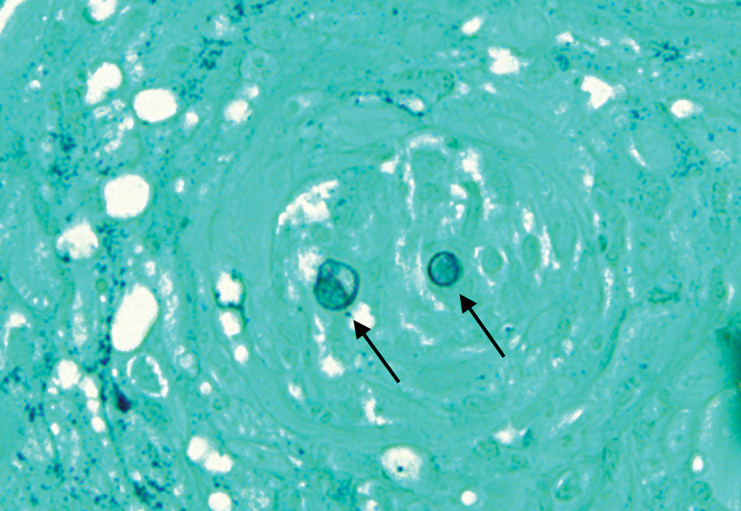
Blastomycosis is a fungal infection caused by Blastomyces dermatitidis, a thermally dimorphic fungus endemic in the soils of the Ohio and Mississippi River valleys and southeastern United States.1 It most commonly manifests as a pulmonary infection following inhalation of spores that are transformed into thick-walled yeasts capable of evading the host's immune system. Unlike other deep fungal infections, blastomycosis occurs in both immunocompetent and immunocompromised hosts. Extrapulmonary disease after hematogenous dissemination from the lungs occurs in approximately 25% to 30% of patients, with the skin as the most common site of dissemination.2 Clinically, cutaneous blastomycosis typically starts as papules that evolve into crusted vegetative plaques, often with central clearing or ulceration. Primary cutaneous blastomycosis is rare and occurs due to direct inoculation after trauma to the skin via an infected animal bite, direct inoculation in laboratory settings, or due to injury during outdoor activities involving contact with soil.3 Given our patient's horticultural hobbies, lack of pulmonary symptoms, and negative radiologic examination, primary cutaneous blastomycosis infection due to direct inoculation from contaminated soil was a possibility; however, definite confirmation was difficult, as the primary pulmonary infection of blastomycosis can be asymptomatic and therefore often goes undetected.
Cutaneous blastomycosis can be mistaken for pemphigus vegetans, leishmaniasis, herpes vegetans, bacterial pyoderma, and other deep fungal infections that also display pseudoepitheliomatous hyperplasia with pyogranulomatous inflammation on histopathology. Direct visualization of the characteristic yeast forms in a histologic specimen or the growth of fungus in culture is essential for a definitive diagnosis. The yeasts are 8 to 15 µm in diameter with thick, double-contoured walls and characteristically display broad-based budding.4 This budding pattern aids in differentiating blastomycosis from other entities with a similar histopathologic appearance. Chromoblastomycosis would show brown, thick-walled fungal cells inside giant cells, while coccidioidomycosis displays large spherules containing endospores, and leishmaniasis demonstrates amastigotes (small oval organisms with a bar-shaped kinetoplast) highlighted with Giemsa staining. Pemphigus vegetans would show intercellular deposition of IgG on direct immunofluorescence. Blastomyces dermatitidis can be difficult to visualize with routine hematoxylin and eosin stains, and it is important to note that a negative result does not exclude the possibility of blastomycosis, as demonstrated in our case.4 Special stains including Grocott-Gomori methenamine-silver and periodic acid-Schiff can aid in examining tissue for the presence of fungal elements, which typically can be found within histiocytes or abscesses in the dermis. Culture is the most sensitive method for detecting and diagnosing blastomycosis. Growth typically is detected in 5 to 10 days but can take up to 30 days if few organisms are present in the specimen.1
Although spontaneous remission can occur, it is recommended that all patients with cutaneous blastomycosis be treated to avoid dissemination and recurrence. Itraconazole currently is the treatment of choice.5 Doses typically are 200 to 400 mg/d for 8 to 12 months.6 Itraconazole-related side effects experienced by our patient during his 6-month treatment course included leg edema, 20-lb weight gain, gastrointestinal upset, blurred vision, and a transient increase in blood pressure, all resolving once the medication was discontinued. Complete resolution of the lesion was noted at the completion of the treatment course. At a 6-month posttreatment follow-up, residual scarring and alopecia were noted in parts of the previously affected areas of the upper cutaneous lip and nasolabial fold.
- Saccente M, Woods GL. Clinical and laboratory update on blastomycosis. Clin Microbiol Rev. 2010;23:367-831.
- Chapman SW, Lin AC, Hendricks KA, et al. Endemic blastomycosis in Mississippi: epidemiological and clinical studies. Semin Respir Infect. 1997;12:219-228.
- Gray NA, Baddour LM. Cutaneous inoculation blastomycosis. Clin Infect Dis. 2002;34:E44-E49.
- Patel AJ, Gattuso P, Reddy VB. Diagnosis of blastomycosis in surgical pathology and cytopathology: correlation with microbiologic culture. Am J Surg Pathol. 2010;34:256-261.
- Chapman SW, Dismukes WE, Proia LA, et al. Clinical practice guidelines for the management of blastomycosis: 2008 update by the Infectious Diseases Society of America. Clin Infect Dis. 2008;46:1801-1812.
- Lomaestro, BM, Piatek MA. Update on drug interactions with azole antifungal agents. Ann Pharmacother. 1998;32:915-928.
The Diagnosis: Blastomycosis
Histopathologic examination of 3 punch biopsies from the left side of the upper lip showed pseudoepitheliomatous hyperplasia with intraepidermal microabscesses and dermal suppurative granulomatous inflammation (Figure 1A). Stains were negative for periodic acid-Schiff, herpes simplex virus, and varicella-zoster virus. Direct and indirect immunofluorescence for skin autoantibodies were negative. Two separate tissue culture specimens showed no bacterial, fungal, or mycobacterial growth. Leishmania polymerase chain reaction and DNA sequencing were negative. An additional punch biopsy revealed yeast forms with broad-based budding and refractile walls (Figures 1B and 1C) that were highlighted with Grocott-Gomori methenamine-silver stain of the tissue (Figure 2). Chest radiography demonstrated no pulmonary involvement. In collaboration with an infectious disease specialist, the patient was started on itraconazole 200 mg twice daily for a total of 6 months.
Blastomycosis is a fungal infection caused by Blastomyces dermatitidis, a thermally dimorphic fungus endemic in the soils of the Ohio and Mississippi River valleys and southeastern United States.1 It most commonly manifests as a pulmonary infection following inhalation of spores that are transformed into thick-walled yeasts capable of evading the host's immune system. Unlike other deep fungal infections, blastomycosis occurs in both immunocompetent and immunocompromised hosts. Extrapulmonary disease after hematogenous dissemination from the lungs occurs in approximately 25% to 30% of patients, with the skin as the most common site of dissemination.2 Clinically, cutaneous blastomycosis typically starts as papules that evolve into crusted vegetative plaques, often with central clearing or ulceration. Primary cutaneous blastomycosis is rare and occurs due to direct inoculation after trauma to the skin via an infected animal bite, direct inoculation in laboratory settings, or due to injury during outdoor activities involving contact with soil.3 Given our patient's horticultural hobbies, lack of pulmonary symptoms, and negative radiologic examination, primary cutaneous blastomycosis infection due to direct inoculation from contaminated soil was a possibility; however, definite confirmation was difficult, as the primary pulmonary infection of blastomycosis can be asymptomatic and therefore often goes undetected.
Cutaneous blastomycosis can be mistaken for pemphigus vegetans, leishmaniasis, herpes vegetans, bacterial pyoderma, and other deep fungal infections that also display pseudoepitheliomatous hyperplasia with pyogranulomatous inflammation on histopathology. Direct visualization of the characteristic yeast forms in a histologic specimen or the growth of fungus in culture is essential for a definitive diagnosis. The yeasts are 8 to 15 µm in diameter with thick, double-contoured walls and characteristically display broad-based budding.4 This budding pattern aids in differentiating blastomycosis from other entities with a similar histopathologic appearance. Chromoblastomycosis would show brown, thick-walled fungal cells inside giant cells, while coccidioidomycosis displays large spherules containing endospores, and leishmaniasis demonstrates amastigotes (small oval organisms with a bar-shaped kinetoplast) highlighted with Giemsa staining. Pemphigus vegetans would show intercellular deposition of IgG on direct immunofluorescence. Blastomyces dermatitidis can be difficult to visualize with routine hematoxylin and eosin stains, and it is important to note that a negative result does not exclude the possibility of blastomycosis, as demonstrated in our case.4 Special stains including Grocott-Gomori methenamine-silver and periodic acid-Schiff can aid in examining tissue for the presence of fungal elements, which typically can be found within histiocytes or abscesses in the dermis. Culture is the most sensitive method for detecting and diagnosing blastomycosis. Growth typically is detected in 5 to 10 days but can take up to 30 days if few organisms are present in the specimen.1
Although spontaneous remission can occur, it is recommended that all patients with cutaneous blastomycosis be treated to avoid dissemination and recurrence. Itraconazole currently is the treatment of choice.5 Doses typically are 200 to 400 mg/d for 8 to 12 months.6 Itraconazole-related side effects experienced by our patient during his 6-month treatment course included leg edema, 20-lb weight gain, gastrointestinal upset, blurred vision, and a transient increase in blood pressure, all resolving once the medication was discontinued. Complete resolution of the lesion was noted at the completion of the treatment course. At a 6-month posttreatment follow-up, residual scarring and alopecia were noted in parts of the previously affected areas of the upper cutaneous lip and nasolabial fold.
The Diagnosis: Blastomycosis
Histopathologic examination of 3 punch biopsies from the left side of the upper lip showed pseudoepitheliomatous hyperplasia with intraepidermal microabscesses and dermal suppurative granulomatous inflammation (Figure 1A). Stains were negative for periodic acid-Schiff, herpes simplex virus, and varicella-zoster virus. Direct and indirect immunofluorescence for skin autoantibodies were negative. Two separate tissue culture specimens showed no bacterial, fungal, or mycobacterial growth. Leishmania polymerase chain reaction and DNA sequencing were negative. An additional punch biopsy revealed yeast forms with broad-based budding and refractile walls (Figures 1B and 1C) that were highlighted with Grocott-Gomori methenamine-silver stain of the tissue (Figure 2). Chest radiography demonstrated no pulmonary involvement. In collaboration with an infectious disease specialist, the patient was started on itraconazole 200 mg twice daily for a total of 6 months.
Blastomycosis is a fungal infection caused by Blastomyces dermatitidis, a thermally dimorphic fungus endemic in the soils of the Ohio and Mississippi River valleys and southeastern United States.1 It most commonly manifests as a pulmonary infection following inhalation of spores that are transformed into thick-walled yeasts capable of evading the host's immune system. Unlike other deep fungal infections, blastomycosis occurs in both immunocompetent and immunocompromised hosts. Extrapulmonary disease after hematogenous dissemination from the lungs occurs in approximately 25% to 30% of patients, with the skin as the most common site of dissemination.2 Clinically, cutaneous blastomycosis typically starts as papules that evolve into crusted vegetative plaques, often with central clearing or ulceration. Primary cutaneous blastomycosis is rare and occurs due to direct inoculation after trauma to the skin via an infected animal bite, direct inoculation in laboratory settings, or due to injury during outdoor activities involving contact with soil.3 Given our patient's horticultural hobbies, lack of pulmonary symptoms, and negative radiologic examination, primary cutaneous blastomycosis infection due to direct inoculation from contaminated soil was a possibility; however, definite confirmation was difficult, as the primary pulmonary infection of blastomycosis can be asymptomatic and therefore often goes undetected.
Cutaneous blastomycosis can be mistaken for pemphigus vegetans, leishmaniasis, herpes vegetans, bacterial pyoderma, and other deep fungal infections that also display pseudoepitheliomatous hyperplasia with pyogranulomatous inflammation on histopathology. Direct visualization of the characteristic yeast forms in a histologic specimen or the growth of fungus in culture is essential for a definitive diagnosis. The yeasts are 8 to 15 µm in diameter with thick, double-contoured walls and characteristically display broad-based budding.4 This budding pattern aids in differentiating blastomycosis from other entities with a similar histopathologic appearance. Chromoblastomycosis would show brown, thick-walled fungal cells inside giant cells, while coccidioidomycosis displays large spherules containing endospores, and leishmaniasis demonstrates amastigotes (small oval organisms with a bar-shaped kinetoplast) highlighted with Giemsa staining. Pemphigus vegetans would show intercellular deposition of IgG on direct immunofluorescence. Blastomyces dermatitidis can be difficult to visualize with routine hematoxylin and eosin stains, and it is important to note that a negative result does not exclude the possibility of blastomycosis, as demonstrated in our case.4 Special stains including Grocott-Gomori methenamine-silver and periodic acid-Schiff can aid in examining tissue for the presence of fungal elements, which typically can be found within histiocytes or abscesses in the dermis. Culture is the most sensitive method for detecting and diagnosing blastomycosis. Growth typically is detected in 5 to 10 days but can take up to 30 days if few organisms are present in the specimen.1
Although spontaneous remission can occur, it is recommended that all patients with cutaneous blastomycosis be treated to avoid dissemination and recurrence. Itraconazole currently is the treatment of choice.5 Doses typically are 200 to 400 mg/d for 8 to 12 months.6 Itraconazole-related side effects experienced by our patient during his 6-month treatment course included leg edema, 20-lb weight gain, gastrointestinal upset, blurred vision, and a transient increase in blood pressure, all resolving once the medication was discontinued. Complete resolution of the lesion was noted at the completion of the treatment course. At a 6-month posttreatment follow-up, residual scarring and alopecia were noted in parts of the previously affected areas of the upper cutaneous lip and nasolabial fold.
- Saccente M, Woods GL. Clinical and laboratory update on blastomycosis. Clin Microbiol Rev. 2010;23:367-831.
- Chapman SW, Lin AC, Hendricks KA, et al. Endemic blastomycosis in Mississippi: epidemiological and clinical studies. Semin Respir Infect. 1997;12:219-228.
- Gray NA, Baddour LM. Cutaneous inoculation blastomycosis. Clin Infect Dis. 2002;34:E44-E49.
- Patel AJ, Gattuso P, Reddy VB. Diagnosis of blastomycosis in surgical pathology and cytopathology: correlation with microbiologic culture. Am J Surg Pathol. 2010;34:256-261.
- Chapman SW, Dismukes WE, Proia LA, et al. Clinical practice guidelines for the management of blastomycosis: 2008 update by the Infectious Diseases Society of America. Clin Infect Dis. 2008;46:1801-1812.
- Lomaestro, BM, Piatek MA. Update on drug interactions with azole antifungal agents. Ann Pharmacother. 1998;32:915-928.
- Saccente M, Woods GL. Clinical and laboratory update on blastomycosis. Clin Microbiol Rev. 2010;23:367-831.
- Chapman SW, Lin AC, Hendricks KA, et al. Endemic blastomycosis in Mississippi: epidemiological and clinical studies. Semin Respir Infect. 1997;12:219-228.
- Gray NA, Baddour LM. Cutaneous inoculation blastomycosis. Clin Infect Dis. 2002;34:E44-E49.
- Patel AJ, Gattuso P, Reddy VB. Diagnosis of blastomycosis in surgical pathology and cytopathology: correlation with microbiologic culture. Am J Surg Pathol. 2010;34:256-261.
- Chapman SW, Dismukes WE, Proia LA, et al. Clinical practice guidelines for the management of blastomycosis: 2008 update by the Infectious Diseases Society of America. Clin Infect Dis. 2008;46:1801-1812.
- Lomaestro, BM, Piatek MA. Update on drug interactions with azole antifungal agents. Ann Pharmacother. 1998;32:915-928.
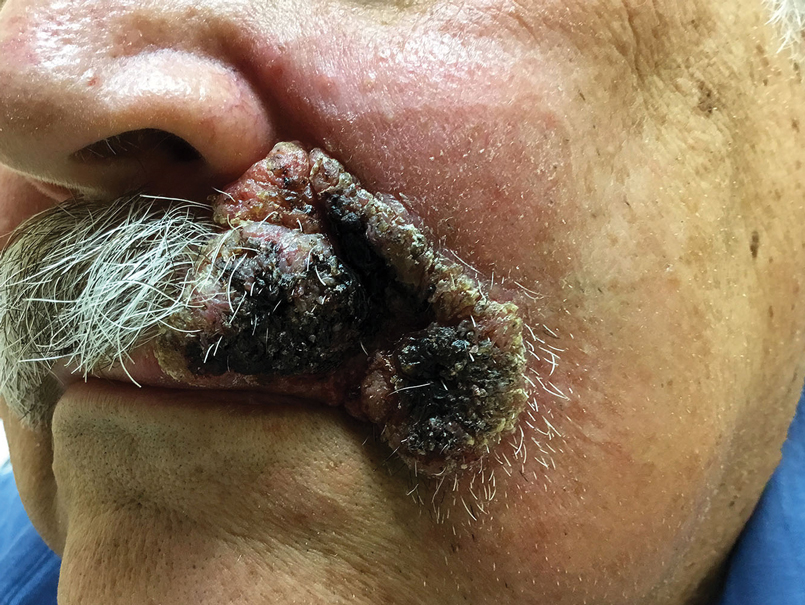
A 69-year-old man presented with a slowly expanding, verrucous plaque on the left side of the upper cutaneous lip of 4 months’ duration. The lesion reportedly began as an abscess and had undergone incision and drainage followed by multiple courses of oral antibiotics that were unsuccessful prior to presentation to our clinic. The patient’s hobbies included gardening near his summer home in the mountains of western North Carolina, where he resided when the lesion appeared. Physical examination revealed an approximately 6×4-cm verrucous plaque with central ulceration on the left side of the upper cutaneous and vermilion lip extending to the nasolabial fold. A review of systems was negative for any systemic symptoms. Routine laboratory tests and computed tomography of the head and neck were normal.
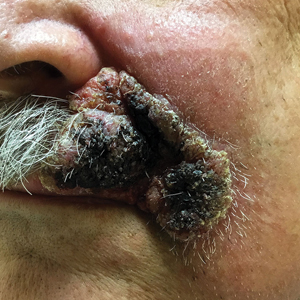
Unilateral Alar Ulceration
The Diagnosis: Trigeminal Trophic Syndrome (Self-induced Trauma)
The patient admitted to manipulation of the ala in response to persistent pain despite resolution of the herpes zoster, for which he recently had completed a course of oral acyclovir. A preliminary diagnosis of trigeminal trophic syndrome (TTS) was made, and a subsequent punch biopsy revealed no evidence of malignancy. Topical antibiotic prophylaxis was prescribed, and he was instructed to avoid manipulation of the affected area. Treatment was initiated in consultation with pain specialists, and over the following 3 years our patient experienced a waxing and waning course of persistent pain complicated by new scalp and oral ulcers as well as alar impetigo. His condition eventually stabilized with tolerable pain on oral gabapentin and doxepin cream 5% applied up to 4 times daily. The alar lesion healed following sufficient abstinence from manipulation, leaving a crescent-shaped rim defect.
Trigeminal trophic syndrome classically is characterized by a triad of cutaneous anesthesia, paresthesia and/or pain, and ulceration secondary to pathology of trigeminal nerve sensory branches. Ulceration arises primarily through excoriation in response to paresthetic pruritus or pain. The differential diagnosis for TTS includes ulcerating cutaneous neoplasms (eg, basal cell carcinoma); mycobacterial, fungal, and viral infections (especially herpetic lesions); and cutaneous involvement of systemic vasculitides (eg, granulomatosis with polyangiitis).1 Biopsy is necessary to exclude malignancy, and ulcers may be scraped for viral diagnosis. Complete blood cell count and serologic testing also may help to exclude immunodeficiencies or disorders. Apart from viral neuropathy, common etiologies of TTS include iatrogenic trigeminal injury (eg, in ablation treatment for trigeminal neuralgia) and stroke (eg, lateral medullary syndrome).
- Khan AU, Khachemoune A. Trigeminal trophic syndrome: an updated review. Int J Dermatol. 2019;58:530-537.
The Diagnosis: Trigeminal Trophic Syndrome (Self-induced Trauma)
The patient admitted to manipulation of the ala in response to persistent pain despite resolution of the herpes zoster, for which he recently had completed a course of oral acyclovir. A preliminary diagnosis of trigeminal trophic syndrome (TTS) was made, and a subsequent punch biopsy revealed no evidence of malignancy. Topical antibiotic prophylaxis was prescribed, and he was instructed to avoid manipulation of the affected area. Treatment was initiated in consultation with pain specialists, and over the following 3 years our patient experienced a waxing and waning course of persistent pain complicated by new scalp and oral ulcers as well as alar impetigo. His condition eventually stabilized with tolerable pain on oral gabapentin and doxepin cream 5% applied up to 4 times daily. The alar lesion healed following sufficient abstinence from manipulation, leaving a crescent-shaped rim defect.
Trigeminal trophic syndrome classically is characterized by a triad of cutaneous anesthesia, paresthesia and/or pain, and ulceration secondary to pathology of trigeminal nerve sensory branches. Ulceration arises primarily through excoriation in response to paresthetic pruritus or pain. The differential diagnosis for TTS includes ulcerating cutaneous neoplasms (eg, basal cell carcinoma); mycobacterial, fungal, and viral infections (especially herpetic lesions); and cutaneous involvement of systemic vasculitides (eg, granulomatosis with polyangiitis).1 Biopsy is necessary to exclude malignancy, and ulcers may be scraped for viral diagnosis. Complete blood cell count and serologic testing also may help to exclude immunodeficiencies or disorders. Apart from viral neuropathy, common etiologies of TTS include iatrogenic trigeminal injury (eg, in ablation treatment for trigeminal neuralgia) and stroke (eg, lateral medullary syndrome).
The Diagnosis: Trigeminal Trophic Syndrome (Self-induced Trauma)
The patient admitted to manipulation of the ala in response to persistent pain despite resolution of the herpes zoster, for which he recently had completed a course of oral acyclovir. A preliminary diagnosis of trigeminal trophic syndrome (TTS) was made, and a subsequent punch biopsy revealed no evidence of malignancy. Topical antibiotic prophylaxis was prescribed, and he was instructed to avoid manipulation of the affected area. Treatment was initiated in consultation with pain specialists, and over the following 3 years our patient experienced a waxing and waning course of persistent pain complicated by new scalp and oral ulcers as well as alar impetigo. His condition eventually stabilized with tolerable pain on oral gabapentin and doxepin cream 5% applied up to 4 times daily. The alar lesion healed following sufficient abstinence from manipulation, leaving a crescent-shaped rim defect.
Trigeminal trophic syndrome classically is characterized by a triad of cutaneous anesthesia, paresthesia and/or pain, and ulceration secondary to pathology of trigeminal nerve sensory branches. Ulceration arises primarily through excoriation in response to paresthetic pruritus or pain. The differential diagnosis for TTS includes ulcerating cutaneous neoplasms (eg, basal cell carcinoma); mycobacterial, fungal, and viral infections (especially herpetic lesions); and cutaneous involvement of systemic vasculitides (eg, granulomatosis with polyangiitis).1 Biopsy is necessary to exclude malignancy, and ulcers may be scraped for viral diagnosis. Complete blood cell count and serologic testing also may help to exclude immunodeficiencies or disorders. Apart from viral neuropathy, common etiologies of TTS include iatrogenic trigeminal injury (eg, in ablation treatment for trigeminal neuralgia) and stroke (eg, lateral medullary syndrome).
- Khan AU, Khachemoune A. Trigeminal trophic syndrome: an updated review. Int J Dermatol. 2019;58:530-537.
- Khan AU, Khachemoune A. Trigeminal trophic syndrome: an updated review. Int J Dermatol. 2019;58:530-537.
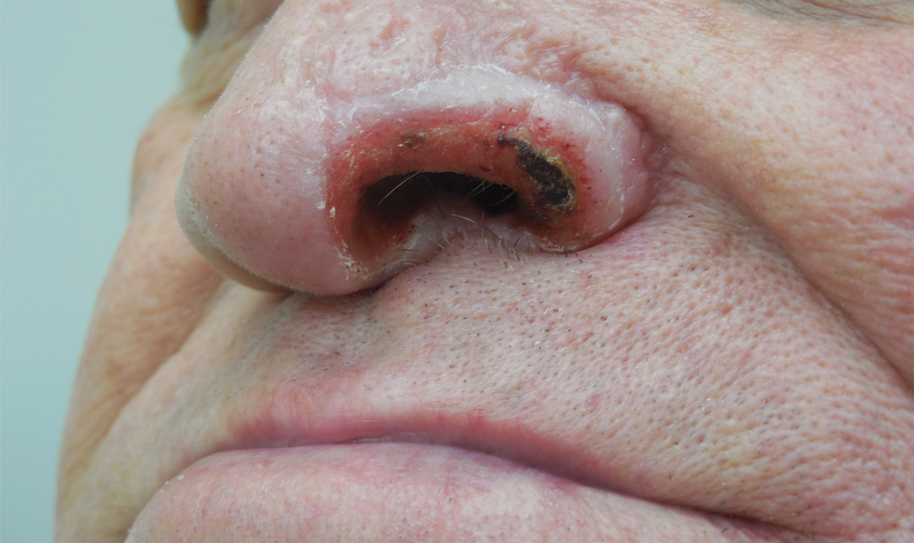
A 68-year-old man presented with a new left nasal alar ulcer following a recent episode of primary herpes zoster. Physical examination revealed erythema, erosion, and necrosis of the left naris with partial loss of the alar rim. Additional erythema was present without vesicles around the left eye and on the forehead.
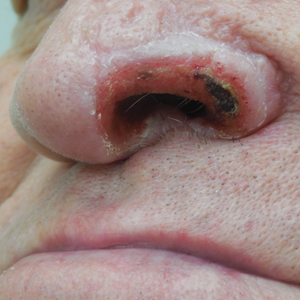
Telangiectatic Patch on the Forehead
The Diagnosis: Cutaneous B-cell Lymphoma
Histopathology was suggestive of cutaneous B-cell lymphoma (Figure). Further immunohistochemical studies including Bcl-6 positivity and Bcl-2 negativity in the large atypical cells supported a diagnosis of primary cutaneous follicle center lymphoma (PCFCL). The designation of primary cutaneous B-cell lymphoma includes several different types of lymphoma, including marginal zone lymphoma, diffuse large B-cell lymphoma, and intravascular lymphoma. To be considered a primary cutaneous lymphoma, there must be evidence of the lymphoma in the skin without concomitant evidence of systemic involvement, as determined through a full staging workup. Primary cutaneous follicle center lymphoma is an indolent lymphoma that most commonly presents as solitary or grouped, pink to plum-colored papules, plaques, nodules, and tumors on the scalp, forehead, or back.1 The lesions often are biopsied as suspected basal cell carcinomas or Merkel cell carcinomas (MCCs). Lesions on the face or scalp may easily evade diagnosis, as they initially may mimic rosacea or insect bites. Less common presentations include infiltrative lesions that cause rhinophymatous changes or scarring alopecia. Multifocal or disseminated lesions rarely can be observed. This case presentation is unique in its patchy appearance that clinically resembled angiosarcoma.2 When identified and treated, the disease-specific 5-year survival rate for PCFCL is greater than 95%.3
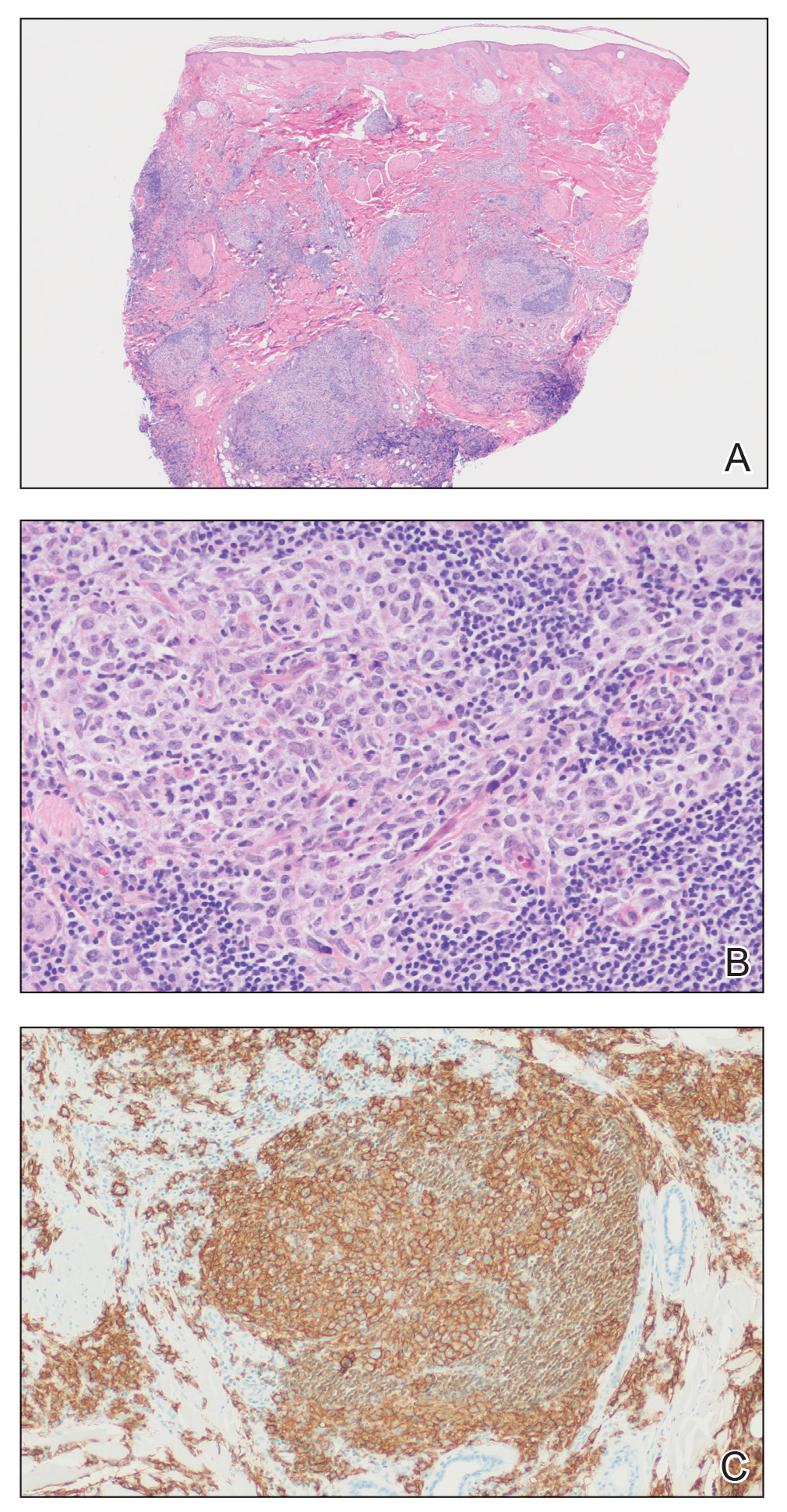
Merkel cell carcinoma was first described in 1972 and has been diagnosed with increasing frequency each year.4 It generally presents as an erythematous or violaceous, tender, indurated nodule on sun-exposed skin of the head or neck in elderly White men. However, other presentations have been reported, including papules, plaques, cystlike structures, pruritic tumors, pedunculated lesions, subcutaneous masses, and telangiectatic papules.5 Histopathologically, MCC is characterized by dermal nests and sheets of basaloid cells with finely granular salt and pepper-like chromatin. The histologic features can resemble other small blue cell tumors; therefore, the differential diagnosis can be broad.5 Immunohistochemistry that can confirm the diagnosis of MCC generally will be positive for cytokeratin 20 and neuroendocrine markers but negative for cytokeratin 7 and thyroid transcription factor 1. Merkel cell carcinoma is an aggressive tumor with a high risk for local recurrence and distant metastasis that carries a generally poor prognosis, especially when there is evidence of metastatic disease at presentation.5,6
Rosacea can appear as telangiectatic patches, though generally not as one discrete patch limited to the forehead, as in our patient. Histologic features vary based on the age of the lesion and clinical variant. In early lesions there is a mild perivascular lymphoplasmacytic infiltrate within the dermis, while older lesions can have a mixed infiltrate crowded around vessels and adnexal structures. Granulomas often are seen near hair follicles and interspersed throughout the dermis with ectatic vessels and dermal edema.7
Angiosarcoma is divided into 3 clinicopathological subtypes: idiopathic angiosarcoma of the head and neck, angiosarcoma in the setting of lymphedema, and postirradiation angiosarcoma.7 Idiopathic angiosarcoma most closely mimics PCFCL, as it can present as single or multifocal nodules, plaques, or patches. Histologically, the 3 groups appear similar with poorly circumscribed, infiltrative, dermal tumors. The neoplastic endothelial cells have large hyperchromatic nuclei that protrude into vascular lumens. The prognosis for idiopathic angiosarcoma of the head and neck is poor, with a 5-year survival rate of 15% to 34%, which often is due to delayed diagnosis.7
Pigmented purpuric dermatoses (PPDs) are chronic skin disorders characterized by purpura due to extravasation of blood from capillaries; the resulting hemosiderin deposition leads to pigmentation.7 There are various forms of PPD, which are classified into groups based on clinical appearance including Schamberg disease, purpura annularis telangiectodes of Majocchi, pigmented purpuric lichenoid dermatosis of Gougerot and Blum, lichen aureus, and others including eczematid and itching variants, which some consider to be distinct entities. Purpura annularis telangiectodes of Majocchi is the specific PPD that should be included in the clinical differential for PCFCL because it presents as annular patches with telangiectasias. Histologically, PPDs are characterized by a CD4+ lymphocytic infiltrate in the upper dermis with extravasated red blood cells and the presence of hemosiderin mostly within macrophages and a lack of true vasculitis. Clonality of the T cells has been shown, and there is some evidence that PPD may overlap with mycosis fungoides. However, this overlap mainly has been seen in patients with widespread lesions and would not apply to this case. In general, patients with PPD can be reassured of the benign process. In cases of widespread PPD, patients should be followed clinically to assess for progression to mycosis fungoides, though the likelihood is low.7
Our patient underwent a full staging workup, which confirmed the diagnosis of PCFCL. He was treated with radiation to the forehead that resulted in clearance of the lesion. Approximately 2 years after the initial diagnosis, the patient was alive and well with no evidence of recurrence of PCFCL.
In conclusion, it is imperative to identify unusual, macular, vascular-appearing patches, especially on the head and neck in older individuals. Because the clinical presentations of PCFCL, angiosarcoma, rosacea, MCC, and PPD can overlap with one another as well as with other entities, it is necessary to have a high level of suspicion and low threshold to biopsy these types of lesions, as outcomes can be drastically different.
- Goyal A, LeBlanc RE, Carter JB. Cutaneous B-cell lymphoma. Hematol Oncol Clin North Am. 2019;33:149-161.
- Massone C, Fink-Puches R, Cerroni L. Atypical clinical presentation of primary and secondary cutaneous follicle center lymphoma (FCL) on the head characterized by macular lesions. J Am Acad Dermatol. 2016;75:1000-1006.
- Wilcox RA. Cutaneous B-cell lymphomas: 2016 update on diagnosis, risk-stratification, and management. Am J Hematol. 2016;91:1052-1055.
- Conic RRZ, Ko J, Saridakis S, et al. Sentinel lymph node biopsy in Merkel cell carcinoma: predictors of sentinel lymph node positivity and association with overall survival. J Am Acad Dermatol. 2019;81:364-372
- Coggshall K, Tello TL, North JP, et al. Merkel cell carcinoma: an update and review: pathogenesis, diagnosis, and staging. J Am Acad Dermatol. 2018;78:433-442.
- Tello TL, Coggshall K, Yom SS, et al. Merkel cell carcinoma: an update and review: current and future therapy. J Am Acad Dermatol. 2018;78:445-454.
- Patterson JW, Hosler GA. Weedon's Skin Pathology. 4th ed. China: Churchill Livingstone Elsevier; 2016.
The Diagnosis: Cutaneous B-cell Lymphoma
Histopathology was suggestive of cutaneous B-cell lymphoma (Figure). Further immunohistochemical studies including Bcl-6 positivity and Bcl-2 negativity in the large atypical cells supported a diagnosis of primary cutaneous follicle center lymphoma (PCFCL). The designation of primary cutaneous B-cell lymphoma includes several different types of lymphoma, including marginal zone lymphoma, diffuse large B-cell lymphoma, and intravascular lymphoma. To be considered a primary cutaneous lymphoma, there must be evidence of the lymphoma in the skin without concomitant evidence of systemic involvement, as determined through a full staging workup. Primary cutaneous follicle center lymphoma is an indolent lymphoma that most commonly presents as solitary or grouped, pink to plum-colored papules, plaques, nodules, and tumors on the scalp, forehead, or back.1 The lesions often are biopsied as suspected basal cell carcinomas or Merkel cell carcinomas (MCCs). Lesions on the face or scalp may easily evade diagnosis, as they initially may mimic rosacea or insect bites. Less common presentations include infiltrative lesions that cause rhinophymatous changes or scarring alopecia. Multifocal or disseminated lesions rarely can be observed. This case presentation is unique in its patchy appearance that clinically resembled angiosarcoma.2 When identified and treated, the disease-specific 5-year survival rate for PCFCL is greater than 95%.3
Merkel cell carcinoma was first described in 1972 and has been diagnosed with increasing frequency each year.4 It generally presents as an erythematous or violaceous, tender, indurated nodule on sun-exposed skin of the head or neck in elderly White men. However, other presentations have been reported, including papules, plaques, cystlike structures, pruritic tumors, pedunculated lesions, subcutaneous masses, and telangiectatic papules.5 Histopathologically, MCC is characterized by dermal nests and sheets of basaloid cells with finely granular salt and pepper-like chromatin. The histologic features can resemble other small blue cell tumors; therefore, the differential diagnosis can be broad.5 Immunohistochemistry that can confirm the diagnosis of MCC generally will be positive for cytokeratin 20 and neuroendocrine markers but negative for cytokeratin 7 and thyroid transcription factor 1. Merkel cell carcinoma is an aggressive tumor with a high risk for local recurrence and distant metastasis that carries a generally poor prognosis, especially when there is evidence of metastatic disease at presentation.5,6
Rosacea can appear as telangiectatic patches, though generally not as one discrete patch limited to the forehead, as in our patient. Histologic features vary based on the age of the lesion and clinical variant. In early lesions there is a mild perivascular lymphoplasmacytic infiltrate within the dermis, while older lesions can have a mixed infiltrate crowded around vessels and adnexal structures. Granulomas often are seen near hair follicles and interspersed throughout the dermis with ectatic vessels and dermal edema.7
Angiosarcoma is divided into 3 clinicopathological subtypes: idiopathic angiosarcoma of the head and neck, angiosarcoma in the setting of lymphedema, and postirradiation angiosarcoma.7 Idiopathic angiosarcoma most closely mimics PCFCL, as it can present as single or multifocal nodules, plaques, or patches. Histologically, the 3 groups appear similar with poorly circumscribed, infiltrative, dermal tumors. The neoplastic endothelial cells have large hyperchromatic nuclei that protrude into vascular lumens. The prognosis for idiopathic angiosarcoma of the head and neck is poor, with a 5-year survival rate of 15% to 34%, which often is due to delayed diagnosis.7
Pigmented purpuric dermatoses (PPDs) are chronic skin disorders characterized by purpura due to extravasation of blood from capillaries; the resulting hemosiderin deposition leads to pigmentation.7 There are various forms of PPD, which are classified into groups based on clinical appearance including Schamberg disease, purpura annularis telangiectodes of Majocchi, pigmented purpuric lichenoid dermatosis of Gougerot and Blum, lichen aureus, and others including eczematid and itching variants, which some consider to be distinct entities. Purpura annularis telangiectodes of Majocchi is the specific PPD that should be included in the clinical differential for PCFCL because it presents as annular patches with telangiectasias. Histologically, PPDs are characterized by a CD4+ lymphocytic infiltrate in the upper dermis with extravasated red blood cells and the presence of hemosiderin mostly within macrophages and a lack of true vasculitis. Clonality of the T cells has been shown, and there is some evidence that PPD may overlap with mycosis fungoides. However, this overlap mainly has been seen in patients with widespread lesions and would not apply to this case. In general, patients with PPD can be reassured of the benign process. In cases of widespread PPD, patients should be followed clinically to assess for progression to mycosis fungoides, though the likelihood is low.7
Our patient underwent a full staging workup, which confirmed the diagnosis of PCFCL. He was treated with radiation to the forehead that resulted in clearance of the lesion. Approximately 2 years after the initial diagnosis, the patient was alive and well with no evidence of recurrence of PCFCL.
In conclusion, it is imperative to identify unusual, macular, vascular-appearing patches, especially on the head and neck in older individuals. Because the clinical presentations of PCFCL, angiosarcoma, rosacea, MCC, and PPD can overlap with one another as well as with other entities, it is necessary to have a high level of suspicion and low threshold to biopsy these types of lesions, as outcomes can be drastically different.
The Diagnosis: Cutaneous B-cell Lymphoma
Histopathology was suggestive of cutaneous B-cell lymphoma (Figure). Further immunohistochemical studies including Bcl-6 positivity and Bcl-2 negativity in the large atypical cells supported a diagnosis of primary cutaneous follicle center lymphoma (PCFCL). The designation of primary cutaneous B-cell lymphoma includes several different types of lymphoma, including marginal zone lymphoma, diffuse large B-cell lymphoma, and intravascular lymphoma. To be considered a primary cutaneous lymphoma, there must be evidence of the lymphoma in the skin without concomitant evidence of systemic involvement, as determined through a full staging workup. Primary cutaneous follicle center lymphoma is an indolent lymphoma that most commonly presents as solitary or grouped, pink to plum-colored papules, plaques, nodules, and tumors on the scalp, forehead, or back.1 The lesions often are biopsied as suspected basal cell carcinomas or Merkel cell carcinomas (MCCs). Lesions on the face or scalp may easily evade diagnosis, as they initially may mimic rosacea or insect bites. Less common presentations include infiltrative lesions that cause rhinophymatous changes or scarring alopecia. Multifocal or disseminated lesions rarely can be observed. This case presentation is unique in its patchy appearance that clinically resembled angiosarcoma.2 When identified and treated, the disease-specific 5-year survival rate for PCFCL is greater than 95%.3
Merkel cell carcinoma was first described in 1972 and has been diagnosed with increasing frequency each year.4 It generally presents as an erythematous or violaceous, tender, indurated nodule on sun-exposed skin of the head or neck in elderly White men. However, other presentations have been reported, including papules, plaques, cystlike structures, pruritic tumors, pedunculated lesions, subcutaneous masses, and telangiectatic papules.5 Histopathologically, MCC is characterized by dermal nests and sheets of basaloid cells with finely granular salt and pepper-like chromatin. The histologic features can resemble other small blue cell tumors; therefore, the differential diagnosis can be broad.5 Immunohistochemistry that can confirm the diagnosis of MCC generally will be positive for cytokeratin 20 and neuroendocrine markers but negative for cytokeratin 7 and thyroid transcription factor 1. Merkel cell carcinoma is an aggressive tumor with a high risk for local recurrence and distant metastasis that carries a generally poor prognosis, especially when there is evidence of metastatic disease at presentation.5,6
Rosacea can appear as telangiectatic patches, though generally not as one discrete patch limited to the forehead, as in our patient. Histologic features vary based on the age of the lesion and clinical variant. In early lesions there is a mild perivascular lymphoplasmacytic infiltrate within the dermis, while older lesions can have a mixed infiltrate crowded around vessels and adnexal structures. Granulomas often are seen near hair follicles and interspersed throughout the dermis with ectatic vessels and dermal edema.7
Angiosarcoma is divided into 3 clinicopathological subtypes: idiopathic angiosarcoma of the head and neck, angiosarcoma in the setting of lymphedema, and postirradiation angiosarcoma.7 Idiopathic angiosarcoma most closely mimics PCFCL, as it can present as single or multifocal nodules, plaques, or patches. Histologically, the 3 groups appear similar with poorly circumscribed, infiltrative, dermal tumors. The neoplastic endothelial cells have large hyperchromatic nuclei that protrude into vascular lumens. The prognosis for idiopathic angiosarcoma of the head and neck is poor, with a 5-year survival rate of 15% to 34%, which often is due to delayed diagnosis.7
Pigmented purpuric dermatoses (PPDs) are chronic skin disorders characterized by purpura due to extravasation of blood from capillaries; the resulting hemosiderin deposition leads to pigmentation.7 There are various forms of PPD, which are classified into groups based on clinical appearance including Schamberg disease, purpura annularis telangiectodes of Majocchi, pigmented purpuric lichenoid dermatosis of Gougerot and Blum, lichen aureus, and others including eczematid and itching variants, which some consider to be distinct entities. Purpura annularis telangiectodes of Majocchi is the specific PPD that should be included in the clinical differential for PCFCL because it presents as annular patches with telangiectasias. Histologically, PPDs are characterized by a CD4+ lymphocytic infiltrate in the upper dermis with extravasated red blood cells and the presence of hemosiderin mostly within macrophages and a lack of true vasculitis. Clonality of the T cells has been shown, and there is some evidence that PPD may overlap with mycosis fungoides. However, this overlap mainly has been seen in patients with widespread lesions and would not apply to this case. In general, patients with PPD can be reassured of the benign process. In cases of widespread PPD, patients should be followed clinically to assess for progression to mycosis fungoides, though the likelihood is low.7
Our patient underwent a full staging workup, which confirmed the diagnosis of PCFCL. He was treated with radiation to the forehead that resulted in clearance of the lesion. Approximately 2 years after the initial diagnosis, the patient was alive and well with no evidence of recurrence of PCFCL.
In conclusion, it is imperative to identify unusual, macular, vascular-appearing patches, especially on the head and neck in older individuals. Because the clinical presentations of PCFCL, angiosarcoma, rosacea, MCC, and PPD can overlap with one another as well as with other entities, it is necessary to have a high level of suspicion and low threshold to biopsy these types of lesions, as outcomes can be drastically different.
- Goyal A, LeBlanc RE, Carter JB. Cutaneous B-cell lymphoma. Hematol Oncol Clin North Am. 2019;33:149-161.
- Massone C, Fink-Puches R, Cerroni L. Atypical clinical presentation of primary and secondary cutaneous follicle center lymphoma (FCL) on the head characterized by macular lesions. J Am Acad Dermatol. 2016;75:1000-1006.
- Wilcox RA. Cutaneous B-cell lymphomas: 2016 update on diagnosis, risk-stratification, and management. Am J Hematol. 2016;91:1052-1055.
- Conic RRZ, Ko J, Saridakis S, et al. Sentinel lymph node biopsy in Merkel cell carcinoma: predictors of sentinel lymph node positivity and association with overall survival. J Am Acad Dermatol. 2019;81:364-372
- Coggshall K, Tello TL, North JP, et al. Merkel cell carcinoma: an update and review: pathogenesis, diagnosis, and staging. J Am Acad Dermatol. 2018;78:433-442.
- Tello TL, Coggshall K, Yom SS, et al. Merkel cell carcinoma: an update and review: current and future therapy. J Am Acad Dermatol. 2018;78:445-454.
- Patterson JW, Hosler GA. Weedon's Skin Pathology. 4th ed. China: Churchill Livingstone Elsevier; 2016.
- Goyal A, LeBlanc RE, Carter JB. Cutaneous B-cell lymphoma. Hematol Oncol Clin North Am. 2019;33:149-161.
- Massone C, Fink-Puches R, Cerroni L. Atypical clinical presentation of primary and secondary cutaneous follicle center lymphoma (FCL) on the head characterized by macular lesions. J Am Acad Dermatol. 2016;75:1000-1006.
- Wilcox RA. Cutaneous B-cell lymphomas: 2016 update on diagnosis, risk-stratification, and management. Am J Hematol. 2016;91:1052-1055.
- Conic RRZ, Ko J, Saridakis S, et al. Sentinel lymph node biopsy in Merkel cell carcinoma: predictors of sentinel lymph node positivity and association with overall survival. J Am Acad Dermatol. 2019;81:364-372
- Coggshall K, Tello TL, North JP, et al. Merkel cell carcinoma: an update and review: pathogenesis, diagnosis, and staging. J Am Acad Dermatol. 2018;78:433-442.
- Tello TL, Coggshall K, Yom SS, et al. Merkel cell carcinoma: an update and review: current and future therapy. J Am Acad Dermatol. 2018;78:445-454.
- Patterson JW, Hosler GA. Weedon's Skin Pathology. 4th ed. China: Churchill Livingstone Elsevier; 2016.
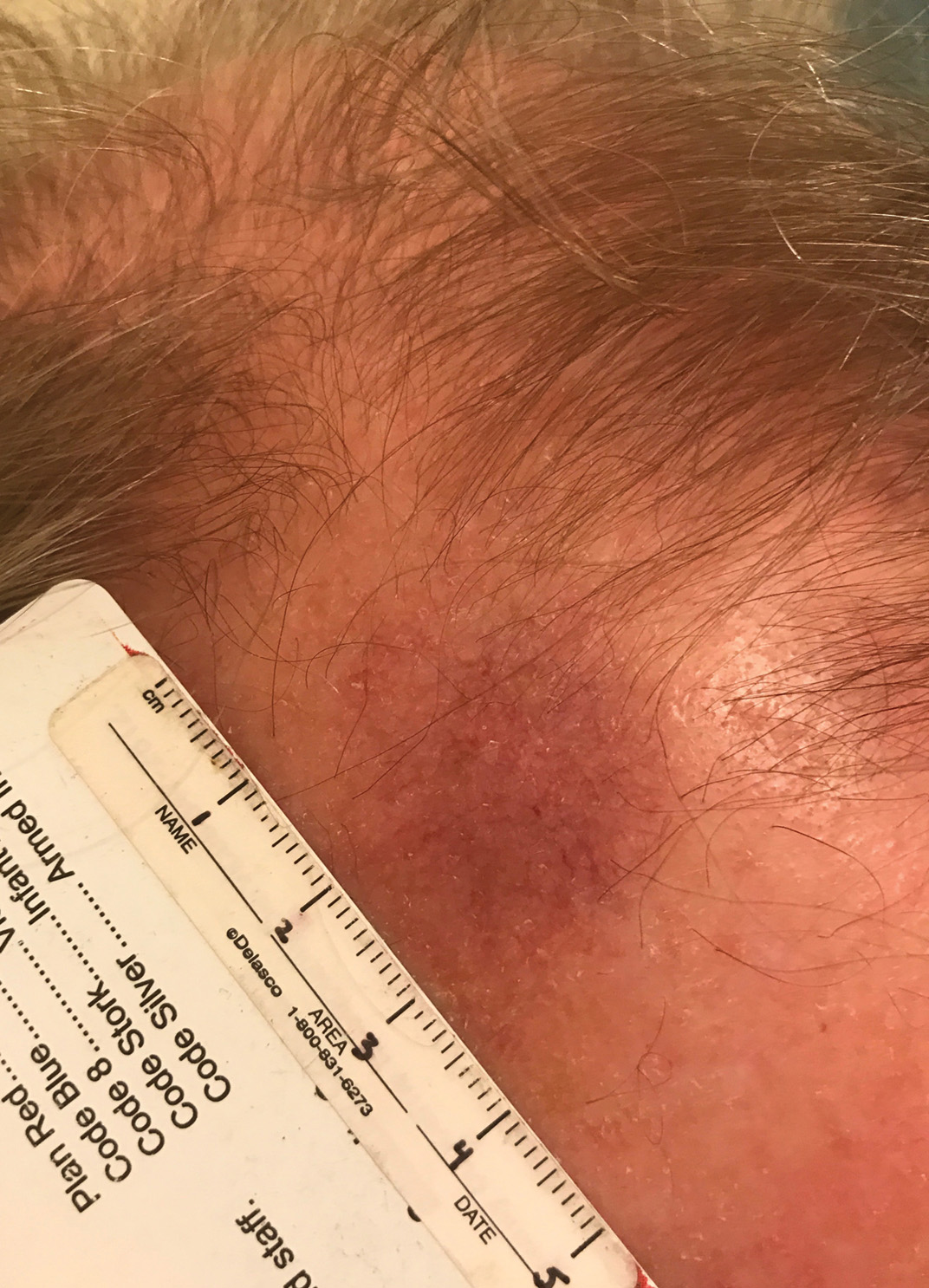
Widespread Purple Plaques
The Diagnosis: Kaposi Sarcoma
On initial presentation, the differential diagnosis included secondary syphilis, Kaposi sarcoma (KS), lichen planus pigmentosus, sarcoidosis, and psoriasis. A laboratory workup was ordered, which included complete blood cell count, comprehensive metabolic panel, antinuclear antibodies, anti-Ro/Sjögren syndrome antigen A and anti-La/Sjögren syndrome antigen B autoantibodies, angiotensin-converting enzyme, rapid plasma reagin, and human immunodeficiency virus (HIV) antibodies. A 4-mm punch biopsy of the rash also was performed from the right upper back. Histology revealed a vascular proliferation that was diffusely positive for human herpesvirus 8 (HHV-8)(Figure 1). The patient was informed of the diagnosis, at which time he revealed he had a history of homosexual relationships, with his last sexual contact being more than 1 year prior to presentation. The laboratory workup confirmed a diagnosis of HIV, and the remainder of the tests were unremarkable.
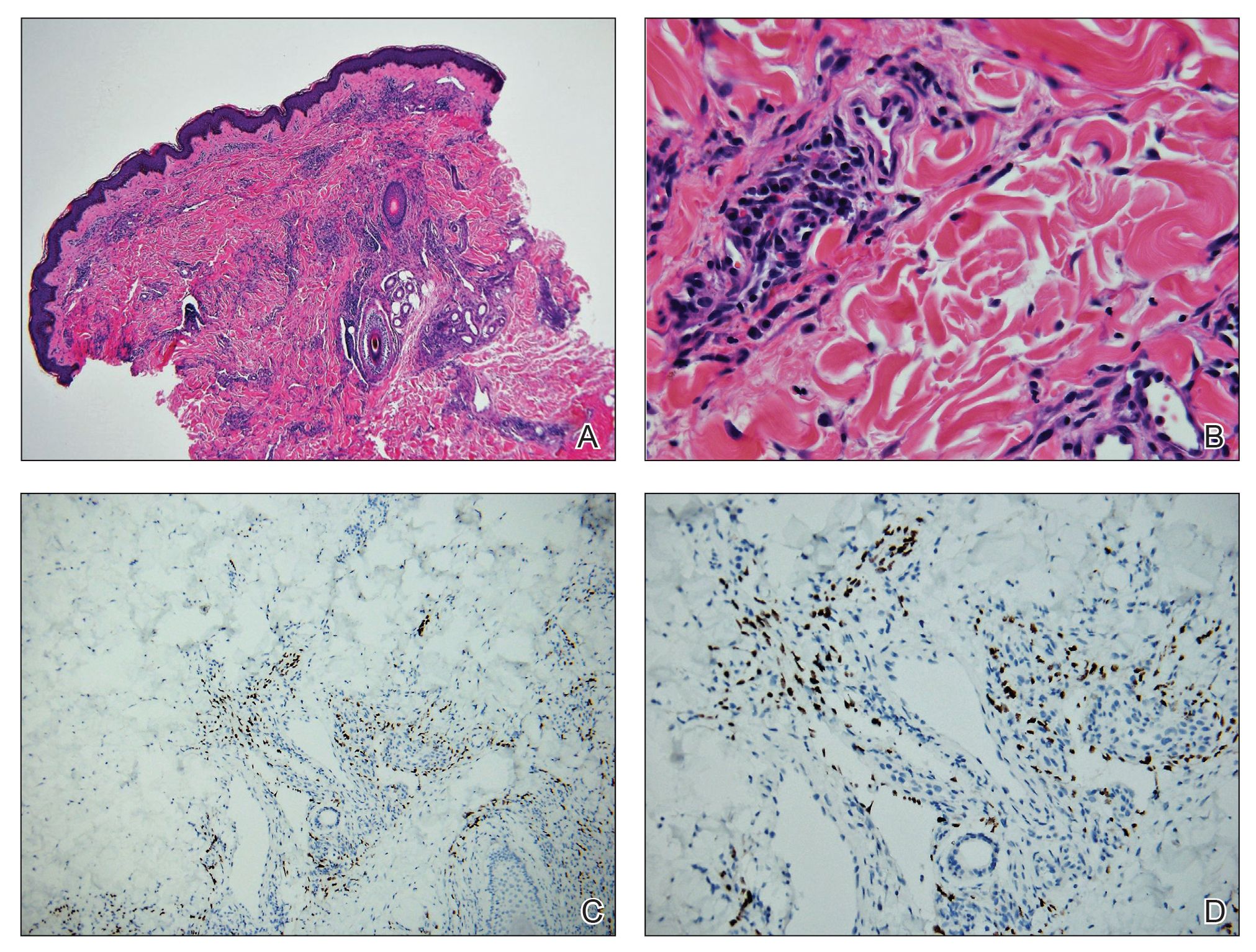
He was referred to our university's HIV clinic where he was started on highly active antiretroviral therapy (HAART). His facial swelling worsened, leading to hospital admission. Computed tomography (CT) of the chest, abdomen, and pelvis showed diffuse lymphadenopathy and lung nodules concerning for visceral involvement of KS. Hematology and oncology was consulted for further evaluation, and he was treated with 6 cycles of doxorubicin 20 mg/m2, which led to resolution of the lung nodules on CT and improvement of the rash burden. He was then started on alitretinoin gel 0.1% twice daily, which led to continued slow improvement (Figure 2).

Kaposi sarcoma is a vascular neoplasm that occurs from infection with HHV-8. It typically presents as painless, reddish to violaceous macules or patches involving the skin and mucosa that often progress to plaques or nodules with possible visceral involvement. Kaposi sarcoma is classified into 4 subtypes based on epidemiology and clinical presentation: classic, endemic, iatrogenic, and AIDS associated.1,2
Classic KS primarily affects elderly males of Mediterranean or Eastern European descent, with a mean age of 64.1 years and a male to female ratio of 3 to 1. It has an indolent course and a strong predilection for the skin of the lower extremities. The endemic form occurs mainly in Africa and has a more aggressive course, especially the lymphadenopathic type that affects children younger than 10 years.3 Iatrogenic KS develops in immunosuppressed patients, such as transplant recipients, and may regress if the immunosuppressive agent is stopped.1 Kaposi sarcoma is an AIDS-defining illness and is the most common malignancy in AIDS patients. It is strongly associated with a low CD4 count, which accounts for the notable decline in its incidence after the widespread introduction of HAART.1 Among HIV patients, KS has the highest incidence in men who have sex with men. This population has a higher seroprevalence of HHV-8, which suggests possible sexual transmission of HHV-8. AIDS-associated KS most commonly involves the lower extremities, face, and oral mucosa. It may have visceral involvement, particularly of the gastrointestinal and respiratory systems, which carries a poor prognosis.4,5
Approximately 40% of patients presenting with KS have gastrointestinal tract involvement.6 Of these patients, up to 80% are asymptomatic, with diagnosis usually being made on endoscopy.7 In contrast, pulmonary KS is less common and typically is symptomatic. It can involve the lung parenchyma, airways, or pleura and is diagnosed by chest radiography or CT scans. Glucocorticoid therapy is a known trigger for pulmonary KS exacerbation.8
All 4 subtypes share the same histopathologic findings consisting of spindled endothelial cell proliferation, inflammation, and angiogenesis. Immunohistochemistry reveals tumor cells that are CD34 and CD31 positive but are factor VIII negative. Staining for HHV-8 antigen is used to confirm the diagnosis. The inflammatory infiltrate predominantly is lymphocytic with scattered plasma cells.9
The laboratory results and histopathologic findings clearly indicated a diagnosis of KS in our patient. Other entities in the clinical differential would have shown notably different histopathologic findings and laboratory results. Lichen planus pigmentosus displays a lichenoid infiltrate and pigment dropout on histology. Histologic findings of psoriasis include psoriasiform acanthosis, dilated vessels in the dermal papillae, thinning of suprapapillary plates, and neutrophilic microabscesses. Sarcoidosis would demonstrate naked granulomas on histopathology. Syphilis displays variable but often psoriasiform or lichenoid findings on histology, and a positive rapid plasma reagin also would be noted.
First-line treatment of AIDS-related KS is HAART. For patients with severe and rapidly progressive KS or with visceral involvement, cytotoxic chemotherapy with doxorubicin or taxanes often is required. Additional therapies include radiotherapy, topical alitretinoin, and cryotherapy.1,10
- Schneider JW, Dittmer DP. Diagnosis and treatment of Kaposi sarcoma. Am J Clin Dermatol. 2017;18:529-539.
- Schwartz RA, Micali G, Nasca MR, et al. Kaposi sarcoma: a continuing conundrum. J Am Acad Dermatol. 2008;59:179-206; quiz 207-208.
- Mohanna S, Maco V, Bravo F, et al. Epidemiology and clinical characteristics of classic Kaposi’s sarcoma, seroprevalence, and variants of human herpesvirus 8 in South America: a critical review of an old disease. Int J Infect Dis. 2005;9:239-250.
- Beral V, Peterman TA, Berkelman RL, et al. Kaposi’s sarcoma among persons with AIDS: a sexually transmitted infection? Lancet. 1990;335:123-128.
- Smith NA, Sabin CA, Gopal R, et al. Serologic evidence of human herpesvirus 8 transmission by homosexual but not heterosexual sex. J Infect Dis. 1999;180:600-606.
- Arora M, Goldberg EM. Kaposi sarcoma involving the gastrointestinal tract. Gastroenterol Hepatol (N Y). 2010;6:459-462.
- Parente F, Cernuschi M, Orlando G, et al. Kaposi’s sarcoma and AIDS: frequency of gastrointestinal involvement and its effect on survival. a prospective study in a heterogeneous population. Scand J Gastroenterol. 1991;26:1007-1012.
- Gasparetto TD, Marchiori E, Lourenco S, et al. Pulmonary involvement in Kaposi sarcoma: correlation between imaging and pathology. Orphanet J Rare Dis. 2009;4:18.
- Radu O, Pantanowitz L. Kaposi sarcoma. Arch Pathol Lab Med. 2013;137:289-294.
- Regnier-Rosencher E, Guillot B, Dupin N. Treatments for classic Kaposi sarcoma: a systematic review of the literature. J Am Acad Dermatol. 2013;68:313-331.
The Diagnosis: Kaposi Sarcoma
On initial presentation, the differential diagnosis included secondary syphilis, Kaposi sarcoma (KS), lichen planus pigmentosus, sarcoidosis, and psoriasis. A laboratory workup was ordered, which included complete blood cell count, comprehensive metabolic panel, antinuclear antibodies, anti-Ro/Sjögren syndrome antigen A and anti-La/Sjögren syndrome antigen B autoantibodies, angiotensin-converting enzyme, rapid plasma reagin, and human immunodeficiency virus (HIV) antibodies. A 4-mm punch biopsy of the rash also was performed from the right upper back. Histology revealed a vascular proliferation that was diffusely positive for human herpesvirus 8 (HHV-8)(Figure 1). The patient was informed of the diagnosis, at which time he revealed he had a history of homosexual relationships, with his last sexual contact being more than 1 year prior to presentation. The laboratory workup confirmed a diagnosis of HIV, and the remainder of the tests were unremarkable.
He was referred to our university's HIV clinic where he was started on highly active antiretroviral therapy (HAART). His facial swelling worsened, leading to hospital admission. Computed tomography (CT) of the chest, abdomen, and pelvis showed diffuse lymphadenopathy and lung nodules concerning for visceral involvement of KS. Hematology and oncology was consulted for further evaluation, and he was treated with 6 cycles of doxorubicin 20 mg/m2, which led to resolution of the lung nodules on CT and improvement of the rash burden. He was then started on alitretinoin gel 0.1% twice daily, which led to continued slow improvement (Figure 2).
Kaposi sarcoma is a vascular neoplasm that occurs from infection with HHV-8. It typically presents as painless, reddish to violaceous macules or patches involving the skin and mucosa that often progress to plaques or nodules with possible visceral involvement. Kaposi sarcoma is classified into 4 subtypes based on epidemiology and clinical presentation: classic, endemic, iatrogenic, and AIDS associated.1,2
Classic KS primarily affects elderly males of Mediterranean or Eastern European descent, with a mean age of 64.1 years and a male to female ratio of 3 to 1. It has an indolent course and a strong predilection for the skin of the lower extremities. The endemic form occurs mainly in Africa and has a more aggressive course, especially the lymphadenopathic type that affects children younger than 10 years.3 Iatrogenic KS develops in immunosuppressed patients, such as transplant recipients, and may regress if the immunosuppressive agent is stopped.1 Kaposi sarcoma is an AIDS-defining illness and is the most common malignancy in AIDS patients. It is strongly associated with a low CD4 count, which accounts for the notable decline in its incidence after the widespread introduction of HAART.1 Among HIV patients, KS has the highest incidence in men who have sex with men. This population has a higher seroprevalence of HHV-8, which suggests possible sexual transmission of HHV-8. AIDS-associated KS most commonly involves the lower extremities, face, and oral mucosa. It may have visceral involvement, particularly of the gastrointestinal and respiratory systems, which carries a poor prognosis.4,5
Approximately 40% of patients presenting with KS have gastrointestinal tract involvement.6 Of these patients, up to 80% are asymptomatic, with diagnosis usually being made on endoscopy.7 In contrast, pulmonary KS is less common and typically is symptomatic. It can involve the lung parenchyma, airways, or pleura and is diagnosed by chest radiography or CT scans. Glucocorticoid therapy is a known trigger for pulmonary KS exacerbation.8
All 4 subtypes share the same histopathologic findings consisting of spindled endothelial cell proliferation, inflammation, and angiogenesis. Immunohistochemistry reveals tumor cells that are CD34 and CD31 positive but are factor VIII negative. Staining for HHV-8 antigen is used to confirm the diagnosis. The inflammatory infiltrate predominantly is lymphocytic with scattered plasma cells.9
The laboratory results and histopathologic findings clearly indicated a diagnosis of KS in our patient. Other entities in the clinical differential would have shown notably different histopathologic findings and laboratory results. Lichen planus pigmentosus displays a lichenoid infiltrate and pigment dropout on histology. Histologic findings of psoriasis include psoriasiform acanthosis, dilated vessels in the dermal papillae, thinning of suprapapillary plates, and neutrophilic microabscesses. Sarcoidosis would demonstrate naked granulomas on histopathology. Syphilis displays variable but often psoriasiform or lichenoid findings on histology, and a positive rapid plasma reagin also would be noted.
First-line treatment of AIDS-related KS is HAART. For patients with severe and rapidly progressive KS or with visceral involvement, cytotoxic chemotherapy with doxorubicin or taxanes often is required. Additional therapies include radiotherapy, topical alitretinoin, and cryotherapy.1,10
The Diagnosis: Kaposi Sarcoma
On initial presentation, the differential diagnosis included secondary syphilis, Kaposi sarcoma (KS), lichen planus pigmentosus, sarcoidosis, and psoriasis. A laboratory workup was ordered, which included complete blood cell count, comprehensive metabolic panel, antinuclear antibodies, anti-Ro/Sjögren syndrome antigen A and anti-La/Sjögren syndrome antigen B autoantibodies, angiotensin-converting enzyme, rapid plasma reagin, and human immunodeficiency virus (HIV) antibodies. A 4-mm punch biopsy of the rash also was performed from the right upper back. Histology revealed a vascular proliferation that was diffusely positive for human herpesvirus 8 (HHV-8)(Figure 1). The patient was informed of the diagnosis, at which time he revealed he had a history of homosexual relationships, with his last sexual contact being more than 1 year prior to presentation. The laboratory workup confirmed a diagnosis of HIV, and the remainder of the tests were unremarkable.
He was referred to our university's HIV clinic where he was started on highly active antiretroviral therapy (HAART). His facial swelling worsened, leading to hospital admission. Computed tomography (CT) of the chest, abdomen, and pelvis showed diffuse lymphadenopathy and lung nodules concerning for visceral involvement of KS. Hematology and oncology was consulted for further evaluation, and he was treated with 6 cycles of doxorubicin 20 mg/m2, which led to resolution of the lung nodules on CT and improvement of the rash burden. He was then started on alitretinoin gel 0.1% twice daily, which led to continued slow improvement (Figure 2).
Kaposi sarcoma is a vascular neoplasm that occurs from infection with HHV-8. It typically presents as painless, reddish to violaceous macules or patches involving the skin and mucosa that often progress to plaques or nodules with possible visceral involvement. Kaposi sarcoma is classified into 4 subtypes based on epidemiology and clinical presentation: classic, endemic, iatrogenic, and AIDS associated.1,2
Classic KS primarily affects elderly males of Mediterranean or Eastern European descent, with a mean age of 64.1 years and a male to female ratio of 3 to 1. It has an indolent course and a strong predilection for the skin of the lower extremities. The endemic form occurs mainly in Africa and has a more aggressive course, especially the lymphadenopathic type that affects children younger than 10 years.3 Iatrogenic KS develops in immunosuppressed patients, such as transplant recipients, and may regress if the immunosuppressive agent is stopped.1 Kaposi sarcoma is an AIDS-defining illness and is the most common malignancy in AIDS patients. It is strongly associated with a low CD4 count, which accounts for the notable decline in its incidence after the widespread introduction of HAART.1 Among HIV patients, KS has the highest incidence in men who have sex with men. This population has a higher seroprevalence of HHV-8, which suggests possible sexual transmission of HHV-8. AIDS-associated KS most commonly involves the lower extremities, face, and oral mucosa. It may have visceral involvement, particularly of the gastrointestinal and respiratory systems, which carries a poor prognosis.4,5
Approximately 40% of patients presenting with KS have gastrointestinal tract involvement.6 Of these patients, up to 80% are asymptomatic, with diagnosis usually being made on endoscopy.7 In contrast, pulmonary KS is less common and typically is symptomatic. It can involve the lung parenchyma, airways, or pleura and is diagnosed by chest radiography or CT scans. Glucocorticoid therapy is a known trigger for pulmonary KS exacerbation.8
All 4 subtypes share the same histopathologic findings consisting of spindled endothelial cell proliferation, inflammation, and angiogenesis. Immunohistochemistry reveals tumor cells that are CD34 and CD31 positive but are factor VIII negative. Staining for HHV-8 antigen is used to confirm the diagnosis. The inflammatory infiltrate predominantly is lymphocytic with scattered plasma cells.9
The laboratory results and histopathologic findings clearly indicated a diagnosis of KS in our patient. Other entities in the clinical differential would have shown notably different histopathologic findings and laboratory results. Lichen planus pigmentosus displays a lichenoid infiltrate and pigment dropout on histology. Histologic findings of psoriasis include psoriasiform acanthosis, dilated vessels in the dermal papillae, thinning of suprapapillary plates, and neutrophilic microabscesses. Sarcoidosis would demonstrate naked granulomas on histopathology. Syphilis displays variable but often psoriasiform or lichenoid findings on histology, and a positive rapid plasma reagin also would be noted.
First-line treatment of AIDS-related KS is HAART. For patients with severe and rapidly progressive KS or with visceral involvement, cytotoxic chemotherapy with doxorubicin or taxanes often is required. Additional therapies include radiotherapy, topical alitretinoin, and cryotherapy.1,10
- Schneider JW, Dittmer DP. Diagnosis and treatment of Kaposi sarcoma. Am J Clin Dermatol. 2017;18:529-539.
- Schwartz RA, Micali G, Nasca MR, et al. Kaposi sarcoma: a continuing conundrum. J Am Acad Dermatol. 2008;59:179-206; quiz 207-208.
- Mohanna S, Maco V, Bravo F, et al. Epidemiology and clinical characteristics of classic Kaposi’s sarcoma, seroprevalence, and variants of human herpesvirus 8 in South America: a critical review of an old disease. Int J Infect Dis. 2005;9:239-250.
- Beral V, Peterman TA, Berkelman RL, et al. Kaposi’s sarcoma among persons with AIDS: a sexually transmitted infection? Lancet. 1990;335:123-128.
- Smith NA, Sabin CA, Gopal R, et al. Serologic evidence of human herpesvirus 8 transmission by homosexual but not heterosexual sex. J Infect Dis. 1999;180:600-606.
- Arora M, Goldberg EM. Kaposi sarcoma involving the gastrointestinal tract. Gastroenterol Hepatol (N Y). 2010;6:459-462.
- Parente F, Cernuschi M, Orlando G, et al. Kaposi’s sarcoma and AIDS: frequency of gastrointestinal involvement and its effect on survival. a prospective study in a heterogeneous population. Scand J Gastroenterol. 1991;26:1007-1012.
- Gasparetto TD, Marchiori E, Lourenco S, et al. Pulmonary involvement in Kaposi sarcoma: correlation between imaging and pathology. Orphanet J Rare Dis. 2009;4:18.
- Radu O, Pantanowitz L. Kaposi sarcoma. Arch Pathol Lab Med. 2013;137:289-294.
- Regnier-Rosencher E, Guillot B, Dupin N. Treatments for classic Kaposi sarcoma: a systematic review of the literature. J Am Acad Dermatol. 2013;68:313-331.
- Schneider JW, Dittmer DP. Diagnosis and treatment of Kaposi sarcoma. Am J Clin Dermatol. 2017;18:529-539.
- Schwartz RA, Micali G, Nasca MR, et al. Kaposi sarcoma: a continuing conundrum. J Am Acad Dermatol. 2008;59:179-206; quiz 207-208.
- Mohanna S, Maco V, Bravo F, et al. Epidemiology and clinical characteristics of classic Kaposi’s sarcoma, seroprevalence, and variants of human herpesvirus 8 in South America: a critical review of an old disease. Int J Infect Dis. 2005;9:239-250.
- Beral V, Peterman TA, Berkelman RL, et al. Kaposi’s sarcoma among persons with AIDS: a sexually transmitted infection? Lancet. 1990;335:123-128.
- Smith NA, Sabin CA, Gopal R, et al. Serologic evidence of human herpesvirus 8 transmission by homosexual but not heterosexual sex. J Infect Dis. 1999;180:600-606.
- Arora M, Goldberg EM. Kaposi sarcoma involving the gastrointestinal tract. Gastroenterol Hepatol (N Y). 2010;6:459-462.
- Parente F, Cernuschi M, Orlando G, et al. Kaposi’s sarcoma and AIDS: frequency of gastrointestinal involvement and its effect on survival. a prospective study in a heterogeneous population. Scand J Gastroenterol. 1991;26:1007-1012.
- Gasparetto TD, Marchiori E, Lourenco S, et al. Pulmonary involvement in Kaposi sarcoma: correlation between imaging and pathology. Orphanet J Rare Dis. 2009;4:18.
- Radu O, Pantanowitz L. Kaposi sarcoma. Arch Pathol Lab Med. 2013;137:289-294.
- Regnier-Rosencher E, Guillot B, Dupin N. Treatments for classic Kaposi sarcoma: a systematic review of the literature. J Am Acad Dermatol. 2013;68:313-331.
A 24-year-old Black man presented for evaluation of an asymptomatic rash on the face, chest, back, and arms that had been progressively spreading over the course of 3 months. He had some swelling of the lips prior to the onset of the rash and was prescribed prednisone 10 mg daily by an outside physician. He had no known medical problems and was taking no medications. Physical examination revealed numerous violaceous plaques scattered symmetrically on the trunk, arms, legs, and face. His family history was negative for autoimmune disease, and a review of systems was unremarkable. He denied any recent sexual contacts.
Umbilicated Keratotic Papule on the Scalp
The Diagnosis: Warty Dyskeratoma
Warty dyskeratoma (WD) is a benign cutaneous tumor that was first described in 1954 as isolated Darier disease (DD). In 1957, Szymanski1 renamed it warty dyskeratoma as a distinct condition from DD. Warty dyskeratoma typically presents as a flesh-colored to brownish, round, well-demarcated, and slightly elevated papule or nodule accompanied by an umbilical invagination at the center. It most commonly arises on the scalp, face, or neck.2 In contrast to DD, familial occurrence is uncommon. It usually is difficult to distinguish WD from other conditions such as seborrheic keratosis, verruca vulgaris, or keratoacanthoma due to its macroscopic features. Therefore, histopathologic investigation is necessary for a precise diagnosis.
In our case, histologic investigation revealed a symmetric cup-shaped invagination filled with acantholytic and dyskeratotic keratinocytes with no atypia or mitotic figures (Figure, A). The bottom of the invagination was occupied with numerous villi covered by a single layer of basal cells (Figure, B). At the edge of the invagination, corps ronds and grains were observed in the granular and cornified layers, respectively (Figure, C).
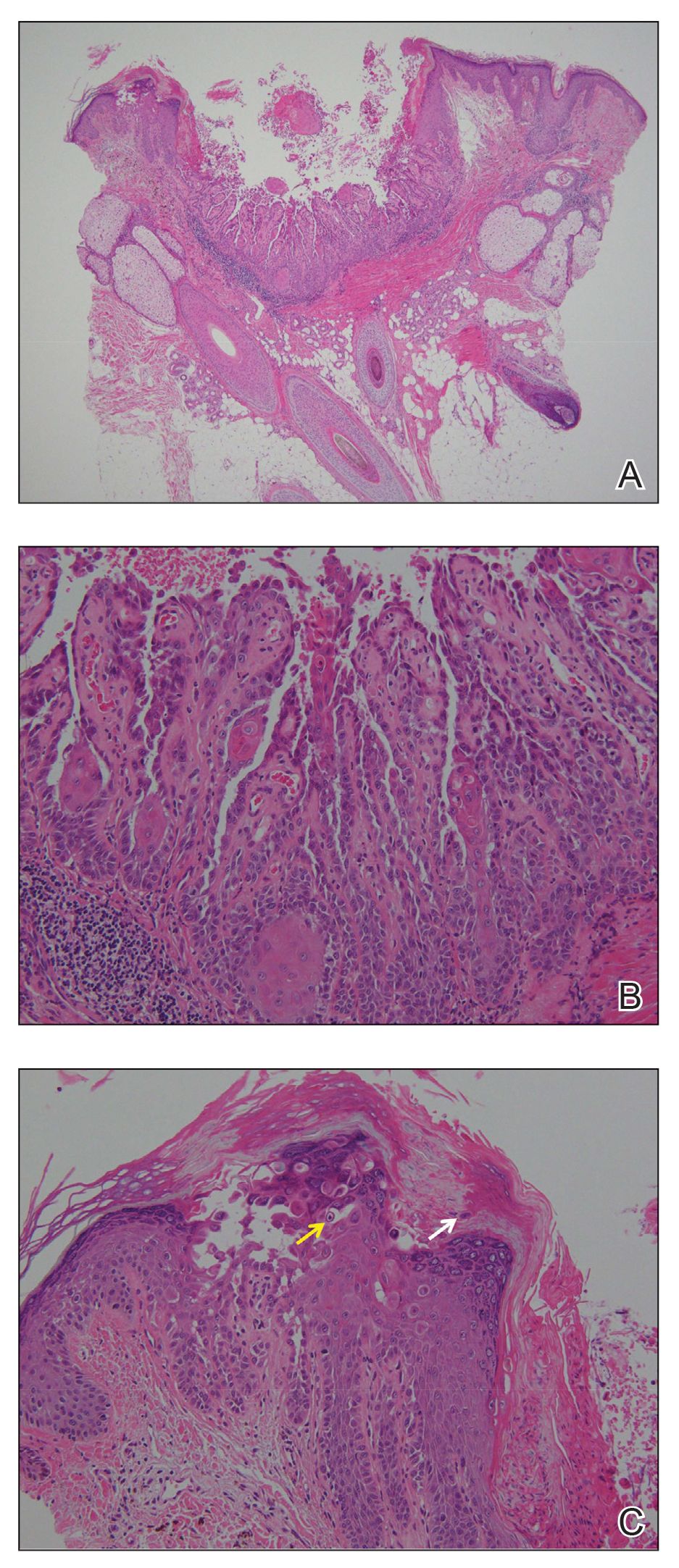
The hallmark histopathologic findings are acantholysis and dyskeratosis just above the basal cell layer, called focal acantholytic dyskeratosis. The differential diagnosis includes other disorders associated with focal acantholytic dyskeratosis, such as DD and acantholytic squamous cell carcinoma.3 Distinguishing WD from DD may be difficult in rare cases with multiple lesions.4 In such cases, an autosomal-dominant inheritance pattern and younger age of onset should prompt clinicians to seek for mutations in the ATPase sarcoplasmic/endoplasmic reticulum Ca2+ transporting 2 gene, ATP2A2, for the diagnosis of DD.5 Additionally, the presence of atypia or mitotic figures will rule out malignant disorders such as squamous cell carcinoma.
Although the pathogenesis of WD is not fully understood, most clinicians consider it a follicular adnexal neoplasm because the lesions often are connected to the pilosebaceous unit on microscopic observation.6 Although WD-like lesions arising from the oral mucosa have been reported,7 their etiology may be different from WD because the oral mucosa lacks hair follicles.8 The term warty leads to speculation of the contribution of human papillomavirus to the pathogenesis of WD, but this has been questioned due to the negative result of viral DNA detection from WD lesions by polymerase chain reaction analysis.2 Therefore, the term follicular dyskeratoma has been suggested as a novel denomination that reflects its etiology more precisely.2
The efficacy of topical treatment has not yet been established. Cryosurgery is another therapeutic option, but it sometimes fails.9 As performed in our patient, excisional biopsy is the most reasonable treatment option to obtain both complete removal and precise diagnosis.
The Diagnosis: Warty Dyskeratoma
Warty dyskeratoma (WD) is a benign cutaneous tumor that was first described in 1954 as isolated Darier disease (DD). In 1957, Szymanski1 renamed it warty dyskeratoma as a distinct condition from DD. Warty dyskeratoma typically presents as a flesh-colored to brownish, round, well-demarcated, and slightly elevated papule or nodule accompanied by an umbilical invagination at the center. It most commonly arises on the scalp, face, or neck.2 In contrast to DD, familial occurrence is uncommon. It usually is difficult to distinguish WD from other conditions such as seborrheic keratosis, verruca vulgaris, or keratoacanthoma due to its macroscopic features. Therefore, histopathologic investigation is necessary for a precise diagnosis.
In our case, histologic investigation revealed a symmetric cup-shaped invagination filled with acantholytic and dyskeratotic keratinocytes with no atypia or mitotic figures (Figure, A). The bottom of the invagination was occupied with numerous villi covered by a single layer of basal cells (Figure, B). At the edge of the invagination, corps ronds and grains were observed in the granular and cornified layers, respectively (Figure, C).
The hallmark histopathologic findings are acantholysis and dyskeratosis just above the basal cell layer, called focal acantholytic dyskeratosis. The differential diagnosis includes other disorders associated with focal acantholytic dyskeratosis, such as DD and acantholytic squamous cell carcinoma.3 Distinguishing WD from DD may be difficult in rare cases with multiple lesions.4 In such cases, an autosomal-dominant inheritance pattern and younger age of onset should prompt clinicians to seek for mutations in the ATPase sarcoplasmic/endoplasmic reticulum Ca2+ transporting 2 gene, ATP2A2, for the diagnosis of DD.5 Additionally, the presence of atypia or mitotic figures will rule out malignant disorders such as squamous cell carcinoma.
Although the pathogenesis of WD is not fully understood, most clinicians consider it a follicular adnexal neoplasm because the lesions often are connected to the pilosebaceous unit on microscopic observation.6 Although WD-like lesions arising from the oral mucosa have been reported,7 their etiology may be different from WD because the oral mucosa lacks hair follicles.8 The term warty leads to speculation of the contribution of human papillomavirus to the pathogenesis of WD, but this has been questioned due to the negative result of viral DNA detection from WD lesions by polymerase chain reaction analysis.2 Therefore, the term follicular dyskeratoma has been suggested as a novel denomination that reflects its etiology more precisely.2
The efficacy of topical treatment has not yet been established. Cryosurgery is another therapeutic option, but it sometimes fails.9 As performed in our patient, excisional biopsy is the most reasonable treatment option to obtain both complete removal and precise diagnosis.
The Diagnosis: Warty Dyskeratoma
Warty dyskeratoma (WD) is a benign cutaneous tumor that was first described in 1954 as isolated Darier disease (DD). In 1957, Szymanski1 renamed it warty dyskeratoma as a distinct condition from DD. Warty dyskeratoma typically presents as a flesh-colored to brownish, round, well-demarcated, and slightly elevated papule or nodule accompanied by an umbilical invagination at the center. It most commonly arises on the scalp, face, or neck.2 In contrast to DD, familial occurrence is uncommon. It usually is difficult to distinguish WD from other conditions such as seborrheic keratosis, verruca vulgaris, or keratoacanthoma due to its macroscopic features. Therefore, histopathologic investigation is necessary for a precise diagnosis.
In our case, histologic investigation revealed a symmetric cup-shaped invagination filled with acantholytic and dyskeratotic keratinocytes with no atypia or mitotic figures (Figure, A). The bottom of the invagination was occupied with numerous villi covered by a single layer of basal cells (Figure, B). At the edge of the invagination, corps ronds and grains were observed in the granular and cornified layers, respectively (Figure, C).
The hallmark histopathologic findings are acantholysis and dyskeratosis just above the basal cell layer, called focal acantholytic dyskeratosis. The differential diagnosis includes other disorders associated with focal acantholytic dyskeratosis, such as DD and acantholytic squamous cell carcinoma.3 Distinguishing WD from DD may be difficult in rare cases with multiple lesions.4 In such cases, an autosomal-dominant inheritance pattern and younger age of onset should prompt clinicians to seek for mutations in the ATPase sarcoplasmic/endoplasmic reticulum Ca2+ transporting 2 gene, ATP2A2, for the diagnosis of DD.5 Additionally, the presence of atypia or mitotic figures will rule out malignant disorders such as squamous cell carcinoma.
Although the pathogenesis of WD is not fully understood, most clinicians consider it a follicular adnexal neoplasm because the lesions often are connected to the pilosebaceous unit on microscopic observation.6 Although WD-like lesions arising from the oral mucosa have been reported,7 their etiology may be different from WD because the oral mucosa lacks hair follicles.8 The term warty leads to speculation of the contribution of human papillomavirus to the pathogenesis of WD, but this has been questioned due to the negative result of viral DNA detection from WD lesions by polymerase chain reaction analysis.2 Therefore, the term follicular dyskeratoma has been suggested as a novel denomination that reflects its etiology more precisely.2
The efficacy of topical treatment has not yet been established. Cryosurgery is another therapeutic option, but it sometimes fails.9 As performed in our patient, excisional biopsy is the most reasonable treatment option to obtain both complete removal and precise diagnosis.
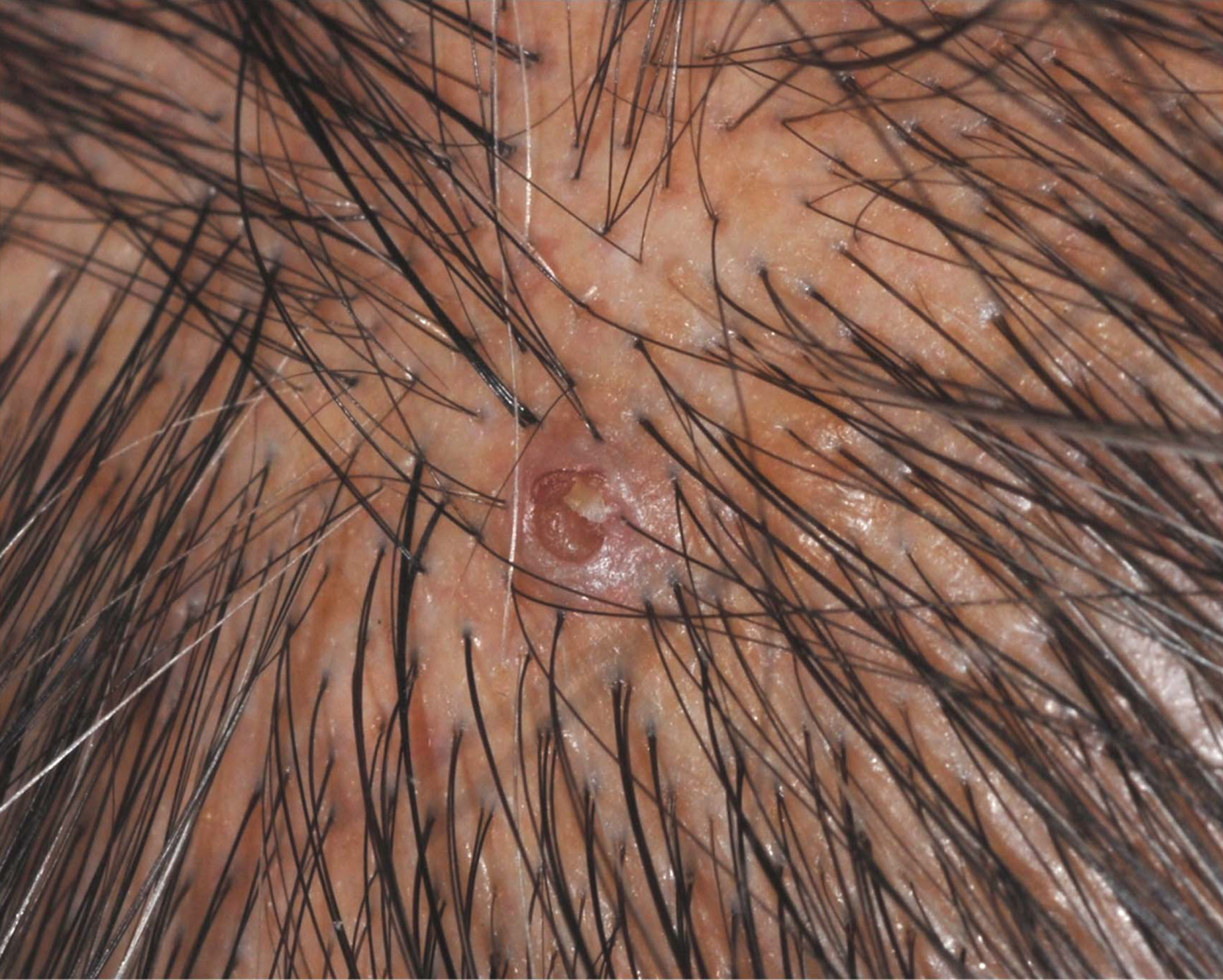
A 72-year-old man was referred to our dermatology clinic for evaluation of a solitary papule on the scalp measuring 3.2 mm in diameter with a keratotic umbilicated center of 1 year’s duration. His medical history included acute appendicitis. Treatment with fusidic acid ointment 2% was unsuccessful. The papule was hard without tenderness on palpation. An excisional biopsy was performed under local anesthesia.
Inverse Distribution of Pink Macules and Patches
Punch biopsies from the right axilla (Figure) and right abdomen as well as a tangential biopsy from the left volar wrist papule showed an interstitial histiocytic infiltrate with focal palisading of histiocytes around central regions with collagen alteration and increased mucin. Grocott-Gomori methenamine-silver stain and acid-fast bacilli smear both were negative for organisms; these findings were consistent with a diagnosis of granuloma annulare (GA).

Granuloma annulare is a noninfectious granulomatous disease of unknown etiology. It most commonly appears as asymptomatic, flesh-colored, pink or violaceous annular patches or thin plaques favoring the trunk and extremities. Granuloma annulare has many documented presentations including generalized, patch, subcutaneous, and perforating forms. It can present as macules, papules, nodules, patches, or plaques. Reported associations include diabetes mellitus, hyperlipidemia, solid organ tumors, systemic infection, and thyroid disease.1 Granuloma annulare can occur in any age group but is most common between the ages of 20 and 40 years.2
Diagnosis most often is made clinically and can be confirmed by histopathology. Histologic examination most commonly shows histiocytes within the dermis that palisade around a central area of mucin deposition between degenerating collagen fibers. The histiocytes of GA stain positive with vimentin, lysozyme, and CD68. The increased mucin stains with colloidal iron and Alcian blue. Multinucleated giant cells and perivascular lymphocytic infiltrate also are commonly seen.3
Cutaneous B-cell lymphoma has a wide range of presentations but usually occurs as hyperpigmented plaques and patches with dermal atrophy. Psoriasis can present in an inverse distribution but will show epidermal changes including scale. Sarcoidosis presents as multiple erythematous plaques and papules and also can be accompanied by erythema nodosum. Tinea corporis likely would have resolved with antifungal treatment.
Many different treatments have been described as effective, including cryosurgery, topical and intralesional corticosteroids, antibiotics, immune modulators, phototherapy, and oral corticosteroids.1 We started our patient on triple-antibiotic therapy with rifampin 600 mg, minocycline 100 mg, and ofloxacin 400 mg all once monthly for 6 months, which has been shown to be efficacious in treating GA.4 The patient returned for follow-up 1 year after the initial presentation. At that time, she had faint pink patches on the waist and medial upper thighs, and the axillary lesions had cleared. In the interim, she developed more classic GA lesions—pink to violaceous smooth papules with no overlying epidermal changes—on the volar wrists and dorsal feet. These lesions were asymptomatic, and she currently is not undergoing any further treatment.
- Piette EW, Rosenbach M. Granuloma annulare: pathogenesis, disease associations and triggers, and therapeutic options. J Am Acad Dermatol. 2016;75:467-479.
- Piette EW, Rosenbach M. Granuloma annulare: clinical and histologic variants, epidemiology, and genetics. J Am Acad Dermatol. 2016;75:457-465.
- Patterson JW, Hosler GA. The granulomatous reaction pattern. Weedon’s Skin Pathology. 4th ed. China: Churchill Livingstone Elsevier; 2016:198-203.
- Marcus DV, Mahmoud BH, Hamzavi IH. Granuloma annulare treated with rifampin, ofloxacin, and minocycline combination therapy. Arch Dermatol. 2009;145:787-789.
Punch biopsies from the right axilla (Figure) and right abdomen as well as a tangential biopsy from the left volar wrist papule showed an interstitial histiocytic infiltrate with focal palisading of histiocytes around central regions with collagen alteration and increased mucin. Grocott-Gomori methenamine-silver stain and acid-fast bacilli smear both were negative for organisms; these findings were consistent with a diagnosis of granuloma annulare (GA).
Granuloma annulare is a noninfectious granulomatous disease of unknown etiology. It most commonly appears as asymptomatic, flesh-colored, pink or violaceous annular patches or thin plaques favoring the trunk and extremities. Granuloma annulare has many documented presentations including generalized, patch, subcutaneous, and perforating forms. It can present as macules, papules, nodules, patches, or plaques. Reported associations include diabetes mellitus, hyperlipidemia, solid organ tumors, systemic infection, and thyroid disease.1 Granuloma annulare can occur in any age group but is most common between the ages of 20 and 40 years.2
Diagnosis most often is made clinically and can be confirmed by histopathology. Histologic examination most commonly shows histiocytes within the dermis that palisade around a central area of mucin deposition between degenerating collagen fibers. The histiocytes of GA stain positive with vimentin, lysozyme, and CD68. The increased mucin stains with colloidal iron and Alcian blue. Multinucleated giant cells and perivascular lymphocytic infiltrate also are commonly seen.3
Cutaneous B-cell lymphoma has a wide range of presentations but usually occurs as hyperpigmented plaques and patches with dermal atrophy. Psoriasis can present in an inverse distribution but will show epidermal changes including scale. Sarcoidosis presents as multiple erythematous plaques and papules and also can be accompanied by erythema nodosum. Tinea corporis likely would have resolved with antifungal treatment.
Many different treatments have been described as effective, including cryosurgery, topical and intralesional corticosteroids, antibiotics, immune modulators, phototherapy, and oral corticosteroids.1 We started our patient on triple-antibiotic therapy with rifampin 600 mg, minocycline 100 mg, and ofloxacin 400 mg all once monthly for 6 months, which has been shown to be efficacious in treating GA.4 The patient returned for follow-up 1 year after the initial presentation. At that time, she had faint pink patches on the waist and medial upper thighs, and the axillary lesions had cleared. In the interim, she developed more classic GA lesions—pink to violaceous smooth papules with no overlying epidermal changes—on the volar wrists and dorsal feet. These lesions were asymptomatic, and she currently is not undergoing any further treatment.
Punch biopsies from the right axilla (Figure) and right abdomen as well as a tangential biopsy from the left volar wrist papule showed an interstitial histiocytic infiltrate with focal palisading of histiocytes around central regions with collagen alteration and increased mucin. Grocott-Gomori methenamine-silver stain and acid-fast bacilli smear both were negative for organisms; these findings were consistent with a diagnosis of granuloma annulare (GA).
Granuloma annulare is a noninfectious granulomatous disease of unknown etiology. It most commonly appears as asymptomatic, flesh-colored, pink or violaceous annular patches or thin plaques favoring the trunk and extremities. Granuloma annulare has many documented presentations including generalized, patch, subcutaneous, and perforating forms. It can present as macules, papules, nodules, patches, or plaques. Reported associations include diabetes mellitus, hyperlipidemia, solid organ tumors, systemic infection, and thyroid disease.1 Granuloma annulare can occur in any age group but is most common between the ages of 20 and 40 years.2
Diagnosis most often is made clinically and can be confirmed by histopathology. Histologic examination most commonly shows histiocytes within the dermis that palisade around a central area of mucin deposition between degenerating collagen fibers. The histiocytes of GA stain positive with vimentin, lysozyme, and CD68. The increased mucin stains with colloidal iron and Alcian blue. Multinucleated giant cells and perivascular lymphocytic infiltrate also are commonly seen.3
Cutaneous B-cell lymphoma has a wide range of presentations but usually occurs as hyperpigmented plaques and patches with dermal atrophy. Psoriasis can present in an inverse distribution but will show epidermal changes including scale. Sarcoidosis presents as multiple erythematous plaques and papules and also can be accompanied by erythema nodosum. Tinea corporis likely would have resolved with antifungal treatment.
Many different treatments have been described as effective, including cryosurgery, topical and intralesional corticosteroids, antibiotics, immune modulators, phototherapy, and oral corticosteroids.1 We started our patient on triple-antibiotic therapy with rifampin 600 mg, minocycline 100 mg, and ofloxacin 400 mg all once monthly for 6 months, which has been shown to be efficacious in treating GA.4 The patient returned for follow-up 1 year after the initial presentation. At that time, she had faint pink patches on the waist and medial upper thighs, and the axillary lesions had cleared. In the interim, she developed more classic GA lesions—pink to violaceous smooth papules with no overlying epidermal changes—on the volar wrists and dorsal feet. These lesions were asymptomatic, and she currently is not undergoing any further treatment.
- Piette EW, Rosenbach M. Granuloma annulare: pathogenesis, disease associations and triggers, and therapeutic options. J Am Acad Dermatol. 2016;75:467-479.
- Piette EW, Rosenbach M. Granuloma annulare: clinical and histologic variants, epidemiology, and genetics. J Am Acad Dermatol. 2016;75:457-465.
- Patterson JW, Hosler GA. The granulomatous reaction pattern. Weedon’s Skin Pathology. 4th ed. China: Churchill Livingstone Elsevier; 2016:198-203.
- Marcus DV, Mahmoud BH, Hamzavi IH. Granuloma annulare treated with rifampin, ofloxacin, and minocycline combination therapy. Arch Dermatol. 2009;145:787-789.
- Piette EW, Rosenbach M. Granuloma annulare: pathogenesis, disease associations and triggers, and therapeutic options. J Am Acad Dermatol. 2016;75:467-479.
- Piette EW, Rosenbach M. Granuloma annulare: clinical and histologic variants, epidemiology, and genetics. J Am Acad Dermatol. 2016;75:457-465.
- Patterson JW, Hosler GA. The granulomatous reaction pattern. Weedon’s Skin Pathology. 4th ed. China: Churchill Livingstone Elsevier; 2016:198-203.
- Marcus DV, Mahmoud BH, Hamzavi IH. Granuloma annulare treated with rifampin, ofloxacin, and minocycline combination therapy. Arch Dermatol. 2009;145:787-789.
A 73-year-old woman presented for evaluation of an asymptomatic progressive rash on the left wrist, waist, groin, and inner thighs of 2 months’ duration. Her primary care provider prescribed clotrimazole and fluconazole with no improvement. Review of systems was negative. Medications included omeprazole, candesartan hydrochlorothiazide, potassium chloride, and levothyroxine. Physical examination revealed many scattered, pink to violaceous macules and patches in the axillae (sparing the vaults) and inguinal folds as well as on the waist and medial upper thighs. The lesions were without scale or other epidermal change. She also had a pink papule on the left volar wrist. A Wood lamp examination was unremarkable, and punch biopsies were performed.
Umbilicated Neoplasm on the Chest
Dermoscopy showed polylobular, whitish yellow, amorphous structures at the center of the lesion surrounded by a crown of vessels (Figure 1). Histopathology revealed hyperplastic crateriform lesions containing large eosinophilic intracytoplasmic inclusion bodies within keratinocytes (Figure 2). At follow-up 2 weeks after the biopsy, the patient presented with approximately 20 more reddish papules of varying sizes on the abdomen and back that presented as dome-shaped papules and had a typical umbilicated center. The clinical manifestations, dermoscopy, and pathology findings were consistent with molluscum contagiosum (MC).
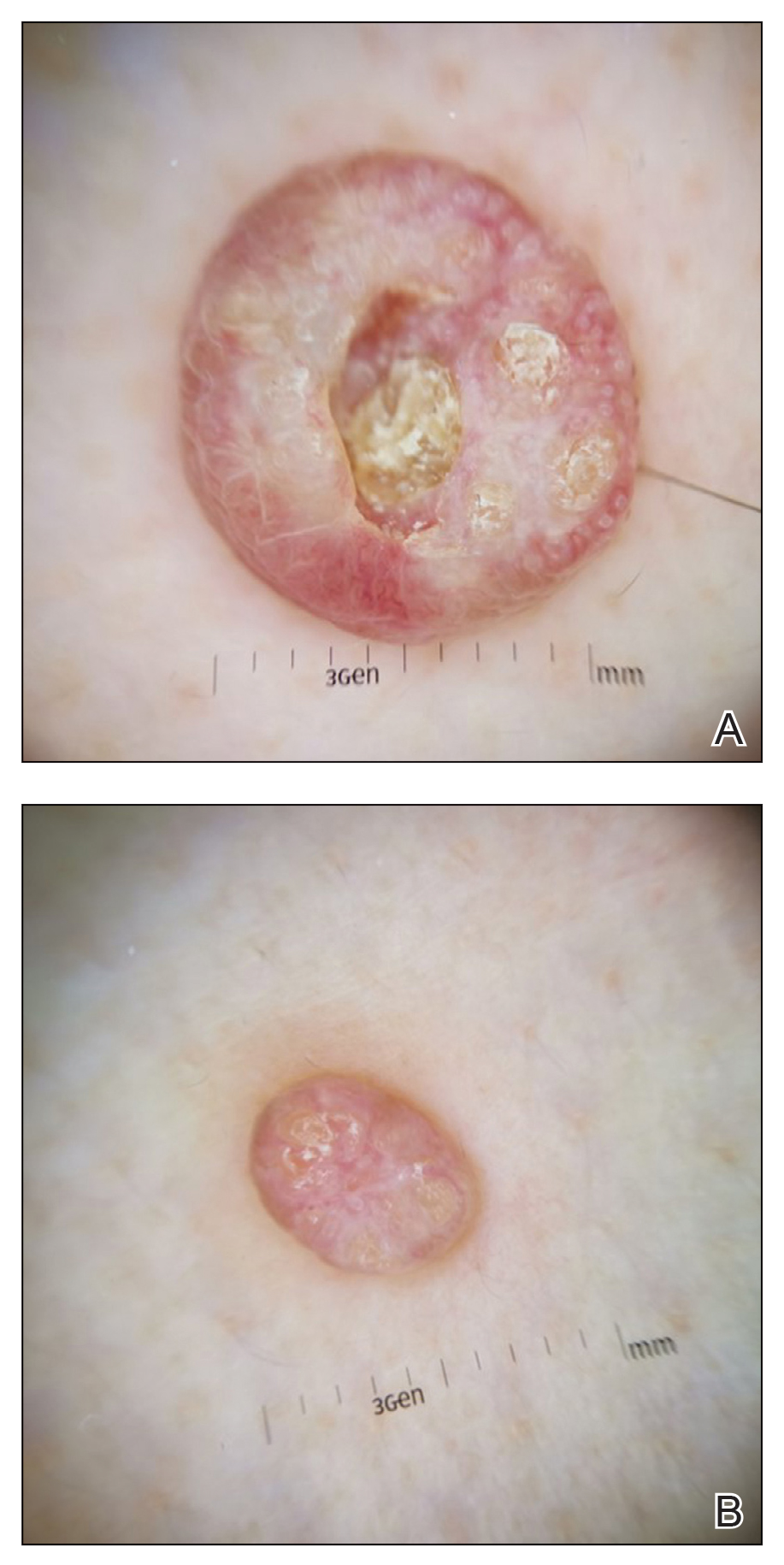
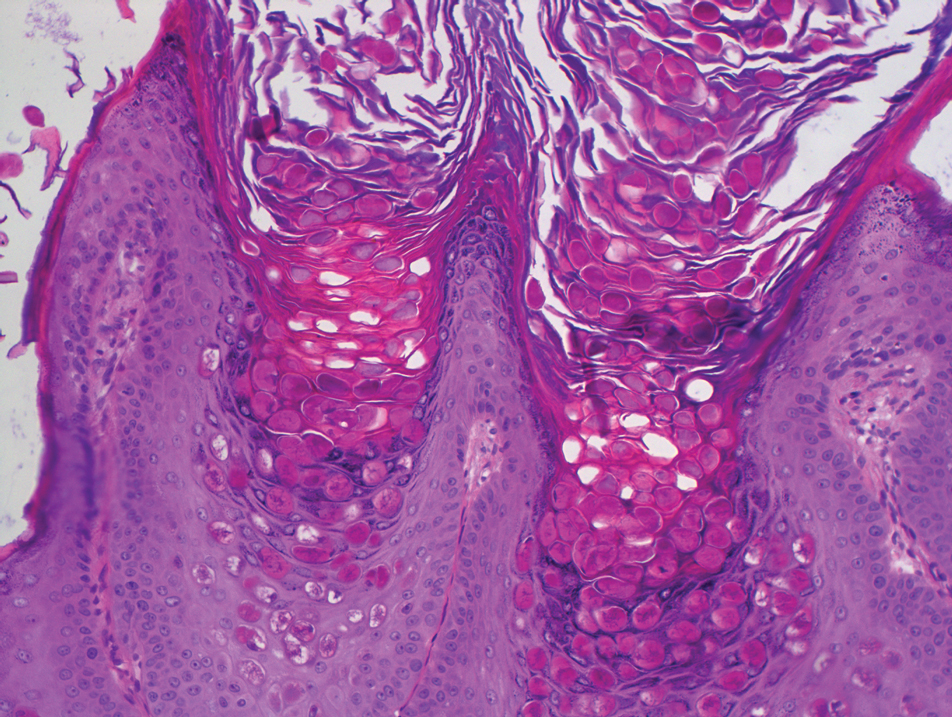
Molluscum contagiosum was first described in 1814. It is a benign cutaneous infectious disease caused by a double-stranded DNA virus of the poxvirus family. Molluscum contagiosum lesions usually manifest clinically as dome-shaped, flesh-colored or translucent, umbilicated papules measuring 1 to 5 mm in diameter that are commonly distributed over the face, trunk, and extremities and usually are self-limiting.1
Giant MC is rare and can be seen either in patients on immunosuppressive therapy or in those with diseases that can cause immunosuppression, such as human immunodeficiency virus, leukemia, atopic dermatitis, Wiskott-Aldrich syndrome, and sarcoidosis. In these instances, MC often is greater than 1 cm in diameter. Atypical variants may have an eczematous presentation or a lesion with secondary abscess formation and also can be spread widely over the body.2 Due to these atypical appearances and large dimensions in immunocompromised patients, other dermatologic diseases should be considered in the differential diagnosis, such as basal cell carcinoma, keratoacanthoma, squamous cell carcinoma, cutaneous horn, cutaneous cryptococcosis, histoplasmosis, and xanthomatosis.3
In our patient, the differential diagnosis included keratoacanthoma, which may present as a solitary, discrete, round to oval, flesh-colored, umbilicated nodule with a central keratin-filled crater and has a rapid clinical evolution, usually regressing within 4 to 6 months.
Squamous cell carcinoma may appear as scaly red patches, open sores, warts, or elevated growths with a central depression and may crust or bleed. Basal cell carcinoma typically may appear as a dome-shaped skin nodule with visible blood vessels or sometimes presents as a red patch similar to eczema. Xanthomatosis often appears as yellow to orange, mostly asymptomatic, supple patches or plaques, usually with sharp and distinctive edges.
Ancillary diagnostic modalities such as dermoscopy may be used to improve diagnostic accuracy. The best known capillaroscopic feature of MC is the peripheral crown of vessels in a radial distribution. A study of 258 MC lesions highlighted that crown and crown plus radial arrangements are the most common vascular structure patterns under dermoscopy. In addition, polylobular amorphous white structures in the center of the lesions tend to be a feature of larger MC papules.4 Histologically, MC shows lobulated crateriform lesions, thickening of the epidermis into the dermis, and the typical appearance of large eosinophilic intracytoplasmic inclusion bodies within keratinocytes.5
There are several treatment options available for MC. Common modalities include liquid nitrogen cryospray, curettage, and electrocauterization. In immunocompromised patients, MC lesions usually are resistant to ordinary therapy. The efficacy of topical agents such as imiquimod, which can induce high levels of IFN-α and other cytokines, has been demonstrated in these patients.6 Cidofovir, a nucleoside analog that has potent antiviral properties, also can be included as a therapeutic option.3 Our patient’s largest MC lesion was treated with surgical excision, the 2 large lesions on the left side of the chest with cryotherapy, and the other small lesions with curettage.
- Hanson D, Diven DG. Molluscum contagiosum. Dermatol Online J. 2003;9:2.
- Singh S, Swain M, Shukla S, et al. An unusual presentation of giant molluscum contagiosum diagnosed on cytology. Diagn Cytopathol. 2018;46:794-796.
- Mansur AT, Goktay F, Gunduz S, et al. Multiple giant molluscum contagiosum in a renal transplant recipient. Transpl Infect Dis. 2004;6:120-123.
- Ku SH, Cho EB, Park EJ, et al. Dermoscopic features of molluscum contagiosum based on white structures and their correlation with histopathological findings. Clin Exp Dermatol. 2015;40:208-210.
- Trčko K, Hošnjak L, Kušar B, et al. Clinical, histopathological, and virological evaluation of 203 patients with a clinical diagnosis of molluscum contagiosum [published online November 12, 2018]. Open Forum Infect Dis. 2018;5.
- Gardner LS, Ormond PJ. Treatment of multiple giant molluscum contagiosum in a renal transplant patient with imiquimod 5% cream. Clin Exp Dermatol. 2010;31:452-453.
Dermoscopy showed polylobular, whitish yellow, amorphous structures at the center of the lesion surrounded by a crown of vessels (Figure 1). Histopathology revealed hyperplastic crateriform lesions containing large eosinophilic intracytoplasmic inclusion bodies within keratinocytes (Figure 2). At follow-up 2 weeks after the biopsy, the patient presented with approximately 20 more reddish papules of varying sizes on the abdomen and back that presented as dome-shaped papules and had a typical umbilicated center. The clinical manifestations, dermoscopy, and pathology findings were consistent with molluscum contagiosum (MC).
Molluscum contagiosum was first described in 1814. It is a benign cutaneous infectious disease caused by a double-stranded DNA virus of the poxvirus family. Molluscum contagiosum lesions usually manifest clinically as dome-shaped, flesh-colored or translucent, umbilicated papules measuring 1 to 5 mm in diameter that are commonly distributed over the face, trunk, and extremities and usually are self-limiting.1
Giant MC is rare and can be seen either in patients on immunosuppressive therapy or in those with diseases that can cause immunosuppression, such as human immunodeficiency virus, leukemia, atopic dermatitis, Wiskott-Aldrich syndrome, and sarcoidosis. In these instances, MC often is greater than 1 cm in diameter. Atypical variants may have an eczematous presentation or a lesion with secondary abscess formation and also can be spread widely over the body.2 Due to these atypical appearances and large dimensions in immunocompromised patients, other dermatologic diseases should be considered in the differential diagnosis, such as basal cell carcinoma, keratoacanthoma, squamous cell carcinoma, cutaneous horn, cutaneous cryptococcosis, histoplasmosis, and xanthomatosis.3
In our patient, the differential diagnosis included keratoacanthoma, which may present as a solitary, discrete, round to oval, flesh-colored, umbilicated nodule with a central keratin-filled crater and has a rapid clinical evolution, usually regressing within 4 to 6 months.
Squamous cell carcinoma may appear as scaly red patches, open sores, warts, or elevated growths with a central depression and may crust or bleed. Basal cell carcinoma typically may appear as a dome-shaped skin nodule with visible blood vessels or sometimes presents as a red patch similar to eczema. Xanthomatosis often appears as yellow to orange, mostly asymptomatic, supple patches or plaques, usually with sharp and distinctive edges.
Ancillary diagnostic modalities such as dermoscopy may be used to improve diagnostic accuracy. The best known capillaroscopic feature of MC is the peripheral crown of vessels in a radial distribution. A study of 258 MC lesions highlighted that crown and crown plus radial arrangements are the most common vascular structure patterns under dermoscopy. In addition, polylobular amorphous white structures in the center of the lesions tend to be a feature of larger MC papules.4 Histologically, MC shows lobulated crateriform lesions, thickening of the epidermis into the dermis, and the typical appearance of large eosinophilic intracytoplasmic inclusion bodies within keratinocytes.5
There are several treatment options available for MC. Common modalities include liquid nitrogen cryospray, curettage, and electrocauterization. In immunocompromised patients, MC lesions usually are resistant to ordinary therapy. The efficacy of topical agents such as imiquimod, which can induce high levels of IFN-α and other cytokines, has been demonstrated in these patients.6 Cidofovir, a nucleoside analog that has potent antiviral properties, also can be included as a therapeutic option.3 Our patient’s largest MC lesion was treated with surgical excision, the 2 large lesions on the left side of the chest with cryotherapy, and the other small lesions with curettage.
Dermoscopy showed polylobular, whitish yellow, amorphous structures at the center of the lesion surrounded by a crown of vessels (Figure 1). Histopathology revealed hyperplastic crateriform lesions containing large eosinophilic intracytoplasmic inclusion bodies within keratinocytes (Figure 2). At follow-up 2 weeks after the biopsy, the patient presented with approximately 20 more reddish papules of varying sizes on the abdomen and back that presented as dome-shaped papules and had a typical umbilicated center. The clinical manifestations, dermoscopy, and pathology findings were consistent with molluscum contagiosum (MC).
Molluscum contagiosum was first described in 1814. It is a benign cutaneous infectious disease caused by a double-stranded DNA virus of the poxvirus family. Molluscum contagiosum lesions usually manifest clinically as dome-shaped, flesh-colored or translucent, umbilicated papules measuring 1 to 5 mm in diameter that are commonly distributed over the face, trunk, and extremities and usually are self-limiting.1
Giant MC is rare and can be seen either in patients on immunosuppressive therapy or in those with diseases that can cause immunosuppression, such as human immunodeficiency virus, leukemia, atopic dermatitis, Wiskott-Aldrich syndrome, and sarcoidosis. In these instances, MC often is greater than 1 cm in diameter. Atypical variants may have an eczematous presentation or a lesion with secondary abscess formation and also can be spread widely over the body.2 Due to these atypical appearances and large dimensions in immunocompromised patients, other dermatologic diseases should be considered in the differential diagnosis, such as basal cell carcinoma, keratoacanthoma, squamous cell carcinoma, cutaneous horn, cutaneous cryptococcosis, histoplasmosis, and xanthomatosis.3
In our patient, the differential diagnosis included keratoacanthoma, which may present as a solitary, discrete, round to oval, flesh-colored, umbilicated nodule with a central keratin-filled crater and has a rapid clinical evolution, usually regressing within 4 to 6 months.
Squamous cell carcinoma may appear as scaly red patches, open sores, warts, or elevated growths with a central depression and may crust or bleed. Basal cell carcinoma typically may appear as a dome-shaped skin nodule with visible blood vessels or sometimes presents as a red patch similar to eczema. Xanthomatosis often appears as yellow to orange, mostly asymptomatic, supple patches or plaques, usually with sharp and distinctive edges.
Ancillary diagnostic modalities such as dermoscopy may be used to improve diagnostic accuracy. The best known capillaroscopic feature of MC is the peripheral crown of vessels in a radial distribution. A study of 258 MC lesions highlighted that crown and crown plus radial arrangements are the most common vascular structure patterns under dermoscopy. In addition, polylobular amorphous white structures in the center of the lesions tend to be a feature of larger MC papules.4 Histologically, MC shows lobulated crateriform lesions, thickening of the epidermis into the dermis, and the typical appearance of large eosinophilic intracytoplasmic inclusion bodies within keratinocytes.5
There are several treatment options available for MC. Common modalities include liquid nitrogen cryospray, curettage, and electrocauterization. In immunocompromised patients, MC lesions usually are resistant to ordinary therapy. The efficacy of topical agents such as imiquimod, which can induce high levels of IFN-α and other cytokines, has been demonstrated in these patients.6 Cidofovir, a nucleoside analog that has potent antiviral properties, also can be included as a therapeutic option.3 Our patient’s largest MC lesion was treated with surgical excision, the 2 large lesions on the left side of the chest with cryotherapy, and the other small lesions with curettage.
- Hanson D, Diven DG. Molluscum contagiosum. Dermatol Online J. 2003;9:2.
- Singh S, Swain M, Shukla S, et al. An unusual presentation of giant molluscum contagiosum diagnosed on cytology. Diagn Cytopathol. 2018;46:794-796.
- Mansur AT, Goktay F, Gunduz S, et al. Multiple giant molluscum contagiosum in a renal transplant recipient. Transpl Infect Dis. 2004;6:120-123.
- Ku SH, Cho EB, Park EJ, et al. Dermoscopic features of molluscum contagiosum based on white structures and their correlation with histopathological findings. Clin Exp Dermatol. 2015;40:208-210.
- Trčko K, Hošnjak L, Kušar B, et al. Clinical, histopathological, and virological evaluation of 203 patients with a clinical diagnosis of molluscum contagiosum [published online November 12, 2018]. Open Forum Infect Dis. 2018;5.
- Gardner LS, Ormond PJ. Treatment of multiple giant molluscum contagiosum in a renal transplant patient with imiquimod 5% cream. Clin Exp Dermatol. 2010;31:452-453.
- Hanson D, Diven DG. Molluscum contagiosum. Dermatol Online J. 2003;9:2.
- Singh S, Swain M, Shukla S, et al. An unusual presentation of giant molluscum contagiosum diagnosed on cytology. Diagn Cytopathol. 2018;46:794-796.
- Mansur AT, Goktay F, Gunduz S, et al. Multiple giant molluscum contagiosum in a renal transplant recipient. Transpl Infect Dis. 2004;6:120-123.
- Ku SH, Cho EB, Park EJ, et al. Dermoscopic features of molluscum contagiosum based on white structures and their correlation with histopathological findings. Clin Exp Dermatol. 2015;40:208-210.
- Trčko K, Hošnjak L, Kušar B, et al. Clinical, histopathological, and virological evaluation of 203 patients with a clinical diagnosis of molluscum contagiosum [published online November 12, 2018]. Open Forum Infect Dis. 2018;5.
- Gardner LS, Ormond PJ. Treatment of multiple giant molluscum contagiosum in a renal transplant patient with imiquimod 5% cream. Clin Exp Dermatol. 2010;31:452-453.
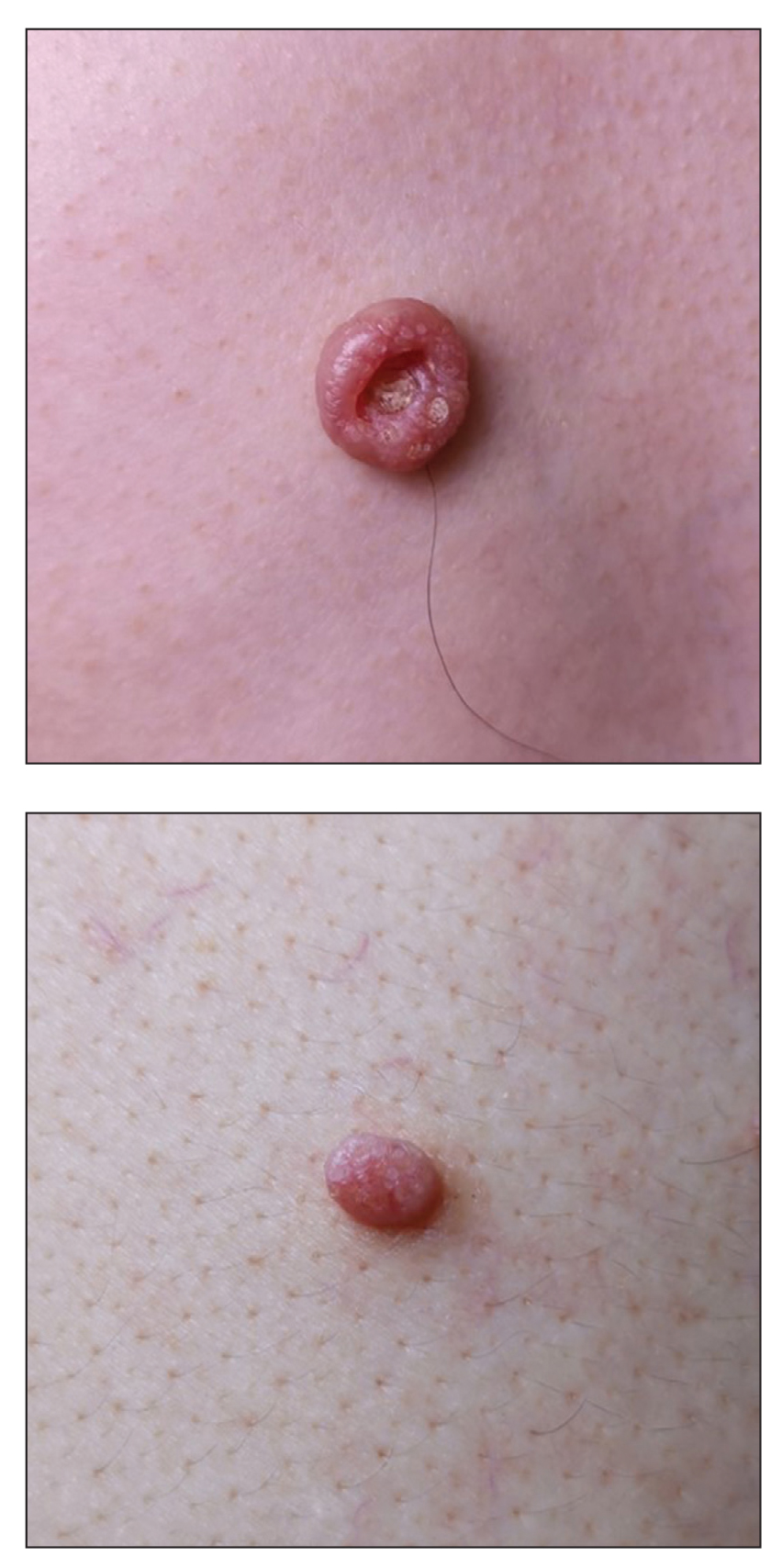
A 49-year-old man presented with a slow-growing mass on the chest of 1 year’s duration. The neoplasm started as a small papule that gradually increased in size. The patient denied pain, itching, bleeding, or discharge. He had a history of end-stage renal disease with a kidney transplant 8 years prior. His medication history included long-term use of oral tacrolimus, mycophenolate mofetil, and prednisone. Physical examination revealed a yellowish red, exogenous, pedunculated neoplasm on the right side of the chest measuring 1 cm in diameter with an umbilicated center and keratotic material (top). There were 2 more yellowish red papules on the left side of the chest measuring 0.5 cm in diameter without an umbilicated center (bottom). Dermoscopy and a biopsy were performed.