User login
Red lesions on face

These small (1-3 mm) grouped and solitary erythematous papules distributed around the mouth and nares were classic presentations of perioral dermatitis. Although perioral dermatitis typically affects the skin around the mouth, a newer term—periorificial dermatitis—is used because the eruption can, as seen in this patient, involve the skin around the mouth, nares, and/or eyes. Pustules also may occur. There also is a granulomatous form of periorificial dermatitis that occurs in children.
Periorificial dermatitis more closely resembles a rosacea-like eruption than a true dermatitis. Patients often report that the affected areas burn or sting, although occasionally they may be pruritic. Like rosacea, the pathogenesis of periorificial dermatitis is not completely understood. A major risk factor for the development of this condition is the use of topical corticosteroids—especially high-potency products—on the face. Therefore, the most important step in treating periorificial dermatitis is the discontinuation of topical corticosteroids (if they were being used).
In adults, oral tetracycline antibiotics are the drug of choice. As in rosacea, oral antibiotics are used not for their antimicrobial effect, but for their anti-inflammatory effect. For this reason, subantimicrobial dosing of doxycycline has become increasingly common. This reduces the likelihood of antibiotic-related adverse effects and bacterial resistance. In children or adults with a contraindication to tetracyclines, erythromycin is often used. Topical alternatives include erythromycin, metronidazole, and pimecrolimus.
In this case, the patient was advised to discontinue the topical corticosteroid and was started on subantimicrobial dosing of delayed-release doxycycline 40 mg. If the delayed-release form is not available, or is prohibitively expensive, doxycycline 20 mg bid may be used. This patient was told that it could take several weeks for the condition to improve, and that tapering the medication might help reduce recurrence, which is common.
Image courtesy of Daniel Stulberg, MD, FAAFP. Text courtesy of D. Alexander Phillips, MD, and Daniel Stulberg, MD, FAAFP Department of Family and Community Medicine, University of New Mexico School of Medicine, Albuquerque.
Wollina U. Subantimicrobial-dose doxycycline monohydrate in dermatology. Wien Med Wochenschr. 2015;165:499-503.
These small (1-3 mm) grouped and solitary erythematous papules distributed around the mouth and nares were classic presentations of perioral dermatitis. Although perioral dermatitis typically affects the skin around the mouth, a newer term—periorificial dermatitis—is used because the eruption can, as seen in this patient, involve the skin around the mouth, nares, and/or eyes. Pustules also may occur. There also is a granulomatous form of periorificial dermatitis that occurs in children.
Periorificial dermatitis more closely resembles a rosacea-like eruption than a true dermatitis. Patients often report that the affected areas burn or sting, although occasionally they may be pruritic. Like rosacea, the pathogenesis of periorificial dermatitis is not completely understood. A major risk factor for the development of this condition is the use of topical corticosteroids—especially high-potency products—on the face. Therefore, the most important step in treating periorificial dermatitis is the discontinuation of topical corticosteroids (if they were being used).
In adults, oral tetracycline antibiotics are the drug of choice. As in rosacea, oral antibiotics are used not for their antimicrobial effect, but for their anti-inflammatory effect. For this reason, subantimicrobial dosing of doxycycline has become increasingly common. This reduces the likelihood of antibiotic-related adverse effects and bacterial resistance. In children or adults with a contraindication to tetracyclines, erythromycin is often used. Topical alternatives include erythromycin, metronidazole, and pimecrolimus.
In this case, the patient was advised to discontinue the topical corticosteroid and was started on subantimicrobial dosing of delayed-release doxycycline 40 mg. If the delayed-release form is not available, or is prohibitively expensive, doxycycline 20 mg bid may be used. This patient was told that it could take several weeks for the condition to improve, and that tapering the medication might help reduce recurrence, which is common.
Image courtesy of Daniel Stulberg, MD, FAAFP. Text courtesy of D. Alexander Phillips, MD, and Daniel Stulberg, MD, FAAFP Department of Family and Community Medicine, University of New Mexico School of Medicine, Albuquerque.
These small (1-3 mm) grouped and solitary erythematous papules distributed around the mouth and nares were classic presentations of perioral dermatitis. Although perioral dermatitis typically affects the skin around the mouth, a newer term—periorificial dermatitis—is used because the eruption can, as seen in this patient, involve the skin around the mouth, nares, and/or eyes. Pustules also may occur. There also is a granulomatous form of periorificial dermatitis that occurs in children.
Periorificial dermatitis more closely resembles a rosacea-like eruption than a true dermatitis. Patients often report that the affected areas burn or sting, although occasionally they may be pruritic. Like rosacea, the pathogenesis of periorificial dermatitis is not completely understood. A major risk factor for the development of this condition is the use of topical corticosteroids—especially high-potency products—on the face. Therefore, the most important step in treating periorificial dermatitis is the discontinuation of topical corticosteroids (if they were being used).
In adults, oral tetracycline antibiotics are the drug of choice. As in rosacea, oral antibiotics are used not for their antimicrobial effect, but for their anti-inflammatory effect. For this reason, subantimicrobial dosing of doxycycline has become increasingly common. This reduces the likelihood of antibiotic-related adverse effects and bacterial resistance. In children or adults with a contraindication to tetracyclines, erythromycin is often used. Topical alternatives include erythromycin, metronidazole, and pimecrolimus.
In this case, the patient was advised to discontinue the topical corticosteroid and was started on subantimicrobial dosing of delayed-release doxycycline 40 mg. If the delayed-release form is not available, or is prohibitively expensive, doxycycline 20 mg bid may be used. This patient was told that it could take several weeks for the condition to improve, and that tapering the medication might help reduce recurrence, which is common.
Image courtesy of Daniel Stulberg, MD, FAAFP. Text courtesy of D. Alexander Phillips, MD, and Daniel Stulberg, MD, FAAFP Department of Family and Community Medicine, University of New Mexico School of Medicine, Albuquerque.
Wollina U. Subantimicrobial-dose doxycycline monohydrate in dermatology. Wien Med Wochenschr. 2015;165:499-503.
Wollina U. Subantimicrobial-dose doxycycline monohydrate in dermatology. Wien Med Wochenschr. 2015;165:499-503.

Pruritic rash on trunk

The patient underwent a skin biopsy, which was consistent with mycosis fungoides (MF) a form of cutaneous T cell lymphoma. Common MF complaints are a persistent pruritic rash that slowly progresses in size and shape. Nodules, papules, alopecia, and excoriations are often seen and can become secondarily infected. Sun spared areas are most affected. Itching and erythroderma can be quite intense, especially in Sezary Syndrome (SS)—a rare subtype of cutaneous T cell lymphoma that has a worse prognosis than localized MF.
It is a common for many patients with MF to go undiagnosed or incorrectly diagnosed for years. The differential on initial presentation can include eczema, dermatitis, psoriasis, or a drug reaction. Clues to this patient’s diagnosis were that the rash involved sun-spared areas and didn’t improve with a change to her oral medications or a course of topical steroids. Other clues that pointed to the diagnosis were that she had no history of prior scaling skin disease or psoriasis, her age (usual age of onset for MF is between 50 and 60 years), and the observation that the rash, although it looked like eczema or tinea, presented in atypical and multiple locations.
MF and SS are the most common cutaneous T cell lymphomas. Although these disorders initially involve the skin, later stages can spread to internal organs. Early MF can be difficult to diagnose on histology and can require multiple biopsies. The most accurate biopsy practice involves stopping topical medications for 4 weeks, then taking multiple biopsies of clinically different areas to confirm the diagnosis and clonality (the same cell line in more than one location). SS typically presents with a larger area of involvement and may be associated with lymphadenopathy.
The patient was seen by Dermatology. Due to the extent of the disease, which was suggestive of SS, she underwent peripheral blood flow cytometry, which revealed > 1000 Sezary cells/mcL. This confirmed the diagnosis of SS. The patient was started on photophoresis, interferon, high-potency topical steroids for the local symptoms, and bexarotene, which blocks abnormal cell growth by binding to retinoid receptors.
Photos and text courtesy of John Durkin, MD, Pigmented Lesions Clinic, University of New Mexico, and Kirill Balatsky, MS II, University of New Mexico School of Medicine.
Submitted for publication by Dr. Daniel Stulberg, MD, Department of Family and Community Medicine, University of New Mexico School of Medicine, Albuquerque.
Hoppe RT, Kim YH. Clinical manifestations, pathologic features, and diagnosis of mycosis fungoides. UpToDate Web site. https://www.uptodate.com/contents/clinical-manifestations-pathologic-features-and-diagnosis-of-mycosis-fungoides. Updated February 27, 2019. Accessed January 29, 2020.
The patient underwent a skin biopsy, which was consistent with mycosis fungoides (MF) a form of cutaneous T cell lymphoma. Common MF complaints are a persistent pruritic rash that slowly progresses in size and shape. Nodules, papules, alopecia, and excoriations are often seen and can become secondarily infected. Sun spared areas are most affected. Itching and erythroderma can be quite intense, especially in Sezary Syndrome (SS)—a rare subtype of cutaneous T cell lymphoma that has a worse prognosis than localized MF.
It is a common for many patients with MF to go undiagnosed or incorrectly diagnosed for years. The differential on initial presentation can include eczema, dermatitis, psoriasis, or a drug reaction. Clues to this patient’s diagnosis were that the rash involved sun-spared areas and didn’t improve with a change to her oral medications or a course of topical steroids. Other clues that pointed to the diagnosis were that she had no history of prior scaling skin disease or psoriasis, her age (usual age of onset for MF is between 50 and 60 years), and the observation that the rash, although it looked like eczema or tinea, presented in atypical and multiple locations.
MF and SS are the most common cutaneous T cell lymphomas. Although these disorders initially involve the skin, later stages can spread to internal organs. Early MF can be difficult to diagnose on histology and can require multiple biopsies. The most accurate biopsy practice involves stopping topical medications for 4 weeks, then taking multiple biopsies of clinically different areas to confirm the diagnosis and clonality (the same cell line in more than one location). SS typically presents with a larger area of involvement and may be associated with lymphadenopathy.
The patient was seen by Dermatology. Due to the extent of the disease, which was suggestive of SS, she underwent peripheral blood flow cytometry, which revealed > 1000 Sezary cells/mcL. This confirmed the diagnosis of SS. The patient was started on photophoresis, interferon, high-potency topical steroids for the local symptoms, and bexarotene, which blocks abnormal cell growth by binding to retinoid receptors.
Photos and text courtesy of John Durkin, MD, Pigmented Lesions Clinic, University of New Mexico, and Kirill Balatsky, MS II, University of New Mexico School of Medicine.
Submitted for publication by Dr. Daniel Stulberg, MD, Department of Family and Community Medicine, University of New Mexico School of Medicine, Albuquerque.
The patient underwent a skin biopsy, which was consistent with mycosis fungoides (MF) a form of cutaneous T cell lymphoma. Common MF complaints are a persistent pruritic rash that slowly progresses in size and shape. Nodules, papules, alopecia, and excoriations are often seen and can become secondarily infected. Sun spared areas are most affected. Itching and erythroderma can be quite intense, especially in Sezary Syndrome (SS)—a rare subtype of cutaneous T cell lymphoma that has a worse prognosis than localized MF.
It is a common for many patients with MF to go undiagnosed or incorrectly diagnosed for years. The differential on initial presentation can include eczema, dermatitis, psoriasis, or a drug reaction. Clues to this patient’s diagnosis were that the rash involved sun-spared areas and didn’t improve with a change to her oral medications or a course of topical steroids. Other clues that pointed to the diagnosis were that she had no history of prior scaling skin disease or psoriasis, her age (usual age of onset for MF is between 50 and 60 years), and the observation that the rash, although it looked like eczema or tinea, presented in atypical and multiple locations.
MF and SS are the most common cutaneous T cell lymphomas. Although these disorders initially involve the skin, later stages can spread to internal organs. Early MF can be difficult to diagnose on histology and can require multiple biopsies. The most accurate biopsy practice involves stopping topical medications for 4 weeks, then taking multiple biopsies of clinically different areas to confirm the diagnosis and clonality (the same cell line in more than one location). SS typically presents with a larger area of involvement and may be associated with lymphadenopathy.
The patient was seen by Dermatology. Due to the extent of the disease, which was suggestive of SS, she underwent peripheral blood flow cytometry, which revealed > 1000 Sezary cells/mcL. This confirmed the diagnosis of SS. The patient was started on photophoresis, interferon, high-potency topical steroids for the local symptoms, and bexarotene, which blocks abnormal cell growth by binding to retinoid receptors.
Photos and text courtesy of John Durkin, MD, Pigmented Lesions Clinic, University of New Mexico, and Kirill Balatsky, MS II, University of New Mexico School of Medicine.
Submitted for publication by Dr. Daniel Stulberg, MD, Department of Family and Community Medicine, University of New Mexico School of Medicine, Albuquerque.
Hoppe RT, Kim YH. Clinical manifestations, pathologic features, and diagnosis of mycosis fungoides. UpToDate Web site. https://www.uptodate.com/contents/clinical-manifestations-pathologic-features-and-diagnosis-of-mycosis-fungoides. Updated February 27, 2019. Accessed January 29, 2020.
Hoppe RT, Kim YH. Clinical manifestations, pathologic features, and diagnosis of mycosis fungoides. UpToDate Web site. https://www.uptodate.com/contents/clinical-manifestations-pathologic-features-and-diagnosis-of-mycosis-fungoides. Updated February 27, 2019. Accessed January 29, 2020.

Toenail thickening

The FP suspected onychauxis, more commonly called hypertrophic nail. The patient’s toenail had the characteristic features of onychauxis, which include discoloration (usually yellow or brown) and a dull appearance. Often, there is an increased curvature or deviation of the nail and a “clam shell” appearance with transverse lines or a lamellar pattern like a ram’s horn. This is in contrast to the longitudinal lines and furrows that one would see with brittle nails associated with old age or the longitudinal melanonychia (hyperpigmented lines) seen in melanoma. Trauma to the nailbed, including trauma from ill-fitting shoes, is the most common cause of onychauxis.
Although nail discoloration and thickening raise the concern for onychomycosis, not all thickened and discolored nails are due to fungal infection. In this case, the thickening of the nail itself (as opposed to the subungal hyperkeratosis typical of onychomycosis) and a lack of improvement with antifungal treatment prompted the FP to consider other causes of nail dystrophy besides onychomycosis.
Nail trimming and filing can minimize discomfort and limit nail margin trauma caused by the nail’s abnormal shape. If this does not provide relief, the curative treatment for onychauxis is toenail removal and matrix ablation. In this case, the patient chose to defer nail removal and resection of the matrix. She said she would consider this treatment option if her nail became more bothersome.
Photos and text courtesy of Sabrina Gill, MD, and Daniel Stulberg, MD, FAAFP, Department of Family and Community Medicine, University of New Mexico School of Medicine, Albuquerque.
Abdullah L, Abba O. Common nail changes and disorders in older people: Diagnosis and management. Can Fam Physician. 2011;57:173–181.
The FP suspected onychauxis, more commonly called hypertrophic nail. The patient’s toenail had the characteristic features of onychauxis, which include discoloration (usually yellow or brown) and a dull appearance. Often, there is an increased curvature or deviation of the nail and a “clam shell” appearance with transverse lines or a lamellar pattern like a ram’s horn. This is in contrast to the longitudinal lines and furrows that one would see with brittle nails associated with old age or the longitudinal melanonychia (hyperpigmented lines) seen in melanoma. Trauma to the nailbed, including trauma from ill-fitting shoes, is the most common cause of onychauxis.
Although nail discoloration and thickening raise the concern for onychomycosis, not all thickened and discolored nails are due to fungal infection. In this case, the thickening of the nail itself (as opposed to the subungal hyperkeratosis typical of onychomycosis) and a lack of improvement with antifungal treatment prompted the FP to consider other causes of nail dystrophy besides onychomycosis.
Nail trimming and filing can minimize discomfort and limit nail margin trauma caused by the nail’s abnormal shape. If this does not provide relief, the curative treatment for onychauxis is toenail removal and matrix ablation. In this case, the patient chose to defer nail removal and resection of the matrix. She said she would consider this treatment option if her nail became more bothersome.
Photos and text courtesy of Sabrina Gill, MD, and Daniel Stulberg, MD, FAAFP, Department of Family and Community Medicine, University of New Mexico School of Medicine, Albuquerque.
The FP suspected onychauxis, more commonly called hypertrophic nail. The patient’s toenail had the characteristic features of onychauxis, which include discoloration (usually yellow or brown) and a dull appearance. Often, there is an increased curvature or deviation of the nail and a “clam shell” appearance with transverse lines or a lamellar pattern like a ram’s horn. This is in contrast to the longitudinal lines and furrows that one would see with brittle nails associated with old age or the longitudinal melanonychia (hyperpigmented lines) seen in melanoma. Trauma to the nailbed, including trauma from ill-fitting shoes, is the most common cause of onychauxis.
Although nail discoloration and thickening raise the concern for onychomycosis, not all thickened and discolored nails are due to fungal infection. In this case, the thickening of the nail itself (as opposed to the subungal hyperkeratosis typical of onychomycosis) and a lack of improvement with antifungal treatment prompted the FP to consider other causes of nail dystrophy besides onychomycosis.
Nail trimming and filing can minimize discomfort and limit nail margin trauma caused by the nail’s abnormal shape. If this does not provide relief, the curative treatment for onychauxis is toenail removal and matrix ablation. In this case, the patient chose to defer nail removal and resection of the matrix. She said she would consider this treatment option if her nail became more bothersome.
Photos and text courtesy of Sabrina Gill, MD, and Daniel Stulberg, MD, FAAFP, Department of Family and Community Medicine, University of New Mexico School of Medicine, Albuquerque.
Abdullah L, Abba O. Common nail changes and disorders in older people: Diagnosis and management. Can Fam Physician. 2011;57:173–181.
Abdullah L, Abba O. Common nail changes and disorders in older people: Diagnosis and management. Can Fam Physician. 2011;57:173–181.

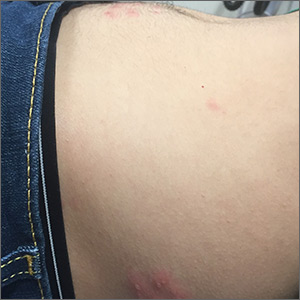
Abdominal rash
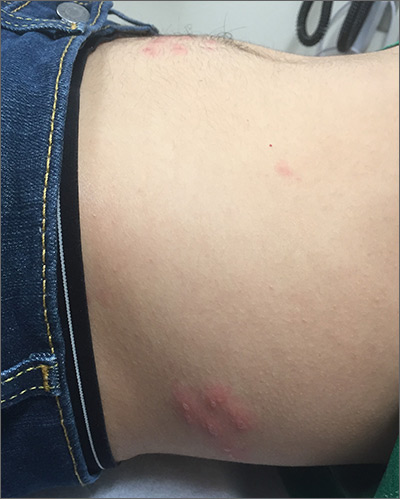
The pruritic prodrome, which evolved into painful vesicles, on an erythematous base in a dermatomal distribution was a classic presentation of herpes zoster.
Although herpes zoster is usually thought of as a disease that strikes older individuals, a study by Kawai et al found that of 8000 patients experiencing an outbreak, 43% were < 50 years of age. Additionally, this study found that the rate of herpes zoster infection had increased 4-fold in the past 60 years, and the rise had not been affected by the introduction of varicella vaccination. In Kawai’s series, 6.6% of the cases were in immunocompromised individuals.
In this case, the patient began antiviral therapy (Valacyclovir 1g tid for 7 days) to treat the active lesions. In further discussion with the patient, he indicated that he was concerned he might have an underlying undiagnosed autoimmune condition or human immunodeficiency virus (HIV) infection. His provider completed routine health care maintenance recommendations including an HIV screen, which was negative. Since he was otherwise asymptomatic, with no history of recurrent infections, no additional testing was warranted. However, since the patient worked with immunocompromised patients at a local hospital, he was placed on medical leave (per the facility’s protocol) until the lesions had crusted over. By Day 10, his lesions had crusted over and he returned to work.
Text courtesy of Brent Gillespie, PAC, University of New Mexico. Photo and editing courtesy of Daniel Stulberg, MD, FAAFP, Department of Family and Community Medicine, University of New Mexico School of Medicine, Albuquerque.
Kawai K, Yawn BP, Wollan P, et al. Increasing incidence of herpes zoster over a 60-year period from a population-based study. Clin Infect Dis. 2016;63:221-226.
The pruritic prodrome, which evolved into painful vesicles, on an erythematous base in a dermatomal distribution was a classic presentation of herpes zoster.
Although herpes zoster is usually thought of as a disease that strikes older individuals, a study by Kawai et al found that of 8000 patients experiencing an outbreak, 43% were < 50 years of age. Additionally, this study found that the rate of herpes zoster infection had increased 4-fold in the past 60 years, and the rise had not been affected by the introduction of varicella vaccination. In Kawai’s series, 6.6% of the cases were in immunocompromised individuals.
In this case, the patient began antiviral therapy (Valacyclovir 1g tid for 7 days) to treat the active lesions. In further discussion with the patient, he indicated that he was concerned he might have an underlying undiagnosed autoimmune condition or human immunodeficiency virus (HIV) infection. His provider completed routine health care maintenance recommendations including an HIV screen, which was negative. Since he was otherwise asymptomatic, with no history of recurrent infections, no additional testing was warranted. However, since the patient worked with immunocompromised patients at a local hospital, he was placed on medical leave (per the facility’s protocol) until the lesions had crusted over. By Day 10, his lesions had crusted over and he returned to work.
Text courtesy of Brent Gillespie, PAC, University of New Mexico. Photo and editing courtesy of Daniel Stulberg, MD, FAAFP, Department of Family and Community Medicine, University of New Mexico School of Medicine, Albuquerque.
The pruritic prodrome, which evolved into painful vesicles, on an erythematous base in a dermatomal distribution was a classic presentation of herpes zoster.
Although herpes zoster is usually thought of as a disease that strikes older individuals, a study by Kawai et al found that of 8000 patients experiencing an outbreak, 43% were < 50 years of age. Additionally, this study found that the rate of herpes zoster infection had increased 4-fold in the past 60 years, and the rise had not been affected by the introduction of varicella vaccination. In Kawai’s series, 6.6% of the cases were in immunocompromised individuals.
In this case, the patient began antiviral therapy (Valacyclovir 1g tid for 7 days) to treat the active lesions. In further discussion with the patient, he indicated that he was concerned he might have an underlying undiagnosed autoimmune condition or human immunodeficiency virus (HIV) infection. His provider completed routine health care maintenance recommendations including an HIV screen, which was negative. Since he was otherwise asymptomatic, with no history of recurrent infections, no additional testing was warranted. However, since the patient worked with immunocompromised patients at a local hospital, he was placed on medical leave (per the facility’s protocol) until the lesions had crusted over. By Day 10, his lesions had crusted over and he returned to work.
Text courtesy of Brent Gillespie, PAC, University of New Mexico. Photo and editing courtesy of Daniel Stulberg, MD, FAAFP, Department of Family and Community Medicine, University of New Mexico School of Medicine, Albuquerque.
Kawai K, Yawn BP, Wollan P, et al. Increasing incidence of herpes zoster over a 60-year period from a population-based study. Clin Infect Dis. 2016;63:221-226.
Kawai K, Yawn BP, Wollan P, et al. Increasing incidence of herpes zoster over a 60-year period from a population-based study. Clin Infect Dis. 2016;63:221-226.
Draining papule on chin

A biopsy of the skin was consistent with folliculitis, but a dental panoramic x-ray revealed an abscess of tooth number 30, which is on the right side. This combination of findings was consistent with a dental sinus.
This unusual diagnosis is most common in adults and arises from dental disease in the mid mandibular pre-molars. It will appear as an abscess or crusted papule and can occur in the lower jaw, neck, or occasionally, the mid cheek. Children are rarely affected. Because the sinus opening relieves pressure from the dental abscess, there is often little to no pain.
It can be challenging to convince a patient that his or her skin lesion derives from dental disease. Often, though, patients are glad it is not cancer. Patients should be referred to an endodontist for a work-up and a root canal, which is the preferred treatment. If a root canal is not feasible, dental extraction will lead to cure.
In this case, the patient promptly underwent a root canal and was cured in 2 weeks. The scar from the papule can have a depressed appearance (FIGURE 2), which can be addressed with excisional scar revision once the underlying abscess is cured.
Photos and text for Photo Rounds Friday courtesy of Jonathan Karnes, MD (copyright retained). Dr. Karnes is the medical director of MDFMR Dermatology Services, Augusta, ME.
Janev E, Redzep E. Managing the cutaneous sinus tract of dental origine. Open Access Maced J Med Sci. 2016;4:489-492.
A biopsy of the skin was consistent with folliculitis, but a dental panoramic x-ray revealed an abscess of tooth number 30, which is on the right side. This combination of findings was consistent with a dental sinus.
This unusual diagnosis is most common in adults and arises from dental disease in the mid mandibular pre-molars. It will appear as an abscess or crusted papule and can occur in the lower jaw, neck, or occasionally, the mid cheek. Children are rarely affected. Because the sinus opening relieves pressure from the dental abscess, there is often little to no pain.
It can be challenging to convince a patient that his or her skin lesion derives from dental disease. Often, though, patients are glad it is not cancer. Patients should be referred to an endodontist for a work-up and a root canal, which is the preferred treatment. If a root canal is not feasible, dental extraction will lead to cure.
In this case, the patient promptly underwent a root canal and was cured in 2 weeks. The scar from the papule can have a depressed appearance (FIGURE 2), which can be addressed with excisional scar revision once the underlying abscess is cured.
Photos and text for Photo Rounds Friday courtesy of Jonathan Karnes, MD (copyright retained). Dr. Karnes is the medical director of MDFMR Dermatology Services, Augusta, ME.
A biopsy of the skin was consistent with folliculitis, but a dental panoramic x-ray revealed an abscess of tooth number 30, which is on the right side. This combination of findings was consistent with a dental sinus.
This unusual diagnosis is most common in adults and arises from dental disease in the mid mandibular pre-molars. It will appear as an abscess or crusted papule and can occur in the lower jaw, neck, or occasionally, the mid cheek. Children are rarely affected. Because the sinus opening relieves pressure from the dental abscess, there is often little to no pain.
It can be challenging to convince a patient that his or her skin lesion derives from dental disease. Often, though, patients are glad it is not cancer. Patients should be referred to an endodontist for a work-up and a root canal, which is the preferred treatment. If a root canal is not feasible, dental extraction will lead to cure.
In this case, the patient promptly underwent a root canal and was cured in 2 weeks. The scar from the papule can have a depressed appearance (FIGURE 2), which can be addressed with excisional scar revision once the underlying abscess is cured.
Photos and text for Photo Rounds Friday courtesy of Jonathan Karnes, MD (copyright retained). Dr. Karnes is the medical director of MDFMR Dermatology Services, Augusta, ME.
Janev E, Redzep E. Managing the cutaneous sinus tract of dental origine. Open Access Maced J Med Sci. 2016;4:489-492.
Janev E, Redzep E. Managing the cutaneous sinus tract of dental origine. Open Access Maced J Med Sci. 2016;4:489-492.

Right hip and pelvic pain
A 65-year-old man with a history of remote colon cancer, peptic ulcer disease, gastroesophageal reflux disease (GERD), and bilateral knee replacements presented with right groin and hip pain of more than a year’s duration. The patient described his hip pain as aching and said that it had worsened over the previous 6 months, interfering with his sleep. He said the pain worsened following activity, and it briefly felt better following an intra-articular corticosteroid injection into his right hip. The patient denied recent trauma or fracture and said he had no scalp pain, hearing loss, or spinal tenderness. Physical examination showed limited range of motion of the right hip and mild tenderness to palpation. Laboratory values were within normal limits. X-rays of the pelvis (Figure 1A) and right hip (Figure 1B) were ordered.

WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Dx: Paget disease of bone
Based on the patient’s clinical history and initial imaging studies, which showed characteristic trabecular thickening with bony enlargement of the right femur, we suspected that he had Paget disease of bone. This was confirmed on subsequent whole-body 99mTc-MDP bone scan (Figure 2), which revealed corresponding diffuse increased radiotracer uptake of the right femur. There was no scintigraphic evidence of osseous involvement of the skull, spine, or pelvis.

Epidemiology/incidence. Paget disease, also known as osteitis deformans, is fairly common in the aging population, with a prevalence ranging from 2% to almost 10%.1,2 Although onset before age 40 is rare, the diagnosis should be considered in younger patients, given the high prevalence. There is a slight male predominance, and the disease is more common in the United Kingdom and Western Europe, as well as in countries settled by European immigrants.3
Both genetic and environmental causes are believed to contribute to the pathogenesis of Paget disease. Mutations in the gene encoding sequestosome 1 (SQSTM1) can be seen in the autosomal dominant familial type (25%-50% of these cases), as well as in sporadic cases.4 Environmental influence has also been postulated as a possible cause, with a viral etiology (eg, chronic measles infection) being the most cited.5
Most patients will be asymptomatic
Paget disease can affect any bone in the body, although the skull, spine, pelvis, and long bones of the lower extremity are the most commonly affected sites.2 Most patients with Paget disease are asymptomatic. When symptoms are present, they either result from direct involvement of the bone or are secondary to bone overgrowth and deformity.
Direct involvement manifests as deep, constant bone pain that is worse at night. Symptoms related to bone overgrowth and deformity include spinal stenosis and related neurologic abnormalities, increased skull size, hearing loss (impingement of cranial nerve VIII), pathologic fracture (most commonly of the femur), and deformity such as protrusio acetabuli or femoral or tibial bowing.6 High-output heart failure and abnormalities in calcium and phosphate balance are uncommon but do occur.
Continue to: Degeneration into osteosarcoma...
Degeneration into osteosarcoma is a rare but almost invariably fatal complication of Paget disease, with an incidence of 0.2% to 1%.7 It clinically manifests as increased bone pain that is poorly responsive to medical therapy, local swelling, and pathologic fracture.8
Radiography is key to the work-up
The diagnosis of Paget disease is primarily radiographic. Early in the disease process, lytic lesions with thinning of the cortex will be noted. Later in the disease, there will be a mixed lytic/sclerotic phase, in which enlargement of the bone, a thickened cortex, and coarsened trabeculae are observed.
Characteristic radiographic findings. Focal lytic lesions in the skull are known as osteoporosis circumscripta. In the sclerotic phase, there is a thickening of the calvaria (termed “cotton wool”). Lesions involving the long bones will begin at the proximal or distal subchondral region and progress toward the diaphysis, with a sharp oblique delineation between involved bone and normal bone; this is described as “blade of grass” or “flame-shaped.”9
Within the pelvis, there will be cortical thickening and sclerosis with enlargement of the iliac wing. Within the spine, there will be enlarged vertebrae with a thickened sclerotic border, resulting in a “picture frame” appearance. Later in the disease, the sclerosis will involve the entire vertebrae (termed “ivory vertebra”).10
Additional testing options include magnetic resonance imaging (MRI), bone scintigraphy, laboratory testing, and biopsy.
Continue to: MRI is recommended...
MRI is recommended when degeneration into osteosarcoma is present—indicated by permeative lesions with cortical breakthrough and a soft-tissue mass. MRI is helpful to further characterize the lesion. Absence of the normal fatty marrow on T1-weighted images would be concerning for tumor involvement.
Bone scintigraphy is used to determine the extent of disease. It will show increased uptake when the lesions are active.
Laboratory testing. Serum alkaline phosphatase (sAP) is frequently elevated in patients with Paget disease (normal range, 20-140 IU/L) and reflects the extent and activity of disease. However, this correlation is not always reliable; it depends on monostotic vs polyostotic involvement, as well as which bones are involved. For example, sAP levels may be markedly elevated when the skull is involved but normal when other bones are involved.11 In patients with elevated sAP, serum calcium and 25-hydroxyvitamin D measurements should be obtained in anticipation of bisphosphonate treatment.
Biopsy. If the radiographic findings are typical for Paget disease, bone biopsy is not indicated. However, the main competing diagnosis to consider is malignancy; in atypical cases when imaging is unable to elucidate an underlying tumor, biopsy would be warranted.
Differentiating Paget disease from sclerotic metastasis is important. In metastasis, there will be no trabecular coarsening or enlargement of the bone.
Continue to: Bisphosphonates are a Tx mainstay
Bisphosphonates are a Tx mainstay
Indications for treatment include symptomatic or asymptomatic disease with any of the following: elevated sAP with pagetic changes at sites where complications could occur; sAP more than 2 to 4 times the upper limit of normal; normal sAP with abnormal bone scintigraphy at a site where complications could occur; planned surgery at an active pagetic site; and hypercalcemia in association with immobilization in patients with polyostotic disease.
Newer generation nitrogen-containing bisphosphonates are the mainstay of treatment; they ease pain, slow bone turnover, and promote deposition of normal lamellar bone, which over time will normalize sAP levels.12 The most frequently used and studied bisphosphonates include oral alendronate, oral risedronate, and intravenous zoledronic acid.13
Prior to treatment initiation, the patient should have documented normal serum levels of calcium, phosphorus, and 25-hydroxyvitamin D, and these levels should be monitored throughout the first year of treatment. All patients should receive supplemental vitamin D and calcium to avoid hypocalcemia. sAP should be measured at 3 to 6 months to assess the initial response to therapy. Once the levels equilibrate, sAP can be measured once or twice a year to asses bone activity.14
Our patient was referred to Endocrinology for management of Paget disease of his right hip and femur. Lab values, including sAP and liver function test results, were normal. The patient was prescribed a zoledronic acid infusion (Reclast). At 4-week follow-up, the patient reported moderate relief of bone pain and improved sleep.
CORRESPONDENCE
Don Nguyen, MD, MHA, Brigham and Women’s Hospital, Department of Radiology, 75 Francis Street, Boston, MA 02115; [email protected]
1. Altman RD, Bloch DA, Hochberg MC, et al. Prevalence of pelvic Paget’s disease of bone in the United States. J Bone Miner Res. 2000;15:461-465.
2. Singer F. Paget’s disease of bone. In: Feingold KR, Anawalt B, Boyce A, et al, eds. Endotext. South Dartmouth, MA: MDText.com, Inc.; 2000.
3. Merashli M, Jawad A. Paget’s disease of bone among various ethnic groups. Sultan Qaboos Univ Med J. 2015;15:E22-E26.
4. Hocking LJ, Lucas GJ, Daroszewska A, et al. Domain-specific mutations in sequestosome 1 (SQSTM1) cause familial and sporadic Paget’s disease. Hum Mol Genet. 2002;11:2735-2739.
5. Reddy SV, Kurihara N, Menaa C, et al. Osteoclasts formed by measles virus-infected osteoclast precursors from hCD46 transgenic mice express characteristics of pagetic osteoclasts. Endocrinology. 2001;142:2898-2905.
6. Moore TE, King AR, Kathol MH, et al. Sarcoma in Paget disease of bone: clinical, radiologic, and pathologic features in 22 cases. AJR Am J Roentgenol. 1991;156:1199-1203.
7. van Staa TP, Selby P, Leufkens HG, et al. Incidence and natural history of Paget’s disease of bone in England and Wales. J Bone Miner Res. 2002;17:465-471.
8. Hansen MF, Seton M, Merchant A. Osteosarcoma in Paget’s disease of bone. J Bone Miner Res. 2006;21(suppl 2):P58-P63.
9. Wittenberg K. The blade of grass sign. Radiology. 2001;221:199-200.
10. Dennis JM. The solitary dense vertebral body. Radiology. 1961;77:618-621.
11. Seton M. Paget’s disease of bone. In: Hochberg MC, Silman AJ, Smolen JS, et al, eds. Rheumatology. 4th ed. Philadelphia, PA: Mosby (Elsevier); 2008:2003.
12. Reid IR, Nicholson GC, Weinstein RS, et al. Biochemical and radiologic improvement in Paget’s disease of bone treated with alendronate: a randomized, placebo-controlled trial. Am J Med. 1996;101:341-348.
13. Siris ES, Lyles KW, Singer FR, et al. Medical management of Paget’s disease of bone: indications for treatment and review of current therapies. J Bone Miner Res. 2006;21(suppl 2):P94-P98.
14. Alvarez L, Peris P, Guañabens N, et al. Long-term biochemical response after bisphosphonate therapy in Paget’s disease of bone: proposed intervals for monitoring treatment. Rheumatology (Oxford). 2004;43:869-874.
A 65-year-old man with a history of remote colon cancer, peptic ulcer disease, gastroesophageal reflux disease (GERD), and bilateral knee replacements presented with right groin and hip pain of more than a year’s duration. The patient described his hip pain as aching and said that it had worsened over the previous 6 months, interfering with his sleep. He said the pain worsened following activity, and it briefly felt better following an intra-articular corticosteroid injection into his right hip. The patient denied recent trauma or fracture and said he had no scalp pain, hearing loss, or spinal tenderness. Physical examination showed limited range of motion of the right hip and mild tenderness to palpation. Laboratory values were within normal limits. X-rays of the pelvis (Figure 1A) and right hip (Figure 1B) were ordered.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Dx: Paget disease of bone
Based on the patient’s clinical history and initial imaging studies, which showed characteristic trabecular thickening with bony enlargement of the right femur, we suspected that he had Paget disease of bone. This was confirmed on subsequent whole-body 99mTc-MDP bone scan (Figure 2), which revealed corresponding diffuse increased radiotracer uptake of the right femur. There was no scintigraphic evidence of osseous involvement of the skull, spine, or pelvis.
Epidemiology/incidence. Paget disease, also known as osteitis deformans, is fairly common in the aging population, with a prevalence ranging from 2% to almost 10%.1,2 Although onset before age 40 is rare, the diagnosis should be considered in younger patients, given the high prevalence. There is a slight male predominance, and the disease is more common in the United Kingdom and Western Europe, as well as in countries settled by European immigrants.3
Both genetic and environmental causes are believed to contribute to the pathogenesis of Paget disease. Mutations in the gene encoding sequestosome 1 (SQSTM1) can be seen in the autosomal dominant familial type (25%-50% of these cases), as well as in sporadic cases.4 Environmental influence has also been postulated as a possible cause, with a viral etiology (eg, chronic measles infection) being the most cited.5
Most patients will be asymptomatic
Paget disease can affect any bone in the body, although the skull, spine, pelvis, and long bones of the lower extremity are the most commonly affected sites.2 Most patients with Paget disease are asymptomatic. When symptoms are present, they either result from direct involvement of the bone or are secondary to bone overgrowth and deformity.
Direct involvement manifests as deep, constant bone pain that is worse at night. Symptoms related to bone overgrowth and deformity include spinal stenosis and related neurologic abnormalities, increased skull size, hearing loss (impingement of cranial nerve VIII), pathologic fracture (most commonly of the femur), and deformity such as protrusio acetabuli or femoral or tibial bowing.6 High-output heart failure and abnormalities in calcium and phosphate balance are uncommon but do occur.
Continue to: Degeneration into osteosarcoma...
Degeneration into osteosarcoma is a rare but almost invariably fatal complication of Paget disease, with an incidence of 0.2% to 1%.7 It clinically manifests as increased bone pain that is poorly responsive to medical therapy, local swelling, and pathologic fracture.8
Radiography is key to the work-up
The diagnosis of Paget disease is primarily radiographic. Early in the disease process, lytic lesions with thinning of the cortex will be noted. Later in the disease, there will be a mixed lytic/sclerotic phase, in which enlargement of the bone, a thickened cortex, and coarsened trabeculae are observed.
Characteristic radiographic findings. Focal lytic lesions in the skull are known as osteoporosis circumscripta. In the sclerotic phase, there is a thickening of the calvaria (termed “cotton wool”). Lesions involving the long bones will begin at the proximal or distal subchondral region and progress toward the diaphysis, with a sharp oblique delineation between involved bone and normal bone; this is described as “blade of grass” or “flame-shaped.”9
Within the pelvis, there will be cortical thickening and sclerosis with enlargement of the iliac wing. Within the spine, there will be enlarged vertebrae with a thickened sclerotic border, resulting in a “picture frame” appearance. Later in the disease, the sclerosis will involve the entire vertebrae (termed “ivory vertebra”).10
Additional testing options include magnetic resonance imaging (MRI), bone scintigraphy, laboratory testing, and biopsy.
Continue to: MRI is recommended...
MRI is recommended when degeneration into osteosarcoma is present—indicated by permeative lesions with cortical breakthrough and a soft-tissue mass. MRI is helpful to further characterize the lesion. Absence of the normal fatty marrow on T1-weighted images would be concerning for tumor involvement.
Bone scintigraphy is used to determine the extent of disease. It will show increased uptake when the lesions are active.
Laboratory testing. Serum alkaline phosphatase (sAP) is frequently elevated in patients with Paget disease (normal range, 20-140 IU/L) and reflects the extent and activity of disease. However, this correlation is not always reliable; it depends on monostotic vs polyostotic involvement, as well as which bones are involved. For example, sAP levels may be markedly elevated when the skull is involved but normal when other bones are involved.11 In patients with elevated sAP, serum calcium and 25-hydroxyvitamin D measurements should be obtained in anticipation of bisphosphonate treatment.
Biopsy. If the radiographic findings are typical for Paget disease, bone biopsy is not indicated. However, the main competing diagnosis to consider is malignancy; in atypical cases when imaging is unable to elucidate an underlying tumor, biopsy would be warranted.
Differentiating Paget disease from sclerotic metastasis is important. In metastasis, there will be no trabecular coarsening or enlargement of the bone.
Continue to: Bisphosphonates are a Tx mainstay
Bisphosphonates are a Tx mainstay
Indications for treatment include symptomatic or asymptomatic disease with any of the following: elevated sAP with pagetic changes at sites where complications could occur; sAP more than 2 to 4 times the upper limit of normal; normal sAP with abnormal bone scintigraphy at a site where complications could occur; planned surgery at an active pagetic site; and hypercalcemia in association with immobilization in patients with polyostotic disease.
Newer generation nitrogen-containing bisphosphonates are the mainstay of treatment; they ease pain, slow bone turnover, and promote deposition of normal lamellar bone, which over time will normalize sAP levels.12 The most frequently used and studied bisphosphonates include oral alendronate, oral risedronate, and intravenous zoledronic acid.13
Prior to treatment initiation, the patient should have documented normal serum levels of calcium, phosphorus, and 25-hydroxyvitamin D, and these levels should be monitored throughout the first year of treatment. All patients should receive supplemental vitamin D and calcium to avoid hypocalcemia. sAP should be measured at 3 to 6 months to assess the initial response to therapy. Once the levels equilibrate, sAP can be measured once or twice a year to asses bone activity.14
Our patient was referred to Endocrinology for management of Paget disease of his right hip and femur. Lab values, including sAP and liver function test results, were normal. The patient was prescribed a zoledronic acid infusion (Reclast). At 4-week follow-up, the patient reported moderate relief of bone pain and improved sleep.
CORRESPONDENCE
Don Nguyen, MD, MHA, Brigham and Women’s Hospital, Department of Radiology, 75 Francis Street, Boston, MA 02115; [email protected]
A 65-year-old man with a history of remote colon cancer, peptic ulcer disease, gastroesophageal reflux disease (GERD), and bilateral knee replacements presented with right groin and hip pain of more than a year’s duration. The patient described his hip pain as aching and said that it had worsened over the previous 6 months, interfering with his sleep. He said the pain worsened following activity, and it briefly felt better following an intra-articular corticosteroid injection into his right hip. The patient denied recent trauma or fracture and said he had no scalp pain, hearing loss, or spinal tenderness. Physical examination showed limited range of motion of the right hip and mild tenderness to palpation. Laboratory values were within normal limits. X-rays of the pelvis (Figure 1A) and right hip (Figure 1B) were ordered.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Dx: Paget disease of bone
Based on the patient’s clinical history and initial imaging studies, which showed characteristic trabecular thickening with bony enlargement of the right femur, we suspected that he had Paget disease of bone. This was confirmed on subsequent whole-body 99mTc-MDP bone scan (Figure 2), which revealed corresponding diffuse increased radiotracer uptake of the right femur. There was no scintigraphic evidence of osseous involvement of the skull, spine, or pelvis.
Epidemiology/incidence. Paget disease, also known as osteitis deformans, is fairly common in the aging population, with a prevalence ranging from 2% to almost 10%.1,2 Although onset before age 40 is rare, the diagnosis should be considered in younger patients, given the high prevalence. There is a slight male predominance, and the disease is more common in the United Kingdom and Western Europe, as well as in countries settled by European immigrants.3
Both genetic and environmental causes are believed to contribute to the pathogenesis of Paget disease. Mutations in the gene encoding sequestosome 1 (SQSTM1) can be seen in the autosomal dominant familial type (25%-50% of these cases), as well as in sporadic cases.4 Environmental influence has also been postulated as a possible cause, with a viral etiology (eg, chronic measles infection) being the most cited.5
Most patients will be asymptomatic
Paget disease can affect any bone in the body, although the skull, spine, pelvis, and long bones of the lower extremity are the most commonly affected sites.2 Most patients with Paget disease are asymptomatic. When symptoms are present, they either result from direct involvement of the bone or are secondary to bone overgrowth and deformity.
Direct involvement manifests as deep, constant bone pain that is worse at night. Symptoms related to bone overgrowth and deformity include spinal stenosis and related neurologic abnormalities, increased skull size, hearing loss (impingement of cranial nerve VIII), pathologic fracture (most commonly of the femur), and deformity such as protrusio acetabuli or femoral or tibial bowing.6 High-output heart failure and abnormalities in calcium and phosphate balance are uncommon but do occur.
Continue to: Degeneration into osteosarcoma...
Degeneration into osteosarcoma is a rare but almost invariably fatal complication of Paget disease, with an incidence of 0.2% to 1%.7 It clinically manifests as increased bone pain that is poorly responsive to medical therapy, local swelling, and pathologic fracture.8
Radiography is key to the work-up
The diagnosis of Paget disease is primarily radiographic. Early in the disease process, lytic lesions with thinning of the cortex will be noted. Later in the disease, there will be a mixed lytic/sclerotic phase, in which enlargement of the bone, a thickened cortex, and coarsened trabeculae are observed.
Characteristic radiographic findings. Focal lytic lesions in the skull are known as osteoporosis circumscripta. In the sclerotic phase, there is a thickening of the calvaria (termed “cotton wool”). Lesions involving the long bones will begin at the proximal or distal subchondral region and progress toward the diaphysis, with a sharp oblique delineation between involved bone and normal bone; this is described as “blade of grass” or “flame-shaped.”9
Within the pelvis, there will be cortical thickening and sclerosis with enlargement of the iliac wing. Within the spine, there will be enlarged vertebrae with a thickened sclerotic border, resulting in a “picture frame” appearance. Later in the disease, the sclerosis will involve the entire vertebrae (termed “ivory vertebra”).10
Additional testing options include magnetic resonance imaging (MRI), bone scintigraphy, laboratory testing, and biopsy.
Continue to: MRI is recommended...
MRI is recommended when degeneration into osteosarcoma is present—indicated by permeative lesions with cortical breakthrough and a soft-tissue mass. MRI is helpful to further characterize the lesion. Absence of the normal fatty marrow on T1-weighted images would be concerning for tumor involvement.
Bone scintigraphy is used to determine the extent of disease. It will show increased uptake when the lesions are active.
Laboratory testing. Serum alkaline phosphatase (sAP) is frequently elevated in patients with Paget disease (normal range, 20-140 IU/L) and reflects the extent and activity of disease. However, this correlation is not always reliable; it depends on monostotic vs polyostotic involvement, as well as which bones are involved. For example, sAP levels may be markedly elevated when the skull is involved but normal when other bones are involved.11 In patients with elevated sAP, serum calcium and 25-hydroxyvitamin D measurements should be obtained in anticipation of bisphosphonate treatment.
Biopsy. If the radiographic findings are typical for Paget disease, bone biopsy is not indicated. However, the main competing diagnosis to consider is malignancy; in atypical cases when imaging is unable to elucidate an underlying tumor, biopsy would be warranted.
Differentiating Paget disease from sclerotic metastasis is important. In metastasis, there will be no trabecular coarsening or enlargement of the bone.
Continue to: Bisphosphonates are a Tx mainstay
Bisphosphonates are a Tx mainstay
Indications for treatment include symptomatic or asymptomatic disease with any of the following: elevated sAP with pagetic changes at sites where complications could occur; sAP more than 2 to 4 times the upper limit of normal; normal sAP with abnormal bone scintigraphy at a site where complications could occur; planned surgery at an active pagetic site; and hypercalcemia in association with immobilization in patients with polyostotic disease.
Newer generation nitrogen-containing bisphosphonates are the mainstay of treatment; they ease pain, slow bone turnover, and promote deposition of normal lamellar bone, which over time will normalize sAP levels.12 The most frequently used and studied bisphosphonates include oral alendronate, oral risedronate, and intravenous zoledronic acid.13
Prior to treatment initiation, the patient should have documented normal serum levels of calcium, phosphorus, and 25-hydroxyvitamin D, and these levels should be monitored throughout the first year of treatment. All patients should receive supplemental vitamin D and calcium to avoid hypocalcemia. sAP should be measured at 3 to 6 months to assess the initial response to therapy. Once the levels equilibrate, sAP can be measured once or twice a year to asses bone activity.14
Our patient was referred to Endocrinology for management of Paget disease of his right hip and femur. Lab values, including sAP and liver function test results, were normal. The patient was prescribed a zoledronic acid infusion (Reclast). At 4-week follow-up, the patient reported moderate relief of bone pain and improved sleep.
CORRESPONDENCE
Don Nguyen, MD, MHA, Brigham and Women’s Hospital, Department of Radiology, 75 Francis Street, Boston, MA 02115; [email protected]
1. Altman RD, Bloch DA, Hochberg MC, et al. Prevalence of pelvic Paget’s disease of bone in the United States. J Bone Miner Res. 2000;15:461-465.
2. Singer F. Paget’s disease of bone. In: Feingold KR, Anawalt B, Boyce A, et al, eds. Endotext. South Dartmouth, MA: MDText.com, Inc.; 2000.
3. Merashli M, Jawad A. Paget’s disease of bone among various ethnic groups. Sultan Qaboos Univ Med J. 2015;15:E22-E26.
4. Hocking LJ, Lucas GJ, Daroszewska A, et al. Domain-specific mutations in sequestosome 1 (SQSTM1) cause familial and sporadic Paget’s disease. Hum Mol Genet. 2002;11:2735-2739.
5. Reddy SV, Kurihara N, Menaa C, et al. Osteoclasts formed by measles virus-infected osteoclast precursors from hCD46 transgenic mice express characteristics of pagetic osteoclasts. Endocrinology. 2001;142:2898-2905.
6. Moore TE, King AR, Kathol MH, et al. Sarcoma in Paget disease of bone: clinical, radiologic, and pathologic features in 22 cases. AJR Am J Roentgenol. 1991;156:1199-1203.
7. van Staa TP, Selby P, Leufkens HG, et al. Incidence and natural history of Paget’s disease of bone in England and Wales. J Bone Miner Res. 2002;17:465-471.
8. Hansen MF, Seton M, Merchant A. Osteosarcoma in Paget’s disease of bone. J Bone Miner Res. 2006;21(suppl 2):P58-P63.
9. Wittenberg K. The blade of grass sign. Radiology. 2001;221:199-200.
10. Dennis JM. The solitary dense vertebral body. Radiology. 1961;77:618-621.
11. Seton M. Paget’s disease of bone. In: Hochberg MC, Silman AJ, Smolen JS, et al, eds. Rheumatology. 4th ed. Philadelphia, PA: Mosby (Elsevier); 2008:2003.
12. Reid IR, Nicholson GC, Weinstein RS, et al. Biochemical and radiologic improvement in Paget’s disease of bone treated with alendronate: a randomized, placebo-controlled trial. Am J Med. 1996;101:341-348.
13. Siris ES, Lyles KW, Singer FR, et al. Medical management of Paget’s disease of bone: indications for treatment and review of current therapies. J Bone Miner Res. 2006;21(suppl 2):P94-P98.
14. Alvarez L, Peris P, Guañabens N, et al. Long-term biochemical response after bisphosphonate therapy in Paget’s disease of bone: proposed intervals for monitoring treatment. Rheumatology (Oxford). 2004;43:869-874.
1. Altman RD, Bloch DA, Hochberg MC, et al. Prevalence of pelvic Paget’s disease of bone in the United States. J Bone Miner Res. 2000;15:461-465.
2. Singer F. Paget’s disease of bone. In: Feingold KR, Anawalt B, Boyce A, et al, eds. Endotext. South Dartmouth, MA: MDText.com, Inc.; 2000.
3. Merashli M, Jawad A. Paget’s disease of bone among various ethnic groups. Sultan Qaboos Univ Med J. 2015;15:E22-E26.
4. Hocking LJ, Lucas GJ, Daroszewska A, et al. Domain-specific mutations in sequestosome 1 (SQSTM1) cause familial and sporadic Paget’s disease. Hum Mol Genet. 2002;11:2735-2739.
5. Reddy SV, Kurihara N, Menaa C, et al. Osteoclasts formed by measles virus-infected osteoclast precursors from hCD46 transgenic mice express characteristics of pagetic osteoclasts. Endocrinology. 2001;142:2898-2905.
6. Moore TE, King AR, Kathol MH, et al. Sarcoma in Paget disease of bone: clinical, radiologic, and pathologic features in 22 cases. AJR Am J Roentgenol. 1991;156:1199-1203.
7. van Staa TP, Selby P, Leufkens HG, et al. Incidence and natural history of Paget’s disease of bone in England and Wales. J Bone Miner Res. 2002;17:465-471.
8. Hansen MF, Seton M, Merchant A. Osteosarcoma in Paget’s disease of bone. J Bone Miner Res. 2006;21(suppl 2):P58-P63.
9. Wittenberg K. The blade of grass sign. Radiology. 2001;221:199-200.
10. Dennis JM. The solitary dense vertebral body. Radiology. 1961;77:618-621.
11. Seton M. Paget’s disease of bone. In: Hochberg MC, Silman AJ, Smolen JS, et al, eds. Rheumatology. 4th ed. Philadelphia, PA: Mosby (Elsevier); 2008:2003.
12. Reid IR, Nicholson GC, Weinstein RS, et al. Biochemical and radiologic improvement in Paget’s disease of bone treated with alendronate: a randomized, placebo-controlled trial. Am J Med. 1996;101:341-348.
13. Siris ES, Lyles KW, Singer FR, et al. Medical management of Paget’s disease of bone: indications for treatment and review of current therapies. J Bone Miner Res. 2006;21(suppl 2):P94-P98.
14. Alvarez L, Peris P, Guañabens N, et al. Long-term biochemical response after bisphosphonate therapy in Paget’s disease of bone: proposed intervals for monitoring treatment. Rheumatology (Oxford). 2004;43:869-874.

Pitting of fingernails

Given the constellation of non-scarring alopecia on the patient’s posterior scalp (with no scale, papules, or plaques), the physician diagnosed alopecia areata (AA) with associated nail pitting in this patient. Although a scalp biopsy could have confirmed the diagnosis, it was not needed because the clinical picture was sufficient.
Nail pitting is commonly associated with psoriasis (although it is often less dense in presentation), but it can occur with alopecia areata. It may occur concurrently or separately from active alopecia. Pitting of the nails may occur in one or multiple fingernails and occurs in up to a third of patients with AA.
The patient’s scalp was treated with intralesional triamcinolone diluted with normal saline to a concentration of 5 mg/mL (0.5%) and injected in dermal blebs over every square centimeter of involvement. Not every 10-year-old can tolerate this modality, and many families prefer observation or topical steroids. Other topical treatments for AA of the scalp include anthralin, minoxidil, and immunotherapy with squaric acid dibutyl ester or diphencyprone. None of these therapies are approved by the Food and Drug Administration for the treatment of nail disease. There are case reports of systemic tofacitinib clearing significant AA associated nail pitting in adults.
The physician counseled the family to observe the nails and not pursue any antifungal therapies for the nails.
Photos and text for Photo Rounds Friday courtesy of Jonathan Karnes, MD (copyright retained). Dr. Karnes is the medical director of MDFMR Dermatology Services, Augusta, ME.
Ferreira SB, Scheinberg M2, Steiner D, et al. Remarkable improvement of nail changes in alopecia areata universalis with 10 Months of treatment with tofacitinib: a case report. Case Rep Dermatol. 2016;8:262-266.
Given the constellation of non-scarring alopecia on the patient’s posterior scalp (with no scale, papules, or plaques), the physician diagnosed alopecia areata (AA) with associated nail pitting in this patient. Although a scalp biopsy could have confirmed the diagnosis, it was not needed because the clinical picture was sufficient.
Nail pitting is commonly associated with psoriasis (although it is often less dense in presentation), but it can occur with alopecia areata. It may occur concurrently or separately from active alopecia. Pitting of the nails may occur in one or multiple fingernails and occurs in up to a third of patients with AA.
The patient’s scalp was treated with intralesional triamcinolone diluted with normal saline to a concentration of 5 mg/mL (0.5%) and injected in dermal blebs over every square centimeter of involvement. Not every 10-year-old can tolerate this modality, and many families prefer observation or topical steroids. Other topical treatments for AA of the scalp include anthralin, minoxidil, and immunotherapy with squaric acid dibutyl ester or diphencyprone. None of these therapies are approved by the Food and Drug Administration for the treatment of nail disease. There are case reports of systemic tofacitinib clearing significant AA associated nail pitting in adults.
The physician counseled the family to observe the nails and not pursue any antifungal therapies for the nails.
Photos and text for Photo Rounds Friday courtesy of Jonathan Karnes, MD (copyright retained). Dr. Karnes is the medical director of MDFMR Dermatology Services, Augusta, ME.
Given the constellation of non-scarring alopecia on the patient’s posterior scalp (with no scale, papules, or plaques), the physician diagnosed alopecia areata (AA) with associated nail pitting in this patient. Although a scalp biopsy could have confirmed the diagnosis, it was not needed because the clinical picture was sufficient.
Nail pitting is commonly associated with psoriasis (although it is often less dense in presentation), but it can occur with alopecia areata. It may occur concurrently or separately from active alopecia. Pitting of the nails may occur in one or multiple fingernails and occurs in up to a third of patients with AA.
The patient’s scalp was treated with intralesional triamcinolone diluted with normal saline to a concentration of 5 mg/mL (0.5%) and injected in dermal blebs over every square centimeter of involvement. Not every 10-year-old can tolerate this modality, and many families prefer observation or topical steroids. Other topical treatments for AA of the scalp include anthralin, minoxidil, and immunotherapy with squaric acid dibutyl ester or diphencyprone. None of these therapies are approved by the Food and Drug Administration for the treatment of nail disease. There are case reports of systemic tofacitinib clearing significant AA associated nail pitting in adults.
The physician counseled the family to observe the nails and not pursue any antifungal therapies for the nails.
Photos and text for Photo Rounds Friday courtesy of Jonathan Karnes, MD (copyright retained). Dr. Karnes is the medical director of MDFMR Dermatology Services, Augusta, ME.
Ferreira SB, Scheinberg M2, Steiner D, et al. Remarkable improvement of nail changes in alopecia areata universalis with 10 Months of treatment with tofacitinib: a case report. Case Rep Dermatol. 2016;8:262-266.
Ferreira SB, Scheinberg M2, Steiner D, et al. Remarkable improvement of nail changes in alopecia areata universalis with 10 Months of treatment with tofacitinib: a case report. Case Rep Dermatol. 2016;8:262-266.

Rash on legs and abdomen

The rash was consistent with nonblanching purpura. Two punch biopsies were performed for hematoxylin and eosin stain and direct immunofluorescence, which were consistent with IgA mediated small vessel vasculitis, or Henoch-Schoenlein purpura.
IgA small vessel vasculitis commonly occurs in children after a transient viral illness or as an allergic reaction to a medication. Nonblanching purpura occurs because small vessels have been cracked open by neutrophils and lymphocytes and leaked blood cells outside of the vascular circulation. Pressure fails to move these cells downstream, and thus, the skin does not blanch.
Joint pain is common, as is a self-resolving IgA mediated nephropathy. Approximately 1% to 3% of children will progress to end stage renal disease. IgA vasculitis also occurs in adults, with a higher portion developing nephropathy. In adults, lesions that present on the abdomen are suspected to correspond with gravity dependency and the total burden of IgA, leading to a higher risk for nephropathy.
The differential diagnosis of purpura is broad, but includes leukocytoclastic vasculitis, antineutrophil cytoplasmic antibodies-associated vasculitis, capillaritis, and disseminated intravascular coagulation.
The patient in this case was given topical triamcinolone 0.1% ointment to treat the rash. She returned 3 weeks later and her blood pressure was 160/105 mm Hg and she had protein and blood in her urine. The FP recommended a renal biopsy, which confirmed severe IgA nephropathy and end stage renal failure. She was given systemic steroids and ultimately received a renal transplant. Her outcome was atypical and unfortunate. Usually IgA vasculitis is benign and self-resolves with rest. Even when nephropathy is present, it typically resolves over weeks to months.
Photos and text for Photo Rounds Friday courtesy of Jonathan Karnes, MD (copyright retained). Dr. Karnes is the medical director of MDFMR Dermatology Services, Augusta, ME.
Audemard-Verger A, Terrier B, Dechartres A, et al. French Vasculitis Study Group. Characteristics and management of IgA vasculitis (Henoch-Schönlein) in adults: data from 260 patients included in a French multicenter retrospective survey. Arthritis Rheumatol. 2017;69:1862-1870.
The rash was consistent with nonblanching purpura. Two punch biopsies were performed for hematoxylin and eosin stain and direct immunofluorescence, which were consistent with IgA mediated small vessel vasculitis, or Henoch-Schoenlein purpura.
IgA small vessel vasculitis commonly occurs in children after a transient viral illness or as an allergic reaction to a medication. Nonblanching purpura occurs because small vessels have been cracked open by neutrophils and lymphocytes and leaked blood cells outside of the vascular circulation. Pressure fails to move these cells downstream, and thus, the skin does not blanch.
Joint pain is common, as is a self-resolving IgA mediated nephropathy. Approximately 1% to 3% of children will progress to end stage renal disease. IgA vasculitis also occurs in adults, with a higher portion developing nephropathy. In adults, lesions that present on the abdomen are suspected to correspond with gravity dependency and the total burden of IgA, leading to a higher risk for nephropathy.
The differential diagnosis of purpura is broad, but includes leukocytoclastic vasculitis, antineutrophil cytoplasmic antibodies-associated vasculitis, capillaritis, and disseminated intravascular coagulation.
The patient in this case was given topical triamcinolone 0.1% ointment to treat the rash. She returned 3 weeks later and her blood pressure was 160/105 mm Hg and she had protein and blood in her urine. The FP recommended a renal biopsy, which confirmed severe IgA nephropathy and end stage renal failure. She was given systemic steroids and ultimately received a renal transplant. Her outcome was atypical and unfortunate. Usually IgA vasculitis is benign and self-resolves with rest. Even when nephropathy is present, it typically resolves over weeks to months.
Photos and text for Photo Rounds Friday courtesy of Jonathan Karnes, MD (copyright retained). Dr. Karnes is the medical director of MDFMR Dermatology Services, Augusta, ME.
The rash was consistent with nonblanching purpura. Two punch biopsies were performed for hematoxylin and eosin stain and direct immunofluorescence, which were consistent with IgA mediated small vessel vasculitis, or Henoch-Schoenlein purpura.
IgA small vessel vasculitis commonly occurs in children after a transient viral illness or as an allergic reaction to a medication. Nonblanching purpura occurs because small vessels have been cracked open by neutrophils and lymphocytes and leaked blood cells outside of the vascular circulation. Pressure fails to move these cells downstream, and thus, the skin does not blanch.
Joint pain is common, as is a self-resolving IgA mediated nephropathy. Approximately 1% to 3% of children will progress to end stage renal disease. IgA vasculitis also occurs in adults, with a higher portion developing nephropathy. In adults, lesions that present on the abdomen are suspected to correspond with gravity dependency and the total burden of IgA, leading to a higher risk for nephropathy.
The differential diagnosis of purpura is broad, but includes leukocytoclastic vasculitis, antineutrophil cytoplasmic antibodies-associated vasculitis, capillaritis, and disseminated intravascular coagulation.
The patient in this case was given topical triamcinolone 0.1% ointment to treat the rash. She returned 3 weeks later and her blood pressure was 160/105 mm Hg and she had protein and blood in her urine. The FP recommended a renal biopsy, which confirmed severe IgA nephropathy and end stage renal failure. She was given systemic steroids and ultimately received a renal transplant. Her outcome was atypical and unfortunate. Usually IgA vasculitis is benign and self-resolves with rest. Even when nephropathy is present, it typically resolves over weeks to months.
Photos and text for Photo Rounds Friday courtesy of Jonathan Karnes, MD (copyright retained). Dr. Karnes is the medical director of MDFMR Dermatology Services, Augusta, ME.
Audemard-Verger A, Terrier B, Dechartres A, et al. French Vasculitis Study Group. Characteristics and management of IgA vasculitis (Henoch-Schönlein) in adults: data from 260 patients included in a French multicenter retrospective survey. Arthritis Rheumatol. 2017;69:1862-1870.
Audemard-Verger A, Terrier B, Dechartres A, et al. French Vasculitis Study Group. Characteristics and management of IgA vasculitis (Henoch-Schönlein) in adults: data from 260 patients included in a French multicenter retrospective survey. Arthritis Rheumatol. 2017;69:1862-1870.

Papules on hands and soles

Although these lesions were consistent with actinic keratosis, the location on the palms and the patient’s history of growing up on a ranch in northern Mexico (where he said he was exposed to high arsenic levels in the water and soil), suggested that this was a case of arsenical keratosis. A biopsy excluded frank squamous cell carcinoma; further testing was not needed to make the diagnosis.
While both actinic and arsenical keratoses are precancerous growths with delayed presentations of 20 to 30 years, arsenical keratoses do not typically appear in sun-exposed areas. Rather, these lesions appear on a patient’s palms or soles. They also may present as plate-like hyperpigmentation of the palms and soles or corn-like keratotic papules.
Arsenic occurs naturally in soil and groundwater worldwide and acts as a toxin and carcinogen. It accumulates in the skin, hair, nails, and teeth—making these sites markers of chronic disease. Chronic exposure leads to skin and lung cancer. Cancer of other organs also is possible. Peripheral neuropathy and pancytopenia are hallmarks of chronic exposure.
If arsenical keratosis is suspected, testing for arsenic levels in water or in the work environment is recommended. That said, arsenic exposure is often remote (given the time that has elapsed) and hard to prove.
A blood count and renal and liver function tests should be considered to assess for organ damage. In this case, these tests were normal and the patient was treated with cryotherapy and monitored for recurrence with annual skin exams. Oral retinoids, such as acitretin, have had success in suppressing the frequency and severity of episodes of new lesions in small case reports.
Photos and text for Photo Rounds Friday courtesy of Jonathan Karnes, MD (copyright retained). Dr. Karnes is the medical director of MDFMR Dermatology Services, Augusta, ME.
US Department of Health and Human Services Agency for Toxic Substances and Disease Registry. Arsenic Toxicity. https://www.atsdr.cdc.gov/csem/csem.asp?csem=1&po=0. Updated January 15, 2010. Accessed January 2, 2020.
Although these lesions were consistent with actinic keratosis, the location on the palms and the patient’s history of growing up on a ranch in northern Mexico (where he said he was exposed to high arsenic levels in the water and soil), suggested that this was a case of arsenical keratosis. A biopsy excluded frank squamous cell carcinoma; further testing was not needed to make the diagnosis.
While both actinic and arsenical keratoses are precancerous growths with delayed presentations of 20 to 30 years, arsenical keratoses do not typically appear in sun-exposed areas. Rather, these lesions appear on a patient’s palms or soles. They also may present as plate-like hyperpigmentation of the palms and soles or corn-like keratotic papules.
Arsenic occurs naturally in soil and groundwater worldwide and acts as a toxin and carcinogen. It accumulates in the skin, hair, nails, and teeth—making these sites markers of chronic disease. Chronic exposure leads to skin and lung cancer. Cancer of other organs also is possible. Peripheral neuropathy and pancytopenia are hallmarks of chronic exposure.
If arsenical keratosis is suspected, testing for arsenic levels in water or in the work environment is recommended. That said, arsenic exposure is often remote (given the time that has elapsed) and hard to prove.
A blood count and renal and liver function tests should be considered to assess for organ damage. In this case, these tests were normal and the patient was treated with cryotherapy and monitored for recurrence with annual skin exams. Oral retinoids, such as acitretin, have had success in suppressing the frequency and severity of episodes of new lesions in small case reports.
Photos and text for Photo Rounds Friday courtesy of Jonathan Karnes, MD (copyright retained). Dr. Karnes is the medical director of MDFMR Dermatology Services, Augusta, ME.
Although these lesions were consistent with actinic keratosis, the location on the palms and the patient’s history of growing up on a ranch in northern Mexico (where he said he was exposed to high arsenic levels in the water and soil), suggested that this was a case of arsenical keratosis. A biopsy excluded frank squamous cell carcinoma; further testing was not needed to make the diagnosis.
While both actinic and arsenical keratoses are precancerous growths with delayed presentations of 20 to 30 years, arsenical keratoses do not typically appear in sun-exposed areas. Rather, these lesions appear on a patient’s palms or soles. They also may present as plate-like hyperpigmentation of the palms and soles or corn-like keratotic papules.
Arsenic occurs naturally in soil and groundwater worldwide and acts as a toxin and carcinogen. It accumulates in the skin, hair, nails, and teeth—making these sites markers of chronic disease. Chronic exposure leads to skin and lung cancer. Cancer of other organs also is possible. Peripheral neuropathy and pancytopenia are hallmarks of chronic exposure.
If arsenical keratosis is suspected, testing for arsenic levels in water or in the work environment is recommended. That said, arsenic exposure is often remote (given the time that has elapsed) and hard to prove.
A blood count and renal and liver function tests should be considered to assess for organ damage. In this case, these tests were normal and the patient was treated with cryotherapy and monitored for recurrence with annual skin exams. Oral retinoids, such as acitretin, have had success in suppressing the frequency and severity of episodes of new lesions in small case reports.
Photos and text for Photo Rounds Friday courtesy of Jonathan Karnes, MD (copyright retained). Dr. Karnes is the medical director of MDFMR Dermatology Services, Augusta, ME.
US Department of Health and Human Services Agency for Toxic Substances and Disease Registry. Arsenic Toxicity. https://www.atsdr.cdc.gov/csem/csem.asp?csem=1&po=0. Updated January 15, 2010. Accessed January 2, 2020.
US Department of Health and Human Services Agency for Toxic Substances and Disease Registry. Arsenic Toxicity. https://www.atsdr.cdc.gov/csem/csem.asp?csem=1&po=0. Updated January 15, 2010. Accessed January 2, 2020.
