User login
Light therapy for nonseasonal major depressive disorder?
Consider treatment with bright light therapy, alone or in combination with fluoxetine, for patients with nonseasonal major depressive disorder (MDD).1
Strength of recommendation
B: Based on a single moderate-quality randomized control trial.
Lam RW, Levitt AJ, Levitan RD, et al. Efficacy of bright light treatment, fluoxetine, and the combination in patients with nonseasonal major depressive disorder: a randomized clinical trial. JAMA Psychiatry. 2016;73:56-63.
Illustrative Case
A 38-year-old woman recently diagnosed with MDD without a seasonal pattern comes to see you for her treatment options. Her Hamilton Depression Rating Scale (HAM-D) is 22, and she is not suicidal. Should you consider bright light therapy in addition to pharmacotherapy?
MDD is one of the most common psychiatric illnesses in the United States, affecting approximately one in 5 adults at some point in their lives.2 Selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors are considered effective first-line pharmacotherapy options for MDD.2,3 Despite their effectiveness, however, studies have shown that only about 40% of patients with MDD achieve remission with first- or second-line drugs.2 In addition, pharmacologic agents have a higher frequency of treatment-associated adverse effects than fluorescent light therapy.4
A Cochrane systematic review of 20 studies (N=620) showed the effectiveness of combined light therapy and pharmacotherapy in treating nonseasonal MDD, but found no benefit to light used as a monotherapy.5 However, the majority of the studies were of poor quality, occurred in the inpatient setting, and lasted fewer than 4 weeks.
In a 5-week, controlled, double-blind trial not included in the Cochrane review, 102 patients with nonseasonal MDD were randomized to receive either active treatment (bright light therapy) plus sertraline 50 mg daily or sham light treatment (using a dim red light) plus sertraline 50 mg daily. The investigators found a statistically significant larger reduction in depression score in the active treatment group than in the sham light group, based on the HAM-D, the Hamilton 6-Item Subscale, the Melancholia Scale, and the 7 atypical items from the Structured Interview Guide for the Seasonal Affective Disorder version of the HAM-D.6,7
Study Summary
Light therapy improves depression without a seasonal component
This latest study was an 8-week randomized, double-blind, placebo- and sham-controlled clinical trial evaluating the benefit of light therapy with and without pharmacotherapy for nonseasonal MDD.1 The investigators enrolled 122 adult patients (ages 19-60 years) from outpatient psychiatry clinics with a diagnosis of MDD (as diagnosed by a psychiatrist) and a HAM-D8 score of at least 20. Subjects had to be off psychotropic medication for at least 2 weeks prior to the first visit and were subsequently monitored for one week to identify spontaneous responders and to give patients time to better regulate their sleep-wake cycle (with the goal of sleeping only between 10:00 pm and 8:00 am daily).
The investigators randomly assigned patients to one of 4 treatment groups: active light monotherapy (10,000-lux fluorescent white light for 30 min/d early in the morning) plus a placebo pill; fluoxetine 20 mg/d plus sham light therapy; placebo pills with sham light therapy; and combined active light therapy with fluoxetine 20 mg daily. Sham light therapy consisted of the use of an inactivated negative ion generator, used in the same fashion as a light box. All patients were analyzed based on modified intention to treat.
The investigators monitored patients for adherence to active and sham treatment by review of their daily logs of device treatment times. Pill counts were used to assess medication adherence. The primary outcome at 8 weeks was the change from baseline in the Montgomery-Asberg Depression Rating Scale (MADRS), a 10-item questionnaire with a worst score of 60.9 Secondary outcomes were treatment response (≥50% MADRS score reduction) and remission (≤10 MADRS score) at the final 8th-week visit. MADRS scoring was used because of its higher sensitivity to treatment-induced changes and its high correlation with the HAM-D scale.
At the end of 8 weeks, the mean (standard deviation [SD]) changes in MADRS scores from baseline were: light monotherapy 13.4 (7.5), fluoxetine monotherapy 8.8 (9.9), combination therapy 16.9 (9.2), and placebo 6.5 (9.6). The improvement was significant in the light monotherapy treatment group vs the placebo group (P=.006), in the combination treatment group vs the vs placebo group (P<.001), and in the combination group vs the fluoxetine treatment group (P=.02), but not for the fluoxetine treatment group vs the placebo group (P=.32). The effect sizes vs placebo were: fluoxetine, d=0.24 (95% confidence interval [CI], −0.27 to 0.74); light monotherapy, 0.80 (95% CI, 0.28 to 1.31); and combination therapy, 1.11 (95% CI, 0.54 to 1.64). Effect sizes of more than 0.8 are often considered large.10
The treatment response (≥50% MADRS improvement) rate was highest in the combination treatment group (75.9%) with response rates to light monotherapy, placebo, and fluoxetine monotherapy of 50%, 33.3%, and 29%, respectively. There was a significant response effect for the combination vs placebo treatment group (P=.005). Similarly, there was a higher remission rate in the combination treatment group (58.6%) than in the placebo, light monotherapy, or fluoxetine treatment groups (30%, 43.8%, and 19.4%, respectively) with a significant effect for the combination vs placebo treatment group (P=.02).
Combination therapy was superior to placebo in treatment response (≥50% reduction in the MADRS score) and remission (MADRS ≤10) with numbers needed to treat of 2.4 (95% CI, 1.6-5.8) and 3.5 (95% CI, 2.0-29.9), respectively.
By the end of the 8-week study period, 16 of 122 patients had dropped out; 2 reported lack of efficacy, 5 reported adverse effects, and the remainder cited administrative reasons, were lost to follow-up, or withdrew consent.
What’s New?
New evidence on a not-so-new treatment
We now have evidence that bright light therapy, either alone or in combination with fluoxetine, is efficacious in increasing the remission rate of nonseasonal MDD.
Caveats
Choice of SSRI, geography, and trial duration may have affected results
A single SSRI (fluoxetine) was used in this study; other more potent SSRIs might work better. This study was conducted in southern Canada, and light therapy may not demonstrate as large a benefit in regions located farther south. The study excluded pregnant and breastfeeding women.
The trial duration was relatively short, and the investigators did not attain their pre-planned sample size for the study, which limited the power to detect clinically significant seasonal treatment effects and differences between the fluoxetine and placebo groups, regardless of whether they received active phototherapy.
Also, it’s worth noting that there were trends for some adverse events (nausea, heartburn, weight gain, agitation, sexual dysfunction, and skin rash) to occur less frequently in the combination group than in the fluoxetine monotherapy group. Possible explanations are that the study had inadequate power, that the sham treatment did not adequately blind patients, or that light therapy can ameliorate some of the adverse effects of fluoxetine.
Challenges to Implementation
Commercial insurance doesn’t usually cover light therapy
Bright light therapy is fairly safe, and some evidence exists supporting its use in the treatment of nonseasonal MDD; however, the data for its use in this area are limited.11 Since only a few studies have tested light therapy for nonseasonal MDD, significant uncertainty remains about patient selection, as well as optimal dose, timing, and duration of light therapy in the management of nonseasonal MDD.12 Although the risks associated with bright light therapy are minimal, the therapy can lead to mania or hypomania,3 so clinicians need to monitor for such effects when initiating therapy.
Lastly, commercial insurance does not usually cover light therapy. The average price of the bright light devices, which can be found in medical supply stores and online outlets, ranges between $118 and $237.4,12 However, such devices are reusable, making the amortized cost almost negligible.13
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
1. Lam RW, Levitt AJ, Levitan RD, et al. Efficacy of bright light treatment, fluoxetine, and the combination in patients with nonseasonal major depressive disorder: a randomized clinical trial. JAMA Psychiatry. 2016;73:56-63.
2. Weihs K, Wert JM. A primary care focus on the treatment of patients with major depressive disorder. Am J Med Sci. 2011;342:324-330.
3. Gelenberg AJ, Freeman CMP, Markowitz JC, et al. Practice guideline for the treatment of patients with major depressive disorder. 3rd edition. 2010. Available at: http://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/mdd.pdf. Accessed April 20, 2016.
4. Lam RW, Tam EM. A Clinician’s Guide to Using Light Therapy. New York, NY: Cambridge University Press; 2009. Available at: http://www.ubcmood.ca/sad/SAD%20resources%20package%202009.pdf. Accessed April 20, 2016.
5. Tuunainen A, Kripke DF, Endo T. Light therapy for non-seasonal depression. Cochrane Database Syst Rev. 2004;2:CD004050.
6. Martiny K. Adjunctive bright light in non-seasonal major depression. Acta Psychiatr Scand Suppl. 2004;425:7-28.
7. Martiny K, Lunde M, Unden M, et al. Adjunctive bright light in non-seasonal major depression: results from clinician-rated depression scales. Acta Psychiatr Scand. 2005;112:117-125.
8. Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry. 1960;23:56-62.
9. Montgomery SA, Asberg M. A new depression scale designed to be sensitive to change. Br J Psychiatry. 1979;134:382-389.
10. Sullivan GM, Feinn R. Using effect size—or why the P value is not enough. J Grad Med Educ. 2012;4:279-282.
11. Oldham MA, Ciraulo DA. Use of bright light therapy among psychiatrists in Massachusetts: an e-mail survey. The Primary Care Companion for CNS Disorders. 2014;16.
12. Sloane PD, Figueiro M, Cohen L. Light as therapy for sleep disorders and depression in older adults. Clin Geriatr. 2008;16:25-31.
13. Kripke DF. A breakthrough treatment for major depression. J Clin Psychiatry. 2015;76:e660-e661.
Consider treatment with bright light therapy, alone or in combination with fluoxetine, for patients with nonseasonal major depressive disorder (MDD).1
Strength of recommendation
B: Based on a single moderate-quality randomized control trial.
Lam RW, Levitt AJ, Levitan RD, et al. Efficacy of bright light treatment, fluoxetine, and the combination in patients with nonseasonal major depressive disorder: a randomized clinical trial. JAMA Psychiatry. 2016;73:56-63.
Illustrative Case
A 38-year-old woman recently diagnosed with MDD without a seasonal pattern comes to see you for her treatment options. Her Hamilton Depression Rating Scale (HAM-D) is 22, and she is not suicidal. Should you consider bright light therapy in addition to pharmacotherapy?
MDD is one of the most common psychiatric illnesses in the United States, affecting approximately one in 5 adults at some point in their lives.2 Selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors are considered effective first-line pharmacotherapy options for MDD.2,3 Despite their effectiveness, however, studies have shown that only about 40% of patients with MDD achieve remission with first- or second-line drugs.2 In addition, pharmacologic agents have a higher frequency of treatment-associated adverse effects than fluorescent light therapy.4
A Cochrane systematic review of 20 studies (N=620) showed the effectiveness of combined light therapy and pharmacotherapy in treating nonseasonal MDD, but found no benefit to light used as a monotherapy.5 However, the majority of the studies were of poor quality, occurred in the inpatient setting, and lasted fewer than 4 weeks.
In a 5-week, controlled, double-blind trial not included in the Cochrane review, 102 patients with nonseasonal MDD were randomized to receive either active treatment (bright light therapy) plus sertraline 50 mg daily or sham light treatment (using a dim red light) plus sertraline 50 mg daily. The investigators found a statistically significant larger reduction in depression score in the active treatment group than in the sham light group, based on the HAM-D, the Hamilton 6-Item Subscale, the Melancholia Scale, and the 7 atypical items from the Structured Interview Guide for the Seasonal Affective Disorder version of the HAM-D.6,7
Study Summary
Light therapy improves depression without a seasonal component
This latest study was an 8-week randomized, double-blind, placebo- and sham-controlled clinical trial evaluating the benefit of light therapy with and without pharmacotherapy for nonseasonal MDD.1 The investigators enrolled 122 adult patients (ages 19-60 years) from outpatient psychiatry clinics with a diagnosis of MDD (as diagnosed by a psychiatrist) and a HAM-D8 score of at least 20. Subjects had to be off psychotropic medication for at least 2 weeks prior to the first visit and were subsequently monitored for one week to identify spontaneous responders and to give patients time to better regulate their sleep-wake cycle (with the goal of sleeping only between 10:00 pm and 8:00 am daily).
The investigators randomly assigned patients to one of 4 treatment groups: active light monotherapy (10,000-lux fluorescent white light for 30 min/d early in the morning) plus a placebo pill; fluoxetine 20 mg/d plus sham light therapy; placebo pills with sham light therapy; and combined active light therapy with fluoxetine 20 mg daily. Sham light therapy consisted of the use of an inactivated negative ion generator, used in the same fashion as a light box. All patients were analyzed based on modified intention to treat.
The investigators monitored patients for adherence to active and sham treatment by review of their daily logs of device treatment times. Pill counts were used to assess medication adherence. The primary outcome at 8 weeks was the change from baseline in the Montgomery-Asberg Depression Rating Scale (MADRS), a 10-item questionnaire with a worst score of 60.9 Secondary outcomes were treatment response (≥50% MADRS score reduction) and remission (≤10 MADRS score) at the final 8th-week visit. MADRS scoring was used because of its higher sensitivity to treatment-induced changes and its high correlation with the HAM-D scale.
At the end of 8 weeks, the mean (standard deviation [SD]) changes in MADRS scores from baseline were: light monotherapy 13.4 (7.5), fluoxetine monotherapy 8.8 (9.9), combination therapy 16.9 (9.2), and placebo 6.5 (9.6). The improvement was significant in the light monotherapy treatment group vs the placebo group (P=.006), in the combination treatment group vs the vs placebo group (P<.001), and in the combination group vs the fluoxetine treatment group (P=.02), but not for the fluoxetine treatment group vs the placebo group (P=.32). The effect sizes vs placebo were: fluoxetine, d=0.24 (95% confidence interval [CI], −0.27 to 0.74); light monotherapy, 0.80 (95% CI, 0.28 to 1.31); and combination therapy, 1.11 (95% CI, 0.54 to 1.64). Effect sizes of more than 0.8 are often considered large.10
The treatment response (≥50% MADRS improvement) rate was highest in the combination treatment group (75.9%) with response rates to light monotherapy, placebo, and fluoxetine monotherapy of 50%, 33.3%, and 29%, respectively. There was a significant response effect for the combination vs placebo treatment group (P=.005). Similarly, there was a higher remission rate in the combination treatment group (58.6%) than in the placebo, light monotherapy, or fluoxetine treatment groups (30%, 43.8%, and 19.4%, respectively) with a significant effect for the combination vs placebo treatment group (P=.02).
Combination therapy was superior to placebo in treatment response (≥50% reduction in the MADRS score) and remission (MADRS ≤10) with numbers needed to treat of 2.4 (95% CI, 1.6-5.8) and 3.5 (95% CI, 2.0-29.9), respectively.
By the end of the 8-week study period, 16 of 122 patients had dropped out; 2 reported lack of efficacy, 5 reported adverse effects, and the remainder cited administrative reasons, were lost to follow-up, or withdrew consent.
What’s New?
New evidence on a not-so-new treatment
We now have evidence that bright light therapy, either alone or in combination with fluoxetine, is efficacious in increasing the remission rate of nonseasonal MDD.
Caveats
Choice of SSRI, geography, and trial duration may have affected results
A single SSRI (fluoxetine) was used in this study; other more potent SSRIs might work better. This study was conducted in southern Canada, and light therapy may not demonstrate as large a benefit in regions located farther south. The study excluded pregnant and breastfeeding women.
The trial duration was relatively short, and the investigators did not attain their pre-planned sample size for the study, which limited the power to detect clinically significant seasonal treatment effects and differences between the fluoxetine and placebo groups, regardless of whether they received active phototherapy.
Also, it’s worth noting that there were trends for some adverse events (nausea, heartburn, weight gain, agitation, sexual dysfunction, and skin rash) to occur less frequently in the combination group than in the fluoxetine monotherapy group. Possible explanations are that the study had inadequate power, that the sham treatment did not adequately blind patients, or that light therapy can ameliorate some of the adverse effects of fluoxetine.
Challenges to Implementation
Commercial insurance doesn’t usually cover light therapy
Bright light therapy is fairly safe, and some evidence exists supporting its use in the treatment of nonseasonal MDD; however, the data for its use in this area are limited.11 Since only a few studies have tested light therapy for nonseasonal MDD, significant uncertainty remains about patient selection, as well as optimal dose, timing, and duration of light therapy in the management of nonseasonal MDD.12 Although the risks associated with bright light therapy are minimal, the therapy can lead to mania or hypomania,3 so clinicians need to monitor for such effects when initiating therapy.
Lastly, commercial insurance does not usually cover light therapy. The average price of the bright light devices, which can be found in medical supply stores and online outlets, ranges between $118 and $237.4,12 However, such devices are reusable, making the amortized cost almost negligible.13
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
Consider treatment with bright light therapy, alone or in combination with fluoxetine, for patients with nonseasonal major depressive disorder (MDD).1
Strength of recommendation
B: Based on a single moderate-quality randomized control trial.
Lam RW, Levitt AJ, Levitan RD, et al. Efficacy of bright light treatment, fluoxetine, and the combination in patients with nonseasonal major depressive disorder: a randomized clinical trial. JAMA Psychiatry. 2016;73:56-63.
Illustrative Case
A 38-year-old woman recently diagnosed with MDD without a seasonal pattern comes to see you for her treatment options. Her Hamilton Depression Rating Scale (HAM-D) is 22, and she is not suicidal. Should you consider bright light therapy in addition to pharmacotherapy?
MDD is one of the most common psychiatric illnesses in the United States, affecting approximately one in 5 adults at some point in their lives.2 Selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors are considered effective first-line pharmacotherapy options for MDD.2,3 Despite their effectiveness, however, studies have shown that only about 40% of patients with MDD achieve remission with first- or second-line drugs.2 In addition, pharmacologic agents have a higher frequency of treatment-associated adverse effects than fluorescent light therapy.4
A Cochrane systematic review of 20 studies (N=620) showed the effectiveness of combined light therapy and pharmacotherapy in treating nonseasonal MDD, but found no benefit to light used as a monotherapy.5 However, the majority of the studies were of poor quality, occurred in the inpatient setting, and lasted fewer than 4 weeks.
In a 5-week, controlled, double-blind trial not included in the Cochrane review, 102 patients with nonseasonal MDD were randomized to receive either active treatment (bright light therapy) plus sertraline 50 mg daily or sham light treatment (using a dim red light) plus sertraline 50 mg daily. The investigators found a statistically significant larger reduction in depression score in the active treatment group than in the sham light group, based on the HAM-D, the Hamilton 6-Item Subscale, the Melancholia Scale, and the 7 atypical items from the Structured Interview Guide for the Seasonal Affective Disorder version of the HAM-D.6,7
Study Summary
Light therapy improves depression without a seasonal component
This latest study was an 8-week randomized, double-blind, placebo- and sham-controlled clinical trial evaluating the benefit of light therapy with and without pharmacotherapy for nonseasonal MDD.1 The investigators enrolled 122 adult patients (ages 19-60 years) from outpatient psychiatry clinics with a diagnosis of MDD (as diagnosed by a psychiatrist) and a HAM-D8 score of at least 20. Subjects had to be off psychotropic medication for at least 2 weeks prior to the first visit and were subsequently monitored for one week to identify spontaneous responders and to give patients time to better regulate their sleep-wake cycle (with the goal of sleeping only between 10:00 pm and 8:00 am daily).
The investigators randomly assigned patients to one of 4 treatment groups: active light monotherapy (10,000-lux fluorescent white light for 30 min/d early in the morning) plus a placebo pill; fluoxetine 20 mg/d plus sham light therapy; placebo pills with sham light therapy; and combined active light therapy with fluoxetine 20 mg daily. Sham light therapy consisted of the use of an inactivated negative ion generator, used in the same fashion as a light box. All patients were analyzed based on modified intention to treat.
The investigators monitored patients for adherence to active and sham treatment by review of their daily logs of device treatment times. Pill counts were used to assess medication adherence. The primary outcome at 8 weeks was the change from baseline in the Montgomery-Asberg Depression Rating Scale (MADRS), a 10-item questionnaire with a worst score of 60.9 Secondary outcomes were treatment response (≥50% MADRS score reduction) and remission (≤10 MADRS score) at the final 8th-week visit. MADRS scoring was used because of its higher sensitivity to treatment-induced changes and its high correlation with the HAM-D scale.
At the end of 8 weeks, the mean (standard deviation [SD]) changes in MADRS scores from baseline were: light monotherapy 13.4 (7.5), fluoxetine monotherapy 8.8 (9.9), combination therapy 16.9 (9.2), and placebo 6.5 (9.6). The improvement was significant in the light monotherapy treatment group vs the placebo group (P=.006), in the combination treatment group vs the vs placebo group (P<.001), and in the combination group vs the fluoxetine treatment group (P=.02), but not for the fluoxetine treatment group vs the placebo group (P=.32). The effect sizes vs placebo were: fluoxetine, d=0.24 (95% confidence interval [CI], −0.27 to 0.74); light monotherapy, 0.80 (95% CI, 0.28 to 1.31); and combination therapy, 1.11 (95% CI, 0.54 to 1.64). Effect sizes of more than 0.8 are often considered large.10
The treatment response (≥50% MADRS improvement) rate was highest in the combination treatment group (75.9%) with response rates to light monotherapy, placebo, and fluoxetine monotherapy of 50%, 33.3%, and 29%, respectively. There was a significant response effect for the combination vs placebo treatment group (P=.005). Similarly, there was a higher remission rate in the combination treatment group (58.6%) than in the placebo, light monotherapy, or fluoxetine treatment groups (30%, 43.8%, and 19.4%, respectively) with a significant effect for the combination vs placebo treatment group (P=.02).
Combination therapy was superior to placebo in treatment response (≥50% reduction in the MADRS score) and remission (MADRS ≤10) with numbers needed to treat of 2.4 (95% CI, 1.6-5.8) and 3.5 (95% CI, 2.0-29.9), respectively.
By the end of the 8-week study period, 16 of 122 patients had dropped out; 2 reported lack of efficacy, 5 reported adverse effects, and the remainder cited administrative reasons, were lost to follow-up, or withdrew consent.
What’s New?
New evidence on a not-so-new treatment
We now have evidence that bright light therapy, either alone or in combination with fluoxetine, is efficacious in increasing the remission rate of nonseasonal MDD.
Caveats
Choice of SSRI, geography, and trial duration may have affected results
A single SSRI (fluoxetine) was used in this study; other more potent SSRIs might work better. This study was conducted in southern Canada, and light therapy may not demonstrate as large a benefit in regions located farther south. The study excluded pregnant and breastfeeding women.
The trial duration was relatively short, and the investigators did not attain their pre-planned sample size for the study, which limited the power to detect clinically significant seasonal treatment effects and differences between the fluoxetine and placebo groups, regardless of whether they received active phototherapy.
Also, it’s worth noting that there were trends for some adverse events (nausea, heartburn, weight gain, agitation, sexual dysfunction, and skin rash) to occur less frequently in the combination group than in the fluoxetine monotherapy group. Possible explanations are that the study had inadequate power, that the sham treatment did not adequately blind patients, or that light therapy can ameliorate some of the adverse effects of fluoxetine.
Challenges to Implementation
Commercial insurance doesn’t usually cover light therapy
Bright light therapy is fairly safe, and some evidence exists supporting its use in the treatment of nonseasonal MDD; however, the data for its use in this area are limited.11 Since only a few studies have tested light therapy for nonseasonal MDD, significant uncertainty remains about patient selection, as well as optimal dose, timing, and duration of light therapy in the management of nonseasonal MDD.12 Although the risks associated with bright light therapy are minimal, the therapy can lead to mania or hypomania,3 so clinicians need to monitor for such effects when initiating therapy.
Lastly, commercial insurance does not usually cover light therapy. The average price of the bright light devices, which can be found in medical supply stores and online outlets, ranges between $118 and $237.4,12 However, such devices are reusable, making the amortized cost almost negligible.13
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
1. Lam RW, Levitt AJ, Levitan RD, et al. Efficacy of bright light treatment, fluoxetine, and the combination in patients with nonseasonal major depressive disorder: a randomized clinical trial. JAMA Psychiatry. 2016;73:56-63.
2. Weihs K, Wert JM. A primary care focus on the treatment of patients with major depressive disorder. Am J Med Sci. 2011;342:324-330.
3. Gelenberg AJ, Freeman CMP, Markowitz JC, et al. Practice guideline for the treatment of patients with major depressive disorder. 3rd edition. 2010. Available at: http://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/mdd.pdf. Accessed April 20, 2016.
4. Lam RW, Tam EM. A Clinician’s Guide to Using Light Therapy. New York, NY: Cambridge University Press; 2009. Available at: http://www.ubcmood.ca/sad/SAD%20resources%20package%202009.pdf. Accessed April 20, 2016.
5. Tuunainen A, Kripke DF, Endo T. Light therapy for non-seasonal depression. Cochrane Database Syst Rev. 2004;2:CD004050.
6. Martiny K. Adjunctive bright light in non-seasonal major depression. Acta Psychiatr Scand Suppl. 2004;425:7-28.
7. Martiny K, Lunde M, Unden M, et al. Adjunctive bright light in non-seasonal major depression: results from clinician-rated depression scales. Acta Psychiatr Scand. 2005;112:117-125.
8. Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry. 1960;23:56-62.
9. Montgomery SA, Asberg M. A new depression scale designed to be sensitive to change. Br J Psychiatry. 1979;134:382-389.
10. Sullivan GM, Feinn R. Using effect size—or why the P value is not enough. J Grad Med Educ. 2012;4:279-282.
11. Oldham MA, Ciraulo DA. Use of bright light therapy among psychiatrists in Massachusetts: an e-mail survey. The Primary Care Companion for CNS Disorders. 2014;16.
12. Sloane PD, Figueiro M, Cohen L. Light as therapy for sleep disorders and depression in older adults. Clin Geriatr. 2008;16:25-31.
13. Kripke DF. A breakthrough treatment for major depression. J Clin Psychiatry. 2015;76:e660-e661.
1. Lam RW, Levitt AJ, Levitan RD, et al. Efficacy of bright light treatment, fluoxetine, and the combination in patients with nonseasonal major depressive disorder: a randomized clinical trial. JAMA Psychiatry. 2016;73:56-63.
2. Weihs K, Wert JM. A primary care focus on the treatment of patients with major depressive disorder. Am J Med Sci. 2011;342:324-330.
3. Gelenberg AJ, Freeman CMP, Markowitz JC, et al. Practice guideline for the treatment of patients with major depressive disorder. 3rd edition. 2010. Available at: http://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/mdd.pdf. Accessed April 20, 2016.
4. Lam RW, Tam EM. A Clinician’s Guide to Using Light Therapy. New York, NY: Cambridge University Press; 2009. Available at: http://www.ubcmood.ca/sad/SAD%20resources%20package%202009.pdf. Accessed April 20, 2016.
5. Tuunainen A, Kripke DF, Endo T. Light therapy for non-seasonal depression. Cochrane Database Syst Rev. 2004;2:CD004050.
6. Martiny K. Adjunctive bright light in non-seasonal major depression. Acta Psychiatr Scand Suppl. 2004;425:7-28.
7. Martiny K, Lunde M, Unden M, et al. Adjunctive bright light in non-seasonal major depression: results from clinician-rated depression scales. Acta Psychiatr Scand. 2005;112:117-125.
8. Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry. 1960;23:56-62.
9. Montgomery SA, Asberg M. A new depression scale designed to be sensitive to change. Br J Psychiatry. 1979;134:382-389.
10. Sullivan GM, Feinn R. Using effect size—or why the P value is not enough. J Grad Med Educ. 2012;4:279-282.
11. Oldham MA, Ciraulo DA. Use of bright light therapy among psychiatrists in Massachusetts: an e-mail survey. The Primary Care Companion for CNS Disorders. 2014;16.
12. Sloane PD, Figueiro M, Cohen L. Light as therapy for sleep disorders and depression in older adults. Clin Geriatr. 2008;16:25-31.
13. Kripke DF. A breakthrough treatment for major depression. J Clin Psychiatry. 2015;76:e660-e661.
Copyright © 2016. The Family Physicians Inquiries Network. All rights reserved.

More Isn’t Better With Acute Low Back Pain Treatment
PRACTICE CHANGER
Consider treating patients with acute low back pain with naproxen only, as adding cyclobenzaprine or oxycodone/acetaminophen to scheduled naproxen increases adverse effects and does not improve functional assessment at seven days or three months.
Strength of Recommendation
B: Based on a high-quality, randomized controlled trial (RCT).1
A 46-year-old man presents to the emergency department (ED) with low back pain (LBP) after helping a friend move a couch three days ago. He denies any direct trauma to his back and describes the pain as a “spasm” in his lumbar spinal region with no radicular symptoms. The pain worsens with prolonged standing and position changes. He has tried acetaminophen with no benefit. You diagnose a lumbar muscular strain. What medications should you prescribe to help relieve his LBP and improve his overall function?
Acute LBP prompts nearly 2.7 million ED visits in the United States each year.2 It leads to persistent subjective impairment and continued analgesic use at seven days (impairment, 70%; analgesic use, 69%) and three months (48% and 46%, respectively) after ED discharge.3 Systematic reviews show that monotherapy with NSAIDs or muscle relaxants is more effective than placebo for pain relief.4,5 A secondary analysis of patients (N = 715) from a prospective cohort study showed worse functioning at six months in those who were prescribed opiates for LBP than in those who were not.6
Monotherapy or combination therapy for LBP?
Because medications used for LBP have different mechanisms of action, clinicians frequently combine them in an attempt to improve symptoms and function.2 Current evidence on combination therapy shows mixed results. A large RCT (N = 867) showed that the combination of cyclobenzaprine and ibuprofen led to lower subjective pain intensity, but it did not result in self-reported pain improvement, compared to cyclobenzaprine alone. However, a small RCT (N = 40) demonstrated improved LBP and spasm with naprozen plus cyclobenzaprine, compared to naproxen alone.7,8
This study sought to determine the benefit of treating acute LBP with cyclobenzaprine or oxycodone/acetaminophen in combination with an NSAID, compared to treatment with an NSAID alone.
Continue for the study summary >>
STUDY SUMMARY
Adding second pain reliever provided no significant benefit
This double-blinded RCT enrolled 323 adults presenting to an ED with two weeks or less of nontraumatic, nonradicular LBP.1 Subjects had a score of > 5 on the Roland-Morris Disability Questionnaire (RMDQ), which measures functional impairment due to LBP (range, 0-24). Patients were excluded if they had radicular pain radiating below the gluteal folds, direct trauma to the back within the previous month, pain lasting > 2 wk, a recent history of multiple LBP episodes per month, or a history of opioid use.
All subjects received 10 days’ worth of naproxen (500 mg bid). They were then randomized to receive either oxycodone/acetaminophen (5 mg/325 mg), cyclobenzaprine (5 mg), or placebo, with instructions to take one to two tablets as needed every eight hours for 10 days. All patients also received a 10-minute educational session emphasizing the role of nonpharmacologic interventions.
The primary outcome was change in the RMDQ between ED discharge and a phone call seven days later; a 5-point improvement in the RMDQ was considered clinically significant. Secondary outcomes included subjective description of worst pain, frequency of LBP, frequency of analgesic use, satisfaction with treatment, median number of days to return to work and usual activities, need for follow-up health care visits, and opioid use. Investigators also asked about any adverse effects.
At seven days, reported RMDQ scores had improved by 9.8 points in patients taking naproxen plus placebo, 10.1 points in those receiving naproxen plus cyclobenzaprine, and 11.1 points in those using naproxen plus oxycodone/acetaminophen. There were no statistically significant between-group differences for placebo vs cyclobenzaprine or oxycodone/acetaminophen (0.3 points and 1.3 points, respectively) or cyclobenzaprine vs oxycodone/acetaminophen (0.9 points).
Secondary outcomes. At seven days, there was no significant difference between study groups in subjective pain assessment, frequency of LBP, or use of as-needed medications in the prior 24 hours. There was also no difference in the median number of days to return to work or need for follow-up health care visits.
Among patients who took more than one dose of the study medication, those who took oxycodone/acetaminophen were more likely to describe their worst pain in the last 24 hours as mild/none, compared to patients taking placebo (number needed to treat, 6). About 72% of all subjects reported that they would choose the same treatment option again, with no difference between groups. At three months, there was no difference between groups in subjective pain assessment, frequency of LBP, use of as-needed medications, or opioid use during the previous 72 hours.
Adverse effects, including drowsiness, dizziness, stomach irritation, and nausea or vomiting, were more common in the oxycodone/acetaminophen and the cyclobenzaprine treatment groups, with a number needed to harm of 5.3 and 7.8, respectively.
Continue for what's new >>
WHAT’S NEW
Second med adds nothing
This RCT found that adding cyclobenzaprine or oxycodone/acetaminophen to naproxen for the treatment of nontraumatic, nonradicular acute LBP did not significantly improve functional assessment at seven days or three months after the initial ED visit. But it did increase adverse effects.
CAVEATS
Specific subset studied
This study was performed in a single urban ED and included a very specific subset of LBP patients, which limits the generalizability of the results. However, patients often present to primary care with similar LBP complaints, and the results of the study should reasonably apply to other settings.
The findings may not generalize to all NSAIDs, but there is no evidence to suggest that other NSAIDs would behave differently when combined with cyclobenzaprine or oxycodone/acetaminophen. In this analysis, only about one-third of patients used the as-needed medication more than once daily; another third used it intermittently or never.
CHALLENGES TO IMPLEMENTATION
Patients may expect more
Patients expect to receive prescriptions, and clinicians are inclined to write them if they believe doing so will help their patients. The evidence, however, does not demonstrate a benefit to these prescription-only medications for LBP.
REFERENCES
1. Friedman BW, Dym AA, Davitt M, et al. Naproxen with cyclobenzaprine, oxycodone/acetaminophen, or placebo for treating acute low back pain: a randomized clinical trial. JAMA. 2015;314:1572-1580.
2. Friedman BW, Chilstrom M, Bijur PE, et al. Diagnostic testing and treatment of low back pain in United States emergency departments: a national perspective. Spine (Phila Pa 1976). 2010;35:E1406-E1411.
3. Friedman BW, O’Mahony S, Mulvey L, et al. One-week and 3-month outcomes after an emergency department visit for undifferentiated musculoskeletal low back pain. Ann Emerg Med. 2012;59:128-133.
4. Roelofs PD, Deyo RA, Koes BW, et al. Nonsteroidal anti-inflammatory drugs for low back pain: an updated Cochrane review. Spine (Phila Pa 1976). 2008;33:1766-1774.
5. van Tulder MW, Touray T, Furlan AD, et al. Muscle relaxants for nonspecific low back pain: a systematic review within the framework of the Cochrane collaboration. Spine (Phila Pa 1976). 2003;28:1978-1992.
6. Ashworth J, Green DJ, Dunn KM, et al. Opioid use among low back pain patients in primary care: is opioid prescription associated with disability at 6-month follow-up? Pain. 2013; 154:1038-1044.
7. Childers MK, Borenstein D, Brown RL, et al. Low-dose cyclobenzaprine versus combination therapy with ibuprofen for acute neck or back pain with muscle spasm: a randomized trial. Curr Med Res Opin. 2005;21:1485-1493.
8. Borenstein DG, Lacks S, Wiesel SW. Cyclobenzaprine and naproxen versus naproxen alone in the treatment of acute low back pain and muscle spasm. Clin Ther. 1990;12:125-131.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
Copyright © 2016. The Family Physicians Inquiries Network. All rights reserved.
Reprinted with permission from the Family Physicians Inquiries Network and The Journal of Family Practice. 2016;65(6):404-406.
PRACTICE CHANGER
Consider treating patients with acute low back pain with naproxen only, as adding cyclobenzaprine or oxycodone/acetaminophen to scheduled naproxen increases adverse effects and does not improve functional assessment at seven days or three months.
Strength of Recommendation
B: Based on a high-quality, randomized controlled trial (RCT).1
A 46-year-old man presents to the emergency department (ED) with low back pain (LBP) after helping a friend move a couch three days ago. He denies any direct trauma to his back and describes the pain as a “spasm” in his lumbar spinal region with no radicular symptoms. The pain worsens with prolonged standing and position changes. He has tried acetaminophen with no benefit. You diagnose a lumbar muscular strain. What medications should you prescribe to help relieve his LBP and improve his overall function?
Acute LBP prompts nearly 2.7 million ED visits in the United States each year.2 It leads to persistent subjective impairment and continued analgesic use at seven days (impairment, 70%; analgesic use, 69%) and three months (48% and 46%, respectively) after ED discharge.3 Systematic reviews show that monotherapy with NSAIDs or muscle relaxants is more effective than placebo for pain relief.4,5 A secondary analysis of patients (N = 715) from a prospective cohort study showed worse functioning at six months in those who were prescribed opiates for LBP than in those who were not.6
Monotherapy or combination therapy for LBP?
Because medications used for LBP have different mechanisms of action, clinicians frequently combine them in an attempt to improve symptoms and function.2 Current evidence on combination therapy shows mixed results. A large RCT (N = 867) showed that the combination of cyclobenzaprine and ibuprofen led to lower subjective pain intensity, but it did not result in self-reported pain improvement, compared to cyclobenzaprine alone. However, a small RCT (N = 40) demonstrated improved LBP and spasm with naprozen plus cyclobenzaprine, compared to naproxen alone.7,8
This study sought to determine the benefit of treating acute LBP with cyclobenzaprine or oxycodone/acetaminophen in combination with an NSAID, compared to treatment with an NSAID alone.
Continue for the study summary >>
STUDY SUMMARY
Adding second pain reliever provided no significant benefit
This double-blinded RCT enrolled 323 adults presenting to an ED with two weeks or less of nontraumatic, nonradicular LBP.1 Subjects had a score of > 5 on the Roland-Morris Disability Questionnaire (RMDQ), which measures functional impairment due to LBP (range, 0-24). Patients were excluded if they had radicular pain radiating below the gluteal folds, direct trauma to the back within the previous month, pain lasting > 2 wk, a recent history of multiple LBP episodes per month, or a history of opioid use.
All subjects received 10 days’ worth of naproxen (500 mg bid). They were then randomized to receive either oxycodone/acetaminophen (5 mg/325 mg), cyclobenzaprine (5 mg), or placebo, with instructions to take one to two tablets as needed every eight hours for 10 days. All patients also received a 10-minute educational session emphasizing the role of nonpharmacologic interventions.
The primary outcome was change in the RMDQ between ED discharge and a phone call seven days later; a 5-point improvement in the RMDQ was considered clinically significant. Secondary outcomes included subjective description of worst pain, frequency of LBP, frequency of analgesic use, satisfaction with treatment, median number of days to return to work and usual activities, need for follow-up health care visits, and opioid use. Investigators also asked about any adverse effects.
At seven days, reported RMDQ scores had improved by 9.8 points in patients taking naproxen plus placebo, 10.1 points in those receiving naproxen plus cyclobenzaprine, and 11.1 points in those using naproxen plus oxycodone/acetaminophen. There were no statistically significant between-group differences for placebo vs cyclobenzaprine or oxycodone/acetaminophen (0.3 points and 1.3 points, respectively) or cyclobenzaprine vs oxycodone/acetaminophen (0.9 points).
Secondary outcomes. At seven days, there was no significant difference between study groups in subjective pain assessment, frequency of LBP, or use of as-needed medications in the prior 24 hours. There was also no difference in the median number of days to return to work or need for follow-up health care visits.
Among patients who took more than one dose of the study medication, those who took oxycodone/acetaminophen were more likely to describe their worst pain in the last 24 hours as mild/none, compared to patients taking placebo (number needed to treat, 6). About 72% of all subjects reported that they would choose the same treatment option again, with no difference between groups. At three months, there was no difference between groups in subjective pain assessment, frequency of LBP, use of as-needed medications, or opioid use during the previous 72 hours.
Adverse effects, including drowsiness, dizziness, stomach irritation, and nausea or vomiting, were more common in the oxycodone/acetaminophen and the cyclobenzaprine treatment groups, with a number needed to harm of 5.3 and 7.8, respectively.
Continue for what's new >>
WHAT’S NEW
Second med adds nothing
This RCT found that adding cyclobenzaprine or oxycodone/acetaminophen to naproxen for the treatment of nontraumatic, nonradicular acute LBP did not significantly improve functional assessment at seven days or three months after the initial ED visit. But it did increase adverse effects.
CAVEATS
Specific subset studied
This study was performed in a single urban ED and included a very specific subset of LBP patients, which limits the generalizability of the results. However, patients often present to primary care with similar LBP complaints, and the results of the study should reasonably apply to other settings.
The findings may not generalize to all NSAIDs, but there is no evidence to suggest that other NSAIDs would behave differently when combined with cyclobenzaprine or oxycodone/acetaminophen. In this analysis, only about one-third of patients used the as-needed medication more than once daily; another third used it intermittently or never.
CHALLENGES TO IMPLEMENTATION
Patients may expect more
Patients expect to receive prescriptions, and clinicians are inclined to write them if they believe doing so will help their patients. The evidence, however, does not demonstrate a benefit to these prescription-only medications for LBP.
REFERENCES
1. Friedman BW, Dym AA, Davitt M, et al. Naproxen with cyclobenzaprine, oxycodone/acetaminophen, or placebo for treating acute low back pain: a randomized clinical trial. JAMA. 2015;314:1572-1580.
2. Friedman BW, Chilstrom M, Bijur PE, et al. Diagnostic testing and treatment of low back pain in United States emergency departments: a national perspective. Spine (Phila Pa 1976). 2010;35:E1406-E1411.
3. Friedman BW, O’Mahony S, Mulvey L, et al. One-week and 3-month outcomes after an emergency department visit for undifferentiated musculoskeletal low back pain. Ann Emerg Med. 2012;59:128-133.
4. Roelofs PD, Deyo RA, Koes BW, et al. Nonsteroidal anti-inflammatory drugs for low back pain: an updated Cochrane review. Spine (Phila Pa 1976). 2008;33:1766-1774.
5. van Tulder MW, Touray T, Furlan AD, et al. Muscle relaxants for nonspecific low back pain: a systematic review within the framework of the Cochrane collaboration. Spine (Phila Pa 1976). 2003;28:1978-1992.
6. Ashworth J, Green DJ, Dunn KM, et al. Opioid use among low back pain patients in primary care: is opioid prescription associated with disability at 6-month follow-up? Pain. 2013; 154:1038-1044.
7. Childers MK, Borenstein D, Brown RL, et al. Low-dose cyclobenzaprine versus combination therapy with ibuprofen for acute neck or back pain with muscle spasm: a randomized trial. Curr Med Res Opin. 2005;21:1485-1493.
8. Borenstein DG, Lacks S, Wiesel SW. Cyclobenzaprine and naproxen versus naproxen alone in the treatment of acute low back pain and muscle spasm. Clin Ther. 1990;12:125-131.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
Copyright © 2016. The Family Physicians Inquiries Network. All rights reserved.
Reprinted with permission from the Family Physicians Inquiries Network and The Journal of Family Practice. 2016;65(6):404-406.
PRACTICE CHANGER
Consider treating patients with acute low back pain with naproxen only, as adding cyclobenzaprine or oxycodone/acetaminophen to scheduled naproxen increases adverse effects and does not improve functional assessment at seven days or three months.
Strength of Recommendation
B: Based on a high-quality, randomized controlled trial (RCT).1
A 46-year-old man presents to the emergency department (ED) with low back pain (LBP) after helping a friend move a couch three days ago. He denies any direct trauma to his back and describes the pain as a “spasm” in his lumbar spinal region with no radicular symptoms. The pain worsens with prolonged standing and position changes. He has tried acetaminophen with no benefit. You diagnose a lumbar muscular strain. What medications should you prescribe to help relieve his LBP and improve his overall function?
Acute LBP prompts nearly 2.7 million ED visits in the United States each year.2 It leads to persistent subjective impairment and continued analgesic use at seven days (impairment, 70%; analgesic use, 69%) and three months (48% and 46%, respectively) after ED discharge.3 Systematic reviews show that monotherapy with NSAIDs or muscle relaxants is more effective than placebo for pain relief.4,5 A secondary analysis of patients (N = 715) from a prospective cohort study showed worse functioning at six months in those who were prescribed opiates for LBP than in those who were not.6
Monotherapy or combination therapy for LBP?
Because medications used for LBP have different mechanisms of action, clinicians frequently combine them in an attempt to improve symptoms and function.2 Current evidence on combination therapy shows mixed results. A large RCT (N = 867) showed that the combination of cyclobenzaprine and ibuprofen led to lower subjective pain intensity, but it did not result in self-reported pain improvement, compared to cyclobenzaprine alone. However, a small RCT (N = 40) demonstrated improved LBP and spasm with naprozen plus cyclobenzaprine, compared to naproxen alone.7,8
This study sought to determine the benefit of treating acute LBP with cyclobenzaprine or oxycodone/acetaminophen in combination with an NSAID, compared to treatment with an NSAID alone.
Continue for the study summary >>
STUDY SUMMARY
Adding second pain reliever provided no significant benefit
This double-blinded RCT enrolled 323 adults presenting to an ED with two weeks or less of nontraumatic, nonradicular LBP.1 Subjects had a score of > 5 on the Roland-Morris Disability Questionnaire (RMDQ), which measures functional impairment due to LBP (range, 0-24). Patients were excluded if they had radicular pain radiating below the gluteal folds, direct trauma to the back within the previous month, pain lasting > 2 wk, a recent history of multiple LBP episodes per month, or a history of opioid use.
All subjects received 10 days’ worth of naproxen (500 mg bid). They were then randomized to receive either oxycodone/acetaminophen (5 mg/325 mg), cyclobenzaprine (5 mg), or placebo, with instructions to take one to two tablets as needed every eight hours for 10 days. All patients also received a 10-minute educational session emphasizing the role of nonpharmacologic interventions.
The primary outcome was change in the RMDQ between ED discharge and a phone call seven days later; a 5-point improvement in the RMDQ was considered clinically significant. Secondary outcomes included subjective description of worst pain, frequency of LBP, frequency of analgesic use, satisfaction with treatment, median number of days to return to work and usual activities, need for follow-up health care visits, and opioid use. Investigators also asked about any adverse effects.
At seven days, reported RMDQ scores had improved by 9.8 points in patients taking naproxen plus placebo, 10.1 points in those receiving naproxen plus cyclobenzaprine, and 11.1 points in those using naproxen plus oxycodone/acetaminophen. There were no statistically significant between-group differences for placebo vs cyclobenzaprine or oxycodone/acetaminophen (0.3 points and 1.3 points, respectively) or cyclobenzaprine vs oxycodone/acetaminophen (0.9 points).
Secondary outcomes. At seven days, there was no significant difference between study groups in subjective pain assessment, frequency of LBP, or use of as-needed medications in the prior 24 hours. There was also no difference in the median number of days to return to work or need for follow-up health care visits.
Among patients who took more than one dose of the study medication, those who took oxycodone/acetaminophen were more likely to describe their worst pain in the last 24 hours as mild/none, compared to patients taking placebo (number needed to treat, 6). About 72% of all subjects reported that they would choose the same treatment option again, with no difference between groups. At three months, there was no difference between groups in subjective pain assessment, frequency of LBP, use of as-needed medications, or opioid use during the previous 72 hours.
Adverse effects, including drowsiness, dizziness, stomach irritation, and nausea or vomiting, were more common in the oxycodone/acetaminophen and the cyclobenzaprine treatment groups, with a number needed to harm of 5.3 and 7.8, respectively.
Continue for what's new >>
WHAT’S NEW
Second med adds nothing
This RCT found that adding cyclobenzaprine or oxycodone/acetaminophen to naproxen for the treatment of nontraumatic, nonradicular acute LBP did not significantly improve functional assessment at seven days or three months after the initial ED visit. But it did increase adverse effects.
CAVEATS
Specific subset studied
This study was performed in a single urban ED and included a very specific subset of LBP patients, which limits the generalizability of the results. However, patients often present to primary care with similar LBP complaints, and the results of the study should reasonably apply to other settings.
The findings may not generalize to all NSAIDs, but there is no evidence to suggest that other NSAIDs would behave differently when combined with cyclobenzaprine or oxycodone/acetaminophen. In this analysis, only about one-third of patients used the as-needed medication more than once daily; another third used it intermittently or never.
CHALLENGES TO IMPLEMENTATION
Patients may expect more
Patients expect to receive prescriptions, and clinicians are inclined to write them if they believe doing so will help their patients. The evidence, however, does not demonstrate a benefit to these prescription-only medications for LBP.
REFERENCES
1. Friedman BW, Dym AA, Davitt M, et al. Naproxen with cyclobenzaprine, oxycodone/acetaminophen, or placebo for treating acute low back pain: a randomized clinical trial. JAMA. 2015;314:1572-1580.
2. Friedman BW, Chilstrom M, Bijur PE, et al. Diagnostic testing and treatment of low back pain in United States emergency departments: a national perspective. Spine (Phila Pa 1976). 2010;35:E1406-E1411.
3. Friedman BW, O’Mahony S, Mulvey L, et al. One-week and 3-month outcomes after an emergency department visit for undifferentiated musculoskeletal low back pain. Ann Emerg Med. 2012;59:128-133.
4. Roelofs PD, Deyo RA, Koes BW, et al. Nonsteroidal anti-inflammatory drugs for low back pain: an updated Cochrane review. Spine (Phila Pa 1976). 2008;33:1766-1774.
5. van Tulder MW, Touray T, Furlan AD, et al. Muscle relaxants for nonspecific low back pain: a systematic review within the framework of the Cochrane collaboration. Spine (Phila Pa 1976). 2003;28:1978-1992.
6. Ashworth J, Green DJ, Dunn KM, et al. Opioid use among low back pain patients in primary care: is opioid prescription associated with disability at 6-month follow-up? Pain. 2013; 154:1038-1044.
7. Childers MK, Borenstein D, Brown RL, et al. Low-dose cyclobenzaprine versus combination therapy with ibuprofen for acute neck or back pain with muscle spasm: a randomized trial. Curr Med Res Opin. 2005;21:1485-1493.
8. Borenstein DG, Lacks S, Wiesel SW. Cyclobenzaprine and naproxen versus naproxen alone in the treatment of acute low back pain and muscle spasm. Clin Ther. 1990;12:125-131.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
Copyright © 2016. The Family Physicians Inquiries Network. All rights reserved.
Reprinted with permission from the Family Physicians Inquiries Network and The Journal of Family Practice. 2016;65(6):404-406.
More isn’t better with acute low back pain treatment
Consider treating patients with acute low back pain with naproxen only, as adding cyclobenzaprine or oxycodone/acetaminophen to scheduled naproxen does not improve functional assessment at 7 days or 3 months and increases adverse effects.
Strength of recommendation
B: Based on a high-quality, randomized controlled trial (RCT).1
Friedman BW, Dym AA, Davitt M, et al. Naproxen with cyclobenzaprine, oxycodone/acetaminophen, or placebo for treating acute low back pain: a randomized clinical trial. JAMA. 2015;314:1572-1580.
Illustrative Case
A 46-year-old man presents to the emergency department (ED) with low back pain (LBP) after helping a friend move a couch 3 days earlier. He denies any direct trauma to his back and describes the pain as a spasm in his lumbar spinal region with no radicular symptoms. The pain worsens with prolonged standing and any position changes. He has tried acetaminophen with no benefit. You diagnose a lumbar muscular strain. What medications should you prescribe to help relieve his LBP and improve his overall function?
Acute LBP prompts close to 2.7 million ED visits annually in the United States.2 It leads to persistent subjective impairment and continued analgesic usage at 7 days (impairment 70%, analgesic use 69%) and at 3 months (48% and 46%, respectively) after ED discharge.3 Systematic reviews show that monotherapy with nonsteroidal anti-inflammatory drugs (NSAIDs) or muscle relaxers is better than placebo for relieving pain.4,5 A secondary analysis of patients (N=715) from a prospective cohort study showed that patients prescribed opiates for LBP had worse functioning at 6 months than those not prescribed opiates.6
Monotherapy or combination therapy for LBP? That is the question
Because medications used for LBP have different mechanisms of action, clinicians frequently combine them in an attempt to improve symptoms and function.2 Current evidence evaluating combination therapy demonstrates mixed results. A large RCT (N=867) showed that the combination of cyclobenzaprine and ibuprofen led to lower subjective pain intensity, but did not result in self-reported pain improvement (based on answers to the Patient Global Impression of Change and the Oswestry Disability Index) than cyclobenzaprine alone. However, a small RCT (N=40) combining naproxen with cyclobenzaprine demonstrated improved LBP and spasm compared to naproxen alone.7,8
This study sought to determine the benefit of treating acute LBP with cyclobenzaprine or oxycodone/acetaminophen in combination with an NSAID compared to treatment with an NSAID alone.
Study Summary
Adding second pain reliever to the NSAID provided no significant benefit
This double-blinded RCT enrolled 323 adult patients presenting to an ED with ≤2 weeks of nontraumatic, nonradicular LBP, which was defined as pain between the lower border of the scapulae and the upper gluteal folds.1 Participants had a score of >5 on the Roland-Morris Disability Questionnaire (RMDQ), which measures functional impairment due to LBP (range: 0-24). Patients were excluded if they had radicular pain radiating below the gluteal folds, direct trauma to the back within the previous month, pain duration >2 weeks, or a recent history of >1 LBP episode per month. Patients with current or past chronic opioid use were also excluded.
All participants received 10 days’ worth of naproxen (500 mg twice daily). They were then randomized to receive either: oxycodone 5 mg/acetaminophen 325 mg; cyclobenzaprine 5 mg; or placebo, with instructions to take one to 2 tablets prn every 8 hours for 10 days. They were told that if one tablet afforded sufficient relief, there was no need to take the second one, but if the first tablet did not provide relief within 30 minutes, they should take the second one. All patients also received a 10-minute educational session emphasizing the role of exercise, stretching, physical/massage therapy, and other non-pharmacologic interventions.
The primary outcome was change in the RMDQ between ED discharge and a phone call 7 days later, with a 5-point improvement in the RMDQ considered clinically significant. Secondary outcomes at 7 days and 3 months after ED discharge included subjective description of worst pain, frequency of LBP pain, frequency of analgesic use, satisfaction with treatment, median number of days to return to work and usual activities, need for follow-up health care visits, and opioid use. Investigators also asked about any adverse effects at 7 days and 3 months.
At 7 days, patients randomized to naproxen plus placebo improved on reported RMDQ scores by a mean of 9.8 points, naproxen plus cyclobenzaprine by 10.1 points, and naproxen plus oxycodone/acetaminophen by 11.1 points. Between group differences in mean RMDQ changes showed no statistically significant differences with placebo vs cyclobenzaprine (0.3 points; P=.77), placebo vs oxycodone/acetaminophen (1.3 points; P=.28), and cyclobenzaprine vs oxycodone/acetaminophen (0.9 points; P=.45).
Secondary outcomes. At 7 days, there was no significant difference between study groups in subjective pain assessment, frequency of LBP, or use of as-needed medications in the prior 24 hours. There was also no difference in the median number of days to return to work or need for follow-up health care visits. In patients who took more than one dose of the study medication, those who took oxycodone/acetaminophen were more likely to describe their worst pain in the last 24 hours as mild/none when compared to those taking placebo (number needed to treat [NNT]=6). About 72% of all subjects reported that they would choose the same treatment option again, with no difference between groups. At 3 months, no difference existed between groups in subjective pain assessment, frequency of LBP, use of as-needed medications, or opioid use during the previous 72 hours.
Adverse effects, including drowsiness, dizziness, stomach irritation, and nausea or vomiting, were more common in the oxycodone/acetaminophen and cyclobenzaprine treatment groups with a number needed to harm (NNH) of 5.3 and 7.8, respectively.
What’s New
A second pain reliever adds nothing—except adverse effects
This RCT found that adding cyclobenzaprine or oxycodone/acetaminophen to naproxen for the treatment of nontraumatic, nonradicular acute LBP did not significantly improve functional assessment based on RMDQ scores or pain measures at 7 days or 3 months after the initial ED visit. It did, however, increase adverse effects.
Caveats
Researchers studied a specific subset of patients
This study was performed in a single-site urban ED and included a very specific subset of LBP patients, which limits the generalizability of the results. However, patients often present to their primary care physician with similar LBP complaints, and the results of the study should reasonably apply to other settings.
The findings may not generalize to all NSAIDs, but there is no evidence to suggest that other NSAIDs would behave differently when combined with cyclobenzaprine or oxycodone/acetaminophen. In this intention-to-treat analysis, only about one-third of patients used the as-needed medication more than once daily; about another third of patients used the as-needed medication intermittently or never.
Challenges to Implementation
Patients may expect more than an NSAID for their back pain
Patients expect to receive prescriptions, and physicians are inclined to write them if they believe they will help their patients. The evidence, however, does not show a benefit to these prescription-only medications for low back pain.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
1. Friedman BW, Dym AA, Davitt M, et al. Naproxen with cyclobenzaprine, oxycodone/acetaminophen, or placebo for treating acute low back pain: a randomized clinical trial. JAMA. 2015;314:1572-1580.
2. Friedman BW, Chilstrom M, Bijur PE, et al. Diagnostic testing and treatment of low back pain in United States emergency departments: a national perspective. Spine (Phila Pa 1976). 2010;35:E1406-E1411.
3. Friedman BW, O’Mahony S, Mulvey L, et al. One-week and 3-month outcomes after an emergency department visit for undifferentiated musculoskeletal low back pain. Ann Emerg Med. 2012;59:128-133.
4. Roelofs PD, Deyo RA, Koes BW, et al. Nonsteroidal anti-inflammatory drugs for low back pain: an updated Cochrane review. Spine (Phila Pa 1976). 2008;33:1766-1774.
5. van Tulder MW, Touray T, Furlan AD, et al. Muscle relaxants for nonspecific low back pain: a systematic review within the framework of the cochrane collaboration. Spine (Phila Pa 1976). 2003;28:1978-1992.
6. Ashworth J, Green DJ, Dunn KM, et al. Opioid use among low back pain patients in primary care: is opioid prescription associated with disability at 6-month follow-up? Pain. 2013;154:1038-1044.
7. Childers MK, Borenstein D, Brown RL, et al. Low-dose cyclobenzaprine versus combination therapy with ibuprofen for acute neck or back pain with muscle spasm: a randomized trial. Curr Med Res Opin. 2005;21:1485-1493.
8. Borenstein DG, Lacks S, Wiesel SW. Cyclobenzaprine and naproxen versus naproxen alone in the treatment of acute low back pain and muscle spasm. Clin Ther. 1990;12:125-131.
Consider treating patients with acute low back pain with naproxen only, as adding cyclobenzaprine or oxycodone/acetaminophen to scheduled naproxen does not improve functional assessment at 7 days or 3 months and increases adverse effects.
Strength of recommendation
B: Based on a high-quality, randomized controlled trial (RCT).1
Friedman BW, Dym AA, Davitt M, et al. Naproxen with cyclobenzaprine, oxycodone/acetaminophen, or placebo for treating acute low back pain: a randomized clinical trial. JAMA. 2015;314:1572-1580.
Illustrative Case
A 46-year-old man presents to the emergency department (ED) with low back pain (LBP) after helping a friend move a couch 3 days earlier. He denies any direct trauma to his back and describes the pain as a spasm in his lumbar spinal region with no radicular symptoms. The pain worsens with prolonged standing and any position changes. He has tried acetaminophen with no benefit. You diagnose a lumbar muscular strain. What medications should you prescribe to help relieve his LBP and improve his overall function?
Acute LBP prompts close to 2.7 million ED visits annually in the United States.2 It leads to persistent subjective impairment and continued analgesic usage at 7 days (impairment 70%, analgesic use 69%) and at 3 months (48% and 46%, respectively) after ED discharge.3 Systematic reviews show that monotherapy with nonsteroidal anti-inflammatory drugs (NSAIDs) or muscle relaxers is better than placebo for relieving pain.4,5 A secondary analysis of patients (N=715) from a prospective cohort study showed that patients prescribed opiates for LBP had worse functioning at 6 months than those not prescribed opiates.6
Monotherapy or combination therapy for LBP? That is the question
Because medications used for LBP have different mechanisms of action, clinicians frequently combine them in an attempt to improve symptoms and function.2 Current evidence evaluating combination therapy demonstrates mixed results. A large RCT (N=867) showed that the combination of cyclobenzaprine and ibuprofen led to lower subjective pain intensity, but did not result in self-reported pain improvement (based on answers to the Patient Global Impression of Change and the Oswestry Disability Index) than cyclobenzaprine alone. However, a small RCT (N=40) combining naproxen with cyclobenzaprine demonstrated improved LBP and spasm compared to naproxen alone.7,8
This study sought to determine the benefit of treating acute LBP with cyclobenzaprine or oxycodone/acetaminophen in combination with an NSAID compared to treatment with an NSAID alone.
Study Summary
Adding second pain reliever to the NSAID provided no significant benefit
This double-blinded RCT enrolled 323 adult patients presenting to an ED with ≤2 weeks of nontraumatic, nonradicular LBP, which was defined as pain between the lower border of the scapulae and the upper gluteal folds.1 Participants had a score of >5 on the Roland-Morris Disability Questionnaire (RMDQ), which measures functional impairment due to LBP (range: 0-24). Patients were excluded if they had radicular pain radiating below the gluteal folds, direct trauma to the back within the previous month, pain duration >2 weeks, or a recent history of >1 LBP episode per month. Patients with current or past chronic opioid use were also excluded.
All participants received 10 days’ worth of naproxen (500 mg twice daily). They were then randomized to receive either: oxycodone 5 mg/acetaminophen 325 mg; cyclobenzaprine 5 mg; or placebo, with instructions to take one to 2 tablets prn every 8 hours for 10 days. They were told that if one tablet afforded sufficient relief, there was no need to take the second one, but if the first tablet did not provide relief within 30 minutes, they should take the second one. All patients also received a 10-minute educational session emphasizing the role of exercise, stretching, physical/massage therapy, and other non-pharmacologic interventions.
The primary outcome was change in the RMDQ between ED discharge and a phone call 7 days later, with a 5-point improvement in the RMDQ considered clinically significant. Secondary outcomes at 7 days and 3 months after ED discharge included subjective description of worst pain, frequency of LBP pain, frequency of analgesic use, satisfaction with treatment, median number of days to return to work and usual activities, need for follow-up health care visits, and opioid use. Investigators also asked about any adverse effects at 7 days and 3 months.
At 7 days, patients randomized to naproxen plus placebo improved on reported RMDQ scores by a mean of 9.8 points, naproxen plus cyclobenzaprine by 10.1 points, and naproxen plus oxycodone/acetaminophen by 11.1 points. Between group differences in mean RMDQ changes showed no statistically significant differences with placebo vs cyclobenzaprine (0.3 points; P=.77), placebo vs oxycodone/acetaminophen (1.3 points; P=.28), and cyclobenzaprine vs oxycodone/acetaminophen (0.9 points; P=.45).
Secondary outcomes. At 7 days, there was no significant difference between study groups in subjective pain assessment, frequency of LBP, or use of as-needed medications in the prior 24 hours. There was also no difference in the median number of days to return to work or need for follow-up health care visits. In patients who took more than one dose of the study medication, those who took oxycodone/acetaminophen were more likely to describe their worst pain in the last 24 hours as mild/none when compared to those taking placebo (number needed to treat [NNT]=6). About 72% of all subjects reported that they would choose the same treatment option again, with no difference between groups. At 3 months, no difference existed between groups in subjective pain assessment, frequency of LBP, use of as-needed medications, or opioid use during the previous 72 hours.
Adverse effects, including drowsiness, dizziness, stomach irritation, and nausea or vomiting, were more common in the oxycodone/acetaminophen and cyclobenzaprine treatment groups with a number needed to harm (NNH) of 5.3 and 7.8, respectively.
What’s New
A second pain reliever adds nothing—except adverse effects
This RCT found that adding cyclobenzaprine or oxycodone/acetaminophen to naproxen for the treatment of nontraumatic, nonradicular acute LBP did not significantly improve functional assessment based on RMDQ scores or pain measures at 7 days or 3 months after the initial ED visit. It did, however, increase adverse effects.
Caveats
Researchers studied a specific subset of patients
This study was performed in a single-site urban ED and included a very specific subset of LBP patients, which limits the generalizability of the results. However, patients often present to their primary care physician with similar LBP complaints, and the results of the study should reasonably apply to other settings.
The findings may not generalize to all NSAIDs, but there is no evidence to suggest that other NSAIDs would behave differently when combined with cyclobenzaprine or oxycodone/acetaminophen. In this intention-to-treat analysis, only about one-third of patients used the as-needed medication more than once daily; about another third of patients used the as-needed medication intermittently or never.
Challenges to Implementation
Patients may expect more than an NSAID for their back pain
Patients expect to receive prescriptions, and physicians are inclined to write them if they believe they will help their patients. The evidence, however, does not show a benefit to these prescription-only medications for low back pain.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
Consider treating patients with acute low back pain with naproxen only, as adding cyclobenzaprine or oxycodone/acetaminophen to scheduled naproxen does not improve functional assessment at 7 days or 3 months and increases adverse effects.
Strength of recommendation
B: Based on a high-quality, randomized controlled trial (RCT).1
Friedman BW, Dym AA, Davitt M, et al. Naproxen with cyclobenzaprine, oxycodone/acetaminophen, or placebo for treating acute low back pain: a randomized clinical trial. JAMA. 2015;314:1572-1580.
Illustrative Case
A 46-year-old man presents to the emergency department (ED) with low back pain (LBP) after helping a friend move a couch 3 days earlier. He denies any direct trauma to his back and describes the pain as a spasm in his lumbar spinal region with no radicular symptoms. The pain worsens with prolonged standing and any position changes. He has tried acetaminophen with no benefit. You diagnose a lumbar muscular strain. What medications should you prescribe to help relieve his LBP and improve his overall function?
Acute LBP prompts close to 2.7 million ED visits annually in the United States.2 It leads to persistent subjective impairment and continued analgesic usage at 7 days (impairment 70%, analgesic use 69%) and at 3 months (48% and 46%, respectively) after ED discharge.3 Systematic reviews show that monotherapy with nonsteroidal anti-inflammatory drugs (NSAIDs) or muscle relaxers is better than placebo for relieving pain.4,5 A secondary analysis of patients (N=715) from a prospective cohort study showed that patients prescribed opiates for LBP had worse functioning at 6 months than those not prescribed opiates.6
Monotherapy or combination therapy for LBP? That is the question
Because medications used for LBP have different mechanisms of action, clinicians frequently combine them in an attempt to improve symptoms and function.2 Current evidence evaluating combination therapy demonstrates mixed results. A large RCT (N=867) showed that the combination of cyclobenzaprine and ibuprofen led to lower subjective pain intensity, but did not result in self-reported pain improvement (based on answers to the Patient Global Impression of Change and the Oswestry Disability Index) than cyclobenzaprine alone. However, a small RCT (N=40) combining naproxen with cyclobenzaprine demonstrated improved LBP and spasm compared to naproxen alone.7,8
This study sought to determine the benefit of treating acute LBP with cyclobenzaprine or oxycodone/acetaminophen in combination with an NSAID compared to treatment with an NSAID alone.
Study Summary
Adding second pain reliever to the NSAID provided no significant benefit
This double-blinded RCT enrolled 323 adult patients presenting to an ED with ≤2 weeks of nontraumatic, nonradicular LBP, which was defined as pain between the lower border of the scapulae and the upper gluteal folds.1 Participants had a score of >5 on the Roland-Morris Disability Questionnaire (RMDQ), which measures functional impairment due to LBP (range: 0-24). Patients were excluded if they had radicular pain radiating below the gluteal folds, direct trauma to the back within the previous month, pain duration >2 weeks, or a recent history of >1 LBP episode per month. Patients with current or past chronic opioid use were also excluded.
All participants received 10 days’ worth of naproxen (500 mg twice daily). They were then randomized to receive either: oxycodone 5 mg/acetaminophen 325 mg; cyclobenzaprine 5 mg; or placebo, with instructions to take one to 2 tablets prn every 8 hours for 10 days. They were told that if one tablet afforded sufficient relief, there was no need to take the second one, but if the first tablet did not provide relief within 30 minutes, they should take the second one. All patients also received a 10-minute educational session emphasizing the role of exercise, stretching, physical/massage therapy, and other non-pharmacologic interventions.
The primary outcome was change in the RMDQ between ED discharge and a phone call 7 days later, with a 5-point improvement in the RMDQ considered clinically significant. Secondary outcomes at 7 days and 3 months after ED discharge included subjective description of worst pain, frequency of LBP pain, frequency of analgesic use, satisfaction with treatment, median number of days to return to work and usual activities, need for follow-up health care visits, and opioid use. Investigators also asked about any adverse effects at 7 days and 3 months.
At 7 days, patients randomized to naproxen plus placebo improved on reported RMDQ scores by a mean of 9.8 points, naproxen plus cyclobenzaprine by 10.1 points, and naproxen plus oxycodone/acetaminophen by 11.1 points. Between group differences in mean RMDQ changes showed no statistically significant differences with placebo vs cyclobenzaprine (0.3 points; P=.77), placebo vs oxycodone/acetaminophen (1.3 points; P=.28), and cyclobenzaprine vs oxycodone/acetaminophen (0.9 points; P=.45).
Secondary outcomes. At 7 days, there was no significant difference between study groups in subjective pain assessment, frequency of LBP, or use of as-needed medications in the prior 24 hours. There was also no difference in the median number of days to return to work or need for follow-up health care visits. In patients who took more than one dose of the study medication, those who took oxycodone/acetaminophen were more likely to describe their worst pain in the last 24 hours as mild/none when compared to those taking placebo (number needed to treat [NNT]=6). About 72% of all subjects reported that they would choose the same treatment option again, with no difference between groups. At 3 months, no difference existed between groups in subjective pain assessment, frequency of LBP, use of as-needed medications, or opioid use during the previous 72 hours.
Adverse effects, including drowsiness, dizziness, stomach irritation, and nausea or vomiting, were more common in the oxycodone/acetaminophen and cyclobenzaprine treatment groups with a number needed to harm (NNH) of 5.3 and 7.8, respectively.
What’s New
A second pain reliever adds nothing—except adverse effects
This RCT found that adding cyclobenzaprine or oxycodone/acetaminophen to naproxen for the treatment of nontraumatic, nonradicular acute LBP did not significantly improve functional assessment based on RMDQ scores or pain measures at 7 days or 3 months after the initial ED visit. It did, however, increase adverse effects.
Caveats
Researchers studied a specific subset of patients
This study was performed in a single-site urban ED and included a very specific subset of LBP patients, which limits the generalizability of the results. However, patients often present to their primary care physician with similar LBP complaints, and the results of the study should reasonably apply to other settings.
The findings may not generalize to all NSAIDs, but there is no evidence to suggest that other NSAIDs would behave differently when combined with cyclobenzaprine or oxycodone/acetaminophen. In this intention-to-treat analysis, only about one-third of patients used the as-needed medication more than once daily; about another third of patients used the as-needed medication intermittently or never.
Challenges to Implementation
Patients may expect more than an NSAID for their back pain
Patients expect to receive prescriptions, and physicians are inclined to write them if they believe they will help their patients. The evidence, however, does not show a benefit to these prescription-only medications for low back pain.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
1. Friedman BW, Dym AA, Davitt M, et al. Naproxen with cyclobenzaprine, oxycodone/acetaminophen, or placebo for treating acute low back pain: a randomized clinical trial. JAMA. 2015;314:1572-1580.
2. Friedman BW, Chilstrom M, Bijur PE, et al. Diagnostic testing and treatment of low back pain in United States emergency departments: a national perspective. Spine (Phila Pa 1976). 2010;35:E1406-E1411.
3. Friedman BW, O’Mahony S, Mulvey L, et al. One-week and 3-month outcomes after an emergency department visit for undifferentiated musculoskeletal low back pain. Ann Emerg Med. 2012;59:128-133.
4. Roelofs PD, Deyo RA, Koes BW, et al. Nonsteroidal anti-inflammatory drugs for low back pain: an updated Cochrane review. Spine (Phila Pa 1976). 2008;33:1766-1774.
5. van Tulder MW, Touray T, Furlan AD, et al. Muscle relaxants for nonspecific low back pain: a systematic review within the framework of the cochrane collaboration. Spine (Phila Pa 1976). 2003;28:1978-1992.
6. Ashworth J, Green DJ, Dunn KM, et al. Opioid use among low back pain patients in primary care: is opioid prescription associated with disability at 6-month follow-up? Pain. 2013;154:1038-1044.
7. Childers MK, Borenstein D, Brown RL, et al. Low-dose cyclobenzaprine versus combination therapy with ibuprofen for acute neck or back pain with muscle spasm: a randomized trial. Curr Med Res Opin. 2005;21:1485-1493.
8. Borenstein DG, Lacks S, Wiesel SW. Cyclobenzaprine and naproxen versus naproxen alone in the treatment of acute low back pain and muscle spasm. Clin Ther. 1990;12:125-131.
1. Friedman BW, Dym AA, Davitt M, et al. Naproxen with cyclobenzaprine, oxycodone/acetaminophen, or placebo for treating acute low back pain: a randomized clinical trial. JAMA. 2015;314:1572-1580.
2. Friedman BW, Chilstrom M, Bijur PE, et al. Diagnostic testing and treatment of low back pain in United States emergency departments: a national perspective. Spine (Phila Pa 1976). 2010;35:E1406-E1411.
3. Friedman BW, O’Mahony S, Mulvey L, et al. One-week and 3-month outcomes after an emergency department visit for undifferentiated musculoskeletal low back pain. Ann Emerg Med. 2012;59:128-133.
4. Roelofs PD, Deyo RA, Koes BW, et al. Nonsteroidal anti-inflammatory drugs for low back pain: an updated Cochrane review. Spine (Phila Pa 1976). 2008;33:1766-1774.
5. van Tulder MW, Touray T, Furlan AD, et al. Muscle relaxants for nonspecific low back pain: a systematic review within the framework of the cochrane collaboration. Spine (Phila Pa 1976). 2003;28:1978-1992.
6. Ashworth J, Green DJ, Dunn KM, et al. Opioid use among low back pain patients in primary care: is opioid prescription associated with disability at 6-month follow-up? Pain. 2013;154:1038-1044.
7. Childers MK, Borenstein D, Brown RL, et al. Low-dose cyclobenzaprine versus combination therapy with ibuprofen for acute neck or back pain with muscle spasm: a randomized trial. Curr Med Res Opin. 2005;21:1485-1493.
8. Borenstein DG, Lacks S, Wiesel SW. Cyclobenzaprine and naproxen versus naproxen alone in the treatment of acute low back pain and muscle spasm. Clin Ther. 1990;12:125-131.
Copyright © 2016. The Family Physicians Inquiries Network. All rights reserved.
“Go Low” or “Say No” to Aggressive Systolic BP Goals?
PRACTICE CHANGER
Consider treating nondiabetic patients ages 50 and older to a systolic blood pressure (SBP) target < 120 mm Hg (as compared to < 140 mm Hg) when the benefits—lower rates of fatal and nonfatal cardiovascular (CV) events and death from any cause—are likely to outweigh the risks from possible additional medication.1
Strength of Recommendation
B: Based on a single, good-quality randomized controlled trial (RCT). 1
A 55-year-old man with hypertension and stage 3 chronic kidney disease (CKD) presents for routine care. His blood pressure is 135/85 mm Hg, and he is currently taking lisinopril 40 mg/d. Should you increase his antihypertensive regimen?
Hypertension is common and leads to significant morbidity and mortality, but pharmacologic treatment reduces incidence of stroke by 35% to 40%, myocardial infarction (MI) by 15% to 25%, and heart failure by up to 64%.2-4 Specific blood pressure targets for defined populations continue to be studied.
The ACCORD (Action to Control Cardiovascular Risk in Diabetes) trial found that more intensive BP targets did not reduce the rate of major CV events in patients with diabetes, but the study may have been underpowered.5 The members of the Eighth Joint National Committee (JNC 8) recommended treating patients older than 60 to BP goals < 150/90 mm Hg.6 This was based on evidence from six RCTs, but there remains debate—even among the JNC 8 committee members—as to appropriate BP goals in patients of any age without CV disease who have BP measurements of 140-159/90-99 mm Hg. 7-13
Continue for the study summary >>
STUDY SUMMARY
Treating to SBP < 120 mm Hg lowers mortality
The Systolic Blood Pressure Intervention Trial (SPRINT) was a multicenter RCT designed to determine if treating to lower SBP targets in nondiabetic patients at high risk for CV events improves outcomes, compared with standard care. Patients were at least 50, had an SBP of 130 to 180 mm Hg, and were at increased CV risk; the last was defined as clinical or subclinical CV disease other than stroke; CKD with a glomerular filtration rate (GFR) of 20 to 60 mL/min/1.73 m2; 10-year risk for CV disease > 15% on Framingham risk score; or age 75 or older. Patients with diabetes, prior stroke, polycystic kidney disease, significant proteinuria or symptomatic heart failure within the past six months, or left ventricular ejection fraction < 35% were excluded.1
Patients (N = 9,361) were randomly assigned to an SBP target < 120 mm Hg in the intensive group or < 140 mm Hg in the standard treatment group, in an open-label design. Allocation was concealed. The study protocol encouraged, but did not require, the use of thiazide-type diuretics, loop diuretics (for those with advanced renal disease), ACE inhibitors or angiotensin receptor blockers, calcium channel blockers, and ß-blockers. Clinicians could add other agents as needed. All major classes of antihypertensives were used.
Medication dosing adjustments were based on the average of three BP measurements taken with an automated measurement system with the patient seated after 5 minutes of quiet rest. Target SBP in the standard therapy group was 135 to 139 mm Hg. Medication dosages were lowered if SBP was < 130 mm Hg at a single visit or < 135 mm Hg at two consecutive visits.1
The primary composite outcome included the first occurrence of MI, acute coronary syndrome, stroke, heart failure, or death from CV causes. Secondary outcomes were the individual components of the primary composite outcome; death from any cause; and the composite of the primary outcome or death from any cause.1
Study halted early. The study was stopped early due to significantly lower rates of the primary outcome in the intensive therapy group versus the standard therapy group (1.65% vs 2.19% per year, respectively; hazard ratio [HR], 0.75 with intensive treatment). The resulting median follow-up time was 3.26 years.1 This corresponds to a 25% lower relative risk for the primary outcome, with a decrease in event rates from 6.8% to 5.2% over the trial period. All-cause mortality was also lower in the intensive therapy group: 3.4% vs 4.5% (HR, 0.73).
The number needed to treat (NNT) over 3.26 years to prevent a primary outcome event, death from any cause, and death from CV causes was 61, 90, and 172, respectively. Serious adverse events occurred more frequently in the intensive therapy group than in the standard therapy group (38.3% vs 37.1%; HR, 1.04), with a number needed to harm (NNH) of 46 over the study period.1
Rates of serious adverse events that were identified as likely associated with the intervention were 4.7% vs 2.5%, respectively. Hypotension, syncope, electrolyte abnormalities, and acute kidney injury/acute renal failure reached statistical significance. The incidence of bradycardia and injurious falls, although higher in the intensive treatment group, did not reach statistical significance. In the subgroup of patients 75 or older, 48% in each study group experienced a serious adverse event.1
Throughout the study, mean SBP was 121.5 mm Hg in the intensive therapy group and 134.6 mm Hg in the standard treatment group. Patients in the intensive therapy group required, on average, one additional BP medication, compared to those in the standard treatment group (2.8 vs 1.8, respectively).1
Continue for what's new >>
WHAT’S NEW
Lower SBP produces mortality benefits in those younger, and older, than 75
This trial builds on a body of evidence that shows the advantages of lowering SBP to < 150 mm Hg7,11,12 by demonstrating benefits, including reduced all-cause mortality, for lower SBP targets in nondiabetic patients at high risk for CV disease. The SPRINT trial also showed that the benefits of intensive therapy remained true in a subgroup of patients 75 or older.
The incidence of the primary outcome in the cohort 75 or older receiving intensive therapy was 7.7%, compared with 10.9% for those receiving standard therapy (HR, 0.67; NNT, 31). All-cause mortality was also lower in the intensive therapy group than in the standard therapy group among patients 75 or older: 5.5% vs 8.04% (HR, 0.68; NNT, 38).1
CAVEATS
Many do not benefit from—or are harmed by—increased medication
The absolute risk reduction for the primary outcome is 1.6%, meaning 98.4% of patients receiving more intensive treatment will not benefit. In a group of 1,000 patients, an estimated 16 patients will benefit, 22 patients will be seriously harmed, and 962 patients will experience neither benefit nor harm.14 The difference between how BP was measured in this trial (an average of three readings after the patient had rested for 5 minutes) and what occurs typically in clinical practice could potentially lead to overtreatment in a “real world” setting.
Also, reducing antihypertensive therapies when the SBP was about 130 to 135 mm Hg in the standard therapy group likely exaggerated the difference in outcomes between the intensive and standard therapy groups; this is neither routine nor recommended in clinical practice.6 Finally, the trial specifically studied nondiabetic patients at high risk for CV disease who were 50 or older, limiting generalizability to other populations.
CHALLENGES TO IMPLEMENTATION
Who will benefit/who can achieve intensive SBP goals?
Identifying patients most likely to benefit from more intensive BP targets remains challenging. The SPRINT trial showed a mortality benefit, but at a cost of increased morbidity.1,14 Caution should be exercised particularly in the subgroup of patients 75 or older. Despite a lower NNT than the rest of the study population, this group experienced serious adverse events more frequently. Also, this particular cohort of volunteers may not be representative of those 75 or older in the general population.
Additionally, achieving intensive SBP goals can be challenging. In the SPRINT trial, only half of the intensive target group achieved an SBP < 120 mm Hg.1 And in a 2011-2012 National Health and Nutrition Examination Survey, only 52% of patients in the general population achieved a BP target < 140/90 mm Hg.15 Lower morbidity and mortality should remain the ultimate goals in the management of hypertension, requiring clinicians to carefully assess an individual patient’s likelihood of benefit versus harm.
REFERENCES
1. Wright JT Jr, Williamson JD, Whelton PK, et al. A randomized trial of intensive versus standard blood-pressure control. N Engl J Med. 2015;373:2103-2116.
2. Chobanian AV, Bakris GL, Black HR, et al. The seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. JAMA. 2003;289:2560-2572.
3. Neal B, MacMahon S, Chapman N. Effects of ACE inhibitors, calcium antagonists, and other blood-pressure-lowering drugs: results of prospectively designed overviews of randomised trials.Lancet. 2000;356:1955-1964.
4. Psaty BM, Smith NL, Siscovick DS, et al. Health outcomes associated with antihypertensive therapies used as first-line agents: a systematic review and meta-analysis. JAMA. 1997;277:739-745.
5. Margolis KL, O’Connor PJ, Morgan TM, et al. Outcomes of combined cardiovascular risk factor management strategies in type 2 diabetes: the ACCORD randomized trial. Diabetes Care. 2014;37:1721-1728.
6. James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8).JAMA. 2014;311:507-520.
7. Beckett NS, Peters R, Fletcher AE, et al. Treatment of hypertension in patients 80 years of age or older.N Engl J Med. 2008;358:1887-1898.
8. Verdecchia P, Staessen JA, Angeli F, et al. Usual versus tight control of systolic blood pressure in non-diabetic patients with hypertension (Cardio-Sis): an open-label randomised trial. Lancet. 2009;374:525-533.
9. JATOS Study Group. Principal results of the Japanese trial to assess optimal systolic blood pressure in elderly hypertensive patients (JATOS). Hypertens Res. 2008;31:2115-2127.
10. Ogihara T, Saruta T, Rakugi H, et al. Target blood pressure for treatment of isolated systolic hypertension in the elderly: valsartan in elderly isolated systolic hypertension study. Hypertension. 2010;56:196-202.
11. Staessen JA, Fagard R, Thijs L, et al; the Systolic Hypertension in Europe (Syst-Eur) Trial Investigators. Randomised double-blind comparison of placebo and active treatment for older patients with isolated systolic hypertension.Lancet. 1997;350:757-764.
12. SHEP Cooperative Research Group. Prevention of stroke by antihypertensive drug treatment in older persons with isolated systolic hypertension: final results of the Systolic Hypertension in the Elderly Program (SHEP). JAMA. 1991;265:3255-3264.
13. Cundiff DK, Gueyffier F, Wright JM. Guidelines for managing high blood pressure. JAMA. 2014;312:294.
14. Ortiz E, James PA. Let’s not SPRINT to judgment about new blood pressure goals. Ann Intern Med. 2016 Feb 23. [Epub ahead of print]
15. Nwankwo T, Yoon SS, Burt V, et al. Hypertension among adults in the United States: National Health and Nutrition Examination Survey, 2011-2012. NCHS Data Brief. 2013;1-8.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
Copyright © 2016. The Family Physicians Inquiries Network. All rights reserved.
Reprinted with permission from the Family Physicians Inquiries Network and The Journal of Family Practice. 2016;65(5):342-344.
PRACTICE CHANGER
Consider treating nondiabetic patients ages 50 and older to a systolic blood pressure (SBP) target < 120 mm Hg (as compared to < 140 mm Hg) when the benefits—lower rates of fatal and nonfatal cardiovascular (CV) events and death from any cause—are likely to outweigh the risks from possible additional medication.1
Strength of Recommendation
B: Based on a single, good-quality randomized controlled trial (RCT). 1
A 55-year-old man with hypertension and stage 3 chronic kidney disease (CKD) presents for routine care. His blood pressure is 135/85 mm Hg, and he is currently taking lisinopril 40 mg/d. Should you increase his antihypertensive regimen?
Hypertension is common and leads to significant morbidity and mortality, but pharmacologic treatment reduces incidence of stroke by 35% to 40%, myocardial infarction (MI) by 15% to 25%, and heart failure by up to 64%.2-4 Specific blood pressure targets for defined populations continue to be studied.
The ACCORD (Action to Control Cardiovascular Risk in Diabetes) trial found that more intensive BP targets did not reduce the rate of major CV events in patients with diabetes, but the study may have been underpowered.5 The members of the Eighth Joint National Committee (JNC 8) recommended treating patients older than 60 to BP goals < 150/90 mm Hg.6 This was based on evidence from six RCTs, but there remains debate—even among the JNC 8 committee members—as to appropriate BP goals in patients of any age without CV disease who have BP measurements of 140-159/90-99 mm Hg. 7-13
Continue for the study summary >>
STUDY SUMMARY
Treating to SBP < 120 mm Hg lowers mortality
The Systolic Blood Pressure Intervention Trial (SPRINT) was a multicenter RCT designed to determine if treating to lower SBP targets in nondiabetic patients at high risk for CV events improves outcomes, compared with standard care. Patients were at least 50, had an SBP of 130 to 180 mm Hg, and were at increased CV risk; the last was defined as clinical or subclinical CV disease other than stroke; CKD with a glomerular filtration rate (GFR) of 20 to 60 mL/min/1.73 m2; 10-year risk for CV disease > 15% on Framingham risk score; or age 75 or older. Patients with diabetes, prior stroke, polycystic kidney disease, significant proteinuria or symptomatic heart failure within the past six months, or left ventricular ejection fraction < 35% were excluded.1
Patients (N = 9,361) were randomly assigned to an SBP target < 120 mm Hg in the intensive group or < 140 mm Hg in the standard treatment group, in an open-label design. Allocation was concealed. The study protocol encouraged, but did not require, the use of thiazide-type diuretics, loop diuretics (for those with advanced renal disease), ACE inhibitors or angiotensin receptor blockers, calcium channel blockers, and ß-blockers. Clinicians could add other agents as needed. All major classes of antihypertensives were used.
Medication dosing adjustments were based on the average of three BP measurements taken with an automated measurement system with the patient seated after 5 minutes of quiet rest. Target SBP in the standard therapy group was 135 to 139 mm Hg. Medication dosages were lowered if SBP was < 130 mm Hg at a single visit or < 135 mm Hg at two consecutive visits.1
The primary composite outcome included the first occurrence of MI, acute coronary syndrome, stroke, heart failure, or death from CV causes. Secondary outcomes were the individual components of the primary composite outcome; death from any cause; and the composite of the primary outcome or death from any cause.1
Study halted early. The study was stopped early due to significantly lower rates of the primary outcome in the intensive therapy group versus the standard therapy group (1.65% vs 2.19% per year, respectively; hazard ratio [HR], 0.75 with intensive treatment). The resulting median follow-up time was 3.26 years.1 This corresponds to a 25% lower relative risk for the primary outcome, with a decrease in event rates from 6.8% to 5.2% over the trial period. All-cause mortality was also lower in the intensive therapy group: 3.4% vs 4.5% (HR, 0.73).
The number needed to treat (NNT) over 3.26 years to prevent a primary outcome event, death from any cause, and death from CV causes was 61, 90, and 172, respectively. Serious adverse events occurred more frequently in the intensive therapy group than in the standard therapy group (38.3% vs 37.1%; HR, 1.04), with a number needed to harm (NNH) of 46 over the study period.1
Rates of serious adverse events that were identified as likely associated with the intervention were 4.7% vs 2.5%, respectively. Hypotension, syncope, electrolyte abnormalities, and acute kidney injury/acute renal failure reached statistical significance. The incidence of bradycardia and injurious falls, although higher in the intensive treatment group, did not reach statistical significance. In the subgroup of patients 75 or older, 48% in each study group experienced a serious adverse event.1
Throughout the study, mean SBP was 121.5 mm Hg in the intensive therapy group and 134.6 mm Hg in the standard treatment group. Patients in the intensive therapy group required, on average, one additional BP medication, compared to those in the standard treatment group (2.8 vs 1.8, respectively).1
Continue for what's new >>
WHAT’S NEW
Lower SBP produces mortality benefits in those younger, and older, than 75
This trial builds on a body of evidence that shows the advantages of lowering SBP to < 150 mm Hg7,11,12 by demonstrating benefits, including reduced all-cause mortality, for lower SBP targets in nondiabetic patients at high risk for CV disease. The SPRINT trial also showed that the benefits of intensive therapy remained true in a subgroup of patients 75 or older.
The incidence of the primary outcome in the cohort 75 or older receiving intensive therapy was 7.7%, compared with 10.9% for those receiving standard therapy (HR, 0.67; NNT, 31). All-cause mortality was also lower in the intensive therapy group than in the standard therapy group among patients 75 or older: 5.5% vs 8.04% (HR, 0.68; NNT, 38).1
CAVEATS
Many do not benefit from—or are harmed by—increased medication
The absolute risk reduction for the primary outcome is 1.6%, meaning 98.4% of patients receiving more intensive treatment will not benefit. In a group of 1,000 patients, an estimated 16 patients will benefit, 22 patients will be seriously harmed, and 962 patients will experience neither benefit nor harm.14 The difference between how BP was measured in this trial (an average of three readings after the patient had rested for 5 minutes) and what occurs typically in clinical practice could potentially lead to overtreatment in a “real world” setting.
Also, reducing antihypertensive therapies when the SBP was about 130 to 135 mm Hg in the standard therapy group likely exaggerated the difference in outcomes between the intensive and standard therapy groups; this is neither routine nor recommended in clinical practice.6 Finally, the trial specifically studied nondiabetic patients at high risk for CV disease who were 50 or older, limiting generalizability to other populations.
CHALLENGES TO IMPLEMENTATION
Who will benefit/who can achieve intensive SBP goals?
Identifying patients most likely to benefit from more intensive BP targets remains challenging. The SPRINT trial showed a mortality benefit, but at a cost of increased morbidity.1,14 Caution should be exercised particularly in the subgroup of patients 75 or older. Despite a lower NNT than the rest of the study population, this group experienced serious adverse events more frequently. Also, this particular cohort of volunteers may not be representative of those 75 or older in the general population.
Additionally, achieving intensive SBP goals can be challenging. In the SPRINT trial, only half of the intensive target group achieved an SBP < 120 mm Hg.1 And in a 2011-2012 National Health and Nutrition Examination Survey, only 52% of patients in the general population achieved a BP target < 140/90 mm Hg.15 Lower morbidity and mortality should remain the ultimate goals in the management of hypertension, requiring clinicians to carefully assess an individual patient’s likelihood of benefit versus harm.
REFERENCES
1. Wright JT Jr, Williamson JD, Whelton PK, et al. A randomized trial of intensive versus standard blood-pressure control. N Engl J Med. 2015;373:2103-2116.
2. Chobanian AV, Bakris GL, Black HR, et al. The seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. JAMA. 2003;289:2560-2572.
3. Neal B, MacMahon S, Chapman N. Effects of ACE inhibitors, calcium antagonists, and other blood-pressure-lowering drugs: results of prospectively designed overviews of randomised trials.Lancet. 2000;356:1955-1964.
4. Psaty BM, Smith NL, Siscovick DS, et al. Health outcomes associated with antihypertensive therapies used as first-line agents: a systematic review and meta-analysis. JAMA. 1997;277:739-745.
5. Margolis KL, O’Connor PJ, Morgan TM, et al. Outcomes of combined cardiovascular risk factor management strategies in type 2 diabetes: the ACCORD randomized trial. Diabetes Care. 2014;37:1721-1728.
6. James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8).JAMA. 2014;311:507-520.
7. Beckett NS, Peters R, Fletcher AE, et al. Treatment of hypertension in patients 80 years of age or older.N Engl J Med. 2008;358:1887-1898.
8. Verdecchia P, Staessen JA, Angeli F, et al. Usual versus tight control of systolic blood pressure in non-diabetic patients with hypertension (Cardio-Sis): an open-label randomised trial. Lancet. 2009;374:525-533.
9. JATOS Study Group. Principal results of the Japanese trial to assess optimal systolic blood pressure in elderly hypertensive patients (JATOS). Hypertens Res. 2008;31:2115-2127.
10. Ogihara T, Saruta T, Rakugi H, et al. Target blood pressure for treatment of isolated systolic hypertension in the elderly: valsartan in elderly isolated systolic hypertension study. Hypertension. 2010;56:196-202.
11. Staessen JA, Fagard R, Thijs L, et al; the Systolic Hypertension in Europe (Syst-Eur) Trial Investigators. Randomised double-blind comparison of placebo and active treatment for older patients with isolated systolic hypertension.Lancet. 1997;350:757-764.
12. SHEP Cooperative Research Group. Prevention of stroke by antihypertensive drug treatment in older persons with isolated systolic hypertension: final results of the Systolic Hypertension in the Elderly Program (SHEP). JAMA. 1991;265:3255-3264.
13. Cundiff DK, Gueyffier F, Wright JM. Guidelines for managing high blood pressure. JAMA. 2014;312:294.
14. Ortiz E, James PA. Let’s not SPRINT to judgment about new blood pressure goals. Ann Intern Med. 2016 Feb 23. [Epub ahead of print]
15. Nwankwo T, Yoon SS, Burt V, et al. Hypertension among adults in the United States: National Health and Nutrition Examination Survey, 2011-2012. NCHS Data Brief. 2013;1-8.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
Copyright © 2016. The Family Physicians Inquiries Network. All rights reserved.
Reprinted with permission from the Family Physicians Inquiries Network and The Journal of Family Practice. 2016;65(5):342-344.
PRACTICE CHANGER
Consider treating nondiabetic patients ages 50 and older to a systolic blood pressure (SBP) target < 120 mm Hg (as compared to < 140 mm Hg) when the benefits—lower rates of fatal and nonfatal cardiovascular (CV) events and death from any cause—are likely to outweigh the risks from possible additional medication.1
Strength of Recommendation
B: Based on a single, good-quality randomized controlled trial (RCT). 1
A 55-year-old man with hypertension and stage 3 chronic kidney disease (CKD) presents for routine care. His blood pressure is 135/85 mm Hg, and he is currently taking lisinopril 40 mg/d. Should you increase his antihypertensive regimen?
Hypertension is common and leads to significant morbidity and mortality, but pharmacologic treatment reduces incidence of stroke by 35% to 40%, myocardial infarction (MI) by 15% to 25%, and heart failure by up to 64%.2-4 Specific blood pressure targets for defined populations continue to be studied.
The ACCORD (Action to Control Cardiovascular Risk in Diabetes) trial found that more intensive BP targets did not reduce the rate of major CV events in patients with diabetes, but the study may have been underpowered.5 The members of the Eighth Joint National Committee (JNC 8) recommended treating patients older than 60 to BP goals < 150/90 mm Hg.6 This was based on evidence from six RCTs, but there remains debate—even among the JNC 8 committee members—as to appropriate BP goals in patients of any age without CV disease who have BP measurements of 140-159/90-99 mm Hg. 7-13
Continue for the study summary >>
STUDY SUMMARY
Treating to SBP < 120 mm Hg lowers mortality
The Systolic Blood Pressure Intervention Trial (SPRINT) was a multicenter RCT designed to determine if treating to lower SBP targets in nondiabetic patients at high risk for CV events improves outcomes, compared with standard care. Patients were at least 50, had an SBP of 130 to 180 mm Hg, and were at increased CV risk; the last was defined as clinical or subclinical CV disease other than stroke; CKD with a glomerular filtration rate (GFR) of 20 to 60 mL/min/1.73 m2; 10-year risk for CV disease > 15% on Framingham risk score; or age 75 or older. Patients with diabetes, prior stroke, polycystic kidney disease, significant proteinuria or symptomatic heart failure within the past six months, or left ventricular ejection fraction < 35% were excluded.1
Patients (N = 9,361) were randomly assigned to an SBP target < 120 mm Hg in the intensive group or < 140 mm Hg in the standard treatment group, in an open-label design. Allocation was concealed. The study protocol encouraged, but did not require, the use of thiazide-type diuretics, loop diuretics (for those with advanced renal disease), ACE inhibitors or angiotensin receptor blockers, calcium channel blockers, and ß-blockers. Clinicians could add other agents as needed. All major classes of antihypertensives were used.
Medication dosing adjustments were based on the average of three BP measurements taken with an automated measurement system with the patient seated after 5 minutes of quiet rest. Target SBP in the standard therapy group was 135 to 139 mm Hg. Medication dosages were lowered if SBP was < 130 mm Hg at a single visit or < 135 mm Hg at two consecutive visits.1
The primary composite outcome included the first occurrence of MI, acute coronary syndrome, stroke, heart failure, or death from CV causes. Secondary outcomes were the individual components of the primary composite outcome; death from any cause; and the composite of the primary outcome or death from any cause.1
Study halted early. The study was stopped early due to significantly lower rates of the primary outcome in the intensive therapy group versus the standard therapy group (1.65% vs 2.19% per year, respectively; hazard ratio [HR], 0.75 with intensive treatment). The resulting median follow-up time was 3.26 years.1 This corresponds to a 25% lower relative risk for the primary outcome, with a decrease in event rates from 6.8% to 5.2% over the trial period. All-cause mortality was also lower in the intensive therapy group: 3.4% vs 4.5% (HR, 0.73).
The number needed to treat (NNT) over 3.26 years to prevent a primary outcome event, death from any cause, and death from CV causes was 61, 90, and 172, respectively. Serious adverse events occurred more frequently in the intensive therapy group than in the standard therapy group (38.3% vs 37.1%; HR, 1.04), with a number needed to harm (NNH) of 46 over the study period.1
Rates of serious adverse events that were identified as likely associated with the intervention were 4.7% vs 2.5%, respectively. Hypotension, syncope, electrolyte abnormalities, and acute kidney injury/acute renal failure reached statistical significance. The incidence of bradycardia and injurious falls, although higher in the intensive treatment group, did not reach statistical significance. In the subgroup of patients 75 or older, 48% in each study group experienced a serious adverse event.1
Throughout the study, mean SBP was 121.5 mm Hg in the intensive therapy group and 134.6 mm Hg in the standard treatment group. Patients in the intensive therapy group required, on average, one additional BP medication, compared to those in the standard treatment group (2.8 vs 1.8, respectively).1
Continue for what's new >>
WHAT’S NEW
Lower SBP produces mortality benefits in those younger, and older, than 75
This trial builds on a body of evidence that shows the advantages of lowering SBP to < 150 mm Hg7,11,12 by demonstrating benefits, including reduced all-cause mortality, for lower SBP targets in nondiabetic patients at high risk for CV disease. The SPRINT trial also showed that the benefits of intensive therapy remained true in a subgroup of patients 75 or older.
The incidence of the primary outcome in the cohort 75 or older receiving intensive therapy was 7.7%, compared with 10.9% for those receiving standard therapy (HR, 0.67; NNT, 31). All-cause mortality was also lower in the intensive therapy group than in the standard therapy group among patients 75 or older: 5.5% vs 8.04% (HR, 0.68; NNT, 38).1
CAVEATS
Many do not benefit from—or are harmed by—increased medication
The absolute risk reduction for the primary outcome is 1.6%, meaning 98.4% of patients receiving more intensive treatment will not benefit. In a group of 1,000 patients, an estimated 16 patients will benefit, 22 patients will be seriously harmed, and 962 patients will experience neither benefit nor harm.14 The difference between how BP was measured in this trial (an average of three readings after the patient had rested for 5 minutes) and what occurs typically in clinical practice could potentially lead to overtreatment in a “real world” setting.
Also, reducing antihypertensive therapies when the SBP was about 130 to 135 mm Hg in the standard therapy group likely exaggerated the difference in outcomes between the intensive and standard therapy groups; this is neither routine nor recommended in clinical practice.6 Finally, the trial specifically studied nondiabetic patients at high risk for CV disease who were 50 or older, limiting generalizability to other populations.
CHALLENGES TO IMPLEMENTATION
Who will benefit/who can achieve intensive SBP goals?
Identifying patients most likely to benefit from more intensive BP targets remains challenging. The SPRINT trial showed a mortality benefit, but at a cost of increased morbidity.1,14 Caution should be exercised particularly in the subgroup of patients 75 or older. Despite a lower NNT than the rest of the study population, this group experienced serious adverse events more frequently. Also, this particular cohort of volunteers may not be representative of those 75 or older in the general population.
Additionally, achieving intensive SBP goals can be challenging. In the SPRINT trial, only half of the intensive target group achieved an SBP < 120 mm Hg.1 And in a 2011-2012 National Health and Nutrition Examination Survey, only 52% of patients in the general population achieved a BP target < 140/90 mm Hg.15 Lower morbidity and mortality should remain the ultimate goals in the management of hypertension, requiring clinicians to carefully assess an individual patient’s likelihood of benefit versus harm.
REFERENCES
1. Wright JT Jr, Williamson JD, Whelton PK, et al. A randomized trial of intensive versus standard blood-pressure control. N Engl J Med. 2015;373:2103-2116.
2. Chobanian AV, Bakris GL, Black HR, et al. The seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. JAMA. 2003;289:2560-2572.
3. Neal B, MacMahon S, Chapman N. Effects of ACE inhibitors, calcium antagonists, and other blood-pressure-lowering drugs: results of prospectively designed overviews of randomised trials.Lancet. 2000;356:1955-1964.
4. Psaty BM, Smith NL, Siscovick DS, et al. Health outcomes associated with antihypertensive therapies used as first-line agents: a systematic review and meta-analysis. JAMA. 1997;277:739-745.
5. Margolis KL, O’Connor PJ, Morgan TM, et al. Outcomes of combined cardiovascular risk factor management strategies in type 2 diabetes: the ACCORD randomized trial. Diabetes Care. 2014;37:1721-1728.
6. James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8).JAMA. 2014;311:507-520.
7. Beckett NS, Peters R, Fletcher AE, et al. Treatment of hypertension in patients 80 years of age or older.N Engl J Med. 2008;358:1887-1898.
8. Verdecchia P, Staessen JA, Angeli F, et al. Usual versus tight control of systolic blood pressure in non-diabetic patients with hypertension (Cardio-Sis): an open-label randomised trial. Lancet. 2009;374:525-533.
9. JATOS Study Group. Principal results of the Japanese trial to assess optimal systolic blood pressure in elderly hypertensive patients (JATOS). Hypertens Res. 2008;31:2115-2127.
10. Ogihara T, Saruta T, Rakugi H, et al. Target blood pressure for treatment of isolated systolic hypertension in the elderly: valsartan in elderly isolated systolic hypertension study. Hypertension. 2010;56:196-202.
11. Staessen JA, Fagard R, Thijs L, et al; the Systolic Hypertension in Europe (Syst-Eur) Trial Investigators. Randomised double-blind comparison of placebo and active treatment for older patients with isolated systolic hypertension.Lancet. 1997;350:757-764.
12. SHEP Cooperative Research Group. Prevention of stroke by antihypertensive drug treatment in older persons with isolated systolic hypertension: final results of the Systolic Hypertension in the Elderly Program (SHEP). JAMA. 1991;265:3255-3264.
13. Cundiff DK, Gueyffier F, Wright JM. Guidelines for managing high blood pressure. JAMA. 2014;312:294.
14. Ortiz E, James PA. Let’s not SPRINT to judgment about new blood pressure goals. Ann Intern Med. 2016 Feb 23. [Epub ahead of print]
15. Nwankwo T, Yoon SS, Burt V, et al. Hypertension among adults in the United States: National Health and Nutrition Examination Survey, 2011-2012. NCHS Data Brief. 2013;1-8.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
Copyright © 2016. The Family Physicians Inquiries Network. All rights reserved.
Reprinted with permission from the Family Physicians Inquiries Network and The Journal of Family Practice. 2016;65(5):342-344.
Resistant Hypertension? Time to Consider This Fourth-line Drug
PRACTICE CHANGER
When a triple regimen (ACE inhibitor or ARB, calcium channel blocker, and thiazide diuretic) fails to achieve the target blood pressure, try adding spironolactone.
Strength of recommendation
C: Based on a high-quality disease-oriented randomized controlled trial.1
Willie S, a 56-year-old man with chronic essential hypertension, has been on an optimally dosed three-drug regimen of an ACE inhibitor, a calcium channel blocker, and a thiazide diuretic for more than three months, but his blood pressure is still not at goal. What is the best antihypertensive agent to add to his regimen?
About 5% to 30% of those being treated for hypertension have resistant hypertension, defined as inadequate blood pressure (BP) control despite a triple regimen of an ACE inhibitor or angiotensin receptor blocker (ARB), calcium channel blocker (CCB), and thiazide diuretic.1,2Guidelines from the Eighth Joint National Committee (JNC-8) on the management of high BP recommend ß-blockers, α-blockers, or aldosterone antagonists (AAs) as equivalent choices for a fourth-line agent. The recommendation is based on expert opinion.3
Earlier hypertension guidelines from the UK’s National Institute for Health and Care Excellence recommend an AA if BP targets have not been met with the triple regimen. But this recommendation is based on lower-quality evidence, without comparison to ß-blockers, α-blockers, or other drug classes.4
More evidence since guideline’s release
A 2015 meta-analysis of 15 studies and a total of more than 1,200 participants (three randomized controlled trials [RCTs], one non-randomized placebo-controlled comparative trial, and 11 single-arm observational studies) demonstrated the effectiveness of the AAs spironolactone and eplerenone on resistant hypertension.5In the four comparative studies, AAs decreased office systolic blood pressure (SBP) by 24.3 mm Hg and diastolic blood pressure (DBP) by 7.8 mm Hg more than placebo. In the 11 single-arm studies, AAs reduced SBP by 22.74 mm Hg and DBP by 10.49 mm Hg.
Another RCT examined the effect of low-dose (25-mg) spironolactone, compared with placebo, in 161 patients with resistant hypertension.6At eight weeks, 73% of those receiving spironolactone reached a goal SBP < 140 mm Hg versus 41% of patients on placebo. The same proportion (73%) achieved a goal DBP < 90 mm Hg in the spironolactone group, compared with 63% of those in the placebo group. Ambulatory BP was also found to be significantly improved among those receiving spironolactone versus placebo, with a decrease in SBP of 9.8 mm Hg and in DSP of 3.2 mm Hg.6
Continue for the study summary >>
STUDY SUMMARY
Spironolactone vs other drugs
The placebo-controlled crossover RCT conducted in the UK by Williams et al was the first to directly compare spironolactone with other medications for the treatment of resistant hypertension in adults already taking triple therapy.1The trial randomized 335 individuals with a mean age of 61.4 (range, 18 to 79), 69% of whom were male; 314 were included in the intention-to-treat analysis.1
Enrollment criteria for resistant hypertension specified a clinic-recorded SBP of ≥ 140 mm Hg (or ≥ 135 mm Hg in those with diabetes) and home SBP (in 18 readings over four days) of ≥ 130 mm Hg.1 To ensure fidelity to treatment protocols, the investigators directly observed therapy, took tablet counts, measured serum ACE activity, and assessed BP measurement technique, with all participants adhering to a minimum of three months on a maximally dosed triple regimen.
Among subjects, 14% had diabetes and 7.8% reported tobacco use. Average weight was 93.5 kg (205.7 lbs).1 Because of the expected inverse relationship between plasma renin and response to AAs, plasma renin was measured at baseline to test whether resistant hypertension was primarily due to sodium retention.1
Four 12-week rotations
All participants began the trial with four weeks of placebo, followed by randomization to 12-week rotations of once-daily oral treatment with (1) spironolactone 25 to 50 mg, (2) doxazosin modified release 4 to 8 mg, (3) bisoprolol 5 to 10 mg, and (4) placebo.1 Six weeks after initiation of each study medication, participants were titrated to the higher dose. There was no washout period between cycles.
The primary outcome was mean SBP measured at home on four consecutive days prior to the study visits in weeks 6 and 12. Participants were required to have at least six BP measurements per each six-week period in order to establish a valid average. Primary endpoints included the difference in home SBP between spironolactone and placebo, the difference in home SBP between spironolactone and the mean of the other two drugs, and the difference in home SBP between spironolactone and each of the other two drugs.
The results. Spironolactone lowered SBP more than placebo, doxazosin, and bisoprolol (see the Table).1 Clinic measurements were consistent with home BP readings.
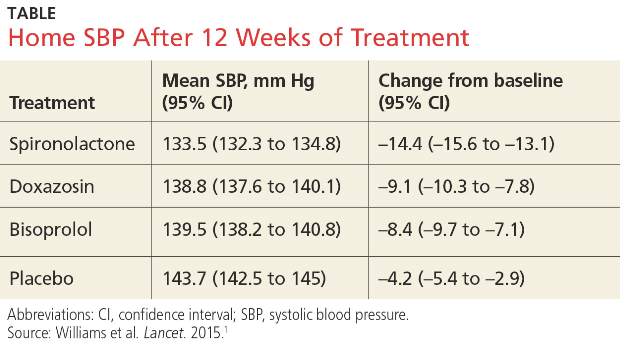
Overall, 58% of participants achieved goal SBP < 135 mm Hg on spironolactone, compared with 42% on doxazosin, 44% on bisoprolol, and 24% on placebo.1 The effectiveness of spironolactone on SBP reduction was shown to exhibit an inverse relationship to plasma renin levels, a finding that was not apparent with the other two study drugs. However, spironolactone had a superior BP-lowering effect throughout nearly the entire renin distribution of the cohort.
The mean difference between spironolactone and placebo was –10.2 mm Hg; compared with the other drugs, spironolactone lowered SBP, on average, by 5.64 mm Hg more than bisoprolol and doxazosin; 5.3 mm Hg more than doxazosin alone; and 5.98 mm Hg more than bisoprolol alone.
Only 1% of trial participants had to discontinue spironolactone due to adverse events—the same proportion of withdrawals as that for bisoprolol and placebo and three times less than for doxazosin.1
Continue for what's new >>
WHAT’S NEW
Evidence of superiority
This is the first RCT to compare spironolactone with two other commonly used fourth-line antihypertensives—bisoprolol and doxazosin—in patients with resistant hypertension. The study demonstrated clear superiority of spironolactone in achieving carefully measured ambulatory and clinic-recorded BP targets versus a ß-blocker or an α-blocker.
CAVEATS
Findings not universal
Spironolactone is contraindicated in patients with severe renal impairment. Although multiple drug trials have demonstrated the medication’s safety and effectiveness, especially in patients with resistant hypertension, we should factor in the need for monitoring electrolytes and renal function within weeks of treatment initiation and periodically thereafter.7,8 In this study, spironolactone increased potassium levels, on average, by 0.45 mmol/L. No gynecomastia (typically seen in about 6% of men) was found in those taking spironolactone for a 12-week cycle.1
This single trial enrolled mostly Caucasian men with a mean age of 61. Although smaller observational studies that included African-American patients have shown promising results for spironolactone, the question of external validity or applicability to a diverse population has yet to be decisively answered.9
CHALLENGES TO IMPLEMENTATION
Potential for adverse reactions
The evidence supporting this change in practice has been accumulating for the past few years. However, clinicians who treat patients with resistant hypertension may have concerns about hyperkalemia, gynecomastia, and effects on renal function. More patient-oriented evidence is likewise needed to assist with the revision of guidelines and wider adoption of AAs by primary care providers.
References
1. Williams B, MacDonald TM, Morant S, et al. Spironolactone versus placebo, bisoprolol, and doxazosin to determine the optimal treatment for drug-resistant hypertension (PATHWAY-2): a randomised, double-blind, crossover trial. Lancet. 2015;386:2059-2068.
2. Rosa J, Widimsky P, Tousek P, et al. Randomized comparison of renal denervation versus intensified pharmacotherapy including spironolactone in true-resistant hypertension: six-month results from the Prague-15 Study. Hypertension. 2015;65:407-413.
3. James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults. JAMA. 2014;311:507-520.
4. National Institute for Health and Care Excellence. Hypertension in adults: diagnosis and management (Clinical Guideline CG127). August 2011. https://www.nice.org.uk/guidance/cg127. Accessed March 4, 2016.
5. Dahal K, Kunwar S, Rijal J, et al. The effects of aldosterone antagonists in patients with resistant hypertension: a meta-analysis of randomized and nonrandomized studies. Am J Hypertens. 2015;28:1376-1385.
6. Václavík J, Sedlák R, Jarkovský J, et al. Effect of spironolactone in resistant arterial hypertension: a randomized, double-blind, placebo-controlled trial (ASPIRANT-EXT). Medicine (Baltimore). 2014;93:e162.
7. Wei L, Struthers AD, Fahey T, et al. Spironolactone use and renal toxicity: population based longitudinal analysis. BMJ. 2010;340:c1768.
8. Oxlund CS, Henriksen JE, Tarnow L, et al. Low dose spironolactone reduces blood pressure in patients with resistant hypertension and type 2 diabetes mellitus. J Hypertens. 2013;31:2094-2102.
9. Nishizaka M, Zaman MA, Calhoun DA. Efficacy of low-dose spironolactone in subjects with resistant hypertension. Am J Hypertens. 2003;16:925-930.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
Copyright © 2016. The Family Physicians Inquiries Network. All rights reserved.
Reprinted with permission from the Family Physicians Inquiries Network and The Journal of Family Practice. 2016;65(4):266-268.
PRACTICE CHANGER
When a triple regimen (ACE inhibitor or ARB, calcium channel blocker, and thiazide diuretic) fails to achieve the target blood pressure, try adding spironolactone.
Strength of recommendation
C: Based on a high-quality disease-oriented randomized controlled trial.1
Willie S, a 56-year-old man with chronic essential hypertension, has been on an optimally dosed three-drug regimen of an ACE inhibitor, a calcium channel blocker, and a thiazide diuretic for more than three months, but his blood pressure is still not at goal. What is the best antihypertensive agent to add to his regimen?
About 5% to 30% of those being treated for hypertension have resistant hypertension, defined as inadequate blood pressure (BP) control despite a triple regimen of an ACE inhibitor or angiotensin receptor blocker (ARB), calcium channel blocker (CCB), and thiazide diuretic.1,2Guidelines from the Eighth Joint National Committee (JNC-8) on the management of high BP recommend ß-blockers, α-blockers, or aldosterone antagonists (AAs) as equivalent choices for a fourth-line agent. The recommendation is based on expert opinion.3
Earlier hypertension guidelines from the UK’s National Institute for Health and Care Excellence recommend an AA if BP targets have not been met with the triple regimen. But this recommendation is based on lower-quality evidence, without comparison to ß-blockers, α-blockers, or other drug classes.4
More evidence since guideline’s release
A 2015 meta-analysis of 15 studies and a total of more than 1,200 participants (three randomized controlled trials [RCTs], one non-randomized placebo-controlled comparative trial, and 11 single-arm observational studies) demonstrated the effectiveness of the AAs spironolactone and eplerenone on resistant hypertension.5In the four comparative studies, AAs decreased office systolic blood pressure (SBP) by 24.3 mm Hg and diastolic blood pressure (DBP) by 7.8 mm Hg more than placebo. In the 11 single-arm studies, AAs reduced SBP by 22.74 mm Hg and DBP by 10.49 mm Hg.
Another RCT examined the effect of low-dose (25-mg) spironolactone, compared with placebo, in 161 patients with resistant hypertension.6At eight weeks, 73% of those receiving spironolactone reached a goal SBP < 140 mm Hg versus 41% of patients on placebo. The same proportion (73%) achieved a goal DBP < 90 mm Hg in the spironolactone group, compared with 63% of those in the placebo group. Ambulatory BP was also found to be significantly improved among those receiving spironolactone versus placebo, with a decrease in SBP of 9.8 mm Hg and in DSP of 3.2 mm Hg.6
Continue for the study summary >>
STUDY SUMMARY
Spironolactone vs other drugs
The placebo-controlled crossover RCT conducted in the UK by Williams et al was the first to directly compare spironolactone with other medications for the treatment of resistant hypertension in adults already taking triple therapy.1The trial randomized 335 individuals with a mean age of 61.4 (range, 18 to 79), 69% of whom were male; 314 were included in the intention-to-treat analysis.1
Enrollment criteria for resistant hypertension specified a clinic-recorded SBP of ≥ 140 mm Hg (or ≥ 135 mm Hg in those with diabetes) and home SBP (in 18 readings over four days) of ≥ 130 mm Hg.1 To ensure fidelity to treatment protocols, the investigators directly observed therapy, took tablet counts, measured serum ACE activity, and assessed BP measurement technique, with all participants adhering to a minimum of three months on a maximally dosed triple regimen.
Among subjects, 14% had diabetes and 7.8% reported tobacco use. Average weight was 93.5 kg (205.7 lbs).1 Because of the expected inverse relationship between plasma renin and response to AAs, plasma renin was measured at baseline to test whether resistant hypertension was primarily due to sodium retention.1
Four 12-week rotations
All participants began the trial with four weeks of placebo, followed by randomization to 12-week rotations of once-daily oral treatment with (1) spironolactone 25 to 50 mg, (2) doxazosin modified release 4 to 8 mg, (3) bisoprolol 5 to 10 mg, and (4) placebo.1 Six weeks after initiation of each study medication, participants were titrated to the higher dose. There was no washout period between cycles.
The primary outcome was mean SBP measured at home on four consecutive days prior to the study visits in weeks 6 and 12. Participants were required to have at least six BP measurements per each six-week period in order to establish a valid average. Primary endpoints included the difference in home SBP between spironolactone and placebo, the difference in home SBP between spironolactone and the mean of the other two drugs, and the difference in home SBP between spironolactone and each of the other two drugs.
The results. Spironolactone lowered SBP more than placebo, doxazosin, and bisoprolol (see the Table).1 Clinic measurements were consistent with home BP readings.
Overall, 58% of participants achieved goal SBP < 135 mm Hg on spironolactone, compared with 42% on doxazosin, 44% on bisoprolol, and 24% on placebo.1 The effectiveness of spironolactone on SBP reduction was shown to exhibit an inverse relationship to plasma renin levels, a finding that was not apparent with the other two study drugs. However, spironolactone had a superior BP-lowering effect throughout nearly the entire renin distribution of the cohort.
The mean difference between spironolactone and placebo was –10.2 mm Hg; compared with the other drugs, spironolactone lowered SBP, on average, by 5.64 mm Hg more than bisoprolol and doxazosin; 5.3 mm Hg more than doxazosin alone; and 5.98 mm Hg more than bisoprolol alone.
Only 1% of trial participants had to discontinue spironolactone due to adverse events—the same proportion of withdrawals as that for bisoprolol and placebo and three times less than for doxazosin.1
Continue for what's new >>
WHAT’S NEW
Evidence of superiority
This is the first RCT to compare spironolactone with two other commonly used fourth-line antihypertensives—bisoprolol and doxazosin—in patients with resistant hypertension. The study demonstrated clear superiority of spironolactone in achieving carefully measured ambulatory and clinic-recorded BP targets versus a ß-blocker or an α-blocker.
CAVEATS
Findings not universal
Spironolactone is contraindicated in patients with severe renal impairment. Although multiple drug trials have demonstrated the medication’s safety and effectiveness, especially in patients with resistant hypertension, we should factor in the need for monitoring electrolytes and renal function within weeks of treatment initiation and periodically thereafter.7,8 In this study, spironolactone increased potassium levels, on average, by 0.45 mmol/L. No gynecomastia (typically seen in about 6% of men) was found in those taking spironolactone for a 12-week cycle.1
This single trial enrolled mostly Caucasian men with a mean age of 61. Although smaller observational studies that included African-American patients have shown promising results for spironolactone, the question of external validity or applicability to a diverse population has yet to be decisively answered.9
CHALLENGES TO IMPLEMENTATION
Potential for adverse reactions
The evidence supporting this change in practice has been accumulating for the past few years. However, clinicians who treat patients with resistant hypertension may have concerns about hyperkalemia, gynecomastia, and effects on renal function. More patient-oriented evidence is likewise needed to assist with the revision of guidelines and wider adoption of AAs by primary care providers.
References
1. Williams B, MacDonald TM, Morant S, et al. Spironolactone versus placebo, bisoprolol, and doxazosin to determine the optimal treatment for drug-resistant hypertension (PATHWAY-2): a randomised, double-blind, crossover trial. Lancet. 2015;386:2059-2068.
2. Rosa J, Widimsky P, Tousek P, et al. Randomized comparison of renal denervation versus intensified pharmacotherapy including spironolactone in true-resistant hypertension: six-month results from the Prague-15 Study. Hypertension. 2015;65:407-413.
3. James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults. JAMA. 2014;311:507-520.
4. National Institute for Health and Care Excellence. Hypertension in adults: diagnosis and management (Clinical Guideline CG127). August 2011. https://www.nice.org.uk/guidance/cg127. Accessed March 4, 2016.
5. Dahal K, Kunwar S, Rijal J, et al. The effects of aldosterone antagonists in patients with resistant hypertension: a meta-analysis of randomized and nonrandomized studies. Am J Hypertens. 2015;28:1376-1385.
6. Václavík J, Sedlák R, Jarkovský J, et al. Effect of spironolactone in resistant arterial hypertension: a randomized, double-blind, placebo-controlled trial (ASPIRANT-EXT). Medicine (Baltimore). 2014;93:e162.
7. Wei L, Struthers AD, Fahey T, et al. Spironolactone use and renal toxicity: population based longitudinal analysis. BMJ. 2010;340:c1768.
8. Oxlund CS, Henriksen JE, Tarnow L, et al. Low dose spironolactone reduces blood pressure in patients with resistant hypertension and type 2 diabetes mellitus. J Hypertens. 2013;31:2094-2102.
9. Nishizaka M, Zaman MA, Calhoun DA. Efficacy of low-dose spironolactone in subjects with resistant hypertension. Am J Hypertens. 2003;16:925-930.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
Copyright © 2016. The Family Physicians Inquiries Network. All rights reserved.
Reprinted with permission from the Family Physicians Inquiries Network and The Journal of Family Practice. 2016;65(4):266-268.
PRACTICE CHANGER
When a triple regimen (ACE inhibitor or ARB, calcium channel blocker, and thiazide diuretic) fails to achieve the target blood pressure, try adding spironolactone.
Strength of recommendation
C: Based on a high-quality disease-oriented randomized controlled trial.1
Willie S, a 56-year-old man with chronic essential hypertension, has been on an optimally dosed three-drug regimen of an ACE inhibitor, a calcium channel blocker, and a thiazide diuretic for more than three months, but his blood pressure is still not at goal. What is the best antihypertensive agent to add to his regimen?
About 5% to 30% of those being treated for hypertension have resistant hypertension, defined as inadequate blood pressure (BP) control despite a triple regimen of an ACE inhibitor or angiotensin receptor blocker (ARB), calcium channel blocker (CCB), and thiazide diuretic.1,2Guidelines from the Eighth Joint National Committee (JNC-8) on the management of high BP recommend ß-blockers, α-blockers, or aldosterone antagonists (AAs) as equivalent choices for a fourth-line agent. The recommendation is based on expert opinion.3
Earlier hypertension guidelines from the UK’s National Institute for Health and Care Excellence recommend an AA if BP targets have not been met with the triple regimen. But this recommendation is based on lower-quality evidence, without comparison to ß-blockers, α-blockers, or other drug classes.4
More evidence since guideline’s release
A 2015 meta-analysis of 15 studies and a total of more than 1,200 participants (three randomized controlled trials [RCTs], one non-randomized placebo-controlled comparative trial, and 11 single-arm observational studies) demonstrated the effectiveness of the AAs spironolactone and eplerenone on resistant hypertension.5In the four comparative studies, AAs decreased office systolic blood pressure (SBP) by 24.3 mm Hg and diastolic blood pressure (DBP) by 7.8 mm Hg more than placebo. In the 11 single-arm studies, AAs reduced SBP by 22.74 mm Hg and DBP by 10.49 mm Hg.
Another RCT examined the effect of low-dose (25-mg) spironolactone, compared with placebo, in 161 patients with resistant hypertension.6At eight weeks, 73% of those receiving spironolactone reached a goal SBP < 140 mm Hg versus 41% of patients on placebo. The same proportion (73%) achieved a goal DBP < 90 mm Hg in the spironolactone group, compared with 63% of those in the placebo group. Ambulatory BP was also found to be significantly improved among those receiving spironolactone versus placebo, with a decrease in SBP of 9.8 mm Hg and in DSP of 3.2 mm Hg.6
Continue for the study summary >>
STUDY SUMMARY
Spironolactone vs other drugs
The placebo-controlled crossover RCT conducted in the UK by Williams et al was the first to directly compare spironolactone with other medications for the treatment of resistant hypertension in adults already taking triple therapy.1The trial randomized 335 individuals with a mean age of 61.4 (range, 18 to 79), 69% of whom were male; 314 were included in the intention-to-treat analysis.1
Enrollment criteria for resistant hypertension specified a clinic-recorded SBP of ≥ 140 mm Hg (or ≥ 135 mm Hg in those with diabetes) and home SBP (in 18 readings over four days) of ≥ 130 mm Hg.1 To ensure fidelity to treatment protocols, the investigators directly observed therapy, took tablet counts, measured serum ACE activity, and assessed BP measurement technique, with all participants adhering to a minimum of three months on a maximally dosed triple regimen.
Among subjects, 14% had diabetes and 7.8% reported tobacco use. Average weight was 93.5 kg (205.7 lbs).1 Because of the expected inverse relationship between plasma renin and response to AAs, plasma renin was measured at baseline to test whether resistant hypertension was primarily due to sodium retention.1
Four 12-week rotations
All participants began the trial with four weeks of placebo, followed by randomization to 12-week rotations of once-daily oral treatment with (1) spironolactone 25 to 50 mg, (2) doxazosin modified release 4 to 8 mg, (3) bisoprolol 5 to 10 mg, and (4) placebo.1 Six weeks after initiation of each study medication, participants were titrated to the higher dose. There was no washout period between cycles.
The primary outcome was mean SBP measured at home on four consecutive days prior to the study visits in weeks 6 and 12. Participants were required to have at least six BP measurements per each six-week period in order to establish a valid average. Primary endpoints included the difference in home SBP between spironolactone and placebo, the difference in home SBP between spironolactone and the mean of the other two drugs, and the difference in home SBP between spironolactone and each of the other two drugs.
The results. Spironolactone lowered SBP more than placebo, doxazosin, and bisoprolol (see the Table).1 Clinic measurements were consistent with home BP readings.
Overall, 58% of participants achieved goal SBP < 135 mm Hg on spironolactone, compared with 42% on doxazosin, 44% on bisoprolol, and 24% on placebo.1 The effectiveness of spironolactone on SBP reduction was shown to exhibit an inverse relationship to plasma renin levels, a finding that was not apparent with the other two study drugs. However, spironolactone had a superior BP-lowering effect throughout nearly the entire renin distribution of the cohort.
The mean difference between spironolactone and placebo was –10.2 mm Hg; compared with the other drugs, spironolactone lowered SBP, on average, by 5.64 mm Hg more than bisoprolol and doxazosin; 5.3 mm Hg more than doxazosin alone; and 5.98 mm Hg more than bisoprolol alone.
Only 1% of trial participants had to discontinue spironolactone due to adverse events—the same proportion of withdrawals as that for bisoprolol and placebo and three times less than for doxazosin.1
Continue for what's new >>
WHAT’S NEW
Evidence of superiority
This is the first RCT to compare spironolactone with two other commonly used fourth-line antihypertensives—bisoprolol and doxazosin—in patients with resistant hypertension. The study demonstrated clear superiority of spironolactone in achieving carefully measured ambulatory and clinic-recorded BP targets versus a ß-blocker or an α-blocker.
CAVEATS
Findings not universal
Spironolactone is contraindicated in patients with severe renal impairment. Although multiple drug trials have demonstrated the medication’s safety and effectiveness, especially in patients with resistant hypertension, we should factor in the need for monitoring electrolytes and renal function within weeks of treatment initiation and periodically thereafter.7,8 In this study, spironolactone increased potassium levels, on average, by 0.45 mmol/L. No gynecomastia (typically seen in about 6% of men) was found in those taking spironolactone for a 12-week cycle.1
This single trial enrolled mostly Caucasian men with a mean age of 61. Although smaller observational studies that included African-American patients have shown promising results for spironolactone, the question of external validity or applicability to a diverse population has yet to be decisively answered.9
CHALLENGES TO IMPLEMENTATION
Potential for adverse reactions
The evidence supporting this change in practice has been accumulating for the past few years. However, clinicians who treat patients with resistant hypertension may have concerns about hyperkalemia, gynecomastia, and effects on renal function. More patient-oriented evidence is likewise needed to assist with the revision of guidelines and wider adoption of AAs by primary care providers.
References
1. Williams B, MacDonald TM, Morant S, et al. Spironolactone versus placebo, bisoprolol, and doxazosin to determine the optimal treatment for drug-resistant hypertension (PATHWAY-2): a randomised, double-blind, crossover trial. Lancet. 2015;386:2059-2068.
2. Rosa J, Widimsky P, Tousek P, et al. Randomized comparison of renal denervation versus intensified pharmacotherapy including spironolactone in true-resistant hypertension: six-month results from the Prague-15 Study. Hypertension. 2015;65:407-413.
3. James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults. JAMA. 2014;311:507-520.
4. National Institute for Health and Care Excellence. Hypertension in adults: diagnosis and management (Clinical Guideline CG127). August 2011. https://www.nice.org.uk/guidance/cg127. Accessed March 4, 2016.
5. Dahal K, Kunwar S, Rijal J, et al. The effects of aldosterone antagonists in patients with resistant hypertension: a meta-analysis of randomized and nonrandomized studies. Am J Hypertens. 2015;28:1376-1385.
6. Václavík J, Sedlák R, Jarkovský J, et al. Effect of spironolactone in resistant arterial hypertension: a randomized, double-blind, placebo-controlled trial (ASPIRANT-EXT). Medicine (Baltimore). 2014;93:e162.
7. Wei L, Struthers AD, Fahey T, et al. Spironolactone use and renal toxicity: population based longitudinal analysis. BMJ. 2010;340:c1768.
8. Oxlund CS, Henriksen JE, Tarnow L, et al. Low dose spironolactone reduces blood pressure in patients with resistant hypertension and type 2 diabetes mellitus. J Hypertens. 2013;31:2094-2102.
9. Nishizaka M, Zaman MA, Calhoun DA. Efficacy of low-dose spironolactone in subjects with resistant hypertension. Am J Hypertens. 2003;16:925-930.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
Copyright © 2016. The Family Physicians Inquiries Network. All rights reserved.
Reprinted with permission from the Family Physicians Inquiries Network and The Journal of Family Practice. 2016;65(4):266-268.
“Go low” or say “No” to aggressive systolic BP goals?
Consider treating non-diabetic patients age ≥50 years to a systolic blood pressure (SBP) target <120 mm Hg as compared to <140 mm Hg when the benefits—lower rates of fatal and nonfatal cardiovascular (CV) events and death from any cause—are likely to outweigh the risks from possible additional medication.1
Strength of recommendation
B: Based on a single, good-quality randomized controlled trial (RCT).
Wright JT Jr, Williamson JD, Whelton PK, et al. A randomized trial of intensive versus standard blood-pressure control. N Engl J Med. 2015;373:2103-2116.
Illustrative Case
A 55-year-old man with hypertension and stage 3 chronic kidney disease (CKD) comes in to your office for routine care. His blood pressure is 135/85 mm Hg, and he is presently taking lisinopril 40 mg daily. Should you increase his antihypertensive regimen?
Hypertension is common and leads to significant morbidity and mortality, but pharmacologic treatment reduces incidence of stroke by 35% to 40%, myocardial infarction (MI) by 15% to 25%, and heart failure by up to 64%.2-4 Specific blood pressure targets for defined populations continue to be studied.
In patients with diabetes, the ACCORD (Action to Control Cardiovascular Risk in Diabetes) trial found that more intensive BP targets did not reduce the rate of major CV events, but the study may have been underpowered.5 The members of The Eighth Joint National Committee recommended treating patients over age 60 years to BP goals <150/90 mm Hg.6 This was based on evidence from 6 randomized controlled trials (RCTs),7-12 but there remains debate—even among the members of the Committee—as to appropriate BP goals in patients of any age without CV disease who have BP measurements of 140-159/90-99 mm Hg.13
Study Summary
Treating to SBP <120 mm Hg lowers mortality
The Systolic Blood Pressure Intervention Trial (SPRINT) was a multicenter RCT designed to determine if treating to lower SBP targets in non-diabetic patients at high risk for CV events improves outcomes as compared to standard care. Patients were at least 50 years of age with SBP of 130 to 180 mm Hg and were at increased CV risk as defined by clinical or subclinical CV disease other than stroke, CKD with glomerular filtration rate (GFR) 20 to 60 mL/min/1.73 m2, 10-year risk of CV disease >15% on Framingham risk score, or age ≥75 years of age. Patients with diabetes; prior stroke; polycystic kidney disease; significant proteinuria within the past 6 months; symptomatic heart failure within the past 6 months; or left ventricular ejection fraction <35% were excluded.1
Patients (N=9361) were randomly assigned to an SBP target <120 mm Hg in the intensive group or <140 mm Hg in the standard treatment group, in an open-label design. Allocation was concealed. The study protocol encouraged, but did not require, the use of thiazide-type diuretics, loop diuretics (for those with advanced renal disease), angiotensin-converting enzyme inhibitors or angiotensin receptor blocker agents, calcium channel blockers, and beta-blockers. Clinicians could add other agents as needed. All major classes of antihypertensives were used.
Medication dosing adjustments were based on the average of 3 BP measurements taken with an automated measurement system (Omron Healthcare, Model 907) with the patient seated after 5 minutes of quiet rest. Target SBP in the standard therapy group was 135 to 139 mm Hg. Medication dosages were lowered if SBP was <130 mm Hg at a single visit or <135 mm Hg at 2 consecutive visits.1
The primary composite outcome included the first occurrence of MI, acute coronary syndrome, stroke, heart failure, or death from CV causes. Secondary outcomes were the individual components of the primary composite outcome, death from any cause, and the composite of the primary outcome or death from any cause.1
Study halted early. The study was stopped early due to significantly lower rates of the primary outcome in the intensive therapy group vs the standard therapy group (1.65% per year vs 2.19% per year, respectively, hazard ratio [HR] with intensive treatment=0.75; 95% confidence interval [CI], 0.64-0.89; P<.001). The resulting median follow-up time was 3.26 years.1 This corresponds to a 25% lower relative risk of the primary outcome, with a decrease in event rates from 6.8% to 5.2% over the trial period. All-cause mortality was also lower in the intensive therapy group: 3.4% vs 4.5% (HR=0.73; 95% CI, 0.60-0.90; P=.003).
The number needed to treat (NNT) over 3.26 years to prevent a primary outcome event, death from any cause, and death from CV causes was 61, 90, and 172, respectively. Serious adverse events occurred more frequently in the intensive therapy group than in the standard therapy group (38.3% vs 37.1%; HR=1.04; P=.25) with a number needed to harm (NNH) of 46 over the study period.1 (When looking at serious adverse events identified as likely associated with the intervention, rates were 4.7% vs 2.5%, respectively [P<.001].) Hypotension, syncope, electrolyte abnormalities, and acute kidney injury/acute renal failure reached statistical significance. The incidence of bradycardia and injurious falls was higher in the intensive treatment group, but did not reach statistical significance. In the subgroup of patients ≥75 years of age, 48% in each study group experienced a serious adverse event.1
Throughout the study, mean SBP was 121.5 mm Hg in the intensive therapy group and 134.6 mm Hg in the standard treatment group. This required an average of one additional BP medication in the intensive therapy group (2.8 vs 1.8, respectively).1
What’s New
Lower SBP produces mortality benefits in those under, and over, age 75
This trial builds on a body of evidence that shows the advantages of lowering SBP to <150 mm Hg7,11,12 by demonstrating benefits, including lower all-cause mortality, for lower SBP targets in non-diabetic patients at high risk of CV disease. The SPRINT trial also showed that the benefits of intensive therapy remained true in a subgroup of patients ≥75 years of age.
The incidence of the primary outcome in the cohort ≥75 years of age receiving intensive therapy was 7.7% vs 10.9% for those receiving standard therapy (HR=0.67; 95% CI, 0.51-0.86; NNT=31). All-cause mortality was also lower in the intensive therapy group than in the standard therapy group among patients ≥75 years of age: 5.5% vs 8.04% (HR=0.68; 95% CI, 0.50-0.92; NNT=38).1
Caveats
Many do not benefit from—or are harmed by—increased medication
The absolute risk reduction for the primary outcome is 1.6%, meaning 98.4% of patients receiving more intensive treatment will not benefit. In a group of 1000 patients, an estimated 16 patients will benefit, 22 patients will be seriously harmed, and 962 patients will experience neither benefit nor harm.14 The difference between how BP was measured in this trial (an average of 3 readings after the patient had rested for 5 minutes) and that which occurs typically in clinical practice could potentially lead to overtreatment in practice.
Also, reducing antihypertensive therapies when the SBP was about 130 to 135 mm Hg in the standard therapy group likely exaggerated the difference in outcomes between the intensive and standard therapy groups, and is neither routine nor recommended in clinical practice.6 Finally, the trial specifically studied non-diabetic patients at high risk of CV disease ≥50 years of age, limiting generalizability to other populations.
Challenges to implementation
Who will benefit/who can achieve intensive SBP goals?
Identifying patients most likely to benefit from more intensive BP targets remains challenging. The SPRINT trial showed a mortality benefit, but at a cost of increased morbidity.1,14 In particular, caution should be exercised in the subgroup of patients ≥75 years. Despite a lower NNT than the rest of the study population, serious adverse events happened more frequently. Also, this particular cohort of volunteers may not be representative of those ≥75 years of age in the general population.
Additionally, achieving intensive SBP goals can be challenging. In the SPRINT trial, only half of the intensive target group achieved an SBP <120 mm Hg.1 And in a 2011-12 National Health and Nutrition Examination Survey, only 52% of patients in the general population achieved a BP target <140/90 mm Hg.15 Lower morbidity and mortality should remain the ultimate goals to the management of hypertension, requiring physicians to carefully assess an individual patient’s likelihood of benefit vs harm.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
1. Wright JT Jr, Williamson JD, Whelton PK, et al. A randomized trial of intensive versus standard blood-pressure control. N Engl J Med. 2015;373:2103-2116.
2. Chobanian AV, Bakris GL, Black HR, et al. The seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. JAMA. 2003;289:2560-2572.
3. Neal B, MacMahon S, Chapman N. Effects of ACE inhibitors, calcium antagonists, and other blood-pressure-lowering drugs: results of prospectively designed overviews of randomised trials. Lancet. 2000;356:1955-1964.
4. Psaty BM, Smith NL, Siscovick DS, et al. Health outcomes associated with antihypertensive therapies used as first-line agents. A systematic review and meta-analysis. JAMA. 1997;277:739-745.
5. Margolis KL, O’Connor PJ, Morgan TM, et al. Outcomes of combined cardiovascular risk factor management strategies in type 2 diabetes: the ACCORD randomized trial. Diabetes Care. 2014;37:1721-1728.
6. James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA. 2014;311:507-520.
7. Beckett NS, Peters R, Fletcher AE, et al. Treatment of hypertension in patients 80 years of age or older. N Engl J Med. 2008;358:1887-1898.
8. Verdecchia P, Staessen JA, Angeli F, et al. Usual versus tight control of systolic blood pressure in non-diabetic patients with hypertension (Cardio-Sis): an open-label randomised trial. Lancet. 2009;374:525-533.
9. JATOS Study Group. Principal results of the Japanese trial to assess optimal systolic blood pressure in elderly hypertensive patients (JATOS). Hypertens Res. 2008;31:2115-2127.
10. Ogihara T, Saruta T, Rakugi H, et al. Target blood pressure for treatment of isolated systolic hypertension in the elderly: valsartan in elderly isolated systolic hypertension study. Hypertension. 2010;56:196-202.
11. Staessen JA, Fagard R, Thijs L, et al. Randomised double-blind comparison of placebo and active treatment for older patients with isolated systolic hypertension. The Systolic Hypertension in Europe (Syst-Eur) Trial Investigators. Lancet. 1997;350:757-764.
12. Prevention of stroke by antihypertensive drug treatment in older persons with isolated systolic hypertension. Final results of the Systolic Hypertension in the Elderly Program (SHEP). SHEP Cooperative Research Group. JAMA. 1991;265:3255-3264.
13. Cundiff DK, Gueyffier F, Wright JM. Guidelines for managing high blood pressure. JAMA. 2014; 312:294.
14. Ortiz E, James PA. Let’s not SPRINT to judgment about new blood pressure goals. Ann Intern Med. 2016.
15. Nwankwo T, Yoon SS, Burt V, et al. Hypertension among adults in the United States: National Health and Nutrition Examination Survey, 2011-2012. NCHS Data Brief. 2013;1-8.
Consider treating non-diabetic patients age ≥50 years to a systolic blood pressure (SBP) target <120 mm Hg as compared to <140 mm Hg when the benefits—lower rates of fatal and nonfatal cardiovascular (CV) events and death from any cause—are likely to outweigh the risks from possible additional medication.1
Strength of recommendation
B: Based on a single, good-quality randomized controlled trial (RCT).
Wright JT Jr, Williamson JD, Whelton PK, et al. A randomized trial of intensive versus standard blood-pressure control. N Engl J Med. 2015;373:2103-2116.
Illustrative Case
A 55-year-old man with hypertension and stage 3 chronic kidney disease (CKD) comes in to your office for routine care. His blood pressure is 135/85 mm Hg, and he is presently taking lisinopril 40 mg daily. Should you increase his antihypertensive regimen?
Hypertension is common and leads to significant morbidity and mortality, but pharmacologic treatment reduces incidence of stroke by 35% to 40%, myocardial infarction (MI) by 15% to 25%, and heart failure by up to 64%.2-4 Specific blood pressure targets for defined populations continue to be studied.
In patients with diabetes, the ACCORD (Action to Control Cardiovascular Risk in Diabetes) trial found that more intensive BP targets did not reduce the rate of major CV events, but the study may have been underpowered.5 The members of The Eighth Joint National Committee recommended treating patients over age 60 years to BP goals <150/90 mm Hg.6 This was based on evidence from 6 randomized controlled trials (RCTs),7-12 but there remains debate—even among the members of the Committee—as to appropriate BP goals in patients of any age without CV disease who have BP measurements of 140-159/90-99 mm Hg.13
Study Summary
Treating to SBP <120 mm Hg lowers mortality
The Systolic Blood Pressure Intervention Trial (SPRINT) was a multicenter RCT designed to determine if treating to lower SBP targets in non-diabetic patients at high risk for CV events improves outcomes as compared to standard care. Patients were at least 50 years of age with SBP of 130 to 180 mm Hg and were at increased CV risk as defined by clinical or subclinical CV disease other than stroke, CKD with glomerular filtration rate (GFR) 20 to 60 mL/min/1.73 m2, 10-year risk of CV disease >15% on Framingham risk score, or age ≥75 years of age. Patients with diabetes; prior stroke; polycystic kidney disease; significant proteinuria within the past 6 months; symptomatic heart failure within the past 6 months; or left ventricular ejection fraction <35% were excluded.1
Patients (N=9361) were randomly assigned to an SBP target <120 mm Hg in the intensive group or <140 mm Hg in the standard treatment group, in an open-label design. Allocation was concealed. The study protocol encouraged, but did not require, the use of thiazide-type diuretics, loop diuretics (for those with advanced renal disease), angiotensin-converting enzyme inhibitors or angiotensin receptor blocker agents, calcium channel blockers, and beta-blockers. Clinicians could add other agents as needed. All major classes of antihypertensives were used.
Medication dosing adjustments were based on the average of 3 BP measurements taken with an automated measurement system (Omron Healthcare, Model 907) with the patient seated after 5 minutes of quiet rest. Target SBP in the standard therapy group was 135 to 139 mm Hg. Medication dosages were lowered if SBP was <130 mm Hg at a single visit or <135 mm Hg at 2 consecutive visits.1
The primary composite outcome included the first occurrence of MI, acute coronary syndrome, stroke, heart failure, or death from CV causes. Secondary outcomes were the individual components of the primary composite outcome, death from any cause, and the composite of the primary outcome or death from any cause.1
Study halted early. The study was stopped early due to significantly lower rates of the primary outcome in the intensive therapy group vs the standard therapy group (1.65% per year vs 2.19% per year, respectively, hazard ratio [HR] with intensive treatment=0.75; 95% confidence interval [CI], 0.64-0.89; P<.001). The resulting median follow-up time was 3.26 years.1 This corresponds to a 25% lower relative risk of the primary outcome, with a decrease in event rates from 6.8% to 5.2% over the trial period. All-cause mortality was also lower in the intensive therapy group: 3.4% vs 4.5% (HR=0.73; 95% CI, 0.60-0.90; P=.003).
The number needed to treat (NNT) over 3.26 years to prevent a primary outcome event, death from any cause, and death from CV causes was 61, 90, and 172, respectively. Serious adverse events occurred more frequently in the intensive therapy group than in the standard therapy group (38.3% vs 37.1%; HR=1.04; P=.25) with a number needed to harm (NNH) of 46 over the study period.1 (When looking at serious adverse events identified as likely associated with the intervention, rates were 4.7% vs 2.5%, respectively [P<.001].) Hypotension, syncope, electrolyte abnormalities, and acute kidney injury/acute renal failure reached statistical significance. The incidence of bradycardia and injurious falls was higher in the intensive treatment group, but did not reach statistical significance. In the subgroup of patients ≥75 years of age, 48% in each study group experienced a serious adverse event.1
Throughout the study, mean SBP was 121.5 mm Hg in the intensive therapy group and 134.6 mm Hg in the standard treatment group. This required an average of one additional BP medication in the intensive therapy group (2.8 vs 1.8, respectively).1
What’s New
Lower SBP produces mortality benefits in those under, and over, age 75
This trial builds on a body of evidence that shows the advantages of lowering SBP to <150 mm Hg7,11,12 by demonstrating benefits, including lower all-cause mortality, for lower SBP targets in non-diabetic patients at high risk of CV disease. The SPRINT trial also showed that the benefits of intensive therapy remained true in a subgroup of patients ≥75 years of age.
The incidence of the primary outcome in the cohort ≥75 years of age receiving intensive therapy was 7.7% vs 10.9% for those receiving standard therapy (HR=0.67; 95% CI, 0.51-0.86; NNT=31). All-cause mortality was also lower in the intensive therapy group than in the standard therapy group among patients ≥75 years of age: 5.5% vs 8.04% (HR=0.68; 95% CI, 0.50-0.92; NNT=38).1
Caveats
Many do not benefit from—or are harmed by—increased medication
The absolute risk reduction for the primary outcome is 1.6%, meaning 98.4% of patients receiving more intensive treatment will not benefit. In a group of 1000 patients, an estimated 16 patients will benefit, 22 patients will be seriously harmed, and 962 patients will experience neither benefit nor harm.14 The difference between how BP was measured in this trial (an average of 3 readings after the patient had rested for 5 minutes) and that which occurs typically in clinical practice could potentially lead to overtreatment in practice.
Also, reducing antihypertensive therapies when the SBP was about 130 to 135 mm Hg in the standard therapy group likely exaggerated the difference in outcomes between the intensive and standard therapy groups, and is neither routine nor recommended in clinical practice.6 Finally, the trial specifically studied non-diabetic patients at high risk of CV disease ≥50 years of age, limiting generalizability to other populations.
Challenges to implementation
Who will benefit/who can achieve intensive SBP goals?
Identifying patients most likely to benefit from more intensive BP targets remains challenging. The SPRINT trial showed a mortality benefit, but at a cost of increased morbidity.1,14 In particular, caution should be exercised in the subgroup of patients ≥75 years. Despite a lower NNT than the rest of the study population, serious adverse events happened more frequently. Also, this particular cohort of volunteers may not be representative of those ≥75 years of age in the general population.
Additionally, achieving intensive SBP goals can be challenging. In the SPRINT trial, only half of the intensive target group achieved an SBP <120 mm Hg.1 And in a 2011-12 National Health and Nutrition Examination Survey, only 52% of patients in the general population achieved a BP target <140/90 mm Hg.15 Lower morbidity and mortality should remain the ultimate goals to the management of hypertension, requiring physicians to carefully assess an individual patient’s likelihood of benefit vs harm.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
Consider treating non-diabetic patients age ≥50 years to a systolic blood pressure (SBP) target <120 mm Hg as compared to <140 mm Hg when the benefits—lower rates of fatal and nonfatal cardiovascular (CV) events and death from any cause—are likely to outweigh the risks from possible additional medication.1
Strength of recommendation
B: Based on a single, good-quality randomized controlled trial (RCT).
Wright JT Jr, Williamson JD, Whelton PK, et al. A randomized trial of intensive versus standard blood-pressure control. N Engl J Med. 2015;373:2103-2116.
Illustrative Case
A 55-year-old man with hypertension and stage 3 chronic kidney disease (CKD) comes in to your office for routine care. His blood pressure is 135/85 mm Hg, and he is presently taking lisinopril 40 mg daily. Should you increase his antihypertensive regimen?
Hypertension is common and leads to significant morbidity and mortality, but pharmacologic treatment reduces incidence of stroke by 35% to 40%, myocardial infarction (MI) by 15% to 25%, and heart failure by up to 64%.2-4 Specific blood pressure targets for defined populations continue to be studied.
In patients with diabetes, the ACCORD (Action to Control Cardiovascular Risk in Diabetes) trial found that more intensive BP targets did not reduce the rate of major CV events, but the study may have been underpowered.5 The members of The Eighth Joint National Committee recommended treating patients over age 60 years to BP goals <150/90 mm Hg.6 This was based on evidence from 6 randomized controlled trials (RCTs),7-12 but there remains debate—even among the members of the Committee—as to appropriate BP goals in patients of any age without CV disease who have BP measurements of 140-159/90-99 mm Hg.13
Study Summary
Treating to SBP <120 mm Hg lowers mortality
The Systolic Blood Pressure Intervention Trial (SPRINT) was a multicenter RCT designed to determine if treating to lower SBP targets in non-diabetic patients at high risk for CV events improves outcomes as compared to standard care. Patients were at least 50 years of age with SBP of 130 to 180 mm Hg and were at increased CV risk as defined by clinical or subclinical CV disease other than stroke, CKD with glomerular filtration rate (GFR) 20 to 60 mL/min/1.73 m2, 10-year risk of CV disease >15% on Framingham risk score, or age ≥75 years of age. Patients with diabetes; prior stroke; polycystic kidney disease; significant proteinuria within the past 6 months; symptomatic heart failure within the past 6 months; or left ventricular ejection fraction <35% were excluded.1
Patients (N=9361) were randomly assigned to an SBP target <120 mm Hg in the intensive group or <140 mm Hg in the standard treatment group, in an open-label design. Allocation was concealed. The study protocol encouraged, but did not require, the use of thiazide-type diuretics, loop diuretics (for those with advanced renal disease), angiotensin-converting enzyme inhibitors or angiotensin receptor blocker agents, calcium channel blockers, and beta-blockers. Clinicians could add other agents as needed. All major classes of antihypertensives were used.
Medication dosing adjustments were based on the average of 3 BP measurements taken with an automated measurement system (Omron Healthcare, Model 907) with the patient seated after 5 minutes of quiet rest. Target SBP in the standard therapy group was 135 to 139 mm Hg. Medication dosages were lowered if SBP was <130 mm Hg at a single visit or <135 mm Hg at 2 consecutive visits.1
The primary composite outcome included the first occurrence of MI, acute coronary syndrome, stroke, heart failure, or death from CV causes. Secondary outcomes were the individual components of the primary composite outcome, death from any cause, and the composite of the primary outcome or death from any cause.1
Study halted early. The study was stopped early due to significantly lower rates of the primary outcome in the intensive therapy group vs the standard therapy group (1.65% per year vs 2.19% per year, respectively, hazard ratio [HR] with intensive treatment=0.75; 95% confidence interval [CI], 0.64-0.89; P<.001). The resulting median follow-up time was 3.26 years.1 This corresponds to a 25% lower relative risk of the primary outcome, with a decrease in event rates from 6.8% to 5.2% over the trial period. All-cause mortality was also lower in the intensive therapy group: 3.4% vs 4.5% (HR=0.73; 95% CI, 0.60-0.90; P=.003).
The number needed to treat (NNT) over 3.26 years to prevent a primary outcome event, death from any cause, and death from CV causes was 61, 90, and 172, respectively. Serious adverse events occurred more frequently in the intensive therapy group than in the standard therapy group (38.3% vs 37.1%; HR=1.04; P=.25) with a number needed to harm (NNH) of 46 over the study period.1 (When looking at serious adverse events identified as likely associated with the intervention, rates were 4.7% vs 2.5%, respectively [P<.001].) Hypotension, syncope, electrolyte abnormalities, and acute kidney injury/acute renal failure reached statistical significance. The incidence of bradycardia and injurious falls was higher in the intensive treatment group, but did not reach statistical significance. In the subgroup of patients ≥75 years of age, 48% in each study group experienced a serious adverse event.1
Throughout the study, mean SBP was 121.5 mm Hg in the intensive therapy group and 134.6 mm Hg in the standard treatment group. This required an average of one additional BP medication in the intensive therapy group (2.8 vs 1.8, respectively).1
What’s New
Lower SBP produces mortality benefits in those under, and over, age 75
This trial builds on a body of evidence that shows the advantages of lowering SBP to <150 mm Hg7,11,12 by demonstrating benefits, including lower all-cause mortality, for lower SBP targets in non-diabetic patients at high risk of CV disease. The SPRINT trial also showed that the benefits of intensive therapy remained true in a subgroup of patients ≥75 years of age.
The incidence of the primary outcome in the cohort ≥75 years of age receiving intensive therapy was 7.7% vs 10.9% for those receiving standard therapy (HR=0.67; 95% CI, 0.51-0.86; NNT=31). All-cause mortality was also lower in the intensive therapy group than in the standard therapy group among patients ≥75 years of age: 5.5% vs 8.04% (HR=0.68; 95% CI, 0.50-0.92; NNT=38).1
Caveats
Many do not benefit from—or are harmed by—increased medication
The absolute risk reduction for the primary outcome is 1.6%, meaning 98.4% of patients receiving more intensive treatment will not benefit. In a group of 1000 patients, an estimated 16 patients will benefit, 22 patients will be seriously harmed, and 962 patients will experience neither benefit nor harm.14 The difference between how BP was measured in this trial (an average of 3 readings after the patient had rested for 5 minutes) and that which occurs typically in clinical practice could potentially lead to overtreatment in practice.
Also, reducing antihypertensive therapies when the SBP was about 130 to 135 mm Hg in the standard therapy group likely exaggerated the difference in outcomes between the intensive and standard therapy groups, and is neither routine nor recommended in clinical practice.6 Finally, the trial specifically studied non-diabetic patients at high risk of CV disease ≥50 years of age, limiting generalizability to other populations.
Challenges to implementation
Who will benefit/who can achieve intensive SBP goals?
Identifying patients most likely to benefit from more intensive BP targets remains challenging. The SPRINT trial showed a mortality benefit, but at a cost of increased morbidity.1,14 In particular, caution should be exercised in the subgroup of patients ≥75 years. Despite a lower NNT than the rest of the study population, serious adverse events happened more frequently. Also, this particular cohort of volunteers may not be representative of those ≥75 years of age in the general population.
Additionally, achieving intensive SBP goals can be challenging. In the SPRINT trial, only half of the intensive target group achieved an SBP <120 mm Hg.1 And in a 2011-12 National Health and Nutrition Examination Survey, only 52% of patients in the general population achieved a BP target <140/90 mm Hg.15 Lower morbidity and mortality should remain the ultimate goals to the management of hypertension, requiring physicians to carefully assess an individual patient’s likelihood of benefit vs harm.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
1. Wright JT Jr, Williamson JD, Whelton PK, et al. A randomized trial of intensive versus standard blood-pressure control. N Engl J Med. 2015;373:2103-2116.
2. Chobanian AV, Bakris GL, Black HR, et al. The seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. JAMA. 2003;289:2560-2572.
3. Neal B, MacMahon S, Chapman N. Effects of ACE inhibitors, calcium antagonists, and other blood-pressure-lowering drugs: results of prospectively designed overviews of randomised trials. Lancet. 2000;356:1955-1964.
4. Psaty BM, Smith NL, Siscovick DS, et al. Health outcomes associated with antihypertensive therapies used as first-line agents. A systematic review and meta-analysis. JAMA. 1997;277:739-745.
5. Margolis KL, O’Connor PJ, Morgan TM, et al. Outcomes of combined cardiovascular risk factor management strategies in type 2 diabetes: the ACCORD randomized trial. Diabetes Care. 2014;37:1721-1728.
6. James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA. 2014;311:507-520.
7. Beckett NS, Peters R, Fletcher AE, et al. Treatment of hypertension in patients 80 years of age or older. N Engl J Med. 2008;358:1887-1898.
8. Verdecchia P, Staessen JA, Angeli F, et al. Usual versus tight control of systolic blood pressure in non-diabetic patients with hypertension (Cardio-Sis): an open-label randomised trial. Lancet. 2009;374:525-533.
9. JATOS Study Group. Principal results of the Japanese trial to assess optimal systolic blood pressure in elderly hypertensive patients (JATOS). Hypertens Res. 2008;31:2115-2127.
10. Ogihara T, Saruta T, Rakugi H, et al. Target blood pressure for treatment of isolated systolic hypertension in the elderly: valsartan in elderly isolated systolic hypertension study. Hypertension. 2010;56:196-202.
11. Staessen JA, Fagard R, Thijs L, et al. Randomised double-blind comparison of placebo and active treatment for older patients with isolated systolic hypertension. The Systolic Hypertension in Europe (Syst-Eur) Trial Investigators. Lancet. 1997;350:757-764.
12. Prevention of stroke by antihypertensive drug treatment in older persons with isolated systolic hypertension. Final results of the Systolic Hypertension in the Elderly Program (SHEP). SHEP Cooperative Research Group. JAMA. 1991;265:3255-3264.
13. Cundiff DK, Gueyffier F, Wright JM. Guidelines for managing high blood pressure. JAMA. 2014; 312:294.
14. Ortiz E, James PA. Let’s not SPRINT to judgment about new blood pressure goals. Ann Intern Med. 2016.
15. Nwankwo T, Yoon SS, Burt V, et al. Hypertension among adults in the United States: National Health and Nutrition Examination Survey, 2011-2012. NCHS Data Brief. 2013;1-8.
1. Wright JT Jr, Williamson JD, Whelton PK, et al. A randomized trial of intensive versus standard blood-pressure control. N Engl J Med. 2015;373:2103-2116.
2. Chobanian AV, Bakris GL, Black HR, et al. The seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. JAMA. 2003;289:2560-2572.
3. Neal B, MacMahon S, Chapman N. Effects of ACE inhibitors, calcium antagonists, and other blood-pressure-lowering drugs: results of prospectively designed overviews of randomised trials. Lancet. 2000;356:1955-1964.
4. Psaty BM, Smith NL, Siscovick DS, et al. Health outcomes associated with antihypertensive therapies used as first-line agents. A systematic review and meta-analysis. JAMA. 1997;277:739-745.
5. Margolis KL, O’Connor PJ, Morgan TM, et al. Outcomes of combined cardiovascular risk factor management strategies in type 2 diabetes: the ACCORD randomized trial. Diabetes Care. 2014;37:1721-1728.
6. James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA. 2014;311:507-520.
7. Beckett NS, Peters R, Fletcher AE, et al. Treatment of hypertension in patients 80 years of age or older. N Engl J Med. 2008;358:1887-1898.
8. Verdecchia P, Staessen JA, Angeli F, et al. Usual versus tight control of systolic blood pressure in non-diabetic patients with hypertension (Cardio-Sis): an open-label randomised trial. Lancet. 2009;374:525-533.
9. JATOS Study Group. Principal results of the Japanese trial to assess optimal systolic blood pressure in elderly hypertensive patients (JATOS). Hypertens Res. 2008;31:2115-2127.
10. Ogihara T, Saruta T, Rakugi H, et al. Target blood pressure for treatment of isolated systolic hypertension in the elderly: valsartan in elderly isolated systolic hypertension study. Hypertension. 2010;56:196-202.
11. Staessen JA, Fagard R, Thijs L, et al. Randomised double-blind comparison of placebo and active treatment for older patients with isolated systolic hypertension. The Systolic Hypertension in Europe (Syst-Eur) Trial Investigators. Lancet. 1997;350:757-764.
12. Prevention of stroke by antihypertensive drug treatment in older persons with isolated systolic hypertension. Final results of the Systolic Hypertension in the Elderly Program (SHEP). SHEP Cooperative Research Group. JAMA. 1991;265:3255-3264.
13. Cundiff DK, Gueyffier F, Wright JM. Guidelines for managing high blood pressure. JAMA. 2014; 312:294.
14. Ortiz E, James PA. Let’s not SPRINT to judgment about new blood pressure goals. Ann Intern Med. 2016.
15. Nwankwo T, Yoon SS, Burt V, et al. Hypertension among adults in the United States: National Health and Nutrition Examination Survey, 2011-2012. NCHS Data Brief. 2013;1-8.
Copyright © 2016. The Family Physicians Inquiries Network. All rights reserved.
Kidney Stones? It’s Time to Rethink Those Meds
PRACTICE CHANGER
Do not prescribe tamsulosin or nifedipine for stone expulsion in patients with ureteral stones that are ≤ 10 mm.1
Strength of recommendation
A: Based on a high-quality randomized controlled trial (RCT).1
Bob Z, age 48, presents to the emergency department (ED) with unspecified groin pain. CT of the kidney, ureter, and bladder (CT KUB) finds evidence of a single ureteral stone measuring 8 mm. He’s prescribed medication for the pain and discharged. The day after his ED visit, he comes to your office to discuss further treatment options. Should you prescribe tamsulosin or nifedipine to help him pass the stone?
The most recent National Health and Nutrition Examination Survey found kidney stones affect 8.8% of the population.2 Outpatient therapy is indicated for patients with ureteric colic secondary to stones ≤ 10 mm who do not have uncontrolled pain, impaired kidney function, or severe infection. Routine outpatient care includes oral hydration, antiemetics, and pain medications.
Medical expulsive therapy (MET) is also used to facilitate stone passage. MET is increasingly becoming part of routine care; use of MET in kidney stone patients in the United States has grown from 14% in 2009 to 64% in 2012.3,4
The joint European Association of Urology/American Urological Association Nephrolithiasis Guideline Panel supports the use of MET.5 Meta-analyses of multiple RCTs suggest that an α-blocker (tamsulosin) or a calcium channel blocker (nifedipine) can reduce pain and lead to quicker stone passage and a higher rate of eventual stone passage when compared to placebo or observation.6,7 However, these reviews included small, heterogeneous studies with a high or unclear risk for bias.
Continue for the study summary >>
STUDY SUMMARY
MET doesn’t increase the rate of stone passage
The SUSPEND (Spontaneous Urinary Stone Passage ENabled by Drugs) trial1 was a multicenter RCT designed to determine the effectiveness of tamsulosin or nifedipine as MET for patients ages 18 to 65 with a single ureteric stone measuring ≤ 10 mm on CT KUB, which has 98% diagnostic accuracy.8 (Stones > 10 mm typically require surgery or lithotripsy.)
In this RCT, 1,167 adults were randomized to take tamsulosin (0.4 mg/d), nifedipine (30 mg/d), or placebo for four weeks or until the stone spontaneously passed, whichever came first. The participants, clinicians, and research staff were blinded to treatment assignment. The primary outcome was the proportion of participants who spontaneously passed their stone, as indicated in patient self-reported questionnaires and case-report forms completed by researchers. Secondary outcomes were time to stone passage and pain as assessed by analgesic use and a visual analogue scale (VAS).
At four weeks, 1,136 (97%) of the randomized participants had data available for analysis. The proportion of participants who passed their stone did not differ between MET and placebo; 80% of the placebo group (303 of 379 participants) passed the stone, compared with 81% (307 of 378) of the tamsulosin group and 80% (304 of 379) of the nifedipine group. The odds ratio (OR) for MET vs placebo was 1.04 (95% confidence interval [CI], 0.77 to 1.43) and the OR for tamsulosin vs nifedipine was 1.07 (95% CI, 0.74 to 1.53). These findings did not change with further subgroup analysis, including by sex, stone size (≤ 5 mm vs > 5 mm), or stone location.
There were no differences between groups in time to stone passage as measured by clinical report and confirmed by imaging. Time to passage of stone was available for 237 (21% of) participants. The mean days to stone passage was 15.9 (n = 84) for placebo, 16.5 (n = 79) for tamsulosin, and 16.2 (n = 74) for nifedipine, with a MET vs placebo difference of 0.5 days (95% CI, –2.9 to 3.9; P = .78). Sensitivity analysis accounting for bias from missing data did not change this outcome.
No differences in analgesic use or pain. Self-reported use of pain medication during the first four weeks was similar between groups: 59% (placebo patients), 56% (tamsulosin), and 56% (nifedipine). The mean days of pain medication use was 10.5 for placebo, 11.6 for tamsulosin, and 10.7 for nifedipine, with a MET vs placebo difference of 0.6 days (95% CI, –1.6 to 2.8; P = .45).
There was no difference between groups in the VAS pain score at four weeks. The MET vs placebo difference was 0.0 (95% CI, –0.4 to 0.4; P = .96) and the mean VAS pain score was 1.2 for placebo, 1.0 for tamsulosin, and 1.3 for nifedipine.
WHAT’S NEW
This large RCT contradicts results from previous meta-analyses
The SUSPEND study is the first large, multicenter RCT of MET with tamsulosin or nifedipine for kidney stones that used patient-oriented outcomes to find no benefit for stone expulsion, analgesic use, or reported pain compared to placebo. The discrepancy with prior meta-analyses is not unusual. Up to one-third of meta-analyses that show positive outcomes of a therapy are subsequently altered by the inclusion of results from a single, large, well-designed, multicenter RCT.9
Continue for caveats >>
CAVEATS
This trial included fewer women than previous studies
The SUSPEND study included a smaller proportion of women than previously published case series due to a need for a diagnostic CT KUB, which excluded more women than men due to radiation concerns. However, the proportion of women was balanced across all groups in this trial, and there was no evidence that sex impacted the efficacy of treatment for the primary outcome.1
CHALLENGES TO IMPLEMENTATION
We see no challenges to the implementation of this recommendation.
References
1. Pickard R, Starr K, MacLennan G, et al. Medical expulsive therapy in adults with ureteric colic: a multicentre, randomised, placebo-controlled trial. Lancet. 2015;386:341-349.
2. Scales CD Jr, Smith AC, Hanley JM, et al. Prevalence of kidney stones in the United States. Eur Urol. 2012;62:160-165.
3. Fwu CW, Eggers PW, Kimmel PL, et al. Emergency department visits, use of imaging, and drugs for urolithiasis have increased in the United States. Kidney Int. 2013;89:479-486.
4. Bagga H, Appa A, Wang R, et al. 2257 medical expulsion therapy is underutilized in women presenting to an emergency department with acute urinary stone disease. J Urol. 2013; 189:e925-e926.
5. Preminger GM, Tiselius HG, Assimos DG, et al; American Urological Association Education and Research, Inc; European Association of Urology. 2007 Guideline for the management of ureteral calculi. Eur Urol. 2007;52:1610-1631.
6. Campschroer T, Zhu Y, Duijvesz D, et al. Alpha-blockers as medical expulsive therapy for ureteral stones. Cochrane Database Syst Rev. 2014;4:CD008509.
7. Seitz C, Liatsikos E, Porpiglia F, et al. Medical therapy to facilitate the passage of stones: what is the evidence? Eur Urol. 2009;56:455-471.
8. Worster A, Preyra I, Weaver B, et al. The accuracy of noncontrast helical computed tomography versus intravenous pyelography in the diagnosis of suspected acute urolithiasis: a meta-analysis. Ann Emerg Med. 2002;40: 280-286.
9. LeLorier J, Gregoire G, Benhaddad A, et al. Discrepancies between meta-analyses and subsequent large randomized, controlled trials. N Engl J Med. 1997;337:536-542.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
Copyright © 2016. The Family Physicians Inquiries Network. All rights reserved.
Reprinted with permission from the Family Physicians Inquiries Network and The Journal of Family Practice. 2016;65(2):118-120.
PRACTICE CHANGER
Do not prescribe tamsulosin or nifedipine for stone expulsion in patients with ureteral stones that are ≤ 10 mm.1
Strength of recommendation
A: Based on a high-quality randomized controlled trial (RCT).1
Bob Z, age 48, presents to the emergency department (ED) with unspecified groin pain. CT of the kidney, ureter, and bladder (CT KUB) finds evidence of a single ureteral stone measuring 8 mm. He’s prescribed medication for the pain and discharged. The day after his ED visit, he comes to your office to discuss further treatment options. Should you prescribe tamsulosin or nifedipine to help him pass the stone?
The most recent National Health and Nutrition Examination Survey found kidney stones affect 8.8% of the population.2 Outpatient therapy is indicated for patients with ureteric colic secondary to stones ≤ 10 mm who do not have uncontrolled pain, impaired kidney function, or severe infection. Routine outpatient care includes oral hydration, antiemetics, and pain medications.
Medical expulsive therapy (MET) is also used to facilitate stone passage. MET is increasingly becoming part of routine care; use of MET in kidney stone patients in the United States has grown from 14% in 2009 to 64% in 2012.3,4
The joint European Association of Urology/American Urological Association Nephrolithiasis Guideline Panel supports the use of MET.5 Meta-analyses of multiple RCTs suggest that an α-blocker (tamsulosin) or a calcium channel blocker (nifedipine) can reduce pain and lead to quicker stone passage and a higher rate of eventual stone passage when compared to placebo or observation.6,7 However, these reviews included small, heterogeneous studies with a high or unclear risk for bias.
Continue for the study summary >>
STUDY SUMMARY
MET doesn’t increase the rate of stone passage
The SUSPEND (Spontaneous Urinary Stone Passage ENabled by Drugs) trial1 was a multicenter RCT designed to determine the effectiveness of tamsulosin or nifedipine as MET for patients ages 18 to 65 with a single ureteric stone measuring ≤ 10 mm on CT KUB, which has 98% diagnostic accuracy.8 (Stones > 10 mm typically require surgery or lithotripsy.)
In this RCT, 1,167 adults were randomized to take tamsulosin (0.4 mg/d), nifedipine (30 mg/d), or placebo for four weeks or until the stone spontaneously passed, whichever came first. The participants, clinicians, and research staff were blinded to treatment assignment. The primary outcome was the proportion of participants who spontaneously passed their stone, as indicated in patient self-reported questionnaires and case-report forms completed by researchers. Secondary outcomes were time to stone passage and pain as assessed by analgesic use and a visual analogue scale (VAS).
At four weeks, 1,136 (97%) of the randomized participants had data available for analysis. The proportion of participants who passed their stone did not differ between MET and placebo; 80% of the placebo group (303 of 379 participants) passed the stone, compared with 81% (307 of 378) of the tamsulosin group and 80% (304 of 379) of the nifedipine group. The odds ratio (OR) for MET vs placebo was 1.04 (95% confidence interval [CI], 0.77 to 1.43) and the OR for tamsulosin vs nifedipine was 1.07 (95% CI, 0.74 to 1.53). These findings did not change with further subgroup analysis, including by sex, stone size (≤ 5 mm vs > 5 mm), or stone location.
There were no differences between groups in time to stone passage as measured by clinical report and confirmed by imaging. Time to passage of stone was available for 237 (21% of) participants. The mean days to stone passage was 15.9 (n = 84) for placebo, 16.5 (n = 79) for tamsulosin, and 16.2 (n = 74) for nifedipine, with a MET vs placebo difference of 0.5 days (95% CI, –2.9 to 3.9; P = .78). Sensitivity analysis accounting for bias from missing data did not change this outcome.
No differences in analgesic use or pain. Self-reported use of pain medication during the first four weeks was similar between groups: 59% (placebo patients), 56% (tamsulosin), and 56% (nifedipine). The mean days of pain medication use was 10.5 for placebo, 11.6 for tamsulosin, and 10.7 for nifedipine, with a MET vs placebo difference of 0.6 days (95% CI, –1.6 to 2.8; P = .45).
There was no difference between groups in the VAS pain score at four weeks. The MET vs placebo difference was 0.0 (95% CI, –0.4 to 0.4; P = .96) and the mean VAS pain score was 1.2 for placebo, 1.0 for tamsulosin, and 1.3 for nifedipine.
WHAT’S NEW
This large RCT contradicts results from previous meta-analyses
The SUSPEND study is the first large, multicenter RCT of MET with tamsulosin or nifedipine for kidney stones that used patient-oriented outcomes to find no benefit for stone expulsion, analgesic use, or reported pain compared to placebo. The discrepancy with prior meta-analyses is not unusual. Up to one-third of meta-analyses that show positive outcomes of a therapy are subsequently altered by the inclusion of results from a single, large, well-designed, multicenter RCT.9
Continue for caveats >>
CAVEATS
This trial included fewer women than previous studies
The SUSPEND study included a smaller proportion of women than previously published case series due to a need for a diagnostic CT KUB, which excluded more women than men due to radiation concerns. However, the proportion of women was balanced across all groups in this trial, and there was no evidence that sex impacted the efficacy of treatment for the primary outcome.1
CHALLENGES TO IMPLEMENTATION
We see no challenges to the implementation of this recommendation.
References
1. Pickard R, Starr K, MacLennan G, et al. Medical expulsive therapy in adults with ureteric colic: a multicentre, randomised, placebo-controlled trial. Lancet. 2015;386:341-349.
2. Scales CD Jr, Smith AC, Hanley JM, et al. Prevalence of kidney stones in the United States. Eur Urol. 2012;62:160-165.
3. Fwu CW, Eggers PW, Kimmel PL, et al. Emergency department visits, use of imaging, and drugs for urolithiasis have increased in the United States. Kidney Int. 2013;89:479-486.
4. Bagga H, Appa A, Wang R, et al. 2257 medical expulsion therapy is underutilized in women presenting to an emergency department with acute urinary stone disease. J Urol. 2013; 189:e925-e926.
5. Preminger GM, Tiselius HG, Assimos DG, et al; American Urological Association Education and Research, Inc; European Association of Urology. 2007 Guideline for the management of ureteral calculi. Eur Urol. 2007;52:1610-1631.
6. Campschroer T, Zhu Y, Duijvesz D, et al. Alpha-blockers as medical expulsive therapy for ureteral stones. Cochrane Database Syst Rev. 2014;4:CD008509.
7. Seitz C, Liatsikos E, Porpiglia F, et al. Medical therapy to facilitate the passage of stones: what is the evidence? Eur Urol. 2009;56:455-471.
8. Worster A, Preyra I, Weaver B, et al. The accuracy of noncontrast helical computed tomography versus intravenous pyelography in the diagnosis of suspected acute urolithiasis: a meta-analysis. Ann Emerg Med. 2002;40: 280-286.
9. LeLorier J, Gregoire G, Benhaddad A, et al. Discrepancies between meta-analyses and subsequent large randomized, controlled trials. N Engl J Med. 1997;337:536-542.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
Copyright © 2016. The Family Physicians Inquiries Network. All rights reserved.
Reprinted with permission from the Family Physicians Inquiries Network and The Journal of Family Practice. 2016;65(2):118-120.
PRACTICE CHANGER
Do not prescribe tamsulosin or nifedipine for stone expulsion in patients with ureteral stones that are ≤ 10 mm.1
Strength of recommendation
A: Based on a high-quality randomized controlled trial (RCT).1
Bob Z, age 48, presents to the emergency department (ED) with unspecified groin pain. CT of the kidney, ureter, and bladder (CT KUB) finds evidence of a single ureteral stone measuring 8 mm. He’s prescribed medication for the pain and discharged. The day after his ED visit, he comes to your office to discuss further treatment options. Should you prescribe tamsulosin or nifedipine to help him pass the stone?
The most recent National Health and Nutrition Examination Survey found kidney stones affect 8.8% of the population.2 Outpatient therapy is indicated for patients with ureteric colic secondary to stones ≤ 10 mm who do not have uncontrolled pain, impaired kidney function, or severe infection. Routine outpatient care includes oral hydration, antiemetics, and pain medications.
Medical expulsive therapy (MET) is also used to facilitate stone passage. MET is increasingly becoming part of routine care; use of MET in kidney stone patients in the United States has grown from 14% in 2009 to 64% in 2012.3,4
The joint European Association of Urology/American Urological Association Nephrolithiasis Guideline Panel supports the use of MET.5 Meta-analyses of multiple RCTs suggest that an α-blocker (tamsulosin) or a calcium channel blocker (nifedipine) can reduce pain and lead to quicker stone passage and a higher rate of eventual stone passage when compared to placebo or observation.6,7 However, these reviews included small, heterogeneous studies with a high or unclear risk for bias.
Continue for the study summary >>
STUDY SUMMARY
MET doesn’t increase the rate of stone passage
The SUSPEND (Spontaneous Urinary Stone Passage ENabled by Drugs) trial1 was a multicenter RCT designed to determine the effectiveness of tamsulosin or nifedipine as MET for patients ages 18 to 65 with a single ureteric stone measuring ≤ 10 mm on CT KUB, which has 98% diagnostic accuracy.8 (Stones > 10 mm typically require surgery or lithotripsy.)
In this RCT, 1,167 adults were randomized to take tamsulosin (0.4 mg/d), nifedipine (30 mg/d), or placebo for four weeks or until the stone spontaneously passed, whichever came first. The participants, clinicians, and research staff were blinded to treatment assignment. The primary outcome was the proportion of participants who spontaneously passed their stone, as indicated in patient self-reported questionnaires and case-report forms completed by researchers. Secondary outcomes were time to stone passage and pain as assessed by analgesic use and a visual analogue scale (VAS).
At four weeks, 1,136 (97%) of the randomized participants had data available for analysis. The proportion of participants who passed their stone did not differ between MET and placebo; 80% of the placebo group (303 of 379 participants) passed the stone, compared with 81% (307 of 378) of the tamsulosin group and 80% (304 of 379) of the nifedipine group. The odds ratio (OR) for MET vs placebo was 1.04 (95% confidence interval [CI], 0.77 to 1.43) and the OR for tamsulosin vs nifedipine was 1.07 (95% CI, 0.74 to 1.53). These findings did not change with further subgroup analysis, including by sex, stone size (≤ 5 mm vs > 5 mm), or stone location.
There were no differences between groups in time to stone passage as measured by clinical report and confirmed by imaging. Time to passage of stone was available for 237 (21% of) participants. The mean days to stone passage was 15.9 (n = 84) for placebo, 16.5 (n = 79) for tamsulosin, and 16.2 (n = 74) for nifedipine, with a MET vs placebo difference of 0.5 days (95% CI, –2.9 to 3.9; P = .78). Sensitivity analysis accounting for bias from missing data did not change this outcome.
No differences in analgesic use or pain. Self-reported use of pain medication during the first four weeks was similar between groups: 59% (placebo patients), 56% (tamsulosin), and 56% (nifedipine). The mean days of pain medication use was 10.5 for placebo, 11.6 for tamsulosin, and 10.7 for nifedipine, with a MET vs placebo difference of 0.6 days (95% CI, –1.6 to 2.8; P = .45).
There was no difference between groups in the VAS pain score at four weeks. The MET vs placebo difference was 0.0 (95% CI, –0.4 to 0.4; P = .96) and the mean VAS pain score was 1.2 for placebo, 1.0 for tamsulosin, and 1.3 for nifedipine.
WHAT’S NEW
This large RCT contradicts results from previous meta-analyses
The SUSPEND study is the first large, multicenter RCT of MET with tamsulosin or nifedipine for kidney stones that used patient-oriented outcomes to find no benefit for stone expulsion, analgesic use, or reported pain compared to placebo. The discrepancy with prior meta-analyses is not unusual. Up to one-third of meta-analyses that show positive outcomes of a therapy are subsequently altered by the inclusion of results from a single, large, well-designed, multicenter RCT.9
Continue for caveats >>
CAVEATS
This trial included fewer women than previous studies
The SUSPEND study included a smaller proportion of women than previously published case series due to a need for a diagnostic CT KUB, which excluded more women than men due to radiation concerns. However, the proportion of women was balanced across all groups in this trial, and there was no evidence that sex impacted the efficacy of treatment for the primary outcome.1
CHALLENGES TO IMPLEMENTATION
We see no challenges to the implementation of this recommendation.
References
1. Pickard R, Starr K, MacLennan G, et al. Medical expulsive therapy in adults with ureteric colic: a multicentre, randomised, placebo-controlled trial. Lancet. 2015;386:341-349.
2. Scales CD Jr, Smith AC, Hanley JM, et al. Prevalence of kidney stones in the United States. Eur Urol. 2012;62:160-165.
3. Fwu CW, Eggers PW, Kimmel PL, et al. Emergency department visits, use of imaging, and drugs for urolithiasis have increased in the United States. Kidney Int. 2013;89:479-486.
4. Bagga H, Appa A, Wang R, et al. 2257 medical expulsion therapy is underutilized in women presenting to an emergency department with acute urinary stone disease. J Urol. 2013; 189:e925-e926.
5. Preminger GM, Tiselius HG, Assimos DG, et al; American Urological Association Education and Research, Inc; European Association of Urology. 2007 Guideline for the management of ureteral calculi. Eur Urol. 2007;52:1610-1631.
6. Campschroer T, Zhu Y, Duijvesz D, et al. Alpha-blockers as medical expulsive therapy for ureteral stones. Cochrane Database Syst Rev. 2014;4:CD008509.
7. Seitz C, Liatsikos E, Porpiglia F, et al. Medical therapy to facilitate the passage of stones: what is the evidence? Eur Urol. 2009;56:455-471.
8. Worster A, Preyra I, Weaver B, et al. The accuracy of noncontrast helical computed tomography versus intravenous pyelography in the diagnosis of suspected acute urolithiasis: a meta-analysis. Ann Emerg Med. 2002;40: 280-286.
9. LeLorier J, Gregoire G, Benhaddad A, et al. Discrepancies between meta-analyses and subsequent large randomized, controlled trials. N Engl J Med. 1997;337:536-542.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
Copyright © 2016. The Family Physicians Inquiries Network. All rights reserved.
Reprinted with permission from the Family Physicians Inquiries Network and The Journal of Family Practice. 2016;65(2):118-120.
Resistant hypertension? Time to consider this fourth-line drug
When a triple regimen of an ACE inhibitor or ARB, calcium channel blocker, and a thiazide diuretic fails to achieve the target blood pressure, try adding spironolactone.
Strength of recommendation
C: Based on a high-quality disease-oriented randomized controlled trial.1
Williams B, MacDonald TM, Morant S, et al. Spironolactone versus placebo, bisoprolol, and doxazosin to determine the optimal treatment for drug-resistant hypertension (PATHWAY-2): a randomised, double-blind, crossover trial. Lancet. 2015;386:2059–2068.
Illustrative case
Willie S, a 56-year-old with chronic essential hypertension, has been on an optimally dosed 3-drug regimen of an ACE inhibitor, a calcium channel blocker, and a thiazide diuretic for more than 3 months, but his blood pressure is still not at goal.
What is the best antihypertensive agent to add to his regimen?
Resistant hypertension—defined as inadequate blood pressure (BP) control despite a triple regimen of angiotensin-converting enzyme (ACE) inhibitor or angiotensin receptor blocker (ARB), calcium channel blocker (CCB), and thiazide diuretic—affects an estimated 5% to 30% of those being treated for hypertension.1,2 Guidelines from the 8th Joint National Committee (JNC-8) on the management of high BP, released in 2014, recommend beta-blockers, alpha-blockers, or aldosterone antagonists (AAs) as equivalent choices for a fourth-line agent. The recommendation is based on expert opinion.3
Hypertension guidelines from the UK’s National Institute for Health and Care Excellence, released in 2011, recommend an AA if BP targets have not been met with the triple regimen. This recommendation, however, is based on lower-quality evidence, without comparison with beta-blockers, alpha-blockers, or other drug classes.4
More evidence since guideline’s release
A 2015 meta-analysis of 15 studies and a total of more than 1200 participants (3 randomized controlled trials [RCTs], one nonrandomized placebo-controlled comparative trial, and 11 single-arm observational studies) demonstrated the effectiveness of the AAs spironolactone and eplerenone on resistant hypertension.5 In the 4 comparative studies, AAs decreased office systolic blood pressure (SBP) by 24.3 mm Hg (95% confidence interval [CI], 8.65-39.87; P=.002) and diastolic blood pressure (DBP) by 7.8 mm Hg (95% CI, 3.79-11.79; P=.0001) more than placebo. In the 11 single arm studies, AAs reduced SBP by 22.74 mm Hg (95% CI, 18.21-27.27; P <.00001), and DBP by 10.49 mm Hg (95% CI, 8.85–12.13; P <.00001).
The previous year, a randomized, placebo-controlled trial examined the effect of low-dose (25 mg) spironolactone compared with placebo in 161 patients with resistant hypertension.6 At 8 weeks, 73% of those receiving spironolactone reached a goal SBP <140 mm Hg vs 41% of patients on placebo (P=.001). The same proportion (73%) achieved a goal DBP <90 mm Hg in the spironolactone group, compared with 63% of those in the placebo group (P=.223).
Ambulatory BP was likewise assessed and found to be significantly improved among those receiving spironolactone vs placebo, with a decrease in SBP of 9.8 mm Hg (95% CI, -14.2 to -5.4; P<.001), and a 3.2 mm Hg decline in DBP (95% CI, -5.9 to -0.5; P=.013).6
STUDY SUMMARY
First study to compare spironolactone with other drugs
The study by Williams et al—a double-blind, randomized placebo-controlled crossover trial conducted in the UK—was the first RCT to directly compare spironolactone with other medications for the treatment of resistant hypertension in adults already on triple therapy with an ACE inhibitor or ARB, a CCB, and a thiazide diuretic.1 The trial randomized 335 individuals with a mean age of 61.4 years (age range 18 to 79), 69% of whom were male; 314 were included in the intention-to-treat analysis.1
Enrollment criteria for resistant hypertension specified a clinic-recorded SBP of ≥140 mm Hg (or ≥135 mm Hg in those with diabetes) and home SBP (in 18 readings over 4 days) of ≥130 mm Hg.1 To ensure fidelity to treatment protocols, the investigators directly observed therapy, took tablet counts, measured serum ACE activity, and assessed BP measurement technique, with all participants adhering to a minimum of 3 months on a maximally dosed triple regimen.
Diabetes prevalence was 14%; tobacco use was 7.8%; and average weight was 93.5 kg (205.7 lbs).1 Because of the expected inverse relationship between plasma renin and response to AAs, plasma renin was measured at baseline to test whether resistant hypertension was primarily due to sodium retention.1
Participants underwent 4, 12-week rotations
All participants began the trial with 4 weeks of placebo, followed by randomization to 12-week rotations of once daily oral treatment with 1) spironolactone 25 to 50 mg, 2) doxazosin modified release 4 to 8 mg, 3) bisoprolol 5 to 10 mg, and 4) placebo.1 Six weeks after initiation of each study medication, participants were titrated to the higher dose. There was no washout period between cycles.
The primary outcome was mean SBP measured at home on 4 consecutive days prior to the study visits on Weeks 6 and 12. Participants were required to have at least 6 BP measurements per each 6-week period in order to establish a valid average. Primary endpoints included: the difference in home SBP between spironolactone and placebo, the difference in home SBP between spironolactone and the mean of the other 2 drugs, and the difference in home SBP between spironolactone and each of the other 2 drugs.
The results: Spironolactone lowered SBP more than placebo, doxazosin, and bisoprolol (TABLE),1 and clinic measurements were consistent with home BP readings.
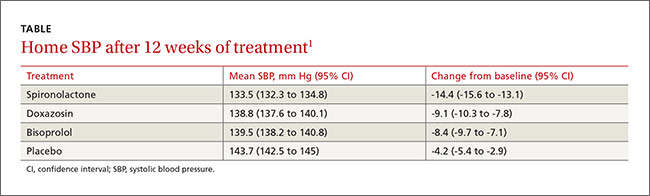
Overall, 58% of participants achieved goal SBP <135 mm Hg on spironolactone, compared with 42% on doxazosin, 44% on bisoprolol, and 24% on placebo.1 The effectiveness of spironolactone on SBP reduction was shown to exhibit an inverse relationship to plasma renin levels, a finding that was not apparent with the other 2 study drugs. However, spironolactone had a superior BP lowering effect throughout nearly the entire renin distribution of the cohort. The mean difference between spironolactone and placebo was -10.2 mm Hg; compared with the other drugs, spironolactone lowered SBP, on average, by 5.64 mm Hg more than bisoprolol and doxazosin; 5.3 mm Hg more than doxazosin alone, and 5.98 mm Hg more than bisoprolol alone.
Only 1% of trial participants had to discontinue spironolactone due to adverse events—the same proportion of withdrawals as that for bisoprolol and placebo and 3 times less than for doxazosin.1
WHAT’S NEW
Evidence of spironolactone’s superiority
This is the first RCT to compare spironolactone with 2 other commonly used fourth-line antihypertensives—bisoprolol and doxazosin—in patients with resistant hypertension. The study demonstrated clear superiority of spironolactone in achieving carefully measured ambulatory and clinic-recorded BP targets vs a beta-blocker or an alpha-blocker.
CAVEATS
Findings do not apply across the board
Spironolactone is contraindicated in patients with severe renal impairment. Although multiple drug trials have demonstrated the drug’s safety and effectiveness, especially in patients with resistant hypertension, we should factor in the need for monitoring electrolytes and renal function within weeks of initiating treatment and periodically thereafter.7,8 In this study, spironolactone increased potassium levels, on average, by 0.45 mmol/L. No gynecomastia (typically seen in about 6% of men) was found in those taking spironolactone for a 12-week cycle.1
This single trial enrolled mostly Caucasian men with a mean age of 61 years. Although smaller observational studies that included African American patients have shown promising results for spironolactone, the question of external validity or applicability to a diverse population has yet to be decisively answered.9
CHALLENGES TO IMPLEMENTATION
Potential for adverse reactions, lack of patient-oriented results
The evidence supporting this change in practice has been accumulating for the past few years. However, physicians treating patients with resistant hypertension may have concerns about hyperkalemia, gynecomastia, and effects on renal function. More patient-oriented evidence is likewise needed to assist with the revision of guidelines and wider adoption of AAs by primary care providers.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
1. Williams B, MacDonald TM, Morant S, et al. Spironolactone versus placebo, bisoprolol, and doxazosin to determine the optimal treatment for drug-resistant hypertension (PATHWAY-2): a randomised, double-blind, crossover trial. Lancet. 2015;386:2059-2068.
2. Rosa J, Widimsky P, Tousek P, et al. Randomized comparison of renal denervation versus intensified pharmacotherapy including spironolactone in true-resistant hypertension: six-month results from the Prague-15 Study. Hypertension. 2015;65:407-413.
3. James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults. JAMA. 2014;311:507-520.
4. Hypertension in adults: diagnosis and management (Clinical Guideline CG127). (NICE), National Institute for Health and Care Excellence. 2011. Available at: https://www.nice.org.uk/guidance/cg127. Accessed March 4, 2016.
5. Dahal K, Kunwar S, Rijal J, et al. The effects of aldosterone antagonists in patients with resistant hypertension: a meta-analysis of randomized and nonrandomized studies. Am J Hypertens. 2015;28:1376-1385.
6. Václavík J, Sedlák R, Jarkovský J, et al. Effect of spironolactone in resistant arterial hypertension: a randomized, double-blind, placebo-controlled trial (ASPIRANT-EXT). Medicine (Baltimore). 2014;93:e162.
7. Wei L, Struthers AD, Fahey T, et al. Spironolactone use and renal toxicity: population based longitudinal analysis. BMJ. 2010;340:c1768.
8. Oxlund CS, Henriksen JE, Tarnow L, et al. Low dose spironolactone reduces blood pressure in patients with resistant hypertension and type 2 diabetes mellitus. J Hypertens. 2013;31:2094-2102.
9. Nishizaka M, Zaman MA, Calhoun DA. Efficacy of low-dose spironolactone in subjects with resistant hypertension. Am J Hypertens. 2003;16:925-930.
When a triple regimen of an ACE inhibitor or ARB, calcium channel blocker, and a thiazide diuretic fails to achieve the target blood pressure, try adding spironolactone.
Strength of recommendation
C: Based on a high-quality disease-oriented randomized controlled trial.1
Williams B, MacDonald TM, Morant S, et al. Spironolactone versus placebo, bisoprolol, and doxazosin to determine the optimal treatment for drug-resistant hypertension (PATHWAY-2): a randomised, double-blind, crossover trial. Lancet. 2015;386:2059–2068.
Illustrative case
Willie S, a 56-year-old with chronic essential hypertension, has been on an optimally dosed 3-drug regimen of an ACE inhibitor, a calcium channel blocker, and a thiazide diuretic for more than 3 months, but his blood pressure is still not at goal.
What is the best antihypertensive agent to add to his regimen?
Resistant hypertension—defined as inadequate blood pressure (BP) control despite a triple regimen of angiotensin-converting enzyme (ACE) inhibitor or angiotensin receptor blocker (ARB), calcium channel blocker (CCB), and thiazide diuretic—affects an estimated 5% to 30% of those being treated for hypertension.1,2 Guidelines from the 8th Joint National Committee (JNC-8) on the management of high BP, released in 2014, recommend beta-blockers, alpha-blockers, or aldosterone antagonists (AAs) as equivalent choices for a fourth-line agent. The recommendation is based on expert opinion.3
Hypertension guidelines from the UK’s National Institute for Health and Care Excellence, released in 2011, recommend an AA if BP targets have not been met with the triple regimen. This recommendation, however, is based on lower-quality evidence, without comparison with beta-blockers, alpha-blockers, or other drug classes.4
More evidence since guideline’s release
A 2015 meta-analysis of 15 studies and a total of more than 1200 participants (3 randomized controlled trials [RCTs], one nonrandomized placebo-controlled comparative trial, and 11 single-arm observational studies) demonstrated the effectiveness of the AAs spironolactone and eplerenone on resistant hypertension.5 In the 4 comparative studies, AAs decreased office systolic blood pressure (SBP) by 24.3 mm Hg (95% confidence interval [CI], 8.65-39.87; P=.002) and diastolic blood pressure (DBP) by 7.8 mm Hg (95% CI, 3.79-11.79; P=.0001) more than placebo. In the 11 single arm studies, AAs reduced SBP by 22.74 mm Hg (95% CI, 18.21-27.27; P <.00001), and DBP by 10.49 mm Hg (95% CI, 8.85–12.13; P <.00001).
The previous year, a randomized, placebo-controlled trial examined the effect of low-dose (25 mg) spironolactone compared with placebo in 161 patients with resistant hypertension.6 At 8 weeks, 73% of those receiving spironolactone reached a goal SBP <140 mm Hg vs 41% of patients on placebo (P=.001). The same proportion (73%) achieved a goal DBP <90 mm Hg in the spironolactone group, compared with 63% of those in the placebo group (P=.223).
Ambulatory BP was likewise assessed and found to be significantly improved among those receiving spironolactone vs placebo, with a decrease in SBP of 9.8 mm Hg (95% CI, -14.2 to -5.4; P<.001), and a 3.2 mm Hg decline in DBP (95% CI, -5.9 to -0.5; P=.013).6
STUDY SUMMARY
First study to compare spironolactone with other drugs
The study by Williams et al—a double-blind, randomized placebo-controlled crossover trial conducted in the UK—was the first RCT to directly compare spironolactone with other medications for the treatment of resistant hypertension in adults already on triple therapy with an ACE inhibitor or ARB, a CCB, and a thiazide diuretic.1 The trial randomized 335 individuals with a mean age of 61.4 years (age range 18 to 79), 69% of whom were male; 314 were included in the intention-to-treat analysis.1
Enrollment criteria for resistant hypertension specified a clinic-recorded SBP of ≥140 mm Hg (or ≥135 mm Hg in those with diabetes) and home SBP (in 18 readings over 4 days) of ≥130 mm Hg.1 To ensure fidelity to treatment protocols, the investigators directly observed therapy, took tablet counts, measured serum ACE activity, and assessed BP measurement technique, with all participants adhering to a minimum of 3 months on a maximally dosed triple regimen.
Diabetes prevalence was 14%; tobacco use was 7.8%; and average weight was 93.5 kg (205.7 lbs).1 Because of the expected inverse relationship between plasma renin and response to AAs, plasma renin was measured at baseline to test whether resistant hypertension was primarily due to sodium retention.1
Participants underwent 4, 12-week rotations
All participants began the trial with 4 weeks of placebo, followed by randomization to 12-week rotations of once daily oral treatment with 1) spironolactone 25 to 50 mg, 2) doxazosin modified release 4 to 8 mg, 3) bisoprolol 5 to 10 mg, and 4) placebo.1 Six weeks after initiation of each study medication, participants were titrated to the higher dose. There was no washout period between cycles.
The primary outcome was mean SBP measured at home on 4 consecutive days prior to the study visits on Weeks 6 and 12. Participants were required to have at least 6 BP measurements per each 6-week period in order to establish a valid average. Primary endpoints included: the difference in home SBP between spironolactone and placebo, the difference in home SBP between spironolactone and the mean of the other 2 drugs, and the difference in home SBP between spironolactone and each of the other 2 drugs.
The results: Spironolactone lowered SBP more than placebo, doxazosin, and bisoprolol (TABLE),1 and clinic measurements were consistent with home BP readings.
Overall, 58% of participants achieved goal SBP <135 mm Hg on spironolactone, compared with 42% on doxazosin, 44% on bisoprolol, and 24% on placebo.1 The effectiveness of spironolactone on SBP reduction was shown to exhibit an inverse relationship to plasma renin levels, a finding that was not apparent with the other 2 study drugs. However, spironolactone had a superior BP lowering effect throughout nearly the entire renin distribution of the cohort. The mean difference between spironolactone and placebo was -10.2 mm Hg; compared with the other drugs, spironolactone lowered SBP, on average, by 5.64 mm Hg more than bisoprolol and doxazosin; 5.3 mm Hg more than doxazosin alone, and 5.98 mm Hg more than bisoprolol alone.
Only 1% of trial participants had to discontinue spironolactone due to adverse events—the same proportion of withdrawals as that for bisoprolol and placebo and 3 times less than for doxazosin.1
WHAT’S NEW
Evidence of spironolactone’s superiority
This is the first RCT to compare spironolactone with 2 other commonly used fourth-line antihypertensives—bisoprolol and doxazosin—in patients with resistant hypertension. The study demonstrated clear superiority of spironolactone in achieving carefully measured ambulatory and clinic-recorded BP targets vs a beta-blocker or an alpha-blocker.
CAVEATS
Findings do not apply across the board
Spironolactone is contraindicated in patients with severe renal impairment. Although multiple drug trials have demonstrated the drug’s safety and effectiveness, especially in patients with resistant hypertension, we should factor in the need for monitoring electrolytes and renal function within weeks of initiating treatment and periodically thereafter.7,8 In this study, spironolactone increased potassium levels, on average, by 0.45 mmol/L. No gynecomastia (typically seen in about 6% of men) was found in those taking spironolactone for a 12-week cycle.1
This single trial enrolled mostly Caucasian men with a mean age of 61 years. Although smaller observational studies that included African American patients have shown promising results for spironolactone, the question of external validity or applicability to a diverse population has yet to be decisively answered.9
CHALLENGES TO IMPLEMENTATION
Potential for adverse reactions, lack of patient-oriented results
The evidence supporting this change in practice has been accumulating for the past few years. However, physicians treating patients with resistant hypertension may have concerns about hyperkalemia, gynecomastia, and effects on renal function. More patient-oriented evidence is likewise needed to assist with the revision of guidelines and wider adoption of AAs by primary care providers.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
When a triple regimen of an ACE inhibitor or ARB, calcium channel blocker, and a thiazide diuretic fails to achieve the target blood pressure, try adding spironolactone.
Strength of recommendation
C: Based on a high-quality disease-oriented randomized controlled trial.1
Williams B, MacDonald TM, Morant S, et al. Spironolactone versus placebo, bisoprolol, and doxazosin to determine the optimal treatment for drug-resistant hypertension (PATHWAY-2): a randomised, double-blind, crossover trial. Lancet. 2015;386:2059–2068.
Illustrative case
Willie S, a 56-year-old with chronic essential hypertension, has been on an optimally dosed 3-drug regimen of an ACE inhibitor, a calcium channel blocker, and a thiazide diuretic for more than 3 months, but his blood pressure is still not at goal.
What is the best antihypertensive agent to add to his regimen?
Resistant hypertension—defined as inadequate blood pressure (BP) control despite a triple regimen of angiotensin-converting enzyme (ACE) inhibitor or angiotensin receptor blocker (ARB), calcium channel blocker (CCB), and thiazide diuretic—affects an estimated 5% to 30% of those being treated for hypertension.1,2 Guidelines from the 8th Joint National Committee (JNC-8) on the management of high BP, released in 2014, recommend beta-blockers, alpha-blockers, or aldosterone antagonists (AAs) as equivalent choices for a fourth-line agent. The recommendation is based on expert opinion.3
Hypertension guidelines from the UK’s National Institute for Health and Care Excellence, released in 2011, recommend an AA if BP targets have not been met with the triple regimen. This recommendation, however, is based on lower-quality evidence, without comparison with beta-blockers, alpha-blockers, or other drug classes.4
More evidence since guideline’s release
A 2015 meta-analysis of 15 studies and a total of more than 1200 participants (3 randomized controlled trials [RCTs], one nonrandomized placebo-controlled comparative trial, and 11 single-arm observational studies) demonstrated the effectiveness of the AAs spironolactone and eplerenone on resistant hypertension.5 In the 4 comparative studies, AAs decreased office systolic blood pressure (SBP) by 24.3 mm Hg (95% confidence interval [CI], 8.65-39.87; P=.002) and diastolic blood pressure (DBP) by 7.8 mm Hg (95% CI, 3.79-11.79; P=.0001) more than placebo. In the 11 single arm studies, AAs reduced SBP by 22.74 mm Hg (95% CI, 18.21-27.27; P <.00001), and DBP by 10.49 mm Hg (95% CI, 8.85–12.13; P <.00001).
The previous year, a randomized, placebo-controlled trial examined the effect of low-dose (25 mg) spironolactone compared with placebo in 161 patients with resistant hypertension.6 At 8 weeks, 73% of those receiving spironolactone reached a goal SBP <140 mm Hg vs 41% of patients on placebo (P=.001). The same proportion (73%) achieved a goal DBP <90 mm Hg in the spironolactone group, compared with 63% of those in the placebo group (P=.223).
Ambulatory BP was likewise assessed and found to be significantly improved among those receiving spironolactone vs placebo, with a decrease in SBP of 9.8 mm Hg (95% CI, -14.2 to -5.4; P<.001), and a 3.2 mm Hg decline in DBP (95% CI, -5.9 to -0.5; P=.013).6
STUDY SUMMARY
First study to compare spironolactone with other drugs
The study by Williams et al—a double-blind, randomized placebo-controlled crossover trial conducted in the UK—was the first RCT to directly compare spironolactone with other medications for the treatment of resistant hypertension in adults already on triple therapy with an ACE inhibitor or ARB, a CCB, and a thiazide diuretic.1 The trial randomized 335 individuals with a mean age of 61.4 years (age range 18 to 79), 69% of whom were male; 314 were included in the intention-to-treat analysis.1
Enrollment criteria for resistant hypertension specified a clinic-recorded SBP of ≥140 mm Hg (or ≥135 mm Hg in those with diabetes) and home SBP (in 18 readings over 4 days) of ≥130 mm Hg.1 To ensure fidelity to treatment protocols, the investigators directly observed therapy, took tablet counts, measured serum ACE activity, and assessed BP measurement technique, with all participants adhering to a minimum of 3 months on a maximally dosed triple regimen.
Diabetes prevalence was 14%; tobacco use was 7.8%; and average weight was 93.5 kg (205.7 lbs).1 Because of the expected inverse relationship between plasma renin and response to AAs, plasma renin was measured at baseline to test whether resistant hypertension was primarily due to sodium retention.1
Participants underwent 4, 12-week rotations
All participants began the trial with 4 weeks of placebo, followed by randomization to 12-week rotations of once daily oral treatment with 1) spironolactone 25 to 50 mg, 2) doxazosin modified release 4 to 8 mg, 3) bisoprolol 5 to 10 mg, and 4) placebo.1 Six weeks after initiation of each study medication, participants were titrated to the higher dose. There was no washout period between cycles.
The primary outcome was mean SBP measured at home on 4 consecutive days prior to the study visits on Weeks 6 and 12. Participants were required to have at least 6 BP measurements per each 6-week period in order to establish a valid average. Primary endpoints included: the difference in home SBP between spironolactone and placebo, the difference in home SBP between spironolactone and the mean of the other 2 drugs, and the difference in home SBP between spironolactone and each of the other 2 drugs.
The results: Spironolactone lowered SBP more than placebo, doxazosin, and bisoprolol (TABLE),1 and clinic measurements were consistent with home BP readings.
Overall, 58% of participants achieved goal SBP <135 mm Hg on spironolactone, compared with 42% on doxazosin, 44% on bisoprolol, and 24% on placebo.1 The effectiveness of spironolactone on SBP reduction was shown to exhibit an inverse relationship to plasma renin levels, a finding that was not apparent with the other 2 study drugs. However, spironolactone had a superior BP lowering effect throughout nearly the entire renin distribution of the cohort. The mean difference between spironolactone and placebo was -10.2 mm Hg; compared with the other drugs, spironolactone lowered SBP, on average, by 5.64 mm Hg more than bisoprolol and doxazosin; 5.3 mm Hg more than doxazosin alone, and 5.98 mm Hg more than bisoprolol alone.
Only 1% of trial participants had to discontinue spironolactone due to adverse events—the same proportion of withdrawals as that for bisoprolol and placebo and 3 times less than for doxazosin.1
WHAT’S NEW
Evidence of spironolactone’s superiority
This is the first RCT to compare spironolactone with 2 other commonly used fourth-line antihypertensives—bisoprolol and doxazosin—in patients with resistant hypertension. The study demonstrated clear superiority of spironolactone in achieving carefully measured ambulatory and clinic-recorded BP targets vs a beta-blocker or an alpha-blocker.
CAVEATS
Findings do not apply across the board
Spironolactone is contraindicated in patients with severe renal impairment. Although multiple drug trials have demonstrated the drug’s safety and effectiveness, especially in patients with resistant hypertension, we should factor in the need for monitoring electrolytes and renal function within weeks of initiating treatment and periodically thereafter.7,8 In this study, spironolactone increased potassium levels, on average, by 0.45 mmol/L. No gynecomastia (typically seen in about 6% of men) was found in those taking spironolactone for a 12-week cycle.1
This single trial enrolled mostly Caucasian men with a mean age of 61 years. Although smaller observational studies that included African American patients have shown promising results for spironolactone, the question of external validity or applicability to a diverse population has yet to be decisively answered.9
CHALLENGES TO IMPLEMENTATION
Potential for adverse reactions, lack of patient-oriented results
The evidence supporting this change in practice has been accumulating for the past few years. However, physicians treating patients with resistant hypertension may have concerns about hyperkalemia, gynecomastia, and effects on renal function. More patient-oriented evidence is likewise needed to assist with the revision of guidelines and wider adoption of AAs by primary care providers.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
1. Williams B, MacDonald TM, Morant S, et al. Spironolactone versus placebo, bisoprolol, and doxazosin to determine the optimal treatment for drug-resistant hypertension (PATHWAY-2): a randomised, double-blind, crossover trial. Lancet. 2015;386:2059-2068.
2. Rosa J, Widimsky P, Tousek P, et al. Randomized comparison of renal denervation versus intensified pharmacotherapy including spironolactone in true-resistant hypertension: six-month results from the Prague-15 Study. Hypertension. 2015;65:407-413.
3. James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults. JAMA. 2014;311:507-520.
4. Hypertension in adults: diagnosis and management (Clinical Guideline CG127). (NICE), National Institute for Health and Care Excellence. 2011. Available at: https://www.nice.org.uk/guidance/cg127. Accessed March 4, 2016.
5. Dahal K, Kunwar S, Rijal J, et al. The effects of aldosterone antagonists in patients with resistant hypertension: a meta-analysis of randomized and nonrandomized studies. Am J Hypertens. 2015;28:1376-1385.
6. Václavík J, Sedlák R, Jarkovský J, et al. Effect of spironolactone in resistant arterial hypertension: a randomized, double-blind, placebo-controlled trial (ASPIRANT-EXT). Medicine (Baltimore). 2014;93:e162.
7. Wei L, Struthers AD, Fahey T, et al. Spironolactone use and renal toxicity: population based longitudinal analysis. BMJ. 2010;340:c1768.
8. Oxlund CS, Henriksen JE, Tarnow L, et al. Low dose spironolactone reduces blood pressure in patients with resistant hypertension and type 2 diabetes mellitus. J Hypertens. 2013;31:2094-2102.
9. Nishizaka M, Zaman MA, Calhoun DA. Efficacy of low-dose spironolactone in subjects with resistant hypertension. Am J Hypertens. 2003;16:925-930.
1. Williams B, MacDonald TM, Morant S, et al. Spironolactone versus placebo, bisoprolol, and doxazosin to determine the optimal treatment for drug-resistant hypertension (PATHWAY-2): a randomised, double-blind, crossover trial. Lancet. 2015;386:2059-2068.
2. Rosa J, Widimsky P, Tousek P, et al. Randomized comparison of renal denervation versus intensified pharmacotherapy including spironolactone in true-resistant hypertension: six-month results from the Prague-15 Study. Hypertension. 2015;65:407-413.
3. James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults. JAMA. 2014;311:507-520.
4. Hypertension in adults: diagnosis and management (Clinical Guideline CG127). (NICE), National Institute for Health and Care Excellence. 2011. Available at: https://www.nice.org.uk/guidance/cg127. Accessed March 4, 2016.
5. Dahal K, Kunwar S, Rijal J, et al. The effects of aldosterone antagonists in patients with resistant hypertension: a meta-analysis of randomized and nonrandomized studies. Am J Hypertens. 2015;28:1376-1385.
6. Václavík J, Sedlák R, Jarkovský J, et al. Effect of spironolactone in resistant arterial hypertension: a randomized, double-blind, placebo-controlled trial (ASPIRANT-EXT). Medicine (Baltimore). 2014;93:e162.
7. Wei L, Struthers AD, Fahey T, et al. Spironolactone use and renal toxicity: population based longitudinal analysis. BMJ. 2010;340:c1768.
8. Oxlund CS, Henriksen JE, Tarnow L, et al. Low dose spironolactone reduces blood pressure in patients with resistant hypertension and type 2 diabetes mellitus. J Hypertens. 2013;31:2094-2102.
9. Nishizaka M, Zaman MA, Calhoun DA. Efficacy of low-dose spironolactone in subjects with resistant hypertension. Am J Hypertens. 2003;16:925-930.
Copyright © 2016. The Family Physicians Inquiries Network. All rights reserved.

Kidney stones? It’s time to rethink those meds
Do not prescribe tamsulosin or nifedipine for stone expulsion in patients with ureteral stones ≤10 mm.1
Strength of recommendation
A: Based on a high-quality randomized controlled trial.
Pickard R, Starr K, MacLennan G, et al. Medical expulsive therapy in adults with ureteric colic: a multicentre, randomised, placebo-controlled trial. Lancet. 2015;386:341-349.
Illustrative case
Bob Z, age 48, presents to the emergency department (ED) with unspecified groin pain. A computed tomography scan of the kidney, ureter, and bladder (CT KUB) finds evidence of a single ureteral stone measuring 8 mm. He’s prescribed medication for the pain and discharged. The day after his ED visit, he comes to your office to discuss further treatment options. Should you prescribe tamsulosin or nifedipine to help him pass the stone?
The most recent National Health and Nutrition Examination Survey found kidney stones affect 8.8% of the population.2 Outpatient therapy is indicated for patients with ureteric colic secondary to stones ≤10 mm who do not have uncontrolled pain, impaired kidney function, or severe infection. Routine outpatient care includes oral hydration, antiemetics, and pain medications. Medical expulsive therapy (MET) is also used to facilitate stone passage. MET is increasingly becoming part of routine care; use of MET in kidney stone patients in the United States has grown from 14% in 2009 to 64% in 2012.3,4
The joint European Association of Urology/American Urological Association Nephrolithiasis Guideline Panel supports the use of MET.5 Meta-analyses of multiple randomized controlled trials (RCTs) suggest that an alpha-blocker (tamsulosin) or a calcium channel blocker (nifedipine) can reduce pain and lead to quicker stone passage and a higher rate of eventual stone passage when compared to placebo or observation.6,7 However, these reviews included small, heterogeneous studies with a high or unclear risk of bias.
STUDY SUMMARY: MET doesn’t increase the rate of stone passage
The SUSPEND (Spontaneous Urinary Stone Passage ENabled by Drugs) trial1 was a multicenter RCT designed to determine the effectiveness of tamsulosin or nifedipine as MET for patients ages 18 to 65 years with a single ureteric stone measuring ≤10 mm on CT KUB, which has 98% diagnostic accuracy.8 (Stones >10 mm typically require surgery or lithotripsy.)
In this RCT, 1167 adults were randomized to take tamsulosin 0.4 mg/d, nifedipine 30 mg/d, or placebo for 4 weeks or until the stone spontaneously passed, whichever came first. The participants, clinicians, and research staff were blinded to treatment assignment. The primary outcome was the proportion of participants who spontaneously passed their stone, as indicated in patient self-reported questionnaires and case-report forms completed by researchers. Secondary outcomes were time to stone passage and pain as assessed by analgesic use and a visual analogue scale (VAS).
At 4 weeks, 1136 (97%) of the randomized participants had data available for analysis. The proportion of participants who passed their stone did not differ between MET and placebo; 80% of the placebo group (303 of 379 participants) passed the stone, compared with 81% (307 of 378) of the tamsulosin group and 80% (304 of 379) of the nifedipine group. The odds ratio (OR) for MET vs placebo was 1.04 (95% confidence interval [CI], 0.77 to 1.43) and the OR for tamsulosin vs nifedipine was 1.07 (95% CI, 0.74 to 1.53). These findings did not change with further subgroup analysis, including by sex, stone size (≤5 mm vs >5 mm), or stone location.
There were no differences between groups in time to stone passage as measured by clinical report and confirmed by imaging. Time to passage of stone was available for 237 (21%) of participants. The mean days to stone passage was 15.9 (n=84) for placebo, 16.5 (n=79) for tamsulosin and 16.2 (n=74) for nifedipine, with a MET vs placebo difference of 0.5 days (95% CI, -2.9 to 3.9; P=.78). Sensitivity analysis accounting for bias from missing data did not change this outcome.
No differences in analgesic use or pain. Self-reported use of pain medication during the first 4 weeks was similar between groups: 59% (placebo patients), 56% (tamsulosin), and 56% (nifedipine). The mean days of pain medication use was 10.5 for placebo, 11.6 for tamsulosin, and 10.7 for nifedipine, with a MET vs placebo difference of 0.6 days (95% CI, -1.6 to 2.8; P=.45).
There was no difference between groups in the VAS pain score at 4 weeks. The MET vs placebo difference was 0.0 (95% CI, -0.4 to 0.4; P=.96) and the mean VAS pain score was 1.2 for placebo, 1.0 for tamsulosin, and 1.3 for nifedipine.
WHAT'S NEW: This large RCT contradicts results from previous meta-analyses
The SUSPEND study is the first large, multicenter RCT of MET with tamsulosin or nifedipine for kidney stones that used patient-oriented outcomes to find no benefit for stone expulsion, analgesic use, or reported pain compared to placebo. The discrepancy with prior meta-analyses is not unusual. Up to one-third of meta-analyses that show positive outcomes of a therapy are subsequently altered by the inclusion of results from a single, large, multicenter, well-designed RCT.9
CAVEATS: This trial included fewer women than previous studies
The SUSPEND study included a smaller proportion of women than previously published case series due to a need for a diagnostic CT KUB, which excluded more women than men due to radiation concerns. However, the proportion of women was balanced across all groups in this trial, and there was no evidence that sex impacted the efficacy of treatment for the primary outcome.1
CHALLENGES TO IMPLEMENTATION
We see no challenges to the implementation of this recommendation.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
Click here to view PURL METHODOLOGY
1. Pickard R, Starr K, MacLennan G, et al. Medical expulsive therapy in adults with ureteric colic: a multicentre, randomised, placebo-controlled trial. Lancet. 2015;386:341-349.
2. Scales CD Jr., Smith AC, Hanley JM, et al. Prevalence of kidney stones in the United States. Eur Urol. 2012;62:160-165.
3. Fwu CU, Eggers PW, Kimmel PL, et al. Emergency department visits, use of imaging, and drugs for urolithiasis have increased in the United States. Kidney Int. 2013;89:479-486.
4. Bagga H, Appa A, Wang R, et al. 2257 medical expulsion therapy is underutilized in women presenting to an emergency department with acute urinary stone disease. J Urol. 2013;189:e925-e926.
5. Preminger GM, Tiselius HG, Assimos DG, et al; American Urological Association Education and Research, Inc; European Association of Urology. 2007 Guideline for the management of ureteral calculi. Eur Urol. 2007;52:1610-1631.
6. Campschroer T, Zhu Y, Duijvesz D, et al. Alpha-blockers as medical expulsive therapy for ureteral stones. Cochrane Database Syst Rev. 2014;4:CD008509.
7. Seitz C, Liatsikos E, Porpiglia F, et al. Medical therapy to facilitate the passage of stones: what is the evidence? Eur Urol. 2009;56:455-471.
8. Worster A, Preyra I, Weaver B, et al. The accuracy of noncontrast helical computed tomography versus intravenous pyelography in the diagnosis of suspected acute urolithiasis: a meta-analysis. Ann Emerg Med. 2002;40:280-286.
9. LeLorier J, Gregoire G, Benhaddad A, et al. Discrepancies between meta-analyses and subsequent large randomized, controlled trials. N Engl J Med. 1997;337:536-542.
Do not prescribe tamsulosin or nifedipine for stone expulsion in patients with ureteral stones ≤10 mm.1
Strength of recommendation
A: Based on a high-quality randomized controlled trial.
Pickard R, Starr K, MacLennan G, et al. Medical expulsive therapy in adults with ureteric colic: a multicentre, randomised, placebo-controlled trial. Lancet. 2015;386:341-349.
Illustrative case
Bob Z, age 48, presents to the emergency department (ED) with unspecified groin pain. A computed tomography scan of the kidney, ureter, and bladder (CT KUB) finds evidence of a single ureteral stone measuring 8 mm. He’s prescribed medication for the pain and discharged. The day after his ED visit, he comes to your office to discuss further treatment options. Should you prescribe tamsulosin or nifedipine to help him pass the stone?
The most recent National Health and Nutrition Examination Survey found kidney stones affect 8.8% of the population.2 Outpatient therapy is indicated for patients with ureteric colic secondary to stones ≤10 mm who do not have uncontrolled pain, impaired kidney function, or severe infection. Routine outpatient care includes oral hydration, antiemetics, and pain medications. Medical expulsive therapy (MET) is also used to facilitate stone passage. MET is increasingly becoming part of routine care; use of MET in kidney stone patients in the United States has grown from 14% in 2009 to 64% in 2012.3,4
The joint European Association of Urology/American Urological Association Nephrolithiasis Guideline Panel supports the use of MET.5 Meta-analyses of multiple randomized controlled trials (RCTs) suggest that an alpha-blocker (tamsulosin) or a calcium channel blocker (nifedipine) can reduce pain and lead to quicker stone passage and a higher rate of eventual stone passage when compared to placebo or observation.6,7 However, these reviews included small, heterogeneous studies with a high or unclear risk of bias.
STUDY SUMMARY: MET doesn’t increase the rate of stone passage
The SUSPEND (Spontaneous Urinary Stone Passage ENabled by Drugs) trial1 was a multicenter RCT designed to determine the effectiveness of tamsulosin or nifedipine as MET for patients ages 18 to 65 years with a single ureteric stone measuring ≤10 mm on CT KUB, which has 98% diagnostic accuracy.8 (Stones >10 mm typically require surgery or lithotripsy.)
In this RCT, 1167 adults were randomized to take tamsulosin 0.4 mg/d, nifedipine 30 mg/d, or placebo for 4 weeks or until the stone spontaneously passed, whichever came first. The participants, clinicians, and research staff were blinded to treatment assignment. The primary outcome was the proportion of participants who spontaneously passed their stone, as indicated in patient self-reported questionnaires and case-report forms completed by researchers. Secondary outcomes were time to stone passage and pain as assessed by analgesic use and a visual analogue scale (VAS).
At 4 weeks, 1136 (97%) of the randomized participants had data available for analysis. The proportion of participants who passed their stone did not differ between MET and placebo; 80% of the placebo group (303 of 379 participants) passed the stone, compared with 81% (307 of 378) of the tamsulosin group and 80% (304 of 379) of the nifedipine group. The odds ratio (OR) for MET vs placebo was 1.04 (95% confidence interval [CI], 0.77 to 1.43) and the OR for tamsulosin vs nifedipine was 1.07 (95% CI, 0.74 to 1.53). These findings did not change with further subgroup analysis, including by sex, stone size (≤5 mm vs >5 mm), or stone location.
There were no differences between groups in time to stone passage as measured by clinical report and confirmed by imaging. Time to passage of stone was available for 237 (21%) of participants. The mean days to stone passage was 15.9 (n=84) for placebo, 16.5 (n=79) for tamsulosin and 16.2 (n=74) for nifedipine, with a MET vs placebo difference of 0.5 days (95% CI, -2.9 to 3.9; P=.78). Sensitivity analysis accounting for bias from missing data did not change this outcome.
No differences in analgesic use or pain. Self-reported use of pain medication during the first 4 weeks was similar between groups: 59% (placebo patients), 56% (tamsulosin), and 56% (nifedipine). The mean days of pain medication use was 10.5 for placebo, 11.6 for tamsulosin, and 10.7 for nifedipine, with a MET vs placebo difference of 0.6 days (95% CI, -1.6 to 2.8; P=.45).
There was no difference between groups in the VAS pain score at 4 weeks. The MET vs placebo difference was 0.0 (95% CI, -0.4 to 0.4; P=.96) and the mean VAS pain score was 1.2 for placebo, 1.0 for tamsulosin, and 1.3 for nifedipine.
WHAT'S NEW: This large RCT contradicts results from previous meta-analyses
The SUSPEND study is the first large, multicenter RCT of MET with tamsulosin or nifedipine for kidney stones that used patient-oriented outcomes to find no benefit for stone expulsion, analgesic use, or reported pain compared to placebo. The discrepancy with prior meta-analyses is not unusual. Up to one-third of meta-analyses that show positive outcomes of a therapy are subsequently altered by the inclusion of results from a single, large, multicenter, well-designed RCT.9
CAVEATS: This trial included fewer women than previous studies
The SUSPEND study included a smaller proportion of women than previously published case series due to a need for a diagnostic CT KUB, which excluded more women than men due to radiation concerns. However, the proportion of women was balanced across all groups in this trial, and there was no evidence that sex impacted the efficacy of treatment for the primary outcome.1
CHALLENGES TO IMPLEMENTATION
We see no challenges to the implementation of this recommendation.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
Click here to view PURL METHODOLOGY
Do not prescribe tamsulosin or nifedipine for stone expulsion in patients with ureteral stones ≤10 mm.1
Strength of recommendation
A: Based on a high-quality randomized controlled trial.
Pickard R, Starr K, MacLennan G, et al. Medical expulsive therapy in adults with ureteric colic: a multicentre, randomised, placebo-controlled trial. Lancet. 2015;386:341-349.
Illustrative case
Bob Z, age 48, presents to the emergency department (ED) with unspecified groin pain. A computed tomography scan of the kidney, ureter, and bladder (CT KUB) finds evidence of a single ureteral stone measuring 8 mm. He’s prescribed medication for the pain and discharged. The day after his ED visit, he comes to your office to discuss further treatment options. Should you prescribe tamsulosin or nifedipine to help him pass the stone?
The most recent National Health and Nutrition Examination Survey found kidney stones affect 8.8% of the population.2 Outpatient therapy is indicated for patients with ureteric colic secondary to stones ≤10 mm who do not have uncontrolled pain, impaired kidney function, or severe infection. Routine outpatient care includes oral hydration, antiemetics, and pain medications. Medical expulsive therapy (MET) is also used to facilitate stone passage. MET is increasingly becoming part of routine care; use of MET in kidney stone patients in the United States has grown from 14% in 2009 to 64% in 2012.3,4
The joint European Association of Urology/American Urological Association Nephrolithiasis Guideline Panel supports the use of MET.5 Meta-analyses of multiple randomized controlled trials (RCTs) suggest that an alpha-blocker (tamsulosin) or a calcium channel blocker (nifedipine) can reduce pain and lead to quicker stone passage and a higher rate of eventual stone passage when compared to placebo or observation.6,7 However, these reviews included small, heterogeneous studies with a high or unclear risk of bias.
STUDY SUMMARY: MET doesn’t increase the rate of stone passage
The SUSPEND (Spontaneous Urinary Stone Passage ENabled by Drugs) trial1 was a multicenter RCT designed to determine the effectiveness of tamsulosin or nifedipine as MET for patients ages 18 to 65 years with a single ureteric stone measuring ≤10 mm on CT KUB, which has 98% diagnostic accuracy.8 (Stones >10 mm typically require surgery or lithotripsy.)
In this RCT, 1167 adults were randomized to take tamsulosin 0.4 mg/d, nifedipine 30 mg/d, or placebo for 4 weeks or until the stone spontaneously passed, whichever came first. The participants, clinicians, and research staff were blinded to treatment assignment. The primary outcome was the proportion of participants who spontaneously passed their stone, as indicated in patient self-reported questionnaires and case-report forms completed by researchers. Secondary outcomes were time to stone passage and pain as assessed by analgesic use and a visual analogue scale (VAS).
At 4 weeks, 1136 (97%) of the randomized participants had data available for analysis. The proportion of participants who passed their stone did not differ between MET and placebo; 80% of the placebo group (303 of 379 participants) passed the stone, compared with 81% (307 of 378) of the tamsulosin group and 80% (304 of 379) of the nifedipine group. The odds ratio (OR) for MET vs placebo was 1.04 (95% confidence interval [CI], 0.77 to 1.43) and the OR for tamsulosin vs nifedipine was 1.07 (95% CI, 0.74 to 1.53). These findings did not change with further subgroup analysis, including by sex, stone size (≤5 mm vs >5 mm), or stone location.
There were no differences between groups in time to stone passage as measured by clinical report and confirmed by imaging. Time to passage of stone was available for 237 (21%) of participants. The mean days to stone passage was 15.9 (n=84) for placebo, 16.5 (n=79) for tamsulosin and 16.2 (n=74) for nifedipine, with a MET vs placebo difference of 0.5 days (95% CI, -2.9 to 3.9; P=.78). Sensitivity analysis accounting for bias from missing data did not change this outcome.
No differences in analgesic use or pain. Self-reported use of pain medication during the first 4 weeks was similar between groups: 59% (placebo patients), 56% (tamsulosin), and 56% (nifedipine). The mean days of pain medication use was 10.5 for placebo, 11.6 for tamsulosin, and 10.7 for nifedipine, with a MET vs placebo difference of 0.6 days (95% CI, -1.6 to 2.8; P=.45).
There was no difference between groups in the VAS pain score at 4 weeks. The MET vs placebo difference was 0.0 (95% CI, -0.4 to 0.4; P=.96) and the mean VAS pain score was 1.2 for placebo, 1.0 for tamsulosin, and 1.3 for nifedipine.
WHAT'S NEW: This large RCT contradicts results from previous meta-analyses
The SUSPEND study is the first large, multicenter RCT of MET with tamsulosin or nifedipine for kidney stones that used patient-oriented outcomes to find no benefit for stone expulsion, analgesic use, or reported pain compared to placebo. The discrepancy with prior meta-analyses is not unusual. Up to one-third of meta-analyses that show positive outcomes of a therapy are subsequently altered by the inclusion of results from a single, large, multicenter, well-designed RCT.9
CAVEATS: This trial included fewer women than previous studies
The SUSPEND study included a smaller proportion of women than previously published case series due to a need for a diagnostic CT KUB, which excluded more women than men due to radiation concerns. However, the proportion of women was balanced across all groups in this trial, and there was no evidence that sex impacted the efficacy of treatment for the primary outcome.1
CHALLENGES TO IMPLEMENTATION
We see no challenges to the implementation of this recommendation.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
Click here to view PURL METHODOLOGY
1. Pickard R, Starr K, MacLennan G, et al. Medical expulsive therapy in adults with ureteric colic: a multicentre, randomised, placebo-controlled trial. Lancet. 2015;386:341-349.
2. Scales CD Jr., Smith AC, Hanley JM, et al. Prevalence of kidney stones in the United States. Eur Urol. 2012;62:160-165.
3. Fwu CU, Eggers PW, Kimmel PL, et al. Emergency department visits, use of imaging, and drugs for urolithiasis have increased in the United States. Kidney Int. 2013;89:479-486.
4. Bagga H, Appa A, Wang R, et al. 2257 medical expulsion therapy is underutilized in women presenting to an emergency department with acute urinary stone disease. J Urol. 2013;189:e925-e926.
5. Preminger GM, Tiselius HG, Assimos DG, et al; American Urological Association Education and Research, Inc; European Association of Urology. 2007 Guideline for the management of ureteral calculi. Eur Urol. 2007;52:1610-1631.
6. Campschroer T, Zhu Y, Duijvesz D, et al. Alpha-blockers as medical expulsive therapy for ureteral stones. Cochrane Database Syst Rev. 2014;4:CD008509.
7. Seitz C, Liatsikos E, Porpiglia F, et al. Medical therapy to facilitate the passage of stones: what is the evidence? Eur Urol. 2009;56:455-471.
8. Worster A, Preyra I, Weaver B, et al. The accuracy of noncontrast helical computed tomography versus intravenous pyelography in the diagnosis of suspected acute urolithiasis: a meta-analysis. Ann Emerg Med. 2002;40:280-286.
9. LeLorier J, Gregoire G, Benhaddad A, et al. Discrepancies between meta-analyses and subsequent large randomized, controlled trials. N Engl J Med. 1997;337:536-542.
1. Pickard R, Starr K, MacLennan G, et al. Medical expulsive therapy in adults with ureteric colic: a multicentre, randomised, placebo-controlled trial. Lancet. 2015;386:341-349.
2. Scales CD Jr., Smith AC, Hanley JM, et al. Prevalence of kidney stones in the United States. Eur Urol. 2012;62:160-165.
3. Fwu CU, Eggers PW, Kimmel PL, et al. Emergency department visits, use of imaging, and drugs for urolithiasis have increased in the United States. Kidney Int. 2013;89:479-486.
4. Bagga H, Appa A, Wang R, et al. 2257 medical expulsion therapy is underutilized in women presenting to an emergency department with acute urinary stone disease. J Urol. 2013;189:e925-e926.
5. Preminger GM, Tiselius HG, Assimos DG, et al; American Urological Association Education and Research, Inc; European Association of Urology. 2007 Guideline for the management of ureteral calculi. Eur Urol. 2007;52:1610-1631.
6. Campschroer T, Zhu Y, Duijvesz D, et al. Alpha-blockers as medical expulsive therapy for ureteral stones. Cochrane Database Syst Rev. 2014;4:CD008509.
7. Seitz C, Liatsikos E, Porpiglia F, et al. Medical therapy to facilitate the passage of stones: what is the evidence? Eur Urol. 2009;56:455-471.
8. Worster A, Preyra I, Weaver B, et al. The accuracy of noncontrast helical computed tomography versus intravenous pyelography in the diagnosis of suspected acute urolithiasis: a meta-analysis. Ann Emerg Med. 2002;40:280-286.
9. LeLorier J, Gregoire G, Benhaddad A, et al. Discrepancies between meta-analyses and subsequent large randomized, controlled trials. N Engl J Med. 1997;337:536-542.
Copyright © 2016. The Family Physicians Inquiries Network. All rights reserved.
Aneuploidy Screening: Newer Noninvasive Test Gains Traction
PRACTICE CHANGER
Discuss cell-free DNA testing when offering fetal aneuploidy screening to pregnant women.1,2
Strength of recommendation
A: Based on multiple large, multicenter cohort studies.1,2
A 28-year-old woman (gravida 2, para 1001) at 10 weeks’ gestation presents to your clinic for a routine first-trimester prenatal visit. Her first child has no known chromosomal abnormalities, and she has no family history of aneuploidy. She asks you which tests are available to screen her fetus for chromosomal abnormalities.
Pregnant women have traditionally been offered some combination of serum biomarkers and nuchal translucency to assess the risk for fetal aneuploidy. Cell-free DNA testing (cfDNA) is a form of noninvasive prenatal testing that uses maternal serum samples to conduct massively parallel sequencing of cell-free fetal DNA fragments.
It has been offered to pregnant women as a screening test to detect fetal chromosomal abnormalities since 2011, after multiple clinical studies found high sensitivities, specificities, and negative predictive values (NPVs) for detecting aneuploidy.3-6 However, until 2015, practice guidelines from the American Congress of Obstetricians and Gynecologists (ACOG) recommended that standard aneuploidy screening or diagnostic testing be offered to all pregnant women and cfDNA be reserved for women with pregnancies at high risk for aneuploidy (strength of recommendation: B).7
CARE (Comparison of Aneuploidy Risk Evaluation) and NEXT (Noninvasive Examination of Trisomy) are two large studies that compared cfDNA and standard aneuploidy screening methods in pregnant women at low risk for fetal aneuploidy. Based on new data from these and other studies, ACOG and the Society for Maternal-Fetal Medicine (SMFM) released a new consensus statement in June 2015 that addressed the use of cfDNA in the general obstetric population. The two groups still recommend conventional first- and second-trimester screening by serum chemical biomarkers and nuchal translucency as the firstline approach for low-risk women who want to pursue aneuploidy screening; however, they also recommend that the risks and benefits of cfDNA be discussed with all patients.8
Continue for study summaries >>
STUDY SUMMARIES
CARE was a prospective, blinded, multicenter (21 US sites across 14 states) study that compared the aneuploidy detection rates of cfDNA to those of standard screening. Standard aneuploidy screening included assays of first- or second-trimester serum biomarkers with or without fetal nuchal translucency measurement.
This study enrolled 2,042 pregnant patients ages 18 to 49 (mean, 29.6) with singleton pregnancies. The population was racially and ethnically diverse (65% white, 22% black, 11% Hispanic, 7% Asian). This study included women with diabetes, thyroid disorders, and other comorbidities. cfDNA testing was done on 1,909 maternal blood samples for trisomy 21 and 1,905 for trisomy 18.
cfDNA and standard aneuploidy screening results were compared to pregnancy outcomes. The presence of aneuploidy was determined by physician-documented newborn physical exam (97%) or karyotype analysis (3%). In both live and nonlive births, the incidence of trisomy 21 was 5 of 1,909 cases (0.3%) and the incidence of trisomy 18 was 2 of 1,905 cases (0.1%).
The NPV of cfDNA in this study was 100% (95% confidence interval, 99.8%-100%) for both trisomy 21 and trisomy 18. The positive predictive value (PPV) was higher with cfDNA compared to standard screening (45.5% vs 4.2% for trisomy 21 and 40% vs 8.3% for trisomy 18). This means that approximately 1 in 25 women with a positive standard aneuploidy screen actually has aneuploidy. In contrast, nearly 1 in 2 women with a positive cfDNA result has aneuploidy.
Similarly, false-positive rates with cfDNA were significantly lower than those with standard screening. For trisomy 21, the cfDNA false-positive rate was 0.3% compared to 3.6% for standard screening (P < .001); for trisomy 18, the cfDNA false-positive rate was 0.2% compared to 0.6% for standard screening (P = .03).
NEXT was a prospective, blinded cohort study that compared cfDNA testing with standard first-trimester screening (with measurements of nuchal translucency and serum biochemical analysis) in a routine prenatal population at 35 centers in six countries.
This study enrolled 18,955 women ages 18 to 48 (mean, 31) who underwent traditional first-trimester screening and cfDNA testing. Eligible patients included pregnant women with a singleton pregnancy with a gestational age between 10 and 14.3 weeks. Prenatal screening results were compared to newborn outcomes using a documented newborn physical examination and, if performed, results of genetic testing. For women who had a miscarriage or stillbirth or chose to terminate the pregnancy, outcomes were determined by diagnostic genetic testing.
The primary outcome was the area under the receiver-operating-characteristic (ROC) curve for trisomy 21. Area under the ROC curve is a measure of a diagnostic test’s accuracy that plots sensitivity against 1 – specificity; < .700 is considered a poor test, whereas 1.00 is a perfect test. A secondary analysis evaluated cfDNA testing in low-risk women (ages < 35).
The area under the ROC curve was 0.999 for cfDNA compared with 0.958 for standard screening (P = .001). For diagnosis of trisomy 21, cfDNA had a higher PPV than standard testing (80.9% vs 3.4%; P < .001) and a lower false-positive rate (0.06% vs 5.4%; P < .001). These findings were consistent in the secondary analysis of low-risk women.
Both the CARE and NEXT trials also evaluated cfDNA testing versus standard screening for diagnosis of trisomy 13 and 18 and found higher PPVs and lower false-positive rates for cfDNA, compared with traditional screening.
WHAT’S NEW
Previously, cfDNA was recommended only for women with high-risk pregnancies. The new data demonstrate that cfDNA has substantially better PPVs and lower false-positive rates than standard fetal aneuploidy screening for the general obstetric population.
So while conventional screening tests remain the most appropriate methods for aneuploidy detection in the general obstetric population, according to ACOG and SMFM, the two groups now recommend that all screening options—including cfDNA—be discussed with every woman. Any woman may choose cfDNA but should be counseled about the risks and benefits.8
Continue for caveats >>
CAVEATS
Both the CARE and NEXT studies had limitations. They compared cfDNA testing with first- or second-trimester screening and did not evaluate integrated screening methods (sequential first- and second-trimester biomarkers plus first-trimester nuchal translucency), which have a slightly higher sensitivity and specificity than first-trimester screening alone.
Multiple companies offer cfDNA, and the test is not subject to FDA approval. The CARE and NEXT studies used tests from companies that provided funding for these studies and employ several of the study authors.
Although cfDNA has increased specificity compared to standard screening, there have been case reports of false-negative results. Further testing has shown that such false-negative results could be caused by mosaicism in either the fetus and/or placenta, vanishing twins, or maternal malignancies.8-10
In the CARE and NEXT trials, cfDNA produced no results in 0.9% and 3% of women, respectively. Patients for whom cfDNA testing yields no results have higher rates of aneuploidy, and therefore require further diagnostic testing.
Because the prevalence of aneuploidy is lower in the general obstetric population than it is among women whose pregnancies are at high risk for aneuploidy, the PPV of cfDNA testing is also lower in the general obstetric population. This means that there are more false-positive results for women at lower risk for aneuploidy. Therefore, it is imperative that women with positive cfDNA tests receive follow-up diagnostic testing, such as chorionic villus sampling or amniocentesis, before making a decision about termination.
All commercially available cfDNA tests have high sensitivity and specificity for trisomy 21, 18, and 13. Some offer testing for sex chromosome abnormalities and microdeletions. However, current cfDNA testing methods are unable to detect up to 17% of other clinically significant chromosomal abnormalities,11 and cfDNA cannot detect neural tube or ventral wall defects. Therefore, ACOG and SMFM recommend that women who choose cfDNA as their aneuploidy screening method also be offered maternal serum alpha-fetoprotein or ultrasound evaluation.
Continue for challenges to implementation >>
CHALLENGES TO IMPLEMENTATION
cfDNA testing is validated only for singleton pregnancies. Clinicians should obtain a baseline fetal ultrasound to confirm the number of fetuses, gestational age, and viability before ordering cfDNA to ensure it is the most appropriate screening test. This may add to the overall number of early pregnancy ultrasounds conducted.
Counseling patients about aneuploidy screening options is time-consuming and requires discussion of the limitations of each screening method and caution that a negative cfDNA result does not guarantee an unaffected fetus, nor does a positive result guarantee an affected fetus. However, aneuploidy screening is well within the scope of care for family practice clinicians who provide prenatal care, and referral to genetic specialists is not necessary or recommended.
Some patients may request cfDNA in order to facilitate earlier identification of fetal sex. In such cases, clinicians should advise patients that cfDNA testing also assesses trisomy risk. Patients who do not wish to assess their risk for aneuploidy should not receive cfDNA testing.
Finally, while cfDNA is routinely recommended for women with pregnancies considered at high risk for aneuploidy, many insurance companies do not cover the cost of cfDNA for women with low-risk pregnancies, and the test may cost up to $1,700.12 The overall cost-effectiveness of cfDNA for aneuploidy screening in low-risk women is unknown.
References
1. Bianchi DW, Parker RL, Wentworth J, et al; CARE Study Group. DNA sequencing versus standard prenatal aneuploidy screening. N Engl J Med. 2014;370:799-808.
2. Norton ME, Jacobsson B, Swamy GK, et al. Cell-free DNA analysis for noninvasive examination of trisomy. N Engl J Med. 2015;372: 1589-1597.
3. Chiu RW, Akolekar R, Zheng YW, et al. Non-invasive prenatal assessment of trisomy 21 by multiplexed maternal plasma DNA sequencing: large scale validity study. BMJ. 2011; 342:c7401.
4. Ehrich M, Deciu C, Zwiefelhofer T, et al. Noninvasive detection of fetal trisomy 21 by sequencing of DNA in maternal blood: a study in a clinical setting. Am J Obstet Gynecol. 2011;204:205.e1-11.
5. Bianchi DW, Platt LD, Goldberg JD, et al; MatERNal BLood IS Source to Accurately diagnose fetal aneuploidy (MELISSA) Study Group. Genome-wide fetal aneuploidy detection by maternal plasma DNA sequencing. Obstet Gynecol. 2012;119:890-901.
6. Norton ME, Brar H, Weiss J, et al. Non-invasive chromosomal evaluation (NICE) study: results of a multicenter prospective cohort study for detection of fetal trisomy 21 and trisomy 18. Am J Obstet Gynecol. 2012;207: 137.e1-e8.
7. American College of Obstetricians and Gynecologists Committee on Genetics. Committee Opinion No. 545: Noninvasive prenatal testing for fetal aneuploidy. Obstet Gynecol. 2012;120:1532-1534.
8. American College of Obstetricians and Gynecologists Committee on Genetics. Committee Opinion No. 640: Cell-free DNA screening for fetal aneuploidy. Obstet Gynecol. 2015;126:e31-e37.
9. Wang Y, Zhu J, Chen Y, et al. Two cases of placental T21 mosaicism: challenging the detection limits of non-invasive prenatal testing. Prenat Diagn. 2013;33:1207-1210.
10. Choi H, Lau TK, Jiang FM, et al. Fetal aneuploidy screening by maternal plasma DNA sequencing: ‘false positive’ due to confined placental mosaicism. Prenat Diagn. 2013; 33:198-200.
11. Norton ME, Jelliffe-Pawlowski LL, Currier RJ. Chromosome abnormalities detected by current prenatal screening and noninvasive prenatal testing. Obstet Gynecol. 2014;124:979-986.
12. Agarwal A, Sayres LC, Cho MK, et al. Commercial landscape of noninvasive prenatal testing in the United States. Prenat Diagn. 2013;33:521-531.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
Copyright © 2016. The Family Physicians Inquiries Network. All rights reserved.
Reprinted with permission from the Family Physicians Inquiries Network and The Journal of Family Practice. 2016;65(1):49-52.
PRACTICE CHANGER
Discuss cell-free DNA testing when offering fetal aneuploidy screening to pregnant women.1,2
Strength of recommendation
A: Based on multiple large, multicenter cohort studies.1,2
A 28-year-old woman (gravida 2, para 1001) at 10 weeks’ gestation presents to your clinic for a routine first-trimester prenatal visit. Her first child has no known chromosomal abnormalities, and she has no family history of aneuploidy. She asks you which tests are available to screen her fetus for chromosomal abnormalities.
Pregnant women have traditionally been offered some combination of serum biomarkers and nuchal translucency to assess the risk for fetal aneuploidy. Cell-free DNA testing (cfDNA) is a form of noninvasive prenatal testing that uses maternal serum samples to conduct massively parallel sequencing of cell-free fetal DNA fragments.
It has been offered to pregnant women as a screening test to detect fetal chromosomal abnormalities since 2011, after multiple clinical studies found high sensitivities, specificities, and negative predictive values (NPVs) for detecting aneuploidy.3-6 However, until 2015, practice guidelines from the American Congress of Obstetricians and Gynecologists (ACOG) recommended that standard aneuploidy screening or diagnostic testing be offered to all pregnant women and cfDNA be reserved for women with pregnancies at high risk for aneuploidy (strength of recommendation: B).7
CARE (Comparison of Aneuploidy Risk Evaluation) and NEXT (Noninvasive Examination of Trisomy) are two large studies that compared cfDNA and standard aneuploidy screening methods in pregnant women at low risk for fetal aneuploidy. Based on new data from these and other studies, ACOG and the Society for Maternal-Fetal Medicine (SMFM) released a new consensus statement in June 2015 that addressed the use of cfDNA in the general obstetric population. The two groups still recommend conventional first- and second-trimester screening by serum chemical biomarkers and nuchal translucency as the firstline approach for low-risk women who want to pursue aneuploidy screening; however, they also recommend that the risks and benefits of cfDNA be discussed with all patients.8
Continue for study summaries >>
STUDY SUMMARIES
CARE was a prospective, blinded, multicenter (21 US sites across 14 states) study that compared the aneuploidy detection rates of cfDNA to those of standard screening. Standard aneuploidy screening included assays of first- or second-trimester serum biomarkers with or without fetal nuchal translucency measurement.
This study enrolled 2,042 pregnant patients ages 18 to 49 (mean, 29.6) with singleton pregnancies. The population was racially and ethnically diverse (65% white, 22% black, 11% Hispanic, 7% Asian). This study included women with diabetes, thyroid disorders, and other comorbidities. cfDNA testing was done on 1,909 maternal blood samples for trisomy 21 and 1,905 for trisomy 18.
cfDNA and standard aneuploidy screening results were compared to pregnancy outcomes. The presence of aneuploidy was determined by physician-documented newborn physical exam (97%) or karyotype analysis (3%). In both live and nonlive births, the incidence of trisomy 21 was 5 of 1,909 cases (0.3%) and the incidence of trisomy 18 was 2 of 1,905 cases (0.1%).
The NPV of cfDNA in this study was 100% (95% confidence interval, 99.8%-100%) for both trisomy 21 and trisomy 18. The positive predictive value (PPV) was higher with cfDNA compared to standard screening (45.5% vs 4.2% for trisomy 21 and 40% vs 8.3% for trisomy 18). This means that approximately 1 in 25 women with a positive standard aneuploidy screen actually has aneuploidy. In contrast, nearly 1 in 2 women with a positive cfDNA result has aneuploidy.
Similarly, false-positive rates with cfDNA were significantly lower than those with standard screening. For trisomy 21, the cfDNA false-positive rate was 0.3% compared to 3.6% for standard screening (P < .001); for trisomy 18, the cfDNA false-positive rate was 0.2% compared to 0.6% for standard screening (P = .03).
NEXT was a prospective, blinded cohort study that compared cfDNA testing with standard first-trimester screening (with measurements of nuchal translucency and serum biochemical analysis) in a routine prenatal population at 35 centers in six countries.
This study enrolled 18,955 women ages 18 to 48 (mean, 31) who underwent traditional first-trimester screening and cfDNA testing. Eligible patients included pregnant women with a singleton pregnancy with a gestational age between 10 and 14.3 weeks. Prenatal screening results were compared to newborn outcomes using a documented newborn physical examination and, if performed, results of genetic testing. For women who had a miscarriage or stillbirth or chose to terminate the pregnancy, outcomes were determined by diagnostic genetic testing.
The primary outcome was the area under the receiver-operating-characteristic (ROC) curve for trisomy 21. Area under the ROC curve is a measure of a diagnostic test’s accuracy that plots sensitivity against 1 – specificity; < .700 is considered a poor test, whereas 1.00 is a perfect test. A secondary analysis evaluated cfDNA testing in low-risk women (ages < 35).
The area under the ROC curve was 0.999 for cfDNA compared with 0.958 for standard screening (P = .001). For diagnosis of trisomy 21, cfDNA had a higher PPV than standard testing (80.9% vs 3.4%; P < .001) and a lower false-positive rate (0.06% vs 5.4%; P < .001). These findings were consistent in the secondary analysis of low-risk women.
Both the CARE and NEXT trials also evaluated cfDNA testing versus standard screening for diagnosis of trisomy 13 and 18 and found higher PPVs and lower false-positive rates for cfDNA, compared with traditional screening.
WHAT’S NEW
Previously, cfDNA was recommended only for women with high-risk pregnancies. The new data demonstrate that cfDNA has substantially better PPVs and lower false-positive rates than standard fetal aneuploidy screening for the general obstetric population.
So while conventional screening tests remain the most appropriate methods for aneuploidy detection in the general obstetric population, according to ACOG and SMFM, the two groups now recommend that all screening options—including cfDNA—be discussed with every woman. Any woman may choose cfDNA but should be counseled about the risks and benefits.8
Continue for caveats >>
CAVEATS
Both the CARE and NEXT studies had limitations. They compared cfDNA testing with first- or second-trimester screening and did not evaluate integrated screening methods (sequential first- and second-trimester biomarkers plus first-trimester nuchal translucency), which have a slightly higher sensitivity and specificity than first-trimester screening alone.
Multiple companies offer cfDNA, and the test is not subject to FDA approval. The CARE and NEXT studies used tests from companies that provided funding for these studies and employ several of the study authors.
Although cfDNA has increased specificity compared to standard screening, there have been case reports of false-negative results. Further testing has shown that such false-negative results could be caused by mosaicism in either the fetus and/or placenta, vanishing twins, or maternal malignancies.8-10
In the CARE and NEXT trials, cfDNA produced no results in 0.9% and 3% of women, respectively. Patients for whom cfDNA testing yields no results have higher rates of aneuploidy, and therefore require further diagnostic testing.
Because the prevalence of aneuploidy is lower in the general obstetric population than it is among women whose pregnancies are at high risk for aneuploidy, the PPV of cfDNA testing is also lower in the general obstetric population. This means that there are more false-positive results for women at lower risk for aneuploidy. Therefore, it is imperative that women with positive cfDNA tests receive follow-up diagnostic testing, such as chorionic villus sampling or amniocentesis, before making a decision about termination.
All commercially available cfDNA tests have high sensitivity and specificity for trisomy 21, 18, and 13. Some offer testing for sex chromosome abnormalities and microdeletions. However, current cfDNA testing methods are unable to detect up to 17% of other clinically significant chromosomal abnormalities,11 and cfDNA cannot detect neural tube or ventral wall defects. Therefore, ACOG and SMFM recommend that women who choose cfDNA as their aneuploidy screening method also be offered maternal serum alpha-fetoprotein or ultrasound evaluation.
Continue for challenges to implementation >>
CHALLENGES TO IMPLEMENTATION
cfDNA testing is validated only for singleton pregnancies. Clinicians should obtain a baseline fetal ultrasound to confirm the number of fetuses, gestational age, and viability before ordering cfDNA to ensure it is the most appropriate screening test. This may add to the overall number of early pregnancy ultrasounds conducted.
Counseling patients about aneuploidy screening options is time-consuming and requires discussion of the limitations of each screening method and caution that a negative cfDNA result does not guarantee an unaffected fetus, nor does a positive result guarantee an affected fetus. However, aneuploidy screening is well within the scope of care for family practice clinicians who provide prenatal care, and referral to genetic specialists is not necessary or recommended.
Some patients may request cfDNA in order to facilitate earlier identification of fetal sex. In such cases, clinicians should advise patients that cfDNA testing also assesses trisomy risk. Patients who do not wish to assess their risk for aneuploidy should not receive cfDNA testing.
Finally, while cfDNA is routinely recommended for women with pregnancies considered at high risk for aneuploidy, many insurance companies do not cover the cost of cfDNA for women with low-risk pregnancies, and the test may cost up to $1,700.12 The overall cost-effectiveness of cfDNA for aneuploidy screening in low-risk women is unknown.
References
1. Bianchi DW, Parker RL, Wentworth J, et al; CARE Study Group. DNA sequencing versus standard prenatal aneuploidy screening. N Engl J Med. 2014;370:799-808.
2. Norton ME, Jacobsson B, Swamy GK, et al. Cell-free DNA analysis for noninvasive examination of trisomy. N Engl J Med. 2015;372: 1589-1597.
3. Chiu RW, Akolekar R, Zheng YW, et al. Non-invasive prenatal assessment of trisomy 21 by multiplexed maternal plasma DNA sequencing: large scale validity study. BMJ. 2011; 342:c7401.
4. Ehrich M, Deciu C, Zwiefelhofer T, et al. Noninvasive detection of fetal trisomy 21 by sequencing of DNA in maternal blood: a study in a clinical setting. Am J Obstet Gynecol. 2011;204:205.e1-11.
5. Bianchi DW, Platt LD, Goldberg JD, et al; MatERNal BLood IS Source to Accurately diagnose fetal aneuploidy (MELISSA) Study Group. Genome-wide fetal aneuploidy detection by maternal plasma DNA sequencing. Obstet Gynecol. 2012;119:890-901.
6. Norton ME, Brar H, Weiss J, et al. Non-invasive chromosomal evaluation (NICE) study: results of a multicenter prospective cohort study for detection of fetal trisomy 21 and trisomy 18. Am J Obstet Gynecol. 2012;207: 137.e1-e8.
7. American College of Obstetricians and Gynecologists Committee on Genetics. Committee Opinion No. 545: Noninvasive prenatal testing for fetal aneuploidy. Obstet Gynecol. 2012;120:1532-1534.
8. American College of Obstetricians and Gynecologists Committee on Genetics. Committee Opinion No. 640: Cell-free DNA screening for fetal aneuploidy. Obstet Gynecol. 2015;126:e31-e37.
9. Wang Y, Zhu J, Chen Y, et al. Two cases of placental T21 mosaicism: challenging the detection limits of non-invasive prenatal testing. Prenat Diagn. 2013;33:1207-1210.
10. Choi H, Lau TK, Jiang FM, et al. Fetal aneuploidy screening by maternal plasma DNA sequencing: ‘false positive’ due to confined placental mosaicism. Prenat Diagn. 2013; 33:198-200.
11. Norton ME, Jelliffe-Pawlowski LL, Currier RJ. Chromosome abnormalities detected by current prenatal screening and noninvasive prenatal testing. Obstet Gynecol. 2014;124:979-986.
12. Agarwal A, Sayres LC, Cho MK, et al. Commercial landscape of noninvasive prenatal testing in the United States. Prenat Diagn. 2013;33:521-531.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
Copyright © 2016. The Family Physicians Inquiries Network. All rights reserved.
Reprinted with permission from the Family Physicians Inquiries Network and The Journal of Family Practice. 2016;65(1):49-52.
PRACTICE CHANGER
Discuss cell-free DNA testing when offering fetal aneuploidy screening to pregnant women.1,2
Strength of recommendation
A: Based on multiple large, multicenter cohort studies.1,2
A 28-year-old woman (gravida 2, para 1001) at 10 weeks’ gestation presents to your clinic for a routine first-trimester prenatal visit. Her first child has no known chromosomal abnormalities, and she has no family history of aneuploidy. She asks you which tests are available to screen her fetus for chromosomal abnormalities.
Pregnant women have traditionally been offered some combination of serum biomarkers and nuchal translucency to assess the risk for fetal aneuploidy. Cell-free DNA testing (cfDNA) is a form of noninvasive prenatal testing that uses maternal serum samples to conduct massively parallel sequencing of cell-free fetal DNA fragments.
It has been offered to pregnant women as a screening test to detect fetal chromosomal abnormalities since 2011, after multiple clinical studies found high sensitivities, specificities, and negative predictive values (NPVs) for detecting aneuploidy.3-6 However, until 2015, practice guidelines from the American Congress of Obstetricians and Gynecologists (ACOG) recommended that standard aneuploidy screening or diagnostic testing be offered to all pregnant women and cfDNA be reserved for women with pregnancies at high risk for aneuploidy (strength of recommendation: B).7
CARE (Comparison of Aneuploidy Risk Evaluation) and NEXT (Noninvasive Examination of Trisomy) are two large studies that compared cfDNA and standard aneuploidy screening methods in pregnant women at low risk for fetal aneuploidy. Based on new data from these and other studies, ACOG and the Society for Maternal-Fetal Medicine (SMFM) released a new consensus statement in June 2015 that addressed the use of cfDNA in the general obstetric population. The two groups still recommend conventional first- and second-trimester screening by serum chemical biomarkers and nuchal translucency as the firstline approach for low-risk women who want to pursue aneuploidy screening; however, they also recommend that the risks and benefits of cfDNA be discussed with all patients.8
Continue for study summaries >>
STUDY SUMMARIES
CARE was a prospective, blinded, multicenter (21 US sites across 14 states) study that compared the aneuploidy detection rates of cfDNA to those of standard screening. Standard aneuploidy screening included assays of first- or second-trimester serum biomarkers with or without fetal nuchal translucency measurement.
This study enrolled 2,042 pregnant patients ages 18 to 49 (mean, 29.6) with singleton pregnancies. The population was racially and ethnically diverse (65% white, 22% black, 11% Hispanic, 7% Asian). This study included women with diabetes, thyroid disorders, and other comorbidities. cfDNA testing was done on 1,909 maternal blood samples for trisomy 21 and 1,905 for trisomy 18.
cfDNA and standard aneuploidy screening results were compared to pregnancy outcomes. The presence of aneuploidy was determined by physician-documented newborn physical exam (97%) or karyotype analysis (3%). In both live and nonlive births, the incidence of trisomy 21 was 5 of 1,909 cases (0.3%) and the incidence of trisomy 18 was 2 of 1,905 cases (0.1%).
The NPV of cfDNA in this study was 100% (95% confidence interval, 99.8%-100%) for both trisomy 21 and trisomy 18. The positive predictive value (PPV) was higher with cfDNA compared to standard screening (45.5% vs 4.2% for trisomy 21 and 40% vs 8.3% for trisomy 18). This means that approximately 1 in 25 women with a positive standard aneuploidy screen actually has aneuploidy. In contrast, nearly 1 in 2 women with a positive cfDNA result has aneuploidy.
Similarly, false-positive rates with cfDNA were significantly lower than those with standard screening. For trisomy 21, the cfDNA false-positive rate was 0.3% compared to 3.6% for standard screening (P < .001); for trisomy 18, the cfDNA false-positive rate was 0.2% compared to 0.6% for standard screening (P = .03).
NEXT was a prospective, blinded cohort study that compared cfDNA testing with standard first-trimester screening (with measurements of nuchal translucency and serum biochemical analysis) in a routine prenatal population at 35 centers in six countries.
This study enrolled 18,955 women ages 18 to 48 (mean, 31) who underwent traditional first-trimester screening and cfDNA testing. Eligible patients included pregnant women with a singleton pregnancy with a gestational age between 10 and 14.3 weeks. Prenatal screening results were compared to newborn outcomes using a documented newborn physical examination and, if performed, results of genetic testing. For women who had a miscarriage or stillbirth or chose to terminate the pregnancy, outcomes were determined by diagnostic genetic testing.
The primary outcome was the area under the receiver-operating-characteristic (ROC) curve for trisomy 21. Area under the ROC curve is a measure of a diagnostic test’s accuracy that plots sensitivity against 1 – specificity; < .700 is considered a poor test, whereas 1.00 is a perfect test. A secondary analysis evaluated cfDNA testing in low-risk women (ages < 35).
The area under the ROC curve was 0.999 for cfDNA compared with 0.958 for standard screening (P = .001). For diagnosis of trisomy 21, cfDNA had a higher PPV than standard testing (80.9% vs 3.4%; P < .001) and a lower false-positive rate (0.06% vs 5.4%; P < .001). These findings were consistent in the secondary analysis of low-risk women.
Both the CARE and NEXT trials also evaluated cfDNA testing versus standard screening for diagnosis of trisomy 13 and 18 and found higher PPVs and lower false-positive rates for cfDNA, compared with traditional screening.
WHAT’S NEW
Previously, cfDNA was recommended only for women with high-risk pregnancies. The new data demonstrate that cfDNA has substantially better PPVs and lower false-positive rates than standard fetal aneuploidy screening for the general obstetric population.
So while conventional screening tests remain the most appropriate methods for aneuploidy detection in the general obstetric population, according to ACOG and SMFM, the two groups now recommend that all screening options—including cfDNA—be discussed with every woman. Any woman may choose cfDNA but should be counseled about the risks and benefits.8
Continue for caveats >>
CAVEATS
Both the CARE and NEXT studies had limitations. They compared cfDNA testing with first- or second-trimester screening and did not evaluate integrated screening methods (sequential first- and second-trimester biomarkers plus first-trimester nuchal translucency), which have a slightly higher sensitivity and specificity than first-trimester screening alone.
Multiple companies offer cfDNA, and the test is not subject to FDA approval. The CARE and NEXT studies used tests from companies that provided funding for these studies and employ several of the study authors.
Although cfDNA has increased specificity compared to standard screening, there have been case reports of false-negative results. Further testing has shown that such false-negative results could be caused by mosaicism in either the fetus and/or placenta, vanishing twins, or maternal malignancies.8-10
In the CARE and NEXT trials, cfDNA produced no results in 0.9% and 3% of women, respectively. Patients for whom cfDNA testing yields no results have higher rates of aneuploidy, and therefore require further diagnostic testing.
Because the prevalence of aneuploidy is lower in the general obstetric population than it is among women whose pregnancies are at high risk for aneuploidy, the PPV of cfDNA testing is also lower in the general obstetric population. This means that there are more false-positive results for women at lower risk for aneuploidy. Therefore, it is imperative that women with positive cfDNA tests receive follow-up diagnostic testing, such as chorionic villus sampling or amniocentesis, before making a decision about termination.
All commercially available cfDNA tests have high sensitivity and specificity for trisomy 21, 18, and 13. Some offer testing for sex chromosome abnormalities and microdeletions. However, current cfDNA testing methods are unable to detect up to 17% of other clinically significant chromosomal abnormalities,11 and cfDNA cannot detect neural tube or ventral wall defects. Therefore, ACOG and SMFM recommend that women who choose cfDNA as their aneuploidy screening method also be offered maternal serum alpha-fetoprotein or ultrasound evaluation.
Continue for challenges to implementation >>
CHALLENGES TO IMPLEMENTATION
cfDNA testing is validated only for singleton pregnancies. Clinicians should obtain a baseline fetal ultrasound to confirm the number of fetuses, gestational age, and viability before ordering cfDNA to ensure it is the most appropriate screening test. This may add to the overall number of early pregnancy ultrasounds conducted.
Counseling patients about aneuploidy screening options is time-consuming and requires discussion of the limitations of each screening method and caution that a negative cfDNA result does not guarantee an unaffected fetus, nor does a positive result guarantee an affected fetus. However, aneuploidy screening is well within the scope of care for family practice clinicians who provide prenatal care, and referral to genetic specialists is not necessary or recommended.
Some patients may request cfDNA in order to facilitate earlier identification of fetal sex. In such cases, clinicians should advise patients that cfDNA testing also assesses trisomy risk. Patients who do not wish to assess their risk for aneuploidy should not receive cfDNA testing.
Finally, while cfDNA is routinely recommended for women with pregnancies considered at high risk for aneuploidy, many insurance companies do not cover the cost of cfDNA for women with low-risk pregnancies, and the test may cost up to $1,700.12 The overall cost-effectiveness of cfDNA for aneuploidy screening in low-risk women is unknown.
References
1. Bianchi DW, Parker RL, Wentworth J, et al; CARE Study Group. DNA sequencing versus standard prenatal aneuploidy screening. N Engl J Med. 2014;370:799-808.
2. Norton ME, Jacobsson B, Swamy GK, et al. Cell-free DNA analysis for noninvasive examination of trisomy. N Engl J Med. 2015;372: 1589-1597.
3. Chiu RW, Akolekar R, Zheng YW, et al. Non-invasive prenatal assessment of trisomy 21 by multiplexed maternal plasma DNA sequencing: large scale validity study. BMJ. 2011; 342:c7401.
4. Ehrich M, Deciu C, Zwiefelhofer T, et al. Noninvasive detection of fetal trisomy 21 by sequencing of DNA in maternal blood: a study in a clinical setting. Am J Obstet Gynecol. 2011;204:205.e1-11.
5. Bianchi DW, Platt LD, Goldberg JD, et al; MatERNal BLood IS Source to Accurately diagnose fetal aneuploidy (MELISSA) Study Group. Genome-wide fetal aneuploidy detection by maternal plasma DNA sequencing. Obstet Gynecol. 2012;119:890-901.
6. Norton ME, Brar H, Weiss J, et al. Non-invasive chromosomal evaluation (NICE) study: results of a multicenter prospective cohort study for detection of fetal trisomy 21 and trisomy 18. Am J Obstet Gynecol. 2012;207: 137.e1-e8.
7. American College of Obstetricians and Gynecologists Committee on Genetics. Committee Opinion No. 545: Noninvasive prenatal testing for fetal aneuploidy. Obstet Gynecol. 2012;120:1532-1534.
8. American College of Obstetricians and Gynecologists Committee on Genetics. Committee Opinion No. 640: Cell-free DNA screening for fetal aneuploidy. Obstet Gynecol. 2015;126:e31-e37.
9. Wang Y, Zhu J, Chen Y, et al. Two cases of placental T21 mosaicism: challenging the detection limits of non-invasive prenatal testing. Prenat Diagn. 2013;33:1207-1210.
10. Choi H, Lau TK, Jiang FM, et al. Fetal aneuploidy screening by maternal plasma DNA sequencing: ‘false positive’ due to confined placental mosaicism. Prenat Diagn. 2013; 33:198-200.
11. Norton ME, Jelliffe-Pawlowski LL, Currier RJ. Chromosome abnormalities detected by current prenatal screening and noninvasive prenatal testing. Obstet Gynecol. 2014;124:979-986.
12. Agarwal A, Sayres LC, Cho MK, et al. Commercial landscape of noninvasive prenatal testing in the United States. Prenat Diagn. 2013;33:521-531.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
Copyright © 2016. The Family Physicians Inquiries Network. All rights reserved.
Reprinted with permission from the Family Physicians Inquiries Network and The Journal of Family Practice. 2016;65(1):49-52.