User login
Pushback on Part B drug payment proposal already beginning
Rheumatologists already are voicing concerns regarding a new proposal to test adjustments to how drugs administered in a physician’s office are paid for.
That proposal, published March 11 in the Federal Register, would test a change to the current reimbursement of average sales price plus 6% for Part B drugs with a lower add-on percentage of 2.5% plus $16.50.

In a fact sheet highlighting the proposals, the Centers for Medicare & Medicaid Services said the change to a lower percentage plus a flat fee “will cover the cost of any drug paid under Medicare Part B.”
However, there are already questions about that.
“While this may seem the way in which CMS will control costs, they fail to recognize the cost to facilities in obtaining approval for these treatments, receiving and storing, and ultimately safely administering these therapies in an environment that provides the best outcomes for patients,” Dr. Norman B. Gaylis, a rheumatologist in private practice in Aventura, Fla., said.
In fact, Dr. Gaylis adds that the focus on lowering drug expenses in the Part B space could have the unintended consequence of raising these expenses because it will force a change of venue.
“It has become so prohibitive that ultimately many patients will be referred to more expensive, less efficient outpatient facilities with, in fact, an increase in overall costs,” he said.
More than 300 provider and patient groups covering a range of specialties and including the American College of Rheumatology, the Coalition of State Rheumatology Organizations, and a number of state rheumatology organizations, are calling on Congress to ask CMS to withdraw the proposal.
In a March 17 letter to the majority and minority leaders in both chambers, the group is challenging the CMS assertion in the proposed rule that the current 6% add-on “may encourage the use of more expensive drugs because the 6% add-on generates more revenues for more expensive drugs.”
“This assumption fails to take into account the fact that providers’ prescribing decisions depend on a variety of factors, including clinical characteristics and the complex needs of the Medicare population,” the letter states. “Most importantly, there is no evidence indicating that the payment changes contemplated by the model will improve quality of care, and may adversely impact those patients that lose access to their most appropriate treatments.”
CMS offered two other pricing models that would be tested: indications-based pricing and reference pricing. The former would set payment rates based on the clinical effectiveness of a drug, while the latter would test the impact of setting a benchmark price for a group of drugs in a similar therapeutic class. Related to that is a proposal that CMS enter into voluntary risk-sharing agreements with drug manufacturers to link outcomes with price adjustments.
Dr. Gaylis suggested that CMS is going after the wrong party if cost containment is the ultimate goal here and should be focusing its efforts on the prices of the drugs themselves rather than how much they spend on physician reimbursement.
“Ironically, the major expense, i.e., the cost of drugs themselves, continues to spiral in the absence of any legitimate mechanism between CMS and pharma to contract prices that could save health care billions of dollars,” he said. “Ultimately, in my opinion, the solution rests in creating a fair and equal price for facilities administering these therapies and creating a pass-through where the drugs are not part of the physician’s risk, cost, or benefit and all payers, including CMS, can negotiate drug costs directly with the manufacturer.”
As part of the proposed rule, CMS also is considering creating feedback and decision-support tools to help, such as offering best practices for prescribing certain medications or providing feedback on prescribing patterns relative to local, regional, and national trends.
On the patient side, CMS is proposing to eliminate any patient cost sharing for office-administered drugs.
Comments on the proposals are due May 9.
Rheumatologists already are voicing concerns regarding a new proposal to test adjustments to how drugs administered in a physician’s office are paid for.
That proposal, published March 11 in the Federal Register, would test a change to the current reimbursement of average sales price plus 6% for Part B drugs with a lower add-on percentage of 2.5% plus $16.50.
In a fact sheet highlighting the proposals, the Centers for Medicare & Medicaid Services said the change to a lower percentage plus a flat fee “will cover the cost of any drug paid under Medicare Part B.”
However, there are already questions about that.
“While this may seem the way in which CMS will control costs, they fail to recognize the cost to facilities in obtaining approval for these treatments, receiving and storing, and ultimately safely administering these therapies in an environment that provides the best outcomes for patients,” Dr. Norman B. Gaylis, a rheumatologist in private practice in Aventura, Fla., said.
In fact, Dr. Gaylis adds that the focus on lowering drug expenses in the Part B space could have the unintended consequence of raising these expenses because it will force a change of venue.
“It has become so prohibitive that ultimately many patients will be referred to more expensive, less efficient outpatient facilities with, in fact, an increase in overall costs,” he said.
More than 300 provider and patient groups covering a range of specialties and including the American College of Rheumatology, the Coalition of State Rheumatology Organizations, and a number of state rheumatology organizations, are calling on Congress to ask CMS to withdraw the proposal.
In a March 17 letter to the majority and minority leaders in both chambers, the group is challenging the CMS assertion in the proposed rule that the current 6% add-on “may encourage the use of more expensive drugs because the 6% add-on generates more revenues for more expensive drugs.”
“This assumption fails to take into account the fact that providers’ prescribing decisions depend on a variety of factors, including clinical characteristics and the complex needs of the Medicare population,” the letter states. “Most importantly, there is no evidence indicating that the payment changes contemplated by the model will improve quality of care, and may adversely impact those patients that lose access to their most appropriate treatments.”
CMS offered two other pricing models that would be tested: indications-based pricing and reference pricing. The former would set payment rates based on the clinical effectiveness of a drug, while the latter would test the impact of setting a benchmark price for a group of drugs in a similar therapeutic class. Related to that is a proposal that CMS enter into voluntary risk-sharing agreements with drug manufacturers to link outcomes with price adjustments.
Dr. Gaylis suggested that CMS is going after the wrong party if cost containment is the ultimate goal here and should be focusing its efforts on the prices of the drugs themselves rather than how much they spend on physician reimbursement.
“Ironically, the major expense, i.e., the cost of drugs themselves, continues to spiral in the absence of any legitimate mechanism between CMS and pharma to contract prices that could save health care billions of dollars,” he said. “Ultimately, in my opinion, the solution rests in creating a fair and equal price for facilities administering these therapies and creating a pass-through where the drugs are not part of the physician’s risk, cost, or benefit and all payers, including CMS, can negotiate drug costs directly with the manufacturer.”
As part of the proposed rule, CMS also is considering creating feedback and decision-support tools to help, such as offering best practices for prescribing certain medications or providing feedback on prescribing patterns relative to local, regional, and national trends.
On the patient side, CMS is proposing to eliminate any patient cost sharing for office-administered drugs.
Comments on the proposals are due May 9.
Rheumatologists already are voicing concerns regarding a new proposal to test adjustments to how drugs administered in a physician’s office are paid for.
That proposal, published March 11 in the Federal Register, would test a change to the current reimbursement of average sales price plus 6% for Part B drugs with a lower add-on percentage of 2.5% plus $16.50.
In a fact sheet highlighting the proposals, the Centers for Medicare & Medicaid Services said the change to a lower percentage plus a flat fee “will cover the cost of any drug paid under Medicare Part B.”
However, there are already questions about that.
“While this may seem the way in which CMS will control costs, they fail to recognize the cost to facilities in obtaining approval for these treatments, receiving and storing, and ultimately safely administering these therapies in an environment that provides the best outcomes for patients,” Dr. Norman B. Gaylis, a rheumatologist in private practice in Aventura, Fla., said.
In fact, Dr. Gaylis adds that the focus on lowering drug expenses in the Part B space could have the unintended consequence of raising these expenses because it will force a change of venue.
“It has become so prohibitive that ultimately many patients will be referred to more expensive, less efficient outpatient facilities with, in fact, an increase in overall costs,” he said.
More than 300 provider and patient groups covering a range of specialties and including the American College of Rheumatology, the Coalition of State Rheumatology Organizations, and a number of state rheumatology organizations, are calling on Congress to ask CMS to withdraw the proposal.
In a March 17 letter to the majority and minority leaders in both chambers, the group is challenging the CMS assertion in the proposed rule that the current 6% add-on “may encourage the use of more expensive drugs because the 6% add-on generates more revenues for more expensive drugs.”
“This assumption fails to take into account the fact that providers’ prescribing decisions depend on a variety of factors, including clinical characteristics and the complex needs of the Medicare population,” the letter states. “Most importantly, there is no evidence indicating that the payment changes contemplated by the model will improve quality of care, and may adversely impact those patients that lose access to their most appropriate treatments.”
CMS offered two other pricing models that would be tested: indications-based pricing and reference pricing. The former would set payment rates based on the clinical effectiveness of a drug, while the latter would test the impact of setting a benchmark price for a group of drugs in a similar therapeutic class. Related to that is a proposal that CMS enter into voluntary risk-sharing agreements with drug manufacturers to link outcomes with price adjustments.
Dr. Gaylis suggested that CMS is going after the wrong party if cost containment is the ultimate goal here and should be focusing its efforts on the prices of the drugs themselves rather than how much they spend on physician reimbursement.
“Ironically, the major expense, i.e., the cost of drugs themselves, continues to spiral in the absence of any legitimate mechanism between CMS and pharma to contract prices that could save health care billions of dollars,” he said. “Ultimately, in my opinion, the solution rests in creating a fair and equal price for facilities administering these therapies and creating a pass-through where the drugs are not part of the physician’s risk, cost, or benefit and all payers, including CMS, can negotiate drug costs directly with the manufacturer.”
As part of the proposed rule, CMS also is considering creating feedback and decision-support tools to help, such as offering best practices for prescribing certain medications or providing feedback on prescribing patterns relative to local, regional, and national trends.
On the patient side, CMS is proposing to eliminate any patient cost sharing for office-administered drugs.
Comments on the proposals are due May 9.

Home infusion policies called out in ACR position statement
Proper administration of intravenous biologics should take place under the close supervision of a physician in a physician’s office, infusion center, or hospital rather than in a patient’s home in order to address potential infusion reactions that can range from mild to life threatening, according to a position statement issued by the American College of Rheumatology’s Committee on Rheumatologic Care.
The “Patient Safety and Site of Service for Infusible Biologics” statement, issued in late February, comes in opposition to “policies that require home infusion” that appear to seek potential cost savings with home infusions rather than meet the standard of care with on-site physician supervision.
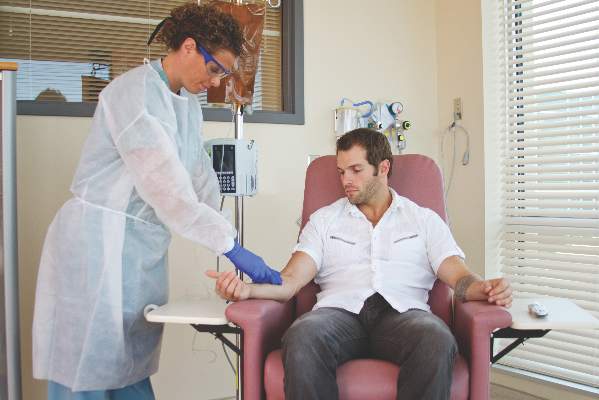
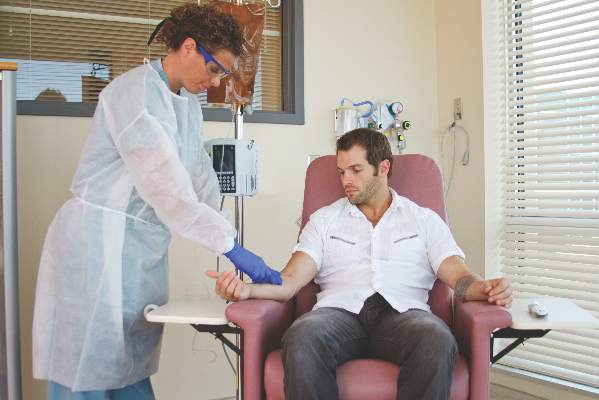
“One observation made by some but not all payers is that infusible biologics are about twice as expensive when infused in a hospital-based infusion center as compared to other locations, such as a clinic-based infusion center or the patient’s home. Thus, some payers are rolling out policies designed to shift patients from hospital-based infusion centers to less expensive sites. The ACR is opposed to policies that would force patients, solely for the purpose of cost containment, to receive infusible biologics in an improperly supervised setting. The purpose of the position statement is to outline that stance,” Dr. Douglas W. White, chair of the ACR’s Committee on Rheumatologic Care, said in an interview.
He noted that he’s “been in on conversations with two payers who are implementing policies to move patients away from hospital-based infusions, but we are aware that others are in various stages of implementing such policies, too. It’s not so much an issue of critical mass for us, rather we’re just trying to keep ahead of the trends, and we think this will be a big trend.”
The potential for adverse reactions is not uncommon during intravenous administration of biologics, the committee wrote, noting, for example, that 10% of patients given infliximab have acute infusion reactions. On-site physicians such as rheumatologists who have experience with the “tremendous heterogeneity of patients with autoimmune disease and the diversity of conditions treated with biologics” can determine the severity of infusion reactions and decide whether or not it is safe to continue a particular biologic agent, in addition to providing reassurance to patients during acute and potentially severe reactions, according to the ACR statement.
Infusion reactions can range in severity from a mild rash to life-threatening anaphylaxis that can involve multiple organ systems leading to respiratory and cardiovascular collapse and requiring immediate treatment with medications such as epinephrine or intravenous glucocorticoids.
The position statement recognizes unusual situations in which home infusion is necessary for a patient to receive treatment because of transportation problems to a medical facility or comorbid conditions in which the risk of no treatment may outweigh the risk of home infusion. In these circumstances, the ACR “encourages providers in such unusual and difficult situations to make the best medical decision based on the individual needs of the patient. Routine home infusion of biologics is considered an unnecessary and dangerous risk to patients and violates our current clinical standards of practice.”
Requirements for using home infusion also threaten “to reduce access to” intravenous biologics, the ACR contends, because “specially trained physicians are less likely to prescribe treatments that are not properly administered in the safest clinical setting [and] patient fear of biologic therapy may lead to noncompliance and inadequate control of disease.”
The ACR noted that home administration of subcutaneous biologics is medically appropriate and the injection site reactions that can occur with their use are often easily managed.
Proper administration of intravenous biologics should take place under the close supervision of a physician in a physician’s office, infusion center, or hospital rather than in a patient’s home in order to address potential infusion reactions that can range from mild to life threatening, according to a position statement issued by the American College of Rheumatology’s Committee on Rheumatologic Care.
The “Patient Safety and Site of Service for Infusible Biologics” statement, issued in late February, comes in opposition to “policies that require home infusion” that appear to seek potential cost savings with home infusions rather than meet the standard of care with on-site physician supervision.
“One observation made by some but not all payers is that infusible biologics are about twice as expensive when infused in a hospital-based infusion center as compared to other locations, such as a clinic-based infusion center or the patient’s home. Thus, some payers are rolling out policies designed to shift patients from hospital-based infusion centers to less expensive sites. The ACR is opposed to policies that would force patients, solely for the purpose of cost containment, to receive infusible biologics in an improperly supervised setting. The purpose of the position statement is to outline that stance,” Dr. Douglas W. White, chair of the ACR’s Committee on Rheumatologic Care, said in an interview.
He noted that he’s “been in on conversations with two payers who are implementing policies to move patients away from hospital-based infusions, but we are aware that others are in various stages of implementing such policies, too. It’s not so much an issue of critical mass for us, rather we’re just trying to keep ahead of the trends, and we think this will be a big trend.”
The potential for adverse reactions is not uncommon during intravenous administration of biologics, the committee wrote, noting, for example, that 10% of patients given infliximab have acute infusion reactions. On-site physicians such as rheumatologists who have experience with the “tremendous heterogeneity of patients with autoimmune disease and the diversity of conditions treated with biologics” can determine the severity of infusion reactions and decide whether or not it is safe to continue a particular biologic agent, in addition to providing reassurance to patients during acute and potentially severe reactions, according to the ACR statement.
Infusion reactions can range in severity from a mild rash to life-threatening anaphylaxis that can involve multiple organ systems leading to respiratory and cardiovascular collapse and requiring immediate treatment with medications such as epinephrine or intravenous glucocorticoids.
The position statement recognizes unusual situations in which home infusion is necessary for a patient to receive treatment because of transportation problems to a medical facility or comorbid conditions in which the risk of no treatment may outweigh the risk of home infusion. In these circumstances, the ACR “encourages providers in such unusual and difficult situations to make the best medical decision based on the individual needs of the patient. Routine home infusion of biologics is considered an unnecessary and dangerous risk to patients and violates our current clinical standards of practice.”
Requirements for using home infusion also threaten “to reduce access to” intravenous biologics, the ACR contends, because “specially trained physicians are less likely to prescribe treatments that are not properly administered in the safest clinical setting [and] patient fear of biologic therapy may lead to noncompliance and inadequate control of disease.”
The ACR noted that home administration of subcutaneous biologics is medically appropriate and the injection site reactions that can occur with their use are often easily managed.
Proper administration of intravenous biologics should take place under the close supervision of a physician in a physician’s office, infusion center, or hospital rather than in a patient’s home in order to address potential infusion reactions that can range from mild to life threatening, according to a position statement issued by the American College of Rheumatology’s Committee on Rheumatologic Care.
The “Patient Safety and Site of Service for Infusible Biologics” statement, issued in late February, comes in opposition to “policies that require home infusion” that appear to seek potential cost savings with home infusions rather than meet the standard of care with on-site physician supervision.
“One observation made by some but not all payers is that infusible biologics are about twice as expensive when infused in a hospital-based infusion center as compared to other locations, such as a clinic-based infusion center or the patient’s home. Thus, some payers are rolling out policies designed to shift patients from hospital-based infusion centers to less expensive sites. The ACR is opposed to policies that would force patients, solely for the purpose of cost containment, to receive infusible biologics in an improperly supervised setting. The purpose of the position statement is to outline that stance,” Dr. Douglas W. White, chair of the ACR’s Committee on Rheumatologic Care, said in an interview.
He noted that he’s “been in on conversations with two payers who are implementing policies to move patients away from hospital-based infusions, but we are aware that others are in various stages of implementing such policies, too. It’s not so much an issue of critical mass for us, rather we’re just trying to keep ahead of the trends, and we think this will be a big trend.”
The potential for adverse reactions is not uncommon during intravenous administration of biologics, the committee wrote, noting, for example, that 10% of patients given infliximab have acute infusion reactions. On-site physicians such as rheumatologists who have experience with the “tremendous heterogeneity of patients with autoimmune disease and the diversity of conditions treated with biologics” can determine the severity of infusion reactions and decide whether or not it is safe to continue a particular biologic agent, in addition to providing reassurance to patients during acute and potentially severe reactions, according to the ACR statement.
Infusion reactions can range in severity from a mild rash to life-threatening anaphylaxis that can involve multiple organ systems leading to respiratory and cardiovascular collapse and requiring immediate treatment with medications such as epinephrine or intravenous glucocorticoids.
The position statement recognizes unusual situations in which home infusion is necessary for a patient to receive treatment because of transportation problems to a medical facility or comorbid conditions in which the risk of no treatment may outweigh the risk of home infusion. In these circumstances, the ACR “encourages providers in such unusual and difficult situations to make the best medical decision based on the individual needs of the patient. Routine home infusion of biologics is considered an unnecessary and dangerous risk to patients and violates our current clinical standards of practice.”
Requirements for using home infusion also threaten “to reduce access to” intravenous biologics, the ACR contends, because “specially trained physicians are less likely to prescribe treatments that are not properly administered in the safest clinical setting [and] patient fear of biologic therapy may lead to noncompliance and inadequate control of disease.”
The ACR noted that home administration of subcutaneous biologics is medically appropriate and the injection site reactions that can occur with their use are often easily managed.
ACR’s 2016-2020 research agenda built through consensus
Therapeutic goals set the tone for the American College of Rheumatology National Research Agenda 2016-2020 by calling for the discovery and development of new therapies for rheumatic disease; finding predictors of response and nonresponse to, and adverse events from therapy; and improving the understanding of how therapies should be used.
Those are the top 3 out of 15 goals facilitated by the ACR’s Committee on Research, which finalized the agenda after seeking input from members of the ACR and Association of Rheumatology Health Professionals (ARHP) living in the United States, and going through several rounds of refining and prioritizing the importance of goals through the input of clinicians, researchers, patients, and stakeholders. The Committee on Research uses the agenda to “set the compass for the organization in terms of research initiatives and facilitate the ACR’s advocacy for the research goals identified.”

Dr. Alexis R. Ogdie-Beatty, who jointly led the development of the agenda for the Committee on Research along with Dr. S. Louis Bridges, said that while the goals for 2016-2020 had a great deal of overlap with those of 2011-2015, “some of the topics that came up were different. Some of the topics were more specific than in the previous agenda. We have some idea how important these issues were to rheumatologists, given that rheumatologists (and patients) rated the importance of the items. Defining new therapeutic targets and developing new therapies for rheumatic diseases was by far the most highly rated goal by rheumatologists. Next most highly rated was to advocate for increased support for rheumatology research and rheumatology investigators – this was included as a supplementary goal that supports the rest of the agenda. Other newer items were those around determining how the changing health care landscape affects rheumatology patients and clinicians. In addition, nonpharmacologic therapy, adult outcomes of pediatric disease, and optimizing patient engagement were topics that were felt to be important. I think these highlight the input of clinicians in identifying research objectives.”
The 2016-2020 agenda is the third set of goals developed by the committee since 2005, and the first to “crowdsource” the important questions to ACR and ARHP members rather than be assembled solely by the committee.

The agenda arose from a multistage process that began with a web-based survey to the ACR/ARHP membership that asked respondents to “list the five most important research questions that need to be addressed over the next 5 years in order to improve the care for patients with rheumatic disease.” A selected group of 100 individuals representing patients, clinicians (academic and community), research (all types with diverse areas/diseases of interest), allied health professionals, pediatric and adult rheumatology, men and women, all career stages, and all regions of the country, used a Delphi exercise to rate 30 statements generated from the survey on a scale from 1 (not important) to 10 (very important). They had the option to provide comments. At a Leadership Summit, stakeholders from various nonprofit foundations associated with rheumatic diseases, the National Institutes of Health, and the president of the Rheumatology Research Foundation gave comments on a draft agenda to the Committee on Research, after which the committee discussed the results and input and then solicited further 1-10 ratings and comments on preliminary agenda goals from the same group of 100 individuals as in the second phase, plus an additional 17 clinicians.
Up next in the rank-ordering after therapeutic goals were three goals about understanding:
• The etiology, pathogenesis, and genetic basis of rheumatic diseases.
• Early disease states to improve early diagnosis, develop biomarkers for early detection, and determine how earlier treatment changes outcomes.
• The immune system and autoimmunity by defining autoimmunity triggers and determining how epigenetics affect disease susceptibility and inflammation.
The 5-year plan proposed developing improved outcome measures that incorporate patient self-reports, imaging, and measures of clinical response and disease activity. The agenda also seeks to gain better understanding of how patients with rheumatic disease, rheumatologists, and rheumatology health professionals are being affected by the changing U.S. health care landscape.
The plan calls for determining the role of nonpharmacologic therapy in the management of rheumatic disease (promoting and improving adherence to physical activity, finding optimal exercise prescriptions, and determining the role of diet on disease activity), as well as evaluating the role of regenerative medicine.
The agenda spells out the need for better engagement of patients in their care as well as for understanding how comorbidities are influenced by rheumatic disease and how pain and fatigue arise in rheumatic disease.
In two separate goals, committee members listed the importance of determining adult outcomes of pediatric rheumatic diseases and the effect of aging on the development, progression, and management of rheumatic diseases.
The Committee on Research identified three supplemental goals that support the others:
• Advocating for increased support for rheumatology research and rheumatology investigators.
• Harmonizing data from existing cohorts and registries to optimize research capabilities.
• Improving patient research partner involvement in research protocols.
Therapeutic goals set the tone for the American College of Rheumatology National Research Agenda 2016-2020 by calling for the discovery and development of new therapies for rheumatic disease; finding predictors of response and nonresponse to, and adverse events from therapy; and improving the understanding of how therapies should be used.
Those are the top 3 out of 15 goals facilitated by the ACR’s Committee on Research, which finalized the agenda after seeking input from members of the ACR and Association of Rheumatology Health Professionals (ARHP) living in the United States, and going through several rounds of refining and prioritizing the importance of goals through the input of clinicians, researchers, patients, and stakeholders. The Committee on Research uses the agenda to “set the compass for the organization in terms of research initiatives and facilitate the ACR’s advocacy for the research goals identified.”
Dr. Alexis R. Ogdie-Beatty, who jointly led the development of the agenda for the Committee on Research along with Dr. S. Louis Bridges, said that while the goals for 2016-2020 had a great deal of overlap with those of 2011-2015, “some of the topics that came up were different. Some of the topics were more specific than in the previous agenda. We have some idea how important these issues were to rheumatologists, given that rheumatologists (and patients) rated the importance of the items. Defining new therapeutic targets and developing new therapies for rheumatic diseases was by far the most highly rated goal by rheumatologists. Next most highly rated was to advocate for increased support for rheumatology research and rheumatology investigators – this was included as a supplementary goal that supports the rest of the agenda. Other newer items were those around determining how the changing health care landscape affects rheumatology patients and clinicians. In addition, nonpharmacologic therapy, adult outcomes of pediatric disease, and optimizing patient engagement were topics that were felt to be important. I think these highlight the input of clinicians in identifying research objectives.”
The 2016-2020 agenda is the third set of goals developed by the committee since 2005, and the first to “crowdsource” the important questions to ACR and ARHP members rather than be assembled solely by the committee.
The agenda arose from a multistage process that began with a web-based survey to the ACR/ARHP membership that asked respondents to “list the five most important research questions that need to be addressed over the next 5 years in order to improve the care for patients with rheumatic disease.” A selected group of 100 individuals representing patients, clinicians (academic and community), research (all types with diverse areas/diseases of interest), allied health professionals, pediatric and adult rheumatology, men and women, all career stages, and all regions of the country, used a Delphi exercise to rate 30 statements generated from the survey on a scale from 1 (not important) to 10 (very important). They had the option to provide comments. At a Leadership Summit, stakeholders from various nonprofit foundations associated with rheumatic diseases, the National Institutes of Health, and the president of the Rheumatology Research Foundation gave comments on a draft agenda to the Committee on Research, after which the committee discussed the results and input and then solicited further 1-10 ratings and comments on preliminary agenda goals from the same group of 100 individuals as in the second phase, plus an additional 17 clinicians.
Up next in the rank-ordering after therapeutic goals were three goals about understanding:
• The etiology, pathogenesis, and genetic basis of rheumatic diseases.
• Early disease states to improve early diagnosis, develop biomarkers for early detection, and determine how earlier treatment changes outcomes.
• The immune system and autoimmunity by defining autoimmunity triggers and determining how epigenetics affect disease susceptibility and inflammation.
The 5-year plan proposed developing improved outcome measures that incorporate patient self-reports, imaging, and measures of clinical response and disease activity. The agenda also seeks to gain better understanding of how patients with rheumatic disease, rheumatologists, and rheumatology health professionals are being affected by the changing U.S. health care landscape.
The plan calls for determining the role of nonpharmacologic therapy in the management of rheumatic disease (promoting and improving adherence to physical activity, finding optimal exercise prescriptions, and determining the role of diet on disease activity), as well as evaluating the role of regenerative medicine.
The agenda spells out the need for better engagement of patients in their care as well as for understanding how comorbidities are influenced by rheumatic disease and how pain and fatigue arise in rheumatic disease.
In two separate goals, committee members listed the importance of determining adult outcomes of pediatric rheumatic diseases and the effect of aging on the development, progression, and management of rheumatic diseases.
The Committee on Research identified three supplemental goals that support the others:
• Advocating for increased support for rheumatology research and rheumatology investigators.
• Harmonizing data from existing cohorts and registries to optimize research capabilities.
• Improving patient research partner involvement in research protocols.
Therapeutic goals set the tone for the American College of Rheumatology National Research Agenda 2016-2020 by calling for the discovery and development of new therapies for rheumatic disease; finding predictors of response and nonresponse to, and adverse events from therapy; and improving the understanding of how therapies should be used.
Those are the top 3 out of 15 goals facilitated by the ACR’s Committee on Research, which finalized the agenda after seeking input from members of the ACR and Association of Rheumatology Health Professionals (ARHP) living in the United States, and going through several rounds of refining and prioritizing the importance of goals through the input of clinicians, researchers, patients, and stakeholders. The Committee on Research uses the agenda to “set the compass for the organization in terms of research initiatives and facilitate the ACR’s advocacy for the research goals identified.”
Dr. Alexis R. Ogdie-Beatty, who jointly led the development of the agenda for the Committee on Research along with Dr. S. Louis Bridges, said that while the goals for 2016-2020 had a great deal of overlap with those of 2011-2015, “some of the topics that came up were different. Some of the topics were more specific than in the previous agenda. We have some idea how important these issues were to rheumatologists, given that rheumatologists (and patients) rated the importance of the items. Defining new therapeutic targets and developing new therapies for rheumatic diseases was by far the most highly rated goal by rheumatologists. Next most highly rated was to advocate for increased support for rheumatology research and rheumatology investigators – this was included as a supplementary goal that supports the rest of the agenda. Other newer items were those around determining how the changing health care landscape affects rheumatology patients and clinicians. In addition, nonpharmacologic therapy, adult outcomes of pediatric disease, and optimizing patient engagement were topics that were felt to be important. I think these highlight the input of clinicians in identifying research objectives.”
The 2016-2020 agenda is the third set of goals developed by the committee since 2005, and the first to “crowdsource” the important questions to ACR and ARHP members rather than be assembled solely by the committee.
The agenda arose from a multistage process that began with a web-based survey to the ACR/ARHP membership that asked respondents to “list the five most important research questions that need to be addressed over the next 5 years in order to improve the care for patients with rheumatic disease.” A selected group of 100 individuals representing patients, clinicians (academic and community), research (all types with diverse areas/diseases of interest), allied health professionals, pediatric and adult rheumatology, men and women, all career stages, and all regions of the country, used a Delphi exercise to rate 30 statements generated from the survey on a scale from 1 (not important) to 10 (very important). They had the option to provide comments. At a Leadership Summit, stakeholders from various nonprofit foundations associated with rheumatic diseases, the National Institutes of Health, and the president of the Rheumatology Research Foundation gave comments on a draft agenda to the Committee on Research, after which the committee discussed the results and input and then solicited further 1-10 ratings and comments on preliminary agenda goals from the same group of 100 individuals as in the second phase, plus an additional 17 clinicians.
Up next in the rank-ordering after therapeutic goals were three goals about understanding:
• The etiology, pathogenesis, and genetic basis of rheumatic diseases.
• Early disease states to improve early diagnosis, develop biomarkers for early detection, and determine how earlier treatment changes outcomes.
• The immune system and autoimmunity by defining autoimmunity triggers and determining how epigenetics affect disease susceptibility and inflammation.
The 5-year plan proposed developing improved outcome measures that incorporate patient self-reports, imaging, and measures of clinical response and disease activity. The agenda also seeks to gain better understanding of how patients with rheumatic disease, rheumatologists, and rheumatology health professionals are being affected by the changing U.S. health care landscape.
The plan calls for determining the role of nonpharmacologic therapy in the management of rheumatic disease (promoting and improving adherence to physical activity, finding optimal exercise prescriptions, and determining the role of diet on disease activity), as well as evaluating the role of regenerative medicine.
The agenda spells out the need for better engagement of patients in their care as well as for understanding how comorbidities are influenced by rheumatic disease and how pain and fatigue arise in rheumatic disease.
In two separate goals, committee members listed the importance of determining adult outcomes of pediatric rheumatic diseases and the effect of aging on the development, progression, and management of rheumatic diseases.
The Committee on Research identified three supplemental goals that support the others:
• Advocating for increased support for rheumatology research and rheumatology investigators.
• Harmonizing data from existing cohorts and registries to optimize research capabilities.
• Improving patient research partner involvement in research protocols.

Anti-Remicade antibodies also cross-react with infliximab biosimilar
Antibodies to the innovator infliximab drug Remicade found in rheumatoid arthritis and spondyloarthritis patients also cross-react with the infliximab biosimilar CT-P13, marketed as Remsima or Inflectra, suggesting that switches from the innovator drug to the biosimilar are not advisable in the presence of anti-infliximab antibodies.
Switching an antibody-positive patient from the innovator drug to the biosimilar could mean that existing infliximab antibodies will “interact with the new drug, enhance clearance, and potentially lead to loss of response and infusion-related reactions,” wrote first author M. Begoña Ruiz-Argüello, Ph.D., an employee of the molecular biology testing company Progenika-Grifols in Derio, Spain, and colleagues (Ann Rheum Dis. 2016 Mar 10. doi: 10.1136/annrheumdis-2015-208684).

In the current study, the investigators set out to discover whether anti-Remicade antibodies cross-reacted with the biosimilar CT-P13, which was approved by the European Medicines Agency in 2013 for the same indications as the originator infliximab biologic Remicade.
They retrospectively selected 250 patients with rheumatoid arthritis (RA) or spondyloarthritis (SpA) who were treated with Remicade and 77 control patients who were infliximab naive.
Antibodies to infliximab were measured at the same time using three bridging ELISA assays: one that used Remicade to detect antibodies (Promonitor-ANTI-IFX kit, Progenika-Grifols, Spain); one that used Remsima (Orion Pharma, Norway); and another that used Inflectra (Hospira, United States).
Overall, 126 (50.4%) patients tested positive for antibodies using the Promonitor-ANTI-IFX kit.
These patients also tested positive for antibodies when the Remsima and Inflectra assays were used. Median antibody concentrations between the assays were not statistically different (P greater than .05). No significant differences were observed between patients with RA and SpA (P greater than .05) or in patients on concomitant immunosuppressive treatment, such as methotrexate.
Contrary to previous research, patients who tested negative for antibodies with the Promonitor-ANTI-IFX kit also tested negative with the Remsima and Inflectra assays. “Although additional epitopes may be present in the biosimilar, results suggest that epitopes influencing the immune response to [infliximab] are also present in the biosimilar,” the researchers said.
The investigators said that their findings also supported the use of therapeutic drug monitoring before considering switching patients between drugs.
Although the researchers recommended not switching between Remicade and Remsima or Inflectra, a small subanalysis in their study suggests it would be okay to switch from adalimumab to the infliximab biosimilar. A control population of 19 patients involved in the study who were anti–adalimumab antibody positive tested negative for antibodies to infliximab across the three assays.
Six of the authors are full-time employees of Progenika Biopharma S.A., maker of the Remicade assay used in the study.
Antibodies to the innovator infliximab drug Remicade found in rheumatoid arthritis and spondyloarthritis patients also cross-react with the infliximab biosimilar CT-P13, marketed as Remsima or Inflectra, suggesting that switches from the innovator drug to the biosimilar are not advisable in the presence of anti-infliximab antibodies.
Switching an antibody-positive patient from the innovator drug to the biosimilar could mean that existing infliximab antibodies will “interact with the new drug, enhance clearance, and potentially lead to loss of response and infusion-related reactions,” wrote first author M. Begoña Ruiz-Argüello, Ph.D., an employee of the molecular biology testing company Progenika-Grifols in Derio, Spain, and colleagues (Ann Rheum Dis. 2016 Mar 10. doi: 10.1136/annrheumdis-2015-208684).
In the current study, the investigators set out to discover whether anti-Remicade antibodies cross-reacted with the biosimilar CT-P13, which was approved by the European Medicines Agency in 2013 for the same indications as the originator infliximab biologic Remicade.
They retrospectively selected 250 patients with rheumatoid arthritis (RA) or spondyloarthritis (SpA) who were treated with Remicade and 77 control patients who were infliximab naive.
Antibodies to infliximab were measured at the same time using three bridging ELISA assays: one that used Remicade to detect antibodies (Promonitor-ANTI-IFX kit, Progenika-Grifols, Spain); one that used Remsima (Orion Pharma, Norway); and another that used Inflectra (Hospira, United States).
Overall, 126 (50.4%) patients tested positive for antibodies using the Promonitor-ANTI-IFX kit.
These patients also tested positive for antibodies when the Remsima and Inflectra assays were used. Median antibody concentrations between the assays were not statistically different (P greater than .05). No significant differences were observed between patients with RA and SpA (P greater than .05) or in patients on concomitant immunosuppressive treatment, such as methotrexate.
Contrary to previous research, patients who tested negative for antibodies with the Promonitor-ANTI-IFX kit also tested negative with the Remsima and Inflectra assays. “Although additional epitopes may be present in the biosimilar, results suggest that epitopes influencing the immune response to [infliximab] are also present in the biosimilar,” the researchers said.
The investigators said that their findings also supported the use of therapeutic drug monitoring before considering switching patients between drugs.
Although the researchers recommended not switching between Remicade and Remsima or Inflectra, a small subanalysis in their study suggests it would be okay to switch from adalimumab to the infliximab biosimilar. A control population of 19 patients involved in the study who were anti–adalimumab antibody positive tested negative for antibodies to infliximab across the three assays.
Six of the authors are full-time employees of Progenika Biopharma S.A., maker of the Remicade assay used in the study.
Antibodies to the innovator infliximab drug Remicade found in rheumatoid arthritis and spondyloarthritis patients also cross-react with the infliximab biosimilar CT-P13, marketed as Remsima or Inflectra, suggesting that switches from the innovator drug to the biosimilar are not advisable in the presence of anti-infliximab antibodies.
Switching an antibody-positive patient from the innovator drug to the biosimilar could mean that existing infliximab antibodies will “interact with the new drug, enhance clearance, and potentially lead to loss of response and infusion-related reactions,” wrote first author M. Begoña Ruiz-Argüello, Ph.D., an employee of the molecular biology testing company Progenika-Grifols in Derio, Spain, and colleagues (Ann Rheum Dis. 2016 Mar 10. doi: 10.1136/annrheumdis-2015-208684).
In the current study, the investigators set out to discover whether anti-Remicade antibodies cross-reacted with the biosimilar CT-P13, which was approved by the European Medicines Agency in 2013 for the same indications as the originator infliximab biologic Remicade.
They retrospectively selected 250 patients with rheumatoid arthritis (RA) or spondyloarthritis (SpA) who were treated with Remicade and 77 control patients who were infliximab naive.
Antibodies to infliximab were measured at the same time using three bridging ELISA assays: one that used Remicade to detect antibodies (Promonitor-ANTI-IFX kit, Progenika-Grifols, Spain); one that used Remsima (Orion Pharma, Norway); and another that used Inflectra (Hospira, United States).
Overall, 126 (50.4%) patients tested positive for antibodies using the Promonitor-ANTI-IFX kit.
These patients also tested positive for antibodies when the Remsima and Inflectra assays were used. Median antibody concentrations between the assays were not statistically different (P greater than .05). No significant differences were observed between patients with RA and SpA (P greater than .05) or in patients on concomitant immunosuppressive treatment, such as methotrexate.
Contrary to previous research, patients who tested negative for antibodies with the Promonitor-ANTI-IFX kit also tested negative with the Remsima and Inflectra assays. “Although additional epitopes may be present in the biosimilar, results suggest that epitopes influencing the immune response to [infliximab] are also present in the biosimilar,” the researchers said.
The investigators said that their findings also supported the use of therapeutic drug monitoring before considering switching patients between drugs.
Although the researchers recommended not switching between Remicade and Remsima or Inflectra, a small subanalysis in their study suggests it would be okay to switch from adalimumab to the infliximab biosimilar. A control population of 19 patients involved in the study who were anti–adalimumab antibody positive tested negative for antibodies to infliximab across the three assays.
Six of the authors are full-time employees of Progenika Biopharma S.A., maker of the Remicade assay used in the study.
FROM ANNALS OF THE RHEUMATIC DISEASES
Key clinical point: Rheumatology patients with positive antibodies to Remicade should not be switched to infliximab biosimilar (Remsima, Inflectra).
Major finding: Antibodies to infliximab in Remicade-treated rheumatology patients showed identical reactivity towards the biosimilar CT-P13.
Data source: A retrospective study of 250 consecutive patients with RA and SpA taking Remicade and 77 infliximab-naive controls.
Disclosures: Six of the authors are full-time employees of Progenika Biopharma S.A., maker of the Remicade assay used in the study.

Expert examines secukinumab’s role in ankylosing spondylitis treatment strategies
MAUI, HAWAII – The most important development within the past year in the treatment of ankylosing spondylitis was the Food and Drug Administration approval of secukinumab (Cosentyx) as the first non-tumor necrosis factor inhibitor biologic for this condition – but the interleukin-17A inhibitor is not going to immediately step into a role as a first-line therapy, Dr. Eric M. Ruderman predicted at the 2016 Rheumatology Winter Clinical Symposium.
“In all likelihood nobody’s going to use this as a first-line drug right out of the gate. It’s a drug you’re going to potentially go to in people who haven’t responded to the things that you’ve been comfortable using for the last 10 or 15 years. So the big practical issue becomes, ‘How does secukinumab perform in TNF inhibitor-naive patients versus prior TNF inhibitor inadequate responders?’ ” according to the rheumatologist, who is professor of medicine at Northwestern University in Chicago.

This question has been addressed in secondary analyses of the pivotal phase III MEASURE 1 and MEASURE 2 trials which have been presented at the annual European League Against Rheumatism and American College of Rheumatology meetings. The bottom line was that the therapeutic response rate in both trials was markedly lower in TNF inhibitor inadequate responders than in TNF inhibitor-naive subjects.
“But there still is a significant response rate in the inadequate responders. It’s clearly better than placebo. So this is a drug that may have a role in your practice at the point where patients have failed on one or two anti-TNF biologics,” according to Dr. Ruderman.
The difference between MEASURE 1 and MEASURE 2 is that MEASURE 1 entailed three intravenous loading doses of the biologic at 2-week intervals before switching to monthly subcutaneous dosing, while MEASURE 2 featured subcutaneous loading doses given weekly for 4 weeks before moving to monthly administration. Interestingly, the FDA approval of secukinumab at 150 mg doesn’t call for a loading dose, even though both pivotal trials relied on them, the rheumatologist observed.
At 16 weeks in MEASURE 1, 66% of TNF inhibitor-naive subjects on secukinumab 150 mg had at least a 20% improvement from baseline in ankylosing spondylitis signs and symptoms, or Assessment of Spondyloarthritis International Society (ASAS) 20, compared with 46% of TNF inhibitor inadequate responders. The week 16 ASAS 20 rate in MEASURE 2 was 68% in TNF inhibitor-naive patients and 50% in those with a prior inadequate response to TNF inhibitor therapy.
How should rheumatologists expect secukinumab to perform in daily clinical practice? In the 181 ankylosing spoindylitis patients who completed 52 weeks in the MEASURE 2 extension study, 74% of those on secukinumab at 150 mg had an ASAS 20 response. In both trials, the secukinumab side effect profile was “reasonably clean,” in Dr. Ruderman’s view, with serious adverse events that were similar to placebo.
Serial MRI scans showed rapid resolution of bone marrow edema and inflammation by 16 weeks, an effect sustained through 52 weeks.
The big unanswered question is whether secukinumab prevents radiographic progression of the disease. Serial cervical and spinal X-rays rated using the modified Stoke Ankylosing Spondylitis Spinal Score showed a mean increase of just 0.30 points at 2 years from a baseline of 10.22, with 80% of patients demonstrating no change over time. But there were no untreated controls for comparison in this analysis, so it’s not possible to say whether the drug actually slowed disease progression or that’s the natural history of disease in those subjects, Dr. Ruderman noted.
Effect of NSAID dosing frequency on progression
On the topic of preventing radiographic progression in ankylosing spondylitis, the rheumatologist highlighted a prospective study presented at last year’s EULAR meeting and published online last summer (Ann Rheum Dis. 2015 Aug 4. doi: 10.1136/annrheumdis-2015-207897) that demonstrated that continuous use of diclofenac didn’t do any better at preventing radiographic spinal disease progression than on-demand use of the nonsteroidal anti-inflammatory drug (NSAID) over the course of 2 years.
“There’s been a lot of noise in the ankylosing spondylitis community about the potential benefit of NSAIDs in preventing structural progression. Previous information suggested that staying on them continuously actually reduced radiographic progression. This diclofenac study has shaken things up a little. It raises the question of whether there is any added benefit for NSAIDs in terms of structural progression,” he commented.
Current ACR/SAA/SPARTAN guidelines, which predate the study, feature a conditional recommendation that patients with active ankylosing spondylitis stay on continuous NSAID therapy.
Secukinumab is also approved for treatment of psoriasis and psoriatic arthritis.
Dr. Ruderman reported serving as a consultant to and/or receiving research grants from numerous pharmaceutical companies, including Novartis, which markets secukinumab.
MAUI, HAWAII – The most important development within the past year in the treatment of ankylosing spondylitis was the Food and Drug Administration approval of secukinumab (Cosentyx) as the first non-tumor necrosis factor inhibitor biologic for this condition – but the interleukin-17A inhibitor is not going to immediately step into a role as a first-line therapy, Dr. Eric M. Ruderman predicted at the 2016 Rheumatology Winter Clinical Symposium.
“In all likelihood nobody’s going to use this as a first-line drug right out of the gate. It’s a drug you’re going to potentially go to in people who haven’t responded to the things that you’ve been comfortable using for the last 10 or 15 years. So the big practical issue becomes, ‘How does secukinumab perform in TNF inhibitor-naive patients versus prior TNF inhibitor inadequate responders?’ ” according to the rheumatologist, who is professor of medicine at Northwestern University in Chicago.
This question has been addressed in secondary analyses of the pivotal phase III MEASURE 1 and MEASURE 2 trials which have been presented at the annual European League Against Rheumatism and American College of Rheumatology meetings. The bottom line was that the therapeutic response rate in both trials was markedly lower in TNF inhibitor inadequate responders than in TNF inhibitor-naive subjects.
“But there still is a significant response rate in the inadequate responders. It’s clearly better than placebo. So this is a drug that may have a role in your practice at the point where patients have failed on one or two anti-TNF biologics,” according to Dr. Ruderman.
The difference between MEASURE 1 and MEASURE 2 is that MEASURE 1 entailed three intravenous loading doses of the biologic at 2-week intervals before switching to monthly subcutaneous dosing, while MEASURE 2 featured subcutaneous loading doses given weekly for 4 weeks before moving to monthly administration. Interestingly, the FDA approval of secukinumab at 150 mg doesn’t call for a loading dose, even though both pivotal trials relied on them, the rheumatologist observed.
At 16 weeks in MEASURE 1, 66% of TNF inhibitor-naive subjects on secukinumab 150 mg had at least a 20% improvement from baseline in ankylosing spondylitis signs and symptoms, or Assessment of Spondyloarthritis International Society (ASAS) 20, compared with 46% of TNF inhibitor inadequate responders. The week 16 ASAS 20 rate in MEASURE 2 was 68% in TNF inhibitor-naive patients and 50% in those with a prior inadequate response to TNF inhibitor therapy.
How should rheumatologists expect secukinumab to perform in daily clinical practice? In the 181 ankylosing spoindylitis patients who completed 52 weeks in the MEASURE 2 extension study, 74% of those on secukinumab at 150 mg had an ASAS 20 response. In both trials, the secukinumab side effect profile was “reasonably clean,” in Dr. Ruderman’s view, with serious adverse events that were similar to placebo.
Serial MRI scans showed rapid resolution of bone marrow edema and inflammation by 16 weeks, an effect sustained through 52 weeks.
The big unanswered question is whether secukinumab prevents radiographic progression of the disease. Serial cervical and spinal X-rays rated using the modified Stoke Ankylosing Spondylitis Spinal Score showed a mean increase of just 0.30 points at 2 years from a baseline of 10.22, with 80% of patients demonstrating no change over time. But there were no untreated controls for comparison in this analysis, so it’s not possible to say whether the drug actually slowed disease progression or that’s the natural history of disease in those subjects, Dr. Ruderman noted.
Effect of NSAID dosing frequency on progression
On the topic of preventing radiographic progression in ankylosing spondylitis, the rheumatologist highlighted a prospective study presented at last year’s EULAR meeting and published online last summer (Ann Rheum Dis. 2015 Aug 4. doi: 10.1136/annrheumdis-2015-207897) that demonstrated that continuous use of diclofenac didn’t do any better at preventing radiographic spinal disease progression than on-demand use of the nonsteroidal anti-inflammatory drug (NSAID) over the course of 2 years.
“There’s been a lot of noise in the ankylosing spondylitis community about the potential benefit of NSAIDs in preventing structural progression. Previous information suggested that staying on them continuously actually reduced radiographic progression. This diclofenac study has shaken things up a little. It raises the question of whether there is any added benefit for NSAIDs in terms of structural progression,” he commented.
Current ACR/SAA/SPARTAN guidelines, which predate the study, feature a conditional recommendation that patients with active ankylosing spondylitis stay on continuous NSAID therapy.
Secukinumab is also approved for treatment of psoriasis and psoriatic arthritis.
Dr. Ruderman reported serving as a consultant to and/or receiving research grants from numerous pharmaceutical companies, including Novartis, which markets secukinumab.
MAUI, HAWAII – The most important development within the past year in the treatment of ankylosing spondylitis was the Food and Drug Administration approval of secukinumab (Cosentyx) as the first non-tumor necrosis factor inhibitor biologic for this condition – but the interleukin-17A inhibitor is not going to immediately step into a role as a first-line therapy, Dr. Eric M. Ruderman predicted at the 2016 Rheumatology Winter Clinical Symposium.
“In all likelihood nobody’s going to use this as a first-line drug right out of the gate. It’s a drug you’re going to potentially go to in people who haven’t responded to the things that you’ve been comfortable using for the last 10 or 15 years. So the big practical issue becomes, ‘How does secukinumab perform in TNF inhibitor-naive patients versus prior TNF inhibitor inadequate responders?’ ” according to the rheumatologist, who is professor of medicine at Northwestern University in Chicago.
This question has been addressed in secondary analyses of the pivotal phase III MEASURE 1 and MEASURE 2 trials which have been presented at the annual European League Against Rheumatism and American College of Rheumatology meetings. The bottom line was that the therapeutic response rate in both trials was markedly lower in TNF inhibitor inadequate responders than in TNF inhibitor-naive subjects.
“But there still is a significant response rate in the inadequate responders. It’s clearly better than placebo. So this is a drug that may have a role in your practice at the point where patients have failed on one or two anti-TNF biologics,” according to Dr. Ruderman.
The difference between MEASURE 1 and MEASURE 2 is that MEASURE 1 entailed three intravenous loading doses of the biologic at 2-week intervals before switching to monthly subcutaneous dosing, while MEASURE 2 featured subcutaneous loading doses given weekly for 4 weeks before moving to monthly administration. Interestingly, the FDA approval of secukinumab at 150 mg doesn’t call for a loading dose, even though both pivotal trials relied on them, the rheumatologist observed.
At 16 weeks in MEASURE 1, 66% of TNF inhibitor-naive subjects on secukinumab 150 mg had at least a 20% improvement from baseline in ankylosing spondylitis signs and symptoms, or Assessment of Spondyloarthritis International Society (ASAS) 20, compared with 46% of TNF inhibitor inadequate responders. The week 16 ASAS 20 rate in MEASURE 2 was 68% in TNF inhibitor-naive patients and 50% in those with a prior inadequate response to TNF inhibitor therapy.
How should rheumatologists expect secukinumab to perform in daily clinical practice? In the 181 ankylosing spoindylitis patients who completed 52 weeks in the MEASURE 2 extension study, 74% of those on secukinumab at 150 mg had an ASAS 20 response. In both trials, the secukinumab side effect profile was “reasonably clean,” in Dr. Ruderman’s view, with serious adverse events that were similar to placebo.
Serial MRI scans showed rapid resolution of bone marrow edema and inflammation by 16 weeks, an effect sustained through 52 weeks.
The big unanswered question is whether secukinumab prevents radiographic progression of the disease. Serial cervical and spinal X-rays rated using the modified Stoke Ankylosing Spondylitis Spinal Score showed a mean increase of just 0.30 points at 2 years from a baseline of 10.22, with 80% of patients demonstrating no change over time. But there were no untreated controls for comparison in this analysis, so it’s not possible to say whether the drug actually slowed disease progression or that’s the natural history of disease in those subjects, Dr. Ruderman noted.
Effect of NSAID dosing frequency on progression
On the topic of preventing radiographic progression in ankylosing spondylitis, the rheumatologist highlighted a prospective study presented at last year’s EULAR meeting and published online last summer (Ann Rheum Dis. 2015 Aug 4. doi: 10.1136/annrheumdis-2015-207897) that demonstrated that continuous use of diclofenac didn’t do any better at preventing radiographic spinal disease progression than on-demand use of the nonsteroidal anti-inflammatory drug (NSAID) over the course of 2 years.
“There’s been a lot of noise in the ankylosing spondylitis community about the potential benefit of NSAIDs in preventing structural progression. Previous information suggested that staying on them continuously actually reduced radiographic progression. This diclofenac study has shaken things up a little. It raises the question of whether there is any added benefit for NSAIDs in terms of structural progression,” he commented.
Current ACR/SAA/SPARTAN guidelines, which predate the study, feature a conditional recommendation that patients with active ankylosing spondylitis stay on continuous NSAID therapy.
Secukinumab is also approved for treatment of psoriasis and psoriatic arthritis.
Dr. Ruderman reported serving as a consultant to and/or receiving research grants from numerous pharmaceutical companies, including Novartis, which markets secukinumab.
EXPERT ANALYSIS FROM RWCS 2016

Expert advises how to use shingles vaccine in rheumatology patients
MAUI, HAWAII – The herpes zoster vaccine is particularly important in patients with rheumatic diseases because their risks of shingles and postherpetic neuralgia are substantially higher than in the general population, Dr. John J. Cush observed at the 2016 Rheumatology Winter Clinical Symposium.
This is a live attenuated virus vaccine, and the rules regarding its use in patients with rheumatic diseases are fairly complicated. Here’s what physicians need to know: the Centers for Disease Control and Prevention and the Advisory Committee on Immunization Practices say the shingles vaccine can safely be given to patients on prednisone at less than 20 mg/day, azathioprine at up to 3 mg/kg/day, or methotrexate at up to 0.4 mg/kg/week, which works out to about 25 mg/week in anyone weighing more than 136 pounds.

However, the shingles vaccine is contraindicated in patients on recombinant biologic agents, including tumor necrosis factor (TNF) inhibitors, abatacept (Orencia), rituximab (Rituxan), or Janus kinase inhibitors, explained Dr. Cush, professor of medicine and rheumatology at Baylor University, Dallas, and director of clinical rheumatology at the Baylor Research Institute.
The lifetime risk of shingles in the general population is roughly one in three. The risk in patients with rheumatoid arthritis is roughly twice that of the age-matched general population, and the risks are substantially greater than that in individuals with other rheumatic diseases, including lupus and granulomatosis with polyangiitis.
Payers cover the vaccine in patients age 60 or older. The vaccine is approved for and has been shown to be effective in 50- to 59-year-olds as well, but that typically entails an out-of-pocket expense of around $200.
Given that close to 60% of all rheumatoid arthritis patients will eventually be placed on biologic therapy, Dr. Cush believes in seizing any opportunity to give the shingles vaccine in age-appropriate patients beforehand. However, he advises against temporarily stopping a biologic for the express purpose of administering the live virus vaccine.
“Find the opportunity: between changes in medication, after they have surgery, during a lapse in therapy,” he suggested.
It’s recommended that Zostavax be deferred until after a patient has been off biologic therapy or high-dose steroids for at least 4 weeks, and that a biologic agent shouldn’t be started for 2-4 weeks after vaccination.
The shingles vaccine can safely be given with multiple inactivated virus vaccines such as an influenza vaccine or pneumococcal vaccine on a single day.
Controversy surrounds the issue of whether TNF inhibitors increase the risk of shingles. Several retrospective studies have reported they do. But the largest retrospective study, involving more than 33,000 new users of anti-TNF agents, found that patients with rheumatoid arthritis, psoriasis, psoriatic arthritis, ankylosing spondylitis, and other inflammatory diseases who initiated anti-TNF therapy weren’t at any higher risk of herpes zoster than those who started on methotrexate or other nonbiologic disease-modifying antirheumatic drugs (JAMA. 2013 Mar 6;309[9]:887-95). The lead investigator in this study, Dr. Kevin L. Winthrop of Oregon Health and Science University, Portland, is currently conducting a prospective study in an effort to confirm these findings.
The recommendation against giving the zoster vaccine to patients while on biologics is based upon the theoretical risk that exposure to the live attenuated virus will trigger an acute shingles attack. However, when Dr. Cush conducted a survey of his fellow rheumatologists, they reported that among more than a collective 200 patients inadvertently given the vaccine while on biologic therapy, not one case of shingles subsequently occurred over the short term.
More persuasively, Dr. Jeffrey R. Curtis of the University of Alabama at Birmingham and coinvestigators conducted a formal retrospective study of close to a half-million Medicare patients and found there were no cases of herpes zoster or varicella within 42 days following inadvertent vaccination of 633 patients while on biologics. During a median 2-year follow-up of Medicare patients with rheumatoid arthritis and other immune-mediated diseases, herpes zoster vaccination was associated with a 39% reduction in the risk of shingles (JAMA. 2012 Jul 4;308[1]:43-9).
“You shouldn’t be vaccinating for herpes zoster while patients are on a biologic, but you know what? If it happens, don’t wig out. Move on and try to avoid it,” Dr. Cush advised.
In any event, this is an issue that is eventually likely to go away. An inactivated virus vaccine for the prevention of shingles is now in clinical trials. It appears to be more effective than the current vaccine, according to the rheumatologist.
He reported having no financial interests relevant to his presentation.
MAUI, HAWAII – The herpes zoster vaccine is particularly important in patients with rheumatic diseases because their risks of shingles and postherpetic neuralgia are substantially higher than in the general population, Dr. John J. Cush observed at the 2016 Rheumatology Winter Clinical Symposium.
This is a live attenuated virus vaccine, and the rules regarding its use in patients with rheumatic diseases are fairly complicated. Here’s what physicians need to know: the Centers for Disease Control and Prevention and the Advisory Committee on Immunization Practices say the shingles vaccine can safely be given to patients on prednisone at less than 20 mg/day, azathioprine at up to 3 mg/kg/day, or methotrexate at up to 0.4 mg/kg/week, which works out to about 25 mg/week in anyone weighing more than 136 pounds.
However, the shingles vaccine is contraindicated in patients on recombinant biologic agents, including tumor necrosis factor (TNF) inhibitors, abatacept (Orencia), rituximab (Rituxan), or Janus kinase inhibitors, explained Dr. Cush, professor of medicine and rheumatology at Baylor University, Dallas, and director of clinical rheumatology at the Baylor Research Institute.
The lifetime risk of shingles in the general population is roughly one in three. The risk in patients with rheumatoid arthritis is roughly twice that of the age-matched general population, and the risks are substantially greater than that in individuals with other rheumatic diseases, including lupus and granulomatosis with polyangiitis.
Payers cover the vaccine in patients age 60 or older. The vaccine is approved for and has been shown to be effective in 50- to 59-year-olds as well, but that typically entails an out-of-pocket expense of around $200.
Given that close to 60% of all rheumatoid arthritis patients will eventually be placed on biologic therapy, Dr. Cush believes in seizing any opportunity to give the shingles vaccine in age-appropriate patients beforehand. However, he advises against temporarily stopping a biologic for the express purpose of administering the live virus vaccine.
“Find the opportunity: between changes in medication, after they have surgery, during a lapse in therapy,” he suggested.
It’s recommended that Zostavax be deferred until after a patient has been off biologic therapy or high-dose steroids for at least 4 weeks, and that a biologic agent shouldn’t be started for 2-4 weeks after vaccination.
The shingles vaccine can safely be given with multiple inactivated virus vaccines such as an influenza vaccine or pneumococcal vaccine on a single day.
Controversy surrounds the issue of whether TNF inhibitors increase the risk of shingles. Several retrospective studies have reported they do. But the largest retrospective study, involving more than 33,000 new users of anti-TNF agents, found that patients with rheumatoid arthritis, psoriasis, psoriatic arthritis, ankylosing spondylitis, and other inflammatory diseases who initiated anti-TNF therapy weren’t at any higher risk of herpes zoster than those who started on methotrexate or other nonbiologic disease-modifying antirheumatic drugs (JAMA. 2013 Mar 6;309[9]:887-95). The lead investigator in this study, Dr. Kevin L. Winthrop of Oregon Health and Science University, Portland, is currently conducting a prospective study in an effort to confirm these findings.
The recommendation against giving the zoster vaccine to patients while on biologics is based upon the theoretical risk that exposure to the live attenuated virus will trigger an acute shingles attack. However, when Dr. Cush conducted a survey of his fellow rheumatologists, they reported that among more than a collective 200 patients inadvertently given the vaccine while on biologic therapy, not one case of shingles subsequently occurred over the short term.
More persuasively, Dr. Jeffrey R. Curtis of the University of Alabama at Birmingham and coinvestigators conducted a formal retrospective study of close to a half-million Medicare patients and found there were no cases of herpes zoster or varicella within 42 days following inadvertent vaccination of 633 patients while on biologics. During a median 2-year follow-up of Medicare patients with rheumatoid arthritis and other immune-mediated diseases, herpes zoster vaccination was associated with a 39% reduction in the risk of shingles (JAMA. 2012 Jul 4;308[1]:43-9).
“You shouldn’t be vaccinating for herpes zoster while patients are on a biologic, but you know what? If it happens, don’t wig out. Move on and try to avoid it,” Dr. Cush advised.
In any event, this is an issue that is eventually likely to go away. An inactivated virus vaccine for the prevention of shingles is now in clinical trials. It appears to be more effective than the current vaccine, according to the rheumatologist.
He reported having no financial interests relevant to his presentation.
MAUI, HAWAII – The herpes zoster vaccine is particularly important in patients with rheumatic diseases because their risks of shingles and postherpetic neuralgia are substantially higher than in the general population, Dr. John J. Cush observed at the 2016 Rheumatology Winter Clinical Symposium.
This is a live attenuated virus vaccine, and the rules regarding its use in patients with rheumatic diseases are fairly complicated. Here’s what physicians need to know: the Centers for Disease Control and Prevention and the Advisory Committee on Immunization Practices say the shingles vaccine can safely be given to patients on prednisone at less than 20 mg/day, azathioprine at up to 3 mg/kg/day, or methotrexate at up to 0.4 mg/kg/week, which works out to about 25 mg/week in anyone weighing more than 136 pounds.
However, the shingles vaccine is contraindicated in patients on recombinant biologic agents, including tumor necrosis factor (TNF) inhibitors, abatacept (Orencia), rituximab (Rituxan), or Janus kinase inhibitors, explained Dr. Cush, professor of medicine and rheumatology at Baylor University, Dallas, and director of clinical rheumatology at the Baylor Research Institute.
The lifetime risk of shingles in the general population is roughly one in three. The risk in patients with rheumatoid arthritis is roughly twice that of the age-matched general population, and the risks are substantially greater than that in individuals with other rheumatic diseases, including lupus and granulomatosis with polyangiitis.
Payers cover the vaccine in patients age 60 or older. The vaccine is approved for and has been shown to be effective in 50- to 59-year-olds as well, but that typically entails an out-of-pocket expense of around $200.
Given that close to 60% of all rheumatoid arthritis patients will eventually be placed on biologic therapy, Dr. Cush believes in seizing any opportunity to give the shingles vaccine in age-appropriate patients beforehand. However, he advises against temporarily stopping a biologic for the express purpose of administering the live virus vaccine.
“Find the opportunity: between changes in medication, after they have surgery, during a lapse in therapy,” he suggested.
It’s recommended that Zostavax be deferred until after a patient has been off biologic therapy or high-dose steroids for at least 4 weeks, and that a biologic agent shouldn’t be started for 2-4 weeks after vaccination.
The shingles vaccine can safely be given with multiple inactivated virus vaccines such as an influenza vaccine or pneumococcal vaccine on a single day.
Controversy surrounds the issue of whether TNF inhibitors increase the risk of shingles. Several retrospective studies have reported they do. But the largest retrospective study, involving more than 33,000 new users of anti-TNF agents, found that patients with rheumatoid arthritis, psoriasis, psoriatic arthritis, ankylosing spondylitis, and other inflammatory diseases who initiated anti-TNF therapy weren’t at any higher risk of herpes zoster than those who started on methotrexate or other nonbiologic disease-modifying antirheumatic drugs (JAMA. 2013 Mar 6;309[9]:887-95). The lead investigator in this study, Dr. Kevin L. Winthrop of Oregon Health and Science University, Portland, is currently conducting a prospective study in an effort to confirm these findings.
The recommendation against giving the zoster vaccine to patients while on biologics is based upon the theoretical risk that exposure to the live attenuated virus will trigger an acute shingles attack. However, when Dr. Cush conducted a survey of his fellow rheumatologists, they reported that among more than a collective 200 patients inadvertently given the vaccine while on biologic therapy, not one case of shingles subsequently occurred over the short term.
More persuasively, Dr. Jeffrey R. Curtis of the University of Alabama at Birmingham and coinvestigators conducted a formal retrospective study of close to a half-million Medicare patients and found there were no cases of herpes zoster or varicella within 42 days following inadvertent vaccination of 633 patients while on biologics. During a median 2-year follow-up of Medicare patients with rheumatoid arthritis and other immune-mediated diseases, herpes zoster vaccination was associated with a 39% reduction in the risk of shingles (JAMA. 2012 Jul 4;308[1]:43-9).
“You shouldn’t be vaccinating for herpes zoster while patients are on a biologic, but you know what? If it happens, don’t wig out. Move on and try to avoid it,” Dr. Cush advised.
In any event, this is an issue that is eventually likely to go away. An inactivated virus vaccine for the prevention of shingles is now in clinical trials. It appears to be more effective than the current vaccine, according to the rheumatologist.
He reported having no financial interests relevant to his presentation.
EXPERT ANALYSIS FROM RWCS 2016

Subtle radiographic progression in axial SpA cannot be reliably distinguished from error
Sacroiliitis observed in patients with axial spondyloarthritis more often regressed rather than progressed on radiography over nearly 5 years of follow-up of the Assessment of SpondyloArthritis international Society (ASAS) cohort, which lead author Dr. Alexandre Sepriano and his colleagues called “strange” and “sobering.”
The findings call into question the reliability of plain pelvic radiographs for detecting subtle change in sacroiliitis and should prompt the evaluation of alternative imaging modalities such as MRI and low-dose CT, according to Dr. Sepriano of Leiden (the Netherlands) University Medical Center and his associates.
Determining the presence of radiographic sacroiliitis is prognostically relevant and can pave the way for treatment with biologics, but ambiguity in making this decision and in tracking progression has been revealed in the large inter-and intrareader variability found in previous studies. Furthermore, previous studies tracking progression of nonradiographic axial spondyloarthritis (axSpA) to radiographic axSpA have addressed only disease progression and ignored regression. While regression is likely to be rare, it cannot be ignored from a methodologic standpoint, the investigators wrote.
The researchers therefore set out in the current study to assess positive and negative changes in sacroiliitis on plain pelvic radiographs over time in 975 patients from the ASAS cohort who had chronic back pain of unknown origin or undiagnosed peripheral symptoms (Ann Rheum Dis. 2016 Feb 22. doi: 10.1136/annrheumdis-2015-208964).
Of the 357 of the patients who had paired plain pelvic radiographs available at baseline and follow-up, 17.4% (62/357) fulfilled the criteria for radiographic axSpA at baseline, as defined by modified New York criteria (mNY). At a mean follow-up of 4.4 years, this figure had risen to 22.4% (80/357), suggesting a net progression of 5%.
However, when the authors cross-tabulated their figures, more than half (36/62) of the patients considered mNY positive at baseline were assessed as mNY negative at follow-up. This would mean that radiographic sacroiliitis would have regressed in 58% of the cases; conversely, only 54 of 295 patients (18.3%) became mNY positive at follow-up.
“If only positive change (progression) is valued and negative change is ignored, one would disregard measurement error and spuriously attribute part of the observed positive change to real progression,” the research team explained. “The most likely explanation of our strange and extreme observation is that subtle radiographic progression (the signal) – if truly present – cannot be reliably distinguished from measurement error (the noise). These sobering data clearly illustrate that more research is needed in visualising progression in axSpA.”
ASAS funded the study. The authors had no competing interests to declare.
Sacroiliitis observed in patients with axial spondyloarthritis more often regressed rather than progressed on radiography over nearly 5 years of follow-up of the Assessment of SpondyloArthritis international Society (ASAS) cohort, which lead author Dr. Alexandre Sepriano and his colleagues called “strange” and “sobering.”
The findings call into question the reliability of plain pelvic radiographs for detecting subtle change in sacroiliitis and should prompt the evaluation of alternative imaging modalities such as MRI and low-dose CT, according to Dr. Sepriano of Leiden (the Netherlands) University Medical Center and his associates.
Determining the presence of radiographic sacroiliitis is prognostically relevant and can pave the way for treatment with biologics, but ambiguity in making this decision and in tracking progression has been revealed in the large inter-and intrareader variability found in previous studies. Furthermore, previous studies tracking progression of nonradiographic axial spondyloarthritis (axSpA) to radiographic axSpA have addressed only disease progression and ignored regression. While regression is likely to be rare, it cannot be ignored from a methodologic standpoint, the investigators wrote.
The researchers therefore set out in the current study to assess positive and negative changes in sacroiliitis on plain pelvic radiographs over time in 975 patients from the ASAS cohort who had chronic back pain of unknown origin or undiagnosed peripheral symptoms (Ann Rheum Dis. 2016 Feb 22. doi: 10.1136/annrheumdis-2015-208964).
Of the 357 of the patients who had paired plain pelvic radiographs available at baseline and follow-up, 17.4% (62/357) fulfilled the criteria for radiographic axSpA at baseline, as defined by modified New York criteria (mNY). At a mean follow-up of 4.4 years, this figure had risen to 22.4% (80/357), suggesting a net progression of 5%.
However, when the authors cross-tabulated their figures, more than half (36/62) of the patients considered mNY positive at baseline were assessed as mNY negative at follow-up. This would mean that radiographic sacroiliitis would have regressed in 58% of the cases; conversely, only 54 of 295 patients (18.3%) became mNY positive at follow-up.
“If only positive change (progression) is valued and negative change is ignored, one would disregard measurement error and spuriously attribute part of the observed positive change to real progression,” the research team explained. “The most likely explanation of our strange and extreme observation is that subtle radiographic progression (the signal) – if truly present – cannot be reliably distinguished from measurement error (the noise). These sobering data clearly illustrate that more research is needed in visualising progression in axSpA.”
ASAS funded the study. The authors had no competing interests to declare.
Sacroiliitis observed in patients with axial spondyloarthritis more often regressed rather than progressed on radiography over nearly 5 years of follow-up of the Assessment of SpondyloArthritis international Society (ASAS) cohort, which lead author Dr. Alexandre Sepriano and his colleagues called “strange” and “sobering.”
The findings call into question the reliability of plain pelvic radiographs for detecting subtle change in sacroiliitis and should prompt the evaluation of alternative imaging modalities such as MRI and low-dose CT, according to Dr. Sepriano of Leiden (the Netherlands) University Medical Center and his associates.
Determining the presence of radiographic sacroiliitis is prognostically relevant and can pave the way for treatment with biologics, but ambiguity in making this decision and in tracking progression has been revealed in the large inter-and intrareader variability found in previous studies. Furthermore, previous studies tracking progression of nonradiographic axial spondyloarthritis (axSpA) to radiographic axSpA have addressed only disease progression and ignored regression. While regression is likely to be rare, it cannot be ignored from a methodologic standpoint, the investigators wrote.
The researchers therefore set out in the current study to assess positive and negative changes in sacroiliitis on plain pelvic radiographs over time in 975 patients from the ASAS cohort who had chronic back pain of unknown origin or undiagnosed peripheral symptoms (Ann Rheum Dis. 2016 Feb 22. doi: 10.1136/annrheumdis-2015-208964).
Of the 357 of the patients who had paired plain pelvic radiographs available at baseline and follow-up, 17.4% (62/357) fulfilled the criteria for radiographic axSpA at baseline, as defined by modified New York criteria (mNY). At a mean follow-up of 4.4 years, this figure had risen to 22.4% (80/357), suggesting a net progression of 5%.
However, when the authors cross-tabulated their figures, more than half (36/62) of the patients considered mNY positive at baseline were assessed as mNY negative at follow-up. This would mean that radiographic sacroiliitis would have regressed in 58% of the cases; conversely, only 54 of 295 patients (18.3%) became mNY positive at follow-up.
“If only positive change (progression) is valued and negative change is ignored, one would disregard measurement error and spuriously attribute part of the observed positive change to real progression,” the research team explained. “The most likely explanation of our strange and extreme observation is that subtle radiographic progression (the signal) – if truly present – cannot be reliably distinguished from measurement error (the noise). These sobering data clearly illustrate that more research is needed in visualising progression in axSpA.”
ASAS funded the study. The authors had no competing interests to declare.
FROM ANNALS OF THE RHEUMATIC DISEASES
Key clinical point: Subtle radiographic progression in axSpA cannot be reliably distinguished from measurement error.
Major finding: Using plain radiographs, more than half of the patients identified as mNY positive for axSpA at baseline were assessed as mNY negative at a mean follow-up of 4.4 years.
Data source: 975 patients with chronic back pain of unknown origin or undiagnosed peripheral symptoms taking part in the Assessment of SpondyloArthritis international Society (ASAS) cohort.
Disclosures: ASAS funded the study. The authors had no competing interests to declare.
NSAIDs effective and safe for axSpA in the short term
The findings of a recent Cochrane review back up guidelines that recommend NSAIDs as an appropriate first-line treatment for people with axial spondyloarthritis (axSpA).
NSAIDs have been associated with a variety of gastrointestinal effects and an increased risk of cardiovascular events, heart failure, and renal toxicity, an international team of researchers led by Dr. Féline P.B. Kroon of the Leiden University Medical Center in the Netherlands noted in the review, published in the Journal of Rheumatology.

“It is therefore crucial to know whether the benefits offset the risks, especially because the therapy is often given for extended periods of time,” the researchers wrote in background information to the article (J Rheumatol. 2016. doi: 10.3899/jrheum.150721).
The investigators analyzed the evidence to assess the benefits and harms of NSAIDs in controlling symptoms, disease activity, and radiographic progression in patients with axSpA.
Reviewing 29 randomized, controlled trials and two “quasi” RCT studies in a pooled analyses, the research team found that, compared with placebo, both traditional and cyclooxygenase-2 (COX-2) NSAIDs were consistently more efficacious at 6 weeks and equally safe after 12 weeks.
The researchers also observed no significant differences in benefits or harms between the two NSAID classes. An increased number of neurologic events were initially observed for indomethacin, but the finding was not statistically significant when studies with high or unclear risk of bias were excluded.
Two single studies included in the review also suggested NSAIDs might retard radiographic progression in the spine in axSpA, especially in certain subgroups of patients such as those with high C-reactive protein. Although this would most likely be best achieved by continuous rather than on-demand use, the researchers said.
“The results of the review are in keeping with current recommendations that NSAIDs are appropriate first-line treatments of patients with axSpA with active disease before tumor necrosis factor inhibitor biologicals are applied,” they concluded.
However, they said it was surprising that they were unable to confirm the safety concerns associated with traditional NSAIDs and COX-2 NSAIDs.
This finding could mean that short-term use of either class of NSAID in this population of patients is not associated with an increased risk of GI or other adverse events.
But it could also be because most patients with ankylosing spondylitis are younger and have fewer comorbidities than patients with other rheumatic diseases.
The finding that people with ankylosing spondylitis have fewer adverse events with biologics, compared with patients with other rheumatic diseases, supported this theory, they said.
“It is technically still possible that lack of statistical power is at the basis of this, but we feel it is more likely that in the studied population and within the studied time frame (i.e., short-term), the risks of GI or cardiovascular toxicity are really not increased,” they wrote.
The review had several limitations, including that many trials were older (61% of the included studies were published before 1990) and there were not sufficient data to draw conclusions on long-term safety.
The findings of a recent Cochrane review back up guidelines that recommend NSAIDs as an appropriate first-line treatment for people with axial spondyloarthritis (axSpA).
NSAIDs have been associated with a variety of gastrointestinal effects and an increased risk of cardiovascular events, heart failure, and renal toxicity, an international team of researchers led by Dr. Féline P.B. Kroon of the Leiden University Medical Center in the Netherlands noted in the review, published in the Journal of Rheumatology.
“It is therefore crucial to know whether the benefits offset the risks, especially because the therapy is often given for extended periods of time,” the researchers wrote in background information to the article (J Rheumatol. 2016. doi: 10.3899/jrheum.150721).
The investigators analyzed the evidence to assess the benefits and harms of NSAIDs in controlling symptoms, disease activity, and radiographic progression in patients with axSpA.
Reviewing 29 randomized, controlled trials and two “quasi” RCT studies in a pooled analyses, the research team found that, compared with placebo, both traditional and cyclooxygenase-2 (COX-2) NSAIDs were consistently more efficacious at 6 weeks and equally safe after 12 weeks.
The researchers also observed no significant differences in benefits or harms between the two NSAID classes. An increased number of neurologic events were initially observed for indomethacin, but the finding was not statistically significant when studies with high or unclear risk of bias were excluded.
Two single studies included in the review also suggested NSAIDs might retard radiographic progression in the spine in axSpA, especially in certain subgroups of patients such as those with high C-reactive protein. Although this would most likely be best achieved by continuous rather than on-demand use, the researchers said.
“The results of the review are in keeping with current recommendations that NSAIDs are appropriate first-line treatments of patients with axSpA with active disease before tumor necrosis factor inhibitor biologicals are applied,” they concluded.
However, they said it was surprising that they were unable to confirm the safety concerns associated with traditional NSAIDs and COX-2 NSAIDs.
This finding could mean that short-term use of either class of NSAID in this population of patients is not associated with an increased risk of GI or other adverse events.
But it could also be because most patients with ankylosing spondylitis are younger and have fewer comorbidities than patients with other rheumatic diseases.
The finding that people with ankylosing spondylitis have fewer adverse events with biologics, compared with patients with other rheumatic diseases, supported this theory, they said.
“It is technically still possible that lack of statistical power is at the basis of this, but we feel it is more likely that in the studied population and within the studied time frame (i.e., short-term), the risks of GI or cardiovascular toxicity are really not increased,” they wrote.
The review had several limitations, including that many trials were older (61% of the included studies were published before 1990) and there were not sufficient data to draw conclusions on long-term safety.
The findings of a recent Cochrane review back up guidelines that recommend NSAIDs as an appropriate first-line treatment for people with axial spondyloarthritis (axSpA).
NSAIDs have been associated with a variety of gastrointestinal effects and an increased risk of cardiovascular events, heart failure, and renal toxicity, an international team of researchers led by Dr. Féline P.B. Kroon of the Leiden University Medical Center in the Netherlands noted in the review, published in the Journal of Rheumatology.
“It is therefore crucial to know whether the benefits offset the risks, especially because the therapy is often given for extended periods of time,” the researchers wrote in background information to the article (J Rheumatol. 2016. doi: 10.3899/jrheum.150721).
The investigators analyzed the evidence to assess the benefits and harms of NSAIDs in controlling symptoms, disease activity, and radiographic progression in patients with axSpA.
Reviewing 29 randomized, controlled trials and two “quasi” RCT studies in a pooled analyses, the research team found that, compared with placebo, both traditional and cyclooxygenase-2 (COX-2) NSAIDs were consistently more efficacious at 6 weeks and equally safe after 12 weeks.
The researchers also observed no significant differences in benefits or harms between the two NSAID classes. An increased number of neurologic events were initially observed for indomethacin, but the finding was not statistically significant when studies with high or unclear risk of bias were excluded.
Two single studies included in the review also suggested NSAIDs might retard radiographic progression in the spine in axSpA, especially in certain subgroups of patients such as those with high C-reactive protein. Although this would most likely be best achieved by continuous rather than on-demand use, the researchers said.
“The results of the review are in keeping with current recommendations that NSAIDs are appropriate first-line treatments of patients with axSpA with active disease before tumor necrosis factor inhibitor biologicals are applied,” they concluded.
However, they said it was surprising that they were unable to confirm the safety concerns associated with traditional NSAIDs and COX-2 NSAIDs.
This finding could mean that short-term use of either class of NSAID in this population of patients is not associated with an increased risk of GI or other adverse events.
But it could also be because most patients with ankylosing spondylitis are younger and have fewer comorbidities than patients with other rheumatic diseases.
The finding that people with ankylosing spondylitis have fewer adverse events with biologics, compared with patients with other rheumatic diseases, supported this theory, they said.
“It is technically still possible that lack of statistical power is at the basis of this, but we feel it is more likely that in the studied population and within the studied time frame (i.e., short-term), the risks of GI or cardiovascular toxicity are really not increased,” they wrote.
The review had several limitations, including that many trials were older (61% of the included studies were published before 1990) and there were not sufficient data to draw conclusions on long-term safety.
FROM JOURNAL OF RHEUMATOLOGY
Key clinical point: Traditional and COX-2 NSAIDs are appropriate first-line treatments of patients with axSpA with active disease.
Major finding: Compared with placebo, both traditional and COX-2 NSAIDs were consistently more efficacious at 6 weeks and equally safe after 12 weeks.
Data source: A Cochrane review of 29 randomized, controlled trials and 2 “quasi” RCTs were included in the meta-analysis.
Disclosures: No conflicts of interest were declared.

Biosimilar program reshapes FDA’s objectivity
The U.S. program to develop biosimilar agents – somewhat akin to generic drugs for complex, biologic molecules that have come off patent protection – is gathering momentum, with the first U.S. biosimilar, Zarxio, approved by the Food and Drug Administration in March 2015 and with the second, a biosimilar to infliximab, recommended by an FDA advisory committee on Feb. 9 of this year.
What’s striking about the burgeoning biosimilar development process, created by the Affordable Care Act, is how it has morphed the FDA from its traditional role as an objective arbiter of a drug’s safety and efficacy into an active partner in shepherding biosimilars onto the market.

As explained on Feb. 4 in testimony before a Congressional committee by Dr. Janet Woodcock, director of the FDA Center for Drug Evaluation and Research, the Biologic Price Competition and Innovation Act that was part of the Affordable Care Act launched a new U.S. drug-development pathway expressly for biosimilars. To implement that law, the FDA created an entirely new infrastructure within the agency – the Biosimilar Product Development Program – to help guide prospective manufacturers (called sponsors) of biosimilars through the regulatory and research hurdles to get a new biosimilar approved and into the hands of U.S. patients.
According to Dr. Woodcock, this program involves many steps where FDA staffers provide “review” and “advice” to sponsors on the studies they need to conduct and the analysis they need to perform to get their new products to market. The sponsor joins this program by paying an upfront fee that the FDA uses to keep the program running. Once a sponsor of a prospective biosimilar is in the program, the FDA’s staff helps guide the biosimilar development to a smooth conclusion.
To some extent, the FDA staff fills a similar role for conventional drug-development enterprises, conferring with manufacturers from the outset on matters such as the types and design of studies needed to insure success. What’s different about the biosimilar program is that conventional-drug development went on well before the FDA (or its predecessor) entered the scene, and the U.S. government created the FDA to police and regulate the drug production industry and protect the public against unscrupulous manufacturers of ineffective or dangerous drugs.
In contrast, the FDA itself created this new biosimilar development structure, and Dr. Woodcock noted that the in-depth review and advice meetings that the FDA offers to prospective biosimilar sponsors “has no counterpart in the Prescription Drug User Fee Act program and is unique” to the biosimilar program.
The consequence of having the FDA create the biosimilar development program from the ground up and structure it to provide such intimate input from the agency to sponsors at every step of the way seems to give the agency a notable and somewhat unnerving investment in the program’s success.
Dr. Woodcock called the approval of Zarxio an “exciting accomplishment,” and in her testimony before Congress she trumpeted the fact that as of January 2016 the biosimilar program was working on 59 proposed products that would mimic 18 different reference-product biologics. She also said that the FDA is “excited about the growing demand” for biosimilar-oriented meetings and marketing applications.
Don’t get me wrong: I think that the biosimilar concept is great, and has the potential to make what have become life-changing treatments more affordable and more available. And making the FDA such an active participant in getting biosimilar drugs created and approved is undoubtedly the most efficient way to accomplish this.
But in the process, the biosimilar program has changed the FDA from its more disengaged role as objective pharmaceutical judge into an active and seemingly not completely neutral codeveloper, risking at least the appearance of lost impartiality. Given that the FDA now wears two very different hats, we need to trust that the integrity and dedication of its staff will keep them from confusing their roles as proponent and gatekeeper.
On Twitter @mitchelzoler
The U.S. program to develop biosimilar agents – somewhat akin to generic drugs for complex, biologic molecules that have come off patent protection – is gathering momentum, with the first U.S. biosimilar, Zarxio, approved by the Food and Drug Administration in March 2015 and with the second, a biosimilar to infliximab, recommended by an FDA advisory committee on Feb. 9 of this year.
What’s striking about the burgeoning biosimilar development process, created by the Affordable Care Act, is how it has morphed the FDA from its traditional role as an objective arbiter of a drug’s safety and efficacy into an active partner in shepherding biosimilars onto the market.
As explained on Feb. 4 in testimony before a Congressional committee by Dr. Janet Woodcock, director of the FDA Center for Drug Evaluation and Research, the Biologic Price Competition and Innovation Act that was part of the Affordable Care Act launched a new U.S. drug-development pathway expressly for biosimilars. To implement that law, the FDA created an entirely new infrastructure within the agency – the Biosimilar Product Development Program – to help guide prospective manufacturers (called sponsors) of biosimilars through the regulatory and research hurdles to get a new biosimilar approved and into the hands of U.S. patients.
According to Dr. Woodcock, this program involves many steps where FDA staffers provide “review” and “advice” to sponsors on the studies they need to conduct and the analysis they need to perform to get their new products to market. The sponsor joins this program by paying an upfront fee that the FDA uses to keep the program running. Once a sponsor of a prospective biosimilar is in the program, the FDA’s staff helps guide the biosimilar development to a smooth conclusion.
To some extent, the FDA staff fills a similar role for conventional drug-development enterprises, conferring with manufacturers from the outset on matters such as the types and design of studies needed to insure success. What’s different about the biosimilar program is that conventional-drug development went on well before the FDA (or its predecessor) entered the scene, and the U.S. government created the FDA to police and regulate the drug production industry and protect the public against unscrupulous manufacturers of ineffective or dangerous drugs.
In contrast, the FDA itself created this new biosimilar development structure, and Dr. Woodcock noted that the in-depth review and advice meetings that the FDA offers to prospective biosimilar sponsors “has no counterpart in the Prescription Drug User Fee Act program and is unique” to the biosimilar program.
The consequence of having the FDA create the biosimilar development program from the ground up and structure it to provide such intimate input from the agency to sponsors at every step of the way seems to give the agency a notable and somewhat unnerving investment in the program’s success.
Dr. Woodcock called the approval of Zarxio an “exciting accomplishment,” and in her testimony before Congress she trumpeted the fact that as of January 2016 the biosimilar program was working on 59 proposed products that would mimic 18 different reference-product biologics. She also said that the FDA is “excited about the growing demand” for biosimilar-oriented meetings and marketing applications.
Don’t get me wrong: I think that the biosimilar concept is great, and has the potential to make what have become life-changing treatments more affordable and more available. And making the FDA such an active participant in getting biosimilar drugs created and approved is undoubtedly the most efficient way to accomplish this.
But in the process, the biosimilar program has changed the FDA from its more disengaged role as objective pharmaceutical judge into an active and seemingly not completely neutral codeveloper, risking at least the appearance of lost impartiality. Given that the FDA now wears two very different hats, we need to trust that the integrity and dedication of its staff will keep them from confusing their roles as proponent and gatekeeper.
On Twitter @mitchelzoler
The U.S. program to develop biosimilar agents – somewhat akin to generic drugs for complex, biologic molecules that have come off patent protection – is gathering momentum, with the first U.S. biosimilar, Zarxio, approved by the Food and Drug Administration in March 2015 and with the second, a biosimilar to infliximab, recommended by an FDA advisory committee on Feb. 9 of this year.
What’s striking about the burgeoning biosimilar development process, created by the Affordable Care Act, is how it has morphed the FDA from its traditional role as an objective arbiter of a drug’s safety and efficacy into an active partner in shepherding biosimilars onto the market.
As explained on Feb. 4 in testimony before a Congressional committee by Dr. Janet Woodcock, director of the FDA Center for Drug Evaluation and Research, the Biologic Price Competition and Innovation Act that was part of the Affordable Care Act launched a new U.S. drug-development pathway expressly for biosimilars. To implement that law, the FDA created an entirely new infrastructure within the agency – the Biosimilar Product Development Program – to help guide prospective manufacturers (called sponsors) of biosimilars through the regulatory and research hurdles to get a new biosimilar approved and into the hands of U.S. patients.
According to Dr. Woodcock, this program involves many steps where FDA staffers provide “review” and “advice” to sponsors on the studies they need to conduct and the analysis they need to perform to get their new products to market. The sponsor joins this program by paying an upfront fee that the FDA uses to keep the program running. Once a sponsor of a prospective biosimilar is in the program, the FDA’s staff helps guide the biosimilar development to a smooth conclusion.
To some extent, the FDA staff fills a similar role for conventional drug-development enterprises, conferring with manufacturers from the outset on matters such as the types and design of studies needed to insure success. What’s different about the biosimilar program is that conventional-drug development went on well before the FDA (or its predecessor) entered the scene, and the U.S. government created the FDA to police and regulate the drug production industry and protect the public against unscrupulous manufacturers of ineffective or dangerous drugs.
In contrast, the FDA itself created this new biosimilar development structure, and Dr. Woodcock noted that the in-depth review and advice meetings that the FDA offers to prospective biosimilar sponsors “has no counterpart in the Prescription Drug User Fee Act program and is unique” to the biosimilar program.
The consequence of having the FDA create the biosimilar development program from the ground up and structure it to provide such intimate input from the agency to sponsors at every step of the way seems to give the agency a notable and somewhat unnerving investment in the program’s success.
Dr. Woodcock called the approval of Zarxio an “exciting accomplishment,” and in her testimony before Congress she trumpeted the fact that as of January 2016 the biosimilar program was working on 59 proposed products that would mimic 18 different reference-product biologics. She also said that the FDA is “excited about the growing demand” for biosimilar-oriented meetings and marketing applications.
Don’t get me wrong: I think that the biosimilar concept is great, and has the potential to make what have become life-changing treatments more affordable and more available. And making the FDA such an active participant in getting biosimilar drugs created and approved is undoubtedly the most efficient way to accomplish this.
But in the process, the biosimilar program has changed the FDA from its more disengaged role as objective pharmaceutical judge into an active and seemingly not completely neutral codeveloper, risking at least the appearance of lost impartiality. Given that the FDA now wears two very different hats, we need to trust that the integrity and dedication of its staff will keep them from confusing their roles as proponent and gatekeeper.
On Twitter @mitchelzoler

Fibromyalgia found in 20% with spondyloarthritis; could affect management decisions
The presence of fibromyalgia in patients who are undergoing treatment of spondyloarthritis (SpA) is associated with higher measures of disease activity and shorter duration of first-time treatment with tumor necrosis factor inhibitors, according to results of a study measuring the impact and prevalence of fibromyalgia coexisting with SpA.
The results confirm “that the existence of concomitant FM [fibromyalgia] in SpA might complicate the evaluation of treatment response and [suggest] that coexistence of FM should be carefully screened when initiating a TNFi [tumor necrosis factor inhibitor] and/or evaluating its treatment effect, especially in the presence of peripheral and/or enthesitic symptoms and in the presence of very severe disease activity and patient-reported scores,” wrote Dr. Natalia Bello and her colleagues at Cochin Hospital, Paris (Arthritis Res Ther. 2016 Feb 9;18:42. doi: 10.1186/s13075-016-0943-z).

They recruited patients from Cochin Hospital, a tertiary care facility, and its rheumatology department’s outpatient clinic. Rather than use the 1990 American College of Rheumatology (ACR) classification criteria of FM or the 2010 ACR or modified 2010 ACR diagnostic criteria, which were developed for research and classification purposes, the investigators diagnosed FM based on a score of 5 or 6 on the six-question, self-reported Fibromyalgia Rapid Screening Tool (FiRST), which has 90.5% sensitivity and 85.7% specificity for FM. Patients’ SpA diagnoses were made by their rheumatologists. Overall, 30% of the cohort was female and had a mean age of 43 years.
The overall FM prevalence in the cohort was 21.4% (42 of 196 patients) and did not differ significantly according to whether the patients met either the clinical or imaging ASAS (Assessment of Spondyloarthritis International Society) criteria (21.3% vs. 18.8%, respectively) or whether they did or did not fulfill the ASAS criteria (21.1% vs. 30.0%, respectively).
Previous studies have shown the prevalence of FM at 12.6%-15.0% in SpA patients. Classifying axial SpA based on the clinical arm criteria alone has been controversial, the investigators said, mainly because it does not require an objective sign of inflammation (abnormal C-reactive protein or presence of inflammatory lesions seen on MRI of the sacroiliac joint) or structural damage in the sacroiliac joint seen on pelvic radiographs. But at least in this study there was no difference in FM prevalence in regard to whether patients met either the imaging and clinical arms of the ASAS classification criteria for axial SpA or both.
The study, according to the best knowledge of the investigators, is the first “to evaluate the prevalence of FM in a population of patients with SpA with regard to the fulfillment of the ASAS classification criteria.”
FM patients had as expected a significantly higher rate of either history of depression, or use of psychotropic drugs or strong opioids, compared with patients without FM (67% vs. 35%; P less than .01). Rates of exposure to treatment with different drug types (nonsteroidal anti-inflammatory drugs or conventional antirheumatic disease-modifying drugs) did not differ between those with and without FM, but FM patients switched significantly more often from their first TNFi (15.2% vs. 4.0%) and used it for a significantly shorter mean duration (1.7 vs. 3.5 years). The percentage of patients still taking their first TNFi after 2 years also was significantly lower among FM patients (28.1% vs. 41.7%).
Within the entire cohort, FM patients more often had enthesitis (59.5% vs. 39.0%, P = .01), a higher total Bath Ankylosing Spondylitis Disease Activity Index (4.7 vs. 2.6; P less than .01), higher global visual analog scale (5.9 vs. 3.0; P less than .01), and higher Bath Ankylosing Spondylitis Functional Index (4.8 vs. 2.0; P less than .01).
The authors suggested that FM patients’ higher rates of peripheral symptoms and enthesitis may warrant the use of the FiRST questionnaire in clinical practice before starting a TNFi in SpA patients to detect potentially coexisting FM.
The authors had no conflicts of interest to declare.
The presence of fibromyalgia in patients who are undergoing treatment of spondyloarthritis (SpA) is associated with higher measures of disease activity and shorter duration of first-time treatment with tumor necrosis factor inhibitors, according to results of a study measuring the impact and prevalence of fibromyalgia coexisting with SpA.
The results confirm “that the existence of concomitant FM [fibromyalgia] in SpA might complicate the evaluation of treatment response and [suggest] that coexistence of FM should be carefully screened when initiating a TNFi [tumor necrosis factor inhibitor] and/or evaluating its treatment effect, especially in the presence of peripheral and/or enthesitic symptoms and in the presence of very severe disease activity and patient-reported scores,” wrote Dr. Natalia Bello and her colleagues at Cochin Hospital, Paris (Arthritis Res Ther. 2016 Feb 9;18:42. doi: 10.1186/s13075-016-0943-z).
They recruited patients from Cochin Hospital, a tertiary care facility, and its rheumatology department’s outpatient clinic. Rather than use the 1990 American College of Rheumatology (ACR) classification criteria of FM or the 2010 ACR or modified 2010 ACR diagnostic criteria, which were developed for research and classification purposes, the investigators diagnosed FM based on a score of 5 or 6 on the six-question, self-reported Fibromyalgia Rapid Screening Tool (FiRST), which has 90.5% sensitivity and 85.7% specificity for FM. Patients’ SpA diagnoses were made by their rheumatologists. Overall, 30% of the cohort was female and had a mean age of 43 years.
The overall FM prevalence in the cohort was 21.4% (42 of 196 patients) and did not differ significantly according to whether the patients met either the clinical or imaging ASAS (Assessment of Spondyloarthritis International Society) criteria (21.3% vs. 18.8%, respectively) or whether they did or did not fulfill the ASAS criteria (21.1% vs. 30.0%, respectively).
Previous studies have shown the prevalence of FM at 12.6%-15.0% in SpA patients. Classifying axial SpA based on the clinical arm criteria alone has been controversial, the investigators said, mainly because it does not require an objective sign of inflammation (abnormal C-reactive protein or presence of inflammatory lesions seen on MRI of the sacroiliac joint) or structural damage in the sacroiliac joint seen on pelvic radiographs. But at least in this study there was no difference in FM prevalence in regard to whether patients met either the imaging and clinical arms of the ASAS classification criteria for axial SpA or both.
The study, according to the best knowledge of the investigators, is the first “to evaluate the prevalence of FM in a population of patients with SpA with regard to the fulfillment of the ASAS classification criteria.”
FM patients had as expected a significantly higher rate of either history of depression, or use of psychotropic drugs or strong opioids, compared with patients without FM (67% vs. 35%; P less than .01). Rates of exposure to treatment with different drug types (nonsteroidal anti-inflammatory drugs or conventional antirheumatic disease-modifying drugs) did not differ between those with and without FM, but FM patients switched significantly more often from their first TNFi (15.2% vs. 4.0%) and used it for a significantly shorter mean duration (1.7 vs. 3.5 years). The percentage of patients still taking their first TNFi after 2 years also was significantly lower among FM patients (28.1% vs. 41.7%).
Within the entire cohort, FM patients more often had enthesitis (59.5% vs. 39.0%, P = .01), a higher total Bath Ankylosing Spondylitis Disease Activity Index (4.7 vs. 2.6; P less than .01), higher global visual analog scale (5.9 vs. 3.0; P less than .01), and higher Bath Ankylosing Spondylitis Functional Index (4.8 vs. 2.0; P less than .01).
The authors suggested that FM patients’ higher rates of peripheral symptoms and enthesitis may warrant the use of the FiRST questionnaire in clinical practice before starting a TNFi in SpA patients to detect potentially coexisting FM.
The authors had no conflicts of interest to declare.
The presence of fibromyalgia in patients who are undergoing treatment of spondyloarthritis (SpA) is associated with higher measures of disease activity and shorter duration of first-time treatment with tumor necrosis factor inhibitors, according to results of a study measuring the impact and prevalence of fibromyalgia coexisting with SpA.
The results confirm “that the existence of concomitant FM [fibromyalgia] in SpA might complicate the evaluation of treatment response and [suggest] that coexistence of FM should be carefully screened when initiating a TNFi [tumor necrosis factor inhibitor] and/or evaluating its treatment effect, especially in the presence of peripheral and/or enthesitic symptoms and in the presence of very severe disease activity and patient-reported scores,” wrote Dr. Natalia Bello and her colleagues at Cochin Hospital, Paris (Arthritis Res Ther. 2016 Feb 9;18:42. doi: 10.1186/s13075-016-0943-z).
They recruited patients from Cochin Hospital, a tertiary care facility, and its rheumatology department’s outpatient clinic. Rather than use the 1990 American College of Rheumatology (ACR) classification criteria of FM or the 2010 ACR or modified 2010 ACR diagnostic criteria, which were developed for research and classification purposes, the investigators diagnosed FM based on a score of 5 or 6 on the six-question, self-reported Fibromyalgia Rapid Screening Tool (FiRST), which has 90.5% sensitivity and 85.7% specificity for FM. Patients’ SpA diagnoses were made by their rheumatologists. Overall, 30% of the cohort was female and had a mean age of 43 years.
The overall FM prevalence in the cohort was 21.4% (42 of 196 patients) and did not differ significantly according to whether the patients met either the clinical or imaging ASAS (Assessment of Spondyloarthritis International Society) criteria (21.3% vs. 18.8%, respectively) or whether they did or did not fulfill the ASAS criteria (21.1% vs. 30.0%, respectively).
Previous studies have shown the prevalence of FM at 12.6%-15.0% in SpA patients. Classifying axial SpA based on the clinical arm criteria alone has been controversial, the investigators said, mainly because it does not require an objective sign of inflammation (abnormal C-reactive protein or presence of inflammatory lesions seen on MRI of the sacroiliac joint) or structural damage in the sacroiliac joint seen on pelvic radiographs. But at least in this study there was no difference in FM prevalence in regard to whether patients met either the imaging and clinical arms of the ASAS classification criteria for axial SpA or both.
The study, according to the best knowledge of the investigators, is the first “to evaluate the prevalence of FM in a population of patients with SpA with regard to the fulfillment of the ASAS classification criteria.”
FM patients had as expected a significantly higher rate of either history of depression, or use of psychotropic drugs or strong opioids, compared with patients without FM (67% vs. 35%; P less than .01). Rates of exposure to treatment with different drug types (nonsteroidal anti-inflammatory drugs or conventional antirheumatic disease-modifying drugs) did not differ between those with and without FM, but FM patients switched significantly more often from their first TNFi (15.2% vs. 4.0%) and used it for a significantly shorter mean duration (1.7 vs. 3.5 years). The percentage of patients still taking their first TNFi after 2 years also was significantly lower among FM patients (28.1% vs. 41.7%).
Within the entire cohort, FM patients more often had enthesitis (59.5% vs. 39.0%, P = .01), a higher total Bath Ankylosing Spondylitis Disease Activity Index (4.7 vs. 2.6; P less than .01), higher global visual analog scale (5.9 vs. 3.0; P less than .01), and higher Bath Ankylosing Spondylitis Functional Index (4.8 vs. 2.0; P less than .01).
The authors suggested that FM patients’ higher rates of peripheral symptoms and enthesitis may warrant the use of the FiRST questionnaire in clinical practice before starting a TNFi in SpA patients to detect potentially coexisting FM.
The authors had no conflicts of interest to declare.
FROM ARTHRITIS RESEARCH & THERAPY
Key clinical point: Coexistence of fibromyalgia in patients diagnosed with spondyloarthritis might be slightly more frequent than previously reported and does not differ according to whether SpA was classified based on imaging or clinical criteria.
Major finding: The overall FM prevalence in the cohort was 21.4% (42 of 196 patients) and did not differ significantly according to whether the patients met either the clinical or imaging ASAS criteria (21.3% vs. 18.8%, respectively).
Data source: A cohort study of 196 patients diagnosed with spondyloarthritis.
Disclosures: The authors had no conflicts of interest to declare.
