User login
Genetic profile flags scleroderma patients with best HSCT responses
CHICAGO – Subgroup categorization of patients with severe scleroderma by their gene-expression profile correlated with responses to a newly proven treatment for the disease that involves myeloablation and autologous hematopoietic stem cell transplantation (HSCT).

Patients who fell into the “fibroproliferative” scleroderma subgroup, roughly one-third of patients enrolled in the treatment study, showed a high level of benefit from myeloablation and autologous HSCT, Michael L. Whitfield, PhD, said at the annual meeting of the American College of Rheumatology.
In contrast, the roughly one-third of patients in the study with a gene-expression profile that placed them into the “normal-like” subgroup had outcomes that closely matched the normal-like patients in the control group, who were treated with cyclophosphamide, which suggests that the normal-like patients are probably not good candidates for HSCT, said Dr. Whitfield, a professor of molecular and systems biology at the Geisel School of Medicine at Dartmouth in Hanover, N.H.
This study starts to get at the question of “How do you do personalized medicine in a disease like scleroderma?” he explained. “HSCT may be a game changer for patients with the fibroproliferative type of scleroderma,” who did relatively poorly in the control group of the trial when they received cyclophosphamide. Categorization of patients by their gene-expression profiles is a way to find order among scleroderma patients in what is otherwise “a very heterogeneous disease, where some patients improve on a treatment and others do not,” Dr Whitfield said.
The study used data collected in the SCOT (Scleroderma: Cyclophosphamide or Transplantation) trial, which enrolled 75 patients with severe scleroderma at any of 26 sites in the United States and Canada. SCOT compared the safety and efficacy of myeloablation followed by autologous HSCT with that of treatment with cyclophosphamide, and it followed patients for a median of 54 months. The results showed that overall HSCT was superior for the primary endpoint and several secondary endpoints, including event-free survival, which was 79% after HSCT and 50% in control patients by the end of follow-up (N Engl J Med. 2018 Jan 4;378[1]:35-47).
Dr. Whitfield’s group took peripheral blood cells from 30 of the patients treated by HSCT and from 33 of the patients treated with cyclophosphamide per protocol and analyzed the gene-expression profiles of the cells to categorize patients into the gene-expression subtypes of scleroderma that had been previously defined by Dr. Whitfield and his associates: fibroproliferative, inflammatory, limited, or normal-like (PLOS One. 2008 Jul 18;3[7]:e2696). The gene-expression analysis, which now looks at the activity of about 1,300 genes, showed that the 33 cyclophosphamide-treated patients included 12 with an inflammatory profile, 12 with a normal-like profile, and 9 with a fibroproliferative profile. Among the 30 patients treated with HSCT, 11 were in the fibroproliferative group, 11 were normal-like, and 8 had an inflammatory pattern.
Analysis of event-free survival out to 6 years following enrollment showed that, in the fibroproliferative subgroup, roughly 90% of patients treated with HSCT remained alive and event free, compared with about 35% of the cyclophosphamide patients, a highly statistically significant difference. In the inflammatory subgroup, event-free survival persisted in about 90% in the HSCT recipients, compared with about 50% of those in the control arm, a difference that did not reach statistical significance. Among patients with normal-like gene expression, the event-free survival rate was about the same regardless of treatment, about 60% in each treatment arm. The results suggest that patients with normal-like disease “are probably not good candidates for a treatment as intensive as HSCT,” Dr. Whitfield said in an interview.
Although the HSCT and cyclophosphamide treatment groups included relatively small numbers of patients, when the researchers subdivided the trial cohort into three different scleroderma types, the analysis remained “powered well enough to see a difference; the difference was very clearly statistically significant,” Dr. Whitfield declared.
“Now that we have a treatment [HSCT] to tie to the [gene-expression analysis], we can think about using this in routine practice,” he concluded.
Dr. Whitfield is a cofounder of Celdara, and he has been a consultant to Bristol-Myers Squibb, Corbus, UCB, and Third Rock Ventures.
SOURCE: Franks J et al. Arthritis Rheumatol. 2018;70(suppl 10): Abstract 1876.
CHICAGO – Subgroup categorization of patients with severe scleroderma by their gene-expression profile correlated with responses to a newly proven treatment for the disease that involves myeloablation and autologous hematopoietic stem cell transplantation (HSCT).
Patients who fell into the “fibroproliferative” scleroderma subgroup, roughly one-third of patients enrolled in the treatment study, showed a high level of benefit from myeloablation and autologous HSCT, Michael L. Whitfield, PhD, said at the annual meeting of the American College of Rheumatology.
In contrast, the roughly one-third of patients in the study with a gene-expression profile that placed them into the “normal-like” subgroup had outcomes that closely matched the normal-like patients in the control group, who were treated with cyclophosphamide, which suggests that the normal-like patients are probably not good candidates for HSCT, said Dr. Whitfield, a professor of molecular and systems biology at the Geisel School of Medicine at Dartmouth in Hanover, N.H.
This study starts to get at the question of “How do you do personalized medicine in a disease like scleroderma?” he explained. “HSCT may be a game changer for patients with the fibroproliferative type of scleroderma,” who did relatively poorly in the control group of the trial when they received cyclophosphamide. Categorization of patients by their gene-expression profiles is a way to find order among scleroderma patients in what is otherwise “a very heterogeneous disease, where some patients improve on a treatment and others do not,” Dr Whitfield said.
The study used data collected in the SCOT (Scleroderma: Cyclophosphamide or Transplantation) trial, which enrolled 75 patients with severe scleroderma at any of 26 sites in the United States and Canada. SCOT compared the safety and efficacy of myeloablation followed by autologous HSCT with that of treatment with cyclophosphamide, and it followed patients for a median of 54 months. The results showed that overall HSCT was superior for the primary endpoint and several secondary endpoints, including event-free survival, which was 79% after HSCT and 50% in control patients by the end of follow-up (N Engl J Med. 2018 Jan 4;378[1]:35-47).
Dr. Whitfield’s group took peripheral blood cells from 30 of the patients treated by HSCT and from 33 of the patients treated with cyclophosphamide per protocol and analyzed the gene-expression profiles of the cells to categorize patients into the gene-expression subtypes of scleroderma that had been previously defined by Dr. Whitfield and his associates: fibroproliferative, inflammatory, limited, or normal-like (PLOS One. 2008 Jul 18;3[7]:e2696). The gene-expression analysis, which now looks at the activity of about 1,300 genes, showed that the 33 cyclophosphamide-treated patients included 12 with an inflammatory profile, 12 with a normal-like profile, and 9 with a fibroproliferative profile. Among the 30 patients treated with HSCT, 11 were in the fibroproliferative group, 11 were normal-like, and 8 had an inflammatory pattern.
Analysis of event-free survival out to 6 years following enrollment showed that, in the fibroproliferative subgroup, roughly 90% of patients treated with HSCT remained alive and event free, compared with about 35% of the cyclophosphamide patients, a highly statistically significant difference. In the inflammatory subgroup, event-free survival persisted in about 90% in the HSCT recipients, compared with about 50% of those in the control arm, a difference that did not reach statistical significance. Among patients with normal-like gene expression, the event-free survival rate was about the same regardless of treatment, about 60% in each treatment arm. The results suggest that patients with normal-like disease “are probably not good candidates for a treatment as intensive as HSCT,” Dr. Whitfield said in an interview.
Although the HSCT and cyclophosphamide treatment groups included relatively small numbers of patients, when the researchers subdivided the trial cohort into three different scleroderma types, the analysis remained “powered well enough to see a difference; the difference was very clearly statistically significant,” Dr. Whitfield declared.
“Now that we have a treatment [HSCT] to tie to the [gene-expression analysis], we can think about using this in routine practice,” he concluded.
Dr. Whitfield is a cofounder of Celdara, and he has been a consultant to Bristol-Myers Squibb, Corbus, UCB, and Third Rock Ventures.
SOURCE: Franks J et al. Arthritis Rheumatol. 2018;70(suppl 10): Abstract 1876.
CHICAGO – Subgroup categorization of patients with severe scleroderma by their gene-expression profile correlated with responses to a newly proven treatment for the disease that involves myeloablation and autologous hematopoietic stem cell transplantation (HSCT).
Patients who fell into the “fibroproliferative” scleroderma subgroup, roughly one-third of patients enrolled in the treatment study, showed a high level of benefit from myeloablation and autologous HSCT, Michael L. Whitfield, PhD, said at the annual meeting of the American College of Rheumatology.
In contrast, the roughly one-third of patients in the study with a gene-expression profile that placed them into the “normal-like” subgroup had outcomes that closely matched the normal-like patients in the control group, who were treated with cyclophosphamide, which suggests that the normal-like patients are probably not good candidates for HSCT, said Dr. Whitfield, a professor of molecular and systems biology at the Geisel School of Medicine at Dartmouth in Hanover, N.H.
This study starts to get at the question of “How do you do personalized medicine in a disease like scleroderma?” he explained. “HSCT may be a game changer for patients with the fibroproliferative type of scleroderma,” who did relatively poorly in the control group of the trial when they received cyclophosphamide. Categorization of patients by their gene-expression profiles is a way to find order among scleroderma patients in what is otherwise “a very heterogeneous disease, where some patients improve on a treatment and others do not,” Dr Whitfield said.
The study used data collected in the SCOT (Scleroderma: Cyclophosphamide or Transplantation) trial, which enrolled 75 patients with severe scleroderma at any of 26 sites in the United States and Canada. SCOT compared the safety and efficacy of myeloablation followed by autologous HSCT with that of treatment with cyclophosphamide, and it followed patients for a median of 54 months. The results showed that overall HSCT was superior for the primary endpoint and several secondary endpoints, including event-free survival, which was 79% after HSCT and 50% in control patients by the end of follow-up (N Engl J Med. 2018 Jan 4;378[1]:35-47).
Dr. Whitfield’s group took peripheral blood cells from 30 of the patients treated by HSCT and from 33 of the patients treated with cyclophosphamide per protocol and analyzed the gene-expression profiles of the cells to categorize patients into the gene-expression subtypes of scleroderma that had been previously defined by Dr. Whitfield and his associates: fibroproliferative, inflammatory, limited, or normal-like (PLOS One. 2008 Jul 18;3[7]:e2696). The gene-expression analysis, which now looks at the activity of about 1,300 genes, showed that the 33 cyclophosphamide-treated patients included 12 with an inflammatory profile, 12 with a normal-like profile, and 9 with a fibroproliferative profile. Among the 30 patients treated with HSCT, 11 were in the fibroproliferative group, 11 were normal-like, and 8 had an inflammatory pattern.
Analysis of event-free survival out to 6 years following enrollment showed that, in the fibroproliferative subgroup, roughly 90% of patients treated with HSCT remained alive and event free, compared with about 35% of the cyclophosphamide patients, a highly statistically significant difference. In the inflammatory subgroup, event-free survival persisted in about 90% in the HSCT recipients, compared with about 50% of those in the control arm, a difference that did not reach statistical significance. Among patients with normal-like gene expression, the event-free survival rate was about the same regardless of treatment, about 60% in each treatment arm. The results suggest that patients with normal-like disease “are probably not good candidates for a treatment as intensive as HSCT,” Dr. Whitfield said in an interview.
Although the HSCT and cyclophosphamide treatment groups included relatively small numbers of patients, when the researchers subdivided the trial cohort into three different scleroderma types, the analysis remained “powered well enough to see a difference; the difference was very clearly statistically significant,” Dr. Whitfield declared.
“Now that we have a treatment [HSCT] to tie to the [gene-expression analysis], we can think about using this in routine practice,” he concluded.
Dr. Whitfield is a cofounder of Celdara, and he has been a consultant to Bristol-Myers Squibb, Corbus, UCB, and Third Rock Ventures.
SOURCE: Franks J et al. Arthritis Rheumatol. 2018;70(suppl 10): Abstract 1876.
REPORTING FROM THE ACR ANNUAL MEETING
Key clinical point:
Major finding: About 90% of fibroproliferative scleroderma patients had prolonged event-free survival after stem cell transplant, compared with about 35% of controls.
Study details: The study used data collected in the SCOT trial of 75 patients with severe scleroderma.
Disclosures: Dr. Whitfield is a cofounder of Celdara, and he has been a consultant to Bristol-Myers Squibb, Corbus, UCB, and Third Rock Ventures.
Source: Franks J et al. Arthritis Rheumatol. 2018;70(Suppl 10): Abstract 1876.
Dueling SLE classification criteria: And the winner is...
CHICAGO – Newer isn’t necessarily better – especially when it comes to the plethora of SLE classification criteria, according to Michelle A. Petri, MD, professor of medicine and director of the Hopkins Lupus Center at Johns Hopkins University, Baltimore.

“We have an embarrassment of criteria for lupus right now, and everyone wants to know if one is better than the others,” the rheumatologist said at the annual meeting of the American College of Rheumatology.
She and several coinvestigators who are members of the Systemic Lupus International Collaborating Clinics (SLICC) set out to learn the answer. They developed a new, modified, weighted version of the 2012 SLICC criteria and compared its sensitivity and specificity for SLE diagnosis by 690 physicians with three other major classification systems: the 1997 update to ACR-11 criteria, the nonweighted SLICC 2012 criteria, and the proposed EULAR/ACR criteria, which uses a differentially weighted approach in which the various possible disease manifestations are each assigned a different point score. In contrast, the revised ACR-11 and SLICC 2012 criteria count each SLE manifestation equally.
Long story short: “The two newly derived weighted classification rules did not perform better than the existing list-based rules in terms of overall agreement,” according to Dr. Petri.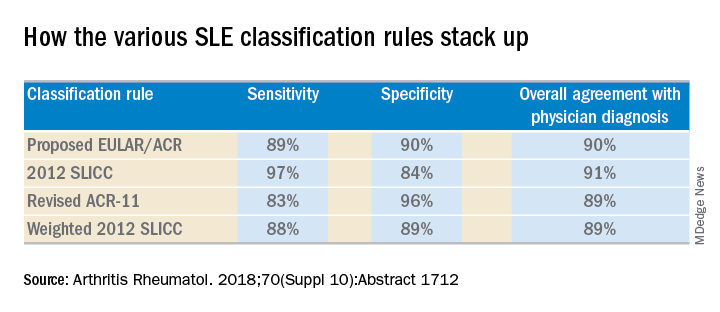
“We don’t think that weighting all the criteria, which is what the EULAR/ACR and weighted SLICC 2012 rules do, adds to the performance of the criteria set, and in fact it makes it much more difficult for clinicians to use when there’s a complicated weighting system, unless it’s web-based or there’s an app for it. And to be honest, clinicians are so busy that they’re probably not going to take time out in a clinic visit to go use the web or an app. Our criteria need to be user friendly,” she continued.
So which of the four classification systems is most user friendly? The EULAR/ACR criteria can be dismissed on that score because they are supposed to be used only for research, according to the rheumatologist.
“I think the SLICC 2012 criteria are very useful for clinicians because they have the highest sensitivity. And what a clinician wants is not to miss a diagnosis and to start treatment early,” Dr. Petri said.
To develop the weighted SLICC criteria, whose future at this point doesn’t look bright, she and her coinvestigators redeployed the same physician-rated patient scenarios used to develop the nonweighted 2012 SLICC classification criteria and assigned each of the potential manifestations of SLE a specific point score. For example, acute cutaneous manifestations received 16 points, serositis 9, oral ulcers 16, thrombocytopenia 15, and so forth. Under this system, a patient with a score of at least 56 points, or lupus nephritis, or least one clinical and one immunologic component of SLE was classified as having the disease.
Dr. Petri reported having no financial conflicts regarding her study, supported by the National Institutes of Health.
SOURCE: Petri MA et al. Arthritis Rheumatol. 2018;70(Suppl 10): Abstract 1712.
CHICAGO – Newer isn’t necessarily better – especially when it comes to the plethora of SLE classification criteria, according to Michelle A. Petri, MD, professor of medicine and director of the Hopkins Lupus Center at Johns Hopkins University, Baltimore.
“We have an embarrassment of criteria for lupus right now, and everyone wants to know if one is better than the others,” the rheumatologist said at the annual meeting of the American College of Rheumatology.
She and several coinvestigators who are members of the Systemic Lupus International Collaborating Clinics (SLICC) set out to learn the answer. They developed a new, modified, weighted version of the 2012 SLICC criteria and compared its sensitivity and specificity for SLE diagnosis by 690 physicians with three other major classification systems: the 1997 update to ACR-11 criteria, the nonweighted SLICC 2012 criteria, and the proposed EULAR/ACR criteria, which uses a differentially weighted approach in which the various possible disease manifestations are each assigned a different point score. In contrast, the revised ACR-11 and SLICC 2012 criteria count each SLE manifestation equally.
Long story short: “The two newly derived weighted classification rules did not perform better than the existing list-based rules in terms of overall agreement,” according to Dr. Petri.
“We don’t think that weighting all the criteria, which is what the EULAR/ACR and weighted SLICC 2012 rules do, adds to the performance of the criteria set, and in fact it makes it much more difficult for clinicians to use when there’s a complicated weighting system, unless it’s web-based or there’s an app for it. And to be honest, clinicians are so busy that they’re probably not going to take time out in a clinic visit to go use the web or an app. Our criteria need to be user friendly,” she continued.
So which of the four classification systems is most user friendly? The EULAR/ACR criteria can be dismissed on that score because they are supposed to be used only for research, according to the rheumatologist.
“I think the SLICC 2012 criteria are very useful for clinicians because they have the highest sensitivity. And what a clinician wants is not to miss a diagnosis and to start treatment early,” Dr. Petri said.
To develop the weighted SLICC criteria, whose future at this point doesn’t look bright, she and her coinvestigators redeployed the same physician-rated patient scenarios used to develop the nonweighted 2012 SLICC classification criteria and assigned each of the potential manifestations of SLE a specific point score. For example, acute cutaneous manifestations received 16 points, serositis 9, oral ulcers 16, thrombocytopenia 15, and so forth. Under this system, a patient with a score of at least 56 points, or lupus nephritis, or least one clinical and one immunologic component of SLE was classified as having the disease.
Dr. Petri reported having no financial conflicts regarding her study, supported by the National Institutes of Health.
SOURCE: Petri MA et al. Arthritis Rheumatol. 2018;70(Suppl 10): Abstract 1712.
CHICAGO – Newer isn’t necessarily better – especially when it comes to the plethora of SLE classification criteria, according to Michelle A. Petri, MD, professor of medicine and director of the Hopkins Lupus Center at Johns Hopkins University, Baltimore.
“We have an embarrassment of criteria for lupus right now, and everyone wants to know if one is better than the others,” the rheumatologist said at the annual meeting of the American College of Rheumatology.
She and several coinvestigators who are members of the Systemic Lupus International Collaborating Clinics (SLICC) set out to learn the answer. They developed a new, modified, weighted version of the 2012 SLICC criteria and compared its sensitivity and specificity for SLE diagnosis by 690 physicians with three other major classification systems: the 1997 update to ACR-11 criteria, the nonweighted SLICC 2012 criteria, and the proposed EULAR/ACR criteria, which uses a differentially weighted approach in which the various possible disease manifestations are each assigned a different point score. In contrast, the revised ACR-11 and SLICC 2012 criteria count each SLE manifestation equally.
Long story short: “The two newly derived weighted classification rules did not perform better than the existing list-based rules in terms of overall agreement,” according to Dr. Petri.
“We don’t think that weighting all the criteria, which is what the EULAR/ACR and weighted SLICC 2012 rules do, adds to the performance of the criteria set, and in fact it makes it much more difficult for clinicians to use when there’s a complicated weighting system, unless it’s web-based or there’s an app for it. And to be honest, clinicians are so busy that they’re probably not going to take time out in a clinic visit to go use the web or an app. Our criteria need to be user friendly,” she continued.
So which of the four classification systems is most user friendly? The EULAR/ACR criteria can be dismissed on that score because they are supposed to be used only for research, according to the rheumatologist.
“I think the SLICC 2012 criteria are very useful for clinicians because they have the highest sensitivity. And what a clinician wants is not to miss a diagnosis and to start treatment early,” Dr. Petri said.
To develop the weighted SLICC criteria, whose future at this point doesn’t look bright, she and her coinvestigators redeployed the same physician-rated patient scenarios used to develop the nonweighted 2012 SLICC classification criteria and assigned each of the potential manifestations of SLE a specific point score. For example, acute cutaneous manifestations received 16 points, serositis 9, oral ulcers 16, thrombocytopenia 15, and so forth. Under this system, a patient with a score of at least 56 points, or lupus nephritis, or least one clinical and one immunologic component of SLE was classified as having the disease.
Dr. Petri reported having no financial conflicts regarding her study, supported by the National Institutes of Health.
SOURCE: Petri MA et al. Arthritis Rheumatol. 2018;70(Suppl 10): Abstract 1712.
REPORTING FROM THE ACR ANNUAL MEETING
Key clinical point: The 2012 SLICC SLE classification criteria are best suited for clinical practice.
Major finding:
Study details: This study compared the sensitivity, specificity, and overall agreement with physician diagnosis of four different sets of SLE classification criteria.
Disclosures: The study was supported by the National Institutes of Health. The presenter reported having no financial conflicts.
Source: Petri MA et al. Arthritis Rheumatol. 2018;70(Suppl 10): Abstract 1712.
Leflunomide-hydroxychloroquine combo shows promise in primary Sjögren’s pilot study
CHICAGO – Combination therapy with leflunomide and hydroxychloroquine met all goals for efficacy, safety, and tolerability among patients with primary Sjögren’s syndrome in a randomized, placebo-controlled pilot study, lending support to evidence suggesting the two drugs have additive benefits.
The combined treatment was associated with a statistically significant decrease in the EULAR Sjögren’s syndrome disease activity index (ESSDAI) over 24 weeks – the primary endpoint of the study – in 21 patients in the treatment group. The ESSDAI score on combination treatment dropped from about 10 at baseline to about 6 at 24 weeks, compared with no change from a baseline of about 10 in eight patients in the placebo group. An ESSDAI decrease of 3 or more points occurred in 11 patients in the combination therapy group, compared with none in the placebo group, Joel A.G. van Roon, PhD, a researcher in the Laboratory of Translational Immunology at the University Medical Center Utrecht, the Netherlands, reported in a late-breaking poster at the annual meeting of the American College of Rheumatology.
Both leflunomide and hydroxychloroquine have been shown to inhibit B-cell hyperactivity, but the clinical benefits have been modest and not statistically significant. Since the two agents have complementary inhibitory properties on different immune cells – including B and T cells and plasmacytoid dendritic cells, and based on in vitro findings of additive benefits with respect to inhibition of T- and B-cell activation and CXCL13 production, Dr. van Roon and his colleagues conducted this double-blind, single-center, proof-of-concept pilot study (REPURpSS-1) to assess the efficacy, safety, and tolerability of combined treatment in primary Sjögren’s syndrome.
In all, 29 patients with clinically active disease, defined by ESSDAI of 5 or greater, were randomized 2:1 to receive either 20 mg of leflunomide daily plus 400 mg of hydroxychloroquine daily or placebo/placebo for 24 weeks.
Secondary endpoints such as oral dryness also improved significantly in the treatment group versus the placebo group. Stimulated whole saliva flow increased from about 800 mcL/5 min to about 1,400 mcL/5 min and decreased from about 1,250 to about 1,000 mcL/5 min in the groups, respectively. Median EULAR Sjögren’s syndrome patient reported index (ESSPRI), ESSPRI pain, and ESSPRI fatigue scores, as well as Physician’s and Patient’s Global Assessment scores each improved significantly in the treatment group (at least P less than .05 in all cases) but not in the placebo groups, said Dr. van Roon.
Additionally, serum IgG, IgM rheumatoid factor, and chemokine CXCL13 – a marker for lymphoid neogenesis – decreased significantly, and complement components 3 and 4 (C3 and C4) increased significantly by 24 weeks in the treatment group, but not in the placebo group. B-cell hyperactivity as measured by serum IgG decreased from about 20 g/L to about 14 g/L versus no change from about 15 g/L at baseline in the placebo group, he noted.
“Overall, combination leflunomide and hydroxychloroquine was safe and well tolerated, but larger randomized, controlled trials are needed to confirm the observed effects and to identify potential biomarkers for response,” he concluded.
This study was supported by ZonMw (the Netherlands Organization for Health Research and Development). Dr. van Roon reported having no relevant disclosures.
SOURCE: van Roon JAG et al. Arthritis Rheumatol. 2018;70(Suppl 10): Abstract L10.
CHICAGO – Combination therapy with leflunomide and hydroxychloroquine met all goals for efficacy, safety, and tolerability among patients with primary Sjögren’s syndrome in a randomized, placebo-controlled pilot study, lending support to evidence suggesting the two drugs have additive benefits.
The combined treatment was associated with a statistically significant decrease in the EULAR Sjögren’s syndrome disease activity index (ESSDAI) over 24 weeks – the primary endpoint of the study – in 21 patients in the treatment group. The ESSDAI score on combination treatment dropped from about 10 at baseline to about 6 at 24 weeks, compared with no change from a baseline of about 10 in eight patients in the placebo group. An ESSDAI decrease of 3 or more points occurred in 11 patients in the combination therapy group, compared with none in the placebo group, Joel A.G. van Roon, PhD, a researcher in the Laboratory of Translational Immunology at the University Medical Center Utrecht, the Netherlands, reported in a late-breaking poster at the annual meeting of the American College of Rheumatology.
Both leflunomide and hydroxychloroquine have been shown to inhibit B-cell hyperactivity, but the clinical benefits have been modest and not statistically significant. Since the two agents have complementary inhibitory properties on different immune cells – including B and T cells and plasmacytoid dendritic cells, and based on in vitro findings of additive benefits with respect to inhibition of T- and B-cell activation and CXCL13 production, Dr. van Roon and his colleagues conducted this double-blind, single-center, proof-of-concept pilot study (REPURpSS-1) to assess the efficacy, safety, and tolerability of combined treatment in primary Sjögren’s syndrome.
In all, 29 patients with clinically active disease, defined by ESSDAI of 5 or greater, were randomized 2:1 to receive either 20 mg of leflunomide daily plus 400 mg of hydroxychloroquine daily or placebo/placebo for 24 weeks.
Secondary endpoints such as oral dryness also improved significantly in the treatment group versus the placebo group. Stimulated whole saliva flow increased from about 800 mcL/5 min to about 1,400 mcL/5 min and decreased from about 1,250 to about 1,000 mcL/5 min in the groups, respectively. Median EULAR Sjögren’s syndrome patient reported index (ESSPRI), ESSPRI pain, and ESSPRI fatigue scores, as well as Physician’s and Patient’s Global Assessment scores each improved significantly in the treatment group (at least P less than .05 in all cases) but not in the placebo groups, said Dr. van Roon.
Additionally, serum IgG, IgM rheumatoid factor, and chemokine CXCL13 – a marker for lymphoid neogenesis – decreased significantly, and complement components 3 and 4 (C3 and C4) increased significantly by 24 weeks in the treatment group, but not in the placebo group. B-cell hyperactivity as measured by serum IgG decreased from about 20 g/L to about 14 g/L versus no change from about 15 g/L at baseline in the placebo group, he noted.
“Overall, combination leflunomide and hydroxychloroquine was safe and well tolerated, but larger randomized, controlled trials are needed to confirm the observed effects and to identify potential biomarkers for response,” he concluded.
This study was supported by ZonMw (the Netherlands Organization for Health Research and Development). Dr. van Roon reported having no relevant disclosures.
SOURCE: van Roon JAG et al. Arthritis Rheumatol. 2018;70(Suppl 10): Abstract L10.
CHICAGO – Combination therapy with leflunomide and hydroxychloroquine met all goals for efficacy, safety, and tolerability among patients with primary Sjögren’s syndrome in a randomized, placebo-controlled pilot study, lending support to evidence suggesting the two drugs have additive benefits.
The combined treatment was associated with a statistically significant decrease in the EULAR Sjögren’s syndrome disease activity index (ESSDAI) over 24 weeks – the primary endpoint of the study – in 21 patients in the treatment group. The ESSDAI score on combination treatment dropped from about 10 at baseline to about 6 at 24 weeks, compared with no change from a baseline of about 10 in eight patients in the placebo group. An ESSDAI decrease of 3 or more points occurred in 11 patients in the combination therapy group, compared with none in the placebo group, Joel A.G. van Roon, PhD, a researcher in the Laboratory of Translational Immunology at the University Medical Center Utrecht, the Netherlands, reported in a late-breaking poster at the annual meeting of the American College of Rheumatology.
Both leflunomide and hydroxychloroquine have been shown to inhibit B-cell hyperactivity, but the clinical benefits have been modest and not statistically significant. Since the two agents have complementary inhibitory properties on different immune cells – including B and T cells and plasmacytoid dendritic cells, and based on in vitro findings of additive benefits with respect to inhibition of T- and B-cell activation and CXCL13 production, Dr. van Roon and his colleagues conducted this double-blind, single-center, proof-of-concept pilot study (REPURpSS-1) to assess the efficacy, safety, and tolerability of combined treatment in primary Sjögren’s syndrome.
In all, 29 patients with clinically active disease, defined by ESSDAI of 5 or greater, were randomized 2:1 to receive either 20 mg of leflunomide daily plus 400 mg of hydroxychloroquine daily or placebo/placebo for 24 weeks.
Secondary endpoints such as oral dryness also improved significantly in the treatment group versus the placebo group. Stimulated whole saliva flow increased from about 800 mcL/5 min to about 1,400 mcL/5 min and decreased from about 1,250 to about 1,000 mcL/5 min in the groups, respectively. Median EULAR Sjögren’s syndrome patient reported index (ESSPRI), ESSPRI pain, and ESSPRI fatigue scores, as well as Physician’s and Patient’s Global Assessment scores each improved significantly in the treatment group (at least P less than .05 in all cases) but not in the placebo groups, said Dr. van Roon.
Additionally, serum IgG, IgM rheumatoid factor, and chemokine CXCL13 – a marker for lymphoid neogenesis – decreased significantly, and complement components 3 and 4 (C3 and C4) increased significantly by 24 weeks in the treatment group, but not in the placebo group. B-cell hyperactivity as measured by serum IgG decreased from about 20 g/L to about 14 g/L versus no change from about 15 g/L at baseline in the placebo group, he noted.
“Overall, combination leflunomide and hydroxychloroquine was safe and well tolerated, but larger randomized, controlled trials are needed to confirm the observed effects and to identify potential biomarkers for response,” he concluded.
This study was supported by ZonMw (the Netherlands Organization for Health Research and Development). Dr. van Roon reported having no relevant disclosures.
SOURCE: van Roon JAG et al. Arthritis Rheumatol. 2018;70(Suppl 10): Abstract L10.
REPORTING FROM THE ACR ANNUAL MEETING
Key clinical point: but larger randomized, controlled trials are needed to confirm the observed effects.
Major finding: Combined treatment was associated with a decline in EULAR Sjögren’s syndrome disease activity index score from about 10 at baseline to about 6 at 24 weeks.
Study details: A randomized, placebo-controlled pilot study of 29 patients.
Disclosures: This study was supported by ZonMw (the Netherlands Organization for Health Research and Development). Dr. van Roon reported having no relevant disclosures.
Source: van Roon JAG et al. Arthritis Rheumatol. 2018;70(Suppl 10): Abstract L10.
SLE low-disease definition receives prospective validation
CHICAGO – The long path toward a validated definition of low disease activity in patients with systemic lupus erythematosus may be nearing an end as a definition first proposed more than 5 years ago received validation with data from more than 1,700 patients in a prospective, multicenter study.

The next step is to test this definition in treat-to-target intervention studies, and to apply the definition in other clinical trials as well as in routine clinical practice, Vera Golder, MD, said at the annual meeting of the American College of Rheumatology.
The tested definition has five elements that a patient needs to achieve to be considered in a lupus low disease activity state (LLDAS):
• A systemic lupus erythematosus (SLE) disease activity index (SLEDAI) score of 4 or less with no major organ involvement.
• No new disease activity.
• A physician’s global assessment of the patient of 1 or less on a 0-3 scale.
• Maintenance on a prednisolone dosage of 7.5 mg/day or less.
• Maintenance on a standard immunosuppressive regimen.
The Asia Pacific Lupus Collaboration (APLC) first proposed this definition of LLDAS in 2013 (Ann Rheum Dis. 2013 June;72[Suppl 3]:THU0298), and was the organization behind the latest test of its validity. The APLC based its LLDAS definition on recommendations made by an international working party (Ann Rheum Dis. 2014 June;73[6]:958-67).
The APLC prospectively collected data from 1,735 SLE patients at 13 centers in eight countries during May 2013–December 2016, with a median follow-up of 2.2 years. During that time, 78% of the patients achieved the LLDAS at least once. Two-thirds of the patients had at least one sustained period of LLDAS of at least 3 months, and overall the enrolled patients spent 69% of the time in the LLDAS, reported Dr. Golder, a rheumatologist at Monash University in Melbourne.
The validation analysis she described focused on examining the correlation between the amount of time that patients spent in the defined LLDAS and their subsequent clinical outcomes. The analysis showed that when patients were in the LLDAS their rate of subsequent flare or damage accrual was substantially reduced.
Patients in the LLDAS for at least half the time had a 51% reduced rate of subsequent flare and a 47% reduced rate of subsequent damage accrual, she reported. Patients with a LLDAS of 3-6 months had a 57% reduced rate of damage accrual. As time spent in continuous LLDAS continued to increase the rate of subsequent damage accrual continued to drop until the duration reached more than 9 months, at which point the rate of subsequent damage fell to nearly 90% lower than that of patients without this amount of sustained LLDAS. Patients with LLDAS sustained for more than 9 months and as long as 12 months had an 86% reduction in subsequent damage accrual. Periods of sustained LLDAS that extended longer than a year continued to maintain a nearly 90% reduced rate of damage accrual, Dr. Golder said.
The Asia Pacific Lupus Collaboration has received grants from AstraZeneca, Bristol-Myers Squibb, GlaxoSmithKline, Janssen, and UCB. Dr. Golder had no disclosures.
SOURCE: Golder V et al. Arthritis Rheumatol. 2018;70(Suppl 10): Abstract 2786.
Having a validated, formal definition of low disease activity in patients with systemic lupus erythematosus will be very helpful to clinicians and patients with this disease. The lack of such a widely accepted definition of a treatment target until now has been a significant issue that had made it more challenging to advise and treat patients.
The concept of a low disease state has been much easier to define in other rheumatic diseases, but lupus has posed a major challenge because of its very heterogeneous presentation. This heterogeneity has led to the creation of several measures of disease activity as well as multiple serologic parameters that also help define disease activity. It’s unrealistic to expect most lupus patients to be in a low disease activity state all the time.

Validation of a reasonable definition of low activity is a great, pragmatic step forward for our field that will help clinicians better care for their patients with systemic lupus erythematosus.
Lisa R. Sammaritano, MD , is a rheumatologist at the Hospital for Special Surgery and Cornell University in New York. She had no disclosures. She made these comments in an interview.
Having a validated, formal definition of low disease activity in patients with systemic lupus erythematosus will be very helpful to clinicians and patients with this disease. The lack of such a widely accepted definition of a treatment target until now has been a significant issue that had made it more challenging to advise and treat patients.
The concept of a low disease state has been much easier to define in other rheumatic diseases, but lupus has posed a major challenge because of its very heterogeneous presentation. This heterogeneity has led to the creation of several measures of disease activity as well as multiple serologic parameters that also help define disease activity. It’s unrealistic to expect most lupus patients to be in a low disease activity state all the time.
Validation of a reasonable definition of low activity is a great, pragmatic step forward for our field that will help clinicians better care for their patients with systemic lupus erythematosus.
Lisa R. Sammaritano, MD , is a rheumatologist at the Hospital for Special Surgery and Cornell University in New York. She had no disclosures. She made these comments in an interview.
Having a validated, formal definition of low disease activity in patients with systemic lupus erythematosus will be very helpful to clinicians and patients with this disease. The lack of such a widely accepted definition of a treatment target until now has been a significant issue that had made it more challenging to advise and treat patients.
The concept of a low disease state has been much easier to define in other rheumatic diseases, but lupus has posed a major challenge because of its very heterogeneous presentation. This heterogeneity has led to the creation of several measures of disease activity as well as multiple serologic parameters that also help define disease activity. It’s unrealistic to expect most lupus patients to be in a low disease activity state all the time.
Validation of a reasonable definition of low activity is a great, pragmatic step forward for our field that will help clinicians better care for their patients with systemic lupus erythematosus.
Lisa R. Sammaritano, MD , is a rheumatologist at the Hospital for Special Surgery and Cornell University in New York. She had no disclosures. She made these comments in an interview.
CHICAGO – The long path toward a validated definition of low disease activity in patients with systemic lupus erythematosus may be nearing an end as a definition first proposed more than 5 years ago received validation with data from more than 1,700 patients in a prospective, multicenter study.
The next step is to test this definition in treat-to-target intervention studies, and to apply the definition in other clinical trials as well as in routine clinical practice, Vera Golder, MD, said at the annual meeting of the American College of Rheumatology.
The tested definition has five elements that a patient needs to achieve to be considered in a lupus low disease activity state (LLDAS):
• A systemic lupus erythematosus (SLE) disease activity index (SLEDAI) score of 4 or less with no major organ involvement.
• No new disease activity.
• A physician’s global assessment of the patient of 1 or less on a 0-3 scale.
• Maintenance on a prednisolone dosage of 7.5 mg/day or less.
• Maintenance on a standard immunosuppressive regimen.
The Asia Pacific Lupus Collaboration (APLC) first proposed this definition of LLDAS in 2013 (Ann Rheum Dis. 2013 June;72[Suppl 3]:THU0298), and was the organization behind the latest test of its validity. The APLC based its LLDAS definition on recommendations made by an international working party (Ann Rheum Dis. 2014 June;73[6]:958-67).
The APLC prospectively collected data from 1,735 SLE patients at 13 centers in eight countries during May 2013–December 2016, with a median follow-up of 2.2 years. During that time, 78% of the patients achieved the LLDAS at least once. Two-thirds of the patients had at least one sustained period of LLDAS of at least 3 months, and overall the enrolled patients spent 69% of the time in the LLDAS, reported Dr. Golder, a rheumatologist at Monash University in Melbourne.
The validation analysis she described focused on examining the correlation between the amount of time that patients spent in the defined LLDAS and their subsequent clinical outcomes. The analysis showed that when patients were in the LLDAS their rate of subsequent flare or damage accrual was substantially reduced.
Patients in the LLDAS for at least half the time had a 51% reduced rate of subsequent flare and a 47% reduced rate of subsequent damage accrual, she reported. Patients with a LLDAS of 3-6 months had a 57% reduced rate of damage accrual. As time spent in continuous LLDAS continued to increase the rate of subsequent damage accrual continued to drop until the duration reached more than 9 months, at which point the rate of subsequent damage fell to nearly 90% lower than that of patients without this amount of sustained LLDAS. Patients with LLDAS sustained for more than 9 months and as long as 12 months had an 86% reduction in subsequent damage accrual. Periods of sustained LLDAS that extended longer than a year continued to maintain a nearly 90% reduced rate of damage accrual, Dr. Golder said.
The Asia Pacific Lupus Collaboration has received grants from AstraZeneca, Bristol-Myers Squibb, GlaxoSmithKline, Janssen, and UCB. Dr. Golder had no disclosures.
SOURCE: Golder V et al. Arthritis Rheumatol. 2018;70(Suppl 10): Abstract 2786.
CHICAGO – The long path toward a validated definition of low disease activity in patients with systemic lupus erythematosus may be nearing an end as a definition first proposed more than 5 years ago received validation with data from more than 1,700 patients in a prospective, multicenter study.
The next step is to test this definition in treat-to-target intervention studies, and to apply the definition in other clinical trials as well as in routine clinical practice, Vera Golder, MD, said at the annual meeting of the American College of Rheumatology.
The tested definition has five elements that a patient needs to achieve to be considered in a lupus low disease activity state (LLDAS):
• A systemic lupus erythematosus (SLE) disease activity index (SLEDAI) score of 4 or less with no major organ involvement.
• No new disease activity.
• A physician’s global assessment of the patient of 1 or less on a 0-3 scale.
• Maintenance on a prednisolone dosage of 7.5 mg/day or less.
• Maintenance on a standard immunosuppressive regimen.
The Asia Pacific Lupus Collaboration (APLC) first proposed this definition of LLDAS in 2013 (Ann Rheum Dis. 2013 June;72[Suppl 3]:THU0298), and was the organization behind the latest test of its validity. The APLC based its LLDAS definition on recommendations made by an international working party (Ann Rheum Dis. 2014 June;73[6]:958-67).
The APLC prospectively collected data from 1,735 SLE patients at 13 centers in eight countries during May 2013–December 2016, with a median follow-up of 2.2 years. During that time, 78% of the patients achieved the LLDAS at least once. Two-thirds of the patients had at least one sustained period of LLDAS of at least 3 months, and overall the enrolled patients spent 69% of the time in the LLDAS, reported Dr. Golder, a rheumatologist at Monash University in Melbourne.
The validation analysis she described focused on examining the correlation between the amount of time that patients spent in the defined LLDAS and their subsequent clinical outcomes. The analysis showed that when patients were in the LLDAS their rate of subsequent flare or damage accrual was substantially reduced.
Patients in the LLDAS for at least half the time had a 51% reduced rate of subsequent flare and a 47% reduced rate of subsequent damage accrual, she reported. Patients with a LLDAS of 3-6 months had a 57% reduced rate of damage accrual. As time spent in continuous LLDAS continued to increase the rate of subsequent damage accrual continued to drop until the duration reached more than 9 months, at which point the rate of subsequent damage fell to nearly 90% lower than that of patients without this amount of sustained LLDAS. Patients with LLDAS sustained for more than 9 months and as long as 12 months had an 86% reduction in subsequent damage accrual. Periods of sustained LLDAS that extended longer than a year continued to maintain a nearly 90% reduced rate of damage accrual, Dr. Golder said.
The Asia Pacific Lupus Collaboration has received grants from AstraZeneca, Bristol-Myers Squibb, GlaxoSmithKline, Janssen, and UCB. Dr. Golder had no disclosures.
SOURCE: Golder V et al. Arthritis Rheumatol. 2018;70(Suppl 10): Abstract 2786.
REPORTING FROM THE ACR ANNUAL MEETING
Key clinical point: A proposed definition of low disease activity in systemic lupus erythematosus received prospective validation.
Major finding: Patients meeting the definition for 9-12 months had an 86% reduced rate of subsequent damage accrual.
Study details: A prospective, multicenter study of 1,735 patients.
Disclosures: The Asia Pacific Lupus Collaboration has received grants from AstraZeneca, Bristol-Myers Squibb, GlaxoSmithKline, Janssen, and UCB. Dr. Golder had no disclosures.
Source: Golder V et al. Arthritis Rheumatol. 2018;70(Suppl 10): Abstract 2786.
Ablation plus transplant for severe scleroderma shows 11-year benefits
CHICAGO – Follow-up out to as long as 11 years from treatment confirmed the long-term efficacy and safety of myeloablative autologous stem cell transplantation for patients with severe scleroderma.
This extended follow-up comprised 43 survivors from the 75 patients originally randomized in a controlled, 6-year trial. Follow-up showed that, among the patients who underwent myeloablation and autologous transplant with hematopoietic stem cells, there were no long-term deaths or cancers, there was an 88% survival rate, and 92% remained off disease-modifying treatment, Keith M. Sullivan, MD, said at the annual meeting of the American College of Rheumatology.
Long-term survival among patients randomized to the study’s control arm, who received treatment with cyclophosphamide, was 53%.
Patients with severe scleroderma with significant internal organ damage who “are improved and off of disease-modifying antirheumatic drugs after 10 or more years from treatment is something new in autoimmune disease,” said Dr. Sullivan, a professor of medicine at Duke University, Durham, N.C.
Based on accumulated data from this randomized trial and other studies, the American Society for Blood and Marrow Transplantation issued a position statement in June 2018 that endorsed autologous hematopoietic stem cell transplantation as “standard of care” for systemic sclerosis (Biol Blood Marrow Transplant. 2018 June 25. doi: 10.1016/j.bbmt.2018.06.025), Dr. Sullivan noted in a video interview.
The SCOT (Scleroderma: Cyclophosphamide or Transplantation) trial randomized 75 patients with severe scleroderma and substantial internal organ involvement to receive treatment with either cyclophosphamide or myeloablative radiation followed by immune reconstitution with an autologous hematopoietic stem cell transplant. The trial’s primary endpoint, the global rank composite score at 54 months, showed the superiority of transplantation over standard treatment (N Engl J Med. 2018 Jan 4;378[1]:35-47).
Dr. Sullivan and his associates ran their long-term follow-up study on 43 of these 75 patients (25 from the transplanted group and 18 controls), excluding 21 patients who died during the original study, 4 additional patients from the control arm who died following the end of the original SCOT protocol, and 7 patients either lost to follow-up or who refused to participate in follow-up. Among the 25 transplanted patients, none died during the extended follow-up, 2 experienced cardiac failure, and 23 remained off of any disease-modifying antirheumatic drugs. Among the 18 survivors in the control arm, 3 had cardiac failure, 3 had respiratory failure, and 7 were on treatment with disease-modifying drugs, Dr. Sullivan reported.
In addition, 23 of the 25 (92%) transplanted patients had normal performance status by the Eastern Cooperative Oncology Group criteria, compared with 11 of the 18 controls (61%). A total of 14 (56%) transplant patients were employed, compared with 6 of the 18 controls (33%).
Patients who were transplanted “have their life back, are doing well, and are off treatment,” Dr. Sullivan noted.
Myeloablation and transplant is appropriate for scleroderma patients with significant internal organ involvement, about half of all patients with this disease. The best gauge of severe organ involvement is a pulmonary function test, with a forced vital capacity of 70% or less of predicted as a flag for patients who should consider transplantation, Dr. Sullivan said. He recommended monitoring lung function every 3 months in scleroderma patients because it can deteriorate very suddenly and quickly.
SCOT received no commercial funding. Dr. Sullivan had no disclosures to report.
SOURCE: Sullivan KM et al. ACR Annual Meeting, Abstract 1820.
CHICAGO – Follow-up out to as long as 11 years from treatment confirmed the long-term efficacy and safety of myeloablative autologous stem cell transplantation for patients with severe scleroderma.
This extended follow-up comprised 43 survivors from the 75 patients originally randomized in a controlled, 6-year trial. Follow-up showed that, among the patients who underwent myeloablation and autologous transplant with hematopoietic stem cells, there were no long-term deaths or cancers, there was an 88% survival rate, and 92% remained off disease-modifying treatment, Keith M. Sullivan, MD, said at the annual meeting of the American College of Rheumatology.
Long-term survival among patients randomized to the study’s control arm, who received treatment with cyclophosphamide, was 53%.
Patients with severe scleroderma with significant internal organ damage who “are improved and off of disease-modifying antirheumatic drugs after 10 or more years from treatment is something new in autoimmune disease,” said Dr. Sullivan, a professor of medicine at Duke University, Durham, N.C.
Based on accumulated data from this randomized trial and other studies, the American Society for Blood and Marrow Transplantation issued a position statement in June 2018 that endorsed autologous hematopoietic stem cell transplantation as “standard of care” for systemic sclerosis (Biol Blood Marrow Transplant. 2018 June 25. doi: 10.1016/j.bbmt.2018.06.025), Dr. Sullivan noted in a video interview.
The SCOT (Scleroderma: Cyclophosphamide or Transplantation) trial randomized 75 patients with severe scleroderma and substantial internal organ involvement to receive treatment with either cyclophosphamide or myeloablative radiation followed by immune reconstitution with an autologous hematopoietic stem cell transplant. The trial’s primary endpoint, the global rank composite score at 54 months, showed the superiority of transplantation over standard treatment (N Engl J Med. 2018 Jan 4;378[1]:35-47).
Dr. Sullivan and his associates ran their long-term follow-up study on 43 of these 75 patients (25 from the transplanted group and 18 controls), excluding 21 patients who died during the original study, 4 additional patients from the control arm who died following the end of the original SCOT protocol, and 7 patients either lost to follow-up or who refused to participate in follow-up. Among the 25 transplanted patients, none died during the extended follow-up, 2 experienced cardiac failure, and 23 remained off of any disease-modifying antirheumatic drugs. Among the 18 survivors in the control arm, 3 had cardiac failure, 3 had respiratory failure, and 7 were on treatment with disease-modifying drugs, Dr. Sullivan reported.
In addition, 23 of the 25 (92%) transplanted patients had normal performance status by the Eastern Cooperative Oncology Group criteria, compared with 11 of the 18 controls (61%). A total of 14 (56%) transplant patients were employed, compared with 6 of the 18 controls (33%).
Patients who were transplanted “have their life back, are doing well, and are off treatment,” Dr. Sullivan noted.
Myeloablation and transplant is appropriate for scleroderma patients with significant internal organ involvement, about half of all patients with this disease. The best gauge of severe organ involvement is a pulmonary function test, with a forced vital capacity of 70% or less of predicted as a flag for patients who should consider transplantation, Dr. Sullivan said. He recommended monitoring lung function every 3 months in scleroderma patients because it can deteriorate very suddenly and quickly.
SCOT received no commercial funding. Dr. Sullivan had no disclosures to report.
SOURCE: Sullivan KM et al. ACR Annual Meeting, Abstract 1820.
CHICAGO – Follow-up out to as long as 11 years from treatment confirmed the long-term efficacy and safety of myeloablative autologous stem cell transplantation for patients with severe scleroderma.
This extended follow-up comprised 43 survivors from the 75 patients originally randomized in a controlled, 6-year trial. Follow-up showed that, among the patients who underwent myeloablation and autologous transplant with hematopoietic stem cells, there were no long-term deaths or cancers, there was an 88% survival rate, and 92% remained off disease-modifying treatment, Keith M. Sullivan, MD, said at the annual meeting of the American College of Rheumatology.
Long-term survival among patients randomized to the study’s control arm, who received treatment with cyclophosphamide, was 53%.
Patients with severe scleroderma with significant internal organ damage who “are improved and off of disease-modifying antirheumatic drugs after 10 or more years from treatment is something new in autoimmune disease,” said Dr. Sullivan, a professor of medicine at Duke University, Durham, N.C.
Based on accumulated data from this randomized trial and other studies, the American Society for Blood and Marrow Transplantation issued a position statement in June 2018 that endorsed autologous hematopoietic stem cell transplantation as “standard of care” for systemic sclerosis (Biol Blood Marrow Transplant. 2018 June 25. doi: 10.1016/j.bbmt.2018.06.025), Dr. Sullivan noted in a video interview.
The SCOT (Scleroderma: Cyclophosphamide or Transplantation) trial randomized 75 patients with severe scleroderma and substantial internal organ involvement to receive treatment with either cyclophosphamide or myeloablative radiation followed by immune reconstitution with an autologous hematopoietic stem cell transplant. The trial’s primary endpoint, the global rank composite score at 54 months, showed the superiority of transplantation over standard treatment (N Engl J Med. 2018 Jan 4;378[1]:35-47).
Dr. Sullivan and his associates ran their long-term follow-up study on 43 of these 75 patients (25 from the transplanted group and 18 controls), excluding 21 patients who died during the original study, 4 additional patients from the control arm who died following the end of the original SCOT protocol, and 7 patients either lost to follow-up or who refused to participate in follow-up. Among the 25 transplanted patients, none died during the extended follow-up, 2 experienced cardiac failure, and 23 remained off of any disease-modifying antirheumatic drugs. Among the 18 survivors in the control arm, 3 had cardiac failure, 3 had respiratory failure, and 7 were on treatment with disease-modifying drugs, Dr. Sullivan reported.
In addition, 23 of the 25 (92%) transplanted patients had normal performance status by the Eastern Cooperative Oncology Group criteria, compared with 11 of the 18 controls (61%). A total of 14 (56%) transplant patients were employed, compared with 6 of the 18 controls (33%).
Patients who were transplanted “have their life back, are doing well, and are off treatment,” Dr. Sullivan noted.
Myeloablation and transplant is appropriate for scleroderma patients with significant internal organ involvement, about half of all patients with this disease. The best gauge of severe organ involvement is a pulmonary function test, with a forced vital capacity of 70% or less of predicted as a flag for patients who should consider transplantation, Dr. Sullivan said. He recommended monitoring lung function every 3 months in scleroderma patients because it can deteriorate very suddenly and quickly.
SCOT received no commercial funding. Dr. Sullivan had no disclosures to report.
SOURCE: Sullivan KM et al. ACR Annual Meeting, Abstract 1820.
REPORTING FROM THE ACR ANNUAL MEETING
Key clinical point:
Major finding: Survival after 11 years was 88% among transplanted patients and 53% among control patients treated with cyclophosphamide.
Study details: A long-term follow-up of 43 of the 75 patients enrolled in the SCOT trial.
Disclosures: SCOT received no commercial funding. Dr. Sullivan had no disclosures to report.
Source: Sullivan KM et al. ACR Annual Meeting, Abstract 1820.

IgA vasculitis may be more common in adults than assumed
LAS VEGAS – IgA vasculitis has a reputation as an illness of childhood, but rheumatologist Alexandra Villa-Forte, MD, MPH, cautioned colleagues that it can strike adults, too, often in a much more severe form. And, she warned, it’s likely not as rare as physicians assume.
“I believe it’s more common in adults than reported. There’s a huge problem with establishing the right way to make the diagnosis in adults, which is why it is missed,” said Dr. Villa-Forte of the Cleveland Clinic, who spoke at the annual Perspectives in Rheumatic Diseases held by Global Academy for Medical Education.
But even if IgA vasculitis (IV) is diagnosed correctly in adults, there are many questions about how to move forward, she said. “Treatment in adults remains a problem since we don’t have data.”
IV is also known as Henoch-Schönlein purpura, or HSP, and “spring fever” because it often appears in children in the spring following an upper respiratory infection.
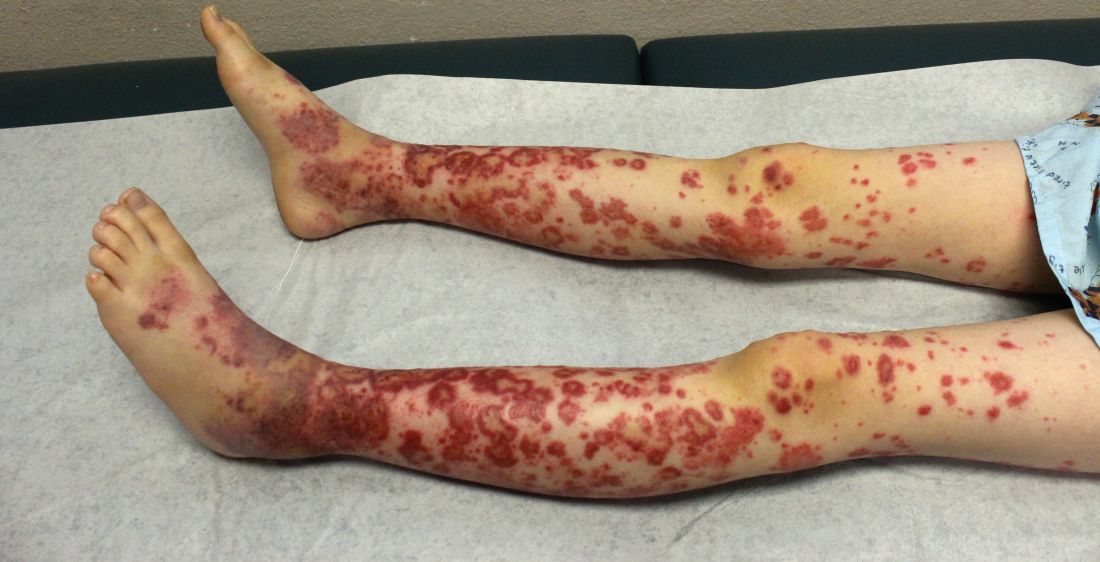
The condition causes vasculitis, the swelling of small blood vessels in organs such as the skin, joints, kidneys, and intestines. Leaking blood vessels can cause skin rashes known as purpura.
The estimated ranges of disease are high, with the annual incidence in children estimated at 3-26 per 100,000 and in adults at 0.1-1.8 per 100,000. Dr. Villa-Forte noted that the male-to-female ratio is 1.5, and she said the condition is less common in African Americans.
“In children, this is a disease that is frequently self-limited. Most of them don’t need treatment, and most of them do not relapse,” Dr. Villa-Forte said. “In adults, it’s more resistant to treatment, frequently chronic, and frequently relapsing over the years.”
There are other differences in IV between children and adults. “In children, there is a clear seasonal pattern of disease that is not seen in adults,” she said, and it’s linked to preceding infections.
“In adults, there are multiple causes, and most of the time they’re not identified,” she said.
As for diagnosis, she suggests looking at clinical presentation and whether tissue biopsy shows cutaneous leukocytoclastic vasculitis with IgA deposits. She cautioned that increased serum IgA is seen in about 50% of adult patients, making it an unreliable indicator.
Prognosis is much better for children than adults. According to a 2014 study, 80% of children completely recover, compared with 40% of adults. Persistent hematuria or proteinuria occurs in 30% of children and 60% of adults, respectively, while chronic renal failure occurs in 2% of children and 10% of adults (J Korean Med Sci. 2014 Feb;29[2]:198-203).
It’s possible that the latter number may be higher, with as many as 30% of adults developing chronic kidney disease (CKD), Dr. Villa-Forte said.
An estimated 97% of nephritis develops within the first 6 months of disease onset in adults, she said, “and in adults, the active renal disease can persist for over 20 years.”
Guidelines suggest that patients be monitored for CKD for 6 months after disease onset, but Dr. Villa-Forte said it’s better to monitor them for 12 months.
What about treatment? “The major challenge in adults is the real absence between correlation between initial presentation and long-term renal outcome,” she said. “That makes for a difficult choice in terms of treatment selection.”
Fever and cutaneous lesions above the waist may predict renal involvement, she said, although that isn’t confirmed, and increasing proteinuria is a probable factor predicting progression/complications.
According to Dr. Villa-Forte, the value of early treatment hasn’t been proved. Due to the risk of kidney problems, she said, “we don’t feel like we can just watch patients – that we need to do something. But there’s not good data supporting that.”
Various protocols involving steroids, early plasmapheresis, rituximab (Rituxan), and other drug regimens lack evidence, she said, although use of steroids in early nephritis may be beneficial in adults.
Dr. Villa-Forte had no relevant disclosures.
Global Academy for Medical Education and this news organization are owned by the same parent company.
LAS VEGAS – IgA vasculitis has a reputation as an illness of childhood, but rheumatologist Alexandra Villa-Forte, MD, MPH, cautioned colleagues that it can strike adults, too, often in a much more severe form. And, she warned, it’s likely not as rare as physicians assume.
“I believe it’s more common in adults than reported. There’s a huge problem with establishing the right way to make the diagnosis in adults, which is why it is missed,” said Dr. Villa-Forte of the Cleveland Clinic, who spoke at the annual Perspectives in Rheumatic Diseases held by Global Academy for Medical Education.
But even if IgA vasculitis (IV) is diagnosed correctly in adults, there are many questions about how to move forward, she said. “Treatment in adults remains a problem since we don’t have data.”
IV is also known as Henoch-Schönlein purpura, or HSP, and “spring fever” because it often appears in children in the spring following an upper respiratory infection.
The condition causes vasculitis, the swelling of small blood vessels in organs such as the skin, joints, kidneys, and intestines. Leaking blood vessels can cause skin rashes known as purpura.
The estimated ranges of disease are high, with the annual incidence in children estimated at 3-26 per 100,000 and in adults at 0.1-1.8 per 100,000. Dr. Villa-Forte noted that the male-to-female ratio is 1.5, and she said the condition is less common in African Americans.
“In children, this is a disease that is frequently self-limited. Most of them don’t need treatment, and most of them do not relapse,” Dr. Villa-Forte said. “In adults, it’s more resistant to treatment, frequently chronic, and frequently relapsing over the years.”
There are other differences in IV between children and adults. “In children, there is a clear seasonal pattern of disease that is not seen in adults,” she said, and it’s linked to preceding infections.
“In adults, there are multiple causes, and most of the time they’re not identified,” she said.
As for diagnosis, she suggests looking at clinical presentation and whether tissue biopsy shows cutaneous leukocytoclastic vasculitis with IgA deposits. She cautioned that increased serum IgA is seen in about 50% of adult patients, making it an unreliable indicator.
Prognosis is much better for children than adults. According to a 2014 study, 80% of children completely recover, compared with 40% of adults. Persistent hematuria or proteinuria occurs in 30% of children and 60% of adults, respectively, while chronic renal failure occurs in 2% of children and 10% of adults (J Korean Med Sci. 2014 Feb;29[2]:198-203).
It’s possible that the latter number may be higher, with as many as 30% of adults developing chronic kidney disease (CKD), Dr. Villa-Forte said.
An estimated 97% of nephritis develops within the first 6 months of disease onset in adults, she said, “and in adults, the active renal disease can persist for over 20 years.”
Guidelines suggest that patients be monitored for CKD for 6 months after disease onset, but Dr. Villa-Forte said it’s better to monitor them for 12 months.
What about treatment? “The major challenge in adults is the real absence between correlation between initial presentation and long-term renal outcome,” she said. “That makes for a difficult choice in terms of treatment selection.”
Fever and cutaneous lesions above the waist may predict renal involvement, she said, although that isn’t confirmed, and increasing proteinuria is a probable factor predicting progression/complications.
According to Dr. Villa-Forte, the value of early treatment hasn’t been proved. Due to the risk of kidney problems, she said, “we don’t feel like we can just watch patients – that we need to do something. But there’s not good data supporting that.”
Various protocols involving steroids, early plasmapheresis, rituximab (Rituxan), and other drug regimens lack evidence, she said, although use of steroids in early nephritis may be beneficial in adults.
Dr. Villa-Forte had no relevant disclosures.
Global Academy for Medical Education and this news organization are owned by the same parent company.
LAS VEGAS – IgA vasculitis has a reputation as an illness of childhood, but rheumatologist Alexandra Villa-Forte, MD, MPH, cautioned colleagues that it can strike adults, too, often in a much more severe form. And, she warned, it’s likely not as rare as physicians assume.
“I believe it’s more common in adults than reported. There’s a huge problem with establishing the right way to make the diagnosis in adults, which is why it is missed,” said Dr. Villa-Forte of the Cleveland Clinic, who spoke at the annual Perspectives in Rheumatic Diseases held by Global Academy for Medical Education.
But even if IgA vasculitis (IV) is diagnosed correctly in adults, there are many questions about how to move forward, she said. “Treatment in adults remains a problem since we don’t have data.”
IV is also known as Henoch-Schönlein purpura, or HSP, and “spring fever” because it often appears in children in the spring following an upper respiratory infection.
The condition causes vasculitis, the swelling of small blood vessels in organs such as the skin, joints, kidneys, and intestines. Leaking blood vessels can cause skin rashes known as purpura.
The estimated ranges of disease are high, with the annual incidence in children estimated at 3-26 per 100,000 and in adults at 0.1-1.8 per 100,000. Dr. Villa-Forte noted that the male-to-female ratio is 1.5, and she said the condition is less common in African Americans.
“In children, this is a disease that is frequently self-limited. Most of them don’t need treatment, and most of them do not relapse,” Dr. Villa-Forte said. “In adults, it’s more resistant to treatment, frequently chronic, and frequently relapsing over the years.”
There are other differences in IV between children and adults. “In children, there is a clear seasonal pattern of disease that is not seen in adults,” she said, and it’s linked to preceding infections.
“In adults, there are multiple causes, and most of the time they’re not identified,” she said.
As for diagnosis, she suggests looking at clinical presentation and whether tissue biopsy shows cutaneous leukocytoclastic vasculitis with IgA deposits. She cautioned that increased serum IgA is seen in about 50% of adult patients, making it an unreliable indicator.
Prognosis is much better for children than adults. According to a 2014 study, 80% of children completely recover, compared with 40% of adults. Persistent hematuria or proteinuria occurs in 30% of children and 60% of adults, respectively, while chronic renal failure occurs in 2% of children and 10% of adults (J Korean Med Sci. 2014 Feb;29[2]:198-203).
It’s possible that the latter number may be higher, with as many as 30% of adults developing chronic kidney disease (CKD), Dr. Villa-Forte said.
An estimated 97% of nephritis develops within the first 6 months of disease onset in adults, she said, “and in adults, the active renal disease can persist for over 20 years.”
Guidelines suggest that patients be monitored for CKD for 6 months after disease onset, but Dr. Villa-Forte said it’s better to monitor them for 12 months.
What about treatment? “The major challenge in adults is the real absence between correlation between initial presentation and long-term renal outcome,” she said. “That makes for a difficult choice in terms of treatment selection.”
Fever and cutaneous lesions above the waist may predict renal involvement, she said, although that isn’t confirmed, and increasing proteinuria is a probable factor predicting progression/complications.
According to Dr. Villa-Forte, the value of early treatment hasn’t been proved. Due to the risk of kidney problems, she said, “we don’t feel like we can just watch patients – that we need to do something. But there’s not good data supporting that.”
Various protocols involving steroids, early plasmapheresis, rituximab (Rituxan), and other drug regimens lack evidence, she said, although use of steroids in early nephritis may be beneficial in adults.
Dr. Villa-Forte had no relevant disclosures.
Global Academy for Medical Education and this news organization are owned by the same parent company.
REPORTING FROM THE ANNUAL PERSPECTIVES IN RHEUMATIC DISEASES
Baseline patient-reported physical function predicts SLE-related mortality
The baseline physical component score of the Medical Outcomes Short Form 36 (SF-36) was an independent predictor of mortality for patients with systemic lupus erythematosus (SLE), according to recent analysis of data from the University of California, San Francisco, (UCSF) Lupus Outcomes Study.

Desiree R. Azizoddin, PsyD, of Stanford Health Care in Palo Alto, Calif., and her colleagues analyzed patient-reported outcomes (PROs) of 728 patients with SLE from the UCSF study, which included measures such as self-rated health, the Center for Epidemiologic Studies Depression scale (CESD), and SF-36 mental and physical component scores between 2007 and 2015. Patients had a mean age of 50.6 years and consisted of mostly whites (68.5%) and women (92.2%), with a mean SLE disease duration of 16.7 years.
“As PROs can be measured easily, quickly, reliably in a systematic manner, without use of many resources (including cost) and experts, PROs can serve as an important tool in community settings that provide care for individuals with SLE,” Dr. Azizoddin and her colleagues wrote in Arthritis Care & Research. “Implementation and integration of PROs may potentially help decrease mortality among patients with SLE in the long run.”
The researchers reported 71 deaths (9.8%) in the study. A univariate analysis showed that self-rated health as fair or poor and all SF-36 subscale scores, except for mental health and role emotional subscales, were predictors of mortality. After the investigators performed a multivariate analysis, they found that baseline SF-36 physical component scores alone were associated with an increased mortality risk (hazard ratio, 0.97; 95% confidence interval, 0.94-0.99; P less than .01), with a 3.5% lower risk for each point-rating increase in the score.
“Insight to predictors of mortality, above and beyond those that require physician resources such as assessments of renal involvement and traditional physician-completed measures of disease activity and damage in this complex disease, affords rheumatologists and SLE providers with additional tools to assess potential patient health trajectories,” Dr. Azizoddin and her colleagues wrote in their study. “Identification of such predictors will expand opportunities for early interventions aimed at the reduction of such risk, including allocation of appropriate medical resources in areas with large numbers of patients with compromised health.”
The researchers noted that bias in self-reporting, a lack of prospective evaluations at each visit, and clinical applicability to only similar cohorts were limitations in the study.
This study was funded by the Robert Wood Johnson Foundation Investigator Awards in Health Policy Research and individually was awarded grants from the National Institute of Arthritis and Musculoskeletal and Skin Diseases.
SOURCE: Azizoddin DR et al. Arthritis Care Res. 2018 Aug 24. doi: 10.1002/acr.23734.
The baseline physical component score of the Medical Outcomes Short Form 36 (SF-36) was an independent predictor of mortality for patients with systemic lupus erythematosus (SLE), according to recent analysis of data from the University of California, San Francisco, (UCSF) Lupus Outcomes Study.
Desiree R. Azizoddin, PsyD, of Stanford Health Care in Palo Alto, Calif., and her colleagues analyzed patient-reported outcomes (PROs) of 728 patients with SLE from the UCSF study, which included measures such as self-rated health, the Center for Epidemiologic Studies Depression scale (CESD), and SF-36 mental and physical component scores between 2007 and 2015. Patients had a mean age of 50.6 years and consisted of mostly whites (68.5%) and women (92.2%), with a mean SLE disease duration of 16.7 years.
“As PROs can be measured easily, quickly, reliably in a systematic manner, without use of many resources (including cost) and experts, PROs can serve as an important tool in community settings that provide care for individuals with SLE,” Dr. Azizoddin and her colleagues wrote in Arthritis Care & Research. “Implementation and integration of PROs may potentially help decrease mortality among patients with SLE in the long run.”
The researchers reported 71 deaths (9.8%) in the study. A univariate analysis showed that self-rated health as fair or poor and all SF-36 subscale scores, except for mental health and role emotional subscales, were predictors of mortality. After the investigators performed a multivariate analysis, they found that baseline SF-36 physical component scores alone were associated with an increased mortality risk (hazard ratio, 0.97; 95% confidence interval, 0.94-0.99; P less than .01), with a 3.5% lower risk for each point-rating increase in the score.
“Insight to predictors of mortality, above and beyond those that require physician resources such as assessments of renal involvement and traditional physician-completed measures of disease activity and damage in this complex disease, affords rheumatologists and SLE providers with additional tools to assess potential patient health trajectories,” Dr. Azizoddin and her colleagues wrote in their study. “Identification of such predictors will expand opportunities for early interventions aimed at the reduction of such risk, including allocation of appropriate medical resources in areas with large numbers of patients with compromised health.”
The researchers noted that bias in self-reporting, a lack of prospective evaluations at each visit, and clinical applicability to only similar cohorts were limitations in the study.
This study was funded by the Robert Wood Johnson Foundation Investigator Awards in Health Policy Research and individually was awarded grants from the National Institute of Arthritis and Musculoskeletal and Skin Diseases.
SOURCE: Azizoddin DR et al. Arthritis Care Res. 2018 Aug 24. doi: 10.1002/acr.23734.
The baseline physical component score of the Medical Outcomes Short Form 36 (SF-36) was an independent predictor of mortality for patients with systemic lupus erythematosus (SLE), according to recent analysis of data from the University of California, San Francisco, (UCSF) Lupus Outcomes Study.
Desiree R. Azizoddin, PsyD, of Stanford Health Care in Palo Alto, Calif., and her colleagues analyzed patient-reported outcomes (PROs) of 728 patients with SLE from the UCSF study, which included measures such as self-rated health, the Center for Epidemiologic Studies Depression scale (CESD), and SF-36 mental and physical component scores between 2007 and 2015. Patients had a mean age of 50.6 years and consisted of mostly whites (68.5%) and women (92.2%), with a mean SLE disease duration of 16.7 years.
“As PROs can be measured easily, quickly, reliably in a systematic manner, without use of many resources (including cost) and experts, PROs can serve as an important tool in community settings that provide care for individuals with SLE,” Dr. Azizoddin and her colleagues wrote in Arthritis Care & Research. “Implementation and integration of PROs may potentially help decrease mortality among patients with SLE in the long run.”
The researchers reported 71 deaths (9.8%) in the study. A univariate analysis showed that self-rated health as fair or poor and all SF-36 subscale scores, except for mental health and role emotional subscales, were predictors of mortality. After the investigators performed a multivariate analysis, they found that baseline SF-36 physical component scores alone were associated with an increased mortality risk (hazard ratio, 0.97; 95% confidence interval, 0.94-0.99; P less than .01), with a 3.5% lower risk for each point-rating increase in the score.
“Insight to predictors of mortality, above and beyond those that require physician resources such as assessments of renal involvement and traditional physician-completed measures of disease activity and damage in this complex disease, affords rheumatologists and SLE providers with additional tools to assess potential patient health trajectories,” Dr. Azizoddin and her colleagues wrote in their study. “Identification of such predictors will expand opportunities for early interventions aimed at the reduction of such risk, including allocation of appropriate medical resources in areas with large numbers of patients with compromised health.”
The researchers noted that bias in self-reporting, a lack of prospective evaluations at each visit, and clinical applicability to only similar cohorts were limitations in the study.
This study was funded by the Robert Wood Johnson Foundation Investigator Awards in Health Policy Research and individually was awarded grants from the National Institute of Arthritis and Musculoskeletal and Skin Diseases.
SOURCE: Azizoddin DR et al. Arthritis Care Res. 2018 Aug 24. doi: 10.1002/acr.23734.
FROM ARTHRITIS CARE & RESEARCH
Key clinical point:
Major finding: There was a 3.5% lower risk of mortality per each increased point rating on the SF-36 physical component score.
Study details: An analysis of 728 patients with SLE from the University of California, San Francisco, Lupus Outcomes Study.
Disclosures: This study was funded by the Robert Wood Johnson Foundation Investigator Awards in Health Policy Research and individually was awarded grants from the National Institute of Arthritis and Musculoskeletal and Skin Diseases.
Source: Azizoddin DR et al. Arthritis Care Res. 2018 Aug 24. doi: 10.1002/acr.23734.
Childhood-onset SLE has major impact in adult life
The majority of adults with childhood-onset systemic lupus erythematosus in a longitudinal Dutch study developed significant damage at a young age, remain on corticosteroids in adulthood, and have an impaired health-related quality of life.

The findings in the study, dubbed Childhood-Onset SLE in the Netherlands (CHILL-NL), highlighted the need for preventive screening measures to be put in place before the age of 30 to facilitate a better outcome for patients, who still face high morbidity from childhood-onset systemic lupus erythematosus (cSLE) despite improved survival. Such information is helpful to “answer questions from children and parents regarding the future course of the disease,” wrote Noortje Groot, PhD, of the department of pediatric rheumatology at Erasmus Medical Center–Sophia Children’s Hospital in Rotterdam, the Netherlands, and her colleagues. The report is in Arthritis & Rheumatology.
The current study included all adult SLE patients treated in any Dutch public hospital during the period of November 2013 to April 2016 who were diagnosed with the autoimmune disease prior to their 18th birthday.
All 111 patients involved in the study were seen for a 1.5-hour visit at Erasmus University Medical Center or a local hospital of their choice. During the appointment, a medical history was taken, a physical examination was performed, and the patients completed questionnaires on health-related quality of life (HRQOL).
The average age of patients at the study visit was 33 years, 91% were female, and 72% were white. Median disease duration was 20 years and disease activity was low (median SLE Disease Activity Index 2000 [SLEDAI-2k] = 4). Low complement (32%), skin rashes (14%), and proteinuria (13%) were the most common SLEDAI items reported.
Overall, 68% of the cohort (n = 76) were taking hydroxychloroquine (HCQ), 29% of whom (n = 22) were taking it as monotherapy.
Furthermore, 68% of patients at the study visit were taking corticosteroids and/or non-HCQ disease-modifying antirheumatic drugs (DMARDs), and just over half (51%) of patients (n = 56) were taking corticosteroids either alone or with a non-HCQ DMARD.
“This [finding] is worrying as corticosteroids are associated with the development of damage. Patients are certainly eager to limit corticosteroid use, as almost all patients in the CHILL-NL cohort reported to have negative experiences with prednisone regarding their physical appearance and or mental well-being,” the study authors wrote.
Results also showed that 62% of the patients had damage, predominantly in the musculoskeletal, neuropsychiatric, and renal systems.
Most organ systems became involved within the first 2 years of diagnosis, but after 5 years of disease the nature of disease manifestations tended to shift to damage such as myocardial infarctions.
Notably, after 10-20 years, when cSLE patients were in their early 20s and 30s, significant damage had occurred in more than half of the patients, the authors noted.
“This shift to damage has also been observed in adult-onset SLE patients and urges for preventative screening measures of such (cardiovascular) damage and healthy lifestyle advice (healthy diet, regular exercise, abstinence from smoking),” they wrote.
Multivariate logistic regression showed that damage accrual was associated with disease duration (odds ratio, 1.15; P less than .001), antiphospholipid‐antibody positivity (OR, 3.56; P = .026), and hypertension (OR, 3.21; P = .043). On the other hand, current HCQ monotherapy was associated with an SLICC-Damage Index score of 0 (OR, 0.16; P = .009).
The HRQOL of the cohort, assessed via the Short Form–36, was also impaired compared with the general population, the researchers discovered. For example, the presence of damage reduced HRQOL in one domain, and high disease activity, defined as SLEDAI-2k of 8 or more, strongly reduced HRQOL in four of eight domains. Changes in physical appearance lowered HRQOL in seven of eight domains.
“HRQOL of adults with cSLE is impaired and affected by other factors than disease activity or damage alone. By identifying and addressing these factors, like physical appearance and potentially coping styles, HRQOL may be improved,” they advised.
The study was supported financially by the Dutch Arthritis Foundation and the Dutch national patient association for lupus, antiphospholipid syndrome, scleroderma, and mixed connective tissue diseases.
SOURCE: Groot N et al. Arthritis Rheumatol. 2018 Aug 27. doi: 10.1002/art.40697
The majority of adults with childhood-onset systemic lupus erythematosus in a longitudinal Dutch study developed significant damage at a young age, remain on corticosteroids in adulthood, and have an impaired health-related quality of life.
The findings in the study, dubbed Childhood-Onset SLE in the Netherlands (CHILL-NL), highlighted the need for preventive screening measures to be put in place before the age of 30 to facilitate a better outcome for patients, who still face high morbidity from childhood-onset systemic lupus erythematosus (cSLE) despite improved survival. Such information is helpful to “answer questions from children and parents regarding the future course of the disease,” wrote Noortje Groot, PhD, of the department of pediatric rheumatology at Erasmus Medical Center–Sophia Children’s Hospital in Rotterdam, the Netherlands, and her colleagues. The report is in Arthritis & Rheumatology.
The current study included all adult SLE patients treated in any Dutch public hospital during the period of November 2013 to April 2016 who were diagnosed with the autoimmune disease prior to their 18th birthday.
All 111 patients involved in the study were seen for a 1.5-hour visit at Erasmus University Medical Center or a local hospital of their choice. During the appointment, a medical history was taken, a physical examination was performed, and the patients completed questionnaires on health-related quality of life (HRQOL).
The average age of patients at the study visit was 33 years, 91% were female, and 72% were white. Median disease duration was 20 years and disease activity was low (median SLE Disease Activity Index 2000 [SLEDAI-2k] = 4). Low complement (32%), skin rashes (14%), and proteinuria (13%) were the most common SLEDAI items reported.
Overall, 68% of the cohort (n = 76) were taking hydroxychloroquine (HCQ), 29% of whom (n = 22) were taking it as monotherapy.
Furthermore, 68% of patients at the study visit were taking corticosteroids and/or non-HCQ disease-modifying antirheumatic drugs (DMARDs), and just over half (51%) of patients (n = 56) were taking corticosteroids either alone or with a non-HCQ DMARD.
“This [finding] is worrying as corticosteroids are associated with the development of damage. Patients are certainly eager to limit corticosteroid use, as almost all patients in the CHILL-NL cohort reported to have negative experiences with prednisone regarding their physical appearance and or mental well-being,” the study authors wrote.
Results also showed that 62% of the patients had damage, predominantly in the musculoskeletal, neuropsychiatric, and renal systems.
Most organ systems became involved within the first 2 years of diagnosis, but after 5 years of disease the nature of disease manifestations tended to shift to damage such as myocardial infarctions.
Notably, after 10-20 years, when cSLE patients were in their early 20s and 30s, significant damage had occurred in more than half of the patients, the authors noted.
“This shift to damage has also been observed in adult-onset SLE patients and urges for preventative screening measures of such (cardiovascular) damage and healthy lifestyle advice (healthy diet, regular exercise, abstinence from smoking),” they wrote.
Multivariate logistic regression showed that damage accrual was associated with disease duration (odds ratio, 1.15; P less than .001), antiphospholipid‐antibody positivity (OR, 3.56; P = .026), and hypertension (OR, 3.21; P = .043). On the other hand, current HCQ monotherapy was associated with an SLICC-Damage Index score of 0 (OR, 0.16; P = .009).
The HRQOL of the cohort, assessed via the Short Form–36, was also impaired compared with the general population, the researchers discovered. For example, the presence of damage reduced HRQOL in one domain, and high disease activity, defined as SLEDAI-2k of 8 or more, strongly reduced HRQOL in four of eight domains. Changes in physical appearance lowered HRQOL in seven of eight domains.
“HRQOL of adults with cSLE is impaired and affected by other factors than disease activity or damage alone. By identifying and addressing these factors, like physical appearance and potentially coping styles, HRQOL may be improved,” they advised.
The study was supported financially by the Dutch Arthritis Foundation and the Dutch national patient association for lupus, antiphospholipid syndrome, scleroderma, and mixed connective tissue diseases.
SOURCE: Groot N et al. Arthritis Rheumatol. 2018 Aug 27. doi: 10.1002/art.40697
The majority of adults with childhood-onset systemic lupus erythematosus in a longitudinal Dutch study developed significant damage at a young age, remain on corticosteroids in adulthood, and have an impaired health-related quality of life.
The findings in the study, dubbed Childhood-Onset SLE in the Netherlands (CHILL-NL), highlighted the need for preventive screening measures to be put in place before the age of 30 to facilitate a better outcome for patients, who still face high morbidity from childhood-onset systemic lupus erythematosus (cSLE) despite improved survival. Such information is helpful to “answer questions from children and parents regarding the future course of the disease,” wrote Noortje Groot, PhD, of the department of pediatric rheumatology at Erasmus Medical Center–Sophia Children’s Hospital in Rotterdam, the Netherlands, and her colleagues. The report is in Arthritis & Rheumatology.
The current study included all adult SLE patients treated in any Dutch public hospital during the period of November 2013 to April 2016 who were diagnosed with the autoimmune disease prior to their 18th birthday.
All 111 patients involved in the study were seen for a 1.5-hour visit at Erasmus University Medical Center or a local hospital of their choice. During the appointment, a medical history was taken, a physical examination was performed, and the patients completed questionnaires on health-related quality of life (HRQOL).
The average age of patients at the study visit was 33 years, 91% were female, and 72% were white. Median disease duration was 20 years and disease activity was low (median SLE Disease Activity Index 2000 [SLEDAI-2k] = 4). Low complement (32%), skin rashes (14%), and proteinuria (13%) were the most common SLEDAI items reported.
Overall, 68% of the cohort (n = 76) were taking hydroxychloroquine (HCQ), 29% of whom (n = 22) were taking it as monotherapy.
Furthermore, 68% of patients at the study visit were taking corticosteroids and/or non-HCQ disease-modifying antirheumatic drugs (DMARDs), and just over half (51%) of patients (n = 56) were taking corticosteroids either alone or with a non-HCQ DMARD.
“This [finding] is worrying as corticosteroids are associated with the development of damage. Patients are certainly eager to limit corticosteroid use, as almost all patients in the CHILL-NL cohort reported to have negative experiences with prednisone regarding their physical appearance and or mental well-being,” the study authors wrote.
Results also showed that 62% of the patients had damage, predominantly in the musculoskeletal, neuropsychiatric, and renal systems.
Most organ systems became involved within the first 2 years of diagnosis, but after 5 years of disease the nature of disease manifestations tended to shift to damage such as myocardial infarctions.
Notably, after 10-20 years, when cSLE patients were in their early 20s and 30s, significant damage had occurred in more than half of the patients, the authors noted.
“This shift to damage has also been observed in adult-onset SLE patients and urges for preventative screening measures of such (cardiovascular) damage and healthy lifestyle advice (healthy diet, regular exercise, abstinence from smoking),” they wrote.
Multivariate logistic regression showed that damage accrual was associated with disease duration (odds ratio, 1.15; P less than .001), antiphospholipid‐antibody positivity (OR, 3.56; P = .026), and hypertension (OR, 3.21; P = .043). On the other hand, current HCQ monotherapy was associated with an SLICC-Damage Index score of 0 (OR, 0.16; P = .009).
The HRQOL of the cohort, assessed via the Short Form–36, was also impaired compared with the general population, the researchers discovered. For example, the presence of damage reduced HRQOL in one domain, and high disease activity, defined as SLEDAI-2k of 8 or more, strongly reduced HRQOL in four of eight domains. Changes in physical appearance lowered HRQOL in seven of eight domains.
“HRQOL of adults with cSLE is impaired and affected by other factors than disease activity or damage alone. By identifying and addressing these factors, like physical appearance and potentially coping styles, HRQOL may be improved,” they advised.
The study was supported financially by the Dutch Arthritis Foundation and the Dutch national patient association for lupus, antiphospholipid syndrome, scleroderma, and mixed connective tissue diseases.
SOURCE: Groot N et al. Arthritis Rheumatol. 2018 Aug 27. doi: 10.1002/art.40697
FROM ARTHRITIS & RHEUMATOLOGY
Key clinical point:
Major finding: Among 111 patients, 62% had damage, predominantly in the musculoskeletal, neuropsychiatric, and renal systems.
Study details: Adult SLE patients diagnosed prior to their 18th birthday who were treated during November 2013 to April 2016 in any Dutch public hospital.
Disclosures: The study was supported financially by the Dutch Arthritis Foundation and the Dutch national patient association for lupus, antiphospholipid syndrome, scleroderma, and mixed connective tissue diseases.
Source: Groot N et al. Arthritis Rheumatol. 2018 Aug 27. doi: 10.1002/art.40697.
Autoimmune connective tissue disease predicted by interferon status, family history
Patients at risk of autoimmune connective tissue disease who progressed to actual disease had elevated interferon scores and a family history of autoimmune rheumatic disease, suggesting the interferon scores could be used to predict disease progression, according to results from a prospective, observational study published in Annals of the Rheumatic Diseases.

“Referrals of ANA-positive individuals to rheumatologists has increased over the last decade. Concerns are that these at-risk individuals may be discharged prematurely or be observed in an inefficient ‘watch and wait’ fashion until the diagnosis is clear, by which time the potential to prevent disease and confer the most benefit may be lost,” Dr. Yusof and his colleagues wrote in their study.
There were 19 of 118 patients who progressed to AI-CTD after 12 months of follow-up; of these patients, 14 developed SLE and 5 developed primary Sjögren’s syndrome. The researchers noted no significant differences among baseline characteristics or findings from ultrasound, compared with other groups. Compared with healthy controls, IFN-Score-A increased in 105 at-risk patients (fold difference = 2.21; 95% confidence interval, 1.22-4.00; P = .005) and in all 114 patients in the SLE group (fold difference = 7.81; 95% CI, 4.33-14.04; P less than .001). IFN-Score-A was also increased in the SLE group, compared with the at-risk group (fold difference = 3.54; 95% CI, 2.22-5.63; P less than .001). For IFN-Score-B, there was no difference between rates in the healthy control group and the at-risk group. However, IFN-Score-B was increased in the SLE group, compared with the healthy control group (fold difference = 3.85; 95% CI, 2.60-5.72; P less than .001) and the at-risk group (fold difference = 3.93; 95% CI, 2.87-5.37; P less than .001).
Independent baseline predictors of AI-CTD progression in a multivariate analysis included family history of autoimmune rheumatic disease (odds ratio, 8.20; P = .012) and IFN-Score-B (OR, 3.79; P = .005), researchers said.
“Although we could not confirm which IFN pathways predominate, our findings suggest that progression to AI-CTD may not be exclusively driven by IFN-I [type I interferon] but by a synergistic activation of [interferon-stimulated genes] induced by a range of IFNs and IFN-Score-B [that] could act as a biomarker for more diverse immune activation,” Dr. Yusof and his colleagues wrote.
Dr. Yusof is a U.K. National Institute for Health Research doctoral fellow at the University of Leeds. Several other authors reported financial support from range of pharmaceutical companies.
SOURCE: Yusof MYM et al. Ann Rheum Dis. 21 Jun 2018. doi: 10.1136/annrheumdis-2018-213386.
Patients at risk of autoimmune connective tissue disease who progressed to actual disease had elevated interferon scores and a family history of autoimmune rheumatic disease, suggesting the interferon scores could be used to predict disease progression, according to results from a prospective, observational study published in Annals of the Rheumatic Diseases.
“Referrals of ANA-positive individuals to rheumatologists has increased over the last decade. Concerns are that these at-risk individuals may be discharged prematurely or be observed in an inefficient ‘watch and wait’ fashion until the diagnosis is clear, by which time the potential to prevent disease and confer the most benefit may be lost,” Dr. Yusof and his colleagues wrote in their study.
There were 19 of 118 patients who progressed to AI-CTD after 12 months of follow-up; of these patients, 14 developed SLE and 5 developed primary Sjögren’s syndrome. The researchers noted no significant differences among baseline characteristics or findings from ultrasound, compared with other groups. Compared with healthy controls, IFN-Score-A increased in 105 at-risk patients (fold difference = 2.21; 95% confidence interval, 1.22-4.00; P = .005) and in all 114 patients in the SLE group (fold difference = 7.81; 95% CI, 4.33-14.04; P less than .001). IFN-Score-A was also increased in the SLE group, compared with the at-risk group (fold difference = 3.54; 95% CI, 2.22-5.63; P less than .001). For IFN-Score-B, there was no difference between rates in the healthy control group and the at-risk group. However, IFN-Score-B was increased in the SLE group, compared with the healthy control group (fold difference = 3.85; 95% CI, 2.60-5.72; P less than .001) and the at-risk group (fold difference = 3.93; 95% CI, 2.87-5.37; P less than .001).
Independent baseline predictors of AI-CTD progression in a multivariate analysis included family history of autoimmune rheumatic disease (odds ratio, 8.20; P = .012) and IFN-Score-B (OR, 3.79; P = .005), researchers said.
“Although we could not confirm which IFN pathways predominate, our findings suggest that progression to AI-CTD may not be exclusively driven by IFN-I [type I interferon] but by a synergistic activation of [interferon-stimulated genes] induced by a range of IFNs and IFN-Score-B [that] could act as a biomarker for more diverse immune activation,” Dr. Yusof and his colleagues wrote.
Dr. Yusof is a U.K. National Institute for Health Research doctoral fellow at the University of Leeds. Several other authors reported financial support from range of pharmaceutical companies.
SOURCE: Yusof MYM et al. Ann Rheum Dis. 21 Jun 2018. doi: 10.1136/annrheumdis-2018-213386.
Patients at risk of autoimmune connective tissue disease who progressed to actual disease had elevated interferon scores and a family history of autoimmune rheumatic disease, suggesting the interferon scores could be used to predict disease progression, according to results from a prospective, observational study published in Annals of the Rheumatic Diseases.
“Referrals of ANA-positive individuals to rheumatologists has increased over the last decade. Concerns are that these at-risk individuals may be discharged prematurely or be observed in an inefficient ‘watch and wait’ fashion until the diagnosis is clear, by which time the potential to prevent disease and confer the most benefit may be lost,” Dr. Yusof and his colleagues wrote in their study.
There were 19 of 118 patients who progressed to AI-CTD after 12 months of follow-up; of these patients, 14 developed SLE and 5 developed primary Sjögren’s syndrome. The researchers noted no significant differences among baseline characteristics or findings from ultrasound, compared with other groups. Compared with healthy controls, IFN-Score-A increased in 105 at-risk patients (fold difference = 2.21; 95% confidence interval, 1.22-4.00; P = .005) and in all 114 patients in the SLE group (fold difference = 7.81; 95% CI, 4.33-14.04; P less than .001). IFN-Score-A was also increased in the SLE group, compared with the at-risk group (fold difference = 3.54; 95% CI, 2.22-5.63; P less than .001). For IFN-Score-B, there was no difference between rates in the healthy control group and the at-risk group. However, IFN-Score-B was increased in the SLE group, compared with the healthy control group (fold difference = 3.85; 95% CI, 2.60-5.72; P less than .001) and the at-risk group (fold difference = 3.93; 95% CI, 2.87-5.37; P less than .001).
Independent baseline predictors of AI-CTD progression in a multivariate analysis included family history of autoimmune rheumatic disease (odds ratio, 8.20; P = .012) and IFN-Score-B (OR, 3.79; P = .005), researchers said.
“Although we could not confirm which IFN pathways predominate, our findings suggest that progression to AI-CTD may not be exclusively driven by IFN-I [type I interferon] but by a synergistic activation of [interferon-stimulated genes] induced by a range of IFNs and IFN-Score-B [that] could act as a biomarker for more diverse immune activation,” Dr. Yusof and his colleagues wrote.
Dr. Yusof is a U.K. National Institute for Health Research doctoral fellow at the University of Leeds. Several other authors reported financial support from range of pharmaceutical companies.
SOURCE: Yusof MYM et al. Ann Rheum Dis. 21 Jun 2018. doi: 10.1136/annrheumdis-2018-213386.
FROM ANNALS OF THE RHEUMATIC DISEASES
Key clinical point: Elevated levels of two different interferon scores as well as family history predicted the likelihood of patients at risk of autoimmune connective tissue disease progressing to actual disease.
Major finding: Of 118 patients at risk, 14 patients developed SLE and 5 patients developed primary Sjögren’s syndrome.
Study details: A prospective, observational study of 118 patients at risk of developing autoimmune connective tissue disease.
Disclosures: Dr. Yusof is a U.K. National Institute for Health Research doctoral fellow at the University of Leeds. Several other authors reported financial support from range of pharmaceutical companies.
Source: Yusof MYM et al. Ann Rheum Dis. 21 Jun 2018. doi: 10.1136/annrheumdis-2018-213386.
SLE flares linked to air temperature, pollution
AMSTERDAM – Both air temperature and air pollution levels affected the likelihood of experiencing a flare in systemic lupus erythematosus (SLE) organ-specific disease activity in a study presented at the European Congress of Rheumatology.
For every 1° F increase in temperature, there was an increase in the odds of experiencing a skin flare (odds ratio, 1.0075), joint flare (OR, 1.0110), or neurologic flare (OR, 1.0096), reported George Stojan, MD, and associates during one of the poster sessions. Conversely, renal flares were less likely to occur with rising temperature (OR, 0.9960). The latter is something previously reported, Dr. Stojan said in an interview, “as we found most renal flares occur in the winter, not in the summer months.”

Furthermore, for every 1 mcg per cubic meter increase in fine particulate matter pollution (PM2.5), there were increases in serositis (OR, 1.0240) and hematologic flares (OR, 1.011).
There were two reasons for looking at the role of these environmental factors in relation to SLE flares, said Dr. Stojan, an assistant professor of medicine at Johns Hopkins University, Baltimore. “The first was a clinical observation – I was getting clusters of patients with certain disease manifestations, for example with serositis or joint flares, and there wasn’t a random distribution.”
The second was a patient observation, added Dr. Stojan, who is also codirector of the Johns Hopkins Lupus Center. Every year, patients treated at the Center have the opportunity to meet each other and hear about the research being done by the team, and it was at this meeting that patients said they felt they were experiencing similar disease flares.
To look at the underlying role of environmental exposures in the development of SLE and possible associations with disease activity, Dr. Stojan and associates used a method known as cluster detection, which is commonly used in public health studies.
The investigators used a 350-km radial zone around the Johns Hopkins Lupus Center for the analysis as this was an area where a uniform number of patients treated by the center were living. They obtained data on 1,261 patients in the Hopkins Lupus Cohort, spanning a 10-year period from 1999 to 2009, and used SaTScan software to identify clusters of disease activity occurring during 3 separate monthly time intervals in different counties. The researchers then linked these clusters to average temperature and PM2.5 data obtained from the Environmental Protection Agency for the 10 days prior to patients’ visits.
“The SaTScan system predicts how many flares per organ system you would expect in a county based on the number of patients and based on the total flares we have in our cohort,” Dr. Stojan explained. Previously, the system helped to identify areas that had a higher flare incidence for each organ system that lasted for about 2-3 years, did not overlap, and could not be explained. So, the next step was to look for potential environmental triggers.
“Basically, the SaTScan adjusts the data that’s inputted for temperature and small particulate pollution. If the cluster moves in space and time then these did affect it,” Dr. Stojan said. “It seems that these do affect certain types of organ flares,” even after adjustment for other variables such as patients’ age, gender, income, ethnicity, and living situation (rural or urban).
Flares in skin symptoms during the summer have been identified before, he acknowledged, but the link to joint flares or neurologic flares have not. The latter includes things like seizures, neuropathy, or abnormal brain imaging rather than mood changes or mild cognitive dysfunction.
These data could have an impact on how clinical trials are designed, Dr. Stojan added, suggesting that factoring in where patients live and how close they are to areas of pollution could ensure a uniform population of patients is studied.
From a more practical perspective, these data might help to develop predictive models to help understand when patients are likely to experience a flare and if any action can be taken to ameliorate the effects of exposure.
The next step is a collaboration with patients to develop software or a mobile application where patients could input information about any disease flares. This would enable a finer view of what could be happening, Dr. Stojan said, as while daily readings are available for the environmental factors studied, disease activity data is only available during 3 separate monthly intervals. It would also allow other environmental factors to be considered.
“I think this is an important step in figuring out environmental factors and their influence on lupus,” he said. “There has been an extensive amount of research into viral causes and potential infectious triggers, but spatial-temporal analysis of environmental variables have never been done before in lupus.”
The study received no commercial funding, and Dr. Stojan reported having no disclosures.
SOURCE: Stojan G et al. Ann Rheum Dis. 2018;77(Suppl 2):1191. Abstract SAT0685.
AMSTERDAM – Both air temperature and air pollution levels affected the likelihood of experiencing a flare in systemic lupus erythematosus (SLE) organ-specific disease activity in a study presented at the European Congress of Rheumatology.
For every 1° F increase in temperature, there was an increase in the odds of experiencing a skin flare (odds ratio, 1.0075), joint flare (OR, 1.0110), or neurologic flare (OR, 1.0096), reported George Stojan, MD, and associates during one of the poster sessions. Conversely, renal flares were less likely to occur with rising temperature (OR, 0.9960). The latter is something previously reported, Dr. Stojan said in an interview, “as we found most renal flares occur in the winter, not in the summer months.”
Furthermore, for every 1 mcg per cubic meter increase in fine particulate matter pollution (PM2.5), there were increases in serositis (OR, 1.0240) and hematologic flares (OR, 1.011).
There were two reasons for looking at the role of these environmental factors in relation to SLE flares, said Dr. Stojan, an assistant professor of medicine at Johns Hopkins University, Baltimore. “The first was a clinical observation – I was getting clusters of patients with certain disease manifestations, for example with serositis or joint flares, and there wasn’t a random distribution.”
The second was a patient observation, added Dr. Stojan, who is also codirector of the Johns Hopkins Lupus Center. Every year, patients treated at the Center have the opportunity to meet each other and hear about the research being done by the team, and it was at this meeting that patients said they felt they were experiencing similar disease flares.
To look at the underlying role of environmental exposures in the development of SLE and possible associations with disease activity, Dr. Stojan and associates used a method known as cluster detection, which is commonly used in public health studies.
The investigators used a 350-km radial zone around the Johns Hopkins Lupus Center for the analysis as this was an area where a uniform number of patients treated by the center were living. They obtained data on 1,261 patients in the Hopkins Lupus Cohort, spanning a 10-year period from 1999 to 2009, and used SaTScan software to identify clusters of disease activity occurring during 3 separate monthly time intervals in different counties. The researchers then linked these clusters to average temperature and PM2.5 data obtained from the Environmental Protection Agency for the 10 days prior to patients’ visits.
“The SaTScan system predicts how many flares per organ system you would expect in a county based on the number of patients and based on the total flares we have in our cohort,” Dr. Stojan explained. Previously, the system helped to identify areas that had a higher flare incidence for each organ system that lasted for about 2-3 years, did not overlap, and could not be explained. So, the next step was to look for potential environmental triggers.
“Basically, the SaTScan adjusts the data that’s inputted for temperature and small particulate pollution. If the cluster moves in space and time then these did affect it,” Dr. Stojan said. “It seems that these do affect certain types of organ flares,” even after adjustment for other variables such as patients’ age, gender, income, ethnicity, and living situation (rural or urban).
Flares in skin symptoms during the summer have been identified before, he acknowledged, but the link to joint flares or neurologic flares have not. The latter includes things like seizures, neuropathy, or abnormal brain imaging rather than mood changes or mild cognitive dysfunction.
These data could have an impact on how clinical trials are designed, Dr. Stojan added, suggesting that factoring in where patients live and how close they are to areas of pollution could ensure a uniform population of patients is studied.
From a more practical perspective, these data might help to develop predictive models to help understand when patients are likely to experience a flare and if any action can be taken to ameliorate the effects of exposure.
The next step is a collaboration with patients to develop software or a mobile application where patients could input information about any disease flares. This would enable a finer view of what could be happening, Dr. Stojan said, as while daily readings are available for the environmental factors studied, disease activity data is only available during 3 separate monthly intervals. It would also allow other environmental factors to be considered.
“I think this is an important step in figuring out environmental factors and their influence on lupus,” he said. “There has been an extensive amount of research into viral causes and potential infectious triggers, but spatial-temporal analysis of environmental variables have never been done before in lupus.”
The study received no commercial funding, and Dr. Stojan reported having no disclosures.
SOURCE: Stojan G et al. Ann Rheum Dis. 2018;77(Suppl 2):1191. Abstract SAT0685.
AMSTERDAM – Both air temperature and air pollution levels affected the likelihood of experiencing a flare in systemic lupus erythematosus (SLE) organ-specific disease activity in a study presented at the European Congress of Rheumatology.
For every 1° F increase in temperature, there was an increase in the odds of experiencing a skin flare (odds ratio, 1.0075), joint flare (OR, 1.0110), or neurologic flare (OR, 1.0096), reported George Stojan, MD, and associates during one of the poster sessions. Conversely, renal flares were less likely to occur with rising temperature (OR, 0.9960). The latter is something previously reported, Dr. Stojan said in an interview, “as we found most renal flares occur in the winter, not in the summer months.”
Furthermore, for every 1 mcg per cubic meter increase in fine particulate matter pollution (PM2.5), there were increases in serositis (OR, 1.0240) and hematologic flares (OR, 1.011).
There were two reasons for looking at the role of these environmental factors in relation to SLE flares, said Dr. Stojan, an assistant professor of medicine at Johns Hopkins University, Baltimore. “The first was a clinical observation – I was getting clusters of patients with certain disease manifestations, for example with serositis or joint flares, and there wasn’t a random distribution.”
The second was a patient observation, added Dr. Stojan, who is also codirector of the Johns Hopkins Lupus Center. Every year, patients treated at the Center have the opportunity to meet each other and hear about the research being done by the team, and it was at this meeting that patients said they felt they were experiencing similar disease flares.
To look at the underlying role of environmental exposures in the development of SLE and possible associations with disease activity, Dr. Stojan and associates used a method known as cluster detection, which is commonly used in public health studies.
The investigators used a 350-km radial zone around the Johns Hopkins Lupus Center for the analysis as this was an area where a uniform number of patients treated by the center were living. They obtained data on 1,261 patients in the Hopkins Lupus Cohort, spanning a 10-year period from 1999 to 2009, and used SaTScan software to identify clusters of disease activity occurring during 3 separate monthly time intervals in different counties. The researchers then linked these clusters to average temperature and PM2.5 data obtained from the Environmental Protection Agency for the 10 days prior to patients’ visits.
“The SaTScan system predicts how many flares per organ system you would expect in a county based on the number of patients and based on the total flares we have in our cohort,” Dr. Stojan explained. Previously, the system helped to identify areas that had a higher flare incidence for each organ system that lasted for about 2-3 years, did not overlap, and could not be explained. So, the next step was to look for potential environmental triggers.
“Basically, the SaTScan adjusts the data that’s inputted for temperature and small particulate pollution. If the cluster moves in space and time then these did affect it,” Dr. Stojan said. “It seems that these do affect certain types of organ flares,” even after adjustment for other variables such as patients’ age, gender, income, ethnicity, and living situation (rural or urban).
Flares in skin symptoms during the summer have been identified before, he acknowledged, but the link to joint flares or neurologic flares have not. The latter includes things like seizures, neuropathy, or abnormal brain imaging rather than mood changes or mild cognitive dysfunction.
These data could have an impact on how clinical trials are designed, Dr. Stojan added, suggesting that factoring in where patients live and how close they are to areas of pollution could ensure a uniform population of patients is studied.
From a more practical perspective, these data might help to develop predictive models to help understand when patients are likely to experience a flare and if any action can be taken to ameliorate the effects of exposure.
The next step is a collaboration with patients to develop software or a mobile application where patients could input information about any disease flares. This would enable a finer view of what could be happening, Dr. Stojan said, as while daily readings are available for the environmental factors studied, disease activity data is only available during 3 separate monthly intervals. It would also allow other environmental factors to be considered.
“I think this is an important step in figuring out environmental factors and their influence on lupus,” he said. “There has been an extensive amount of research into viral causes and potential infectious triggers, but spatial-temporal analysis of environmental variables have never been done before in lupus.”
The study received no commercial funding, and Dr. Stojan reported having no disclosures.
SOURCE: Stojan G et al. Ann Rheum Dis. 2018;77(Suppl 2):1191. Abstract SAT0685.
REPORTING FROM THE EULAR 2018 CONGRESS
Key clinical point:
Major finding: For every 1° F increase in temperature, the risk for skin, joint, or neurologic flares increased.
Study details: A spatial-time cluster analysis of 1,261 patients in the Hopkins Lupus Cohort linking disease activity to temperature changes and fine particulate matter pollution.
Disclosures: The study received no commercial funding, and Dr. Stojan reported having no disclosures.
Source: Stojan G et al. Ann Rheum Dis. 2018;77(Suppl 2):1191. Abstract SAT0685.