User login
Inherited thrombocytopenia type is a risk factor for hematologic cancers
Copenhagen – Investigators in Italy have found that a recently identified form of inherited thrombocytopenia related to mutations in the gene ETV6 (ETV6-RT) appears to be largely asymptomatic, but puts patients at significantly increased risk for hematologic malignancies, particularly childhood B-cell acute lymphoblastic leukemia (ALL).
“When we are talking about ETV6-related thrombocytopenia patients, we must pay more attention to this hidden but very dangerous risk of developing blood cancer – so that we can define prognosis and plan a personalized follow-up – [rather] than to bleeding complications, which are rare and usually do not affect the quality of life,” said Dr. Federica Melazzini from the University of Pavia, Italy.

At a briefing at the annual congress of the European Hematology Association, Dr. Melazzini described a prospective study of the clinical and laboratory features of the newly identified disorder, with a focus on its association with hematologic cancers.
They enrolled 130 unrelated consecutive patients with inherited thrombocytopenias (IT) who did not have definitive diagnoses because they did exhibit criteria for any of the known forms of IT.
The investigators used whole exome sequencing or Sanger sequencing to look for mutations in ETV6, and when a mutation was identified, all available relatives of the affected participant (proband) also were evaluated. The authors also included data on five patients from two families known to have ETV6-RT from previous studies.
The participants were evaluated with complete blood counts, platelet sizing, flow cytometry studies of platelet membrane glycoproteins, as well as platelet aggregation, activation, adhesion and spreading. Other evaluations included differentiation of human megakaryocytes, morphological analysis of megakaryocytes, megakaryocytes flow cytometry, and evaluation of pro-platelet formation by megakaryocytes differentiated in vitro.
The investigators identified a total of 20 patients from 7 families bearing 5 different mutations in ETV6. Although these patients had only mild thrombocytopenias and a low bleeding tendency, 4 of the 20 had childhood ALL, supporting the association between ETV6 mutations and early leukemic transformation.
This translated into an incidence rate for hematologic malignancies of nearly 1 in 100 persons per year, compared with 1 in 10,000 per year in the general population, Dr. Melazzini said.
The investigators also noted that one patient developed Janus kinase 2 (JAK2)-positive polycythemia vera at age 37 years, “suggesting that this disease should be added to the list of malignancies to which the ETV6-RT predisposes.”
Dr. Melazzini said that clinicians should suspect ETV6-RT when a patient presents with an autosomal inherited thrombocytopenia without platelet macrocytosis, and should confirm the diagnosis with genetic analysis.
Patients also should be followed with whole blood cell counts and peripheral blood smear evaluations every 6-12 months, she recommended.
Dr Anton Hagenbeek, professor of hematology at the University of Amsterdam, who moderated the briefing but was not involved in the study, asked how clinicians should communicate the increased risk of hematologic malignancies to the patients.
Dr. Melazzini replied that patients need counseling about the risks of developing ALL or of passing on the trait if they plan to have children. Additionally, if patients develop ALL, the family should be cautioned against using a related donor for any allogeneic stem cell transplants, she said.
The study funding source was not disclosed. Dr. Melazzini and Dr. Hagenbeek reported no relevant disclosures.
Copenhagen – Investigators in Italy have found that a recently identified form of inherited thrombocytopenia related to mutations in the gene ETV6 (ETV6-RT) appears to be largely asymptomatic, but puts patients at significantly increased risk for hematologic malignancies, particularly childhood B-cell acute lymphoblastic leukemia (ALL).
“When we are talking about ETV6-related thrombocytopenia patients, we must pay more attention to this hidden but very dangerous risk of developing blood cancer – so that we can define prognosis and plan a personalized follow-up – [rather] than to bleeding complications, which are rare and usually do not affect the quality of life,” said Dr. Federica Melazzini from the University of Pavia, Italy.
At a briefing at the annual congress of the European Hematology Association, Dr. Melazzini described a prospective study of the clinical and laboratory features of the newly identified disorder, with a focus on its association with hematologic cancers.
They enrolled 130 unrelated consecutive patients with inherited thrombocytopenias (IT) who did not have definitive diagnoses because they did exhibit criteria for any of the known forms of IT.
The investigators used whole exome sequencing or Sanger sequencing to look for mutations in ETV6, and when a mutation was identified, all available relatives of the affected participant (proband) also were evaluated. The authors also included data on five patients from two families known to have ETV6-RT from previous studies.
The participants were evaluated with complete blood counts, platelet sizing, flow cytometry studies of platelet membrane glycoproteins, as well as platelet aggregation, activation, adhesion and spreading. Other evaluations included differentiation of human megakaryocytes, morphological analysis of megakaryocytes, megakaryocytes flow cytometry, and evaluation of pro-platelet formation by megakaryocytes differentiated in vitro.
The investigators identified a total of 20 patients from 7 families bearing 5 different mutations in ETV6. Although these patients had only mild thrombocytopenias and a low bleeding tendency, 4 of the 20 had childhood ALL, supporting the association between ETV6 mutations and early leukemic transformation.
This translated into an incidence rate for hematologic malignancies of nearly 1 in 100 persons per year, compared with 1 in 10,000 per year in the general population, Dr. Melazzini said.
The investigators also noted that one patient developed Janus kinase 2 (JAK2)-positive polycythemia vera at age 37 years, “suggesting that this disease should be added to the list of malignancies to which the ETV6-RT predisposes.”
Dr. Melazzini said that clinicians should suspect ETV6-RT when a patient presents with an autosomal inherited thrombocytopenia without platelet macrocytosis, and should confirm the diagnosis with genetic analysis.
Patients also should be followed with whole blood cell counts and peripheral blood smear evaluations every 6-12 months, she recommended.
Dr Anton Hagenbeek, professor of hematology at the University of Amsterdam, who moderated the briefing but was not involved in the study, asked how clinicians should communicate the increased risk of hematologic malignancies to the patients.
Dr. Melazzini replied that patients need counseling about the risks of developing ALL or of passing on the trait if they plan to have children. Additionally, if patients develop ALL, the family should be cautioned against using a related donor for any allogeneic stem cell transplants, she said.
The study funding source was not disclosed. Dr. Melazzini and Dr. Hagenbeek reported no relevant disclosures.
Copenhagen – Investigators in Italy have found that a recently identified form of inherited thrombocytopenia related to mutations in the gene ETV6 (ETV6-RT) appears to be largely asymptomatic, but puts patients at significantly increased risk for hematologic malignancies, particularly childhood B-cell acute lymphoblastic leukemia (ALL).
“When we are talking about ETV6-related thrombocytopenia patients, we must pay more attention to this hidden but very dangerous risk of developing blood cancer – so that we can define prognosis and plan a personalized follow-up – [rather] than to bleeding complications, which are rare and usually do not affect the quality of life,” said Dr. Federica Melazzini from the University of Pavia, Italy.
At a briefing at the annual congress of the European Hematology Association, Dr. Melazzini described a prospective study of the clinical and laboratory features of the newly identified disorder, with a focus on its association with hematologic cancers.
They enrolled 130 unrelated consecutive patients with inherited thrombocytopenias (IT) who did not have definitive diagnoses because they did exhibit criteria for any of the known forms of IT.
The investigators used whole exome sequencing or Sanger sequencing to look for mutations in ETV6, and when a mutation was identified, all available relatives of the affected participant (proband) also were evaluated. The authors also included data on five patients from two families known to have ETV6-RT from previous studies.
The participants were evaluated with complete blood counts, platelet sizing, flow cytometry studies of platelet membrane glycoproteins, as well as platelet aggregation, activation, adhesion and spreading. Other evaluations included differentiation of human megakaryocytes, morphological analysis of megakaryocytes, megakaryocytes flow cytometry, and evaluation of pro-platelet formation by megakaryocytes differentiated in vitro.
The investigators identified a total of 20 patients from 7 families bearing 5 different mutations in ETV6. Although these patients had only mild thrombocytopenias and a low bleeding tendency, 4 of the 20 had childhood ALL, supporting the association between ETV6 mutations and early leukemic transformation.
This translated into an incidence rate for hematologic malignancies of nearly 1 in 100 persons per year, compared with 1 in 10,000 per year in the general population, Dr. Melazzini said.
The investigators also noted that one patient developed Janus kinase 2 (JAK2)-positive polycythemia vera at age 37 years, “suggesting that this disease should be added to the list of malignancies to which the ETV6-RT predisposes.”
Dr. Melazzini said that clinicians should suspect ETV6-RT when a patient presents with an autosomal inherited thrombocytopenia without platelet macrocytosis, and should confirm the diagnosis with genetic analysis.
Patients also should be followed with whole blood cell counts and peripheral blood smear evaluations every 6-12 months, she recommended.
Dr Anton Hagenbeek, professor of hematology at the University of Amsterdam, who moderated the briefing but was not involved in the study, asked how clinicians should communicate the increased risk of hematologic malignancies to the patients.
Dr. Melazzini replied that patients need counseling about the risks of developing ALL or of passing on the trait if they plan to have children. Additionally, if patients develop ALL, the family should be cautioned against using a related donor for any allogeneic stem cell transplants, she said.
The study funding source was not disclosed. Dr. Melazzini and Dr. Hagenbeek reported no relevant disclosures.
At THE EHA CONGRESS
Key clinical point: ETV6-related thrombocytopenia (EV6-RT) appears to predispose carriers to acute lymphoblastic leukemia and other hematologic cancers.
Major finding: Four of 20 patients with ETV6-RT had childhood ALL, and one developed JAK2-positive polycythemia vera at age 37.
Data source: Study of clinical and laboratory findings in 20 patients with EV6-RT.
Disclosures: The study funding source was not disclosed. Dr. Melazzini and Dr. Hagenbeek reported no relevant disclosures.

Antibodies cut factor VIII half-life in patients with hemophilia A
Non-neutralizing, factor VIII–specific IgG antibodies can contribute significantly to reductions in factor VIII half-life in patients with hemophilia A, according to a study published online in Blood.
Screening for factor VIII–specific IgG may aid in tailoring factor VIII prophylactic regimens for hemophilia A patients, said Christoph J. Hofbauer, Ph.D., of the biopharmaceutical company Baxalta in Vienna, and his associates at the Medical University of Vienna.

The researchers examined the effect of factor VIII–specific IgG antibodies on factor VIII half-life in 42 adult patients with hemophilia A without inhibitors. Patients ranged in age from 18-61 years, and 37 of them had severe disease. Of the cohort, 31 received recombinant factor VIII concentrates and 11 received plasma-derived factor VIII concentrates (Blood. 2016 May 23 [Epub ahead of print])
In the initial antibody screen, 15 patients tested positive for factor VIII–binding IgG with titers of at least 1:20. Factor VIII–specific antibodies were found at titers of at least 1:40 in 9 of the 15 subjects. Most had low- to moderate-affinity IgG1 and IgG3 antibodies. One patient with high-affinity IgG4 antibodies went on to develop low titers of factor VIII inhibitors.
Patients with factor VIII–specific antibodies had a shorter factor VIII half-life (median 7.8 hours) than did patients without antibodies (median 10.4 hours).
Dr. Hofbauer is employed by Baxalta, which makes a variety of antihemophilic factors.
On Twitter @maryjodales
Non-neutralizing, factor VIII–specific IgG antibodies can contribute significantly to reductions in factor VIII half-life in patients with hemophilia A, according to a study published online in Blood.
Screening for factor VIII–specific IgG may aid in tailoring factor VIII prophylactic regimens for hemophilia A patients, said Christoph J. Hofbauer, Ph.D., of the biopharmaceutical company Baxalta in Vienna, and his associates at the Medical University of Vienna.
The researchers examined the effect of factor VIII–specific IgG antibodies on factor VIII half-life in 42 adult patients with hemophilia A without inhibitors. Patients ranged in age from 18-61 years, and 37 of them had severe disease. Of the cohort, 31 received recombinant factor VIII concentrates and 11 received plasma-derived factor VIII concentrates (Blood. 2016 May 23 [Epub ahead of print])
In the initial antibody screen, 15 patients tested positive for factor VIII–binding IgG with titers of at least 1:20. Factor VIII–specific antibodies were found at titers of at least 1:40 in 9 of the 15 subjects. Most had low- to moderate-affinity IgG1 and IgG3 antibodies. One patient with high-affinity IgG4 antibodies went on to develop low titers of factor VIII inhibitors.
Patients with factor VIII–specific antibodies had a shorter factor VIII half-life (median 7.8 hours) than did patients without antibodies (median 10.4 hours).
Dr. Hofbauer is employed by Baxalta, which makes a variety of antihemophilic factors.
On Twitter @maryjodales
Non-neutralizing, factor VIII–specific IgG antibodies can contribute significantly to reductions in factor VIII half-life in patients with hemophilia A, according to a study published online in Blood.
Screening for factor VIII–specific IgG may aid in tailoring factor VIII prophylactic regimens for hemophilia A patients, said Christoph J. Hofbauer, Ph.D., of the biopharmaceutical company Baxalta in Vienna, and his associates at the Medical University of Vienna.
The researchers examined the effect of factor VIII–specific IgG antibodies on factor VIII half-life in 42 adult patients with hemophilia A without inhibitors. Patients ranged in age from 18-61 years, and 37 of them had severe disease. Of the cohort, 31 received recombinant factor VIII concentrates and 11 received plasma-derived factor VIII concentrates (Blood. 2016 May 23 [Epub ahead of print])
In the initial antibody screen, 15 patients tested positive for factor VIII–binding IgG with titers of at least 1:20. Factor VIII–specific antibodies were found at titers of at least 1:40 in 9 of the 15 subjects. Most had low- to moderate-affinity IgG1 and IgG3 antibodies. One patient with high-affinity IgG4 antibodies went on to develop low titers of factor VIII inhibitors.
Patients with factor VIII–specific antibodies had a shorter factor VIII half-life (median 7.8 hours) than did patients without antibodies (median 10.4 hours).
Dr. Hofbauer is employed by Baxalta, which makes a variety of antihemophilic factors.
On Twitter @maryjodales
FROM BLOOD
Key clinical point: Non-neutralizing, factor VIII–specific IgG antibodies can contribute significantly to reductions in factor VIII half-life in patients with hemophilia A.
Major finding: Patients with factor VIII–specific antibodies had a shorter factor VIII half-life (median 7.8 hours) than did patients without antibodies (median 10.4 hours).
Data source: 42 adult patients, ranging in age from 18-61 years, with hemophilia A without inhibitors, 37 of whom had severe disease.
Disclosures: Dr. Hofbauer is employed by Baxalta, which makes a variety of antihemophilic factors.

In hemophilia A, emicizumab cuts bleeding; plasma-derived factor VIII less likely to trigger antibodies
Patients with severe hemophilia A treated with plasma-derived factor VIII, as compared with recombinant factor VIII, had a lower risk of developing neutralizing antibodies, based on a recent report from the randomized SIPPET trial published in the New England Journal of Medicine.
In a second study, also published in the journal, prophylactic treatment with the bispecific antibody emicizumab decreased bleeding in patients with or without neutralizing antibodies.

Of 125 patients with severe hemophilia A who were treated with plasma-derived factor VIII with von Willebrand factor, 29 (23.2%) developed inhibitors, compared with 47 of 126 patients (37.3%) treated with recombinant factor VIII. In the plasma-derived group, 20 (16.0%) had high-titer inhibitors compared with 30 (23.8%) in the recombinant group. Cox regression models showed an 87% higher rate of inhibitor development with recombinant factor VIII (hazard ratio, 1.87; 95% confidence interval, 1.17-2.96). For development of high-titer inhibitors, the HR was 1.69 (95% CI, 0.96-2.98).
Previous observational studies have suggested greater immunogenicity of recombinant vs. plasm-derived factor VIII, but the results remained inconclusive. Meta-analyses of studies have been hindered by differences in design, enrollment criteria, definition of hemophilia A, sample size, method of inhibitor detection, and intervals of follow-up testing, according to Dr. Flora Peyvandi of the IRCCS Maggiore Hospital, University of Milan and colleagues. “Our trial was specifically designed to compare the immunogenicity of factor VIII products. As a result of randomization, the main risk factors for inhibitor development were evenly distributed between the two factor VIII classes,” they wrote, adding, “The finding that native factor VIII products from human plasma are less immunogenic than those engineered by recombinant DNA technology in animal cell lines has the potential to affect treatment strategies and open new investigations to better understand the mechanisms of the immunogenicity of various factor VIII preparations.” (N Engl J Med. 2016 May 25. doi: 10.1200/JCO.2015.64.0730).
The Survey of Inhibitors in Plasma-Product Exposed Toddlers (SIPPET) clinical trial randomized 251 patients with severe hemophilia A who received infusions of plasma-derived or recombinant factor VIII. The investigators found no association between risk of inhibitor development and race, intensity of treatment, or age at first treatment.
The second study, a 12-week dose-escalation trial of emicizumab, included 18 patients with severe hemophilia A, aged 12-59 years. Patients were assigned to one of three cohorts and received a once-weekly subcutaneous dose of 0.3, 1, or 3 mg per kilogram of body weight. During prophylactic emicizumab treatment, median annualized bleeding rates decreased from 32.5 (range, 8.1-77.1) to 4.4 (0.0-59.5) in cohort 1, from 18.3 (range, 10.1-38.6) to 0.0 (range, 0.0-4.3) in cohort 2, and from 15.2 (range, 0.0-32.5) to 0.0 (range, 0.0-4.2) in cohort 3. The annualized bleeding rate decreased regardless of factor VIII inhibitor status. Of 11 patients with factor VIII inhibitors, 8 (73%) had no bleeding episodes; 5 of 7 patients without factor VIII inhibitors (71%) had no bleeding episodes.
“This study showed that once-weekly subcutaneous administration of emicizumab as prophylaxis is safe and has the potential to reduce or prevent bleeding episodes in patients who have severe hemophilia A with or without factor VIII inhibitors,” wrote Dr. Midori Shima of Nara Medical University, Kashihara, Japan, and colleagues (N. Engl J Med. 2016 May 25. doi: 10.1056/NEJMoa1511769).
Emicizumab had an acceptable safety profile. In total, 43 adverse events were reported in 15 of 18 patients. All were mild, except two moderate events – an upper respiratory tract infection and a headache. Events thought to be treatment related included malaise, injection-site erythema, injection-site rash, diarrhea, increased C-reactive protein level, and increased blood creatine kinase level.
Dr. Peyvandi and coauthors disclosed consulting, advising, research funding and other relationships with several industry sources, including Grifols, Kedrion, and LFB. Dr. Shima and coauthors disclosed consulting, advising, research funding and other relationships with several industry sources, including Chugai Pharmaceuticals, which supported the study.
The studies by Peyvandi et al. and Shima et al. bring exciting news to patients with hemophilia. Peyvandi et al. report the influence of factor VIII product source on the cumulative incidence of inhibitors in children with severe hemophilia. Exclusive use of recombinant factor VIII through the highest-risk period increased the cumulative incidence of all inhibitors, compared with plasma-derived factor VIII. Factor VIII protein glycosylation and von Willebrand factor association play a role in endocytosis, clearance, and antigen presentation and are altered by recombinant technology, which lends plausibility to the implication of product source affecting inhibitor generation. The results may lead to valuable mechanistic insight into factor VIII immunogenicity.
In the second study, Shima et al. evaluate once-weekly administration of emicizumab in patients with severe hemophilia. The bifunctional antibody functions as a conformation replica of factor VIII, forming a thrombin-generating complex with factors IX and X. Emicizumab would likely not induce or be inhibited by factor VIII–neutralizing antibodies, and its subcutaneous bioavailability permits a once-weekly treatment regimen instead of intravenous infusion. The study reports impressive short-term decreases in annualized bleeding rates in both inhibitor-positive and inhibitor-negative patients. The next steps in testing more substantial and sustained thrombin generation required for the treatment of acute hemorrhage will be important for emicizumab.
If proven feasible as a therapeutic agent, emicizumab may play a role in mediating factor VIII immunogenicity. Early sustained treatment with emicizumab in children could ultimately affect the epidemiology of neutralizing antibodies.
The clinical and scientific ramifications of these two studies will take time to be fully realized but offer exciting potential as therapeutic options where new approaches to bypassing the factor VIII requirement have so far fallen short (N Engl J Med. 2016 May 25. doi: 10.1056/NEJMe1603419).
Dr. Donna DiMichele is the deputy director of the Division of Blood Diseases and Resources at the National Heart, Lung, and Blood Institute, part of the National Institutes of Health. These remarks were part of an editorial accompanying two reports in the New England Journal of Medicine. Dr. DiMichele reports personal fees from Chugai Pharmaceuticals outside the submitted work. She was an early founding member of the SIPPET study Steering Committee but resigned when she came to NIH in 2010.
The studies by Peyvandi et al. and Shima et al. bring exciting news to patients with hemophilia. Peyvandi et al. report the influence of factor VIII product source on the cumulative incidence of inhibitors in children with severe hemophilia. Exclusive use of recombinant factor VIII through the highest-risk period increased the cumulative incidence of all inhibitors, compared with plasma-derived factor VIII. Factor VIII protein glycosylation and von Willebrand factor association play a role in endocytosis, clearance, and antigen presentation and are altered by recombinant technology, which lends plausibility to the implication of product source affecting inhibitor generation. The results may lead to valuable mechanistic insight into factor VIII immunogenicity.
In the second study, Shima et al. evaluate once-weekly administration of emicizumab in patients with severe hemophilia. The bifunctional antibody functions as a conformation replica of factor VIII, forming a thrombin-generating complex with factors IX and X. Emicizumab would likely not induce or be inhibited by factor VIII–neutralizing antibodies, and its subcutaneous bioavailability permits a once-weekly treatment regimen instead of intravenous infusion. The study reports impressive short-term decreases in annualized bleeding rates in both inhibitor-positive and inhibitor-negative patients. The next steps in testing more substantial and sustained thrombin generation required for the treatment of acute hemorrhage will be important for emicizumab.
If proven feasible as a therapeutic agent, emicizumab may play a role in mediating factor VIII immunogenicity. Early sustained treatment with emicizumab in children could ultimately affect the epidemiology of neutralizing antibodies.
The clinical and scientific ramifications of these two studies will take time to be fully realized but offer exciting potential as therapeutic options where new approaches to bypassing the factor VIII requirement have so far fallen short (N Engl J Med. 2016 May 25. doi: 10.1056/NEJMe1603419).
Dr. Donna DiMichele is the deputy director of the Division of Blood Diseases and Resources at the National Heart, Lung, and Blood Institute, part of the National Institutes of Health. These remarks were part of an editorial accompanying two reports in the New England Journal of Medicine. Dr. DiMichele reports personal fees from Chugai Pharmaceuticals outside the submitted work. She was an early founding member of the SIPPET study Steering Committee but resigned when she came to NIH in 2010.
The studies by Peyvandi et al. and Shima et al. bring exciting news to patients with hemophilia. Peyvandi et al. report the influence of factor VIII product source on the cumulative incidence of inhibitors in children with severe hemophilia. Exclusive use of recombinant factor VIII through the highest-risk period increased the cumulative incidence of all inhibitors, compared with plasma-derived factor VIII. Factor VIII protein glycosylation and von Willebrand factor association play a role in endocytosis, clearance, and antigen presentation and are altered by recombinant technology, which lends plausibility to the implication of product source affecting inhibitor generation. The results may lead to valuable mechanistic insight into factor VIII immunogenicity.
In the second study, Shima et al. evaluate once-weekly administration of emicizumab in patients with severe hemophilia. The bifunctional antibody functions as a conformation replica of factor VIII, forming a thrombin-generating complex with factors IX and X. Emicizumab would likely not induce or be inhibited by factor VIII–neutralizing antibodies, and its subcutaneous bioavailability permits a once-weekly treatment regimen instead of intravenous infusion. The study reports impressive short-term decreases in annualized bleeding rates in both inhibitor-positive and inhibitor-negative patients. The next steps in testing more substantial and sustained thrombin generation required for the treatment of acute hemorrhage will be important for emicizumab.
If proven feasible as a therapeutic agent, emicizumab may play a role in mediating factor VIII immunogenicity. Early sustained treatment with emicizumab in children could ultimately affect the epidemiology of neutralizing antibodies.
The clinical and scientific ramifications of these two studies will take time to be fully realized but offer exciting potential as therapeutic options where new approaches to bypassing the factor VIII requirement have so far fallen short (N Engl J Med. 2016 May 25. doi: 10.1056/NEJMe1603419).
Dr. Donna DiMichele is the deputy director of the Division of Blood Diseases and Resources at the National Heart, Lung, and Blood Institute, part of the National Institutes of Health. These remarks were part of an editorial accompanying two reports in the New England Journal of Medicine. Dr. DiMichele reports personal fees from Chugai Pharmaceuticals outside the submitted work. She was an early founding member of the SIPPET study Steering Committee but resigned when she came to NIH in 2010.
Patients with severe hemophilia A treated with plasma-derived factor VIII, as compared with recombinant factor VIII, had a lower risk of developing neutralizing antibodies, based on a recent report from the randomized SIPPET trial published in the New England Journal of Medicine.
In a second study, also published in the journal, prophylactic treatment with the bispecific antibody emicizumab decreased bleeding in patients with or without neutralizing antibodies.
Of 125 patients with severe hemophilia A who were treated with plasma-derived factor VIII with von Willebrand factor, 29 (23.2%) developed inhibitors, compared with 47 of 126 patients (37.3%) treated with recombinant factor VIII. In the plasma-derived group, 20 (16.0%) had high-titer inhibitors compared with 30 (23.8%) in the recombinant group. Cox regression models showed an 87% higher rate of inhibitor development with recombinant factor VIII (hazard ratio, 1.87; 95% confidence interval, 1.17-2.96). For development of high-titer inhibitors, the HR was 1.69 (95% CI, 0.96-2.98).
Previous observational studies have suggested greater immunogenicity of recombinant vs. plasm-derived factor VIII, but the results remained inconclusive. Meta-analyses of studies have been hindered by differences in design, enrollment criteria, definition of hemophilia A, sample size, method of inhibitor detection, and intervals of follow-up testing, according to Dr. Flora Peyvandi of the IRCCS Maggiore Hospital, University of Milan and colleagues. “Our trial was specifically designed to compare the immunogenicity of factor VIII products. As a result of randomization, the main risk factors for inhibitor development were evenly distributed between the two factor VIII classes,” they wrote, adding, “The finding that native factor VIII products from human plasma are less immunogenic than those engineered by recombinant DNA technology in animal cell lines has the potential to affect treatment strategies and open new investigations to better understand the mechanisms of the immunogenicity of various factor VIII preparations.” (N Engl J Med. 2016 May 25. doi: 10.1200/JCO.2015.64.0730).
The Survey of Inhibitors in Plasma-Product Exposed Toddlers (SIPPET) clinical trial randomized 251 patients with severe hemophilia A who received infusions of plasma-derived or recombinant factor VIII. The investigators found no association between risk of inhibitor development and race, intensity of treatment, or age at first treatment.
The second study, a 12-week dose-escalation trial of emicizumab, included 18 patients with severe hemophilia A, aged 12-59 years. Patients were assigned to one of three cohorts and received a once-weekly subcutaneous dose of 0.3, 1, or 3 mg per kilogram of body weight. During prophylactic emicizumab treatment, median annualized bleeding rates decreased from 32.5 (range, 8.1-77.1) to 4.4 (0.0-59.5) in cohort 1, from 18.3 (range, 10.1-38.6) to 0.0 (range, 0.0-4.3) in cohort 2, and from 15.2 (range, 0.0-32.5) to 0.0 (range, 0.0-4.2) in cohort 3. The annualized bleeding rate decreased regardless of factor VIII inhibitor status. Of 11 patients with factor VIII inhibitors, 8 (73%) had no bleeding episodes; 5 of 7 patients without factor VIII inhibitors (71%) had no bleeding episodes.
“This study showed that once-weekly subcutaneous administration of emicizumab as prophylaxis is safe and has the potential to reduce or prevent bleeding episodes in patients who have severe hemophilia A with or without factor VIII inhibitors,” wrote Dr. Midori Shima of Nara Medical University, Kashihara, Japan, and colleagues (N. Engl J Med. 2016 May 25. doi: 10.1056/NEJMoa1511769).
Emicizumab had an acceptable safety profile. In total, 43 adverse events were reported in 15 of 18 patients. All were mild, except two moderate events – an upper respiratory tract infection and a headache. Events thought to be treatment related included malaise, injection-site erythema, injection-site rash, diarrhea, increased C-reactive protein level, and increased blood creatine kinase level.
Dr. Peyvandi and coauthors disclosed consulting, advising, research funding and other relationships with several industry sources, including Grifols, Kedrion, and LFB. Dr. Shima and coauthors disclosed consulting, advising, research funding and other relationships with several industry sources, including Chugai Pharmaceuticals, which supported the study.
Patients with severe hemophilia A treated with plasma-derived factor VIII, as compared with recombinant factor VIII, had a lower risk of developing neutralizing antibodies, based on a recent report from the randomized SIPPET trial published in the New England Journal of Medicine.
In a second study, also published in the journal, prophylactic treatment with the bispecific antibody emicizumab decreased bleeding in patients with or without neutralizing antibodies.
Of 125 patients with severe hemophilia A who were treated with plasma-derived factor VIII with von Willebrand factor, 29 (23.2%) developed inhibitors, compared with 47 of 126 patients (37.3%) treated with recombinant factor VIII. In the plasma-derived group, 20 (16.0%) had high-titer inhibitors compared with 30 (23.8%) in the recombinant group. Cox regression models showed an 87% higher rate of inhibitor development with recombinant factor VIII (hazard ratio, 1.87; 95% confidence interval, 1.17-2.96). For development of high-titer inhibitors, the HR was 1.69 (95% CI, 0.96-2.98).
Previous observational studies have suggested greater immunogenicity of recombinant vs. plasm-derived factor VIII, but the results remained inconclusive. Meta-analyses of studies have been hindered by differences in design, enrollment criteria, definition of hemophilia A, sample size, method of inhibitor detection, and intervals of follow-up testing, according to Dr. Flora Peyvandi of the IRCCS Maggiore Hospital, University of Milan and colleagues. “Our trial was specifically designed to compare the immunogenicity of factor VIII products. As a result of randomization, the main risk factors for inhibitor development were evenly distributed between the two factor VIII classes,” they wrote, adding, “The finding that native factor VIII products from human plasma are less immunogenic than those engineered by recombinant DNA technology in animal cell lines has the potential to affect treatment strategies and open new investigations to better understand the mechanisms of the immunogenicity of various factor VIII preparations.” (N Engl J Med. 2016 May 25. doi: 10.1200/JCO.2015.64.0730).
The Survey of Inhibitors in Plasma-Product Exposed Toddlers (SIPPET) clinical trial randomized 251 patients with severe hemophilia A who received infusions of plasma-derived or recombinant factor VIII. The investigators found no association between risk of inhibitor development and race, intensity of treatment, or age at first treatment.
The second study, a 12-week dose-escalation trial of emicizumab, included 18 patients with severe hemophilia A, aged 12-59 years. Patients were assigned to one of three cohorts and received a once-weekly subcutaneous dose of 0.3, 1, or 3 mg per kilogram of body weight. During prophylactic emicizumab treatment, median annualized bleeding rates decreased from 32.5 (range, 8.1-77.1) to 4.4 (0.0-59.5) in cohort 1, from 18.3 (range, 10.1-38.6) to 0.0 (range, 0.0-4.3) in cohort 2, and from 15.2 (range, 0.0-32.5) to 0.0 (range, 0.0-4.2) in cohort 3. The annualized bleeding rate decreased regardless of factor VIII inhibitor status. Of 11 patients with factor VIII inhibitors, 8 (73%) had no bleeding episodes; 5 of 7 patients without factor VIII inhibitors (71%) had no bleeding episodes.
“This study showed that once-weekly subcutaneous administration of emicizumab as prophylaxis is safe and has the potential to reduce or prevent bleeding episodes in patients who have severe hemophilia A with or without factor VIII inhibitors,” wrote Dr. Midori Shima of Nara Medical University, Kashihara, Japan, and colleagues (N. Engl J Med. 2016 May 25. doi: 10.1056/NEJMoa1511769).
Emicizumab had an acceptable safety profile. In total, 43 adverse events were reported in 15 of 18 patients. All were mild, except two moderate events – an upper respiratory tract infection and a headache. Events thought to be treatment related included malaise, injection-site erythema, injection-site rash, diarrhea, increased C-reactive protein level, and increased blood creatine kinase level.
Dr. Peyvandi and coauthors disclosed consulting, advising, research funding and other relationships with several industry sources, including Grifols, Kedrion, and LFB. Dr. Shima and coauthors disclosed consulting, advising, research funding and other relationships with several industry sources, including Chugai Pharmaceuticals, which supported the study.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Key clinical points: Plasma-derived factor VIII carries lower risk of developing neutralizing antibodies than recombinant factor VIII; once-weekly emicizumab decreased bleeding in hemophilia A patients with or without neutralizing antibodies.
Major findings: Inhibitors developed in 23% of patients treated with plasma-derived factor VIII (16% high titer), compared with 37% treated with recombinant factor VIII (24% high titer); during prophylactic treatment with emicizumab, bleeding rates were markedly reduced, and more than 70% of patients had no bleeding episodes.
Data sources: The Survey of Inhibitors in Plasma-Product Exposed Toddlers (SIPPET) randomized trial evaluated 251 patients with severe hemophilia A who received plasma-derived or recombinant factor VIII; emicizumab was evaluated in a 12-week, open-label, nonrandomized dose-escalation study of 18 patients.
Disclosures: Dr. Peyvandi and coauthors disclosed consulting, advising, research funding and other relationships with several industry sources, including Grifols, Kedrion, and LFB. Dr. Shima and coauthors disclosed consulting, advising, research funding and other relationships with several industry sources, including Chugai Pharmaceuticals, which supported the study.
Search is on for cases of aggressive, ruxolitinib-associated skin cancers
ORLANDO – The hematologic cancer drug ruxolitinib seems to be associated with cases of aggressive nonmelanoma skin cancer.
After treating a very aggressive squamous cell carcinoma in a 55-year-old man treated with ruxolitinib for polycythemia vera, and hearing firsthand of three other similar cases, Dr. Fiona Zwald is collecting additional data on the association. She intends to publish these cases in a monograph as a warning to dermatologists, hematologists, oncologists, and other physicians who manage patients with hematologic malignancies, she said at the annual meeting of the American College of Mohs Surgery.
The prescribing information for ruxolitinib (Jakafi, Incyte Pharmaceuticals; Jakavi, Novartis) was updated in 2014 to warn that patients taking the drug face an increased risk of nonmelanoma skin cancers. The label also recommends that physicians inspect the skin regularly and urge patients to be alert for and report any new or changing lesions.
Despite the warnings and recommendations, cases are occurring – and some are quite serious, said Dr. Zwald, a Mohs surgeon in Atlanta.
“People should know this is actually happening. If you have experience with this medication, please let us know so we can compile this report. We are trying to assess the number of skin cancers before and after initiating this medication,” she said.
Ruxolitinib is an inhibitor of Janus kinase with a special affinity for the JAK1 and JAK2 subtypes. Like other cytokine-signaling molecules, their function depends on cell context; it may inhibit cell growth in one setting, and, in another, stimulate it. Ruxolitinib was initially approved in 2011 for the treatment of intermediate- and high-risk myelofibrosis, including primary myelofibrosis, post–polycythemia vera myelofibrosis, and post–essential thrombocythemia myelofibrosis.
In 2014, indications for ruxolitinib were expanded to include treatment of patients with polycythemia vera who have had an inadequate response to or are intolerant of hydroxyurea.
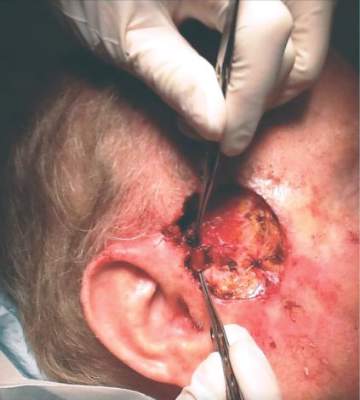
Dr. Zwald’s patient had a 10-year history of polycythemia vera. He was initially well controlled on the standard hydroxyurea treatment. In the meantime, he began working as a caddy at a major U.S. golf club. He developed many facial squamous cell carcinomas that were treated with excision and radiation. A year before he presented to Dr. Zwald, he stopped responding to hydroxyurea and was placed on ruxolitinib.
The patient presented with a 4-cm ulcerated lesion over part of his right temple and to the right helical crus; the lesion had developed over 3 months. Dr. Zwald consulted with the patient’s medical oncologist; treatment with ruxolitinib continued, albeit at a reduced dosage in light of recent events.
She performed Mohs surgery on the patient. It was a challenging case, she said, not the least because adequate anesthesia could not be achieved with local anesthetic. Preoperative staging showed no nodal spread.
“He did, unfortunately demonstrate a large, indurated mass located over one branch of the superficial temporal artery. At the helical crus there was an area of bound-down, fixed tumor. Knowing that I would not be able to fully resect this, I passed him on to the operating room,” Dr. Zwald said. “This tumor was found to extend down to the parotid capsule, but margins were clear.” The surgical defect was successfully repaired with a split-thickness skin graft.
The tumor recurred about 3 months later, and the patient underwent another surgery.
“This time we could not get clear surgical margins, and the tumor was approaching the external auditory meatus. Surgery was abandoned due to fears of complications to that area,” she said.
She presented the case at tumor board, during which she and her colleagues discussed adjuvant radiation. They initially abandoned this idea because he had already had so much radiation to his face. After the second surgery, they decide to proceed with radiation. “The next conversation we have will be whether to add another adjuvant therapy to treatment.”
She sent out the case and requests for feedback to the International Transplant Skin Cancer Collaborative, an 800-member consortium of dermatologists and Mohs surgeons who take care of transplant patients. She received information on three additional cases of aggressive squamous cell carcinoma (SCC) associated with ruxolitinib treatment:
• A patient with myelodysplastic syndrome with aggressive scalp SCC with cutaneous metastases.
• A patient with undifferentiated pleomorphic sarcoma of the scalp, several cutaneous SCCs.
• A patient with a myelodysplastic syndrome with in-transit metastases and explosive cutaneous SCCs. The patient has had the ruxolitinib dose reduced and may be switched to capecitabine.
Dr. Zwald noted that her patient was at risk for aggressive skin cancers for reasons in addition to ruxolitinib treatment.
“He was already immunosuppressed from his malignancy. He was on hydroxyurea, a drug that’s a cumulative phototoxin, and he’s out in the sun playing golf every day, and then was put on ruxolitinib. But the question we face now is how to try and stop this medication so we can get better treatment for him which will, of course, be very difficult.”
To contribute to Dr. Zwald’s case series, please email her at [email protected].
She had no relevant financial disclosures.
On Twitter @Alz_Gal
ORLANDO – The hematologic cancer drug ruxolitinib seems to be associated with cases of aggressive nonmelanoma skin cancer.
After treating a very aggressive squamous cell carcinoma in a 55-year-old man treated with ruxolitinib for polycythemia vera, and hearing firsthand of three other similar cases, Dr. Fiona Zwald is collecting additional data on the association. She intends to publish these cases in a monograph as a warning to dermatologists, hematologists, oncologists, and other physicians who manage patients with hematologic malignancies, she said at the annual meeting of the American College of Mohs Surgery.
The prescribing information for ruxolitinib (Jakafi, Incyte Pharmaceuticals; Jakavi, Novartis) was updated in 2014 to warn that patients taking the drug face an increased risk of nonmelanoma skin cancers. The label also recommends that physicians inspect the skin regularly and urge patients to be alert for and report any new or changing lesions.
Despite the warnings and recommendations, cases are occurring – and some are quite serious, said Dr. Zwald, a Mohs surgeon in Atlanta.
“People should know this is actually happening. If you have experience with this medication, please let us know so we can compile this report. We are trying to assess the number of skin cancers before and after initiating this medication,” she said.
Ruxolitinib is an inhibitor of Janus kinase with a special affinity for the JAK1 and JAK2 subtypes. Like other cytokine-signaling molecules, their function depends on cell context; it may inhibit cell growth in one setting, and, in another, stimulate it. Ruxolitinib was initially approved in 2011 for the treatment of intermediate- and high-risk myelofibrosis, including primary myelofibrosis, post–polycythemia vera myelofibrosis, and post–essential thrombocythemia myelofibrosis.
In 2014, indications for ruxolitinib were expanded to include treatment of patients with polycythemia vera who have had an inadequate response to or are intolerant of hydroxyurea.
Dr. Zwald’s patient had a 10-year history of polycythemia vera. He was initially well controlled on the standard hydroxyurea treatment. In the meantime, he began working as a caddy at a major U.S. golf club. He developed many facial squamous cell carcinomas that were treated with excision and radiation. A year before he presented to Dr. Zwald, he stopped responding to hydroxyurea and was placed on ruxolitinib.
The patient presented with a 4-cm ulcerated lesion over part of his right temple and to the right helical crus; the lesion had developed over 3 months. Dr. Zwald consulted with the patient’s medical oncologist; treatment with ruxolitinib continued, albeit at a reduced dosage in light of recent events.
She performed Mohs surgery on the patient. It was a challenging case, she said, not the least because adequate anesthesia could not be achieved with local anesthetic. Preoperative staging showed no nodal spread.
“He did, unfortunately demonstrate a large, indurated mass located over one branch of the superficial temporal artery. At the helical crus there was an area of bound-down, fixed tumor. Knowing that I would not be able to fully resect this, I passed him on to the operating room,” Dr. Zwald said. “This tumor was found to extend down to the parotid capsule, but margins were clear.” The surgical defect was successfully repaired with a split-thickness skin graft.
The tumor recurred about 3 months later, and the patient underwent another surgery.
“This time we could not get clear surgical margins, and the tumor was approaching the external auditory meatus. Surgery was abandoned due to fears of complications to that area,” she said.
She presented the case at tumor board, during which she and her colleagues discussed adjuvant radiation. They initially abandoned this idea because he had already had so much radiation to his face. After the second surgery, they decide to proceed with radiation. “The next conversation we have will be whether to add another adjuvant therapy to treatment.”
She sent out the case and requests for feedback to the International Transplant Skin Cancer Collaborative, an 800-member consortium of dermatologists and Mohs surgeons who take care of transplant patients. She received information on three additional cases of aggressive squamous cell carcinoma (SCC) associated with ruxolitinib treatment:
• A patient with myelodysplastic syndrome with aggressive scalp SCC with cutaneous metastases.
• A patient with undifferentiated pleomorphic sarcoma of the scalp, several cutaneous SCCs.
• A patient with a myelodysplastic syndrome with in-transit metastases and explosive cutaneous SCCs. The patient has had the ruxolitinib dose reduced and may be switched to capecitabine.
Dr. Zwald noted that her patient was at risk for aggressive skin cancers for reasons in addition to ruxolitinib treatment.
“He was already immunosuppressed from his malignancy. He was on hydroxyurea, a drug that’s a cumulative phototoxin, and he’s out in the sun playing golf every day, and then was put on ruxolitinib. But the question we face now is how to try and stop this medication so we can get better treatment for him which will, of course, be very difficult.”
To contribute to Dr. Zwald’s case series, please email her at [email protected].
She had no relevant financial disclosures.
On Twitter @Alz_Gal
ORLANDO – The hematologic cancer drug ruxolitinib seems to be associated with cases of aggressive nonmelanoma skin cancer.
After treating a very aggressive squamous cell carcinoma in a 55-year-old man treated with ruxolitinib for polycythemia vera, and hearing firsthand of three other similar cases, Dr. Fiona Zwald is collecting additional data on the association. She intends to publish these cases in a monograph as a warning to dermatologists, hematologists, oncologists, and other physicians who manage patients with hematologic malignancies, she said at the annual meeting of the American College of Mohs Surgery.
The prescribing information for ruxolitinib (Jakafi, Incyte Pharmaceuticals; Jakavi, Novartis) was updated in 2014 to warn that patients taking the drug face an increased risk of nonmelanoma skin cancers. The label also recommends that physicians inspect the skin regularly and urge patients to be alert for and report any new or changing lesions.
Despite the warnings and recommendations, cases are occurring – and some are quite serious, said Dr. Zwald, a Mohs surgeon in Atlanta.
“People should know this is actually happening. If you have experience with this medication, please let us know so we can compile this report. We are trying to assess the number of skin cancers before and after initiating this medication,” she said.
Ruxolitinib is an inhibitor of Janus kinase with a special affinity for the JAK1 and JAK2 subtypes. Like other cytokine-signaling molecules, their function depends on cell context; it may inhibit cell growth in one setting, and, in another, stimulate it. Ruxolitinib was initially approved in 2011 for the treatment of intermediate- and high-risk myelofibrosis, including primary myelofibrosis, post–polycythemia vera myelofibrosis, and post–essential thrombocythemia myelofibrosis.
In 2014, indications for ruxolitinib were expanded to include treatment of patients with polycythemia vera who have had an inadequate response to or are intolerant of hydroxyurea.
Dr. Zwald’s patient had a 10-year history of polycythemia vera. He was initially well controlled on the standard hydroxyurea treatment. In the meantime, he began working as a caddy at a major U.S. golf club. He developed many facial squamous cell carcinomas that were treated with excision and radiation. A year before he presented to Dr. Zwald, he stopped responding to hydroxyurea and was placed on ruxolitinib.
The patient presented with a 4-cm ulcerated lesion over part of his right temple and to the right helical crus; the lesion had developed over 3 months. Dr. Zwald consulted with the patient’s medical oncologist; treatment with ruxolitinib continued, albeit at a reduced dosage in light of recent events.
She performed Mohs surgery on the patient. It was a challenging case, she said, not the least because adequate anesthesia could not be achieved with local anesthetic. Preoperative staging showed no nodal spread.
“He did, unfortunately demonstrate a large, indurated mass located over one branch of the superficial temporal artery. At the helical crus there was an area of bound-down, fixed tumor. Knowing that I would not be able to fully resect this, I passed him on to the operating room,” Dr. Zwald said. “This tumor was found to extend down to the parotid capsule, but margins were clear.” The surgical defect was successfully repaired with a split-thickness skin graft.
The tumor recurred about 3 months later, and the patient underwent another surgery.
“This time we could not get clear surgical margins, and the tumor was approaching the external auditory meatus. Surgery was abandoned due to fears of complications to that area,” she said.
She presented the case at tumor board, during which she and her colleagues discussed adjuvant radiation. They initially abandoned this idea because he had already had so much radiation to his face. After the second surgery, they decide to proceed with radiation. “The next conversation we have will be whether to add another adjuvant therapy to treatment.”
She sent out the case and requests for feedback to the International Transplant Skin Cancer Collaborative, an 800-member consortium of dermatologists and Mohs surgeons who take care of transplant patients. She received information on three additional cases of aggressive squamous cell carcinoma (SCC) associated with ruxolitinib treatment:
• A patient with myelodysplastic syndrome with aggressive scalp SCC with cutaneous metastases.
• A patient with undifferentiated pleomorphic sarcoma of the scalp, several cutaneous SCCs.
• A patient with a myelodysplastic syndrome with in-transit metastases and explosive cutaneous SCCs. The patient has had the ruxolitinib dose reduced and may be switched to capecitabine.
Dr. Zwald noted that her patient was at risk for aggressive skin cancers for reasons in addition to ruxolitinib treatment.
“He was already immunosuppressed from his malignancy. He was on hydroxyurea, a drug that’s a cumulative phototoxin, and he’s out in the sun playing golf every day, and then was put on ruxolitinib. But the question we face now is how to try and stop this medication so we can get better treatment for him which will, of course, be very difficult.”
To contribute to Dr. Zwald’s case series, please email her at [email protected].
She had no relevant financial disclosures.
On Twitter @Alz_Gal
AT THE ACMS ANNUAL MEETING
In severe hemophilia B, rIX-FP prophylaxis gets good results with less frequent dosing
Recombinant fusion protein linking coagulation factor IX with albumin (rIX-FP) could result in a “paradigm shift” in prophylaxis regimens for patients with hemophilia B, according to researchers reporting results from a phase III trial.
At dosing intervals of up to 14 days, rIX-FP was a safe and effective factor IX (FIX) replacement product for preventing and treating bleeding episodes in an open-label study of 63 previously treated adolescents and adults with severe hemophilia B.
Compared to standard FIX products, rIX-FP is more active, has a longer half-life, and has better clearance. A study is now underway to see whether a 21-day prophylaxis regimen provides even better results than the 14-day regimen, reported Dr. Elena Santagostino of Istituto di Ricovero e Cura a Carattere Scientifico Ca’ Granda Foundation, Maggiore Hospital Policlinico, Milan, and her associates in the PROLONG-9FP Investigators Study Group.
For prophylaxis, standard FIX products are administered two times per week. Patients are at increased risk of bleeding when their FIX activity is low, particularly just before their next scheduled dose. With the prolonged FIX activity of rIX-FP, the dosing is less frequent and that could potentially improve adherence, the researchers wrote (Blood. 2016;127[14]:1761-9).
In addition, rIX-FP was associated with a reduced risk of spontaneous and trauma-related bleeding episodes.
The findings came from a study of patients who had FIX activity levels of 2% or less and were assigned to one of two groups. The first group received routine prophylaxis once every 7 days for 26 weeks; they were then placed on either a 7-, 10-, or 14-day prophylaxis regimen for a mean of 50, 38, or 51 weeks, respectively. The second group received on-demand treatment of bleeding episodes for 26 weeks and were then switched to a 7-day prophylaxis regimen for a mean of 45 weeks. Only patients who were previously receiving on-demand treatment were eligible for group 2 assignment.
Patients self-administered rIX-FP for routine prophylaxis and on-demand treatment of bleeding episodes; all home administrations were recorded in an electronic diary. A second dose of rIX-FP was administered at least 24 hours after the first injection, if needed, to achieve hemostasis. Efficacy and safety assessments were performed at study sites on a monthly basis.
The study design controlled for the variability of bleeding frequency within the hemophilia B patient population, as group 2 patients started rIX-FP treatment on-demand and continued on 7-day prophylaxis. During the on-demand phase, rIX-FP controlled 98.6% of bleeding episodes, and 93.6% of bleeds were controlled with one infusion.
Once the group was switched to 7-day prophylaxis, the median annualized spontaneous bleeding rate dropped to 0.0 and all target joints resolved.(P less than .0001). No patient developed an inhibitor, and no safety concerns were identified.
The annualized bleeding rate decreased slightly in group 2 patients during on-demand treatment with rIX-FP, compared with the rate in the 12-month period before they entered the study. The researchers speculated that on-demand treatment with rIX-FP “might provide, to a certain extent, some protection against a subsequent bleeding episode. Therefore, patients with a severe bleeding phenotype treated on-demand might experience fewer bleeding episodes if switched to rIX-FP. Furthermore, such on-demand patients who require more than one infusion per month may have the benefit of a reduction of ABR by administering a similar number of infusions according to a 14-day prophylaxis regimen with rIX-FP.”
The sample size was small, however, and differences of care in the clinical study could have affected the results.
Compared to other FIX products, rIX-FP had an extended terminal half-life of 102 hours, 4.3-fold longer than seen with the patients’ previous FIX treatment. Other pharmacokinetic measures such as area under the curve (7176 h × IU/dL), clearance (0.77 mL/h per kg), and incremental recovery (1.27 IU/dL per IU/kg) also were improved. Patients maintained a mean trough of 20 and 12 IU/dL FIX activity on prophylaxis with rIX-FP 40 IU/kg every 7 days and 75 IU/kg every 14 days, respectively. Rather than half-life alone, a slower clearance may be a factor in the success of prophylaxis regimens, the researchers said.
The principal investigators received research support from CSL Behring, which sponsored the study and was responsible for trial operations and data analysis.
On Twitter @maryjodales
Recombinant fusion protein linking coagulation factor IX with albumin (rIX-FP) could result in a “paradigm shift” in prophylaxis regimens for patients with hemophilia B, according to researchers reporting results from a phase III trial.
At dosing intervals of up to 14 days, rIX-FP was a safe and effective factor IX (FIX) replacement product for preventing and treating bleeding episodes in an open-label study of 63 previously treated adolescents and adults with severe hemophilia B.
Compared to standard FIX products, rIX-FP is more active, has a longer half-life, and has better clearance. A study is now underway to see whether a 21-day prophylaxis regimen provides even better results than the 14-day regimen, reported Dr. Elena Santagostino of Istituto di Ricovero e Cura a Carattere Scientifico Ca’ Granda Foundation, Maggiore Hospital Policlinico, Milan, and her associates in the PROLONG-9FP Investigators Study Group.
For prophylaxis, standard FIX products are administered two times per week. Patients are at increased risk of bleeding when their FIX activity is low, particularly just before their next scheduled dose. With the prolonged FIX activity of rIX-FP, the dosing is less frequent and that could potentially improve adherence, the researchers wrote (Blood. 2016;127[14]:1761-9).
In addition, rIX-FP was associated with a reduced risk of spontaneous and trauma-related bleeding episodes.
The findings came from a study of patients who had FIX activity levels of 2% or less and were assigned to one of two groups. The first group received routine prophylaxis once every 7 days for 26 weeks; they were then placed on either a 7-, 10-, or 14-day prophylaxis regimen for a mean of 50, 38, or 51 weeks, respectively. The second group received on-demand treatment of bleeding episodes for 26 weeks and were then switched to a 7-day prophylaxis regimen for a mean of 45 weeks. Only patients who were previously receiving on-demand treatment were eligible for group 2 assignment.
Patients self-administered rIX-FP for routine prophylaxis and on-demand treatment of bleeding episodes; all home administrations were recorded in an electronic diary. A second dose of rIX-FP was administered at least 24 hours after the first injection, if needed, to achieve hemostasis. Efficacy and safety assessments were performed at study sites on a monthly basis.
The study design controlled for the variability of bleeding frequency within the hemophilia B patient population, as group 2 patients started rIX-FP treatment on-demand and continued on 7-day prophylaxis. During the on-demand phase, rIX-FP controlled 98.6% of bleeding episodes, and 93.6% of bleeds were controlled with one infusion.
Once the group was switched to 7-day prophylaxis, the median annualized spontaneous bleeding rate dropped to 0.0 and all target joints resolved.(P less than .0001). No patient developed an inhibitor, and no safety concerns were identified.
The annualized bleeding rate decreased slightly in group 2 patients during on-demand treatment with rIX-FP, compared with the rate in the 12-month period before they entered the study. The researchers speculated that on-demand treatment with rIX-FP “might provide, to a certain extent, some protection against a subsequent bleeding episode. Therefore, patients with a severe bleeding phenotype treated on-demand might experience fewer bleeding episodes if switched to rIX-FP. Furthermore, such on-demand patients who require more than one infusion per month may have the benefit of a reduction of ABR by administering a similar number of infusions according to a 14-day prophylaxis regimen with rIX-FP.”
The sample size was small, however, and differences of care in the clinical study could have affected the results.
Compared to other FIX products, rIX-FP had an extended terminal half-life of 102 hours, 4.3-fold longer than seen with the patients’ previous FIX treatment. Other pharmacokinetic measures such as area under the curve (7176 h × IU/dL), clearance (0.77 mL/h per kg), and incremental recovery (1.27 IU/dL per IU/kg) also were improved. Patients maintained a mean trough of 20 and 12 IU/dL FIX activity on prophylaxis with rIX-FP 40 IU/kg every 7 days and 75 IU/kg every 14 days, respectively. Rather than half-life alone, a slower clearance may be a factor in the success of prophylaxis regimens, the researchers said.
The principal investigators received research support from CSL Behring, which sponsored the study and was responsible for trial operations and data analysis.
On Twitter @maryjodales
Recombinant fusion protein linking coagulation factor IX with albumin (rIX-FP) could result in a “paradigm shift” in prophylaxis regimens for patients with hemophilia B, according to researchers reporting results from a phase III trial.
At dosing intervals of up to 14 days, rIX-FP was a safe and effective factor IX (FIX) replacement product for preventing and treating bleeding episodes in an open-label study of 63 previously treated adolescents and adults with severe hemophilia B.
Compared to standard FIX products, rIX-FP is more active, has a longer half-life, and has better clearance. A study is now underway to see whether a 21-day prophylaxis regimen provides even better results than the 14-day regimen, reported Dr. Elena Santagostino of Istituto di Ricovero e Cura a Carattere Scientifico Ca’ Granda Foundation, Maggiore Hospital Policlinico, Milan, and her associates in the PROLONG-9FP Investigators Study Group.
For prophylaxis, standard FIX products are administered two times per week. Patients are at increased risk of bleeding when their FIX activity is low, particularly just before their next scheduled dose. With the prolonged FIX activity of rIX-FP, the dosing is less frequent and that could potentially improve adherence, the researchers wrote (Blood. 2016;127[14]:1761-9).
In addition, rIX-FP was associated with a reduced risk of spontaneous and trauma-related bleeding episodes.
The findings came from a study of patients who had FIX activity levels of 2% or less and were assigned to one of two groups. The first group received routine prophylaxis once every 7 days for 26 weeks; they were then placed on either a 7-, 10-, or 14-day prophylaxis regimen for a mean of 50, 38, or 51 weeks, respectively. The second group received on-demand treatment of bleeding episodes for 26 weeks and were then switched to a 7-day prophylaxis regimen for a mean of 45 weeks. Only patients who were previously receiving on-demand treatment were eligible for group 2 assignment.
Patients self-administered rIX-FP for routine prophylaxis and on-demand treatment of bleeding episodes; all home administrations were recorded in an electronic diary. A second dose of rIX-FP was administered at least 24 hours after the first injection, if needed, to achieve hemostasis. Efficacy and safety assessments were performed at study sites on a monthly basis.
The study design controlled for the variability of bleeding frequency within the hemophilia B patient population, as group 2 patients started rIX-FP treatment on-demand and continued on 7-day prophylaxis. During the on-demand phase, rIX-FP controlled 98.6% of bleeding episodes, and 93.6% of bleeds were controlled with one infusion.
Once the group was switched to 7-day prophylaxis, the median annualized spontaneous bleeding rate dropped to 0.0 and all target joints resolved.(P less than .0001). No patient developed an inhibitor, and no safety concerns were identified.
The annualized bleeding rate decreased slightly in group 2 patients during on-demand treatment with rIX-FP, compared with the rate in the 12-month period before they entered the study. The researchers speculated that on-demand treatment with rIX-FP “might provide, to a certain extent, some protection against a subsequent bleeding episode. Therefore, patients with a severe bleeding phenotype treated on-demand might experience fewer bleeding episodes if switched to rIX-FP. Furthermore, such on-demand patients who require more than one infusion per month may have the benefit of a reduction of ABR by administering a similar number of infusions according to a 14-day prophylaxis regimen with rIX-FP.”
The sample size was small, however, and differences of care in the clinical study could have affected the results.
Compared to other FIX products, rIX-FP had an extended terminal half-life of 102 hours, 4.3-fold longer than seen with the patients’ previous FIX treatment. Other pharmacokinetic measures such as area under the curve (7176 h × IU/dL), clearance (0.77 mL/h per kg), and incremental recovery (1.27 IU/dL per IU/kg) also were improved. Patients maintained a mean trough of 20 and 12 IU/dL FIX activity on prophylaxis with rIX-FP 40 IU/kg every 7 days and 75 IU/kg every 14 days, respectively. Rather than half-life alone, a slower clearance may be a factor in the success of prophylaxis regimens, the researchers said.
The principal investigators received research support from CSL Behring, which sponsored the study and was responsible for trial operations and data analysis.
On Twitter @maryjodales
FROM BLOOD
Key clinical point: Weekly and 14-day prophylaxis regimens with rIX-FP were well tolerated and provided low bleeding rates and target joint improvement.
Major finding: On 7-day prophylaxis, the median annualized spontaneous bleeding rate dropped to 0.0 and all target joints resolved (P less than .0001).
Data source: An open-label study of 63 previously treated adolescents and adults with severe hemophilia B.
Disclosures: The principal investigators received research support from CSL Behring, which sponsored the study and was responsible for trial operations and data analysis.
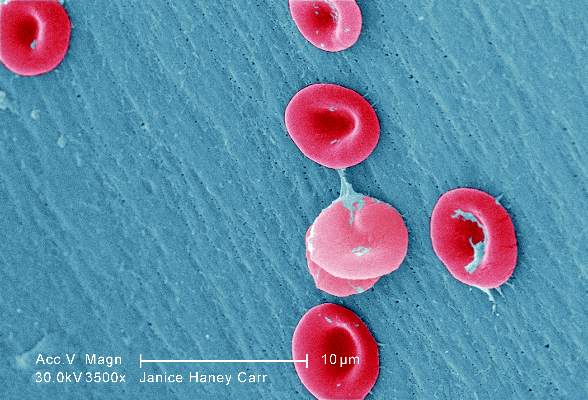
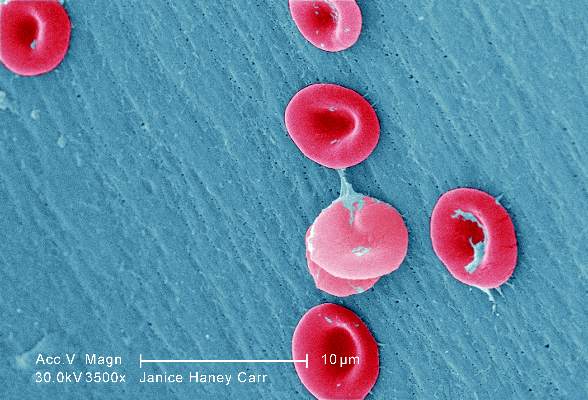
Sickle cell anemia: Stroke screening still underused
Children and adolescents with sickle cell anemia still are not being screened for stroke risk using transcranial Doppler, despite clinical guidelines that strongly recommend annual screening and despite these patients’ frequent health care encounters, according to a report published online April 11 in JAMA Pediatrics.
Approximately 10% of children and adolescents with sickle cell anemia experience stroke before the age of 20 years, unless those at high risk are identified and treated preemptively with blood transfusions, which reduces stroke risk by 92%. The National Heart, Lung, and Blood Institute clinical practice guideline on treating sickle cell disease calls for patients aged 2-16 years to undergo transcranial Doppler every year to detect any elevated velocity of cerebral blood flow, which indicates high stroke risk, said Sarah L. Reeves, Ph.D., of the Child Health Evaluation and Research Unit, University of Michigan, Ann Arbor, and her associates.
To assess screening rates, the investigators performed a retrospective cross-sectional analysis of administrative claims data for 4,775 affected children and adolescents treated during a 5-year period in Florida, Illinois, Louisiana, Michigan, South Carolina, and Texas. This yielded 10,787 person-years of data.
Overall, screening rates increased somewhat across all six states during the study period – from 22% to 44% – but “even the highest rates we report are suboptimal,” Dr. Reeves and her associates noted (JAMA Ped. 2016 Apr 11. doi: 10.1001/jamapediatrics.2015.4859).
This is especially true given that the average patient had 20.0 disease-related outpatient visits, 2.1 disease-related hospitalizations, 3.7 emergency department visits, and 1 well-child visit each year – numerous missed opportunities when they could have been referred for screening.
One way to improve screening rates would be to integrate transcranial Doppler exams into comprehensive sickle-cell healthcare, rather than requiring separate scheduled appointments at imaging facilities, they added.
This study was funded by the Agency for Healthcare Research and Quality and the Centers for Medicare & Medicaid Services. Dr. Reeves and her associates reported having no relevant financial disclosures.
Children and adolescents with sickle cell anemia still are not being screened for stroke risk using transcranial Doppler, despite clinical guidelines that strongly recommend annual screening and despite these patients’ frequent health care encounters, according to a report published online April 11 in JAMA Pediatrics.
Approximately 10% of children and adolescents with sickle cell anemia experience stroke before the age of 20 years, unless those at high risk are identified and treated preemptively with blood transfusions, which reduces stroke risk by 92%. The National Heart, Lung, and Blood Institute clinical practice guideline on treating sickle cell disease calls for patients aged 2-16 years to undergo transcranial Doppler every year to detect any elevated velocity of cerebral blood flow, which indicates high stroke risk, said Sarah L. Reeves, Ph.D., of the Child Health Evaluation and Research Unit, University of Michigan, Ann Arbor, and her associates.
To assess screening rates, the investigators performed a retrospective cross-sectional analysis of administrative claims data for 4,775 affected children and adolescents treated during a 5-year period in Florida, Illinois, Louisiana, Michigan, South Carolina, and Texas. This yielded 10,787 person-years of data.
Overall, screening rates increased somewhat across all six states during the study period – from 22% to 44% – but “even the highest rates we report are suboptimal,” Dr. Reeves and her associates noted (JAMA Ped. 2016 Apr 11. doi: 10.1001/jamapediatrics.2015.4859).
This is especially true given that the average patient had 20.0 disease-related outpatient visits, 2.1 disease-related hospitalizations, 3.7 emergency department visits, and 1 well-child visit each year – numerous missed opportunities when they could have been referred for screening.
One way to improve screening rates would be to integrate transcranial Doppler exams into comprehensive sickle-cell healthcare, rather than requiring separate scheduled appointments at imaging facilities, they added.
This study was funded by the Agency for Healthcare Research and Quality and the Centers for Medicare & Medicaid Services. Dr. Reeves and her associates reported having no relevant financial disclosures.
Children and adolescents with sickle cell anemia still are not being screened for stroke risk using transcranial Doppler, despite clinical guidelines that strongly recommend annual screening and despite these patients’ frequent health care encounters, according to a report published online April 11 in JAMA Pediatrics.
Approximately 10% of children and adolescents with sickle cell anemia experience stroke before the age of 20 years, unless those at high risk are identified and treated preemptively with blood transfusions, which reduces stroke risk by 92%. The National Heart, Lung, and Blood Institute clinical practice guideline on treating sickle cell disease calls for patients aged 2-16 years to undergo transcranial Doppler every year to detect any elevated velocity of cerebral blood flow, which indicates high stroke risk, said Sarah L. Reeves, Ph.D., of the Child Health Evaluation and Research Unit, University of Michigan, Ann Arbor, and her associates.
To assess screening rates, the investigators performed a retrospective cross-sectional analysis of administrative claims data for 4,775 affected children and adolescents treated during a 5-year period in Florida, Illinois, Louisiana, Michigan, South Carolina, and Texas. This yielded 10,787 person-years of data.
Overall, screening rates increased somewhat across all six states during the study period – from 22% to 44% – but “even the highest rates we report are suboptimal,” Dr. Reeves and her associates noted (JAMA Ped. 2016 Apr 11. doi: 10.1001/jamapediatrics.2015.4859).
This is especially true given that the average patient had 20.0 disease-related outpatient visits, 2.1 disease-related hospitalizations, 3.7 emergency department visits, and 1 well-child visit each year – numerous missed opportunities when they could have been referred for screening.
One way to improve screening rates would be to integrate transcranial Doppler exams into comprehensive sickle-cell healthcare, rather than requiring separate scheduled appointments at imaging facilities, they added.
This study was funded by the Agency for Healthcare Research and Quality and the Centers for Medicare & Medicaid Services. Dr. Reeves and her associates reported having no relevant financial disclosures.
FROM JAMA PEDIATRICS
Key clinical point: Transcranial Doppler screening for stroke risk is still underused among children and adolescents with sickle cell anemia, despite clinical guidelines that strongly recommend annual screening.
Major finding: Screening rates increased somewhat across all six states studied, from 22% to 44%, but even the highest rates were suboptimal.
Data source: A retrospective cross-sectional analysis of administrative claims data for 4,775 pediatric patients treated in a 5-year period.
Disclosures: This study was funded by the Agency for Healthcare Research and Quality and the Centers for Medicare & Medicaid Services. Dr. Reeves and her associates reported having no relevant financial disclosures.
Colombia reports first Zika deaths, all in medically compromised patients
AMSTERDAM – Five people with confirmed Zika virus infections have died in Colombia, and all had medical comorbidities, including leukemia, diabetes, sickle cell anemia, and hypertension.
All of the deaths occurred last October in northern and central Colombia, Dr. Alfonso Rodriguez-Morales said at the European Society of Clinical Microbiology and Infectious Diseases annual congress.
Four of the cases were simultaneously published April 7 in the Lancet Infectious Diseases (2016 Apr 7. doi: 10.1016/S1473-3099[16]30006-8). The fifth case occurred in northern Colombia, and was reported in Emerging Infectious Diseases (2016 May. doi: 10.3201/eid2205.151934).
Reports of confirmed Zika-related deaths are rare. Brazil, the only other country to disclose them, has now reported three, said Dr. Rodriguez-Morales of the Universidad Tecnológica de Pereira, Colombia.

“Before the current outbreak in Latin America, Zika virus was not linked to deaths,” he noted. But the eight confirmed Zika-related deaths in South America “call attention to the need for evidence-based guidelines for clinical management of Zika, as well as the possible occurrence of atypical and severe cases, including possibly congenitally related microcephaly.”
Because they all occurred in medically compromised patients, Dr. Rodriguez-Morales also urged clinicians to cast a wary eye on such patients who present with arbovirus-type symptoms, including fever and rash.
From September 2015 to March 2016, Colombia had 58,838 reported cases of Zika. Of those, only 2,361 were lab confirmed. The rest were either diagnosed clinically or were suspected cases, Dr. Rodriguez-Morales said. Although Colombia has a much smaller population than Brazil (49 million vs. 210 million), its Zika case rate is much higher, 120 cases per 100,000 people vs. 34 cases per 100,000 people.
The group of four deaths occurred in central Colombia, and included a 2-year-old girl, a 30-year-old woman, a 61-year-old man, and a 72-year-old woman. All presented with 2-6 days of fever. All were initially suspected to have dengue fever or chikungunya. None tested positive for dengue, but the man was coinfected with chikungunya.
All patients presented with anemia. All but the older man also had severe thrombocytopenia.
The toddler presented with hepatomegaly, mucosal hemorrhage, progressive respiratory collapse, progressive thrombocytopenia, and intravascular coagulation. She died 5 days after symptom onset and was found to have had unrecognized lymphoblastic leukemia.
The 30-year-old woman presented with a severe rash on both arms. She also exhibited coagulation dysfunction, including severe thrombocytopenia and leukopenia that progressed to intracerebral and subarachnoid hemorrhage. She died 12 days after symptom onset. She was determined to have had unrecognized acute myeloid leukemia.
The elderly man had a history of medically controlled hypertension. He experienced mucosal hemorrhage and respiratory distress. He died 7 days after symptom onset. On autopsy, his liver showed necrotic areas, and his spleen indicated a systemic inflammatory response.
The elderly woman had a history of insulin-controlled type 2 diabetes. Her symptoms included gastrointestinal distress, thrombocytopenia, and acute respiratory failure. She died 48 hours after symptom onset; her brain showed edema and ischemic lesions.
The 15-year-old girl in northern Colombia had a 5-year history of sickle cell disease, which, Dr. Rodriguez-Morales pointed out, is a risk factor for arbovirus diseases. However, the patient had never been hospitalized for a vasoocclusive crisis. She presented with a high fever; joint, muscle, and abdominal pain; and jaundice. She was assumed to have dengue virus. Within another day, she had progressed into respiratory failure and was on a ventilator. She died less than 2 days later.
Her autopsy showed hepatic necrosis and severe decrease of splenic lymphoid tissue with splenic sequestration. Systemic inflammation probably triggered a fatal vasoocclusive crisis and splenic sequestration.
Dr. Rodriguez-Morales had no financial disclosures.
AMSTERDAM – Five people with confirmed Zika virus infections have died in Colombia, and all had medical comorbidities, including leukemia, diabetes, sickle cell anemia, and hypertension.
All of the deaths occurred last October in northern and central Colombia, Dr. Alfonso Rodriguez-Morales said at the European Society of Clinical Microbiology and Infectious Diseases annual congress.
Four of the cases were simultaneously published April 7 in the Lancet Infectious Diseases (2016 Apr 7. doi: 10.1016/S1473-3099[16]30006-8). The fifth case occurred in northern Colombia, and was reported in Emerging Infectious Diseases (2016 May. doi: 10.3201/eid2205.151934).
Reports of confirmed Zika-related deaths are rare. Brazil, the only other country to disclose them, has now reported three, said Dr. Rodriguez-Morales of the Universidad Tecnológica de Pereira, Colombia.
“Before the current outbreak in Latin America, Zika virus was not linked to deaths,” he noted. But the eight confirmed Zika-related deaths in South America “call attention to the need for evidence-based guidelines for clinical management of Zika, as well as the possible occurrence of atypical and severe cases, including possibly congenitally related microcephaly.”
Because they all occurred in medically compromised patients, Dr. Rodriguez-Morales also urged clinicians to cast a wary eye on such patients who present with arbovirus-type symptoms, including fever and rash.
From September 2015 to March 2016, Colombia had 58,838 reported cases of Zika. Of those, only 2,361 were lab confirmed. The rest were either diagnosed clinically or were suspected cases, Dr. Rodriguez-Morales said. Although Colombia has a much smaller population than Brazil (49 million vs. 210 million), its Zika case rate is much higher, 120 cases per 100,000 people vs. 34 cases per 100,000 people.
The group of four deaths occurred in central Colombia, and included a 2-year-old girl, a 30-year-old woman, a 61-year-old man, and a 72-year-old woman. All presented with 2-6 days of fever. All were initially suspected to have dengue fever or chikungunya. None tested positive for dengue, but the man was coinfected with chikungunya.
All patients presented with anemia. All but the older man also had severe thrombocytopenia.
The toddler presented with hepatomegaly, mucosal hemorrhage, progressive respiratory collapse, progressive thrombocytopenia, and intravascular coagulation. She died 5 days after symptom onset and was found to have had unrecognized lymphoblastic leukemia.
The 30-year-old woman presented with a severe rash on both arms. She also exhibited coagulation dysfunction, including severe thrombocytopenia and leukopenia that progressed to intracerebral and subarachnoid hemorrhage. She died 12 days after symptom onset. She was determined to have had unrecognized acute myeloid leukemia.
The elderly man had a history of medically controlled hypertension. He experienced mucosal hemorrhage and respiratory distress. He died 7 days after symptom onset. On autopsy, his liver showed necrotic areas, and his spleen indicated a systemic inflammatory response.
The elderly woman had a history of insulin-controlled type 2 diabetes. Her symptoms included gastrointestinal distress, thrombocytopenia, and acute respiratory failure. She died 48 hours after symptom onset; her brain showed edema and ischemic lesions.
The 15-year-old girl in northern Colombia had a 5-year history of sickle cell disease, which, Dr. Rodriguez-Morales pointed out, is a risk factor for arbovirus diseases. However, the patient had never been hospitalized for a vasoocclusive crisis. She presented with a high fever; joint, muscle, and abdominal pain; and jaundice. She was assumed to have dengue virus. Within another day, she had progressed into respiratory failure and was on a ventilator. She died less than 2 days later.
Her autopsy showed hepatic necrosis and severe decrease of splenic lymphoid tissue with splenic sequestration. Systemic inflammation probably triggered a fatal vasoocclusive crisis and splenic sequestration.
Dr. Rodriguez-Morales had no financial disclosures.
AMSTERDAM – Five people with confirmed Zika virus infections have died in Colombia, and all had medical comorbidities, including leukemia, diabetes, sickle cell anemia, and hypertension.
All of the deaths occurred last October in northern and central Colombia, Dr. Alfonso Rodriguez-Morales said at the European Society of Clinical Microbiology and Infectious Diseases annual congress.
Four of the cases were simultaneously published April 7 in the Lancet Infectious Diseases (2016 Apr 7. doi: 10.1016/S1473-3099[16]30006-8). The fifth case occurred in northern Colombia, and was reported in Emerging Infectious Diseases (2016 May. doi: 10.3201/eid2205.151934).
Reports of confirmed Zika-related deaths are rare. Brazil, the only other country to disclose them, has now reported three, said Dr. Rodriguez-Morales of the Universidad Tecnológica de Pereira, Colombia.
“Before the current outbreak in Latin America, Zika virus was not linked to deaths,” he noted. But the eight confirmed Zika-related deaths in South America “call attention to the need for evidence-based guidelines for clinical management of Zika, as well as the possible occurrence of atypical and severe cases, including possibly congenitally related microcephaly.”
Because they all occurred in medically compromised patients, Dr. Rodriguez-Morales also urged clinicians to cast a wary eye on such patients who present with arbovirus-type symptoms, including fever and rash.
From September 2015 to March 2016, Colombia had 58,838 reported cases of Zika. Of those, only 2,361 were lab confirmed. The rest were either diagnosed clinically or were suspected cases, Dr. Rodriguez-Morales said. Although Colombia has a much smaller population than Brazil (49 million vs. 210 million), its Zika case rate is much higher, 120 cases per 100,000 people vs. 34 cases per 100,000 people.
The group of four deaths occurred in central Colombia, and included a 2-year-old girl, a 30-year-old woman, a 61-year-old man, and a 72-year-old woman. All presented with 2-6 days of fever. All were initially suspected to have dengue fever or chikungunya. None tested positive for dengue, but the man was coinfected with chikungunya.
All patients presented with anemia. All but the older man also had severe thrombocytopenia.
The toddler presented with hepatomegaly, mucosal hemorrhage, progressive respiratory collapse, progressive thrombocytopenia, and intravascular coagulation. She died 5 days after symptom onset and was found to have had unrecognized lymphoblastic leukemia.
The 30-year-old woman presented with a severe rash on both arms. She also exhibited coagulation dysfunction, including severe thrombocytopenia and leukopenia that progressed to intracerebral and subarachnoid hemorrhage. She died 12 days after symptom onset. She was determined to have had unrecognized acute myeloid leukemia.
The elderly man had a history of medically controlled hypertension. He experienced mucosal hemorrhage and respiratory distress. He died 7 days after symptom onset. On autopsy, his liver showed necrotic areas, and his spleen indicated a systemic inflammatory response.
The elderly woman had a history of insulin-controlled type 2 diabetes. Her symptoms included gastrointestinal distress, thrombocytopenia, and acute respiratory failure. She died 48 hours after symptom onset; her brain showed edema and ischemic lesions.
The 15-year-old girl in northern Colombia had a 5-year history of sickle cell disease, which, Dr. Rodriguez-Morales pointed out, is a risk factor for arbovirus diseases. However, the patient had never been hospitalized for a vasoocclusive crisis. She presented with a high fever; joint, muscle, and abdominal pain; and jaundice. She was assumed to have dengue virus. Within another day, she had progressed into respiratory failure and was on a ventilator. She died less than 2 days later.
Her autopsy showed hepatic necrosis and severe decrease of splenic lymphoid tissue with splenic sequestration. Systemic inflammation probably triggered a fatal vasoocclusive crisis and splenic sequestration.
Dr. Rodriguez-Morales had no financial disclosures.
AT ECCMID 2016

Factor VIII–mimetic antibody effective in hemophilia A
The bispecific factor VIII–mimetic antibody ACE910 has a longer half-life than current hemophilia A treatments, potentially offering a more convenient once-weekly, subcutaneous injection, based on a study published in Blood.
In a phase I study, ACE910 was well tolerated at doses up to 1 mg/kg, with an average half life of 28 to 34 days. Based on tests in FVIII-depleted plasma, ACE910 shortened activated partial thromboplastin time (APTT) and increased peak height of thrombin generation, and exhibited a long-lasting response throughout the 24-week study period. ACE910 at 1 mg/kg resulted in APTT similar to that seen in normal plasma, although the peak height of thrombin generation did not reach normal levels.
The findings suggest “that ACE910 has the potential to reduce bleeding frequency in patients with severe hemophilia A to that of patients with mild hemophilia A, even at less frequent dosing, compared with existing FVIII and bypassing drugs. Furthermore, ACE910 may change the treatment paradigm from the current approach of maintaining trough levels of FVIII:C greater than 1% to a new approach of maintaining a constant hemostatic activity corresponding to a mild hemophilia A level,” wrote Dr. Naoki Uchida of Showa University Clinical Research Institute for Clinical Pharmacology and Therapeutics, Tokyo, and colleagues (Blood. 2016 Apr 7. doi: 10.1182/blood-2015-06-650226).
Adverse events were comparable with those of placebo; 13 of 48 subjects who received ACE910 reported 15 adverse events, compared with 6 adverse events reported by 4 of 16 subjects who received placebo. Except for moderate nasopharyngitis reported in one subject, all adverse events were mild, were not dose dependent, and were similar for Japanese and white subjects. Clinical and laboratory findings showed normal coagulability with ACE910 administered at any dose.
An anti-drug antibody response was observed in 2 of 48 patients (1 Japanese, 1 white) both at 0.1 mg/kg ACE910. The anti-drug antibody responses were not IgE mediated, and no allergic symptoms were observed.
This new nonsubstitutive therapy for hemophilia A has potential to become a disruptive technology that displaces the current therapeutic approach.
By binding to factor IXa and factor X, ACE910 bypasses factor VIII in the generation of factor Xa. Since ACE910 is a distinct entity from factor VIII, anti-FVIII antibodies do not neutralize the agent.
Infusion of recombinant factor VIII has an average half-life of 8 to 12 hours, or 1.5 to 1.7 hours longer with the recent introduction of extended half-life products. Patients who develop factor VIII allo- or autoantibodies require frequent, less effective intravenous therapies. ACE910 switches up the paradigm, with repeat doses given subcutaneously once per week.
Of concern is the potential for thrombosis and anti-drug antibodies (ADAs). Despite the long half-life of ACE910 and that the phase I study was conducted on healthy volunteers with normal coagulation systems, investigators observed no rise in D-dimer formation. Similarly, no rise in thrombin-antithrombin complex was observed.
Two of 48 subjects had anti-ACE910 antibodies, and in one of these subjects ACE910 half-life was reduced, as was APTT correction and thrombin generation. These findings suggest a functional ADA. A key issue for future studies will be the number of individuals who develop ADAs after repeated exposure to ACE910.
Dr. Michael Makris is professor of haemostasis and thrombosis at the University of Sheffield (England). These remarks were part of an editorial accompanying a report in Blood (2016 Apr 7. doi: 10.1182/blood-2016-01-691469).
This new nonsubstitutive therapy for hemophilia A has potential to become a disruptive technology that displaces the current therapeutic approach.
By binding to factor IXa and factor X, ACE910 bypasses factor VIII in the generation of factor Xa. Since ACE910 is a distinct entity from factor VIII, anti-FVIII antibodies do not neutralize the agent.
Infusion of recombinant factor VIII has an average half-life of 8 to 12 hours, or 1.5 to 1.7 hours longer with the recent introduction of extended half-life products. Patients who develop factor VIII allo- or autoantibodies require frequent, less effective intravenous therapies. ACE910 switches up the paradigm, with repeat doses given subcutaneously once per week.
Of concern is the potential for thrombosis and anti-drug antibodies (ADAs). Despite the long half-life of ACE910 and that the phase I study was conducted on healthy volunteers with normal coagulation systems, investigators observed no rise in D-dimer formation. Similarly, no rise in thrombin-antithrombin complex was observed.
Two of 48 subjects had anti-ACE910 antibodies, and in one of these subjects ACE910 half-life was reduced, as was APTT correction and thrombin generation. These findings suggest a functional ADA. A key issue for future studies will be the number of individuals who develop ADAs after repeated exposure to ACE910.
Dr. Michael Makris is professor of haemostasis and thrombosis at the University of Sheffield (England). These remarks were part of an editorial accompanying a report in Blood (2016 Apr 7. doi: 10.1182/blood-2016-01-691469).
This new nonsubstitutive therapy for hemophilia A has potential to become a disruptive technology that displaces the current therapeutic approach.
By binding to factor IXa and factor X, ACE910 bypasses factor VIII in the generation of factor Xa. Since ACE910 is a distinct entity from factor VIII, anti-FVIII antibodies do not neutralize the agent.
Infusion of recombinant factor VIII has an average half-life of 8 to 12 hours, or 1.5 to 1.7 hours longer with the recent introduction of extended half-life products. Patients who develop factor VIII allo- or autoantibodies require frequent, less effective intravenous therapies. ACE910 switches up the paradigm, with repeat doses given subcutaneously once per week.
Of concern is the potential for thrombosis and anti-drug antibodies (ADAs). Despite the long half-life of ACE910 and that the phase I study was conducted on healthy volunteers with normal coagulation systems, investigators observed no rise in D-dimer formation. Similarly, no rise in thrombin-antithrombin complex was observed.
Two of 48 subjects had anti-ACE910 antibodies, and in one of these subjects ACE910 half-life was reduced, as was APTT correction and thrombin generation. These findings suggest a functional ADA. A key issue for future studies will be the number of individuals who develop ADAs after repeated exposure to ACE910.
Dr. Michael Makris is professor of haemostasis and thrombosis at the University of Sheffield (England). These remarks were part of an editorial accompanying a report in Blood (2016 Apr 7. doi: 10.1182/blood-2016-01-691469).
The bispecific factor VIII–mimetic antibody ACE910 has a longer half-life than current hemophilia A treatments, potentially offering a more convenient once-weekly, subcutaneous injection, based on a study published in Blood.
In a phase I study, ACE910 was well tolerated at doses up to 1 mg/kg, with an average half life of 28 to 34 days. Based on tests in FVIII-depleted plasma, ACE910 shortened activated partial thromboplastin time (APTT) and increased peak height of thrombin generation, and exhibited a long-lasting response throughout the 24-week study period. ACE910 at 1 mg/kg resulted in APTT similar to that seen in normal plasma, although the peak height of thrombin generation did not reach normal levels.
The findings suggest “that ACE910 has the potential to reduce bleeding frequency in patients with severe hemophilia A to that of patients with mild hemophilia A, even at less frequent dosing, compared with existing FVIII and bypassing drugs. Furthermore, ACE910 may change the treatment paradigm from the current approach of maintaining trough levels of FVIII:C greater than 1% to a new approach of maintaining a constant hemostatic activity corresponding to a mild hemophilia A level,” wrote Dr. Naoki Uchida of Showa University Clinical Research Institute for Clinical Pharmacology and Therapeutics, Tokyo, and colleagues (Blood. 2016 Apr 7. doi: 10.1182/blood-2015-06-650226).
Adverse events were comparable with those of placebo; 13 of 48 subjects who received ACE910 reported 15 adverse events, compared with 6 adverse events reported by 4 of 16 subjects who received placebo. Except for moderate nasopharyngitis reported in one subject, all adverse events were mild, were not dose dependent, and were similar for Japanese and white subjects. Clinical and laboratory findings showed normal coagulability with ACE910 administered at any dose.
An anti-drug antibody response was observed in 2 of 48 patients (1 Japanese, 1 white) both at 0.1 mg/kg ACE910. The anti-drug antibody responses were not IgE mediated, and no allergic symptoms were observed.
The bispecific factor VIII–mimetic antibody ACE910 has a longer half-life than current hemophilia A treatments, potentially offering a more convenient once-weekly, subcutaneous injection, based on a study published in Blood.
In a phase I study, ACE910 was well tolerated at doses up to 1 mg/kg, with an average half life of 28 to 34 days. Based on tests in FVIII-depleted plasma, ACE910 shortened activated partial thromboplastin time (APTT) and increased peak height of thrombin generation, and exhibited a long-lasting response throughout the 24-week study period. ACE910 at 1 mg/kg resulted in APTT similar to that seen in normal plasma, although the peak height of thrombin generation did not reach normal levels.
The findings suggest “that ACE910 has the potential to reduce bleeding frequency in patients with severe hemophilia A to that of patients with mild hemophilia A, even at less frequent dosing, compared with existing FVIII and bypassing drugs. Furthermore, ACE910 may change the treatment paradigm from the current approach of maintaining trough levels of FVIII:C greater than 1% to a new approach of maintaining a constant hemostatic activity corresponding to a mild hemophilia A level,” wrote Dr. Naoki Uchida of Showa University Clinical Research Institute for Clinical Pharmacology and Therapeutics, Tokyo, and colleagues (Blood. 2016 Apr 7. doi: 10.1182/blood-2015-06-650226).
Adverse events were comparable with those of placebo; 13 of 48 subjects who received ACE910 reported 15 adverse events, compared with 6 adverse events reported by 4 of 16 subjects who received placebo. Except for moderate nasopharyngitis reported in one subject, all adverse events were mild, were not dose dependent, and were similar for Japanese and white subjects. Clinical and laboratory findings showed normal coagulability with ACE910 administered at any dose.
An anti-drug antibody response was observed in 2 of 48 patients (1 Japanese, 1 white) both at 0.1 mg/kg ACE910. The anti-drug antibody responses were not IgE mediated, and no allergic symptoms were observed.
FROM BLOOD
Key clinical point: The bispecific antibody ACE910 was safe and effective as a long-acting hemostatic drug for hemophilia A in a phase I trial of healthy subjects.
Major finding: ACE910 was well tolerated up to 1 mg/kg with an average half-life of 28 to 34 days; in factor VIII–depleted plasma, ACE910 at 1 mg/kg resulted in an APTT similar to normal plasma.
Data sources: The first-in-human, double-blind, randomized, placebo-controlled dose-escalation study included 40 Japanese men and 24 white men.
Disclosures: The study was sponsored by Chugai Pharmaceutical. Dr. Uchida reported having no disclosures. His coauthors reported ties to Chugai Pharmaceutical and F. Hoffmann-La Roche, as well as holding patents related to anti-FIXa/X bispecific antibodies.
LMWH doesn’t reduce late pregnancy loss in women with thrombophilias
Prophylactic-dose low molecular weight heparin (LMWH), with or without aspirin, did not reduce the risk of pregnancy loss in women with inherited thrombophilia and a history of prior late or recurrent early pregnancy loss, based on a meta-analysis of randomized, controlled trials.
“To our knowledge, this is the largest study published to date that evaluates LMWH in women with inherited thrombophilia and previous pregnancy loss,” Dr. Leslie Skeith, of the University of Ottawa, and her colleagues wrote in Blood (2016 Mar 31;127[13]:1650-55). A recent Cochrane Review (2014 Jul 4;7:CD004734) similarly found no difference in live birth rates in women with or without inherited thrombophilia treated with LMWH and aspirin, compared with women given no treatment. Additionally, the Effects of Aspirin in Gestation and Reproduction [EAGeR] trial found no difference in live birth rates in women with previous pregnancy loss given aspirin or placebo (Lancet. 2014;384[9937]:29-36).
Based on a literature search, 8 publications and 483 participants met eligibility criteria as randomized, controlled trials for the meta-analysis. Four trials included an LMWH-plus-aspirin arm, and five trials included an LMWH-only arm. The control groups included four trials with an aspirin arm, and five trials with a placebo or no-treatment arm. The data indicated no difference in the treated groups and controls (relative risk of 0.81; 95% confidence interval, 0.55-1.19; P = .28).
As there is the potential for adverse side effects and significant cost with LMWH, the researchers advise against the use of LMWH to prevent recurrent and prior late pregnancy loss (greater than 10 weeks gestation) in women with inherited thrombophilia (Grade 1B, strong recommendation with moderate-quality evidence) and suggest against LMWH to prevent recurrent pregnancy loss in women with inherited thrombophilia and prior recurrent early (less than 10 weeks) pregnancy loss. (Grade 2B, weak recommendation with moderate-quality evidence.)
Given that the analysis included just 66 women with thrombophilia and prior recurrent early pregnancy loss, the researchers could not exclude a beneficial effect of LMWH in this subgroup. An ongoing randomized controlled trial, ALIFE2 (Netherlands Trial Registration Identifier: NTR3361), “is evaluating LMWH in women with inherited thrombophilia and a history of two or more miscarriages and/or intrauterine fetal death, which we hope will provide definitive answers to this question,” the researchers wrote.
They also suggest not testing for inherited thrombophilia in women with prior late or recurrent early pregnancy loss (Grade 2B, weak recommendation with moderate-quality evidence).
The study was supported by a series of university and institutional investigator research awards. Dr. Skeith received a Thrombosis Canada CanVECTOR Research Fellowship award.
On Twitter @maryjodales
Prophylactic-dose low molecular weight heparin (LMWH), with or without aspirin, did not reduce the risk of pregnancy loss in women with inherited thrombophilia and a history of prior late or recurrent early pregnancy loss, based on a meta-analysis of randomized, controlled trials.
“To our knowledge, this is the largest study published to date that evaluates LMWH in women with inherited thrombophilia and previous pregnancy loss,” Dr. Leslie Skeith, of the University of Ottawa, and her colleagues wrote in Blood (2016 Mar 31;127[13]:1650-55). A recent Cochrane Review (2014 Jul 4;7:CD004734) similarly found no difference in live birth rates in women with or without inherited thrombophilia treated with LMWH and aspirin, compared with women given no treatment. Additionally, the Effects of Aspirin in Gestation and Reproduction [EAGeR] trial found no difference in live birth rates in women with previous pregnancy loss given aspirin or placebo (Lancet. 2014;384[9937]:29-36).
Based on a literature search, 8 publications and 483 participants met eligibility criteria as randomized, controlled trials for the meta-analysis. Four trials included an LMWH-plus-aspirin arm, and five trials included an LMWH-only arm. The control groups included four trials with an aspirin arm, and five trials with a placebo or no-treatment arm. The data indicated no difference in the treated groups and controls (relative risk of 0.81; 95% confidence interval, 0.55-1.19; P = .28).
As there is the potential for adverse side effects and significant cost with LMWH, the researchers advise against the use of LMWH to prevent recurrent and prior late pregnancy loss (greater than 10 weeks gestation) in women with inherited thrombophilia (Grade 1B, strong recommendation with moderate-quality evidence) and suggest against LMWH to prevent recurrent pregnancy loss in women with inherited thrombophilia and prior recurrent early (less than 10 weeks) pregnancy loss. (Grade 2B, weak recommendation with moderate-quality evidence.)
Given that the analysis included just 66 women with thrombophilia and prior recurrent early pregnancy loss, the researchers could not exclude a beneficial effect of LMWH in this subgroup. An ongoing randomized controlled trial, ALIFE2 (Netherlands Trial Registration Identifier: NTR3361), “is evaluating LMWH in women with inherited thrombophilia and a history of two or more miscarriages and/or intrauterine fetal death, which we hope will provide definitive answers to this question,” the researchers wrote.
They also suggest not testing for inherited thrombophilia in women with prior late or recurrent early pregnancy loss (Grade 2B, weak recommendation with moderate-quality evidence).
The study was supported by a series of university and institutional investigator research awards. Dr. Skeith received a Thrombosis Canada CanVECTOR Research Fellowship award.
On Twitter @maryjodales
Prophylactic-dose low molecular weight heparin (LMWH), with or without aspirin, did not reduce the risk of pregnancy loss in women with inherited thrombophilia and a history of prior late or recurrent early pregnancy loss, based on a meta-analysis of randomized, controlled trials.
“To our knowledge, this is the largest study published to date that evaluates LMWH in women with inherited thrombophilia and previous pregnancy loss,” Dr. Leslie Skeith, of the University of Ottawa, and her colleagues wrote in Blood (2016 Mar 31;127[13]:1650-55). A recent Cochrane Review (2014 Jul 4;7:CD004734) similarly found no difference in live birth rates in women with or without inherited thrombophilia treated with LMWH and aspirin, compared with women given no treatment. Additionally, the Effects of Aspirin in Gestation and Reproduction [EAGeR] trial found no difference in live birth rates in women with previous pregnancy loss given aspirin or placebo (Lancet. 2014;384[9937]:29-36).
Based on a literature search, 8 publications and 483 participants met eligibility criteria as randomized, controlled trials for the meta-analysis. Four trials included an LMWH-plus-aspirin arm, and five trials included an LMWH-only arm. The control groups included four trials with an aspirin arm, and five trials with a placebo or no-treatment arm. The data indicated no difference in the treated groups and controls (relative risk of 0.81; 95% confidence interval, 0.55-1.19; P = .28).
As there is the potential for adverse side effects and significant cost with LMWH, the researchers advise against the use of LMWH to prevent recurrent and prior late pregnancy loss (greater than 10 weeks gestation) in women with inherited thrombophilia (Grade 1B, strong recommendation with moderate-quality evidence) and suggest against LMWH to prevent recurrent pregnancy loss in women with inherited thrombophilia and prior recurrent early (less than 10 weeks) pregnancy loss. (Grade 2B, weak recommendation with moderate-quality evidence.)
Given that the analysis included just 66 women with thrombophilia and prior recurrent early pregnancy loss, the researchers could not exclude a beneficial effect of LMWH in this subgroup. An ongoing randomized controlled trial, ALIFE2 (Netherlands Trial Registration Identifier: NTR3361), “is evaluating LMWH in women with inherited thrombophilia and a history of two or more miscarriages and/or intrauterine fetal death, which we hope will provide definitive answers to this question,” the researchers wrote.
They also suggest not testing for inherited thrombophilia in women with prior late or recurrent early pregnancy loss (Grade 2B, weak recommendation with moderate-quality evidence).
The study was supported by a series of university and institutional investigator research awards. Dr. Skeith received a Thrombosis Canada CanVECTOR Research Fellowship award.
On Twitter @maryjodales
FROM BLOOD
Key clinical point: Given the potential for adverse side effects and significant cost, the researchers advise against the use of LMWH to prevent recurrent and prior late pregnancy loss (greater than 10 weeks gestation) in women with inherited thrombophilia.
Major finding: The data indicated no difference in the treated groups and controls (relative risk of 0.81; 95% confidence interval, 0.55-1.19; P = .28).
Data source: Based on a literature search, 8 publications and 483 participants met eligibility criteria as randomized, controlled trials for the meta-analysis.
Disclosures: The study was supported by a series of university and institutional investigator research awards. Dr. Skeith received a Thrombosis Canada CanVECTOR Research Fellowship award.
Hormonal birth control doesn’t induce VTE recurrence
Hormonal contraceptives don’t appear to increase the risk of recurrence of venous thromboembolism among women taking anticoagulants, according to a report published in Blood.
Clinicians are often reluctant to prescribe hormonal contraceptives for women who develop venous thromboembolism (VTE) because the drugs are known to raise the risk of VTE and are considered to be contraindicated in either active or previous VTE. But effective contraception is necessary for women of childbearing age who are taking anticoagulants, because these drugs cross the placenta and could cause fetal bleeding and other adverse events, wrote Dr. Ida Martinelli of the A. Bianchi Bonomi Hemophilia and Thrombosis Center, Fondazione IRCCS Ca’ Granda-Ospedale Maggiore Policlinico, Milan, and her associates (Blood 2016;127[11]:1417-25).
Adding further to the confusion, World Health Organization guidelines state that estrogen-containing contraceptives confer “an unacceptable health risk” during anticoagulant therapy for VTE, but the International Society on Thrombosis and Haemostasis recommends that women diagnosed with VTE continue oral contraceptive and estrogen-replacement hormonal therapy until they discontinue anticoagulant therapy “because any prothrombotic effect of hormonal therapy is likely to be suppressed by therapeutic-intensity anticoagulation,” the investigators noted.
In the current study, the investigators performed a secondary analysis of data accrued in two large trials evaluating rivaroxaban versus enoxaparin plus vitamin K antagonists, which involved 1,888 women younger than age 60 (mean age, 41 years) who were being treated for acute deep vein thrombosis or acute pulmonary embolism. A total of 402 of these women used hormonal therapy during the 4-year studies.
There were 7 VTE recurrences during hormonal contraceptive use and 38 without hormonal contraceptive use. The incidence densities were 3.7% per year with hormonal therapy and 4.7% per year without it, for a hazard ratio of 0.56.
This indicates that hormonal contraceptive use did not make a clinically important difference in the rate of VTE recurrence. Moreover, these findings were consistent regardless of whether the contraceptives contained estrogen (incidence density, 3.7% per year) or progestin only (incidence density, 3.8% per year), and remained consistent in sensitivity analyses.
“These results challenge the WHO guidelines and instead support the International Society on Thrombosis and Haemostasis recommendations,” the investigators wrote.
“Our finding of similar risks of recurrent VTE for women who did or did not receive hormonal therapy, whether progestin-only or estrogen-containing therapy, supports a treatment selection that incorporates patient preference, including the choice of estrogen-containing contraception,” they added.
The study was funded in part by Bayer Healthcare Pharmaceuticals, which also provided editorial assistance. Dr. Martinelli reported having no relevant financial disclosures; her associates reported ties to numerous industry sources.
Martinelli et al. provide reassurance that women taking anticoagulants for VTE may safely use estrogen- or progestin-containing hormonal therapy, although it is important to note that their conclusions are based on only seven events (four with estrogen-containing and three with progestin-containing drugs).
Another important finding was that excessive uterine bleeding was more than twice as common among women taking rivaroxaban as among those taking enoxaparin plus vitamin K antagonists. Patients should be informed of this when they initiate anticoagulation.
Dr. Sam Schulman is in the division of hematology and thromboembolism at McMaster University, Hamilton, Ont. He reported receiving honoraria from Bayer, Boehringer Ingelheim, Bristol-Myers Squibb, and Daiichi. These comments are adapted from an accompanying editorial (Blood 2016;127[11]:1378-9).
Martinelli et al. provide reassurance that women taking anticoagulants for VTE may safely use estrogen- or progestin-containing hormonal therapy, although it is important to note that their conclusions are based on only seven events (four with estrogen-containing and three with progestin-containing drugs).
Another important finding was that excessive uterine bleeding was more than twice as common among women taking rivaroxaban as among those taking enoxaparin plus vitamin K antagonists. Patients should be informed of this when they initiate anticoagulation.
Dr. Sam Schulman is in the division of hematology and thromboembolism at McMaster University, Hamilton, Ont. He reported receiving honoraria from Bayer, Boehringer Ingelheim, Bristol-Myers Squibb, and Daiichi. These comments are adapted from an accompanying editorial (Blood 2016;127[11]:1378-9).
Martinelli et al. provide reassurance that women taking anticoagulants for VTE may safely use estrogen- or progestin-containing hormonal therapy, although it is important to note that their conclusions are based on only seven events (four with estrogen-containing and three with progestin-containing drugs).
Another important finding was that excessive uterine bleeding was more than twice as common among women taking rivaroxaban as among those taking enoxaparin plus vitamin K antagonists. Patients should be informed of this when they initiate anticoagulation.
Dr. Sam Schulman is in the division of hematology and thromboembolism at McMaster University, Hamilton, Ont. He reported receiving honoraria from Bayer, Boehringer Ingelheim, Bristol-Myers Squibb, and Daiichi. These comments are adapted from an accompanying editorial (Blood 2016;127[11]:1378-9).
Hormonal contraceptives don’t appear to increase the risk of recurrence of venous thromboembolism among women taking anticoagulants, according to a report published in Blood.
Clinicians are often reluctant to prescribe hormonal contraceptives for women who develop venous thromboembolism (VTE) because the drugs are known to raise the risk of VTE and are considered to be contraindicated in either active or previous VTE. But effective contraception is necessary for women of childbearing age who are taking anticoagulants, because these drugs cross the placenta and could cause fetal bleeding and other adverse events, wrote Dr. Ida Martinelli of the A. Bianchi Bonomi Hemophilia and Thrombosis Center, Fondazione IRCCS Ca’ Granda-Ospedale Maggiore Policlinico, Milan, and her associates (Blood 2016;127[11]:1417-25).
Adding further to the confusion, World Health Organization guidelines state that estrogen-containing contraceptives confer “an unacceptable health risk” during anticoagulant therapy for VTE, but the International Society on Thrombosis and Haemostasis recommends that women diagnosed with VTE continue oral contraceptive and estrogen-replacement hormonal therapy until they discontinue anticoagulant therapy “because any prothrombotic effect of hormonal therapy is likely to be suppressed by therapeutic-intensity anticoagulation,” the investigators noted.
In the current study, the investigators performed a secondary analysis of data accrued in two large trials evaluating rivaroxaban versus enoxaparin plus vitamin K antagonists, which involved 1,888 women younger than age 60 (mean age, 41 years) who were being treated for acute deep vein thrombosis or acute pulmonary embolism. A total of 402 of these women used hormonal therapy during the 4-year studies.
There were 7 VTE recurrences during hormonal contraceptive use and 38 without hormonal contraceptive use. The incidence densities were 3.7% per year with hormonal therapy and 4.7% per year without it, for a hazard ratio of 0.56.
This indicates that hormonal contraceptive use did not make a clinically important difference in the rate of VTE recurrence. Moreover, these findings were consistent regardless of whether the contraceptives contained estrogen (incidence density, 3.7% per year) or progestin only (incidence density, 3.8% per year), and remained consistent in sensitivity analyses.
“These results challenge the WHO guidelines and instead support the International Society on Thrombosis and Haemostasis recommendations,” the investigators wrote.
“Our finding of similar risks of recurrent VTE for women who did or did not receive hormonal therapy, whether progestin-only or estrogen-containing therapy, supports a treatment selection that incorporates patient preference, including the choice of estrogen-containing contraception,” they added.
The study was funded in part by Bayer Healthcare Pharmaceuticals, which also provided editorial assistance. Dr. Martinelli reported having no relevant financial disclosures; her associates reported ties to numerous industry sources.
Hormonal contraceptives don’t appear to increase the risk of recurrence of venous thromboembolism among women taking anticoagulants, according to a report published in Blood.
Clinicians are often reluctant to prescribe hormonal contraceptives for women who develop venous thromboembolism (VTE) because the drugs are known to raise the risk of VTE and are considered to be contraindicated in either active or previous VTE. But effective contraception is necessary for women of childbearing age who are taking anticoagulants, because these drugs cross the placenta and could cause fetal bleeding and other adverse events, wrote Dr. Ida Martinelli of the A. Bianchi Bonomi Hemophilia and Thrombosis Center, Fondazione IRCCS Ca’ Granda-Ospedale Maggiore Policlinico, Milan, and her associates (Blood 2016;127[11]:1417-25).
Adding further to the confusion, World Health Organization guidelines state that estrogen-containing contraceptives confer “an unacceptable health risk” during anticoagulant therapy for VTE, but the International Society on Thrombosis and Haemostasis recommends that women diagnosed with VTE continue oral contraceptive and estrogen-replacement hormonal therapy until they discontinue anticoagulant therapy “because any prothrombotic effect of hormonal therapy is likely to be suppressed by therapeutic-intensity anticoagulation,” the investigators noted.
In the current study, the investigators performed a secondary analysis of data accrued in two large trials evaluating rivaroxaban versus enoxaparin plus vitamin K antagonists, which involved 1,888 women younger than age 60 (mean age, 41 years) who were being treated for acute deep vein thrombosis or acute pulmonary embolism. A total of 402 of these women used hormonal therapy during the 4-year studies.
There were 7 VTE recurrences during hormonal contraceptive use and 38 without hormonal contraceptive use. The incidence densities were 3.7% per year with hormonal therapy and 4.7% per year without it, for a hazard ratio of 0.56.
This indicates that hormonal contraceptive use did not make a clinically important difference in the rate of VTE recurrence. Moreover, these findings were consistent regardless of whether the contraceptives contained estrogen (incidence density, 3.7% per year) or progestin only (incidence density, 3.8% per year), and remained consistent in sensitivity analyses.
“These results challenge the WHO guidelines and instead support the International Society on Thrombosis and Haemostasis recommendations,” the investigators wrote.
“Our finding of similar risks of recurrent VTE for women who did or did not receive hormonal therapy, whether progestin-only or estrogen-containing therapy, supports a treatment selection that incorporates patient preference, including the choice of estrogen-containing contraception,” they added.
The study was funded in part by Bayer Healthcare Pharmaceuticals, which also provided editorial assistance. Dr. Martinelli reported having no relevant financial disclosures; her associates reported ties to numerous industry sources.
FROM BLOOD
Key clinical point: Hormonal contraceptives don’t appear to affect VTE recurrence among women taking anticoagulants for acute VTE.
Major finding: There were 7 VTE recurrences with hormonal contraception use and 38 without, for incidence densities of 3.7% per year with hormonal use and 4.7% per year without it (HR, 0.56).
Data source: A secondary analysis of data from two open-label randomized trials involving 1,888 women younger than age 60 with acute VTE.
Disclosures: The study was funded in part by Bayer Healthcare Pharmaceuticals, which also provided editorial assistance. Dr. Martinelli reported having no relevant financial disclosures; her associates reported ties to numerous industry sources.