User login
Sickle cell trait raises exertional rhabdomyolysis risk
Sickle cell trait doesn’t raise the risk of death but does raise the risk of exertional rhabdomyolysis among black soldiers, “a population that is known to engage consistently in regular and strenuous exercise while protected by exertional-injury precautions,” investigators reported. The study was published online Aug. 4 in the New England Journal of Medicine.
A number of high-profile deaths among athletes and military personnel involving exertional rhabdomyolysis have been attributed to sickle cell trait. The National Collegiate Athletic Association, the U.S. Air Force, and the U.S. Navy all require universal screening for sickle cell trait.
“However, concerns have been raised by the American Society of Hematology and other professional organizations about the possibility of stigmatization and discrimination resulting from the mandated screening for sickle cell trait,” which predominantly affects people of African descent and some of Hispanic ancestry, but usually not whites, wrote D. Alan Nelson, PhD, of Stanford (Calif.) University, and his associates.
“These concerns warrant consideration, especially given the absence of published evidence that such screening is effective in preventing exertion-related events,” added the researchers.
The U.S. Army primarily screens for sickle cell trait among personnel deployed for combat and certain specialists whose work at high altitudes puts them at risk. To study a large enough population to generate accurate data about this rare blood abnormality, the investigators analyzed data for 47,944 black soldiers who had been tested for sickle cell trait and who served on active duty during a recent 3-year period.
The researchers assessed all inpatient and outpatient health care visits at military and civilian facilities. There were 391 exertional rhabdomyolysis events and 96 deaths from all causes during 1.61 million person-months of observation. A total of 7.4% of the study cohort carried sickle cell trait. These participants were similar to noncarriers in demographic characteristics and health predictors.
Soldiers with sickle cell trait did not have a greater risk of death than did those without sickle cell trait (hazard ratio, 0.99), and there also was no difference between carriers and noncarriers in either battle-related or non–battle-related mortality rates.
However, sickle cell trait was associated with a 54% greater risk of exertional rhabdomyolysis (HR, 1.54), Dr. Nelson and his associates noted (N Engl J Med. 2016 Aug 4. doi: 10.1056/NEJMoa1516257).
It is important to note that factors other than sickle cell trait raised the risk of exertional rhabdomyolysis to a similar or even greater degree, including obesity (HR, 1.39) and tobacco use (HR, 1.54). Recent use of antipsychotic medication tripled the risk for exertional rhabdomyolysis (HR, 3.02), and statin use nearly tripled it (HR, 2.89).
“These findings are compelling because case reports dominate the relevant literature and emphasize the presence of sickle cell trait as a risk factor for adverse outcomes, including exertional rhabdomyolysis and sudden death,” the researchers wrote. “A large longitudinal study involving a population fully tested for sickle cell trait that has formally investigated [these outcomes] while also examining other known major risk factors, such as medications, has been lacking.”
The study found that women were at much lower risk for exertional rhabdomyolysis than were men (HR, 0.51), and that risk increased with increasing age, so that soldiers aged 36 years and older had a 57% greater risk compared with those in the youngest age group.
The National Heart, Lung, and Blood Institute and the Uniformed Services University of the Health Sciences supported the study. Dr. Nelson and his associates reported having no relevant financial disclosures.
Sickle cell trait doesn’t raise the risk of death but does raise the risk of exertional rhabdomyolysis among black soldiers, “a population that is known to engage consistently in regular and strenuous exercise while protected by exertional-injury precautions,” investigators reported. The study was published online Aug. 4 in the New England Journal of Medicine.
A number of high-profile deaths among athletes and military personnel involving exertional rhabdomyolysis have been attributed to sickle cell trait. The National Collegiate Athletic Association, the U.S. Air Force, and the U.S. Navy all require universal screening for sickle cell trait.
“However, concerns have been raised by the American Society of Hematology and other professional organizations about the possibility of stigmatization and discrimination resulting from the mandated screening for sickle cell trait,” which predominantly affects people of African descent and some of Hispanic ancestry, but usually not whites, wrote D. Alan Nelson, PhD, of Stanford (Calif.) University, and his associates.
“These concerns warrant consideration, especially given the absence of published evidence that such screening is effective in preventing exertion-related events,” added the researchers.
The U.S. Army primarily screens for sickle cell trait among personnel deployed for combat and certain specialists whose work at high altitudes puts them at risk. To study a large enough population to generate accurate data about this rare blood abnormality, the investigators analyzed data for 47,944 black soldiers who had been tested for sickle cell trait and who served on active duty during a recent 3-year period.
The researchers assessed all inpatient and outpatient health care visits at military and civilian facilities. There were 391 exertional rhabdomyolysis events and 96 deaths from all causes during 1.61 million person-months of observation. A total of 7.4% of the study cohort carried sickle cell trait. These participants were similar to noncarriers in demographic characteristics and health predictors.
Soldiers with sickle cell trait did not have a greater risk of death than did those without sickle cell trait (hazard ratio, 0.99), and there also was no difference between carriers and noncarriers in either battle-related or non–battle-related mortality rates.
However, sickle cell trait was associated with a 54% greater risk of exertional rhabdomyolysis (HR, 1.54), Dr. Nelson and his associates noted (N Engl J Med. 2016 Aug 4. doi: 10.1056/NEJMoa1516257).
It is important to note that factors other than sickle cell trait raised the risk of exertional rhabdomyolysis to a similar or even greater degree, including obesity (HR, 1.39) and tobacco use (HR, 1.54). Recent use of antipsychotic medication tripled the risk for exertional rhabdomyolysis (HR, 3.02), and statin use nearly tripled it (HR, 2.89).
“These findings are compelling because case reports dominate the relevant literature and emphasize the presence of sickle cell trait as a risk factor for adverse outcomes, including exertional rhabdomyolysis and sudden death,” the researchers wrote. “A large longitudinal study involving a population fully tested for sickle cell trait that has formally investigated [these outcomes] while also examining other known major risk factors, such as medications, has been lacking.”
The study found that women were at much lower risk for exertional rhabdomyolysis than were men (HR, 0.51), and that risk increased with increasing age, so that soldiers aged 36 years and older had a 57% greater risk compared with those in the youngest age group.
The National Heart, Lung, and Blood Institute and the Uniformed Services University of the Health Sciences supported the study. Dr. Nelson and his associates reported having no relevant financial disclosures.
Sickle cell trait doesn’t raise the risk of death but does raise the risk of exertional rhabdomyolysis among black soldiers, “a population that is known to engage consistently in regular and strenuous exercise while protected by exertional-injury precautions,” investigators reported. The study was published online Aug. 4 in the New England Journal of Medicine.
A number of high-profile deaths among athletes and military personnel involving exertional rhabdomyolysis have been attributed to sickle cell trait. The National Collegiate Athletic Association, the U.S. Air Force, and the U.S. Navy all require universal screening for sickle cell trait.
“However, concerns have been raised by the American Society of Hematology and other professional organizations about the possibility of stigmatization and discrimination resulting from the mandated screening for sickle cell trait,” which predominantly affects people of African descent and some of Hispanic ancestry, but usually not whites, wrote D. Alan Nelson, PhD, of Stanford (Calif.) University, and his associates.
“These concerns warrant consideration, especially given the absence of published evidence that such screening is effective in preventing exertion-related events,” added the researchers.
The U.S. Army primarily screens for sickle cell trait among personnel deployed for combat and certain specialists whose work at high altitudes puts them at risk. To study a large enough population to generate accurate data about this rare blood abnormality, the investigators analyzed data for 47,944 black soldiers who had been tested for sickle cell trait and who served on active duty during a recent 3-year period.
The researchers assessed all inpatient and outpatient health care visits at military and civilian facilities. There were 391 exertional rhabdomyolysis events and 96 deaths from all causes during 1.61 million person-months of observation. A total of 7.4% of the study cohort carried sickle cell trait. These participants were similar to noncarriers in demographic characteristics and health predictors.
Soldiers with sickle cell trait did not have a greater risk of death than did those without sickle cell trait (hazard ratio, 0.99), and there also was no difference between carriers and noncarriers in either battle-related or non–battle-related mortality rates.
However, sickle cell trait was associated with a 54% greater risk of exertional rhabdomyolysis (HR, 1.54), Dr. Nelson and his associates noted (N Engl J Med. 2016 Aug 4. doi: 10.1056/NEJMoa1516257).
It is important to note that factors other than sickle cell trait raised the risk of exertional rhabdomyolysis to a similar or even greater degree, including obesity (HR, 1.39) and tobacco use (HR, 1.54). Recent use of antipsychotic medication tripled the risk for exertional rhabdomyolysis (HR, 3.02), and statin use nearly tripled it (HR, 2.89).
“These findings are compelling because case reports dominate the relevant literature and emphasize the presence of sickle cell trait as a risk factor for adverse outcomes, including exertional rhabdomyolysis and sudden death,” the researchers wrote. “A large longitudinal study involving a population fully tested for sickle cell trait that has formally investigated [these outcomes] while also examining other known major risk factors, such as medications, has been lacking.”
The study found that women were at much lower risk for exertional rhabdomyolysis than were men (HR, 0.51), and that risk increased with increasing age, so that soldiers aged 36 years and older had a 57% greater risk compared with those in the youngest age group.
The National Heart, Lung, and Blood Institute and the Uniformed Services University of the Health Sciences supported the study. Dr. Nelson and his associates reported having no relevant financial disclosures.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point: Sickle cell trait doesn’t raise the risk of death but does raise the risk of exertional rhabdomyolysis among black soldiers.
Major finding: Soldiers with sickle cell trait did not have a greater risk of death than did those without sickle cell trait (hazard ratio, 0.99) but did have a higher risk of exertional rhabdomyolysis (HR, 1.54).
Data source: A retrospective longitudinal cohort study involving 47,944 black U.S. soldiers on active duty during 2011-2014.
Disclosures: The National Heart, Lung, and Blood Institute and the Uniformed Services University of the Health Sciences supported the study. Dr. Nelson and his associates reported having no relevant financial disclosures.
Children under 6 with factor XIII deficiency had no major bleeds with recombinant product
ORLANDO –A recombinant form of factor XIII was effective at preventing serious bleeding episodes in young children with factor XIII-A subunit deficiency, a rare and serious bleeding disorder.
In a small international phase III trial, there were no major bleeding episodes among six young children treated for at least 1 year with recombinant factor XIII (rFXIII; trade name Tretten), reported Susan L. Kearney, MD, of Children’s Hospitals and Clinics of Minnesota in Minneapolis.
“Prophylaxis was effective. The annualized bleeding rate was zero and the mean trough [FXIII activity] was greater than 10%,” she said at a moderated poster session at the World Federation of Hemophilia World Congress. “We feel that recombinant factor XIII is safe and effective in pediatric subjects less than 6 years of age with congenital factor XIII-A subunit deficiency, similar to the older age cohort.”
Factor XIII-A subunit deficiency is a rare and serious heritable bleeding disorder associated with spontaneous intracranial hemorrhage and other unpredictable types of serious bleeding.
In a previous phase III trial, 77 patients, ranging in age from 7 to 60 years, received rFXIII for bleeding prophylaxis. When given monthly, the recombinant factor was effective at preventing serious bleeding in 90% of patients. The most commonly reported adverse events were headache, pain in the extremities, and injection site pain.
Based on these results, the Food and Drug Administration granted rFXIII orphan-drug designation for treatment of patients 6 and older with factor XIII-A subunit deficiency.
In the trial reported here, investigators from the United States, United Kingdom, Israel, and Denmark enrolled three boys and three girls under age 6 who had previously completed a single dose efficacy and safety study of rFXIII. The patients received intravenous rFXIII at a dose of 35 IU/kg every 28 days for a minimum of 52 weeks.
The total treatment duration ranged from 1.8 to 3.5 years, for a total of 16.6 patient years.
There were no thromboembolic events or systemic allergic reactions, the primary safety endpoint of the study. One patient experienced three incidences of atopic dermatitis, however; two serious adverse events related to head injuries from falls during play occurred in one patient, who did not experience intracranial hemorrhage.
Two adverse events were deemed to be probably or possibly related to rFXIII: a case of viral gastroenteritis affected one patient who recovered without a change in dose, and mild fluctuating lymphocytopenia seen at baseline persisted in another patient throughout the trial.
There were no inhibitory or noninhibitory antibodies to rFXIII detected in any patient during the trial, and there were no bleeding episodes requiring additional treatment. The 14 minor bleeding episodes seen in five patients did not require treatment with an FXIII-containing product, the authors noted.
“It’s a very rare disorder, but ... the phenotype is quite severe and patients are severely affected. So this product is very useful,” said Lakshmi Srivaths, MD, a pediatric hematologist at Texas Children’s Hospital in Houston. She was not involved in the study. Unlike patients with hemophilia A or B, who require frequent factor infusions, the long half-life of this product means patients need just once-a-month infusions “that change the phenotype very significantly.”
Dr. Kearney disclosed grant/research support from Novo Nordisk, which funded the study. Some coauthors reported consulting or employment with the company.
ORLANDO –A recombinant form of factor XIII was effective at preventing serious bleeding episodes in young children with factor XIII-A subunit deficiency, a rare and serious bleeding disorder.
In a small international phase III trial, there were no major bleeding episodes among six young children treated for at least 1 year with recombinant factor XIII (rFXIII; trade name Tretten), reported Susan L. Kearney, MD, of Children’s Hospitals and Clinics of Minnesota in Minneapolis.
“Prophylaxis was effective. The annualized bleeding rate was zero and the mean trough [FXIII activity] was greater than 10%,” she said at a moderated poster session at the World Federation of Hemophilia World Congress. “We feel that recombinant factor XIII is safe and effective in pediatric subjects less than 6 years of age with congenital factor XIII-A subunit deficiency, similar to the older age cohort.”
Factor XIII-A subunit deficiency is a rare and serious heritable bleeding disorder associated with spontaneous intracranial hemorrhage and other unpredictable types of serious bleeding.
In a previous phase III trial, 77 patients, ranging in age from 7 to 60 years, received rFXIII for bleeding prophylaxis. When given monthly, the recombinant factor was effective at preventing serious bleeding in 90% of patients. The most commonly reported adverse events were headache, pain in the extremities, and injection site pain.
Based on these results, the Food and Drug Administration granted rFXIII orphan-drug designation for treatment of patients 6 and older with factor XIII-A subunit deficiency.
In the trial reported here, investigators from the United States, United Kingdom, Israel, and Denmark enrolled three boys and three girls under age 6 who had previously completed a single dose efficacy and safety study of rFXIII. The patients received intravenous rFXIII at a dose of 35 IU/kg every 28 days for a minimum of 52 weeks.
The total treatment duration ranged from 1.8 to 3.5 years, for a total of 16.6 patient years.
There were no thromboembolic events or systemic allergic reactions, the primary safety endpoint of the study. One patient experienced three incidences of atopic dermatitis, however; two serious adverse events related to head injuries from falls during play occurred in one patient, who did not experience intracranial hemorrhage.
Two adverse events were deemed to be probably or possibly related to rFXIII: a case of viral gastroenteritis affected one patient who recovered without a change in dose, and mild fluctuating lymphocytopenia seen at baseline persisted in another patient throughout the trial.
There were no inhibitory or noninhibitory antibodies to rFXIII detected in any patient during the trial, and there were no bleeding episodes requiring additional treatment. The 14 minor bleeding episodes seen in five patients did not require treatment with an FXIII-containing product, the authors noted.
“It’s a very rare disorder, but ... the phenotype is quite severe and patients are severely affected. So this product is very useful,” said Lakshmi Srivaths, MD, a pediatric hematologist at Texas Children’s Hospital in Houston. She was not involved in the study. Unlike patients with hemophilia A or B, who require frequent factor infusions, the long half-life of this product means patients need just once-a-month infusions “that change the phenotype very significantly.”
Dr. Kearney disclosed grant/research support from Novo Nordisk, which funded the study. Some coauthors reported consulting or employment with the company.
ORLANDO –A recombinant form of factor XIII was effective at preventing serious bleeding episodes in young children with factor XIII-A subunit deficiency, a rare and serious bleeding disorder.
In a small international phase III trial, there were no major bleeding episodes among six young children treated for at least 1 year with recombinant factor XIII (rFXIII; trade name Tretten), reported Susan L. Kearney, MD, of Children’s Hospitals and Clinics of Minnesota in Minneapolis.
“Prophylaxis was effective. The annualized bleeding rate was zero and the mean trough [FXIII activity] was greater than 10%,” she said at a moderated poster session at the World Federation of Hemophilia World Congress. “We feel that recombinant factor XIII is safe and effective in pediatric subjects less than 6 years of age with congenital factor XIII-A subunit deficiency, similar to the older age cohort.”
Factor XIII-A subunit deficiency is a rare and serious heritable bleeding disorder associated with spontaneous intracranial hemorrhage and other unpredictable types of serious bleeding.
In a previous phase III trial, 77 patients, ranging in age from 7 to 60 years, received rFXIII for bleeding prophylaxis. When given monthly, the recombinant factor was effective at preventing serious bleeding in 90% of patients. The most commonly reported adverse events were headache, pain in the extremities, and injection site pain.
Based on these results, the Food and Drug Administration granted rFXIII orphan-drug designation for treatment of patients 6 and older with factor XIII-A subunit deficiency.
In the trial reported here, investigators from the United States, United Kingdom, Israel, and Denmark enrolled three boys and three girls under age 6 who had previously completed a single dose efficacy and safety study of rFXIII. The patients received intravenous rFXIII at a dose of 35 IU/kg every 28 days for a minimum of 52 weeks.
The total treatment duration ranged from 1.8 to 3.5 years, for a total of 16.6 patient years.
There were no thromboembolic events or systemic allergic reactions, the primary safety endpoint of the study. One patient experienced three incidences of atopic dermatitis, however; two serious adverse events related to head injuries from falls during play occurred in one patient, who did not experience intracranial hemorrhage.
Two adverse events were deemed to be probably or possibly related to rFXIII: a case of viral gastroenteritis affected one patient who recovered without a change in dose, and mild fluctuating lymphocytopenia seen at baseline persisted in another patient throughout the trial.
There were no inhibitory or noninhibitory antibodies to rFXIII detected in any patient during the trial, and there were no bleeding episodes requiring additional treatment. The 14 minor bleeding episodes seen in five patients did not require treatment with an FXIII-containing product, the authors noted.
“It’s a very rare disorder, but ... the phenotype is quite severe and patients are severely affected. So this product is very useful,” said Lakshmi Srivaths, MD, a pediatric hematologist at Texas Children’s Hospital in Houston. She was not involved in the study. Unlike patients with hemophilia A or B, who require frequent factor infusions, the long half-life of this product means patients need just once-a-month infusions “that change the phenotype very significantly.”
Dr. Kearney disclosed grant/research support from Novo Nordisk, which funded the study. Some coauthors reported consulting or employment with the company.
AT WFH 2016 WORLD CONGRESS
Key clinical point: A recombinant form of factor XIII was effective at preventing serious bleeding episodes in young children with factor XIII-A subunit deficiency.
Major finding: No bleeds occurred within a year in children with factor XIII-A subunit deficiency.
Data source: Open-label international phase III trial in three boys and three girls under age 6.
Disclosures: Dr. Kearney disclosed grant/research support from Novo Nordisk, which funded the study. Some coauthors reported consulting or employment with the company.
Hemophilia guideline recommends integrated care model
ORLANDO – An integrated care model that includes a hematologist, a specialized hematology nurse, a physical therapist, a social worker, and 24/7 access to a specialized coagulation laboratory is recommended in a new hemophilia care guideline jointly developed by the National Hemophilia Foundation and McMaster University in Hamilton, Ontario.
The guideline has been formally accepted for inclusion in the National Guideline Clearinghouse (NGC), the National Hemophilia Foundation announced at its annual meeting, held immediately before the World Hemophilia Foundation World Congress here.

“The integrated care model, as is utilized within the U.S. federally funded network of hemophilia treatment centers (HTCs), should be advocated for optimal care of persons with hemophilia,” wrote guideline coauthors Steven W. Pipe, MD, from the University of Michigan School of Medicine in Ann Arbor, and Craig M. Kessler, MD, from Georgetown University in Washington, in an introduction to the guideline, published in the journal Hemophilia.
Developed according to evidence-based principles, the guideline is hoped to “promote harmonization of care delivery and reduce practice variations within the U.S. HTC network. This guideline will inform the HTC network how best to prioritize additional ‘high-value’ research to fill data gaps or strengthen the evidence base as outlined in the manuscript,” they added.
The guideline authors recognized three basic models of care in use in the United States:
The integrated care, comprehensive care, or HTC model, which generally assumes that all aspects of care will be delivered in a specialized center.
Specialist-based care, under which a hematologist may or may not have training in the management of patients with hemophilia, is provided in a hospital or medical office, but not in a specialized center.
Non-specialist care, delivered in an emergency department or primary care practitioners office.
“We believe that the ‘No Care’ model, theoretically indicating complete absence of care, does not currently operate in the U.S. Yet, this is likely the de facto model of care for many individuals with hemophilia who do not have access to care due to profound resource constraints, particularly in developing countries or underserved minorities,” the authors wrote.
The guideline’s main recommendation – that the integrated-care model is preferable to the non-integrated care model – is conditional, with moderate certainty in the evidence.
For persons with hemophilia with inhibitors to clotting factors, however, the integrated-care model recommendation is considered to be strong, with moderate certainty.
The guideline development panel found that there were significant gaps in evidence for the benefits of integrated care for specific populations such as older patients and populations with poor access to care, and called for additional studies to clarify these questions.
Additionally, the panelists call for study into which specific interventions or aspects of care should be included in the integrated model, and for more in-depth studies into the effects on patient-important outcomes.
“This collaborative project constitutes an important milestone on a critical component of evidence-based guideline methodology,” Alfonso Iorio, MD, PhD, from the Department of Clinical Epidemiology and Biostatistics at McMaster University, said in a press statement. “It demonstrates how a patient advocacy organization can promote and support a guideline process, in the true spirit of patient involvement in research and care process, without compromising a rigorous and transparent conflict of interest management process.”
The guidelines were funded by the National Hemophilia Foundation. Several guideline panelists reported financial relationships with companies that make clotting factors and other products for persons with hemophilia.
ORLANDO – An integrated care model that includes a hematologist, a specialized hematology nurse, a physical therapist, a social worker, and 24/7 access to a specialized coagulation laboratory is recommended in a new hemophilia care guideline jointly developed by the National Hemophilia Foundation and McMaster University in Hamilton, Ontario.
The guideline has been formally accepted for inclusion in the National Guideline Clearinghouse (NGC), the National Hemophilia Foundation announced at its annual meeting, held immediately before the World Hemophilia Foundation World Congress here.
“The integrated care model, as is utilized within the U.S. federally funded network of hemophilia treatment centers (HTCs), should be advocated for optimal care of persons with hemophilia,” wrote guideline coauthors Steven W. Pipe, MD, from the University of Michigan School of Medicine in Ann Arbor, and Craig M. Kessler, MD, from Georgetown University in Washington, in an introduction to the guideline, published in the journal Hemophilia.
Developed according to evidence-based principles, the guideline is hoped to “promote harmonization of care delivery and reduce practice variations within the U.S. HTC network. This guideline will inform the HTC network how best to prioritize additional ‘high-value’ research to fill data gaps or strengthen the evidence base as outlined in the manuscript,” they added.
The guideline authors recognized three basic models of care in use in the United States:
The integrated care, comprehensive care, or HTC model, which generally assumes that all aspects of care will be delivered in a specialized center.
Specialist-based care, under which a hematologist may or may not have training in the management of patients with hemophilia, is provided in a hospital or medical office, but not in a specialized center.
Non-specialist care, delivered in an emergency department or primary care practitioners office.
“We believe that the ‘No Care’ model, theoretically indicating complete absence of care, does not currently operate in the U.S. Yet, this is likely the de facto model of care for many individuals with hemophilia who do not have access to care due to profound resource constraints, particularly in developing countries or underserved minorities,” the authors wrote.
The guideline’s main recommendation – that the integrated-care model is preferable to the non-integrated care model – is conditional, with moderate certainty in the evidence.
For persons with hemophilia with inhibitors to clotting factors, however, the integrated-care model recommendation is considered to be strong, with moderate certainty.
The guideline development panel found that there were significant gaps in evidence for the benefits of integrated care for specific populations such as older patients and populations with poor access to care, and called for additional studies to clarify these questions.
Additionally, the panelists call for study into which specific interventions or aspects of care should be included in the integrated model, and for more in-depth studies into the effects on patient-important outcomes.
“This collaborative project constitutes an important milestone on a critical component of evidence-based guideline methodology,” Alfonso Iorio, MD, PhD, from the Department of Clinical Epidemiology and Biostatistics at McMaster University, said in a press statement. “It demonstrates how a patient advocacy organization can promote and support a guideline process, in the true spirit of patient involvement in research and care process, without compromising a rigorous and transparent conflict of interest management process.”
The guidelines were funded by the National Hemophilia Foundation. Several guideline panelists reported financial relationships with companies that make clotting factors and other products for persons with hemophilia.
ORLANDO – An integrated care model that includes a hematologist, a specialized hematology nurse, a physical therapist, a social worker, and 24/7 access to a specialized coagulation laboratory is recommended in a new hemophilia care guideline jointly developed by the National Hemophilia Foundation and McMaster University in Hamilton, Ontario.
The guideline has been formally accepted for inclusion in the National Guideline Clearinghouse (NGC), the National Hemophilia Foundation announced at its annual meeting, held immediately before the World Hemophilia Foundation World Congress here.
“The integrated care model, as is utilized within the U.S. federally funded network of hemophilia treatment centers (HTCs), should be advocated for optimal care of persons with hemophilia,” wrote guideline coauthors Steven W. Pipe, MD, from the University of Michigan School of Medicine in Ann Arbor, and Craig M. Kessler, MD, from Georgetown University in Washington, in an introduction to the guideline, published in the journal Hemophilia.
Developed according to evidence-based principles, the guideline is hoped to “promote harmonization of care delivery and reduce practice variations within the U.S. HTC network. This guideline will inform the HTC network how best to prioritize additional ‘high-value’ research to fill data gaps or strengthen the evidence base as outlined in the manuscript,” they added.
The guideline authors recognized three basic models of care in use in the United States:
The integrated care, comprehensive care, or HTC model, which generally assumes that all aspects of care will be delivered in a specialized center.
Specialist-based care, under which a hematologist may or may not have training in the management of patients with hemophilia, is provided in a hospital or medical office, but not in a specialized center.
Non-specialist care, delivered in an emergency department or primary care practitioners office.
“We believe that the ‘No Care’ model, theoretically indicating complete absence of care, does not currently operate in the U.S. Yet, this is likely the de facto model of care for many individuals with hemophilia who do not have access to care due to profound resource constraints, particularly in developing countries or underserved minorities,” the authors wrote.
The guideline’s main recommendation – that the integrated-care model is preferable to the non-integrated care model – is conditional, with moderate certainty in the evidence.
For persons with hemophilia with inhibitors to clotting factors, however, the integrated-care model recommendation is considered to be strong, with moderate certainty.
The guideline development panel found that there were significant gaps in evidence for the benefits of integrated care for specific populations such as older patients and populations with poor access to care, and called for additional studies to clarify these questions.
Additionally, the panelists call for study into which specific interventions or aspects of care should be included in the integrated model, and for more in-depth studies into the effects on patient-important outcomes.
“This collaborative project constitutes an important milestone on a critical component of evidence-based guideline methodology,” Alfonso Iorio, MD, PhD, from the Department of Clinical Epidemiology and Biostatistics at McMaster University, said in a press statement. “It demonstrates how a patient advocacy organization can promote and support a guideline process, in the true spirit of patient involvement in research and care process, without compromising a rigorous and transparent conflict of interest management process.”
The guidelines were funded by the National Hemophilia Foundation. Several guideline panelists reported financial relationships with companies that make clotting factors and other products for persons with hemophilia.
AT WFH 2016 WORLD CONGRESS
Key clinical point: An integrated care model is recommended as optimal for management of persons with hemophilia.
Major finding: The joint National Hemophilia Foundation/McMaster University guidelines have been accepted by the National Guideline Clearinghouse.
Data source: Evidence-based recommendations on models of care.
Disclosures: The guidelines were funded by the National Hemophilia Foundation. Several guideline panelists reported financial relationships with companies that make clotting factors and other products for persons with hemophilia.

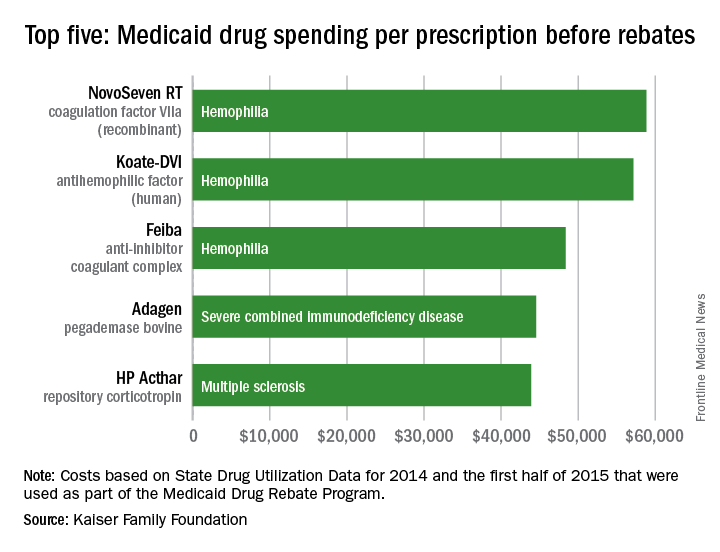
Hemophilia drugs top Medicaid spending per prescription
Medicaid’s three most expensive drugs by spending per prescription were for treatment of hemophilia, with the cost leader coming in at almost $59,000, according to an analysis by the Kaiser Family Foundation covering 2014 and the first half of 2015.
The trio of hemophilia drugs was topped by NovoSeven RT (coagulation factor VIIa [recombinant]), with Koate-DVI (antihemophilic factor [human]) second at $57,000 per prescription and Feiba (anti-inhibitor coagulant complex) third at $48,000 for each prescription.
None of the Medicaid costs include rebates since those data are unavailable to the public, Kaiser noted.
The fourth and fifth most expensive drugs were Adagen (pegademase bovine), which is used in the treatment of severe combined immunodeficiency disease associated with a deficiency of adenosine deaminase and cost Medicaid $45,000 per prescription, and the multiple sclerosis drug HP Acthar (repository corticotropin), which went for almost $44,000 a prescription, Kaiser said in its analysis, which used State Drug Utilization Data that are part of the Medicaid Drug Rebate Program.

Medicaid’s three most expensive drugs by spending per prescription were for treatment of hemophilia, with the cost leader coming in at almost $59,000, according to an analysis by the Kaiser Family Foundation covering 2014 and the first half of 2015.
The trio of hemophilia drugs was topped by NovoSeven RT (coagulation factor VIIa [recombinant]), with Koate-DVI (antihemophilic factor [human]) second at $57,000 per prescription and Feiba (anti-inhibitor coagulant complex) third at $48,000 for each prescription.
None of the Medicaid costs include rebates since those data are unavailable to the public, Kaiser noted.
The fourth and fifth most expensive drugs were Adagen (pegademase bovine), which is used in the treatment of severe combined immunodeficiency disease associated with a deficiency of adenosine deaminase and cost Medicaid $45,000 per prescription, and the multiple sclerosis drug HP Acthar (repository corticotropin), which went for almost $44,000 a prescription, Kaiser said in its analysis, which used State Drug Utilization Data that are part of the Medicaid Drug Rebate Program.
Medicaid’s three most expensive drugs by spending per prescription were for treatment of hemophilia, with the cost leader coming in at almost $59,000, according to an analysis by the Kaiser Family Foundation covering 2014 and the first half of 2015.
The trio of hemophilia drugs was topped by NovoSeven RT (coagulation factor VIIa [recombinant]), with Koate-DVI (antihemophilic factor [human]) second at $57,000 per prescription and Feiba (anti-inhibitor coagulant complex) third at $48,000 for each prescription.
None of the Medicaid costs include rebates since those data are unavailable to the public, Kaiser noted.
The fourth and fifth most expensive drugs were Adagen (pegademase bovine), which is used in the treatment of severe combined immunodeficiency disease associated with a deficiency of adenosine deaminase and cost Medicaid $45,000 per prescription, and the multiple sclerosis drug HP Acthar (repository corticotropin), which went for almost $44,000 a prescription, Kaiser said in its analysis, which used State Drug Utilization Data that are part of the Medicaid Drug Rebate Program.
The liver drives RBC elimination, iron recycling
“The liver, not the spleen, is the major on-demand site of red blood cell elimination and iron recycling,” according to Filip Swirski, PhD, of the Massachusetts General Hospital Center for Systems Biology, and his colleagues.
The liver relies on a buffer system consisting of bone marrow–derived monocytes that consume damaged red blood cells (RBCs) in the blood and settle in the liver, where they become the transient macrophages capable of iron recycling, the researchers concluded in a study published in Nature Medicine.
The study was designed to examine how the body clears old and damaged RBCs without releasing toxic levels of free iron. Damaged RBCs can release unbound forms of iron-carrying hemoglobin, which can cause kidney injury and can lead to anemia.
The researchers used several different models of RBC damage to examine RBC clearance and iron recycling in a mouse model. Damaged RBCs in the bloodstream prompted an increase in monocytes that took up the damaged cells and traveled to both the liver and the spleen. Within hours, almost all of the damaged RBCs were located within specialized macrophages seen only in the liver. Chemokines drew the monocytes to the liver, resulting in the accumulation of the iron-recycling macrophages.
Monocytes that express high levels of lymphocyte antigen 6 complex, locus C1 ingest stressed and senescent erythrocytes, accumulate in the liver via coordinated chemotactic cues, and differentiate into ferroportin 1–expressing macrophages that can deliver iron to hepatocytes. Monocyte-derived FPN1+Tim-4neg macrophages are transient, reside alongside embryonically derived T-cell immunoglobulin and mucin domain containing 4high Kupffer cells, and depend on the growth factor Csf1 and the transcription factor Nrf2, the researchers wrote.
Blocking that process resulted in impaired RBC clearance, toxic levels of free iron and hemoglobin, and signs of liver and kidney damage.
“If overactive, (the mechanism) could remove too many RBCs, but if (the mechanism is) sluggish or otherwise impaired, it could lead to iron toxicity. Further study could provide us with details of how this mechanism occurs in the first place and help us understand how to harness or suppress it in various conditions,” Dr. Swirski said in a press release.
The researchers had no financial conflicts of interest. The study was funded by the National Institutes of Health.
On Twitter @maryjodales
“The liver, not the spleen, is the major on-demand site of red blood cell elimination and iron recycling,” according to Filip Swirski, PhD, of the Massachusetts General Hospital Center for Systems Biology, and his colleagues.
The liver relies on a buffer system consisting of bone marrow–derived monocytes that consume damaged red blood cells (RBCs) in the blood and settle in the liver, where they become the transient macrophages capable of iron recycling, the researchers concluded in a study published in Nature Medicine.
The study was designed to examine how the body clears old and damaged RBCs without releasing toxic levels of free iron. Damaged RBCs can release unbound forms of iron-carrying hemoglobin, which can cause kidney injury and can lead to anemia.
The researchers used several different models of RBC damage to examine RBC clearance and iron recycling in a mouse model. Damaged RBCs in the bloodstream prompted an increase in monocytes that took up the damaged cells and traveled to both the liver and the spleen. Within hours, almost all of the damaged RBCs were located within specialized macrophages seen only in the liver. Chemokines drew the monocytes to the liver, resulting in the accumulation of the iron-recycling macrophages.
Monocytes that express high levels of lymphocyte antigen 6 complex, locus C1 ingest stressed and senescent erythrocytes, accumulate in the liver via coordinated chemotactic cues, and differentiate into ferroportin 1–expressing macrophages that can deliver iron to hepatocytes. Monocyte-derived FPN1+Tim-4neg macrophages are transient, reside alongside embryonically derived T-cell immunoglobulin and mucin domain containing 4high Kupffer cells, and depend on the growth factor Csf1 and the transcription factor Nrf2, the researchers wrote.
Blocking that process resulted in impaired RBC clearance, toxic levels of free iron and hemoglobin, and signs of liver and kidney damage.
“If overactive, (the mechanism) could remove too many RBCs, but if (the mechanism is) sluggish or otherwise impaired, it could lead to iron toxicity. Further study could provide us with details of how this mechanism occurs in the first place and help us understand how to harness or suppress it in various conditions,” Dr. Swirski said in a press release.
The researchers had no financial conflicts of interest. The study was funded by the National Institutes of Health.
On Twitter @maryjodales
“The liver, not the spleen, is the major on-demand site of red blood cell elimination and iron recycling,” according to Filip Swirski, PhD, of the Massachusetts General Hospital Center for Systems Biology, and his colleagues.
The liver relies on a buffer system consisting of bone marrow–derived monocytes that consume damaged red blood cells (RBCs) in the blood and settle in the liver, where they become the transient macrophages capable of iron recycling, the researchers concluded in a study published in Nature Medicine.
The study was designed to examine how the body clears old and damaged RBCs without releasing toxic levels of free iron. Damaged RBCs can release unbound forms of iron-carrying hemoglobin, which can cause kidney injury and can lead to anemia.
The researchers used several different models of RBC damage to examine RBC clearance and iron recycling in a mouse model. Damaged RBCs in the bloodstream prompted an increase in monocytes that took up the damaged cells and traveled to both the liver and the spleen. Within hours, almost all of the damaged RBCs were located within specialized macrophages seen only in the liver. Chemokines drew the monocytes to the liver, resulting in the accumulation of the iron-recycling macrophages.
Monocytes that express high levels of lymphocyte antigen 6 complex, locus C1 ingest stressed and senescent erythrocytes, accumulate in the liver via coordinated chemotactic cues, and differentiate into ferroportin 1–expressing macrophages that can deliver iron to hepatocytes. Monocyte-derived FPN1+Tim-4neg macrophages are transient, reside alongside embryonically derived T-cell immunoglobulin and mucin domain containing 4high Kupffer cells, and depend on the growth factor Csf1 and the transcription factor Nrf2, the researchers wrote.
Blocking that process resulted in impaired RBC clearance, toxic levels of free iron and hemoglobin, and signs of liver and kidney damage.
“If overactive, (the mechanism) could remove too many RBCs, but if (the mechanism is) sluggish or otherwise impaired, it could lead to iron toxicity. Further study could provide us with details of how this mechanism occurs in the first place and help us understand how to harness or suppress it in various conditions,” Dr. Swirski said in a press release.
The researchers had no financial conflicts of interest. The study was funded by the National Institutes of Health.
On Twitter @maryjodales
FROM NATURE MEDICINE
COMP recommends orphan status for drug to treat PNH

The European Medicines Agency’s Committee for Orphan Medicinal Products (COMP) has issued a positive opinion recommending orphan designation for Coversin for the treatment of paroxysmal nocturnal hemoglobinuria (PNH).
Coversin is a second-generation complement inhibitor that acts on complement component-C5, preventing release of C5a and formation of C5b-9 (also known as the membrane attack complex).
Coversin is a recombinant small protein (16,740 Da) derived from a native protein found in the saliva of the Ornithodoros moubata tick.
The drug is being developed by Akari Therapeutics.
In vitro experiments have shown that Coversin inhibits red blood cell lysis in PNH, and the drug can achieve full complement inhibition in the blood of PNH patients who are resistant to the drug eculizumab.
In a phase 1a trial of healthy volunteers, Coversin completely inhibited complement C5 activity within 12 hours of administration.
Akari Therapeutics is currently conducting a phase 1b study of Coversin in healthy volunteers and is administering the drug to a patient with eculizumab-resistant PNH. Thus far, Coversin has prevented hemolytic episodes and improved disease symptoms in this patient. And the only drug-related adverse event has been occasional local and transient irritation at the injection site.
Coversin is also being studied in atypical hemolytic uremic syndrome and Guillain Barré syndrome.
About orphan designation
The COMP adopts an opinion on the granting of orphan drug designation, and that opinion is submitted to the European Commission for a final decision.
Orphan designation provides regulatory and financial incentives for companies to develop and market therapies that treat a life-threatening or chronically debilitating condition affecting no more than 5 in 10,000 people in the European Union, and where no satisfactory treatment is available.
Orphan designation provides a 10-year period of marketing exclusivity in the European Union if the drug receives regulatory approval.
The designation also provides incentives for companies seeking protocol assistance from the European Medicines Agency during the product development phase and direct access to the centralized authorization procedure. ![]()
The European Medicines Agency’s Committee for Orphan Medicinal Products (COMP) has issued a positive opinion recommending orphan designation for Coversin for the treatment of paroxysmal nocturnal hemoglobinuria (PNH).
Coversin is a second-generation complement inhibitor that acts on complement component-C5, preventing release of C5a and formation of C5b-9 (also known as the membrane attack complex).
Coversin is a recombinant small protein (16,740 Da) derived from a native protein found in the saliva of the Ornithodoros moubata tick.
The drug is being developed by Akari Therapeutics.
In vitro experiments have shown that Coversin inhibits red blood cell lysis in PNH, and the drug can achieve full complement inhibition in the blood of PNH patients who are resistant to the drug eculizumab.
In a phase 1a trial of healthy volunteers, Coversin completely inhibited complement C5 activity within 12 hours of administration.
Akari Therapeutics is currently conducting a phase 1b study of Coversin in healthy volunteers and is administering the drug to a patient with eculizumab-resistant PNH. Thus far, Coversin has prevented hemolytic episodes and improved disease symptoms in this patient. And the only drug-related adverse event has been occasional local and transient irritation at the injection site.
Coversin is also being studied in atypical hemolytic uremic syndrome and Guillain Barré syndrome.
About orphan designation
The COMP adopts an opinion on the granting of orphan drug designation, and that opinion is submitted to the European Commission for a final decision.
Orphan designation provides regulatory and financial incentives for companies to develop and market therapies that treat a life-threatening or chronically debilitating condition affecting no more than 5 in 10,000 people in the European Union, and where no satisfactory treatment is available.
Orphan designation provides a 10-year period of marketing exclusivity in the European Union if the drug receives regulatory approval.
The designation also provides incentives for companies seeking protocol assistance from the European Medicines Agency during the product development phase and direct access to the centralized authorization procedure. ![]()
The European Medicines Agency’s Committee for Orphan Medicinal Products (COMP) has issued a positive opinion recommending orphan designation for Coversin for the treatment of paroxysmal nocturnal hemoglobinuria (PNH).
Coversin is a second-generation complement inhibitor that acts on complement component-C5, preventing release of C5a and formation of C5b-9 (also known as the membrane attack complex).
Coversin is a recombinant small protein (16,740 Da) derived from a native protein found in the saliva of the Ornithodoros moubata tick.
The drug is being developed by Akari Therapeutics.
In vitro experiments have shown that Coversin inhibits red blood cell lysis in PNH, and the drug can achieve full complement inhibition in the blood of PNH patients who are resistant to the drug eculizumab.
In a phase 1a trial of healthy volunteers, Coversin completely inhibited complement C5 activity within 12 hours of administration.
Akari Therapeutics is currently conducting a phase 1b study of Coversin in healthy volunteers and is administering the drug to a patient with eculizumab-resistant PNH. Thus far, Coversin has prevented hemolytic episodes and improved disease symptoms in this patient. And the only drug-related adverse event has been occasional local and transient irritation at the injection site.
Coversin is also being studied in atypical hemolytic uremic syndrome and Guillain Barré syndrome.
About orphan designation
The COMP adopts an opinion on the granting of orphan drug designation, and that opinion is submitted to the European Commission for a final decision.
Orphan designation provides regulatory and financial incentives for companies to develop and market therapies that treat a life-threatening or chronically debilitating condition affecting no more than 5 in 10,000 people in the European Union, and where no satisfactory treatment is available.
Orphan designation provides a 10-year period of marketing exclusivity in the European Union if the drug receives regulatory approval.
The designation also provides incentives for companies seeking protocol assistance from the European Medicines Agency during the product development phase and direct access to the centralized authorization procedure. ![]()
Mechanism proposed for microvascular thrombosis in thrombotic thrombocytopenic purpura
In patients with acquired autoimmune thrombotic thrombocytopenic purpura, elevated plasma levels of human neutrophil proteins 1-3 inhibit proteolytic cleavage of von Willebrand factor by ADAMTS13, Vikram G. Pillai, PhD, of the University of Alabama at Birmingham, and colleagues reported.
The finding may explain how inflammation triggers microvascular thrombosis in these patients and potentially others with immune thrombotic disorders, according to the researchers (Blood 2016;128:110-9).
They performed enzyme-linked immunosorbent assays and found markedly increased levels of plasma human neutrophil proteins (HNPs) 1-3 in most of the patients with acquired autoimmune thrombotic thrombocytopenic purpura (TTP). The median levels in the 19 patients were 170 ng/mL, compared with 23 ng/mL in 18 healthy controls, a statistically significant difference (P less than .0001).
Liquid chromatography plus tandem mass spectrometry similarly confirmed statistically significant increases in HNP1, HNP2, and HNP3 in patient samples (P less than .001).
Measures of HNPs 1-3 by both methods correlated well, and the researchers concluded that HNPs 1-3 likely inhibit ADAMTS13 activity by binding to the central A2 domain of von Willebrand factor and physically blocking ADAMTS13 binding.
The researchers had no relevant financial disclosures.
On Twitter @maryjodales
In patients with acquired autoimmune thrombotic thrombocytopenic purpura, elevated plasma levels of human neutrophil proteins 1-3 inhibit proteolytic cleavage of von Willebrand factor by ADAMTS13, Vikram G. Pillai, PhD, of the University of Alabama at Birmingham, and colleagues reported.
The finding may explain how inflammation triggers microvascular thrombosis in these patients and potentially others with immune thrombotic disorders, according to the researchers (Blood 2016;128:110-9).
They performed enzyme-linked immunosorbent assays and found markedly increased levels of plasma human neutrophil proteins (HNPs) 1-3 in most of the patients with acquired autoimmune thrombotic thrombocytopenic purpura (TTP). The median levels in the 19 patients were 170 ng/mL, compared with 23 ng/mL in 18 healthy controls, a statistically significant difference (P less than .0001).
Liquid chromatography plus tandem mass spectrometry similarly confirmed statistically significant increases in HNP1, HNP2, and HNP3 in patient samples (P less than .001).
Measures of HNPs 1-3 by both methods correlated well, and the researchers concluded that HNPs 1-3 likely inhibit ADAMTS13 activity by binding to the central A2 domain of von Willebrand factor and physically blocking ADAMTS13 binding.
The researchers had no relevant financial disclosures.
On Twitter @maryjodales
In patients with acquired autoimmune thrombotic thrombocytopenic purpura, elevated plasma levels of human neutrophil proteins 1-3 inhibit proteolytic cleavage of von Willebrand factor by ADAMTS13, Vikram G. Pillai, PhD, of the University of Alabama at Birmingham, and colleagues reported.
The finding may explain how inflammation triggers microvascular thrombosis in these patients and potentially others with immune thrombotic disorders, according to the researchers (Blood 2016;128:110-9).
They performed enzyme-linked immunosorbent assays and found markedly increased levels of plasma human neutrophil proteins (HNPs) 1-3 in most of the patients with acquired autoimmune thrombotic thrombocytopenic purpura (TTP). The median levels in the 19 patients were 170 ng/mL, compared with 23 ng/mL in 18 healthy controls, a statistically significant difference (P less than .0001).
Liquid chromatography plus tandem mass spectrometry similarly confirmed statistically significant increases in HNP1, HNP2, and HNP3 in patient samples (P less than .001).
Measures of HNPs 1-3 by both methods correlated well, and the researchers concluded that HNPs 1-3 likely inhibit ADAMTS13 activity by binding to the central A2 domain of von Willebrand factor and physically blocking ADAMTS13 binding.
The researchers had no relevant financial disclosures.
On Twitter @maryjodales
FROM BLOOD
Key clinical point: In patients with acquired autoimmune thrombotic thrombocytopenic purpura, elevated plasma levels of human neutrophil proteins 1-3 inhibit proteolytic cleavage of von Willebrand factor by ADAMTS13.
Major finding: The median levels of plasma human neutrophil proteins 1-3 in patients with acquired autoimmune TTP were 170 ng/mL, compared with 23 ng/mL in healthy controls, a statistically significant difference (P less than .0001).
Data source: Studies in 19 patients with TTP and 18 control subjects.
Disclosures: The researchers had no relevant financial disclosures.
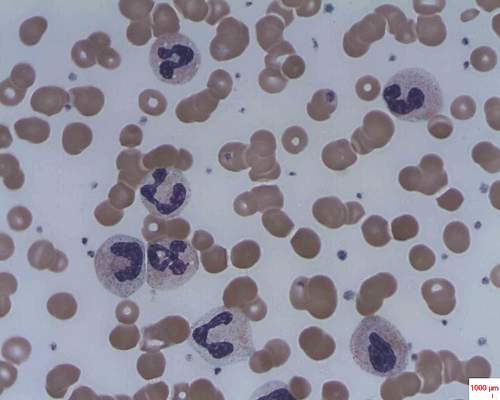
Dematin key to erythrocyte membrane stability in mice
Dematin is newly recognized as a protein that is crucial to red blood cell (RBC) membrane integrity, and dematin’s absence in mice resulted in severe abnormalities of erythrocyte shape, membrane stability, and hemolytic anemia, Yunzhe Lu of Tufts University, Boston, and her colleagues reported in the journal Blood.
The finding indicates that dematin is the major determinant of membrane stability within the junctional protein complex.
The researchers defined the role of dematin by designing a mouse model that lacked the protein. Affected mice developed severe anemia and had abnormally shaped erythrocytes with unstable membranes.
They examined the mechanism behind erythrocyte membrane instability in the mice by using membrane protein analysis, domain mapping, electron microscopy, and dynamic deformability measurements. Although many membrane and cytoskeletal proteins remained at their normal levels, spectrin, adducin, and actin were greatly reduced in these erythrocytes. The findings indicate that dematin plays a critical role in maintaining the fundamental properties of the erythrocyte’s membrane cytoskeleton complex, the researchers wrote (Blood 2016;128:93-103).
On Twitter @maryjodales
Can these findings in the erythrocytes of genetically altered mice be extrapolated to humans?
While similar, membrane composition differs in mouse and human erythrocytes. The junctional complex contains Rh polypeptides in mice but does not in humans. Glucose transporter 1 (Glut1), which associates with dematin and the adducins in humans, is not expressed in the mature erythrocytes of mice. The authors propose a model in which adducin stabilized by dematin provides linkage to the plasma membrane via band 3; however, the relatively mild phenotype seen in the alpha adducin knockout mouse argues for additional linkages, likely via dematin.
It will be important to determine the role of dematin and the effect of its deficiency in junctional complex assembly, in regulation of membrane deformability and stability in human erythrocytes, and in the context of its identified association with Glut1. Given the importance of phosphorylation in regulation of dematin-binding function and interactions, and in light of the gross disruptive effects of dematin absence reported in the study by Ms. Lu and her colleagues, investigation of the role of dematin modification in junctional protein complex assembly, enucleation and cytoskeletal remodeling, and response to malaria invasion of the red blood cell will all represent important areas of future research.
Timothy J. Satchwell, PhD, and Ashley M. Toye, PhD, of the University of Bristol, England, made their comments in an accompanying editorial (Blood. 2016;128:11-12).
Can these findings in the erythrocytes of genetically altered mice be extrapolated to humans?
While similar, membrane composition differs in mouse and human erythrocytes. The junctional complex contains Rh polypeptides in mice but does not in humans. Glucose transporter 1 (Glut1), which associates with dematin and the adducins in humans, is not expressed in the mature erythrocytes of mice. The authors propose a model in which adducin stabilized by dematin provides linkage to the plasma membrane via band 3; however, the relatively mild phenotype seen in the alpha adducin knockout mouse argues for additional linkages, likely via dematin.
It will be important to determine the role of dematin and the effect of its deficiency in junctional complex assembly, in regulation of membrane deformability and stability in human erythrocytes, and in the context of its identified association with Glut1. Given the importance of phosphorylation in regulation of dematin-binding function and interactions, and in light of the gross disruptive effects of dematin absence reported in the study by Ms. Lu and her colleagues, investigation of the role of dematin modification in junctional protein complex assembly, enucleation and cytoskeletal remodeling, and response to malaria invasion of the red blood cell will all represent important areas of future research.
Timothy J. Satchwell, PhD, and Ashley M. Toye, PhD, of the University of Bristol, England, made their comments in an accompanying editorial (Blood. 2016;128:11-12).
Can these findings in the erythrocytes of genetically altered mice be extrapolated to humans?
While similar, membrane composition differs in mouse and human erythrocytes. The junctional complex contains Rh polypeptides in mice but does not in humans. Glucose transporter 1 (Glut1), which associates with dematin and the adducins in humans, is not expressed in the mature erythrocytes of mice. The authors propose a model in which adducin stabilized by dematin provides linkage to the plasma membrane via band 3; however, the relatively mild phenotype seen in the alpha adducin knockout mouse argues for additional linkages, likely via dematin.
It will be important to determine the role of dematin and the effect of its deficiency in junctional complex assembly, in regulation of membrane deformability and stability in human erythrocytes, and in the context of its identified association with Glut1. Given the importance of phosphorylation in regulation of dematin-binding function and interactions, and in light of the gross disruptive effects of dematin absence reported in the study by Ms. Lu and her colleagues, investigation of the role of dematin modification in junctional protein complex assembly, enucleation and cytoskeletal remodeling, and response to malaria invasion of the red blood cell will all represent important areas of future research.
Timothy J. Satchwell, PhD, and Ashley M. Toye, PhD, of the University of Bristol, England, made their comments in an accompanying editorial (Blood. 2016;128:11-12).
Dematin is newly recognized as a protein that is crucial to red blood cell (RBC) membrane integrity, and dematin’s absence in mice resulted in severe abnormalities of erythrocyte shape, membrane stability, and hemolytic anemia, Yunzhe Lu of Tufts University, Boston, and her colleagues reported in the journal Blood.
The finding indicates that dematin is the major determinant of membrane stability within the junctional protein complex.
The researchers defined the role of dematin by designing a mouse model that lacked the protein. Affected mice developed severe anemia and had abnormally shaped erythrocytes with unstable membranes.
They examined the mechanism behind erythrocyte membrane instability in the mice by using membrane protein analysis, domain mapping, electron microscopy, and dynamic deformability measurements. Although many membrane and cytoskeletal proteins remained at their normal levels, spectrin, adducin, and actin were greatly reduced in these erythrocytes. The findings indicate that dematin plays a critical role in maintaining the fundamental properties of the erythrocyte’s membrane cytoskeleton complex, the researchers wrote (Blood 2016;128:93-103).
On Twitter @maryjodales
Dematin is newly recognized as a protein that is crucial to red blood cell (RBC) membrane integrity, and dematin’s absence in mice resulted in severe abnormalities of erythrocyte shape, membrane stability, and hemolytic anemia, Yunzhe Lu of Tufts University, Boston, and her colleagues reported in the journal Blood.
The finding indicates that dematin is the major determinant of membrane stability within the junctional protein complex.
The researchers defined the role of dematin by designing a mouse model that lacked the protein. Affected mice developed severe anemia and had abnormally shaped erythrocytes with unstable membranes.
They examined the mechanism behind erythrocyte membrane instability in the mice by using membrane protein analysis, domain mapping, electron microscopy, and dynamic deformability measurements. Although many membrane and cytoskeletal proteins remained at their normal levels, spectrin, adducin, and actin were greatly reduced in these erythrocytes. The findings indicate that dematin plays a critical role in maintaining the fundamental properties of the erythrocyte’s membrane cytoskeleton complex, the researchers wrote (Blood 2016;128:93-103).
On Twitter @maryjodales
FROM BLOOD
Key clinical point: Dematin is newly recognized as a protein crucial to the integrity of red blood cell membranes.
Major finding: Dematin’s absence in mice resulted in severe abnormalities of erythrocyte shape, membrane stability, and hemolytic anemia.
Data source: Studies in a newly created mouse model designed to lack dematin.
Disclosures: The researchers had no relevant financial disclosures.
Genetic therapy lowers joint bleeding in hemophilia B
COPENHAGEN – It’s early days yet, but results look highly promising for the ability of an experimental gene-transfer therapy to improve coagulation parameters in patients with severe hemophilia B.
In a phase I/II dose-escalation study, a single 1-hour infusion of the gene-transfer product, labeled SPK-9001, resulted in factor IX activity levels ranging from 25% to 39% of normal in four men with severe or moderately severe hemophilia B, reported Dr. Katherine A High, president and chief scientific officer of Spark Therapeutics, maker of the product.

“One of the most remarkable features of the data in my mind has been a very consistent performance,” she said at a briefing at the annual congress of the European Hematology Association.
The product consists of a vector containing a novel bio-engineered adeno-associated virus (AAV) capsid with tropism for liver, and a factor IX cassette that carries a strong liver-specific promoter to drive the expression of the factor IX variant, dubbed factor IX Padua.
“The hypothesis of the work was that if we could engineer a vector efficient enough, we would be able to infuse it at a dose low enough that it would drive loads of expression greater than 12% of normal, which in previous work has been shown to be associated with an absence of joint bleeds in natural history studies of people with mild disease, and that infusion at a low dose would eliminate the need for any type of immune suppression with steroids,” Dr. High said.
Other attempts at genetic engineering in patients with hemophilia B have been hampered by the need to use high doses of vector that can induce an immune response, thereby negating the benefit of therapy.
In this ongoing study, conducted in Mississippi, Pennsylvania, and California, males 18 and older with a confirmed diagnosis of hemophilia B (defined as equal to or less than 2 IU/dL or 2% endogenous factor IX) who have received 50 or more days of exposure to factor IX products are enrolled. The patients must have a minimum average of four bleeding events per year requiring episodic treatment of factor IX infusions or prophylactic factor IX infusions, no measurable factor IX inhibitor as assessed by the central laboratory, and no prior history of inhibitors to factor IX protein.
In an oral session at the congress, Dr. Spencer Sullivan, assistant professor of pediatrics and medicine at the University of Mississippi Medical Center in Jackson, presented data on four men whose age ranges from 18 to 47 years.
In the dose-escalation phase of the study, the patients received single infusions of SPK-9001 at an initial starting dose of 5 x 1011 vector genomes of body weight. They were followed for 7-26 weeks after gene transfer for factor IX activity levels, liver enzymes, bleeding episode, and factor usage. As of May 22, 2016, the first four patients showed factor IX activity levels of 32%, 39%, 25%, and 27% of normal, respectively.
All subjects are currently off prophylactic factor IX infusions. During the course of follow-up, one patient infused himself with factor IX once, treating himself 2 days after vector infusion for a suspected ankle bleed.
Asked by a reporter how durable the effect was, Dr. High replied that in dog models of hemophilia B the effect of the gene transfer has been durable.
The best evidence to date of durability in humans, she said, comes from investigators at University College in London (England), who found that if patients can make it past the first 8-10 weeks without developing an immune response to the transfer product, they are likely to do well, and to have a durable effect, she said.
Dr. Anton Hagenbeek, from the Academic Medical Center, University of Amsterdam, who moderated the briefing but was not involved in the study, said that Dr. High was “to be congratulated for these best data ever seen.”
He asked, facetiously, whether she thought that “thousands of patients would buy a ticket to Philadelphia.” The “City of Brotherly Love” is home to one of the trial sites and to Spark headquarters.
The study is sponsored by Spark Therapeutics and Pfizer. Dr. High is president and chief scientific officer of Spark. Dr. Sullivan and Dr. Hagenbeek reported no relevant disclosures.
COPENHAGEN – It’s early days yet, but results look highly promising for the ability of an experimental gene-transfer therapy to improve coagulation parameters in patients with severe hemophilia B.
In a phase I/II dose-escalation study, a single 1-hour infusion of the gene-transfer product, labeled SPK-9001, resulted in factor IX activity levels ranging from 25% to 39% of normal in four men with severe or moderately severe hemophilia B, reported Dr. Katherine A High, president and chief scientific officer of Spark Therapeutics, maker of the product.
“One of the most remarkable features of the data in my mind has been a very consistent performance,” she said at a briefing at the annual congress of the European Hematology Association.
The product consists of a vector containing a novel bio-engineered adeno-associated virus (AAV) capsid with tropism for liver, and a factor IX cassette that carries a strong liver-specific promoter to drive the expression of the factor IX variant, dubbed factor IX Padua.
“The hypothesis of the work was that if we could engineer a vector efficient enough, we would be able to infuse it at a dose low enough that it would drive loads of expression greater than 12% of normal, which in previous work has been shown to be associated with an absence of joint bleeds in natural history studies of people with mild disease, and that infusion at a low dose would eliminate the need for any type of immune suppression with steroids,” Dr. High said.
Other attempts at genetic engineering in patients with hemophilia B have been hampered by the need to use high doses of vector that can induce an immune response, thereby negating the benefit of therapy.
In this ongoing study, conducted in Mississippi, Pennsylvania, and California, males 18 and older with a confirmed diagnosis of hemophilia B (defined as equal to or less than 2 IU/dL or 2% endogenous factor IX) who have received 50 or more days of exposure to factor IX products are enrolled. The patients must have a minimum average of four bleeding events per year requiring episodic treatment of factor IX infusions or prophylactic factor IX infusions, no measurable factor IX inhibitor as assessed by the central laboratory, and no prior history of inhibitors to factor IX protein.
In an oral session at the congress, Dr. Spencer Sullivan, assistant professor of pediatrics and medicine at the University of Mississippi Medical Center in Jackson, presented data on four men whose age ranges from 18 to 47 years.
In the dose-escalation phase of the study, the patients received single infusions of SPK-9001 at an initial starting dose of 5 x 1011 vector genomes of body weight. They were followed for 7-26 weeks after gene transfer for factor IX activity levels, liver enzymes, bleeding episode, and factor usage. As of May 22, 2016, the first four patients showed factor IX activity levels of 32%, 39%, 25%, and 27% of normal, respectively.
All subjects are currently off prophylactic factor IX infusions. During the course of follow-up, one patient infused himself with factor IX once, treating himself 2 days after vector infusion for a suspected ankle bleed.
Asked by a reporter how durable the effect was, Dr. High replied that in dog models of hemophilia B the effect of the gene transfer has been durable.
The best evidence to date of durability in humans, she said, comes from investigators at University College in London (England), who found that if patients can make it past the first 8-10 weeks without developing an immune response to the transfer product, they are likely to do well, and to have a durable effect, she said.
Dr. Anton Hagenbeek, from the Academic Medical Center, University of Amsterdam, who moderated the briefing but was not involved in the study, said that Dr. High was “to be congratulated for these best data ever seen.”
He asked, facetiously, whether she thought that “thousands of patients would buy a ticket to Philadelphia.” The “City of Brotherly Love” is home to one of the trial sites and to Spark headquarters.
The study is sponsored by Spark Therapeutics and Pfizer. Dr. High is president and chief scientific officer of Spark. Dr. Sullivan and Dr. Hagenbeek reported no relevant disclosures.
COPENHAGEN – It’s early days yet, but results look highly promising for the ability of an experimental gene-transfer therapy to improve coagulation parameters in patients with severe hemophilia B.
In a phase I/II dose-escalation study, a single 1-hour infusion of the gene-transfer product, labeled SPK-9001, resulted in factor IX activity levels ranging from 25% to 39% of normal in four men with severe or moderately severe hemophilia B, reported Dr. Katherine A High, president and chief scientific officer of Spark Therapeutics, maker of the product.
“One of the most remarkable features of the data in my mind has been a very consistent performance,” she said at a briefing at the annual congress of the European Hematology Association.
The product consists of a vector containing a novel bio-engineered adeno-associated virus (AAV) capsid with tropism for liver, and a factor IX cassette that carries a strong liver-specific promoter to drive the expression of the factor IX variant, dubbed factor IX Padua.
“The hypothesis of the work was that if we could engineer a vector efficient enough, we would be able to infuse it at a dose low enough that it would drive loads of expression greater than 12% of normal, which in previous work has been shown to be associated with an absence of joint bleeds in natural history studies of people with mild disease, and that infusion at a low dose would eliminate the need for any type of immune suppression with steroids,” Dr. High said.
Other attempts at genetic engineering in patients with hemophilia B have been hampered by the need to use high doses of vector that can induce an immune response, thereby negating the benefit of therapy.
In this ongoing study, conducted in Mississippi, Pennsylvania, and California, males 18 and older with a confirmed diagnosis of hemophilia B (defined as equal to or less than 2 IU/dL or 2% endogenous factor IX) who have received 50 or more days of exposure to factor IX products are enrolled. The patients must have a minimum average of four bleeding events per year requiring episodic treatment of factor IX infusions or prophylactic factor IX infusions, no measurable factor IX inhibitor as assessed by the central laboratory, and no prior history of inhibitors to factor IX protein.
In an oral session at the congress, Dr. Spencer Sullivan, assistant professor of pediatrics and medicine at the University of Mississippi Medical Center in Jackson, presented data on four men whose age ranges from 18 to 47 years.
In the dose-escalation phase of the study, the patients received single infusions of SPK-9001 at an initial starting dose of 5 x 1011 vector genomes of body weight. They were followed for 7-26 weeks after gene transfer for factor IX activity levels, liver enzymes, bleeding episode, and factor usage. As of May 22, 2016, the first four patients showed factor IX activity levels of 32%, 39%, 25%, and 27% of normal, respectively.
All subjects are currently off prophylactic factor IX infusions. During the course of follow-up, one patient infused himself with factor IX once, treating himself 2 days after vector infusion for a suspected ankle bleed.
Asked by a reporter how durable the effect was, Dr. High replied that in dog models of hemophilia B the effect of the gene transfer has been durable.
The best evidence to date of durability in humans, she said, comes from investigators at University College in London (England), who found that if patients can make it past the first 8-10 weeks without developing an immune response to the transfer product, they are likely to do well, and to have a durable effect, she said.
Dr. Anton Hagenbeek, from the Academic Medical Center, University of Amsterdam, who moderated the briefing but was not involved in the study, said that Dr. High was “to be congratulated for these best data ever seen.”
He asked, facetiously, whether she thought that “thousands of patients would buy a ticket to Philadelphia.” The “City of Brotherly Love” is home to one of the trial sites and to Spark headquarters.
The study is sponsored by Spark Therapeutics and Pfizer. Dr. High is president and chief scientific officer of Spark. Dr. Sullivan and Dr. Hagenbeek reported no relevant disclosures.
At THE EHA CONGRESS
Key clinical point:. The experimental SPK-9001 is a gene transfer product carrying a strong factor IX promoter.
Major finding: A single one-hour infusion of SPK-9001 was associated with factor IX activity levels ranging from 25% to 39% of normal.
Data source: Dose-escalation cohort of four adult males in a phase I/II study.
Disclosures: The study is sponsored by Spark Therapeutics and Pfizer. Dr. High is president and chief scientific officer of Spark. Dr. Sullivan and Dr. Hagenbeek reported no relevant disclosures.

Novel anticoagulants linked to lower intracranial hemorrhages, higher GI bleeds
Copenhagen – A real-life comparison of major bleeding risk with anticoagulants showed that in-hospital mortality was lower with novel oral agents than with vitamin K antagonists, but the risk varied by bleeding site.
Novel oral anticoagulants (NOACs) such as dabigatran (Pradaxa), rivaroxaban (Xarelto), and apixaban (Eliquis) were associated in randomized phase III clinical trials with lower incidences of major bleeding than vitamin K antagonists (VKA) such as warfarin, and the newer, heavily advertised agents are widely used in clinical practice, noted Dr. Laura Franco of the Stroke Unit at the University of Perugia, Italy.
“But there is [little] information about the management and outcomes of major bleeding with NOACs in real life,” she said at a briefing at the annual congress of the European Hematology Association.

To rectify this situation, Dr. Franco and her colleagues at nine Italian hospitals and the NOAC registry based in Dresden, Germany, looked at 30-day mortality and other outcomes among 874 consecutive patients admitted for a major bleeding episode from September 2013 through May 2016. In all, 220 of the patients were on NOACs, and 654 were on VKAs.
Intracranial bleeds occurred in 44% of all patients, but were significantly less common among patients on NOACs than on VKAs (22% vs, 52%, respectively, odds ratio 0.26, P less than .001), whereas gastrointestinal bleeds, which occurred in 30% of all patients, were more frequent with the newer oral agents than with the older VKAs (46% vs. 30%, OR 2.69, P less than .001).
Deaths within 30 days of emergency department admission were less frequent with NOACs (10% vs. 17%, hazard ratio 0.56, P = .012).
“But when we analyzed the subpopulation of patients with different sites of bleeding, we saw that among intracranial hemorrhage, the rate was equal among NOAC and VKAs patients, 25% in both groups,” Dr. Franco said.
Deaths from gastrointestinal bleeding were numerically lower among patients on NOACs (7% vs. 10%), but this difference was not statistically significant.
“The great advantages of the new oral anticoagulants are the reduced risk in intracranial hemorrhage, so we showed that the lower mortality is not due to an intrinsic capacity to reduce deaths by NOACs as compared to VKAs, but to a different pattern of bleeding sites,” Dr. Franco said.
The study funding source was not disclosed. Dr. Franco did not report relevant disclosures.
Copenhagen – A real-life comparison of major bleeding risk with anticoagulants showed that in-hospital mortality was lower with novel oral agents than with vitamin K antagonists, but the risk varied by bleeding site.
Novel oral anticoagulants (NOACs) such as dabigatran (Pradaxa), rivaroxaban (Xarelto), and apixaban (Eliquis) were associated in randomized phase III clinical trials with lower incidences of major bleeding than vitamin K antagonists (VKA) such as warfarin, and the newer, heavily advertised agents are widely used in clinical practice, noted Dr. Laura Franco of the Stroke Unit at the University of Perugia, Italy.
“But there is [little] information about the management and outcomes of major bleeding with NOACs in real life,” she said at a briefing at the annual congress of the European Hematology Association.
To rectify this situation, Dr. Franco and her colleagues at nine Italian hospitals and the NOAC registry based in Dresden, Germany, looked at 30-day mortality and other outcomes among 874 consecutive patients admitted for a major bleeding episode from September 2013 through May 2016. In all, 220 of the patients were on NOACs, and 654 were on VKAs.
Intracranial bleeds occurred in 44% of all patients, but were significantly less common among patients on NOACs than on VKAs (22% vs, 52%, respectively, odds ratio 0.26, P less than .001), whereas gastrointestinal bleeds, which occurred in 30% of all patients, were more frequent with the newer oral agents than with the older VKAs (46% vs. 30%, OR 2.69, P less than .001).
Deaths within 30 days of emergency department admission were less frequent with NOACs (10% vs. 17%, hazard ratio 0.56, P = .012).
“But when we analyzed the subpopulation of patients with different sites of bleeding, we saw that among intracranial hemorrhage, the rate was equal among NOAC and VKAs patients, 25% in both groups,” Dr. Franco said.
Deaths from gastrointestinal bleeding were numerically lower among patients on NOACs (7% vs. 10%), but this difference was not statistically significant.
“The great advantages of the new oral anticoagulants are the reduced risk in intracranial hemorrhage, so we showed that the lower mortality is not due to an intrinsic capacity to reduce deaths by NOACs as compared to VKAs, but to a different pattern of bleeding sites,” Dr. Franco said.
The study funding source was not disclosed. Dr. Franco did not report relevant disclosures.
Copenhagen – A real-life comparison of major bleeding risk with anticoagulants showed that in-hospital mortality was lower with novel oral agents than with vitamin K antagonists, but the risk varied by bleeding site.
Novel oral anticoagulants (NOACs) such as dabigatran (Pradaxa), rivaroxaban (Xarelto), and apixaban (Eliquis) were associated in randomized phase III clinical trials with lower incidences of major bleeding than vitamin K antagonists (VKA) such as warfarin, and the newer, heavily advertised agents are widely used in clinical practice, noted Dr. Laura Franco of the Stroke Unit at the University of Perugia, Italy.
“But there is [little] information about the management and outcomes of major bleeding with NOACs in real life,” she said at a briefing at the annual congress of the European Hematology Association.
To rectify this situation, Dr. Franco and her colleagues at nine Italian hospitals and the NOAC registry based in Dresden, Germany, looked at 30-day mortality and other outcomes among 874 consecutive patients admitted for a major bleeding episode from September 2013 through May 2016. In all, 220 of the patients were on NOACs, and 654 were on VKAs.
Intracranial bleeds occurred in 44% of all patients, but were significantly less common among patients on NOACs than on VKAs (22% vs, 52%, respectively, odds ratio 0.26, P less than .001), whereas gastrointestinal bleeds, which occurred in 30% of all patients, were more frequent with the newer oral agents than with the older VKAs (46% vs. 30%, OR 2.69, P less than .001).
Deaths within 30 days of emergency department admission were less frequent with NOACs (10% vs. 17%, hazard ratio 0.56, P = .012).
“But when we analyzed the subpopulation of patients with different sites of bleeding, we saw that among intracranial hemorrhage, the rate was equal among NOAC and VKAs patients, 25% in both groups,” Dr. Franco said.
Deaths from gastrointestinal bleeding were numerically lower among patients on NOACs (7% vs. 10%), but this difference was not statistically significant.
“The great advantages of the new oral anticoagulants are the reduced risk in intracranial hemorrhage, so we showed that the lower mortality is not due to an intrinsic capacity to reduce deaths by NOACs as compared to VKAs, but to a different pattern of bleeding sites,” Dr. Franco said.
The study funding source was not disclosed. Dr. Franco did not report relevant disclosures.
At THE EHA CONGRESS
Key clinical point:. Lower rates of 30-day mortality from major bleeding with novel oral anticoagulants (NOACs) may be due to different patterns of bleeding sites.
Major finding: 30-day mortality rates were 10% with NOACs vs. 17% with vitamin K antagonists (hazard ratio 0.56, P = .012)
Data source: Study of 874 consecutive patients admitted to hospitals in Italy and Germany for major bleeding episodes.Disclosures: The study funding source was not disclosed. Dr. Franco did not report relevant disclosures.
