User login
ISCHEMIA substudy data don’t add up, cardiac surgeons say
A recent ISCHEMIA trial substudy is under scrutiny from surgeons for a data discrepancy, rekindling concerns about reliance on the landmark trial data in the latest coronary revascularization guidelines.
As previously reported, the main ISCHEMIA findings showed no significant benefit for an initial strategy of percutaneous coronary intervention (PCI) or coronary bypass graft surgery (CABG) over medical therapy in patients with stable moderate to severe ischemic heart disease.
The 2021 substudy by Reynolds et al. showed that coronary artery disease (CAD) severity, classified using the modified Duke Prognostic Index score, predicted 4-year mortality and myocardial infarction in the trial, whereas ischemia severity did not.

Cardiac surgeons Joseph F. Sabik III, MD, and Faisal Bakaeen, MD, however, spotted that only 40 patients are in the Duke category 6 group (three-vessel severe stenosis of at least 70% or two-vessel severe stenosis with a proximal left anterior descending lesion) in Supplemental tables 1 and 2, whereas 659 are in the main paper.
In addition, the Supplemental tables list the following:
- 659 patients in Duke group 5, not 894 as in the paper.
- 894 patients in Duke group 4, not 743 as in the paper.
- 743 patients in Duke group 3, not 179 as in the paper.
The surgeons penned a letter to Circulation early in April flagging the discrepancies, but say it was rejected April 15 because it was submitted outside the journal’s 6-week window for letters. They posted a public comment on the Remarq research platform, as advised by Circulation’s editorial office, and reached out directly to the authors and ISCHEMIA leadership.
“They just keep saying it’s a simple formatting error. Well, if it is a simple formatting error, then fix it,” Dr. Sabik, chair of surgery at University Hospitals Cleveland Medical Center, said in an interview. “But here we are now, a month later, and they still haven’t published our letter. Why? We’re the ones who identified the problem.”
Dr. Sabik said the accuracy of the data has important implications because the recent AHA/ACC/SCAI coronary revascularization guidelines used the ISCHEMIA data to downgrade the CABG recommendation for complex multivessel disease from class 1 to class 2B. Patients with a Duke 6 score are also typically the ones referred for CABG by today’s heart teams.
Several surgical societies have contested the guidelines, questioning whether the ISCHEMIA patients are truly reflective of those seen in clinical practice and questioning the decision to treat PCI and surgery as equivalent strategies to decrease ischemic events.
Dr. Bakaeen, from the Cleveland Clinic, told this news organization they don’t want a public battle over the data like the one that befell the EXCEL trial, and that it’s entirely possible the investigators might have inadvertently upgraded all the Duke score assignments by 1.
A systematic error, however, is more plausible than a formatting error, he said, because Supplemental tables 1 and 2 correspond exactly to the Duke 1 to Duke 7 sequence, suggesting the tables are correct and that the error might have occurred downstream, including in the manuscript.
The numbers should be consistent across all the ISCHEMIA manuscripts, Dr. Bakaeen added, but currently “don’t add up,” even after adjustment for different denominators, and especially for participants with left main disease.
They hope that publication of their letter, he said, will convince the authors to publicly share the data for patients in each of the seven modified Duke categories.
Lead author of the ISCHEMIA substudy, Harmony Reynolds, MD, New York (N.Y.) University Langone Health, told this news organization via email that as a result of a “formatting error in the transfer of data from the statistical output file to a Word document, data in Supplemental tables 1 and 2 were incorrect.”

She explained that they planned to present six, not seven, rows for the Duke score in the tables, collapsing the first two categories of nonobstructive disease (Duke 1-2), as they were in all other tables and figures. However, the Supplemental tables had incorrect row headings and because the Word program is designed to fill all available rows, it inserted the data from the output file into a seven-row table shell, duplicating the values for row 1 in the last row for left main disease of at least 50%.
“The data were correctly presented in the main manuscript tables and figures and in the remainder of the supplement, with a total of 659 patients in the subset with modified Duke prognostic index category 6 on coronary CT angiography,” Dr. Reynolds said.
She noted that Circulation will issue a correction. In addition, “we are in the process of preparing the data for public sharing soon. The data will include the Duke prognostic score at all levels.”
Circulation editor-in-chief Joseph A. Hill, MD, PhD, chief of cardiology at UT Southwestern Medical Center, Dallas, declined to be interviewed but confirmed via email that Dr. Bakaeen and Dr. Sabik’s letter and the correction will be published the week of May 16.
As for the delay, he said, “I received their reach-out just over 1 week ago, and per protocol, we conducted an internal evaluation of their allegations, which took a bit of time.”
A version of this article first appeared on Medscape.com.
A recent ISCHEMIA trial substudy is under scrutiny from surgeons for a data discrepancy, rekindling concerns about reliance on the landmark trial data in the latest coronary revascularization guidelines.
As previously reported, the main ISCHEMIA findings showed no significant benefit for an initial strategy of percutaneous coronary intervention (PCI) or coronary bypass graft surgery (CABG) over medical therapy in patients with stable moderate to severe ischemic heart disease.
The 2021 substudy by Reynolds et al. showed that coronary artery disease (CAD) severity, classified using the modified Duke Prognostic Index score, predicted 4-year mortality and myocardial infarction in the trial, whereas ischemia severity did not.
Cardiac surgeons Joseph F. Sabik III, MD, and Faisal Bakaeen, MD, however, spotted that only 40 patients are in the Duke category 6 group (three-vessel severe stenosis of at least 70% or two-vessel severe stenosis with a proximal left anterior descending lesion) in Supplemental tables 1 and 2, whereas 659 are in the main paper.
In addition, the Supplemental tables list the following:
- 659 patients in Duke group 5, not 894 as in the paper.
- 894 patients in Duke group 4, not 743 as in the paper.
- 743 patients in Duke group 3, not 179 as in the paper.
The surgeons penned a letter to Circulation early in April flagging the discrepancies, but say it was rejected April 15 because it was submitted outside the journal’s 6-week window for letters. They posted a public comment on the Remarq research platform, as advised by Circulation’s editorial office, and reached out directly to the authors and ISCHEMIA leadership.
“They just keep saying it’s a simple formatting error. Well, if it is a simple formatting error, then fix it,” Dr. Sabik, chair of surgery at University Hospitals Cleveland Medical Center, said in an interview. “But here we are now, a month later, and they still haven’t published our letter. Why? We’re the ones who identified the problem.”
Dr. Sabik said the accuracy of the data has important implications because the recent AHA/ACC/SCAI coronary revascularization guidelines used the ISCHEMIA data to downgrade the CABG recommendation for complex multivessel disease from class 1 to class 2B. Patients with a Duke 6 score are also typically the ones referred for CABG by today’s heart teams.
Several surgical societies have contested the guidelines, questioning whether the ISCHEMIA patients are truly reflective of those seen in clinical practice and questioning the decision to treat PCI and surgery as equivalent strategies to decrease ischemic events.
Dr. Bakaeen, from the Cleveland Clinic, told this news organization they don’t want a public battle over the data like the one that befell the EXCEL trial, and that it’s entirely possible the investigators might have inadvertently upgraded all the Duke score assignments by 1.
A systematic error, however, is more plausible than a formatting error, he said, because Supplemental tables 1 and 2 correspond exactly to the Duke 1 to Duke 7 sequence, suggesting the tables are correct and that the error might have occurred downstream, including in the manuscript.
The numbers should be consistent across all the ISCHEMIA manuscripts, Dr. Bakaeen added, but currently “don’t add up,” even after adjustment for different denominators, and especially for participants with left main disease.
They hope that publication of their letter, he said, will convince the authors to publicly share the data for patients in each of the seven modified Duke categories.
Lead author of the ISCHEMIA substudy, Harmony Reynolds, MD, New York (N.Y.) University Langone Health, told this news organization via email that as a result of a “formatting error in the transfer of data from the statistical output file to a Word document, data in Supplemental tables 1 and 2 were incorrect.”
She explained that they planned to present six, not seven, rows for the Duke score in the tables, collapsing the first two categories of nonobstructive disease (Duke 1-2), as they were in all other tables and figures. However, the Supplemental tables had incorrect row headings and because the Word program is designed to fill all available rows, it inserted the data from the output file into a seven-row table shell, duplicating the values for row 1 in the last row for left main disease of at least 50%.
“The data were correctly presented in the main manuscript tables and figures and in the remainder of the supplement, with a total of 659 patients in the subset with modified Duke prognostic index category 6 on coronary CT angiography,” Dr. Reynolds said.
She noted that Circulation will issue a correction. In addition, “we are in the process of preparing the data for public sharing soon. The data will include the Duke prognostic score at all levels.”
Circulation editor-in-chief Joseph A. Hill, MD, PhD, chief of cardiology at UT Southwestern Medical Center, Dallas, declined to be interviewed but confirmed via email that Dr. Bakaeen and Dr. Sabik’s letter and the correction will be published the week of May 16.
As for the delay, he said, “I received their reach-out just over 1 week ago, and per protocol, we conducted an internal evaluation of their allegations, which took a bit of time.”
A version of this article first appeared on Medscape.com.
A recent ISCHEMIA trial substudy is under scrutiny from surgeons for a data discrepancy, rekindling concerns about reliance on the landmark trial data in the latest coronary revascularization guidelines.
As previously reported, the main ISCHEMIA findings showed no significant benefit for an initial strategy of percutaneous coronary intervention (PCI) or coronary bypass graft surgery (CABG) over medical therapy in patients with stable moderate to severe ischemic heart disease.
The 2021 substudy by Reynolds et al. showed that coronary artery disease (CAD) severity, classified using the modified Duke Prognostic Index score, predicted 4-year mortality and myocardial infarction in the trial, whereas ischemia severity did not.
Cardiac surgeons Joseph F. Sabik III, MD, and Faisal Bakaeen, MD, however, spotted that only 40 patients are in the Duke category 6 group (three-vessel severe stenosis of at least 70% or two-vessel severe stenosis with a proximal left anterior descending lesion) in Supplemental tables 1 and 2, whereas 659 are in the main paper.
In addition, the Supplemental tables list the following:
- 659 patients in Duke group 5, not 894 as in the paper.
- 894 patients in Duke group 4, not 743 as in the paper.
- 743 patients in Duke group 3, not 179 as in the paper.
The surgeons penned a letter to Circulation early in April flagging the discrepancies, but say it was rejected April 15 because it was submitted outside the journal’s 6-week window for letters. They posted a public comment on the Remarq research platform, as advised by Circulation’s editorial office, and reached out directly to the authors and ISCHEMIA leadership.
“They just keep saying it’s a simple formatting error. Well, if it is a simple formatting error, then fix it,” Dr. Sabik, chair of surgery at University Hospitals Cleveland Medical Center, said in an interview. “But here we are now, a month later, and they still haven’t published our letter. Why? We’re the ones who identified the problem.”
Dr. Sabik said the accuracy of the data has important implications because the recent AHA/ACC/SCAI coronary revascularization guidelines used the ISCHEMIA data to downgrade the CABG recommendation for complex multivessel disease from class 1 to class 2B. Patients with a Duke 6 score are also typically the ones referred for CABG by today’s heart teams.
Several surgical societies have contested the guidelines, questioning whether the ISCHEMIA patients are truly reflective of those seen in clinical practice and questioning the decision to treat PCI and surgery as equivalent strategies to decrease ischemic events.
Dr. Bakaeen, from the Cleveland Clinic, told this news organization they don’t want a public battle over the data like the one that befell the EXCEL trial, and that it’s entirely possible the investigators might have inadvertently upgraded all the Duke score assignments by 1.
A systematic error, however, is more plausible than a formatting error, he said, because Supplemental tables 1 and 2 correspond exactly to the Duke 1 to Duke 7 sequence, suggesting the tables are correct and that the error might have occurred downstream, including in the manuscript.
The numbers should be consistent across all the ISCHEMIA manuscripts, Dr. Bakaeen added, but currently “don’t add up,” even after adjustment for different denominators, and especially for participants with left main disease.
They hope that publication of their letter, he said, will convince the authors to publicly share the data for patients in each of the seven modified Duke categories.
Lead author of the ISCHEMIA substudy, Harmony Reynolds, MD, New York (N.Y.) University Langone Health, told this news organization via email that as a result of a “formatting error in the transfer of data from the statistical output file to a Word document, data in Supplemental tables 1 and 2 were incorrect.”
She explained that they planned to present six, not seven, rows for the Duke score in the tables, collapsing the first two categories of nonobstructive disease (Duke 1-2), as they were in all other tables and figures. However, the Supplemental tables had incorrect row headings and because the Word program is designed to fill all available rows, it inserted the data from the output file into a seven-row table shell, duplicating the values for row 1 in the last row for left main disease of at least 50%.
“The data were correctly presented in the main manuscript tables and figures and in the remainder of the supplement, with a total of 659 patients in the subset with modified Duke prognostic index category 6 on coronary CT angiography,” Dr. Reynolds said.
She noted that Circulation will issue a correction. In addition, “we are in the process of preparing the data for public sharing soon. The data will include the Duke prognostic score at all levels.”
Circulation editor-in-chief Joseph A. Hill, MD, PhD, chief of cardiology at UT Southwestern Medical Center, Dallas, declined to be interviewed but confirmed via email that Dr. Bakaeen and Dr. Sabik’s letter and the correction will be published the week of May 16.
As for the delay, he said, “I received their reach-out just over 1 week ago, and per protocol, we conducted an internal evaluation of their allegations, which took a bit of time.”
A version of this article first appeared on Medscape.com.
Bronchoscopic lung reduction boosts survival in severe COPD
Bronchoscopic lung volume reduction significantly increased survival in patients with severe chronic obstructive pulmonary disease, based on data from more than 1,400 individuals.
Previous studies have shown that patients with severe chronic obstructive pulmonary disease (COPD) can benefit from treatment with bronchoscopic lung volume reduction (BLVR) involving lung volume reduction coils or endobronchial valves (EBVs) in terms of improved pulmonary function, lung volume, exercise capacity, and quality of life.
However, data on the impact of the procedure on patient survival are limited, and most previous studies have been small, wrote Jorine E. Hartman, MD, of the University of Groningen, the Netherlands, and colleagues.
In a study published in Respiratory Medicine, the researchers reviewed data from 1,471 patients with severe COPD who had consultations for BLVR at a single center between June 2006 and July 2019. Of these, 483 (33%) underwent a BLVR treatment.
The follow-up period ranged from 633 days to 5,401 days. During this time, 531 patients died (35%); 165 of these (34%) were in the BLVR group.
Overall, the median survival of BLVR patients was significantly longer, compared with those who did not have the procedure, for a difference of approximately 1.7 years (3,133 days vs. 2,503 days, P < .001). No significant differences in survival were noted in BLVR patients treated with coils or EBVs.
The average age of the study population at baseline was 61 years, and 63% were women. Overall, patients treated with BLVR were more likely to be younger and female, with fewer COPD exacerbations but worse pulmonary function, as well as lower body mass index and more evidence of emphysema than the untreated patients, the researchers noted. Patients treated with BLVR also were more likely than untreated patients to have a history of myocardial infarction, percutaneous coronary intervention, or stroke.
However, BLVR was a significant independent predictor of survival after controlling for multiple variables, including age, sex, and disease severity, the researchers noted.
The current study supports existing literature on the value of BLVR for severe COPD but stands out from previous studies by comparing patients who underwent BLVR with those who did not, the researchers noted in their discussion of the findings.
The study findings were limited by several factors, including the fact that the non-treated patients were not eligible for treatment for various reasons that might have impacted survival, the researchers noted. Another limitation was the lack of data on cause of death and other medical events and treatments during the follow-up period, they said.
However, the results were strengthened by the large sample size and long-term follow-up and suggest that “reducing lung volume in patients with COPD and severe hyperinflation and reduced life expectancy may lead to a survival benefit,” they concluded.
The study received no outside funding. Dr. Hartman had no financial conflicts to disclose.
Bronchoscopic lung volume reduction significantly increased survival in patients with severe chronic obstructive pulmonary disease, based on data from more than 1,400 individuals.
Previous studies have shown that patients with severe chronic obstructive pulmonary disease (COPD) can benefit from treatment with bronchoscopic lung volume reduction (BLVR) involving lung volume reduction coils or endobronchial valves (EBVs) in terms of improved pulmonary function, lung volume, exercise capacity, and quality of life.
However, data on the impact of the procedure on patient survival are limited, and most previous studies have been small, wrote Jorine E. Hartman, MD, of the University of Groningen, the Netherlands, and colleagues.
In a study published in Respiratory Medicine, the researchers reviewed data from 1,471 patients with severe COPD who had consultations for BLVR at a single center between June 2006 and July 2019. Of these, 483 (33%) underwent a BLVR treatment.
The follow-up period ranged from 633 days to 5,401 days. During this time, 531 patients died (35%); 165 of these (34%) were in the BLVR group.
Overall, the median survival of BLVR patients was significantly longer, compared with those who did not have the procedure, for a difference of approximately 1.7 years (3,133 days vs. 2,503 days, P < .001). No significant differences in survival were noted in BLVR patients treated with coils or EBVs.
The average age of the study population at baseline was 61 years, and 63% were women. Overall, patients treated with BLVR were more likely to be younger and female, with fewer COPD exacerbations but worse pulmonary function, as well as lower body mass index and more evidence of emphysema than the untreated patients, the researchers noted. Patients treated with BLVR also were more likely than untreated patients to have a history of myocardial infarction, percutaneous coronary intervention, or stroke.
However, BLVR was a significant independent predictor of survival after controlling for multiple variables, including age, sex, and disease severity, the researchers noted.
The current study supports existing literature on the value of BLVR for severe COPD but stands out from previous studies by comparing patients who underwent BLVR with those who did not, the researchers noted in their discussion of the findings.
The study findings were limited by several factors, including the fact that the non-treated patients were not eligible for treatment for various reasons that might have impacted survival, the researchers noted. Another limitation was the lack of data on cause of death and other medical events and treatments during the follow-up period, they said.
However, the results were strengthened by the large sample size and long-term follow-up and suggest that “reducing lung volume in patients with COPD and severe hyperinflation and reduced life expectancy may lead to a survival benefit,” they concluded.
The study received no outside funding. Dr. Hartman had no financial conflicts to disclose.
Bronchoscopic lung volume reduction significantly increased survival in patients with severe chronic obstructive pulmonary disease, based on data from more than 1,400 individuals.
Previous studies have shown that patients with severe chronic obstructive pulmonary disease (COPD) can benefit from treatment with bronchoscopic lung volume reduction (BLVR) involving lung volume reduction coils or endobronchial valves (EBVs) in terms of improved pulmonary function, lung volume, exercise capacity, and quality of life.
However, data on the impact of the procedure on patient survival are limited, and most previous studies have been small, wrote Jorine E. Hartman, MD, of the University of Groningen, the Netherlands, and colleagues.
In a study published in Respiratory Medicine, the researchers reviewed data from 1,471 patients with severe COPD who had consultations for BLVR at a single center between June 2006 and July 2019. Of these, 483 (33%) underwent a BLVR treatment.
The follow-up period ranged from 633 days to 5,401 days. During this time, 531 patients died (35%); 165 of these (34%) were in the BLVR group.
Overall, the median survival of BLVR patients was significantly longer, compared with those who did not have the procedure, for a difference of approximately 1.7 years (3,133 days vs. 2,503 days, P < .001). No significant differences in survival were noted in BLVR patients treated with coils or EBVs.
The average age of the study population at baseline was 61 years, and 63% were women. Overall, patients treated with BLVR were more likely to be younger and female, with fewer COPD exacerbations but worse pulmonary function, as well as lower body mass index and more evidence of emphysema than the untreated patients, the researchers noted. Patients treated with BLVR also were more likely than untreated patients to have a history of myocardial infarction, percutaneous coronary intervention, or stroke.
However, BLVR was a significant independent predictor of survival after controlling for multiple variables, including age, sex, and disease severity, the researchers noted.
The current study supports existing literature on the value of BLVR for severe COPD but stands out from previous studies by comparing patients who underwent BLVR with those who did not, the researchers noted in their discussion of the findings.
The study findings were limited by several factors, including the fact that the non-treated patients were not eligible for treatment for various reasons that might have impacted survival, the researchers noted. Another limitation was the lack of data on cause of death and other medical events and treatments during the follow-up period, they said.
However, the results were strengthened by the large sample size and long-term follow-up and suggest that “reducing lung volume in patients with COPD and severe hyperinflation and reduced life expectancy may lead to a survival benefit,” they concluded.
The study received no outside funding. Dr. Hartman had no financial conflicts to disclose.
FROM RESPIRATORY MEDICINE
Porcine virus a suspect in man’s death after pig heart transplant
A porcine cytomegalovirus (PCMV) in the heart had gone undetected before the operation and may or may not have been instrumental in David Bennett’s death 2 months later, according to a report published in MIT Technology Review.
“The issue is now a subject of wide discussion among specialists, who think the infection was a potential contributor to Mr. Bennett’s death and a possible reason why the heart did not last longer,” states the article, written by staff journalist Antonio Regalado.
As described in the story, the xenotransplant saga’s new twist comes from the surgeon who performed the operation, Bartley P. Griffith, MD, University of Maryland, Baltimore, who related the PCMV finding in an April 20 online presentation hosted by the American Society of Transplantation.
Mr. Bennett’s initially promising but later turbulent clinical course, described by his surgeons and widely reported upon his death, included repeated skirmishes with infection and retaliatory adjustments to his immunosuppressant regimen. Those episodes were thought to have contributed to his death, the actual cause of which is undetermined or at least not yet reported.
“We are beginning to learn why he passed on,” Dr. Griffith said in Mr. Regalado’s article, acknowledging further that the porcine virus “maybe was the actor, or could be the actor,” that set off the events leading to Bennett’s death.
Xenotransplant specialists know that PCMV is a potential problem with pig organs and know to test for it before attempting the procedure in animal models, notes the article. It refers to a published series of pig-heart transplants to baboons in Germany. The hearts “lasted only a couple of weeks if the virus was present, while organs free from the infection could survive more than half a year.”
The heart Mr. Bennett received had been extensively screened for bacteria, viruses, and other issues that could have threatened the organ and Mr. Bennett, but the effort apparently fell short. In the MIT Technology Review story, the first author of the German baboon series speculates on how the University of Maryland team might have missed PCMV.
“The U.S. team appears to have tested the pig’s snout for the virus, but often it is lurking deeper in the tissues,” Joachim Denner, PhD, Institute of Virology, Free University of Berlin, said in the article. The virus, he contended, “can be detected and easily removed from pig populations, but unfortunately they didn’t use a good assay and didn’t detect the virus.”
That PCMV escaped detection before the operation “could now factor into some people’s questions over whether the experiment should have taken place at all,” the MIT Technology Review article proposes. “It’s a big red flag,” bioethicist Arthur Caplan, PhD, New York University, said in a quote, adding: “If doctors can’t prevent or control infection, ‘then such experiments are tough to justify.’ ”
A version of this article first appeared on Medscape.com.
A porcine cytomegalovirus (PCMV) in the heart had gone undetected before the operation and may or may not have been instrumental in David Bennett’s death 2 months later, according to a report published in MIT Technology Review.
“The issue is now a subject of wide discussion among specialists, who think the infection was a potential contributor to Mr. Bennett’s death and a possible reason why the heart did not last longer,” states the article, written by staff journalist Antonio Regalado.
As described in the story, the xenotransplant saga’s new twist comes from the surgeon who performed the operation, Bartley P. Griffith, MD, University of Maryland, Baltimore, who related the PCMV finding in an April 20 online presentation hosted by the American Society of Transplantation.
Mr. Bennett’s initially promising but later turbulent clinical course, described by his surgeons and widely reported upon his death, included repeated skirmishes with infection and retaliatory adjustments to his immunosuppressant regimen. Those episodes were thought to have contributed to his death, the actual cause of which is undetermined or at least not yet reported.
“We are beginning to learn why he passed on,” Dr. Griffith said in Mr. Regalado’s article, acknowledging further that the porcine virus “maybe was the actor, or could be the actor,” that set off the events leading to Bennett’s death.
Xenotransplant specialists know that PCMV is a potential problem with pig organs and know to test for it before attempting the procedure in animal models, notes the article. It refers to a published series of pig-heart transplants to baboons in Germany. The hearts “lasted only a couple of weeks if the virus was present, while organs free from the infection could survive more than half a year.”
The heart Mr. Bennett received had been extensively screened for bacteria, viruses, and other issues that could have threatened the organ and Mr. Bennett, but the effort apparently fell short. In the MIT Technology Review story, the first author of the German baboon series speculates on how the University of Maryland team might have missed PCMV.
“The U.S. team appears to have tested the pig’s snout for the virus, but often it is lurking deeper in the tissues,” Joachim Denner, PhD, Institute of Virology, Free University of Berlin, said in the article. The virus, he contended, “can be detected and easily removed from pig populations, but unfortunately they didn’t use a good assay and didn’t detect the virus.”
That PCMV escaped detection before the operation “could now factor into some people’s questions over whether the experiment should have taken place at all,” the MIT Technology Review article proposes. “It’s a big red flag,” bioethicist Arthur Caplan, PhD, New York University, said in a quote, adding: “If doctors can’t prevent or control infection, ‘then such experiments are tough to justify.’ ”
A version of this article first appeared on Medscape.com.
A porcine cytomegalovirus (PCMV) in the heart had gone undetected before the operation and may or may not have been instrumental in David Bennett’s death 2 months later, according to a report published in MIT Technology Review.
“The issue is now a subject of wide discussion among specialists, who think the infection was a potential contributor to Mr. Bennett’s death and a possible reason why the heart did not last longer,” states the article, written by staff journalist Antonio Regalado.
As described in the story, the xenotransplant saga’s new twist comes from the surgeon who performed the operation, Bartley P. Griffith, MD, University of Maryland, Baltimore, who related the PCMV finding in an April 20 online presentation hosted by the American Society of Transplantation.
Mr. Bennett’s initially promising but later turbulent clinical course, described by his surgeons and widely reported upon his death, included repeated skirmishes with infection and retaliatory adjustments to his immunosuppressant regimen. Those episodes were thought to have contributed to his death, the actual cause of which is undetermined or at least not yet reported.
“We are beginning to learn why he passed on,” Dr. Griffith said in Mr. Regalado’s article, acknowledging further that the porcine virus “maybe was the actor, or could be the actor,” that set off the events leading to Bennett’s death.
Xenotransplant specialists know that PCMV is a potential problem with pig organs and know to test for it before attempting the procedure in animal models, notes the article. It refers to a published series of pig-heart transplants to baboons in Germany. The hearts “lasted only a couple of weeks if the virus was present, while organs free from the infection could survive more than half a year.”
The heart Mr. Bennett received had been extensively screened for bacteria, viruses, and other issues that could have threatened the organ and Mr. Bennett, but the effort apparently fell short. In the MIT Technology Review story, the first author of the German baboon series speculates on how the University of Maryland team might have missed PCMV.
“The U.S. team appears to have tested the pig’s snout for the virus, but often it is lurking deeper in the tissues,” Joachim Denner, PhD, Institute of Virology, Free University of Berlin, said in the article. The virus, he contended, “can be detected and easily removed from pig populations, but unfortunately they didn’t use a good assay and didn’t detect the virus.”
That PCMV escaped detection before the operation “could now factor into some people’s questions over whether the experiment should have taken place at all,” the MIT Technology Review article proposes. “It’s a big red flag,” bioethicist Arthur Caplan, PhD, New York University, said in a quote, adding: “If doctors can’t prevent or control infection, ‘then such experiments are tough to justify.’ ”
A version of this article first appeared on Medscape.com.
FROM MIT TECHNOLOGY REVIEW
First-ever best practices for percutaneous axillary access
The Society for Cardiovascular Angiography and Interventions (SCAI) has issued the first statement on best practices for percutaneous axillary arterial access and training.
The position statement helps fill a gap amid increasing use of transaxillary access as an alternative to the femoral route for large-bore transcatheter aortic valve replacement (TAVR), endovascular aortic repair (EVAR), and mechanical circulatory support.
“The need for alternative access has increased as we are using more and more TAVR for our elderly population, and EVAR has also increased,” writing committee chair Arnold H. Seto, MD, Long Beach VA Health Care System (California) said in an interview. “There’s also a set of patients who require balloon pumps for a prolonged period, and people were using balloon pumps from the axillary approach, which were not custom-designed for that purpose.”
He noted that the evidence base leans heavily on case reports and case series, and that they were approached for guidance by a vendor developing a balloon pump specific to axillary access. “So that helped spur all of us to get together and decide to write up something on this topic, which was developing, but was certainly picking up steam rapidly.”
The statement was published in the Journal of the Society for Cardiovascular Angiography and Interventions, and it reflects the consensus of experts in heart failure, interventional cardiology and radiology, and cardiothoracic and vascular surgery. It reviews anatomic considerations and risks for percutaneous axillary access and suggests techniques for insertion, closure, and complication management.
Although the femoral artery is the most frequent access site for percutaneous large-bore procedures, the document notes that this approach may be limited in 13%-20% of patients because of prior surgeries or severe aortoiliac and/or iliofemoral atherosclerotic disease, tortuosity, or calcification.
“Absolutely, the femoral should be the predominant access site,” Dr. Seto said. Whenever there is a compromised femoral artery, “the axillary artery, which is rarely involved with atherosclerosis, makes for the most optimal alternative access. Other forms of alternative access, including transcaval and transcarotid, are possible but have their own issues and difficulties.”
Axillary access has traditionally been done through an open surgical approach, which allows for direct puncture, primary arterial repair, or placement of a sidearm conduit. Percutaneous transaxillary access avoids a surgical incision and general anesthesia and, theoretically, reduces the risk of infection, he said. It also allows for better mobility for patients, for example, who may have a balloon pump in place for weeks or even a month when waiting for a bridge to transplant.
In terms of technique, key recommendations include:
- Gaining access preferably through the left axillary
- Inserting the needle directly through the pectoralis minor into the second segment of the axillary artery
- Using a shallow-needle angle of 25-30 degrees to improve access success and decrease sheath malformation, kinking, bleeding, or vessel perforation
- Using micropuncture needles to minimize trauma to adjacent tissues
- Abducting the patient’s arm to 45-90 degrees to reduce tortuosity
- Using angiographic and ultrasound techniques to optimize vascular access
The latter point was the one area of debate among the writing committee, Dr. Seto observed. “That is one of the controversies: Should we make ultrasound mandatory? ... Everybody agreed that it can be quite useful and was likely to be useful because of its success in every other access area,” he said. “But in the absence of randomized evidence, we couldn’t make it mandatory or a strong recommendation. We just had to make it one of several options for the operator.”
The document highlights the need for familiarity with potential axillary artery complications and their management, noting that the axillary is more fragile than the femoral artery and, thus, potentially more prone to complications during instrumentation.
Data from the ARMS study in 102 patients undergoing transaxillary access for mechanical hemodynamic support reported 17 procedural complications, including 10 minor access site bleeding events, one stroke, and one pseudoaneurysm. A small study of 25 complex EVAR procedures reported a perioperative access complication rate of 8%, including one axillary artery dissection and one stenosis.
“Despite the brachial plexus being around there, there’s actually rare reports of neurologic injury and certainly none that have been permanent,” Dr. Seto said. “Also, stroke risk is probably more related to your device size and type of device rather than the approach itself.”
A significant amount of the paper is also devoted to training and privileging suggestions with an emphasis on a multidisciplinary team. The writing group recommends graduate medical education programs develop training curricula in percutaneous axillary artery access.
Those already in practice should participate in a formal training program that focuses on axillary artery anatomy, training in large bore access and closure devices, and didactic training in imaging modalities as applied to the axillary artery. Training can occur hands-on or using online simulations.
They also recommend outlining the potential need or role for proctoring and call for ongoing formal professional monitoring programs to evaluate operator outcomes using local or registry data.
“From a privileging standpoint, it was important for hospitals to be equally fair, regardless of the specialty that a requesting practitioner came from,” Dr. Seto said. “In other words, treat the vascular surgeons and interventional cardiologists and radiologists equally in terms of who has the privilege to do transaxillary access.”
The SCAI position statement has been endorsed by the American College of Cardiology, the Heart Failure Society of America, the Society of Interventional Radiology, and the Vascular & Endovascular Surgery Society.
Dr. Seto reported receiving honoraria from Getinge prior to initiation of the document. Disclosures for the rest of the writing group are available with the original article.
A version of this article first appeared on Medscape.com.
The Society for Cardiovascular Angiography and Interventions (SCAI) has issued the first statement on best practices for percutaneous axillary arterial access and training.
The position statement helps fill a gap amid increasing use of transaxillary access as an alternative to the femoral route for large-bore transcatheter aortic valve replacement (TAVR), endovascular aortic repair (EVAR), and mechanical circulatory support.
“The need for alternative access has increased as we are using more and more TAVR for our elderly population, and EVAR has also increased,” writing committee chair Arnold H. Seto, MD, Long Beach VA Health Care System (California) said in an interview. “There’s also a set of patients who require balloon pumps for a prolonged period, and people were using balloon pumps from the axillary approach, which were not custom-designed for that purpose.”
He noted that the evidence base leans heavily on case reports and case series, and that they were approached for guidance by a vendor developing a balloon pump specific to axillary access. “So that helped spur all of us to get together and decide to write up something on this topic, which was developing, but was certainly picking up steam rapidly.”
The statement was published in the Journal of the Society for Cardiovascular Angiography and Interventions, and it reflects the consensus of experts in heart failure, interventional cardiology and radiology, and cardiothoracic and vascular surgery. It reviews anatomic considerations and risks for percutaneous axillary access and suggests techniques for insertion, closure, and complication management.
Although the femoral artery is the most frequent access site for percutaneous large-bore procedures, the document notes that this approach may be limited in 13%-20% of patients because of prior surgeries or severe aortoiliac and/or iliofemoral atherosclerotic disease, tortuosity, or calcification.
“Absolutely, the femoral should be the predominant access site,” Dr. Seto said. Whenever there is a compromised femoral artery, “the axillary artery, which is rarely involved with atherosclerosis, makes for the most optimal alternative access. Other forms of alternative access, including transcaval and transcarotid, are possible but have their own issues and difficulties.”
Axillary access has traditionally been done through an open surgical approach, which allows for direct puncture, primary arterial repair, or placement of a sidearm conduit. Percutaneous transaxillary access avoids a surgical incision and general anesthesia and, theoretically, reduces the risk of infection, he said. It also allows for better mobility for patients, for example, who may have a balloon pump in place for weeks or even a month when waiting for a bridge to transplant.
In terms of technique, key recommendations include:
- Gaining access preferably through the left axillary
- Inserting the needle directly through the pectoralis minor into the second segment of the axillary artery
- Using a shallow-needle angle of 25-30 degrees to improve access success and decrease sheath malformation, kinking, bleeding, or vessel perforation
- Using micropuncture needles to minimize trauma to adjacent tissues
- Abducting the patient’s arm to 45-90 degrees to reduce tortuosity
- Using angiographic and ultrasound techniques to optimize vascular access
The latter point was the one area of debate among the writing committee, Dr. Seto observed. “That is one of the controversies: Should we make ultrasound mandatory? ... Everybody agreed that it can be quite useful and was likely to be useful because of its success in every other access area,” he said. “But in the absence of randomized evidence, we couldn’t make it mandatory or a strong recommendation. We just had to make it one of several options for the operator.”
The document highlights the need for familiarity with potential axillary artery complications and their management, noting that the axillary is more fragile than the femoral artery and, thus, potentially more prone to complications during instrumentation.
Data from the ARMS study in 102 patients undergoing transaxillary access for mechanical hemodynamic support reported 17 procedural complications, including 10 minor access site bleeding events, one stroke, and one pseudoaneurysm. A small study of 25 complex EVAR procedures reported a perioperative access complication rate of 8%, including one axillary artery dissection and one stenosis.
“Despite the brachial plexus being around there, there’s actually rare reports of neurologic injury and certainly none that have been permanent,” Dr. Seto said. “Also, stroke risk is probably more related to your device size and type of device rather than the approach itself.”
A significant amount of the paper is also devoted to training and privileging suggestions with an emphasis on a multidisciplinary team. The writing group recommends graduate medical education programs develop training curricula in percutaneous axillary artery access.
Those already in practice should participate in a formal training program that focuses on axillary artery anatomy, training in large bore access and closure devices, and didactic training in imaging modalities as applied to the axillary artery. Training can occur hands-on or using online simulations.
They also recommend outlining the potential need or role for proctoring and call for ongoing formal professional monitoring programs to evaluate operator outcomes using local or registry data.
“From a privileging standpoint, it was important for hospitals to be equally fair, regardless of the specialty that a requesting practitioner came from,” Dr. Seto said. “In other words, treat the vascular surgeons and interventional cardiologists and radiologists equally in terms of who has the privilege to do transaxillary access.”
The SCAI position statement has been endorsed by the American College of Cardiology, the Heart Failure Society of America, the Society of Interventional Radiology, and the Vascular & Endovascular Surgery Society.
Dr. Seto reported receiving honoraria from Getinge prior to initiation of the document. Disclosures for the rest of the writing group are available with the original article.
A version of this article first appeared on Medscape.com.
The Society for Cardiovascular Angiography and Interventions (SCAI) has issued the first statement on best practices for percutaneous axillary arterial access and training.
The position statement helps fill a gap amid increasing use of transaxillary access as an alternative to the femoral route for large-bore transcatheter aortic valve replacement (TAVR), endovascular aortic repair (EVAR), and mechanical circulatory support.
“The need for alternative access has increased as we are using more and more TAVR for our elderly population, and EVAR has also increased,” writing committee chair Arnold H. Seto, MD, Long Beach VA Health Care System (California) said in an interview. “There’s also a set of patients who require balloon pumps for a prolonged period, and people were using balloon pumps from the axillary approach, which were not custom-designed for that purpose.”
He noted that the evidence base leans heavily on case reports and case series, and that they were approached for guidance by a vendor developing a balloon pump specific to axillary access. “So that helped spur all of us to get together and decide to write up something on this topic, which was developing, but was certainly picking up steam rapidly.”
The statement was published in the Journal of the Society for Cardiovascular Angiography and Interventions, and it reflects the consensus of experts in heart failure, interventional cardiology and radiology, and cardiothoracic and vascular surgery. It reviews anatomic considerations and risks for percutaneous axillary access and suggests techniques for insertion, closure, and complication management.
Although the femoral artery is the most frequent access site for percutaneous large-bore procedures, the document notes that this approach may be limited in 13%-20% of patients because of prior surgeries or severe aortoiliac and/or iliofemoral atherosclerotic disease, tortuosity, or calcification.
“Absolutely, the femoral should be the predominant access site,” Dr. Seto said. Whenever there is a compromised femoral artery, “the axillary artery, which is rarely involved with atherosclerosis, makes for the most optimal alternative access. Other forms of alternative access, including transcaval and transcarotid, are possible but have their own issues and difficulties.”
Axillary access has traditionally been done through an open surgical approach, which allows for direct puncture, primary arterial repair, or placement of a sidearm conduit. Percutaneous transaxillary access avoids a surgical incision and general anesthesia and, theoretically, reduces the risk of infection, he said. It also allows for better mobility for patients, for example, who may have a balloon pump in place for weeks or even a month when waiting for a bridge to transplant.
In terms of technique, key recommendations include:
- Gaining access preferably through the left axillary
- Inserting the needle directly through the pectoralis minor into the second segment of the axillary artery
- Using a shallow-needle angle of 25-30 degrees to improve access success and decrease sheath malformation, kinking, bleeding, or vessel perforation
- Using micropuncture needles to minimize trauma to adjacent tissues
- Abducting the patient’s arm to 45-90 degrees to reduce tortuosity
- Using angiographic and ultrasound techniques to optimize vascular access
The latter point was the one area of debate among the writing committee, Dr. Seto observed. “That is one of the controversies: Should we make ultrasound mandatory? ... Everybody agreed that it can be quite useful and was likely to be useful because of its success in every other access area,” he said. “But in the absence of randomized evidence, we couldn’t make it mandatory or a strong recommendation. We just had to make it one of several options for the operator.”
The document highlights the need for familiarity with potential axillary artery complications and their management, noting that the axillary is more fragile than the femoral artery and, thus, potentially more prone to complications during instrumentation.
Data from the ARMS study in 102 patients undergoing transaxillary access for mechanical hemodynamic support reported 17 procedural complications, including 10 minor access site bleeding events, one stroke, and one pseudoaneurysm. A small study of 25 complex EVAR procedures reported a perioperative access complication rate of 8%, including one axillary artery dissection and one stenosis.
“Despite the brachial plexus being around there, there’s actually rare reports of neurologic injury and certainly none that have been permanent,” Dr. Seto said. “Also, stroke risk is probably more related to your device size and type of device rather than the approach itself.”
A significant amount of the paper is also devoted to training and privileging suggestions with an emphasis on a multidisciplinary team. The writing group recommends graduate medical education programs develop training curricula in percutaneous axillary artery access.
Those already in practice should participate in a formal training program that focuses on axillary artery anatomy, training in large bore access and closure devices, and didactic training in imaging modalities as applied to the axillary artery. Training can occur hands-on or using online simulations.
They also recommend outlining the potential need or role for proctoring and call for ongoing formal professional monitoring programs to evaluate operator outcomes using local or registry data.
“From a privileging standpoint, it was important for hospitals to be equally fair, regardless of the specialty that a requesting practitioner came from,” Dr. Seto said. “In other words, treat the vascular surgeons and interventional cardiologists and radiologists equally in terms of who has the privilege to do transaxillary access.”
The SCAI position statement has been endorsed by the American College of Cardiology, the Heart Failure Society of America, the Society of Interventional Radiology, and the Vascular & Endovascular Surgery Society.
Dr. Seto reported receiving honoraria from Getinge prior to initiation of the document. Disclosures for the rest of the writing group are available with the original article.
A version of this article first appeared on Medscape.com.
FROM THE JOURNAL OF THE SOCIETY FOR CARDIOVASCULAR ANGIOGRAPHY AND INTERVENTIONS
FDA clears mavacamten (Camzyos) for obstructive hypertrophic cardiomyopathy
The U.S. Food and Drug Administration has approved mavacamten (Camzyos, Bristol Myers Squibb) to improve functional capacity and symptoms in adults with symptomatic New York Heart Association (NYHA) class II-III obstructive hypertrophic cardiomyopathy (oHCM).
Mavacamten is the first FDA-approved allosteric and reversible inhibitor selective for cardiac myosin that targets the underlying pathophysiology of the genetic disorder. It’s available in 2.5-mg, 5-mg, 10-mg, and 15-mg capsules.
“The approval of Camzyos represents a significant milestone for appropriate symptomatic obstructive HCM patients and their families, who have long awaited a new treatment option for this chronic and progressive disease,” Anjali T. Owens, MD, medical director of the Center for Inherited Cardiac Disease and assistant professor of medicine, University of Pennsylvania, Philadelphia, said in a news release.
‘Revolutionary’ change
The approval of mavacamten was based on data from the pivotal EXPLORER-HCM and EXPLORER-LTE (long-term extension) trial of adults with symptomatic NYHA class II-III oHCM.
In EXPLORER-HCM, treatment with mavacamten over 30 weeks led to significant improvement in exercise capacity, left ventricular outflow tract (LVOT) obstruction, NYHA functional class, and health status, as reported by this news organization.
The safety and efficacy findings seen at the end of the blinded, randomized, initial 30-week phase of EXPLORER-LTE were maintained in patients who continued treatment for a median of about 62 weeks.
Mavacamten represents “an almost revolutionary change” for the treatment of oHCM, Maya E. Guglin, MD, professor of clinical medicine and an advanced heart failure physician at Indiana University, Indianapolis, said during a press briefing earlier this month at the American College of Cardiology 2022 Scientific Session earlier this month.
“Until now, there was no good medical treatment for symptomatic oHCM. This will change the landscape, and without question it will change guidelines for treating oHCM,” Dr. Guglin said.
The product information for mavacamten includes a boxed warning citing a risk for heart failure.
Echocardiogram assessments of left ventricular ejection fraction (LVEF) are required before and during treatment.
Starting mavacamten in patients with LVEF below 55% is not recommended and the drug should be interrupted if LVEF falls below 50% at any visit or if the patient experiences heart failure symptoms or worsening clinical status.
Concomitant use of mavacamten with certain cytochrome P450 inhibitors or discontinuation of certain cytochrome P450 inducers can increase the risk for heart failure attributable to systolic dysfunction. Therefore, its use is contraindicated in patients using moderate to strong CYP2C19 inhibitors or strong CYP3A4 inhibitors, and moderate to strong CYP2C19 inducers or moderate to strong CYP3A4 inducers.
Because of the risk for heart failure attributable to systolic dysfunction, mavacamten is only available through the Camzyos Risk Evaluation and Mitigation Strategy (REMS) Program.
Full prescribing information is available online.
A version of this article first appeared on Medscape.com.
The U.S. Food and Drug Administration has approved mavacamten (Camzyos, Bristol Myers Squibb) to improve functional capacity and symptoms in adults with symptomatic New York Heart Association (NYHA) class II-III obstructive hypertrophic cardiomyopathy (oHCM).
Mavacamten is the first FDA-approved allosteric and reversible inhibitor selective for cardiac myosin that targets the underlying pathophysiology of the genetic disorder. It’s available in 2.5-mg, 5-mg, 10-mg, and 15-mg capsules.
“The approval of Camzyos represents a significant milestone for appropriate symptomatic obstructive HCM patients and their families, who have long awaited a new treatment option for this chronic and progressive disease,” Anjali T. Owens, MD, medical director of the Center for Inherited Cardiac Disease and assistant professor of medicine, University of Pennsylvania, Philadelphia, said in a news release.
‘Revolutionary’ change
The approval of mavacamten was based on data from the pivotal EXPLORER-HCM and EXPLORER-LTE (long-term extension) trial of adults with symptomatic NYHA class II-III oHCM.
In EXPLORER-HCM, treatment with mavacamten over 30 weeks led to significant improvement in exercise capacity, left ventricular outflow tract (LVOT) obstruction, NYHA functional class, and health status, as reported by this news organization.
The safety and efficacy findings seen at the end of the blinded, randomized, initial 30-week phase of EXPLORER-LTE were maintained in patients who continued treatment for a median of about 62 weeks.
Mavacamten represents “an almost revolutionary change” for the treatment of oHCM, Maya E. Guglin, MD, professor of clinical medicine and an advanced heart failure physician at Indiana University, Indianapolis, said during a press briefing earlier this month at the American College of Cardiology 2022 Scientific Session earlier this month.
“Until now, there was no good medical treatment for symptomatic oHCM. This will change the landscape, and without question it will change guidelines for treating oHCM,” Dr. Guglin said.
The product information for mavacamten includes a boxed warning citing a risk for heart failure.
Echocardiogram assessments of left ventricular ejection fraction (LVEF) are required before and during treatment.
Starting mavacamten in patients with LVEF below 55% is not recommended and the drug should be interrupted if LVEF falls below 50% at any visit or if the patient experiences heart failure symptoms or worsening clinical status.
Concomitant use of mavacamten with certain cytochrome P450 inhibitors or discontinuation of certain cytochrome P450 inducers can increase the risk for heart failure attributable to systolic dysfunction. Therefore, its use is contraindicated in patients using moderate to strong CYP2C19 inhibitors or strong CYP3A4 inhibitors, and moderate to strong CYP2C19 inducers or moderate to strong CYP3A4 inducers.
Because of the risk for heart failure attributable to systolic dysfunction, mavacamten is only available through the Camzyos Risk Evaluation and Mitigation Strategy (REMS) Program.
Full prescribing information is available online.
A version of this article first appeared on Medscape.com.
The U.S. Food and Drug Administration has approved mavacamten (Camzyos, Bristol Myers Squibb) to improve functional capacity and symptoms in adults with symptomatic New York Heart Association (NYHA) class II-III obstructive hypertrophic cardiomyopathy (oHCM).
Mavacamten is the first FDA-approved allosteric and reversible inhibitor selective for cardiac myosin that targets the underlying pathophysiology of the genetic disorder. It’s available in 2.5-mg, 5-mg, 10-mg, and 15-mg capsules.
“The approval of Camzyos represents a significant milestone for appropriate symptomatic obstructive HCM patients and their families, who have long awaited a new treatment option for this chronic and progressive disease,” Anjali T. Owens, MD, medical director of the Center for Inherited Cardiac Disease and assistant professor of medicine, University of Pennsylvania, Philadelphia, said in a news release.
‘Revolutionary’ change
The approval of mavacamten was based on data from the pivotal EXPLORER-HCM and EXPLORER-LTE (long-term extension) trial of adults with symptomatic NYHA class II-III oHCM.
In EXPLORER-HCM, treatment with mavacamten over 30 weeks led to significant improvement in exercise capacity, left ventricular outflow tract (LVOT) obstruction, NYHA functional class, and health status, as reported by this news organization.
The safety and efficacy findings seen at the end of the blinded, randomized, initial 30-week phase of EXPLORER-LTE were maintained in patients who continued treatment for a median of about 62 weeks.
Mavacamten represents “an almost revolutionary change” for the treatment of oHCM, Maya E. Guglin, MD, professor of clinical medicine and an advanced heart failure physician at Indiana University, Indianapolis, said during a press briefing earlier this month at the American College of Cardiology 2022 Scientific Session earlier this month.
“Until now, there was no good medical treatment for symptomatic oHCM. This will change the landscape, and without question it will change guidelines for treating oHCM,” Dr. Guglin said.
The product information for mavacamten includes a boxed warning citing a risk for heart failure.
Echocardiogram assessments of left ventricular ejection fraction (LVEF) are required before and during treatment.
Starting mavacamten in patients with LVEF below 55% is not recommended and the drug should be interrupted if LVEF falls below 50% at any visit or if the patient experiences heart failure symptoms or worsening clinical status.
Concomitant use of mavacamten with certain cytochrome P450 inhibitors or discontinuation of certain cytochrome P450 inducers can increase the risk for heart failure attributable to systolic dysfunction. Therefore, its use is contraindicated in patients using moderate to strong CYP2C19 inhibitors or strong CYP3A4 inhibitors, and moderate to strong CYP2C19 inducers or moderate to strong CYP3A4 inducers.
Because of the risk for heart failure attributable to systolic dysfunction, mavacamten is only available through the Camzyos Risk Evaluation and Mitigation Strategy (REMS) Program.
Full prescribing information is available online.
A version of this article first appeared on Medscape.com.
FDA warns of pump defect with Medtronic HVAD system
Patients implanted with the Medtronic HeartWare ventricular assist device (HVAD) System who develop pump thrombosis could have a welding defect in the internal pump causing the pump to malfunction, the Food and Drug Administration said in a letter to health care professionals.
Medtronic has sent providers an urgent medical device notice about the pump weld defect and is trying to identify which HVAD pumps are affected.
The Medtronic HVAD System was approved as a bridge to heart transplantation in 2012. Since then, it has been fraught with problems.
This past June, the company announced it was stopping all sales of the device and advised physicians to stop implanting it, as reported by this news organization.
Pump thrombosis
Medtronic has received complaints of suspected pump thrombosis in three patients with the HVAD System.
All three patients presented with one or more of the following signs or symptoms: grinding sound, transient power spikes on log files and high watt alarms, elevated lactate dehydrogenase, and low motor speed resulting in low perfusion or dizziness or lightheadedness.
Inspection of the returned pumps in these three cases identified a malfunction of the internal pump. The pumps were exchanged in all three patients. Two patients died after the pump exchange.
The FDA does not recommend the elective removal of properly functioning systems.
“Decisions about removing or exchanging the Medtronic HVAD System should be made by health care providers and patients on a case-by-case basis, considering the patient’s clinical status and surgical risks,” the agency advised.
Patients who present with one or more of the signs or symptoms of pump thrombosis should be first treated for pump thrombosis.
If symptoms fail to resolve, providers may consider whether the patient is a candidate for pump exchange, heart transplant, or pump explant for recovery, taking into account the patient’s clinical condition and surgical risks.
For patients with any of the signs and symptoms of pump thrombosis, logfiles from the controller should be uploaded to Medtronic.
The FDA is working with Medtronic to monitor for any adverse events related to pump weld defects and ensure patients with the HVAD implant continue to receive appropriate follow-up monitoring.
Problems related to the Medtronic HVAD System should be reported to the FDA’s MedWatch program.
A version of this article first appeared on Medscape.com.
Patients implanted with the Medtronic HeartWare ventricular assist device (HVAD) System who develop pump thrombosis could have a welding defect in the internal pump causing the pump to malfunction, the Food and Drug Administration said in a letter to health care professionals.
Medtronic has sent providers an urgent medical device notice about the pump weld defect and is trying to identify which HVAD pumps are affected.
The Medtronic HVAD System was approved as a bridge to heart transplantation in 2012. Since then, it has been fraught with problems.
This past June, the company announced it was stopping all sales of the device and advised physicians to stop implanting it, as reported by this news organization.
Pump thrombosis
Medtronic has received complaints of suspected pump thrombosis in three patients with the HVAD System.
All three patients presented with one or more of the following signs or symptoms: grinding sound, transient power spikes on log files and high watt alarms, elevated lactate dehydrogenase, and low motor speed resulting in low perfusion or dizziness or lightheadedness.
Inspection of the returned pumps in these three cases identified a malfunction of the internal pump. The pumps were exchanged in all three patients. Two patients died after the pump exchange.
The FDA does not recommend the elective removal of properly functioning systems.
“Decisions about removing or exchanging the Medtronic HVAD System should be made by health care providers and patients on a case-by-case basis, considering the patient’s clinical status and surgical risks,” the agency advised.
Patients who present with one or more of the signs or symptoms of pump thrombosis should be first treated for pump thrombosis.
If symptoms fail to resolve, providers may consider whether the patient is a candidate for pump exchange, heart transplant, or pump explant for recovery, taking into account the patient’s clinical condition and surgical risks.
For patients with any of the signs and symptoms of pump thrombosis, logfiles from the controller should be uploaded to Medtronic.
The FDA is working with Medtronic to monitor for any adverse events related to pump weld defects and ensure patients with the HVAD implant continue to receive appropriate follow-up monitoring.
Problems related to the Medtronic HVAD System should be reported to the FDA’s MedWatch program.
A version of this article first appeared on Medscape.com.
Patients implanted with the Medtronic HeartWare ventricular assist device (HVAD) System who develop pump thrombosis could have a welding defect in the internal pump causing the pump to malfunction, the Food and Drug Administration said in a letter to health care professionals.
Medtronic has sent providers an urgent medical device notice about the pump weld defect and is trying to identify which HVAD pumps are affected.
The Medtronic HVAD System was approved as a bridge to heart transplantation in 2012. Since then, it has been fraught with problems.
This past June, the company announced it was stopping all sales of the device and advised physicians to stop implanting it, as reported by this news organization.
Pump thrombosis
Medtronic has received complaints of suspected pump thrombosis in three patients with the HVAD System.
All three patients presented with one or more of the following signs or symptoms: grinding sound, transient power spikes on log files and high watt alarms, elevated lactate dehydrogenase, and low motor speed resulting in low perfusion or dizziness or lightheadedness.
Inspection of the returned pumps in these three cases identified a malfunction of the internal pump. The pumps were exchanged in all three patients. Two patients died after the pump exchange.
The FDA does not recommend the elective removal of properly functioning systems.
“Decisions about removing or exchanging the Medtronic HVAD System should be made by health care providers and patients on a case-by-case basis, considering the patient’s clinical status and surgical risks,” the agency advised.
Patients who present with one or more of the signs or symptoms of pump thrombosis should be first treated for pump thrombosis.
If symptoms fail to resolve, providers may consider whether the patient is a candidate for pump exchange, heart transplant, or pump explant for recovery, taking into account the patient’s clinical condition and surgical risks.
For patients with any of the signs and symptoms of pump thrombosis, logfiles from the controller should be uploaded to Medtronic.
The FDA is working with Medtronic to monitor for any adverse events related to pump weld defects and ensure patients with the HVAD implant continue to receive appropriate follow-up monitoring.
Problems related to the Medtronic HVAD System should be reported to the FDA’s MedWatch program.
A version of this article first appeared on Medscape.com.
TAVI device shows less deterioration than surgery 5 years out
Structural aortic valve deterioration (SVD) at 5 years is lower following repair with a contemporary transcatheter implantation (TAVI) device than with surgery, according to a pooled analysis of major trials.
For healthier patients with a relatively long life expectancy, this is important information for deciding whether to undergo TAVI or surgical aortic valve repair (SAVR), Michael J. Reardon, MD, said at the annual scientific sessions of the American College of Cardiology.

“Every week I get this question about which repair is more durable,” said Dr. Reardon, whose study was not only designed to compare device deterioration but to evaluate the effect of SVD on major outcomes.
In this analysis, the rates of SVD were compared for the self-expanding supra-annular CoreValve Evolut device and SAVR. The SVD curves separated within the first year. At 5 years, the differences were highly significant favoring TAVI (2.57% vs. 4.38%; P = .0095).
As part of this analysis, the impact of SVD was also assessed independent of type of repair. At 5 years, those with SVD relative to those without had an approximately twofold increase in all-cause mortality, cardiovascular mortality, and hospitalization of aortic valve worsening. These risks were elevated regardless of type of valve repair.
The data presented by Dr. Reardon can be considered device specific. The earlier PARTNER 2A study comparing older- and newer-generation TAVI devices with SAVR produced a different result. When a second-generation balloon-expandable SAPIEN XT device and a third-generation SAPIEN 3 device were compared with surgery, neither device achieved lower SVD rates relative to SAVR.
In PARTNER 2A, the SVD rate for the older device was nearly three times greater than SAVR (1.61 vs. 0.58 per 100 patient-years). The numerically higher SVD rates for the newer device (0.68 vs. 0.58 per 100 patient-years) was not statistically different, but the TAVI device was not superior.
More than 4,000 patients evaluated at 5 years
In the analysis presented by Dr. Reardon, data were pooled from the randomized CoreValve U.S. High-Risk Pivotal Trial and the SURTAVI Intermediate Risk Trial. Together, these studies randomized 971 patients to surgery and 1,128 patients to TAVI. Data on an additional 2,663 patients treated with the Evolut valve in two registries were added to the randomized trial data, providing data on 4,762 total patients with 5-year follow-up.
SVD was defined by two criteria. The first was a mean gradient increase of at least 10 mm Hg plus a mean overall gradient of at least 20 mm Hg as measured with echocardiography and assessed, when possible, by an independent core laboratory. The second was new-onset or increased intraprosthetic aortic regurgitation of at least moderate severity.
When graphed over time, the SVD curves separated in favor of TAVI after about 6 months of follow-up. The shape of the curves also differed. Unlike the steady rise in SVD observed in the surgery group, the SVD rate in the TAVI group remained below 1% for almost 4 years before beginning to climb.
There was greater relative benefit for the TAVI device in patients with annular diameters of 23 mm or less. Unlike the rise in SVD rates that began about 6 months after SAVR, the SVD rates in the TAVI patients remained at 0% for more than 2 years. At 5 years, the differences remained significant favoring TAVI (1.39% vs. 5.86%; P = .049).
In those with larger annular diameters, there was still a consistently lower SVD rate over time for TAVI relative to SAVR, but the trend for an advantage at 5 years fell just short of significance (2.48% vs. 3.96%; P = .067).
SVD linked to doubling of mortality
SVD worsened outcomes. When all data surgery and TAVI data were pooled, the hazard ratios corresponded with about a doubling of risk for major adverse outcomes, including all-cause mortality (HR, 1.98; P < .001), cardiovascular mortality (HR, 1.82; P = .008), and hospitalization for aortic valve disease or worsening heart failure (HR, 2.11; P = .01). The relative risks were similar in the two treatment groups, including the risk of all-cause mortality (HR, 2.24; P < .001 for TAVI vs. HR, 2.45; P = .002 for SAVR).
The predictors for SVD on multivariate analysis included female sex, increased body surface area, prior percutaneous coronary intervention, and a prior diagnosis of atrial fibrillation.
Design improvements in TAVI devices are likely to explain these results, said Dr. Reardon, chair of cardiovascular research at Houston Methodist Hospital.
“The CoreValve/Evolut supra-annular, self-expanding bioprosthesis is the first and only transcatheter bioprosthesis to demonstrate lower rates of SVD, compared with surgery,” Dr. Reardon said.
This analysis validated the risks posed by the definition of SVD applied in this study, which appears to be a practical tool for tracking valve function and patient risk. Dr. Reardon also said that the study confirms the value of serial Doppler transthoracic echocardiography as a tool for monitoring SVD.
Several experts agreed that this is important new information.
“This is a remarkable series of findings,” said James McClurken, MD, who is a cardiovascular surgeon affiliated with Temple University, Philadelphia, and practices in Doylestown, Penn. By both demonstrating the prognostic importance of SVD and showing differences between the study device and SAVR, this trial will yield practical data to inform patients about relative risks and benefits.

Athena Poppas, MD, the new president of the ACC and a professor of medicine at Brown University, Providence, R.I., called this study “practice changing” for the same reasons. She also thinks it has valuable data for guiding choice of intervention.
Overall, the data are likely to change thinking about the role of TAVI and surgery in younger, fit patients, according to Megan Coylewright, MD, chief of cardiology at Erlanger Cardiology, Chattanooga, Tenn.
“There are patients [in need of aortic valve repair] with a long life expectancy who have been told you have to have a surgical repair because we know they last longer,” she said. Although she said that relative outcomes after longer follow-up remain unknown, “I think this does throw that comment into question.”
Dr. Reardon has financial relationships with Abbott, Boston Scientific, Medtronic, and Gore Medical. Dr. Poppas and McClurken reported no potential financial conflicts of interest. Dr. Coylewright reported financial relationships with Abbott, Alleviant, Boston Scientific, Cardiosmart, Edwards Lifesciences, Medtronic, and Occlutech. The study received financial support from Medtronic.
Structural aortic valve deterioration (SVD) at 5 years is lower following repair with a contemporary transcatheter implantation (TAVI) device than with surgery, according to a pooled analysis of major trials.
For healthier patients with a relatively long life expectancy, this is important information for deciding whether to undergo TAVI or surgical aortic valve repair (SAVR), Michael J. Reardon, MD, said at the annual scientific sessions of the American College of Cardiology.
“Every week I get this question about which repair is more durable,” said Dr. Reardon, whose study was not only designed to compare device deterioration but to evaluate the effect of SVD on major outcomes.
In this analysis, the rates of SVD were compared for the self-expanding supra-annular CoreValve Evolut device and SAVR. The SVD curves separated within the first year. At 5 years, the differences were highly significant favoring TAVI (2.57% vs. 4.38%; P = .0095).
As part of this analysis, the impact of SVD was also assessed independent of type of repair. At 5 years, those with SVD relative to those without had an approximately twofold increase in all-cause mortality, cardiovascular mortality, and hospitalization of aortic valve worsening. These risks were elevated regardless of type of valve repair.
The data presented by Dr. Reardon can be considered device specific. The earlier PARTNER 2A study comparing older- and newer-generation TAVI devices with SAVR produced a different result. When a second-generation balloon-expandable SAPIEN XT device and a third-generation SAPIEN 3 device were compared with surgery, neither device achieved lower SVD rates relative to SAVR.
In PARTNER 2A, the SVD rate for the older device was nearly three times greater than SAVR (1.61 vs. 0.58 per 100 patient-years). The numerically higher SVD rates for the newer device (0.68 vs. 0.58 per 100 patient-years) was not statistically different, but the TAVI device was not superior.
More than 4,000 patients evaluated at 5 years
In the analysis presented by Dr. Reardon, data were pooled from the randomized CoreValve U.S. High-Risk Pivotal Trial and the SURTAVI Intermediate Risk Trial. Together, these studies randomized 971 patients to surgery and 1,128 patients to TAVI. Data on an additional 2,663 patients treated with the Evolut valve in two registries were added to the randomized trial data, providing data on 4,762 total patients with 5-year follow-up.
SVD was defined by two criteria. The first was a mean gradient increase of at least 10 mm Hg plus a mean overall gradient of at least 20 mm Hg as measured with echocardiography and assessed, when possible, by an independent core laboratory. The second was new-onset or increased intraprosthetic aortic regurgitation of at least moderate severity.
When graphed over time, the SVD curves separated in favor of TAVI after about 6 months of follow-up. The shape of the curves also differed. Unlike the steady rise in SVD observed in the surgery group, the SVD rate in the TAVI group remained below 1% for almost 4 years before beginning to climb.
There was greater relative benefit for the TAVI device in patients with annular diameters of 23 mm or less. Unlike the rise in SVD rates that began about 6 months after SAVR, the SVD rates in the TAVI patients remained at 0% for more than 2 years. At 5 years, the differences remained significant favoring TAVI (1.39% vs. 5.86%; P = .049).
In those with larger annular diameters, there was still a consistently lower SVD rate over time for TAVI relative to SAVR, but the trend for an advantage at 5 years fell just short of significance (2.48% vs. 3.96%; P = .067).
SVD linked to doubling of mortality
SVD worsened outcomes. When all data surgery and TAVI data were pooled, the hazard ratios corresponded with about a doubling of risk for major adverse outcomes, including all-cause mortality (HR, 1.98; P < .001), cardiovascular mortality (HR, 1.82; P = .008), and hospitalization for aortic valve disease or worsening heart failure (HR, 2.11; P = .01). The relative risks were similar in the two treatment groups, including the risk of all-cause mortality (HR, 2.24; P < .001 for TAVI vs. HR, 2.45; P = .002 for SAVR).
The predictors for SVD on multivariate analysis included female sex, increased body surface area, prior percutaneous coronary intervention, and a prior diagnosis of atrial fibrillation.
Design improvements in TAVI devices are likely to explain these results, said Dr. Reardon, chair of cardiovascular research at Houston Methodist Hospital.
“The CoreValve/Evolut supra-annular, self-expanding bioprosthesis is the first and only transcatheter bioprosthesis to demonstrate lower rates of SVD, compared with surgery,” Dr. Reardon said.
This analysis validated the risks posed by the definition of SVD applied in this study, which appears to be a practical tool for tracking valve function and patient risk. Dr. Reardon also said that the study confirms the value of serial Doppler transthoracic echocardiography as a tool for monitoring SVD.
Several experts agreed that this is important new information.
“This is a remarkable series of findings,” said James McClurken, MD, who is a cardiovascular surgeon affiliated with Temple University, Philadelphia, and practices in Doylestown, Penn. By both demonstrating the prognostic importance of SVD and showing differences between the study device and SAVR, this trial will yield practical data to inform patients about relative risks and benefits.
Athena Poppas, MD, the new president of the ACC and a professor of medicine at Brown University, Providence, R.I., called this study “practice changing” for the same reasons. She also thinks it has valuable data for guiding choice of intervention.
Overall, the data are likely to change thinking about the role of TAVI and surgery in younger, fit patients, according to Megan Coylewright, MD, chief of cardiology at Erlanger Cardiology, Chattanooga, Tenn.
“There are patients [in need of aortic valve repair] with a long life expectancy who have been told you have to have a surgical repair because we know they last longer,” she said. Although she said that relative outcomes after longer follow-up remain unknown, “I think this does throw that comment into question.”
Dr. Reardon has financial relationships with Abbott, Boston Scientific, Medtronic, and Gore Medical. Dr. Poppas and McClurken reported no potential financial conflicts of interest. Dr. Coylewright reported financial relationships with Abbott, Alleviant, Boston Scientific, Cardiosmart, Edwards Lifesciences, Medtronic, and Occlutech. The study received financial support from Medtronic.
Structural aortic valve deterioration (SVD) at 5 years is lower following repair with a contemporary transcatheter implantation (TAVI) device than with surgery, according to a pooled analysis of major trials.
For healthier patients with a relatively long life expectancy, this is important information for deciding whether to undergo TAVI or surgical aortic valve repair (SAVR), Michael J. Reardon, MD, said at the annual scientific sessions of the American College of Cardiology.
“Every week I get this question about which repair is more durable,” said Dr. Reardon, whose study was not only designed to compare device deterioration but to evaluate the effect of SVD on major outcomes.
In this analysis, the rates of SVD were compared for the self-expanding supra-annular CoreValve Evolut device and SAVR. The SVD curves separated within the first year. At 5 years, the differences were highly significant favoring TAVI (2.57% vs. 4.38%; P = .0095).
As part of this analysis, the impact of SVD was also assessed independent of type of repair. At 5 years, those with SVD relative to those without had an approximately twofold increase in all-cause mortality, cardiovascular mortality, and hospitalization of aortic valve worsening. These risks were elevated regardless of type of valve repair.
The data presented by Dr. Reardon can be considered device specific. The earlier PARTNER 2A study comparing older- and newer-generation TAVI devices with SAVR produced a different result. When a second-generation balloon-expandable SAPIEN XT device and a third-generation SAPIEN 3 device were compared with surgery, neither device achieved lower SVD rates relative to SAVR.
In PARTNER 2A, the SVD rate for the older device was nearly three times greater than SAVR (1.61 vs. 0.58 per 100 patient-years). The numerically higher SVD rates for the newer device (0.68 vs. 0.58 per 100 patient-years) was not statistically different, but the TAVI device was not superior.
More than 4,000 patients evaluated at 5 years
In the analysis presented by Dr. Reardon, data were pooled from the randomized CoreValve U.S. High-Risk Pivotal Trial and the SURTAVI Intermediate Risk Trial. Together, these studies randomized 971 patients to surgery and 1,128 patients to TAVI. Data on an additional 2,663 patients treated with the Evolut valve in two registries were added to the randomized trial data, providing data on 4,762 total patients with 5-year follow-up.
SVD was defined by two criteria. The first was a mean gradient increase of at least 10 mm Hg plus a mean overall gradient of at least 20 mm Hg as measured with echocardiography and assessed, when possible, by an independent core laboratory. The second was new-onset or increased intraprosthetic aortic regurgitation of at least moderate severity.
When graphed over time, the SVD curves separated in favor of TAVI after about 6 months of follow-up. The shape of the curves also differed. Unlike the steady rise in SVD observed in the surgery group, the SVD rate in the TAVI group remained below 1% for almost 4 years before beginning to climb.
There was greater relative benefit for the TAVI device in patients with annular diameters of 23 mm or less. Unlike the rise in SVD rates that began about 6 months after SAVR, the SVD rates in the TAVI patients remained at 0% for more than 2 years. At 5 years, the differences remained significant favoring TAVI (1.39% vs. 5.86%; P = .049).
In those with larger annular diameters, there was still a consistently lower SVD rate over time for TAVI relative to SAVR, but the trend for an advantage at 5 years fell just short of significance (2.48% vs. 3.96%; P = .067).
SVD linked to doubling of mortality
SVD worsened outcomes. When all data surgery and TAVI data were pooled, the hazard ratios corresponded with about a doubling of risk for major adverse outcomes, including all-cause mortality (HR, 1.98; P < .001), cardiovascular mortality (HR, 1.82; P = .008), and hospitalization for aortic valve disease or worsening heart failure (HR, 2.11; P = .01). The relative risks were similar in the two treatment groups, including the risk of all-cause mortality (HR, 2.24; P < .001 for TAVI vs. HR, 2.45; P = .002 for SAVR).
The predictors for SVD on multivariate analysis included female sex, increased body surface area, prior percutaneous coronary intervention, and a prior diagnosis of atrial fibrillation.
Design improvements in TAVI devices are likely to explain these results, said Dr. Reardon, chair of cardiovascular research at Houston Methodist Hospital.
“The CoreValve/Evolut supra-annular, self-expanding bioprosthesis is the first and only transcatheter bioprosthesis to demonstrate lower rates of SVD, compared with surgery,” Dr. Reardon said.
This analysis validated the risks posed by the definition of SVD applied in this study, which appears to be a practical tool for tracking valve function and patient risk. Dr. Reardon also said that the study confirms the value of serial Doppler transthoracic echocardiography as a tool for monitoring SVD.
Several experts agreed that this is important new information.
“This is a remarkable series of findings,” said James McClurken, MD, who is a cardiovascular surgeon affiliated with Temple University, Philadelphia, and practices in Doylestown, Penn. By both demonstrating the prognostic importance of SVD and showing differences between the study device and SAVR, this trial will yield practical data to inform patients about relative risks and benefits.
Athena Poppas, MD, the new president of the ACC and a professor of medicine at Brown University, Providence, R.I., called this study “practice changing” for the same reasons. She also thinks it has valuable data for guiding choice of intervention.
Overall, the data are likely to change thinking about the role of TAVI and surgery in younger, fit patients, according to Megan Coylewright, MD, chief of cardiology at Erlanger Cardiology, Chattanooga, Tenn.
“There are patients [in need of aortic valve repair] with a long life expectancy who have been told you have to have a surgical repair because we know they last longer,” she said. Although she said that relative outcomes after longer follow-up remain unknown, “I think this does throw that comment into question.”
Dr. Reardon has financial relationships with Abbott, Boston Scientific, Medtronic, and Gore Medical. Dr. Poppas and McClurken reported no potential financial conflicts of interest. Dr. Coylewright reported financial relationships with Abbott, Alleviant, Boston Scientific, Cardiosmart, Edwards Lifesciences, Medtronic, and Occlutech. The study received financial support from Medtronic.
FROM ACC 2022
Extraction of infected implanted cardiac devices rare, despite guidelines
The rates of infection involving cardiac implanted electronic devices (CIEDs), like pacemakers and cardioverter defibrillators (ICDs), are substantial, but only a minority of patients in the United States receive the guideline-directed recommendation of device removal, according to data from a Medicare population.
The study was conducted on the hypothesis that adherence to guidelines were low, “but we were surprised by how low the extraction rates turned out to be,” Sean D. Pokorney, MD, an electrophysiologist at the Duke Clinical Research Institute, Durham, N.C., reported at the annual scientific sessions of the American College of Cardiology.
The major U.S. and European guidelines are uniform in recommending complete extraction for a CIED infection. The American Heart Association and the Heart Rhythm Society and two out of the three other guidelines cited by Dr. Pokorney not only recommend extraction but specify prompt extraction.
Neither complete extraction nor prompt extraction are typical.
Of the 11,619 CIED infection cases identified in the Medicare database, 18.2% underwent extraction within 30 days of diagnosis. Only 13% were extracted within 6 days.
Lack of extraction may cause avoidable mortality
The result is likely to be avoidable mortality. Among those with extraction within 30 days, 80% were still alive 1 year later. Survival at 1 year fell to 67.6% in those without an extraction within this time frame.
This translated to a 22% lower rate of death at 1 year (hazard ratio, 0.78; P = .008) in those who underwent extraction within 30 days.
For those in whom the device was extracted within 7 days, the associated HR for death at 1 year was more than 40% lower (HR, 0.59; P < .001), reported Dr. Pokorney, who characterized these reductions as occurring in “a dose-response fashion.”
The very high risk of relapse despite antibiotics is the reason that “there is a class 1 indication for complete hardware removal,” Dr. Pokorney. He cited five studies that addressed this question. With partial device removal or medical therapy alone, relapse was consistently 50% or greater. In one study, it was 67%. In another it was 100%.
With complete removal, the rate of infection relapse was 1% or lower in four. In the fifth, the rate was 4.2%.
Infections can occur early or late after implantation, but cases accumulate over time. In the Medicare data sample, infection rates climbed from 0.3% at 1 year to 0.6% at 2 years and then to 1.1% at 3 years, Dr. Pokorney reported.
Other studies have also shown a steady increase in the proportion of implanted devices associated with infection over time. In a cohort study conducted in Olmstead County, Minnesota, the cumulative probability of a CIED infection reached 6.2% after 15 years and 11.7% after 25 years. While about half of these were infections localized to the device pocket, the others were potentially life-threatening systemic infections, according to Dr. Pokorney, who cited this study.
In his analysis of the Medicare data, all fee-for-service patients receiving a first CIED implant over a period of 14 years were included. The 14-year period ended just before the COVID-19 epidemic.
The more than 11,000 CIED infections were identified in 1,065,549 total CIED patients. Most (72%) had received a pacemaker. Of the others , more than half received an ICD and the others received a cardiac resynchronization device. The median age was 78 years.
Female and Black patients even less likely to undergo extraction
About half (49.1%) of the overall study population was female, but females represented only about 40% of those who developed an infection. Blacks represented just under 8% of the population but nearly 16% of the CIED infections. Both females and Blacks were significantly less likely than the overall study population to undergo extraction for their infection (P < .001 for both).
Perhaps predictably, patients with comorbidities were more likely to develop CIED infections. For example, 87% of those with infection, versus only 64.9% of the overall population, were in heart failure at the time of implantation. Diabetes (68.3% vs. 49.3%), ischemic heart disease (91.9% vs. 79.4%), renal disease (70.5% vs. 37.9%), and chronic obstructive pulmonary disease (70.6% vs. 55.0%) were also more common at baseline in those who went on to a CIED infection than in the overall population.
Based on the evidence that there is a large unmet need to improve adherence to the guidelines, Dr. Pokorney called for care pathways and other quality initiatives to address the problem.
The reasons that so many patients are not undergoing prompt device extraction at the time of infection is unclear, but Dr. Pokorney offered some hypotheses.
“There appears to be a false belief in the efficacy of antibiotics for treating CIED infections,” Dr. Pokorney said.
Comorbidities shouldn’t delay extraction
It is also possible that clinicians are concerned about performing extractions in patients with multiple comorbidities. If clinicians are delaying extractions for this reason, Dr. Pokorney suggested this behavior is misdirected given the fact that delays appear to increase mortality risk.
Several experts, including Rachel Lambert, MD, an electrophysiologist and professor of medicine at Yale University, New Haven, Conn., agreed that these data deserve a response.
“I was not surprised by the mortality data, but I was surprised at this low extraction rate,” said Dr. Lambert, who concurs with the guidelines. She indicated this study provides teeth to prompt action.
“It is great to have these data about the increased mortality risk to back up the guidelines,” she said.
More information is needed to understand exactly why CIED infection is not now leading to guideline-directed care. Dr. Pokorney said: “Where do we go from here is a key question.”
While several different types of initiatives might be needed, Dr. Pokorney called for regionalization of care to address the fact that not every center that places CIEDs has the capability to perform extractions.
“Extraction is not available at every center, and it probably should not be available at every center, so mechanisms are need to get patients with infection to the specialized centers that provide care,” he said.
Dr. Pokorney has financial relationships with Boston Scientific, Bristol-Myers Squibb, Gilead, Janssen, Medtronic, Pfizer, and Philips. Dr. Lambert reported financial relationships with Abbott, Amgen, and Medtronic.
The rates of infection involving cardiac implanted electronic devices (CIEDs), like pacemakers and cardioverter defibrillators (ICDs), are substantial, but only a minority of patients in the United States receive the guideline-directed recommendation of device removal, according to data from a Medicare population.
The study was conducted on the hypothesis that adherence to guidelines were low, “but we were surprised by how low the extraction rates turned out to be,” Sean D. Pokorney, MD, an electrophysiologist at the Duke Clinical Research Institute, Durham, N.C., reported at the annual scientific sessions of the American College of Cardiology.
The major U.S. and European guidelines are uniform in recommending complete extraction for a CIED infection. The American Heart Association and the Heart Rhythm Society and two out of the three other guidelines cited by Dr. Pokorney not only recommend extraction but specify prompt extraction.
Neither complete extraction nor prompt extraction are typical.
Of the 11,619 CIED infection cases identified in the Medicare database, 18.2% underwent extraction within 30 days of diagnosis. Only 13% were extracted within 6 days.
Lack of extraction may cause avoidable mortality
The result is likely to be avoidable mortality. Among those with extraction within 30 days, 80% were still alive 1 year later. Survival at 1 year fell to 67.6% in those without an extraction within this time frame.
This translated to a 22% lower rate of death at 1 year (hazard ratio, 0.78; P = .008) in those who underwent extraction within 30 days.
For those in whom the device was extracted within 7 days, the associated HR for death at 1 year was more than 40% lower (HR, 0.59; P < .001), reported Dr. Pokorney, who characterized these reductions as occurring in “a dose-response fashion.”
The very high risk of relapse despite antibiotics is the reason that “there is a class 1 indication for complete hardware removal,” Dr. Pokorney. He cited five studies that addressed this question. With partial device removal or medical therapy alone, relapse was consistently 50% or greater. In one study, it was 67%. In another it was 100%.
With complete removal, the rate of infection relapse was 1% or lower in four. In the fifth, the rate was 4.2%.
Infections can occur early or late after implantation, but cases accumulate over time. In the Medicare data sample, infection rates climbed from 0.3% at 1 year to 0.6% at 2 years and then to 1.1% at 3 years, Dr. Pokorney reported.
Other studies have also shown a steady increase in the proportion of implanted devices associated with infection over time. In a cohort study conducted in Olmstead County, Minnesota, the cumulative probability of a CIED infection reached 6.2% after 15 years and 11.7% after 25 years. While about half of these were infections localized to the device pocket, the others were potentially life-threatening systemic infections, according to Dr. Pokorney, who cited this study.
In his analysis of the Medicare data, all fee-for-service patients receiving a first CIED implant over a period of 14 years were included. The 14-year period ended just before the COVID-19 epidemic.
The more than 11,000 CIED infections were identified in 1,065,549 total CIED patients. Most (72%) had received a pacemaker. Of the others , more than half received an ICD and the others received a cardiac resynchronization device. The median age was 78 years.
Female and Black patients even less likely to undergo extraction
About half (49.1%) of the overall study population was female, but females represented only about 40% of those who developed an infection. Blacks represented just under 8% of the population but nearly 16% of the CIED infections. Both females and Blacks were significantly less likely than the overall study population to undergo extraction for their infection (P < .001 for both).
Perhaps predictably, patients with comorbidities were more likely to develop CIED infections. For example, 87% of those with infection, versus only 64.9% of the overall population, were in heart failure at the time of implantation. Diabetes (68.3% vs. 49.3%), ischemic heart disease (91.9% vs. 79.4%), renal disease (70.5% vs. 37.9%), and chronic obstructive pulmonary disease (70.6% vs. 55.0%) were also more common at baseline in those who went on to a CIED infection than in the overall population.
Based on the evidence that there is a large unmet need to improve adherence to the guidelines, Dr. Pokorney called for care pathways and other quality initiatives to address the problem.
The reasons that so many patients are not undergoing prompt device extraction at the time of infection is unclear, but Dr. Pokorney offered some hypotheses.
“There appears to be a false belief in the efficacy of antibiotics for treating CIED infections,” Dr. Pokorney said.
Comorbidities shouldn’t delay extraction
It is also possible that clinicians are concerned about performing extractions in patients with multiple comorbidities. If clinicians are delaying extractions for this reason, Dr. Pokorney suggested this behavior is misdirected given the fact that delays appear to increase mortality risk.
Several experts, including Rachel Lambert, MD, an electrophysiologist and professor of medicine at Yale University, New Haven, Conn., agreed that these data deserve a response.
“I was not surprised by the mortality data, but I was surprised at this low extraction rate,” said Dr. Lambert, who concurs with the guidelines. She indicated this study provides teeth to prompt action.
“It is great to have these data about the increased mortality risk to back up the guidelines,” she said.
More information is needed to understand exactly why CIED infection is not now leading to guideline-directed care. Dr. Pokorney said: “Where do we go from here is a key question.”
While several different types of initiatives might be needed, Dr. Pokorney called for regionalization of care to address the fact that not every center that places CIEDs has the capability to perform extractions.
“Extraction is not available at every center, and it probably should not be available at every center, so mechanisms are need to get patients with infection to the specialized centers that provide care,” he said.
Dr. Pokorney has financial relationships with Boston Scientific, Bristol-Myers Squibb, Gilead, Janssen, Medtronic, Pfizer, and Philips. Dr. Lambert reported financial relationships with Abbott, Amgen, and Medtronic.
The rates of infection involving cardiac implanted electronic devices (CIEDs), like pacemakers and cardioverter defibrillators (ICDs), are substantial, but only a minority of patients in the United States receive the guideline-directed recommendation of device removal, according to data from a Medicare population.
The study was conducted on the hypothesis that adherence to guidelines were low, “but we were surprised by how low the extraction rates turned out to be,” Sean D. Pokorney, MD, an electrophysiologist at the Duke Clinical Research Institute, Durham, N.C., reported at the annual scientific sessions of the American College of Cardiology.
The major U.S. and European guidelines are uniform in recommending complete extraction for a CIED infection. The American Heart Association and the Heart Rhythm Society and two out of the three other guidelines cited by Dr. Pokorney not only recommend extraction but specify prompt extraction.
Neither complete extraction nor prompt extraction are typical.
Of the 11,619 CIED infection cases identified in the Medicare database, 18.2% underwent extraction within 30 days of diagnosis. Only 13% were extracted within 6 days.
Lack of extraction may cause avoidable mortality
The result is likely to be avoidable mortality. Among those with extraction within 30 days, 80% were still alive 1 year later. Survival at 1 year fell to 67.6% in those without an extraction within this time frame.
This translated to a 22% lower rate of death at 1 year (hazard ratio, 0.78; P = .008) in those who underwent extraction within 30 days.
For those in whom the device was extracted within 7 days, the associated HR for death at 1 year was more than 40% lower (HR, 0.59; P < .001), reported Dr. Pokorney, who characterized these reductions as occurring in “a dose-response fashion.”
The very high risk of relapse despite antibiotics is the reason that “there is a class 1 indication for complete hardware removal,” Dr. Pokorney. He cited five studies that addressed this question. With partial device removal or medical therapy alone, relapse was consistently 50% or greater. In one study, it was 67%. In another it was 100%.
With complete removal, the rate of infection relapse was 1% or lower in four. In the fifth, the rate was 4.2%.
Infections can occur early or late after implantation, but cases accumulate over time. In the Medicare data sample, infection rates climbed from 0.3% at 1 year to 0.6% at 2 years and then to 1.1% at 3 years, Dr. Pokorney reported.
Other studies have also shown a steady increase in the proportion of implanted devices associated with infection over time. In a cohort study conducted in Olmstead County, Minnesota, the cumulative probability of a CIED infection reached 6.2% after 15 years and 11.7% after 25 years. While about half of these were infections localized to the device pocket, the others were potentially life-threatening systemic infections, according to Dr. Pokorney, who cited this study.
In his analysis of the Medicare data, all fee-for-service patients receiving a first CIED implant over a period of 14 years were included. The 14-year period ended just before the COVID-19 epidemic.
The more than 11,000 CIED infections were identified in 1,065,549 total CIED patients. Most (72%) had received a pacemaker. Of the others , more than half received an ICD and the others received a cardiac resynchronization device. The median age was 78 years.
Female and Black patients even less likely to undergo extraction
About half (49.1%) of the overall study population was female, but females represented only about 40% of those who developed an infection. Blacks represented just under 8% of the population but nearly 16% of the CIED infections. Both females and Blacks were significantly less likely than the overall study population to undergo extraction for their infection (P < .001 for both).
Perhaps predictably, patients with comorbidities were more likely to develop CIED infections. For example, 87% of those with infection, versus only 64.9% of the overall population, were in heart failure at the time of implantation. Diabetes (68.3% vs. 49.3%), ischemic heart disease (91.9% vs. 79.4%), renal disease (70.5% vs. 37.9%), and chronic obstructive pulmonary disease (70.6% vs. 55.0%) were also more common at baseline in those who went on to a CIED infection than in the overall population.
Based on the evidence that there is a large unmet need to improve adherence to the guidelines, Dr. Pokorney called for care pathways and other quality initiatives to address the problem.
The reasons that so many patients are not undergoing prompt device extraction at the time of infection is unclear, but Dr. Pokorney offered some hypotheses.
“There appears to be a false belief in the efficacy of antibiotics for treating CIED infections,” Dr. Pokorney said.
Comorbidities shouldn’t delay extraction
It is also possible that clinicians are concerned about performing extractions in patients with multiple comorbidities. If clinicians are delaying extractions for this reason, Dr. Pokorney suggested this behavior is misdirected given the fact that delays appear to increase mortality risk.
Several experts, including Rachel Lambert, MD, an electrophysiologist and professor of medicine at Yale University, New Haven, Conn., agreed that these data deserve a response.
“I was not surprised by the mortality data, but I was surprised at this low extraction rate,” said Dr. Lambert, who concurs with the guidelines. She indicated this study provides teeth to prompt action.
“It is great to have these data about the increased mortality risk to back up the guidelines,” she said.
More information is needed to understand exactly why CIED infection is not now leading to guideline-directed care. Dr. Pokorney said: “Where do we go from here is a key question.”
While several different types of initiatives might be needed, Dr. Pokorney called for regionalization of care to address the fact that not every center that places CIEDs has the capability to perform extractions.
“Extraction is not available at every center, and it probably should not be available at every center, so mechanisms are need to get patients with infection to the specialized centers that provide care,” he said.
Dr. Pokorney has financial relationships with Boston Scientific, Bristol-Myers Squibb, Gilead, Janssen, Medtronic, Pfizer, and Philips. Dr. Lambert reported financial relationships with Abbott, Amgen, and Medtronic.
FROM ACC 2022
VALOR-HCM: Novel drug may delay, avert invasive therapy in OHCM
Treatment with a novel myosin-inhibiting agent may improve symptoms and hemodynamics enough in patients with obstructive hypertrophic cardiomyopathy (OHCM) so that they can avoid or at least delay septal reduction therapy (SRT), suggests a randomized trial of modest size and duration.
Of 112 patients with OHCM who were sick enough while receiving standard medications to qualify for SRT, those assigned to take mavacamten (MyoKardia) instead of placebo were far less likely to still be eligible for SRT 16 weeks later.
In other words, their OHCM had improved enough during therapy with mavacamten such that SRT, either surgical septal myectomy or transcatheter alcohol septal ablation, could no longer be recommended per guidelines.
Mavacamten, which lessens myocardial contractility by selective inhibition of cardiac myosin, is the first agent tested in prospective trials to appear as a viable medical option in patients with severe, symptomatic OHCM, observed principal investigator Milind Y. Desai, MD, MBA, of the Cleveland Clinic.
“There’s clearly an unmet need for noninvasive therapies, medical therapies, that work in OHCM,” he said in an interview. Mavacamten “adds to the armamentarium” of OHCM management options and may give patients with symptoms despite conventional medications an alternative to SRT, which is considered definitive but has drawbacks.
The goal of SRT is to alleviate obstruction of the left ventricular outflow tract (LVOT), but surgical SRT requires a sternotomy, with all the risks and recovery time that entails. Catheter-based alcohol septal ablation is a less common alternative for some patients with suitable anatomy, Dr. Desai noted.
But those procedures “are not uniformly available, and even when available, the outcomes are fairly heterogeneous,” he said. “The guidelines recommend that you should go to a center with a mortality rate of less than 1% with these procedures. Centers like that are very few across the world,” and procedural mortality can be much higher at centers with less SRT experience.
Dr. Desai presented the results of VALOR-HCM at the annual scientific sessions of the American College of Cardiology. Of the 56 patients assigned to mavacamten, 10 (17.9%) decided to undergo SRT by the end of the trial, or otherwise still met guideline-recommended criteria for receiving SRT, the primary endpoint. In comparison, 43 of the 56 patients (76.8%) in the control group (P < .0001) met that endpoint.
More patients receiving mavacamten improved by at least one New York Heart Association (NYHA) functional class during the trial’s 16 weeks: 63% versus 21% for those assigned to placebo. And 27% and 2%, respectively, improved by at least two NYHA classes, Dr. Desai said.
Guidelines recommend that SRT be reserved for patients in NYHA class III or IV heart failure with a resting or provoked LVOT gradient of at least 50 mm Hg.
Of note, Desai said, only two patients in each group elected to undergo SRT during the study. “The primary endpoint was driven by reduction in guideline eligibility for SRT, but 95% of patients in the study chose to continue with medical therapy.”
Speaking as a panelist after Dr. Desai’s presentation, Lynne W. Stevenson, MD, lauded the phase 3 trial’s “brave design,” which featured a highly unusual subjective primary endpoint and framed it as an advantage.
That the trial showed a significant mavacamten effect for that endpoint “answered, in one step, the question of what does this actually mean to the patient – which often takes much longer,” observed Dr. Stevenson, from Vanderbilt University, Nashville, Tenn.
Even so, she added, whether patients still qualified for SRT in the trial at least had to be supported by objective measures of LVOT gradient and NT-proBNP levels.
“My perspective is that of a cardiac surgeon who performs septal myectomies,” said John Cleveland, MD, University of Colorado at Denver, Aurora, who said he was impressed at how few patients receiving mavacamten went on to undergo SRT, while the rest were able to at least defer that decision.
Current recommendations are that patients who go to SRT “should be maximally medically treated and still symptomatic,” Dr. Cleveland observed at a press conference on VALOR-HCM. Should mavacamten be added to the list of agents to use before resorting to invasive therapy? “My answer would be yes,” he said, and patients who remain symptomatic even while receiving the myosin inhibitor and other medications should proceed to SRT.
The trial’s patients had documented OHCM, severe symptoms, and a resting or provoked LVOT gradient of at least 50 mm Hg despite maximally tolerated medications – which could include disopyramide, beta-blockers, and calcium channel blockers. About half the study population was female, and 89% were White. All had been referred for SRT.
Active therapy consisted of mavacamten initiated at 5 mg/day, with up-titrations at 8 and 12 weeks as tolerated, guided by echocardiographic left ventricular ejection fraction and LVOT gradient.
Most secondary endpoints improved significantly in patients receiving the drug, compared with placebo. They included measures of quality of life, symptom status, ventricular function, natriuretic peptides, and troponin I.
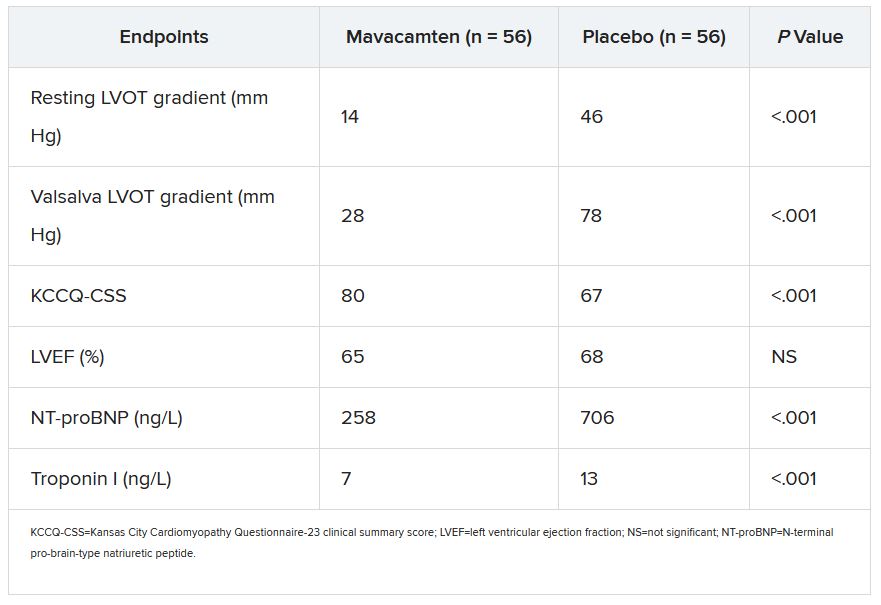
The secondary outcomes are consistent with what was observed in the EXPLORER-HCM trial, which in 2020 suggested that mavacamten could improve measures of quality of life, NYHA functional class, LVOT gradient, peak VO2, and other metrics in patients with OHCM.
Dr. Desai said mavacamten was well tolerated. “There were two patients who had a transient drop in ejection fraction to less than 50%, so the drug was temporarily discontinued, but resumed at a lower dose and they were able to complete the study.”
Dr. Stevenson commented on the “pretty quick” up-titration of mavacamten dosages in a study lasting only 4 months, which could have been a concern given the drug’s limited track record and its mechanism of action targeting contractility. “Fortunately, no serious safety signals” were observed.
Dr. Desai emphasized that mavacamten up-titrations were strictly guided by regular echocardiographic monitoring and assessment of LVOT gradients, in addition to clinical responses. And that, he said, is likely how up-titrations should be carried out if mavacamten is approved for OHCM.
VALOR-HCM was supported by MyoKardia. Dr. Desai disclosed receiving honoraria or consulting fees from Caristo Diagnostics, Medtronic, and MyoKardia. Dr. Stevenson disclosed receiving honoraria or consulting fees from Novartis; serving on a data safety monitoring board for Livanova; and other relationships with Abbott Medical, Biotronik, Boston Scientific, Bristol-Myers Squibb, Endotronic, Gore Medical, and Johnson & Johnson. Dr. Cleveland had no disclosures.
A version of this article first appeared on Medscape.com.
Treatment with a novel myosin-inhibiting agent may improve symptoms and hemodynamics enough in patients with obstructive hypertrophic cardiomyopathy (OHCM) so that they can avoid or at least delay septal reduction therapy (SRT), suggests a randomized trial of modest size and duration.
Of 112 patients with OHCM who were sick enough while receiving standard medications to qualify for SRT, those assigned to take mavacamten (MyoKardia) instead of placebo were far less likely to still be eligible for SRT 16 weeks later.
In other words, their OHCM had improved enough during therapy with mavacamten such that SRT, either surgical septal myectomy or transcatheter alcohol septal ablation, could no longer be recommended per guidelines.
Mavacamten, which lessens myocardial contractility by selective inhibition of cardiac myosin, is the first agent tested in prospective trials to appear as a viable medical option in patients with severe, symptomatic OHCM, observed principal investigator Milind Y. Desai, MD, MBA, of the Cleveland Clinic.
“There’s clearly an unmet need for noninvasive therapies, medical therapies, that work in OHCM,” he said in an interview. Mavacamten “adds to the armamentarium” of OHCM management options and may give patients with symptoms despite conventional medications an alternative to SRT, which is considered definitive but has drawbacks.
The goal of SRT is to alleviate obstruction of the left ventricular outflow tract (LVOT), but surgical SRT requires a sternotomy, with all the risks and recovery time that entails. Catheter-based alcohol septal ablation is a less common alternative for some patients with suitable anatomy, Dr. Desai noted.
But those procedures “are not uniformly available, and even when available, the outcomes are fairly heterogeneous,” he said. “The guidelines recommend that you should go to a center with a mortality rate of less than 1% with these procedures. Centers like that are very few across the world,” and procedural mortality can be much higher at centers with less SRT experience.
Dr. Desai presented the results of VALOR-HCM at the annual scientific sessions of the American College of Cardiology. Of the 56 patients assigned to mavacamten, 10 (17.9%) decided to undergo SRT by the end of the trial, or otherwise still met guideline-recommended criteria for receiving SRT, the primary endpoint. In comparison, 43 of the 56 patients (76.8%) in the control group (P < .0001) met that endpoint.
More patients receiving mavacamten improved by at least one New York Heart Association (NYHA) functional class during the trial’s 16 weeks: 63% versus 21% for those assigned to placebo. And 27% and 2%, respectively, improved by at least two NYHA classes, Dr. Desai said.
Guidelines recommend that SRT be reserved for patients in NYHA class III or IV heart failure with a resting or provoked LVOT gradient of at least 50 mm Hg.
Of note, Desai said, only two patients in each group elected to undergo SRT during the study. “The primary endpoint was driven by reduction in guideline eligibility for SRT, but 95% of patients in the study chose to continue with medical therapy.”
Speaking as a panelist after Dr. Desai’s presentation, Lynne W. Stevenson, MD, lauded the phase 3 trial’s “brave design,” which featured a highly unusual subjective primary endpoint and framed it as an advantage.
That the trial showed a significant mavacamten effect for that endpoint “answered, in one step, the question of what does this actually mean to the patient – which often takes much longer,” observed Dr. Stevenson, from Vanderbilt University, Nashville, Tenn.
Even so, she added, whether patients still qualified for SRT in the trial at least had to be supported by objective measures of LVOT gradient and NT-proBNP levels.
“My perspective is that of a cardiac surgeon who performs septal myectomies,” said John Cleveland, MD, University of Colorado at Denver, Aurora, who said he was impressed at how few patients receiving mavacamten went on to undergo SRT, while the rest were able to at least defer that decision.
Current recommendations are that patients who go to SRT “should be maximally medically treated and still symptomatic,” Dr. Cleveland observed at a press conference on VALOR-HCM. Should mavacamten be added to the list of agents to use before resorting to invasive therapy? “My answer would be yes,” he said, and patients who remain symptomatic even while receiving the myosin inhibitor and other medications should proceed to SRT.
The trial’s patients had documented OHCM, severe symptoms, and a resting or provoked LVOT gradient of at least 50 mm Hg despite maximally tolerated medications – which could include disopyramide, beta-blockers, and calcium channel blockers. About half the study population was female, and 89% were White. All had been referred for SRT.
Active therapy consisted of mavacamten initiated at 5 mg/day, with up-titrations at 8 and 12 weeks as tolerated, guided by echocardiographic left ventricular ejection fraction and LVOT gradient.
Most secondary endpoints improved significantly in patients receiving the drug, compared with placebo. They included measures of quality of life, symptom status, ventricular function, natriuretic peptides, and troponin I.
The secondary outcomes are consistent with what was observed in the EXPLORER-HCM trial, which in 2020 suggested that mavacamten could improve measures of quality of life, NYHA functional class, LVOT gradient, peak VO2, and other metrics in patients with OHCM.
Dr. Desai said mavacamten was well tolerated. “There were two patients who had a transient drop in ejection fraction to less than 50%, so the drug was temporarily discontinued, but resumed at a lower dose and they were able to complete the study.”
Dr. Stevenson commented on the “pretty quick” up-titration of mavacamten dosages in a study lasting only 4 months, which could have been a concern given the drug’s limited track record and its mechanism of action targeting contractility. “Fortunately, no serious safety signals” were observed.
Dr. Desai emphasized that mavacamten up-titrations were strictly guided by regular echocardiographic monitoring and assessment of LVOT gradients, in addition to clinical responses. And that, he said, is likely how up-titrations should be carried out if mavacamten is approved for OHCM.
VALOR-HCM was supported by MyoKardia. Dr. Desai disclosed receiving honoraria or consulting fees from Caristo Diagnostics, Medtronic, and MyoKardia. Dr. Stevenson disclosed receiving honoraria or consulting fees from Novartis; serving on a data safety monitoring board for Livanova; and other relationships with Abbott Medical, Biotronik, Boston Scientific, Bristol-Myers Squibb, Endotronic, Gore Medical, and Johnson & Johnson. Dr. Cleveland had no disclosures.
A version of this article first appeared on Medscape.com.
Treatment with a novel myosin-inhibiting agent may improve symptoms and hemodynamics enough in patients with obstructive hypertrophic cardiomyopathy (OHCM) so that they can avoid or at least delay septal reduction therapy (SRT), suggests a randomized trial of modest size and duration.
Of 112 patients with OHCM who were sick enough while receiving standard medications to qualify for SRT, those assigned to take mavacamten (MyoKardia) instead of placebo were far less likely to still be eligible for SRT 16 weeks later.
In other words, their OHCM had improved enough during therapy with mavacamten such that SRT, either surgical septal myectomy or transcatheter alcohol septal ablation, could no longer be recommended per guidelines.
Mavacamten, which lessens myocardial contractility by selective inhibition of cardiac myosin, is the first agent tested in prospective trials to appear as a viable medical option in patients with severe, symptomatic OHCM, observed principal investigator Milind Y. Desai, MD, MBA, of the Cleveland Clinic.
“There’s clearly an unmet need for noninvasive therapies, medical therapies, that work in OHCM,” he said in an interview. Mavacamten “adds to the armamentarium” of OHCM management options and may give patients with symptoms despite conventional medications an alternative to SRT, which is considered definitive but has drawbacks.
The goal of SRT is to alleviate obstruction of the left ventricular outflow tract (LVOT), but surgical SRT requires a sternotomy, with all the risks and recovery time that entails. Catheter-based alcohol septal ablation is a less common alternative for some patients with suitable anatomy, Dr. Desai noted.
But those procedures “are not uniformly available, and even when available, the outcomes are fairly heterogeneous,” he said. “The guidelines recommend that you should go to a center with a mortality rate of less than 1% with these procedures. Centers like that are very few across the world,” and procedural mortality can be much higher at centers with less SRT experience.
Dr. Desai presented the results of VALOR-HCM at the annual scientific sessions of the American College of Cardiology. Of the 56 patients assigned to mavacamten, 10 (17.9%) decided to undergo SRT by the end of the trial, or otherwise still met guideline-recommended criteria for receiving SRT, the primary endpoint. In comparison, 43 of the 56 patients (76.8%) in the control group (P < .0001) met that endpoint.
More patients receiving mavacamten improved by at least one New York Heart Association (NYHA) functional class during the trial’s 16 weeks: 63% versus 21% for those assigned to placebo. And 27% and 2%, respectively, improved by at least two NYHA classes, Dr. Desai said.
Guidelines recommend that SRT be reserved for patients in NYHA class III or IV heart failure with a resting or provoked LVOT gradient of at least 50 mm Hg.
Of note, Desai said, only two patients in each group elected to undergo SRT during the study. “The primary endpoint was driven by reduction in guideline eligibility for SRT, but 95% of patients in the study chose to continue with medical therapy.”
Speaking as a panelist after Dr. Desai’s presentation, Lynne W. Stevenson, MD, lauded the phase 3 trial’s “brave design,” which featured a highly unusual subjective primary endpoint and framed it as an advantage.
That the trial showed a significant mavacamten effect for that endpoint “answered, in one step, the question of what does this actually mean to the patient – which often takes much longer,” observed Dr. Stevenson, from Vanderbilt University, Nashville, Tenn.
Even so, she added, whether patients still qualified for SRT in the trial at least had to be supported by objective measures of LVOT gradient and NT-proBNP levels.
“My perspective is that of a cardiac surgeon who performs septal myectomies,” said John Cleveland, MD, University of Colorado at Denver, Aurora, who said he was impressed at how few patients receiving mavacamten went on to undergo SRT, while the rest were able to at least defer that decision.
Current recommendations are that patients who go to SRT “should be maximally medically treated and still symptomatic,” Dr. Cleveland observed at a press conference on VALOR-HCM. Should mavacamten be added to the list of agents to use before resorting to invasive therapy? “My answer would be yes,” he said, and patients who remain symptomatic even while receiving the myosin inhibitor and other medications should proceed to SRT.
The trial’s patients had documented OHCM, severe symptoms, and a resting or provoked LVOT gradient of at least 50 mm Hg despite maximally tolerated medications – which could include disopyramide, beta-blockers, and calcium channel blockers. About half the study population was female, and 89% were White. All had been referred for SRT.
Active therapy consisted of mavacamten initiated at 5 mg/day, with up-titrations at 8 and 12 weeks as tolerated, guided by echocardiographic left ventricular ejection fraction and LVOT gradient.
Most secondary endpoints improved significantly in patients receiving the drug, compared with placebo. They included measures of quality of life, symptom status, ventricular function, natriuretic peptides, and troponin I.
The secondary outcomes are consistent with what was observed in the EXPLORER-HCM trial, which in 2020 suggested that mavacamten could improve measures of quality of life, NYHA functional class, LVOT gradient, peak VO2, and other metrics in patients with OHCM.
Dr. Desai said mavacamten was well tolerated. “There were two patients who had a transient drop in ejection fraction to less than 50%, so the drug was temporarily discontinued, but resumed at a lower dose and they were able to complete the study.”
Dr. Stevenson commented on the “pretty quick” up-titration of mavacamten dosages in a study lasting only 4 months, which could have been a concern given the drug’s limited track record and its mechanism of action targeting contractility. “Fortunately, no serious safety signals” were observed.
Dr. Desai emphasized that mavacamten up-titrations were strictly guided by regular echocardiographic monitoring and assessment of LVOT gradients, in addition to clinical responses. And that, he said, is likely how up-titrations should be carried out if mavacamten is approved for OHCM.
VALOR-HCM was supported by MyoKardia. Dr. Desai disclosed receiving honoraria or consulting fees from Caristo Diagnostics, Medtronic, and MyoKardia. Dr. Stevenson disclosed receiving honoraria or consulting fees from Novartis; serving on a data safety monitoring board for Livanova; and other relationships with Abbott Medical, Biotronik, Boston Scientific, Bristol-Myers Squibb, Endotronic, Gore Medical, and Johnson & Johnson. Dr. Cleveland had no disclosures.
A version of this article first appeared on Medscape.com.
FROM ACC 2022
FAME 3 subanalysis adds twist to negative primary results
A new subanalysis of the FAME 3 trial, which failed to show that percutaneous intervention (PCI) guided by fractional flow reserve (FFR) is noninferior to coronary artery bypass grafting (CABG) for treating three-vessel coronary artery disease, has associated PCI with early quality of life (QOL) advantages, according to findings presented at the annual scientific sessions of the American College of Cardiology.
Despite a modestly greater risk of major adverse cardiac events (MACE) at the end of 12 months’ follow-up among those treated with FFR-guided PCI, the greater QOL early after the procedure might be relevant to patients weighing these options, according to Frederik M. Zimmerman, MD, of Catharina Hospital in Eindhoven, the Netherlands.
“FFR-guided PCI results in a faster improvement in quality of life than CABG during the first year after revascularization, and it improved working status in patients younger than 65 years of age,” Dr. Zimmermann said.
The primary results of FAME 3 were presented at the 2021 Transcatheter Cardiovascular Therapeutics annual meeting by lead author William F. Fearon, MD, of Stanford (Calif.) University and published simultaneously in the New England Journal of Medicine.
Rather than confirming the hypothesis that FFR-guided PCI is comparable with CABG for the primary composite MACE outcome death from any cause, myocardial infarction, stroke, or revascularization, the incidence of MACE at 12 months was 10.6% in those randomized to PCI and 6.9% in the group assigned to CABG.
This translated into a hazard ratio for MACE of 1.5, signifying a 50% increase in risk for FFR-guided PCI relative to CABG for the primary outcome, a difference that negated the study definition of noninferiority (P = .35).
In this new health-related subanalysis, which was published simultaneously with his ACC presentation, the groups were compared over 12 months for QOL as measured with European Quality of Life–5 dimensions (EQ-5D) scale, angina as measured with the Canadian Cardiovascular Classification (CCC) system, and employment.
Outcomes data available in >85% of patients
Of the 1,500 patients enrolled and randomized in FAME 3 (757 to FFR-guided PCI and 743 to CABG), this health outcomes subanalysis was performed with complete data at 12 months from 89% of those in the PCI group and 88% of those in the CABG group.
Ultimately, the study did not show differences in any of these measures at the end of 12 months, but there were significant differences in QOL and employment at earlier time points. In particular, the significantly different (P < .001) trajectory for QOL improvement at 1 and 6 months favored FFR-guided PCI whether evaluated with the EQ-5D instrument or an EQ visual analog scale.
Rates of angina defined by as CCC class of at least 2 were low after revascularization in both arms of the study, negating any opportunity for differences, but patients aged younger than 65 years were almost twice as likely to have returned to full- or part-time work 1 month after revascularization (60.2% vs. 33.1%), and they remained at higher odds for working at 12 months (68.1% vs. 57.4%).
In patients aged older than 65 years, return-to-work rates did not differ significantly at any time point.
These results suggest potentially clinically meaningful early advantages for FFR-guided PCI, but some experts questioned the rationale for reporting positive secondary findings from a negative trial.
“This subanalysis is curious,” said Allen Jeremias, MD, director of interventional cardiology research, Saint Francis Hospital, Roslyn, N.Y. He pointed out that reporting these data is an anomaly.
Subanalyses uncommon in negative trials
“CABG was found to be better, so why look at QOL,” said Dr. Jeremias, who was an ACC-invited expert to discuss the results. However, he went on to say, “this could be an exception to the rule.”
The reason, according to Dr. Jeremias, is that the absolute difference at 12 months between FFR-guided PCI and CABG for the MACE events of greatest concern – death, MI, or stroke – was only about 2% greater in the FFR-guided PCI group (7.3% vs. 5.2%). The biggest contributor to the difference in MACE in FAME 3 at 12 months was the higher rate of repeat revascularization (5.9% vs. 3.9%).
Moreover, patients randomized to FFR-guided PCI had lower rates of many adverse events. This included risk of bleeding (1.6% vs. 3.8%; P = .009 as defined by type ≥3 Bleeding Academic Research Consortium , acute kidney injury (0.1% vs. 0.9%; P = .04), atrial fibrillation (2.4% vs. 14.1%; P < .001) and rehospitalization within 30 days (5.5% vs. 10.2%; P < .001).
In the context of a modest increase in risk of MACE and the lower rate of several important treatment-related adverse events, the QOL advantages identified in this subanalysis “might be a reasonable topic for patient-shared decision-making,” Dr. Jeremias suggested.
New data might inform patient decision-making
He granted the possibility that well-informed patients might accept the modestly increased risk of MACE for one or more of the outcomes, such as a higher likelihood of an early return to work, that favored FFR-guided PCI.
This is the point of this subanalysis, agreed Dr. Zimmermann.
“It is all about shared decision-making,” he said. Also emphasizing that the negative trial endpoint of FAME 3 “was driven largely by an increased risk of revascularization,” he believes that these new data might be a basis for discussions with patients weighing relative risks and benefits.
There are more data to come, according to Dr. Zimmermann, who said that follow-up of up to 5 years is planned. The 3-year data will be made available in 2023.
Dr. Zimmermann reported no potential conflicts of interest. Dr. Jeremias reported financial relationships with Abbott, ACIST, Boston Scientific, and Volcano. The investigator-initiated trial received research grants from Abbott Vascular and Medtronic.
A new subanalysis of the FAME 3 trial, which failed to show that percutaneous intervention (PCI) guided by fractional flow reserve (FFR) is noninferior to coronary artery bypass grafting (CABG) for treating three-vessel coronary artery disease, has associated PCI with early quality of life (QOL) advantages, according to findings presented at the annual scientific sessions of the American College of Cardiology.
Despite a modestly greater risk of major adverse cardiac events (MACE) at the end of 12 months’ follow-up among those treated with FFR-guided PCI, the greater QOL early after the procedure might be relevant to patients weighing these options, according to Frederik M. Zimmerman, MD, of Catharina Hospital in Eindhoven, the Netherlands.
“FFR-guided PCI results in a faster improvement in quality of life than CABG during the first year after revascularization, and it improved working status in patients younger than 65 years of age,” Dr. Zimmermann said.
The primary results of FAME 3 were presented at the 2021 Transcatheter Cardiovascular Therapeutics annual meeting by lead author William F. Fearon, MD, of Stanford (Calif.) University and published simultaneously in the New England Journal of Medicine.
Rather than confirming the hypothesis that FFR-guided PCI is comparable with CABG for the primary composite MACE outcome death from any cause, myocardial infarction, stroke, or revascularization, the incidence of MACE at 12 months was 10.6% in those randomized to PCI and 6.9% in the group assigned to CABG.
This translated into a hazard ratio for MACE of 1.5, signifying a 50% increase in risk for FFR-guided PCI relative to CABG for the primary outcome, a difference that negated the study definition of noninferiority (P = .35).
In this new health-related subanalysis, which was published simultaneously with his ACC presentation, the groups were compared over 12 months for QOL as measured with European Quality of Life–5 dimensions (EQ-5D) scale, angina as measured with the Canadian Cardiovascular Classification (CCC) system, and employment.
Outcomes data available in >85% of patients
Of the 1,500 patients enrolled and randomized in FAME 3 (757 to FFR-guided PCI and 743 to CABG), this health outcomes subanalysis was performed with complete data at 12 months from 89% of those in the PCI group and 88% of those in the CABG group.
Ultimately, the study did not show differences in any of these measures at the end of 12 months, but there were significant differences in QOL and employment at earlier time points. In particular, the significantly different (P < .001) trajectory for QOL improvement at 1 and 6 months favored FFR-guided PCI whether evaluated with the EQ-5D instrument or an EQ visual analog scale.
Rates of angina defined by as CCC class of at least 2 were low after revascularization in both arms of the study, negating any opportunity for differences, but patients aged younger than 65 years were almost twice as likely to have returned to full- or part-time work 1 month after revascularization (60.2% vs. 33.1%), and they remained at higher odds for working at 12 months (68.1% vs. 57.4%).
In patients aged older than 65 years, return-to-work rates did not differ significantly at any time point.
These results suggest potentially clinically meaningful early advantages for FFR-guided PCI, but some experts questioned the rationale for reporting positive secondary findings from a negative trial.
“This subanalysis is curious,” said Allen Jeremias, MD, director of interventional cardiology research, Saint Francis Hospital, Roslyn, N.Y. He pointed out that reporting these data is an anomaly.
Subanalyses uncommon in negative trials
“CABG was found to be better, so why look at QOL,” said Dr. Jeremias, who was an ACC-invited expert to discuss the results. However, he went on to say, “this could be an exception to the rule.”
The reason, according to Dr. Jeremias, is that the absolute difference at 12 months between FFR-guided PCI and CABG for the MACE events of greatest concern – death, MI, or stroke – was only about 2% greater in the FFR-guided PCI group (7.3% vs. 5.2%). The biggest contributor to the difference in MACE in FAME 3 at 12 months was the higher rate of repeat revascularization (5.9% vs. 3.9%).
Moreover, patients randomized to FFR-guided PCI had lower rates of many adverse events. This included risk of bleeding (1.6% vs. 3.8%; P = .009 as defined by type ≥3 Bleeding Academic Research Consortium , acute kidney injury (0.1% vs. 0.9%; P = .04), atrial fibrillation (2.4% vs. 14.1%; P < .001) and rehospitalization within 30 days (5.5% vs. 10.2%; P < .001).
In the context of a modest increase in risk of MACE and the lower rate of several important treatment-related adverse events, the QOL advantages identified in this subanalysis “might be a reasonable topic for patient-shared decision-making,” Dr. Jeremias suggested.
New data might inform patient decision-making
He granted the possibility that well-informed patients might accept the modestly increased risk of MACE for one or more of the outcomes, such as a higher likelihood of an early return to work, that favored FFR-guided PCI.
This is the point of this subanalysis, agreed Dr. Zimmermann.
“It is all about shared decision-making,” he said. Also emphasizing that the negative trial endpoint of FAME 3 “was driven largely by an increased risk of revascularization,” he believes that these new data might be a basis for discussions with patients weighing relative risks and benefits.
There are more data to come, according to Dr. Zimmermann, who said that follow-up of up to 5 years is planned. The 3-year data will be made available in 2023.
Dr. Zimmermann reported no potential conflicts of interest. Dr. Jeremias reported financial relationships with Abbott, ACIST, Boston Scientific, and Volcano. The investigator-initiated trial received research grants from Abbott Vascular and Medtronic.
A new subanalysis of the FAME 3 trial, which failed to show that percutaneous intervention (PCI) guided by fractional flow reserve (FFR) is noninferior to coronary artery bypass grafting (CABG) for treating three-vessel coronary artery disease, has associated PCI with early quality of life (QOL) advantages, according to findings presented at the annual scientific sessions of the American College of Cardiology.
Despite a modestly greater risk of major adverse cardiac events (MACE) at the end of 12 months’ follow-up among those treated with FFR-guided PCI, the greater QOL early after the procedure might be relevant to patients weighing these options, according to Frederik M. Zimmerman, MD, of Catharina Hospital in Eindhoven, the Netherlands.
“FFR-guided PCI results in a faster improvement in quality of life than CABG during the first year after revascularization, and it improved working status in patients younger than 65 years of age,” Dr. Zimmermann said.
The primary results of FAME 3 were presented at the 2021 Transcatheter Cardiovascular Therapeutics annual meeting by lead author William F. Fearon, MD, of Stanford (Calif.) University and published simultaneously in the New England Journal of Medicine.
Rather than confirming the hypothesis that FFR-guided PCI is comparable with CABG for the primary composite MACE outcome death from any cause, myocardial infarction, stroke, or revascularization, the incidence of MACE at 12 months was 10.6% in those randomized to PCI and 6.9% in the group assigned to CABG.
This translated into a hazard ratio for MACE of 1.5, signifying a 50% increase in risk for FFR-guided PCI relative to CABG for the primary outcome, a difference that negated the study definition of noninferiority (P = .35).
In this new health-related subanalysis, which was published simultaneously with his ACC presentation, the groups were compared over 12 months for QOL as measured with European Quality of Life–5 dimensions (EQ-5D) scale, angina as measured with the Canadian Cardiovascular Classification (CCC) system, and employment.
Outcomes data available in >85% of patients
Of the 1,500 patients enrolled and randomized in FAME 3 (757 to FFR-guided PCI and 743 to CABG), this health outcomes subanalysis was performed with complete data at 12 months from 89% of those in the PCI group and 88% of those in the CABG group.
Ultimately, the study did not show differences in any of these measures at the end of 12 months, but there were significant differences in QOL and employment at earlier time points. In particular, the significantly different (P < .001) trajectory for QOL improvement at 1 and 6 months favored FFR-guided PCI whether evaluated with the EQ-5D instrument or an EQ visual analog scale.
Rates of angina defined by as CCC class of at least 2 were low after revascularization in both arms of the study, negating any opportunity for differences, but patients aged younger than 65 years were almost twice as likely to have returned to full- or part-time work 1 month after revascularization (60.2% vs. 33.1%), and they remained at higher odds for working at 12 months (68.1% vs. 57.4%).
In patients aged older than 65 years, return-to-work rates did not differ significantly at any time point.
These results suggest potentially clinically meaningful early advantages for FFR-guided PCI, but some experts questioned the rationale for reporting positive secondary findings from a negative trial.
“This subanalysis is curious,” said Allen Jeremias, MD, director of interventional cardiology research, Saint Francis Hospital, Roslyn, N.Y. He pointed out that reporting these data is an anomaly.
Subanalyses uncommon in negative trials
“CABG was found to be better, so why look at QOL,” said Dr. Jeremias, who was an ACC-invited expert to discuss the results. However, he went on to say, “this could be an exception to the rule.”
The reason, according to Dr. Jeremias, is that the absolute difference at 12 months between FFR-guided PCI and CABG for the MACE events of greatest concern – death, MI, or stroke – was only about 2% greater in the FFR-guided PCI group (7.3% vs. 5.2%). The biggest contributor to the difference in MACE in FAME 3 at 12 months was the higher rate of repeat revascularization (5.9% vs. 3.9%).
Moreover, patients randomized to FFR-guided PCI had lower rates of many adverse events. This included risk of bleeding (1.6% vs. 3.8%; P = .009 as defined by type ≥3 Bleeding Academic Research Consortium , acute kidney injury (0.1% vs. 0.9%; P = .04), atrial fibrillation (2.4% vs. 14.1%; P < .001) and rehospitalization within 30 days (5.5% vs. 10.2%; P < .001).
In the context of a modest increase in risk of MACE and the lower rate of several important treatment-related adverse events, the QOL advantages identified in this subanalysis “might be a reasonable topic for patient-shared decision-making,” Dr. Jeremias suggested.
New data might inform patient decision-making
He granted the possibility that well-informed patients might accept the modestly increased risk of MACE for one or more of the outcomes, such as a higher likelihood of an early return to work, that favored FFR-guided PCI.
This is the point of this subanalysis, agreed Dr. Zimmermann.
“It is all about shared decision-making,” he said. Also emphasizing that the negative trial endpoint of FAME 3 “was driven largely by an increased risk of revascularization,” he believes that these new data might be a basis for discussions with patients weighing relative risks and benefits.
There are more data to come, according to Dr. Zimmermann, who said that follow-up of up to 5 years is planned. The 3-year data will be made available in 2023.
Dr. Zimmermann reported no potential conflicts of interest. Dr. Jeremias reported financial relationships with Abbott, ACIST, Boston Scientific, and Volcano. The investigator-initiated trial received research grants from Abbott Vascular and Medtronic.
FROM ACC 2021