User login
Shifting Demographics: A Temporal Analysis of the Alarming Rise in Rectal Adenocarcinoma Among Young Adults
Background
Rectal adenocarcinoma has long been associated with older adults, with routine screening typically beginning at age 45 or older. However, recent data reveal a concerning rise in rectal cancer incidence among adults under 40. These early-onset cases often present at later stages and may have distinct biological features. While some research attributes this trend to genetic or environmental factors, the contribution of socioeconomic disparities and healthcare access has not been fully explored. Identifying these influences is essential to shaping targeted prevention and early detection strategies for younger populations.
Objective
To evaluate temporal trends in rectal adenocarcinoma among young adults and assess demographic and socioeconomic predictors of early-onset diagnosis.
Methods
Data were drawn from the National Cancer Database (NCDB) for patients diagnosed with rectal adenocarcinoma from 2004 to 2022. Among 440,316 cases, 17,842 (4.1%) occurred in individuals under 40. Linear regression assessed temporal trends, while logistic regression evaluated associations between early-onset diagnosis and variables including sex, race, insurance status, income level, Charlson-Deyo comorbidity score, and tumor stage. Statistical significance was defined as α = 0.05.
Results
The number of young adults diagnosed rose from 424 in 2004 to 937 in 2022—an increase of over 120%. Each year was associated with a 1.7% rise in odds of early diagnosis (OR = 1.017, p < 0.001). Male patients had 24.7% higher odds (OR = 1.247, p < 0.001), and Black patients had 59.3% higher odds compared to White patients (OR = 1.593, p < 0.001). Non-private insurance was linked to a 41.6% decrease in early diagnosis (OR = 0.584, p < 0.001). Income level was not significant (p = 0.426). Lower Charlson-Deyo scores and higher tumor stages were also associated with early-onset cases.
Conclusions
Rectal adenocarcinoma is increasingly affecting younger adults, with significant associations across demographic and insurance variables. These findings call for improved awareness, early diagnostic strategies, and further research into underlying causes to mitigate this growing public health concern.
Background
Rectal adenocarcinoma has long been associated with older adults, with routine screening typically beginning at age 45 or older. However, recent data reveal a concerning rise in rectal cancer incidence among adults under 40. These early-onset cases often present at later stages and may have distinct biological features. While some research attributes this trend to genetic or environmental factors, the contribution of socioeconomic disparities and healthcare access has not been fully explored. Identifying these influences is essential to shaping targeted prevention and early detection strategies for younger populations.
Objective
To evaluate temporal trends in rectal adenocarcinoma among young adults and assess demographic and socioeconomic predictors of early-onset diagnosis.
Methods
Data were drawn from the National Cancer Database (NCDB) for patients diagnosed with rectal adenocarcinoma from 2004 to 2022. Among 440,316 cases, 17,842 (4.1%) occurred in individuals under 40. Linear regression assessed temporal trends, while logistic regression evaluated associations between early-onset diagnosis and variables including sex, race, insurance status, income level, Charlson-Deyo comorbidity score, and tumor stage. Statistical significance was defined as α = 0.05.
Results
The number of young adults diagnosed rose from 424 in 2004 to 937 in 2022—an increase of over 120%. Each year was associated with a 1.7% rise in odds of early diagnosis (OR = 1.017, p < 0.001). Male patients had 24.7% higher odds (OR = 1.247, p < 0.001), and Black patients had 59.3% higher odds compared to White patients (OR = 1.593, p < 0.001). Non-private insurance was linked to a 41.6% decrease in early diagnosis (OR = 0.584, p < 0.001). Income level was not significant (p = 0.426). Lower Charlson-Deyo scores and higher tumor stages were also associated with early-onset cases.
Conclusions
Rectal adenocarcinoma is increasingly affecting younger adults, with significant associations across demographic and insurance variables. These findings call for improved awareness, early diagnostic strategies, and further research into underlying causes to mitigate this growing public health concern.
Background
Rectal adenocarcinoma has long been associated with older adults, with routine screening typically beginning at age 45 or older. However, recent data reveal a concerning rise in rectal cancer incidence among adults under 40. These early-onset cases often present at later stages and may have distinct biological features. While some research attributes this trend to genetic or environmental factors, the contribution of socioeconomic disparities and healthcare access has not been fully explored. Identifying these influences is essential to shaping targeted prevention and early detection strategies for younger populations.
Objective
To evaluate temporal trends in rectal adenocarcinoma among young adults and assess demographic and socioeconomic predictors of early-onset diagnosis.
Methods
Data were drawn from the National Cancer Database (NCDB) for patients diagnosed with rectal adenocarcinoma from 2004 to 2022. Among 440,316 cases, 17,842 (4.1%) occurred in individuals under 40. Linear regression assessed temporal trends, while logistic regression evaluated associations between early-onset diagnosis and variables including sex, race, insurance status, income level, Charlson-Deyo comorbidity score, and tumor stage. Statistical significance was defined as α = 0.05.
Results
The number of young adults diagnosed rose from 424 in 2004 to 937 in 2022—an increase of over 120%. Each year was associated with a 1.7% rise in odds of early diagnosis (OR = 1.017, p < 0.001). Male patients had 24.7% higher odds (OR = 1.247, p < 0.001), and Black patients had 59.3% higher odds compared to White patients (OR = 1.593, p < 0.001). Non-private insurance was linked to a 41.6% decrease in early diagnosis (OR = 0.584, p < 0.001). Income level was not significant (p = 0.426). Lower Charlson-Deyo scores and higher tumor stages were also associated with early-onset cases.
Conclusions
Rectal adenocarcinoma is increasingly affecting younger adults, with significant associations across demographic and insurance variables. These findings call for improved awareness, early diagnostic strategies, and further research into underlying causes to mitigate this growing public health concern.
An Unusual Metastasis of Anal Squamous Cell Carcinoma
Background
Anal squamous cell carcinoma is a rare cancer which usually has locoregional spread. We report a case of distant metastasis of primary anal squamous cell carcinoma to the posterior mediastinal lymph node without lung involvement.
Case Presentation
A 63-year-old female presented with a painful anal mass, bleeding, and fluid leakage for around six months. The patient was found to have a near-circumferential fungating anal mass with bilateral inguinal lymphadenopathy. MR imaging revealed an 8.7 x 5.9 cm anal mass extending beyond the mesorectal fascia, with lymphadenopathy involving inguinal, pelvic sidewall, and iliac regions. A biopsy of the mass confirmed anal squamous cell carcinoma (ASCC). Initial treatment included diverting colostomy followed by definitive chemoradiotherapy with Mitomycin and 5-Fluorouracil. Colonoscopy post-treatment revealed tubular adenomas and a hyperplastic polyp, with no malignancy detected. The patient demonstrated a strong therapeutic response, with resolution of the anal mass and improved symptoms. However, one year later, new FDG-avid mediastinal lymph node were detected on the CT/PET scan with no pulmonary involvement. Metastatic ASCC of the Mediastinal lymph node was confirmed by biopsy. Salvage chemotherapy with Carboplatin and Paclitaxel every three weeks for six cycles achieved complete resolution of metastases.
Conclusions
This case underscores the importance of a multidisciplinary approach in managing advanced ASCC and highlights the efficacy of salvage chemotherapy in addressing metastases. Close monitoring of disease progression following surgery and chemotherapy is crucial due to the risk of recurrence.
Background
Anal squamous cell carcinoma is a rare cancer which usually has locoregional spread. We report a case of distant metastasis of primary anal squamous cell carcinoma to the posterior mediastinal lymph node without lung involvement.
Case Presentation
A 63-year-old female presented with a painful anal mass, bleeding, and fluid leakage for around six months. The patient was found to have a near-circumferential fungating anal mass with bilateral inguinal lymphadenopathy. MR imaging revealed an 8.7 x 5.9 cm anal mass extending beyond the mesorectal fascia, with lymphadenopathy involving inguinal, pelvic sidewall, and iliac regions. A biopsy of the mass confirmed anal squamous cell carcinoma (ASCC). Initial treatment included diverting colostomy followed by definitive chemoradiotherapy with Mitomycin and 5-Fluorouracil. Colonoscopy post-treatment revealed tubular adenomas and a hyperplastic polyp, with no malignancy detected. The patient demonstrated a strong therapeutic response, with resolution of the anal mass and improved symptoms. However, one year later, new FDG-avid mediastinal lymph node were detected on the CT/PET scan with no pulmonary involvement. Metastatic ASCC of the Mediastinal lymph node was confirmed by biopsy. Salvage chemotherapy with Carboplatin and Paclitaxel every three weeks for six cycles achieved complete resolution of metastases.
Conclusions
This case underscores the importance of a multidisciplinary approach in managing advanced ASCC and highlights the efficacy of salvage chemotherapy in addressing metastases. Close monitoring of disease progression following surgery and chemotherapy is crucial due to the risk of recurrence.
Background
Anal squamous cell carcinoma is a rare cancer which usually has locoregional spread. We report a case of distant metastasis of primary anal squamous cell carcinoma to the posterior mediastinal lymph node without lung involvement.
Case Presentation
A 63-year-old female presented with a painful anal mass, bleeding, and fluid leakage for around six months. The patient was found to have a near-circumferential fungating anal mass with bilateral inguinal lymphadenopathy. MR imaging revealed an 8.7 x 5.9 cm anal mass extending beyond the mesorectal fascia, with lymphadenopathy involving inguinal, pelvic sidewall, and iliac regions. A biopsy of the mass confirmed anal squamous cell carcinoma (ASCC). Initial treatment included diverting colostomy followed by definitive chemoradiotherapy with Mitomycin and 5-Fluorouracil. Colonoscopy post-treatment revealed tubular adenomas and a hyperplastic polyp, with no malignancy detected. The patient demonstrated a strong therapeutic response, with resolution of the anal mass and improved symptoms. However, one year later, new FDG-avid mediastinal lymph node were detected on the CT/PET scan with no pulmonary involvement. Metastatic ASCC of the Mediastinal lymph node was confirmed by biopsy. Salvage chemotherapy with Carboplatin and Paclitaxel every three weeks for six cycles achieved complete resolution of metastases.
Conclusions
This case underscores the importance of a multidisciplinary approach in managing advanced ASCC and highlights the efficacy of salvage chemotherapy in addressing metastases. Close monitoring of disease progression following surgery and chemotherapy is crucial due to the risk of recurrence.
Eating More Cruciferous Vegetables May Cut Colon Cancer Risk
TOPLINE:
A higher consumption of cruciferous vegetables such as broccoli and cauliflower was associated with a notably reduced risk for colon cancer (CC), with an optimal intake of 40-60 g/d providing a risk reduction of 20% to 26%.
METHODOLOGY:
- Previous meta-analyses have studied the association between the intake of cruciferous vegetables and the risk for CC; however, the quantitative dose-response relationship remained uncharacterized, limiting insights for dietary guidance.
- Researchers performed a systematic review and meta-analysis of 17 studies (seven cohort and 10 case-control studies) to analyze the dose-response association between the consumption of cruciferous vegetables and CC risk.
- Studies were included if they enrolled adults without CC at baseline (cohort studies) or adults with diagnosed cases who were matched with control individuals (case-control studies), quantified the dietary intake of cruciferous vegetables through standardized questionnaires, and included comparator groups with lower or no intake of such vegetables.
- The studies included 639,539 participants, of whom 97,595 had CC. Incident cases of CC were confirmed via medical records, pathology, registries, or validated self-report.
TAKEAWAY:
- A pooled analysis revealed that people who consumed the largest amounts of cruciferous vegetables had a 20% lower risk for CC than those who consumed the lowest amounts.
- A dose-response analysis showed that risk reduction was near maximal at an intake of 40-60 g/d (odds ratio, 0.74-0.8), with benefits plateauing beyond this range.
- The peak protective effect per gram occurred at an intake of 20-40 g/d of cruciferous vegetables and fell after 60 g/d.
IN PRACTICE:
“The pathophysiology of CC has been linked to dietary factors, specifically inadequate intake of vegetables and dietary fiber, as well as excessive alcohol and caffeine use. These empirical findings lend credence to our results, suggesting a potential chemopreventive role of CV [cruciferous vegetables] against CC development,” the authors wrote.
SOURCE:
This study, led by Bo Lai, Department of Interventional Radiology, The Second Clinical Medical School of Inner Mongolia University for the Nationalities, Yakeshi, China, was published online in BMC Gastroenterology.
LIMITATIONS:
The inclusion of both case-control and cohort studies and variations in the assessment of cruciferous vegetable intake across studies may have introduced methodological heterogeneity and measurement error, respectively. This study did not measure factors such as pesticide exposure and genetic susceptibility. The predominance of studies from North America and Asia — regions with an elevated incidence of CC — may have limited generalizability to other populations.
DISCLOSURES:
This study received no financial support. The authors declared having no competing interests.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication.
A version of this article first appeared on Medscape.com.
TOPLINE:
A higher consumption of cruciferous vegetables such as broccoli and cauliflower was associated with a notably reduced risk for colon cancer (CC), with an optimal intake of 40-60 g/d providing a risk reduction of 20% to 26%.
METHODOLOGY:
- Previous meta-analyses have studied the association between the intake of cruciferous vegetables and the risk for CC; however, the quantitative dose-response relationship remained uncharacterized, limiting insights for dietary guidance.
- Researchers performed a systematic review and meta-analysis of 17 studies (seven cohort and 10 case-control studies) to analyze the dose-response association between the consumption of cruciferous vegetables and CC risk.
- Studies were included if they enrolled adults without CC at baseline (cohort studies) or adults with diagnosed cases who were matched with control individuals (case-control studies), quantified the dietary intake of cruciferous vegetables through standardized questionnaires, and included comparator groups with lower or no intake of such vegetables.
- The studies included 639,539 participants, of whom 97,595 had CC. Incident cases of CC were confirmed via medical records, pathology, registries, or validated self-report.
TAKEAWAY:
- A pooled analysis revealed that people who consumed the largest amounts of cruciferous vegetables had a 20% lower risk for CC than those who consumed the lowest amounts.
- A dose-response analysis showed that risk reduction was near maximal at an intake of 40-60 g/d (odds ratio, 0.74-0.8), with benefits plateauing beyond this range.
- The peak protective effect per gram occurred at an intake of 20-40 g/d of cruciferous vegetables and fell after 60 g/d.
IN PRACTICE:
“The pathophysiology of CC has been linked to dietary factors, specifically inadequate intake of vegetables and dietary fiber, as well as excessive alcohol and caffeine use. These empirical findings lend credence to our results, suggesting a potential chemopreventive role of CV [cruciferous vegetables] against CC development,” the authors wrote.
SOURCE:
This study, led by Bo Lai, Department of Interventional Radiology, The Second Clinical Medical School of Inner Mongolia University for the Nationalities, Yakeshi, China, was published online in BMC Gastroenterology.
LIMITATIONS:
The inclusion of both case-control and cohort studies and variations in the assessment of cruciferous vegetable intake across studies may have introduced methodological heterogeneity and measurement error, respectively. This study did not measure factors such as pesticide exposure and genetic susceptibility. The predominance of studies from North America and Asia — regions with an elevated incidence of CC — may have limited generalizability to other populations.
DISCLOSURES:
This study received no financial support. The authors declared having no competing interests.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication.
A version of this article first appeared on Medscape.com.
TOPLINE:
A higher consumption of cruciferous vegetables such as broccoli and cauliflower was associated with a notably reduced risk for colon cancer (CC), with an optimal intake of 40-60 g/d providing a risk reduction of 20% to 26%.
METHODOLOGY:
- Previous meta-analyses have studied the association between the intake of cruciferous vegetables and the risk for CC; however, the quantitative dose-response relationship remained uncharacterized, limiting insights for dietary guidance.
- Researchers performed a systematic review and meta-analysis of 17 studies (seven cohort and 10 case-control studies) to analyze the dose-response association between the consumption of cruciferous vegetables and CC risk.
- Studies were included if they enrolled adults without CC at baseline (cohort studies) or adults with diagnosed cases who were matched with control individuals (case-control studies), quantified the dietary intake of cruciferous vegetables through standardized questionnaires, and included comparator groups with lower or no intake of such vegetables.
- The studies included 639,539 participants, of whom 97,595 had CC. Incident cases of CC were confirmed via medical records, pathology, registries, or validated self-report.
TAKEAWAY:
- A pooled analysis revealed that people who consumed the largest amounts of cruciferous vegetables had a 20% lower risk for CC than those who consumed the lowest amounts.
- A dose-response analysis showed that risk reduction was near maximal at an intake of 40-60 g/d (odds ratio, 0.74-0.8), with benefits plateauing beyond this range.
- The peak protective effect per gram occurred at an intake of 20-40 g/d of cruciferous vegetables and fell after 60 g/d.
IN PRACTICE:
“The pathophysiology of CC has been linked to dietary factors, specifically inadequate intake of vegetables and dietary fiber, as well as excessive alcohol and caffeine use. These empirical findings lend credence to our results, suggesting a potential chemopreventive role of CV [cruciferous vegetables] against CC development,” the authors wrote.
SOURCE:
This study, led by Bo Lai, Department of Interventional Radiology, The Second Clinical Medical School of Inner Mongolia University for the Nationalities, Yakeshi, China, was published online in BMC Gastroenterology.
LIMITATIONS:
The inclusion of both case-control and cohort studies and variations in the assessment of cruciferous vegetable intake across studies may have introduced methodological heterogeneity and measurement error, respectively. This study did not measure factors such as pesticide exposure and genetic susceptibility. The predominance of studies from North America and Asia — regions with an elevated incidence of CC — may have limited generalizability to other populations.
DISCLOSURES:
This study received no financial support. The authors declared having no competing interests.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication.
A version of this article first appeared on Medscape.com.

Associations Between Prescreening Dietary Patterns and Longitudinal Colonoscopy Outcomes in Veterans
Associations Between Prescreening Dietary Patterns and Longitudinal Colonoscopy Outcomes in Veterans
Screening for colorectal cancer (CRC) with colonoscopy enables the identification and removal of CRC precursors (colonic adenomas) and has been associated with reduced risk of CRC incidence and mortality.1-3 Furthermore, there is consensus that diet and lifestyle may be associated with forestalling CRC pathogenesis at the intermediate adenoma stages.4-7 However, studies have shown that US veterans have poorer diet quality and a higher risk for neoplasia compared with nonveterans, reinforcing the need for tailored clinical approaches.8,9 Combining screening with conversations about modifiable environmental and lifestyle risk factors, such as poor diet, is a highly relevant and possibly easily leveraged prevention for those at high risk. However, there is limited evidence for any particular dietary patterns or dietary features that are most important over time.7
Several dietary components have been shown to be associated with CRC risk,10 either as potentially chemopreventive (fiber, fruits and vegetables,11 dairy,12 supplemental vitamin D,13 calcium,14 and multivitamins15) or carcinogenic (red meat16 and alcohol17). Previous studies of veterans have similarly shown that higher intake of fiber and vitamin D reduced risk, and red meat is associated with an increased risk for finding CRC precursors during colonoscopy.18 However, these dietary categories are often analyzed in isolation. Studying healthy dietary patterns in aggregate may be more clinically relevant and easier to implement for prevention of CRC and its precursors.19-21 Healthy dietary patterns, such as the US Dietary Guidelines for Americans represented by the Healthy Eating Index (HEI), the Mediterranean diet (MD), and the Dietary Approaches to Stop Hypertension (DASH) diet, have been associated with lower risk for chronic disease.22-24 Despite the extant literature, no known studies have compared these dietary patterns for associations with risk of CRC precursor or CRC development among US veterans undergoing long-term screening and follow-up after a baseline colonoscopy.
The objective of this study was to test for associations between baseline scores of healthy dietary patterns and the most severe colonoscopy findings (MSCFs) over ≥ 10 years following a baseline screening colonoscopy in veterans.
Methods
Participants in the Cooperative Studies Program (CSP) #380 cohort study included 3121 asymptomatic veterans aged 50 to 75 years at baseline who had consented to initial screening colonoscopy between 1994 and 1997, with subsequent follow-up and surveillance.25 Prior to their colonoscopy, all participants completed a baseline study survey that included questions about cancer risk factors including family history of CRC, diet, physical activity, and medication use.
Included in this cross-sectional analysis were data from a sample of veteran participants of the CSP #380 cohort with 1 baseline colonoscopy, follow-up surveillance through 2009, a cancer risk factor survey collected at baseline, and complete demographic and clinical indicator data. Excluded from the analysis were 67 participants with insufficient responses to the dietary food frequency questionnaire (FFQ) and 31 participants with missing body mass index (BMI), 3023 veterans.
Measures
MSCF. The outcome of interest in this study was the MSCF recorded across all participant colonoscopies during the study period. MSCF was categorized as either (1) no neoplasia; (2) < 2 nonadvanced adenomas, including small adenomas (diameter < 10 mm) with tubular histology; or (3) advanced neoplasia (AN), which is characterized by adenomas > 10 mm in diameter, with villous histology, with high-grade dysplasia, or CRC.
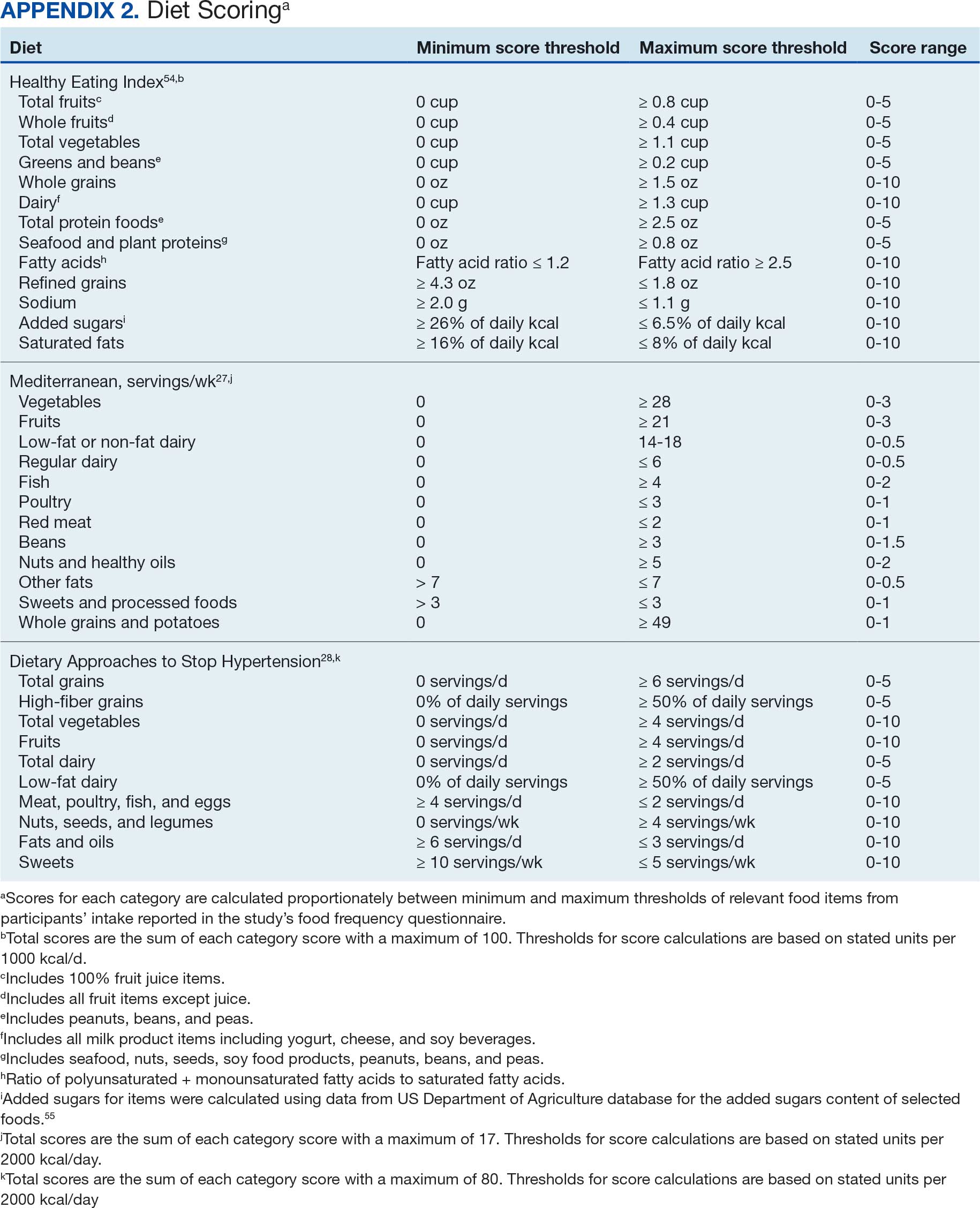
Dietary patterns. Dietary pattern scores representing dietary quality and calculated based on recommendations of the US Dietary Guidelines for Americans using the HEI, MD, and DASH diets were independent variables.26-28 These 3 dietary patterns were chosen for their hypothesized relationship with CRC risk, but each weighs food categories differently (Appendix 1).22-24,29 Dietary pattern scores were calculated using the CSP #380 self-reported responses to 129 baseline survey questions adapted from a well-established and previously validated semiquantitative FFQ.30 The form was administered by mail twice to a sample of 127 participants at baseline and at 1 year. During this interval, men completed 1-week diet records twice, spaced about 6 months apart. Mean values for intake of most nutrients assessed by the 2 methods were similar. Intraclass correlation coefficients for the baseline and 1-year FFQ-assessed nutrient intakes that ranged from 0.47 for vitamin E (without supplements) to 0.80 for vitamin C (with supplements). Correlation coefficients between the energy-adjusted nutrient intakes were measured by diet records and the 1-year FFQ, which asked about diet during the year encompassing the diet records. Higher raw and percent scores indicated better alignment with recommendations from each respective dietary pattern. Percent scores were calculated as a standardizing method and used in analyses for ease of comparing the dietary patterns. Scoring can be found in Appendix 2.
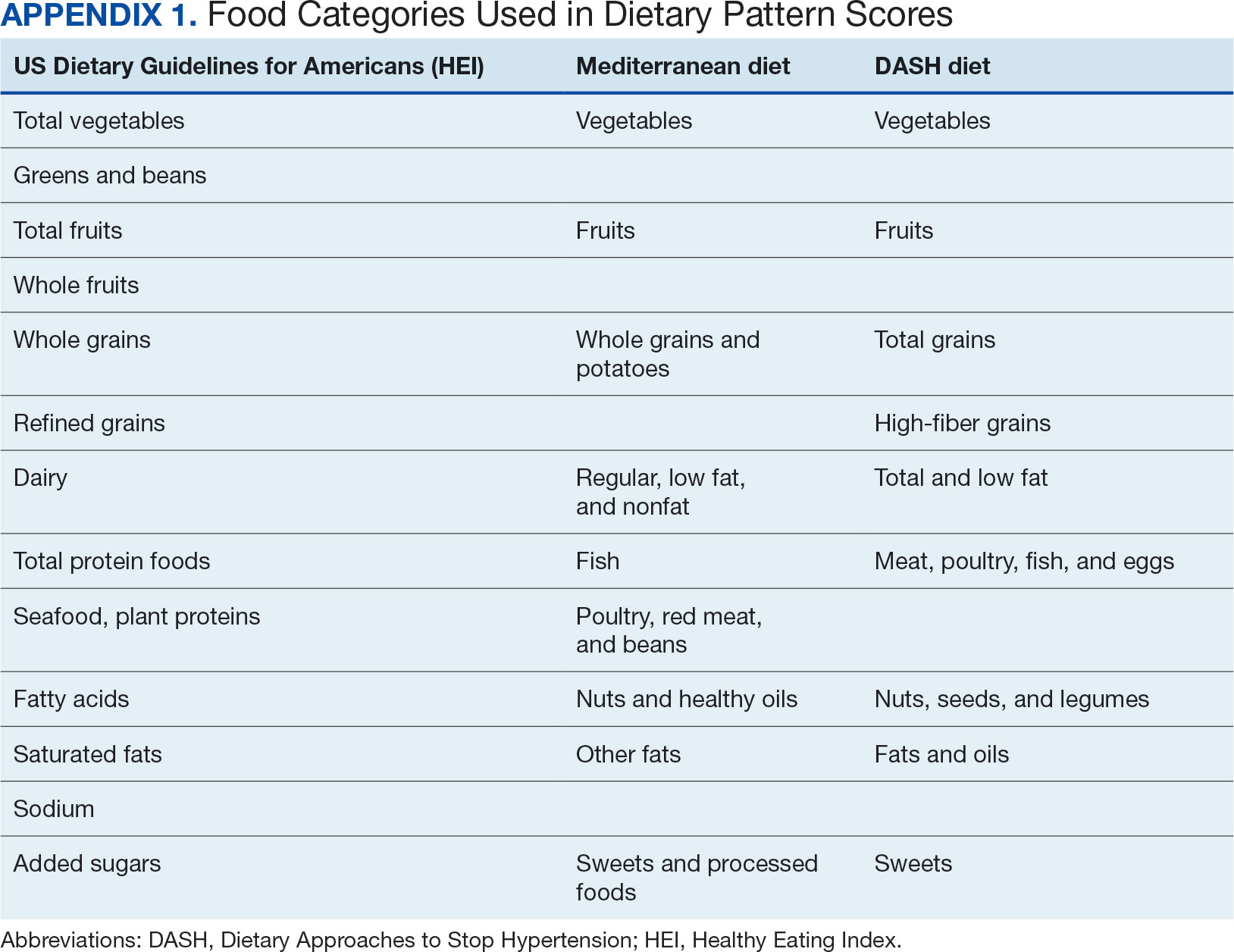
Demographic characteristics and clinical indicators. Demographic characteristics included age categories, sex, and race/ethnicity. Clinical indicators included BMI, the number of comorbid conditions used to calculate the Charlson Comorbidity Index, family history of CRC in first-degree relatives, number of follow-up colonoscopies across the study period, and food-based vitamin D intake.31 These variables were chosen for their applicability found in previous CSP #380 cohort studies.18,32,33 Self-reported race and ethnicity were collapsed due to small numbers in some groups. The authors acknowledge these are distinct concepts and the variable has limited utility other than for controlling for systemic racism in the model.
Statistical Analyses
Descriptive statistics were used to describe distributional assumptions for all variables, including demographics, clinical indicators, colonoscopy results, and dietary patterns. Pairwise correlations between the total dietary pattern scores and food category scores were calculated with Pearson correlation (r).
Multinomial logistic regression models were created using SAS procedure LOGISTIC with the outcome of the categorical MSCF (no neoplasia, nonadvanced adenoma, or AN).34 A model was created for each independent predictor variable of interest (ie, the HEI, MD, or DASH percentage-standardized dietary pattern score and each food category comprising each dietary pattern score). All models were adjusted for age, sex, race/ethnicity, BMI, number of comorbidities, family history of CRC, number of follow-up colonoscopies, and estimated daily food-derived vitamin D intake. The demographic and clinical indicators were included in the models as they are known to be associated with CRC risk.18 The number of colonoscopies was included to control for surveillance intensity presuming risk for AN is reduced as polyps are removed. Because colonoscopy findings from an initial screening have unique clinical implications compared with follow- up and surveillance, MSCF was observed in 2 ways in sensitivity analyses: (1) baseline and (2) aggregate follow-up and surveillance only, excluding baseline findings.
Adjusted odds ratios (aORs) and 95% CIs for each of the MSCF outcomes with a reference finding of no neoplasia for the models are presented. We chose not to adjust for multiple comparisons across the different dietary patterns given the correlation between dietary pattern total and category scores but did adjust for multiple comparisons for dietary categories within each dietary pattern. Tests for statistical significance used α= .05 for the dietary pattern total scores and P values for the dietary category scores for each dietary pattern controlled for false discovery rate using the MULTTEST SAS procedure.35 All data manipulations and analyses were performed using SAS version 9.4.
Results
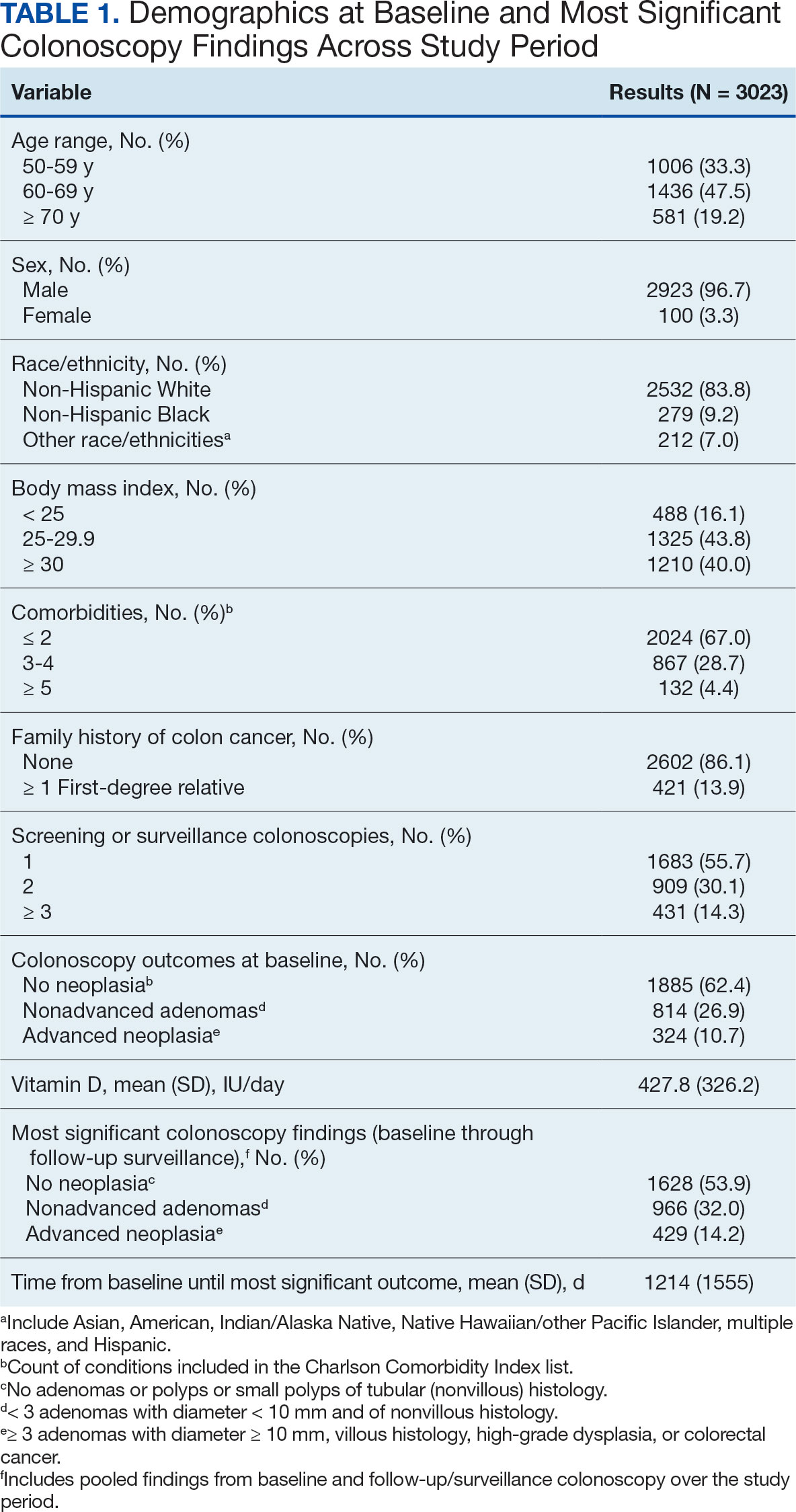
The study included 3023 patients. All were aged 50 to 75 years, 2923 (96.7%) were male and 2532 (83.8%) were non-Hispanic White (Table 1). Most participants were overweight or obese (n = 2535 [83.8%]), 2024 (67.0%) had ≤ 2 comorbidities, and 2602 (86.1%) had no family history of CRC. The MSCF for 1628 patients (53.9%) was no neoplasia, 966 patients (32.0%) was nonadvanced adenoma, and 429 participants (14.2%) had AN.
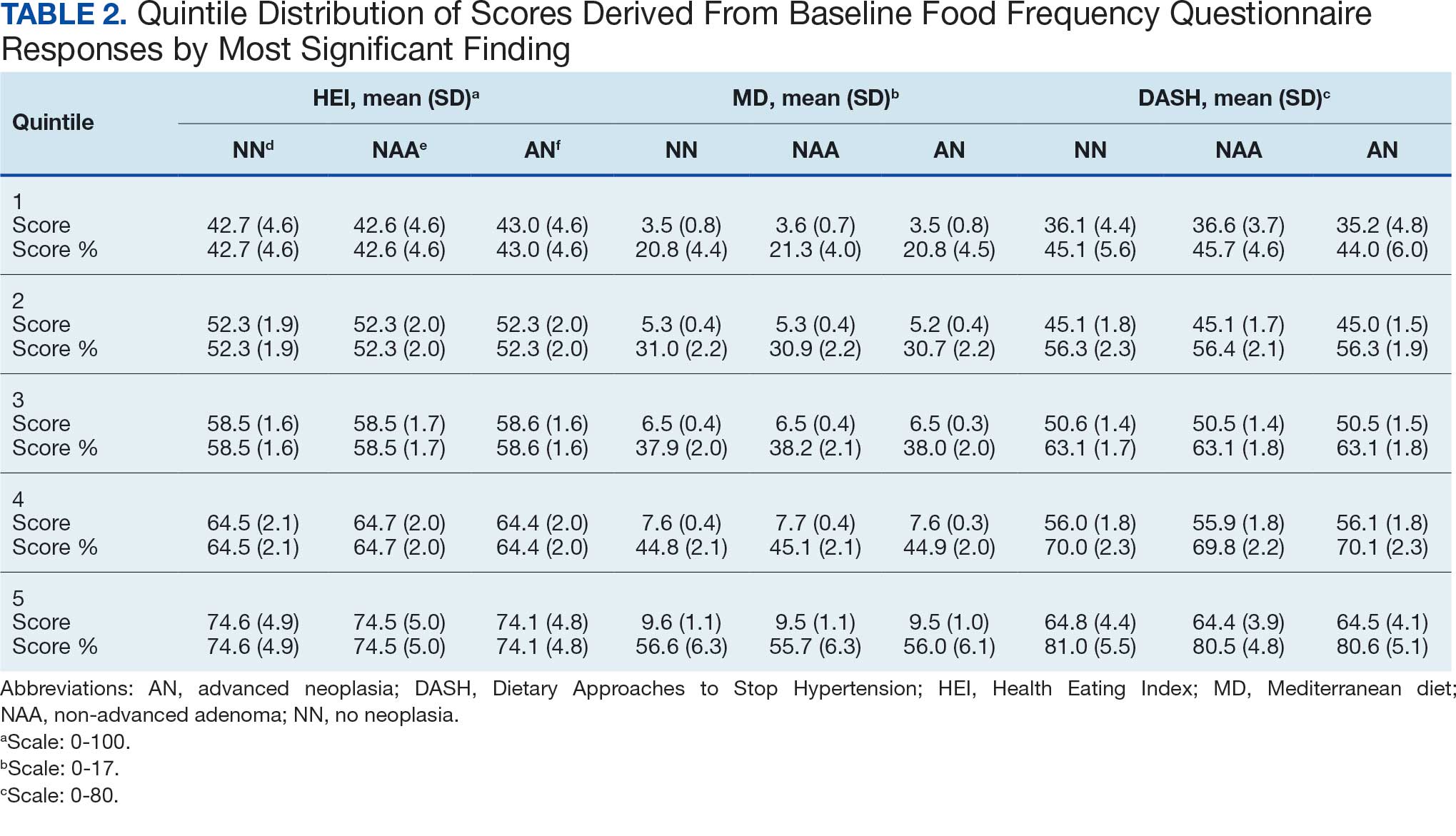
Mean percent scores were 58.5% for HEI, 38.2% for MD, and 63.1% for the DASH diet, with higher percentages indicating greater alignment with the recommendations for each diet (Table 2). All 3 dietary patterns scores standardized to percentages were strongly and significantly correlated in pairwise comparisons: HEI:MD, r = 0.62 (P < .001); HEI:DASH, r = 0.60 (P < .001); and MD:DASH, r = 0.72 (P < .001). Likewise, food category scores were significantly correlated across dietary patterns. For example, whole grain and fiber values from each dietary score were strongly correlated in pairwise comparisons: HEI Whole Grain:MD Grain, r = 0.64 (P < .001); HEI Whole Grain:DASH Fiber, r = 0.71 (P < .001); and MD Grain:DASH Fiber, r = 0.70 (P < .001).
Associations between individual participants' dietary pattern scores and the outcome of their pooled MSCF from baseline screening and ≥ 10 years of surveillance are presented in Table 3. For each single-point increases in dietary pattern scores (reflecting better dietary quality), aORs for nonadvanced adenoma vs no neoplasia were slightly lower but not statistically significantly: HEI, aOR, 1.00 (95% CI, 0.99-1.01); MD, aOR, 0.98 (95% CI, 0.94-1.02); and DASH, aOR, 0.99 (95% CI, 0.99-1.00). aORs for AN vs no neoplasia were slightly lower for each dietary pattern assessed, and only the MD and DASH scores were significantly different from 1.00: HEI, aOR, 1.00 (95% CI, 0.99-1.01); MD, aOR, 0.95 (95% CI, 0.90-1.00); and DASH, aOR, 0.99 (95% CI, 0.98-1.00).
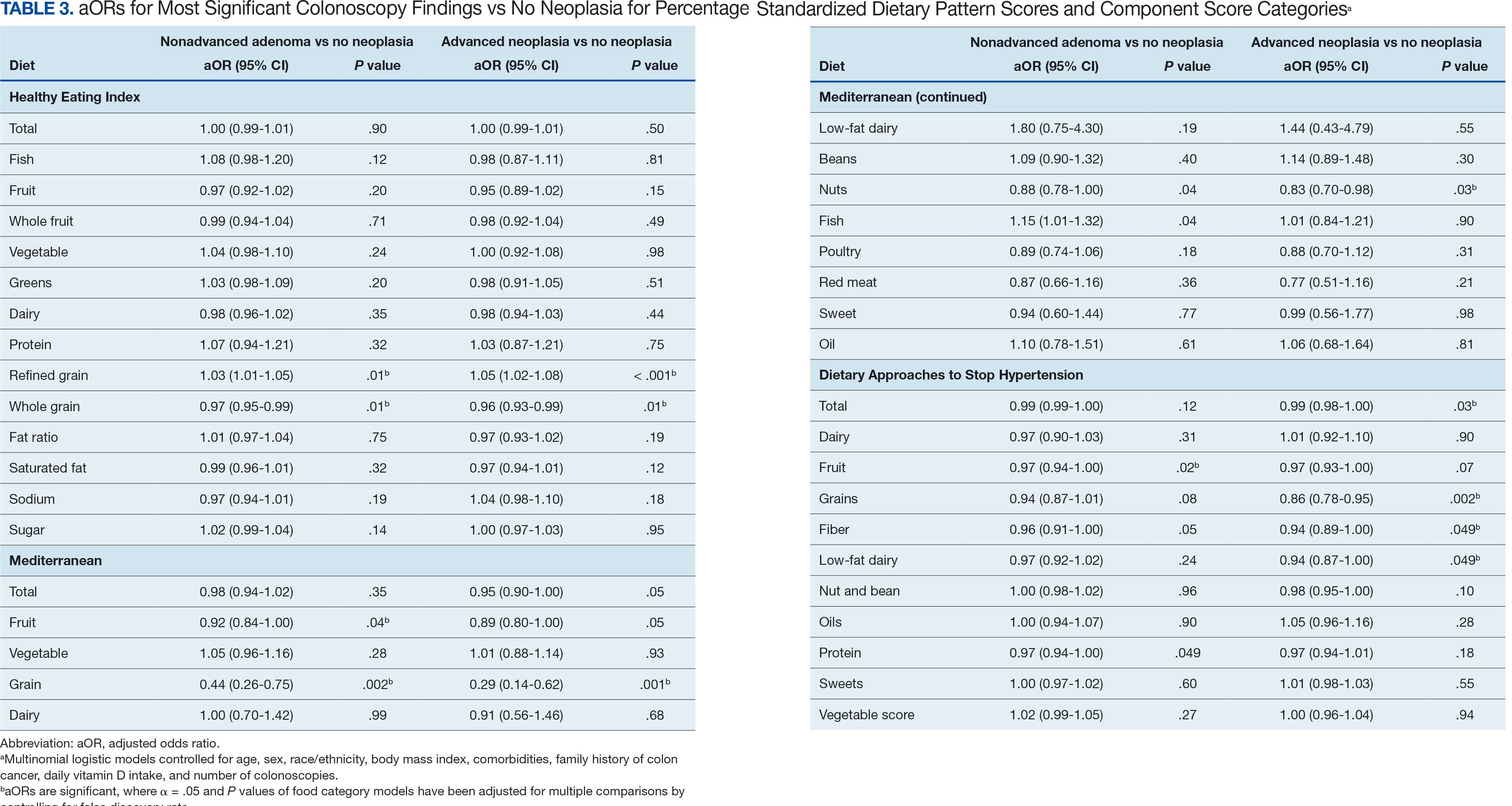
We observed lower odds for nonadvanced adenoma and AN among all these dietary patterns when there was greater alignment with the recommended intake of whole grains and fiber. In separate models conducted using food categories comprising the dietary patterns as independent variables and after correcting for multiple tests, higher scores for the HEI Refined Grain category were associated with higher odds for nonadvanced adenoma (aOR, 1.03 [95% CI, 1.01-1.05]; P = .01) and AN (aOR, 1.05 [95% CI, 1.02-1.08]; P < .001). Higher scores for the HEI Whole Grain category were associated with lower odds for nonadvanced adenoma (aOR, 0.97 [95% CI, 0.95-0.99]; P = .01) and AN (aOR, 0.96 [95% CI, 0.93-0.99]; P = .01). Higher scores for the MD Grain category were significantly associated with lower odds for nonadvanced adenoma (aOR, 0.44 [95% CI, 0.26-0.75]; P = .002) and AN (aOR, 0.29 [95% CI, 0.14-0.62]; P = .001). The DASH Grains category also was significantly associated with lower odds for AN (aOR, 0.86 [95% CI, 0.78-0.95]; P = .002).
Discussion
In this study of 3023 veterans undergoing first-time screening colonoscopy and ≥ 10 years of surveillance, we found that healthy dietary patterns, as assessed by the MD and DASH diet, were significantly associated with lower risk of AN. Additionally, we identified lower odds for AN and nonadvanced adenoma compared with no neoplasia for higher grain scores for all the dietary patterns studied. Other food categories that comprise the dietary pattern scores had mixed associations with the MSCF outcomes. Several other studies have examined associations between dietary patterns and risk for CRC but to our knowledge, no studies have explored these associations among US veterans.
These results also indicate study participants had better than average (based on a 50% threshold) dietary quality according to the HEI and DASH diet scoring methods we used, but poor dietary quality according to the MD scoring method. The mean HEI scores for the present study were higher than a US Department of Agriculture study by Dong et al that compared dietary quality between veterans and nonveterans using the HEI, for which veterans’ expected HEI score was 45.6 of 100.8 This could be explained by the fact that the participants needed to be healthy to be eligible and those with healthier behaviors overall may have self-selected into the study due to motivation for screening during a time when screening was not yet commonplace. 36 Similarly, participants of the present study had higher adherence to the DASH diet (63.1%) than adolescents with diabetes in a study by Günther et al. Conversely, firefighters who were coached to use a Mediterranean-style dietary pattern and dietary had higher adherence to MD than did participants in this study.27
A closer examination of specific food category component scores that comprise the 3 distinct dietary patterns revealed mixed results from the multinomial modeling, which may have to do with the guideline thresholds used to calculate the dietary scores. When analyzed separately in the logistic regression models for their associations with nonadvanced adenomas and AN compared with no neoplasia, higher MD and DASH fruit scores (but not HEI fruit scores) were found to be significant. Other studies have had mixed findings when attempting to test for associations of fruit intake with adenoma recurrence.10,37
This study had some unexpected findings. Vegetable intake was not associated with nonadvanced adenomas or AN risk. Studies of food categories have consistently found vegetable (specifically cruciferous ones) intake to be linked with lower odds for cancers.38 Likewise, the red meat category, which was only a unique food category in the MD score, was not associated with nonadvanced adenomas or AN. Despite consistent literature suggesting higher intake of red meat and processed meats increases CRC risk, in 2019 the Nutritional Recommendations Consortium indicated that the evidence was weak.39,40 This study showed higher DASH diet scores for low-fat dairy, which were maximized when participants reported at least 50% of their dairy servings per day as being low-fat, had lower odds for AN. Yet, the MD scores for low-fat dairy had no association with either outcome; their calculation was based on total number of servings per week. This difference in findings suggests the fat intake ratio may be more relevant to CRC risk than intake quantity.
The literature is mixed regarding fatty acid intake and CRC risk, which may be relevant to both dairy and meat intake. One systematic review and meta-analysis found dietary fat and types of fatty acid intake had no association with CRC risk.41 However, a more recent meta-analysis that assessed both dietary intake and plasma levels of fatty acids did find some statistically significant differences for various types of fatty acids and CRC risk.42
The findings in the present study that grain intake is associated with lower odds for more severe colonoscopy findings among veterans are notable.43 Lieberman et al, using the CSP #380 data, found that cereal fiber intake was associated with a lower odds for AN compared with hyperplastic polyps (OR, 0.98 [95% CI, 0.96- 1.00]).18 Similarly, Hullings et al determined that older adults in the highest quintile of cereal fiber intake had significantly lower odds of CRC than those in lower odds for CRC when compared with lowest quintile (OR, 0.89 [95% CI, 0.83- 0.96]; P < .001).44 These findings support existing guidance that prioritizes whole grains as a key source of dietary fiber for CRC prevention.
A recent literature review on fiber, fat, and CRC risk suggested a consensus regarding one protective mechanism: dietary fiber from grains modulates the gut microbiota by promoting butyrate synthesis.45 Butyrate is a short-chain fatty acid that supports energy production in colonocytes and has tumor-suppressing properties.46 Our findings suggest there could be more to learn about the relationship between butyrate production and reduction of CRC risk through metabolomic studies that use measurements of plasma butyrate. These studies may examine associations between not just a singular food or food category, but rather food patterns that include fruits, vegetables, nuts and seeds, and whole grains known to promote butyrate production and plasma butyrate.47
Improved understanding of mechanisms and risk-modifying lifestyle factors such as dietary patterns may enhance prevention strategies. Identifying the collective chemopreventive characteristics of a specific dietary pattern (eg, MD) will be helpful to clinicians and health care staff to promote healthy eating to reduce cancer risk. More studies are needed to understand whether such promotion is more clinically applicable and effective for patients, as compared with eating more or less of specific foods (eg, more whole grains, less red meat). Furthermore, considering important environmental factors collectively beyond dietary patterns may offer a way to better tailor screening and implement a variety of lifestyle interventions. In the literature, this is often referred to as a teachable moment when patients’ attentions are captured and may position them to be more receptive to guidance.48
Limitations
This study has several important limitations and leaves opportunities for future studies that explore the role of dietary patterns and AN or CRC risk. First, the FFQ data used to calculate dietary pattern scores used in analysis were only captured at baseline, and there are nearly 3 decades across the study period. However, it is widely assumed that the diets of older adults, like those included in this study, remain stable over time which is appropriate given our sample population was aged 50 to 75 years when the baseline FFQ data were collected.49-51 Additionally, while the HEI is a well-documented, standard scoring method for dietary quality, there are multitudes of dietary pattern scoring approaches for MD and DASH.23,52,53 Finally, findings from this study using the sample of veterans may not be generalizable to a broader population. Future longitudinal studies that test for a clinically significant change threshold are warranted.
Conclusion
Results of this study suggest future research should further explore the effects of dietary patterns, particularly intake of specific food groups in combination, as opposed to individual nutrients or food items, on AN and CRC risk. Possible studies might explore these dietary patterns for their mechanistic role in altering the microbiome metabolism, which may influence CRC outcomes or include diet in a more comprehensive, holistic risk score that could be used to predict colonic neoplasia risk or in intervention studies that assess the effects of dietary changes on long-term CRC prevention. We suggest there are differences in people’s dietary intake patterns that might be important to consider when implementing tailored approaches to CRC risk mitigation.
- Zauber AG, Winawer SJ, O’Brien MJ, et al. Colonoscopic polypectomy and long-term prevention of colorectalcancer deaths. N Engl J Med. 2012;366(8):687-696. doi:10.1056/NEJMoa1100370
- Nishihara R, Wu K, Lochhead P, et al. Long-term colorectal-cancer incidence and mortality after lower endoscopy. N Engl J Med. 2013;369(12):1095-1105. doi:10.1056/NEJMoa1301969
- Bretthauer M, Løberg M, Wieszczy P, et al. Effect of colonoscopy screening on risks of colorectal cancer and related death. N Engl J Med. 2022;387(17):1547-1556. doi:10.1056/NEJMoa2208375
- Cottet V, Bonithon-Kopp C, Kronborg O, et al. Dietary patterns and the risk of colorectal adenoma recurrence in a European intervention trial. Eur J Cancer Prev. 2005;14(1):21.
- Miller PE, Lesko SM, Muscat JE, Lazarus P, Hartman TJ. Dietary patterns and colorectal adenoma and cancer risk: a review of the epidemiological evidence. Nutr Cancer. 2010;62(4):413-424. doi:10.1080/01635580903407114
- Godos J, Bella F, Torrisi A, Sciacca S, Galvano F, Grosso G. Dietary patterns and risk of colorectal adenoma: a systematic review and meta-analysis of observational studies. J Hum Nutr Diet Off J Br Diet Assoc. 2016;29(6):757-767. doi:10.1111/jhn.12395
- Haggar FA, Boushey RP. Colorectal cancer epidemiology: incidence, mortality, survival, and risk factors. Clin Colon Rectal Surg. 2009;22(4):191-197. doi:10.1055/s-0029-1242458
- Dong D, Stewart H, Carlson AC. An Examination of Veterans’ Diet Quality. U.S. Department of Agriculture, Economic Research Service; 2019:32.
- El-Halabi MM, Rex DK, Saito A, Eckert GJ, Kahi CJ. Defining adenoma detection rate benchmarks in average-risk male veterans. Gastrointest Endosc. 2019;89(1):137-143. doi:10.1016/j.gie.2018.08.021
- Alberts DS, Hess LM, eds. Fundamentals of Cancer Prevention. Springer International Publishing; 2019. doi:10.1007/978-3-030-15935-1
- Dahm CC, Keogh RH, Spencer EA, et al. Dietary fiber and colorectal cancer risk: a nested case-control study using food diaries. J Natl Cancer Inst. 2010;102(9):614-626. doi:10.1093/jnci/djq092
- Aune D, Lau R, Chan DSM, et al. Dairy products and colorectal cancer risk: a systematic review and metaanalysis of cohort studies. Ann Oncol. 2012;23(1):37-45. doi:10.1093/annonc/mdr269
- Lee JE, Li H, Chan AT, et al. Circulating levels of vitamin D and colon and rectal cancer: the Physicians’ Health Study and a meta-analysis of prospective studies. Cancer Prev Res Phila Pa. 2011;4(5):735-743. doi:10.1158/1940-6207.CAPR-10-0289
- Carroll C, Cooper K, Papaioannou D, Hind D, Pilgrim H, Tappenden P. Supplemental calcium in the chemoprevention of colorectal cancer: a systematic review and meta-analysis. Clin Ther. 2010;32(5):789-803. doi:10.1016/j.clinthera.2010.04.024
- Park Y, Spiegelman D, Hunter DJ, et al. Intakes of vitamins A, C, and E and use of multiple vitamin supplements and risk of colon cancer: a pooled analysis of prospective cohort studies. Cancer Causes Control CCC. 2010;21(11):1745- 1757. doi:10.1007/s10552-010-9549-y
- Alexander DD, Weed DL, Miller PE, Mohamed MA. Red meat and colorectal cancer: a quantitative update on the state of the epidemiologic science. J Am Coll Nutr. 2015;34(6):521-543. doi:10.1080/07315724.2014.992553
- Park SY, Wilkens LR, Setiawan VW, Monroe KR, Haiman CA, Le Marchand L. Alcohol intake and colorectal cancer risk in the multiethnic cohort study. Am J Epidemiol. 2019;188(1):67-76. doi:10.1093/aje/kwy208
- Lieberman DA. Risk Factors for advanced colonic neoplasia and hyperplastic polyps in asymptomatic individuals. JAMA. 2003;290(22):2959. doi:10.1001/jama.290.22.2959
- Archambault AN, Jeon J, Lin Y, et al. Risk stratification for early-onset colorectal cancer using a combination of genetic and environmental risk scores: an international multi-center study. J Natl Cancer Inst. 2022;114(4):528-539. doi:10.1093/jnci/djac003
- Carr PR, Weigl K, Edelmann D, et al. Estimation of absolute risk of colorectal cancer based on healthy lifestyle, genetic risk, and colonoscopy status in a populationbased study. Gastroenterology. 2020;159(1):129-138.e9. doi:10.1053/j.gastro.2020.03.016
- Sullivan BA, Qin X, Miller C, et al. Screening colonoscopy findings are associated with noncolorectal cancer mortality. Clin Transl Gastroenterol. 2022;13(4):e00479. doi:10.14309/ctg.0000000000000479
- Erben V, Carr PR, Holleczek B, Stegmaier C, Hoffmeister M, Brenner H. Dietary patterns and risk of advanced colorectal neoplasms: A large population based screening study in Germany. Prev Med. 2018;111:101-109. doi:10.1016/j.ypmed.2018.02.025
- Donovan MG, Selmin OI, Doetschman TC, Romagnolo DF. Mediterranean diet: prevention of colorectal cancer. Front Nutr. 2017;4:59. doi:10.3389/fnut.2017.00059
- Mohseni R, Mohseni F, Alizadeh S, Abbasi S. The Association of Dietary Approaches to Stop Hypertension (DASH) diet with the risk of colorectal cancer: a meta-analysis of observational studies.Nutr Cancer. 2020;72(5):778-790. doi:10.1080/01635581.2019.1651880
- Lieberman DA, Weiss DG, Bond JH, Ahnen DJ, Garewal H, Chejfec G. Use of colonoscopy to screen asymptomatic adults for colorectal cancer. Veterans Affairs Cooperative Study Group 380. N Engl J Med. 2000;343(3):162-168. doi:10.1056/NEJM200007203430301
- Developing the Healthy Eating Index (HEI) | EGRP/ DCCPS/NCI/NIH. Accessed July 22, 2025. https://epi.grants.cancer.gov/hei/developing.html#2015c
- Reeve E, Piccici F, Feairheller DL. Validation of a Mediterranean diet scoring system for intervention based research. J Nutr Med Diet Care. 2021;7(1):053. doi:10.23937/2572-3278/1510053
- Günther AL, Liese AD, Bell RA, et al. ASSOCIATION BETWEEN THE DIETARY APPROACHES TO HYPERTENSION (DASH) DIET AND HYPERTENSION IN YOUTH WITH DIABETES. Hypertens Dallas Tex 1979. 2009;53(1):6-12. doi:10.1161/HYPERTENSIONAHA.108.116665
- Buckland G, Agudo A, Luján L, et al. Adherence to a Mediterranean diet and risk of gastric adenocarcinoma within the European Prospective Investigation into Cancer and Nutrition (EPIC) cohort study. Am J Clin Nutr. 2010;91(2):381- 390. doi:10.3945/ajcn.2009.28209
- Rimm EB, Giovannucci EL, Stampfer MJ, Colditz GA, Litin LB, Willett WC. Reproducibility and validity of an expanded self-administered semiquantitative food frequency questionnaire among male health professionals. Am J Epidemiol. 1992;135(10):1114-1126. doi:10.1093/oxfordjournals.aje.a116211
- Charlson ME, Pompei P, Ales KL, MacKenzie CR. A new method of classifying prognostic comorbidity in longitudinal studies: development and validation. J Chronic Dis. 1987;40(5):373-383. doi:10.1016/0021-9681(87)90171-8
- Lieberman DA, Weiss DG, Harford WV, et al. Fiveyear colon surveillance after screening colonoscopy. Gastroenterology. 2007;133(4):1077-1085. doi:10.1053/j.gastro.2007.07.006
- Lieberman D, Sullivan BA, Hauser ER, et al. Baseline colonoscopy findings associated with 10-year outcomes in a screening cohort undergoing colonoscopy surveillance. Gastroenterology. 2020;158(4):862-874.e8. doi:10.1053/j.gastro.2019.07.052
- PROC LOGISTIC: PROC LOGISTIC Statement : SAS/STAT(R) 9.22 User’s Guide. Accessed July 22, 2025. https://support.sas.com/documentation/cdl/en/statug/63347/HTML/default/viewer.htm#statug_logistic_sect004.htm
- PROC MULTTEST: PROC MULTTEST Statement : SAS/ STAT(R) 9.22 User’s Guide. Accessed July 22, 2025. https://support.sas.com/documentation/cdl/en/statug/63347/HTML/default/viewer.htm#statug_multtest_sect005.htm
- Elston DM. Participation bias, self-selection bias, and response bias. J Am Acad Dermatol. Published online June 18, 2021. doi:10.1016/j.jaad.2021.06.025
- Sansbury LB, Wanke K, Albert PS, et al. The effect of strict adherence to a high-fiber, high-fruit and -vegetable, and low-fat eating pattern on adenoma recurrence. Am J Epidemiol. 2009;170(5):576-584. doi:10.1093/aje/kwp169
- Borgas P, Gonzalez G, Veselkov K, Mirnezami R. Phytochemically rich dietary components and the risk of colorectal cancer: A systematic review and meta-analysis of observational studies. World J Clin Oncol. 2021;12(6):482- 499. doi:10.5306/wjco.v12.i6.482
- Papadimitriou N, Markozannes G, Kanellopoulou A, et al. An umbrella review of the evidence associating diet and cancer risk at 11 anatomical sites. Nat Commun. 2021;12(1):4579. doi:10.1038/s41467-021-24861-8
- Johnston BC, Zeraatkar D, Han MA, et al. Unprocessed red meat and processed meat consumption: dietary guideline recommendations from the nutritional recommendations (NutriRECS) Consortium. Ann Intern Med. 2019;171(10):756-764. doi:10.7326/M19-1621
- Kim M, Park K. Dietary fat intake and risk of colorectal cancer: a systematic review and meta-analysis of prospective studies. Nutrients. 2018;10(12):1963. doi:10.3390/nu10121963
- Lu Y, Li D, Wang L, et al. Comprehensive investigation on associations between dietary intake and blood levels of fatty acids and colorectal cancer risk. Nutrients. 2023;15(3):730. doi:10.3390/nu15030730
- Gherasim A, Arhire LI, Ni.a O, Popa AD, Graur M, Mihalache L. The relationship between lifestyle components and dietary patterns. Proc Nutr Soc. 2020;79(3):311-323. doi:10.1017/S0029665120006898
- Hullings AG, Sinha R, Liao LM, Freedman ND, Graubard BI, Loftfield E. Whole grain and dietary fiber intake and risk of colorectal cancer in the NIH-AARP Diet and Health Study cohort. Am J Clin Nutr. 2020;112(3):603- 612. doi:10.1093/ajcn/nqaa161
- Ocvirk S, Wilson AS, Appolonia CN, Thomas TK, O’Keefe SJD. Fiber, fat, and colorectal cancer: new insight into modifiable dietary risk factors. Curr Gastroenterol Rep. 2019;21(11):62. doi:10.1007/s11894-019-0725-2
- O’Keefe SJD. Diet, microorganisms and their metabolites, and colon cancer. Nat Rev Gastroenterol Hepatol. 2016;13(12):691-706. doi:10.1038/nrgastro.2016.165
- The health benefits and side effects of Butyrate Cleveland Clinic. July 11, 2022. Accessed July 22, 2025. https://health.clevelandclinic.org/butyrate-benefits/
- Knudsen MD, Wang L, Wang K, et al. Changes in lifestyle factors after endoscopic screening: a prospective study in the United States. Clin Gastroenterol Hepatol Off ClinPract J Am Gastroenterol Assoc. 2022;20(6):e1240-e1249. doi:10.1016/j.cgh.2021.07.014
- Thorpe MG, Milte CM, Crawford D, McNaughton SA. Education and lifestyle predict change in dietary patterns and diet quality of adults 55 years and over. Nutr J. 2019;18(1):67. doi:10.1186/s12937-019-0495-6
- Chapman K, Ogden J. How do people change their diet?: an exploration into mechanisms of dietary change. J Health Psychol. 2009;14(8):1229-1242. doi:10.1177/1359105309342289
- Djoussé L, Petrone AB, Weir NL, et al. Repeated versus single measurement of plasma omega-3 fatty acids and risk of heart failure. Eur J Nutr. 2014;53(6):1403-1408. doi:10.1007/s00394-013-0642-3
- Bach-Faig A, Berry EM, Lairon D, et al. Mediterranean diet pyramid today. Science and cultural updates. Public Health Nutr. 2011;14(12A):2274-2284. doi:10.1017/S1368980011002515
- Miller PE, Cross AJ, Subar AF, et al. Comparison of 4 established DASH diet indexes: examining associations of index scores and colorectal cancer123. Am J Clin Nutr. 2013;98(3):794-803. doi:10.3945/ajcn.113.063602
- Krebs-Smith SM, Pannucci TE, Subar AF, et al. Update of the Healthy Eating Index: HEI-2015. J Acad Nutr Diet. 2018;118(9):1591-1602. doi:10.1016/j.jand.2018.05.021
- P.R. Pehrsson, Cutrufelli RL, Gebhardt SE, et al. USDA Database for the Added Sugars Content of Selected Foods. USDA; 2005. www.ars.usda.gov/nutrientdata
Screening for colorectal cancer (CRC) with colonoscopy enables the identification and removal of CRC precursors (colonic adenomas) and has been associated with reduced risk of CRC incidence and mortality.1-3 Furthermore, there is consensus that diet and lifestyle may be associated with forestalling CRC pathogenesis at the intermediate adenoma stages.4-7 However, studies have shown that US veterans have poorer diet quality and a higher risk for neoplasia compared with nonveterans, reinforcing the need for tailored clinical approaches.8,9 Combining screening with conversations about modifiable environmental and lifestyle risk factors, such as poor diet, is a highly relevant and possibly easily leveraged prevention for those at high risk. However, there is limited evidence for any particular dietary patterns or dietary features that are most important over time.7
Several dietary components have been shown to be associated with CRC risk,10 either as potentially chemopreventive (fiber, fruits and vegetables,11 dairy,12 supplemental vitamin D,13 calcium,14 and multivitamins15) or carcinogenic (red meat16 and alcohol17). Previous studies of veterans have similarly shown that higher intake of fiber and vitamin D reduced risk, and red meat is associated with an increased risk for finding CRC precursors during colonoscopy.18 However, these dietary categories are often analyzed in isolation. Studying healthy dietary patterns in aggregate may be more clinically relevant and easier to implement for prevention of CRC and its precursors.19-21 Healthy dietary patterns, such as the US Dietary Guidelines for Americans represented by the Healthy Eating Index (HEI), the Mediterranean diet (MD), and the Dietary Approaches to Stop Hypertension (DASH) diet, have been associated with lower risk for chronic disease.22-24 Despite the extant literature, no known studies have compared these dietary patterns for associations with risk of CRC precursor or CRC development among US veterans undergoing long-term screening and follow-up after a baseline colonoscopy.
The objective of this study was to test for associations between baseline scores of healthy dietary patterns and the most severe colonoscopy findings (MSCFs) over ≥ 10 years following a baseline screening colonoscopy in veterans.
Methods
Participants in the Cooperative Studies Program (CSP) #380 cohort study included 3121 asymptomatic veterans aged 50 to 75 years at baseline who had consented to initial screening colonoscopy between 1994 and 1997, with subsequent follow-up and surveillance.25 Prior to their colonoscopy, all participants completed a baseline study survey that included questions about cancer risk factors including family history of CRC, diet, physical activity, and medication use.
Included in this cross-sectional analysis were data from a sample of veteran participants of the CSP #380 cohort with 1 baseline colonoscopy, follow-up surveillance through 2009, a cancer risk factor survey collected at baseline, and complete demographic and clinical indicator data. Excluded from the analysis were 67 participants with insufficient responses to the dietary food frequency questionnaire (FFQ) and 31 participants with missing body mass index (BMI), 3023 veterans.
Measures
MSCF. The outcome of interest in this study was the MSCF recorded across all participant colonoscopies during the study period. MSCF was categorized as either (1) no neoplasia; (2) < 2 nonadvanced adenomas, including small adenomas (diameter < 10 mm) with tubular histology; or (3) advanced neoplasia (AN), which is characterized by adenomas > 10 mm in diameter, with villous histology, with high-grade dysplasia, or CRC.
Dietary patterns. Dietary pattern scores representing dietary quality and calculated based on recommendations of the US Dietary Guidelines for Americans using the HEI, MD, and DASH diets were independent variables.26-28 These 3 dietary patterns were chosen for their hypothesized relationship with CRC risk, but each weighs food categories differently (Appendix 1).22-24,29 Dietary pattern scores were calculated using the CSP #380 self-reported responses to 129 baseline survey questions adapted from a well-established and previously validated semiquantitative FFQ.30 The form was administered by mail twice to a sample of 127 participants at baseline and at 1 year. During this interval, men completed 1-week diet records twice, spaced about 6 months apart. Mean values for intake of most nutrients assessed by the 2 methods were similar. Intraclass correlation coefficients for the baseline and 1-year FFQ-assessed nutrient intakes that ranged from 0.47 for vitamin E (without supplements) to 0.80 for vitamin C (with supplements). Correlation coefficients between the energy-adjusted nutrient intakes were measured by diet records and the 1-year FFQ, which asked about diet during the year encompassing the diet records. Higher raw and percent scores indicated better alignment with recommendations from each respective dietary pattern. Percent scores were calculated as a standardizing method and used in analyses for ease of comparing the dietary patterns. Scoring can be found in Appendix 2.
Demographic characteristics and clinical indicators. Demographic characteristics included age categories, sex, and race/ethnicity. Clinical indicators included BMI, the number of comorbid conditions used to calculate the Charlson Comorbidity Index, family history of CRC in first-degree relatives, number of follow-up colonoscopies across the study period, and food-based vitamin D intake.31 These variables were chosen for their applicability found in previous CSP #380 cohort studies.18,32,33 Self-reported race and ethnicity were collapsed due to small numbers in some groups. The authors acknowledge these are distinct concepts and the variable has limited utility other than for controlling for systemic racism in the model.
Statistical Analyses
Descriptive statistics were used to describe distributional assumptions for all variables, including demographics, clinical indicators, colonoscopy results, and dietary patterns. Pairwise correlations between the total dietary pattern scores and food category scores were calculated with Pearson correlation (r).
Multinomial logistic regression models were created using SAS procedure LOGISTIC with the outcome of the categorical MSCF (no neoplasia, nonadvanced adenoma, or AN).34 A model was created for each independent predictor variable of interest (ie, the HEI, MD, or DASH percentage-standardized dietary pattern score and each food category comprising each dietary pattern score). All models were adjusted for age, sex, race/ethnicity, BMI, number of comorbidities, family history of CRC, number of follow-up colonoscopies, and estimated daily food-derived vitamin D intake. The demographic and clinical indicators were included in the models as they are known to be associated with CRC risk.18 The number of colonoscopies was included to control for surveillance intensity presuming risk for AN is reduced as polyps are removed. Because colonoscopy findings from an initial screening have unique clinical implications compared with follow- up and surveillance, MSCF was observed in 2 ways in sensitivity analyses: (1) baseline and (2) aggregate follow-up and surveillance only, excluding baseline findings.
Adjusted odds ratios (aORs) and 95% CIs for each of the MSCF outcomes with a reference finding of no neoplasia for the models are presented. We chose not to adjust for multiple comparisons across the different dietary patterns given the correlation between dietary pattern total and category scores but did adjust for multiple comparisons for dietary categories within each dietary pattern. Tests for statistical significance used α= .05 for the dietary pattern total scores and P values for the dietary category scores for each dietary pattern controlled for false discovery rate using the MULTTEST SAS procedure.35 All data manipulations and analyses were performed using SAS version 9.4.
Results
The study included 3023 patients. All were aged 50 to 75 years, 2923 (96.7%) were male and 2532 (83.8%) were non-Hispanic White (Table 1). Most participants were overweight or obese (n = 2535 [83.8%]), 2024 (67.0%) had ≤ 2 comorbidities, and 2602 (86.1%) had no family history of CRC. The MSCF for 1628 patients (53.9%) was no neoplasia, 966 patients (32.0%) was nonadvanced adenoma, and 429 participants (14.2%) had AN.
Mean percent scores were 58.5% for HEI, 38.2% for MD, and 63.1% for the DASH diet, with higher percentages indicating greater alignment with the recommendations for each diet (Table 2). All 3 dietary patterns scores standardized to percentages were strongly and significantly correlated in pairwise comparisons: HEI:MD, r = 0.62 (P < .001); HEI:DASH, r = 0.60 (P < .001); and MD:DASH, r = 0.72 (P < .001). Likewise, food category scores were significantly correlated across dietary patterns. For example, whole grain and fiber values from each dietary score were strongly correlated in pairwise comparisons: HEI Whole Grain:MD Grain, r = 0.64 (P < .001); HEI Whole Grain:DASH Fiber, r = 0.71 (P < .001); and MD Grain:DASH Fiber, r = 0.70 (P < .001).
Associations between individual participants' dietary pattern scores and the outcome of their pooled MSCF from baseline screening and ≥ 10 years of surveillance are presented in Table 3. For each single-point increases in dietary pattern scores (reflecting better dietary quality), aORs for nonadvanced adenoma vs no neoplasia were slightly lower but not statistically significantly: HEI, aOR, 1.00 (95% CI, 0.99-1.01); MD, aOR, 0.98 (95% CI, 0.94-1.02); and DASH, aOR, 0.99 (95% CI, 0.99-1.00). aORs for AN vs no neoplasia were slightly lower for each dietary pattern assessed, and only the MD and DASH scores were significantly different from 1.00: HEI, aOR, 1.00 (95% CI, 0.99-1.01); MD, aOR, 0.95 (95% CI, 0.90-1.00); and DASH, aOR, 0.99 (95% CI, 0.98-1.00).
We observed lower odds for nonadvanced adenoma and AN among all these dietary patterns when there was greater alignment with the recommended intake of whole grains and fiber. In separate models conducted using food categories comprising the dietary patterns as independent variables and after correcting for multiple tests, higher scores for the HEI Refined Grain category were associated with higher odds for nonadvanced adenoma (aOR, 1.03 [95% CI, 1.01-1.05]; P = .01) and AN (aOR, 1.05 [95% CI, 1.02-1.08]; P < .001). Higher scores for the HEI Whole Grain category were associated with lower odds for nonadvanced adenoma (aOR, 0.97 [95% CI, 0.95-0.99]; P = .01) and AN (aOR, 0.96 [95% CI, 0.93-0.99]; P = .01). Higher scores for the MD Grain category were significantly associated with lower odds for nonadvanced adenoma (aOR, 0.44 [95% CI, 0.26-0.75]; P = .002) and AN (aOR, 0.29 [95% CI, 0.14-0.62]; P = .001). The DASH Grains category also was significantly associated with lower odds for AN (aOR, 0.86 [95% CI, 0.78-0.95]; P = .002).
Discussion
In this study of 3023 veterans undergoing first-time screening colonoscopy and ≥ 10 years of surveillance, we found that healthy dietary patterns, as assessed by the MD and DASH diet, were significantly associated with lower risk of AN. Additionally, we identified lower odds for AN and nonadvanced adenoma compared with no neoplasia for higher grain scores for all the dietary patterns studied. Other food categories that comprise the dietary pattern scores had mixed associations with the MSCF outcomes. Several other studies have examined associations between dietary patterns and risk for CRC but to our knowledge, no studies have explored these associations among US veterans.
These results also indicate study participants had better than average (based on a 50% threshold) dietary quality according to the HEI and DASH diet scoring methods we used, but poor dietary quality according to the MD scoring method. The mean HEI scores for the present study were higher than a US Department of Agriculture study by Dong et al that compared dietary quality between veterans and nonveterans using the HEI, for which veterans’ expected HEI score was 45.6 of 100.8 This could be explained by the fact that the participants needed to be healthy to be eligible and those with healthier behaviors overall may have self-selected into the study due to motivation for screening during a time when screening was not yet commonplace. 36 Similarly, participants of the present study had higher adherence to the DASH diet (63.1%) than adolescents with diabetes in a study by Günther et al. Conversely, firefighters who were coached to use a Mediterranean-style dietary pattern and dietary had higher adherence to MD than did participants in this study.27
A closer examination of specific food category component scores that comprise the 3 distinct dietary patterns revealed mixed results from the multinomial modeling, which may have to do with the guideline thresholds used to calculate the dietary scores. When analyzed separately in the logistic regression models for their associations with nonadvanced adenomas and AN compared with no neoplasia, higher MD and DASH fruit scores (but not HEI fruit scores) were found to be significant. Other studies have had mixed findings when attempting to test for associations of fruit intake with adenoma recurrence.10,37
This study had some unexpected findings. Vegetable intake was not associated with nonadvanced adenomas or AN risk. Studies of food categories have consistently found vegetable (specifically cruciferous ones) intake to be linked with lower odds for cancers.38 Likewise, the red meat category, which was only a unique food category in the MD score, was not associated with nonadvanced adenomas or AN. Despite consistent literature suggesting higher intake of red meat and processed meats increases CRC risk, in 2019 the Nutritional Recommendations Consortium indicated that the evidence was weak.39,40 This study showed higher DASH diet scores for low-fat dairy, which were maximized when participants reported at least 50% of their dairy servings per day as being low-fat, had lower odds for AN. Yet, the MD scores for low-fat dairy had no association with either outcome; their calculation was based on total number of servings per week. This difference in findings suggests the fat intake ratio may be more relevant to CRC risk than intake quantity.
The literature is mixed regarding fatty acid intake and CRC risk, which may be relevant to both dairy and meat intake. One systematic review and meta-analysis found dietary fat and types of fatty acid intake had no association with CRC risk.41 However, a more recent meta-analysis that assessed both dietary intake and plasma levels of fatty acids did find some statistically significant differences for various types of fatty acids and CRC risk.42
The findings in the present study that grain intake is associated with lower odds for more severe colonoscopy findings among veterans are notable.43 Lieberman et al, using the CSP #380 data, found that cereal fiber intake was associated with a lower odds for AN compared with hyperplastic polyps (OR, 0.98 [95% CI, 0.96- 1.00]).18 Similarly, Hullings et al determined that older adults in the highest quintile of cereal fiber intake had significantly lower odds of CRC than those in lower odds for CRC when compared with lowest quintile (OR, 0.89 [95% CI, 0.83- 0.96]; P < .001).44 These findings support existing guidance that prioritizes whole grains as a key source of dietary fiber for CRC prevention.
A recent literature review on fiber, fat, and CRC risk suggested a consensus regarding one protective mechanism: dietary fiber from grains modulates the gut microbiota by promoting butyrate synthesis.45 Butyrate is a short-chain fatty acid that supports energy production in colonocytes and has tumor-suppressing properties.46 Our findings suggest there could be more to learn about the relationship between butyrate production and reduction of CRC risk through metabolomic studies that use measurements of plasma butyrate. These studies may examine associations between not just a singular food or food category, but rather food patterns that include fruits, vegetables, nuts and seeds, and whole grains known to promote butyrate production and plasma butyrate.47
Improved understanding of mechanisms and risk-modifying lifestyle factors such as dietary patterns may enhance prevention strategies. Identifying the collective chemopreventive characteristics of a specific dietary pattern (eg, MD) will be helpful to clinicians and health care staff to promote healthy eating to reduce cancer risk. More studies are needed to understand whether such promotion is more clinically applicable and effective for patients, as compared with eating more or less of specific foods (eg, more whole grains, less red meat). Furthermore, considering important environmental factors collectively beyond dietary patterns may offer a way to better tailor screening and implement a variety of lifestyle interventions. In the literature, this is often referred to as a teachable moment when patients’ attentions are captured and may position them to be more receptive to guidance.48
Limitations
This study has several important limitations and leaves opportunities for future studies that explore the role of dietary patterns and AN or CRC risk. First, the FFQ data used to calculate dietary pattern scores used in analysis were only captured at baseline, and there are nearly 3 decades across the study period. However, it is widely assumed that the diets of older adults, like those included in this study, remain stable over time which is appropriate given our sample population was aged 50 to 75 years when the baseline FFQ data were collected.49-51 Additionally, while the HEI is a well-documented, standard scoring method for dietary quality, there are multitudes of dietary pattern scoring approaches for MD and DASH.23,52,53 Finally, findings from this study using the sample of veterans may not be generalizable to a broader population. Future longitudinal studies that test for a clinically significant change threshold are warranted.
Conclusion
Results of this study suggest future research should further explore the effects of dietary patterns, particularly intake of specific food groups in combination, as opposed to individual nutrients or food items, on AN and CRC risk. Possible studies might explore these dietary patterns for their mechanistic role in altering the microbiome metabolism, which may influence CRC outcomes or include diet in a more comprehensive, holistic risk score that could be used to predict colonic neoplasia risk or in intervention studies that assess the effects of dietary changes on long-term CRC prevention. We suggest there are differences in people’s dietary intake patterns that might be important to consider when implementing tailored approaches to CRC risk mitigation.
Screening for colorectal cancer (CRC) with colonoscopy enables the identification and removal of CRC precursors (colonic adenomas) and has been associated with reduced risk of CRC incidence and mortality.1-3 Furthermore, there is consensus that diet and lifestyle may be associated with forestalling CRC pathogenesis at the intermediate adenoma stages.4-7 However, studies have shown that US veterans have poorer diet quality and a higher risk for neoplasia compared with nonveterans, reinforcing the need for tailored clinical approaches.8,9 Combining screening with conversations about modifiable environmental and lifestyle risk factors, such as poor diet, is a highly relevant and possibly easily leveraged prevention for those at high risk. However, there is limited evidence for any particular dietary patterns or dietary features that are most important over time.7
Several dietary components have been shown to be associated with CRC risk,10 either as potentially chemopreventive (fiber, fruits and vegetables,11 dairy,12 supplemental vitamin D,13 calcium,14 and multivitamins15) or carcinogenic (red meat16 and alcohol17). Previous studies of veterans have similarly shown that higher intake of fiber and vitamin D reduced risk, and red meat is associated with an increased risk for finding CRC precursors during colonoscopy.18 However, these dietary categories are often analyzed in isolation. Studying healthy dietary patterns in aggregate may be more clinically relevant and easier to implement for prevention of CRC and its precursors.19-21 Healthy dietary patterns, such as the US Dietary Guidelines for Americans represented by the Healthy Eating Index (HEI), the Mediterranean diet (MD), and the Dietary Approaches to Stop Hypertension (DASH) diet, have been associated with lower risk for chronic disease.22-24 Despite the extant literature, no known studies have compared these dietary patterns for associations with risk of CRC precursor or CRC development among US veterans undergoing long-term screening and follow-up after a baseline colonoscopy.
The objective of this study was to test for associations between baseline scores of healthy dietary patterns and the most severe colonoscopy findings (MSCFs) over ≥ 10 years following a baseline screening colonoscopy in veterans.
Methods
Participants in the Cooperative Studies Program (CSP) #380 cohort study included 3121 asymptomatic veterans aged 50 to 75 years at baseline who had consented to initial screening colonoscopy between 1994 and 1997, with subsequent follow-up and surveillance.25 Prior to their colonoscopy, all participants completed a baseline study survey that included questions about cancer risk factors including family history of CRC, diet, physical activity, and medication use.
Included in this cross-sectional analysis were data from a sample of veteran participants of the CSP #380 cohort with 1 baseline colonoscopy, follow-up surveillance through 2009, a cancer risk factor survey collected at baseline, and complete demographic and clinical indicator data. Excluded from the analysis were 67 participants with insufficient responses to the dietary food frequency questionnaire (FFQ) and 31 participants with missing body mass index (BMI), 3023 veterans.
Measures
MSCF. The outcome of interest in this study was the MSCF recorded across all participant colonoscopies during the study period. MSCF was categorized as either (1) no neoplasia; (2) < 2 nonadvanced adenomas, including small adenomas (diameter < 10 mm) with tubular histology; or (3) advanced neoplasia (AN), which is characterized by adenomas > 10 mm in diameter, with villous histology, with high-grade dysplasia, or CRC.
Dietary patterns. Dietary pattern scores representing dietary quality and calculated based on recommendations of the US Dietary Guidelines for Americans using the HEI, MD, and DASH diets were independent variables.26-28 These 3 dietary patterns were chosen for their hypothesized relationship with CRC risk, but each weighs food categories differently (Appendix 1).22-24,29 Dietary pattern scores were calculated using the CSP #380 self-reported responses to 129 baseline survey questions adapted from a well-established and previously validated semiquantitative FFQ.30 The form was administered by mail twice to a sample of 127 participants at baseline and at 1 year. During this interval, men completed 1-week diet records twice, spaced about 6 months apart. Mean values for intake of most nutrients assessed by the 2 methods were similar. Intraclass correlation coefficients for the baseline and 1-year FFQ-assessed nutrient intakes that ranged from 0.47 for vitamin E (without supplements) to 0.80 for vitamin C (with supplements). Correlation coefficients between the energy-adjusted nutrient intakes were measured by diet records and the 1-year FFQ, which asked about diet during the year encompassing the diet records. Higher raw and percent scores indicated better alignment with recommendations from each respective dietary pattern. Percent scores were calculated as a standardizing method and used in analyses for ease of comparing the dietary patterns. Scoring can be found in Appendix 2.
Demographic characteristics and clinical indicators. Demographic characteristics included age categories, sex, and race/ethnicity. Clinical indicators included BMI, the number of comorbid conditions used to calculate the Charlson Comorbidity Index, family history of CRC in first-degree relatives, number of follow-up colonoscopies across the study period, and food-based vitamin D intake.31 These variables were chosen for their applicability found in previous CSP #380 cohort studies.18,32,33 Self-reported race and ethnicity were collapsed due to small numbers in some groups. The authors acknowledge these are distinct concepts and the variable has limited utility other than for controlling for systemic racism in the model.
Statistical Analyses
Descriptive statistics were used to describe distributional assumptions for all variables, including demographics, clinical indicators, colonoscopy results, and dietary patterns. Pairwise correlations between the total dietary pattern scores and food category scores were calculated with Pearson correlation (r).
Multinomial logistic regression models were created using SAS procedure LOGISTIC with the outcome of the categorical MSCF (no neoplasia, nonadvanced adenoma, or AN).34 A model was created for each independent predictor variable of interest (ie, the HEI, MD, or DASH percentage-standardized dietary pattern score and each food category comprising each dietary pattern score). All models were adjusted for age, sex, race/ethnicity, BMI, number of comorbidities, family history of CRC, number of follow-up colonoscopies, and estimated daily food-derived vitamin D intake. The demographic and clinical indicators were included in the models as they are known to be associated with CRC risk.18 The number of colonoscopies was included to control for surveillance intensity presuming risk for AN is reduced as polyps are removed. Because colonoscopy findings from an initial screening have unique clinical implications compared with follow- up and surveillance, MSCF was observed in 2 ways in sensitivity analyses: (1) baseline and (2) aggregate follow-up and surveillance only, excluding baseline findings.
Adjusted odds ratios (aORs) and 95% CIs for each of the MSCF outcomes with a reference finding of no neoplasia for the models are presented. We chose not to adjust for multiple comparisons across the different dietary patterns given the correlation between dietary pattern total and category scores but did adjust for multiple comparisons for dietary categories within each dietary pattern. Tests for statistical significance used α= .05 for the dietary pattern total scores and P values for the dietary category scores for each dietary pattern controlled for false discovery rate using the MULTTEST SAS procedure.35 All data manipulations and analyses were performed using SAS version 9.4.
Results
The study included 3023 patients. All were aged 50 to 75 years, 2923 (96.7%) were male and 2532 (83.8%) were non-Hispanic White (Table 1). Most participants were overweight or obese (n = 2535 [83.8%]), 2024 (67.0%) had ≤ 2 comorbidities, and 2602 (86.1%) had no family history of CRC. The MSCF for 1628 patients (53.9%) was no neoplasia, 966 patients (32.0%) was nonadvanced adenoma, and 429 participants (14.2%) had AN.
Mean percent scores were 58.5% for HEI, 38.2% for MD, and 63.1% for the DASH diet, with higher percentages indicating greater alignment with the recommendations for each diet (Table 2). All 3 dietary patterns scores standardized to percentages were strongly and significantly correlated in pairwise comparisons: HEI:MD, r = 0.62 (P < .001); HEI:DASH, r = 0.60 (P < .001); and MD:DASH, r = 0.72 (P < .001). Likewise, food category scores were significantly correlated across dietary patterns. For example, whole grain and fiber values from each dietary score were strongly correlated in pairwise comparisons: HEI Whole Grain:MD Grain, r = 0.64 (P < .001); HEI Whole Grain:DASH Fiber, r = 0.71 (P < .001); and MD Grain:DASH Fiber, r = 0.70 (P < .001).
Associations between individual participants' dietary pattern scores and the outcome of their pooled MSCF from baseline screening and ≥ 10 years of surveillance are presented in Table 3. For each single-point increases in dietary pattern scores (reflecting better dietary quality), aORs for nonadvanced adenoma vs no neoplasia were slightly lower but not statistically significantly: HEI, aOR, 1.00 (95% CI, 0.99-1.01); MD, aOR, 0.98 (95% CI, 0.94-1.02); and DASH, aOR, 0.99 (95% CI, 0.99-1.00). aORs for AN vs no neoplasia were slightly lower for each dietary pattern assessed, and only the MD and DASH scores were significantly different from 1.00: HEI, aOR, 1.00 (95% CI, 0.99-1.01); MD, aOR, 0.95 (95% CI, 0.90-1.00); and DASH, aOR, 0.99 (95% CI, 0.98-1.00).
We observed lower odds for nonadvanced adenoma and AN among all these dietary patterns when there was greater alignment with the recommended intake of whole grains and fiber. In separate models conducted using food categories comprising the dietary patterns as independent variables and after correcting for multiple tests, higher scores for the HEI Refined Grain category were associated with higher odds for nonadvanced adenoma (aOR, 1.03 [95% CI, 1.01-1.05]; P = .01) and AN (aOR, 1.05 [95% CI, 1.02-1.08]; P < .001). Higher scores for the HEI Whole Grain category were associated with lower odds for nonadvanced adenoma (aOR, 0.97 [95% CI, 0.95-0.99]; P = .01) and AN (aOR, 0.96 [95% CI, 0.93-0.99]; P = .01). Higher scores for the MD Grain category were significantly associated with lower odds for nonadvanced adenoma (aOR, 0.44 [95% CI, 0.26-0.75]; P = .002) and AN (aOR, 0.29 [95% CI, 0.14-0.62]; P = .001). The DASH Grains category also was significantly associated with lower odds for AN (aOR, 0.86 [95% CI, 0.78-0.95]; P = .002).
Discussion
In this study of 3023 veterans undergoing first-time screening colonoscopy and ≥ 10 years of surveillance, we found that healthy dietary patterns, as assessed by the MD and DASH diet, were significantly associated with lower risk of AN. Additionally, we identified lower odds for AN and nonadvanced adenoma compared with no neoplasia for higher grain scores for all the dietary patterns studied. Other food categories that comprise the dietary pattern scores had mixed associations with the MSCF outcomes. Several other studies have examined associations between dietary patterns and risk for CRC but to our knowledge, no studies have explored these associations among US veterans.
These results also indicate study participants had better than average (based on a 50% threshold) dietary quality according to the HEI and DASH diet scoring methods we used, but poor dietary quality according to the MD scoring method. The mean HEI scores for the present study were higher than a US Department of Agriculture study by Dong et al that compared dietary quality between veterans and nonveterans using the HEI, for which veterans’ expected HEI score was 45.6 of 100.8 This could be explained by the fact that the participants needed to be healthy to be eligible and those with healthier behaviors overall may have self-selected into the study due to motivation for screening during a time when screening was not yet commonplace. 36 Similarly, participants of the present study had higher adherence to the DASH diet (63.1%) than adolescents with diabetes in a study by Günther et al. Conversely, firefighters who were coached to use a Mediterranean-style dietary pattern and dietary had higher adherence to MD than did participants in this study.27
A closer examination of specific food category component scores that comprise the 3 distinct dietary patterns revealed mixed results from the multinomial modeling, which may have to do with the guideline thresholds used to calculate the dietary scores. When analyzed separately in the logistic regression models for their associations with nonadvanced adenomas and AN compared with no neoplasia, higher MD and DASH fruit scores (but not HEI fruit scores) were found to be significant. Other studies have had mixed findings when attempting to test for associations of fruit intake with adenoma recurrence.10,37
This study had some unexpected findings. Vegetable intake was not associated with nonadvanced adenomas or AN risk. Studies of food categories have consistently found vegetable (specifically cruciferous ones) intake to be linked with lower odds for cancers.38 Likewise, the red meat category, which was only a unique food category in the MD score, was not associated with nonadvanced adenomas or AN. Despite consistent literature suggesting higher intake of red meat and processed meats increases CRC risk, in 2019 the Nutritional Recommendations Consortium indicated that the evidence was weak.39,40 This study showed higher DASH diet scores for low-fat dairy, which were maximized when participants reported at least 50% of their dairy servings per day as being low-fat, had lower odds for AN. Yet, the MD scores for low-fat dairy had no association with either outcome; their calculation was based on total number of servings per week. This difference in findings suggests the fat intake ratio may be more relevant to CRC risk than intake quantity.
The literature is mixed regarding fatty acid intake and CRC risk, which may be relevant to both dairy and meat intake. One systematic review and meta-analysis found dietary fat and types of fatty acid intake had no association with CRC risk.41 However, a more recent meta-analysis that assessed both dietary intake and plasma levels of fatty acids did find some statistically significant differences for various types of fatty acids and CRC risk.42
The findings in the present study that grain intake is associated with lower odds for more severe colonoscopy findings among veterans are notable.43 Lieberman et al, using the CSP #380 data, found that cereal fiber intake was associated with a lower odds for AN compared with hyperplastic polyps (OR, 0.98 [95% CI, 0.96- 1.00]).18 Similarly, Hullings et al determined that older adults in the highest quintile of cereal fiber intake had significantly lower odds of CRC than those in lower odds for CRC when compared with lowest quintile (OR, 0.89 [95% CI, 0.83- 0.96]; P < .001).44 These findings support existing guidance that prioritizes whole grains as a key source of dietary fiber for CRC prevention.
A recent literature review on fiber, fat, and CRC risk suggested a consensus regarding one protective mechanism: dietary fiber from grains modulates the gut microbiota by promoting butyrate synthesis.45 Butyrate is a short-chain fatty acid that supports energy production in colonocytes and has tumor-suppressing properties.46 Our findings suggest there could be more to learn about the relationship between butyrate production and reduction of CRC risk through metabolomic studies that use measurements of plasma butyrate. These studies may examine associations between not just a singular food or food category, but rather food patterns that include fruits, vegetables, nuts and seeds, and whole grains known to promote butyrate production and plasma butyrate.47
Improved understanding of mechanisms and risk-modifying lifestyle factors such as dietary patterns may enhance prevention strategies. Identifying the collective chemopreventive characteristics of a specific dietary pattern (eg, MD) will be helpful to clinicians and health care staff to promote healthy eating to reduce cancer risk. More studies are needed to understand whether such promotion is more clinically applicable and effective for patients, as compared with eating more or less of specific foods (eg, more whole grains, less red meat). Furthermore, considering important environmental factors collectively beyond dietary patterns may offer a way to better tailor screening and implement a variety of lifestyle interventions. In the literature, this is often referred to as a teachable moment when patients’ attentions are captured and may position them to be more receptive to guidance.48
Limitations
This study has several important limitations and leaves opportunities for future studies that explore the role of dietary patterns and AN or CRC risk. First, the FFQ data used to calculate dietary pattern scores used in analysis were only captured at baseline, and there are nearly 3 decades across the study period. However, it is widely assumed that the diets of older adults, like those included in this study, remain stable over time which is appropriate given our sample population was aged 50 to 75 years when the baseline FFQ data were collected.49-51 Additionally, while the HEI is a well-documented, standard scoring method for dietary quality, there are multitudes of dietary pattern scoring approaches for MD and DASH.23,52,53 Finally, findings from this study using the sample of veterans may not be generalizable to a broader population. Future longitudinal studies that test for a clinically significant change threshold are warranted.
Conclusion
Results of this study suggest future research should further explore the effects of dietary patterns, particularly intake of specific food groups in combination, as opposed to individual nutrients or food items, on AN and CRC risk. Possible studies might explore these dietary patterns for their mechanistic role in altering the microbiome metabolism, which may influence CRC outcomes or include diet in a more comprehensive, holistic risk score that could be used to predict colonic neoplasia risk or in intervention studies that assess the effects of dietary changes on long-term CRC prevention. We suggest there are differences in people’s dietary intake patterns that might be important to consider when implementing tailored approaches to CRC risk mitigation.
- Zauber AG, Winawer SJ, O’Brien MJ, et al. Colonoscopic polypectomy and long-term prevention of colorectalcancer deaths. N Engl J Med. 2012;366(8):687-696. doi:10.1056/NEJMoa1100370
- Nishihara R, Wu K, Lochhead P, et al. Long-term colorectal-cancer incidence and mortality after lower endoscopy. N Engl J Med. 2013;369(12):1095-1105. doi:10.1056/NEJMoa1301969
- Bretthauer M, Løberg M, Wieszczy P, et al. Effect of colonoscopy screening on risks of colorectal cancer and related death. N Engl J Med. 2022;387(17):1547-1556. doi:10.1056/NEJMoa2208375
- Cottet V, Bonithon-Kopp C, Kronborg O, et al. Dietary patterns and the risk of colorectal adenoma recurrence in a European intervention trial. Eur J Cancer Prev. 2005;14(1):21.
- Miller PE, Lesko SM, Muscat JE, Lazarus P, Hartman TJ. Dietary patterns and colorectal adenoma and cancer risk: a review of the epidemiological evidence. Nutr Cancer. 2010;62(4):413-424. doi:10.1080/01635580903407114
- Godos J, Bella F, Torrisi A, Sciacca S, Galvano F, Grosso G. Dietary patterns and risk of colorectal adenoma: a systematic review and meta-analysis of observational studies. J Hum Nutr Diet Off J Br Diet Assoc. 2016;29(6):757-767. doi:10.1111/jhn.12395
- Haggar FA, Boushey RP. Colorectal cancer epidemiology: incidence, mortality, survival, and risk factors. Clin Colon Rectal Surg. 2009;22(4):191-197. doi:10.1055/s-0029-1242458
- Dong D, Stewart H, Carlson AC. An Examination of Veterans’ Diet Quality. U.S. Department of Agriculture, Economic Research Service; 2019:32.
- El-Halabi MM, Rex DK, Saito A, Eckert GJ, Kahi CJ. Defining adenoma detection rate benchmarks in average-risk male veterans. Gastrointest Endosc. 2019;89(1):137-143. doi:10.1016/j.gie.2018.08.021
- Alberts DS, Hess LM, eds. Fundamentals of Cancer Prevention. Springer International Publishing; 2019. doi:10.1007/978-3-030-15935-1
- Dahm CC, Keogh RH, Spencer EA, et al. Dietary fiber and colorectal cancer risk: a nested case-control study using food diaries. J Natl Cancer Inst. 2010;102(9):614-626. doi:10.1093/jnci/djq092
- Aune D, Lau R, Chan DSM, et al. Dairy products and colorectal cancer risk: a systematic review and metaanalysis of cohort studies. Ann Oncol. 2012;23(1):37-45. doi:10.1093/annonc/mdr269
- Lee JE, Li H, Chan AT, et al. Circulating levels of vitamin D and colon and rectal cancer: the Physicians’ Health Study and a meta-analysis of prospective studies. Cancer Prev Res Phila Pa. 2011;4(5):735-743. doi:10.1158/1940-6207.CAPR-10-0289
- Carroll C, Cooper K, Papaioannou D, Hind D, Pilgrim H, Tappenden P. Supplemental calcium in the chemoprevention of colorectal cancer: a systematic review and meta-analysis. Clin Ther. 2010;32(5):789-803. doi:10.1016/j.clinthera.2010.04.024
- Park Y, Spiegelman D, Hunter DJ, et al. Intakes of vitamins A, C, and E and use of multiple vitamin supplements and risk of colon cancer: a pooled analysis of prospective cohort studies. Cancer Causes Control CCC. 2010;21(11):1745- 1757. doi:10.1007/s10552-010-9549-y
- Alexander DD, Weed DL, Miller PE, Mohamed MA. Red meat and colorectal cancer: a quantitative update on the state of the epidemiologic science. J Am Coll Nutr. 2015;34(6):521-543. doi:10.1080/07315724.2014.992553
- Park SY, Wilkens LR, Setiawan VW, Monroe KR, Haiman CA, Le Marchand L. Alcohol intake and colorectal cancer risk in the multiethnic cohort study. Am J Epidemiol. 2019;188(1):67-76. doi:10.1093/aje/kwy208
- Lieberman DA. Risk Factors for advanced colonic neoplasia and hyperplastic polyps in asymptomatic individuals. JAMA. 2003;290(22):2959. doi:10.1001/jama.290.22.2959
- Archambault AN, Jeon J, Lin Y, et al. Risk stratification for early-onset colorectal cancer using a combination of genetic and environmental risk scores: an international multi-center study. J Natl Cancer Inst. 2022;114(4):528-539. doi:10.1093/jnci/djac003
- Carr PR, Weigl K, Edelmann D, et al. Estimation of absolute risk of colorectal cancer based on healthy lifestyle, genetic risk, and colonoscopy status in a populationbased study. Gastroenterology. 2020;159(1):129-138.e9. doi:10.1053/j.gastro.2020.03.016
- Sullivan BA, Qin X, Miller C, et al. Screening colonoscopy findings are associated with noncolorectal cancer mortality. Clin Transl Gastroenterol. 2022;13(4):e00479. doi:10.14309/ctg.0000000000000479
- Erben V, Carr PR, Holleczek B, Stegmaier C, Hoffmeister M, Brenner H. Dietary patterns and risk of advanced colorectal neoplasms: A large population based screening study in Germany. Prev Med. 2018;111:101-109. doi:10.1016/j.ypmed.2018.02.025
- Donovan MG, Selmin OI, Doetschman TC, Romagnolo DF. Mediterranean diet: prevention of colorectal cancer. Front Nutr. 2017;4:59. doi:10.3389/fnut.2017.00059
- Mohseni R, Mohseni F, Alizadeh S, Abbasi S. The Association of Dietary Approaches to Stop Hypertension (DASH) diet with the risk of colorectal cancer: a meta-analysis of observational studies.Nutr Cancer. 2020;72(5):778-790. doi:10.1080/01635581.2019.1651880
- Lieberman DA, Weiss DG, Bond JH, Ahnen DJ, Garewal H, Chejfec G. Use of colonoscopy to screen asymptomatic adults for colorectal cancer. Veterans Affairs Cooperative Study Group 380. N Engl J Med. 2000;343(3):162-168. doi:10.1056/NEJM200007203430301
- Developing the Healthy Eating Index (HEI) | EGRP/ DCCPS/NCI/NIH. Accessed July 22, 2025. https://epi.grants.cancer.gov/hei/developing.html#2015c
- Reeve E, Piccici F, Feairheller DL. Validation of a Mediterranean diet scoring system for intervention based research. J Nutr Med Diet Care. 2021;7(1):053. doi:10.23937/2572-3278/1510053
- Günther AL, Liese AD, Bell RA, et al. ASSOCIATION BETWEEN THE DIETARY APPROACHES TO HYPERTENSION (DASH) DIET AND HYPERTENSION IN YOUTH WITH DIABETES. Hypertens Dallas Tex 1979. 2009;53(1):6-12. doi:10.1161/HYPERTENSIONAHA.108.116665
- Buckland G, Agudo A, Luján L, et al. Adherence to a Mediterranean diet and risk of gastric adenocarcinoma within the European Prospective Investigation into Cancer and Nutrition (EPIC) cohort study. Am J Clin Nutr. 2010;91(2):381- 390. doi:10.3945/ajcn.2009.28209
- Rimm EB, Giovannucci EL, Stampfer MJ, Colditz GA, Litin LB, Willett WC. Reproducibility and validity of an expanded self-administered semiquantitative food frequency questionnaire among male health professionals. Am J Epidemiol. 1992;135(10):1114-1126. doi:10.1093/oxfordjournals.aje.a116211
- Charlson ME, Pompei P, Ales KL, MacKenzie CR. A new method of classifying prognostic comorbidity in longitudinal studies: development and validation. J Chronic Dis. 1987;40(5):373-383. doi:10.1016/0021-9681(87)90171-8
- Lieberman DA, Weiss DG, Harford WV, et al. Fiveyear colon surveillance after screening colonoscopy. Gastroenterology. 2007;133(4):1077-1085. doi:10.1053/j.gastro.2007.07.006
- Lieberman D, Sullivan BA, Hauser ER, et al. Baseline colonoscopy findings associated with 10-year outcomes in a screening cohort undergoing colonoscopy surveillance. Gastroenterology. 2020;158(4):862-874.e8. doi:10.1053/j.gastro.2019.07.052
- PROC LOGISTIC: PROC LOGISTIC Statement : SAS/STAT(R) 9.22 User’s Guide. Accessed July 22, 2025. https://support.sas.com/documentation/cdl/en/statug/63347/HTML/default/viewer.htm#statug_logistic_sect004.htm
- PROC MULTTEST: PROC MULTTEST Statement : SAS/ STAT(R) 9.22 User’s Guide. Accessed July 22, 2025. https://support.sas.com/documentation/cdl/en/statug/63347/HTML/default/viewer.htm#statug_multtest_sect005.htm
- Elston DM. Participation bias, self-selection bias, and response bias. J Am Acad Dermatol. Published online June 18, 2021. doi:10.1016/j.jaad.2021.06.025
- Sansbury LB, Wanke K, Albert PS, et al. The effect of strict adherence to a high-fiber, high-fruit and -vegetable, and low-fat eating pattern on adenoma recurrence. Am J Epidemiol. 2009;170(5):576-584. doi:10.1093/aje/kwp169
- Borgas P, Gonzalez G, Veselkov K, Mirnezami R. Phytochemically rich dietary components and the risk of colorectal cancer: A systematic review and meta-analysis of observational studies. World J Clin Oncol. 2021;12(6):482- 499. doi:10.5306/wjco.v12.i6.482
- Papadimitriou N, Markozannes G, Kanellopoulou A, et al. An umbrella review of the evidence associating diet and cancer risk at 11 anatomical sites. Nat Commun. 2021;12(1):4579. doi:10.1038/s41467-021-24861-8
- Johnston BC, Zeraatkar D, Han MA, et al. Unprocessed red meat and processed meat consumption: dietary guideline recommendations from the nutritional recommendations (NutriRECS) Consortium. Ann Intern Med. 2019;171(10):756-764. doi:10.7326/M19-1621
- Kim M, Park K. Dietary fat intake and risk of colorectal cancer: a systematic review and meta-analysis of prospective studies. Nutrients. 2018;10(12):1963. doi:10.3390/nu10121963
- Lu Y, Li D, Wang L, et al. Comprehensive investigation on associations between dietary intake and blood levels of fatty acids and colorectal cancer risk. Nutrients. 2023;15(3):730. doi:10.3390/nu15030730
- Gherasim A, Arhire LI, Ni.a O, Popa AD, Graur M, Mihalache L. The relationship between lifestyle components and dietary patterns. Proc Nutr Soc. 2020;79(3):311-323. doi:10.1017/S0029665120006898
- Hullings AG, Sinha R, Liao LM, Freedman ND, Graubard BI, Loftfield E. Whole grain and dietary fiber intake and risk of colorectal cancer in the NIH-AARP Diet and Health Study cohort. Am J Clin Nutr. 2020;112(3):603- 612. doi:10.1093/ajcn/nqaa161
- Ocvirk S, Wilson AS, Appolonia CN, Thomas TK, O’Keefe SJD. Fiber, fat, and colorectal cancer: new insight into modifiable dietary risk factors. Curr Gastroenterol Rep. 2019;21(11):62. doi:10.1007/s11894-019-0725-2
- O’Keefe SJD. Diet, microorganisms and their metabolites, and colon cancer. Nat Rev Gastroenterol Hepatol. 2016;13(12):691-706. doi:10.1038/nrgastro.2016.165
- The health benefits and side effects of Butyrate Cleveland Clinic. July 11, 2022. Accessed July 22, 2025. https://health.clevelandclinic.org/butyrate-benefits/
- Knudsen MD, Wang L, Wang K, et al. Changes in lifestyle factors after endoscopic screening: a prospective study in the United States. Clin Gastroenterol Hepatol Off ClinPract J Am Gastroenterol Assoc. 2022;20(6):e1240-e1249. doi:10.1016/j.cgh.2021.07.014
- Thorpe MG, Milte CM, Crawford D, McNaughton SA. Education and lifestyle predict change in dietary patterns and diet quality of adults 55 years and over. Nutr J. 2019;18(1):67. doi:10.1186/s12937-019-0495-6
- Chapman K, Ogden J. How do people change their diet?: an exploration into mechanisms of dietary change. J Health Psychol. 2009;14(8):1229-1242. doi:10.1177/1359105309342289
- Djoussé L, Petrone AB, Weir NL, et al. Repeated versus single measurement of plasma omega-3 fatty acids and risk of heart failure. Eur J Nutr. 2014;53(6):1403-1408. doi:10.1007/s00394-013-0642-3
- Bach-Faig A, Berry EM, Lairon D, et al. Mediterranean diet pyramid today. Science and cultural updates. Public Health Nutr. 2011;14(12A):2274-2284. doi:10.1017/S1368980011002515
- Miller PE, Cross AJ, Subar AF, et al. Comparison of 4 established DASH diet indexes: examining associations of index scores and colorectal cancer123. Am J Clin Nutr. 2013;98(3):794-803. doi:10.3945/ajcn.113.063602
- Krebs-Smith SM, Pannucci TE, Subar AF, et al. Update of the Healthy Eating Index: HEI-2015. J Acad Nutr Diet. 2018;118(9):1591-1602. doi:10.1016/j.jand.2018.05.021
- P.R. Pehrsson, Cutrufelli RL, Gebhardt SE, et al. USDA Database for the Added Sugars Content of Selected Foods. USDA; 2005. www.ars.usda.gov/nutrientdata
- Zauber AG, Winawer SJ, O’Brien MJ, et al. Colonoscopic polypectomy and long-term prevention of colorectalcancer deaths. N Engl J Med. 2012;366(8):687-696. doi:10.1056/NEJMoa1100370
- Nishihara R, Wu K, Lochhead P, et al. Long-term colorectal-cancer incidence and mortality after lower endoscopy. N Engl J Med. 2013;369(12):1095-1105. doi:10.1056/NEJMoa1301969
- Bretthauer M, Løberg M, Wieszczy P, et al. Effect of colonoscopy screening on risks of colorectal cancer and related death. N Engl J Med. 2022;387(17):1547-1556. doi:10.1056/NEJMoa2208375
- Cottet V, Bonithon-Kopp C, Kronborg O, et al. Dietary patterns and the risk of colorectal adenoma recurrence in a European intervention trial. Eur J Cancer Prev. 2005;14(1):21.
- Miller PE, Lesko SM, Muscat JE, Lazarus P, Hartman TJ. Dietary patterns and colorectal adenoma and cancer risk: a review of the epidemiological evidence. Nutr Cancer. 2010;62(4):413-424. doi:10.1080/01635580903407114
- Godos J, Bella F, Torrisi A, Sciacca S, Galvano F, Grosso G. Dietary patterns and risk of colorectal adenoma: a systematic review and meta-analysis of observational studies. J Hum Nutr Diet Off J Br Diet Assoc. 2016;29(6):757-767. doi:10.1111/jhn.12395
- Haggar FA, Boushey RP. Colorectal cancer epidemiology: incidence, mortality, survival, and risk factors. Clin Colon Rectal Surg. 2009;22(4):191-197. doi:10.1055/s-0029-1242458
- Dong D, Stewart H, Carlson AC. An Examination of Veterans’ Diet Quality. U.S. Department of Agriculture, Economic Research Service; 2019:32.
- El-Halabi MM, Rex DK, Saito A, Eckert GJ, Kahi CJ. Defining adenoma detection rate benchmarks in average-risk male veterans. Gastrointest Endosc. 2019;89(1):137-143. doi:10.1016/j.gie.2018.08.021
- Alberts DS, Hess LM, eds. Fundamentals of Cancer Prevention. Springer International Publishing; 2019. doi:10.1007/978-3-030-15935-1
- Dahm CC, Keogh RH, Spencer EA, et al. Dietary fiber and colorectal cancer risk: a nested case-control study using food diaries. J Natl Cancer Inst. 2010;102(9):614-626. doi:10.1093/jnci/djq092
- Aune D, Lau R, Chan DSM, et al. Dairy products and colorectal cancer risk: a systematic review and metaanalysis of cohort studies. Ann Oncol. 2012;23(1):37-45. doi:10.1093/annonc/mdr269
- Lee JE, Li H, Chan AT, et al. Circulating levels of vitamin D and colon and rectal cancer: the Physicians’ Health Study and a meta-analysis of prospective studies. Cancer Prev Res Phila Pa. 2011;4(5):735-743. doi:10.1158/1940-6207.CAPR-10-0289
- Carroll C, Cooper K, Papaioannou D, Hind D, Pilgrim H, Tappenden P. Supplemental calcium in the chemoprevention of colorectal cancer: a systematic review and meta-analysis. Clin Ther. 2010;32(5):789-803. doi:10.1016/j.clinthera.2010.04.024
- Park Y, Spiegelman D, Hunter DJ, et al. Intakes of vitamins A, C, and E and use of multiple vitamin supplements and risk of colon cancer: a pooled analysis of prospective cohort studies. Cancer Causes Control CCC. 2010;21(11):1745- 1757. doi:10.1007/s10552-010-9549-y
- Alexander DD, Weed DL, Miller PE, Mohamed MA. Red meat and colorectal cancer: a quantitative update on the state of the epidemiologic science. J Am Coll Nutr. 2015;34(6):521-543. doi:10.1080/07315724.2014.992553
- Park SY, Wilkens LR, Setiawan VW, Monroe KR, Haiman CA, Le Marchand L. Alcohol intake and colorectal cancer risk in the multiethnic cohort study. Am J Epidemiol. 2019;188(1):67-76. doi:10.1093/aje/kwy208
- Lieberman DA. Risk Factors for advanced colonic neoplasia and hyperplastic polyps in asymptomatic individuals. JAMA. 2003;290(22):2959. doi:10.1001/jama.290.22.2959
- Archambault AN, Jeon J, Lin Y, et al. Risk stratification for early-onset colorectal cancer using a combination of genetic and environmental risk scores: an international multi-center study. J Natl Cancer Inst. 2022;114(4):528-539. doi:10.1093/jnci/djac003
- Carr PR, Weigl K, Edelmann D, et al. Estimation of absolute risk of colorectal cancer based on healthy lifestyle, genetic risk, and colonoscopy status in a populationbased study. Gastroenterology. 2020;159(1):129-138.e9. doi:10.1053/j.gastro.2020.03.016
- Sullivan BA, Qin X, Miller C, et al. Screening colonoscopy findings are associated with noncolorectal cancer mortality. Clin Transl Gastroenterol. 2022;13(4):e00479. doi:10.14309/ctg.0000000000000479
- Erben V, Carr PR, Holleczek B, Stegmaier C, Hoffmeister M, Brenner H. Dietary patterns and risk of advanced colorectal neoplasms: A large population based screening study in Germany. Prev Med. 2018;111:101-109. doi:10.1016/j.ypmed.2018.02.025
- Donovan MG, Selmin OI, Doetschman TC, Romagnolo DF. Mediterranean diet: prevention of colorectal cancer. Front Nutr. 2017;4:59. doi:10.3389/fnut.2017.00059
- Mohseni R, Mohseni F, Alizadeh S, Abbasi S. The Association of Dietary Approaches to Stop Hypertension (DASH) diet with the risk of colorectal cancer: a meta-analysis of observational studies.Nutr Cancer. 2020;72(5):778-790. doi:10.1080/01635581.2019.1651880
- Lieberman DA, Weiss DG, Bond JH, Ahnen DJ, Garewal H, Chejfec G. Use of colonoscopy to screen asymptomatic adults for colorectal cancer. Veterans Affairs Cooperative Study Group 380. N Engl J Med. 2000;343(3):162-168. doi:10.1056/NEJM200007203430301
- Developing the Healthy Eating Index (HEI) | EGRP/ DCCPS/NCI/NIH. Accessed July 22, 2025. https://epi.grants.cancer.gov/hei/developing.html#2015c
- Reeve E, Piccici F, Feairheller DL. Validation of a Mediterranean diet scoring system for intervention based research. J Nutr Med Diet Care. 2021;7(1):053. doi:10.23937/2572-3278/1510053
- Günther AL, Liese AD, Bell RA, et al. ASSOCIATION BETWEEN THE DIETARY APPROACHES TO HYPERTENSION (DASH) DIET AND HYPERTENSION IN YOUTH WITH DIABETES. Hypertens Dallas Tex 1979. 2009;53(1):6-12. doi:10.1161/HYPERTENSIONAHA.108.116665
- Buckland G, Agudo A, Luján L, et al. Adherence to a Mediterranean diet and risk of gastric adenocarcinoma within the European Prospective Investigation into Cancer and Nutrition (EPIC) cohort study. Am J Clin Nutr. 2010;91(2):381- 390. doi:10.3945/ajcn.2009.28209
- Rimm EB, Giovannucci EL, Stampfer MJ, Colditz GA, Litin LB, Willett WC. Reproducibility and validity of an expanded self-administered semiquantitative food frequency questionnaire among male health professionals. Am J Epidemiol. 1992;135(10):1114-1126. doi:10.1093/oxfordjournals.aje.a116211
- Charlson ME, Pompei P, Ales KL, MacKenzie CR. A new method of classifying prognostic comorbidity in longitudinal studies: development and validation. J Chronic Dis. 1987;40(5):373-383. doi:10.1016/0021-9681(87)90171-8
- Lieberman DA, Weiss DG, Harford WV, et al. Fiveyear colon surveillance after screening colonoscopy. Gastroenterology. 2007;133(4):1077-1085. doi:10.1053/j.gastro.2007.07.006
- Lieberman D, Sullivan BA, Hauser ER, et al. Baseline colonoscopy findings associated with 10-year outcomes in a screening cohort undergoing colonoscopy surveillance. Gastroenterology. 2020;158(4):862-874.e8. doi:10.1053/j.gastro.2019.07.052
- PROC LOGISTIC: PROC LOGISTIC Statement : SAS/STAT(R) 9.22 User’s Guide. Accessed July 22, 2025. https://support.sas.com/documentation/cdl/en/statug/63347/HTML/default/viewer.htm#statug_logistic_sect004.htm
- PROC MULTTEST: PROC MULTTEST Statement : SAS/ STAT(R) 9.22 User’s Guide. Accessed July 22, 2025. https://support.sas.com/documentation/cdl/en/statug/63347/HTML/default/viewer.htm#statug_multtest_sect005.htm
- Elston DM. Participation bias, self-selection bias, and response bias. J Am Acad Dermatol. Published online June 18, 2021. doi:10.1016/j.jaad.2021.06.025
- Sansbury LB, Wanke K, Albert PS, et al. The effect of strict adherence to a high-fiber, high-fruit and -vegetable, and low-fat eating pattern on adenoma recurrence. Am J Epidemiol. 2009;170(5):576-584. doi:10.1093/aje/kwp169
- Borgas P, Gonzalez G, Veselkov K, Mirnezami R. Phytochemically rich dietary components and the risk of colorectal cancer: A systematic review and meta-analysis of observational studies. World J Clin Oncol. 2021;12(6):482- 499. doi:10.5306/wjco.v12.i6.482
- Papadimitriou N, Markozannes G, Kanellopoulou A, et al. An umbrella review of the evidence associating diet and cancer risk at 11 anatomical sites. Nat Commun. 2021;12(1):4579. doi:10.1038/s41467-021-24861-8
- Johnston BC, Zeraatkar D, Han MA, et al. Unprocessed red meat and processed meat consumption: dietary guideline recommendations from the nutritional recommendations (NutriRECS) Consortium. Ann Intern Med. 2019;171(10):756-764. doi:10.7326/M19-1621
- Kim M, Park K. Dietary fat intake and risk of colorectal cancer: a systematic review and meta-analysis of prospective studies. Nutrients. 2018;10(12):1963. doi:10.3390/nu10121963
- Lu Y, Li D, Wang L, et al. Comprehensive investigation on associations between dietary intake and blood levels of fatty acids and colorectal cancer risk. Nutrients. 2023;15(3):730. doi:10.3390/nu15030730
- Gherasim A, Arhire LI, Ni.a O, Popa AD, Graur M, Mihalache L. The relationship between lifestyle components and dietary patterns. Proc Nutr Soc. 2020;79(3):311-323. doi:10.1017/S0029665120006898
- Hullings AG, Sinha R, Liao LM, Freedman ND, Graubard BI, Loftfield E. Whole grain and dietary fiber intake and risk of colorectal cancer in the NIH-AARP Diet and Health Study cohort. Am J Clin Nutr. 2020;112(3):603- 612. doi:10.1093/ajcn/nqaa161
- Ocvirk S, Wilson AS, Appolonia CN, Thomas TK, O’Keefe SJD. Fiber, fat, and colorectal cancer: new insight into modifiable dietary risk factors. Curr Gastroenterol Rep. 2019;21(11):62. doi:10.1007/s11894-019-0725-2
- O’Keefe SJD. Diet, microorganisms and their metabolites, and colon cancer. Nat Rev Gastroenterol Hepatol. 2016;13(12):691-706. doi:10.1038/nrgastro.2016.165
- The health benefits and side effects of Butyrate Cleveland Clinic. July 11, 2022. Accessed July 22, 2025. https://health.clevelandclinic.org/butyrate-benefits/
- Knudsen MD, Wang L, Wang K, et al. Changes in lifestyle factors after endoscopic screening: a prospective study in the United States. Clin Gastroenterol Hepatol Off ClinPract J Am Gastroenterol Assoc. 2022;20(6):e1240-e1249. doi:10.1016/j.cgh.2021.07.014
- Thorpe MG, Milte CM, Crawford D, McNaughton SA. Education and lifestyle predict change in dietary patterns and diet quality of adults 55 years and over. Nutr J. 2019;18(1):67. doi:10.1186/s12937-019-0495-6
- Chapman K, Ogden J. How do people change their diet?: an exploration into mechanisms of dietary change. J Health Psychol. 2009;14(8):1229-1242. doi:10.1177/1359105309342289
- Djoussé L, Petrone AB, Weir NL, et al. Repeated versus single measurement of plasma omega-3 fatty acids and risk of heart failure. Eur J Nutr. 2014;53(6):1403-1408. doi:10.1007/s00394-013-0642-3
- Bach-Faig A, Berry EM, Lairon D, et al. Mediterranean diet pyramid today. Science and cultural updates. Public Health Nutr. 2011;14(12A):2274-2284. doi:10.1017/S1368980011002515
- Miller PE, Cross AJ, Subar AF, et al. Comparison of 4 established DASH diet indexes: examining associations of index scores and colorectal cancer123. Am J Clin Nutr. 2013;98(3):794-803. doi:10.3945/ajcn.113.063602
- Krebs-Smith SM, Pannucci TE, Subar AF, et al. Update of the Healthy Eating Index: HEI-2015. J Acad Nutr Diet. 2018;118(9):1591-1602. doi:10.1016/j.jand.2018.05.021
- P.R. Pehrsson, Cutrufelli RL, Gebhardt SE, et al. USDA Database for the Added Sugars Content of Selected Foods. USDA; 2005. www.ars.usda.gov/nutrientdata
Associations Between Prescreening Dietary Patterns and Longitudinal Colonoscopy Outcomes in Veterans
Associations Between Prescreening Dietary Patterns and Longitudinal Colonoscopy Outcomes in Veterans

These Two Simple Interventions May Cut Colorectal Cancer Recurrence Risk
This transcript has been edited for clarity.
New guidelines have lowered the age to begin screening for colon cancer to 45 years old. Although this change is positive, we’re still seeing advanced cancer in younger patients who haven’t been screened in time.
Once diagnosed, these patients undergo surgery and chemotherapy and often return to us asking, “What can I do now to help myself?”
Two recent studies highlight interventions that are simple, affordable, and actionable today: exercise and aspirin. Let’s take a closer look at the results.
Exercise’s Risk Reduction Potential
The idea that exercise reduces cancer recurrence and mortality is supported by observational data. The mechanistic effects behind this have been ascribed to metabolic growth factors, inflammatory changes, immune function changes, and perhaps even positive impact on sleep.
A study just published in The New England Journal of Medicine examined structured exercise after adjuvant chemotherapy for colon cancer. The phase 3 randomized CHALLENGE trial, mostly conducted at Canadian and Australian centers, recruited patients with resected stage II or III colon cancer (9.8% and 90.2%, respectively) who had completed adjuvant chemotherapy. Patients with recurrences within a year of diagnosis were excluded, as they were more likely to have highly aggressive, biologically active disease.
Patients were randomized to receive healthcare education materials alone or in conjunction with a structured exercise program over a 3-year follow-up period.
The exercise intervention, delivered in person or virtually, focused on increasing recreational aerobic activity over baseline by at least 10 metabolic equivalent task (MET). An increment of 10 MET hours per week is not too vigorous. It is essentially the equivalent of adding about 45-60 minutes of brisk walking or 25-30 minutes of jogging 3-4 times a week.
Patients were asked to increase MET over the first 6 months and then maintain or further increase the amount over the next 2.5 years. They were permitted to structure their own exercise program by choosing the type, frequency, intensity, and duration of aerobic exercise.
The primary endpoint was disease-free survival, with secondary endpoints assessing overall survival, patient-reported outcomes, and other outcomes. Although designed to detect differences at 3 years, follow-up was also performed out to 5 and 8 years.
At a median follow-up of 7.9 years, exercise reduced the relative risk of disease recurrence, new primary cancer, or death by 28% (P = .02). This benefit persisted — and even strengthened — over time, with disease-free survival increasing by 6.4 and 7.1 percentage points at 5 and 8 years, respectively.
Musculoskeletal adverse events were slightly higher in the exercise group compared with the health education group (18.5% vs 11.5%, respectively), but only 10% were directly attributed to the exercise.
There are considerations when interpreting these results. First, there was an attrition over time for compliance and training. It would be interesting to see whether that impacted the results. Second, it’s unclear whether patient pedigree or a genomic pathway may predispose to a benefit here for the exercise group.
But overall, this phase 3 trial provides class 1 evidence supporting exercise as a low-cost, high-impact intervention to reduce cancer recurrence.
Adjuvant Aspirin in Colon Cancer Subset
That’s a perfect segue into another recent study looking at the effects of adjuvant aspirin on the prevention of recurrence.
The ALASCCA trial— conducted across centers in Sweden, Denmark, Finland, and Norway — assessed patients with stage I-III rectal cancer or stage II-III colon cancer. It focused on a subset of patients with an oncogenic abnormality called PIK3CA (phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha).
PIK3CA occurs in approximately a third of colon cancers and is associated with significant chemotherapy resistance and a higher rate of disease progression.
Of the included patients, 1103 (37%) had alterations in the PIK3CA pathway. Researchers randomized patients to receive either 160 mg of aspirin or placebo daily for 3 years, starting within 3 months of surgery.
Among patients with PIK3CA mutations, aspirin dramatically reduced the risk for time to recurrence by nearly 50% at 3 years (P = .044). Adverse events associated with aspirin were minimal, including one case each of gastrointestinal bleeding, hematoma, and allergic reaction.
There is no evidence that higher aspirin doses provide greater prevention of colorectal cancer recurrence. The 160-mg use in the current study is fairly normal, roughly equivalent to two low-dose (81-mg) aspirin tablets.
Now, it’s worth noting that the use of aspirin for the primary prevention of cardiovascular disease was initially recommended by the US Preventive Services Task Force in 2016. This recommendation was then recanted in 2022, when the same group reported limited net benefit to this approach.
Two Proactive Actions
These studies highlight 2 interventions — exercise and aspirin — that are low cost, accessible, and appeal to patients eager to help prevent their cancer from recurring.
Exercise is broadly beneficial and can be recommended immediately.
For aspirin, patients should work with their oncologist to determine their PIK3CA mutation status, as this subgroup appears to benefit the most.
These findings offer patients meaningful, proactive interventions they can apply to support their recovery and reduce the risk of recurrence. Hopefully these new findings will help guide your clinical conversations.
Johnson is a regular contributor to Medscape. He is professor of medicine and chief of gastroenterology at Eastern Virginia Medical School in Norfolk, and a past president of the American College of Gastroenterology. His primary focus is the clinical practice of gastroenterology. He has published extensively in the internal medicine/gastroenterology literature, with principal research interests in esophageal and colon disease, and more recently in sleep and microbiome effects on gastrointestinal health and disease. He disclosed that he is an adviser for ISOThrive.
A version of this article appeared on Medscape.com.
This transcript has been edited for clarity.
New guidelines have lowered the age to begin screening for colon cancer to 45 years old. Although this change is positive, we’re still seeing advanced cancer in younger patients who haven’t been screened in time.
Once diagnosed, these patients undergo surgery and chemotherapy and often return to us asking, “What can I do now to help myself?”
Two recent studies highlight interventions that are simple, affordable, and actionable today: exercise and aspirin. Let’s take a closer look at the results.
Exercise’s Risk Reduction Potential
The idea that exercise reduces cancer recurrence and mortality is supported by observational data. The mechanistic effects behind this have been ascribed to metabolic growth factors, inflammatory changes, immune function changes, and perhaps even positive impact on sleep.
A study just published in The New England Journal of Medicine examined structured exercise after adjuvant chemotherapy for colon cancer. The phase 3 randomized CHALLENGE trial, mostly conducted at Canadian and Australian centers, recruited patients with resected stage II or III colon cancer (9.8% and 90.2%, respectively) who had completed adjuvant chemotherapy. Patients with recurrences within a year of diagnosis were excluded, as they were more likely to have highly aggressive, biologically active disease.
Patients were randomized to receive healthcare education materials alone or in conjunction with a structured exercise program over a 3-year follow-up period.
The exercise intervention, delivered in person or virtually, focused on increasing recreational aerobic activity over baseline by at least 10 metabolic equivalent task (MET). An increment of 10 MET hours per week is not too vigorous. It is essentially the equivalent of adding about 45-60 minutes of brisk walking or 25-30 minutes of jogging 3-4 times a week.
Patients were asked to increase MET over the first 6 months and then maintain or further increase the amount over the next 2.5 years. They were permitted to structure their own exercise program by choosing the type, frequency, intensity, and duration of aerobic exercise.
The primary endpoint was disease-free survival, with secondary endpoints assessing overall survival, patient-reported outcomes, and other outcomes. Although designed to detect differences at 3 years, follow-up was also performed out to 5 and 8 years.
At a median follow-up of 7.9 years, exercise reduced the relative risk of disease recurrence, new primary cancer, or death by 28% (P = .02). This benefit persisted — and even strengthened — over time, with disease-free survival increasing by 6.4 and 7.1 percentage points at 5 and 8 years, respectively.
Musculoskeletal adverse events were slightly higher in the exercise group compared with the health education group (18.5% vs 11.5%, respectively), but only 10% were directly attributed to the exercise.
There are considerations when interpreting these results. First, there was an attrition over time for compliance and training. It would be interesting to see whether that impacted the results. Second, it’s unclear whether patient pedigree or a genomic pathway may predispose to a benefit here for the exercise group.
But overall, this phase 3 trial provides class 1 evidence supporting exercise as a low-cost, high-impact intervention to reduce cancer recurrence.
Adjuvant Aspirin in Colon Cancer Subset
That’s a perfect segue into another recent study looking at the effects of adjuvant aspirin on the prevention of recurrence.
The ALASCCA trial— conducted across centers in Sweden, Denmark, Finland, and Norway — assessed patients with stage I-III rectal cancer or stage II-III colon cancer. It focused on a subset of patients with an oncogenic abnormality called PIK3CA (phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha).
PIK3CA occurs in approximately a third of colon cancers and is associated with significant chemotherapy resistance and a higher rate of disease progression.
Of the included patients, 1103 (37%) had alterations in the PIK3CA pathway. Researchers randomized patients to receive either 160 mg of aspirin or placebo daily for 3 years, starting within 3 months of surgery.
Among patients with PIK3CA mutations, aspirin dramatically reduced the risk for time to recurrence by nearly 50% at 3 years (P = .044). Adverse events associated with aspirin were minimal, including one case each of gastrointestinal bleeding, hematoma, and allergic reaction.
There is no evidence that higher aspirin doses provide greater prevention of colorectal cancer recurrence. The 160-mg use in the current study is fairly normal, roughly equivalent to two low-dose (81-mg) aspirin tablets.
Now, it’s worth noting that the use of aspirin for the primary prevention of cardiovascular disease was initially recommended by the US Preventive Services Task Force in 2016. This recommendation was then recanted in 2022, when the same group reported limited net benefit to this approach.
Two Proactive Actions
These studies highlight 2 interventions — exercise and aspirin — that are low cost, accessible, and appeal to patients eager to help prevent their cancer from recurring.
Exercise is broadly beneficial and can be recommended immediately.
For aspirin, patients should work with their oncologist to determine their PIK3CA mutation status, as this subgroup appears to benefit the most.
These findings offer patients meaningful, proactive interventions they can apply to support their recovery and reduce the risk of recurrence. Hopefully these new findings will help guide your clinical conversations.
Johnson is a regular contributor to Medscape. He is professor of medicine and chief of gastroenterology at Eastern Virginia Medical School in Norfolk, and a past president of the American College of Gastroenterology. His primary focus is the clinical practice of gastroenterology. He has published extensively in the internal medicine/gastroenterology literature, with principal research interests in esophageal and colon disease, and more recently in sleep and microbiome effects on gastrointestinal health and disease. He disclosed that he is an adviser for ISOThrive.
A version of this article appeared on Medscape.com.
This transcript has been edited for clarity.
New guidelines have lowered the age to begin screening for colon cancer to 45 years old. Although this change is positive, we’re still seeing advanced cancer in younger patients who haven’t been screened in time.
Once diagnosed, these patients undergo surgery and chemotherapy and often return to us asking, “What can I do now to help myself?”
Two recent studies highlight interventions that are simple, affordable, and actionable today: exercise and aspirin. Let’s take a closer look at the results.
Exercise’s Risk Reduction Potential
The idea that exercise reduces cancer recurrence and mortality is supported by observational data. The mechanistic effects behind this have been ascribed to metabolic growth factors, inflammatory changes, immune function changes, and perhaps even positive impact on sleep.
A study just published in The New England Journal of Medicine examined structured exercise after adjuvant chemotherapy for colon cancer. The phase 3 randomized CHALLENGE trial, mostly conducted at Canadian and Australian centers, recruited patients with resected stage II or III colon cancer (9.8% and 90.2%, respectively) who had completed adjuvant chemotherapy. Patients with recurrences within a year of diagnosis were excluded, as they were more likely to have highly aggressive, biologically active disease.
Patients were randomized to receive healthcare education materials alone or in conjunction with a structured exercise program over a 3-year follow-up period.
The exercise intervention, delivered in person or virtually, focused on increasing recreational aerobic activity over baseline by at least 10 metabolic equivalent task (MET). An increment of 10 MET hours per week is not too vigorous. It is essentially the equivalent of adding about 45-60 minutes of brisk walking or 25-30 minutes of jogging 3-4 times a week.
Patients were asked to increase MET over the first 6 months and then maintain or further increase the amount over the next 2.5 years. They were permitted to structure their own exercise program by choosing the type, frequency, intensity, and duration of aerobic exercise.
The primary endpoint was disease-free survival, with secondary endpoints assessing overall survival, patient-reported outcomes, and other outcomes. Although designed to detect differences at 3 years, follow-up was also performed out to 5 and 8 years.
At a median follow-up of 7.9 years, exercise reduced the relative risk of disease recurrence, new primary cancer, or death by 28% (P = .02). This benefit persisted — and even strengthened — over time, with disease-free survival increasing by 6.4 and 7.1 percentage points at 5 and 8 years, respectively.
Musculoskeletal adverse events were slightly higher in the exercise group compared with the health education group (18.5% vs 11.5%, respectively), but only 10% were directly attributed to the exercise.
There are considerations when interpreting these results. First, there was an attrition over time for compliance and training. It would be interesting to see whether that impacted the results. Second, it’s unclear whether patient pedigree or a genomic pathway may predispose to a benefit here for the exercise group.
But overall, this phase 3 trial provides class 1 evidence supporting exercise as a low-cost, high-impact intervention to reduce cancer recurrence.
Adjuvant Aspirin in Colon Cancer Subset
That’s a perfect segue into another recent study looking at the effects of adjuvant aspirin on the prevention of recurrence.
The ALASCCA trial— conducted across centers in Sweden, Denmark, Finland, and Norway — assessed patients with stage I-III rectal cancer or stage II-III colon cancer. It focused on a subset of patients with an oncogenic abnormality called PIK3CA (phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha).
PIK3CA occurs in approximately a third of colon cancers and is associated with significant chemotherapy resistance and a higher rate of disease progression.
Of the included patients, 1103 (37%) had alterations in the PIK3CA pathway. Researchers randomized patients to receive either 160 mg of aspirin or placebo daily for 3 years, starting within 3 months of surgery.
Among patients with PIK3CA mutations, aspirin dramatically reduced the risk for time to recurrence by nearly 50% at 3 years (P = .044). Adverse events associated with aspirin were minimal, including one case each of gastrointestinal bleeding, hematoma, and allergic reaction.
There is no evidence that higher aspirin doses provide greater prevention of colorectal cancer recurrence. The 160-mg use in the current study is fairly normal, roughly equivalent to two low-dose (81-mg) aspirin tablets.
Now, it’s worth noting that the use of aspirin for the primary prevention of cardiovascular disease was initially recommended by the US Preventive Services Task Force in 2016. This recommendation was then recanted in 2022, when the same group reported limited net benefit to this approach.
Two Proactive Actions
These studies highlight 2 interventions — exercise and aspirin — that are low cost, accessible, and appeal to patients eager to help prevent their cancer from recurring.
Exercise is broadly beneficial and can be recommended immediately.
For aspirin, patients should work with their oncologist to determine their PIK3CA mutation status, as this subgroup appears to benefit the most.
These findings offer patients meaningful, proactive interventions they can apply to support their recovery and reduce the risk of recurrence. Hopefully these new findings will help guide your clinical conversations.
Johnson is a regular contributor to Medscape. He is professor of medicine and chief of gastroenterology at Eastern Virginia Medical School in Norfolk, and a past president of the American College of Gastroenterology. His primary focus is the clinical practice of gastroenterology. He has published extensively in the internal medicine/gastroenterology literature, with principal research interests in esophageal and colon disease, and more recently in sleep and microbiome effects on gastrointestinal health and disease. He disclosed that he is an adviser for ISOThrive.
A version of this article appeared on Medscape.com.
Can Lifestyle Changes Save Lives in Colon Cancer?
Can Lifestyle Changes Save Lives in Colon Cancer?
Can exercise “therapy” and diet improve survival in patients with colon cancer? It appears so, according to two pivotal studies presented at American Society of Clinical Oncology (ASCO) 2025 annual meeting.
In the CHALLENGE trial, a structured exercise program after surgery and adjuvant chemotherapy cut the risk for colon cancer recurrence in patients with stage III and high-risk stage II disease by more than one quarter and the risk for death by more than one third.
“The magnitude of benefit with exercise is substantial. In fact, it is comparable, and in some cases exceeds the magnitude of benefit of many of our very good standard medical therapies in oncology,” study presenter Christopher Booth, MD, with Queen’s University, Kingston, Ontario, Canada, told attendees.
Results of the study were published online in The New England Journal of Medicine to coincide with the presentation at the meeting.
The findings are “nothing short of a major milestone,” said study discussant Peter Campbell, PhD, with Montefiore Einstein Comprehensive Cancer Center, Bronx, New York.
The other study showed that eating a less inflammatory diet may reduce the risk for death in patients with colon cancer, with the greatest benefits seen in those who embraced anti-inflammatory foods and exercised regularly.
“Putting these two abstracts into perspective, we as physicians need to be essentially prescribing healthy diet and exercise. The combination of the two are synergistic,” Julie Gralow, MD, ASCO chief medical officer and executive vice president, told attendees.
Despite the benefits of these lifestyle changes, exercise and diet are meant to supplement, not replace, established colon cancer treatments.
It would be a false binary to frame this as lifestyle vs cancer treatment, explained Mark Lewis, MD, director of Gastrointestinal Oncology at Intermountain Healthcare in Salt Lake City, Utah. With exercise, for instance, “the key is giving enough chemo to protect against recurrence and eliminate micrometastases but not so much that we cause neuropathy and reduce function and ability to follow the CHALLENGE structured program,” Lewis said.
Exercise and Survival
Colon cancer remains the second-leading cause of cancer death worldwide. Even with surgery and chemotherapy, roughly 30% of patients with stage III and high-risk stage II colon cancer will experience disease recurrence.
“As oncologists, one of the most common questions we get asked by patients is — what else can I do to improve my outcome?” Booth said.
Observational studies published nearly two decades ago hinted that physically active cancer survivors fare better, but no randomized trial has definitively tested whether exercise could alter disease course. That knowledge gap prompted the Canadian Cancer Trials Group to launch the CHALLENGE trial.
Between 2009 and 2023, the phase 3 study enrolled 889 adults (median age, 61 years; 51% women) who had completed surgery and adjuvant chemotherapy for stage III (90%) or high-risk stage II (10%) colon cancer. Most patients were from Canada and Australia and were enrolled 2-6 months after completing chemotherapy.
Half of study participants were randomly allocated to a structured exercise program (n = 445) and half to receive standard health education materials promoting physical activity and healthy eating (control individuals, n = 444).
As part of the structured exercise intervention, patients met with a physical activity consultant twice a month for the first 6 months. These sessions included exercise coaching and supervised exercise. Patients could choose their preferred aerobic exercise and most picked brisk walking.
The consultants gave each patient an “exercise prescription” to hit a specific amount of exercise. The target was an additional 10 metabolic equivalent (MET)–hours of aerobic activity per week — about three to four brisk walks each lasting 45-60 minutes. After 6 months, patients met with their consultants once a month, with additional sessions available for extra support if needed.
Structured exercise led to “substantial and sustained” increases in the amount of exercise participants did, as well as physiologic measures of their fitness, with “highly relevant” improvements in VO2 max, 6-minute walk test, and patient-reported physical function, underscoring that participants were not only exercising more but also getting fitter, Booth said.
Exercise was associated with a clinically meaningful and statistically significant 28% reduction in the risk for recurrent or new cancer (hazard ratio [HR], 0.72; P = .017), with a 5-year disease free survival rate of 80% in the exercise group and 74% in the control group.
In other words, “for every 16 patients that went on the exercise program, exercise prevented 1 person from recurrent or new cancer” at 5 years, Booth reported.
Overall survival results were “even more impressive,” he said.
At 8 years, 90% of patients in the exercise program were alive vs 83% of those in the control group, which translated to a 37% lower risk for death (HR, 0.63; P = .022).
“For every 14 patients who went on the exercise program, exercise prevented 1 person from dying” at the 8-year mark, Booth noted.
“Notably, this difference in survival was not driven by difference in cardiovascular deaths but by a reduction in the risk of death from colon cancer,” he said.
Besides a slight uptick in musculoskeletal aches, no major safety signals emerged in the exercise group.
It’s important to note that the survival benefit associated with exercise came after patients had received surgery followed by chemotherapy — in other words, exercise did not replace established cancer treatments. It’s also unclear whether initiating an exercise intervention earlier in the treatment trajectory — before surgery or during chemotherapy, instead of after chemotherapy — could further improve cancer outcomes, the authors noted.
Still, “exercise as an intervention is a no brainer and should be implemented broadly,” said ASCO expert Pamela Kunz, MD, with Yale School of Medicine, New Haven, Connecticut.
Marco Gerlinger, MD, with Barts Cancer Institute, London, England, agreed.
“Oncologists can now make a very clear evidence-based recommendation for patients who just completed their chemotherapy for bowel cancer and are fit enough for such an exercise program,” Gerlinger said in a statement from the nonprofit UK Science Media Centre.
Booth noted that knowledge alone will not be sufficient to allow most patients to change their lifestyle and realize the health benefits.
“The policy implementation piece of this is really key, and we need health systems, hospitals, and payers to invest in these behavior support programs so that patients have access to a physical activity consultant and can realize the health benefits,” he said.
“This intervention is empowering and achievable for patients and with much, much lower cost than many of our therapies. It is also sustainable for health systems,” he concluded.
Diet and Survival
Diet can also affect outcomes in patients with colon cancer.
In the same session describing the CHALLENGE results, Sara Char, MD, with Dana-Farber Cancer Institute in Boston, reported findings showing that consuming a diet high in proinflammatory foods was associated with worse overall survival in patients with stage III colon cancer. A proinflammatory diet includes red and processed meats, sugary drinks, and refined grains, while an anti-inflammatory diet focuses on fruits, vegetables, whole grains, fish, and olive oil.
Chronic systemic inflammation has been implicated in both colon cancer development and in its progression, and elevated levels of inflammatory markers in the blood have previously been associated with worse survival outcomes in patients with stage III colon cancer.
Char and colleagues analyzed dietary patterns of a subset of 1625 patients (mean age, 61 years) with resected stage III colon cancer enrolled in the phase 3 CALGB/SWOG 80702 (Alliance) clinical trial, which compared 3 months of adjuvant chemotherapy with 6 months of adjuvant chemotherapy, with or without the anti-inflammatory medication celecoxib.
As part of the trial, participants reported their diet and exercise habits at various timepoints. Their diets were scored using the validated empirical dietary inflammatory pattern (EDIP) tool, which is a weighted sum of 18 food groups — nine proinflammatory and nine anti-inflammatory. A high EDIP score marks a proinflammatory diet, and a low EDIP score indicates a less inflammatory diet.
During median follow-up of nearly 4 years, researchers noted a trend toward worse disease-free survival in patients with high proinflammatory diets (HR, 1.46), but this association was not significant in the multivariable adjusted model (HR, 1.36; P = .22), Char reported.
However, higher intake of proinflammatory foods was associated with significantly worse overall survival.
Patients who consumed the most proinflammatory foods (top 20%) had an 87% higher risk for death compared with those who consumed the least (bottom 20%; HR, 1.87). The median overall survival in the highest quintile was 7.7 years and was not reached in the lowest quintile.
Combine Exercise and Diet for Best Results
To examine the joint effect of physical activity and diet on overall survival, patients were divided into higher and lower levels of physical activity using a cut-off of 9 MET hours per week, which roughly correlates to 30 minutes of vigorous walking five days a week with a little bit of light yoga, Char explained.
In this analysis, patients with less proinflammatory diets and higher physical activity levels had the best overall survival outcomes, with a 63% lower risk for death compared with peers who consumed more pro-inflammatory diets and exercised less (HR, 0.37; P < .0001).
Daily celecoxib use and low-dose aspirin use (< 100 mg/d) did not affect the association between inflammatory diet and survival.
Char cautioned, that while the EDIP tool is useful to measure the inflammatory potential of a diet, “this is not a dietary recommendation, and we need further studies to be able to tailor our findings into dietary recommendations that can be provided to patients at the bedside.”
Gralow said this “early but promising observational study suggests a powerful synergy: Patients with stage III colon cancer who embraced anti-inflammatory foods and exercised regularly showed the best overall survival compared to those with inflammatory diets and limited exercise.”
The CHALLENGE trial was funded by the Canadian Cancer Society, the National Health and Medical Research Council, Cancer Research UK, and the University of Sydney Cancer Research Fund. Booth had no disclosures. The diet study was funded by the National Institutes of Health, Pfizer, and the Project P Fund. Char disclosed an advisory/consultant role with Goodpath. Kunz, Gralow and Campbell had no relevant disclosures.
A version of this article first appeared on Medscape.com.
Can exercise “therapy” and diet improve survival in patients with colon cancer? It appears so, according to two pivotal studies presented at American Society of Clinical Oncology (ASCO) 2025 annual meeting.
In the CHALLENGE trial, a structured exercise program after surgery and adjuvant chemotherapy cut the risk for colon cancer recurrence in patients with stage III and high-risk stage II disease by more than one quarter and the risk for death by more than one third.
“The magnitude of benefit with exercise is substantial. In fact, it is comparable, and in some cases exceeds the magnitude of benefit of many of our very good standard medical therapies in oncology,” study presenter Christopher Booth, MD, with Queen’s University, Kingston, Ontario, Canada, told attendees.
Results of the study were published online in The New England Journal of Medicine to coincide with the presentation at the meeting.
The findings are “nothing short of a major milestone,” said study discussant Peter Campbell, PhD, with Montefiore Einstein Comprehensive Cancer Center, Bronx, New York.
The other study showed that eating a less inflammatory diet may reduce the risk for death in patients with colon cancer, with the greatest benefits seen in those who embraced anti-inflammatory foods and exercised regularly.
“Putting these two abstracts into perspective, we as physicians need to be essentially prescribing healthy diet and exercise. The combination of the two are synergistic,” Julie Gralow, MD, ASCO chief medical officer and executive vice president, told attendees.
Despite the benefits of these lifestyle changes, exercise and diet are meant to supplement, not replace, established colon cancer treatments.
It would be a false binary to frame this as lifestyle vs cancer treatment, explained Mark Lewis, MD, director of Gastrointestinal Oncology at Intermountain Healthcare in Salt Lake City, Utah. With exercise, for instance, “the key is giving enough chemo to protect against recurrence and eliminate micrometastases but not so much that we cause neuropathy and reduce function and ability to follow the CHALLENGE structured program,” Lewis said.
Exercise and Survival
Colon cancer remains the second-leading cause of cancer death worldwide. Even with surgery and chemotherapy, roughly 30% of patients with stage III and high-risk stage II colon cancer will experience disease recurrence.
“As oncologists, one of the most common questions we get asked by patients is — what else can I do to improve my outcome?” Booth said.
Observational studies published nearly two decades ago hinted that physically active cancer survivors fare better, but no randomized trial has definitively tested whether exercise could alter disease course. That knowledge gap prompted the Canadian Cancer Trials Group to launch the CHALLENGE trial.
Between 2009 and 2023, the phase 3 study enrolled 889 adults (median age, 61 years; 51% women) who had completed surgery and adjuvant chemotherapy for stage III (90%) or high-risk stage II (10%) colon cancer. Most patients were from Canada and Australia and were enrolled 2-6 months after completing chemotherapy.
Half of study participants were randomly allocated to a structured exercise program (n = 445) and half to receive standard health education materials promoting physical activity and healthy eating (control individuals, n = 444).
As part of the structured exercise intervention, patients met with a physical activity consultant twice a month for the first 6 months. These sessions included exercise coaching and supervised exercise. Patients could choose their preferred aerobic exercise and most picked brisk walking.
The consultants gave each patient an “exercise prescription” to hit a specific amount of exercise. The target was an additional 10 metabolic equivalent (MET)–hours of aerobic activity per week — about three to four brisk walks each lasting 45-60 minutes. After 6 months, patients met with their consultants once a month, with additional sessions available for extra support if needed.
Structured exercise led to “substantial and sustained” increases in the amount of exercise participants did, as well as physiologic measures of their fitness, with “highly relevant” improvements in VO2 max, 6-minute walk test, and patient-reported physical function, underscoring that participants were not only exercising more but also getting fitter, Booth said.
Exercise was associated with a clinically meaningful and statistically significant 28% reduction in the risk for recurrent or new cancer (hazard ratio [HR], 0.72; P = .017), with a 5-year disease free survival rate of 80% in the exercise group and 74% in the control group.
In other words, “for every 16 patients that went on the exercise program, exercise prevented 1 person from recurrent or new cancer” at 5 years, Booth reported.
Overall survival results were “even more impressive,” he said.
At 8 years, 90% of patients in the exercise program were alive vs 83% of those in the control group, which translated to a 37% lower risk for death (HR, 0.63; P = .022).
“For every 14 patients who went on the exercise program, exercise prevented 1 person from dying” at the 8-year mark, Booth noted.
“Notably, this difference in survival was not driven by difference in cardiovascular deaths but by a reduction in the risk of death from colon cancer,” he said.
Besides a slight uptick in musculoskeletal aches, no major safety signals emerged in the exercise group.
It’s important to note that the survival benefit associated with exercise came after patients had received surgery followed by chemotherapy — in other words, exercise did not replace established cancer treatments. It’s also unclear whether initiating an exercise intervention earlier in the treatment trajectory — before surgery or during chemotherapy, instead of after chemotherapy — could further improve cancer outcomes, the authors noted.
Still, “exercise as an intervention is a no brainer and should be implemented broadly,” said ASCO expert Pamela Kunz, MD, with Yale School of Medicine, New Haven, Connecticut.
Marco Gerlinger, MD, with Barts Cancer Institute, London, England, agreed.
“Oncologists can now make a very clear evidence-based recommendation for patients who just completed their chemotherapy for bowel cancer and are fit enough for such an exercise program,” Gerlinger said in a statement from the nonprofit UK Science Media Centre.
Booth noted that knowledge alone will not be sufficient to allow most patients to change their lifestyle and realize the health benefits.
“The policy implementation piece of this is really key, and we need health systems, hospitals, and payers to invest in these behavior support programs so that patients have access to a physical activity consultant and can realize the health benefits,” he said.
“This intervention is empowering and achievable for patients and with much, much lower cost than many of our therapies. It is also sustainable for health systems,” he concluded.
Diet and Survival
Diet can also affect outcomes in patients with colon cancer.
In the same session describing the CHALLENGE results, Sara Char, MD, with Dana-Farber Cancer Institute in Boston, reported findings showing that consuming a diet high in proinflammatory foods was associated with worse overall survival in patients with stage III colon cancer. A proinflammatory diet includes red and processed meats, sugary drinks, and refined grains, while an anti-inflammatory diet focuses on fruits, vegetables, whole grains, fish, and olive oil.
Chronic systemic inflammation has been implicated in both colon cancer development and in its progression, and elevated levels of inflammatory markers in the blood have previously been associated with worse survival outcomes in patients with stage III colon cancer.
Char and colleagues analyzed dietary patterns of a subset of 1625 patients (mean age, 61 years) with resected stage III colon cancer enrolled in the phase 3 CALGB/SWOG 80702 (Alliance) clinical trial, which compared 3 months of adjuvant chemotherapy with 6 months of adjuvant chemotherapy, with or without the anti-inflammatory medication celecoxib.
As part of the trial, participants reported their diet and exercise habits at various timepoints. Their diets were scored using the validated empirical dietary inflammatory pattern (EDIP) tool, which is a weighted sum of 18 food groups — nine proinflammatory and nine anti-inflammatory. A high EDIP score marks a proinflammatory diet, and a low EDIP score indicates a less inflammatory diet.
During median follow-up of nearly 4 years, researchers noted a trend toward worse disease-free survival in patients with high proinflammatory diets (HR, 1.46), but this association was not significant in the multivariable adjusted model (HR, 1.36; P = .22), Char reported.
However, higher intake of proinflammatory foods was associated with significantly worse overall survival.
Patients who consumed the most proinflammatory foods (top 20%) had an 87% higher risk for death compared with those who consumed the least (bottom 20%; HR, 1.87). The median overall survival in the highest quintile was 7.7 years and was not reached in the lowest quintile.
Combine Exercise and Diet for Best Results
To examine the joint effect of physical activity and diet on overall survival, patients were divided into higher and lower levels of physical activity using a cut-off of 9 MET hours per week, which roughly correlates to 30 minutes of vigorous walking five days a week with a little bit of light yoga, Char explained.
In this analysis, patients with less proinflammatory diets and higher physical activity levels had the best overall survival outcomes, with a 63% lower risk for death compared with peers who consumed more pro-inflammatory diets and exercised less (HR, 0.37; P < .0001).
Daily celecoxib use and low-dose aspirin use (< 100 mg/d) did not affect the association between inflammatory diet and survival.
Char cautioned, that while the EDIP tool is useful to measure the inflammatory potential of a diet, “this is not a dietary recommendation, and we need further studies to be able to tailor our findings into dietary recommendations that can be provided to patients at the bedside.”
Gralow said this “early but promising observational study suggests a powerful synergy: Patients with stage III colon cancer who embraced anti-inflammatory foods and exercised regularly showed the best overall survival compared to those with inflammatory diets and limited exercise.”
The CHALLENGE trial was funded by the Canadian Cancer Society, the National Health and Medical Research Council, Cancer Research UK, and the University of Sydney Cancer Research Fund. Booth had no disclosures. The diet study was funded by the National Institutes of Health, Pfizer, and the Project P Fund. Char disclosed an advisory/consultant role with Goodpath. Kunz, Gralow and Campbell had no relevant disclosures.
A version of this article first appeared on Medscape.com.
Can exercise “therapy” and diet improve survival in patients with colon cancer? It appears so, according to two pivotal studies presented at American Society of Clinical Oncology (ASCO) 2025 annual meeting.
In the CHALLENGE trial, a structured exercise program after surgery and adjuvant chemotherapy cut the risk for colon cancer recurrence in patients with stage III and high-risk stage II disease by more than one quarter and the risk for death by more than one third.
“The magnitude of benefit with exercise is substantial. In fact, it is comparable, and in some cases exceeds the magnitude of benefit of many of our very good standard medical therapies in oncology,” study presenter Christopher Booth, MD, with Queen’s University, Kingston, Ontario, Canada, told attendees.
Results of the study were published online in The New England Journal of Medicine to coincide with the presentation at the meeting.
The findings are “nothing short of a major milestone,” said study discussant Peter Campbell, PhD, with Montefiore Einstein Comprehensive Cancer Center, Bronx, New York.
The other study showed that eating a less inflammatory diet may reduce the risk for death in patients with colon cancer, with the greatest benefits seen in those who embraced anti-inflammatory foods and exercised regularly.
“Putting these two abstracts into perspective, we as physicians need to be essentially prescribing healthy diet and exercise. The combination of the two are synergistic,” Julie Gralow, MD, ASCO chief medical officer and executive vice president, told attendees.
Despite the benefits of these lifestyle changes, exercise and diet are meant to supplement, not replace, established colon cancer treatments.
It would be a false binary to frame this as lifestyle vs cancer treatment, explained Mark Lewis, MD, director of Gastrointestinal Oncology at Intermountain Healthcare in Salt Lake City, Utah. With exercise, for instance, “the key is giving enough chemo to protect against recurrence and eliminate micrometastases but not so much that we cause neuropathy and reduce function and ability to follow the CHALLENGE structured program,” Lewis said.
Exercise and Survival
Colon cancer remains the second-leading cause of cancer death worldwide. Even with surgery and chemotherapy, roughly 30% of patients with stage III and high-risk stage II colon cancer will experience disease recurrence.
“As oncologists, one of the most common questions we get asked by patients is — what else can I do to improve my outcome?” Booth said.
Observational studies published nearly two decades ago hinted that physically active cancer survivors fare better, but no randomized trial has definitively tested whether exercise could alter disease course. That knowledge gap prompted the Canadian Cancer Trials Group to launch the CHALLENGE trial.
Between 2009 and 2023, the phase 3 study enrolled 889 adults (median age, 61 years; 51% women) who had completed surgery and adjuvant chemotherapy for stage III (90%) or high-risk stage II (10%) colon cancer. Most patients were from Canada and Australia and were enrolled 2-6 months after completing chemotherapy.
Half of study participants were randomly allocated to a structured exercise program (n = 445) and half to receive standard health education materials promoting physical activity and healthy eating (control individuals, n = 444).
As part of the structured exercise intervention, patients met with a physical activity consultant twice a month for the first 6 months. These sessions included exercise coaching and supervised exercise. Patients could choose their preferred aerobic exercise and most picked brisk walking.
The consultants gave each patient an “exercise prescription” to hit a specific amount of exercise. The target was an additional 10 metabolic equivalent (MET)–hours of aerobic activity per week — about three to four brisk walks each lasting 45-60 minutes. After 6 months, patients met with their consultants once a month, with additional sessions available for extra support if needed.
Structured exercise led to “substantial and sustained” increases in the amount of exercise participants did, as well as physiologic measures of their fitness, with “highly relevant” improvements in VO2 max, 6-minute walk test, and patient-reported physical function, underscoring that participants were not only exercising more but also getting fitter, Booth said.
Exercise was associated with a clinically meaningful and statistically significant 28% reduction in the risk for recurrent or new cancer (hazard ratio [HR], 0.72; P = .017), with a 5-year disease free survival rate of 80% in the exercise group and 74% in the control group.
In other words, “for every 16 patients that went on the exercise program, exercise prevented 1 person from recurrent or new cancer” at 5 years, Booth reported.
Overall survival results were “even more impressive,” he said.
At 8 years, 90% of patients in the exercise program were alive vs 83% of those in the control group, which translated to a 37% lower risk for death (HR, 0.63; P = .022).
“For every 14 patients who went on the exercise program, exercise prevented 1 person from dying” at the 8-year mark, Booth noted.
“Notably, this difference in survival was not driven by difference in cardiovascular deaths but by a reduction in the risk of death from colon cancer,” he said.
Besides a slight uptick in musculoskeletal aches, no major safety signals emerged in the exercise group.
It’s important to note that the survival benefit associated with exercise came after patients had received surgery followed by chemotherapy — in other words, exercise did not replace established cancer treatments. It’s also unclear whether initiating an exercise intervention earlier in the treatment trajectory — before surgery or during chemotherapy, instead of after chemotherapy — could further improve cancer outcomes, the authors noted.
Still, “exercise as an intervention is a no brainer and should be implemented broadly,” said ASCO expert Pamela Kunz, MD, with Yale School of Medicine, New Haven, Connecticut.
Marco Gerlinger, MD, with Barts Cancer Institute, London, England, agreed.
“Oncologists can now make a very clear evidence-based recommendation for patients who just completed their chemotherapy for bowel cancer and are fit enough for such an exercise program,” Gerlinger said in a statement from the nonprofit UK Science Media Centre.
Booth noted that knowledge alone will not be sufficient to allow most patients to change their lifestyle and realize the health benefits.
“The policy implementation piece of this is really key, and we need health systems, hospitals, and payers to invest in these behavior support programs so that patients have access to a physical activity consultant and can realize the health benefits,” he said.
“This intervention is empowering and achievable for patients and with much, much lower cost than many of our therapies. It is also sustainable for health systems,” he concluded.
Diet and Survival
Diet can also affect outcomes in patients with colon cancer.
In the same session describing the CHALLENGE results, Sara Char, MD, with Dana-Farber Cancer Institute in Boston, reported findings showing that consuming a diet high in proinflammatory foods was associated with worse overall survival in patients with stage III colon cancer. A proinflammatory diet includes red and processed meats, sugary drinks, and refined grains, while an anti-inflammatory diet focuses on fruits, vegetables, whole grains, fish, and olive oil.
Chronic systemic inflammation has been implicated in both colon cancer development and in its progression, and elevated levels of inflammatory markers in the blood have previously been associated with worse survival outcomes in patients with stage III colon cancer.
Char and colleagues analyzed dietary patterns of a subset of 1625 patients (mean age, 61 years) with resected stage III colon cancer enrolled in the phase 3 CALGB/SWOG 80702 (Alliance) clinical trial, which compared 3 months of adjuvant chemotherapy with 6 months of adjuvant chemotherapy, with or without the anti-inflammatory medication celecoxib.
As part of the trial, participants reported their diet and exercise habits at various timepoints. Their diets were scored using the validated empirical dietary inflammatory pattern (EDIP) tool, which is a weighted sum of 18 food groups — nine proinflammatory and nine anti-inflammatory. A high EDIP score marks a proinflammatory diet, and a low EDIP score indicates a less inflammatory diet.
During median follow-up of nearly 4 years, researchers noted a trend toward worse disease-free survival in patients with high proinflammatory diets (HR, 1.46), but this association was not significant in the multivariable adjusted model (HR, 1.36; P = .22), Char reported.
However, higher intake of proinflammatory foods was associated with significantly worse overall survival.
Patients who consumed the most proinflammatory foods (top 20%) had an 87% higher risk for death compared with those who consumed the least (bottom 20%; HR, 1.87). The median overall survival in the highest quintile was 7.7 years and was not reached in the lowest quintile.
Combine Exercise and Diet for Best Results
To examine the joint effect of physical activity and diet on overall survival, patients were divided into higher and lower levels of physical activity using a cut-off of 9 MET hours per week, which roughly correlates to 30 minutes of vigorous walking five days a week with a little bit of light yoga, Char explained.
In this analysis, patients with less proinflammatory diets and higher physical activity levels had the best overall survival outcomes, with a 63% lower risk for death compared with peers who consumed more pro-inflammatory diets and exercised less (HR, 0.37; P < .0001).
Daily celecoxib use and low-dose aspirin use (< 100 mg/d) did not affect the association between inflammatory diet and survival.
Char cautioned, that while the EDIP tool is useful to measure the inflammatory potential of a diet, “this is not a dietary recommendation, and we need further studies to be able to tailor our findings into dietary recommendations that can be provided to patients at the bedside.”
Gralow said this “early but promising observational study suggests a powerful synergy: Patients with stage III colon cancer who embraced anti-inflammatory foods and exercised regularly showed the best overall survival compared to those with inflammatory diets and limited exercise.”
The CHALLENGE trial was funded by the Canadian Cancer Society, the National Health and Medical Research Council, Cancer Research UK, and the University of Sydney Cancer Research Fund. Booth had no disclosures. The diet study was funded by the National Institutes of Health, Pfizer, and the Project P Fund. Char disclosed an advisory/consultant role with Goodpath. Kunz, Gralow and Campbell had no relevant disclosures.
A version of this article first appeared on Medscape.com.
Can Lifestyle Changes Save Lives in Colon Cancer?
Can Lifestyle Changes Save Lives in Colon Cancer?

Measuring Fecal Hemoglobin Levels in Negative FIT Tests May Enhance CRC Screening Strategies
The risk of detecting colorectal cancer (CRC) increases by up to 13-fold in the presence of prior fecal hemoglobin (f-Hb) concentrations in fecal immunochemical tests (FIT), especially negative ones, according to a large international dose-response meta-analysis.
Although the association with neoplasia decreased as f-Hb levels rose, the findings support the development of risk-stratified screening strategies based on these concentrations, according to researchers led by Danica M.N. van den Berg, MSc, a PhD candidate and econometrics researcher in the department of public health at Erasmus University Medical Center in Rotterdam, the Netherlands.
Higher f-Hb concentrations in prior negative screening tests are strongly associated with an increased risk of detecting colorectal neoplasia in subsequent screenings, van den Berg said in an interview. “Gastroenterologists and other clinicians should consider the value of f-Hb concentrations in refining screening protocols and personalizing patient care to detect colorectal neoplasia earlier and more accurately.”
Published in Gastroenterology, the study was prompted by prior research showing individuals with f-Hb concentrations just below the positivity cutoff had an elevated CRC risk vs those with low or no f-Hb. “However, global variations in FIT positivity cutoffs and f-Hb category definitions complicated cross-study comparisons,” van den Berg said.
Given the lack of an established dose-response relationship, the study aimed to clarify how f-Hb levels in previous screenings correlate with colorectal neoplasia detection. “Understanding this relationship is crucial for developing risk-stratified colorectal cancer screening strategies based on prior FIT results, which could improve the harm-benefit balance of screening,” she said.
According to van den Berg, f-Hb concentrations could help determine optimal CRC screening intervals by identifying higher-risk individuals who could benefit from more frequent testing, while those with lower concentrations could be screened less frequently.
Study Details
The systematic review and meta-analysis are the first to focus on the dose-response relationship between f-Hb levels in prior FIT screenings and colorectal neoplasia detection, van den Berg said. It included 13 ethnically diverse studies published during 2011-2023 with 4,493,223 individuals from Spain, France, the Netherlands, Taiwan, Denmark, Scotland, Ireland, Korea, Italy, and Norway. Most studies were cohort-based, and one was a randomized controlled trial.
All studies demonstrated a positive association between f-Hb in previous screenings and colorectal neoplasia detection. Almost all reported the f-Hb concentration measured in the prior screening round, while one study combined the f-Hb concentration of two previous screening rounds by using the cumulative f-Hb value. There was, however, wide variability in the stool positivity cut-offs in the included studies, ranging from 10 μg f-Hb/g to 80 μg f-Hb/g.
With an overall effect size of 0.69 (95% CI, 0.59-0.79), pooled analysis revealed that in the next screening round, individuals with f-Hb concentrations in stool of 5, 10, 20, and 40 μg/g had a threefold, fivefold, eightfold, and 13-fold higher risk for colorectal neoplasia, respectively, vs individuals showing 0 μg/g. Although there was significant study heterogeneity (I2 = 97.5%, P < .001), sensitivity analyses confirmed the consistency of findings. Interestingly, subgroup analyses indicated that f-Hb concentrations from a previous negative test were especially predictive of advanced neoplasia in subsequent screenings.
“This is a strategy worth pursuing and evaluating in the United States,” said gastroenterologist Theodore R. Levin, MD, a research scientist at Kaiser Permanente Division of Research in Northern California, commenting on the study but not involved in it. “However, there is no currently available FIT brand in the US that reports f-Hb concentration. All FITs in the United States report as a qualitative positive-negative result.”
The Dutch investigation aligns with prior studies demonstrating a positive association between f-Hb concentrations in previous screenings and the detection of colorectal neoplasia. “Our working hypothesis was that risk increases in a decreasing manner as f-Hb concentrations rise, and the findings supported this hypothesis,” van den Berg said.
Other research has projected f-Hb level risk stratification to be effective and perhaps cost-effective in reducing delayed diagnosis of CRC.
Feasibility of Implementation
In large national screening programs in Europe, Asia, and Australia, as well as those of Kaiser Permanente and the Veterans Health Administration in the United States, information on f-Hb concentrations is already available.
“Therefore, incorporating an Hb-based approach should be relatively easy and affordable,” van den Berg said, and may help to optimize resource use while maintaining high detection rates. “However, the more critical question is whether such an approach would be acceptable to the target population.” To that end, randomized controlled trials in Italy and the Netherlands are offering tailored invitation intervals based on prior f-Hb concentrations and may provide insight into the real-world application of risk-stratified screening.
Among the many variables to be considered in the context of population-wide screening are cost-effectiveness, acceptability, and practicality, as well as invitation intervals, positivity cut-off levels, and start and stop ages for screening. “A key focus will be understanding the acceptability of risk-stratified colorectal cancer screening based on f-Hb among the target population and addressing any information needs they may have, as these are critical factors for successful implementation,” said van den Berg. Her group is currently studying the most effective and cost-effective risk-based strategy for CRC screening based on f-Hb levels.
The authors cautioned that since individuals with undetectable f-Hb levels make up the majority of those with negative FIT results, care must be taken that reducing screening frequency for this low-risk group does not lead to unfavorable outcomes at the population level.
This study was funded by the Dutch Organization for Scientific Research, which had no role in study design, data collection, analysis, interpretation, or writing. The authors declared no competing interests. Levin disclosed no competing interests relevant to his comments.
A version of this article first appeared on Medscape.com.
The risk of detecting colorectal cancer (CRC) increases by up to 13-fold in the presence of prior fecal hemoglobin (f-Hb) concentrations in fecal immunochemical tests (FIT), especially negative ones, according to a large international dose-response meta-analysis.
Although the association with neoplasia decreased as f-Hb levels rose, the findings support the development of risk-stratified screening strategies based on these concentrations, according to researchers led by Danica M.N. van den Berg, MSc, a PhD candidate and econometrics researcher in the department of public health at Erasmus University Medical Center in Rotterdam, the Netherlands.
Higher f-Hb concentrations in prior negative screening tests are strongly associated with an increased risk of detecting colorectal neoplasia in subsequent screenings, van den Berg said in an interview. “Gastroenterologists and other clinicians should consider the value of f-Hb concentrations in refining screening protocols and personalizing patient care to detect colorectal neoplasia earlier and more accurately.”
Published in Gastroenterology, the study was prompted by prior research showing individuals with f-Hb concentrations just below the positivity cutoff had an elevated CRC risk vs those with low or no f-Hb. “However, global variations in FIT positivity cutoffs and f-Hb category definitions complicated cross-study comparisons,” van den Berg said.
Given the lack of an established dose-response relationship, the study aimed to clarify how f-Hb levels in previous screenings correlate with colorectal neoplasia detection. “Understanding this relationship is crucial for developing risk-stratified colorectal cancer screening strategies based on prior FIT results, which could improve the harm-benefit balance of screening,” she said.
According to van den Berg, f-Hb concentrations could help determine optimal CRC screening intervals by identifying higher-risk individuals who could benefit from more frequent testing, while those with lower concentrations could be screened less frequently.
Study Details
The systematic review and meta-analysis are the first to focus on the dose-response relationship between f-Hb levels in prior FIT screenings and colorectal neoplasia detection, van den Berg said. It included 13 ethnically diverse studies published during 2011-2023 with 4,493,223 individuals from Spain, France, the Netherlands, Taiwan, Denmark, Scotland, Ireland, Korea, Italy, and Norway. Most studies were cohort-based, and one was a randomized controlled trial.
All studies demonstrated a positive association between f-Hb in previous screenings and colorectal neoplasia detection. Almost all reported the f-Hb concentration measured in the prior screening round, while one study combined the f-Hb concentration of two previous screening rounds by using the cumulative f-Hb value. There was, however, wide variability in the stool positivity cut-offs in the included studies, ranging from 10 μg f-Hb/g to 80 μg f-Hb/g.
With an overall effect size of 0.69 (95% CI, 0.59-0.79), pooled analysis revealed that in the next screening round, individuals with f-Hb concentrations in stool of 5, 10, 20, and 40 μg/g had a threefold, fivefold, eightfold, and 13-fold higher risk for colorectal neoplasia, respectively, vs individuals showing 0 μg/g. Although there was significant study heterogeneity (I2 = 97.5%, P < .001), sensitivity analyses confirmed the consistency of findings. Interestingly, subgroup analyses indicated that f-Hb concentrations from a previous negative test were especially predictive of advanced neoplasia in subsequent screenings.
“This is a strategy worth pursuing and evaluating in the United States,” said gastroenterologist Theodore R. Levin, MD, a research scientist at Kaiser Permanente Division of Research in Northern California, commenting on the study but not involved in it. “However, there is no currently available FIT brand in the US that reports f-Hb concentration. All FITs in the United States report as a qualitative positive-negative result.”
The Dutch investigation aligns with prior studies demonstrating a positive association between f-Hb concentrations in previous screenings and the detection of colorectal neoplasia. “Our working hypothesis was that risk increases in a decreasing manner as f-Hb concentrations rise, and the findings supported this hypothesis,” van den Berg said.
Other research has projected f-Hb level risk stratification to be effective and perhaps cost-effective in reducing delayed diagnosis of CRC.
Feasibility of Implementation
In large national screening programs in Europe, Asia, and Australia, as well as those of Kaiser Permanente and the Veterans Health Administration in the United States, information on f-Hb concentrations is already available.
“Therefore, incorporating an Hb-based approach should be relatively easy and affordable,” van den Berg said, and may help to optimize resource use while maintaining high detection rates. “However, the more critical question is whether such an approach would be acceptable to the target population.” To that end, randomized controlled trials in Italy and the Netherlands are offering tailored invitation intervals based on prior f-Hb concentrations and may provide insight into the real-world application of risk-stratified screening.
Among the many variables to be considered in the context of population-wide screening are cost-effectiveness, acceptability, and practicality, as well as invitation intervals, positivity cut-off levels, and start and stop ages for screening. “A key focus will be understanding the acceptability of risk-stratified colorectal cancer screening based on f-Hb among the target population and addressing any information needs they may have, as these are critical factors for successful implementation,” said van den Berg. Her group is currently studying the most effective and cost-effective risk-based strategy for CRC screening based on f-Hb levels.
The authors cautioned that since individuals with undetectable f-Hb levels make up the majority of those with negative FIT results, care must be taken that reducing screening frequency for this low-risk group does not lead to unfavorable outcomes at the population level.
This study was funded by the Dutch Organization for Scientific Research, which had no role in study design, data collection, analysis, interpretation, or writing. The authors declared no competing interests. Levin disclosed no competing interests relevant to his comments.
A version of this article first appeared on Medscape.com.
The risk of detecting colorectal cancer (CRC) increases by up to 13-fold in the presence of prior fecal hemoglobin (f-Hb) concentrations in fecal immunochemical tests (FIT), especially negative ones, according to a large international dose-response meta-analysis.
Although the association with neoplasia decreased as f-Hb levels rose, the findings support the development of risk-stratified screening strategies based on these concentrations, according to researchers led by Danica M.N. van den Berg, MSc, a PhD candidate and econometrics researcher in the department of public health at Erasmus University Medical Center in Rotterdam, the Netherlands.
Higher f-Hb concentrations in prior negative screening tests are strongly associated with an increased risk of detecting colorectal neoplasia in subsequent screenings, van den Berg said in an interview. “Gastroenterologists and other clinicians should consider the value of f-Hb concentrations in refining screening protocols and personalizing patient care to detect colorectal neoplasia earlier and more accurately.”
Published in Gastroenterology, the study was prompted by prior research showing individuals with f-Hb concentrations just below the positivity cutoff had an elevated CRC risk vs those with low or no f-Hb. “However, global variations in FIT positivity cutoffs and f-Hb category definitions complicated cross-study comparisons,” van den Berg said.
Given the lack of an established dose-response relationship, the study aimed to clarify how f-Hb levels in previous screenings correlate with colorectal neoplasia detection. “Understanding this relationship is crucial for developing risk-stratified colorectal cancer screening strategies based on prior FIT results, which could improve the harm-benefit balance of screening,” she said.
According to van den Berg, f-Hb concentrations could help determine optimal CRC screening intervals by identifying higher-risk individuals who could benefit from more frequent testing, while those with lower concentrations could be screened less frequently.
Study Details
The systematic review and meta-analysis are the first to focus on the dose-response relationship between f-Hb levels in prior FIT screenings and colorectal neoplasia detection, van den Berg said. It included 13 ethnically diverse studies published during 2011-2023 with 4,493,223 individuals from Spain, France, the Netherlands, Taiwan, Denmark, Scotland, Ireland, Korea, Italy, and Norway. Most studies were cohort-based, and one was a randomized controlled trial.
All studies demonstrated a positive association between f-Hb in previous screenings and colorectal neoplasia detection. Almost all reported the f-Hb concentration measured in the prior screening round, while one study combined the f-Hb concentration of two previous screening rounds by using the cumulative f-Hb value. There was, however, wide variability in the stool positivity cut-offs in the included studies, ranging from 10 μg f-Hb/g to 80 μg f-Hb/g.
With an overall effect size of 0.69 (95% CI, 0.59-0.79), pooled analysis revealed that in the next screening round, individuals with f-Hb concentrations in stool of 5, 10, 20, and 40 μg/g had a threefold, fivefold, eightfold, and 13-fold higher risk for colorectal neoplasia, respectively, vs individuals showing 0 μg/g. Although there was significant study heterogeneity (I2 = 97.5%, P < .001), sensitivity analyses confirmed the consistency of findings. Interestingly, subgroup analyses indicated that f-Hb concentrations from a previous negative test were especially predictive of advanced neoplasia in subsequent screenings.
“This is a strategy worth pursuing and evaluating in the United States,” said gastroenterologist Theodore R. Levin, MD, a research scientist at Kaiser Permanente Division of Research in Northern California, commenting on the study but not involved in it. “However, there is no currently available FIT brand in the US that reports f-Hb concentration. All FITs in the United States report as a qualitative positive-negative result.”
The Dutch investigation aligns with prior studies demonstrating a positive association between f-Hb concentrations in previous screenings and the detection of colorectal neoplasia. “Our working hypothesis was that risk increases in a decreasing manner as f-Hb concentrations rise, and the findings supported this hypothesis,” van den Berg said.
Other research has projected f-Hb level risk stratification to be effective and perhaps cost-effective in reducing delayed diagnosis of CRC.
Feasibility of Implementation
In large national screening programs in Europe, Asia, and Australia, as well as those of Kaiser Permanente and the Veterans Health Administration in the United States, information on f-Hb concentrations is already available.
“Therefore, incorporating an Hb-based approach should be relatively easy and affordable,” van den Berg said, and may help to optimize resource use while maintaining high detection rates. “However, the more critical question is whether such an approach would be acceptable to the target population.” To that end, randomized controlled trials in Italy and the Netherlands are offering tailored invitation intervals based on prior f-Hb concentrations and may provide insight into the real-world application of risk-stratified screening.
Among the many variables to be considered in the context of population-wide screening are cost-effectiveness, acceptability, and practicality, as well as invitation intervals, positivity cut-off levels, and start and stop ages for screening. “A key focus will be understanding the acceptability of risk-stratified colorectal cancer screening based on f-Hb among the target population and addressing any information needs they may have, as these are critical factors for successful implementation,” said van den Berg. Her group is currently studying the most effective and cost-effective risk-based strategy for CRC screening based on f-Hb levels.
The authors cautioned that since individuals with undetectable f-Hb levels make up the majority of those with negative FIT results, care must be taken that reducing screening frequency for this low-risk group does not lead to unfavorable outcomes at the population level.
This study was funded by the Dutch Organization for Scientific Research, which had no role in study design, data collection, analysis, interpretation, or writing. The authors declared no competing interests. Levin disclosed no competing interests relevant to his comments.
A version of this article first appeared on Medscape.com.
FROM GASTROENTEROLOGY
Oral Microbiome Dysbiosis: Biomarker for Upper GI Disorders?
TOPLINE:
Dysbiosis of the oral microbiome is associated with various upper gastrointestinal (UGI) disorders and precancerous lesions, with specific microbial signatures varying by disease and oral site, research shows.
METHODOLOGY:
- Emerging evidence suggests that the oral microbiota may contribute to the development of gastrointestinal malignancies, leading to efforts to identify biomarkers for early detection and progress of disease.
- In this population-based cross-sectional study, researchers studied the association between the microbiome of saliva, subgingival, and buccal mucosa and UGI disorders, particularly precancerous lesions.
- Participants included 388 adults who underwent upper endoscopy with biopsies for histopathologic analysis.
- UGI symptoms were evaluated using a validated tool, and 16S ribosomal RNA sequencing was used to characterize microbial diversity and composition of 380 saliva, 200 subgingival, and 267 buccal mucosa samples.
TAKEAWAY:
- Saliva dysbiosis was associated with several UGI disorders, including gastroesophageal reflux symptoms alone, symptomatic esophagitis, combined esophagitis and Barrett’s esophagus (BE), Helicobacter pylori–positive histology, chemical reactive gastritis, atrophic H pylori gastritis, and intestinal metaplasia.
- In contrast, dysbiosis in subgingival and buccal mucosa was more specifically associated with BE and atrophic H pylori gastritis.
- Among several identified genera, Prevotella and Fusobacterium in saliva were associated with gastric atrophy and intestinal metaplasia, and in subgingival samples, there was a notable link between Fretibacterium in BE and Fusobacterium in gastric atrophy and intestinal metaplasia.
IN PRACTICE:
“Our study for the first time suggests that microbiota in the subgingival and buccal regions may serve as more specific biomarkers for detecting precancerous lesions in asymptomatic patients, particularly for Barrett’s esophagus,” the authors wrote. “Saliva might be more appropriate for monitoring any UGI disorders at the population level.”
SOURCE:
The study, with first author Fatemeh Sadeghi, PhD, with Karolinska Institutet, Stockholm, Sweden, was published online in the American Journal of Gastroenterology.
LIMITATIONS:
The study used bacterial DNA, which cannot distinguish metabolically active bacteria. Data on diet and probiotic use were not collected. The cross-sectional design precludes conclusions about causality.
DISCLOSURES:
The authors declared no conflicts of interest. The study was funded by the Swedish Cancer Society and the Swedish Research Council.
A version of this article first appeared on Medscape.com.
TOPLINE:
Dysbiosis of the oral microbiome is associated with various upper gastrointestinal (UGI) disorders and precancerous lesions, with specific microbial signatures varying by disease and oral site, research shows.
METHODOLOGY:
- Emerging evidence suggests that the oral microbiota may contribute to the development of gastrointestinal malignancies, leading to efforts to identify biomarkers for early detection and progress of disease.
- In this population-based cross-sectional study, researchers studied the association between the microbiome of saliva, subgingival, and buccal mucosa and UGI disorders, particularly precancerous lesions.
- Participants included 388 adults who underwent upper endoscopy with biopsies for histopathologic analysis.
- UGI symptoms were evaluated using a validated tool, and 16S ribosomal RNA sequencing was used to characterize microbial diversity and composition of 380 saliva, 200 subgingival, and 267 buccal mucosa samples.
TAKEAWAY:
- Saliva dysbiosis was associated with several UGI disorders, including gastroesophageal reflux symptoms alone, symptomatic esophagitis, combined esophagitis and Barrett’s esophagus (BE), Helicobacter pylori–positive histology, chemical reactive gastritis, atrophic H pylori gastritis, and intestinal metaplasia.
- In contrast, dysbiosis in subgingival and buccal mucosa was more specifically associated with BE and atrophic H pylori gastritis.
- Among several identified genera, Prevotella and Fusobacterium in saliva were associated with gastric atrophy and intestinal metaplasia, and in subgingival samples, there was a notable link between Fretibacterium in BE and Fusobacterium in gastric atrophy and intestinal metaplasia.
IN PRACTICE:
“Our study for the first time suggests that microbiota in the subgingival and buccal regions may serve as more specific biomarkers for detecting precancerous lesions in asymptomatic patients, particularly for Barrett’s esophagus,” the authors wrote. “Saliva might be more appropriate for monitoring any UGI disorders at the population level.”
SOURCE:
The study, with first author Fatemeh Sadeghi, PhD, with Karolinska Institutet, Stockholm, Sweden, was published online in the American Journal of Gastroenterology.
LIMITATIONS:
The study used bacterial DNA, which cannot distinguish metabolically active bacteria. Data on diet and probiotic use were not collected. The cross-sectional design precludes conclusions about causality.
DISCLOSURES:
The authors declared no conflicts of interest. The study was funded by the Swedish Cancer Society and the Swedish Research Council.
A version of this article first appeared on Medscape.com.
TOPLINE:
Dysbiosis of the oral microbiome is associated with various upper gastrointestinal (UGI) disorders and precancerous lesions, with specific microbial signatures varying by disease and oral site, research shows.
METHODOLOGY:
- Emerging evidence suggests that the oral microbiota may contribute to the development of gastrointestinal malignancies, leading to efforts to identify biomarkers for early detection and progress of disease.
- In this population-based cross-sectional study, researchers studied the association between the microbiome of saliva, subgingival, and buccal mucosa and UGI disorders, particularly precancerous lesions.
- Participants included 388 adults who underwent upper endoscopy with biopsies for histopathologic analysis.
- UGI symptoms were evaluated using a validated tool, and 16S ribosomal RNA sequencing was used to characterize microbial diversity and composition of 380 saliva, 200 subgingival, and 267 buccal mucosa samples.
TAKEAWAY:
- Saliva dysbiosis was associated with several UGI disorders, including gastroesophageal reflux symptoms alone, symptomatic esophagitis, combined esophagitis and Barrett’s esophagus (BE), Helicobacter pylori–positive histology, chemical reactive gastritis, atrophic H pylori gastritis, and intestinal metaplasia.
- In contrast, dysbiosis in subgingival and buccal mucosa was more specifically associated with BE and atrophic H pylori gastritis.
- Among several identified genera, Prevotella and Fusobacterium in saliva were associated with gastric atrophy and intestinal metaplasia, and in subgingival samples, there was a notable link between Fretibacterium in BE and Fusobacterium in gastric atrophy and intestinal metaplasia.
IN PRACTICE:
“Our study for the first time suggests that microbiota in the subgingival and buccal regions may serve as more specific biomarkers for detecting precancerous lesions in asymptomatic patients, particularly for Barrett’s esophagus,” the authors wrote. “Saliva might be more appropriate for monitoring any UGI disorders at the population level.”
SOURCE:
The study, with first author Fatemeh Sadeghi, PhD, with Karolinska Institutet, Stockholm, Sweden, was published online in the American Journal of Gastroenterology.
LIMITATIONS:
The study used bacterial DNA, which cannot distinguish metabolically active bacteria. Data on diet and probiotic use were not collected. The cross-sectional design precludes conclusions about causality.
DISCLOSURES:
The authors declared no conflicts of interest. The study was funded by the Swedish Cancer Society and the Swedish Research Council.
A version of this article first appeared on Medscape.com.
Emergency Presentations for Vets with CRC Linked to Higher Mortality
TOPLINE: More than 28% of US Department of Veterans Affairs (VA) patients with colorectal cancer were diagnosed through emergency presentations, which were associated with a higher mortality risk. Emergency presentations increased during COVID-19 from prepandemic rates.
METHODOLOGY:
- A retrospective cohort study analyzed 9096 incident colorectal cancer cancer cases diagnosed in the Veterans Health Administration from 2017 to 2021.
- Researchers applied a validated algorithm to identify emergency presentations, defined as cancer diagnoses within 30 days following emergency care episodes or unplanned hospital admissions.
- Analysis utilized multivariable logistic regression and Cox proportional hazards models to examine associations between emergency presentations and cancer stage, treatment, and mortality.
TAKEAWAY:
- Patients with emergency presentations were more likely to have advanced stage disease (odds ratio [OR], 1.70; 95% CI, 1.53-1.88) compared to those without emergency presentations.
- Emergency presentations were associated with lower likelihood of receiving cancer treatment (OR, 0.65; 95% CI, 0.56-0.75) and higher mortality risk (hazard ratio [HR], 1.70; 95% CI, 1.56-1.84).
- The proportion of emergency presentations increased from 26.4% in 2017-2019 to 31.4% during the COVID-19 pandemic years 2020-2021 (P < .0001).
IN PRACTICE: " Our findings from one of the largest studies within a US population to examine emergency presentations among patients with colorectal cancer show that emergency presentations are common and an important negative predictor of cancer outcomes…Our study findings highlight the need for continued research and implementation efforts focused on measurement and mitigation of emergency presentations among patients with colorectal cancer.”
SOURCE: The study was led by the Center for Innovations in Quality, Effectiveness and Safety at Michael E. DeBakey Veterans Affairs Medical Center and Baylor College of Medicine in Houston. It was published online on December 11 in Digestive Diseases and Sciences.
LIMITATIONS: The study's findings are limited by the predominantly male veteran population with lower socioeconomic status, which may affect generalizability. The equal access health care model used by the VA and its and strong screening programs may result in emergency presentation rates that differ from the private sector.
TOPLINE: More than 28% of US Department of Veterans Affairs (VA) patients with colorectal cancer were diagnosed through emergency presentations, which were associated with a higher mortality risk. Emergency presentations increased during COVID-19 from prepandemic rates.
METHODOLOGY:
- A retrospective cohort study analyzed 9096 incident colorectal cancer cancer cases diagnosed in the Veterans Health Administration from 2017 to 2021.
- Researchers applied a validated algorithm to identify emergency presentations, defined as cancer diagnoses within 30 days following emergency care episodes or unplanned hospital admissions.
- Analysis utilized multivariable logistic regression and Cox proportional hazards models to examine associations between emergency presentations and cancer stage, treatment, and mortality.
TAKEAWAY:
- Patients with emergency presentations were more likely to have advanced stage disease (odds ratio [OR], 1.70; 95% CI, 1.53-1.88) compared to those without emergency presentations.
- Emergency presentations were associated with lower likelihood of receiving cancer treatment (OR, 0.65; 95% CI, 0.56-0.75) and higher mortality risk (hazard ratio [HR], 1.70; 95% CI, 1.56-1.84).
- The proportion of emergency presentations increased from 26.4% in 2017-2019 to 31.4% during the COVID-19 pandemic years 2020-2021 (P < .0001).
IN PRACTICE: " Our findings from one of the largest studies within a US population to examine emergency presentations among patients with colorectal cancer show that emergency presentations are common and an important negative predictor of cancer outcomes…Our study findings highlight the need for continued research and implementation efforts focused on measurement and mitigation of emergency presentations among patients with colorectal cancer.”
SOURCE: The study was led by the Center for Innovations in Quality, Effectiveness and Safety at Michael E. DeBakey Veterans Affairs Medical Center and Baylor College of Medicine in Houston. It was published online on December 11 in Digestive Diseases and Sciences.
LIMITATIONS: The study's findings are limited by the predominantly male veteran population with lower socioeconomic status, which may affect generalizability. The equal access health care model used by the VA and its and strong screening programs may result in emergency presentation rates that differ from the private sector.
TOPLINE: More than 28% of US Department of Veterans Affairs (VA) patients with colorectal cancer were diagnosed through emergency presentations, which were associated with a higher mortality risk. Emergency presentations increased during COVID-19 from prepandemic rates.
METHODOLOGY:
- A retrospective cohort study analyzed 9096 incident colorectal cancer cancer cases diagnosed in the Veterans Health Administration from 2017 to 2021.
- Researchers applied a validated algorithm to identify emergency presentations, defined as cancer diagnoses within 30 days following emergency care episodes or unplanned hospital admissions.
- Analysis utilized multivariable logistic regression and Cox proportional hazards models to examine associations between emergency presentations and cancer stage, treatment, and mortality.
TAKEAWAY:
- Patients with emergency presentations were more likely to have advanced stage disease (odds ratio [OR], 1.70; 95% CI, 1.53-1.88) compared to those without emergency presentations.
- Emergency presentations were associated with lower likelihood of receiving cancer treatment (OR, 0.65; 95% CI, 0.56-0.75) and higher mortality risk (hazard ratio [HR], 1.70; 95% CI, 1.56-1.84).
- The proportion of emergency presentations increased from 26.4% in 2017-2019 to 31.4% during the COVID-19 pandemic years 2020-2021 (P < .0001).
IN PRACTICE: " Our findings from one of the largest studies within a US population to examine emergency presentations among patients with colorectal cancer show that emergency presentations are common and an important negative predictor of cancer outcomes…Our study findings highlight the need for continued research and implementation efforts focused on measurement and mitigation of emergency presentations among patients with colorectal cancer.”
SOURCE: The study was led by the Center for Innovations in Quality, Effectiveness and Safety at Michael E. DeBakey Veterans Affairs Medical Center and Baylor College of Medicine in Houston. It was published online on December 11 in Digestive Diseases and Sciences.
LIMITATIONS: The study's findings are limited by the predominantly male veteran population with lower socioeconomic status, which may affect generalizability. The equal access health care model used by the VA and its and strong screening programs may result in emergency presentation rates that differ from the private sector.
FDA Approves Sotorasib + Panitumumab for mCRC
The US Food and Drug Administration (FDA) has approved sotorasib (Lumakras, Amgen Inc.) with panitumumab (Vectibix, Amgen Inc.) for the treatment of certain adult patients with metastatic colorectal cancer (mCRC).
Specifically, the combination therapy is indicated for those with KRAS G12C-mutated mCRC, as determined using an FDA-approved test, who have received prior treatment with fluoropyrimidine-, oxaliplatin-, and irinotecan-based chemotherapy, according to the FDA notice. The FDA also approved the therascreen KRAS RGQ PCR Kit (QIAGEN GmbH) as a companion diagnostic device for identifying eligible patients.
Approval of sotorasib with panitumumab was based on findings from the randomized, open-label, controlled CodeBreaK 300 trial showing improved overall response rates (ORR) and progression-free survival (PFS) with sotorasib and panitumumab vs investigator’s choice of trifluridine/tipiracil or regorafenib, which are current standard-of-care options.
Median PFS was 5.6 months in 53 patients randomized to receive 960 mg of oral sotorasib once daily plus 6 mg/kg of intravenous (IV) panitumumab every 2 weeks, and 2 months in 54 patients randomized to receive standard-of-care therapy (hazard ratio, 0.48). The ORR was 26% vs 0% in the arms, respectively, and the duration of response in the sotorasib/panitumumab arm was 4.4 months. No significant difference in PFS was observed between the standard-of-care arm and a third arm with 53 patients who received 240 mg of oral sotorasib daily plus 6 mg/kg of IV panitumumab every 2 weeks.
Overall survival (OS) did not differ significantly between the treatment arms in the final analysis, but the study was not statistically powered for OS.
Adverse reactions occurring in at least 20% of patients receiving sotorasib/panitumumab were rash, dry skin, diarrhea, stomatitis, fatigue, and musculoskeletal pain. Common grade 3-4 laboratory abnormalities, which occurred in two or more patients, included decreased magnesium, decreased potassium, decreased corrected calcium, and increased potassium.
The recommended dose of sotorasib is 960 mg given orally once daily and administered before the first panitumumab infusion. The recommended panitumumab dose is 6 mg/kg as an IV infusion every 14 days until disease progression, unacceptable toxicity, or until sotorasib is withheld or discontinued, according to the full prescribing information.
A version of this article first appeared on Medscape.com.
The US Food and Drug Administration (FDA) has approved sotorasib (Lumakras, Amgen Inc.) with panitumumab (Vectibix, Amgen Inc.) for the treatment of certain adult patients with metastatic colorectal cancer (mCRC).
Specifically, the combination therapy is indicated for those with KRAS G12C-mutated mCRC, as determined using an FDA-approved test, who have received prior treatment with fluoropyrimidine-, oxaliplatin-, and irinotecan-based chemotherapy, according to the FDA notice. The FDA also approved the therascreen KRAS RGQ PCR Kit (QIAGEN GmbH) as a companion diagnostic device for identifying eligible patients.
Approval of sotorasib with panitumumab was based on findings from the randomized, open-label, controlled CodeBreaK 300 trial showing improved overall response rates (ORR) and progression-free survival (PFS) with sotorasib and panitumumab vs investigator’s choice of trifluridine/tipiracil or regorafenib, which are current standard-of-care options.
Median PFS was 5.6 months in 53 patients randomized to receive 960 mg of oral sotorasib once daily plus 6 mg/kg of intravenous (IV) panitumumab every 2 weeks, and 2 months in 54 patients randomized to receive standard-of-care therapy (hazard ratio, 0.48). The ORR was 26% vs 0% in the arms, respectively, and the duration of response in the sotorasib/panitumumab arm was 4.4 months. No significant difference in PFS was observed between the standard-of-care arm and a third arm with 53 patients who received 240 mg of oral sotorasib daily plus 6 mg/kg of IV panitumumab every 2 weeks.
Overall survival (OS) did not differ significantly between the treatment arms in the final analysis, but the study was not statistically powered for OS.
Adverse reactions occurring in at least 20% of patients receiving sotorasib/panitumumab were rash, dry skin, diarrhea, stomatitis, fatigue, and musculoskeletal pain. Common grade 3-4 laboratory abnormalities, which occurred in two or more patients, included decreased magnesium, decreased potassium, decreased corrected calcium, and increased potassium.
The recommended dose of sotorasib is 960 mg given orally once daily and administered before the first panitumumab infusion. The recommended panitumumab dose is 6 mg/kg as an IV infusion every 14 days until disease progression, unacceptable toxicity, or until sotorasib is withheld or discontinued, according to the full prescribing information.
A version of this article first appeared on Medscape.com.
The US Food and Drug Administration (FDA) has approved sotorasib (Lumakras, Amgen Inc.) with panitumumab (Vectibix, Amgen Inc.) for the treatment of certain adult patients with metastatic colorectal cancer (mCRC).
Specifically, the combination therapy is indicated for those with KRAS G12C-mutated mCRC, as determined using an FDA-approved test, who have received prior treatment with fluoropyrimidine-, oxaliplatin-, and irinotecan-based chemotherapy, according to the FDA notice. The FDA also approved the therascreen KRAS RGQ PCR Kit (QIAGEN GmbH) as a companion diagnostic device for identifying eligible patients.
Approval of sotorasib with panitumumab was based on findings from the randomized, open-label, controlled CodeBreaK 300 trial showing improved overall response rates (ORR) and progression-free survival (PFS) with sotorasib and panitumumab vs investigator’s choice of trifluridine/tipiracil or regorafenib, which are current standard-of-care options.
Median PFS was 5.6 months in 53 patients randomized to receive 960 mg of oral sotorasib once daily plus 6 mg/kg of intravenous (IV) panitumumab every 2 weeks, and 2 months in 54 patients randomized to receive standard-of-care therapy (hazard ratio, 0.48). The ORR was 26% vs 0% in the arms, respectively, and the duration of response in the sotorasib/panitumumab arm was 4.4 months. No significant difference in PFS was observed between the standard-of-care arm and a third arm with 53 patients who received 240 mg of oral sotorasib daily plus 6 mg/kg of IV panitumumab every 2 weeks.
Overall survival (OS) did not differ significantly between the treatment arms in the final analysis, but the study was not statistically powered for OS.
Adverse reactions occurring in at least 20% of patients receiving sotorasib/panitumumab were rash, dry skin, diarrhea, stomatitis, fatigue, and musculoskeletal pain. Common grade 3-4 laboratory abnormalities, which occurred in two or more patients, included decreased magnesium, decreased potassium, decreased corrected calcium, and increased potassium.
The recommended dose of sotorasib is 960 mg given orally once daily and administered before the first panitumumab infusion. The recommended panitumumab dose is 6 mg/kg as an IV infusion every 14 days until disease progression, unacceptable toxicity, or until sotorasib is withheld or discontinued, according to the full prescribing information.
A version of this article first appeared on Medscape.com.