User login
NIH director updates study enrolling one million participants
NEW ORLEANS – It is not too late to enroll your patients or yourself into the largest longitudinal cohort study ever initiated, according to Francis S. Collins, MD, PhD, who is director of the National Institutes of Health (NIH).
Since May 2018, when it was initiated, the NIH-funded All of Us Research Program has already enrolled 200,000 of the planned goal of one million participants in the United States. Of these, approximately half have already provided baseline demographics and health information as well as their consent to use the slew of health data that is being collected.
“The only way to do this kind of thing is to have data – a lot of it,” said Dr. Collins, explaining the premise of the All of Us Research Program in an interview conducted at the annual meeting of the Endocrine Society.
The data are not limited to medical records: Blood samples, whole genome sequencing, wearable activity monitors, and subject-completed questionnaires are among a long list of sources of information to be collected from participants, who are expected to be followed indefinitely.
According to Dr. Collins, who delivered a plenary address at the meeting, these data will become more valuable over time, one of the most important goals of this study is to prepare the way for precision medicine. As opposed to the traditional one-size-fits-all approach to treating disease, he believes that this large dataset will allow researchers to understand differences in common diseases at the individual level.
In relation to endocrinology, Dr. Collins said that a cohort of one million participants would be expected to have close to 100,000 individuals with diabetes mellitus.
“This is going to be transformative,” said Dr. Collins, who emphasized that the enrollment is specifically designed to capture participants from diverse ethnic and racial groups.
All of the data collected will be made broadly available to research initiatives of all kinds, many of which have not yet even been envisioned.
Information on enrollment is available on line: joinallofus.org.
NEW ORLEANS – It is not too late to enroll your patients or yourself into the largest longitudinal cohort study ever initiated, according to Francis S. Collins, MD, PhD, who is director of the National Institutes of Health (NIH).
Since May 2018, when it was initiated, the NIH-funded All of Us Research Program has already enrolled 200,000 of the planned goal of one million participants in the United States. Of these, approximately half have already provided baseline demographics and health information as well as their consent to use the slew of health data that is being collected.
“The only way to do this kind of thing is to have data – a lot of it,” said Dr. Collins, explaining the premise of the All of Us Research Program in an interview conducted at the annual meeting of the Endocrine Society.
The data are not limited to medical records: Blood samples, whole genome sequencing, wearable activity monitors, and subject-completed questionnaires are among a long list of sources of information to be collected from participants, who are expected to be followed indefinitely.
According to Dr. Collins, who delivered a plenary address at the meeting, these data will become more valuable over time, one of the most important goals of this study is to prepare the way for precision medicine. As opposed to the traditional one-size-fits-all approach to treating disease, he believes that this large dataset will allow researchers to understand differences in common diseases at the individual level.
In relation to endocrinology, Dr. Collins said that a cohort of one million participants would be expected to have close to 100,000 individuals with diabetes mellitus.
“This is going to be transformative,” said Dr. Collins, who emphasized that the enrollment is specifically designed to capture participants from diverse ethnic and racial groups.
All of the data collected will be made broadly available to research initiatives of all kinds, many of which have not yet even been envisioned.
Information on enrollment is available on line: joinallofus.org.
NEW ORLEANS – It is not too late to enroll your patients or yourself into the largest longitudinal cohort study ever initiated, according to Francis S. Collins, MD, PhD, who is director of the National Institutes of Health (NIH).
Since May 2018, when it was initiated, the NIH-funded All of Us Research Program has already enrolled 200,000 of the planned goal of one million participants in the United States. Of these, approximately half have already provided baseline demographics and health information as well as their consent to use the slew of health data that is being collected.
“The only way to do this kind of thing is to have data – a lot of it,” said Dr. Collins, explaining the premise of the All of Us Research Program in an interview conducted at the annual meeting of the Endocrine Society.
The data are not limited to medical records: Blood samples, whole genome sequencing, wearable activity monitors, and subject-completed questionnaires are among a long list of sources of information to be collected from participants, who are expected to be followed indefinitely.
According to Dr. Collins, who delivered a plenary address at the meeting, these data will become more valuable over time, one of the most important goals of this study is to prepare the way for precision medicine. As opposed to the traditional one-size-fits-all approach to treating disease, he believes that this large dataset will allow researchers to understand differences in common diseases at the individual level.
In relation to endocrinology, Dr. Collins said that a cohort of one million participants would be expected to have close to 100,000 individuals with diabetes mellitus.
“This is going to be transformative,” said Dr. Collins, who emphasized that the enrollment is specifically designed to capture participants from diverse ethnic and racial groups.
All of the data collected will be made broadly available to research initiatives of all kinds, many of which have not yet even been envisioned.
Information on enrollment is available on line: joinallofus.org.
REPORTING FROM ENDO 2019

No birth rate gains from levothyroxine in pregnancy
Treatment with levothyroxine does not improve the live birth rate in women with thyroid peroxidase antibodies before conception, according to data presented at the annual meeting of the Endocrine Society.

Until now, the evidence for the use of levothyroxine in pregnant women with thyroid peroxidase antibodies but normal thyroid function has been inconclusive, Rima K. Dhillon-Smith, MBChB, PhD, of the University of Birmingham (England), and her coauthors said in a paper published simultaneously with the meeting presentation March 23 in the New England Journal of Medicine.
Previous studies have shown that women with thyroid peroxidase antibodies but normal thyroid function have a nearly fourfold higher risk of miscarriage and twofold higher risk of preterm birth, compared with women who don’t have the antibodies.
In the new double-blind study, 952 women with thyroid peroxidase antibodies, normal thyroid function, and a history of miscarriage or infertility were randomized either to daily 50 mcg levothyroxine or placebo, taken from conception to the end of pregnancy.
The rate of pregnancy was similar in the levothyroxine and placebo groups (56.6% vs. 58.3%, respectively), as was the live birth rate (37.4% vs. 37.9%), despite the observation that the levothyroxine group had consistently lower serum thyrotropin and higher free T4 concentrations than did the placebo group.
There were also no significant differences between the two groups in secondary outcomes of miscarriage, preterm birth, or neonatal outcomes such as birth weight.
Researchers also saw no statistically significant differences in the rate of serious adverse events or in the number of women who showed abnormal results on thyroid function tests.
The authors noted that the dosage of levothyroxine used in the study was fixed, leaving the possibility that “the dose may need to be adjusted depending on the participant’s body weight, thyroid peroxidase antibody level, or thyrotropin concentration.”
Existing guidelines from the American Thyroid Association acknowledge the lack of evidence in favor of levothyroxine decreasing the risk of pregnancy loss. However, the guidelines also state that it can be considered in antibody-positive, euthyroid women with a history of loss, “given its potential benefits in comparison with its minimal risk.”
The study was supported by the National Institute for Health Research. No conflicts of interest were declared.
SOURCE: Dhillon-Smith R et al. N Engl J Med. 2019 March 23. doi: 10.1056/NEJMoa1812537
Treatment with levothyroxine does not improve the live birth rate in women with thyroid peroxidase antibodies before conception, according to data presented at the annual meeting of the Endocrine Society.
Until now, the evidence for the use of levothyroxine in pregnant women with thyroid peroxidase antibodies but normal thyroid function has been inconclusive, Rima K. Dhillon-Smith, MBChB, PhD, of the University of Birmingham (England), and her coauthors said in a paper published simultaneously with the meeting presentation March 23 in the New England Journal of Medicine.
Previous studies have shown that women with thyroid peroxidase antibodies but normal thyroid function have a nearly fourfold higher risk of miscarriage and twofold higher risk of preterm birth, compared with women who don’t have the antibodies.
In the new double-blind study, 952 women with thyroid peroxidase antibodies, normal thyroid function, and a history of miscarriage or infertility were randomized either to daily 50 mcg levothyroxine or placebo, taken from conception to the end of pregnancy.
The rate of pregnancy was similar in the levothyroxine and placebo groups (56.6% vs. 58.3%, respectively), as was the live birth rate (37.4% vs. 37.9%), despite the observation that the levothyroxine group had consistently lower serum thyrotropin and higher free T4 concentrations than did the placebo group.
There were also no significant differences between the two groups in secondary outcomes of miscarriage, preterm birth, or neonatal outcomes such as birth weight.
Researchers also saw no statistically significant differences in the rate of serious adverse events or in the number of women who showed abnormal results on thyroid function tests.
The authors noted that the dosage of levothyroxine used in the study was fixed, leaving the possibility that “the dose may need to be adjusted depending on the participant’s body weight, thyroid peroxidase antibody level, or thyrotropin concentration.”
Existing guidelines from the American Thyroid Association acknowledge the lack of evidence in favor of levothyroxine decreasing the risk of pregnancy loss. However, the guidelines also state that it can be considered in antibody-positive, euthyroid women with a history of loss, “given its potential benefits in comparison with its minimal risk.”
The study was supported by the National Institute for Health Research. No conflicts of interest were declared.
SOURCE: Dhillon-Smith R et al. N Engl J Med. 2019 March 23. doi: 10.1056/NEJMoa1812537
Treatment with levothyroxine does not improve the live birth rate in women with thyroid peroxidase antibodies before conception, according to data presented at the annual meeting of the Endocrine Society.
Until now, the evidence for the use of levothyroxine in pregnant women with thyroid peroxidase antibodies but normal thyroid function has been inconclusive, Rima K. Dhillon-Smith, MBChB, PhD, of the University of Birmingham (England), and her coauthors said in a paper published simultaneously with the meeting presentation March 23 in the New England Journal of Medicine.
Previous studies have shown that women with thyroid peroxidase antibodies but normal thyroid function have a nearly fourfold higher risk of miscarriage and twofold higher risk of preterm birth, compared with women who don’t have the antibodies.
In the new double-blind study, 952 women with thyroid peroxidase antibodies, normal thyroid function, and a history of miscarriage or infertility were randomized either to daily 50 mcg levothyroxine or placebo, taken from conception to the end of pregnancy.
The rate of pregnancy was similar in the levothyroxine and placebo groups (56.6% vs. 58.3%, respectively), as was the live birth rate (37.4% vs. 37.9%), despite the observation that the levothyroxine group had consistently lower serum thyrotropin and higher free T4 concentrations than did the placebo group.
There were also no significant differences between the two groups in secondary outcomes of miscarriage, preterm birth, or neonatal outcomes such as birth weight.
Researchers also saw no statistically significant differences in the rate of serious adverse events or in the number of women who showed abnormal results on thyroid function tests.
The authors noted that the dosage of levothyroxine used in the study was fixed, leaving the possibility that “the dose may need to be adjusted depending on the participant’s body weight, thyroid peroxidase antibody level, or thyrotropin concentration.”
Existing guidelines from the American Thyroid Association acknowledge the lack of evidence in favor of levothyroxine decreasing the risk of pregnancy loss. However, the guidelines also state that it can be considered in antibody-positive, euthyroid women with a history of loss, “given its potential benefits in comparison with its minimal risk.”
The study was supported by the National Institute for Health Research. No conflicts of interest were declared.
SOURCE: Dhillon-Smith R et al. N Engl J Med. 2019 March 23. doi: 10.1056/NEJMoa1812537
FROM ENDO 2019
Key clinical point:
Major finding: Pregnancy rates and outcomes were similar in women treated with levothyroxine and those treated with placebo.
Study details: Double-blind, randomized, placebo-controlled trial in 952 women.
Disclosures: The study was supported by the National Institute for Health Research. No conflicts of interest were declared.
Source: Dhillon-Smith R et al. N Engl J Med. 2019 March 23. doi: 10.1056/NEJMoa1812537
Newer antihyperglycemic drugs have distinctive CV, kidney benefits
The two newer classes of antihyperglycemic drugs that lower cardiovascular risk have different effects on specific cardiovascular and kidney disease outcomes in patients with type 2 diabetes, results of a meta-analysis suggest. Sodium-glucose contransporter-2 (SGLT2) inhibitors significantly reduced hospitalization from heart failure, whereas glucagon-like peptide-1 receptor agonists (GLP-1 RAs) did not, according to the reported results.
The GLP-1–RA class reduced risk of kidney disease progression, largely driven by a reduction in macroalbuminuria, according to the authors, whereas only the SGLT2 inhibitors reduced adverse kidney disease outcomes in a composite excluding that biomarker.
“The prevention of heart failure and progression of kidney disease by SGLT2 [inhibitors] should be considered in the decision-making process when treating patients with type 2 diabetes,” study senior author Marc S. Sabatine, MD, MPH, of Brigham and Women’s Hospital, Boston, and his coauthors wrote in a report on the study appearing in Circulation.
Both GLP-1 RAs and SGLT2 inhibitors significantly reduced major adverse cardiovascular events (MACE) and, as shown in other recent findings, their benefits were confined to patients with established atherosclerotic cardiovascular disease, Dr. Sabatine and his colleagues wrote.
The systematic review and meta-analysis of eight cardiovascular outcomes trials included 77,242 patients, of whom about 56% participated in GLP-1–RA studies and 44% in SGLT2-inhibitor trials. Just under three-quarters of the patients had established atherosclerotic cardiovascular disease, while the remainder had multiple risk factors for it.
Relative risk of hospitalization for heart failure was reduced by 31% with SGLT2 inhibitors, but it was not significantly reduced by GLP-1 RAs, the authors noted.
Risk of kidney disease progression was reduced by 38% with SGLT2 inhibitors and by 18% with GLP-1 RAs when the researchers used a broad composite endpoint including macroalbuminuria, estimated glomerular filtration rate (eGFR), end-stage kidney disease, and death due to renal causes.
By contrast, SGLT2 inhibitors reduced by 45% the relative risk of a narrower kidney outcome that excluded macroalbuminuria, whereas GLP-1 RAs had only a nonsignificant effect on the risk of doubling serum creatinine. That suggests the relative risk reduction of the kidney composite with GLP-1 RAs was driven mainly by a reduction in macroalbuminuria, the authors wrote.
Although albuminuria is an established biomarker for kidney and cardiovascular disease, it is a surrogate marker and can even be absent in patients with reduced eGFR, they said.
“Reduction in eGFR has emerged as a more meaningful endpoint of greater importance and is used in ongoing diabetes trials for kidney outcomes,” the authors said in a discussion of their results.
Relative risk of the composite MACE endpoint, including myocardial infarction, stroke, and cardiovascular death, was reduced by 12% for GLP-1 RAs and by 11% for SGLT2 [inhibitors], according to results of the analysis. However, the benefit was confined to patients with established cardiovascular disease, who had a 14% reduction of risk, compared with no treatment effect in patients who had multiple risk factors only.
Looking at individual MACE components, investigators found that both drug classes significantly reduced relative risk of myocardial infarction and of cardiovascular death, whereas only GLP-1 RAs significantly reduced relative risk of stroke.
Study authors provided disclosures related to AstraZeneca, Amgen, Daiichi-Sankyo, Eisai, GlaxoSmithKline, Intarcia, Janssen Research and Development, and Medimmune, among others.
SOURCE: Zelniker TA et al. Circulation. 2019 Feb 21. doi: 10.1161/CIRCULATIONAHA.118.038868.
The two newer classes of antihyperglycemic drugs that lower cardiovascular risk have different effects on specific cardiovascular and kidney disease outcomes in patients with type 2 diabetes, results of a meta-analysis suggest. Sodium-glucose contransporter-2 (SGLT2) inhibitors significantly reduced hospitalization from heart failure, whereas glucagon-like peptide-1 receptor agonists (GLP-1 RAs) did not, according to the reported results.
The GLP-1–RA class reduced risk of kidney disease progression, largely driven by a reduction in macroalbuminuria, according to the authors, whereas only the SGLT2 inhibitors reduced adverse kidney disease outcomes in a composite excluding that biomarker.
“The prevention of heart failure and progression of kidney disease by SGLT2 [inhibitors] should be considered in the decision-making process when treating patients with type 2 diabetes,” study senior author Marc S. Sabatine, MD, MPH, of Brigham and Women’s Hospital, Boston, and his coauthors wrote in a report on the study appearing in Circulation.
Both GLP-1 RAs and SGLT2 inhibitors significantly reduced major adverse cardiovascular events (MACE) and, as shown in other recent findings, their benefits were confined to patients with established atherosclerotic cardiovascular disease, Dr. Sabatine and his colleagues wrote.
The systematic review and meta-analysis of eight cardiovascular outcomes trials included 77,242 patients, of whom about 56% participated in GLP-1–RA studies and 44% in SGLT2-inhibitor trials. Just under three-quarters of the patients had established atherosclerotic cardiovascular disease, while the remainder had multiple risk factors for it.
Relative risk of hospitalization for heart failure was reduced by 31% with SGLT2 inhibitors, but it was not significantly reduced by GLP-1 RAs, the authors noted.
Risk of kidney disease progression was reduced by 38% with SGLT2 inhibitors and by 18% with GLP-1 RAs when the researchers used a broad composite endpoint including macroalbuminuria, estimated glomerular filtration rate (eGFR), end-stage kidney disease, and death due to renal causes.
By contrast, SGLT2 inhibitors reduced by 45% the relative risk of a narrower kidney outcome that excluded macroalbuminuria, whereas GLP-1 RAs had only a nonsignificant effect on the risk of doubling serum creatinine. That suggests the relative risk reduction of the kidney composite with GLP-1 RAs was driven mainly by a reduction in macroalbuminuria, the authors wrote.
Although albuminuria is an established biomarker for kidney and cardiovascular disease, it is a surrogate marker and can even be absent in patients with reduced eGFR, they said.
“Reduction in eGFR has emerged as a more meaningful endpoint of greater importance and is used in ongoing diabetes trials for kidney outcomes,” the authors said in a discussion of their results.
Relative risk of the composite MACE endpoint, including myocardial infarction, stroke, and cardiovascular death, was reduced by 12% for GLP-1 RAs and by 11% for SGLT2 [inhibitors], according to results of the analysis. However, the benefit was confined to patients with established cardiovascular disease, who had a 14% reduction of risk, compared with no treatment effect in patients who had multiple risk factors only.
Looking at individual MACE components, investigators found that both drug classes significantly reduced relative risk of myocardial infarction and of cardiovascular death, whereas only GLP-1 RAs significantly reduced relative risk of stroke.
Study authors provided disclosures related to AstraZeneca, Amgen, Daiichi-Sankyo, Eisai, GlaxoSmithKline, Intarcia, Janssen Research and Development, and Medimmune, among others.
SOURCE: Zelniker TA et al. Circulation. 2019 Feb 21. doi: 10.1161/CIRCULATIONAHA.118.038868.
The two newer classes of antihyperglycemic drugs that lower cardiovascular risk have different effects on specific cardiovascular and kidney disease outcomes in patients with type 2 diabetes, results of a meta-analysis suggest. Sodium-glucose contransporter-2 (SGLT2) inhibitors significantly reduced hospitalization from heart failure, whereas glucagon-like peptide-1 receptor agonists (GLP-1 RAs) did not, according to the reported results.
The GLP-1–RA class reduced risk of kidney disease progression, largely driven by a reduction in macroalbuminuria, according to the authors, whereas only the SGLT2 inhibitors reduced adverse kidney disease outcomes in a composite excluding that biomarker.
“The prevention of heart failure and progression of kidney disease by SGLT2 [inhibitors] should be considered in the decision-making process when treating patients with type 2 diabetes,” study senior author Marc S. Sabatine, MD, MPH, of Brigham and Women’s Hospital, Boston, and his coauthors wrote in a report on the study appearing in Circulation.
Both GLP-1 RAs and SGLT2 inhibitors significantly reduced major adverse cardiovascular events (MACE) and, as shown in other recent findings, their benefits were confined to patients with established atherosclerotic cardiovascular disease, Dr. Sabatine and his colleagues wrote.
The systematic review and meta-analysis of eight cardiovascular outcomes trials included 77,242 patients, of whom about 56% participated in GLP-1–RA studies and 44% in SGLT2-inhibitor trials. Just under three-quarters of the patients had established atherosclerotic cardiovascular disease, while the remainder had multiple risk factors for it.
Relative risk of hospitalization for heart failure was reduced by 31% with SGLT2 inhibitors, but it was not significantly reduced by GLP-1 RAs, the authors noted.
Risk of kidney disease progression was reduced by 38% with SGLT2 inhibitors and by 18% with GLP-1 RAs when the researchers used a broad composite endpoint including macroalbuminuria, estimated glomerular filtration rate (eGFR), end-stage kidney disease, and death due to renal causes.
By contrast, SGLT2 inhibitors reduced by 45% the relative risk of a narrower kidney outcome that excluded macroalbuminuria, whereas GLP-1 RAs had only a nonsignificant effect on the risk of doubling serum creatinine. That suggests the relative risk reduction of the kidney composite with GLP-1 RAs was driven mainly by a reduction in macroalbuminuria, the authors wrote.
Although albuminuria is an established biomarker for kidney and cardiovascular disease, it is a surrogate marker and can even be absent in patients with reduced eGFR, they said.
“Reduction in eGFR has emerged as a more meaningful endpoint of greater importance and is used in ongoing diabetes trials for kidney outcomes,” the authors said in a discussion of their results.
Relative risk of the composite MACE endpoint, including myocardial infarction, stroke, and cardiovascular death, was reduced by 12% for GLP-1 RAs and by 11% for SGLT2 [inhibitors], according to results of the analysis. However, the benefit was confined to patients with established cardiovascular disease, who had a 14% reduction of risk, compared with no treatment effect in patients who had multiple risk factors only.
Looking at individual MACE components, investigators found that both drug classes significantly reduced relative risk of myocardial infarction and of cardiovascular death, whereas only GLP-1 RAs significantly reduced relative risk of stroke.
Study authors provided disclosures related to AstraZeneca, Amgen, Daiichi-Sankyo, Eisai, GlaxoSmithKline, Intarcia, Janssen Research and Development, and Medimmune, among others.
SOURCE: Zelniker TA et al. Circulation. 2019 Feb 21. doi: 10.1161/CIRCULATIONAHA.118.038868.
FROM CIRCULATION
A paraneoplastic potassium and acid-base disturbance
NOTE: The scenario presented here is partly based on cases reported elsewhere by Martínez-Valles et al1 and Fernández-Rodríguez et al.2
A 55-year-old man is admitted to the hospital with generalized malaise, paresthesias, and severe hypertension. He says he had experienced agitation along with weakness on exertion 24 hours before presentation to the emergency department, with subsequent onset of paresthesias in his lower extremities and perioral area.
He is already known to have mild chronic obstructive pulmonary disease, with a ratio of forced expiratory volume in 1 second (FEV1)to forced vital capacity (FVC) of less than 70% and an FEV1 85% of predicted. In addition, he was recently diagnosed with diabetes, resistant hypertension requiring maximum doses of 3 agents (a calcium channel blocker, an angiotensin-converting enzyme inhibitor, and a loop diuretic), and hyperlipidemia.
He is a current smoker with a 30-pack-year smoking history. He does not use alcohol. His family history is noncontributory.

ASSESSING ACID-BASE DISORDERS
1. What type of acid-base disorder does this patient have?
- Metabolic acidosis
- Respiratory acidosis
- Metabolic alkalosis
- Respiratory alkalosis
The patient has metabolic alkalosis.
A 5-step approach

1. Acidosis or alkalosis? The patient’s arterial pH is 7.5, which is alkalemic because it is higher than 7.44.
2. Metabolic or respiratory? The primary process in our patient is overwhelmingly metabolic, as his partial pressure of carbon dioxide (Pco2) is slightly elevated, a direction that would cause acidosis, not alkalosis.
3. The anion gap (the serum sodium concentration minus the sum of the chloride and bicarbonate concentrations) is normal at 8 mmol/L (DRG:HYBRiD-XL Immunoassay and Clinical Chemistry Analyzer, reference range 8–16).
4. Is the disturbance compensated? We have determined that this patient has a metabolic alkalemia; the question now is whether there is any compensation for the primary disturbance.
In metabolic alkalosis, the Pco2 may increase by approximately 0.6 mm Hg (range 0.5–0.8) above the nominal normal level of 40 mm Hg for each 1-mmol/L increase in bicarbonate above the nominal normal level of 25 mmol/L.4 If the patient requires oxygen, the calculation may be unreliable, however, as hypoxemia may have an overriding influence on respiratory drive.
Patients with chronically high Pco2 levels such as those with chronic obstructive pulmonary disease can become accustomed to high carbon dioxide levels and lose their hyper-
capnic respiratory drive. Giving oxygen supplementation is thought to decrease respiratory drive in these patients, so that they will breathe slower and retain more carbon dioxide. There is some degree of respiratory compensation for metabolic alkalosis that occurs by breathing less, though it is limited overall—even in very alkalotic patients, breathing less results in CO2 retention, which, by displacing O2 molecules in the alveoli, will in turn result in hypoxia. The brain then senses the hypoxia and makes one breathe faster, thereby limiting this compensation.
This patient’s serum bicarbonate level is 40 mmol/L, or 15 mmol/L higher than the nominal normal level. If he is compensating, his Pco2 should be 40 + (15 × 0.6) = 49 mm Hg, and in fact it is 51 mm Hg, which is within the normal range of expected compensation (47.5–52 mm Hg). Therefore, yes, he is compensating for the primary disturbance.
5. In metabolic acidosis, is there a delta gap? As our patient has metabolic alkalosis, not acidosis, this question does not apply in this case.
WHICH TEST TO FIND THE CAUSE?
2. Which is the best test to order next to determine the cause of this patient’s hypokalemic metabolic alkalosis?
- Serum magnesium level
- Spot urine chloride
- Renal ultrasonography
- 24-hour urine collection for sodium, potassium, and chloride

The patient’s loop diuretic is withheld for 12 hours and a spot urine chloride is obtained, which is reported as 44 mmol/L. This high value suggests that a volume-independent hypokalemic metabolic alkalosis is present with potassium depletion.
As for the other answer choices:
Serum magnesium. Though hypomagnesemia can cause hypokalemia due to lack of inhibition of renal outer medullary potassium channels and subsequent increased excretion of potassium in the apical tubular membrane, it is not independently associated with acid-base disturbances.5
Renal ultrasonography gives information about structural kidney disease but is of limited utility in identifying the cause of hypokalemic metabolic alkalosis.
A 24-hour urine collection is unnecessary in this setting and would ultimately result in delay in diagnosis, as spot urine chloride is a more efficient means of rapidly distinguishing volume-responsive vs volume-independent causes of hypokalemic metabolic alkalosis.6
IS HIS HYPERTENSION SECONDARY? IF SO, WHAT IS THE CAUSE?
Several features of this case suggest that the patient’s hypertension is secondary rather than primary. It is of recent onset. The patient’s family history is noncontributory, and his hypertension is resistant to the use of maximum doses of 3 antihypertensive agents.
3. Which of the following causes of secondary hypertension is not commonly associated with hypokalemia and metabolic alkalosis?
- Hyperaldosteronism
- Liddle syndrome
- Cushing syndrome
- Renal parenchymal disease
- Chronic licorice ingestion
Renal parenchymal disease is a cause of resistant hypertension, but it is not characterized by metabolic alkalosis, hypokalemia, and elevated urine chloride,7 while the others listed here—hyperaldosteronism, Liddle syndrome, Cushing syndrome, and chronic licorice ingestion—are. Other common causes of resistant hypertension without these metabolic abnormalities include obstructive sleep apnea, alcohol abuse, and nonadherence to treatment.
While treatment of hypertension with loop diuretics can result in hypokalemia and metabolic alkalosis due to the effect of these drugs on potassium reabsorption in the loop of Henle, the patient’s hypokalemia persisted after this agent was withdrawn.8
Causes of hypokalemic metabolic alkalosis with and without hypertension are further delineated in Figure 1.
Additional diagnostic testing: Plasma renin and plasma aldosterone
At this juncture, the differential diagnosis for this patient’s potassium depletion, metabolic alkalosis, high urine chloride, and hypertension has been narrowed to primary or secondary hyperaldosteronism, surreptitious mineralocorticoid ingestion, Cushing syndrome, licorice ingestion, Liddle syndrome, or one of the 3 hydroxylase deficiencies (11-, 17-, and 21-) (Figure 1).

Although clues in the history, physical examination, and imaging may suggest a specific cause of his abnormal laboratory values, the next step in the diagnostic workup is to measure the plasma renin and aldosterone levels (Table 3).
HYPERALDOSTERONISM
4. Hyperaldosteronism is associated with which of the following patterns of renin and aldosterone values?
- High renin, high aldosterone, normal ratio of plasma aldosterone concentration (PAC) to plasma renin activity (PRA)
- Low renin, low aldosterone, normal PAC–PRA ratio
- Low renin, high aldosterone, high PAC–PRA ratio
- High renin, low aldosterone, low PAC–PRA ratio
The pattern of low renin, high aldosterone, and high PAC–PRA ratio is associated with hyperaldosteronism.
Primary hyperaldosteronism
Primary hyperaldosteronism is one of the most common causes of resistant hypertension and is underappreciated, being diagnosed in up to 20% of patients referred to hypertension specialty clinics.7 Potassium levels may be normal, likely contributing to its lack of recognition in this target population.
Primary hyperaldosteronism should be suspected in patients who have a plasma aldosterone PAC–PRA ratio greater than 20 with elevated plasma aldosterone concentrations
(> 15 ng/dL).
Persistently elevated aldosterone levels in the setting of elevated plasma volume is proof that aldosterone secretion is independent of the renin-angiotensin-aldosterone axis, and therefore is autonomous (secondary to adrenal tumor or hyperplasia). Further testing in the form of oral salt loading, saline infusion, or fludrocortisone (a sodium-retaining steroid) administration is thus required to confirm inappropriate, autonomous aldosterone secretion.9
After establishing the diagnosis of primary hyperaldosteronism, one should determine the subtype (ie, due to an adrenal carcinoma, unilateral hypersecreting adenoma, or unilateral or bilateral hyperplasia). Further testing includes adrenal computed tomography (CT) to rule out adrenal carcinomas, which are suspected with adenomas larger than 4 cm. Though part of the diagnostic workup, CT as a means of confirmational testing alone does not preclude the possibility of bilateral adrenal hyperplasia in some patients, even in the presence of an adrenal adenoma. For this reason, adrenal venous sampling is required to definitively determine whether the condition is due to a hypersecreting adrenal adenoma or unilateral or bilateral hyperplasia.9,10
Treatment of primary hyperaldosteronism depends on the subtype of the disease and involves salt restriction in addition to an aldosterone antagonist (spironolactone or eplerenone in the case of bilateral disease) or surgery (unilateral disease).9,11,12
Secondary hyperaldosteronism
Secondary hyperaldosteronism should be suspected when plasma renin and aldosterone levels are both elevated with a PAC–PRA ratio less than 10.
This pattern is most commonly seen with diuretic use but can also be a consequence of renal artery stenosis or, rarely, a renin-secreting tumor.13 Renal artery stenosis is a common finding in patients with hypertension undergoing cardiac catheterization, which is not surprising as more than 90% of such stenoses are atherosclerotic.7 Renin-secreting tumors are exceedingly rare, with fewer than 100 cases reported in the literature, and are more common in younger individuals.13
Our patient has low-normal aldosterone and plasma renin
On further testing, this patient’s plasma aldosterone level is 2.55 ng/dL (normal < 15 ng/dL), his plasma renin activity is 0.53 ng/mL/hour (normal 0.2–2.8 ng/mL/hour), and his PAC–PRA ratio is therefore 4.81.
The categories discussed thus far have included primary and secondary hyperaldosteronism, which typically do not present with low to normal levels of both renin and aldosterone. Surreptitious mineralocorticoid use could present in this manner, but is unlikely in this patient, whose medications do not include fludrocortisone.
The low-normal values thus lead to consideration of a third category: apparent mineralocorticoid excess. Diseases in this category such as Cushing disease or adrenocorticotropic hormone (ACTH) excess are characterized by increases in corticosteroids so that the potassium depletion, metabolic alkalosis, and hypertension are not a consequence of renin and aldosterone but rather the excess corticosteroids.14
Causes of apparent mineralocorticoid excess
There are several possible causes of mineralocorticoid excess associated with hypertension and hypokalemic metabolic alkalosis not due to renin and aldosterone.
Chronic licorice ingestion in high volumes is one such cause and is thought to result in inhibition of 11B-hydroxysteroid dehydrogenase or possibly cortisol oxidase by licorice’s active component, glycyrrhetinic acid. This inhibition results in an inability to convert cortisol to cortisone. The cortisol excess binds to mineralocorticoid receptors, and acting like aldosterone, results in hypertension and hypokalemic metabolic alkalosis as well as feedback inhibition of renin and aldosterone levels.15
Partial hydroxylase deficiencies, though rare, should also be considered as a cause of hypokalemic metabolic alkalosis, hypertension, and, potentially, hirsutism and clitoromegaly in women. They can be diagnosed with elevated levels of 17-ketosteroids and dehydroepiandrosterone sulfate, both of which, in excess, may act on aldosterone receptors in a manner similar to cortisol.16
Liddle syndrome, a rare autosomal dominant condition, may also present with suppressed levels of both renin and aldosterone. In contrast to the disorders of nonaldosterone mineralocorticoid excess, however, the sodium channel defect in Liddle syndrome is characterized by a primary increase in sodium reabsorption in the collecting tubule and potassium wasting. The resultant volume expansion leads to suppressed renin and aldosterone levels and hypertension with low potassium and elevated bicarbonate concentrations.17
Liddle syndrome is commonly diagnosed in childhood but may go unrecognized due to occasional absence of hypokalemia at presentation. Potassium-sparing diuretics such as amiloride or triamterene are the mainstays of treatment.18


Rates of cardiovascular and all-cause mortality are increased in patients with long-term hypercortisolism, even after plasma concentrations of cortisol are normalized.21
Figure 2 shows the cascade of the hypothalamic-pituitary-adrenal axis.
TESTING FOR HYPERCORTISOLISM IN OUR PATIENT
Given the patient’s clinical presentation and laboratory and imaging findings with normal plasma renin and aldosterone levels, a workup for suspected hypercortisolism is initiated.
Initial diagnostic testing for hypercortisolism depends on the degree of clinical suspicion. In those with low probability of the disease, testing should consist of 1 of the following, as a single negative test may be sufficient to rule out the disease:
- 24-hour urinary cortisol levels
- Overnight dexamethasone suppression testing
- Late-night salivary cortisol measurements.
In those with a high index of suspicion, 2 of the aforementioned tests should be performed, as 1 normal result may not be sufficient to exclude the diagnosis.22,23
A 24-hour urinary cortisol collection and overnight dexamethasone suppression test are obtained. His 24-hour urinary free cortisol level is elevated at 6,600 µg (normal 4–100), and suppression testing with 8 mg of dexamethasone (a form of “high-dose” testing)demonstrates only an 8% decline in serum cortisol levels. Cortisol should generally drop more than 90%.
Morning serum cortisol concentration is less than 5 µg/dL (140 nmol/L) in most patients with Cushing disease (ie, a pituitary tumor), and is usually undetectable in normal subjects. Only about 50% of neuroendocrine ACTH-secreting tumors will suppress with this test.
The patient’s clinical presentation, in conjunction with his diagnostic testing, are thus consistent with Cushing syndrome.
CUSHING SYNDROME
Cushing syndrome is most often exogenous or iatrogenic, ie, a result of supraphysiologic doses of glucocorticoids used to treat a variety of inflammatory, autoimmune, and neoplastic conditions.
Endogenous Cushing syndrome, on the other hand, is rare, with an estimated prevalence of 0.7 to 2.4 cases per million per year. ACTH-dependent causes account for 80% of endogenous Cushing syndrome cases, with ACTH-secreting pituitary adenomas (Cushing disease) accounting for 75% to 80% and ectopic ACTH secretion accounting for 15% to 20%. Less than 1% of cases are due to tumors that produce corticotropin-releasing hormone (CRH).
ACTH-independent Cushing syndrome is diagnosed in 20% of endogenous cases and is most commonly caused by a unilateral adrenal tumor. Rare causes of ACTH-independent disease include adrenal carcinoma, McCune-Albright syndrome, and adrenal hyperplasia.24
The patient’s ACTH is high
To determine whether this is an ACTH-dependent or independent process, the next step is to order an ACTH level. His ACTH level is high at 107 pg/mL (normal < 46 pg/mL), confirming the diagnosis of ACTH-dependent Cushing syndrome.
To find out if this ACTH-dependent process is due to a pituitary adenoma, magnetic resonance imaging (MRI) of the pituitary is obtained but is normal.
Large masses (> 6 mm) strongly suggest Cushing disease, but these tumors are often small and may be missed even with more advanced imaging techniques. Corticotropin-secreting adenomas arising from normal cells in the pituitary retain some sensitivity to glucocorticoid negative feedback and CRH stimulation, and thus high-dose dexamethasone suppression testing in conjunction with CRH stimulation testing can be used to differentiate Cushing disease from ectopic ACTH secretion.24,25 Both of these tests have poor diagnostic accuracy, however, and thus inferior petrosal sampling remains the gold standard for the diagnosis of Cushing disease.

ACTH-SECRETING TUMORS
5. Cushing syndrome due to ectopic ACTH secretion is most commonly attributed to which of the following tumors?
- Small-cell lung carcinoma
- Pancreatic carcinoma
- Medullary thyroid carcinoma
- Gastrinoma
Severe cases of Cushing syndrome are often attributable to ectopic ACTH secretion due to an underlying malignancy, most commonly small-cell lung carcinoma or neuroendocrine tumors of pulmonary origin. Other causes include pancreatic and thymic neuroendocrine tumors, gastrinomas, and medullary thyroid carcinoma.25,26
Because most ACTH-producing tumors are intrathoracic, initial imaging in cases of suspected ectopic ACTH secretion should focus on the chest, with CT the usual first choice. Octreotide scintigraphy can also be useful in localizing disease, as many neuroendocrine tumors express somatostatin receptors. Specialized positron-emission tomography scans may also be helpful in tumor identification.24
TREATMENT OF CUSHING SYNDROME DUE TO ECTOPIC ACTH SECRETION
6. Which of the following is most appropriate medical therapy for suppression of cortisol secretion in Cushing syndrome due to ectopic ACTH secretion?
- Spironolactone
- Dexamethasone
- Somatostatin
- Estrogen
- Ketoconazole
Hyperglycemia, hypokalemia, hypertension, psychiatric disturbances, venous thromboembolism, and systemic infections appear to be common in ectopic ACTH syndrome and often correlate with the degree of hypercortisolemia. Severe Cushing syndrome due to ectopic ACTH secretion is an emergency requiring prompt control of cortisol secretion.
First-line treatments include steroidogenesis inhibitors (ketoconazole, metyrapone, etomidate, mitotane) and glucocorticoid receptor antagonists (mifepristone). High-dose spironolactone and eplerenone can also be used to treat the hypertension and hypokalemia associated with mineralocorticoid receptor stimulation. Definitive treatment involves surgical resection, chemotherapy, or radiotherapy when applicable.24,25
After confirmation of the diagnosis, the patient is prescribed ketoconazole and spironolactone, with substantial improvement. He subsequently is started on combination chemotherapy and radiation therapy for his small-cell lung carcinoma.
DISCUSSION
The differential diagnosis for hypokalemia is broad and relies on information obtained during the history and physical examination, followed by interpretation of selected laboratory results. Myriad pathologies in diverse organ systems, eg, diarrhea, renal tubular acidosis, and adrenal disease, may be responsible for a low serum potassium. Further categorizing potassium depletion on the basis of an associated acid-base disturbance, such as metabolic alkalosis, allows one to use an algorithmic approach that can identify specific etiologies responsible for both the potassium and the acid-base disturbances.
Using the spot urine chloride in the setting of hypokalemic metabolic alkalosis with or without hypertension can narrow the differential diagnosis and allow additional clinical findings to guide clinical problem-solving and decision-making, even for conditions not commonly encountered in routine medical practice.
Obtaining renin and aldosterone measurements in patients with potassium depletion, metabolic alkalosis, high urine chloride excretion, and hypertension permits further categorization into 3 clinical groups: elevated aldosterone and renin (secondary hyperaldosteronism), elevated aldosterone and low renin (primary hyperaldosteronism), or apparent mineralocorticoid excess wherein neither renin nor aldosterone are responsible for the syndrome.
The patient in our case had apparent mineralocorticoid excess as a consequence of an ACTH-producing small-cell carcinoma.
- Martínez-Valles MA, Palafox-Cazarez A, Paredes-Avina JA. Severe hypokalemia, metabolic alkalosis and hypertension in a 54 year old male with ectopic ACTH syndrome: a case report. Cases J 2009; 2:6174. doi:10.4076/1757-1626-2-6174
- Fernández-Rodríguez E, Villar-Taibo R, Pinal-Osorio I, et al. Severe hypertension and hypokalemia as first clinical manifestations in ectopic Cushing’s syndrome. Arq Bras Endocrinol Metabol 2008; 52(6):1066–1070. pmid:18820819
- Mani S, Rutecki GW. A patient with altered mental status and an acid-base disturbance. Cleve Clin J Med 2017; 84(1):27–34. doi:10.3949/ccjm.84a.16042
- Adrogué HJ, Madias NE. Secondary responses to altered acid-base status: the rules of engagement. J Am Soc Nephrol 2010; 21(6):920–923. doi:10.1681/ASN.2009121211
- Huang CL, Kuo E. Mechanism of hypokalemia in magnesium deficiency. J Am Soc Nephrol 2007; 18(10):2649–2652. doi:10.1681/ASN.2007070792
- Rose BD. Metabolic alkalosis. In: Clinical Physiology of Acid-Base and Electrolyte Disorders. 4th ed. New York, NY: McGraw-Hill, Health Professions Division; 1994:515.
- Calhoun DA, Jones D, Textor S, et al; American Heart Association Professional Education Committee. Resistant hypertension: diagnosis, evaluation, and treatment: a scientific statement from the American Heart Association Professional Education Committee of the Council for High Blood Pressure Research. Circulation 2008; 117(25):e510–e526. doi:10.1161/CIRCULATIONAHA.108.189141
- Koeppen BM, Stanton BA. Physiology of diuretic action. In: Renal Physiology. 5th ed. Philadelphia, PA: Elsevier Inc; 2013:167–178.
- Blumenfeld JD, Sealey JE, Schlussel Y, et al. Diagnosis and treatment of primary hyperaldosteronism. Ann Intern Med 1994; 121(11):877–885. pmid:7978702
- Kempers MJ, Lenders JW, van Outheusden L, et al. Systematic review: diagnostic procedures to differentiate unilateral from bilateral adrenal abnormality in primary aldosteronism. Ann Intern Med 2009; 151(5):329–337. pmid:19721021
- Karagiannis A, Tziomalos K, Papageorgiou A, et al. Spironolactone versus eplerenone for the treatment of idiopathic hyperaldosteronism. Expert Opin Pharmacother 2008; 9(4):509–515. doi:10.1517/14656566.9.4.509
- Sawka AM, Young WF, Thompson GB, et al. Primary aldosteronism: factors associated with normalization of blood pressure after surgery. Ann Intern Med 2001; 135(4):258–261. pmid:11511140
- Haab F, Duclos JM, Guyenne T, Plouin PF, Corvol P. Renin secreting tumors: diagnosis, conservative surgical approach and long-term results. J Urol 1995; 153(6):1781–1784. pmid:7752315
- Sabbadin C, Armanini D. Syndromes that mimic an excess of mineralocorticoids. High Blood Press Cardiovasc Prev 2016; 23(3):231–235. doi:10.1007/s40292-016-0160-5
- Apostolakos JM, Caines LC. Apparent mineralocorticoid excess syndrome: a case of resistant hypertension from licorice tea consumption. J Clin Hypertens (Greenwich) 2016; 18(10):991–993. doi:10.1111/jch.12841
- Glatt K, Garzon DL, Popovic J. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Spec Pediatr Nurs 2005; 10(3):104–114. doi:10.1111/j.1744-6155.2005.00022.x
- Findling JW, Raff H, Hansson JH, Lifton RP. Liddle’s syndrome: prospective genetic screening and suppressed aldosterone secretion in an extended kindred. J Clin Endocrinol Metab 1997; 82(4):1071–1074. doi:10.1210/jcem.82.4.3862
- Wang C, Chan TK, Yeung RT, Coghlan JP, Scoggins BA, Stockigt JR. The effect of triamterene and sodium intake on renin, aldosterone, and erythrocyte sodium transport in Liddle’s syndrome. J Clin Endocrinol Metab 1981; 52(5):1027–1032. doi:10.1210/jcem-52-5-1027
- Torpy DJ, Mullen N, Ilias I, Nieman LK. Association of hypertension and hypokalemia with Cushing’s syndrome caused by ectopic ACTH secretion: a series of 58 cases. Ann N Y Acad Sci 2002; 970:134–144. pmid:12381548
- Saruta T, Suzuki H, Handa M, Igarashi Y, Kondo K, Senba S. Multiple factors contribute to the pathogenesis of hypertension in Cushing’s syndrome. J Clin Endocrinol Metab 1986; 62(2):275–279. doi:10.1210/jcem-62-2-275
- Clayton RN, Jones PW, Reulen RC, et al. Mortality in patients with Cushing’s disease more than 10 years after remission: a multicentre, multinational, retrospective cohort study. Lancet Diabetes Endocrinol 2016; 4(7):569–576. doi:10.1016/S2213-8587(16)30005-5
- Baid SK, Rubino D, Sinaii N, Ramsey S, Frank A, Nieman LK. Specificity of screening tests for Cushing’s syndrome in an overweight and obese population. J Clin Endocrinol Metab 2009; 94(10):3857–3864. doi:10.1210/jc.2008-2766
- Nieman LK, Biller BM, Findling JW, et al. The diagnosis of Cushing’s syndrome: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab 2008; 93(5):1526–1540. doi:10.1210/jc.2008-0125
- Sharma ST, Nieman LK, Feelders RA. Cushing’s syndrome: epidemiology and developments in disease management. Clin Epidemiol 2015; 7:281–293. doi:10.2147/CLEP.S44336
- Tavares Bello C, van der Poest Clement E, Feelders R. Severe Cushing’s syndrome and bilateral pulmonary nodules: beyond ectopic ACTH. Endocrinol Diabetes Metab Case Rep 2017; pii:17–0100. doi:10.1530/EDM-17-0100
- Sathyakumar S, Paul TV, Asha HS, et al. Ectopic Cushing syndrome: a 10-year experience from a tertiary care center in southern India. Endocr Pract 2017; 23(8):907–914. doi:10.4158/EP161677.OR
NOTE: The scenario presented here is partly based on cases reported elsewhere by Martínez-Valles et al1 and Fernández-Rodríguez et al.2
A 55-year-old man is admitted to the hospital with generalized malaise, paresthesias, and severe hypertension. He says he had experienced agitation along with weakness on exertion 24 hours before presentation to the emergency department, with subsequent onset of paresthesias in his lower extremities and perioral area.
He is already known to have mild chronic obstructive pulmonary disease, with a ratio of forced expiratory volume in 1 second (FEV1)to forced vital capacity (FVC) of less than 70% and an FEV1 85% of predicted. In addition, he was recently diagnosed with diabetes, resistant hypertension requiring maximum doses of 3 agents (a calcium channel blocker, an angiotensin-converting enzyme inhibitor, and a loop diuretic), and hyperlipidemia.
He is a current smoker with a 30-pack-year smoking history. He does not use alcohol. His family history is noncontributory.
ASSESSING ACID-BASE DISORDERS
1. What type of acid-base disorder does this patient have?
- Metabolic acidosis
- Respiratory acidosis
- Metabolic alkalosis
- Respiratory alkalosis
The patient has metabolic alkalosis.
A 5-step approach
1. Acidosis or alkalosis? The patient’s arterial pH is 7.5, which is alkalemic because it is higher than 7.44.
2. Metabolic or respiratory? The primary process in our patient is overwhelmingly metabolic, as his partial pressure of carbon dioxide (Pco2) is slightly elevated, a direction that would cause acidosis, not alkalosis.
3. The anion gap (the serum sodium concentration minus the sum of the chloride and bicarbonate concentrations) is normal at 8 mmol/L (DRG:HYBRiD-XL Immunoassay and Clinical Chemistry Analyzer, reference range 8–16).
4. Is the disturbance compensated? We have determined that this patient has a metabolic alkalemia; the question now is whether there is any compensation for the primary disturbance.
In metabolic alkalosis, the Pco2 may increase by approximately 0.6 mm Hg (range 0.5–0.8) above the nominal normal level of 40 mm Hg for each 1-mmol/L increase in bicarbonate above the nominal normal level of 25 mmol/L.4 If the patient requires oxygen, the calculation may be unreliable, however, as hypoxemia may have an overriding influence on respiratory drive.
Patients with chronically high Pco2 levels such as those with chronic obstructive pulmonary disease can become accustomed to high carbon dioxide levels and lose their hyper-
capnic respiratory drive. Giving oxygen supplementation is thought to decrease respiratory drive in these patients, so that they will breathe slower and retain more carbon dioxide. There is some degree of respiratory compensation for metabolic alkalosis that occurs by breathing less, though it is limited overall—even in very alkalotic patients, breathing less results in CO2 retention, which, by displacing O2 molecules in the alveoli, will in turn result in hypoxia. The brain then senses the hypoxia and makes one breathe faster, thereby limiting this compensation.
This patient’s serum bicarbonate level is 40 mmol/L, or 15 mmol/L higher than the nominal normal level. If he is compensating, his Pco2 should be 40 + (15 × 0.6) = 49 mm Hg, and in fact it is 51 mm Hg, which is within the normal range of expected compensation (47.5–52 mm Hg). Therefore, yes, he is compensating for the primary disturbance.
5. In metabolic acidosis, is there a delta gap? As our patient has metabolic alkalosis, not acidosis, this question does not apply in this case.
WHICH TEST TO FIND THE CAUSE?
2. Which is the best test to order next to determine the cause of this patient’s hypokalemic metabolic alkalosis?
- Serum magnesium level
- Spot urine chloride
- Renal ultrasonography
- 24-hour urine collection for sodium, potassium, and chloride
The patient’s loop diuretic is withheld for 12 hours and a spot urine chloride is obtained, which is reported as 44 mmol/L. This high value suggests that a volume-independent hypokalemic metabolic alkalosis is present with potassium depletion.
As for the other answer choices:
Serum magnesium. Though hypomagnesemia can cause hypokalemia due to lack of inhibition of renal outer medullary potassium channels and subsequent increased excretion of potassium in the apical tubular membrane, it is not independently associated with acid-base disturbances.5
Renal ultrasonography gives information about structural kidney disease but is of limited utility in identifying the cause of hypokalemic metabolic alkalosis.
A 24-hour urine collection is unnecessary in this setting and would ultimately result in delay in diagnosis, as spot urine chloride is a more efficient means of rapidly distinguishing volume-responsive vs volume-independent causes of hypokalemic metabolic alkalosis.6
IS HIS HYPERTENSION SECONDARY? IF SO, WHAT IS THE CAUSE?
Several features of this case suggest that the patient’s hypertension is secondary rather than primary. It is of recent onset. The patient’s family history is noncontributory, and his hypertension is resistant to the use of maximum doses of 3 antihypertensive agents.
3. Which of the following causes of secondary hypertension is not commonly associated with hypokalemia and metabolic alkalosis?
- Hyperaldosteronism
- Liddle syndrome
- Cushing syndrome
- Renal parenchymal disease
- Chronic licorice ingestion
Renal parenchymal disease is a cause of resistant hypertension, but it is not characterized by metabolic alkalosis, hypokalemia, and elevated urine chloride,7 while the others listed here—hyperaldosteronism, Liddle syndrome, Cushing syndrome, and chronic licorice ingestion—are. Other common causes of resistant hypertension without these metabolic abnormalities include obstructive sleep apnea, alcohol abuse, and nonadherence to treatment.
While treatment of hypertension with loop diuretics can result in hypokalemia and metabolic alkalosis due to the effect of these drugs on potassium reabsorption in the loop of Henle, the patient’s hypokalemia persisted after this agent was withdrawn.8
Causes of hypokalemic metabolic alkalosis with and without hypertension are further delineated in Figure 1.
Additional diagnostic testing: Plasma renin and plasma aldosterone
At this juncture, the differential diagnosis for this patient’s potassium depletion, metabolic alkalosis, high urine chloride, and hypertension has been narrowed to primary or secondary hyperaldosteronism, surreptitious mineralocorticoid ingestion, Cushing syndrome, licorice ingestion, Liddle syndrome, or one of the 3 hydroxylase deficiencies (11-, 17-, and 21-) (Figure 1).
Although clues in the history, physical examination, and imaging may suggest a specific cause of his abnormal laboratory values, the next step in the diagnostic workup is to measure the plasma renin and aldosterone levels (Table 3).
HYPERALDOSTERONISM
4. Hyperaldosteronism is associated with which of the following patterns of renin and aldosterone values?
- High renin, high aldosterone, normal ratio of plasma aldosterone concentration (PAC) to plasma renin activity (PRA)
- Low renin, low aldosterone, normal PAC–PRA ratio
- Low renin, high aldosterone, high PAC–PRA ratio
- High renin, low aldosterone, low PAC–PRA ratio
The pattern of low renin, high aldosterone, and high PAC–PRA ratio is associated with hyperaldosteronism.
Primary hyperaldosteronism
Primary hyperaldosteronism is one of the most common causes of resistant hypertension and is underappreciated, being diagnosed in up to 20% of patients referred to hypertension specialty clinics.7 Potassium levels may be normal, likely contributing to its lack of recognition in this target population.
Primary hyperaldosteronism should be suspected in patients who have a plasma aldosterone PAC–PRA ratio greater than 20 with elevated plasma aldosterone concentrations
(> 15 ng/dL).
Persistently elevated aldosterone levels in the setting of elevated plasma volume is proof that aldosterone secretion is independent of the renin-angiotensin-aldosterone axis, and therefore is autonomous (secondary to adrenal tumor or hyperplasia). Further testing in the form of oral salt loading, saline infusion, or fludrocortisone (a sodium-retaining steroid) administration is thus required to confirm inappropriate, autonomous aldosterone secretion.9
After establishing the diagnosis of primary hyperaldosteronism, one should determine the subtype (ie, due to an adrenal carcinoma, unilateral hypersecreting adenoma, or unilateral or bilateral hyperplasia). Further testing includes adrenal computed tomography (CT) to rule out adrenal carcinomas, which are suspected with adenomas larger than 4 cm. Though part of the diagnostic workup, CT as a means of confirmational testing alone does not preclude the possibility of bilateral adrenal hyperplasia in some patients, even in the presence of an adrenal adenoma. For this reason, adrenal venous sampling is required to definitively determine whether the condition is due to a hypersecreting adrenal adenoma or unilateral or bilateral hyperplasia.9,10
Treatment of primary hyperaldosteronism depends on the subtype of the disease and involves salt restriction in addition to an aldosterone antagonist (spironolactone or eplerenone in the case of bilateral disease) or surgery (unilateral disease).9,11,12
Secondary hyperaldosteronism
Secondary hyperaldosteronism should be suspected when plasma renin and aldosterone levels are both elevated with a PAC–PRA ratio less than 10.
This pattern is most commonly seen with diuretic use but can also be a consequence of renal artery stenosis or, rarely, a renin-secreting tumor.13 Renal artery stenosis is a common finding in patients with hypertension undergoing cardiac catheterization, which is not surprising as more than 90% of such stenoses are atherosclerotic.7 Renin-secreting tumors are exceedingly rare, with fewer than 100 cases reported in the literature, and are more common in younger individuals.13
Our patient has low-normal aldosterone and plasma renin
On further testing, this patient’s plasma aldosterone level is 2.55 ng/dL (normal < 15 ng/dL), his plasma renin activity is 0.53 ng/mL/hour (normal 0.2–2.8 ng/mL/hour), and his PAC–PRA ratio is therefore 4.81.
The categories discussed thus far have included primary and secondary hyperaldosteronism, which typically do not present with low to normal levels of both renin and aldosterone. Surreptitious mineralocorticoid use could present in this manner, but is unlikely in this patient, whose medications do not include fludrocortisone.
The low-normal values thus lead to consideration of a third category: apparent mineralocorticoid excess. Diseases in this category such as Cushing disease or adrenocorticotropic hormone (ACTH) excess are characterized by increases in corticosteroids so that the potassium depletion, metabolic alkalosis, and hypertension are not a consequence of renin and aldosterone but rather the excess corticosteroids.14
Causes of apparent mineralocorticoid excess
There are several possible causes of mineralocorticoid excess associated with hypertension and hypokalemic metabolic alkalosis not due to renin and aldosterone.
Chronic licorice ingestion in high volumes is one such cause and is thought to result in inhibition of 11B-hydroxysteroid dehydrogenase or possibly cortisol oxidase by licorice’s active component, glycyrrhetinic acid. This inhibition results in an inability to convert cortisol to cortisone. The cortisol excess binds to mineralocorticoid receptors, and acting like aldosterone, results in hypertension and hypokalemic metabolic alkalosis as well as feedback inhibition of renin and aldosterone levels.15
Partial hydroxylase deficiencies, though rare, should also be considered as a cause of hypokalemic metabolic alkalosis, hypertension, and, potentially, hirsutism and clitoromegaly in women. They can be diagnosed with elevated levels of 17-ketosteroids and dehydroepiandrosterone sulfate, both of which, in excess, may act on aldosterone receptors in a manner similar to cortisol.16
Liddle syndrome, a rare autosomal dominant condition, may also present with suppressed levels of both renin and aldosterone. In contrast to the disorders of nonaldosterone mineralocorticoid excess, however, the sodium channel defect in Liddle syndrome is characterized by a primary increase in sodium reabsorption in the collecting tubule and potassium wasting. The resultant volume expansion leads to suppressed renin and aldosterone levels and hypertension with low potassium and elevated bicarbonate concentrations.17
Liddle syndrome is commonly diagnosed in childhood but may go unrecognized due to occasional absence of hypokalemia at presentation. Potassium-sparing diuretics such as amiloride or triamterene are the mainstays of treatment.18
Rates of cardiovascular and all-cause mortality are increased in patients with long-term hypercortisolism, even after plasma concentrations of cortisol are normalized.21
Figure 2 shows the cascade of the hypothalamic-pituitary-adrenal axis.
TESTING FOR HYPERCORTISOLISM IN OUR PATIENT
Given the patient’s clinical presentation and laboratory and imaging findings with normal plasma renin and aldosterone levels, a workup for suspected hypercortisolism is initiated.
Initial diagnostic testing for hypercortisolism depends on the degree of clinical suspicion. In those with low probability of the disease, testing should consist of 1 of the following, as a single negative test may be sufficient to rule out the disease:
- 24-hour urinary cortisol levels
- Overnight dexamethasone suppression testing
- Late-night salivary cortisol measurements.
In those with a high index of suspicion, 2 of the aforementioned tests should be performed, as 1 normal result may not be sufficient to exclude the diagnosis.22,23
A 24-hour urinary cortisol collection and overnight dexamethasone suppression test are obtained. His 24-hour urinary free cortisol level is elevated at 6,600 µg (normal 4–100), and suppression testing with 8 mg of dexamethasone (a form of “high-dose” testing)demonstrates only an 8% decline in serum cortisol levels. Cortisol should generally drop more than 90%.
Morning serum cortisol concentration is less than 5 µg/dL (140 nmol/L) in most patients with Cushing disease (ie, a pituitary tumor), and is usually undetectable in normal subjects. Only about 50% of neuroendocrine ACTH-secreting tumors will suppress with this test.
The patient’s clinical presentation, in conjunction with his diagnostic testing, are thus consistent with Cushing syndrome.
CUSHING SYNDROME
Cushing syndrome is most often exogenous or iatrogenic, ie, a result of supraphysiologic doses of glucocorticoids used to treat a variety of inflammatory, autoimmune, and neoplastic conditions.
Endogenous Cushing syndrome, on the other hand, is rare, with an estimated prevalence of 0.7 to 2.4 cases per million per year. ACTH-dependent causes account for 80% of endogenous Cushing syndrome cases, with ACTH-secreting pituitary adenomas (Cushing disease) accounting for 75% to 80% and ectopic ACTH secretion accounting for 15% to 20%. Less than 1% of cases are due to tumors that produce corticotropin-releasing hormone (CRH).
ACTH-independent Cushing syndrome is diagnosed in 20% of endogenous cases and is most commonly caused by a unilateral adrenal tumor. Rare causes of ACTH-independent disease include adrenal carcinoma, McCune-Albright syndrome, and adrenal hyperplasia.24
The patient’s ACTH is high
To determine whether this is an ACTH-dependent or independent process, the next step is to order an ACTH level. His ACTH level is high at 107 pg/mL (normal < 46 pg/mL), confirming the diagnosis of ACTH-dependent Cushing syndrome.
To find out if this ACTH-dependent process is due to a pituitary adenoma, magnetic resonance imaging (MRI) of the pituitary is obtained but is normal.
Large masses (> 6 mm) strongly suggest Cushing disease, but these tumors are often small and may be missed even with more advanced imaging techniques. Corticotropin-secreting adenomas arising from normal cells in the pituitary retain some sensitivity to glucocorticoid negative feedback and CRH stimulation, and thus high-dose dexamethasone suppression testing in conjunction with CRH stimulation testing can be used to differentiate Cushing disease from ectopic ACTH secretion.24,25 Both of these tests have poor diagnostic accuracy, however, and thus inferior petrosal sampling remains the gold standard for the diagnosis of Cushing disease.
ACTH-SECRETING TUMORS
5. Cushing syndrome due to ectopic ACTH secretion is most commonly attributed to which of the following tumors?
- Small-cell lung carcinoma
- Pancreatic carcinoma
- Medullary thyroid carcinoma
- Gastrinoma
Severe cases of Cushing syndrome are often attributable to ectopic ACTH secretion due to an underlying malignancy, most commonly small-cell lung carcinoma or neuroendocrine tumors of pulmonary origin. Other causes include pancreatic and thymic neuroendocrine tumors, gastrinomas, and medullary thyroid carcinoma.25,26
Because most ACTH-producing tumors are intrathoracic, initial imaging in cases of suspected ectopic ACTH secretion should focus on the chest, with CT the usual first choice. Octreotide scintigraphy can also be useful in localizing disease, as many neuroendocrine tumors express somatostatin receptors. Specialized positron-emission tomography scans may also be helpful in tumor identification.24
TREATMENT OF CUSHING SYNDROME DUE TO ECTOPIC ACTH SECRETION
6. Which of the following is most appropriate medical therapy for suppression of cortisol secretion in Cushing syndrome due to ectopic ACTH secretion?
- Spironolactone
- Dexamethasone
- Somatostatin
- Estrogen
- Ketoconazole
Hyperglycemia, hypokalemia, hypertension, psychiatric disturbances, venous thromboembolism, and systemic infections appear to be common in ectopic ACTH syndrome and often correlate with the degree of hypercortisolemia. Severe Cushing syndrome due to ectopic ACTH secretion is an emergency requiring prompt control of cortisol secretion.
First-line treatments include steroidogenesis inhibitors (ketoconazole, metyrapone, etomidate, mitotane) and glucocorticoid receptor antagonists (mifepristone). High-dose spironolactone and eplerenone can also be used to treat the hypertension and hypokalemia associated with mineralocorticoid receptor stimulation. Definitive treatment involves surgical resection, chemotherapy, or radiotherapy when applicable.24,25
After confirmation of the diagnosis, the patient is prescribed ketoconazole and spironolactone, with substantial improvement. He subsequently is started on combination chemotherapy and radiation therapy for his small-cell lung carcinoma.
DISCUSSION
The differential diagnosis for hypokalemia is broad and relies on information obtained during the history and physical examination, followed by interpretation of selected laboratory results. Myriad pathologies in diverse organ systems, eg, diarrhea, renal tubular acidosis, and adrenal disease, may be responsible for a low serum potassium. Further categorizing potassium depletion on the basis of an associated acid-base disturbance, such as metabolic alkalosis, allows one to use an algorithmic approach that can identify specific etiologies responsible for both the potassium and the acid-base disturbances.
Using the spot urine chloride in the setting of hypokalemic metabolic alkalosis with or without hypertension can narrow the differential diagnosis and allow additional clinical findings to guide clinical problem-solving and decision-making, even for conditions not commonly encountered in routine medical practice.
Obtaining renin and aldosterone measurements in patients with potassium depletion, metabolic alkalosis, high urine chloride excretion, and hypertension permits further categorization into 3 clinical groups: elevated aldosterone and renin (secondary hyperaldosteronism), elevated aldosterone and low renin (primary hyperaldosteronism), or apparent mineralocorticoid excess wherein neither renin nor aldosterone are responsible for the syndrome.
The patient in our case had apparent mineralocorticoid excess as a consequence of an ACTH-producing small-cell carcinoma.
NOTE: The scenario presented here is partly based on cases reported elsewhere by Martínez-Valles et al1 and Fernández-Rodríguez et al.2
A 55-year-old man is admitted to the hospital with generalized malaise, paresthesias, and severe hypertension. He says he had experienced agitation along with weakness on exertion 24 hours before presentation to the emergency department, with subsequent onset of paresthesias in his lower extremities and perioral area.
He is already known to have mild chronic obstructive pulmonary disease, with a ratio of forced expiratory volume in 1 second (FEV1)to forced vital capacity (FVC) of less than 70% and an FEV1 85% of predicted. In addition, he was recently diagnosed with diabetes, resistant hypertension requiring maximum doses of 3 agents (a calcium channel blocker, an angiotensin-converting enzyme inhibitor, and a loop diuretic), and hyperlipidemia.
He is a current smoker with a 30-pack-year smoking history. He does not use alcohol. His family history is noncontributory.
ASSESSING ACID-BASE DISORDERS
1. What type of acid-base disorder does this patient have?
- Metabolic acidosis
- Respiratory acidosis
- Metabolic alkalosis
- Respiratory alkalosis
The patient has metabolic alkalosis.
A 5-step approach
1. Acidosis or alkalosis? The patient’s arterial pH is 7.5, which is alkalemic because it is higher than 7.44.
2. Metabolic or respiratory? The primary process in our patient is overwhelmingly metabolic, as his partial pressure of carbon dioxide (Pco2) is slightly elevated, a direction that would cause acidosis, not alkalosis.
3. The anion gap (the serum sodium concentration minus the sum of the chloride and bicarbonate concentrations) is normal at 8 mmol/L (DRG:HYBRiD-XL Immunoassay and Clinical Chemistry Analyzer, reference range 8–16).
4. Is the disturbance compensated? We have determined that this patient has a metabolic alkalemia; the question now is whether there is any compensation for the primary disturbance.
In metabolic alkalosis, the Pco2 may increase by approximately 0.6 mm Hg (range 0.5–0.8) above the nominal normal level of 40 mm Hg for each 1-mmol/L increase in bicarbonate above the nominal normal level of 25 mmol/L.4 If the patient requires oxygen, the calculation may be unreliable, however, as hypoxemia may have an overriding influence on respiratory drive.
Patients with chronically high Pco2 levels such as those with chronic obstructive pulmonary disease can become accustomed to high carbon dioxide levels and lose their hyper-
capnic respiratory drive. Giving oxygen supplementation is thought to decrease respiratory drive in these patients, so that they will breathe slower and retain more carbon dioxide. There is some degree of respiratory compensation for metabolic alkalosis that occurs by breathing less, though it is limited overall—even in very alkalotic patients, breathing less results in CO2 retention, which, by displacing O2 molecules in the alveoli, will in turn result in hypoxia. The brain then senses the hypoxia and makes one breathe faster, thereby limiting this compensation.
This patient’s serum bicarbonate level is 40 mmol/L, or 15 mmol/L higher than the nominal normal level. If he is compensating, his Pco2 should be 40 + (15 × 0.6) = 49 mm Hg, and in fact it is 51 mm Hg, which is within the normal range of expected compensation (47.5–52 mm Hg). Therefore, yes, he is compensating for the primary disturbance.
5. In metabolic acidosis, is there a delta gap? As our patient has metabolic alkalosis, not acidosis, this question does not apply in this case.
WHICH TEST TO FIND THE CAUSE?
2. Which is the best test to order next to determine the cause of this patient’s hypokalemic metabolic alkalosis?
- Serum magnesium level
- Spot urine chloride
- Renal ultrasonography
- 24-hour urine collection for sodium, potassium, and chloride
The patient’s loop diuretic is withheld for 12 hours and a spot urine chloride is obtained, which is reported as 44 mmol/L. This high value suggests that a volume-independent hypokalemic metabolic alkalosis is present with potassium depletion.
As for the other answer choices:
Serum magnesium. Though hypomagnesemia can cause hypokalemia due to lack of inhibition of renal outer medullary potassium channels and subsequent increased excretion of potassium in the apical tubular membrane, it is not independently associated with acid-base disturbances.5
Renal ultrasonography gives information about structural kidney disease but is of limited utility in identifying the cause of hypokalemic metabolic alkalosis.
A 24-hour urine collection is unnecessary in this setting and would ultimately result in delay in diagnosis, as spot urine chloride is a more efficient means of rapidly distinguishing volume-responsive vs volume-independent causes of hypokalemic metabolic alkalosis.6
IS HIS HYPERTENSION SECONDARY? IF SO, WHAT IS THE CAUSE?
Several features of this case suggest that the patient’s hypertension is secondary rather than primary. It is of recent onset. The patient’s family history is noncontributory, and his hypertension is resistant to the use of maximum doses of 3 antihypertensive agents.
3. Which of the following causes of secondary hypertension is not commonly associated with hypokalemia and metabolic alkalosis?
- Hyperaldosteronism
- Liddle syndrome
- Cushing syndrome
- Renal parenchymal disease
- Chronic licorice ingestion
Renal parenchymal disease is a cause of resistant hypertension, but it is not characterized by metabolic alkalosis, hypokalemia, and elevated urine chloride,7 while the others listed here—hyperaldosteronism, Liddle syndrome, Cushing syndrome, and chronic licorice ingestion—are. Other common causes of resistant hypertension without these metabolic abnormalities include obstructive sleep apnea, alcohol abuse, and nonadherence to treatment.
While treatment of hypertension with loop diuretics can result in hypokalemia and metabolic alkalosis due to the effect of these drugs on potassium reabsorption in the loop of Henle, the patient’s hypokalemia persisted after this agent was withdrawn.8
Causes of hypokalemic metabolic alkalosis with and without hypertension are further delineated in Figure 1.
Additional diagnostic testing: Plasma renin and plasma aldosterone
At this juncture, the differential diagnosis for this patient’s potassium depletion, metabolic alkalosis, high urine chloride, and hypertension has been narrowed to primary or secondary hyperaldosteronism, surreptitious mineralocorticoid ingestion, Cushing syndrome, licorice ingestion, Liddle syndrome, or one of the 3 hydroxylase deficiencies (11-, 17-, and 21-) (Figure 1).
Although clues in the history, physical examination, and imaging may suggest a specific cause of his abnormal laboratory values, the next step in the diagnostic workup is to measure the plasma renin and aldosterone levels (Table 3).
HYPERALDOSTERONISM
4. Hyperaldosteronism is associated with which of the following patterns of renin and aldosterone values?
- High renin, high aldosterone, normal ratio of plasma aldosterone concentration (PAC) to plasma renin activity (PRA)
- Low renin, low aldosterone, normal PAC–PRA ratio
- Low renin, high aldosterone, high PAC–PRA ratio
- High renin, low aldosterone, low PAC–PRA ratio
The pattern of low renin, high aldosterone, and high PAC–PRA ratio is associated with hyperaldosteronism.
Primary hyperaldosteronism
Primary hyperaldosteronism is one of the most common causes of resistant hypertension and is underappreciated, being diagnosed in up to 20% of patients referred to hypertension specialty clinics.7 Potassium levels may be normal, likely contributing to its lack of recognition in this target population.
Primary hyperaldosteronism should be suspected in patients who have a plasma aldosterone PAC–PRA ratio greater than 20 with elevated plasma aldosterone concentrations
(> 15 ng/dL).
Persistently elevated aldosterone levels in the setting of elevated plasma volume is proof that aldosterone secretion is independent of the renin-angiotensin-aldosterone axis, and therefore is autonomous (secondary to adrenal tumor or hyperplasia). Further testing in the form of oral salt loading, saline infusion, or fludrocortisone (a sodium-retaining steroid) administration is thus required to confirm inappropriate, autonomous aldosterone secretion.9
After establishing the diagnosis of primary hyperaldosteronism, one should determine the subtype (ie, due to an adrenal carcinoma, unilateral hypersecreting adenoma, or unilateral or bilateral hyperplasia). Further testing includes adrenal computed tomography (CT) to rule out adrenal carcinomas, which are suspected with adenomas larger than 4 cm. Though part of the diagnostic workup, CT as a means of confirmational testing alone does not preclude the possibility of bilateral adrenal hyperplasia in some patients, even in the presence of an adrenal adenoma. For this reason, adrenal venous sampling is required to definitively determine whether the condition is due to a hypersecreting adrenal adenoma or unilateral or bilateral hyperplasia.9,10
Treatment of primary hyperaldosteronism depends on the subtype of the disease and involves salt restriction in addition to an aldosterone antagonist (spironolactone or eplerenone in the case of bilateral disease) or surgery (unilateral disease).9,11,12
Secondary hyperaldosteronism
Secondary hyperaldosteronism should be suspected when plasma renin and aldosterone levels are both elevated with a PAC–PRA ratio less than 10.
This pattern is most commonly seen with diuretic use but can also be a consequence of renal artery stenosis or, rarely, a renin-secreting tumor.13 Renal artery stenosis is a common finding in patients with hypertension undergoing cardiac catheterization, which is not surprising as more than 90% of such stenoses are atherosclerotic.7 Renin-secreting tumors are exceedingly rare, with fewer than 100 cases reported in the literature, and are more common in younger individuals.13
Our patient has low-normal aldosterone and plasma renin
On further testing, this patient’s plasma aldosterone level is 2.55 ng/dL (normal < 15 ng/dL), his plasma renin activity is 0.53 ng/mL/hour (normal 0.2–2.8 ng/mL/hour), and his PAC–PRA ratio is therefore 4.81.
The categories discussed thus far have included primary and secondary hyperaldosteronism, which typically do not present with low to normal levels of both renin and aldosterone. Surreptitious mineralocorticoid use could present in this manner, but is unlikely in this patient, whose medications do not include fludrocortisone.
The low-normal values thus lead to consideration of a third category: apparent mineralocorticoid excess. Diseases in this category such as Cushing disease or adrenocorticotropic hormone (ACTH) excess are characterized by increases in corticosteroids so that the potassium depletion, metabolic alkalosis, and hypertension are not a consequence of renin and aldosterone but rather the excess corticosteroids.14
Causes of apparent mineralocorticoid excess
There are several possible causes of mineralocorticoid excess associated with hypertension and hypokalemic metabolic alkalosis not due to renin and aldosterone.
Chronic licorice ingestion in high volumes is one such cause and is thought to result in inhibition of 11B-hydroxysteroid dehydrogenase or possibly cortisol oxidase by licorice’s active component, glycyrrhetinic acid. This inhibition results in an inability to convert cortisol to cortisone. The cortisol excess binds to mineralocorticoid receptors, and acting like aldosterone, results in hypertension and hypokalemic metabolic alkalosis as well as feedback inhibition of renin and aldosterone levels.15
Partial hydroxylase deficiencies, though rare, should also be considered as a cause of hypokalemic metabolic alkalosis, hypertension, and, potentially, hirsutism and clitoromegaly in women. They can be diagnosed with elevated levels of 17-ketosteroids and dehydroepiandrosterone sulfate, both of which, in excess, may act on aldosterone receptors in a manner similar to cortisol.16
Liddle syndrome, a rare autosomal dominant condition, may also present with suppressed levels of both renin and aldosterone. In contrast to the disorders of nonaldosterone mineralocorticoid excess, however, the sodium channel defect in Liddle syndrome is characterized by a primary increase in sodium reabsorption in the collecting tubule and potassium wasting. The resultant volume expansion leads to suppressed renin and aldosterone levels and hypertension with low potassium and elevated bicarbonate concentrations.17
Liddle syndrome is commonly diagnosed in childhood but may go unrecognized due to occasional absence of hypokalemia at presentation. Potassium-sparing diuretics such as amiloride or triamterene are the mainstays of treatment.18
Rates of cardiovascular and all-cause mortality are increased in patients with long-term hypercortisolism, even after plasma concentrations of cortisol are normalized.21
Figure 2 shows the cascade of the hypothalamic-pituitary-adrenal axis.
TESTING FOR HYPERCORTISOLISM IN OUR PATIENT
Given the patient’s clinical presentation and laboratory and imaging findings with normal plasma renin and aldosterone levels, a workup for suspected hypercortisolism is initiated.
Initial diagnostic testing for hypercortisolism depends on the degree of clinical suspicion. In those with low probability of the disease, testing should consist of 1 of the following, as a single negative test may be sufficient to rule out the disease:
- 24-hour urinary cortisol levels
- Overnight dexamethasone suppression testing
- Late-night salivary cortisol measurements.
In those with a high index of suspicion, 2 of the aforementioned tests should be performed, as 1 normal result may not be sufficient to exclude the diagnosis.22,23
A 24-hour urinary cortisol collection and overnight dexamethasone suppression test are obtained. His 24-hour urinary free cortisol level is elevated at 6,600 µg (normal 4–100), and suppression testing with 8 mg of dexamethasone (a form of “high-dose” testing)demonstrates only an 8% decline in serum cortisol levels. Cortisol should generally drop more than 90%.
Morning serum cortisol concentration is less than 5 µg/dL (140 nmol/L) in most patients with Cushing disease (ie, a pituitary tumor), and is usually undetectable in normal subjects. Only about 50% of neuroendocrine ACTH-secreting tumors will suppress with this test.
The patient’s clinical presentation, in conjunction with his diagnostic testing, are thus consistent with Cushing syndrome.
CUSHING SYNDROME
Cushing syndrome is most often exogenous or iatrogenic, ie, a result of supraphysiologic doses of glucocorticoids used to treat a variety of inflammatory, autoimmune, and neoplastic conditions.
Endogenous Cushing syndrome, on the other hand, is rare, with an estimated prevalence of 0.7 to 2.4 cases per million per year. ACTH-dependent causes account for 80% of endogenous Cushing syndrome cases, with ACTH-secreting pituitary adenomas (Cushing disease) accounting for 75% to 80% and ectopic ACTH secretion accounting for 15% to 20%. Less than 1% of cases are due to tumors that produce corticotropin-releasing hormone (CRH).
ACTH-independent Cushing syndrome is diagnosed in 20% of endogenous cases and is most commonly caused by a unilateral adrenal tumor. Rare causes of ACTH-independent disease include adrenal carcinoma, McCune-Albright syndrome, and adrenal hyperplasia.24
The patient’s ACTH is high
To determine whether this is an ACTH-dependent or independent process, the next step is to order an ACTH level. His ACTH level is high at 107 pg/mL (normal < 46 pg/mL), confirming the diagnosis of ACTH-dependent Cushing syndrome.
To find out if this ACTH-dependent process is due to a pituitary adenoma, magnetic resonance imaging (MRI) of the pituitary is obtained but is normal.
Large masses (> 6 mm) strongly suggest Cushing disease, but these tumors are often small and may be missed even with more advanced imaging techniques. Corticotropin-secreting adenomas arising from normal cells in the pituitary retain some sensitivity to glucocorticoid negative feedback and CRH stimulation, and thus high-dose dexamethasone suppression testing in conjunction with CRH stimulation testing can be used to differentiate Cushing disease from ectopic ACTH secretion.24,25 Both of these tests have poor diagnostic accuracy, however, and thus inferior petrosal sampling remains the gold standard for the diagnosis of Cushing disease.
ACTH-SECRETING TUMORS
5. Cushing syndrome due to ectopic ACTH secretion is most commonly attributed to which of the following tumors?
- Small-cell lung carcinoma
- Pancreatic carcinoma
- Medullary thyroid carcinoma
- Gastrinoma
Severe cases of Cushing syndrome are often attributable to ectopic ACTH secretion due to an underlying malignancy, most commonly small-cell lung carcinoma or neuroendocrine tumors of pulmonary origin. Other causes include pancreatic and thymic neuroendocrine tumors, gastrinomas, and medullary thyroid carcinoma.25,26
Because most ACTH-producing tumors are intrathoracic, initial imaging in cases of suspected ectopic ACTH secretion should focus on the chest, with CT the usual first choice. Octreotide scintigraphy can also be useful in localizing disease, as many neuroendocrine tumors express somatostatin receptors. Specialized positron-emission tomography scans may also be helpful in tumor identification.24
TREATMENT OF CUSHING SYNDROME DUE TO ECTOPIC ACTH SECRETION
6. Which of the following is most appropriate medical therapy for suppression of cortisol secretion in Cushing syndrome due to ectopic ACTH secretion?
- Spironolactone
- Dexamethasone
- Somatostatin
- Estrogen
- Ketoconazole
Hyperglycemia, hypokalemia, hypertension, psychiatric disturbances, venous thromboembolism, and systemic infections appear to be common in ectopic ACTH syndrome and often correlate with the degree of hypercortisolemia. Severe Cushing syndrome due to ectopic ACTH secretion is an emergency requiring prompt control of cortisol secretion.
First-line treatments include steroidogenesis inhibitors (ketoconazole, metyrapone, etomidate, mitotane) and glucocorticoid receptor antagonists (mifepristone). High-dose spironolactone and eplerenone can also be used to treat the hypertension and hypokalemia associated with mineralocorticoid receptor stimulation. Definitive treatment involves surgical resection, chemotherapy, or radiotherapy when applicable.24,25
After confirmation of the diagnosis, the patient is prescribed ketoconazole and spironolactone, with substantial improvement. He subsequently is started on combination chemotherapy and radiation therapy for his small-cell lung carcinoma.
DISCUSSION
The differential diagnosis for hypokalemia is broad and relies on information obtained during the history and physical examination, followed by interpretation of selected laboratory results. Myriad pathologies in diverse organ systems, eg, diarrhea, renal tubular acidosis, and adrenal disease, may be responsible for a low serum potassium. Further categorizing potassium depletion on the basis of an associated acid-base disturbance, such as metabolic alkalosis, allows one to use an algorithmic approach that can identify specific etiologies responsible for both the potassium and the acid-base disturbances.
Using the spot urine chloride in the setting of hypokalemic metabolic alkalosis with or without hypertension can narrow the differential diagnosis and allow additional clinical findings to guide clinical problem-solving and decision-making, even for conditions not commonly encountered in routine medical practice.
Obtaining renin and aldosterone measurements in patients with potassium depletion, metabolic alkalosis, high urine chloride excretion, and hypertension permits further categorization into 3 clinical groups: elevated aldosterone and renin (secondary hyperaldosteronism), elevated aldosterone and low renin (primary hyperaldosteronism), or apparent mineralocorticoid excess wherein neither renin nor aldosterone are responsible for the syndrome.
The patient in our case had apparent mineralocorticoid excess as a consequence of an ACTH-producing small-cell carcinoma.
- Martínez-Valles MA, Palafox-Cazarez A, Paredes-Avina JA. Severe hypokalemia, metabolic alkalosis and hypertension in a 54 year old male with ectopic ACTH syndrome: a case report. Cases J 2009; 2:6174. doi:10.4076/1757-1626-2-6174
- Fernández-Rodríguez E, Villar-Taibo R, Pinal-Osorio I, et al. Severe hypertension and hypokalemia as first clinical manifestations in ectopic Cushing’s syndrome. Arq Bras Endocrinol Metabol 2008; 52(6):1066–1070. pmid:18820819
- Mani S, Rutecki GW. A patient with altered mental status and an acid-base disturbance. Cleve Clin J Med 2017; 84(1):27–34. doi:10.3949/ccjm.84a.16042
- Adrogué HJ, Madias NE. Secondary responses to altered acid-base status: the rules of engagement. J Am Soc Nephrol 2010; 21(6):920–923. doi:10.1681/ASN.2009121211
- Huang CL, Kuo E. Mechanism of hypokalemia in magnesium deficiency. J Am Soc Nephrol 2007; 18(10):2649–2652. doi:10.1681/ASN.2007070792
- Rose BD. Metabolic alkalosis. In: Clinical Physiology of Acid-Base and Electrolyte Disorders. 4th ed. New York, NY: McGraw-Hill, Health Professions Division; 1994:515.
- Calhoun DA, Jones D, Textor S, et al; American Heart Association Professional Education Committee. Resistant hypertension: diagnosis, evaluation, and treatment: a scientific statement from the American Heart Association Professional Education Committee of the Council for High Blood Pressure Research. Circulation 2008; 117(25):e510–e526. doi:10.1161/CIRCULATIONAHA.108.189141
- Koeppen BM, Stanton BA. Physiology of diuretic action. In: Renal Physiology. 5th ed. Philadelphia, PA: Elsevier Inc; 2013:167–178.
- Blumenfeld JD, Sealey JE, Schlussel Y, et al. Diagnosis and treatment of primary hyperaldosteronism. Ann Intern Med 1994; 121(11):877–885. pmid:7978702
- Kempers MJ, Lenders JW, van Outheusden L, et al. Systematic review: diagnostic procedures to differentiate unilateral from bilateral adrenal abnormality in primary aldosteronism. Ann Intern Med 2009; 151(5):329–337. pmid:19721021
- Karagiannis A, Tziomalos K, Papageorgiou A, et al. Spironolactone versus eplerenone for the treatment of idiopathic hyperaldosteronism. Expert Opin Pharmacother 2008; 9(4):509–515. doi:10.1517/14656566.9.4.509
- Sawka AM, Young WF, Thompson GB, et al. Primary aldosteronism: factors associated with normalization of blood pressure after surgery. Ann Intern Med 2001; 135(4):258–261. pmid:11511140
- Haab F, Duclos JM, Guyenne T, Plouin PF, Corvol P. Renin secreting tumors: diagnosis, conservative surgical approach and long-term results. J Urol 1995; 153(6):1781–1784. pmid:7752315
- Sabbadin C, Armanini D. Syndromes that mimic an excess of mineralocorticoids. High Blood Press Cardiovasc Prev 2016; 23(3):231–235. doi:10.1007/s40292-016-0160-5
- Apostolakos JM, Caines LC. Apparent mineralocorticoid excess syndrome: a case of resistant hypertension from licorice tea consumption. J Clin Hypertens (Greenwich) 2016; 18(10):991–993. doi:10.1111/jch.12841
- Glatt K, Garzon DL, Popovic J. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Spec Pediatr Nurs 2005; 10(3):104–114. doi:10.1111/j.1744-6155.2005.00022.x
- Findling JW, Raff H, Hansson JH, Lifton RP. Liddle’s syndrome: prospective genetic screening and suppressed aldosterone secretion in an extended kindred. J Clin Endocrinol Metab 1997; 82(4):1071–1074. doi:10.1210/jcem.82.4.3862
- Wang C, Chan TK, Yeung RT, Coghlan JP, Scoggins BA, Stockigt JR. The effect of triamterene and sodium intake on renin, aldosterone, and erythrocyte sodium transport in Liddle’s syndrome. J Clin Endocrinol Metab 1981; 52(5):1027–1032. doi:10.1210/jcem-52-5-1027
- Torpy DJ, Mullen N, Ilias I, Nieman LK. Association of hypertension and hypokalemia with Cushing’s syndrome caused by ectopic ACTH secretion: a series of 58 cases. Ann N Y Acad Sci 2002; 970:134–144. pmid:12381548
- Saruta T, Suzuki H, Handa M, Igarashi Y, Kondo K, Senba S. Multiple factors contribute to the pathogenesis of hypertension in Cushing’s syndrome. J Clin Endocrinol Metab 1986; 62(2):275–279. doi:10.1210/jcem-62-2-275
- Clayton RN, Jones PW, Reulen RC, et al. Mortality in patients with Cushing’s disease more than 10 years after remission: a multicentre, multinational, retrospective cohort study. Lancet Diabetes Endocrinol 2016; 4(7):569–576. doi:10.1016/S2213-8587(16)30005-5
- Baid SK, Rubino D, Sinaii N, Ramsey S, Frank A, Nieman LK. Specificity of screening tests for Cushing’s syndrome in an overweight and obese population. J Clin Endocrinol Metab 2009; 94(10):3857–3864. doi:10.1210/jc.2008-2766
- Nieman LK, Biller BM, Findling JW, et al. The diagnosis of Cushing’s syndrome: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab 2008; 93(5):1526–1540. doi:10.1210/jc.2008-0125
- Sharma ST, Nieman LK, Feelders RA. Cushing’s syndrome: epidemiology and developments in disease management. Clin Epidemiol 2015; 7:281–293. doi:10.2147/CLEP.S44336
- Tavares Bello C, van der Poest Clement E, Feelders R. Severe Cushing’s syndrome and bilateral pulmonary nodules: beyond ectopic ACTH. Endocrinol Diabetes Metab Case Rep 2017; pii:17–0100. doi:10.1530/EDM-17-0100
- Sathyakumar S, Paul TV, Asha HS, et al. Ectopic Cushing syndrome: a 10-year experience from a tertiary care center in southern India. Endocr Pract 2017; 23(8):907–914. doi:10.4158/EP161677.OR
- Martínez-Valles MA, Palafox-Cazarez A, Paredes-Avina JA. Severe hypokalemia, metabolic alkalosis and hypertension in a 54 year old male with ectopic ACTH syndrome: a case report. Cases J 2009; 2:6174. doi:10.4076/1757-1626-2-6174
- Fernández-Rodríguez E, Villar-Taibo R, Pinal-Osorio I, et al. Severe hypertension and hypokalemia as first clinical manifestations in ectopic Cushing’s syndrome. Arq Bras Endocrinol Metabol 2008; 52(6):1066–1070. pmid:18820819
- Mani S, Rutecki GW. A patient with altered mental status and an acid-base disturbance. Cleve Clin J Med 2017; 84(1):27–34. doi:10.3949/ccjm.84a.16042
- Adrogué HJ, Madias NE. Secondary responses to altered acid-base status: the rules of engagement. J Am Soc Nephrol 2010; 21(6):920–923. doi:10.1681/ASN.2009121211
- Huang CL, Kuo E. Mechanism of hypokalemia in magnesium deficiency. J Am Soc Nephrol 2007; 18(10):2649–2652. doi:10.1681/ASN.2007070792
- Rose BD. Metabolic alkalosis. In: Clinical Physiology of Acid-Base and Electrolyte Disorders. 4th ed. New York, NY: McGraw-Hill, Health Professions Division; 1994:515.
- Calhoun DA, Jones D, Textor S, et al; American Heart Association Professional Education Committee. Resistant hypertension: diagnosis, evaluation, and treatment: a scientific statement from the American Heart Association Professional Education Committee of the Council for High Blood Pressure Research. Circulation 2008; 117(25):e510–e526. doi:10.1161/CIRCULATIONAHA.108.189141
- Koeppen BM, Stanton BA. Physiology of diuretic action. In: Renal Physiology. 5th ed. Philadelphia, PA: Elsevier Inc; 2013:167–178.
- Blumenfeld JD, Sealey JE, Schlussel Y, et al. Diagnosis and treatment of primary hyperaldosteronism. Ann Intern Med 1994; 121(11):877–885. pmid:7978702
- Kempers MJ, Lenders JW, van Outheusden L, et al. Systematic review: diagnostic procedures to differentiate unilateral from bilateral adrenal abnormality in primary aldosteronism. Ann Intern Med 2009; 151(5):329–337. pmid:19721021
- Karagiannis A, Tziomalos K, Papageorgiou A, et al. Spironolactone versus eplerenone for the treatment of idiopathic hyperaldosteronism. Expert Opin Pharmacother 2008; 9(4):509–515. doi:10.1517/14656566.9.4.509
- Sawka AM, Young WF, Thompson GB, et al. Primary aldosteronism: factors associated with normalization of blood pressure after surgery. Ann Intern Med 2001; 135(4):258–261. pmid:11511140
- Haab F, Duclos JM, Guyenne T, Plouin PF, Corvol P. Renin secreting tumors: diagnosis, conservative surgical approach and long-term results. J Urol 1995; 153(6):1781–1784. pmid:7752315
- Sabbadin C, Armanini D. Syndromes that mimic an excess of mineralocorticoids. High Blood Press Cardiovasc Prev 2016; 23(3):231–235. doi:10.1007/s40292-016-0160-5
- Apostolakos JM, Caines LC. Apparent mineralocorticoid excess syndrome: a case of resistant hypertension from licorice tea consumption. J Clin Hypertens (Greenwich) 2016; 18(10):991–993. doi:10.1111/jch.12841
- Glatt K, Garzon DL, Popovic J. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Spec Pediatr Nurs 2005; 10(3):104–114. doi:10.1111/j.1744-6155.2005.00022.x
- Findling JW, Raff H, Hansson JH, Lifton RP. Liddle’s syndrome: prospective genetic screening and suppressed aldosterone secretion in an extended kindred. J Clin Endocrinol Metab 1997; 82(4):1071–1074. doi:10.1210/jcem.82.4.3862
- Wang C, Chan TK, Yeung RT, Coghlan JP, Scoggins BA, Stockigt JR. The effect of triamterene and sodium intake on renin, aldosterone, and erythrocyte sodium transport in Liddle’s syndrome. J Clin Endocrinol Metab 1981; 52(5):1027–1032. doi:10.1210/jcem-52-5-1027
- Torpy DJ, Mullen N, Ilias I, Nieman LK. Association of hypertension and hypokalemia with Cushing’s syndrome caused by ectopic ACTH secretion: a series of 58 cases. Ann N Y Acad Sci 2002; 970:134–144. pmid:12381548
- Saruta T, Suzuki H, Handa M, Igarashi Y, Kondo K, Senba S. Multiple factors contribute to the pathogenesis of hypertension in Cushing’s syndrome. J Clin Endocrinol Metab 1986; 62(2):275–279. doi:10.1210/jcem-62-2-275
- Clayton RN, Jones PW, Reulen RC, et al. Mortality in patients with Cushing’s disease more than 10 years after remission: a multicentre, multinational, retrospective cohort study. Lancet Diabetes Endocrinol 2016; 4(7):569–576. doi:10.1016/S2213-8587(16)30005-5
- Baid SK, Rubino D, Sinaii N, Ramsey S, Frank A, Nieman LK. Specificity of screening tests for Cushing’s syndrome in an overweight and obese population. J Clin Endocrinol Metab 2009; 94(10):3857–3864. doi:10.1210/jc.2008-2766
- Nieman LK, Biller BM, Findling JW, et al. The diagnosis of Cushing’s syndrome: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab 2008; 93(5):1526–1540. doi:10.1210/jc.2008-0125
- Sharma ST, Nieman LK, Feelders RA. Cushing’s syndrome: epidemiology and developments in disease management. Clin Epidemiol 2015; 7:281–293. doi:10.2147/CLEP.S44336
- Tavares Bello C, van der Poest Clement E, Feelders R. Severe Cushing’s syndrome and bilateral pulmonary nodules: beyond ectopic ACTH. Endocrinol Diabetes Metab Case Rep 2017; pii:17–0100. doi:10.1530/EDM-17-0100
- Sathyakumar S, Paul TV, Asha HS, et al. Ectopic Cushing syndrome: a 10-year experience from a tertiary care center in southern India. Endocr Pract 2017; 23(8):907–914. doi:10.4158/EP161677.OR
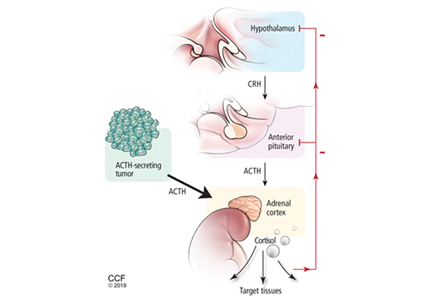
FDA approves first interoperable insulin infusion pump
, for insulin delivery in children and adults with diabetes.

The pump delivers insulin under the skin at a variable or fixed rate. It can function on its own, or it can be digitally connected to automatically communicate with and receive drug-dosing commands from other diabetes management devices, such as automated insulin-dosing systems, the agency announced.
The approval was based on a review of performance data demonstrating that the device can deliver insulin accurately and reliably and at the rates and volumes programmed by the user. The agency also assessed the pump’s ability to connect reliably with other devices, as well as its cybersecurity and fail-safe modes.
Risks associated with the device were similar to those of other infusion pumps and include infection, bleeding, pain, or skin irritations. Blockages and air bubbles can occur in the tubing, which will affect drug delivery. Risks associated with incorrect drug delivery include hypo- and hyperglycemia as well as diabetic ketoacidosis.
“The marketing authorization of the [pump] has the potential to aid patients who seek more individualized diabetes therapy systems and opens the door for developers of future connected diabetes devices to get other safe and effective products to patients more efficiently,” FDA Commissioner Scott Gottlieb, MD, said in the announcement.
, for insulin delivery in children and adults with diabetes.
The pump delivers insulin under the skin at a variable or fixed rate. It can function on its own, or it can be digitally connected to automatically communicate with and receive drug-dosing commands from other diabetes management devices, such as automated insulin-dosing systems, the agency announced.
The approval was based on a review of performance data demonstrating that the device can deliver insulin accurately and reliably and at the rates and volumes programmed by the user. The agency also assessed the pump’s ability to connect reliably with other devices, as well as its cybersecurity and fail-safe modes.
Risks associated with the device were similar to those of other infusion pumps and include infection, bleeding, pain, or skin irritations. Blockages and air bubbles can occur in the tubing, which will affect drug delivery. Risks associated with incorrect drug delivery include hypo- and hyperglycemia as well as diabetic ketoacidosis.
“The marketing authorization of the [pump] has the potential to aid patients who seek more individualized diabetes therapy systems and opens the door for developers of future connected diabetes devices to get other safe and effective products to patients more efficiently,” FDA Commissioner Scott Gottlieb, MD, said in the announcement.
, for insulin delivery in children and adults with diabetes.
The pump delivers insulin under the skin at a variable or fixed rate. It can function on its own, or it can be digitally connected to automatically communicate with and receive drug-dosing commands from other diabetes management devices, such as automated insulin-dosing systems, the agency announced.
The approval was based on a review of performance data demonstrating that the device can deliver insulin accurately and reliably and at the rates and volumes programmed by the user. The agency also assessed the pump’s ability to connect reliably with other devices, as well as its cybersecurity and fail-safe modes.
Risks associated with the device were similar to those of other infusion pumps and include infection, bleeding, pain, or skin irritations. Blockages and air bubbles can occur in the tubing, which will affect drug delivery. Risks associated with incorrect drug delivery include hypo- and hyperglycemia as well as diabetic ketoacidosis.
“The marketing authorization of the [pump] has the potential to aid patients who seek more individualized diabetes therapy systems and opens the door for developers of future connected diabetes devices to get other safe and effective products to patients more efficiently,” FDA Commissioner Scott Gottlieb, MD, said in the announcement.
Subclinical hypothyroidism: When to treat
Whether subclinical hypothyroidism is clinically important and should be treated remains controversial. Studies have differed in their findings, and although most have found this condition to be associated with a variety of adverse outcomes, large randomized controlled trials are needed to clearly demonstrate its clinical impact in various age groups and the benefit of levothyroxine therapy.
Currently, the best practical approach is to base treatment decisions on the magnitude of elevation of thyroid-stimulating hormone (TSH) and whether the patient has thyroid autoantibodies and associated comorbid conditions.
HIGH TSH, NORMAL FREE T4 LEVELS
Subclinical hypothyroidism is defined by elevated TSH along with a normal free thyroxine (T4).1
The hypothalamic-pituitary-thyroid axis is a balanced homeostatic system, and TSH and thyroid hormone levels have an inverse log-linear relation: if free T4 and triiodothyronine (T3) levels go down even a little, TSH levels go up a lot.2
TSH secretion is pulsatile and has a circadian rhythm: serum TSH levels are 50% higher at night and early in the morning than during the rest of the day. Thus, repeated measurements in the same patient can vary by as much as half of the reference range.3
WHAT IS THE UPPER LIMIT OF NORMAL FOR TSH?
The upper limit of normal for TSH, defined as the 97.5th percentile, is approximately 4 or 5 mIU/L depending on the laboratory and the population, but some experts believe it should be lower.3
In favor of a lower upper limit: the distribution of serum TSH levels in the healthy general population does not seem to be a typical bell-shaped Gaussian curve, but rather has a tail at the high end. Some argue that some of the individuals with values in the upper end of the normal range may actually have undiagnosed hypothyroidism and that the upper 97.5th percentile cutoff would be 2.5 mIU/L if these people were excluded.4 Also, TSH levels higher than 2.5 mIU/L have been associated with a higher prevalence of antithyroid antibodies and a higher risk of clinical hypothyroidism.5
On the other hand, lowering the upper limit of normal to 2.5 mIU/L would result in 4 times as many people receiving a diagnosis of subclinical hypothyroidism, or 22 to 28 million people in the United States.4,6 Thus, lowering the cutoff may lead to unnecessary therapy and could even harm from overtreatment.
Another argument against lowering the upper limit of normal for TSH is that, with age, serum TSH levels shift higher.7 The third National Health and Nutrition Education Survey (NHANES III) found that the 97.5th percentile for serum TSH was 3.56 mIU/L for age group 20 to 29 but 7.49 mIU/L for octogenarians.7,8
It has been suggested that the upper limit of normal for TSH be adjusted for age, race, sex, and iodine intake.3 Currently available TSH reference ranges are not adjusted for these variables, and there is not enough evidence to suggest age-appropriate ranges,9 although higher TSH cutoffs for treatment are advised in elderly patients.10 Interestingly, higher TSH in older people has been linked to lower mortality rates in some studies.11
Authors of the NHANES III8 and Hanford Thyroid Disease study12 have proposed a cutoff of 4.1 mIU/L for the upper limit of normal for serum TSH in patients with negative antithyroid antibodies and normal findings on thyroid ultrasonography.
SUBCLINICAL HYPOTHYROIDISM IS COMMON
In different studies, the prevalence of subclinical hypothyroidism has been as low as 4% and as high as 20%.1,8,13 The prevalence is higher in women and increases with age.8 It is higher in iodine-sufficient areas, and it increases in iodine-deficient areas with iodine supplementation.14 Genetics also plays a role, as subclinical hypothyroidism is more common in white people than in African Americans.8
A difficulty in estimating the prevalence is the disagreement about the cutoff for TSH, which may differ from that in the general population in certain subgroups such as adolescents, the elderly, and pregnant women.10,15
A VARIETY OF CAUSES
The most common cause of subclinical hypothyroidism, accounting for 60% to 80% of cases, is Hashimoto (autoimmune) thyroiditis,2 in which thyroid peroxidase antibodies are usually present.2,16

Also important to rule out are false-positive elevations due to substances that interfere with TSH assays (eg, heterophile antibodies, rheumatoid factor, biotin, macro-TSH); reversible causes such as the recovery phase of euthyroid sick syndrome; subacute, painless, or postpartum thyroiditis; central hypo- or hyperthyroidism; and thyroid hormone resistance.
SUBCLINICAL HYPOTHYROIDISM CAN RESOLVE OR PROGRESS
“Subclinical” suggests that the disease is in its early stage, with changes in TSH already apparent but decreases in thyroid hormone levels yet to come.17 And indeed, subclinical hypothyroidism can progress to overt hypothyroidism,18 although it has been reported to resolve spontaneously in half of cases within 2 years,19 typically in patients with TSH values of 4 to 6 mIU/L.20 The rate of progression to overt hypothyroidism is estimated to be 33% to 55% over 10 to 20 years of follow-up.18

Figure 1 shows the natural history of subclinical hypothyroidism.1
GUIDELINES FOR SCREENING DIFFER
Guidelines differ on screening for thyroid disease in the general population, owing to lack of large-scale randomized controlled trials showing treatment benefit in otherwise-healthy people with mildly elevated TSH values.
Various professional societies have adopted different criteria for aggressive case-finding in patients at risk of thyroid disease. Risk factors include family history of thyroid disease, neck irradiation, partial thyroidectomy, dyslipidemia, atrial fibrillation, unexplained weight loss, hyperprolactinemia, autoimmune disorders, and use of medications affecting thyroid function.23
The US Preventive Services Task Force in 2014 found insufficient evidence on the benefits and harms of screening.24
The American Thyroid Association (ATA) recommends screening adults starting at age 35, with repeat testing every 5 years in patients who have no signs or symptoms of hypothyroidism, and more frequently in those who do.25
The American Association of Clinical Endocrinologists recommends screening in women and older patients. Their guidelines and those of the ATA also suggest screening people at high risk of thyroid disease due to risk factors such as history of autoimmune diseases, neck irradiation, or medications affecting thyroid function.26
The American Academy of Family Physicians recommends screening after age 60.18
The American College of Physicians recommends screening patients over age 50 who have symptoms.18
Our approach. Although evidence is lacking to recommend routine screening in adults, aggressive case-finding and treatment in patients at risk of thyroid disease can, we believe, offset the risks associated with subclinical hypothyroidism.24
CLINICAL PRESENTATION
About 70% of patients with subclinical hypothyroidism have no symptoms.13
Tiredness was more common in subclinical hypothyroid patients with TSH levels lower than 10 mIU/L compared with euthyroid controls in 1 study, but other studies have been unable to replicate this finding.27,28
Other frequently reported symptoms include dry skin, cognitive slowing, poor memory, muscle weakness, cold intolerance, constipation, puffy eyes, and hoarseness.13
The evidence in favor of levothyroxine therapy to improve symptoms in subclinical hypothyroidism has varied, with some studies showing an improvement in symptom scores compared with placebo, while others have not shown any benefit.29–31
In one study, the average TSH value for patients whose symptoms did not improve with therapy was 4.6 mIU/L.31 An explanation for the lack of effect in this group may be that the TSH values for these patients were in the high-normal range. Also, because most subclinical hypothyroid patients have no symptoms, it is difficult to ascertain symptomatic improvement. Though it is possible to conclude that levothyroxine therapy has a limited role in this group, it is important to also consider the suggestive evidence that untreated subclinical hypothyroidism may lead to increased morbidity and mortality.
ADVERSE EFFECTS OF SUBCLINICAL HYPOTHYROIDISM, EFFECTS OF THERAPY

INDIVIDUALIZED MANAGEMENT AND SHARED DECISION-MAKING
The management of subclinical hypothyroidism should be individualized on the basis of extent of thyroid dysfunction, comorbid conditions, risk factors, and patient preference.118 Shared decision-making is key, weighing the risks and benefits of levothyroxine treatment and the patient’s goals.

The risks of treatment should be kept in mind and explained to the patient. Levothyroxine has a narrow therapeutic range, causing a possibility of overreplacement, and a half-life of 7 days that can cause dosing errors to have longer effect.118,119
Adherence can be a challenge. The drug needs to be taken on an empty stomach because foods and supplements interfere with its absorption.118,120 In addition, the cost of medication, frequent biochemical monitoring, and possible need for titration can add to financial burden.
When choosing the dose, one should consider the degree of hypothyroidism or TSH elevation and the patient’s weight, and adjust the dose gently.
If the TSH is high-normal
It is proposed that a TSH range of 3 to 5 mIU/L overlaps with normal thyroid function in a great segment of the population, and at this level it is probably not associated with clinically significant consequences. For these reasons, levothyroxine therapy is not thought to be beneficial for those with TSH in this range.
Pollock et al121 found that, in patients with symptoms suggesting hypothyroidism and TSH values in the upper end of the normal range, there was no improvement in cognitive function or psychological well-being after 12 weeks of levothyroxine therapy.
However, due to the concern for possible adverse maternal and fetal outcomes and low IQ in children of pregnant patients with subclinical hypothyroidism, levothyroxine therapy is advised in those who are pregnant or planning pregnancy who have TSH levels higher than 2.5 mIU/L, especially if they have thyroid peroxidase antibody. Levothyroxine therapy is not recommended for pregnant patients with negative thyroid peroxidase antibody and TSH within the pregnancy-specific range or less than 4 mIU/L if the reference ranges are unavailable.
Keep in mind that, even at these TSH values, there is risk of progression to overt hypothyroidism, especially in the presence of thyroid peroxidase antibody, so patients in this group should be monitored closely.
If TSH is mildly elevated
The evidence to support levothyroxine therapy in patients with subclinical hypothyroidism with TSH levels less than 10 mIU/L remains inconclusive, and the decision to treat should be based on clinical judgment.2 The studies that have looked at the benefit of treating subclinical hypothyroidism in terms of cardiac, neuromuscular, cognitive, and neuropsychiatric outcomes have included patients with a wide range of TSH levels, and some of these studies were not stratified on the basis of degree of TSH elevation.
The risk that subclinical hypothyroidism will progress to overt hypothyroidism in patients with TSH higher than 8 mIU/L is high, and in 70% of these patients, the TSH level rises to more than 10 mIU/L within 4 years. Early treatment should be considered if the TSH is higher than 7 or 8 mIU/L.
If TSH is higher than 10 mIU/L

Figure 2 outlines an algorithmic approach to subclinical hypothyroidism in nonpregnant patients as suggested by Peeters.122
- Cooper DS, Biondi B. Subclinical thyroid disease. Lancet 2012; 379(9821):1142–1154. doi:10.1016/S0140-6736(11)60276-6
- Fatourechi V. Subclinical hypothyroidism: an update for primary care physicians. Mayo Clin Proc 2009; 84(1):65–71. doi:10.4065/84.1.65
- Laurberg P, Andersen S, Carle A, Karmisholt J, Knudsen N, Pedersen IB. The TSH upper reference limit: where are we at? Nat Rev Endocrinol 2011; 7(4):232–239. doi:10.1038/nrendo.2011.13
- Wartofsky L, Dickey RA. The evidence for a narrower thyrotropin reference range is compelling. J Clin Endocrinol Metab 2005; 90(9):5483–5488. doi:10.1210/jc.2005-0455
- Spencer CA, Hollowell JG, Kazarosyan M, Braverman LE. National Health and Nutrition Examination Survey III thyroid-stimulating hormone (TSH)-thyroperoxidase antibody relationships demonstrate that TSH upper reference limits may be skewed by occult thyroid dysfunction. J Clin Endocrinol Metab 2007; 92(11):4236–4240. doi:10.1210/jc.2007-0287
- Fatourechi V, Klee GG, Grebe SK, et al. Effects of reducing the upper limit of normal TSH values. JAMA 2003; 290(24):3195–3196. doi:10.1001/jama.290.24.3195-a
- Surks MI, Hollowell JG. Age-specific distribution of serum thyrotropin and antithyroid antibodies in the US population: implications for the prevalence of subclinical hypothyroidism. J Clin Endocrinol Metab 2007; 92(12):4575–4582. doi:10.1210/jc.2007-1499
- Hollowell JG, Staehling NW, Flanders WD, et al. Serum TSH, T(4), and thyroid antibodies in the United States population (1988 to 1994): National Health and Nutrition Examination Survey (NHANES III). J Clin Endocrinol Metab 2002; 87(2):489–499. doi:10.1210/jcem.87.2.8182
- Jonklaas J, Bianco AC, Bauer AJ, et al; American Thyroid Association Task Force on Thyroid Hormone Replacement. Guidelines for the treatment of hypothyroidism: prepared by the American Thyroid Association Task Force on Thyroid Hormone Replacement. Thyroid 2014; 24(12):1670–1751. doi:10.1089/thy.2014.0028
- Hennessey JV, Espaillat R. Diagnosis and management of subclinical hypothyroidism in elderly adults: a review of the literature. J Am Geriatr Soc 2015; 63(8):1663–1673. doi:10.1111/jgs.13532
- Razvi S, Shakoor A, Vanderpump M, Weaver JU, Pearce SH. The influence of age on the relationship between subclinical hypothyroidism and ischemic heart disease: a metaanalysis. J Clin Endocrinol Metab 2008; 93(8):2998–3007. doi:10.1210/jc.2008-0167
- Hamilton TE, Davis S, Onstad L, Kopecky KJ. Thyrotropin levels in a population with no clinical, autoantibody, or ultrasonographic evidence of thyroid disease: implications for the diagnosis of subclinical hypothyroidism. J Clin Endocrinol Metab 2008; 93(4):1224–1230. doi:10.1210/jc.2006-2300
- Canaris GJ, Manowitz NR, Mayor G, Ridgway EC. The Colorado thyroid disease prevalence study. Arch Intern Med 2000; 160(4):526–534. pmid:10695693
- Teng W, Shan Z, Teng X, et al. Effect of iodine intake on thyroid diseases in China. N Engl J Med 2006; 354(26):2783–2793. doi:10.1056/NEJMoa054022
- Negro R, Stagnaro-Green A. Diagnosis and management of subclinical hypothyroidism in pregnancy. BMJ 2014; 349:g4929. doi:10.1136/bmj.g4929
- Baumgartner C, Blum MR, Rodondi N. Subclinical hypothyroidism: summary of evidence in 2014. Swiss Med Wkly 2014; 144:w14058. doi:10.4414/smw.2014.14058
- Stedman TL. Stedman’s Medical Dictionary. 28th ed. Baltimore, MD: Lippincott Williams and Wilkins; 2006.
- Raza SA, Mahmood N. Subclinical hypothyroidism: controversies to consensus. Indian J Endocrinol Metab 2013; 17(suppl 3):S636–S642. doi:10.4103/2230-8210.123555
- Huber G, Staub JJ, Meier C, et al. Prospective study of the spontaneous course of subclinical hypothyroidism: prognostic value of thyrotropin, thyroid reserve, and thyroid antibodies. J Clin Endocrinol Metab 2002; 87(7):3221–3226. doi:10.1210/jcem.87.7.8678
- Diez JJ, Iglesias P, Burman KD. Spontaneous normalization of thyrotropin concentrations in patients with subclinical hypothyroidism. J Clin Endocrinol Metab 2005; 90(7):4124–4127. doi:10.1210/jc.2005-0375
- Vanderpump MP, Tunbridge WM, French JM, et al. The incidence of thyroid disorders in the community: a twenty-year follow-up of the Whickham survey. Clin Endocrinol (Oxf) 1995; 43(1):55–68. pmid:7641412
- Li Y, Teng D, Shan Z, et al. Antithyroperoxidase and antithyroglobulin antibodies in a five-year follow-up survey of populations with different iodine intakes. J Clin Endocrinol Metab 2008; 93(5):1751–1757. doi:10.1210/jc.2007-2368
- Hennessey JV, Klein I, Woeber KA, Cobin R, Garber JR. Aggressive case finding: a clinical strategy for the documentation of thyroid dysfunction. Ann Intern Med 2015; 163(4):311–312. doi:10.7326/M15-0762
- Rugge JB, Bougatsos C, Chou R. Screening and treatment of thyroid dysfunction: an evidence review for the US.Preventive Services Task Force. Ann Intern Med 2015; 162(1):35–45. doi:10.7326/M14-1456
- Ladenson PW, Singer PA, Ain KB, et al. American Thyroid Association guidelines for detection of thyroid dysfunction. Arch Intern Med 2000; 160(11):1573–1575. pmid:10847249
- Garber JR, Cobin RH, Gharib H, et al; American Association of Clinical Endocrinologists and American Thyroid Association Taskforce on Hypothyroidism in Adults. Clinical practice guidelines for hypothyroidism in adults: cosponsored by the American Association of Clinical Endocrinologists and the American Thyroid Association. Endocr Pract 2012; 18(6):988–1028. doi:10.4158/EP12280.GL
- Jorde R, Waterloo K, Storhaug H, Nyrnes A, Sundsfjord J, Jenssen TG. Neuropsychological function and symptoms in subjects with subclinical hypothyroidism and the effect of thyroxine treatment. J Clin Endocrinol Metab 2006; 91(1):145–153. doi:10.1210/jc.2005-1775
- Joffe RT, Pearce EN, Hennessey JV, Ryan JJ, Stern RA. Subclinical hypothyroidism, mood, and cognition in older adults: a review. Int J Geriatr Psychiatry 2013; 28(2):111–118. doi:10.1002/gps.3796
- Cooper DS, Halpern R, Wood LC, Levin AA, Ridgway EC. L-thyroxine therapy in subclinical hypothyroidism. A double-blind, placebo-controlled trial. Ann Intern Med 1984; 101(1):18–24. pmid:6428290
- Nystrom E, Caidahl K, Fager G, Wikkelso C, Lundberg PA, Lindstedt G. A double-blind cross-over 12-month study of L-thyroxine treatment of women with ‘subclinical’ hypothyroidism. Clin Endocrinol (Oxf) 1988; 29(1):63–75. pmid:3073880
- Monzani F, Del Guerra P, Caraccio N, et al. Subclinical hypothyroidism: neurobehavioral features and beneficial effect of L-thyroxine treatment. Clin Investig 1993; 71(5):367–371. pmid:8508006
- Biondi B. Thyroid and obesity: an intriguing relationship. J Clin Endocrinol Metab 2010; 95(8):3614–3617. doi:10.1210/jc.2010-1245
- Erdogan M, Canataroglu A, Ganidagli S, Kulaksizoglu M. Metabolic syndrome prevalence in subclinic and overt hypothyroid patients and the relation among metabolic syndrome parameters. J Endocrinol Invest 2011; 34(7):488–492. doi:10.3275/7202
- Javed Z, Sathyapalan T. Levothyroxine treatment of mild subclinical hypothyroidism: a review of potential risks and benefits. Ther Adv Endocrinol Metab 2016; 7(1):12–23. doi:10.1177/2042018815616543
- Pearce SH, Brabant G, Duntas LH, et al. 2013 ETA guideline: management of subclinical hypothyroidism. Eur Thyroid J 2013; 2(4):215–228. doi:10.1159/000356507
- Wang C. The relationship between type 2 diabetes mellitus and related thyroid diseases. J Diabetes Res 2013; 2013:390534. doi:10.1155/2013/390534
- Razvi S, Weaver JU, Vanderpump MP, Pearce SH. The incidence of ischemic heart disease and mortality in people with subclinical hypothyroidism: reanalysis of the Whickham survey cohort. J Clin Endocrinol Metab 2010; 95(4):1734–1740. doi:10.1210/jc.2009-1749
- Bindels AJ, Westendorp RG, Frolich M, Seidell JC, Blokstra A, Smelt AH. The prevalence of subclinical hypothyroidism at different total plasma cholesterol levels in middle aged men and women: a need for case-finding? Clin Endocrinol (Oxf) 1999; 50(2):217–220. pmid:10396365
- Pearce EN. Hypothyroidism and dyslipidemia: modern concepts and approaches. Curr Cardiol Rep 2004; 6(6):451–456. pmid:15485607
- Pearce EN. Update in lipid alterations in subclinical hypothyroidism. J Clin Endocrinol Metab 2012; 97(2):326–333. doi:10.1210/jc.2011-2532
- Rizos CV, Elisaf MS, Liberopoulos EN. Effects of thyroid dysfunction on lipid profile. Open Cardiovasc Med J 2011; 5:76–84. doi:10.2174/1874192401105010076
- Peppa M, Betsi G, Dimitriadis G. Lipid abnormalities and cardiometabolic risk in patients with overt and subclinical thyroid disease. J Lipids 2011; 2011:575840. doi:10.1155/2011/575840
- Asvold BO, Vatten LJ, Nilsen TI, Bjoro T. The association between TSH within the reference range and serum lipid concentrations in a population-based study. the HUNT study. Eur J Endocrinol 2007;1 56(2):181–186. doi:10.1530/eje.1.02333
- Danese MD, Ladenson PW, Meinert CL, Powe NR. Clinical review 115: effect of thyroxine therapy on serum lipoproteins in patients with mild thyroid failure: a quantitative review of the literature. J Clin Endocrinol Metab 2000; 85(9):2993–3001. doi:10.1210/jcem.85.9.6841
- Razvi S, Ingoe L, Keeka G, Oates C, McMillan C, Weaver JU. The beneficial effect of L-thyroxine on cardiovascular risk factors, endothelial function, and quality of life in subclinical hypothyroidism: randomized, crossover trial. J Clin Endocrinol Metab 2007; 92(5):1715–1723. doi:10.1210/jc.2006-1869
- Abreu IM, Lau E, de Sousa Pinto B, Carvalho D. Subclinical hypothyroidism: to treat or not to treat, that is the question! A systematic review with meta-analysis on lipid profile. Endocr Connect 2017; 6(3):188–199. doi:10.1530/EC-17-0028
- Robison CD, Bair TL, Horne BD, et al. Hypothyroidism as a risk factor for statin intolerance. J Clin Lipidol 2014; 8(4):401–407. doi:10.1016/j.jacl.2014.05.005
- Hak AE, Pols HA, Visser TJ, Drexhage HA, Hofman A, Witteman JC. Subclinical hypothyroidism is an independent risk factor for atherosclerosis and myocardial infarction in elderly women: the Rotterdam study. Ann Intern Med 2000; 132(4):270–278. pmid:10681281
- Boekholdt SM, Titan SM, Wiersinga WM, et al. Initial thyroid status and cardiovascular risk factors: the EPIC-Norfolk prospective population study. Clin Endocrinol (Oxf) 2010; 72(3):404–410. doi:10.1111/j.1365-2265.2009.03640.x
- Andersen MN, Olsen AM, Madsen JC, et al. Levothyroxine substitution in patients with subclinical hypothyroidism and the risk of myocardial infarction and mortality. PLoS One 2015; 10(6):e0129793. doi:10.1371/journal.pone.0129793
- Biondi B. Cardiovascular effects of mild hypothyroidism. Thyroid 2007; 17(7):625–630. doi:10.1089/thy.2007.0158
- Brenta G, Mutti LA, Schnitman M, Fretes O, Perrone A, Matute ML. Assessment of left ventricular diastolic function by radionuclide ventriculography at rest and exercise in subclinical hypothyroidism, and its response to L-thyroxine therapy. Am J Cardiol 2003; 91(11):1327–1330. pmid:12767425
- Taddei S, Caraccio N, Virdis A, et al. Impaired endothelium-dependent vasodilatation in subclinical hypothyroidism: beneficial effect of levothyroxine therapy. J Clin Endocrinol Metab 2003; 88(8):3731–3737. doi:10.1210/jc.2003-030039
- Gao N, Zhang W, Zhang YZ, Yang Q, Chen SH. Carotid intima-media thickness in patients with subclinical hypothyroidism: a meta-analysis. Atherosclerosis 2013; 227(1):18–25. doi:10.1016/j.atherosclerosis.2012.10.070
- Biondi B, Cooper DS. The clinical significance of subclinical thyroid dysfunction. Endocr Rev 2008; 29(1):76–131. doi:10.1210/er.2006-0043
- Chaker L, Baumgartner C, den Elzen WP, et al; Thyroid Studies Collaboration. Subclinical hypothyroidism and the risk of stroke events and fatal stroke: an individual participant data analysis. J Clin Endocrinol Metab 2015; 100(6):2181–2191. doi:10.1210/jc.2015-1438
- Monzani F, Di Bello V, Caraccio N, et al. Effect of levothyroxine on cardiac function and structure in subclinical hypothyroidism: a double blind, placebo-controlled study. J Clin Endocrinol Metab 2001; 86(3):1110–1115. doi:10.1210/jcem.86.3.7291
- Parle JV, Maisonneuve P, Sheppard MC, Boyle P, Franklyn JA. Prediction of all-cause and cardiovascular mortality in elderly people from one low serum thyrotropin result: a 10-year cohort study. Lancet 2001; 358(9285):861-865. doi:10.1016/S0140-6736(01)06067-6
- Razvi S, Weaver JU, Butler TJ, Pearce SH. Levothyroxine treatment of subclinical hypothyroidism, fatal and nonfatal cardiovascular events, and mortality. Arch Intern Med 2012; 172(10):811–817. doi:10.1001/archinternmed.2012.1159
- Pasqualetti G, Tognini S, Polini A, Caraccio N, Monzani F. Is subclinical hypothyroidism a cardiovascular risk factor in the elderly? J Clin Endocrinol Metab 2013; 98(6):2256–2266. doi:10.1210/jc.2012-3818
- Mooijaart SP, IEMO 80-plus Thyroid Trial Collaboration. Subclinical thyroid disorders. Lancet 2012; 380(9839):335. doi:10.1016/S0140-6736(12)61241-0
- Rodondi N, Bauer DC. Subclinical hypothyroidism and cardiovascular risk: how to end the controversy. J Clin Endocrinol Metab 2013; 98(6):2267–2269. doi:10.1210/jc.2013-1875
- Rodondi N, Newman AB, Vittinghoff E, et al. Subclinical hypothyroidism and the risk of heart failure, other cardiovascular events, and death. Arch Intern Med 2005; 165(21):2460–2466. doi:10.1001/archinte.165.21.2460
- Rodondi N, Bauer DC, Cappola AR, et al. Subclinical thyroid dysfunction, cardiac function, and the risk of heart failure. The Cardiovascular Health study. J Am Coll Cardiol 2008; 52(14):1152–1159. doi:10.1016/j.jacc.2008.07.009
- Haggerty JJ Jr, Garbutt JC, Evans DL, et al. Subclinical hypothyroidism: a review of neuropsychiatric aspects. Int J Psychiatry Med 1990; 20(2):193–208. doi:10.2190/ADLY-1UU0-1A8L-HPXY
- Baldini IM, Vita A, Mauri MC, et al. Psychopathological and cognitive features in subclinical hypothyroidism. Prog Neuropsychopharmacol Biol Psychiatry 1997; 21(6):925–935. pmid:9380789
- del Ser Quijano T, Delgado C, Martinez Espinosa S, Vazquez C. Cognitive deficiency in mild hypothyroidism. Neurologia 2000; 15(5):193–198. Spanish. pmid:10850118
- Correia N, Mullally S, Cooke G, et al. Evidence for a specific defect in hippocampal memory in overt and subclinical hypothyroidism. J Clin Endocrinol Metab 2009; 94(10):3789–3797. doi:10.1210/jc.2008-2702
- Aghili R, Khamseh ME, Malek M, et al. Changes of subtests of Wechsler memory scale and cognitive function in subjects with subclinical hypothyroidism following treatment with levothyroxine. Arch Med Sci 2012; 8(6):1096–1101. doi:10.5114/aoms.2012.32423
- Pasqualetti G, Pagano G, Rengo G, Ferrara N, Monzani F. Subclinical hypothyroidism and cognitive impairment: systematic review and meta-analysis. J Clin Endocrinol Metab 2015; 100(11):4240–4248. doi:10.1210/jc.2015-2046
- Christ-Crain M, Meier C, Huber PR, Staub J, Muller B. Effect of L-thyroxine replacement therapy on surrogate markers of skeletal and cardiac function in subclinical hypothyroidism. Endocrinologist 2004; 14(3):161–166. doi:10.1097/01.ten.0000127932.31710.4f
- Brennan MD, Powell C, Kaufman KR, Sun PC, Bahn RS, Nair KS. The impact of overt and subclinical hyperthyroidism on skeletal muscle. Thyroid 2006; 16(4):375–380. doi:10.1089/thy.2006.16.375
- Reuters VS, Teixeira Pde F, Vigario PS, et al. Functional capacity and muscular abnormalities in subclinical hypothyroidism. Am J Med Sci 2009; 338(4):259–263. doi:10.1097/MAJ.0b013e3181af7c7c
- Mainenti MR, Vigario PS, Teixeira PF, Maia MD, Oliveira FP, Vaisman M. Effect of levothyroxine replacement on exercise performance in subclinical hypothyroidism. J Endocrinol Invest 2009; 32(5):470–473. doi:10.3275/6106
- Lankhaar JA, de Vries WR, Jansen JA, Zelissen PM, Backx FJ. Impact of overt and subclinical hypothyroidism on exercise tolerance: a systematic review. Res Q Exerc Sport 2014; 85(3):365–389. doi:10.1080/02701367.2014.930405
- Lee JS, Buzkova P, Fink HA, et al. Subclinical thyroid dysfunction and incident hip fracture in older adults. Arch Intern Med 2010; 170(21):1876–1883. doi:10.1001/archinternmed.2010.424
- Svare A, Nilsen TI, Asvold BO, et al. Does thyroid function influence fracture risk? Prospective data from the HUNT2 study, Norway. Eur J Endocrinol 2013; 169(6):845–852. doi:10.1530/EJE-13-0546
- Di Mase R, Cerbone M, Improda N, et al. Bone health in children with long-term idiopathic subclinical hypothyroidism. Ital J Pediatr 2012; 38:56. doi:10.1186/1824-7288-38-56
- Boelaert K. The association between serum TSH concentration and thyroid cancer. Endocr Relat Cancer 2009; 16(4):1065–1072. doi:10.1677/ERC-09-0150
- Haymart MR, Glinberg SL, Liu J, Sippel RS, Jaume JC, Chen H. Higher serum TSH in thyroid cancer patients occurs independent of age and correlates with extrathyroidal extension. Clin Endocrinol (Oxf) 2009; 71(3):434–439. doi:10.1111/j.1365-2265.2008.03489.x
- Fiore E, Vitti P. Serum TSH and risk of papillary thyroid cancer in nodular thyroid disease. J Clin Endocrinol Metab 2012; 97(4):1134–1145. doi:10.1210/jc.2011-2735
- Fiore E, Rago T, Provenzale MA, et al. L-thyroxine-treated patients with nodular goiter have lower serum TSH and lower frequency of papillary thyroid cancer: results of a cross-sectional study on 27,914 patients. Endocr Relat Cancer 2010; 17(1):231–239. doi:10.1677/ERC-09-0251
- Hercbergs AH, Ashur-Fabian O, Garfield D. Thyroid hormones and cancer: clinical studies of hypothyroidism in oncology. Curr Opin Endocrinol Diabetes Obes 2010; 17(5):432–436. doi:10.1097/MED.0b013e32833d9710
- Thvilum M, Brandt F, Brix TH, Hegedus L. A review of the evidence for and against increased mortality in hypothyroidism. Nat Rev Endocrinol 2012; 8(7):417–424. doi:10.1038/nrendo.2012.29
- Stott DJ, Rodondi N, Kearney PM, et al; TRUST Study Group. Thyroid hormone therapy for older adults with subclinical hypothyroidism. N Engl J Med 2017; 376(26):2534–2544. doi:10.1056/NEJMoa1603825
- Practice Committee of the American Society for Reproductive Medicine. Subclinical hypothyroidism in the infertile female population: a guideline. Fertil Steril 2015; 104(3):545–753. doi:10.1016/j.fertnstert.2015.05.028
- Stagnaro-Green A, Abalovich M, Alexander E, et al; American Thyroid Association Taskforce on Thyroid Disease During Pregnancy and Postpartum. Guidelines of the American Thyroid Association for the diagnosis and management of thyroid disease during pregnancy and postpartum. Thyroid 2011; 21(10):1081–1125. doi:10.1089/thy.2011.0087
- Goldsmith RE, Sturgis SH, Lerman J, Stanbury JB. The menstrual pattern in thyroid disease. J Clin Endocrinol Metab. 1952; 12(7):846-855. doi:10.1210/jcem-12-7-846
- Plowden TC, Schisterman EF, Sjaarda LA, et al. Subclinical hypothyroidism and thyroid autoimmunity are not associated with fecundity, pregnancy loss, or live birth. J Clin Endocrinol Metab 2016; 101(6):2358–2365. doi:10.1210/jc.2016-1049
- Alexander EK, Pearce EN, Brent GA, et al. 2017 Guidelines of the American Thyroid Association for the diagnosis and management of thyroid disease during pregnancy and the postpartum. Thyroid 2017; 27(3):315–389. doi:10.1089/thy.2016.0457
- Negro R, Formoso G, Mangieri T, Pezzarossa A, Dazzi D, Hassan H. Levothyroxine treatment in euthyroid pregnant women with autoimmune thyroid disease: effects on obstetrical complications. J Clin Endocrinol Metab 2006; 91(7):2587–2591. doi:10.1210/jc.2005-1603
- Panesar NS, Li CY, Rogers MS. Reference intervals for thyroid hormones in pregnant Chinese women. Ann Clin Biochem 2001; 38(pt 4):329–332. doi:10.1258/0004563011900830
- Lepoutre T, Debieve F, Gruson D, Daumerie C. Reduction of miscarriages through universal screening and treatment of thyroid autoimmune diseases. Gynecol Obstet Invest 2012; 74(4):265–273. doi:10.1159/000343759
- De Groot L, Abalovich M, Alexander EK, et al. Management of thyroid dysfunction during pregnancy and postpartum: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2012; 97(8):2543–2565. doi:10.1210/jc.2011-2803
- Crawford NM, Steiner AZ. Thyroid autoimmunity and reproductive function. Semin Reprod Med 2016; 34(6):343–350. doi:10.1055/s-0036-1593485
- Maraka S, Ospina NM, O’Keeffe DT, et al. Subclinical hypothyroidism in pregnancy: a systematic review and meta-analysis. Thyroid 2016; 26(4):580–590. doi:10.1089/thy.2015.0418
- Wiles KS, Jarvis S, Nelson-Piercy C. Are we overtreating subclinical hypothyroidism in pregnancy? BMJ 2015; 351:h4726. doi:10.1136/bmj.h4726
- Tudela CM, Casey BM, McIntire DD, Cunningham FG. Relationship of subclinical thyroid disease to the incidence of gestational diabetes. Obstet Gynecol 2012; 119(5):983–988. doi:10.1097/AOG.0b013e318250aeeb
- Lazarus J, Brown RS, Daumerie C, Hubalewska-Dydejczyk A, Negro R, Vaidya B. 2014 European Thyroid Association guidelines for the management of subclinical hypothyroidism in pregnancy and in children. Eur Thyroid J 2014; 3(2):76–94. doi:10.1159/000362597
- Karakosta P, Alegakis D, Georgiou V, et al. Thyroid dysfunction and autoantibodies in early pregnancy are associated with increased risk of gestational diabetes and adverse birth outcomes. J Clin Endocrinol Metab 2012; 97(12):4464–4472. doi:10.1210/jc.2012-2540
- Toulis KA, Stagnaro-Green A, Negro R. Maternal subclinical hypothyroidsm and gestational diabetes mellitus: a meta-analysis. Endocr Pract 2014; 20(7):703–714. doi:10.4158/EP13440.RA
- van den Boogaard E, Vissenberg R, Land JA, et al. Significance of subclinical thyroid dysfunction and thyroid autoimmunity before conception and in early pregnancy: a systematic review. Hum Reprod Update 2011; 17(5):605–619. doi:10.1093/humupd/dmr024
- Wilson KL, Casey BM, McIntire DD, Halvorson LM, Cunningham FG. Subclinical thyroid disease and the incidence of hypertension in pregnancy. Obstet Gynecol 2012; 119(2 Pt 1):315–320. doi:10.1097/AOG.0b013e318240de6a
- Ashoor G, Maiz N, Rotas M, Jawdat F, Nicolaides KH. Maternal thyroid function at 11 to 13 weeks of gestation and subsequent fetal death. Thyroid 2010; 20(9):989–993. doi:10.1089/thy.2010.0058
- Casey BM, Dashe JS, Wells CE, et al. Subclinical hypothyroidism and pregnancy outcomes. Obstet Gynecol 2005; 105(2):239–245. doi:10.1097/01.AOG.0000152345.99421.22
- Negro R, Schwartz A, Gismondi R, Tinelli A, Mangieri T, Stagnaro-Green A. Increased pregnancy loss rate in thyroid antibody negative women with TSH levels between 2.5 and 5.0 in the first trimester of pregnancy. J Clin Endocrinol Metab 2010; 95(9):E44–E48. doi:10.1210/jc.2010-0340
- Su PY, Huang K, Hao JH, et al. Maternal thyroid function in the first twenty weeks of pregnancy and subsequent fetal and infant development: a prospective population-based cohort study in China. J Clin Endocrinol Metab 2011; 96(10):3234–3241. doi:10.1210/jc.2011-0274
- Allan WC, Haddow JE, Palomaki GE, et al. Maternal thyroid deficiency and pregnancy complications: implications for population screening. J Med Screen 2000; 7(3):127–130. doi:10.1136/jms.7.3.127
- Benhadi N, Wiersinga WM, Reitsma JB, Vrijkotte TG, Bonsel GJ. Higher maternal TSH levels in pregnancy are associated with increased risk for miscarriage, fetal or neonatal death. Eur J Endocrinol 2009; 160(6):985–991. doi:10.1530/EJE-08-0953
- Korevaar TI, Medici M, de Rijke YB, et al. Ethnic differences in maternal thyroid parameters during pregnancy: the generation R study. J Clin Endocrinol Metab 2013; 98(9):3678–3686. doi:10.1210/jc.2013-2005
- Cleary-Goldman J, Malone FD, Lambert-Messerlian G, et al. Maternal thyroid hypofunction and pregnancy outcome. Obstet Gynecol 2008; 112(1):85–92. doi:10.1097/AOG.0b013e3181788dd7
- Li Y, Shan Z, Teng W, et al. Abnormalities of maternal thyroid function during pregnancy affect neuropsychological development of their children at 25-30 months. Clin Endocrinol (Oxf) 2010; 72(6):825–829. doi:10.1111/j.1365-2265.2009.03743.x
- Haddow JE, Palomaki GE, Allan WC, et al. Maternal thyroid deficiency during pregnancy and subsequent neuropsychological development of the child. N Engl J Med 1999; 341(8):549–555. doi:10.1056/NEJM199908193410801
- Henrichs J, Bongers-Schokking JJ, Schenk JJ, et al. Maternal thyroid function during early pregnancy and cognitive functioning in early childhood: the generation R study. J Clin Endocrinol Metab 2010; 95(9):4227–4234. doi:10.1210/jc.2010-0415
- Behrooz HG, Tohidi M, Mehrabi Y, Behrooz EG, Tehranidoost M, Azizi F. Subclinical hypothyroidism in pregnancy: intellectual development of offspring. Thyroid 2011; 21(10):1143–1147. doi:10.1089/thy.2011.0053
- Julvez J, Alvarez-Pedrerol M, Rebagliato M, et al. Thyroxine levels during pregnancy in healthy women and early child neurodevelopment. Epidemiology 2013; 24(1):150–157. doi:10.1097/EDE.0b013e318276ccd3
- Casey BM, Thom EA, Peaceman AM, et al; Eunice Kennedy Shriver National Institute of Child Health and Human Development Maternal–Fetal Medicine Units Network. Treatment of subclinical hypothyroidism or hypothyroxinemia in pregnancy. N Engl J Med 2017; 376(9):815–825. doi:10.1056/NEJMoa1606205
- Burns RB, Bates CK, Hartzband P, Smetana GW. Should we treat for subclinical hypothyroidism?: Grand rounds discussion from Beth Israel Deaconess Medical Center. Ann Intern Med 2016; 164(11):764–770. doi:10.7326/M16-0857
- Kucukler FK, Akbaba G, Arduc A, Simsek Y, Guler S. Evaluation of the common mistakes made by patients in the use of levothyroxine. Eur J Intern Med 2014; 25(9):e107–e108. doi:10.1016/j.ejim.2014.09.002
- McMillan M, Rotenberg KS, Vora K, et al. Comorbidities, concomitant medications, and diet as factors affecting levothyroxine therapy: results of the CONTROL surveillance project. Drugs R D 2016; 16(1):53–68. doi:10.1007/s40268-015-0116-6
- Pollock MA, Sturrock A, Marshall K, et al. Thyroxine treatment in patients with symptoms of hypothyroidism but thyroid function tests within the reference range: Randomised double blind placebo controlled crossover trial. BMJ 2001; 323(7318):891–895. pmid:11668132
- Peeters RP. Subclinical hypothyroidism. N Engl J Med 2017; 376(26):2556–2565. doi:10.1056/NEJMcp1611144
Whether subclinical hypothyroidism is clinically important and should be treated remains controversial. Studies have differed in their findings, and although most have found this condition to be associated with a variety of adverse outcomes, large randomized controlled trials are needed to clearly demonstrate its clinical impact in various age groups and the benefit of levothyroxine therapy.
Currently, the best practical approach is to base treatment decisions on the magnitude of elevation of thyroid-stimulating hormone (TSH) and whether the patient has thyroid autoantibodies and associated comorbid conditions.
HIGH TSH, NORMAL FREE T4 LEVELS
Subclinical hypothyroidism is defined by elevated TSH along with a normal free thyroxine (T4).1
The hypothalamic-pituitary-thyroid axis is a balanced homeostatic system, and TSH and thyroid hormone levels have an inverse log-linear relation: if free T4 and triiodothyronine (T3) levels go down even a little, TSH levels go up a lot.2
TSH secretion is pulsatile and has a circadian rhythm: serum TSH levels are 50% higher at night and early in the morning than during the rest of the day. Thus, repeated measurements in the same patient can vary by as much as half of the reference range.3
WHAT IS THE UPPER LIMIT OF NORMAL FOR TSH?
The upper limit of normal for TSH, defined as the 97.5th percentile, is approximately 4 or 5 mIU/L depending on the laboratory and the population, but some experts believe it should be lower.3
In favor of a lower upper limit: the distribution of serum TSH levels in the healthy general population does not seem to be a typical bell-shaped Gaussian curve, but rather has a tail at the high end. Some argue that some of the individuals with values in the upper end of the normal range may actually have undiagnosed hypothyroidism and that the upper 97.5th percentile cutoff would be 2.5 mIU/L if these people were excluded.4 Also, TSH levels higher than 2.5 mIU/L have been associated with a higher prevalence of antithyroid antibodies and a higher risk of clinical hypothyroidism.5
On the other hand, lowering the upper limit of normal to 2.5 mIU/L would result in 4 times as many people receiving a diagnosis of subclinical hypothyroidism, or 22 to 28 million people in the United States.4,6 Thus, lowering the cutoff may lead to unnecessary therapy and could even harm from overtreatment.
Another argument against lowering the upper limit of normal for TSH is that, with age, serum TSH levels shift higher.7 The third National Health and Nutrition Education Survey (NHANES III) found that the 97.5th percentile for serum TSH was 3.56 mIU/L for age group 20 to 29 but 7.49 mIU/L for octogenarians.7,8
It has been suggested that the upper limit of normal for TSH be adjusted for age, race, sex, and iodine intake.3 Currently available TSH reference ranges are not adjusted for these variables, and there is not enough evidence to suggest age-appropriate ranges,9 although higher TSH cutoffs for treatment are advised in elderly patients.10 Interestingly, higher TSH in older people has been linked to lower mortality rates in some studies.11
Authors of the NHANES III8 and Hanford Thyroid Disease study12 have proposed a cutoff of 4.1 mIU/L for the upper limit of normal for serum TSH in patients with negative antithyroid antibodies and normal findings on thyroid ultrasonography.
SUBCLINICAL HYPOTHYROIDISM IS COMMON
In different studies, the prevalence of subclinical hypothyroidism has been as low as 4% and as high as 20%.1,8,13 The prevalence is higher in women and increases with age.8 It is higher in iodine-sufficient areas, and it increases in iodine-deficient areas with iodine supplementation.14 Genetics also plays a role, as subclinical hypothyroidism is more common in white people than in African Americans.8
A difficulty in estimating the prevalence is the disagreement about the cutoff for TSH, which may differ from that in the general population in certain subgroups such as adolescents, the elderly, and pregnant women.10,15
A VARIETY OF CAUSES
The most common cause of subclinical hypothyroidism, accounting for 60% to 80% of cases, is Hashimoto (autoimmune) thyroiditis,2 in which thyroid peroxidase antibodies are usually present.2,16
Also important to rule out are false-positive elevations due to substances that interfere with TSH assays (eg, heterophile antibodies, rheumatoid factor, biotin, macro-TSH); reversible causes such as the recovery phase of euthyroid sick syndrome; subacute, painless, or postpartum thyroiditis; central hypo- or hyperthyroidism; and thyroid hormone resistance.
SUBCLINICAL HYPOTHYROIDISM CAN RESOLVE OR PROGRESS
“Subclinical” suggests that the disease is in its early stage, with changes in TSH already apparent but decreases in thyroid hormone levels yet to come.17 And indeed, subclinical hypothyroidism can progress to overt hypothyroidism,18 although it has been reported to resolve spontaneously in half of cases within 2 years,19 typically in patients with TSH values of 4 to 6 mIU/L.20 The rate of progression to overt hypothyroidism is estimated to be 33% to 55% over 10 to 20 years of follow-up.18
Figure 1 shows the natural history of subclinical hypothyroidism.1
GUIDELINES FOR SCREENING DIFFER
Guidelines differ on screening for thyroid disease in the general population, owing to lack of large-scale randomized controlled trials showing treatment benefit in otherwise-healthy people with mildly elevated TSH values.
Various professional societies have adopted different criteria for aggressive case-finding in patients at risk of thyroid disease. Risk factors include family history of thyroid disease, neck irradiation, partial thyroidectomy, dyslipidemia, atrial fibrillation, unexplained weight loss, hyperprolactinemia, autoimmune disorders, and use of medications affecting thyroid function.23
The US Preventive Services Task Force in 2014 found insufficient evidence on the benefits and harms of screening.24
The American Thyroid Association (ATA) recommends screening adults starting at age 35, with repeat testing every 5 years in patients who have no signs or symptoms of hypothyroidism, and more frequently in those who do.25
The American Association of Clinical Endocrinologists recommends screening in women and older patients. Their guidelines and those of the ATA also suggest screening people at high risk of thyroid disease due to risk factors such as history of autoimmune diseases, neck irradiation, or medications affecting thyroid function.26
The American Academy of Family Physicians recommends screening after age 60.18
The American College of Physicians recommends screening patients over age 50 who have symptoms.18
Our approach. Although evidence is lacking to recommend routine screening in adults, aggressive case-finding and treatment in patients at risk of thyroid disease can, we believe, offset the risks associated with subclinical hypothyroidism.24
CLINICAL PRESENTATION
About 70% of patients with subclinical hypothyroidism have no symptoms.13
Tiredness was more common in subclinical hypothyroid patients with TSH levels lower than 10 mIU/L compared with euthyroid controls in 1 study, but other studies have been unable to replicate this finding.27,28
Other frequently reported symptoms include dry skin, cognitive slowing, poor memory, muscle weakness, cold intolerance, constipation, puffy eyes, and hoarseness.13
The evidence in favor of levothyroxine therapy to improve symptoms in subclinical hypothyroidism has varied, with some studies showing an improvement in symptom scores compared with placebo, while others have not shown any benefit.29–31
In one study, the average TSH value for patients whose symptoms did not improve with therapy was 4.6 mIU/L.31 An explanation for the lack of effect in this group may be that the TSH values for these patients were in the high-normal range. Also, because most subclinical hypothyroid patients have no symptoms, it is difficult to ascertain symptomatic improvement. Though it is possible to conclude that levothyroxine therapy has a limited role in this group, it is important to also consider the suggestive evidence that untreated subclinical hypothyroidism may lead to increased morbidity and mortality.
ADVERSE EFFECTS OF SUBCLINICAL HYPOTHYROIDISM, EFFECTS OF THERAPY
INDIVIDUALIZED MANAGEMENT AND SHARED DECISION-MAKING
The management of subclinical hypothyroidism should be individualized on the basis of extent of thyroid dysfunction, comorbid conditions, risk factors, and patient preference.118 Shared decision-making is key, weighing the risks and benefits of levothyroxine treatment and the patient’s goals.
The risks of treatment should be kept in mind and explained to the patient. Levothyroxine has a narrow therapeutic range, causing a possibility of overreplacement, and a half-life of 7 days that can cause dosing errors to have longer effect.118,119
Adherence can be a challenge. The drug needs to be taken on an empty stomach because foods and supplements interfere with its absorption.118,120 In addition, the cost of medication, frequent biochemical monitoring, and possible need for titration can add to financial burden.
When choosing the dose, one should consider the degree of hypothyroidism or TSH elevation and the patient’s weight, and adjust the dose gently.
If the TSH is high-normal
It is proposed that a TSH range of 3 to 5 mIU/L overlaps with normal thyroid function in a great segment of the population, and at this level it is probably not associated with clinically significant consequences. For these reasons, levothyroxine therapy is not thought to be beneficial for those with TSH in this range.
Pollock et al121 found that, in patients with symptoms suggesting hypothyroidism and TSH values in the upper end of the normal range, there was no improvement in cognitive function or psychological well-being after 12 weeks of levothyroxine therapy.
However, due to the concern for possible adverse maternal and fetal outcomes and low IQ in children of pregnant patients with subclinical hypothyroidism, levothyroxine therapy is advised in those who are pregnant or planning pregnancy who have TSH levels higher than 2.5 mIU/L, especially if they have thyroid peroxidase antibody. Levothyroxine therapy is not recommended for pregnant patients with negative thyroid peroxidase antibody and TSH within the pregnancy-specific range or less than 4 mIU/L if the reference ranges are unavailable.
Keep in mind that, even at these TSH values, there is risk of progression to overt hypothyroidism, especially in the presence of thyroid peroxidase antibody, so patients in this group should be monitored closely.
If TSH is mildly elevated
The evidence to support levothyroxine therapy in patients with subclinical hypothyroidism with TSH levels less than 10 mIU/L remains inconclusive, and the decision to treat should be based on clinical judgment.2 The studies that have looked at the benefit of treating subclinical hypothyroidism in terms of cardiac, neuromuscular, cognitive, and neuropsychiatric outcomes have included patients with a wide range of TSH levels, and some of these studies were not stratified on the basis of degree of TSH elevation.
The risk that subclinical hypothyroidism will progress to overt hypothyroidism in patients with TSH higher than 8 mIU/L is high, and in 70% of these patients, the TSH level rises to more than 10 mIU/L within 4 years. Early treatment should be considered if the TSH is higher than 7 or 8 mIU/L.
If TSH is higher than 10 mIU/L
Figure 2 outlines an algorithmic approach to subclinical hypothyroidism in nonpregnant patients as suggested by Peeters.122
Whether subclinical hypothyroidism is clinically important and should be treated remains controversial. Studies have differed in their findings, and although most have found this condition to be associated with a variety of adverse outcomes, large randomized controlled trials are needed to clearly demonstrate its clinical impact in various age groups and the benefit of levothyroxine therapy.
Currently, the best practical approach is to base treatment decisions on the magnitude of elevation of thyroid-stimulating hormone (TSH) and whether the patient has thyroid autoantibodies and associated comorbid conditions.
HIGH TSH, NORMAL FREE T4 LEVELS
Subclinical hypothyroidism is defined by elevated TSH along with a normal free thyroxine (T4).1
The hypothalamic-pituitary-thyroid axis is a balanced homeostatic system, and TSH and thyroid hormone levels have an inverse log-linear relation: if free T4 and triiodothyronine (T3) levels go down even a little, TSH levels go up a lot.2
TSH secretion is pulsatile and has a circadian rhythm: serum TSH levels are 50% higher at night and early in the morning than during the rest of the day. Thus, repeated measurements in the same patient can vary by as much as half of the reference range.3
WHAT IS THE UPPER LIMIT OF NORMAL FOR TSH?
The upper limit of normal for TSH, defined as the 97.5th percentile, is approximately 4 or 5 mIU/L depending on the laboratory and the population, but some experts believe it should be lower.3
In favor of a lower upper limit: the distribution of serum TSH levels in the healthy general population does not seem to be a typical bell-shaped Gaussian curve, but rather has a tail at the high end. Some argue that some of the individuals with values in the upper end of the normal range may actually have undiagnosed hypothyroidism and that the upper 97.5th percentile cutoff would be 2.5 mIU/L if these people were excluded.4 Also, TSH levels higher than 2.5 mIU/L have been associated with a higher prevalence of antithyroid antibodies and a higher risk of clinical hypothyroidism.5
On the other hand, lowering the upper limit of normal to 2.5 mIU/L would result in 4 times as many people receiving a diagnosis of subclinical hypothyroidism, or 22 to 28 million people in the United States.4,6 Thus, lowering the cutoff may lead to unnecessary therapy and could even harm from overtreatment.
Another argument against lowering the upper limit of normal for TSH is that, with age, serum TSH levels shift higher.7 The third National Health and Nutrition Education Survey (NHANES III) found that the 97.5th percentile for serum TSH was 3.56 mIU/L for age group 20 to 29 but 7.49 mIU/L for octogenarians.7,8
It has been suggested that the upper limit of normal for TSH be adjusted for age, race, sex, and iodine intake.3 Currently available TSH reference ranges are not adjusted for these variables, and there is not enough evidence to suggest age-appropriate ranges,9 although higher TSH cutoffs for treatment are advised in elderly patients.10 Interestingly, higher TSH in older people has been linked to lower mortality rates in some studies.11
Authors of the NHANES III8 and Hanford Thyroid Disease study12 have proposed a cutoff of 4.1 mIU/L for the upper limit of normal for serum TSH in patients with negative antithyroid antibodies and normal findings on thyroid ultrasonography.
SUBCLINICAL HYPOTHYROIDISM IS COMMON
In different studies, the prevalence of subclinical hypothyroidism has been as low as 4% and as high as 20%.1,8,13 The prevalence is higher in women and increases with age.8 It is higher in iodine-sufficient areas, and it increases in iodine-deficient areas with iodine supplementation.14 Genetics also plays a role, as subclinical hypothyroidism is more common in white people than in African Americans.8
A difficulty in estimating the prevalence is the disagreement about the cutoff for TSH, which may differ from that in the general population in certain subgroups such as adolescents, the elderly, and pregnant women.10,15
A VARIETY OF CAUSES
The most common cause of subclinical hypothyroidism, accounting for 60% to 80% of cases, is Hashimoto (autoimmune) thyroiditis,2 in which thyroid peroxidase antibodies are usually present.2,16
Also important to rule out are false-positive elevations due to substances that interfere with TSH assays (eg, heterophile antibodies, rheumatoid factor, biotin, macro-TSH); reversible causes such as the recovery phase of euthyroid sick syndrome; subacute, painless, or postpartum thyroiditis; central hypo- or hyperthyroidism; and thyroid hormone resistance.
SUBCLINICAL HYPOTHYROIDISM CAN RESOLVE OR PROGRESS
“Subclinical” suggests that the disease is in its early stage, with changes in TSH already apparent but decreases in thyroid hormone levels yet to come.17 And indeed, subclinical hypothyroidism can progress to overt hypothyroidism,18 although it has been reported to resolve spontaneously in half of cases within 2 years,19 typically in patients with TSH values of 4 to 6 mIU/L.20 The rate of progression to overt hypothyroidism is estimated to be 33% to 55% over 10 to 20 years of follow-up.18
Figure 1 shows the natural history of subclinical hypothyroidism.1
GUIDELINES FOR SCREENING DIFFER
Guidelines differ on screening for thyroid disease in the general population, owing to lack of large-scale randomized controlled trials showing treatment benefit in otherwise-healthy people with mildly elevated TSH values.
Various professional societies have adopted different criteria for aggressive case-finding in patients at risk of thyroid disease. Risk factors include family history of thyroid disease, neck irradiation, partial thyroidectomy, dyslipidemia, atrial fibrillation, unexplained weight loss, hyperprolactinemia, autoimmune disorders, and use of medications affecting thyroid function.23
The US Preventive Services Task Force in 2014 found insufficient evidence on the benefits and harms of screening.24
The American Thyroid Association (ATA) recommends screening adults starting at age 35, with repeat testing every 5 years in patients who have no signs or symptoms of hypothyroidism, and more frequently in those who do.25
The American Association of Clinical Endocrinologists recommends screening in women and older patients. Their guidelines and those of the ATA also suggest screening people at high risk of thyroid disease due to risk factors such as history of autoimmune diseases, neck irradiation, or medications affecting thyroid function.26
The American Academy of Family Physicians recommends screening after age 60.18
The American College of Physicians recommends screening patients over age 50 who have symptoms.18
Our approach. Although evidence is lacking to recommend routine screening in adults, aggressive case-finding and treatment in patients at risk of thyroid disease can, we believe, offset the risks associated with subclinical hypothyroidism.24
CLINICAL PRESENTATION
About 70% of patients with subclinical hypothyroidism have no symptoms.13
Tiredness was more common in subclinical hypothyroid patients with TSH levels lower than 10 mIU/L compared with euthyroid controls in 1 study, but other studies have been unable to replicate this finding.27,28
Other frequently reported symptoms include dry skin, cognitive slowing, poor memory, muscle weakness, cold intolerance, constipation, puffy eyes, and hoarseness.13
The evidence in favor of levothyroxine therapy to improve symptoms in subclinical hypothyroidism has varied, with some studies showing an improvement in symptom scores compared with placebo, while others have not shown any benefit.29–31
In one study, the average TSH value for patients whose symptoms did not improve with therapy was 4.6 mIU/L.31 An explanation for the lack of effect in this group may be that the TSH values for these patients were in the high-normal range. Also, because most subclinical hypothyroid patients have no symptoms, it is difficult to ascertain symptomatic improvement. Though it is possible to conclude that levothyroxine therapy has a limited role in this group, it is important to also consider the suggestive evidence that untreated subclinical hypothyroidism may lead to increased morbidity and mortality.
ADVERSE EFFECTS OF SUBCLINICAL HYPOTHYROIDISM, EFFECTS OF THERAPY
INDIVIDUALIZED MANAGEMENT AND SHARED DECISION-MAKING
The management of subclinical hypothyroidism should be individualized on the basis of extent of thyroid dysfunction, comorbid conditions, risk factors, and patient preference.118 Shared decision-making is key, weighing the risks and benefits of levothyroxine treatment and the patient’s goals.
The risks of treatment should be kept in mind and explained to the patient. Levothyroxine has a narrow therapeutic range, causing a possibility of overreplacement, and a half-life of 7 days that can cause dosing errors to have longer effect.118,119
Adherence can be a challenge. The drug needs to be taken on an empty stomach because foods and supplements interfere with its absorption.118,120 In addition, the cost of medication, frequent biochemical monitoring, and possible need for titration can add to financial burden.
When choosing the dose, one should consider the degree of hypothyroidism or TSH elevation and the patient’s weight, and adjust the dose gently.
If the TSH is high-normal
It is proposed that a TSH range of 3 to 5 mIU/L overlaps with normal thyroid function in a great segment of the population, and at this level it is probably not associated with clinically significant consequences. For these reasons, levothyroxine therapy is not thought to be beneficial for those with TSH in this range.
Pollock et al121 found that, in patients with symptoms suggesting hypothyroidism and TSH values in the upper end of the normal range, there was no improvement in cognitive function or psychological well-being after 12 weeks of levothyroxine therapy.
However, due to the concern for possible adverse maternal and fetal outcomes and low IQ in children of pregnant patients with subclinical hypothyroidism, levothyroxine therapy is advised in those who are pregnant or planning pregnancy who have TSH levels higher than 2.5 mIU/L, especially if they have thyroid peroxidase antibody. Levothyroxine therapy is not recommended for pregnant patients with negative thyroid peroxidase antibody and TSH within the pregnancy-specific range or less than 4 mIU/L if the reference ranges are unavailable.
Keep in mind that, even at these TSH values, there is risk of progression to overt hypothyroidism, especially in the presence of thyroid peroxidase antibody, so patients in this group should be monitored closely.
If TSH is mildly elevated
The evidence to support levothyroxine therapy in patients with subclinical hypothyroidism with TSH levels less than 10 mIU/L remains inconclusive, and the decision to treat should be based on clinical judgment.2 The studies that have looked at the benefit of treating subclinical hypothyroidism in terms of cardiac, neuromuscular, cognitive, and neuropsychiatric outcomes have included patients with a wide range of TSH levels, and some of these studies were not stratified on the basis of degree of TSH elevation.
The risk that subclinical hypothyroidism will progress to overt hypothyroidism in patients with TSH higher than 8 mIU/L is high, and in 70% of these patients, the TSH level rises to more than 10 mIU/L within 4 years. Early treatment should be considered if the TSH is higher than 7 or 8 mIU/L.
If TSH is higher than 10 mIU/L
Figure 2 outlines an algorithmic approach to subclinical hypothyroidism in nonpregnant patients as suggested by Peeters.122
- Cooper DS, Biondi B. Subclinical thyroid disease. Lancet 2012; 379(9821):1142–1154. doi:10.1016/S0140-6736(11)60276-6
- Fatourechi V. Subclinical hypothyroidism: an update for primary care physicians. Mayo Clin Proc 2009; 84(1):65–71. doi:10.4065/84.1.65
- Laurberg P, Andersen S, Carle A, Karmisholt J, Knudsen N, Pedersen IB. The TSH upper reference limit: where are we at? Nat Rev Endocrinol 2011; 7(4):232–239. doi:10.1038/nrendo.2011.13
- Wartofsky L, Dickey RA. The evidence for a narrower thyrotropin reference range is compelling. J Clin Endocrinol Metab 2005; 90(9):5483–5488. doi:10.1210/jc.2005-0455
- Spencer CA, Hollowell JG, Kazarosyan M, Braverman LE. National Health and Nutrition Examination Survey III thyroid-stimulating hormone (TSH)-thyroperoxidase antibody relationships demonstrate that TSH upper reference limits may be skewed by occult thyroid dysfunction. J Clin Endocrinol Metab 2007; 92(11):4236–4240. doi:10.1210/jc.2007-0287
- Fatourechi V, Klee GG, Grebe SK, et al. Effects of reducing the upper limit of normal TSH values. JAMA 2003; 290(24):3195–3196. doi:10.1001/jama.290.24.3195-a
- Surks MI, Hollowell JG. Age-specific distribution of serum thyrotropin and antithyroid antibodies in the US population: implications for the prevalence of subclinical hypothyroidism. J Clin Endocrinol Metab 2007; 92(12):4575–4582. doi:10.1210/jc.2007-1499
- Hollowell JG, Staehling NW, Flanders WD, et al. Serum TSH, T(4), and thyroid antibodies in the United States population (1988 to 1994): National Health and Nutrition Examination Survey (NHANES III). J Clin Endocrinol Metab 2002; 87(2):489–499. doi:10.1210/jcem.87.2.8182
- Jonklaas J, Bianco AC, Bauer AJ, et al; American Thyroid Association Task Force on Thyroid Hormone Replacement. Guidelines for the treatment of hypothyroidism: prepared by the American Thyroid Association Task Force on Thyroid Hormone Replacement. Thyroid 2014; 24(12):1670–1751. doi:10.1089/thy.2014.0028
- Hennessey JV, Espaillat R. Diagnosis and management of subclinical hypothyroidism in elderly adults: a review of the literature. J Am Geriatr Soc 2015; 63(8):1663–1673. doi:10.1111/jgs.13532
- Razvi S, Shakoor A, Vanderpump M, Weaver JU, Pearce SH. The influence of age on the relationship between subclinical hypothyroidism and ischemic heart disease: a metaanalysis. J Clin Endocrinol Metab 2008; 93(8):2998–3007. doi:10.1210/jc.2008-0167
- Hamilton TE, Davis S, Onstad L, Kopecky KJ. Thyrotropin levels in a population with no clinical, autoantibody, or ultrasonographic evidence of thyroid disease: implications for the diagnosis of subclinical hypothyroidism. J Clin Endocrinol Metab 2008; 93(4):1224–1230. doi:10.1210/jc.2006-2300
- Canaris GJ, Manowitz NR, Mayor G, Ridgway EC. The Colorado thyroid disease prevalence study. Arch Intern Med 2000; 160(4):526–534. pmid:10695693
- Teng W, Shan Z, Teng X, et al. Effect of iodine intake on thyroid diseases in China. N Engl J Med 2006; 354(26):2783–2793. doi:10.1056/NEJMoa054022
- Negro R, Stagnaro-Green A. Diagnosis and management of subclinical hypothyroidism in pregnancy. BMJ 2014; 349:g4929. doi:10.1136/bmj.g4929
- Baumgartner C, Blum MR, Rodondi N. Subclinical hypothyroidism: summary of evidence in 2014. Swiss Med Wkly 2014; 144:w14058. doi:10.4414/smw.2014.14058
- Stedman TL. Stedman’s Medical Dictionary. 28th ed. Baltimore, MD: Lippincott Williams and Wilkins; 2006.
- Raza SA, Mahmood N. Subclinical hypothyroidism: controversies to consensus. Indian J Endocrinol Metab 2013; 17(suppl 3):S636–S642. doi:10.4103/2230-8210.123555
- Huber G, Staub JJ, Meier C, et al. Prospective study of the spontaneous course of subclinical hypothyroidism: prognostic value of thyrotropin, thyroid reserve, and thyroid antibodies. J Clin Endocrinol Metab 2002; 87(7):3221–3226. doi:10.1210/jcem.87.7.8678
- Diez JJ, Iglesias P, Burman KD. Spontaneous normalization of thyrotropin concentrations in patients with subclinical hypothyroidism. J Clin Endocrinol Metab 2005; 90(7):4124–4127. doi:10.1210/jc.2005-0375
- Vanderpump MP, Tunbridge WM, French JM, et al. The incidence of thyroid disorders in the community: a twenty-year follow-up of the Whickham survey. Clin Endocrinol (Oxf) 1995; 43(1):55–68. pmid:7641412
- Li Y, Teng D, Shan Z, et al. Antithyroperoxidase and antithyroglobulin antibodies in a five-year follow-up survey of populations with different iodine intakes. J Clin Endocrinol Metab 2008; 93(5):1751–1757. doi:10.1210/jc.2007-2368
- Hennessey JV, Klein I, Woeber KA, Cobin R, Garber JR. Aggressive case finding: a clinical strategy for the documentation of thyroid dysfunction. Ann Intern Med 2015; 163(4):311–312. doi:10.7326/M15-0762
- Rugge JB, Bougatsos C, Chou R. Screening and treatment of thyroid dysfunction: an evidence review for the US.Preventive Services Task Force. Ann Intern Med 2015; 162(1):35–45. doi:10.7326/M14-1456
- Ladenson PW, Singer PA, Ain KB, et al. American Thyroid Association guidelines for detection of thyroid dysfunction. Arch Intern Med 2000; 160(11):1573–1575. pmid:10847249
- Garber JR, Cobin RH, Gharib H, et al; American Association of Clinical Endocrinologists and American Thyroid Association Taskforce on Hypothyroidism in Adults. Clinical practice guidelines for hypothyroidism in adults: cosponsored by the American Association of Clinical Endocrinologists and the American Thyroid Association. Endocr Pract 2012; 18(6):988–1028. doi:10.4158/EP12280.GL
- Jorde R, Waterloo K, Storhaug H, Nyrnes A, Sundsfjord J, Jenssen TG. Neuropsychological function and symptoms in subjects with subclinical hypothyroidism and the effect of thyroxine treatment. J Clin Endocrinol Metab 2006; 91(1):145–153. doi:10.1210/jc.2005-1775
- Joffe RT, Pearce EN, Hennessey JV, Ryan JJ, Stern RA. Subclinical hypothyroidism, mood, and cognition in older adults: a review. Int J Geriatr Psychiatry 2013; 28(2):111–118. doi:10.1002/gps.3796
- Cooper DS, Halpern R, Wood LC, Levin AA, Ridgway EC. L-thyroxine therapy in subclinical hypothyroidism. A double-blind, placebo-controlled trial. Ann Intern Med 1984; 101(1):18–24. pmid:6428290
- Nystrom E, Caidahl K, Fager G, Wikkelso C, Lundberg PA, Lindstedt G. A double-blind cross-over 12-month study of L-thyroxine treatment of women with ‘subclinical’ hypothyroidism. Clin Endocrinol (Oxf) 1988; 29(1):63–75. pmid:3073880
- Monzani F, Del Guerra P, Caraccio N, et al. Subclinical hypothyroidism: neurobehavioral features and beneficial effect of L-thyroxine treatment. Clin Investig 1993; 71(5):367–371. pmid:8508006
- Biondi B. Thyroid and obesity: an intriguing relationship. J Clin Endocrinol Metab 2010; 95(8):3614–3617. doi:10.1210/jc.2010-1245
- Erdogan M, Canataroglu A, Ganidagli S, Kulaksizoglu M. Metabolic syndrome prevalence in subclinic and overt hypothyroid patients and the relation among metabolic syndrome parameters. J Endocrinol Invest 2011; 34(7):488–492. doi:10.3275/7202
- Javed Z, Sathyapalan T. Levothyroxine treatment of mild subclinical hypothyroidism: a review of potential risks and benefits. Ther Adv Endocrinol Metab 2016; 7(1):12–23. doi:10.1177/2042018815616543
- Pearce SH, Brabant G, Duntas LH, et al. 2013 ETA guideline: management of subclinical hypothyroidism. Eur Thyroid J 2013; 2(4):215–228. doi:10.1159/000356507
- Wang C. The relationship between type 2 diabetes mellitus and related thyroid diseases. J Diabetes Res 2013; 2013:390534. doi:10.1155/2013/390534
- Razvi S, Weaver JU, Vanderpump MP, Pearce SH. The incidence of ischemic heart disease and mortality in people with subclinical hypothyroidism: reanalysis of the Whickham survey cohort. J Clin Endocrinol Metab 2010; 95(4):1734–1740. doi:10.1210/jc.2009-1749
- Bindels AJ, Westendorp RG, Frolich M, Seidell JC, Blokstra A, Smelt AH. The prevalence of subclinical hypothyroidism at different total plasma cholesterol levels in middle aged men and women: a need for case-finding? Clin Endocrinol (Oxf) 1999; 50(2):217–220. pmid:10396365
- Pearce EN. Hypothyroidism and dyslipidemia: modern concepts and approaches. Curr Cardiol Rep 2004; 6(6):451–456. pmid:15485607
- Pearce EN. Update in lipid alterations in subclinical hypothyroidism. J Clin Endocrinol Metab 2012; 97(2):326–333. doi:10.1210/jc.2011-2532
- Rizos CV, Elisaf MS, Liberopoulos EN. Effects of thyroid dysfunction on lipid profile. Open Cardiovasc Med J 2011; 5:76–84. doi:10.2174/1874192401105010076
- Peppa M, Betsi G, Dimitriadis G. Lipid abnormalities and cardiometabolic risk in patients with overt and subclinical thyroid disease. J Lipids 2011; 2011:575840. doi:10.1155/2011/575840
- Asvold BO, Vatten LJ, Nilsen TI, Bjoro T. The association between TSH within the reference range and serum lipid concentrations in a population-based study. the HUNT study. Eur J Endocrinol 2007;1 56(2):181–186. doi:10.1530/eje.1.02333
- Danese MD, Ladenson PW, Meinert CL, Powe NR. Clinical review 115: effect of thyroxine therapy on serum lipoproteins in patients with mild thyroid failure: a quantitative review of the literature. J Clin Endocrinol Metab 2000; 85(9):2993–3001. doi:10.1210/jcem.85.9.6841
- Razvi S, Ingoe L, Keeka G, Oates C, McMillan C, Weaver JU. The beneficial effect of L-thyroxine on cardiovascular risk factors, endothelial function, and quality of life in subclinical hypothyroidism: randomized, crossover trial. J Clin Endocrinol Metab 2007; 92(5):1715–1723. doi:10.1210/jc.2006-1869
- Abreu IM, Lau E, de Sousa Pinto B, Carvalho D. Subclinical hypothyroidism: to treat or not to treat, that is the question! A systematic review with meta-analysis on lipid profile. Endocr Connect 2017; 6(3):188–199. doi:10.1530/EC-17-0028
- Robison CD, Bair TL, Horne BD, et al. Hypothyroidism as a risk factor for statin intolerance. J Clin Lipidol 2014; 8(4):401–407. doi:10.1016/j.jacl.2014.05.005
- Hak AE, Pols HA, Visser TJ, Drexhage HA, Hofman A, Witteman JC. Subclinical hypothyroidism is an independent risk factor for atherosclerosis and myocardial infarction in elderly women: the Rotterdam study. Ann Intern Med 2000; 132(4):270–278. pmid:10681281
- Boekholdt SM, Titan SM, Wiersinga WM, et al. Initial thyroid status and cardiovascular risk factors: the EPIC-Norfolk prospective population study. Clin Endocrinol (Oxf) 2010; 72(3):404–410. doi:10.1111/j.1365-2265.2009.03640.x
- Andersen MN, Olsen AM, Madsen JC, et al. Levothyroxine substitution in patients with subclinical hypothyroidism and the risk of myocardial infarction and mortality. PLoS One 2015; 10(6):e0129793. doi:10.1371/journal.pone.0129793
- Biondi B. Cardiovascular effects of mild hypothyroidism. Thyroid 2007; 17(7):625–630. doi:10.1089/thy.2007.0158
- Brenta G, Mutti LA, Schnitman M, Fretes O, Perrone A, Matute ML. Assessment of left ventricular diastolic function by radionuclide ventriculography at rest and exercise in subclinical hypothyroidism, and its response to L-thyroxine therapy. Am J Cardiol 2003; 91(11):1327–1330. pmid:12767425
- Taddei S, Caraccio N, Virdis A, et al. Impaired endothelium-dependent vasodilatation in subclinical hypothyroidism: beneficial effect of levothyroxine therapy. J Clin Endocrinol Metab 2003; 88(8):3731–3737. doi:10.1210/jc.2003-030039
- Gao N, Zhang W, Zhang YZ, Yang Q, Chen SH. Carotid intima-media thickness in patients with subclinical hypothyroidism: a meta-analysis. Atherosclerosis 2013; 227(1):18–25. doi:10.1016/j.atherosclerosis.2012.10.070
- Biondi B, Cooper DS. The clinical significance of subclinical thyroid dysfunction. Endocr Rev 2008; 29(1):76–131. doi:10.1210/er.2006-0043
- Chaker L, Baumgartner C, den Elzen WP, et al; Thyroid Studies Collaboration. Subclinical hypothyroidism and the risk of stroke events and fatal stroke: an individual participant data analysis. J Clin Endocrinol Metab 2015; 100(6):2181–2191. doi:10.1210/jc.2015-1438
- Monzani F, Di Bello V, Caraccio N, et al. Effect of levothyroxine on cardiac function and structure in subclinical hypothyroidism: a double blind, placebo-controlled study. J Clin Endocrinol Metab 2001; 86(3):1110–1115. doi:10.1210/jcem.86.3.7291
- Parle JV, Maisonneuve P, Sheppard MC, Boyle P, Franklyn JA. Prediction of all-cause and cardiovascular mortality in elderly people from one low serum thyrotropin result: a 10-year cohort study. Lancet 2001; 358(9285):861-865. doi:10.1016/S0140-6736(01)06067-6
- Razvi S, Weaver JU, Butler TJ, Pearce SH. Levothyroxine treatment of subclinical hypothyroidism, fatal and nonfatal cardiovascular events, and mortality. Arch Intern Med 2012; 172(10):811–817. doi:10.1001/archinternmed.2012.1159
- Pasqualetti G, Tognini S, Polini A, Caraccio N, Monzani F. Is subclinical hypothyroidism a cardiovascular risk factor in the elderly? J Clin Endocrinol Metab 2013; 98(6):2256–2266. doi:10.1210/jc.2012-3818
- Mooijaart SP, IEMO 80-plus Thyroid Trial Collaboration. Subclinical thyroid disorders. Lancet 2012; 380(9839):335. doi:10.1016/S0140-6736(12)61241-0
- Rodondi N, Bauer DC. Subclinical hypothyroidism and cardiovascular risk: how to end the controversy. J Clin Endocrinol Metab 2013; 98(6):2267–2269. doi:10.1210/jc.2013-1875
- Rodondi N, Newman AB, Vittinghoff E, et al. Subclinical hypothyroidism and the risk of heart failure, other cardiovascular events, and death. Arch Intern Med 2005; 165(21):2460–2466. doi:10.1001/archinte.165.21.2460
- Rodondi N, Bauer DC, Cappola AR, et al. Subclinical thyroid dysfunction, cardiac function, and the risk of heart failure. The Cardiovascular Health study. J Am Coll Cardiol 2008; 52(14):1152–1159. doi:10.1016/j.jacc.2008.07.009
- Haggerty JJ Jr, Garbutt JC, Evans DL, et al. Subclinical hypothyroidism: a review of neuropsychiatric aspects. Int J Psychiatry Med 1990; 20(2):193–208. doi:10.2190/ADLY-1UU0-1A8L-HPXY
- Baldini IM, Vita A, Mauri MC, et al. Psychopathological and cognitive features in subclinical hypothyroidism. Prog Neuropsychopharmacol Biol Psychiatry 1997; 21(6):925–935. pmid:9380789
- del Ser Quijano T, Delgado C, Martinez Espinosa S, Vazquez C. Cognitive deficiency in mild hypothyroidism. Neurologia 2000; 15(5):193–198. Spanish. pmid:10850118
- Correia N, Mullally S, Cooke G, et al. Evidence for a specific defect in hippocampal memory in overt and subclinical hypothyroidism. J Clin Endocrinol Metab 2009; 94(10):3789–3797. doi:10.1210/jc.2008-2702
- Aghili R, Khamseh ME, Malek M, et al. Changes of subtests of Wechsler memory scale and cognitive function in subjects with subclinical hypothyroidism following treatment with levothyroxine. Arch Med Sci 2012; 8(6):1096–1101. doi:10.5114/aoms.2012.32423
- Pasqualetti G, Pagano G, Rengo G, Ferrara N, Monzani F. Subclinical hypothyroidism and cognitive impairment: systematic review and meta-analysis. J Clin Endocrinol Metab 2015; 100(11):4240–4248. doi:10.1210/jc.2015-2046
- Christ-Crain M, Meier C, Huber PR, Staub J, Muller B. Effect of L-thyroxine replacement therapy on surrogate markers of skeletal and cardiac function in subclinical hypothyroidism. Endocrinologist 2004; 14(3):161–166. doi:10.1097/01.ten.0000127932.31710.4f
- Brennan MD, Powell C, Kaufman KR, Sun PC, Bahn RS, Nair KS. The impact of overt and subclinical hyperthyroidism on skeletal muscle. Thyroid 2006; 16(4):375–380. doi:10.1089/thy.2006.16.375
- Reuters VS, Teixeira Pde F, Vigario PS, et al. Functional capacity and muscular abnormalities in subclinical hypothyroidism. Am J Med Sci 2009; 338(4):259–263. doi:10.1097/MAJ.0b013e3181af7c7c
- Mainenti MR, Vigario PS, Teixeira PF, Maia MD, Oliveira FP, Vaisman M. Effect of levothyroxine replacement on exercise performance in subclinical hypothyroidism. J Endocrinol Invest 2009; 32(5):470–473. doi:10.3275/6106
- Lankhaar JA, de Vries WR, Jansen JA, Zelissen PM, Backx FJ. Impact of overt and subclinical hypothyroidism on exercise tolerance: a systematic review. Res Q Exerc Sport 2014; 85(3):365–389. doi:10.1080/02701367.2014.930405
- Lee JS, Buzkova P, Fink HA, et al. Subclinical thyroid dysfunction and incident hip fracture in older adults. Arch Intern Med 2010; 170(21):1876–1883. doi:10.1001/archinternmed.2010.424
- Svare A, Nilsen TI, Asvold BO, et al. Does thyroid function influence fracture risk? Prospective data from the HUNT2 study, Norway. Eur J Endocrinol 2013; 169(6):845–852. doi:10.1530/EJE-13-0546
- Di Mase R, Cerbone M, Improda N, et al. Bone health in children with long-term idiopathic subclinical hypothyroidism. Ital J Pediatr 2012; 38:56. doi:10.1186/1824-7288-38-56
- Boelaert K. The association between serum TSH concentration and thyroid cancer. Endocr Relat Cancer 2009; 16(4):1065–1072. doi:10.1677/ERC-09-0150
- Haymart MR, Glinberg SL, Liu J, Sippel RS, Jaume JC, Chen H. Higher serum TSH in thyroid cancer patients occurs independent of age and correlates with extrathyroidal extension. Clin Endocrinol (Oxf) 2009; 71(3):434–439. doi:10.1111/j.1365-2265.2008.03489.x
- Fiore E, Vitti P. Serum TSH and risk of papillary thyroid cancer in nodular thyroid disease. J Clin Endocrinol Metab 2012; 97(4):1134–1145. doi:10.1210/jc.2011-2735
- Fiore E, Rago T, Provenzale MA, et al. L-thyroxine-treated patients with nodular goiter have lower serum TSH and lower frequency of papillary thyroid cancer: results of a cross-sectional study on 27,914 patients. Endocr Relat Cancer 2010; 17(1):231–239. doi:10.1677/ERC-09-0251
- Hercbergs AH, Ashur-Fabian O, Garfield D. Thyroid hormones and cancer: clinical studies of hypothyroidism in oncology. Curr Opin Endocrinol Diabetes Obes 2010; 17(5):432–436. doi:10.1097/MED.0b013e32833d9710
- Thvilum M, Brandt F, Brix TH, Hegedus L. A review of the evidence for and against increased mortality in hypothyroidism. Nat Rev Endocrinol 2012; 8(7):417–424. doi:10.1038/nrendo.2012.29
- Stott DJ, Rodondi N, Kearney PM, et al; TRUST Study Group. Thyroid hormone therapy for older adults with subclinical hypothyroidism. N Engl J Med 2017; 376(26):2534–2544. doi:10.1056/NEJMoa1603825
- Practice Committee of the American Society for Reproductive Medicine. Subclinical hypothyroidism in the infertile female population: a guideline. Fertil Steril 2015; 104(3):545–753. doi:10.1016/j.fertnstert.2015.05.028
- Stagnaro-Green A, Abalovich M, Alexander E, et al; American Thyroid Association Taskforce on Thyroid Disease During Pregnancy and Postpartum. Guidelines of the American Thyroid Association for the diagnosis and management of thyroid disease during pregnancy and postpartum. Thyroid 2011; 21(10):1081–1125. doi:10.1089/thy.2011.0087
- Goldsmith RE, Sturgis SH, Lerman J, Stanbury JB. The menstrual pattern in thyroid disease. J Clin Endocrinol Metab. 1952; 12(7):846-855. doi:10.1210/jcem-12-7-846
- Plowden TC, Schisterman EF, Sjaarda LA, et al. Subclinical hypothyroidism and thyroid autoimmunity are not associated with fecundity, pregnancy loss, or live birth. J Clin Endocrinol Metab 2016; 101(6):2358–2365. doi:10.1210/jc.2016-1049
- Alexander EK, Pearce EN, Brent GA, et al. 2017 Guidelines of the American Thyroid Association for the diagnosis and management of thyroid disease during pregnancy and the postpartum. Thyroid 2017; 27(3):315–389. doi:10.1089/thy.2016.0457
- Negro R, Formoso G, Mangieri T, Pezzarossa A, Dazzi D, Hassan H. Levothyroxine treatment in euthyroid pregnant women with autoimmune thyroid disease: effects on obstetrical complications. J Clin Endocrinol Metab 2006; 91(7):2587–2591. doi:10.1210/jc.2005-1603
- Panesar NS, Li CY, Rogers MS. Reference intervals for thyroid hormones in pregnant Chinese women. Ann Clin Biochem 2001; 38(pt 4):329–332. doi:10.1258/0004563011900830
- Lepoutre T, Debieve F, Gruson D, Daumerie C. Reduction of miscarriages through universal screening and treatment of thyroid autoimmune diseases. Gynecol Obstet Invest 2012; 74(4):265–273. doi:10.1159/000343759
- De Groot L, Abalovich M, Alexander EK, et al. Management of thyroid dysfunction during pregnancy and postpartum: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2012; 97(8):2543–2565. doi:10.1210/jc.2011-2803
- Crawford NM, Steiner AZ. Thyroid autoimmunity and reproductive function. Semin Reprod Med 2016; 34(6):343–350. doi:10.1055/s-0036-1593485
- Maraka S, Ospina NM, O’Keeffe DT, et al. Subclinical hypothyroidism in pregnancy: a systematic review and meta-analysis. Thyroid 2016; 26(4):580–590. doi:10.1089/thy.2015.0418
- Wiles KS, Jarvis S, Nelson-Piercy C. Are we overtreating subclinical hypothyroidism in pregnancy? BMJ 2015; 351:h4726. doi:10.1136/bmj.h4726
- Tudela CM, Casey BM, McIntire DD, Cunningham FG. Relationship of subclinical thyroid disease to the incidence of gestational diabetes. Obstet Gynecol 2012; 119(5):983–988. doi:10.1097/AOG.0b013e318250aeeb
- Lazarus J, Brown RS, Daumerie C, Hubalewska-Dydejczyk A, Negro R, Vaidya B. 2014 European Thyroid Association guidelines for the management of subclinical hypothyroidism in pregnancy and in children. Eur Thyroid J 2014; 3(2):76–94. doi:10.1159/000362597
- Karakosta P, Alegakis D, Georgiou V, et al. Thyroid dysfunction and autoantibodies in early pregnancy are associated with increased risk of gestational diabetes and adverse birth outcomes. J Clin Endocrinol Metab 2012; 97(12):4464–4472. doi:10.1210/jc.2012-2540
- Toulis KA, Stagnaro-Green A, Negro R. Maternal subclinical hypothyroidsm and gestational diabetes mellitus: a meta-analysis. Endocr Pract 2014; 20(7):703–714. doi:10.4158/EP13440.RA
- van den Boogaard E, Vissenberg R, Land JA, et al. Significance of subclinical thyroid dysfunction and thyroid autoimmunity before conception and in early pregnancy: a systematic review. Hum Reprod Update 2011; 17(5):605–619. doi:10.1093/humupd/dmr024
- Wilson KL, Casey BM, McIntire DD, Halvorson LM, Cunningham FG. Subclinical thyroid disease and the incidence of hypertension in pregnancy. Obstet Gynecol 2012; 119(2 Pt 1):315–320. doi:10.1097/AOG.0b013e318240de6a
- Ashoor G, Maiz N, Rotas M, Jawdat F, Nicolaides KH. Maternal thyroid function at 11 to 13 weeks of gestation and subsequent fetal death. Thyroid 2010; 20(9):989–993. doi:10.1089/thy.2010.0058
- Casey BM, Dashe JS, Wells CE, et al. Subclinical hypothyroidism and pregnancy outcomes. Obstet Gynecol 2005; 105(2):239–245. doi:10.1097/01.AOG.0000152345.99421.22
- Negro R, Schwartz A, Gismondi R, Tinelli A, Mangieri T, Stagnaro-Green A. Increased pregnancy loss rate in thyroid antibody negative women with TSH levels between 2.5 and 5.0 in the first trimester of pregnancy. J Clin Endocrinol Metab 2010; 95(9):E44–E48. doi:10.1210/jc.2010-0340
- Su PY, Huang K, Hao JH, et al. Maternal thyroid function in the first twenty weeks of pregnancy and subsequent fetal and infant development: a prospective population-based cohort study in China. J Clin Endocrinol Metab 2011; 96(10):3234–3241. doi:10.1210/jc.2011-0274
- Allan WC, Haddow JE, Palomaki GE, et al. Maternal thyroid deficiency and pregnancy complications: implications for population screening. J Med Screen 2000; 7(3):127–130. doi:10.1136/jms.7.3.127
- Benhadi N, Wiersinga WM, Reitsma JB, Vrijkotte TG, Bonsel GJ. Higher maternal TSH levels in pregnancy are associated with increased risk for miscarriage, fetal or neonatal death. Eur J Endocrinol 2009; 160(6):985–991. doi:10.1530/EJE-08-0953
- Korevaar TI, Medici M, de Rijke YB, et al. Ethnic differences in maternal thyroid parameters during pregnancy: the generation R study. J Clin Endocrinol Metab 2013; 98(9):3678–3686. doi:10.1210/jc.2013-2005
- Cleary-Goldman J, Malone FD, Lambert-Messerlian G, et al. Maternal thyroid hypofunction and pregnancy outcome. Obstet Gynecol 2008; 112(1):85–92. doi:10.1097/AOG.0b013e3181788dd7
- Li Y, Shan Z, Teng W, et al. Abnormalities of maternal thyroid function during pregnancy affect neuropsychological development of their children at 25-30 months. Clin Endocrinol (Oxf) 2010; 72(6):825–829. doi:10.1111/j.1365-2265.2009.03743.x
- Haddow JE, Palomaki GE, Allan WC, et al. Maternal thyroid deficiency during pregnancy and subsequent neuropsychological development of the child. N Engl J Med 1999; 341(8):549–555. doi:10.1056/NEJM199908193410801
- Henrichs J, Bongers-Schokking JJ, Schenk JJ, et al. Maternal thyroid function during early pregnancy and cognitive functioning in early childhood: the generation R study. J Clin Endocrinol Metab 2010; 95(9):4227–4234. doi:10.1210/jc.2010-0415
- Behrooz HG, Tohidi M, Mehrabi Y, Behrooz EG, Tehranidoost M, Azizi F. Subclinical hypothyroidism in pregnancy: intellectual development of offspring. Thyroid 2011; 21(10):1143–1147. doi:10.1089/thy.2011.0053
- Julvez J, Alvarez-Pedrerol M, Rebagliato M, et al. Thyroxine levels during pregnancy in healthy women and early child neurodevelopment. Epidemiology 2013; 24(1):150–157. doi:10.1097/EDE.0b013e318276ccd3
- Casey BM, Thom EA, Peaceman AM, et al; Eunice Kennedy Shriver National Institute of Child Health and Human Development Maternal–Fetal Medicine Units Network. Treatment of subclinical hypothyroidism or hypothyroxinemia in pregnancy. N Engl J Med 2017; 376(9):815–825. doi:10.1056/NEJMoa1606205
- Burns RB, Bates CK, Hartzband P, Smetana GW. Should we treat for subclinical hypothyroidism?: Grand rounds discussion from Beth Israel Deaconess Medical Center. Ann Intern Med 2016; 164(11):764–770. doi:10.7326/M16-0857
- Kucukler FK, Akbaba G, Arduc A, Simsek Y, Guler S. Evaluation of the common mistakes made by patients in the use of levothyroxine. Eur J Intern Med 2014; 25(9):e107–e108. doi:10.1016/j.ejim.2014.09.002
- McMillan M, Rotenberg KS, Vora K, et al. Comorbidities, concomitant medications, and diet as factors affecting levothyroxine therapy: results of the CONTROL surveillance project. Drugs R D 2016; 16(1):53–68. doi:10.1007/s40268-015-0116-6
- Pollock MA, Sturrock A, Marshall K, et al. Thyroxine treatment in patients with symptoms of hypothyroidism but thyroid function tests within the reference range: Randomised double blind placebo controlled crossover trial. BMJ 2001; 323(7318):891–895. pmid:11668132
- Peeters RP. Subclinical hypothyroidism. N Engl J Med 2017; 376(26):2556–2565. doi:10.1056/NEJMcp1611144
- Cooper DS, Biondi B. Subclinical thyroid disease. Lancet 2012; 379(9821):1142–1154. doi:10.1016/S0140-6736(11)60276-6
- Fatourechi V. Subclinical hypothyroidism: an update for primary care physicians. Mayo Clin Proc 2009; 84(1):65–71. doi:10.4065/84.1.65
- Laurberg P, Andersen S, Carle A, Karmisholt J, Knudsen N, Pedersen IB. The TSH upper reference limit: where are we at? Nat Rev Endocrinol 2011; 7(4):232–239. doi:10.1038/nrendo.2011.13
- Wartofsky L, Dickey RA. The evidence for a narrower thyrotropin reference range is compelling. J Clin Endocrinol Metab 2005; 90(9):5483–5488. doi:10.1210/jc.2005-0455
- Spencer CA, Hollowell JG, Kazarosyan M, Braverman LE. National Health and Nutrition Examination Survey III thyroid-stimulating hormone (TSH)-thyroperoxidase antibody relationships demonstrate that TSH upper reference limits may be skewed by occult thyroid dysfunction. J Clin Endocrinol Metab 2007; 92(11):4236–4240. doi:10.1210/jc.2007-0287
- Fatourechi V, Klee GG, Grebe SK, et al. Effects of reducing the upper limit of normal TSH values. JAMA 2003; 290(24):3195–3196. doi:10.1001/jama.290.24.3195-a
- Surks MI, Hollowell JG. Age-specific distribution of serum thyrotropin and antithyroid antibodies in the US population: implications for the prevalence of subclinical hypothyroidism. J Clin Endocrinol Metab 2007; 92(12):4575–4582. doi:10.1210/jc.2007-1499
- Hollowell JG, Staehling NW, Flanders WD, et al. Serum TSH, T(4), and thyroid antibodies in the United States population (1988 to 1994): National Health and Nutrition Examination Survey (NHANES III). J Clin Endocrinol Metab 2002; 87(2):489–499. doi:10.1210/jcem.87.2.8182
- Jonklaas J, Bianco AC, Bauer AJ, et al; American Thyroid Association Task Force on Thyroid Hormone Replacement. Guidelines for the treatment of hypothyroidism: prepared by the American Thyroid Association Task Force on Thyroid Hormone Replacement. Thyroid 2014; 24(12):1670–1751. doi:10.1089/thy.2014.0028
- Hennessey JV, Espaillat R. Diagnosis and management of subclinical hypothyroidism in elderly adults: a review of the literature. J Am Geriatr Soc 2015; 63(8):1663–1673. doi:10.1111/jgs.13532
- Razvi S, Shakoor A, Vanderpump M, Weaver JU, Pearce SH. The influence of age on the relationship between subclinical hypothyroidism and ischemic heart disease: a metaanalysis. J Clin Endocrinol Metab 2008; 93(8):2998–3007. doi:10.1210/jc.2008-0167
- Hamilton TE, Davis S, Onstad L, Kopecky KJ. Thyrotropin levels in a population with no clinical, autoantibody, or ultrasonographic evidence of thyroid disease: implications for the diagnosis of subclinical hypothyroidism. J Clin Endocrinol Metab 2008; 93(4):1224–1230. doi:10.1210/jc.2006-2300
- Canaris GJ, Manowitz NR, Mayor G, Ridgway EC. The Colorado thyroid disease prevalence study. Arch Intern Med 2000; 160(4):526–534. pmid:10695693
- Teng W, Shan Z, Teng X, et al. Effect of iodine intake on thyroid diseases in China. N Engl J Med 2006; 354(26):2783–2793. doi:10.1056/NEJMoa054022
- Negro R, Stagnaro-Green A. Diagnosis and management of subclinical hypothyroidism in pregnancy. BMJ 2014; 349:g4929. doi:10.1136/bmj.g4929
- Baumgartner C, Blum MR, Rodondi N. Subclinical hypothyroidism: summary of evidence in 2014. Swiss Med Wkly 2014; 144:w14058. doi:10.4414/smw.2014.14058
- Stedman TL. Stedman’s Medical Dictionary. 28th ed. Baltimore, MD: Lippincott Williams and Wilkins; 2006.
- Raza SA, Mahmood N. Subclinical hypothyroidism: controversies to consensus. Indian J Endocrinol Metab 2013; 17(suppl 3):S636–S642. doi:10.4103/2230-8210.123555
- Huber G, Staub JJ, Meier C, et al. Prospective study of the spontaneous course of subclinical hypothyroidism: prognostic value of thyrotropin, thyroid reserve, and thyroid antibodies. J Clin Endocrinol Metab 2002; 87(7):3221–3226. doi:10.1210/jcem.87.7.8678
- Diez JJ, Iglesias P, Burman KD. Spontaneous normalization of thyrotropin concentrations in patients with subclinical hypothyroidism. J Clin Endocrinol Metab 2005; 90(7):4124–4127. doi:10.1210/jc.2005-0375
- Vanderpump MP, Tunbridge WM, French JM, et al. The incidence of thyroid disorders in the community: a twenty-year follow-up of the Whickham survey. Clin Endocrinol (Oxf) 1995; 43(1):55–68. pmid:7641412
- Li Y, Teng D, Shan Z, et al. Antithyroperoxidase and antithyroglobulin antibodies in a five-year follow-up survey of populations with different iodine intakes. J Clin Endocrinol Metab 2008; 93(5):1751–1757. doi:10.1210/jc.2007-2368
- Hennessey JV, Klein I, Woeber KA, Cobin R, Garber JR. Aggressive case finding: a clinical strategy for the documentation of thyroid dysfunction. Ann Intern Med 2015; 163(4):311–312. doi:10.7326/M15-0762
- Rugge JB, Bougatsos C, Chou R. Screening and treatment of thyroid dysfunction: an evidence review for the US.Preventive Services Task Force. Ann Intern Med 2015; 162(1):35–45. doi:10.7326/M14-1456
- Ladenson PW, Singer PA, Ain KB, et al. American Thyroid Association guidelines for detection of thyroid dysfunction. Arch Intern Med 2000; 160(11):1573–1575. pmid:10847249
- Garber JR, Cobin RH, Gharib H, et al; American Association of Clinical Endocrinologists and American Thyroid Association Taskforce on Hypothyroidism in Adults. Clinical practice guidelines for hypothyroidism in adults: cosponsored by the American Association of Clinical Endocrinologists and the American Thyroid Association. Endocr Pract 2012; 18(6):988–1028. doi:10.4158/EP12280.GL
- Jorde R, Waterloo K, Storhaug H, Nyrnes A, Sundsfjord J, Jenssen TG. Neuropsychological function and symptoms in subjects with subclinical hypothyroidism and the effect of thyroxine treatment. J Clin Endocrinol Metab 2006; 91(1):145–153. doi:10.1210/jc.2005-1775
- Joffe RT, Pearce EN, Hennessey JV, Ryan JJ, Stern RA. Subclinical hypothyroidism, mood, and cognition in older adults: a review. Int J Geriatr Psychiatry 2013; 28(2):111–118. doi:10.1002/gps.3796
- Cooper DS, Halpern R, Wood LC, Levin AA, Ridgway EC. L-thyroxine therapy in subclinical hypothyroidism. A double-blind, placebo-controlled trial. Ann Intern Med 1984; 101(1):18–24. pmid:6428290
- Nystrom E, Caidahl K, Fager G, Wikkelso C, Lundberg PA, Lindstedt G. A double-blind cross-over 12-month study of L-thyroxine treatment of women with ‘subclinical’ hypothyroidism. Clin Endocrinol (Oxf) 1988; 29(1):63–75. pmid:3073880
- Monzani F, Del Guerra P, Caraccio N, et al. Subclinical hypothyroidism: neurobehavioral features and beneficial effect of L-thyroxine treatment. Clin Investig 1993; 71(5):367–371. pmid:8508006
- Biondi B. Thyroid and obesity: an intriguing relationship. J Clin Endocrinol Metab 2010; 95(8):3614–3617. doi:10.1210/jc.2010-1245
- Erdogan M, Canataroglu A, Ganidagli S, Kulaksizoglu M. Metabolic syndrome prevalence in subclinic and overt hypothyroid patients and the relation among metabolic syndrome parameters. J Endocrinol Invest 2011; 34(7):488–492. doi:10.3275/7202
- Javed Z, Sathyapalan T. Levothyroxine treatment of mild subclinical hypothyroidism: a review of potential risks and benefits. Ther Adv Endocrinol Metab 2016; 7(1):12–23. doi:10.1177/2042018815616543
- Pearce SH, Brabant G, Duntas LH, et al. 2013 ETA guideline: management of subclinical hypothyroidism. Eur Thyroid J 2013; 2(4):215–228. doi:10.1159/000356507
- Wang C. The relationship between type 2 diabetes mellitus and related thyroid diseases. J Diabetes Res 2013; 2013:390534. doi:10.1155/2013/390534
- Razvi S, Weaver JU, Vanderpump MP, Pearce SH. The incidence of ischemic heart disease and mortality in people with subclinical hypothyroidism: reanalysis of the Whickham survey cohort. J Clin Endocrinol Metab 2010; 95(4):1734–1740. doi:10.1210/jc.2009-1749
- Bindels AJ, Westendorp RG, Frolich M, Seidell JC, Blokstra A, Smelt AH. The prevalence of subclinical hypothyroidism at different total plasma cholesterol levels in middle aged men and women: a need for case-finding? Clin Endocrinol (Oxf) 1999; 50(2):217–220. pmid:10396365
- Pearce EN. Hypothyroidism and dyslipidemia: modern concepts and approaches. Curr Cardiol Rep 2004; 6(6):451–456. pmid:15485607
- Pearce EN. Update in lipid alterations in subclinical hypothyroidism. J Clin Endocrinol Metab 2012; 97(2):326–333. doi:10.1210/jc.2011-2532
- Rizos CV, Elisaf MS, Liberopoulos EN. Effects of thyroid dysfunction on lipid profile. Open Cardiovasc Med J 2011; 5:76–84. doi:10.2174/1874192401105010076
- Peppa M, Betsi G, Dimitriadis G. Lipid abnormalities and cardiometabolic risk in patients with overt and subclinical thyroid disease. J Lipids 2011; 2011:575840. doi:10.1155/2011/575840
- Asvold BO, Vatten LJ, Nilsen TI, Bjoro T. The association between TSH within the reference range and serum lipid concentrations in a population-based study. the HUNT study. Eur J Endocrinol 2007;1 56(2):181–186. doi:10.1530/eje.1.02333
- Danese MD, Ladenson PW, Meinert CL, Powe NR. Clinical review 115: effect of thyroxine therapy on serum lipoproteins in patients with mild thyroid failure: a quantitative review of the literature. J Clin Endocrinol Metab 2000; 85(9):2993–3001. doi:10.1210/jcem.85.9.6841
- Razvi S, Ingoe L, Keeka G, Oates C, McMillan C, Weaver JU. The beneficial effect of L-thyroxine on cardiovascular risk factors, endothelial function, and quality of life in subclinical hypothyroidism: randomized, crossover trial. J Clin Endocrinol Metab 2007; 92(5):1715–1723. doi:10.1210/jc.2006-1869
- Abreu IM, Lau E, de Sousa Pinto B, Carvalho D. Subclinical hypothyroidism: to treat or not to treat, that is the question! A systematic review with meta-analysis on lipid profile. Endocr Connect 2017; 6(3):188–199. doi:10.1530/EC-17-0028
- Robison CD, Bair TL, Horne BD, et al. Hypothyroidism as a risk factor for statin intolerance. J Clin Lipidol 2014; 8(4):401–407. doi:10.1016/j.jacl.2014.05.005
- Hak AE, Pols HA, Visser TJ, Drexhage HA, Hofman A, Witteman JC. Subclinical hypothyroidism is an independent risk factor for atherosclerosis and myocardial infarction in elderly women: the Rotterdam study. Ann Intern Med 2000; 132(4):270–278. pmid:10681281
- Boekholdt SM, Titan SM, Wiersinga WM, et al. Initial thyroid status and cardiovascular risk factors: the EPIC-Norfolk prospective population study. Clin Endocrinol (Oxf) 2010; 72(3):404–410. doi:10.1111/j.1365-2265.2009.03640.x
- Andersen MN, Olsen AM, Madsen JC, et al. Levothyroxine substitution in patients with subclinical hypothyroidism and the risk of myocardial infarction and mortality. PLoS One 2015; 10(6):e0129793. doi:10.1371/journal.pone.0129793
- Biondi B. Cardiovascular effects of mild hypothyroidism. Thyroid 2007; 17(7):625–630. doi:10.1089/thy.2007.0158
- Brenta G, Mutti LA, Schnitman M, Fretes O, Perrone A, Matute ML. Assessment of left ventricular diastolic function by radionuclide ventriculography at rest and exercise in subclinical hypothyroidism, and its response to L-thyroxine therapy. Am J Cardiol 2003; 91(11):1327–1330. pmid:12767425
- Taddei S, Caraccio N, Virdis A, et al. Impaired endothelium-dependent vasodilatation in subclinical hypothyroidism: beneficial effect of levothyroxine therapy. J Clin Endocrinol Metab 2003; 88(8):3731–3737. doi:10.1210/jc.2003-030039
- Gao N, Zhang W, Zhang YZ, Yang Q, Chen SH. Carotid intima-media thickness in patients with subclinical hypothyroidism: a meta-analysis. Atherosclerosis 2013; 227(1):18–25. doi:10.1016/j.atherosclerosis.2012.10.070
- Biondi B, Cooper DS. The clinical significance of subclinical thyroid dysfunction. Endocr Rev 2008; 29(1):76–131. doi:10.1210/er.2006-0043
- Chaker L, Baumgartner C, den Elzen WP, et al; Thyroid Studies Collaboration. Subclinical hypothyroidism and the risk of stroke events and fatal stroke: an individual participant data analysis. J Clin Endocrinol Metab 2015; 100(6):2181–2191. doi:10.1210/jc.2015-1438
- Monzani F, Di Bello V, Caraccio N, et al. Effect of levothyroxine on cardiac function and structure in subclinical hypothyroidism: a double blind, placebo-controlled study. J Clin Endocrinol Metab 2001; 86(3):1110–1115. doi:10.1210/jcem.86.3.7291
- Parle JV, Maisonneuve P, Sheppard MC, Boyle P, Franklyn JA. Prediction of all-cause and cardiovascular mortality in elderly people from one low serum thyrotropin result: a 10-year cohort study. Lancet 2001; 358(9285):861-865. doi:10.1016/S0140-6736(01)06067-6
- Razvi S, Weaver JU, Butler TJ, Pearce SH. Levothyroxine treatment of subclinical hypothyroidism, fatal and nonfatal cardiovascular events, and mortality. Arch Intern Med 2012; 172(10):811–817. doi:10.1001/archinternmed.2012.1159
- Pasqualetti G, Tognini S, Polini A, Caraccio N, Monzani F. Is subclinical hypothyroidism a cardiovascular risk factor in the elderly? J Clin Endocrinol Metab 2013; 98(6):2256–2266. doi:10.1210/jc.2012-3818
- Mooijaart SP, IEMO 80-plus Thyroid Trial Collaboration. Subclinical thyroid disorders. Lancet 2012; 380(9839):335. doi:10.1016/S0140-6736(12)61241-0
- Rodondi N, Bauer DC. Subclinical hypothyroidism and cardiovascular risk: how to end the controversy. J Clin Endocrinol Metab 2013; 98(6):2267–2269. doi:10.1210/jc.2013-1875
- Rodondi N, Newman AB, Vittinghoff E, et al. Subclinical hypothyroidism and the risk of heart failure, other cardiovascular events, and death. Arch Intern Med 2005; 165(21):2460–2466. doi:10.1001/archinte.165.21.2460
- Rodondi N, Bauer DC, Cappola AR, et al. Subclinical thyroid dysfunction, cardiac function, and the risk of heart failure. The Cardiovascular Health study. J Am Coll Cardiol 2008; 52(14):1152–1159. doi:10.1016/j.jacc.2008.07.009
- Haggerty JJ Jr, Garbutt JC, Evans DL, et al. Subclinical hypothyroidism: a review of neuropsychiatric aspects. Int J Psychiatry Med 1990; 20(2):193–208. doi:10.2190/ADLY-1UU0-1A8L-HPXY
- Baldini IM, Vita A, Mauri MC, et al. Psychopathological and cognitive features in subclinical hypothyroidism. Prog Neuropsychopharmacol Biol Psychiatry 1997; 21(6):925–935. pmid:9380789
- del Ser Quijano T, Delgado C, Martinez Espinosa S, Vazquez C. Cognitive deficiency in mild hypothyroidism. Neurologia 2000; 15(5):193–198. Spanish. pmid:10850118
- Correia N, Mullally S, Cooke G, et al. Evidence for a specific defect in hippocampal memory in overt and subclinical hypothyroidism. J Clin Endocrinol Metab 2009; 94(10):3789–3797. doi:10.1210/jc.2008-2702
- Aghili R, Khamseh ME, Malek M, et al. Changes of subtests of Wechsler memory scale and cognitive function in subjects with subclinical hypothyroidism following treatment with levothyroxine. Arch Med Sci 2012; 8(6):1096–1101. doi:10.5114/aoms.2012.32423
- Pasqualetti G, Pagano G, Rengo G, Ferrara N, Monzani F. Subclinical hypothyroidism and cognitive impairment: systematic review and meta-analysis. J Clin Endocrinol Metab 2015; 100(11):4240–4248. doi:10.1210/jc.2015-2046
- Christ-Crain M, Meier C, Huber PR, Staub J, Muller B. Effect of L-thyroxine replacement therapy on surrogate markers of skeletal and cardiac function in subclinical hypothyroidism. Endocrinologist 2004; 14(3):161–166. doi:10.1097/01.ten.0000127932.31710.4f
- Brennan MD, Powell C, Kaufman KR, Sun PC, Bahn RS, Nair KS. The impact of overt and subclinical hyperthyroidism on skeletal muscle. Thyroid 2006; 16(4):375–380. doi:10.1089/thy.2006.16.375
- Reuters VS, Teixeira Pde F, Vigario PS, et al. Functional capacity and muscular abnormalities in subclinical hypothyroidism. Am J Med Sci 2009; 338(4):259–263. doi:10.1097/MAJ.0b013e3181af7c7c
- Mainenti MR, Vigario PS, Teixeira PF, Maia MD, Oliveira FP, Vaisman M. Effect of levothyroxine replacement on exercise performance in subclinical hypothyroidism. J Endocrinol Invest 2009; 32(5):470–473. doi:10.3275/6106
- Lankhaar JA, de Vries WR, Jansen JA, Zelissen PM, Backx FJ. Impact of overt and subclinical hypothyroidism on exercise tolerance: a systematic review. Res Q Exerc Sport 2014; 85(3):365–389. doi:10.1080/02701367.2014.930405
- Lee JS, Buzkova P, Fink HA, et al. Subclinical thyroid dysfunction and incident hip fracture in older adults. Arch Intern Med 2010; 170(21):1876–1883. doi:10.1001/archinternmed.2010.424
- Svare A, Nilsen TI, Asvold BO, et al. Does thyroid function influence fracture risk? Prospective data from the HUNT2 study, Norway. Eur J Endocrinol 2013; 169(6):845–852. doi:10.1530/EJE-13-0546
- Di Mase R, Cerbone M, Improda N, et al. Bone health in children with long-term idiopathic subclinical hypothyroidism. Ital J Pediatr 2012; 38:56. doi:10.1186/1824-7288-38-56
- Boelaert K. The association between serum TSH concentration and thyroid cancer. Endocr Relat Cancer 2009; 16(4):1065–1072. doi:10.1677/ERC-09-0150
- Haymart MR, Glinberg SL, Liu J, Sippel RS, Jaume JC, Chen H. Higher serum TSH in thyroid cancer patients occurs independent of age and correlates with extrathyroidal extension. Clin Endocrinol (Oxf) 2009; 71(3):434–439. doi:10.1111/j.1365-2265.2008.03489.x
- Fiore E, Vitti P. Serum TSH and risk of papillary thyroid cancer in nodular thyroid disease. J Clin Endocrinol Metab 2012; 97(4):1134–1145. doi:10.1210/jc.2011-2735
- Fiore E, Rago T, Provenzale MA, et al. L-thyroxine-treated patients with nodular goiter have lower serum TSH and lower frequency of papillary thyroid cancer: results of a cross-sectional study on 27,914 patients. Endocr Relat Cancer 2010; 17(1):231–239. doi:10.1677/ERC-09-0251
- Hercbergs AH, Ashur-Fabian O, Garfield D. Thyroid hormones and cancer: clinical studies of hypothyroidism in oncology. Curr Opin Endocrinol Diabetes Obes 2010; 17(5):432–436. doi:10.1097/MED.0b013e32833d9710
- Thvilum M, Brandt F, Brix TH, Hegedus L. A review of the evidence for and against increased mortality in hypothyroidism. Nat Rev Endocrinol 2012; 8(7):417–424. doi:10.1038/nrendo.2012.29
- Stott DJ, Rodondi N, Kearney PM, et al; TRUST Study Group. Thyroid hormone therapy for older adults with subclinical hypothyroidism. N Engl J Med 2017; 376(26):2534–2544. doi:10.1056/NEJMoa1603825
- Practice Committee of the American Society for Reproductive Medicine. Subclinical hypothyroidism in the infertile female population: a guideline. Fertil Steril 2015; 104(3):545–753. doi:10.1016/j.fertnstert.2015.05.028
- Stagnaro-Green A, Abalovich M, Alexander E, et al; American Thyroid Association Taskforce on Thyroid Disease During Pregnancy and Postpartum. Guidelines of the American Thyroid Association for the diagnosis and management of thyroid disease during pregnancy and postpartum. Thyroid 2011; 21(10):1081–1125. doi:10.1089/thy.2011.0087
- Goldsmith RE, Sturgis SH, Lerman J, Stanbury JB. The menstrual pattern in thyroid disease. J Clin Endocrinol Metab. 1952; 12(7):846-855. doi:10.1210/jcem-12-7-846
- Plowden TC, Schisterman EF, Sjaarda LA, et al. Subclinical hypothyroidism and thyroid autoimmunity are not associated with fecundity, pregnancy loss, or live birth. J Clin Endocrinol Metab 2016; 101(6):2358–2365. doi:10.1210/jc.2016-1049
- Alexander EK, Pearce EN, Brent GA, et al. 2017 Guidelines of the American Thyroid Association for the diagnosis and management of thyroid disease during pregnancy and the postpartum. Thyroid 2017; 27(3):315–389. doi:10.1089/thy.2016.0457
- Negro R, Formoso G, Mangieri T, Pezzarossa A, Dazzi D, Hassan H. Levothyroxine treatment in euthyroid pregnant women with autoimmune thyroid disease: effects on obstetrical complications. J Clin Endocrinol Metab 2006; 91(7):2587–2591. doi:10.1210/jc.2005-1603
- Panesar NS, Li CY, Rogers MS. Reference intervals for thyroid hormones in pregnant Chinese women. Ann Clin Biochem 2001; 38(pt 4):329–332. doi:10.1258/0004563011900830
- Lepoutre T, Debieve F, Gruson D, Daumerie C. Reduction of miscarriages through universal screening and treatment of thyroid autoimmune diseases. Gynecol Obstet Invest 2012; 74(4):265–273. doi:10.1159/000343759
- De Groot L, Abalovich M, Alexander EK, et al. Management of thyroid dysfunction during pregnancy and postpartum: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2012; 97(8):2543–2565. doi:10.1210/jc.2011-2803
- Crawford NM, Steiner AZ. Thyroid autoimmunity and reproductive function. Semin Reprod Med 2016; 34(6):343–350. doi:10.1055/s-0036-1593485
- Maraka S, Ospina NM, O’Keeffe DT, et al. Subclinical hypothyroidism in pregnancy: a systematic review and meta-analysis. Thyroid 2016; 26(4):580–590. doi:10.1089/thy.2015.0418
- Wiles KS, Jarvis S, Nelson-Piercy C. Are we overtreating subclinical hypothyroidism in pregnancy? BMJ 2015; 351:h4726. doi:10.1136/bmj.h4726
- Tudela CM, Casey BM, McIntire DD, Cunningham FG. Relationship of subclinical thyroid disease to the incidence of gestational diabetes. Obstet Gynecol 2012; 119(5):983–988. doi:10.1097/AOG.0b013e318250aeeb
- Lazarus J, Brown RS, Daumerie C, Hubalewska-Dydejczyk A, Negro R, Vaidya B. 2014 European Thyroid Association guidelines for the management of subclinical hypothyroidism in pregnancy and in children. Eur Thyroid J 2014; 3(2):76–94. doi:10.1159/000362597
- Karakosta P, Alegakis D, Georgiou V, et al. Thyroid dysfunction and autoantibodies in early pregnancy are associated with increased risk of gestational diabetes and adverse birth outcomes. J Clin Endocrinol Metab 2012; 97(12):4464–4472. doi:10.1210/jc.2012-2540
- Toulis KA, Stagnaro-Green A, Negro R. Maternal subclinical hypothyroidsm and gestational diabetes mellitus: a meta-analysis. Endocr Pract 2014; 20(7):703–714. doi:10.4158/EP13440.RA
- van den Boogaard E, Vissenberg R, Land JA, et al. Significance of subclinical thyroid dysfunction and thyroid autoimmunity before conception and in early pregnancy: a systematic review. Hum Reprod Update 2011; 17(5):605–619. doi:10.1093/humupd/dmr024
- Wilson KL, Casey BM, McIntire DD, Halvorson LM, Cunningham FG. Subclinical thyroid disease and the incidence of hypertension in pregnancy. Obstet Gynecol 2012; 119(2 Pt 1):315–320. doi:10.1097/AOG.0b013e318240de6a
- Ashoor G, Maiz N, Rotas M, Jawdat F, Nicolaides KH. Maternal thyroid function at 11 to 13 weeks of gestation and subsequent fetal death. Thyroid 2010; 20(9):989–993. doi:10.1089/thy.2010.0058
- Casey BM, Dashe JS, Wells CE, et al. Subclinical hypothyroidism and pregnancy outcomes. Obstet Gynecol 2005; 105(2):239–245. doi:10.1097/01.AOG.0000152345.99421.22
- Negro R, Schwartz A, Gismondi R, Tinelli A, Mangieri T, Stagnaro-Green A. Increased pregnancy loss rate in thyroid antibody negative women with TSH levels between 2.5 and 5.0 in the first trimester of pregnancy. J Clin Endocrinol Metab 2010; 95(9):E44–E48. doi:10.1210/jc.2010-0340
- Su PY, Huang K, Hao JH, et al. Maternal thyroid function in the first twenty weeks of pregnancy and subsequent fetal and infant development: a prospective population-based cohort study in China. J Clin Endocrinol Metab 2011; 96(10):3234–3241. doi:10.1210/jc.2011-0274
- Allan WC, Haddow JE, Palomaki GE, et al. Maternal thyroid deficiency and pregnancy complications: implications for population screening. J Med Screen 2000; 7(3):127–130. doi:10.1136/jms.7.3.127
- Benhadi N, Wiersinga WM, Reitsma JB, Vrijkotte TG, Bonsel GJ. Higher maternal TSH levels in pregnancy are associated with increased risk for miscarriage, fetal or neonatal death. Eur J Endocrinol 2009; 160(6):985–991. doi:10.1530/EJE-08-0953
- Korevaar TI, Medici M, de Rijke YB, et al. Ethnic differences in maternal thyroid parameters during pregnancy: the generation R study. J Clin Endocrinol Metab 2013; 98(9):3678–3686. doi:10.1210/jc.2013-2005
- Cleary-Goldman J, Malone FD, Lambert-Messerlian G, et al. Maternal thyroid hypofunction and pregnancy outcome. Obstet Gynecol 2008; 112(1):85–92. doi:10.1097/AOG.0b013e3181788dd7
- Li Y, Shan Z, Teng W, et al. Abnormalities of maternal thyroid function during pregnancy affect neuropsychological development of their children at 25-30 months. Clin Endocrinol (Oxf) 2010; 72(6):825–829. doi:10.1111/j.1365-2265.2009.03743.x
- Haddow JE, Palomaki GE, Allan WC, et al. Maternal thyroid deficiency during pregnancy and subsequent neuropsychological development of the child. N Engl J Med 1999; 341(8):549–555. doi:10.1056/NEJM199908193410801
- Henrichs J, Bongers-Schokking JJ, Schenk JJ, et al. Maternal thyroid function during early pregnancy and cognitive functioning in early childhood: the generation R study. J Clin Endocrinol Metab 2010; 95(9):4227–4234. doi:10.1210/jc.2010-0415
- Behrooz HG, Tohidi M, Mehrabi Y, Behrooz EG, Tehranidoost M, Azizi F. Subclinical hypothyroidism in pregnancy: intellectual development of offspring. Thyroid 2011; 21(10):1143–1147. doi:10.1089/thy.2011.0053
- Julvez J, Alvarez-Pedrerol M, Rebagliato M, et al. Thyroxine levels during pregnancy in healthy women and early child neurodevelopment. Epidemiology 2013; 24(1):150–157. doi:10.1097/EDE.0b013e318276ccd3
- Casey BM, Thom EA, Peaceman AM, et al; Eunice Kennedy Shriver National Institute of Child Health and Human Development Maternal–Fetal Medicine Units Network. Treatment of subclinical hypothyroidism or hypothyroxinemia in pregnancy. N Engl J Med 2017; 376(9):815–825. doi:10.1056/NEJMoa1606205
- Burns RB, Bates CK, Hartzband P, Smetana GW. Should we treat for subclinical hypothyroidism?: Grand rounds discussion from Beth Israel Deaconess Medical Center. Ann Intern Med 2016; 164(11):764–770. doi:10.7326/M16-0857
- Kucukler FK, Akbaba G, Arduc A, Simsek Y, Guler S. Evaluation of the common mistakes made by patients in the use of levothyroxine. Eur J Intern Med 2014; 25(9):e107–e108. doi:10.1016/j.ejim.2014.09.002
- McMillan M, Rotenberg KS, Vora K, et al. Comorbidities, concomitant medications, and diet as factors affecting levothyroxine therapy: results of the CONTROL surveillance project. Drugs R D 2016; 16(1):53–68. doi:10.1007/s40268-015-0116-6
- Pollock MA, Sturrock A, Marshall K, et al. Thyroxine treatment in patients with symptoms of hypothyroidism but thyroid function tests within the reference range: Randomised double blind placebo controlled crossover trial. BMJ 2001; 323(7318):891–895. pmid:11668132
- Peeters RP. Subclinical hypothyroidism. N Engl J Med 2017; 376(26):2556–2565. doi:10.1056/NEJMcp1611144
KEY POINTS
- From 4% to 20% of adults have subclinical hypothyroidism, with a higher prevalence in women, older people, and those with thyroid autoimmunity.
- Subclinical hypothyroidism can progress to overt hypothyroidism, especially if antithyroid antibodies are present, and has been associated with adverse metabolic, cardiovascular, reproductive, maternal-fetal, neuromuscular, and cognitive abnormalities and lower quality of life.
- Some studies have suggested that levothyroxine therapy is beneficial, but others have not, possibly owing to variability in study designs, sample sizes, and patient populations.
- Further trials are needed to clearly demonstrate the clinical impact of subclinical hypothyroidism and the effect of levothyroxine therapy.

There is more to the TSH than a number

At a quick read, the messages from these articles may seem contradictory. But the biology is more complex in the setting of endogenous production of T4 by the thyroid gland, which is regulated by TSH, which in turn is regulated in a feedback loop by the thyroid-produced T4. In the setting of a fixed replacement dose of exogenous levothyroxine, the provided hormone affects the pituitary production of TSH, which likely will have no significant subsequent effect on the T4 level. Thus, the feedback control loop is far simpler.
There has not been a definitive study demonstrating that thyroxine supplementation in patients with subclinical hypothyroidism results in a superior clinical outcome. There are hints that this may be the case, and Azim and Nasr cite some of these studies. Recognizing a few markedly different physiologic reasons why the TSH can be slightly elevated and the T4 normal helps explain the lack of uniform clinical success with supplementation therapy and provides rationales for some management strategies.
Any biological variability in the responsiveness of the thyroid gland to TSH may affect the relationship between the levels of TSH and thyroid gland-released T4. In theory, if the thyroid receptor has decreased affinity for TSH, a higher TSH concentration will be needed to get the thyroid gland to secrete the level of T4 that the pituitary sensing mechanism deems normal for that individual. If the receptor affinity was decreased due to a gene polymorphism, this relationship between TSH and T4 may be stable, and providing exogenous T4 will result in a lower, “normalized” TSH level but may disrupt the thyroid-pituitary crosstalk and may even produce clinical hyperthyroidism.
A similar scenario exists in the setting of early thyroid gland failure, such as in Hashimoto thyroiditis. But in the latter scenario, the TSH-to-T4 production relationship may be unstable over time, for as additional thyroid gland is destroyed, T4 production will continue to decrease, the TSH will increase, and the thyroid gland may ultimately fail and hypothyroidism will occur. Hence the recommendation that in the setting of subclinical hypothyroidism and antiperoxidase antibodies, T4 and TSH levels should be monitored regularly in order to detect early true thyroid gland failure when the T4 level can no longer be maintained despite the increased stimulation of the gland by the elevated TSH. Analogous to this may be subclinical hypothyroidism in the elderly, in whom thyroid gland failure may develop, despite an increased TSH, from senescence rather than autoimmunity. What I am suggesting is that the natural history of all patients with subclinical hypothyroidism is not alike, and it thus should not be surprising that there does not seem to be a one-size-fits-all approach to management.
Symptoms in patients with subclinical hypothyroidism have not uniformly improved with T4 treatment compared with placebo. Notably, most patients with subclinical hypothyroidism experience no symptoms. But consider the extremely common symptom of fatigue, which can be present for a myriad of defined and undefined reasons. This symptom may often lead physicians to check the TSH and, if that is even slightly elevated, to also check the T4. It may also lead some physicians to routinely check the T4. Subclinical hypothyroidism is also quite common; thus, by chance alone or because of the circadian timing of checking the TSH, a slightly elevated TSH and fatigue may coexist and yet be unrelated.
Additionally, a positive biochemical response to thyroxine supplementation, such as a lowering of cholesterol, does not prove that the patient was clinically hypothyroid prior to supplementation, any more than lowering a patient’s blood glucose with insulin proves that the patient was diabetic. The management of subclinical hypothyroidism should be nuanced and based on both clinical and laboratory parameters.
- Nasr C. Is a serum TSH measurement sufficient to monitor the treatment of primary hypothyroidism? Cleve Clin J Med 2016; 83(8):571–573. doi:10.3949/ccjm.83a.15165
- Mandell BF. Trust the thyroid thermostat. Cleve Clin J Med 2016; 83(8):552–553. doi:10.3949/ccjm.83b.08016
At a quick read, the messages from these articles may seem contradictory. But the biology is more complex in the setting of endogenous production of T4 by the thyroid gland, which is regulated by TSH, which in turn is regulated in a feedback loop by the thyroid-produced T4. In the setting of a fixed replacement dose of exogenous levothyroxine, the provided hormone affects the pituitary production of TSH, which likely will have no significant subsequent effect on the T4 level. Thus, the feedback control loop is far simpler.
There has not been a definitive study demonstrating that thyroxine supplementation in patients with subclinical hypothyroidism results in a superior clinical outcome. There are hints that this may be the case, and Azim and Nasr cite some of these studies. Recognizing a few markedly different physiologic reasons why the TSH can be slightly elevated and the T4 normal helps explain the lack of uniform clinical success with supplementation therapy and provides rationales for some management strategies.
Any biological variability in the responsiveness of the thyroid gland to TSH may affect the relationship between the levels of TSH and thyroid gland-released T4. In theory, if the thyroid receptor has decreased affinity for TSH, a higher TSH concentration will be needed to get the thyroid gland to secrete the level of T4 that the pituitary sensing mechanism deems normal for that individual. If the receptor affinity was decreased due to a gene polymorphism, this relationship between TSH and T4 may be stable, and providing exogenous T4 will result in a lower, “normalized” TSH level but may disrupt the thyroid-pituitary crosstalk and may even produce clinical hyperthyroidism.
A similar scenario exists in the setting of early thyroid gland failure, such as in Hashimoto thyroiditis. But in the latter scenario, the TSH-to-T4 production relationship may be unstable over time, for as additional thyroid gland is destroyed, T4 production will continue to decrease, the TSH will increase, and the thyroid gland may ultimately fail and hypothyroidism will occur. Hence the recommendation that in the setting of subclinical hypothyroidism and antiperoxidase antibodies, T4 and TSH levels should be monitored regularly in order to detect early true thyroid gland failure when the T4 level can no longer be maintained despite the increased stimulation of the gland by the elevated TSH. Analogous to this may be subclinical hypothyroidism in the elderly, in whom thyroid gland failure may develop, despite an increased TSH, from senescence rather than autoimmunity. What I am suggesting is that the natural history of all patients with subclinical hypothyroidism is not alike, and it thus should not be surprising that there does not seem to be a one-size-fits-all approach to management.
Symptoms in patients with subclinical hypothyroidism have not uniformly improved with T4 treatment compared with placebo. Notably, most patients with subclinical hypothyroidism experience no symptoms. But consider the extremely common symptom of fatigue, which can be present for a myriad of defined and undefined reasons. This symptom may often lead physicians to check the TSH and, if that is even slightly elevated, to also check the T4. It may also lead some physicians to routinely check the T4. Subclinical hypothyroidism is also quite common; thus, by chance alone or because of the circadian timing of checking the TSH, a slightly elevated TSH and fatigue may coexist and yet be unrelated.
Additionally, a positive biochemical response to thyroxine supplementation, such as a lowering of cholesterol, does not prove that the patient was clinically hypothyroid prior to supplementation, any more than lowering a patient’s blood glucose with insulin proves that the patient was diabetic. The management of subclinical hypothyroidism should be nuanced and based on both clinical and laboratory parameters.
At a quick read, the messages from these articles may seem contradictory. But the biology is more complex in the setting of endogenous production of T4 by the thyroid gland, which is regulated by TSH, which in turn is regulated in a feedback loop by the thyroid-produced T4. In the setting of a fixed replacement dose of exogenous levothyroxine, the provided hormone affects the pituitary production of TSH, which likely will have no significant subsequent effect on the T4 level. Thus, the feedback control loop is far simpler.
There has not been a definitive study demonstrating that thyroxine supplementation in patients with subclinical hypothyroidism results in a superior clinical outcome. There are hints that this may be the case, and Azim and Nasr cite some of these studies. Recognizing a few markedly different physiologic reasons why the TSH can be slightly elevated and the T4 normal helps explain the lack of uniform clinical success with supplementation therapy and provides rationales for some management strategies.
Any biological variability in the responsiveness of the thyroid gland to TSH may affect the relationship between the levels of TSH and thyroid gland-released T4. In theory, if the thyroid receptor has decreased affinity for TSH, a higher TSH concentration will be needed to get the thyroid gland to secrete the level of T4 that the pituitary sensing mechanism deems normal for that individual. If the receptor affinity was decreased due to a gene polymorphism, this relationship between TSH and T4 may be stable, and providing exogenous T4 will result in a lower, “normalized” TSH level but may disrupt the thyroid-pituitary crosstalk and may even produce clinical hyperthyroidism.
A similar scenario exists in the setting of early thyroid gland failure, such as in Hashimoto thyroiditis. But in the latter scenario, the TSH-to-T4 production relationship may be unstable over time, for as additional thyroid gland is destroyed, T4 production will continue to decrease, the TSH will increase, and the thyroid gland may ultimately fail and hypothyroidism will occur. Hence the recommendation that in the setting of subclinical hypothyroidism and antiperoxidase antibodies, T4 and TSH levels should be monitored regularly in order to detect early true thyroid gland failure when the T4 level can no longer be maintained despite the increased stimulation of the gland by the elevated TSH. Analogous to this may be subclinical hypothyroidism in the elderly, in whom thyroid gland failure may develop, despite an increased TSH, from senescence rather than autoimmunity. What I am suggesting is that the natural history of all patients with subclinical hypothyroidism is not alike, and it thus should not be surprising that there does not seem to be a one-size-fits-all approach to management.
Symptoms in patients with subclinical hypothyroidism have not uniformly improved with T4 treatment compared with placebo. Notably, most patients with subclinical hypothyroidism experience no symptoms. But consider the extremely common symptom of fatigue, which can be present for a myriad of defined and undefined reasons. This symptom may often lead physicians to check the TSH and, if that is even slightly elevated, to also check the T4. It may also lead some physicians to routinely check the T4. Subclinical hypothyroidism is also quite common; thus, by chance alone or because of the circadian timing of checking the TSH, a slightly elevated TSH and fatigue may coexist and yet be unrelated.
Additionally, a positive biochemical response to thyroxine supplementation, such as a lowering of cholesterol, does not prove that the patient was clinically hypothyroid prior to supplementation, any more than lowering a patient’s blood glucose with insulin proves that the patient was diabetic. The management of subclinical hypothyroidism should be nuanced and based on both clinical and laboratory parameters.
- Nasr C. Is a serum TSH measurement sufficient to monitor the treatment of primary hypothyroidism? Cleve Clin J Med 2016; 83(8):571–573. doi:10.3949/ccjm.83a.15165
- Mandell BF. Trust the thyroid thermostat. Cleve Clin J Med 2016; 83(8):552–553. doi:10.3949/ccjm.83b.08016
- Nasr C. Is a serum TSH measurement sufficient to monitor the treatment of primary hypothyroidism? Cleve Clin J Med 2016; 83(8):571–573. doi:10.3949/ccjm.83a.15165
- Mandell BF. Trust the thyroid thermostat. Cleve Clin J Med 2016; 83(8):552–553. doi:10.3949/ccjm.83b.08016
Subclinical hypothyroidism boosts immediate risk of heart failure
CHICAGO – The short-term risk of developing heart failure in patients with newly identified hypothyroidism, be it overt or subclinical, is double that of euthyroid individuals, Caroline H. Noergaard, MD, reported at the American Heart Association scientific sessions.

“This is really important clinically. The association with heart failure has previously been shown in both overt and subclinical hyperthyroidism, but it’s actually new knowledge that hypothyroidism is associated with immediate risk of heart failure. And a lot of people have subclinical hypothyroidism,” said Dr. Noergaard, a PhD student in epidemiology at Aalborg (Denmark) University.
Also at the meeting, Jeffrey L. Anderson, MD, reported that free thyroxine levels within the normal reference range were associated in graded fashion with an increased prevalence and incidence of atrial fibrillation in a large Utah study, a finding that provides independent confirmation of an earlier report by investigators from the population-based Rotterdam Study.
“These findings validate those of the Rotterdam Study in a much larger dataset and may have important clinical implications, including a redefinition of the reference range and the target-free T4 levels for thyroxine replacement therapy,” observed Dr. Anderson, professor of internal medicine at the University of Utah, Salt Lake City, and a research cardiologist at the Intermountain Medical Center Heart Institute.
Hypothyroidism and heart failure
Dr. Noergaard presented a retrospective study of over 1 million Copenhagen-area adults (mean age, 50 years) with no history of heart failure, who had their first thyroid function test. She and her coinvestigators turned to comprehensive Danish national health care registries to determine how many of these individuals were diagnosed with new-onset heart failure within 90 days after their thyroid function test.
Subclinical hypothyroidism was defined by a thyroid-stimulating hormone level greater than 5 mIU/L and a free T4 of 9-22 pmol/L. Overt hypothyroidism required a TSH greater than 5 mIU/L with a free T4 less than 9 pmol/L.
Free T4 predicts atrial fibrillation risk
Dr. Anderson presented a retrospective analysis of 174,914 adult patients in the Intermountain Healthcare EMR database, none of whom were on thyroid replacement at entry. The patients, who were a mean age of 64 years and 65% women, were followed for an average of 6.3 years. Of these, 88.4% had a free T4 within the normal reference range of 0.75-1.5 ng/dL, 7.4% had a value below the cutoff for normal, and 4.2% had a free T4 above the reference range.
Upon dividing the patients within the normal range into quartiles based upon their free T4 level, he and his coinvestigators found that the baseline prevalence of atrial fibrillation was 8.7% in those in quartile 1, 9.3% in quartile 2, 10.5% in quartile 3, and 12.6% in quartile 4. In a multivariate analysis adjusted for potential confounders, the risk of prevalent atrial fibrillation was increased by 11% for patients in quartile 2, compared with those in the first quartile, by 22% in quartile 3, and by 40% in quartile 4.
The incidence of new-onset atrial fibrillation during 3 years of follow-up was 4.1% in patients in normal-range quartile 1, 4.3% in quartile 2, 4.5% in quartile 3, and 5.2% in the top normal-range quartile. The odds of developing atrial fibrillation were increased by 8% and 16% in quartiles 3 and 4, compared with quartile 1.
Serum TSH and free T3 levels showed no consistent relationship with atrial fibrillation.
The Utah findings confirm in a large U.S. population the earlier report from the Rotterdam Study (J Clin Endocrinol Metab. 2015 Oct;100(10):3718-24).
Dr. Noergaard and Dr. Anderson reported having no financial conflicts regarding their studies, which were carried out free of commercial support.
CHICAGO – The short-term risk of developing heart failure in patients with newly identified hypothyroidism, be it overt or subclinical, is double that of euthyroid individuals, Caroline H. Noergaard, MD, reported at the American Heart Association scientific sessions.
“This is really important clinically. The association with heart failure has previously been shown in both overt and subclinical hyperthyroidism, but it’s actually new knowledge that hypothyroidism is associated with immediate risk of heart failure. And a lot of people have subclinical hypothyroidism,” said Dr. Noergaard, a PhD student in epidemiology at Aalborg (Denmark) University.
Also at the meeting, Jeffrey L. Anderson, MD, reported that free thyroxine levels within the normal reference range were associated in graded fashion with an increased prevalence and incidence of atrial fibrillation in a large Utah study, a finding that provides independent confirmation of an earlier report by investigators from the population-based Rotterdam Study.
“These findings validate those of the Rotterdam Study in a much larger dataset and may have important clinical implications, including a redefinition of the reference range and the target-free T4 levels for thyroxine replacement therapy,” observed Dr. Anderson, professor of internal medicine at the University of Utah, Salt Lake City, and a research cardiologist at the Intermountain Medical Center Heart Institute.
Hypothyroidism and heart failure
Dr. Noergaard presented a retrospective study of over 1 million Copenhagen-area adults (mean age, 50 years) with no history of heart failure, who had their first thyroid function test. She and her coinvestigators turned to comprehensive Danish national health care registries to determine how many of these individuals were diagnosed with new-onset heart failure within 90 days after their thyroid function test.
Subclinical hypothyroidism was defined by a thyroid-stimulating hormone level greater than 5 mIU/L and a free T4 of 9-22 pmol/L. Overt hypothyroidism required a TSH greater than 5 mIU/L with a free T4 less than 9 pmol/L.
Free T4 predicts atrial fibrillation risk
Dr. Anderson presented a retrospective analysis of 174,914 adult patients in the Intermountain Healthcare EMR database, none of whom were on thyroid replacement at entry. The patients, who were a mean age of 64 years and 65% women, were followed for an average of 6.3 years. Of these, 88.4% had a free T4 within the normal reference range of 0.75-1.5 ng/dL, 7.4% had a value below the cutoff for normal, and 4.2% had a free T4 above the reference range.
Upon dividing the patients within the normal range into quartiles based upon their free T4 level, he and his coinvestigators found that the baseline prevalence of atrial fibrillation was 8.7% in those in quartile 1, 9.3% in quartile 2, 10.5% in quartile 3, and 12.6% in quartile 4. In a multivariate analysis adjusted for potential confounders, the risk of prevalent atrial fibrillation was increased by 11% for patients in quartile 2, compared with those in the first quartile, by 22% in quartile 3, and by 40% in quartile 4.
The incidence of new-onset atrial fibrillation during 3 years of follow-up was 4.1% in patients in normal-range quartile 1, 4.3% in quartile 2, 4.5% in quartile 3, and 5.2% in the top normal-range quartile. The odds of developing atrial fibrillation were increased by 8% and 16% in quartiles 3 and 4, compared with quartile 1.
Serum TSH and free T3 levels showed no consistent relationship with atrial fibrillation.
The Utah findings confirm in a large U.S. population the earlier report from the Rotterdam Study (J Clin Endocrinol Metab. 2015 Oct;100(10):3718-24).
Dr. Noergaard and Dr. Anderson reported having no financial conflicts regarding their studies, which were carried out free of commercial support.
CHICAGO – The short-term risk of developing heart failure in patients with newly identified hypothyroidism, be it overt or subclinical, is double that of euthyroid individuals, Caroline H. Noergaard, MD, reported at the American Heart Association scientific sessions.
“This is really important clinically. The association with heart failure has previously been shown in both overt and subclinical hyperthyroidism, but it’s actually new knowledge that hypothyroidism is associated with immediate risk of heart failure. And a lot of people have subclinical hypothyroidism,” said Dr. Noergaard, a PhD student in epidemiology at Aalborg (Denmark) University.
Also at the meeting, Jeffrey L. Anderson, MD, reported that free thyroxine levels within the normal reference range were associated in graded fashion with an increased prevalence and incidence of atrial fibrillation in a large Utah study, a finding that provides independent confirmation of an earlier report by investigators from the population-based Rotterdam Study.
“These findings validate those of the Rotterdam Study in a much larger dataset and may have important clinical implications, including a redefinition of the reference range and the target-free T4 levels for thyroxine replacement therapy,” observed Dr. Anderson, professor of internal medicine at the University of Utah, Salt Lake City, and a research cardiologist at the Intermountain Medical Center Heart Institute.
Hypothyroidism and heart failure
Dr. Noergaard presented a retrospective study of over 1 million Copenhagen-area adults (mean age, 50 years) with no history of heart failure, who had their first thyroid function test. She and her coinvestigators turned to comprehensive Danish national health care registries to determine how many of these individuals were diagnosed with new-onset heart failure within 90 days after their thyroid function test.
Subclinical hypothyroidism was defined by a thyroid-stimulating hormone level greater than 5 mIU/L and a free T4 of 9-22 pmol/L. Overt hypothyroidism required a TSH greater than 5 mIU/L with a free T4 less than 9 pmol/L.
Free T4 predicts atrial fibrillation risk
Dr. Anderson presented a retrospective analysis of 174,914 adult patients in the Intermountain Healthcare EMR database, none of whom were on thyroid replacement at entry. The patients, who were a mean age of 64 years and 65% women, were followed for an average of 6.3 years. Of these, 88.4% had a free T4 within the normal reference range of 0.75-1.5 ng/dL, 7.4% had a value below the cutoff for normal, and 4.2% had a free T4 above the reference range.
Upon dividing the patients within the normal range into quartiles based upon their free T4 level, he and his coinvestigators found that the baseline prevalence of atrial fibrillation was 8.7% in those in quartile 1, 9.3% in quartile 2, 10.5% in quartile 3, and 12.6% in quartile 4. In a multivariate analysis adjusted for potential confounders, the risk of prevalent atrial fibrillation was increased by 11% for patients in quartile 2, compared with those in the first quartile, by 22% in quartile 3, and by 40% in quartile 4.
The incidence of new-onset atrial fibrillation during 3 years of follow-up was 4.1% in patients in normal-range quartile 1, 4.3% in quartile 2, 4.5% in quartile 3, and 5.2% in the top normal-range quartile. The odds of developing atrial fibrillation were increased by 8% and 16% in quartiles 3 and 4, compared with quartile 1.
Serum TSH and free T3 levels showed no consistent relationship with atrial fibrillation.
The Utah findings confirm in a large U.S. population the earlier report from the Rotterdam Study (J Clin Endocrinol Metab. 2015 Oct;100(10):3718-24).
Dr. Noergaard and Dr. Anderson reported having no financial conflicts regarding their studies, which were carried out free of commercial support.
REPORTING FROM THE AHA SCIENTIFIC SESSIONS
Key clinical point:
Major finding: Both subclinical and overt hypothyroidism are associated with a 100% increased risk of being diagnosed with heart failure, compared with euthyroid individuals.
Study details: This was a retrospective study of the association between free thyroxine levels and short-term risk of developing heart failure in more than 1 million Copenhagen-area patients.
Disclosures: The presenter reported having no financial conflicts regarding the Danish study, conducted free of commercial support.
FDA committee votes yes on romosozumab for osteoporosis
In an 18-1 vote, the FDA’s Bone, Reproductive, and Urologic Drugs Advisory Committee agreed that the risk-benefit profile of romosozumab, to be marketed by Amgen as Evenity, was favorable enough to support approval. Relative risk reductions of up to 75%, compared with placebo, and 36%, compared with alendronate, were seen in pivotal clinical trials.
A signal for increased major adverse cardiovascular events (MACE) among those receiving romosozumab had been seen in just one of the clinical trials, with a hazard ratio for MACE of 1.87 for those taking romosozumab, compared with those taking alendronate (95% confidence interval, 1.11-3.14).
Committee members were enthusiastic about the efficacy of the monoclonal antibody, which binds to sclerostin and prevents its inhibiting effect, allowing robust new bone formation. “I want to emphasize the remarkable skeletal efficacy of this drug; truly, it’s better than anything we’ve seen before,” said committee member Sundeep Khosla, MD, professor of medicine and physiology at the Mayo Clinic, Rochester, Minn.

In its application, the sponsor relied on two clinical trials. In the first, 7,180 women with osteoporosis aged 55-90 years were randomized 1:1 to receive romosozumab or placebo for 12 months in a double-blind trial. After this time, participants in each arm received follow-on treatment with denosumab (Prolia) for another 12 months. This study, dubbed Trial 337, followed morphometric vertebral fractures at 12 and 24 months. Morphometric fractures included both symptomatic and asymptomatic fractures.
Those treated with romosozumab had relative risk reductions of new vertebral fractures of 73% and 75%, compared with those given placebo at 12 and 24 months. Absolute risk reductions for vertebral fractures were 1.30% and 1.89% at 1 and 2 years (P less than .001 for both).
The second study, Trial 142, was a double-blind, active-controlled study that included 4,093 women aged 55-90 years with osteoporosis and a history of prior fragility fracture. Participants were randomized 1:1 to receive either romosozumab or alendronate for 12 months, with an additional variable period of alendronate follow-on of at least 12 months for both arms.
For Trial 142, one primary endpoint was morphometric vertebral fractures at month 24. An additional endpoint, clinical fracture, was a composite of symptomatic vertebral fractures and nonvertebral fractures. This second endpoint was assessed at the time of primary analysis, an event-driven cut point that occurred when at least 330 participants experienced a clinical fracture and all participants had completed the 24-month visit.
Hip fractures were less common among those given romosozumab, and bone mineral density increased significantly as well.
Vertebral fractures were reduced by 36% in the romosozumab group relative to the alendronate group, and clinical fractures by 27% (P less than .001 for both).
Overall, the number of adverse events for the more than 7,500 patients in the safety population was similar between those receiving romosozumab and either placebo or alendronate, said Scott Wasserman, MD, vice president of global development for Amgen.
However, in Trial 142, which included patients who were slightly older and on more cardiovascular medications at baseline than in Trial 337, MACE – defined as cardiovascular death, MI, and stroke – occurred more frequently among those taking romosozumab, driven primarily by increased cardiac and cerebral ischemic events occurring within the first 12 months of beginning the study drug. At 12 months, the romosozumab arm saw 41 instances of MACE, compared with 22 in the alendronate arm to produce the HR of 1.87 (2.0% vs. 1.1%).
With regard to the imbalance in MACE seen in Trial 142, both the FDA and presenters for the sponsor entertained the notion that alendronate may have been somewhat protective for cardiovascular events. Although there is some biologic plausibility for a cardioprotective event for bisphosphonates, alendronate is highly specific for bone activity and the preponderance of previous studies have not shown such cardioprotection, the FDA, sponsors, and committee members all agreed.
Marc Sabatine, MD, the Lewis Dexter, MD Distinguished Chair in Cardiovascular Medicine at Harvard Medical School, Boston, was available to answer questions on behalf of the study’s sponsor. He noted at several points during the meeting just how few total cardiovascular events were seen overall in the romosozumab trial. The overall small numbers, he said, made it very difficult to distinguish whether the smaller number of MACE seen in the alendronate arm of Trial 142 were a true safety signal or just “a play of chance.”

Almost all the committee’s questioning and discussion centered on this potential increased cardiovascular risk. Amgen, in discussion with the FDA, had agreed to a black box warning designed to wave off prescribing romosozumab to those at increased risk for cardiovascular disease, focusing on those with a history of MI or stroke.
Additionally, the sponsor proposed a postmarketing real-world observational study to track incidence of MACE in those receiving romosozumab, comparing them with those receiving standard of care for osteoporosis.
Several committee members pointed out a problem with the proposed safety mitigation scheme: By conducting a postmarketing observational study for a drug that has a black-box warning to exclude those at high risk for MACE, the chance of detecting an actual cardiovascular safety problem plummets. “This is the textbook example of when observational studies struggle or are virtually guaranteed to fail,” noted Tobias Gerhard, PhD, a pharmacoepidemiologist at Rutgers University, New Brunswick, N.J.
On the other hand, noted several committee members, pausing to conduct a premarketing randomized, controlled trial would keep a beneficial drug away from a population in need. A postmarketing randomized, controlled trial, even a simple trial, still presents challenges, some of the committee acknowledged. Dr. Khosla voiced the opinion that “A randomized, controlled trial is virtually impossible.”
The committee, which was charged with discussing, but not voting on, what additional data should be obtained – and when – to sort out the cardiovascular safety question, was approximately evenly divided in the matter of whether an observational or registry-based trial, or a controlled trial, would be the best path forward.
Committee member Robert A. Adler, MD, put a realistic frame around the debate. “As an endocrinologist, I deal with nuances every day. I really think the kind of clinician who is going to be using this drug is used to dealing with benefits and risks and trying to tailor treatment to a given patient,” said Dr. Adler, professor of internal medicine and epidemiology at Virginia Commonwealth University, Richmond.
In its proposed indication, Amgen defined the population of menopausal women at high risk of fracture as those with a history of osteoporotic fracture, multiple risk factors for fracture, or patients who have failed or are intolerant to other available osteoporosis therapy. Romosozumab would be given as a once-monthly subcutaneous injection of 210 mg for a period of 12 months, to be followed by antiresorptive therapy.
Most of the participants in the clinical trials resided outside the United States, primarily because of the difficulty of recruiting clinical trial participants in the United States, Amgen officials said during their presentation. However, neither the time to the first positively adjudicated MACE nor bone mineral density responses at month 12 differed significantly across the various geographic regions where clinical trial sites were located, said Rachel Wagman, MD, the executive medical director of global clinical development for Amgen. Still, several committee members called for postmarketing data to focus on U.S. patients.
Romosozumab was approved for marketing in Japan on Jan. 8, 2019; approval is also being sought in Europe .
The FDA usually follows the recommendations of its advisory panels.
In an 18-1 vote, the FDA’s Bone, Reproductive, and Urologic Drugs Advisory Committee agreed that the risk-benefit profile of romosozumab, to be marketed by Amgen as Evenity, was favorable enough to support approval. Relative risk reductions of up to 75%, compared with placebo, and 36%, compared with alendronate, were seen in pivotal clinical trials.
A signal for increased major adverse cardiovascular events (MACE) among those receiving romosozumab had been seen in just one of the clinical trials, with a hazard ratio for MACE of 1.87 for those taking romosozumab, compared with those taking alendronate (95% confidence interval, 1.11-3.14).
Committee members were enthusiastic about the efficacy of the monoclonal antibody, which binds to sclerostin and prevents its inhibiting effect, allowing robust new bone formation. “I want to emphasize the remarkable skeletal efficacy of this drug; truly, it’s better than anything we’ve seen before,” said committee member Sundeep Khosla, MD, professor of medicine and physiology at the Mayo Clinic, Rochester, Minn.
In its application, the sponsor relied on two clinical trials. In the first, 7,180 women with osteoporosis aged 55-90 years were randomized 1:1 to receive romosozumab or placebo for 12 months in a double-blind trial. After this time, participants in each arm received follow-on treatment with denosumab (Prolia) for another 12 months. This study, dubbed Trial 337, followed morphometric vertebral fractures at 12 and 24 months. Morphometric fractures included both symptomatic and asymptomatic fractures.
Those treated with romosozumab had relative risk reductions of new vertebral fractures of 73% and 75%, compared with those given placebo at 12 and 24 months. Absolute risk reductions for vertebral fractures were 1.30% and 1.89% at 1 and 2 years (P less than .001 for both).
The second study, Trial 142, was a double-blind, active-controlled study that included 4,093 women aged 55-90 years with osteoporosis and a history of prior fragility fracture. Participants were randomized 1:1 to receive either romosozumab or alendronate for 12 months, with an additional variable period of alendronate follow-on of at least 12 months for both arms.
For Trial 142, one primary endpoint was morphometric vertebral fractures at month 24. An additional endpoint, clinical fracture, was a composite of symptomatic vertebral fractures and nonvertebral fractures. This second endpoint was assessed at the time of primary analysis, an event-driven cut point that occurred when at least 330 participants experienced a clinical fracture and all participants had completed the 24-month visit.
Hip fractures were less common among those given romosozumab, and bone mineral density increased significantly as well.
Vertebral fractures were reduced by 36% in the romosozumab group relative to the alendronate group, and clinical fractures by 27% (P less than .001 for both).
Overall, the number of adverse events for the more than 7,500 patients in the safety population was similar between those receiving romosozumab and either placebo or alendronate, said Scott Wasserman, MD, vice president of global development for Amgen.
However, in Trial 142, which included patients who were slightly older and on more cardiovascular medications at baseline than in Trial 337, MACE – defined as cardiovascular death, MI, and stroke – occurred more frequently among those taking romosozumab, driven primarily by increased cardiac and cerebral ischemic events occurring within the first 12 months of beginning the study drug. At 12 months, the romosozumab arm saw 41 instances of MACE, compared with 22 in the alendronate arm to produce the HR of 1.87 (2.0% vs. 1.1%).
With regard to the imbalance in MACE seen in Trial 142, both the FDA and presenters for the sponsor entertained the notion that alendronate may have been somewhat protective for cardiovascular events. Although there is some biologic plausibility for a cardioprotective event for bisphosphonates, alendronate is highly specific for bone activity and the preponderance of previous studies have not shown such cardioprotection, the FDA, sponsors, and committee members all agreed.
Marc Sabatine, MD, the Lewis Dexter, MD Distinguished Chair in Cardiovascular Medicine at Harvard Medical School, Boston, was available to answer questions on behalf of the study’s sponsor. He noted at several points during the meeting just how few total cardiovascular events were seen overall in the romosozumab trial. The overall small numbers, he said, made it very difficult to distinguish whether the smaller number of MACE seen in the alendronate arm of Trial 142 were a true safety signal or just “a play of chance.”
Almost all the committee’s questioning and discussion centered on this potential increased cardiovascular risk. Amgen, in discussion with the FDA, had agreed to a black box warning designed to wave off prescribing romosozumab to those at increased risk for cardiovascular disease, focusing on those with a history of MI or stroke.
Additionally, the sponsor proposed a postmarketing real-world observational study to track incidence of MACE in those receiving romosozumab, comparing them with those receiving standard of care for osteoporosis.
Several committee members pointed out a problem with the proposed safety mitigation scheme: By conducting a postmarketing observational study for a drug that has a black-box warning to exclude those at high risk for MACE, the chance of detecting an actual cardiovascular safety problem plummets. “This is the textbook example of when observational studies struggle or are virtually guaranteed to fail,” noted Tobias Gerhard, PhD, a pharmacoepidemiologist at Rutgers University, New Brunswick, N.J.
On the other hand, noted several committee members, pausing to conduct a premarketing randomized, controlled trial would keep a beneficial drug away from a population in need. A postmarketing randomized, controlled trial, even a simple trial, still presents challenges, some of the committee acknowledged. Dr. Khosla voiced the opinion that “A randomized, controlled trial is virtually impossible.”
The committee, which was charged with discussing, but not voting on, what additional data should be obtained – and when – to sort out the cardiovascular safety question, was approximately evenly divided in the matter of whether an observational or registry-based trial, or a controlled trial, would be the best path forward.
Committee member Robert A. Adler, MD, put a realistic frame around the debate. “As an endocrinologist, I deal with nuances every day. I really think the kind of clinician who is going to be using this drug is used to dealing with benefits and risks and trying to tailor treatment to a given patient,” said Dr. Adler, professor of internal medicine and epidemiology at Virginia Commonwealth University, Richmond.
In its proposed indication, Amgen defined the population of menopausal women at high risk of fracture as those with a history of osteoporotic fracture, multiple risk factors for fracture, or patients who have failed or are intolerant to other available osteoporosis therapy. Romosozumab would be given as a once-monthly subcutaneous injection of 210 mg for a period of 12 months, to be followed by antiresorptive therapy.
Most of the participants in the clinical trials resided outside the United States, primarily because of the difficulty of recruiting clinical trial participants in the United States, Amgen officials said during their presentation. However, neither the time to the first positively adjudicated MACE nor bone mineral density responses at month 12 differed significantly across the various geographic regions where clinical trial sites were located, said Rachel Wagman, MD, the executive medical director of global clinical development for Amgen. Still, several committee members called for postmarketing data to focus on U.S. patients.
Romosozumab was approved for marketing in Japan on Jan. 8, 2019; approval is also being sought in Europe .
The FDA usually follows the recommendations of its advisory panels.
In an 18-1 vote, the FDA’s Bone, Reproductive, and Urologic Drugs Advisory Committee agreed that the risk-benefit profile of romosozumab, to be marketed by Amgen as Evenity, was favorable enough to support approval. Relative risk reductions of up to 75%, compared with placebo, and 36%, compared with alendronate, were seen in pivotal clinical trials.
A signal for increased major adverse cardiovascular events (MACE) among those receiving romosozumab had been seen in just one of the clinical trials, with a hazard ratio for MACE of 1.87 for those taking romosozumab, compared with those taking alendronate (95% confidence interval, 1.11-3.14).
Committee members were enthusiastic about the efficacy of the monoclonal antibody, which binds to sclerostin and prevents its inhibiting effect, allowing robust new bone formation. “I want to emphasize the remarkable skeletal efficacy of this drug; truly, it’s better than anything we’ve seen before,” said committee member Sundeep Khosla, MD, professor of medicine and physiology at the Mayo Clinic, Rochester, Minn.
In its application, the sponsor relied on two clinical trials. In the first, 7,180 women with osteoporosis aged 55-90 years were randomized 1:1 to receive romosozumab or placebo for 12 months in a double-blind trial. After this time, participants in each arm received follow-on treatment with denosumab (Prolia) for another 12 months. This study, dubbed Trial 337, followed morphometric vertebral fractures at 12 and 24 months. Morphometric fractures included both symptomatic and asymptomatic fractures.
Those treated with romosozumab had relative risk reductions of new vertebral fractures of 73% and 75%, compared with those given placebo at 12 and 24 months. Absolute risk reductions for vertebral fractures were 1.30% and 1.89% at 1 and 2 years (P less than .001 for both).
The second study, Trial 142, was a double-blind, active-controlled study that included 4,093 women aged 55-90 years with osteoporosis and a history of prior fragility fracture. Participants were randomized 1:1 to receive either romosozumab or alendronate for 12 months, with an additional variable period of alendronate follow-on of at least 12 months for both arms.
For Trial 142, one primary endpoint was morphometric vertebral fractures at month 24. An additional endpoint, clinical fracture, was a composite of symptomatic vertebral fractures and nonvertebral fractures. This second endpoint was assessed at the time of primary analysis, an event-driven cut point that occurred when at least 330 participants experienced a clinical fracture and all participants had completed the 24-month visit.
Hip fractures were less common among those given romosozumab, and bone mineral density increased significantly as well.
Vertebral fractures were reduced by 36% in the romosozumab group relative to the alendronate group, and clinical fractures by 27% (P less than .001 for both).
Overall, the number of adverse events for the more than 7,500 patients in the safety population was similar between those receiving romosozumab and either placebo or alendronate, said Scott Wasserman, MD, vice president of global development for Amgen.
However, in Trial 142, which included patients who were slightly older and on more cardiovascular medications at baseline than in Trial 337, MACE – defined as cardiovascular death, MI, and stroke – occurred more frequently among those taking romosozumab, driven primarily by increased cardiac and cerebral ischemic events occurring within the first 12 months of beginning the study drug. At 12 months, the romosozumab arm saw 41 instances of MACE, compared with 22 in the alendronate arm to produce the HR of 1.87 (2.0% vs. 1.1%).
With regard to the imbalance in MACE seen in Trial 142, both the FDA and presenters for the sponsor entertained the notion that alendronate may have been somewhat protective for cardiovascular events. Although there is some biologic plausibility for a cardioprotective event for bisphosphonates, alendronate is highly specific for bone activity and the preponderance of previous studies have not shown such cardioprotection, the FDA, sponsors, and committee members all agreed.
Marc Sabatine, MD, the Lewis Dexter, MD Distinguished Chair in Cardiovascular Medicine at Harvard Medical School, Boston, was available to answer questions on behalf of the study’s sponsor. He noted at several points during the meeting just how few total cardiovascular events were seen overall in the romosozumab trial. The overall small numbers, he said, made it very difficult to distinguish whether the smaller number of MACE seen in the alendronate arm of Trial 142 were a true safety signal or just “a play of chance.”
Almost all the committee’s questioning and discussion centered on this potential increased cardiovascular risk. Amgen, in discussion with the FDA, had agreed to a black box warning designed to wave off prescribing romosozumab to those at increased risk for cardiovascular disease, focusing on those with a history of MI or stroke.
Additionally, the sponsor proposed a postmarketing real-world observational study to track incidence of MACE in those receiving romosozumab, comparing them with those receiving standard of care for osteoporosis.
Several committee members pointed out a problem with the proposed safety mitigation scheme: By conducting a postmarketing observational study for a drug that has a black-box warning to exclude those at high risk for MACE, the chance of detecting an actual cardiovascular safety problem plummets. “This is the textbook example of when observational studies struggle or are virtually guaranteed to fail,” noted Tobias Gerhard, PhD, a pharmacoepidemiologist at Rutgers University, New Brunswick, N.J.
On the other hand, noted several committee members, pausing to conduct a premarketing randomized, controlled trial would keep a beneficial drug away from a population in need. A postmarketing randomized, controlled trial, even a simple trial, still presents challenges, some of the committee acknowledged. Dr. Khosla voiced the opinion that “A randomized, controlled trial is virtually impossible.”
The committee, which was charged with discussing, but not voting on, what additional data should be obtained – and when – to sort out the cardiovascular safety question, was approximately evenly divided in the matter of whether an observational or registry-based trial, or a controlled trial, would be the best path forward.
Committee member Robert A. Adler, MD, put a realistic frame around the debate. “As an endocrinologist, I deal with nuances every day. I really think the kind of clinician who is going to be using this drug is used to dealing with benefits and risks and trying to tailor treatment to a given patient,” said Dr. Adler, professor of internal medicine and epidemiology at Virginia Commonwealth University, Richmond.
In its proposed indication, Amgen defined the population of menopausal women at high risk of fracture as those with a history of osteoporotic fracture, multiple risk factors for fracture, or patients who have failed or are intolerant to other available osteoporosis therapy. Romosozumab would be given as a once-monthly subcutaneous injection of 210 mg for a period of 12 months, to be followed by antiresorptive therapy.
Most of the participants in the clinical trials resided outside the United States, primarily because of the difficulty of recruiting clinical trial participants in the United States, Amgen officials said during their presentation. However, neither the time to the first positively adjudicated MACE nor bone mineral density responses at month 12 differed significantly across the various geographic regions where clinical trial sites were located, said Rachel Wagman, MD, the executive medical director of global clinical development for Amgen. Still, several committee members called for postmarketing data to focus on U.S. patients.
Romosozumab was approved for marketing in Japan on Jan. 8, 2019; approval is also being sought in Europe .
The FDA usually follows the recommendations of its advisory panels.
SGLT-2 inhibitors promising for heart failure prevention, not treatment
LOS ANGELES –

They also may play a role in the treatment of patients with known heart failure (HF), but further studies are required to prove definite treatment benefit.
“These trials enrolled a minority of patients with known heart failure, and, in those subgroups, the drugs seems to reduce the risk for hospitalization, opening the possibility of treatment benefit,” Javed Butler, MD, said at the World Congress on Insulin Resistance, Diabetes & Cardiovascular Disease. “But there were not enough patients to conclude this. If you are treating diabetes with these agents in patients with heart failure, more power to you. But don’t think you are treating heart failure per se until the results of the dedicated heart failure trials come out.”
Good glycemic control has not been shown to affect heart failure outcomes per se, said Dr. Butler, professor and chairman of the department of medicine at the University of Mississippi Medical Center, Jackson.
“People seem to mix the concepts of prevention and treatment together,” he said. “We have now very good evidence across all trials with SGLT-2 inhibitors for prevention of heart failure. But for treatment, we need more data despite favorable early signals.
“Also, these trials include most patients with ischemic heart disease, but we don’t have data on nonischemic etiology for the development of heart failure from these trials,” Dr. Butler added.
The best available data from clinical trials suggest that patients with American College of Cardiology Foundation/American Heart Association heart failure classification stages A and B benefit the most from aggressive treatment to prevent HF.
“Either they have diseases like high blood pressure or diabetes, but their hearts are normal, or, perhaps, their hearts are abnormal, and they develop left ventricular hypertrophy or atrial fibrillation,” he said. “However, if someone is stage C – manifest heart failure – or stage D – advanced heart failure – we need further data on novel therapies to improve their outcomes.”
Dr. Butler emphasized that not all heart failure is associated with atherosclerotic vascular disease. In fact, the Health, Aging, and Body Composition Study showed that the incidence of heart failure increased progressively across age groups, both for those with and without a preceding vascular event (P = .03 and P less than .001, respectively; Eur J Heart Fail. 2014 May;16[5]:526-34). “There’s a whole other world of nonischemic heart failure that we also need to worry about,” he said. “There is a lot of microvascular endothelial dysfunction.”
The combination of heart failure and diabetes is especially lethal. “If you put them together, you’re looking at about a 10-fold higher risk of mortality, which is a horrible prognosis,” Dr. Butler said. “That means that we need to think about prevention and treatment separately.”
Data from the SAVOR-TIMI 53, EXAMINE, and TECOS trials show there is no protective effect of dipeptidyl peptidase–4 inhibitors when it comes to hospitalization for heart failure.
“The other classes of drugs either increase the risk, or we don’t have very good data,” Dr. Butler said. “So far, across the spectrum of therapies for diabetes, the effect on heart failure is neutral and perhaps confers some risk.”
SGLT-2 inhibitors convey a different story.
In the EMPA-REG OUTCOME Trial, one inclusion criterion was established cardiovascular disease (CVD) in the form of a prior MI, coronary artery disease, stroke, unstable angina, or occlusive peripheral artery disease, but not heart failure alone (N Engl J Med. 2015 Nov 26; 373[22]:2117-28). “This was not a heart failure study, so we don’t know what their New York Heart Association class was, or the details of their baseline HF treatment in the minority of patients who were enrolled who had a history of HF,” Dr. Butler cautioned.
However, the trial found that empagliflozin conferred an overall cardiovascular death risk reduction of 38%, compared with placebo. When the researchers assessed the impact of treatment on all modes of cardiovascular death, they found that death from heart failure benefited the most (hazard ratio, 0.32; P = .0008), while sudden death benefited as well. Empagliflozin also had a significant impact on reduced hospitalization for heart failure, compared with placebo (HR, 0.65).
“This is a large enough cohort that you should feel comfortable that this drug is preventing heart failure in those with HF at baseline,” said Dr. Butler, who was not involved with the study. “We can have a debate about whether this is a treatment for heart failure or not, but for prevention of heart failure, I feel comfortable that these drugs do that.”
A subsequent study of canagliflozin and cardiovascular and renal events in type 2 diabetes showed the same result (N Engl J Med. 2017 Aug 17; 377[7]:644-57). It reduced hospitalization for heart failure by 33% (HR, 0.67).
Then came the CVD-REAL study, which found low rates of hospitalization for heart failure and all-cause death in new users of SGLT-2 inhibitors. More recently, DECLARE-TIMI 58 yielded similar results.
“One of the criticisms of these findings is that heart failure characteristics were not well phenotyped in these studies,” Dr. Butler said. “I say it really does not matter. Heart failure hospitalizations are associated with a poor prognosis irrespective of whether the hospitalization occurred in patients without heart failure or in a patient with previously diagnosed heart failure, or whether the patient has reduced or preserved ejection fraction.
“Framingham and other classic studies show us that 5-year mortality for heart failure is about 50%,” he noted. “If you can prevent a disease that has a 5-year mortality of 50%, doesn’t that sound like a really good deal?”
A contemporary appraisal of the heart failure epidemic in Olmstead County, Minn., during 2000-2010 found that the mortality was 20.2% at 1 year after diagnosis, and 52.6% at 5 years after diagnosis. The data include new-onset HF in both inpatient and outpatient settings.
Specifically, new-onset HF hospitalization was associated with a 1-year post discharge mortality of 21.1% (JAMA Intern Med. 2015;175[6]:996-1004). “We cannot ignore prevention of heart failure,” Dr. Butler said. “Also, for treatment, once you get hospitalized for heart failure, the fundamental natural history of the disease changes. There is a 30% cumulative incremental death risk between the second and third hospitalizations.”
Dr. Butler concluded his presentation by noting that five randomized, controlled trials evaluating SGLT-2 inhibitors in HF have been launched, and should help elucidate any effects the drugs may have in treating the condition. They include EMPEROR-Preserved (NCT03057951), EMPEROR-Reduced (NCT03057977), Dapa-HF (NCT03036124), and SOLOIST-WHF (NCT03521934) and DELIVER (NCT03619213).
Dr. Butler disclosed that he has received research support from the National Institutes of Health, the European Union, and the Patient-Centered Outcomes Research Institute. He has also been a consultant for numerous pharmaceutical companies, including Boehringer Ingelheim, Janssen, and AstraZeneca, which sponsored the EMPA-REG, CANVAS, and DECLARE TIMI 58 trials.
LOS ANGELES –
They also may play a role in the treatment of patients with known heart failure (HF), but further studies are required to prove definite treatment benefit.
“These trials enrolled a minority of patients with known heart failure, and, in those subgroups, the drugs seems to reduce the risk for hospitalization, opening the possibility of treatment benefit,” Javed Butler, MD, said at the World Congress on Insulin Resistance, Diabetes & Cardiovascular Disease. “But there were not enough patients to conclude this. If you are treating diabetes with these agents in patients with heart failure, more power to you. But don’t think you are treating heart failure per se until the results of the dedicated heart failure trials come out.”
Good glycemic control has not been shown to affect heart failure outcomes per se, said Dr. Butler, professor and chairman of the department of medicine at the University of Mississippi Medical Center, Jackson.
“People seem to mix the concepts of prevention and treatment together,” he said. “We have now very good evidence across all trials with SGLT-2 inhibitors for prevention of heart failure. But for treatment, we need more data despite favorable early signals.
“Also, these trials include most patients with ischemic heart disease, but we don’t have data on nonischemic etiology for the development of heart failure from these trials,” Dr. Butler added.
The best available data from clinical trials suggest that patients with American College of Cardiology Foundation/American Heart Association heart failure classification stages A and B benefit the most from aggressive treatment to prevent HF.
“Either they have diseases like high blood pressure or diabetes, but their hearts are normal, or, perhaps, their hearts are abnormal, and they develop left ventricular hypertrophy or atrial fibrillation,” he said. “However, if someone is stage C – manifest heart failure – or stage D – advanced heart failure – we need further data on novel therapies to improve their outcomes.”
Dr. Butler emphasized that not all heart failure is associated with atherosclerotic vascular disease. In fact, the Health, Aging, and Body Composition Study showed that the incidence of heart failure increased progressively across age groups, both for those with and without a preceding vascular event (P = .03 and P less than .001, respectively; Eur J Heart Fail. 2014 May;16[5]:526-34). “There’s a whole other world of nonischemic heart failure that we also need to worry about,” he said. “There is a lot of microvascular endothelial dysfunction.”
The combination of heart failure and diabetes is especially lethal. “If you put them together, you’re looking at about a 10-fold higher risk of mortality, which is a horrible prognosis,” Dr. Butler said. “That means that we need to think about prevention and treatment separately.”
Data from the SAVOR-TIMI 53, EXAMINE, and TECOS trials show there is no protective effect of dipeptidyl peptidase–4 inhibitors when it comes to hospitalization for heart failure.
“The other classes of drugs either increase the risk, or we don’t have very good data,” Dr. Butler said. “So far, across the spectrum of therapies for diabetes, the effect on heart failure is neutral and perhaps confers some risk.”
SGLT-2 inhibitors convey a different story.
In the EMPA-REG OUTCOME Trial, one inclusion criterion was established cardiovascular disease (CVD) in the form of a prior MI, coronary artery disease, stroke, unstable angina, or occlusive peripheral artery disease, but not heart failure alone (N Engl J Med. 2015 Nov 26; 373[22]:2117-28). “This was not a heart failure study, so we don’t know what their New York Heart Association class was, or the details of their baseline HF treatment in the minority of patients who were enrolled who had a history of HF,” Dr. Butler cautioned.
However, the trial found that empagliflozin conferred an overall cardiovascular death risk reduction of 38%, compared with placebo. When the researchers assessed the impact of treatment on all modes of cardiovascular death, they found that death from heart failure benefited the most (hazard ratio, 0.32; P = .0008), while sudden death benefited as well. Empagliflozin also had a significant impact on reduced hospitalization for heart failure, compared with placebo (HR, 0.65).
“This is a large enough cohort that you should feel comfortable that this drug is preventing heart failure in those with HF at baseline,” said Dr. Butler, who was not involved with the study. “We can have a debate about whether this is a treatment for heart failure or not, but for prevention of heart failure, I feel comfortable that these drugs do that.”
A subsequent study of canagliflozin and cardiovascular and renal events in type 2 diabetes showed the same result (N Engl J Med. 2017 Aug 17; 377[7]:644-57). It reduced hospitalization for heart failure by 33% (HR, 0.67).
Then came the CVD-REAL study, which found low rates of hospitalization for heart failure and all-cause death in new users of SGLT-2 inhibitors. More recently, DECLARE-TIMI 58 yielded similar results.
“One of the criticisms of these findings is that heart failure characteristics were not well phenotyped in these studies,” Dr. Butler said. “I say it really does not matter. Heart failure hospitalizations are associated with a poor prognosis irrespective of whether the hospitalization occurred in patients without heart failure or in a patient with previously diagnosed heart failure, or whether the patient has reduced or preserved ejection fraction.
“Framingham and other classic studies show us that 5-year mortality for heart failure is about 50%,” he noted. “If you can prevent a disease that has a 5-year mortality of 50%, doesn’t that sound like a really good deal?”
A contemporary appraisal of the heart failure epidemic in Olmstead County, Minn., during 2000-2010 found that the mortality was 20.2% at 1 year after diagnosis, and 52.6% at 5 years after diagnosis. The data include new-onset HF in both inpatient and outpatient settings.
Specifically, new-onset HF hospitalization was associated with a 1-year post discharge mortality of 21.1% (JAMA Intern Med. 2015;175[6]:996-1004). “We cannot ignore prevention of heart failure,” Dr. Butler said. “Also, for treatment, once you get hospitalized for heart failure, the fundamental natural history of the disease changes. There is a 30% cumulative incremental death risk between the second and third hospitalizations.”
Dr. Butler concluded his presentation by noting that five randomized, controlled trials evaluating SGLT-2 inhibitors in HF have been launched, and should help elucidate any effects the drugs may have in treating the condition. They include EMPEROR-Preserved (NCT03057951), EMPEROR-Reduced (NCT03057977), Dapa-HF (NCT03036124), and SOLOIST-WHF (NCT03521934) and DELIVER (NCT03619213).
Dr. Butler disclosed that he has received research support from the National Institutes of Health, the European Union, and the Patient-Centered Outcomes Research Institute. He has also been a consultant for numerous pharmaceutical companies, including Boehringer Ingelheim, Janssen, and AstraZeneca, which sponsored the EMPA-REG, CANVAS, and DECLARE TIMI 58 trials.
LOS ANGELES –
They also may play a role in the treatment of patients with known heart failure (HF), but further studies are required to prove definite treatment benefit.
“These trials enrolled a minority of patients with known heart failure, and, in those subgroups, the drugs seems to reduce the risk for hospitalization, opening the possibility of treatment benefit,” Javed Butler, MD, said at the World Congress on Insulin Resistance, Diabetes & Cardiovascular Disease. “But there were not enough patients to conclude this. If you are treating diabetes with these agents in patients with heart failure, more power to you. But don’t think you are treating heart failure per se until the results of the dedicated heart failure trials come out.”
Good glycemic control has not been shown to affect heart failure outcomes per se, said Dr. Butler, professor and chairman of the department of medicine at the University of Mississippi Medical Center, Jackson.
“People seem to mix the concepts of prevention and treatment together,” he said. “We have now very good evidence across all trials with SGLT-2 inhibitors for prevention of heart failure. But for treatment, we need more data despite favorable early signals.
“Also, these trials include most patients with ischemic heart disease, but we don’t have data on nonischemic etiology for the development of heart failure from these trials,” Dr. Butler added.
The best available data from clinical trials suggest that patients with American College of Cardiology Foundation/American Heart Association heart failure classification stages A and B benefit the most from aggressive treatment to prevent HF.
“Either they have diseases like high blood pressure or diabetes, but their hearts are normal, or, perhaps, their hearts are abnormal, and they develop left ventricular hypertrophy or atrial fibrillation,” he said. “However, if someone is stage C – manifest heart failure – or stage D – advanced heart failure – we need further data on novel therapies to improve their outcomes.”
Dr. Butler emphasized that not all heart failure is associated with atherosclerotic vascular disease. In fact, the Health, Aging, and Body Composition Study showed that the incidence of heart failure increased progressively across age groups, both for those with and without a preceding vascular event (P = .03 and P less than .001, respectively; Eur J Heart Fail. 2014 May;16[5]:526-34). “There’s a whole other world of nonischemic heart failure that we also need to worry about,” he said. “There is a lot of microvascular endothelial dysfunction.”
The combination of heart failure and diabetes is especially lethal. “If you put them together, you’re looking at about a 10-fold higher risk of mortality, which is a horrible prognosis,” Dr. Butler said. “That means that we need to think about prevention and treatment separately.”
Data from the SAVOR-TIMI 53, EXAMINE, and TECOS trials show there is no protective effect of dipeptidyl peptidase–4 inhibitors when it comes to hospitalization for heart failure.
“The other classes of drugs either increase the risk, or we don’t have very good data,” Dr. Butler said. “So far, across the spectrum of therapies for diabetes, the effect on heart failure is neutral and perhaps confers some risk.”
SGLT-2 inhibitors convey a different story.
In the EMPA-REG OUTCOME Trial, one inclusion criterion was established cardiovascular disease (CVD) in the form of a prior MI, coronary artery disease, stroke, unstable angina, or occlusive peripheral artery disease, but not heart failure alone (N Engl J Med. 2015 Nov 26; 373[22]:2117-28). “This was not a heart failure study, so we don’t know what their New York Heart Association class was, or the details of their baseline HF treatment in the minority of patients who were enrolled who had a history of HF,” Dr. Butler cautioned.
However, the trial found that empagliflozin conferred an overall cardiovascular death risk reduction of 38%, compared with placebo. When the researchers assessed the impact of treatment on all modes of cardiovascular death, they found that death from heart failure benefited the most (hazard ratio, 0.32; P = .0008), while sudden death benefited as well. Empagliflozin also had a significant impact on reduced hospitalization for heart failure, compared with placebo (HR, 0.65).
“This is a large enough cohort that you should feel comfortable that this drug is preventing heart failure in those with HF at baseline,” said Dr. Butler, who was not involved with the study. “We can have a debate about whether this is a treatment for heart failure or not, but for prevention of heart failure, I feel comfortable that these drugs do that.”
A subsequent study of canagliflozin and cardiovascular and renal events in type 2 diabetes showed the same result (N Engl J Med. 2017 Aug 17; 377[7]:644-57). It reduced hospitalization for heart failure by 33% (HR, 0.67).
Then came the CVD-REAL study, which found low rates of hospitalization for heart failure and all-cause death in new users of SGLT-2 inhibitors. More recently, DECLARE-TIMI 58 yielded similar results.
“One of the criticisms of these findings is that heart failure characteristics were not well phenotyped in these studies,” Dr. Butler said. “I say it really does not matter. Heart failure hospitalizations are associated with a poor prognosis irrespective of whether the hospitalization occurred in patients without heart failure or in a patient with previously diagnosed heart failure, or whether the patient has reduced or preserved ejection fraction.
“Framingham and other classic studies show us that 5-year mortality for heart failure is about 50%,” he noted. “If you can prevent a disease that has a 5-year mortality of 50%, doesn’t that sound like a really good deal?”
A contemporary appraisal of the heart failure epidemic in Olmstead County, Minn., during 2000-2010 found that the mortality was 20.2% at 1 year after diagnosis, and 52.6% at 5 years after diagnosis. The data include new-onset HF in both inpatient and outpatient settings.
Specifically, new-onset HF hospitalization was associated with a 1-year post discharge mortality of 21.1% (JAMA Intern Med. 2015;175[6]:996-1004). “We cannot ignore prevention of heart failure,” Dr. Butler said. “Also, for treatment, once you get hospitalized for heart failure, the fundamental natural history of the disease changes. There is a 30% cumulative incremental death risk between the second and third hospitalizations.”
Dr. Butler concluded his presentation by noting that five randomized, controlled trials evaluating SGLT-2 inhibitors in HF have been launched, and should help elucidate any effects the drugs may have in treating the condition. They include EMPEROR-Preserved (NCT03057951), EMPEROR-Reduced (NCT03057977), Dapa-HF (NCT03036124), and SOLOIST-WHF (NCT03521934) and DELIVER (NCT03619213).
Dr. Butler disclosed that he has received research support from the National Institutes of Health, the European Union, and the Patient-Centered Outcomes Research Institute. He has also been a consultant for numerous pharmaceutical companies, including Boehringer Ingelheim, Janssen, and AstraZeneca, which sponsored the EMPA-REG, CANVAS, and DECLARE TIMI 58 trials.
EXPERT ANALYSIS FROM WCIRDC 2018