User login
‘Forward-Oriented’ Vector Holds Potential for Sickle Cell Cure
About 100,000 people in America have sickle cell disease. Of those, an estimated 27 people have undergone experimental gene therapy using conventional vectors—virus-based vehicles for delivering “therapeutic genes.” Now National Institutes of Health researchers have taken the vector idea and revved it up, bringing the possibility of curing sickle cell disease a bit closer.
With gene therapy, doctors add a normal copy of the β-globin gene to the patient’s hematopoietic stem cells, then reinfuse the modified stem cells into the patient to produce normal disc-shaped red blood cells. In animal studies, the new vector was up to 10 times more efficient at incorporating corrective genes into bone marrow stem cells with a carrying capacity of up to 6 times greater viral load than current vectors. The new vectors also can be produced in much higher amounts, saving time and lowering costs.
The researchers call it a “forward-oriented” vector because it changes the usual direction of how gene sequences in globin-containing vectors are read: from right to left. That backward orientation—globin-containing vectors are the only therapeutic vectors in clinical development that use it—the researchers say, “has remained unchallenged for decades despite its negative impacts on efficiency.”
The right-to-left orientation was dictated by the need to prevent the loss of a key molecular component, intron 2, by RNA splicing during the vector preparation. The redesigned forward-reading method crucially leaves intron 2 intact and makes the gene-translation approach less complicated, says John Tisdale, MD, chief of the Cellular and Molecular Therapeutic Branch at the National Heart, Lung, and Blood Institute, who, with Naoya Uchida, MD, PhD, came up with the idea.
In testing, the new vectors also proved longer lasting, remaining in place 4 years after transplantation.
National Institutes of Health is working to accelerate research and development through the Cure Sickle Cell Initiative, launched by NHLBI in 2018 to identify and support the most promising genetic therapies for the more than 20 million people worldwide who have sickle cell disease. NIH holds the patent for the new vector, which still will need clinical testing in humans. Clinical trials are actively enrolling.
About 100,000 people in America have sickle cell disease. Of those, an estimated 27 people have undergone experimental gene therapy using conventional vectors—virus-based vehicles for delivering “therapeutic genes.” Now National Institutes of Health researchers have taken the vector idea and revved it up, bringing the possibility of curing sickle cell disease a bit closer.
With gene therapy, doctors add a normal copy of the β-globin gene to the patient’s hematopoietic stem cells, then reinfuse the modified stem cells into the patient to produce normal disc-shaped red blood cells. In animal studies, the new vector was up to 10 times more efficient at incorporating corrective genes into bone marrow stem cells with a carrying capacity of up to 6 times greater viral load than current vectors. The new vectors also can be produced in much higher amounts, saving time and lowering costs.
The researchers call it a “forward-oriented” vector because it changes the usual direction of how gene sequences in globin-containing vectors are read: from right to left. That backward orientation—globin-containing vectors are the only therapeutic vectors in clinical development that use it—the researchers say, “has remained unchallenged for decades despite its negative impacts on efficiency.”
The right-to-left orientation was dictated by the need to prevent the loss of a key molecular component, intron 2, by RNA splicing during the vector preparation. The redesigned forward-reading method crucially leaves intron 2 intact and makes the gene-translation approach less complicated, says John Tisdale, MD, chief of the Cellular and Molecular Therapeutic Branch at the National Heart, Lung, and Blood Institute, who, with Naoya Uchida, MD, PhD, came up with the idea.
In testing, the new vectors also proved longer lasting, remaining in place 4 years after transplantation.
National Institutes of Health is working to accelerate research and development through the Cure Sickle Cell Initiative, launched by NHLBI in 2018 to identify and support the most promising genetic therapies for the more than 20 million people worldwide who have sickle cell disease. NIH holds the patent for the new vector, which still will need clinical testing in humans. Clinical trials are actively enrolling.
About 100,000 people in America have sickle cell disease. Of those, an estimated 27 people have undergone experimental gene therapy using conventional vectors—virus-based vehicles for delivering “therapeutic genes.” Now National Institutes of Health researchers have taken the vector idea and revved it up, bringing the possibility of curing sickle cell disease a bit closer.
With gene therapy, doctors add a normal copy of the β-globin gene to the patient’s hematopoietic stem cells, then reinfuse the modified stem cells into the patient to produce normal disc-shaped red blood cells. In animal studies, the new vector was up to 10 times more efficient at incorporating corrective genes into bone marrow stem cells with a carrying capacity of up to 6 times greater viral load than current vectors. The new vectors also can be produced in much higher amounts, saving time and lowering costs.
The researchers call it a “forward-oriented” vector because it changes the usual direction of how gene sequences in globin-containing vectors are read: from right to left. That backward orientation—globin-containing vectors are the only therapeutic vectors in clinical development that use it—the researchers say, “has remained unchallenged for decades despite its negative impacts on efficiency.”
The right-to-left orientation was dictated by the need to prevent the loss of a key molecular component, intron 2, by RNA splicing during the vector preparation. The redesigned forward-reading method crucially leaves intron 2 intact and makes the gene-translation approach less complicated, says John Tisdale, MD, chief of the Cellular and Molecular Therapeutic Branch at the National Heart, Lung, and Blood Institute, who, with Naoya Uchida, MD, PhD, came up with the idea.
In testing, the new vectors also proved longer lasting, remaining in place 4 years after transplantation.
National Institutes of Health is working to accelerate research and development through the Cure Sickle Cell Initiative, launched by NHLBI in 2018 to identify and support the most promising genetic therapies for the more than 20 million people worldwide who have sickle cell disease. NIH holds the patent for the new vector, which still will need clinical testing in humans. Clinical trials are actively enrolling.
Stem cells gene edited to be HIV resistant treat ALL, but not HIV
Gene editing of donor stem cells prior to transplantation into a patient with both HIV infection and acute lymphoblastic leukemia (ALL) was safe and effectively treated the patient’s leukemia, but failed to resolve his HIV, investigators reported.

The 27-year-old man received an HLA-matched transplant of hematopoietic stem and progenitor cells (HSPCs) that had been genetically engineered to lack CCR5, a key gateway for HIV entry into cells.
Although the transplant resulted in complete remission of leukemia with full donor chimerism, only about 9% of the posttransplant lymphocytes showed disruption of CCR5, and during a brief trial of antiretroviral therapy interruption his HIV viral load rebounded, reported Hongkui Deng, PhD, and colleagues from Peking University in China.
Although the experiment did not meet its goal of a drug-free HIV remission, it serves as a proof of concept for the use of CRISPR-Cas9 (clustered regularly interspaced palindromic repeats/CRISPR-associated protein 9) gene editing to treat HIV infection, the authors contend.
“These results show the proof of principle that transplantation and long-term engraftment of CRISPR-edited allogeneic HSPCs can be achieved; however, the efficiency of the response was not adequate to achieve the target of cure of HIV-1 infection,” they wrote in a brief report published in the New England Journal of Medicine.
As previously reported, other research groups have investigated genetic editing to mimic a naturally occurring mutation that effectively disables the CCR5 HIV coreceptor, preventing the retrovirus from entering healthy cells. The mutation was first identified in a man named Timothy Brown who came to be known as “the Berlin patient” after he was apparently cured of HIV infection after a bone marrow transplant from a donor who had the mutation.
Dr. Deng and colleagues took advantage of HSPC transplantation, a standard therapy for ALL to see whether it could also have beneficial effects on concomitant HIV infection.
They treated donor HSPCs with CRISPR-Cas9 to ablate CCR5 and then delivered them to the patient along with additional CD34-depleted donor cells from mobilized peripheral blood.
The transplant was a success, with neutrophil engraftment on day 13 and platelet engraftment on day 27, and the leukemia was in morphologic complete remission at week 4 following transplantation. The patient remained in complete remission from leukemia throughout the 19-month follow-up period, with full donor chimerism .
However, when a planned interruption of antiretroviral therapy was carried out at 7 months post transplant, the serum viral load increased to 3 × 107 copies/ml at week 4 following interruption, and the patient was restarted on the drug. His viral levels gradually decreased to undetectable level during the subsequent months.
The investigators noted that 2 weeks after the drug interruption trial was started there was a small increase in the percentage of CCR5 insertion/deletions.
“The low efficiency of gene editing in the patient may be due to the competitive engraftment of the coinfused HSPCs in CD34-depleted cells and the persistence of donor T cells. To further clarify the anti-HIV effect of CCR5-ablated HSPCs, it will be essential to increase the gene-editing efficiency of our CRISPR-Cas9 system and improve the transplantation protocol,” they wrote.
The study was funded by the Beijing Municipal Science and Technology Commission and others (unspecified). All authors reported having nothing to disclose.
SOURCE: Xu L et al. N Engl J Med. 2019. doi: 10.1056/NEJMoa1817426.
Gene editing of donor stem cells prior to transplantation into a patient with both HIV infection and acute lymphoblastic leukemia (ALL) was safe and effectively treated the patient’s leukemia, but failed to resolve his HIV, investigators reported.
The 27-year-old man received an HLA-matched transplant of hematopoietic stem and progenitor cells (HSPCs) that had been genetically engineered to lack CCR5, a key gateway for HIV entry into cells.
Although the transplant resulted in complete remission of leukemia with full donor chimerism, only about 9% of the posttransplant lymphocytes showed disruption of CCR5, and during a brief trial of antiretroviral therapy interruption his HIV viral load rebounded, reported Hongkui Deng, PhD, and colleagues from Peking University in China.
Although the experiment did not meet its goal of a drug-free HIV remission, it serves as a proof of concept for the use of CRISPR-Cas9 (clustered regularly interspaced palindromic repeats/CRISPR-associated protein 9) gene editing to treat HIV infection, the authors contend.
“These results show the proof of principle that transplantation and long-term engraftment of CRISPR-edited allogeneic HSPCs can be achieved; however, the efficiency of the response was not adequate to achieve the target of cure of HIV-1 infection,” they wrote in a brief report published in the New England Journal of Medicine.
As previously reported, other research groups have investigated genetic editing to mimic a naturally occurring mutation that effectively disables the CCR5 HIV coreceptor, preventing the retrovirus from entering healthy cells. The mutation was first identified in a man named Timothy Brown who came to be known as “the Berlin patient” after he was apparently cured of HIV infection after a bone marrow transplant from a donor who had the mutation.
Dr. Deng and colleagues took advantage of HSPC transplantation, a standard therapy for ALL to see whether it could also have beneficial effects on concomitant HIV infection.
They treated donor HSPCs with CRISPR-Cas9 to ablate CCR5 and then delivered them to the patient along with additional CD34-depleted donor cells from mobilized peripheral blood.
The transplant was a success, with neutrophil engraftment on day 13 and platelet engraftment on day 27, and the leukemia was in morphologic complete remission at week 4 following transplantation. The patient remained in complete remission from leukemia throughout the 19-month follow-up period, with full donor chimerism .
However, when a planned interruption of antiretroviral therapy was carried out at 7 months post transplant, the serum viral load increased to 3 × 107 copies/ml at week 4 following interruption, and the patient was restarted on the drug. His viral levels gradually decreased to undetectable level during the subsequent months.
The investigators noted that 2 weeks after the drug interruption trial was started there was a small increase in the percentage of CCR5 insertion/deletions.
“The low efficiency of gene editing in the patient may be due to the competitive engraftment of the coinfused HSPCs in CD34-depleted cells and the persistence of donor T cells. To further clarify the anti-HIV effect of CCR5-ablated HSPCs, it will be essential to increase the gene-editing efficiency of our CRISPR-Cas9 system and improve the transplantation protocol,” they wrote.
The study was funded by the Beijing Municipal Science and Technology Commission and others (unspecified). All authors reported having nothing to disclose.
SOURCE: Xu L et al. N Engl J Med. 2019. doi: 10.1056/NEJMoa1817426.
Gene editing of donor stem cells prior to transplantation into a patient with both HIV infection and acute lymphoblastic leukemia (ALL) was safe and effectively treated the patient’s leukemia, but failed to resolve his HIV, investigators reported.
The 27-year-old man received an HLA-matched transplant of hematopoietic stem and progenitor cells (HSPCs) that had been genetically engineered to lack CCR5, a key gateway for HIV entry into cells.
Although the transplant resulted in complete remission of leukemia with full donor chimerism, only about 9% of the posttransplant lymphocytes showed disruption of CCR5, and during a brief trial of antiretroviral therapy interruption his HIV viral load rebounded, reported Hongkui Deng, PhD, and colleagues from Peking University in China.
Although the experiment did not meet its goal of a drug-free HIV remission, it serves as a proof of concept for the use of CRISPR-Cas9 (clustered regularly interspaced palindromic repeats/CRISPR-associated protein 9) gene editing to treat HIV infection, the authors contend.
“These results show the proof of principle that transplantation and long-term engraftment of CRISPR-edited allogeneic HSPCs can be achieved; however, the efficiency of the response was not adequate to achieve the target of cure of HIV-1 infection,” they wrote in a brief report published in the New England Journal of Medicine.
As previously reported, other research groups have investigated genetic editing to mimic a naturally occurring mutation that effectively disables the CCR5 HIV coreceptor, preventing the retrovirus from entering healthy cells. The mutation was first identified in a man named Timothy Brown who came to be known as “the Berlin patient” after he was apparently cured of HIV infection after a bone marrow transplant from a donor who had the mutation.
Dr. Deng and colleagues took advantage of HSPC transplantation, a standard therapy for ALL to see whether it could also have beneficial effects on concomitant HIV infection.
They treated donor HSPCs with CRISPR-Cas9 to ablate CCR5 and then delivered them to the patient along with additional CD34-depleted donor cells from mobilized peripheral blood.
The transplant was a success, with neutrophil engraftment on day 13 and platelet engraftment on day 27, and the leukemia was in morphologic complete remission at week 4 following transplantation. The patient remained in complete remission from leukemia throughout the 19-month follow-up period, with full donor chimerism .
However, when a planned interruption of antiretroviral therapy was carried out at 7 months post transplant, the serum viral load increased to 3 × 107 copies/ml at week 4 following interruption, and the patient was restarted on the drug. His viral levels gradually decreased to undetectable level during the subsequent months.
The investigators noted that 2 weeks after the drug interruption trial was started there was a small increase in the percentage of CCR5 insertion/deletions.
“The low efficiency of gene editing in the patient may be due to the competitive engraftment of the coinfused HSPCs in CD34-depleted cells and the persistence of donor T cells. To further clarify the anti-HIV effect of CCR5-ablated HSPCs, it will be essential to increase the gene-editing efficiency of our CRISPR-Cas9 system and improve the transplantation protocol,” they wrote.
The study was funded by the Beijing Municipal Science and Technology Commission and others (unspecified). All authors reported having nothing to disclose.
SOURCE: Xu L et al. N Engl J Med. 2019. doi: 10.1056/NEJMoa1817426.
FROM NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point: Donor cells depleted of the HIV coreceptor CCR5 effectively treated ALL, but not HIV.
Major finding: The patient had a sustained complete remission of ALL, but HIV persisted after transplantation.
Study details: Case report of a 27-year-old man with ALL and HIV.
Disclosures: The study was funded by the Beijing Municipal Science and Technology Commission and others (unspecified). All authors reported having nothing to disclose.
Source: Xu L et al. N Engl J Med. 2019. doi: 10.1056/NEJMoa1817426.
Genomic Medicine and Genetic Counseling in the Department of Veterans Affairs and Department of Defense (FULL)
Vickie Venne, MS. What is the Genomic Medicine Service (GMS) at the US Department of Veterans Affairs (VA)?
Renee Rider, JD, MS, LCGC. GMS is a telehealth service. We are part of central office and field stationed at the George E. Wahlen VA Medical Center (VAMC) in Salt Lake City, Utah. We provide care to about 90 VAMCs and their associated clinics. Veterans are referred to us by entering an interfacility consult in the VA Computerized Patient Record System (CPRS). We review the consult to determine whether the patient needs to be seen, whether we can answer with an e-consult, or whether we need more information. For the patients who need an appointment, the telehealth department at the veteran’s VA facility will contact the patient to arrange a visit with us. At the time of the appointment, the facility has a staff member available to seat the patient and connect them to us using video equipment.
We provide genetic care for all specialties, including cancer, women’s health, cardiology and neurology. In today’s discussion, we are focusing on cancer care.
Vickie Venne. What do patients do at facilities that don’t get care through GMS?
Renee Rider. There are a handful of facilities that provide their own genetic care in-house. For example, VA Boston Healthcare System in Massachusetts and the Michael E. DeBakey VAMC in Houston, Texas each have their own programs. For veterans who are not at a VA facility that has an agreement with GMS and do not have a different genetics program, their providers need to make referrals to community care.
Vickie Venne. How do patients get referred and what happens at their facility when the patients return to the specialty and primary care providers (PCP)? Ishta, who do you refer to GMS and how do you define them initially?
Ishta Thakar, MD, FACP. Referrals can come at a couple of points during a veteran’s journey at the VA. The VA covers obstetrics care for women veterans. Whenever a PCP or a women’s health provider is doing the initial history and physical on a new patient, if the female veteran has an extensive family history of breast, ovarian, colon, or endometrial cancer, then we take more history and we send a consult to GMS. The second instance would be if she tells us that she has had a personal history of breast, ovarian, or endometrial cancer and she has never had genetic testing. The third instance would be whenever we have a female veteran who is diagnosed with breast, ovarian, endometrial, or colon cancer. We would definitely talk to her about genetic counseling and send a referral to GMS. We would ask for a GMS consult for a patient with advanced maternal age, with exposure to some kind of teratogens, with an abnormal ultrasound, a family history of chromosomal disorders, or if she’s seeing an obstetrician who wants her to be tested. And finally, if a patient has a constellation of multiple cancers in the family and we don’t know what’s going on, we would also refer the patient to GMS.
Vickie Venne. That would be why GMS fields over 150 referrals every week. It is a large list. We also see veterans with personal or family histories of neurologic or cardiologic concerns as well.
Renee, as somebody who fields many of these referrals from unaffected individuals, what is the family history process?
Renee Rider. We don’t expect the referring provider to be a genetic expert. When a provider is seeing a constellation of several different cancers and he or she doesn’t know if there’s anything going on genetically or even if it’s possible, absolutely they should put in a referral to GMS. We have a triage counselor who reviews every consult that comes into our service within 24 hours.
Many cancers are due to exposures that are not concerning for a genetic etiology. We can let you know that it is not concerning, and the PCP can counsel the patient that it is very unlikely to be genetic in nature. We still give feedback even if it’s not someone who is appropriate for genetic counseling and testing. It is important to reach out to GMS even if you don’t know whether a cancer is genetic in nature.
It also is important to take your time when gathering family histories. We get a lot of patients who say, “There’s a lot of cancer in my family. I have no idea who had cancer, but I know a lot of people had cancer.” That’s not the day to put in a referral to GMS. At that point, providers should tell the patient to get as much information as they can about the family history and then reassess. It’s important for us to have accurate information. We’ve had several times where we receive a referral because the veteran says that their sister had ovarian cancer. And then when our staff calls, they later find out it was cervical cancer. That’s not a good use of the veteran’s time, and it’s not a good use of VA resources.
The other important thing about family histories is keeping the questions open-ended. Often a PCP or specialist will ask about a certain type of cancer: “Does anyone in your family have breast cancer, ovarian cancer?” Or if the veteran
is getting a colonoscopy, they ask, “Does anybody have colon cancer?” Where really, we need to be a little bit more open-ended. We prefer questions like, “Has anyone in your family
had cancer?” because that’s the question that prompts a response of, “Yes, 3 people in my family have had thyroid cancer.” That’s very important for us to know, too.
If you do get a positive response, probe a little bit more: what kind of cancer did someone have, how old were they when they had their cancer? And how are they related? Is this an aunt on your mom’s side or on your dad’s side? Those are the types of information that we need to figure out if that person needs a referral.
Vickie Venne. It’s a different story when people already have a cancer diagnosis. Which hematology or oncology patients are good referrals and why?
Lisa Arfons, MD. When patients come in with newly diagnosed cancer, breast for example, it is an emotional diagnosis and psychologicallydistressing. Oftentimes, they want to know why this happened to them. The issues surrounding
genetic testing also becomes very emotional. They want to know whether their children are at risk as well.
Genetic discussions take a long time. I rarely do that on the first visit. I always record for myself in my clinic note if something strikes me regarding the patient’s diagnosis. I quickly run through the National Comprehensive Cancer Network (NCCN) guidelines to remind myself of what I need to go over with the patient at our next meeting. Most patients don’t need to be referred to GMS, and most patients don’t need to be tested once they’re seen.
I often save the referral discussion for after I have established a rapport with a patient, we have a treatment plan, or they already have had their first surgery. Therefore, we are not making decisions about their first surgery based on the genetic medicine results.
If I’m considering a referral, I do a deeper dive with the patient. Is the patient older or younger than 45 years? I pull up NCCN guidelines and we go through the entire checklist.
We have male breast cancer patients at the VA—probably more than the community—so we refer those patients. At the Louis Stokes Cleveland VAMC in Ohio, we have had some in-depth discussions about referring male breast cancer patients for genetic testing and whether it was beneficial to older patients with male breast cancer. Ultimately, we decided that it was important for our male veterans to be tested because it empowered them to have better understanding of their medical conditions that may not just have effect on them but on their offspring, and that that can be a source of psychological and emotional support.
I don’t refer most people to GMS once I go through the checklist. I appreciate the action for an e-consult within the CPRS telemedicine consult itself, as Renee noted. If it is not necessary, GMS makes it an e-consult. I try to communicate that I don’t know whether it is necessary or not so that GMS understands where I’m coming from.
Vickie Venne. In the US Department of Defense (DoD) the process is quite different. Mauricio, can you explain the clinical referral process, who is referred, and how that works from a laboratory perspective?
Maj De Castro, MD, FACMG, USAF. The VA has led the way in demonstrating how to best provide for the medical genetic needs of a large, decentralized population distributed all over the country. Over the last 5 to 10 years, the DoD has made strides in recognizing the role genetics plays in the practice of everyday medicine and redoubling efforts to meet the needs of servicemembers.
The way that it traditionally has worked in the DoD is that military treatment facilities (MTFs) that have dedicated geneticists and genetic counselors: Kessler Medical Center in Mississippi, Walter Reed National Military Medical
Center in Maryland, Tripler Army Medical Center in Hawaii, Madigan Army Medical Center in Washington, Brooke Army Medical Center in Texas, Naval Medical Center San Diego in California, and Naval Medical Center Portsmouth in Virginia. A patient seeking genetic evaluation, counseling, or testing in those larger facilities would be referred to the genetics service by their primary care manager. Wait times vary, but it would usually be weeks, maybe months. However, the great majority of MTFs do not have dedicated genetics support. Most of the time, those patients would have to be referred to the local civilian community—there was no process for them to be seen in in the military healthcare system—with wait times that exceed 6 to 8 months in some cases. This is due to just not a military but a national shortage of genetics professionals (counselors and physicians).
Last year we started the telegenetics initiative, which is small compared to the VA—it is comprised of 2 geneticists and 1 genetic counselor—but with the full intent of growing it over time. Its purpose is to extend the resources we
had to other MTFs. Genetics professionals stationed state-side can provide care to remote facilities with limited access to local genetics support such as Cannon Air Force Base (AFB) or overseas facilities such as Spangdahlem AFB in Germany.
We recognize there are military-specific needs for the DoD regarding the genetic counseling process that have to take into account readiness, genetic discrimination, continued ability to serve and fitness for duty. For this important reason, we are seeking to expand our telegenetics initiative. The goal is to be able to provide 100% of all genetic counseling in-house, so to speak.
Currently, providers at the 4 pilot sites (Cannon AFB, Fort Bragg, Spangdahlem AFB, and Guantanamo Bay) send us referrals. We triage them and assign the patient to see a geneticist or a counselor depending on the indication.
On the laboratory side, it has been a very interesting experience. Because we provide comprehensive germline cancer testing at very little cost to the provider at any MTF, we have had high numbers of test requests over the years.
In addition to saving the DoD millions of dollars in testing, we have learned some interesting lessons in the process. For instance, we have worked closely with several different groups to better understand how to educate providers on the genetic counseling and testing process. This has allowed us to craft a thorough and inclusive consent form that addresses the needs of the DoD. We have also learned valuable lessons about population-based screening vs evidence-based testing, and lessons surrounding narrow-based testing (BRCA1 and BRCA2 only testing) vs ordering a more comprehensive panel that includes other genes supported by strong evidence (such as PALB2, CHEK2, or TP53).
For example, we have found that in a significant proportion of individuals with and without family history, there are clinically relevant variants in genes other than BRCA1 or BRCA2. And so, we have made part of our consent process,
a statement on secondary findings. If the patient consents, we will report pathogenic variants in other genes known to be associated with cancer (with strong evidence) even if the provider ordered a narrow panel such as BRCA1 and BRCA2 testing only. In about 1% to 4% of patients that would otherwise not meet NCCN guidelines, we’ve reported variants that were clinically actionable and changed the medical management of that patient.
We feel strongly that this is a conversation that we need to have in our field, and we realize it’s a complex issue, maybe we need to expand who gets testing. Guideline based testing is missing some patients out there that could benefit from it.
Vickie Venne. There certainly are many sides to the conversation of population-based vs evidence-based genetic testing. Genetic testing policies are changing rapidly. There are teams exploring comprehensive gene sequencing for
newborns and how that potential 1-time test can provide information will be reinterpreted as a person goes from cradle to grave. However, unlike the current DoD process, in the VA there are patients who we don’t see.
Renee Rider. I want to talk about money. When we order a genetic test, that test is paid for by the pathology department at the patient’s VAMC. Most of the pathology departments we work with are clear that they only can provide
genetic testing that is considered medically necessary. Thus, we review each test to make sure it meets established guidelines for testing. We don’t do population genetic screening as there isn’t evidence or guidelines to support offering it. We are strict about who does and does not get genetic testing, partly because we have a responsibility to pathology departments and to the taxpayers.
GMS focuses on conditions that are inherited, that is to say, we deal with germline genetics. Therefore, we discontinue referrals for somatic requests, such as when an OncotypeDX test is requested. It is my understanding that pharmacogenetic referrals may be sent to the new PHASeR initiative, which is a joint collaboration between the VA and Sanford Health and is headed by Deepak Voora, MD.

We generally don’t see patients who still are having diagnostic procedures done. For example, if a veteran has a suspicious breast mass, we recommend that the provider workup the mass before referring to GMS. Regardless of a genetic test result, a suspicious mass needs to be worked up. And, knowing if the mass is cancerous could change how we would proceed with the genetic workup. For example, if the mass were not cancerous, we may recommend that an affected relative have the first genetic evaluation. Furthermore, knowing if the patient has cancer changes how we interpret negative test results.
Another group of patients we don’t see are those who already had genetic testing done by the referring provider. It’s a VA directive that if you order a test, you’re the person who is responsible for giving the results. We agree with
this directive. If you don’t feel comfortable giving back test results, don’t order the test. Often, when a provider sends a patient to us after the test was done, we discover that the patient didn’t have appropriate pretest counseling. A test result, such as a variant of uncertain significance (VUS), should never be a surprise to either the provider or the patient.
Ishta Thakar. For newly diagnosed cancers, the first call is to the patient to inform them that they have cancer. We usually bring up genetic counseling or testing, if applicable, when they are ready to accept the diagnosis and have a conversation about it. All our consults are via telehealth, so none of our patients physically come to GMS in Salt Lake City. All the consults are done virtually.
For newly diagnosed patients, we would send a consult in within a couple of weeks. For patients who had a family history, the referral would not be urgent: They can be seen within about 3 months. The turnaround times for GMS are so much better than what we have available in the community where it’s often at least 6 months, as previously noted.
Vickie Venne. Thank you. We continue to work on that. One of the interesting things that we’ve done, which is the brainchild of Renee, is shared medical appointments.
Renee Rider. We have now created 4 group appointments for people who have concerns surrounding cancer. One group is for people who don’t have cancer but have family members who have cancer who may be the best testing candidate. For example, that might be a 30-year old who tells you that her mother had breast cancer at age 45 years. Her mother is still living, but she’s never had genetic testing. We would put her in a group where we discuss the importance of talking to the family members and encouraging them to go get that first genetic evaluation in the family.
Our second group is for people who don’t have cancer themselves, but have a family history of cancer and those affected relatives have passed away. The family needs a genetic evaluation, and the veteran is the best living testing candidate.
That group is geared towards education about the test and informed consent.
The third group is for people with cancer who qualify for genetic testing. We provide all of the information that they need to make an informed decision on having (or not having) genetic testing.
The final group is for people who have family histories of known genetic mutations in cancer genes. Again, we provide them with all of the information that they need to make an informed decision regarding genetic testing.
With the shared medical appointments, we have been able to greatly increase the number of patients that we can see. Our first 3 groups all meet once a week and can have 10 or 12 veterans. Our last group meets every other week and has a maximum of 6 veterans. Wait times for our groups are generally ≤ 2 weeks. All veterans can choose to have an individual appointment if they prefer. We regularly get unsolicited feedback from veterans that they learn a lot during our groups and appreciate it.
Our group appointments have lowered the wait time for the people in the groups. And, they’ve lowered the wait time for the people who are seen individually. They’ve allowed us to address the backlog of patients waiting to see us in a more timely manner. Our wait time for individual appointment had been approaching 6 months, and it is now about 1.5 months.
We also think that being in a group normalizes the experience. Most people don’t know anyone who has had genetic testing. Now, they are in a group with others going through the same experience. In one of my groups, a male veteran talked about his breast cancer being really rare. Another male in the group volunteer that he had breast cancer, too. They both seemed to appreciate not feeling alone.
Vickie Venne. I want to move to our final piece. What do the referring providers tell the patients about a genetics referral and what should they expect?
Lisa Arfons. First and foremost, I tell the patient that it is a discussion with a genetic counselor. I make it clear that they understand that it is a discussion. They then can agree or not agree to accept genetic testing if it’s recommended.
I talk in general terms about why I think it can be important for them to have the discussion, but that we don’t have great data for decisionmaking. We understand that there are more options for preventive measures but then it ultimately will be a discussion between the PCP, the patient, and their family members about how they proceed about the preventive measures. I want them to start thinking about how the genetic test results, regardless of if they are positive, negative, or a variant that is not yet understood, can impact their offspring.
Probably I am biased, as my mom had breast cancer and she underwent genetic testing. So, I have a bit of an offspring focus as well. I already mentioned that you must discuss about whether or not it’s worth screening or doing any preventive measures on contralateral breast, or screening for things like prostate cancer at age 75 years. And so I focus more on the family members.
I try to stay in my lane. I am extremely uncomfortable when I hear about someone in our facility sending off a blood test and then asking someone else to interpret the results and discuss it with the patient. Just because it’s a blood test and it’s easy to order doesn’t mean that it is easy to know what to do with it, and it needs to be respected as such.
Ishta Thakar. Our PCPs let the patients know that GMS will contact the patient to schedule a video appointment and that if they want to bring any family members along with them, they’re welcome to. We also explain that certain cancers are genetically based and that if they have a genetic mutation, it can be passed on to their offspring. I also explain that if they have certain mutations, then we would be more vigilant in screening them for other kinds of cancers. That’s the reason that we refer that they get counseled. After counseling if they’re ready for the testing, then the counselor orders the test and does the posttest discussion with the patient.
Vickie Venne. In the VA, people are invited to attend a genetic counseling session but can certainly decline. Does the the DoD have a different approach?
Maj De Castro. I would say that the great majority of active duty patients have limited knowledge of what to expect out of a genetics appointment. One of the main things we do is educate them on their rights and protections and the potential risks associated with performing genetic testing, in particular when it comes to their continued ability to serve. Genetic testing for clinical purposes is not mandatory in the DoD, patients can certainly decline testing. Because genetic testing has the potential to alter someone’s career, it is critical we have a very thorough and comprehensive pre- and posttest counseling sessions that includes everything from career implications to the Genetic Information Nondiscrimination Act (GINA) and genetic discrimination in the military, in addition to the standard of care medical information.
Scenarios in which a servicemember is negatively impacted by pursuing a genetic diagnosis are very rare. More than 90% of the time, genetic counseling and/or testing has no adverse career effect. When they do, it is out of concern for the safety and wellbeing of a servicemember. For instance, if we diagnosis a patient with a genetic form of some arrhythmogenic disorder, part of the treatment plan can be to limit that person’s level of exertion, because it could potentially lead to death. We don’t want to put someone in a situation that may trigger that.
Vickie Venne. We also have a certain number of veterans who ask us about their service disability pay and the impact of genetic testing on it. One example is veterans with prostate cancer who were exposed to Agent Orange, which has been associated with increased risk for developing prostate cancer. I have had men who have been referred for genetic evaluation ask, “Well, if I have an identifiable mutation, how will that impact my service disability?” So we discuss the carcinogenic process that may include an inherited component as well as the environmental risk factors. I think that’s a unique issue for a population we’re honored to be able to serve.
Renee Rider. When we are talking about how the population of veterans is unique, I think it is also important to acknowledge mental health. I’ve had several patients tell me that they have posttraumatic stress disorder or anxiety and the idea of getting an indeterminant test result, such as VUS, would really weigh on them.
In the community, a lot of providers order the biggest panel they can, but for these patients who are worried about getting those indeterminant test results, I’ve been able to work with them to limit the size of the panel. I order a small panel that only has genes that have implications for that veteran’s clinical management. For example, in a patient with ductal breast cancer, I remove the genes that cause lobular breast cancer. This takes a bit of knowledge and critical thinking that our VA genetic counselors have because they have experience with veterans and their needs.
As our time draws to a close, I have one final thought. This has been a heartwarming conversation today. It is really nice to hear that GMS services are appreciated. We in GMS want to partner with our referring providers. Help us help you! When you enter a referral, please let us know how we can help you. The more we understand why you are sending your veteran to GMS, the more we can help meet your needs. If there are any questions or problems, feel free to send us an email or pick up the phone and call us.
Vickie Venne, MS. What is the Genomic Medicine Service (GMS) at the US Department of Veterans Affairs (VA)?
Renee Rider, JD, MS, LCGC. GMS is a telehealth service. We are part of central office and field stationed at the George E. Wahlen VA Medical Center (VAMC) in Salt Lake City, Utah. We provide care to about 90 VAMCs and their associated clinics. Veterans are referred to us by entering an interfacility consult in the VA Computerized Patient Record System (CPRS). We review the consult to determine whether the patient needs to be seen, whether we can answer with an e-consult, or whether we need more information. For the patients who need an appointment, the telehealth department at the veteran’s VA facility will contact the patient to arrange a visit with us. At the time of the appointment, the facility has a staff member available to seat the patient and connect them to us using video equipment.
We provide genetic care for all specialties, including cancer, women’s health, cardiology and neurology. In today’s discussion, we are focusing on cancer care.
Vickie Venne. What do patients do at facilities that don’t get care through GMS?
Renee Rider. There are a handful of facilities that provide their own genetic care in-house. For example, VA Boston Healthcare System in Massachusetts and the Michael E. DeBakey VAMC in Houston, Texas each have their own programs. For veterans who are not at a VA facility that has an agreement with GMS and do not have a different genetics program, their providers need to make referrals to community care.
Vickie Venne. How do patients get referred and what happens at their facility when the patients return to the specialty and primary care providers (PCP)? Ishta, who do you refer to GMS and how do you define them initially?
Ishta Thakar, MD, FACP. Referrals can come at a couple of points during a veteran’s journey at the VA. The VA covers obstetrics care for women veterans. Whenever a PCP or a women’s health provider is doing the initial history and physical on a new patient, if the female veteran has an extensive family history of breast, ovarian, colon, or endometrial cancer, then we take more history and we send a consult to GMS. The second instance would be if she tells us that she has had a personal history of breast, ovarian, or endometrial cancer and she has never had genetic testing. The third instance would be whenever we have a female veteran who is diagnosed with breast, ovarian, endometrial, or colon cancer. We would definitely talk to her about genetic counseling and send a referral to GMS. We would ask for a GMS consult for a patient with advanced maternal age, with exposure to some kind of teratogens, with an abnormal ultrasound, a family history of chromosomal disorders, or if she’s seeing an obstetrician who wants her to be tested. And finally, if a patient has a constellation of multiple cancers in the family and we don’t know what’s going on, we would also refer the patient to GMS.
Vickie Venne. That would be why GMS fields over 150 referrals every week. It is a large list. We also see veterans with personal or family histories of neurologic or cardiologic concerns as well.
Renee, as somebody who fields many of these referrals from unaffected individuals, what is the family history process?
Renee Rider. We don’t expect the referring provider to be a genetic expert. When a provider is seeing a constellation of several different cancers and he or she doesn’t know if there’s anything going on genetically or even if it’s possible, absolutely they should put in a referral to GMS. We have a triage counselor who reviews every consult that comes into our service within 24 hours.
Many cancers are due to exposures that are not concerning for a genetic etiology. We can let you know that it is not concerning, and the PCP can counsel the patient that it is very unlikely to be genetic in nature. We still give feedback even if it’s not someone who is appropriate for genetic counseling and testing. It is important to reach out to GMS even if you don’t know whether a cancer is genetic in nature.
It also is important to take your time when gathering family histories. We get a lot of patients who say, “There’s a lot of cancer in my family. I have no idea who had cancer, but I know a lot of people had cancer.” That’s not the day to put in a referral to GMS. At that point, providers should tell the patient to get as much information as they can about the family history and then reassess. It’s important for us to have accurate information. We’ve had several times where we receive a referral because the veteran says that their sister had ovarian cancer. And then when our staff calls, they later find out it was cervical cancer. That’s not a good use of the veteran’s time, and it’s not a good use of VA resources.
The other important thing about family histories is keeping the questions open-ended. Often a PCP or specialist will ask about a certain type of cancer: “Does anyone in your family have breast cancer, ovarian cancer?” Or if the veteran
is getting a colonoscopy, they ask, “Does anybody have colon cancer?” Where really, we need to be a little bit more open-ended. We prefer questions like, “Has anyone in your family
had cancer?” because that’s the question that prompts a response of, “Yes, 3 people in my family have had thyroid cancer.” That’s very important for us to know, too.
If you do get a positive response, probe a little bit more: what kind of cancer did someone have, how old were they when they had their cancer? And how are they related? Is this an aunt on your mom’s side or on your dad’s side? Those are the types of information that we need to figure out if that person needs a referral.
Vickie Venne. It’s a different story when people already have a cancer diagnosis. Which hematology or oncology patients are good referrals and why?
Lisa Arfons, MD. When patients come in with newly diagnosed cancer, breast for example, it is an emotional diagnosis and psychologicallydistressing. Oftentimes, they want to know why this happened to them. The issues surrounding
genetic testing also becomes very emotional. They want to know whether their children are at risk as well.
Genetic discussions take a long time. I rarely do that on the first visit. I always record for myself in my clinic note if something strikes me regarding the patient’s diagnosis. I quickly run through the National Comprehensive Cancer Network (NCCN) guidelines to remind myself of what I need to go over with the patient at our next meeting. Most patients don’t need to be referred to GMS, and most patients don’t need to be tested once they’re seen.
I often save the referral discussion for after I have established a rapport with a patient, we have a treatment plan, or they already have had their first surgery. Therefore, we are not making decisions about their first surgery based on the genetic medicine results.
If I’m considering a referral, I do a deeper dive with the patient. Is the patient older or younger than 45 years? I pull up NCCN guidelines and we go through the entire checklist.
We have male breast cancer patients at the VA—probably more than the community—so we refer those patients. At the Louis Stokes Cleveland VAMC in Ohio, we have had some in-depth discussions about referring male breast cancer patients for genetic testing and whether it was beneficial to older patients with male breast cancer. Ultimately, we decided that it was important for our male veterans to be tested because it empowered them to have better understanding of their medical conditions that may not just have effect on them but on their offspring, and that that can be a source of psychological and emotional support.
I don’t refer most people to GMS once I go through the checklist. I appreciate the action for an e-consult within the CPRS telemedicine consult itself, as Renee noted. If it is not necessary, GMS makes it an e-consult. I try to communicate that I don’t know whether it is necessary or not so that GMS understands where I’m coming from.
Vickie Venne. In the US Department of Defense (DoD) the process is quite different. Mauricio, can you explain the clinical referral process, who is referred, and how that works from a laboratory perspective?
Maj De Castro, MD, FACMG, USAF. The VA has led the way in demonstrating how to best provide for the medical genetic needs of a large, decentralized population distributed all over the country. Over the last 5 to 10 years, the DoD has made strides in recognizing the role genetics plays in the practice of everyday medicine and redoubling efforts to meet the needs of servicemembers.
The way that it traditionally has worked in the DoD is that military treatment facilities (MTFs) that have dedicated geneticists and genetic counselors: Kessler Medical Center in Mississippi, Walter Reed National Military Medical
Center in Maryland, Tripler Army Medical Center in Hawaii, Madigan Army Medical Center in Washington, Brooke Army Medical Center in Texas, Naval Medical Center San Diego in California, and Naval Medical Center Portsmouth in Virginia. A patient seeking genetic evaluation, counseling, or testing in those larger facilities would be referred to the genetics service by their primary care manager. Wait times vary, but it would usually be weeks, maybe months. However, the great majority of MTFs do not have dedicated genetics support. Most of the time, those patients would have to be referred to the local civilian community—there was no process for them to be seen in in the military healthcare system—with wait times that exceed 6 to 8 months in some cases. This is due to just not a military but a national shortage of genetics professionals (counselors and physicians).
Last year we started the telegenetics initiative, which is small compared to the VA—it is comprised of 2 geneticists and 1 genetic counselor—but with the full intent of growing it over time. Its purpose is to extend the resources we
had to other MTFs. Genetics professionals stationed state-side can provide care to remote facilities with limited access to local genetics support such as Cannon Air Force Base (AFB) or overseas facilities such as Spangdahlem AFB in Germany.
We recognize there are military-specific needs for the DoD regarding the genetic counseling process that have to take into account readiness, genetic discrimination, continued ability to serve and fitness for duty. For this important reason, we are seeking to expand our telegenetics initiative. The goal is to be able to provide 100% of all genetic counseling in-house, so to speak.
Currently, providers at the 4 pilot sites (Cannon AFB, Fort Bragg, Spangdahlem AFB, and Guantanamo Bay) send us referrals. We triage them and assign the patient to see a geneticist or a counselor depending on the indication.
On the laboratory side, it has been a very interesting experience. Because we provide comprehensive germline cancer testing at very little cost to the provider at any MTF, we have had high numbers of test requests over the years.
In addition to saving the DoD millions of dollars in testing, we have learned some interesting lessons in the process. For instance, we have worked closely with several different groups to better understand how to educate providers on the genetic counseling and testing process. This has allowed us to craft a thorough and inclusive consent form that addresses the needs of the DoD. We have also learned valuable lessons about population-based screening vs evidence-based testing, and lessons surrounding narrow-based testing (BRCA1 and BRCA2 only testing) vs ordering a more comprehensive panel that includes other genes supported by strong evidence (such as PALB2, CHEK2, or TP53).
For example, we have found that in a significant proportion of individuals with and without family history, there are clinically relevant variants in genes other than BRCA1 or BRCA2. And so, we have made part of our consent process,
a statement on secondary findings. If the patient consents, we will report pathogenic variants in other genes known to be associated with cancer (with strong evidence) even if the provider ordered a narrow panel such as BRCA1 and BRCA2 testing only. In about 1% to 4% of patients that would otherwise not meet NCCN guidelines, we’ve reported variants that were clinically actionable and changed the medical management of that patient.
We feel strongly that this is a conversation that we need to have in our field, and we realize it’s a complex issue, maybe we need to expand who gets testing. Guideline based testing is missing some patients out there that could benefit from it.
Vickie Venne. There certainly are many sides to the conversation of population-based vs evidence-based genetic testing. Genetic testing policies are changing rapidly. There are teams exploring comprehensive gene sequencing for
newborns and how that potential 1-time test can provide information will be reinterpreted as a person goes from cradle to grave. However, unlike the current DoD process, in the VA there are patients who we don’t see.
Renee Rider. I want to talk about money. When we order a genetic test, that test is paid for by the pathology department at the patient’s VAMC. Most of the pathology departments we work with are clear that they only can provide
genetic testing that is considered medically necessary. Thus, we review each test to make sure it meets established guidelines for testing. We don’t do population genetic screening as there isn’t evidence or guidelines to support offering it. We are strict about who does and does not get genetic testing, partly because we have a responsibility to pathology departments and to the taxpayers.
GMS focuses on conditions that are inherited, that is to say, we deal with germline genetics. Therefore, we discontinue referrals for somatic requests, such as when an OncotypeDX test is requested. It is my understanding that pharmacogenetic referrals may be sent to the new PHASeR initiative, which is a joint collaboration between the VA and Sanford Health and is headed by Deepak Voora, MD.
We generally don’t see patients who still are having diagnostic procedures done. For example, if a veteran has a suspicious breast mass, we recommend that the provider workup the mass before referring to GMS. Regardless of a genetic test result, a suspicious mass needs to be worked up. And, knowing if the mass is cancerous could change how we would proceed with the genetic workup. For example, if the mass were not cancerous, we may recommend that an affected relative have the first genetic evaluation. Furthermore, knowing if the patient has cancer changes how we interpret negative test results.
Another group of patients we don’t see are those who already had genetic testing done by the referring provider. It’s a VA directive that if you order a test, you’re the person who is responsible for giving the results. We agree with
this directive. If you don’t feel comfortable giving back test results, don’t order the test. Often, when a provider sends a patient to us after the test was done, we discover that the patient didn’t have appropriate pretest counseling. A test result, such as a variant of uncertain significance (VUS), should never be a surprise to either the provider or the patient.
Ishta Thakar. For newly diagnosed cancers, the first call is to the patient to inform them that they have cancer. We usually bring up genetic counseling or testing, if applicable, when they are ready to accept the diagnosis and have a conversation about it. All our consults are via telehealth, so none of our patients physically come to GMS in Salt Lake City. All the consults are done virtually.
For newly diagnosed patients, we would send a consult in within a couple of weeks. For patients who had a family history, the referral would not be urgent: They can be seen within about 3 months. The turnaround times for GMS are so much better than what we have available in the community where it’s often at least 6 months, as previously noted.
Vickie Venne. Thank you. We continue to work on that. One of the interesting things that we’ve done, which is the brainchild of Renee, is shared medical appointments.
Renee Rider. We have now created 4 group appointments for people who have concerns surrounding cancer. One group is for people who don’t have cancer but have family members who have cancer who may be the best testing candidate. For example, that might be a 30-year old who tells you that her mother had breast cancer at age 45 years. Her mother is still living, but she’s never had genetic testing. We would put her in a group where we discuss the importance of talking to the family members and encouraging them to go get that first genetic evaluation in the family.
Our second group is for people who don’t have cancer themselves, but have a family history of cancer and those affected relatives have passed away. The family needs a genetic evaluation, and the veteran is the best living testing candidate.
That group is geared towards education about the test and informed consent.
The third group is for people with cancer who qualify for genetic testing. We provide all of the information that they need to make an informed decision on having (or not having) genetic testing.
The final group is for people who have family histories of known genetic mutations in cancer genes. Again, we provide them with all of the information that they need to make an informed decision regarding genetic testing.
With the shared medical appointments, we have been able to greatly increase the number of patients that we can see. Our first 3 groups all meet once a week and can have 10 or 12 veterans. Our last group meets every other week and has a maximum of 6 veterans. Wait times for our groups are generally ≤ 2 weeks. All veterans can choose to have an individual appointment if they prefer. We regularly get unsolicited feedback from veterans that they learn a lot during our groups and appreciate it.
Our group appointments have lowered the wait time for the people in the groups. And, they’ve lowered the wait time for the people who are seen individually. They’ve allowed us to address the backlog of patients waiting to see us in a more timely manner. Our wait time for individual appointment had been approaching 6 months, and it is now about 1.5 months.
We also think that being in a group normalizes the experience. Most people don’t know anyone who has had genetic testing. Now, they are in a group with others going through the same experience. In one of my groups, a male veteran talked about his breast cancer being really rare. Another male in the group volunteer that he had breast cancer, too. They both seemed to appreciate not feeling alone.
Vickie Venne. I want to move to our final piece. What do the referring providers tell the patients about a genetics referral and what should they expect?
Lisa Arfons. First and foremost, I tell the patient that it is a discussion with a genetic counselor. I make it clear that they understand that it is a discussion. They then can agree or not agree to accept genetic testing if it’s recommended.
I talk in general terms about why I think it can be important for them to have the discussion, but that we don’t have great data for decisionmaking. We understand that there are more options for preventive measures but then it ultimately will be a discussion between the PCP, the patient, and their family members about how they proceed about the preventive measures. I want them to start thinking about how the genetic test results, regardless of if they are positive, negative, or a variant that is not yet understood, can impact their offspring.
Probably I am biased, as my mom had breast cancer and she underwent genetic testing. So, I have a bit of an offspring focus as well. I already mentioned that you must discuss about whether or not it’s worth screening or doing any preventive measures on contralateral breast, or screening for things like prostate cancer at age 75 years. And so I focus more on the family members.
I try to stay in my lane. I am extremely uncomfortable when I hear about someone in our facility sending off a blood test and then asking someone else to interpret the results and discuss it with the patient. Just because it’s a blood test and it’s easy to order doesn’t mean that it is easy to know what to do with it, and it needs to be respected as such.
Ishta Thakar. Our PCPs let the patients know that GMS will contact the patient to schedule a video appointment and that if they want to bring any family members along with them, they’re welcome to. We also explain that certain cancers are genetically based and that if they have a genetic mutation, it can be passed on to their offspring. I also explain that if they have certain mutations, then we would be more vigilant in screening them for other kinds of cancers. That’s the reason that we refer that they get counseled. After counseling if they’re ready for the testing, then the counselor orders the test and does the posttest discussion with the patient.
Vickie Venne. In the VA, people are invited to attend a genetic counseling session but can certainly decline. Does the the DoD have a different approach?
Maj De Castro. I would say that the great majority of active duty patients have limited knowledge of what to expect out of a genetics appointment. One of the main things we do is educate them on their rights and protections and the potential risks associated with performing genetic testing, in particular when it comes to their continued ability to serve. Genetic testing for clinical purposes is not mandatory in the DoD, patients can certainly decline testing. Because genetic testing has the potential to alter someone’s career, it is critical we have a very thorough and comprehensive pre- and posttest counseling sessions that includes everything from career implications to the Genetic Information Nondiscrimination Act (GINA) and genetic discrimination in the military, in addition to the standard of care medical information.
Scenarios in which a servicemember is negatively impacted by pursuing a genetic diagnosis are very rare. More than 90% of the time, genetic counseling and/or testing has no adverse career effect. When they do, it is out of concern for the safety and wellbeing of a servicemember. For instance, if we diagnosis a patient with a genetic form of some arrhythmogenic disorder, part of the treatment plan can be to limit that person’s level of exertion, because it could potentially lead to death. We don’t want to put someone in a situation that may trigger that.
Vickie Venne. We also have a certain number of veterans who ask us about their service disability pay and the impact of genetic testing on it. One example is veterans with prostate cancer who were exposed to Agent Orange, which has been associated with increased risk for developing prostate cancer. I have had men who have been referred for genetic evaluation ask, “Well, if I have an identifiable mutation, how will that impact my service disability?” So we discuss the carcinogenic process that may include an inherited component as well as the environmental risk factors. I think that’s a unique issue for a population we’re honored to be able to serve.
Renee Rider. When we are talking about how the population of veterans is unique, I think it is also important to acknowledge mental health. I’ve had several patients tell me that they have posttraumatic stress disorder or anxiety and the idea of getting an indeterminant test result, such as VUS, would really weigh on them.
In the community, a lot of providers order the biggest panel they can, but for these patients who are worried about getting those indeterminant test results, I’ve been able to work with them to limit the size of the panel. I order a small panel that only has genes that have implications for that veteran’s clinical management. For example, in a patient with ductal breast cancer, I remove the genes that cause lobular breast cancer. This takes a bit of knowledge and critical thinking that our VA genetic counselors have because they have experience with veterans and their needs.
As our time draws to a close, I have one final thought. This has been a heartwarming conversation today. It is really nice to hear that GMS services are appreciated. We in GMS want to partner with our referring providers. Help us help you! When you enter a referral, please let us know how we can help you. The more we understand why you are sending your veteran to GMS, the more we can help meet your needs. If there are any questions or problems, feel free to send us an email or pick up the phone and call us.
Vickie Venne, MS. What is the Genomic Medicine Service (GMS) at the US Department of Veterans Affairs (VA)?
Renee Rider, JD, MS, LCGC. GMS is a telehealth service. We are part of central office and field stationed at the George E. Wahlen VA Medical Center (VAMC) in Salt Lake City, Utah. We provide care to about 90 VAMCs and their associated clinics. Veterans are referred to us by entering an interfacility consult in the VA Computerized Patient Record System (CPRS). We review the consult to determine whether the patient needs to be seen, whether we can answer with an e-consult, or whether we need more information. For the patients who need an appointment, the telehealth department at the veteran’s VA facility will contact the patient to arrange a visit with us. At the time of the appointment, the facility has a staff member available to seat the patient and connect them to us using video equipment.
We provide genetic care for all specialties, including cancer, women’s health, cardiology and neurology. In today’s discussion, we are focusing on cancer care.
Vickie Venne. What do patients do at facilities that don’t get care through GMS?
Renee Rider. There are a handful of facilities that provide their own genetic care in-house. For example, VA Boston Healthcare System in Massachusetts and the Michael E. DeBakey VAMC in Houston, Texas each have their own programs. For veterans who are not at a VA facility that has an agreement with GMS and do not have a different genetics program, their providers need to make referrals to community care.
Vickie Venne. How do patients get referred and what happens at their facility when the patients return to the specialty and primary care providers (PCP)? Ishta, who do you refer to GMS and how do you define them initially?
Ishta Thakar, MD, FACP. Referrals can come at a couple of points during a veteran’s journey at the VA. The VA covers obstetrics care for women veterans. Whenever a PCP or a women’s health provider is doing the initial history and physical on a new patient, if the female veteran has an extensive family history of breast, ovarian, colon, or endometrial cancer, then we take more history and we send a consult to GMS. The second instance would be if she tells us that she has had a personal history of breast, ovarian, or endometrial cancer and she has never had genetic testing. The third instance would be whenever we have a female veteran who is diagnosed with breast, ovarian, endometrial, or colon cancer. We would definitely talk to her about genetic counseling and send a referral to GMS. We would ask for a GMS consult for a patient with advanced maternal age, with exposure to some kind of teratogens, with an abnormal ultrasound, a family history of chromosomal disorders, or if she’s seeing an obstetrician who wants her to be tested. And finally, if a patient has a constellation of multiple cancers in the family and we don’t know what’s going on, we would also refer the patient to GMS.
Vickie Venne. That would be why GMS fields over 150 referrals every week. It is a large list. We also see veterans with personal or family histories of neurologic or cardiologic concerns as well.
Renee, as somebody who fields many of these referrals from unaffected individuals, what is the family history process?
Renee Rider. We don’t expect the referring provider to be a genetic expert. When a provider is seeing a constellation of several different cancers and he or she doesn’t know if there’s anything going on genetically or even if it’s possible, absolutely they should put in a referral to GMS. We have a triage counselor who reviews every consult that comes into our service within 24 hours.
Many cancers are due to exposures that are not concerning for a genetic etiology. We can let you know that it is not concerning, and the PCP can counsel the patient that it is very unlikely to be genetic in nature. We still give feedback even if it’s not someone who is appropriate for genetic counseling and testing. It is important to reach out to GMS even if you don’t know whether a cancer is genetic in nature.
It also is important to take your time when gathering family histories. We get a lot of patients who say, “There’s a lot of cancer in my family. I have no idea who had cancer, but I know a lot of people had cancer.” That’s not the day to put in a referral to GMS. At that point, providers should tell the patient to get as much information as they can about the family history and then reassess. It’s important for us to have accurate information. We’ve had several times where we receive a referral because the veteran says that their sister had ovarian cancer. And then when our staff calls, they later find out it was cervical cancer. That’s not a good use of the veteran’s time, and it’s not a good use of VA resources.
The other important thing about family histories is keeping the questions open-ended. Often a PCP or specialist will ask about a certain type of cancer: “Does anyone in your family have breast cancer, ovarian cancer?” Or if the veteran
is getting a colonoscopy, they ask, “Does anybody have colon cancer?” Where really, we need to be a little bit more open-ended. We prefer questions like, “Has anyone in your family
had cancer?” because that’s the question that prompts a response of, “Yes, 3 people in my family have had thyroid cancer.” That’s very important for us to know, too.
If you do get a positive response, probe a little bit more: what kind of cancer did someone have, how old were they when they had their cancer? And how are they related? Is this an aunt on your mom’s side or on your dad’s side? Those are the types of information that we need to figure out if that person needs a referral.
Vickie Venne. It’s a different story when people already have a cancer diagnosis. Which hematology or oncology patients are good referrals and why?
Lisa Arfons, MD. When patients come in with newly diagnosed cancer, breast for example, it is an emotional diagnosis and psychologicallydistressing. Oftentimes, they want to know why this happened to them. The issues surrounding
genetic testing also becomes very emotional. They want to know whether their children are at risk as well.
Genetic discussions take a long time. I rarely do that on the first visit. I always record for myself in my clinic note if something strikes me regarding the patient’s diagnosis. I quickly run through the National Comprehensive Cancer Network (NCCN) guidelines to remind myself of what I need to go over with the patient at our next meeting. Most patients don’t need to be referred to GMS, and most patients don’t need to be tested once they’re seen.
I often save the referral discussion for after I have established a rapport with a patient, we have a treatment plan, or they already have had their first surgery. Therefore, we are not making decisions about their first surgery based on the genetic medicine results.
If I’m considering a referral, I do a deeper dive with the patient. Is the patient older or younger than 45 years? I pull up NCCN guidelines and we go through the entire checklist.
We have male breast cancer patients at the VA—probably more than the community—so we refer those patients. At the Louis Stokes Cleveland VAMC in Ohio, we have had some in-depth discussions about referring male breast cancer patients for genetic testing and whether it was beneficial to older patients with male breast cancer. Ultimately, we decided that it was important for our male veterans to be tested because it empowered them to have better understanding of their medical conditions that may not just have effect on them but on their offspring, and that that can be a source of psychological and emotional support.
I don’t refer most people to GMS once I go through the checklist. I appreciate the action for an e-consult within the CPRS telemedicine consult itself, as Renee noted. If it is not necessary, GMS makes it an e-consult. I try to communicate that I don’t know whether it is necessary or not so that GMS understands where I’m coming from.
Vickie Venne. In the US Department of Defense (DoD) the process is quite different. Mauricio, can you explain the clinical referral process, who is referred, and how that works from a laboratory perspective?
Maj De Castro, MD, FACMG, USAF. The VA has led the way in demonstrating how to best provide for the medical genetic needs of a large, decentralized population distributed all over the country. Over the last 5 to 10 years, the DoD has made strides in recognizing the role genetics plays in the practice of everyday medicine and redoubling efforts to meet the needs of servicemembers.
The way that it traditionally has worked in the DoD is that military treatment facilities (MTFs) that have dedicated geneticists and genetic counselors: Kessler Medical Center in Mississippi, Walter Reed National Military Medical
Center in Maryland, Tripler Army Medical Center in Hawaii, Madigan Army Medical Center in Washington, Brooke Army Medical Center in Texas, Naval Medical Center San Diego in California, and Naval Medical Center Portsmouth in Virginia. A patient seeking genetic evaluation, counseling, or testing in those larger facilities would be referred to the genetics service by their primary care manager. Wait times vary, but it would usually be weeks, maybe months. However, the great majority of MTFs do not have dedicated genetics support. Most of the time, those patients would have to be referred to the local civilian community—there was no process for them to be seen in in the military healthcare system—with wait times that exceed 6 to 8 months in some cases. This is due to just not a military but a national shortage of genetics professionals (counselors and physicians).
Last year we started the telegenetics initiative, which is small compared to the VA—it is comprised of 2 geneticists and 1 genetic counselor—but with the full intent of growing it over time. Its purpose is to extend the resources we
had to other MTFs. Genetics professionals stationed state-side can provide care to remote facilities with limited access to local genetics support such as Cannon Air Force Base (AFB) or overseas facilities such as Spangdahlem AFB in Germany.
We recognize there are military-specific needs for the DoD regarding the genetic counseling process that have to take into account readiness, genetic discrimination, continued ability to serve and fitness for duty. For this important reason, we are seeking to expand our telegenetics initiative. The goal is to be able to provide 100% of all genetic counseling in-house, so to speak.
Currently, providers at the 4 pilot sites (Cannon AFB, Fort Bragg, Spangdahlem AFB, and Guantanamo Bay) send us referrals. We triage them and assign the patient to see a geneticist or a counselor depending on the indication.
On the laboratory side, it has been a very interesting experience. Because we provide comprehensive germline cancer testing at very little cost to the provider at any MTF, we have had high numbers of test requests over the years.
In addition to saving the DoD millions of dollars in testing, we have learned some interesting lessons in the process. For instance, we have worked closely with several different groups to better understand how to educate providers on the genetic counseling and testing process. This has allowed us to craft a thorough and inclusive consent form that addresses the needs of the DoD. We have also learned valuable lessons about population-based screening vs evidence-based testing, and lessons surrounding narrow-based testing (BRCA1 and BRCA2 only testing) vs ordering a more comprehensive panel that includes other genes supported by strong evidence (such as PALB2, CHEK2, or TP53).
For example, we have found that in a significant proportion of individuals with and without family history, there are clinically relevant variants in genes other than BRCA1 or BRCA2. And so, we have made part of our consent process,
a statement on secondary findings. If the patient consents, we will report pathogenic variants in other genes known to be associated with cancer (with strong evidence) even if the provider ordered a narrow panel such as BRCA1 and BRCA2 testing only. In about 1% to 4% of patients that would otherwise not meet NCCN guidelines, we’ve reported variants that were clinically actionable and changed the medical management of that patient.
We feel strongly that this is a conversation that we need to have in our field, and we realize it’s a complex issue, maybe we need to expand who gets testing. Guideline based testing is missing some patients out there that could benefit from it.
Vickie Venne. There certainly are many sides to the conversation of population-based vs evidence-based genetic testing. Genetic testing policies are changing rapidly. There are teams exploring comprehensive gene sequencing for
newborns and how that potential 1-time test can provide information will be reinterpreted as a person goes from cradle to grave. However, unlike the current DoD process, in the VA there are patients who we don’t see.
Renee Rider. I want to talk about money. When we order a genetic test, that test is paid for by the pathology department at the patient’s VAMC. Most of the pathology departments we work with are clear that they only can provide
genetic testing that is considered medically necessary. Thus, we review each test to make sure it meets established guidelines for testing. We don’t do population genetic screening as there isn’t evidence or guidelines to support offering it. We are strict about who does and does not get genetic testing, partly because we have a responsibility to pathology departments and to the taxpayers.
GMS focuses on conditions that are inherited, that is to say, we deal with germline genetics. Therefore, we discontinue referrals for somatic requests, such as when an OncotypeDX test is requested. It is my understanding that pharmacogenetic referrals may be sent to the new PHASeR initiative, which is a joint collaboration between the VA and Sanford Health and is headed by Deepak Voora, MD.
We generally don’t see patients who still are having diagnostic procedures done. For example, if a veteran has a suspicious breast mass, we recommend that the provider workup the mass before referring to GMS. Regardless of a genetic test result, a suspicious mass needs to be worked up. And, knowing if the mass is cancerous could change how we would proceed with the genetic workup. For example, if the mass were not cancerous, we may recommend that an affected relative have the first genetic evaluation. Furthermore, knowing if the patient has cancer changes how we interpret negative test results.
Another group of patients we don’t see are those who already had genetic testing done by the referring provider. It’s a VA directive that if you order a test, you’re the person who is responsible for giving the results. We agree with
this directive. If you don’t feel comfortable giving back test results, don’t order the test. Often, when a provider sends a patient to us after the test was done, we discover that the patient didn’t have appropriate pretest counseling. A test result, such as a variant of uncertain significance (VUS), should never be a surprise to either the provider or the patient.
Ishta Thakar. For newly diagnosed cancers, the first call is to the patient to inform them that they have cancer. We usually bring up genetic counseling or testing, if applicable, when they are ready to accept the diagnosis and have a conversation about it. All our consults are via telehealth, so none of our patients physically come to GMS in Salt Lake City. All the consults are done virtually.
For newly diagnosed patients, we would send a consult in within a couple of weeks. For patients who had a family history, the referral would not be urgent: They can be seen within about 3 months. The turnaround times for GMS are so much better than what we have available in the community where it’s often at least 6 months, as previously noted.
Vickie Venne. Thank you. We continue to work on that. One of the interesting things that we’ve done, which is the brainchild of Renee, is shared medical appointments.
Renee Rider. We have now created 4 group appointments for people who have concerns surrounding cancer. One group is for people who don’t have cancer but have family members who have cancer who may be the best testing candidate. For example, that might be a 30-year old who tells you that her mother had breast cancer at age 45 years. Her mother is still living, but she’s never had genetic testing. We would put her in a group where we discuss the importance of talking to the family members and encouraging them to go get that first genetic evaluation in the family.
Our second group is for people who don’t have cancer themselves, but have a family history of cancer and those affected relatives have passed away. The family needs a genetic evaluation, and the veteran is the best living testing candidate.
That group is geared towards education about the test and informed consent.
The third group is for people with cancer who qualify for genetic testing. We provide all of the information that they need to make an informed decision on having (or not having) genetic testing.
The final group is for people who have family histories of known genetic mutations in cancer genes. Again, we provide them with all of the information that they need to make an informed decision regarding genetic testing.
With the shared medical appointments, we have been able to greatly increase the number of patients that we can see. Our first 3 groups all meet once a week and can have 10 or 12 veterans. Our last group meets every other week and has a maximum of 6 veterans. Wait times for our groups are generally ≤ 2 weeks. All veterans can choose to have an individual appointment if they prefer. We regularly get unsolicited feedback from veterans that they learn a lot during our groups and appreciate it.
Our group appointments have lowered the wait time for the people in the groups. And, they’ve lowered the wait time for the people who are seen individually. They’ve allowed us to address the backlog of patients waiting to see us in a more timely manner. Our wait time for individual appointment had been approaching 6 months, and it is now about 1.5 months.
We also think that being in a group normalizes the experience. Most people don’t know anyone who has had genetic testing. Now, they are in a group with others going through the same experience. In one of my groups, a male veteran talked about his breast cancer being really rare. Another male in the group volunteer that he had breast cancer, too. They both seemed to appreciate not feeling alone.
Vickie Venne. I want to move to our final piece. What do the referring providers tell the patients about a genetics referral and what should they expect?
Lisa Arfons. First and foremost, I tell the patient that it is a discussion with a genetic counselor. I make it clear that they understand that it is a discussion. They then can agree or not agree to accept genetic testing if it’s recommended.
I talk in general terms about why I think it can be important for them to have the discussion, but that we don’t have great data for decisionmaking. We understand that there are more options for preventive measures but then it ultimately will be a discussion between the PCP, the patient, and their family members about how they proceed about the preventive measures. I want them to start thinking about how the genetic test results, regardless of if they are positive, negative, or a variant that is not yet understood, can impact their offspring.
Probably I am biased, as my mom had breast cancer and she underwent genetic testing. So, I have a bit of an offspring focus as well. I already mentioned that you must discuss about whether or not it’s worth screening or doing any preventive measures on contralateral breast, or screening for things like prostate cancer at age 75 years. And so I focus more on the family members.
I try to stay in my lane. I am extremely uncomfortable when I hear about someone in our facility sending off a blood test and then asking someone else to interpret the results and discuss it with the patient. Just because it’s a blood test and it’s easy to order doesn’t mean that it is easy to know what to do with it, and it needs to be respected as such.
Ishta Thakar. Our PCPs let the patients know that GMS will contact the patient to schedule a video appointment and that if they want to bring any family members along with them, they’re welcome to. We also explain that certain cancers are genetically based and that if they have a genetic mutation, it can be passed on to their offspring. I also explain that if they have certain mutations, then we would be more vigilant in screening them for other kinds of cancers. That’s the reason that we refer that they get counseled. After counseling if they’re ready for the testing, then the counselor orders the test and does the posttest discussion with the patient.
Vickie Venne. In the VA, people are invited to attend a genetic counseling session but can certainly decline. Does the the DoD have a different approach?
Maj De Castro. I would say that the great majority of active duty patients have limited knowledge of what to expect out of a genetics appointment. One of the main things we do is educate them on their rights and protections and the potential risks associated with performing genetic testing, in particular when it comes to their continued ability to serve. Genetic testing for clinical purposes is not mandatory in the DoD, patients can certainly decline testing. Because genetic testing has the potential to alter someone’s career, it is critical we have a very thorough and comprehensive pre- and posttest counseling sessions that includes everything from career implications to the Genetic Information Nondiscrimination Act (GINA) and genetic discrimination in the military, in addition to the standard of care medical information.
Scenarios in which a servicemember is negatively impacted by pursuing a genetic diagnosis are very rare. More than 90% of the time, genetic counseling and/or testing has no adverse career effect. When they do, it is out of concern for the safety and wellbeing of a servicemember. For instance, if we diagnosis a patient with a genetic form of some arrhythmogenic disorder, part of the treatment plan can be to limit that person’s level of exertion, because it could potentially lead to death. We don’t want to put someone in a situation that may trigger that.
Vickie Venne. We also have a certain number of veterans who ask us about their service disability pay and the impact of genetic testing on it. One example is veterans with prostate cancer who were exposed to Agent Orange, which has been associated with increased risk for developing prostate cancer. I have had men who have been referred for genetic evaluation ask, “Well, if I have an identifiable mutation, how will that impact my service disability?” So we discuss the carcinogenic process that may include an inherited component as well as the environmental risk factors. I think that’s a unique issue for a population we’re honored to be able to serve.
Renee Rider. When we are talking about how the population of veterans is unique, I think it is also important to acknowledge mental health. I’ve had several patients tell me that they have posttraumatic stress disorder or anxiety and the idea of getting an indeterminant test result, such as VUS, would really weigh on them.
In the community, a lot of providers order the biggest panel they can, but for these patients who are worried about getting those indeterminant test results, I’ve been able to work with them to limit the size of the panel. I order a small panel that only has genes that have implications for that veteran’s clinical management. For example, in a patient with ductal breast cancer, I remove the genes that cause lobular breast cancer. This takes a bit of knowledge and critical thinking that our VA genetic counselors have because they have experience with veterans and their needs.
As our time draws to a close, I have one final thought. This has been a heartwarming conversation today. It is really nice to hear that GMS services are appreciated. We in GMS want to partner with our referring providers. Help us help you! When you enter a referral, please let us know how we can help you. The more we understand why you are sending your veteran to GMS, the more we can help meet your needs. If there are any questions or problems, feel free to send us an email or pick up the phone and call us.
SC daratumumab deemed feasible for every multiple myeloma patient
CHICAGO – Subcutaneous (SC) daratumumab is noninferior to intravenous (IV) daratumumab for patients with relapsed or refractory multiple myeloma (MM), according findings from a phase 3 trial.
In the COLUMBA trial, SC daratumumab proved noninferior to IV daratumumab with regard to overall response rate and maximum trough concentration (Ctrough).
The safety profiles of the two formulations were similar, although patients who received SC daratumumab had a lower rate of infusion-related reactions. SC daratumumab also had a lower treatment burden.
“The COLUMBA study shows that [SC daratumumab] can be used in every myeloma patient [as a] single agent or, maybe in the future, in combination with the different backbones,” said Maria-Victoria Mateos, MD, PhD, of University Hospital of Salamanca (Spain).
Dr. Mateos presented results from the COLUMBA trial at the annual meeting of the American Society of Clinical Oncology.
Dr. Mateos cited a previous phase 1b study that had suggested that SC daratumumab might produce similar results as IV daratumumab (Blood. 2017;130:838) while providing a more convenient delivery method. She pointed out that infusions of IV daratumumab can last hours, while the SC formulation can be delivered in minutes.
The aim of the phase 3 COLUMBA study was to compare the IV and SC formulations head-to-head. The trial enrolled 522 patients with relapsed/refractory multiple myeloma. They were randomized to receive daratumumab SC (n = 263) or IV (n = 259).
The median patient age was 68 years (range, 33-92 years) in the IV arm and 65 years (range, 42-84 years) in the SC arm. Patients had received a median of four prior lines of therapy (range, 1-15 in the IV arm and 2-12 in the SC arm). Most patients were refractory to their last line of therapy – 85% in the IV arm and 80% in the SC arm – and most patients had standard-risk cytogenetics – 83% and 74%, respectively.
Treatment
Patients received SC daratumumab at 1,800 mg and IV daratumumab at 16 mg/kg. Both were given weekly for cycles 1-2, every 2 weeks for cycles 3-6, and every 4 weeks thereafter until disease progression.
The median duration of the first infusion was 421 minutes in the IV arm and 5 minutes in the SC arm. The median duration of the second infusion was 255 minutes and 5 minutes, respectively, and the median duration of subsequent infusions was 205 minutes and 5 minutes, respectively.
At a median follow-up of 7.46 months, 57% of patients in each arm had discontinued the study treatment. The most common reasons for discontinuation were progression – 44% of the IV arm and 43% of the SC arm – and adverse events (AEs) – 8% and 7%, respectively.
Safety
Dr. Mateos said the safety profiles of IV and SC daratumumab were comparable. However, infusion-related reactions were significantly less likely in the SC arm, occurring in 12.7% of those patients and 34.5% of patients in the IV arm (P less than .0001).
Grade 3 or higher treatment-emergent AEs occurred in 49% of patients in the IV arm and 46% of those in the SC arm. Rates of grade 5 AEs were 7% and 5%, respectively. The most common grade 3/4 AEs (in the IV and SC arms, respectively) were anemia (14% and 13%), thrombocytopenia (14% for both), neutropenia (8% and 13%), lymphopenia (6% and 5%), and hypertension (6% and 3%).
Efficacy
One of the study’s primary endpoints was overall response rate, which was 37.1% in the IV arm and 41.1% in the SC arm (relative risk, 1.11; 95% CI, 0.89-1.37; P less than .0001). This met the criteria for noninferiority, and overall response rates were comparable across all patient subgroups, Dr. Mateos noted.
The rates of complete response or stringent complete response were also comparable at 2.7% in the IV arm and 1.9% in the SC arm. Rates of very good partial response were 17.0% and 19.0%, respectively.
The study’s other primary endpoint was maximum Ctrough predose on day 1 of cycle 3. The ratio of maximum Ctrough for daratumumab SC over IV was 107.93% (90% CI, 95.74%-121.67%), which met the noninferiority criterion.
Survival outcomes were also similar between the IV and SC arms. The median progression-free survival was 6.1 months and 5.6 months, respectively (P = .9258). The rate of overall survival at 6 months was 83.0% and 87.5%, respectively (P = .6032).
Considering these results together, Dr. Mateos and colleagues concluded that SC daratumumab is noninferior to IV daratumumab.
“[SC daratumumab] has a reduced treatment burden due to a considerably shorter administration duration, and patients treated with [SC daratumumab] reported higher satisfaction with therapy,” Dr. Mateos said.
The results support the use of flat-dose 1,800-mg SC daratumumab, which is comparable with the IV formulation, she said.
The COLUMBA trial was sponsored by Janssen Research & Development. Dr. Mateos reported relationships with Amgen, Celgene, Janssen-Cilag, and Takeda.
SOURCE: Mateos MV et al. ASCO 2019, Abstract 8005.
CHICAGO – Subcutaneous (SC) daratumumab is noninferior to intravenous (IV) daratumumab for patients with relapsed or refractory multiple myeloma (MM), according findings from a phase 3 trial.
In the COLUMBA trial, SC daratumumab proved noninferior to IV daratumumab with regard to overall response rate and maximum trough concentration (Ctrough).
The safety profiles of the two formulations were similar, although patients who received SC daratumumab had a lower rate of infusion-related reactions. SC daratumumab also had a lower treatment burden.
“The COLUMBA study shows that [SC daratumumab] can be used in every myeloma patient [as a] single agent or, maybe in the future, in combination with the different backbones,” said Maria-Victoria Mateos, MD, PhD, of University Hospital of Salamanca (Spain).
Dr. Mateos presented results from the COLUMBA trial at the annual meeting of the American Society of Clinical Oncology.
Dr. Mateos cited a previous phase 1b study that had suggested that SC daratumumab might produce similar results as IV daratumumab (Blood. 2017;130:838) while providing a more convenient delivery method. She pointed out that infusions of IV daratumumab can last hours, while the SC formulation can be delivered in minutes.
The aim of the phase 3 COLUMBA study was to compare the IV and SC formulations head-to-head. The trial enrolled 522 patients with relapsed/refractory multiple myeloma. They were randomized to receive daratumumab SC (n = 263) or IV (n = 259).
The median patient age was 68 years (range, 33-92 years) in the IV arm and 65 years (range, 42-84 years) in the SC arm. Patients had received a median of four prior lines of therapy (range, 1-15 in the IV arm and 2-12 in the SC arm). Most patients were refractory to their last line of therapy – 85% in the IV arm and 80% in the SC arm – and most patients had standard-risk cytogenetics – 83% and 74%, respectively.
Treatment
Patients received SC daratumumab at 1,800 mg and IV daratumumab at 16 mg/kg. Both were given weekly for cycles 1-2, every 2 weeks for cycles 3-6, and every 4 weeks thereafter until disease progression.
The median duration of the first infusion was 421 minutes in the IV arm and 5 minutes in the SC arm. The median duration of the second infusion was 255 minutes and 5 minutes, respectively, and the median duration of subsequent infusions was 205 minutes and 5 minutes, respectively.
At a median follow-up of 7.46 months, 57% of patients in each arm had discontinued the study treatment. The most common reasons for discontinuation were progression – 44% of the IV arm and 43% of the SC arm – and adverse events (AEs) – 8% and 7%, respectively.
Safety
Dr. Mateos said the safety profiles of IV and SC daratumumab were comparable. However, infusion-related reactions were significantly less likely in the SC arm, occurring in 12.7% of those patients and 34.5% of patients in the IV arm (P less than .0001).
Grade 3 or higher treatment-emergent AEs occurred in 49% of patients in the IV arm and 46% of those in the SC arm. Rates of grade 5 AEs were 7% and 5%, respectively. The most common grade 3/4 AEs (in the IV and SC arms, respectively) were anemia (14% and 13%), thrombocytopenia (14% for both), neutropenia (8% and 13%), lymphopenia (6% and 5%), and hypertension (6% and 3%).
Efficacy
One of the study’s primary endpoints was overall response rate, which was 37.1% in the IV arm and 41.1% in the SC arm (relative risk, 1.11; 95% CI, 0.89-1.37; P less than .0001). This met the criteria for noninferiority, and overall response rates were comparable across all patient subgroups, Dr. Mateos noted.
The rates of complete response or stringent complete response were also comparable at 2.7% in the IV arm and 1.9% in the SC arm. Rates of very good partial response were 17.0% and 19.0%, respectively.
The study’s other primary endpoint was maximum Ctrough predose on day 1 of cycle 3. The ratio of maximum Ctrough for daratumumab SC over IV was 107.93% (90% CI, 95.74%-121.67%), which met the noninferiority criterion.
Survival outcomes were also similar between the IV and SC arms. The median progression-free survival was 6.1 months and 5.6 months, respectively (P = .9258). The rate of overall survival at 6 months was 83.0% and 87.5%, respectively (P = .6032).
Considering these results together, Dr. Mateos and colleagues concluded that SC daratumumab is noninferior to IV daratumumab.
“[SC daratumumab] has a reduced treatment burden due to a considerably shorter administration duration, and patients treated with [SC daratumumab] reported higher satisfaction with therapy,” Dr. Mateos said.
The results support the use of flat-dose 1,800-mg SC daratumumab, which is comparable with the IV formulation, she said.
The COLUMBA trial was sponsored by Janssen Research & Development. Dr. Mateos reported relationships with Amgen, Celgene, Janssen-Cilag, and Takeda.
SOURCE: Mateos MV et al. ASCO 2019, Abstract 8005.
CHICAGO – Subcutaneous (SC) daratumumab is noninferior to intravenous (IV) daratumumab for patients with relapsed or refractory multiple myeloma (MM), according findings from a phase 3 trial.
In the COLUMBA trial, SC daratumumab proved noninferior to IV daratumumab with regard to overall response rate and maximum trough concentration (Ctrough).
The safety profiles of the two formulations were similar, although patients who received SC daratumumab had a lower rate of infusion-related reactions. SC daratumumab also had a lower treatment burden.
“The COLUMBA study shows that [SC daratumumab] can be used in every myeloma patient [as a] single agent or, maybe in the future, in combination with the different backbones,” said Maria-Victoria Mateos, MD, PhD, of University Hospital of Salamanca (Spain).
Dr. Mateos presented results from the COLUMBA trial at the annual meeting of the American Society of Clinical Oncology.
Dr. Mateos cited a previous phase 1b study that had suggested that SC daratumumab might produce similar results as IV daratumumab (Blood. 2017;130:838) while providing a more convenient delivery method. She pointed out that infusions of IV daratumumab can last hours, while the SC formulation can be delivered in minutes.
The aim of the phase 3 COLUMBA study was to compare the IV and SC formulations head-to-head. The trial enrolled 522 patients with relapsed/refractory multiple myeloma. They were randomized to receive daratumumab SC (n = 263) or IV (n = 259).
The median patient age was 68 years (range, 33-92 years) in the IV arm and 65 years (range, 42-84 years) in the SC arm. Patients had received a median of four prior lines of therapy (range, 1-15 in the IV arm and 2-12 in the SC arm). Most patients were refractory to their last line of therapy – 85% in the IV arm and 80% in the SC arm – and most patients had standard-risk cytogenetics – 83% and 74%, respectively.
Treatment
Patients received SC daratumumab at 1,800 mg and IV daratumumab at 16 mg/kg. Both were given weekly for cycles 1-2, every 2 weeks for cycles 3-6, and every 4 weeks thereafter until disease progression.
The median duration of the first infusion was 421 minutes in the IV arm and 5 minutes in the SC arm. The median duration of the second infusion was 255 minutes and 5 minutes, respectively, and the median duration of subsequent infusions was 205 minutes and 5 minutes, respectively.
At a median follow-up of 7.46 months, 57% of patients in each arm had discontinued the study treatment. The most common reasons for discontinuation were progression – 44% of the IV arm and 43% of the SC arm – and adverse events (AEs) – 8% and 7%, respectively.
Safety
Dr. Mateos said the safety profiles of IV and SC daratumumab were comparable. However, infusion-related reactions were significantly less likely in the SC arm, occurring in 12.7% of those patients and 34.5% of patients in the IV arm (P less than .0001).
Grade 3 or higher treatment-emergent AEs occurred in 49% of patients in the IV arm and 46% of those in the SC arm. Rates of grade 5 AEs were 7% and 5%, respectively. The most common grade 3/4 AEs (in the IV and SC arms, respectively) were anemia (14% and 13%), thrombocytopenia (14% for both), neutropenia (8% and 13%), lymphopenia (6% and 5%), and hypertension (6% and 3%).
Efficacy
One of the study’s primary endpoints was overall response rate, which was 37.1% in the IV arm and 41.1% in the SC arm (relative risk, 1.11; 95% CI, 0.89-1.37; P less than .0001). This met the criteria for noninferiority, and overall response rates were comparable across all patient subgroups, Dr. Mateos noted.
The rates of complete response or stringent complete response were also comparable at 2.7% in the IV arm and 1.9% in the SC arm. Rates of very good partial response were 17.0% and 19.0%, respectively.
The study’s other primary endpoint was maximum Ctrough predose on day 1 of cycle 3. The ratio of maximum Ctrough for daratumumab SC over IV was 107.93% (90% CI, 95.74%-121.67%), which met the noninferiority criterion.
Survival outcomes were also similar between the IV and SC arms. The median progression-free survival was 6.1 months and 5.6 months, respectively (P = .9258). The rate of overall survival at 6 months was 83.0% and 87.5%, respectively (P = .6032).
Considering these results together, Dr. Mateos and colleagues concluded that SC daratumumab is noninferior to IV daratumumab.
“[SC daratumumab] has a reduced treatment burden due to a considerably shorter administration duration, and patients treated with [SC daratumumab] reported higher satisfaction with therapy,” Dr. Mateos said.
The results support the use of flat-dose 1,800-mg SC daratumumab, which is comparable with the IV formulation, she said.
The COLUMBA trial was sponsored by Janssen Research & Development. Dr. Mateos reported relationships with Amgen, Celgene, Janssen-Cilag, and Takeda.
SOURCE: Mateos MV et al. ASCO 2019, Abstract 8005.
REPORTING FROM ASCO 2019

Another study supports safety of 2-cm margins for thick melanomas
based on data from a randomized, multicenter trial of 936 patients.
“Over time, and in light of the findings of several randomized studies, less extensive surgery for primary melanoma with tumor thickness greater than 2 mm has become more established,” and most recent guidelines recommend a 2-cm margin for these tumors, wrote Deborah Utjés, MD, of the Karolinska Institute in Stockholm and colleagues.
To reinforce the safety and effectiveness of the 2-cm margin, the researchers conducted an open-label, randomized trial of clinically staged melanoma patients aged 75 years and younger with localized cutaneous melanomas thicker than 2 mm, from January 1992 to May 2004. Patients were treated in Denmark, Estonia, Norway, and Sweden. The findings were published in the Lancet.
Patients were randomized to treatment with a 2-cm (471) or 4-cm excision margin (465). The melanomas were located on the trunk, upper extremities, or lower extremities.
The primary outcome of overall survival was similar between the groups. Over a median 20-year follow-up period, the death rate was approximately 50% in each group (49% in the 2-cm group and 51% in the 4-cm group). Disease-specific survival rates were similar as well. Of the 621 reported deaths, 397 were attributed to melanoma: 192 (48%) in the 2-cm group and 205 (52%) in the 4-cm group.
The study findings were limited by several factors, including a lower-than-expected number of patients, lack of nodal staging during the study period, and a focus only on the surgical margin without recording data on pathological excision margins.
However, the extended follow-up supports the safe use of the 2-cm margin for the treatment of melanomas thicker than 2 mm, the investigators wrote. In addition, results from an ongoing trial comparing 1-cm and 2-cm margins for melanomas at least 1 mm thick may yield more evidence to support still narrower surgical margins for some cutaneous melanomas.
The study notes that guidelines from organizations that include the American National Comprehensive Cancer Network and the American Academy of Dermatology recommend the 2-cm margin for tumors that are thicker than 2 mm.
The study was supported by the Swedish Cancer Society, Stockholm Cancer Society, Swedish Society for Medical Research, and the Stockholm County Council, and by funds from Radiumhemmet Research and Wallström. The authors reported no disclosures.
SOURCE: Utjés D et al. Lancet. 2019 Jul 4. doi: 10.1016/S0140-6736(19)31132-8.
based on data from a randomized, multicenter trial of 936 patients.
“Over time, and in light of the findings of several randomized studies, less extensive surgery for primary melanoma with tumor thickness greater than 2 mm has become more established,” and most recent guidelines recommend a 2-cm margin for these tumors, wrote Deborah Utjés, MD, of the Karolinska Institute in Stockholm and colleagues.
To reinforce the safety and effectiveness of the 2-cm margin, the researchers conducted an open-label, randomized trial of clinically staged melanoma patients aged 75 years and younger with localized cutaneous melanomas thicker than 2 mm, from January 1992 to May 2004. Patients were treated in Denmark, Estonia, Norway, and Sweden. The findings were published in the Lancet.
Patients were randomized to treatment with a 2-cm (471) or 4-cm excision margin (465). The melanomas were located on the trunk, upper extremities, or lower extremities.
The primary outcome of overall survival was similar between the groups. Over a median 20-year follow-up period, the death rate was approximately 50% in each group (49% in the 2-cm group and 51% in the 4-cm group). Disease-specific survival rates were similar as well. Of the 621 reported deaths, 397 were attributed to melanoma: 192 (48%) in the 2-cm group and 205 (52%) in the 4-cm group.
The study findings were limited by several factors, including a lower-than-expected number of patients, lack of nodal staging during the study period, and a focus only on the surgical margin without recording data on pathological excision margins.
However, the extended follow-up supports the safe use of the 2-cm margin for the treatment of melanomas thicker than 2 mm, the investigators wrote. In addition, results from an ongoing trial comparing 1-cm and 2-cm margins for melanomas at least 1 mm thick may yield more evidence to support still narrower surgical margins for some cutaneous melanomas.
The study notes that guidelines from organizations that include the American National Comprehensive Cancer Network and the American Academy of Dermatology recommend the 2-cm margin for tumors that are thicker than 2 mm.
The study was supported by the Swedish Cancer Society, Stockholm Cancer Society, Swedish Society for Medical Research, and the Stockholm County Council, and by funds from Radiumhemmet Research and Wallström. The authors reported no disclosures.
SOURCE: Utjés D et al. Lancet. 2019 Jul 4. doi: 10.1016/S0140-6736(19)31132-8.
based on data from a randomized, multicenter trial of 936 patients.
“Over time, and in light of the findings of several randomized studies, less extensive surgery for primary melanoma with tumor thickness greater than 2 mm has become more established,” and most recent guidelines recommend a 2-cm margin for these tumors, wrote Deborah Utjés, MD, of the Karolinska Institute in Stockholm and colleagues.
To reinforce the safety and effectiveness of the 2-cm margin, the researchers conducted an open-label, randomized trial of clinically staged melanoma patients aged 75 years and younger with localized cutaneous melanomas thicker than 2 mm, from January 1992 to May 2004. Patients were treated in Denmark, Estonia, Norway, and Sweden. The findings were published in the Lancet.
Patients were randomized to treatment with a 2-cm (471) or 4-cm excision margin (465). The melanomas were located on the trunk, upper extremities, or lower extremities.
The primary outcome of overall survival was similar between the groups. Over a median 20-year follow-up period, the death rate was approximately 50% in each group (49% in the 2-cm group and 51% in the 4-cm group). Disease-specific survival rates were similar as well. Of the 621 reported deaths, 397 were attributed to melanoma: 192 (48%) in the 2-cm group and 205 (52%) in the 4-cm group.
The study findings were limited by several factors, including a lower-than-expected number of patients, lack of nodal staging during the study period, and a focus only on the surgical margin without recording data on pathological excision margins.
However, the extended follow-up supports the safe use of the 2-cm margin for the treatment of melanomas thicker than 2 mm, the investigators wrote. In addition, results from an ongoing trial comparing 1-cm and 2-cm margins for melanomas at least 1 mm thick may yield more evidence to support still narrower surgical margins for some cutaneous melanomas.
The study notes that guidelines from organizations that include the American National Comprehensive Cancer Network and the American Academy of Dermatology recommend the 2-cm margin for tumors that are thicker than 2 mm.
The study was supported by the Swedish Cancer Society, Stockholm Cancer Society, Swedish Society for Medical Research, and the Stockholm County Council, and by funds from Radiumhemmet Research and Wallström. The authors reported no disclosures.
SOURCE: Utjés D et al. Lancet. 2019 Jul 4. doi: 10.1016/S0140-6736(19)31132-8.
FROM THE LANCET
Ibrutinib tops chlorambucil against CLL
AMSTERDAM – After 5 years, a large majority of patients with chronic lymphocytic leukemia treated with front-line ibrutinib (Imbruvica) have not experienced disease progression, and the median progression-free survival has still not been reached, long-term follow-up from the RESONATE-2 shows.

The 5-year estimated progression-free survival (PFS) rates were 70% for patients who had been randomized to receive ibrutinib monotherapy, compared with 12% for patients randomized to chlorambucil, reported Alessandra Tedeschi, MD, from Azienda Ospedaliera Niguarda Ca’ Granda in Milan.
Ibrutinib was also associated with a halving of risk for death, compared with chlorambucil, she said at the annual congress of the European Hematology Association.
“Importantly, the rate of progression during ibrutinib treatment was very low; only 8 – that is, 6% of patients” – experienced disease progression while receiving ibrutinib, she noted.
In the RESONATE-2 (PCYC-1115) trial, investigators enrolled 269 adults aged 65 years and older with previously untreated CLL/small lymphocytic lymphoma (SLL). Patients at the younger end of the age range (65-69 years) had to have comorbidities that would have made them ineligible for the FCR chemotherapy regimen (fludarabine, cyclophosphamide, and rituximab). Additionally, patients with the deleterious 17p deletion were excluded.
Patients were stratified by performance status and Rai stage and then randomized to receive either ibrutinib 420 mg once daily until disease progression or unacceptable toxicity (136 patients) or chlorambucil 0.5 mg/kg to a maximum of 0.8 mg/kg for up to 12 cycles (133 patients). The trial also had an extension study for patients who had disease progression as confirmed by an independent review committee or who had completed the RESONATE-2 trial. Of the 133 patients in the chlorambucil arm, 76 (57% of the intention-to-treat population) were crossed over to ibrutinib following disease progression.
The median duration of ibrutinib treatment was 57.1 months, with 73% of patients being on it for more than 3 years, 65% for more than 4 years, and 27% for more than 5 years. As of the data cutoff, 79 patients (58%) were continuing with ibrutinib on study.
At 5 years, 70% of ibrutinib-treated patients and 12% of chlorambucil-treated patients were estimated to be progression-free and alive (hazard ratio for PFS with ibrutinib 0.146 (95% confidence interval, 0.10-0.22). The benefit of ibrutinib was consistent for patients with high-risk genomic features, including the 11q deletion and unmutated immunoglobulin heavy-chain variable genes.
Estimated 5-year overall survival was also better with ibrutinib, at 83% vs. 68% (hazard ratio, 0.45; 95% CI, 0.266-0.761).
The most common grade 3 or greater adverse events occurring with ibrutinib were neutropenia (13%), pneumonia (12%), hypertension (8%), anemia (7%), hyponatremia (6%), atrial fibrillation (5%), and cataract (5%). The rates of most adverse events decreased over time, and dose reductions because of adverse events also diminished over time, from 5% of patients in the first year down to zero in years 4 through 5.
Patients responded to subsequent CLL therapies following ibrutinib discontinuation, including chemoimmunotherapy and other kinase inhibitors, Dr. Tedeschi said.
The trial was sponsored by Pharmacyclics with collaboration from Janssen Research & Development. Dr. Tedeschi reported advisory board activities with Janssen, AbbVie, and BeiGene.
SOURCE: Tedeschi A et al. EHA Congress, Abstract S107.
AMSTERDAM – After 5 years, a large majority of patients with chronic lymphocytic leukemia treated with front-line ibrutinib (Imbruvica) have not experienced disease progression, and the median progression-free survival has still not been reached, long-term follow-up from the RESONATE-2 shows.
The 5-year estimated progression-free survival (PFS) rates were 70% for patients who had been randomized to receive ibrutinib monotherapy, compared with 12% for patients randomized to chlorambucil, reported Alessandra Tedeschi, MD, from Azienda Ospedaliera Niguarda Ca’ Granda in Milan.
Ibrutinib was also associated with a halving of risk for death, compared with chlorambucil, she said at the annual congress of the European Hematology Association.
“Importantly, the rate of progression during ibrutinib treatment was very low; only 8 – that is, 6% of patients” – experienced disease progression while receiving ibrutinib, she noted.
In the RESONATE-2 (PCYC-1115) trial, investigators enrolled 269 adults aged 65 years and older with previously untreated CLL/small lymphocytic lymphoma (SLL). Patients at the younger end of the age range (65-69 years) had to have comorbidities that would have made them ineligible for the FCR chemotherapy regimen (fludarabine, cyclophosphamide, and rituximab). Additionally, patients with the deleterious 17p deletion were excluded.
Patients were stratified by performance status and Rai stage and then randomized to receive either ibrutinib 420 mg once daily until disease progression or unacceptable toxicity (136 patients) or chlorambucil 0.5 mg/kg to a maximum of 0.8 mg/kg for up to 12 cycles (133 patients). The trial also had an extension study for patients who had disease progression as confirmed by an independent review committee or who had completed the RESONATE-2 trial. Of the 133 patients in the chlorambucil arm, 76 (57% of the intention-to-treat population) were crossed over to ibrutinib following disease progression.
The median duration of ibrutinib treatment was 57.1 months, with 73% of patients being on it for more than 3 years, 65% for more than 4 years, and 27% for more than 5 years. As of the data cutoff, 79 patients (58%) were continuing with ibrutinib on study.
At 5 years, 70% of ibrutinib-treated patients and 12% of chlorambucil-treated patients were estimated to be progression-free and alive (hazard ratio for PFS with ibrutinib 0.146 (95% confidence interval, 0.10-0.22). The benefit of ibrutinib was consistent for patients with high-risk genomic features, including the 11q deletion and unmutated immunoglobulin heavy-chain variable genes.
Estimated 5-year overall survival was also better with ibrutinib, at 83% vs. 68% (hazard ratio, 0.45; 95% CI, 0.266-0.761).
The most common grade 3 or greater adverse events occurring with ibrutinib were neutropenia (13%), pneumonia (12%), hypertension (8%), anemia (7%), hyponatremia (6%), atrial fibrillation (5%), and cataract (5%). The rates of most adverse events decreased over time, and dose reductions because of adverse events also diminished over time, from 5% of patients in the first year down to zero in years 4 through 5.
Patients responded to subsequent CLL therapies following ibrutinib discontinuation, including chemoimmunotherapy and other kinase inhibitors, Dr. Tedeschi said.
The trial was sponsored by Pharmacyclics with collaboration from Janssen Research & Development. Dr. Tedeschi reported advisory board activities with Janssen, AbbVie, and BeiGene.
SOURCE: Tedeschi A et al. EHA Congress, Abstract S107.
AMSTERDAM – After 5 years, a large majority of patients with chronic lymphocytic leukemia treated with front-line ibrutinib (Imbruvica) have not experienced disease progression, and the median progression-free survival has still not been reached, long-term follow-up from the RESONATE-2 shows.
The 5-year estimated progression-free survival (PFS) rates were 70% for patients who had been randomized to receive ibrutinib monotherapy, compared with 12% for patients randomized to chlorambucil, reported Alessandra Tedeschi, MD, from Azienda Ospedaliera Niguarda Ca’ Granda in Milan.
Ibrutinib was also associated with a halving of risk for death, compared with chlorambucil, she said at the annual congress of the European Hematology Association.
“Importantly, the rate of progression during ibrutinib treatment was very low; only 8 – that is, 6% of patients” – experienced disease progression while receiving ibrutinib, she noted.
In the RESONATE-2 (PCYC-1115) trial, investigators enrolled 269 adults aged 65 years and older with previously untreated CLL/small lymphocytic lymphoma (SLL). Patients at the younger end of the age range (65-69 years) had to have comorbidities that would have made them ineligible for the FCR chemotherapy regimen (fludarabine, cyclophosphamide, and rituximab). Additionally, patients with the deleterious 17p deletion were excluded.
Patients were stratified by performance status and Rai stage and then randomized to receive either ibrutinib 420 mg once daily until disease progression or unacceptable toxicity (136 patients) or chlorambucil 0.5 mg/kg to a maximum of 0.8 mg/kg for up to 12 cycles (133 patients). The trial also had an extension study for patients who had disease progression as confirmed by an independent review committee or who had completed the RESONATE-2 trial. Of the 133 patients in the chlorambucil arm, 76 (57% of the intention-to-treat population) were crossed over to ibrutinib following disease progression.
The median duration of ibrutinib treatment was 57.1 months, with 73% of patients being on it for more than 3 years, 65% for more than 4 years, and 27% for more than 5 years. As of the data cutoff, 79 patients (58%) were continuing with ibrutinib on study.
At 5 years, 70% of ibrutinib-treated patients and 12% of chlorambucil-treated patients were estimated to be progression-free and alive (hazard ratio for PFS with ibrutinib 0.146 (95% confidence interval, 0.10-0.22). The benefit of ibrutinib was consistent for patients with high-risk genomic features, including the 11q deletion and unmutated immunoglobulin heavy-chain variable genes.
Estimated 5-year overall survival was also better with ibrutinib, at 83% vs. 68% (hazard ratio, 0.45; 95% CI, 0.266-0.761).
The most common grade 3 or greater adverse events occurring with ibrutinib were neutropenia (13%), pneumonia (12%), hypertension (8%), anemia (7%), hyponatremia (6%), atrial fibrillation (5%), and cataract (5%). The rates of most adverse events decreased over time, and dose reductions because of adverse events also diminished over time, from 5% of patients in the first year down to zero in years 4 through 5.
Patients responded to subsequent CLL therapies following ibrutinib discontinuation, including chemoimmunotherapy and other kinase inhibitors, Dr. Tedeschi said.
The trial was sponsored by Pharmacyclics with collaboration from Janssen Research & Development. Dr. Tedeschi reported advisory board activities with Janssen, AbbVie, and BeiGene.
SOURCE: Tedeschi A et al. EHA Congress, Abstract S107.
REPORTING FROM EHA CONGRESS
Recombinant vaccine cut herpes zoster rate in immunocompromised patients
Two doses of recombinant zoster vaccine significantly reduced incidence of herpes zoster in adults who had undergone autologous hematopoietic stem cell transplantation (HSCT), results of a randomized, placebo-controlled trial indicate.
The incidence of herpes zoster was 30 per 1,000 person-years for patients who received the adjuvanted recombinant zoster vaccine (Shingrix) versus 94 per 1,000 person-years for those who received placebo, according to study results.
Recombinant zoster vaccine induced humoral and cellular responses that were strong and occurring at a rate higher than what was seen in the placebo group, said senior author Keith M. Sullivan, MD, of Duke University Medical Center, Durham, N.C., and coauthors, who reported findings on behalf of the Zoster Efficacy Study in Patients Undergoing HSCT (ZOE-HSCT) Study Group.
“The vaccinations were generally well tolerated, and most symptoms were mild and transient and did not substantially deter participants from receiving their second dose,” Dr. Sullivan and colleagues wrote in JAMA.
The risk of herpes zoster is increased for 2-3 years after autologous HSCT because of diminished T-cell immunity, according to the authors.
“Antiviral prophylaxis is commonly administered to patients after HSCT to prevent such complications, but the efficacy depends on adherence to treatment,” they said.
While vaccines could provide long-term protection, immunocompromised individuals receiving live attenuated vaccine would be at increased risk of varicella caused by spread of the vaccine strain, they added.
There have been a few encouraging recent studies of non-live vaccines in this setting, including one large phase 3 trial of a heat-inactivated varicella-zoster virus vaccine that showed patients undergoing autologous HSCT had a 63.8% estimated efficacy in preventing herpes zoster, investigators from that study said in The Lancet (2018 May 26;391[10135]:2116-27).
A phase 1/2a study of the adjuvanted recombinant zoster vaccine in patients undergoing HSCT demonstrated strong humoral and cell-mediated immunity responses, which provided the rationale for studying the vaccine further in the randomized ZOE-HSCT study, according to Dr. Sullivan and coauthors.
Their study included a total of 1,846 adults who had undergone autologous HSCT. They were randomized to receive two doses of the recombinant zoster vaccine, the first at 50-70 days after the procedure and the second 1-2 months later.
Herpes zoster cases were seen in 49 and 136 individuals in the vaccine and placebo groups, respectively, which resulted in overall incidences of 30 and 94 per 1,000 person-years.
The incidence rate ratio of a first episode of herpes zoster was 0.36 for individuals receiving at least one dose, which authors said was equivalent to a vaccine efficacy of 63.7%.
That efficacy rate is “very similar” to the estimated efficacy reported for the heat-inactivated varicella-zoster virus vaccine reported in The Lancet, said Dr. Sullivan and coauthors.
However, the heat-inactivated vaccine achieved that level of protection with a four-dose schedule, including one dose given prior to autologous HSCT.
“An advantage of the short 2-dose posttransplantation schedule is that more patients might complete the vaccination program,” they said in a discussion of the results, noting that 94.7% of the recombinant zoster vaccine recipients completed two doses, compared with 81.9% of recipients who received the heat-inactivated herpes zoster vaccine in the previous report.
The study was funded and sponsored by GlaxoSmithKline Biologicals SA. Dr. Sullivan reported disclosures related to GlaxoSmithKline (GSK), Kiadis Pharmaceutical, Roche Genentech, and the National Institute of Allergy and Infectious Diseases. Coauthors provided disclosures related to GSK, AbbVie, Roche, Gilead, Janssen, Pharmacyclics, Morphosys, Helsinn, Celgene, and others.
SOURCE: Bastidas A et al. JAMA. 2019 July 9. doi: 10.1001/jama.2019.9053.
Two doses of recombinant zoster vaccine significantly reduced incidence of herpes zoster in adults who had undergone autologous hematopoietic stem cell transplantation (HSCT), results of a randomized, placebo-controlled trial indicate.
The incidence of herpes zoster was 30 per 1,000 person-years for patients who received the adjuvanted recombinant zoster vaccine (Shingrix) versus 94 per 1,000 person-years for those who received placebo, according to study results.
Recombinant zoster vaccine induced humoral and cellular responses that were strong and occurring at a rate higher than what was seen in the placebo group, said senior author Keith M. Sullivan, MD, of Duke University Medical Center, Durham, N.C., and coauthors, who reported findings on behalf of the Zoster Efficacy Study in Patients Undergoing HSCT (ZOE-HSCT) Study Group.
“The vaccinations were generally well tolerated, and most symptoms were mild and transient and did not substantially deter participants from receiving their second dose,” Dr. Sullivan and colleagues wrote in JAMA.
The risk of herpes zoster is increased for 2-3 years after autologous HSCT because of diminished T-cell immunity, according to the authors.
“Antiviral prophylaxis is commonly administered to patients after HSCT to prevent such complications, but the efficacy depends on adherence to treatment,” they said.
While vaccines could provide long-term protection, immunocompromised individuals receiving live attenuated vaccine would be at increased risk of varicella caused by spread of the vaccine strain, they added.
There have been a few encouraging recent studies of non-live vaccines in this setting, including one large phase 3 trial of a heat-inactivated varicella-zoster virus vaccine that showed patients undergoing autologous HSCT had a 63.8% estimated efficacy in preventing herpes zoster, investigators from that study said in The Lancet (2018 May 26;391[10135]:2116-27).
A phase 1/2a study of the adjuvanted recombinant zoster vaccine in patients undergoing HSCT demonstrated strong humoral and cell-mediated immunity responses, which provided the rationale for studying the vaccine further in the randomized ZOE-HSCT study, according to Dr. Sullivan and coauthors.
Their study included a total of 1,846 adults who had undergone autologous HSCT. They were randomized to receive two doses of the recombinant zoster vaccine, the first at 50-70 days after the procedure and the second 1-2 months later.
Herpes zoster cases were seen in 49 and 136 individuals in the vaccine and placebo groups, respectively, which resulted in overall incidences of 30 and 94 per 1,000 person-years.
The incidence rate ratio of a first episode of herpes zoster was 0.36 for individuals receiving at least one dose, which authors said was equivalent to a vaccine efficacy of 63.7%.
That efficacy rate is “very similar” to the estimated efficacy reported for the heat-inactivated varicella-zoster virus vaccine reported in The Lancet, said Dr. Sullivan and coauthors.
However, the heat-inactivated vaccine achieved that level of protection with a four-dose schedule, including one dose given prior to autologous HSCT.
“An advantage of the short 2-dose posttransplantation schedule is that more patients might complete the vaccination program,” they said in a discussion of the results, noting that 94.7% of the recombinant zoster vaccine recipients completed two doses, compared with 81.9% of recipients who received the heat-inactivated herpes zoster vaccine in the previous report.
The study was funded and sponsored by GlaxoSmithKline Biologicals SA. Dr. Sullivan reported disclosures related to GlaxoSmithKline (GSK), Kiadis Pharmaceutical, Roche Genentech, and the National Institute of Allergy and Infectious Diseases. Coauthors provided disclosures related to GSK, AbbVie, Roche, Gilead, Janssen, Pharmacyclics, Morphosys, Helsinn, Celgene, and others.
SOURCE: Bastidas A et al. JAMA. 2019 July 9. doi: 10.1001/jama.2019.9053.
Two doses of recombinant zoster vaccine significantly reduced incidence of herpes zoster in adults who had undergone autologous hematopoietic stem cell transplantation (HSCT), results of a randomized, placebo-controlled trial indicate.
The incidence of herpes zoster was 30 per 1,000 person-years for patients who received the adjuvanted recombinant zoster vaccine (Shingrix) versus 94 per 1,000 person-years for those who received placebo, according to study results.
Recombinant zoster vaccine induced humoral and cellular responses that were strong and occurring at a rate higher than what was seen in the placebo group, said senior author Keith M. Sullivan, MD, of Duke University Medical Center, Durham, N.C., and coauthors, who reported findings on behalf of the Zoster Efficacy Study in Patients Undergoing HSCT (ZOE-HSCT) Study Group.
“The vaccinations were generally well tolerated, and most symptoms were mild and transient and did not substantially deter participants from receiving their second dose,” Dr. Sullivan and colleagues wrote in JAMA.
The risk of herpes zoster is increased for 2-3 years after autologous HSCT because of diminished T-cell immunity, according to the authors.
“Antiviral prophylaxis is commonly administered to patients after HSCT to prevent such complications, but the efficacy depends on adherence to treatment,” they said.
While vaccines could provide long-term protection, immunocompromised individuals receiving live attenuated vaccine would be at increased risk of varicella caused by spread of the vaccine strain, they added.
There have been a few encouraging recent studies of non-live vaccines in this setting, including one large phase 3 trial of a heat-inactivated varicella-zoster virus vaccine that showed patients undergoing autologous HSCT had a 63.8% estimated efficacy in preventing herpes zoster, investigators from that study said in The Lancet (2018 May 26;391[10135]:2116-27).
A phase 1/2a study of the adjuvanted recombinant zoster vaccine in patients undergoing HSCT demonstrated strong humoral and cell-mediated immunity responses, which provided the rationale for studying the vaccine further in the randomized ZOE-HSCT study, according to Dr. Sullivan and coauthors.
Their study included a total of 1,846 adults who had undergone autologous HSCT. They were randomized to receive two doses of the recombinant zoster vaccine, the first at 50-70 days after the procedure and the second 1-2 months later.
Herpes zoster cases were seen in 49 and 136 individuals in the vaccine and placebo groups, respectively, which resulted in overall incidences of 30 and 94 per 1,000 person-years.
The incidence rate ratio of a first episode of herpes zoster was 0.36 for individuals receiving at least one dose, which authors said was equivalent to a vaccine efficacy of 63.7%.
That efficacy rate is “very similar” to the estimated efficacy reported for the heat-inactivated varicella-zoster virus vaccine reported in The Lancet, said Dr. Sullivan and coauthors.
However, the heat-inactivated vaccine achieved that level of protection with a four-dose schedule, including one dose given prior to autologous HSCT.
“An advantage of the short 2-dose posttransplantation schedule is that more patients might complete the vaccination program,” they said in a discussion of the results, noting that 94.7% of the recombinant zoster vaccine recipients completed two doses, compared with 81.9% of recipients who received the heat-inactivated herpes zoster vaccine in the previous report.
The study was funded and sponsored by GlaxoSmithKline Biologicals SA. Dr. Sullivan reported disclosures related to GlaxoSmithKline (GSK), Kiadis Pharmaceutical, Roche Genentech, and the National Institute of Allergy and Infectious Diseases. Coauthors provided disclosures related to GSK, AbbVie, Roche, Gilead, Janssen, Pharmacyclics, Morphosys, Helsinn, Celgene, and others.
SOURCE: Bastidas A et al. JAMA. 2019 July 9. doi: 10.1001/jama.2019.9053.
FROM JAMA
Key clinical point: Two doses of recombinant zoster vaccine significantly reduced incidence of herpes zoster versus placebo in adults who had undergone autologous hematopoietic stem cell transplantation (HSCT).
Major finding: Herpes zoster cases were seen in 49 and 136 individuals in the vaccine and placebo groups, respectively, resulting in overall incidences of 30 and 94 per 1,000 person-years.
Study details: A randomized clinical trial (ZOE-HSCT) including 1,846 adults who had undergone autologous HSCT.
Disclosures: The study was funded and sponsored by GlaxoSmithKline Biologicals SA. Study authors reported disclosures related to GlaxoSmithKline, Kiadis Pharmaceutical, Roche Genentech, AbbVie, Roche, Gilead, Janssen, Pharmacyclics, Morphosys, Helsinn, Celgene, and others.
Source: Bastidas A et al. JAMA. 2019 July 9. doi: 10.1001/jama.2019.9053.
FDA approves Xpovio for relapsed/refractory multiple myeloma

The oral therapy was approved for patients who have received at least four prior therapies and whose disease is resistant to several other forms of treatment, including at least two proteasome inhibitors, at least two immunomodulatory agents, and an anti-CD38 monoclonal antibody, according to the FDA.
The approval provides a “treatment option for patients with multiple myeloma with no (other) available therapy,” said Richard Pazdur, MD, director of the FDA Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA Center for Drug Evaluation and Research.
The approval was based on a study that included 83 patients with RRMM who had an overall response rate of 25.3% to Xpovio in combination with dexamethasone.
“The median time to first response was 4 weeks, with a range of 1-10 weeks. The median duration of response was 3.8 months. The efficacy evaluation was supported by additional information from an ongoing, randomized trial in patients with multiple myeloma,” according to the statement.
Common side effects seen in patients taking Xpovio in combination with dexamethasone include leukopenia, neutropenia, thrombocytopenia, and anemia. Patients also reported vomiting, nausea, fatigue, diarrhea, fever, decreased appetite and weight, constipation, upper respiratory tract infections, and hyponatremia.
Patients taking Xpovio should be monitored for low blood counts, platelets, and sodium levels, and should avoid other medications that may cause dizziness or confusion. Patients’ hydration status, blood counts, and other medications should be optimized to avoid dizziness or confusion. Females of reproductive age and males with a female partner of reproductive potential must use effective contraception during treatment with Xpovio. Women who are pregnant or breastfeeding should not take Xpovio.
Xpovio must be dispensed with a patient Medication Guide that describes important information about the drug’s uses and risks.
Xpovio in combination with dexamethasone was granted accelerated approval, and further clinical trials are required to verify and describe the drug’s clinical benefit.
The FDA granted the approval of Xpovio to Karyopharm Therapeutics.
The oral therapy was approved for patients who have received at least four prior therapies and whose disease is resistant to several other forms of treatment, including at least two proteasome inhibitors, at least two immunomodulatory agents, and an anti-CD38 monoclonal antibody, according to the FDA.
The approval provides a “treatment option for patients with multiple myeloma with no (other) available therapy,” said Richard Pazdur, MD, director of the FDA Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA Center for Drug Evaluation and Research.
The approval was based on a study that included 83 patients with RRMM who had an overall response rate of 25.3% to Xpovio in combination with dexamethasone.
“The median time to first response was 4 weeks, with a range of 1-10 weeks. The median duration of response was 3.8 months. The efficacy evaluation was supported by additional information from an ongoing, randomized trial in patients with multiple myeloma,” according to the statement.
Common side effects seen in patients taking Xpovio in combination with dexamethasone include leukopenia, neutropenia, thrombocytopenia, and anemia. Patients also reported vomiting, nausea, fatigue, diarrhea, fever, decreased appetite and weight, constipation, upper respiratory tract infections, and hyponatremia.
Patients taking Xpovio should be monitored for low blood counts, platelets, and sodium levels, and should avoid other medications that may cause dizziness or confusion. Patients’ hydration status, blood counts, and other medications should be optimized to avoid dizziness or confusion. Females of reproductive age and males with a female partner of reproductive potential must use effective contraception during treatment with Xpovio. Women who are pregnant or breastfeeding should not take Xpovio.
Xpovio must be dispensed with a patient Medication Guide that describes important information about the drug’s uses and risks.
Xpovio in combination with dexamethasone was granted accelerated approval, and further clinical trials are required to verify and describe the drug’s clinical benefit.
The FDA granted the approval of Xpovio to Karyopharm Therapeutics.
The oral therapy was approved for patients who have received at least four prior therapies and whose disease is resistant to several other forms of treatment, including at least two proteasome inhibitors, at least two immunomodulatory agents, and an anti-CD38 monoclonal antibody, according to the FDA.
The approval provides a “treatment option for patients with multiple myeloma with no (other) available therapy,” said Richard Pazdur, MD, director of the FDA Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA Center for Drug Evaluation and Research.
The approval was based on a study that included 83 patients with RRMM who had an overall response rate of 25.3% to Xpovio in combination with dexamethasone.
“The median time to first response was 4 weeks, with a range of 1-10 weeks. The median duration of response was 3.8 months. The efficacy evaluation was supported by additional information from an ongoing, randomized trial in patients with multiple myeloma,” according to the statement.
Common side effects seen in patients taking Xpovio in combination with dexamethasone include leukopenia, neutropenia, thrombocytopenia, and anemia. Patients also reported vomiting, nausea, fatigue, diarrhea, fever, decreased appetite and weight, constipation, upper respiratory tract infections, and hyponatremia.
Patients taking Xpovio should be monitored for low blood counts, platelets, and sodium levels, and should avoid other medications that may cause dizziness or confusion. Patients’ hydration status, blood counts, and other medications should be optimized to avoid dizziness or confusion. Females of reproductive age and males with a female partner of reproductive potential must use effective contraception during treatment with Xpovio. Women who are pregnant or breastfeeding should not take Xpovio.
Xpovio must be dispensed with a patient Medication Guide that describes important information about the drug’s uses and risks.
Xpovio in combination with dexamethasone was granted accelerated approval, and further clinical trials are required to verify and describe the drug’s clinical benefit.
The FDA granted the approval of Xpovio to Karyopharm Therapeutics.
Researchers propose new risk groups for NK-AML
NEWPORT BEACH, CALIF. – New research suggests patients with normal karyotype acute myeloid leukemia (NK-AML) can be divided into four risk groups associated with overall survival.
Investigators used machine learning algorithms to study the association between mutations and overall survival in 1,352 patients with NK-AML. The analysis revealed combinations of mutations that could be used to classify NK-AML patients into favorable, intermediate-1, intermediate-2, and unfavorable risk groups.
For example, patients who had NPM1 mutations but wild-type FLT3-ITD and DNMT3A, had a median overall survival of 99.1 months and could be classified as favorable risk. Conversely, patients who had NPM1, FLT3-ITD, and DNMT3A mutations, had a median overall survival of 13.4 months and could be classified as unfavorable risk.
Aziz Nazha, MD, of the Cleveland Clinic, and his colleagues conducted this research and presented the findings at the Acute Leukemia Forum of Hemedicus.
The investigators looked at genomic and clinical data from 1,352 patients with NK-AML. The patients were a median age of 55 years and had a median white blood cell count of 21.3 x 109/L, a median hemoglobin of 9.1 g/dL, and a median platelet count of 61 x 109/L. More than half of patients (57.3%) were male.
The patients were screened for 35 genes that are commonly mutated in AML and other myeloid malignancies. The investigators used machine learning algorithms, including random survival forest and recommender system algorithms, to study the association between mutations and overall survival in an “unbiased” way.
Dr. Nazha said there were a median of three mutations per patient sample, and “there are some competing interests between those mutations to impact the prognosis of the patient.”
The investigators used the mutations and their associations with overall survival to classify patients into the risk groups outlined in the table below.
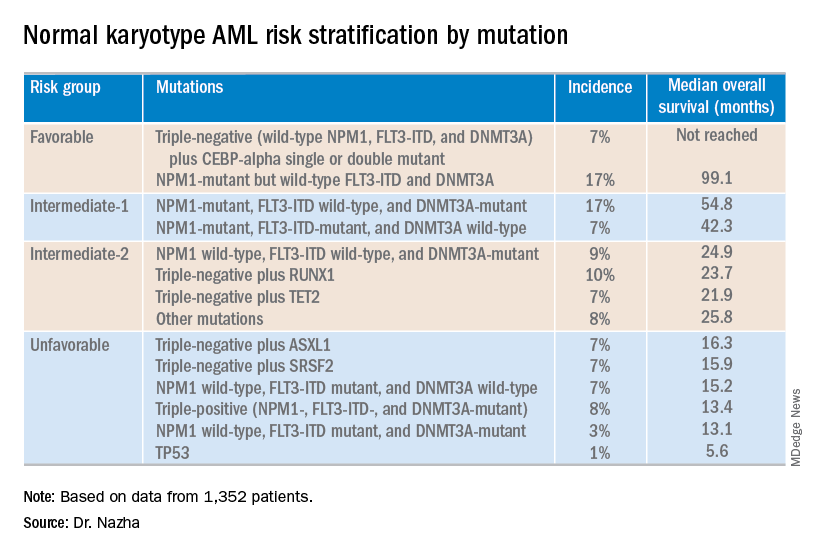
These findings can improve the risk stratification of NK-AML and may aid physicians in making treatment decisions, according to Dr. Nazha and his colleagues. To move this work forward, the investigators are attempting to develop a personalized model that can make predictions specific to an individual patient based on that patient’s mutation information.
Dr. Nazha reported having no financial disclosures relevant to this research. Other investigators reported relationships with the Munich Leukemia Laboratory.
The Acute Leukemia Forum is held by Hemedicus, which is owned by the same company as this news organization.
NEWPORT BEACH, CALIF. – New research suggests patients with normal karyotype acute myeloid leukemia (NK-AML) can be divided into four risk groups associated with overall survival.
Investigators used machine learning algorithms to study the association between mutations and overall survival in 1,352 patients with NK-AML. The analysis revealed combinations of mutations that could be used to classify NK-AML patients into favorable, intermediate-1, intermediate-2, and unfavorable risk groups.
For example, patients who had NPM1 mutations but wild-type FLT3-ITD and DNMT3A, had a median overall survival of 99.1 months and could be classified as favorable risk. Conversely, patients who had NPM1, FLT3-ITD, and DNMT3A mutations, had a median overall survival of 13.4 months and could be classified as unfavorable risk.
Aziz Nazha, MD, of the Cleveland Clinic, and his colleagues conducted this research and presented the findings at the Acute Leukemia Forum of Hemedicus.
The investigators looked at genomic and clinical data from 1,352 patients with NK-AML. The patients were a median age of 55 years and had a median white blood cell count of 21.3 x 109/L, a median hemoglobin of 9.1 g/dL, and a median platelet count of 61 x 109/L. More than half of patients (57.3%) were male.
The patients were screened for 35 genes that are commonly mutated in AML and other myeloid malignancies. The investigators used machine learning algorithms, including random survival forest and recommender system algorithms, to study the association between mutations and overall survival in an “unbiased” way.
Dr. Nazha said there were a median of three mutations per patient sample, and “there are some competing interests between those mutations to impact the prognosis of the patient.”
The investigators used the mutations and their associations with overall survival to classify patients into the risk groups outlined in the table below.
These findings can improve the risk stratification of NK-AML and may aid physicians in making treatment decisions, according to Dr. Nazha and his colleagues. To move this work forward, the investigators are attempting to develop a personalized model that can make predictions specific to an individual patient based on that patient’s mutation information.
Dr. Nazha reported having no financial disclosures relevant to this research. Other investigators reported relationships with the Munich Leukemia Laboratory.
The Acute Leukemia Forum is held by Hemedicus, which is owned by the same company as this news organization.
NEWPORT BEACH, CALIF. – New research suggests patients with normal karyotype acute myeloid leukemia (NK-AML) can be divided into four risk groups associated with overall survival.
Investigators used machine learning algorithms to study the association between mutations and overall survival in 1,352 patients with NK-AML. The analysis revealed combinations of mutations that could be used to classify NK-AML patients into favorable, intermediate-1, intermediate-2, and unfavorable risk groups.
For example, patients who had NPM1 mutations but wild-type FLT3-ITD and DNMT3A, had a median overall survival of 99.1 months and could be classified as favorable risk. Conversely, patients who had NPM1, FLT3-ITD, and DNMT3A mutations, had a median overall survival of 13.4 months and could be classified as unfavorable risk.
Aziz Nazha, MD, of the Cleveland Clinic, and his colleagues conducted this research and presented the findings at the Acute Leukemia Forum of Hemedicus.
The investigators looked at genomic and clinical data from 1,352 patients with NK-AML. The patients were a median age of 55 years and had a median white blood cell count of 21.3 x 109/L, a median hemoglobin of 9.1 g/dL, and a median platelet count of 61 x 109/L. More than half of patients (57.3%) were male.
The patients were screened for 35 genes that are commonly mutated in AML and other myeloid malignancies. The investigators used machine learning algorithms, including random survival forest and recommender system algorithms, to study the association between mutations and overall survival in an “unbiased” way.
Dr. Nazha said there were a median of three mutations per patient sample, and “there are some competing interests between those mutations to impact the prognosis of the patient.”
The investigators used the mutations and their associations with overall survival to classify patients into the risk groups outlined in the table below.
These findings can improve the risk stratification of NK-AML and may aid physicians in making treatment decisions, according to Dr. Nazha and his colleagues. To move this work forward, the investigators are attempting to develop a personalized model that can make predictions specific to an individual patient based on that patient’s mutation information.
Dr. Nazha reported having no financial disclosures relevant to this research. Other investigators reported relationships with the Munich Leukemia Laboratory.
The Acute Leukemia Forum is held by Hemedicus, which is owned by the same company as this news organization.
REPORTING FROM ALF 2019
CAR T-cell therapy bb2121 performs well in phase 1 trial of refractory multiple myeloma
Chimeric antigen receptor (CAR) T-cell therapy bb2121, which targets B-cell maturation agent (BCMA), appears safe and effective for treating patients with refractory multiple myeloma, according to results of a phase 1 trial.
The objective response rate of 85% among 33 heavily pretreated patients suggests “promising efficacy,” reported lead author Noopur Raje, MD, of Massachusetts General Hospital Cancer Center in Boston and colleagues.
“Although comparisons among studies are complicated by differences in patient populations, CAR constructs, administered doses, and grading scales of toxic effects, the results observed with bb2121 indicate a favorable safety profile,” the investigators wrote in a study published in the New England Journal of Medicine.
The study initially involved 36 patients with refractory multiple myeloma who had received at least three lines of prior therapy, including an immunomodulatory agent and a proteasome inhibitor. Although leukapheresis and therapy manufacturing were successful in all patients, three patients were excluded from receiving the infusion because of disease progression.
The 33 remaining patients were lymphodepleted with fludarabine and cyclophosphamide. Bridging therapy was allowed during the manufacturing process but was stopped at least 2 weeks prior to infusion. In the dose-escalation phase of the study, b2121 was delivered as a single infusion at one of four dose levels: 50 × 106, 150 × 106, 450 × 106, or 800 × 106 CAR T cells. In the expansion phase, the treatment was given at either 150 x 106 or 450 x 106 CAR T cells. The primary endpoint was safety; the secondary endpoints were response rate and duration of response.
After a median follow-up of 11.3 months, most patients (85%) had responded to therapy, and almost half (45%) had achieved a complete response. Of the 15 complete responders, 6 relapsed. The median progression-free survival was 11.8 months; stated differently, two out of five patients (40%) had not experienced disease progression after 1 year. CAR T cells were detectable 1 month after infusion in 96% of patients; however, this value dropped to 86% at 3 months, 57% at 6 months, and 20% at 12 months. The investigators noted that CAR T-cell persistence was associated with treatment response.
All patients had adverse events. Most (85%) had grade 3 or higher hematologic toxicity, which the investigators considered to be the “expected toxic effects of lymphodepleting chemotherapy.” Although other adverse events occurred in the majority of patients, these were generally mild to moderate. Cytokine release syndrome occurred in 25 patients (76%), including two instances of grade 3 toxicity but none of grade 4 or higher. Fourteen patients (42%) developed neurologic toxicities: Most were grade 1 or 2, but one patient had a grade 4 toxicity that resolved after a month. Infections occurred at the same rate (42%), although, again, most were grade 1 or 2.
The study was funded by Bluebird Bio and Celgene. The investigators disclosed financial relationships with Bluebird and other drug companies.
SOURCE: Raje N et al. NEJM. 1 May 2019. doi: 10.1056/NEJMoa1817226.
Chimeric antigen receptor (CAR) T-cell therapy bb2121, which targets B-cell maturation agent (BCMA), appears safe and effective for treating patients with refractory multiple myeloma, according to results of a phase 1 trial.
The objective response rate of 85% among 33 heavily pretreated patients suggests “promising efficacy,” reported lead author Noopur Raje, MD, of Massachusetts General Hospital Cancer Center in Boston and colleagues.
“Although comparisons among studies are complicated by differences in patient populations, CAR constructs, administered doses, and grading scales of toxic effects, the results observed with bb2121 indicate a favorable safety profile,” the investigators wrote in a study published in the New England Journal of Medicine.
The study initially involved 36 patients with refractory multiple myeloma who had received at least three lines of prior therapy, including an immunomodulatory agent and a proteasome inhibitor. Although leukapheresis and therapy manufacturing were successful in all patients, three patients were excluded from receiving the infusion because of disease progression.
The 33 remaining patients were lymphodepleted with fludarabine and cyclophosphamide. Bridging therapy was allowed during the manufacturing process but was stopped at least 2 weeks prior to infusion. In the dose-escalation phase of the study, b2121 was delivered as a single infusion at one of four dose levels: 50 × 106, 150 × 106, 450 × 106, or 800 × 106 CAR T cells. In the expansion phase, the treatment was given at either 150 x 106 or 450 x 106 CAR T cells. The primary endpoint was safety; the secondary endpoints were response rate and duration of response.
After a median follow-up of 11.3 months, most patients (85%) had responded to therapy, and almost half (45%) had achieved a complete response. Of the 15 complete responders, 6 relapsed. The median progression-free survival was 11.8 months; stated differently, two out of five patients (40%) had not experienced disease progression after 1 year. CAR T cells were detectable 1 month after infusion in 96% of patients; however, this value dropped to 86% at 3 months, 57% at 6 months, and 20% at 12 months. The investigators noted that CAR T-cell persistence was associated with treatment response.
All patients had adverse events. Most (85%) had grade 3 or higher hematologic toxicity, which the investigators considered to be the “expected toxic effects of lymphodepleting chemotherapy.” Although other adverse events occurred in the majority of patients, these were generally mild to moderate. Cytokine release syndrome occurred in 25 patients (76%), including two instances of grade 3 toxicity but none of grade 4 or higher. Fourteen patients (42%) developed neurologic toxicities: Most were grade 1 or 2, but one patient had a grade 4 toxicity that resolved after a month. Infections occurred at the same rate (42%), although, again, most were grade 1 or 2.
The study was funded by Bluebird Bio and Celgene. The investigators disclosed financial relationships with Bluebird and other drug companies.
SOURCE: Raje N et al. NEJM. 1 May 2019. doi: 10.1056/NEJMoa1817226.
Chimeric antigen receptor (CAR) T-cell therapy bb2121, which targets B-cell maturation agent (BCMA), appears safe and effective for treating patients with refractory multiple myeloma, according to results of a phase 1 trial.
The objective response rate of 85% among 33 heavily pretreated patients suggests “promising efficacy,” reported lead author Noopur Raje, MD, of Massachusetts General Hospital Cancer Center in Boston and colleagues.
“Although comparisons among studies are complicated by differences in patient populations, CAR constructs, administered doses, and grading scales of toxic effects, the results observed with bb2121 indicate a favorable safety profile,” the investigators wrote in a study published in the New England Journal of Medicine.
The study initially involved 36 patients with refractory multiple myeloma who had received at least three lines of prior therapy, including an immunomodulatory agent and a proteasome inhibitor. Although leukapheresis and therapy manufacturing were successful in all patients, three patients were excluded from receiving the infusion because of disease progression.
The 33 remaining patients were lymphodepleted with fludarabine and cyclophosphamide. Bridging therapy was allowed during the manufacturing process but was stopped at least 2 weeks prior to infusion. In the dose-escalation phase of the study, b2121 was delivered as a single infusion at one of four dose levels: 50 × 106, 150 × 106, 450 × 106, or 800 × 106 CAR T cells. In the expansion phase, the treatment was given at either 150 x 106 or 450 x 106 CAR T cells. The primary endpoint was safety; the secondary endpoints were response rate and duration of response.
After a median follow-up of 11.3 months, most patients (85%) had responded to therapy, and almost half (45%) had achieved a complete response. Of the 15 complete responders, 6 relapsed. The median progression-free survival was 11.8 months; stated differently, two out of five patients (40%) had not experienced disease progression after 1 year. CAR T cells were detectable 1 month after infusion in 96% of patients; however, this value dropped to 86% at 3 months, 57% at 6 months, and 20% at 12 months. The investigators noted that CAR T-cell persistence was associated with treatment response.
All patients had adverse events. Most (85%) had grade 3 or higher hematologic toxicity, which the investigators considered to be the “expected toxic effects of lymphodepleting chemotherapy.” Although other adverse events occurred in the majority of patients, these were generally mild to moderate. Cytokine release syndrome occurred in 25 patients (76%), including two instances of grade 3 toxicity but none of grade 4 or higher. Fourteen patients (42%) developed neurologic toxicities: Most were grade 1 or 2, but one patient had a grade 4 toxicity that resolved after a month. Infections occurred at the same rate (42%), although, again, most were grade 1 or 2.
The study was funded by Bluebird Bio and Celgene. The investigators disclosed financial relationships with Bluebird and other drug companies.
SOURCE: Raje N et al. NEJM. 1 May 2019. doi: 10.1056/NEJMoa1817226.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE