User login
CAR T-cell therapy neurotoxicity linked to NfL elevations
“This is the first study to show NfL levels are elevated even before CAR T treatment is given,” first author Omar H. Butt, MD, PhD, of the Siteman Cancer Center at Barnes-Jewish Hospital and Washington University in St. Louis, said in an interview.

“While unlikely to be the sole driver of [the neurotoxicity], neural injury reflected by NfL may aid in identifying a high-risk subset of patients undergoing cellular therapy,” the authors concluded in the study, published in JAMA Oncology.
CAR T-cell therapy has gained favor for virtually revolutionizing the treatment of some leukemias and lymphomas, however, as many as 40%-60% of patients develop the neurotoxicity side effect, called immune effector cell–associated neurotoxicity syndrome (ICANS), which, though usually low grade, in more severe cases can cause substantial morbidity and even mortality.
Hence, “the early identification of patients at risk for ICANS is critical for preemptive management,” the authors noted.
NfL, an established marker of neuroaxonal injury in neurodegenerative diseases including multiple sclerosis and Alzheimer’s disease, has been shown in previous studies to be elevated following the development of ICANS and up to 5 days prior to its peak symptoms.
To further evaluate NfL elevations in relation to ICANS, Dr. Butt and colleagues identified 30 patients undergoing CD19 CART-cell therapy, including 77% for diffuse large B-cell lymphoma, at two U.S. centers: Washington University in St. Louis and Case Western Reserve University, Cleveland.
The patients had a median age of 64 and were 40% female.
Among them, four developed low-grade ICANS grade 1-2, and 7 developed ICANS grade 3 or higher.
Of those developing any-grade ICANS, baseline elevations of NfL prior to the CAR T-cell treatment, were significantly higher, compared with those who did not develop ICANs (mean 87.6 pg/mL vs. 29.4 pg/mL, P < .001), with no significant differences between the low-grade (1 and 2) and higher-grade (3 or higher) ICANS groups.
A receiver operating characteristic analysis showed baseline NfL levels significantly predicted the development of ICANS with high accuracy (area under the ROC curve, 0.96), as well as sensitivity (AUROC, 0.91) and specificity (AUROC, 0.95).
Notably, baseline NfL levels were associated with ICANS severity, but did not correlate with other factors including demographic, oncologic history, nononcologic neurologic history, or history of exposure to neurotoxic therapies.
However, Dr. Butt added, “it is important to note that our study was insufficiently powered to examine those relationships in earnest. Therefore, [a correlation between NfL and those factors] remains possible,” he said.
The elevated NfL levels observed prior to the development of ICANS remained high across the study’s seven time points, up to day 30 post infusion.
Interest in NfL levels on the rise
NfL assessment is currently only clinically validated in amyotrophic lateral sclerosis, where it is used to assess neuroaxonal health and integrity. However, testing is available as interest and evidence of NfL’s potential role in other settings grows.
Meanwhile, Dr. Butt and associates are themselves developing an assay to predict the development of ICANS, which will likely include NfL, if the role is validated in further studies.
“Future studies will explore validating NfL for ICANS and additional indications,” he said.
ICANS symptoms can range from headaches and confusion to seizures or strokes in more severe cases.
The current gold standard for treatment includes early intervention with high-dose steroids and careful monitoring, but there is reluctance to use such therapies because of concerns about their blunting the anticancer effects of the CAR T cells.
Importantly, if validated, elevations in NfL could signal the need for more precautionary measures with CAR T-cell therapy, Dr. Butt noted.
“Our data suggests patients with high NfL levels at baseline would benefit most from perhaps closer monitoring with frequent checks and possible early intervention at the first sign of symptoms, a period of time when it may be hard to distinguish ICANS from other causes of confusion, such as delirium,” he explained.
Limitations: Validation, preventive measures needed
Commenting on the study, Sattva S. Neelapu, MD, a professor and deputy chair of the department of lymphoma and myeloma at the University of Texas MD Anderson Cancer Center, Houston, agreed that the findings have potentially important implications.
“I think this is a very intriguing and novel finding that needs to be investigated further prospectively in a larger cohort and across different CAR T products in patients with lymphoma, leukemia, and myeloma,” Dr. Neelapu said in an interview.
The NfL elevations observed even before CAR T-cell therapy among those who went on to develop ICANS are notable, he added.
“This is the surprising finding in the study,” Dr. Neelapu said. “It raises the question whether neurologic injury is caused by prior therapies that these patients received or whether it is an age-related phenomenon, as we do see higher incidence and severity of ICANS in older patients or some other mechanisms.”
A key caveat, however, is that even if a risk is identified, options to prevent ICANS are currently limited, Dr. Neelapu noted.
“I think it is too early to implement this into clinical practice,” he said. In addition to needing further validation, “assessing NfL levels would be useful when there is an effective prophylactic or therapeutic strategy – both of which also need to be investigated.”
Dr. Butt and colleagues are developing a clinical assay for ICANS and reported a provisional patent pending on the use of plasma NfL as a predictive biomarker for ICANS. The study received support from the Washington University in St. Louis, the Paula and Rodger O. Riney Fund, the Daniel J. Brennan MD Fund, the Fred Simmons and Olga Mohan Fund; the National Cancer Institute, the National Multiple Sclerosis Society, and the National Institute of Neurological Disorders and Stroke. Dr. Neelapu reported conflicts of interest with numerous pharmaceutical companies.
“This is the first study to show NfL levels are elevated even before CAR T treatment is given,” first author Omar H. Butt, MD, PhD, of the Siteman Cancer Center at Barnes-Jewish Hospital and Washington University in St. Louis, said in an interview.
“While unlikely to be the sole driver of [the neurotoxicity], neural injury reflected by NfL may aid in identifying a high-risk subset of patients undergoing cellular therapy,” the authors concluded in the study, published in JAMA Oncology.
CAR T-cell therapy has gained favor for virtually revolutionizing the treatment of some leukemias and lymphomas, however, as many as 40%-60% of patients develop the neurotoxicity side effect, called immune effector cell–associated neurotoxicity syndrome (ICANS), which, though usually low grade, in more severe cases can cause substantial morbidity and even mortality.
Hence, “the early identification of patients at risk for ICANS is critical for preemptive management,” the authors noted.
NfL, an established marker of neuroaxonal injury in neurodegenerative diseases including multiple sclerosis and Alzheimer’s disease, has been shown in previous studies to be elevated following the development of ICANS and up to 5 days prior to its peak symptoms.
To further evaluate NfL elevations in relation to ICANS, Dr. Butt and colleagues identified 30 patients undergoing CD19 CART-cell therapy, including 77% for diffuse large B-cell lymphoma, at two U.S. centers: Washington University in St. Louis and Case Western Reserve University, Cleveland.
The patients had a median age of 64 and were 40% female.
Among them, four developed low-grade ICANS grade 1-2, and 7 developed ICANS grade 3 or higher.
Of those developing any-grade ICANS, baseline elevations of NfL prior to the CAR T-cell treatment, were significantly higher, compared with those who did not develop ICANs (mean 87.6 pg/mL vs. 29.4 pg/mL, P < .001), with no significant differences between the low-grade (1 and 2) and higher-grade (3 or higher) ICANS groups.
A receiver operating characteristic analysis showed baseline NfL levels significantly predicted the development of ICANS with high accuracy (area under the ROC curve, 0.96), as well as sensitivity (AUROC, 0.91) and specificity (AUROC, 0.95).
Notably, baseline NfL levels were associated with ICANS severity, but did not correlate with other factors including demographic, oncologic history, nononcologic neurologic history, or history of exposure to neurotoxic therapies.
However, Dr. Butt added, “it is important to note that our study was insufficiently powered to examine those relationships in earnest. Therefore, [a correlation between NfL and those factors] remains possible,” he said.
The elevated NfL levels observed prior to the development of ICANS remained high across the study’s seven time points, up to day 30 post infusion.
Interest in NfL levels on the rise
NfL assessment is currently only clinically validated in amyotrophic lateral sclerosis, where it is used to assess neuroaxonal health and integrity. However, testing is available as interest and evidence of NfL’s potential role in other settings grows.
Meanwhile, Dr. Butt and associates are themselves developing an assay to predict the development of ICANS, which will likely include NfL, if the role is validated in further studies.
“Future studies will explore validating NfL for ICANS and additional indications,” he said.
ICANS symptoms can range from headaches and confusion to seizures or strokes in more severe cases.
The current gold standard for treatment includes early intervention with high-dose steroids and careful monitoring, but there is reluctance to use such therapies because of concerns about their blunting the anticancer effects of the CAR T cells.
Importantly, if validated, elevations in NfL could signal the need for more precautionary measures with CAR T-cell therapy, Dr. Butt noted.
“Our data suggests patients with high NfL levels at baseline would benefit most from perhaps closer monitoring with frequent checks and possible early intervention at the first sign of symptoms, a period of time when it may be hard to distinguish ICANS from other causes of confusion, such as delirium,” he explained.
Limitations: Validation, preventive measures needed
Commenting on the study, Sattva S. Neelapu, MD, a professor and deputy chair of the department of lymphoma and myeloma at the University of Texas MD Anderson Cancer Center, Houston, agreed that the findings have potentially important implications.
“I think this is a very intriguing and novel finding that needs to be investigated further prospectively in a larger cohort and across different CAR T products in patients with lymphoma, leukemia, and myeloma,” Dr. Neelapu said in an interview.
The NfL elevations observed even before CAR T-cell therapy among those who went on to develop ICANS are notable, he added.
“This is the surprising finding in the study,” Dr. Neelapu said. “It raises the question whether neurologic injury is caused by prior therapies that these patients received or whether it is an age-related phenomenon, as we do see higher incidence and severity of ICANS in older patients or some other mechanisms.”
A key caveat, however, is that even if a risk is identified, options to prevent ICANS are currently limited, Dr. Neelapu noted.
“I think it is too early to implement this into clinical practice,” he said. In addition to needing further validation, “assessing NfL levels would be useful when there is an effective prophylactic or therapeutic strategy – both of which also need to be investigated.”
Dr. Butt and colleagues are developing a clinical assay for ICANS and reported a provisional patent pending on the use of plasma NfL as a predictive biomarker for ICANS. The study received support from the Washington University in St. Louis, the Paula and Rodger O. Riney Fund, the Daniel J. Brennan MD Fund, the Fred Simmons and Olga Mohan Fund; the National Cancer Institute, the National Multiple Sclerosis Society, and the National Institute of Neurological Disorders and Stroke. Dr. Neelapu reported conflicts of interest with numerous pharmaceutical companies.
“This is the first study to show NfL levels are elevated even before CAR T treatment is given,” first author Omar H. Butt, MD, PhD, of the Siteman Cancer Center at Barnes-Jewish Hospital and Washington University in St. Louis, said in an interview.
“While unlikely to be the sole driver of [the neurotoxicity], neural injury reflected by NfL may aid in identifying a high-risk subset of patients undergoing cellular therapy,” the authors concluded in the study, published in JAMA Oncology.
CAR T-cell therapy has gained favor for virtually revolutionizing the treatment of some leukemias and lymphomas, however, as many as 40%-60% of patients develop the neurotoxicity side effect, called immune effector cell–associated neurotoxicity syndrome (ICANS), which, though usually low grade, in more severe cases can cause substantial morbidity and even mortality.
Hence, “the early identification of patients at risk for ICANS is critical for preemptive management,” the authors noted.
NfL, an established marker of neuroaxonal injury in neurodegenerative diseases including multiple sclerosis and Alzheimer’s disease, has been shown in previous studies to be elevated following the development of ICANS and up to 5 days prior to its peak symptoms.
To further evaluate NfL elevations in relation to ICANS, Dr. Butt and colleagues identified 30 patients undergoing CD19 CART-cell therapy, including 77% for diffuse large B-cell lymphoma, at two U.S. centers: Washington University in St. Louis and Case Western Reserve University, Cleveland.
The patients had a median age of 64 and were 40% female.
Among them, four developed low-grade ICANS grade 1-2, and 7 developed ICANS grade 3 or higher.
Of those developing any-grade ICANS, baseline elevations of NfL prior to the CAR T-cell treatment, were significantly higher, compared with those who did not develop ICANs (mean 87.6 pg/mL vs. 29.4 pg/mL, P < .001), with no significant differences between the low-grade (1 and 2) and higher-grade (3 or higher) ICANS groups.
A receiver operating characteristic analysis showed baseline NfL levels significantly predicted the development of ICANS with high accuracy (area under the ROC curve, 0.96), as well as sensitivity (AUROC, 0.91) and specificity (AUROC, 0.95).
Notably, baseline NfL levels were associated with ICANS severity, but did not correlate with other factors including demographic, oncologic history, nononcologic neurologic history, or history of exposure to neurotoxic therapies.
However, Dr. Butt added, “it is important to note that our study was insufficiently powered to examine those relationships in earnest. Therefore, [a correlation between NfL and those factors] remains possible,” he said.
The elevated NfL levels observed prior to the development of ICANS remained high across the study’s seven time points, up to day 30 post infusion.
Interest in NfL levels on the rise
NfL assessment is currently only clinically validated in amyotrophic lateral sclerosis, where it is used to assess neuroaxonal health and integrity. However, testing is available as interest and evidence of NfL’s potential role in other settings grows.
Meanwhile, Dr. Butt and associates are themselves developing an assay to predict the development of ICANS, which will likely include NfL, if the role is validated in further studies.
“Future studies will explore validating NfL for ICANS and additional indications,” he said.
ICANS symptoms can range from headaches and confusion to seizures or strokes in more severe cases.
The current gold standard for treatment includes early intervention with high-dose steroids and careful monitoring, but there is reluctance to use such therapies because of concerns about their blunting the anticancer effects of the CAR T cells.
Importantly, if validated, elevations in NfL could signal the need for more precautionary measures with CAR T-cell therapy, Dr. Butt noted.
“Our data suggests patients with high NfL levels at baseline would benefit most from perhaps closer monitoring with frequent checks and possible early intervention at the first sign of symptoms, a period of time when it may be hard to distinguish ICANS from other causes of confusion, such as delirium,” he explained.
Limitations: Validation, preventive measures needed
Commenting on the study, Sattva S. Neelapu, MD, a professor and deputy chair of the department of lymphoma and myeloma at the University of Texas MD Anderson Cancer Center, Houston, agreed that the findings have potentially important implications.
“I think this is a very intriguing and novel finding that needs to be investigated further prospectively in a larger cohort and across different CAR T products in patients with lymphoma, leukemia, and myeloma,” Dr. Neelapu said in an interview.
The NfL elevations observed even before CAR T-cell therapy among those who went on to develop ICANS are notable, he added.
“This is the surprising finding in the study,” Dr. Neelapu said. “It raises the question whether neurologic injury is caused by prior therapies that these patients received or whether it is an age-related phenomenon, as we do see higher incidence and severity of ICANS in older patients or some other mechanisms.”
A key caveat, however, is that even if a risk is identified, options to prevent ICANS are currently limited, Dr. Neelapu noted.
“I think it is too early to implement this into clinical practice,” he said. In addition to needing further validation, “assessing NfL levels would be useful when there is an effective prophylactic or therapeutic strategy – both of which also need to be investigated.”
Dr. Butt and colleagues are developing a clinical assay for ICANS and reported a provisional patent pending on the use of plasma NfL as a predictive biomarker for ICANS. The study received support from the Washington University in St. Louis, the Paula and Rodger O. Riney Fund, the Daniel J. Brennan MD Fund, the Fred Simmons and Olga Mohan Fund; the National Cancer Institute, the National Multiple Sclerosis Society, and the National Institute of Neurological Disorders and Stroke. Dr. Neelapu reported conflicts of interest with numerous pharmaceutical companies.
FROM JAMA ONCOLOGY
Drug combo holds promise of better AML outcomes
Adding venetoclax (Venclexta) to a gilteritinib (Xospata) regimen appeared to improve outcomes in refractory/relapsed FLT3-mutated acute myeloid leukemia (AML), a new industry-funded phase 1b study reported.
“.
Outcomes in AML are poor. As the study notes, most patients relapse and face a median overall survival of 4-7 months even with standard chemotherapy. Gilteritinib, a selective oral FLT3 inhibitor, is Food and Drug Administration–approved for the 30% of relapsed/refractory patients with AML who have FLT3 mutations.
“The general sentiment is that, although some patients have great benefit from gilteritinib monotherapy, there is room to improve the quality, frequency, and duration of responses with combinations,” said hematologist Andrew Brunner, MD, of Massachusetts General Hospital in Boston, in an interview. He was not involved with the study research.
For the new open-label, dose-escalation/dose-expansion study, led by hematologist Naval Daver, MD, of the University of Texas MD Anderson Cancer Center, Houston, researchers enrolled 61 patients (56 with FLT3 mutations) from 2018 to 2020. The median age was 63 years (range 21-85).
The subjects were assigned to get a recommended phase 2 dose of 400 mg venetoclax once daily and 120 mg gilteritinib once daily.
Over a median follow-up of 17.5 months, the median remission time was 4.9 months (95% confidence interval, 3.4-6.6), and the patients with FLT3 mutations survived a median of 10 months.
“The combination of venetoclax and gilteritinib was tolerable at standard doses of each drug, generated remarkably high response rates, and markedly reduced FLT3-internal tandem duplications mutation burden. … Early mortality was similar to gilteritinib monotherapy,” the authors wrote.
Eighty percent of patients experienced cytopenias, and “adverse events prompted venetoclax and gilteritinib dose interruptions in 51% and 48%, respectively.”
About 60% of patients who went on to receive allogeneic hematopoietic stem cell transplantation were alive at the end of follow-up, “suggesting that VenGilt [the combo treatment] could be an effective bridge to transplant in young/fit patients with relapsed FLT3mut AML,” the researchers wrote.
All patients withdrew from the study by November 2021 for several reasons such as death (n=42), adverse events (n=10), and disease progression (29); some had multiple reasons.
Dr. Brunner said the study is “an important step toward evaluating a new potential regimen.”
The remission duration, FLT3 molecular response, and median overall survival “seem quite encouraging for a severe disease like AML in relapse,” he said. However, he added that the drug combo “would need to be evaluated in a randomized and, ideally, placebo-controlled setting to know if this is a significant improvement.”
He also highlighted the high number of severe cyptopenias with associated complications such as death. “Whether this is acceptable depends on the patient and circumstances,” he said. “But it does suggest that this regimen would potentially be for more robust patients, particularly since the group that did best were those who went to transplant later.”
Pending more research, Dr. Brunner said, “I am not sure I would use [the combination treatment] over gilteritinib monotherapy, for instance. But there may be settings where no other options are available, and this could be considered, particularly if a transplant option is a next step.”
The study was funded by AbbVie, Genentech, and Astellas. The study authors report multiple disclosures; some are employed by Astellas, AbbVie, and Genentech/Roche.
Dr. Bronner reports running clinical trials, advisory board service and/or consultation for Acceleron, Agios, Abbvie, BMS/Celgene, Keros Therapeutics, Novartis, Takeda, GSK, AstraZeneca, Janssen, and Gilead.
Adding venetoclax (Venclexta) to a gilteritinib (Xospata) regimen appeared to improve outcomes in refractory/relapsed FLT3-mutated acute myeloid leukemia (AML), a new industry-funded phase 1b study reported.
“.
Outcomes in AML are poor. As the study notes, most patients relapse and face a median overall survival of 4-7 months even with standard chemotherapy. Gilteritinib, a selective oral FLT3 inhibitor, is Food and Drug Administration–approved for the 30% of relapsed/refractory patients with AML who have FLT3 mutations.
“The general sentiment is that, although some patients have great benefit from gilteritinib monotherapy, there is room to improve the quality, frequency, and duration of responses with combinations,” said hematologist Andrew Brunner, MD, of Massachusetts General Hospital in Boston, in an interview. He was not involved with the study research.
For the new open-label, dose-escalation/dose-expansion study, led by hematologist Naval Daver, MD, of the University of Texas MD Anderson Cancer Center, Houston, researchers enrolled 61 patients (56 with FLT3 mutations) from 2018 to 2020. The median age was 63 years (range 21-85).
The subjects were assigned to get a recommended phase 2 dose of 400 mg venetoclax once daily and 120 mg gilteritinib once daily.
Over a median follow-up of 17.5 months, the median remission time was 4.9 months (95% confidence interval, 3.4-6.6), and the patients with FLT3 mutations survived a median of 10 months.
“The combination of venetoclax and gilteritinib was tolerable at standard doses of each drug, generated remarkably high response rates, and markedly reduced FLT3-internal tandem duplications mutation burden. … Early mortality was similar to gilteritinib monotherapy,” the authors wrote.
Eighty percent of patients experienced cytopenias, and “adverse events prompted venetoclax and gilteritinib dose interruptions in 51% and 48%, respectively.”
About 60% of patients who went on to receive allogeneic hematopoietic stem cell transplantation were alive at the end of follow-up, “suggesting that VenGilt [the combo treatment] could be an effective bridge to transplant in young/fit patients with relapsed FLT3mut AML,” the researchers wrote.
All patients withdrew from the study by November 2021 for several reasons such as death (n=42), adverse events (n=10), and disease progression (29); some had multiple reasons.
Dr. Brunner said the study is “an important step toward evaluating a new potential regimen.”
The remission duration, FLT3 molecular response, and median overall survival “seem quite encouraging for a severe disease like AML in relapse,” he said. However, he added that the drug combo “would need to be evaluated in a randomized and, ideally, placebo-controlled setting to know if this is a significant improvement.”
He also highlighted the high number of severe cyptopenias with associated complications such as death. “Whether this is acceptable depends on the patient and circumstances,” he said. “But it does suggest that this regimen would potentially be for more robust patients, particularly since the group that did best were those who went to transplant later.”
Pending more research, Dr. Brunner said, “I am not sure I would use [the combination treatment] over gilteritinib monotherapy, for instance. But there may be settings where no other options are available, and this could be considered, particularly if a transplant option is a next step.”
The study was funded by AbbVie, Genentech, and Astellas. The study authors report multiple disclosures; some are employed by Astellas, AbbVie, and Genentech/Roche.
Dr. Bronner reports running clinical trials, advisory board service and/or consultation for Acceleron, Agios, Abbvie, BMS/Celgene, Keros Therapeutics, Novartis, Takeda, GSK, AstraZeneca, Janssen, and Gilead.
Adding venetoclax (Venclexta) to a gilteritinib (Xospata) regimen appeared to improve outcomes in refractory/relapsed FLT3-mutated acute myeloid leukemia (AML), a new industry-funded phase 1b study reported.
“.
Outcomes in AML are poor. As the study notes, most patients relapse and face a median overall survival of 4-7 months even with standard chemotherapy. Gilteritinib, a selective oral FLT3 inhibitor, is Food and Drug Administration–approved for the 30% of relapsed/refractory patients with AML who have FLT3 mutations.
“The general sentiment is that, although some patients have great benefit from gilteritinib monotherapy, there is room to improve the quality, frequency, and duration of responses with combinations,” said hematologist Andrew Brunner, MD, of Massachusetts General Hospital in Boston, in an interview. He was not involved with the study research.
For the new open-label, dose-escalation/dose-expansion study, led by hematologist Naval Daver, MD, of the University of Texas MD Anderson Cancer Center, Houston, researchers enrolled 61 patients (56 with FLT3 mutations) from 2018 to 2020. The median age was 63 years (range 21-85).
The subjects were assigned to get a recommended phase 2 dose of 400 mg venetoclax once daily and 120 mg gilteritinib once daily.
Over a median follow-up of 17.5 months, the median remission time was 4.9 months (95% confidence interval, 3.4-6.6), and the patients with FLT3 mutations survived a median of 10 months.
“The combination of venetoclax and gilteritinib was tolerable at standard doses of each drug, generated remarkably high response rates, and markedly reduced FLT3-internal tandem duplications mutation burden. … Early mortality was similar to gilteritinib monotherapy,” the authors wrote.
Eighty percent of patients experienced cytopenias, and “adverse events prompted venetoclax and gilteritinib dose interruptions in 51% and 48%, respectively.”
About 60% of patients who went on to receive allogeneic hematopoietic stem cell transplantation were alive at the end of follow-up, “suggesting that VenGilt [the combo treatment] could be an effective bridge to transplant in young/fit patients with relapsed FLT3mut AML,” the researchers wrote.
All patients withdrew from the study by November 2021 for several reasons such as death (n=42), adverse events (n=10), and disease progression (29); some had multiple reasons.
Dr. Brunner said the study is “an important step toward evaluating a new potential regimen.”
The remission duration, FLT3 molecular response, and median overall survival “seem quite encouraging for a severe disease like AML in relapse,” he said. However, he added that the drug combo “would need to be evaluated in a randomized and, ideally, placebo-controlled setting to know if this is a significant improvement.”
He also highlighted the high number of severe cyptopenias with associated complications such as death. “Whether this is acceptable depends on the patient and circumstances,” he said. “But it does suggest that this regimen would potentially be for more robust patients, particularly since the group that did best were those who went to transplant later.”
Pending more research, Dr. Brunner said, “I am not sure I would use [the combination treatment] over gilteritinib monotherapy, for instance. But there may be settings where no other options are available, and this could be considered, particularly if a transplant option is a next step.”
The study was funded by AbbVie, Genentech, and Astellas. The study authors report multiple disclosures; some are employed by Astellas, AbbVie, and Genentech/Roche.
Dr. Bronner reports running clinical trials, advisory board service and/or consultation for Acceleron, Agios, Abbvie, BMS/Celgene, Keros Therapeutics, Novartis, Takeda, GSK, AstraZeneca, Janssen, and Gilead.
FROM JOURNAL OF CLINICAL ONCOLOGY
From B to T: a Case of Concurrent B-Cell and T-Cell Lymphomas Successfully Palliated With Targeted Therapies
Background
Diffuse large B-cell lymphoma (DLBCL) is the most common aggressive type of non- Hodgkin lymphoma (NHL), comprising 30% of all NHL. Due to a decreased state of immunosurveillance resulting from the disease itself and its associated therapies, patients are at increased risk of developing a secondary malignancy. Multiple primary malignancies have been reported to occur in up to 15% of patients with DLBCL, retrospectively.
Herein, we review a case of a man with DLBCL who concomitantly developed ALK negative anaplastic large cell lymphoma (ALCL) distinctly of T-cell lineage who was successfully treated with palliative therapy for both diagnoses despite his advanced age and diagnosis associated with a poor prognosis with continued effect and sustained quality of life.
Case Report
An 88-year-old man presented with stage III DLBCL, diagnosed in 12/2018, was deemed not to be an aggressive therapy candidate. As such, he was treated with Rituximab monotherapy for 6 cycles, ending in 02/2019, with remarkably good effect. He remained in a PR with stable disease on serial PET/CTs until 09/2021, at which time he was noted to have Horner’s Syndrome in clinic. CT chest demonstrated a right apical lung mass, not previously seen on prior scans measuring 4.2 x 2.7 cm. Other sites of nodal disease remained stable on PET/CT.
Biopsy of the lesion revealed CD30+ ALK-negative ALCL with distinct T-cell marker positivity on immunohistochemistry and the absence of B-cell lineage markers. After discussion at our treatment planning conference, we decided to treat with brentuximab-vedotin (Bv) monotherapy for 6 cycles. End of treatment PET/CT demonstrated a PR with near resolution in background PET avidity at the lesion. His symptoms of Horner syndrome also improved.
Conclusion
A diagnosis of aggressive lymphoma increases the risk of developing a secondary malignancy and providers should remain vigilant of this. Elderly individuals in whom aggressive therapies may be precluded can still greatly benefit from palliative targeted therapy even in the setting of diseases historically associated with a poor prognosis.
Background
Diffuse large B-cell lymphoma (DLBCL) is the most common aggressive type of non- Hodgkin lymphoma (NHL), comprising 30% of all NHL. Due to a decreased state of immunosurveillance resulting from the disease itself and its associated therapies, patients are at increased risk of developing a secondary malignancy. Multiple primary malignancies have been reported to occur in up to 15% of patients with DLBCL, retrospectively.
Herein, we review a case of a man with DLBCL who concomitantly developed ALK negative anaplastic large cell lymphoma (ALCL) distinctly of T-cell lineage who was successfully treated with palliative therapy for both diagnoses despite his advanced age and diagnosis associated with a poor prognosis with continued effect and sustained quality of life.
Case Report
An 88-year-old man presented with stage III DLBCL, diagnosed in 12/2018, was deemed not to be an aggressive therapy candidate. As such, he was treated with Rituximab monotherapy for 6 cycles, ending in 02/2019, with remarkably good effect. He remained in a PR with stable disease on serial PET/CTs until 09/2021, at which time he was noted to have Horner’s Syndrome in clinic. CT chest demonstrated a right apical lung mass, not previously seen on prior scans measuring 4.2 x 2.7 cm. Other sites of nodal disease remained stable on PET/CT.
Biopsy of the lesion revealed CD30+ ALK-negative ALCL with distinct T-cell marker positivity on immunohistochemistry and the absence of B-cell lineage markers. After discussion at our treatment planning conference, we decided to treat with brentuximab-vedotin (Bv) monotherapy for 6 cycles. End of treatment PET/CT demonstrated a PR with near resolution in background PET avidity at the lesion. His symptoms of Horner syndrome also improved.
Conclusion
A diagnosis of aggressive lymphoma increases the risk of developing a secondary malignancy and providers should remain vigilant of this. Elderly individuals in whom aggressive therapies may be precluded can still greatly benefit from palliative targeted therapy even in the setting of diseases historically associated with a poor prognosis.
Background
Diffuse large B-cell lymphoma (DLBCL) is the most common aggressive type of non- Hodgkin lymphoma (NHL), comprising 30% of all NHL. Due to a decreased state of immunosurveillance resulting from the disease itself and its associated therapies, patients are at increased risk of developing a secondary malignancy. Multiple primary malignancies have been reported to occur in up to 15% of patients with DLBCL, retrospectively.
Herein, we review a case of a man with DLBCL who concomitantly developed ALK negative anaplastic large cell lymphoma (ALCL) distinctly of T-cell lineage who was successfully treated with palliative therapy for both diagnoses despite his advanced age and diagnosis associated with a poor prognosis with continued effect and sustained quality of life.
Case Report
An 88-year-old man presented with stage III DLBCL, diagnosed in 12/2018, was deemed not to be an aggressive therapy candidate. As such, he was treated with Rituximab monotherapy for 6 cycles, ending in 02/2019, with remarkably good effect. He remained in a PR with stable disease on serial PET/CTs until 09/2021, at which time he was noted to have Horner’s Syndrome in clinic. CT chest demonstrated a right apical lung mass, not previously seen on prior scans measuring 4.2 x 2.7 cm. Other sites of nodal disease remained stable on PET/CT.
Biopsy of the lesion revealed CD30+ ALK-negative ALCL with distinct T-cell marker positivity on immunohistochemistry and the absence of B-cell lineage markers. After discussion at our treatment planning conference, we decided to treat with brentuximab-vedotin (Bv) monotherapy for 6 cycles. End of treatment PET/CT demonstrated a PR with near resolution in background PET avidity at the lesion. His symptoms of Horner syndrome also improved.
Conclusion
A diagnosis of aggressive lymphoma increases the risk of developing a secondary malignancy and providers should remain vigilant of this. Elderly individuals in whom aggressive therapies may be precluded can still greatly benefit from palliative targeted therapy even in the setting of diseases historically associated with a poor prognosis.
Impact of Pharmacist-Driven Telemedicine Services in Hematopoietic Stem Cell Transplant (HSCT) Long-term Care Clinic in a Veteran Population
Background
Patients undergoing allogeneic hematopoietic stem cell transplant (HSCT) are high-risk patients with complex medication regimens, including anti-rejection medications, infection prophylaxis, other post-transplant complication prophylaxis in addition to their chronic medications for co-morbid conditions. At the VA Tennessee Valley Healthcare System (TVHS), there are 3 stages of care once a patient receives an allogeneic transplant: inpatient transplant (through engraftment), outpatient posttransplant (through day +100), and long-term care (LTC) transplant (post-departure from the transplant facility). Currently, TVHS has 2 Clinical Pharmacist Practitioners (CPP) involved in the inpatient and outpatient settings. The purpose of this quality improvement initiative was to evaluate the impact of pharmacist services on continuity of care for longterm HSCT patients, vaccine completion rates, and immunosuppression/chemotherapy monitoring.
Methods
Patients were identified for enrollment based on a referral from a CPP, nurse practitioner (NP), or physician (MD). Patients with a history of allogeneic transplant were automatically referred from the CPP at departure and scheduled for a 2-week and 6-week post-departure visit. During these visits, the pharmacist conducted a medication reconciliation, assessed for medication errors or lapses in therapy, and provided medication counseling deemed necessary by clinical judgement. In addition to these 2 medication reconciliation visits, patients were also automatically scheduled for a vaccine assessment 6-months post-transplant. Pharmacy interventions from these visits were recorded in pre-specified categories. In addition to these predetermined visits, patients with complex medication regimens or undergoing significant changes could also be referred by either the NP or MD.
Results
A total of 18 patients were enrolled in the CPP clinic from October 2021 through May 2022. During this period, 42 visits were completed as each patient was seen multiple times (mean number of visits 1.8). A total of 16 medication errors/lapses were identified and addressed. The most common types of interventions included medication reconciliation (42), adherence counseling (39), general medication interventions (26), and vaccine interventions (20).
Conclusions
This pharmacist-driven telemedicine service incorporated into the long-term care HSCT clinic demonstrated benefit in identifying and addressing medication errors/lapses. Further study including the impact on patient outcomes such as hospital readmissions post-transplant, could strengthen the importance of pharmacy involvement in this setting.
Background
Patients undergoing allogeneic hematopoietic stem cell transplant (HSCT) are high-risk patients with complex medication regimens, including anti-rejection medications, infection prophylaxis, other post-transplant complication prophylaxis in addition to their chronic medications for co-morbid conditions. At the VA Tennessee Valley Healthcare System (TVHS), there are 3 stages of care once a patient receives an allogeneic transplant: inpatient transplant (through engraftment), outpatient posttransplant (through day +100), and long-term care (LTC) transplant (post-departure from the transplant facility). Currently, TVHS has 2 Clinical Pharmacist Practitioners (CPP) involved in the inpatient and outpatient settings. The purpose of this quality improvement initiative was to evaluate the impact of pharmacist services on continuity of care for longterm HSCT patients, vaccine completion rates, and immunosuppression/chemotherapy monitoring.
Methods
Patients were identified for enrollment based on a referral from a CPP, nurse practitioner (NP), or physician (MD). Patients with a history of allogeneic transplant were automatically referred from the CPP at departure and scheduled for a 2-week and 6-week post-departure visit. During these visits, the pharmacist conducted a medication reconciliation, assessed for medication errors or lapses in therapy, and provided medication counseling deemed necessary by clinical judgement. In addition to these 2 medication reconciliation visits, patients were also automatically scheduled for a vaccine assessment 6-months post-transplant. Pharmacy interventions from these visits were recorded in pre-specified categories. In addition to these predetermined visits, patients with complex medication regimens or undergoing significant changes could also be referred by either the NP or MD.
Results
A total of 18 patients were enrolled in the CPP clinic from October 2021 through May 2022. During this period, 42 visits were completed as each patient was seen multiple times (mean number of visits 1.8). A total of 16 medication errors/lapses were identified and addressed. The most common types of interventions included medication reconciliation (42), adherence counseling (39), general medication interventions (26), and vaccine interventions (20).
Conclusions
This pharmacist-driven telemedicine service incorporated into the long-term care HSCT clinic demonstrated benefit in identifying and addressing medication errors/lapses. Further study including the impact on patient outcomes such as hospital readmissions post-transplant, could strengthen the importance of pharmacy involvement in this setting.
Background
Patients undergoing allogeneic hematopoietic stem cell transplant (HSCT) are high-risk patients with complex medication regimens, including anti-rejection medications, infection prophylaxis, other post-transplant complication prophylaxis in addition to their chronic medications for co-morbid conditions. At the VA Tennessee Valley Healthcare System (TVHS), there are 3 stages of care once a patient receives an allogeneic transplant: inpatient transplant (through engraftment), outpatient posttransplant (through day +100), and long-term care (LTC) transplant (post-departure from the transplant facility). Currently, TVHS has 2 Clinical Pharmacist Practitioners (CPP) involved in the inpatient and outpatient settings. The purpose of this quality improvement initiative was to evaluate the impact of pharmacist services on continuity of care for longterm HSCT patients, vaccine completion rates, and immunosuppression/chemotherapy monitoring.
Methods
Patients were identified for enrollment based on a referral from a CPP, nurse practitioner (NP), or physician (MD). Patients with a history of allogeneic transplant were automatically referred from the CPP at departure and scheduled for a 2-week and 6-week post-departure visit. During these visits, the pharmacist conducted a medication reconciliation, assessed for medication errors or lapses in therapy, and provided medication counseling deemed necessary by clinical judgement. In addition to these 2 medication reconciliation visits, patients were also automatically scheduled for a vaccine assessment 6-months post-transplant. Pharmacy interventions from these visits were recorded in pre-specified categories. In addition to these predetermined visits, patients with complex medication regimens or undergoing significant changes could also be referred by either the NP or MD.
Results
A total of 18 patients were enrolled in the CPP clinic from October 2021 through May 2022. During this period, 42 visits were completed as each patient was seen multiple times (mean number of visits 1.8). A total of 16 medication errors/lapses were identified and addressed. The most common types of interventions included medication reconciliation (42), adherence counseling (39), general medication interventions (26), and vaccine interventions (20).
Conclusions
This pharmacist-driven telemedicine service incorporated into the long-term care HSCT clinic demonstrated benefit in identifying and addressing medication errors/lapses. Further study including the impact on patient outcomes such as hospital readmissions post-transplant, could strengthen the importance of pharmacy involvement in this setting.
Evaluating the Incidence of Febrile Neutropenia and the Appropriate Use of Prophylactic Granulocyte Colony Stimulating Factors in Veterans Who Received Treatment for Non- Hodgkin’s Lymphoma
Introduction
Febrile neutropenia (FN) is one of the most concerning complications associated with chemotherapy treatment, often leading to hospitalizations and delays in chemotherapy. The NCCN Guideline recommends primary prophylaxis with G-CSFs for patients receiving chemotherapy regimens that have an intermediate risk for FN if the patients have risk factors. A common intermediate risk for FN regimen is CHOP plus an anti-CD20 monoclonal antibody (mAb) for the treatment of non-Hodgkin’s lymphoma (NHL). At VASDHCS, an evaluation of the appropriate use of prophylactic GCSFs in this risk group would allow better optimization of patient care.
Objective
To evaluate the incidence of FN in correlation with the appropriate use of G-CSFs in patients who received CHOP plus an anti-CD20 mAb for the treatment of NHL
Methods
This is a retrospective study at VA San Diego of adult veterans with a confirmed diagnosis of NHL who received the first cycle of CHOP plus an anti- CD20 mAb between January 1, 2006, to October 1, 2021. Patients were categorized based on whether they received prophylactic G-CSF during the first cycle. The primary outcome measured was the incidence of FN in veterans with risk factor(s) who received CHOP plus an anti-CD20 mAb. The secondary outcome was the percentage of patients with risk factors who received G-CSF regardless of FN incidence. Primary outcome was analyzed using 2-tailed Fisher exact test.
Results
57 patients were included in the final analysis. In patients with at least one risk factor for FN, 26 (60%) received prophylactic G-CSF and 17 (40%) did not. There is 1 case of FN in the group that received G-CSF and 2 cases of FN in the group without G-CSF (RR, 0.33; P = .55; 95% CI, 0.03-3.33).
Conculsions
In patients receiving treatment for NHL with CHOP plus an anti-CD20 mAb, most of the patients with at least 1 risk factor for FN were initiated on G-CSF. Based on the results of the study, our veteran population does not appear to have an increased risk for FN without G-CSF. A larger study is warranted to further evaluate the significance of FN in correlation with prophylactic G-CSF.
Introduction
Febrile neutropenia (FN) is one of the most concerning complications associated with chemotherapy treatment, often leading to hospitalizations and delays in chemotherapy. The NCCN Guideline recommends primary prophylaxis with G-CSFs for patients receiving chemotherapy regimens that have an intermediate risk for FN if the patients have risk factors. A common intermediate risk for FN regimen is CHOP plus an anti-CD20 monoclonal antibody (mAb) for the treatment of non-Hodgkin’s lymphoma (NHL). At VASDHCS, an evaluation of the appropriate use of prophylactic GCSFs in this risk group would allow better optimization of patient care.
Objective
To evaluate the incidence of FN in correlation with the appropriate use of G-CSFs in patients who received CHOP plus an anti-CD20 mAb for the treatment of NHL
Methods
This is a retrospective study at VA San Diego of adult veterans with a confirmed diagnosis of NHL who received the first cycle of CHOP plus an anti- CD20 mAb between January 1, 2006, to October 1, 2021. Patients were categorized based on whether they received prophylactic G-CSF during the first cycle. The primary outcome measured was the incidence of FN in veterans with risk factor(s) who received CHOP plus an anti-CD20 mAb. The secondary outcome was the percentage of patients with risk factors who received G-CSF regardless of FN incidence. Primary outcome was analyzed using 2-tailed Fisher exact test.
Results
57 patients were included in the final analysis. In patients with at least one risk factor for FN, 26 (60%) received prophylactic G-CSF and 17 (40%) did not. There is 1 case of FN in the group that received G-CSF and 2 cases of FN in the group without G-CSF (RR, 0.33; P = .55; 95% CI, 0.03-3.33).
Conculsions
In patients receiving treatment for NHL with CHOP plus an anti-CD20 mAb, most of the patients with at least 1 risk factor for FN were initiated on G-CSF. Based on the results of the study, our veteran population does not appear to have an increased risk for FN without G-CSF. A larger study is warranted to further evaluate the significance of FN in correlation with prophylactic G-CSF.
Introduction
Febrile neutropenia (FN) is one of the most concerning complications associated with chemotherapy treatment, often leading to hospitalizations and delays in chemotherapy. The NCCN Guideline recommends primary prophylaxis with G-CSFs for patients receiving chemotherapy regimens that have an intermediate risk for FN if the patients have risk factors. A common intermediate risk for FN regimen is CHOP plus an anti-CD20 monoclonal antibody (mAb) for the treatment of non-Hodgkin’s lymphoma (NHL). At VASDHCS, an evaluation of the appropriate use of prophylactic GCSFs in this risk group would allow better optimization of patient care.
Objective
To evaluate the incidence of FN in correlation with the appropriate use of G-CSFs in patients who received CHOP plus an anti-CD20 mAb for the treatment of NHL
Methods
This is a retrospective study at VA San Diego of adult veterans with a confirmed diagnosis of NHL who received the first cycle of CHOP plus an anti- CD20 mAb between January 1, 2006, to October 1, 2021. Patients were categorized based on whether they received prophylactic G-CSF during the first cycle. The primary outcome measured was the incidence of FN in veterans with risk factor(s) who received CHOP plus an anti-CD20 mAb. The secondary outcome was the percentage of patients with risk factors who received G-CSF regardless of FN incidence. Primary outcome was analyzed using 2-tailed Fisher exact test.
Results
57 patients were included in the final analysis. In patients with at least one risk factor for FN, 26 (60%) received prophylactic G-CSF and 17 (40%) did not. There is 1 case of FN in the group that received G-CSF and 2 cases of FN in the group without G-CSF (RR, 0.33; P = .55; 95% CI, 0.03-3.33).
Conculsions
In patients receiving treatment for NHL with CHOP plus an anti-CD20 mAb, most of the patients with at least 1 risk factor for FN were initiated on G-CSF. Based on the results of the study, our veteran population does not appear to have an increased risk for FN without G-CSF. A larger study is warranted to further evaluate the significance of FN in correlation with prophylactic G-CSF.
A Rare Case of HHV8+ Multicentric Castleman Disease Presenting as Dermatitis
Introduction
Castleman disease (CD) is a rare non-neoplastic disorder presenting as lymphadenopathy. Skin involvement and progression to lymphomas are uncommon, and such presentation can pose a diagnostic challenge. We describe an interesting case of multicentric CD presenting as a rash.
Case Description
A 79-year-old male presented with a 1-year history of blanchable maculopapular rash and new onset dyspnea in the absence of fever, fatigue or weight loss. Examination revealed axillary, cervical and inguinal lymphadenopathy, and firm splenomegaly. Initial labs were notable for leukocytosis, occasional lymphoplasmacytic cells, anemia, thrombocytopenia, negative HIV screen, and elevated ESR and LDH. Further testing identified polyclonal hypergammaglobulinemia. CT scans revealed generalized lymphadenopathy, splenomegaly with infarcts and unilateral pleural effusion. An inguinal lymph node needle biopsy, skin biopsy and pleural fluid cytology were concerning for lymphoplasmacytic, so he was started on rituximab and bendamustine. However, B cell clonality could not be demonstrated, making these findings concerning for Castleman disease.
Results
Human herpesvirus 8 (HHV-8) testing performed on the inguinal lymph node sample came out positive, and he was diagnosed with HHV-8 positive multicentric Castleman disease and continued on weekly rituximab. He demonstrated an excellent response with complete resolution of rash, palpable lymphadenopathy and anemia after 4 cycles of treatment.
Discussion
Castleman disease (CD) is a rare disorder of polyclonal B cell proliferation classically presenting as lymphadenopathy with constitutional symptoms. Cutaneous presentations include eruptive angiomas or petechial rash but can be variable. Intrinsic or viral IL-6 play a key role in the pathogenesis of the disease. CD can be localised or multicentric (related to HHV-8 +/- HIV or idiopathic), and these subtypes differ in prognosis and management, with HIV and HHV-8 co-positivity indicating worse outcomes. While human IL-6 in unicentric and idiopathic multicentric disease respond well to IL-6 receptor antagonists, viral IL-6 in HHV-8 associated cases has a limited response. This is the rationale for preferring anti-CD20 therapy with rituximab in these patients.
Conculsions
Correct biopsy specimen, keen analysis of distinct pathologic features, and HHV-8 testing on tissue sample guide the diagnosis as HHV-8 serology can be falsely negative.
Introduction
Castleman disease (CD) is a rare non-neoplastic disorder presenting as lymphadenopathy. Skin involvement and progression to lymphomas are uncommon, and such presentation can pose a diagnostic challenge. We describe an interesting case of multicentric CD presenting as a rash.
Case Description
A 79-year-old male presented with a 1-year history of blanchable maculopapular rash and new onset dyspnea in the absence of fever, fatigue or weight loss. Examination revealed axillary, cervical and inguinal lymphadenopathy, and firm splenomegaly. Initial labs were notable for leukocytosis, occasional lymphoplasmacytic cells, anemia, thrombocytopenia, negative HIV screen, and elevated ESR and LDH. Further testing identified polyclonal hypergammaglobulinemia. CT scans revealed generalized lymphadenopathy, splenomegaly with infarcts and unilateral pleural effusion. An inguinal lymph node needle biopsy, skin biopsy and pleural fluid cytology were concerning for lymphoplasmacytic, so he was started on rituximab and bendamustine. However, B cell clonality could not be demonstrated, making these findings concerning for Castleman disease.
Results
Human herpesvirus 8 (HHV-8) testing performed on the inguinal lymph node sample came out positive, and he was diagnosed with HHV-8 positive multicentric Castleman disease and continued on weekly rituximab. He demonstrated an excellent response with complete resolution of rash, palpable lymphadenopathy and anemia after 4 cycles of treatment.
Discussion
Castleman disease (CD) is a rare disorder of polyclonal B cell proliferation classically presenting as lymphadenopathy with constitutional symptoms. Cutaneous presentations include eruptive angiomas or petechial rash but can be variable. Intrinsic or viral IL-6 play a key role in the pathogenesis of the disease. CD can be localised or multicentric (related to HHV-8 +/- HIV or idiopathic), and these subtypes differ in prognosis and management, with HIV and HHV-8 co-positivity indicating worse outcomes. While human IL-6 in unicentric and idiopathic multicentric disease respond well to IL-6 receptor antagonists, viral IL-6 in HHV-8 associated cases has a limited response. This is the rationale for preferring anti-CD20 therapy with rituximab in these patients.
Conculsions
Correct biopsy specimen, keen analysis of distinct pathologic features, and HHV-8 testing on tissue sample guide the diagnosis as HHV-8 serology can be falsely negative.
Introduction
Castleman disease (CD) is a rare non-neoplastic disorder presenting as lymphadenopathy. Skin involvement and progression to lymphomas are uncommon, and such presentation can pose a diagnostic challenge. We describe an interesting case of multicentric CD presenting as a rash.
Case Description
A 79-year-old male presented with a 1-year history of blanchable maculopapular rash and new onset dyspnea in the absence of fever, fatigue or weight loss. Examination revealed axillary, cervical and inguinal lymphadenopathy, and firm splenomegaly. Initial labs were notable for leukocytosis, occasional lymphoplasmacytic cells, anemia, thrombocytopenia, negative HIV screen, and elevated ESR and LDH. Further testing identified polyclonal hypergammaglobulinemia. CT scans revealed generalized lymphadenopathy, splenomegaly with infarcts and unilateral pleural effusion. An inguinal lymph node needle biopsy, skin biopsy and pleural fluid cytology were concerning for lymphoplasmacytic, so he was started on rituximab and bendamustine. However, B cell clonality could not be demonstrated, making these findings concerning for Castleman disease.
Results
Human herpesvirus 8 (HHV-8) testing performed on the inguinal lymph node sample came out positive, and he was diagnosed with HHV-8 positive multicentric Castleman disease and continued on weekly rituximab. He demonstrated an excellent response with complete resolution of rash, palpable lymphadenopathy and anemia after 4 cycles of treatment.
Discussion
Castleman disease (CD) is a rare disorder of polyclonal B cell proliferation classically presenting as lymphadenopathy with constitutional symptoms. Cutaneous presentations include eruptive angiomas or petechial rash but can be variable. Intrinsic or viral IL-6 play a key role in the pathogenesis of the disease. CD can be localised or multicentric (related to HHV-8 +/- HIV or idiopathic), and these subtypes differ in prognosis and management, with HIV and HHV-8 co-positivity indicating worse outcomes. While human IL-6 in unicentric and idiopathic multicentric disease respond well to IL-6 receptor antagonists, viral IL-6 in HHV-8 associated cases has a limited response. This is the rationale for preferring anti-CD20 therapy with rituximab in these patients.
Conculsions
Correct biopsy specimen, keen analysis of distinct pathologic features, and HHV-8 testing on tissue sample guide the diagnosis as HHV-8 serology can be falsely negative.
Agent Orange Exposure, Transformation From MGUS to Multiple Myeloma, and Outcomes in Veterans
Multiple myeloma (MM) accounts for 1% to 2% of all cancers and slightly more than 17% of hematologic malignancies in the United States.1 MM is characterized by the neoplastic proliferation of immunoglobulin (Ig)-producing plasma cells with ≥ 10% clonal plasma cells in the bone marrow or biopsy-proven bony or soft tissue plasmacytoma, plus presence of related organ or tissue impairment or presence of a biomarker associated with near-inevitable progression to end-organ damage.2
Background
Up to 97% of patients with MM will have a monoclonal (M) protein produced and secreted by the malignant plasma cells, which can be detected by protein electrophoresis of the serum and an aliquot of urine from a 24-hour collection combined with immunofixation of the serum and urine. The M protein in MM usually consists of IgG 50% of the time and light chains 16% of the time. Patients who lack detectable M protein are considered to have nonsecretory myeloma. MM presents with end-organ damage, which includes hypercalcemia, renal dysfunction, anemia, or lytic bone lesions. Patients with MM frequently present with renal insufficiency due to cast nephropathy or light chain deposition disease.3
MM is thought to evolve from monoclonal gammopathy of uncertain significance (MGUS), an asymptomatic premalignant stage of clonal plasma cell proliferation with a risk of progression to active myeloma at 1% per year.4,5 Epidemiologic data suggest that people who develop MM have a genetic predisposition, but risk factors may develop or be acquired, such as age, immunosuppression, and environmental exposures. To better assess what causes transformation from MGUS to MM, it is important to identify agents that may cause this second hit.6
In November 1961, President John F. Kennedy authorized the start of Operation Ranch Hand, the US Air Force’s herbicide program during the Vietnam War. Twenty million gallons of various chemicals were sprayed in Vietnam, eastern Laos, and parts of Cambodia to defoliate rural land, depriving guerillas of their support base. Agent Orange (AO) was one of these chemicals; it is a mixed herbicide with traces of dioxin, a compound that has been associated with major health problems among exposed individuals.7 Several studies have evaluated exposure to AO and its potential harmful repercussions. Studies have assessed the link between AO and MGUS as well as AO to various leukemias, such as chronic lymphocytic leukemia.8,9 Other studies have shown the relationship between AO exposure and worse outcomes in persons with MM.10 To date, only a single abstract from a US Department of Veterans Affairs (VA) medical center has investigated the relationships between AO exposure and MGUS, MM, and the rate of transformation. The VA study of patients seen from 2005 to 2015 in Detroit, Michigan, found that AO exposure led to an increase in cumulative incidence rate of MGUS/MM, suggesting possible changes in disease biology and genetics.11
In this study, we aimed to determine the incidence of transformation of MGUS to MM in patients with and without exposure to AO. We then analyzed survival as a function of AO exposure, transformation, and clinical and sociodemographic variables. We also explored the impact of psychosocial variables and hematopoietic stem cell transplantation (HSCT), a standard of treatment for MM.
Methods
This retrospective cohort study assembled electronic health record (EHR) data from the Veterans Health Administration Corporate Data Warehouse (CDW). The VA Central Texas Veterans Healthcare System Institutional Review Board granted a waiver of consent for this record review. Eligible patients were Vietnam-era veterans who were in the military during the time that AO was used (1961-1971). Veterans were included if they were being cared for and received a diagnosis for MGUS or MM between October 1, 2009, and September 30, 2015 (all prevalent cases fiscal years 2010-2015). Cases were excluded if there was illogical death data or if age, race, ethnicity, body mass index (BMI), or prior-year diagnostic data were missing.
Measures
Patients were followed through April 2020. Presence of MGUS was defined by the International Classification of Diseases, Ninth Revision (ICD-9) diagnosis code 273.1. MM was identified by ICD-9 diagnosis codes 203.00, 203.01, and 203.02. The study index date was the earliest date of diagnosis of MGUS or MM in fiscal years 2010-2015. It was suspected that some patients with MM may have had a history of MGUS prior to this period. Therefore, for patients with MM, historical diagnosis of MGUS was extracted going back through the earliest data in the CDW (October 1999). Patients diagnosed with both MGUS and MM were considered transformation patients.
Other measures included age at index date, sex, race, ethnicity, VA priority status (a value 1 to 8 summarizing why the veteran qualified for VA care, such as military service-connected disability or very low income), and AO exposure authenticated per VA enrollment files and disability records. Service years were separated into 1961 to 1968 and 1969 to 1971 to match a change in the formulation of AO associated with decreased carcinogenic effect. Comorbidity data from the year prior to first MGUS/MM diagnosis in the observation period were extracted. Lifestyle factors associated with development of MGUS/MM were determined using the following codes: obesity per BMI calculation or diagnosis (ICD-9, 278.0), tobacco use per diagnosis (ICD-9, 305.1, V15.82), and survival from MGUS/MM diagnosis index date to date of death from any cause. Comorbidity was assessed using ICD-9 diagnosis codes to calculate the Charlson Comorbidity Index (CCI), which includes cardiovascular diseases, diabetes mellitus, liver and kidney diseases, cancers, and metastatic solid tumors. Cancers were omitted from our adapted CCI to avoid collinearity in the multivariable models. The theoretical maximum CCI score in this study was 25.12,13 Additional conditions known to be associated with variation in outcomes among veterans using the VA were indicated, including major depressive disorder, posttraumatic stress disorder (PTSD), alcohol use disorder (AUD), substance use disorder (SUD), and common chronic disease (hypertension, lipid disorders).14
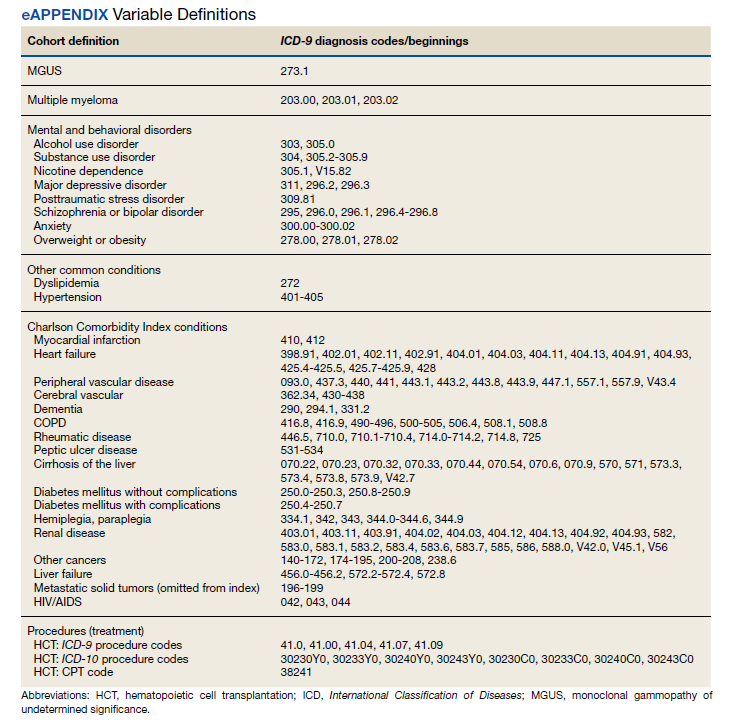
Treatment with autologous HSCT was defined by Current Procedural Terminology and ICD-9 Clinical Modification procedure codes for bone marrow and autologous HSCT occurring at any time in the CDW (eAppendix). Days elapsed from MM diagnosis to HSCT were calculated.
Statistical Analysis
Sample characteristics were represented by frequencies and percentages for categorical variables and means and SDs (or medians and ranges where appropriate) for continuous variables. A χ2 test (or Fisher exact test when cell counts were low) assessed associations in bivariate comparisons. A 2-sample t test (or Wilcoxon rank sum test as appropriate) assessed differences in continuous variables between 2 groups. Kaplan-Meier curves depicted the unadjusted relationship of AO exposure to survival. Cox proportional hazards survival models examined an unadjusted model containing only the AO exposure indicator as a predictor and adjusted models were used for demographic and clinical factors for MGUS and patients with MM separately.
Predictors were age in decades, sex, Hispanic ethnicity, race, nicotine dependence, obesity, overweight, AUD, SUD, major depressive disorder, PTSD, and the adapted CCI. When modeling patients with MM, MGUS was added to the model to identify the transformation group. The interaction of AO with transformation was also analyzed for patients with MM. Results were reported as hazard ratios (HR) with their 95% CI.
Results
We identified 18,215 veterans diagnosed with either MGUS or MM during fiscal years 2010-2015 with 16,366 meeting inclusion criteria. Patients were excluded for missing data on exposure (n = 334), age (n = 12), race (n = 1058), ethnicity (n = 164), diagnosis (n = 47), treatment (n = 56), and BMI (n = 178). All were Vietnam War era veterans; 14 also served in other eras.
The cohort was 98.5% male (Table 1). Twenty-nine percent were Black veterans, 65% were White veterans, and 4% of individuals reported Hispanic ethnicity. Patients had a mean (SD) age of 66.7 (5.9) years (range, 52-96). Most patients were married (58%) or divorced/separated (27%). All were VA priority 1 to 5 (no 6, 7, or 8); 50% were priority 1 with 50% to 100% service-connected disability. Another 29% were eligible for VA care by reason of low income, 17% had 10% to 40% service-connected disability, and 4% were otherwise disabled.
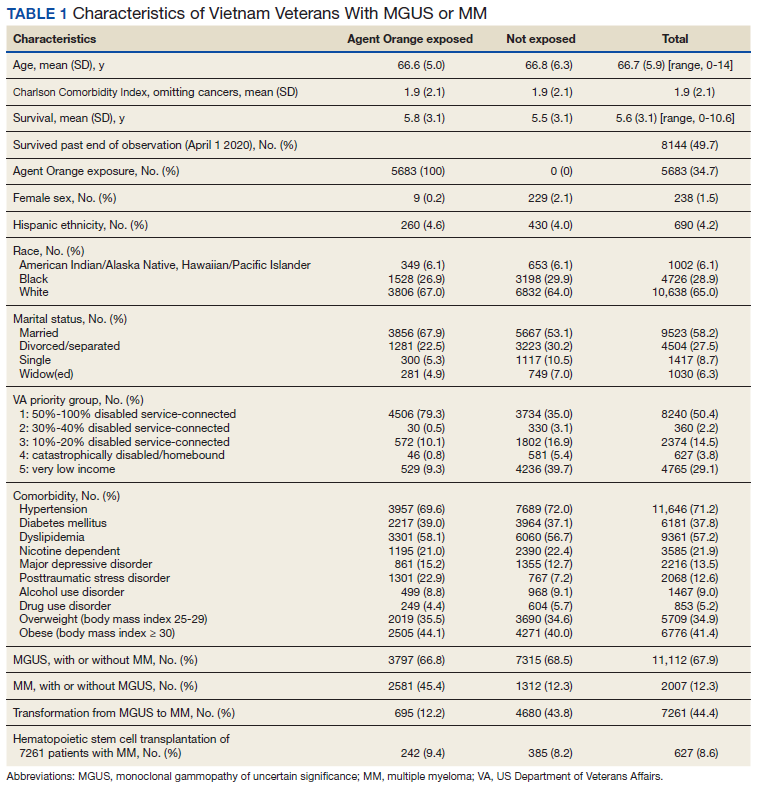
During fiscal years 2010 to 2015, 68% of our cohort had a diagnosis of MGUS (n = 11,112; 9105 had MGUS only), 44% had MM (n = 7261; 5254 had MM only), and 12% of these were transformation patients (n = 2007). AO exposure characterized 3102 MGUS-only patients (34%), 1886 MM-only patients (36%), and 695 transformation patients (35%) (χ2 = 4.92, P = .09). Among 5683 AO-exposed patients, 695 (12.2%) underwent MGUS-to-MM transformation. Among 10,683 nonexposed veterans, 1312 (12.3%) experienced transformation.
Comorbidity in the year leading up to the index MGUS/MM date determined using CCI was a mean (SD) of 1.9 (2.1) (range, 0-14). Among disorders not included in the CCI, 71% were diagnosed with hypertension, 57% with lipid disorders, 22% with nicotine dependence, 14% with major depressive disorder, 13% with PTSD, and 9% with AUD. Overweight (BMI 25 to < 30) and obesity (BMI ≥ 30) were common (35% and 41%, respectively). For 98% of patients, weight was measured within 90 days of their index MGUS/MM date. Most of the cohort (70%) were in Vietnam in 1961 to 1968.
HSCT was provided to 632 patients with MM (8.7%), including 441 patients who were treated after their index date and 219 patients treated before their index date. From fiscal years 2010 to 2015, the median (IQR) number of days from MM index date to HSCT receipt was 349 (243-650) days. Historical HSCT occurred a median (IQR) of 857 (353-1592) days before the index date, per data available back to October 1999; this median suggests long histories of MM in this cohort.
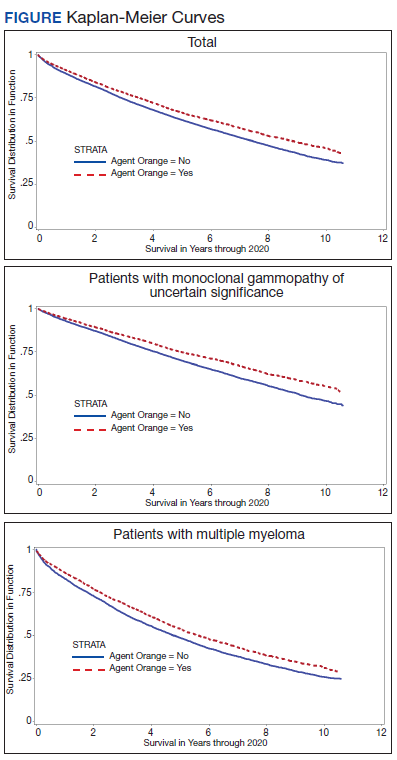
The unadjusted survival model found a very small inverse association of mortality with AO exposure in the total sample, meaning patients with documented AO exposure lived longer (HR, 0.85; 95% CI, 0.81-0.89; Table 2; Figure). Among 11,112 MGUS patients, AO was similarly associated with mortality (HR, 0.79; 95% CI, 0.74-0.84). The effect was also seen among 7269 patients with MM (HR, 0.86; 95% CI, 0.81-0.91).
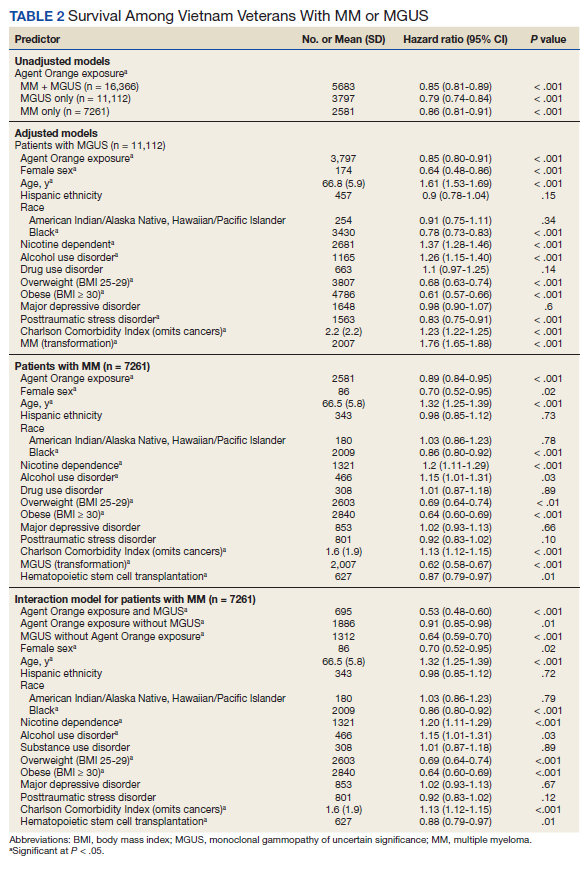
In the adjusted model of the total sample, the mortality hazard was greater for veterans who were older, with AUD and nicotine dependence, greater comorbidity per the CCI, diagnosis of MM, and transformation from MGUS to MM. Protective effects were noted for AO exposure, female sex, Black race, obesity, overweight, PTSD, and HSCT.
After adjusting for covariates, AO exposure was still associated with lower mortality among 11,112 patients with MGUS (HR, 0.85; 95% CI, 0.80-0.91). Risk factors were older age, nicotine dependence, AUD, the adapted CCI score (HR, 1.23 per point increase in the index; 95% CI, 1.22-1.25), and transformation to MM (HR, 1.76; 95% CI, 1.65-1.88). Additional protective factors were female sex, Black race, obesity, overweight, and PTSD.
After adjusting for covariates and limiting the analytic cohort to MM patients, the effect of AO exposure persisted (HR, 0.89; 95% CI, 0.84-0.95). Mortality risk factors were older age, nicotine dependence, AUD, and higher CCI score. Also protective were female sex, Black race, obesity, overweight, diagnosis of MGUS (transformation), and HSCT.
In the final model on patients with MM, the interaction term of AO exposure with transformation was significant. The combination of AO exposure with MGUS transformation had a greater protective effect than either AO exposure alone or MGUS without prior AO exposure. Additional protective factors were female sex, Black race, obesity, overweight, and HSCT. Older age, AUD, nicotine dependence, and greater comorbidity increased mortality risk.
Disscussion
Elucidating the pathophysiology and risk of transformation from MGUS to MM is an ongoing endeavor, even 35 years after the end of US involvement in the Vietnam War. Our study sought to understand a relationship between AO exposure, risk of MGUS transforming to MM, and associated mortality in US Vietnam War veterans. The rate of transformation (MGUS progressing to active MM) is well cited at 1% per year.15 Here, we found 12% of our cohort had undergone this transformation over 10 years.
Vietnam War era veterans who were exposed to AO during the Operation Ranch Hand period had 2.4 times greater risk of developing MGUS compared with veterans not exposed to AO.8 Our study was not designed to look at this association of AO exposure and MGUS/MM as this was a retrospective review to assess the difference in outcomes based on AO exposure. We found that AO exposure is associated with a decrease in mortality in contrast to a prior study showing worse survival with individuals with AO exposure.10 Another single center study found no association between AO exposure and overall survival, but it did identify an increased risk of progression from MGUS to MM.11 Our study did not show increased risk of transformation but did show positive effect on survival.
Black individuals have twice the risk of developing MM compared with White individuals and are diagnosed at a younger age (66 vs 70 years, respectively).16 Interestingly, Black race was a protective factor in our study. Given the length of time (35 years) elapsed since the Vietnam War ended, it is likely that most vulnerable Black veterans did not survive until our observation period.
HSCT, as expected, was a protective factor for veterans undergoing this treatment modality, but it is unclear why such a small number (8%) underwent HSCT as this is a standard of care in the management of MM. Obesity was also found to be a protective factor in a prior study, which was also seen in our study cohort.8
Limitations
This study was limited by its retrospective review of survivors among the Vietnam-era cohort several decades after the exposure of concern. Clinician notes and full historical data, such as date of onset for any disorder, were unavailable. These data also relied on the practitioners caring for the veterans to make the correct diagnosis with the associated code so that the data could be captured. Neither AO exposure nor diagnoses codes were verified against other sources of data; however, validation studies over the years have supported the accuracy of the diagnosis codes recorded in the VA EHR.
Conclusions
Because AO exposure is a nonmodifiable risk factor, focus should be placed on modifiable risk factors (eg, nicotine dependence, alcohol and substance use disorders, underlying comorbid conditions) as these were associated with worse outcomes. Future studies will look at the correlation of AO exposure, cytogenetics, and clinical outcomes in these veterans to learn how best to identify their disease course and optimize their care in the latter part of their life.
Acknowledgments
This research was supported by the Central Texas Veterans Health Care System and Baylor Scott and White Health, both in Temple and Veterans Affairs Central Western Massachusetts Healthcare System, Leeds.
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7-30. doi:10.3322/caac.21442
2. Rajkumar SV, Dimopoulos MA, Palumbo A, et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014;15(12):e538-e548. doi:10.1016/S1470-2045(14)70442-5
3. Kyle RA, Gertz MA, Witzig TE, et al. Review of 1027 patients with newly diagnosed multiple myeloma. Mayo Clin Proc. 2003;78(1):21-33. doi:10.4065/78.1.21
4. Kyle RA, Therneau TM, Rajkumar SV, et al. A long-term study of prognosis in monoclonal gammopathy of undetermined significance. N Engl J Med. 2002;346(8):564- 569. doi:10.1056/NEJMoa01133202
5. International Myeloma Foundation. What Are MGUS, smoldering and active myeloma? Updated June 6, 2021. Accessed June 20, 2022. https://www.myeloma .org/what-are-mgus-smm-mm
6. Riedel DA, Pottern LM. The epidemiology of multiple myeloma. Hematol Oncol Clin North Am. 1992;6(2):225-247. doi:10.1016/S0889-8588(18)30341-1
7. Buckingham Jr WA. Operation Ranch Hand: The Air Force and herbicides in southeast Asia, 1961-1971. Washington, DC: Office of Air Force History, United States Air Force; 1982. Accessed June 20, 2022. https://apps.dtic.mil/sti /pdfs/ADA121709.pdf
8. Landgren O, Shim YK, Michalek J, et al. Agent Orange exposure and monoclonal gammopathy of undetermined significance: an Operation Ranch Hand veteran cohort study. JAMA Oncol. 2015;1(8):1061-1068. doi:10.1001/jamaoncol.2015.2938
9. Mescher C, Gilbertson D, Randall NM, et al. The impact of Agent Orange exposure on prognosis and management in patients with chronic lymphocytic leukemia: a National Veteran Affairs Tumor Registry Study. Leuk Lymphoma. 2018;59(6):1348-1355. doi:10.1080/10428194.2017.1375109
10. Callander NS, Freytes CO, Luo S, Carson KR. Previous Agent Orange exposure is correlated with worse outcome in patients with multiple myeloma (MM) [abstract]. Blood. 2015;126(23):4194. doi:10.1182/blood.V126.23.4194.4194
11. Bumma N, Nagasaka M, Kim S, Vankayala HM, Ahmed S, Jasti P. Incidence of monoclonal gammopathy of undetermined significance (MGUS) and subsequent transformation to multiple myeloma (MM) and effect of exposure to Agent Orange (AO): a single center experience from VA Detroit [abstract]. Blood. 2017;130(suppl 1):5383. doi:10.1182/blood.V130.Suppl_1.5383.5383
12. Charlson ME, Pompei P, Ales KL, MacKenzie CR. A new method of classifying prognostic comorbidity in longitudinal studies: development and validation. J Chronic Dis. 1987;40(5):373-383. doi:10.1016/0021-9681(87)90171-8
13. Deyo RA, Cherkin DC, Ciol MA. Adapting a clinical comorbidity index for use with ICD-9-CM administrative databases. J Clin Epidemiol. 1992;45(6):613-619. doi:10.1016/0895-4356(92)90133-8
14. Copeland LA, Zeber JE, Sako EY, et al. Serious mental illnesses associated with receipt of surgery in retrospective analysis of patients in the Veterans Health Administration. BMC Surg. 2015;15:74. doi:10.1186/s12893-015-0064-7
15. Younes MA, Perez JD, Alirhayim Z, Ochoa C, Patel R, Dabak VS. MGUS Transformation into multiple myeloma in patients with solid organ transplantation [Abstract presented at American Society of Hematology Annual Meeting, November 15, 2013]. Blood. 2013;122(21):5325. doi:10.1182/blood.V122.21.5325.5325
16. Waxman AJ, Mink PJ, Devesa SS, et al. Racial disparities in incidence and outcome in multiple myeloma: a population- based study. Blood. 2010 Dec 16;116(25):5501-5506. doi:10.1182/blood-2010-07-298760
Multiple myeloma (MM) accounts for 1% to 2% of all cancers and slightly more than 17% of hematologic malignancies in the United States.1 MM is characterized by the neoplastic proliferation of immunoglobulin (Ig)-producing plasma cells with ≥ 10% clonal plasma cells in the bone marrow or biopsy-proven bony or soft tissue plasmacytoma, plus presence of related organ or tissue impairment or presence of a biomarker associated with near-inevitable progression to end-organ damage.2
Background
Up to 97% of patients with MM will have a monoclonal (M) protein produced and secreted by the malignant plasma cells, which can be detected by protein electrophoresis of the serum and an aliquot of urine from a 24-hour collection combined with immunofixation of the serum and urine. The M protein in MM usually consists of IgG 50% of the time and light chains 16% of the time. Patients who lack detectable M protein are considered to have nonsecretory myeloma. MM presents with end-organ damage, which includes hypercalcemia, renal dysfunction, anemia, or lytic bone lesions. Patients with MM frequently present with renal insufficiency due to cast nephropathy or light chain deposition disease.3
MM is thought to evolve from monoclonal gammopathy of uncertain significance (MGUS), an asymptomatic premalignant stage of clonal plasma cell proliferation with a risk of progression to active myeloma at 1% per year.4,5 Epidemiologic data suggest that people who develop MM have a genetic predisposition, but risk factors may develop or be acquired, such as age, immunosuppression, and environmental exposures. To better assess what causes transformation from MGUS to MM, it is important to identify agents that may cause this second hit.6
In November 1961, President John F. Kennedy authorized the start of Operation Ranch Hand, the US Air Force’s herbicide program during the Vietnam War. Twenty million gallons of various chemicals were sprayed in Vietnam, eastern Laos, and parts of Cambodia to defoliate rural land, depriving guerillas of their support base. Agent Orange (AO) was one of these chemicals; it is a mixed herbicide with traces of dioxin, a compound that has been associated with major health problems among exposed individuals.7 Several studies have evaluated exposure to AO and its potential harmful repercussions. Studies have assessed the link between AO and MGUS as well as AO to various leukemias, such as chronic lymphocytic leukemia.8,9 Other studies have shown the relationship between AO exposure and worse outcomes in persons with MM.10 To date, only a single abstract from a US Department of Veterans Affairs (VA) medical center has investigated the relationships between AO exposure and MGUS, MM, and the rate of transformation. The VA study of patients seen from 2005 to 2015 in Detroit, Michigan, found that AO exposure led to an increase in cumulative incidence rate of MGUS/MM, suggesting possible changes in disease biology and genetics.11
In this study, we aimed to determine the incidence of transformation of MGUS to MM in patients with and without exposure to AO. We then analyzed survival as a function of AO exposure, transformation, and clinical and sociodemographic variables. We also explored the impact of psychosocial variables and hematopoietic stem cell transplantation (HSCT), a standard of treatment for MM.
Methods
This retrospective cohort study assembled electronic health record (EHR) data from the Veterans Health Administration Corporate Data Warehouse (CDW). The VA Central Texas Veterans Healthcare System Institutional Review Board granted a waiver of consent for this record review. Eligible patients were Vietnam-era veterans who were in the military during the time that AO was used (1961-1971). Veterans were included if they were being cared for and received a diagnosis for MGUS or MM between October 1, 2009, and September 30, 2015 (all prevalent cases fiscal years 2010-2015). Cases were excluded if there was illogical death data or if age, race, ethnicity, body mass index (BMI), or prior-year diagnostic data were missing.
Measures
Patients were followed through April 2020. Presence of MGUS was defined by the International Classification of Diseases, Ninth Revision (ICD-9) diagnosis code 273.1. MM was identified by ICD-9 diagnosis codes 203.00, 203.01, and 203.02. The study index date was the earliest date of diagnosis of MGUS or MM in fiscal years 2010-2015. It was suspected that some patients with MM may have had a history of MGUS prior to this period. Therefore, for patients with MM, historical diagnosis of MGUS was extracted going back through the earliest data in the CDW (October 1999). Patients diagnosed with both MGUS and MM were considered transformation patients.
Other measures included age at index date, sex, race, ethnicity, VA priority status (a value 1 to 8 summarizing why the veteran qualified for VA care, such as military service-connected disability or very low income), and AO exposure authenticated per VA enrollment files and disability records. Service years were separated into 1961 to 1968 and 1969 to 1971 to match a change in the formulation of AO associated with decreased carcinogenic effect. Comorbidity data from the year prior to first MGUS/MM diagnosis in the observation period were extracted. Lifestyle factors associated with development of MGUS/MM were determined using the following codes: obesity per BMI calculation or diagnosis (ICD-9, 278.0), tobacco use per diagnosis (ICD-9, 305.1, V15.82), and survival from MGUS/MM diagnosis index date to date of death from any cause. Comorbidity was assessed using ICD-9 diagnosis codes to calculate the Charlson Comorbidity Index (CCI), which includes cardiovascular diseases, diabetes mellitus, liver and kidney diseases, cancers, and metastatic solid tumors. Cancers were omitted from our adapted CCI to avoid collinearity in the multivariable models. The theoretical maximum CCI score in this study was 25.12,13 Additional conditions known to be associated with variation in outcomes among veterans using the VA were indicated, including major depressive disorder, posttraumatic stress disorder (PTSD), alcohol use disorder (AUD), substance use disorder (SUD), and common chronic disease (hypertension, lipid disorders).14
Treatment with autologous HSCT was defined by Current Procedural Terminology and ICD-9 Clinical Modification procedure codes for bone marrow and autologous HSCT occurring at any time in the CDW (eAppendix). Days elapsed from MM diagnosis to HSCT were calculated.
Statistical Analysis
Sample characteristics were represented by frequencies and percentages for categorical variables and means and SDs (or medians and ranges where appropriate) for continuous variables. A χ2 test (or Fisher exact test when cell counts were low) assessed associations in bivariate comparisons. A 2-sample t test (or Wilcoxon rank sum test as appropriate) assessed differences in continuous variables between 2 groups. Kaplan-Meier curves depicted the unadjusted relationship of AO exposure to survival. Cox proportional hazards survival models examined an unadjusted model containing only the AO exposure indicator as a predictor and adjusted models were used for demographic and clinical factors for MGUS and patients with MM separately.
Predictors were age in decades, sex, Hispanic ethnicity, race, nicotine dependence, obesity, overweight, AUD, SUD, major depressive disorder, PTSD, and the adapted CCI. When modeling patients with MM, MGUS was added to the model to identify the transformation group. The interaction of AO with transformation was also analyzed for patients with MM. Results were reported as hazard ratios (HR) with their 95% CI.
Results
We identified 18,215 veterans diagnosed with either MGUS or MM during fiscal years 2010-2015 with 16,366 meeting inclusion criteria. Patients were excluded for missing data on exposure (n = 334), age (n = 12), race (n = 1058), ethnicity (n = 164), diagnosis (n = 47), treatment (n = 56), and BMI (n = 178). All were Vietnam War era veterans; 14 also served in other eras.
The cohort was 98.5% male (Table 1). Twenty-nine percent were Black veterans, 65% were White veterans, and 4% of individuals reported Hispanic ethnicity. Patients had a mean (SD) age of 66.7 (5.9) years (range, 52-96). Most patients were married (58%) or divorced/separated (27%). All were VA priority 1 to 5 (no 6, 7, or 8); 50% were priority 1 with 50% to 100% service-connected disability. Another 29% were eligible for VA care by reason of low income, 17% had 10% to 40% service-connected disability, and 4% were otherwise disabled.
During fiscal years 2010 to 2015, 68% of our cohort had a diagnosis of MGUS (n = 11,112; 9105 had MGUS only), 44% had MM (n = 7261; 5254 had MM only), and 12% of these were transformation patients (n = 2007). AO exposure characterized 3102 MGUS-only patients (34%), 1886 MM-only patients (36%), and 695 transformation patients (35%) (χ2 = 4.92, P = .09). Among 5683 AO-exposed patients, 695 (12.2%) underwent MGUS-to-MM transformation. Among 10,683 nonexposed veterans, 1312 (12.3%) experienced transformation.
Comorbidity in the year leading up to the index MGUS/MM date determined using CCI was a mean (SD) of 1.9 (2.1) (range, 0-14). Among disorders not included in the CCI, 71% were diagnosed with hypertension, 57% with lipid disorders, 22% with nicotine dependence, 14% with major depressive disorder, 13% with PTSD, and 9% with AUD. Overweight (BMI 25 to < 30) and obesity (BMI ≥ 30) were common (35% and 41%, respectively). For 98% of patients, weight was measured within 90 days of their index MGUS/MM date. Most of the cohort (70%) were in Vietnam in 1961 to 1968.
HSCT was provided to 632 patients with MM (8.7%), including 441 patients who were treated after their index date and 219 patients treated before their index date. From fiscal years 2010 to 2015, the median (IQR) number of days from MM index date to HSCT receipt was 349 (243-650) days. Historical HSCT occurred a median (IQR) of 857 (353-1592) days before the index date, per data available back to October 1999; this median suggests long histories of MM in this cohort.
The unadjusted survival model found a very small inverse association of mortality with AO exposure in the total sample, meaning patients with documented AO exposure lived longer (HR, 0.85; 95% CI, 0.81-0.89; Table 2; Figure). Among 11,112 MGUS patients, AO was similarly associated with mortality (HR, 0.79; 95% CI, 0.74-0.84). The effect was also seen among 7269 patients with MM (HR, 0.86; 95% CI, 0.81-0.91).
In the adjusted model of the total sample, the mortality hazard was greater for veterans who were older, with AUD and nicotine dependence, greater comorbidity per the CCI, diagnosis of MM, and transformation from MGUS to MM. Protective effects were noted for AO exposure, female sex, Black race, obesity, overweight, PTSD, and HSCT.
After adjusting for covariates, AO exposure was still associated with lower mortality among 11,112 patients with MGUS (HR, 0.85; 95% CI, 0.80-0.91). Risk factors were older age, nicotine dependence, AUD, the adapted CCI score (HR, 1.23 per point increase in the index; 95% CI, 1.22-1.25), and transformation to MM (HR, 1.76; 95% CI, 1.65-1.88). Additional protective factors were female sex, Black race, obesity, overweight, and PTSD.
After adjusting for covariates and limiting the analytic cohort to MM patients, the effect of AO exposure persisted (HR, 0.89; 95% CI, 0.84-0.95). Mortality risk factors were older age, nicotine dependence, AUD, and higher CCI score. Also protective were female sex, Black race, obesity, overweight, diagnosis of MGUS (transformation), and HSCT.
In the final model on patients with MM, the interaction term of AO exposure with transformation was significant. The combination of AO exposure with MGUS transformation had a greater protective effect than either AO exposure alone or MGUS without prior AO exposure. Additional protective factors were female sex, Black race, obesity, overweight, and HSCT. Older age, AUD, nicotine dependence, and greater comorbidity increased mortality risk.
Disscussion
Elucidating the pathophysiology and risk of transformation from MGUS to MM is an ongoing endeavor, even 35 years after the end of US involvement in the Vietnam War. Our study sought to understand a relationship between AO exposure, risk of MGUS transforming to MM, and associated mortality in US Vietnam War veterans. The rate of transformation (MGUS progressing to active MM) is well cited at 1% per year.15 Here, we found 12% of our cohort had undergone this transformation over 10 years.
Vietnam War era veterans who were exposed to AO during the Operation Ranch Hand period had 2.4 times greater risk of developing MGUS compared with veterans not exposed to AO.8 Our study was not designed to look at this association of AO exposure and MGUS/MM as this was a retrospective review to assess the difference in outcomes based on AO exposure. We found that AO exposure is associated with a decrease in mortality in contrast to a prior study showing worse survival with individuals with AO exposure.10 Another single center study found no association between AO exposure and overall survival, but it did identify an increased risk of progression from MGUS to MM.11 Our study did not show increased risk of transformation but did show positive effect on survival.
Black individuals have twice the risk of developing MM compared with White individuals and are diagnosed at a younger age (66 vs 70 years, respectively).16 Interestingly, Black race was a protective factor in our study. Given the length of time (35 years) elapsed since the Vietnam War ended, it is likely that most vulnerable Black veterans did not survive until our observation period.
HSCT, as expected, was a protective factor for veterans undergoing this treatment modality, but it is unclear why such a small number (8%) underwent HSCT as this is a standard of care in the management of MM. Obesity was also found to be a protective factor in a prior study, which was also seen in our study cohort.8
Limitations
This study was limited by its retrospective review of survivors among the Vietnam-era cohort several decades after the exposure of concern. Clinician notes and full historical data, such as date of onset for any disorder, were unavailable. These data also relied on the practitioners caring for the veterans to make the correct diagnosis with the associated code so that the data could be captured. Neither AO exposure nor diagnoses codes were verified against other sources of data; however, validation studies over the years have supported the accuracy of the diagnosis codes recorded in the VA EHR.
Conclusions
Because AO exposure is a nonmodifiable risk factor, focus should be placed on modifiable risk factors (eg, nicotine dependence, alcohol and substance use disorders, underlying comorbid conditions) as these were associated with worse outcomes. Future studies will look at the correlation of AO exposure, cytogenetics, and clinical outcomes in these veterans to learn how best to identify their disease course and optimize their care in the latter part of their life.
Acknowledgments
This research was supported by the Central Texas Veterans Health Care System and Baylor Scott and White Health, both in Temple and Veterans Affairs Central Western Massachusetts Healthcare System, Leeds.
Multiple myeloma (MM) accounts for 1% to 2% of all cancers and slightly more than 17% of hematologic malignancies in the United States.1 MM is characterized by the neoplastic proliferation of immunoglobulin (Ig)-producing plasma cells with ≥ 10% clonal plasma cells in the bone marrow or biopsy-proven bony or soft tissue plasmacytoma, plus presence of related organ or tissue impairment or presence of a biomarker associated with near-inevitable progression to end-organ damage.2
Background
Up to 97% of patients with MM will have a monoclonal (M) protein produced and secreted by the malignant plasma cells, which can be detected by protein electrophoresis of the serum and an aliquot of urine from a 24-hour collection combined with immunofixation of the serum and urine. The M protein in MM usually consists of IgG 50% of the time and light chains 16% of the time. Patients who lack detectable M protein are considered to have nonsecretory myeloma. MM presents with end-organ damage, which includes hypercalcemia, renal dysfunction, anemia, or lytic bone lesions. Patients with MM frequently present with renal insufficiency due to cast nephropathy or light chain deposition disease.3
MM is thought to evolve from monoclonal gammopathy of uncertain significance (MGUS), an asymptomatic premalignant stage of clonal plasma cell proliferation with a risk of progression to active myeloma at 1% per year.4,5 Epidemiologic data suggest that people who develop MM have a genetic predisposition, but risk factors may develop or be acquired, such as age, immunosuppression, and environmental exposures. To better assess what causes transformation from MGUS to MM, it is important to identify agents that may cause this second hit.6
In November 1961, President John F. Kennedy authorized the start of Operation Ranch Hand, the US Air Force’s herbicide program during the Vietnam War. Twenty million gallons of various chemicals were sprayed in Vietnam, eastern Laos, and parts of Cambodia to defoliate rural land, depriving guerillas of their support base. Agent Orange (AO) was one of these chemicals; it is a mixed herbicide with traces of dioxin, a compound that has been associated with major health problems among exposed individuals.7 Several studies have evaluated exposure to AO and its potential harmful repercussions. Studies have assessed the link between AO and MGUS as well as AO to various leukemias, such as chronic lymphocytic leukemia.8,9 Other studies have shown the relationship between AO exposure and worse outcomes in persons with MM.10 To date, only a single abstract from a US Department of Veterans Affairs (VA) medical center has investigated the relationships between AO exposure and MGUS, MM, and the rate of transformation. The VA study of patients seen from 2005 to 2015 in Detroit, Michigan, found that AO exposure led to an increase in cumulative incidence rate of MGUS/MM, suggesting possible changes in disease biology and genetics.11
In this study, we aimed to determine the incidence of transformation of MGUS to MM in patients with and without exposure to AO. We then analyzed survival as a function of AO exposure, transformation, and clinical and sociodemographic variables. We also explored the impact of psychosocial variables and hematopoietic stem cell transplantation (HSCT), a standard of treatment for MM.
Methods
This retrospective cohort study assembled electronic health record (EHR) data from the Veterans Health Administration Corporate Data Warehouse (CDW). The VA Central Texas Veterans Healthcare System Institutional Review Board granted a waiver of consent for this record review. Eligible patients were Vietnam-era veterans who were in the military during the time that AO was used (1961-1971). Veterans were included if they were being cared for and received a diagnosis for MGUS or MM between October 1, 2009, and September 30, 2015 (all prevalent cases fiscal years 2010-2015). Cases were excluded if there was illogical death data or if age, race, ethnicity, body mass index (BMI), or prior-year diagnostic data were missing.
Measures
Patients were followed through April 2020. Presence of MGUS was defined by the International Classification of Diseases, Ninth Revision (ICD-9) diagnosis code 273.1. MM was identified by ICD-9 diagnosis codes 203.00, 203.01, and 203.02. The study index date was the earliest date of diagnosis of MGUS or MM in fiscal years 2010-2015. It was suspected that some patients with MM may have had a history of MGUS prior to this period. Therefore, for patients with MM, historical diagnosis of MGUS was extracted going back through the earliest data in the CDW (October 1999). Patients diagnosed with both MGUS and MM were considered transformation patients.
Other measures included age at index date, sex, race, ethnicity, VA priority status (a value 1 to 8 summarizing why the veteran qualified for VA care, such as military service-connected disability or very low income), and AO exposure authenticated per VA enrollment files and disability records. Service years were separated into 1961 to 1968 and 1969 to 1971 to match a change in the formulation of AO associated with decreased carcinogenic effect. Comorbidity data from the year prior to first MGUS/MM diagnosis in the observation period were extracted. Lifestyle factors associated with development of MGUS/MM were determined using the following codes: obesity per BMI calculation or diagnosis (ICD-9, 278.0), tobacco use per diagnosis (ICD-9, 305.1, V15.82), and survival from MGUS/MM diagnosis index date to date of death from any cause. Comorbidity was assessed using ICD-9 diagnosis codes to calculate the Charlson Comorbidity Index (CCI), which includes cardiovascular diseases, diabetes mellitus, liver and kidney diseases, cancers, and metastatic solid tumors. Cancers were omitted from our adapted CCI to avoid collinearity in the multivariable models. The theoretical maximum CCI score in this study was 25.12,13 Additional conditions known to be associated with variation in outcomes among veterans using the VA were indicated, including major depressive disorder, posttraumatic stress disorder (PTSD), alcohol use disorder (AUD), substance use disorder (SUD), and common chronic disease (hypertension, lipid disorders).14
Treatment with autologous HSCT was defined by Current Procedural Terminology and ICD-9 Clinical Modification procedure codes for bone marrow and autologous HSCT occurring at any time in the CDW (eAppendix). Days elapsed from MM diagnosis to HSCT were calculated.
Statistical Analysis
Sample characteristics were represented by frequencies and percentages for categorical variables and means and SDs (or medians and ranges where appropriate) for continuous variables. A χ2 test (or Fisher exact test when cell counts were low) assessed associations in bivariate comparisons. A 2-sample t test (or Wilcoxon rank sum test as appropriate) assessed differences in continuous variables between 2 groups. Kaplan-Meier curves depicted the unadjusted relationship of AO exposure to survival. Cox proportional hazards survival models examined an unadjusted model containing only the AO exposure indicator as a predictor and adjusted models were used for demographic and clinical factors for MGUS and patients with MM separately.
Predictors were age in decades, sex, Hispanic ethnicity, race, nicotine dependence, obesity, overweight, AUD, SUD, major depressive disorder, PTSD, and the adapted CCI. When modeling patients with MM, MGUS was added to the model to identify the transformation group. The interaction of AO with transformation was also analyzed for patients with MM. Results were reported as hazard ratios (HR) with their 95% CI.
Results
We identified 18,215 veterans diagnosed with either MGUS or MM during fiscal years 2010-2015 with 16,366 meeting inclusion criteria. Patients were excluded for missing data on exposure (n = 334), age (n = 12), race (n = 1058), ethnicity (n = 164), diagnosis (n = 47), treatment (n = 56), and BMI (n = 178). All were Vietnam War era veterans; 14 also served in other eras.
The cohort was 98.5% male (Table 1). Twenty-nine percent were Black veterans, 65% were White veterans, and 4% of individuals reported Hispanic ethnicity. Patients had a mean (SD) age of 66.7 (5.9) years (range, 52-96). Most patients were married (58%) or divorced/separated (27%). All were VA priority 1 to 5 (no 6, 7, or 8); 50% were priority 1 with 50% to 100% service-connected disability. Another 29% were eligible for VA care by reason of low income, 17% had 10% to 40% service-connected disability, and 4% were otherwise disabled.
During fiscal years 2010 to 2015, 68% of our cohort had a diagnosis of MGUS (n = 11,112; 9105 had MGUS only), 44% had MM (n = 7261; 5254 had MM only), and 12% of these were transformation patients (n = 2007). AO exposure characterized 3102 MGUS-only patients (34%), 1886 MM-only patients (36%), and 695 transformation patients (35%) (χ2 = 4.92, P = .09). Among 5683 AO-exposed patients, 695 (12.2%) underwent MGUS-to-MM transformation. Among 10,683 nonexposed veterans, 1312 (12.3%) experienced transformation.
Comorbidity in the year leading up to the index MGUS/MM date determined using CCI was a mean (SD) of 1.9 (2.1) (range, 0-14). Among disorders not included in the CCI, 71% were diagnosed with hypertension, 57% with lipid disorders, 22% with nicotine dependence, 14% with major depressive disorder, 13% with PTSD, and 9% with AUD. Overweight (BMI 25 to < 30) and obesity (BMI ≥ 30) were common (35% and 41%, respectively). For 98% of patients, weight was measured within 90 days of their index MGUS/MM date. Most of the cohort (70%) were in Vietnam in 1961 to 1968.
HSCT was provided to 632 patients with MM (8.7%), including 441 patients who were treated after their index date and 219 patients treated before their index date. From fiscal years 2010 to 2015, the median (IQR) number of days from MM index date to HSCT receipt was 349 (243-650) days. Historical HSCT occurred a median (IQR) of 857 (353-1592) days before the index date, per data available back to October 1999; this median suggests long histories of MM in this cohort.
The unadjusted survival model found a very small inverse association of mortality with AO exposure in the total sample, meaning patients with documented AO exposure lived longer (HR, 0.85; 95% CI, 0.81-0.89; Table 2; Figure). Among 11,112 MGUS patients, AO was similarly associated with mortality (HR, 0.79; 95% CI, 0.74-0.84). The effect was also seen among 7269 patients with MM (HR, 0.86; 95% CI, 0.81-0.91).
In the adjusted model of the total sample, the mortality hazard was greater for veterans who were older, with AUD and nicotine dependence, greater comorbidity per the CCI, diagnosis of MM, and transformation from MGUS to MM. Protective effects were noted for AO exposure, female sex, Black race, obesity, overweight, PTSD, and HSCT.
After adjusting for covariates, AO exposure was still associated with lower mortality among 11,112 patients with MGUS (HR, 0.85; 95% CI, 0.80-0.91). Risk factors were older age, nicotine dependence, AUD, the adapted CCI score (HR, 1.23 per point increase in the index; 95% CI, 1.22-1.25), and transformation to MM (HR, 1.76; 95% CI, 1.65-1.88). Additional protective factors were female sex, Black race, obesity, overweight, and PTSD.
After adjusting for covariates and limiting the analytic cohort to MM patients, the effect of AO exposure persisted (HR, 0.89; 95% CI, 0.84-0.95). Mortality risk factors were older age, nicotine dependence, AUD, and higher CCI score. Also protective were female sex, Black race, obesity, overweight, diagnosis of MGUS (transformation), and HSCT.
In the final model on patients with MM, the interaction term of AO exposure with transformation was significant. The combination of AO exposure with MGUS transformation had a greater protective effect than either AO exposure alone or MGUS without prior AO exposure. Additional protective factors were female sex, Black race, obesity, overweight, and HSCT. Older age, AUD, nicotine dependence, and greater comorbidity increased mortality risk.
Disscussion
Elucidating the pathophysiology and risk of transformation from MGUS to MM is an ongoing endeavor, even 35 years after the end of US involvement in the Vietnam War. Our study sought to understand a relationship between AO exposure, risk of MGUS transforming to MM, and associated mortality in US Vietnam War veterans. The rate of transformation (MGUS progressing to active MM) is well cited at 1% per year.15 Here, we found 12% of our cohort had undergone this transformation over 10 years.
Vietnam War era veterans who were exposed to AO during the Operation Ranch Hand period had 2.4 times greater risk of developing MGUS compared with veterans not exposed to AO.8 Our study was not designed to look at this association of AO exposure and MGUS/MM as this was a retrospective review to assess the difference in outcomes based on AO exposure. We found that AO exposure is associated with a decrease in mortality in contrast to a prior study showing worse survival with individuals with AO exposure.10 Another single center study found no association between AO exposure and overall survival, but it did identify an increased risk of progression from MGUS to MM.11 Our study did not show increased risk of transformation but did show positive effect on survival.
Black individuals have twice the risk of developing MM compared with White individuals and are diagnosed at a younger age (66 vs 70 years, respectively).16 Interestingly, Black race was a protective factor in our study. Given the length of time (35 years) elapsed since the Vietnam War ended, it is likely that most vulnerable Black veterans did not survive until our observation period.
HSCT, as expected, was a protective factor for veterans undergoing this treatment modality, but it is unclear why such a small number (8%) underwent HSCT as this is a standard of care in the management of MM. Obesity was also found to be a protective factor in a prior study, which was also seen in our study cohort.8
Limitations
This study was limited by its retrospective review of survivors among the Vietnam-era cohort several decades after the exposure of concern. Clinician notes and full historical data, such as date of onset for any disorder, were unavailable. These data also relied on the practitioners caring for the veterans to make the correct diagnosis with the associated code so that the data could be captured. Neither AO exposure nor diagnoses codes were verified against other sources of data; however, validation studies over the years have supported the accuracy of the diagnosis codes recorded in the VA EHR.
Conclusions
Because AO exposure is a nonmodifiable risk factor, focus should be placed on modifiable risk factors (eg, nicotine dependence, alcohol and substance use disorders, underlying comorbid conditions) as these were associated with worse outcomes. Future studies will look at the correlation of AO exposure, cytogenetics, and clinical outcomes in these veterans to learn how best to identify their disease course and optimize their care in the latter part of their life.
Acknowledgments
This research was supported by the Central Texas Veterans Health Care System and Baylor Scott and White Health, both in Temple and Veterans Affairs Central Western Massachusetts Healthcare System, Leeds.
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7-30. doi:10.3322/caac.21442
2. Rajkumar SV, Dimopoulos MA, Palumbo A, et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014;15(12):e538-e548. doi:10.1016/S1470-2045(14)70442-5
3. Kyle RA, Gertz MA, Witzig TE, et al. Review of 1027 patients with newly diagnosed multiple myeloma. Mayo Clin Proc. 2003;78(1):21-33. doi:10.4065/78.1.21
4. Kyle RA, Therneau TM, Rajkumar SV, et al. A long-term study of prognosis in monoclonal gammopathy of undetermined significance. N Engl J Med. 2002;346(8):564- 569. doi:10.1056/NEJMoa01133202
5. International Myeloma Foundation. What Are MGUS, smoldering and active myeloma? Updated June 6, 2021. Accessed June 20, 2022. https://www.myeloma .org/what-are-mgus-smm-mm
6. Riedel DA, Pottern LM. The epidemiology of multiple myeloma. Hematol Oncol Clin North Am. 1992;6(2):225-247. doi:10.1016/S0889-8588(18)30341-1
7. Buckingham Jr WA. Operation Ranch Hand: The Air Force and herbicides in southeast Asia, 1961-1971. Washington, DC: Office of Air Force History, United States Air Force; 1982. Accessed June 20, 2022. https://apps.dtic.mil/sti /pdfs/ADA121709.pdf
8. Landgren O, Shim YK, Michalek J, et al. Agent Orange exposure and monoclonal gammopathy of undetermined significance: an Operation Ranch Hand veteran cohort study. JAMA Oncol. 2015;1(8):1061-1068. doi:10.1001/jamaoncol.2015.2938
9. Mescher C, Gilbertson D, Randall NM, et al. The impact of Agent Orange exposure on prognosis and management in patients with chronic lymphocytic leukemia: a National Veteran Affairs Tumor Registry Study. Leuk Lymphoma. 2018;59(6):1348-1355. doi:10.1080/10428194.2017.1375109
10. Callander NS, Freytes CO, Luo S, Carson KR. Previous Agent Orange exposure is correlated with worse outcome in patients with multiple myeloma (MM) [abstract]. Blood. 2015;126(23):4194. doi:10.1182/blood.V126.23.4194.4194
11. Bumma N, Nagasaka M, Kim S, Vankayala HM, Ahmed S, Jasti P. Incidence of monoclonal gammopathy of undetermined significance (MGUS) and subsequent transformation to multiple myeloma (MM) and effect of exposure to Agent Orange (AO): a single center experience from VA Detroit [abstract]. Blood. 2017;130(suppl 1):5383. doi:10.1182/blood.V130.Suppl_1.5383.5383
12. Charlson ME, Pompei P, Ales KL, MacKenzie CR. A new method of classifying prognostic comorbidity in longitudinal studies: development and validation. J Chronic Dis. 1987;40(5):373-383. doi:10.1016/0021-9681(87)90171-8
13. Deyo RA, Cherkin DC, Ciol MA. Adapting a clinical comorbidity index for use with ICD-9-CM administrative databases. J Clin Epidemiol. 1992;45(6):613-619. doi:10.1016/0895-4356(92)90133-8
14. Copeland LA, Zeber JE, Sako EY, et al. Serious mental illnesses associated with receipt of surgery in retrospective analysis of patients in the Veterans Health Administration. BMC Surg. 2015;15:74. doi:10.1186/s12893-015-0064-7
15. Younes MA, Perez JD, Alirhayim Z, Ochoa C, Patel R, Dabak VS. MGUS Transformation into multiple myeloma in patients with solid organ transplantation [Abstract presented at American Society of Hematology Annual Meeting, November 15, 2013]. Blood. 2013;122(21):5325. doi:10.1182/blood.V122.21.5325.5325
16. Waxman AJ, Mink PJ, Devesa SS, et al. Racial disparities in incidence and outcome in multiple myeloma: a population- based study. Blood. 2010 Dec 16;116(25):5501-5506. doi:10.1182/blood-2010-07-298760
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7-30. doi:10.3322/caac.21442
2. Rajkumar SV, Dimopoulos MA, Palumbo A, et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014;15(12):e538-e548. doi:10.1016/S1470-2045(14)70442-5
3. Kyle RA, Gertz MA, Witzig TE, et al. Review of 1027 patients with newly diagnosed multiple myeloma. Mayo Clin Proc. 2003;78(1):21-33. doi:10.4065/78.1.21
4. Kyle RA, Therneau TM, Rajkumar SV, et al. A long-term study of prognosis in monoclonal gammopathy of undetermined significance. N Engl J Med. 2002;346(8):564- 569. doi:10.1056/NEJMoa01133202
5. International Myeloma Foundation. What Are MGUS, smoldering and active myeloma? Updated June 6, 2021. Accessed June 20, 2022. https://www.myeloma .org/what-are-mgus-smm-mm
6. Riedel DA, Pottern LM. The epidemiology of multiple myeloma. Hematol Oncol Clin North Am. 1992;6(2):225-247. doi:10.1016/S0889-8588(18)30341-1
7. Buckingham Jr WA. Operation Ranch Hand: The Air Force and herbicides in southeast Asia, 1961-1971. Washington, DC: Office of Air Force History, United States Air Force; 1982. Accessed June 20, 2022. https://apps.dtic.mil/sti /pdfs/ADA121709.pdf
8. Landgren O, Shim YK, Michalek J, et al. Agent Orange exposure and monoclonal gammopathy of undetermined significance: an Operation Ranch Hand veteran cohort study. JAMA Oncol. 2015;1(8):1061-1068. doi:10.1001/jamaoncol.2015.2938
9. Mescher C, Gilbertson D, Randall NM, et al. The impact of Agent Orange exposure on prognosis and management in patients with chronic lymphocytic leukemia: a National Veteran Affairs Tumor Registry Study. Leuk Lymphoma. 2018;59(6):1348-1355. doi:10.1080/10428194.2017.1375109
10. Callander NS, Freytes CO, Luo S, Carson KR. Previous Agent Orange exposure is correlated with worse outcome in patients with multiple myeloma (MM) [abstract]. Blood. 2015;126(23):4194. doi:10.1182/blood.V126.23.4194.4194
11. Bumma N, Nagasaka M, Kim S, Vankayala HM, Ahmed S, Jasti P. Incidence of monoclonal gammopathy of undetermined significance (MGUS) and subsequent transformation to multiple myeloma (MM) and effect of exposure to Agent Orange (AO): a single center experience from VA Detroit [abstract]. Blood. 2017;130(suppl 1):5383. doi:10.1182/blood.V130.Suppl_1.5383.5383
12. Charlson ME, Pompei P, Ales KL, MacKenzie CR. A new method of classifying prognostic comorbidity in longitudinal studies: development and validation. J Chronic Dis. 1987;40(5):373-383. doi:10.1016/0021-9681(87)90171-8
13. Deyo RA, Cherkin DC, Ciol MA. Adapting a clinical comorbidity index for use with ICD-9-CM administrative databases. J Clin Epidemiol. 1992;45(6):613-619. doi:10.1016/0895-4356(92)90133-8
14. Copeland LA, Zeber JE, Sako EY, et al. Serious mental illnesses associated with receipt of surgery in retrospective analysis of patients in the Veterans Health Administration. BMC Surg. 2015;15:74. doi:10.1186/s12893-015-0064-7
15. Younes MA, Perez JD, Alirhayim Z, Ochoa C, Patel R, Dabak VS. MGUS Transformation into multiple myeloma in patients with solid organ transplantation [Abstract presented at American Society of Hematology Annual Meeting, November 15, 2013]. Blood. 2013;122(21):5325. doi:10.1182/blood.V122.21.5325.5325
16. Waxman AJ, Mink PJ, Devesa SS, et al. Racial disparities in incidence and outcome in multiple myeloma: a population- based study. Blood. 2010 Dec 16;116(25):5501-5506. doi:10.1182/blood-2010-07-298760
Fewer transplants for MM with quadruplet therapy?
“It is not a big leap of faith to imagine that, in the near future, with the availability of quadruplets and T-cell therapies, the role of high-dose melphalan and autologous stem cell transplant will be diminished,” said Dickran Kazandjian, MD, and Ola Landgren, MD, PhD, of the myeloma division, Sylvester Comprehensive Cancer Center, University of Miami.
They commented in a editorial in JAMA Oncology, prompted by a paper describing new results with a novel quadruple combination of therapies. These treatments included the monoclonal antibody elotuzumab (Empliciti) added onto the established backbone of carfilzomib (Kyprolis), lenalidomide (Revlimid), and dexamethasone (known as KRd).
“Regardless of what the future holds for elotuzumab-based combinations, it is clear that the new treatment paradigm of newly diagnosed MM will incorporate antibody-based quadruplet regimens,” the editorialists commented.
“Novel immunotherapies are here to stay,” they added, “as they are already transforming the lives of patients with multiple MM and bringing a bright horizon to the treatment landscape.”
Study details
The trial of the novel quadruplet regimen was a multicenter, single-arm, phase 2 study that involved 46 patients with newly diagnosed multiple myeloma, explain first author Benjamin A. Derman, MD, of the University of Chicago Medical Center, and colleagues.
These patients had a median age of 62; more than two-thirds were male (72%) and White (70%). About half (48%) had high-risk cytogenetic abnormalities.
All patients were treated with 12 cycles of the quadruple therapy Elo-KRd regimen. They underwent bone marrow assessment of measurable residual disease (MRD; with 10-5 sensitivity) after cycle 8 and cycle 12.
“An MRD-adapted treatment approach is rational because it may identify which patients can be administered shorter courses of intensive therapy without compromising efficacy,” the authors explained.
Patients who had MRD negativity at both time points did not receive further Elo-KRd, while patients who converted from MRD positivity to negativity in between cycles 8 and 12 received 6 additional cycles of Elo-KRd. Those who remained MRD positive or converted to positivity after 12 cycles received an additional 12 cycles of Elo-KRd.
Following Elo-KRd treatment, all patients transitioned to triple therapy with Elo-Rd (with no carfilzomib), for indefinite maintenance therapy or until disease progression.
For the primary endpoint, the rate of stringent complete response and/or MRD-negativity after cycle 8 was 58% (26 of 45), meeting the predefined definition of efficacy.
Importantly, 26% of patients converted from MRD positivity after cycle 8 to negativity at a later time point, while 50% of patients reached 1-year sustained MRD negativity.
Overall, the estimated 3-year, progression-free survival was 72%, and the rate was 92% for patients with MRD-negativity at cycle 8. The overall survival rate was 78%.
The most common grade 3 or 4 adverse events were lung and nonpulmonary infections (13% and 11%, respectively), and one patient had a grade 5 MI. Three patients discontinued the treatment because of intolerance.
“An MRD-adapted design using elotuzumab and weekly KRd without autologous stem cell transplantation showed a high rate of stringent complete response (sCR) and/or MRD-negativity and durable responses,” the authors wrote.
“This approach provides support for further evaluation of MRD-guided de-escalation of therapy to decrease treatment exposure while sustaining deep responses.”
To better assess the difference of the therapy versus treatment including stem cell transplantation, a phase 3, randomized trial is currently underway to compare the Elo-KRd regimen against KRd with autologous stem cell transplant in newly diagnosed MM.
“If Elo-KRd proves superior, a randomized comparison of Elo versus anti-CD38 mAb-based quadruplets would help determine the optimal combination of therapies in the frontline setting,” the authors noted.
Randomized trial anticipated to clarify benefit
In their editorial, Dr. Kazandjian and Dr. Landgren agreed with the authors that the role of elotuzumab needs to be better clarified in a randomized trial setting.
Elotuzumab received FDA approval in 2015 based on results from the ELOQUENT-2 study, which showed improved progression-free survival and overall survival with the addition of elotuzumab to lenalidomide and dexamethasone in patients with multiple myeloma who have previously received one to three other therapies.
However, the editorialists pointed out that recently published results from the randomized ELOQUENT-1 trial of lenalidomide and dexamethasone with and without elotuzumab showed the addition of elotuzumab was not associated with a statistically significant difference in progression-free survival.
The editorialists also pointed out that, in the setting of newly diagnosed multiple myeloma, another recent, similarly designed study found that the backbone regimen of carfilzomib, lenalidomide, and dexamethasone – on its own – was also associated with a favorable MRD-negative rate of 62%.
In addition, several studies involving novel quadruple treatments with the monoclonal antibody daratumumab (Darzalex) instead of elotuzumab, have also shown benefit in newly diagnosed multiple myeloma, resulting in high rates of MRD negativity.
Collectively, the findings bode well for the quadruple regimens in the treatment of MM, the editorialists emphasized.
“Importantly, with the rate of deep remissions observed with antibody-based quadruplet therapies, one may question the role of using early high-dose melphalan and autologous stem cell transplant in every patient, especially in those who have achieved MRD negativity with the quadruplet alone,” they added.
The study was sponsored in part by Amgen, Bristol-Myers Squibb, and the Multiple Myeloma Research Consortium. Dr. Derman reported advisory board fees from Sanofi, Janssen, and COTA Healthcare; honoraria from PleXus Communications and MJH Life Sciences. Dr. Kazandjian declares receiving advisory board or consulting fees from Bristol-Myers Squibb, Sanofi, and Arcellx outside the submitted work. Dr. Landgren has received grant support from numerous organizations and pharmaceutical companies. Dr. Landgren has also received honoraria for scientific talks/participated in advisory boards for Adaptive Biotech, Amgen, Binding Site, Bristol-Myers Squibb, Celgene, Cellectis, Glenmark, Janssen, Juno, and Pfizer, and served on independent data monitoring committees for international randomized trials by Takeda, Merck, Janssen, and Theradex.
A version of this article first appeared on Medscape.com.
“It is not a big leap of faith to imagine that, in the near future, with the availability of quadruplets and T-cell therapies, the role of high-dose melphalan and autologous stem cell transplant will be diminished,” said Dickran Kazandjian, MD, and Ola Landgren, MD, PhD, of the myeloma division, Sylvester Comprehensive Cancer Center, University of Miami.
They commented in a editorial in JAMA Oncology, prompted by a paper describing new results with a novel quadruple combination of therapies. These treatments included the monoclonal antibody elotuzumab (Empliciti) added onto the established backbone of carfilzomib (Kyprolis), lenalidomide (Revlimid), and dexamethasone (known as KRd).
“Regardless of what the future holds for elotuzumab-based combinations, it is clear that the new treatment paradigm of newly diagnosed MM will incorporate antibody-based quadruplet regimens,” the editorialists commented.
“Novel immunotherapies are here to stay,” they added, “as they are already transforming the lives of patients with multiple MM and bringing a bright horizon to the treatment landscape.”
Study details
The trial of the novel quadruplet regimen was a multicenter, single-arm, phase 2 study that involved 46 patients with newly diagnosed multiple myeloma, explain first author Benjamin A. Derman, MD, of the University of Chicago Medical Center, and colleagues.
These patients had a median age of 62; more than two-thirds were male (72%) and White (70%). About half (48%) had high-risk cytogenetic abnormalities.
All patients were treated with 12 cycles of the quadruple therapy Elo-KRd regimen. They underwent bone marrow assessment of measurable residual disease (MRD; with 10-5 sensitivity) after cycle 8 and cycle 12.
“An MRD-adapted treatment approach is rational because it may identify which patients can be administered shorter courses of intensive therapy without compromising efficacy,” the authors explained.
Patients who had MRD negativity at both time points did not receive further Elo-KRd, while patients who converted from MRD positivity to negativity in between cycles 8 and 12 received 6 additional cycles of Elo-KRd. Those who remained MRD positive or converted to positivity after 12 cycles received an additional 12 cycles of Elo-KRd.
Following Elo-KRd treatment, all patients transitioned to triple therapy with Elo-Rd (with no carfilzomib), for indefinite maintenance therapy or until disease progression.
For the primary endpoint, the rate of stringent complete response and/or MRD-negativity after cycle 8 was 58% (26 of 45), meeting the predefined definition of efficacy.
Importantly, 26% of patients converted from MRD positivity after cycle 8 to negativity at a later time point, while 50% of patients reached 1-year sustained MRD negativity.
Overall, the estimated 3-year, progression-free survival was 72%, and the rate was 92% for patients with MRD-negativity at cycle 8. The overall survival rate was 78%.
The most common grade 3 or 4 adverse events were lung and nonpulmonary infections (13% and 11%, respectively), and one patient had a grade 5 MI. Three patients discontinued the treatment because of intolerance.
“An MRD-adapted design using elotuzumab and weekly KRd without autologous stem cell transplantation showed a high rate of stringent complete response (sCR) and/or MRD-negativity and durable responses,” the authors wrote.
“This approach provides support for further evaluation of MRD-guided de-escalation of therapy to decrease treatment exposure while sustaining deep responses.”
To better assess the difference of the therapy versus treatment including stem cell transplantation, a phase 3, randomized trial is currently underway to compare the Elo-KRd regimen against KRd with autologous stem cell transplant in newly diagnosed MM.
“If Elo-KRd proves superior, a randomized comparison of Elo versus anti-CD38 mAb-based quadruplets would help determine the optimal combination of therapies in the frontline setting,” the authors noted.
Randomized trial anticipated to clarify benefit
In their editorial, Dr. Kazandjian and Dr. Landgren agreed with the authors that the role of elotuzumab needs to be better clarified in a randomized trial setting.
Elotuzumab received FDA approval in 2015 based on results from the ELOQUENT-2 study, which showed improved progression-free survival and overall survival with the addition of elotuzumab to lenalidomide and dexamethasone in patients with multiple myeloma who have previously received one to three other therapies.
However, the editorialists pointed out that recently published results from the randomized ELOQUENT-1 trial of lenalidomide and dexamethasone with and without elotuzumab showed the addition of elotuzumab was not associated with a statistically significant difference in progression-free survival.
The editorialists also pointed out that, in the setting of newly diagnosed multiple myeloma, another recent, similarly designed study found that the backbone regimen of carfilzomib, lenalidomide, and dexamethasone – on its own – was also associated with a favorable MRD-negative rate of 62%.
In addition, several studies involving novel quadruple treatments with the monoclonal antibody daratumumab (Darzalex) instead of elotuzumab, have also shown benefit in newly diagnosed multiple myeloma, resulting in high rates of MRD negativity.
Collectively, the findings bode well for the quadruple regimens in the treatment of MM, the editorialists emphasized.
“Importantly, with the rate of deep remissions observed with antibody-based quadruplet therapies, one may question the role of using early high-dose melphalan and autologous stem cell transplant in every patient, especially in those who have achieved MRD negativity with the quadruplet alone,” they added.
The study was sponsored in part by Amgen, Bristol-Myers Squibb, and the Multiple Myeloma Research Consortium. Dr. Derman reported advisory board fees from Sanofi, Janssen, and COTA Healthcare; honoraria from PleXus Communications and MJH Life Sciences. Dr. Kazandjian declares receiving advisory board or consulting fees from Bristol-Myers Squibb, Sanofi, and Arcellx outside the submitted work. Dr. Landgren has received grant support from numerous organizations and pharmaceutical companies. Dr. Landgren has also received honoraria for scientific talks/participated in advisory boards for Adaptive Biotech, Amgen, Binding Site, Bristol-Myers Squibb, Celgene, Cellectis, Glenmark, Janssen, Juno, and Pfizer, and served on independent data monitoring committees for international randomized trials by Takeda, Merck, Janssen, and Theradex.
A version of this article first appeared on Medscape.com.
“It is not a big leap of faith to imagine that, in the near future, with the availability of quadruplets and T-cell therapies, the role of high-dose melphalan and autologous stem cell transplant will be diminished,” said Dickran Kazandjian, MD, and Ola Landgren, MD, PhD, of the myeloma division, Sylvester Comprehensive Cancer Center, University of Miami.
They commented in a editorial in JAMA Oncology, prompted by a paper describing new results with a novel quadruple combination of therapies. These treatments included the monoclonal antibody elotuzumab (Empliciti) added onto the established backbone of carfilzomib (Kyprolis), lenalidomide (Revlimid), and dexamethasone (known as KRd).
“Regardless of what the future holds for elotuzumab-based combinations, it is clear that the new treatment paradigm of newly diagnosed MM will incorporate antibody-based quadruplet regimens,” the editorialists commented.
“Novel immunotherapies are here to stay,” they added, “as they are already transforming the lives of patients with multiple MM and bringing a bright horizon to the treatment landscape.”
Study details
The trial of the novel quadruplet regimen was a multicenter, single-arm, phase 2 study that involved 46 patients with newly diagnosed multiple myeloma, explain first author Benjamin A. Derman, MD, of the University of Chicago Medical Center, and colleagues.
These patients had a median age of 62; more than two-thirds were male (72%) and White (70%). About half (48%) had high-risk cytogenetic abnormalities.
All patients were treated with 12 cycles of the quadruple therapy Elo-KRd regimen. They underwent bone marrow assessment of measurable residual disease (MRD; with 10-5 sensitivity) after cycle 8 and cycle 12.
“An MRD-adapted treatment approach is rational because it may identify which patients can be administered shorter courses of intensive therapy without compromising efficacy,” the authors explained.
Patients who had MRD negativity at both time points did not receive further Elo-KRd, while patients who converted from MRD positivity to negativity in between cycles 8 and 12 received 6 additional cycles of Elo-KRd. Those who remained MRD positive or converted to positivity after 12 cycles received an additional 12 cycles of Elo-KRd.
Following Elo-KRd treatment, all patients transitioned to triple therapy with Elo-Rd (with no carfilzomib), for indefinite maintenance therapy or until disease progression.
For the primary endpoint, the rate of stringent complete response and/or MRD-negativity after cycle 8 was 58% (26 of 45), meeting the predefined definition of efficacy.
Importantly, 26% of patients converted from MRD positivity after cycle 8 to negativity at a later time point, while 50% of patients reached 1-year sustained MRD negativity.
Overall, the estimated 3-year, progression-free survival was 72%, and the rate was 92% for patients with MRD-negativity at cycle 8. The overall survival rate was 78%.
The most common grade 3 or 4 adverse events were lung and nonpulmonary infections (13% and 11%, respectively), and one patient had a grade 5 MI. Three patients discontinued the treatment because of intolerance.
“An MRD-adapted design using elotuzumab and weekly KRd without autologous stem cell transplantation showed a high rate of stringent complete response (sCR) and/or MRD-negativity and durable responses,” the authors wrote.
“This approach provides support for further evaluation of MRD-guided de-escalation of therapy to decrease treatment exposure while sustaining deep responses.”
To better assess the difference of the therapy versus treatment including stem cell transplantation, a phase 3, randomized trial is currently underway to compare the Elo-KRd regimen against KRd with autologous stem cell transplant in newly diagnosed MM.
“If Elo-KRd proves superior, a randomized comparison of Elo versus anti-CD38 mAb-based quadruplets would help determine the optimal combination of therapies in the frontline setting,” the authors noted.
Randomized trial anticipated to clarify benefit
In their editorial, Dr. Kazandjian and Dr. Landgren agreed with the authors that the role of elotuzumab needs to be better clarified in a randomized trial setting.
Elotuzumab received FDA approval in 2015 based on results from the ELOQUENT-2 study, which showed improved progression-free survival and overall survival with the addition of elotuzumab to lenalidomide and dexamethasone in patients with multiple myeloma who have previously received one to three other therapies.
However, the editorialists pointed out that recently published results from the randomized ELOQUENT-1 trial of lenalidomide and dexamethasone with and without elotuzumab showed the addition of elotuzumab was not associated with a statistically significant difference in progression-free survival.
The editorialists also pointed out that, in the setting of newly diagnosed multiple myeloma, another recent, similarly designed study found that the backbone regimen of carfilzomib, lenalidomide, and dexamethasone – on its own – was also associated with a favorable MRD-negative rate of 62%.
In addition, several studies involving novel quadruple treatments with the monoclonal antibody daratumumab (Darzalex) instead of elotuzumab, have also shown benefit in newly diagnosed multiple myeloma, resulting in high rates of MRD negativity.
Collectively, the findings bode well for the quadruple regimens in the treatment of MM, the editorialists emphasized.
“Importantly, with the rate of deep remissions observed with antibody-based quadruplet therapies, one may question the role of using early high-dose melphalan and autologous stem cell transplant in every patient, especially in those who have achieved MRD negativity with the quadruplet alone,” they added.
The study was sponsored in part by Amgen, Bristol-Myers Squibb, and the Multiple Myeloma Research Consortium. Dr. Derman reported advisory board fees from Sanofi, Janssen, and COTA Healthcare; honoraria from PleXus Communications and MJH Life Sciences. Dr. Kazandjian declares receiving advisory board or consulting fees from Bristol-Myers Squibb, Sanofi, and Arcellx outside the submitted work. Dr. Landgren has received grant support from numerous organizations and pharmaceutical companies. Dr. Landgren has also received honoraria for scientific talks/participated in advisory boards for Adaptive Biotech, Amgen, Binding Site, Bristol-Myers Squibb, Celgene, Cellectis, Glenmark, Janssen, Juno, and Pfizer, and served on independent data monitoring committees for international randomized trials by Takeda, Merck, Janssen, and Theradex.
A version of this article first appeared on Medscape.com.
FROM JAMA ONCOLOGY
Nodular Sclerosing Hodgkin Lymphoma With Paraneoplastic Cerebellar Degeneration
Paraneoplastic syndrome is a rare disorder involving manifestations of immune dysregulation triggered by malignancy. The immune system develops antibodies to the malignancy, which can cause cross reactivation with various tissues in the body, resulting in an autoimmune response. Paraneoplastic cerebellar degeneration (PCD) is a rare condition caused by immune-mediated damage to the Purkinje cells of the cerebellar tract. Symptoms may include gait instability, double vision, decreased fine motor skills, and ataxia, with progression to brainstem-associated symptoms, such as nystagmus, dysarthria, and dysphagia. Early detection and treatment of the underlying malignancy is critical to halt the progression of autoimmune-mediated destruction. We present a case of a young adult female patient with PCD caused by Purkinje cell cytoplasmic–Tr (PCA-Tr) antibody with Hodgkin lymphoma.
Case Presentation
A 20-year-old previously healthy active-duty female patient presented to the emergency department with acute worsening of chronic intermittent, recurrent episodes of lightheadedness and vertigo. Symptoms persisted for 9 months until acutely worsening over the 2 weeks prior to presentation. She reported left eye double vision but did not report seeing spots, photophobia, tinnitus, or headache. She felt off-balance, leaning on nearby objects to remain standing. Symptoms primarily occurred during ambulation; however, occasionally they happened at rest. Episodes lasted up to several minutes and occurred up to 15 times a day. The patient reported no fever, night sweats, unexplained weight loss, muscle aches, weakness, numbness or tingling, loss of bowel or bladder function, or rash. She had no recent illnesses, changes to medications, or recent travel. Oral intake to include food and water was adequate and unchanged. The patient had a remote history of mild concussions without loss of consciousness while playing sports 4 years previously. She reported no recent trauma. Nine months before, she received treatment for benign paroxysmal positional vertigo (BPPV) with the Epley maneuver with full resolution of symptoms lasting several days. She reported no prescription or over-the-counter medications, herbal remedies, or supplements. She reported no other medical or surgical history and no pertinent social or family history.
Physical examination revealed a nontoxic-appearing female patient with intermittent conversational dysarthria, saccadic pursuits, horizontal nystagmus with lateral gaze, and vertical nystagmus with vertical gaze. The patient exhibited dysdiadochokinesia, or impaired ability to perform rapid alternating hand movements with repetition. Finger-to-nose testing was impaired and heel-to-shin motion remained intact. A Romberg test was positive, and the patient had tandem gait instability. Strength testing, sensation, reflexes, and cranial nerves were otherwise intact. Initial laboratory testing was unremarkable except for mild normocytic anemia. Her infectious workup, including testing for venereal disease, HIV, COVID-19, and Coccidioidies was negative. Heavy metals analysis and urine drug screen were negative. Ophthalmology was consulted and workup revealed small amplitude downbeat nystagmus in primary gaze, sustained gaze evoked lateral beating jerk nystagmus with rebound nystagmus R>L gaze, but there was no evidence of afferent package defect and optic nerve function remained intact. Magnetic resonance imaging of the brain demonstrated cerebellar vermis hypoplasia with prominence of the superior cerebellar folia. Due to concerns for autoimmune encephalitis, a lumbar puncture was performed. Antibody testing revealed PCA-Tr antibodies, which is commonly associated with Hodgkin lymphoma, prompting further evaluation for malignancy.
Computed tomography (CT) of the chest with contrast demonstrated multiple mediastinal masses with a conglomeration of lymph nodes along the right paratracheal region. Further evaluation was performed with a positron emission tomography (PET)–CT, revealing a large conglomeration of hypermetabolic pretracheal, mediastinal, and right supraclavicular lymph that were suggestive of lymphoma. Mediastinoscopy with excisional lymph node biopsy was performed with immunohistochemical staining confirming diagnosis of a nodular sclerosing variant of Hodgkin lymphoma. The patient was treated with IV immunoglobulin at 0.4g/kg daily for 5 days. A central venous catheter was placed into the patient’s right internal jugular vein and a chemotherapy regimen of doxorubicin 46 mg, vinblastine 11 mg, bleomycin 19 units, and dacarbazine 700 mg was initiated. The patient’s symptoms improved with resolution of dysarthria; however, her visual impairment and gait instability persisted. Repeat PET-CT imaging 2 months later revealed interval improvement with decreased intensity and extent of the hypermetabolic lymph nodes and no new hypermetabolic foci.
Discussion
PCA-Tr antibodies affect the delta/notchlike epidermal growth factor–related receptor, expressed on the dendrites of cerebellar Purkinje cells.1 These fibers are the only output neurons of the cerebellar cortex and are critical to the coordination of motor movements, accounting for the ataxia experienced by patients with this subtype of PCD.2 The link between Hodgkin lymphoma and PCA-Tr antibodies has been established; however, most reports involve men with a median age of 61 years with lymphoma-associated symptoms (such as lymphadenopathy) or systemic symptoms (fever, night sweats, or weight loss) preceding neurologic manifestations in 80% of cases.3
Our patient was a young, previously healthy adult female who initially presented with vertigo, a common concern with frequently benign origins. Although there was temporary resolution of symptoms after Epley maneuvers, symptoms recurred and progressed over several months to include brainstem manifestations of nystagmus, diplopia, and dysarthria. Previous reports indicate that after remission of the Hodgkin lymphoma, PCA-Tr antibodies disappear and symptoms can improve or resolve.4,5 Treatment has just begun for our patient and although there has been initial clinical improvement, given the chronicity of symptoms, it is unclear if complete resolution will be achieved.
Conclusions
PCD can result in debilitating neurologic dysfunction and may be associated with malignancy such as Hodgkin lymphoma. This case offers unique insight due to the patient’s demographics and presentation, which involved brainstem pathology typically associated with late-onset disease and preceded by constitutional symptoms. Clinical suspicion of this rare disorder should be considered in all ages, especially if symptoms are progressive or neurologic manifestations arise, as early detection and treatment of the underlying malignancy are paramount to the prevention of significant disability.
1. de Graaff E, Maat P, Hulsenboom E, et al. Identification of delta/notch-like epidermal growth factor-related receptor as the Tr antigen in paraneoplastic cerebellar degeneration. Ann Neurol. 2012;71(6):815-824. doi:10.1002/ana.23550
2. MacKenzie-Graham A, Tiwari-Woodruff SK, Sharma G, et al. Purkinje cell loss in experimental autoimmune encephalomyelitis. Neuroimage. 2009;48(4):637-651. doi:10.1016/j.neuroimage.2009.06.073
3. Bernal F, Shams’ili S, Rojas I, et al. Anti-Tr antibodies as markers of paraneoplastic cerebellar degeneration and Hodgkin’s disease. Neurology. 2003;60(2):230-234. doi:10.1212/01.wnl.0000041495.87539.98
4. Graus F, Ariño H, Dalmau J. Paraneoplastic neurological syndromes in Hodgkin and non-Hodgkin lymphomas. Blood. 2014;123(21):3230-3238. doi:10.1182/blood-2014-03-537506
5. Aly R, Emmady PD. Paraneoplastic cerebellar degeneration. Updated May 8, 2022. Accessed March 30, 2022. https://www.ncbi.nlm.nih.gov/books/NBK560638
Paraneoplastic syndrome is a rare disorder involving manifestations of immune dysregulation triggered by malignancy. The immune system develops antibodies to the malignancy, which can cause cross reactivation with various tissues in the body, resulting in an autoimmune response. Paraneoplastic cerebellar degeneration (PCD) is a rare condition caused by immune-mediated damage to the Purkinje cells of the cerebellar tract. Symptoms may include gait instability, double vision, decreased fine motor skills, and ataxia, with progression to brainstem-associated symptoms, such as nystagmus, dysarthria, and dysphagia. Early detection and treatment of the underlying malignancy is critical to halt the progression of autoimmune-mediated destruction. We present a case of a young adult female patient with PCD caused by Purkinje cell cytoplasmic–Tr (PCA-Tr) antibody with Hodgkin lymphoma.
Case Presentation
A 20-year-old previously healthy active-duty female patient presented to the emergency department with acute worsening of chronic intermittent, recurrent episodes of lightheadedness and vertigo. Symptoms persisted for 9 months until acutely worsening over the 2 weeks prior to presentation. She reported left eye double vision but did not report seeing spots, photophobia, tinnitus, or headache. She felt off-balance, leaning on nearby objects to remain standing. Symptoms primarily occurred during ambulation; however, occasionally they happened at rest. Episodes lasted up to several minutes and occurred up to 15 times a day. The patient reported no fever, night sweats, unexplained weight loss, muscle aches, weakness, numbness or tingling, loss of bowel or bladder function, or rash. She had no recent illnesses, changes to medications, or recent travel. Oral intake to include food and water was adequate and unchanged. The patient had a remote history of mild concussions without loss of consciousness while playing sports 4 years previously. She reported no recent trauma. Nine months before, she received treatment for benign paroxysmal positional vertigo (BPPV) with the Epley maneuver with full resolution of symptoms lasting several days. She reported no prescription or over-the-counter medications, herbal remedies, or supplements. She reported no other medical or surgical history and no pertinent social or family history.
Physical examination revealed a nontoxic-appearing female patient with intermittent conversational dysarthria, saccadic pursuits, horizontal nystagmus with lateral gaze, and vertical nystagmus with vertical gaze. The patient exhibited dysdiadochokinesia, or impaired ability to perform rapid alternating hand movements with repetition. Finger-to-nose testing was impaired and heel-to-shin motion remained intact. A Romberg test was positive, and the patient had tandem gait instability. Strength testing, sensation, reflexes, and cranial nerves were otherwise intact. Initial laboratory testing was unremarkable except for mild normocytic anemia. Her infectious workup, including testing for venereal disease, HIV, COVID-19, and Coccidioidies was negative. Heavy metals analysis and urine drug screen were negative. Ophthalmology was consulted and workup revealed small amplitude downbeat nystagmus in primary gaze, sustained gaze evoked lateral beating jerk nystagmus with rebound nystagmus R>L gaze, but there was no evidence of afferent package defect and optic nerve function remained intact. Magnetic resonance imaging of the brain demonstrated cerebellar vermis hypoplasia with prominence of the superior cerebellar folia. Due to concerns for autoimmune encephalitis, a lumbar puncture was performed. Antibody testing revealed PCA-Tr antibodies, which is commonly associated with Hodgkin lymphoma, prompting further evaluation for malignancy.
Computed tomography (CT) of the chest with contrast demonstrated multiple mediastinal masses with a conglomeration of lymph nodes along the right paratracheal region. Further evaluation was performed with a positron emission tomography (PET)–CT, revealing a large conglomeration of hypermetabolic pretracheal, mediastinal, and right supraclavicular lymph that were suggestive of lymphoma. Mediastinoscopy with excisional lymph node biopsy was performed with immunohistochemical staining confirming diagnosis of a nodular sclerosing variant of Hodgkin lymphoma. The patient was treated with IV immunoglobulin at 0.4g/kg daily for 5 days. A central venous catheter was placed into the patient’s right internal jugular vein and a chemotherapy regimen of doxorubicin 46 mg, vinblastine 11 mg, bleomycin 19 units, and dacarbazine 700 mg was initiated. The patient’s symptoms improved with resolution of dysarthria; however, her visual impairment and gait instability persisted. Repeat PET-CT imaging 2 months later revealed interval improvement with decreased intensity and extent of the hypermetabolic lymph nodes and no new hypermetabolic foci.
Discussion
PCA-Tr antibodies affect the delta/notchlike epidermal growth factor–related receptor, expressed on the dendrites of cerebellar Purkinje cells.1 These fibers are the only output neurons of the cerebellar cortex and are critical to the coordination of motor movements, accounting for the ataxia experienced by patients with this subtype of PCD.2 The link between Hodgkin lymphoma and PCA-Tr antibodies has been established; however, most reports involve men with a median age of 61 years with lymphoma-associated symptoms (such as lymphadenopathy) or systemic symptoms (fever, night sweats, or weight loss) preceding neurologic manifestations in 80% of cases.3
Our patient was a young, previously healthy adult female who initially presented with vertigo, a common concern with frequently benign origins. Although there was temporary resolution of symptoms after Epley maneuvers, symptoms recurred and progressed over several months to include brainstem manifestations of nystagmus, diplopia, and dysarthria. Previous reports indicate that after remission of the Hodgkin lymphoma, PCA-Tr antibodies disappear and symptoms can improve or resolve.4,5 Treatment has just begun for our patient and although there has been initial clinical improvement, given the chronicity of symptoms, it is unclear if complete resolution will be achieved.
Conclusions
PCD can result in debilitating neurologic dysfunction and may be associated with malignancy such as Hodgkin lymphoma. This case offers unique insight due to the patient’s demographics and presentation, which involved brainstem pathology typically associated with late-onset disease and preceded by constitutional symptoms. Clinical suspicion of this rare disorder should be considered in all ages, especially if symptoms are progressive or neurologic manifestations arise, as early detection and treatment of the underlying malignancy are paramount to the prevention of significant disability.
Paraneoplastic syndrome is a rare disorder involving manifestations of immune dysregulation triggered by malignancy. The immune system develops antibodies to the malignancy, which can cause cross reactivation with various tissues in the body, resulting in an autoimmune response. Paraneoplastic cerebellar degeneration (PCD) is a rare condition caused by immune-mediated damage to the Purkinje cells of the cerebellar tract. Symptoms may include gait instability, double vision, decreased fine motor skills, and ataxia, with progression to brainstem-associated symptoms, such as nystagmus, dysarthria, and dysphagia. Early detection and treatment of the underlying malignancy is critical to halt the progression of autoimmune-mediated destruction. We present a case of a young adult female patient with PCD caused by Purkinje cell cytoplasmic–Tr (PCA-Tr) antibody with Hodgkin lymphoma.
Case Presentation
A 20-year-old previously healthy active-duty female patient presented to the emergency department with acute worsening of chronic intermittent, recurrent episodes of lightheadedness and vertigo. Symptoms persisted for 9 months until acutely worsening over the 2 weeks prior to presentation. She reported left eye double vision but did not report seeing spots, photophobia, tinnitus, or headache. She felt off-balance, leaning on nearby objects to remain standing. Symptoms primarily occurred during ambulation; however, occasionally they happened at rest. Episodes lasted up to several minutes and occurred up to 15 times a day. The patient reported no fever, night sweats, unexplained weight loss, muscle aches, weakness, numbness or tingling, loss of bowel or bladder function, or rash. She had no recent illnesses, changes to medications, or recent travel. Oral intake to include food and water was adequate and unchanged. The patient had a remote history of mild concussions without loss of consciousness while playing sports 4 years previously. She reported no recent trauma. Nine months before, she received treatment for benign paroxysmal positional vertigo (BPPV) with the Epley maneuver with full resolution of symptoms lasting several days. She reported no prescription or over-the-counter medications, herbal remedies, or supplements. She reported no other medical or surgical history and no pertinent social or family history.
Physical examination revealed a nontoxic-appearing female patient with intermittent conversational dysarthria, saccadic pursuits, horizontal nystagmus with lateral gaze, and vertical nystagmus with vertical gaze. The patient exhibited dysdiadochokinesia, or impaired ability to perform rapid alternating hand movements with repetition. Finger-to-nose testing was impaired and heel-to-shin motion remained intact. A Romberg test was positive, and the patient had tandem gait instability. Strength testing, sensation, reflexes, and cranial nerves were otherwise intact. Initial laboratory testing was unremarkable except for mild normocytic anemia. Her infectious workup, including testing for venereal disease, HIV, COVID-19, and Coccidioidies was negative. Heavy metals analysis and urine drug screen were negative. Ophthalmology was consulted and workup revealed small amplitude downbeat nystagmus in primary gaze, sustained gaze evoked lateral beating jerk nystagmus with rebound nystagmus R>L gaze, but there was no evidence of afferent package defect and optic nerve function remained intact. Magnetic resonance imaging of the brain demonstrated cerebellar vermis hypoplasia with prominence of the superior cerebellar folia. Due to concerns for autoimmune encephalitis, a lumbar puncture was performed. Antibody testing revealed PCA-Tr antibodies, which is commonly associated with Hodgkin lymphoma, prompting further evaluation for malignancy.
Computed tomography (CT) of the chest with contrast demonstrated multiple mediastinal masses with a conglomeration of lymph nodes along the right paratracheal region. Further evaluation was performed with a positron emission tomography (PET)–CT, revealing a large conglomeration of hypermetabolic pretracheal, mediastinal, and right supraclavicular lymph that were suggestive of lymphoma. Mediastinoscopy with excisional lymph node biopsy was performed with immunohistochemical staining confirming diagnosis of a nodular sclerosing variant of Hodgkin lymphoma. The patient was treated with IV immunoglobulin at 0.4g/kg daily for 5 days. A central venous catheter was placed into the patient’s right internal jugular vein and a chemotherapy regimen of doxorubicin 46 mg, vinblastine 11 mg, bleomycin 19 units, and dacarbazine 700 mg was initiated. The patient’s symptoms improved with resolution of dysarthria; however, her visual impairment and gait instability persisted. Repeat PET-CT imaging 2 months later revealed interval improvement with decreased intensity and extent of the hypermetabolic lymph nodes and no new hypermetabolic foci.
Discussion
PCA-Tr antibodies affect the delta/notchlike epidermal growth factor–related receptor, expressed on the dendrites of cerebellar Purkinje cells.1 These fibers are the only output neurons of the cerebellar cortex and are critical to the coordination of motor movements, accounting for the ataxia experienced by patients with this subtype of PCD.2 The link between Hodgkin lymphoma and PCA-Tr antibodies has been established; however, most reports involve men with a median age of 61 years with lymphoma-associated symptoms (such as lymphadenopathy) or systemic symptoms (fever, night sweats, or weight loss) preceding neurologic manifestations in 80% of cases.3
Our patient was a young, previously healthy adult female who initially presented with vertigo, a common concern with frequently benign origins. Although there was temporary resolution of symptoms after Epley maneuvers, symptoms recurred and progressed over several months to include brainstem manifestations of nystagmus, diplopia, and dysarthria. Previous reports indicate that after remission of the Hodgkin lymphoma, PCA-Tr antibodies disappear and symptoms can improve or resolve.4,5 Treatment has just begun for our patient and although there has been initial clinical improvement, given the chronicity of symptoms, it is unclear if complete resolution will be achieved.
Conclusions
PCD can result in debilitating neurologic dysfunction and may be associated with malignancy such as Hodgkin lymphoma. This case offers unique insight due to the patient’s demographics and presentation, which involved brainstem pathology typically associated with late-onset disease and preceded by constitutional symptoms. Clinical suspicion of this rare disorder should be considered in all ages, especially if symptoms are progressive or neurologic manifestations arise, as early detection and treatment of the underlying malignancy are paramount to the prevention of significant disability.
1. de Graaff E, Maat P, Hulsenboom E, et al. Identification of delta/notch-like epidermal growth factor-related receptor as the Tr antigen in paraneoplastic cerebellar degeneration. Ann Neurol. 2012;71(6):815-824. doi:10.1002/ana.23550
2. MacKenzie-Graham A, Tiwari-Woodruff SK, Sharma G, et al. Purkinje cell loss in experimental autoimmune encephalomyelitis. Neuroimage. 2009;48(4):637-651. doi:10.1016/j.neuroimage.2009.06.073
3. Bernal F, Shams’ili S, Rojas I, et al. Anti-Tr antibodies as markers of paraneoplastic cerebellar degeneration and Hodgkin’s disease. Neurology. 2003;60(2):230-234. doi:10.1212/01.wnl.0000041495.87539.98
4. Graus F, Ariño H, Dalmau J. Paraneoplastic neurological syndromes in Hodgkin and non-Hodgkin lymphomas. Blood. 2014;123(21):3230-3238. doi:10.1182/blood-2014-03-537506
5. Aly R, Emmady PD. Paraneoplastic cerebellar degeneration. Updated May 8, 2022. Accessed March 30, 2022. https://www.ncbi.nlm.nih.gov/books/NBK560638
1. de Graaff E, Maat P, Hulsenboom E, et al. Identification of delta/notch-like epidermal growth factor-related receptor as the Tr antigen in paraneoplastic cerebellar degeneration. Ann Neurol. 2012;71(6):815-824. doi:10.1002/ana.23550
2. MacKenzie-Graham A, Tiwari-Woodruff SK, Sharma G, et al. Purkinje cell loss in experimental autoimmune encephalomyelitis. Neuroimage. 2009;48(4):637-651. doi:10.1016/j.neuroimage.2009.06.073
3. Bernal F, Shams’ili S, Rojas I, et al. Anti-Tr antibodies as markers of paraneoplastic cerebellar degeneration and Hodgkin’s disease. Neurology. 2003;60(2):230-234. doi:10.1212/01.wnl.0000041495.87539.98
4. Graus F, Ariño H, Dalmau J. Paraneoplastic neurological syndromes in Hodgkin and non-Hodgkin lymphomas. Blood. 2014;123(21):3230-3238. doi:10.1182/blood-2014-03-537506
5. Aly R, Emmady PD. Paraneoplastic cerebellar degeneration. Updated May 8, 2022. Accessed March 30, 2022. https://www.ncbi.nlm.nih.gov/books/NBK560638
Approach to Pancytopenia in a Deployed Service Member
Pancytopenia is a condition in which all 3 hematologic cell lines are lower than expected in the blood, often representing either an increase in cellular destruction or decrease in bone marrow production. Destruction often occurs in the setting of autoimmune conditions (eg, systemic lupus erythematosus, rheumatoid arthritis) or splenic sequestration, often affecting erythrocytes and platelets more than leukocytes. Decreased production represents central etiologies, which are often due to nutritional deficiencies, infections, drug toxicities, or malabsorption.1 Pancytopenia secondary to vitamin B12 deficiency is rare, accounting for about 5% of the hematologic manifestations of symptomatic vitamin B12 deficient patients.2
Pernicious anemia, named for a once lethal disease, is a form of vitamin B12 (cobalamin) deficiency that results from an autoimmune (type II hypersensitivity) reaction to gastric parietal cells or intrinsic factor. Antibodies bind to gastric parietal cells and reduce gastric acid production, leading to atrophic gastritis, or they bind intrinsic factor and block the binding and absorption of vitamin B12 in the gastrointestinal tract. While first described in the 1820s, it was not until a century later when scientists were studying hematopoiesis in response to the heavy casualty burden from battlefield exsanguination in World War I that dogs fed raw liver were noted to have significantly better blood regeneration response than those fed cooked liver. This discovery led physicians Minot and Murphy to use raw liver to treat pernicious anemia and found that jaundice improved, reticulocyte counts increased, and hemoglobin (Hb) concentration improved, resulting in the duo becoming the first American recipients of the Nobel Prize in physiology or medicine.3 It was ultimately determined in 1948 by chemists Folkers and Todd that the active ingredient in raw liver responsible for this phenomenon was vitamin B12.4

Patients with pernicious anemia typically present with macrocytic anemia, low reticulocyte count, hypersegmented neutrophils, as well as mild leukopenia and/or thrombocytopenia, distinguishable from folate deficiency by an elevated serum methylmalonic acid level. World Health Organization cytopenia thresholds are listed in Table 1.5 Treatment consists of lifelong vitamin B12 supplementation, and endoscopic screening is often recommended after diagnosis due to increased risk of gastrointestinal malignancy.6 Pernicious anemia can be difficult to distinguish from thrombotic thrombocytopenia purpura (TTP), a microangiopathic hemolytic anemia that can cause rapid end-organ failure and death if treatment is delayed.7 While pernicious anemia is not typically hemolytic, case reports of hemolysis in severe deficiency have been reported.7 Adequate bone marrow response to hemolysis in TTP results in an elevated reticulocyte count, which can be useful in differentiating from pernicious anemia where there is typically an inadequate bone marrow response and low reticulocyte count.8,9
The approach to working up pancytopenia begins with a detailed history inquiring about medications, exposures (benzenes, pesticides), alcohol use, and infection history. A thorough physical examination may help point the health care practitioner (HCP) toward a certain etiology, as the differential for pancytopenia is broad. In the deployed soldier downrange, resources are often limited, and the history/physical are crucial in preventing an expensive and unnecessary workup.
Case Presentation
A 24-year-old active-duty female patient presented in late December 2020 to a theater hospital in Djibouti after a witnessed syncopal episode. She had a history of Hashimoto thyroiditis and was taking levothyroxine sodium 75 mcg daily. The patient reported gluten intolerance, which was never formally evaluated. The syncopal episode lasted a few seconds and was not associated with any prodromal or postictal symptoms. No seizure activity was observed, and she had no history of syncopal episodes. She reported that she had been feeling ill 24 to 48 hours prior, with nausea, fatigue, decreased oral intake, decreased urine output, and 2 episodes of nonbilious, nonbloody emesis.
When the patient arrived, she was tachycardic with heart rate in the 130s beats per minute (baseline, 100-110 beats per minute), febrile (103 °F), and had systolic blood pressure (SBP) in the low 100s (baseline, SBP 120s-130s). An electrocardiogram and chest radiographs were unremarkable. Her complete blood count (CBC) could not be processed due to Hb and platelet levels too low to detect on assay (Table 2). Lactate dehydrogenase (LDH) was elevated at > 1000 U/L with mild elevation in liver enzymes (aspartate aminotransferase, 98 U/L; alanine aminotransferase, 51 U/L) and prolonged partial thromboplastin time 70 seconds. She did not report any increased bleeding or bruising. The peripheral blood smear demonstrated pancytopenia, without any schistocytes, and she was started on broad-spectrum antibiotics for presumed sepsis from urinary source and possible TTP.
The patient received 5 units of packed red blood cells, transfusion of platelets, and 2 doses of vitamin B12 in Djibouti with clinical improvement and resolution of orthostasis, hypotension, tachycardia, and fever. Her final posttransfusion CBC showed a Hb level of 11.2 g/dL, white blood cell (WBC) count of 1.7 K/µL, and platelet count of 23 K/µL (Table 3). Two days later her Hb level was 9.0 g/dL, WBC count 1.8 K/µL, and platelet count was 12 K/µL. She was evacuated via air to Landstuhl Regional Medical Center (LRMC) in Germany within 48 hours of presentation, given limited testing capabilities and persistent anemia and thrombocytopenia, refractory to transfusion, concerning for aplastic anemia or acute leukemia.
On arrival at LRMC, she was transfused 1 unit of platelets and given 3 doses of intramuscular vitamin B12 for undetectable levels (< 50 pg/mL) at presentation. An extensive infectious workup was obtained, which did not reveal any viral, bacterial, or parasitic causes. The patient also had a bone marrow biopsy performed at a civilian site, which revealed hypocellular bone marrow. She was transferred to Walter Reed National Military Medical Center (WRNMMC) for further workup and evaluation, given the infectious workup, which was negative. Concern for hematologic malignancy remained. At the time of her arrival, the laboratory values had drastically improved with vitamin supplementation. The patient’s absolute reticulocyte count indicated adequate bone marrow response and because of her improvement, a repeat bone marrow biopsy was not performed.
Intrinsic factor antibodies were elevated (34.5 AU/mL; reference range, 0.0-1.1), which confirmed that this patient’s underlying etiology was secondary to pernicious anemia. The patient continued to improve and repeat vitamin B12 and folate levels revealed that she was responding to therapy. At discharge, intramuscular vitamin B12 injections were planned to continue monthly, indefinitely per guidelines. Oral supplementation is typically avoided due to poor absorption.
Of note, during her inpatient admission at WRNMMC, further evaluation of reported gluten intolerance was performed, which revealed a negative celiac disease panel (IgG/IgA tissue transglutaminase antibodies). On discharge, she was to establish care with gastroenterology for further evaluation, likely including endoscopic evaluation, at her next duty station. She was able to resume full travel and duty functions on discharge from WRNMMC.
Discussion
We highlight a complex case of pancytopenia secondary to pernicious anemia in a deployed service member. With limited resources downrange, the workup of pancytopenia can be resource intensive, expensive, and time sensitive, which can have detrimental impacts on medical readiness. Additionally, undiagnosed coagulopathies can have lethal consequences in a deployed service member where bleeding risk may be elevated depending on the mission. The differential for pancytopenia is vast, and given its relative rarity in pernicious anemia, the HCP must use key components of the history and laboratory results to narrow the differential (eAppendix).10
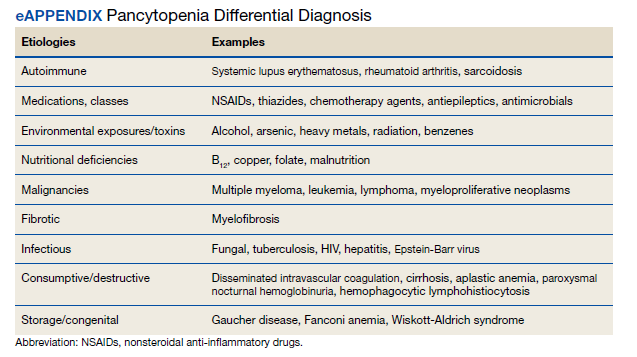
Pernicious anemia commonly presents as an isolated anemia. In a study looking at the hematologic manifestations of 201 cohort patients with well-documented vitamin B12 deficiency, 5% had symptomatic pancytopenia and 1.5% had a hemolytic anemia.2 The majority (> 67%) of hematologic abnormalities were correctable with cobalamin replacement.2 In our case, the solider presented with symptomatic anemia, manifesting as syncope, and was found to have transfusion-resistant pancytopenia.She had a hemolytic anemia with an LDH > 1000 U/L, haptoglobin < 3 mg/dL, and mild transaminitis with hyperbilirubinemia (1.8 mg/dL). No schistocytes were observed on peripheral smear, suggesting intramedullary hemolysis, which is believed to be due to the destruction of megaloblastic cells by macrophages in bone marrow.11 A French study found high LDH levels and low reticulocyte counts to be strongly suggestive of vitamin B12 deficiency and helpful in differentiating pernicious anemia from TTP, given that bone marrow response to anemia in TTP is preserved.8
While vitamin B12 deficiency is not often associated with hemolytic anemia, multiple cases have been reported in the literature.6 Screening for vitamin B12 deficiency may have shortened this patient’s clinical course and limited the need for air evacuation to a stateside quaternary medical center. However, testing for cobalamin levels in overseas deployed environments is difficult, timely, and costly. New technologies, such as optical sensors, can detect vitamin B12 levels in the blood in < 1 minute and offer portable, low-cost options that may be useful in the deployed military setting.12
Diet plays a key role in this case, since the patient had a reported history of gluten intolerance, although it was never documented or evaluated prior to this presentation. Prior to deployment, the patient ate mostly rice, potatoes, and vegetables. While deployed in an austere environment, food options were limited. These conditions forced her to intermittently consume gluten products, which led to gastrointestinal issues, exacerbating her nutritional deficiencies. In the 2 months before her first syncopal episode, she reported worsening fatigue that impacted her ability to exercise. Vitamin B12 stores often take years to deplete, suggesting that she had a chronic nutritional deficiency before deployment. Another possibility was that she developed an autoimmune gastritis that acutely worsened in the setting of poor nutritional intake. Her history of Hashimoto thyroiditis is also important, as up to one-third of patients with autoimmune thyroid disease have been associated with pernicious anemia (range, 3%-32%) with certain shared human leukocyte antigen alleles implicated in autoimmune gastritis.13,14
Conclusions
This rare case of pernicious anemia presenting as pancytopenia illustrates the challenge in working up pancytopenia, especially in austere military environments with limited testing capabilities. Screening for chronic dietary and nutritional deficiency is important in a service member, raising the question of what role predeployment screening may have and what dietary accommodations may be available during overseas deployments, which can potentially dampen inflammation of the gastrointestinal tract, especially for those with preexisting autoimmune gastrointestinal conditions. Also, newer technology allows portable, low-cost testing of cobalamin and may aid in its diagnosis. In patients who are anemic with low vitamin B12, HCPs can begin vitamin B12 supplementation while continuing the workup (eg, antibody testing, endoscopy). If the patient responds appropriately, further workup becomes less urgent, therefore, decreasing resource use and increasing military readiness. When hemolysis is present, a low reticulocyte count can be beneficial to help differentiate this condition from TTP, a life-threatening condition that must also be ruled out or treated. Pernicious anemia should be on the differential in any patients with autoimmune conditions presenting with cytopenias, especially in those with a history of autoimmune thyroid disorders.
1. Takeshima M, Ishikawa H, Kitadate A, et al. Anorexia nervosa-associated pancytopenia mimicking idiopathic aplastic anemia: a case report. BMC Psychiatry. 2018;18(1):150. doi:10.1186/s12888-018-1743-6
2. Andrès E, Affenberger S, Zimmer J, et al. Current hematological findings in cobalamin deficiency. A study of 201 consecutive patients with documented cobalamin deficiency. Clin Lab Haematol. 2006;28(1):50-56. doi:10.1111/j.1365-2257.2006.00755.x
3. Sinclair L. Recognizing, treating and understanding pernicious anaemia. J R Soc Med. 2008;101(5):262-264. doi:10.1258/jrsm.2008.081006
4. Shampo MA, Kyle RA, Steensma DP. William Murphy—Nobel Prize for the treatment of pernicious anemia. Mayo Clin Proc. 2006;81(6):726. doi:10.4065/81.6.726
5. Hong M, He G. The 2016 revision to the World Health Organization classification of myelodysplastic syndromes. J Transl Int Med. 2017;5(3):139-143. doi:10.1515/jtim-2017-0002
6. Tunio NA, Sheriff MZ, Cooper G. Prevalence of gastric cancer in patients with pernicious anemia: a population-based study. Am J Gastroenterol. 2020;115:S665. doi:10.14309/01.ajg.0000707332.16739.72
7. Bailey M, Maestas T, Betancourt R, Mikhael D, Babiker HM. A rare cause of thrombotic thrombocytopenic purpura- (TTP-) like syndrome, vitamin B12 deficiency: interpretation of significant pathological findings. Case Rep Hematol. 2019;2019:1529306. doi:10.1155/2019/1529306
8. Stanley M, Michalski JM. Thrombotic Thrombocytopenic Purpura. StatPearls Publishing LLC; 2021.
9. Noël N, Maigné G, Tertian G, et al. Hemolysis and schistocytosis in the emergency department: consider pseudothrombotic microangiopathy related to vitamin B12 deficiency. QJM. 2013;106(11):1017-1022. doi:10.1093/qjmed/hct142
10. Chiravuri S, De Jesus O. Pancytopenia. StatPearls Publishing LLC; 2021.
11. Gladstone E. Pernicious anemia presenting with pancytopenia and hemolysis: a case report. February 8, 2019. Accessed June 9, 2022. https://www.journalmc.org/index.php/JMC/article/view/3269/2563
12. ScienceDaily. Developing a sensor for vitamin B12 deficiency. October 17, 2016. Accessed June 9, 2022. https://www.sciencedaily.com/releases/2016/10/161017103221.htm
13. Rodriguez NM, Shackelford K. Pernicious Anemia. StatPearls Publishing LLC; 2021.
14. Fernando MM, Stevens CR, Walsh EC, et al. Defining the role of the MHC in autoimmunity: a review and pooled analysis. PLoS Genet. 2008;4(4):e1000024. doi:10.1371/journal.pgen.1000024
Pancytopenia is a condition in which all 3 hematologic cell lines are lower than expected in the blood, often representing either an increase in cellular destruction or decrease in bone marrow production. Destruction often occurs in the setting of autoimmune conditions (eg, systemic lupus erythematosus, rheumatoid arthritis) or splenic sequestration, often affecting erythrocytes and platelets more than leukocytes. Decreased production represents central etiologies, which are often due to nutritional deficiencies, infections, drug toxicities, or malabsorption.1 Pancytopenia secondary to vitamin B12 deficiency is rare, accounting for about 5% of the hematologic manifestations of symptomatic vitamin B12 deficient patients.2
Pernicious anemia, named for a once lethal disease, is a form of vitamin B12 (cobalamin) deficiency that results from an autoimmune (type II hypersensitivity) reaction to gastric parietal cells or intrinsic factor. Antibodies bind to gastric parietal cells and reduce gastric acid production, leading to atrophic gastritis, or they bind intrinsic factor and block the binding and absorption of vitamin B12 in the gastrointestinal tract. While first described in the 1820s, it was not until a century later when scientists were studying hematopoiesis in response to the heavy casualty burden from battlefield exsanguination in World War I that dogs fed raw liver were noted to have significantly better blood regeneration response than those fed cooked liver. This discovery led physicians Minot and Murphy to use raw liver to treat pernicious anemia and found that jaundice improved, reticulocyte counts increased, and hemoglobin (Hb) concentration improved, resulting in the duo becoming the first American recipients of the Nobel Prize in physiology or medicine.3 It was ultimately determined in 1948 by chemists Folkers and Todd that the active ingredient in raw liver responsible for this phenomenon was vitamin B12.4
Patients with pernicious anemia typically present with macrocytic anemia, low reticulocyte count, hypersegmented neutrophils, as well as mild leukopenia and/or thrombocytopenia, distinguishable from folate deficiency by an elevated serum methylmalonic acid level. World Health Organization cytopenia thresholds are listed in Table 1.5 Treatment consists of lifelong vitamin B12 supplementation, and endoscopic screening is often recommended after diagnosis due to increased risk of gastrointestinal malignancy.6 Pernicious anemia can be difficult to distinguish from thrombotic thrombocytopenia purpura (TTP), a microangiopathic hemolytic anemia that can cause rapid end-organ failure and death if treatment is delayed.7 While pernicious anemia is not typically hemolytic, case reports of hemolysis in severe deficiency have been reported.7 Adequate bone marrow response to hemolysis in TTP results in an elevated reticulocyte count, which can be useful in differentiating from pernicious anemia where there is typically an inadequate bone marrow response and low reticulocyte count.8,9
The approach to working up pancytopenia begins with a detailed history inquiring about medications, exposures (benzenes, pesticides), alcohol use, and infection history. A thorough physical examination may help point the health care practitioner (HCP) toward a certain etiology, as the differential for pancytopenia is broad. In the deployed soldier downrange, resources are often limited, and the history/physical are crucial in preventing an expensive and unnecessary workup.
Case Presentation
A 24-year-old active-duty female patient presented in late December 2020 to a theater hospital in Djibouti after a witnessed syncopal episode. She had a history of Hashimoto thyroiditis and was taking levothyroxine sodium 75 mcg daily. The patient reported gluten intolerance, which was never formally evaluated. The syncopal episode lasted a few seconds and was not associated with any prodromal or postictal symptoms. No seizure activity was observed, and she had no history of syncopal episodes. She reported that she had been feeling ill 24 to 48 hours prior, with nausea, fatigue, decreased oral intake, decreased urine output, and 2 episodes of nonbilious, nonbloody emesis.
When the patient arrived, she was tachycardic with heart rate in the 130s beats per minute (baseline, 100-110 beats per minute), febrile (103 °F), and had systolic blood pressure (SBP) in the low 100s (baseline, SBP 120s-130s). An electrocardiogram and chest radiographs were unremarkable. Her complete blood count (CBC) could not be processed due to Hb and platelet levels too low to detect on assay (Table 2). Lactate dehydrogenase (LDH) was elevated at > 1000 U/L with mild elevation in liver enzymes (aspartate aminotransferase, 98 U/L; alanine aminotransferase, 51 U/L) and prolonged partial thromboplastin time 70 seconds. She did not report any increased bleeding or bruising. The peripheral blood smear demonstrated pancytopenia, without any schistocytes, and she was started on broad-spectrum antibiotics for presumed sepsis from urinary source and possible TTP.
The patient received 5 units of packed red blood cells, transfusion of platelets, and 2 doses of vitamin B12 in Djibouti with clinical improvement and resolution of orthostasis, hypotension, tachycardia, and fever. Her final posttransfusion CBC showed a Hb level of 11.2 g/dL, white blood cell (WBC) count of 1.7 K/µL, and platelet count of 23 K/µL (Table 3). Two days later her Hb level was 9.0 g/dL, WBC count 1.8 K/µL, and platelet count was 12 K/µL. She was evacuated via air to Landstuhl Regional Medical Center (LRMC) in Germany within 48 hours of presentation, given limited testing capabilities and persistent anemia and thrombocytopenia, refractory to transfusion, concerning for aplastic anemia or acute leukemia.
On arrival at LRMC, she was transfused 1 unit of platelets and given 3 doses of intramuscular vitamin B12 for undetectable levels (< 50 pg/mL) at presentation. An extensive infectious workup was obtained, which did not reveal any viral, bacterial, or parasitic causes. The patient also had a bone marrow biopsy performed at a civilian site, which revealed hypocellular bone marrow. She was transferred to Walter Reed National Military Medical Center (WRNMMC) for further workup and evaluation, given the infectious workup, which was negative. Concern for hematologic malignancy remained. At the time of her arrival, the laboratory values had drastically improved with vitamin supplementation. The patient’s absolute reticulocyte count indicated adequate bone marrow response and because of her improvement, a repeat bone marrow biopsy was not performed.
Intrinsic factor antibodies were elevated (34.5 AU/mL; reference range, 0.0-1.1), which confirmed that this patient’s underlying etiology was secondary to pernicious anemia. The patient continued to improve and repeat vitamin B12 and folate levels revealed that she was responding to therapy. At discharge, intramuscular vitamin B12 injections were planned to continue monthly, indefinitely per guidelines. Oral supplementation is typically avoided due to poor absorption.
Of note, during her inpatient admission at WRNMMC, further evaluation of reported gluten intolerance was performed, which revealed a negative celiac disease panel (IgG/IgA tissue transglutaminase antibodies). On discharge, she was to establish care with gastroenterology for further evaluation, likely including endoscopic evaluation, at her next duty station. She was able to resume full travel and duty functions on discharge from WRNMMC.
Discussion
We highlight a complex case of pancytopenia secondary to pernicious anemia in a deployed service member. With limited resources downrange, the workup of pancytopenia can be resource intensive, expensive, and time sensitive, which can have detrimental impacts on medical readiness. Additionally, undiagnosed coagulopathies can have lethal consequences in a deployed service member where bleeding risk may be elevated depending on the mission. The differential for pancytopenia is vast, and given its relative rarity in pernicious anemia, the HCP must use key components of the history and laboratory results to narrow the differential (eAppendix).10
Pernicious anemia commonly presents as an isolated anemia. In a study looking at the hematologic manifestations of 201 cohort patients with well-documented vitamin B12 deficiency, 5% had symptomatic pancytopenia and 1.5% had a hemolytic anemia.2 The majority (> 67%) of hematologic abnormalities were correctable with cobalamin replacement.2 In our case, the solider presented with symptomatic anemia, manifesting as syncope, and was found to have transfusion-resistant pancytopenia.She had a hemolytic anemia with an LDH > 1000 U/L, haptoglobin < 3 mg/dL, and mild transaminitis with hyperbilirubinemia (1.8 mg/dL). No schistocytes were observed on peripheral smear, suggesting intramedullary hemolysis, which is believed to be due to the destruction of megaloblastic cells by macrophages in bone marrow.11 A French study found high LDH levels and low reticulocyte counts to be strongly suggestive of vitamin B12 deficiency and helpful in differentiating pernicious anemia from TTP, given that bone marrow response to anemia in TTP is preserved.8
While vitamin B12 deficiency is not often associated with hemolytic anemia, multiple cases have been reported in the literature.6 Screening for vitamin B12 deficiency may have shortened this patient’s clinical course and limited the need for air evacuation to a stateside quaternary medical center. However, testing for cobalamin levels in overseas deployed environments is difficult, timely, and costly. New technologies, such as optical sensors, can detect vitamin B12 levels in the blood in < 1 minute and offer portable, low-cost options that may be useful in the deployed military setting.12
Diet plays a key role in this case, since the patient had a reported history of gluten intolerance, although it was never documented or evaluated prior to this presentation. Prior to deployment, the patient ate mostly rice, potatoes, and vegetables. While deployed in an austere environment, food options were limited. These conditions forced her to intermittently consume gluten products, which led to gastrointestinal issues, exacerbating her nutritional deficiencies. In the 2 months before her first syncopal episode, she reported worsening fatigue that impacted her ability to exercise. Vitamin B12 stores often take years to deplete, suggesting that she had a chronic nutritional deficiency before deployment. Another possibility was that she developed an autoimmune gastritis that acutely worsened in the setting of poor nutritional intake. Her history of Hashimoto thyroiditis is also important, as up to one-third of patients with autoimmune thyroid disease have been associated with pernicious anemia (range, 3%-32%) with certain shared human leukocyte antigen alleles implicated in autoimmune gastritis.13,14
Conclusions
This rare case of pernicious anemia presenting as pancytopenia illustrates the challenge in working up pancytopenia, especially in austere military environments with limited testing capabilities. Screening for chronic dietary and nutritional deficiency is important in a service member, raising the question of what role predeployment screening may have and what dietary accommodations may be available during overseas deployments, which can potentially dampen inflammation of the gastrointestinal tract, especially for those with preexisting autoimmune gastrointestinal conditions. Also, newer technology allows portable, low-cost testing of cobalamin and may aid in its diagnosis. In patients who are anemic with low vitamin B12, HCPs can begin vitamin B12 supplementation while continuing the workup (eg, antibody testing, endoscopy). If the patient responds appropriately, further workup becomes less urgent, therefore, decreasing resource use and increasing military readiness. When hemolysis is present, a low reticulocyte count can be beneficial to help differentiate this condition from TTP, a life-threatening condition that must also be ruled out or treated. Pernicious anemia should be on the differential in any patients with autoimmune conditions presenting with cytopenias, especially in those with a history of autoimmune thyroid disorders.
Pancytopenia is a condition in which all 3 hematologic cell lines are lower than expected in the blood, often representing either an increase in cellular destruction or decrease in bone marrow production. Destruction often occurs in the setting of autoimmune conditions (eg, systemic lupus erythematosus, rheumatoid arthritis) or splenic sequestration, often affecting erythrocytes and platelets more than leukocytes. Decreased production represents central etiologies, which are often due to nutritional deficiencies, infections, drug toxicities, or malabsorption.1 Pancytopenia secondary to vitamin B12 deficiency is rare, accounting for about 5% of the hematologic manifestations of symptomatic vitamin B12 deficient patients.2
Pernicious anemia, named for a once lethal disease, is a form of vitamin B12 (cobalamin) deficiency that results from an autoimmune (type II hypersensitivity) reaction to gastric parietal cells or intrinsic factor. Antibodies bind to gastric parietal cells and reduce gastric acid production, leading to atrophic gastritis, or they bind intrinsic factor and block the binding and absorption of vitamin B12 in the gastrointestinal tract. While first described in the 1820s, it was not until a century later when scientists were studying hematopoiesis in response to the heavy casualty burden from battlefield exsanguination in World War I that dogs fed raw liver were noted to have significantly better blood regeneration response than those fed cooked liver. This discovery led physicians Minot and Murphy to use raw liver to treat pernicious anemia and found that jaundice improved, reticulocyte counts increased, and hemoglobin (Hb) concentration improved, resulting in the duo becoming the first American recipients of the Nobel Prize in physiology or medicine.3 It was ultimately determined in 1948 by chemists Folkers and Todd that the active ingredient in raw liver responsible for this phenomenon was vitamin B12.4
Patients with pernicious anemia typically present with macrocytic anemia, low reticulocyte count, hypersegmented neutrophils, as well as mild leukopenia and/or thrombocytopenia, distinguishable from folate deficiency by an elevated serum methylmalonic acid level. World Health Organization cytopenia thresholds are listed in Table 1.5 Treatment consists of lifelong vitamin B12 supplementation, and endoscopic screening is often recommended after diagnosis due to increased risk of gastrointestinal malignancy.6 Pernicious anemia can be difficult to distinguish from thrombotic thrombocytopenia purpura (TTP), a microangiopathic hemolytic anemia that can cause rapid end-organ failure and death if treatment is delayed.7 While pernicious anemia is not typically hemolytic, case reports of hemolysis in severe deficiency have been reported.7 Adequate bone marrow response to hemolysis in TTP results in an elevated reticulocyte count, which can be useful in differentiating from pernicious anemia where there is typically an inadequate bone marrow response and low reticulocyte count.8,9
The approach to working up pancytopenia begins with a detailed history inquiring about medications, exposures (benzenes, pesticides), alcohol use, and infection history. A thorough physical examination may help point the health care practitioner (HCP) toward a certain etiology, as the differential for pancytopenia is broad. In the deployed soldier downrange, resources are often limited, and the history/physical are crucial in preventing an expensive and unnecessary workup.
Case Presentation
A 24-year-old active-duty female patient presented in late December 2020 to a theater hospital in Djibouti after a witnessed syncopal episode. She had a history of Hashimoto thyroiditis and was taking levothyroxine sodium 75 mcg daily. The patient reported gluten intolerance, which was never formally evaluated. The syncopal episode lasted a few seconds and was not associated with any prodromal or postictal symptoms. No seizure activity was observed, and she had no history of syncopal episodes. She reported that she had been feeling ill 24 to 48 hours prior, with nausea, fatigue, decreased oral intake, decreased urine output, and 2 episodes of nonbilious, nonbloody emesis.
When the patient arrived, she was tachycardic with heart rate in the 130s beats per minute (baseline, 100-110 beats per minute), febrile (103 °F), and had systolic blood pressure (SBP) in the low 100s (baseline, SBP 120s-130s). An electrocardiogram and chest radiographs were unremarkable. Her complete blood count (CBC) could not be processed due to Hb and platelet levels too low to detect on assay (Table 2). Lactate dehydrogenase (LDH) was elevated at > 1000 U/L with mild elevation in liver enzymes (aspartate aminotransferase, 98 U/L; alanine aminotransferase, 51 U/L) and prolonged partial thromboplastin time 70 seconds. She did not report any increased bleeding or bruising. The peripheral blood smear demonstrated pancytopenia, without any schistocytes, and she was started on broad-spectrum antibiotics for presumed sepsis from urinary source and possible TTP.
The patient received 5 units of packed red blood cells, transfusion of platelets, and 2 doses of vitamin B12 in Djibouti with clinical improvement and resolution of orthostasis, hypotension, tachycardia, and fever. Her final posttransfusion CBC showed a Hb level of 11.2 g/dL, white blood cell (WBC) count of 1.7 K/µL, and platelet count of 23 K/µL (Table 3). Two days later her Hb level was 9.0 g/dL, WBC count 1.8 K/µL, and platelet count was 12 K/µL. She was evacuated via air to Landstuhl Regional Medical Center (LRMC) in Germany within 48 hours of presentation, given limited testing capabilities and persistent anemia and thrombocytopenia, refractory to transfusion, concerning for aplastic anemia or acute leukemia.
On arrival at LRMC, she was transfused 1 unit of platelets and given 3 doses of intramuscular vitamin B12 for undetectable levels (< 50 pg/mL) at presentation. An extensive infectious workup was obtained, which did not reveal any viral, bacterial, or parasitic causes. The patient also had a bone marrow biopsy performed at a civilian site, which revealed hypocellular bone marrow. She was transferred to Walter Reed National Military Medical Center (WRNMMC) for further workup and evaluation, given the infectious workup, which was negative. Concern for hematologic malignancy remained. At the time of her arrival, the laboratory values had drastically improved with vitamin supplementation. The patient’s absolute reticulocyte count indicated adequate bone marrow response and because of her improvement, a repeat bone marrow biopsy was not performed.
Intrinsic factor antibodies were elevated (34.5 AU/mL; reference range, 0.0-1.1), which confirmed that this patient’s underlying etiology was secondary to pernicious anemia. The patient continued to improve and repeat vitamin B12 and folate levels revealed that she was responding to therapy. At discharge, intramuscular vitamin B12 injections were planned to continue monthly, indefinitely per guidelines. Oral supplementation is typically avoided due to poor absorption.
Of note, during her inpatient admission at WRNMMC, further evaluation of reported gluten intolerance was performed, which revealed a negative celiac disease panel (IgG/IgA tissue transglutaminase antibodies). On discharge, she was to establish care with gastroenterology for further evaluation, likely including endoscopic evaluation, at her next duty station. She was able to resume full travel and duty functions on discharge from WRNMMC.
Discussion
We highlight a complex case of pancytopenia secondary to pernicious anemia in a deployed service member. With limited resources downrange, the workup of pancytopenia can be resource intensive, expensive, and time sensitive, which can have detrimental impacts on medical readiness. Additionally, undiagnosed coagulopathies can have lethal consequences in a deployed service member where bleeding risk may be elevated depending on the mission. The differential for pancytopenia is vast, and given its relative rarity in pernicious anemia, the HCP must use key components of the history and laboratory results to narrow the differential (eAppendix).10
Pernicious anemia commonly presents as an isolated anemia. In a study looking at the hematologic manifestations of 201 cohort patients with well-documented vitamin B12 deficiency, 5% had symptomatic pancytopenia and 1.5% had a hemolytic anemia.2 The majority (> 67%) of hematologic abnormalities were correctable with cobalamin replacement.2 In our case, the solider presented with symptomatic anemia, manifesting as syncope, and was found to have transfusion-resistant pancytopenia.She had a hemolytic anemia with an LDH > 1000 U/L, haptoglobin < 3 mg/dL, and mild transaminitis with hyperbilirubinemia (1.8 mg/dL). No schistocytes were observed on peripheral smear, suggesting intramedullary hemolysis, which is believed to be due to the destruction of megaloblastic cells by macrophages in bone marrow.11 A French study found high LDH levels and low reticulocyte counts to be strongly suggestive of vitamin B12 deficiency and helpful in differentiating pernicious anemia from TTP, given that bone marrow response to anemia in TTP is preserved.8
While vitamin B12 deficiency is not often associated with hemolytic anemia, multiple cases have been reported in the literature.6 Screening for vitamin B12 deficiency may have shortened this patient’s clinical course and limited the need for air evacuation to a stateside quaternary medical center. However, testing for cobalamin levels in overseas deployed environments is difficult, timely, and costly. New technologies, such as optical sensors, can detect vitamin B12 levels in the blood in < 1 minute and offer portable, low-cost options that may be useful in the deployed military setting.12
Diet plays a key role in this case, since the patient had a reported history of gluten intolerance, although it was never documented or evaluated prior to this presentation. Prior to deployment, the patient ate mostly rice, potatoes, and vegetables. While deployed in an austere environment, food options were limited. These conditions forced her to intermittently consume gluten products, which led to gastrointestinal issues, exacerbating her nutritional deficiencies. In the 2 months before her first syncopal episode, she reported worsening fatigue that impacted her ability to exercise. Vitamin B12 stores often take years to deplete, suggesting that she had a chronic nutritional deficiency before deployment. Another possibility was that she developed an autoimmune gastritis that acutely worsened in the setting of poor nutritional intake. Her history of Hashimoto thyroiditis is also important, as up to one-third of patients with autoimmune thyroid disease have been associated with pernicious anemia (range, 3%-32%) with certain shared human leukocyte antigen alleles implicated in autoimmune gastritis.13,14
Conclusions
This rare case of pernicious anemia presenting as pancytopenia illustrates the challenge in working up pancytopenia, especially in austere military environments with limited testing capabilities. Screening for chronic dietary and nutritional deficiency is important in a service member, raising the question of what role predeployment screening may have and what dietary accommodations may be available during overseas deployments, which can potentially dampen inflammation of the gastrointestinal tract, especially for those with preexisting autoimmune gastrointestinal conditions. Also, newer technology allows portable, low-cost testing of cobalamin and may aid in its diagnosis. In patients who are anemic with low vitamin B12, HCPs can begin vitamin B12 supplementation while continuing the workup (eg, antibody testing, endoscopy). If the patient responds appropriately, further workup becomes less urgent, therefore, decreasing resource use and increasing military readiness. When hemolysis is present, a low reticulocyte count can be beneficial to help differentiate this condition from TTP, a life-threatening condition that must also be ruled out or treated. Pernicious anemia should be on the differential in any patients with autoimmune conditions presenting with cytopenias, especially in those with a history of autoimmune thyroid disorders.
1. Takeshima M, Ishikawa H, Kitadate A, et al. Anorexia nervosa-associated pancytopenia mimicking idiopathic aplastic anemia: a case report. BMC Psychiatry. 2018;18(1):150. doi:10.1186/s12888-018-1743-6
2. Andrès E, Affenberger S, Zimmer J, et al. Current hematological findings in cobalamin deficiency. A study of 201 consecutive patients with documented cobalamin deficiency. Clin Lab Haematol. 2006;28(1):50-56. doi:10.1111/j.1365-2257.2006.00755.x
3. Sinclair L. Recognizing, treating and understanding pernicious anaemia. J R Soc Med. 2008;101(5):262-264. doi:10.1258/jrsm.2008.081006
4. Shampo MA, Kyle RA, Steensma DP. William Murphy—Nobel Prize for the treatment of pernicious anemia. Mayo Clin Proc. 2006;81(6):726. doi:10.4065/81.6.726
5. Hong M, He G. The 2016 revision to the World Health Organization classification of myelodysplastic syndromes. J Transl Int Med. 2017;5(3):139-143. doi:10.1515/jtim-2017-0002
6. Tunio NA, Sheriff MZ, Cooper G. Prevalence of gastric cancer in patients with pernicious anemia: a population-based study. Am J Gastroenterol. 2020;115:S665. doi:10.14309/01.ajg.0000707332.16739.72
7. Bailey M, Maestas T, Betancourt R, Mikhael D, Babiker HM. A rare cause of thrombotic thrombocytopenic purpura- (TTP-) like syndrome, vitamin B12 deficiency: interpretation of significant pathological findings. Case Rep Hematol. 2019;2019:1529306. doi:10.1155/2019/1529306
8. Stanley M, Michalski JM. Thrombotic Thrombocytopenic Purpura. StatPearls Publishing LLC; 2021.
9. Noël N, Maigné G, Tertian G, et al. Hemolysis and schistocytosis in the emergency department: consider pseudothrombotic microangiopathy related to vitamin B12 deficiency. QJM. 2013;106(11):1017-1022. doi:10.1093/qjmed/hct142
10. Chiravuri S, De Jesus O. Pancytopenia. StatPearls Publishing LLC; 2021.
11. Gladstone E. Pernicious anemia presenting with pancytopenia and hemolysis: a case report. February 8, 2019. Accessed June 9, 2022. https://www.journalmc.org/index.php/JMC/article/view/3269/2563
12. ScienceDaily. Developing a sensor for vitamin B12 deficiency. October 17, 2016. Accessed June 9, 2022. https://www.sciencedaily.com/releases/2016/10/161017103221.htm
13. Rodriguez NM, Shackelford K. Pernicious Anemia. StatPearls Publishing LLC; 2021.
14. Fernando MM, Stevens CR, Walsh EC, et al. Defining the role of the MHC in autoimmunity: a review and pooled analysis. PLoS Genet. 2008;4(4):e1000024. doi:10.1371/journal.pgen.1000024
1. Takeshima M, Ishikawa H, Kitadate A, et al. Anorexia nervosa-associated pancytopenia mimicking idiopathic aplastic anemia: a case report. BMC Psychiatry. 2018;18(1):150. doi:10.1186/s12888-018-1743-6
2. Andrès E, Affenberger S, Zimmer J, et al. Current hematological findings in cobalamin deficiency. A study of 201 consecutive patients with documented cobalamin deficiency. Clin Lab Haematol. 2006;28(1):50-56. doi:10.1111/j.1365-2257.2006.00755.x
3. Sinclair L. Recognizing, treating and understanding pernicious anaemia. J R Soc Med. 2008;101(5):262-264. doi:10.1258/jrsm.2008.081006
4. Shampo MA, Kyle RA, Steensma DP. William Murphy—Nobel Prize for the treatment of pernicious anemia. Mayo Clin Proc. 2006;81(6):726. doi:10.4065/81.6.726
5. Hong M, He G. The 2016 revision to the World Health Organization classification of myelodysplastic syndromes. J Transl Int Med. 2017;5(3):139-143. doi:10.1515/jtim-2017-0002
6. Tunio NA, Sheriff MZ, Cooper G. Prevalence of gastric cancer in patients with pernicious anemia: a population-based study. Am J Gastroenterol. 2020;115:S665. doi:10.14309/01.ajg.0000707332.16739.72
7. Bailey M, Maestas T, Betancourt R, Mikhael D, Babiker HM. A rare cause of thrombotic thrombocytopenic purpura- (TTP-) like syndrome, vitamin B12 deficiency: interpretation of significant pathological findings. Case Rep Hematol. 2019;2019:1529306. doi:10.1155/2019/1529306
8. Stanley M, Michalski JM. Thrombotic Thrombocytopenic Purpura. StatPearls Publishing LLC; 2021.
9. Noël N, Maigné G, Tertian G, et al. Hemolysis and schistocytosis in the emergency department: consider pseudothrombotic microangiopathy related to vitamin B12 deficiency. QJM. 2013;106(11):1017-1022. doi:10.1093/qjmed/hct142
10. Chiravuri S, De Jesus O. Pancytopenia. StatPearls Publishing LLC; 2021.
11. Gladstone E. Pernicious anemia presenting with pancytopenia and hemolysis: a case report. February 8, 2019. Accessed June 9, 2022. https://www.journalmc.org/index.php/JMC/article/view/3269/2563
12. ScienceDaily. Developing a sensor for vitamin B12 deficiency. October 17, 2016. Accessed June 9, 2022. https://www.sciencedaily.com/releases/2016/10/161017103221.htm
13. Rodriguez NM, Shackelford K. Pernicious Anemia. StatPearls Publishing LLC; 2021.
14. Fernando MM, Stevens CR, Walsh EC, et al. Defining the role of the MHC in autoimmunity: a review and pooled analysis. PLoS Genet. 2008;4(4):e1000024. doi:10.1371/journal.pgen.1000024