User login
Nonpharmacologic AD therapy: Strongest evidence supports moisturizers
BOSTON – Moisturizers are “a cornerstone” of therapy for children with atopic dermatitis, according to Julie V. Schaffer, MD.
Moisturizers improve skin hydration, increase the time between flares, and reduce xerosis and pruritus, Dr. Schaffer of Hackensack (N.J.) University Medical Group said at the American Academy of Dermatology summer meeting.

In 2014, the AAD released guidelines that “very strongly” recommended moisturizers as an important nonpharmacologic intervention for patients with AD, stating that moisturizer use decreases disease severity and can reduce the need for pharmacologic intervention, she said.
In fact, the recommendation for moisturizer was based on “strength A, level 1 evidence,” she noted.
The role of bathing is a bit less clear; bathing is suggested as part of treatment and maintenance, but no standard exists with respect to frequency or duration for those with AD (evidence level: III, strength of recommendation: C). In general, the AAD recommends daily or less frequent bathing in warm water for 5-10 minutes, but surveys suggest that bathing recommendations vary widely among specialists and primary care providers, Dr. Schaffer said.
She noted that she sometimes sees children who have been told to bathe only once a week.
“They will come in just covered with disgusting gunk and it can’t be good for them,” she said. Bathing, especially if they have crusting and scaling, removes irritants and potential allergens, and provides hydration. It can also improve penetration of topical medications, as well as tolerance of those medications so that they burn less.
“So I give a thumbs up to daily bathing,” she said.
It is generally agreed that moisturizers should be applied soon after bathing (after applying medication) to improve skin hydration in patients with AD, Dr. Schaffer said.
The AAD says that moisturizers should be applied liberally and frequently, but the ideal frequency and type of moisturizer remains “a bit of an art form rather than a precise science,” she added.
The ideal moisturizer is one that is safe, effective, and free of fragrance, irritants, and potential sensitizers, she said, noting that “an individualized approach to moisturizer and vehicle selection can be very helpful.”
For young children, it is important that the product doesn’t sting; an ointment may be preferable in this population. Preteens and teenagers may dislike greasiness, so that is an important consideration, she said.
Dr. Schaffer pointed out that lotion formulations typically have water content that is too high to be helpful for patients with substantial xerosis. Creams or ointments may be a better bet, but take care to avoid contamination in large jars of such products, she advised.
“I’ve had a couple times when patients were getting recurrent infections, and we traced it down to a nasty jar that had a little too much bacteria in it,” she said, noting that using a clean scoop or pump can help prevent contamination.
As for cleansers, the “pretty clear winner” is a nonsoap cleanser, Dr. Schaffer said.
The AAD recommends limited use of hypoallergenic, fragrance-free, nonsoap cleansers with neutral to low pH, but the evidence is insufficient for recommending the addition of bath oils, emollients, oatmeal, and most other additives to bath water, as well as for the use of acidic spring water, she said (evidence level: III, strength of recommendation: C). An exception is bleach baths, as adding a small amount of bleach to bath water has been shown to improve symptoms, but the other products have not been shown to be beneficial.
The AAD notes that wet wrap therapy, either with or without a topical corticosteroid, can be recommended for patients with moderate to severe AD, as this can decrease disease severity and water loss during flares (evidence level: II, strength of recommendation: B).
Use moisturizer in newborns at risk for AD
Moisturizers don’t just help improve atopic dermatitis in children, they may also prevent the condition in at risk newborns.
Parents of a child with eczema who are concerned about the condition developing in their next child may find hope in the findings from two studies published in 2014, Dr. Schaffer said.
In a study of 124 newborns at high risk for AD who were randomized to daily emollient therapy or usual infant skin care started by age 3 weeks, the incidence of AD over 6 months was 43% in the control group, vs. 22% in the emollient group, a relative risk reduction of 50% (J Allergy Clin Immunol. 2014 Oct;134[4]:818-23). Parents in the emollient therapy group were allowed to choose between sunflower oil, Cetaphil cream, or Aquaphor Healing Ointment.
In a similar Japanese study of 118 high risk infants who were randomized to daily treatment with an emulsion-type emollient or usual skin care starting the first week of life, the AD/eczema rates at 32 weeks were 47% and 32% in the control and emollient groups, respectively (J Allergy Clin Immunol. 2014 Oct;134[4], 824-30). Both groups were allowed to use petroleum jelly.
“So that is something you can potentially make a recommendation for,” she said.
Dr. Schaffer reported having no conflicts of interest.
BOSTON – Moisturizers are “a cornerstone” of therapy for children with atopic dermatitis, according to Julie V. Schaffer, MD.
Moisturizers improve skin hydration, increase the time between flares, and reduce xerosis and pruritus, Dr. Schaffer of Hackensack (N.J.) University Medical Group said at the American Academy of Dermatology summer meeting.
In 2014, the AAD released guidelines that “very strongly” recommended moisturizers as an important nonpharmacologic intervention for patients with AD, stating that moisturizer use decreases disease severity and can reduce the need for pharmacologic intervention, she said.
In fact, the recommendation for moisturizer was based on “strength A, level 1 evidence,” she noted.
The role of bathing is a bit less clear; bathing is suggested as part of treatment and maintenance, but no standard exists with respect to frequency or duration for those with AD (evidence level: III, strength of recommendation: C). In general, the AAD recommends daily or less frequent bathing in warm water for 5-10 minutes, but surveys suggest that bathing recommendations vary widely among specialists and primary care providers, Dr. Schaffer said.
She noted that she sometimes sees children who have been told to bathe only once a week.
“They will come in just covered with disgusting gunk and it can’t be good for them,” she said. Bathing, especially if they have crusting and scaling, removes irritants and potential allergens, and provides hydration. It can also improve penetration of topical medications, as well as tolerance of those medications so that they burn less.
“So I give a thumbs up to daily bathing,” she said.
It is generally agreed that moisturizers should be applied soon after bathing (after applying medication) to improve skin hydration in patients with AD, Dr. Schaffer said.
The AAD says that moisturizers should be applied liberally and frequently, but the ideal frequency and type of moisturizer remains “a bit of an art form rather than a precise science,” she added.
The ideal moisturizer is one that is safe, effective, and free of fragrance, irritants, and potential sensitizers, she said, noting that “an individualized approach to moisturizer and vehicle selection can be very helpful.”
For young children, it is important that the product doesn’t sting; an ointment may be preferable in this population. Preteens and teenagers may dislike greasiness, so that is an important consideration, she said.
Dr. Schaffer pointed out that lotion formulations typically have water content that is too high to be helpful for patients with substantial xerosis. Creams or ointments may be a better bet, but take care to avoid contamination in large jars of such products, she advised.
“I’ve had a couple times when patients were getting recurrent infections, and we traced it down to a nasty jar that had a little too much bacteria in it,” she said, noting that using a clean scoop or pump can help prevent contamination.
As for cleansers, the “pretty clear winner” is a nonsoap cleanser, Dr. Schaffer said.
The AAD recommends limited use of hypoallergenic, fragrance-free, nonsoap cleansers with neutral to low pH, but the evidence is insufficient for recommending the addition of bath oils, emollients, oatmeal, and most other additives to bath water, as well as for the use of acidic spring water, she said (evidence level: III, strength of recommendation: C). An exception is bleach baths, as adding a small amount of bleach to bath water has been shown to improve symptoms, but the other products have not been shown to be beneficial.
The AAD notes that wet wrap therapy, either with or without a topical corticosteroid, can be recommended for patients with moderate to severe AD, as this can decrease disease severity and water loss during flares (evidence level: II, strength of recommendation: B).
Use moisturizer in newborns at risk for AD
Moisturizers don’t just help improve atopic dermatitis in children, they may also prevent the condition in at risk newborns.
Parents of a child with eczema who are concerned about the condition developing in their next child may find hope in the findings from two studies published in 2014, Dr. Schaffer said.
In a study of 124 newborns at high risk for AD who were randomized to daily emollient therapy or usual infant skin care started by age 3 weeks, the incidence of AD over 6 months was 43% in the control group, vs. 22% in the emollient group, a relative risk reduction of 50% (J Allergy Clin Immunol. 2014 Oct;134[4]:818-23). Parents in the emollient therapy group were allowed to choose between sunflower oil, Cetaphil cream, or Aquaphor Healing Ointment.
In a similar Japanese study of 118 high risk infants who were randomized to daily treatment with an emulsion-type emollient or usual skin care starting the first week of life, the AD/eczema rates at 32 weeks were 47% and 32% in the control and emollient groups, respectively (J Allergy Clin Immunol. 2014 Oct;134[4], 824-30). Both groups were allowed to use petroleum jelly.
“So that is something you can potentially make a recommendation for,” she said.
Dr. Schaffer reported having no conflicts of interest.
BOSTON – Moisturizers are “a cornerstone” of therapy for children with atopic dermatitis, according to Julie V. Schaffer, MD.
Moisturizers improve skin hydration, increase the time between flares, and reduce xerosis and pruritus, Dr. Schaffer of Hackensack (N.J.) University Medical Group said at the American Academy of Dermatology summer meeting.
In 2014, the AAD released guidelines that “very strongly” recommended moisturizers as an important nonpharmacologic intervention for patients with AD, stating that moisturizer use decreases disease severity and can reduce the need for pharmacologic intervention, she said.
In fact, the recommendation for moisturizer was based on “strength A, level 1 evidence,” she noted.
The role of bathing is a bit less clear; bathing is suggested as part of treatment and maintenance, but no standard exists with respect to frequency or duration for those with AD (evidence level: III, strength of recommendation: C). In general, the AAD recommends daily or less frequent bathing in warm water for 5-10 minutes, but surveys suggest that bathing recommendations vary widely among specialists and primary care providers, Dr. Schaffer said.
She noted that she sometimes sees children who have been told to bathe only once a week.
“They will come in just covered with disgusting gunk and it can’t be good for them,” she said. Bathing, especially if they have crusting and scaling, removes irritants and potential allergens, and provides hydration. It can also improve penetration of topical medications, as well as tolerance of those medications so that they burn less.
“So I give a thumbs up to daily bathing,” she said.
It is generally agreed that moisturizers should be applied soon after bathing (after applying medication) to improve skin hydration in patients with AD, Dr. Schaffer said.
The AAD says that moisturizers should be applied liberally and frequently, but the ideal frequency and type of moisturizer remains “a bit of an art form rather than a precise science,” she added.
The ideal moisturizer is one that is safe, effective, and free of fragrance, irritants, and potential sensitizers, she said, noting that “an individualized approach to moisturizer and vehicle selection can be very helpful.”
For young children, it is important that the product doesn’t sting; an ointment may be preferable in this population. Preteens and teenagers may dislike greasiness, so that is an important consideration, she said.
Dr. Schaffer pointed out that lotion formulations typically have water content that is too high to be helpful for patients with substantial xerosis. Creams or ointments may be a better bet, but take care to avoid contamination in large jars of such products, she advised.
“I’ve had a couple times when patients were getting recurrent infections, and we traced it down to a nasty jar that had a little too much bacteria in it,” she said, noting that using a clean scoop or pump can help prevent contamination.
As for cleansers, the “pretty clear winner” is a nonsoap cleanser, Dr. Schaffer said.
The AAD recommends limited use of hypoallergenic, fragrance-free, nonsoap cleansers with neutral to low pH, but the evidence is insufficient for recommending the addition of bath oils, emollients, oatmeal, and most other additives to bath water, as well as for the use of acidic spring water, she said (evidence level: III, strength of recommendation: C). An exception is bleach baths, as adding a small amount of bleach to bath water has been shown to improve symptoms, but the other products have not been shown to be beneficial.
The AAD notes that wet wrap therapy, either with or without a topical corticosteroid, can be recommended for patients with moderate to severe AD, as this can decrease disease severity and water loss during flares (evidence level: II, strength of recommendation: B).
Use moisturizer in newborns at risk for AD
Moisturizers don’t just help improve atopic dermatitis in children, they may also prevent the condition in at risk newborns.
Parents of a child with eczema who are concerned about the condition developing in their next child may find hope in the findings from two studies published in 2014, Dr. Schaffer said.
In a study of 124 newborns at high risk for AD who were randomized to daily emollient therapy or usual infant skin care started by age 3 weeks, the incidence of AD over 6 months was 43% in the control group, vs. 22% in the emollient group, a relative risk reduction of 50% (J Allergy Clin Immunol. 2014 Oct;134[4]:818-23). Parents in the emollient therapy group were allowed to choose between sunflower oil, Cetaphil cream, or Aquaphor Healing Ointment.
In a similar Japanese study of 118 high risk infants who were randomized to daily treatment with an emulsion-type emollient or usual skin care starting the first week of life, the AD/eczema rates at 32 weeks were 47% and 32% in the control and emollient groups, respectively (J Allergy Clin Immunol. 2014 Oct;134[4], 824-30). Both groups were allowed to use petroleum jelly.
“So that is something you can potentially make a recommendation for,” she said.
Dr. Schaffer reported having no conflicts of interest.
EXPERT ANALYSIS FROM THE AAD SUMMER ACADEMY 2016

Food allergy testing only rarely needed for AD patients
BOSTON – Between 15% and 30% of children with moderate to severe atopic dermatitis also have food allergies, but the allergies are a trigger for AD in only a small subset of patients, according to Mercedes E. Gonzalez, MD.
In most cases, allergy testing is not indicated, she said at the American Academy of Dermatology summer meeting.

She described a scenario involving a parent who is concerned that a food allergy is causing her child’s AD. The child has had no hives, no lip swelling, and no other signs of immediate hypersensitivity. In such a case, the best approach is to treat with topical therapies and follow the patient clinically.
“Allergy testing independent of history is not recommended,” she said.
However, in cases involving a significant concern about food allergy, such as the presence of hives or urticaria, or when the child has severe dermatitis that is not improving with optimized topical therapies, an assessment can be undertaken, said Dr. Gonzalez of the University of Miami.
She recommended limited food allergy testing – for common culprits such as cow’s milk, eggs, wheat, soy, and peanuts – in children younger than age 5 years with moderate to severe AD, if the AD persists despite optimized topical treatment and/or a history of immediate and reproducible reaction after ingestion of a specific food.
Food elimination diets based solely on the findings of food allergy test results are not recommended for managing AD, she noted.
If a patient has true immunoglobulin E–mediated allergy they should practice avoidance to prevent potential serious health sequelae, Dr. Gonzalez said.
When testing is done, keep in mind that skin prick tests and serum-specific IgE levels have high negative predictive values above 95%, but low specificity and positive predictive values of 40%-60%, she pointed out. Positive tests should be verified with a food elimination diet or oral food challenge.
Also, most children develop tolerance to the foods over time and should be retested, Dr. Gonzalez said.
Early peanut introduction advised in infants with AD
There is no need to delay the introduction of peanuts into the diet of an infant at high risk for atopic dermatitis, Dr. Gonzalez said.
A 2015 consensus communication from the American Academy of Pediatrics and numerous other organizations, including the American Academy of Allergy, Asthma & Immunology and the Society of Pediatric Dermatology, offering interim guidance on the topic calls for introduction of peanut products into the diets of high-risk infants in countries where peanut allergy is present, she said.
High-risk infants were defined in the study as those with egg allergy and/or severe eczema.
The guidance, which the AAP “endorses and accepts as its policy” pending more formal guidelines currently in development, was based largely on findings from the LEAP (Learn Early About Peanut Allergy) trial – a 5-year randomized, controlled trial of 640 high-risk infants aged 4-11 months. The trial showed that 17.2% of infants who avoided peanuts had peanut allergy at 5 years, compared with 3.2% of those with peanut consumption three times weekly, a relative risk reduction of 81% (N Engl J Med. 2015; 372:803-13).
In infants with egg allergy or severe eczema, an evaluation by an allergist or dermatologist familiar with the guidance may be warranted to assist in implementing the suggestions, Dr. Gonzalez said.
Dr. Gonzalez reported receiving honoraria for serving as a speaker and/or advisory board member for Pierre Fabre Dermatologie, Anacor Pharmaceuticals, Encore Dermatology, and PuraCap Pharmaceutical.
BOSTON – Between 15% and 30% of children with moderate to severe atopic dermatitis also have food allergies, but the allergies are a trigger for AD in only a small subset of patients, according to Mercedes E. Gonzalez, MD.
In most cases, allergy testing is not indicated, she said at the American Academy of Dermatology summer meeting.
She described a scenario involving a parent who is concerned that a food allergy is causing her child’s AD. The child has had no hives, no lip swelling, and no other signs of immediate hypersensitivity. In such a case, the best approach is to treat with topical therapies and follow the patient clinically.
“Allergy testing independent of history is not recommended,” she said.
However, in cases involving a significant concern about food allergy, such as the presence of hives or urticaria, or when the child has severe dermatitis that is not improving with optimized topical therapies, an assessment can be undertaken, said Dr. Gonzalez of the University of Miami.
She recommended limited food allergy testing – for common culprits such as cow’s milk, eggs, wheat, soy, and peanuts – in children younger than age 5 years with moderate to severe AD, if the AD persists despite optimized topical treatment and/or a history of immediate and reproducible reaction after ingestion of a specific food.
Food elimination diets based solely on the findings of food allergy test results are not recommended for managing AD, she noted.
If a patient has true immunoglobulin E–mediated allergy they should practice avoidance to prevent potential serious health sequelae, Dr. Gonzalez said.
When testing is done, keep in mind that skin prick tests and serum-specific IgE levels have high negative predictive values above 95%, but low specificity and positive predictive values of 40%-60%, she pointed out. Positive tests should be verified with a food elimination diet or oral food challenge.
Also, most children develop tolerance to the foods over time and should be retested, Dr. Gonzalez said.
Early peanut introduction advised in infants with AD
There is no need to delay the introduction of peanuts into the diet of an infant at high risk for atopic dermatitis, Dr. Gonzalez said.
A 2015 consensus communication from the American Academy of Pediatrics and numerous other organizations, including the American Academy of Allergy, Asthma & Immunology and the Society of Pediatric Dermatology, offering interim guidance on the topic calls for introduction of peanut products into the diets of high-risk infants in countries where peanut allergy is present, she said.
High-risk infants were defined in the study as those with egg allergy and/or severe eczema.
The guidance, which the AAP “endorses and accepts as its policy” pending more formal guidelines currently in development, was based largely on findings from the LEAP (Learn Early About Peanut Allergy) trial – a 5-year randomized, controlled trial of 640 high-risk infants aged 4-11 months. The trial showed that 17.2% of infants who avoided peanuts had peanut allergy at 5 years, compared with 3.2% of those with peanut consumption three times weekly, a relative risk reduction of 81% (N Engl J Med. 2015; 372:803-13).
In infants with egg allergy or severe eczema, an evaluation by an allergist or dermatologist familiar with the guidance may be warranted to assist in implementing the suggestions, Dr. Gonzalez said.
Dr. Gonzalez reported receiving honoraria for serving as a speaker and/or advisory board member for Pierre Fabre Dermatologie, Anacor Pharmaceuticals, Encore Dermatology, and PuraCap Pharmaceutical.
BOSTON – Between 15% and 30% of children with moderate to severe atopic dermatitis also have food allergies, but the allergies are a trigger for AD in only a small subset of patients, according to Mercedes E. Gonzalez, MD.
In most cases, allergy testing is not indicated, she said at the American Academy of Dermatology summer meeting.
She described a scenario involving a parent who is concerned that a food allergy is causing her child’s AD. The child has had no hives, no lip swelling, and no other signs of immediate hypersensitivity. In such a case, the best approach is to treat with topical therapies and follow the patient clinically.
“Allergy testing independent of history is not recommended,” she said.
However, in cases involving a significant concern about food allergy, such as the presence of hives or urticaria, or when the child has severe dermatitis that is not improving with optimized topical therapies, an assessment can be undertaken, said Dr. Gonzalez of the University of Miami.
She recommended limited food allergy testing – for common culprits such as cow’s milk, eggs, wheat, soy, and peanuts – in children younger than age 5 years with moderate to severe AD, if the AD persists despite optimized topical treatment and/or a history of immediate and reproducible reaction after ingestion of a specific food.
Food elimination diets based solely on the findings of food allergy test results are not recommended for managing AD, she noted.
If a patient has true immunoglobulin E–mediated allergy they should practice avoidance to prevent potential serious health sequelae, Dr. Gonzalez said.
When testing is done, keep in mind that skin prick tests and serum-specific IgE levels have high negative predictive values above 95%, but low specificity and positive predictive values of 40%-60%, she pointed out. Positive tests should be verified with a food elimination diet or oral food challenge.
Also, most children develop tolerance to the foods over time and should be retested, Dr. Gonzalez said.
Early peanut introduction advised in infants with AD
There is no need to delay the introduction of peanuts into the diet of an infant at high risk for atopic dermatitis, Dr. Gonzalez said.
A 2015 consensus communication from the American Academy of Pediatrics and numerous other organizations, including the American Academy of Allergy, Asthma & Immunology and the Society of Pediatric Dermatology, offering interim guidance on the topic calls for introduction of peanut products into the diets of high-risk infants in countries where peanut allergy is present, she said.
High-risk infants were defined in the study as those with egg allergy and/or severe eczema.
The guidance, which the AAP “endorses and accepts as its policy” pending more formal guidelines currently in development, was based largely on findings from the LEAP (Learn Early About Peanut Allergy) trial – a 5-year randomized, controlled trial of 640 high-risk infants aged 4-11 months. The trial showed that 17.2% of infants who avoided peanuts had peanut allergy at 5 years, compared with 3.2% of those with peanut consumption three times weekly, a relative risk reduction of 81% (N Engl J Med. 2015; 372:803-13).
In infants with egg allergy or severe eczema, an evaluation by an allergist or dermatologist familiar with the guidance may be warranted to assist in implementing the suggestions, Dr. Gonzalez said.
Dr. Gonzalez reported receiving honoraria for serving as a speaker and/or advisory board member for Pierre Fabre Dermatologie, Anacor Pharmaceuticals, Encore Dermatology, and PuraCap Pharmaceutical.
EXPERT ANALYSIS FROM AAD SUMMER ACADEMY 2016

Pilot program helps children better understand food allergies
Elementary school students had improved attitudes toward food allergies and felt more confident in taking action during a food allergy emergency after completing an education program on the subject, results of a pilot study in Japan suggest.
At present there is no standard school curriculum for children regarding food allergy, wrote Dr. Kiwako Yamamoto-Hanada of the National Center for Child Health and Development, Tokyo, and associates, so they developed such a program consisting of two 60-minute sessions. A total of 36 elementary school children, 8 of whom had a history of food allergies, filled out questionnaires before and after participating in the program.
After completing the program, 79% of the students stated that it should be included in the school curriculum. Students also demonstrated improved knowledge about food allergies, with a greater percentage knowing what an EpiPen is (100% vs. 0%), understanding that food allergy is related to death (100% vs. 43%), and feeling confident that they could take immediate action if they saw a food allergy emergency (61% vs. 4%). “This is the first report to find that a [food allergy] program for elementary schoolchildren was well tolerated and that perceptions and attitudes toward [food allergies] improved,” the investigators wrote.
The authors stated that they had no financial conflicts of interest.
Read the full story here: http://dx.doi.org/10.1016/j.anai.2016.06.018
Elementary school students had improved attitudes toward food allergies and felt more confident in taking action during a food allergy emergency after completing an education program on the subject, results of a pilot study in Japan suggest.
At present there is no standard school curriculum for children regarding food allergy, wrote Dr. Kiwako Yamamoto-Hanada of the National Center for Child Health and Development, Tokyo, and associates, so they developed such a program consisting of two 60-minute sessions. A total of 36 elementary school children, 8 of whom had a history of food allergies, filled out questionnaires before and after participating in the program.
After completing the program, 79% of the students stated that it should be included in the school curriculum. Students also demonstrated improved knowledge about food allergies, with a greater percentage knowing what an EpiPen is (100% vs. 0%), understanding that food allergy is related to death (100% vs. 43%), and feeling confident that they could take immediate action if they saw a food allergy emergency (61% vs. 4%). “This is the first report to find that a [food allergy] program for elementary schoolchildren was well tolerated and that perceptions and attitudes toward [food allergies] improved,” the investigators wrote.
The authors stated that they had no financial conflicts of interest.
Read the full story here: http://dx.doi.org/10.1016/j.anai.2016.06.018
Elementary school students had improved attitudes toward food allergies and felt more confident in taking action during a food allergy emergency after completing an education program on the subject, results of a pilot study in Japan suggest.
At present there is no standard school curriculum for children regarding food allergy, wrote Dr. Kiwako Yamamoto-Hanada of the National Center for Child Health and Development, Tokyo, and associates, so they developed such a program consisting of two 60-minute sessions. A total of 36 elementary school children, 8 of whom had a history of food allergies, filled out questionnaires before and after participating in the program.
After completing the program, 79% of the students stated that it should be included in the school curriculum. Students also demonstrated improved knowledge about food allergies, with a greater percentage knowing what an EpiPen is (100% vs. 0%), understanding that food allergy is related to death (100% vs. 43%), and feeling confident that they could take immediate action if they saw a food allergy emergency (61% vs. 4%). “This is the first report to find that a [food allergy] program for elementary schoolchildren was well tolerated and that perceptions and attitudes toward [food allergies] improved,” the investigators wrote.
The authors stated that they had no financial conflicts of interest.
Read the full story here: http://dx.doi.org/10.1016/j.anai.2016.06.018
FROM ANNALS OF ALLERGY, ASTHMA & IMMUNOLOGY
Routine screening unwarranted in siblings of food-allergic children
FROM THE JOURNAL OF ALLERGY AND CLINICAL IMMUNOLOGY: IN PRACTICE
Siblings of children with food allergy should not be screened routinely for such allergies before food introduction, results of a large cohort study suggest.
In a study of 478 food-allergic children and 642 of their siblings, 53% of the siblings were sensitized without clinical reactivity and only 13.6% were found to have a clinically reactive food allergy, wrote Ruchi S. Gupta, MD, of Northwestern University, Chicago, and her associates.

The investigators noted that their findings support current guidelines from the National Institute of Allergy and Infectious Diseases to not screen siblings based on another sibling having a food allergy.
“Given the lack of a dramatically increased risk of food allergy in siblings, compared with that of the general population, as well as the high rate of what are falsely positive diagnostic test results among siblings of a food allergic child, [these siblings] should not have routine screening for food allergy before food introduction,” the investigators concluded. “Such siblings are likely to be mislabeled as allergic when they are actually tolerant to the food, which may lead to an increased risk of developing allergy via avoidance,” and both quality of life and nutrition may be adversely impacted.
Dr. Gupta has received research support from Mylan, Food Allergy Research and Education, and United Health Care.
Read the full study here (http://dx.doi.org/10.1016/j.jaip.2016.04.009)
FROM THE JOURNAL OF ALLERGY AND CLINICAL IMMUNOLOGY: IN PRACTICE
Siblings of children with food allergy should not be screened routinely for such allergies before food introduction, results of a large cohort study suggest.
In a study of 478 food-allergic children and 642 of their siblings, 53% of the siblings were sensitized without clinical reactivity and only 13.6% were found to have a clinically reactive food allergy, wrote Ruchi S. Gupta, MD, of Northwestern University, Chicago, and her associates.
The investigators noted that their findings support current guidelines from the National Institute of Allergy and Infectious Diseases to not screen siblings based on another sibling having a food allergy.
“Given the lack of a dramatically increased risk of food allergy in siblings, compared with that of the general population, as well as the high rate of what are falsely positive diagnostic test results among siblings of a food allergic child, [these siblings] should not have routine screening for food allergy before food introduction,” the investigators concluded. “Such siblings are likely to be mislabeled as allergic when they are actually tolerant to the food, which may lead to an increased risk of developing allergy via avoidance,” and both quality of life and nutrition may be adversely impacted.
Dr. Gupta has received research support from Mylan, Food Allergy Research and Education, and United Health Care.
Read the full study here (http://dx.doi.org/10.1016/j.jaip.2016.04.009)
FROM THE JOURNAL OF ALLERGY AND CLINICAL IMMUNOLOGY: IN PRACTICE
Siblings of children with food allergy should not be screened routinely for such allergies before food introduction, results of a large cohort study suggest.
In a study of 478 food-allergic children and 642 of their siblings, 53% of the siblings were sensitized without clinical reactivity and only 13.6% were found to have a clinically reactive food allergy, wrote Ruchi S. Gupta, MD, of Northwestern University, Chicago, and her associates.
The investigators noted that their findings support current guidelines from the National Institute of Allergy and Infectious Diseases to not screen siblings based on another sibling having a food allergy.
“Given the lack of a dramatically increased risk of food allergy in siblings, compared with that of the general population, as well as the high rate of what are falsely positive diagnostic test results among siblings of a food allergic child, [these siblings] should not have routine screening for food allergy before food introduction,” the investigators concluded. “Such siblings are likely to be mislabeled as allergic when they are actually tolerant to the food, which may lead to an increased risk of developing allergy via avoidance,” and both quality of life and nutrition may be adversely impacted.
Dr. Gupta has received research support from Mylan, Food Allergy Research and Education, and United Health Care.
Read the full study here (http://dx.doi.org/10.1016/j.jaip.2016.04.009)

Common Allergic Dermatitis Culprits Are Hiding in Plain Sight
MINNEAPOLIS – When it comes to allergic contact dermatitis in children, the answer is sometimes hiding in plain sight. Cleansers, moisturizers, shampoos, detergents – all can contain ingredients that provoke significant reactions, yet many of these ingredients are not on the most common testing panels.
Erin Warshaw, MD, professor of dermatology at the University of Minnesota, reviewed common but often unsuspected causes of allergic dermatitis in the pediatric population at the annual meeting of the Society for Pediatric Dermatology.

Even some hypoallergenic and frequently recommended products can contain preservatives and other ingredients that provoke allergic reactions, according to Dr. Warshaw. A chief culprit is methylisothiazolinone (MI), a preservative that came into common use as formaldehyde has been gradually phased out.
“If there’s anything I could emphasize from this talk, it’s MI, MI, MI. This is the major epidemic of our time in the contact dermatitis world,” Dr. Warshaw said. Upcoming publications, she added, will place MI in the top five most common contact allergens. “MI is in everything, including things you would think would be hypoallergenic,” she said. She recommended looking at ingredient labels with a keen eye when making testing decisions.
Despite MI’s status as a frequent culprit, it’s not an allergen that appears on common test kits, Dr. Warshaw pointed out. For example, it’s absent from one of the most commonly used test kits, the Thin-Layer Rapid Use Epicutaneous Patch (T.R.U.E. test).
The T.R.U.E. test, said Dr. Warshaw, has reasonable sensitivity – it can detect 71% of relevant positive patch tests (RPPTs) in children. However, she added, a recent study showed that about 23% of children reacted to a supplemental allergen. “That’s significant. One quarter of these individuals only reacted to a preservative … or a sunscreen, or an acrylate. These aren’t on the T.R.U.E. test.”
Decyl glucoside is another frequent culprit that is not included in commercial patch test kits. “It’s really an important emerging allergen,” said Dr. Warshaw, noting that it commonly cross-reacts with coco and lauryl glucoside, frequently found in fragrance-free products. “It’s always humbling when we find the allergen in the product we’ve recommended to our patients.”
Other important allergens not on the T.R.U.E. test include propolis, tocopherol, oxybenzone, and many surfactants and botanicals.
In order to avoid a confounding reaction to aluminum, Dr. Warshaw recommends testing using plastic-backed test chambers, such as IQ chambers, rather than Finn chambers, which are aluminum backed.
When working with families to track down allergens in the pediatric population, Dr. Warshaw adjusts her approach from what she would use for adults.
“What do I do differently in kids? First of all, I set expectations for children and parents,” she said. Some of the most frequent parental questions deal with food allergies, so she allots time to explain the rationale for not testing for food allergens when allergic contact dermatitis is suspected.
For many patients, “I try and frame that there is probably baseline eczema, and our goal is to try to figure out if there is an allergy in addition to that that is contributing to the flares,” she said. She makes sure to convey that “all it takes is one exposure every 3 weeks; that will keep this reaction going.”
However, she’s judicious in interpreting equivocal results. “I feel a responsibility not to label children with an allergy” if results are unclear. Finally, providing enough time is key, said Dr. Warshaw, who allots an hour for reviewing final testing results.
The take-home points? It’s worthwhile to patch test children, since over half of children will have at least one RPPT. Also, contact dermatitis can be an overlay on preexisting allergic dermatitis, so patch testing can still be helpful for these children. Supplemental allergens are important in patch testing, “especially in children with a negative test to a screening series,” Dr. Warshaw said.
She recommended accessing the Contact Allergen Management Program (CAMP) database, found on the American Contact Dermatitis Society website. The list is a searchable database that generates a list of “safe” products that don’t contain a given allergen. This resource is available for society members, but a member’s access code can be shared among faculty members at academic institutions, she said. Patients can also be given unique codes that will give them access for life, so they can use the CAMP database on a computer or via a smartphone app.
Dr. Warshaw reported no relevant financial disclosures.
MINNEAPOLIS – When it comes to allergic contact dermatitis in children, the answer is sometimes hiding in plain sight. Cleansers, moisturizers, shampoos, detergents – all can contain ingredients that provoke significant reactions, yet many of these ingredients are not on the most common testing panels.
Erin Warshaw, MD, professor of dermatology at the University of Minnesota, reviewed common but often unsuspected causes of allergic dermatitis in the pediatric population at the annual meeting of the Society for Pediatric Dermatology.
Even some hypoallergenic and frequently recommended products can contain preservatives and other ingredients that provoke allergic reactions, according to Dr. Warshaw. A chief culprit is methylisothiazolinone (MI), a preservative that came into common use as formaldehyde has been gradually phased out.
“If there’s anything I could emphasize from this talk, it’s MI, MI, MI. This is the major epidemic of our time in the contact dermatitis world,” Dr. Warshaw said. Upcoming publications, she added, will place MI in the top five most common contact allergens. “MI is in everything, including things you would think would be hypoallergenic,” she said. She recommended looking at ingredient labels with a keen eye when making testing decisions.
Despite MI’s status as a frequent culprit, it’s not an allergen that appears on common test kits, Dr. Warshaw pointed out. For example, it’s absent from one of the most commonly used test kits, the Thin-Layer Rapid Use Epicutaneous Patch (T.R.U.E. test).
The T.R.U.E. test, said Dr. Warshaw, has reasonable sensitivity – it can detect 71% of relevant positive patch tests (RPPTs) in children. However, she added, a recent study showed that about 23% of children reacted to a supplemental allergen. “That’s significant. One quarter of these individuals only reacted to a preservative … or a sunscreen, or an acrylate. These aren’t on the T.R.U.E. test.”
Decyl glucoside is another frequent culprit that is not included in commercial patch test kits. “It’s really an important emerging allergen,” said Dr. Warshaw, noting that it commonly cross-reacts with coco and lauryl glucoside, frequently found in fragrance-free products. “It’s always humbling when we find the allergen in the product we’ve recommended to our patients.”
Other important allergens not on the T.R.U.E. test include propolis, tocopherol, oxybenzone, and many surfactants and botanicals.
In order to avoid a confounding reaction to aluminum, Dr. Warshaw recommends testing using plastic-backed test chambers, such as IQ chambers, rather than Finn chambers, which are aluminum backed.
When working with families to track down allergens in the pediatric population, Dr. Warshaw adjusts her approach from what she would use for adults.
“What do I do differently in kids? First of all, I set expectations for children and parents,” she said. Some of the most frequent parental questions deal with food allergies, so she allots time to explain the rationale for not testing for food allergens when allergic contact dermatitis is suspected.
For many patients, “I try and frame that there is probably baseline eczema, and our goal is to try to figure out if there is an allergy in addition to that that is contributing to the flares,” she said. She makes sure to convey that “all it takes is one exposure every 3 weeks; that will keep this reaction going.”
However, she’s judicious in interpreting equivocal results. “I feel a responsibility not to label children with an allergy” if results are unclear. Finally, providing enough time is key, said Dr. Warshaw, who allots an hour for reviewing final testing results.
The take-home points? It’s worthwhile to patch test children, since over half of children will have at least one RPPT. Also, contact dermatitis can be an overlay on preexisting allergic dermatitis, so patch testing can still be helpful for these children. Supplemental allergens are important in patch testing, “especially in children with a negative test to a screening series,” Dr. Warshaw said.
She recommended accessing the Contact Allergen Management Program (CAMP) database, found on the American Contact Dermatitis Society website. The list is a searchable database that generates a list of “safe” products that don’t contain a given allergen. This resource is available for society members, but a member’s access code can be shared among faculty members at academic institutions, she said. Patients can also be given unique codes that will give them access for life, so they can use the CAMP database on a computer or via a smartphone app.
Dr. Warshaw reported no relevant financial disclosures.
MINNEAPOLIS – When it comes to allergic contact dermatitis in children, the answer is sometimes hiding in plain sight. Cleansers, moisturizers, shampoos, detergents – all can contain ingredients that provoke significant reactions, yet many of these ingredients are not on the most common testing panels.
Erin Warshaw, MD, professor of dermatology at the University of Minnesota, reviewed common but often unsuspected causes of allergic dermatitis in the pediatric population at the annual meeting of the Society for Pediatric Dermatology.
Even some hypoallergenic and frequently recommended products can contain preservatives and other ingredients that provoke allergic reactions, according to Dr. Warshaw. A chief culprit is methylisothiazolinone (MI), a preservative that came into common use as formaldehyde has been gradually phased out.
“If there’s anything I could emphasize from this talk, it’s MI, MI, MI. This is the major epidemic of our time in the contact dermatitis world,” Dr. Warshaw said. Upcoming publications, she added, will place MI in the top five most common contact allergens. “MI is in everything, including things you would think would be hypoallergenic,” she said. She recommended looking at ingredient labels with a keen eye when making testing decisions.
Despite MI’s status as a frequent culprit, it’s not an allergen that appears on common test kits, Dr. Warshaw pointed out. For example, it’s absent from one of the most commonly used test kits, the Thin-Layer Rapid Use Epicutaneous Patch (T.R.U.E. test).
The T.R.U.E. test, said Dr. Warshaw, has reasonable sensitivity – it can detect 71% of relevant positive patch tests (RPPTs) in children. However, she added, a recent study showed that about 23% of children reacted to a supplemental allergen. “That’s significant. One quarter of these individuals only reacted to a preservative … or a sunscreen, or an acrylate. These aren’t on the T.R.U.E. test.”
Decyl glucoside is another frequent culprit that is not included in commercial patch test kits. “It’s really an important emerging allergen,” said Dr. Warshaw, noting that it commonly cross-reacts with coco and lauryl glucoside, frequently found in fragrance-free products. “It’s always humbling when we find the allergen in the product we’ve recommended to our patients.”
Other important allergens not on the T.R.U.E. test include propolis, tocopherol, oxybenzone, and many surfactants and botanicals.
In order to avoid a confounding reaction to aluminum, Dr. Warshaw recommends testing using plastic-backed test chambers, such as IQ chambers, rather than Finn chambers, which are aluminum backed.
When working with families to track down allergens in the pediatric population, Dr. Warshaw adjusts her approach from what she would use for adults.
“What do I do differently in kids? First of all, I set expectations for children and parents,” she said. Some of the most frequent parental questions deal with food allergies, so she allots time to explain the rationale for not testing for food allergens when allergic contact dermatitis is suspected.
For many patients, “I try and frame that there is probably baseline eczema, and our goal is to try to figure out if there is an allergy in addition to that that is contributing to the flares,” she said. She makes sure to convey that “all it takes is one exposure every 3 weeks; that will keep this reaction going.”
However, she’s judicious in interpreting equivocal results. “I feel a responsibility not to label children with an allergy” if results are unclear. Finally, providing enough time is key, said Dr. Warshaw, who allots an hour for reviewing final testing results.
The take-home points? It’s worthwhile to patch test children, since over half of children will have at least one RPPT. Also, contact dermatitis can be an overlay on preexisting allergic dermatitis, so patch testing can still be helpful for these children. Supplemental allergens are important in patch testing, “especially in children with a negative test to a screening series,” Dr. Warshaw said.
She recommended accessing the Contact Allergen Management Program (CAMP) database, found on the American Contact Dermatitis Society website. The list is a searchable database that generates a list of “safe” products that don’t contain a given allergen. This resource is available for society members, but a member’s access code can be shared among faculty members at academic institutions, she said. Patients can also be given unique codes that will give them access for life, so they can use the CAMP database on a computer or via a smartphone app.
Dr. Warshaw reported no relevant financial disclosures.
EXPERT ANALYSIS FROM THE SPD ANNUAL MEETING

LABA achieves better asthma control when combined with FDC inhaler
Long-acting beta-2 agonists achieve better asthma control when added to inhaled corticosteroids in a fixed-dose combination, compared with use of a LABA as a separate inhaler, according to Steve Turner, MD, and his associates.
At baseline, 35% of children in the FDC ICS (fixed-dose combination inhaled corticosteroids)/LABA cohort and in the separate ICS+LABA cohort had achieved overall asthma control. After 2 years, 43% of children in the FDC ICS/LABA cohort had achieved overall asthma control, compared with 37% of children in the separate ICS+LABA cohort. The adjusted odds ratio for overall asthma control in the separate ICS+LABA cohort was 0.77.
The adjusted relative risk of acute respiratory events for the separate ICS+LABA cohort was 1.21, compared with the FDC ICS/LABA cohort, and the aRR for severe exacerbations was 1.31 for the separate ICS+LABA cohort. More children in the separate ICS+LABA cohort were treated with antibiotics; however, the incidence of thrush was higher in the FDC ICS/LABA cohort.
“This small effect may be partly explained by improvement in all outcomes in both groups as the children became older. An additional factor may be that adherence was relatively poor for all participants (22%-33%), and poor adherence is associated with poor control. This may have led to the decision to step up and also to a relatively disappointing response to treatment,” the investigators wrote.
Find the full study in the Journal of Allergy and Clinical Immunology (doi:10.1016/j.jaip.2016.06.009).
Long-acting beta-2 agonists achieve better asthma control when added to inhaled corticosteroids in a fixed-dose combination, compared with use of a LABA as a separate inhaler, according to Steve Turner, MD, and his associates.
At baseline, 35% of children in the FDC ICS (fixed-dose combination inhaled corticosteroids)/LABA cohort and in the separate ICS+LABA cohort had achieved overall asthma control. After 2 years, 43% of children in the FDC ICS/LABA cohort had achieved overall asthma control, compared with 37% of children in the separate ICS+LABA cohort. The adjusted odds ratio for overall asthma control in the separate ICS+LABA cohort was 0.77.
The adjusted relative risk of acute respiratory events for the separate ICS+LABA cohort was 1.21, compared with the FDC ICS/LABA cohort, and the aRR for severe exacerbations was 1.31 for the separate ICS+LABA cohort. More children in the separate ICS+LABA cohort were treated with antibiotics; however, the incidence of thrush was higher in the FDC ICS/LABA cohort.
“This small effect may be partly explained by improvement in all outcomes in both groups as the children became older. An additional factor may be that adherence was relatively poor for all participants (22%-33%), and poor adherence is associated with poor control. This may have led to the decision to step up and also to a relatively disappointing response to treatment,” the investigators wrote.
Find the full study in the Journal of Allergy and Clinical Immunology (doi:10.1016/j.jaip.2016.06.009).
Long-acting beta-2 agonists achieve better asthma control when added to inhaled corticosteroids in a fixed-dose combination, compared with use of a LABA as a separate inhaler, according to Steve Turner, MD, and his associates.
At baseline, 35% of children in the FDC ICS (fixed-dose combination inhaled corticosteroids)/LABA cohort and in the separate ICS+LABA cohort had achieved overall asthma control. After 2 years, 43% of children in the FDC ICS/LABA cohort had achieved overall asthma control, compared with 37% of children in the separate ICS+LABA cohort. The adjusted odds ratio for overall asthma control in the separate ICS+LABA cohort was 0.77.
The adjusted relative risk of acute respiratory events for the separate ICS+LABA cohort was 1.21, compared with the FDC ICS/LABA cohort, and the aRR for severe exacerbations was 1.31 for the separate ICS+LABA cohort. More children in the separate ICS+LABA cohort were treated with antibiotics; however, the incidence of thrush was higher in the FDC ICS/LABA cohort.
“This small effect may be partly explained by improvement in all outcomes in both groups as the children became older. An additional factor may be that adherence was relatively poor for all participants (22%-33%), and poor adherence is associated with poor control. This may have led to the decision to step up and also to a relatively disappointing response to treatment,” the investigators wrote.
Find the full study in the Journal of Allergy and Clinical Immunology (doi:10.1016/j.jaip.2016.06.009).
FROM THE JOURNAL OF ALLERGY AND CLINICAL IMMUNOLOGY
Serum vitamin D levels, atopy not significantly linked
SCOTTSDALE, ARIZ. – Serum vitamin D level was not significantly associated with atopic dermatitis or disease severity in a single-center study of more than 600 children and adolescents.
However, “we did observe a strong correlation between average serum vitamin D levels and skin type, as well as body mass index,” said Kavita Darji, a medical student at Saint Louis (Mo.) University, who presented the findings in a poster at the annual meeting of the Society for Investigative Dermatology. Those findings challenge the logic of following universal definitions of vitamin D deficiency, especially given the phenotypic heterogeneity of patients in the United States, she added in an interview.

Serum vitamin D testing is one of most common laboratory assays in this country, but clinicians still debate the risks and benefits of supplementing children and adolescents who test below the Endocrine Society’s threshold for sufficiency (30.0 ng/mL).
To identify factors affecting vitamin D levels, Ms. Darji and her associates reviewed electronic medical charts for patients under age 22 years at Saint Louis University medical centers between 2009 and 2014. The cohort of 655 patients was primarily white (64%) or black (29%), and was nearly equally balanced by gender; their average age was 10 years. The researchers analyzed only the first vitamin D serum measurement for each patient, and defined deficiency as a level under 20 ng/mL, insufficiency as a level between 20 and 29.9 ng/mL, and sufficiency as a level of at least 30 ng/mL.
Serum vitamin D levels were slightly lower among atopic patients, compared with those without atopy, but the difference did not reach statistical significance (about 25 ng/mL vs. about 38 ng/mL; P greater than .05). “We also did not find an association between AD severity and vitamin D level,” Ms. Darji reported. Instead, race and body mass index were the most significant predictors of vitamin D deficiency, probably because these factors directly affect cutaneous photo-induced vitamin D synthesis and the sequestration of fat-soluble vitamins in adipose tissue, she said.
Using the standard definitions, more than 50% of black patients were vitamin D deficient, while less than 30% had sufficient vitamin D levels. In contrast, about 25% of white patients were vitamin D deficient, while nearly 40% had sufficient vitamin D levels (P less than .0001 for proportions of deficiency by race). Furthermore, only about 10% of obese children (those who exceeded the 99th percentile of BMI for age) had sufficient vitamin D levels, compared with more than 40% of underweight children and about 30% of normal-weight children (P less than .00001).
Since vitamin D deficiency was more common among black and obese patients, “maybe they could benefit from a different cut-off value than the standard 30 ng per mL that we used,” Ms. Darji said. “The question is, do they really require these supplements? It may be beneficial to look at the unique characteristics of each patient before supplementing, because the risks of supplementation are considerable in terms of bone health and cardiovascular disease.”
Vitamin D levels did not vary significantly by gender or by month or season measured, Ms. Darji noted. She reported no funding sources and had no disclosures.
SCOTTSDALE, ARIZ. – Serum vitamin D level was not significantly associated with atopic dermatitis or disease severity in a single-center study of more than 600 children and adolescents.
However, “we did observe a strong correlation between average serum vitamin D levels and skin type, as well as body mass index,” said Kavita Darji, a medical student at Saint Louis (Mo.) University, who presented the findings in a poster at the annual meeting of the Society for Investigative Dermatology. Those findings challenge the logic of following universal definitions of vitamin D deficiency, especially given the phenotypic heterogeneity of patients in the United States, she added in an interview.
Serum vitamin D testing is one of most common laboratory assays in this country, but clinicians still debate the risks and benefits of supplementing children and adolescents who test below the Endocrine Society’s threshold for sufficiency (30.0 ng/mL).
To identify factors affecting vitamin D levels, Ms. Darji and her associates reviewed electronic medical charts for patients under age 22 years at Saint Louis University medical centers between 2009 and 2014. The cohort of 655 patients was primarily white (64%) or black (29%), and was nearly equally balanced by gender; their average age was 10 years. The researchers analyzed only the first vitamin D serum measurement for each patient, and defined deficiency as a level under 20 ng/mL, insufficiency as a level between 20 and 29.9 ng/mL, and sufficiency as a level of at least 30 ng/mL.
Serum vitamin D levels were slightly lower among atopic patients, compared with those without atopy, but the difference did not reach statistical significance (about 25 ng/mL vs. about 38 ng/mL; P greater than .05). “We also did not find an association between AD severity and vitamin D level,” Ms. Darji reported. Instead, race and body mass index were the most significant predictors of vitamin D deficiency, probably because these factors directly affect cutaneous photo-induced vitamin D synthesis and the sequestration of fat-soluble vitamins in adipose tissue, she said.
Using the standard definitions, more than 50% of black patients were vitamin D deficient, while less than 30% had sufficient vitamin D levels. In contrast, about 25% of white patients were vitamin D deficient, while nearly 40% had sufficient vitamin D levels (P less than .0001 for proportions of deficiency by race). Furthermore, only about 10% of obese children (those who exceeded the 99th percentile of BMI for age) had sufficient vitamin D levels, compared with more than 40% of underweight children and about 30% of normal-weight children (P less than .00001).
Since vitamin D deficiency was more common among black and obese patients, “maybe they could benefit from a different cut-off value than the standard 30 ng per mL that we used,” Ms. Darji said. “The question is, do they really require these supplements? It may be beneficial to look at the unique characteristics of each patient before supplementing, because the risks of supplementation are considerable in terms of bone health and cardiovascular disease.”
Vitamin D levels did not vary significantly by gender or by month or season measured, Ms. Darji noted. She reported no funding sources and had no disclosures.
SCOTTSDALE, ARIZ. – Serum vitamin D level was not significantly associated with atopic dermatitis or disease severity in a single-center study of more than 600 children and adolescents.
However, “we did observe a strong correlation between average serum vitamin D levels and skin type, as well as body mass index,” said Kavita Darji, a medical student at Saint Louis (Mo.) University, who presented the findings in a poster at the annual meeting of the Society for Investigative Dermatology. Those findings challenge the logic of following universal definitions of vitamin D deficiency, especially given the phenotypic heterogeneity of patients in the United States, she added in an interview.
Serum vitamin D testing is one of most common laboratory assays in this country, but clinicians still debate the risks and benefits of supplementing children and adolescents who test below the Endocrine Society’s threshold for sufficiency (30.0 ng/mL).
To identify factors affecting vitamin D levels, Ms. Darji and her associates reviewed electronic medical charts for patients under age 22 years at Saint Louis University medical centers between 2009 and 2014. The cohort of 655 patients was primarily white (64%) or black (29%), and was nearly equally balanced by gender; their average age was 10 years. The researchers analyzed only the first vitamin D serum measurement for each patient, and defined deficiency as a level under 20 ng/mL, insufficiency as a level between 20 and 29.9 ng/mL, and sufficiency as a level of at least 30 ng/mL.
Serum vitamin D levels were slightly lower among atopic patients, compared with those without atopy, but the difference did not reach statistical significance (about 25 ng/mL vs. about 38 ng/mL; P greater than .05). “We also did not find an association between AD severity and vitamin D level,” Ms. Darji reported. Instead, race and body mass index were the most significant predictors of vitamin D deficiency, probably because these factors directly affect cutaneous photo-induced vitamin D synthesis and the sequestration of fat-soluble vitamins in adipose tissue, she said.
Using the standard definitions, more than 50% of black patients were vitamin D deficient, while less than 30% had sufficient vitamin D levels. In contrast, about 25% of white patients were vitamin D deficient, while nearly 40% had sufficient vitamin D levels (P less than .0001 for proportions of deficiency by race). Furthermore, only about 10% of obese children (those who exceeded the 99th percentile of BMI for age) had sufficient vitamin D levels, compared with more than 40% of underweight children and about 30% of normal-weight children (P less than .00001).
Since vitamin D deficiency was more common among black and obese patients, “maybe they could benefit from a different cut-off value than the standard 30 ng per mL that we used,” Ms. Darji said. “The question is, do they really require these supplements? It may be beneficial to look at the unique characteristics of each patient before supplementing, because the risks of supplementation are considerable in terms of bone health and cardiovascular disease.”
Vitamin D levels did not vary significantly by gender or by month or season measured, Ms. Darji noted. She reported no funding sources and had no disclosures.
AT THE 2016 SID ANNUAL MEETING
Key clinical point: Serum vitamin D was not a significant marker for pediatric atopic dermatitis or disease severity.
Major finding: The average serum vitamin D level was lower among patients with atopic dermatitis than healthy children, but the difference did not reach statistical significance.
Data source: A single-center retrospective review of electronic medical records from 655 patients aged 21 years and younger (average age, 10 years).
Disclosures: Ms. Darji reported no funding sources and had no disclosures.

Trust the thyroid thermostat

Primary hypothyroidism is common. Most patients acquire it when the thyroid gland is damaged by autoimmune inflammation. It is readily and reliably treated with the orally administered synthetic hormone levothyroxine, or less reliably with thyroid gland extracts. Absorption of either medication is significantly influenced by food, so patients need to pay attention to the timing of ingestion. But occasional blood testing can be used to easily monitor the sufficiency of replacement therapy.
The predominant active thyroid hormone is triiodothyronine (T3), most of which is converted from thyroxine (T4) by deiodination outside of the thyroid gland. Circulating thyroid-binding globulins tie up significant amounts of these hormones in the blood, and this protein binding is affected by a number of factors. Free T3 and T4—not bound—are the substances that exert physiologic effects on target organs and also give feedback information to the pituitary gland which, completing the loop, releases thyroid-stimulating hormone (TSH) and ultimately controls the healthy thyroid gland’s production and release of its hormones. Hence, the total circulating thyroid hormone levels are not as biologically relevant as the free T3 and T4 levels. Even in the absence of a functioning thyroid gland, the TSH level reliably reflects the bioactivity of circulating thyroid hormones so long as the pituitary gland is functioning normally.
Routine tracking of the biologic effects of thyroid hormone, such as the metabolic rate, is unreasonable, and other biologic effects such as the cholesterol level are influenced by so many factors in addition to T3 as to be unreliable indicators of thyroid hormone levels. Assuming the patient’s hypothalamic-pituitary axis is normal, the most reasonable and reliable way to track the biologic effect of thyroid hormone is to follow the TSH level. Although the exact relationship between free thyroid hormone and TSH levels is slightly different between patients with normal thyroid glands and those with damaged glands receiving replacement therapy, TSH measurement is an excellent indicator of the level that the brain wants thyroid function to be. Other than in specific nonhomeostatic circumstances, the pituitary gland is usually a superb thermostat for thyroid hormone activity.
In this issue of the Journal, Dr. Christian Nasr discusses the rationale for routinely using TSH measurement alone to direct exogenous thyroid replacement, explaining why it is cost-effective and clinically appropriate.
While following T3 and T4 may occasionally be useful in a few patients, the wealth of clinical data does not support this practice. As a routine practice it is certainly financially wasteful, and may lead to inappropriate clinical decisions.
Why, then, do some physicians persist in regularly following T3 and T4 levels in addition to TSH? There is no single answer. Although some patients may feel “better” if they take a little more rather than a little less levothyroxine, whether this benefit outweighs the metabolic price in the long run is not at all clear. Plus, in the published experience with treating subclinical hypothyroidism,1 patients did not generally feel better or experience desired weight loss when they received slightly more exogenous thyroid hormone. Somewhat analogously, if the TSH level is already normal, increasing thyroid replacement to attain a free T3 or T4 level in the high-normal range is unlikely to improve clinical outcomes in a meaningful way and may well be detrimental in the long term.
Despite a lot of chatter on Internet blogs regarding the multiple benefits of selective T3 replacement and higher T3 levels, akin to testosterone supplementation above what the (normal functioning) hypothalamic-pituitary axis has determined to be biologically appropriate, there is limited clinical evidence to support this practice. When replacing the output of a diseased or absent thyroid gland, it is reasonable clinical practice to trust the pituitary readings of the thyroid thermostat.
- Rugge JB, Bougatsos C, Chou R. Screening and treatment of thyroid dysfunction: an evidence review for the U.S. Preventive Services Task Force. Ann Intern Med 2015; 162:35–45.
Primary hypothyroidism is common. Most patients acquire it when the thyroid gland is damaged by autoimmune inflammation. It is readily and reliably treated with the orally administered synthetic hormone levothyroxine, or less reliably with thyroid gland extracts. Absorption of either medication is significantly influenced by food, so patients need to pay attention to the timing of ingestion. But occasional blood testing can be used to easily monitor the sufficiency of replacement therapy.
The predominant active thyroid hormone is triiodothyronine (T3), most of which is converted from thyroxine (T4) by deiodination outside of the thyroid gland. Circulating thyroid-binding globulins tie up significant amounts of these hormones in the blood, and this protein binding is affected by a number of factors. Free T3 and T4—not bound—are the substances that exert physiologic effects on target organs and also give feedback information to the pituitary gland which, completing the loop, releases thyroid-stimulating hormone (TSH) and ultimately controls the healthy thyroid gland’s production and release of its hormones. Hence, the total circulating thyroid hormone levels are not as biologically relevant as the free T3 and T4 levels. Even in the absence of a functioning thyroid gland, the TSH level reliably reflects the bioactivity of circulating thyroid hormones so long as the pituitary gland is functioning normally.
Routine tracking of the biologic effects of thyroid hormone, such as the metabolic rate, is unreasonable, and other biologic effects such as the cholesterol level are influenced by so many factors in addition to T3 as to be unreliable indicators of thyroid hormone levels. Assuming the patient’s hypothalamic-pituitary axis is normal, the most reasonable and reliable way to track the biologic effect of thyroid hormone is to follow the TSH level. Although the exact relationship between free thyroid hormone and TSH levels is slightly different between patients with normal thyroid glands and those with damaged glands receiving replacement therapy, TSH measurement is an excellent indicator of the level that the brain wants thyroid function to be. Other than in specific nonhomeostatic circumstances, the pituitary gland is usually a superb thermostat for thyroid hormone activity.
In this issue of the Journal, Dr. Christian Nasr discusses the rationale for routinely using TSH measurement alone to direct exogenous thyroid replacement, explaining why it is cost-effective and clinically appropriate.
While following T3 and T4 may occasionally be useful in a few patients, the wealth of clinical data does not support this practice. As a routine practice it is certainly financially wasteful, and may lead to inappropriate clinical decisions.
Why, then, do some physicians persist in regularly following T3 and T4 levels in addition to TSH? There is no single answer. Although some patients may feel “better” if they take a little more rather than a little less levothyroxine, whether this benefit outweighs the metabolic price in the long run is not at all clear. Plus, in the published experience with treating subclinical hypothyroidism,1 patients did not generally feel better or experience desired weight loss when they received slightly more exogenous thyroid hormone. Somewhat analogously, if the TSH level is already normal, increasing thyroid replacement to attain a free T3 or T4 level in the high-normal range is unlikely to improve clinical outcomes in a meaningful way and may well be detrimental in the long term.
Despite a lot of chatter on Internet blogs regarding the multiple benefits of selective T3 replacement and higher T3 levels, akin to testosterone supplementation above what the (normal functioning) hypothalamic-pituitary axis has determined to be biologically appropriate, there is limited clinical evidence to support this practice. When replacing the output of a diseased or absent thyroid gland, it is reasonable clinical practice to trust the pituitary readings of the thyroid thermostat.
Primary hypothyroidism is common. Most patients acquire it when the thyroid gland is damaged by autoimmune inflammation. It is readily and reliably treated with the orally administered synthetic hormone levothyroxine, or less reliably with thyroid gland extracts. Absorption of either medication is significantly influenced by food, so patients need to pay attention to the timing of ingestion. But occasional blood testing can be used to easily monitor the sufficiency of replacement therapy.
The predominant active thyroid hormone is triiodothyronine (T3), most of which is converted from thyroxine (T4) by deiodination outside of the thyroid gland. Circulating thyroid-binding globulins tie up significant amounts of these hormones in the blood, and this protein binding is affected by a number of factors. Free T3 and T4—not bound—are the substances that exert physiologic effects on target organs and also give feedback information to the pituitary gland which, completing the loop, releases thyroid-stimulating hormone (TSH) and ultimately controls the healthy thyroid gland’s production and release of its hormones. Hence, the total circulating thyroid hormone levels are not as biologically relevant as the free T3 and T4 levels. Even in the absence of a functioning thyroid gland, the TSH level reliably reflects the bioactivity of circulating thyroid hormones so long as the pituitary gland is functioning normally.
Routine tracking of the biologic effects of thyroid hormone, such as the metabolic rate, is unreasonable, and other biologic effects such as the cholesterol level are influenced by so many factors in addition to T3 as to be unreliable indicators of thyroid hormone levels. Assuming the patient’s hypothalamic-pituitary axis is normal, the most reasonable and reliable way to track the biologic effect of thyroid hormone is to follow the TSH level. Although the exact relationship between free thyroid hormone and TSH levels is slightly different between patients with normal thyroid glands and those with damaged glands receiving replacement therapy, TSH measurement is an excellent indicator of the level that the brain wants thyroid function to be. Other than in specific nonhomeostatic circumstances, the pituitary gland is usually a superb thermostat for thyroid hormone activity.
In this issue of the Journal, Dr. Christian Nasr discusses the rationale for routinely using TSH measurement alone to direct exogenous thyroid replacement, explaining why it is cost-effective and clinically appropriate.
While following T3 and T4 may occasionally be useful in a few patients, the wealth of clinical data does not support this practice. As a routine practice it is certainly financially wasteful, and may lead to inappropriate clinical decisions.
Why, then, do some physicians persist in regularly following T3 and T4 levels in addition to TSH? There is no single answer. Although some patients may feel “better” if they take a little more rather than a little less levothyroxine, whether this benefit outweighs the metabolic price in the long run is not at all clear. Plus, in the published experience with treating subclinical hypothyroidism,1 patients did not generally feel better or experience desired weight loss when they received slightly more exogenous thyroid hormone. Somewhat analogously, if the TSH level is already normal, increasing thyroid replacement to attain a free T3 or T4 level in the high-normal range is unlikely to improve clinical outcomes in a meaningful way and may well be detrimental in the long term.
Despite a lot of chatter on Internet blogs regarding the multiple benefits of selective T3 replacement and higher T3 levels, akin to testosterone supplementation above what the (normal functioning) hypothalamic-pituitary axis has determined to be biologically appropriate, there is limited clinical evidence to support this practice. When replacing the output of a diseased or absent thyroid gland, it is reasonable clinical practice to trust the pituitary readings of the thyroid thermostat.
- Rugge JB, Bougatsos C, Chou R. Screening and treatment of thyroid dysfunction: an evidence review for the U.S. Preventive Services Task Force. Ann Intern Med 2015; 162:35–45.
- Rugge JB, Bougatsos C, Chou R. Screening and treatment of thyroid dysfunction: an evidence review for the U.S. Preventive Services Task Force. Ann Intern Med 2015; 162:35–45.
Systemic Lupus Erythematosus: The Devastatingly Deceptive Disease
CE/CME No: CR-1608
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest and evaluation. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Describe the pathophysiology and explain the various clinical manifestations of systemic lupus erythematosus (SLE).
• Define the differential diagnosis for SLE.
• List the elements of the laboratory work-up used in the diagnosis of lupus.
• Describe the therapeutic options for patients with SLE.
FACULTY
Michael Felz is an Assistant Professor at Augusta University (formerly Georgia Regents University) in Augusta, Georgia. Mary Bailey Wickham is a PA student in her final year at Augusta University.
The authors have no financial relationships to disclose.
ACCREDITATION STATEMENT
![]()
This program has been reviewed and is approved for a maximum of 1.0 hour of American Academy of Physician Assistants (AAPA) Category 1 CME credit by the Physician Assistant Review Panel. [NPs: Both ANCC and the AANP Certification Program recognize AAPA as an approved provider of Category 1 credit.] Approval is valid for one year from the issue date of August 2016.
Article begins on next page >>
Systemic lupus erythematosus (SLE) is a chronic autoimmune disease that often goes undiagnosed initially. Timely detection of SLE is important, because prompt treatment can prevent its many major complications—notably, end organ damage. Here’s how to distinguish SLE from other illnesses with similar presentations and how to recognize the complications of undiagnosed SLE, which can progress rapidly and fatally.
Systemic lupus erythematosus (SLE) is a chronic inflammatory disorder that can involve multiple organ systems. The presence of antinuclear antibodies (ANA) is a common marker for this disease. In autoimmune diseases such as SLE, the immune system attacks the cells of healthy tissues throughout the body. Genetic, hormonal, and environmental factors (eg, ultraviolet light, infectious viruses, and even use of certain medications) have been implicated in the pathogenesis.1-3
It is estimated that 1.5 million people in the United States and up to 5 million people worldwide have SLE.4 It is nine to 10 times more prevalent in women—especially those of reproductive age—than menand occurs more frequently in African-American, Hispanic, and Asian women than in non-Hispanic Caucasian women.1,2,4-6 Siblings of SLE patients are 30 times more likely to develop the disease, compared to individuals without an affected relative.2 Increased mortality in persons with SLE is attributed to accelerated atherosclerosis, infection, malignancy, and target organ damage, particularly end-stage renal disease.3 Women ages 33 to 45 with SLE are at increased risk (50x greater) for myocardial infarction due to premature atherosclerosis than age-matched women in the general population.7 The life expectancy of SLE patients with renal damage is 23.7 years less than that of the general population.8
Increased awareness of SLE has led to drastic improvements in associated mortality over the past five decades. The survival rate in the 1950s was 50% at 2 years, while current rates are about 95% at 5 years and about 90% at 10 years.3,9 These improvements likely reflect earlier diagnosis and treatment on the part of well-informed clinicians, as well as more effective treatment.
SLE MANIFESTATIONS
SLE can affect any organ in the body with a broad spectrum of clinical manifestations, making it a devastatingly deceptive disease. Disease severity may vary by age, by organ involvement, and over time. Onset may be gradual and mild or rapidly progressive with severe organ involvement. Constitutional manifestations such as fatigue, weight loss, anorexia, and low-grade fever often serve as initial complaints. However, these features are common to a variety of infectious and inflammatory conditions, making early SLE easily overlooked and frequently misdiagnosed. 2
A mix of manifestations involving the joints, skin, mouth, kidneys, lungs, heart, and nervous system offers clues to the diagnosis of SLE (see Table 1). Arthritis is the most common symptom, occurring in 85% to 90% of SLE cases.1,10 It is typically nonerosive, inflammatory, symmetric or asymmetric, and polyarticular (involving five or more joints)and may be accompanied by constitutional symptoms.1,2,11 The joints most commonly affected are the proximal interphalangeals, metacarpophalangeals (MCP), knees, and wrists.2 Morning stiffness is a common complaint.1,11 Jaccoud arthropathy, which is characterized by reducible, nonerosive joint subluxations (eg, swan neck deformities, ulnar deviation, boutonniere deformities, and z-shaped thumbs), can be seen in SLE patients.3 When patients present with articular and constitutional symptoms but lack other typical manifestations of SLE, such as skin rash, appropriate measures—for example, arthrocentesis—should be taken to evaluate for infection.11
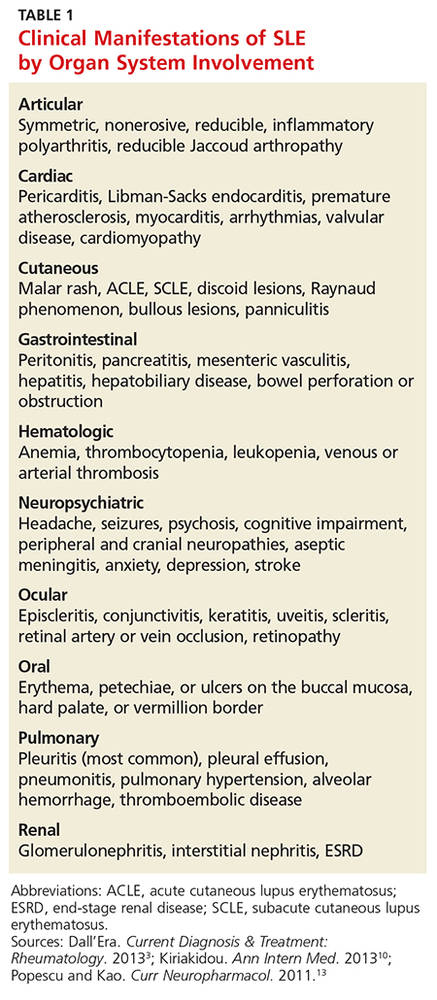
Cutaneous manifestations are the second most common feature at disease onset, with photosensitivity and malar rash being the most prevalent.10 Nearly all patients experience skin lesions at some point during the disease course.1 Diagnostic, or lupus-specific, lesions can be classified into three types: acute, subacute, and chronic.
Acute cutaneous lupus erythematosus (ACLE) is almost always associated with SLE, while subacute cutaneous lupus erythematosus (SCLE) is seen in about 50% of SLE patients.12 ACLE is usually precipitated by sunlight exposure and includes the classic erythematous, macular, “butterfly” rash located on the malar regions of the face, which may remain for days to weeks.2,12 Diffuse or discoid alopecia also may develop in ACLE, along with oral ulcers arising in purpuric necrotic lesions on the palate, buccal mucosa, or gums. Generalized erythematous, papular, or urticarial lesions may affect the face, arms, dorsa of the hands, or “V” of the neck.12
SCLE tends to be sudden in onset, with annular lesions or psoriasiform plaques on the upper trunk, arms, and dorsa of the hands that often coalesce into polycyclic lesions.12 These subacute rashes are often associated with anti-SSA/Ro antibodies.
Chronic cutaneous lupus erythematosus is usually characterized by skin disease alone.12 Discoid lupus is the most common type, with circular scaly plaques with erythematous, hyperpigmented rims and atrophic hypopigmented centers that leave scars.2,12 It is commonly seen on the face, neck, and scalp.
During the course of SLE, mucous membrane involvement—typically painless oral or nasal ulcers—occurs in 25% to 45% of patients.2 Oral lesions are most commonly found on the hard palate and buccal mucosa.3,12
Lupus nephritis, perhaps the most dangerous manifestation of SLE, conveys high risk for organ failure, a higher mortality rate compared to patients without renal involvement, and lower life expectancy.8,11 Up to 60% of Asians, African Americans, and Hispanics develop renal disease during the course of their illness.8 The dominant feature is proteinuria, typically accompanied by microscopic hematuria.2
Neuropsychiatric SLE (NPSLE) is a clinical manifestation that is poorly understood.13 An estimated 28% to 40% of NPSLE manifestations develop prior to or synchronous with the diagnosis, and 63% arise within the first year of diagnosis.13 Mild cognitive impairment is the most common manifestation,reported in up to 20% to 30% of SLE patients.2,13 Seizures and psychosis are reported in 7% to 10% of SLE patients, and psychosis—characterized by hallucinations or delusions—in 3.5%.2
Cardiac findings are common among SLE patients, with an estimated prevalence of 50%, but are rarely the presenting manifestation.14 Pericarditis with effusion is the most common cardiac manifestation, occurring in 25% of SLE patients.2 Advancing atherosclerosis due to chronic inflammation becomes a major cause of mortality in the later years for SLE patients.1 Compared to the general population, the incidence of myocardial infarction in SLE patients is increased fivefold.1 Pleuritis is the most common pleuropulmonary manifestation in SLE.11 Pleuritic chest pain with or without a pleural effusion occurs in 45% to 60% of SLE patients.2
Continue for differential diagnoses >>
DIFFERENTIAL DIAGNOSES
The differential diagnosis for SLE includes rheumatoid arthritis (RA), septic arthritis, mixed connective tissue disease (MCTD), Sjögren syndrome, systemic sclerosis (SSc), polymyositis (PM), fibromyalgia, and drug-induced lupus. Symmetrical, inflammatory, polyarticular arthritis with a predilection for the wrist and MCP joints occurs in both RA and SLE.1,15 And, because the initial articular features of SLE are symmetric arthralgias, patients with SLE are frequently misdiagnosed with RA. The absence of destructive bony erosions on radiographs and large joint effusions, along with the joint reducibility in SLE, can help distinguish it from RA.16 Asymmetric arthritis, which can be a presenting feature in both RA and SLE, is more commonly seen in the latter. ANA and rheumatoid factor test results can be positive in both disorders, but antibodies to anti-cyclic citrullinated peptides, with a 95% specificity for RA but absent in SLE, distinguish RA from SLE.1,16
Patients with MCTD display an array of overlapping features of SLE, PM, and SSc, making the diagnosis difficult.17 Although MCTD can evolve into other connective tissue diseases, such as SLE, it is nonetheless considered a distinct entity.17 High titers of anti-U1 ribonucleoprotein (anti-U1RNP) antibodies are indicative of MCTD. Anti-U1RNP is rarely detected in SLE and almost never seen in other rheumatic diseases.17 Typical manifestations of MCTD are Raynaud phenomenon, swollen fingers (referred to as “sausage digits”), and protuberant polyarthritis.17
Anti-SSA/Ro and anti-SSB/La antibodies, although detectable in SLE patients, are more commonly associated with Sjögren syndrome. In addition, patients with Sjögren syndrome frequently demonstrate signs of keratoconjunctivitis sicca and xerostomia.16
The clinical features of fibromyalgia include diffuse musculoskeletal pain that readily mimics SLE arthralgias. The 2011 modification of the 2010 American College of Rheumatology (ACR) preliminary diagnostic criteria for fibromyalgia serves as a reliable tool for diagnosing patients with nonspecific, diffuse pain.18 This 2011 modification includes 19 pain locations and the six self-reported symptoms: fatigue, impaired sleep, headaches, depression, poor cognition, and abdominal pain.18
SSc, also known as scleroderma, is characterized by skin thickening and/or CREST syndrome (calcinosis, Raynaud phenomenon, esophageal dysmotility, sclerodactyly, telangiectasia). The presence of anti-Scl-70 and anti-centromere antibodies are noted as well.16
Finally, a suspicion of SLE mandates an evaluation for drug-induced lupus by assessing the patient’s exposure to culprit medications, such as hydralazine, procainamide, isoniazid, methyldopa, chlorpromazine, quinidine, minocycline, and tumor necrosis factor inhibitiors.1,11 Four key features point toward drug-induced lupus:
• The female-to-male ratio is nearly equivalent.
• Nephritis and central nervous system (CNS) manifestations are not commonly present.
• Anti–double-stranded DNA (anti-dsDNA) antibodies and hypocomplementemia are absent.
• The clinical features and laboratory abnormalities return to baseline once the offending agent is removed.1
Anti-histone antibodies are present in approximately 75% of patients with drug-induced lupus but can also be seen in patients with SLE.11
Continue for laboratory work-up >>
LABORATORY WORK-UP
Laboratory abnormalities associated with SLE include anemia, leukopenia, lymphopenia, thrombocytopenia, hypocomplementemia, and proteinuria. A typical work-up includes a routine complete blood count (CBC) with differential, serum creatinine, erythrocyte sedimentation rate (ESR), C-reactive protein (CRP), urinalysis with microscopy, and serologic ANA titer.1,16,19 A CBC with differential may reveal hematologic abnormalities, such as anemia of chronic disease (most commonly) or autoimmune hemolytic anemia, as well as leukopenia and thrombocytopenia due to circulating autoantibodies.3 An elevated ESR and CRP indicate the severity of the systemic inflammation and/or infection. Urinalysis is effective for detecting lupus with renal diseaseand may reveal proteinuria due to renal dysfunction.2
A positive ANA titer indicates widespread activation of the immune system targeted against nuclear and cytoplasmic subparticles. The vast majority of patients with SLE will develop a positive ANA with a high titer at some point during the course of their disease.16 The ANA is highly sensitive for SLE (93% to 95%) but lacks specificity (57%).20The most common tests for ANA are enzyme-linked immunosorbent assay (ELISA) and indirect immunofluorescence assay (IFA). ELISA is more sensitive in detecting ANA, while IFA is the gold standard due to its high specificity.21 Some laboratories may use immunoassay as a screening tool for ANA and then use IFA to confirm positive or equivocal results.21 Positive ANA results can be seen in patients with other rheumatologic diseases and in up to 15% of all healthy persons, but with low or borderline titers.22 For these reasons, ANA testing alone is a poor predictor of SLE.
When either the ANA test results are positive or are negative but a strong clinical suspicion for SLE remains, clinicians should order tests for antibodies to extractable nuclear antigens (ENA panel; see Table 2).3,16 Anti-dsDNA and anti-Smith (anti-Sm) antibodies are both specific for SLE, and levels of anti-dsDNA reflect disease activity in many patients.1,19 In contrast, anti-dsDNA antibodies are found in fewer than 0.5% of healthy individuals and patients with other autoimmune conditions.19 Among patients with high levels of anti-dsDNA antibodies and clinically inactive disease, 80% will have active disease within five years after elevated antibodies are detected.19

Autoantibodies, including ANA, anti-SSA/Ro, anti-SSB/La, and antiphospholipid antibodies, are usually detectable for many years prior to the onset of symptomatic SLE, while others, such as anti-Sm and anti-U1RNP, appear just months before the diagnosis.23 Patients with positive ANA results who do not meet criteria for SLE are still at risk for lupus and other autoimmune diseases, because complex autoimmune changes occur years before the diagnosis of SLE.23 These patients should be followed closely.
Continue for making the diagnosis >>
MAKING THE DIAGNOSIS
Diagnosing SLE may prove problematic because of the remarkable variety of relapsing and remitting clinical features, mimicry of similar conditions, and lack of a simple, definitive diagnostic test. Initial diagnosis of SLE depends on the disease manifestation, published criteria, and exclusion of alternative diagnoses. Confirmation requires careful clinical assessment, based on a thorough medical history and complete physical examination, along with specific laboratory testing.1,16 Biopsy results indicative of lupus nephritis in the presence of ANA or anti-dsDNA antibodies also confirm the diagnosis of SLE.24
Although created for research purposes, ACR classification criteria for SLE, published in 1982 and revised in 1997, have been used for more than 30 years to diagnose lupus (see www.rheumatology.org/Practice-Quality/Clinical-Support/Criteria/ACR-Endorsed-Criteria). In 2012, the Systemic Lupus International Collaborating Clinics (SLICC) group revised the 1997 ACR classification criteria to address major flaws and to improve clinical precision.24 According to SLICC, a definitive diagnosis requires the presence of at least four of 17 criteria, including at least one clinical and one immunologic criterion.24 The SLICC revisions have resulted in fewer misclassifications and provide greater sensitivity but lower specificity in the identification of SLE in comparison to the 1997 ACR criteria.24 To date, no one set of criteria allows for early diagnosis of SLE.
MANAGEMENT OPTIONS
Treatment must be tailored to the patient’s specific organ system involvement. Effective therapy hinges on controlling symptoms and reducing underlying inflammation.25 Four classes of drugs are used: NSAIDs, antimalarial drugs, corticosteroids, and cytotoxic drugs (see Table 3). Most patients benefit from NSAIDs to alleviate minor arthritis and arthralgia symptoms, but the risk for peptic ulcers and nephrotoxicity should be addressed; this may require the concomitant use of gastroprotective agents such as proton pump inhibitors.25 Antimalarials are effective for musculoskeletal symptoms that do not respond to NSAIDs and for cutaneous rashes.1 The current antimalarial drug of choice is hydroxychloroquine (200 to 400 mg/d po), which has been shown to control SLE manifestations by reducing and preventing disease flares.1,11,26 It is well tolerated and can be used for the duration of treatment.11,26 Patients should be informed that this drug’s onset of action is one month.26 In rare cases, this drug can cause retinal toxicity; therefore, SLE patients receiving hydroxychloroquine should be referred to an ophthalmologist for a baseline eye examination and yearly assessments to monitor for this rare adverse effect.25,26
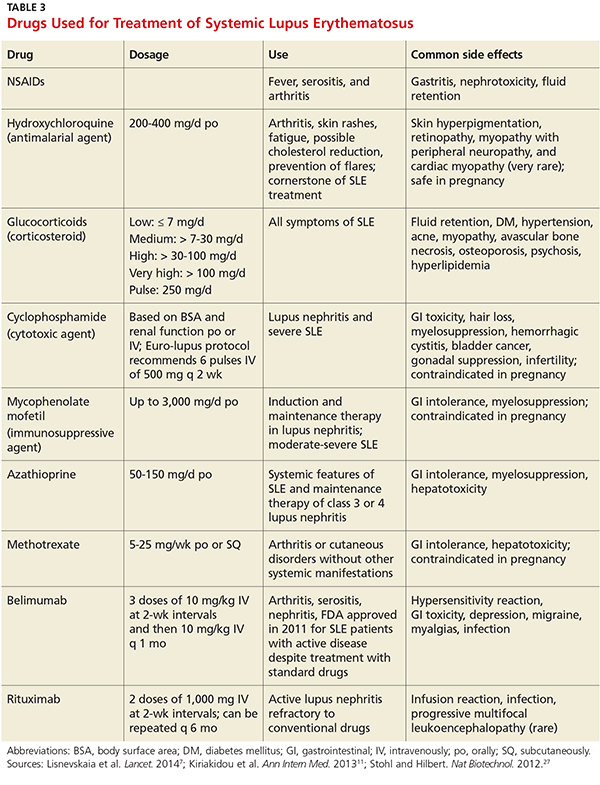
Low-dose corticosteroids, such as oral prednisolone or methylprednisolone, are employed when NSAIDs and antimalarials fail to control arthritis or cutaneous SLE eruptions.25 Major systemic manifestations that occur during a disease flare—such as severe arthritis, hemolytic anemia, glomerulonephritis, alveolar hemorrhage, pericarditis, pleurisy, or CNS involvement—necessitate high-dose IV corticosteroids in conjunction with immunosuppressive agents.1,11,25 These high-dose glucocorticoids should be gradually withdrawn as soon as remission is achieved.11 Long-term suppressive therapy with oral corticosteroids in addition to other agents is often needed to preserve organ function.25
The major adverse effects of long-term glucocorticoids are osteoporosis, hypertension, hyperlipidemia, glucose intolerance, and susceptibility to infection. It is recommended that patients taking prednisolone 7.5 mg/d or more undergo a bone mineral density scan every two years.25 Those with T scores below –2.5 should be prescribed bisphosphonates.25
Immunosuppressive agents, such as cyclophosphamide, mycophenolate mofetil, and azathioprine, are used in conjunction with corticosteroids or when syndromes are resistant to corticosteroids.1 Collaboration between primary care, rheumatology, and nephrology is advisable for patients requiring immunosuppressive or disease-modifying pharmacologic agents.
Two new treatments for SLE are the immunologic agents belimumab and rituximab.7 Belimumab, a monoclonal human antibody, is the first medication in the past 50 years that has been approved by the FDA for antibody-positive SLE patients with active lupus unresponsive to standard treatment.7,27 Rituximab is an anti-CD20 monoclonal antibody, approved by the FDA for non-Hodgkin lymphoma, chronic lymphocytic leukemia, and RA, and is now considered an option for SLE refractory to conventional treatment regimens.7,27 The efficacy of belimumab and rituximab, and the spectrum of indications for their use, are still under study, but these new therapeutic agents hold promise for the treatment of patients with refractory SLE.
Continue for helping patients live with SLE >>
HELPING PATIENTs LIVE WITH SLE
Patients with SLE have a higher mortality rate, as well as a lower quality of life, compared to the general population.28 The major contributors to a decreased quality of life are fatigue, mood disturbances (eg, depression), and chronic pain.28 Practitioners should advise SLE patients to participate in support groups and psychotherapy to alleviate the anxiety and depression associated with this chronic disease.
For patients with long-standing disease, accelerated atherosclerotic cardiovascular disease adds to morbidity and mortality. For this reason, obesity, hypertension, hyperlipidemia, and smoking are targets for intervention. Lifestyle modifications—such as exercise, smoking cessation, a healthy diet with low saturated fat, stress avoidance, and adequate rest—are recommended.26
Avoiding overexposure to sunlight, by using sunscreen with an SPF of at least 30 and wearing sun-protective clothing, is essential for management of cutaneous lupus.25,26 Yearly influenza vaccination is appropriate, as are other immunizations (eg, pneumococcal vaccine).26
Advise women of childbearing age with SLE that lupus flares result in a high risk for miscarriage. All women should undergo yearly cervical cancer screening.26
Patients taking long-term glucocorticoids should adopt bone-protective behaviors, including quitting smoking, limiting alcohol intake, partaking in weight-bearing exercise, and consuming dietary calcium and vitamin D.25 Patients taking these drugs should avoid live virus vaccines. Those on immunosuppressive therapy should be warned about the hazardous adverse effects of glucocorticoids.
MONITORING AND FOLLOW-UP
Collaborative efforts between primary care providers and several types of specialty providers can facilitate coordinated interventions in the long-term management of lupus. Rheumatologists are experts in making therapeutic decisions for SLE.
Patients being treated for SLE require routine monitoring to assess disease activity and detect flares. The European League Against Rheumatism (EULAR) guidelines recommend that monitoring include assessment for new clinical manifestations, routine laboratory tests, and immunologic assays, chiefly anti-dsDNA, anti-Sm, and serum complement levels, coupled with one of the validated global activity indices, such as the Systemic Lupus Erythematosus Disease Activity Index (SLEDAI).29
A routine office visit with a physical examination and laboratory testing for CBC with differential, basic metabolic panel, and urinalysis every three months is recommended for patients with stable disease; patients with uncontrolled SLE may require weekly visits.11,29 Patients taking immunosuppressive drugs should be provided with adverse-effect profiles alerting them to toxicity symptoms and require frequent laboratory monitoring for potential toxicity.11
CONCLUSION
Advances in immunologically targeted serologic tests have shed more light on the underlying pathogenesis of SLE, which in turn has led to improvements in disease detection and monitoring of complications, as well as advances in therapy. Although SLE cannot be cured, emerging therapies targeting different mechanisms of SLE offer hope for patients diagnosed with this complex disease.
1. Hellmann DB, Imboden JB. Rheumatologic & immunologic disorders. In: Papadakis M, McPhee SJ, Rabow MW, eds. Current Medical Diagnosis & Treatment. 53rd ed. New York, NY: McGraw-Hill; 2014:786-836.
2. Bertsias G, Cervera R, Boumpas DT. Systemic lupus erythematosus: pathogenesis and clinical features. In: Bijlsma JWJ, ed. EULAR Textbook on Rheumatic Diseases. London: BMJ Group; 2012:476-505.
3. Dall’Era M. Chapter 21. Systemic lupus erythematosus. In: Imboden JB, Hellmann DB, Stone JH, eds. Current Diagnosis & Treatment: Rheumatology. 3rd ed. New York, NY: McGraw-Hill; 2013.
4. Lupus Foundation of America. What is lupus? www.lupus.org/answers/entry/what-is-lupus. Accessed July 19, 2016.
5. Furst DE, Clarke AE, Fernandes AW, et al. Incidence and prevalence of adult systemic lupus erythematosus in a large US managed-care population. Lupus. 2012;22(1):99-105.
6. Pons-Estel GL, Alarcón GS, Scofield L, et al. Understanding the epidemiology and progression of systemic lupus erythematosus. Semin Arthritis Rheum. 2010;39(4):257-268.
7. Lisnevskaia L, Murphy G, Isenberg D. Systemic lupus erythematosus. Lancet. 2014;384(9957):1878-1888.
8. Mok CC, Kwok RC, Yip PS. Effect of renal disease on the standardized mortality ratio and life expectancy of patients with systemic lupus erythematosus. Arthritis Rheum. 2013;65(8):2154-2160.
9. Merola JF, Bermas B, Lu B, et al. Clinical manifestations and survival among adults with SLE according to age at diagnosis. Lupus. 2014;23(8):778-784.
10. Font J, Cervera R, Ramos-Casals M, et al. Clusters of clinical and immunologic features in systemic lupus erythematosus: analysis of 600 patients from a single center. Semin Arthritis Rheum. 2004;33(4):217-230.
11. Kiriakidou M, Cotton D, Taichman D, Williams S. Systemic lupus erythematosus.Ann Intern Med. 2013;159(7):2-16.
12. Wolff K, Johnson R, Saavedra A. Fitzpatrick’s Color Atlas and Synopsis of Clinical Dermatology. 7th ed. New York, NY: McGraw-Hill; 2013:334-342.
13. Popescu A, Kao AH. Neuropsychiatric systemic lupus erythematosus. Curr Neuropharmacol. 2011;9(3):449-457.
14. Chen PY, Chang CH, Hsu CC, et al. Systemic lupus erythematosus presenting with cardiac symptoms. Am J Emerg Med. 2014;32(9):1117-1119.
15. Hahn BHH. Chapter 378: Systemic lupus erythematosus. In: Kasper DL, Fauci AS, Hauser SL, et al, eds. Harrison’s Principles of Internal Medicine. 19th ed. New York, NY: McGraw-Hill; 2015.
16. Wallace DJ. Diagnosis and differential diagnosis of systemic lupus erythematosus in adults. UpToDate. www.uptodate.com/contents/diagnosis-and-differential-diagnosis-of-systemic-lupus-erythematosus-in-adults. Accessed July 19, 2016.
17. Cappelli S, Bellando Randone S, Martinovic D, et al. “To be or not to be,” ten years after: evidence for mixed connective tissue disease as a distinct entity. Semin Arthritis Rheum. 2012;41(4):589-598.
18. Bennett RM, Friend R, Marcus D, et al. Criteria for the diagnosis of fibromyalgia: validation of the modified 2010 preliminary American College of Rheumatology criteria and the development of alternative criteria. Arthritis Care Res (Hoboken). 2014;66(9):1364-1373.
19. Rahman A, Isenberg DA. Systemic lupus erythematosus. N Engl J Med. 2008;358(9):929-939.
20. Magrey M, Abelson A. Laboratory evaluation of rheumatic diseases. Cleveland Clinic Center for Continuing Education 2010. www.cleveland clinicmeded.com/medicalpubs/diseasemanagement/rheumatology/laboratory-evaluation-rheumatic-diseases/. Accessed July 19, 2016.
21. Copple SS, Sawitzke AD, Wilson AM, et al. Enzyme-linked immunosorbent assay screening then indirect immunofluorescence confirmation of antinuclear antibodies: a statistical analysis. Am J Clin Pathol. 2011;135(5):678-684.
22. Von Feld JM; American College of Rheumatology. Antinuclear antibodies (ANA). 2015. www.rheumatology.org/I-Am-A/Patient-Caregiver/Diseases-Conditions/Antinuclear-Antibodies-ANA. Accessed July 19, 2016.
23. Arbuckle MR, McClain MT, Rubertone MV, et al. Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N Engl J Med. 2003;349(16):1526-1533.
24. Petri M, Orbai A, Alarcón G, et al. Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum. 2012;64(8):2677-2686.
25. Ioannou Y, Isenberg DA. Current concepts for the management of systemic lupus erythematosus in adults: a therapeutic challenge. Postgrad Med J. 2002;78:599-606.
26. Dall’Era M, Wofsy D. Treatment of systemic lupus erythematosus. In: Imboden JB, Hellmann DB, Stone JH, eds. Current Diagnosis & Treatment: Rheumatology. 3rd ed. New York, NY: McGraw-Hill; 2013.
27. Stohl W, Hilbert DM. The discovery and development of belimumab: the anti-BLyS–lupus connection. Nat Biotechnol. 2012;30(1):69-77.
28. Lateef A, Petri M. Unmet medical needs in systemic lupus erythematosus. Arthritis Research & Ther. 2012;14(suppl 4):S4.
29. Bertsias G, Ioannidis JP, Boletis J, et al. EULAR recommendations for the management of systemic lupus erythematosus. Report of a task force of the EULAR standing committee for international clinical studies including therapeutics. Ann Rheum Dis. 2008;67(2):195-205.
CE/CME No: CR-1608
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest and evaluation. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Describe the pathophysiology and explain the various clinical manifestations of systemic lupus erythematosus (SLE).
• Define the differential diagnosis for SLE.
• List the elements of the laboratory work-up used in the diagnosis of lupus.
• Describe the therapeutic options for patients with SLE.
FACULTY
Michael Felz is an Assistant Professor at Augusta University (formerly Georgia Regents University) in Augusta, Georgia. Mary Bailey Wickham is a PA student in her final year at Augusta University.
The authors have no financial relationships to disclose.
ACCREDITATION STATEMENT
![]()
This program has been reviewed and is approved for a maximum of 1.0 hour of American Academy of Physician Assistants (AAPA) Category 1 CME credit by the Physician Assistant Review Panel. [NPs: Both ANCC and the AANP Certification Program recognize AAPA as an approved provider of Category 1 credit.] Approval is valid for one year from the issue date of August 2016.
Article begins on next page >>
Systemic lupus erythematosus (SLE) is a chronic autoimmune disease that often goes undiagnosed initially. Timely detection of SLE is important, because prompt treatment can prevent its many major complications—notably, end organ damage. Here’s how to distinguish SLE from other illnesses with similar presentations and how to recognize the complications of undiagnosed SLE, which can progress rapidly and fatally.
Systemic lupus erythematosus (SLE) is a chronic inflammatory disorder that can involve multiple organ systems. The presence of antinuclear antibodies (ANA) is a common marker for this disease. In autoimmune diseases such as SLE, the immune system attacks the cells of healthy tissues throughout the body. Genetic, hormonal, and environmental factors (eg, ultraviolet light, infectious viruses, and even use of certain medications) have been implicated in the pathogenesis.1-3
It is estimated that 1.5 million people in the United States and up to 5 million people worldwide have SLE.4 It is nine to 10 times more prevalent in women—especially those of reproductive age—than menand occurs more frequently in African-American, Hispanic, and Asian women than in non-Hispanic Caucasian women.1,2,4-6 Siblings of SLE patients are 30 times more likely to develop the disease, compared to individuals without an affected relative.2 Increased mortality in persons with SLE is attributed to accelerated atherosclerosis, infection, malignancy, and target organ damage, particularly end-stage renal disease.3 Women ages 33 to 45 with SLE are at increased risk (50x greater) for myocardial infarction due to premature atherosclerosis than age-matched women in the general population.7 The life expectancy of SLE patients with renal damage is 23.7 years less than that of the general population.8
Increased awareness of SLE has led to drastic improvements in associated mortality over the past five decades. The survival rate in the 1950s was 50% at 2 years, while current rates are about 95% at 5 years and about 90% at 10 years.3,9 These improvements likely reflect earlier diagnosis and treatment on the part of well-informed clinicians, as well as more effective treatment.
SLE MANIFESTATIONS
SLE can affect any organ in the body with a broad spectrum of clinical manifestations, making it a devastatingly deceptive disease. Disease severity may vary by age, by organ involvement, and over time. Onset may be gradual and mild or rapidly progressive with severe organ involvement. Constitutional manifestations such as fatigue, weight loss, anorexia, and low-grade fever often serve as initial complaints. However, these features are common to a variety of infectious and inflammatory conditions, making early SLE easily overlooked and frequently misdiagnosed. 2
A mix of manifestations involving the joints, skin, mouth, kidneys, lungs, heart, and nervous system offers clues to the diagnosis of SLE (see Table 1). Arthritis is the most common symptom, occurring in 85% to 90% of SLE cases.1,10 It is typically nonerosive, inflammatory, symmetric or asymmetric, and polyarticular (involving five or more joints)and may be accompanied by constitutional symptoms.1,2,11 The joints most commonly affected are the proximal interphalangeals, metacarpophalangeals (MCP), knees, and wrists.2 Morning stiffness is a common complaint.1,11 Jaccoud arthropathy, which is characterized by reducible, nonerosive joint subluxations (eg, swan neck deformities, ulnar deviation, boutonniere deformities, and z-shaped thumbs), can be seen in SLE patients.3 When patients present with articular and constitutional symptoms but lack other typical manifestations of SLE, such as skin rash, appropriate measures—for example, arthrocentesis—should be taken to evaluate for infection.11
Cutaneous manifestations are the second most common feature at disease onset, with photosensitivity and malar rash being the most prevalent.10 Nearly all patients experience skin lesions at some point during the disease course.1 Diagnostic, or lupus-specific, lesions can be classified into three types: acute, subacute, and chronic.
Acute cutaneous lupus erythematosus (ACLE) is almost always associated with SLE, while subacute cutaneous lupus erythematosus (SCLE) is seen in about 50% of SLE patients.12 ACLE is usually precipitated by sunlight exposure and includes the classic erythematous, macular, “butterfly” rash located on the malar regions of the face, which may remain for days to weeks.2,12 Diffuse or discoid alopecia also may develop in ACLE, along with oral ulcers arising in purpuric necrotic lesions on the palate, buccal mucosa, or gums. Generalized erythematous, papular, or urticarial lesions may affect the face, arms, dorsa of the hands, or “V” of the neck.12
SCLE tends to be sudden in onset, with annular lesions or psoriasiform plaques on the upper trunk, arms, and dorsa of the hands that often coalesce into polycyclic lesions.12 These subacute rashes are often associated with anti-SSA/Ro antibodies.
Chronic cutaneous lupus erythematosus is usually characterized by skin disease alone.12 Discoid lupus is the most common type, with circular scaly plaques with erythematous, hyperpigmented rims and atrophic hypopigmented centers that leave scars.2,12 It is commonly seen on the face, neck, and scalp.
During the course of SLE, mucous membrane involvement—typically painless oral or nasal ulcers—occurs in 25% to 45% of patients.2 Oral lesions are most commonly found on the hard palate and buccal mucosa.3,12
Lupus nephritis, perhaps the most dangerous manifestation of SLE, conveys high risk for organ failure, a higher mortality rate compared to patients without renal involvement, and lower life expectancy.8,11 Up to 60% of Asians, African Americans, and Hispanics develop renal disease during the course of their illness.8 The dominant feature is proteinuria, typically accompanied by microscopic hematuria.2
Neuropsychiatric SLE (NPSLE) is a clinical manifestation that is poorly understood.13 An estimated 28% to 40% of NPSLE manifestations develop prior to or synchronous with the diagnosis, and 63% arise within the first year of diagnosis.13 Mild cognitive impairment is the most common manifestation,reported in up to 20% to 30% of SLE patients.2,13 Seizures and psychosis are reported in 7% to 10% of SLE patients, and psychosis—characterized by hallucinations or delusions—in 3.5%.2
Cardiac findings are common among SLE patients, with an estimated prevalence of 50%, but are rarely the presenting manifestation.14 Pericarditis with effusion is the most common cardiac manifestation, occurring in 25% of SLE patients.2 Advancing atherosclerosis due to chronic inflammation becomes a major cause of mortality in the later years for SLE patients.1 Compared to the general population, the incidence of myocardial infarction in SLE patients is increased fivefold.1 Pleuritis is the most common pleuropulmonary manifestation in SLE.11 Pleuritic chest pain with or without a pleural effusion occurs in 45% to 60% of SLE patients.2
Continue for differential diagnoses >>
DIFFERENTIAL DIAGNOSES
The differential diagnosis for SLE includes rheumatoid arthritis (RA), septic arthritis, mixed connective tissue disease (MCTD), Sjögren syndrome, systemic sclerosis (SSc), polymyositis (PM), fibromyalgia, and drug-induced lupus. Symmetrical, inflammatory, polyarticular arthritis with a predilection for the wrist and MCP joints occurs in both RA and SLE.1,15 And, because the initial articular features of SLE are symmetric arthralgias, patients with SLE are frequently misdiagnosed with RA. The absence of destructive bony erosions on radiographs and large joint effusions, along with the joint reducibility in SLE, can help distinguish it from RA.16 Asymmetric arthritis, which can be a presenting feature in both RA and SLE, is more commonly seen in the latter. ANA and rheumatoid factor test results can be positive in both disorders, but antibodies to anti-cyclic citrullinated peptides, with a 95% specificity for RA but absent in SLE, distinguish RA from SLE.1,16
Patients with MCTD display an array of overlapping features of SLE, PM, and SSc, making the diagnosis difficult.17 Although MCTD can evolve into other connective tissue diseases, such as SLE, it is nonetheless considered a distinct entity.17 High titers of anti-U1 ribonucleoprotein (anti-U1RNP) antibodies are indicative of MCTD. Anti-U1RNP is rarely detected in SLE and almost never seen in other rheumatic diseases.17 Typical manifestations of MCTD are Raynaud phenomenon, swollen fingers (referred to as “sausage digits”), and protuberant polyarthritis.17
Anti-SSA/Ro and anti-SSB/La antibodies, although detectable in SLE patients, are more commonly associated with Sjögren syndrome. In addition, patients with Sjögren syndrome frequently demonstrate signs of keratoconjunctivitis sicca and xerostomia.16
The clinical features of fibromyalgia include diffuse musculoskeletal pain that readily mimics SLE arthralgias. The 2011 modification of the 2010 American College of Rheumatology (ACR) preliminary diagnostic criteria for fibromyalgia serves as a reliable tool for diagnosing patients with nonspecific, diffuse pain.18 This 2011 modification includes 19 pain locations and the six self-reported symptoms: fatigue, impaired sleep, headaches, depression, poor cognition, and abdominal pain.18
SSc, also known as scleroderma, is characterized by skin thickening and/or CREST syndrome (calcinosis, Raynaud phenomenon, esophageal dysmotility, sclerodactyly, telangiectasia). The presence of anti-Scl-70 and anti-centromere antibodies are noted as well.16
Finally, a suspicion of SLE mandates an evaluation for drug-induced lupus by assessing the patient’s exposure to culprit medications, such as hydralazine, procainamide, isoniazid, methyldopa, chlorpromazine, quinidine, minocycline, and tumor necrosis factor inhibitiors.1,11 Four key features point toward drug-induced lupus:
• The female-to-male ratio is nearly equivalent.
• Nephritis and central nervous system (CNS) manifestations are not commonly present.
• Anti–double-stranded DNA (anti-dsDNA) antibodies and hypocomplementemia are absent.
• The clinical features and laboratory abnormalities return to baseline once the offending agent is removed.1
Anti-histone antibodies are present in approximately 75% of patients with drug-induced lupus but can also be seen in patients with SLE.11
Continue for laboratory work-up >>
LABORATORY WORK-UP
Laboratory abnormalities associated with SLE include anemia, leukopenia, lymphopenia, thrombocytopenia, hypocomplementemia, and proteinuria. A typical work-up includes a routine complete blood count (CBC) with differential, serum creatinine, erythrocyte sedimentation rate (ESR), C-reactive protein (CRP), urinalysis with microscopy, and serologic ANA titer.1,16,19 A CBC with differential may reveal hematologic abnormalities, such as anemia of chronic disease (most commonly) or autoimmune hemolytic anemia, as well as leukopenia and thrombocytopenia due to circulating autoantibodies.3 An elevated ESR and CRP indicate the severity of the systemic inflammation and/or infection. Urinalysis is effective for detecting lupus with renal diseaseand may reveal proteinuria due to renal dysfunction.2
A positive ANA titer indicates widespread activation of the immune system targeted against nuclear and cytoplasmic subparticles. The vast majority of patients with SLE will develop a positive ANA with a high titer at some point during the course of their disease.16 The ANA is highly sensitive for SLE (93% to 95%) but lacks specificity (57%).20The most common tests for ANA are enzyme-linked immunosorbent assay (ELISA) and indirect immunofluorescence assay (IFA). ELISA is more sensitive in detecting ANA, while IFA is the gold standard due to its high specificity.21 Some laboratories may use immunoassay as a screening tool for ANA and then use IFA to confirm positive or equivocal results.21 Positive ANA results can be seen in patients with other rheumatologic diseases and in up to 15% of all healthy persons, but with low or borderline titers.22 For these reasons, ANA testing alone is a poor predictor of SLE.
When either the ANA test results are positive or are negative but a strong clinical suspicion for SLE remains, clinicians should order tests for antibodies to extractable nuclear antigens (ENA panel; see Table 2).3,16 Anti-dsDNA and anti-Smith (anti-Sm) antibodies are both specific for SLE, and levels of anti-dsDNA reflect disease activity in many patients.1,19 In contrast, anti-dsDNA antibodies are found in fewer than 0.5% of healthy individuals and patients with other autoimmune conditions.19 Among patients with high levels of anti-dsDNA antibodies and clinically inactive disease, 80% will have active disease within five years after elevated antibodies are detected.19
Autoantibodies, including ANA, anti-SSA/Ro, anti-SSB/La, and antiphospholipid antibodies, are usually detectable for many years prior to the onset of symptomatic SLE, while others, such as anti-Sm and anti-U1RNP, appear just months before the diagnosis.23 Patients with positive ANA results who do not meet criteria for SLE are still at risk for lupus and other autoimmune diseases, because complex autoimmune changes occur years before the diagnosis of SLE.23 These patients should be followed closely.
Continue for making the diagnosis >>
MAKING THE DIAGNOSIS
Diagnosing SLE may prove problematic because of the remarkable variety of relapsing and remitting clinical features, mimicry of similar conditions, and lack of a simple, definitive diagnostic test. Initial diagnosis of SLE depends on the disease manifestation, published criteria, and exclusion of alternative diagnoses. Confirmation requires careful clinical assessment, based on a thorough medical history and complete physical examination, along with specific laboratory testing.1,16 Biopsy results indicative of lupus nephritis in the presence of ANA or anti-dsDNA antibodies also confirm the diagnosis of SLE.24
Although created for research purposes, ACR classification criteria for SLE, published in 1982 and revised in 1997, have been used for more than 30 years to diagnose lupus (see www.rheumatology.org/Practice-Quality/Clinical-Support/Criteria/ACR-Endorsed-Criteria). In 2012, the Systemic Lupus International Collaborating Clinics (SLICC) group revised the 1997 ACR classification criteria to address major flaws and to improve clinical precision.24 According to SLICC, a definitive diagnosis requires the presence of at least four of 17 criteria, including at least one clinical and one immunologic criterion.24 The SLICC revisions have resulted in fewer misclassifications and provide greater sensitivity but lower specificity in the identification of SLE in comparison to the 1997 ACR criteria.24 To date, no one set of criteria allows for early diagnosis of SLE.
MANAGEMENT OPTIONS
Treatment must be tailored to the patient’s specific organ system involvement. Effective therapy hinges on controlling symptoms and reducing underlying inflammation.25 Four classes of drugs are used: NSAIDs, antimalarial drugs, corticosteroids, and cytotoxic drugs (see Table 3). Most patients benefit from NSAIDs to alleviate minor arthritis and arthralgia symptoms, but the risk for peptic ulcers and nephrotoxicity should be addressed; this may require the concomitant use of gastroprotective agents such as proton pump inhibitors.25 Antimalarials are effective for musculoskeletal symptoms that do not respond to NSAIDs and for cutaneous rashes.1 The current antimalarial drug of choice is hydroxychloroquine (200 to 400 mg/d po), which has been shown to control SLE manifestations by reducing and preventing disease flares.1,11,26 It is well tolerated and can be used for the duration of treatment.11,26 Patients should be informed that this drug’s onset of action is one month.26 In rare cases, this drug can cause retinal toxicity; therefore, SLE patients receiving hydroxychloroquine should be referred to an ophthalmologist for a baseline eye examination and yearly assessments to monitor for this rare adverse effect.25,26
Low-dose corticosteroids, such as oral prednisolone or methylprednisolone, are employed when NSAIDs and antimalarials fail to control arthritis or cutaneous SLE eruptions.25 Major systemic manifestations that occur during a disease flare—such as severe arthritis, hemolytic anemia, glomerulonephritis, alveolar hemorrhage, pericarditis, pleurisy, or CNS involvement—necessitate high-dose IV corticosteroids in conjunction with immunosuppressive agents.1,11,25 These high-dose glucocorticoids should be gradually withdrawn as soon as remission is achieved.11 Long-term suppressive therapy with oral corticosteroids in addition to other agents is often needed to preserve organ function.25
The major adverse effects of long-term glucocorticoids are osteoporosis, hypertension, hyperlipidemia, glucose intolerance, and susceptibility to infection. It is recommended that patients taking prednisolone 7.5 mg/d or more undergo a bone mineral density scan every two years.25 Those with T scores below –2.5 should be prescribed bisphosphonates.25
Immunosuppressive agents, such as cyclophosphamide, mycophenolate mofetil, and azathioprine, are used in conjunction with corticosteroids or when syndromes are resistant to corticosteroids.1 Collaboration between primary care, rheumatology, and nephrology is advisable for patients requiring immunosuppressive or disease-modifying pharmacologic agents.
Two new treatments for SLE are the immunologic agents belimumab and rituximab.7 Belimumab, a monoclonal human antibody, is the first medication in the past 50 years that has been approved by the FDA for antibody-positive SLE patients with active lupus unresponsive to standard treatment.7,27 Rituximab is an anti-CD20 monoclonal antibody, approved by the FDA for non-Hodgkin lymphoma, chronic lymphocytic leukemia, and RA, and is now considered an option for SLE refractory to conventional treatment regimens.7,27 The efficacy of belimumab and rituximab, and the spectrum of indications for their use, are still under study, but these new therapeutic agents hold promise for the treatment of patients with refractory SLE.
Continue for helping patients live with SLE >>
HELPING PATIENTs LIVE WITH SLE
Patients with SLE have a higher mortality rate, as well as a lower quality of life, compared to the general population.28 The major contributors to a decreased quality of life are fatigue, mood disturbances (eg, depression), and chronic pain.28 Practitioners should advise SLE patients to participate in support groups and psychotherapy to alleviate the anxiety and depression associated with this chronic disease.
For patients with long-standing disease, accelerated atherosclerotic cardiovascular disease adds to morbidity and mortality. For this reason, obesity, hypertension, hyperlipidemia, and smoking are targets for intervention. Lifestyle modifications—such as exercise, smoking cessation, a healthy diet with low saturated fat, stress avoidance, and adequate rest—are recommended.26
Avoiding overexposure to sunlight, by using sunscreen with an SPF of at least 30 and wearing sun-protective clothing, is essential for management of cutaneous lupus.25,26 Yearly influenza vaccination is appropriate, as are other immunizations (eg, pneumococcal vaccine).26
Advise women of childbearing age with SLE that lupus flares result in a high risk for miscarriage. All women should undergo yearly cervical cancer screening.26
Patients taking long-term glucocorticoids should adopt bone-protective behaviors, including quitting smoking, limiting alcohol intake, partaking in weight-bearing exercise, and consuming dietary calcium and vitamin D.25 Patients taking these drugs should avoid live virus vaccines. Those on immunosuppressive therapy should be warned about the hazardous adverse effects of glucocorticoids.
MONITORING AND FOLLOW-UP
Collaborative efforts between primary care providers and several types of specialty providers can facilitate coordinated interventions in the long-term management of lupus. Rheumatologists are experts in making therapeutic decisions for SLE.
Patients being treated for SLE require routine monitoring to assess disease activity and detect flares. The European League Against Rheumatism (EULAR) guidelines recommend that monitoring include assessment for new clinical manifestations, routine laboratory tests, and immunologic assays, chiefly anti-dsDNA, anti-Sm, and serum complement levels, coupled with one of the validated global activity indices, such as the Systemic Lupus Erythematosus Disease Activity Index (SLEDAI).29
A routine office visit with a physical examination and laboratory testing for CBC with differential, basic metabolic panel, and urinalysis every three months is recommended for patients with stable disease; patients with uncontrolled SLE may require weekly visits.11,29 Patients taking immunosuppressive drugs should be provided with adverse-effect profiles alerting them to toxicity symptoms and require frequent laboratory monitoring for potential toxicity.11
CONCLUSION
Advances in immunologically targeted serologic tests have shed more light on the underlying pathogenesis of SLE, which in turn has led to improvements in disease detection and monitoring of complications, as well as advances in therapy. Although SLE cannot be cured, emerging therapies targeting different mechanisms of SLE offer hope for patients diagnosed with this complex disease.
CE/CME No: CR-1608
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest and evaluation. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Describe the pathophysiology and explain the various clinical manifestations of systemic lupus erythematosus (SLE).
• Define the differential diagnosis for SLE.
• List the elements of the laboratory work-up used in the diagnosis of lupus.
• Describe the therapeutic options for patients with SLE.
FACULTY
Michael Felz is an Assistant Professor at Augusta University (formerly Georgia Regents University) in Augusta, Georgia. Mary Bailey Wickham is a PA student in her final year at Augusta University.
The authors have no financial relationships to disclose.
ACCREDITATION STATEMENT
![]()
This program has been reviewed and is approved for a maximum of 1.0 hour of American Academy of Physician Assistants (AAPA) Category 1 CME credit by the Physician Assistant Review Panel. [NPs: Both ANCC and the AANP Certification Program recognize AAPA as an approved provider of Category 1 credit.] Approval is valid for one year from the issue date of August 2016.
Article begins on next page >>
Systemic lupus erythematosus (SLE) is a chronic autoimmune disease that often goes undiagnosed initially. Timely detection of SLE is important, because prompt treatment can prevent its many major complications—notably, end organ damage. Here’s how to distinguish SLE from other illnesses with similar presentations and how to recognize the complications of undiagnosed SLE, which can progress rapidly and fatally.
Systemic lupus erythematosus (SLE) is a chronic inflammatory disorder that can involve multiple organ systems. The presence of antinuclear antibodies (ANA) is a common marker for this disease. In autoimmune diseases such as SLE, the immune system attacks the cells of healthy tissues throughout the body. Genetic, hormonal, and environmental factors (eg, ultraviolet light, infectious viruses, and even use of certain medications) have been implicated in the pathogenesis.1-3
It is estimated that 1.5 million people in the United States and up to 5 million people worldwide have SLE.4 It is nine to 10 times more prevalent in women—especially those of reproductive age—than menand occurs more frequently in African-American, Hispanic, and Asian women than in non-Hispanic Caucasian women.1,2,4-6 Siblings of SLE patients are 30 times more likely to develop the disease, compared to individuals without an affected relative.2 Increased mortality in persons with SLE is attributed to accelerated atherosclerosis, infection, malignancy, and target organ damage, particularly end-stage renal disease.3 Women ages 33 to 45 with SLE are at increased risk (50x greater) for myocardial infarction due to premature atherosclerosis than age-matched women in the general population.7 The life expectancy of SLE patients with renal damage is 23.7 years less than that of the general population.8
Increased awareness of SLE has led to drastic improvements in associated mortality over the past five decades. The survival rate in the 1950s was 50% at 2 years, while current rates are about 95% at 5 years and about 90% at 10 years.3,9 These improvements likely reflect earlier diagnosis and treatment on the part of well-informed clinicians, as well as more effective treatment.
SLE MANIFESTATIONS
SLE can affect any organ in the body with a broad spectrum of clinical manifestations, making it a devastatingly deceptive disease. Disease severity may vary by age, by organ involvement, and over time. Onset may be gradual and mild or rapidly progressive with severe organ involvement. Constitutional manifestations such as fatigue, weight loss, anorexia, and low-grade fever often serve as initial complaints. However, these features are common to a variety of infectious and inflammatory conditions, making early SLE easily overlooked and frequently misdiagnosed. 2
A mix of manifestations involving the joints, skin, mouth, kidneys, lungs, heart, and nervous system offers clues to the diagnosis of SLE (see Table 1). Arthritis is the most common symptom, occurring in 85% to 90% of SLE cases.1,10 It is typically nonerosive, inflammatory, symmetric or asymmetric, and polyarticular (involving five or more joints)and may be accompanied by constitutional symptoms.1,2,11 The joints most commonly affected are the proximal interphalangeals, metacarpophalangeals (MCP), knees, and wrists.2 Morning stiffness is a common complaint.1,11 Jaccoud arthropathy, which is characterized by reducible, nonerosive joint subluxations (eg, swan neck deformities, ulnar deviation, boutonniere deformities, and z-shaped thumbs), can be seen in SLE patients.3 When patients present with articular and constitutional symptoms but lack other typical manifestations of SLE, such as skin rash, appropriate measures—for example, arthrocentesis—should be taken to evaluate for infection.11
Cutaneous manifestations are the second most common feature at disease onset, with photosensitivity and malar rash being the most prevalent.10 Nearly all patients experience skin lesions at some point during the disease course.1 Diagnostic, or lupus-specific, lesions can be classified into three types: acute, subacute, and chronic.
Acute cutaneous lupus erythematosus (ACLE) is almost always associated with SLE, while subacute cutaneous lupus erythematosus (SCLE) is seen in about 50% of SLE patients.12 ACLE is usually precipitated by sunlight exposure and includes the classic erythematous, macular, “butterfly” rash located on the malar regions of the face, which may remain for days to weeks.2,12 Diffuse or discoid alopecia also may develop in ACLE, along with oral ulcers arising in purpuric necrotic lesions on the palate, buccal mucosa, or gums. Generalized erythematous, papular, or urticarial lesions may affect the face, arms, dorsa of the hands, or “V” of the neck.12
SCLE tends to be sudden in onset, with annular lesions or psoriasiform plaques on the upper trunk, arms, and dorsa of the hands that often coalesce into polycyclic lesions.12 These subacute rashes are often associated with anti-SSA/Ro antibodies.
Chronic cutaneous lupus erythematosus is usually characterized by skin disease alone.12 Discoid lupus is the most common type, with circular scaly plaques with erythematous, hyperpigmented rims and atrophic hypopigmented centers that leave scars.2,12 It is commonly seen on the face, neck, and scalp.
During the course of SLE, mucous membrane involvement—typically painless oral or nasal ulcers—occurs in 25% to 45% of patients.2 Oral lesions are most commonly found on the hard palate and buccal mucosa.3,12
Lupus nephritis, perhaps the most dangerous manifestation of SLE, conveys high risk for organ failure, a higher mortality rate compared to patients without renal involvement, and lower life expectancy.8,11 Up to 60% of Asians, African Americans, and Hispanics develop renal disease during the course of their illness.8 The dominant feature is proteinuria, typically accompanied by microscopic hematuria.2
Neuropsychiatric SLE (NPSLE) is a clinical manifestation that is poorly understood.13 An estimated 28% to 40% of NPSLE manifestations develop prior to or synchronous with the diagnosis, and 63% arise within the first year of diagnosis.13 Mild cognitive impairment is the most common manifestation,reported in up to 20% to 30% of SLE patients.2,13 Seizures and psychosis are reported in 7% to 10% of SLE patients, and psychosis—characterized by hallucinations or delusions—in 3.5%.2
Cardiac findings are common among SLE patients, with an estimated prevalence of 50%, but are rarely the presenting manifestation.14 Pericarditis with effusion is the most common cardiac manifestation, occurring in 25% of SLE patients.2 Advancing atherosclerosis due to chronic inflammation becomes a major cause of mortality in the later years for SLE patients.1 Compared to the general population, the incidence of myocardial infarction in SLE patients is increased fivefold.1 Pleuritis is the most common pleuropulmonary manifestation in SLE.11 Pleuritic chest pain with or without a pleural effusion occurs in 45% to 60% of SLE patients.2
Continue for differential diagnoses >>
DIFFERENTIAL DIAGNOSES
The differential diagnosis for SLE includes rheumatoid arthritis (RA), septic arthritis, mixed connective tissue disease (MCTD), Sjögren syndrome, systemic sclerosis (SSc), polymyositis (PM), fibromyalgia, and drug-induced lupus. Symmetrical, inflammatory, polyarticular arthritis with a predilection for the wrist and MCP joints occurs in both RA and SLE.1,15 And, because the initial articular features of SLE are symmetric arthralgias, patients with SLE are frequently misdiagnosed with RA. The absence of destructive bony erosions on radiographs and large joint effusions, along with the joint reducibility in SLE, can help distinguish it from RA.16 Asymmetric arthritis, which can be a presenting feature in both RA and SLE, is more commonly seen in the latter. ANA and rheumatoid factor test results can be positive in both disorders, but antibodies to anti-cyclic citrullinated peptides, with a 95% specificity for RA but absent in SLE, distinguish RA from SLE.1,16
Patients with MCTD display an array of overlapping features of SLE, PM, and SSc, making the diagnosis difficult.17 Although MCTD can evolve into other connective tissue diseases, such as SLE, it is nonetheless considered a distinct entity.17 High titers of anti-U1 ribonucleoprotein (anti-U1RNP) antibodies are indicative of MCTD. Anti-U1RNP is rarely detected in SLE and almost never seen in other rheumatic diseases.17 Typical manifestations of MCTD are Raynaud phenomenon, swollen fingers (referred to as “sausage digits”), and protuberant polyarthritis.17
Anti-SSA/Ro and anti-SSB/La antibodies, although detectable in SLE patients, are more commonly associated with Sjögren syndrome. In addition, patients with Sjögren syndrome frequently demonstrate signs of keratoconjunctivitis sicca and xerostomia.16
The clinical features of fibromyalgia include diffuse musculoskeletal pain that readily mimics SLE arthralgias. The 2011 modification of the 2010 American College of Rheumatology (ACR) preliminary diagnostic criteria for fibromyalgia serves as a reliable tool for diagnosing patients with nonspecific, diffuse pain.18 This 2011 modification includes 19 pain locations and the six self-reported symptoms: fatigue, impaired sleep, headaches, depression, poor cognition, and abdominal pain.18
SSc, also known as scleroderma, is characterized by skin thickening and/or CREST syndrome (calcinosis, Raynaud phenomenon, esophageal dysmotility, sclerodactyly, telangiectasia). The presence of anti-Scl-70 and anti-centromere antibodies are noted as well.16
Finally, a suspicion of SLE mandates an evaluation for drug-induced lupus by assessing the patient’s exposure to culprit medications, such as hydralazine, procainamide, isoniazid, methyldopa, chlorpromazine, quinidine, minocycline, and tumor necrosis factor inhibitiors.1,11 Four key features point toward drug-induced lupus:
• The female-to-male ratio is nearly equivalent.
• Nephritis and central nervous system (CNS) manifestations are not commonly present.
• Anti–double-stranded DNA (anti-dsDNA) antibodies and hypocomplementemia are absent.
• The clinical features and laboratory abnormalities return to baseline once the offending agent is removed.1
Anti-histone antibodies are present in approximately 75% of patients with drug-induced lupus but can also be seen in patients with SLE.11
Continue for laboratory work-up >>
LABORATORY WORK-UP
Laboratory abnormalities associated with SLE include anemia, leukopenia, lymphopenia, thrombocytopenia, hypocomplementemia, and proteinuria. A typical work-up includes a routine complete blood count (CBC) with differential, serum creatinine, erythrocyte sedimentation rate (ESR), C-reactive protein (CRP), urinalysis with microscopy, and serologic ANA titer.1,16,19 A CBC with differential may reveal hematologic abnormalities, such as anemia of chronic disease (most commonly) or autoimmune hemolytic anemia, as well as leukopenia and thrombocytopenia due to circulating autoantibodies.3 An elevated ESR and CRP indicate the severity of the systemic inflammation and/or infection. Urinalysis is effective for detecting lupus with renal diseaseand may reveal proteinuria due to renal dysfunction.2
A positive ANA titer indicates widespread activation of the immune system targeted against nuclear and cytoplasmic subparticles. The vast majority of patients with SLE will develop a positive ANA with a high titer at some point during the course of their disease.16 The ANA is highly sensitive for SLE (93% to 95%) but lacks specificity (57%).20The most common tests for ANA are enzyme-linked immunosorbent assay (ELISA) and indirect immunofluorescence assay (IFA). ELISA is more sensitive in detecting ANA, while IFA is the gold standard due to its high specificity.21 Some laboratories may use immunoassay as a screening tool for ANA and then use IFA to confirm positive or equivocal results.21 Positive ANA results can be seen in patients with other rheumatologic diseases and in up to 15% of all healthy persons, but with low or borderline titers.22 For these reasons, ANA testing alone is a poor predictor of SLE.
When either the ANA test results are positive or are negative but a strong clinical suspicion for SLE remains, clinicians should order tests for antibodies to extractable nuclear antigens (ENA panel; see Table 2).3,16 Anti-dsDNA and anti-Smith (anti-Sm) antibodies are both specific for SLE, and levels of anti-dsDNA reflect disease activity in many patients.1,19 In contrast, anti-dsDNA antibodies are found in fewer than 0.5% of healthy individuals and patients with other autoimmune conditions.19 Among patients with high levels of anti-dsDNA antibodies and clinically inactive disease, 80% will have active disease within five years after elevated antibodies are detected.19
Autoantibodies, including ANA, anti-SSA/Ro, anti-SSB/La, and antiphospholipid antibodies, are usually detectable for many years prior to the onset of symptomatic SLE, while others, such as anti-Sm and anti-U1RNP, appear just months before the diagnosis.23 Patients with positive ANA results who do not meet criteria for SLE are still at risk for lupus and other autoimmune diseases, because complex autoimmune changes occur years before the diagnosis of SLE.23 These patients should be followed closely.
Continue for making the diagnosis >>
MAKING THE DIAGNOSIS
Diagnosing SLE may prove problematic because of the remarkable variety of relapsing and remitting clinical features, mimicry of similar conditions, and lack of a simple, definitive diagnostic test. Initial diagnosis of SLE depends on the disease manifestation, published criteria, and exclusion of alternative diagnoses. Confirmation requires careful clinical assessment, based on a thorough medical history and complete physical examination, along with specific laboratory testing.1,16 Biopsy results indicative of lupus nephritis in the presence of ANA or anti-dsDNA antibodies also confirm the diagnosis of SLE.24
Although created for research purposes, ACR classification criteria for SLE, published in 1982 and revised in 1997, have been used for more than 30 years to diagnose lupus (see www.rheumatology.org/Practice-Quality/Clinical-Support/Criteria/ACR-Endorsed-Criteria). In 2012, the Systemic Lupus International Collaborating Clinics (SLICC) group revised the 1997 ACR classification criteria to address major flaws and to improve clinical precision.24 According to SLICC, a definitive diagnosis requires the presence of at least four of 17 criteria, including at least one clinical and one immunologic criterion.24 The SLICC revisions have resulted in fewer misclassifications and provide greater sensitivity but lower specificity in the identification of SLE in comparison to the 1997 ACR criteria.24 To date, no one set of criteria allows for early diagnosis of SLE.
MANAGEMENT OPTIONS
Treatment must be tailored to the patient’s specific organ system involvement. Effective therapy hinges on controlling symptoms and reducing underlying inflammation.25 Four classes of drugs are used: NSAIDs, antimalarial drugs, corticosteroids, and cytotoxic drugs (see Table 3). Most patients benefit from NSAIDs to alleviate minor arthritis and arthralgia symptoms, but the risk for peptic ulcers and nephrotoxicity should be addressed; this may require the concomitant use of gastroprotective agents such as proton pump inhibitors.25 Antimalarials are effective for musculoskeletal symptoms that do not respond to NSAIDs and for cutaneous rashes.1 The current antimalarial drug of choice is hydroxychloroquine (200 to 400 mg/d po), which has been shown to control SLE manifestations by reducing and preventing disease flares.1,11,26 It is well tolerated and can be used for the duration of treatment.11,26 Patients should be informed that this drug’s onset of action is one month.26 In rare cases, this drug can cause retinal toxicity; therefore, SLE patients receiving hydroxychloroquine should be referred to an ophthalmologist for a baseline eye examination and yearly assessments to monitor for this rare adverse effect.25,26
Low-dose corticosteroids, such as oral prednisolone or methylprednisolone, are employed when NSAIDs and antimalarials fail to control arthritis or cutaneous SLE eruptions.25 Major systemic manifestations that occur during a disease flare—such as severe arthritis, hemolytic anemia, glomerulonephritis, alveolar hemorrhage, pericarditis, pleurisy, or CNS involvement—necessitate high-dose IV corticosteroids in conjunction with immunosuppressive agents.1,11,25 These high-dose glucocorticoids should be gradually withdrawn as soon as remission is achieved.11 Long-term suppressive therapy with oral corticosteroids in addition to other agents is often needed to preserve organ function.25
The major adverse effects of long-term glucocorticoids are osteoporosis, hypertension, hyperlipidemia, glucose intolerance, and susceptibility to infection. It is recommended that patients taking prednisolone 7.5 mg/d or more undergo a bone mineral density scan every two years.25 Those with T scores below –2.5 should be prescribed bisphosphonates.25
Immunosuppressive agents, such as cyclophosphamide, mycophenolate mofetil, and azathioprine, are used in conjunction with corticosteroids or when syndromes are resistant to corticosteroids.1 Collaboration between primary care, rheumatology, and nephrology is advisable for patients requiring immunosuppressive or disease-modifying pharmacologic agents.
Two new treatments for SLE are the immunologic agents belimumab and rituximab.7 Belimumab, a monoclonal human antibody, is the first medication in the past 50 years that has been approved by the FDA for antibody-positive SLE patients with active lupus unresponsive to standard treatment.7,27 Rituximab is an anti-CD20 monoclonal antibody, approved by the FDA for non-Hodgkin lymphoma, chronic lymphocytic leukemia, and RA, and is now considered an option for SLE refractory to conventional treatment regimens.7,27 The efficacy of belimumab and rituximab, and the spectrum of indications for their use, are still under study, but these new therapeutic agents hold promise for the treatment of patients with refractory SLE.
Continue for helping patients live with SLE >>
HELPING PATIENTs LIVE WITH SLE
Patients with SLE have a higher mortality rate, as well as a lower quality of life, compared to the general population.28 The major contributors to a decreased quality of life are fatigue, mood disturbances (eg, depression), and chronic pain.28 Practitioners should advise SLE patients to participate in support groups and psychotherapy to alleviate the anxiety and depression associated with this chronic disease.
For patients with long-standing disease, accelerated atherosclerotic cardiovascular disease adds to morbidity and mortality. For this reason, obesity, hypertension, hyperlipidemia, and smoking are targets for intervention. Lifestyle modifications—such as exercise, smoking cessation, a healthy diet with low saturated fat, stress avoidance, and adequate rest—are recommended.26
Avoiding overexposure to sunlight, by using sunscreen with an SPF of at least 30 and wearing sun-protective clothing, is essential for management of cutaneous lupus.25,26 Yearly influenza vaccination is appropriate, as are other immunizations (eg, pneumococcal vaccine).26
Advise women of childbearing age with SLE that lupus flares result in a high risk for miscarriage. All women should undergo yearly cervical cancer screening.26
Patients taking long-term glucocorticoids should adopt bone-protective behaviors, including quitting smoking, limiting alcohol intake, partaking in weight-bearing exercise, and consuming dietary calcium and vitamin D.25 Patients taking these drugs should avoid live virus vaccines. Those on immunosuppressive therapy should be warned about the hazardous adverse effects of glucocorticoids.
MONITORING AND FOLLOW-UP
Collaborative efforts between primary care providers and several types of specialty providers can facilitate coordinated interventions in the long-term management of lupus. Rheumatologists are experts in making therapeutic decisions for SLE.
Patients being treated for SLE require routine monitoring to assess disease activity and detect flares. The European League Against Rheumatism (EULAR) guidelines recommend that monitoring include assessment for new clinical manifestations, routine laboratory tests, and immunologic assays, chiefly anti-dsDNA, anti-Sm, and serum complement levels, coupled with one of the validated global activity indices, such as the Systemic Lupus Erythematosus Disease Activity Index (SLEDAI).29
A routine office visit with a physical examination and laboratory testing for CBC with differential, basic metabolic panel, and urinalysis every three months is recommended for patients with stable disease; patients with uncontrolled SLE may require weekly visits.11,29 Patients taking immunosuppressive drugs should be provided with adverse-effect profiles alerting them to toxicity symptoms and require frequent laboratory monitoring for potential toxicity.11
CONCLUSION
Advances in immunologically targeted serologic tests have shed more light on the underlying pathogenesis of SLE, which in turn has led to improvements in disease detection and monitoring of complications, as well as advances in therapy. Although SLE cannot be cured, emerging therapies targeting different mechanisms of SLE offer hope for patients diagnosed with this complex disease.
1. Hellmann DB, Imboden JB. Rheumatologic & immunologic disorders. In: Papadakis M, McPhee SJ, Rabow MW, eds. Current Medical Diagnosis & Treatment. 53rd ed. New York, NY: McGraw-Hill; 2014:786-836.
2. Bertsias G, Cervera R, Boumpas DT. Systemic lupus erythematosus: pathogenesis and clinical features. In: Bijlsma JWJ, ed. EULAR Textbook on Rheumatic Diseases. London: BMJ Group; 2012:476-505.
3. Dall’Era M. Chapter 21. Systemic lupus erythematosus. In: Imboden JB, Hellmann DB, Stone JH, eds. Current Diagnosis & Treatment: Rheumatology. 3rd ed. New York, NY: McGraw-Hill; 2013.
4. Lupus Foundation of America. What is lupus? www.lupus.org/answers/entry/what-is-lupus. Accessed July 19, 2016.
5. Furst DE, Clarke AE, Fernandes AW, et al. Incidence and prevalence of adult systemic lupus erythematosus in a large US managed-care population. Lupus. 2012;22(1):99-105.
6. Pons-Estel GL, Alarcón GS, Scofield L, et al. Understanding the epidemiology and progression of systemic lupus erythematosus. Semin Arthritis Rheum. 2010;39(4):257-268.
7. Lisnevskaia L, Murphy G, Isenberg D. Systemic lupus erythematosus. Lancet. 2014;384(9957):1878-1888.
8. Mok CC, Kwok RC, Yip PS. Effect of renal disease on the standardized mortality ratio and life expectancy of patients with systemic lupus erythematosus. Arthritis Rheum. 2013;65(8):2154-2160.
9. Merola JF, Bermas B, Lu B, et al. Clinical manifestations and survival among adults with SLE according to age at diagnosis. Lupus. 2014;23(8):778-784.
10. Font J, Cervera R, Ramos-Casals M, et al. Clusters of clinical and immunologic features in systemic lupus erythematosus: analysis of 600 patients from a single center. Semin Arthritis Rheum. 2004;33(4):217-230.
11. Kiriakidou M, Cotton D, Taichman D, Williams S. Systemic lupus erythematosus.Ann Intern Med. 2013;159(7):2-16.
12. Wolff K, Johnson R, Saavedra A. Fitzpatrick’s Color Atlas and Synopsis of Clinical Dermatology. 7th ed. New York, NY: McGraw-Hill; 2013:334-342.
13. Popescu A, Kao AH. Neuropsychiatric systemic lupus erythematosus. Curr Neuropharmacol. 2011;9(3):449-457.
14. Chen PY, Chang CH, Hsu CC, et al. Systemic lupus erythematosus presenting with cardiac symptoms. Am J Emerg Med. 2014;32(9):1117-1119.
15. Hahn BHH. Chapter 378: Systemic lupus erythematosus. In: Kasper DL, Fauci AS, Hauser SL, et al, eds. Harrison’s Principles of Internal Medicine. 19th ed. New York, NY: McGraw-Hill; 2015.
16. Wallace DJ. Diagnosis and differential diagnosis of systemic lupus erythematosus in adults. UpToDate. www.uptodate.com/contents/diagnosis-and-differential-diagnosis-of-systemic-lupus-erythematosus-in-adults. Accessed July 19, 2016.
17. Cappelli S, Bellando Randone S, Martinovic D, et al. “To be or not to be,” ten years after: evidence for mixed connective tissue disease as a distinct entity. Semin Arthritis Rheum. 2012;41(4):589-598.
18. Bennett RM, Friend R, Marcus D, et al. Criteria for the diagnosis of fibromyalgia: validation of the modified 2010 preliminary American College of Rheumatology criteria and the development of alternative criteria. Arthritis Care Res (Hoboken). 2014;66(9):1364-1373.
19. Rahman A, Isenberg DA. Systemic lupus erythematosus. N Engl J Med. 2008;358(9):929-939.
20. Magrey M, Abelson A. Laboratory evaluation of rheumatic diseases. Cleveland Clinic Center for Continuing Education 2010. www.cleveland clinicmeded.com/medicalpubs/diseasemanagement/rheumatology/laboratory-evaluation-rheumatic-diseases/. Accessed July 19, 2016.
21. Copple SS, Sawitzke AD, Wilson AM, et al. Enzyme-linked immunosorbent assay screening then indirect immunofluorescence confirmation of antinuclear antibodies: a statistical analysis. Am J Clin Pathol. 2011;135(5):678-684.
22. Von Feld JM; American College of Rheumatology. Antinuclear antibodies (ANA). 2015. www.rheumatology.org/I-Am-A/Patient-Caregiver/Diseases-Conditions/Antinuclear-Antibodies-ANA. Accessed July 19, 2016.
23. Arbuckle MR, McClain MT, Rubertone MV, et al. Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N Engl J Med. 2003;349(16):1526-1533.
24. Petri M, Orbai A, Alarcón G, et al. Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum. 2012;64(8):2677-2686.
25. Ioannou Y, Isenberg DA. Current concepts for the management of systemic lupus erythematosus in adults: a therapeutic challenge. Postgrad Med J. 2002;78:599-606.
26. Dall’Era M, Wofsy D. Treatment of systemic lupus erythematosus. In: Imboden JB, Hellmann DB, Stone JH, eds. Current Diagnosis & Treatment: Rheumatology. 3rd ed. New York, NY: McGraw-Hill; 2013.
27. Stohl W, Hilbert DM. The discovery and development of belimumab: the anti-BLyS–lupus connection. Nat Biotechnol. 2012;30(1):69-77.
28. Lateef A, Petri M. Unmet medical needs in systemic lupus erythematosus. Arthritis Research & Ther. 2012;14(suppl 4):S4.
29. Bertsias G, Ioannidis JP, Boletis J, et al. EULAR recommendations for the management of systemic lupus erythematosus. Report of a task force of the EULAR standing committee for international clinical studies including therapeutics. Ann Rheum Dis. 2008;67(2):195-205.
1. Hellmann DB, Imboden JB. Rheumatologic & immunologic disorders. In: Papadakis M, McPhee SJ, Rabow MW, eds. Current Medical Diagnosis & Treatment. 53rd ed. New York, NY: McGraw-Hill; 2014:786-836.
2. Bertsias G, Cervera R, Boumpas DT. Systemic lupus erythematosus: pathogenesis and clinical features. In: Bijlsma JWJ, ed. EULAR Textbook on Rheumatic Diseases. London: BMJ Group; 2012:476-505.
3. Dall’Era M. Chapter 21. Systemic lupus erythematosus. In: Imboden JB, Hellmann DB, Stone JH, eds. Current Diagnosis & Treatment: Rheumatology. 3rd ed. New York, NY: McGraw-Hill; 2013.
4. Lupus Foundation of America. What is lupus? www.lupus.org/answers/entry/what-is-lupus. Accessed July 19, 2016.
5. Furst DE, Clarke AE, Fernandes AW, et al. Incidence and prevalence of adult systemic lupus erythematosus in a large US managed-care population. Lupus. 2012;22(1):99-105.
6. Pons-Estel GL, Alarcón GS, Scofield L, et al. Understanding the epidemiology and progression of systemic lupus erythematosus. Semin Arthritis Rheum. 2010;39(4):257-268.
7. Lisnevskaia L, Murphy G, Isenberg D. Systemic lupus erythematosus. Lancet. 2014;384(9957):1878-1888.
8. Mok CC, Kwok RC, Yip PS. Effect of renal disease on the standardized mortality ratio and life expectancy of patients with systemic lupus erythematosus. Arthritis Rheum. 2013;65(8):2154-2160.
9. Merola JF, Bermas B, Lu B, et al. Clinical manifestations and survival among adults with SLE according to age at diagnosis. Lupus. 2014;23(8):778-784.
10. Font J, Cervera R, Ramos-Casals M, et al. Clusters of clinical and immunologic features in systemic lupus erythematosus: analysis of 600 patients from a single center. Semin Arthritis Rheum. 2004;33(4):217-230.
11. Kiriakidou M, Cotton D, Taichman D, Williams S. Systemic lupus erythematosus.Ann Intern Med. 2013;159(7):2-16.
12. Wolff K, Johnson R, Saavedra A. Fitzpatrick’s Color Atlas and Synopsis of Clinical Dermatology. 7th ed. New York, NY: McGraw-Hill; 2013:334-342.
13. Popescu A, Kao AH. Neuropsychiatric systemic lupus erythematosus. Curr Neuropharmacol. 2011;9(3):449-457.
14. Chen PY, Chang CH, Hsu CC, et al. Systemic lupus erythematosus presenting with cardiac symptoms. Am J Emerg Med. 2014;32(9):1117-1119.
15. Hahn BHH. Chapter 378: Systemic lupus erythematosus. In: Kasper DL, Fauci AS, Hauser SL, et al, eds. Harrison’s Principles of Internal Medicine. 19th ed. New York, NY: McGraw-Hill; 2015.
16. Wallace DJ. Diagnosis and differential diagnosis of systemic lupus erythematosus in adults. UpToDate. www.uptodate.com/contents/diagnosis-and-differential-diagnosis-of-systemic-lupus-erythematosus-in-adults. Accessed July 19, 2016.
17. Cappelli S, Bellando Randone S, Martinovic D, et al. “To be or not to be,” ten years after: evidence for mixed connective tissue disease as a distinct entity. Semin Arthritis Rheum. 2012;41(4):589-598.
18. Bennett RM, Friend R, Marcus D, et al. Criteria for the diagnosis of fibromyalgia: validation of the modified 2010 preliminary American College of Rheumatology criteria and the development of alternative criteria. Arthritis Care Res (Hoboken). 2014;66(9):1364-1373.
19. Rahman A, Isenberg DA. Systemic lupus erythematosus. N Engl J Med. 2008;358(9):929-939.
20. Magrey M, Abelson A. Laboratory evaluation of rheumatic diseases. Cleveland Clinic Center for Continuing Education 2010. www.cleveland clinicmeded.com/medicalpubs/diseasemanagement/rheumatology/laboratory-evaluation-rheumatic-diseases/. Accessed July 19, 2016.
21. Copple SS, Sawitzke AD, Wilson AM, et al. Enzyme-linked immunosorbent assay screening then indirect immunofluorescence confirmation of antinuclear antibodies: a statistical analysis. Am J Clin Pathol. 2011;135(5):678-684.
22. Von Feld JM; American College of Rheumatology. Antinuclear antibodies (ANA). 2015. www.rheumatology.org/I-Am-A/Patient-Caregiver/Diseases-Conditions/Antinuclear-Antibodies-ANA. Accessed July 19, 2016.
23. Arbuckle MR, McClain MT, Rubertone MV, et al. Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N Engl J Med. 2003;349(16):1526-1533.
24. Petri M, Orbai A, Alarcón G, et al. Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum. 2012;64(8):2677-2686.
25. Ioannou Y, Isenberg DA. Current concepts for the management of systemic lupus erythematosus in adults: a therapeutic challenge. Postgrad Med J. 2002;78:599-606.
26. Dall’Era M, Wofsy D. Treatment of systemic lupus erythematosus. In: Imboden JB, Hellmann DB, Stone JH, eds. Current Diagnosis & Treatment: Rheumatology. 3rd ed. New York, NY: McGraw-Hill; 2013.
27. Stohl W, Hilbert DM. The discovery and development of belimumab: the anti-BLyS–lupus connection. Nat Biotechnol. 2012;30(1):69-77.
28. Lateef A, Petri M. Unmet medical needs in systemic lupus erythematosus. Arthritis Research & Ther. 2012;14(suppl 4):S4.
29. Bertsias G, Ioannidis JP, Boletis J, et al. EULAR recommendations for the management of systemic lupus erythematosus. Report of a task force of the EULAR standing committee for international clinical studies including therapeutics. Ann Rheum Dis. 2008;67(2):195-205.
Hospitalization costs unaffected by Medicaid status for children with asthma
Medicaid status did not significantly affect costs for children who were hospitalized because of asthma, according to Jeffrey H. Silber, MD, and his associates.
In a study of 17,739 matched pairs of children with and without Medicaid who were hospitalized because of asthma, the median cost for Medicaid patients was $4,263; for non-Medicaid patients, it was $4,160. The median difference in cost between Medicaid and non-Medicaid patients was $84, and the mean difference in cost was $49.
Both Medicaid and non-Medicaid patients had similar lengths of stay, with a median of 1 day for both groups. Intensive care unit use was similar, with 10.1% of Medicaid patients visiting the ICU, compared with 10.6% of non-Medicaid patients.
“Our study should serve to provide potential benchmarks for use and reimbursement standards, with implications for care and payment even when children are hospitalized outside the [Pediatric Hospital Information System],” the investigators wrote.
Find the full study in Pediatrics (doi: 10.1542/peds.2016-0371).
Medicaid status did not significantly affect costs for children who were hospitalized because of asthma, according to Jeffrey H. Silber, MD, and his associates.
In a study of 17,739 matched pairs of children with and without Medicaid who were hospitalized because of asthma, the median cost for Medicaid patients was $4,263; for non-Medicaid patients, it was $4,160. The median difference in cost between Medicaid and non-Medicaid patients was $84, and the mean difference in cost was $49.
Both Medicaid and non-Medicaid patients had similar lengths of stay, with a median of 1 day for both groups. Intensive care unit use was similar, with 10.1% of Medicaid patients visiting the ICU, compared with 10.6% of non-Medicaid patients.
“Our study should serve to provide potential benchmarks for use and reimbursement standards, with implications for care and payment even when children are hospitalized outside the [Pediatric Hospital Information System],” the investigators wrote.
Find the full study in Pediatrics (doi: 10.1542/peds.2016-0371).
Medicaid status did not significantly affect costs for children who were hospitalized because of asthma, according to Jeffrey H. Silber, MD, and his associates.
In a study of 17,739 matched pairs of children with and without Medicaid who were hospitalized because of asthma, the median cost for Medicaid patients was $4,263; for non-Medicaid patients, it was $4,160. The median difference in cost between Medicaid and non-Medicaid patients was $84, and the mean difference in cost was $49.
Both Medicaid and non-Medicaid patients had similar lengths of stay, with a median of 1 day for both groups. Intensive care unit use was similar, with 10.1% of Medicaid patients visiting the ICU, compared with 10.6% of non-Medicaid patients.
“Our study should serve to provide potential benchmarks for use and reimbursement standards, with implications for care and payment even when children are hospitalized outside the [Pediatric Hospital Information System],” the investigators wrote.
Find the full study in Pediatrics (doi: 10.1542/peds.2016-0371).
FROM PEDIATRICS