User login
Peripartum probiotics linked to lower atopic dermatitis risk in offspring
Peripartum consumption of probiotic milk was associated with a decline in the incidence of atopic dermatitis in offspring, but other allergy-related diseases were not reduced in the children, the results of a randomized study indicate.
In a randomized study, 415 pregnant women drank eiher probiotic or placebo milk from 36 weeks’ gestation to 3 months postpartum. At 6 years follow-up, there was a lower cumulative incidence of AD in the probiotic group compared with the placebo group (OR = 0.64; 95% CI 0.39-1.07; p = .086; NNT = 10), Dr. Melanie Rae Simpson and coauthors from the Department of Public Health and General Practice Faculty of Medicine at the Norwegian University of Science and Technology in Trondheim, reported.
The children’s incidence of other allergy related diseases, including asthma, atopic sensitization, and allergic rhinoconjunctivitis, were not significantly changed by probiotic use, the investigators said in the report.
The findings suggest that perinatal probiotics prevent, rather than merely delay, the onset of childhood atopic dermatitis, the authors concluded.
Read the full article in BMC Dermatology.
Peripartum consumption of probiotic milk was associated with a decline in the incidence of atopic dermatitis in offspring, but other allergy-related diseases were not reduced in the children, the results of a randomized study indicate.
In a randomized study, 415 pregnant women drank eiher probiotic or placebo milk from 36 weeks’ gestation to 3 months postpartum. At 6 years follow-up, there was a lower cumulative incidence of AD in the probiotic group compared with the placebo group (OR = 0.64; 95% CI 0.39-1.07; p = .086; NNT = 10), Dr. Melanie Rae Simpson and coauthors from the Department of Public Health and General Practice Faculty of Medicine at the Norwegian University of Science and Technology in Trondheim, reported.
The children’s incidence of other allergy related diseases, including asthma, atopic sensitization, and allergic rhinoconjunctivitis, were not significantly changed by probiotic use, the investigators said in the report.
The findings suggest that perinatal probiotics prevent, rather than merely delay, the onset of childhood atopic dermatitis, the authors concluded.
Read the full article in BMC Dermatology.
Peripartum consumption of probiotic milk was associated with a decline in the incidence of atopic dermatitis in offspring, but other allergy-related diseases were not reduced in the children, the results of a randomized study indicate.
In a randomized study, 415 pregnant women drank eiher probiotic or placebo milk from 36 weeks’ gestation to 3 months postpartum. At 6 years follow-up, there was a lower cumulative incidence of AD in the probiotic group compared with the placebo group (OR = 0.64; 95% CI 0.39-1.07; p = .086; NNT = 10), Dr. Melanie Rae Simpson and coauthors from the Department of Public Health and General Practice Faculty of Medicine at the Norwegian University of Science and Technology in Trondheim, reported.
The children’s incidence of other allergy related diseases, including asthma, atopic sensitization, and allergic rhinoconjunctivitis, were not significantly changed by probiotic use, the investigators said in the report.
The findings suggest that perinatal probiotics prevent, rather than merely delay, the onset of childhood atopic dermatitis, the authors concluded.
Read the full article in BMC Dermatology.

Ankle pain in a young woman with Gaucher disease
A 20-year-old woman with Gaucher disease presents with pain in her right ankle and in her back. She has had the ankle pain for the past 12 months and the back pain for the past 2 years. She describes the ankle pain as stabbing and moderately severe. It is constant, present both at rest and during physical activity, but aggravated by walking and twisting movements. She has noticed grinding and clicking sounds as she moves her ankle. The ankle pain has worsened over the past several months.
She says her back pain is similar to her ankle pain but less severe. She also reports generalized mild aches and bone pain. No other joints are involved. She has no history of fever, chills, or trauma.
A COMPLICATED MEDICAL HISTORY
Her Gaucher disease was diagnosed at age 4 when she presented with failure to thrive and with thrombocytopenia and splenomegaly. She and was found to have an N370S/IVS2+1 mutation of the GBA gene. She underwent removal of 90% of her spleen at the time of diagnosis and was on enzyme replacement therapy with imiglucerase until 3 years ago, when the treatment was stopped because the drug had become unavailable (because of a temporary closure of the manufacturing facility), and because she had developed neutralizing antibodies to it. Despite a dosage as high as 120 U/kg every 2 weeks (the recommended range is 2.5 U/kg three times a week up to 60 U/kg every 2 weeks), her anemia and thrombocytopenia worsened to the point that she became dependent on transfusion of red blood cells and platelets. She has also taken glucocorticoids at various times in the past as a premedication before enzyme replacement therapy.
About 3 years ago, she developed dryness of the skin, pruritus, shiny skin, hardening of the skin, and decreased oral aperture, which was diagnosed as scleroderma.
During the past 5 years, she has had multiple episodes of pale coloration of her skin on exposure to cold, suggestive of Raynaud phenomenon. And for the past 5 months, she has noticed a burning sensation in her throat and retrosternal pain, suggestive of gastroesophageal reflux disease.
She is a college student, with no history of smoking or use of alcohol or recreational drugs. She is sexually active, with no history of sexually transmitted disease, and she uses condoms and oral contraceptives for contraception.
Her father and mother are both carriers of Gaucher disease. She is not of Ashkenazi Jewish descent.
FINDINGS ON PHYSICAL EXAMINATION
On physical examination, her temperature, blood pressure, pulse, and respiratory rate are within normal limits. She has extensive tattooing on her upper chest to hide scarring from previous cannulation ports. The right ankle joint is moderately swollen but shows no other signs of inflammation; its range of motion is limited by severe pain. She has tenderness of the spinous processes and paraspinal area, in addition to multiple tender points in the thoracolumbar area. Palpation of the right hip reveals tenderness of the groin and trochanteric bursa.
No lymphadenopathy, hepatomegaly, splenomegaly, or abdominal masses are noted. Neurologic examination is essentially nonfocal.
Her current medications include omeprazole, ergocalciferol, calcium carbonate, gabapentin, citalopram, and celecoxib. She also takes a multivitamin daily.
1. Which is the most likely underlying cause of her ankle pain?
- Rheumatoid arthritis
- Gaucher disease
- Septic arthritis
- Avascular necrosis secondary to steroid use
Rheumatoid arthritis varies in its presentation. It is usually insidious in onset, migratory, and intermittent, with polyarticular or even monoarticular involvement, and it presents with pain, stiffness, and swelling of the joint.1 Most often affected are the metacarpophalangeal, proximal interphalangeal, wrist, and metatarsophalangeal joints. Involvement of large joints of the upper and lower limbs is also common.2 This is not the most likely cause of this patient’s symptoms, based on the history and the current presentation.
Gaucher disease is a lipidosis caused by accumulation of cellular glycolipids, especially glucocerebrosides, due to deficiency of the enzyme beta-glucosidase. Clinical manifestations include hepatomegaly, splenomegaly, and bone marrow disease presenting as anemia, thrombocytopenia, or skeletal disease.3 Skeletal involvement in Gaucher disease includes bone pain, bone infarcts, and lytic lesions.
Whether splenectomy predisposes the patient to bone manifestations is controversial. Some believe that splenectomy decreases the total body reservoir for the storage of glycolipids and predisposes to their deposition in bone, which in turn results in cortical thinning, impaired remodeling, and decreased intraosseous blood flow, leading to osteonecrosis and fractures.4 This is more common in patients with type 1 Gaucher disease who have undergone splenectomy. (Types 2 and 3 are much rarer, occurring mainly in children; central nervous system involvement is a key feature. A discussion of these types is beyond the focus of this paper.) However, some studies suggest that the increase in bone manifestations after splenectomy may be simply because of severe disease.5 It should be noted that, since the advent of enzyme replacement therapy for Gaucher disease, splenectomy is now rarely performed.6
Anemia is also considered an independent risk factor for the development of avascular necrosis in type 1 Gaucher disease.7 Osteonecrosis due to Gaucher disease is relatively common in the femur, tibia, and humerus and uncommon in the ankle joints.8
Septic arthritis is unlikely in this patient in the absence of fever or signs of inflammation of the joint. Her long-standing history of ankle pain would also be unusual for infection, but a superimposed infectious process should always be suspected in an arthritic joint.
Avascular necrosis secondary to steroid use. Glucocorticoids are notorious for their adverse effects on bone. They induce osteocyte apoptosis and a decrease in bone remodeling, potentially predisposing to osteonecrosis.9 There is a high incidence of osteoporosis, osteonecrosis, and fracture risk with glucocorticoid therapy, and the incidence is dose-dependent. Discontinuation of the drug only partially restores fracture risk to baseline levels.10,11
A meta-analysis of cohort studies with a total sample size of about 42,000 reported an increased risk of fracture at all ages with the use of glucocorticoids.12 Because the minimum dosage and duration of therapy to prevent glucocorticoid-induced osteoporosis are not known, the only recommendation is to keep the dosage as low as possible.13
Glucocorticoid therapy is the most common cause of nontraumatic avascular necrosis. The risk of osteonecrosis in patients on long-term glucocorticoid therapy may be as high as 40%.14 The risk is increased with prolonged treatment and with high doses, but it can also occur with short-term exposure to high doses. The increased risk has been shown to persist for as long as 2 years after the drugs are discontinued.15 Glucocorticoid-induced bone disease commonly affects the hip and vertebrae.
At this stage of the workup, we cannot completely rule out glucocorticoid use as the cause. However, after considering this patient’s presentation and the key features of the other diagnoses, her ankle pain and back pain are more likely caused by her preexisting Gaucher disease.
CONTINUED EVALUATION
Initial laboratory tests (Table 1) reveal severe anemia and thrombocytopenia. Bone marrow biopsy of the iliac crest done as part of the workup for these conditions shows extensive bone marrow space replacement by histiocytic infiltrate, consistent with Gaucher disease. No other marrow process is observed.
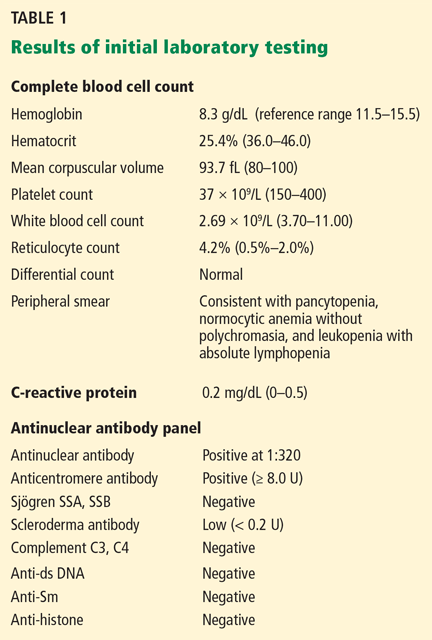
Radiography of the ankle (Figure 1) shows a subtle lucency in the talar dome with minimal subarticular collapse seen on the lateral view, suggestive of avascular necrosis and diffuse osteopenia. Joint spaces are maintained.
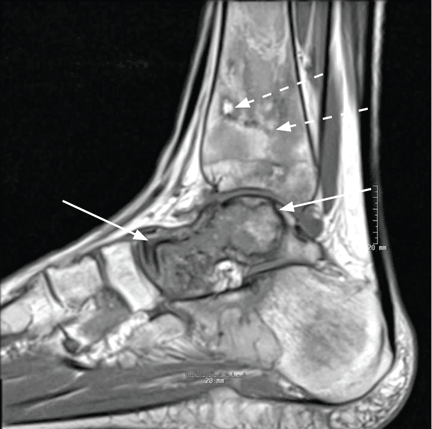
Magnetic resonance imaging (MRI) of the ankle shows numerous bone infarcts with an approximately 15-mm region of mild articular surface collapse in the central and lateral aspect of the talar dome.
MRI of the back shows extensive abnormal bone marrow signal intensity throughout the spine, compatible with a marrow replacement process. Patchy nonexpansile T2/stir hyperintensity with serpiginous enhancement within the T9, T11, T12, L2, and L3 vertebral bodies as well as throughout the entire sacrum is consistent with bone infarct.
2. Based on the results of radiographic studies, which is most likely the immediate cause of her ankle pain?
- Talar avascular necrosis secondary to rheumatoid arthritis
- Talar avascular necrosis secondary to Gaucher disease
- Trauma-induced fracture of the talus
- Plantar fasciitis
Of the bones of the feet, the talus is unique. It is the second largest of the tarsal bones and does not have muscular or tendinous attachments. Sixty percent of the talus bone is covered by articular cartilage,16 so only a limited area is available for penetration of blood vessels. Also, small nutrient vessels and variations of intraosseous anastomoses with a lack of collateral circulation predispose the talus to osteonecrosis when the vascular supply is compromised.16
Radiographic evidence of avascular necrosis is the presence of bone that is more radiopaque than normal bone; this is necrotic bone surrounded by osteopenic bone. Avascular necrosis causes hyperemia and resorption of bone. The resorption does not take place in necrotic bone because of the lack of a vascular supply, and so it appears radiopaque, whereas the bone surrounding the necrotic bone becomes osteopenic and radiolucent.
The sclerotic rim of a bone infarct is also enhanced by an attempted healing process in which new bone forms on the surface of necrotic trabeculae, a process known as “creeping substitution.” This gives a typical sclerotic picture of the talus.
MRI is the most sensitive technique for detecting osteonecrosis. A characteristic radiographic pattern is seen with osteonecrosis of the talus starting with talar dome opacity, followed by deformity and, in severe cases, articular collapse and bone fragmentation.17
The radiograph in our patient’s case is not consistent with features of rheumatoid arthritis or traumatic fracture of the talus. In plantar fasciitis, radiographs are used to rule out other pathologies of the foot, and the only finding may be a bone spur seen at the site of pain. The bone spur is not the cause of pain in plantar fasciitis but may be a result of the plantar fasciitis itself.
Therefore, avascular necrosis secondary to Gaucher disease is most likely the immediate cause of her ankle pain.
THE COURSE OF TREATMENT
The patient is started on enzyme replacement therapy with taliglucerase alfa (see discussion of enzyme replacement below). For the ankle pain, conservative management is prescribed, with application of a splint and a boot.
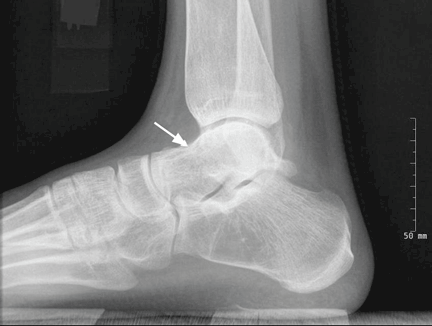
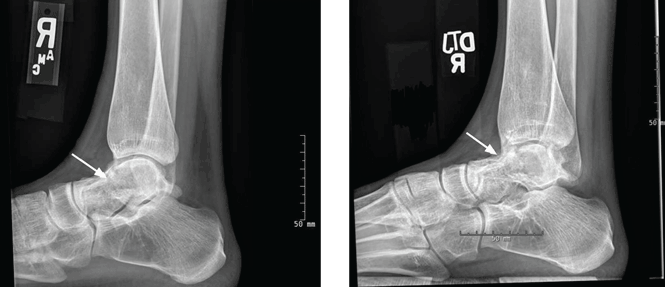
After 4 months of conservative management, radiography (Figure 2) and magnetic resonance imaging (Figure 3) show progressive deterioration of the talus body, and her ankle pain has worsened. A 6-week trial of an ankle brace also proves futile. Her pain continues to worsen and is not controllable with high doses of pain medication. She requests below-the-knee amputation.
Given the complexity of this patient’s medical condition, fusion of the ankle and hindfoot—which in some patients is preferable to amputation—is not considered because of her extensive bone involvement and ongoing thrombocytopenia, which would impede healing after the procedure. Below-the-knee amputation is performed without complications.
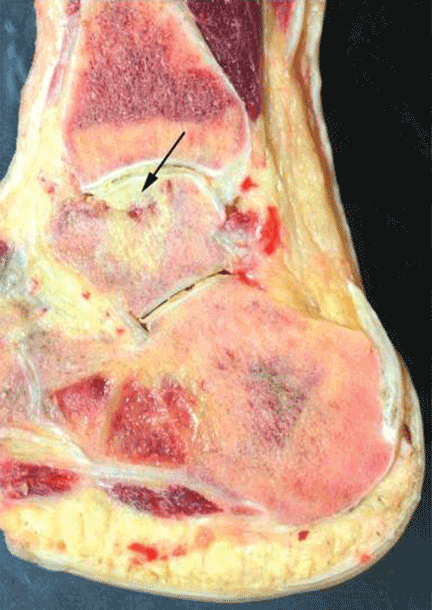
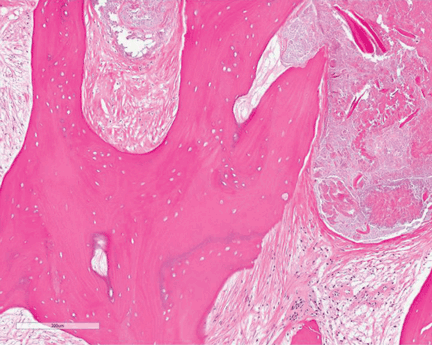
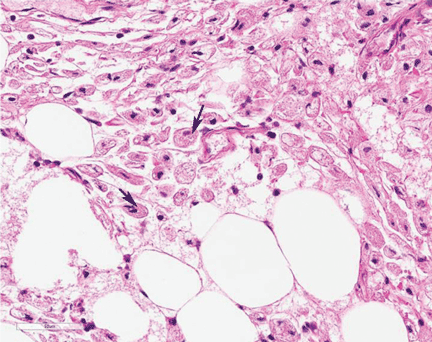
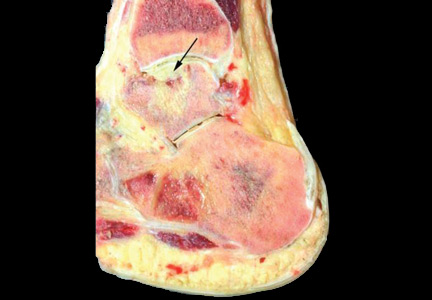
Study of the specimen after amputation reveals talar bone necrosis and bone marrow infiltration by foamy macrophages, consistent with Gaucher disease (Figures 4–6).
GAUCHER DISEASE
Pharmacologic treatments, effective only for type 1 Gaucher disease, target hepatosplenomegaly, cytopenia, and bone manifestations. Two approaches are enzyme replacement therapy—ie, to replace the defective enzyme—and substrate reduction therapy—ie, to reduce the production and thus the accumulation of glucocerebroside. Enzyme replacement is the first choice of therapy; substrate reduction is reserved for patients unable to tolerate enzyme replacement therapy.
Enzyme replacement
Current drugs for enzyme replacement therapy are imiglucerase, taliglucerase alfa, and velaglucerase alfa. The drugs are given by intravenous infusion over 1 to 2 hours in an outpatient clinic or office every 2 weeks.
These drugs are extremely expensive. Currently, the estimated cost of therapy for 1 year would be $432,978 for imiglucerase, $324,870 for taliglucerase alfa, and $368,550 for velaglucerase alfa. (The estimated costs are for 1 year of treatment for a 70-kg patient at 60 U/kg every 2 weeks.)18 Taliglucerase alfa is less expensive than the other two because it is plant-derived and thus can be more readily produced on a large scale.19
Substrate reduction
Current drugs for substrate reduction therapy are eliglustat and miglustat. They are given orally. Eliglustat is the first oral drug approved as a first-line treatment for Gaucher disease.20 Miglustat is approved only for mild to moderate disease when enzyme replacement fails or is not tolerated.
Patients can develop antibodies to any of the enzyme replacement drugs. It is not known whether this antibody response differs among the three drugs.21
Avascular necrosis of bone can occur in many clinical settings especially after a fracture, particularly of the head of the femur, which leads to interruption of blood supply to the area. Patients with sickle cell disease, those on corticosteroids or bisphosphonates (the latter causing osteonecrosis of the jaw), and those who have pancreatitis or human immunodeficiency virus infection are more prone to this bone complication.
In Gaucher disease, osteonecrosis is associated with splenectomy and severe disease and tends to occur at a younger age than in patients with other diagnoses.8 The plasma chitotriosidase activity and pulmonary and activation-regulated chemokines (PARC/CCL18), which are 10 to 40 times higher than normal in symptomatic patients with Gaucher disease, can be used as a biomarker of disease activity.8 Only plasma chitotriosidase is clinically available and used on a routine basis.
Bone involvement is seen in approximately 75% of the patients with type 1 Gaucher disease,22 and osteonecrosis is a severe form of bone involvement. Monitoring of patients for bone involvement is recommended. Enzyme replacement therapy for Gaucher disease needs to be started even if visceral disease is absent if the patient has evidence of bone involvement in the form of avascular necrosis.7 Prospective studies have shown that enzyme replacement therapy reduces the incidence of osteonecrosis.23
FOLLOW-UP MANAGEMENT OF OUR PATIENT
Avascular necrosis in Gaucher disease more typically involves the hips and shoulders. In the case of our patient, the talus was the most affected bone. Other contributing factors may have been the use of steroids as a premedication (often unnecessary) for her enzyme replacement therapy, as well as the coexistent scleroderma.24
The decision to switch from imiglucerase, to which she developed antibodies, to taliglucerase was made in the hope that the antibodies would not cross-react. After she started taliglucerase, her complete blood count values improved steadily. She did not require transfusions for more than 1 year. Her platelet count rose to 90 × 109/L, and her hemoglobin to 12 g/dL.
A multidisciplinary approach with regular monitoring and appropriate initiation of therapy is necessary to prevent disastrous complications in patients with Gaucher disease.
- Lee DM, Weinblatt ME. Rheumatoid arthritis. Lancet 2001; 358:903–911.
- Fleming A, Crown JM, Corbett M. Early rheumatoid disease. I. Onset. Ann Rheum Dis 1976; 35:357–360.
- Grabowski GA, Andria G, Baldellou A, et al. Pediatric non-neuronopathic Gaucher disease: presentation, diagnosis, and assessment. Consensus statements. Eur J Pediatr 2004; 163:58–66.
- Rodrigue SW, Rosenthal DI, Barton NW, Zurakowski D, Mankin HJ. Risk factors for osteonecrosis in patients with type 1 Gaucher’s disease. Clin Orthop Relat Res 1999; May (362):201–207.
- Lee RE. The pathology of Gaucher disease. Prog Clin Biol Res 1982; 95:177–217.
- Cox TM, Aerts JM, Belmatoug N, et al. Management of non-neuronopathic Gaucher disease with special reference to pregnancy, splenectomy, bisphosphonate therapy, use of biomarkers and bone disease monitoring. J Inherit Metab Dis 2008; 31:319–336.
- Khan A, Hangartner T, Weinreb NJ, Taylor JS, Mistry PK. Risk factors for fractures and avascular osteonecrosis in type 1 Gaucher disease: a study from the International Collaborative Gaucher Group (ICGG) Gaucher Registry. J Bone Miner Res 2012; 27:1839–1848.
- Deegan PB, Pavlova E, Tindall J, et al. Osseous manifestations of adult Gaucher disease in the era of enzyme replacement therapy. Medicine (Baltimore) 2011; 90:52–60.
- Weinstein RS. Glucocorticoid-induced osteonecrosis. Endocrine 2012; 41:183–190.
- Compston J. Management of glucocorticoid-induced osteoporosis. Nat Rev Rheumatol 2010; 6:82–88.
- Van Staa TP, Laan RF, Barton IP, Cohen S, Reid DM, Cooper C. Bone density threshold and other predictors of vertebral fracture in patients receiving oral glucocorticoid therapy. Arthritis Rheum 2003; 48:3224–3229.
- Kanis JA, Johnell O, Oden A, et al. The risk and burden of vertebral fractures in Sweden. Osteoporos Int 2004; 15:20–26.
- Seguro LP, Rosario C, Shoenfeld Y. Long-term complications of past glucocorticoid use. Autoimmun Rev 2013; 12:629–632.
- Weinstein RS. Glucocorticoid-induced osteoporosis and osteonecrosis. Endocrinol Metab Clin North Am 2012; 41:595–611.
- Cooper C, Steinbuch M, Stevenson R, Miday R, Watts NB. The epidemiology of osteonecrosis: findings from the GPRD and THIN databases in the UK. Osteoporos Int 2010; 21:569–577.
- Mulfinger GL, Trueta J. The blood supply of the talus. J Bone Joint Surg Br 1970; 52:160–167.
- Pearce DH, Mongiardi CN, Fornasier VL, Daniels TR. Avascular necrosis of the talus: a pictoral essay. Radiographics 2005; 25:399–410.
- In brief: Taliglucerase (Elelyso) for Gaucher disease. Med Lett Drugs Ther 2012 Jul 9; 54(1394):56.
- Hollak CE. An evidence-based review of the potential benefits of taliglucerase alfa in the treatment of patients with Gaucher disease. Core Evid 2012; 7:15–20.
- Poole RM. Eliglustat: first global approval. Drugs 2014; 74:1829–1836.
- Bennett LL, Mohan D. Gaucher disease and its treatment options. Ann Pharmacother 2013; 47:1182–1193.
- Germain DP. Gaucher’s disease: a paradigm for interventional genetics. Clin Genet 2004; 65:77–86.
- Sims KB, Pastores GM, Weinreb NJ, et al. Improvement of bone disease by imiglucerase (Cerezyme) therapy in patients with skeletal manifestations of type 1 Gaucher disease: results of a 48-month longitudinal cohort study. Clin Genet 2008; 73:430–440.
- Rennie C, Britton J, Prouse P. Bilateral avascular necrosis of the lunate in a patient with severe Raynaud’s phenomenon and scleroderma. J Clin Rheumatol 1999; 5:165–168.
A 20-year-old woman with Gaucher disease presents with pain in her right ankle and in her back. She has had the ankle pain for the past 12 months and the back pain for the past 2 years. She describes the ankle pain as stabbing and moderately severe. It is constant, present both at rest and during physical activity, but aggravated by walking and twisting movements. She has noticed grinding and clicking sounds as she moves her ankle. The ankle pain has worsened over the past several months.
She says her back pain is similar to her ankle pain but less severe. She also reports generalized mild aches and bone pain. No other joints are involved. She has no history of fever, chills, or trauma.
A COMPLICATED MEDICAL HISTORY
Her Gaucher disease was diagnosed at age 4 when she presented with failure to thrive and with thrombocytopenia and splenomegaly. She and was found to have an N370S/IVS2+1 mutation of the GBA gene. She underwent removal of 90% of her spleen at the time of diagnosis and was on enzyme replacement therapy with imiglucerase until 3 years ago, when the treatment was stopped because the drug had become unavailable (because of a temporary closure of the manufacturing facility), and because she had developed neutralizing antibodies to it. Despite a dosage as high as 120 U/kg every 2 weeks (the recommended range is 2.5 U/kg three times a week up to 60 U/kg every 2 weeks), her anemia and thrombocytopenia worsened to the point that she became dependent on transfusion of red blood cells and platelets. She has also taken glucocorticoids at various times in the past as a premedication before enzyme replacement therapy.
About 3 years ago, she developed dryness of the skin, pruritus, shiny skin, hardening of the skin, and decreased oral aperture, which was diagnosed as scleroderma.
During the past 5 years, she has had multiple episodes of pale coloration of her skin on exposure to cold, suggestive of Raynaud phenomenon. And for the past 5 months, she has noticed a burning sensation in her throat and retrosternal pain, suggestive of gastroesophageal reflux disease.
She is a college student, with no history of smoking or use of alcohol or recreational drugs. She is sexually active, with no history of sexually transmitted disease, and she uses condoms and oral contraceptives for contraception.
Her father and mother are both carriers of Gaucher disease. She is not of Ashkenazi Jewish descent.
FINDINGS ON PHYSICAL EXAMINATION
On physical examination, her temperature, blood pressure, pulse, and respiratory rate are within normal limits. She has extensive tattooing on her upper chest to hide scarring from previous cannulation ports. The right ankle joint is moderately swollen but shows no other signs of inflammation; its range of motion is limited by severe pain. She has tenderness of the spinous processes and paraspinal area, in addition to multiple tender points in the thoracolumbar area. Palpation of the right hip reveals tenderness of the groin and trochanteric bursa.
No lymphadenopathy, hepatomegaly, splenomegaly, or abdominal masses are noted. Neurologic examination is essentially nonfocal.
Her current medications include omeprazole, ergocalciferol, calcium carbonate, gabapentin, citalopram, and celecoxib. She also takes a multivitamin daily.
1. Which is the most likely underlying cause of her ankle pain?
- Rheumatoid arthritis
- Gaucher disease
- Septic arthritis
- Avascular necrosis secondary to steroid use
Rheumatoid arthritis varies in its presentation. It is usually insidious in onset, migratory, and intermittent, with polyarticular or even monoarticular involvement, and it presents with pain, stiffness, and swelling of the joint.1 Most often affected are the metacarpophalangeal, proximal interphalangeal, wrist, and metatarsophalangeal joints. Involvement of large joints of the upper and lower limbs is also common.2 This is not the most likely cause of this patient’s symptoms, based on the history and the current presentation.
Gaucher disease is a lipidosis caused by accumulation of cellular glycolipids, especially glucocerebrosides, due to deficiency of the enzyme beta-glucosidase. Clinical manifestations include hepatomegaly, splenomegaly, and bone marrow disease presenting as anemia, thrombocytopenia, or skeletal disease.3 Skeletal involvement in Gaucher disease includes bone pain, bone infarcts, and lytic lesions.
Whether splenectomy predisposes the patient to bone manifestations is controversial. Some believe that splenectomy decreases the total body reservoir for the storage of glycolipids and predisposes to their deposition in bone, which in turn results in cortical thinning, impaired remodeling, and decreased intraosseous blood flow, leading to osteonecrosis and fractures.4 This is more common in patients with type 1 Gaucher disease who have undergone splenectomy. (Types 2 and 3 are much rarer, occurring mainly in children; central nervous system involvement is a key feature. A discussion of these types is beyond the focus of this paper.) However, some studies suggest that the increase in bone manifestations after splenectomy may be simply because of severe disease.5 It should be noted that, since the advent of enzyme replacement therapy for Gaucher disease, splenectomy is now rarely performed.6
Anemia is also considered an independent risk factor for the development of avascular necrosis in type 1 Gaucher disease.7 Osteonecrosis due to Gaucher disease is relatively common in the femur, tibia, and humerus and uncommon in the ankle joints.8
Septic arthritis is unlikely in this patient in the absence of fever or signs of inflammation of the joint. Her long-standing history of ankle pain would also be unusual for infection, but a superimposed infectious process should always be suspected in an arthritic joint.
Avascular necrosis secondary to steroid use. Glucocorticoids are notorious for their adverse effects on bone. They induce osteocyte apoptosis and a decrease in bone remodeling, potentially predisposing to osteonecrosis.9 There is a high incidence of osteoporosis, osteonecrosis, and fracture risk with glucocorticoid therapy, and the incidence is dose-dependent. Discontinuation of the drug only partially restores fracture risk to baseline levels.10,11
A meta-analysis of cohort studies with a total sample size of about 42,000 reported an increased risk of fracture at all ages with the use of glucocorticoids.12 Because the minimum dosage and duration of therapy to prevent glucocorticoid-induced osteoporosis are not known, the only recommendation is to keep the dosage as low as possible.13
Glucocorticoid therapy is the most common cause of nontraumatic avascular necrosis. The risk of osteonecrosis in patients on long-term glucocorticoid therapy may be as high as 40%.14 The risk is increased with prolonged treatment and with high doses, but it can also occur with short-term exposure to high doses. The increased risk has been shown to persist for as long as 2 years after the drugs are discontinued.15 Glucocorticoid-induced bone disease commonly affects the hip and vertebrae.
At this stage of the workup, we cannot completely rule out glucocorticoid use as the cause. However, after considering this patient’s presentation and the key features of the other diagnoses, her ankle pain and back pain are more likely caused by her preexisting Gaucher disease.
CONTINUED EVALUATION
Initial laboratory tests (Table 1) reveal severe anemia and thrombocytopenia. Bone marrow biopsy of the iliac crest done as part of the workup for these conditions shows extensive bone marrow space replacement by histiocytic infiltrate, consistent with Gaucher disease. No other marrow process is observed.
Radiography of the ankle (Figure 1) shows a subtle lucency in the talar dome with minimal subarticular collapse seen on the lateral view, suggestive of avascular necrosis and diffuse osteopenia. Joint spaces are maintained.
Magnetic resonance imaging (MRI) of the ankle shows numerous bone infarcts with an approximately 15-mm region of mild articular surface collapse in the central and lateral aspect of the talar dome.
MRI of the back shows extensive abnormal bone marrow signal intensity throughout the spine, compatible with a marrow replacement process. Patchy nonexpansile T2/stir hyperintensity with serpiginous enhancement within the T9, T11, T12, L2, and L3 vertebral bodies as well as throughout the entire sacrum is consistent with bone infarct.
2. Based on the results of radiographic studies, which is most likely the immediate cause of her ankle pain?
- Talar avascular necrosis secondary to rheumatoid arthritis
- Talar avascular necrosis secondary to Gaucher disease
- Trauma-induced fracture of the talus
- Plantar fasciitis
Of the bones of the feet, the talus is unique. It is the second largest of the tarsal bones and does not have muscular or tendinous attachments. Sixty percent of the talus bone is covered by articular cartilage,16 so only a limited area is available for penetration of blood vessels. Also, small nutrient vessels and variations of intraosseous anastomoses with a lack of collateral circulation predispose the talus to osteonecrosis when the vascular supply is compromised.16
Radiographic evidence of avascular necrosis is the presence of bone that is more radiopaque than normal bone; this is necrotic bone surrounded by osteopenic bone. Avascular necrosis causes hyperemia and resorption of bone. The resorption does not take place in necrotic bone because of the lack of a vascular supply, and so it appears radiopaque, whereas the bone surrounding the necrotic bone becomes osteopenic and radiolucent.
The sclerotic rim of a bone infarct is also enhanced by an attempted healing process in which new bone forms on the surface of necrotic trabeculae, a process known as “creeping substitution.” This gives a typical sclerotic picture of the talus.
MRI is the most sensitive technique for detecting osteonecrosis. A characteristic radiographic pattern is seen with osteonecrosis of the talus starting with talar dome opacity, followed by deformity and, in severe cases, articular collapse and bone fragmentation.17
The radiograph in our patient’s case is not consistent with features of rheumatoid arthritis or traumatic fracture of the talus. In plantar fasciitis, radiographs are used to rule out other pathologies of the foot, and the only finding may be a bone spur seen at the site of pain. The bone spur is not the cause of pain in plantar fasciitis but may be a result of the plantar fasciitis itself.
Therefore, avascular necrosis secondary to Gaucher disease is most likely the immediate cause of her ankle pain.
THE COURSE OF TREATMENT
The patient is started on enzyme replacement therapy with taliglucerase alfa (see discussion of enzyme replacement below). For the ankle pain, conservative management is prescribed, with application of a splint and a boot.
After 4 months of conservative management, radiography (Figure 2) and magnetic resonance imaging (Figure 3) show progressive deterioration of the talus body, and her ankle pain has worsened. A 6-week trial of an ankle brace also proves futile. Her pain continues to worsen and is not controllable with high doses of pain medication. She requests below-the-knee amputation.
Given the complexity of this patient’s medical condition, fusion of the ankle and hindfoot—which in some patients is preferable to amputation—is not considered because of her extensive bone involvement and ongoing thrombocytopenia, which would impede healing after the procedure. Below-the-knee amputation is performed without complications.
Study of the specimen after amputation reveals talar bone necrosis and bone marrow infiltration by foamy macrophages, consistent with Gaucher disease (Figures 4–6).
GAUCHER DISEASE
Pharmacologic treatments, effective only for type 1 Gaucher disease, target hepatosplenomegaly, cytopenia, and bone manifestations. Two approaches are enzyme replacement therapy—ie, to replace the defective enzyme—and substrate reduction therapy—ie, to reduce the production and thus the accumulation of glucocerebroside. Enzyme replacement is the first choice of therapy; substrate reduction is reserved for patients unable to tolerate enzyme replacement therapy.
Enzyme replacement
Current drugs for enzyme replacement therapy are imiglucerase, taliglucerase alfa, and velaglucerase alfa. The drugs are given by intravenous infusion over 1 to 2 hours in an outpatient clinic or office every 2 weeks.
These drugs are extremely expensive. Currently, the estimated cost of therapy for 1 year would be $432,978 for imiglucerase, $324,870 for taliglucerase alfa, and $368,550 for velaglucerase alfa. (The estimated costs are for 1 year of treatment for a 70-kg patient at 60 U/kg every 2 weeks.)18 Taliglucerase alfa is less expensive than the other two because it is plant-derived and thus can be more readily produced on a large scale.19
Substrate reduction
Current drugs for substrate reduction therapy are eliglustat and miglustat. They are given orally. Eliglustat is the first oral drug approved as a first-line treatment for Gaucher disease.20 Miglustat is approved only for mild to moderate disease when enzyme replacement fails or is not tolerated.
Patients can develop antibodies to any of the enzyme replacement drugs. It is not known whether this antibody response differs among the three drugs.21
Avascular necrosis of bone can occur in many clinical settings especially after a fracture, particularly of the head of the femur, which leads to interruption of blood supply to the area. Patients with sickle cell disease, those on corticosteroids or bisphosphonates (the latter causing osteonecrosis of the jaw), and those who have pancreatitis or human immunodeficiency virus infection are more prone to this bone complication.
In Gaucher disease, osteonecrosis is associated with splenectomy and severe disease and tends to occur at a younger age than in patients with other diagnoses.8 The plasma chitotriosidase activity and pulmonary and activation-regulated chemokines (PARC/CCL18), which are 10 to 40 times higher than normal in symptomatic patients with Gaucher disease, can be used as a biomarker of disease activity.8 Only plasma chitotriosidase is clinically available and used on a routine basis.
Bone involvement is seen in approximately 75% of the patients with type 1 Gaucher disease,22 and osteonecrosis is a severe form of bone involvement. Monitoring of patients for bone involvement is recommended. Enzyme replacement therapy for Gaucher disease needs to be started even if visceral disease is absent if the patient has evidence of bone involvement in the form of avascular necrosis.7 Prospective studies have shown that enzyme replacement therapy reduces the incidence of osteonecrosis.23
FOLLOW-UP MANAGEMENT OF OUR PATIENT
Avascular necrosis in Gaucher disease more typically involves the hips and shoulders. In the case of our patient, the talus was the most affected bone. Other contributing factors may have been the use of steroids as a premedication (often unnecessary) for her enzyme replacement therapy, as well as the coexistent scleroderma.24
The decision to switch from imiglucerase, to which she developed antibodies, to taliglucerase was made in the hope that the antibodies would not cross-react. After she started taliglucerase, her complete blood count values improved steadily. She did not require transfusions for more than 1 year. Her platelet count rose to 90 × 109/L, and her hemoglobin to 12 g/dL.
A multidisciplinary approach with regular monitoring and appropriate initiation of therapy is necessary to prevent disastrous complications in patients with Gaucher disease.
A 20-year-old woman with Gaucher disease presents with pain in her right ankle and in her back. She has had the ankle pain for the past 12 months and the back pain for the past 2 years. She describes the ankle pain as stabbing and moderately severe. It is constant, present both at rest and during physical activity, but aggravated by walking and twisting movements. She has noticed grinding and clicking sounds as she moves her ankle. The ankle pain has worsened over the past several months.
She says her back pain is similar to her ankle pain but less severe. She also reports generalized mild aches and bone pain. No other joints are involved. She has no history of fever, chills, or trauma.
A COMPLICATED MEDICAL HISTORY
Her Gaucher disease was diagnosed at age 4 when she presented with failure to thrive and with thrombocytopenia and splenomegaly. She and was found to have an N370S/IVS2+1 mutation of the GBA gene. She underwent removal of 90% of her spleen at the time of diagnosis and was on enzyme replacement therapy with imiglucerase until 3 years ago, when the treatment was stopped because the drug had become unavailable (because of a temporary closure of the manufacturing facility), and because she had developed neutralizing antibodies to it. Despite a dosage as high as 120 U/kg every 2 weeks (the recommended range is 2.5 U/kg three times a week up to 60 U/kg every 2 weeks), her anemia and thrombocytopenia worsened to the point that she became dependent on transfusion of red blood cells and platelets. She has also taken glucocorticoids at various times in the past as a premedication before enzyme replacement therapy.
About 3 years ago, she developed dryness of the skin, pruritus, shiny skin, hardening of the skin, and decreased oral aperture, which was diagnosed as scleroderma.
During the past 5 years, she has had multiple episodes of pale coloration of her skin on exposure to cold, suggestive of Raynaud phenomenon. And for the past 5 months, she has noticed a burning sensation in her throat and retrosternal pain, suggestive of gastroesophageal reflux disease.
She is a college student, with no history of smoking or use of alcohol or recreational drugs. She is sexually active, with no history of sexually transmitted disease, and she uses condoms and oral contraceptives for contraception.
Her father and mother are both carriers of Gaucher disease. She is not of Ashkenazi Jewish descent.
FINDINGS ON PHYSICAL EXAMINATION
On physical examination, her temperature, blood pressure, pulse, and respiratory rate are within normal limits. She has extensive tattooing on her upper chest to hide scarring from previous cannulation ports. The right ankle joint is moderately swollen but shows no other signs of inflammation; its range of motion is limited by severe pain. She has tenderness of the spinous processes and paraspinal area, in addition to multiple tender points in the thoracolumbar area. Palpation of the right hip reveals tenderness of the groin and trochanteric bursa.
No lymphadenopathy, hepatomegaly, splenomegaly, or abdominal masses are noted. Neurologic examination is essentially nonfocal.
Her current medications include omeprazole, ergocalciferol, calcium carbonate, gabapentin, citalopram, and celecoxib. She also takes a multivitamin daily.
1. Which is the most likely underlying cause of her ankle pain?
- Rheumatoid arthritis
- Gaucher disease
- Septic arthritis
- Avascular necrosis secondary to steroid use
Rheumatoid arthritis varies in its presentation. It is usually insidious in onset, migratory, and intermittent, with polyarticular or even monoarticular involvement, and it presents with pain, stiffness, and swelling of the joint.1 Most often affected are the metacarpophalangeal, proximal interphalangeal, wrist, and metatarsophalangeal joints. Involvement of large joints of the upper and lower limbs is also common.2 This is not the most likely cause of this patient’s symptoms, based on the history and the current presentation.
Gaucher disease is a lipidosis caused by accumulation of cellular glycolipids, especially glucocerebrosides, due to deficiency of the enzyme beta-glucosidase. Clinical manifestations include hepatomegaly, splenomegaly, and bone marrow disease presenting as anemia, thrombocytopenia, or skeletal disease.3 Skeletal involvement in Gaucher disease includes bone pain, bone infarcts, and lytic lesions.
Whether splenectomy predisposes the patient to bone manifestations is controversial. Some believe that splenectomy decreases the total body reservoir for the storage of glycolipids and predisposes to their deposition in bone, which in turn results in cortical thinning, impaired remodeling, and decreased intraosseous blood flow, leading to osteonecrosis and fractures.4 This is more common in patients with type 1 Gaucher disease who have undergone splenectomy. (Types 2 and 3 are much rarer, occurring mainly in children; central nervous system involvement is a key feature. A discussion of these types is beyond the focus of this paper.) However, some studies suggest that the increase in bone manifestations after splenectomy may be simply because of severe disease.5 It should be noted that, since the advent of enzyme replacement therapy for Gaucher disease, splenectomy is now rarely performed.6
Anemia is also considered an independent risk factor for the development of avascular necrosis in type 1 Gaucher disease.7 Osteonecrosis due to Gaucher disease is relatively common in the femur, tibia, and humerus and uncommon in the ankle joints.8
Septic arthritis is unlikely in this patient in the absence of fever or signs of inflammation of the joint. Her long-standing history of ankle pain would also be unusual for infection, but a superimposed infectious process should always be suspected in an arthritic joint.
Avascular necrosis secondary to steroid use. Glucocorticoids are notorious for their adverse effects on bone. They induce osteocyte apoptosis and a decrease in bone remodeling, potentially predisposing to osteonecrosis.9 There is a high incidence of osteoporosis, osteonecrosis, and fracture risk with glucocorticoid therapy, and the incidence is dose-dependent. Discontinuation of the drug only partially restores fracture risk to baseline levels.10,11
A meta-analysis of cohort studies with a total sample size of about 42,000 reported an increased risk of fracture at all ages with the use of glucocorticoids.12 Because the minimum dosage and duration of therapy to prevent glucocorticoid-induced osteoporosis are not known, the only recommendation is to keep the dosage as low as possible.13
Glucocorticoid therapy is the most common cause of nontraumatic avascular necrosis. The risk of osteonecrosis in patients on long-term glucocorticoid therapy may be as high as 40%.14 The risk is increased with prolonged treatment and with high doses, but it can also occur with short-term exposure to high doses. The increased risk has been shown to persist for as long as 2 years after the drugs are discontinued.15 Glucocorticoid-induced bone disease commonly affects the hip and vertebrae.
At this stage of the workup, we cannot completely rule out glucocorticoid use as the cause. However, after considering this patient’s presentation and the key features of the other diagnoses, her ankle pain and back pain are more likely caused by her preexisting Gaucher disease.
CONTINUED EVALUATION
Initial laboratory tests (Table 1) reveal severe anemia and thrombocytopenia. Bone marrow biopsy of the iliac crest done as part of the workup for these conditions shows extensive bone marrow space replacement by histiocytic infiltrate, consistent with Gaucher disease. No other marrow process is observed.
Radiography of the ankle (Figure 1) shows a subtle lucency in the talar dome with minimal subarticular collapse seen on the lateral view, suggestive of avascular necrosis and diffuse osteopenia. Joint spaces are maintained.
Magnetic resonance imaging (MRI) of the ankle shows numerous bone infarcts with an approximately 15-mm region of mild articular surface collapse in the central and lateral aspect of the talar dome.
MRI of the back shows extensive abnormal bone marrow signal intensity throughout the spine, compatible with a marrow replacement process. Patchy nonexpansile T2/stir hyperintensity with serpiginous enhancement within the T9, T11, T12, L2, and L3 vertebral bodies as well as throughout the entire sacrum is consistent with bone infarct.
2. Based on the results of radiographic studies, which is most likely the immediate cause of her ankle pain?
- Talar avascular necrosis secondary to rheumatoid arthritis
- Talar avascular necrosis secondary to Gaucher disease
- Trauma-induced fracture of the talus
- Plantar fasciitis
Of the bones of the feet, the talus is unique. It is the second largest of the tarsal bones and does not have muscular or tendinous attachments. Sixty percent of the talus bone is covered by articular cartilage,16 so only a limited area is available for penetration of blood vessels. Also, small nutrient vessels and variations of intraosseous anastomoses with a lack of collateral circulation predispose the talus to osteonecrosis when the vascular supply is compromised.16
Radiographic evidence of avascular necrosis is the presence of bone that is more radiopaque than normal bone; this is necrotic bone surrounded by osteopenic bone. Avascular necrosis causes hyperemia and resorption of bone. The resorption does not take place in necrotic bone because of the lack of a vascular supply, and so it appears radiopaque, whereas the bone surrounding the necrotic bone becomes osteopenic and radiolucent.
The sclerotic rim of a bone infarct is also enhanced by an attempted healing process in which new bone forms on the surface of necrotic trabeculae, a process known as “creeping substitution.” This gives a typical sclerotic picture of the talus.
MRI is the most sensitive technique for detecting osteonecrosis. A characteristic radiographic pattern is seen with osteonecrosis of the talus starting with talar dome opacity, followed by deformity and, in severe cases, articular collapse and bone fragmentation.17
The radiograph in our patient’s case is not consistent with features of rheumatoid arthritis or traumatic fracture of the talus. In plantar fasciitis, radiographs are used to rule out other pathologies of the foot, and the only finding may be a bone spur seen at the site of pain. The bone spur is not the cause of pain in plantar fasciitis but may be a result of the plantar fasciitis itself.
Therefore, avascular necrosis secondary to Gaucher disease is most likely the immediate cause of her ankle pain.
THE COURSE OF TREATMENT
The patient is started on enzyme replacement therapy with taliglucerase alfa (see discussion of enzyme replacement below). For the ankle pain, conservative management is prescribed, with application of a splint and a boot.
After 4 months of conservative management, radiography (Figure 2) and magnetic resonance imaging (Figure 3) show progressive deterioration of the talus body, and her ankle pain has worsened. A 6-week trial of an ankle brace also proves futile. Her pain continues to worsen and is not controllable with high doses of pain medication. She requests below-the-knee amputation.
Given the complexity of this patient’s medical condition, fusion of the ankle and hindfoot—which in some patients is preferable to amputation—is not considered because of her extensive bone involvement and ongoing thrombocytopenia, which would impede healing after the procedure. Below-the-knee amputation is performed without complications.
Study of the specimen after amputation reveals talar bone necrosis and bone marrow infiltration by foamy macrophages, consistent with Gaucher disease (Figures 4–6).
GAUCHER DISEASE
Pharmacologic treatments, effective only for type 1 Gaucher disease, target hepatosplenomegaly, cytopenia, and bone manifestations. Two approaches are enzyme replacement therapy—ie, to replace the defective enzyme—and substrate reduction therapy—ie, to reduce the production and thus the accumulation of glucocerebroside. Enzyme replacement is the first choice of therapy; substrate reduction is reserved for patients unable to tolerate enzyme replacement therapy.
Enzyme replacement
Current drugs for enzyme replacement therapy are imiglucerase, taliglucerase alfa, and velaglucerase alfa. The drugs are given by intravenous infusion over 1 to 2 hours in an outpatient clinic or office every 2 weeks.
These drugs are extremely expensive. Currently, the estimated cost of therapy for 1 year would be $432,978 for imiglucerase, $324,870 for taliglucerase alfa, and $368,550 for velaglucerase alfa. (The estimated costs are for 1 year of treatment for a 70-kg patient at 60 U/kg every 2 weeks.)18 Taliglucerase alfa is less expensive than the other two because it is plant-derived and thus can be more readily produced on a large scale.19
Substrate reduction
Current drugs for substrate reduction therapy are eliglustat and miglustat. They are given orally. Eliglustat is the first oral drug approved as a first-line treatment for Gaucher disease.20 Miglustat is approved only for mild to moderate disease when enzyme replacement fails or is not tolerated.
Patients can develop antibodies to any of the enzyme replacement drugs. It is not known whether this antibody response differs among the three drugs.21
Avascular necrosis of bone can occur in many clinical settings especially after a fracture, particularly of the head of the femur, which leads to interruption of blood supply to the area. Patients with sickle cell disease, those on corticosteroids or bisphosphonates (the latter causing osteonecrosis of the jaw), and those who have pancreatitis or human immunodeficiency virus infection are more prone to this bone complication.
In Gaucher disease, osteonecrosis is associated with splenectomy and severe disease and tends to occur at a younger age than in patients with other diagnoses.8 The plasma chitotriosidase activity and pulmonary and activation-regulated chemokines (PARC/CCL18), which are 10 to 40 times higher than normal in symptomatic patients with Gaucher disease, can be used as a biomarker of disease activity.8 Only plasma chitotriosidase is clinically available and used on a routine basis.
Bone involvement is seen in approximately 75% of the patients with type 1 Gaucher disease,22 and osteonecrosis is a severe form of bone involvement. Monitoring of patients for bone involvement is recommended. Enzyme replacement therapy for Gaucher disease needs to be started even if visceral disease is absent if the patient has evidence of bone involvement in the form of avascular necrosis.7 Prospective studies have shown that enzyme replacement therapy reduces the incidence of osteonecrosis.23
FOLLOW-UP MANAGEMENT OF OUR PATIENT
Avascular necrosis in Gaucher disease more typically involves the hips and shoulders. In the case of our patient, the talus was the most affected bone. Other contributing factors may have been the use of steroids as a premedication (often unnecessary) for her enzyme replacement therapy, as well as the coexistent scleroderma.24
The decision to switch from imiglucerase, to which she developed antibodies, to taliglucerase was made in the hope that the antibodies would not cross-react. After she started taliglucerase, her complete blood count values improved steadily. She did not require transfusions for more than 1 year. Her platelet count rose to 90 × 109/L, and her hemoglobin to 12 g/dL.
A multidisciplinary approach with regular monitoring and appropriate initiation of therapy is necessary to prevent disastrous complications in patients with Gaucher disease.
- Lee DM, Weinblatt ME. Rheumatoid arthritis. Lancet 2001; 358:903–911.
- Fleming A, Crown JM, Corbett M. Early rheumatoid disease. I. Onset. Ann Rheum Dis 1976; 35:357–360.
- Grabowski GA, Andria G, Baldellou A, et al. Pediatric non-neuronopathic Gaucher disease: presentation, diagnosis, and assessment. Consensus statements. Eur J Pediatr 2004; 163:58–66.
- Rodrigue SW, Rosenthal DI, Barton NW, Zurakowski D, Mankin HJ. Risk factors for osteonecrosis in patients with type 1 Gaucher’s disease. Clin Orthop Relat Res 1999; May (362):201–207.
- Lee RE. The pathology of Gaucher disease. Prog Clin Biol Res 1982; 95:177–217.
- Cox TM, Aerts JM, Belmatoug N, et al. Management of non-neuronopathic Gaucher disease with special reference to pregnancy, splenectomy, bisphosphonate therapy, use of biomarkers and bone disease monitoring. J Inherit Metab Dis 2008; 31:319–336.
- Khan A, Hangartner T, Weinreb NJ, Taylor JS, Mistry PK. Risk factors for fractures and avascular osteonecrosis in type 1 Gaucher disease: a study from the International Collaborative Gaucher Group (ICGG) Gaucher Registry. J Bone Miner Res 2012; 27:1839–1848.
- Deegan PB, Pavlova E, Tindall J, et al. Osseous manifestations of adult Gaucher disease in the era of enzyme replacement therapy. Medicine (Baltimore) 2011; 90:52–60.
- Weinstein RS. Glucocorticoid-induced osteonecrosis. Endocrine 2012; 41:183–190.
- Compston J. Management of glucocorticoid-induced osteoporosis. Nat Rev Rheumatol 2010; 6:82–88.
- Van Staa TP, Laan RF, Barton IP, Cohen S, Reid DM, Cooper C. Bone density threshold and other predictors of vertebral fracture in patients receiving oral glucocorticoid therapy. Arthritis Rheum 2003; 48:3224–3229.
- Kanis JA, Johnell O, Oden A, et al. The risk and burden of vertebral fractures in Sweden. Osteoporos Int 2004; 15:20–26.
- Seguro LP, Rosario C, Shoenfeld Y. Long-term complications of past glucocorticoid use. Autoimmun Rev 2013; 12:629–632.
- Weinstein RS. Glucocorticoid-induced osteoporosis and osteonecrosis. Endocrinol Metab Clin North Am 2012; 41:595–611.
- Cooper C, Steinbuch M, Stevenson R, Miday R, Watts NB. The epidemiology of osteonecrosis: findings from the GPRD and THIN databases in the UK. Osteoporos Int 2010; 21:569–577.
- Mulfinger GL, Trueta J. The blood supply of the talus. J Bone Joint Surg Br 1970; 52:160–167.
- Pearce DH, Mongiardi CN, Fornasier VL, Daniels TR. Avascular necrosis of the talus: a pictoral essay. Radiographics 2005; 25:399–410.
- In brief: Taliglucerase (Elelyso) for Gaucher disease. Med Lett Drugs Ther 2012 Jul 9; 54(1394):56.
- Hollak CE. An evidence-based review of the potential benefits of taliglucerase alfa in the treatment of patients with Gaucher disease. Core Evid 2012; 7:15–20.
- Poole RM. Eliglustat: first global approval. Drugs 2014; 74:1829–1836.
- Bennett LL, Mohan D. Gaucher disease and its treatment options. Ann Pharmacother 2013; 47:1182–1193.
- Germain DP. Gaucher’s disease: a paradigm for interventional genetics. Clin Genet 2004; 65:77–86.
- Sims KB, Pastores GM, Weinreb NJ, et al. Improvement of bone disease by imiglucerase (Cerezyme) therapy in patients with skeletal manifestations of type 1 Gaucher disease: results of a 48-month longitudinal cohort study. Clin Genet 2008; 73:430–440.
- Rennie C, Britton J, Prouse P. Bilateral avascular necrosis of the lunate in a patient with severe Raynaud’s phenomenon and scleroderma. J Clin Rheumatol 1999; 5:165–168.
- Lee DM, Weinblatt ME. Rheumatoid arthritis. Lancet 2001; 358:903–911.
- Fleming A, Crown JM, Corbett M. Early rheumatoid disease. I. Onset. Ann Rheum Dis 1976; 35:357–360.
- Grabowski GA, Andria G, Baldellou A, et al. Pediatric non-neuronopathic Gaucher disease: presentation, diagnosis, and assessment. Consensus statements. Eur J Pediatr 2004; 163:58–66.
- Rodrigue SW, Rosenthal DI, Barton NW, Zurakowski D, Mankin HJ. Risk factors for osteonecrosis in patients with type 1 Gaucher’s disease. Clin Orthop Relat Res 1999; May (362):201–207.
- Lee RE. The pathology of Gaucher disease. Prog Clin Biol Res 1982; 95:177–217.
- Cox TM, Aerts JM, Belmatoug N, et al. Management of non-neuronopathic Gaucher disease with special reference to pregnancy, splenectomy, bisphosphonate therapy, use of biomarkers and bone disease monitoring. J Inherit Metab Dis 2008; 31:319–336.
- Khan A, Hangartner T, Weinreb NJ, Taylor JS, Mistry PK. Risk factors for fractures and avascular osteonecrosis in type 1 Gaucher disease: a study from the International Collaborative Gaucher Group (ICGG) Gaucher Registry. J Bone Miner Res 2012; 27:1839–1848.
- Deegan PB, Pavlova E, Tindall J, et al. Osseous manifestations of adult Gaucher disease in the era of enzyme replacement therapy. Medicine (Baltimore) 2011; 90:52–60.
- Weinstein RS. Glucocorticoid-induced osteonecrosis. Endocrine 2012; 41:183–190.
- Compston J. Management of glucocorticoid-induced osteoporosis. Nat Rev Rheumatol 2010; 6:82–88.
- Van Staa TP, Laan RF, Barton IP, Cohen S, Reid DM, Cooper C. Bone density threshold and other predictors of vertebral fracture in patients receiving oral glucocorticoid therapy. Arthritis Rheum 2003; 48:3224–3229.
- Kanis JA, Johnell O, Oden A, et al. The risk and burden of vertebral fractures in Sweden. Osteoporos Int 2004; 15:20–26.
- Seguro LP, Rosario C, Shoenfeld Y. Long-term complications of past glucocorticoid use. Autoimmun Rev 2013; 12:629–632.
- Weinstein RS. Glucocorticoid-induced osteoporosis and osteonecrosis. Endocrinol Metab Clin North Am 2012; 41:595–611.
- Cooper C, Steinbuch M, Stevenson R, Miday R, Watts NB. The epidemiology of osteonecrosis: findings from the GPRD and THIN databases in the UK. Osteoporos Int 2010; 21:569–577.
- Mulfinger GL, Trueta J. The blood supply of the talus. J Bone Joint Surg Br 1970; 52:160–167.
- Pearce DH, Mongiardi CN, Fornasier VL, Daniels TR. Avascular necrosis of the talus: a pictoral essay. Radiographics 2005; 25:399–410.
- In brief: Taliglucerase (Elelyso) for Gaucher disease. Med Lett Drugs Ther 2012 Jul 9; 54(1394):56.
- Hollak CE. An evidence-based review of the potential benefits of taliglucerase alfa in the treatment of patients with Gaucher disease. Core Evid 2012; 7:15–20.
- Poole RM. Eliglustat: first global approval. Drugs 2014; 74:1829–1836.
- Bennett LL, Mohan D. Gaucher disease and its treatment options. Ann Pharmacother 2013; 47:1182–1193.
- Germain DP. Gaucher’s disease: a paradigm for interventional genetics. Clin Genet 2004; 65:77–86.
- Sims KB, Pastores GM, Weinreb NJ, et al. Improvement of bone disease by imiglucerase (Cerezyme) therapy in patients with skeletal manifestations of type 1 Gaucher disease: results of a 48-month longitudinal cohort study. Clin Genet 2008; 73:430–440.
- Rennie C, Britton J, Prouse P. Bilateral avascular necrosis of the lunate in a patient with severe Raynaud’s phenomenon and scleroderma. J Clin Rheumatol 1999; 5:165–168.
AAP: Give peanut products to high-risk infants to cut allergy risk
Infants at high risk for peanut allergy should start a peanut-based diet by age 4-11 months, experts from the American Academy of Pediatrics and nine other medical groups advised in the September issue of Pediatrics.*
The consensus communication upends traditional views about preventing childhood peanut allergy and highlights the landmark LEAP study in which high-risk infants fed peanut-based foods had about an 80% lower risk of developing peanut allergy, compared with those fed a peanut-free diet.

“Early intervention will prevent peanut allergy, and the pediatrician’s involvement is absolutely essential to the success of this approach,” said Dr. Hugh Sampson, who contributed to the guidance and is at the Icahn School of Medicine at Mount Sinai, New York. “Without very early evaluation and implementation, we won’t change anything.”
LEAP investigators defined “high risk” for peanut allergy as severe eczema with or without egg allergy, “but many other infants are likely at risk, and thus would benefit from early peanut introduction,” added Dr. David Fleischer, who also contributed to the guidance and is at the University of Colorado at Denver, Aurora. “Many feel that given the potential benefit, all infants, regardless of risk level, should have peanut introduced early into the diet,” he said.
Peanut allergy affects more than 2% of American children and is about twice as prevalent in Western countries as it was a decade ago. It’s not clear why rates have increased, but pediatricians can help stem the rising tide, Dr. Sampson said. “When a pediatrician suspects a ‘high-risk’ baby, he or she needs to explain to parents the risks involved in their baby developing peanut allergy, and the benefits of early evaluation and introduction. Once peanut allergy is established, the vast majority of young children will retain the allergy for life.”
Because “high-risk” children might already be allergic to peanuts, they could benefit from evaluation by an allergist, according to the consensus communication (Pediatrics 2015;136[3]:601-4). Expert consultation might also benefit those who feel reluctant to introduce peanuts for other reasons, Dr. Fleischer said.
Dr. Sampson recommends peanut-based skin prick testing for high-risk infants aged 4-8 months. Patients with a negative result should receive 2 grams of peanut protein three times a week for the next 3 years. Those who are mildly sensitive (wheal diameter less than 4 mm) should undergo a peanut challenge observed by an experienced physician. Infants who do not react can start the peanut-based diet.
The LEAP study randomized 640 high-risk infants to either avoid peanuts or consume at least 6 grams per week of the allergen in foods such as smooth peanut butter mixed with mashed fruit, peanut soup, and ground peanuts in other foods. Five-year-olds in the peanut group had significantly lower rates of peanut allergy, regardless of whether their skin prick test had been positive at baseline (N. Engl. J. Med. 2015;372[9]:803-13). While the consensus communication provides interim guidance, a panel sponsored by the National Institute of Allergy and Infectious Diseases is reviewing food allergy data in preparation for updating its guidelines, Dr. Sampson noted. “Several major questions remain,” he said. “Do we need to give such large amounts of peanut to induce tolerance? Is it necessary to give this amount of peanut for such an extended period? What happens if parents don’t give peanut to their infants on a regular basis, as done in the LEAP trial? Could this put them at higher risk? And will this approach apply to other foods?”
Another key knowledge gap is whether the results from one single-center study can be applied elsewhere, said Dr. Fleischer. “We do not know the effects of early peanut introduction in other risk populations.”
Authors of the consensus communication reported no funding sources or conflicts of interest.
*Correction, 8/31/2015: An earlier version of this article misstated the journal in which the study was published.
Infants at high risk for peanut allergy should start a peanut-based diet by age 4-11 months, experts from the American Academy of Pediatrics and nine other medical groups advised in the September issue of Pediatrics.*
The consensus communication upends traditional views about preventing childhood peanut allergy and highlights the landmark LEAP study in which high-risk infants fed peanut-based foods had about an 80% lower risk of developing peanut allergy, compared with those fed a peanut-free diet.
“Early intervention will prevent peanut allergy, and the pediatrician’s involvement is absolutely essential to the success of this approach,” said Dr. Hugh Sampson, who contributed to the guidance and is at the Icahn School of Medicine at Mount Sinai, New York. “Without very early evaluation and implementation, we won’t change anything.”
LEAP investigators defined “high risk” for peanut allergy as severe eczema with or without egg allergy, “but many other infants are likely at risk, and thus would benefit from early peanut introduction,” added Dr. David Fleischer, who also contributed to the guidance and is at the University of Colorado at Denver, Aurora. “Many feel that given the potential benefit, all infants, regardless of risk level, should have peanut introduced early into the diet,” he said.
Peanut allergy affects more than 2% of American children and is about twice as prevalent in Western countries as it was a decade ago. It’s not clear why rates have increased, but pediatricians can help stem the rising tide, Dr. Sampson said. “When a pediatrician suspects a ‘high-risk’ baby, he or she needs to explain to parents the risks involved in their baby developing peanut allergy, and the benefits of early evaluation and introduction. Once peanut allergy is established, the vast majority of young children will retain the allergy for life.”
Because “high-risk” children might already be allergic to peanuts, they could benefit from evaluation by an allergist, according to the consensus communication (Pediatrics 2015;136[3]:601-4). Expert consultation might also benefit those who feel reluctant to introduce peanuts for other reasons, Dr. Fleischer said.
Dr. Sampson recommends peanut-based skin prick testing for high-risk infants aged 4-8 months. Patients with a negative result should receive 2 grams of peanut protein three times a week for the next 3 years. Those who are mildly sensitive (wheal diameter less than 4 mm) should undergo a peanut challenge observed by an experienced physician. Infants who do not react can start the peanut-based diet.
The LEAP study randomized 640 high-risk infants to either avoid peanuts or consume at least 6 grams per week of the allergen in foods such as smooth peanut butter mixed with mashed fruit, peanut soup, and ground peanuts in other foods. Five-year-olds in the peanut group had significantly lower rates of peanut allergy, regardless of whether their skin prick test had been positive at baseline (N. Engl. J. Med. 2015;372[9]:803-13). While the consensus communication provides interim guidance, a panel sponsored by the National Institute of Allergy and Infectious Diseases is reviewing food allergy data in preparation for updating its guidelines, Dr. Sampson noted. “Several major questions remain,” he said. “Do we need to give such large amounts of peanut to induce tolerance? Is it necessary to give this amount of peanut for such an extended period? What happens if parents don’t give peanut to their infants on a regular basis, as done in the LEAP trial? Could this put them at higher risk? And will this approach apply to other foods?”
Another key knowledge gap is whether the results from one single-center study can be applied elsewhere, said Dr. Fleischer. “We do not know the effects of early peanut introduction in other risk populations.”
Authors of the consensus communication reported no funding sources or conflicts of interest.
*Correction, 8/31/2015: An earlier version of this article misstated the journal in which the study was published.
Infants at high risk for peanut allergy should start a peanut-based diet by age 4-11 months, experts from the American Academy of Pediatrics and nine other medical groups advised in the September issue of Pediatrics.*
The consensus communication upends traditional views about preventing childhood peanut allergy and highlights the landmark LEAP study in which high-risk infants fed peanut-based foods had about an 80% lower risk of developing peanut allergy, compared with those fed a peanut-free diet.
“Early intervention will prevent peanut allergy, and the pediatrician’s involvement is absolutely essential to the success of this approach,” said Dr. Hugh Sampson, who contributed to the guidance and is at the Icahn School of Medicine at Mount Sinai, New York. “Without very early evaluation and implementation, we won’t change anything.”
LEAP investigators defined “high risk” for peanut allergy as severe eczema with or without egg allergy, “but many other infants are likely at risk, and thus would benefit from early peanut introduction,” added Dr. David Fleischer, who also contributed to the guidance and is at the University of Colorado at Denver, Aurora. “Many feel that given the potential benefit, all infants, regardless of risk level, should have peanut introduced early into the diet,” he said.
Peanut allergy affects more than 2% of American children and is about twice as prevalent in Western countries as it was a decade ago. It’s not clear why rates have increased, but pediatricians can help stem the rising tide, Dr. Sampson said. “When a pediatrician suspects a ‘high-risk’ baby, he or she needs to explain to parents the risks involved in their baby developing peanut allergy, and the benefits of early evaluation and introduction. Once peanut allergy is established, the vast majority of young children will retain the allergy for life.”
Because “high-risk” children might already be allergic to peanuts, they could benefit from evaluation by an allergist, according to the consensus communication (Pediatrics 2015;136[3]:601-4). Expert consultation might also benefit those who feel reluctant to introduce peanuts for other reasons, Dr. Fleischer said.
Dr. Sampson recommends peanut-based skin prick testing for high-risk infants aged 4-8 months. Patients with a negative result should receive 2 grams of peanut protein three times a week for the next 3 years. Those who are mildly sensitive (wheal diameter less than 4 mm) should undergo a peanut challenge observed by an experienced physician. Infants who do not react can start the peanut-based diet.
The LEAP study randomized 640 high-risk infants to either avoid peanuts or consume at least 6 grams per week of the allergen in foods such as smooth peanut butter mixed with mashed fruit, peanut soup, and ground peanuts in other foods. Five-year-olds in the peanut group had significantly lower rates of peanut allergy, regardless of whether their skin prick test had been positive at baseline (N. Engl. J. Med. 2015;372[9]:803-13). While the consensus communication provides interim guidance, a panel sponsored by the National Institute of Allergy and Infectious Diseases is reviewing food allergy data in preparation for updating its guidelines, Dr. Sampson noted. “Several major questions remain,” he said. “Do we need to give such large amounts of peanut to induce tolerance? Is it necessary to give this amount of peanut for such an extended period? What happens if parents don’t give peanut to their infants on a regular basis, as done in the LEAP trial? Could this put them at higher risk? And will this approach apply to other foods?”
Another key knowledge gap is whether the results from one single-center study can be applied elsewhere, said Dr. Fleischer. “We do not know the effects of early peanut introduction in other risk populations.”
Authors of the consensus communication reported no funding sources or conflicts of interest.
*Correction, 8/31/2015: An earlier version of this article misstated the journal in which the study was published.
FROM PEDIATRICS

ACIP Releases 2015-2016 Flu Vaccine Recommendations
Influenza vaccination is recommended for all patients aged 6 months and older, as long as they don’t have a contraindication, according to a report Aug. 7 in Morbidity and Mortality Weekly Report.
Trivalent influenza vaccines for the 2015-2016 season will contain hemagglutinin (HA) derived from an H1N1-like virus, an H3N2-like virus, and a B/Phuket/3073/2013-like (Yamagata lineage) virus. Quadrivalent vaccines will contain those components, as well as a B/Brisbane/60/2008-like (Victoria lineage) virus, the same virus recommended for quadrivalent formulations in the 2013-14 and 2014-15 seasons, ACIP said in a statement.
New FDA-approved vaccines include Afluria, the Fluzone Intradermal Quadrivalent vaccine (both approved in 2014 for adults aged 18-64 years), and an expanded age indication for Flublok, which is now indicated for adults aged 18 years and older.
The live attenuated influenza vaccine (LAIV) should not be used in certain populations, including those aged less than 2 years or greater than 49 years; children aged 2-17 years taking aspirin; pateints with severe allergic reactions to the vaccine; pregnant women; and those with egg allergies, among others. Either the LAIV or the inactivated influenza vaccine (IIV) is appropriate for administration in healthy children aged 2-8 years, ACIP said.
For a detailed explanation of the recommendations, see MMWR.
Influenza vaccination is recommended for all patients aged 6 months and older, as long as they don’t have a contraindication, according to a report Aug. 7 in Morbidity and Mortality Weekly Report.
Trivalent influenza vaccines for the 2015-2016 season will contain hemagglutinin (HA) derived from an H1N1-like virus, an H3N2-like virus, and a B/Phuket/3073/2013-like (Yamagata lineage) virus. Quadrivalent vaccines will contain those components, as well as a B/Brisbane/60/2008-like (Victoria lineage) virus, the same virus recommended for quadrivalent formulations in the 2013-14 and 2014-15 seasons, ACIP said in a statement.
New FDA-approved vaccines include Afluria, the Fluzone Intradermal Quadrivalent vaccine (both approved in 2014 for adults aged 18-64 years), and an expanded age indication for Flublok, which is now indicated for adults aged 18 years and older.
The live attenuated influenza vaccine (LAIV) should not be used in certain populations, including those aged less than 2 years or greater than 49 years; children aged 2-17 years taking aspirin; pateints with severe allergic reactions to the vaccine; pregnant women; and those with egg allergies, among others. Either the LAIV or the inactivated influenza vaccine (IIV) is appropriate for administration in healthy children aged 2-8 years, ACIP said.
For a detailed explanation of the recommendations, see MMWR.
Influenza vaccination is recommended for all patients aged 6 months and older, as long as they don’t have a contraindication, according to a report Aug. 7 in Morbidity and Mortality Weekly Report.
Trivalent influenza vaccines for the 2015-2016 season will contain hemagglutinin (HA) derived from an H1N1-like virus, an H3N2-like virus, and a B/Phuket/3073/2013-like (Yamagata lineage) virus. Quadrivalent vaccines will contain those components, as well as a B/Brisbane/60/2008-like (Victoria lineage) virus, the same virus recommended for quadrivalent formulations in the 2013-14 and 2014-15 seasons, ACIP said in a statement.
New FDA-approved vaccines include Afluria, the Fluzone Intradermal Quadrivalent vaccine (both approved in 2014 for adults aged 18-64 years), and an expanded age indication for Flublok, which is now indicated for adults aged 18 years and older.
The live attenuated influenza vaccine (LAIV) should not be used in certain populations, including those aged less than 2 years or greater than 49 years; children aged 2-17 years taking aspirin; pateints with severe allergic reactions to the vaccine; pregnant women; and those with egg allergies, among others. Either the LAIV or the inactivated influenza vaccine (IIV) is appropriate for administration in healthy children aged 2-8 years, ACIP said.
For a detailed explanation of the recommendations, see MMWR.
New HPV vaccine safe with meningitis, Tdap vaccines
Children and adolescents receiving the 9-valent human papillomavirus vaccine Gardasil 9 along with the Menactra meningococcal vaccine and a Tdap booster at the same visit do not experience decreased effectiveness or safety from any of the vaccines, a study showed.
The titers from all three vaccines were noninferior when administered together, compared with administration of the Tdap (tetanus, diphtheria, and acellular pertussis) and the meningococcal vaccine a month after the first HPV-9 dose. Adverse events also were similar across both groups, except injection-site reactions were more common in those receiving all three vaccines together initially.
“Providing vaccinations to adolescents is challenging because they make infrequent health care visits,” wrote Dr. Andrea Schilling of Alemana-Universidad del Desarrollo in Santiago, Chile, and her associates. “Concomitant administration would minimize the number of visits required to deliver each vaccine individually and therefore facilitate adherence to recommended vaccination regimens.”
Boys and girls aged 11-15 who self-reported as sexually naive in Chile, Colombia, Mexico, Peru, and the United States received recommended vaccines between October 2009 and February 2011. Among the 1,241 participants who received the 9-valent HPV vaccine, 621 of them received the Menactra meningococcal vaccine, covering Neisseria meningitidis serotypes A, C, Y, and W-135 (MCV4), and Tdap vaccines at the same visit. The other 620 participants received the MCV4 and Tdap vaccines 1 month after the HPV vaccine dose (Pediatrics. doi: 10.1542/peds.2014-4199).
Both groups showed similar levels of geometric mean titers for all nine HPV types included in Gardasil 9, all four N. meningitidis serotypes included in MCV4, and diphtheria, tetanus, and pertussis 4 weeks after the third dose of Gardasil 9. Both groups had 100% seroconversion for all HPV types at 7 months after the first dose. In addition, at least 75% of participants in both groups showed at least a fourfold increase in titers to N. meningitidis serogroup A, and at least 89% in both groups showed a fourfold increase for serogroups C, Y, and W-135. Finally, at least 99.8% of participants across both groups had at least 0.1 IU/mL diphtheria and tetanus titers 4 weeks after the Tdap booster. Antibodies to pertussis proteins were likewise noninferior in the concomitant group, compared with the nonconcomitant group.
The most commonly reported adverse events were pain, redness, or swelling at the injection site, reported by 85.3% of those receiving all three vaccines in the same visit and by 85.1% of those receiving the Tdap and MCV4 after the 9-valent HPV vaccine. However, 14.4% of those who received all the vaccines together reported swelling at the injection site, compared with 9.4% of those who received the MCV4 and Tdap a month after the HPV-9. The authors said that the higher rate for combined administration was attributable to the concomitant administration of the Tdap with the HPV vaccine. Otherwise, 0.8% of participants in both groups reported serious adverse events, all deemed unrelated to the vaccine.
“In the United States, coverage for the first dose of HPV vaccine remains substantially lower (by 20-25 percentage points) than coverage for other vaccines recommended by the Advisory Committee on Immunization Practices for children 11-12 years of age,” the authors wrote. “It is estimated that coadministration of HPV vaccine with other vaccines, such as diphtheria, tetanus, pertussis, meningococcal conjugate, and influenza vaccines, could increase coverage for the first dose of HPV vaccine to more than 90%.”
The investigators said the main limitation of the study was that it was not blinded. “As such, safety assessment could have been biased toward an overestimation of [adverse events],” they wrote.
The research was funded by Merck & Co. Dr. Schilling reported receiving past research support from Merck and honoraria from Merck-Chile, Grunenthal-Chile, and Grunenthal-L.A. Her associates reported financial relationships with numerous pharmaceutical companies.
Children and adolescents receiving the 9-valent human papillomavirus vaccine Gardasil 9 along with the Menactra meningococcal vaccine and a Tdap booster at the same visit do not experience decreased effectiveness or safety from any of the vaccines, a study showed.
The titers from all three vaccines were noninferior when administered together, compared with administration of the Tdap (tetanus, diphtheria, and acellular pertussis) and the meningococcal vaccine a month after the first HPV-9 dose. Adverse events also were similar across both groups, except injection-site reactions were more common in those receiving all three vaccines together initially.
“Providing vaccinations to adolescents is challenging because they make infrequent health care visits,” wrote Dr. Andrea Schilling of Alemana-Universidad del Desarrollo in Santiago, Chile, and her associates. “Concomitant administration would minimize the number of visits required to deliver each vaccine individually and therefore facilitate adherence to recommended vaccination regimens.”
Boys and girls aged 11-15 who self-reported as sexually naive in Chile, Colombia, Mexico, Peru, and the United States received recommended vaccines between October 2009 and February 2011. Among the 1,241 participants who received the 9-valent HPV vaccine, 621 of them received the Menactra meningococcal vaccine, covering Neisseria meningitidis serotypes A, C, Y, and W-135 (MCV4), and Tdap vaccines at the same visit. The other 620 participants received the MCV4 and Tdap vaccines 1 month after the HPV vaccine dose (Pediatrics. doi: 10.1542/peds.2014-4199).
Both groups showed similar levels of geometric mean titers for all nine HPV types included in Gardasil 9, all four N. meningitidis serotypes included in MCV4, and diphtheria, tetanus, and pertussis 4 weeks after the third dose of Gardasil 9. Both groups had 100% seroconversion for all HPV types at 7 months after the first dose. In addition, at least 75% of participants in both groups showed at least a fourfold increase in titers to N. meningitidis serogroup A, and at least 89% in both groups showed a fourfold increase for serogroups C, Y, and W-135. Finally, at least 99.8% of participants across both groups had at least 0.1 IU/mL diphtheria and tetanus titers 4 weeks after the Tdap booster. Antibodies to pertussis proteins were likewise noninferior in the concomitant group, compared with the nonconcomitant group.
The most commonly reported adverse events were pain, redness, or swelling at the injection site, reported by 85.3% of those receiving all three vaccines in the same visit and by 85.1% of those receiving the Tdap and MCV4 after the 9-valent HPV vaccine. However, 14.4% of those who received all the vaccines together reported swelling at the injection site, compared with 9.4% of those who received the MCV4 and Tdap a month after the HPV-9. The authors said that the higher rate for combined administration was attributable to the concomitant administration of the Tdap with the HPV vaccine. Otherwise, 0.8% of participants in both groups reported serious adverse events, all deemed unrelated to the vaccine.
“In the United States, coverage for the first dose of HPV vaccine remains substantially lower (by 20-25 percentage points) than coverage for other vaccines recommended by the Advisory Committee on Immunization Practices for children 11-12 years of age,” the authors wrote. “It is estimated that coadministration of HPV vaccine with other vaccines, such as diphtheria, tetanus, pertussis, meningococcal conjugate, and influenza vaccines, could increase coverage for the first dose of HPV vaccine to more than 90%.”
The investigators said the main limitation of the study was that it was not blinded. “As such, safety assessment could have been biased toward an overestimation of [adverse events],” they wrote.
The research was funded by Merck & Co. Dr. Schilling reported receiving past research support from Merck and honoraria from Merck-Chile, Grunenthal-Chile, and Grunenthal-L.A. Her associates reported financial relationships with numerous pharmaceutical companies.
Children and adolescents receiving the 9-valent human papillomavirus vaccine Gardasil 9 along with the Menactra meningococcal vaccine and a Tdap booster at the same visit do not experience decreased effectiveness or safety from any of the vaccines, a study showed.
The titers from all three vaccines were noninferior when administered together, compared with administration of the Tdap (tetanus, diphtheria, and acellular pertussis) and the meningococcal vaccine a month after the first HPV-9 dose. Adverse events also were similar across both groups, except injection-site reactions were more common in those receiving all three vaccines together initially.
“Providing vaccinations to adolescents is challenging because they make infrequent health care visits,” wrote Dr. Andrea Schilling of Alemana-Universidad del Desarrollo in Santiago, Chile, and her associates. “Concomitant administration would minimize the number of visits required to deliver each vaccine individually and therefore facilitate adherence to recommended vaccination regimens.”
Boys and girls aged 11-15 who self-reported as sexually naive in Chile, Colombia, Mexico, Peru, and the United States received recommended vaccines between October 2009 and February 2011. Among the 1,241 participants who received the 9-valent HPV vaccine, 621 of them received the Menactra meningococcal vaccine, covering Neisseria meningitidis serotypes A, C, Y, and W-135 (MCV4), and Tdap vaccines at the same visit. The other 620 participants received the MCV4 and Tdap vaccines 1 month after the HPV vaccine dose (Pediatrics. doi: 10.1542/peds.2014-4199).
Both groups showed similar levels of geometric mean titers for all nine HPV types included in Gardasil 9, all four N. meningitidis serotypes included in MCV4, and diphtheria, tetanus, and pertussis 4 weeks after the third dose of Gardasil 9. Both groups had 100% seroconversion for all HPV types at 7 months after the first dose. In addition, at least 75% of participants in both groups showed at least a fourfold increase in titers to N. meningitidis serogroup A, and at least 89% in both groups showed a fourfold increase for serogroups C, Y, and W-135. Finally, at least 99.8% of participants across both groups had at least 0.1 IU/mL diphtheria and tetanus titers 4 weeks after the Tdap booster. Antibodies to pertussis proteins were likewise noninferior in the concomitant group, compared with the nonconcomitant group.
The most commonly reported adverse events were pain, redness, or swelling at the injection site, reported by 85.3% of those receiving all three vaccines in the same visit and by 85.1% of those receiving the Tdap and MCV4 after the 9-valent HPV vaccine. However, 14.4% of those who received all the vaccines together reported swelling at the injection site, compared with 9.4% of those who received the MCV4 and Tdap a month after the HPV-9. The authors said that the higher rate for combined administration was attributable to the concomitant administration of the Tdap with the HPV vaccine. Otherwise, 0.8% of participants in both groups reported serious adverse events, all deemed unrelated to the vaccine.
“In the United States, coverage for the first dose of HPV vaccine remains substantially lower (by 20-25 percentage points) than coverage for other vaccines recommended by the Advisory Committee on Immunization Practices for children 11-12 years of age,” the authors wrote. “It is estimated that coadministration of HPV vaccine with other vaccines, such as diphtheria, tetanus, pertussis, meningococcal conjugate, and influenza vaccines, could increase coverage for the first dose of HPV vaccine to more than 90%.”
The investigators said the main limitation of the study was that it was not blinded. “As such, safety assessment could have been biased toward an overestimation of [adverse events],” they wrote.
The research was funded by Merck & Co. Dr. Schilling reported receiving past research support from Merck and honoraria from Merck-Chile, Grunenthal-Chile, and Grunenthal-L.A. Her associates reported financial relationships with numerous pharmaceutical companies.
FROM PEDIATRICS
Key clinical point: The new 9-valent HPV vaccine can be given with other standard vaccines in one visit.
Major finding: No difference in safety or effectiveness was seen with coadministration of HPV-9, Tdap booster, and Menactra vaccines.
Data source: An open-label, randomized, multicenter comparative study involving 1,241 boys and girls, aged 11-15, who received the 9-valent HPV vaccine along with the Tdap booster and Menactra vaccines.
Disclosures: The research was funded by Merck & Co. Dr. Schilling reported receiving past research support from Merck and honoraria from Merck-Chile, Grunenthal-Chile, and Grunenthal-L.A. Her associates reported financial relationships with numerous pharmaceutical companies.
Allergy & Immunology
DOES PEANUT EXPOSURE INCREASE ALLERGY RISK?
Du Toit G, Roberts G, Sayre PH, et al; LEAP Study Team. Randomized trial of peanut consumption in infants at risk for peanut allergy. N Engl J Med. 2015;372(9):803-813. doi: 10.1056/NEJMoa1414850.
Among children at high risk for peanut allergy, early introduction of peanuts significantly reduced the risk for allergy, according to a randomized controlled trial of 640 infants with severe eczema, egg allergy, or both.
Participants ages 4 to 11 months who were high risk—based on severe atopy or allergy to eggs—were tested for sensitivity to peanut extract at baseline and then assigned to either avoid or consume peanuts.
At 60 months of age, skin allergy testing was repeated; the resulting prevalence of peanut allergy was as follows

A greater percentage of the consumption group showed an increase in levels of peanut-specific IgG4 antibody; the avoidance group, elevated titers of peanut specific IgE antibody.
COMMENTARY
The prevalence of peanut allergy in the United States has increased from 0.4% in 1997 to more than 2% in 2010.1 In 2000, the American Academy of Pediatrics recommended peanut avoidance until age 3 in children at high risk for atopic disease; then in 2008, based on emerging evidence that early introduction of allergenic foods, including peanuts, may decrease the development of allergy, the recommendations were retracted.2,3 The present study, using a randomized trial design, confirms the paradigm-changing hypothesis that early introduction of allergenic foods decreases the subsequent development of food allergies. The authors of the accompanying editorial state that these data are incontrovertible and that children at high risk for peanut allergy should, under supervision of an allergist, be tested and if skin-prick–negative for peanut allergy, be started on a peanut protein–containing diet.1—NS
1. Gruchalla RS, Sampson HA. Preventing peanut allergy through early consumption ready for prime time? N Engl J Med. 2015;372:875-876.
2. Du Toit G, Katz Y, Sasieni P, et al. Early consumption of peanuts in infancy is associated with a low prevalence of peanut allergy. J Allergy Clin Immunol. 2008;122:984-991.
3. Katz Y, Rajuan N, Goldberg MR, et al. Early exposure to cow’s milk protein is protective against IgE-mediated cow’s milk protein allergy. J Allergy Clin Immunol. 2010;126:77-82.
Continue for Anaphylaxis guideline from AAAAI/ACAAI >>
ANAPHYLAXIS GUIDELINE FROM AAAAI/ACAAI
Campbell RL, Li JT, Nicklas RA, Sadosty AT. Emergency department diagnosis and treatment of anaphylaxis: a practice parameter. Ann Allergy Asthma Immunol. 2014;113(6):599-608. doi: 10.1016/j.anai.2014.10.007.
The American Academy of Allergy, Asthma & Immunology and the American College of Allergy, Asthma, and Immunology guideline for emergency department diagnosis and treatment of anaphylaxis includes the following recommendations
• Carefully and immediately triage and monitor patients who have signs and symptoms of anaphylaxis in preparation for epinephrine administration.
• First-line treatment for patients experiencing anaphylaxis is epinephrine. It should be administered intramuscularly in the anterolateral thigh immediately after the diagnosis is made. Epinephrine can be administered every 5 to 15 minutes as needed to control symptoms.
• Do not substitute for epinephrine in the treatment of anaphylaxis. Antihistamines and corticosteroids can be administered in conjunction with epinephrine, not in place of it.
• Determine whether the patient has risk factors for severe and potentially fatal anaphylaxis, such as delayed administration of epinephrine, asthma, a history of biphasic reactions, or cardiovascular disease.
• For anaphylaxis patients with bronchospasms, administer a β-agonist.
• Patients should be observed for 4 to 8 hours (longer for those with a history of risk factors for severe anaphylaxis).
• Refer patients to an allergist–immunologist upon discharge.
COMMENTARY
Identification of anaphylaxis requires judgment. Abbreviated criteria for anaphylaxis include essentially two organ systems of involvement: skin manifestations of pruritus, flushing, hives, or angioedema; respiratory manifestations of wheezing or stridor; decreased blood pressure; and GI symptoms of vomiting, cramping abdominal pain, or diarrhea. Quick identification and treatment of anaphylaxis is important, as the median time to respiratory or cardiac arrest in food-induced anaphylaxis is only 30 minutes. All offices should stock epinephrine in an obvious place; you may consider use of a prefilled pen, so it is easy to find and to give the correct dose. —NS
Continue for Updated practice parameter: Diagnosing and treating food allergies >>
UPDATED PRACTICE PARAMETER: DIAGNOSING AND TREATING FOOD ALLERGIES
Sampson HA, Aceves S, Bock SA, et al. Food allergy: a practice parameter update—2014. J Allergy Clin Immunol. 2014. pii: S0091-6749(14)00672-1. doi: 10.1016/j.jaci.2014.05.013. [Epub ahead of print]
Over the past decade, health care providers have been confronted with a growing number of patients with suspected food allergies, but data supporting an increase in confirmed allergy cases is limited.
The 2014 practice parameter update on food allergies from the American Academy of Allergy, Asthma & Immunology advises clinicians to keep in mind that self-reported food allergy is more common than proven food allergy; that allergy is more common in children and in patients with other atopic conditions; and that the majority of allergic reactions are from peanuts, tree nuts, fish, shellfish, milk, eggs, wheat, soy, and seeds.
The update of the 2006 guidelines includes 64 new summary statements. Highlights include
• Clinicians should advise patients about the risk for cross-reactions from foods similar to their allergens, such as other tree nuts, vertebrate fish, crustaceans, or milk from cows, goats, or other mammals.
• Patients with a seafood allergy should be advised that they are not at increased risk for a reaction to radiocontrast media.
• Patients with food allergies do not need to be concerned about eating genetically modified foods (GMO), due to current FDA screening requirements to rule out allergenicity.
• Patients with chronic idiopathic urticaria or hyperactivity/attention-deficit disorder should not routinely be advised to avoid food additives.
• Patients with asthma should not routinely be advised to avoid sulfates, unless they have had a previous reaction to them.
The guidelines also include a new section on diagnosing and treating non-IgE-mediated food allergies, such as food-protein–induced enterocolitis syndrome, allergic proctocolitis, enteropathy, eosinophilic esophagitis, and gastroenteritis.
COMMENTARY
Food allergies are a commonly encountered and difficult area of practice for those of us in primary care. They can be life threatening; yet many patients who are concerned about the possibility of food allergy do not actually have a food allergy, so making an accurate diagnosis is important. Allergic evaluation starts with a careful history, and then laboratory testing can begin with specific IgE testing to foods suspected to have caused the clinical reaction of concern. The IgE results need to be carefully interpreted in light of the clinical context in which they were ordered. Oral food challenge can be helpful in addition to IgE testing. Consultation with an allergy-immunology specialist is often helpful as well. —NS
DOES PEANUT EXPOSURE INCREASE ALLERGY RISK?
Du Toit G, Roberts G, Sayre PH, et al; LEAP Study Team. Randomized trial of peanut consumption in infants at risk for peanut allergy. N Engl J Med. 2015;372(9):803-813. doi: 10.1056/NEJMoa1414850.
Among children at high risk for peanut allergy, early introduction of peanuts significantly reduced the risk for allergy, according to a randomized controlled trial of 640 infants with severe eczema, egg allergy, or both.
Participants ages 4 to 11 months who were high risk—based on severe atopy or allergy to eggs—were tested for sensitivity to peanut extract at baseline and then assigned to either avoid or consume peanuts.
At 60 months of age, skin allergy testing was repeated; the resulting prevalence of peanut allergy was as follows
A greater percentage of the consumption group showed an increase in levels of peanut-specific IgG4 antibody; the avoidance group, elevated titers of peanut specific IgE antibody.
COMMENTARY
The prevalence of peanut allergy in the United States has increased from 0.4% in 1997 to more than 2% in 2010.1 In 2000, the American Academy of Pediatrics recommended peanut avoidance until age 3 in children at high risk for atopic disease; then in 2008, based on emerging evidence that early introduction of allergenic foods, including peanuts, may decrease the development of allergy, the recommendations were retracted.2,3 The present study, using a randomized trial design, confirms the paradigm-changing hypothesis that early introduction of allergenic foods decreases the subsequent development of food allergies. The authors of the accompanying editorial state that these data are incontrovertible and that children at high risk for peanut allergy should, under supervision of an allergist, be tested and if skin-prick–negative for peanut allergy, be started on a peanut protein–containing diet.1—NS
1. Gruchalla RS, Sampson HA. Preventing peanut allergy through early consumption ready for prime time? N Engl J Med. 2015;372:875-876.
2. Du Toit G, Katz Y, Sasieni P, et al. Early consumption of peanuts in infancy is associated with a low prevalence of peanut allergy. J Allergy Clin Immunol. 2008;122:984-991.
3. Katz Y, Rajuan N, Goldberg MR, et al. Early exposure to cow’s milk protein is protective against IgE-mediated cow’s milk protein allergy. J Allergy Clin Immunol. 2010;126:77-82.
Continue for Anaphylaxis guideline from AAAAI/ACAAI >>
ANAPHYLAXIS GUIDELINE FROM AAAAI/ACAAI
Campbell RL, Li JT, Nicklas RA, Sadosty AT. Emergency department diagnosis and treatment of anaphylaxis: a practice parameter. Ann Allergy Asthma Immunol. 2014;113(6):599-608. doi: 10.1016/j.anai.2014.10.007.
The American Academy of Allergy, Asthma & Immunology and the American College of Allergy, Asthma, and Immunology guideline for emergency department diagnosis and treatment of anaphylaxis includes the following recommendations
• Carefully and immediately triage and monitor patients who have signs and symptoms of anaphylaxis in preparation for epinephrine administration.
• First-line treatment for patients experiencing anaphylaxis is epinephrine. It should be administered intramuscularly in the anterolateral thigh immediately after the diagnosis is made. Epinephrine can be administered every 5 to 15 minutes as needed to control symptoms.
• Do not substitute for epinephrine in the treatment of anaphylaxis. Antihistamines and corticosteroids can be administered in conjunction with epinephrine, not in place of it.
• Determine whether the patient has risk factors for severe and potentially fatal anaphylaxis, such as delayed administration of epinephrine, asthma, a history of biphasic reactions, or cardiovascular disease.
• For anaphylaxis patients with bronchospasms, administer a β-agonist.
• Patients should be observed for 4 to 8 hours (longer for those with a history of risk factors for severe anaphylaxis).
• Refer patients to an allergist–immunologist upon discharge.
COMMENTARY
Identification of anaphylaxis requires judgment. Abbreviated criteria for anaphylaxis include essentially two organ systems of involvement: skin manifestations of pruritus, flushing, hives, or angioedema; respiratory manifestations of wheezing or stridor; decreased blood pressure; and GI symptoms of vomiting, cramping abdominal pain, or diarrhea. Quick identification and treatment of anaphylaxis is important, as the median time to respiratory or cardiac arrest in food-induced anaphylaxis is only 30 minutes. All offices should stock epinephrine in an obvious place; you may consider use of a prefilled pen, so it is easy to find and to give the correct dose. —NS
Continue for Updated practice parameter: Diagnosing and treating food allergies >>
UPDATED PRACTICE PARAMETER: DIAGNOSING AND TREATING FOOD ALLERGIES
Sampson HA, Aceves S, Bock SA, et al. Food allergy: a practice parameter update—2014. J Allergy Clin Immunol. 2014. pii: S0091-6749(14)00672-1. doi: 10.1016/j.jaci.2014.05.013. [Epub ahead of print]
Over the past decade, health care providers have been confronted with a growing number of patients with suspected food allergies, but data supporting an increase in confirmed allergy cases is limited.
The 2014 practice parameter update on food allergies from the American Academy of Allergy, Asthma & Immunology advises clinicians to keep in mind that self-reported food allergy is more common than proven food allergy; that allergy is more common in children and in patients with other atopic conditions; and that the majority of allergic reactions are from peanuts, tree nuts, fish, shellfish, milk, eggs, wheat, soy, and seeds.
The update of the 2006 guidelines includes 64 new summary statements. Highlights include
• Clinicians should advise patients about the risk for cross-reactions from foods similar to their allergens, such as other tree nuts, vertebrate fish, crustaceans, or milk from cows, goats, or other mammals.
• Patients with a seafood allergy should be advised that they are not at increased risk for a reaction to radiocontrast media.
• Patients with food allergies do not need to be concerned about eating genetically modified foods (GMO), due to current FDA screening requirements to rule out allergenicity.
• Patients with chronic idiopathic urticaria or hyperactivity/attention-deficit disorder should not routinely be advised to avoid food additives.
• Patients with asthma should not routinely be advised to avoid sulfates, unless they have had a previous reaction to them.
The guidelines also include a new section on diagnosing and treating non-IgE-mediated food allergies, such as food-protein–induced enterocolitis syndrome, allergic proctocolitis, enteropathy, eosinophilic esophagitis, and gastroenteritis.
COMMENTARY
Food allergies are a commonly encountered and difficult area of practice for those of us in primary care. They can be life threatening; yet many patients who are concerned about the possibility of food allergy do not actually have a food allergy, so making an accurate diagnosis is important. Allergic evaluation starts with a careful history, and then laboratory testing can begin with specific IgE testing to foods suspected to have caused the clinical reaction of concern. The IgE results need to be carefully interpreted in light of the clinical context in which they were ordered. Oral food challenge can be helpful in addition to IgE testing. Consultation with an allergy-immunology specialist is often helpful as well. —NS
DOES PEANUT EXPOSURE INCREASE ALLERGY RISK?
Du Toit G, Roberts G, Sayre PH, et al; LEAP Study Team. Randomized trial of peanut consumption in infants at risk for peanut allergy. N Engl J Med. 2015;372(9):803-813. doi: 10.1056/NEJMoa1414850.
Among children at high risk for peanut allergy, early introduction of peanuts significantly reduced the risk for allergy, according to a randomized controlled trial of 640 infants with severe eczema, egg allergy, or both.
Participants ages 4 to 11 months who were high risk—based on severe atopy or allergy to eggs—were tested for sensitivity to peanut extract at baseline and then assigned to either avoid or consume peanuts.
At 60 months of age, skin allergy testing was repeated; the resulting prevalence of peanut allergy was as follows
A greater percentage of the consumption group showed an increase in levels of peanut-specific IgG4 antibody; the avoidance group, elevated titers of peanut specific IgE antibody.
COMMENTARY
The prevalence of peanut allergy in the United States has increased from 0.4% in 1997 to more than 2% in 2010.1 In 2000, the American Academy of Pediatrics recommended peanut avoidance until age 3 in children at high risk for atopic disease; then in 2008, based on emerging evidence that early introduction of allergenic foods, including peanuts, may decrease the development of allergy, the recommendations were retracted.2,3 The present study, using a randomized trial design, confirms the paradigm-changing hypothesis that early introduction of allergenic foods decreases the subsequent development of food allergies. The authors of the accompanying editorial state that these data are incontrovertible and that children at high risk for peanut allergy should, under supervision of an allergist, be tested and if skin-prick–negative for peanut allergy, be started on a peanut protein–containing diet.1—NS
1. Gruchalla RS, Sampson HA. Preventing peanut allergy through early consumption ready for prime time? N Engl J Med. 2015;372:875-876.
2. Du Toit G, Katz Y, Sasieni P, et al. Early consumption of peanuts in infancy is associated with a low prevalence of peanut allergy. J Allergy Clin Immunol. 2008;122:984-991.
3. Katz Y, Rajuan N, Goldberg MR, et al. Early exposure to cow’s milk protein is protective against IgE-mediated cow’s milk protein allergy. J Allergy Clin Immunol. 2010;126:77-82.
Continue for Anaphylaxis guideline from AAAAI/ACAAI >>
ANAPHYLAXIS GUIDELINE FROM AAAAI/ACAAI
Campbell RL, Li JT, Nicklas RA, Sadosty AT. Emergency department diagnosis and treatment of anaphylaxis: a practice parameter. Ann Allergy Asthma Immunol. 2014;113(6):599-608. doi: 10.1016/j.anai.2014.10.007.
The American Academy of Allergy, Asthma & Immunology and the American College of Allergy, Asthma, and Immunology guideline for emergency department diagnosis and treatment of anaphylaxis includes the following recommendations
• Carefully and immediately triage and monitor patients who have signs and symptoms of anaphylaxis in preparation for epinephrine administration.
• First-line treatment for patients experiencing anaphylaxis is epinephrine. It should be administered intramuscularly in the anterolateral thigh immediately after the diagnosis is made. Epinephrine can be administered every 5 to 15 minutes as needed to control symptoms.
• Do not substitute for epinephrine in the treatment of anaphylaxis. Antihistamines and corticosteroids can be administered in conjunction with epinephrine, not in place of it.
• Determine whether the patient has risk factors for severe and potentially fatal anaphylaxis, such as delayed administration of epinephrine, asthma, a history of biphasic reactions, or cardiovascular disease.
• For anaphylaxis patients with bronchospasms, administer a β-agonist.
• Patients should be observed for 4 to 8 hours (longer for those with a history of risk factors for severe anaphylaxis).
• Refer patients to an allergist–immunologist upon discharge.
COMMENTARY
Identification of anaphylaxis requires judgment. Abbreviated criteria for anaphylaxis include essentially two organ systems of involvement: skin manifestations of pruritus, flushing, hives, or angioedema; respiratory manifestations of wheezing or stridor; decreased blood pressure; and GI symptoms of vomiting, cramping abdominal pain, or diarrhea. Quick identification and treatment of anaphylaxis is important, as the median time to respiratory or cardiac arrest in food-induced anaphylaxis is only 30 minutes. All offices should stock epinephrine in an obvious place; you may consider use of a prefilled pen, so it is easy to find and to give the correct dose. —NS
Continue for Updated practice parameter: Diagnosing and treating food allergies >>
UPDATED PRACTICE PARAMETER: DIAGNOSING AND TREATING FOOD ALLERGIES
Sampson HA, Aceves S, Bock SA, et al. Food allergy: a practice parameter update—2014. J Allergy Clin Immunol. 2014. pii: S0091-6749(14)00672-1. doi: 10.1016/j.jaci.2014.05.013. [Epub ahead of print]
Over the past decade, health care providers have been confronted with a growing number of patients with suspected food allergies, but data supporting an increase in confirmed allergy cases is limited.
The 2014 practice parameter update on food allergies from the American Academy of Allergy, Asthma & Immunology advises clinicians to keep in mind that self-reported food allergy is more common than proven food allergy; that allergy is more common in children and in patients with other atopic conditions; and that the majority of allergic reactions are from peanuts, tree nuts, fish, shellfish, milk, eggs, wheat, soy, and seeds.
The update of the 2006 guidelines includes 64 new summary statements. Highlights include
• Clinicians should advise patients about the risk for cross-reactions from foods similar to their allergens, such as other tree nuts, vertebrate fish, crustaceans, or milk from cows, goats, or other mammals.
• Patients with a seafood allergy should be advised that they are not at increased risk for a reaction to radiocontrast media.
• Patients with food allergies do not need to be concerned about eating genetically modified foods (GMO), due to current FDA screening requirements to rule out allergenicity.
• Patients with chronic idiopathic urticaria or hyperactivity/attention-deficit disorder should not routinely be advised to avoid food additives.
• Patients with asthma should not routinely be advised to avoid sulfates, unless they have had a previous reaction to them.
The guidelines also include a new section on diagnosing and treating non-IgE-mediated food allergies, such as food-protein–induced enterocolitis syndrome, allergic proctocolitis, enteropathy, eosinophilic esophagitis, and gastroenteritis.
COMMENTARY
Food allergies are a commonly encountered and difficult area of practice for those of us in primary care. They can be life threatening; yet many patients who are concerned about the possibility of food allergy do not actually have a food allergy, so making an accurate diagnosis is important. Allergic evaluation starts with a careful history, and then laboratory testing can begin with specific IgE testing to foods suspected to have caused the clinical reaction of concern. The IgE results need to be carefully interpreted in light of the clinical context in which they were ordered. Oral food challenge can be helpful in addition to IgE testing. Consultation with an allergy-immunology specialist is often helpful as well. —NS
Easy Bruising, Low Platelets, Recent Coldlike Illness
A 6-year-old girl was brought to the emergency department (ED) by her mother after the child bumped her head while playing. While the physician examined the child’s head, the mother remarked that her daughter had recently developed bruises that appeared suddenly and only after minor, if any, known trauma. The ED physician determined that the child’s bump to the head was nothing to worry about, attributed the bruising to the child being a “healthy, active 6-year-old,” and sent her home.
Two days later, the child was brought to our office because the mother was still concerned about her daughter’s easy bruising. The mother pointed out ecchymosis scattered across her daughter’s extremities and torso. The child denied any pain or other complaints, including any active or recurrent bleeding. Upon further questioning, the mother mentioned that her daughter had recovered from a coldlike illness several weeks earlier.
THE DIAGNOSIS
We ordered a complete blood count (CBC) and peripheral smear, which were normal except for the platelet count, which was 7,000/μL (normal, 150,000-450,000/μL). Based on the child’s easy bruising and isolated thrombocytopenia, we diagnosed immune thrombocytopenia, which is also known as idiopathic thrombocytopenic purpura (ITP).
DISCUSSION
In ITP, autoantibodies are directed against platelets, leading to their sequestration and destruction in the spleen and a resultant drop in platelet count.1 Children with ITP typically present between the ages of 2 and 10, with a peak incidence between 2 and 5.2 The incidence is estimated to be as high as 8 per 100,000 children.3 However, this estimate primarily reflects symptomatic children, and the true incidence of childhood ITP may be much higher because asymptomatic children may not be brought in to see a doctor.
For the majority of patients, ITP resolves within three months. However, for 20% to 30% of patients, thrombocytopenia will last beyond six months, with or without treatment.4 In 1% of cases, patients will have a recurrence of ITP.3
In addition to easy bruising, nearly all patients who present with possible ITP will complain of cutaneous bleeding, typically a nose bleed or bleeding in the oral cavity.2 Upon questioning, 60% of patients will report a history of recent infection.4 Not surprisingly, bleeding severity correlates inversely with platelet count; severe bleeding is seen in patients with a platelet count < 10,000/μL.
While rare, the more worrisome complications include intracranial hemorrhage (incidence, 0.1% to 0.8%) and other serious hemorrhages that would require transfusion (estimated incidence, 2.9%).2
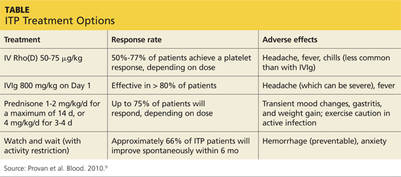
Vast differential seen in child bruising
When a child presents with bruising, perform a thorough history, including birth and prenatal course, as well as a physical to exclude other potential causes, such as physical abuse, use of herbal remedies or other natural supplements that may not be disclosed as medication, or even environmental exposure. When bruising is present in a child who has isolated thrombocytopenia, the diagnosis of ITP may be straightforward. However, many conditions share thrombocytopenia in their disease process and should be considered in the differential diagnosis of a child who you suspect may have ITP.
• Suspect physical abuse in a bruised child who does not have thrombocytopenia, whose mood is flat or depressed, or who has experienced recurrent injuries or bruising.
• Leukemia, particularly acute lymphoblastic leukemia (ALL), the predominant leukemia found in children, should be ruled out as well. Symptoms that may distinguish a child with ALL from one with ITP include fever, weight loss, and joint pain, as well as signs such as lymphadenopathy, hepatosplenomegaly, anemia, and leukocytosis. A peripheral smear may be ordered to help confirm or exclude a diagnosis of ALL, should any of the above be present in a child with thrombocytopenia.5 It may show lymphoblasts and/or atypical cells in a patient with ALL.5
• Infections should also be included in a differential when a patient is suspected of having ITP, particularly if he or she has systemic symptoms. Viral infections that may cause thrombocytopenia include mononucleosis, dengue virus, human herpesvirus-6, and HIV.6,7
ITP often follows an infection, and the incidence of ITP may be higher during winter months, when infections are more common. However, infection may not always be the cause of ITP. Sepsis may also lead to thrombocytopenia, but a child with sepsis would present very differently from one who has only ITP. A septic child would present with acute illness and signs and symptoms of severe systemic illness, such as high fever, altered mental status, tachycardia, pallor, diaphoresis, and hypotension.
• Drug-induced thrombocytopenia (DIT) should be considered in any child who is taking or recently took a medication that may cause thrombocytopenia. Medications that can cause thrombocytopenia include heparin, quinine, vancomycin, trimethoprim-sulfamethoxazole, rifampin, carbamazepine, phenytoin, piperacillin, linezolid, and valproic acid.8 The measles, mumps, and rubella vaccine also can cause thrombocytopenia.8 A careful medication history may determine if the child is at risk for DIT.
• To narrow the differential, obtain a CBC and peripheral smear when evaluating a patient you suspect may have ITP5 (strength of recommendation [SOR]: A). A CBC will determine the patient’s platelet count, and a peripheral smear should be obtained to exclude other possible diagnoses.5
If there are any questions regarding the results of a peripheral smear, it may be necessary to perform a bone marrow aspiration. This, however, is not usually necessary in an otherwise typical case of ITP.9 Bone marrow aspiration may, however, be necessary to reevaluate the initial diagnosis for a child who does not respond to treatment for ITP.
Continue for corticosteroids, IVIg are usually effective >>
Corticosteroids, IVIg are usually effective
The first step in treating a patient with ITP is to limit the risk for further injury or bleeding by stopping NSAIDs or ending participation in contact sports2,9 (SOR: C). The next step is to determine if pharmacologic therapy is warranted.
Medication, if necessary, is the mainstay of treatment for patients with ITP, particularly those experiencing significant bleeding.2 Corticosteroids, intravenous (IV) immunoglobulin (IVIg), and IV Rho(D) immune globulin (also known as anti-D) are the medications typically used to treat a child with ITP, depending on availability of the drugs, bleeding or bleeding risk, and convenience of dosing. For example, corticosteroids can be used orally or IV, whereas IVIg and IV Rho(D) may not be readily available in some treatment settings.
Corticosteroids have been shown to more rapidly increase platelet count compared to placebo and appear to have a dose-related effect.10,11 Oral prednisone can be dosed at 1 to 2 mg/kg/d for 14 days and then tapered over the course of one week10,11 or one may prescribe 4 mg/kg/d for four days.10,11 IV methylprednisolone typically is given at 30 mg/kg/d for three to four days.9
IVIg may have greater efficacy than corticosteroids in treating ITP, but it may also cause adverse effects, including nausea, headache, and fever. IVIg can be administered as a single 800 to 1,000 mg/kg dose, or as a daily 400 mg/kg dose form five days; higher doses should be reserved for patients with severe bleeding.12
If ITP persists despite the use of corticosteroids or IVIg, IV Rho(D) Ig may be used in patients with Rho(D)-positive blood at a single dose of 25 to 50 μg/kg, with additional doses administered on separate days as required to elevate platelet count. However, only Rho(D)-positive patients are eligible for anti-D treatment.
The response rates/times and adverse effects of common treatments for ITP are summarized in the Table.9 A small randomized study found that oral methylprednisolone 30 mg/kg/d for three days followed by 20 mg/kg/d for an additional four days was comparable to IVIg 0.4 g/kg/d for five days.11 A different study that compared oral methylprednisolone (30 mg/kg/d or 50 mg/kg/d for seven days) and IVIg (0.5 g/kg/d for five days) found no difference in outcomes among the three treatments.13 One advantage, though, of IVIg is that it can be administered as a single IV dose, rather than multiple doses over several weeks, as is the case with oral prednisone.9,11-13
• Follow platelet counts closely. Patients with ITP should have their platelet counts monitored at least once weekly and as often as twice weekly. The frequency of monitoring may be tapered depending on an individual patient’s response to treatment and the severity of the thrombocytopenia.14
• We referred our patient to a nearby children’s hospital, where a repeat CBC showed her platelets had decreased to 3,000/μL. She received a six-hour infusion of IVIg and was discharged with instructions to have her CBC closely monitored. Her platelets remained stable until four weeks later, when they decreased from 102,000/μL to 71,000/μL. She received a second infusion of IVIg as an outpatient.
Soon after, she presented to our ED with a headache, nausea, and fever of 102°F. CT of her head was normal; a repeat CBC showed no elevation in white blood cells, but her hemoglobin had decreased from 11.9 g/dL to 9.7 g/dL. (Her platelets were 254,000/μL.) The patient’s complaints were likely adverse effects of the IVIg. The CBC abnormalities, fever, headache, and malaise resolved shortly thereafter, and the patient remains asymptomatic with no recurrence of ITP.
Continue for the takeaway >>
THE TAKEAWAY
Suspect ITP in a child who bruises easily and who also has thrombocytopenia. Order a CBC and peripheral blood smear to rule out other potential illnesses. Pharmacotherapy, if needed, typically consists of an oral or IV corticosteroid or IVIg; IV Rho(D) Ig may be used in Rho(D)-positive patients who don’t respond to other treatments. Patients with ITP should have their platelet count monitored at least once weekly until platelets have increased to 150,000/μL or higher. Frequency of monitoring may be reduced as the clinical picture improves and the patient remains stable. More frequent monitoring may be necessary based on severity, complications, and response to treatment.
REFERENCES
1. Johnsen J. Pathogenesis in immune thrombocytopenia: new insights. Hematology Am Soc Hematol Educ Program. 2012;2012:306-312.
2. Kühne T, Buchanan GR, Zimmerman S, et al; Intercontinental Childhood ITP Study Group. A prospective comparative study of 2540 infants and children with newly diagnosed idiopathic thrombocytopenic purpura (ITP) from the Intercontinental Childhood ITP Study Group. J Pediatr. 2003;143:605-608.
3. Kurtzberg J, Stockman JA III. Idiopathic autoimmune thrombocytopenic purpura. Adv Pediatr. 1994;41:111-134.
4. Zeller B, Rajantie J, Hedlund-Treutiger I, et al. Childhood idiopathic thrombocytopenic purpura in the Nordic countries: epidemiology and predictors of chronic disease. Acta Paediatr. 2005;94:178-184.
5. Margolin JF, Steuber CP, Poplack DG. Acute lymphoblastic leukemia. In: Pizzo PA, Poplack DG, eds. Principles and Practice of Pediatric Oncology. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2001:317-321.
6. Hashimoto H, Maruyama H, Fujimoto K, et al. Hematologic findings associated with thrombocytopenia during the acute phase of exanthem subitum confirmed by primary human herpesvirus-6 infection. J Pediatr Hematol Oncol. 2002;24:211-214.
7. La Russa VF, Innis BL. Mechanisms of dengue virus-induced bone marrow suppression. Baillieres Clin Haematol. 1995;8:249-270.
8. Aster RH, Curtis BR, McFarland JG, et al. Drug-induced immune thrombocytopenia: pathogenesis, diagnosis, and management. Thromb Haemost. 2009;7:911-918.
9. Provan D, Stasi R, Newland AC, et al. International consensus report on the investigation and management of primary immune thrombocytopenia. Blood. 2010;115:168-186.
10. Bellucci S, Charpak Y, Chastang C, et al. Low doses v conventional doses of corticoids in immune thrombocytopenic purpura (ITP): results of a randomized clinical trial in 160 children, 223 adults. Blood. 1988;71:1165-1169.
11. Ozsoylu S, Sayli TR, Oztürk G. Oral megadose methylprednisolone versus intravenous immunoglobulin for acute childhood idiopathic thrombocytopenic purpura. Pediatr Hematol Oncol. 1993;10:317-321.
12. Beck CE, Nathan PC, Parkin PC, et al. Corticosteroids versus intravenous immune globulin for the treatment of acute immune thrombocytopenic purpura in children: a systematic review and meta-analysis of randomized controlled trials. J Pediatr. 2005;147:521-527.
13. Albayrak D, Islek I, Kalaycí AG, et al. Acute immune thrombocytopenic purpura: a comparative study of very high oral doses of methylprednisolone and intravenously administered immune globulin. J Pediatr. 1994;125(6 pt 1):1004-1007.
14. Tarantino MD, Madden RM, Fennewald DL, et al. Treatment of childhood acute immune thrombocytopenic purpura with anti-D immune globulin or pooled immune globulin. J Pediatr. 1999;134:21-26.
A 6-year-old girl was brought to the emergency department (ED) by her mother after the child bumped her head while playing. While the physician examined the child’s head, the mother remarked that her daughter had recently developed bruises that appeared suddenly and only after minor, if any, known trauma. The ED physician determined that the child’s bump to the head was nothing to worry about, attributed the bruising to the child being a “healthy, active 6-year-old,” and sent her home.
Two days later, the child was brought to our office because the mother was still concerned about her daughter’s easy bruising. The mother pointed out ecchymosis scattered across her daughter’s extremities and torso. The child denied any pain or other complaints, including any active or recurrent bleeding. Upon further questioning, the mother mentioned that her daughter had recovered from a coldlike illness several weeks earlier.
THE DIAGNOSIS
We ordered a complete blood count (CBC) and peripheral smear, which were normal except for the platelet count, which was 7,000/μL (normal, 150,000-450,000/μL). Based on the child’s easy bruising and isolated thrombocytopenia, we diagnosed immune thrombocytopenia, which is also known as idiopathic thrombocytopenic purpura (ITP).
DISCUSSION
In ITP, autoantibodies are directed against platelets, leading to their sequestration and destruction in the spleen and a resultant drop in platelet count.1 Children with ITP typically present between the ages of 2 and 10, with a peak incidence between 2 and 5.2 The incidence is estimated to be as high as 8 per 100,000 children.3 However, this estimate primarily reflects symptomatic children, and the true incidence of childhood ITP may be much higher because asymptomatic children may not be brought in to see a doctor.
For the majority of patients, ITP resolves within three months. However, for 20% to 30% of patients, thrombocytopenia will last beyond six months, with or without treatment.4 In 1% of cases, patients will have a recurrence of ITP.3
In addition to easy bruising, nearly all patients who present with possible ITP will complain of cutaneous bleeding, typically a nose bleed or bleeding in the oral cavity.2 Upon questioning, 60% of patients will report a history of recent infection.4 Not surprisingly, bleeding severity correlates inversely with platelet count; severe bleeding is seen in patients with a platelet count < 10,000/μL.
While rare, the more worrisome complications include intracranial hemorrhage (incidence, 0.1% to 0.8%) and other serious hemorrhages that would require transfusion (estimated incidence, 2.9%).2
Vast differential seen in child bruising
When a child presents with bruising, perform a thorough history, including birth and prenatal course, as well as a physical to exclude other potential causes, such as physical abuse, use of herbal remedies or other natural supplements that may not be disclosed as medication, or even environmental exposure. When bruising is present in a child who has isolated thrombocytopenia, the diagnosis of ITP may be straightforward. However, many conditions share thrombocytopenia in their disease process and should be considered in the differential diagnosis of a child who you suspect may have ITP.
• Suspect physical abuse in a bruised child who does not have thrombocytopenia, whose mood is flat or depressed, or who has experienced recurrent injuries or bruising.
• Leukemia, particularly acute lymphoblastic leukemia (ALL), the predominant leukemia found in children, should be ruled out as well. Symptoms that may distinguish a child with ALL from one with ITP include fever, weight loss, and joint pain, as well as signs such as lymphadenopathy, hepatosplenomegaly, anemia, and leukocytosis. A peripheral smear may be ordered to help confirm or exclude a diagnosis of ALL, should any of the above be present in a child with thrombocytopenia.5 It may show lymphoblasts and/or atypical cells in a patient with ALL.5
• Infections should also be included in a differential when a patient is suspected of having ITP, particularly if he or she has systemic symptoms. Viral infections that may cause thrombocytopenia include mononucleosis, dengue virus, human herpesvirus-6, and HIV.6,7
ITP often follows an infection, and the incidence of ITP may be higher during winter months, when infections are more common. However, infection may not always be the cause of ITP. Sepsis may also lead to thrombocytopenia, but a child with sepsis would present very differently from one who has only ITP. A septic child would present with acute illness and signs and symptoms of severe systemic illness, such as high fever, altered mental status, tachycardia, pallor, diaphoresis, and hypotension.
• Drug-induced thrombocytopenia (DIT) should be considered in any child who is taking or recently took a medication that may cause thrombocytopenia. Medications that can cause thrombocytopenia include heparin, quinine, vancomycin, trimethoprim-sulfamethoxazole, rifampin, carbamazepine, phenytoin, piperacillin, linezolid, and valproic acid.8 The measles, mumps, and rubella vaccine also can cause thrombocytopenia.8 A careful medication history may determine if the child is at risk for DIT.
• To narrow the differential, obtain a CBC and peripheral smear when evaluating a patient you suspect may have ITP5 (strength of recommendation [SOR]: A). A CBC will determine the patient’s platelet count, and a peripheral smear should be obtained to exclude other possible diagnoses.5
If there are any questions regarding the results of a peripheral smear, it may be necessary to perform a bone marrow aspiration. This, however, is not usually necessary in an otherwise typical case of ITP.9 Bone marrow aspiration may, however, be necessary to reevaluate the initial diagnosis for a child who does not respond to treatment for ITP.
Continue for corticosteroids, IVIg are usually effective >>
Corticosteroids, IVIg are usually effective
The first step in treating a patient with ITP is to limit the risk for further injury or bleeding by stopping NSAIDs or ending participation in contact sports2,9 (SOR: C). The next step is to determine if pharmacologic therapy is warranted.
Medication, if necessary, is the mainstay of treatment for patients with ITP, particularly those experiencing significant bleeding.2 Corticosteroids, intravenous (IV) immunoglobulin (IVIg), and IV Rho(D) immune globulin (also known as anti-D) are the medications typically used to treat a child with ITP, depending on availability of the drugs, bleeding or bleeding risk, and convenience of dosing. For example, corticosteroids can be used orally or IV, whereas IVIg and IV Rho(D) may not be readily available in some treatment settings.
Corticosteroids have been shown to more rapidly increase platelet count compared to placebo and appear to have a dose-related effect.10,11 Oral prednisone can be dosed at 1 to 2 mg/kg/d for 14 days and then tapered over the course of one week10,11 or one may prescribe 4 mg/kg/d for four days.10,11 IV methylprednisolone typically is given at 30 mg/kg/d for three to four days.9
IVIg may have greater efficacy than corticosteroids in treating ITP, but it may also cause adverse effects, including nausea, headache, and fever. IVIg can be administered as a single 800 to 1,000 mg/kg dose, or as a daily 400 mg/kg dose form five days; higher doses should be reserved for patients with severe bleeding.12
If ITP persists despite the use of corticosteroids or IVIg, IV Rho(D) Ig may be used in patients with Rho(D)-positive blood at a single dose of 25 to 50 μg/kg, with additional doses administered on separate days as required to elevate platelet count. However, only Rho(D)-positive patients are eligible for anti-D treatment.
The response rates/times and adverse effects of common treatments for ITP are summarized in the Table.9 A small randomized study found that oral methylprednisolone 30 mg/kg/d for three days followed by 20 mg/kg/d for an additional four days was comparable to IVIg 0.4 g/kg/d for five days.11 A different study that compared oral methylprednisolone (30 mg/kg/d or 50 mg/kg/d for seven days) and IVIg (0.5 g/kg/d for five days) found no difference in outcomes among the three treatments.13 One advantage, though, of IVIg is that it can be administered as a single IV dose, rather than multiple doses over several weeks, as is the case with oral prednisone.9,11-13
• Follow platelet counts closely. Patients with ITP should have their platelet counts monitored at least once weekly and as often as twice weekly. The frequency of monitoring may be tapered depending on an individual patient’s response to treatment and the severity of the thrombocytopenia.14
• We referred our patient to a nearby children’s hospital, where a repeat CBC showed her platelets had decreased to 3,000/μL. She received a six-hour infusion of IVIg and was discharged with instructions to have her CBC closely monitored. Her platelets remained stable until four weeks later, when they decreased from 102,000/μL to 71,000/μL. She received a second infusion of IVIg as an outpatient.
Soon after, she presented to our ED with a headache, nausea, and fever of 102°F. CT of her head was normal; a repeat CBC showed no elevation in white blood cells, but her hemoglobin had decreased from 11.9 g/dL to 9.7 g/dL. (Her platelets were 254,000/μL.) The patient’s complaints were likely adverse effects of the IVIg. The CBC abnormalities, fever, headache, and malaise resolved shortly thereafter, and the patient remains asymptomatic with no recurrence of ITP.
Continue for the takeaway >>
THE TAKEAWAY
Suspect ITP in a child who bruises easily and who also has thrombocytopenia. Order a CBC and peripheral blood smear to rule out other potential illnesses. Pharmacotherapy, if needed, typically consists of an oral or IV corticosteroid or IVIg; IV Rho(D) Ig may be used in Rho(D)-positive patients who don’t respond to other treatments. Patients with ITP should have their platelet count monitored at least once weekly until platelets have increased to 150,000/μL or higher. Frequency of monitoring may be reduced as the clinical picture improves and the patient remains stable. More frequent monitoring may be necessary based on severity, complications, and response to treatment.
REFERENCES
1. Johnsen J. Pathogenesis in immune thrombocytopenia: new insights. Hematology Am Soc Hematol Educ Program. 2012;2012:306-312.
2. Kühne T, Buchanan GR, Zimmerman S, et al; Intercontinental Childhood ITP Study Group. A prospective comparative study of 2540 infants and children with newly diagnosed idiopathic thrombocytopenic purpura (ITP) from the Intercontinental Childhood ITP Study Group. J Pediatr. 2003;143:605-608.
3. Kurtzberg J, Stockman JA III. Idiopathic autoimmune thrombocytopenic purpura. Adv Pediatr. 1994;41:111-134.
4. Zeller B, Rajantie J, Hedlund-Treutiger I, et al. Childhood idiopathic thrombocytopenic purpura in the Nordic countries: epidemiology and predictors of chronic disease. Acta Paediatr. 2005;94:178-184.
5. Margolin JF, Steuber CP, Poplack DG. Acute lymphoblastic leukemia. In: Pizzo PA, Poplack DG, eds. Principles and Practice of Pediatric Oncology. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2001:317-321.
6. Hashimoto H, Maruyama H, Fujimoto K, et al. Hematologic findings associated with thrombocytopenia during the acute phase of exanthem subitum confirmed by primary human herpesvirus-6 infection. J Pediatr Hematol Oncol. 2002;24:211-214.
7. La Russa VF, Innis BL. Mechanisms of dengue virus-induced bone marrow suppression. Baillieres Clin Haematol. 1995;8:249-270.
8. Aster RH, Curtis BR, McFarland JG, et al. Drug-induced immune thrombocytopenia: pathogenesis, diagnosis, and management. Thromb Haemost. 2009;7:911-918.
9. Provan D, Stasi R, Newland AC, et al. International consensus report on the investigation and management of primary immune thrombocytopenia. Blood. 2010;115:168-186.
10. Bellucci S, Charpak Y, Chastang C, et al. Low doses v conventional doses of corticoids in immune thrombocytopenic purpura (ITP): results of a randomized clinical trial in 160 children, 223 adults. Blood. 1988;71:1165-1169.
11. Ozsoylu S, Sayli TR, Oztürk G. Oral megadose methylprednisolone versus intravenous immunoglobulin for acute childhood idiopathic thrombocytopenic purpura. Pediatr Hematol Oncol. 1993;10:317-321.
12. Beck CE, Nathan PC, Parkin PC, et al. Corticosteroids versus intravenous immune globulin for the treatment of acute immune thrombocytopenic purpura in children: a systematic review and meta-analysis of randomized controlled trials. J Pediatr. 2005;147:521-527.
13. Albayrak D, Islek I, Kalaycí AG, et al. Acute immune thrombocytopenic purpura: a comparative study of very high oral doses of methylprednisolone and intravenously administered immune globulin. J Pediatr. 1994;125(6 pt 1):1004-1007.
14. Tarantino MD, Madden RM, Fennewald DL, et al. Treatment of childhood acute immune thrombocytopenic purpura with anti-D immune globulin or pooled immune globulin. J Pediatr. 1999;134:21-26.
A 6-year-old girl was brought to the emergency department (ED) by her mother after the child bumped her head while playing. While the physician examined the child’s head, the mother remarked that her daughter had recently developed bruises that appeared suddenly and only after minor, if any, known trauma. The ED physician determined that the child’s bump to the head was nothing to worry about, attributed the bruising to the child being a “healthy, active 6-year-old,” and sent her home.
Two days later, the child was brought to our office because the mother was still concerned about her daughter’s easy bruising. The mother pointed out ecchymosis scattered across her daughter’s extremities and torso. The child denied any pain or other complaints, including any active or recurrent bleeding. Upon further questioning, the mother mentioned that her daughter had recovered from a coldlike illness several weeks earlier.
THE DIAGNOSIS
We ordered a complete blood count (CBC) and peripheral smear, which were normal except for the platelet count, which was 7,000/μL (normal, 150,000-450,000/μL). Based on the child’s easy bruising and isolated thrombocytopenia, we diagnosed immune thrombocytopenia, which is also known as idiopathic thrombocytopenic purpura (ITP).
DISCUSSION
In ITP, autoantibodies are directed against platelets, leading to their sequestration and destruction in the spleen and a resultant drop in platelet count.1 Children with ITP typically present between the ages of 2 and 10, with a peak incidence between 2 and 5.2 The incidence is estimated to be as high as 8 per 100,000 children.3 However, this estimate primarily reflects symptomatic children, and the true incidence of childhood ITP may be much higher because asymptomatic children may not be brought in to see a doctor.
For the majority of patients, ITP resolves within three months. However, for 20% to 30% of patients, thrombocytopenia will last beyond six months, with or without treatment.4 In 1% of cases, patients will have a recurrence of ITP.3
In addition to easy bruising, nearly all patients who present with possible ITP will complain of cutaneous bleeding, typically a nose bleed or bleeding in the oral cavity.2 Upon questioning, 60% of patients will report a history of recent infection.4 Not surprisingly, bleeding severity correlates inversely with platelet count; severe bleeding is seen in patients with a platelet count < 10,000/μL.
While rare, the more worrisome complications include intracranial hemorrhage (incidence, 0.1% to 0.8%) and other serious hemorrhages that would require transfusion (estimated incidence, 2.9%).2
Vast differential seen in child bruising
When a child presents with bruising, perform a thorough history, including birth and prenatal course, as well as a physical to exclude other potential causes, such as physical abuse, use of herbal remedies or other natural supplements that may not be disclosed as medication, or even environmental exposure. When bruising is present in a child who has isolated thrombocytopenia, the diagnosis of ITP may be straightforward. However, many conditions share thrombocytopenia in their disease process and should be considered in the differential diagnosis of a child who you suspect may have ITP.
• Suspect physical abuse in a bruised child who does not have thrombocytopenia, whose mood is flat or depressed, or who has experienced recurrent injuries or bruising.
• Leukemia, particularly acute lymphoblastic leukemia (ALL), the predominant leukemia found in children, should be ruled out as well. Symptoms that may distinguish a child with ALL from one with ITP include fever, weight loss, and joint pain, as well as signs such as lymphadenopathy, hepatosplenomegaly, anemia, and leukocytosis. A peripheral smear may be ordered to help confirm or exclude a diagnosis of ALL, should any of the above be present in a child with thrombocytopenia.5 It may show lymphoblasts and/or atypical cells in a patient with ALL.5
• Infections should also be included in a differential when a patient is suspected of having ITP, particularly if he or she has systemic symptoms. Viral infections that may cause thrombocytopenia include mononucleosis, dengue virus, human herpesvirus-6, and HIV.6,7
ITP often follows an infection, and the incidence of ITP may be higher during winter months, when infections are more common. However, infection may not always be the cause of ITP. Sepsis may also lead to thrombocytopenia, but a child with sepsis would present very differently from one who has only ITP. A septic child would present with acute illness and signs and symptoms of severe systemic illness, such as high fever, altered mental status, tachycardia, pallor, diaphoresis, and hypotension.
• Drug-induced thrombocytopenia (DIT) should be considered in any child who is taking or recently took a medication that may cause thrombocytopenia. Medications that can cause thrombocytopenia include heparin, quinine, vancomycin, trimethoprim-sulfamethoxazole, rifampin, carbamazepine, phenytoin, piperacillin, linezolid, and valproic acid.8 The measles, mumps, and rubella vaccine also can cause thrombocytopenia.8 A careful medication history may determine if the child is at risk for DIT.
• To narrow the differential, obtain a CBC and peripheral smear when evaluating a patient you suspect may have ITP5 (strength of recommendation [SOR]: A). A CBC will determine the patient’s platelet count, and a peripheral smear should be obtained to exclude other possible diagnoses.5
If there are any questions regarding the results of a peripheral smear, it may be necessary to perform a bone marrow aspiration. This, however, is not usually necessary in an otherwise typical case of ITP.9 Bone marrow aspiration may, however, be necessary to reevaluate the initial diagnosis for a child who does not respond to treatment for ITP.
Continue for corticosteroids, IVIg are usually effective >>
Corticosteroids, IVIg are usually effective
The first step in treating a patient with ITP is to limit the risk for further injury or bleeding by stopping NSAIDs or ending participation in contact sports2,9 (SOR: C). The next step is to determine if pharmacologic therapy is warranted.
Medication, if necessary, is the mainstay of treatment for patients with ITP, particularly those experiencing significant bleeding.2 Corticosteroids, intravenous (IV) immunoglobulin (IVIg), and IV Rho(D) immune globulin (also known as anti-D) are the medications typically used to treat a child with ITP, depending on availability of the drugs, bleeding or bleeding risk, and convenience of dosing. For example, corticosteroids can be used orally or IV, whereas IVIg and IV Rho(D) may not be readily available in some treatment settings.
Corticosteroids have been shown to more rapidly increase platelet count compared to placebo and appear to have a dose-related effect.10,11 Oral prednisone can be dosed at 1 to 2 mg/kg/d for 14 days and then tapered over the course of one week10,11 or one may prescribe 4 mg/kg/d for four days.10,11 IV methylprednisolone typically is given at 30 mg/kg/d for three to four days.9
IVIg may have greater efficacy than corticosteroids in treating ITP, but it may also cause adverse effects, including nausea, headache, and fever. IVIg can be administered as a single 800 to 1,000 mg/kg dose, or as a daily 400 mg/kg dose form five days; higher doses should be reserved for patients with severe bleeding.12
If ITP persists despite the use of corticosteroids or IVIg, IV Rho(D) Ig may be used in patients with Rho(D)-positive blood at a single dose of 25 to 50 μg/kg, with additional doses administered on separate days as required to elevate platelet count. However, only Rho(D)-positive patients are eligible for anti-D treatment.
The response rates/times and adverse effects of common treatments for ITP are summarized in the Table.9 A small randomized study found that oral methylprednisolone 30 mg/kg/d for three days followed by 20 mg/kg/d for an additional four days was comparable to IVIg 0.4 g/kg/d for five days.11 A different study that compared oral methylprednisolone (30 mg/kg/d or 50 mg/kg/d for seven days) and IVIg (0.5 g/kg/d for five days) found no difference in outcomes among the three treatments.13 One advantage, though, of IVIg is that it can be administered as a single IV dose, rather than multiple doses over several weeks, as is the case with oral prednisone.9,11-13
• Follow platelet counts closely. Patients with ITP should have their platelet counts monitored at least once weekly and as often as twice weekly. The frequency of monitoring may be tapered depending on an individual patient’s response to treatment and the severity of the thrombocytopenia.14
• We referred our patient to a nearby children’s hospital, where a repeat CBC showed her platelets had decreased to 3,000/μL. She received a six-hour infusion of IVIg and was discharged with instructions to have her CBC closely monitored. Her platelets remained stable until four weeks later, when they decreased from 102,000/μL to 71,000/μL. She received a second infusion of IVIg as an outpatient.
Soon after, she presented to our ED with a headache, nausea, and fever of 102°F. CT of her head was normal; a repeat CBC showed no elevation in white blood cells, but her hemoglobin had decreased from 11.9 g/dL to 9.7 g/dL. (Her platelets were 254,000/μL.) The patient’s complaints were likely adverse effects of the IVIg. The CBC abnormalities, fever, headache, and malaise resolved shortly thereafter, and the patient remains asymptomatic with no recurrence of ITP.
Continue for the takeaway >>
THE TAKEAWAY
Suspect ITP in a child who bruises easily and who also has thrombocytopenia. Order a CBC and peripheral blood smear to rule out other potential illnesses. Pharmacotherapy, if needed, typically consists of an oral or IV corticosteroid or IVIg; IV Rho(D) Ig may be used in Rho(D)-positive patients who don’t respond to other treatments. Patients with ITP should have their platelet count monitored at least once weekly until platelets have increased to 150,000/μL or higher. Frequency of monitoring may be reduced as the clinical picture improves and the patient remains stable. More frequent monitoring may be necessary based on severity, complications, and response to treatment.
REFERENCES
1. Johnsen J. Pathogenesis in immune thrombocytopenia: new insights. Hematology Am Soc Hematol Educ Program. 2012;2012:306-312.
2. Kühne T, Buchanan GR, Zimmerman S, et al; Intercontinental Childhood ITP Study Group. A prospective comparative study of 2540 infants and children with newly diagnosed idiopathic thrombocytopenic purpura (ITP) from the Intercontinental Childhood ITP Study Group. J Pediatr. 2003;143:605-608.
3. Kurtzberg J, Stockman JA III. Idiopathic autoimmune thrombocytopenic purpura. Adv Pediatr. 1994;41:111-134.
4. Zeller B, Rajantie J, Hedlund-Treutiger I, et al. Childhood idiopathic thrombocytopenic purpura in the Nordic countries: epidemiology and predictors of chronic disease. Acta Paediatr. 2005;94:178-184.
5. Margolin JF, Steuber CP, Poplack DG. Acute lymphoblastic leukemia. In: Pizzo PA, Poplack DG, eds. Principles and Practice of Pediatric Oncology. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2001:317-321.
6. Hashimoto H, Maruyama H, Fujimoto K, et al. Hematologic findings associated with thrombocytopenia during the acute phase of exanthem subitum confirmed by primary human herpesvirus-6 infection. J Pediatr Hematol Oncol. 2002;24:211-214.
7. La Russa VF, Innis BL. Mechanisms of dengue virus-induced bone marrow suppression. Baillieres Clin Haematol. 1995;8:249-270.
8. Aster RH, Curtis BR, McFarland JG, et al. Drug-induced immune thrombocytopenia: pathogenesis, diagnosis, and management. Thromb Haemost. 2009;7:911-918.
9. Provan D, Stasi R, Newland AC, et al. International consensus report on the investigation and management of primary immune thrombocytopenia. Blood. 2010;115:168-186.
10. Bellucci S, Charpak Y, Chastang C, et al. Low doses v conventional doses of corticoids in immune thrombocytopenic purpura (ITP): results of a randomized clinical trial in 160 children, 223 adults. Blood. 1988;71:1165-1169.
11. Ozsoylu S, Sayli TR, Oztürk G. Oral megadose methylprednisolone versus intravenous immunoglobulin for acute childhood idiopathic thrombocytopenic purpura. Pediatr Hematol Oncol. 1993;10:317-321.
12. Beck CE, Nathan PC, Parkin PC, et al. Corticosteroids versus intravenous immune globulin for the treatment of acute immune thrombocytopenic purpura in children: a systematic review and meta-analysis of randomized controlled trials. J Pediatr. 2005;147:521-527.
13. Albayrak D, Islek I, Kalaycí AG, et al. Acute immune thrombocytopenic purpura: a comparative study of very high oral doses of methylprednisolone and intravenously administered immune globulin. J Pediatr. 1994;125(6 pt 1):1004-1007.
14. Tarantino MD, Madden RM, Fennewald DL, et al. Treatment of childhood acute immune thrombocytopenic purpura with anti-D immune globulin or pooled immune globulin. J Pediatr. 1999;134:21-26.
WCD: Pramocaine is a common contact sensitizer
VANCOUVER – Pramocaine, a topical anesthetic long used in many OTC anti-itch preparations, turns out to itself be a common cause of contact sensitization, Dr. Mariam Abbas reported at the World Congress of Dermatology.
Pramocaine, also known as pramoxine, is an ingredient in Gold Bond cream, some forms of calamine lotion, hemorrhoid relief products, and other OTC antipruritic agents. It’s not available in commercial patch test kits, so Dr. Abbas created a test material comprised of 2% pramocaine in petrolatum.
Seven of 232 patients (3%) referred to a university patch test clinic and who underwent testing to pramocaine along with recognized contact sensitizers showed a clear positive response to pramocaine at 96 hours. The reactions were clinically relevant, according to Dr. Abbas of the University of Alberta, Edmonton.
That 3% positive rate is in the range of published figures for such well-known problem contact sensitizers as neomycin, thiuram mix, and Balsam of Peru.
Dr. Mariam Abbas reported having no financial conflicts regarding this study, conducted free of commercial support.
VANCOUVER – Pramocaine, a topical anesthetic long used in many OTC anti-itch preparations, turns out to itself be a common cause of contact sensitization, Dr. Mariam Abbas reported at the World Congress of Dermatology.
Pramocaine, also known as pramoxine, is an ingredient in Gold Bond cream, some forms of calamine lotion, hemorrhoid relief products, and other OTC antipruritic agents. It’s not available in commercial patch test kits, so Dr. Abbas created a test material comprised of 2% pramocaine in petrolatum.
Seven of 232 patients (3%) referred to a university patch test clinic and who underwent testing to pramocaine along with recognized contact sensitizers showed a clear positive response to pramocaine at 96 hours. The reactions were clinically relevant, according to Dr. Abbas of the University of Alberta, Edmonton.
That 3% positive rate is in the range of published figures for such well-known problem contact sensitizers as neomycin, thiuram mix, and Balsam of Peru.
Dr. Mariam Abbas reported having no financial conflicts regarding this study, conducted free of commercial support.
VANCOUVER – Pramocaine, a topical anesthetic long used in many OTC anti-itch preparations, turns out to itself be a common cause of contact sensitization, Dr. Mariam Abbas reported at the World Congress of Dermatology.
Pramocaine, also known as pramoxine, is an ingredient in Gold Bond cream, some forms of calamine lotion, hemorrhoid relief products, and other OTC antipruritic agents. It’s not available in commercial patch test kits, so Dr. Abbas created a test material comprised of 2% pramocaine in petrolatum.
Seven of 232 patients (3%) referred to a university patch test clinic and who underwent testing to pramocaine along with recognized contact sensitizers showed a clear positive response to pramocaine at 96 hours. The reactions were clinically relevant, according to Dr. Abbas of the University of Alberta, Edmonton.
That 3% positive rate is in the range of published figures for such well-known problem contact sensitizers as neomycin, thiuram mix, and Balsam of Peru.
Dr. Mariam Abbas reported having no financial conflicts regarding this study, conducted free of commercial support.
AT WCD 2015
Key clinical point: Pramocaine, a topical anesthetic that’s an ingredient in many widely used OTC anti-itch products, turns out to be a common cause of contact sensitization.
Major finding: Seven of 232 patients (3%) referred to a patch test clinic showed a clinically relevant positive test to pramocaine.
Data source: A prospective study of 232 patients referred to a patch test clinic.
Disclosures: Dr. Mariam Abbas reported having no financial conflicts regarding this study, conducted free of commercial support.
Severe Anaphylaxis in Children Predicts Biphasic Reactions
Children who present with a severe anaphylactic reaction are two to three times more likely to develop a second delayed, or late-phase, reaction, Dr. Waleed Alqurashi and his colleagues reported.
Their retrospective study found that of 484 children who came to the emergency department for anaphylaxis, 15% developed a biphasic reaction. Most of these (69%) were respiratory and/or cardiovascular; half required epinephrine (Ann. Allerg. Asth. Immunol. 2015 [doi.org/10.1016/j.anai.2015.05.013]).

Dr. Alqurashi of the pediatric department at the University of Ottawa and his associates identified five independent predictors of a biphasic reaction: age 6-9 years (odds ratio, 3.6); a delay in presentation of more than 90 minutes after the onset of the initial reaction (OR, 2.58); wide pulse pressure at triage (OR, 2.92); requiring more than one epinephrine dose for initial reaction (OR, 2.7); and needing inhaled beta-agonists at the ED (OR, 2.39).
Children with these characteristics should stay for observation, rather than being discharged when their initial reactions are stabilized, the authors wrote.
“This period should be individualized and based primarily on the degree of severity of the initial anaphylactic reaction. Given the relatively high sensitivity of the present predictors, children with mild anaphylaxis who do not match any of these predictors could be considered for early disposition from the ED (less than 6 hours timed from the onset of the anaphylactic reaction) as long as an appropriate counseling has been provided,” the investigators said. Nonetheless, Dr. Alqurashi and his associates think large-scale prospective pediatric studies are necessary to validate these predictors.
Dr Alqurashi and his associates have no financial disclosures.
Children who present with a severe anaphylactic reaction are two to three times more likely to develop a second delayed, or late-phase, reaction, Dr. Waleed Alqurashi and his colleagues reported.
Their retrospective study found that of 484 children who came to the emergency department for anaphylaxis, 15% developed a biphasic reaction. Most of these (69%) were respiratory and/or cardiovascular; half required epinephrine (Ann. Allerg. Asth. Immunol. 2015 [doi.org/10.1016/j.anai.2015.05.013]).
Dr. Alqurashi of the pediatric department at the University of Ottawa and his associates identified five independent predictors of a biphasic reaction: age 6-9 years (odds ratio, 3.6); a delay in presentation of more than 90 minutes after the onset of the initial reaction (OR, 2.58); wide pulse pressure at triage (OR, 2.92); requiring more than one epinephrine dose for initial reaction (OR, 2.7); and needing inhaled beta-agonists at the ED (OR, 2.39).
Children with these characteristics should stay for observation, rather than being discharged when their initial reactions are stabilized, the authors wrote.
“This period should be individualized and based primarily on the degree of severity of the initial anaphylactic reaction. Given the relatively high sensitivity of the present predictors, children with mild anaphylaxis who do not match any of these predictors could be considered for early disposition from the ED (less than 6 hours timed from the onset of the anaphylactic reaction) as long as an appropriate counseling has been provided,” the investigators said. Nonetheless, Dr. Alqurashi and his associates think large-scale prospective pediatric studies are necessary to validate these predictors.
Dr Alqurashi and his associates have no financial disclosures.
Children who present with a severe anaphylactic reaction are two to three times more likely to develop a second delayed, or late-phase, reaction, Dr. Waleed Alqurashi and his colleagues reported.
Their retrospective study found that of 484 children who came to the emergency department for anaphylaxis, 15% developed a biphasic reaction. Most of these (69%) were respiratory and/or cardiovascular; half required epinephrine (Ann. Allerg. Asth. Immunol. 2015 [doi.org/10.1016/j.anai.2015.05.013]).
Dr. Alqurashi of the pediatric department at the University of Ottawa and his associates identified five independent predictors of a biphasic reaction: age 6-9 years (odds ratio, 3.6); a delay in presentation of more than 90 minutes after the onset of the initial reaction (OR, 2.58); wide pulse pressure at triage (OR, 2.92); requiring more than one epinephrine dose for initial reaction (OR, 2.7); and needing inhaled beta-agonists at the ED (OR, 2.39).
Children with these characteristics should stay for observation, rather than being discharged when their initial reactions are stabilized, the authors wrote.
“This period should be individualized and based primarily on the degree of severity of the initial anaphylactic reaction. Given the relatively high sensitivity of the present predictors, children with mild anaphylaxis who do not match any of these predictors could be considered for early disposition from the ED (less than 6 hours timed from the onset of the anaphylactic reaction) as long as an appropriate counseling has been provided,” the investigators said. Nonetheless, Dr. Alqurashi and his associates think large-scale prospective pediatric studies are necessary to validate these predictors.
Dr Alqurashi and his associates have no financial disclosures.
FROM THE ANNALS OF ALLERGY, ASTHMA, AND IMMUNOLOGY

Severe anaphylaxis in children predicts biphasic reactions
Children who present with a severe anaphylactic reaction are two to three times more likely to develop a second delayed, or late-phase, reaction, Dr. Waleed Alqurashi and his colleagues reported.
Their retrospective study found that of 484 children who came to the emergency department for anaphylaxis, 15% developed a biphasic reaction. Most of these (69%) were respiratory and/or cardiovascular; half required epinephrine (Ann. Allerg. Asth. Immunol. 2015 [doi.org/10.1016/j.anai.2015.05.013]).

Dr. Alqurashi of the pediatric department at the University of Ottawa and his associates identified five independent predictors of a biphasic reaction: age 6-9 years (odds ratio, 3.6); a delay in presentation of more than 90 minutes after the onset of the initial reaction (OR, 2.58); wide pulse pressure at triage (OR, 2.92); requiring more than one epinephrine dose for initial reaction (OR, 2.7); and needing inhaled beta-agonists at the ED (OR, 2.39).
Children with these characteristics should stay for observation, rather than being discharged when their initial reactions are stabilized, the authors wrote.
“This period should be individualized and based primarily on the degree of severity of the initial anaphylactic reaction. Given the relatively high sensitivity of the present predictors, children with mild anaphylaxis who do not match any of these predictors could be considered for early disposition from the ED (less than 6 hours timed from the onset of the anaphylactic reaction) as long as an appropriate counseling has been provided,” the investigators said. Nonetheless, Dr. Alqurashi and his associates think large-scale prospective pediatric studies are necessary to validate these predictors.
Dr Alqurashi and his associates have no financial disclosures.
On Twitter @Alz_Gal
Children who present with a severe anaphylactic reaction are two to three times more likely to develop a second delayed, or late-phase, reaction, Dr. Waleed Alqurashi and his colleagues reported.
Their retrospective study found that of 484 children who came to the emergency department for anaphylaxis, 15% developed a biphasic reaction. Most of these (69%) were respiratory and/or cardiovascular; half required epinephrine (Ann. Allerg. Asth. Immunol. 2015 [doi.org/10.1016/j.anai.2015.05.013]).
Dr. Alqurashi of the pediatric department at the University of Ottawa and his associates identified five independent predictors of a biphasic reaction: age 6-9 years (odds ratio, 3.6); a delay in presentation of more than 90 minutes after the onset of the initial reaction (OR, 2.58); wide pulse pressure at triage (OR, 2.92); requiring more than one epinephrine dose for initial reaction (OR, 2.7); and needing inhaled beta-agonists at the ED (OR, 2.39).
Children with these characteristics should stay for observation, rather than being discharged when their initial reactions are stabilized, the authors wrote.
“This period should be individualized and based primarily on the degree of severity of the initial anaphylactic reaction. Given the relatively high sensitivity of the present predictors, children with mild anaphylaxis who do not match any of these predictors could be considered for early disposition from the ED (less than 6 hours timed from the onset of the anaphylactic reaction) as long as an appropriate counseling has been provided,” the investigators said. Nonetheless, Dr. Alqurashi and his associates think large-scale prospective pediatric studies are necessary to validate these predictors.
Dr Alqurashi and his associates have no financial disclosures.
On Twitter @Alz_Gal
Children who present with a severe anaphylactic reaction are two to three times more likely to develop a second delayed, or late-phase, reaction, Dr. Waleed Alqurashi and his colleagues reported.
Their retrospective study found that of 484 children who came to the emergency department for anaphylaxis, 15% developed a biphasic reaction. Most of these (69%) were respiratory and/or cardiovascular; half required epinephrine (Ann. Allerg. Asth. Immunol. 2015 [doi.org/10.1016/j.anai.2015.05.013]).
Dr. Alqurashi of the pediatric department at the University of Ottawa and his associates identified five independent predictors of a biphasic reaction: age 6-9 years (odds ratio, 3.6); a delay in presentation of more than 90 minutes after the onset of the initial reaction (OR, 2.58); wide pulse pressure at triage (OR, 2.92); requiring more than one epinephrine dose for initial reaction (OR, 2.7); and needing inhaled beta-agonists at the ED (OR, 2.39).
Children with these characteristics should stay for observation, rather than being discharged when their initial reactions are stabilized, the authors wrote.
“This period should be individualized and based primarily on the degree of severity of the initial anaphylactic reaction. Given the relatively high sensitivity of the present predictors, children with mild anaphylaxis who do not match any of these predictors could be considered for early disposition from the ED (less than 6 hours timed from the onset of the anaphylactic reaction) as long as an appropriate counseling has been provided,” the investigators said. Nonetheless, Dr. Alqurashi and his associates think large-scale prospective pediatric studies are necessary to validate these predictors.
Dr Alqurashi and his associates have no financial disclosures.
On Twitter @Alz_Gal
FROM THE ANNALS OF ALLERGY, ASTHMA, AND IMMUNOLOGY
Key clinical point: In children, severe anaphylactic reactions predict biphasic reactions.
Major finding: Children who present with a severe anaphylactic reaction, or delayed treatment, were two to three times times more likely to develop a secondary reaction.
Data source: The retrospective study comprised 484 children, 15% of whom had a biphasic reaction.
Disclosures: Dr. Alqurashi and his associates have no financial disclosures.
