User login
Company withdraws application for eryaspase in ALL
ERYTECH Pharma has announced its decision to withdraw the European marketing authorization application (MAA) for eryaspase (GRASPA®) as a treatment for acute lymphoblastic leukemia (ALL).
The European Medicines Agency’s (EMA’s) Committee for Medicinal Products for Human Use (CHMP) asked for additional data on eryaspase, but ERYTECH said the time allowed by the EMA’s approval process is not sufficient to provide the data requested.
Therefore, the company decided to withdraw the MAA and resubmit it next year.
About eryaspase
Eryaspase consists of L-asparaginase encapsulated inside donor-derived red blood cells. These enzyme-loaded red blood cells function as bioreactors to eliminate circulating asparagine and “starve” cancer cells, thereby inducing their death.
Research has suggested this delivery system provides improved pharmacodynamics, protecting L-asparaginase from circulating proteolytic enzymes and preventing early liver or renal clearance.
The system also appears to reduce the risk of adverse events compared to native L-asparaginase.
The EMA and the US Food and Drug Administration have granted orphan drug designations for eryaspase for the treatment of ALL, acute myeloid
leukemia, and pancreatic cancer.
About the MAA
ERYTECH submitted an MAA for eryaspase in September 2015, based on positive results from a phase 2/3 study in which researchers compared eryaspase and native L-asparaginase in patients with relapsed and refractory ALL.
One year later (September 2016), the company received the CHMP’s Day 180 List of Outstanding Issues, which highlighted the need for additional data.
Specifically, the CHMP asked for data regarding the comparability between the old and new form of asparaginase encapsulated in eryaspase and the development of a new immunogenicity assay, as well as the pharmacodynamic effects of eryaspase.
Given the fact that the generation of these data will require more time than allowed in the EMA’s approval procedures, ERYTECH has notified the CHMP of the withdrawal of the MAA.
The company intends to resubmit the MAA in mid-2017, as soon as the newly generated data are available.
ERYTECH stressed that there have been no safety issues with eryaspase, and the withdrawal of this MAA will not affect any ongoing trials.
“We are committed to pursuing regulatory approval for GRASPA and intend to work closely with our investigators and advisors to generate the additional information requested and to resubmit an MAA next year,” said Iman El-Hariry, chief medical officer of ERYTECH.
“We believe we have generated strong clinical data in our different programs of eryaspase, and we continue to execute our plans towards making the product available to patients with aggressive forms of cancer, such as acute lymphoblastic leukemia, acute myeloid leukemia, and pancreatic cancer,” added Gil Beyen, ERYTECH’s chairman and chief executive officer. ![]()
ERYTECH Pharma has announced its decision to withdraw the European marketing authorization application (MAA) for eryaspase (GRASPA®) as a treatment for acute lymphoblastic leukemia (ALL).
The European Medicines Agency’s (EMA’s) Committee for Medicinal Products for Human Use (CHMP) asked for additional data on eryaspase, but ERYTECH said the time allowed by the EMA’s approval process is not sufficient to provide the data requested.
Therefore, the company decided to withdraw the MAA and resubmit it next year.
About eryaspase
Eryaspase consists of L-asparaginase encapsulated inside donor-derived red blood cells. These enzyme-loaded red blood cells function as bioreactors to eliminate circulating asparagine and “starve” cancer cells, thereby inducing their death.
Research has suggested this delivery system provides improved pharmacodynamics, protecting L-asparaginase from circulating proteolytic enzymes and preventing early liver or renal clearance.
The system also appears to reduce the risk of adverse events compared to native L-asparaginase.
The EMA and the US Food and Drug Administration have granted orphan drug designations for eryaspase for the treatment of ALL, acute myeloid
leukemia, and pancreatic cancer.
About the MAA
ERYTECH submitted an MAA for eryaspase in September 2015, based on positive results from a phase 2/3 study in which researchers compared eryaspase and native L-asparaginase in patients with relapsed and refractory ALL.
One year later (September 2016), the company received the CHMP’s Day 180 List of Outstanding Issues, which highlighted the need for additional data.
Specifically, the CHMP asked for data regarding the comparability between the old and new form of asparaginase encapsulated in eryaspase and the development of a new immunogenicity assay, as well as the pharmacodynamic effects of eryaspase.
Given the fact that the generation of these data will require more time than allowed in the EMA’s approval procedures, ERYTECH has notified the CHMP of the withdrawal of the MAA.
The company intends to resubmit the MAA in mid-2017, as soon as the newly generated data are available.
ERYTECH stressed that there have been no safety issues with eryaspase, and the withdrawal of this MAA will not affect any ongoing trials.
“We are committed to pursuing regulatory approval for GRASPA and intend to work closely with our investigators and advisors to generate the additional information requested and to resubmit an MAA next year,” said Iman El-Hariry, chief medical officer of ERYTECH.
“We believe we have generated strong clinical data in our different programs of eryaspase, and we continue to execute our plans towards making the product available to patients with aggressive forms of cancer, such as acute lymphoblastic leukemia, acute myeloid leukemia, and pancreatic cancer,” added Gil Beyen, ERYTECH’s chairman and chief executive officer. ![]()
ERYTECH Pharma has announced its decision to withdraw the European marketing authorization application (MAA) for eryaspase (GRASPA®) as a treatment for acute lymphoblastic leukemia (ALL).
The European Medicines Agency’s (EMA’s) Committee for Medicinal Products for Human Use (CHMP) asked for additional data on eryaspase, but ERYTECH said the time allowed by the EMA’s approval process is not sufficient to provide the data requested.
Therefore, the company decided to withdraw the MAA and resubmit it next year.
About eryaspase
Eryaspase consists of L-asparaginase encapsulated inside donor-derived red blood cells. These enzyme-loaded red blood cells function as bioreactors to eliminate circulating asparagine and “starve” cancer cells, thereby inducing their death.
Research has suggested this delivery system provides improved pharmacodynamics, protecting L-asparaginase from circulating proteolytic enzymes and preventing early liver or renal clearance.
The system also appears to reduce the risk of adverse events compared to native L-asparaginase.
The EMA and the US Food and Drug Administration have granted orphan drug designations for eryaspase for the treatment of ALL, acute myeloid
leukemia, and pancreatic cancer.
About the MAA
ERYTECH submitted an MAA for eryaspase in September 2015, based on positive results from a phase 2/3 study in which researchers compared eryaspase and native L-asparaginase in patients with relapsed and refractory ALL.
One year later (September 2016), the company received the CHMP’s Day 180 List of Outstanding Issues, which highlighted the need for additional data.
Specifically, the CHMP asked for data regarding the comparability between the old and new form of asparaginase encapsulated in eryaspase and the development of a new immunogenicity assay, as well as the pharmacodynamic effects of eryaspase.
Given the fact that the generation of these data will require more time than allowed in the EMA’s approval procedures, ERYTECH has notified the CHMP of the withdrawal of the MAA.
The company intends to resubmit the MAA in mid-2017, as soon as the newly generated data are available.
ERYTECH stressed that there have been no safety issues with eryaspase, and the withdrawal of this MAA will not affect any ongoing trials.
“We are committed to pursuing regulatory approval for GRASPA and intend to work closely with our investigators and advisors to generate the additional information requested and to resubmit an MAA next year,” said Iman El-Hariry, chief medical officer of ERYTECH.
“We believe we have generated strong clinical data in our different programs of eryaspase, and we continue to execute our plans towards making the product available to patients with aggressive forms of cancer, such as acute lymphoblastic leukemia, acute myeloid leukemia, and pancreatic cancer,” added Gil Beyen, ERYTECH’s chairman and chief executive officer. ![]()
FDA grants priority review for midostaurin
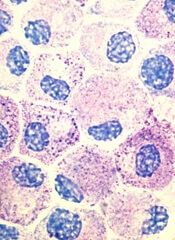
The US Food and Drug Administration (FDA) has granted priority review for the new drug application for midostaurin (PKC412) as a treatment for advanced systemic mastocytosis (SM) and newly diagnosed, FLT3-mutated acute myeloid leukemia (AML).
The FDA has also accepted for review the premarket approval application for the midostaurin FLT3 companion diagnostic, which is designed to help identify patients who may have a FLT3 mutation and could potentially benefit from treatment with midostaurin.
Midostaurin is being developed by Novartis. The companion diagnostic is being developed by Novartis and Invivoscribe Technologies, Inc.
About priority review
The FDA grants priority review to applications for therapies that may provide significant improvements in the treatment, diagnosis, or prevention of serious conditions.
The agency’s goal is to take action on a priority review application within 6 months of receiving it. The goal in the standard review process is to take action within 10 months.
About midostaurin
Midostaurin is an oral, multi-targeted kinase inhibitor. The drug was granted breakthrough therapy designation by the FDA earlier this year for newly diagnosed, FLT3-mutated AML.
According to Novartis, the new drug application submission for midostaurin includes data from the largest clinical trials conducted to date in advanced SM and newly diagnosed, FLT3-mutated AML.
Midostaurin in AML
In the phase 3 RATIFY trial, researchers compared midostaurin plus standard chemotherapy to placebo plus standard chemotherapy in adults younger than 60 with FLT3-mutated AML. Results from this trial were presented at the 2015 ASH Annual Meeting.
Patients in the midostaurin arm experienced a statistically significant improvement in overall survival, with a 23% reduction in risk of death compared to the placebo arm (hazard ratio=0.77, P=0.0074).
There was no significant difference in the overall rate of grade 3 or higher hematologic and non-hematologic adverse events in midostaurin arm and the placebo arm. Similarly, there was no significant difference in treatment-related deaths between the arms.
Midostaurin in SM
Data from the phase 2 study of midostaurin in patients with advanced SM were published in NEJM in June.
The drug produced a 60% overall response rate, and the median duration of response was 24.1 months.
Fifty-six percent of patients required dose reductions due to toxic effects, but 32% of these patients were able to return to the starting dose of midostaurin.
Access to midostaurin
Since midostaurin remains investigational, both within the US and globally, Novartis opened a Global Individual Patient Program (compassionate use program) and, in the US, an Expanded Treatment Protocol, to provide access to midostaurin for eligible patients with newly diagnosed AML and advanced SM.
Physicians who want to request midostaurin for eligible patients can contact a Novartis medical representative in their respective countries. In the US, physicians can call 1-888-NOW-NOVA (1-888-669-6682) for more information. ![]()
The US Food and Drug Administration (FDA) has granted priority review for the new drug application for midostaurin (PKC412) as a treatment for advanced systemic mastocytosis (SM) and newly diagnosed, FLT3-mutated acute myeloid leukemia (AML).
The FDA has also accepted for review the premarket approval application for the midostaurin FLT3 companion diagnostic, which is designed to help identify patients who may have a FLT3 mutation and could potentially benefit from treatment with midostaurin.
Midostaurin is being developed by Novartis. The companion diagnostic is being developed by Novartis and Invivoscribe Technologies, Inc.
About priority review
The FDA grants priority review to applications for therapies that may provide significant improvements in the treatment, diagnosis, or prevention of serious conditions.
The agency’s goal is to take action on a priority review application within 6 months of receiving it. The goal in the standard review process is to take action within 10 months.
About midostaurin
Midostaurin is an oral, multi-targeted kinase inhibitor. The drug was granted breakthrough therapy designation by the FDA earlier this year for newly diagnosed, FLT3-mutated AML.
According to Novartis, the new drug application submission for midostaurin includes data from the largest clinical trials conducted to date in advanced SM and newly diagnosed, FLT3-mutated AML.
Midostaurin in AML
In the phase 3 RATIFY trial, researchers compared midostaurin plus standard chemotherapy to placebo plus standard chemotherapy in adults younger than 60 with FLT3-mutated AML. Results from this trial were presented at the 2015 ASH Annual Meeting.
Patients in the midostaurin arm experienced a statistically significant improvement in overall survival, with a 23% reduction in risk of death compared to the placebo arm (hazard ratio=0.77, P=0.0074).
There was no significant difference in the overall rate of grade 3 or higher hematologic and non-hematologic adverse events in midostaurin arm and the placebo arm. Similarly, there was no significant difference in treatment-related deaths between the arms.
Midostaurin in SM
Data from the phase 2 study of midostaurin in patients with advanced SM were published in NEJM in June.
The drug produced a 60% overall response rate, and the median duration of response was 24.1 months.
Fifty-six percent of patients required dose reductions due to toxic effects, but 32% of these patients were able to return to the starting dose of midostaurin.
Access to midostaurin
Since midostaurin remains investigational, both within the US and globally, Novartis opened a Global Individual Patient Program (compassionate use program) and, in the US, an Expanded Treatment Protocol, to provide access to midostaurin for eligible patients with newly diagnosed AML and advanced SM.
Physicians who want to request midostaurin for eligible patients can contact a Novartis medical representative in their respective countries. In the US, physicians can call 1-888-NOW-NOVA (1-888-669-6682) for more information. ![]()
The US Food and Drug Administration (FDA) has granted priority review for the new drug application for midostaurin (PKC412) as a treatment for advanced systemic mastocytosis (SM) and newly diagnosed, FLT3-mutated acute myeloid leukemia (AML).
The FDA has also accepted for review the premarket approval application for the midostaurin FLT3 companion diagnostic, which is designed to help identify patients who may have a FLT3 mutation and could potentially benefit from treatment with midostaurin.
Midostaurin is being developed by Novartis. The companion diagnostic is being developed by Novartis and Invivoscribe Technologies, Inc.
About priority review
The FDA grants priority review to applications for therapies that may provide significant improvements in the treatment, diagnosis, or prevention of serious conditions.
The agency’s goal is to take action on a priority review application within 6 months of receiving it. The goal in the standard review process is to take action within 10 months.
About midostaurin
Midostaurin is an oral, multi-targeted kinase inhibitor. The drug was granted breakthrough therapy designation by the FDA earlier this year for newly diagnosed, FLT3-mutated AML.
According to Novartis, the new drug application submission for midostaurin includes data from the largest clinical trials conducted to date in advanced SM and newly diagnosed, FLT3-mutated AML.
Midostaurin in AML
In the phase 3 RATIFY trial, researchers compared midostaurin plus standard chemotherapy to placebo plus standard chemotherapy in adults younger than 60 with FLT3-mutated AML. Results from this trial were presented at the 2015 ASH Annual Meeting.
Patients in the midostaurin arm experienced a statistically significant improvement in overall survival, with a 23% reduction in risk of death compared to the placebo arm (hazard ratio=0.77, P=0.0074).
There was no significant difference in the overall rate of grade 3 or higher hematologic and non-hematologic adverse events in midostaurin arm and the placebo arm. Similarly, there was no significant difference in treatment-related deaths between the arms.
Midostaurin in SM
Data from the phase 2 study of midostaurin in patients with advanced SM were published in NEJM in June.
The drug produced a 60% overall response rate, and the median duration of response was 24.1 months.
Fifty-six percent of patients required dose reductions due to toxic effects, but 32% of these patients were able to return to the starting dose of midostaurin.
Access to midostaurin
Since midostaurin remains investigational, both within the US and globally, Novartis opened a Global Individual Patient Program (compassionate use program) and, in the US, an Expanded Treatment Protocol, to provide access to midostaurin for eligible patients with newly diagnosed AML and advanced SM.
Physicians who want to request midostaurin for eligible patients can contact a Novartis medical representative in their respective countries. In the US, physicians can call 1-888-NOW-NOVA (1-888-669-6682) for more information. ![]()
EMA recommends orphan status for drug in AML

The European Medicines Agency’s (EMA) Committee for Orphan Medicinal Products (COMP) has recommended that BP1001 receive orphan designation as a treatment for acute myeloid leukemia (AML).
BP1001 (liposomal Grb2 antisense) is a neutral-charge, liposome-incorporated, antisense drug designed to inhibit protein synthesis of growth factor receptor bound protein 2 (Grb2).
BP1001 is being developed by Bio-Path Holdings, Inc.
According to Bio-Path, inhibition of Grb2 by BP1001 represents a significant advance in treating cancers with activated tyrosine kinases using a target not druggable with small molecule inhibitors.
Research has suggested that Grb2 plays an essential role in cancer cell activation via the RAS pathway. Grb2 bridges signals between activated and mutated tyrosine kinases, such as Flt3, c-Kit, and Bcr-Abl, and the Ras pathway, leading to activation of the ERK and AKT proteins.
About orphan designation
The EMA’s COMP adopts an opinion on the granting of orphan drug designation, and that opinion is submitted to the European Commission for a final decision. The European Commission typically makes a decision within 30 days.
Orphan designation provides regulatory and financial incentives for companies to develop and market therapies that treat life-threatening or chronically debilitating conditions affecting no more than 5 in 10,000 people in the European Union, and where no satisfactory treatment is available.
Orphan designation provides a 10-year period of marketing exclusivity if the drug receives regulatory approval. The designation also provides incentives for companies seeking protocol assistance from the EMA during the product development phase and direct access to the centralized authorization procedure.
Trials of BP1001
Bio-Path has completed a phase 1 trial of BP1001 in patients with relapsed/refractory AML, chronic myeloid leukemia, and myelodysplastic syndromes.
The company has also completed the safety segment of a phase 2 trial in which BP1001 is being investigated in combination with low-dose ara-C to treat AML.
Bio-Path recently released data from these studies.
The phase 1 study included patients who had received an average of 6 prior therapies.
The patients received 8 doses of BP1001 over 4 weeks, escalating to a maximum dose of 90 mg/m2. There were no dose-limiting toxicities, and Bio-Path said the drug was well tolerated.
Of the 18 evaluable patients with circulating blasts, 83% responded to BP1001. The average reduction in circulating blasts was 67%.
The phase 2 trial included patients with relapsed/refractory AML. There were 3 evaluable patients in each of 2 dosing cohorts—60 mg/m2 and 90 mg/m2. Patients received BP1001 twice a week for 4 weeks.
Five of the patients responded—3 with a complete response and 2 with a partial response. There were no adverse events attributed to BP1001, and the maximum-tolerated dose was not reached. ![]()
The European Medicines Agency’s (EMA) Committee for Orphan Medicinal Products (COMP) has recommended that BP1001 receive orphan designation as a treatment for acute myeloid leukemia (AML).
BP1001 (liposomal Grb2 antisense) is a neutral-charge, liposome-incorporated, antisense drug designed to inhibit protein synthesis of growth factor receptor bound protein 2 (Grb2).
BP1001 is being developed by Bio-Path Holdings, Inc.
According to Bio-Path, inhibition of Grb2 by BP1001 represents a significant advance in treating cancers with activated tyrosine kinases using a target not druggable with small molecule inhibitors.
Research has suggested that Grb2 plays an essential role in cancer cell activation via the RAS pathway. Grb2 bridges signals between activated and mutated tyrosine kinases, such as Flt3, c-Kit, and Bcr-Abl, and the Ras pathway, leading to activation of the ERK and AKT proteins.
About orphan designation
The EMA’s COMP adopts an opinion on the granting of orphan drug designation, and that opinion is submitted to the European Commission for a final decision. The European Commission typically makes a decision within 30 days.
Orphan designation provides regulatory and financial incentives for companies to develop and market therapies that treat life-threatening or chronically debilitating conditions affecting no more than 5 in 10,000 people in the European Union, and where no satisfactory treatment is available.
Orphan designation provides a 10-year period of marketing exclusivity if the drug receives regulatory approval. The designation also provides incentives for companies seeking protocol assistance from the EMA during the product development phase and direct access to the centralized authorization procedure.
Trials of BP1001
Bio-Path has completed a phase 1 trial of BP1001 in patients with relapsed/refractory AML, chronic myeloid leukemia, and myelodysplastic syndromes.
The company has also completed the safety segment of a phase 2 trial in which BP1001 is being investigated in combination with low-dose ara-C to treat AML.
Bio-Path recently released data from these studies.
The phase 1 study included patients who had received an average of 6 prior therapies.
The patients received 8 doses of BP1001 over 4 weeks, escalating to a maximum dose of 90 mg/m2. There were no dose-limiting toxicities, and Bio-Path said the drug was well tolerated.
Of the 18 evaluable patients with circulating blasts, 83% responded to BP1001. The average reduction in circulating blasts was 67%.
The phase 2 trial included patients with relapsed/refractory AML. There were 3 evaluable patients in each of 2 dosing cohorts—60 mg/m2 and 90 mg/m2. Patients received BP1001 twice a week for 4 weeks.
Five of the patients responded—3 with a complete response and 2 with a partial response. There were no adverse events attributed to BP1001, and the maximum-tolerated dose was not reached. ![]()
The European Medicines Agency’s (EMA) Committee for Orphan Medicinal Products (COMP) has recommended that BP1001 receive orphan designation as a treatment for acute myeloid leukemia (AML).
BP1001 (liposomal Grb2 antisense) is a neutral-charge, liposome-incorporated, antisense drug designed to inhibit protein synthesis of growth factor receptor bound protein 2 (Grb2).
BP1001 is being developed by Bio-Path Holdings, Inc.
According to Bio-Path, inhibition of Grb2 by BP1001 represents a significant advance in treating cancers with activated tyrosine kinases using a target not druggable with small molecule inhibitors.
Research has suggested that Grb2 plays an essential role in cancer cell activation via the RAS pathway. Grb2 bridges signals between activated and mutated tyrosine kinases, such as Flt3, c-Kit, and Bcr-Abl, and the Ras pathway, leading to activation of the ERK and AKT proteins.
About orphan designation
The EMA’s COMP adopts an opinion on the granting of orphan drug designation, and that opinion is submitted to the European Commission for a final decision. The European Commission typically makes a decision within 30 days.
Orphan designation provides regulatory and financial incentives for companies to develop and market therapies that treat life-threatening or chronically debilitating conditions affecting no more than 5 in 10,000 people in the European Union, and where no satisfactory treatment is available.
Orphan designation provides a 10-year period of marketing exclusivity if the drug receives regulatory approval. The designation also provides incentives for companies seeking protocol assistance from the EMA during the product development phase and direct access to the centralized authorization procedure.
Trials of BP1001
Bio-Path has completed a phase 1 trial of BP1001 in patients with relapsed/refractory AML, chronic myeloid leukemia, and myelodysplastic syndromes.
The company has also completed the safety segment of a phase 2 trial in which BP1001 is being investigated in combination with low-dose ara-C to treat AML.
Bio-Path recently released data from these studies.
The phase 1 study included patients who had received an average of 6 prior therapies.
The patients received 8 doses of BP1001 over 4 weeks, escalating to a maximum dose of 90 mg/m2. There were no dose-limiting toxicities, and Bio-Path said the drug was well tolerated.
Of the 18 evaluable patients with circulating blasts, 83% responded to BP1001. The average reduction in circulating blasts was 67%.
The phase 2 trial included patients with relapsed/refractory AML. There were 3 evaluable patients in each of 2 dosing cohorts—60 mg/m2 and 90 mg/m2. Patients received BP1001 twice a week for 4 weeks.
Five of the patients responded—3 with a complete response and 2 with a partial response. There were no adverse events attributed to BP1001, and the maximum-tolerated dose was not reached. ![]()
NCCN: Deliver vincristine by mini IV drip bag
Always dilute chemotherapy agent vincristine and administer it by mini IV-drip bag, instead of syringe, urges the National Comprehensive Cancer Network in a new campaign.
The goal of “Just Bag It” is to prevent a rare but uniformly fatal medical error – administering vincristine to the spinal fluid. When syringes are side by side – one with vincristine for IV push, another with a chemotherapeutic agent meant for push into the spinal fluid – it is just too easy to make a mistake. When administered intrathecally, vincristine causes ascending paralysis, neurological defects, and eventually death.
Despite all the warning labels and checks, “this still happens,” Marc Stewart, MD, cochair of the National Comprehensive Cancer Network (NCCN) Best Practices Committee, as well as medical director of the Seattle Cancer Care Alliance and professor of medicine at the University of Washington, said at a press conference.

Mini IV-drip bag administration will make it “virtually impossible. No physician would hook the bag up to a needle in someone’s spine” and even if they did, there wouldn’t be enough pressure in the bag to push vincristine in, he said.
The group has encouraged drip-bag delivery of vincristine for years, but only about half of hospitals have adopted the policy. The mistake happens so rarely – about 125 cases since the 1960s – “that the motivation for change is just not there.” Until somebody like NCCN calls it out in a high-profile campaign, “it’s not high on the radar screen,” Dr. Stewart said. It should be a relatively easy fix because bagging vincristine is not more costly. In general, the cost difference versus syringe “is going to be pennies,” he said.
“We challenge all medical centers, hospitals, and oncology practices around the nation and the world to implement this medication safety policy so this error never occurs again,” NCCN Chief Executive Officer Robert Carlson, MD, said in a press release. A medical oncologist, he witnessed the death of a 21-year-old patient after an intrathecal vincristine injection in 2005.
“Some health care providers may associate the use of an IV bag with a heightened risk of extravasation, but research shows that the risk of extravasation is extremely low (less than 0.05%) regardless of how vincristine is administered,” the press release noted.
Vincristine is widely used in treating patients with leukemia or lymphoma.
The safety of intravenous administration of vincristine has been a long-standing concern for anyone who participates in the management of patients with hematologic malignancies. As we all know, accidental intrathecal administration of vincristine is uniformly fatal.

At many centers, including ours, policies related to intravenous infusion of vesicants via a peripheral line have made the implementation of the safety recommendations difficult. It is not surprising that only 50% of hospitals surveyed by NCCN have fully implemented the mini-bag recommendation given the concern for extravasation. However, the newest ONS guidelines for vesicant administration allow for short-term infusions via a peripheral line. For our center, this support has been instrumental in allowing us to move to a practice with the recommended mini-bags. The NCCN “Just Bag It” campaign will likely help to move institutions such as ours to be in compliance with this important safety initiative.
Donna Capozzi, PharmD, is associate director of ambulatory services in the department of pharmacy at the Hospital of the University of Pennsylvania Perelman Center for Advanced Medicine in Philadelphia. She is on the editorial advisory board of Hematology News, a publication of this news company.
The safety of intravenous administration of vincristine has been a long-standing concern for anyone who participates in the management of patients with hematologic malignancies. As we all know, accidental intrathecal administration of vincristine is uniformly fatal.
At many centers, including ours, policies related to intravenous infusion of vesicants via a peripheral line have made the implementation of the safety recommendations difficult. It is not surprising that only 50% of hospitals surveyed by NCCN have fully implemented the mini-bag recommendation given the concern for extravasation. However, the newest ONS guidelines for vesicant administration allow for short-term infusions via a peripheral line. For our center, this support has been instrumental in allowing us to move to a practice with the recommended mini-bags. The NCCN “Just Bag It” campaign will likely help to move institutions such as ours to be in compliance with this important safety initiative.
Donna Capozzi, PharmD, is associate director of ambulatory services in the department of pharmacy at the Hospital of the University of Pennsylvania Perelman Center for Advanced Medicine in Philadelphia. She is on the editorial advisory board of Hematology News, a publication of this news company.
The safety of intravenous administration of vincristine has been a long-standing concern for anyone who participates in the management of patients with hematologic malignancies. As we all know, accidental intrathecal administration of vincristine is uniformly fatal.
At many centers, including ours, policies related to intravenous infusion of vesicants via a peripheral line have made the implementation of the safety recommendations difficult. It is not surprising that only 50% of hospitals surveyed by NCCN have fully implemented the mini-bag recommendation given the concern for extravasation. However, the newest ONS guidelines for vesicant administration allow for short-term infusions via a peripheral line. For our center, this support has been instrumental in allowing us to move to a practice with the recommended mini-bags. The NCCN “Just Bag It” campaign will likely help to move institutions such as ours to be in compliance with this important safety initiative.
Donna Capozzi, PharmD, is associate director of ambulatory services in the department of pharmacy at the Hospital of the University of Pennsylvania Perelman Center for Advanced Medicine in Philadelphia. She is on the editorial advisory board of Hematology News, a publication of this news company.
Always dilute chemotherapy agent vincristine and administer it by mini IV-drip bag, instead of syringe, urges the National Comprehensive Cancer Network in a new campaign.
The goal of “Just Bag It” is to prevent a rare but uniformly fatal medical error – administering vincristine to the spinal fluid. When syringes are side by side – one with vincristine for IV push, another with a chemotherapeutic agent meant for push into the spinal fluid – it is just too easy to make a mistake. When administered intrathecally, vincristine causes ascending paralysis, neurological defects, and eventually death.
Despite all the warning labels and checks, “this still happens,” Marc Stewart, MD, cochair of the National Comprehensive Cancer Network (NCCN) Best Practices Committee, as well as medical director of the Seattle Cancer Care Alliance and professor of medicine at the University of Washington, said at a press conference.
Mini IV-drip bag administration will make it “virtually impossible. No physician would hook the bag up to a needle in someone’s spine” and even if they did, there wouldn’t be enough pressure in the bag to push vincristine in, he said.
The group has encouraged drip-bag delivery of vincristine for years, but only about half of hospitals have adopted the policy. The mistake happens so rarely – about 125 cases since the 1960s – “that the motivation for change is just not there.” Until somebody like NCCN calls it out in a high-profile campaign, “it’s not high on the radar screen,” Dr. Stewart said. It should be a relatively easy fix because bagging vincristine is not more costly. In general, the cost difference versus syringe “is going to be pennies,” he said.
“We challenge all medical centers, hospitals, and oncology practices around the nation and the world to implement this medication safety policy so this error never occurs again,” NCCN Chief Executive Officer Robert Carlson, MD, said in a press release. A medical oncologist, he witnessed the death of a 21-year-old patient after an intrathecal vincristine injection in 2005.
“Some health care providers may associate the use of an IV bag with a heightened risk of extravasation, but research shows that the risk of extravasation is extremely low (less than 0.05%) regardless of how vincristine is administered,” the press release noted.
Vincristine is widely used in treating patients with leukemia or lymphoma.
Always dilute chemotherapy agent vincristine and administer it by mini IV-drip bag, instead of syringe, urges the National Comprehensive Cancer Network in a new campaign.
The goal of “Just Bag It” is to prevent a rare but uniformly fatal medical error – administering vincristine to the spinal fluid. When syringes are side by side – one with vincristine for IV push, another with a chemotherapeutic agent meant for push into the spinal fluid – it is just too easy to make a mistake. When administered intrathecally, vincristine causes ascending paralysis, neurological defects, and eventually death.
Despite all the warning labels and checks, “this still happens,” Marc Stewart, MD, cochair of the National Comprehensive Cancer Network (NCCN) Best Practices Committee, as well as medical director of the Seattle Cancer Care Alliance and professor of medicine at the University of Washington, said at a press conference.
Mini IV-drip bag administration will make it “virtually impossible. No physician would hook the bag up to a needle in someone’s spine” and even if they did, there wouldn’t be enough pressure in the bag to push vincristine in, he said.
The group has encouraged drip-bag delivery of vincristine for years, but only about half of hospitals have adopted the policy. The mistake happens so rarely – about 125 cases since the 1960s – “that the motivation for change is just not there.” Until somebody like NCCN calls it out in a high-profile campaign, “it’s not high on the radar screen,” Dr. Stewart said. It should be a relatively easy fix because bagging vincristine is not more costly. In general, the cost difference versus syringe “is going to be pennies,” he said.
“We challenge all medical centers, hospitals, and oncology practices around the nation and the world to implement this medication safety policy so this error never occurs again,” NCCN Chief Executive Officer Robert Carlson, MD, said in a press release. A medical oncologist, he witnessed the death of a 21-year-old patient after an intrathecal vincristine injection in 2005.
“Some health care providers may associate the use of an IV bag with a heightened risk of extravasation, but research shows that the risk of extravasation is extremely low (less than 0.05%) regardless of how vincristine is administered,” the press release noted.
Vincristine is widely used in treating patients with leukemia or lymphoma.
CHMP recommends expanding use of drug in CLL

Photo courtesy of GSK
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended expanding the approved indication for ofatumumab (Arzerra®).
The CHMP is recommending the drug be approved for use in combination with fludarabine and cyclophosphamide to treat adults with relapsed chronic lymphocytic leukemia (CLL).
The CHMP’s recommendation will be reviewed by the European Commission.
A final decision is expected in the coming months.
Ofatumumab is a monoclonal antibody designed to target CD20. The drug is marketed under a collaboration agreement between Genmab and Novartis.
The European Commission has already approved ofatumumab for the following indications:
- As a single-agent to treat CLL patients who are refractory to fludarabine and alemtuzumab
- For use in combination with chlorambucil or bendamustine in CLL patients who
have not received prior therapy and are not eligible for
fludarabine-based therapy.
COMPLEMENT 2 trial
The CHMP’s recommendation to approve ofatumumab in combination with fludarabine and cyclophosphamide was based on results from the phase 3 COMPLEMENT 2 study. Novartis reported top-line results from this study last April.
The trial enrolled 365 patients with relapsed CLL. The patients were randomized 1:1 to receive up to 6 cycles of ofatumumab in combination with fludarabine and cyclophosphamide or up to 6 cycles of fludarabine and cyclophosphamide alone.
The primary endpoint was progression-free survival, as assessed by an independent review committee.
The median progression-free survival was 28.9 months for patients receiving ofatumumab plus fludarabine and cyclophosphamide, compared to 18.8 months for patients receiving fludarabine and cyclophosphamide alone (hazard ratio=0.67, P=0.0032).
Novartis said the safety profile observed in this study was consistent with other trials of ofatumumab, and no new safety signals were observed. ![]()
Photo courtesy of GSK
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended expanding the approved indication for ofatumumab (Arzerra®).
The CHMP is recommending the drug be approved for use in combination with fludarabine and cyclophosphamide to treat adults with relapsed chronic lymphocytic leukemia (CLL).
The CHMP’s recommendation will be reviewed by the European Commission.
A final decision is expected in the coming months.
Ofatumumab is a monoclonal antibody designed to target CD20. The drug is marketed under a collaboration agreement between Genmab and Novartis.
The European Commission has already approved ofatumumab for the following indications:
- As a single-agent to treat CLL patients who are refractory to fludarabine and alemtuzumab
- For use in combination with chlorambucil or bendamustine in CLL patients who
have not received prior therapy and are not eligible for
fludarabine-based therapy.
COMPLEMENT 2 trial
The CHMP’s recommendation to approve ofatumumab in combination with fludarabine and cyclophosphamide was based on results from the phase 3 COMPLEMENT 2 study. Novartis reported top-line results from this study last April.
The trial enrolled 365 patients with relapsed CLL. The patients were randomized 1:1 to receive up to 6 cycles of ofatumumab in combination with fludarabine and cyclophosphamide or up to 6 cycles of fludarabine and cyclophosphamide alone.
The primary endpoint was progression-free survival, as assessed by an independent review committee.
The median progression-free survival was 28.9 months for patients receiving ofatumumab plus fludarabine and cyclophosphamide, compared to 18.8 months for patients receiving fludarabine and cyclophosphamide alone (hazard ratio=0.67, P=0.0032).
Novartis said the safety profile observed in this study was consistent with other trials of ofatumumab, and no new safety signals were observed. ![]()
Photo courtesy of GSK
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended expanding the approved indication for ofatumumab (Arzerra®).
The CHMP is recommending the drug be approved for use in combination with fludarabine and cyclophosphamide to treat adults with relapsed chronic lymphocytic leukemia (CLL).
The CHMP’s recommendation will be reviewed by the European Commission.
A final decision is expected in the coming months.
Ofatumumab is a monoclonal antibody designed to target CD20. The drug is marketed under a collaboration agreement between Genmab and Novartis.
The European Commission has already approved ofatumumab for the following indications:
- As a single-agent to treat CLL patients who are refractory to fludarabine and alemtuzumab
- For use in combination with chlorambucil or bendamustine in CLL patients who
have not received prior therapy and are not eligible for
fludarabine-based therapy.
COMPLEMENT 2 trial
The CHMP’s recommendation to approve ofatumumab in combination with fludarabine and cyclophosphamide was based on results from the phase 3 COMPLEMENT 2 study. Novartis reported top-line results from this study last April.
The trial enrolled 365 patients with relapsed CLL. The patients were randomized 1:1 to receive up to 6 cycles of ofatumumab in combination with fludarabine and cyclophosphamide or up to 6 cycles of fludarabine and cyclophosphamide alone.
The primary endpoint was progression-free survival, as assessed by an independent review committee.
The median progression-free survival was 28.9 months for patients receiving ofatumumab plus fludarabine and cyclophosphamide, compared to 18.8 months for patients receiving fludarabine and cyclophosphamide alone (hazard ratio=0.67, P=0.0032).
Novartis said the safety profile observed in this study was consistent with other trials of ofatumumab, and no new safety signals were observed. ![]()
NCCN issues challenge to ‘bag’ vincristine
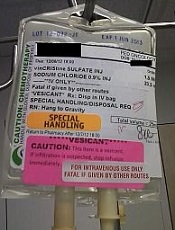
Photo courtesy of ISMP
PHILADELPHIA—To ensure proper administration of vincristine, the National Comprehensive Cancer Network (NCCN) has issued a challenge to hospitals, medical centers, and oncology practices as part of its “Just Bag It!” campaign.
Vincristine—the “O” for Oncovin in the CHOP regimen—is widely used to treat patients with leukemia or lymphoma.
It is considered an important chemotherapeutic agent. However, if administered incorrectly, vincristine is uniformly fatal, usually within a week, according to the NCCN.
Vincristine is highly neurotoxic and should always be administered intravenously. If it is mistakenly given intrathecally along with other chemotherapy drugs, it causes ascending paralysis, neurological defects, and death.
Therefore, the NCCN recommends always diluting and administering vincristine in a mini IV-drip bag, never through a syringe.
This precaution decreases the chances of improper dosage and makes it impossible to accidentally administer vincristine into the spinal fluid.
The NCCN initiated the safe-handling campaign in response to the death 11 years ago of a 21-year-old patient who received vincristine incorrectly administered into his spinal fluid. He was referred to Robert W. Carlson, MD, NCCN’s chief executive officer, who, at the time, was at Stanford Hospital, not the hospital where the error occurred.
The patient, Christopher Wibeto, had a “likely curable” non-Hodgkin lymphoma and died 4 days later.
“When I first met Christopher, he was doing well,” Dr Carlson said. “He was a delightful young gentleman, very articulate. He was funny. Even in the ICU, he had me chuckling and laughing at what he was saying.”
“But we knew that the medical error would almost certainly lead to his death. Shortly thereafter, I met his parents, Debra and Robin, . . . and had to tell them what the consequences of that medical error were likely to be. And they joined me in Christopher’s room while we talked with him about what the consequences of that medical error were likely to be.”
Making the situation even more painful, Dr Carlson, at that time, was the father of a young son who is now almost the age Christopher was then.
Dr Carlson said he realized that “we needed to come up with systems to assure that this did not happen, not today or tomorrow or ever again.”
Motivated by the tragedy, Dr Carlson spearheaded a national effort to address this mistake when he joined NCCN as CEO, enlisting the help of NCCN’s Best Practices Committee.
The NCCN developed and issued guidelines, and all 27 member institutions have adopted policies in line with the guidelines.
The Institute for Safe Medication Practices (ISMP) has undertaken efforts over more than a decade to implement procedures for safe vincristine administration.
ISMP conducted surveys and follow-up self-assessments regarding use of IV bags for vincristine at oncology practice sites. They found that only about half the institutions surveyed dilute IV vincristine for administration in a small-volume bag.
Some practitioners associate the use of an IV bag with an increased risk of extravasation (when the chemotherapy agent leaks into the tissue surrounding the administration site). Research shows, however, that the risk of extravasation is extremely low (less than 0.5%), regardless of how vincristine is administered.
And cost is not an issue when implementing the mini-bag policy, according to the president of ISMP, Michael R. Cohen, RPh.
“It cost a few pennies more,” he said. “And I mean pennies. I think probably what is an issue is just the age-old habit of putting vincristine in a syringe and being able to change that habit.”
Since the introduction of vincristine use in the 1960s, 125 documented cases of accidental death in the US and abroad have been reported. While the error is relatively rare, it is preventable and unique in its level of mortality.
“It’s hard to understand why this idea of ‘Just Bag It’ hasn’t permeated healthcare at this point,” Cohen said. “Because it is a sure-fire way to prevent this type of error.”
The ISMP, the Joint Commission, the World Health Organization, and the Oncology Nursing Society also recommend the bag-it policy. ![]()
Photo courtesy of ISMP
PHILADELPHIA—To ensure proper administration of vincristine, the National Comprehensive Cancer Network (NCCN) has issued a challenge to hospitals, medical centers, and oncology practices as part of its “Just Bag It!” campaign.
Vincristine—the “O” for Oncovin in the CHOP regimen—is widely used to treat patients with leukemia or lymphoma.
It is considered an important chemotherapeutic agent. However, if administered incorrectly, vincristine is uniformly fatal, usually within a week, according to the NCCN.
Vincristine is highly neurotoxic and should always be administered intravenously. If it is mistakenly given intrathecally along with other chemotherapy drugs, it causes ascending paralysis, neurological defects, and death.
Therefore, the NCCN recommends always diluting and administering vincristine in a mini IV-drip bag, never through a syringe.
This precaution decreases the chances of improper dosage and makes it impossible to accidentally administer vincristine into the spinal fluid.
The NCCN initiated the safe-handling campaign in response to the death 11 years ago of a 21-year-old patient who received vincristine incorrectly administered into his spinal fluid. He was referred to Robert W. Carlson, MD, NCCN’s chief executive officer, who, at the time, was at Stanford Hospital, not the hospital where the error occurred.
The patient, Christopher Wibeto, had a “likely curable” non-Hodgkin lymphoma and died 4 days later.
“When I first met Christopher, he was doing well,” Dr Carlson said. “He was a delightful young gentleman, very articulate. He was funny. Even in the ICU, he had me chuckling and laughing at what he was saying.”
“But we knew that the medical error would almost certainly lead to his death. Shortly thereafter, I met his parents, Debra and Robin, . . . and had to tell them what the consequences of that medical error were likely to be. And they joined me in Christopher’s room while we talked with him about what the consequences of that medical error were likely to be.”
Making the situation even more painful, Dr Carlson, at that time, was the father of a young son who is now almost the age Christopher was then.
Dr Carlson said he realized that “we needed to come up with systems to assure that this did not happen, not today or tomorrow or ever again.”
Motivated by the tragedy, Dr Carlson spearheaded a national effort to address this mistake when he joined NCCN as CEO, enlisting the help of NCCN’s Best Practices Committee.
The NCCN developed and issued guidelines, and all 27 member institutions have adopted policies in line with the guidelines.
The Institute for Safe Medication Practices (ISMP) has undertaken efforts over more than a decade to implement procedures for safe vincristine administration.
ISMP conducted surveys and follow-up self-assessments regarding use of IV bags for vincristine at oncology practice sites. They found that only about half the institutions surveyed dilute IV vincristine for administration in a small-volume bag.
Some practitioners associate the use of an IV bag with an increased risk of extravasation (when the chemotherapy agent leaks into the tissue surrounding the administration site). Research shows, however, that the risk of extravasation is extremely low (less than 0.5%), regardless of how vincristine is administered.
And cost is not an issue when implementing the mini-bag policy, according to the president of ISMP, Michael R. Cohen, RPh.
“It cost a few pennies more,” he said. “And I mean pennies. I think probably what is an issue is just the age-old habit of putting vincristine in a syringe and being able to change that habit.”
Since the introduction of vincristine use in the 1960s, 125 documented cases of accidental death in the US and abroad have been reported. While the error is relatively rare, it is preventable and unique in its level of mortality.
“It’s hard to understand why this idea of ‘Just Bag It’ hasn’t permeated healthcare at this point,” Cohen said. “Because it is a sure-fire way to prevent this type of error.”
The ISMP, the Joint Commission, the World Health Organization, and the Oncology Nursing Society also recommend the bag-it policy. ![]()
Photo courtesy of ISMP
PHILADELPHIA—To ensure proper administration of vincristine, the National Comprehensive Cancer Network (NCCN) has issued a challenge to hospitals, medical centers, and oncology practices as part of its “Just Bag It!” campaign.
Vincristine—the “O” for Oncovin in the CHOP regimen—is widely used to treat patients with leukemia or lymphoma.
It is considered an important chemotherapeutic agent. However, if administered incorrectly, vincristine is uniformly fatal, usually within a week, according to the NCCN.
Vincristine is highly neurotoxic and should always be administered intravenously. If it is mistakenly given intrathecally along with other chemotherapy drugs, it causes ascending paralysis, neurological defects, and death.
Therefore, the NCCN recommends always diluting and administering vincristine in a mini IV-drip bag, never through a syringe.
This precaution decreases the chances of improper dosage and makes it impossible to accidentally administer vincristine into the spinal fluid.
The NCCN initiated the safe-handling campaign in response to the death 11 years ago of a 21-year-old patient who received vincristine incorrectly administered into his spinal fluid. He was referred to Robert W. Carlson, MD, NCCN’s chief executive officer, who, at the time, was at Stanford Hospital, not the hospital where the error occurred.
The patient, Christopher Wibeto, had a “likely curable” non-Hodgkin lymphoma and died 4 days later.
“When I first met Christopher, he was doing well,” Dr Carlson said. “He was a delightful young gentleman, very articulate. He was funny. Even in the ICU, he had me chuckling and laughing at what he was saying.”
“But we knew that the medical error would almost certainly lead to his death. Shortly thereafter, I met his parents, Debra and Robin, . . . and had to tell them what the consequences of that medical error were likely to be. And they joined me in Christopher’s room while we talked with him about what the consequences of that medical error were likely to be.”
Making the situation even more painful, Dr Carlson, at that time, was the father of a young son who is now almost the age Christopher was then.
Dr Carlson said he realized that “we needed to come up with systems to assure that this did not happen, not today or tomorrow or ever again.”
Motivated by the tragedy, Dr Carlson spearheaded a national effort to address this mistake when he joined NCCN as CEO, enlisting the help of NCCN’s Best Practices Committee.
The NCCN developed and issued guidelines, and all 27 member institutions have adopted policies in line with the guidelines.
The Institute for Safe Medication Practices (ISMP) has undertaken efforts over more than a decade to implement procedures for safe vincristine administration.
ISMP conducted surveys and follow-up self-assessments regarding use of IV bags for vincristine at oncology practice sites. They found that only about half the institutions surveyed dilute IV vincristine for administration in a small-volume bag.
Some practitioners associate the use of an IV bag with an increased risk of extravasation (when the chemotherapy agent leaks into the tissue surrounding the administration site). Research shows, however, that the risk of extravasation is extremely low (less than 0.5%), regardless of how vincristine is administered.
And cost is not an issue when implementing the mini-bag policy, according to the president of ISMP, Michael R. Cohen, RPh.
“It cost a few pennies more,” he said. “And I mean pennies. I think probably what is an issue is just the age-old habit of putting vincristine in a syringe and being able to change that habit.”
Since the introduction of vincristine use in the 1960s, 125 documented cases of accidental death in the US and abroad have been reported. While the error is relatively rare, it is preventable and unique in its level of mortality.
“It’s hard to understand why this idea of ‘Just Bag It’ hasn’t permeated healthcare at this point,” Cohen said. “Because it is a sure-fire way to prevent this type of error.”
The ISMP, the Joint Commission, the World Health Organization, and the Oncology Nursing Society also recommend the bag-it policy. ![]()
Secondary reference panel expands access for CML monitoring
A cell-based BCR-ABL1 secondary reference panel traceable to the World Health Organization BCR-ABL1 International Genetic Reference Panel – with an additional MR4.5 level – provides easier access to International Scale calibration and can act as a tool for assay optimization, validation, and quality assurance for molecular monitoring in chronic myeloid leukemia patients.
Such monitoring is important to disease management, especially for decision making with respect to treatment cessation. The new secondary reference panel would allow laboratories to circumvent the oft-used and time-consuming sample exchange process with reference laboratories for International Scale (IS) calibration, Nicholas C. P. Cross, PhD, of the University of Southampton (England) and colleagues wrote in an article published in Leukemia (2016 Jun 3;30:1844-52).
“The development of BCR-ABL1 tyrosine kinase inhibitors ... has enabled progressively deeper molecular responses in CML patients undergoing tyrosine kinase inhibitor therapy,” the authors wrote, noting that deeper molecular responses are “important milestones for patients considering treatment cessation,” and that “other landmarks on the IS also represent different treatment decision thresholds and prognostic outcomes.”
For this reason, regular molecular monitoring using real-time reverse-transcription quantitative PCR (RqPCR) is recommended for optimal disease management, they said.
“As treatment decisions are directly impacted by test results, accuracy and precision of BCR-ABL1 assays across the entire measurement range is crucial for patient management, especially in patients with deep molecular responses when considering possible treatment cessation,” they wrote.
Access to the material for the WHO BCR-ABL1 reference panel (MR1-MR4), which was developed in 2010 as a primary standard for BCR-ABL1 assay IS calibration, is limited, particularly for smaller laboratories, the authors said.
The secondary reference panel they developed, however, is “traceable to and faithfully replicates the WHO panel in both raw materials (lyopholized.K562 and HL-60 cell mixes) and manufacturing process, with the addition of a MR4.5 level.” The secondary panel was calibrated to IS using digital PCR against ABL1, BCR, and GUSB as reference genes, and was successfully evaluated by 45 different BCR-ABL1 assays at 44 different clinical laboratories in a multinational evaluation study.
The MR4.5 level was added to allow for more accurate IS calibration “as CML patients reaching this deep molecular response are increasingly being considered for treatment cessation,” the authors noted.
“Quality-control assessments indicated that the secondary panel had minimal residual moisture, excellent vial-to-vial homogeneity and greater than 2.5 years of real-time stability,” they said.
Further, the panel was successfully processed by all of the laboratories, indicating that it is compatible with many different BCR-ABL1 test configurations, they added.
Of note, the number of assays that achieved good precision and sensitivity exceeded the number that achieved good IS accuracy, suggesting an unmet need for “a simple and broadly available calibration mechanism, such as this secondary panel, to ensure IS accuracy is maintained in laboratories over time,” they wrote, concluding that such a panel “can provide easier access to IS calibration, as well as act as a tool for assay optimization, validation and quality assurance.”
In a letter to the editor in regard to the findings by Cross et al., Maria Sol Ruiz of Instituto Alexander Fleming in Buenos Aires, and colleagues wrote about their own development and validation of “secondary reference materials calibrated to the IS through the WHO primary standards in order to facilitate standardization of molecular monitoring in Latin America,” (Leukemia. 2016 Aug 19. doi: 10.1038/leu.2016.197).
In their study, the letter’s authors demonstrated that secondary reference biological calibrators anchored to the WHO primary standards can decrease inter-laboratory variability.
“Our results, together with those recently reported by Cross et al., substantiate the objective initially set during the establishment of the WHO primary standards, that is, to facilitate worldwide diffusion of the IS. For the first time in Latin America, this study provides a platform on which to assess the performance of distinct clinical BCR-ABL1 tests and confirm the utility of secondary reference materials to further improve IS accuracy and inter-laboratory precision,” they wrote, noting that their efforts will continue through provision of secondary reference material to centers involved in their project, as well as to potential new participants.
“Moreover, due to its higher precision and absolute quantification capability, we are evaluating the possibility of including digital PCR as the calibration method for the future,” they said.
The study discussed in the letter to the editor was supported by a grant from Novartis to one of the authors. Novartis also paid speaking fees to some of the authors. The study by Dr. Cross et al. was also funded by Novartis and some authors are employed by Novartis. The remaining authors, including Dr. Cross, reported having no disclosures.
Harmonizing BCR-ABL1 real time quantitative PCR (RqPCR) is extremely important for precisely interpreting therapeutic response in CML patients and for being able to compare results from various laboratories.
Levels of BCR-ABL1 RNA transcripts are expected to progressively decline with successful response to tyrosine kinase inhibitor therapy. A rise in those levels indicates a loss of response to therapy and typically prompts dose modification or a change in therapy.
Initially, only a qualitative test was available and it measured only the presence or absence of the transcript. The International Standard (IS) allowed the development of a quantitative test. The test is identical to that used in the International Randomized Study of Interferons and STI571 (IRIS), which has a standard baseline of 100% of BCR-ABL1 and major molecular response is defined as a 3 log reduction relative to standard baseline or 0.1% of BCR-ABL1 IS.
However, access became limited – especially for smaller laboratories – to the material for the WHO BCR-ABL1 reference panel (MR1-MR4), a primary standard for BCR-ABL1 assay IS calibration. Calibrated, accredited, primary reference reagents for BCR-ABL1 RqPCR were often too expensive for emergent economies. In response, some laboratories developed conversion factors – a laborious process – and other alternative methods.
By using locally produced secondary cellular calibrators anchored to the WHO primary standards, the elegant work of Cross et al. and Ruiz et al. provided standardization. With this type of initiative, more regional laboratories can assess the performance of their tests and improve their accuracy. Indeed, it is of utmost importance that the exchange of reference standards and quality control samples becomes a common practice in such regions to maximize the reliability of this test. The authors of these studies have started down that path, but there is still a long way to go to achieve higher sensitivity that would permit the detection of even deeper responses and the introduction of digitalized PCR testing.
Maria de Lourdes Chauffaille, MD, and Daniella Kerbauy, MD, are with Fleury Medicina Diagnostica, Sao Paulo, Brazil. Both reported having no disclosures.
Harmonizing BCR-ABL1 real time quantitative PCR (RqPCR) is extremely important for precisely interpreting therapeutic response in CML patients and for being able to compare results from various laboratories.
Levels of BCR-ABL1 RNA transcripts are expected to progressively decline with successful response to tyrosine kinase inhibitor therapy. A rise in those levels indicates a loss of response to therapy and typically prompts dose modification or a change in therapy.
Initially, only a qualitative test was available and it measured only the presence or absence of the transcript. The International Standard (IS) allowed the development of a quantitative test. The test is identical to that used in the International Randomized Study of Interferons and STI571 (IRIS), which has a standard baseline of 100% of BCR-ABL1 and major molecular response is defined as a 3 log reduction relative to standard baseline or 0.1% of BCR-ABL1 IS.
However, access became limited – especially for smaller laboratories – to the material for the WHO BCR-ABL1 reference panel (MR1-MR4), a primary standard for BCR-ABL1 assay IS calibration. Calibrated, accredited, primary reference reagents for BCR-ABL1 RqPCR were often too expensive for emergent economies. In response, some laboratories developed conversion factors – a laborious process – and other alternative methods.
By using locally produced secondary cellular calibrators anchored to the WHO primary standards, the elegant work of Cross et al. and Ruiz et al. provided standardization. With this type of initiative, more regional laboratories can assess the performance of their tests and improve their accuracy. Indeed, it is of utmost importance that the exchange of reference standards and quality control samples becomes a common practice in such regions to maximize the reliability of this test. The authors of these studies have started down that path, but there is still a long way to go to achieve higher sensitivity that would permit the detection of even deeper responses and the introduction of digitalized PCR testing.
Maria de Lourdes Chauffaille, MD, and Daniella Kerbauy, MD, are with Fleury Medicina Diagnostica, Sao Paulo, Brazil. Both reported having no disclosures.
Harmonizing BCR-ABL1 real time quantitative PCR (RqPCR) is extremely important for precisely interpreting therapeutic response in CML patients and for being able to compare results from various laboratories.
Levels of BCR-ABL1 RNA transcripts are expected to progressively decline with successful response to tyrosine kinase inhibitor therapy. A rise in those levels indicates a loss of response to therapy and typically prompts dose modification or a change in therapy.
Initially, only a qualitative test was available and it measured only the presence or absence of the transcript. The International Standard (IS) allowed the development of a quantitative test. The test is identical to that used in the International Randomized Study of Interferons and STI571 (IRIS), which has a standard baseline of 100% of BCR-ABL1 and major molecular response is defined as a 3 log reduction relative to standard baseline or 0.1% of BCR-ABL1 IS.
However, access became limited – especially for smaller laboratories – to the material for the WHO BCR-ABL1 reference panel (MR1-MR4), a primary standard for BCR-ABL1 assay IS calibration. Calibrated, accredited, primary reference reagents for BCR-ABL1 RqPCR were often too expensive for emergent economies. In response, some laboratories developed conversion factors – a laborious process – and other alternative methods.
By using locally produced secondary cellular calibrators anchored to the WHO primary standards, the elegant work of Cross et al. and Ruiz et al. provided standardization. With this type of initiative, more regional laboratories can assess the performance of their tests and improve their accuracy. Indeed, it is of utmost importance that the exchange of reference standards and quality control samples becomes a common practice in such regions to maximize the reliability of this test. The authors of these studies have started down that path, but there is still a long way to go to achieve higher sensitivity that would permit the detection of even deeper responses and the introduction of digitalized PCR testing.
Maria de Lourdes Chauffaille, MD, and Daniella Kerbauy, MD, are with Fleury Medicina Diagnostica, Sao Paulo, Brazil. Both reported having no disclosures.
A cell-based BCR-ABL1 secondary reference panel traceable to the World Health Organization BCR-ABL1 International Genetic Reference Panel – with an additional MR4.5 level – provides easier access to International Scale calibration and can act as a tool for assay optimization, validation, and quality assurance for molecular monitoring in chronic myeloid leukemia patients.
Such monitoring is important to disease management, especially for decision making with respect to treatment cessation. The new secondary reference panel would allow laboratories to circumvent the oft-used and time-consuming sample exchange process with reference laboratories for International Scale (IS) calibration, Nicholas C. P. Cross, PhD, of the University of Southampton (England) and colleagues wrote in an article published in Leukemia (2016 Jun 3;30:1844-52).
“The development of BCR-ABL1 tyrosine kinase inhibitors ... has enabled progressively deeper molecular responses in CML patients undergoing tyrosine kinase inhibitor therapy,” the authors wrote, noting that deeper molecular responses are “important milestones for patients considering treatment cessation,” and that “other landmarks on the IS also represent different treatment decision thresholds and prognostic outcomes.”
For this reason, regular molecular monitoring using real-time reverse-transcription quantitative PCR (RqPCR) is recommended for optimal disease management, they said.
“As treatment decisions are directly impacted by test results, accuracy and precision of BCR-ABL1 assays across the entire measurement range is crucial for patient management, especially in patients with deep molecular responses when considering possible treatment cessation,” they wrote.
Access to the material for the WHO BCR-ABL1 reference panel (MR1-MR4), which was developed in 2010 as a primary standard for BCR-ABL1 assay IS calibration, is limited, particularly for smaller laboratories, the authors said.
The secondary reference panel they developed, however, is “traceable to and faithfully replicates the WHO panel in both raw materials (lyopholized.K562 and HL-60 cell mixes) and manufacturing process, with the addition of a MR4.5 level.” The secondary panel was calibrated to IS using digital PCR against ABL1, BCR, and GUSB as reference genes, and was successfully evaluated by 45 different BCR-ABL1 assays at 44 different clinical laboratories in a multinational evaluation study.
The MR4.5 level was added to allow for more accurate IS calibration “as CML patients reaching this deep molecular response are increasingly being considered for treatment cessation,” the authors noted.
“Quality-control assessments indicated that the secondary panel had minimal residual moisture, excellent vial-to-vial homogeneity and greater than 2.5 years of real-time stability,” they said.
Further, the panel was successfully processed by all of the laboratories, indicating that it is compatible with many different BCR-ABL1 test configurations, they added.
Of note, the number of assays that achieved good precision and sensitivity exceeded the number that achieved good IS accuracy, suggesting an unmet need for “a simple and broadly available calibration mechanism, such as this secondary panel, to ensure IS accuracy is maintained in laboratories over time,” they wrote, concluding that such a panel “can provide easier access to IS calibration, as well as act as a tool for assay optimization, validation and quality assurance.”
In a letter to the editor in regard to the findings by Cross et al., Maria Sol Ruiz of Instituto Alexander Fleming in Buenos Aires, and colleagues wrote about their own development and validation of “secondary reference materials calibrated to the IS through the WHO primary standards in order to facilitate standardization of molecular monitoring in Latin America,” (Leukemia. 2016 Aug 19. doi: 10.1038/leu.2016.197).
In their study, the letter’s authors demonstrated that secondary reference biological calibrators anchored to the WHO primary standards can decrease inter-laboratory variability.
“Our results, together with those recently reported by Cross et al., substantiate the objective initially set during the establishment of the WHO primary standards, that is, to facilitate worldwide diffusion of the IS. For the first time in Latin America, this study provides a platform on which to assess the performance of distinct clinical BCR-ABL1 tests and confirm the utility of secondary reference materials to further improve IS accuracy and inter-laboratory precision,” they wrote, noting that their efforts will continue through provision of secondary reference material to centers involved in their project, as well as to potential new participants.
“Moreover, due to its higher precision and absolute quantification capability, we are evaluating the possibility of including digital PCR as the calibration method for the future,” they said.
The study discussed in the letter to the editor was supported by a grant from Novartis to one of the authors. Novartis also paid speaking fees to some of the authors. The study by Dr. Cross et al. was also funded by Novartis and some authors are employed by Novartis. The remaining authors, including Dr. Cross, reported having no disclosures.
A cell-based BCR-ABL1 secondary reference panel traceable to the World Health Organization BCR-ABL1 International Genetic Reference Panel – with an additional MR4.5 level – provides easier access to International Scale calibration and can act as a tool for assay optimization, validation, and quality assurance for molecular monitoring in chronic myeloid leukemia patients.
Such monitoring is important to disease management, especially for decision making with respect to treatment cessation. The new secondary reference panel would allow laboratories to circumvent the oft-used and time-consuming sample exchange process with reference laboratories for International Scale (IS) calibration, Nicholas C. P. Cross, PhD, of the University of Southampton (England) and colleagues wrote in an article published in Leukemia (2016 Jun 3;30:1844-52).
“The development of BCR-ABL1 tyrosine kinase inhibitors ... has enabled progressively deeper molecular responses in CML patients undergoing tyrosine kinase inhibitor therapy,” the authors wrote, noting that deeper molecular responses are “important milestones for patients considering treatment cessation,” and that “other landmarks on the IS also represent different treatment decision thresholds and prognostic outcomes.”
For this reason, regular molecular monitoring using real-time reverse-transcription quantitative PCR (RqPCR) is recommended for optimal disease management, they said.
“As treatment decisions are directly impacted by test results, accuracy and precision of BCR-ABL1 assays across the entire measurement range is crucial for patient management, especially in patients with deep molecular responses when considering possible treatment cessation,” they wrote.
Access to the material for the WHO BCR-ABL1 reference panel (MR1-MR4), which was developed in 2010 as a primary standard for BCR-ABL1 assay IS calibration, is limited, particularly for smaller laboratories, the authors said.
The secondary reference panel they developed, however, is “traceable to and faithfully replicates the WHO panel in both raw materials (lyopholized.K562 and HL-60 cell mixes) and manufacturing process, with the addition of a MR4.5 level.” The secondary panel was calibrated to IS using digital PCR against ABL1, BCR, and GUSB as reference genes, and was successfully evaluated by 45 different BCR-ABL1 assays at 44 different clinical laboratories in a multinational evaluation study.
The MR4.5 level was added to allow for more accurate IS calibration “as CML patients reaching this deep molecular response are increasingly being considered for treatment cessation,” the authors noted.
“Quality-control assessments indicated that the secondary panel had minimal residual moisture, excellent vial-to-vial homogeneity and greater than 2.5 years of real-time stability,” they said.
Further, the panel was successfully processed by all of the laboratories, indicating that it is compatible with many different BCR-ABL1 test configurations, they added.
Of note, the number of assays that achieved good precision and sensitivity exceeded the number that achieved good IS accuracy, suggesting an unmet need for “a simple and broadly available calibration mechanism, such as this secondary panel, to ensure IS accuracy is maintained in laboratories over time,” they wrote, concluding that such a panel “can provide easier access to IS calibration, as well as act as a tool for assay optimization, validation and quality assurance.”
In a letter to the editor in regard to the findings by Cross et al., Maria Sol Ruiz of Instituto Alexander Fleming in Buenos Aires, and colleagues wrote about their own development and validation of “secondary reference materials calibrated to the IS through the WHO primary standards in order to facilitate standardization of molecular monitoring in Latin America,” (Leukemia. 2016 Aug 19. doi: 10.1038/leu.2016.197).
In their study, the letter’s authors demonstrated that secondary reference biological calibrators anchored to the WHO primary standards can decrease inter-laboratory variability.
“Our results, together with those recently reported by Cross et al., substantiate the objective initially set during the establishment of the WHO primary standards, that is, to facilitate worldwide diffusion of the IS. For the first time in Latin America, this study provides a platform on which to assess the performance of distinct clinical BCR-ABL1 tests and confirm the utility of secondary reference materials to further improve IS accuracy and inter-laboratory precision,” they wrote, noting that their efforts will continue through provision of secondary reference material to centers involved in their project, as well as to potential new participants.
“Moreover, due to its higher precision and absolute quantification capability, we are evaluating the possibility of including digital PCR as the calibration method for the future,” they said.
The study discussed in the letter to the editor was supported by a grant from Novartis to one of the authors. Novartis also paid speaking fees to some of the authors. The study by Dr. Cross et al. was also funded by Novartis and some authors are employed by Novartis. The remaining authors, including Dr. Cross, reported having no disclosures.
FROM LEUKEMIA
Key clinical point:
Major finding: The secondary panel was calibrated to IS using digital PCR against ABL1, BCR, and GUSB as reference genes, and was successfully evaluated by 45 different BCR-ABL1 assays at 44 different clinical laboratories.
Data source: A multicenter evaluation study of a secondary reference panel for BCR-ABL1 quantification on the IS.
Disclosures: The study discussed in the letter to the editor was supported by a grant from Novartis to one of the authors. Novartis also paid speaking fees to some of the authors. The study by Dr. Cross et al. was also funded by Novartis and some authors are employed by Novartis. The remaining authors, including Dr. Cross, reported having no disclosures.
Tool provides info for cancer patients, survivors

receiving treatment
Photo by Rhoda Baer
The American Cancer Society and National Cancer Institute have launched an online tool for cancer patients and survivors.
The tool, Springboard Beyond Cancer, was designed to help these individuals address medical, psychosocial, and wellness needs during and after treatment.
Springboard Beyond Cancer provides information to help cancer patients and survivors manage ongoing cancer-related symptoms, deal with stress, ensure healthy behavior, communicate better with healthcare teams, and seek support from friends and family.
“With Springboard Beyond Cancer, we want to empower cancer survivors by giving them the information they need to help identify issues, set goals, and create a plan to more smoothly navigate the cancer journey and take control of their health,” said Corinne Leach, PhD, a behavioral scientist and strategic director in the Behavioral Research Center at the American Cancer Society.
“We hope that Springboard Beyond Cancer, along with the close collaboration of their medical team, can help cancer survivors reduce their disease burden and improve their overall wellbeing,” added Erik Augustson, PhD, program director at the National Cancer Institute. ![]()
receiving treatment
Photo by Rhoda Baer
The American Cancer Society and National Cancer Institute have launched an online tool for cancer patients and survivors.
The tool, Springboard Beyond Cancer, was designed to help these individuals address medical, psychosocial, and wellness needs during and after treatment.
Springboard Beyond Cancer provides information to help cancer patients and survivors manage ongoing cancer-related symptoms, deal with stress, ensure healthy behavior, communicate better with healthcare teams, and seek support from friends and family.
“With Springboard Beyond Cancer, we want to empower cancer survivors by giving them the information they need to help identify issues, set goals, and create a plan to more smoothly navigate the cancer journey and take control of their health,” said Corinne Leach, PhD, a behavioral scientist and strategic director in the Behavioral Research Center at the American Cancer Society.
“We hope that Springboard Beyond Cancer, along with the close collaboration of their medical team, can help cancer survivors reduce their disease burden and improve their overall wellbeing,” added Erik Augustson, PhD, program director at the National Cancer Institute. ![]()
receiving treatment
Photo by Rhoda Baer
The American Cancer Society and National Cancer Institute have launched an online tool for cancer patients and survivors.
The tool, Springboard Beyond Cancer, was designed to help these individuals address medical, psychosocial, and wellness needs during and after treatment.
Springboard Beyond Cancer provides information to help cancer patients and survivors manage ongoing cancer-related symptoms, deal with stress, ensure healthy behavior, communicate better with healthcare teams, and seek support from friends and family.
“With Springboard Beyond Cancer, we want to empower cancer survivors by giving them the information they need to help identify issues, set goals, and create a plan to more smoothly navigate the cancer journey and take control of their health,” said Corinne Leach, PhD, a behavioral scientist and strategic director in the Behavioral Research Center at the American Cancer Society.
“We hope that Springboard Beyond Cancer, along with the close collaboration of their medical team, can help cancer survivors reduce their disease burden and improve their overall wellbeing,” added Erik Augustson, PhD, program director at the National Cancer Institute. ![]()
Investigational AML drugs boosted remission rates
Two investigational drugs appear to be improving outcomes for patients with acute myeloid leukemia (AML), based on results reported in separate abstracts of studies that will be featured at press conferences to be held during the annual meeting of the American Society of Hematology.
In the first study, induction therapy with the investigational drug CPX-351 (Vyxeos), a liposomal formulation of cytarabine and daunorubicin, allowed a higher proportion of patients over age 60 with secondary AML to qualify for allogeneic hematopoietic cell transplants. Those patients went on to have improved survival, compared with patients who received standard 7+3 cytarabine and daunorubicin, Jeffrey E. Lancet, MD, of the H. Lee Moffitt Cancer Center and Research Institute, Tampa, and his colleagues reported in abstract 906.

The finding that CPX-351 may be an effective bridge to successful transplant for older patients with newly diagnosed secondary AML comes from an exploratory analysis of a phase III study comparing induction therapy with CPX-351 and standard cytarabine and daunorubicin. Initial data from the randomized open-label study, reported last June at the annual meeting of the American Society of Clinical Oncology, indicated CPX-351 significantly improved overall survival, event-free survival, and treatment response without an increase in 60-day mortality or in the frequency and severity of adverse events, compared with the standard 7+3 regimen of cytarabine and daunorubicin.
The data to be presented at ASH 2016 will examine the outcomes of 52 patients in the CPX-351 arm and 39 patients in the standard cytarabine and daunorubicin arm who underwent allogeneic hematopoietic cell transplantation (HCT) after induction. Data reported in the abstract indicate that 18 of the 52 patients in the CPX-351 arm and 26 of the 39 patients in the standard cytarabine and daunorubicin arm have died. The median survival time was 10.25 months with standard therapy; median survival has not yet been reached in the CPX-351 arm. The results indicate 53% fewer deaths occurred within 100 days of transplant in the CPX-351 group.
Newly diagnosed secondary AML was defined as having a history of prior cytotoxic treatment, antecedent myelodysplastic syndrome (MDS) with or without prior treatment with hypomethylating agents, or AML with World Health Organization–defined MDS-related cytogenetic abnormalities.
For the trial, conducted over 2 years at 39 U.S. and Canadian sites, 153 patients were randomized to the CPX-351 arm and 156 randomized to the standard therapy arm. Of 125 patients who had a complete response (CR) or a CR with incomplete (CRi) platelet or neutrophil recovery, 91 underwent allogeneic HCT: 52 (34%) from the CPX-351 arm and 39 (25%) from the standard therapy arm. Each arm had a similar percentage of patients who underwent transplant in CR/CRi status; however, the CPX-351 arm contained a higher percentage of patients age 70 and older (31% vs. 15%). Mortality at 100 days after transplant was 9.6% for patients in the CPX-351 arm and 20.5% for patients in the standard therapy arm. Deaths that occurred within 100 days after allogeneic HCT were due to refractory AML (CPX-351, 3.8%; standard therapy, 7.7%); graft vs. host disease (CPX-351, 3.8%; standard therapy, 2.6%); or renal, respiratory, or multiorgan failure, or septic shock (CPX-351, 0 for each; standard therapy, 2.6% for each), or the cause of death was unknown (CPX-351, 1.9%; standard therapy, 0).
For the 91 patients who had transplants, those in the CPX-351 arm had markedly better overall survival (hazard ratio, 0.46; P = .0046). The time-dependent Cox hazard ratio for overall survival in the CPX-351 arm vs. the 7+3 arm was 0.51 (95% confidence interval, 0.35–0.75; P = .0007).
In the phase Ib trial of vadastuximab talirine, 42 patients received the drug on days 1 and 4 of standard 7+3 cytarabine and daunorubicin induction therapy. Most patients had intermediate (40%) or adverse (43%) cytogenetic risk by Medical Research Council criteria, and 17% of patients had secondary AML. Response was assessed on days 15 and 28; MRD was assessed centrally by bone marrow examination using a multiparametric flow cytometric assay. The investigator chose whether to do a second induction regimen and any postremission therapies, which did not include additional administration of vadastuximab talirine.
Of the 40 patients who could be evaluated for efficacy, 24 (60%) had a CR and 7 (18%) had a CRi, and 4 (10%) reached a morphologic leukemia-free state. Nearly all (94%) of CR and CRi responses occurred after one cycle of induction therapy, and 23 of the 31 patients who reached CR or CRi achieved MRD-negative status.
Extramedullary adverse events, including hepatic toxicity, and induction mortality rates were similar to reported rates for 7+3 cytarabine and daunorubicin alone. All patients had grade 4 myelosuppression. In patients who achieved CR or CRi, the estimated median time to count recovery from day 1 of therapy was 33 days for neutrophils and 35 days for platelets. The 30- and 60-day mortality rates were 0% and 7%, respectively.
An alternative schedule of single-day dosing on day 1 is under investigation, and enrollment continues.
The CPX-351 (Vyxeos) study was supported by the drug’s maker, Celator Pharmaceuticals, which is a subsidiary of Jazz Pharmaceuticals. Dr. Lancet is a consultant to Celator as well as numerous other drug companies. Several of his colleagues disclosed a wide variety of relationships with drug companies, including Celator. Two of the study investigators disclosed employment by and equity ownership in Celator.
The vadastuximab talirine study was sponsored by the drug’s maker, Seattle Genetics. Dr. Erba disclosed a wide variety of relationships with drug companies, including research funding from Seattle Genetics. His colleagues had a similar wide variety of relationships, and two disclosed employment by and equity ownership in Seattle Genetics.
Abstract 906 Survival Following Allogeneic Hematopoietic Cell Transplantation in Older High-Risk Acute Myeloid Leukemia Patients Initially Treated With CPX-351 Liposome Injection Versus Standard Cytarabine and Daunorubicin: Subgroup Analysis of a Large Phase III Trial, will be presented in session 616 at 4:00 p.m. on Monday, Dec. 5.
Abstract 211 A Phase Ib Study of Vadastuximab Talirine in Combination With 7+3 Induction Therapy for Patients With Newly Diagnosed Acute Myeloid Leukemia (AML) will be presented in session 613 at 4:00 p.m. on Saturday, Dec. 3.
[email protected]
On Twitter @maryjodales
Two investigational drugs appear to be improving outcomes for patients with acute myeloid leukemia (AML), based on results reported in separate abstracts of studies that will be featured at press conferences to be held during the annual meeting of the American Society of Hematology.
In the first study, induction therapy with the investigational drug CPX-351 (Vyxeos), a liposomal formulation of cytarabine and daunorubicin, allowed a higher proportion of patients over age 60 with secondary AML to qualify for allogeneic hematopoietic cell transplants. Those patients went on to have improved survival, compared with patients who received standard 7+3 cytarabine and daunorubicin, Jeffrey E. Lancet, MD, of the H. Lee Moffitt Cancer Center and Research Institute, Tampa, and his colleagues reported in abstract 906.
The finding that CPX-351 may be an effective bridge to successful transplant for older patients with newly diagnosed secondary AML comes from an exploratory analysis of a phase III study comparing induction therapy with CPX-351 and standard cytarabine and daunorubicin. Initial data from the randomized open-label study, reported last June at the annual meeting of the American Society of Clinical Oncology, indicated CPX-351 significantly improved overall survival, event-free survival, and treatment response without an increase in 60-day mortality or in the frequency and severity of adverse events, compared with the standard 7+3 regimen of cytarabine and daunorubicin.
The data to be presented at ASH 2016 will examine the outcomes of 52 patients in the CPX-351 arm and 39 patients in the standard cytarabine and daunorubicin arm who underwent allogeneic hematopoietic cell transplantation (HCT) after induction. Data reported in the abstract indicate that 18 of the 52 patients in the CPX-351 arm and 26 of the 39 patients in the standard cytarabine and daunorubicin arm have died. The median survival time was 10.25 months with standard therapy; median survival has not yet been reached in the CPX-351 arm. The results indicate 53% fewer deaths occurred within 100 days of transplant in the CPX-351 group.
Newly diagnosed secondary AML was defined as having a history of prior cytotoxic treatment, antecedent myelodysplastic syndrome (MDS) with or without prior treatment with hypomethylating agents, or AML with World Health Organization–defined MDS-related cytogenetic abnormalities.
For the trial, conducted over 2 years at 39 U.S. and Canadian sites, 153 patients were randomized to the CPX-351 arm and 156 randomized to the standard therapy arm. Of 125 patients who had a complete response (CR) or a CR with incomplete (CRi) platelet or neutrophil recovery, 91 underwent allogeneic HCT: 52 (34%) from the CPX-351 arm and 39 (25%) from the standard therapy arm. Each arm had a similar percentage of patients who underwent transplant in CR/CRi status; however, the CPX-351 arm contained a higher percentage of patients age 70 and older (31% vs. 15%). Mortality at 100 days after transplant was 9.6% for patients in the CPX-351 arm and 20.5% for patients in the standard therapy arm. Deaths that occurred within 100 days after allogeneic HCT were due to refractory AML (CPX-351, 3.8%; standard therapy, 7.7%); graft vs. host disease (CPX-351, 3.8%; standard therapy, 2.6%); or renal, respiratory, or multiorgan failure, or septic shock (CPX-351, 0 for each; standard therapy, 2.6% for each), or the cause of death was unknown (CPX-351, 1.9%; standard therapy, 0).
For the 91 patients who had transplants, those in the CPX-351 arm had markedly better overall survival (hazard ratio, 0.46; P = .0046). The time-dependent Cox hazard ratio for overall survival in the CPX-351 arm vs. the 7+3 arm was 0.51 (95% confidence interval, 0.35–0.75; P = .0007).
In the phase Ib trial of vadastuximab talirine, 42 patients received the drug on days 1 and 4 of standard 7+3 cytarabine and daunorubicin induction therapy. Most patients had intermediate (40%) or adverse (43%) cytogenetic risk by Medical Research Council criteria, and 17% of patients had secondary AML. Response was assessed on days 15 and 28; MRD was assessed centrally by bone marrow examination using a multiparametric flow cytometric assay. The investigator chose whether to do a second induction regimen and any postremission therapies, which did not include additional administration of vadastuximab talirine.
Of the 40 patients who could be evaluated for efficacy, 24 (60%) had a CR and 7 (18%) had a CRi, and 4 (10%) reached a morphologic leukemia-free state. Nearly all (94%) of CR and CRi responses occurred after one cycle of induction therapy, and 23 of the 31 patients who reached CR or CRi achieved MRD-negative status.
Extramedullary adverse events, including hepatic toxicity, and induction mortality rates were similar to reported rates for 7+3 cytarabine and daunorubicin alone. All patients had grade 4 myelosuppression. In patients who achieved CR or CRi, the estimated median time to count recovery from day 1 of therapy was 33 days for neutrophils and 35 days for platelets. The 30- and 60-day mortality rates were 0% and 7%, respectively.
An alternative schedule of single-day dosing on day 1 is under investigation, and enrollment continues.
The CPX-351 (Vyxeos) study was supported by the drug’s maker, Celator Pharmaceuticals, which is a subsidiary of Jazz Pharmaceuticals. Dr. Lancet is a consultant to Celator as well as numerous other drug companies. Several of his colleagues disclosed a wide variety of relationships with drug companies, including Celator. Two of the study investigators disclosed employment by and equity ownership in Celator.
The vadastuximab talirine study was sponsored by the drug’s maker, Seattle Genetics. Dr. Erba disclosed a wide variety of relationships with drug companies, including research funding from Seattle Genetics. His colleagues had a similar wide variety of relationships, and two disclosed employment by and equity ownership in Seattle Genetics.
Abstract 906 Survival Following Allogeneic Hematopoietic Cell Transplantation in Older High-Risk Acute Myeloid Leukemia Patients Initially Treated With CPX-351 Liposome Injection Versus Standard Cytarabine and Daunorubicin: Subgroup Analysis of a Large Phase III Trial, will be presented in session 616 at 4:00 p.m. on Monday, Dec. 5.
Abstract 211 A Phase Ib Study of Vadastuximab Talirine in Combination With 7+3 Induction Therapy for Patients With Newly Diagnosed Acute Myeloid Leukemia (AML) will be presented in session 613 at 4:00 p.m. on Saturday, Dec. 3.
[email protected]
On Twitter @maryjodales
Two investigational drugs appear to be improving outcomes for patients with acute myeloid leukemia (AML), based on results reported in separate abstracts of studies that will be featured at press conferences to be held during the annual meeting of the American Society of Hematology.
In the first study, induction therapy with the investigational drug CPX-351 (Vyxeos), a liposomal formulation of cytarabine and daunorubicin, allowed a higher proportion of patients over age 60 with secondary AML to qualify for allogeneic hematopoietic cell transplants. Those patients went on to have improved survival, compared with patients who received standard 7+3 cytarabine and daunorubicin, Jeffrey E. Lancet, MD, of the H. Lee Moffitt Cancer Center and Research Institute, Tampa, and his colleagues reported in abstract 906.
The finding that CPX-351 may be an effective bridge to successful transplant for older patients with newly diagnosed secondary AML comes from an exploratory analysis of a phase III study comparing induction therapy with CPX-351 and standard cytarabine and daunorubicin. Initial data from the randomized open-label study, reported last June at the annual meeting of the American Society of Clinical Oncology, indicated CPX-351 significantly improved overall survival, event-free survival, and treatment response without an increase in 60-day mortality or in the frequency and severity of adverse events, compared with the standard 7+3 regimen of cytarabine and daunorubicin.
The data to be presented at ASH 2016 will examine the outcomes of 52 patients in the CPX-351 arm and 39 patients in the standard cytarabine and daunorubicin arm who underwent allogeneic hematopoietic cell transplantation (HCT) after induction. Data reported in the abstract indicate that 18 of the 52 patients in the CPX-351 arm and 26 of the 39 patients in the standard cytarabine and daunorubicin arm have died. The median survival time was 10.25 months with standard therapy; median survival has not yet been reached in the CPX-351 arm. The results indicate 53% fewer deaths occurred within 100 days of transplant in the CPX-351 group.
Newly diagnosed secondary AML was defined as having a history of prior cytotoxic treatment, antecedent myelodysplastic syndrome (MDS) with or without prior treatment with hypomethylating agents, or AML with World Health Organization–defined MDS-related cytogenetic abnormalities.
For the trial, conducted over 2 years at 39 U.S. and Canadian sites, 153 patients were randomized to the CPX-351 arm and 156 randomized to the standard therapy arm. Of 125 patients who had a complete response (CR) or a CR with incomplete (CRi) platelet or neutrophil recovery, 91 underwent allogeneic HCT: 52 (34%) from the CPX-351 arm and 39 (25%) from the standard therapy arm. Each arm had a similar percentage of patients who underwent transplant in CR/CRi status; however, the CPX-351 arm contained a higher percentage of patients age 70 and older (31% vs. 15%). Mortality at 100 days after transplant was 9.6% for patients in the CPX-351 arm and 20.5% for patients in the standard therapy arm. Deaths that occurred within 100 days after allogeneic HCT were due to refractory AML (CPX-351, 3.8%; standard therapy, 7.7%); graft vs. host disease (CPX-351, 3.8%; standard therapy, 2.6%); or renal, respiratory, or multiorgan failure, or septic shock (CPX-351, 0 for each; standard therapy, 2.6% for each), or the cause of death was unknown (CPX-351, 1.9%; standard therapy, 0).
For the 91 patients who had transplants, those in the CPX-351 arm had markedly better overall survival (hazard ratio, 0.46; P = .0046). The time-dependent Cox hazard ratio for overall survival in the CPX-351 arm vs. the 7+3 arm was 0.51 (95% confidence interval, 0.35–0.75; P = .0007).
In the phase Ib trial of vadastuximab talirine, 42 patients received the drug on days 1 and 4 of standard 7+3 cytarabine and daunorubicin induction therapy. Most patients had intermediate (40%) or adverse (43%) cytogenetic risk by Medical Research Council criteria, and 17% of patients had secondary AML. Response was assessed on days 15 and 28; MRD was assessed centrally by bone marrow examination using a multiparametric flow cytometric assay. The investigator chose whether to do a second induction regimen and any postremission therapies, which did not include additional administration of vadastuximab talirine.
Of the 40 patients who could be evaluated for efficacy, 24 (60%) had a CR and 7 (18%) had a CRi, and 4 (10%) reached a morphologic leukemia-free state. Nearly all (94%) of CR and CRi responses occurred after one cycle of induction therapy, and 23 of the 31 patients who reached CR or CRi achieved MRD-negative status.
Extramedullary adverse events, including hepatic toxicity, and induction mortality rates were similar to reported rates for 7+3 cytarabine and daunorubicin alone. All patients had grade 4 myelosuppression. In patients who achieved CR or CRi, the estimated median time to count recovery from day 1 of therapy was 33 days for neutrophils and 35 days for platelets. The 30- and 60-day mortality rates were 0% and 7%, respectively.
An alternative schedule of single-day dosing on day 1 is under investigation, and enrollment continues.
The CPX-351 (Vyxeos) study was supported by the drug’s maker, Celator Pharmaceuticals, which is a subsidiary of Jazz Pharmaceuticals. Dr. Lancet is a consultant to Celator as well as numerous other drug companies. Several of his colleagues disclosed a wide variety of relationships with drug companies, including Celator. Two of the study investigators disclosed employment by and equity ownership in Celator.
The vadastuximab talirine study was sponsored by the drug’s maker, Seattle Genetics. Dr. Erba disclosed a wide variety of relationships with drug companies, including research funding from Seattle Genetics. His colleagues had a similar wide variety of relationships, and two disclosed employment by and equity ownership in Seattle Genetics.
Abstract 906 Survival Following Allogeneic Hematopoietic Cell Transplantation in Older High-Risk Acute Myeloid Leukemia Patients Initially Treated With CPX-351 Liposome Injection Versus Standard Cytarabine and Daunorubicin: Subgroup Analysis of a Large Phase III Trial, will be presented in session 616 at 4:00 p.m. on Monday, Dec. 5.
Abstract 211 A Phase Ib Study of Vadastuximab Talirine in Combination With 7+3 Induction Therapy for Patients With Newly Diagnosed Acute Myeloid Leukemia (AML) will be presented in session 613 at 4:00 p.m. on Saturday, Dec. 3.
[email protected]
On Twitter @maryjodales
FROM ASH 2016
Panobinostat might treat high-risk ALL subtype
Photo courtesy of Novartis
Researchers say they have identified a high-risk subtype of acute lymphoblastic leukemia (ALL) that may respond to treatment with the histone deacetylase (HDAC) inhibitor panobinostat.
The ALL subtype is characterized by chromosomal rearrangements that involve the MEF2D gene and 1 of 6 partner genes, most often BCL9.
The researchers described this subtype, known as MEF2D-rearranged ALL, in Nature Communications.
“MEF2D is a transcription factor that switches on expression of other genes during normal development,” said study author Charles Mullighan, MD, MBBS, of the St. Jude Children’s Research Hospital in Memphis, Tennessee.
“We found that MEF2D chromosomal rearrangements disrupt expression of those genes and create a vulnerability to at least one targeted therapy, the drug panobinostat.”
Dr Mullighan and his colleagues performed genomic analyses on samples from a total of 1724 children, adolescents, and adults with ALL. This revealed 52 patients with MEF2D rearrangements.
MEF2D-rearranged ALL
The researchers calculated that MEF2D-rearranged ALL accounted for 5.3% of the ALL cases whose genetic basis was unknown.
The team also noted that MEF2D-rearranged ALL occurred most frequently in adolescents. Although, overall, ALL occurs most often in children between 3 and 5 years old, the average patient with MEF2D-rearranged ALL was 14.
In addition, MEF2D-rearranged ALL was associated with reduced survival when compared to some other ALL subtypes. The 5-year cancer-free survival for MEF2D-rearranged ALL patients was 71.6%.
The researchers also found that a fusion protein resulting from the MEF2D rearrangement led to sustained growth of mouse cells when compared to wild-type MEF2D or other proteins.
“That indicates the MEF2D fusion is a key step in transforming a normal white blood cell with a finite lifespan into a leukemic cell that is immortal,” Dr Mullighan said.
Role for panobinostat
MEF2D-rearranged leukemic cells produced high levels of HDAC9, which is targeted by panobinostat.
The researchers tested panobinostat in the lab and found the drug stopped proliferation of human MEF2D-rearranged leukemic cells.
Dr Mullighan said MEF2D-rearranged leukemic cells were exquisitely sensitive to panobinostat, which suggested the drug might function in a more targeted manner against cells with the rearrangement.
“If further testing of panobinostat, either alone or in combination therapy, confirms the anti-proliferative activity, that would lay the foundation for a clinical trial in patients, particularly patients with high-risk disease or those who have relapsed,” he said. ![]()
Photo courtesy of Novartis
Researchers say they have identified a high-risk subtype of acute lymphoblastic leukemia (ALL) that may respond to treatment with the histone deacetylase (HDAC) inhibitor panobinostat.
The ALL subtype is characterized by chromosomal rearrangements that involve the MEF2D gene and 1 of 6 partner genes, most often BCL9.
The researchers described this subtype, known as MEF2D-rearranged ALL, in Nature Communications.
“MEF2D is a transcription factor that switches on expression of other genes during normal development,” said study author Charles Mullighan, MD, MBBS, of the St. Jude Children’s Research Hospital in Memphis, Tennessee.
“We found that MEF2D chromosomal rearrangements disrupt expression of those genes and create a vulnerability to at least one targeted therapy, the drug panobinostat.”
Dr Mullighan and his colleagues performed genomic analyses on samples from a total of 1724 children, adolescents, and adults with ALL. This revealed 52 patients with MEF2D rearrangements.
MEF2D-rearranged ALL
The researchers calculated that MEF2D-rearranged ALL accounted for 5.3% of the ALL cases whose genetic basis was unknown.
The team also noted that MEF2D-rearranged ALL occurred most frequently in adolescents. Although, overall, ALL occurs most often in children between 3 and 5 years old, the average patient with MEF2D-rearranged ALL was 14.
In addition, MEF2D-rearranged ALL was associated with reduced survival when compared to some other ALL subtypes. The 5-year cancer-free survival for MEF2D-rearranged ALL patients was 71.6%.
The researchers also found that a fusion protein resulting from the MEF2D rearrangement led to sustained growth of mouse cells when compared to wild-type MEF2D or other proteins.
“That indicates the MEF2D fusion is a key step in transforming a normal white blood cell with a finite lifespan into a leukemic cell that is immortal,” Dr Mullighan said.
Role for panobinostat
MEF2D-rearranged leukemic cells produced high levels of HDAC9, which is targeted by panobinostat.
The researchers tested panobinostat in the lab and found the drug stopped proliferation of human MEF2D-rearranged leukemic cells.
Dr Mullighan said MEF2D-rearranged leukemic cells were exquisitely sensitive to panobinostat, which suggested the drug might function in a more targeted manner against cells with the rearrangement.
“If further testing of panobinostat, either alone or in combination therapy, confirms the anti-proliferative activity, that would lay the foundation for a clinical trial in patients, particularly patients with high-risk disease or those who have relapsed,” he said. ![]()
Photo courtesy of Novartis
Researchers say they have identified a high-risk subtype of acute lymphoblastic leukemia (ALL) that may respond to treatment with the histone deacetylase (HDAC) inhibitor panobinostat.
The ALL subtype is characterized by chromosomal rearrangements that involve the MEF2D gene and 1 of 6 partner genes, most often BCL9.
The researchers described this subtype, known as MEF2D-rearranged ALL, in Nature Communications.
“MEF2D is a transcription factor that switches on expression of other genes during normal development,” said study author Charles Mullighan, MD, MBBS, of the St. Jude Children’s Research Hospital in Memphis, Tennessee.
“We found that MEF2D chromosomal rearrangements disrupt expression of those genes and create a vulnerability to at least one targeted therapy, the drug panobinostat.”
Dr Mullighan and his colleagues performed genomic analyses on samples from a total of 1724 children, adolescents, and adults with ALL. This revealed 52 patients with MEF2D rearrangements.
MEF2D-rearranged ALL
The researchers calculated that MEF2D-rearranged ALL accounted for 5.3% of the ALL cases whose genetic basis was unknown.
The team also noted that MEF2D-rearranged ALL occurred most frequently in adolescents. Although, overall, ALL occurs most often in children between 3 and 5 years old, the average patient with MEF2D-rearranged ALL was 14.
In addition, MEF2D-rearranged ALL was associated with reduced survival when compared to some other ALL subtypes. The 5-year cancer-free survival for MEF2D-rearranged ALL patients was 71.6%.
The researchers also found that a fusion protein resulting from the MEF2D rearrangement led to sustained growth of mouse cells when compared to wild-type MEF2D or other proteins.
“That indicates the MEF2D fusion is a key step in transforming a normal white blood cell with a finite lifespan into a leukemic cell that is immortal,” Dr Mullighan said.
Role for panobinostat
MEF2D-rearranged leukemic cells produced high levels of HDAC9, which is targeted by panobinostat.
The researchers tested panobinostat in the lab and found the drug stopped proliferation of human MEF2D-rearranged leukemic cells.
Dr Mullighan said MEF2D-rearranged leukemic cells were exquisitely sensitive to panobinostat, which suggested the drug might function in a more targeted manner against cells with the rearrangement.
“If further testing of panobinostat, either alone or in combination therapy, confirms the anti-proliferative activity, that would lay the foundation for a clinical trial in patients, particularly patients with high-risk disease or those who have relapsed,” he said.