User login
Treatment guidelines for CAR T-cell therapy

Researchers have developed treatment guidelines for pediatric patients receiving chimeric antigen receptor (CAR) T-cell therapy.
The guidelines include recommendations for patient selection and consent, treatment details, and advice on managing cytokine release syndrome (CRS) and other adverse events associated with CAR T-cell therapy.
The guidelines were published in Nature Reviews Clinical Oncology.
“CAR T-cell therapy has been associated with remarkable response rates for children and young adults with ALL [acute lymphoblastic leukemia], yet this innovative form of cellular immunotherapy has resulted in unique and severe toxicities which can lead to rapid cardiorespiratory and/or neurological deterioration,” said guidelines author Kris Mahadeo, MD, of The University of Texas MD Anderson Cancer Center in Houston.
“This novel therapy requires the medical vigilance of a diverse multi-disciplinary team and associated clinical infrastructure to ensure optimal patient outcomes.”
Pediatric patient selection and consent
The guidelines state that providers of CAR T-cell therapies should adhere to product information labels and guidance from risk evaluation and mitigation strategy programs (level of evidence: IV, grade: D).
In addition, patient selection should be based on the indications approved by the US Food and Drug Administration and criteria used in pivotal studies. However, this can change as new information becomes available (level of evidence: IV, grade: D).
Informed consent should include descriptions of the risks and benefits associated with leukapheresis, lymphodepletion, CRS, CAR T-cell-related encephalopathy syndrome (CRES), bridging chemotherapy, intensive care support, and anti-IL-6 therapy (level of evidence: IIA, grade: B).
Providers should obtain child assent when appropriate and may benefit from incorporating child life and psychological services in assent discussions (level of evidence: IV, grade: D).
Treatment specifics
The guidelines recommend cyclophosphamide–fludarabine regimens for lymphodepletion, although exceptions can be considered in cases of hemorrhagic cystitis and/or resistance to a prior cyclophosphamide-based regimen (level of evidence: IIA, grade: B).
Providers should consider inpatient admission for a minimum of 3 to 7 days after receipt of tisagenlecleucel. This was based on the experience in pediatric and young adult patients with CD19+ relapsed and/or refractory B-cell acute lymphoblastic leukemia (level of evidence: IIA, grade: B).
Patients should be closely monitored for hypotension, hypocalcemia, and catheter-related pain during leukapheresis (level of evidence: IIA, grade: B).
For patients receiving tocilizumab, those weighing <30 kg should receive 12 mg/kg, and those weighing ≥30 kg should receive 8 mg/kg (level of evidence: IIA, grade: B).
Adverse events
The guidelines say parent and/or caregiver concerns should be addressed as these individuals may be best equipped to recognize early signs or symptoms of CRS (level of evidence: III, grade: C).
When CAR T-cell therapy is administered in an outpatient setting, there should be a low threshold for patient admission upon the development of signs or symptoms suggestive of CRS and/or CRES (level of evidence: IIA, grade: B).
CRS grading should be performed at least once every 12 hours (level of evidence: IIA, grade: B). Detailed information on grading is provided in the guidelines.
Providers should suspect CRS if any of the following signs/symptoms are present within the first 2 weeks of CAR T-cell infusion:
- Fever ≥38 °C
- Hypotension
- Hypoxia with an arterial oxygen saturation of <90% on room air
- Evidence of organ toxicity as determined by the most recent CTCAE grading system and considerations detailed in the guidelines (level of evidence: IIA, grade: C).
The guidelines also recommend “high vigilance” for sinus tachycardia as an early sign of CRS (level of evidence: IIA, grade: B) as well as application of the PALICC (Pediatric Acute Lung Injury Consensus Conference) at-risk P-ARDS (pediatric acute respiratory distress syndrome) criteria for the CRS grading of hypoxia (level of evidence: IIA, grade: B).
Hemophagocytic lymphohistiocytosis and/or macrophage-activation syndrome can be treated with anti-IL-6 therapy and corticosteroids. However, refractory cases may require systemic and/or intrathecal therapy or use of the IL-1 receptor antagonist anakinra (level of evidence: IIA, grade: C).
The guidelines recommend that delirium screening be performed at least twice per 24-hour period among admitted patients and at least daily among outpatients during the high-risk periods for CRES (level of evidence: IIA, grade: C). Delirium screening should be performed with the CAPD (Cornell Assessment of Pediatric Delirium) tool or CARTOX-10 (CAR T-Cell Therapy-Associated Toxicity 10-point assessment scale) for patients age 12 and older who have sufficient cognitive abilities.
Acute kidney injury in children can be graded according to the CTCAE (Common Terminology Criteria for Adverse Events) using pRIFLE (Pediatric Risk, Injury, Failure, Loss, End-Stage Renal Disease) and KDIGO (Kidney Disease: Improving Global Outcomes) definitions of oliguria (level of evidence: IIA, grade: B).
Other considerations
The guidelines “strongly encourage” consideration of quality-adjusted life-years gained for pediatric patients who might achieve long-term remission from CAR T-cell therapy and encourage efforts to reduce the cost of care (level of evidence: IV, grade: D).
The guidelines also recommend that CAR T-cell programs seek FACT IEC (Foundation for the Accreditation of Cellular Therapy for Immune Effector Cells) accreditation to ensure adherence to quality standards (level of evidence: IV, grade: D).
Finally, the guidelines suggest the possibility of a prospective collaboration with intensive-care registries, which could allow accurate data entry of cell therapy variables into the CIBMTR registry with concurrent entry of intensive-care variables into an appropriate registry by pediatric critical care teams (level of evidence: IV, grade: D).
Researchers have developed treatment guidelines for pediatric patients receiving chimeric antigen receptor (CAR) T-cell therapy.
The guidelines include recommendations for patient selection and consent, treatment details, and advice on managing cytokine release syndrome (CRS) and other adverse events associated with CAR T-cell therapy.
The guidelines were published in Nature Reviews Clinical Oncology.
“CAR T-cell therapy has been associated with remarkable response rates for children and young adults with ALL [acute lymphoblastic leukemia], yet this innovative form of cellular immunotherapy has resulted in unique and severe toxicities which can lead to rapid cardiorespiratory and/or neurological deterioration,” said guidelines author Kris Mahadeo, MD, of The University of Texas MD Anderson Cancer Center in Houston.
“This novel therapy requires the medical vigilance of a diverse multi-disciplinary team and associated clinical infrastructure to ensure optimal patient outcomes.”
Pediatric patient selection and consent
The guidelines state that providers of CAR T-cell therapies should adhere to product information labels and guidance from risk evaluation and mitigation strategy programs (level of evidence: IV, grade: D).
In addition, patient selection should be based on the indications approved by the US Food and Drug Administration and criteria used in pivotal studies. However, this can change as new information becomes available (level of evidence: IV, grade: D).
Informed consent should include descriptions of the risks and benefits associated with leukapheresis, lymphodepletion, CRS, CAR T-cell-related encephalopathy syndrome (CRES), bridging chemotherapy, intensive care support, and anti-IL-6 therapy (level of evidence: IIA, grade: B).
Providers should obtain child assent when appropriate and may benefit from incorporating child life and psychological services in assent discussions (level of evidence: IV, grade: D).
Treatment specifics
The guidelines recommend cyclophosphamide–fludarabine regimens for lymphodepletion, although exceptions can be considered in cases of hemorrhagic cystitis and/or resistance to a prior cyclophosphamide-based regimen (level of evidence: IIA, grade: B).
Providers should consider inpatient admission for a minimum of 3 to 7 days after receipt of tisagenlecleucel. This was based on the experience in pediatric and young adult patients with CD19+ relapsed and/or refractory B-cell acute lymphoblastic leukemia (level of evidence: IIA, grade: B).
Patients should be closely monitored for hypotension, hypocalcemia, and catheter-related pain during leukapheresis (level of evidence: IIA, grade: B).
For patients receiving tocilizumab, those weighing <30 kg should receive 12 mg/kg, and those weighing ≥30 kg should receive 8 mg/kg (level of evidence: IIA, grade: B).
Adverse events
The guidelines say parent and/or caregiver concerns should be addressed as these individuals may be best equipped to recognize early signs or symptoms of CRS (level of evidence: III, grade: C).
When CAR T-cell therapy is administered in an outpatient setting, there should be a low threshold for patient admission upon the development of signs or symptoms suggestive of CRS and/or CRES (level of evidence: IIA, grade: B).
CRS grading should be performed at least once every 12 hours (level of evidence: IIA, grade: B). Detailed information on grading is provided in the guidelines.
Providers should suspect CRS if any of the following signs/symptoms are present within the first 2 weeks of CAR T-cell infusion:
- Fever ≥38 °C
- Hypotension
- Hypoxia with an arterial oxygen saturation of <90% on room air
- Evidence of organ toxicity as determined by the most recent CTCAE grading system and considerations detailed in the guidelines (level of evidence: IIA, grade: C).
The guidelines also recommend “high vigilance” for sinus tachycardia as an early sign of CRS (level of evidence: IIA, grade: B) as well as application of the PALICC (Pediatric Acute Lung Injury Consensus Conference) at-risk P-ARDS (pediatric acute respiratory distress syndrome) criteria for the CRS grading of hypoxia (level of evidence: IIA, grade: B).
Hemophagocytic lymphohistiocytosis and/or macrophage-activation syndrome can be treated with anti-IL-6 therapy and corticosteroids. However, refractory cases may require systemic and/or intrathecal therapy or use of the IL-1 receptor antagonist anakinra (level of evidence: IIA, grade: C).
The guidelines recommend that delirium screening be performed at least twice per 24-hour period among admitted patients and at least daily among outpatients during the high-risk periods for CRES (level of evidence: IIA, grade: C). Delirium screening should be performed with the CAPD (Cornell Assessment of Pediatric Delirium) tool or CARTOX-10 (CAR T-Cell Therapy-Associated Toxicity 10-point assessment scale) for patients age 12 and older who have sufficient cognitive abilities.
Acute kidney injury in children can be graded according to the CTCAE (Common Terminology Criteria for Adverse Events) using pRIFLE (Pediatric Risk, Injury, Failure, Loss, End-Stage Renal Disease) and KDIGO (Kidney Disease: Improving Global Outcomes) definitions of oliguria (level of evidence: IIA, grade: B).
Other considerations
The guidelines “strongly encourage” consideration of quality-adjusted life-years gained for pediatric patients who might achieve long-term remission from CAR T-cell therapy and encourage efforts to reduce the cost of care (level of evidence: IV, grade: D).
The guidelines also recommend that CAR T-cell programs seek FACT IEC (Foundation for the Accreditation of Cellular Therapy for Immune Effector Cells) accreditation to ensure adherence to quality standards (level of evidence: IV, grade: D).
Finally, the guidelines suggest the possibility of a prospective collaboration with intensive-care registries, which could allow accurate data entry of cell therapy variables into the CIBMTR registry with concurrent entry of intensive-care variables into an appropriate registry by pediatric critical care teams (level of evidence: IV, grade: D).
Researchers have developed treatment guidelines for pediatric patients receiving chimeric antigen receptor (CAR) T-cell therapy.
The guidelines include recommendations for patient selection and consent, treatment details, and advice on managing cytokine release syndrome (CRS) and other adverse events associated with CAR T-cell therapy.
The guidelines were published in Nature Reviews Clinical Oncology.
“CAR T-cell therapy has been associated with remarkable response rates for children and young adults with ALL [acute lymphoblastic leukemia], yet this innovative form of cellular immunotherapy has resulted in unique and severe toxicities which can lead to rapid cardiorespiratory and/or neurological deterioration,” said guidelines author Kris Mahadeo, MD, of The University of Texas MD Anderson Cancer Center in Houston.
“This novel therapy requires the medical vigilance of a diverse multi-disciplinary team and associated clinical infrastructure to ensure optimal patient outcomes.”
Pediatric patient selection and consent
The guidelines state that providers of CAR T-cell therapies should adhere to product information labels and guidance from risk evaluation and mitigation strategy programs (level of evidence: IV, grade: D).
In addition, patient selection should be based on the indications approved by the US Food and Drug Administration and criteria used in pivotal studies. However, this can change as new information becomes available (level of evidence: IV, grade: D).
Informed consent should include descriptions of the risks and benefits associated with leukapheresis, lymphodepletion, CRS, CAR T-cell-related encephalopathy syndrome (CRES), bridging chemotherapy, intensive care support, and anti-IL-6 therapy (level of evidence: IIA, grade: B).
Providers should obtain child assent when appropriate and may benefit from incorporating child life and psychological services in assent discussions (level of evidence: IV, grade: D).
Treatment specifics
The guidelines recommend cyclophosphamide–fludarabine regimens for lymphodepletion, although exceptions can be considered in cases of hemorrhagic cystitis and/or resistance to a prior cyclophosphamide-based regimen (level of evidence: IIA, grade: B).
Providers should consider inpatient admission for a minimum of 3 to 7 days after receipt of tisagenlecleucel. This was based on the experience in pediatric and young adult patients with CD19+ relapsed and/or refractory B-cell acute lymphoblastic leukemia (level of evidence: IIA, grade: B).
Patients should be closely monitored for hypotension, hypocalcemia, and catheter-related pain during leukapheresis (level of evidence: IIA, grade: B).
For patients receiving tocilizumab, those weighing <30 kg should receive 12 mg/kg, and those weighing ≥30 kg should receive 8 mg/kg (level of evidence: IIA, grade: B).
Adverse events
The guidelines say parent and/or caregiver concerns should be addressed as these individuals may be best equipped to recognize early signs or symptoms of CRS (level of evidence: III, grade: C).
When CAR T-cell therapy is administered in an outpatient setting, there should be a low threshold for patient admission upon the development of signs or symptoms suggestive of CRS and/or CRES (level of evidence: IIA, grade: B).
CRS grading should be performed at least once every 12 hours (level of evidence: IIA, grade: B). Detailed information on grading is provided in the guidelines.
Providers should suspect CRS if any of the following signs/symptoms are present within the first 2 weeks of CAR T-cell infusion:
- Fever ≥38 °C
- Hypotension
- Hypoxia with an arterial oxygen saturation of <90% on room air
- Evidence of organ toxicity as determined by the most recent CTCAE grading system and considerations detailed in the guidelines (level of evidence: IIA, grade: C).
The guidelines also recommend “high vigilance” for sinus tachycardia as an early sign of CRS (level of evidence: IIA, grade: B) as well as application of the PALICC (Pediatric Acute Lung Injury Consensus Conference) at-risk P-ARDS (pediatric acute respiratory distress syndrome) criteria for the CRS grading of hypoxia (level of evidence: IIA, grade: B).
Hemophagocytic lymphohistiocytosis and/or macrophage-activation syndrome can be treated with anti-IL-6 therapy and corticosteroids. However, refractory cases may require systemic and/or intrathecal therapy or use of the IL-1 receptor antagonist anakinra (level of evidence: IIA, grade: C).
The guidelines recommend that delirium screening be performed at least twice per 24-hour period among admitted patients and at least daily among outpatients during the high-risk periods for CRES (level of evidence: IIA, grade: C). Delirium screening should be performed with the CAPD (Cornell Assessment of Pediatric Delirium) tool or CARTOX-10 (CAR T-Cell Therapy-Associated Toxicity 10-point assessment scale) for patients age 12 and older who have sufficient cognitive abilities.
Acute kidney injury in children can be graded according to the CTCAE (Common Terminology Criteria for Adverse Events) using pRIFLE (Pediatric Risk, Injury, Failure, Loss, End-Stage Renal Disease) and KDIGO (Kidney Disease: Improving Global Outcomes) definitions of oliguria (level of evidence: IIA, grade: B).
Other considerations
The guidelines “strongly encourage” consideration of quality-adjusted life-years gained for pediatric patients who might achieve long-term remission from CAR T-cell therapy and encourage efforts to reduce the cost of care (level of evidence: IV, grade: D).
The guidelines also recommend that CAR T-cell programs seek FACT IEC (Foundation for the Accreditation of Cellular Therapy for Immune Effector Cells) accreditation to ensure adherence to quality standards (level of evidence: IV, grade: D).
Finally, the guidelines suggest the possibility of a prospective collaboration with intensive-care registries, which could allow accurate data entry of cell therapy variables into the CIBMTR registry with concurrent entry of intensive-care variables into an appropriate registry by pediatric critical care teams (level of evidence: IV, grade: D).
Insurance is a matter of life or death for lymphoma patients
Having health insurance can mean the difference between life and death for patients with follicular lymphoma, suggest results of a study showing that patients with private health insurance had nearly twofold better survival outcomes than patients without insurance or those who were covered by Medicare or Medicaid.

A review of records on more than 43,000 patients with follicular lymphoma (FL) in a national cancer registry showed that, compared with patients under age 65 with private insurance, the hazard ratios (HR) for death among patients in the same age bracket with either no insurance, Medicaid, or Medicare were, respectively, 1.96, 1.83, and 1.96 (P less than .0001 for each comparison).
“Our study finds that insurance status contributes to survival disparities in FL. Future studies on outcomes in FL should include insurance status as an important predictor,” Christopher R. Flowers, MD, of Emory University in Atlanta and his colleagues wrote in Blood.
“Further research on prognosis for FL should examine the impact of public policy, such as the passage of the [Affordable Care Act], on FL outcomes, as well as examine other factors that influence access to care, such as individual-level socioeconomic status, regular primary care visits, access to prescription medications, and care affordability,” they added.
The investigators noted that earlier research found that patients with Medicaid or no insurance were more likely than privately-insured patients to be diagnosed with cancers at advanced stages, and that some patients with aggressive non-Hodgkin lymphomas have been shown to have insurance-related disparities in treatments and outcomes.
To see whether the same could be true for patients with indolent-histology lymphomas such as FL, they extracted data from the National Cancer Database, a nationwide hospital-based cancer registry sponsored jointly by the American College of Surgeons and the American Cancer Society.
They identified a total of 43,648 patients aged 18 years or older who were diagnosed with FL from 2004 through 2014. They looked at both patients 18-64 years and those 65 years and older to account for changes in insurance with Medicare eligibility.
Overall survival among patients younger than age 65 was significantly worse for patients with public insurance (Medicaid or Medicare) or no insurance in Cox proportional hazard models controlling for available data on sociodemographic factors and prognostic indicators.
However, compared with patients aged 65 and older with private insurance, only patients with Medicare as their sole source of insurance had significantly worse overall survival (HR, 1.28; P less than .0001).
Patients who were uninsured or had Medicaid were more likely than others to have lower socioeconomic status, present with advanced-stage disease, have systemic symptoms, and have multiple comorbidities that persisted after controlling for known sociodemographic and prognostic factors.
The investigators found that, among patients under age 65, those with a comorbidity score of 1 had an HR for death of 1.71, compared with patients with no comorbidities, and that patients with a score of 2 or greater had a HR of 3.1 (P less than .0001 for each comparison).
“The findings of the study indicate that improving access to affordable, quality health care may reduce disparities in survival for those currently lacking coverage,” the investigators wrote.
The study was supported by Emory University, the National Institutes of Health, and the National Center for Advancing Translational Sciences. Dr. Flowers reported financial relationships with AbbVie, Spectrum, Celgene, and several other companies. The other authors reported having nothing to disclose.
SOURCE: Goldstein JS et al. Blood. 2018 Jul 24. doi: 10.1182/blood-2018-03-839035.
Having health insurance can mean the difference between life and death for patients with follicular lymphoma, suggest results of a study showing that patients with private health insurance had nearly twofold better survival outcomes than patients without insurance or those who were covered by Medicare or Medicaid.
A review of records on more than 43,000 patients with follicular lymphoma (FL) in a national cancer registry showed that, compared with patients under age 65 with private insurance, the hazard ratios (HR) for death among patients in the same age bracket with either no insurance, Medicaid, or Medicare were, respectively, 1.96, 1.83, and 1.96 (P less than .0001 for each comparison).
“Our study finds that insurance status contributes to survival disparities in FL. Future studies on outcomes in FL should include insurance status as an important predictor,” Christopher R. Flowers, MD, of Emory University in Atlanta and his colleagues wrote in Blood.
“Further research on prognosis for FL should examine the impact of public policy, such as the passage of the [Affordable Care Act], on FL outcomes, as well as examine other factors that influence access to care, such as individual-level socioeconomic status, regular primary care visits, access to prescription medications, and care affordability,” they added.
The investigators noted that earlier research found that patients with Medicaid or no insurance were more likely than privately-insured patients to be diagnosed with cancers at advanced stages, and that some patients with aggressive non-Hodgkin lymphomas have been shown to have insurance-related disparities in treatments and outcomes.
To see whether the same could be true for patients with indolent-histology lymphomas such as FL, they extracted data from the National Cancer Database, a nationwide hospital-based cancer registry sponsored jointly by the American College of Surgeons and the American Cancer Society.
They identified a total of 43,648 patients aged 18 years or older who were diagnosed with FL from 2004 through 2014. They looked at both patients 18-64 years and those 65 years and older to account for changes in insurance with Medicare eligibility.
Overall survival among patients younger than age 65 was significantly worse for patients with public insurance (Medicaid or Medicare) or no insurance in Cox proportional hazard models controlling for available data on sociodemographic factors and prognostic indicators.
However, compared with patients aged 65 and older with private insurance, only patients with Medicare as their sole source of insurance had significantly worse overall survival (HR, 1.28; P less than .0001).
Patients who were uninsured or had Medicaid were more likely than others to have lower socioeconomic status, present with advanced-stage disease, have systemic symptoms, and have multiple comorbidities that persisted after controlling for known sociodemographic and prognostic factors.
The investigators found that, among patients under age 65, those with a comorbidity score of 1 had an HR for death of 1.71, compared with patients with no comorbidities, and that patients with a score of 2 or greater had a HR of 3.1 (P less than .0001 for each comparison).
“The findings of the study indicate that improving access to affordable, quality health care may reduce disparities in survival for those currently lacking coverage,” the investigators wrote.
The study was supported by Emory University, the National Institutes of Health, and the National Center for Advancing Translational Sciences. Dr. Flowers reported financial relationships with AbbVie, Spectrum, Celgene, and several other companies. The other authors reported having nothing to disclose.
SOURCE: Goldstein JS et al. Blood. 2018 Jul 24. doi: 10.1182/blood-2018-03-839035.
Having health insurance can mean the difference between life and death for patients with follicular lymphoma, suggest results of a study showing that patients with private health insurance had nearly twofold better survival outcomes than patients without insurance or those who were covered by Medicare or Medicaid.
A review of records on more than 43,000 patients with follicular lymphoma (FL) in a national cancer registry showed that, compared with patients under age 65 with private insurance, the hazard ratios (HR) for death among patients in the same age bracket with either no insurance, Medicaid, or Medicare were, respectively, 1.96, 1.83, and 1.96 (P less than .0001 for each comparison).
“Our study finds that insurance status contributes to survival disparities in FL. Future studies on outcomes in FL should include insurance status as an important predictor,” Christopher R. Flowers, MD, of Emory University in Atlanta and his colleagues wrote in Blood.
“Further research on prognosis for FL should examine the impact of public policy, such as the passage of the [Affordable Care Act], on FL outcomes, as well as examine other factors that influence access to care, such as individual-level socioeconomic status, regular primary care visits, access to prescription medications, and care affordability,” they added.
The investigators noted that earlier research found that patients with Medicaid or no insurance were more likely than privately-insured patients to be diagnosed with cancers at advanced stages, and that some patients with aggressive non-Hodgkin lymphomas have been shown to have insurance-related disparities in treatments and outcomes.
To see whether the same could be true for patients with indolent-histology lymphomas such as FL, they extracted data from the National Cancer Database, a nationwide hospital-based cancer registry sponsored jointly by the American College of Surgeons and the American Cancer Society.
They identified a total of 43,648 patients aged 18 years or older who were diagnosed with FL from 2004 through 2014. They looked at both patients 18-64 years and those 65 years and older to account for changes in insurance with Medicare eligibility.
Overall survival among patients younger than age 65 was significantly worse for patients with public insurance (Medicaid or Medicare) or no insurance in Cox proportional hazard models controlling for available data on sociodemographic factors and prognostic indicators.
However, compared with patients aged 65 and older with private insurance, only patients with Medicare as their sole source of insurance had significantly worse overall survival (HR, 1.28; P less than .0001).
Patients who were uninsured or had Medicaid were more likely than others to have lower socioeconomic status, present with advanced-stage disease, have systemic symptoms, and have multiple comorbidities that persisted after controlling for known sociodemographic and prognostic factors.
The investigators found that, among patients under age 65, those with a comorbidity score of 1 had an HR for death of 1.71, compared with patients with no comorbidities, and that patients with a score of 2 or greater had a HR of 3.1 (P less than .0001 for each comparison).
“The findings of the study indicate that improving access to affordable, quality health care may reduce disparities in survival for those currently lacking coverage,” the investigators wrote.
The study was supported by Emory University, the National Institutes of Health, and the National Center for Advancing Translational Sciences. Dr. Flowers reported financial relationships with AbbVie, Spectrum, Celgene, and several other companies. The other authors reported having nothing to disclose.
SOURCE: Goldstein JS et al. Blood. 2018 Jul 24. doi: 10.1182/blood-2018-03-839035.
FROM BLOOD
Key clinical point:
Major finding: The risk for death among patients under age 65 with no insurance, Medicaid, or Medicare was nearly twice that of similar patients with private health insurance.
Study details: Review of data on 43,648 patients with follicular lymphoma in the National Cancer Database.
Disclosures: The study was supported by Emory University, the National Institutes of Health, and the National Center for Advancing Translational Sciences. Dr. Flowers reported financial relationships with AbbVie, Spectrum, Celgene, and several other companies. The other authors reported having nothing to disclose.
Source: Goldstein JS et al. Blood. 2018 Jul 24. doi: 10.1182/blood-2018-03-839035.
Company narrows focus of development for tazemetostat

Epizyme, Inc., has announced its decision to stop developing tazemetostat for use as monotherapy or in combination with prednisolone for patients with diffuse large B-cell lymphoma (DLBCL).
However, tazemetostat is still under investigation as a potential treatment for DLBCL as part of other combination regimens.
Tazemetostat is an EZH2 inhibitor being developed to treat multiple hematologic and solid tumor malignancies.
Epizyme has been conducting a phase 1/2 trial of tazemetostat in patients with relapsed and/or refractory DLBCL as well as other B-cell lymphomas and solid tumors (NCT01897571).
The trial includes DLBCL patients with and without EZH2 activating mutations. Some patients were assigned to receive tazemetostat monotherapy, and some were assigned to tazemetostat in combination with prednisolone.
Epizyme has conducted an interim assessment of data from this trial and concluded that the clinical activity observed “is not sufficient to warrant further development of tazemetostat in DLBCL as a monotherapy or in combination with prednisolone.”
Epizyme said it plans to present data from this trial at a medical meeting in the second half of 2018.
The company is still conducting other studies of tazemetostat in patients with DLBCL.
In one study (NCT02889523), Epizyme and the Lymphoma Academic Research Organisation are evaluating tazemetostat in combination with R-CHOP (rituximab, cyclophosphamide, vincristine, doxorubicin, and prednisolone) in patients with newly diagnosed DLBCL.
In another study (NCT03028103), Epizyme is evaluating tazemetostat in combination with fluconazole or omeprazole and repaglinide in patients with relapsed/refractory DLBCL, other B-cell lymphomas, or solid tumor malignancies.
Epizyme, Inc., has announced its decision to stop developing tazemetostat for use as monotherapy or in combination with prednisolone for patients with diffuse large B-cell lymphoma (DLBCL).
However, tazemetostat is still under investigation as a potential treatment for DLBCL as part of other combination regimens.
Tazemetostat is an EZH2 inhibitor being developed to treat multiple hematologic and solid tumor malignancies.
Epizyme has been conducting a phase 1/2 trial of tazemetostat in patients with relapsed and/or refractory DLBCL as well as other B-cell lymphomas and solid tumors (NCT01897571).
The trial includes DLBCL patients with and without EZH2 activating mutations. Some patients were assigned to receive tazemetostat monotherapy, and some were assigned to tazemetostat in combination with prednisolone.
Epizyme has conducted an interim assessment of data from this trial and concluded that the clinical activity observed “is not sufficient to warrant further development of tazemetostat in DLBCL as a monotherapy or in combination with prednisolone.”
Epizyme said it plans to present data from this trial at a medical meeting in the second half of 2018.
The company is still conducting other studies of tazemetostat in patients with DLBCL.
In one study (NCT02889523), Epizyme and the Lymphoma Academic Research Organisation are evaluating tazemetostat in combination with R-CHOP (rituximab, cyclophosphamide, vincristine, doxorubicin, and prednisolone) in patients with newly diagnosed DLBCL.
In another study (NCT03028103), Epizyme is evaluating tazemetostat in combination with fluconazole or omeprazole and repaglinide in patients with relapsed/refractory DLBCL, other B-cell lymphomas, or solid tumor malignancies.
Epizyme, Inc., has announced its decision to stop developing tazemetostat for use as monotherapy or in combination with prednisolone for patients with diffuse large B-cell lymphoma (DLBCL).
However, tazemetostat is still under investigation as a potential treatment for DLBCL as part of other combination regimens.
Tazemetostat is an EZH2 inhibitor being developed to treat multiple hematologic and solid tumor malignancies.
Epizyme has been conducting a phase 1/2 trial of tazemetostat in patients with relapsed and/or refractory DLBCL as well as other B-cell lymphomas and solid tumors (NCT01897571).
The trial includes DLBCL patients with and without EZH2 activating mutations. Some patients were assigned to receive tazemetostat monotherapy, and some were assigned to tazemetostat in combination with prednisolone.
Epizyme has conducted an interim assessment of data from this trial and concluded that the clinical activity observed “is not sufficient to warrant further development of tazemetostat in DLBCL as a monotherapy or in combination with prednisolone.”
Epizyme said it plans to present data from this trial at a medical meeting in the second half of 2018.
The company is still conducting other studies of tazemetostat in patients with DLBCL.
In one study (NCT02889523), Epizyme and the Lymphoma Academic Research Organisation are evaluating tazemetostat in combination with R-CHOP (rituximab, cyclophosphamide, vincristine, doxorubicin, and prednisolone) in patients with newly diagnosed DLBCL.
In another study (NCT03028103), Epizyme is evaluating tazemetostat in combination with fluconazole or omeprazole and repaglinide in patients with relapsed/refractory DLBCL, other B-cell lymphomas, or solid tumor malignancies.
PET/CT accurately predicts MCL stage
Bone marrow involvement in mantle cell lymphoma could be assessed using just 18fluorodeoxyglucose (FDG)–PET/CT, according to findings from a small, retrospective study published in Clinical Lymphoma, Myeloma & Leukemia.
Rustain Morgan, MD, of the University of Colorado, Aurora, and his colleagues found that, at a certain threshold of bone marrow voxels in standard uptake value (SUV), there was 100% sensitivity and 80% specificity in determining bone marrow involvement in mantle cell lymphoma (MCL).
Currently, National Comprehensive Cancer Network guidelines call for bone marrow biopsy and whole body FDG PET/CT scan to complete an initial diagnosis of MCL.
“One of the most important factors for correct staging is the identification of bone marrow involvement, occurring in approximately 55% of patients with MCL, which classifies patients as advanced stage. However, accurate analysis of bone marrow involvement can be challenging due to sampling error,” the researchers wrote. “While bone marrow biopsy remains the gold standard, it is not a perfect standard given unilateral variability.”
In previous studies, FDG PET/CT was not considered sensitive enough to detect gastrointestinal or bone marrow involvement. However, these earlier studies used SUV maximum or mean or a visual assessment of the bone marrow activity, compared with hepatic uptake. To address this issue, the researchers developed a new method of examining SUV distribution throughout the pelvic bones by analyzing thousands of bone marrow voxels within the bilateral iliacs.
During the developmental phase, an institutional dataset of 11 patients with MCL was used to define the voxel-based analysis. These patients had undergone both unilateral iliac bone marrow biopsy and FDG PET/CT at the initial diagnosis. Then, FDG PET/CT scans from another 12 patients with MCL from a different institution were used to validate the developmental phase findings. Finally, a control group of 5 people with no known malignancy were referred for FDG PET/CT pulmonary nodule evaluation.
“The hypothesis of the study was that, if the bone marrow was involved by lymphoma, then there would be a small increase in the SUV of each voxel, reflecting involvement by the lymphoma. In order to capture such changes, we analyzed the percent of total voxels in SUV ranging from 0.75 to 1.20, in increments of 0.05, as this is where the greatest divergence was visually identified,” the researchers wrote. “The goal was to identify if a percentage of voxels at a set SUV could detect lymphomatous involvement.”
The researchers identified 10 candidate thresholds in the developmental phase; 4 of these performed better than the others in the validation phase. Using those thresholds, 10 of the 12 patients in the validation cohort could be correctly staged using FDG PET/CT.
Further analysis identified a single threshold that performed best: If greater than 38% of the voxels (averaging 1,734 voxels) demonstrated an SUV of less than 0.95, the sensitivity was 100% and the specificity was 80%.
The researchers acknowledged that the findings are limited because of the study’s small sample size and said the results should be validated in a larger trial.
There was no external funding for the study and the researchers reported having no financial disclosures.
SOURCE: Morgan R et al. Clin Lymphoma Myeloma Leuk. 2018 Jul 4. doi: 10.1016/j.clml.2018.06.024.
Bone marrow involvement in mantle cell lymphoma could be assessed using just 18fluorodeoxyglucose (FDG)–PET/CT, according to findings from a small, retrospective study published in Clinical Lymphoma, Myeloma & Leukemia.
Rustain Morgan, MD, of the University of Colorado, Aurora, and his colleagues found that, at a certain threshold of bone marrow voxels in standard uptake value (SUV), there was 100% sensitivity and 80% specificity in determining bone marrow involvement in mantle cell lymphoma (MCL).
Currently, National Comprehensive Cancer Network guidelines call for bone marrow biopsy and whole body FDG PET/CT scan to complete an initial diagnosis of MCL.
“One of the most important factors for correct staging is the identification of bone marrow involvement, occurring in approximately 55% of patients with MCL, which classifies patients as advanced stage. However, accurate analysis of bone marrow involvement can be challenging due to sampling error,” the researchers wrote. “While bone marrow biopsy remains the gold standard, it is not a perfect standard given unilateral variability.”
In previous studies, FDG PET/CT was not considered sensitive enough to detect gastrointestinal or bone marrow involvement. However, these earlier studies used SUV maximum or mean or a visual assessment of the bone marrow activity, compared with hepatic uptake. To address this issue, the researchers developed a new method of examining SUV distribution throughout the pelvic bones by analyzing thousands of bone marrow voxels within the bilateral iliacs.
During the developmental phase, an institutional dataset of 11 patients with MCL was used to define the voxel-based analysis. These patients had undergone both unilateral iliac bone marrow biopsy and FDG PET/CT at the initial diagnosis. Then, FDG PET/CT scans from another 12 patients with MCL from a different institution were used to validate the developmental phase findings. Finally, a control group of 5 people with no known malignancy were referred for FDG PET/CT pulmonary nodule evaluation.
“The hypothesis of the study was that, if the bone marrow was involved by lymphoma, then there would be a small increase in the SUV of each voxel, reflecting involvement by the lymphoma. In order to capture such changes, we analyzed the percent of total voxels in SUV ranging from 0.75 to 1.20, in increments of 0.05, as this is where the greatest divergence was visually identified,” the researchers wrote. “The goal was to identify if a percentage of voxels at a set SUV could detect lymphomatous involvement.”
The researchers identified 10 candidate thresholds in the developmental phase; 4 of these performed better than the others in the validation phase. Using those thresholds, 10 of the 12 patients in the validation cohort could be correctly staged using FDG PET/CT.
Further analysis identified a single threshold that performed best: If greater than 38% of the voxels (averaging 1,734 voxels) demonstrated an SUV of less than 0.95, the sensitivity was 100% and the specificity was 80%.
The researchers acknowledged that the findings are limited because of the study’s small sample size and said the results should be validated in a larger trial.
There was no external funding for the study and the researchers reported having no financial disclosures.
SOURCE: Morgan R et al. Clin Lymphoma Myeloma Leuk. 2018 Jul 4. doi: 10.1016/j.clml.2018.06.024.
Bone marrow involvement in mantle cell lymphoma could be assessed using just 18fluorodeoxyglucose (FDG)–PET/CT, according to findings from a small, retrospective study published in Clinical Lymphoma, Myeloma & Leukemia.
Rustain Morgan, MD, of the University of Colorado, Aurora, and his colleagues found that, at a certain threshold of bone marrow voxels in standard uptake value (SUV), there was 100% sensitivity and 80% specificity in determining bone marrow involvement in mantle cell lymphoma (MCL).
Currently, National Comprehensive Cancer Network guidelines call for bone marrow biopsy and whole body FDG PET/CT scan to complete an initial diagnosis of MCL.
“One of the most important factors for correct staging is the identification of bone marrow involvement, occurring in approximately 55% of patients with MCL, which classifies patients as advanced stage. However, accurate analysis of bone marrow involvement can be challenging due to sampling error,” the researchers wrote. “While bone marrow biopsy remains the gold standard, it is not a perfect standard given unilateral variability.”
In previous studies, FDG PET/CT was not considered sensitive enough to detect gastrointestinal or bone marrow involvement. However, these earlier studies used SUV maximum or mean or a visual assessment of the bone marrow activity, compared with hepatic uptake. To address this issue, the researchers developed a new method of examining SUV distribution throughout the pelvic bones by analyzing thousands of bone marrow voxels within the bilateral iliacs.
During the developmental phase, an institutional dataset of 11 patients with MCL was used to define the voxel-based analysis. These patients had undergone both unilateral iliac bone marrow biopsy and FDG PET/CT at the initial diagnosis. Then, FDG PET/CT scans from another 12 patients with MCL from a different institution were used to validate the developmental phase findings. Finally, a control group of 5 people with no known malignancy were referred for FDG PET/CT pulmonary nodule evaluation.
“The hypothesis of the study was that, if the bone marrow was involved by lymphoma, then there would be a small increase in the SUV of each voxel, reflecting involvement by the lymphoma. In order to capture such changes, we analyzed the percent of total voxels in SUV ranging from 0.75 to 1.20, in increments of 0.05, as this is where the greatest divergence was visually identified,” the researchers wrote. “The goal was to identify if a percentage of voxels at a set SUV could detect lymphomatous involvement.”
The researchers identified 10 candidate thresholds in the developmental phase; 4 of these performed better than the others in the validation phase. Using those thresholds, 10 of the 12 patients in the validation cohort could be correctly staged using FDG PET/CT.
Further analysis identified a single threshold that performed best: If greater than 38% of the voxels (averaging 1,734 voxels) demonstrated an SUV of less than 0.95, the sensitivity was 100% and the specificity was 80%.
The researchers acknowledged that the findings are limited because of the study’s small sample size and said the results should be validated in a larger trial.
There was no external funding for the study and the researchers reported having no financial disclosures.
SOURCE: Morgan R et al. Clin Lymphoma Myeloma Leuk. 2018 Jul 4. doi: 10.1016/j.clml.2018.06.024.
REPORTING FROM CLINICAL LYMPHOMA, MYELOMA & LEUKEMIA
Key clinical point:
Major finding: If greater than 38% of the voxels demonstrated an standard uptake value of less than 0.95, there was a sensitivity of 100% and a specificity of 80%.
Study details: A retrospective cohort study of 23 patients with mantle cell leukemia and 5 controls.
Disclosures: There was no external funding for the study and the researchers reported having no financial disclosures.
Source: Morgan R et al. Clin Lymphoma Myeloma Leuk. 2018 Jul 4. doi: 10.1016/j.clml.2018.06.024.
Lenalidomide becomes standard of care for multiple myeloma in the maintenance setting
The treatment of multiple myeloma has been revolutionized in the past few decades, with the introduction of numerous novel drug classes that have more than doubled median survival times. The immunomodulatory drug (IMiD), lenalidomide, forms the backbone of the majority of treatment paradigms, first receiving US Food and Drug Administration approval in 2006 for use in combination with dexamethasone in previously treated patients with multiple myeloma. Since then, approved indications for lenalidomide in multiple myeloma have continued to expand.
Most recently, on February 22, 2017, lenalidomide was approved for use as maintenance therapy following autologous stem cell transplant (ASCT), making it the first and only treatment available in this setting. This approval was based on 2 randomized, controlled trials that evaluated the efficacy and safety of lenalidomide in more than 1,000 patients in this setting and demonstrated a significant advantage in progression-free survival (PFS) compared with patients receiving placebo.
CALGB 1001041 and IFM 2005-022 were randomized, double-blind phase 3 trials conducted at 47 locations across the United States and 78 centers in France, Belgium, and Switzerland, respectively. In the CALGB trial, eligible patients were 18-70 years of age, with a European Cooperative Oncology Group (ECOG) performance status of 0 or 1, symptomatic disease requiring treatment (Durie-Salmon stage ≥1), and who received any induction therapy of 2-12 months duration. In the IFM trial, eligible patients were younger than 65 years, with multiple myeloma that had not progressed in the interval between first-line ASCT, performed within the previous 6 months, and randomization, and who had normal liver function tests and blood cell counts.
In CALGB 100104, after undergoing ASCT, 460 patients were randomly assigned to lenalidomide (starting at a dose of 10 mg/day) or placebo between day 100 and day 110 after transplantation. In IFM 2005-02, after undergoing ASCT, 614 patients were randomized 1:1 to receive either consolidation treatment with lenalidomide (at a dose of 25 mg/day on days 1-21 of each 28-day cycle for 2 cycles) followed by maintenance with lenalidomide (10 mg/day for the first 3 months, increasing to 15 mg if tolerated), or the same consolidation treatment followed by maintenance therapy with placebo.
The primary endpoint of CALGB 100104 was time to progression (TTP) and lenalidomide was associated with a significantly longer TTP. Median PFS was also improved by around 15 months (hazard ratio [HR], 0.38; P < .001). In a more recent long-term PFS analysis, median PFS was 5.7 years in the lenalidomide arm compared with 1.9 years with placebo, a difference of 3.8 years (HR, 0.38).3
The primary endpoint for IFM 2005-02 was PFS and lenalidomide maintenance therapy resulted in a significant improvement in PFS in both the originally published study (18-month PFS advantage) and long-term follow-up. The most recent PFS analysis demonstrated a PFS of 3.9 years for lenalidomide, compared with 2 years for no maintenance, a difference of 1.9 years (HR, 0.53). Although the studies were not powered for an overall survival (OS) endpoint, a descriptive analysis showed a median OS of 9.3 years, compared with 7 years in CALGB 100104, and 8.8 years compared with 7.3 years in IFM 2005-02.
In a meta-analysis of data pooled from these 2 studies and a third randomized trial (GIMEMA-RVMM-PI-209),4 which was presented at the 2016 annual meeting of the American Society of Clinical Oncology, maintenance therapy with lenalidomide following frontline treatment with high-dose melphalan and ASCT reduced the risk of death by 26% compared with placebo or no maintenance therapy, prompting suggestions that lenalidomide become standard of care in this setting.
The safety profile of lenalidomide in this setting was similar to that previously described in other studies. The most frequently reported adverse events (AEs), across both studies, were neutropenia, thrombocytopenia, leukopenia, anemia, upper respiratory tract infection, bronchitis, nasopharyngitis, cough, gastroenteritis, diarrhea, rash, fatigue, asthenia, muscle spasm, and pyrexia. The most common grade 3/4 AEs included neutropenia, thrombocytopenia, and leukopenia. AEs were generally most common in the first 6 months of treatment and subsequently declined in frequency over time or remained stable.
The prescribing information carries warnings and precautions about embryo-fetal toxicity, hematologic toxicity, venous/arterial thromboembolic events, secondary primary malignancies, hepatotoxicity, allergic reactions, tumor lysis syndrome, and thyroid disorders.5 Given its teratogenic effects, lenalidomide is only available through a restricted program under a risk evaluation mitigation strategy.
Patients with neutropenia should be monitored for signs of infections, patients advised to look for signs of bleeding or bruising, and weekly complete blood count performed for the first 2 cycles, on days 1 and 15 of cycle 3 and every 4 weeks thereafter.
Action should be taken to try to reduce the risk of venous and arterial thromboembolic events where possible and thrombophylaxis is recommended, based on the assessment of the underlying risk. Since lenalidomide can increase the risk of secondary primary malignancies, each case should be evaluated for risk-to-benefit ratio.
Liver enzymes should be monitored periodically and treatment interrupted upon their elevation, resuming at a lower dose if levels return to baseline values. Patients who have a history of grade 4 rash following thalidomide treatment should not receive lenalidomide. If grade 2-3 skin rash occurs, treatment interruption or discontinuation should be considered and lenalidomide should be discontinued in the event of angioedema, grade 4 rash, exfoliative or bullous rash, or if Stevens-Johnson s
Patients with high tumor burden prior to treatment are at highest risk of tumor lysis syndrome and should be monitored closely and appropriate precautions taken, and thyroid function should be measured before and during lenalidomide treatment to address potential thyroid disorders. Lenalidomide is marketed as Revlimid by Celgene Corporation.
The treatment of multiple myeloma has been revolutionized in the past few decades, with the introduction of numerous novel drug classes that have more than doubled median survival times. The immunomodulatory drug (IMiD), lenalidomide, forms the backbone of the majority of treatment paradigms, first receiving US Food and Drug Administration approval in 2006 for use in combination with dexamethasone in previously treated patients with multiple myeloma. Since then, approved indications for lenalidomide in multiple myeloma have continued to expand.
Most recently, on February 22, 2017, lenalidomide was approved for use as maintenance therapy following autologous stem cell transplant (ASCT), making it the first and only treatment available in this setting. This approval was based on 2 randomized, controlled trials that evaluated the efficacy and safety of lenalidomide in more than 1,000 patients in this setting and demonstrated a significant advantage in progression-free survival (PFS) compared with patients receiving placebo.
CALGB 1001041 and IFM 2005-022 were randomized, double-blind phase 3 trials conducted at 47 locations across the United States and 78 centers in France, Belgium, and Switzerland, respectively. In the CALGB trial, eligible patients were 18-70 years of age, with a European Cooperative Oncology Group (ECOG) performance status of 0 or 1, symptomatic disease requiring treatment (Durie-Salmon stage ≥1), and who received any induction therapy of 2-12 months duration. In the IFM trial, eligible patients were younger than 65 years, with multiple myeloma that had not progressed in the interval between first-line ASCT, performed within the previous 6 months, and randomization, and who had normal liver function tests and blood cell counts.
In CALGB 100104, after undergoing ASCT, 460 patients were randomly assigned to lenalidomide (starting at a dose of 10 mg/day) or placebo between day 100 and day 110 after transplantation. In IFM 2005-02, after undergoing ASCT, 614 patients were randomized 1:1 to receive either consolidation treatment with lenalidomide (at a dose of 25 mg/day on days 1-21 of each 28-day cycle for 2 cycles) followed by maintenance with lenalidomide (10 mg/day for the first 3 months, increasing to 15 mg if tolerated), or the same consolidation treatment followed by maintenance therapy with placebo.
The primary endpoint of CALGB 100104 was time to progression (TTP) and lenalidomide was associated with a significantly longer TTP. Median PFS was also improved by around 15 months (hazard ratio [HR], 0.38; P < .001). In a more recent long-term PFS analysis, median PFS was 5.7 years in the lenalidomide arm compared with 1.9 years with placebo, a difference of 3.8 years (HR, 0.38).3
The primary endpoint for IFM 2005-02 was PFS and lenalidomide maintenance therapy resulted in a significant improvement in PFS in both the originally published study (18-month PFS advantage) and long-term follow-up. The most recent PFS analysis demonstrated a PFS of 3.9 years for lenalidomide, compared with 2 years for no maintenance, a difference of 1.9 years (HR, 0.53). Although the studies were not powered for an overall survival (OS) endpoint, a descriptive analysis showed a median OS of 9.3 years, compared with 7 years in CALGB 100104, and 8.8 years compared with 7.3 years in IFM 2005-02.
In a meta-analysis of data pooled from these 2 studies and a third randomized trial (GIMEMA-RVMM-PI-209),4 which was presented at the 2016 annual meeting of the American Society of Clinical Oncology, maintenance therapy with lenalidomide following frontline treatment with high-dose melphalan and ASCT reduced the risk of death by 26% compared with placebo or no maintenance therapy, prompting suggestions that lenalidomide become standard of care in this setting.
The safety profile of lenalidomide in this setting was similar to that previously described in other studies. The most frequently reported adverse events (AEs), across both studies, were neutropenia, thrombocytopenia, leukopenia, anemia, upper respiratory tract infection, bronchitis, nasopharyngitis, cough, gastroenteritis, diarrhea, rash, fatigue, asthenia, muscle spasm, and pyrexia. The most common grade 3/4 AEs included neutropenia, thrombocytopenia, and leukopenia. AEs were generally most common in the first 6 months of treatment and subsequently declined in frequency over time or remained stable.
The prescribing information carries warnings and precautions about embryo-fetal toxicity, hematologic toxicity, venous/arterial thromboembolic events, secondary primary malignancies, hepatotoxicity, allergic reactions, tumor lysis syndrome, and thyroid disorders.5 Given its teratogenic effects, lenalidomide is only available through a restricted program under a risk evaluation mitigation strategy.
Patients with neutropenia should be monitored for signs of infections, patients advised to look for signs of bleeding or bruising, and weekly complete blood count performed for the first 2 cycles, on days 1 and 15 of cycle 3 and every 4 weeks thereafter.
Action should be taken to try to reduce the risk of venous and arterial thromboembolic events where possible and thrombophylaxis is recommended, based on the assessment of the underlying risk. Since lenalidomide can increase the risk of secondary primary malignancies, each case should be evaluated for risk-to-benefit ratio.
Liver enzymes should be monitored periodically and treatment interrupted upon their elevation, resuming at a lower dose if levels return to baseline values. Patients who have a history of grade 4 rash following thalidomide treatment should not receive lenalidomide. If grade 2-3 skin rash occurs, treatment interruption or discontinuation should be considered and lenalidomide should be discontinued in the event of angioedema, grade 4 rash, exfoliative or bullous rash, or if Stevens-Johnson s
Patients with high tumor burden prior to treatment are at highest risk of tumor lysis syndrome and should be monitored closely and appropriate precautions taken, and thyroid function should be measured before and during lenalidomide treatment to address potential thyroid disorders. Lenalidomide is marketed as Revlimid by Celgene Corporation.
The treatment of multiple myeloma has been revolutionized in the past few decades, with the introduction of numerous novel drug classes that have more than doubled median survival times. The immunomodulatory drug (IMiD), lenalidomide, forms the backbone of the majority of treatment paradigms, first receiving US Food and Drug Administration approval in 2006 for use in combination with dexamethasone in previously treated patients with multiple myeloma. Since then, approved indications for lenalidomide in multiple myeloma have continued to expand.
Most recently, on February 22, 2017, lenalidomide was approved for use as maintenance therapy following autologous stem cell transplant (ASCT), making it the first and only treatment available in this setting. This approval was based on 2 randomized, controlled trials that evaluated the efficacy and safety of lenalidomide in more than 1,000 patients in this setting and demonstrated a significant advantage in progression-free survival (PFS) compared with patients receiving placebo.
CALGB 1001041 and IFM 2005-022 were randomized, double-blind phase 3 trials conducted at 47 locations across the United States and 78 centers in France, Belgium, and Switzerland, respectively. In the CALGB trial, eligible patients were 18-70 years of age, with a European Cooperative Oncology Group (ECOG) performance status of 0 or 1, symptomatic disease requiring treatment (Durie-Salmon stage ≥1), and who received any induction therapy of 2-12 months duration. In the IFM trial, eligible patients were younger than 65 years, with multiple myeloma that had not progressed in the interval between first-line ASCT, performed within the previous 6 months, and randomization, and who had normal liver function tests and blood cell counts.
In CALGB 100104, after undergoing ASCT, 460 patients were randomly assigned to lenalidomide (starting at a dose of 10 mg/day) or placebo between day 100 and day 110 after transplantation. In IFM 2005-02, after undergoing ASCT, 614 patients were randomized 1:1 to receive either consolidation treatment with lenalidomide (at a dose of 25 mg/day on days 1-21 of each 28-day cycle for 2 cycles) followed by maintenance with lenalidomide (10 mg/day for the first 3 months, increasing to 15 mg if tolerated), or the same consolidation treatment followed by maintenance therapy with placebo.
The primary endpoint of CALGB 100104 was time to progression (TTP) and lenalidomide was associated with a significantly longer TTP. Median PFS was also improved by around 15 months (hazard ratio [HR], 0.38; P < .001). In a more recent long-term PFS analysis, median PFS was 5.7 years in the lenalidomide arm compared with 1.9 years with placebo, a difference of 3.8 years (HR, 0.38).3
The primary endpoint for IFM 2005-02 was PFS and lenalidomide maintenance therapy resulted in a significant improvement in PFS in both the originally published study (18-month PFS advantage) and long-term follow-up. The most recent PFS analysis demonstrated a PFS of 3.9 years for lenalidomide, compared with 2 years for no maintenance, a difference of 1.9 years (HR, 0.53). Although the studies were not powered for an overall survival (OS) endpoint, a descriptive analysis showed a median OS of 9.3 years, compared with 7 years in CALGB 100104, and 8.8 years compared with 7.3 years in IFM 2005-02.
In a meta-analysis of data pooled from these 2 studies and a third randomized trial (GIMEMA-RVMM-PI-209),4 which was presented at the 2016 annual meeting of the American Society of Clinical Oncology, maintenance therapy with lenalidomide following frontline treatment with high-dose melphalan and ASCT reduced the risk of death by 26% compared with placebo or no maintenance therapy, prompting suggestions that lenalidomide become standard of care in this setting.
The safety profile of lenalidomide in this setting was similar to that previously described in other studies. The most frequently reported adverse events (AEs), across both studies, were neutropenia, thrombocytopenia, leukopenia, anemia, upper respiratory tract infection, bronchitis, nasopharyngitis, cough, gastroenteritis, diarrhea, rash, fatigue, asthenia, muscle spasm, and pyrexia. The most common grade 3/4 AEs included neutropenia, thrombocytopenia, and leukopenia. AEs were generally most common in the first 6 months of treatment and subsequently declined in frequency over time or remained stable.
The prescribing information carries warnings and precautions about embryo-fetal toxicity, hematologic toxicity, venous/arterial thromboembolic events, secondary primary malignancies, hepatotoxicity, allergic reactions, tumor lysis syndrome, and thyroid disorders.5 Given its teratogenic effects, lenalidomide is only available through a restricted program under a risk evaluation mitigation strategy.
Patients with neutropenia should be monitored for signs of infections, patients advised to look for signs of bleeding or bruising, and weekly complete blood count performed for the first 2 cycles, on days 1 and 15 of cycle 3 and every 4 weeks thereafter.
Action should be taken to try to reduce the risk of venous and arterial thromboembolic events where possible and thrombophylaxis is recommended, based on the assessment of the underlying risk. Since lenalidomide can increase the risk of secondary primary malignancies, each case should be evaluated for risk-to-benefit ratio.
Liver enzymes should be monitored periodically and treatment interrupted upon their elevation, resuming at a lower dose if levels return to baseline values. Patients who have a history of grade 4 rash following thalidomide treatment should not receive lenalidomide. If grade 2-3 skin rash occurs, treatment interruption or discontinuation should be considered and lenalidomide should be discontinued in the event of angioedema, grade 4 rash, exfoliative or bullous rash, or if Stevens-Johnson s
Patients with high tumor burden prior to treatment are at highest risk of tumor lysis syndrome and should be monitored closely and appropriate precautions taken, and thyroid function should be measured before and during lenalidomide treatment to address potential thyroid disorders. Lenalidomide is marketed as Revlimid by Celgene Corporation.
Expanded approval for daratumumab in multiple myeloma
In November 2016, the US Food and Drug Administration expanded the approval of daratumumab for patients with multiple myeloma. The monoclonal antibody, which targets CD38, a protein that is highly expressed on the surface of multiple myeloma cells, was previously granted approval by the agency as a single agent for the treatment of patients who had received at least three previous therapies.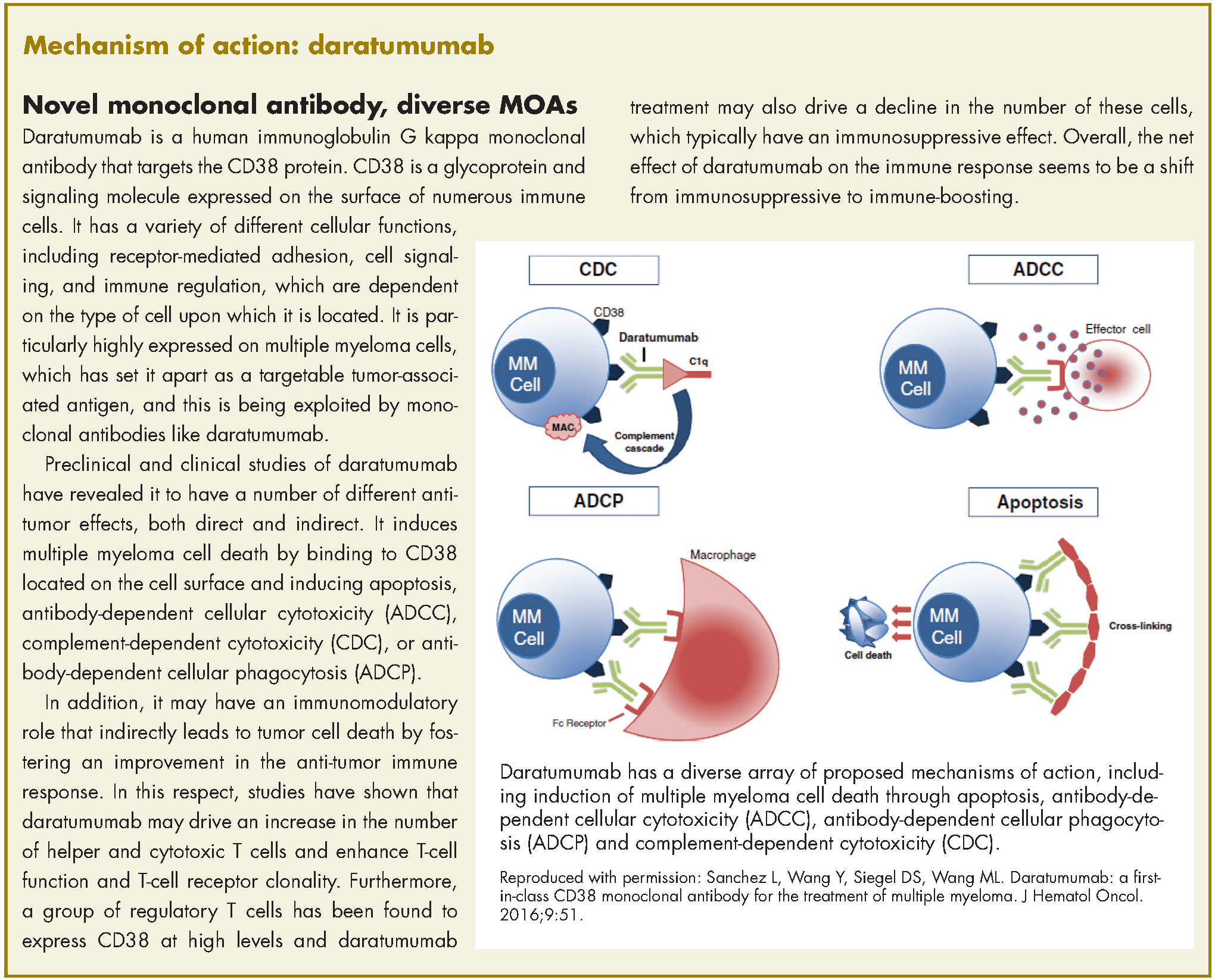
The current approval was for the use of daratumumab in two different combination regimens for the treatment of patients who have received one previous line of treatment. On the basis of improved progression-free survival (PFS), demonstrated in two randomized, open-label, phase 3 trials, daratumumab can now be used in combination with the immunomodulatory agent lenalidomide and dexamethasone, or the proteasome inhibitor bortezomib and dexamethasone, both standard therapies for the treatment of multiple myeloma.
In the POLLUX trial, 569 patients with relapsed/refractory multiple myeloma were randomized 1:1 to receive daratumumab in combination with lenalidomide-dexamethasone or lenalidomide-dexamethasone alone. The CASTOR trial randomized 498 patients with relapsed/refractory multiple myeloma 1:1 to daratumumab in combination with bortezomib-dexamethasone, or bortezomib-dexamethasone alone.
The eligibility and exclusion criteria for both trials were similar; patients had received at least one previous line of therapy, had documented progressive disease according to International Myeloma Working Group criteria, and had measurable disease on the basis of urine and/or serum assessments or serum-free, light-chain assay.
Patients with a neutrophil count of ≤1,000 cells/mm3, hemoglobin level of ≤7.5 g/dL, platelet count of <75,000 cells/mm3, creatinine clearance of ≤20 mL/min per 1.73m2 body surface area (or <30 mL/min in the POLLUX trial), alanine aminotransferase or aspartate aminotransferase level ≥2.5 times the upper limit of normal (ULN) range, bilirubin level of ≥1.5 or more times the ULN range, disease refractory to bortezomib or lenalidomide, and unacceptable side effects from bortezomib or lenalidomide, were ineligible for these studies. In addition, patients with grade 2 or higher peripheral neuropathy or neuropathic pain, were excluded from the CASTOR study.
Randomization was stratified according to International Staging System disease stage at the time of screening (stage I, II or III, with higher stage indicating more severe disease), number of previous lines of therapy (1 vs 2, or 3 vs >3), and previous receipt of lenalidomide or bortezomib.
In the CASTOR trial, patients received up to eight 21-day cycles of bortezomib, administered subcutaneously at a dose of 1.3 mg/m2 on days 1, 4, 8, and 11 of cycles 1-8, and dexamethasone, administered orally or intravenously at a dose of 20 mg on days 1, 2, 4, 5, 8, 9, 11, and 12 for a total dose of 160 mg per cycle. Daratumumab was administered at a dose of 16 mg/kg intravenously once weekly on days 1, 8, and 15 during cycles 1 to 3, once every 3 weeks on day 1 of cycles 4-8, and once every 4 weeks thereafter.
In the POLLUX trial, patients were treated in 28-day cycles. Daratumumab was administered at the same dose as in the CASTOR trial, but on days 1, 8, 15 and 22 for 8 weeks during cycles 1 and 2, every 2 weeks on days 1 and 15 for 16 weeks during cycles 3 through 7, and every 4 weeks from then onwards. Lenalidomide was administered at a dose of 25 mg orally on days 1-21 of each cycle, and dexamethasone at a dose of 20 mg before infusion and 20 mg the following day.
The combination of daratumumab with lenalidomide-dexamethasone demonstrated a substantial improvement in PFS, compared with lenalidomide-dexamethasone alone (estimated PFS not yet reached vs 18.4 months, respectively; HR, 0.37; P < .0001), representing a 63% reduction in the risk of disease progression or death. Meanwhile, there was a 61% reduction in the risk of disease progression or death for the combination of daratumumab with bortezomib-dexamethasone in the CASTOR trial (estimated PFS not yet reached vs 7.2 months; HR: 0.39; P < .0001). The PFS benefit was observed across all prespecified subgroups in both studies.
In the CASTOR trial, over a median follow-up of 7.4 months, the overall response rate (ORR) was 82.9% for the combination arm, compared with 63.2% for the bortezomib-dexamethasone arm (P < .001), with a very good partial response (VGPR) or better rate of 59.2% compared with 29.1%, and a complete response (CR) rate of 19.2% compared with 9%. In the POLLUX trial, over a median follow-up of 13.5 months, ORR was 92.9% for the combination arm, compared with 76.4% for lenalidomide-dexamethasone, with a VGPR or better rate of 75.8% versus 44% and a CR rate of 43.1% versus 19.2%.
Overall, the safety profile for both combinations was consistent with what is usually observed with daratumumab monotherapy and lenalidomide-dexamethasone or bortezomib-dexamethasone combinations. The most frequently reported adverse events (AEs) were similar in both studies and included infusion reactions, diarrhea, and upper respiratory tract infection. In the POLLUX trial they also included nausea, fatigue, pyrexia, muscle spasm, cough, and dyspnea, whereas in the CASTOR trial patients also frequently experienced peripheral edema.
The most common grade 3/4 AEs in both trials were neutropenia (51.9% vs 37% in the POLLUX trial and 12.8 vs 4.2% in the CASTOR trial), thrombocytopenia (12.7% vs 13.5% and 45.3% vs 32.9%, respectively), and anemia (12.4% vs 19.6% and 14.4% vs 16%, respectively). The percentage of patients who discontinued treatment due to AEs was similar in both groups across the two studies; in the CASTOR trial discontinuations resulted most commonly from peripheral sensory neuropathy and pneumonia, while in the POLLUX trial, from pneumonia, pulmonary embolism and deterioration in general physical health.
The recommended dose for daratumumab in both combination regimens is 16 mg/kg intravenously, calculated on actual body weight. The dosing schedules begin with weekly administration during weeks 1-8 (when used in combination with lenalidomide-dexamethasone) and weeks 1-9 (for use with the bortezomib-dexamethasone combination), decreasing to every 2 weeks between weeks 9 and 24 or 10 and 24, respectively, and progressing to every 4 weeks from week 25 onward until disease progression and unacceptable toxicity.
Daratumumab is marketed as Darzalex by Janssen Biotech Inc. Neutropenia and thrombocytopenia have been added to the list of warnings and precautions for the prescribing information for these new indications. Complete blood cell count should be monitored periodically during treatment and daratumumab administration delayed to allow recovery of neutrophils or platelets. Supportive care with growth factors or transfusion should be considered in the event of neutropenia or thrombocytopenia, respectively.
1. Darzalex (daratumumab) injection, for intravenous use. Prescribing information. Janssen Biotech Inc. https://www.darzalexhcp.com/shared/product/darzalex/darzalex-prescribing-information.pdf. Released November 2016. Accessed January 8, 2017.
2. Palumbo A, Chanan-Khan A, Weisel K, et al. Daratumumab, bortezomib, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375:754-766.
3. Dimopoulos MA, Oriol A, Nahi H, et al. Daratumumab, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375:1319-1331.
In November 2016, the US Food and Drug Administration expanded the approval of daratumumab for patients with multiple myeloma. The monoclonal antibody, which targets CD38, a protein that is highly expressed on the surface of multiple myeloma cells, was previously granted approval by the agency as a single agent for the treatment of patients who had received at least three previous therapies.
The current approval was for the use of daratumumab in two different combination regimens for the treatment of patients who have received one previous line of treatment. On the basis of improved progression-free survival (PFS), demonstrated in two randomized, open-label, phase 3 trials, daratumumab can now be used in combination with the immunomodulatory agent lenalidomide and dexamethasone, or the proteasome inhibitor bortezomib and dexamethasone, both standard therapies for the treatment of multiple myeloma.
In the POLLUX trial, 569 patients with relapsed/refractory multiple myeloma were randomized 1:1 to receive daratumumab in combination with lenalidomide-dexamethasone or lenalidomide-dexamethasone alone. The CASTOR trial randomized 498 patients with relapsed/refractory multiple myeloma 1:1 to daratumumab in combination with bortezomib-dexamethasone, or bortezomib-dexamethasone alone.
The eligibility and exclusion criteria for both trials were similar; patients had received at least one previous line of therapy, had documented progressive disease according to International Myeloma Working Group criteria, and had measurable disease on the basis of urine and/or serum assessments or serum-free, light-chain assay.
Patients with a neutrophil count of ≤1,000 cells/mm3, hemoglobin level of ≤7.5 g/dL, platelet count of <75,000 cells/mm3, creatinine clearance of ≤20 mL/min per 1.73m2 body surface area (or <30 mL/min in the POLLUX trial), alanine aminotransferase or aspartate aminotransferase level ≥2.5 times the upper limit of normal (ULN) range, bilirubin level of ≥1.5 or more times the ULN range, disease refractory to bortezomib or lenalidomide, and unacceptable side effects from bortezomib or lenalidomide, were ineligible for these studies. In addition, patients with grade 2 or higher peripheral neuropathy or neuropathic pain, were excluded from the CASTOR study.
Randomization was stratified according to International Staging System disease stage at the time of screening (stage I, II or III, with higher stage indicating more severe disease), number of previous lines of therapy (1 vs 2, or 3 vs >3), and previous receipt of lenalidomide or bortezomib.
In the CASTOR trial, patients received up to eight 21-day cycles of bortezomib, administered subcutaneously at a dose of 1.3 mg/m2 on days 1, 4, 8, and 11 of cycles 1-8, and dexamethasone, administered orally or intravenously at a dose of 20 mg on days 1, 2, 4, 5, 8, 9, 11, and 12 for a total dose of 160 mg per cycle. Daratumumab was administered at a dose of 16 mg/kg intravenously once weekly on days 1, 8, and 15 during cycles 1 to 3, once every 3 weeks on day 1 of cycles 4-8, and once every 4 weeks thereafter.
In the POLLUX trial, patients were treated in 28-day cycles. Daratumumab was administered at the same dose as in the CASTOR trial, but on days 1, 8, 15 and 22 for 8 weeks during cycles 1 and 2, every 2 weeks on days 1 and 15 for 16 weeks during cycles 3 through 7, and every 4 weeks from then onwards. Lenalidomide was administered at a dose of 25 mg orally on days 1-21 of each cycle, and dexamethasone at a dose of 20 mg before infusion and 20 mg the following day.
The combination of daratumumab with lenalidomide-dexamethasone demonstrated a substantial improvement in PFS, compared with lenalidomide-dexamethasone alone (estimated PFS not yet reached vs 18.4 months, respectively; HR, 0.37; P < .0001), representing a 63% reduction in the risk of disease progression or death. Meanwhile, there was a 61% reduction in the risk of disease progression or death for the combination of daratumumab with bortezomib-dexamethasone in the CASTOR trial (estimated PFS not yet reached vs 7.2 months; HR: 0.39; P < .0001). The PFS benefit was observed across all prespecified subgroups in both studies.
In the CASTOR trial, over a median follow-up of 7.4 months, the overall response rate (ORR) was 82.9% for the combination arm, compared with 63.2% for the bortezomib-dexamethasone arm (P < .001), with a very good partial response (VGPR) or better rate of 59.2% compared with 29.1%, and a complete response (CR) rate of 19.2% compared with 9%. In the POLLUX trial, over a median follow-up of 13.5 months, ORR was 92.9% for the combination arm, compared with 76.4% for lenalidomide-dexamethasone, with a VGPR or better rate of 75.8% versus 44% and a CR rate of 43.1% versus 19.2%.
Overall, the safety profile for both combinations was consistent with what is usually observed with daratumumab monotherapy and lenalidomide-dexamethasone or bortezomib-dexamethasone combinations. The most frequently reported adverse events (AEs) were similar in both studies and included infusion reactions, diarrhea, and upper respiratory tract infection. In the POLLUX trial they also included nausea, fatigue, pyrexia, muscle spasm, cough, and dyspnea, whereas in the CASTOR trial patients also frequently experienced peripheral edema.
The most common grade 3/4 AEs in both trials were neutropenia (51.9% vs 37% in the POLLUX trial and 12.8 vs 4.2% in the CASTOR trial), thrombocytopenia (12.7% vs 13.5% and 45.3% vs 32.9%, respectively), and anemia (12.4% vs 19.6% and 14.4% vs 16%, respectively). The percentage of patients who discontinued treatment due to AEs was similar in both groups across the two studies; in the CASTOR trial discontinuations resulted most commonly from peripheral sensory neuropathy and pneumonia, while in the POLLUX trial, from pneumonia, pulmonary embolism and deterioration in general physical health.
The recommended dose for daratumumab in both combination regimens is 16 mg/kg intravenously, calculated on actual body weight. The dosing schedules begin with weekly administration during weeks 1-8 (when used in combination with lenalidomide-dexamethasone) and weeks 1-9 (for use with the bortezomib-dexamethasone combination), decreasing to every 2 weeks between weeks 9 and 24 or 10 and 24, respectively, and progressing to every 4 weeks from week 25 onward until disease progression and unacceptable toxicity.
Daratumumab is marketed as Darzalex by Janssen Biotech Inc. Neutropenia and thrombocytopenia have been added to the list of warnings and precautions for the prescribing information for these new indications. Complete blood cell count should be monitored periodically during treatment and daratumumab administration delayed to allow recovery of neutrophils or platelets. Supportive care with growth factors or transfusion should be considered in the event of neutropenia or thrombocytopenia, respectively.
In November 2016, the US Food and Drug Administration expanded the approval of daratumumab for patients with multiple myeloma. The monoclonal antibody, which targets CD38, a protein that is highly expressed on the surface of multiple myeloma cells, was previously granted approval by the agency as a single agent for the treatment of patients who had received at least three previous therapies.
The current approval was for the use of daratumumab in two different combination regimens for the treatment of patients who have received one previous line of treatment. On the basis of improved progression-free survival (PFS), demonstrated in two randomized, open-label, phase 3 trials, daratumumab can now be used in combination with the immunomodulatory agent lenalidomide and dexamethasone, or the proteasome inhibitor bortezomib and dexamethasone, both standard therapies for the treatment of multiple myeloma.
In the POLLUX trial, 569 patients with relapsed/refractory multiple myeloma were randomized 1:1 to receive daratumumab in combination with lenalidomide-dexamethasone or lenalidomide-dexamethasone alone. The CASTOR trial randomized 498 patients with relapsed/refractory multiple myeloma 1:1 to daratumumab in combination with bortezomib-dexamethasone, or bortezomib-dexamethasone alone.
The eligibility and exclusion criteria for both trials were similar; patients had received at least one previous line of therapy, had documented progressive disease according to International Myeloma Working Group criteria, and had measurable disease on the basis of urine and/or serum assessments or serum-free, light-chain assay.
Patients with a neutrophil count of ≤1,000 cells/mm3, hemoglobin level of ≤7.5 g/dL, platelet count of <75,000 cells/mm3, creatinine clearance of ≤20 mL/min per 1.73m2 body surface area (or <30 mL/min in the POLLUX trial), alanine aminotransferase or aspartate aminotransferase level ≥2.5 times the upper limit of normal (ULN) range, bilirubin level of ≥1.5 or more times the ULN range, disease refractory to bortezomib or lenalidomide, and unacceptable side effects from bortezomib or lenalidomide, were ineligible for these studies. In addition, patients with grade 2 or higher peripheral neuropathy or neuropathic pain, were excluded from the CASTOR study.
Randomization was stratified according to International Staging System disease stage at the time of screening (stage I, II or III, with higher stage indicating more severe disease), number of previous lines of therapy (1 vs 2, or 3 vs >3), and previous receipt of lenalidomide or bortezomib.
In the CASTOR trial, patients received up to eight 21-day cycles of bortezomib, administered subcutaneously at a dose of 1.3 mg/m2 on days 1, 4, 8, and 11 of cycles 1-8, and dexamethasone, administered orally or intravenously at a dose of 20 mg on days 1, 2, 4, 5, 8, 9, 11, and 12 for a total dose of 160 mg per cycle. Daratumumab was administered at a dose of 16 mg/kg intravenously once weekly on days 1, 8, and 15 during cycles 1 to 3, once every 3 weeks on day 1 of cycles 4-8, and once every 4 weeks thereafter.
In the POLLUX trial, patients were treated in 28-day cycles. Daratumumab was administered at the same dose as in the CASTOR trial, but on days 1, 8, 15 and 22 for 8 weeks during cycles 1 and 2, every 2 weeks on days 1 and 15 for 16 weeks during cycles 3 through 7, and every 4 weeks from then onwards. Lenalidomide was administered at a dose of 25 mg orally on days 1-21 of each cycle, and dexamethasone at a dose of 20 mg before infusion and 20 mg the following day.
The combination of daratumumab with lenalidomide-dexamethasone demonstrated a substantial improvement in PFS, compared with lenalidomide-dexamethasone alone (estimated PFS not yet reached vs 18.4 months, respectively; HR, 0.37; P < .0001), representing a 63% reduction in the risk of disease progression or death. Meanwhile, there was a 61% reduction in the risk of disease progression or death for the combination of daratumumab with bortezomib-dexamethasone in the CASTOR trial (estimated PFS not yet reached vs 7.2 months; HR: 0.39; P < .0001). The PFS benefit was observed across all prespecified subgroups in both studies.
In the CASTOR trial, over a median follow-up of 7.4 months, the overall response rate (ORR) was 82.9% for the combination arm, compared with 63.2% for the bortezomib-dexamethasone arm (P < .001), with a very good partial response (VGPR) or better rate of 59.2% compared with 29.1%, and a complete response (CR) rate of 19.2% compared with 9%. In the POLLUX trial, over a median follow-up of 13.5 months, ORR was 92.9% for the combination arm, compared with 76.4% for lenalidomide-dexamethasone, with a VGPR or better rate of 75.8% versus 44% and a CR rate of 43.1% versus 19.2%.
Overall, the safety profile for both combinations was consistent with what is usually observed with daratumumab monotherapy and lenalidomide-dexamethasone or bortezomib-dexamethasone combinations. The most frequently reported adverse events (AEs) were similar in both studies and included infusion reactions, diarrhea, and upper respiratory tract infection. In the POLLUX trial they also included nausea, fatigue, pyrexia, muscle spasm, cough, and dyspnea, whereas in the CASTOR trial patients also frequently experienced peripheral edema.
The most common grade 3/4 AEs in both trials were neutropenia (51.9% vs 37% in the POLLUX trial and 12.8 vs 4.2% in the CASTOR trial), thrombocytopenia (12.7% vs 13.5% and 45.3% vs 32.9%, respectively), and anemia (12.4% vs 19.6% and 14.4% vs 16%, respectively). The percentage of patients who discontinued treatment due to AEs was similar in both groups across the two studies; in the CASTOR trial discontinuations resulted most commonly from peripheral sensory neuropathy and pneumonia, while in the POLLUX trial, from pneumonia, pulmonary embolism and deterioration in general physical health.
The recommended dose for daratumumab in both combination regimens is 16 mg/kg intravenously, calculated on actual body weight. The dosing schedules begin with weekly administration during weeks 1-8 (when used in combination with lenalidomide-dexamethasone) and weeks 1-9 (for use with the bortezomib-dexamethasone combination), decreasing to every 2 weeks between weeks 9 and 24 or 10 and 24, respectively, and progressing to every 4 weeks from week 25 onward until disease progression and unacceptable toxicity.
Daratumumab is marketed as Darzalex by Janssen Biotech Inc. Neutropenia and thrombocytopenia have been added to the list of warnings and precautions for the prescribing information for these new indications. Complete blood cell count should be monitored periodically during treatment and daratumumab administration delayed to allow recovery of neutrophils or platelets. Supportive care with growth factors or transfusion should be considered in the event of neutropenia or thrombocytopenia, respectively.
1. Darzalex (daratumumab) injection, for intravenous use. Prescribing information. Janssen Biotech Inc. https://www.darzalexhcp.com/shared/product/darzalex/darzalex-prescribing-information.pdf. Released November 2016. Accessed January 8, 2017.
2. Palumbo A, Chanan-Khan A, Weisel K, et al. Daratumumab, bortezomib, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375:754-766.
3. Dimopoulos MA, Oriol A, Nahi H, et al. Daratumumab, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375:1319-1331.
1. Darzalex (daratumumab) injection, for intravenous use. Prescribing information. Janssen Biotech Inc. https://www.darzalexhcp.com/shared/product/darzalex/darzalex-prescribing-information.pdf. Released November 2016. Accessed January 8, 2017.
2. Palumbo A, Chanan-Khan A, Weisel K, et al. Daratumumab, bortezomib, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375:754-766.
3. Dimopoulos MA, Oriol A, Nahi H, et al. Daratumumab, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375:1319-1331.
Treatment improves PFS in early stage FL

A multidrug regimen can improve upon involved-field radiotherapy (IFRT) in patients with early stage follicular lymphoma (FL), according to research published in the Journal of Clinical Oncology.
FL patients who received IFRT plus cyclophosphamide, vincristine, and prednisolone (CVP)—with or without rituximab—had a significant improvement in progression-free survival (PFS) compared to patients who received standard treatment with IFRT alone.
However, there was no significant difference in overall survival (OS) between the treatment arms.
“This is the first successful randomized study ever to be conducted in early stage follicular lymphoma comparing standard therapy to standard therapy plus effective chemotherapy or immunochemotherapy,” said Michael MacManus, MBBCh, of Peter MacCallum Cancer Centre in Melbourne, Victoria, Australia.
“It shows that the initial treatment received by patients can significantly affect their long-term chance of staying free from disease. Moving forward, we are interested in determining whether there is a benefit in overall long-term survival for patients treated with the combination with further follow-up, and if there is any way to predict if a person will benefit from combined treatment based on analyses of blood or biopsy specimens.”
Dr MacManus and his colleagues studied 150 patients with stage I to II, low-grade FL who were enrolled in this trial between 2000 and 2012.
At randomization, the patients’ median age was 57, 52% were male, 75% had stage I disease, and 48% had PET staging.
Half of patients (n=75) were randomized to receive IFRT (30-36 Gy) alone, and half were randomized to IFRT (30-36 Gy) plus 6 cycles of CVP. From 2006 on, patients in the CVP arm received rituximab (R) as well (n=31).
Baseline characteristics were well-balanced between the treatment arms.
Efficacy
The median follow-up was 9.6 years (range, 3.1 to 15.8 years).
PFS was significantly better among patients randomized to receive CVP±R (hazard ratio [HR]=0.57; P=0.033). The estimated 10-year PFS rate was 41% in the IFRT arm and 59% in the CVP±R arm.
Patients randomized to receive CVP plus R (n=31) had significantly better PFS than patients randomized to receive IFRT alone (n=31) over the same time period (HR=0.26; P=0.045).
There were 10 deaths in the IRFT arm and 5 in the CVP±R arm, but there was no significant difference in OS between the arms (HR=0.62; P=0.40). The 10-year OS rate was 86% in the IFRT arm and 95% in the CVP±R arm.
There was no significant between-arm difference in transformation to aggressive lymphoma (P=0.1). Transformation occurred in 10 patients in the IFRT arm and 4 in the CVP±R arm.
Safety
There were 148 patients from both arms who ultimately received IFRT, and 69 patients who received CVP±R.
Grade 2 toxicities occurring in more than 10% of IFRT recipients included upper gastrointestinal (n=27; 18%), skin (n=21; 14%), and mucous membrane (n=19; 12%) toxicity. One IFRT recipient had grade 3 mucositis, and 1 had grade 4 esophageal/pharyngeal mucosal toxicity.
Grade 3 toxicities occurring in at least 2 patients in the CVP±R arm included neutropenia (n=10; 14%), infection (n=8; 12%), diarrhea (n=3; 4%), elevated gamma-glutamyl transferase (n=3; 4%), fatigue (n=3; 4%), and febrile neutropenia (n=3; 4%).
Three patients (4%) in the CVP±R arm had acute grade 3 neuropathy related to vincristine. Ten patients (14%) had grade 4 neutropenia.
The most common late toxicities for the entire patient cohort were salivary gland (n=8; 5%) and skin (n=4; 3%) toxicities.
Grade 3 lung and menopausal toxicities occurred in 1 patient each. Two patients had late grade 3 vincristine neuropathy. One patient who had grade 3 neuropathy during chemotherapy progressed to grade 4.
A multidrug regimen can improve upon involved-field radiotherapy (IFRT) in patients with early stage follicular lymphoma (FL), according to research published in the Journal of Clinical Oncology.
FL patients who received IFRT plus cyclophosphamide, vincristine, and prednisolone (CVP)—with or without rituximab—had a significant improvement in progression-free survival (PFS) compared to patients who received standard treatment with IFRT alone.
However, there was no significant difference in overall survival (OS) between the treatment arms.
“This is the first successful randomized study ever to be conducted in early stage follicular lymphoma comparing standard therapy to standard therapy plus effective chemotherapy or immunochemotherapy,” said Michael MacManus, MBBCh, of Peter MacCallum Cancer Centre in Melbourne, Victoria, Australia.
“It shows that the initial treatment received by patients can significantly affect their long-term chance of staying free from disease. Moving forward, we are interested in determining whether there is a benefit in overall long-term survival for patients treated with the combination with further follow-up, and if there is any way to predict if a person will benefit from combined treatment based on analyses of blood or biopsy specimens.”
Dr MacManus and his colleagues studied 150 patients with stage I to II, low-grade FL who were enrolled in this trial between 2000 and 2012.
At randomization, the patients’ median age was 57, 52% were male, 75% had stage I disease, and 48% had PET staging.
Half of patients (n=75) were randomized to receive IFRT (30-36 Gy) alone, and half were randomized to IFRT (30-36 Gy) plus 6 cycles of CVP. From 2006 on, patients in the CVP arm received rituximab (R) as well (n=31).
Baseline characteristics were well-balanced between the treatment arms.
Efficacy
The median follow-up was 9.6 years (range, 3.1 to 15.8 years).
PFS was significantly better among patients randomized to receive CVP±R (hazard ratio [HR]=0.57; P=0.033). The estimated 10-year PFS rate was 41% in the IFRT arm and 59% in the CVP±R arm.
Patients randomized to receive CVP plus R (n=31) had significantly better PFS than patients randomized to receive IFRT alone (n=31) over the same time period (HR=0.26; P=0.045).
There were 10 deaths in the IRFT arm and 5 in the CVP±R arm, but there was no significant difference in OS between the arms (HR=0.62; P=0.40). The 10-year OS rate was 86% in the IFRT arm and 95% in the CVP±R arm.
There was no significant between-arm difference in transformation to aggressive lymphoma (P=0.1). Transformation occurred in 10 patients in the IFRT arm and 4 in the CVP±R arm.
Safety
There were 148 patients from both arms who ultimately received IFRT, and 69 patients who received CVP±R.
Grade 2 toxicities occurring in more than 10% of IFRT recipients included upper gastrointestinal (n=27; 18%), skin (n=21; 14%), and mucous membrane (n=19; 12%) toxicity. One IFRT recipient had grade 3 mucositis, and 1 had grade 4 esophageal/pharyngeal mucosal toxicity.
Grade 3 toxicities occurring in at least 2 patients in the CVP±R arm included neutropenia (n=10; 14%), infection (n=8; 12%), diarrhea (n=3; 4%), elevated gamma-glutamyl transferase (n=3; 4%), fatigue (n=3; 4%), and febrile neutropenia (n=3; 4%).
Three patients (4%) in the CVP±R arm had acute grade 3 neuropathy related to vincristine. Ten patients (14%) had grade 4 neutropenia.
The most common late toxicities for the entire patient cohort were salivary gland (n=8; 5%) and skin (n=4; 3%) toxicities.
Grade 3 lung and menopausal toxicities occurred in 1 patient each. Two patients had late grade 3 vincristine neuropathy. One patient who had grade 3 neuropathy during chemotherapy progressed to grade 4.
A multidrug regimen can improve upon involved-field radiotherapy (IFRT) in patients with early stage follicular lymphoma (FL), according to research published in the Journal of Clinical Oncology.
FL patients who received IFRT plus cyclophosphamide, vincristine, and prednisolone (CVP)—with or without rituximab—had a significant improvement in progression-free survival (PFS) compared to patients who received standard treatment with IFRT alone.
However, there was no significant difference in overall survival (OS) between the treatment arms.
“This is the first successful randomized study ever to be conducted in early stage follicular lymphoma comparing standard therapy to standard therapy plus effective chemotherapy or immunochemotherapy,” said Michael MacManus, MBBCh, of Peter MacCallum Cancer Centre in Melbourne, Victoria, Australia.
“It shows that the initial treatment received by patients can significantly affect their long-term chance of staying free from disease. Moving forward, we are interested in determining whether there is a benefit in overall long-term survival for patients treated with the combination with further follow-up, and if there is any way to predict if a person will benefit from combined treatment based on analyses of blood or biopsy specimens.”
Dr MacManus and his colleagues studied 150 patients with stage I to II, low-grade FL who were enrolled in this trial between 2000 and 2012.
At randomization, the patients’ median age was 57, 52% were male, 75% had stage I disease, and 48% had PET staging.
Half of patients (n=75) were randomized to receive IFRT (30-36 Gy) alone, and half were randomized to IFRT (30-36 Gy) plus 6 cycles of CVP. From 2006 on, patients in the CVP arm received rituximab (R) as well (n=31).
Baseline characteristics were well-balanced between the treatment arms.
Efficacy
The median follow-up was 9.6 years (range, 3.1 to 15.8 years).
PFS was significantly better among patients randomized to receive CVP±R (hazard ratio [HR]=0.57; P=0.033). The estimated 10-year PFS rate was 41% in the IFRT arm and 59% in the CVP±R arm.
Patients randomized to receive CVP plus R (n=31) had significantly better PFS than patients randomized to receive IFRT alone (n=31) over the same time period (HR=0.26; P=0.045).
There were 10 deaths in the IRFT arm and 5 in the CVP±R arm, but there was no significant difference in OS between the arms (HR=0.62; P=0.40). The 10-year OS rate was 86% in the IFRT arm and 95% in the CVP±R arm.
There was no significant between-arm difference in transformation to aggressive lymphoma (P=0.1). Transformation occurred in 10 patients in the IFRT arm and 4 in the CVP±R arm.
Safety
There were 148 patients from both arms who ultimately received IFRT, and 69 patients who received CVP±R.
Grade 2 toxicities occurring in more than 10% of IFRT recipients included upper gastrointestinal (n=27; 18%), skin (n=21; 14%), and mucous membrane (n=19; 12%) toxicity. One IFRT recipient had grade 3 mucositis, and 1 had grade 4 esophageal/pharyngeal mucosal toxicity.
Grade 3 toxicities occurring in at least 2 patients in the CVP±R arm included neutropenia (n=10; 14%), infection (n=8; 12%), diarrhea (n=3; 4%), elevated gamma-glutamyl transferase (n=3; 4%), fatigue (n=3; 4%), and febrile neutropenia (n=3; 4%).
Three patients (4%) in the CVP±R arm had acute grade 3 neuropathy related to vincristine. Ten patients (14%) had grade 4 neutropenia.
The most common late toxicities for the entire patient cohort were salivary gland (n=8; 5%) and skin (n=4; 3%) toxicities.
Grade 3 lung and menopausal toxicities occurred in 1 patient each. Two patients had late grade 3 vincristine neuropathy. One patient who had grade 3 neuropathy during chemotherapy progressed to grade 4.
Adult CCSs report financial hardships

Health-related financial hardship is common among adult survivors of childhood cancer, according to a study published in the Journal of the National Cancer Institute.
Researchers analyzed more than 2800 long-term childhood cancer survivors (CCSs) and found that 65% had financial challenges related to their cancer diagnosis.
“These findings suggest primary care doctors and oncologists should routinely screen childhood cancer survivors for possible financial hardship,” said I-Chan Huang, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
Specifically, Dr Huang recommends that healthcare providers routinely ask CCSs if they are unable to purchase medications, ever skip appointments for economic reasons, or worry about how to pay their medical bills.
For this study, Dr Huang and his colleagues analyzed data from 2811 CCSs. The subjects had a mean age of 31.8 (range, 18 to 65) and were a mean of 23.6 years from cancer diagnosis. Most (57.8%) had been diagnosed with hematologic malignancies, 32.0% with solid tumors, and 10.1% with central nervous system malignancies.
All subjects had been treated at St. Jude and enrolled in the St. Jude LIFE study. Participants return to St. Jude periodically for several days of clinical and functional assessments. Data for this study were collected during the CCSs’ first St. Jude LIFE evaluations.
Assessing hardship
The researchers measured 3 types of financial hardship—material, psychological, and coping/behavioral.
About 1 in 5 CCSs (22.4%) reported material financial hardship. In other words, their cancer had an impact on their financial situation.
More than half of CCSs (51.1%) reported psychological hardship—concern about their ability to pay for medical expenses.
And 33% of CCSs reported coping/behavioral hardship—an inability to see a doctor or go to the hospital due to finances.
Roughly 65% of CCSs reported at least 1 type of financial hardship.
All 3 types of hardship were significantly associated with somatization (all P<0.001), anxiety (all P<0.001), depression (all P<0.001), suicidal thoughts (all P<0.05), and difficulty in retirement planning (all P<0.001).
Furthermore, CCSs who reported financial hardship had significantly lower health-related quality of life (P<0.001 for all 3 domains), sensation abnormality (all P<0.001), pulmonary symptoms (all P<0.05), and cardiac symptoms (all P<0.05).
Predicting hardship
Intensive cancer treatment, chronic health conditions, second cancers, age at the time of study evaluation, education level, and annual household income were all significantly associated with a greater risk of financial hardship.
CCSs age 40 and older had an increased risk of psychological and coping/behavioral hardship (P<0.001 for both domains).
CCSs with an annual household income of less than $40,000 had an increased risk of material, psychological, and coping/behavioral hardship, compared to CCSs with an income of $80,000 or more (P<0.001 for all domains).
CCSs who did not obtain a high school diploma had an increased risk of material (P<0.001), psychological (P<0.01), and coping/behavioral hardship (P<0.001) compared to college graduates.
CCSs who received cancer treatments associated with a high-risk disease burden (vs low-risk) had an increased risk of material (P=0.01) and psychological (P=0.004) hardship.
Health conditions associated with material financial hardship included grade 2-4 myocardial infarction (P<0.001), peripheral neuropathy (P<0.001), subsequent neoplasm (P<0.001), seizure (P=0.007), reproductive disorders (P=0.01), stroke (P=0.02), amputation (P=0.02), upper gastrointestinal disease (P=0.04), and hearing loss (P=0.05).
Grade 2-4 myocardial infarction and reproductive disorders were significantly associated with psychological financial hardship (P=0.02 for both).
“Severe late effects that emerge early in life and disrupt education and training opportunities are a double hit for survivors,” Dr Huang said. “These health problems decrease the survivors’ earning mobility and financial security later in life. The phenomenon leaves them at risk for poor health and psychological outcomes compared to healthier survivors.”
Health-related financial hardship is common among adult survivors of childhood cancer, according to a study published in the Journal of the National Cancer Institute.
Researchers analyzed more than 2800 long-term childhood cancer survivors (CCSs) and found that 65% had financial challenges related to their cancer diagnosis.
“These findings suggest primary care doctors and oncologists should routinely screen childhood cancer survivors for possible financial hardship,” said I-Chan Huang, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
Specifically, Dr Huang recommends that healthcare providers routinely ask CCSs if they are unable to purchase medications, ever skip appointments for economic reasons, or worry about how to pay their medical bills.
For this study, Dr Huang and his colleagues analyzed data from 2811 CCSs. The subjects had a mean age of 31.8 (range, 18 to 65) and were a mean of 23.6 years from cancer diagnosis. Most (57.8%) had been diagnosed with hematologic malignancies, 32.0% with solid tumors, and 10.1% with central nervous system malignancies.
All subjects had been treated at St. Jude and enrolled in the St. Jude LIFE study. Participants return to St. Jude periodically for several days of clinical and functional assessments. Data for this study were collected during the CCSs’ first St. Jude LIFE evaluations.
Assessing hardship
The researchers measured 3 types of financial hardship—material, psychological, and coping/behavioral.
About 1 in 5 CCSs (22.4%) reported material financial hardship. In other words, their cancer had an impact on their financial situation.
More than half of CCSs (51.1%) reported psychological hardship—concern about their ability to pay for medical expenses.
And 33% of CCSs reported coping/behavioral hardship—an inability to see a doctor or go to the hospital due to finances.
Roughly 65% of CCSs reported at least 1 type of financial hardship.
All 3 types of hardship were significantly associated with somatization (all P<0.001), anxiety (all P<0.001), depression (all P<0.001), suicidal thoughts (all P<0.05), and difficulty in retirement planning (all P<0.001).
Furthermore, CCSs who reported financial hardship had significantly lower health-related quality of life (P<0.001 for all 3 domains), sensation abnormality (all P<0.001), pulmonary symptoms (all P<0.05), and cardiac symptoms (all P<0.05).
Predicting hardship
Intensive cancer treatment, chronic health conditions, second cancers, age at the time of study evaluation, education level, and annual household income were all significantly associated with a greater risk of financial hardship.
CCSs age 40 and older had an increased risk of psychological and coping/behavioral hardship (P<0.001 for both domains).
CCSs with an annual household income of less than $40,000 had an increased risk of material, psychological, and coping/behavioral hardship, compared to CCSs with an income of $80,000 or more (P<0.001 for all domains).
CCSs who did not obtain a high school diploma had an increased risk of material (P<0.001), psychological (P<0.01), and coping/behavioral hardship (P<0.001) compared to college graduates.
CCSs who received cancer treatments associated with a high-risk disease burden (vs low-risk) had an increased risk of material (P=0.01) and psychological (P=0.004) hardship.
Health conditions associated with material financial hardship included grade 2-4 myocardial infarction (P<0.001), peripheral neuropathy (P<0.001), subsequent neoplasm (P<0.001), seizure (P=0.007), reproductive disorders (P=0.01), stroke (P=0.02), amputation (P=0.02), upper gastrointestinal disease (P=0.04), and hearing loss (P=0.05).
Grade 2-4 myocardial infarction and reproductive disorders were significantly associated with psychological financial hardship (P=0.02 for both).
“Severe late effects that emerge early in life and disrupt education and training opportunities are a double hit for survivors,” Dr Huang said. “These health problems decrease the survivors’ earning mobility and financial security later in life. The phenomenon leaves them at risk for poor health and psychological outcomes compared to healthier survivors.”
Health-related financial hardship is common among adult survivors of childhood cancer, according to a study published in the Journal of the National Cancer Institute.
Researchers analyzed more than 2800 long-term childhood cancer survivors (CCSs) and found that 65% had financial challenges related to their cancer diagnosis.
“These findings suggest primary care doctors and oncologists should routinely screen childhood cancer survivors for possible financial hardship,” said I-Chan Huang, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
Specifically, Dr Huang recommends that healthcare providers routinely ask CCSs if they are unable to purchase medications, ever skip appointments for economic reasons, or worry about how to pay their medical bills.
For this study, Dr Huang and his colleagues analyzed data from 2811 CCSs. The subjects had a mean age of 31.8 (range, 18 to 65) and were a mean of 23.6 years from cancer diagnosis. Most (57.8%) had been diagnosed with hematologic malignancies, 32.0% with solid tumors, and 10.1% with central nervous system malignancies.
All subjects had been treated at St. Jude and enrolled in the St. Jude LIFE study. Participants return to St. Jude periodically for several days of clinical and functional assessments. Data for this study were collected during the CCSs’ first St. Jude LIFE evaluations.
Assessing hardship
The researchers measured 3 types of financial hardship—material, psychological, and coping/behavioral.
About 1 in 5 CCSs (22.4%) reported material financial hardship. In other words, their cancer had an impact on their financial situation.
More than half of CCSs (51.1%) reported psychological hardship—concern about their ability to pay for medical expenses.
And 33% of CCSs reported coping/behavioral hardship—an inability to see a doctor or go to the hospital due to finances.
Roughly 65% of CCSs reported at least 1 type of financial hardship.
All 3 types of hardship were significantly associated with somatization (all P<0.001), anxiety (all P<0.001), depression (all P<0.001), suicidal thoughts (all P<0.05), and difficulty in retirement planning (all P<0.001).
Furthermore, CCSs who reported financial hardship had significantly lower health-related quality of life (P<0.001 for all 3 domains), sensation abnormality (all P<0.001), pulmonary symptoms (all P<0.05), and cardiac symptoms (all P<0.05).
Predicting hardship
Intensive cancer treatment, chronic health conditions, second cancers, age at the time of study evaluation, education level, and annual household income were all significantly associated with a greater risk of financial hardship.
CCSs age 40 and older had an increased risk of psychological and coping/behavioral hardship (P<0.001 for both domains).
CCSs with an annual household income of less than $40,000 had an increased risk of material, psychological, and coping/behavioral hardship, compared to CCSs with an income of $80,000 or more (P<0.001 for all domains).
CCSs who did not obtain a high school diploma had an increased risk of material (P<0.001), psychological (P<0.01), and coping/behavioral hardship (P<0.001) compared to college graduates.
CCSs who received cancer treatments associated with a high-risk disease burden (vs low-risk) had an increased risk of material (P=0.01) and psychological (P=0.004) hardship.
Health conditions associated with material financial hardship included grade 2-4 myocardial infarction (P<0.001), peripheral neuropathy (P<0.001), subsequent neoplasm (P<0.001), seizure (P=0.007), reproductive disorders (P=0.01), stroke (P=0.02), amputation (P=0.02), upper gastrointestinal disease (P=0.04), and hearing loss (P=0.05).
Grade 2-4 myocardial infarction and reproductive disorders were significantly associated with psychological financial hardship (P=0.02 for both).
“Severe late effects that emerge early in life and disrupt education and training opportunities are a double hit for survivors,” Dr Huang said. “These health problems decrease the survivors’ earning mobility and financial security later in life. The phenomenon leaves them at risk for poor health and psychological outcomes compared to healthier survivors.”
Treatment simulation could help personalize myeloma therapy
With the help of gene expression signatures, a simulated treatment learning model identified which patients with multiple myeloma would benefit most from treatment with bortezomib or lenalidomide, researchers reported in Nature Communications.
The study included 910 participants across three phase 3 trials. In all, 20% would have a 100% greater-than-average progression-free survival (PFS) benefit from bortezomib, while 31% would have a 200% greater-than-average PFS benefit from lenalidomide, wrote Joske Ubels of University Center Utrecht, the Netherlands, and her colleagues.
The genetic heterogeneity of cancer and risk of treatment necessitate tools that “predict – at the moment of diagnosis – which patients will benefit most from a certain treatment,” the researchers wrote. While gene expression signatures can predict a favorable or adverse prognosis, they do not account for the effect of treatment on survival.
“The key idea of simulated treatment learning is that a patient’s treatment benefit can be estimated by comparing [his or her] survival to a set of genetically similar patients [who] received the comparator treatment,” they noted.
To do so, the researchers applied an algorithm called GESTURE to combined data from the TT2 (Total Therapy 2 for Multiple Myeloma), TT3, and HOVON-65/GMMG-HD4 trials. These trials compared bortezomib or lenalidomide with conventional therapies for multiple myeloma. The model identified 180 patients (20%) for whom bortezomib would produce a 100% greater PFS benefit than in the study population as a whole. Conversely, lenalidomide would produce a 200% greater PFS benefit in 31% of patients.
The simulated treatment learning model “can derive clinically actionable gene expression signatures that enable a more personalized approach to treatment,” the researchers concluded. The method requires a large dataset but could be useful for trials that have missed their primary endpoint, such as the CheckMate-026 trial of nivolumab. The next step is to see if the model makes useful treatment predictions for other cancers. The code needed to train and validate the model is available at github.com/jubels/GESTURE.
The Van Herk Fellowship provided support. The lenalidomide dataset was created as part of the Multiple Myeloma Research Foundation Personalized Medicine Initiative. Dr. Ubels and one coinvestigator are employees of SkylineDx; another coinvestigator served on its advisory board. The others reported having no relevant conflicts of interest.
SOURCE: Ubels J et al. Nat Commun. 2018 Jul 27. doi: 10.1038/s41467-018-05348-5.
With the help of gene expression signatures, a simulated treatment learning model identified which patients with multiple myeloma would benefit most from treatment with bortezomib or lenalidomide, researchers reported in Nature Communications.
The study included 910 participants across three phase 3 trials. In all, 20% would have a 100% greater-than-average progression-free survival (PFS) benefit from bortezomib, while 31% would have a 200% greater-than-average PFS benefit from lenalidomide, wrote Joske Ubels of University Center Utrecht, the Netherlands, and her colleagues.
The genetic heterogeneity of cancer and risk of treatment necessitate tools that “predict – at the moment of diagnosis – which patients will benefit most from a certain treatment,” the researchers wrote. While gene expression signatures can predict a favorable or adverse prognosis, they do not account for the effect of treatment on survival.
“The key idea of simulated treatment learning is that a patient’s treatment benefit can be estimated by comparing [his or her] survival to a set of genetically similar patients [who] received the comparator treatment,” they noted.
To do so, the researchers applied an algorithm called GESTURE to combined data from the TT2 (Total Therapy 2 for Multiple Myeloma), TT3, and HOVON-65/GMMG-HD4 trials. These trials compared bortezomib or lenalidomide with conventional therapies for multiple myeloma. The model identified 180 patients (20%) for whom bortezomib would produce a 100% greater PFS benefit than in the study population as a whole. Conversely, lenalidomide would produce a 200% greater PFS benefit in 31% of patients.
The simulated treatment learning model “can derive clinically actionable gene expression signatures that enable a more personalized approach to treatment,” the researchers concluded. The method requires a large dataset but could be useful for trials that have missed their primary endpoint, such as the CheckMate-026 trial of nivolumab. The next step is to see if the model makes useful treatment predictions for other cancers. The code needed to train and validate the model is available at github.com/jubels/GESTURE.
The Van Herk Fellowship provided support. The lenalidomide dataset was created as part of the Multiple Myeloma Research Foundation Personalized Medicine Initiative. Dr. Ubels and one coinvestigator are employees of SkylineDx; another coinvestigator served on its advisory board. The others reported having no relevant conflicts of interest.
SOURCE: Ubels J et al. Nat Commun. 2018 Jul 27. doi: 10.1038/s41467-018-05348-5.
With the help of gene expression signatures, a simulated treatment learning model identified which patients with multiple myeloma would benefit most from treatment with bortezomib or lenalidomide, researchers reported in Nature Communications.
The study included 910 participants across three phase 3 trials. In all, 20% would have a 100% greater-than-average progression-free survival (PFS) benefit from bortezomib, while 31% would have a 200% greater-than-average PFS benefit from lenalidomide, wrote Joske Ubels of University Center Utrecht, the Netherlands, and her colleagues.
The genetic heterogeneity of cancer and risk of treatment necessitate tools that “predict – at the moment of diagnosis – which patients will benefit most from a certain treatment,” the researchers wrote. While gene expression signatures can predict a favorable or adverse prognosis, they do not account for the effect of treatment on survival.
“The key idea of simulated treatment learning is that a patient’s treatment benefit can be estimated by comparing [his or her] survival to a set of genetically similar patients [who] received the comparator treatment,” they noted.
To do so, the researchers applied an algorithm called GESTURE to combined data from the TT2 (Total Therapy 2 for Multiple Myeloma), TT3, and HOVON-65/GMMG-HD4 trials. These trials compared bortezomib or lenalidomide with conventional therapies for multiple myeloma. The model identified 180 patients (20%) for whom bortezomib would produce a 100% greater PFS benefit than in the study population as a whole. Conversely, lenalidomide would produce a 200% greater PFS benefit in 31% of patients.
The simulated treatment learning model “can derive clinically actionable gene expression signatures that enable a more personalized approach to treatment,” the researchers concluded. The method requires a large dataset but could be useful for trials that have missed their primary endpoint, such as the CheckMate-026 trial of nivolumab. The next step is to see if the model makes useful treatment predictions for other cancers. The code needed to train and validate the model is available at github.com/jubels/GESTURE.
The Van Herk Fellowship provided support. The lenalidomide dataset was created as part of the Multiple Myeloma Research Foundation Personalized Medicine Initiative. Dr. Ubels and one coinvestigator are employees of SkylineDx; another coinvestigator served on its advisory board. The others reported having no relevant conflicts of interest.
SOURCE: Ubels J et al. Nat Commun. 2018 Jul 27. doi: 10.1038/s41467-018-05348-5.
FROM NATURE COMMUNICATIONS
Key clinical point:
Major finding: Bortezomib would yield a 100% greater-than-average progression-free survival benefit in 20% of patients; lenalidomide would yield a 200% greater-than-average PFS benefit in 31% of patients.
Study details: Three randomized, phase 3 clinical trials of 910 patients with multiple myeloma were used for the simulation.
Disclosures: The Van Herk Fellowship provided support. The lenalidomide dataset was created as part of the Multiple Myeloma Research Foundation Personalized Medicine Initiative. Dr. Ubels and one coinvestigator are employees of SkylineDx; another coinvestigator served on its advisory board. The others reported having no relevant conflicts of interest.
Source: Ubels J et al. Nat Commun. 2018 Jul 27. doi: 10.1038/s41467-018-05348-5.
CHMP backs 2 biosimilar pegfilgrastim products
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended approval for 2 pegfilgrastim biosimilar candidates—Udenyca and Pelgraz.
Both products have been deemed highly similar to the reference product, Neulasta, a growth-colony-stimulating factor intended to reduce the duration of neutropenia and the incidence of febrile neutropenia due to chemotherapy.
The CHMP’s recommendations for Pelgraz and Udenyca will be reviewed by the European Commission, which has the authority to approve medicines for use in the European Union, Norway, Iceland, and Liechtenstein.
The European Commission usually makes a decision within 67 days of the CHMP’s recommendation.
If approved, Udenyca and Pelgraz will be available as 6 mg solutions for injection.
The full indication for both products will be to reduce the duration of neutropenia and the incidence of febrile neutropenia in adults receiving cytotoxic chemotherapy for malignancies, except chronic myeloid leukemia and myelodysplastic syndromes.
The CHMP said data have shown that Pelgraz and Udenyca both have comparable quality, safety, and efficacy to Neulasta.
Pelgraz’s marketing authorization application is supported by data from a phase 1 pharmacokinetic (PK) and pharmacodynamic (PD) study in healthy volunteers and a phase 3 study of breast cancer patients receiving docetaxel, doxorubicin, and cyclophosphamide.
Results from the phase 1 study were published in Clinical Pharmacology in Drug Development in 2016.
Udenyca’s marketing authorization application is supported by data from an immunogenicity study as well as a PK/PD study comparing Udenyca (formerly CHS-1701) and Neulasta in healthy subjects.
Results from the PK/PD trial were presented at the 2017 ASCO Annual Meeting.
The applicant for Udenyca is ERA Consulting GmbH. The applicant for Pelgraz is Accord Healthcare Limited (the international arm of Intas Pharmaceuticals Ltd).
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended approval for 2 pegfilgrastim biosimilar candidates—Udenyca and Pelgraz.
Both products have been deemed highly similar to the reference product, Neulasta, a growth-colony-stimulating factor intended to reduce the duration of neutropenia and the incidence of febrile neutropenia due to chemotherapy.
The CHMP’s recommendations for Pelgraz and Udenyca will be reviewed by the European Commission, which has the authority to approve medicines for use in the European Union, Norway, Iceland, and Liechtenstein.
The European Commission usually makes a decision within 67 days of the CHMP’s recommendation.
If approved, Udenyca and Pelgraz will be available as 6 mg solutions for injection.
The full indication for both products will be to reduce the duration of neutropenia and the incidence of febrile neutropenia in adults receiving cytotoxic chemotherapy for malignancies, except chronic myeloid leukemia and myelodysplastic syndromes.
The CHMP said data have shown that Pelgraz and Udenyca both have comparable quality, safety, and efficacy to Neulasta.
Pelgraz’s marketing authorization application is supported by data from a phase 1 pharmacokinetic (PK) and pharmacodynamic (PD) study in healthy volunteers and a phase 3 study of breast cancer patients receiving docetaxel, doxorubicin, and cyclophosphamide.
Results from the phase 1 study were published in Clinical Pharmacology in Drug Development in 2016.
Udenyca’s marketing authorization application is supported by data from an immunogenicity study as well as a PK/PD study comparing Udenyca (formerly CHS-1701) and Neulasta in healthy subjects.
Results from the PK/PD trial were presented at the 2017 ASCO Annual Meeting.
The applicant for Udenyca is ERA Consulting GmbH. The applicant for Pelgraz is Accord Healthcare Limited (the international arm of Intas Pharmaceuticals Ltd).
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended approval for 2 pegfilgrastim biosimilar candidates—Udenyca and Pelgraz.
Both products have been deemed highly similar to the reference product, Neulasta, a growth-colony-stimulating factor intended to reduce the duration of neutropenia and the incidence of febrile neutropenia due to chemotherapy.
The CHMP’s recommendations for Pelgraz and Udenyca will be reviewed by the European Commission, which has the authority to approve medicines for use in the European Union, Norway, Iceland, and Liechtenstein.
The European Commission usually makes a decision within 67 days of the CHMP’s recommendation.
If approved, Udenyca and Pelgraz will be available as 6 mg solutions for injection.
The full indication for both products will be to reduce the duration of neutropenia and the incidence of febrile neutropenia in adults receiving cytotoxic chemotherapy for malignancies, except chronic myeloid leukemia and myelodysplastic syndromes.
The CHMP said data have shown that Pelgraz and Udenyca both have comparable quality, safety, and efficacy to Neulasta.
Pelgraz’s marketing authorization application is supported by data from a phase 1 pharmacokinetic (PK) and pharmacodynamic (PD) study in healthy volunteers and a phase 3 study of breast cancer patients receiving docetaxel, doxorubicin, and cyclophosphamide.
Results from the phase 1 study were published in Clinical Pharmacology in Drug Development in 2016.
Udenyca’s marketing authorization application is supported by data from an immunogenicity study as well as a PK/PD study comparing Udenyca (formerly CHS-1701) and Neulasta in healthy subjects.
Results from the PK/PD trial were presented at the 2017 ASCO Annual Meeting.
The applicant for Udenyca is ERA Consulting GmbH. The applicant for Pelgraz is Accord Healthcare Limited (the international arm of Intas Pharmaceuticals Ltd).