User login
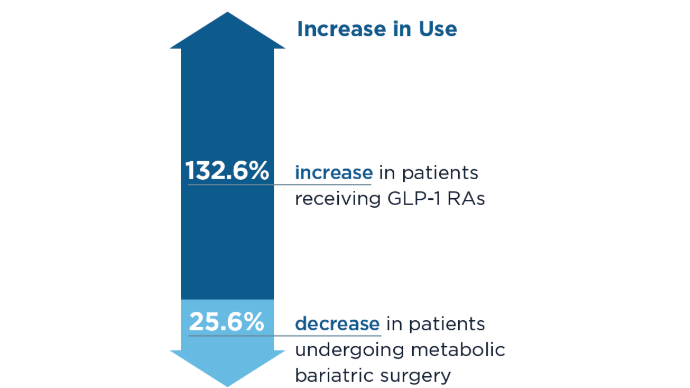
Obesity Management in the Era of GLP-1 RAs: The Role of GLP-1 RAs
Obesity Management in the Era of GLP-1 RAs: The Role of GLP-1 RAs
Click to view more from Gastroenterology Data Trends 2025.
- Lin K, Mehrotra A, Tsai TC. Metabolic Bariatric Surgery in the Era of GLP-1 Receptor Agonists for Obesity Management. JAMA Netw Open. 2024;7(10):e2441380. doi:10.1001/jamanetworkopen.2024.41380
- Camilleri M, El-Omar EM. Ten reasons gastroenterologists and hepatologists should be treating obesity. Gut. 2023;72(6):1033-1038. doi:10.1136/gutjnl-2023-329639
- Camilleri M. Definite benefits of GLP-1 receptor agonists: what is the risk of gastroparesis and lung aspiration? Gut. 2024. doi:10.1136/gutjnl-2024-333036
- Camilleri M, Carlson P, Dilmaghani S. Letter to the Editor. Prevalence and variations in gastric emptying delay in response to GLP-1 receptor agonist liraglutide. Obesity (Silver Spring). 2024;32(2):232-233. doi:10.1002/oby.23941
- Camilleri, M. Incretin impact on gastric function in obesity: physiology, and pharmacological, surgical and endoscopic treatments. J Physiol. 2024.doi:10.1113/JP287535
- Kindel TL, Wang AY, Wadhwa A, et al; American Gastroenterological Association; American Society for Metabolic and Bariatric Surgery; American Society of Anesthesiologists; International Society of Perioperative Care of Patients with Obesity;
Society of American Gastrointestinal and Endoscopic Surgeons. Multisociety Clinical Practice Guidance for the Safe Use of Glucagon-like Peptide-1 Receptor Agonists in the Perioperative Period. Clin Gastroenterol Hepatol. 2024:S1542-3565(24)00910-8. doi:10.1016/j.cgh.2024.10.003
Click to view more from Gastroenterology Data Trends 2025.
Click to view more from Gastroenterology Data Trends 2025.
- Lin K, Mehrotra A, Tsai TC. Metabolic Bariatric Surgery in the Era of GLP-1 Receptor Agonists for Obesity Management. JAMA Netw Open. 2024;7(10):e2441380. doi:10.1001/jamanetworkopen.2024.41380
- Camilleri M, El-Omar EM. Ten reasons gastroenterologists and hepatologists should be treating obesity. Gut. 2023;72(6):1033-1038. doi:10.1136/gutjnl-2023-329639
- Camilleri M. Definite benefits of GLP-1 receptor agonists: what is the risk of gastroparesis and lung aspiration? Gut. 2024. doi:10.1136/gutjnl-2024-333036
- Camilleri M, Carlson P, Dilmaghani S. Letter to the Editor. Prevalence and variations in gastric emptying delay in response to GLP-1 receptor agonist liraglutide. Obesity (Silver Spring). 2024;32(2):232-233. doi:10.1002/oby.23941
- Camilleri, M. Incretin impact on gastric function in obesity: physiology, and pharmacological, surgical and endoscopic treatments. J Physiol. 2024.doi:10.1113/JP287535
- Kindel TL, Wang AY, Wadhwa A, et al; American Gastroenterological Association; American Society for Metabolic and Bariatric Surgery; American Society of Anesthesiologists; International Society of Perioperative Care of Patients with Obesity;
Society of American Gastrointestinal and Endoscopic Surgeons. Multisociety Clinical Practice Guidance for the Safe Use of Glucagon-like Peptide-1 Receptor Agonists in the Perioperative Period. Clin Gastroenterol Hepatol. 2024:S1542-3565(24)00910-8. doi:10.1016/j.cgh.2024.10.003
- Lin K, Mehrotra A, Tsai TC. Metabolic Bariatric Surgery in the Era of GLP-1 Receptor Agonists for Obesity Management. JAMA Netw Open. 2024;7(10):e2441380. doi:10.1001/jamanetworkopen.2024.41380
- Camilleri M, El-Omar EM. Ten reasons gastroenterologists and hepatologists should be treating obesity. Gut. 2023;72(6):1033-1038. doi:10.1136/gutjnl-2023-329639
- Camilleri M. Definite benefits of GLP-1 receptor agonists: what is the risk of gastroparesis and lung aspiration? Gut. 2024. doi:10.1136/gutjnl-2024-333036
- Camilleri M, Carlson P, Dilmaghani S. Letter to the Editor. Prevalence and variations in gastric emptying delay in response to GLP-1 receptor agonist liraglutide. Obesity (Silver Spring). 2024;32(2):232-233. doi:10.1002/oby.23941
- Camilleri, M. Incretin impact on gastric function in obesity: physiology, and pharmacological, surgical and endoscopic treatments. J Physiol. 2024.doi:10.1113/JP287535
- Kindel TL, Wang AY, Wadhwa A, et al; American Gastroenterological Association; American Society for Metabolic and Bariatric Surgery; American Society of Anesthesiologists; International Society of Perioperative Care of Patients with Obesity;
Society of American Gastrointestinal and Endoscopic Surgeons. Multisociety Clinical Practice Guidance for the Safe Use of Glucagon-like Peptide-1 Receptor Agonists in the Perioperative Period. Clin Gastroenterol Hepatol. 2024:S1542-3565(24)00910-8. doi:10.1016/j.cgh.2024.10.003
Obesity Management in the Era of GLP-1 RAs: The Role of GLP-1 RAs
Obesity Management in the Era of GLP-1 RAs: The Role of GLP-1 RAs




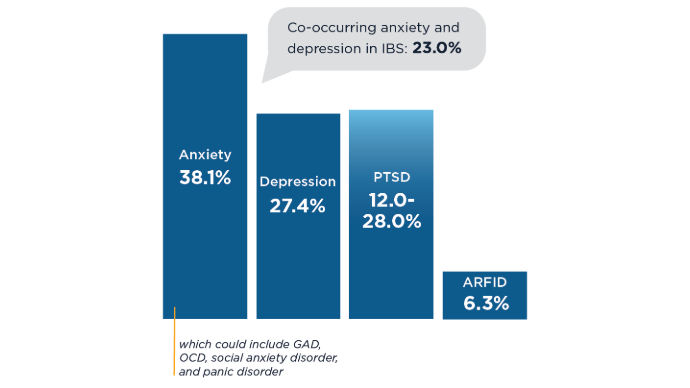
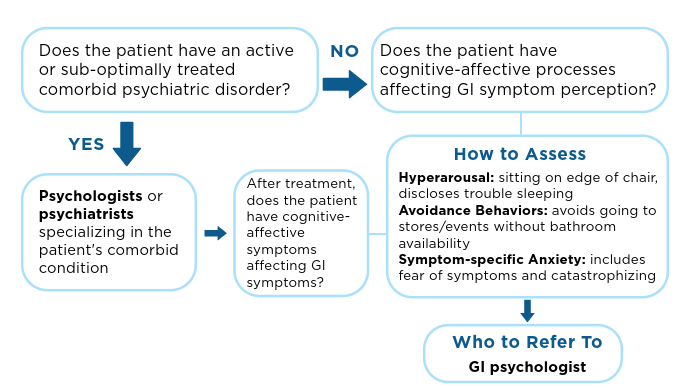
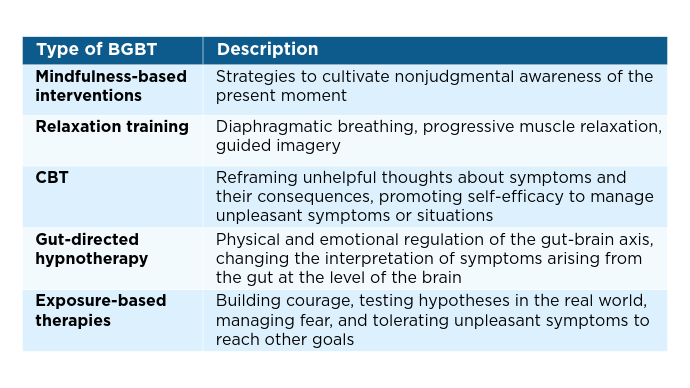
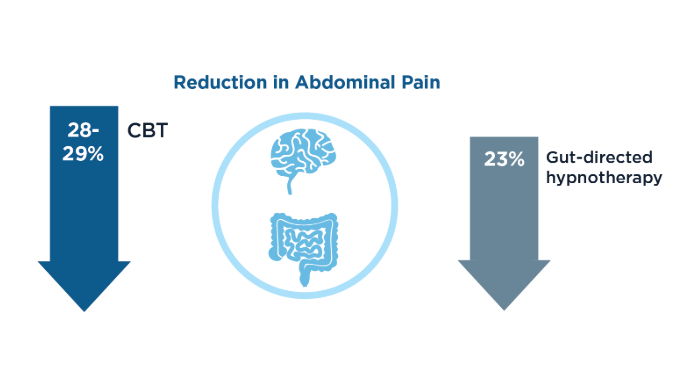
IBS: Mental Health Factors and Comorbidities
IBS: Mental Health Factors and Comorbidities
Click to view more from Gastroenterology Data Trends 2025.
- Staudacher HM, Black CJ, Teasdale SB, Mikocka-Walus A, Keefer L. Irritable bowel syndrome and mental health comorbidity - approach to multidisciplinary management. Nat Rev Gastroenterol Hepatol. 2023;20(9):582-596. doi:10.1038/s41575-023-00794-z
- Ballou S, Vasant DH, Guadagnoli L, et al. A primer for the gastroenterology provider on psychosocial assessment of patients with disorders of gut-brain interaction. Neurogastroenterol Motil. 2024;36(12):e14894. doi:10.1111/nmo.14894
- Keefer L, Ballou SK, Drossman DA, Ringstrom G, Elsenbruch S, Ljótsson B. A Rome Working Team Report on Brain-Gut Behavior Therapies for Disorders of Gut-Brain Interaction. Gastroenterology. 2022;162(1):300-315. doi:10.1053/j.gastro.2021.09.015
- Goodoory VC, Khasawneh M, Thakur ER, et al. Effect of Brain-Gut Behavioral Treatments on Abdominal Pain in Irritable Bowel Syndrome: Systematic Review and Network Meta-Analysis. Gastroenterology. 2024;167(5):934-943.e5. doi:10.1053/j.gastro.2024.05.010
- Chang L, Sultan S, Lembo A, Verne GN, Smalley W, Heidelbaugh JJ. AGA Clinical Practice Guideline on the Pharmacological Management of Irritable Bowel Syndrome With Constipation. Gastroenterology. 2022;163(1):118-136. doi:10.1053/j.gastro.2022.04.016
- Drossman DA, Tack J, Ford AC, Szigethy E, Törnblom H, Van Oudenhove L. Neuromodulators for Functional Gastrointestinal Disorders (Disorders of Gut-Brain Interaction): A Rome Foundation Working Team Report. Gastroenterology. 2018;154(4):1140-1171.e1. doi:10.1053/j.gastro.2017.11.279
- Hasan SS, Ballou S, Keefer L, Vasant DH. Improving access to gut-directed hypnotherapy for irritable bowel syndrome in the digital therapeutics’ era: Are mobile applications a “smart” solution? Neurogastroenterol Motil. 2023;35(4):e14554. doi:10.1111/nmo.14554
- Tarar ZI, Farooq U, Zafar Y, et al. Burden of anxiety and depression among hospitalized patients with irritable bowel syndrome: a nationwide analysis. Ir J Med Sci. 2023;192(5):2159-2166. doi:10.1007/s11845-022-03258-6
- Barbara G, Aziz I, Ballou S, et al. Rome Foundation Working Team Report on overlap in disorders of gut-brain interaction. Nat Rev Gastroenterol Hepatol. doi:10.1038/s41575-024-01033-9
- Thakur ER, Kunik M, Jarbrink-Sehgal ME, Lackner J, Dindo L, El-Serag H. Behavior Medicine Management of Irritable Bowel Syndrome: A Referral Toolkit for Gastroenterology Providers. 2018. Accessed February 19, 2025. https://www.mirecc.va.gov/VISN16/docs/ibs-referral-toolkit.pdf
- Irritable bowel syndrome (IBS). Johns Hopkins Medicine website. Accessed February 19, 2025. https://www.hopkinsmedicine.org/health/conditionsanddiseases/irritable-bowel-syndrome-ibs
- Burton-Murray H, Guadagnoli L, Kamp K, et al. Rome Foundation Working Team Report: Consensus Statement on the Design and Conduct of Behavioural Clinical Trials for Disorders of Gut-Brain Interaction. Aliment Pharmacol Ther. 2025;61(5):787-802. doi:10.1111/apt.18482
- Rome GastroPsych. Rome Foundation website. Accessed February 19, 2025. https://romegipsych.org/
- Scarlata K, Riehl M. Mind Your Gut: The Science-based, Whole-body Guide to Living Well with IBS. Hachette Book Group, 2025.
- IFFGD International Foundation for Gastrointestinal Disorders. IFFGD website. January 10, 2025. Accessed February 19, 2025. https://iffgd.org/
- GI Psychology: Mind Your Gut website. April 2, 2024. Accessed February 19, 2025. https://www.gipsychology.com/
- Drossman DA, Ruddy J. Gut Feelings: Disorders of the Gut-Brain Interaction (DGBI) and the Patient-Doctor Relationship. DrossmanCare Chapel Hill, 2020.
- Tuesday Night IBS website. Accessed February 19, 2025. https://www.tuesdaynightibs.com/
Click to view more from Gastroenterology Data Trends 2025.
Click to view more from Gastroenterology Data Trends 2025.
- Staudacher HM, Black CJ, Teasdale SB, Mikocka-Walus A, Keefer L. Irritable bowel syndrome and mental health comorbidity - approach to multidisciplinary management. Nat Rev Gastroenterol Hepatol. 2023;20(9):582-596. doi:10.1038/s41575-023-00794-z
- Ballou S, Vasant DH, Guadagnoli L, et al. A primer for the gastroenterology provider on psychosocial assessment of patients with disorders of gut-brain interaction. Neurogastroenterol Motil. 2024;36(12):e14894. doi:10.1111/nmo.14894
- Keefer L, Ballou SK, Drossman DA, Ringstrom G, Elsenbruch S, Ljótsson B. A Rome Working Team Report on Brain-Gut Behavior Therapies for Disorders of Gut-Brain Interaction. Gastroenterology. 2022;162(1):300-315. doi:10.1053/j.gastro.2021.09.015
- Goodoory VC, Khasawneh M, Thakur ER, et al. Effect of Brain-Gut Behavioral Treatments on Abdominal Pain in Irritable Bowel Syndrome: Systematic Review and Network Meta-Analysis. Gastroenterology. 2024;167(5):934-943.e5. doi:10.1053/j.gastro.2024.05.010
- Chang L, Sultan S, Lembo A, Verne GN, Smalley W, Heidelbaugh JJ. AGA Clinical Practice Guideline on the Pharmacological Management of Irritable Bowel Syndrome With Constipation. Gastroenterology. 2022;163(1):118-136. doi:10.1053/j.gastro.2022.04.016
- Drossman DA, Tack J, Ford AC, Szigethy E, Törnblom H, Van Oudenhove L. Neuromodulators for Functional Gastrointestinal Disorders (Disorders of Gut-Brain Interaction): A Rome Foundation Working Team Report. Gastroenterology. 2018;154(4):1140-1171.e1. doi:10.1053/j.gastro.2017.11.279
- Hasan SS, Ballou S, Keefer L, Vasant DH. Improving access to gut-directed hypnotherapy for irritable bowel syndrome in the digital therapeutics’ era: Are mobile applications a “smart” solution? Neurogastroenterol Motil. 2023;35(4):e14554. doi:10.1111/nmo.14554
- Tarar ZI, Farooq U, Zafar Y, et al. Burden of anxiety and depression among hospitalized patients with irritable bowel syndrome: a nationwide analysis. Ir J Med Sci. 2023;192(5):2159-2166. doi:10.1007/s11845-022-03258-6
- Barbara G, Aziz I, Ballou S, et al. Rome Foundation Working Team Report on overlap in disorders of gut-brain interaction. Nat Rev Gastroenterol Hepatol. doi:10.1038/s41575-024-01033-9
- Thakur ER, Kunik M, Jarbrink-Sehgal ME, Lackner J, Dindo L, El-Serag H. Behavior Medicine Management of Irritable Bowel Syndrome: A Referral Toolkit for Gastroenterology Providers. 2018. Accessed February 19, 2025. https://www.mirecc.va.gov/VISN16/docs/ibs-referral-toolkit.pdf
- Irritable bowel syndrome (IBS). Johns Hopkins Medicine website. Accessed February 19, 2025. https://www.hopkinsmedicine.org/health/conditionsanddiseases/irritable-bowel-syndrome-ibs
- Burton-Murray H, Guadagnoli L, Kamp K, et al. Rome Foundation Working Team Report: Consensus Statement on the Design and Conduct of Behavioural Clinical Trials for Disorders of Gut-Brain Interaction. Aliment Pharmacol Ther. 2025;61(5):787-802. doi:10.1111/apt.18482
- Rome GastroPsych. Rome Foundation website. Accessed February 19, 2025. https://romegipsych.org/
- Scarlata K, Riehl M. Mind Your Gut: The Science-based, Whole-body Guide to Living Well with IBS. Hachette Book Group, 2025.
- IFFGD International Foundation for Gastrointestinal Disorders. IFFGD website. January 10, 2025. Accessed February 19, 2025. https://iffgd.org/
- GI Psychology: Mind Your Gut website. April 2, 2024. Accessed February 19, 2025. https://www.gipsychology.com/
- Drossman DA, Ruddy J. Gut Feelings: Disorders of the Gut-Brain Interaction (DGBI) and the Patient-Doctor Relationship. DrossmanCare Chapel Hill, 2020.
- Tuesday Night IBS website. Accessed February 19, 2025. https://www.tuesdaynightibs.com/
- Staudacher HM, Black CJ, Teasdale SB, Mikocka-Walus A, Keefer L. Irritable bowel syndrome and mental health comorbidity - approach to multidisciplinary management. Nat Rev Gastroenterol Hepatol. 2023;20(9):582-596. doi:10.1038/s41575-023-00794-z
- Ballou S, Vasant DH, Guadagnoli L, et al. A primer for the gastroenterology provider on psychosocial assessment of patients with disorders of gut-brain interaction. Neurogastroenterol Motil. 2024;36(12):e14894. doi:10.1111/nmo.14894
- Keefer L, Ballou SK, Drossman DA, Ringstrom G, Elsenbruch S, Ljótsson B. A Rome Working Team Report on Brain-Gut Behavior Therapies for Disorders of Gut-Brain Interaction. Gastroenterology. 2022;162(1):300-315. doi:10.1053/j.gastro.2021.09.015
- Goodoory VC, Khasawneh M, Thakur ER, et al. Effect of Brain-Gut Behavioral Treatments on Abdominal Pain in Irritable Bowel Syndrome: Systematic Review and Network Meta-Analysis. Gastroenterology. 2024;167(5):934-943.e5. doi:10.1053/j.gastro.2024.05.010
- Chang L, Sultan S, Lembo A, Verne GN, Smalley W, Heidelbaugh JJ. AGA Clinical Practice Guideline on the Pharmacological Management of Irritable Bowel Syndrome With Constipation. Gastroenterology. 2022;163(1):118-136. doi:10.1053/j.gastro.2022.04.016
- Drossman DA, Tack J, Ford AC, Szigethy E, Törnblom H, Van Oudenhove L. Neuromodulators for Functional Gastrointestinal Disorders (Disorders of Gut-Brain Interaction): A Rome Foundation Working Team Report. Gastroenterology. 2018;154(4):1140-1171.e1. doi:10.1053/j.gastro.2017.11.279
- Hasan SS, Ballou S, Keefer L, Vasant DH. Improving access to gut-directed hypnotherapy for irritable bowel syndrome in the digital therapeutics’ era: Are mobile applications a “smart” solution? Neurogastroenterol Motil. 2023;35(4):e14554. doi:10.1111/nmo.14554
- Tarar ZI, Farooq U, Zafar Y, et al. Burden of anxiety and depression among hospitalized patients with irritable bowel syndrome: a nationwide analysis. Ir J Med Sci. 2023;192(5):2159-2166. doi:10.1007/s11845-022-03258-6
- Barbara G, Aziz I, Ballou S, et al. Rome Foundation Working Team Report on overlap in disorders of gut-brain interaction. Nat Rev Gastroenterol Hepatol. doi:10.1038/s41575-024-01033-9
- Thakur ER, Kunik M, Jarbrink-Sehgal ME, Lackner J, Dindo L, El-Serag H. Behavior Medicine Management of Irritable Bowel Syndrome: A Referral Toolkit for Gastroenterology Providers. 2018. Accessed February 19, 2025. https://www.mirecc.va.gov/VISN16/docs/ibs-referral-toolkit.pdf
- Irritable bowel syndrome (IBS). Johns Hopkins Medicine website. Accessed February 19, 2025. https://www.hopkinsmedicine.org/health/conditionsanddiseases/irritable-bowel-syndrome-ibs
- Burton-Murray H, Guadagnoli L, Kamp K, et al. Rome Foundation Working Team Report: Consensus Statement on the Design and Conduct of Behavioural Clinical Trials for Disorders of Gut-Brain Interaction. Aliment Pharmacol Ther. 2025;61(5):787-802. doi:10.1111/apt.18482
- Rome GastroPsych. Rome Foundation website. Accessed February 19, 2025. https://romegipsych.org/
- Scarlata K, Riehl M. Mind Your Gut: The Science-based, Whole-body Guide to Living Well with IBS. Hachette Book Group, 2025.
- IFFGD International Foundation for Gastrointestinal Disorders. IFFGD website. January 10, 2025. Accessed February 19, 2025. https://iffgd.org/
- GI Psychology: Mind Your Gut website. April 2, 2024. Accessed February 19, 2025. https://www.gipsychology.com/
- Drossman DA, Ruddy J. Gut Feelings: Disorders of the Gut-Brain Interaction (DGBI) and the Patient-Doctor Relationship. DrossmanCare Chapel Hill, 2020.
- Tuesday Night IBS website. Accessed February 19, 2025. https://www.tuesdaynightibs.com/
IBS: Mental Health Factors and Comorbidities
IBS: Mental Health Factors and Comorbidities


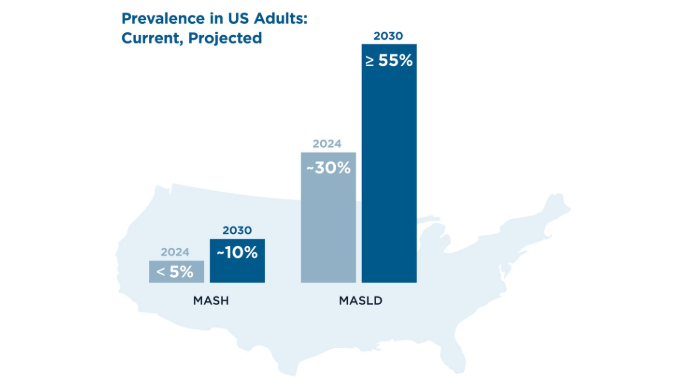
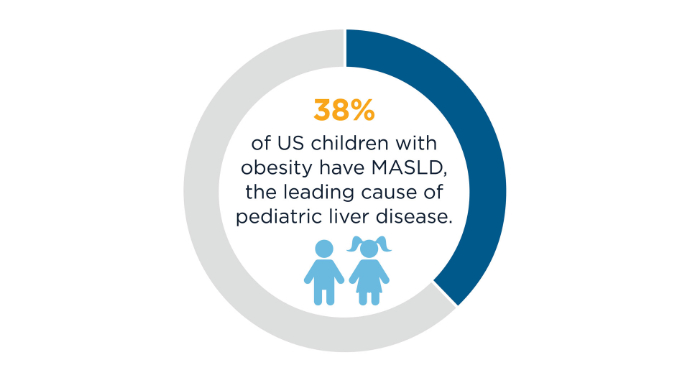
AGA Data Trends 2025: MASLD
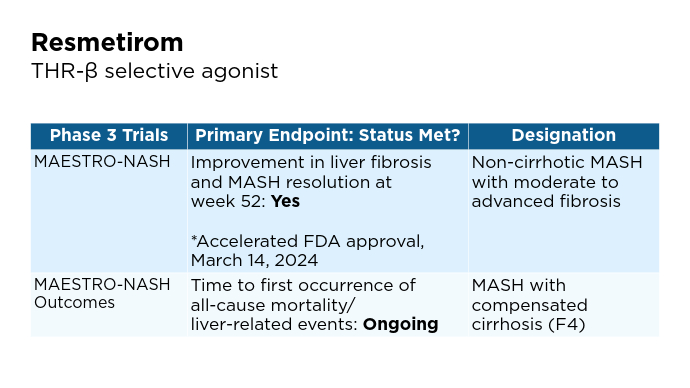
New and Emerging Treatments for MASLD/MASH
- Hu Y, Sun C, Chen Y, Liu Y-D, Fan J-G. Pipeline of New Drug Treatment for Non-alcoholic Fatty Liver Disease/Metabolic Dysfunction-associated Steatotic Liver Disease. J Clin Transl Hepatol. 2024;12(9):802-814. doi:10.14218/JCTH.2024.00123
- Petta S, Targher G, Romeo S, et al. The first MASH drug therapy on the horizon: Current perspectives of resmetirom. Liver Int. 2024;44(7):1526-1536. doi:10.14218/JCTH.2024.00123doi:10.1111/liv.15930
- Ciardullo S, Muraca E, Vergani M, Invernizzi P, Perseghin G. Advancements in pharmacological treatment of NAFLD/MASLD: a focus on metabolic and liver-targeted interventions. Gastroenterol Rep (Oxf). 2024;12:goae029. doi:10.1093/gastro/goae029
- Chen VL, Morgan TR, Rotman Y, et al. Resmetirom therapy for metabolic dysfunction-associated steatotic liver disease: October 2024 updates to AASLD Practice Guidance. Hepatology. 2025;81(1):312-320. doi:10.1097/HEP.0000000000001112
- Economist Impact 2024. MASLD/MASH in the US: A liver disease country profile. Published 2024. Accessed January 22, 2025. https://impact.economist.com/perspectives/sites/default/files/download/liver-disease-country-profile_united_states_final.pdf
- Tincopa MA, Anstee QM, Loomba R. New and emerging treatments for metabolic dysfunction-associated steatohepatitis. Cell Metab. 2024;36(5):912-926. doi:10.1016/j.cmet.2024.03.011
- Carpi S, Daniele S, de Almeida JFM, Gabbia D. Recent Advances in miRNA-Based Therapy for MASLD/MASH and MASH-Associated HCC. Int J Mol Sci. 2024;25(22):12229. https://www.mdpi.com/1422-0067/25/22/1222
- Wong RJ. Epidemiology of metabolic dysfunction-associated steatotic liver disease (MASLD) and alcohol-related liver disease (ALD). Metab Target Organ Damage. 2024;4:35. http://dx.doi.org/10.20517/mtod.2024.57
- Younossi ZM, Kalligeros M, Henry L. Epidemiology of Metabolic Dysfunction-Associated Steatotic Liver Disease. Clin Mol Hepatol. 2024. doi:10.3350/cmh.2024.0431
- Jozst L. Estimating the True Prevalence of MASH and MASLD in the US. AJMC. Published October 17, 2024. Accessed January 22, 2025. https://www.ajmc.com/view/estimating-the-true-prevalence-of-mash-and-masld-in-the-us
- Mayo Clinic website. Pediatric metabolic dysfunction-associated steatotic liver disease (MASLD), formerly known as nonalcoholic fatty liver disease (NAFLD). Published October 4, 2023. Accessed January 22, 2025. https://www.mayoclinic.org/medical-professionals/pediatrics/news/pediatric-metabolic-dysfunction-associated-steatotic-liver-disease-masld-formerly-known-as-nonalcoholic-fatty-liver-disease-nafld/mac-20555493
- Younossi ZM. Economic burden of MASLD/MASH. Conference report for NATAP. EASL 2024. Published June 5-8, 2024. Accessed January 22, 2025. https://www.natap.org/2024/EASL/EASL_41.htm
- Loomba R, Noureddin M, Kowdley KV, et al. Combination Therapies Including Cilofexor and Firsocostat for Bridging Fibrosis and Cirrhosis Attributable to NASH. Hepatol. 202;73(2):625-643. doi:10.1002/hep.31622
- Nicastro E. D’Antiga L. Nutritional Interventions, Probiotics, Synbiotics and Fecal Microbiota Transplantation in Steatoic Liver Disease. Advances in experimental medicine and Biology. Published online January 1. 202:113-133. doi:https://doi.org.10.1007/978-3-031-58572-2_7
- Shera S, Katzka W, Yang JC, et al. Bariatric-induced microbiome changes alter MASLD development in association with changes in the innate immune system. Front Microbiol. 2024;15:1407555. doi:10.3389/fmicb.2024.1407555
- Globe Newswire website. Akero Therapeutics Reports Second Quarter 2024 Financial Results and Provides Business Update [press release]. Published August 9, 2024. Accessed January 22, 2025. https://www.globenewswire.com/en/news-release/2024/08/09/2927685/0/en/Akero-Therapeutics-Reports-Second-Quarter-2024-Financial-Results-and-Provides-Business-Update.html
- Akero website. Clinical Trials Overview. We are currently enrolling three clinical trials as part of a Phase 3 SYNCHRONY program evaluating EFX for the treatment of pre-cirrhotic MASH (F2-F3) and compensated cirrhosis (F4) due to MASH [press release]. Published 2024. Accessed January 22, 2025. https://akerotx.com/clinical-trials/
- 89bio website. 89bio Initiates Phase 3 ENLIGHTEN-Fibrosis Trial of Pegozafermin in Non-Cirrhotic Metabolic Dysfunction-Associated Steatohepatitis (MASH) Patients with Fibrosis [press release]. Published March 12, 2024. Accessed January 22, 2025. https://www.89bio.com/news/89bio-initiates-phase-3-enlighten-fibrosis-trial-of-pegozafermin-in-non-cirrhotic-metabolic-dysfunction-associated-steatohepatitis-mash-patients-with-fibrosis/
- 89bio website. 89bio Reaches Alignment with the FDA and EMA on Phase 3 Program for Pegozafermin in Nonalcoholic Steatohepatitis (NASH); Program Initiation Planned in the First Half of 2024 [press release]. Published December 4, 2023. Accessed January 22, 2025. https://www.89bio.com/news/89bio-reaches-alignment-with-the-fda-and-ema-on-phase-3-program-for-pegozafermin-in-nonalcoholic-steatohepatitis-nash-program-initiation-planned-in-the-first-half-of-2024/
- Boehringer Ingelheim website. Boehringer receives U.S. FDA Breakthrough Therapy designation and initiates two phase III trials in MASH for survodutide [press release]. Published October 8, 2024. Accessed January 22, 2025. https://www.boehringer-ingelheim.com/human-health/metabolic-diseases/survodutide-us-fda-breakthrough-therapy-phase-3-trials-mash
- Hu Y, Sun C, Chen Y, Liu Y-D, Fan J-G. Pipeline of New Drug Treatment for Non-alcoholic Fatty Liver Disease/Metabolic Dysfunction-associated Steatotic Liver Disease. J Clin Transl Hepatol. 2024;12(9):802-814. doi:10.14218/JCTH.2024.00123
- Petta S, Targher G, Romeo S, et al. The first MASH drug therapy on the horizon: Current perspectives of resmetirom. Liver Int. 2024;44(7):1526-1536. doi:10.14218/JCTH.2024.00123doi:10.1111/liv.15930
- Ciardullo S, Muraca E, Vergani M, Invernizzi P, Perseghin G. Advancements in pharmacological treatment of NAFLD/MASLD: a focus on metabolic and liver-targeted interventions. Gastroenterol Rep (Oxf). 2024;12:goae029. doi:10.1093/gastro/goae029
- Chen VL, Morgan TR, Rotman Y, et al. Resmetirom therapy for metabolic dysfunction-associated steatotic liver disease: October 2024 updates to AASLD Practice Guidance. Hepatology. 2025;81(1):312-320. doi:10.1097/HEP.0000000000001112
- Economist Impact 2024. MASLD/MASH in the US: A liver disease country profile. Published 2024. Accessed January 22, 2025. https://impact.economist.com/perspectives/sites/default/files/download/liver-disease-country-profile_united_states_final.pdf
- Tincopa MA, Anstee QM, Loomba R. New and emerging treatments for metabolic dysfunction-associated steatohepatitis. Cell Metab. 2024;36(5):912-926. doi:10.1016/j.cmet.2024.03.011
- Carpi S, Daniele S, de Almeida JFM, Gabbia D. Recent Advances in miRNA-Based Therapy for MASLD/MASH and MASH-Associated HCC. Int J Mol Sci. 2024;25(22):12229. https://www.mdpi.com/1422-0067/25/22/1222
- Wong RJ. Epidemiology of metabolic dysfunction-associated steatotic liver disease (MASLD) and alcohol-related liver disease (ALD). Metab Target Organ Damage. 2024;4:35. http://dx.doi.org/10.20517/mtod.2024.57
- Younossi ZM, Kalligeros M, Henry L. Epidemiology of Metabolic Dysfunction-Associated Steatotic Liver Disease. Clin Mol Hepatol. 2024. doi:10.3350/cmh.2024.0431
- Jozst L. Estimating the True Prevalence of MASH and MASLD in the US. AJMC. Published October 17, 2024. Accessed January 22, 2025. https://www.ajmc.com/view/estimating-the-true-prevalence-of-mash-and-masld-in-the-us
- Mayo Clinic website. Pediatric metabolic dysfunction-associated steatotic liver disease (MASLD), formerly known as nonalcoholic fatty liver disease (NAFLD). Published October 4, 2023. Accessed January 22, 2025. https://www.mayoclinic.org/medical-professionals/pediatrics/news/pediatric-metabolic-dysfunction-associated-steatotic-liver-disease-masld-formerly-known-as-nonalcoholic-fatty-liver-disease-nafld/mac-20555493
- Younossi ZM. Economic burden of MASLD/MASH. Conference report for NATAP. EASL 2024. Published June 5-8, 2024. Accessed January 22, 2025. https://www.natap.org/2024/EASL/EASL_41.htm
- Loomba R, Noureddin M, Kowdley KV, et al. Combination Therapies Including Cilofexor and Firsocostat for Bridging Fibrosis and Cirrhosis Attributable to NASH. Hepatol. 202;73(2):625-643. doi:10.1002/hep.31622
- Nicastro E. D’Antiga L. Nutritional Interventions, Probiotics, Synbiotics and Fecal Microbiota Transplantation in Steatoic Liver Disease. Advances in experimental medicine and Biology. Published online January 1. 202:113-133. doi:https://doi.org.10.1007/978-3-031-58572-2_7
- Shera S, Katzka W, Yang JC, et al. Bariatric-induced microbiome changes alter MASLD development in association with changes in the innate immune system. Front Microbiol. 2024;15:1407555. doi:10.3389/fmicb.2024.1407555
- Globe Newswire website. Akero Therapeutics Reports Second Quarter 2024 Financial Results and Provides Business Update [press release]. Published August 9, 2024. Accessed January 22, 2025. https://www.globenewswire.com/en/news-release/2024/08/09/2927685/0/en/Akero-Therapeutics-Reports-Second-Quarter-2024-Financial-Results-and-Provides-Business-Update.html
- Akero website. Clinical Trials Overview. We are currently enrolling three clinical trials as part of a Phase 3 SYNCHRONY program evaluating EFX for the treatment of pre-cirrhotic MASH (F2-F3) and compensated cirrhosis (F4) due to MASH [press release]. Published 2024. Accessed January 22, 2025. https://akerotx.com/clinical-trials/
- 89bio website. 89bio Initiates Phase 3 ENLIGHTEN-Fibrosis Trial of Pegozafermin in Non-Cirrhotic Metabolic Dysfunction-Associated Steatohepatitis (MASH) Patients with Fibrosis [press release]. Published March 12, 2024. Accessed January 22, 2025. https://www.89bio.com/news/89bio-initiates-phase-3-enlighten-fibrosis-trial-of-pegozafermin-in-non-cirrhotic-metabolic-dysfunction-associated-steatohepatitis-mash-patients-with-fibrosis/
- 89bio website. 89bio Reaches Alignment with the FDA and EMA on Phase 3 Program for Pegozafermin in Nonalcoholic Steatohepatitis (NASH); Program Initiation Planned in the First Half of 2024 [press release]. Published December 4, 2023. Accessed January 22, 2025. https://www.89bio.com/news/89bio-reaches-alignment-with-the-fda-and-ema-on-phase-3-program-for-pegozafermin-in-nonalcoholic-steatohepatitis-nash-program-initiation-planned-in-the-first-half-of-2024/
- Boehringer Ingelheim website. Boehringer receives U.S. FDA Breakthrough Therapy designation and initiates two phase III trials in MASH for survodutide [press release]. Published October 8, 2024. Accessed January 22, 2025. https://www.boehringer-ingelheim.com/human-health/metabolic-diseases/survodutide-us-fda-breakthrough-therapy-phase-3-trials-mash
- Hu Y, Sun C, Chen Y, Liu Y-D, Fan J-G. Pipeline of New Drug Treatment for Non-alcoholic Fatty Liver Disease/Metabolic Dysfunction-associated Steatotic Liver Disease. J Clin Transl Hepatol. 2024;12(9):802-814. doi:10.14218/JCTH.2024.00123
- Petta S, Targher G, Romeo S, et al. The first MASH drug therapy on the horizon: Current perspectives of resmetirom. Liver Int. 2024;44(7):1526-1536. doi:10.14218/JCTH.2024.00123doi:10.1111/liv.15930
- Ciardullo S, Muraca E, Vergani M, Invernizzi P, Perseghin G. Advancements in pharmacological treatment of NAFLD/MASLD: a focus on metabolic and liver-targeted interventions. Gastroenterol Rep (Oxf). 2024;12:goae029. doi:10.1093/gastro/goae029
- Chen VL, Morgan TR, Rotman Y, et al. Resmetirom therapy for metabolic dysfunction-associated steatotic liver disease: October 2024 updates to AASLD Practice Guidance. Hepatology. 2025;81(1):312-320. doi:10.1097/HEP.0000000000001112
- Economist Impact 2024. MASLD/MASH in the US: A liver disease country profile. Published 2024. Accessed January 22, 2025. https://impact.economist.com/perspectives/sites/default/files/download/liver-disease-country-profile_united_states_final.pdf
- Tincopa MA, Anstee QM, Loomba R. New and emerging treatments for metabolic dysfunction-associated steatohepatitis. Cell Metab. 2024;36(5):912-926. doi:10.1016/j.cmet.2024.03.011
- Carpi S, Daniele S, de Almeida JFM, Gabbia D. Recent Advances in miRNA-Based Therapy for MASLD/MASH and MASH-Associated HCC. Int J Mol Sci. 2024;25(22):12229. https://www.mdpi.com/1422-0067/25/22/1222
- Wong RJ. Epidemiology of metabolic dysfunction-associated steatotic liver disease (MASLD) and alcohol-related liver disease (ALD). Metab Target Organ Damage. 2024;4:35. http://dx.doi.org/10.20517/mtod.2024.57
- Younossi ZM, Kalligeros M, Henry L. Epidemiology of Metabolic Dysfunction-Associated Steatotic Liver Disease. Clin Mol Hepatol. 2024. doi:10.3350/cmh.2024.0431
- Jozst L. Estimating the True Prevalence of MASH and MASLD in the US. AJMC. Published October 17, 2024. Accessed January 22, 2025. https://www.ajmc.com/view/estimating-the-true-prevalence-of-mash-and-masld-in-the-us
- Mayo Clinic website. Pediatric metabolic dysfunction-associated steatotic liver disease (MASLD), formerly known as nonalcoholic fatty liver disease (NAFLD). Published October 4, 2023. Accessed January 22, 2025. https://www.mayoclinic.org/medical-professionals/pediatrics/news/pediatric-metabolic-dysfunction-associated-steatotic-liver-disease-masld-formerly-known-as-nonalcoholic-fatty-liver-disease-nafld/mac-20555493
- Younossi ZM. Economic burden of MASLD/MASH. Conference report for NATAP. EASL 2024. Published June 5-8, 2024. Accessed January 22, 2025. https://www.natap.org/2024/EASL/EASL_41.htm
- Loomba R, Noureddin M, Kowdley KV, et al. Combination Therapies Including Cilofexor and Firsocostat for Bridging Fibrosis and Cirrhosis Attributable to NASH. Hepatol. 202;73(2):625-643. doi:10.1002/hep.31622
- Nicastro E. D’Antiga L. Nutritional Interventions, Probiotics, Synbiotics and Fecal Microbiota Transplantation in Steatoic Liver Disease. Advances in experimental medicine and Biology. Published online January 1. 202:113-133. doi:https://doi.org.10.1007/978-3-031-58572-2_7
- Shera S, Katzka W, Yang JC, et al. Bariatric-induced microbiome changes alter MASLD development in association with changes in the innate immune system. Front Microbiol. 2024;15:1407555. doi:10.3389/fmicb.2024.1407555
- Globe Newswire website. Akero Therapeutics Reports Second Quarter 2024 Financial Results and Provides Business Update [press release]. Published August 9, 2024. Accessed January 22, 2025. https://www.globenewswire.com/en/news-release/2024/08/09/2927685/0/en/Akero-Therapeutics-Reports-Second-Quarter-2024-Financial-Results-and-Provides-Business-Update.html
- Akero website. Clinical Trials Overview. We are currently enrolling three clinical trials as part of a Phase 3 SYNCHRONY program evaluating EFX for the treatment of pre-cirrhotic MASH (F2-F3) and compensated cirrhosis (F4) due to MASH [press release]. Published 2024. Accessed January 22, 2025. https://akerotx.com/clinical-trials/
- 89bio website. 89bio Initiates Phase 3 ENLIGHTEN-Fibrosis Trial of Pegozafermin in Non-Cirrhotic Metabolic Dysfunction-Associated Steatohepatitis (MASH) Patients with Fibrosis [press release]. Published March 12, 2024. Accessed January 22, 2025. https://www.89bio.com/news/89bio-initiates-phase-3-enlighten-fibrosis-trial-of-pegozafermin-in-non-cirrhotic-metabolic-dysfunction-associated-steatohepatitis-mash-patients-with-fibrosis/
- 89bio website. 89bio Reaches Alignment with the FDA and EMA on Phase 3 Program for Pegozafermin in Nonalcoholic Steatohepatitis (NASH); Program Initiation Planned in the First Half of 2024 [press release]. Published December 4, 2023. Accessed January 22, 2025. https://www.89bio.com/news/89bio-reaches-alignment-with-the-fda-and-ema-on-phase-3-program-for-pegozafermin-in-nonalcoholic-steatohepatitis-nash-program-initiation-planned-in-the-first-half-of-2024/
- Boehringer Ingelheim website. Boehringer receives U.S. FDA Breakthrough Therapy designation and initiates two phase III trials in MASH for survodutide [press release]. Published October 8, 2024. Accessed January 22, 2025. https://www.boehringer-ingelheim.com/human-health/metabolic-diseases/survodutide-us-fda-breakthrough-therapy-phase-3-trials-mash
New and Emerging Treatments for MASLD/MASH
New and Emerging Treatments for MASLD/MASH








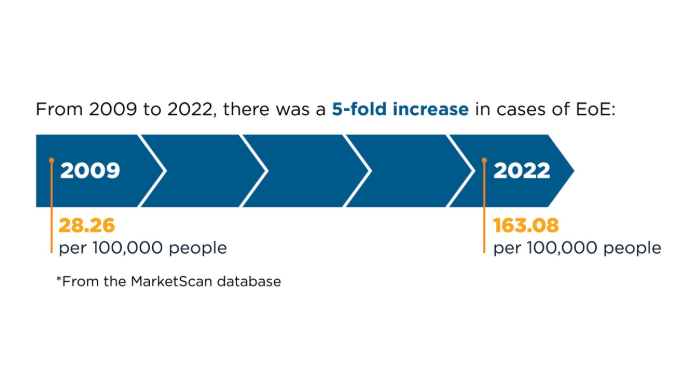
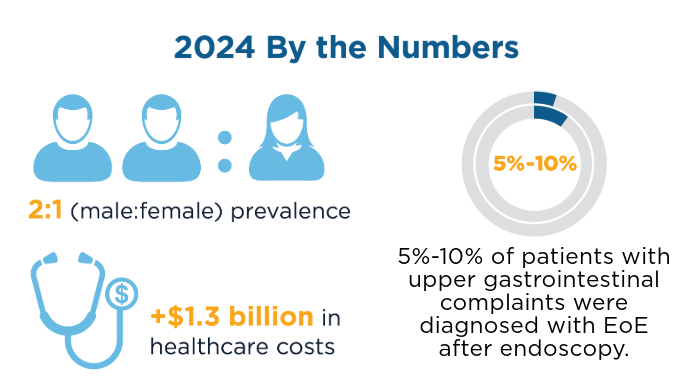
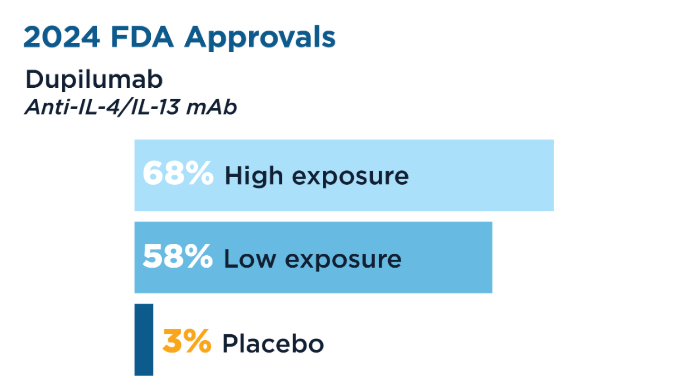
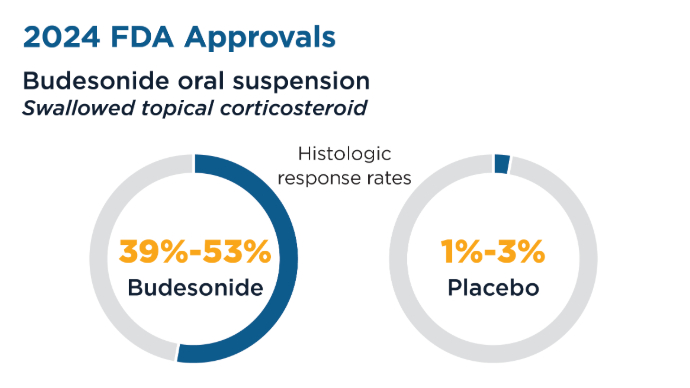
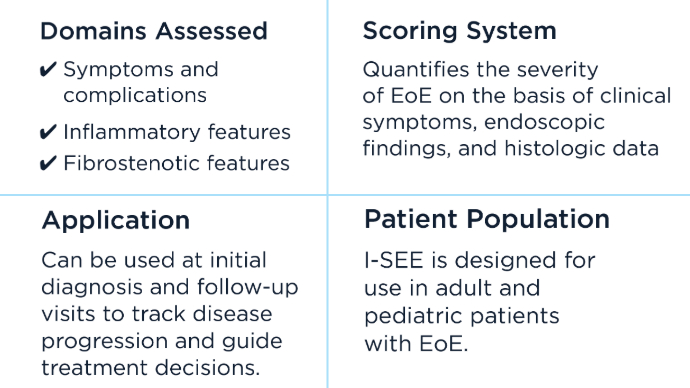
New Therapeutic Frontiers in the Treatment of Eosinophilic Esophagitis
New Therapeutic Frontiers in the Treatment of Eosinophilic Esophagitis
Rossi CM, Santacroce G, Lenti MV, di Sabatino A. Eosinophilic esophagitis in the era of biologics. Expert Rev Gastroenterol Hepatol. 2024;18(6):271-281. doi:10.1080/17474124.2024.2374471
Dellon ES, Liacouras CA, Molina-Infante J, et al. Updated International Consensus Diagnostic Criteria for Eosinophilic Esophagitis: Proceedings of the AGREE Conference. Gastroenterology. 2018;155(4):1022-1033.e10. doi:10.1053/j.gastro.2018.07.009
Geow R, Arena G, Siah C, Picardo S. A retrospective real-world study on the safety and efficacy of budesonide orodispersible tablets for the induction therapy of eosinophilic oesophagitis. Therap Adv Gastroenterol. 2024;17:17562848241290346. doi:10.1177/17562848241290346
Sato H, Dellon ES, Aceves SS, et al. Clinical and molecular correlates of the Index of Severity for Eosinophilic Esophagitis. J Allergy Clin Immunol. 2024;154(2):375-386.e4. doi:10.1016/j.jaci.2024.04.025
Dellon ES, Khoury P, Muir AB, et al. A Clinical Severity Index for Eosinophilic Esophagitis: Development, Consensus, and Future Directions. Gastroenterology. 2022;163(1):59-76. doi:10.1053/j.gastro.2022.03.025
Thel HL, Anderson C, Xue AZ, Jensen ET, Dellon ES. Prevalence and Costs of Eosinophilic Esophagitis in the United States. Clin Gastroenterol Hepatol. 2025;23(2)272-280. doi:10.1016/j.cgh.2024.09.031
Yang E-J, Jung KW. Role of endoscopy in eosinophilic esophagitis. Clin Endosc. 2025;58(1):1-9. doi.org/10.5946/ce.2024.023
Chehade M, Dellon ES, Spergel JM, et al. Dupilumab for Eosinophilic Esophagitis in Patients 1 to 11 Years of Age. N Engl J Med. 2024;390(24):2239-2251. doi:10.1056/NEJMoa2312282
Hirano I, Collins MH, Katzka DA, et al; ORBIT1/SHP621-301 Investigators. Budesonide Oral Suspension Improves Outcomes in Patients With Eosinophilic Esophagitis: Results from a Phase 3 Trial. Clin Gastroenterol Hepatol. 2022;20(3):525-534.e10. doi:10.1016/j.cgh.2021.04.022
Dellon ES, Katzka DA, Collins MH, et al; MP-101-06 Investigators. Budesonide Oral Suspension Improves Symptomatic, Endoscopic, and Histologic Parameters Compared With Placebo in Patients With Eosinophilic Esophagitis. Gastroenterology. 2017;152(4):776-786.e5. doi:10.1053/j.gastro.2016.11.021
Dellon ES, Charriez CM, Zhang S, et al. Cendakimab efficacy and safety in adult and adolescent patients with eosinophilic esophagitis 48-week results from the randomized, placebo-controlled, phase 3 study (late-breaking abstract). Paper presented at: ACG 2024 Annual Scientific Meeting. Philadelphia, Pennsylvania. October 25-30, 2024.
National Institutes of Health, National Library of Medicine, Cinicaltrials.gov website. Efficacy and Safety of Tezepelumab in Patients With Eosinophilic Esophagitis (CROSSING). ClinicalTrials.gov ID NCT05583227. Published December 2024. Accessed February 3, 2025. https://clinicaltrials.gov/study/NCT05583227
National Institutes of Health, National Library of Medicine, Cinicaltrials.gov website. A Study to Investigate the Efficacy and Safety of NSI-8226 in Adults with Eosinophilic Esophagitis. ClinicalTrials.gov ID NCT06598462. Published November 2024. Accessed February 3, 2025. https://clinicaltrials.gov/study/NCT06598462
National Institutes of Health, National Library of Medicine, Cinicaltrials.gov website. A Study of CDX-0519 in Patients With Eosinophilic Esophagitis (EvolvE). ClinicalTrials.gov ID NCT05774184. Published June 2024. Accessed February 3, 2025. https://clinicaltrials.gov/study/NCT05774184
Rossi CM, Santacroce G, Lenti MV, di Sabatino A. Eosinophilic esophagitis in the era of biologics. Expert Rev Gastroenterol Hepatol. 2024;18(6):271-281. doi:10.1080/17474124.2024.2374471
Dellon ES, Liacouras CA, Molina-Infante J, et al. Updated International Consensus Diagnostic Criteria for Eosinophilic Esophagitis: Proceedings of the AGREE Conference. Gastroenterology. 2018;155(4):1022-1033.e10. doi:10.1053/j.gastro.2018.07.009
Geow R, Arena G, Siah C, Picardo S. A retrospective real-world study on the safety and efficacy of budesonide orodispersible tablets for the induction therapy of eosinophilic oesophagitis. Therap Adv Gastroenterol. 2024;17:17562848241290346. doi:10.1177/17562848241290346
Sato H, Dellon ES, Aceves SS, et al. Clinical and molecular correlates of the Index of Severity for Eosinophilic Esophagitis. J Allergy Clin Immunol. 2024;154(2):375-386.e4. doi:10.1016/j.jaci.2024.04.025
Dellon ES, Khoury P, Muir AB, et al. A Clinical Severity Index for Eosinophilic Esophagitis: Development, Consensus, and Future Directions. Gastroenterology. 2022;163(1):59-76. doi:10.1053/j.gastro.2022.03.025
Thel HL, Anderson C, Xue AZ, Jensen ET, Dellon ES. Prevalence and Costs of Eosinophilic Esophagitis in the United States. Clin Gastroenterol Hepatol. 2025;23(2)272-280. doi:10.1016/j.cgh.2024.09.031
Yang E-J, Jung KW. Role of endoscopy in eosinophilic esophagitis. Clin Endosc. 2025;58(1):1-9. doi.org/10.5946/ce.2024.023
Chehade M, Dellon ES, Spergel JM, et al. Dupilumab for Eosinophilic Esophagitis in Patients 1 to 11 Years of Age. N Engl J Med. 2024;390(24):2239-2251. doi:10.1056/NEJMoa2312282
Hirano I, Collins MH, Katzka DA, et al; ORBIT1/SHP621-301 Investigators. Budesonide Oral Suspension Improves Outcomes in Patients With Eosinophilic Esophagitis: Results from a Phase 3 Trial. Clin Gastroenterol Hepatol. 2022;20(3):525-534.e10. doi:10.1016/j.cgh.2021.04.022
Dellon ES, Katzka DA, Collins MH, et al; MP-101-06 Investigators. Budesonide Oral Suspension Improves Symptomatic, Endoscopic, and Histologic Parameters Compared With Placebo in Patients With Eosinophilic Esophagitis. Gastroenterology. 2017;152(4):776-786.e5. doi:10.1053/j.gastro.2016.11.021
Dellon ES, Charriez CM, Zhang S, et al. Cendakimab efficacy and safety in adult and adolescent patients with eosinophilic esophagitis 48-week results from the randomized, placebo-controlled, phase 3 study (late-breaking abstract). Paper presented at: ACG 2024 Annual Scientific Meeting. Philadelphia, Pennsylvania. October 25-30, 2024.
National Institutes of Health, National Library of Medicine, Cinicaltrials.gov website. Efficacy and Safety of Tezepelumab in Patients With Eosinophilic Esophagitis (CROSSING). ClinicalTrials.gov ID NCT05583227. Published December 2024. Accessed February 3, 2025. https://clinicaltrials.gov/study/NCT05583227
National Institutes of Health, National Library of Medicine, Cinicaltrials.gov website. A Study to Investigate the Efficacy and Safety of NSI-8226 in Adults with Eosinophilic Esophagitis. ClinicalTrials.gov ID NCT06598462. Published November 2024. Accessed February 3, 2025. https://clinicaltrials.gov/study/NCT06598462
National Institutes of Health, National Library of Medicine, Cinicaltrials.gov website. A Study of CDX-0519 in Patients With Eosinophilic Esophagitis (EvolvE). ClinicalTrials.gov ID NCT05774184. Published June 2024. Accessed February 3, 2025. https://clinicaltrials.gov/study/NCT05774184
Rossi CM, Santacroce G, Lenti MV, di Sabatino A. Eosinophilic esophagitis in the era of biologics. Expert Rev Gastroenterol Hepatol. 2024;18(6):271-281. doi:10.1080/17474124.2024.2374471
Dellon ES, Liacouras CA, Molina-Infante J, et al. Updated International Consensus Diagnostic Criteria for Eosinophilic Esophagitis: Proceedings of the AGREE Conference. Gastroenterology. 2018;155(4):1022-1033.e10. doi:10.1053/j.gastro.2018.07.009
Geow R, Arena G, Siah C, Picardo S. A retrospective real-world study on the safety and efficacy of budesonide orodispersible tablets for the induction therapy of eosinophilic oesophagitis. Therap Adv Gastroenterol. 2024;17:17562848241290346. doi:10.1177/17562848241290346
Sato H, Dellon ES, Aceves SS, et al. Clinical and molecular correlates of the Index of Severity for Eosinophilic Esophagitis. J Allergy Clin Immunol. 2024;154(2):375-386.e4. doi:10.1016/j.jaci.2024.04.025
Dellon ES, Khoury P, Muir AB, et al. A Clinical Severity Index for Eosinophilic Esophagitis: Development, Consensus, and Future Directions. Gastroenterology. 2022;163(1):59-76. doi:10.1053/j.gastro.2022.03.025
Thel HL, Anderson C, Xue AZ, Jensen ET, Dellon ES. Prevalence and Costs of Eosinophilic Esophagitis in the United States. Clin Gastroenterol Hepatol. 2025;23(2)272-280. doi:10.1016/j.cgh.2024.09.031
Yang E-J, Jung KW. Role of endoscopy in eosinophilic esophagitis. Clin Endosc. 2025;58(1):1-9. doi.org/10.5946/ce.2024.023
Chehade M, Dellon ES, Spergel JM, et al. Dupilumab for Eosinophilic Esophagitis in Patients 1 to 11 Years of Age. N Engl J Med. 2024;390(24):2239-2251. doi:10.1056/NEJMoa2312282
Hirano I, Collins MH, Katzka DA, et al; ORBIT1/SHP621-301 Investigators. Budesonide Oral Suspension Improves Outcomes in Patients With Eosinophilic Esophagitis: Results from a Phase 3 Trial. Clin Gastroenterol Hepatol. 2022;20(3):525-534.e10. doi:10.1016/j.cgh.2021.04.022
Dellon ES, Katzka DA, Collins MH, et al; MP-101-06 Investigators. Budesonide Oral Suspension Improves Symptomatic, Endoscopic, and Histologic Parameters Compared With Placebo in Patients With Eosinophilic Esophagitis. Gastroenterology. 2017;152(4):776-786.e5. doi:10.1053/j.gastro.2016.11.021
Dellon ES, Charriez CM, Zhang S, et al. Cendakimab efficacy and safety in adult and adolescent patients with eosinophilic esophagitis 48-week results from the randomized, placebo-controlled, phase 3 study (late-breaking abstract). Paper presented at: ACG 2024 Annual Scientific Meeting. Philadelphia, Pennsylvania. October 25-30, 2024.
National Institutes of Health, National Library of Medicine, Cinicaltrials.gov website. Efficacy and Safety of Tezepelumab in Patients With Eosinophilic Esophagitis (CROSSING). ClinicalTrials.gov ID NCT05583227. Published December 2024. Accessed February 3, 2025. https://clinicaltrials.gov/study/NCT05583227
National Institutes of Health, National Library of Medicine, Cinicaltrials.gov website. A Study to Investigate the Efficacy and Safety of NSI-8226 in Adults with Eosinophilic Esophagitis. ClinicalTrials.gov ID NCT06598462. Published November 2024. Accessed February 3, 2025. https://clinicaltrials.gov/study/NCT06598462
National Institutes of Health, National Library of Medicine, Cinicaltrials.gov website. A Study of CDX-0519 in Patients With Eosinophilic Esophagitis (EvolvE). ClinicalTrials.gov ID NCT05774184. Published June 2024. Accessed February 3, 2025. https://clinicaltrials.gov/study/NCT05774184
New Therapeutic Frontiers in the Treatment of Eosinophilic Esophagitis
New Therapeutic Frontiers in the Treatment of Eosinophilic Esophagitis




AGA Data Trends 2025: IBD
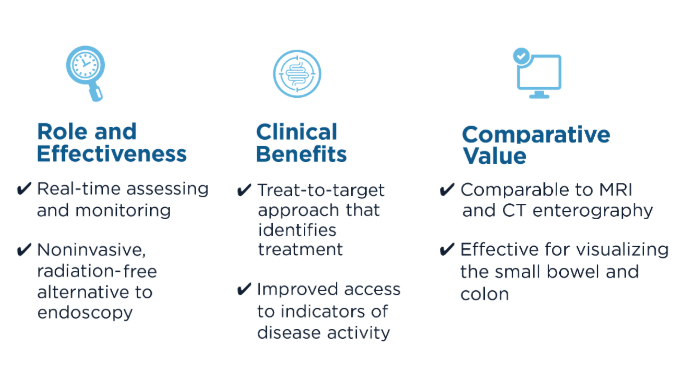
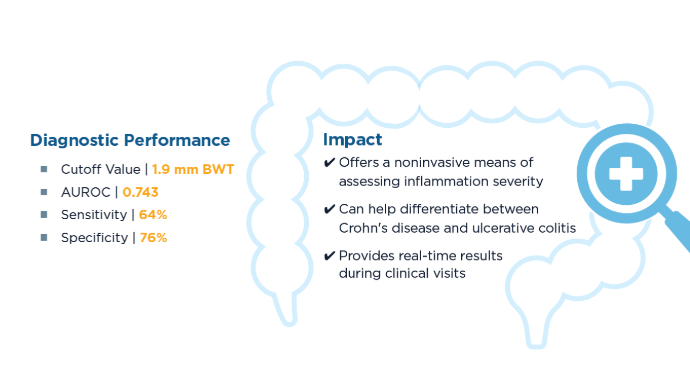
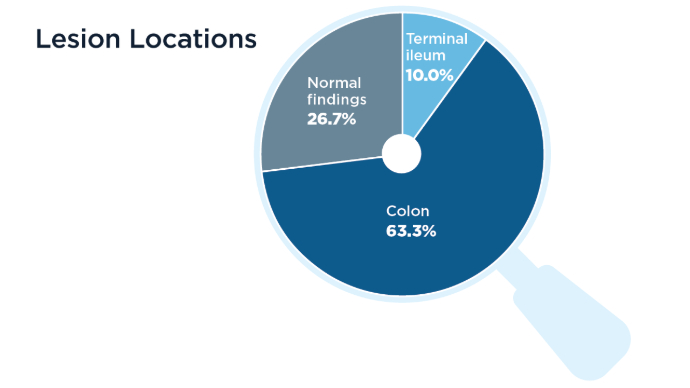
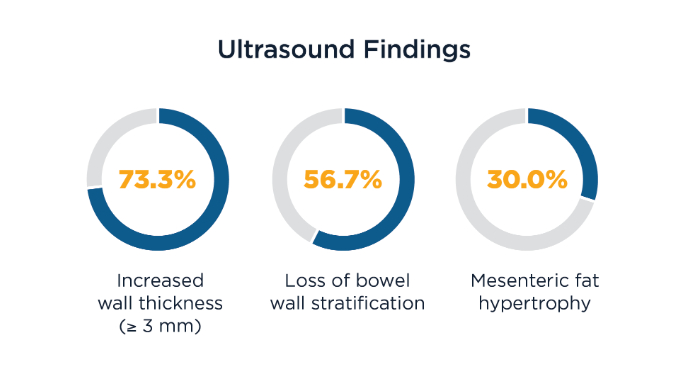
The Role of Bedside Intestinal Ultrasound in IBD Management
- Chavannes M, Dolinger MT, Cohen-Mekelburg S, Abraham B. AGA Clinical Practice update on the Role of Intestinal Ultrasound in Inflammatory Bowel Disease: Commentary. Clin Gastroenterol Hepatol. 2024;22(9):1790-1795.e1. doi:10.1016/j.cgh.2024.04.039
- El-Nakeep S. The intestinal ultrasound role in inflammatory bowel disease in clinical practice and a critical appraisal of the current guidelines (mini-review). Egypt J Intern Med. 2024;36:51. doi:10.1186/s43162-024-00316-6
- Chavannes M, Hart L, Hayati Rezvan P, Dillman JR, Polk DB. Bedside Intestinal Ultrasound Predicts Disease Severity and the Disease Distribution of Pediatric Patients With Inflammatory Bowel Disease: A Pilot Cross-sectional Study. Inflamm Bowel Dis. 2024;30(3):402-409. doi:10.1093/ibd/izad083
- St-Pierre J, Delisle M, Kheirkhahrahimabadi H, et al; International Bowel Ultrasound Group. Bedside Intestinal Ultrasound Performed in an Inflammatory Bowel Disease Urgent Assessment Clinic Improves Clinical Decision-making and Resource Utilization. Crohns Colitis 360. 2023;5(4):otad050. doi:10.1093/crocol/otad050
- Jevdokimova N, Jevdokimov D, Teterina I, Pokrotnieks J, Puķītis A, Mokricka V. Correlation of Intestinal Ultrasound Data With Laboratory Markers of Inflammation for Patients With Inflammatory Bowel Disease. Proc Latv
- Chavannes M, Dolinger MT, Cohen-Mekelburg S, Abraham B. AGA Clinical Practice update on the Role of Intestinal Ultrasound in Inflammatory Bowel Disease: Commentary. Clin Gastroenterol Hepatol. 2024;22(9):1790-1795.e1. doi:10.1016/j.cgh.2024.04.039
- El-Nakeep S. The intestinal ultrasound role in inflammatory bowel disease in clinical practice and a critical appraisal of the current guidelines (mini-review). Egypt J Intern Med. 2024;36:51. doi:10.1186/s43162-024-00316-6
- Chavannes M, Hart L, Hayati Rezvan P, Dillman JR, Polk DB. Bedside Intestinal Ultrasound Predicts Disease Severity and the Disease Distribution of Pediatric Patients With Inflammatory Bowel Disease: A Pilot Cross-sectional Study. Inflamm Bowel Dis. 2024;30(3):402-409. doi:10.1093/ibd/izad083
- St-Pierre J, Delisle M, Kheirkhahrahimabadi H, et al; International Bowel Ultrasound Group. Bedside Intestinal Ultrasound Performed in an Inflammatory Bowel Disease Urgent Assessment Clinic Improves Clinical Decision-making and Resource Utilization. Crohns Colitis 360. 2023;5(4):otad050. doi:10.1093/crocol/otad050
- Jevdokimova N, Jevdokimov D, Teterina I, Pokrotnieks J, Puķītis A, Mokricka V. Correlation of Intestinal Ultrasound Data With Laboratory Markers of Inflammation for Patients With Inflammatory Bowel Disease. Proc Latv
- Chavannes M, Dolinger MT, Cohen-Mekelburg S, Abraham B. AGA Clinical Practice update on the Role of Intestinal Ultrasound in Inflammatory Bowel Disease: Commentary. Clin Gastroenterol Hepatol. 2024;22(9):1790-1795.e1. doi:10.1016/j.cgh.2024.04.039
- El-Nakeep S. The intestinal ultrasound role in inflammatory bowel disease in clinical practice and a critical appraisal of the current guidelines (mini-review). Egypt J Intern Med. 2024;36:51. doi:10.1186/s43162-024-00316-6
- Chavannes M, Hart L, Hayati Rezvan P, Dillman JR, Polk DB. Bedside Intestinal Ultrasound Predicts Disease Severity and the Disease Distribution of Pediatric Patients With Inflammatory Bowel Disease: A Pilot Cross-sectional Study. Inflamm Bowel Dis. 2024;30(3):402-409. doi:10.1093/ibd/izad083
- St-Pierre J, Delisle M, Kheirkhahrahimabadi H, et al; International Bowel Ultrasound Group. Bedside Intestinal Ultrasound Performed in an Inflammatory Bowel Disease Urgent Assessment Clinic Improves Clinical Decision-making and Resource Utilization. Crohns Colitis 360. 2023;5(4):otad050. doi:10.1093/crocol/otad050
- Jevdokimova N, Jevdokimov D, Teterina I, Pokrotnieks J, Puķītis A, Mokricka V. Correlation of Intestinal Ultrasound Data With Laboratory Markers of Inflammation for Patients With Inflammatory Bowel Disease. Proc Latv
The Role of Bedside Intestinal Ultrasound in IBD Management
The Role of Bedside Intestinal Ultrasound in IBD Management


Bridging the Knowledge-Action Gap in Skin Cancer Prevention Among US Military Personnel
Bridging the Knowledge-Action Gap in Skin Cancer Prevention Among US Military Personnel
Skin cancer is a major health concern for military service members, who experience notably higher incidence rates than the general population.1 Active-duty military personnel are particularly vulnerable to prolonged sun exposure due to deployments, specialized training, and everyday outdoor duties.1 Despite skin cancer being the most commonly diagnosed malignancy in active-duty service members,2 tracking and documenting the quantity and diversity of these risk factors remain limited. This knowledge gap comes at high cost, simultaneously impairing military medicine preventive measures while burdening the military health care system with substantial expenditures.3 These findings underscore the critical need for targeted surveillance, early-detection programs, and policy-driven interventions to mitigate these medical and economic concerns.
Skin cancer has been recognized as a major health risk to the military population for decades, yet incidence and prevalence remain high. This phenomenon is closely linked to the inherent responsibilities and expectations of active-duty military members, including outdoor physical training, field exercises, standing in formation, and outdoor working environments—all of which can occur during peak sunlight hours. These risks are further elevated at duty stations in geographic regions with high levels of UV exposure, such as those in tropical and arid regions of the world. Certain military occupational specialties and missions may further introduce unique risk factors; for instance, pilots with frequent high-altitude missions experience heightened UV exposure and melanoma risk.4 Secondary to compounding determinants, the aviation, diving, and nuclear subgroups of the military community are particularly vulnerable to skin cancer.5
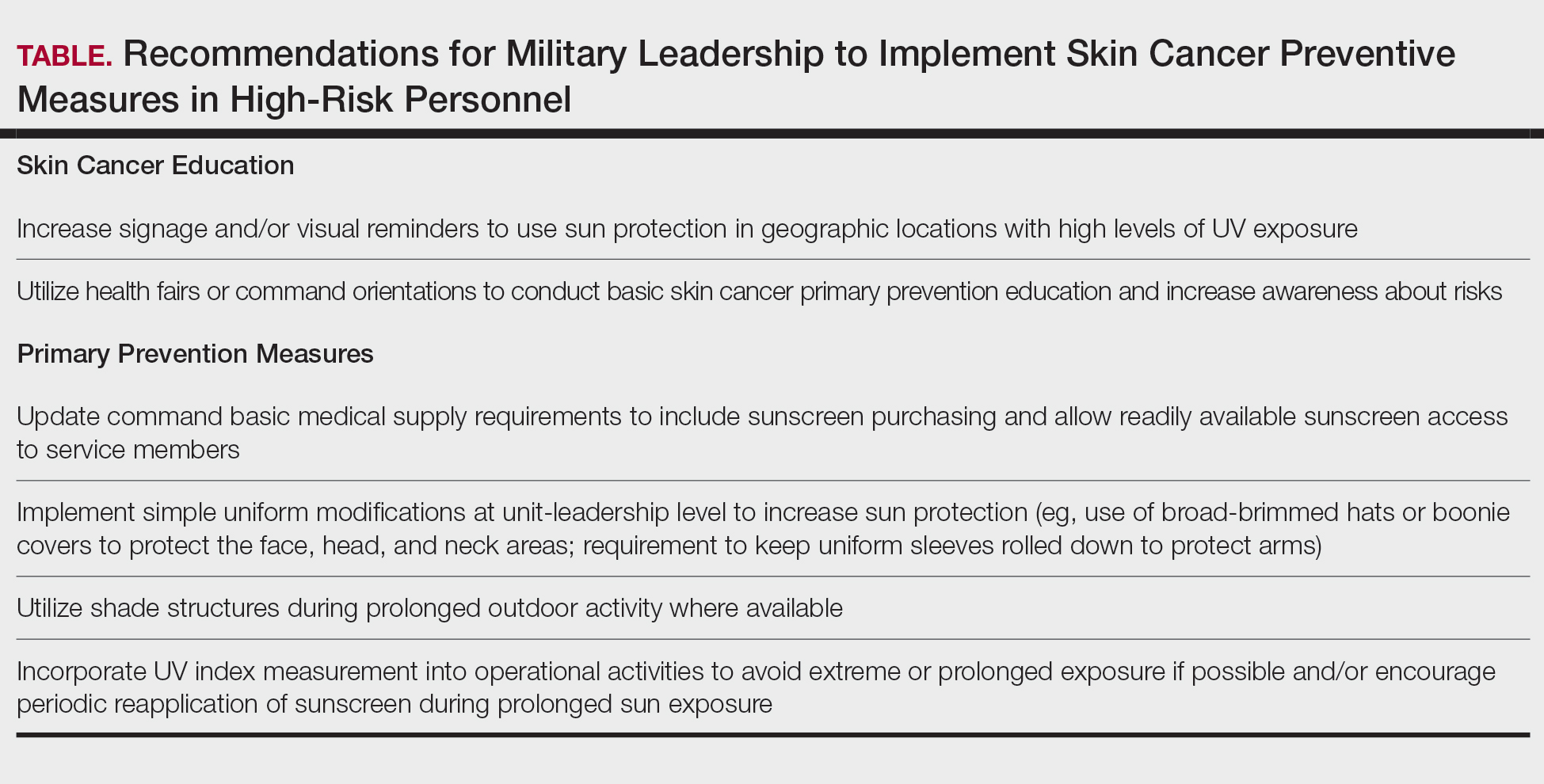
Despite well-documented risks, considerable gaps remain in quantifying and analyzing variations in UV exposure across military occupations, duty locations, and operational roles. Factors such as the existence of over 150 distinct military occupational specialties, frequent geographic relocations, and routine work in austere environments contribute to a wide range of UV exposure profiles that remain insufficiently characterized. This lack of comprehensive exposure data hinders the development of large-scale, targeted skin cancer prevention strategies. Initial approaches to addressing these challenges include enhanced surveillance, education, and policy initiatives. The Table presents practical recommendations for military leadership to consider in implementing preventive measures for skin cancer. Herein, we outline broader systemic strategies to bridge knowledge gaps and address underrecognized occupational risk factors for skin cancer in military service members; these elements include proposed modifications to the electronic Periodic Health Assessment (ePHA) and the development of standardized, military-specific screening and prevention guidelines to support early detection and resource optimization.
Skin Cancer Education for Service Members
Sunscreen and Signage—Diligent primary prevention offers a promising avenue for mitigating skin cancer incidence in military service members. Basic education and precautionary messaging on photoprotection can be widely implemented to simultaneously educate service members on the dangers of sun exposure while reinforcing healthy behaviors in real time. Simple low-cost initiatives such as strategically placed visual signage reminding service members to apply sunscreen in high UV environments can support consistent sun-safe practices. Educational efforts also should emphasize proper sunscreen use, including application on high-risk anatomic sites (eg, the face, neck, scalp, dorsal hands, and ears) and the essentiality of using sufficient quantities of broad-spectrum sunscreen for effective protection. Incorporating this guidance into training materials, briefings, and visual reminders allow seamless integration of photoprotection into service members’ daily routines without compromising operational efficiency.6 Younger service members, who may be less likely to prioritize preventive behaviors, may be particularly responsive to sun safety reminders in training areas, bases, and deployment zones.7 Health fairs and orientation briefs in high-UV regions also offer potential opportunities for targeted education.
Resources for Sun Protection in the Military
Sunscreen—Although sunscreen is critical in minimizing the risk for UV-induced skin cancer, its widespread use in the military is hindered by practical challenges related to accessibility and the need for consistent reapplication; for instance, providing free sunscreen dispensers at institutions for staff working under intense or prolonged UV exposure may improve sunscreen accessibility and use.8 Including sunscreen in standard-issue gear offers another logical way to embed its use into operational readiness as part of the routine protective measures.
Uniform Modifications—Adapting military uniforms and practices to improve sun protection plays a critical role in reducing skin cancer risk. Targeted protective gear for commonly sun-exposed areas can help mitigate UV exposure. One practical option is the use of wide-brimmed headgear (eg, boonie hats), which provide more face and neck coverage than standard-issue military caps, or covers. The wide-brimmed headgear currently is only selectively authorized during specific scenarios, such as field operations and training exercises, or at the discretion of unit-level leadership. Wide-brimmed headgear, already used by many service members, has been associated with up to a 17% reduction in UV exposure to inadequately protected areas, potentially lowering skin cancer risk.9,10 Similarly, a “sleeves-down” policy—requiring sleeves to remain unrolled and covering the forearms during outdoor activities—offers a simple way to minimize sun exposure without necessitating additional gear. Other specialized clothing items, including UV-blocking neck gaiters, photoprotective clothing, and lightweight gloves, also may be appropriate for high-risk groups and can be implemented in a relatively straightforward manner.
Shade Structures and UV Index Monitoring—Aside from uniform adaptation, physical barrier intervention can further complement skin cancer prevention efforts in the military. Shade structures offer a straightforward way to reduce UV exposure during prolonged outdoor activities. Incorporating daily UV index monitoring into operational guidance can help inform adjustments to training schedules and guide the implementation of additional sun protection measures, such as mandatory sunscreen application, use of wide-brimmed hats, or increased access to shaded rest areas during heavy sunlight hours. Currently, outdoor physical training is restricted during periods of high heat index, measured via Wet Bulb Globe Temperature, to reduce heat-related injuries. We argue that avoidance of nonoperational outdoor activity during peak UV index hours also should be incorporated into standardized policies. This intervention is of particular benefit to service members stationed in regions with a high UV index year-round, such as those stationed in the Middle East, Guam, Okinawa, and southern coastal United States bases.
Policy Changes to Support Photoprotective Measures
Annual Risk Factor Screening‐Screening—Effective secondary prevention efforts by military dermatologists remain an important measure in reducing the burden of skin cancer among military personnel; however, these efforts have become increasingly challenging due to 2 main factors—the diversity of military occupational specialties and their associated unique occupational risks as well as the limited availability of military dermatologists across all branches (approximately 100 active-duty dermatologists for nearly 3 million service members).11 Therefore, targeted interventions that enhance risk assessment, refined screening protocols, and leveraging of existing military health networks can improve early skin cancer detection while optimizing resource allocation.
The ePHA is an online screening tool used annually by all service members to evaluate their overall health. Presently, the ePHA lacks specific questions to assess sun exposure and skin cancer risks. Integrating annual skin cancer risk factor assessments into the ePHA would offer a practical and straightforward approach to identifying at-risk individuals, as suggested by Newnam et al12 in 2022. Skin cancer risk factor assessments allow for targeted data collection related to sun exposure history, family history, and personal risk factors, which can be used to determine individualized risk stratification to assess the need for early secondary prevention measures and specialist referral. These ePHA data can also support population-based analyses to inform preventive strategies and address knowledge gaps related to high-risk exposures, such as extended field exercises or assignments in high-UV regions, that may impede effective skin cancer prevention.
Development of Military-Specific Screening Guidelines—Given the limited number of military dermatologists, a standardized risk-assessment tool could enhance early detection of skin cancer and streamline the referral process. We propose a military-specific skin cancer screening algorithm or risk nomogram that could help to consolidate risk factors into a clear and actionable framework for more efficient triage and appropriate allocation of dermatologic resources and manpower. This nomogram could be developed by military dermatologists and then implemented on a command level, affording primary care providers a useful tool to expedite evaluation of individuals at higher risk for skin cancer while simultaneously promoting judicious use of limited dermatology resources.
Although the United States Preventive Services Task Force does not universally recommend routine skin cancer screenings for asymptomatic adults, military service members are exposed to higher occupational risks than the general population, as previously mentioned. Currently, there is no standardized screening guideline across all military services due to the unique nature and exposure risks for each branch of service and their varied occupations; however, we propose the development of basic standardized screening guidelines by adapting the framework of the United States Preventive Services Task Force and adjusting for military-specific UV exposure and occupational risks to improve early detection of skin cancer. These guidelines could be updated and tailored appropriately when additional population-based data are collected and analyzed through ePHA.
Critiques and Limitations of Implementation
Several challenges and limitations must be considered when attempting to integrate large-scale preventive measures for skin cancer within the US military. A primary concern is the extent to which military resources should be allocated to prevention when off-duty sun exposure remains largely beyond institutional control. Although military health initiatives can address workplace risk through education and policy, individual decisions during both work and leisure time remain a major variable that cannot be feasibly controlled. Cultural and operational barriers also pose challenges; for instance, the US Marine Corps maintains a strong cultural identity tied to uniform appearance, making it difficult to implement widespread changes to clothing-based sun-protection measures. Institutional changes, particularly those involving uniforms, likely will face substantial administrative resistance and potential operational limitations. When broad uniform modifications are unattainable, a more feasible approach may be to encourage unit-level leadership to authorize and promote the frequent use of nonuniform protective measures.
Furthermore, integrating additional skin cancer risk questions into the already extensive ePHA means extra time required to complete the assessment; this adds to service members’ administrative burden, potentially leading to reduced timely compliance, rushed responses, and survey fatigue, which threaten data quality. If new items are to be included, they should be carefully selected for efficiency and clinical relevance. Existing validated questionnaires such as those from the study by Lyford et al7 published in 2021 can serve as a foundation.
Another critical limitation is access to dermatologic care for active-duty service members. Raising awareness of skin cancer risk without ensuring adequate resources may create ethical concerns, particularly in high-risk environments such as the Middle East and Indo-Pacific. Additionally, because skin cancer often develops years or decades after exposure, securing early buy-in from service members and their leaders can be challenging. These concerns make it clear that, while skin cancer prevention is important, implementing widespread measures is not straightforward and requires a practical and balanced approach.
Final Thoughts
Implementing prevention strategies for skin cancer in the military requires balancing evidence-based recommendations with the practical realities of military culture, resource limitations, and operational demands. Challenges remain for dermatologists in providing targeted recommendations due to the multifaceted nature of military roles, including over 150 Navy Military Occupational Specialties, limited familiarity with the unique UV exposure risks associated with each occupation, and variability in local and regional policies on uniform wear, physical training requirements, and other operational practices. Although targeted prevention measures are difficult to establish in the setting of these knowledge gaps, leveraging unit-level leadership to align with existing screening guidelines and optimizing primary prevention measures can be meaningful steps toward reducing skin cancer risk for military service members while maintaining mission readiness.
- Riemenschneider K, Liu J, Powers JG. Skin cancer in the military: a systematic review of melanoma and nonmelanoma skin cancerincidence, prevention, and screening among active duty and veteran personnel. J Am Acad Dermatol. 2018;78:1185-1192. doi:10.1016/j.jaad.2017.11.062
- Lee T, Taubman SB, Williams VF. Incident diagnoses of non-melanoma skin cancer, active component, U.S. Armed Forces, 2005-2014. MSMR. 2016;23:2-6.
- Krivda KR, Watson NL, Lyford WH, et al. The burden of skin cancer in the military health system, 2017-2022. Cutis. 2024;113:200-215. doi:10.12788/cutis.1015
- Sanlorenzo M, Wehner MR, Linos E, et al. The risk of melanoma in airline pilots and cabin crew: a meta-analysis. JAMA Dermatol. 2015;151:51-58. doi:10.1001/jamadermatol.2014.1077
- Brundage JF, Williams VF, Stahlman S, et al. Incidence rates of malignant melanoma in relation to years of military service, overall and in selected military occupational groups, active component, U.S. Armed Forces, 2001-2015. MMSR. 2017;24:8-14.
- Subramaniam P, Olsen CM, Thompson BS, et al, for the QSkin Sun and Health Study Investigators. Anatomical distributions of basal cell carcinoma and squamous cell carcinoma in a population-based study in Queensland, Australia. JAMA Dermatol. 2017;153:175-182. doi:10.1001/jamadermatol.2016.4070
- Lyford WH, Crotty A, Logemann NF. Sun exposure prevention practices within U.S. naval aviation. Mil Med. 2021;186:1169-1175. doi:10.1093/milmed/usab099
- Wood M, Raisanen T, Polcari I. Observational study of free public sunscreen dispenser use at a major US outdoor event. J Am Acad Dermatol. 2017;77:164-166.
- Schissel D. Operation shadow warrior: a quantitative analysis of the ultraviolet radiation protection demonstrated by various headgear. Mil Med. 2001;166:783-785.
- Milch JM, Logemann NF. Photoprotection prevents skin cancer: let’s make it fashionable to wear sun-protective clothing. Cutis. 2017;99:89-92.
- Association of Military Dermatologists. (n.d.). Military dermatology. https://militaryderm.org/military-dermatology/
- Newnam R, Le-Jenkins U, Rutledge C, et al. The association of skin cancer prevention knowledge, sun-protective attitudes, and sunprotective behaviors in a Navy population. Mil Med. 2024;189:1-7. doi:10.1093/milmed/usac285
Skin cancer is a major health concern for military service members, who experience notably higher incidence rates than the general population.1 Active-duty military personnel are particularly vulnerable to prolonged sun exposure due to deployments, specialized training, and everyday outdoor duties.1 Despite skin cancer being the most commonly diagnosed malignancy in active-duty service members,2 tracking and documenting the quantity and diversity of these risk factors remain limited. This knowledge gap comes at high cost, simultaneously impairing military medicine preventive measures while burdening the military health care system with substantial expenditures.3 These findings underscore the critical need for targeted surveillance, early-detection programs, and policy-driven interventions to mitigate these medical and economic concerns.
Skin cancer has been recognized as a major health risk to the military population for decades, yet incidence and prevalence remain high. This phenomenon is closely linked to the inherent responsibilities and expectations of active-duty military members, including outdoor physical training, field exercises, standing in formation, and outdoor working environments—all of which can occur during peak sunlight hours. These risks are further elevated at duty stations in geographic regions with high levels of UV exposure, such as those in tropical and arid regions of the world. Certain military occupational specialties and missions may further introduce unique risk factors; for instance, pilots with frequent high-altitude missions experience heightened UV exposure and melanoma risk.4 Secondary to compounding determinants, the aviation, diving, and nuclear subgroups of the military community are particularly vulnerable to skin cancer.5
Despite well-documented risks, considerable gaps remain in quantifying and analyzing variations in UV exposure across military occupations, duty locations, and operational roles. Factors such as the existence of over 150 distinct military occupational specialties, frequent geographic relocations, and routine work in austere environments contribute to a wide range of UV exposure profiles that remain insufficiently characterized. This lack of comprehensive exposure data hinders the development of large-scale, targeted skin cancer prevention strategies. Initial approaches to addressing these challenges include enhanced surveillance, education, and policy initiatives. The Table presents practical recommendations for military leadership to consider in implementing preventive measures for skin cancer. Herein, we outline broader systemic strategies to bridge knowledge gaps and address underrecognized occupational risk factors for skin cancer in military service members; these elements include proposed modifications to the electronic Periodic Health Assessment (ePHA) and the development of standardized, military-specific screening and prevention guidelines to support early detection and resource optimization.
Skin Cancer Education for Service Members
Sunscreen and Signage—Diligent primary prevention offers a promising avenue for mitigating skin cancer incidence in military service members. Basic education and precautionary messaging on photoprotection can be widely implemented to simultaneously educate service members on the dangers of sun exposure while reinforcing healthy behaviors in real time. Simple low-cost initiatives such as strategically placed visual signage reminding service members to apply sunscreen in high UV environments can support consistent sun-safe practices. Educational efforts also should emphasize proper sunscreen use, including application on high-risk anatomic sites (eg, the face, neck, scalp, dorsal hands, and ears) and the essentiality of using sufficient quantities of broad-spectrum sunscreen for effective protection. Incorporating this guidance into training materials, briefings, and visual reminders allow seamless integration of photoprotection into service members’ daily routines without compromising operational efficiency.6 Younger service members, who may be less likely to prioritize preventive behaviors, may be particularly responsive to sun safety reminders in training areas, bases, and deployment zones.7 Health fairs and orientation briefs in high-UV regions also offer potential opportunities for targeted education.
Resources for Sun Protection in the Military
Sunscreen—Although sunscreen is critical in minimizing the risk for UV-induced skin cancer, its widespread use in the military is hindered by practical challenges related to accessibility and the need for consistent reapplication; for instance, providing free sunscreen dispensers at institutions for staff working under intense or prolonged UV exposure may improve sunscreen accessibility and use.8 Including sunscreen in standard-issue gear offers another logical way to embed its use into operational readiness as part of the routine protective measures.
Uniform Modifications—Adapting military uniforms and practices to improve sun protection plays a critical role in reducing skin cancer risk. Targeted protective gear for commonly sun-exposed areas can help mitigate UV exposure. One practical option is the use of wide-brimmed headgear (eg, boonie hats), which provide more face and neck coverage than standard-issue military caps, or covers. The wide-brimmed headgear currently is only selectively authorized during specific scenarios, such as field operations and training exercises, or at the discretion of unit-level leadership. Wide-brimmed headgear, already used by many service members, has been associated with up to a 17% reduction in UV exposure to inadequately protected areas, potentially lowering skin cancer risk.9,10 Similarly, a “sleeves-down” policy—requiring sleeves to remain unrolled and covering the forearms during outdoor activities—offers a simple way to minimize sun exposure without necessitating additional gear. Other specialized clothing items, including UV-blocking neck gaiters, photoprotective clothing, and lightweight gloves, also may be appropriate for high-risk groups and can be implemented in a relatively straightforward manner.
Shade Structures and UV Index Monitoring—Aside from uniform adaptation, physical barrier intervention can further complement skin cancer prevention efforts in the military. Shade structures offer a straightforward way to reduce UV exposure during prolonged outdoor activities. Incorporating daily UV index monitoring into operational guidance can help inform adjustments to training schedules and guide the implementation of additional sun protection measures, such as mandatory sunscreen application, use of wide-brimmed hats, or increased access to shaded rest areas during heavy sunlight hours. Currently, outdoor physical training is restricted during periods of high heat index, measured via Wet Bulb Globe Temperature, to reduce heat-related injuries. We argue that avoidance of nonoperational outdoor activity during peak UV index hours also should be incorporated into standardized policies. This intervention is of particular benefit to service members stationed in regions with a high UV index year-round, such as those stationed in the Middle East, Guam, Okinawa, and southern coastal United States bases.
Policy Changes to Support Photoprotective Measures
Annual Risk Factor Screening‐Screening—Effective secondary prevention efforts by military dermatologists remain an important measure in reducing the burden of skin cancer among military personnel; however, these efforts have become increasingly challenging due to 2 main factors—the diversity of military occupational specialties and their associated unique occupational risks as well as the limited availability of military dermatologists across all branches (approximately 100 active-duty dermatologists for nearly 3 million service members).11 Therefore, targeted interventions that enhance risk assessment, refined screening protocols, and leveraging of existing military health networks can improve early skin cancer detection while optimizing resource allocation.
The ePHA is an online screening tool used annually by all service members to evaluate their overall health. Presently, the ePHA lacks specific questions to assess sun exposure and skin cancer risks. Integrating annual skin cancer risk factor assessments into the ePHA would offer a practical and straightforward approach to identifying at-risk individuals, as suggested by Newnam et al12 in 2022. Skin cancer risk factor assessments allow for targeted data collection related to sun exposure history, family history, and personal risk factors, which can be used to determine individualized risk stratification to assess the need for early secondary prevention measures and specialist referral. These ePHA data can also support population-based analyses to inform preventive strategies and address knowledge gaps related to high-risk exposures, such as extended field exercises or assignments in high-UV regions, that may impede effective skin cancer prevention.
Development of Military-Specific Screening Guidelines—Given the limited number of military dermatologists, a standardized risk-assessment tool could enhance early detection of skin cancer and streamline the referral process. We propose a military-specific skin cancer screening algorithm or risk nomogram that could help to consolidate risk factors into a clear and actionable framework for more efficient triage and appropriate allocation of dermatologic resources and manpower. This nomogram could be developed by military dermatologists and then implemented on a command level, affording primary care providers a useful tool to expedite evaluation of individuals at higher risk for skin cancer while simultaneously promoting judicious use of limited dermatology resources.
Although the United States Preventive Services Task Force does not universally recommend routine skin cancer screenings for asymptomatic adults, military service members are exposed to higher occupational risks than the general population, as previously mentioned. Currently, there is no standardized screening guideline across all military services due to the unique nature and exposure risks for each branch of service and their varied occupations; however, we propose the development of basic standardized screening guidelines by adapting the framework of the United States Preventive Services Task Force and adjusting for military-specific UV exposure and occupational risks to improve early detection of skin cancer. These guidelines could be updated and tailored appropriately when additional population-based data are collected and analyzed through ePHA.
Critiques and Limitations of Implementation
Several challenges and limitations must be considered when attempting to integrate large-scale preventive measures for skin cancer within the US military. A primary concern is the extent to which military resources should be allocated to prevention when off-duty sun exposure remains largely beyond institutional control. Although military health initiatives can address workplace risk through education and policy, individual decisions during both work and leisure time remain a major variable that cannot be feasibly controlled. Cultural and operational barriers also pose challenges; for instance, the US Marine Corps maintains a strong cultural identity tied to uniform appearance, making it difficult to implement widespread changes to clothing-based sun-protection measures. Institutional changes, particularly those involving uniforms, likely will face substantial administrative resistance and potential operational limitations. When broad uniform modifications are unattainable, a more feasible approach may be to encourage unit-level leadership to authorize and promote the frequent use of nonuniform protective measures.
Furthermore, integrating additional skin cancer risk questions into the already extensive ePHA means extra time required to complete the assessment; this adds to service members’ administrative burden, potentially leading to reduced timely compliance, rushed responses, and survey fatigue, which threaten data quality. If new items are to be included, they should be carefully selected for efficiency and clinical relevance. Existing validated questionnaires such as those from the study by Lyford et al7 published in 2021 can serve as a foundation.
Another critical limitation is access to dermatologic care for active-duty service members. Raising awareness of skin cancer risk without ensuring adequate resources may create ethical concerns, particularly in high-risk environments such as the Middle East and Indo-Pacific. Additionally, because skin cancer often develops years or decades after exposure, securing early buy-in from service members and their leaders can be challenging. These concerns make it clear that, while skin cancer prevention is important, implementing widespread measures is not straightforward and requires a practical and balanced approach.
Final Thoughts
Implementing prevention strategies for skin cancer in the military requires balancing evidence-based recommendations with the practical realities of military culture, resource limitations, and operational demands. Challenges remain for dermatologists in providing targeted recommendations due to the multifaceted nature of military roles, including over 150 Navy Military Occupational Specialties, limited familiarity with the unique UV exposure risks associated with each occupation, and variability in local and regional policies on uniform wear, physical training requirements, and other operational practices. Although targeted prevention measures are difficult to establish in the setting of these knowledge gaps, leveraging unit-level leadership to align with existing screening guidelines and optimizing primary prevention measures can be meaningful steps toward reducing skin cancer risk for military service members while maintaining mission readiness.
Skin cancer is a major health concern for military service members, who experience notably higher incidence rates than the general population.1 Active-duty military personnel are particularly vulnerable to prolonged sun exposure due to deployments, specialized training, and everyday outdoor duties.1 Despite skin cancer being the most commonly diagnosed malignancy in active-duty service members,2 tracking and documenting the quantity and diversity of these risk factors remain limited. This knowledge gap comes at high cost, simultaneously impairing military medicine preventive measures while burdening the military health care system with substantial expenditures.3 These findings underscore the critical need for targeted surveillance, early-detection programs, and policy-driven interventions to mitigate these medical and economic concerns.
Skin cancer has been recognized as a major health risk to the military population for decades, yet incidence and prevalence remain high. This phenomenon is closely linked to the inherent responsibilities and expectations of active-duty military members, including outdoor physical training, field exercises, standing in formation, and outdoor working environments—all of which can occur during peak sunlight hours. These risks are further elevated at duty stations in geographic regions with high levels of UV exposure, such as those in tropical and arid regions of the world. Certain military occupational specialties and missions may further introduce unique risk factors; for instance, pilots with frequent high-altitude missions experience heightened UV exposure and melanoma risk.4 Secondary to compounding determinants, the aviation, diving, and nuclear subgroups of the military community are particularly vulnerable to skin cancer.5
Despite well-documented risks, considerable gaps remain in quantifying and analyzing variations in UV exposure across military occupations, duty locations, and operational roles. Factors such as the existence of over 150 distinct military occupational specialties, frequent geographic relocations, and routine work in austere environments contribute to a wide range of UV exposure profiles that remain insufficiently characterized. This lack of comprehensive exposure data hinders the development of large-scale, targeted skin cancer prevention strategies. Initial approaches to addressing these challenges include enhanced surveillance, education, and policy initiatives. The Table presents practical recommendations for military leadership to consider in implementing preventive measures for skin cancer. Herein, we outline broader systemic strategies to bridge knowledge gaps and address underrecognized occupational risk factors for skin cancer in military service members; these elements include proposed modifications to the electronic Periodic Health Assessment (ePHA) and the development of standardized, military-specific screening and prevention guidelines to support early detection and resource optimization.
Skin Cancer Education for Service Members
Sunscreen and Signage—Diligent primary prevention offers a promising avenue for mitigating skin cancer incidence in military service members. Basic education and precautionary messaging on photoprotection can be widely implemented to simultaneously educate service members on the dangers of sun exposure while reinforcing healthy behaviors in real time. Simple low-cost initiatives such as strategically placed visual signage reminding service members to apply sunscreen in high UV environments can support consistent sun-safe practices. Educational efforts also should emphasize proper sunscreen use, including application on high-risk anatomic sites (eg, the face, neck, scalp, dorsal hands, and ears) and the essentiality of using sufficient quantities of broad-spectrum sunscreen for effective protection. Incorporating this guidance into training materials, briefings, and visual reminders allow seamless integration of photoprotection into service members’ daily routines without compromising operational efficiency.6 Younger service members, who may be less likely to prioritize preventive behaviors, may be particularly responsive to sun safety reminders in training areas, bases, and deployment zones.7 Health fairs and orientation briefs in high-UV regions also offer potential opportunities for targeted education.
Resources for Sun Protection in the Military
Sunscreen—Although sunscreen is critical in minimizing the risk for UV-induced skin cancer, its widespread use in the military is hindered by practical challenges related to accessibility and the need for consistent reapplication; for instance, providing free sunscreen dispensers at institutions for staff working under intense or prolonged UV exposure may improve sunscreen accessibility and use.8 Including sunscreen in standard-issue gear offers another logical way to embed its use into operational readiness as part of the routine protective measures.
Uniform Modifications—Adapting military uniforms and practices to improve sun protection plays a critical role in reducing skin cancer risk. Targeted protective gear for commonly sun-exposed areas can help mitigate UV exposure. One practical option is the use of wide-brimmed headgear (eg, boonie hats), which provide more face and neck coverage than standard-issue military caps, or covers. The wide-brimmed headgear currently is only selectively authorized during specific scenarios, such as field operations and training exercises, or at the discretion of unit-level leadership. Wide-brimmed headgear, already used by many service members, has been associated with up to a 17% reduction in UV exposure to inadequately protected areas, potentially lowering skin cancer risk.9,10 Similarly, a “sleeves-down” policy—requiring sleeves to remain unrolled and covering the forearms during outdoor activities—offers a simple way to minimize sun exposure without necessitating additional gear. Other specialized clothing items, including UV-blocking neck gaiters, photoprotective clothing, and lightweight gloves, also may be appropriate for high-risk groups and can be implemented in a relatively straightforward manner.
Shade Structures and UV Index Monitoring—Aside from uniform adaptation, physical barrier intervention can further complement skin cancer prevention efforts in the military. Shade structures offer a straightforward way to reduce UV exposure during prolonged outdoor activities. Incorporating daily UV index monitoring into operational guidance can help inform adjustments to training schedules and guide the implementation of additional sun protection measures, such as mandatory sunscreen application, use of wide-brimmed hats, or increased access to shaded rest areas during heavy sunlight hours. Currently, outdoor physical training is restricted during periods of high heat index, measured via Wet Bulb Globe Temperature, to reduce heat-related injuries. We argue that avoidance of nonoperational outdoor activity during peak UV index hours also should be incorporated into standardized policies. This intervention is of particular benefit to service members stationed in regions with a high UV index year-round, such as those stationed in the Middle East, Guam, Okinawa, and southern coastal United States bases.
Policy Changes to Support Photoprotective Measures
Annual Risk Factor Screening‐Screening—Effective secondary prevention efforts by military dermatologists remain an important measure in reducing the burden of skin cancer among military personnel; however, these efforts have become increasingly challenging due to 2 main factors—the diversity of military occupational specialties and their associated unique occupational risks as well as the limited availability of military dermatologists across all branches (approximately 100 active-duty dermatologists for nearly 3 million service members).11 Therefore, targeted interventions that enhance risk assessment, refined screening protocols, and leveraging of existing military health networks can improve early skin cancer detection while optimizing resource allocation.
The ePHA is an online screening tool used annually by all service members to evaluate their overall health. Presently, the ePHA lacks specific questions to assess sun exposure and skin cancer risks. Integrating annual skin cancer risk factor assessments into the ePHA would offer a practical and straightforward approach to identifying at-risk individuals, as suggested by Newnam et al12 in 2022. Skin cancer risk factor assessments allow for targeted data collection related to sun exposure history, family history, and personal risk factors, which can be used to determine individualized risk stratification to assess the need for early secondary prevention measures and specialist referral. These ePHA data can also support population-based analyses to inform preventive strategies and address knowledge gaps related to high-risk exposures, such as extended field exercises or assignments in high-UV regions, that may impede effective skin cancer prevention.
Development of Military-Specific Screening Guidelines—Given the limited number of military dermatologists, a standardized risk-assessment tool could enhance early detection of skin cancer and streamline the referral process. We propose a military-specific skin cancer screening algorithm or risk nomogram that could help to consolidate risk factors into a clear and actionable framework for more efficient triage and appropriate allocation of dermatologic resources and manpower. This nomogram could be developed by military dermatologists and then implemented on a command level, affording primary care providers a useful tool to expedite evaluation of individuals at higher risk for skin cancer while simultaneously promoting judicious use of limited dermatology resources.
Although the United States Preventive Services Task Force does not universally recommend routine skin cancer screenings for asymptomatic adults, military service members are exposed to higher occupational risks than the general population, as previously mentioned. Currently, there is no standardized screening guideline across all military services due to the unique nature and exposure risks for each branch of service and their varied occupations; however, we propose the development of basic standardized screening guidelines by adapting the framework of the United States Preventive Services Task Force and adjusting for military-specific UV exposure and occupational risks to improve early detection of skin cancer. These guidelines could be updated and tailored appropriately when additional population-based data are collected and analyzed through ePHA.
Critiques and Limitations of Implementation
Several challenges and limitations must be considered when attempting to integrate large-scale preventive measures for skin cancer within the US military. A primary concern is the extent to which military resources should be allocated to prevention when off-duty sun exposure remains largely beyond institutional control. Although military health initiatives can address workplace risk through education and policy, individual decisions during both work and leisure time remain a major variable that cannot be feasibly controlled. Cultural and operational barriers also pose challenges; for instance, the US Marine Corps maintains a strong cultural identity tied to uniform appearance, making it difficult to implement widespread changes to clothing-based sun-protection measures. Institutional changes, particularly those involving uniforms, likely will face substantial administrative resistance and potential operational limitations. When broad uniform modifications are unattainable, a more feasible approach may be to encourage unit-level leadership to authorize and promote the frequent use of nonuniform protective measures.
Furthermore, integrating additional skin cancer risk questions into the already extensive ePHA means extra time required to complete the assessment; this adds to service members’ administrative burden, potentially leading to reduced timely compliance, rushed responses, and survey fatigue, which threaten data quality. If new items are to be included, they should be carefully selected for efficiency and clinical relevance. Existing validated questionnaires such as those from the study by Lyford et al7 published in 2021 can serve as a foundation.
Another critical limitation is access to dermatologic care for active-duty service members. Raising awareness of skin cancer risk without ensuring adequate resources may create ethical concerns, particularly in high-risk environments such as the Middle East and Indo-Pacific. Additionally, because skin cancer often develops years or decades after exposure, securing early buy-in from service members and their leaders can be challenging. These concerns make it clear that, while skin cancer prevention is important, implementing widespread measures is not straightforward and requires a practical and balanced approach.
Final Thoughts
Implementing prevention strategies for skin cancer in the military requires balancing evidence-based recommendations with the practical realities of military culture, resource limitations, and operational demands. Challenges remain for dermatologists in providing targeted recommendations due to the multifaceted nature of military roles, including over 150 Navy Military Occupational Specialties, limited familiarity with the unique UV exposure risks associated with each occupation, and variability in local and regional policies on uniform wear, physical training requirements, and other operational practices. Although targeted prevention measures are difficult to establish in the setting of these knowledge gaps, leveraging unit-level leadership to align with existing screening guidelines and optimizing primary prevention measures can be meaningful steps toward reducing skin cancer risk for military service members while maintaining mission readiness.
- Riemenschneider K, Liu J, Powers JG. Skin cancer in the military: a systematic review of melanoma and nonmelanoma skin cancerincidence, prevention, and screening among active duty and veteran personnel. J Am Acad Dermatol. 2018;78:1185-1192. doi:10.1016/j.jaad.2017.11.062
- Lee T, Taubman SB, Williams VF. Incident diagnoses of non-melanoma skin cancer, active component, U.S. Armed Forces, 2005-2014. MSMR. 2016;23:2-6.
- Krivda KR, Watson NL, Lyford WH, et al. The burden of skin cancer in the military health system, 2017-2022. Cutis. 2024;113:200-215. doi:10.12788/cutis.1015
- Sanlorenzo M, Wehner MR, Linos E, et al. The risk of melanoma in airline pilots and cabin crew: a meta-analysis. JAMA Dermatol. 2015;151:51-58. doi:10.1001/jamadermatol.2014.1077
- Brundage JF, Williams VF, Stahlman S, et al. Incidence rates of malignant melanoma in relation to years of military service, overall and in selected military occupational groups, active component, U.S. Armed Forces, 2001-2015. MMSR. 2017;24:8-14.
- Subramaniam P, Olsen CM, Thompson BS, et al, for the QSkin Sun and Health Study Investigators. Anatomical distributions of basal cell carcinoma and squamous cell carcinoma in a population-based study in Queensland, Australia. JAMA Dermatol. 2017;153:175-182. doi:10.1001/jamadermatol.2016.4070
- Lyford WH, Crotty A, Logemann NF. Sun exposure prevention practices within U.S. naval aviation. Mil Med. 2021;186:1169-1175. doi:10.1093/milmed/usab099
- Wood M, Raisanen T, Polcari I. Observational study of free public sunscreen dispenser use at a major US outdoor event. J Am Acad Dermatol. 2017;77:164-166.
- Schissel D. Operation shadow warrior: a quantitative analysis of the ultraviolet radiation protection demonstrated by various headgear. Mil Med. 2001;166:783-785.
- Milch JM, Logemann NF. Photoprotection prevents skin cancer: let’s make it fashionable to wear sun-protective clothing. Cutis. 2017;99:89-92.
- Association of Military Dermatologists. (n.d.). Military dermatology. https://militaryderm.org/military-dermatology/
- Newnam R, Le-Jenkins U, Rutledge C, et al. The association of skin cancer prevention knowledge, sun-protective attitudes, and sunprotective behaviors in a Navy population. Mil Med. 2024;189:1-7. doi:10.1093/milmed/usac285
- Riemenschneider K, Liu J, Powers JG. Skin cancer in the military: a systematic review of melanoma and nonmelanoma skin cancerincidence, prevention, and screening among active duty and veteran personnel. J Am Acad Dermatol. 2018;78:1185-1192. doi:10.1016/j.jaad.2017.11.062
- Lee T, Taubman SB, Williams VF. Incident diagnoses of non-melanoma skin cancer, active component, U.S. Armed Forces, 2005-2014. MSMR. 2016;23:2-6.
- Krivda KR, Watson NL, Lyford WH, et al. The burden of skin cancer in the military health system, 2017-2022. Cutis. 2024;113:200-215. doi:10.12788/cutis.1015
- Sanlorenzo M, Wehner MR, Linos E, et al. The risk of melanoma in airline pilots and cabin crew: a meta-analysis. JAMA Dermatol. 2015;151:51-58. doi:10.1001/jamadermatol.2014.1077
- Brundage JF, Williams VF, Stahlman S, et al. Incidence rates of malignant melanoma in relation to years of military service, overall and in selected military occupational groups, active component, U.S. Armed Forces, 2001-2015. MMSR. 2017;24:8-14.
- Subramaniam P, Olsen CM, Thompson BS, et al, for the QSkin Sun and Health Study Investigators. Anatomical distributions of basal cell carcinoma and squamous cell carcinoma in a population-based study in Queensland, Australia. JAMA Dermatol. 2017;153:175-182. doi:10.1001/jamadermatol.2016.4070
- Lyford WH, Crotty A, Logemann NF. Sun exposure prevention practices within U.S. naval aviation. Mil Med. 2021;186:1169-1175. doi:10.1093/milmed/usab099
- Wood M, Raisanen T, Polcari I. Observational study of free public sunscreen dispenser use at a major US outdoor event. J Am Acad Dermatol. 2017;77:164-166.
- Schissel D. Operation shadow warrior: a quantitative analysis of the ultraviolet radiation protection demonstrated by various headgear. Mil Med. 2001;166:783-785.
- Milch JM, Logemann NF. Photoprotection prevents skin cancer: let’s make it fashionable to wear sun-protective clothing. Cutis. 2017;99:89-92.
- Association of Military Dermatologists. (n.d.). Military dermatology. https://militaryderm.org/military-dermatology/
- Newnam R, Le-Jenkins U, Rutledge C, et al. The association of skin cancer prevention knowledge, sun-protective attitudes, and sunprotective behaviors in a Navy population. Mil Med. 2024;189:1-7. doi:10.1093/milmed/usac285
Bridging the Knowledge-Action Gap in Skin Cancer Prevention Among US Military Personnel
Bridging the Knowledge-Action Gap in Skin Cancer Prevention Among US Military Personnel
PRACTICE POINTS
- Military personnel face elevated skin cancer risks due to prolonged occupational UV exposure.
- Medical providers can partner with unit-level leadership to implement low-cost interventions such as shade structures and uniform modifications.
- Annual sun exposure risk assessments should be integrated into the military Electronic Periodic Health Assessment for targeted screening and early intervention of risk factors.
- Photoprotective gear and signage in high—UV index areas can improve service member awareness and adherence to preventive measures.
Alagille Syndrome: Epidemiology and Management of a Rare Genetic Disease
Alagille Syndrome: Epidemiology and Management of a Rare Genetic Disease
Yan J, Huang Y, Cao L, et al. Clinical, pathological and genetic characteristics of 17 unrelated children with Alagille Syndrome. BMC Pediatr. 2024;24(1):532. doi:10.1186/s12887-024-04973-y
Cheng K, Rosenthal P. Diagnosis and management of Alagille and progressive familial intrahepatic cholestasis. Hepatol Commun. 2023 Dec 7;7(12):e0314. doi:10.1097/HC9.0000000000000314
Global Allagile Alliance, GALA, website. The GALA Study. Published 2022. Accessed January 27, 2025. https://www.galastudy.com/
Karim F, Hiremath G, Samayoa JC, Said SM. Complex Pulmonary Artery Rehabilitation in Children with Alagille Syndrome: An Early Single-Center Experience of a Successful Collaborative Work. J Cardiovasc Dev Dis. 2024;11(8):232. doi:10.3390/jcdd11080232
Vandriel SM, Loomes K, Sokal E, et al. Surgical biliary diversion is associated with an increased risk of liver transplantation or death in Alagille syndrome. AASLD Liver Meeting (Boston, USA. 09/11/2023 to 13/11/2023).
Hansen BE, Vandriel SM, Vig P, et al; the Global ALagille Alliance (GALA) Study Group. Event-free survival of maralixibat-treated patients with Alagille syndrome compared to a real-world cohort from GALA. Hepatology. 2024;79(6):1279-1292. doi:10.1097/HEP.0000000000000727
Vandriel SM, Li L-T, She H, et al; the Global ALagille Alliance (GALA) Study Group. Natural history of liver disease in a large international cohort of children with Alagille syndrome: Results from the GALA study. Hepatology. 2023;77(2):512-529. doi:10.1002/hep.32761
Murillo Perez CF, Vandriel SM, Sokal E, et al. Serum bile acids are associated with native liver survival in patients with Alagille syndrome: Results from the GALA Study Group. AASLD Liver Meeting (Boston, USA, 09/11/2023 to 13/11/2023).
Yan J, Huang Y, Cao L, et al. Clinical, pathological and genetic characteristics of 17 unrelated children with Alagille Syndrome. BMC Pediatr. 2024;24(1):532. doi:10.1186/s12887-024-04973-y
Cheng K, Rosenthal P. Diagnosis and management of Alagille and progressive familial intrahepatic cholestasis. Hepatol Commun. 2023 Dec 7;7(12):e0314. doi:10.1097/HC9.0000000000000314
Global Allagile Alliance, GALA, website. The GALA Study. Published 2022. Accessed January 27, 2025. https://www.galastudy.com/
Karim F, Hiremath G, Samayoa JC, Said SM. Complex Pulmonary Artery Rehabilitation in Children with Alagille Syndrome: An Early Single-Center Experience of a Successful Collaborative Work. J Cardiovasc Dev Dis. 2024;11(8):232. doi:10.3390/jcdd11080232
Vandriel SM, Loomes K, Sokal E, et al. Surgical biliary diversion is associated with an increased risk of liver transplantation or death in Alagille syndrome. AASLD Liver Meeting (Boston, USA. 09/11/2023 to 13/11/2023).
Hansen BE, Vandriel SM, Vig P, et al; the Global ALagille Alliance (GALA) Study Group. Event-free survival of maralixibat-treated patients with Alagille syndrome compared to a real-world cohort from GALA. Hepatology. 2024;79(6):1279-1292. doi:10.1097/HEP.0000000000000727
Vandriel SM, Li L-T, She H, et al; the Global ALagille Alliance (GALA) Study Group. Natural history of liver disease in a large international cohort of children with Alagille syndrome: Results from the GALA study. Hepatology. 2023;77(2):512-529. doi:10.1002/hep.32761
Murillo Perez CF, Vandriel SM, Sokal E, et al. Serum bile acids are associated with native liver survival in patients with Alagille syndrome: Results from the GALA Study Group. AASLD Liver Meeting (Boston, USA, 09/11/2023 to 13/11/2023).
Yan J, Huang Y, Cao L, et al. Clinical, pathological and genetic characteristics of 17 unrelated children with Alagille Syndrome. BMC Pediatr. 2024;24(1):532. doi:10.1186/s12887-024-04973-y
Cheng K, Rosenthal P. Diagnosis and management of Alagille and progressive familial intrahepatic cholestasis. Hepatol Commun. 2023 Dec 7;7(12):e0314. doi:10.1097/HC9.0000000000000314
Global Allagile Alliance, GALA, website. The GALA Study. Published 2022. Accessed January 27, 2025. https://www.galastudy.com/
Karim F, Hiremath G, Samayoa JC, Said SM. Complex Pulmonary Artery Rehabilitation in Children with Alagille Syndrome: An Early Single-Center Experience of a Successful Collaborative Work. J Cardiovasc Dev Dis. 2024;11(8):232. doi:10.3390/jcdd11080232
Vandriel SM, Loomes K, Sokal E, et al. Surgical biliary diversion is associated with an increased risk of liver transplantation or death in Alagille syndrome. AASLD Liver Meeting (Boston, USA. 09/11/2023 to 13/11/2023).
Hansen BE, Vandriel SM, Vig P, et al; the Global ALagille Alliance (GALA) Study Group. Event-free survival of maralixibat-treated patients with Alagille syndrome compared to a real-world cohort from GALA. Hepatology. 2024;79(6):1279-1292. doi:10.1097/HEP.0000000000000727
Vandriel SM, Li L-T, She H, et al; the Global ALagille Alliance (GALA) Study Group. Natural history of liver disease in a large international cohort of children with Alagille syndrome: Results from the GALA study. Hepatology. 2023;77(2):512-529. doi:10.1002/hep.32761
Murillo Perez CF, Vandriel SM, Sokal E, et al. Serum bile acids are associated with native liver survival in patients with Alagille syndrome: Results from the GALA Study Group. AASLD Liver Meeting (Boston, USA, 09/11/2023 to 13/11/2023).
Alagille Syndrome: Epidemiology and Management of a Rare Genetic Disease
Alagille Syndrome: Epidemiology and Management of a Rare Genetic Disease

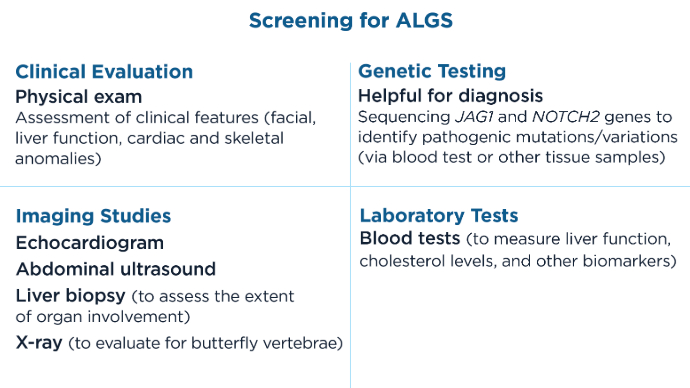


Gastroenterology Data Trends 2025
Gastroenterology Data Trends 2025

GI & Hepatology News and the American Gastroenterological Association (AGA) present Gastroenterology Data Trends 2025, a special report on hot topics in GI told through original infographics and visual storytelling.
In this issue:
The Role of Bedside Intestinal Ultrasound in IBD Management
Bincy Abraham, MD, MS
Obesity Management in the Era of GLP-1: The Role of GLP-1 RAs
Michael Camilleri, MD, MPhil, DSc
Ergonomics in Endoscopy
Amandeep K. Shergill, MD, MS
Optimizing the Delivery of GI Care in Transgender and Gender-Diverse Communities
Kira Newman, MD, PhD
New Therapeutic Frontiers in the Treatment of Eosinophilic Esophagitis
Evan S. Dellon, MD, MPH
New and Emerging Treatments for MASLD/MASH
Naim Alkhouri, MD
Advances in Screening for Barrett’s Esophagus and Esophageal Adenocarcinoma
Joel Rubenstein, MD, MS
Alagille Syndrome: Epidemiology and Management of a Rare Genetic Disease
Alisha Mavis, MD
IBS: Mental Health Factors and Comorbidities
Lin Chang, MD, and Laurie A. Keefer, PhD
GI & Hepatology News and the American Gastroenterological Association (AGA) present Gastroenterology Data Trends 2025, a special report on hot topics in GI told through original infographics and visual storytelling.
In this issue:
The Role of Bedside Intestinal Ultrasound in IBD Management
Bincy Abraham, MD, MS
Obesity Management in the Era of GLP-1: The Role of GLP-1 RAs
Michael Camilleri, MD, MPhil, DSc
Ergonomics in Endoscopy
Amandeep K. Shergill, MD, MS
Optimizing the Delivery of GI Care in Transgender and Gender-Diverse Communities
Kira Newman, MD, PhD
New Therapeutic Frontiers in the Treatment of Eosinophilic Esophagitis
Evan S. Dellon, MD, MPH
New and Emerging Treatments for MASLD/MASH
Naim Alkhouri, MD
Advances in Screening for Barrett’s Esophagus and Esophageal Adenocarcinoma
Joel Rubenstein, MD, MS
Alagille Syndrome: Epidemiology and Management of a Rare Genetic Disease
Alisha Mavis, MD
IBS: Mental Health Factors and Comorbidities
Lin Chang, MD, and Laurie A. Keefer, PhD
GI & Hepatology News and the American Gastroenterological Association (AGA) present Gastroenterology Data Trends 2025, a special report on hot topics in GI told through original infographics and visual storytelling.
In this issue:
The Role of Bedside Intestinal Ultrasound in IBD Management
Bincy Abraham, MD, MS
Obesity Management in the Era of GLP-1: The Role of GLP-1 RAs
Michael Camilleri, MD, MPhil, DSc
Ergonomics in Endoscopy
Amandeep K. Shergill, MD, MS
Optimizing the Delivery of GI Care in Transgender and Gender-Diverse Communities
Kira Newman, MD, PhD
New Therapeutic Frontiers in the Treatment of Eosinophilic Esophagitis
Evan S. Dellon, MD, MPH
New and Emerging Treatments for MASLD/MASH
Naim Alkhouri, MD
Advances in Screening for Barrett’s Esophagus and Esophageal Adenocarcinoma
Joel Rubenstein, MD, MS
Alagille Syndrome: Epidemiology and Management of a Rare Genetic Disease
Alisha Mavis, MD
IBS: Mental Health Factors and Comorbidities
Lin Chang, MD, and Laurie A. Keefer, PhD
Gastroenterology Data Trends 2025
Gastroenterology Data Trends 2025
Ergonomics in Endoscopy
Ergonomics in Endoscopy
Click to view more from Gastroenterology Data Trends 2025.
- Ridtitid W, Cote GA, Leung W, et al. Prevalence and risk factors for musculoskeletal injuries related to endoscopy. Gastrointest Endosc. 2015;81(2):294-302 e294.
- Mohan N, Singla M, Pawa S, et al. Gastroenterologists’ goals for ergonomic colonoscopes: results of a national survey. Gastrointest Endosc. 2025;s0016-5107(25)00051-3. doi:10.1016/j.gie.2025.01.027
- Pawa S, Kwon RS, Fishman DS, et al. American Society for Gastrointestinal Endoscopy guideline on the role of ergonomics for prevention of endoscopy-related injury: summary and recommendations. Gastrointest Endosc. 2023;98(4):482-491. Accessed: February 1, 2025. https://www.asge.org/docs/default-source/guidelines/asge-guideline-on-the-role-of-ergonomics-summary.pdf
- Austin K, Schoenberger H, Sesto M, Gaumnitz E, Teo Broman A, Saha S. Musculoskeletal injuries are commonly reported among gastroenterology trainees: Results of a national survey. Dig Dis Sci. 2019;64(6):1439-1447.
- Shergill AK, Rempel D, Barr A, et al. Biomechanical risk factors associated with distal upper extremity musculoskeletal disorders in endoscopists performing colonoscopy. Gastrointest Endosc. 2021;93(3):704–711.e3. doi:10.1016/j.gie.2020.11.001
- Lipowska A, Shergill A. Coping with burnout and repetitive injuries -The hazards of endoscopy: Ergonomics guide the way. GI & Hepatology News. September 1, 2023. Accessed: February 1, 2025. https://www.mdedge.com/gihepnews/article/264737/practice-management/coping-burnout-and-repetitive-injuries/page/0/2
- Taking Care of You: Ergonomic Essentials for Your Practice (DV074). American Society for Gastrointestinal Endoscopy. May 2017. Accessed: February 1, 2025. https://learn.asge.org/Listing/Taking-Care-of-You-Ergonomic-Essentials-for-Your-Practice-DV074-231
- Shergill A, Shin E, Woods K, et al. “MYSELF” - A novel and easy-to-implement pre-procedure ergonomic time-out that reduces endoscopists’ risk of musculoskeletal injury. Gastrointest Endosc. 2024;99(6) Supplement AB154. Accessed: February 1, 2025. https://www.giejournal.org/article/S0016-5107(24)01207-0/abstract
Click to view more from Gastroenterology Data Trends 2025.
Click to view more from Gastroenterology Data Trends 2025.
- Ridtitid W, Cote GA, Leung W, et al. Prevalence and risk factors for musculoskeletal injuries related to endoscopy. Gastrointest Endosc. 2015;81(2):294-302 e294.
- Mohan N, Singla M, Pawa S, et al. Gastroenterologists’ goals for ergonomic colonoscopes: results of a national survey. Gastrointest Endosc. 2025;s0016-5107(25)00051-3. doi:10.1016/j.gie.2025.01.027
- Pawa S, Kwon RS, Fishman DS, et al. American Society for Gastrointestinal Endoscopy guideline on the role of ergonomics for prevention of endoscopy-related injury: summary and recommendations. Gastrointest Endosc. 2023;98(4):482-491. Accessed: February 1, 2025. https://www.asge.org/docs/default-source/guidelines/asge-guideline-on-the-role-of-ergonomics-summary.pdf
- Austin K, Schoenberger H, Sesto M, Gaumnitz E, Teo Broman A, Saha S. Musculoskeletal injuries are commonly reported among gastroenterology trainees: Results of a national survey. Dig Dis Sci. 2019;64(6):1439-1447.
- Shergill AK, Rempel D, Barr A, et al. Biomechanical risk factors associated with distal upper extremity musculoskeletal disorders in endoscopists performing colonoscopy. Gastrointest Endosc. 2021;93(3):704–711.e3. doi:10.1016/j.gie.2020.11.001
- Lipowska A, Shergill A. Coping with burnout and repetitive injuries -The hazards of endoscopy: Ergonomics guide the way. GI & Hepatology News. September 1, 2023. Accessed: February 1, 2025. https://www.mdedge.com/gihepnews/article/264737/practice-management/coping-burnout-and-repetitive-injuries/page/0/2
- Taking Care of You: Ergonomic Essentials for Your Practice (DV074). American Society for Gastrointestinal Endoscopy. May 2017. Accessed: February 1, 2025. https://learn.asge.org/Listing/Taking-Care-of-You-Ergonomic-Essentials-for-Your-Practice-DV074-231
- Shergill A, Shin E, Woods K, et al. “MYSELF” - A novel and easy-to-implement pre-procedure ergonomic time-out that reduces endoscopists’ risk of musculoskeletal injury. Gastrointest Endosc. 2024;99(6) Supplement AB154. Accessed: February 1, 2025. https://www.giejournal.org/article/S0016-5107(24)01207-0/abstract
- Ridtitid W, Cote GA, Leung W, et al. Prevalence and risk factors for musculoskeletal injuries related to endoscopy. Gastrointest Endosc. 2015;81(2):294-302 e294.
- Mohan N, Singla M, Pawa S, et al. Gastroenterologists’ goals for ergonomic colonoscopes: results of a national survey. Gastrointest Endosc. 2025;s0016-5107(25)00051-3. doi:10.1016/j.gie.2025.01.027
- Pawa S, Kwon RS, Fishman DS, et al. American Society for Gastrointestinal Endoscopy guideline on the role of ergonomics for prevention of endoscopy-related injury: summary and recommendations. Gastrointest Endosc. 2023;98(4):482-491. Accessed: February 1, 2025. https://www.asge.org/docs/default-source/guidelines/asge-guideline-on-the-role-of-ergonomics-summary.pdf
- Austin K, Schoenberger H, Sesto M, Gaumnitz E, Teo Broman A, Saha S. Musculoskeletal injuries are commonly reported among gastroenterology trainees: Results of a national survey. Dig Dis Sci. 2019;64(6):1439-1447.
- Shergill AK, Rempel D, Barr A, et al. Biomechanical risk factors associated with distal upper extremity musculoskeletal disorders in endoscopists performing colonoscopy. Gastrointest Endosc. 2021;93(3):704–711.e3. doi:10.1016/j.gie.2020.11.001
- Lipowska A, Shergill A. Coping with burnout and repetitive injuries -The hazards of endoscopy: Ergonomics guide the way. GI & Hepatology News. September 1, 2023. Accessed: February 1, 2025. https://www.mdedge.com/gihepnews/article/264737/practice-management/coping-burnout-and-repetitive-injuries/page/0/2
- Taking Care of You: Ergonomic Essentials for Your Practice (DV074). American Society for Gastrointestinal Endoscopy. May 2017. Accessed: February 1, 2025. https://learn.asge.org/Listing/Taking-Care-of-You-Ergonomic-Essentials-for-Your-Practice-DV074-231
- Shergill A, Shin E, Woods K, et al. “MYSELF” - A novel and easy-to-implement pre-procedure ergonomic time-out that reduces endoscopists’ risk of musculoskeletal injury. Gastrointest Endosc. 2024;99(6) Supplement AB154. Accessed: February 1, 2025. https://www.giejournal.org/article/S0016-5107(24)01207-0/abstract
Ergonomics in Endoscopy
Ergonomics in Endoscopy
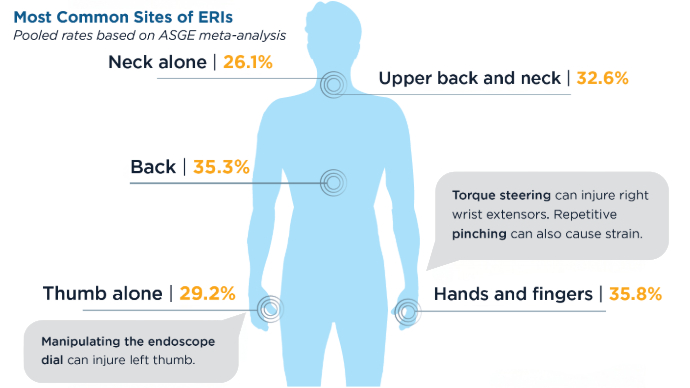







The Rise of Antifungal-Resistant Dermatophyte Infections: What Dermatologists Need to Know
The Rise of Antifungal-Resistant Dermatophyte Infections: What Dermatologists Need to Know
Worldwide, it is estimated that up to 1 in 5 individuals will experience a dermatophyte infection (commonly called ringworm or tinea infection) in their lifetime.1 Historically, dermatophyte infections have been considered relatively minor conditions usually treated with short courses of topical antifungals.2 Oral antifungals historically were needed only for patients with nail or hair shaft infections or extensive cutaneous fungal infections, which typically occurred in immunosuppressed patients.2 However, the landscape is changing rapidly due to the global emergence of severe dermatophyte infections that frequently are resistant to first-line antifungal medications.3-5 In this article, we aimed to review the epidemiology of emerging dermatophyte infections and provide dermatologists with information needed for effective diagnosis and management.
Emergence of Trichophyton indotineae
In recent decades, public health officials and dermatologists have noted with concern the spread of the recently emerged dermatophyte species Trichophyton indotineae in South Asia.3,6 This species (previously known as Trichophyton mentagrophytes genotype VIII) usually is transmitted from person to person, either through direct skin-to-skin contact or by fomites.4,6 Potential sexual transmission of T indotineae infections also has been reported,7 and it is possible that animals may serve as reservoirs for this pathogen, although there are no known reports of direct spread from animals to humans.8,9 Major outbreaks of T indotineae are ongoing in South Asia, and cases have been documented in 6 continents.10-12 In the United States, most but not all cases have occurred in immigrants from or recently returned travelers to South Asia.6,13 The emergence and spread of T indotineae is hypothesized to be promoted by the misuse and overuse of topical antifungal products, particularly those containing combinations of potent corticosteroids with other antimicrobial drugs.14,15
Cutaneous manifestations of T indotineae infections tend to cover large body surface areas, recur frequently, and pose substantial treatment challenges.6,13,16 Several clinical presentations have been documented, including erythematous, scaly concentric plaques; papulosquamous lesions; pustular forms; and corticosteroid-modified disease (Figure 1).6,16 Affected patients seldom are immunocompromised and often have a history of multiple failed courses of topical or oral antifungals, including oral terbinafine.13 Many also have been prescribed topical corticosteroids or have used over-the-counter topical corticosteroids, which worsen the rash.17
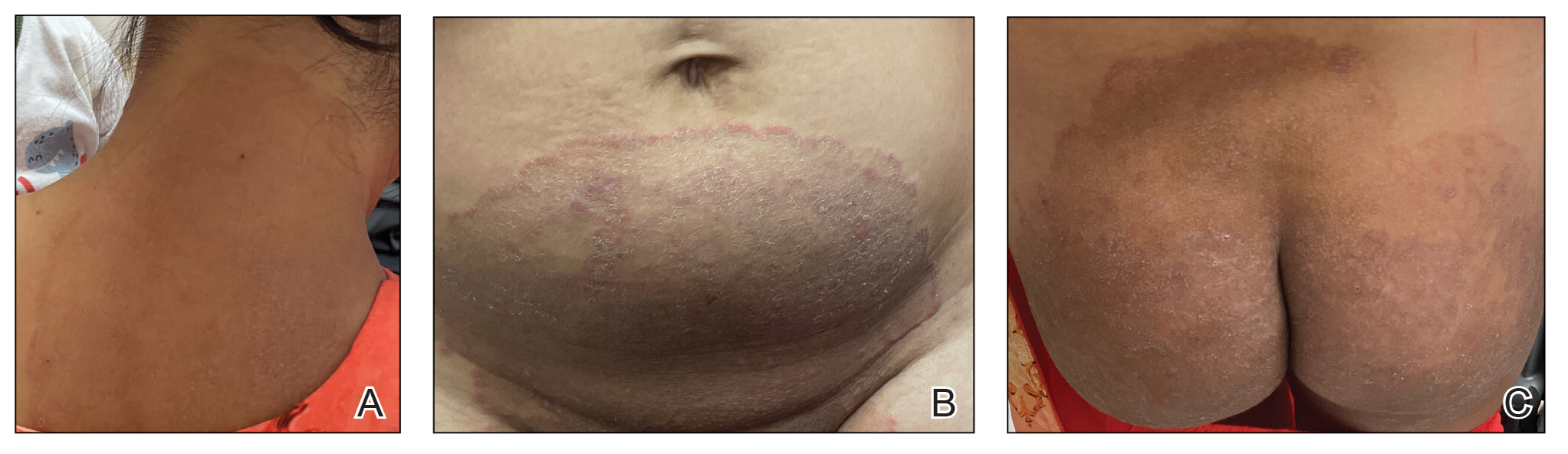
Direct microscopy with potassium hydroxide could be used to confirm the diagnosis of dermatophyte infection, but it does not distinguish T indotineae from other dermatophyte species.2,6 Importantly, culture-based testing usually will misidentify T indotineae as other Trichophyton species such as the more common T mentagrophytes or Trichophyton interdigitale. Definitive identification of T indotineae requires advanced molecular techniques that are available only at select laboratories.6 Unfortunately, availability of such testing is limited (Table), and results may take several weeks; therefore, it is suggested that dermatologists who suspect T indotineae infections based on the patient’s history and clinical presentation begin antifungal treatment after confirmation of dermatophyte infection but not wait for definitive confirmation of the causative organism.16
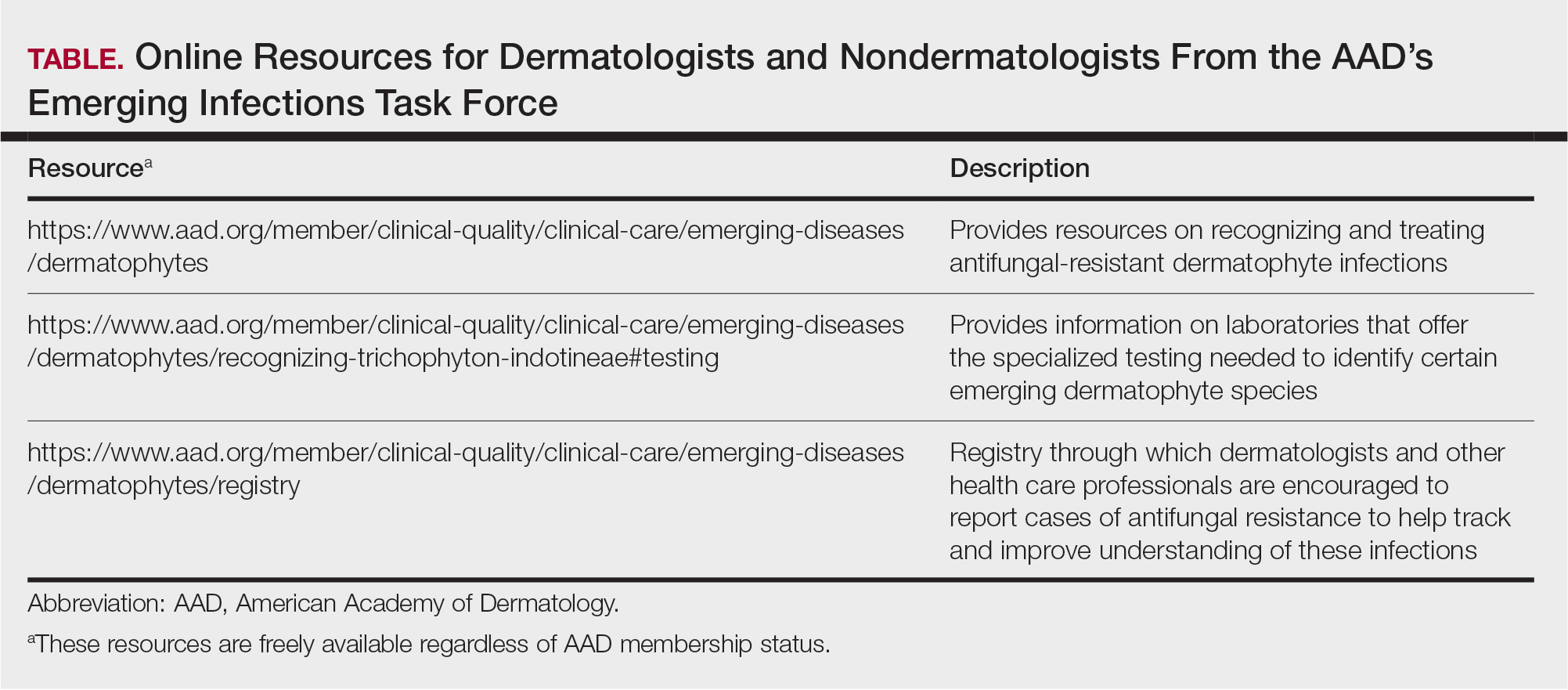
Itraconazole is considered the first-line therapy for T indotineae infection, as terbinafine usually is ineffective due to mutations in the squalene epoxidase gene.16 Dermatologists should be aware that itraconazole is available in different formulations that can affect absorption. The oral solution has greater bioavailability and should be taken on an empty stomach, whereas the capsules are required to be taken with food for effective absorption; the capsules also should be taken with an acidic beverage such as orange juice. Dermatologists should carefully assess for drug-drug interactions when prescribing itraconazole, given its extensive interaction profile with numerous other medications. Patients may require treatment with itraconazole (100 mg/d or 200 mg/d) for a minimum of 6 to 8 weeks until complete clearance has been achieved and ideally a negative potassium hydroxide preparation of skin scrapings has been obtained. A longer treatment period (eg, ≥3 months) frequently is needed, and relapses are common.6,16,18 Regular follow-up is needed to monitor for infection clearance and recurrences. It is important to note that cases of itraconazole resistance have been reported, although this currently appears to be uncommon.19,20
Other Emerging Dermatophytes to Watch
Trichophyton rubrum is the most common cause of dermatophyte infections among humans,21 and cases of terbinafine-resistant T rubrum infections have been reported increasingly in the United States and Canada.5,22-24 Onychomycosis caused by terbinafine-resistant T rubrum has been documented, and patients may have infections that do not respond to terbinafine given at the standard dose and duration.22,23 Case reports have indicated successful treatment using itraconazole 200 mg/d and posaconazole 300 mg/d.5,23
Trichophyton mentagrophytes genotype VII (TMVII) is an emerging dermatophyte that recently has been reported as a cause of sexually transmitted dermatophyte infections in Europe and the United States primarily affecting men who have sex with men.25-27 Patients may present with pruritic, annular, scaly patches and plaques involving the trunk, groin, genital region, or face (Figure 2). Although closely related to T indotineae, TMVII differs in that it more often affects the genital region, generally is susceptible to terbinafine, and in the United States and Europe usually is not related to travel or immigration involving South Asia.26 Although TMVII has not been associated with antifungal resistance, awareness among dermatologists is important because patients may experience inflamed, painful, and persistent rashes that can lead to secondary bacterial infection or scarring, and physicians might mistake it for mimics including eczema or psoriasis.25,26
Importance of Judicious Antifungal Use
Optimizing the use of antifungals is critical to improving patient outcomes and preserving available treatment options.28,29 A retrospective analysis of commercial health insurance data estimated that topical antifungal prescriptions were potentially unnecessary for more than half of the more than 560,000 patients who were prescribed these medications in 2023. In this study, it also was observed that only 16% of patients prescribed a topical antifungal had received diagnostic testing, with low rates across specialties.30 This is concerning because even among board-certified dermatologists, incorrect diagnosis of suspected fungal skin infections can occur; in one survey-based study of board-certified dermatologists who were presented with dermatomycosis images, respondents categorized cases with greater than 75% accuracy in only 31% (4/13) of instances.31 Clotrimazole-betamethasone is among the most commonly prescribed topical antifungals in the United States,14,32 and 2 recent retrospective analyses highlighted that the majority of patients prescribed this medication did not receive any fungal diagnostic testing.33,34
Final Thoughts
In an era of emerging antifungal-resistant dermatophyte infections, it is important for dermatologists to educate nondermatologists about the importance of using diagnostic testing for suspected dermatophyte infections.14,28 Dermatologists also can educate nondermatologist colleagues on the importance of avoiding the use of topical combination antifungal/corticosteroid medications and referring for dermatologic evaluation when diagnoses are uncertain.33,34 Strategies for education by dermatologists could include giving workshops, creating educational materials, and fostering open communication about optimal treatment practices and referral parameters for suspected dermatophyte infections.
- Noble SL, Forbes RC, Stamm PL. Diagnosis and management of common tinea infections. Am Fam Physician. 1998;58:163-174, 177-168.
- Ely JW, Rosenfeld S, Seabury Stone M. Diagnosis and management of tinea infections. Am Fam Physician. 2014;90:702-710.
- Uhrlaß S, Verma SB, Gräser Y, et al. Trichophyton indotineae—an emerging pathogen causing recalcitrant dermatophytoses in India and worldwide—a multidimensional perspective. J Fungi (Basel). 2022;8:757. doi:10.3390/jof8070757
- Verma SB, Panda S, Nenoff P, et al. The unprecedented epidemic-like scenario of dermatophytosis in India: I. epidemiology, risk factors and clinical features. Indian J Dermatol Venereol Leprol. 2021;87:154-175.
- Chen E, Ghannoum M, Elewski BE. Treatment]resistant tinea corporis, a potential public health issue. Br J Dermatol. 2021;184:164-165.
- Caplan AS. Notes from the field: first reported US cases of tinea caused by Trichophyton indotineae—New York City, December 2021–March 2023. MMWR Morbidity and Mortality Weekly Report. 2023;72:536-537. doi:10.15585/mmwr.mm7219a4
- Spivack S, Gold JA, Lockhart SR, et al. Potential sexual transmission of antifungal-resistant Trichophyton indotineae. Emerg Infect Dis. 2024;30:807.
- Jabet A, Brun S, Normand AC, et al. Extensive dermatophytosis caused by terbinafine-resistant Trichophyton indotineae, France. Emerg Infect Dis. 2022;28:229-233.
- Thakur S, Spruijtenburg B, Abhishek, et al. Whole genome sequence analysis of terbinafine resistant and susceptible Trichophyton isolates from human and animal origin. Mycopathologia. 2025;190:13.
- Lockhart SR, Chowdhary A, Gold JA. The rapid emergence of antifungal-resistant human-pathogenic fungi. Nat Rev Microbiol. 2023;21:818-832.
- Mosam A, Shuping L, Naicker S, et al. A case of antifungal-resistant ringworm infection in KwaZulu-Natal Province, South Africa, caused by Trichophyton indotineae. Public Health Bulletin South Africa. Accessed April 4, 2025. https://www.phbsa.ac.za/wp-content/uploads/2023/12PHBSA-Ringworm-Article-2023.pdf
- Cañete-Gibas CF, Mele J, Patterson HP, et al. Terbinafine-resistant dermatophytes and the presence of Trichophyton indotineae in North America. J Clin Microbiol. 2023;61:E0056223
- Caplan AS, Todd GC, Zhu Y, et al. Clinical course, antifungal susceptibility, and genomic sequencing of Trichophyton indotineae. JAMA Dermatol. 2024;160:701-709. doi:10.1001/jamadermatol.2024.1126
- Benedict K. Topical antifungal prescribing for Medicare Part D beneficiaries—United States, 2021. MMWR Morb Mortal Wkly Rep. 2024;73:1-5.
- Verma SB. Emergence of recalcitrant dermatophytosis in India. Lancet Infect Dis. 2018;18:718-719.
- Khurana A, Sharath S, Sardana K, et al. Clinico-mycological and therapeutic updates on cutaneous dermatophytic infections in the era of Trichophyton indotineae. J Am Acad Dermatol. 2024;91:315-323. doi:10.1016/j.jaad.2024.03.024
- Verma S. Steroid modified tinea. BMJ. 2017;356:j973.
- Khurana A, Agarwal A, Agrawal D, et al. Effect of different itraconazole dosing regimens on cure rates, treatment duration, safety, and relapse rates in adult patients with tinea corporis/cruris: a randomized clinical trial. JAMA Dermatol. 2022;158:1269-1278.
- Burmester A, Hipler UC, Uhrlaß S, et al. Indian Trichophyton mentagrophytes squalene epoxidase erg1 double mutants show high proportion of combined fluconazole and terbinafine resistance. Mycoses. 2020;63:1175-1180.
- Bhuiyan MSI, Verma SB, Illigner GM, et al. Trichophyton mentagrophytes ITS genotype VIII/Trichophyton indotineae infection and antifungal resistance in Bangladesh. J Fungi (Basel). 2024;10:768. doi:10.3390 /jof10110768
- Hay RJ. Chapter 82: superficial mycoses. In: Ryan ET, Hill DR, Solomon T, et al, eds. Hunter’s Tropical Medicine and Emerging Infectious Diseases. 10th ed. Elsevier; 2020:648-652.
- Gupta AK, Cooper EA, Wang T, et al. Detection of squalene epoxidase mutations in United States patients with onychomycosis: implications for management. J Invest Dermatol. 2023;143:2476-2483.E2477.
- Hwang JK, Bakotic WL, Gold JA, et al. Isolation of terbinafine-resistant Trichophyton rubrum from onychomycosis patients who failed treatment at an academic center in New York, United States. J Fungi. 2023;9:710.
- Gu D, Hatch M, Ghannoum M, et al. Treatment-resistant dermatophytosis: a representative case highlighting an emerging public health threat. JAAD Case Rep. 2020;6:1153-1155.
- Jabet A, Dellière S, Seang S, et al. Sexually transmitted Trichophyton mentagrophytes genotype VII infection among men who have sex with men. Emerg Infect Dis. 2023;29:1411-1414.
- Zucker J, Caplan AS, Gunaratne SH, et al. Notes from the field: Trichophyton mentagrophytes genotype VII—New York City, April-July 2024. MMWR Morb Mortal Wkly Rep. 2024;73:985-988.
- Jabet A, Bérot V, Chiarabini T, et al. Trichophyton mentagrophytes ITS genotype VII infections among men who have sex with men in France: an ongoing phenomenon. J Eur Acad Dermatol Venereol. 2025;39:407-415.
- Caplan AS, Gold JA, Smith DJ, et al. Improving antifungal stewardship in dermatology in an era of emerging dermatophyte resistance. JAAD International. 2024;15:168-169.
- Elewski B. A call for antifungal stewardship. Br J Dermatol. 2020; 183:798-799.
- Gold JAW, Benedict K, Caplan AS, et al. High rates of potentially unnecessary topical antifungal prescribing in a large commercial health insurance claims database, United States. J Am Acad Dermatol. 2025:S0190-9622(25)00098-2. doi:10.1016/j.jaad.2025.01.022
- Yadgar RJ, Bhatia N, Friedman A. Cutaneous fungal infections are commonly misdiagnosed: a survey-based study. J Am Acad Dermatol. 2017;76:562-563.
- Flint ND, Rhoads JLW, Carlisle R, et al. The continued inappropriate use and overuse of combination topical clotrimazole-betamethasone. Dermatol Online J. 2021;27. doi:10.5070/D327854686
- Currie DW, Caplan AS, Benedict K, et al. Prescribing of clotrimazolebetamethasone dipropionate, a topical combination corticosteroidantifungal product, for Medicare part D beneficiaries, United States, 2016–2022. Antimicrob Steward Healthc Epidemiol. 2024;4:E174.
- Gold JA, Caplan AS, Benedict K, et al. Clotrimazole-betamethasone dipropionate prescribing for nonfungal skin conditions. JAMA Network Open. 2024;7:E2411721-E2411721.
Worldwide, it is estimated that up to 1 in 5 individuals will experience a dermatophyte infection (commonly called ringworm or tinea infection) in their lifetime.1 Historically, dermatophyte infections have been considered relatively minor conditions usually treated with short courses of topical antifungals.2 Oral antifungals historically were needed only for patients with nail or hair shaft infections or extensive cutaneous fungal infections, which typically occurred in immunosuppressed patients.2 However, the landscape is changing rapidly due to the global emergence of severe dermatophyte infections that frequently are resistant to first-line antifungal medications.3-5 In this article, we aimed to review the epidemiology of emerging dermatophyte infections and provide dermatologists with information needed for effective diagnosis and management.
Emergence of Trichophyton indotineae
In recent decades, public health officials and dermatologists have noted with concern the spread of the recently emerged dermatophyte species Trichophyton indotineae in South Asia.3,6 This species (previously known as Trichophyton mentagrophytes genotype VIII) usually is transmitted from person to person, either through direct skin-to-skin contact or by fomites.4,6 Potential sexual transmission of T indotineae infections also has been reported,7 and it is possible that animals may serve as reservoirs for this pathogen, although there are no known reports of direct spread from animals to humans.8,9 Major outbreaks of T indotineae are ongoing in South Asia, and cases have been documented in 6 continents.10-12 In the United States, most but not all cases have occurred in immigrants from or recently returned travelers to South Asia.6,13 The emergence and spread of T indotineae is hypothesized to be promoted by the misuse and overuse of topical antifungal products, particularly those containing combinations of potent corticosteroids with other antimicrobial drugs.14,15
Cutaneous manifestations of T indotineae infections tend to cover large body surface areas, recur frequently, and pose substantial treatment challenges.6,13,16 Several clinical presentations have been documented, including erythematous, scaly concentric plaques; papulosquamous lesions; pustular forms; and corticosteroid-modified disease (Figure 1).6,16 Affected patients seldom are immunocompromised and often have a history of multiple failed courses of topical or oral antifungals, including oral terbinafine.13 Many also have been prescribed topical corticosteroids or have used over-the-counter topical corticosteroids, which worsen the rash.17
Direct microscopy with potassium hydroxide could be used to confirm the diagnosis of dermatophyte infection, but it does not distinguish T indotineae from other dermatophyte species.2,6 Importantly, culture-based testing usually will misidentify T indotineae as other Trichophyton species such as the more common T mentagrophytes or Trichophyton interdigitale. Definitive identification of T indotineae requires advanced molecular techniques that are available only at select laboratories.6 Unfortunately, availability of such testing is limited (Table), and results may take several weeks; therefore, it is suggested that dermatologists who suspect T indotineae infections based on the patient’s history and clinical presentation begin antifungal treatment after confirmation of dermatophyte infection but not wait for definitive confirmation of the causative organism.16
Itraconazole is considered the first-line therapy for T indotineae infection, as terbinafine usually is ineffective due to mutations in the squalene epoxidase gene.16 Dermatologists should be aware that itraconazole is available in different formulations that can affect absorption. The oral solution has greater bioavailability and should be taken on an empty stomach, whereas the capsules are required to be taken with food for effective absorption; the capsules also should be taken with an acidic beverage such as orange juice. Dermatologists should carefully assess for drug-drug interactions when prescribing itraconazole, given its extensive interaction profile with numerous other medications. Patients may require treatment with itraconazole (100 mg/d or 200 mg/d) for a minimum of 6 to 8 weeks until complete clearance has been achieved and ideally a negative potassium hydroxide preparation of skin scrapings has been obtained. A longer treatment period (eg, ≥3 months) frequently is needed, and relapses are common.6,16,18 Regular follow-up is needed to monitor for infection clearance and recurrences. It is important to note that cases of itraconazole resistance have been reported, although this currently appears to be uncommon.19,20
Other Emerging Dermatophytes to Watch
Trichophyton rubrum is the most common cause of dermatophyte infections among humans,21 and cases of terbinafine-resistant T rubrum infections have been reported increasingly in the United States and Canada.5,22-24 Onychomycosis caused by terbinafine-resistant T rubrum has been documented, and patients may have infections that do not respond to terbinafine given at the standard dose and duration.22,23 Case reports have indicated successful treatment using itraconazole 200 mg/d and posaconazole 300 mg/d.5,23
Trichophyton mentagrophytes genotype VII (TMVII) is an emerging dermatophyte that recently has been reported as a cause of sexually transmitted dermatophyte infections in Europe and the United States primarily affecting men who have sex with men.25-27 Patients may present with pruritic, annular, scaly patches and plaques involving the trunk, groin, genital region, or face (Figure 2). Although closely related to T indotineae, TMVII differs in that it more often affects the genital region, generally is susceptible to terbinafine, and in the United States and Europe usually is not related to travel or immigration involving South Asia.26 Although TMVII has not been associated with antifungal resistance, awareness among dermatologists is important because patients may experience inflamed, painful, and persistent rashes that can lead to secondary bacterial infection or scarring, and physicians might mistake it for mimics including eczema or psoriasis.25,26
Importance of Judicious Antifungal Use
Optimizing the use of antifungals is critical to improving patient outcomes and preserving available treatment options.28,29 A retrospective analysis of commercial health insurance data estimated that topical antifungal prescriptions were potentially unnecessary for more than half of the more than 560,000 patients who were prescribed these medications in 2023. In this study, it also was observed that only 16% of patients prescribed a topical antifungal had received diagnostic testing, with low rates across specialties.30 This is concerning because even among board-certified dermatologists, incorrect diagnosis of suspected fungal skin infections can occur; in one survey-based study of board-certified dermatologists who were presented with dermatomycosis images, respondents categorized cases with greater than 75% accuracy in only 31% (4/13) of instances.31 Clotrimazole-betamethasone is among the most commonly prescribed topical antifungals in the United States,14,32 and 2 recent retrospective analyses highlighted that the majority of patients prescribed this medication did not receive any fungal diagnostic testing.33,34
Final Thoughts
In an era of emerging antifungal-resistant dermatophyte infections, it is important for dermatologists to educate nondermatologists about the importance of using diagnostic testing for suspected dermatophyte infections.14,28 Dermatologists also can educate nondermatologist colleagues on the importance of avoiding the use of topical combination antifungal/corticosteroid medications and referring for dermatologic evaluation when diagnoses are uncertain.33,34 Strategies for education by dermatologists could include giving workshops, creating educational materials, and fostering open communication about optimal treatment practices and referral parameters for suspected dermatophyte infections.
Worldwide, it is estimated that up to 1 in 5 individuals will experience a dermatophyte infection (commonly called ringworm or tinea infection) in their lifetime.1 Historically, dermatophyte infections have been considered relatively minor conditions usually treated with short courses of topical antifungals.2 Oral antifungals historically were needed only for patients with nail or hair shaft infections or extensive cutaneous fungal infections, which typically occurred in immunosuppressed patients.2 However, the landscape is changing rapidly due to the global emergence of severe dermatophyte infections that frequently are resistant to first-line antifungal medications.3-5 In this article, we aimed to review the epidemiology of emerging dermatophyte infections and provide dermatologists with information needed for effective diagnosis and management.
Emergence of Trichophyton indotineae
In recent decades, public health officials and dermatologists have noted with concern the spread of the recently emerged dermatophyte species Trichophyton indotineae in South Asia.3,6 This species (previously known as Trichophyton mentagrophytes genotype VIII) usually is transmitted from person to person, either through direct skin-to-skin contact or by fomites.4,6 Potential sexual transmission of T indotineae infections also has been reported,7 and it is possible that animals may serve as reservoirs for this pathogen, although there are no known reports of direct spread from animals to humans.8,9 Major outbreaks of T indotineae are ongoing in South Asia, and cases have been documented in 6 continents.10-12 In the United States, most but not all cases have occurred in immigrants from or recently returned travelers to South Asia.6,13 The emergence and spread of T indotineae is hypothesized to be promoted by the misuse and overuse of topical antifungal products, particularly those containing combinations of potent corticosteroids with other antimicrobial drugs.14,15
Cutaneous manifestations of T indotineae infections tend to cover large body surface areas, recur frequently, and pose substantial treatment challenges.6,13,16 Several clinical presentations have been documented, including erythematous, scaly concentric plaques; papulosquamous lesions; pustular forms; and corticosteroid-modified disease (Figure 1).6,16 Affected patients seldom are immunocompromised and often have a history of multiple failed courses of topical or oral antifungals, including oral terbinafine.13 Many also have been prescribed topical corticosteroids or have used over-the-counter topical corticosteroids, which worsen the rash.17
Direct microscopy with potassium hydroxide could be used to confirm the diagnosis of dermatophyte infection, but it does not distinguish T indotineae from other dermatophyte species.2,6 Importantly, culture-based testing usually will misidentify T indotineae as other Trichophyton species such as the more common T mentagrophytes or Trichophyton interdigitale. Definitive identification of T indotineae requires advanced molecular techniques that are available only at select laboratories.6 Unfortunately, availability of such testing is limited (Table), and results may take several weeks; therefore, it is suggested that dermatologists who suspect T indotineae infections based on the patient’s history and clinical presentation begin antifungal treatment after confirmation of dermatophyte infection but not wait for definitive confirmation of the causative organism.16
Itraconazole is considered the first-line therapy for T indotineae infection, as terbinafine usually is ineffective due to mutations in the squalene epoxidase gene.16 Dermatologists should be aware that itraconazole is available in different formulations that can affect absorption. The oral solution has greater bioavailability and should be taken on an empty stomach, whereas the capsules are required to be taken with food for effective absorption; the capsules also should be taken with an acidic beverage such as orange juice. Dermatologists should carefully assess for drug-drug interactions when prescribing itraconazole, given its extensive interaction profile with numerous other medications. Patients may require treatment with itraconazole (100 mg/d or 200 mg/d) for a minimum of 6 to 8 weeks until complete clearance has been achieved and ideally a negative potassium hydroxide preparation of skin scrapings has been obtained. A longer treatment period (eg, ≥3 months) frequently is needed, and relapses are common.6,16,18 Regular follow-up is needed to monitor for infection clearance and recurrences. It is important to note that cases of itraconazole resistance have been reported, although this currently appears to be uncommon.19,20
Other Emerging Dermatophytes to Watch
Trichophyton rubrum is the most common cause of dermatophyte infections among humans,21 and cases of terbinafine-resistant T rubrum infections have been reported increasingly in the United States and Canada.5,22-24 Onychomycosis caused by terbinafine-resistant T rubrum has been documented, and patients may have infections that do not respond to terbinafine given at the standard dose and duration.22,23 Case reports have indicated successful treatment using itraconazole 200 mg/d and posaconazole 300 mg/d.5,23
Trichophyton mentagrophytes genotype VII (TMVII) is an emerging dermatophyte that recently has been reported as a cause of sexually transmitted dermatophyte infections in Europe and the United States primarily affecting men who have sex with men.25-27 Patients may present with pruritic, annular, scaly patches and plaques involving the trunk, groin, genital region, or face (Figure 2). Although closely related to T indotineae, TMVII differs in that it more often affects the genital region, generally is susceptible to terbinafine, and in the United States and Europe usually is not related to travel or immigration involving South Asia.26 Although TMVII has not been associated with antifungal resistance, awareness among dermatologists is important because patients may experience inflamed, painful, and persistent rashes that can lead to secondary bacterial infection or scarring, and physicians might mistake it for mimics including eczema or psoriasis.25,26
Importance of Judicious Antifungal Use
Optimizing the use of antifungals is critical to improving patient outcomes and preserving available treatment options.28,29 A retrospective analysis of commercial health insurance data estimated that topical antifungal prescriptions were potentially unnecessary for more than half of the more than 560,000 patients who were prescribed these medications in 2023. In this study, it also was observed that only 16% of patients prescribed a topical antifungal had received diagnostic testing, with low rates across specialties.30 This is concerning because even among board-certified dermatologists, incorrect diagnosis of suspected fungal skin infections can occur; in one survey-based study of board-certified dermatologists who were presented with dermatomycosis images, respondents categorized cases with greater than 75% accuracy in only 31% (4/13) of instances.31 Clotrimazole-betamethasone is among the most commonly prescribed topical antifungals in the United States,14,32 and 2 recent retrospective analyses highlighted that the majority of patients prescribed this medication did not receive any fungal diagnostic testing.33,34
Final Thoughts
In an era of emerging antifungal-resistant dermatophyte infections, it is important for dermatologists to educate nondermatologists about the importance of using diagnostic testing for suspected dermatophyte infections.14,28 Dermatologists also can educate nondermatologist colleagues on the importance of avoiding the use of topical combination antifungal/corticosteroid medications and referring for dermatologic evaluation when diagnoses are uncertain.33,34 Strategies for education by dermatologists could include giving workshops, creating educational materials, and fostering open communication about optimal treatment practices and referral parameters for suspected dermatophyte infections.
- Noble SL, Forbes RC, Stamm PL. Diagnosis and management of common tinea infections. Am Fam Physician. 1998;58:163-174, 177-168.
- Ely JW, Rosenfeld S, Seabury Stone M. Diagnosis and management of tinea infections. Am Fam Physician. 2014;90:702-710.
- Uhrlaß S, Verma SB, Gräser Y, et al. Trichophyton indotineae—an emerging pathogen causing recalcitrant dermatophytoses in India and worldwide—a multidimensional perspective. J Fungi (Basel). 2022;8:757. doi:10.3390/jof8070757
- Verma SB, Panda S, Nenoff P, et al. The unprecedented epidemic-like scenario of dermatophytosis in India: I. epidemiology, risk factors and clinical features. Indian J Dermatol Venereol Leprol. 2021;87:154-175.
- Chen E, Ghannoum M, Elewski BE. Treatment]resistant tinea corporis, a potential public health issue. Br J Dermatol. 2021;184:164-165.
- Caplan AS. Notes from the field: first reported US cases of tinea caused by Trichophyton indotineae—New York City, December 2021–March 2023. MMWR Morbidity and Mortality Weekly Report. 2023;72:536-537. doi:10.15585/mmwr.mm7219a4
- Spivack S, Gold JA, Lockhart SR, et al. Potential sexual transmission of antifungal-resistant Trichophyton indotineae. Emerg Infect Dis. 2024;30:807.
- Jabet A, Brun S, Normand AC, et al. Extensive dermatophytosis caused by terbinafine-resistant Trichophyton indotineae, France. Emerg Infect Dis. 2022;28:229-233.
- Thakur S, Spruijtenburg B, Abhishek, et al. Whole genome sequence analysis of terbinafine resistant and susceptible Trichophyton isolates from human and animal origin. Mycopathologia. 2025;190:13.
- Lockhart SR, Chowdhary A, Gold JA. The rapid emergence of antifungal-resistant human-pathogenic fungi. Nat Rev Microbiol. 2023;21:818-832.
- Mosam A, Shuping L, Naicker S, et al. A case of antifungal-resistant ringworm infection in KwaZulu-Natal Province, South Africa, caused by Trichophyton indotineae. Public Health Bulletin South Africa. Accessed April 4, 2025. https://www.phbsa.ac.za/wp-content/uploads/2023/12PHBSA-Ringworm-Article-2023.pdf
- Cañete-Gibas CF, Mele J, Patterson HP, et al. Terbinafine-resistant dermatophytes and the presence of Trichophyton indotineae in North America. J Clin Microbiol. 2023;61:E0056223
- Caplan AS, Todd GC, Zhu Y, et al. Clinical course, antifungal susceptibility, and genomic sequencing of Trichophyton indotineae. JAMA Dermatol. 2024;160:701-709. doi:10.1001/jamadermatol.2024.1126
- Benedict K. Topical antifungal prescribing for Medicare Part D beneficiaries—United States, 2021. MMWR Morb Mortal Wkly Rep. 2024;73:1-5.
- Verma SB. Emergence of recalcitrant dermatophytosis in India. Lancet Infect Dis. 2018;18:718-719.
- Khurana A, Sharath S, Sardana K, et al. Clinico-mycological and therapeutic updates on cutaneous dermatophytic infections in the era of Trichophyton indotineae. J Am Acad Dermatol. 2024;91:315-323. doi:10.1016/j.jaad.2024.03.024
- Verma S. Steroid modified tinea. BMJ. 2017;356:j973.
- Khurana A, Agarwal A, Agrawal D, et al. Effect of different itraconazole dosing regimens on cure rates, treatment duration, safety, and relapse rates in adult patients with tinea corporis/cruris: a randomized clinical trial. JAMA Dermatol. 2022;158:1269-1278.
- Burmester A, Hipler UC, Uhrlaß S, et al. Indian Trichophyton mentagrophytes squalene epoxidase erg1 double mutants show high proportion of combined fluconazole and terbinafine resistance. Mycoses. 2020;63:1175-1180.
- Bhuiyan MSI, Verma SB, Illigner GM, et al. Trichophyton mentagrophytes ITS genotype VIII/Trichophyton indotineae infection and antifungal resistance in Bangladesh. J Fungi (Basel). 2024;10:768. doi:10.3390 /jof10110768
- Hay RJ. Chapter 82: superficial mycoses. In: Ryan ET, Hill DR, Solomon T, et al, eds. Hunter’s Tropical Medicine and Emerging Infectious Diseases. 10th ed. Elsevier; 2020:648-652.
- Gupta AK, Cooper EA, Wang T, et al. Detection of squalene epoxidase mutations in United States patients with onychomycosis: implications for management. J Invest Dermatol. 2023;143:2476-2483.E2477.
- Hwang JK, Bakotic WL, Gold JA, et al. Isolation of terbinafine-resistant Trichophyton rubrum from onychomycosis patients who failed treatment at an academic center in New York, United States. J Fungi. 2023;9:710.
- Gu D, Hatch M, Ghannoum M, et al. Treatment-resistant dermatophytosis: a representative case highlighting an emerging public health threat. JAAD Case Rep. 2020;6:1153-1155.
- Jabet A, Dellière S, Seang S, et al. Sexually transmitted Trichophyton mentagrophytes genotype VII infection among men who have sex with men. Emerg Infect Dis. 2023;29:1411-1414.
- Zucker J, Caplan AS, Gunaratne SH, et al. Notes from the field: Trichophyton mentagrophytes genotype VII—New York City, April-July 2024. MMWR Morb Mortal Wkly Rep. 2024;73:985-988.
- Jabet A, Bérot V, Chiarabini T, et al. Trichophyton mentagrophytes ITS genotype VII infections among men who have sex with men in France: an ongoing phenomenon. J Eur Acad Dermatol Venereol. 2025;39:407-415.
- Caplan AS, Gold JA, Smith DJ, et al. Improving antifungal stewardship in dermatology in an era of emerging dermatophyte resistance. JAAD International. 2024;15:168-169.
- Elewski B. A call for antifungal stewardship. Br J Dermatol. 2020; 183:798-799.
- Gold JAW, Benedict K, Caplan AS, et al. High rates of potentially unnecessary topical antifungal prescribing in a large commercial health insurance claims database, United States. J Am Acad Dermatol. 2025:S0190-9622(25)00098-2. doi:10.1016/j.jaad.2025.01.022
- Yadgar RJ, Bhatia N, Friedman A. Cutaneous fungal infections are commonly misdiagnosed: a survey-based study. J Am Acad Dermatol. 2017;76:562-563.
- Flint ND, Rhoads JLW, Carlisle R, et al. The continued inappropriate use and overuse of combination topical clotrimazole-betamethasone. Dermatol Online J. 2021;27. doi:10.5070/D327854686
- Currie DW, Caplan AS, Benedict K, et al. Prescribing of clotrimazolebetamethasone dipropionate, a topical combination corticosteroidantifungal product, for Medicare part D beneficiaries, United States, 2016–2022. Antimicrob Steward Healthc Epidemiol. 2024;4:E174.
- Gold JA, Caplan AS, Benedict K, et al. Clotrimazole-betamethasone dipropionate prescribing for nonfungal skin conditions. JAMA Network Open. 2024;7:E2411721-E2411721.
- Noble SL, Forbes RC, Stamm PL. Diagnosis and management of common tinea infections. Am Fam Physician. 1998;58:163-174, 177-168.
- Ely JW, Rosenfeld S, Seabury Stone M. Diagnosis and management of tinea infections. Am Fam Physician. 2014;90:702-710.
- Uhrlaß S, Verma SB, Gräser Y, et al. Trichophyton indotineae—an emerging pathogen causing recalcitrant dermatophytoses in India and worldwide—a multidimensional perspective. J Fungi (Basel). 2022;8:757. doi:10.3390/jof8070757
- Verma SB, Panda S, Nenoff P, et al. The unprecedented epidemic-like scenario of dermatophytosis in India: I. epidemiology, risk factors and clinical features. Indian J Dermatol Venereol Leprol. 2021;87:154-175.
- Chen E, Ghannoum M, Elewski BE. Treatment]resistant tinea corporis, a potential public health issue. Br J Dermatol. 2021;184:164-165.
- Caplan AS. Notes from the field: first reported US cases of tinea caused by Trichophyton indotineae—New York City, December 2021–March 2023. MMWR Morbidity and Mortality Weekly Report. 2023;72:536-537. doi:10.15585/mmwr.mm7219a4
- Spivack S, Gold JA, Lockhart SR, et al. Potential sexual transmission of antifungal-resistant Trichophyton indotineae. Emerg Infect Dis. 2024;30:807.
- Jabet A, Brun S, Normand AC, et al. Extensive dermatophytosis caused by terbinafine-resistant Trichophyton indotineae, France. Emerg Infect Dis. 2022;28:229-233.
- Thakur S, Spruijtenburg B, Abhishek, et al. Whole genome sequence analysis of terbinafine resistant and susceptible Trichophyton isolates from human and animal origin. Mycopathologia. 2025;190:13.
- Lockhart SR, Chowdhary A, Gold JA. The rapid emergence of antifungal-resistant human-pathogenic fungi. Nat Rev Microbiol. 2023;21:818-832.
- Mosam A, Shuping L, Naicker S, et al. A case of antifungal-resistant ringworm infection in KwaZulu-Natal Province, South Africa, caused by Trichophyton indotineae. Public Health Bulletin South Africa. Accessed April 4, 2025. https://www.phbsa.ac.za/wp-content/uploads/2023/12PHBSA-Ringworm-Article-2023.pdf
- Cañete-Gibas CF, Mele J, Patterson HP, et al. Terbinafine-resistant dermatophytes and the presence of Trichophyton indotineae in North America. J Clin Microbiol. 2023;61:E0056223
- Caplan AS, Todd GC, Zhu Y, et al. Clinical course, antifungal susceptibility, and genomic sequencing of Trichophyton indotineae. JAMA Dermatol. 2024;160:701-709. doi:10.1001/jamadermatol.2024.1126
- Benedict K. Topical antifungal prescribing for Medicare Part D beneficiaries—United States, 2021. MMWR Morb Mortal Wkly Rep. 2024;73:1-5.
- Verma SB. Emergence of recalcitrant dermatophytosis in India. Lancet Infect Dis. 2018;18:718-719.
- Khurana A, Sharath S, Sardana K, et al. Clinico-mycological and therapeutic updates on cutaneous dermatophytic infections in the era of Trichophyton indotineae. J Am Acad Dermatol. 2024;91:315-323. doi:10.1016/j.jaad.2024.03.024
- Verma S. Steroid modified tinea. BMJ. 2017;356:j973.
- Khurana A, Agarwal A, Agrawal D, et al. Effect of different itraconazole dosing regimens on cure rates, treatment duration, safety, and relapse rates in adult patients with tinea corporis/cruris: a randomized clinical trial. JAMA Dermatol. 2022;158:1269-1278.
- Burmester A, Hipler UC, Uhrlaß S, et al. Indian Trichophyton mentagrophytes squalene epoxidase erg1 double mutants show high proportion of combined fluconazole and terbinafine resistance. Mycoses. 2020;63:1175-1180.
- Bhuiyan MSI, Verma SB, Illigner GM, et al. Trichophyton mentagrophytes ITS genotype VIII/Trichophyton indotineae infection and antifungal resistance in Bangladesh. J Fungi (Basel). 2024;10:768. doi:10.3390 /jof10110768
- Hay RJ. Chapter 82: superficial mycoses. In: Ryan ET, Hill DR, Solomon T, et al, eds. Hunter’s Tropical Medicine and Emerging Infectious Diseases. 10th ed. Elsevier; 2020:648-652.
- Gupta AK, Cooper EA, Wang T, et al. Detection of squalene epoxidase mutations in United States patients with onychomycosis: implications for management. J Invest Dermatol. 2023;143:2476-2483.E2477.
- Hwang JK, Bakotic WL, Gold JA, et al. Isolation of terbinafine-resistant Trichophyton rubrum from onychomycosis patients who failed treatment at an academic center in New York, United States. J Fungi. 2023;9:710.
- Gu D, Hatch M, Ghannoum M, et al. Treatment-resistant dermatophytosis: a representative case highlighting an emerging public health threat. JAAD Case Rep. 2020;6:1153-1155.
- Jabet A, Dellière S, Seang S, et al. Sexually transmitted Trichophyton mentagrophytes genotype VII infection among men who have sex with men. Emerg Infect Dis. 2023;29:1411-1414.
- Zucker J, Caplan AS, Gunaratne SH, et al. Notes from the field: Trichophyton mentagrophytes genotype VII—New York City, April-July 2024. MMWR Morb Mortal Wkly Rep. 2024;73:985-988.
- Jabet A, Bérot V, Chiarabini T, et al. Trichophyton mentagrophytes ITS genotype VII infections among men who have sex with men in France: an ongoing phenomenon. J Eur Acad Dermatol Venereol. 2025;39:407-415.
- Caplan AS, Gold JA, Smith DJ, et al. Improving antifungal stewardship in dermatology in an era of emerging dermatophyte resistance. JAAD International. 2024;15:168-169.
- Elewski B. A call for antifungal stewardship. Br J Dermatol. 2020; 183:798-799.
- Gold JAW, Benedict K, Caplan AS, et al. High rates of potentially unnecessary topical antifungal prescribing in a large commercial health insurance claims database, United States. J Am Acad Dermatol. 2025:S0190-9622(25)00098-2. doi:10.1016/j.jaad.2025.01.022
- Yadgar RJ, Bhatia N, Friedman A. Cutaneous fungal infections are commonly misdiagnosed: a survey-based study. J Am Acad Dermatol. 2017;76:562-563.
- Flint ND, Rhoads JLW, Carlisle R, et al. The continued inappropriate use and overuse of combination topical clotrimazole-betamethasone. Dermatol Online J. 2021;27. doi:10.5070/D327854686
- Currie DW, Caplan AS, Benedict K, et al. Prescribing of clotrimazolebetamethasone dipropionate, a topical combination corticosteroidantifungal product, for Medicare part D beneficiaries, United States, 2016–2022. Antimicrob Steward Healthc Epidemiol. 2024;4:E174.
- Gold JA, Caplan AS, Benedict K, et al. Clotrimazole-betamethasone dipropionate prescribing for nonfungal skin conditions. JAMA Network Open. 2024;7:E2411721-E2411721.
The Rise of Antifungal-Resistant Dermatophyte Infections: What Dermatologists Need to Know
The Rise of Antifungal-Resistant Dermatophyte Infections: What Dermatologists Need to Know
PRACTICE POINTS
- Recently emerged dermatophyte species pose a global public health concern because of infection severity, frequent resistance to terbinafine, and easy person-to-person transmission.
- Prolonged itraconazole therapy is considered the firstline treatment for infections caused by Trichophyton indotineae, a globally emerging and frequently terbinafine-resistant dermatophyte.
- Dermatologists can educate nondermatologists on the importance of mycologic confirmation and avoidance of the use of topical antifungal/ corticosteroid products, which are hypothesized to contribute to emergence and spread of resistance.
