User login
Myopathy for the general internist: Statins and much more
Myopathies can present with a wide variety of symptoms, so patients with muscle weakness are often seen initially by a general practitioner. Nonrheumatologists should be able to evaluate a patient presenting with muscle weakness or myalgia and be aware of red flags indicating potentially dangerous syndromes that require a prompt, thorough investigation.
This article reviews selected causes of muscle weakness, such as statin-induced and autoimmune disorders, and systemic features of inflammatory myopathies beyond myositis, such as dermatologic and pulmonary manifestations.
FOCUSING THE EVALUATION
The evaluation of a patient presenting with muscle weakness should include several assessments:
Temporal progression. Was the onset of symptoms rapid or insidious? Patterns of onset may give clues to etiology, including the possibility of an associated autoimmune condition.
Location of muscle weakness. Are symptoms global or localized? And if localized, are they proximal or distal? Proximal weakness can be manifested by difficulty rising from a chair (hip muscles) or combing one’s hair (shoulder muscles), whereas distal weakness can involve difficulty standing on toes (gastrocnemius and soleus muscles) or performing fine motor activities (intrinsic hand muscles).
Symmetry. A focal or asymmetric pattern often has a neurologic etiology, but this could also be consistent with inclusion body myositis.
Other symptoms. Arthritis, rash, and swallowing problems point to a possible underlying rheumatologic disease. Weight gain or loss may indicate a thyroid disorder.
Family history. Some patients report that others in their family have this pattern of weakness, indicating a likely genetic myopathy. If the patient reports a relative with multiple sclerosis, lupus erythematosus, rheumatoid arthritis, or another autoimmune disease, then an immune-mediated myopathy should be considered.
Medications should be reviewed, particularly statins.
CASE 1: SLOWLY PROGRESSIVE WEAKNESS
A 65-year-old man presented with the insidious onset of muscle weakness and episodes of falling. On review of his medical record, his serum creatine kinase (CK) levels were elevated at various periods at 2 to 4 times the upper limit of normal. Electromyography (EMG) previously showed a myopathic pattern, and a muscle biopsy was abnormal, consistent with endomysial inflammation (term is consistent with “polymyositis”). He was treated for polymyositis for several years with prednisone alone, with steroids plus methotrexate, and with combined immunosuppression including methotrexate and azathioprine, but with no improvement. Eventually, another muscle biopsy revealed inclusion bodies with rimmed vacuoles, consistent with inclusion body myositis.
Inclusion body myositis
Inclusion body myositis is the most common myopathy in middle-aged to elderly people, especially men. These patients are often told “You are just getting old,” but they have a defined condition. It should also be considered in patients failing to respond to treatment or with those with “refractory” polymyositis.
The onset of muscle weakness is insidious and painless, and the weakness progresses slowly. The pattern is distal and asymmetric (eg, foot drop), and muscle atrophy typically affects the forearm flexors, quadriceps, and intrinsic muscles of the hands.1
Magnetic resonance imaging may show marked muscle atrophy. Unfortunately, no treatment has shown efficacy, and most neuromuscular and rheumatology experts do not treat inclusion body myositis with immunosuppressive drugs.
CASE 2: MILD MYALGIA WITHOUT WEAKNESS
A black 52-year-old man was referred because of myalgia and a CK level of 862 U/L (reference range < 200). His physician wanted to start him on a statin but was hesitant to do so without first consulting a rheumatologist.
The patient had a long history of mild arthralgias and myalgias without muscle weakness. He had dyslipidemia and hypertension. He reported no family history of myopathy and no illicit drug use. He was formerly an athlete. Medications included a thiazide diuretic and a beta-blocker. On examination, his muscles were strong (rated 5 on a scale of 5) in the upper and lower extremities, without atrophy.
His records showed that his CK levels had risen and fallen repeatedly over the past few years, ranging from 600 to 1,100 U/L. On further questioning, he reported that when he had joined the army 30 years previously, a physician had recommended he undergo a liver biopsy in view of elevated liver function tests, but that he had refused because he felt fine.
Currently, his gamma-glutamyl transpeptidase levels were normal.
Idiopathic ‘hyperCKemia’
So-called idiopathic hyperCKemia is not a form of myositis but merely a laboratory result outside the “normal” range. Reference ranges are based predominantly on measurements in white people and on an assumption that the distribution is Gaussian (bell-shaped). A normal CK level is usually defined as less than 200 U/L. Using this standard, up to 20% of men and 5% of women have hyperCKemia.2
However, CK levels vary by sex and ethnicity, with mean levels highest in black men, followed by black women, white men, and white women. The mean level in black men is higher than the standard cutoff point for normal, and especially in this population, there is wide fluctuation around the mean, leading to hyperCKemia quite frequently in black men. Exercise and manual labor also drive up CK levels.3–5
Idiopathic hyperCKemia is benign. D’Adda et al6 followed 55 patients for a mean of 7.5 years. CK levels normalized in 12 patients or at least decreased in 24. Most remained symptom-free or had minimal symptoms.
Idiopathic hyperCKemia: Bottom line
Before prescribing a statin, determine the baseline CK level. If slightly elevated (ie, up to 3 to 5 times the upper limit of normal, or even higher) in the setting of normal muscle strength, there is no need for electromyography or muscle biopsy, and the patient can certainly receive a statin. Most of these patients do not need to see a rheumatologist but can simply have their CK and muscle strength monitored.
CLASSIFYING MYOSITIS
Myositis (idiopathic inflammatory myopathy) is a heterogeneous group of autoimmune syndromes of unknown cause characterized by chronic muscle weakness and inflammation of striated muscle. These syndromes likely arise as a result of genetic predisposition and an environmental or infectious “hit.”
Myositis is rare, with an incidence of 5 to 10 cases per million per year and an estimated prevalence of 50 to 90 cases per million. It has 2 incidence peaks: 1 in childhood (age 5–15) and another in adult midlife (age 30–50). Women are affected 2 to 3 times more often than men, with black women most commonly affected.
Myositis is traditionally classified as follows:
- Adult polymyositis
- Adult dermatomyositis
- Juvenile myositis (dermatomyositis much more frequent than polymyositis)
- Malignancy-associated myositis (usually dermatomyositis)
- Myositis overlapping with another autoimmune disease
- Inclusion body myositis.
However, polymyositis is less common than we originally thought, and the term necrotizing myopathy is now used in many patients, as noted in the case studies below. Further, myositis overlap syndromes are being increasingly diagnosed, likely related to the emergence of autoantibodies and clinical “syndromes” associated with these autoantibody subsets (discussed in cases below).
Dermatomyositis
Dermatomyositis is characterized by muscle weakness and a rash that can be obvious or subtle. Classic skin lesions are Gottron papules, which are raised, flat-topped red or purplish lesions over the knuckles, elbows, or knees.
Lesions may be confused with those of psoriasis. There can also be a V-neck rash over the anterior chest or upper back (“shawl sign”) or a rash over the lateral thigh (“holster sign”). A facial rash may occur, but unlike lupus, dermatomyositis does not spare the nasolabial area. However, the V-neck rash can be similar to that seen in lupus.
Dermatomyositis may cause muscle pain, perhaps related to muscle ischemia, whereas polymyositis and necrotizing myopathy are often painless. However, pain is also associated with fibromyalgia, which may be seen in many autoimmune conditions. It is important not to overtreat rheumatologic diseases with immunosuppression to try to control pain if the pain is actually caused by fibromyalgia.
Polymyositis mimics

Hypothyroid myopathy can present as classic polymyositis. The serum CK may be elevated, and there may be myalgias, muscle hypertrophy with stiffness, weakness, cramps, and even features of a proximal myopathy, and rhabdomyolysis. The electromyogram can be normal or myopathic. Results of muscle biopsy are often normal but may show focal necrosis and mild inflammatory infiltrates, thus mimicking that seen with inflammatory myopathy.7
Drug-induced or toxic myopathies can also mimic polymyositis. Statins are among the most commonly prescribed drugs in the United States, with more than 35 million people taking them. Statins are generally well tolerated but have a broad spectrum of toxicity, ranging from myalgias to life-threatening rhabdomyolysis. Myalgias lead to about 5% to 10% of patients refusing to take a statin or stopping it on their own.
Myalgias affect up to 20% of statin users in clinical practice.8,9 A small cross-sectional study10 of 1,000 patients in a primary care setting found that the risk of muscle complaints in statin users was 1.5 times higher than in nonstatin users, similar to findings in other studies.

My strategy for managing a patient with possible statin-induced myopathy is illustrated in Figure 1.
CASE 3: WEAKNESS, VERY HIGH CK ON A STATIN
In March 2010, a 67-year-old woman presented with muscle weakness. She had a history of hypertension, hyperlipidemia, and, more than 10 years previously, uterine cancer. In 2004, she was given atorvastatin for dyslipidemia. Four years later, she developed lower-extremity weakness, which her doctor attributed to normal aging. A year after that, she found it difficult to walk up steps and lift her arms overhead. In June 2009, she stopped taking the atorvastatin on her own, but the weakness did not improve.
In September 2009, she returned to her doctor, who found her CK level was 6,473 U/L but believed it to be an error, so the test was repeated, with a result of 9,375 U/L. She had no rash or joint involvement.
She was admitted to the hospital and underwent muscle biopsy, which showed myonecrosis with no inflammation or vasculitis. She was treated with prednisone 60 mg/day, and her elevated CK level and weakness improved.
Immune-mediated necrotizing myopathy associated with statins
The hallmark of necrotizing myopathy is myonecrosis without significant inflammation.12 This pattern contrasts with that of polymyositis, which is characterized by lymphocytic inflammation.
Although statins became available in the United States in 1987, immune-mediated necrotizing myopathy associated with statins was first described only in 2010. In that report, Grable-Esposito et al13 described 25 patients from 2 neuromuscular centers seen between 2000 and 2008 who had elevated CK and proximal weakness during or after statin use, both of which persisted despite stopping the statin. Patients improved with immunosuppressive agents but had a relapse when steroids were stopped or tapered, a pattern typical in autoimmune disease.
Autoantibody defines subgroup of necrotizing myopathy
Also in 2010, Christopher-Stine et al14 reported an antibody associated with necrotizing myopathy. Of 38 patients with the condition, 16 were found to have an abnormal “doublet” autoantibody recognizing 200- and 100-kDa proteins. All patients had weakness and a high CK level, and 63% had statin exposure before the weakness (this percentage increased to 83% in patients older than 50). All responded to immunosuppressive therapy, and many had a relapse when it was withdrawn.
Statins lower cholesterol by inhibiting 3-hydroxy-3-methylglutaryl-Co A reductase (HMGCR), and paradoxically, they also upregulate it. HMGCR has a molecular weight of 97 kDa. Mammen et al15 identified HMGCR as the 100-kDa target of the identified antibody and developed an enzyme-linked immunosorbent assay for it. Of 750 patients presenting to one center, only 45 (6%) had anti-HMGCR autoantibodies, but all 16 patients who had the abnormal doublet antibody tested positive for anti-HMGCR. Regenerating muscle cells express high levels of HMGCR, which may sustain the immune response after statins are discontinued.
Case 3 continued: Intravenous immunoglobulin brings improvement
In March 2010, when the 67-year-old patient presented to our myositis center, her CK level was 5,800 U/L, which increased as prednisone was tapered. She still felt weak. On examination, her muscle strength findings were deltoids 4+/5, neck flexors 4/5, and iliopsoas 3+/5. She was treated with methotrexate and azathioprine without benefit. She was next treated with intravenous immunoglobulin, and after 3 months, her strength normalized for the first time in years. Her CK level decreased but did not normalize. Testing showed that she was positive for anti-HMGCR autoantibody, as this test had become commercially available.
In 2015, Mammen and Tiniakou16 suggested using intravenous immunoglobulin as first-line therapy for statin-associated autoimmune necrotizing myopathy, based on experience at a single center with 3 patients who declined glucocorticoid treatment.
Necrotizing myopathy: Bottom line

Myositis overlap syndromes
Heterogeneity is the rule in myositis, and it can present with a wide variety of signs and symptoms as outlined in Table 2.
CASE 4: FEVER, NEW ‘RHEUMATOID ARTHRITIS,’ AND LUNG DISEASE
A 52-year-old woman with knee osteoarthritis saw her primary care physician in November 2013 for dyspnea and low-grade fever. The next month, she presented with polyarthritis, muscle weakness, and Raynaud phenomenon.
In January 2014, she developed acrocyanosis of her fingers. Examination revealed hyperkeratotic, cracked areas of her fingers. Her oxygen saturation by pulse oximetry was low. She was admitted to the hospital. Her doctor suspected new onset of rheumatoid arthritis, but blood tests revealed a negative antinuclear antibody, so an autoimmune condition was deemed unlikely. Her CK was mildly elevated at 350 U/L.
Because of her dyspnea, an open-lung biopsy was performed. High-resolution computed tomography (CT) revealed infiltrates and ground-glass opacities, leading to the diagnosis of nonspecific interstitial pneumonia. A rheumatologist was consulted and recommended pulse methylprednisolone, followed by prednisone 60 mg/day and mycophenolate mofetil. Testing for Jo-1 antibodies was positive.
Antisynthetase syndrome
The antisynthetase syndrome is a clinically heterogeneous condition that can occur with any or all of the following:
- Fever
- Myositis
- Arthritis (often misdiagnosed as rheumatoid arthritis)
- Raynaud phenomenon
- Mechanic’s hands (hyperkeratotic roughness with fissures on the lateral aspects of the fingers and finger pads)
- Interstitial lung disease.
The skin rashes and myositis may be subtle, making the presentation “lung-dominant,” and nonrheumatologists should be aware of this syndrome. Although in our patient the condition developed in a classic manner, with all of the aforementioned features of the antisynthetase syndrome, some patients will manifest one or a few of the features.

Clinically, patients with the Jo-1 antisynthetase syndrome often present differently than those with non-Jo-1 antisynthetase autoantibodies. When we compared 122 patients with Jo-1 vs 80 patients with a non-Jo-1 antisynthetase autoantibody, patients with Jo-1 antibodies were more likely to have initially received a diagnosis of myositis (83%), while myositis was the original diagnosis in only 17% of those possessing non-Jo-1 antisynthetase autoantibodies. In fact, many patients (approximately 50%) were diagnosed as having undifferentiated connective tissue disease or an overlap syndrome, and 13% had scleroderma as their first diagnosis.17
We also found that the survival rate was higher in patients with Jo-1 syndrome compared with patients with non-Jo-1 antisynthetase syndromes. We attributed the difference in survival rates to a delayed diagnosis in the non-Jo-1 group, perhaps due to their “nonclassic” presentations of the antisynthetase syndrome, delaying appropriate treatment. Patients received a diagnosis of Jo-1 antibody syndrome after a mean of 0.4 year (range 0.2–0.8), while those with a non-Jo-1 antisynthetase autoantibody had a delay in diagnosis of 1.0 year (range 0.4–5.1) (P < .01).17
In nearly half the cases in this cohort, pulmonary fibrosis was the cause of death, with primary pulmonary hypertension being the second leading cause (11%).
Antisynthetase syndrome: Bottom line
Antisynthetase syndrome is an often fatal disease that does not always present in a typical fashion with symptoms of myositis, as lung disease may be the predominant feature. A negative antinuclear antibody test result does not imply antibody negativity, as the autoantigen in these diseases is not located in the nucleus. Prompt diagnosis and appropriate immunosuppressive therapy are critical to improving outcomes.
CASE 5: FEVER, UNDIAGNOSED LUNG DISEASE, NO MYOSITIS
In January 2001, a 39-year-old woman was admitted to the hospital after 5 weeks of fever (temperatures 103°–104°F) and myalgias. An extensive workup was negative except for low-titer antinuclear antibody and for mild basilar fibrosis noted on chest radiography. She left the hospital against medical advice because of frustration with a lack of a specific diagnosis (“fever of unknown origin”).
Two months later, at a follow-up rheumatology consult, she reported more myalgias and arthralgias, as well as fever. Chest radiography now showed pleural effusions. Her fingers had color changes consistent with Raynaud phenomenon. At that time, I diagnosed an undifferentiated connective tissue disease and told her that I suspected an autoimmune condition that would need time to reveal itself. In the meantime, I treated her empirically with prednisone.
In April, she returned, much more short of breath and with more prominent diffuse pulmonary infiltrates. Physical examination revealed subtle Gottron changes. Testing revealed poor pulmonary function: forced vital capacity (FVC) 56%, forced expiratory volume in 1 second (FEV1) 52%, and diffusing capacity for carbon monoxide (Dlco) 40%. Blood testing was positive for anti-PL-12 antibody, one of the non-Jo-1 antisynthetase antibodies. At this time, we treated her with glucocorticoids and tacrolimus.
More than 15 years later, this patient is doing well. Her skin rash, joint symptoms, and fever have not returned, and interestingly, she never developed myositis. Her Raynaud symptoms are mild. Her most recent pulmonary function test results (January 2018) were FVC 75%, FEV1 87%, and Dlco 78%. Although these results are not normal, they are much improved and allow her to be completely functional without supplemental oxygen. Echocardiography showed normal pulmonary artery systolic pressure (25 mm Hg). She was still taking tacrolimus and prednisone. When we tried to stop tacrolimus after she had done well for many years, her condition flared.
Non-Jo-1 antisynthetase syndrome: Bottom line
Patients with a non-Jo-1 antisynthetase syndrome often present without myositis symptoms and may never manifest myositis symptoms. Likely because of this presentation, diagnosis of a specific connective tissue disorder is delayed, perhaps leading to increased mortality risk from pulmonary disease. Chronic immunosuppression is often required for these autoimmune conditions.
CASE 6: DERMATOMYOSITIS, RAPIDLY PROGRESSIVE INTERSTITIAL LUNG DISEASE
A 58-year-old woman presented in the summer of 2012 with a photosensitive rash. The following January, she returned with polyarthritis, mild muscle weakness, and a dermatomyositis-pattern rash. Her CK level was normal, and her antinuclear antibody and Sjögren syndrome antibody test results were negative. She improved on low-dose prednisone and methotrexate.
She was originally referred to me in May of that year for worsening rash and mild weakness. She denied pulmonary symptoms, but examination revealed faint basilar crackles. I increased her prednisone dosage to 20 mg/day and started mycophenolate mofetil mainly for the mild cutaneous and myositis features. I also recommended high-resolution CT of the lungs and pulmonary function tests, which she underwent in early June. High-resolution CT showed nonspecific mild infiltrates with minimal ground-glass opacities.
On July 1, she presented to her local emergency department with severe shortness of breath, requiring oxygen 12 L/min. She had a palmar rash. Repeat high-resolution CT showed dramatic worsening compared with the scan the previous month. Because of continued inadequate oxygenation, she was transferred to our center. A blood test later was positive for antimelanoma differentiation-associated gene 5 (MDA-5) autoantibody, previously known as anticlinically amyopathic dermatomyositis (anti-CADM)-140 antibody (based on immunoprecipitation results).
She died on the third day after transfer, just 2 months after I had originally seen her, at which time she had had no pulmonary symptoms.
Clinically amyopathic dermatomyositis
Anti-CADM-140, first reported from Asia,18–20 is an autoantibody-associated disease but not an antisynthetase. It is associated with dermatomyositis; patients often have a “vasculopathy” with cutaneous ulcerations and palmar papules.
MDA-5 is a cytoplasmic protein that “senses” viral RNA and induces production of type 1 interferon. It is involved in the innate immune defense against viruses.
Anti-MDA-5 positivity is associated with a poor pulmonary outcome.21 In our cohort from the University of Pittsburgh, many patients died within 3 years, compared with about a 40% survival rate in patients with dermatomyositis who tested negative for this antibody. That being said, many patients with anti-MDA-5 do not develop rapidly progressive interstitial lung disease.
Autoimmune interstitial lung disease: Bottom line
Autoimmune interstitial lung disease is easy to miss, especially in the case of a non-Jo-1 syndrome, for 3 important reasons:
- The autoimmune features may initially be subtle (eg, Raynaud phenomena, mild dermatomyositis rash, undifferentiated connective tissue disease)
- Autoantibody testing is not often ordered, is not standardized, or may be unavailable
- Providers are mistakenly reassured that a patient who tests negative for antinuclear antibody does not have an autoimmune condition.
To emphasize the last point, in a cohort of 202 patients who tested positive for an antisynthetase antibody, only half were antinuclear antibody-positive, but nearly three-quarters demonstrated anticytoplasmic staining on indirect immunofluorescence (due to the location of the autoantigen in the cytoplasm), making the latter a better screening test for an antisynthetase antibody. For scleroderma, 99% were antinculear antibody-positive, but for myositis, this test is much less sensitive.22
- Felice KJ, North WA. Inclusion body myositis in Connecticut: observations in 35 patients during an 8-year period. Medicine (Baltimore) 2001; 80(5):320–327. doi:10.1097/00005792-200109000-00006
- Lev EI, Tur-Kaspa I, Ashkenazy I, et al. Distribution of serum creatine kinase activity in young healthy persons. Clin Chim Acta 1999; 279(1-2):107–115. doi:10.1016/S0009-8981(98)00180-6
- Lilleng H, Abeler K, Johnsen SH, et al. Variation of serum creatine kinase (CK) levels and prevalence of persistent hyperCKemia in a Norwegian normal population. The Tromsø Study. Neuromuscul Disord 2011; 21(7):494–500. doi:10.1016/j.nmd.2011.04.007
- Johnston JD, Lloyd M, Mathews JA, Hawthorne SW. Racial variation in serum creatine kinase levels. J R Soc Med 1996; 89(8):462-464. pmid:8795501
- Prelle A, Tancredi L, Sciacco M, et al. Retrospective study of a large population of patients with asymptomatic or minimally symptomatic raised serum creatine kinase levels. J Neurol 2002; 249(3):305–311. pmid:11993531
- D’Adda E, Sciacco M, Fruguglietti ME, et al. Follow-up of a large population of asymptomatic/oligosymptomatic hyperckemic subjects. J Neurol 2006; 253(11):1399–1403. doi:10.1007/s00415-006-0223-y
- Madariaga MG. Polymyositis-like syndrome in hypothyroidism: review of cases reported over the past twenty-five years. Thyroid 2002; 12(4):331–336. doi:10.1089/10507250252949478
- de Sauvage Nolting PR, Buirma RJ, Hutten BA, Kastelein JJ; Dutch ExPRESS Investigator Group. Two-year efficacy and safety of simvastatin 80 mg in familial hypercholesterolemia (the Examination of Probands and Relatives in Statin Studies With Familial Hypercholesterolemia [ExPRESS FH]). Am J Cardiol 2002; 90(2):181–184. doi:10.1016/s0002-9149(02)02449-9
- Bruckert E, Hayem G, Dejager S, Yau C, Bégaud B. Mild to moderate muscular symptoms with high-dosage statin therapy in hyperlipidemic patients--the PRIMO study. Cardiovasc Drugs Ther 2005; 19(6):403–414. doi:10.1007/s10557-005-5686-z
- Mosshammer D, Lorenz G, Meznaric S, Schwarz J, Muche R, Mörike K. Statin use and its association with musculoskeletal symptoms—a cross-sectional study in primary care settings. Fam Pract 2009; 26(2):88–95. doi:10.1093/fampra/cmp006
- Nichols GA, Koro CE. Does statin therapy initiation increase the risk for myopathy? An observational study of 32,225 diabetic and nondiabetic patients. Clin Ther 2007; 29(8):1761–1770. doi:10.1016/j.clinthera.2007.08.022
- Kassardjian CD, Lennon VA, Alfugham NB, Mahler M, Milone M. Clinical features and treatment outcomes of necrotizing autoimmune myopathy. JAMA Neurol 2015; 72(9):996–1003. doi:10.1001/jamaneurol.2015.1207
- Grable-Esposito P, Katzberg HD, Greenberg SA, Srinivasan J, Katz J, Amato AA. Immune-mediated necrotizing myopathy associated with statins. Muscle Nerve 2010; 41(2):185–190. doi:10.1002/mus.21486
- Christopher-Stine L, Casciola-Rosen LA, Hong G, Chung T, Corse AM, Mammen AL. A novel autoantibody recognizing 200-kd and 100-kd proteins is associated with an immune-mediated necrotizing myopathy. Arthritis Rheum 2010; 62(9):2757–2766. doi:10.1002/art.27572
- Mammen AL, Chung T, Christopher-Stine L, et al. Autoantibodies against 3-hydroxy-3-methylglutaryl-coenzyme A reductase in patients with statin-associated autoimmune myopathy. Arthritis Rheum 2011; 63(3):713–721. doi:10.1002/art.30156
- Mammen AL, Tiniakou E. Intravenous immune globulin for statin-triggered autoimmune myopathy. N Engl J Med 2015; 373(17):1680–1682. doi:10.1056/NEJMc1506163
- Aggarwal R, Cassidy E, Fertig N, et al. Patients with non-Jo-1 anti-tRNA-synthetase autoantibodies have worse survival than Jo-1 positive patients. Ann Rheum Dis 2014; 73(1):227–232. doi:10.1136/annrheumdis-2012-201800
- Sato S, Hirakata M, Kuwana M, et al. Autoantibodies to a 140-kd polypeptide, CADM-140, in Japanese patients with clinically amyopathic dermatomyositis. Arthritis Rheum 2005; 52(5):1571–1576. doi:10.1002/art.21023
- Sato S, Hoshino K, Satoh T, et al. RNA helicase encoded by melanoma differentiation-associated gene 5 is a major autoantigen in patients with clinically amyopathic dermatomyositis: association with rapidly progressive interstitial lung disease. Arthritis Rheum 2009; 60(7):2193–2200. doi:10.1002/art.24621
- Chen F, Wang D, Shu X, Nakashima R, Wang G. Anti-MDA5 antibody is associated with A/SIP and decreased T cells in peripheral blood and predicts poor prognosis of ILD in Chinese patients with dermatomyositis. Rheumatol Int 2012; 32(12):3909–3915. doi:10.1007/s00296-011-2323-y
- Moghadam-Kia S, Oddis CV, Sato S, Kuwana M, Aggarwal R. Anti-melanoma differentiation-associated gene 5 is associated with rapidly progressive lung disease and poor survival in US patients with amyopathic and myopathic dermatomyositis. Arthritis Care Res (Hoboken) 2016; 68(5):689–694. doi:10.1002/acr.22728
- Aggarwal R, Dhillon N, Fertig N, Koontz D, Qi Z, Oddis CV. A negative antinuclear antibody does not indicate autoantibody negativity in myositis: role of anticytoplasmic antibody as a screening test for antisynthetase syndrome. J Rheumatol 2017; 44(2):223–229. doi:10.3899/jrheum.160618
Myopathies can present with a wide variety of symptoms, so patients with muscle weakness are often seen initially by a general practitioner. Nonrheumatologists should be able to evaluate a patient presenting with muscle weakness or myalgia and be aware of red flags indicating potentially dangerous syndromes that require a prompt, thorough investigation.
This article reviews selected causes of muscle weakness, such as statin-induced and autoimmune disorders, and systemic features of inflammatory myopathies beyond myositis, such as dermatologic and pulmonary manifestations.
FOCUSING THE EVALUATION
The evaluation of a patient presenting with muscle weakness should include several assessments:
Temporal progression. Was the onset of symptoms rapid or insidious? Patterns of onset may give clues to etiology, including the possibility of an associated autoimmune condition.
Location of muscle weakness. Are symptoms global or localized? And if localized, are they proximal or distal? Proximal weakness can be manifested by difficulty rising from a chair (hip muscles) or combing one’s hair (shoulder muscles), whereas distal weakness can involve difficulty standing on toes (gastrocnemius and soleus muscles) or performing fine motor activities (intrinsic hand muscles).
Symmetry. A focal or asymmetric pattern often has a neurologic etiology, but this could also be consistent with inclusion body myositis.
Other symptoms. Arthritis, rash, and swallowing problems point to a possible underlying rheumatologic disease. Weight gain or loss may indicate a thyroid disorder.
Family history. Some patients report that others in their family have this pattern of weakness, indicating a likely genetic myopathy. If the patient reports a relative with multiple sclerosis, lupus erythematosus, rheumatoid arthritis, or another autoimmune disease, then an immune-mediated myopathy should be considered.
Medications should be reviewed, particularly statins.
CASE 1: SLOWLY PROGRESSIVE WEAKNESS
A 65-year-old man presented with the insidious onset of muscle weakness and episodes of falling. On review of his medical record, his serum creatine kinase (CK) levels were elevated at various periods at 2 to 4 times the upper limit of normal. Electromyography (EMG) previously showed a myopathic pattern, and a muscle biopsy was abnormal, consistent with endomysial inflammation (term is consistent with “polymyositis”). He was treated for polymyositis for several years with prednisone alone, with steroids plus methotrexate, and with combined immunosuppression including methotrexate and azathioprine, but with no improvement. Eventually, another muscle biopsy revealed inclusion bodies with rimmed vacuoles, consistent with inclusion body myositis.
Inclusion body myositis
Inclusion body myositis is the most common myopathy in middle-aged to elderly people, especially men. These patients are often told “You are just getting old,” but they have a defined condition. It should also be considered in patients failing to respond to treatment or with those with “refractory” polymyositis.
The onset of muscle weakness is insidious and painless, and the weakness progresses slowly. The pattern is distal and asymmetric (eg, foot drop), and muscle atrophy typically affects the forearm flexors, quadriceps, and intrinsic muscles of the hands.1
Magnetic resonance imaging may show marked muscle atrophy. Unfortunately, no treatment has shown efficacy, and most neuromuscular and rheumatology experts do not treat inclusion body myositis with immunosuppressive drugs.
CASE 2: MILD MYALGIA WITHOUT WEAKNESS
A black 52-year-old man was referred because of myalgia and a CK level of 862 U/L (reference range < 200). His physician wanted to start him on a statin but was hesitant to do so without first consulting a rheumatologist.
The patient had a long history of mild arthralgias and myalgias without muscle weakness. He had dyslipidemia and hypertension. He reported no family history of myopathy and no illicit drug use. He was formerly an athlete. Medications included a thiazide diuretic and a beta-blocker. On examination, his muscles were strong (rated 5 on a scale of 5) in the upper and lower extremities, without atrophy.
His records showed that his CK levels had risen and fallen repeatedly over the past few years, ranging from 600 to 1,100 U/L. On further questioning, he reported that when he had joined the army 30 years previously, a physician had recommended he undergo a liver biopsy in view of elevated liver function tests, but that he had refused because he felt fine.
Currently, his gamma-glutamyl transpeptidase levels were normal.
Idiopathic ‘hyperCKemia’
So-called idiopathic hyperCKemia is not a form of myositis but merely a laboratory result outside the “normal” range. Reference ranges are based predominantly on measurements in white people and on an assumption that the distribution is Gaussian (bell-shaped). A normal CK level is usually defined as less than 200 U/L. Using this standard, up to 20% of men and 5% of women have hyperCKemia.2
However, CK levels vary by sex and ethnicity, with mean levels highest in black men, followed by black women, white men, and white women. The mean level in black men is higher than the standard cutoff point for normal, and especially in this population, there is wide fluctuation around the mean, leading to hyperCKemia quite frequently in black men. Exercise and manual labor also drive up CK levels.3–5
Idiopathic hyperCKemia is benign. D’Adda et al6 followed 55 patients for a mean of 7.5 years. CK levels normalized in 12 patients or at least decreased in 24. Most remained symptom-free or had minimal symptoms.
Idiopathic hyperCKemia: Bottom line
Before prescribing a statin, determine the baseline CK level. If slightly elevated (ie, up to 3 to 5 times the upper limit of normal, or even higher) in the setting of normal muscle strength, there is no need for electromyography or muscle biopsy, and the patient can certainly receive a statin. Most of these patients do not need to see a rheumatologist but can simply have their CK and muscle strength monitored.
CLASSIFYING MYOSITIS
Myositis (idiopathic inflammatory myopathy) is a heterogeneous group of autoimmune syndromes of unknown cause characterized by chronic muscle weakness and inflammation of striated muscle. These syndromes likely arise as a result of genetic predisposition and an environmental or infectious “hit.”
Myositis is rare, with an incidence of 5 to 10 cases per million per year and an estimated prevalence of 50 to 90 cases per million. It has 2 incidence peaks: 1 in childhood (age 5–15) and another in adult midlife (age 30–50). Women are affected 2 to 3 times more often than men, with black women most commonly affected.
Myositis is traditionally classified as follows:
- Adult polymyositis
- Adult dermatomyositis
- Juvenile myositis (dermatomyositis much more frequent than polymyositis)
- Malignancy-associated myositis (usually dermatomyositis)
- Myositis overlapping with another autoimmune disease
- Inclusion body myositis.
However, polymyositis is less common than we originally thought, and the term necrotizing myopathy is now used in many patients, as noted in the case studies below. Further, myositis overlap syndromes are being increasingly diagnosed, likely related to the emergence of autoantibodies and clinical “syndromes” associated with these autoantibody subsets (discussed in cases below).
Dermatomyositis
Dermatomyositis is characterized by muscle weakness and a rash that can be obvious or subtle. Classic skin lesions are Gottron papules, which are raised, flat-topped red or purplish lesions over the knuckles, elbows, or knees.
Lesions may be confused with those of psoriasis. There can also be a V-neck rash over the anterior chest or upper back (“shawl sign”) or a rash over the lateral thigh (“holster sign”). A facial rash may occur, but unlike lupus, dermatomyositis does not spare the nasolabial area. However, the V-neck rash can be similar to that seen in lupus.
Dermatomyositis may cause muscle pain, perhaps related to muscle ischemia, whereas polymyositis and necrotizing myopathy are often painless. However, pain is also associated with fibromyalgia, which may be seen in many autoimmune conditions. It is important not to overtreat rheumatologic diseases with immunosuppression to try to control pain if the pain is actually caused by fibromyalgia.
Polymyositis mimics
Hypothyroid myopathy can present as classic polymyositis. The serum CK may be elevated, and there may be myalgias, muscle hypertrophy with stiffness, weakness, cramps, and even features of a proximal myopathy, and rhabdomyolysis. The electromyogram can be normal or myopathic. Results of muscle biopsy are often normal but may show focal necrosis and mild inflammatory infiltrates, thus mimicking that seen with inflammatory myopathy.7
Drug-induced or toxic myopathies can also mimic polymyositis. Statins are among the most commonly prescribed drugs in the United States, with more than 35 million people taking them. Statins are generally well tolerated but have a broad spectrum of toxicity, ranging from myalgias to life-threatening rhabdomyolysis. Myalgias lead to about 5% to 10% of patients refusing to take a statin or stopping it on their own.
Myalgias affect up to 20% of statin users in clinical practice.8,9 A small cross-sectional study10 of 1,000 patients in a primary care setting found that the risk of muscle complaints in statin users was 1.5 times higher than in nonstatin users, similar to findings in other studies.
My strategy for managing a patient with possible statin-induced myopathy is illustrated in Figure 1.
CASE 3: WEAKNESS, VERY HIGH CK ON A STATIN
In March 2010, a 67-year-old woman presented with muscle weakness. She had a history of hypertension, hyperlipidemia, and, more than 10 years previously, uterine cancer. In 2004, she was given atorvastatin for dyslipidemia. Four years later, she developed lower-extremity weakness, which her doctor attributed to normal aging. A year after that, she found it difficult to walk up steps and lift her arms overhead. In June 2009, she stopped taking the atorvastatin on her own, but the weakness did not improve.
In September 2009, she returned to her doctor, who found her CK level was 6,473 U/L but believed it to be an error, so the test was repeated, with a result of 9,375 U/L. She had no rash or joint involvement.
She was admitted to the hospital and underwent muscle biopsy, which showed myonecrosis with no inflammation or vasculitis. She was treated with prednisone 60 mg/day, and her elevated CK level and weakness improved.
Immune-mediated necrotizing myopathy associated with statins
The hallmark of necrotizing myopathy is myonecrosis without significant inflammation.12 This pattern contrasts with that of polymyositis, which is characterized by lymphocytic inflammation.
Although statins became available in the United States in 1987, immune-mediated necrotizing myopathy associated with statins was first described only in 2010. In that report, Grable-Esposito et al13 described 25 patients from 2 neuromuscular centers seen between 2000 and 2008 who had elevated CK and proximal weakness during or after statin use, both of which persisted despite stopping the statin. Patients improved with immunosuppressive agents but had a relapse when steroids were stopped or tapered, a pattern typical in autoimmune disease.
Autoantibody defines subgroup of necrotizing myopathy
Also in 2010, Christopher-Stine et al14 reported an antibody associated with necrotizing myopathy. Of 38 patients with the condition, 16 were found to have an abnormal “doublet” autoantibody recognizing 200- and 100-kDa proteins. All patients had weakness and a high CK level, and 63% had statin exposure before the weakness (this percentage increased to 83% in patients older than 50). All responded to immunosuppressive therapy, and many had a relapse when it was withdrawn.
Statins lower cholesterol by inhibiting 3-hydroxy-3-methylglutaryl-Co A reductase (HMGCR), and paradoxically, they also upregulate it. HMGCR has a molecular weight of 97 kDa. Mammen et al15 identified HMGCR as the 100-kDa target of the identified antibody and developed an enzyme-linked immunosorbent assay for it. Of 750 patients presenting to one center, only 45 (6%) had anti-HMGCR autoantibodies, but all 16 patients who had the abnormal doublet antibody tested positive for anti-HMGCR. Regenerating muscle cells express high levels of HMGCR, which may sustain the immune response after statins are discontinued.
Case 3 continued: Intravenous immunoglobulin brings improvement
In March 2010, when the 67-year-old patient presented to our myositis center, her CK level was 5,800 U/L, which increased as prednisone was tapered. She still felt weak. On examination, her muscle strength findings were deltoids 4+/5, neck flexors 4/5, and iliopsoas 3+/5. She was treated with methotrexate and azathioprine without benefit. She was next treated with intravenous immunoglobulin, and after 3 months, her strength normalized for the first time in years. Her CK level decreased but did not normalize. Testing showed that she was positive for anti-HMGCR autoantibody, as this test had become commercially available.
In 2015, Mammen and Tiniakou16 suggested using intravenous immunoglobulin as first-line therapy for statin-associated autoimmune necrotizing myopathy, based on experience at a single center with 3 patients who declined glucocorticoid treatment.
Necrotizing myopathy: Bottom line
Myositis overlap syndromes
Heterogeneity is the rule in myositis, and it can present with a wide variety of signs and symptoms as outlined in Table 2.
CASE 4: FEVER, NEW ‘RHEUMATOID ARTHRITIS,’ AND LUNG DISEASE
A 52-year-old woman with knee osteoarthritis saw her primary care physician in November 2013 for dyspnea and low-grade fever. The next month, she presented with polyarthritis, muscle weakness, and Raynaud phenomenon.
In January 2014, she developed acrocyanosis of her fingers. Examination revealed hyperkeratotic, cracked areas of her fingers. Her oxygen saturation by pulse oximetry was low. She was admitted to the hospital. Her doctor suspected new onset of rheumatoid arthritis, but blood tests revealed a negative antinuclear antibody, so an autoimmune condition was deemed unlikely. Her CK was mildly elevated at 350 U/L.
Because of her dyspnea, an open-lung biopsy was performed. High-resolution computed tomography (CT) revealed infiltrates and ground-glass opacities, leading to the diagnosis of nonspecific interstitial pneumonia. A rheumatologist was consulted and recommended pulse methylprednisolone, followed by prednisone 60 mg/day and mycophenolate mofetil. Testing for Jo-1 antibodies was positive.
Antisynthetase syndrome
The antisynthetase syndrome is a clinically heterogeneous condition that can occur with any or all of the following:
- Fever
- Myositis
- Arthritis (often misdiagnosed as rheumatoid arthritis)
- Raynaud phenomenon
- Mechanic’s hands (hyperkeratotic roughness with fissures on the lateral aspects of the fingers and finger pads)
- Interstitial lung disease.
The skin rashes and myositis may be subtle, making the presentation “lung-dominant,” and nonrheumatologists should be aware of this syndrome. Although in our patient the condition developed in a classic manner, with all of the aforementioned features of the antisynthetase syndrome, some patients will manifest one or a few of the features.
Clinically, patients with the Jo-1 antisynthetase syndrome often present differently than those with non-Jo-1 antisynthetase autoantibodies. When we compared 122 patients with Jo-1 vs 80 patients with a non-Jo-1 antisynthetase autoantibody, patients with Jo-1 antibodies were more likely to have initially received a diagnosis of myositis (83%), while myositis was the original diagnosis in only 17% of those possessing non-Jo-1 antisynthetase autoantibodies. In fact, many patients (approximately 50%) were diagnosed as having undifferentiated connective tissue disease or an overlap syndrome, and 13% had scleroderma as their first diagnosis.17
We also found that the survival rate was higher in patients with Jo-1 syndrome compared with patients with non-Jo-1 antisynthetase syndromes. We attributed the difference in survival rates to a delayed diagnosis in the non-Jo-1 group, perhaps due to their “nonclassic” presentations of the antisynthetase syndrome, delaying appropriate treatment. Patients received a diagnosis of Jo-1 antibody syndrome after a mean of 0.4 year (range 0.2–0.8), while those with a non-Jo-1 antisynthetase autoantibody had a delay in diagnosis of 1.0 year (range 0.4–5.1) (P < .01).17
In nearly half the cases in this cohort, pulmonary fibrosis was the cause of death, with primary pulmonary hypertension being the second leading cause (11%).
Antisynthetase syndrome: Bottom line
Antisynthetase syndrome is an often fatal disease that does not always present in a typical fashion with symptoms of myositis, as lung disease may be the predominant feature. A negative antinuclear antibody test result does not imply antibody negativity, as the autoantigen in these diseases is not located in the nucleus. Prompt diagnosis and appropriate immunosuppressive therapy are critical to improving outcomes.
CASE 5: FEVER, UNDIAGNOSED LUNG DISEASE, NO MYOSITIS
In January 2001, a 39-year-old woman was admitted to the hospital after 5 weeks of fever (temperatures 103°–104°F) and myalgias. An extensive workup was negative except for low-titer antinuclear antibody and for mild basilar fibrosis noted on chest radiography. She left the hospital against medical advice because of frustration with a lack of a specific diagnosis (“fever of unknown origin”).
Two months later, at a follow-up rheumatology consult, she reported more myalgias and arthralgias, as well as fever. Chest radiography now showed pleural effusions. Her fingers had color changes consistent with Raynaud phenomenon. At that time, I diagnosed an undifferentiated connective tissue disease and told her that I suspected an autoimmune condition that would need time to reveal itself. In the meantime, I treated her empirically with prednisone.
In April, she returned, much more short of breath and with more prominent diffuse pulmonary infiltrates. Physical examination revealed subtle Gottron changes. Testing revealed poor pulmonary function: forced vital capacity (FVC) 56%, forced expiratory volume in 1 second (FEV1) 52%, and diffusing capacity for carbon monoxide (Dlco) 40%. Blood testing was positive for anti-PL-12 antibody, one of the non-Jo-1 antisynthetase antibodies. At this time, we treated her with glucocorticoids and tacrolimus.
More than 15 years later, this patient is doing well. Her skin rash, joint symptoms, and fever have not returned, and interestingly, she never developed myositis. Her Raynaud symptoms are mild. Her most recent pulmonary function test results (January 2018) were FVC 75%, FEV1 87%, and Dlco 78%. Although these results are not normal, they are much improved and allow her to be completely functional without supplemental oxygen. Echocardiography showed normal pulmonary artery systolic pressure (25 mm Hg). She was still taking tacrolimus and prednisone. When we tried to stop tacrolimus after she had done well for many years, her condition flared.
Non-Jo-1 antisynthetase syndrome: Bottom line
Patients with a non-Jo-1 antisynthetase syndrome often present without myositis symptoms and may never manifest myositis symptoms. Likely because of this presentation, diagnosis of a specific connective tissue disorder is delayed, perhaps leading to increased mortality risk from pulmonary disease. Chronic immunosuppression is often required for these autoimmune conditions.
CASE 6: DERMATOMYOSITIS, RAPIDLY PROGRESSIVE INTERSTITIAL LUNG DISEASE
A 58-year-old woman presented in the summer of 2012 with a photosensitive rash. The following January, she returned with polyarthritis, mild muscle weakness, and a dermatomyositis-pattern rash. Her CK level was normal, and her antinuclear antibody and Sjögren syndrome antibody test results were negative. She improved on low-dose prednisone and methotrexate.
She was originally referred to me in May of that year for worsening rash and mild weakness. She denied pulmonary symptoms, but examination revealed faint basilar crackles. I increased her prednisone dosage to 20 mg/day and started mycophenolate mofetil mainly for the mild cutaneous and myositis features. I also recommended high-resolution CT of the lungs and pulmonary function tests, which she underwent in early June. High-resolution CT showed nonspecific mild infiltrates with minimal ground-glass opacities.
On July 1, she presented to her local emergency department with severe shortness of breath, requiring oxygen 12 L/min. She had a palmar rash. Repeat high-resolution CT showed dramatic worsening compared with the scan the previous month. Because of continued inadequate oxygenation, she was transferred to our center. A blood test later was positive for antimelanoma differentiation-associated gene 5 (MDA-5) autoantibody, previously known as anticlinically amyopathic dermatomyositis (anti-CADM)-140 antibody (based on immunoprecipitation results).
She died on the third day after transfer, just 2 months after I had originally seen her, at which time she had had no pulmonary symptoms.
Clinically amyopathic dermatomyositis
Anti-CADM-140, first reported from Asia,18–20 is an autoantibody-associated disease but not an antisynthetase. It is associated with dermatomyositis; patients often have a “vasculopathy” with cutaneous ulcerations and palmar papules.
MDA-5 is a cytoplasmic protein that “senses” viral RNA and induces production of type 1 interferon. It is involved in the innate immune defense against viruses.
Anti-MDA-5 positivity is associated with a poor pulmonary outcome.21 In our cohort from the University of Pittsburgh, many patients died within 3 years, compared with about a 40% survival rate in patients with dermatomyositis who tested negative for this antibody. That being said, many patients with anti-MDA-5 do not develop rapidly progressive interstitial lung disease.
Autoimmune interstitial lung disease: Bottom line
Autoimmune interstitial lung disease is easy to miss, especially in the case of a non-Jo-1 syndrome, for 3 important reasons:
- The autoimmune features may initially be subtle (eg, Raynaud phenomena, mild dermatomyositis rash, undifferentiated connective tissue disease)
- Autoantibody testing is not often ordered, is not standardized, or may be unavailable
- Providers are mistakenly reassured that a patient who tests negative for antinuclear antibody does not have an autoimmune condition.
To emphasize the last point, in a cohort of 202 patients who tested positive for an antisynthetase antibody, only half were antinuclear antibody-positive, but nearly three-quarters demonstrated anticytoplasmic staining on indirect immunofluorescence (due to the location of the autoantigen in the cytoplasm), making the latter a better screening test for an antisynthetase antibody. For scleroderma, 99% were antinculear antibody-positive, but for myositis, this test is much less sensitive.22
Myopathies can present with a wide variety of symptoms, so patients with muscle weakness are often seen initially by a general practitioner. Nonrheumatologists should be able to evaluate a patient presenting with muscle weakness or myalgia and be aware of red flags indicating potentially dangerous syndromes that require a prompt, thorough investigation.
This article reviews selected causes of muscle weakness, such as statin-induced and autoimmune disorders, and systemic features of inflammatory myopathies beyond myositis, such as dermatologic and pulmonary manifestations.
FOCUSING THE EVALUATION
The evaluation of a patient presenting with muscle weakness should include several assessments:
Temporal progression. Was the onset of symptoms rapid or insidious? Patterns of onset may give clues to etiology, including the possibility of an associated autoimmune condition.
Location of muscle weakness. Are symptoms global or localized? And if localized, are they proximal or distal? Proximal weakness can be manifested by difficulty rising from a chair (hip muscles) or combing one’s hair (shoulder muscles), whereas distal weakness can involve difficulty standing on toes (gastrocnemius and soleus muscles) or performing fine motor activities (intrinsic hand muscles).
Symmetry. A focal or asymmetric pattern often has a neurologic etiology, but this could also be consistent with inclusion body myositis.
Other symptoms. Arthritis, rash, and swallowing problems point to a possible underlying rheumatologic disease. Weight gain or loss may indicate a thyroid disorder.
Family history. Some patients report that others in their family have this pattern of weakness, indicating a likely genetic myopathy. If the patient reports a relative with multiple sclerosis, lupus erythematosus, rheumatoid arthritis, or another autoimmune disease, then an immune-mediated myopathy should be considered.
Medications should be reviewed, particularly statins.
CASE 1: SLOWLY PROGRESSIVE WEAKNESS
A 65-year-old man presented with the insidious onset of muscle weakness and episodes of falling. On review of his medical record, his serum creatine kinase (CK) levels were elevated at various periods at 2 to 4 times the upper limit of normal. Electromyography (EMG) previously showed a myopathic pattern, and a muscle biopsy was abnormal, consistent with endomysial inflammation (term is consistent with “polymyositis”). He was treated for polymyositis for several years with prednisone alone, with steroids plus methotrexate, and with combined immunosuppression including methotrexate and azathioprine, but with no improvement. Eventually, another muscle biopsy revealed inclusion bodies with rimmed vacuoles, consistent with inclusion body myositis.
Inclusion body myositis
Inclusion body myositis is the most common myopathy in middle-aged to elderly people, especially men. These patients are often told “You are just getting old,” but they have a defined condition. It should also be considered in patients failing to respond to treatment or with those with “refractory” polymyositis.
The onset of muscle weakness is insidious and painless, and the weakness progresses slowly. The pattern is distal and asymmetric (eg, foot drop), and muscle atrophy typically affects the forearm flexors, quadriceps, and intrinsic muscles of the hands.1
Magnetic resonance imaging may show marked muscle atrophy. Unfortunately, no treatment has shown efficacy, and most neuromuscular and rheumatology experts do not treat inclusion body myositis with immunosuppressive drugs.
CASE 2: MILD MYALGIA WITHOUT WEAKNESS
A black 52-year-old man was referred because of myalgia and a CK level of 862 U/L (reference range < 200). His physician wanted to start him on a statin but was hesitant to do so without first consulting a rheumatologist.
The patient had a long history of mild arthralgias and myalgias without muscle weakness. He had dyslipidemia and hypertension. He reported no family history of myopathy and no illicit drug use. He was formerly an athlete. Medications included a thiazide diuretic and a beta-blocker. On examination, his muscles were strong (rated 5 on a scale of 5) in the upper and lower extremities, without atrophy.
His records showed that his CK levels had risen and fallen repeatedly over the past few years, ranging from 600 to 1,100 U/L. On further questioning, he reported that when he had joined the army 30 years previously, a physician had recommended he undergo a liver biopsy in view of elevated liver function tests, but that he had refused because he felt fine.
Currently, his gamma-glutamyl transpeptidase levels were normal.
Idiopathic ‘hyperCKemia’
So-called idiopathic hyperCKemia is not a form of myositis but merely a laboratory result outside the “normal” range. Reference ranges are based predominantly on measurements in white people and on an assumption that the distribution is Gaussian (bell-shaped). A normal CK level is usually defined as less than 200 U/L. Using this standard, up to 20% of men and 5% of women have hyperCKemia.2
However, CK levels vary by sex and ethnicity, with mean levels highest in black men, followed by black women, white men, and white women. The mean level in black men is higher than the standard cutoff point for normal, and especially in this population, there is wide fluctuation around the mean, leading to hyperCKemia quite frequently in black men. Exercise and manual labor also drive up CK levels.3–5
Idiopathic hyperCKemia is benign. D’Adda et al6 followed 55 patients for a mean of 7.5 years. CK levels normalized in 12 patients or at least decreased in 24. Most remained symptom-free or had minimal symptoms.
Idiopathic hyperCKemia: Bottom line
Before prescribing a statin, determine the baseline CK level. If slightly elevated (ie, up to 3 to 5 times the upper limit of normal, or even higher) in the setting of normal muscle strength, there is no need for electromyography or muscle biopsy, and the patient can certainly receive a statin. Most of these patients do not need to see a rheumatologist but can simply have their CK and muscle strength monitored.
CLASSIFYING MYOSITIS
Myositis (idiopathic inflammatory myopathy) is a heterogeneous group of autoimmune syndromes of unknown cause characterized by chronic muscle weakness and inflammation of striated muscle. These syndromes likely arise as a result of genetic predisposition and an environmental or infectious “hit.”
Myositis is rare, with an incidence of 5 to 10 cases per million per year and an estimated prevalence of 50 to 90 cases per million. It has 2 incidence peaks: 1 in childhood (age 5–15) and another in adult midlife (age 30–50). Women are affected 2 to 3 times more often than men, with black women most commonly affected.
Myositis is traditionally classified as follows:
- Adult polymyositis
- Adult dermatomyositis
- Juvenile myositis (dermatomyositis much more frequent than polymyositis)
- Malignancy-associated myositis (usually dermatomyositis)
- Myositis overlapping with another autoimmune disease
- Inclusion body myositis.
However, polymyositis is less common than we originally thought, and the term necrotizing myopathy is now used in many patients, as noted in the case studies below. Further, myositis overlap syndromes are being increasingly diagnosed, likely related to the emergence of autoantibodies and clinical “syndromes” associated with these autoantibody subsets (discussed in cases below).
Dermatomyositis
Dermatomyositis is characterized by muscle weakness and a rash that can be obvious or subtle. Classic skin lesions are Gottron papules, which are raised, flat-topped red or purplish lesions over the knuckles, elbows, or knees.
Lesions may be confused with those of psoriasis. There can also be a V-neck rash over the anterior chest or upper back (“shawl sign”) or a rash over the lateral thigh (“holster sign”). A facial rash may occur, but unlike lupus, dermatomyositis does not spare the nasolabial area. However, the V-neck rash can be similar to that seen in lupus.
Dermatomyositis may cause muscle pain, perhaps related to muscle ischemia, whereas polymyositis and necrotizing myopathy are often painless. However, pain is also associated with fibromyalgia, which may be seen in many autoimmune conditions. It is important not to overtreat rheumatologic diseases with immunosuppression to try to control pain if the pain is actually caused by fibromyalgia.
Polymyositis mimics
Hypothyroid myopathy can present as classic polymyositis. The serum CK may be elevated, and there may be myalgias, muscle hypertrophy with stiffness, weakness, cramps, and even features of a proximal myopathy, and rhabdomyolysis. The electromyogram can be normal or myopathic. Results of muscle biopsy are often normal but may show focal necrosis and mild inflammatory infiltrates, thus mimicking that seen with inflammatory myopathy.7
Drug-induced or toxic myopathies can also mimic polymyositis. Statins are among the most commonly prescribed drugs in the United States, with more than 35 million people taking them. Statins are generally well tolerated but have a broad spectrum of toxicity, ranging from myalgias to life-threatening rhabdomyolysis. Myalgias lead to about 5% to 10% of patients refusing to take a statin or stopping it on their own.
Myalgias affect up to 20% of statin users in clinical practice.8,9 A small cross-sectional study10 of 1,000 patients in a primary care setting found that the risk of muscle complaints in statin users was 1.5 times higher than in nonstatin users, similar to findings in other studies.
My strategy for managing a patient with possible statin-induced myopathy is illustrated in Figure 1.
CASE 3: WEAKNESS, VERY HIGH CK ON A STATIN
In March 2010, a 67-year-old woman presented with muscle weakness. She had a history of hypertension, hyperlipidemia, and, more than 10 years previously, uterine cancer. In 2004, she was given atorvastatin for dyslipidemia. Four years later, she developed lower-extremity weakness, which her doctor attributed to normal aging. A year after that, she found it difficult to walk up steps and lift her arms overhead. In June 2009, she stopped taking the atorvastatin on her own, but the weakness did not improve.
In September 2009, she returned to her doctor, who found her CK level was 6,473 U/L but believed it to be an error, so the test was repeated, with a result of 9,375 U/L. She had no rash or joint involvement.
She was admitted to the hospital and underwent muscle biopsy, which showed myonecrosis with no inflammation or vasculitis. She was treated with prednisone 60 mg/day, and her elevated CK level and weakness improved.
Immune-mediated necrotizing myopathy associated with statins
The hallmark of necrotizing myopathy is myonecrosis without significant inflammation.12 This pattern contrasts with that of polymyositis, which is characterized by lymphocytic inflammation.
Although statins became available in the United States in 1987, immune-mediated necrotizing myopathy associated with statins was first described only in 2010. In that report, Grable-Esposito et al13 described 25 patients from 2 neuromuscular centers seen between 2000 and 2008 who had elevated CK and proximal weakness during or after statin use, both of which persisted despite stopping the statin. Patients improved with immunosuppressive agents but had a relapse when steroids were stopped or tapered, a pattern typical in autoimmune disease.
Autoantibody defines subgroup of necrotizing myopathy
Also in 2010, Christopher-Stine et al14 reported an antibody associated with necrotizing myopathy. Of 38 patients with the condition, 16 were found to have an abnormal “doublet” autoantibody recognizing 200- and 100-kDa proteins. All patients had weakness and a high CK level, and 63% had statin exposure before the weakness (this percentage increased to 83% in patients older than 50). All responded to immunosuppressive therapy, and many had a relapse when it was withdrawn.
Statins lower cholesterol by inhibiting 3-hydroxy-3-methylglutaryl-Co A reductase (HMGCR), and paradoxically, they also upregulate it. HMGCR has a molecular weight of 97 kDa. Mammen et al15 identified HMGCR as the 100-kDa target of the identified antibody and developed an enzyme-linked immunosorbent assay for it. Of 750 patients presenting to one center, only 45 (6%) had anti-HMGCR autoantibodies, but all 16 patients who had the abnormal doublet antibody tested positive for anti-HMGCR. Regenerating muscle cells express high levels of HMGCR, which may sustain the immune response after statins are discontinued.
Case 3 continued: Intravenous immunoglobulin brings improvement
In March 2010, when the 67-year-old patient presented to our myositis center, her CK level was 5,800 U/L, which increased as prednisone was tapered. She still felt weak. On examination, her muscle strength findings were deltoids 4+/5, neck flexors 4/5, and iliopsoas 3+/5. She was treated with methotrexate and azathioprine without benefit. She was next treated with intravenous immunoglobulin, and after 3 months, her strength normalized for the first time in years. Her CK level decreased but did not normalize. Testing showed that she was positive for anti-HMGCR autoantibody, as this test had become commercially available.
In 2015, Mammen and Tiniakou16 suggested using intravenous immunoglobulin as first-line therapy for statin-associated autoimmune necrotizing myopathy, based on experience at a single center with 3 patients who declined glucocorticoid treatment.
Necrotizing myopathy: Bottom line
Myositis overlap syndromes
Heterogeneity is the rule in myositis, and it can present with a wide variety of signs and symptoms as outlined in Table 2.
CASE 4: FEVER, NEW ‘RHEUMATOID ARTHRITIS,’ AND LUNG DISEASE
A 52-year-old woman with knee osteoarthritis saw her primary care physician in November 2013 for dyspnea and low-grade fever. The next month, she presented with polyarthritis, muscle weakness, and Raynaud phenomenon.
In January 2014, she developed acrocyanosis of her fingers. Examination revealed hyperkeratotic, cracked areas of her fingers. Her oxygen saturation by pulse oximetry was low. She was admitted to the hospital. Her doctor suspected new onset of rheumatoid arthritis, but blood tests revealed a negative antinuclear antibody, so an autoimmune condition was deemed unlikely. Her CK was mildly elevated at 350 U/L.
Because of her dyspnea, an open-lung biopsy was performed. High-resolution computed tomography (CT) revealed infiltrates and ground-glass opacities, leading to the diagnosis of nonspecific interstitial pneumonia. A rheumatologist was consulted and recommended pulse methylprednisolone, followed by prednisone 60 mg/day and mycophenolate mofetil. Testing for Jo-1 antibodies was positive.
Antisynthetase syndrome
The antisynthetase syndrome is a clinically heterogeneous condition that can occur with any or all of the following:
- Fever
- Myositis
- Arthritis (often misdiagnosed as rheumatoid arthritis)
- Raynaud phenomenon
- Mechanic’s hands (hyperkeratotic roughness with fissures on the lateral aspects of the fingers and finger pads)
- Interstitial lung disease.
The skin rashes and myositis may be subtle, making the presentation “lung-dominant,” and nonrheumatologists should be aware of this syndrome. Although in our patient the condition developed in a classic manner, with all of the aforementioned features of the antisynthetase syndrome, some patients will manifest one or a few of the features.
Clinically, patients with the Jo-1 antisynthetase syndrome often present differently than those with non-Jo-1 antisynthetase autoantibodies. When we compared 122 patients with Jo-1 vs 80 patients with a non-Jo-1 antisynthetase autoantibody, patients with Jo-1 antibodies were more likely to have initially received a diagnosis of myositis (83%), while myositis was the original diagnosis in only 17% of those possessing non-Jo-1 antisynthetase autoantibodies. In fact, many patients (approximately 50%) were diagnosed as having undifferentiated connective tissue disease or an overlap syndrome, and 13% had scleroderma as their first diagnosis.17
We also found that the survival rate was higher in patients with Jo-1 syndrome compared with patients with non-Jo-1 antisynthetase syndromes. We attributed the difference in survival rates to a delayed diagnosis in the non-Jo-1 group, perhaps due to their “nonclassic” presentations of the antisynthetase syndrome, delaying appropriate treatment. Patients received a diagnosis of Jo-1 antibody syndrome after a mean of 0.4 year (range 0.2–0.8), while those with a non-Jo-1 antisynthetase autoantibody had a delay in diagnosis of 1.0 year (range 0.4–5.1) (P < .01).17
In nearly half the cases in this cohort, pulmonary fibrosis was the cause of death, with primary pulmonary hypertension being the second leading cause (11%).
Antisynthetase syndrome: Bottom line
Antisynthetase syndrome is an often fatal disease that does not always present in a typical fashion with symptoms of myositis, as lung disease may be the predominant feature. A negative antinuclear antibody test result does not imply antibody negativity, as the autoantigen in these diseases is not located in the nucleus. Prompt diagnosis and appropriate immunosuppressive therapy are critical to improving outcomes.
CASE 5: FEVER, UNDIAGNOSED LUNG DISEASE, NO MYOSITIS
In January 2001, a 39-year-old woman was admitted to the hospital after 5 weeks of fever (temperatures 103°–104°F) and myalgias. An extensive workup was negative except for low-titer antinuclear antibody and for mild basilar fibrosis noted on chest radiography. She left the hospital against medical advice because of frustration with a lack of a specific diagnosis (“fever of unknown origin”).
Two months later, at a follow-up rheumatology consult, she reported more myalgias and arthralgias, as well as fever. Chest radiography now showed pleural effusions. Her fingers had color changes consistent with Raynaud phenomenon. At that time, I diagnosed an undifferentiated connective tissue disease and told her that I suspected an autoimmune condition that would need time to reveal itself. In the meantime, I treated her empirically with prednisone.
In April, she returned, much more short of breath and with more prominent diffuse pulmonary infiltrates. Physical examination revealed subtle Gottron changes. Testing revealed poor pulmonary function: forced vital capacity (FVC) 56%, forced expiratory volume in 1 second (FEV1) 52%, and diffusing capacity for carbon monoxide (Dlco) 40%. Blood testing was positive for anti-PL-12 antibody, one of the non-Jo-1 antisynthetase antibodies. At this time, we treated her with glucocorticoids and tacrolimus.
More than 15 years later, this patient is doing well. Her skin rash, joint symptoms, and fever have not returned, and interestingly, she never developed myositis. Her Raynaud symptoms are mild. Her most recent pulmonary function test results (January 2018) were FVC 75%, FEV1 87%, and Dlco 78%. Although these results are not normal, they are much improved and allow her to be completely functional without supplemental oxygen. Echocardiography showed normal pulmonary artery systolic pressure (25 mm Hg). She was still taking tacrolimus and prednisone. When we tried to stop tacrolimus after she had done well for many years, her condition flared.
Non-Jo-1 antisynthetase syndrome: Bottom line
Patients with a non-Jo-1 antisynthetase syndrome often present without myositis symptoms and may never manifest myositis symptoms. Likely because of this presentation, diagnosis of a specific connective tissue disorder is delayed, perhaps leading to increased mortality risk from pulmonary disease. Chronic immunosuppression is often required for these autoimmune conditions.
CASE 6: DERMATOMYOSITIS, RAPIDLY PROGRESSIVE INTERSTITIAL LUNG DISEASE
A 58-year-old woman presented in the summer of 2012 with a photosensitive rash. The following January, she returned with polyarthritis, mild muscle weakness, and a dermatomyositis-pattern rash. Her CK level was normal, and her antinuclear antibody and Sjögren syndrome antibody test results were negative. She improved on low-dose prednisone and methotrexate.
She was originally referred to me in May of that year for worsening rash and mild weakness. She denied pulmonary symptoms, but examination revealed faint basilar crackles. I increased her prednisone dosage to 20 mg/day and started mycophenolate mofetil mainly for the mild cutaneous and myositis features. I also recommended high-resolution CT of the lungs and pulmonary function tests, which she underwent in early June. High-resolution CT showed nonspecific mild infiltrates with minimal ground-glass opacities.
On July 1, she presented to her local emergency department with severe shortness of breath, requiring oxygen 12 L/min. She had a palmar rash. Repeat high-resolution CT showed dramatic worsening compared with the scan the previous month. Because of continued inadequate oxygenation, she was transferred to our center. A blood test later was positive for antimelanoma differentiation-associated gene 5 (MDA-5) autoantibody, previously known as anticlinically amyopathic dermatomyositis (anti-CADM)-140 antibody (based on immunoprecipitation results).
She died on the third day after transfer, just 2 months after I had originally seen her, at which time she had had no pulmonary symptoms.
Clinically amyopathic dermatomyositis
Anti-CADM-140, first reported from Asia,18–20 is an autoantibody-associated disease but not an antisynthetase. It is associated with dermatomyositis; patients often have a “vasculopathy” with cutaneous ulcerations and palmar papules.
MDA-5 is a cytoplasmic protein that “senses” viral RNA and induces production of type 1 interferon. It is involved in the innate immune defense against viruses.
Anti-MDA-5 positivity is associated with a poor pulmonary outcome.21 In our cohort from the University of Pittsburgh, many patients died within 3 years, compared with about a 40% survival rate in patients with dermatomyositis who tested negative for this antibody. That being said, many patients with anti-MDA-5 do not develop rapidly progressive interstitial lung disease.
Autoimmune interstitial lung disease: Bottom line
Autoimmune interstitial lung disease is easy to miss, especially in the case of a non-Jo-1 syndrome, for 3 important reasons:
- The autoimmune features may initially be subtle (eg, Raynaud phenomena, mild dermatomyositis rash, undifferentiated connective tissue disease)
- Autoantibody testing is not often ordered, is not standardized, or may be unavailable
- Providers are mistakenly reassured that a patient who tests negative for antinuclear antibody does not have an autoimmune condition.
To emphasize the last point, in a cohort of 202 patients who tested positive for an antisynthetase antibody, only half were antinuclear antibody-positive, but nearly three-quarters demonstrated anticytoplasmic staining on indirect immunofluorescence (due to the location of the autoantigen in the cytoplasm), making the latter a better screening test for an antisynthetase antibody. For scleroderma, 99% were antinculear antibody-positive, but for myositis, this test is much less sensitive.22
- Felice KJ, North WA. Inclusion body myositis in Connecticut: observations in 35 patients during an 8-year period. Medicine (Baltimore) 2001; 80(5):320–327. doi:10.1097/00005792-200109000-00006
- Lev EI, Tur-Kaspa I, Ashkenazy I, et al. Distribution of serum creatine kinase activity in young healthy persons. Clin Chim Acta 1999; 279(1-2):107–115. doi:10.1016/S0009-8981(98)00180-6
- Lilleng H, Abeler K, Johnsen SH, et al. Variation of serum creatine kinase (CK) levels and prevalence of persistent hyperCKemia in a Norwegian normal population. The Tromsø Study. Neuromuscul Disord 2011; 21(7):494–500. doi:10.1016/j.nmd.2011.04.007
- Johnston JD, Lloyd M, Mathews JA, Hawthorne SW. Racial variation in serum creatine kinase levels. J R Soc Med 1996; 89(8):462-464. pmid:8795501
- Prelle A, Tancredi L, Sciacco M, et al. Retrospective study of a large population of patients with asymptomatic or minimally symptomatic raised serum creatine kinase levels. J Neurol 2002; 249(3):305–311. pmid:11993531
- D’Adda E, Sciacco M, Fruguglietti ME, et al. Follow-up of a large population of asymptomatic/oligosymptomatic hyperckemic subjects. J Neurol 2006; 253(11):1399–1403. doi:10.1007/s00415-006-0223-y
- Madariaga MG. Polymyositis-like syndrome in hypothyroidism: review of cases reported over the past twenty-five years. Thyroid 2002; 12(4):331–336. doi:10.1089/10507250252949478
- de Sauvage Nolting PR, Buirma RJ, Hutten BA, Kastelein JJ; Dutch ExPRESS Investigator Group. Two-year efficacy and safety of simvastatin 80 mg in familial hypercholesterolemia (the Examination of Probands and Relatives in Statin Studies With Familial Hypercholesterolemia [ExPRESS FH]). Am J Cardiol 2002; 90(2):181–184. doi:10.1016/s0002-9149(02)02449-9
- Bruckert E, Hayem G, Dejager S, Yau C, Bégaud B. Mild to moderate muscular symptoms with high-dosage statin therapy in hyperlipidemic patients--the PRIMO study. Cardiovasc Drugs Ther 2005; 19(6):403–414. doi:10.1007/s10557-005-5686-z
- Mosshammer D, Lorenz G, Meznaric S, Schwarz J, Muche R, Mörike K. Statin use and its association with musculoskeletal symptoms—a cross-sectional study in primary care settings. Fam Pract 2009; 26(2):88–95. doi:10.1093/fampra/cmp006
- Nichols GA, Koro CE. Does statin therapy initiation increase the risk for myopathy? An observational study of 32,225 diabetic and nondiabetic patients. Clin Ther 2007; 29(8):1761–1770. doi:10.1016/j.clinthera.2007.08.022
- Kassardjian CD, Lennon VA, Alfugham NB, Mahler M, Milone M. Clinical features and treatment outcomes of necrotizing autoimmune myopathy. JAMA Neurol 2015; 72(9):996–1003. doi:10.1001/jamaneurol.2015.1207
- Grable-Esposito P, Katzberg HD, Greenberg SA, Srinivasan J, Katz J, Amato AA. Immune-mediated necrotizing myopathy associated with statins. Muscle Nerve 2010; 41(2):185–190. doi:10.1002/mus.21486
- Christopher-Stine L, Casciola-Rosen LA, Hong G, Chung T, Corse AM, Mammen AL. A novel autoantibody recognizing 200-kd and 100-kd proteins is associated with an immune-mediated necrotizing myopathy. Arthritis Rheum 2010; 62(9):2757–2766. doi:10.1002/art.27572
- Mammen AL, Chung T, Christopher-Stine L, et al. Autoantibodies against 3-hydroxy-3-methylglutaryl-coenzyme A reductase in patients with statin-associated autoimmune myopathy. Arthritis Rheum 2011; 63(3):713–721. doi:10.1002/art.30156
- Mammen AL, Tiniakou E. Intravenous immune globulin for statin-triggered autoimmune myopathy. N Engl J Med 2015; 373(17):1680–1682. doi:10.1056/NEJMc1506163
- Aggarwal R, Cassidy E, Fertig N, et al. Patients with non-Jo-1 anti-tRNA-synthetase autoantibodies have worse survival than Jo-1 positive patients. Ann Rheum Dis 2014; 73(1):227–232. doi:10.1136/annrheumdis-2012-201800
- Sato S, Hirakata M, Kuwana M, et al. Autoantibodies to a 140-kd polypeptide, CADM-140, in Japanese patients with clinically amyopathic dermatomyositis. Arthritis Rheum 2005; 52(5):1571–1576. doi:10.1002/art.21023
- Sato S, Hoshino K, Satoh T, et al. RNA helicase encoded by melanoma differentiation-associated gene 5 is a major autoantigen in patients with clinically amyopathic dermatomyositis: association with rapidly progressive interstitial lung disease. Arthritis Rheum 2009; 60(7):2193–2200. doi:10.1002/art.24621
- Chen F, Wang D, Shu X, Nakashima R, Wang G. Anti-MDA5 antibody is associated with A/SIP and decreased T cells in peripheral blood and predicts poor prognosis of ILD in Chinese patients with dermatomyositis. Rheumatol Int 2012; 32(12):3909–3915. doi:10.1007/s00296-011-2323-y
- Moghadam-Kia S, Oddis CV, Sato S, Kuwana M, Aggarwal R. Anti-melanoma differentiation-associated gene 5 is associated with rapidly progressive lung disease and poor survival in US patients with amyopathic and myopathic dermatomyositis. Arthritis Care Res (Hoboken) 2016; 68(5):689–694. doi:10.1002/acr.22728
- Aggarwal R, Dhillon N, Fertig N, Koontz D, Qi Z, Oddis CV. A negative antinuclear antibody does not indicate autoantibody negativity in myositis: role of anticytoplasmic antibody as a screening test for antisynthetase syndrome. J Rheumatol 2017; 44(2):223–229. doi:10.3899/jrheum.160618
- Felice KJ, North WA. Inclusion body myositis in Connecticut: observations in 35 patients during an 8-year period. Medicine (Baltimore) 2001; 80(5):320–327. doi:10.1097/00005792-200109000-00006
- Lev EI, Tur-Kaspa I, Ashkenazy I, et al. Distribution of serum creatine kinase activity in young healthy persons. Clin Chim Acta 1999; 279(1-2):107–115. doi:10.1016/S0009-8981(98)00180-6
- Lilleng H, Abeler K, Johnsen SH, et al. Variation of serum creatine kinase (CK) levels and prevalence of persistent hyperCKemia in a Norwegian normal population. The Tromsø Study. Neuromuscul Disord 2011; 21(7):494–500. doi:10.1016/j.nmd.2011.04.007
- Johnston JD, Lloyd M, Mathews JA, Hawthorne SW. Racial variation in serum creatine kinase levels. J R Soc Med 1996; 89(8):462-464. pmid:8795501
- Prelle A, Tancredi L, Sciacco M, et al. Retrospective study of a large population of patients with asymptomatic or minimally symptomatic raised serum creatine kinase levels. J Neurol 2002; 249(3):305–311. pmid:11993531
- D’Adda E, Sciacco M, Fruguglietti ME, et al. Follow-up of a large population of asymptomatic/oligosymptomatic hyperckemic subjects. J Neurol 2006; 253(11):1399–1403. doi:10.1007/s00415-006-0223-y
- Madariaga MG. Polymyositis-like syndrome in hypothyroidism: review of cases reported over the past twenty-five years. Thyroid 2002; 12(4):331–336. doi:10.1089/10507250252949478
- de Sauvage Nolting PR, Buirma RJ, Hutten BA, Kastelein JJ; Dutch ExPRESS Investigator Group. Two-year efficacy and safety of simvastatin 80 mg in familial hypercholesterolemia (the Examination of Probands and Relatives in Statin Studies With Familial Hypercholesterolemia [ExPRESS FH]). Am J Cardiol 2002; 90(2):181–184. doi:10.1016/s0002-9149(02)02449-9
- Bruckert E, Hayem G, Dejager S, Yau C, Bégaud B. Mild to moderate muscular symptoms with high-dosage statin therapy in hyperlipidemic patients--the PRIMO study. Cardiovasc Drugs Ther 2005; 19(6):403–414. doi:10.1007/s10557-005-5686-z
- Mosshammer D, Lorenz G, Meznaric S, Schwarz J, Muche R, Mörike K. Statin use and its association with musculoskeletal symptoms—a cross-sectional study in primary care settings. Fam Pract 2009; 26(2):88–95. doi:10.1093/fampra/cmp006
- Nichols GA, Koro CE. Does statin therapy initiation increase the risk for myopathy? An observational study of 32,225 diabetic and nondiabetic patients. Clin Ther 2007; 29(8):1761–1770. doi:10.1016/j.clinthera.2007.08.022
- Kassardjian CD, Lennon VA, Alfugham NB, Mahler M, Milone M. Clinical features and treatment outcomes of necrotizing autoimmune myopathy. JAMA Neurol 2015; 72(9):996–1003. doi:10.1001/jamaneurol.2015.1207
- Grable-Esposito P, Katzberg HD, Greenberg SA, Srinivasan J, Katz J, Amato AA. Immune-mediated necrotizing myopathy associated with statins. Muscle Nerve 2010; 41(2):185–190. doi:10.1002/mus.21486
- Christopher-Stine L, Casciola-Rosen LA, Hong G, Chung T, Corse AM, Mammen AL. A novel autoantibody recognizing 200-kd and 100-kd proteins is associated with an immune-mediated necrotizing myopathy. Arthritis Rheum 2010; 62(9):2757–2766. doi:10.1002/art.27572
- Mammen AL, Chung T, Christopher-Stine L, et al. Autoantibodies against 3-hydroxy-3-methylglutaryl-coenzyme A reductase in patients with statin-associated autoimmune myopathy. Arthritis Rheum 2011; 63(3):713–721. doi:10.1002/art.30156
- Mammen AL, Tiniakou E. Intravenous immune globulin for statin-triggered autoimmune myopathy. N Engl J Med 2015; 373(17):1680–1682. doi:10.1056/NEJMc1506163
- Aggarwal R, Cassidy E, Fertig N, et al. Patients with non-Jo-1 anti-tRNA-synthetase autoantibodies have worse survival than Jo-1 positive patients. Ann Rheum Dis 2014; 73(1):227–232. doi:10.1136/annrheumdis-2012-201800
- Sato S, Hirakata M, Kuwana M, et al. Autoantibodies to a 140-kd polypeptide, CADM-140, in Japanese patients with clinically amyopathic dermatomyositis. Arthritis Rheum 2005; 52(5):1571–1576. doi:10.1002/art.21023
- Sato S, Hoshino K, Satoh T, et al. RNA helicase encoded by melanoma differentiation-associated gene 5 is a major autoantigen in patients with clinically amyopathic dermatomyositis: association with rapidly progressive interstitial lung disease. Arthritis Rheum 2009; 60(7):2193–2200. doi:10.1002/art.24621
- Chen F, Wang D, Shu X, Nakashima R, Wang G. Anti-MDA5 antibody is associated with A/SIP and decreased T cells in peripheral blood and predicts poor prognosis of ILD in Chinese patients with dermatomyositis. Rheumatol Int 2012; 32(12):3909–3915. doi:10.1007/s00296-011-2323-y
- Moghadam-Kia S, Oddis CV, Sato S, Kuwana M, Aggarwal R. Anti-melanoma differentiation-associated gene 5 is associated with rapidly progressive lung disease and poor survival in US patients with amyopathic and myopathic dermatomyositis. Arthritis Care Res (Hoboken) 2016; 68(5):689–694. doi:10.1002/acr.22728
- Aggarwal R, Dhillon N, Fertig N, Koontz D, Qi Z, Oddis CV. A negative antinuclear antibody does not indicate autoantibody negativity in myositis: role of anticytoplasmic antibody as a screening test for antisynthetase syndrome. J Rheumatol 2017; 44(2):223–229. doi:10.3899/jrheum.160618
KEY POINTS
- Inclusion body myositis affects older men more than women and is characterized by slowly progressive, asymmetric, distal and proximal weakness and atrophy.
- Statin-associated muscle complaints are common, whereas necrotizing myopathy, characterized by a very high CK plus weakness, is rare but must be recognized.
- Elevated CK does not necessarily indicate myositis, especially in African Americans or after heavy exercise.
- Dermatomyositis is characterized by muscle weakness and raised red or purple Gottron papules over the knuckles, elbows, or knees.
- Autoimmune interstitial lung disease may be caused by a variety of antibodies, the most common being anti-Jo-1 (directed against histidyl tRNA synthetase).
- The rarer non-Jo-1 antisynthetase autoantibodies may be associated with rapidly progressive interstitial lung disease, which is a challenge to recognize because associated rheumatologic symptoms may be minimal.
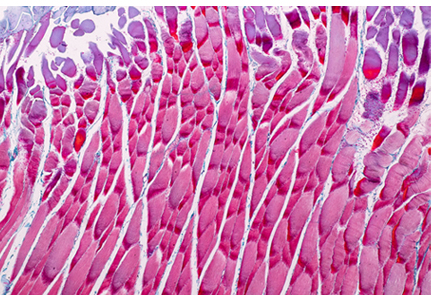
Approach to asymptomatic creatine kinase elevation
Measuring serum creatine kinase (CK) is an important part of the evaluation of patients with muscle weakness or myalgia, and of assessing patients with myopathies or rhabdomyolysis. But elevated CK sometimes is an incidental finding in a patient without muscle-related symptoms or with only minimal nonspecific muscle symptoms (eg, cramps, spasms, fatigue) that do not significantly interfere with activities of daily living. This condition is sometimes referred to as “asymptomatic hyper-CK-emia.” Four other muscle enzymes that may also be elevated are aspartate aminotransferase, alanine aminotransferase, lactate dehydrogenase, and aldolase.
This review focuses on the evaluation of patients with elevated CK without significant muscle-related symptoms and proposes an algorithm for this purpose (Figure 1).
CURRENT THRESHOLDS MAY BE LOW
What appears to be an elevated CK level may in fact be normal, and it is important to determine in the initial assessment whether a CK value is truly abnormal.

Most laboratories use the central 95% of observations in white people as a reference range for serum CK, assuming that levels have a gaussian (bell-shaped) distribution, which is usually about 0 to 200 IU/L. Using these parameters, an abnormal CK level was observed in 19% of men and 5% of women in a study of nearly 1,000 healthy young people,1 leading to overdiagnosis.
The actual distribution of serum CK levels in a healthy population is markedly skewed toward higher values and is nongaussian.1–3 A 97.5% normal threshold is associated with a much lower false-positive rate and is recommended by the European Federation of Neurological Societies (now the European Academy of Neurology).4 This group also recommends pursuing further investigation only for patients whose level is at least 1.5 times the upper limit of normal; this threshold results in only a small reduction in sensitivity.
CK levels vary significantly by sex and race.5 Possible reasons include differences in muscle mass or total body mass and inherited differences in the permeability of the sarcolemma to CK.6 There is also a small reduction in CK levels as people age.2
The European Federation of Neurological Societies suggests redefining elevated CK as values 1.5 times beyond the upper limit of normal. Based on a 97.5% threshold and normal values determined by Brewster et al3 for black and white men and women, the following thresholds can be used to help decide whether to pursue further evaluation4:
- White women—325 IU/L
- White men—504 IU/L
- Black women—621 IU/L
- Black men—1,200 IU/L
PHYSICAL ACTIVITY RAISES CK
CK levels transiently rise after exercise or heavy manual labor. Serum CK levels may increase to as much as 30 times the upper limit of normal within 24 hours of strenuous physical activity, then slowly decline over the next 7 days. The degree of CK elevation depends on the type and duration of exercise, with greater elevation in those who are untrained.2,4
In assessing asymptomatic or minimally symptomatic CK elevation, the test should be repeated after 7 days without exercise. A large community study in Norway found that repeat CK levels in people with incidentally discovered elevated CK were normal after 3 days of rest in 70% of cases.2
NONNEUROMUSCULAR CAUSES
NEED TO BE INVESTIGATED
Asymptomatic or minimally symptomatic elevated CK can be due to a primary neuromuscular disease or a variety of nonneuromuscular causes.

Patients who still have elevated CK after taking into account the 97.5% threshold, repeat testing after a week of rest, and a level more than 1.5 times the upper limit of normal for sex and race should first be evaluated for the many nonneuromuscular conditions that can cause elevated CK (Table 1).7–9
Cardiac causes should be evaluated by history and physical examination, electrocardiography, and possibly testing for cardiac troponins.
Drugs commonly elevate CK
Prescription drugs and supplements are an important and common cause of CK elevation, so it is important to carefully review medications the patient is taking.
Statins can cause myalgia, muscle weakness, and rhabdomyolysis. Up to 5% of users develop CK elevation, typically 2 to 10 times the upper limit of normal.10 CK usually drops after stopping statins but may require weeks to months to normalize. Rarely, statin users develop a serious immune-mediated necrotizing myopathy.11–13
The diversity of response to statin therapy appears to have a genetic basis. The SEARCH Collaborative Group14 conducted a genome-wide association study of 300,000 markers in 85 patients with definite or incipient myopathy and in 90 controls, all of whom were taking simvastatin 80 mg daily. They identified a single-nucleotide polymorphism in the SLCO1B1 gene on chromosome 12 that was strongly associated with a higher risk of statin-induced myopathy.
Patients with statin-related myopathy seem to have a higher frequency of occult metabolic muscle disease than the general population, also suggesting genetic susceptibility, although ascertainment bias could be a factor.14
Mechanisms of CK elevation in response to statins include increased muscle membrane fragility due to decreased cholesterol content, inhibition of isoprenoid production (a necessary step in the synthesis of membrane proteins), and depletion of ubiquinone, leading to mitochondrial dysfunction.
Macro CK: An abnormal enzyme complex
About 4% of patients with asymptomatic or minimally symptomatic elevated CK have “macro CK,” an enzyme complex with an atypically high molecular mass and reduced clearance, resulting in abnormally high blood levels of CK. Macro CK type 1 is more common and is found in up to 1.2% of the general population: complexes are composed of CK and immunoglobulin and are associated with autoimmune diseases.9,15 Macro CK type 2 complexes consist of CK and an undetermined protein and are associated with malignancies.
CK electrophoresis is required to detect macro CK. Types 1 and 2 can be distinguished by protein G affinity chromatography.9,15
Endocrine disorders
Muscle involvement in endocrine disorders often presents with muscle weakness in addition to muscle enzyme abnormalities.
Hypothyroidism often causes weakness, cramps, myalgia, and a mild to moderate serum CK elevation.16 Severe CK elevation has been reported to occur after vigorous exercise.17 Thyroid replacement usually results in normalization of serum CK levels in 1 to 2 months.18
Hyperthyroidism is typically associated with normal serum CK concentrations, but in rare cases it can cause rhabdomyolysis.19
NEUROMUSCULAR CAUSES ARE NOT ALWAYS WORTH PURSUING

Only after the nonneuromuscular causes of elevated CK have been ruled out should neuromuscular disorders be considered (Table 2). Evaluation with electromyography, nerve conduction studies, and muscle biopsy may lead to the diagnosis of a specific neuromuscular disorder: patients may be in the presymptomatic stage of disease and may or may not eventually develop muscle weakness or other symptoms.20,21
Is testing needed?
Most adult dystrophies and metabolic myopathies have no available treatment and their course is often benign, particularly if they present only with asymptomatic elevated CK. The value of a potentially extensive, expensive, and invasive evaluation for a specific neuromuscular cause should be weighed against the limited yield and treatment options. Moreover, specialized testing such as biochemical muscle enzyme analysis, sarcolemmal protein staining, and genetic testing are not available at all centers.
The European Federation of Neurological Societies guidelines recommend biopsy for patients with asymptomatic elevated CK who also have any of the following:
- Abnormal (myopathic) findings on electromyography
- CK more than three times the upper limit of normal
- Age less than 25
- Exercise intolerance.4
Idiopathic inflammatory myopathies rarely present with asymptomatic elevated CK.22–26 In one study,27 they were found in just 5% of patients with asymptomatic elevated CK.
Hypomyopathic dermatomyositis and inclusion body myositis can present with mild CK elevations with normal muscle strength, especially early in the disease course. A myositis subset of antisynthetase syndrome can present with mildly elevated CK and interstitial lung disease.27 Many of the inflammatory myopathies respond to treatment so are worth investigating.
In view of complexities in diagnosis of these conditions, one should proceed with testing only after discussing it with patients. Referral to a rheumatology specialist is preferred.
MUSCLE BIOPSY, ELECTROMYOGRAPHY, AND NERVE CONDUCTION STUDIES
Electromyography, nerve conduction studies, or muscle biopsy, or a combination of these tests, is usually needed to investigate neuromuscular causes of elevated CK.
Muscle biopsy abnormalities are found in about two-thirds of cases of asymptomatic elevated CK, but most abnormalities include nonspecific myopathic changes that are not diagnostic. A muscle biopsy that may include special stains for sarcolemmal proteins for muscular dystrophy and biochemical muscle enzyme analysis for metabolic myopathies is diagnostic in only 20% to 25% cases of asymptomatic elevated CK on average, with a variation between different series of 0% to 79%.7,21,27–33
Electromyography and nerve conduction studies alone add little to the workup of asymptomatic elevated CK apart from a modest negative predictive value and as a guide for muscle biopsy. For a very few neuromuscular disorders causing an elevated CK (eg, motor neuron disease, Charcot-Marie-Tooth disease, myotonic dystrophy), electromyography and nerve conduction studies could suffice to make the diagnosis.
Electromyography and nerve conduction studies detect abnormalities in nearly half of cases of asymptomatic CK elevation,7,21,27,28,30,31,33 but, as with biopsy, most changes are nonspecific. Although electromyography and nerve conduction studies can help distinguish primary neuropathic from myopathic disorders, the sensitivity and specificity are low for diagnosis. Normal studies do not rule out a condition, and abnormal studies are not diagnostic of a particular condition, although completely normal studies provide strong evidence against a severe neuromuscular disorder.
Combined testing
Using combined muscle biopsy, electromyography, and nerve conduction studies, the likelihood of making a diagnosis in patients with asymptomatic elevated CK is 28% on average (range of studies 4%–79%),2,7,21,26–28,30–32 and findings are nonspecific in 30% to 40% of cases. Findings are normal in about 30% to 40% of cases, which are thus diagnosed as idiopathic asymptomatic elevated CK.28–31,34
Prelle et al31 retrospectively reviewed the cases of 114 patients, ages 3 to 70, with incidentally discovered elevated CK and few or no symptoms, who underwent muscle biopsy, electromyography, and nerve conduction studies after nonneuromuscular causes were ruled out. Although muscle biopsy findings were abnormal in 39% of cases, a diagnosis was established in only 18% of cases after an extensive workup: the diagnosis was definitive in only 10% and included dystrophinopathies, metabolic myopathies, and rare noninflammatory myopathies. For the remaining 8%, the diagnosis was probable and included four cases of partial carnitine palmitoyl transferase deficiency, three cases of malignant hyperthermia, and two rare inherited disorders.
DNA testing
In women with a serum CK less than three times the upper limit of normal who have a family history of Duchenne or Becker muscular dystrophy, DNA analysis of blood lymphocytes identifies 70% of carriers.4
IDIOPATHIC ELEVATED SERUM CK
Rowland et al35 first coined the term “idiopathic hyper-CK-emia” and defined it as persistent elevation of serum CK despite a normal neurologic examination and testing, including electromyography, nerve conduction studies, and muscle biopsy.35,36 To receive this diagnosis, patients must also have no family history or clinical evidence of neuromuscular disease.
Idiopathic elevated serum CK is sometimes familial. In one study,37 elevated CK was found in family members of 13 of 28 unrelated probands. In the 13 families, 41 individuals had elevated CK. Genetic studies revealed that the condition is genetically heterogeneous and autosomal dominant in at least 60% of cases, with higher penetrance in men.
D’Adda et al26 followed 55 people with idiopathic elevated CK for 7 years. Ten percent were eventually diagnosed with a neuromuscular disorder, 10% developed malignancy, and the remaining 80% developed no new condition. The CK level normalized or decreased in many patients, but most continued to have persistent CK elevations with minimal or no symptoms.
- Lev EI, Tur-Kaspa I, Ashkenazy I, et al. Distribution of serum creatine kinase activity in young healthy persons. Clin Chim Acta 1999; 279:107–115.
- Lilleng H, Abeler K, Johnsen SH, et al. Variation of serum creatine kinase (CK) levels and prevalence of persistent hyperCKemia in a Norwegian normal population. The Tromsø Study. Neuromuscul Disord 2011; 21:494–500.
- Brewster LM, Mairuhu G, Sturk A, van Montfrans GA. Distribution of creatine kinase in the general population: implications for statin therapy. Am Heart J 2007; 154:655–661.
- Kyriakides T, Angelini C, Schaefer J, et al; European Federation of Neurological Societies. EFNS guidelines on the diagnostic approach to pauci- or asymptomatic hyperCKemia. Eur J Neurol 2010; 17:767–773.
- Prisant LM, Downton M, Watkins LO, et al. Efficacy and tolerability of lovastatin in 459 African-Americans with hypercholesterolemia. Am J Cardiol 1996; 78:420–444.
- Wong ET, Cobb C, Umehara MK, et al. Heterogeneity of serum creatine kinase activity among racial and gender groups of the population. Am J Clin Pathol 1983; 79:582–586.
- Brewster LM, de Visser M. Persistent hyperCKemia: fourteen patients studied in retrospect. Acta Neurol Scand 1988; 77:60–63.
- Weglinski MR, Wedel DJ, Engel AG. Malignant hyperthermia testing in patients with persistently increased serum creatine kinase levels. Anesth Analg 1997; 84:1038–1041.
- Galarraga B, Sinclair D, Fahie-Wilson MN, McCrae FC, Hull RG, Ledingham JM. A rare but important cause for a raised serum creatine kinase concentration: two case reports and a literature review. Rheumatology (Oxford) 2003; 42:186–188.
- Mancini GB, Tashakkor AY, Baker S, et al. Diagnosis, prevention, and management of statin adverse effects and intolerance: Canadian Working Group Consensus update. Can J Cardiol 2013; 29:1553–1568.
- Arora R, Liebo M, Maldonado F. Statin-induced myopathy: the two faces of Janus. J Cardiovasc Pharmacol Ther 2006; 11:105–112.
- Joy TR, Hegele RA. Narrative review: statin-related myopathy. Ann Intern Med 2009; 150:858–868.
- Talbert RL. Safety issues with statin therapy. J Am Pharm Assoc (2003) 2006; 46:479–490.
- SEARCH Collaborative Group; Link E, Parish S, Armitage J, et al. SLCO1B1 variants and statin-induced myopathy—a genomewide study. N Engl J Med 2008; 359:789–799.
- Wyness SP, Hunsaker JJ, La’ulu SL, Rao LV, Roberts WL. Detection of macro-creatine kinase and macroamylase by polyethylene glycol precipitation and ultrafiltration methods. Clin Chim Acta 2011; 412:2052–2057.
- Duyff RF, Van den Bosch J, Laman DM, van Loon BJ, Linssen WH. Neuromuscular findings in thyroid dysfunction: a prospective clinical and electrodiagnostic study. J Neurol Neurosurg Psychiatry 2000; 68:750–755.
- Riggs JE. Acute exertional rhabdomyolysis in hypothyroidism: the result of a reversible defect in glycogenolysis? Mil Med 1990; 155:171–172.
- Mastaglia FL, Ojeda VJ, Sarnat HB, Kakulas BA. Myopathies associated with hypothyroidism: a review based upon 13 cases. Aust N Z J Med 1988; 18:799–806.
- Alshanti M, Eledrisi MS, Jones E. Rhabdomyolysis associated with hyperthyroidism. Am J Emerg Med 2001; 19:317.
- Rosalki SB. Serum enzymes in disease of skeletal muscle. Clin Lab Med 1989; 9:767–781.
- Joy JL, Oh SJ. Asymptomatic hyper-CK-emia: an electrophysiologic and histopathologic study. Muscle Nerve 1989; 12:206–209.
- Merlini L, Sabatelli P, Columbaro M, et al. Hyper-CK-emia as the sole manifestation of myotonic dystrophy type 2. Muscle Nerve 2005; 31:764–767.
- Eeg-Olofsson O, Kalimo H, Eeg-Olofsson KE, et al. Duchenne muscular dystrophy and idiopathic hyperCKemia in the same family. Eur J Paediatr Neurol 2008; 12:404–407.
- Dwianingsih EK, Takeshima Y, Itoh K, et al. A Japanese child with asymptomatic elevation of serum creatine kinase shows PTRF-CAVIN mutation matching with congenital generalized lipodystrophy type 4. Mol Genet Metab 2010; 101:233–237.
- Carbone I, Bruno C, Sotgia F, et al. Mutation in the CAV3 gene causes partial caveolin-3 deficiency and hyperCKemia. Neurology 2000; 54:1373–1376.
- D’Adda E, Sciacco M, Fruguglietti ME, et al. Follow-up of a large population of asymptomatic/oligosymptomatic hyperckemic subjects. J Neurol 2006; 253:1399–1403.
- Fernandez C, de Paula AM, Figarella-Branger D, et al. Diagnostic evaluation of clinically normal subjects with chronic hyperCKemia. Neurology 2006; 66:1585–1587.
- Simmons Z, Peterlin BL, Boyer PJ, Towfighi J. Muscle biopsy in the evaluation of patients with modestly elevated creatine kinase levels. Muscle Nerve 2003; 27:242–244.
- Filosto M, Tonin P, Vattemi G, et al. The role of muscle biopsy in investigating isolated muscle pain. Neurology 2007; 68:181–186.
- Malandrini A, Orrico A, Gaudiano C, et al. Muscle biopsy and in vitro contracture test in subjects with idiopathic hyperCKemia. Anesthesiology 2008; 109:625–628.
- Prelle A, Tancredi L, Sciacco M, et al. Retrospective study of a large population of patients with asymptomatic or minimally symptomatic raised serum creatine kinase levels. J Neurol 2002; 249:305–311.
- Dabby R, Sadeh M, Herman O, et al. Asymptomatic or minimally symptomatic hyperCKemia: histopathologic correlates. Isr Med Assoc J 2006; 8:110–113.
- Reijneveld JC, Notermans NC, Linssen WH, Wokke JH. Benign prognosis in idiopathic hyper-CK-emia. Muscle Nerve 2000; 23:575–579.
- Restivo DA, Pavone V, Nicotra A. Single-fiber electromyography in hyperCKemia: the value of fiber density. Neurol Sci 2012; 33:819–824.
- Rowland LP, Willner J, Cerri C, DiMauro S, Miranda A. Approaches to the membrane theory of Duchenne muscular dystrophy. In: Angelini C, Danielli GA, Fontanari D, editors. Muscular Dystrophy Research: Advances and New Trends, Amsterdam: Excerpta Medica; 1980:3–13.
- Reijneveld JC, Notermans NC, Linssen WH, Bär PR, Wokke JH. Hyper-CK-aemia revisited. Neuromuscul Disord 2001; 11:163–164.
- Capasso M, De Angelis MV, Di Muzio A, et al. Familial idiopathic hyper-CK-emia: an underrecognized condition. Muscle Nerve 2006; 33:760–765.
Measuring serum creatine kinase (CK) is an important part of the evaluation of patients with muscle weakness or myalgia, and of assessing patients with myopathies or rhabdomyolysis. But elevated CK sometimes is an incidental finding in a patient without muscle-related symptoms or with only minimal nonspecific muscle symptoms (eg, cramps, spasms, fatigue) that do not significantly interfere with activities of daily living. This condition is sometimes referred to as “asymptomatic hyper-CK-emia.” Four other muscle enzymes that may also be elevated are aspartate aminotransferase, alanine aminotransferase, lactate dehydrogenase, and aldolase.
This review focuses on the evaluation of patients with elevated CK without significant muscle-related symptoms and proposes an algorithm for this purpose (Figure 1).
CURRENT THRESHOLDS MAY BE LOW
What appears to be an elevated CK level may in fact be normal, and it is important to determine in the initial assessment whether a CK value is truly abnormal.
Most laboratories use the central 95% of observations in white people as a reference range for serum CK, assuming that levels have a gaussian (bell-shaped) distribution, which is usually about 0 to 200 IU/L. Using these parameters, an abnormal CK level was observed in 19% of men and 5% of women in a study of nearly 1,000 healthy young people,1 leading to overdiagnosis.
The actual distribution of serum CK levels in a healthy population is markedly skewed toward higher values and is nongaussian.1–3 A 97.5% normal threshold is associated with a much lower false-positive rate and is recommended by the European Federation of Neurological Societies (now the European Academy of Neurology).4 This group also recommends pursuing further investigation only for patients whose level is at least 1.5 times the upper limit of normal; this threshold results in only a small reduction in sensitivity.
CK levels vary significantly by sex and race.5 Possible reasons include differences in muscle mass or total body mass and inherited differences in the permeability of the sarcolemma to CK.6 There is also a small reduction in CK levels as people age.2
The European Federation of Neurological Societies suggests redefining elevated CK as values 1.5 times beyond the upper limit of normal. Based on a 97.5% threshold and normal values determined by Brewster et al3 for black and white men and women, the following thresholds can be used to help decide whether to pursue further evaluation4:
- White women—325 IU/L
- White men—504 IU/L
- Black women—621 IU/L
- Black men—1,200 IU/L
PHYSICAL ACTIVITY RAISES CK
CK levels transiently rise after exercise or heavy manual labor. Serum CK levels may increase to as much as 30 times the upper limit of normal within 24 hours of strenuous physical activity, then slowly decline over the next 7 days. The degree of CK elevation depends on the type and duration of exercise, with greater elevation in those who are untrained.2,4
In assessing asymptomatic or minimally symptomatic CK elevation, the test should be repeated after 7 days without exercise. A large community study in Norway found that repeat CK levels in people with incidentally discovered elevated CK were normal after 3 days of rest in 70% of cases.2
NONNEUROMUSCULAR CAUSES
NEED TO BE INVESTIGATED
Asymptomatic or minimally symptomatic elevated CK can be due to a primary neuromuscular disease or a variety of nonneuromuscular causes.
Patients who still have elevated CK after taking into account the 97.5% threshold, repeat testing after a week of rest, and a level more than 1.5 times the upper limit of normal for sex and race should first be evaluated for the many nonneuromuscular conditions that can cause elevated CK (Table 1).7–9
Cardiac causes should be evaluated by history and physical examination, electrocardiography, and possibly testing for cardiac troponins.
Drugs commonly elevate CK
Prescription drugs and supplements are an important and common cause of CK elevation, so it is important to carefully review medications the patient is taking.
Statins can cause myalgia, muscle weakness, and rhabdomyolysis. Up to 5% of users develop CK elevation, typically 2 to 10 times the upper limit of normal.10 CK usually drops after stopping statins but may require weeks to months to normalize. Rarely, statin users develop a serious immune-mediated necrotizing myopathy.11–13
The diversity of response to statin therapy appears to have a genetic basis. The SEARCH Collaborative Group14 conducted a genome-wide association study of 300,000 markers in 85 patients with definite or incipient myopathy and in 90 controls, all of whom were taking simvastatin 80 mg daily. They identified a single-nucleotide polymorphism in the SLCO1B1 gene on chromosome 12 that was strongly associated with a higher risk of statin-induced myopathy.
Patients with statin-related myopathy seem to have a higher frequency of occult metabolic muscle disease than the general population, also suggesting genetic susceptibility, although ascertainment bias could be a factor.14
Mechanisms of CK elevation in response to statins include increased muscle membrane fragility due to decreased cholesterol content, inhibition of isoprenoid production (a necessary step in the synthesis of membrane proteins), and depletion of ubiquinone, leading to mitochondrial dysfunction.
Macro CK: An abnormal enzyme complex
About 4% of patients with asymptomatic or minimally symptomatic elevated CK have “macro CK,” an enzyme complex with an atypically high molecular mass and reduced clearance, resulting in abnormally high blood levels of CK. Macro CK type 1 is more common and is found in up to 1.2% of the general population: complexes are composed of CK and immunoglobulin and are associated with autoimmune diseases.9,15 Macro CK type 2 complexes consist of CK and an undetermined protein and are associated with malignancies.
CK electrophoresis is required to detect macro CK. Types 1 and 2 can be distinguished by protein G affinity chromatography.9,15
Endocrine disorders
Muscle involvement in endocrine disorders often presents with muscle weakness in addition to muscle enzyme abnormalities.
Hypothyroidism often causes weakness, cramps, myalgia, and a mild to moderate serum CK elevation.16 Severe CK elevation has been reported to occur after vigorous exercise.17 Thyroid replacement usually results in normalization of serum CK levels in 1 to 2 months.18
Hyperthyroidism is typically associated with normal serum CK concentrations, but in rare cases it can cause rhabdomyolysis.19
NEUROMUSCULAR CAUSES ARE NOT ALWAYS WORTH PURSUING
Only after the nonneuromuscular causes of elevated CK have been ruled out should neuromuscular disorders be considered (Table 2). Evaluation with electromyography, nerve conduction studies, and muscle biopsy may lead to the diagnosis of a specific neuromuscular disorder: patients may be in the presymptomatic stage of disease and may or may not eventually develop muscle weakness or other symptoms.20,21
Is testing needed?
Most adult dystrophies and metabolic myopathies have no available treatment and their course is often benign, particularly if they present only with asymptomatic elevated CK. The value of a potentially extensive, expensive, and invasive evaluation for a specific neuromuscular cause should be weighed against the limited yield and treatment options. Moreover, specialized testing such as biochemical muscle enzyme analysis, sarcolemmal protein staining, and genetic testing are not available at all centers.
The European Federation of Neurological Societies guidelines recommend biopsy for patients with asymptomatic elevated CK who also have any of the following:
- Abnormal (myopathic) findings on electromyography
- CK more than three times the upper limit of normal
- Age less than 25
- Exercise intolerance.4
Idiopathic inflammatory myopathies rarely present with asymptomatic elevated CK.22–26 In one study,27 they were found in just 5% of patients with asymptomatic elevated CK.
Hypomyopathic dermatomyositis and inclusion body myositis can present with mild CK elevations with normal muscle strength, especially early in the disease course. A myositis subset of antisynthetase syndrome can present with mildly elevated CK and interstitial lung disease.27 Many of the inflammatory myopathies respond to treatment so are worth investigating.
In view of complexities in diagnosis of these conditions, one should proceed with testing only after discussing it with patients. Referral to a rheumatology specialist is preferred.
MUSCLE BIOPSY, ELECTROMYOGRAPHY, AND NERVE CONDUCTION STUDIES
Electromyography, nerve conduction studies, or muscle biopsy, or a combination of these tests, is usually needed to investigate neuromuscular causes of elevated CK.
Muscle biopsy abnormalities are found in about two-thirds of cases of asymptomatic elevated CK, but most abnormalities include nonspecific myopathic changes that are not diagnostic. A muscle biopsy that may include special stains for sarcolemmal proteins for muscular dystrophy and biochemical muscle enzyme analysis for metabolic myopathies is diagnostic in only 20% to 25% cases of asymptomatic elevated CK on average, with a variation between different series of 0% to 79%.7,21,27–33
Electromyography and nerve conduction studies alone add little to the workup of asymptomatic elevated CK apart from a modest negative predictive value and as a guide for muscle biopsy. For a very few neuromuscular disorders causing an elevated CK (eg, motor neuron disease, Charcot-Marie-Tooth disease, myotonic dystrophy), electromyography and nerve conduction studies could suffice to make the diagnosis.
Electromyography and nerve conduction studies detect abnormalities in nearly half of cases of asymptomatic CK elevation,7,21,27,28,30,31,33 but, as with biopsy, most changes are nonspecific. Although electromyography and nerve conduction studies can help distinguish primary neuropathic from myopathic disorders, the sensitivity and specificity are low for diagnosis. Normal studies do not rule out a condition, and abnormal studies are not diagnostic of a particular condition, although completely normal studies provide strong evidence against a severe neuromuscular disorder.
Combined testing
Using combined muscle biopsy, electromyography, and nerve conduction studies, the likelihood of making a diagnosis in patients with asymptomatic elevated CK is 28% on average (range of studies 4%–79%),2,7,21,26–28,30–32 and findings are nonspecific in 30% to 40% of cases. Findings are normal in about 30% to 40% of cases, which are thus diagnosed as idiopathic asymptomatic elevated CK.28–31,34
Prelle et al31 retrospectively reviewed the cases of 114 patients, ages 3 to 70, with incidentally discovered elevated CK and few or no symptoms, who underwent muscle biopsy, electromyography, and nerve conduction studies after nonneuromuscular causes were ruled out. Although muscle biopsy findings were abnormal in 39% of cases, a diagnosis was established in only 18% of cases after an extensive workup: the diagnosis was definitive in only 10% and included dystrophinopathies, metabolic myopathies, and rare noninflammatory myopathies. For the remaining 8%, the diagnosis was probable and included four cases of partial carnitine palmitoyl transferase deficiency, three cases of malignant hyperthermia, and two rare inherited disorders.
DNA testing
In women with a serum CK less than three times the upper limit of normal who have a family history of Duchenne or Becker muscular dystrophy, DNA analysis of blood lymphocytes identifies 70% of carriers.4
IDIOPATHIC ELEVATED SERUM CK
Rowland et al35 first coined the term “idiopathic hyper-CK-emia” and defined it as persistent elevation of serum CK despite a normal neurologic examination and testing, including electromyography, nerve conduction studies, and muscle biopsy.35,36 To receive this diagnosis, patients must also have no family history or clinical evidence of neuromuscular disease.
Idiopathic elevated serum CK is sometimes familial. In one study,37 elevated CK was found in family members of 13 of 28 unrelated probands. In the 13 families, 41 individuals had elevated CK. Genetic studies revealed that the condition is genetically heterogeneous and autosomal dominant in at least 60% of cases, with higher penetrance in men.
D’Adda et al26 followed 55 people with idiopathic elevated CK for 7 years. Ten percent were eventually diagnosed with a neuromuscular disorder, 10% developed malignancy, and the remaining 80% developed no new condition. The CK level normalized or decreased in many patients, but most continued to have persistent CK elevations with minimal or no symptoms.
Measuring serum creatine kinase (CK) is an important part of the evaluation of patients with muscle weakness or myalgia, and of assessing patients with myopathies or rhabdomyolysis. But elevated CK sometimes is an incidental finding in a patient without muscle-related symptoms or with only minimal nonspecific muscle symptoms (eg, cramps, spasms, fatigue) that do not significantly interfere with activities of daily living. This condition is sometimes referred to as “asymptomatic hyper-CK-emia.” Four other muscle enzymes that may also be elevated are aspartate aminotransferase, alanine aminotransferase, lactate dehydrogenase, and aldolase.
This review focuses on the evaluation of patients with elevated CK without significant muscle-related symptoms and proposes an algorithm for this purpose (Figure 1).
CURRENT THRESHOLDS MAY BE LOW
What appears to be an elevated CK level may in fact be normal, and it is important to determine in the initial assessment whether a CK value is truly abnormal.
Most laboratories use the central 95% of observations in white people as a reference range for serum CK, assuming that levels have a gaussian (bell-shaped) distribution, which is usually about 0 to 200 IU/L. Using these parameters, an abnormal CK level was observed in 19% of men and 5% of women in a study of nearly 1,000 healthy young people,1 leading to overdiagnosis.
The actual distribution of serum CK levels in a healthy population is markedly skewed toward higher values and is nongaussian.1–3 A 97.5% normal threshold is associated with a much lower false-positive rate and is recommended by the European Federation of Neurological Societies (now the European Academy of Neurology).4 This group also recommends pursuing further investigation only for patients whose level is at least 1.5 times the upper limit of normal; this threshold results in only a small reduction in sensitivity.
CK levels vary significantly by sex and race.5 Possible reasons include differences in muscle mass or total body mass and inherited differences in the permeability of the sarcolemma to CK.6 There is also a small reduction in CK levels as people age.2
The European Federation of Neurological Societies suggests redefining elevated CK as values 1.5 times beyond the upper limit of normal. Based on a 97.5% threshold and normal values determined by Brewster et al3 for black and white men and women, the following thresholds can be used to help decide whether to pursue further evaluation4:
- White women—325 IU/L
- White men—504 IU/L
- Black women—621 IU/L
- Black men—1,200 IU/L
PHYSICAL ACTIVITY RAISES CK
CK levels transiently rise after exercise or heavy manual labor. Serum CK levels may increase to as much as 30 times the upper limit of normal within 24 hours of strenuous physical activity, then slowly decline over the next 7 days. The degree of CK elevation depends on the type and duration of exercise, with greater elevation in those who are untrained.2,4
In assessing asymptomatic or minimally symptomatic CK elevation, the test should be repeated after 7 days without exercise. A large community study in Norway found that repeat CK levels in people with incidentally discovered elevated CK were normal after 3 days of rest in 70% of cases.2
NONNEUROMUSCULAR CAUSES
NEED TO BE INVESTIGATED
Asymptomatic or minimally symptomatic elevated CK can be due to a primary neuromuscular disease or a variety of nonneuromuscular causes.
Patients who still have elevated CK after taking into account the 97.5% threshold, repeat testing after a week of rest, and a level more than 1.5 times the upper limit of normal for sex and race should first be evaluated for the many nonneuromuscular conditions that can cause elevated CK (Table 1).7–9
Cardiac causes should be evaluated by history and physical examination, electrocardiography, and possibly testing for cardiac troponins.
Drugs commonly elevate CK
Prescription drugs and supplements are an important and common cause of CK elevation, so it is important to carefully review medications the patient is taking.
Statins can cause myalgia, muscle weakness, and rhabdomyolysis. Up to 5% of users develop CK elevation, typically 2 to 10 times the upper limit of normal.10 CK usually drops after stopping statins but may require weeks to months to normalize. Rarely, statin users develop a serious immune-mediated necrotizing myopathy.11–13
The diversity of response to statin therapy appears to have a genetic basis. The SEARCH Collaborative Group14 conducted a genome-wide association study of 300,000 markers in 85 patients with definite or incipient myopathy and in 90 controls, all of whom were taking simvastatin 80 mg daily. They identified a single-nucleotide polymorphism in the SLCO1B1 gene on chromosome 12 that was strongly associated with a higher risk of statin-induced myopathy.
Patients with statin-related myopathy seem to have a higher frequency of occult metabolic muscle disease than the general population, also suggesting genetic susceptibility, although ascertainment bias could be a factor.14
Mechanisms of CK elevation in response to statins include increased muscle membrane fragility due to decreased cholesterol content, inhibition of isoprenoid production (a necessary step in the synthesis of membrane proteins), and depletion of ubiquinone, leading to mitochondrial dysfunction.
Macro CK: An abnormal enzyme complex
About 4% of patients with asymptomatic or minimally symptomatic elevated CK have “macro CK,” an enzyme complex with an atypically high molecular mass and reduced clearance, resulting in abnormally high blood levels of CK. Macro CK type 1 is more common and is found in up to 1.2% of the general population: complexes are composed of CK and immunoglobulin and are associated with autoimmune diseases.9,15 Macro CK type 2 complexes consist of CK and an undetermined protein and are associated with malignancies.
CK electrophoresis is required to detect macro CK. Types 1 and 2 can be distinguished by protein G affinity chromatography.9,15
Endocrine disorders
Muscle involvement in endocrine disorders often presents with muscle weakness in addition to muscle enzyme abnormalities.
Hypothyroidism often causes weakness, cramps, myalgia, and a mild to moderate serum CK elevation.16 Severe CK elevation has been reported to occur after vigorous exercise.17 Thyroid replacement usually results in normalization of serum CK levels in 1 to 2 months.18
Hyperthyroidism is typically associated with normal serum CK concentrations, but in rare cases it can cause rhabdomyolysis.19
NEUROMUSCULAR CAUSES ARE NOT ALWAYS WORTH PURSUING
Only after the nonneuromuscular causes of elevated CK have been ruled out should neuromuscular disorders be considered (Table 2). Evaluation with electromyography, nerve conduction studies, and muscle biopsy may lead to the diagnosis of a specific neuromuscular disorder: patients may be in the presymptomatic stage of disease and may or may not eventually develop muscle weakness or other symptoms.20,21
Is testing needed?
Most adult dystrophies and metabolic myopathies have no available treatment and their course is often benign, particularly if they present only with asymptomatic elevated CK. The value of a potentially extensive, expensive, and invasive evaluation for a specific neuromuscular cause should be weighed against the limited yield and treatment options. Moreover, specialized testing such as biochemical muscle enzyme analysis, sarcolemmal protein staining, and genetic testing are not available at all centers.
The European Federation of Neurological Societies guidelines recommend biopsy for patients with asymptomatic elevated CK who also have any of the following:
- Abnormal (myopathic) findings on electromyography
- CK more than three times the upper limit of normal
- Age less than 25
- Exercise intolerance.4
Idiopathic inflammatory myopathies rarely present with asymptomatic elevated CK.22–26 In one study,27 they were found in just 5% of patients with asymptomatic elevated CK.
Hypomyopathic dermatomyositis and inclusion body myositis can present with mild CK elevations with normal muscle strength, especially early in the disease course. A myositis subset of antisynthetase syndrome can present with mildly elevated CK and interstitial lung disease.27 Many of the inflammatory myopathies respond to treatment so are worth investigating.
In view of complexities in diagnosis of these conditions, one should proceed with testing only after discussing it with patients. Referral to a rheumatology specialist is preferred.
MUSCLE BIOPSY, ELECTROMYOGRAPHY, AND NERVE CONDUCTION STUDIES
Electromyography, nerve conduction studies, or muscle biopsy, or a combination of these tests, is usually needed to investigate neuromuscular causes of elevated CK.
Muscle biopsy abnormalities are found in about two-thirds of cases of asymptomatic elevated CK, but most abnormalities include nonspecific myopathic changes that are not diagnostic. A muscle biopsy that may include special stains for sarcolemmal proteins for muscular dystrophy and biochemical muscle enzyme analysis for metabolic myopathies is diagnostic in only 20% to 25% cases of asymptomatic elevated CK on average, with a variation between different series of 0% to 79%.7,21,27–33
Electromyography and nerve conduction studies alone add little to the workup of asymptomatic elevated CK apart from a modest negative predictive value and as a guide for muscle biopsy. For a very few neuromuscular disorders causing an elevated CK (eg, motor neuron disease, Charcot-Marie-Tooth disease, myotonic dystrophy), electromyography and nerve conduction studies could suffice to make the diagnosis.
Electromyography and nerve conduction studies detect abnormalities in nearly half of cases of asymptomatic CK elevation,7,21,27,28,30,31,33 but, as with biopsy, most changes are nonspecific. Although electromyography and nerve conduction studies can help distinguish primary neuropathic from myopathic disorders, the sensitivity and specificity are low for diagnosis. Normal studies do not rule out a condition, and abnormal studies are not diagnostic of a particular condition, although completely normal studies provide strong evidence against a severe neuromuscular disorder.
Combined testing
Using combined muscle biopsy, electromyography, and nerve conduction studies, the likelihood of making a diagnosis in patients with asymptomatic elevated CK is 28% on average (range of studies 4%–79%),2,7,21,26–28,30–32 and findings are nonspecific in 30% to 40% of cases. Findings are normal in about 30% to 40% of cases, which are thus diagnosed as idiopathic asymptomatic elevated CK.28–31,34
Prelle et al31 retrospectively reviewed the cases of 114 patients, ages 3 to 70, with incidentally discovered elevated CK and few or no symptoms, who underwent muscle biopsy, electromyography, and nerve conduction studies after nonneuromuscular causes were ruled out. Although muscle biopsy findings were abnormal in 39% of cases, a diagnosis was established in only 18% of cases after an extensive workup: the diagnosis was definitive in only 10% and included dystrophinopathies, metabolic myopathies, and rare noninflammatory myopathies. For the remaining 8%, the diagnosis was probable and included four cases of partial carnitine palmitoyl transferase deficiency, three cases of malignant hyperthermia, and two rare inherited disorders.
DNA testing
In women with a serum CK less than three times the upper limit of normal who have a family history of Duchenne or Becker muscular dystrophy, DNA analysis of blood lymphocytes identifies 70% of carriers.4
IDIOPATHIC ELEVATED SERUM CK
Rowland et al35 first coined the term “idiopathic hyper-CK-emia” and defined it as persistent elevation of serum CK despite a normal neurologic examination and testing, including electromyography, nerve conduction studies, and muscle biopsy.35,36 To receive this diagnosis, patients must also have no family history or clinical evidence of neuromuscular disease.
Idiopathic elevated serum CK is sometimes familial. In one study,37 elevated CK was found in family members of 13 of 28 unrelated probands. In the 13 families, 41 individuals had elevated CK. Genetic studies revealed that the condition is genetically heterogeneous and autosomal dominant in at least 60% of cases, with higher penetrance in men.
D’Adda et al26 followed 55 people with idiopathic elevated CK for 7 years. Ten percent were eventually diagnosed with a neuromuscular disorder, 10% developed malignancy, and the remaining 80% developed no new condition. The CK level normalized or decreased in many patients, but most continued to have persistent CK elevations with minimal or no symptoms.
- Lev EI, Tur-Kaspa I, Ashkenazy I, et al. Distribution of serum creatine kinase activity in young healthy persons. Clin Chim Acta 1999; 279:107–115.
- Lilleng H, Abeler K, Johnsen SH, et al. Variation of serum creatine kinase (CK) levels and prevalence of persistent hyperCKemia in a Norwegian normal population. The Tromsø Study. Neuromuscul Disord 2011; 21:494–500.
- Brewster LM, Mairuhu G, Sturk A, van Montfrans GA. Distribution of creatine kinase in the general population: implications for statin therapy. Am Heart J 2007; 154:655–661.
- Kyriakides T, Angelini C, Schaefer J, et al; European Federation of Neurological Societies. EFNS guidelines on the diagnostic approach to pauci- or asymptomatic hyperCKemia. Eur J Neurol 2010; 17:767–773.
- Prisant LM, Downton M, Watkins LO, et al. Efficacy and tolerability of lovastatin in 459 African-Americans with hypercholesterolemia. Am J Cardiol 1996; 78:420–444.
- Wong ET, Cobb C, Umehara MK, et al. Heterogeneity of serum creatine kinase activity among racial and gender groups of the population. Am J Clin Pathol 1983; 79:582–586.
- Brewster LM, de Visser M. Persistent hyperCKemia: fourteen patients studied in retrospect. Acta Neurol Scand 1988; 77:60–63.
- Weglinski MR, Wedel DJ, Engel AG. Malignant hyperthermia testing in patients with persistently increased serum creatine kinase levels. Anesth Analg 1997; 84:1038–1041.
- Galarraga B, Sinclair D, Fahie-Wilson MN, McCrae FC, Hull RG, Ledingham JM. A rare but important cause for a raised serum creatine kinase concentration: two case reports and a literature review. Rheumatology (Oxford) 2003; 42:186–188.
- Mancini GB, Tashakkor AY, Baker S, et al. Diagnosis, prevention, and management of statin adverse effects and intolerance: Canadian Working Group Consensus update. Can J Cardiol 2013; 29:1553–1568.
- Arora R, Liebo M, Maldonado F. Statin-induced myopathy: the two faces of Janus. J Cardiovasc Pharmacol Ther 2006; 11:105–112.
- Joy TR, Hegele RA. Narrative review: statin-related myopathy. Ann Intern Med 2009; 150:858–868.
- Talbert RL. Safety issues with statin therapy. J Am Pharm Assoc (2003) 2006; 46:479–490.
- SEARCH Collaborative Group; Link E, Parish S, Armitage J, et al. SLCO1B1 variants and statin-induced myopathy—a genomewide study. N Engl J Med 2008; 359:789–799.
- Wyness SP, Hunsaker JJ, La’ulu SL, Rao LV, Roberts WL. Detection of macro-creatine kinase and macroamylase by polyethylene glycol precipitation and ultrafiltration methods. Clin Chim Acta 2011; 412:2052–2057.
- Duyff RF, Van den Bosch J, Laman DM, van Loon BJ, Linssen WH. Neuromuscular findings in thyroid dysfunction: a prospective clinical and electrodiagnostic study. J Neurol Neurosurg Psychiatry 2000; 68:750–755.
- Riggs JE. Acute exertional rhabdomyolysis in hypothyroidism: the result of a reversible defect in glycogenolysis? Mil Med 1990; 155:171–172.
- Mastaglia FL, Ojeda VJ, Sarnat HB, Kakulas BA. Myopathies associated with hypothyroidism: a review based upon 13 cases. Aust N Z J Med 1988; 18:799–806.
- Alshanti M, Eledrisi MS, Jones E. Rhabdomyolysis associated with hyperthyroidism. Am J Emerg Med 2001; 19:317.
- Rosalki SB. Serum enzymes in disease of skeletal muscle. Clin Lab Med 1989; 9:767–781.
- Joy JL, Oh SJ. Asymptomatic hyper-CK-emia: an electrophysiologic and histopathologic study. Muscle Nerve 1989; 12:206–209.
- Merlini L, Sabatelli P, Columbaro M, et al. Hyper-CK-emia as the sole manifestation of myotonic dystrophy type 2. Muscle Nerve 2005; 31:764–767.
- Eeg-Olofsson O, Kalimo H, Eeg-Olofsson KE, et al. Duchenne muscular dystrophy and idiopathic hyperCKemia in the same family. Eur J Paediatr Neurol 2008; 12:404–407.
- Dwianingsih EK, Takeshima Y, Itoh K, et al. A Japanese child with asymptomatic elevation of serum creatine kinase shows PTRF-CAVIN mutation matching with congenital generalized lipodystrophy type 4. Mol Genet Metab 2010; 101:233–237.
- Carbone I, Bruno C, Sotgia F, et al. Mutation in the CAV3 gene causes partial caveolin-3 deficiency and hyperCKemia. Neurology 2000; 54:1373–1376.
- D’Adda E, Sciacco M, Fruguglietti ME, et al. Follow-up of a large population of asymptomatic/oligosymptomatic hyperckemic subjects. J Neurol 2006; 253:1399–1403.
- Fernandez C, de Paula AM, Figarella-Branger D, et al. Diagnostic evaluation of clinically normal subjects with chronic hyperCKemia. Neurology 2006; 66:1585–1587.
- Simmons Z, Peterlin BL, Boyer PJ, Towfighi J. Muscle biopsy in the evaluation of patients with modestly elevated creatine kinase levels. Muscle Nerve 2003; 27:242–244.
- Filosto M, Tonin P, Vattemi G, et al. The role of muscle biopsy in investigating isolated muscle pain. Neurology 2007; 68:181–186.
- Malandrini A, Orrico A, Gaudiano C, et al. Muscle biopsy and in vitro contracture test in subjects with idiopathic hyperCKemia. Anesthesiology 2008; 109:625–628.
- Prelle A, Tancredi L, Sciacco M, et al. Retrospective study of a large population of patients with asymptomatic or minimally symptomatic raised serum creatine kinase levels. J Neurol 2002; 249:305–311.
- Dabby R, Sadeh M, Herman O, et al. Asymptomatic or minimally symptomatic hyperCKemia: histopathologic correlates. Isr Med Assoc J 2006; 8:110–113.
- Reijneveld JC, Notermans NC, Linssen WH, Wokke JH. Benign prognosis in idiopathic hyper-CK-emia. Muscle Nerve 2000; 23:575–579.
- Restivo DA, Pavone V, Nicotra A. Single-fiber electromyography in hyperCKemia: the value of fiber density. Neurol Sci 2012; 33:819–824.
- Rowland LP, Willner J, Cerri C, DiMauro S, Miranda A. Approaches to the membrane theory of Duchenne muscular dystrophy. In: Angelini C, Danielli GA, Fontanari D, editors. Muscular Dystrophy Research: Advances and New Trends, Amsterdam: Excerpta Medica; 1980:3–13.
- Reijneveld JC, Notermans NC, Linssen WH, Bär PR, Wokke JH. Hyper-CK-aemia revisited. Neuromuscul Disord 2001; 11:163–164.
- Capasso M, De Angelis MV, Di Muzio A, et al. Familial idiopathic hyper-CK-emia: an underrecognized condition. Muscle Nerve 2006; 33:760–765.
- Lev EI, Tur-Kaspa I, Ashkenazy I, et al. Distribution of serum creatine kinase activity in young healthy persons. Clin Chim Acta 1999; 279:107–115.
- Lilleng H, Abeler K, Johnsen SH, et al. Variation of serum creatine kinase (CK) levels and prevalence of persistent hyperCKemia in a Norwegian normal population. The Tromsø Study. Neuromuscul Disord 2011; 21:494–500.
- Brewster LM, Mairuhu G, Sturk A, van Montfrans GA. Distribution of creatine kinase in the general population: implications for statin therapy. Am Heart J 2007; 154:655–661.
- Kyriakides T, Angelini C, Schaefer J, et al; European Federation of Neurological Societies. EFNS guidelines on the diagnostic approach to pauci- or asymptomatic hyperCKemia. Eur J Neurol 2010; 17:767–773.
- Prisant LM, Downton M, Watkins LO, et al. Efficacy and tolerability of lovastatin in 459 African-Americans with hypercholesterolemia. Am J Cardiol 1996; 78:420–444.
- Wong ET, Cobb C, Umehara MK, et al. Heterogeneity of serum creatine kinase activity among racial and gender groups of the population. Am J Clin Pathol 1983; 79:582–586.
- Brewster LM, de Visser M. Persistent hyperCKemia: fourteen patients studied in retrospect. Acta Neurol Scand 1988; 77:60–63.
- Weglinski MR, Wedel DJ, Engel AG. Malignant hyperthermia testing in patients with persistently increased serum creatine kinase levels. Anesth Analg 1997; 84:1038–1041.
- Galarraga B, Sinclair D, Fahie-Wilson MN, McCrae FC, Hull RG, Ledingham JM. A rare but important cause for a raised serum creatine kinase concentration: two case reports and a literature review. Rheumatology (Oxford) 2003; 42:186–188.
- Mancini GB, Tashakkor AY, Baker S, et al. Diagnosis, prevention, and management of statin adverse effects and intolerance: Canadian Working Group Consensus update. Can J Cardiol 2013; 29:1553–1568.
- Arora R, Liebo M, Maldonado F. Statin-induced myopathy: the two faces of Janus. J Cardiovasc Pharmacol Ther 2006; 11:105–112.
- Joy TR, Hegele RA. Narrative review: statin-related myopathy. Ann Intern Med 2009; 150:858–868.
- Talbert RL. Safety issues with statin therapy. J Am Pharm Assoc (2003) 2006; 46:479–490.
- SEARCH Collaborative Group; Link E, Parish S, Armitage J, et al. SLCO1B1 variants and statin-induced myopathy—a genomewide study. N Engl J Med 2008; 359:789–799.
- Wyness SP, Hunsaker JJ, La’ulu SL, Rao LV, Roberts WL. Detection of macro-creatine kinase and macroamylase by polyethylene glycol precipitation and ultrafiltration methods. Clin Chim Acta 2011; 412:2052–2057.
- Duyff RF, Van den Bosch J, Laman DM, van Loon BJ, Linssen WH. Neuromuscular findings in thyroid dysfunction: a prospective clinical and electrodiagnostic study. J Neurol Neurosurg Psychiatry 2000; 68:750–755.
- Riggs JE. Acute exertional rhabdomyolysis in hypothyroidism: the result of a reversible defect in glycogenolysis? Mil Med 1990; 155:171–172.
- Mastaglia FL, Ojeda VJ, Sarnat HB, Kakulas BA. Myopathies associated with hypothyroidism: a review based upon 13 cases. Aust N Z J Med 1988; 18:799–806.
- Alshanti M, Eledrisi MS, Jones E. Rhabdomyolysis associated with hyperthyroidism. Am J Emerg Med 2001; 19:317.
- Rosalki SB. Serum enzymes in disease of skeletal muscle. Clin Lab Med 1989; 9:767–781.
- Joy JL, Oh SJ. Asymptomatic hyper-CK-emia: an electrophysiologic and histopathologic study. Muscle Nerve 1989; 12:206–209.
- Merlini L, Sabatelli P, Columbaro M, et al. Hyper-CK-emia as the sole manifestation of myotonic dystrophy type 2. Muscle Nerve 2005; 31:764–767.
- Eeg-Olofsson O, Kalimo H, Eeg-Olofsson KE, et al. Duchenne muscular dystrophy and idiopathic hyperCKemia in the same family. Eur J Paediatr Neurol 2008; 12:404–407.
- Dwianingsih EK, Takeshima Y, Itoh K, et al. A Japanese child with asymptomatic elevation of serum creatine kinase shows PTRF-CAVIN mutation matching with congenital generalized lipodystrophy type 4. Mol Genet Metab 2010; 101:233–237.
- Carbone I, Bruno C, Sotgia F, et al. Mutation in the CAV3 gene causes partial caveolin-3 deficiency and hyperCKemia. Neurology 2000; 54:1373–1376.
- D’Adda E, Sciacco M, Fruguglietti ME, et al. Follow-up of a large population of asymptomatic/oligosymptomatic hyperckemic subjects. J Neurol 2006; 253:1399–1403.
- Fernandez C, de Paula AM, Figarella-Branger D, et al. Diagnostic evaluation of clinically normal subjects with chronic hyperCKemia. Neurology 2006; 66:1585–1587.
- Simmons Z, Peterlin BL, Boyer PJ, Towfighi J. Muscle biopsy in the evaluation of patients with modestly elevated creatine kinase levels. Muscle Nerve 2003; 27:242–244.
- Filosto M, Tonin P, Vattemi G, et al. The role of muscle biopsy in investigating isolated muscle pain. Neurology 2007; 68:181–186.
- Malandrini A, Orrico A, Gaudiano C, et al. Muscle biopsy and in vitro contracture test in subjects with idiopathic hyperCKemia. Anesthesiology 2008; 109:625–628.
- Prelle A, Tancredi L, Sciacco M, et al. Retrospective study of a large population of patients with asymptomatic or minimally symptomatic raised serum creatine kinase levels. J Neurol 2002; 249:305–311.
- Dabby R, Sadeh M, Herman O, et al. Asymptomatic or minimally symptomatic hyperCKemia: histopathologic correlates. Isr Med Assoc J 2006; 8:110–113.
- Reijneveld JC, Notermans NC, Linssen WH, Wokke JH. Benign prognosis in idiopathic hyper-CK-emia. Muscle Nerve 2000; 23:575–579.
- Restivo DA, Pavone V, Nicotra A. Single-fiber electromyography in hyperCKemia: the value of fiber density. Neurol Sci 2012; 33:819–824.
- Rowland LP, Willner J, Cerri C, DiMauro S, Miranda A. Approaches to the membrane theory of Duchenne muscular dystrophy. In: Angelini C, Danielli GA, Fontanari D, editors. Muscular Dystrophy Research: Advances and New Trends, Amsterdam: Excerpta Medica; 1980:3–13.
- Reijneveld JC, Notermans NC, Linssen WH, Bär PR, Wokke JH. Hyper-CK-aemia revisited. Neuromuscul Disord 2001; 11:163–164.
- Capasso M, De Angelis MV, Di Muzio A, et al. Familial idiopathic hyper-CK-emia: an underrecognized condition. Muscle Nerve 2006; 33:760–765.
KEY POINTS
- Standard reference ranges for serum CK levels used by most laboratories are too low and lead to overdiagnosis of abnormal values.
- Serum CK levels are strongly affected by race, sex, and physical activity.
- A patient with truly elevated levels should be evaluated for a variety of nonneuromuscular causes, including endocrine disorders, metabolic disturbances, drug effects, and malignancy.
- Neuromuscular causes should be investigated only after ruling out nonneuromuscular causes and after considering whether potential benefits of a diagnosis outweigh the risks and expense of extensive testing.