User login
Apixaban approved for preventing DVT and pulmonary embolism after hip/knee surgery
The oral anticoagulant apixaban has been approved for prophylaxis of deep vein thrombosis, which may lead to pulmonary embolism, in patients who have undergone hip or knee replacement surgery.
Approval was based on the results of three studies in more than 11,000 patients randomized to apixaban or enoxaparin, according to a March 14 statement issued by Bristol-Myers Squibb, which is developing and commercializing apixaban with Pfizer.
The studies, the ADVANCE-1, ADVANCE-2, and ADVANCE-3 studies, compared apixaban (2.5 mg twice a day starting after surgery) to enoxaparin (30 mg or 40 mg administered subcutaneously once a day starting before surgery) in 11,569 patients undergoing hip or knee replacement surgery.
Apixaban, marketed as Eliquis, is a selective Factor Xa inhibitor, approved in 2012 for reducing the risk of stroke and systemic embolism in patients with nonvalvular atrial fibrillation, at a recommended dose of 5 mg twice a day. The recommended dose for the new indication is 2.5 mg twice a day.
The apixaban prescribing information includes a boxed warning about the increased risk of spinal or epidural hematoma, "which may cause long-term or permanent paralysis" in patients treated with apixaban who are undergoing spinal epidural anesthesia or spinal puncture.
An application for approval of apixaban for the treatment of deep vein thrombosis and pulmonary embolism, and for the reduction in the risk of recurrent DVT and PE, is currently being reviewed at the Food and Drug Administration, according to BMS.
The oral anticoagulant apixaban has been approved for prophylaxis of deep vein thrombosis, which may lead to pulmonary embolism, in patients who have undergone hip or knee replacement surgery.
Approval was based on the results of three studies in more than 11,000 patients randomized to apixaban or enoxaparin, according to a March 14 statement issued by Bristol-Myers Squibb, which is developing and commercializing apixaban with Pfizer.
The studies, the ADVANCE-1, ADVANCE-2, and ADVANCE-3 studies, compared apixaban (2.5 mg twice a day starting after surgery) to enoxaparin (30 mg or 40 mg administered subcutaneously once a day starting before surgery) in 11,569 patients undergoing hip or knee replacement surgery.
Apixaban, marketed as Eliquis, is a selective Factor Xa inhibitor, approved in 2012 for reducing the risk of stroke and systemic embolism in patients with nonvalvular atrial fibrillation, at a recommended dose of 5 mg twice a day. The recommended dose for the new indication is 2.5 mg twice a day.
The apixaban prescribing information includes a boxed warning about the increased risk of spinal or epidural hematoma, "which may cause long-term or permanent paralysis" in patients treated with apixaban who are undergoing spinal epidural anesthesia or spinal puncture.
An application for approval of apixaban for the treatment of deep vein thrombosis and pulmonary embolism, and for the reduction in the risk of recurrent DVT and PE, is currently being reviewed at the Food and Drug Administration, according to BMS.
The oral anticoagulant apixaban has been approved for prophylaxis of deep vein thrombosis, which may lead to pulmonary embolism, in patients who have undergone hip or knee replacement surgery.
Approval was based on the results of three studies in more than 11,000 patients randomized to apixaban or enoxaparin, according to a March 14 statement issued by Bristol-Myers Squibb, which is developing and commercializing apixaban with Pfizer.
The studies, the ADVANCE-1, ADVANCE-2, and ADVANCE-3 studies, compared apixaban (2.5 mg twice a day starting after surgery) to enoxaparin (30 mg or 40 mg administered subcutaneously once a day starting before surgery) in 11,569 patients undergoing hip or knee replacement surgery.
Apixaban, marketed as Eliquis, is a selective Factor Xa inhibitor, approved in 2012 for reducing the risk of stroke and systemic embolism in patients with nonvalvular atrial fibrillation, at a recommended dose of 5 mg twice a day. The recommended dose for the new indication is 2.5 mg twice a day.
The apixaban prescribing information includes a boxed warning about the increased risk of spinal or epidural hematoma, "which may cause long-term or permanent paralysis" in patients treated with apixaban who are undergoing spinal epidural anesthesia or spinal puncture.
An application for approval of apixaban for the treatment of deep vein thrombosis and pulmonary embolism, and for the reduction in the risk of recurrent DVT and PE, is currently being reviewed at the Food and Drug Administration, according to BMS.
FDA Panel Backs Cobas HPV Test as Primary Screening Tool
COLLEGE PARK, MD. – A Food and Drug Administration advisory panel has unanimously supported expanding the approval of the cobas human papilloma virus test to include its use as a first-line primary cervical cancer screening test in women aged 25 years and older.
At a meeting on March 12, the FDA’s Microbiology Devices Advisory Committee voted 13-0 that the benefits of the cobas human papillomavirus (HPV) test, manufactured by Roche, outweighed the risks for use in women aged 25 years and older, for use as "a first-line primary cervical screening test to detect high-risk HPV, including genotyping for [HPV] 16 and 18." The proposed indication also states that women who test negative for high-risk HPV types "should be followed-up in accordance with the physician’s assessment of screening and medical history, other risk factors, and professional guidelines," and that women who test positive for HPV genotypes 16 and/or 18 "should be referred to colposcopy." Those who are positive for the other high-risk HPV types, but negative for 16/18 with this test, "should be evaluated by cervical cytology to determine the need for referral to colposcopy."
Despite some concerns about the age and the potential for confusion and off-label use of the test, panelists also voted 13-0 that there was reasonable assurance that the test was safe and effective for these proposed indications, with several panelists commenting on the task ahead for professional societies once the FDA approves the test for this use.
The cobas HPV test is an in vitro diagnostic test that detects 14 high-risk HPV types, including types 16 and 18, which are associated with about 70% of invasive cervical cancers. It was approved by the FDA in 2011 for screening women aged 21 years and older with abnormal cervical cytology results and for use adjunctively with normal cervical cytology in women aged 30 years and older to assess the presence or absence of high-risk HPV genotypes.
For approval, Roche submitted results of the ATHENA trial, a prospective study of 47,208 women in the United States, which was the basis of the 2011 approval, including 3-year follow-up data. The results show that HPV testing alone provides greater protection against CIN3 (cervical intraepithelial neoplasia grade 3) and invasive cervical cancer than does cytology alone, and that primary HPV testing provides a similar level of protection against CIN3 and invasive cervical cancer as cotesting with HPV testing and cytology, according to Roche.
Based on the proposed use, in women aged 25 years and older, the sensitivity of HPV testing for detecting CIN3 or higher was 58.26% vs. 42.63% for the comparator cytology alone, according to the FDA summary of the data. The positive predictive value for detecting CIN3 or greater among women referred to colposcopy with HPV testing was 12.25% vs. 6.47% for women referred to colposcopy with the cytology, a statistically significant difference. The risk of CIN3 or greater among women not referred to colposcopy with HPV testing was 0.42% vs. 0.59% among the women not referred to colposcopy with cytology. The false positive rate for CIN3 or greater was 4.09% among those in the HPV testing group, vs. 6.04% among those in the comparator.
Panelist Alan Waxman, professor in the department of obstetrics and gynecology, University of New Mexico, Albuquerque, commended the way the study was conducted, the results of which left the panel little choice on how to vote, and said that women will be "well served by having more choices." Clinicians will have different options for screening women, with potentially different intervals and triage trees, and the challenge will be for the professional societies "to put together data-driven, evidence-based algorithms," and then provide education for health care providers and patients, Dr. Waxman said.
"The balance of safety and effectiveness depends critically on the screening interval," added another panelist, Dr. L. Stewart Massad, professor of obstetrics and gynecology in the division of gynecologic oncology, Washington University, St. Louis. "This is a powerful test with a very strong negative predictive value, and the likelihood of disease will be substantially less than in the data presented today in women who are screened at too short an interval after an initial negative screen," which he said he hoped would be considered by professional societies in the development of guidelines.
About 1% of the women in the ATHENA study had been vaccinated against HPV, and panelists noted that as HPV vaccination increases, the effectiveness of screening would be lower because there will be less disease to detect. The oncologist on the panel, Dr. Joanna Cain, professor of obstetrics and gynecology at the University of Massachusetts, Worcester, said that patient registries could help understand how these proposals will need to modified over time as a result of HPV vaccination.
The FDA usually follows the recommendations of its advisory panels.
Advisory panel members have been cleared of potential conflicts related to the meeting topic. Occasionally, a panelist is given a waiver, but no waivers were given at this meeting.
COLLEGE PARK, MD. – A Food and Drug Administration advisory panel has unanimously supported expanding the approval of the cobas human papilloma virus test to include its use as a first-line primary cervical cancer screening test in women aged 25 years and older.
At a meeting on March 12, the FDA’s Microbiology Devices Advisory Committee voted 13-0 that the benefits of the cobas human papillomavirus (HPV) test, manufactured by Roche, outweighed the risks for use in women aged 25 years and older, for use as "a first-line primary cervical screening test to detect high-risk HPV, including genotyping for [HPV] 16 and 18." The proposed indication also states that women who test negative for high-risk HPV types "should be followed-up in accordance with the physician’s assessment of screening and medical history, other risk factors, and professional guidelines," and that women who test positive for HPV genotypes 16 and/or 18 "should be referred to colposcopy." Those who are positive for the other high-risk HPV types, but negative for 16/18 with this test, "should be evaluated by cervical cytology to determine the need for referral to colposcopy."
Despite some concerns about the age and the potential for confusion and off-label use of the test, panelists also voted 13-0 that there was reasonable assurance that the test was safe and effective for these proposed indications, with several panelists commenting on the task ahead for professional societies once the FDA approves the test for this use.
The cobas HPV test is an in vitro diagnostic test that detects 14 high-risk HPV types, including types 16 and 18, which are associated with about 70% of invasive cervical cancers. It was approved by the FDA in 2011 for screening women aged 21 years and older with abnormal cervical cytology results and for use adjunctively with normal cervical cytology in women aged 30 years and older to assess the presence or absence of high-risk HPV genotypes.
For approval, Roche submitted results of the ATHENA trial, a prospective study of 47,208 women in the United States, which was the basis of the 2011 approval, including 3-year follow-up data. The results show that HPV testing alone provides greater protection against CIN3 (cervical intraepithelial neoplasia grade 3) and invasive cervical cancer than does cytology alone, and that primary HPV testing provides a similar level of protection against CIN3 and invasive cervical cancer as cotesting with HPV testing and cytology, according to Roche.
Based on the proposed use, in women aged 25 years and older, the sensitivity of HPV testing for detecting CIN3 or higher was 58.26% vs. 42.63% for the comparator cytology alone, according to the FDA summary of the data. The positive predictive value for detecting CIN3 or greater among women referred to colposcopy with HPV testing was 12.25% vs. 6.47% for women referred to colposcopy with the cytology, a statistically significant difference. The risk of CIN3 or greater among women not referred to colposcopy with HPV testing was 0.42% vs. 0.59% among the women not referred to colposcopy with cytology. The false positive rate for CIN3 or greater was 4.09% among those in the HPV testing group, vs. 6.04% among those in the comparator.
Panelist Alan Waxman, professor in the department of obstetrics and gynecology, University of New Mexico, Albuquerque, commended the way the study was conducted, the results of which left the panel little choice on how to vote, and said that women will be "well served by having more choices." Clinicians will have different options for screening women, with potentially different intervals and triage trees, and the challenge will be for the professional societies "to put together data-driven, evidence-based algorithms," and then provide education for health care providers and patients, Dr. Waxman said.
"The balance of safety and effectiveness depends critically on the screening interval," added another panelist, Dr. L. Stewart Massad, professor of obstetrics and gynecology in the division of gynecologic oncology, Washington University, St. Louis. "This is a powerful test with a very strong negative predictive value, and the likelihood of disease will be substantially less than in the data presented today in women who are screened at too short an interval after an initial negative screen," which he said he hoped would be considered by professional societies in the development of guidelines.
About 1% of the women in the ATHENA study had been vaccinated against HPV, and panelists noted that as HPV vaccination increases, the effectiveness of screening would be lower because there will be less disease to detect. The oncologist on the panel, Dr. Joanna Cain, professor of obstetrics and gynecology at the University of Massachusetts, Worcester, said that patient registries could help understand how these proposals will need to modified over time as a result of HPV vaccination.
The FDA usually follows the recommendations of its advisory panels.
Advisory panel members have been cleared of potential conflicts related to the meeting topic. Occasionally, a panelist is given a waiver, but no waivers were given at this meeting.
COLLEGE PARK, MD. – A Food and Drug Administration advisory panel has unanimously supported expanding the approval of the cobas human papilloma virus test to include its use as a first-line primary cervical cancer screening test in women aged 25 years and older.
At a meeting on March 12, the FDA’s Microbiology Devices Advisory Committee voted 13-0 that the benefits of the cobas human papillomavirus (HPV) test, manufactured by Roche, outweighed the risks for use in women aged 25 years and older, for use as "a first-line primary cervical screening test to detect high-risk HPV, including genotyping for [HPV] 16 and 18." The proposed indication also states that women who test negative for high-risk HPV types "should be followed-up in accordance with the physician’s assessment of screening and medical history, other risk factors, and professional guidelines," and that women who test positive for HPV genotypes 16 and/or 18 "should be referred to colposcopy." Those who are positive for the other high-risk HPV types, but negative for 16/18 with this test, "should be evaluated by cervical cytology to determine the need for referral to colposcopy."
Despite some concerns about the age and the potential for confusion and off-label use of the test, panelists also voted 13-0 that there was reasonable assurance that the test was safe and effective for these proposed indications, with several panelists commenting on the task ahead for professional societies once the FDA approves the test for this use.
The cobas HPV test is an in vitro diagnostic test that detects 14 high-risk HPV types, including types 16 and 18, which are associated with about 70% of invasive cervical cancers. It was approved by the FDA in 2011 for screening women aged 21 years and older with abnormal cervical cytology results and for use adjunctively with normal cervical cytology in women aged 30 years and older to assess the presence or absence of high-risk HPV genotypes.
For approval, Roche submitted results of the ATHENA trial, a prospective study of 47,208 women in the United States, which was the basis of the 2011 approval, including 3-year follow-up data. The results show that HPV testing alone provides greater protection against CIN3 (cervical intraepithelial neoplasia grade 3) and invasive cervical cancer than does cytology alone, and that primary HPV testing provides a similar level of protection against CIN3 and invasive cervical cancer as cotesting with HPV testing and cytology, according to Roche.
Based on the proposed use, in women aged 25 years and older, the sensitivity of HPV testing for detecting CIN3 or higher was 58.26% vs. 42.63% for the comparator cytology alone, according to the FDA summary of the data. The positive predictive value for detecting CIN3 or greater among women referred to colposcopy with HPV testing was 12.25% vs. 6.47% for women referred to colposcopy with the cytology, a statistically significant difference. The risk of CIN3 or greater among women not referred to colposcopy with HPV testing was 0.42% vs. 0.59% among the women not referred to colposcopy with cytology. The false positive rate for CIN3 or greater was 4.09% among those in the HPV testing group, vs. 6.04% among those in the comparator.
Panelist Alan Waxman, professor in the department of obstetrics and gynecology, University of New Mexico, Albuquerque, commended the way the study was conducted, the results of which left the panel little choice on how to vote, and said that women will be "well served by having more choices." Clinicians will have different options for screening women, with potentially different intervals and triage trees, and the challenge will be for the professional societies "to put together data-driven, evidence-based algorithms," and then provide education for health care providers and patients, Dr. Waxman said.
"The balance of safety and effectiveness depends critically on the screening interval," added another panelist, Dr. L. Stewart Massad, professor of obstetrics and gynecology in the division of gynecologic oncology, Washington University, St. Louis. "This is a powerful test with a very strong negative predictive value, and the likelihood of disease will be substantially less than in the data presented today in women who are screened at too short an interval after an initial negative screen," which he said he hoped would be considered by professional societies in the development of guidelines.
About 1% of the women in the ATHENA study had been vaccinated against HPV, and panelists noted that as HPV vaccination increases, the effectiveness of screening would be lower because there will be less disease to detect. The oncologist on the panel, Dr. Joanna Cain, professor of obstetrics and gynecology at the University of Massachusetts, Worcester, said that patient registries could help understand how these proposals will need to modified over time as a result of HPV vaccination.
The FDA usually follows the recommendations of its advisory panels.
Advisory panel members have been cleared of potential conflicts related to the meeting topic. Occasionally, a panelist is given a waiver, but no waivers were given at this meeting.
AT AN FDA ADVISORY COMMITTEE MEETING
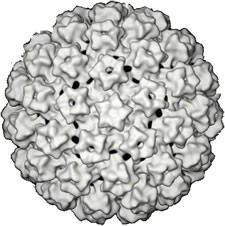
FDA panel unanimously backs cobas HPV test as primary screening tool
COLLEGE PARK, MD. – A Food and Drug Administration advisory panel has unanimously supported expanding the approval of the cobas human papilloma virus test to include its use as a first-line primary cervical cancer screening test in women aged 25 years and older.
At a meeting on March 12, the FDA’s Microbiology Devices Advisory Committee voted 13-0 that the benefits of the cobas human papillomavirus (HPV) test, manufactured by Roche, outweighed the risks for use in women aged 25 years and older, for use as "a first-line primary cervical screening test to detect high-risk HPV, including genotyping for [HPV] 16 and 18." The proposed indication also states that women who test negative for high-risk HPV types "should be followed-up in accordance with the physician’s assessment of screening and medical history, other risk factors, and professional guidelines," and that women who test positive for HPV genotypes 16 and/or 18 "should be referred to colposcopy." Those who are positive for the other high-risk HPV types, but negative for 16/18 with this test, "should be evaluated by cervical cytology to determine the need for referral to colposcopy."
Despite some concerns about the age and the potential for confusion and off-label use of the test, panelists also voted 13-0 that there was reasonable assurance that the test was safe and effective for these proposed indications, with several panelists commenting on the task ahead for professional societies once the FDA approves the test for this use.
The cobas HPV test is an in vitro diagnostic test that detects 14 high-risk HPV types, including types 16 and 18, which are associated with about 70% of invasive cervical cancers. It was approved by the FDA in 2011 for screening women aged 21 years and older with abnormal cervical cytology results and for use adjunctively with normal cervical cytology in women aged 30 years and older to assess the presence or absence of high-risk HPV genotypes.
For approval, Roche submitted results of the ATHENA trial, a prospective study of 47,208 women in the United States, which was the basis of the 2011 approval, including 3-year follow-up data. The results show that HPV testing alone provides greater protection against CIN3 (cervical intraepithelial neoplasia grade 3) and invasive cervical cancer than does cytology alone, and that primary HPV testing provides a similar level of protection against CIN3 and invasive cervical cancer as cotesting with HPV testing and cytology, according to Roche.
Based on the proposed use, in women aged 25 years and older, the sensitivity of HPV testing for detecting CIN3 or higher was 58.26% vs. 42.63% for the comparator cytology alone, according to the FDA summary of the data. The positive predictive value for detecting CIN3 or greater among women referred to colposcopy with HPV testing was 12.25% vs. 6.47% for women referred to colposcopy with the cytology, a statistically significant difference. The risk of CIN3 or greater among women not referred to colposcopy with HPV testing was 0.42% vs. 0.59% among the women not referred to colposcopy with cytology. The false positive rate for CIN3 or greater was 4.09% among those in the HPV testing group, vs. 6.04% among those in the comparator.
Panelist Alan Waxman, professor in the department of obstetrics and gynecology, University of New Mexico, Albuquerque, commended the way the study was conducted, the results of which left the panel little choice on how to vote, and said that women will be "well served by having more choices." Clinicians will have different options for screening women, with potentially different intervals and triage trees, and the challenge will be for the professional societies "to put together data-driven, evidence-based algorithms," and then provide education for health care providers and patients, Dr. Waxman said.
"The balance of safety and effectiveness depends critically on the screening interval," added another panelist, Dr. L. Stewart Massad, professor of obstetrics and gynecology in the division of gynecologic oncology, Washington University, St. Louis. "This is a powerful test with a very strong negative predictive value, and the likelihood of disease will be substantially less than in the data presented today in women who are screened at too short an interval after an initial negative screen," which he said he hoped would be considered by professional societies in the development of guidelines.
About 1% of the women in the ATHENA study had been vaccinated against HPV, and panelists noted that as HPV vaccination increases, the effectiveness of screening would be lower because there will be less disease to detect. The oncologist on the panel, Dr. Joanna Cain, professor of obstetrics and gynecology at the University of Massachusetts, Worcester, said that patient registries could help understand how these proposals will need to modified over time as a result of HPV vaccination.
The FDA usually follows the recommendations of its advisory panels.
Advisory panel members have been cleared of potential conflicts related to the meeting topic. Occasionally, a panelist is given a waiver, but no waivers were given at this meeting.
COLLEGE PARK, MD. – A Food and Drug Administration advisory panel has unanimously supported expanding the approval of the cobas human papilloma virus test to include its use as a first-line primary cervical cancer screening test in women aged 25 years and older.
At a meeting on March 12, the FDA’s Microbiology Devices Advisory Committee voted 13-0 that the benefits of the cobas human papillomavirus (HPV) test, manufactured by Roche, outweighed the risks for use in women aged 25 years and older, for use as "a first-line primary cervical screening test to detect high-risk HPV, including genotyping for [HPV] 16 and 18." The proposed indication also states that women who test negative for high-risk HPV types "should be followed-up in accordance with the physician’s assessment of screening and medical history, other risk factors, and professional guidelines," and that women who test positive for HPV genotypes 16 and/or 18 "should be referred to colposcopy." Those who are positive for the other high-risk HPV types, but negative for 16/18 with this test, "should be evaluated by cervical cytology to determine the need for referral to colposcopy."
Despite some concerns about the age and the potential for confusion and off-label use of the test, panelists also voted 13-0 that there was reasonable assurance that the test was safe and effective for these proposed indications, with several panelists commenting on the task ahead for professional societies once the FDA approves the test for this use.
The cobas HPV test is an in vitro diagnostic test that detects 14 high-risk HPV types, including types 16 and 18, which are associated with about 70% of invasive cervical cancers. It was approved by the FDA in 2011 for screening women aged 21 years and older with abnormal cervical cytology results and for use adjunctively with normal cervical cytology in women aged 30 years and older to assess the presence or absence of high-risk HPV genotypes.
For approval, Roche submitted results of the ATHENA trial, a prospective study of 47,208 women in the United States, which was the basis of the 2011 approval, including 3-year follow-up data. The results show that HPV testing alone provides greater protection against CIN3 (cervical intraepithelial neoplasia grade 3) and invasive cervical cancer than does cytology alone, and that primary HPV testing provides a similar level of protection against CIN3 and invasive cervical cancer as cotesting with HPV testing and cytology, according to Roche.
Based on the proposed use, in women aged 25 years and older, the sensitivity of HPV testing for detecting CIN3 or higher was 58.26% vs. 42.63% for the comparator cytology alone, according to the FDA summary of the data. The positive predictive value for detecting CIN3 or greater among women referred to colposcopy with HPV testing was 12.25% vs. 6.47% for women referred to colposcopy with the cytology, a statistically significant difference. The risk of CIN3 or greater among women not referred to colposcopy with HPV testing was 0.42% vs. 0.59% among the women not referred to colposcopy with cytology. The false positive rate for CIN3 or greater was 4.09% among those in the HPV testing group, vs. 6.04% among those in the comparator.
Panelist Alan Waxman, professor in the department of obstetrics and gynecology, University of New Mexico, Albuquerque, commended the way the study was conducted, the results of which left the panel little choice on how to vote, and said that women will be "well served by having more choices." Clinicians will have different options for screening women, with potentially different intervals and triage trees, and the challenge will be for the professional societies "to put together data-driven, evidence-based algorithms," and then provide education for health care providers and patients, Dr. Waxman said.
"The balance of safety and effectiveness depends critically on the screening interval," added another panelist, Dr. L. Stewart Massad, professor of obstetrics and gynecology in the division of gynecologic oncology, Washington University, St. Louis. "This is a powerful test with a very strong negative predictive value, and the likelihood of disease will be substantially less than in the data presented today in women who are screened at too short an interval after an initial negative screen," which he said he hoped would be considered by professional societies in the development of guidelines.
About 1% of the women in the ATHENA study had been vaccinated against HPV, and panelists noted that as HPV vaccination increases, the effectiveness of screening would be lower because there will be less disease to detect. The oncologist on the panel, Dr. Joanna Cain, professor of obstetrics and gynecology at the University of Massachusetts, Worcester, said that patient registries could help understand how these proposals will need to modified over time as a result of HPV vaccination.
The FDA usually follows the recommendations of its advisory panels.
Advisory panel members have been cleared of potential conflicts related to the meeting topic. Occasionally, a panelist is given a waiver, but no waivers were given at this meeting.
COLLEGE PARK, MD. – A Food and Drug Administration advisory panel has unanimously supported expanding the approval of the cobas human papilloma virus test to include its use as a first-line primary cervical cancer screening test in women aged 25 years and older.
At a meeting on March 12, the FDA’s Microbiology Devices Advisory Committee voted 13-0 that the benefits of the cobas human papillomavirus (HPV) test, manufactured by Roche, outweighed the risks for use in women aged 25 years and older, for use as "a first-line primary cervical screening test to detect high-risk HPV, including genotyping for [HPV] 16 and 18." The proposed indication also states that women who test negative for high-risk HPV types "should be followed-up in accordance with the physician’s assessment of screening and medical history, other risk factors, and professional guidelines," and that women who test positive for HPV genotypes 16 and/or 18 "should be referred to colposcopy." Those who are positive for the other high-risk HPV types, but negative for 16/18 with this test, "should be evaluated by cervical cytology to determine the need for referral to colposcopy."
Despite some concerns about the age and the potential for confusion and off-label use of the test, panelists also voted 13-0 that there was reasonable assurance that the test was safe and effective for these proposed indications, with several panelists commenting on the task ahead for professional societies once the FDA approves the test for this use.
The cobas HPV test is an in vitro diagnostic test that detects 14 high-risk HPV types, including types 16 and 18, which are associated with about 70% of invasive cervical cancers. It was approved by the FDA in 2011 for screening women aged 21 years and older with abnormal cervical cytology results and for use adjunctively with normal cervical cytology in women aged 30 years and older to assess the presence or absence of high-risk HPV genotypes.
For approval, Roche submitted results of the ATHENA trial, a prospective study of 47,208 women in the United States, which was the basis of the 2011 approval, including 3-year follow-up data. The results show that HPV testing alone provides greater protection against CIN3 (cervical intraepithelial neoplasia grade 3) and invasive cervical cancer than does cytology alone, and that primary HPV testing provides a similar level of protection against CIN3 and invasive cervical cancer as cotesting with HPV testing and cytology, according to Roche.
Based on the proposed use, in women aged 25 years and older, the sensitivity of HPV testing for detecting CIN3 or higher was 58.26% vs. 42.63% for the comparator cytology alone, according to the FDA summary of the data. The positive predictive value for detecting CIN3 or greater among women referred to colposcopy with HPV testing was 12.25% vs. 6.47% for women referred to colposcopy with the cytology, a statistically significant difference. The risk of CIN3 or greater among women not referred to colposcopy with HPV testing was 0.42% vs. 0.59% among the women not referred to colposcopy with cytology. The false positive rate for CIN3 or greater was 4.09% among those in the HPV testing group, vs. 6.04% among those in the comparator.
Panelist Alan Waxman, professor in the department of obstetrics and gynecology, University of New Mexico, Albuquerque, commended the way the study was conducted, the results of which left the panel little choice on how to vote, and said that women will be "well served by having more choices." Clinicians will have different options for screening women, with potentially different intervals and triage trees, and the challenge will be for the professional societies "to put together data-driven, evidence-based algorithms," and then provide education for health care providers and patients, Dr. Waxman said.
"The balance of safety and effectiveness depends critically on the screening interval," added another panelist, Dr. L. Stewart Massad, professor of obstetrics and gynecology in the division of gynecologic oncology, Washington University, St. Louis. "This is a powerful test with a very strong negative predictive value, and the likelihood of disease will be substantially less than in the data presented today in women who are screened at too short an interval after an initial negative screen," which he said he hoped would be considered by professional societies in the development of guidelines.
About 1% of the women in the ATHENA study had been vaccinated against HPV, and panelists noted that as HPV vaccination increases, the effectiveness of screening would be lower because there will be less disease to detect. The oncologist on the panel, Dr. Joanna Cain, professor of obstetrics and gynecology at the University of Massachusetts, Worcester, said that patient registries could help understand how these proposals will need to modified over time as a result of HPV vaccination.
The FDA usually follows the recommendations of its advisory panels.
Advisory panel members have been cleared of potential conflicts related to the meeting topic. Occasionally, a panelist is given a waiver, but no waivers were given at this meeting.
AT AN FDA ADVISORY COMMITTEE MEETING

Generic atorvastatin recall applies to 90-count bottles of 10-mg tablets
More than 64,000 bottles of 10-mg generic atorvastatin tablets have been recalled, according to a notice on the Food and Drug Administration website.
The nationwide recall applies to 64,626 bottles that contain 90 tablets, and are manufactured by Ranbaxy Pharmaceuticals. The discovery of a 20-mg atorvastatin tablet in a sealed 90-count bottle of atorvastatin 10-mg tablets was the basis of the recall.
The recall is voluntary and was initiated by Ranbaxy in January 2014.
The affected bottles are pharmacy dispensing bottles, a Ranbaxy spokesperson said.
Lipitor is the brand-name version of atorvastatin.
More than 64,000 bottles of 10-mg generic atorvastatin tablets have been recalled, according to a notice on the Food and Drug Administration website.
The nationwide recall applies to 64,626 bottles that contain 90 tablets, and are manufactured by Ranbaxy Pharmaceuticals. The discovery of a 20-mg atorvastatin tablet in a sealed 90-count bottle of atorvastatin 10-mg tablets was the basis of the recall.
The recall is voluntary and was initiated by Ranbaxy in January 2014.
The affected bottles are pharmacy dispensing bottles, a Ranbaxy spokesperson said.
Lipitor is the brand-name version of atorvastatin.
More than 64,000 bottles of 10-mg generic atorvastatin tablets have been recalled, according to a notice on the Food and Drug Administration website.
The nationwide recall applies to 64,626 bottles that contain 90 tablets, and are manufactured by Ranbaxy Pharmaceuticals. The discovery of a 20-mg atorvastatin tablet in a sealed 90-count bottle of atorvastatin 10-mg tablets was the basis of the recall.
The recall is voluntary and was initiated by Ranbaxy in January 2014.
The affected bottles are pharmacy dispensing bottles, a Ranbaxy spokesperson said.
Lipitor is the brand-name version of atorvastatin.
FDA issues warning about using doripenem for ventilator-associated pneumonia
When used to treat ventilator-associated pneumonia, the antibacterial drug doripenem is linked to a greater risk of mortality and lower clinical cure rates compared with imipenem and cilastatin for injection, according to a drug safety communication issued by the Food and Drug Administration.
Doripenem, an intravenous penem antibacterial drug marketed as Doribax, is not approved to treat any type of pneumonia. A warning about increased mortality in patients with ventilator-associated pneumonia (VAP), for which the use of doripenem is unapproved, has been added to the drug’s label. In a statement posted on March 6, the FDA recommended that health care professionals "consider whether the benefits of Doribax treatment are likely to exceed its potential risks in patients who develop pneumonia while on ventilators."
The safety communication is based on an FDA analysis of a 3-year study comparing 7 days of treatment with doripenem to 10 days of treatment with imipenem and cilastatin in patients with VAP. All-cause mortality over 28 days was 23% among those treated with doripenem, compared with 16.7% among those treated with imipenem and cilastatin. Clinical cure rates also were lower in the doripenem-treated patients in the study, which was stopped prematurely in 2011.
The statement adds that doripenem, approved in 2007, "is still considered safe and effective" for treating adults with complicated intra-abdominal infections and complicated urinary tract infections, including pyelonephritis, which are FDA-approved indications.
In January 2012, the FDA issued a statement about the prematurely terminated study.
The revised doripenem label is available on the FDA website. The label change was made in January. Doripenem is manufactured by Shionogi.
Imipenem and cilastatin for injection is marketed as Primaxin in the United States.
Serious adverse events associated with doripenem should be reported to the FDA’s MedWatch program at 800-332-1088 or www.fda.gov/medwatch/.
When used to treat ventilator-associated pneumonia, the antibacterial drug doripenem is linked to a greater risk of mortality and lower clinical cure rates compared with imipenem and cilastatin for injection, according to a drug safety communication issued by the Food and Drug Administration.
Doripenem, an intravenous penem antibacterial drug marketed as Doribax, is not approved to treat any type of pneumonia. A warning about increased mortality in patients with ventilator-associated pneumonia (VAP), for which the use of doripenem is unapproved, has been added to the drug’s label. In a statement posted on March 6, the FDA recommended that health care professionals "consider whether the benefits of Doribax treatment are likely to exceed its potential risks in patients who develop pneumonia while on ventilators."
The safety communication is based on an FDA analysis of a 3-year study comparing 7 days of treatment with doripenem to 10 days of treatment with imipenem and cilastatin in patients with VAP. All-cause mortality over 28 days was 23% among those treated with doripenem, compared with 16.7% among those treated with imipenem and cilastatin. Clinical cure rates also were lower in the doripenem-treated patients in the study, which was stopped prematurely in 2011.
The statement adds that doripenem, approved in 2007, "is still considered safe and effective" for treating adults with complicated intra-abdominal infections and complicated urinary tract infections, including pyelonephritis, which are FDA-approved indications.
In January 2012, the FDA issued a statement about the prematurely terminated study.
The revised doripenem label is available on the FDA website. The label change was made in January. Doripenem is manufactured by Shionogi.
Imipenem and cilastatin for injection is marketed as Primaxin in the United States.
Serious adverse events associated with doripenem should be reported to the FDA’s MedWatch program at 800-332-1088 or www.fda.gov/medwatch/.
When used to treat ventilator-associated pneumonia, the antibacterial drug doripenem is linked to a greater risk of mortality and lower clinical cure rates compared with imipenem and cilastatin for injection, according to a drug safety communication issued by the Food and Drug Administration.
Doripenem, an intravenous penem antibacterial drug marketed as Doribax, is not approved to treat any type of pneumonia. A warning about increased mortality in patients with ventilator-associated pneumonia (VAP), for which the use of doripenem is unapproved, has been added to the drug’s label. In a statement posted on March 6, the FDA recommended that health care professionals "consider whether the benefits of Doribax treatment are likely to exceed its potential risks in patients who develop pneumonia while on ventilators."
The safety communication is based on an FDA analysis of a 3-year study comparing 7 days of treatment with doripenem to 10 days of treatment with imipenem and cilastatin in patients with VAP. All-cause mortality over 28 days was 23% among those treated with doripenem, compared with 16.7% among those treated with imipenem and cilastatin. Clinical cure rates also were lower in the doripenem-treated patients in the study, which was stopped prematurely in 2011.
The statement adds that doripenem, approved in 2007, "is still considered safe and effective" for treating adults with complicated intra-abdominal infections and complicated urinary tract infections, including pyelonephritis, which are FDA-approved indications.
In January 2012, the FDA issued a statement about the prematurely terminated study.
The revised doripenem label is available on the FDA website. The label change was made in January. Doripenem is manufactured by Shionogi.
Imipenem and cilastatin for injection is marketed as Primaxin in the United States.
Serious adverse events associated with doripenem should be reported to the FDA’s MedWatch program at 800-332-1088 or www.fda.gov/medwatch/.
Injectable testosterone approved for hypogonadism, with REMS addressing risks
A long-acting intramuscular formulation of testosterone has been approved by the Food and Drug Administration for treating hypogonadism, with a label that includes a boxed warning about the risks of pulmonary oil microembolism and anaphylaxis associated with treatment.
The depot formulation of testosterone undecanoate (TU), in castor oil and benzoyl benzoate, has been approved for treating adult men with primary hypogonadism or hypogonadotropic hypogonadism, congenital or acquired. The indications section includes the statement that it "should only be used in patients who require testosterone replacement therapy and in whom the benefits of the product outweigh the serious risks of pulmonary oil microembolism and anaphylaxis."
The drug will be available, with restrictions, through the Aveed Risk Evaluation and Mitigation Strategy (REMS), according to the manufacturer, Endo Pharmaceuticals, which is marketing the product as Aveed. Under the REMS, prescriber education and certification will be required and distribution of the product will be restricted, according to the company.
The product is available in single-use vials; the recommended dosing is 3 mL (750 mg) at the start of treatment, at 4 weeks, and then at 10-week intervals. After each injection, patients are observed for symptoms of pulmonary oil microembolism (POME) or anaphylaxis for 30 minutes in the physician’s office, clinic, or hospital, the only places where the drug can be administered. Symptoms of serious POME reactions include an urge to cough, dyspnea, throat tightening, chest pain, dizziness, and syncope.
Since Endo filed for approval in 2007, approval of TU has been held up for safety reasons, namely reports of anaphylaxis and POME during or shortly after injections were administered, in clinical and postmarketing studies in countries where the product was approved. At a meeting in April 2013, the FDA’s Advisory Committee for Reproductive Health Drugs and the Drug Safety and Risk Management Advisory Committee split on the safety issue, voting 9-9 on whether they believed the drug had an acceptable safety profile for the proposed use.
Approval was based on the results of a phase III, 84-week, single-arm study of 130 hypogonadal men (mean age, 54 years), which determined that TU was an effective testosterone replacement therapy, based on serum testosterone levels. There was one case of a patient who had mild coughing after the third injection, which was later attributed to POME, according to the prescribing information.
But more cases have been reported after approval in countries outside the United States and in a review of 18 clinical studies; where possible cases of POME were adjudicated, of about 3,500 patients there were 9 cases of POME in 8 patients and 2 cases of anaphylaxis.
The product has been available since 2003 outside the United States, where it is marketed by Bayer Pharma and its subsidiaries.
The company is providing information about the REMS at www.AveedREMS.com and 855-755-0494.
Serious adverse events associated with testosterone undecanoate should be reported to the FDA’s MedWatch program at 800-332-1088 or www.fda.gov/medwatch.
A long-acting intramuscular formulation of testosterone has been approved by the Food and Drug Administration for treating hypogonadism, with a label that includes a boxed warning about the risks of pulmonary oil microembolism and anaphylaxis associated with treatment.
The depot formulation of testosterone undecanoate (TU), in castor oil and benzoyl benzoate, has been approved for treating adult men with primary hypogonadism or hypogonadotropic hypogonadism, congenital or acquired. The indications section includes the statement that it "should only be used in patients who require testosterone replacement therapy and in whom the benefits of the product outweigh the serious risks of pulmonary oil microembolism and anaphylaxis."
The drug will be available, with restrictions, through the Aveed Risk Evaluation and Mitigation Strategy (REMS), according to the manufacturer, Endo Pharmaceuticals, which is marketing the product as Aveed. Under the REMS, prescriber education and certification will be required and distribution of the product will be restricted, according to the company.
The product is available in single-use vials; the recommended dosing is 3 mL (750 mg) at the start of treatment, at 4 weeks, and then at 10-week intervals. After each injection, patients are observed for symptoms of pulmonary oil microembolism (POME) or anaphylaxis for 30 minutes in the physician’s office, clinic, or hospital, the only places where the drug can be administered. Symptoms of serious POME reactions include an urge to cough, dyspnea, throat tightening, chest pain, dizziness, and syncope.
Since Endo filed for approval in 2007, approval of TU has been held up for safety reasons, namely reports of anaphylaxis and POME during or shortly after injections were administered, in clinical and postmarketing studies in countries where the product was approved. At a meeting in April 2013, the FDA’s Advisory Committee for Reproductive Health Drugs and the Drug Safety and Risk Management Advisory Committee split on the safety issue, voting 9-9 on whether they believed the drug had an acceptable safety profile for the proposed use.
Approval was based on the results of a phase III, 84-week, single-arm study of 130 hypogonadal men (mean age, 54 years), which determined that TU was an effective testosterone replacement therapy, based on serum testosterone levels. There was one case of a patient who had mild coughing after the third injection, which was later attributed to POME, according to the prescribing information.
But more cases have been reported after approval in countries outside the United States and in a review of 18 clinical studies; where possible cases of POME were adjudicated, of about 3,500 patients there were 9 cases of POME in 8 patients and 2 cases of anaphylaxis.
The product has been available since 2003 outside the United States, where it is marketed by Bayer Pharma and its subsidiaries.
The company is providing information about the REMS at www.AveedREMS.com and 855-755-0494.
Serious adverse events associated with testosterone undecanoate should be reported to the FDA’s MedWatch program at 800-332-1088 or www.fda.gov/medwatch.
A long-acting intramuscular formulation of testosterone has been approved by the Food and Drug Administration for treating hypogonadism, with a label that includes a boxed warning about the risks of pulmonary oil microembolism and anaphylaxis associated with treatment.
The depot formulation of testosterone undecanoate (TU), in castor oil and benzoyl benzoate, has been approved for treating adult men with primary hypogonadism or hypogonadotropic hypogonadism, congenital or acquired. The indications section includes the statement that it "should only be used in patients who require testosterone replacement therapy and in whom the benefits of the product outweigh the serious risks of pulmonary oil microembolism and anaphylaxis."
The drug will be available, with restrictions, through the Aveed Risk Evaluation and Mitigation Strategy (REMS), according to the manufacturer, Endo Pharmaceuticals, which is marketing the product as Aveed. Under the REMS, prescriber education and certification will be required and distribution of the product will be restricted, according to the company.
The product is available in single-use vials; the recommended dosing is 3 mL (750 mg) at the start of treatment, at 4 weeks, and then at 10-week intervals. After each injection, patients are observed for symptoms of pulmonary oil microembolism (POME) or anaphylaxis for 30 minutes in the physician’s office, clinic, or hospital, the only places where the drug can be administered. Symptoms of serious POME reactions include an urge to cough, dyspnea, throat tightening, chest pain, dizziness, and syncope.
Since Endo filed for approval in 2007, approval of TU has been held up for safety reasons, namely reports of anaphylaxis and POME during or shortly after injections were administered, in clinical and postmarketing studies in countries where the product was approved. At a meeting in April 2013, the FDA’s Advisory Committee for Reproductive Health Drugs and the Drug Safety and Risk Management Advisory Committee split on the safety issue, voting 9-9 on whether they believed the drug had an acceptable safety profile for the proposed use.
Approval was based on the results of a phase III, 84-week, single-arm study of 130 hypogonadal men (mean age, 54 years), which determined that TU was an effective testosterone replacement therapy, based on serum testosterone levels. There was one case of a patient who had mild coughing after the third injection, which was later attributed to POME, according to the prescribing information.
But more cases have been reported after approval in countries outside the United States and in a review of 18 clinical studies; where possible cases of POME were adjudicated, of about 3,500 patients there were 9 cases of POME in 8 patients and 2 cases of anaphylaxis.
The product has been available since 2003 outside the United States, where it is marketed by Bayer Pharma and its subsidiaries.
The company is providing information about the REMS at www.AveedREMS.com and 855-755-0494.
Serious adverse events associated with testosterone undecanoate should be reported to the FDA’s MedWatch program at 800-332-1088 or www.fda.gov/medwatch.
Pharmacist discovery spurs recall of extended-release venlafaxine
Different lots of brand-name and generic versions of extended-release venlafaxine are being recalled because of a report that one bottle contained a capsule of the antiarrhythmic drug dofetilide, according to the Food and Drug Administration.
A statement posted March 7 on the agency’s MedWatch site said that Pfizer has issued a voluntary recall of one lot of 30-count venlafaxine 150-mg extended-release capsules (marketed as Effexor XR), one lot of 90-count Effexor XR 150-mg capsules, and one lot of 90-count Greenstone LLC brand of venlafaxine 150-mg extended release capsules.
The recall was spurred by a pharmacist’s report that a 0.25-mg capsule of dofetilide (Tikosyn) was found in a bottle of Effexor XR.
"The use of Tikosyn by an Effexor XR/Venlafaxine HCl patient, where the contraindications and drug-drug interactions with Tikosyn have not been considered by the prescribing physician, could cause serious adverse health consequences that could be fatal," the notice said.
Tikosyn, also manufactured by Pfizer, is a class III antiarrhythmic drug that is approved for treating atrial fibrillation/atrial flutter. The drug’s label includes a black box warning that recommends patients start treatment in a facility where they can be closely monitored, to minimize the risk of a dofetilide-induced arrhythmia. There is also a Risk Evaluation and Mitigation Strategy (REMS) in place that addresses this risk. The affected venlafaxine XR products are Pfizer lot numbers V130142 and V130140, which both expire in October 2015; and Greenstone lot number V130014, which expires in August 2015. Patients who have the affected product are being advised to call their physicians and/or return the product to their pharmacies. The FDA advises patients with questions to call Stericycle at 888-345-0481 (Mon.-Fri., 8 a.m. to 5 p.m., Eastern time); or Pfizer, at 800-438-1985 (Mon.-Thur., 9 a.m. to 8 p.m., Eastern time or Fri., 9 a.m. to 5 p.m., Eastern time).
Adverse events associated with the use of these products should be reported to MedWatch at or 800-332-1088.
Different lots of brand-name and generic versions of extended-release venlafaxine are being recalled because of a report that one bottle contained a capsule of the antiarrhythmic drug dofetilide, according to the Food and Drug Administration.
A statement posted March 7 on the agency’s MedWatch site said that Pfizer has issued a voluntary recall of one lot of 30-count venlafaxine 150-mg extended-release capsules (marketed as Effexor XR), one lot of 90-count Effexor XR 150-mg capsules, and one lot of 90-count Greenstone LLC brand of venlafaxine 150-mg extended release capsules.
The recall was spurred by a pharmacist’s report that a 0.25-mg capsule of dofetilide (Tikosyn) was found in a bottle of Effexor XR.
"The use of Tikosyn by an Effexor XR/Venlafaxine HCl patient, where the contraindications and drug-drug interactions with Tikosyn have not been considered by the prescribing physician, could cause serious adverse health consequences that could be fatal," the notice said.
Tikosyn, also manufactured by Pfizer, is a class III antiarrhythmic drug that is approved for treating atrial fibrillation/atrial flutter. The drug’s label includes a black box warning that recommends patients start treatment in a facility where they can be closely monitored, to minimize the risk of a dofetilide-induced arrhythmia. There is also a Risk Evaluation and Mitigation Strategy (REMS) in place that addresses this risk. The affected venlafaxine XR products are Pfizer lot numbers V130142 and V130140, which both expire in October 2015; and Greenstone lot number V130014, which expires in August 2015. Patients who have the affected product are being advised to call their physicians and/or return the product to their pharmacies. The FDA advises patients with questions to call Stericycle at 888-345-0481 (Mon.-Fri., 8 a.m. to 5 p.m., Eastern time); or Pfizer, at 800-438-1985 (Mon.-Thur., 9 a.m. to 8 p.m., Eastern time or Fri., 9 a.m. to 5 p.m., Eastern time).
Adverse events associated with the use of these products should be reported to MedWatch at or 800-332-1088.
Different lots of brand-name and generic versions of extended-release venlafaxine are being recalled because of a report that one bottle contained a capsule of the antiarrhythmic drug dofetilide, according to the Food and Drug Administration.
A statement posted March 7 on the agency’s MedWatch site said that Pfizer has issued a voluntary recall of one lot of 30-count venlafaxine 150-mg extended-release capsules (marketed as Effexor XR), one lot of 90-count Effexor XR 150-mg capsules, and one lot of 90-count Greenstone LLC brand of venlafaxine 150-mg extended release capsules.
The recall was spurred by a pharmacist’s report that a 0.25-mg capsule of dofetilide (Tikosyn) was found in a bottle of Effexor XR.
"The use of Tikosyn by an Effexor XR/Venlafaxine HCl patient, where the contraindications and drug-drug interactions with Tikosyn have not been considered by the prescribing physician, could cause serious adverse health consequences that could be fatal," the notice said.
Tikosyn, also manufactured by Pfizer, is a class III antiarrhythmic drug that is approved for treating atrial fibrillation/atrial flutter. The drug’s label includes a black box warning that recommends patients start treatment in a facility where they can be closely monitored, to minimize the risk of a dofetilide-induced arrhythmia. There is also a Risk Evaluation and Mitigation Strategy (REMS) in place that addresses this risk. The affected venlafaxine XR products are Pfizer lot numbers V130142 and V130140, which both expire in October 2015; and Greenstone lot number V130014, which expires in August 2015. Patients who have the affected product are being advised to call their physicians and/or return the product to their pharmacies. The FDA advises patients with questions to call Stericycle at 888-345-0481 (Mon.-Fri., 8 a.m. to 5 p.m., Eastern time); or Pfizer, at 800-438-1985 (Mon.-Thur., 9 a.m. to 8 p.m., Eastern time or Fri., 9 a.m. to 5 p.m., Eastern time).
Adverse events associated with the use of these products should be reported to MedWatch at or 800-332-1088.
Different age restrictions will apply to OTC emergency contraceptives
Generic versions of the emergency contraceptive Plan B One-Step will be allowed on pharmacy shelves, but unlike the brand-name version manufactured by Teva Pharmaceuticals, it will include an age restriction in its label.
The Food and Drug Administration is allowing generic versions of the 1.5 mg levonorgestrel pill to be available for women aged 17 years and older without point-of-sale restrictions, "but with labeling that carves out Teva’s protected uses," according to a Feb. 25 letter written by Dr. Kathleen Uhl, acting director of the FDA’s Office of Generic Drugs, to manufacturers of generic versions of Plan B One Step.
As a result, Plan B One-Step will be allowed on pharmacy shelves with no age restrictions. But purchasing generic versions will require an ID documenting the purchaser is aged 17 years or older.
This action is a result of the FDA’s decision to grant "exclusivity" to Teva to market the product with no age restrictions as a result of the company’s "actual use" study evaluating the ability of females aged 16 years and younger to correctly choose the product. The study was conducted as part of the company’s applications for approval of the one pill, over the counter (OTC) product with no age or point-of-sale restrictions. The FDA has determined that this study can be considered a new clinical study and qualifies for exclusivity, according to the letter. Exclusivity lasts for 3 years.
The FDA decided, however, that Teva did not have exclusivity "for all nonprescription use" of Plan B One-Step, "nor for its location on the retail shelf," the letter said.
In 2013, the FDA approved Plan B One-Step for OTC sale for all ages, with no point-of-sale restrictions, after years of litigation involving the FDA, reproductive rights groups, and the department of Health and Human Services.
Plan B One-Step generally costs about $40-$50, and generic versions generally cost about $35-$45, according to the Emergency Contraception information website, operated by Princeton (N.J.) University’s Office of Population Research and the Association of Reproductive Health Professionals.
Generic versions of the emergency contraceptive Plan B One-Step will be allowed on pharmacy shelves, but unlike the brand-name version manufactured by Teva Pharmaceuticals, it will include an age restriction in its label.
The Food and Drug Administration is allowing generic versions of the 1.5 mg levonorgestrel pill to be available for women aged 17 years and older without point-of-sale restrictions, "but with labeling that carves out Teva’s protected uses," according to a Feb. 25 letter written by Dr. Kathleen Uhl, acting director of the FDA’s Office of Generic Drugs, to manufacturers of generic versions of Plan B One Step.
As a result, Plan B One-Step will be allowed on pharmacy shelves with no age restrictions. But purchasing generic versions will require an ID documenting the purchaser is aged 17 years or older.
This action is a result of the FDA’s decision to grant "exclusivity" to Teva to market the product with no age restrictions as a result of the company’s "actual use" study evaluating the ability of females aged 16 years and younger to correctly choose the product. The study was conducted as part of the company’s applications for approval of the one pill, over the counter (OTC) product with no age or point-of-sale restrictions. The FDA has determined that this study can be considered a new clinical study and qualifies for exclusivity, according to the letter. Exclusivity lasts for 3 years.
The FDA decided, however, that Teva did not have exclusivity "for all nonprescription use" of Plan B One-Step, "nor for its location on the retail shelf," the letter said.
In 2013, the FDA approved Plan B One-Step for OTC sale for all ages, with no point-of-sale restrictions, after years of litigation involving the FDA, reproductive rights groups, and the department of Health and Human Services.
Plan B One-Step generally costs about $40-$50, and generic versions generally cost about $35-$45, according to the Emergency Contraception information website, operated by Princeton (N.J.) University’s Office of Population Research and the Association of Reproductive Health Professionals.
Generic versions of the emergency contraceptive Plan B One-Step will be allowed on pharmacy shelves, but unlike the brand-name version manufactured by Teva Pharmaceuticals, it will include an age restriction in its label.
The Food and Drug Administration is allowing generic versions of the 1.5 mg levonorgestrel pill to be available for women aged 17 years and older without point-of-sale restrictions, "but with labeling that carves out Teva’s protected uses," according to a Feb. 25 letter written by Dr. Kathleen Uhl, acting director of the FDA’s Office of Generic Drugs, to manufacturers of generic versions of Plan B One Step.
As a result, Plan B One-Step will be allowed on pharmacy shelves with no age restrictions. But purchasing generic versions will require an ID documenting the purchaser is aged 17 years or older.
This action is a result of the FDA’s decision to grant "exclusivity" to Teva to market the product with no age restrictions as a result of the company’s "actual use" study evaluating the ability of females aged 16 years and younger to correctly choose the product. The study was conducted as part of the company’s applications for approval of the one pill, over the counter (OTC) product with no age or point-of-sale restrictions. The FDA has determined that this study can be considered a new clinical study and qualifies for exclusivity, according to the letter. Exclusivity lasts for 3 years.
The FDA decided, however, that Teva did not have exclusivity "for all nonprescription use" of Plan B One-Step, "nor for its location on the retail shelf," the letter said.
In 2013, the FDA approved Plan B One-Step for OTC sale for all ages, with no point-of-sale restrictions, after years of litigation involving the FDA, reproductive rights groups, and the department of Health and Human Services.
Plan B One-Step generally costs about $40-$50, and generic versions generally cost about $35-$45, according to the Emergency Contraception information website, operated by Princeton (N.J.) University’s Office of Population Research and the Association of Reproductive Health Professionals.
FDA panel votes against OTC Primatene Mist replacement for asthma
The majority of a Food and Drug Administration advisory panel rejected approval of a newly formulated replacement for Primatene Mist as an over-the-counter treatment for asthma, at a recent meeting.
The FDA’s Nonprescription Drugs and Pulmonary-Allergy Drugs advisory committees voted 18 to 6 that the epinephrine metered-dose inhaler for the temporary relief of mild symptoms of intermittent asthma, as an OTC product, did not have a favorable risk-benefit profile.
On Feb. 25, the panel voted 14 to 10 that the efficacy data provided "substantial evidence" that treatment with the device was effective for the proposed indication, "the temporary relief of mild symptoms of intermittent asthma, including wheezing, tightness of chest, and shortness of breath," in adults and children aged 12 years and older. But in a 17 to 7 vote, the majority of the panel voted that safety had not been adequately demonstrated.
Armstrong Pharmaceuticals bought the rights to the Primatene Mist trademark from Wyeth and developed the new chlorofluorocarbon (CFC)-free version, which uses hydrofluoroalkane (HFA) as a propellant and has a proposed tradename of Primatene HFA. Primatene Mist, approved for treating asthma symptoms in 1967, was phased out as part of the Montreal Protocol on Substances that Deplete the Ozone Layer and taken off the market in 2011, because it used CFCs as a propellant. Primatene Mist delivered 200 mcg of epinephrine per actuation; Primatene HFA delivers 125 mcg per actuation. Epinephrine is a short-acting beta2-agonist bronchodilator.
Although the 12-week phase III study showed that treatment with the product showed statistically significant differences in the area under the curve (AUC) change in percent forced expiratory volume in 1 second (%FEV1) at week 12, over placebo, the primary endpoint, the FDA briefing documents said that concerns with the OTC availability of the product include the potential for overuse and off-label use, as well as the reliability of the device.
The agency sought the advisory panel’s input on the risk-benefit issue, noting that the public had raised concerns "both for and against the need for an OTC asthma treatment option," according to the FDA documents. "On the one hand, it has been stated that a quick-relief medication available OTC is needed for use in low-income, elderly, and uninsured individuals who might otherwise not have access to treatment or be able to see a health care practitioner. In contrast, there is also a concern that because asthma is a potentially life-threatening condition that should be diagnosed and treated by a health care professional, availability of an OTC bronchodilator product may discourage consumers from seeking appropriate care, resulting in worse asthma outcomes."
The FDA usually follows the recommendations of its advisory panels. There are no metered-dose inhaler bronchodilators currently available as OTC products in the United States.
The majority of a Food and Drug Administration advisory panel rejected approval of a newly formulated replacement for Primatene Mist as an over-the-counter treatment for asthma, at a recent meeting.
The FDA’s Nonprescription Drugs and Pulmonary-Allergy Drugs advisory committees voted 18 to 6 that the epinephrine metered-dose inhaler for the temporary relief of mild symptoms of intermittent asthma, as an OTC product, did not have a favorable risk-benefit profile.
On Feb. 25, the panel voted 14 to 10 that the efficacy data provided "substantial evidence" that treatment with the device was effective for the proposed indication, "the temporary relief of mild symptoms of intermittent asthma, including wheezing, tightness of chest, and shortness of breath," in adults and children aged 12 years and older. But in a 17 to 7 vote, the majority of the panel voted that safety had not been adequately demonstrated.
Armstrong Pharmaceuticals bought the rights to the Primatene Mist trademark from Wyeth and developed the new chlorofluorocarbon (CFC)-free version, which uses hydrofluoroalkane (HFA) as a propellant and has a proposed tradename of Primatene HFA. Primatene Mist, approved for treating asthma symptoms in 1967, was phased out as part of the Montreal Protocol on Substances that Deplete the Ozone Layer and taken off the market in 2011, because it used CFCs as a propellant. Primatene Mist delivered 200 mcg of epinephrine per actuation; Primatene HFA delivers 125 mcg per actuation. Epinephrine is a short-acting beta2-agonist bronchodilator.
Although the 12-week phase III study showed that treatment with the product showed statistically significant differences in the area under the curve (AUC) change in percent forced expiratory volume in 1 second (%FEV1) at week 12, over placebo, the primary endpoint, the FDA briefing documents said that concerns with the OTC availability of the product include the potential for overuse and off-label use, as well as the reliability of the device.
The agency sought the advisory panel’s input on the risk-benefit issue, noting that the public had raised concerns "both for and against the need for an OTC asthma treatment option," according to the FDA documents. "On the one hand, it has been stated that a quick-relief medication available OTC is needed for use in low-income, elderly, and uninsured individuals who might otherwise not have access to treatment or be able to see a health care practitioner. In contrast, there is also a concern that because asthma is a potentially life-threatening condition that should be diagnosed and treated by a health care professional, availability of an OTC bronchodilator product may discourage consumers from seeking appropriate care, resulting in worse asthma outcomes."
The FDA usually follows the recommendations of its advisory panels. There are no metered-dose inhaler bronchodilators currently available as OTC products in the United States.
The majority of a Food and Drug Administration advisory panel rejected approval of a newly formulated replacement for Primatene Mist as an over-the-counter treatment for asthma, at a recent meeting.
The FDA’s Nonprescription Drugs and Pulmonary-Allergy Drugs advisory committees voted 18 to 6 that the epinephrine metered-dose inhaler for the temporary relief of mild symptoms of intermittent asthma, as an OTC product, did not have a favorable risk-benefit profile.
On Feb. 25, the panel voted 14 to 10 that the efficacy data provided "substantial evidence" that treatment with the device was effective for the proposed indication, "the temporary relief of mild symptoms of intermittent asthma, including wheezing, tightness of chest, and shortness of breath," in adults and children aged 12 years and older. But in a 17 to 7 vote, the majority of the panel voted that safety had not been adequately demonstrated.
Armstrong Pharmaceuticals bought the rights to the Primatene Mist trademark from Wyeth and developed the new chlorofluorocarbon (CFC)-free version, which uses hydrofluoroalkane (HFA) as a propellant and has a proposed tradename of Primatene HFA. Primatene Mist, approved for treating asthma symptoms in 1967, was phased out as part of the Montreal Protocol on Substances that Deplete the Ozone Layer and taken off the market in 2011, because it used CFCs as a propellant. Primatene Mist delivered 200 mcg of epinephrine per actuation; Primatene HFA delivers 125 mcg per actuation. Epinephrine is a short-acting beta2-agonist bronchodilator.
Although the 12-week phase III study showed that treatment with the product showed statistically significant differences in the area under the curve (AUC) change in percent forced expiratory volume in 1 second (%FEV1) at week 12, over placebo, the primary endpoint, the FDA briefing documents said that concerns with the OTC availability of the product include the potential for overuse and off-label use, as well as the reliability of the device.
The agency sought the advisory panel’s input on the risk-benefit issue, noting that the public had raised concerns "both for and against the need for an OTC asthma treatment option," according to the FDA documents. "On the one hand, it has been stated that a quick-relief medication available OTC is needed for use in low-income, elderly, and uninsured individuals who might otherwise not have access to treatment or be able to see a health care practitioner. In contrast, there is also a concern that because asthma is a potentially life-threatening condition that should be diagnosed and treated by a health care professional, availability of an OTC bronchodilator product may discourage consumers from seeking appropriate care, resulting in worse asthma outcomes."
The FDA usually follows the recommendations of its advisory panels. There are no metered-dose inhaler bronchodilators currently available as OTC products in the United States.
FROM AN FDA ADVISORY COMMITTEE MEETING
FDA vaccines panel: Retain current strains in next season’s influenza vaccines
SILVER SPRING, MD. – The trivalent and quadrivalent influenza vaccines for the 2014-2015 influenza season in the United States should retain the same strains that are included in the current season’s vaccines, according to a Food and Drug Administration expert panel.
At a meeting, the FDA’s Vaccines and Related Biological Products Advisory Committee voted 16 to 0 that the 2014-2015 trivalent influenza vaccine should retain the three strains included in the current vaccine: an A/California/7/2009 (H1N1)-like virus and an A/Texas/50/2012 (H3N2)-like virus, the two influenza A strains; and a B/Massachusetts/2/2012-like virus, a B/Yamagata lineage strain, as the influenza B strain.
For the quadrivalent vaccine, the panel unanimously recommended that the B/Victoria lineage strain contained in the current quadrivalent vaccine, a B/Brisbane/60/2008-like virus, be retained as the second influenza B strain.
The 2014-15 season will be the second season that quadrivalent influenza vaccines – which contain both a B/Victoria lineage and a B/Yamagata lineage vaccine virus strain – will be available.
During the current season, the quadrivalent vaccine was widely available, according to Sam Lee, Ph.D., senior director of pandemic influenza strategy at Sanofi Pasteur. Dr. Lee, who represented the vaccine manufacturers at the meeting, said that through January 2014, about 134 million doses of influenza vaccine were distributed in the United States, about the same as during the 2012-2013 season. The quadrivalent vaccine accounted for about 20% of the total vaccine doses distributed, and is expected to account for more than half of the doses distributed during next season, he said.
So far, influenza vaccine manufacturers do not anticipate any problems with the availability of the strains to be included in the vaccines, Dr. Lee told the panel.
Providing an update on influenza activity during the current season, Dr. Lisa Grohskopf, of the influenza division in the Centers for Disease Control & Prevention’s National Center for Immunization and Respiratory Diseases, said that influenza activity peaked in late December and early January and that influenza A (H1N1) viruses have predominated. As of Feb. 15, there have been 52 influenza-associated pediatric deaths reported, which is less than the number reported last season (171) but more than during the 2011-2012 influenza season (35), she noted.
The FDA panel meets at the same time each year to review influenza surveillance and epidemiology data in North America and globally, antigenic characteristics of influenza virus strains currently circulating in human populations, and serologic responses to circulating influenza viruses of people immunized with the current approved vaccine.
The panel’s recommendations are the same as the World Health Organization’s recommendation for next season’s northern hemisphere influenza vaccine, which was made the week before the FDA meeting.
The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting. Occasionally, a panelist may be given a waiver, but not at this meeting.
SILVER SPRING, MD. – The trivalent and quadrivalent influenza vaccines for the 2014-2015 influenza season in the United States should retain the same strains that are included in the current season’s vaccines, according to a Food and Drug Administration expert panel.
At a meeting, the FDA’s Vaccines and Related Biological Products Advisory Committee voted 16 to 0 that the 2014-2015 trivalent influenza vaccine should retain the three strains included in the current vaccine: an A/California/7/2009 (H1N1)-like virus and an A/Texas/50/2012 (H3N2)-like virus, the two influenza A strains; and a B/Massachusetts/2/2012-like virus, a B/Yamagata lineage strain, as the influenza B strain.
For the quadrivalent vaccine, the panel unanimously recommended that the B/Victoria lineage strain contained in the current quadrivalent vaccine, a B/Brisbane/60/2008-like virus, be retained as the second influenza B strain.
The 2014-15 season will be the second season that quadrivalent influenza vaccines – which contain both a B/Victoria lineage and a B/Yamagata lineage vaccine virus strain – will be available.
During the current season, the quadrivalent vaccine was widely available, according to Sam Lee, Ph.D., senior director of pandemic influenza strategy at Sanofi Pasteur. Dr. Lee, who represented the vaccine manufacturers at the meeting, said that through January 2014, about 134 million doses of influenza vaccine were distributed in the United States, about the same as during the 2012-2013 season. The quadrivalent vaccine accounted for about 20% of the total vaccine doses distributed, and is expected to account for more than half of the doses distributed during next season, he said.
So far, influenza vaccine manufacturers do not anticipate any problems with the availability of the strains to be included in the vaccines, Dr. Lee told the panel.
Providing an update on influenza activity during the current season, Dr. Lisa Grohskopf, of the influenza division in the Centers for Disease Control & Prevention’s National Center for Immunization and Respiratory Diseases, said that influenza activity peaked in late December and early January and that influenza A (H1N1) viruses have predominated. As of Feb. 15, there have been 52 influenza-associated pediatric deaths reported, which is less than the number reported last season (171) but more than during the 2011-2012 influenza season (35), she noted.
The FDA panel meets at the same time each year to review influenza surveillance and epidemiology data in North America and globally, antigenic characteristics of influenza virus strains currently circulating in human populations, and serologic responses to circulating influenza viruses of people immunized with the current approved vaccine.
The panel’s recommendations are the same as the World Health Organization’s recommendation for next season’s northern hemisphere influenza vaccine, which was made the week before the FDA meeting.
The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting. Occasionally, a panelist may be given a waiver, but not at this meeting.
SILVER SPRING, MD. – The trivalent and quadrivalent influenza vaccines for the 2014-2015 influenza season in the United States should retain the same strains that are included in the current season’s vaccines, according to a Food and Drug Administration expert panel.
At a meeting, the FDA’s Vaccines and Related Biological Products Advisory Committee voted 16 to 0 that the 2014-2015 trivalent influenza vaccine should retain the three strains included in the current vaccine: an A/California/7/2009 (H1N1)-like virus and an A/Texas/50/2012 (H3N2)-like virus, the two influenza A strains; and a B/Massachusetts/2/2012-like virus, a B/Yamagata lineage strain, as the influenza B strain.
For the quadrivalent vaccine, the panel unanimously recommended that the B/Victoria lineage strain contained in the current quadrivalent vaccine, a B/Brisbane/60/2008-like virus, be retained as the second influenza B strain.
The 2014-15 season will be the second season that quadrivalent influenza vaccines – which contain both a B/Victoria lineage and a B/Yamagata lineage vaccine virus strain – will be available.
During the current season, the quadrivalent vaccine was widely available, according to Sam Lee, Ph.D., senior director of pandemic influenza strategy at Sanofi Pasteur. Dr. Lee, who represented the vaccine manufacturers at the meeting, said that through January 2014, about 134 million doses of influenza vaccine were distributed in the United States, about the same as during the 2012-2013 season. The quadrivalent vaccine accounted for about 20% of the total vaccine doses distributed, and is expected to account for more than half of the doses distributed during next season, he said.
So far, influenza vaccine manufacturers do not anticipate any problems with the availability of the strains to be included in the vaccines, Dr. Lee told the panel.
Providing an update on influenza activity during the current season, Dr. Lisa Grohskopf, of the influenza division in the Centers for Disease Control & Prevention’s National Center for Immunization and Respiratory Diseases, said that influenza activity peaked in late December and early January and that influenza A (H1N1) viruses have predominated. As of Feb. 15, there have been 52 influenza-associated pediatric deaths reported, which is less than the number reported last season (171) but more than during the 2011-2012 influenza season (35), she noted.
The FDA panel meets at the same time each year to review influenza surveillance and epidemiology data in North America and globally, antigenic characteristics of influenza virus strains currently circulating in human populations, and serologic responses to circulating influenza viruses of people immunized with the current approved vaccine.
The panel’s recommendations are the same as the World Health Organization’s recommendation for next season’s northern hemisphere influenza vaccine, which was made the week before the FDA meeting.
The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting. Occasionally, a panelist may be given a waiver, but not at this meeting.
AT AN FDA ADVISORY PANEL MEETING