User login
Psoriasis Flares Rapidly Postpartum
Managing psoriasis during pregnancy includes consideration for quality of life and medication safety issues, according to Dr. Alan Menter.
"Treatment of pregnant women with psoriasis should take into consideration the benefit of the therapy to her and the safety of her fetus. The full spectrum of safe and effective therapies during pregnancy needs to be reviewed," Dr. Menter said at the Women’s & Pediatric Dermatology Seminar, sponsored by the Skin Disease Education Foundation (SDEF).

"Most people present with psoriasis before age 40 years. The disease affects women and men equally, so many women with psoriasis are of reproductive age and are considering pregnancy," said Dr. Menter, chairman of the dermatology division at Baylor University Medical Center, Dallas.
"Maintaining quality of life is particularly important for females with psoriasis who are considering becoming pregnant, as well as during pregnancy and while breastfeeding."
Treatment Options
Patients with mild psoriasis can be treated safely with topical agents during pregnancy, most commonly with steroid and vitamin D preparations. For women with psoriasis unresponsive to topicals, a course of phototherapy with narrow-band UVB is safe and effective before conception, during pregnancy, and during the lactation period, Dr. Menter said.
Methotrexate, which should be stopped 3 months before conceiving, and the retinoid acitretin (Soriatane), which is contraindicated in females of childbearing potential, are pregnancy category X drugs and are not options during pregnancy.
As for cyclosporine, more than 20 years of pregnancy registry data on organ transplant recipients treated with the drug – who are on higher doses than used in psoriasis – indicate that cyclosporine is a "viable drug to be used during pregnancy if absolutely necessary," he said.
For moderate to severe psoriasis, "cyclosporine works very well at relatively low dosages, but has to be discontinued before the baby is born because it is excreted in breast milk," he added. A potential adverse effect of cyclosporine is that it may aggravate hypertension of pregnancy, and, in a small percentage of patients, it produces a pregnancy that is 3-4 weeks shorter than normal.
The biologic treatments etanercept (Enbrel), adalimumab (Humira), infliximab (Remicade), and ustekinumab (Stelara), which are all category B drugs, could interfere with normal embryonic development and affect fetal and newborn immunity. However, there are numerous pregnancy registries for both psoriasis and other inflammatory diseases, such as rheumatoid arthritis, Crohn’s disease, and ulcerative colitis, which show that biologics have had no long-term implications for the normal development of the infant immune system.
Psoriasis improves spontaneously in about 60% of women during pregnancy, although they are at an increased risk for postpartum flares. After delivery, "psoriasis can flare up very quickly and can spread from head to toe within days or weeks ... [and is] sometimes a lot worse than what they had previously," he said.
Patients should be advised to alert their physicians at the first sign of a flare after delivery. Phototherapy and topical treatments are options, but cyclosporine should not be used in breastfeeding patients. There is almost no excretion of the four injectable biologics into breast milk, which can thus be considered safe in lactating women.
Case Studies
Among the cases he presented at the conference was a 26-year-old female with a 5-year history of severe psoriasis who started treatment with cyclosporine. At 12 months, she was 90% clear and the dose was tapered. She became pregnant at 16 weeks. Cyclosporine was discontinued at 27 weeks gestation, and she remained relatively clear for the rest of her pregnancy and delivered a healthy baby at term.
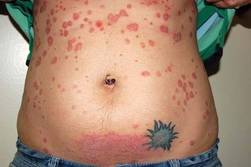
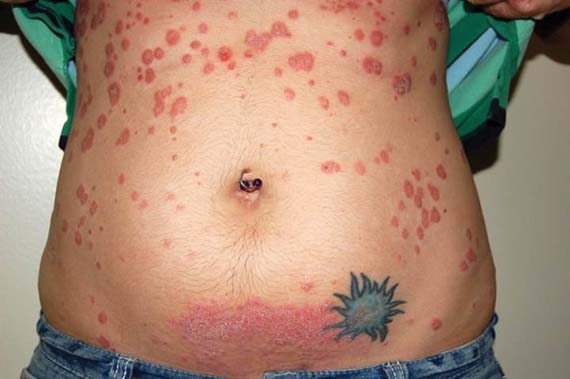
She had a severe postpartum flare, with only a moderate response obtained with the reintroduction of cyclosporine after she stopped breastfeeding. She was then started on a 12-week course of the biologic agent alefacept and UVB treatment, which produced a moderate response. During the second month of alefacept treatment, she became pregnant again. The moderate response achieved with the alefacept was maintained throughout the pregnancy, even after the 12-week course of alefacept was stopped. She delivered a normal, healthy baby.
The same patient was started on infliximab for a significant flare of psoriasis a month after her second delivery. She was 95% clear after three induction infusions and was maintained on infliximab for 5 years. She became pregnant again during treatment with infliximab and had a healthy baby, with a normal delivery. She remains 90% clear to date on infliximab at a dose of 7.5 mg/kg every 8 weeks.
Another patient Dr. Menter described became pregnant while on methotrexate, and had a healthy baby despite the risk of miscarriage and fetal abnormalities with methotrexate. She stopped methotrexate treatment at week 13 of pregnancy, but developed a significant flare and started treatment with cyclosporine, which was tapered and discontinued at 8 months of pregnancy when the patient’s ustekinumab therapy was initiated. Three weeks later, she delivered a healthy baby who is "thriving" at age 3 years; her psoriasis remains 75% clear on a dose of 45 mg of ustekinumab every 12 weeks, Dr. Menter said.
Clinicians should be aware that women with psoriasis are at a slightly increased risk for spontaneous abortion and premature rupture of the membranes, he said, referring to the results of a case-control study that found an increased risk for these pregnancy complications, as well as macrosomia and induced abortions in women with psoriasis (J. Eur. Acad. Dermatol. Venereol. 2011:25;1041-7).
Dr. Menter has received research support from, been a consultant to, or served as a lecturer for Abbott, Amgen, and other companies.
SDEF and this news organization are owned by Frontline Medical Communications.
Managing psoriasis during pregnancy includes consideration for quality of life and medication safety issues, according to Dr. Alan Menter.
"Treatment of pregnant women with psoriasis should take into consideration the benefit of the therapy to her and the safety of her fetus. The full spectrum of safe and effective therapies during pregnancy needs to be reviewed," Dr. Menter said at the Women’s & Pediatric Dermatology Seminar, sponsored by the Skin Disease Education Foundation (SDEF).
"Most people present with psoriasis before age 40 years. The disease affects women and men equally, so many women with psoriasis are of reproductive age and are considering pregnancy," said Dr. Menter, chairman of the dermatology division at Baylor University Medical Center, Dallas.
"Maintaining quality of life is particularly important for females with psoriasis who are considering becoming pregnant, as well as during pregnancy and while breastfeeding."
Treatment Options
Patients with mild psoriasis can be treated safely with topical agents during pregnancy, most commonly with steroid and vitamin D preparations. For women with psoriasis unresponsive to topicals, a course of phototherapy with narrow-band UVB is safe and effective before conception, during pregnancy, and during the lactation period, Dr. Menter said.
Methotrexate, which should be stopped 3 months before conceiving, and the retinoid acitretin (Soriatane), which is contraindicated in females of childbearing potential, are pregnancy category X drugs and are not options during pregnancy.
As for cyclosporine, more than 20 years of pregnancy registry data on organ transplant recipients treated with the drug – who are on higher doses than used in psoriasis – indicate that cyclosporine is a "viable drug to be used during pregnancy if absolutely necessary," he said.
For moderate to severe psoriasis, "cyclosporine works very well at relatively low dosages, but has to be discontinued before the baby is born because it is excreted in breast milk," he added. A potential adverse effect of cyclosporine is that it may aggravate hypertension of pregnancy, and, in a small percentage of patients, it produces a pregnancy that is 3-4 weeks shorter than normal.
The biologic treatments etanercept (Enbrel), adalimumab (Humira), infliximab (Remicade), and ustekinumab (Stelara), which are all category B drugs, could interfere with normal embryonic development and affect fetal and newborn immunity. However, there are numerous pregnancy registries for both psoriasis and other inflammatory diseases, such as rheumatoid arthritis, Crohn’s disease, and ulcerative colitis, which show that biologics have had no long-term implications for the normal development of the infant immune system.
Psoriasis improves spontaneously in about 60% of women during pregnancy, although they are at an increased risk for postpartum flares. After delivery, "psoriasis can flare up very quickly and can spread from head to toe within days or weeks ... [and is] sometimes a lot worse than what they had previously," he said.
Patients should be advised to alert their physicians at the first sign of a flare after delivery. Phototherapy and topical treatments are options, but cyclosporine should not be used in breastfeeding patients. There is almost no excretion of the four injectable biologics into breast milk, which can thus be considered safe in lactating women.
Case Studies
Among the cases he presented at the conference was a 26-year-old female with a 5-year history of severe psoriasis who started treatment with cyclosporine. At 12 months, she was 90% clear and the dose was tapered. She became pregnant at 16 weeks. Cyclosporine was discontinued at 27 weeks gestation, and she remained relatively clear for the rest of her pregnancy and delivered a healthy baby at term.
She had a severe postpartum flare, with only a moderate response obtained with the reintroduction of cyclosporine after she stopped breastfeeding. She was then started on a 12-week course of the biologic agent alefacept and UVB treatment, which produced a moderate response. During the second month of alefacept treatment, she became pregnant again. The moderate response achieved with the alefacept was maintained throughout the pregnancy, even after the 12-week course of alefacept was stopped. She delivered a normal, healthy baby.
The same patient was started on infliximab for a significant flare of psoriasis a month after her second delivery. She was 95% clear after three induction infusions and was maintained on infliximab for 5 years. She became pregnant again during treatment with infliximab and had a healthy baby, with a normal delivery. She remains 90% clear to date on infliximab at a dose of 7.5 mg/kg every 8 weeks.
Another patient Dr. Menter described became pregnant while on methotrexate, and had a healthy baby despite the risk of miscarriage and fetal abnormalities with methotrexate. She stopped methotrexate treatment at week 13 of pregnancy, but developed a significant flare and started treatment with cyclosporine, which was tapered and discontinued at 8 months of pregnancy when the patient’s ustekinumab therapy was initiated. Three weeks later, she delivered a healthy baby who is "thriving" at age 3 years; her psoriasis remains 75% clear on a dose of 45 mg of ustekinumab every 12 weeks, Dr. Menter said.
Clinicians should be aware that women with psoriasis are at a slightly increased risk for spontaneous abortion and premature rupture of the membranes, he said, referring to the results of a case-control study that found an increased risk for these pregnancy complications, as well as macrosomia and induced abortions in women with psoriasis (J. Eur. Acad. Dermatol. Venereol. 2011:25;1041-7).
Dr. Menter has received research support from, been a consultant to, or served as a lecturer for Abbott, Amgen, and other companies.
SDEF and this news organization are owned by Frontline Medical Communications.
Managing psoriasis during pregnancy includes consideration for quality of life and medication safety issues, according to Dr. Alan Menter.
"Treatment of pregnant women with psoriasis should take into consideration the benefit of the therapy to her and the safety of her fetus. The full spectrum of safe and effective therapies during pregnancy needs to be reviewed," Dr. Menter said at the Women’s & Pediatric Dermatology Seminar, sponsored by the Skin Disease Education Foundation (SDEF).
"Most people present with psoriasis before age 40 years. The disease affects women and men equally, so many women with psoriasis are of reproductive age and are considering pregnancy," said Dr. Menter, chairman of the dermatology division at Baylor University Medical Center, Dallas.
"Maintaining quality of life is particularly important for females with psoriasis who are considering becoming pregnant, as well as during pregnancy and while breastfeeding."
Treatment Options
Patients with mild psoriasis can be treated safely with topical agents during pregnancy, most commonly with steroid and vitamin D preparations. For women with psoriasis unresponsive to topicals, a course of phototherapy with narrow-band UVB is safe and effective before conception, during pregnancy, and during the lactation period, Dr. Menter said.
Methotrexate, which should be stopped 3 months before conceiving, and the retinoid acitretin (Soriatane), which is contraindicated in females of childbearing potential, are pregnancy category X drugs and are not options during pregnancy.
As for cyclosporine, more than 20 years of pregnancy registry data on organ transplant recipients treated with the drug – who are on higher doses than used in psoriasis – indicate that cyclosporine is a "viable drug to be used during pregnancy if absolutely necessary," he said.
For moderate to severe psoriasis, "cyclosporine works very well at relatively low dosages, but has to be discontinued before the baby is born because it is excreted in breast milk," he added. A potential adverse effect of cyclosporine is that it may aggravate hypertension of pregnancy, and, in a small percentage of patients, it produces a pregnancy that is 3-4 weeks shorter than normal.
The biologic treatments etanercept (Enbrel), adalimumab (Humira), infliximab (Remicade), and ustekinumab (Stelara), which are all category B drugs, could interfere with normal embryonic development and affect fetal and newborn immunity. However, there are numerous pregnancy registries for both psoriasis and other inflammatory diseases, such as rheumatoid arthritis, Crohn’s disease, and ulcerative colitis, which show that biologics have had no long-term implications for the normal development of the infant immune system.
Psoriasis improves spontaneously in about 60% of women during pregnancy, although they are at an increased risk for postpartum flares. After delivery, "psoriasis can flare up very quickly and can spread from head to toe within days or weeks ... [and is] sometimes a lot worse than what they had previously," he said.
Patients should be advised to alert their physicians at the first sign of a flare after delivery. Phototherapy and topical treatments are options, but cyclosporine should not be used in breastfeeding patients. There is almost no excretion of the four injectable biologics into breast milk, which can thus be considered safe in lactating women.
Case Studies
Among the cases he presented at the conference was a 26-year-old female with a 5-year history of severe psoriasis who started treatment with cyclosporine. At 12 months, she was 90% clear and the dose was tapered. She became pregnant at 16 weeks. Cyclosporine was discontinued at 27 weeks gestation, and she remained relatively clear for the rest of her pregnancy and delivered a healthy baby at term.
She had a severe postpartum flare, with only a moderate response obtained with the reintroduction of cyclosporine after she stopped breastfeeding. She was then started on a 12-week course of the biologic agent alefacept and UVB treatment, which produced a moderate response. During the second month of alefacept treatment, she became pregnant again. The moderate response achieved with the alefacept was maintained throughout the pregnancy, even after the 12-week course of alefacept was stopped. She delivered a normal, healthy baby.
The same patient was started on infliximab for a significant flare of psoriasis a month after her second delivery. She was 95% clear after three induction infusions and was maintained on infliximab for 5 years. She became pregnant again during treatment with infliximab and had a healthy baby, with a normal delivery. She remains 90% clear to date on infliximab at a dose of 7.5 mg/kg every 8 weeks.
Another patient Dr. Menter described became pregnant while on methotrexate, and had a healthy baby despite the risk of miscarriage and fetal abnormalities with methotrexate. She stopped methotrexate treatment at week 13 of pregnancy, but developed a significant flare and started treatment with cyclosporine, which was tapered and discontinued at 8 months of pregnancy when the patient’s ustekinumab therapy was initiated. Three weeks later, she delivered a healthy baby who is "thriving" at age 3 years; her psoriasis remains 75% clear on a dose of 45 mg of ustekinumab every 12 weeks, Dr. Menter said.
Clinicians should be aware that women with psoriasis are at a slightly increased risk for spontaneous abortion and premature rupture of the membranes, he said, referring to the results of a case-control study that found an increased risk for these pregnancy complications, as well as macrosomia and induced abortions in women with psoriasis (J. Eur. Acad. Dermatol. Venereol. 2011:25;1041-7).
Dr. Menter has received research support from, been a consultant to, or served as a lecturer for Abbott, Amgen, and other companies.
SDEF and this news organization are owned by Frontline Medical Communications.
EXPERT ANALYSIS FROM THE SDEF WOMEN'S AND PEDIATRIC DERMATOLOGY SEMINAR
Simple Surgical Risk Tool Useful for Resource-Limited Areas
A risk-adjusted tool based on three preoperative variables from the National Surgical Quality Improvement Program had a high rate of efficacy in predicting inpatient mortality, which suggests it may be useful in resource-limited settings such as small rural hospitals or low- and middle-income countries.
"By offering a simplified risk-adjustment tool, we can compare surgical outcomes among hospitals on a global scale, regardless of the spectrum of surgical procedures offered or hospital resources," Jamie E. Anderson and associates in the department of surgery, University of California, San Diego, wrote in Archives of Surgery.
"Although participation in programs such as the NSQIP offers administrative support and comparison of outcomes among participating hospitals, the low-cost options reported can expand the number of hospitals that participate in risk-adjustment outcomes analysis and quality improvement programs," they added.
The American College of Surgeons’ NSQIP risk-adjusted tool uses more than 130 variables plus a 30-day patient follow-up, and thus is not affordable for use in settings where resources are limited, including small, rural hospitals, the authors pointed out (Arch. Surg. 2012;147:798-803).
Using data from more than 600,000 patients in the 2005-2009 NSQIP database, they developed different models to predict inpatient mortality and validated the models based on data on 239 patients from a 110-bed hospital in California with a level IV trauma center. They calculated that the "area under the receiver operator characteristic curve (AUROC)" for each model as a measure of how well the model separated the two groups of interest (survivors vs. nonsurvivors) with a value of 1.0 (or 100%) would mean that the model was able to completely separate the two groups.
The model using three preoperative NSQIP variables – age, American Society of Anesthesiologists (ASA) physical status classification, and functional status – had AUROC values that were more than 0.90, or more than 90%, which was similar to the value achieved for the model that used four or six variables. The model that used 66 variables was about the same as the value achieved with the model that used 4 variables (about 91%).
Considering that an AUROC value of 0.5 indicates that the model cannot distinguish between two groups any better than chance and that an AUROC value of 1.0 indicates that the model completely discriminates between the two groups, the authors said that an AUROC value of greater than 90% is substantial. Therefore, based on their results, "3 or 4 variables may be sufficient for adequate risk adjustment to measure surgical outcomes," they added.
"Future risk-adjustment tools [should] be based on 6 or fewer variables to allow for surgical outcomes to be measured and compared within and among hospitals in resource-limited settings," they concluded.
The authors had no disclosures.
In an accompanying editorial titled "NSQIP Lite," Dr. Diana L. Farmer wrote that, while the statistical methods used were "dense," the significance of this study should not be overlooked and that "the development of a simple and inexpensive tool that could be used in the most resource-poor settings in this country and around the world is of enormous importance." The tool, which needs to be validated further in diverse settings, "stands as a powerful potential mechanism for answering the most compelling question of in-hospital mortality," she added (Arch. Surg. 2012;147:803-4).
Dr. Farmer is chair of the department of surgery at UC Davis Health Systems, Sacramento, Calif. She had no disclosures to report.
In an accompanying editorial titled "NSQIP Lite," Dr. Diana L. Farmer wrote that, while the statistical methods used were "dense," the significance of this study should not be overlooked and that "the development of a simple and inexpensive tool that could be used in the most resource-poor settings in this country and around the world is of enormous importance." The tool, which needs to be validated further in diverse settings, "stands as a powerful potential mechanism for answering the most compelling question of in-hospital mortality," she added (Arch. Surg. 2012;147:803-4).
Dr. Farmer is chair of the department of surgery at UC Davis Health Systems, Sacramento, Calif. She had no disclosures to report.
In an accompanying editorial titled "NSQIP Lite," Dr. Diana L. Farmer wrote that, while the statistical methods used were "dense," the significance of this study should not be overlooked and that "the development of a simple and inexpensive tool that could be used in the most resource-poor settings in this country and around the world is of enormous importance." The tool, which needs to be validated further in diverse settings, "stands as a powerful potential mechanism for answering the most compelling question of in-hospital mortality," she added (Arch. Surg. 2012;147:803-4).
Dr. Farmer is chair of the department of surgery at UC Davis Health Systems, Sacramento, Calif. She had no disclosures to report.
A risk-adjusted tool based on three preoperative variables from the National Surgical Quality Improvement Program had a high rate of efficacy in predicting inpatient mortality, which suggests it may be useful in resource-limited settings such as small rural hospitals or low- and middle-income countries.
"By offering a simplified risk-adjustment tool, we can compare surgical outcomes among hospitals on a global scale, regardless of the spectrum of surgical procedures offered or hospital resources," Jamie E. Anderson and associates in the department of surgery, University of California, San Diego, wrote in Archives of Surgery.
"Although participation in programs such as the NSQIP offers administrative support and comparison of outcomes among participating hospitals, the low-cost options reported can expand the number of hospitals that participate in risk-adjustment outcomes analysis and quality improvement programs," they added.
The American College of Surgeons’ NSQIP risk-adjusted tool uses more than 130 variables plus a 30-day patient follow-up, and thus is not affordable for use in settings where resources are limited, including small, rural hospitals, the authors pointed out (Arch. Surg. 2012;147:798-803).
Using data from more than 600,000 patients in the 2005-2009 NSQIP database, they developed different models to predict inpatient mortality and validated the models based on data on 239 patients from a 110-bed hospital in California with a level IV trauma center. They calculated that the "area under the receiver operator characteristic curve (AUROC)" for each model as a measure of how well the model separated the two groups of interest (survivors vs. nonsurvivors) with a value of 1.0 (or 100%) would mean that the model was able to completely separate the two groups.
The model using three preoperative NSQIP variables – age, American Society of Anesthesiologists (ASA) physical status classification, and functional status – had AUROC values that were more than 0.90, or more than 90%, which was similar to the value achieved for the model that used four or six variables. The model that used 66 variables was about the same as the value achieved with the model that used 4 variables (about 91%).
Considering that an AUROC value of 0.5 indicates that the model cannot distinguish between two groups any better than chance and that an AUROC value of 1.0 indicates that the model completely discriminates between the two groups, the authors said that an AUROC value of greater than 90% is substantial. Therefore, based on their results, "3 or 4 variables may be sufficient for adequate risk adjustment to measure surgical outcomes," they added.
"Future risk-adjustment tools [should] be based on 6 or fewer variables to allow for surgical outcomes to be measured and compared within and among hospitals in resource-limited settings," they concluded.
The authors had no disclosures.
A risk-adjusted tool based on three preoperative variables from the National Surgical Quality Improvement Program had a high rate of efficacy in predicting inpatient mortality, which suggests it may be useful in resource-limited settings such as small rural hospitals or low- and middle-income countries.
"By offering a simplified risk-adjustment tool, we can compare surgical outcomes among hospitals on a global scale, regardless of the spectrum of surgical procedures offered or hospital resources," Jamie E. Anderson and associates in the department of surgery, University of California, San Diego, wrote in Archives of Surgery.
"Although participation in programs such as the NSQIP offers administrative support and comparison of outcomes among participating hospitals, the low-cost options reported can expand the number of hospitals that participate in risk-adjustment outcomes analysis and quality improvement programs," they added.
The American College of Surgeons’ NSQIP risk-adjusted tool uses more than 130 variables plus a 30-day patient follow-up, and thus is not affordable for use in settings where resources are limited, including small, rural hospitals, the authors pointed out (Arch. Surg. 2012;147:798-803).
Using data from more than 600,000 patients in the 2005-2009 NSQIP database, they developed different models to predict inpatient mortality and validated the models based on data on 239 patients from a 110-bed hospital in California with a level IV trauma center. They calculated that the "area under the receiver operator characteristic curve (AUROC)" for each model as a measure of how well the model separated the two groups of interest (survivors vs. nonsurvivors) with a value of 1.0 (or 100%) would mean that the model was able to completely separate the two groups.
The model using three preoperative NSQIP variables – age, American Society of Anesthesiologists (ASA) physical status classification, and functional status – had AUROC values that were more than 0.90, or more than 90%, which was similar to the value achieved for the model that used four or six variables. The model that used 66 variables was about the same as the value achieved with the model that used 4 variables (about 91%).
Considering that an AUROC value of 0.5 indicates that the model cannot distinguish between two groups any better than chance and that an AUROC value of 1.0 indicates that the model completely discriminates between the two groups, the authors said that an AUROC value of greater than 90% is substantial. Therefore, based on their results, "3 or 4 variables may be sufficient for adequate risk adjustment to measure surgical outcomes," they added.
"Future risk-adjustment tools [should] be based on 6 or fewer variables to allow for surgical outcomes to be measured and compared within and among hospitals in resource-limited settings," they concluded.
The authors had no disclosures.
FROM THE ARCHIVES OF SURGERY
Major Finding: A risk-adjusted tool using three preoperative variables from the National Surgical Quality Improvement Program (NSQIP) database was more than 90% effective at predicting inpatient mortality – an efficacy rate comparable with that of tools using many more variables.
Data Source: The study entailed developing the tool with a different number of variables from the NSQIP database between 2005 and 2009 to predict inpatient mortality and validating the tool using patient data from a 110-bed community hospital in California.
Disclosures: The authors had no disclosures.
Hepatic Concerns Influence Panel's Vote on ApoB Inhibitor
SILVER SPRING, MD. – In as many days, two potential treatments for a rare lipid disorder have gained the support of a Food and Drug Administration advisory panel.
At a meeting on Oct. 18, the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee voted 9:6 to support approval of mipomersen as a treatment for homozygous familial hypercholesterolemia (HoFH), but panelists voting on both sides expressed concerns about the drug’s safety profile and its modest, variable effects on lowering low density lipoprotein cholesterol.
One day earlier, the panel voted 13:2 that lomitapide, a microsomal triglyceride transfer protein inhibitor, should be approved for the same condition.
During the Oct. 18 meeting, the panel reviewed the safety and efficacy data on mipomersen, an apolipoprotein B synthesis inhibitorthat reduces the production of apolipoprotein B (ApoB). The proposed indication for mipomersen is as an adjunctive treatment to maximally tolerated lipid-lowering medications and diet to reduceLDL cholesterol, ApoB, total cholesterol (TC), and non-HDL cholesterol at a dose of 200 mg administered in a subcutaneous injection once a week. Genzyme Corp. is partnering with Isis Pharmaceuticals to develop the drug.
The main safety concern was the potential for drug-induced liver injury because of increased transaminase levels associated with treatment, and the potential for steatohepatitis with chronic use because of cases of hepatic steatosis associated with treatment. Panelists also raised issues about other safety issues, including the development of anti-mipomersen antibodies in a significant portion of treated patients, and the high dropout rate (55%) in the open-label extension study of 141 patients. Most of the patients who discontinued due to adverse effects did so because of injection site reactions and flu-like symptoms associated with treatment.
Mipomersen was studied in a pivotal phase III study and three supportive phase III studies of almost 400 patients with HoFH; all were randomized, double-blind studies comparing the once weekly dose of mipomersen to placebo added to the maximally tolerated lipid-lowering treatments for 26 weeks. Almost all patients were on a statin; none was being treated with lipid apheresis.
The pivotal study enrolled 51 patients, aged 12-53 years (mean age was 31). At week 28, 2 weeks after the last dose, the mean LDL level was reduced by a mean of 113 mg/dL (from a baseline of 439 mg/dL) in those on mipomersen, compared with a mean reduction of 12 mg/dL (from 400 mg/dL at baseline) in those on placebo, a significant difference. The mean percent change at 28 weeks in LDL cholesterol from baseline, the primary end point, was almost 25% among those on mipomerson, compared with 3.3% lower in those on placebo, a 21.4% absolute difference. Statistically significant reductions were also seen in ApoB, TC, and non-HDL cholesterol levels, and "nominally significant" reductions were seen in lipoprotein A, triglycerides, and the LDL/HDL ratio.
Panelists agreed that the studies showed that mipomersen was effective in lowering LDL cholesterol, but they described the reductions as relatively modest and variable, noting that few patients achieved an LDL level below 100 mg/dL. And while they agreed that reduction of LDL cholesterol is an appropriate surrogate for reduced cardiovascular morbidity and mortality, several panelists said that it was not clear whether the reductions in LDL of the magnitude seen in this study would have meaningful beneficial effects.
Genzyme has proposed a risk evaluation and mitigation strategy (REMS) that includes certification of prescribers and pharmacies, who can only dispense the drug if the prescriber is certified and attests that the treatment is medically appropriate and that the patient will be monitored for hepatoxicity.
If approved, the drug will be marketed as Kynamro, according to Genzyme. The company is planning a trial where mipomersen is combined with apheresis in patients with HoFH, and has started to enrolled patients with heterozygous FH in another study of mipomersen.
The FDA usually follows the recommendations of its advisory panels, which are not binding. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting. Occasionally, a panelist may be given a waiver, but not at this meeting.
SILVER SPRING, MD. – In as many days, two potential treatments for a rare lipid disorder have gained the support of a Food and Drug Administration advisory panel.
At a meeting on Oct. 18, the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee voted 9:6 to support approval of mipomersen as a treatment for homozygous familial hypercholesterolemia (HoFH), but panelists voting on both sides expressed concerns about the drug’s safety profile and its modest, variable effects on lowering low density lipoprotein cholesterol.
One day earlier, the panel voted 13:2 that lomitapide, a microsomal triglyceride transfer protein inhibitor, should be approved for the same condition.
During the Oct. 18 meeting, the panel reviewed the safety and efficacy data on mipomersen, an apolipoprotein B synthesis inhibitorthat reduces the production of apolipoprotein B (ApoB). The proposed indication for mipomersen is as an adjunctive treatment to maximally tolerated lipid-lowering medications and diet to reduceLDL cholesterol, ApoB, total cholesterol (TC), and non-HDL cholesterol at a dose of 200 mg administered in a subcutaneous injection once a week. Genzyme Corp. is partnering with Isis Pharmaceuticals to develop the drug.
The main safety concern was the potential for drug-induced liver injury because of increased transaminase levels associated with treatment, and the potential for steatohepatitis with chronic use because of cases of hepatic steatosis associated with treatment. Panelists also raised issues about other safety issues, including the development of anti-mipomersen antibodies in a significant portion of treated patients, and the high dropout rate (55%) in the open-label extension study of 141 patients. Most of the patients who discontinued due to adverse effects did so because of injection site reactions and flu-like symptoms associated with treatment.
Mipomersen was studied in a pivotal phase III study and three supportive phase III studies of almost 400 patients with HoFH; all were randomized, double-blind studies comparing the once weekly dose of mipomersen to placebo added to the maximally tolerated lipid-lowering treatments for 26 weeks. Almost all patients were on a statin; none was being treated with lipid apheresis.
The pivotal study enrolled 51 patients, aged 12-53 years (mean age was 31). At week 28, 2 weeks after the last dose, the mean LDL level was reduced by a mean of 113 mg/dL (from a baseline of 439 mg/dL) in those on mipomersen, compared with a mean reduction of 12 mg/dL (from 400 mg/dL at baseline) in those on placebo, a significant difference. The mean percent change at 28 weeks in LDL cholesterol from baseline, the primary end point, was almost 25% among those on mipomerson, compared with 3.3% lower in those on placebo, a 21.4% absolute difference. Statistically significant reductions were also seen in ApoB, TC, and non-HDL cholesterol levels, and "nominally significant" reductions were seen in lipoprotein A, triglycerides, and the LDL/HDL ratio.
Panelists agreed that the studies showed that mipomersen was effective in lowering LDL cholesterol, but they described the reductions as relatively modest and variable, noting that few patients achieved an LDL level below 100 mg/dL. And while they agreed that reduction of LDL cholesterol is an appropriate surrogate for reduced cardiovascular morbidity and mortality, several panelists said that it was not clear whether the reductions in LDL of the magnitude seen in this study would have meaningful beneficial effects.
Genzyme has proposed a risk evaluation and mitigation strategy (REMS) that includes certification of prescribers and pharmacies, who can only dispense the drug if the prescriber is certified and attests that the treatment is medically appropriate and that the patient will be monitored for hepatoxicity.
If approved, the drug will be marketed as Kynamro, according to Genzyme. The company is planning a trial where mipomersen is combined with apheresis in patients with HoFH, and has started to enrolled patients with heterozygous FH in another study of mipomersen.
The FDA usually follows the recommendations of its advisory panels, which are not binding. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting. Occasionally, a panelist may be given a waiver, but not at this meeting.
SILVER SPRING, MD. – In as many days, two potential treatments for a rare lipid disorder have gained the support of a Food and Drug Administration advisory panel.
At a meeting on Oct. 18, the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee voted 9:6 to support approval of mipomersen as a treatment for homozygous familial hypercholesterolemia (HoFH), but panelists voting on both sides expressed concerns about the drug’s safety profile and its modest, variable effects on lowering low density lipoprotein cholesterol.
One day earlier, the panel voted 13:2 that lomitapide, a microsomal triglyceride transfer protein inhibitor, should be approved for the same condition.
During the Oct. 18 meeting, the panel reviewed the safety and efficacy data on mipomersen, an apolipoprotein B synthesis inhibitorthat reduces the production of apolipoprotein B (ApoB). The proposed indication for mipomersen is as an adjunctive treatment to maximally tolerated lipid-lowering medications and diet to reduceLDL cholesterol, ApoB, total cholesterol (TC), and non-HDL cholesterol at a dose of 200 mg administered in a subcutaneous injection once a week. Genzyme Corp. is partnering with Isis Pharmaceuticals to develop the drug.
The main safety concern was the potential for drug-induced liver injury because of increased transaminase levels associated with treatment, and the potential for steatohepatitis with chronic use because of cases of hepatic steatosis associated with treatment. Panelists also raised issues about other safety issues, including the development of anti-mipomersen antibodies in a significant portion of treated patients, and the high dropout rate (55%) in the open-label extension study of 141 patients. Most of the patients who discontinued due to adverse effects did so because of injection site reactions and flu-like symptoms associated with treatment.
Mipomersen was studied in a pivotal phase III study and three supportive phase III studies of almost 400 patients with HoFH; all were randomized, double-blind studies comparing the once weekly dose of mipomersen to placebo added to the maximally tolerated lipid-lowering treatments for 26 weeks. Almost all patients were on a statin; none was being treated with lipid apheresis.
The pivotal study enrolled 51 patients, aged 12-53 years (mean age was 31). At week 28, 2 weeks after the last dose, the mean LDL level was reduced by a mean of 113 mg/dL (from a baseline of 439 mg/dL) in those on mipomersen, compared with a mean reduction of 12 mg/dL (from 400 mg/dL at baseline) in those on placebo, a significant difference. The mean percent change at 28 weeks in LDL cholesterol from baseline, the primary end point, was almost 25% among those on mipomerson, compared with 3.3% lower in those on placebo, a 21.4% absolute difference. Statistically significant reductions were also seen in ApoB, TC, and non-HDL cholesterol levels, and "nominally significant" reductions were seen in lipoprotein A, triglycerides, and the LDL/HDL ratio.
Panelists agreed that the studies showed that mipomersen was effective in lowering LDL cholesterol, but they described the reductions as relatively modest and variable, noting that few patients achieved an LDL level below 100 mg/dL. And while they agreed that reduction of LDL cholesterol is an appropriate surrogate for reduced cardiovascular morbidity and mortality, several panelists said that it was not clear whether the reductions in LDL of the magnitude seen in this study would have meaningful beneficial effects.
Genzyme has proposed a risk evaluation and mitigation strategy (REMS) that includes certification of prescribers and pharmacies, who can only dispense the drug if the prescriber is certified and attests that the treatment is medically appropriate and that the patient will be monitored for hepatoxicity.
If approved, the drug will be marketed as Kynamro, according to Genzyme. The company is planning a trial where mipomersen is combined with apheresis in patients with HoFH, and has started to enrolled patients with heterozygous FH in another study of mipomersen.
The FDA usually follows the recommendations of its advisory panels, which are not binding. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting. Occasionally, a panelist may be given a waiver, but not at this meeting.
FROM A MEETING OF THE FDA'S ENDOCRINOLOGIC AND METABOLIC DRUGS ADVISORY COMMITTEE
Panel Supports Approval of Cholesterol Drug for Rare Disorder
SILVER SPRING, MD. – A Food and Drug Administration advisory panel voted 13:2 that lomitapide, a first-in-class cholesterol-lowering drug, should be approved for adults with homozygous familial hypercholesterolemia, but cautioned against its use beyond this small group of patients, at a meeting on Oct. 17.
The panelists agreed that treatment with lomitapide, a microsomal triglyceride transfer protein (MTP) inhibitor, was highly effective in reducing LDL cholesterol after 26 weeks, in the pivotal study of 29 adults with homozygous familial hypercholesterolemia (HoFH), acknowledging the reduction in LDL cholesterol is a surrogate end point for a reduction in cardiovascular morbidity and mortality. But the panel was concerned about the potential risk of hepatotoxicity with long-term treatment, and about off-label use in children and adolescents with HoFH, patients with heterozygous FH, as well as patients with difficult-to-treat hypercholesterolemia.
Like other panelists, Dr. William Hiatt, professor of medicine in the division of cardiology at the University of Colorado, Aurora, said that despite a clear signal for hepatic risk and an undefined absolute risk of hepatotoxicity, he voted in favor of approval because the benefits outweigh the risks for patients with this rare condition. Although there were individual variations in responses, the 50% reduction in LDL cholesterol (the highest response achieved in the pivotal trial), "if sustained, is a pretty impressive benefit and one that is probably worth the risk of liver toxicity," he said.
Panelist Dr. Caleb Alexander of Johns Hopkins Bloomberg School of Public Health, Baltimore, who supported approval, said that said despite plans to discourage off-label use, substantial use of the drug in patients who do not have HoFH was highly likely, which changes the risk-benefit balance. "For someone who has a high likelihood of having a coronary bypass by their 20th birthday, I could certainly see a very favorable risk-benefit profile," but that profile would be different "for someone who simply has recalcitrant hyperlipidemia and an LDL of 380 whose primary care physician is comfortable using this therapy," he said. "The elephant in the room is how this will be used in the real world," he added.
The two pediatric gastroenterologists on the panel voted against approval, because the question posed by the FDA did not specify the adult population.
Lomitapide, administered once a day at doses up to 60 mg, inhibits MTP, which is required for the secretion of very low-density lipoprotein, reducing the production of VLDL, the precursor to LDL, according to the manufacturer, Aegerion Pharmaceuticals. If approved, it would be the first MTP inhibitor to become available in the United States. The proposed indication for approval is as an adjunct to a low-fat diet and other lipid-lowering drugs with or without LDL apheresis, to reduce LDL cholesterol, total cholesterol (TC), apolipoprotein B (apoB), and triglycerides (TG) in patients with HoFH.
In the phase III international study of 29 patients with HoFH (most were white, 55% were male, and mean age was aged 31 years), whose mean LDL level was 336 mg/dL, despite intensive lipid-lowering treatment (including apheresis in 62%), were treated with lomitapide at a starting dose of 5 mg/day titrated to 60 mg/day, as tolerated. At 26 weeks, the mean LDL level had dropped to 190 mg/dL, a 40% drop from baseline that was highly significant. The reductions from baseline LDL-C level ranged from 28% to 52%. LDL-C reductions were maintained through the 56-week extension study.
Of the 23 patients who completed the treatment period, 8 (35%) had LDL-C levels below 100 mg/dL at week 26, including 1 patient whose LDL level had dropped below 70 mg/dL. There were also significant reductions in total cholesterol, apoB, non-HDL cholesterol, triglycerides, and VLDL cholesterol from baseline; HDL and apoA1 levels dropped from baseline to the 26th week of treatment, but in the extension study, increased to baseline by the 56th week of treatment. Six patients discontinued treatment for reasons that included gastrointestinal symptoms.
The main safety concern was the potential for hepatotoxicity with treatment. In the study, four patients developed elevated transaminase levels, which responded to dose reductions or interruption in treatment, and were reversible, according to the company. Patients also had modest increases in hepatic fat while the dose was increased, which then stabilized. No malignancies have been reported in patients treated with lomitapide; however, mice developed hepatocellular adenomas and carcinomas at two to nine times the human doses.
Aegerion has proposed a risk evaluation and management plan (REMS), which would require prescribers to enroll in the program and confirm that they have been trained with the educational materials, and that they understand the indication and associated risks and the need to monitor hepatic transaminases. Under the REMS, distribution of the drug would be available only through a specialty pharmacy. The company also proposed a registry study that would follow long-term safety of the drug including serious hepatic effects, malignancies, cardiac events, and pregnancy outcomes – as well as long-term effectiveness in at least 300 patients on lomitapide for 5 years.
Because it is intended to treat a disease that affects fewer than 200,000 people in the United States at any one time, lomitapide was designated an orphan drug for the treatment of HoFH, and in June 2011, the FDA agreed that the data from the pivotal study would be adequate to support the submission of the approval application. Dr. James Smith, a medical officer in the FDA’s division of metabolism and endocrinology products, pointed out that lomitapide is one of the first lipid-lowering drugs that is being considered for approval for an orphan disease, "without the benefit of having a large phase III safety database derived from a broader population."
If approved, Aegerion plans to study the drug in a pediatric population. The company has submitted an application for approval of lomitapide for the adult indication in the European Union.
The FDA is expected to make a decision on approval by Dec. 29, according to the company. The agency usually follows the recommendations of its advisory panels, which are not binding. Panelists have been cleared of potential financial conflicts of interest related to the topic of the meeting. Occasionally, a panelist may be given a waiver, but not at this meeting.
SILVER SPRING, MD. – A Food and Drug Administration advisory panel voted 13:2 that lomitapide, a first-in-class cholesterol-lowering drug, should be approved for adults with homozygous familial hypercholesterolemia, but cautioned against its use beyond this small group of patients, at a meeting on Oct. 17.
The panelists agreed that treatment with lomitapide, a microsomal triglyceride transfer protein (MTP) inhibitor, was highly effective in reducing LDL cholesterol after 26 weeks, in the pivotal study of 29 adults with homozygous familial hypercholesterolemia (HoFH), acknowledging the reduction in LDL cholesterol is a surrogate end point for a reduction in cardiovascular morbidity and mortality. But the panel was concerned about the potential risk of hepatotoxicity with long-term treatment, and about off-label use in children and adolescents with HoFH, patients with heterozygous FH, as well as patients with difficult-to-treat hypercholesterolemia.
Like other panelists, Dr. William Hiatt, professor of medicine in the division of cardiology at the University of Colorado, Aurora, said that despite a clear signal for hepatic risk and an undefined absolute risk of hepatotoxicity, he voted in favor of approval because the benefits outweigh the risks for patients with this rare condition. Although there were individual variations in responses, the 50% reduction in LDL cholesterol (the highest response achieved in the pivotal trial), "if sustained, is a pretty impressive benefit and one that is probably worth the risk of liver toxicity," he said.
Panelist Dr. Caleb Alexander of Johns Hopkins Bloomberg School of Public Health, Baltimore, who supported approval, said that said despite plans to discourage off-label use, substantial use of the drug in patients who do not have HoFH was highly likely, which changes the risk-benefit balance. "For someone who has a high likelihood of having a coronary bypass by their 20th birthday, I could certainly see a very favorable risk-benefit profile," but that profile would be different "for someone who simply has recalcitrant hyperlipidemia and an LDL of 380 whose primary care physician is comfortable using this therapy," he said. "The elephant in the room is how this will be used in the real world," he added.
The two pediatric gastroenterologists on the panel voted against approval, because the question posed by the FDA did not specify the adult population.
Lomitapide, administered once a day at doses up to 60 mg, inhibits MTP, which is required for the secretion of very low-density lipoprotein, reducing the production of VLDL, the precursor to LDL, according to the manufacturer, Aegerion Pharmaceuticals. If approved, it would be the first MTP inhibitor to become available in the United States. The proposed indication for approval is as an adjunct to a low-fat diet and other lipid-lowering drugs with or without LDL apheresis, to reduce LDL cholesterol, total cholesterol (TC), apolipoprotein B (apoB), and triglycerides (TG) in patients with HoFH.
In the phase III international study of 29 patients with HoFH (most were white, 55% were male, and mean age was aged 31 years), whose mean LDL level was 336 mg/dL, despite intensive lipid-lowering treatment (including apheresis in 62%), were treated with lomitapide at a starting dose of 5 mg/day titrated to 60 mg/day, as tolerated. At 26 weeks, the mean LDL level had dropped to 190 mg/dL, a 40% drop from baseline that was highly significant. The reductions from baseline LDL-C level ranged from 28% to 52%. LDL-C reductions were maintained through the 56-week extension study.
Of the 23 patients who completed the treatment period, 8 (35%) had LDL-C levels below 100 mg/dL at week 26, including 1 patient whose LDL level had dropped below 70 mg/dL. There were also significant reductions in total cholesterol, apoB, non-HDL cholesterol, triglycerides, and VLDL cholesterol from baseline; HDL and apoA1 levels dropped from baseline to the 26th week of treatment, but in the extension study, increased to baseline by the 56th week of treatment. Six patients discontinued treatment for reasons that included gastrointestinal symptoms.
The main safety concern was the potential for hepatotoxicity with treatment. In the study, four patients developed elevated transaminase levels, which responded to dose reductions or interruption in treatment, and were reversible, according to the company. Patients also had modest increases in hepatic fat while the dose was increased, which then stabilized. No malignancies have been reported in patients treated with lomitapide; however, mice developed hepatocellular adenomas and carcinomas at two to nine times the human doses.
Aegerion has proposed a risk evaluation and management plan (REMS), which would require prescribers to enroll in the program and confirm that they have been trained with the educational materials, and that they understand the indication and associated risks and the need to monitor hepatic transaminases. Under the REMS, distribution of the drug would be available only through a specialty pharmacy. The company also proposed a registry study that would follow long-term safety of the drug including serious hepatic effects, malignancies, cardiac events, and pregnancy outcomes – as well as long-term effectiveness in at least 300 patients on lomitapide for 5 years.
Because it is intended to treat a disease that affects fewer than 200,000 people in the United States at any one time, lomitapide was designated an orphan drug for the treatment of HoFH, and in June 2011, the FDA agreed that the data from the pivotal study would be adequate to support the submission of the approval application. Dr. James Smith, a medical officer in the FDA’s division of metabolism and endocrinology products, pointed out that lomitapide is one of the first lipid-lowering drugs that is being considered for approval for an orphan disease, "without the benefit of having a large phase III safety database derived from a broader population."
If approved, Aegerion plans to study the drug in a pediatric population. The company has submitted an application for approval of lomitapide for the adult indication in the European Union.
The FDA is expected to make a decision on approval by Dec. 29, according to the company. The agency usually follows the recommendations of its advisory panels, which are not binding. Panelists have been cleared of potential financial conflicts of interest related to the topic of the meeting. Occasionally, a panelist may be given a waiver, but not at this meeting.
SILVER SPRING, MD. – A Food and Drug Administration advisory panel voted 13:2 that lomitapide, a first-in-class cholesterol-lowering drug, should be approved for adults with homozygous familial hypercholesterolemia, but cautioned against its use beyond this small group of patients, at a meeting on Oct. 17.
The panelists agreed that treatment with lomitapide, a microsomal triglyceride transfer protein (MTP) inhibitor, was highly effective in reducing LDL cholesterol after 26 weeks, in the pivotal study of 29 adults with homozygous familial hypercholesterolemia (HoFH), acknowledging the reduction in LDL cholesterol is a surrogate end point for a reduction in cardiovascular morbidity and mortality. But the panel was concerned about the potential risk of hepatotoxicity with long-term treatment, and about off-label use in children and adolescents with HoFH, patients with heterozygous FH, as well as patients with difficult-to-treat hypercholesterolemia.
Like other panelists, Dr. William Hiatt, professor of medicine in the division of cardiology at the University of Colorado, Aurora, said that despite a clear signal for hepatic risk and an undefined absolute risk of hepatotoxicity, he voted in favor of approval because the benefits outweigh the risks for patients with this rare condition. Although there were individual variations in responses, the 50% reduction in LDL cholesterol (the highest response achieved in the pivotal trial), "if sustained, is a pretty impressive benefit and one that is probably worth the risk of liver toxicity," he said.
Panelist Dr. Caleb Alexander of Johns Hopkins Bloomberg School of Public Health, Baltimore, who supported approval, said that said despite plans to discourage off-label use, substantial use of the drug in patients who do not have HoFH was highly likely, which changes the risk-benefit balance. "For someone who has a high likelihood of having a coronary bypass by their 20th birthday, I could certainly see a very favorable risk-benefit profile," but that profile would be different "for someone who simply has recalcitrant hyperlipidemia and an LDL of 380 whose primary care physician is comfortable using this therapy," he said. "The elephant in the room is how this will be used in the real world," he added.
The two pediatric gastroenterologists on the panel voted against approval, because the question posed by the FDA did not specify the adult population.
Lomitapide, administered once a day at doses up to 60 mg, inhibits MTP, which is required for the secretion of very low-density lipoprotein, reducing the production of VLDL, the precursor to LDL, according to the manufacturer, Aegerion Pharmaceuticals. If approved, it would be the first MTP inhibitor to become available in the United States. The proposed indication for approval is as an adjunct to a low-fat diet and other lipid-lowering drugs with or without LDL apheresis, to reduce LDL cholesterol, total cholesterol (TC), apolipoprotein B (apoB), and triglycerides (TG) in patients with HoFH.
In the phase III international study of 29 patients with HoFH (most were white, 55% were male, and mean age was aged 31 years), whose mean LDL level was 336 mg/dL, despite intensive lipid-lowering treatment (including apheresis in 62%), were treated with lomitapide at a starting dose of 5 mg/day titrated to 60 mg/day, as tolerated. At 26 weeks, the mean LDL level had dropped to 190 mg/dL, a 40% drop from baseline that was highly significant. The reductions from baseline LDL-C level ranged from 28% to 52%. LDL-C reductions were maintained through the 56-week extension study.
Of the 23 patients who completed the treatment period, 8 (35%) had LDL-C levels below 100 mg/dL at week 26, including 1 patient whose LDL level had dropped below 70 mg/dL. There were also significant reductions in total cholesterol, apoB, non-HDL cholesterol, triglycerides, and VLDL cholesterol from baseline; HDL and apoA1 levels dropped from baseline to the 26th week of treatment, but in the extension study, increased to baseline by the 56th week of treatment. Six patients discontinued treatment for reasons that included gastrointestinal symptoms.
The main safety concern was the potential for hepatotoxicity with treatment. In the study, four patients developed elevated transaminase levels, which responded to dose reductions or interruption in treatment, and were reversible, according to the company. Patients also had modest increases in hepatic fat while the dose was increased, which then stabilized. No malignancies have been reported in patients treated with lomitapide; however, mice developed hepatocellular adenomas and carcinomas at two to nine times the human doses.
Aegerion has proposed a risk evaluation and management plan (REMS), which would require prescribers to enroll in the program and confirm that they have been trained with the educational materials, and that they understand the indication and associated risks and the need to monitor hepatic transaminases. Under the REMS, distribution of the drug would be available only through a specialty pharmacy. The company also proposed a registry study that would follow long-term safety of the drug including serious hepatic effects, malignancies, cardiac events, and pregnancy outcomes – as well as long-term effectiveness in at least 300 patients on lomitapide for 5 years.
Because it is intended to treat a disease that affects fewer than 200,000 people in the United States at any one time, lomitapide was designated an orphan drug for the treatment of HoFH, and in June 2011, the FDA agreed that the data from the pivotal study would be adequate to support the submission of the approval application. Dr. James Smith, a medical officer in the FDA’s division of metabolism and endocrinology products, pointed out that lomitapide is one of the first lipid-lowering drugs that is being considered for approval for an orphan disease, "without the benefit of having a large phase III safety database derived from a broader population."
If approved, Aegerion plans to study the drug in a pediatric population. The company has submitted an application for approval of lomitapide for the adult indication in the European Union.
The FDA is expected to make a decision on approval by Dec. 29, according to the company. The agency usually follows the recommendations of its advisory panels, which are not binding. Panelists have been cleared of potential financial conflicts of interest related to the topic of the meeting. Occasionally, a panelist may be given a waiver, but not at this meeting.
AT A MEETING OF THE FDA'S ENDOCRINOLOGIC AND METABOLIC DRUGS ADVISORY COMMITTEE
Panel Advises Approving Short Bowel Syndrome Drug
SILVER SPRING, MD. – A Food and Drug Administration advisory panel unanimously supported the approval of teduglutide, a recombinant analogue of human glucagon-like peptide-2 (GLP-2) administered subcutaneously, as a treatment to improve the intestinal absorption of fluid and nutrients in adults with short bowel syndrome, at a meeting on Oct. 16.
The FDA’s Gastrointestinal Drugs Advisory Committee voted 12 to 0 that the benefits of teduglutide outweighed the potential risks in patients with short bowel syndrome (SBS). Clinical studies of adults with SBS, who are highly dependent on parenteral nutrition, showed that teduglutide significantly reduced the volume of parenteral nutrition and IV fluids they needed after 6 months of treatment, compared with placebo. While the panelists felt comfortable with the drug’s safety profile, they recommended that more safety data are needed. They also advised following the long-term safety and potential risks of the treatment after approval, including determining whether the risk of colorectal cancer is increased with treatment.
Produced in the small intestine and the proximal large intestine, GLP-2 is "an intestinotrophic peptide that stimulates mucosal epithelium," increasing absorption of fluids and nutrients, according to the manufacturer, NPS Pharmaceuticals. In studies, patients treated with teduglutide had evidence of increased villus height after 21 days of treatment, according to the company.
In a phase III study of 86 adults with SBS, 63% of those treated with 0.05 mg/kg daily had at least a 20% reduction in the volume of parenteral nutrition and IV fluids (the primary end point) required after 24 weeks of treatment, compared with 30% of those on placebo; this was a statistically significant difference. An extension study indicated that this effect is maintained through 1 year of treatment.
If teduglutide is approved, the company will market it as Gattex. About 10,000-15,000 adults in the United States with SBS are dependent on parenteral nutrition and IV fluids for fluid and nutrient replacement, according to NPS. Teduglutide was recently approved in Europe for the same indication. The FDA is expected to make a decision on approval by Dec. 30; if the drug is approved, the company plans to pursue studies in pediatric patients with SBS.
The two drugs currently approved by the FDA for SBS are somatropin rhGH (Zorbtive), a growth hormone approved in 2003, and L-glutamine powder for oral solution (Nutrestore), an adjunctive treatment approved in 2004.
The FDA usually follows the recommendations of its advisory panels, which are not binding. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting. Occasionally, a panelist may be given a waiver, but not at this meeting.
SILVER SPRING, MD. – A Food and Drug Administration advisory panel unanimously supported the approval of teduglutide, a recombinant analogue of human glucagon-like peptide-2 (GLP-2) administered subcutaneously, as a treatment to improve the intestinal absorption of fluid and nutrients in adults with short bowel syndrome, at a meeting on Oct. 16.
The FDA’s Gastrointestinal Drugs Advisory Committee voted 12 to 0 that the benefits of teduglutide outweighed the potential risks in patients with short bowel syndrome (SBS). Clinical studies of adults with SBS, who are highly dependent on parenteral nutrition, showed that teduglutide significantly reduced the volume of parenteral nutrition and IV fluids they needed after 6 months of treatment, compared with placebo. While the panelists felt comfortable with the drug’s safety profile, they recommended that more safety data are needed. They also advised following the long-term safety and potential risks of the treatment after approval, including determining whether the risk of colorectal cancer is increased with treatment.
Produced in the small intestine and the proximal large intestine, GLP-2 is "an intestinotrophic peptide that stimulates mucosal epithelium," increasing absorption of fluids and nutrients, according to the manufacturer, NPS Pharmaceuticals. In studies, patients treated with teduglutide had evidence of increased villus height after 21 days of treatment, according to the company.
In a phase III study of 86 adults with SBS, 63% of those treated with 0.05 mg/kg daily had at least a 20% reduction in the volume of parenteral nutrition and IV fluids (the primary end point) required after 24 weeks of treatment, compared with 30% of those on placebo; this was a statistically significant difference. An extension study indicated that this effect is maintained through 1 year of treatment.
If teduglutide is approved, the company will market it as Gattex. About 10,000-15,000 adults in the United States with SBS are dependent on parenteral nutrition and IV fluids for fluid and nutrient replacement, according to NPS. Teduglutide was recently approved in Europe for the same indication. The FDA is expected to make a decision on approval by Dec. 30; if the drug is approved, the company plans to pursue studies in pediatric patients with SBS.
The two drugs currently approved by the FDA for SBS are somatropin rhGH (Zorbtive), a growth hormone approved in 2003, and L-glutamine powder for oral solution (Nutrestore), an adjunctive treatment approved in 2004.
The FDA usually follows the recommendations of its advisory panels, which are not binding. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting. Occasionally, a panelist may be given a waiver, but not at this meeting.
SILVER SPRING, MD. – A Food and Drug Administration advisory panel unanimously supported the approval of teduglutide, a recombinant analogue of human glucagon-like peptide-2 (GLP-2) administered subcutaneously, as a treatment to improve the intestinal absorption of fluid and nutrients in adults with short bowel syndrome, at a meeting on Oct. 16.
The FDA’s Gastrointestinal Drugs Advisory Committee voted 12 to 0 that the benefits of teduglutide outweighed the potential risks in patients with short bowel syndrome (SBS). Clinical studies of adults with SBS, who are highly dependent on parenteral nutrition, showed that teduglutide significantly reduced the volume of parenteral nutrition and IV fluids they needed after 6 months of treatment, compared with placebo. While the panelists felt comfortable with the drug’s safety profile, they recommended that more safety data are needed. They also advised following the long-term safety and potential risks of the treatment after approval, including determining whether the risk of colorectal cancer is increased with treatment.
Produced in the small intestine and the proximal large intestine, GLP-2 is "an intestinotrophic peptide that stimulates mucosal epithelium," increasing absorption of fluids and nutrients, according to the manufacturer, NPS Pharmaceuticals. In studies, patients treated with teduglutide had evidence of increased villus height after 21 days of treatment, according to the company.
In a phase III study of 86 adults with SBS, 63% of those treated with 0.05 mg/kg daily had at least a 20% reduction in the volume of parenteral nutrition and IV fluids (the primary end point) required after 24 weeks of treatment, compared with 30% of those on placebo; this was a statistically significant difference. An extension study indicated that this effect is maintained through 1 year of treatment.
If teduglutide is approved, the company will market it as Gattex. About 10,000-15,000 adults in the United States with SBS are dependent on parenteral nutrition and IV fluids for fluid and nutrient replacement, according to NPS. Teduglutide was recently approved in Europe for the same indication. The FDA is expected to make a decision on approval by Dec. 30; if the drug is approved, the company plans to pursue studies in pediatric patients with SBS.
The two drugs currently approved by the FDA for SBS are somatropin rhGH (Zorbtive), a growth hormone approved in 2003, and L-glutamine powder for oral solution (Nutrestore), an adjunctive treatment approved in 2004.
The FDA usually follows the recommendations of its advisory panels, which are not binding. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting. Occasionally, a panelist may be given a waiver, but not at this meeting.
AT A MEETING OF THE GASTROINTESTINAL DRUGS ADVISORY COMMITTEE
FDA Approves Abraxane for Non-Small Cell Lung Cancer
The Food and Drug Administration has approved the albumin-bound formulation of paclitaxel for injectible suspension as first-line treatment of locally advanced or metastatic non–small cell lung cancer, the agency announced on Oct. 12.
Marketed as Abraxane, nanoparticle albumin-bound (nab)-paclitaxel was developed as an alternative to Taxol, which delivers paclitaxel in a highly toxic cremophor solvent. The FDA said it based the new approval on the previous approval of Taxol for this indication and a supportive study.
Abraxis Bioscience, a subsidiary of Celgene Corp., markets Abraxane. Nab-paclitaxel was initially approved in 2005 as a treatment for breast cancer after failure of combination chemotherapy for metastatic disease or relapse within 6 months of adjuvant chemotherapy.
The new indication is for use in combination with carboplatin for the initial treatment of patients with locally advanced or metastatic non–small cell lung cancer (NSCLC) who are not candidates for curative surgery or radiation therapy. The recommended dose and schedule is 100 mg/m2 administered as an intravenous infusion over 30 minutes on days 1, 8, and 15 of each 21-day cycle. The recommended dose of carboplatin is AUC = 6 mg/min per mL on day 1 of each 21-day cycle after the Abraxane infusion is completed.
Approval was based on the previous approval of Taxol injection for the same indication, plus a randomized open-label, multinational study "establishing that Abraxane was as at least as active (as determined by overall response rate) as paclitaxel when both agents are used in combination with carboplatin," the FDA said.
In the study – the CA-031 trial – 521 patients with locally advanced or metastatic NSCLC were treated with Abraxane (at a dose of 100 mg/m2 as a weekly infusion) and 531 were treated with paclitaxel injection (at a dose of 200 mg/m2 as an IV infusion 3 three weeks). All patients received the same dose and schedule of carboplatin every 3 weeks.
Premedication with corticosteroids and an antihistamine was used in all patients receiving paclitaxel, but use was discretionary in the Abraxane arm.
The primary end point of the study, the overall response rate (the proportion of patients who achieved a durable complete or partial response, as determined by blinded radiological reviewers), was 33% among those in the Abraxane arm, compared with 25% of those in the paclitaxel arm, a statistically significant difference.
Among the responders in both groups, the durability of the responses was statistically similar, with median response duration reaching 6.9 months among those on Abraxane and 6.0 months among those on paclitaxel. There was no significant difference in overall survival between the two groups.
Celgene noted in a written statement that Abraxane demonstrated a higher overall response rate for squamous cell carcinoma (41% vs. 24%) and large cell carcinoma (33% vs. 15%) but was similar in patients with carcinoma/adenocarcinoma (26% vs. 27%).
The rate of serious adverse reactions was 18% in both groups: anemia (4%) and thrombocytopenia (3%) were the most common serious adverse reactions reported among those on Abraxane, according to the FDA. The safety evaluation was based on 1,038 patients who received at least one dose of their planned treatments.
The Abraxane label carries a neutropenia warning that it should not be administered in patients with baseline neutrophil counts of less than 1,500 cells/mm3, and that frequent peripheral blood cell counts should be performed to monitor occurrence of bone marrow suppression. The agency also warns that Abraxane should not be substituted "for or with other paclitaxel formulations."
Abraxane is currently being studied for the treatment of pancreatic, metastatic melanoma, bladder, and ovarian cancers, and for expanded applications for breast cancer, according to Celgene.
The Food and Drug Administration has approved the albumin-bound formulation of paclitaxel for injectible suspension as first-line treatment of locally advanced or metastatic non–small cell lung cancer, the agency announced on Oct. 12.
Marketed as Abraxane, nanoparticle albumin-bound (nab)-paclitaxel was developed as an alternative to Taxol, which delivers paclitaxel in a highly toxic cremophor solvent. The FDA said it based the new approval on the previous approval of Taxol for this indication and a supportive study.
Abraxis Bioscience, a subsidiary of Celgene Corp., markets Abraxane. Nab-paclitaxel was initially approved in 2005 as a treatment for breast cancer after failure of combination chemotherapy for metastatic disease or relapse within 6 months of adjuvant chemotherapy.
The new indication is for use in combination with carboplatin for the initial treatment of patients with locally advanced or metastatic non–small cell lung cancer (NSCLC) who are not candidates for curative surgery or radiation therapy. The recommended dose and schedule is 100 mg/m2 administered as an intravenous infusion over 30 minutes on days 1, 8, and 15 of each 21-day cycle. The recommended dose of carboplatin is AUC = 6 mg/min per mL on day 1 of each 21-day cycle after the Abraxane infusion is completed.
Approval was based on the previous approval of Taxol injection for the same indication, plus a randomized open-label, multinational study "establishing that Abraxane was as at least as active (as determined by overall response rate) as paclitaxel when both agents are used in combination with carboplatin," the FDA said.
In the study – the CA-031 trial – 521 patients with locally advanced or metastatic NSCLC were treated with Abraxane (at a dose of 100 mg/m2 as a weekly infusion) and 531 were treated with paclitaxel injection (at a dose of 200 mg/m2 as an IV infusion 3 three weeks). All patients received the same dose and schedule of carboplatin every 3 weeks.
Premedication with corticosteroids and an antihistamine was used in all patients receiving paclitaxel, but use was discretionary in the Abraxane arm.
The primary end point of the study, the overall response rate (the proportion of patients who achieved a durable complete or partial response, as determined by blinded radiological reviewers), was 33% among those in the Abraxane arm, compared with 25% of those in the paclitaxel arm, a statistically significant difference.
Among the responders in both groups, the durability of the responses was statistically similar, with median response duration reaching 6.9 months among those on Abraxane and 6.0 months among those on paclitaxel. There was no significant difference in overall survival between the two groups.
Celgene noted in a written statement that Abraxane demonstrated a higher overall response rate for squamous cell carcinoma (41% vs. 24%) and large cell carcinoma (33% vs. 15%) but was similar in patients with carcinoma/adenocarcinoma (26% vs. 27%).
The rate of serious adverse reactions was 18% in both groups: anemia (4%) and thrombocytopenia (3%) were the most common serious adverse reactions reported among those on Abraxane, according to the FDA. The safety evaluation was based on 1,038 patients who received at least one dose of their planned treatments.
The Abraxane label carries a neutropenia warning that it should not be administered in patients with baseline neutrophil counts of less than 1,500 cells/mm3, and that frequent peripheral blood cell counts should be performed to monitor occurrence of bone marrow suppression. The agency also warns that Abraxane should not be substituted "for or with other paclitaxel formulations."
Abraxane is currently being studied for the treatment of pancreatic, metastatic melanoma, bladder, and ovarian cancers, and for expanded applications for breast cancer, according to Celgene.
The Food and Drug Administration has approved the albumin-bound formulation of paclitaxel for injectible suspension as first-line treatment of locally advanced or metastatic non–small cell lung cancer, the agency announced on Oct. 12.
Marketed as Abraxane, nanoparticle albumin-bound (nab)-paclitaxel was developed as an alternative to Taxol, which delivers paclitaxel in a highly toxic cremophor solvent. The FDA said it based the new approval on the previous approval of Taxol for this indication and a supportive study.
Abraxis Bioscience, a subsidiary of Celgene Corp., markets Abraxane. Nab-paclitaxel was initially approved in 2005 as a treatment for breast cancer after failure of combination chemotherapy for metastatic disease or relapse within 6 months of adjuvant chemotherapy.
The new indication is for use in combination with carboplatin for the initial treatment of patients with locally advanced or metastatic non–small cell lung cancer (NSCLC) who are not candidates for curative surgery or radiation therapy. The recommended dose and schedule is 100 mg/m2 administered as an intravenous infusion over 30 minutes on days 1, 8, and 15 of each 21-day cycle. The recommended dose of carboplatin is AUC = 6 mg/min per mL on day 1 of each 21-day cycle after the Abraxane infusion is completed.
Approval was based on the previous approval of Taxol injection for the same indication, plus a randomized open-label, multinational study "establishing that Abraxane was as at least as active (as determined by overall response rate) as paclitaxel when both agents are used in combination with carboplatin," the FDA said.
In the study – the CA-031 trial – 521 patients with locally advanced or metastatic NSCLC were treated with Abraxane (at a dose of 100 mg/m2 as a weekly infusion) and 531 were treated with paclitaxel injection (at a dose of 200 mg/m2 as an IV infusion 3 three weeks). All patients received the same dose and schedule of carboplatin every 3 weeks.
Premedication with corticosteroids and an antihistamine was used in all patients receiving paclitaxel, but use was discretionary in the Abraxane arm.
The primary end point of the study, the overall response rate (the proportion of patients who achieved a durable complete or partial response, as determined by blinded radiological reviewers), was 33% among those in the Abraxane arm, compared with 25% of those in the paclitaxel arm, a statistically significant difference.
Among the responders in both groups, the durability of the responses was statistically similar, with median response duration reaching 6.9 months among those on Abraxane and 6.0 months among those on paclitaxel. There was no significant difference in overall survival between the two groups.
Celgene noted in a written statement that Abraxane demonstrated a higher overall response rate for squamous cell carcinoma (41% vs. 24%) and large cell carcinoma (33% vs. 15%) but was similar in patients with carcinoma/adenocarcinoma (26% vs. 27%).
The rate of serious adverse reactions was 18% in both groups: anemia (4%) and thrombocytopenia (3%) were the most common serious adverse reactions reported among those on Abraxane, according to the FDA. The safety evaluation was based on 1,038 patients who received at least one dose of their planned treatments.
The Abraxane label carries a neutropenia warning that it should not be administered in patients with baseline neutrophil counts of less than 1,500 cells/mm3, and that frequent peripheral blood cell counts should be performed to monitor occurrence of bone marrow suppression. The agency also warns that Abraxane should not be substituted "for or with other paclitaxel formulations."
Abraxane is currently being studied for the treatment of pancreatic, metastatic melanoma, bladder, and ovarian cancers, and for expanded applications for breast cancer, according to Celgene.
High Remediation, Attrition Rates Among Surgery Residents
Almost one-third of general surgery residents required remediation over an 11-year period, most often because of a deficiency in medical knowledge, judging from findings in a retrospective study of remediation and attrition rates among general surgery residents at six academic surgical residency programs in California.
The high remediation rate identified in this study "begs the question of whether we are falling short in the education of surgical residents," said Dr. Arezou Yaghoubian of the department of surgery, Harbor-UCLA Medical Center, Los Angeles, and associates. The study was published in the September issue of Archives of Surgery (2012;147:829-33).
They conducted the study to determine which of the six Accreditation Council for Graduate Medical Education (ACGME) competencies (patient care, medical knowledge, practice-based learning, interpersonal and communication skills, professionalism, and system-based practice), most often require remediation and to identify predictors of remediation. There is a scarcity of data on how well surgical residency programs have been achieving these competencies, and this information may provide insight into how to modify the surgical curriculum more effectively in this new era of limited hours, they said.
In the study of 348 general surgery residents at the six training programs between 1999 and 2010, the most common reason for remediation was medical knowledge in 74%, followed by interpersonal and communication skills in 24%, patient care in 22%, professionalism in 18%, system-based practice in 14%, and practice-based learning in 8%. Of the 107 residents who required remediation, 27 required remediation more than once. Almost 16% of the residents left their programs, but most (53 of 55 residents) left voluntarily. The other two failed remediation and had to leave the program.
Monthly meetings with faculty was the most common form of remediation, in 79%, followed by specific reading assignments (72%), required attendance at review courses and/or conferences (27%), evaluation by a therapist, psychologist, or psychiatrist (12%), and having to repeat a clinical year (6.5%).
More than half of the remediations were initiated during the first 2 postgraduate years (25% in the first and 35% in the second year), followed by 21% during the third year, 16% during the fourth year, and 4% during the fifth year.
A predictor of remediation was having received honors during the third-year surgery clerkship (58% of those who were subject to remediation vs. 45% of those who were not remediated, a statistically significant difference), which, the authors noted, was counterintuitive.
United States Medical Licensing Examination (USMLE) step 1 and/or step 2 scores and American Board of Surgery In-Training Examination (ABSITE) scores at postgraduate years 1 through 4 were also predictive of remediation. The median USMLE step 1 and step 2 scores were 225 and 223 among the residents subject to remediation, vs. 232 for step 1 and step 2 scores among those who were not remediated, statistically significant differences. The ABSITE scores during postgraduate years 1 through 4 were significantly lower among those who were subject to remediation, but the differences in median scores in years 5 through 7 were not significantly different.
But remediation was not a predictor of attrition. The only predictor of attrition was the ABSITE score at the third postgraduate year, which was a median of 34 among those who left the program and 62 among those who stayed.
Possible explanations for the high remediation rate is that residents are not well prepared for the demands of a surgical residency, they need to be more efficient with their time because of the 80-hour work week, and they may not necessarily be spending their increased time outside of the hospital studying at home, the authors said. Possible reasons for the attrition rate among the residents, an "ongoing concern" in general surgery, may be marital, family, and personal issues and a need for a less stressful environment, they added.
Acknowledging the study’s limitations, including the retrospective design and lack of information on how many residents passed the American Board of Surgery boards, the authors concluded that the high remediation rate "should give surgical educators pause as we should closely examine the potential sources of these deficiencies." They called on surgical societies to "take the initiative to encourage the restructuring of medical school education, such that future surgeons are better prepared to enter surgical residencies," and for residency programs to "determine whether current educational methods are adequate to prepare future surgeons."
The authors had no financial disclosures.
The surgical residents in this study were intelligent high achievers and the mean USMLE step 1 score of the residents who were remediated was a "quite respectable 225," Dr. Karen Deveney said in an editorial.
"A substantial portion of our very bright residents who have a history of great success in everything they do may have difficulty keeping up with the fast pace and high workload demands," while some may opt for a less stressful career path, and others may need more help from faculty to meet expectations and will "persevere," she said. "It is incumbent on those of us in more senior positions to create educational systems that eliminate nonessential tasks so that residents can devote more attention during the compressed work hours to learning what they need to become competent surgeons," she wrote. "Only then can we have a better chance of training and retaining the best and the brightest" (Arch. Surg. 2012;147; 833).
Dr. Deveney is professor of surgery and vice chair of education and program director, department of surgery, Oregon Health and Science University, Portland. She had no financial disclosures to report.
The surgical residents in this study were intelligent high achievers and the mean USMLE step 1 score of the residents who were remediated was a "quite respectable 225," Dr. Karen Deveney said in an editorial.
"A substantial portion of our very bright residents who have a history of great success in everything they do may have difficulty keeping up with the fast pace and high workload demands," while some may opt for a less stressful career path, and others may need more help from faculty to meet expectations and will "persevere," she said. "It is incumbent on those of us in more senior positions to create educational systems that eliminate nonessential tasks so that residents can devote more attention during the compressed work hours to learning what they need to become competent surgeons," she wrote. "Only then can we have a better chance of training and retaining the best and the brightest" (Arch. Surg. 2012;147; 833).
Dr. Deveney is professor of surgery and vice chair of education and program director, department of surgery, Oregon Health and Science University, Portland. She had no financial disclosures to report.
The surgical residents in this study were intelligent high achievers and the mean USMLE step 1 score of the residents who were remediated was a "quite respectable 225," Dr. Karen Deveney said in an editorial.
"A substantial portion of our very bright residents who have a history of great success in everything they do may have difficulty keeping up with the fast pace and high workload demands," while some may opt for a less stressful career path, and others may need more help from faculty to meet expectations and will "persevere," she said. "It is incumbent on those of us in more senior positions to create educational systems that eliminate nonessential tasks so that residents can devote more attention during the compressed work hours to learning what they need to become competent surgeons," she wrote. "Only then can we have a better chance of training and retaining the best and the brightest" (Arch. Surg. 2012;147; 833).
Dr. Deveney is professor of surgery and vice chair of education and program director, department of surgery, Oregon Health and Science University, Portland. She had no financial disclosures to report.
Almost one-third of general surgery residents required remediation over an 11-year period, most often because of a deficiency in medical knowledge, judging from findings in a retrospective study of remediation and attrition rates among general surgery residents at six academic surgical residency programs in California.
The high remediation rate identified in this study "begs the question of whether we are falling short in the education of surgical residents," said Dr. Arezou Yaghoubian of the department of surgery, Harbor-UCLA Medical Center, Los Angeles, and associates. The study was published in the September issue of Archives of Surgery (2012;147:829-33).
They conducted the study to determine which of the six Accreditation Council for Graduate Medical Education (ACGME) competencies (patient care, medical knowledge, practice-based learning, interpersonal and communication skills, professionalism, and system-based practice), most often require remediation and to identify predictors of remediation. There is a scarcity of data on how well surgical residency programs have been achieving these competencies, and this information may provide insight into how to modify the surgical curriculum more effectively in this new era of limited hours, they said.
In the study of 348 general surgery residents at the six training programs between 1999 and 2010, the most common reason for remediation was medical knowledge in 74%, followed by interpersonal and communication skills in 24%, patient care in 22%, professionalism in 18%, system-based practice in 14%, and practice-based learning in 8%. Of the 107 residents who required remediation, 27 required remediation more than once. Almost 16% of the residents left their programs, but most (53 of 55 residents) left voluntarily. The other two failed remediation and had to leave the program.
Monthly meetings with faculty was the most common form of remediation, in 79%, followed by specific reading assignments (72%), required attendance at review courses and/or conferences (27%), evaluation by a therapist, psychologist, or psychiatrist (12%), and having to repeat a clinical year (6.5%).
More than half of the remediations were initiated during the first 2 postgraduate years (25% in the first and 35% in the second year), followed by 21% during the third year, 16% during the fourth year, and 4% during the fifth year.
A predictor of remediation was having received honors during the third-year surgery clerkship (58% of those who were subject to remediation vs. 45% of those who were not remediated, a statistically significant difference), which, the authors noted, was counterintuitive.
United States Medical Licensing Examination (USMLE) step 1 and/or step 2 scores and American Board of Surgery In-Training Examination (ABSITE) scores at postgraduate years 1 through 4 were also predictive of remediation. The median USMLE step 1 and step 2 scores were 225 and 223 among the residents subject to remediation, vs. 232 for step 1 and step 2 scores among those who were not remediated, statistically significant differences. The ABSITE scores during postgraduate years 1 through 4 were significantly lower among those who were subject to remediation, but the differences in median scores in years 5 through 7 were not significantly different.
But remediation was not a predictor of attrition. The only predictor of attrition was the ABSITE score at the third postgraduate year, which was a median of 34 among those who left the program and 62 among those who stayed.
Possible explanations for the high remediation rate is that residents are not well prepared for the demands of a surgical residency, they need to be more efficient with their time because of the 80-hour work week, and they may not necessarily be spending their increased time outside of the hospital studying at home, the authors said. Possible reasons for the attrition rate among the residents, an "ongoing concern" in general surgery, may be marital, family, and personal issues and a need for a less stressful environment, they added.
Acknowledging the study’s limitations, including the retrospective design and lack of information on how many residents passed the American Board of Surgery boards, the authors concluded that the high remediation rate "should give surgical educators pause as we should closely examine the potential sources of these deficiencies." They called on surgical societies to "take the initiative to encourage the restructuring of medical school education, such that future surgeons are better prepared to enter surgical residencies," and for residency programs to "determine whether current educational methods are adequate to prepare future surgeons."
The authors had no financial disclosures.
Almost one-third of general surgery residents required remediation over an 11-year period, most often because of a deficiency in medical knowledge, judging from findings in a retrospective study of remediation and attrition rates among general surgery residents at six academic surgical residency programs in California.
The high remediation rate identified in this study "begs the question of whether we are falling short in the education of surgical residents," said Dr. Arezou Yaghoubian of the department of surgery, Harbor-UCLA Medical Center, Los Angeles, and associates. The study was published in the September issue of Archives of Surgery (2012;147:829-33).
They conducted the study to determine which of the six Accreditation Council for Graduate Medical Education (ACGME) competencies (patient care, medical knowledge, practice-based learning, interpersonal and communication skills, professionalism, and system-based practice), most often require remediation and to identify predictors of remediation. There is a scarcity of data on how well surgical residency programs have been achieving these competencies, and this information may provide insight into how to modify the surgical curriculum more effectively in this new era of limited hours, they said.
In the study of 348 general surgery residents at the six training programs between 1999 and 2010, the most common reason for remediation was medical knowledge in 74%, followed by interpersonal and communication skills in 24%, patient care in 22%, professionalism in 18%, system-based practice in 14%, and practice-based learning in 8%. Of the 107 residents who required remediation, 27 required remediation more than once. Almost 16% of the residents left their programs, but most (53 of 55 residents) left voluntarily. The other two failed remediation and had to leave the program.
Monthly meetings with faculty was the most common form of remediation, in 79%, followed by specific reading assignments (72%), required attendance at review courses and/or conferences (27%), evaluation by a therapist, psychologist, or psychiatrist (12%), and having to repeat a clinical year (6.5%).
More than half of the remediations were initiated during the first 2 postgraduate years (25% in the first and 35% in the second year), followed by 21% during the third year, 16% during the fourth year, and 4% during the fifth year.
A predictor of remediation was having received honors during the third-year surgery clerkship (58% of those who were subject to remediation vs. 45% of those who were not remediated, a statistically significant difference), which, the authors noted, was counterintuitive.
United States Medical Licensing Examination (USMLE) step 1 and/or step 2 scores and American Board of Surgery In-Training Examination (ABSITE) scores at postgraduate years 1 through 4 were also predictive of remediation. The median USMLE step 1 and step 2 scores were 225 and 223 among the residents subject to remediation, vs. 232 for step 1 and step 2 scores among those who were not remediated, statistically significant differences. The ABSITE scores during postgraduate years 1 through 4 were significantly lower among those who were subject to remediation, but the differences in median scores in years 5 through 7 were not significantly different.
But remediation was not a predictor of attrition. The only predictor of attrition was the ABSITE score at the third postgraduate year, which was a median of 34 among those who left the program and 62 among those who stayed.
Possible explanations for the high remediation rate is that residents are not well prepared for the demands of a surgical residency, they need to be more efficient with their time because of the 80-hour work week, and they may not necessarily be spending their increased time outside of the hospital studying at home, the authors said. Possible reasons for the attrition rate among the residents, an "ongoing concern" in general surgery, may be marital, family, and personal issues and a need for a less stressful environment, they added.
Acknowledging the study’s limitations, including the retrospective design and lack of information on how many residents passed the American Board of Surgery boards, the authors concluded that the high remediation rate "should give surgical educators pause as we should closely examine the potential sources of these deficiencies." They called on surgical societies to "take the initiative to encourage the restructuring of medical school education, such that future surgeons are better prepared to enter surgical residencies," and for residency programs to "determine whether current educational methods are adequate to prepare future surgeons."
The authors had no financial disclosures.
FROM ARCHIVES OF SURGERY
Bioequivalence Problem Results in Generic Antidepressant Withdrawal
One of the generic forms of the 300-mg dose of extended-release bupropion is being taken off the market because of evidence indicating that it is not therapeutically equivalent to the brand name version of the antidepressant, according to a Food and Drug Administration notice report posted on Oct. 3.
The affected product is Budeprion XL 300 mg, manufactured by Impax Laboratories and marketed by Teva Pharmaceuticals USA – one of the five generic versions of Wellbutrin XL 300-mg tablets that have been approved by the FDA. The planned withdrawal of the product is the result of studies conducted by the company and FDA aimed at addressing reports of reduced efficacy from patients who were switched to generic forms of Wellbutrin XL 300 mg made more than 5 years ago. These reports were made to the agency "soon after" the Impax/Teva product was approved in December 2006, and the FDA subsequently determined that the complaints "appeared to be linked to the Impax/Teva products," the statement said.
None of the other generic versions of the 300-mg dose or the Impax 150-mg bupropion product are affected, the FDA said.
However, the agency recently has requested that the four other manufacturers of generic versions of Wellbutrin XL 300 mg conduct bioequivalence studies of their products and to submit the results to the FDA by March 2013. (Those manufacturers are Anchen, Actavis, Watson, and Mylan).
Approval of all the generic versions of 300 mg extended-release bupropion was based on studies establishing that the 150-mg strengths were bioequivalent to the 150-mg dose of the brand-name product, according to the FDA guidance at that time. But the FDA "has determined that this approach is no longer appropriate" to establish bioequivalence and "is revising its guidance to industry for how to conduct premarket bioequivalence studies in generic bupropion products."
The bioequivalence problem was confirmed in an FDA-sponsored study conducted in 2010, comparing the bioequivalence of the Impax/Teva product to Wellbutrin XL 300 mg in 24 healthy adults, which determined that the generic tablets "fail to release bupropion into the blood at the same rate and to the same extent as Wellbutrin XL 300 mg," according to the statement. At the request of the FDA, Impax/Teva had started a study comparing the bioequivalence of the 300-mg product with the brand-name product, including patients who had reported the problem of reduced efficacy after the switch to the generic product. But the company was unable to recruit enough patients and terminated the study in late 2011.
Impax and Teva have stopped shipping this product.
Health care professionals and patients should report problems associated with drugs by visiting the FDA MedWatch program or calling 800-332-1088.
One of the generic forms of the 300-mg dose of extended-release bupropion is being taken off the market because of evidence indicating that it is not therapeutically equivalent to the brand name version of the antidepressant, according to a Food and Drug Administration notice report posted on Oct. 3.
The affected product is Budeprion XL 300 mg, manufactured by Impax Laboratories and marketed by Teva Pharmaceuticals USA – one of the five generic versions of Wellbutrin XL 300-mg tablets that have been approved by the FDA. The planned withdrawal of the product is the result of studies conducted by the company and FDA aimed at addressing reports of reduced efficacy from patients who were switched to generic forms of Wellbutrin XL 300 mg made more than 5 years ago. These reports were made to the agency "soon after" the Impax/Teva product was approved in December 2006, and the FDA subsequently determined that the complaints "appeared to be linked to the Impax/Teva products," the statement said.
None of the other generic versions of the 300-mg dose or the Impax 150-mg bupropion product are affected, the FDA said.
However, the agency recently has requested that the four other manufacturers of generic versions of Wellbutrin XL 300 mg conduct bioequivalence studies of their products and to submit the results to the FDA by March 2013. (Those manufacturers are Anchen, Actavis, Watson, and Mylan).
Approval of all the generic versions of 300 mg extended-release bupropion was based on studies establishing that the 150-mg strengths were bioequivalent to the 150-mg dose of the brand-name product, according to the FDA guidance at that time. But the FDA "has determined that this approach is no longer appropriate" to establish bioequivalence and "is revising its guidance to industry for how to conduct premarket bioequivalence studies in generic bupropion products."
The bioequivalence problem was confirmed in an FDA-sponsored study conducted in 2010, comparing the bioequivalence of the Impax/Teva product to Wellbutrin XL 300 mg in 24 healthy adults, which determined that the generic tablets "fail to release bupropion into the blood at the same rate and to the same extent as Wellbutrin XL 300 mg," according to the statement. At the request of the FDA, Impax/Teva had started a study comparing the bioequivalence of the 300-mg product with the brand-name product, including patients who had reported the problem of reduced efficacy after the switch to the generic product. But the company was unable to recruit enough patients and terminated the study in late 2011.
Impax and Teva have stopped shipping this product.
Health care professionals and patients should report problems associated with drugs by visiting the FDA MedWatch program or calling 800-332-1088.
One of the generic forms of the 300-mg dose of extended-release bupropion is being taken off the market because of evidence indicating that it is not therapeutically equivalent to the brand name version of the antidepressant, according to a Food and Drug Administration notice report posted on Oct. 3.
The affected product is Budeprion XL 300 mg, manufactured by Impax Laboratories and marketed by Teva Pharmaceuticals USA – one of the five generic versions of Wellbutrin XL 300-mg tablets that have been approved by the FDA. The planned withdrawal of the product is the result of studies conducted by the company and FDA aimed at addressing reports of reduced efficacy from patients who were switched to generic forms of Wellbutrin XL 300 mg made more than 5 years ago. These reports were made to the agency "soon after" the Impax/Teva product was approved in December 2006, and the FDA subsequently determined that the complaints "appeared to be linked to the Impax/Teva products," the statement said.
None of the other generic versions of the 300-mg dose or the Impax 150-mg bupropion product are affected, the FDA said.
However, the agency recently has requested that the four other manufacturers of generic versions of Wellbutrin XL 300 mg conduct bioequivalence studies of their products and to submit the results to the FDA by March 2013. (Those manufacturers are Anchen, Actavis, Watson, and Mylan).
Approval of all the generic versions of 300 mg extended-release bupropion was based on studies establishing that the 150-mg strengths were bioequivalent to the 150-mg dose of the brand-name product, according to the FDA guidance at that time. But the FDA "has determined that this approach is no longer appropriate" to establish bioequivalence and "is revising its guidance to industry for how to conduct premarket bioequivalence studies in generic bupropion products."
The bioequivalence problem was confirmed in an FDA-sponsored study conducted in 2010, comparing the bioequivalence of the Impax/Teva product to Wellbutrin XL 300 mg in 24 healthy adults, which determined that the generic tablets "fail to release bupropion into the blood at the same rate and to the same extent as Wellbutrin XL 300 mg," according to the statement. At the request of the FDA, Impax/Teva had started a study comparing the bioequivalence of the 300-mg product with the brand-name product, including patients who had reported the problem of reduced efficacy after the switch to the generic product. But the company was unable to recruit enough patients and terminated the study in late 2011.
Impax and Teva have stopped shipping this product.
Health care professionals and patients should report problems associated with drugs by visiting the FDA MedWatch program or calling 800-332-1088.
Liquid, Extended-Release Drug Approved for ADHD
An extended-release liquid formulation of methylphenidate, taken once a day, has been approved by the Food and Drug Administration for treating attention-deficit/hyperactivity disorder.
The concentration of the oral suspension is 5 mg/mL. The recommended dose for children aged 6 years and older is a starting dose of 20 mg given orally once a day, in the morning, and increasing the dose weekly by 10-20 mg per day. Exceeding a daily dosage of 60 mg is not recommended, according to the prescribing information.
Efficacy for the product was established in a 2-week, placebo-controlled crossover study of children who diagnosed with ADHD, aged 6-12 years, in a laboratory classroom, according to the prescribing information.
The FDA, which approved the drug in late September, said it is the first liquid formulation extended-release product to treat ADHD. It is manufactured by NextWave Pharmaceuticals, and will be marketed as Quillivant XR. The company website says it will be available in pharmacies in January 2013.
Click here to see the Quillivant XR medication guide.
An extended-release liquid formulation of methylphenidate, taken once a day, has been approved by the Food and Drug Administration for treating attention-deficit/hyperactivity disorder.
The concentration of the oral suspension is 5 mg/mL. The recommended dose for children aged 6 years and older is a starting dose of 20 mg given orally once a day, in the morning, and increasing the dose weekly by 10-20 mg per day. Exceeding a daily dosage of 60 mg is not recommended, according to the prescribing information.
Efficacy for the product was established in a 2-week, placebo-controlled crossover study of children who diagnosed with ADHD, aged 6-12 years, in a laboratory classroom, according to the prescribing information.
The FDA, which approved the drug in late September, said it is the first liquid formulation extended-release product to treat ADHD. It is manufactured by NextWave Pharmaceuticals, and will be marketed as Quillivant XR. The company website says it will be available in pharmacies in January 2013.
Click here to see the Quillivant XR medication guide.
An extended-release liquid formulation of methylphenidate, taken once a day, has been approved by the Food and Drug Administration for treating attention-deficit/hyperactivity disorder.
The concentration of the oral suspension is 5 mg/mL. The recommended dose for children aged 6 years and older is a starting dose of 20 mg given orally once a day, in the morning, and increasing the dose weekly by 10-20 mg per day. Exceeding a daily dosage of 60 mg is not recommended, according to the prescribing information.
Efficacy for the product was established in a 2-week, placebo-controlled crossover study of children who diagnosed with ADHD, aged 6-12 years, in a laboratory classroom, according to the prescribing information.
The FDA, which approved the drug in late September, said it is the first liquid formulation extended-release product to treat ADHD. It is manufactured by NextWave Pharmaceuticals, and will be marketed as Quillivant XR. The company website says it will be available in pharmacies in January 2013.
Click here to see the Quillivant XR medication guide.
Second TNF-Blocker Approved for Refractory Ulcerative Colitis
Adalimumab, a subcutaneously administered tumor necrosis factor blocker, has been approved for treating adults with moderately to severely active ulcerative colitis who have not had an adequate response with conventional treatments, the Food and Drug Administration announced on Sept. 28.
The safety and effectiveness of adalimumab for this patient population was established in two clinical studies of 908 patients with moderately to severely active ulcerative colitis (UC).
Adalimumab, marketed as Humira by Abbott Laboratories, was first approved for treating rheumatoid arthritis in 2002, followed by psoriatic arthritis in 2005, ankylosing spondylitis in 2006, Crohn’s disease in 2007, and plaque psoriasis and juvenile idiopathic arthritis in 2008.
Adalimumab is the second TNF blocker to be approved for ulcerative colitis; infliximab (Remicade), an intravenous TNF blocker, was previously approved for treating UC.
Clinical remission rates in the two studies were significantly greater among patients treated with infliximab than among those who received placebo: In an 8-week study, which did not include patients who had previously been treated with a TNF blocker, the clinical remission rate at 8 weeks was 18.5% among those on adalimumab vs. 9.2% in those on placebo, a 9.3% difference. In the second study, which followed patients for 1 year and included some who had been treated with infliximab, the clinical remission rate at 8 weeks was 16.5% among those on adalimumab, vs. 9.3% among those on placebo, a 7.2% difference.
At a meeting on Aug. 28 held to review these data, the majority of the FDA’s Gastrointestinal Drugs Advisory Committee agreed that these differences represented clinically meaningful benefits and supported approval of adalimumab for this indication. Panelists cited the need for more treatments for UC and for a subcutaneous TNF blocker for these patients, as well as its potential steroid-sparing effects.
In the studies, no new side effects were identified, the agency said. The FDA statement points out that the effectiveness of adalimumab "has not been established in patients with ulcerative colitis who have lost response to or were intolerant to TNF blockers."
The approved dosing regimen for adalimumab is a starting dose of 160 mg, followed by a second dose of 80 mg 2 weeks later and then a maintenance dose of 40 mg every other week. "The drug should only continue to be used in patients who have shown evidence of clinical remission by 8 weeks of therapy," according to the FDA statement.
Adalimumab is the first self-administered biologic treatment for ulcerative colitis to be approved.
Adalimumab, a subcutaneously administered tumor necrosis factor blocker, has been approved for treating adults with moderately to severely active ulcerative colitis who have not had an adequate response with conventional treatments, the Food and Drug Administration announced on Sept. 28.
The safety and effectiveness of adalimumab for this patient population was established in two clinical studies of 908 patients with moderately to severely active ulcerative colitis (UC).
Adalimumab, marketed as Humira by Abbott Laboratories, was first approved for treating rheumatoid arthritis in 2002, followed by psoriatic arthritis in 2005, ankylosing spondylitis in 2006, Crohn’s disease in 2007, and plaque psoriasis and juvenile idiopathic arthritis in 2008.
Adalimumab is the second TNF blocker to be approved for ulcerative colitis; infliximab (Remicade), an intravenous TNF blocker, was previously approved for treating UC.
Clinical remission rates in the two studies were significantly greater among patients treated with infliximab than among those who received placebo: In an 8-week study, which did not include patients who had previously been treated with a TNF blocker, the clinical remission rate at 8 weeks was 18.5% among those on adalimumab vs. 9.2% in those on placebo, a 9.3% difference. In the second study, which followed patients for 1 year and included some who had been treated with infliximab, the clinical remission rate at 8 weeks was 16.5% among those on adalimumab, vs. 9.3% among those on placebo, a 7.2% difference.
At a meeting on Aug. 28 held to review these data, the majority of the FDA’s Gastrointestinal Drugs Advisory Committee agreed that these differences represented clinically meaningful benefits and supported approval of adalimumab for this indication. Panelists cited the need for more treatments for UC and for a subcutaneous TNF blocker for these patients, as well as its potential steroid-sparing effects.
In the studies, no new side effects were identified, the agency said. The FDA statement points out that the effectiveness of adalimumab "has not been established in patients with ulcerative colitis who have lost response to or were intolerant to TNF blockers."
The approved dosing regimen for adalimumab is a starting dose of 160 mg, followed by a second dose of 80 mg 2 weeks later and then a maintenance dose of 40 mg every other week. "The drug should only continue to be used in patients who have shown evidence of clinical remission by 8 weeks of therapy," according to the FDA statement.
Adalimumab is the first self-administered biologic treatment for ulcerative colitis to be approved.
Adalimumab, a subcutaneously administered tumor necrosis factor blocker, has been approved for treating adults with moderately to severely active ulcerative colitis who have not had an adequate response with conventional treatments, the Food and Drug Administration announced on Sept. 28.
The safety and effectiveness of adalimumab for this patient population was established in two clinical studies of 908 patients with moderately to severely active ulcerative colitis (UC).
Adalimumab, marketed as Humira by Abbott Laboratories, was first approved for treating rheumatoid arthritis in 2002, followed by psoriatic arthritis in 2005, ankylosing spondylitis in 2006, Crohn’s disease in 2007, and plaque psoriasis and juvenile idiopathic arthritis in 2008.
Adalimumab is the second TNF blocker to be approved for ulcerative colitis; infliximab (Remicade), an intravenous TNF blocker, was previously approved for treating UC.
Clinical remission rates in the two studies were significantly greater among patients treated with infliximab than among those who received placebo: In an 8-week study, which did not include patients who had previously been treated with a TNF blocker, the clinical remission rate at 8 weeks was 18.5% among those on adalimumab vs. 9.2% in those on placebo, a 9.3% difference. In the second study, which followed patients for 1 year and included some who had been treated with infliximab, the clinical remission rate at 8 weeks was 16.5% among those on adalimumab, vs. 9.3% among those on placebo, a 7.2% difference.
At a meeting on Aug. 28 held to review these data, the majority of the FDA’s Gastrointestinal Drugs Advisory Committee agreed that these differences represented clinically meaningful benefits and supported approval of adalimumab for this indication. Panelists cited the need for more treatments for UC and for a subcutaneous TNF blocker for these patients, as well as its potential steroid-sparing effects.
In the studies, no new side effects were identified, the agency said. The FDA statement points out that the effectiveness of adalimumab "has not been established in patients with ulcerative colitis who have lost response to or were intolerant to TNF blockers."
The approved dosing regimen for adalimumab is a starting dose of 160 mg, followed by a second dose of 80 mg 2 weeks later and then a maintenance dose of 40 mg every other week. "The drug should only continue to be used in patients who have shown evidence of clinical remission by 8 weeks of therapy," according to the FDA statement.
Adalimumab is the first self-administered biologic treatment for ulcerative colitis to be approved.