User login
Guideline authors inconsistently disclose conflicts
Financial conflicts are often underreported by authors of clinical practice guidelines (CPGs) in several specialties including oncology, rheumatology, and gastroenterology, according to a pair of research letters published in JAMA Internal Medicine. The Institute of Medicine recommends that guideline authors include no more than 50% individuals with financial conflicts.
In one research letter, Rishad Khan, BSc, of the University of Toronto in Ontario and his colleagues reviewed data on undeclared financial conflicts of interest among authors of guidelines related to high-revenue medications.
The researchers identified CPGs via the National Guideline Clearinghouse and selected 18 CPGs for 10 high-revenue medications published between 2013 and 2017. Financial conflicts of interest were based on the Centers for Medicare & Medicaid Services Open Payments.
Of the 160 authors involved in the various guidelines, 79 (49.4%) disclosed a payment in the CPG or supplemental materials, and 50 (31.3%) disclosed payments from companies marketing 1 of the 10 high-revenue medications in the related guidelines.
Another 41 authors (25.6%) received but did not disclose payments from companies marketing 1 of the 10 high-revenue medications in CPGs.
Overall, 91 authors (56.9%) were found to have financial conflicts of interest that involved 1 of the 10 high-revenue medications, and “the median value of undeclared payments from companies marketing 1 of the 10 high-revenue medications recommended in the CPGs was $522 (interquartile range, $0-$40,444) from two companies,” the researchers said.
The study findings were limited by several factors including “potential inaccuracies in CMS-OP reporting, which are rarely corrected, and lack of generalizability outside the United States” and by the limited time frame for data collection, which may have led to underestimation of conflicts for the guidelines, the researchers noted. In addition, “we did not have access to guideline voting records and thus did not know when conflicted panel members recommended against a medication or recused themselves from voting,” they said.
Mr. Khan disclosed research funding from AbbVie and Ferring Pharmaceuticals.
In a second research letter, half of the authors of gastroenterology guidelines received payments from industry, wrote Tyler Combs, BS, of Oklahoma State University, Tulsa, and his colleagues. Previous studies have reviewed the financial conflicts of interest in specialties including oncology, dermatology, and otolaryngology, but financial conflicts of interest among authors of gastroenterology guidelines have not been examined, the researchers said.
Mr. Combs and his colleagues identified 15 CPGs published by the American College of Gastroenterology between 2014 and 2016. They identified 83 authors, with an average of 4 authors for each guideline. Overall, 53% of the authors received industry payments, according to based on data from the 2014 to 2016 Centers for Medicare & Medicaid Services Open Payments database (OPD).
However, OPD information was not always consistent with information published with the guidelines, the researchers noted. They found that 16 (19%) of the 83 authors both disclosed financial conflicts of interests in the CPGs and had received payments according to OPD or had disclosed no financial conflicts of interest and had received no payments according to OPD. In addition, 49 (34%) of 146 cumulative financial conflicts of interest disclosed in the CPGs and 148 relationships identified on OPD were both disclosed as financial conflicts of interest and evidenced by OPD payment records. In this review, the median total payment was $1,000, with an interquartile range from $0 to $39,938.
The study findings were limited by a relatively short 12-month time frame, the researchers noted. However, “our finding that FCOI [financial conflicts of interest] disclosure only corroborates with OPD payment records between 19% and 34% of the time also suggests that guidance from the ACG [American College of Gastroenterology] may be needed to improve FCOI disclosure efforts in future iterations of gastroenterology CPGs,” they said.
The researchers had no financial conflicts to disclose.
SOURCE: Combs T et al. JAMA Intern Med. 2018 Oct 29. doi: 10.1001/jamainternmed.2018.4730; Khan R et al. JAMA Intern Med. 2018 Oct 29. doi: 10.1001/jamainternmed.2018.5106.
Statement from the AGA on the integrity of AGA’s clinical guideline process
The American Gastroenterological Association (AGA) understands how important it is for AGA members, patients, and the public at large to have access to the most trustworthy, actionable, and evidence-based guidelines in order to achieve the highest possible quality of patient care. In developing guidelines, our goal is to maintain a high level of methodologic rigor through the utilization of an evidence-based approach that is very transparent.
However, not all clinical guidelines are created with equal rigor. Clinicians should examine guidelines closely and consider whether or not they follow the Academy of Medicine’s (formerly the Institute of Medicine’s) standards for trustworthy clinical guidelines. The guideline should be based on a systematic review of the evidence, focus on transparency, have a rigorous conflict of interest system in place, include the involvement of an unconflicted Grading of Recommendations Assessment, Development and Evaluation (GRADE) system-trained methodologist, ideally as a cochair, and the recommendations should be concise and actionable. AGA follows a transparent, independent guideline development process that is not subject to company influence or bias and fully complies with the Academy of Medicine’s criteria for trustworthy guidelines.
AGA has been proactive in developing policies to minimize bias in our guidelines. AGA requires that the Chair of the Guideline Development Group, and a majority of Guideline (and other clinical practice documents) Development Group members are free of conflicts of interest relevant to the subject matter of the guideline. At the time of invitation, we ask our panel members to disclose any and all potential conflicts. Furthermore, all author disclosures are verified by means of accessing publicly available sources (such as the Centers for Medicare and Medicaid Services’ Open Payment database) prior to their involvement on the panel.
AGA strives to be transparent in reporting commercial bias and independent of any industry influence in the development of our clinical practice documents. Our goal is to produce the most trustworthy, actionable, and evidence-based guidelines possible for our members.
Learn more about AGA’s clinical guideline process (https://www.gastro.org/guidelines).
Yngve T. Falck-Ytter, MD, AGAF, is chair, and Shahnaz Sultan, MD, MHSc, AGAF, is chair-elect, AGA Institute Clinical Guidelines Committee.
Financial conflicts are often underreported by authors of clinical practice guidelines (CPGs) in several specialties including oncology, rheumatology, and gastroenterology, according to a pair of research letters published in JAMA Internal Medicine. The Institute of Medicine recommends that guideline authors include no more than 50% individuals with financial conflicts.
In one research letter, Rishad Khan, BSc, of the University of Toronto in Ontario and his colleagues reviewed data on undeclared financial conflicts of interest among authors of guidelines related to high-revenue medications.
The researchers identified CPGs via the National Guideline Clearinghouse and selected 18 CPGs for 10 high-revenue medications published between 2013 and 2017. Financial conflicts of interest were based on the Centers for Medicare & Medicaid Services Open Payments.
Of the 160 authors involved in the various guidelines, 79 (49.4%) disclosed a payment in the CPG or supplemental materials, and 50 (31.3%) disclosed payments from companies marketing 1 of the 10 high-revenue medications in the related guidelines.
Another 41 authors (25.6%) received but did not disclose payments from companies marketing 1 of the 10 high-revenue medications in CPGs.
Overall, 91 authors (56.9%) were found to have financial conflicts of interest that involved 1 of the 10 high-revenue medications, and “the median value of undeclared payments from companies marketing 1 of the 10 high-revenue medications recommended in the CPGs was $522 (interquartile range, $0-$40,444) from two companies,” the researchers said.
The study findings were limited by several factors including “potential inaccuracies in CMS-OP reporting, which are rarely corrected, and lack of generalizability outside the United States” and by the limited time frame for data collection, which may have led to underestimation of conflicts for the guidelines, the researchers noted. In addition, “we did not have access to guideline voting records and thus did not know when conflicted panel members recommended against a medication or recused themselves from voting,” they said.
Mr. Khan disclosed research funding from AbbVie and Ferring Pharmaceuticals.
In a second research letter, half of the authors of gastroenterology guidelines received payments from industry, wrote Tyler Combs, BS, of Oklahoma State University, Tulsa, and his colleagues. Previous studies have reviewed the financial conflicts of interest in specialties including oncology, dermatology, and otolaryngology, but financial conflicts of interest among authors of gastroenterology guidelines have not been examined, the researchers said.
Mr. Combs and his colleagues identified 15 CPGs published by the American College of Gastroenterology between 2014 and 2016. They identified 83 authors, with an average of 4 authors for each guideline. Overall, 53% of the authors received industry payments, according to based on data from the 2014 to 2016 Centers for Medicare & Medicaid Services Open Payments database (OPD).
However, OPD information was not always consistent with information published with the guidelines, the researchers noted. They found that 16 (19%) of the 83 authors both disclosed financial conflicts of interests in the CPGs and had received payments according to OPD or had disclosed no financial conflicts of interest and had received no payments according to OPD. In addition, 49 (34%) of 146 cumulative financial conflicts of interest disclosed in the CPGs and 148 relationships identified on OPD were both disclosed as financial conflicts of interest and evidenced by OPD payment records. In this review, the median total payment was $1,000, with an interquartile range from $0 to $39,938.
The study findings were limited by a relatively short 12-month time frame, the researchers noted. However, “our finding that FCOI [financial conflicts of interest] disclosure only corroborates with OPD payment records between 19% and 34% of the time also suggests that guidance from the ACG [American College of Gastroenterology] may be needed to improve FCOI disclosure efforts in future iterations of gastroenterology CPGs,” they said.
The researchers had no financial conflicts to disclose.
SOURCE: Combs T et al. JAMA Intern Med. 2018 Oct 29. doi: 10.1001/jamainternmed.2018.4730; Khan R et al. JAMA Intern Med. 2018 Oct 29. doi: 10.1001/jamainternmed.2018.5106.
Statement from the AGA on the integrity of AGA’s clinical guideline process
The American Gastroenterological Association (AGA) understands how important it is for AGA members, patients, and the public at large to have access to the most trustworthy, actionable, and evidence-based guidelines in order to achieve the highest possible quality of patient care. In developing guidelines, our goal is to maintain a high level of methodologic rigor through the utilization of an evidence-based approach that is very transparent.
However, not all clinical guidelines are created with equal rigor. Clinicians should examine guidelines closely and consider whether or not they follow the Academy of Medicine’s (formerly the Institute of Medicine’s) standards for trustworthy clinical guidelines. The guideline should be based on a systematic review of the evidence, focus on transparency, have a rigorous conflict of interest system in place, include the involvement of an unconflicted Grading of Recommendations Assessment, Development and Evaluation (GRADE) system-trained methodologist, ideally as a cochair, and the recommendations should be concise and actionable. AGA follows a transparent, independent guideline development process that is not subject to company influence or bias and fully complies with the Academy of Medicine’s criteria for trustworthy guidelines.
AGA has been proactive in developing policies to minimize bias in our guidelines. AGA requires that the Chair of the Guideline Development Group, and a majority of Guideline (and other clinical practice documents) Development Group members are free of conflicts of interest relevant to the subject matter of the guideline. At the time of invitation, we ask our panel members to disclose any and all potential conflicts. Furthermore, all author disclosures are verified by means of accessing publicly available sources (such as the Centers for Medicare and Medicaid Services’ Open Payment database) prior to their involvement on the panel.
AGA strives to be transparent in reporting commercial bias and independent of any industry influence in the development of our clinical practice documents. Our goal is to produce the most trustworthy, actionable, and evidence-based guidelines possible for our members.
Learn more about AGA’s clinical guideline process (https://www.gastro.org/guidelines).
Yngve T. Falck-Ytter, MD, AGAF, is chair, and Shahnaz Sultan, MD, MHSc, AGAF, is chair-elect, AGA Institute Clinical Guidelines Committee.
Financial conflicts are often underreported by authors of clinical practice guidelines (CPGs) in several specialties including oncology, rheumatology, and gastroenterology, according to a pair of research letters published in JAMA Internal Medicine. The Institute of Medicine recommends that guideline authors include no more than 50% individuals with financial conflicts.
In one research letter, Rishad Khan, BSc, of the University of Toronto in Ontario and his colleagues reviewed data on undeclared financial conflicts of interest among authors of guidelines related to high-revenue medications.
The researchers identified CPGs via the National Guideline Clearinghouse and selected 18 CPGs for 10 high-revenue medications published between 2013 and 2017. Financial conflicts of interest were based on the Centers for Medicare & Medicaid Services Open Payments.
Of the 160 authors involved in the various guidelines, 79 (49.4%) disclosed a payment in the CPG or supplemental materials, and 50 (31.3%) disclosed payments from companies marketing 1 of the 10 high-revenue medications in the related guidelines.
Another 41 authors (25.6%) received but did not disclose payments from companies marketing 1 of the 10 high-revenue medications in CPGs.
Overall, 91 authors (56.9%) were found to have financial conflicts of interest that involved 1 of the 10 high-revenue medications, and “the median value of undeclared payments from companies marketing 1 of the 10 high-revenue medications recommended in the CPGs was $522 (interquartile range, $0-$40,444) from two companies,” the researchers said.
The study findings were limited by several factors including “potential inaccuracies in CMS-OP reporting, which are rarely corrected, and lack of generalizability outside the United States” and by the limited time frame for data collection, which may have led to underestimation of conflicts for the guidelines, the researchers noted. In addition, “we did not have access to guideline voting records and thus did not know when conflicted panel members recommended against a medication or recused themselves from voting,” they said.
Mr. Khan disclosed research funding from AbbVie and Ferring Pharmaceuticals.
In a second research letter, half of the authors of gastroenterology guidelines received payments from industry, wrote Tyler Combs, BS, of Oklahoma State University, Tulsa, and his colleagues. Previous studies have reviewed the financial conflicts of interest in specialties including oncology, dermatology, and otolaryngology, but financial conflicts of interest among authors of gastroenterology guidelines have not been examined, the researchers said.
Mr. Combs and his colleagues identified 15 CPGs published by the American College of Gastroenterology between 2014 and 2016. They identified 83 authors, with an average of 4 authors for each guideline. Overall, 53% of the authors received industry payments, according to based on data from the 2014 to 2016 Centers for Medicare & Medicaid Services Open Payments database (OPD).
However, OPD information was not always consistent with information published with the guidelines, the researchers noted. They found that 16 (19%) of the 83 authors both disclosed financial conflicts of interests in the CPGs and had received payments according to OPD or had disclosed no financial conflicts of interest and had received no payments according to OPD. In addition, 49 (34%) of 146 cumulative financial conflicts of interest disclosed in the CPGs and 148 relationships identified on OPD were both disclosed as financial conflicts of interest and evidenced by OPD payment records. In this review, the median total payment was $1,000, with an interquartile range from $0 to $39,938.
The study findings were limited by a relatively short 12-month time frame, the researchers noted. However, “our finding that FCOI [financial conflicts of interest] disclosure only corroborates with OPD payment records between 19% and 34% of the time also suggests that guidance from the ACG [American College of Gastroenterology] may be needed to improve FCOI disclosure efforts in future iterations of gastroenterology CPGs,” they said.
The researchers had no financial conflicts to disclose.
SOURCE: Combs T et al. JAMA Intern Med. 2018 Oct 29. doi: 10.1001/jamainternmed.2018.4730; Khan R et al. JAMA Intern Med. 2018 Oct 29. doi: 10.1001/jamainternmed.2018.5106.
Statement from the AGA on the integrity of AGA’s clinical guideline process
The American Gastroenterological Association (AGA) understands how important it is for AGA members, patients, and the public at large to have access to the most trustworthy, actionable, and evidence-based guidelines in order to achieve the highest possible quality of patient care. In developing guidelines, our goal is to maintain a high level of methodologic rigor through the utilization of an evidence-based approach that is very transparent.
However, not all clinical guidelines are created with equal rigor. Clinicians should examine guidelines closely and consider whether or not they follow the Academy of Medicine’s (formerly the Institute of Medicine’s) standards for trustworthy clinical guidelines. The guideline should be based on a systematic review of the evidence, focus on transparency, have a rigorous conflict of interest system in place, include the involvement of an unconflicted Grading of Recommendations Assessment, Development and Evaluation (GRADE) system-trained methodologist, ideally as a cochair, and the recommendations should be concise and actionable. AGA follows a transparent, independent guideline development process that is not subject to company influence or bias and fully complies with the Academy of Medicine’s criteria for trustworthy guidelines.
AGA has been proactive in developing policies to minimize bias in our guidelines. AGA requires that the Chair of the Guideline Development Group, and a majority of Guideline (and other clinical practice documents) Development Group members are free of conflicts of interest relevant to the subject matter of the guideline. At the time of invitation, we ask our panel members to disclose any and all potential conflicts. Furthermore, all author disclosures are verified by means of accessing publicly available sources (such as the Centers for Medicare and Medicaid Services’ Open Payment database) prior to their involvement on the panel.
AGA strives to be transparent in reporting commercial bias and independent of any industry influence in the development of our clinical practice documents. Our goal is to produce the most trustworthy, actionable, and evidence-based guidelines possible for our members.
Learn more about AGA’s clinical guideline process (https://www.gastro.org/guidelines).
Yngve T. Falck-Ytter, MD, AGAF, is chair, and Shahnaz Sultan, MD, MHSc, AGAF, is chair-elect, AGA Institute Clinical Guidelines Committee.
Stroke, arterial dissection events reported with Lemtrada, FDA says
Instances of stroke and arterial dissection in the head and neck have been reported in some multiple sclerosis patients soon after an infusion of alemtuzumab (Lemtrada), according to a safety announcement issued by the Food and Drug Administration on Nov. 29.

Since the FDA approved alemtuzumab in 2014 for relapsing forms of MS, 13 cases of ischemic and hemorrhagic stroke or arterial dissection have been reported worldwide via the FDA Adverse Event Reporting System, but “additional cases we are unaware of may have occurred,” the FDA said in the announcement.
Most of the patients who developed stroke or arterial lining tears showed symptoms within a day of taking the medication, although one patient reported symptoms three days after treatment. The drug is given via intravenous infusion and is generally reserved for patients with relapsing MS who have not responded adequately to other approved MS medications, according to the FDA.
Symptoms include sudden onset of the following: severe headache or neck pain; numbness or weakness in the arms or legs, especially on only one side of the body; confusion or trouble speaking or understanding speech; vision problems in one or both eyes; and dizziness, loss of balance, or difficulty walking.
As a result of the reports, the FDA has updated the drug label prescribing information and the patient Medication Guide to reflect these risks, and added the risk of stroke to the medication’s existing boxed warning.
Health care providers should remind patients of the potential for stroke and arterial dissection at each treatment visit and advise them to seek immediate medical attention if they experience any of the symptoms reported in previous cases. “The diagnosis is often complicated because early symptoms such as headache and neck pain are not specific,” according to the agency, but patients complaining of such symptoms should be evaluated immediately.
Alemtuzumab was also approved in May 2001 for treating B-cell chronic lymphocytic leukemia (B-CLL) under the brand name Campath. The FDA will update the Campath label to reflect the new warnings and risks.
Instances of stroke and arterial dissection in the head and neck have been reported in some multiple sclerosis patients soon after an infusion of alemtuzumab (Lemtrada), according to a safety announcement issued by the Food and Drug Administration on Nov. 29.
Since the FDA approved alemtuzumab in 2014 for relapsing forms of MS, 13 cases of ischemic and hemorrhagic stroke or arterial dissection have been reported worldwide via the FDA Adverse Event Reporting System, but “additional cases we are unaware of may have occurred,” the FDA said in the announcement.
Most of the patients who developed stroke or arterial lining tears showed symptoms within a day of taking the medication, although one patient reported symptoms three days after treatment. The drug is given via intravenous infusion and is generally reserved for patients with relapsing MS who have not responded adequately to other approved MS medications, according to the FDA.
Symptoms include sudden onset of the following: severe headache or neck pain; numbness or weakness in the arms or legs, especially on only one side of the body; confusion or trouble speaking or understanding speech; vision problems in one or both eyes; and dizziness, loss of balance, or difficulty walking.
As a result of the reports, the FDA has updated the drug label prescribing information and the patient Medication Guide to reflect these risks, and added the risk of stroke to the medication’s existing boxed warning.
Health care providers should remind patients of the potential for stroke and arterial dissection at each treatment visit and advise them to seek immediate medical attention if they experience any of the symptoms reported in previous cases. “The diagnosis is often complicated because early symptoms such as headache and neck pain are not specific,” according to the agency, but patients complaining of such symptoms should be evaluated immediately.
Alemtuzumab was also approved in May 2001 for treating B-cell chronic lymphocytic leukemia (B-CLL) under the brand name Campath. The FDA will update the Campath label to reflect the new warnings and risks.
Instances of stroke and arterial dissection in the head and neck have been reported in some multiple sclerosis patients soon after an infusion of alemtuzumab (Lemtrada), according to a safety announcement issued by the Food and Drug Administration on Nov. 29.
Since the FDA approved alemtuzumab in 2014 for relapsing forms of MS, 13 cases of ischemic and hemorrhagic stroke or arterial dissection have been reported worldwide via the FDA Adverse Event Reporting System, but “additional cases we are unaware of may have occurred,” the FDA said in the announcement.
Most of the patients who developed stroke or arterial lining tears showed symptoms within a day of taking the medication, although one patient reported symptoms three days after treatment. The drug is given via intravenous infusion and is generally reserved for patients with relapsing MS who have not responded adequately to other approved MS medications, according to the FDA.
Symptoms include sudden onset of the following: severe headache or neck pain; numbness or weakness in the arms or legs, especially on only one side of the body; confusion or trouble speaking or understanding speech; vision problems in one or both eyes; and dizziness, loss of balance, or difficulty walking.
As a result of the reports, the FDA has updated the drug label prescribing information and the patient Medication Guide to reflect these risks, and added the risk of stroke to the medication’s existing boxed warning.
Health care providers should remind patients of the potential for stroke and arterial dissection at each treatment visit and advise them to seek immediate medical attention if they experience any of the symptoms reported in previous cases. “The diagnosis is often complicated because early symptoms such as headache and neck pain are not specific,” according to the agency, but patients complaining of such symptoms should be evaluated immediately.
Alemtuzumab was also approved in May 2001 for treating B-cell chronic lymphocytic leukemia (B-CLL) under the brand name Campath. The FDA will update the Campath label to reflect the new warnings and risks.
Less-distressed patients driving increase in outpatient services
Adults with less-severe psychological distress contributed to most of the recent increase in outpatient mental health services, based on survey data from nearly 140,000 adults.
“Rising national rates of suicide, opioid misuse, and opioid-related deaths further suggest increasing psychological distress,” wrote Mark Olfson, MD, MPH, of Columbia University, New York, and his colleagues. “However, it is not known whether or to what extent an increase in mental health treatment has occurred in response to rising rates of psychological distress.”
Dr. Olfson and his colleagues reviewed data from the Medical Expenditure Panel Surveys for the years 2004-2005, 2009-2010, and 2014-2015. Overall, 19% of adults received outpatient mental health services in 2004-2005; the percentage increased to 23% in 2014-2015. About half of the study subjects were women, 67% were white, and the average age was 46 years.
The total percentage of adults with serious psychological distress decreased from 5% in 2004-2005 to 4% in 2014-2015, the researchers noted, although those with serious psychological distress had a greater proportionate increase in the use of outpatient services during the study period, from 54% to 68%.
The number of adults with less-serious distress or no distress who were treated with outpatient mental health services increased from 35 million in 2004-2005 to 48 million in 2014-2015, the researchers wrote in JAMA Psychiatry.
The study results were limited by several factors, including the partial reliance on self-reports of mental health care use and on the limitations of the Kessler 6 scale as an assessment of psychological distress. Other limitations included an absence of data on the specific services used and on the effectiveness of treatments. However, the results suggest that, despite increases in outpatient mental health treatment, many adults with serious psychological distress received no mental health care, they wrote. Individuals with more-severe distress might view mental health care less favorably. In addition, the investigators emphasized the need for continued improvement in general medical settings for detecting and treating or referring adults for mental health service.
Dr. Olfson reported no disclosures. One of the coauthors, Steven C. Marcus, PhD, reported receiving consulting fees from several pharmaceutical companies. The study was supported in part by the National Institutes of Health and the New York State Psychiatric Institute. The Medical Expenditure Panel Surveys are sponsored by the Agency for Healthcare Research and Quality.
SOURCE: Olfson M et al. JAMA Psychiatry. 2018 Nov 28. doi: 10.1001/jamapsychiatry.2018.3550.
Adults with less-severe psychological distress contributed to most of the recent increase in outpatient mental health services, based on survey data from nearly 140,000 adults.
“Rising national rates of suicide, opioid misuse, and opioid-related deaths further suggest increasing psychological distress,” wrote Mark Olfson, MD, MPH, of Columbia University, New York, and his colleagues. “However, it is not known whether or to what extent an increase in mental health treatment has occurred in response to rising rates of psychological distress.”
Dr. Olfson and his colleagues reviewed data from the Medical Expenditure Panel Surveys for the years 2004-2005, 2009-2010, and 2014-2015. Overall, 19% of adults received outpatient mental health services in 2004-2005; the percentage increased to 23% in 2014-2015. About half of the study subjects were women, 67% were white, and the average age was 46 years.
The total percentage of adults with serious psychological distress decreased from 5% in 2004-2005 to 4% in 2014-2015, the researchers noted, although those with serious psychological distress had a greater proportionate increase in the use of outpatient services during the study period, from 54% to 68%.
The number of adults with less-serious distress or no distress who were treated with outpatient mental health services increased from 35 million in 2004-2005 to 48 million in 2014-2015, the researchers wrote in JAMA Psychiatry.
The study results were limited by several factors, including the partial reliance on self-reports of mental health care use and on the limitations of the Kessler 6 scale as an assessment of psychological distress. Other limitations included an absence of data on the specific services used and on the effectiveness of treatments. However, the results suggest that, despite increases in outpatient mental health treatment, many adults with serious psychological distress received no mental health care, they wrote. Individuals with more-severe distress might view mental health care less favorably. In addition, the investigators emphasized the need for continued improvement in general medical settings for detecting and treating or referring adults for mental health service.
Dr. Olfson reported no disclosures. One of the coauthors, Steven C. Marcus, PhD, reported receiving consulting fees from several pharmaceutical companies. The study was supported in part by the National Institutes of Health and the New York State Psychiatric Institute. The Medical Expenditure Panel Surveys are sponsored by the Agency for Healthcare Research and Quality.
SOURCE: Olfson M et al. JAMA Psychiatry. 2018 Nov 28. doi: 10.1001/jamapsychiatry.2018.3550.
Adults with less-severe psychological distress contributed to most of the recent increase in outpatient mental health services, based on survey data from nearly 140,000 adults.
“Rising national rates of suicide, opioid misuse, and opioid-related deaths further suggest increasing psychological distress,” wrote Mark Olfson, MD, MPH, of Columbia University, New York, and his colleagues. “However, it is not known whether or to what extent an increase in mental health treatment has occurred in response to rising rates of psychological distress.”
Dr. Olfson and his colleagues reviewed data from the Medical Expenditure Panel Surveys for the years 2004-2005, 2009-2010, and 2014-2015. Overall, 19% of adults received outpatient mental health services in 2004-2005; the percentage increased to 23% in 2014-2015. About half of the study subjects were women, 67% were white, and the average age was 46 years.
The total percentage of adults with serious psychological distress decreased from 5% in 2004-2005 to 4% in 2014-2015, the researchers noted, although those with serious psychological distress had a greater proportionate increase in the use of outpatient services during the study period, from 54% to 68%.
The number of adults with less-serious distress or no distress who were treated with outpatient mental health services increased from 35 million in 2004-2005 to 48 million in 2014-2015, the researchers wrote in JAMA Psychiatry.
The study results were limited by several factors, including the partial reliance on self-reports of mental health care use and on the limitations of the Kessler 6 scale as an assessment of psychological distress. Other limitations included an absence of data on the specific services used and on the effectiveness of treatments. However, the results suggest that, despite increases in outpatient mental health treatment, many adults with serious psychological distress received no mental health care, they wrote. Individuals with more-severe distress might view mental health care less favorably. In addition, the investigators emphasized the need for continued improvement in general medical settings for detecting and treating or referring adults for mental health service.
Dr. Olfson reported no disclosures. One of the coauthors, Steven C. Marcus, PhD, reported receiving consulting fees from several pharmaceutical companies. The study was supported in part by the National Institutes of Health and the New York State Psychiatric Institute. The Medical Expenditure Panel Surveys are sponsored by the Agency for Healthcare Research and Quality.
SOURCE: Olfson M et al. JAMA Psychiatry. 2018 Nov 28. doi: 10.1001/jamapsychiatry.2018.3550.
FROM JAMA PSYCHIATRY
Key clinical point: Overall use of outpatient mental health services is increasing, but most patients report less-severe or no psychological distress.
Major finding: The percentage of U.S. adults receiving outpatient mental health services increased from 19% in 2004-2005 to 23% in 2014-2015.
Study details: The data come from a review of nationally representative surveys taken in 2004-2005, 2009-2010, and 2014-2015 for a total of 139,862 adults aged 18 years and older.
Disclosures: Dr. Olson reported no disclosures. One of the coauthors, Steven C. Marcus, PhD, reported receiving consulting fees from several pharmaceutical companies. The study was supported in part by the National Institutes of Health and the New York State Psychiatric Institute. The Medical Expenditure Panel Surveys are sponsored by the Agency for Healthcare Research and Quality.
Source: Olfson M et al. JAMA Psychiatry. 2018 Nov 28. doi: 10.1001/jamapsychiatry.2018.3550.
Draft guidelines advise HIV screening for most teens and adults
Individuals aged 15-65 years, including pregnant women, should be screened for HIV infection, and those at risk should be given prophylaxis, according to draft recommendations issued by the U.S. Preventive Services Task Force. The screening recommendation extends to younger adolescents and older adults at increased risk for HIV infection. The recommendations are level A.
HIV remains a significant public health issue in the United States, with rates rising among individuals aged 25-29 years, although the overall number of cases has dropped slightly, according to the USPSTF report.
HIV prevention is a multistep process that includes not only screening but also wearing condoms during sex and using clean needles and syringes if injecting drugs, the researchers noted.
However, those at high risk for HIV, such as intravenous drug users, can help reduce their risk by taking a daily pill, the researchers wrote.
In an evidence report submitted to the Agency for Healthcare Research and Quality, researchers reviewed the Cochrane databases, MEDLINE, and Embase for studies up to June 2018. Based on data from 11 trials, pre-exposure prophylaxis (PrEP) consisting of antiretroviral therapy was associated with decreased risk of HIV infection, compared with placebo or no PrEP, with consistent effects across risk categories, the investigators noted.
The most common HIV risk factors include man-to-man sexual contact, injection drug use, having sex without a condom, exchanging sex for drugs or money, and having sex with an HIV-infected partner, according to the USPSTF report.
Although PrEP was associated with renal and gastrointestinal adverse effects, most were mild and resolved when the therapy either ended or continued long term. The use of PrEP does not absolve high-risk individuals from observing safety in sex activity and intravenous drug use, the researchers noted.
The Task Force’s draft recommendation statements and draft evidence reviews are available for public comment and are posted on the Task Force website at www.uspreventiveservicestaskforce.org. Comments can be submitted from Nov. 20, 2018, to Dec. 26, 2018, at www.uspreventiveservicestaskforce.org/tfcomment.htm.
Individuals aged 15-65 years, including pregnant women, should be screened for HIV infection, and those at risk should be given prophylaxis, according to draft recommendations issued by the U.S. Preventive Services Task Force. The screening recommendation extends to younger adolescents and older adults at increased risk for HIV infection. The recommendations are level A.
HIV remains a significant public health issue in the United States, with rates rising among individuals aged 25-29 years, although the overall number of cases has dropped slightly, according to the USPSTF report.
HIV prevention is a multistep process that includes not only screening but also wearing condoms during sex and using clean needles and syringes if injecting drugs, the researchers noted.
However, those at high risk for HIV, such as intravenous drug users, can help reduce their risk by taking a daily pill, the researchers wrote.
In an evidence report submitted to the Agency for Healthcare Research and Quality, researchers reviewed the Cochrane databases, MEDLINE, and Embase for studies up to June 2018. Based on data from 11 trials, pre-exposure prophylaxis (PrEP) consisting of antiretroviral therapy was associated with decreased risk of HIV infection, compared with placebo or no PrEP, with consistent effects across risk categories, the investigators noted.
The most common HIV risk factors include man-to-man sexual contact, injection drug use, having sex without a condom, exchanging sex for drugs or money, and having sex with an HIV-infected partner, according to the USPSTF report.
Although PrEP was associated with renal and gastrointestinal adverse effects, most were mild and resolved when the therapy either ended or continued long term. The use of PrEP does not absolve high-risk individuals from observing safety in sex activity and intravenous drug use, the researchers noted.
The Task Force’s draft recommendation statements and draft evidence reviews are available for public comment and are posted on the Task Force website at www.uspreventiveservicestaskforce.org. Comments can be submitted from Nov. 20, 2018, to Dec. 26, 2018, at www.uspreventiveservicestaskforce.org/tfcomment.htm.
Individuals aged 15-65 years, including pregnant women, should be screened for HIV infection, and those at risk should be given prophylaxis, according to draft recommendations issued by the U.S. Preventive Services Task Force. The screening recommendation extends to younger adolescents and older adults at increased risk for HIV infection. The recommendations are level A.
HIV remains a significant public health issue in the United States, with rates rising among individuals aged 25-29 years, although the overall number of cases has dropped slightly, according to the USPSTF report.
HIV prevention is a multistep process that includes not only screening but also wearing condoms during sex and using clean needles and syringes if injecting drugs, the researchers noted.
However, those at high risk for HIV, such as intravenous drug users, can help reduce their risk by taking a daily pill, the researchers wrote.
In an evidence report submitted to the Agency for Healthcare Research and Quality, researchers reviewed the Cochrane databases, MEDLINE, and Embase for studies up to June 2018. Based on data from 11 trials, pre-exposure prophylaxis (PrEP) consisting of antiretroviral therapy was associated with decreased risk of HIV infection, compared with placebo or no PrEP, with consistent effects across risk categories, the investigators noted.
The most common HIV risk factors include man-to-man sexual contact, injection drug use, having sex without a condom, exchanging sex for drugs or money, and having sex with an HIV-infected partner, according to the USPSTF report.
Although PrEP was associated with renal and gastrointestinal adverse effects, most were mild and resolved when the therapy either ended or continued long term. The use of PrEP does not absolve high-risk individuals from observing safety in sex activity and intravenous drug use, the researchers noted.
The Task Force’s draft recommendation statements and draft evidence reviews are available for public comment and are posted on the Task Force website at www.uspreventiveservicestaskforce.org. Comments can be submitted from Nov. 20, 2018, to Dec. 26, 2018, at www.uspreventiveservicestaskforce.org/tfcomment.htm.
Experts advise risk stratification for newborn early-onset sepsis
according to a pair of clinical reports published in Pediatrics.
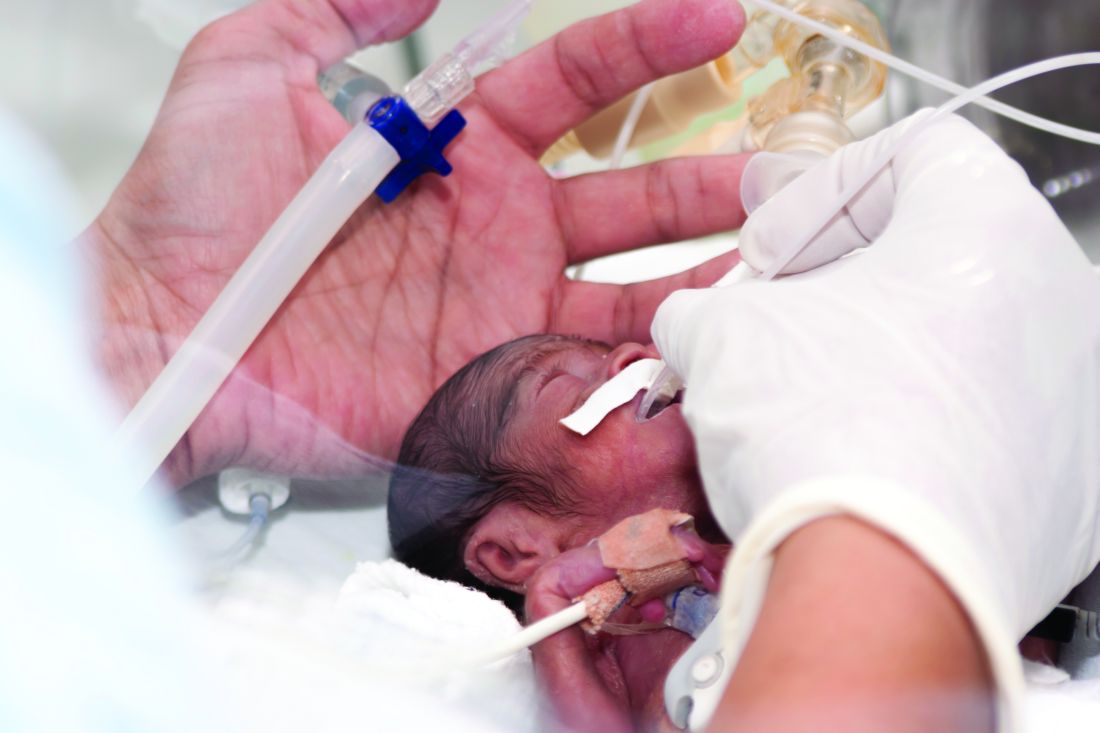
Early-onset sepsis usually begins during labor in term infants, but in preterm infants, “the pathogenesis of preterm EOS likely begins before the onset of labor in many cases of preterm labor and/or PROM [premature rupture of membranes],” wrote Karen M. Puopolo, MD, of the University of Pennsylvania, Philadelphia, and her colleagues.
In the report on preterm infants, the researchers noted that EOS risk assessment using gestational age can be useful for term infants, but not for preterm infants. Instead, they advised categorizing preterm infants as low risk based on birth circumstances. Low-risk preterm infants were defined as those born by cesarean delivery because of maternal noninfectious illness or placental insufficiency in the absence of labor, attempts to induce labor, or rupture of membranes before delivery. Consider the risk/benefit balance of performing an EOS laboratory evaluation and empirical antibiotics, depending on the neonate’s clinical condition, the researchers said.
Preterm infants at high risk for EOS are those born preterm because of maternal cervical incompetence, preterm labor, premature rupture of membranes, clinical concerns for intra-amniotic infection, or acute onset of “unexplained nonreassuring fetal status,” Dr. Puopolo and her associates said. These infants should be managed with a blood culture and empirical antibiotics.
“The combination of ampicillin and gentamicin is the most appropriate empirical antibiotic regimen for infants at risk for EOS,” they noted. “Empirical administration of additional broad-spectrum antibiotics may be indicated in preterm infants who are severely ill and at a high risk for EOS, particularly after prolonged antepartum maternal antibiotic treatment,” they said. Antibiotics should be discontinued by 36-48 hours of incubation unless the infant shows signs of site-specific infection.
In the second report, again with Dr. Puopolo as the primary author, management of EOS was addressed for full-term infants, defined as those born at 35 weeks’ gestation or later.
Infants born at 35 weeks’ gestation or later can be stratified for EOS risk based on algorithms for intrapartum risk factors as well as risk assessments based on these risk factors and infant examinations, the researchers said.
There are a variety of acceptable approaches to risk stratification: categorical algorithms with threshold values for intrapartum risk factors; multivariate risk assessment based on both intrapartum risk factors (such as maternal chorioamnionitis, group B streptococcus colonization, adequacy of intrapartum antibiotic prophylaxis, and duration of ROM); and serial infant examination to detect clinical signs of illness after birth, Dr. Puopolo and her associates wrote.
They recommended that birth centers choose which type of EOS risk assessment to use and tailor it to their own situation. Once local guidelines are developed, ongoing surveillance is suggested.
The same recommendations apply to term infants as preterm infants regarding first-choice use of ampicillin and gentamicin when necessary, to be discontinued when blood cultures are sterile at 36-48 hours of incubation in the absence of site-specific infection, they said.
The reports do “not indicate an exclusive course of treatment or serve as a standard of medical care. Variations, taking into account individual circumstances, may be appropriate,” Dr. Puopolo and her associates noted.
The researchers had no financial conflicts to disclose, and there was no external funding.
SOURCE: Puopolo KM et al. Pediatrics. 2018 Nov. doi: 10.1542/peds.2018-2896; Puopolo KM et al. Pediatrics. 2018 Nov. doi: 10.1542/peds.2018-2894.
according to a pair of clinical reports published in Pediatrics.
Early-onset sepsis usually begins during labor in term infants, but in preterm infants, “the pathogenesis of preterm EOS likely begins before the onset of labor in many cases of preterm labor and/or PROM [premature rupture of membranes],” wrote Karen M. Puopolo, MD, of the University of Pennsylvania, Philadelphia, and her colleagues.
In the report on preterm infants, the researchers noted that EOS risk assessment using gestational age can be useful for term infants, but not for preterm infants. Instead, they advised categorizing preterm infants as low risk based on birth circumstances. Low-risk preterm infants were defined as those born by cesarean delivery because of maternal noninfectious illness or placental insufficiency in the absence of labor, attempts to induce labor, or rupture of membranes before delivery. Consider the risk/benefit balance of performing an EOS laboratory evaluation and empirical antibiotics, depending on the neonate’s clinical condition, the researchers said.
Preterm infants at high risk for EOS are those born preterm because of maternal cervical incompetence, preterm labor, premature rupture of membranes, clinical concerns for intra-amniotic infection, or acute onset of “unexplained nonreassuring fetal status,” Dr. Puopolo and her associates said. These infants should be managed with a blood culture and empirical antibiotics.
“The combination of ampicillin and gentamicin is the most appropriate empirical antibiotic regimen for infants at risk for EOS,” they noted. “Empirical administration of additional broad-spectrum antibiotics may be indicated in preterm infants who are severely ill and at a high risk for EOS, particularly after prolonged antepartum maternal antibiotic treatment,” they said. Antibiotics should be discontinued by 36-48 hours of incubation unless the infant shows signs of site-specific infection.
In the second report, again with Dr. Puopolo as the primary author, management of EOS was addressed for full-term infants, defined as those born at 35 weeks’ gestation or later.
Infants born at 35 weeks’ gestation or later can be stratified for EOS risk based on algorithms for intrapartum risk factors as well as risk assessments based on these risk factors and infant examinations, the researchers said.
There are a variety of acceptable approaches to risk stratification: categorical algorithms with threshold values for intrapartum risk factors; multivariate risk assessment based on both intrapartum risk factors (such as maternal chorioamnionitis, group B streptococcus colonization, adequacy of intrapartum antibiotic prophylaxis, and duration of ROM); and serial infant examination to detect clinical signs of illness after birth, Dr. Puopolo and her associates wrote.
They recommended that birth centers choose which type of EOS risk assessment to use and tailor it to their own situation. Once local guidelines are developed, ongoing surveillance is suggested.
The same recommendations apply to term infants as preterm infants regarding first-choice use of ampicillin and gentamicin when necessary, to be discontinued when blood cultures are sterile at 36-48 hours of incubation in the absence of site-specific infection, they said.
The reports do “not indicate an exclusive course of treatment or serve as a standard of medical care. Variations, taking into account individual circumstances, may be appropriate,” Dr. Puopolo and her associates noted.
The researchers had no financial conflicts to disclose, and there was no external funding.
SOURCE: Puopolo KM et al. Pediatrics. 2018 Nov. doi: 10.1542/peds.2018-2896; Puopolo KM et al. Pediatrics. 2018 Nov. doi: 10.1542/peds.2018-2894.
according to a pair of clinical reports published in Pediatrics.
Early-onset sepsis usually begins during labor in term infants, but in preterm infants, “the pathogenesis of preterm EOS likely begins before the onset of labor in many cases of preterm labor and/or PROM [premature rupture of membranes],” wrote Karen M. Puopolo, MD, of the University of Pennsylvania, Philadelphia, and her colleagues.
In the report on preterm infants, the researchers noted that EOS risk assessment using gestational age can be useful for term infants, but not for preterm infants. Instead, they advised categorizing preterm infants as low risk based on birth circumstances. Low-risk preterm infants were defined as those born by cesarean delivery because of maternal noninfectious illness or placental insufficiency in the absence of labor, attempts to induce labor, or rupture of membranes before delivery. Consider the risk/benefit balance of performing an EOS laboratory evaluation and empirical antibiotics, depending on the neonate’s clinical condition, the researchers said.
Preterm infants at high risk for EOS are those born preterm because of maternal cervical incompetence, preterm labor, premature rupture of membranes, clinical concerns for intra-amniotic infection, or acute onset of “unexplained nonreassuring fetal status,” Dr. Puopolo and her associates said. These infants should be managed with a blood culture and empirical antibiotics.
“The combination of ampicillin and gentamicin is the most appropriate empirical antibiotic regimen for infants at risk for EOS,” they noted. “Empirical administration of additional broad-spectrum antibiotics may be indicated in preterm infants who are severely ill and at a high risk for EOS, particularly after prolonged antepartum maternal antibiotic treatment,” they said. Antibiotics should be discontinued by 36-48 hours of incubation unless the infant shows signs of site-specific infection.
In the second report, again with Dr. Puopolo as the primary author, management of EOS was addressed for full-term infants, defined as those born at 35 weeks’ gestation or later.
Infants born at 35 weeks’ gestation or later can be stratified for EOS risk based on algorithms for intrapartum risk factors as well as risk assessments based on these risk factors and infant examinations, the researchers said.
There are a variety of acceptable approaches to risk stratification: categorical algorithms with threshold values for intrapartum risk factors; multivariate risk assessment based on both intrapartum risk factors (such as maternal chorioamnionitis, group B streptococcus colonization, adequacy of intrapartum antibiotic prophylaxis, and duration of ROM); and serial infant examination to detect clinical signs of illness after birth, Dr. Puopolo and her associates wrote.
They recommended that birth centers choose which type of EOS risk assessment to use and tailor it to their own situation. Once local guidelines are developed, ongoing surveillance is suggested.
The same recommendations apply to term infants as preterm infants regarding first-choice use of ampicillin and gentamicin when necessary, to be discontinued when blood cultures are sterile at 36-48 hours of incubation in the absence of site-specific infection, they said.
The reports do “not indicate an exclusive course of treatment or serve as a standard of medical care. Variations, taking into account individual circumstances, may be appropriate,” Dr. Puopolo and her associates noted.
The researchers had no financial conflicts to disclose, and there was no external funding.
SOURCE: Puopolo KM et al. Pediatrics. 2018 Nov. doi: 10.1542/peds.2018-2896; Puopolo KM et al. Pediatrics. 2018 Nov. doi: 10.1542/peds.2018-2894.
FROM PEDIATRICS
Two-stage preeclampsia screening cuts cost, preserves effectiveness
Using maternal characteristics to identify high-risk pregnant women for second-stage preeclampsia screening achieved similar detection rates as screening all pregnant women, based on data from a prospective study of 61,174 singleton pregnancies.
About 90% of preeclampsia cases can be predicted using the “triple test” of mean arterial pressure (MAP), uterine artery pulsatility index (UtA-PI) and serum placental growth factor (PlGF) plus a combination of maternal factors at 11 to 13 weeks’ gestation, wrote Alan Wright, PhD, of the University of Exeter, United Kingdom, and colleagues. The need for detection of preeclampsia is important, as high-risk pregnant women who take aspirin before 16 weeks’ gestation can reduce early preeclampsia by 90% and preterm preeclampsia by 60%, they said.
However, “measurements of serum PlGF and UtA-PI are not part of routine care and would be associated with an additional cost,” Dr. Wright and his colleagues noted in the American Journal of Obstetrics & Gynecology.
In the study, the researchers used a competing risks model to combine MAP, UtA-PI, and PlGF and maternal factors to compare detection rates if the triple test were used for a subset of high-risk women. Most of the women in the study were white.
Overall, Dr. Wright and his colleagues found, if first-stage screening method is maternal factors, then measurements of MAP, UtA-PI, and PlGF can be reserved for 70% of the pregnant population.
When the researchers compared various components of the triple test, they found that, if the first stage of screening included maternal factors along with MAP and UtA-PI, measurement of PlGF could be saved for 30%-40% of pregnant women. Similarly, if first-stage screening included maternal factors, MAP, and PlGF, measurement of UtA-PI can be saved for 20%-30% of the population, they said.
The study findings were limited by several factors, including the homogeneity of the population studied, the researchers noted. “The risk for development of [preeclampsia] is higher in women of black or South Asian racial origin than in white women,” Dr. Wright and his colleagues wrote. “Consequently, in screening in a population of mixed racial origins, for a given risk cut-off, the [detection rate] and [screen positive rate] would be higher in black and South Asian than white women and the overall performance would be dependent on the proportion of the various racial groups within that population.”
However, the results support the effectiveness of using only certain tests paired with maternal factors to identify high-risk patients for further screening.
“Inevitably, ,” the researchers said.
Dr. Wright and his colleagues reported no conflicts of interest. The study was supported in part by the Fetal Medicine Foundation, and the reagents and equipment used for the measurement of serum placental growth factor were provided by PerkinElmer Life and Analytical Sciences.
SOURCE: Wright A et al. Am J Obstet Gynecol. 2018. doi: 10.1016/j.ajog.2018.10.092.
Using maternal characteristics to identify high-risk pregnant women for second-stage preeclampsia screening achieved similar detection rates as screening all pregnant women, based on data from a prospective study of 61,174 singleton pregnancies.
About 90% of preeclampsia cases can be predicted using the “triple test” of mean arterial pressure (MAP), uterine artery pulsatility index (UtA-PI) and serum placental growth factor (PlGF) plus a combination of maternal factors at 11 to 13 weeks’ gestation, wrote Alan Wright, PhD, of the University of Exeter, United Kingdom, and colleagues. The need for detection of preeclampsia is important, as high-risk pregnant women who take aspirin before 16 weeks’ gestation can reduce early preeclampsia by 90% and preterm preeclampsia by 60%, they said.
However, “measurements of serum PlGF and UtA-PI are not part of routine care and would be associated with an additional cost,” Dr. Wright and his colleagues noted in the American Journal of Obstetrics & Gynecology.
In the study, the researchers used a competing risks model to combine MAP, UtA-PI, and PlGF and maternal factors to compare detection rates if the triple test were used for a subset of high-risk women. Most of the women in the study were white.
Overall, Dr. Wright and his colleagues found, if first-stage screening method is maternal factors, then measurements of MAP, UtA-PI, and PlGF can be reserved for 70% of the pregnant population.
When the researchers compared various components of the triple test, they found that, if the first stage of screening included maternal factors along with MAP and UtA-PI, measurement of PlGF could be saved for 30%-40% of pregnant women. Similarly, if first-stage screening included maternal factors, MAP, and PlGF, measurement of UtA-PI can be saved for 20%-30% of the population, they said.
The study findings were limited by several factors, including the homogeneity of the population studied, the researchers noted. “The risk for development of [preeclampsia] is higher in women of black or South Asian racial origin than in white women,” Dr. Wright and his colleagues wrote. “Consequently, in screening in a population of mixed racial origins, for a given risk cut-off, the [detection rate] and [screen positive rate] would be higher in black and South Asian than white women and the overall performance would be dependent on the proportion of the various racial groups within that population.”
However, the results support the effectiveness of using only certain tests paired with maternal factors to identify high-risk patients for further screening.
“Inevitably, ,” the researchers said.
Dr. Wright and his colleagues reported no conflicts of interest. The study was supported in part by the Fetal Medicine Foundation, and the reagents and equipment used for the measurement of serum placental growth factor were provided by PerkinElmer Life and Analytical Sciences.
SOURCE: Wright A et al. Am J Obstet Gynecol. 2018. doi: 10.1016/j.ajog.2018.10.092.
Using maternal characteristics to identify high-risk pregnant women for second-stage preeclampsia screening achieved similar detection rates as screening all pregnant women, based on data from a prospective study of 61,174 singleton pregnancies.
About 90% of preeclampsia cases can be predicted using the “triple test” of mean arterial pressure (MAP), uterine artery pulsatility index (UtA-PI) and serum placental growth factor (PlGF) plus a combination of maternal factors at 11 to 13 weeks’ gestation, wrote Alan Wright, PhD, of the University of Exeter, United Kingdom, and colleagues. The need for detection of preeclampsia is important, as high-risk pregnant women who take aspirin before 16 weeks’ gestation can reduce early preeclampsia by 90% and preterm preeclampsia by 60%, they said.
However, “measurements of serum PlGF and UtA-PI are not part of routine care and would be associated with an additional cost,” Dr. Wright and his colleagues noted in the American Journal of Obstetrics & Gynecology.
In the study, the researchers used a competing risks model to combine MAP, UtA-PI, and PlGF and maternal factors to compare detection rates if the triple test were used for a subset of high-risk women. Most of the women in the study were white.
Overall, Dr. Wright and his colleagues found, if first-stage screening method is maternal factors, then measurements of MAP, UtA-PI, and PlGF can be reserved for 70% of the pregnant population.
When the researchers compared various components of the triple test, they found that, if the first stage of screening included maternal factors along with MAP and UtA-PI, measurement of PlGF could be saved for 30%-40% of pregnant women. Similarly, if first-stage screening included maternal factors, MAP, and PlGF, measurement of UtA-PI can be saved for 20%-30% of the population, they said.
The study findings were limited by several factors, including the homogeneity of the population studied, the researchers noted. “The risk for development of [preeclampsia] is higher in women of black or South Asian racial origin than in white women,” Dr. Wright and his colleagues wrote. “Consequently, in screening in a population of mixed racial origins, for a given risk cut-off, the [detection rate] and [screen positive rate] would be higher in black and South Asian than white women and the overall performance would be dependent on the proportion of the various racial groups within that population.”
However, the results support the effectiveness of using only certain tests paired with maternal factors to identify high-risk patients for further screening.
“Inevitably, ,” the researchers said.
Dr. Wright and his colleagues reported no conflicts of interest. The study was supported in part by the Fetal Medicine Foundation, and the reagents and equipment used for the measurement of serum placental growth factor were provided by PerkinElmer Life and Analytical Sciences.
SOURCE: Wright A et al. Am J Obstet Gynecol. 2018. doi: 10.1016/j.ajog.2018.10.092.
FROM THE AMERICAN JOURNAL OF OBSTETRICS & GYNECOLOGY
Key clinical point: Reserving two-stage testing for preeclampsia to high-risk subgroups can cut costs without cutting effectiveness.
Major finding: The more costly measurements of MAP, UtA-PI, and PlGF can be reserved for 70% of pregnant women with similar preeclampsia detection rates as testing the entire population.
Study details: The data come from a prospective study of 61,174 singleton pregnancies.
Disclosures: The researchers reported no conflicts of interest. The study was supported in part by the Fetal Medicine Foundation; the reagents and equipment used for the measurement of serum placental growth factor were provided by PerkinElmer Life and Analytical Sciences.
Source: Wright A et al. Am J Obstet Gynecol. 2018. doi: 10.1016/j.ajog.2018.10.092.
AAP advises moderate physical, cognitive activity after sports concussion
according to a new clinical report from the American Academy of Pediatrics.

The update to the 2010 guidelines was needed to reflect the latest research “and it was necessary to provide this new information to guide pediatricians in evaluating and treating concussions they may see in their practice,” Mark Halstead, MD, of Washington University, St. Louis, said in an interview.
The biggest changes to the guidelines involve management of concussion, noted Dr. Halstead, who was a coauthor of the AAP clinical report. “The previous recommendation called for cognitive and physical rest, which unfortunately was interpreted as complete removal from all physical activity and limiting many other things including electronic use.
“Because of research that has been conducted since the original report, it has been shown that starting some light physical activity to increase heart rate, provided it does not worsen symptoms, can be beneficial in recovery. Also, the recommendation for complete removal of electronics and computer use has unfortunately created some issues with kids getting socially isolated,” he added.
“For better or for worse, kids are connected through their electronic devices. Removing them, with no evidence that it worsens the concussion, essentially punishes kids for their injury. We also are trying to discourage prolonged removal of kids from school,” Dr. Halstead emphasized.
The new recommendations emphasize the unique nature of sports-related concussion (SRC) from one individual to another, and the need for individualized management.
Symptoms of SRC fall into five categories, according to the guidelines: somatic, vestibular, oculomotor, cognitive, and emotional/sleep. Pediatric health care providers should rule out more severe head injuries and recognize that concussion symptoms are nonspecific and may reflect preexisting conditions, such as migraine or headache disorders, learning disorders, ADHD, mental health conditions, or sleep disorders.
Use of assessments such as the Sport Concussion Management Tool (SCAT5 for 13 years and older or Child SCAT5 for 5-12 years) can help guide clinicians, but should not be used in isolation to diagnose a concussion, the guideline authors wrote.
Strategies for injury prevention are included in the guidelines as well, such as the use of appropriate headgear. As for management, computerized neurocognitive testing can play a role in decisions regarding return to play, but should not be used in isolation.

“The biggest thing we are lacking is an objective diagnostic test to determine the presence of a concussion or its resolution,” coauthor Kody A. Moffatt, MD, of Creighton University, Omaha, Nebraska, said in an interview.
“Mandatory baseline and postinjury computerized neurocognitive testing is not recommended,” he added.
Clinicians can best manage SRC with prompt recognition and diagnosis using the available tools, followed by relative rest and return to school, then noncontact physical activities, and eventually a return to sport if appropriate.
“Most concussions in children and adolescents will resolve within 4 weeks as long as there is not additional injury to the brain during that time,” Dr. Moffat said.
More research is needed in particular about concussions in elementary and middle school children, Dr. Halstead added.
In the meantime, the take-home message to pediatricians for managing SRC is one of common sense. “Extremes of removing all stimulus from a child is not likely to get them better sooner and research suggests may take them longer to get better,” Dr. Halstead noted. “That doesn’t mean they don’t have to reduce anything, as it is important to reduce physical activity and modify school workload while recovering but we should be avoiding the blanket recommendation to ‘stay home and do nothing until you are better’ approach to concussion management.”
Dr. Halstead and Dr. Moffatt reported no relevant financial conflicts to disclose; the same was true for the other report coauthors. There was no external funding for the report.
SOURCE: Halstead M et al. Pediatrics. 2018 Nov 12. doi: 10.1542/peds.2018-3074.
according to a new clinical report from the American Academy of Pediatrics.
The update to the 2010 guidelines was needed to reflect the latest research “and it was necessary to provide this new information to guide pediatricians in evaluating and treating concussions they may see in their practice,” Mark Halstead, MD, of Washington University, St. Louis, said in an interview.
The biggest changes to the guidelines involve management of concussion, noted Dr. Halstead, who was a coauthor of the AAP clinical report. “The previous recommendation called for cognitive and physical rest, which unfortunately was interpreted as complete removal from all physical activity and limiting many other things including electronic use.
“Because of research that has been conducted since the original report, it has been shown that starting some light physical activity to increase heart rate, provided it does not worsen symptoms, can be beneficial in recovery. Also, the recommendation for complete removal of electronics and computer use has unfortunately created some issues with kids getting socially isolated,” he added.
“For better or for worse, kids are connected through their electronic devices. Removing them, with no evidence that it worsens the concussion, essentially punishes kids for their injury. We also are trying to discourage prolonged removal of kids from school,” Dr. Halstead emphasized.
The new recommendations emphasize the unique nature of sports-related concussion (SRC) from one individual to another, and the need for individualized management.
Symptoms of SRC fall into five categories, according to the guidelines: somatic, vestibular, oculomotor, cognitive, and emotional/sleep. Pediatric health care providers should rule out more severe head injuries and recognize that concussion symptoms are nonspecific and may reflect preexisting conditions, such as migraine or headache disorders, learning disorders, ADHD, mental health conditions, or sleep disorders.
Use of assessments such as the Sport Concussion Management Tool (SCAT5 for 13 years and older or Child SCAT5 for 5-12 years) can help guide clinicians, but should not be used in isolation to diagnose a concussion, the guideline authors wrote.
Strategies for injury prevention are included in the guidelines as well, such as the use of appropriate headgear. As for management, computerized neurocognitive testing can play a role in decisions regarding return to play, but should not be used in isolation.
“The biggest thing we are lacking is an objective diagnostic test to determine the presence of a concussion or its resolution,” coauthor Kody A. Moffatt, MD, of Creighton University, Omaha, Nebraska, said in an interview.
“Mandatory baseline and postinjury computerized neurocognitive testing is not recommended,” he added.
Clinicians can best manage SRC with prompt recognition and diagnosis using the available tools, followed by relative rest and return to school, then noncontact physical activities, and eventually a return to sport if appropriate.
“Most concussions in children and adolescents will resolve within 4 weeks as long as there is not additional injury to the brain during that time,” Dr. Moffat said.
More research is needed in particular about concussions in elementary and middle school children, Dr. Halstead added.
In the meantime, the take-home message to pediatricians for managing SRC is one of common sense. “Extremes of removing all stimulus from a child is not likely to get them better sooner and research suggests may take them longer to get better,” Dr. Halstead noted. “That doesn’t mean they don’t have to reduce anything, as it is important to reduce physical activity and modify school workload while recovering but we should be avoiding the blanket recommendation to ‘stay home and do nothing until you are better’ approach to concussion management.”
Dr. Halstead and Dr. Moffatt reported no relevant financial conflicts to disclose; the same was true for the other report coauthors. There was no external funding for the report.
SOURCE: Halstead M et al. Pediatrics. 2018 Nov 12. doi: 10.1542/peds.2018-3074.
according to a new clinical report from the American Academy of Pediatrics.
The update to the 2010 guidelines was needed to reflect the latest research “and it was necessary to provide this new information to guide pediatricians in evaluating and treating concussions they may see in their practice,” Mark Halstead, MD, of Washington University, St. Louis, said in an interview.
The biggest changes to the guidelines involve management of concussion, noted Dr. Halstead, who was a coauthor of the AAP clinical report. “The previous recommendation called for cognitive and physical rest, which unfortunately was interpreted as complete removal from all physical activity and limiting many other things including electronic use.
“Because of research that has been conducted since the original report, it has been shown that starting some light physical activity to increase heart rate, provided it does not worsen symptoms, can be beneficial in recovery. Also, the recommendation for complete removal of electronics and computer use has unfortunately created some issues with kids getting socially isolated,” he added.
“For better or for worse, kids are connected through their electronic devices. Removing them, with no evidence that it worsens the concussion, essentially punishes kids for their injury. We also are trying to discourage prolonged removal of kids from school,” Dr. Halstead emphasized.
The new recommendations emphasize the unique nature of sports-related concussion (SRC) from one individual to another, and the need for individualized management.
Symptoms of SRC fall into five categories, according to the guidelines: somatic, vestibular, oculomotor, cognitive, and emotional/sleep. Pediatric health care providers should rule out more severe head injuries and recognize that concussion symptoms are nonspecific and may reflect preexisting conditions, such as migraine or headache disorders, learning disorders, ADHD, mental health conditions, or sleep disorders.
Use of assessments such as the Sport Concussion Management Tool (SCAT5 for 13 years and older or Child SCAT5 for 5-12 years) can help guide clinicians, but should not be used in isolation to diagnose a concussion, the guideline authors wrote.
Strategies for injury prevention are included in the guidelines as well, such as the use of appropriate headgear. As for management, computerized neurocognitive testing can play a role in decisions regarding return to play, but should not be used in isolation.
“The biggest thing we are lacking is an objective diagnostic test to determine the presence of a concussion or its resolution,” coauthor Kody A. Moffatt, MD, of Creighton University, Omaha, Nebraska, said in an interview.
“Mandatory baseline and postinjury computerized neurocognitive testing is not recommended,” he added.
Clinicians can best manage SRC with prompt recognition and diagnosis using the available tools, followed by relative rest and return to school, then noncontact physical activities, and eventually a return to sport if appropriate.
“Most concussions in children and adolescents will resolve within 4 weeks as long as there is not additional injury to the brain during that time,” Dr. Moffat said.
More research is needed in particular about concussions in elementary and middle school children, Dr. Halstead added.
In the meantime, the take-home message to pediatricians for managing SRC is one of common sense. “Extremes of removing all stimulus from a child is not likely to get them better sooner and research suggests may take them longer to get better,” Dr. Halstead noted. “That doesn’t mean they don’t have to reduce anything, as it is important to reduce physical activity and modify school workload while recovering but we should be avoiding the blanket recommendation to ‘stay home and do nothing until you are better’ approach to concussion management.”
Dr. Halstead and Dr. Moffatt reported no relevant financial conflicts to disclose; the same was true for the other report coauthors. There was no external funding for the report.
SOURCE: Halstead M et al. Pediatrics. 2018 Nov 12. doi: 10.1542/peds.2018-3074.
FROM PEDIATRICS
USPSTF advises primary care to screen for unhealthy alcohol use
All adults aged 18 years and older, including pregnant women, should be screened in primary care settings for unhealthy alcohol use and offered behavioral counseling if needed, according to recommendations from the U.S. Preventive Services Task Force.

Adults who meet the criteria for unhealthy alcohol use should be offered brief behavioral counseling interventions, the task force concluded with a B recommendation.
However, the task force also concluded that evidence is insufficient to recommend screening for alcohol use in adolescents aged 12-17 years in primary care settings (an I statement), wrote Susan J. Curry, PhD, of the University of Iowa, Iowa City, and colleagues. The recommendations were published in JAMA as an update of the USPSTF 2013 recommendation on screening for unhealthy alcohol use in primary care settings.
Approximately 88,000 deaths occurred each year in the United States between 2006 and 2010, the task force noted. Those deaths include death by acute causes, such as alcohol-related injuries, and chronic causes, such as alcoholic liver disease. In addition, alcohol use during pregnancy is a major preventable cause of birth defects and developmental disabilities, the task force wrote.
After reviewing the evidence, the USPSTF concluded that brief behavioral counseling offered moderate net benefits for adults 18 years and older, including pregnant women, who met criteria for unhealthy alcohol use.
Unhealthy alcohol used was defined as exceeding the National Institute on Alcohol Abuse and Alcoholism (NIAAA) recommended limits of 4 drinks per day, or 14 drinks per week, for men aged 21-64 years, and 3 drinks per day, or 7 drinks per week, for women aged 21-64 years.
In the evidence review accompanying the recommendations, Elizabeth A. O’Connor, PhD, of Kaiser Permanente in Portland, Ore., and colleagues analyzed data from 113 studies, including 314,466 individuals; 10 studies included adolescents.
In 68 studies including 36,528 individuals, brief counseling was associated with fewer drinks per week, fewer individuals exceeding recommended limits for alcohol consumption, fewer drinkers reporting a heavy drinking episode, and a greater proportion of pregnant women reporting alcohol abstinence after 6-12 months.
None of the studies assessed benefits or harms, but no evidence suggested that the interventions could be harmful.
The USPSTF is supported by the Agency for Healthcare Research and Quality. The researchers had no financial conflicts to disclose.
SOURCES: Curry S et al. JAMA. 2018;320(18):1899-1909; O’Connor E et al. JAMA. 2018;320(18):1910-28.
The USPSTF recommendations to screen adults for unhealthy alcohol use acknowledge the serious public health problem it presents, wrote E. Jennifer Edelman, MD, and Jeanette M. Tetrault, MD, in an accompanying editorial.
The recommendations are similar to those issued in 2013 that endorsed screening and brief behavioral interventions for patients with at-risk alcohol use, they said. “Notably, the 2018 recommendations replace alcohol misuse with unhealthy alcohol use and explicitly recommend screening in all pregnant women,” they said.
In clinical practice, most patients with alcohol problems are seen for issues that are consequences of unhealthy alcohol use, such as poorly controlled hypertension, rather than the alcohol use itself, they noted. “Although patients are treated for their immediate problem, they often leave without clear plans to cut back or abstain from alcohol use and thus improve their health.”
Although the recommendations are based on studies showing the effectiveness of brief intervention in primary care, the interventions’ components tend not to be standardized in terms of content, delivery, dose, or duration, the editorialists noted. The terminology used in studies and in clinical practice is inconsistent as well and can cause confusion for doctors and stigma for patients, Dr. Edelman and Dr. Tetrault said.
In addition, they noted that the new USPSTF recommendations don’t incorporate guidance against any alcohol use while taking medications that may interact with it, such as sedating drugs and medications for opioid use disorders.
“Nonetheless, primary care physicians should focus on prevention of alcohol-related harms across the spectrum of alcohol use, including prescribing medications for alcohol use disorder when appropriate,” they noted. “Medications such as naltrexone, acamprosate, and disulfiram can easily be prescribed in primary care and do not require specific training” (JAMA. 2018 Nov 13. doi: 10.1001/jamainternmed.2018.6125).
Dr. Edelman and Dr. Tetrault are affiliated with Yale School of Medicine in New Haven, Conn. They had no financial conflicts to disclose.
The USPSTF recommendations to screen adults for unhealthy alcohol use acknowledge the serious public health problem it presents, wrote E. Jennifer Edelman, MD, and Jeanette M. Tetrault, MD, in an accompanying editorial.
The recommendations are similar to those issued in 2013 that endorsed screening and brief behavioral interventions for patients with at-risk alcohol use, they said. “Notably, the 2018 recommendations replace alcohol misuse with unhealthy alcohol use and explicitly recommend screening in all pregnant women,” they said.
In clinical practice, most patients with alcohol problems are seen for issues that are consequences of unhealthy alcohol use, such as poorly controlled hypertension, rather than the alcohol use itself, they noted. “Although patients are treated for their immediate problem, they often leave without clear plans to cut back or abstain from alcohol use and thus improve their health.”
Although the recommendations are based on studies showing the effectiveness of brief intervention in primary care, the interventions’ components tend not to be standardized in terms of content, delivery, dose, or duration, the editorialists noted. The terminology used in studies and in clinical practice is inconsistent as well and can cause confusion for doctors and stigma for patients, Dr. Edelman and Dr. Tetrault said.
In addition, they noted that the new USPSTF recommendations don’t incorporate guidance against any alcohol use while taking medications that may interact with it, such as sedating drugs and medications for opioid use disorders.
“Nonetheless, primary care physicians should focus on prevention of alcohol-related harms across the spectrum of alcohol use, including prescribing medications for alcohol use disorder when appropriate,” they noted. “Medications such as naltrexone, acamprosate, and disulfiram can easily be prescribed in primary care and do not require specific training” (JAMA. 2018 Nov 13. doi: 10.1001/jamainternmed.2018.6125).
Dr. Edelman and Dr. Tetrault are affiliated with Yale School of Medicine in New Haven, Conn. They had no financial conflicts to disclose.
The USPSTF recommendations to screen adults for unhealthy alcohol use acknowledge the serious public health problem it presents, wrote E. Jennifer Edelman, MD, and Jeanette M. Tetrault, MD, in an accompanying editorial.
The recommendations are similar to those issued in 2013 that endorsed screening and brief behavioral interventions for patients with at-risk alcohol use, they said. “Notably, the 2018 recommendations replace alcohol misuse with unhealthy alcohol use and explicitly recommend screening in all pregnant women,” they said.
In clinical practice, most patients with alcohol problems are seen for issues that are consequences of unhealthy alcohol use, such as poorly controlled hypertension, rather than the alcohol use itself, they noted. “Although patients are treated for their immediate problem, they often leave without clear plans to cut back or abstain from alcohol use and thus improve their health.”
Although the recommendations are based on studies showing the effectiveness of brief intervention in primary care, the interventions’ components tend not to be standardized in terms of content, delivery, dose, or duration, the editorialists noted. The terminology used in studies and in clinical practice is inconsistent as well and can cause confusion for doctors and stigma for patients, Dr. Edelman and Dr. Tetrault said.
In addition, they noted that the new USPSTF recommendations don’t incorporate guidance against any alcohol use while taking medications that may interact with it, such as sedating drugs and medications for opioid use disorders.
“Nonetheless, primary care physicians should focus on prevention of alcohol-related harms across the spectrum of alcohol use, including prescribing medications for alcohol use disorder when appropriate,” they noted. “Medications such as naltrexone, acamprosate, and disulfiram can easily be prescribed in primary care and do not require specific training” (JAMA. 2018 Nov 13. doi: 10.1001/jamainternmed.2018.6125).
Dr. Edelman and Dr. Tetrault are affiliated with Yale School of Medicine in New Haven, Conn. They had no financial conflicts to disclose.
All adults aged 18 years and older, including pregnant women, should be screened in primary care settings for unhealthy alcohol use and offered behavioral counseling if needed, according to recommendations from the U.S. Preventive Services Task Force.
Adults who meet the criteria for unhealthy alcohol use should be offered brief behavioral counseling interventions, the task force concluded with a B recommendation.
However, the task force also concluded that evidence is insufficient to recommend screening for alcohol use in adolescents aged 12-17 years in primary care settings (an I statement), wrote Susan J. Curry, PhD, of the University of Iowa, Iowa City, and colleagues. The recommendations were published in JAMA as an update of the USPSTF 2013 recommendation on screening for unhealthy alcohol use in primary care settings.
Approximately 88,000 deaths occurred each year in the United States between 2006 and 2010, the task force noted. Those deaths include death by acute causes, such as alcohol-related injuries, and chronic causes, such as alcoholic liver disease. In addition, alcohol use during pregnancy is a major preventable cause of birth defects and developmental disabilities, the task force wrote.
After reviewing the evidence, the USPSTF concluded that brief behavioral counseling offered moderate net benefits for adults 18 years and older, including pregnant women, who met criteria for unhealthy alcohol use.
Unhealthy alcohol used was defined as exceeding the National Institute on Alcohol Abuse and Alcoholism (NIAAA) recommended limits of 4 drinks per day, or 14 drinks per week, for men aged 21-64 years, and 3 drinks per day, or 7 drinks per week, for women aged 21-64 years.
In the evidence review accompanying the recommendations, Elizabeth A. O’Connor, PhD, of Kaiser Permanente in Portland, Ore., and colleagues analyzed data from 113 studies, including 314,466 individuals; 10 studies included adolescents.
In 68 studies including 36,528 individuals, brief counseling was associated with fewer drinks per week, fewer individuals exceeding recommended limits for alcohol consumption, fewer drinkers reporting a heavy drinking episode, and a greater proportion of pregnant women reporting alcohol abstinence after 6-12 months.
None of the studies assessed benefits or harms, but no evidence suggested that the interventions could be harmful.
The USPSTF is supported by the Agency for Healthcare Research and Quality. The researchers had no financial conflicts to disclose.
SOURCES: Curry S et al. JAMA. 2018;320(18):1899-1909; O’Connor E et al. JAMA. 2018;320(18):1910-28.
All adults aged 18 years and older, including pregnant women, should be screened in primary care settings for unhealthy alcohol use and offered behavioral counseling if needed, according to recommendations from the U.S. Preventive Services Task Force.
Adults who meet the criteria for unhealthy alcohol use should be offered brief behavioral counseling interventions, the task force concluded with a B recommendation.
However, the task force also concluded that evidence is insufficient to recommend screening for alcohol use in adolescents aged 12-17 years in primary care settings (an I statement), wrote Susan J. Curry, PhD, of the University of Iowa, Iowa City, and colleagues. The recommendations were published in JAMA as an update of the USPSTF 2013 recommendation on screening for unhealthy alcohol use in primary care settings.
Approximately 88,000 deaths occurred each year in the United States between 2006 and 2010, the task force noted. Those deaths include death by acute causes, such as alcohol-related injuries, and chronic causes, such as alcoholic liver disease. In addition, alcohol use during pregnancy is a major preventable cause of birth defects and developmental disabilities, the task force wrote.
After reviewing the evidence, the USPSTF concluded that brief behavioral counseling offered moderate net benefits for adults 18 years and older, including pregnant women, who met criteria for unhealthy alcohol use.
Unhealthy alcohol used was defined as exceeding the National Institute on Alcohol Abuse and Alcoholism (NIAAA) recommended limits of 4 drinks per day, or 14 drinks per week, for men aged 21-64 years, and 3 drinks per day, or 7 drinks per week, for women aged 21-64 years.
In the evidence review accompanying the recommendations, Elizabeth A. O’Connor, PhD, of Kaiser Permanente in Portland, Ore., and colleagues analyzed data from 113 studies, including 314,466 individuals; 10 studies included adolescents.
In 68 studies including 36,528 individuals, brief counseling was associated with fewer drinks per week, fewer individuals exceeding recommended limits for alcohol consumption, fewer drinkers reporting a heavy drinking episode, and a greater proportion of pregnant women reporting alcohol abstinence after 6-12 months.
None of the studies assessed benefits or harms, but no evidence suggested that the interventions could be harmful.
The USPSTF is supported by the Agency for Healthcare Research and Quality. The researchers had no financial conflicts to disclose.
SOURCES: Curry S et al. JAMA. 2018;320(18):1899-1909; O’Connor E et al. JAMA. 2018;320(18):1910-28.
FROM JAMA
FDA accepts priority review of dupilumab for adolescent atopic dermatitis
The Food and Drug Administration has accepted the who have not been well controlled with topical therapies or who are unable to use topical therapies.
In a statement, dupilumab manufacturers Regeneron and Sanofi announced that the target action data for an FDA decision on dupilumab for adolescents is March 11, 2019. “Currently, there are no FDA-approved systemic biologic medicines to treat adolescents with moderate to severe atopic dermatitis,” the companies said in the statement.
The sBLA for dupilumab use in teens is based on data from a phase 3 study presented at the annual congress of European Academy of Dermatology and Venereology in September 2018. In that study, the proportion of patients who achieved a 75% or greater improvement in the Eczema Area and Severity Index at 16 weeks was 38.1% with monthly dupilumab, 41.5% with dupilumab every 2 weeks, and 8.2% with placebo.
According to the companies, the most common adverse events included injection site reactions, oropharyngeal pain, and cold sores. Conjunctivitis has also been reported in some patients.
Dupilumab (Dupixent), which inhibits interleukin-4 and interleukin-13 signaling, is currently approved for treating uncontrolled moderate to severe AD in adults and, more recently, as an add-on maintenance treatment in patients with moderate to severe asthma aged 12 years and older with an eosinophilic phenotype or with oral corticosteroid–dependent asthma.
The FDA granted Breakthrough Therapy designation for dupilumab in 2016 for the treatment of moderate to severe AD in adolescents and severe AD in children aged 6 months to 11 years who are insufficiently controlled with topical medications.
The Food and Drug Administration has accepted the who have not been well controlled with topical therapies or who are unable to use topical therapies.
In a statement, dupilumab manufacturers Regeneron and Sanofi announced that the target action data for an FDA decision on dupilumab for adolescents is March 11, 2019. “Currently, there are no FDA-approved systemic biologic medicines to treat adolescents with moderate to severe atopic dermatitis,” the companies said in the statement.
The sBLA for dupilumab use in teens is based on data from a phase 3 study presented at the annual congress of European Academy of Dermatology and Venereology in September 2018. In that study, the proportion of patients who achieved a 75% or greater improvement in the Eczema Area and Severity Index at 16 weeks was 38.1% with monthly dupilumab, 41.5% with dupilumab every 2 weeks, and 8.2% with placebo.
According to the companies, the most common adverse events included injection site reactions, oropharyngeal pain, and cold sores. Conjunctivitis has also been reported in some patients.
Dupilumab (Dupixent), which inhibits interleukin-4 and interleukin-13 signaling, is currently approved for treating uncontrolled moderate to severe AD in adults and, more recently, as an add-on maintenance treatment in patients with moderate to severe asthma aged 12 years and older with an eosinophilic phenotype or with oral corticosteroid–dependent asthma.
The FDA granted Breakthrough Therapy designation for dupilumab in 2016 for the treatment of moderate to severe AD in adolescents and severe AD in children aged 6 months to 11 years who are insufficiently controlled with topical medications.
The Food and Drug Administration has accepted the who have not been well controlled with topical therapies or who are unable to use topical therapies.
In a statement, dupilumab manufacturers Regeneron and Sanofi announced that the target action data for an FDA decision on dupilumab for adolescents is March 11, 2019. “Currently, there are no FDA-approved systemic biologic medicines to treat adolescents with moderate to severe atopic dermatitis,” the companies said in the statement.
The sBLA for dupilumab use in teens is based on data from a phase 3 study presented at the annual congress of European Academy of Dermatology and Venereology in September 2018. In that study, the proportion of patients who achieved a 75% or greater improvement in the Eczema Area and Severity Index at 16 weeks was 38.1% with monthly dupilumab, 41.5% with dupilumab every 2 weeks, and 8.2% with placebo.
According to the companies, the most common adverse events included injection site reactions, oropharyngeal pain, and cold sores. Conjunctivitis has also been reported in some patients.
Dupilumab (Dupixent), which inhibits interleukin-4 and interleukin-13 signaling, is currently approved for treating uncontrolled moderate to severe AD in adults and, more recently, as an add-on maintenance treatment in patients with moderate to severe asthma aged 12 years and older with an eosinophilic phenotype or with oral corticosteroid–dependent asthma.
The FDA granted Breakthrough Therapy designation for dupilumab in 2016 for the treatment of moderate to severe AD in adolescents and severe AD in children aged 6 months to 11 years who are insufficiently controlled with topical medications.
Guideline authors inconsistently disclose conflicts
Financial conflicts are often underreported by authors of clinical practice guidelines (CPGs) in several specialties including oncology, rheumatology, and gastroenterology, according to a pair of research letters published in JAMA Internal Medicine. The Institute of Medicine recommends that guideline authors include no more than 50% individuals with financial conflicts.
In one research letter, Rishad Khan, BSc, of the University of Toronto in Ontario and his colleagues reviewed data on undeclared financial conflicts of interest among authors of guidelines related to high-revenue medications.
The researchers identified CPGs via the National Guideline Clearinghouse and selected 18 CPGs for 10 high-revenue medications published between 2013 and 2017. Financial conflicts of interest were based on the Centers for Medicare & Medicaid Services Open Payments.
Of the 160 authors involved in the various guidelines, 79 (49.4%) disclosed a payment in the CPG or supplemental materials, and 50 (31.3%) disclosed payments from companies marketing 1 of the 10 high-revenue medications in the related guidelines.
Another 41 authors (25.6%) received but did not disclose payments from companies marketing 1 of the 10 high-revenue medications in CPGs.
Overall, 91 authors (56.9%) were found to have financial conflicts of interest that involved 1 of the 10 high-revenue medications, and “the median value of undeclared payments from companies marketing 1 of the 10 high-revenue medications recommended in the CPGs was $522 (interquartile range, $0-$40,444) from two companies,” the researchers said.
The study findings were limited by several factors including “potential inaccuracies in CMS-OP reporting, which are rarely corrected, and lack of generalizability outside the United States” and by the limited time frame for data collection, which may have led to underestimation of conflicts for the guidelines, the researchers noted. In addition, “we did not have access to guideline voting records and thus did not know when conflicted panel members recommended against a medication or recused themselves from voting,” they said.
Mr. Khan disclosed research funding from AbbVie and Ferring Pharmaceuticals.
In a second research letter, half of the authors of gastroenterology guidelines received payments from industry, wrote Tyler Combs, BS, of Oklahoma State University, Tulsa, and his colleagues. Previous studies have reviewed the financial conflicts of interest in specialties including oncology, dermatology, and otolaryngology, but financial conflicts of interest among authors of gastroenterology guidelines have not been examined, the researchers said.
Mr. Combs and his colleagues identified 15 CPGs published by the American College of Gastroenterology between 2014 and 2016. They identified 83 authors, with an average of 4 authors for each guideline. Overall, 53% of the authors received industry payments, according to based on data from the 2014 to 2016 Centers for Medicare and Medicaid Services Open Payments database (OPD).
However, OPD information was not always consistent with information published with the guidelines, the researchers noted. They found that 16 (19%) of the 83 authors both disclosed financial conflicts of interests in the CPGs and had received payments according to OPD or had disclosed no financial conflicts of interest and had received no payments according to OPD. In addition, 49 (34%) of 146 cumulative financial conflicts of interest disclosed in the CPGs and 148 relationships identified on OPD were both disclosed as financial conflicts of interest and evidenced by OPD payment records. In this review, the median total payment was $1,000, with an interquartile range from $0 to $39,938.
The study findings were limited by a relatively short 12-month time frame, the researchers noted. However, “our finding that FCOI [financial conflicts of interest] disclosure only corroborates with OPD payment records between 19% and 34% of the time also suggests that guidance from the ACG [American College of Gastroenterology] may be needed to improve FCOI disclosure efforts in future iterations of gastroenterology CPGs,” they said.
The researchers had no financial conflicts to disclose.
SOURCE: Combs T et al. JAMA Intern Med. 2018 Oct 29. doi: 10.1001/jamainternmed.2018.4730. Khan R et al. JAMA Intern Med. 2018 Oct 29. doi: 10.1001/jamainternmed.2018.5106.
None of the guidelines included in either study was fully compliant with National Academy of Medicine standards, which include written disclosure, appointing committee chairs or cochairs with no conflicts of interest, and keeping committee members with conflicts to a minority of the committee membership, wrote Colette DeJong, MD, and Robert Steinbrook, MD, in an accompanying editorial. In the study by Khan et al., “Notably, 14 of the 18 panels had chairs with industry payments, and 10 had a majority of members with payments,” they wrote.
However, the federal government has so far shown no interest in supporting a fully independent entity to develop clinical practice guidelines, as occurs in the United Kingdom via the National Institute for Health and Care Excellence. “Preparation of guidelines by an independent public body with assured funding and independence could be an effective approach, not only for eliminating issues related to financial conflicts of interest but also for assuring the use of rigorous methodologies and avoiding the wasteful duplication of efforts by multiple committees,” they wrote.
Financial conflicts in clinical practice guidelines persist in the United States in part because many professional societies have financial conflicts with industry, the editorialists wrote.
“Robust, objective, and unbiased clinical practice guidelines support improvements in patient care; the best interests of patients are the paramount consideration,” they emphasized (JAMA Intern Med. 2018 Oct 29. doi: 10.1001/jamainternmed.2018.4974).
Dr. DeJong is affiliated with the University of California, San Francisco; Dr. Steinbrook is Editor at Large for JAMA Internal Medicine. They had no financial conflicts to disclose.
None of the guidelines included in either study was fully compliant with National Academy of Medicine standards, which include written disclosure, appointing committee chairs or cochairs with no conflicts of interest, and keeping committee members with conflicts to a minority of the committee membership, wrote Colette DeJong, MD, and Robert Steinbrook, MD, in an accompanying editorial. In the study by Khan et al., “Notably, 14 of the 18 panels had chairs with industry payments, and 10 had a majority of members with payments,” they wrote.
However, the federal government has so far shown no interest in supporting a fully independent entity to develop clinical practice guidelines, as occurs in the United Kingdom via the National Institute for Health and Care Excellence. “Preparation of guidelines by an independent public body with assured funding and independence could be an effective approach, not only for eliminating issues related to financial conflicts of interest but also for assuring the use of rigorous methodologies and avoiding the wasteful duplication of efforts by multiple committees,” they wrote.
Financial conflicts in clinical practice guidelines persist in the United States in part because many professional societies have financial conflicts with industry, the editorialists wrote.
“Robust, objective, and unbiased clinical practice guidelines support improvements in patient care; the best interests of patients are the paramount consideration,” they emphasized (JAMA Intern Med. 2018 Oct 29. doi: 10.1001/jamainternmed.2018.4974).
Dr. DeJong is affiliated with the University of California, San Francisco; Dr. Steinbrook is Editor at Large for JAMA Internal Medicine. They had no financial conflicts to disclose.
None of the guidelines included in either study was fully compliant with National Academy of Medicine standards, which include written disclosure, appointing committee chairs or cochairs with no conflicts of interest, and keeping committee members with conflicts to a minority of the committee membership, wrote Colette DeJong, MD, and Robert Steinbrook, MD, in an accompanying editorial. In the study by Khan et al., “Notably, 14 of the 18 panels had chairs with industry payments, and 10 had a majority of members with payments,” they wrote.
However, the federal government has so far shown no interest in supporting a fully independent entity to develop clinical practice guidelines, as occurs in the United Kingdom via the National Institute for Health and Care Excellence. “Preparation of guidelines by an independent public body with assured funding and independence could be an effective approach, not only for eliminating issues related to financial conflicts of interest but also for assuring the use of rigorous methodologies and avoiding the wasteful duplication of efforts by multiple committees,” they wrote.
Financial conflicts in clinical practice guidelines persist in the United States in part because many professional societies have financial conflicts with industry, the editorialists wrote.
“Robust, objective, and unbiased clinical practice guidelines support improvements in patient care; the best interests of patients are the paramount consideration,” they emphasized (JAMA Intern Med. 2018 Oct 29. doi: 10.1001/jamainternmed.2018.4974).
Dr. DeJong is affiliated with the University of California, San Francisco; Dr. Steinbrook is Editor at Large for JAMA Internal Medicine. They had no financial conflicts to disclose.
Financial conflicts are often underreported by authors of clinical practice guidelines (CPGs) in several specialties including oncology, rheumatology, and gastroenterology, according to a pair of research letters published in JAMA Internal Medicine. The Institute of Medicine recommends that guideline authors include no more than 50% individuals with financial conflicts.
In one research letter, Rishad Khan, BSc, of the University of Toronto in Ontario and his colleagues reviewed data on undeclared financial conflicts of interest among authors of guidelines related to high-revenue medications.
The researchers identified CPGs via the National Guideline Clearinghouse and selected 18 CPGs for 10 high-revenue medications published between 2013 and 2017. Financial conflicts of interest were based on the Centers for Medicare & Medicaid Services Open Payments.
Of the 160 authors involved in the various guidelines, 79 (49.4%) disclosed a payment in the CPG or supplemental materials, and 50 (31.3%) disclosed payments from companies marketing 1 of the 10 high-revenue medications in the related guidelines.
Another 41 authors (25.6%) received but did not disclose payments from companies marketing 1 of the 10 high-revenue medications in CPGs.
Overall, 91 authors (56.9%) were found to have financial conflicts of interest that involved 1 of the 10 high-revenue medications, and “the median value of undeclared payments from companies marketing 1 of the 10 high-revenue medications recommended in the CPGs was $522 (interquartile range, $0-$40,444) from two companies,” the researchers said.
The study findings were limited by several factors including “potential inaccuracies in CMS-OP reporting, which are rarely corrected, and lack of generalizability outside the United States” and by the limited time frame for data collection, which may have led to underestimation of conflicts for the guidelines, the researchers noted. In addition, “we did not have access to guideline voting records and thus did not know when conflicted panel members recommended against a medication or recused themselves from voting,” they said.
Mr. Khan disclosed research funding from AbbVie and Ferring Pharmaceuticals.
In a second research letter, half of the authors of gastroenterology guidelines received payments from industry, wrote Tyler Combs, BS, of Oklahoma State University, Tulsa, and his colleagues. Previous studies have reviewed the financial conflicts of interest in specialties including oncology, dermatology, and otolaryngology, but financial conflicts of interest among authors of gastroenterology guidelines have not been examined, the researchers said.
Mr. Combs and his colleagues identified 15 CPGs published by the American College of Gastroenterology between 2014 and 2016. They identified 83 authors, with an average of 4 authors for each guideline. Overall, 53% of the authors received industry payments, according to based on data from the 2014 to 2016 Centers for Medicare and Medicaid Services Open Payments database (OPD).
However, OPD information was not always consistent with information published with the guidelines, the researchers noted. They found that 16 (19%) of the 83 authors both disclosed financial conflicts of interests in the CPGs and had received payments according to OPD or had disclosed no financial conflicts of interest and had received no payments according to OPD. In addition, 49 (34%) of 146 cumulative financial conflicts of interest disclosed in the CPGs and 148 relationships identified on OPD were both disclosed as financial conflicts of interest and evidenced by OPD payment records. In this review, the median total payment was $1,000, with an interquartile range from $0 to $39,938.
The study findings were limited by a relatively short 12-month time frame, the researchers noted. However, “our finding that FCOI [financial conflicts of interest] disclosure only corroborates with OPD payment records between 19% and 34% of the time also suggests that guidance from the ACG [American College of Gastroenterology] may be needed to improve FCOI disclosure efforts in future iterations of gastroenterology CPGs,” they said.
The researchers had no financial conflicts to disclose.
SOURCE: Combs T et al. JAMA Intern Med. 2018 Oct 29. doi: 10.1001/jamainternmed.2018.4730. Khan R et al. JAMA Intern Med. 2018 Oct 29. doi: 10.1001/jamainternmed.2018.5106.
Financial conflicts are often underreported by authors of clinical practice guidelines (CPGs) in several specialties including oncology, rheumatology, and gastroenterology, according to a pair of research letters published in JAMA Internal Medicine. The Institute of Medicine recommends that guideline authors include no more than 50% individuals with financial conflicts.
In one research letter, Rishad Khan, BSc, of the University of Toronto in Ontario and his colleagues reviewed data on undeclared financial conflicts of interest among authors of guidelines related to high-revenue medications.
The researchers identified CPGs via the National Guideline Clearinghouse and selected 18 CPGs for 10 high-revenue medications published between 2013 and 2017. Financial conflicts of interest were based on the Centers for Medicare & Medicaid Services Open Payments.
Of the 160 authors involved in the various guidelines, 79 (49.4%) disclosed a payment in the CPG or supplemental materials, and 50 (31.3%) disclosed payments from companies marketing 1 of the 10 high-revenue medications in the related guidelines.
Another 41 authors (25.6%) received but did not disclose payments from companies marketing 1 of the 10 high-revenue medications in CPGs.
Overall, 91 authors (56.9%) were found to have financial conflicts of interest that involved 1 of the 10 high-revenue medications, and “the median value of undeclared payments from companies marketing 1 of the 10 high-revenue medications recommended in the CPGs was $522 (interquartile range, $0-$40,444) from two companies,” the researchers said.
The study findings were limited by several factors including “potential inaccuracies in CMS-OP reporting, which are rarely corrected, and lack of generalizability outside the United States” and by the limited time frame for data collection, which may have led to underestimation of conflicts for the guidelines, the researchers noted. In addition, “we did not have access to guideline voting records and thus did not know when conflicted panel members recommended against a medication or recused themselves from voting,” they said.
Mr. Khan disclosed research funding from AbbVie and Ferring Pharmaceuticals.
In a second research letter, half of the authors of gastroenterology guidelines received payments from industry, wrote Tyler Combs, BS, of Oklahoma State University, Tulsa, and his colleagues. Previous studies have reviewed the financial conflicts of interest in specialties including oncology, dermatology, and otolaryngology, but financial conflicts of interest among authors of gastroenterology guidelines have not been examined, the researchers said.
Mr. Combs and his colleagues identified 15 CPGs published by the American College of Gastroenterology between 2014 and 2016. They identified 83 authors, with an average of 4 authors for each guideline. Overall, 53% of the authors received industry payments, according to based on data from the 2014 to 2016 Centers for Medicare and Medicaid Services Open Payments database (OPD).
However, OPD information was not always consistent with information published with the guidelines, the researchers noted. They found that 16 (19%) of the 83 authors both disclosed financial conflicts of interests in the CPGs and had received payments according to OPD or had disclosed no financial conflicts of interest and had received no payments according to OPD. In addition, 49 (34%) of 146 cumulative financial conflicts of interest disclosed in the CPGs and 148 relationships identified on OPD were both disclosed as financial conflicts of interest and evidenced by OPD payment records. In this review, the median total payment was $1,000, with an interquartile range from $0 to $39,938.
The study findings were limited by a relatively short 12-month time frame, the researchers noted. However, “our finding that FCOI [financial conflicts of interest] disclosure only corroborates with OPD payment records between 19% and 34% of the time also suggests that guidance from the ACG [American College of Gastroenterology] may be needed to improve FCOI disclosure efforts in future iterations of gastroenterology CPGs,” they said.
The researchers had no financial conflicts to disclose.
SOURCE: Combs T et al. JAMA Intern Med. 2018 Oct 29. doi: 10.1001/jamainternmed.2018.4730. Khan R et al. JAMA Intern Med. 2018 Oct 29. doi: 10.1001/jamainternmed.2018.5106.
FROM JAMA INTERNAL MEDICINE
Key clinical point: Financial conflicts of interest in the development of clinical guidelines persist in the United States.
Major finding: Approximately half of the committee members of guidelines in both studies had financial relationships; many were undisclosed and involved substantial payments.
Study details: The data come from two research letters, including 15 gastroenterology guidelines and 18 guidelines from multiple specialties.
Disclosures: Mr. Khan disclosed research funding from AbbVie and Ferring Pharmaceuticals. Mr. Combs had no financial conflicts to disclose.
Source: Combs T et al. JAMA Intern Med. 2018 Oct 29. doi: 10.1001/jamainternmed.2018.4730. Khan R et al. JAMA Intern Med. 2018 Oct 29. doi: 10.1001/jamainternmed.2018.5106.