User login
Bleeding esophageal varices: Who should receive a shunt?
A transjugular intrahepatic portosystemic shunt (TIPS) has been shown in randomized controlled trials to be effective for:
- Secondary prevention of variceal bleeding
- Controlling refractory ascites in patients with liver cirrhosis.
In addition, findings from retrospective case series have suggested that it helps in cases of:
- Acute variceal bleeding refractory to endoscopic therapy
- Gastropathy due to portal hypertension
- Bleeding gastric varices
- Refractory hepatic hydrothorax
- Hepatorenal syndrome
- Budd-Chiari syndrome
- Veno-occlusive disease
- Hepatopulmonary syndrome.
Here, we discuss the indications for a TIPS in cirrhotic patients with esophageal variceal bleeding.
CIRRHOSIS CAN LEAD TO PORTAL HYPERTENSION, BLEEDING
Cirrhosis of the liver alters the hepatic architecture. Development of regenerating nodules and deposition of connective tissue between these nodules increase the resistance to portal blood flow, which can lead to portal hypertension.1
Esophageal variceal bleeding is a complication of portal hypertension and a major cause of death in patients with liver cirrhosis. Combined treatment with vasoactive drugs, prophylactic antibiotics, and endoscopic band ligation is the standard of care for patients with acute bleeding. However, this treatment fails in about 10% to 15% of these patients. A TIPS creates a connection between the portal and hepatic veins, resulting in portal decompression and homeostasis.2
PRE-TIPS EVALUATION
Patients being considered for a TIPS should be medically assessed before the procedure. The workup should include the following:
- Routine blood tests, including blood type and screen (indirect Coombs test), complete blood cell count, basic metabolic panel, liver function tests, prothrombin time, and partial thromboplastin time
- Doppler ultrasonography of the liver to ensure that the portal and hepatic veins are patent
- Echocardiography to assess pulmonary arterial pressure and right-side heart function
- The hepatic venous pressure gradient, which is measured at the time of TIPS placement, reflects the degree of portal hypertension. A hepatic vein is catheterized, and the right atrial pressure or the free hepatic venous pressure is subtracted from the wedged hepatic venous pressure. The gradient is normally 1 to 5 mm Hg. A gradient greater than 5 mm Hg indicates portal hypertension, and esophageal varices may start to bleed when the gradient is greater than 12 mm Hg. The goal of TIPS placement is to reduce the gradient to less than 12 mm Hg, or at least by 50%.
Heart failure is a contraindication
Pulmonary hypertension may follow TIPS placement because the shunt increases venous return to the heart. Additionally, systemic vascular resistance decreases in patients who have a shunt. This further worsens the hyperdynamic circulatory state already present in patients with cirrhosis. Cardiac output increases in response to these changes. When the heart’s ability to handle this “volume overload” is exceeded, pulmonary venous pressures rise, with increasing ventilation-perfusion mismatch, hypoxia, and pulmonary vasoconstriction; pulmonary edema may ensue.
Congestive heart failure, severe tricuspid regurgitation, and severe pulmonary hypertension (mean pulmonary pressures > 45 mm Hg) are therefore considered absolute contraindications to TIPS placement.3,4 This is why echocardiography is recommended to assess pulmonary pressure along with the size and function of the right side of the heart before proceeding with TIPS insertion.
Other considerations
TIPS insertion is not recommended in patients with active hepatic encephalopathy, which should be adequately controlled before insertion of a TIPS. This can be achieved with lactulose and rifaximin. Lactulose is a laxative; the recommended target is 3 to 4 bowel movements daily. Rifaximin is a poorly absorbed antibiotic that has a wide spectrum of coverage, affecting gram-negative and gram-positive aerobes and anaerobes. It wipes out the gut bacteria and so decreases the production of ammonia by the gut.
Paracentesis is recommended before TIPS placement if a large volume of ascites is present. Draining the fluid allows the liver to drop down and makes it easier to access the portal vein from the hepatic vein.
WHEN TO CONSIDER A TIPS IN ESOPHAGEAL VARICEAL BLEEDING
Acute bleeding refractory to endoscopic therapy

A TIPS remains the only choice to control acute variceal bleeding refractory to medical and endoscopic therapy (Figure 1), with a success rate of 90% to 100%.5 The urgency of TIPS placement is an independent predictor of early mortality.
Esophageal variceal rebleeding
Once varices bleed, the risk of rebleeding is higher than 50%, and rebleeding is associated with a high mortality rate. TIPS should be considered if nonselective beta-blockers and surveillance with upper endoscopy and banding fail to prevent rebleeding, with many studies showing a TIPS to be superior to pharmacologic and endoscopic therapies.6
A meta-analysis in 1999 by Papatheodoridis et al6 found that variceal rebleeding was significantly more frequent with endoscopic therapies, at 47% vs 19% with a TIPS, but the incidence of hepatic encephalopathy was higher with TIPS (34% vs 19%; P < .001), and there was no difference in mortality rates.
Hepatic encephalopathy occurs in 15% to 25% of patients after TIPS procedures. Risk factors include advanced age, poor renal function, and a history of hepatic encephalopathy. Hepatic encephalopathy can be managed with lactulose or rifaximin, or both (see above). Narcotics, antihistamines, and benzodiazepines should be avoided. In rare cases (5%) when hepatic encephalopathy is refractory to medical therapy, liver transplant should be considered.
A surgical distal splenorenal shunt is another option for patients with refractory or recurrent variceal bleeding. In a large randomized controlled trial,7 140 cirrhotic patients with recurrent variceal bleeding were randomized to receive either a distal splenorenal shunt or a TIPS. At a mean follow-up of 48 months, there was no difference in the rates of rebleeding between the two groups (5.5% with a surgical shunt vs 10.5% with a TIPS, P = .29) or in hepatic encephalopathy (50% in both groups). Survival rates were comparable between the two groups at 2 years (81% with a surgical shunt vs 88% with a TIPS) and 5 years (62% vs 61%).
Early use of TIPS after first variceal bleeding
In a 2010 randomized controlled trial,8 63 patients with cirrhosis (Child-Pugh class B or C) and acute variceal bleeding who had received standard medical and endoscopic therapy were randomized to receive either a TIPS within 72 hours of admission or long-term conservative treatment with nonselective beta-blockers and endoscopic band ligation. The 1-year actuarial probability of remaining free of rebleeding or failure to control bleeding was 50% in the conservative treatment group vs 97% in the early-TIPS group (P < .001). The 1-year actuarial survival rate was 61% in the conservative treatment group vs 86% in the early-TIPS group (P < .001).
The authors8 concluded that early use of TIPS in patients with cirrhosis and Child-Pugh scores of 7 to 13 who were hospitalized for acute variceal bleeding was associated with significant reductions in rates of treatment failure and mortality.
- Brenner D, Rippe RA. Pathogenesis of hepatic fibrosis. In: Yamada T, Alpers DH, Laine L, Kaplowitz N, Owyang C, Powell DW, editors. Textbook of Gastroenterology. 4th edition. Philadelphia, PA: Lippincott Williams & Wilkins; 2003.
- Bhogal HK, Sanyal AJ. Using transjugular intrahepatic portosystemic shunts for complications of cirrhosis. Clin Gastroenterol Hepatol 2011; 9:936–946.
- Garcia-Tsao G, Sanyal AJ, Grace ND, Carey WD; Practice Guidelines Committee of American Association for Study of Liver Diseases; Practice Parameters Committee of American College of Gastroenterology. Prevention and management of gastroesophageal varices and variceal hemorrhage in cirrhosis. Am J Gastroenterol 2007; 102:2086–2102.
- Azoulay D, Castaing D, Dennison A, Martino W, Eyraud D, Bismuth H. Transjugular intrahepatic portosystemic shunt worsens the hyperdynamic circulatory state of the cirrhotic patient: preliminary report of a prospective study. Hepatology 1994; 19:129–132.
- Rodríguez-Laiz JM, Bañares R, Echenagusia A, et al. Effects of transjugular intrahepatic portasystemic shunt (TIPS) on splanchnic and systemic hemodynamics, and hepatic function in patients with portal hypertension. Preliminary results. Dig Dis Sci 1995; 40:2121–2127.
- Papatheodoridis GV, Goulis J, Leandro G, Patch D, Burroughs AK. Transjugular intrahepatic portosystemic shunt compared with endoscopic treatment for prevention of variceal rebleeding: a meta-analysis. Hepatology 1999; 30:612–622.
- Henderson JM, Boyer TD, Kutner MH, et al; DIVERT Study Group. Distal splenorenal shunt versus transjugular intrahepatic portal systemic shunt for variceal bleeding: a randomized trial. Gastroenterology 2006; 130:1643–1651.
- García-Pagán JC, Caca K, Bureau C, et al; Early TIPS (Transjugular Intrahepatic Portosystemic Shunt) Cooperative Study Group. Early use of TIPS in patients with cirrhosis and variceal bleeding. N Engl J Med 2010; 362:2370–2379.
A transjugular intrahepatic portosystemic shunt (TIPS) has been shown in randomized controlled trials to be effective for:
- Secondary prevention of variceal bleeding
- Controlling refractory ascites in patients with liver cirrhosis.
In addition, findings from retrospective case series have suggested that it helps in cases of:
- Acute variceal bleeding refractory to endoscopic therapy
- Gastropathy due to portal hypertension
- Bleeding gastric varices
- Refractory hepatic hydrothorax
- Hepatorenal syndrome
- Budd-Chiari syndrome
- Veno-occlusive disease
- Hepatopulmonary syndrome.
Here, we discuss the indications for a TIPS in cirrhotic patients with esophageal variceal bleeding.
CIRRHOSIS CAN LEAD TO PORTAL HYPERTENSION, BLEEDING
Cirrhosis of the liver alters the hepatic architecture. Development of regenerating nodules and deposition of connective tissue between these nodules increase the resistance to portal blood flow, which can lead to portal hypertension.1
Esophageal variceal bleeding is a complication of portal hypertension and a major cause of death in patients with liver cirrhosis. Combined treatment with vasoactive drugs, prophylactic antibiotics, and endoscopic band ligation is the standard of care for patients with acute bleeding. However, this treatment fails in about 10% to 15% of these patients. A TIPS creates a connection between the portal and hepatic veins, resulting in portal decompression and homeostasis.2
PRE-TIPS EVALUATION
Patients being considered for a TIPS should be medically assessed before the procedure. The workup should include the following:
- Routine blood tests, including blood type and screen (indirect Coombs test), complete blood cell count, basic metabolic panel, liver function tests, prothrombin time, and partial thromboplastin time
- Doppler ultrasonography of the liver to ensure that the portal and hepatic veins are patent
- Echocardiography to assess pulmonary arterial pressure and right-side heart function
- The hepatic venous pressure gradient, which is measured at the time of TIPS placement, reflects the degree of portal hypertension. A hepatic vein is catheterized, and the right atrial pressure or the free hepatic venous pressure is subtracted from the wedged hepatic venous pressure. The gradient is normally 1 to 5 mm Hg. A gradient greater than 5 mm Hg indicates portal hypertension, and esophageal varices may start to bleed when the gradient is greater than 12 mm Hg. The goal of TIPS placement is to reduce the gradient to less than 12 mm Hg, or at least by 50%.
Heart failure is a contraindication
Pulmonary hypertension may follow TIPS placement because the shunt increases venous return to the heart. Additionally, systemic vascular resistance decreases in patients who have a shunt. This further worsens the hyperdynamic circulatory state already present in patients with cirrhosis. Cardiac output increases in response to these changes. When the heart’s ability to handle this “volume overload” is exceeded, pulmonary venous pressures rise, with increasing ventilation-perfusion mismatch, hypoxia, and pulmonary vasoconstriction; pulmonary edema may ensue.
Congestive heart failure, severe tricuspid regurgitation, and severe pulmonary hypertension (mean pulmonary pressures > 45 mm Hg) are therefore considered absolute contraindications to TIPS placement.3,4 This is why echocardiography is recommended to assess pulmonary pressure along with the size and function of the right side of the heart before proceeding with TIPS insertion.
Other considerations
TIPS insertion is not recommended in patients with active hepatic encephalopathy, which should be adequately controlled before insertion of a TIPS. This can be achieved with lactulose and rifaximin. Lactulose is a laxative; the recommended target is 3 to 4 bowel movements daily. Rifaximin is a poorly absorbed antibiotic that has a wide spectrum of coverage, affecting gram-negative and gram-positive aerobes and anaerobes. It wipes out the gut bacteria and so decreases the production of ammonia by the gut.
Paracentesis is recommended before TIPS placement if a large volume of ascites is present. Draining the fluid allows the liver to drop down and makes it easier to access the portal vein from the hepatic vein.
WHEN TO CONSIDER A TIPS IN ESOPHAGEAL VARICEAL BLEEDING
Acute bleeding refractory to endoscopic therapy
A TIPS remains the only choice to control acute variceal bleeding refractory to medical and endoscopic therapy (Figure 1), with a success rate of 90% to 100%.5 The urgency of TIPS placement is an independent predictor of early mortality.
Esophageal variceal rebleeding
Once varices bleed, the risk of rebleeding is higher than 50%, and rebleeding is associated with a high mortality rate. TIPS should be considered if nonselective beta-blockers and surveillance with upper endoscopy and banding fail to prevent rebleeding, with many studies showing a TIPS to be superior to pharmacologic and endoscopic therapies.6
A meta-analysis in 1999 by Papatheodoridis et al6 found that variceal rebleeding was significantly more frequent with endoscopic therapies, at 47% vs 19% with a TIPS, but the incidence of hepatic encephalopathy was higher with TIPS (34% vs 19%; P < .001), and there was no difference in mortality rates.
Hepatic encephalopathy occurs in 15% to 25% of patients after TIPS procedures. Risk factors include advanced age, poor renal function, and a history of hepatic encephalopathy. Hepatic encephalopathy can be managed with lactulose or rifaximin, or both (see above). Narcotics, antihistamines, and benzodiazepines should be avoided. In rare cases (5%) when hepatic encephalopathy is refractory to medical therapy, liver transplant should be considered.
A surgical distal splenorenal shunt is another option for patients with refractory or recurrent variceal bleeding. In a large randomized controlled trial,7 140 cirrhotic patients with recurrent variceal bleeding were randomized to receive either a distal splenorenal shunt or a TIPS. At a mean follow-up of 48 months, there was no difference in the rates of rebleeding between the two groups (5.5% with a surgical shunt vs 10.5% with a TIPS, P = .29) or in hepatic encephalopathy (50% in both groups). Survival rates were comparable between the two groups at 2 years (81% with a surgical shunt vs 88% with a TIPS) and 5 years (62% vs 61%).
Early use of TIPS after first variceal bleeding
In a 2010 randomized controlled trial,8 63 patients with cirrhosis (Child-Pugh class B or C) and acute variceal bleeding who had received standard medical and endoscopic therapy were randomized to receive either a TIPS within 72 hours of admission or long-term conservative treatment with nonselective beta-blockers and endoscopic band ligation. The 1-year actuarial probability of remaining free of rebleeding or failure to control bleeding was 50% in the conservative treatment group vs 97% in the early-TIPS group (P < .001). The 1-year actuarial survival rate was 61% in the conservative treatment group vs 86% in the early-TIPS group (P < .001).
The authors8 concluded that early use of TIPS in patients with cirrhosis and Child-Pugh scores of 7 to 13 who were hospitalized for acute variceal bleeding was associated with significant reductions in rates of treatment failure and mortality.
A transjugular intrahepatic portosystemic shunt (TIPS) has been shown in randomized controlled trials to be effective for:
- Secondary prevention of variceal bleeding
- Controlling refractory ascites in patients with liver cirrhosis.
In addition, findings from retrospective case series have suggested that it helps in cases of:
- Acute variceal bleeding refractory to endoscopic therapy
- Gastropathy due to portal hypertension
- Bleeding gastric varices
- Refractory hepatic hydrothorax
- Hepatorenal syndrome
- Budd-Chiari syndrome
- Veno-occlusive disease
- Hepatopulmonary syndrome.
Here, we discuss the indications for a TIPS in cirrhotic patients with esophageal variceal bleeding.
CIRRHOSIS CAN LEAD TO PORTAL HYPERTENSION, BLEEDING
Cirrhosis of the liver alters the hepatic architecture. Development of regenerating nodules and deposition of connective tissue between these nodules increase the resistance to portal blood flow, which can lead to portal hypertension.1
Esophageal variceal bleeding is a complication of portal hypertension and a major cause of death in patients with liver cirrhosis. Combined treatment with vasoactive drugs, prophylactic antibiotics, and endoscopic band ligation is the standard of care for patients with acute bleeding. However, this treatment fails in about 10% to 15% of these patients. A TIPS creates a connection between the portal and hepatic veins, resulting in portal decompression and homeostasis.2
PRE-TIPS EVALUATION
Patients being considered for a TIPS should be medically assessed before the procedure. The workup should include the following:
- Routine blood tests, including blood type and screen (indirect Coombs test), complete blood cell count, basic metabolic panel, liver function tests, prothrombin time, and partial thromboplastin time
- Doppler ultrasonography of the liver to ensure that the portal and hepatic veins are patent
- Echocardiography to assess pulmonary arterial pressure and right-side heart function
- The hepatic venous pressure gradient, which is measured at the time of TIPS placement, reflects the degree of portal hypertension. A hepatic vein is catheterized, and the right atrial pressure or the free hepatic venous pressure is subtracted from the wedged hepatic venous pressure. The gradient is normally 1 to 5 mm Hg. A gradient greater than 5 mm Hg indicates portal hypertension, and esophageal varices may start to bleed when the gradient is greater than 12 mm Hg. The goal of TIPS placement is to reduce the gradient to less than 12 mm Hg, or at least by 50%.
Heart failure is a contraindication
Pulmonary hypertension may follow TIPS placement because the shunt increases venous return to the heart. Additionally, systemic vascular resistance decreases in patients who have a shunt. This further worsens the hyperdynamic circulatory state already present in patients with cirrhosis. Cardiac output increases in response to these changes. When the heart’s ability to handle this “volume overload” is exceeded, pulmonary venous pressures rise, with increasing ventilation-perfusion mismatch, hypoxia, and pulmonary vasoconstriction; pulmonary edema may ensue.
Congestive heart failure, severe tricuspid regurgitation, and severe pulmonary hypertension (mean pulmonary pressures > 45 mm Hg) are therefore considered absolute contraindications to TIPS placement.3,4 This is why echocardiography is recommended to assess pulmonary pressure along with the size and function of the right side of the heart before proceeding with TIPS insertion.
Other considerations
TIPS insertion is not recommended in patients with active hepatic encephalopathy, which should be adequately controlled before insertion of a TIPS. This can be achieved with lactulose and rifaximin. Lactulose is a laxative; the recommended target is 3 to 4 bowel movements daily. Rifaximin is a poorly absorbed antibiotic that has a wide spectrum of coverage, affecting gram-negative and gram-positive aerobes and anaerobes. It wipes out the gut bacteria and so decreases the production of ammonia by the gut.
Paracentesis is recommended before TIPS placement if a large volume of ascites is present. Draining the fluid allows the liver to drop down and makes it easier to access the portal vein from the hepatic vein.
WHEN TO CONSIDER A TIPS IN ESOPHAGEAL VARICEAL BLEEDING
Acute bleeding refractory to endoscopic therapy
A TIPS remains the only choice to control acute variceal bleeding refractory to medical and endoscopic therapy (Figure 1), with a success rate of 90% to 100%.5 The urgency of TIPS placement is an independent predictor of early mortality.
Esophageal variceal rebleeding
Once varices bleed, the risk of rebleeding is higher than 50%, and rebleeding is associated with a high mortality rate. TIPS should be considered if nonselective beta-blockers and surveillance with upper endoscopy and banding fail to prevent rebleeding, with many studies showing a TIPS to be superior to pharmacologic and endoscopic therapies.6
A meta-analysis in 1999 by Papatheodoridis et al6 found that variceal rebleeding was significantly more frequent with endoscopic therapies, at 47% vs 19% with a TIPS, but the incidence of hepatic encephalopathy was higher with TIPS (34% vs 19%; P < .001), and there was no difference in mortality rates.
Hepatic encephalopathy occurs in 15% to 25% of patients after TIPS procedures. Risk factors include advanced age, poor renal function, and a history of hepatic encephalopathy. Hepatic encephalopathy can be managed with lactulose or rifaximin, or both (see above). Narcotics, antihistamines, and benzodiazepines should be avoided. In rare cases (5%) when hepatic encephalopathy is refractory to medical therapy, liver transplant should be considered.
A surgical distal splenorenal shunt is another option for patients with refractory or recurrent variceal bleeding. In a large randomized controlled trial,7 140 cirrhotic patients with recurrent variceal bleeding were randomized to receive either a distal splenorenal shunt or a TIPS. At a mean follow-up of 48 months, there was no difference in the rates of rebleeding between the two groups (5.5% with a surgical shunt vs 10.5% with a TIPS, P = .29) or in hepatic encephalopathy (50% in both groups). Survival rates were comparable between the two groups at 2 years (81% with a surgical shunt vs 88% with a TIPS) and 5 years (62% vs 61%).
Early use of TIPS after first variceal bleeding
In a 2010 randomized controlled trial,8 63 patients with cirrhosis (Child-Pugh class B or C) and acute variceal bleeding who had received standard medical and endoscopic therapy were randomized to receive either a TIPS within 72 hours of admission or long-term conservative treatment with nonselective beta-blockers and endoscopic band ligation. The 1-year actuarial probability of remaining free of rebleeding or failure to control bleeding was 50% in the conservative treatment group vs 97% in the early-TIPS group (P < .001). The 1-year actuarial survival rate was 61% in the conservative treatment group vs 86% in the early-TIPS group (P < .001).
The authors8 concluded that early use of TIPS in patients with cirrhosis and Child-Pugh scores of 7 to 13 who were hospitalized for acute variceal bleeding was associated with significant reductions in rates of treatment failure and mortality.
- Brenner D, Rippe RA. Pathogenesis of hepatic fibrosis. In: Yamada T, Alpers DH, Laine L, Kaplowitz N, Owyang C, Powell DW, editors. Textbook of Gastroenterology. 4th edition. Philadelphia, PA: Lippincott Williams & Wilkins; 2003.
- Bhogal HK, Sanyal AJ. Using transjugular intrahepatic portosystemic shunts for complications of cirrhosis. Clin Gastroenterol Hepatol 2011; 9:936–946.
- Garcia-Tsao G, Sanyal AJ, Grace ND, Carey WD; Practice Guidelines Committee of American Association for Study of Liver Diseases; Practice Parameters Committee of American College of Gastroenterology. Prevention and management of gastroesophageal varices and variceal hemorrhage in cirrhosis. Am J Gastroenterol 2007; 102:2086–2102.
- Azoulay D, Castaing D, Dennison A, Martino W, Eyraud D, Bismuth H. Transjugular intrahepatic portosystemic shunt worsens the hyperdynamic circulatory state of the cirrhotic patient: preliminary report of a prospective study. Hepatology 1994; 19:129–132.
- Rodríguez-Laiz JM, Bañares R, Echenagusia A, et al. Effects of transjugular intrahepatic portasystemic shunt (TIPS) on splanchnic and systemic hemodynamics, and hepatic function in patients with portal hypertension. Preliminary results. Dig Dis Sci 1995; 40:2121–2127.
- Papatheodoridis GV, Goulis J, Leandro G, Patch D, Burroughs AK. Transjugular intrahepatic portosystemic shunt compared with endoscopic treatment for prevention of variceal rebleeding: a meta-analysis. Hepatology 1999; 30:612–622.
- Henderson JM, Boyer TD, Kutner MH, et al; DIVERT Study Group. Distal splenorenal shunt versus transjugular intrahepatic portal systemic shunt for variceal bleeding: a randomized trial. Gastroenterology 2006; 130:1643–1651.
- García-Pagán JC, Caca K, Bureau C, et al; Early TIPS (Transjugular Intrahepatic Portosystemic Shunt) Cooperative Study Group. Early use of TIPS in patients with cirrhosis and variceal bleeding. N Engl J Med 2010; 362:2370–2379.
- Brenner D, Rippe RA. Pathogenesis of hepatic fibrosis. In: Yamada T, Alpers DH, Laine L, Kaplowitz N, Owyang C, Powell DW, editors. Textbook of Gastroenterology. 4th edition. Philadelphia, PA: Lippincott Williams & Wilkins; 2003.
- Bhogal HK, Sanyal AJ. Using transjugular intrahepatic portosystemic shunts for complications of cirrhosis. Clin Gastroenterol Hepatol 2011; 9:936–946.
- Garcia-Tsao G, Sanyal AJ, Grace ND, Carey WD; Practice Guidelines Committee of American Association for Study of Liver Diseases; Practice Parameters Committee of American College of Gastroenterology. Prevention and management of gastroesophageal varices and variceal hemorrhage in cirrhosis. Am J Gastroenterol 2007; 102:2086–2102.
- Azoulay D, Castaing D, Dennison A, Martino W, Eyraud D, Bismuth H. Transjugular intrahepatic portosystemic shunt worsens the hyperdynamic circulatory state of the cirrhotic patient: preliminary report of a prospective study. Hepatology 1994; 19:129–132.
- Rodríguez-Laiz JM, Bañares R, Echenagusia A, et al. Effects of transjugular intrahepatic portasystemic shunt (TIPS) on splanchnic and systemic hemodynamics, and hepatic function in patients with portal hypertension. Preliminary results. Dig Dis Sci 1995; 40:2121–2127.
- Papatheodoridis GV, Goulis J, Leandro G, Patch D, Burroughs AK. Transjugular intrahepatic portosystemic shunt compared with endoscopic treatment for prevention of variceal rebleeding: a meta-analysis. Hepatology 1999; 30:612–622.
- Henderson JM, Boyer TD, Kutner MH, et al; DIVERT Study Group. Distal splenorenal shunt versus transjugular intrahepatic portal systemic shunt for variceal bleeding: a randomized trial. Gastroenterology 2006; 130:1643–1651.
- García-Pagán JC, Caca K, Bureau C, et al; Early TIPS (Transjugular Intrahepatic Portosystemic Shunt) Cooperative Study Group. Early use of TIPS in patients with cirrhosis and variceal bleeding. N Engl J Med 2010; 362:2370–2379.

A female liver transplant recipient asks: Can I become pregnant?
Yes, pregnancy is possible, but not immediately after transplant, and it involves risks. Appropriate management and multidisciplinary care are necessary to optimize the outcomes.
HOW LONG SHOULD PREGNANCY BE POSTPONED?
Hypogonadism and amenorrhea are common and multifactorial in women with end-stage liver disease. Hypogonadotrophic hypogonadism, elevated estrogen level, and malnutrition all contribute to the problem.1 However, most premenopausal women experience a return of their menstrual cycle, and possibly of fertility, within the first 10 months after liver transplant,2,3 after which pregnancy is possible.
In transplant recipients of childbearing age, the need for preconception counseling and family planning should be emphasized. The timing, potential risks, and outcomes of pregnancy, and the importance of coordinated prenatal and perinatal care should be addressed.4 The National Transplant Pregnancy Registry guidelines recommend postponing conception until:
- At least 1 year has elapsed after transplant
- Graft function is stable
- Medical comorbidities such as diabetes and hypertension are well controlled
- Immunosuppression is at a low maintenance level.3
Strong evidence suggests that an appropriate liver transplant-conception interval reduces adverse maternal and fetal outcomes. In particular, the risks of a low birth weight, graft rejection, and loss during pregnancy are significantly decreased.3 Therefore, contraception must be initiated after transplant before any sexual activity, with no preference as to the form of protection used.
Limited data demonstrate the safety and efficacy of combined oral contraceptives and transdermal contraceptive patches in stable solid-organ recipients.5,6 Estrogen-containing contraceptives should, however, be avoided in recurrent liver disease after transplant because of the risk of increased hepatic toxicity.
MANAGING RISKS ASSOCIATED WITH PREGNANCY
Physicians should be alert to the possibility of a pregnancy. Early diagnosis allows the optimization of management and outcomes, as complications are increased in this population of expectant mothers.7
Well-known risks to the expectant liver transplant recipient include hypertension and preeclampsia.8 Moreover, infants born to these patients have a higher risk of prematurity and low birth weight.3,7,9 However, rates of neonatal or maternal deaths and birth defects do not differ significantly from those seen in the general population. Graft rejection is a potential complication, with rates varying between 0% and 20% in different studies.3
Multidisciplinary care is therefore crucial during these high-risk pregnancies.10 An obstetrician, a hepatologist, and a perinatalogist should collaborate to maximize outcomes.11 Frequent evaluations, preferably 2 weeks apart, are suggested for the serial assessment of fetal growth.
Furthermore, daily monitoring of the blood pressure and aggressive management of hypertension are recommended. Methyldopa appears to be the drug treatment of choice.12
Close monitoring of graft function and liver biopsy in suspected graft rejection are of essence as well.3 Routine screening for urinary tract infection, cytomegalovirus and toxoplasmosis infections, gestational diabetes, and preeclampsia should also be undertaken.
MANAGING IMMUNOSUPPRESSION IN THE PREGNANT PATIENT
The choice of immunosuppression is ideally made before pregnancy. All immunosuppressive drugs cross the placenta. Thus, in theory, all agents carry risks of teratogenicity and fetal loss. However, immunosuppression is crucial in avoiding rejection. Furthermore, the use of appropriate immunosuppressive regimens prevents negative outcomes. Drugs are classified as class A (safest to use in pregnancy), through classes B, C, D, and X.
Tacrolimus (class C) monotherapy appears to be safe, with attention to the maintenance of therapeutic levels throughout pregnancy. Allograft function and tacrolimus serum levels need to be monitored because of the change in the volume of drug distribution. Cyclosporine (a pregnancy class C drug), prednisone (class B), and azathioprine (class D) are also reasonable options and may also be used if judged necessary.13
Mycophenolic acid and mTOR (mammalian target of rapamycin) inhibitors such as sirolimus and everolimus are significantly teratogenic and should be avoided in pregnant women. They are more commonly associated with spontaneous abortion, structural abnormalities, and birth defects than other immunosuppressive drugs, especially if taken in the early stages of pregnancy. Cleft lip and palate, absent auditory canals, and microtia have been reported.2,13
- Bell H, Raknerud N, Falch JA, Haug E. Inappropriately low levels of gonadotrophins in amenorrhoeic women with alcoholic and non-alcoholic cirrhosis. Eur J Endocrinol 1995; 132:444–449.
- Mass K, Quint EH, Punch MR, Merion RM. Gynecological and reproductive function after liver transplantation. Transplantation 1996; 62:476–479.
- Coscia LA, Constantinescu S, Moritz MJ, et al. Report from the National Transplantation Pregnancy Registry (NTPR): outcomes of pregnancy after transplantation. Clin Transpl 2009; 103–122.
- Parolin MB, Coelho JC, Urbanetz AA, Pampuch M. Contraception and pregnancy after liver transplantation: an update overview. Arq Gastroenterol 2009; 46:154–158. In Portuguese.
- Paulen ME, Folger SG, Curtis KM, Jamieson DJ. Contraceptive use among solid organ transplant patients: a systematic review. Contraception 2010; 82:102–112.
- Jabiry-Zieniewicz Z, Bobrowska K, Kaminski P, Wielgos M, Zieniewicz K, Krawczyk M. Low-dose hormonal contraception after liver transplantation. Transplant Proc 2007; 39:1530–1532.
- Coffin CS, Shaheen AA, Burak KW, Myers RP. Pregnancy outcomes among liver transplant recipients in the United States: a nationwide case-control analysis. Liver Transpl 2010; 16:56–63.
- Heneghan MA, Selzner M, Yoshida EM, Mullhaupt B. Pregnancy and sexual function in liver transplantation. J Hepatol 2008; 49:507–519.
- Ho JK, Ko HH, Schaeffer DF, et al. Sexual health after orthotopic liver transplantation. Liver Transpl 2006; 12:1478–1484.
- Jabiry-Zieniewicz Z, Dabrowski FA, Pietrzak B, Wielgos M. Pregnancy complications after liver transplantation. Int J Gynaecol Obstet 2015; 128:27–29.
- Parhar KS, Gibson PS, Coffin CS. Pregnancy following liver transplantation: review of outcomes and recommendations for management. Can J Gastroenterol 2012; 26:621–626.
- McKay DB, Josephson MA, Armenti VT, et al; Women’s Health Committee of the American Society of Transplantation. Reproduction and transplantation: report on the AST Consensus Conference on Reproductive Issues and Transplantation. Am J Transplant 2005; 5:1592–1599.
- Sifontis NM, Coscia LA, Constantinescu S, Lavelanet AF, Moritz MJ, Armenti VT. Pregnancy outcomes in solid organ transplant recipients with exposure to mycophenolate mofetil or sirolimus. Transplantation 2006; 82:1698–1702.
Yes, pregnancy is possible, but not immediately after transplant, and it involves risks. Appropriate management and multidisciplinary care are necessary to optimize the outcomes.
HOW LONG SHOULD PREGNANCY BE POSTPONED?
Hypogonadism and amenorrhea are common and multifactorial in women with end-stage liver disease. Hypogonadotrophic hypogonadism, elevated estrogen level, and malnutrition all contribute to the problem.1 However, most premenopausal women experience a return of their menstrual cycle, and possibly of fertility, within the first 10 months after liver transplant,2,3 after which pregnancy is possible.
In transplant recipients of childbearing age, the need for preconception counseling and family planning should be emphasized. The timing, potential risks, and outcomes of pregnancy, and the importance of coordinated prenatal and perinatal care should be addressed.4 The National Transplant Pregnancy Registry guidelines recommend postponing conception until:
- At least 1 year has elapsed after transplant
- Graft function is stable
- Medical comorbidities such as diabetes and hypertension are well controlled
- Immunosuppression is at a low maintenance level.3
Strong evidence suggests that an appropriate liver transplant-conception interval reduces adverse maternal and fetal outcomes. In particular, the risks of a low birth weight, graft rejection, and loss during pregnancy are significantly decreased.3 Therefore, contraception must be initiated after transplant before any sexual activity, with no preference as to the form of protection used.
Limited data demonstrate the safety and efficacy of combined oral contraceptives and transdermal contraceptive patches in stable solid-organ recipients.5,6 Estrogen-containing contraceptives should, however, be avoided in recurrent liver disease after transplant because of the risk of increased hepatic toxicity.
MANAGING RISKS ASSOCIATED WITH PREGNANCY
Physicians should be alert to the possibility of a pregnancy. Early diagnosis allows the optimization of management and outcomes, as complications are increased in this population of expectant mothers.7
Well-known risks to the expectant liver transplant recipient include hypertension and preeclampsia.8 Moreover, infants born to these patients have a higher risk of prematurity and low birth weight.3,7,9 However, rates of neonatal or maternal deaths and birth defects do not differ significantly from those seen in the general population. Graft rejection is a potential complication, with rates varying between 0% and 20% in different studies.3
Multidisciplinary care is therefore crucial during these high-risk pregnancies.10 An obstetrician, a hepatologist, and a perinatalogist should collaborate to maximize outcomes.11 Frequent evaluations, preferably 2 weeks apart, are suggested for the serial assessment of fetal growth.
Furthermore, daily monitoring of the blood pressure and aggressive management of hypertension are recommended. Methyldopa appears to be the drug treatment of choice.12
Close monitoring of graft function and liver biopsy in suspected graft rejection are of essence as well.3 Routine screening for urinary tract infection, cytomegalovirus and toxoplasmosis infections, gestational diabetes, and preeclampsia should also be undertaken.
MANAGING IMMUNOSUPPRESSION IN THE PREGNANT PATIENT
The choice of immunosuppression is ideally made before pregnancy. All immunosuppressive drugs cross the placenta. Thus, in theory, all agents carry risks of teratogenicity and fetal loss. However, immunosuppression is crucial in avoiding rejection. Furthermore, the use of appropriate immunosuppressive regimens prevents negative outcomes. Drugs are classified as class A (safest to use in pregnancy), through classes B, C, D, and X.
Tacrolimus (class C) monotherapy appears to be safe, with attention to the maintenance of therapeutic levels throughout pregnancy. Allograft function and tacrolimus serum levels need to be monitored because of the change in the volume of drug distribution. Cyclosporine (a pregnancy class C drug), prednisone (class B), and azathioprine (class D) are also reasonable options and may also be used if judged necessary.13
Mycophenolic acid and mTOR (mammalian target of rapamycin) inhibitors such as sirolimus and everolimus are significantly teratogenic and should be avoided in pregnant women. They are more commonly associated with spontaneous abortion, structural abnormalities, and birth defects than other immunosuppressive drugs, especially if taken in the early stages of pregnancy. Cleft lip and palate, absent auditory canals, and microtia have been reported.2,13
Yes, pregnancy is possible, but not immediately after transplant, and it involves risks. Appropriate management and multidisciplinary care are necessary to optimize the outcomes.
HOW LONG SHOULD PREGNANCY BE POSTPONED?
Hypogonadism and amenorrhea are common and multifactorial in women with end-stage liver disease. Hypogonadotrophic hypogonadism, elevated estrogen level, and malnutrition all contribute to the problem.1 However, most premenopausal women experience a return of their menstrual cycle, and possibly of fertility, within the first 10 months after liver transplant,2,3 after which pregnancy is possible.
In transplant recipients of childbearing age, the need for preconception counseling and family planning should be emphasized. The timing, potential risks, and outcomes of pregnancy, and the importance of coordinated prenatal and perinatal care should be addressed.4 The National Transplant Pregnancy Registry guidelines recommend postponing conception until:
- At least 1 year has elapsed after transplant
- Graft function is stable
- Medical comorbidities such as diabetes and hypertension are well controlled
- Immunosuppression is at a low maintenance level.3
Strong evidence suggests that an appropriate liver transplant-conception interval reduces adverse maternal and fetal outcomes. In particular, the risks of a low birth weight, graft rejection, and loss during pregnancy are significantly decreased.3 Therefore, contraception must be initiated after transplant before any sexual activity, with no preference as to the form of protection used.
Limited data demonstrate the safety and efficacy of combined oral contraceptives and transdermal contraceptive patches in stable solid-organ recipients.5,6 Estrogen-containing contraceptives should, however, be avoided in recurrent liver disease after transplant because of the risk of increased hepatic toxicity.
MANAGING RISKS ASSOCIATED WITH PREGNANCY
Physicians should be alert to the possibility of a pregnancy. Early diagnosis allows the optimization of management and outcomes, as complications are increased in this population of expectant mothers.7
Well-known risks to the expectant liver transplant recipient include hypertension and preeclampsia.8 Moreover, infants born to these patients have a higher risk of prematurity and low birth weight.3,7,9 However, rates of neonatal or maternal deaths and birth defects do not differ significantly from those seen in the general population. Graft rejection is a potential complication, with rates varying between 0% and 20% in different studies.3
Multidisciplinary care is therefore crucial during these high-risk pregnancies.10 An obstetrician, a hepatologist, and a perinatalogist should collaborate to maximize outcomes.11 Frequent evaluations, preferably 2 weeks apart, are suggested for the serial assessment of fetal growth.
Furthermore, daily monitoring of the blood pressure and aggressive management of hypertension are recommended. Methyldopa appears to be the drug treatment of choice.12
Close monitoring of graft function and liver biopsy in suspected graft rejection are of essence as well.3 Routine screening for urinary tract infection, cytomegalovirus and toxoplasmosis infections, gestational diabetes, and preeclampsia should also be undertaken.
MANAGING IMMUNOSUPPRESSION IN THE PREGNANT PATIENT
The choice of immunosuppression is ideally made before pregnancy. All immunosuppressive drugs cross the placenta. Thus, in theory, all agents carry risks of teratogenicity and fetal loss. However, immunosuppression is crucial in avoiding rejection. Furthermore, the use of appropriate immunosuppressive regimens prevents negative outcomes. Drugs are classified as class A (safest to use in pregnancy), through classes B, C, D, and X.
Tacrolimus (class C) monotherapy appears to be safe, with attention to the maintenance of therapeutic levels throughout pregnancy. Allograft function and tacrolimus serum levels need to be monitored because of the change in the volume of drug distribution. Cyclosporine (a pregnancy class C drug), prednisone (class B), and azathioprine (class D) are also reasonable options and may also be used if judged necessary.13
Mycophenolic acid and mTOR (mammalian target of rapamycin) inhibitors such as sirolimus and everolimus are significantly teratogenic and should be avoided in pregnant women. They are more commonly associated with spontaneous abortion, structural abnormalities, and birth defects than other immunosuppressive drugs, especially if taken in the early stages of pregnancy. Cleft lip and palate, absent auditory canals, and microtia have been reported.2,13
- Bell H, Raknerud N, Falch JA, Haug E. Inappropriately low levels of gonadotrophins in amenorrhoeic women with alcoholic and non-alcoholic cirrhosis. Eur J Endocrinol 1995; 132:444–449.
- Mass K, Quint EH, Punch MR, Merion RM. Gynecological and reproductive function after liver transplantation. Transplantation 1996; 62:476–479.
- Coscia LA, Constantinescu S, Moritz MJ, et al. Report from the National Transplantation Pregnancy Registry (NTPR): outcomes of pregnancy after transplantation. Clin Transpl 2009; 103–122.
- Parolin MB, Coelho JC, Urbanetz AA, Pampuch M. Contraception and pregnancy after liver transplantation: an update overview. Arq Gastroenterol 2009; 46:154–158. In Portuguese.
- Paulen ME, Folger SG, Curtis KM, Jamieson DJ. Contraceptive use among solid organ transplant patients: a systematic review. Contraception 2010; 82:102–112.
- Jabiry-Zieniewicz Z, Bobrowska K, Kaminski P, Wielgos M, Zieniewicz K, Krawczyk M. Low-dose hormonal contraception after liver transplantation. Transplant Proc 2007; 39:1530–1532.
- Coffin CS, Shaheen AA, Burak KW, Myers RP. Pregnancy outcomes among liver transplant recipients in the United States: a nationwide case-control analysis. Liver Transpl 2010; 16:56–63.
- Heneghan MA, Selzner M, Yoshida EM, Mullhaupt B. Pregnancy and sexual function in liver transplantation. J Hepatol 2008; 49:507–519.
- Ho JK, Ko HH, Schaeffer DF, et al. Sexual health after orthotopic liver transplantation. Liver Transpl 2006; 12:1478–1484.
- Jabiry-Zieniewicz Z, Dabrowski FA, Pietrzak B, Wielgos M. Pregnancy complications after liver transplantation. Int J Gynaecol Obstet 2015; 128:27–29.
- Parhar KS, Gibson PS, Coffin CS. Pregnancy following liver transplantation: review of outcomes and recommendations for management. Can J Gastroenterol 2012; 26:621–626.
- McKay DB, Josephson MA, Armenti VT, et al; Women’s Health Committee of the American Society of Transplantation. Reproduction and transplantation: report on the AST Consensus Conference on Reproductive Issues and Transplantation. Am J Transplant 2005; 5:1592–1599.
- Sifontis NM, Coscia LA, Constantinescu S, Lavelanet AF, Moritz MJ, Armenti VT. Pregnancy outcomes in solid organ transplant recipients with exposure to mycophenolate mofetil or sirolimus. Transplantation 2006; 82:1698–1702.
- Bell H, Raknerud N, Falch JA, Haug E. Inappropriately low levels of gonadotrophins in amenorrhoeic women with alcoholic and non-alcoholic cirrhosis. Eur J Endocrinol 1995; 132:444–449.
- Mass K, Quint EH, Punch MR, Merion RM. Gynecological and reproductive function after liver transplantation. Transplantation 1996; 62:476–479.
- Coscia LA, Constantinescu S, Moritz MJ, et al. Report from the National Transplantation Pregnancy Registry (NTPR): outcomes of pregnancy after transplantation. Clin Transpl 2009; 103–122.
- Parolin MB, Coelho JC, Urbanetz AA, Pampuch M. Contraception and pregnancy after liver transplantation: an update overview. Arq Gastroenterol 2009; 46:154–158. In Portuguese.
- Paulen ME, Folger SG, Curtis KM, Jamieson DJ. Contraceptive use among solid organ transplant patients: a systematic review. Contraception 2010; 82:102–112.
- Jabiry-Zieniewicz Z, Bobrowska K, Kaminski P, Wielgos M, Zieniewicz K, Krawczyk M. Low-dose hormonal contraception after liver transplantation. Transplant Proc 2007; 39:1530–1532.
- Coffin CS, Shaheen AA, Burak KW, Myers RP. Pregnancy outcomes among liver transplant recipients in the United States: a nationwide case-control analysis. Liver Transpl 2010; 16:56–63.
- Heneghan MA, Selzner M, Yoshida EM, Mullhaupt B. Pregnancy and sexual function in liver transplantation. J Hepatol 2008; 49:507–519.
- Ho JK, Ko HH, Schaeffer DF, et al. Sexual health after orthotopic liver transplantation. Liver Transpl 2006; 12:1478–1484.
- Jabiry-Zieniewicz Z, Dabrowski FA, Pietrzak B, Wielgos M. Pregnancy complications after liver transplantation. Int J Gynaecol Obstet 2015; 128:27–29.
- Parhar KS, Gibson PS, Coffin CS. Pregnancy following liver transplantation: review of outcomes and recommendations for management. Can J Gastroenterol 2012; 26:621–626.
- McKay DB, Josephson MA, Armenti VT, et al; Women’s Health Committee of the American Society of Transplantation. Reproduction and transplantation: report on the AST Consensus Conference on Reproductive Issues and Transplantation. Am J Transplant 2005; 5:1592–1599.
- Sifontis NM, Coscia LA, Constantinescu S, Lavelanet AF, Moritz MJ, Armenti VT. Pregnancy outcomes in solid organ transplant recipients with exposure to mycophenolate mofetil or sirolimus. Transplantation 2006; 82:1698–1702.
In reply: Wilson disease
In Reply: We thank Dr. Mirrakhimov and colleagues for bringing important questions to our attention.
In terms of the differential diagnosis of cholestatic liver injury, we agree that pathologic processes such choledocholithiasis, cholangitis, primary biliary cirrhosis, and primary sclerosing cholangitis should be generally considered. However, in the case we described, the patient had no abdominal pain or fever, which makes choledocholithiasis or cholangitis very unlikely. Primary biliary cirrhosis and primary sclerosing cholangitis can cause chronic liver disease but should not be considered in the differential diagnosis of acute liver injury (acute hepatitis), such as in the case we described.
We agree that the hemolytic anemia typically seen in patients with Wilson disease is Coombs-negative, and that Coombs testing and a peripheral smear should be performed. Both were negative in our patient.
We also agree with Dr. Mirrakhimov and colleagues that Kayser-Fleischer rings are not necessarily specific for Wilson disease and can be seen in patients with other forms of cholestatic liver disease such as primary biliary cirrhosis. However, Kayser-Fleischer rings are pathognomonic for acute liver failure from Wilson disease. In other words, when Kayser-Fleischer rings are seen in a patient with acute liver failure, the diagnosis is Wilson disease until proven otherwise.
We discussed on page 112 of our article other treatments such as plasmapheresis as adjunctive therapy to bridge patients with acute liver failure secondary to Wilson disease to transplant. However, liver transplant is still the only definitive and potentially curative treatment.
In Reply: We thank Dr. Mirrakhimov and colleagues for bringing important questions to our attention.
In terms of the differential diagnosis of cholestatic liver injury, we agree that pathologic processes such choledocholithiasis, cholangitis, primary biliary cirrhosis, and primary sclerosing cholangitis should be generally considered. However, in the case we described, the patient had no abdominal pain or fever, which makes choledocholithiasis or cholangitis very unlikely. Primary biliary cirrhosis and primary sclerosing cholangitis can cause chronic liver disease but should not be considered in the differential diagnosis of acute liver injury (acute hepatitis), such as in the case we described.
We agree that the hemolytic anemia typically seen in patients with Wilson disease is Coombs-negative, and that Coombs testing and a peripheral smear should be performed. Both were negative in our patient.
We also agree with Dr. Mirrakhimov and colleagues that Kayser-Fleischer rings are not necessarily specific for Wilson disease and can be seen in patients with other forms of cholestatic liver disease such as primary biliary cirrhosis. However, Kayser-Fleischer rings are pathognomonic for acute liver failure from Wilson disease. In other words, when Kayser-Fleischer rings are seen in a patient with acute liver failure, the diagnosis is Wilson disease until proven otherwise.
We discussed on page 112 of our article other treatments such as plasmapheresis as adjunctive therapy to bridge patients with acute liver failure secondary to Wilson disease to transplant. However, liver transplant is still the only definitive and potentially curative treatment.
In Reply: We thank Dr. Mirrakhimov and colleagues for bringing important questions to our attention.
In terms of the differential diagnosis of cholestatic liver injury, we agree that pathologic processes such choledocholithiasis, cholangitis, primary biliary cirrhosis, and primary sclerosing cholangitis should be generally considered. However, in the case we described, the patient had no abdominal pain or fever, which makes choledocholithiasis or cholangitis very unlikely. Primary biliary cirrhosis and primary sclerosing cholangitis can cause chronic liver disease but should not be considered in the differential diagnosis of acute liver injury (acute hepatitis), such as in the case we described.
We agree that the hemolytic anemia typically seen in patients with Wilson disease is Coombs-negative, and that Coombs testing and a peripheral smear should be performed. Both were negative in our patient.
We also agree with Dr. Mirrakhimov and colleagues that Kayser-Fleischer rings are not necessarily specific for Wilson disease and can be seen in patients with other forms of cholestatic liver disease such as primary biliary cirrhosis. However, Kayser-Fleischer rings are pathognomonic for acute liver failure from Wilson disease. In other words, when Kayser-Fleischer rings are seen in a patient with acute liver failure, the diagnosis is Wilson disease until proven otherwise.
We discussed on page 112 of our article other treatments such as plasmapheresis as adjunctive therapy to bridge patients with acute liver failure secondary to Wilson disease to transplant. However, liver transplant is still the only definitive and potentially curative treatment.
A guide to managing acute liver failure
When the liver fails, it usually fails gradually. The sudden (acute) onset of liver failure, while less common, demands prompt management, with transfer to an intensive care unit, specific treatment depending on the cause, and consideration of liver transplant, without which the mortality rate is high.
This article reviews the definition, epidemiology, etiology, and management of acute liver failure.
DEFINITIONS
Acute liver failure is defined as a syndrome of acute hepatitis with evidence of abnormal coagulation (eg, an international normalized ratio > 1.5) complicated by the development of mental alteration (encephalopathy) within 26 weeks of the onset of illness in a patient without a history of liver disease.1 In general, patients have no evidence of underlying chronic liver disease, but there are exceptions; patients with Wilson disease, vertically acquired hepatitis B virus infection, or autoimmune hepatitis can present with acute liver failure superimposed on chronic liver disease or even cirrhosis.
The term acute liver failure has replaced older terms such as fulminant hepatic failure, hyperacute liver failure, and subacute liver failure, which were used for prognostic purposes. Patients with hyperacute liver failure (defined as development of encephalopathy within 7 days of onset of illness) generally have a good prognosis with medical management, whereas those with subacute liver failure (defined as development of encephalopathy within 5 to 26 weeks of onset of illness) have a poor prognosis without liver transplant.2,3
NEARLY 2,000 CASES A YEAR
There are nearly 2,000 cases of acute liver failure each year in the United States, and it accounts for 6% of all deaths due to liver disease.4 It is more common in women than in men, and more common in white people than in other races. The peak incidence is at a fairly young age, ie, 35 to 45 years.
CAUSES
The most common cause of acute liver failure in the United States and other Western countries is acetaminophen toxicity, followed by viral hepatitis. In contrast, viral hepatitis is the most common cause in developing countries.5
Acetaminophen toxicity
Patients with acetaminophen-induced liver failure tend to be younger than other patients with acute liver failure.1 Nearly half of them present after intentionally taking a single large dose, while the rest present with unintentional toxicity while taking acetaminophen for pain relief on a long-term basis and ingesting more than the recommended dose.6
After ingestion, 52% to 57% of acetaminophen is converted to glucuronide conjugates, and 30% to 44% is converted to sulfate conjugates. These compounds are nontoxic, water-soluble, and rapidly excreted in the urine.
However, about 5% to 10% of ingested acetaminophen is shunted to the cytochrome P450 system. P450 2E1 is the main isoenzyme involved in acetaminophen metabolism, but 1A2, 3A4, and 2A6 also contribute.7,8 P450 2E1 is the same isoenzyme responsible for ethanol metabolism and is inducible. Thus, regular alcohol consumption can increase P450 2E1 activity, setting the stage under certain circumstances for increased acetaminophen metabolism through this pathway.

Metabolism of acetaminophen through the cytochrome P450 pathway results in production of N-acetyl-p-benzoquinone imine (NAPQI), the compound that damages the liver. NAPQI is rendered nontoxic by binding to glutathione, forming NAPQI-glutathione adducts. Glutathione capacity is limited, however. With too much acetaminophen, glutathione becomes depleted and NAPQI accumulates, binds with proteins to form adducts, and leads to necrosis of hepatocytes (Figure 1).9,10
Acetylcysteine, used in treating acetaminophen toxicity, is a substrate for glutathione synthesis and ultimately increases the amount of glutathione available to bind NAPQI and prevent damage to hepatocytes.11
Acetaminophen is a dose-related toxin. Most ingestions leading to acute liver failure exceed 10 g/day (> 150 mg/kg/day). Moderate chronic ingestion, eg, 4 g/day, usually leads to transient mild elevation of liver enzymes in healthy individuals12 but can in rare cases cause acute liver failure.13

Whitcomb and Block14 retrospectively identified 49 patients who presented with acetaminophen-induced hepatotoxicity in 1987 through 1993; 21 (43%) had been taking acetaminophen for therapeutic purposes. All 49 patients took more than the recommended limit of 4 g/day, many of them while fasting and some while using alcohol. Acute liver failure was seen with ingestion of more than 12 g/day—or more than 10 g/day in alcohol users. The authors attributed the increased risk to activation of cytochrome P450 2E1 by alcohol and depletion of glutathione stores by starvation or alcohol abuse.
Advice to patients taking acetaminophen is given in Table 1.
Other drugs and supplements

A number of other drugs and herbal supplements can also cause acute liver failure (Table 2), the most common being antimicrobial and antiepileptic drugs.15 Of the antimicrobials, antitubercular drugs (especially isoniazid) are believed to be the most common causes, followed by trimethoprim-sulfamethoxazole. Phenytoin is the antiepileptic drug most often implicated in acute liver failure.
Statins can also cause acute liver failure, especially when combined with other hepatotoxic agents.16
The herbal supplements and weight-loss agents Hydroxycut and Herbalife have both been reported to cause acute liver failure, with patients presenting with either the hepatocellular or the cholestatic pattern of liver injury.17 The exact chemical in these supplements that causes liver injury has not yet been determined.
The National Institutes of Health maintains a database of cases of liver failure due to medications and supplements at livertox.nih.gov. The database includes the pattern of hepatic injury, mechanism of injury, management, and outcomes.
Viral hepatitis
Hepatitis B virus is the most common viral cause of acute liver failure and is responsible for about 8% of cases.18
Patients with chronic hepatitis B virus infection—as evidenced by positive hepatitis B surface antigen—can develop acute liver failure if the infection is reactivated by the use of immunosuppressive drugs for solid-organ or bone-marrow transplant or medications such as anti-tumor necrosis agents, rituximab, or chemotherapy. These patients should be treated prophylactically with a nucleoside analogue, which should be continued for 6 months after immunosuppressive therapy is completed.
Hepatitis A virus is responsible for about 4% of cases.18
Hepatitis C virus rarely causes acute liver failure, especially in the absence of hepatitis A and hepatitis B.3,19
Hepatitis E virus, which is endemic in areas of Asia and Africa, can cause liver disease in pregnant women and in young adults who have concomitant liver disease from another cause. It tends to cause acute liver failure more frequently in pregnant women than in the rest of the population and carries a mortality rate of more than 20% in this subgroup.
TT (transfusion-transmitted) virus was reported in the 1990s to cause acute liver failure in about 27% of patients in whom no other cause could be found.20
Other rare viral causes of acute liver failure include Epstein-Barr virus, cytomegalovirus, and herpes simplex virus types 1, 2, and 6.
Other causes
Other causes of acute liver failure include ischemic hepatitis, autoimmune hepatitis, Wilson disease, Budd-Chiari syndrome, and HELLP (hemolysis, elevated liver enzymes and low platelets) syndrome.
MANY PATIENTS NEED LIVER TRANSPLANT

Many patients with acute liver failure ultimately require orthotopic liver transplant,21 especially if they present with severe encephalopathy. Other aspects of treatment vary according to the cause of liver failure (Table 3).
SPECIFIC MANAGEMENT
Management of acetaminophen toxicity
If the time of ingestion is known, checking the acetaminophen level can help determine the cause of acute liver failure and also predict the risk of hepatotoxicity, based on the work of Rumack and Matthew.22 Calculators are available, eg, http://reference.medscape.com/calculator/acetaminophen-toxicity.
If a patient presents with acute liver failure several days after ingesting acetaminophen, the level can be in the nontoxic range, however. In this scenario, measuring acetaminophen-protein adducts can help establish acetaminophen toxicity as the cause, as the adducts last longer in the serum and provide 100% sensitivity and specificity.23 While most laboratories can rapidly measure acetaminophen levels, only a few can measure acetaminophen-protein adducts, and thus this test is not used clinically.
Acetylcysteine is the main drug used for acetaminophen toxicity. Ideally, it should be given within 8 hours of acetaminophen ingestion, but giving it later is also useful.1
Acetylcysteine is available in oral and intravenous forms, the latter for patients who have encephalopathy or cannot tolerate oral intake due to repeated episodes of vomiting.24,25 The oral form is much less costly and is thus preferred over intravenous acetylcysteine in patients who can tolerate oral intake. Intravenous acetylcysteine should be given in a loading dose of 150 mg/kg in 5% dextrose over 15 minutes, followed by a maintenance dose of 50 mg/kg over 4 hours and then 100 mg/kg given over 16 hours.1 No dose adjustment is needed in patients who have renal toxicity (acetaminophen can also be toxic to the kidneys).
Most patients with acetaminophen-induced liver failure survive with medical management alone and do not need a liver transplant.3,26 Cirrhosis does not occur in these patients.
Management of viral acute liver failure
When patients present with acute liver failure, it is necessary to look for a viral cause by serologic testing, including hepatitis A virus IgM antibody, hepatitis B surface antigen, and hepatitis B core IgM antibody.
Hepatitis B can become reactivated in immunocompromised patients, and therefore the hepatitis B virus DNA level should be checked. Detection of hepatitis B virus DNA in a patient previously known to have undetectable hepatitis B virus DNA confirms hepatitis B reactivation.
Patients with hepatitis B-induced acute liver failure should be treated with entecavir or tenofovir. Although this treatment may not change the course of acute liver failure or accelerate the recovery, it can prevent reinfection in the transplanted liver if liver transplant becomes indicated.27–29
Herpes simplex virus should be suspected in patients presenting with anicteric hepatitis with fever. Polymerase chain reaction testing for herpes simplex virus should be done,30 and if positive, patients should be given intravenous acyclovir.31 Despite treatment, herpes simplex virus disease is associated with a very poor prognosis without liver transplant.
Autoimmune hepatitis
The autoantibodies usually seen in autoimmune hepatitis are antinuclear antibody, antismooth muscle antibody, and anti-liver-kidney microsomal antibody, and patients need to be tested for them.
The diagnosis of autoimmune hepatitis can be challenging, as these autoimmune markers can be negative in 5% of patients. Liver biopsy becomes essential to establish the diagnosis in that setting.32
Guidelines advise starting prednisone 40 to 60 mg/day and placing the patient on the liver transplant list.1
Wilson disease
Although it is an uncommon cause of liver failure, Wilson disease needs special attention because it has a poor prognosis. The mortality rate in acute liver failure from Wilson disease reaches 100% without liver transplant.
Wilson disease is caused by a genetic defect that allows copper to accumulate in the liver and other organs. However, diagnosing Wilson disease as the cause of acute liver failure can be challenging because elevated serum and urine copper levels are not specific to Wilson disease and can be seen in patients with acute liver failure from any cause. In addition, the ceruloplasmin level is usually normal or high because it is an acute-phase reactant. Accumulation of copper in the liver parenchyma is usually patchy; therefore, qualitative copper staining on random liver biopsy samples provides low diagnostic yield. Quantitative copper on liver biopsy is the gold standard test to establish the diagnosis, but the test is time-consuming. Kayser-Fleischer rings around the iris are considered pathognomic for Wilson disease when seen with acute liver failure, but they are seen in only about 50% of patients.33
A unique feature of acute Wilson disease is that most patients have very high bilirubin levels and low alkaline phosphatase levels. An alkaline phosphatase-to-bilirubin ratio less than 2 in patients with acute liver failure is highly suggestive of Wilson disease.34
Another clue to the diagnosis is that patients with Wilson disease tend to develop Coombs-negative hemolytic anemia, which leads to a disproportionate elevation in aminotransferase levels, with aspartate aminotransferase being higher than alanine aminotransferase.
Once Wilson disease is suspected, the patient should be listed for liver transplant because death is almost certain without it. For patients awaiting liver transplant, the American Association for the Study of Liver Diseases guidelines recommend certain measures to lower the serum copper level such as albumin dialysis, continuous hemofiltration, plasmapheresis, and plasma exchange,1 but the evidence supporting their use is limited.
NONSPECIFIC MANAGEMENT
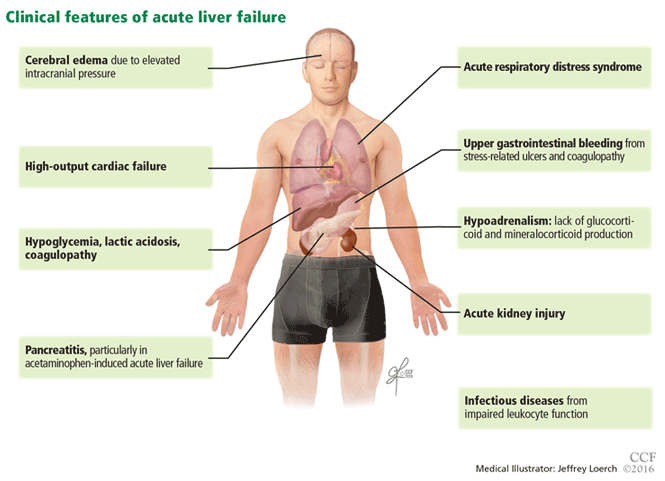
Acute liver failure can affect a number of organs and systems in addition to the liver (Figure 2).
General considerations
Because their condition can rapidly deteriorate, patients with acute liver failure are best managed in intensive care.
Patients who present to a center that does not have the facilities for liver transplant should be transferred to a transplant center as soon as possible, preferably by air. If the patient may not be able to protect the airway, endotracheal intubation should be performed before transfer.
The major causes of death in patients with acute liver failure are cerebral edema and infection. Gastrointestinal bleeding was a major cause of death in the past, but with prophylactic use of histamine H2 receptor blockers and proton pump inhibitors, the incidence of gastrointestinal bleeding has been significantly reduced.
Although initially used only in patients with acetaminophen-induced liver failure, acetylcysteine has also shown benefit in patients with acute liver failure from other causes. In patients with grade 1 or 2 encephalopathy on a scale of 0 (minimal) to 4 (comatose), the transplant-free survival rate is higher when acetylcysteine is given compared with placebo, but this benefit does not extend to patients with a higher grade of encephalopathy.35
Cerebral edema and intracranial hypertension
Cerebral edema is the leading cause of death in patients with acute liver failure, and it develops in nearly 40% of patients.36
The mechanism by which cerebral edema develops is not well understood. Some have proposed that ammonia is converted to glutamine, which causes cerebral edema either directly by its osmotic effect37,38 or indirectly by decreasing other osmolytes, thereby promoting water retention.39
Cerebral edema leads to intracranial hypertension, which can ultimately cause cerebral herniation and death. Because of the high mortality rate associated with cerebral edema, invasive devices were extensively used in the past to monitor intracranial pressure. However, in light of known complications of these devices, including bleeding,40 and lack of evidence of long-term benefit in terms of mortality rates, their use has come under debate.
Treatments. Many treatments are available for cerebral edema and intracranial hypertension. The first step is to elevate the head of the bed about 30 degrees. In addition, hyponatremia should be corrected, as it can worsen cerebral edema.41 If patients are intubated, maintaining a hypercapneic state is advisable to decrease the intracranial pressure.
Of the two pharmacologic options, mannitol is more often used.42 It is given as a bolus dose of 0.5 to 1 g/kg intravenously if the serum osmolality is less than 320 mOsm/L.1 Given the risk of fluid overload with mannitol, caution must be exercised in patients with renal dysfunction. The other pharmacologic option is 3% hypertonic saline.
Therapeutic hypothermia is a newer treatment for cerebral edema. Lowering the body temperature to 32 to 33°C (89.6 to 91.4°F) using cooling blankets decreases intracranial pressure and cerebral blood flow and improves the cerebral perfusion pressure.43 With this treatment, patients should be closely monitored for side effects of infection, coagulopathy, and cardiac arrythmias.1
l-ornithine l-aspartate was successfully used to prevent brain edema in rats, but in humans, no benefit was seen compared with placebo.44,45 The underlying basis for this experimental treatment is that supplemental ornithine and aspartate should increase glutamate synthesis, which should increase the activity of enzyme glutamine synthetase in skeletal muscles. With the increase in enzyme activity, conversion of ammonia to glutamine should increase, thereby decreasing ammonia circulation and thus decreasing cerebral edema.
Patients with cerebral edema have a high incidence of seizures, but prophylactic antiseizure medications such as phenytoin have not been proven to be beneficial.46
Infection
Nearly 80% of patients with acute liver failure develop an infectious complication, which can be attributed to a state of immunodeficiency.47
The respiratory and urinary tracts are the most common sources of infection.48 In patients with bacteremia, Enterococcus species and coagulase-negative Staphylococcus species49 are the commonly isolated organisms. Also, in patients with acute liver failure, fungal infections account for 30% of all infections.50
Infected patients often develop worsening of their encephalopathy51 without fever or elevated white blood cell count.49,52 Thus, in any patient in whom encephalopathy is worsening, an evaluation must be done to rule out infection. In these patients, systemic inflammatory response syndrome is an independent risk factor for death.53
Despite the high mortality rate with infection, whether using antibiotics prophylactically in acute liver failure is beneficial is controversial.54,55
Gastrointestinal bleeding
The current prevalence of upper gastrointestinal bleeding in acute liver failure patients is about 1.5%.56 Coagulopathy and endotracheal intubation are the main risk factors for upper gastrointestinal bleeding in these patients.57 The most common source of bleeding is stress ulcers in the stomach. The ulcers develop from a combination of factors, including decreased blood flow to the mucosa causing ischemia and hypoperfusion-reperfusion injury.
Pharmacologic inhibition of gastric acid secretion has been shown to reduce upper gastrointestinal bleeding in acute liver failure. A histamine H2 receptor blocker or proton pump inhibitor should be given to prevent gastrointestinal bleeding in patients with acute liver failure.1,58
EXPERIMENTAL TREATMENTS
Artificial liver support systems
Membranes and dialysate solutions have been developed to remove toxic substances that are normally metabolized by the liver. Two of these—the molecular adsorbent recycling system (MARS) and the extracorporeal liver assist device (ELAD)—were developed in the late 1990s. MARS consisted of a highly permeable hollow fiber membrane mixed with albumin, and ELAD consisted of porcine hepatocytes attached to microcarriers in the extracapillary space of the hollow fiber membrane. Both systems allowed for transfer of water-soluble and protein-bound toxins in the blood across the membrane and into the dialysate.59 The clinical benefit offered by these devices is controversial,60–62 thus limiting their use to experimental purposes only.
Hepatocyte transplant
Use of hepatocyte transplant as a bridge to liver transplant was tested in 1970s, first in rats and later in humans.63 By reducing the blood ammonia level and improving cerebral perfusion pressure and cardiac function, replacement of 1% to 2% of the total liver cell mass by transplanted hepatocytes acts as a bridge to orthotopic liver transplant.64,65
PROGNOSIS
Different criteria have been used to identify patients with poor prognosis who may eventually need to undergo liver transplant.
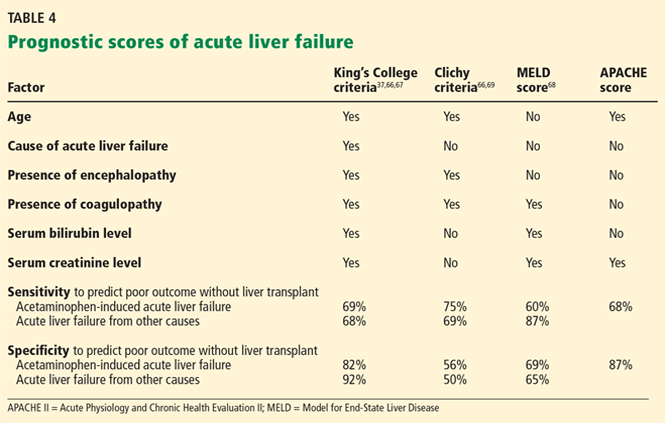
The King’s College criteria system is the most commonly used for prognosis (Table 4).37,66–69 Its main drawback is that it is applicable only in patients with encephalopathy, and when patients reach this stage, their condition often deteriorates rapidly, and they die while awaiting liver transplant.37,66,67
The Model for End-Stage Liver Disease (MELD) score is an alternative to the King’s College criteria. A high MELD score on admission signifies advanced disease, and patients with a high MELD score tend to have a worse prognosis than those with a low score.68
The Acute Physiology and Chronic Health Evaluation (APACHE) II score can also be used, as it is more sensitive than the King’s College criteria.6
The Clichy criteria66,69 can also be used.
Liver biopsy. In addition to helping establish the cause of acute liver failure, liver biopsy can also be used as a prognostic tool. Hepatocellular necrosis greater than 70% on the biopsy predicts death with a specificity of 90% and a sensitivity of 56%.70
Hypophosphatemia has been reported to indicate recovering liver function in patients with acute liver failure.71 As the liver regenerates, its energy requirement increases. To supply the energy, adenosine triphosphate production increases, and phosphorus shifts from the extracellular to the intracellular compartment to meet the need for extra phosphorus during this process. A serum phosphorus level of 2.9 mg/dL or higher appears to indicate a poor prognosis in patients with acute liver failure, as it signifies that adequate hepatocyte regeneration is not occurring.
- Polson J, Lee WM; American Association for the Study of Liver Disease. AASLD position paper: the management of acute liver failure. Hepatology 2005; 41:1179–1197.
- O’Grady JG, Schalm SW, Williams R. Acute liver failure: redefining the syndromes. Lancet 1993; 342:273–275.
- Ostapowicz G, Fontana RJ, Schiodt FV, et al; US Acute Liver Failure Study Group. Results of a prospective study of acute liver failure at 17 tertiary care centers in the United States. Ann Intern Med 2002; 137:947–954.
- Lee WM, Squires RH Jr, Nyberg SL, Doo E, Hoofnagle JH. Acute liver failure: summary of a workshop. Hepatology 2008; 47:1401–1415.
- Acharya SK, Panda SK, Saxena A, Gupta SD. Acute hepatic failure in India: a perspective from the East. J Gastroenterol Hepatol 2000; 15:473–479.
- Larson AM, Polson J, Fontana RJ, et al; Acute Liver Failure Study Group. Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology 2005; 42:1364–1372.
- Patten CJ, Thomas PE, Guy RL, et al. Cytochrome P450 enzymes involved in acetaminophen activation by rat and human liver microsomes and their kinetics. Chem Res Toxicol 1993; 6:511–518.
- Chen W, Koenigs LL, Thompson SJ, et al. Oxidation of acetaminophen to its toxic quinone imine and nontoxic catechol metabolites by baculovirus-expressed and purified human cytochromes P450 2E1 and 2A6. Chem Res Toxicol 1998; 11:295-301.
- Mitchell JR, Jollow DJ, Potter WZ, Gillette JR, Brodie BB. Acetaminophen-induced hepatic necrosis. IV. Protective role of glutathione. J Pharmacol Exp Ther 1973; 187:211–217.
- Schilling A, Corey R, Leonard M, Eghtesad B. Acetaminophen: old drug, new warnings. Cleve Clin J Med 2010; 77:19–27.
- Lauterburg BH, Corcoran GB, Mitchell JR. Mechanism of action of N-acetylcysteine in the protection against the hepatotoxicity of acetaminophen in rats in vivo. J Clin Invest 1983; 71:980–991.
- Watkins PB, Kaplowitz N, Slattery JT, et al. Aminotransferase elevations in healthy adults receiving 4 grams of acetaminophen daily: a randomized controlled trial. JAMA 2006; 296:87–93.
- Schiødt FV, Rochling FA, Casey DL, Lee WM. Acetaminophen toxicity in an urban county hospital. N Engl J Med 1997; 337:1112–1117.
- Whitcomb DC, Block GD. Association of acetaminophen hepatotoxicity with fasting and ethanol use. JAMA 1994; 272:1845–1850.
- Chalasani N, Fontana RJ, Bonkovsky HL, et al; Drug Induced Liver Injury Network (DILIN). Causes, clinical features, and outcomes from a prospective study of drug-induced liver injury in the United States. Gastroenterology 2008; 135:1924–1934 e1–4
- Reuben A, Koch DG, Lee WM; Acute Liver Failure Study Group. Drug-induced acute liver failure: results of a US multicenter, prospective study. Hepatology 2010; 52:2065–2076.
- Stevens T, Qadri A, Zein NN. Two patients with acute liver injury associated with use of the herbal weight-loss supplement hydroxycut. Ann Intern Med 2005; 142:477–478.
- Bernal W, Lee WM, Wendon J, Larsen FS, Williams R. Acute liver failure: a curable disease by 2024? J Hepatol 2015; 62(suppl 1):S112–S120.
- Schiodt FV, Davern TJ, Shakil AO, McGuire B, Samuel G, Lee WM. Viral hepatitis-related acute liver failure. Am J Gastroenterol 2003; 98:448–453.
- Charlton M, Adjei P, Poterucha J, et al. TT-virus infection in North American blood donors, patients with fulminant hepatic failure, and cryptogenic cirrhosis. Hepatology 1998; 28:839–842.
- Bismuth H, Samuel D, Gugenheim J, et al. Emergency liver transplantation for fulminant hepatitis. Ann Intern Med 1987; 107:337–341.
- Rumack BH, Matthew H. Acetaminophen poisoning and toxicity. Pediatrics 1975; 55:871–876.
- Davern TJ 2nd, James LP, Hinson JA, et al; Acute Liver Failure Study Group. Measurement of serum acetaminophen-protein adducts in patients with acute liver failure. Gastroenterology 2006; 130:687–694.
- Perry HE, Shannon MW. Efficacy of oral versus intravenous N-acetylcysteine in acetaminophen overdose: results of an open-label, clinical trial. J Pediatr 1998; 132:149–152.
- Smilkstein MJ, Knapp GL, Kulig KW, Rumack BH. Efficacy of oral N-acetylcysteine in the treatment of acetaminophen overdose. Analysis of the national multicenter study (1976 to 1985). N Engl J Med 1988; 319:1557–1562.
- Makin AJ, Wendon J, Williams R. A 7-year experience of severe acetaminophen-induced hepatotoxicity (1987-1993). Gastroenterology 1995; 109:1907–1916.
- Tsang SW, Chan HL, Leung NW, et al. Lamivudine treatment for fulminant hepatic failure due to acute exacerbation of chronic hepatitis B infection. Aliment Pharmacol Ther 2001; 15:1737–1744.
- Yu JW, Sun LJ, Yan BZ, Kang P, Zhao YH. Lamivudine treatment is associated with improved survival in fulminant hepatitis B. Liver Int 2011; 31:499–506.
- Garg H, Sarin SK, Kumar M, Garg V, Sharma BC, Kumar A. Tenofovir improves the outcome in patients with spontaneous reactivation of hepatitis B presenting as acute-on-chronic liver failure. Hepatology 2011; 53:774–780.
- Pinna AD, Rakela J, Demetris AJ, Fung JJ. Five cases of fulminant hepatitis due to herpes simplex virus in adults. Dig Dis Sci 2002; 47:750–754.
- Farr RW, Short S, Weissman D. Fulminant hepatitis during herpes simplex virus infection in apparently immunocompetent adults: report of two cases and review of the literature. Clin Infect Dis 1997; 24:1191–1194.
- Czaja AJ, Freese DK; American Association for the Study of Liver Disease. Diagnosis and treatment of autoimmune hepatitis. Hepatology 2002; 36:479–497.
- Roberts EA, Schilsky ML. A practice guideline on Wilson disease. Hepatology 2003; 37:1475–1492.
- Berman DH, Leventhal RI, Gavaler JS, Cadoff EM, Van Thiel DH. Clinical differentiation of fulminant Wilsonian hepatitis from other causes of hepatic failure. Gastroenterology 1991; 100:1129–1134.
- Lee WM, Hynan LS, Rossaro L, et al. Intravenous N-acetylcysteine improves transplant-free survival in early stage non-acetaminophen acute liver failure. Gastroenterology 2009; 137:856–864.
- O’Grady JG, Alexander GJ, Hayllar KM, Williams R. Early indicators of prognosis in fulminant hepatic failure. Gastroenterology 1989; 97:439–445.
- Clemmesen JO, Larsen FS, Kondrup J, Hansen BA, Ott P. Cerebral herniation in patients with acute liver failure is correlated with arterial ammonia concentration. Hepatology 1999; 29:648–653.
- Swain M, Butterworth RF, Blei AT. Ammonia and related amino acids in the pathogenesis of brain edema in acute ischemic liver failure in rats. Hepatology 1992; 15:449–453.
- Haussinger D, Laubenberger J, vom Dahl S, et al. Proton magnetic resonance spectroscopy studies on human brain myo-inositol in hypo-osmolarity and hepatic encephalopathy. Gastroenterology 1994; 107:1475–1480.
- Blei AT, Olafsson S, Webster S, Levy R. Complications of intracranial pressure monitoring in fulminant hepatic failure. Lancet 1993; 341:157–158.
- Cordoba J, Gottstein J, Blei AT. Chronic hyponatremia exacerbates ammonia-induced brain edema in rats after portacaval anastomosis. J Hepatol 1998; 29:589–594.
- Canalese J, Gimson AE, Davis C, Mellon PJ, Davis M, Williams R. Controlled trial of dexamethasone and mannitol for the cerebral oedema of fulminant hepatic failure. Gut 1982; 23:625–629.
- Jalan R, SW OD, Deutz NE, Lee A, Hayes PC. Moderate hypothermia for uncontrolled intracranial hypertension in acute liver failure. Lancet 1999; 354:1164–1168.
- Rose C, Michalak A, Rao KV, Quack G, Kircheis G, Butterworth RF. L-ornithine-L-aspartate lowers plasma and cerebrospinal fluid ammonia and prevents brain edema in rats with acute liver failure. Hepatology 1999; 30:636–640.
- Acharya SK, Bhatia V, Sreenivas V, Khanal S, Panda SK. Efficacy of L-ornithine L-aspartate in acute liver failure: a double-blind, randomized, placebo-controlled study. Gastroenterology 2009; 136:2159–2168.
- Bhatia V, Batra Y, Acharya SK. Prophylactic phenytoin does not improve cerebral edema or survival in acute liver failure—a controlled clinical trial. J Hepatol 2004; 41:89–96.
- Canalese J, Gove CD, Gimson AE, Wilkinson SP, Wardle EN, Williams R. Reticuloendothelial system and hepatocytic function in fulminant hepatic failure. Gut 1982; 23:265–269.
- Rolando N, Harvey F, Brahm J, et al. Prospective study of bacterial infection in acute liver failure: an analysis of fifty patients. Hepatology 1990; 11:49–53.
- Rolando N, Wade JJ, Stangou A, et al. Prospective study comparing the efficacy of prophylactic parenteral antimicrobials, with or without enteral decontamination, in patients with acute liver failure. Liver Transpl Surg 1996; 2:8–13.
- Rolando N, Harvey F, Brahm J, et al. Fungal infection: a common, unrecognised complication of acute liver failure. J Hepatol 1991; 12:1–9.
- Vaquero J, Polson J, Chung C, et al. Infection and the progression of hepatic encephalopathy in acute liver failure. Gastroenterology 2003; 125:755–764.
- Rolando N, Philpott-Howard J, Williams R. Bacterial and fungal infection in acute liver failure. Semin Liver Dis 1996; 16:389–402.
- Rolando N, Wade J, Davalos M, Wendon J, Philpott-Howard J, Williams R. The systemic inflammatory response syndrome in acute liver failure. Hepatology 2000; 32:734–739.
- Rolando N, Gimson A, Wade J, Philpott- Howard J, Casewell M, Williams R. Prospective controlled trial of selective parenteral and enteral antimicrobial regimen in fulminant liver failure. Hepatology 1993; 17:196–201.
- Karvellas CJ, Cavazos J, Battenhouse H, et al; US Acute Liver Failure Study Group. Effects of antimicrobial prophylaxis and blood stream infections in patients with acute liver failure: a retrospective cohort study. Clin Gastroenterol Hepatol 2014; 12:1942–1949.
- Acharya SK, Dasarathy S, Kumer TL, et al. Fulminant hepatitis in a tropical population: clinical course, cause, and early predictors of outcome. Hepatology 1996; 23:1148–1155.
- Cook DJ, Fuller HD, Guyatt GH, et al. Risk factors for gastrointestinal bleeding in critically ill patients. Canadian Critical Care Trials Group. N Engl J Med 1994; 330:377–381.
- MacDougall BR, Williams R. H2-receptor antagonist in the prevention of acute gastrointestinal hemorrhage in fulminant hepatic failure: a controlled trial. Gastroenterology 1978; 74:464–465.
- Stange J, Mitzner SR, Risler T, et al. Molecular adsorbent recycling system (MARS): clinical results of a new membrane-based blood purification system for bioartificial liver support. Artif Organs 1999; 23:319–330.
- Vaid A, Chewich H, Balk EM, Jaber BL. Molecular adsorbent recirculating system as artificial support therapy for liver failure: a meta-analysis. ASAIO J 2012; 58:51–59.
- Khuroo MS, Khuroo MS, Farahat KL. Molecular adsorbent recirculating system for acute and acute-on-chronic liver failure: a meta-analysis. Liver Transpl 2004; 10:1099–1106.
- Kjaergard LL, Liu J, Als-Nielsen B, Gluud C. Artificial and bioartificial support systems for acute and acute-on-chronic liver failure: a systematic review. JAMA 2003; 289:217–222.
- Sommer BG, Sutherland DE, Matas AJ, Simmons RL, Najarian JS. Hepatocellular transplantation for treatment of D-galactosamine-induced acute liver failure in rats. Transplant Proc 1979; 11:578–584.
- Demetriou AA, Reisner A, Sanchez J, Levenson SM, Moscioni AD, Chowdhury JR. Transplantation of microcarrier-attached hepatocytes into 90% partially hepatectomized rats. Hepatology 1988; 8:1006–1009.
- Strom SC, Fisher RA, Thompson MT, et al. Hepatocyte transplantation as a bridge to orthotopic liver transplantation in terminal liver failure. Transplantation 1997; 63:559–569.
- Pauwels A, Mostefa-Kara N, Florent C, Levy VG. Emergency liver transplantation for acute liver failure. Evaluation of London and Clichy criteria. J Hepatol 1993; 17:124–127.
- Anand AC, Nightingale P, Neuberger JM. Early indicators of prognosis in fulminant hepatic failure: an assessment of the King's criteria. J Hepatol 1997; 26:62–68.
- Schmidt LE, Larsen FS. MELD score as a predictor of liver failure and death in patients with acetaminophen-induced liver injury. Hepatology 2007; 45:789–796.
- Bernuau J, Goudeau A, Poynard T, et al. Multivariate analysis of prognostic factors in fulminant hepatitis B. Hepatology 1986; 6:648–651.
- Donaldson BW, Gopinath R, Wanless IR, et al. The role of transjugular liver biopsy in fulminant liver failure: relation to other prognostic indicators. Hepatology 1993; 18:1370–1376.
- Schmidt LE, Dalhoff K. Serum phosphate is an early predictor of outcome in severe acetaminophen-induced hepatotoxicity. Hepatology 2002; 36:659–665.
When the liver fails, it usually fails gradually. The sudden (acute) onset of liver failure, while less common, demands prompt management, with transfer to an intensive care unit, specific treatment depending on the cause, and consideration of liver transplant, without which the mortality rate is high.
This article reviews the definition, epidemiology, etiology, and management of acute liver failure.
DEFINITIONS
Acute liver failure is defined as a syndrome of acute hepatitis with evidence of abnormal coagulation (eg, an international normalized ratio > 1.5) complicated by the development of mental alteration (encephalopathy) within 26 weeks of the onset of illness in a patient without a history of liver disease.1 In general, patients have no evidence of underlying chronic liver disease, but there are exceptions; patients with Wilson disease, vertically acquired hepatitis B virus infection, or autoimmune hepatitis can present with acute liver failure superimposed on chronic liver disease or even cirrhosis.
The term acute liver failure has replaced older terms such as fulminant hepatic failure, hyperacute liver failure, and subacute liver failure, which were used for prognostic purposes. Patients with hyperacute liver failure (defined as development of encephalopathy within 7 days of onset of illness) generally have a good prognosis with medical management, whereas those with subacute liver failure (defined as development of encephalopathy within 5 to 26 weeks of onset of illness) have a poor prognosis without liver transplant.2,3
NEARLY 2,000 CASES A YEAR
There are nearly 2,000 cases of acute liver failure each year in the United States, and it accounts for 6% of all deaths due to liver disease.4 It is more common in women than in men, and more common in white people than in other races. The peak incidence is at a fairly young age, ie, 35 to 45 years.
CAUSES
The most common cause of acute liver failure in the United States and other Western countries is acetaminophen toxicity, followed by viral hepatitis. In contrast, viral hepatitis is the most common cause in developing countries.5
Acetaminophen toxicity
Patients with acetaminophen-induced liver failure tend to be younger than other patients with acute liver failure.1 Nearly half of them present after intentionally taking a single large dose, while the rest present with unintentional toxicity while taking acetaminophen for pain relief on a long-term basis and ingesting more than the recommended dose.6
After ingestion, 52% to 57% of acetaminophen is converted to glucuronide conjugates, and 30% to 44% is converted to sulfate conjugates. These compounds are nontoxic, water-soluble, and rapidly excreted in the urine.
However, about 5% to 10% of ingested acetaminophen is shunted to the cytochrome P450 system. P450 2E1 is the main isoenzyme involved in acetaminophen metabolism, but 1A2, 3A4, and 2A6 also contribute.7,8 P450 2E1 is the same isoenzyme responsible for ethanol metabolism and is inducible. Thus, regular alcohol consumption can increase P450 2E1 activity, setting the stage under certain circumstances for increased acetaminophen metabolism through this pathway.
Metabolism of acetaminophen through the cytochrome P450 pathway results in production of N-acetyl-p-benzoquinone imine (NAPQI), the compound that damages the liver. NAPQI is rendered nontoxic by binding to glutathione, forming NAPQI-glutathione adducts. Glutathione capacity is limited, however. With too much acetaminophen, glutathione becomes depleted and NAPQI accumulates, binds with proteins to form adducts, and leads to necrosis of hepatocytes (Figure 1).9,10
Acetylcysteine, used in treating acetaminophen toxicity, is a substrate for glutathione synthesis and ultimately increases the amount of glutathione available to bind NAPQI and prevent damage to hepatocytes.11
Acetaminophen is a dose-related toxin. Most ingestions leading to acute liver failure exceed 10 g/day (> 150 mg/kg/day). Moderate chronic ingestion, eg, 4 g/day, usually leads to transient mild elevation of liver enzymes in healthy individuals12 but can in rare cases cause acute liver failure.13
Whitcomb and Block14 retrospectively identified 49 patients who presented with acetaminophen-induced hepatotoxicity in 1987 through 1993; 21 (43%) had been taking acetaminophen for therapeutic purposes. All 49 patients took more than the recommended limit of 4 g/day, many of them while fasting and some while using alcohol. Acute liver failure was seen with ingestion of more than 12 g/day—or more than 10 g/day in alcohol users. The authors attributed the increased risk to activation of cytochrome P450 2E1 by alcohol and depletion of glutathione stores by starvation or alcohol abuse.
Advice to patients taking acetaminophen is given in Table 1.
Other drugs and supplements
A number of other drugs and herbal supplements can also cause acute liver failure (Table 2), the most common being antimicrobial and antiepileptic drugs.15 Of the antimicrobials, antitubercular drugs (especially isoniazid) are believed to be the most common causes, followed by trimethoprim-sulfamethoxazole. Phenytoin is the antiepileptic drug most often implicated in acute liver failure.
Statins can also cause acute liver failure, especially when combined with other hepatotoxic agents.16
The herbal supplements and weight-loss agents Hydroxycut and Herbalife have both been reported to cause acute liver failure, with patients presenting with either the hepatocellular or the cholestatic pattern of liver injury.17 The exact chemical in these supplements that causes liver injury has not yet been determined.
The National Institutes of Health maintains a database of cases of liver failure due to medications and supplements at livertox.nih.gov. The database includes the pattern of hepatic injury, mechanism of injury, management, and outcomes.
Viral hepatitis
Hepatitis B virus is the most common viral cause of acute liver failure and is responsible for about 8% of cases.18
Patients with chronic hepatitis B virus infection—as evidenced by positive hepatitis B surface antigen—can develop acute liver failure if the infection is reactivated by the use of immunosuppressive drugs for solid-organ or bone-marrow transplant or medications such as anti-tumor necrosis agents, rituximab, or chemotherapy. These patients should be treated prophylactically with a nucleoside analogue, which should be continued for 6 months after immunosuppressive therapy is completed.
Hepatitis A virus is responsible for about 4% of cases.18
Hepatitis C virus rarely causes acute liver failure, especially in the absence of hepatitis A and hepatitis B.3,19
Hepatitis E virus, which is endemic in areas of Asia and Africa, can cause liver disease in pregnant women and in young adults who have concomitant liver disease from another cause. It tends to cause acute liver failure more frequently in pregnant women than in the rest of the population and carries a mortality rate of more than 20% in this subgroup.
TT (transfusion-transmitted) virus was reported in the 1990s to cause acute liver failure in about 27% of patients in whom no other cause could be found.20
Other rare viral causes of acute liver failure include Epstein-Barr virus, cytomegalovirus, and herpes simplex virus types 1, 2, and 6.
Other causes
Other causes of acute liver failure include ischemic hepatitis, autoimmune hepatitis, Wilson disease, Budd-Chiari syndrome, and HELLP (hemolysis, elevated liver enzymes and low platelets) syndrome.
MANY PATIENTS NEED LIVER TRANSPLANT
Many patients with acute liver failure ultimately require orthotopic liver transplant,21 especially if they present with severe encephalopathy. Other aspects of treatment vary according to the cause of liver failure (Table 3).
SPECIFIC MANAGEMENT
Management of acetaminophen toxicity
If the time of ingestion is known, checking the acetaminophen level can help determine the cause of acute liver failure and also predict the risk of hepatotoxicity, based on the work of Rumack and Matthew.22 Calculators are available, eg, http://reference.medscape.com/calculator/acetaminophen-toxicity.
If a patient presents with acute liver failure several days after ingesting acetaminophen, the level can be in the nontoxic range, however. In this scenario, measuring acetaminophen-protein adducts can help establish acetaminophen toxicity as the cause, as the adducts last longer in the serum and provide 100% sensitivity and specificity.23 While most laboratories can rapidly measure acetaminophen levels, only a few can measure acetaminophen-protein adducts, and thus this test is not used clinically.
Acetylcysteine is the main drug used for acetaminophen toxicity. Ideally, it should be given within 8 hours of acetaminophen ingestion, but giving it later is also useful.1
Acetylcysteine is available in oral and intravenous forms, the latter for patients who have encephalopathy or cannot tolerate oral intake due to repeated episodes of vomiting.24,25 The oral form is much less costly and is thus preferred over intravenous acetylcysteine in patients who can tolerate oral intake. Intravenous acetylcysteine should be given in a loading dose of 150 mg/kg in 5% dextrose over 15 minutes, followed by a maintenance dose of 50 mg/kg over 4 hours and then 100 mg/kg given over 16 hours.1 No dose adjustment is needed in patients who have renal toxicity (acetaminophen can also be toxic to the kidneys).
Most patients with acetaminophen-induced liver failure survive with medical management alone and do not need a liver transplant.3,26 Cirrhosis does not occur in these patients.
Management of viral acute liver failure
When patients present with acute liver failure, it is necessary to look for a viral cause by serologic testing, including hepatitis A virus IgM antibody, hepatitis B surface antigen, and hepatitis B core IgM antibody.
Hepatitis B can become reactivated in immunocompromised patients, and therefore the hepatitis B virus DNA level should be checked. Detection of hepatitis B virus DNA in a patient previously known to have undetectable hepatitis B virus DNA confirms hepatitis B reactivation.
Patients with hepatitis B-induced acute liver failure should be treated with entecavir or tenofovir. Although this treatment may not change the course of acute liver failure or accelerate the recovery, it can prevent reinfection in the transplanted liver if liver transplant becomes indicated.27–29
Herpes simplex virus should be suspected in patients presenting with anicteric hepatitis with fever. Polymerase chain reaction testing for herpes simplex virus should be done,30 and if positive, patients should be given intravenous acyclovir.31 Despite treatment, herpes simplex virus disease is associated with a very poor prognosis without liver transplant.
Autoimmune hepatitis
The autoantibodies usually seen in autoimmune hepatitis are antinuclear antibody, antismooth muscle antibody, and anti-liver-kidney microsomal antibody, and patients need to be tested for them.
The diagnosis of autoimmune hepatitis can be challenging, as these autoimmune markers can be negative in 5% of patients. Liver biopsy becomes essential to establish the diagnosis in that setting.32
Guidelines advise starting prednisone 40 to 60 mg/day and placing the patient on the liver transplant list.1
Wilson disease
Although it is an uncommon cause of liver failure, Wilson disease needs special attention because it has a poor prognosis. The mortality rate in acute liver failure from Wilson disease reaches 100% without liver transplant.
Wilson disease is caused by a genetic defect that allows copper to accumulate in the liver and other organs. However, diagnosing Wilson disease as the cause of acute liver failure can be challenging because elevated serum and urine copper levels are not specific to Wilson disease and can be seen in patients with acute liver failure from any cause. In addition, the ceruloplasmin level is usually normal or high because it is an acute-phase reactant. Accumulation of copper in the liver parenchyma is usually patchy; therefore, qualitative copper staining on random liver biopsy samples provides low diagnostic yield. Quantitative copper on liver biopsy is the gold standard test to establish the diagnosis, but the test is time-consuming. Kayser-Fleischer rings around the iris are considered pathognomic for Wilson disease when seen with acute liver failure, but they are seen in only about 50% of patients.33
A unique feature of acute Wilson disease is that most patients have very high bilirubin levels and low alkaline phosphatase levels. An alkaline phosphatase-to-bilirubin ratio less than 2 in patients with acute liver failure is highly suggestive of Wilson disease.34
Another clue to the diagnosis is that patients with Wilson disease tend to develop Coombs-negative hemolytic anemia, which leads to a disproportionate elevation in aminotransferase levels, with aspartate aminotransferase being higher than alanine aminotransferase.
Once Wilson disease is suspected, the patient should be listed for liver transplant because death is almost certain without it. For patients awaiting liver transplant, the American Association for the Study of Liver Diseases guidelines recommend certain measures to lower the serum copper level such as albumin dialysis, continuous hemofiltration, plasmapheresis, and plasma exchange,1 but the evidence supporting their use is limited.
NONSPECIFIC MANAGEMENT
Acute liver failure can affect a number of organs and systems in addition to the liver (Figure 2).
General considerations
Because their condition can rapidly deteriorate, patients with acute liver failure are best managed in intensive care.
Patients who present to a center that does not have the facilities for liver transplant should be transferred to a transplant center as soon as possible, preferably by air. If the patient may not be able to protect the airway, endotracheal intubation should be performed before transfer.
The major causes of death in patients with acute liver failure are cerebral edema and infection. Gastrointestinal bleeding was a major cause of death in the past, but with prophylactic use of histamine H2 receptor blockers and proton pump inhibitors, the incidence of gastrointestinal bleeding has been significantly reduced.
Although initially used only in patients with acetaminophen-induced liver failure, acetylcysteine has also shown benefit in patients with acute liver failure from other causes. In patients with grade 1 or 2 encephalopathy on a scale of 0 (minimal) to 4 (comatose), the transplant-free survival rate is higher when acetylcysteine is given compared with placebo, but this benefit does not extend to patients with a higher grade of encephalopathy.35
Cerebral edema and intracranial hypertension
Cerebral edema is the leading cause of death in patients with acute liver failure, and it develops in nearly 40% of patients.36
The mechanism by which cerebral edema develops is not well understood. Some have proposed that ammonia is converted to glutamine, which causes cerebral edema either directly by its osmotic effect37,38 or indirectly by decreasing other osmolytes, thereby promoting water retention.39
Cerebral edema leads to intracranial hypertension, which can ultimately cause cerebral herniation and death. Because of the high mortality rate associated with cerebral edema, invasive devices were extensively used in the past to monitor intracranial pressure. However, in light of known complications of these devices, including bleeding,40 and lack of evidence of long-term benefit in terms of mortality rates, their use has come under debate.
Treatments. Many treatments are available for cerebral edema and intracranial hypertension. The first step is to elevate the head of the bed about 30 degrees. In addition, hyponatremia should be corrected, as it can worsen cerebral edema.41 If patients are intubated, maintaining a hypercapneic state is advisable to decrease the intracranial pressure.
Of the two pharmacologic options, mannitol is more often used.42 It is given as a bolus dose of 0.5 to 1 g/kg intravenously if the serum osmolality is less than 320 mOsm/L.1 Given the risk of fluid overload with mannitol, caution must be exercised in patients with renal dysfunction. The other pharmacologic option is 3% hypertonic saline.
Therapeutic hypothermia is a newer treatment for cerebral edema. Lowering the body temperature to 32 to 33°C (89.6 to 91.4°F) using cooling blankets decreases intracranial pressure and cerebral blood flow and improves the cerebral perfusion pressure.43 With this treatment, patients should be closely monitored for side effects of infection, coagulopathy, and cardiac arrythmias.1
l-ornithine l-aspartate was successfully used to prevent brain edema in rats, but in humans, no benefit was seen compared with placebo.44,45 The underlying basis for this experimental treatment is that supplemental ornithine and aspartate should increase glutamate synthesis, which should increase the activity of enzyme glutamine synthetase in skeletal muscles. With the increase in enzyme activity, conversion of ammonia to glutamine should increase, thereby decreasing ammonia circulation and thus decreasing cerebral edema.
Patients with cerebral edema have a high incidence of seizures, but prophylactic antiseizure medications such as phenytoin have not been proven to be beneficial.46
Infection
Nearly 80% of patients with acute liver failure develop an infectious complication, which can be attributed to a state of immunodeficiency.47
The respiratory and urinary tracts are the most common sources of infection.48 In patients with bacteremia, Enterococcus species and coagulase-negative Staphylococcus species49 are the commonly isolated organisms. Also, in patients with acute liver failure, fungal infections account for 30% of all infections.50
Infected patients often develop worsening of their encephalopathy51 without fever or elevated white blood cell count.49,52 Thus, in any patient in whom encephalopathy is worsening, an evaluation must be done to rule out infection. In these patients, systemic inflammatory response syndrome is an independent risk factor for death.53
Despite the high mortality rate with infection, whether using antibiotics prophylactically in acute liver failure is beneficial is controversial.54,55
Gastrointestinal bleeding
The current prevalence of upper gastrointestinal bleeding in acute liver failure patients is about 1.5%.56 Coagulopathy and endotracheal intubation are the main risk factors for upper gastrointestinal bleeding in these patients.57 The most common source of bleeding is stress ulcers in the stomach. The ulcers develop from a combination of factors, including decreased blood flow to the mucosa causing ischemia and hypoperfusion-reperfusion injury.
Pharmacologic inhibition of gastric acid secretion has been shown to reduce upper gastrointestinal bleeding in acute liver failure. A histamine H2 receptor blocker or proton pump inhibitor should be given to prevent gastrointestinal bleeding in patients with acute liver failure.1,58
EXPERIMENTAL TREATMENTS
Artificial liver support systems
Membranes and dialysate solutions have been developed to remove toxic substances that are normally metabolized by the liver. Two of these—the molecular adsorbent recycling system (MARS) and the extracorporeal liver assist device (ELAD)—were developed in the late 1990s. MARS consisted of a highly permeable hollow fiber membrane mixed with albumin, and ELAD consisted of porcine hepatocytes attached to microcarriers in the extracapillary space of the hollow fiber membrane. Both systems allowed for transfer of water-soluble and protein-bound toxins in the blood across the membrane and into the dialysate.59 The clinical benefit offered by these devices is controversial,60–62 thus limiting their use to experimental purposes only.
Hepatocyte transplant
Use of hepatocyte transplant as a bridge to liver transplant was tested in 1970s, first in rats and later in humans.63 By reducing the blood ammonia level and improving cerebral perfusion pressure and cardiac function, replacement of 1% to 2% of the total liver cell mass by transplanted hepatocytes acts as a bridge to orthotopic liver transplant.64,65
PROGNOSIS
Different criteria have been used to identify patients with poor prognosis who may eventually need to undergo liver transplant.
The King’s College criteria system is the most commonly used for prognosis (Table 4).37,66–69 Its main drawback is that it is applicable only in patients with encephalopathy, and when patients reach this stage, their condition often deteriorates rapidly, and they die while awaiting liver transplant.37,66,67
The Model for End-Stage Liver Disease (MELD) score is an alternative to the King’s College criteria. A high MELD score on admission signifies advanced disease, and patients with a high MELD score tend to have a worse prognosis than those with a low score.68
The Acute Physiology and Chronic Health Evaluation (APACHE) II score can also be used, as it is more sensitive than the King’s College criteria.6
The Clichy criteria66,69 can also be used.
Liver biopsy. In addition to helping establish the cause of acute liver failure, liver biopsy can also be used as a prognostic tool. Hepatocellular necrosis greater than 70% on the biopsy predicts death with a specificity of 90% and a sensitivity of 56%.70
Hypophosphatemia has been reported to indicate recovering liver function in patients with acute liver failure.71 As the liver regenerates, its energy requirement increases. To supply the energy, adenosine triphosphate production increases, and phosphorus shifts from the extracellular to the intracellular compartment to meet the need for extra phosphorus during this process. A serum phosphorus level of 2.9 mg/dL or higher appears to indicate a poor prognosis in patients with acute liver failure, as it signifies that adequate hepatocyte regeneration is not occurring.
When the liver fails, it usually fails gradually. The sudden (acute) onset of liver failure, while less common, demands prompt management, with transfer to an intensive care unit, specific treatment depending on the cause, and consideration of liver transplant, without which the mortality rate is high.
This article reviews the definition, epidemiology, etiology, and management of acute liver failure.
DEFINITIONS
Acute liver failure is defined as a syndrome of acute hepatitis with evidence of abnormal coagulation (eg, an international normalized ratio > 1.5) complicated by the development of mental alteration (encephalopathy) within 26 weeks of the onset of illness in a patient without a history of liver disease.1 In general, patients have no evidence of underlying chronic liver disease, but there are exceptions; patients with Wilson disease, vertically acquired hepatitis B virus infection, or autoimmune hepatitis can present with acute liver failure superimposed on chronic liver disease or even cirrhosis.
The term acute liver failure has replaced older terms such as fulminant hepatic failure, hyperacute liver failure, and subacute liver failure, which were used for prognostic purposes. Patients with hyperacute liver failure (defined as development of encephalopathy within 7 days of onset of illness) generally have a good prognosis with medical management, whereas those with subacute liver failure (defined as development of encephalopathy within 5 to 26 weeks of onset of illness) have a poor prognosis without liver transplant.2,3
NEARLY 2,000 CASES A YEAR
There are nearly 2,000 cases of acute liver failure each year in the United States, and it accounts for 6% of all deaths due to liver disease.4 It is more common in women than in men, and more common in white people than in other races. The peak incidence is at a fairly young age, ie, 35 to 45 years.
CAUSES
The most common cause of acute liver failure in the United States and other Western countries is acetaminophen toxicity, followed by viral hepatitis. In contrast, viral hepatitis is the most common cause in developing countries.5
Acetaminophen toxicity
Patients with acetaminophen-induced liver failure tend to be younger than other patients with acute liver failure.1 Nearly half of them present after intentionally taking a single large dose, while the rest present with unintentional toxicity while taking acetaminophen for pain relief on a long-term basis and ingesting more than the recommended dose.6
After ingestion, 52% to 57% of acetaminophen is converted to glucuronide conjugates, and 30% to 44% is converted to sulfate conjugates. These compounds are nontoxic, water-soluble, and rapidly excreted in the urine.
However, about 5% to 10% of ingested acetaminophen is shunted to the cytochrome P450 system. P450 2E1 is the main isoenzyme involved in acetaminophen metabolism, but 1A2, 3A4, and 2A6 also contribute.7,8 P450 2E1 is the same isoenzyme responsible for ethanol metabolism and is inducible. Thus, regular alcohol consumption can increase P450 2E1 activity, setting the stage under certain circumstances for increased acetaminophen metabolism through this pathway.
Metabolism of acetaminophen through the cytochrome P450 pathway results in production of N-acetyl-p-benzoquinone imine (NAPQI), the compound that damages the liver. NAPQI is rendered nontoxic by binding to glutathione, forming NAPQI-glutathione adducts. Glutathione capacity is limited, however. With too much acetaminophen, glutathione becomes depleted and NAPQI accumulates, binds with proteins to form adducts, and leads to necrosis of hepatocytes (Figure 1).9,10
Acetylcysteine, used in treating acetaminophen toxicity, is a substrate for glutathione synthesis and ultimately increases the amount of glutathione available to bind NAPQI and prevent damage to hepatocytes.11
Acetaminophen is a dose-related toxin. Most ingestions leading to acute liver failure exceed 10 g/day (> 150 mg/kg/day). Moderate chronic ingestion, eg, 4 g/day, usually leads to transient mild elevation of liver enzymes in healthy individuals12 but can in rare cases cause acute liver failure.13
Whitcomb and Block14 retrospectively identified 49 patients who presented with acetaminophen-induced hepatotoxicity in 1987 through 1993; 21 (43%) had been taking acetaminophen for therapeutic purposes. All 49 patients took more than the recommended limit of 4 g/day, many of them while fasting and some while using alcohol. Acute liver failure was seen with ingestion of more than 12 g/day—or more than 10 g/day in alcohol users. The authors attributed the increased risk to activation of cytochrome P450 2E1 by alcohol and depletion of glutathione stores by starvation or alcohol abuse.
Advice to patients taking acetaminophen is given in Table 1.
Other drugs and supplements
A number of other drugs and herbal supplements can also cause acute liver failure (Table 2), the most common being antimicrobial and antiepileptic drugs.15 Of the antimicrobials, antitubercular drugs (especially isoniazid) are believed to be the most common causes, followed by trimethoprim-sulfamethoxazole. Phenytoin is the antiepileptic drug most often implicated in acute liver failure.
Statins can also cause acute liver failure, especially when combined with other hepatotoxic agents.16
The herbal supplements and weight-loss agents Hydroxycut and Herbalife have both been reported to cause acute liver failure, with patients presenting with either the hepatocellular or the cholestatic pattern of liver injury.17 The exact chemical in these supplements that causes liver injury has not yet been determined.
The National Institutes of Health maintains a database of cases of liver failure due to medications and supplements at livertox.nih.gov. The database includes the pattern of hepatic injury, mechanism of injury, management, and outcomes.
Viral hepatitis
Hepatitis B virus is the most common viral cause of acute liver failure and is responsible for about 8% of cases.18
Patients with chronic hepatitis B virus infection—as evidenced by positive hepatitis B surface antigen—can develop acute liver failure if the infection is reactivated by the use of immunosuppressive drugs for solid-organ or bone-marrow transplant or medications such as anti-tumor necrosis agents, rituximab, or chemotherapy. These patients should be treated prophylactically with a nucleoside analogue, which should be continued for 6 months after immunosuppressive therapy is completed.
Hepatitis A virus is responsible for about 4% of cases.18
Hepatitis C virus rarely causes acute liver failure, especially in the absence of hepatitis A and hepatitis B.3,19
Hepatitis E virus, which is endemic in areas of Asia and Africa, can cause liver disease in pregnant women and in young adults who have concomitant liver disease from another cause. It tends to cause acute liver failure more frequently in pregnant women than in the rest of the population and carries a mortality rate of more than 20% in this subgroup.
TT (transfusion-transmitted) virus was reported in the 1990s to cause acute liver failure in about 27% of patients in whom no other cause could be found.20
Other rare viral causes of acute liver failure include Epstein-Barr virus, cytomegalovirus, and herpes simplex virus types 1, 2, and 6.
Other causes
Other causes of acute liver failure include ischemic hepatitis, autoimmune hepatitis, Wilson disease, Budd-Chiari syndrome, and HELLP (hemolysis, elevated liver enzymes and low platelets) syndrome.
MANY PATIENTS NEED LIVER TRANSPLANT
Many patients with acute liver failure ultimately require orthotopic liver transplant,21 especially if they present with severe encephalopathy. Other aspects of treatment vary according to the cause of liver failure (Table 3).
SPECIFIC MANAGEMENT
Management of acetaminophen toxicity
If the time of ingestion is known, checking the acetaminophen level can help determine the cause of acute liver failure and also predict the risk of hepatotoxicity, based on the work of Rumack and Matthew.22 Calculators are available, eg, http://reference.medscape.com/calculator/acetaminophen-toxicity.
If a patient presents with acute liver failure several days after ingesting acetaminophen, the level can be in the nontoxic range, however. In this scenario, measuring acetaminophen-protein adducts can help establish acetaminophen toxicity as the cause, as the adducts last longer in the serum and provide 100% sensitivity and specificity.23 While most laboratories can rapidly measure acetaminophen levels, only a few can measure acetaminophen-protein adducts, and thus this test is not used clinically.
Acetylcysteine is the main drug used for acetaminophen toxicity. Ideally, it should be given within 8 hours of acetaminophen ingestion, but giving it later is also useful.1
Acetylcysteine is available in oral and intravenous forms, the latter for patients who have encephalopathy or cannot tolerate oral intake due to repeated episodes of vomiting.24,25 The oral form is much less costly and is thus preferred over intravenous acetylcysteine in patients who can tolerate oral intake. Intravenous acetylcysteine should be given in a loading dose of 150 mg/kg in 5% dextrose over 15 minutes, followed by a maintenance dose of 50 mg/kg over 4 hours and then 100 mg/kg given over 16 hours.1 No dose adjustment is needed in patients who have renal toxicity (acetaminophen can also be toxic to the kidneys).
Most patients with acetaminophen-induced liver failure survive with medical management alone and do not need a liver transplant.3,26 Cirrhosis does not occur in these patients.
Management of viral acute liver failure
When patients present with acute liver failure, it is necessary to look for a viral cause by serologic testing, including hepatitis A virus IgM antibody, hepatitis B surface antigen, and hepatitis B core IgM antibody.
Hepatitis B can become reactivated in immunocompromised patients, and therefore the hepatitis B virus DNA level should be checked. Detection of hepatitis B virus DNA in a patient previously known to have undetectable hepatitis B virus DNA confirms hepatitis B reactivation.
Patients with hepatitis B-induced acute liver failure should be treated with entecavir or tenofovir. Although this treatment may not change the course of acute liver failure or accelerate the recovery, it can prevent reinfection in the transplanted liver if liver transplant becomes indicated.27–29
Herpes simplex virus should be suspected in patients presenting with anicteric hepatitis with fever. Polymerase chain reaction testing for herpes simplex virus should be done,30 and if positive, patients should be given intravenous acyclovir.31 Despite treatment, herpes simplex virus disease is associated with a very poor prognosis without liver transplant.
Autoimmune hepatitis
The autoantibodies usually seen in autoimmune hepatitis are antinuclear antibody, antismooth muscle antibody, and anti-liver-kidney microsomal antibody, and patients need to be tested for them.
The diagnosis of autoimmune hepatitis can be challenging, as these autoimmune markers can be negative in 5% of patients. Liver biopsy becomes essential to establish the diagnosis in that setting.32
Guidelines advise starting prednisone 40 to 60 mg/day and placing the patient on the liver transplant list.1
Wilson disease
Although it is an uncommon cause of liver failure, Wilson disease needs special attention because it has a poor prognosis. The mortality rate in acute liver failure from Wilson disease reaches 100% without liver transplant.
Wilson disease is caused by a genetic defect that allows copper to accumulate in the liver and other organs. However, diagnosing Wilson disease as the cause of acute liver failure can be challenging because elevated serum and urine copper levels are not specific to Wilson disease and can be seen in patients with acute liver failure from any cause. In addition, the ceruloplasmin level is usually normal or high because it is an acute-phase reactant. Accumulation of copper in the liver parenchyma is usually patchy; therefore, qualitative copper staining on random liver biopsy samples provides low diagnostic yield. Quantitative copper on liver biopsy is the gold standard test to establish the diagnosis, but the test is time-consuming. Kayser-Fleischer rings around the iris are considered pathognomic for Wilson disease when seen with acute liver failure, but they are seen in only about 50% of patients.33
A unique feature of acute Wilson disease is that most patients have very high bilirubin levels and low alkaline phosphatase levels. An alkaline phosphatase-to-bilirubin ratio less than 2 in patients with acute liver failure is highly suggestive of Wilson disease.34
Another clue to the diagnosis is that patients with Wilson disease tend to develop Coombs-negative hemolytic anemia, which leads to a disproportionate elevation in aminotransferase levels, with aspartate aminotransferase being higher than alanine aminotransferase.
Once Wilson disease is suspected, the patient should be listed for liver transplant because death is almost certain without it. For patients awaiting liver transplant, the American Association for the Study of Liver Diseases guidelines recommend certain measures to lower the serum copper level such as albumin dialysis, continuous hemofiltration, plasmapheresis, and plasma exchange,1 but the evidence supporting their use is limited.
NONSPECIFIC MANAGEMENT
Acute liver failure can affect a number of organs and systems in addition to the liver (Figure 2).
General considerations
Because their condition can rapidly deteriorate, patients with acute liver failure are best managed in intensive care.
Patients who present to a center that does not have the facilities for liver transplant should be transferred to a transplant center as soon as possible, preferably by air. If the patient may not be able to protect the airway, endotracheal intubation should be performed before transfer.
The major causes of death in patients with acute liver failure are cerebral edema and infection. Gastrointestinal bleeding was a major cause of death in the past, but with prophylactic use of histamine H2 receptor blockers and proton pump inhibitors, the incidence of gastrointestinal bleeding has been significantly reduced.
Although initially used only in patients with acetaminophen-induced liver failure, acetylcysteine has also shown benefit in patients with acute liver failure from other causes. In patients with grade 1 or 2 encephalopathy on a scale of 0 (minimal) to 4 (comatose), the transplant-free survival rate is higher when acetylcysteine is given compared with placebo, but this benefit does not extend to patients with a higher grade of encephalopathy.35
Cerebral edema and intracranial hypertension
Cerebral edema is the leading cause of death in patients with acute liver failure, and it develops in nearly 40% of patients.36
The mechanism by which cerebral edema develops is not well understood. Some have proposed that ammonia is converted to glutamine, which causes cerebral edema either directly by its osmotic effect37,38 or indirectly by decreasing other osmolytes, thereby promoting water retention.39
Cerebral edema leads to intracranial hypertension, which can ultimately cause cerebral herniation and death. Because of the high mortality rate associated with cerebral edema, invasive devices were extensively used in the past to monitor intracranial pressure. However, in light of known complications of these devices, including bleeding,40 and lack of evidence of long-term benefit in terms of mortality rates, their use has come under debate.
Treatments. Many treatments are available for cerebral edema and intracranial hypertension. The first step is to elevate the head of the bed about 30 degrees. In addition, hyponatremia should be corrected, as it can worsen cerebral edema.41 If patients are intubated, maintaining a hypercapneic state is advisable to decrease the intracranial pressure.
Of the two pharmacologic options, mannitol is more often used.42 It is given as a bolus dose of 0.5 to 1 g/kg intravenously if the serum osmolality is less than 320 mOsm/L.1 Given the risk of fluid overload with mannitol, caution must be exercised in patients with renal dysfunction. The other pharmacologic option is 3% hypertonic saline.
Therapeutic hypothermia is a newer treatment for cerebral edema. Lowering the body temperature to 32 to 33°C (89.6 to 91.4°F) using cooling blankets decreases intracranial pressure and cerebral blood flow and improves the cerebral perfusion pressure.43 With this treatment, patients should be closely monitored for side effects of infection, coagulopathy, and cardiac arrythmias.1
l-ornithine l-aspartate was successfully used to prevent brain edema in rats, but in humans, no benefit was seen compared with placebo.44,45 The underlying basis for this experimental treatment is that supplemental ornithine and aspartate should increase glutamate synthesis, which should increase the activity of enzyme glutamine synthetase in skeletal muscles. With the increase in enzyme activity, conversion of ammonia to glutamine should increase, thereby decreasing ammonia circulation and thus decreasing cerebral edema.
Patients with cerebral edema have a high incidence of seizures, but prophylactic antiseizure medications such as phenytoin have not been proven to be beneficial.46
Infection
Nearly 80% of patients with acute liver failure develop an infectious complication, which can be attributed to a state of immunodeficiency.47
The respiratory and urinary tracts are the most common sources of infection.48 In patients with bacteremia, Enterococcus species and coagulase-negative Staphylococcus species49 are the commonly isolated organisms. Also, in patients with acute liver failure, fungal infections account for 30% of all infections.50
Infected patients often develop worsening of their encephalopathy51 without fever or elevated white blood cell count.49,52 Thus, in any patient in whom encephalopathy is worsening, an evaluation must be done to rule out infection. In these patients, systemic inflammatory response syndrome is an independent risk factor for death.53
Despite the high mortality rate with infection, whether using antibiotics prophylactically in acute liver failure is beneficial is controversial.54,55
Gastrointestinal bleeding
The current prevalence of upper gastrointestinal bleeding in acute liver failure patients is about 1.5%.56 Coagulopathy and endotracheal intubation are the main risk factors for upper gastrointestinal bleeding in these patients.57 The most common source of bleeding is stress ulcers in the stomach. The ulcers develop from a combination of factors, including decreased blood flow to the mucosa causing ischemia and hypoperfusion-reperfusion injury.
Pharmacologic inhibition of gastric acid secretion has been shown to reduce upper gastrointestinal bleeding in acute liver failure. A histamine H2 receptor blocker or proton pump inhibitor should be given to prevent gastrointestinal bleeding in patients with acute liver failure.1,58
EXPERIMENTAL TREATMENTS
Artificial liver support systems
Membranes and dialysate solutions have been developed to remove toxic substances that are normally metabolized by the liver. Two of these—the molecular adsorbent recycling system (MARS) and the extracorporeal liver assist device (ELAD)—were developed in the late 1990s. MARS consisted of a highly permeable hollow fiber membrane mixed with albumin, and ELAD consisted of porcine hepatocytes attached to microcarriers in the extracapillary space of the hollow fiber membrane. Both systems allowed for transfer of water-soluble and protein-bound toxins in the blood across the membrane and into the dialysate.59 The clinical benefit offered by these devices is controversial,60–62 thus limiting their use to experimental purposes only.
Hepatocyte transplant
Use of hepatocyte transplant as a bridge to liver transplant was tested in 1970s, first in rats and later in humans.63 By reducing the blood ammonia level and improving cerebral perfusion pressure and cardiac function, replacement of 1% to 2% of the total liver cell mass by transplanted hepatocytes acts as a bridge to orthotopic liver transplant.64,65
PROGNOSIS
Different criteria have been used to identify patients with poor prognosis who may eventually need to undergo liver transplant.
The King’s College criteria system is the most commonly used for prognosis (Table 4).37,66–69 Its main drawback is that it is applicable only in patients with encephalopathy, and when patients reach this stage, their condition often deteriorates rapidly, and they die while awaiting liver transplant.37,66,67
The Model for End-Stage Liver Disease (MELD) score is an alternative to the King’s College criteria. A high MELD score on admission signifies advanced disease, and patients with a high MELD score tend to have a worse prognosis than those with a low score.68
The Acute Physiology and Chronic Health Evaluation (APACHE) II score can also be used, as it is more sensitive than the King’s College criteria.6
The Clichy criteria66,69 can also be used.
Liver biopsy. In addition to helping establish the cause of acute liver failure, liver biopsy can also be used as a prognostic tool. Hepatocellular necrosis greater than 70% on the biopsy predicts death with a specificity of 90% and a sensitivity of 56%.70
Hypophosphatemia has been reported to indicate recovering liver function in patients with acute liver failure.71 As the liver regenerates, its energy requirement increases. To supply the energy, adenosine triphosphate production increases, and phosphorus shifts from the extracellular to the intracellular compartment to meet the need for extra phosphorus during this process. A serum phosphorus level of 2.9 mg/dL or higher appears to indicate a poor prognosis in patients with acute liver failure, as it signifies that adequate hepatocyte regeneration is not occurring.
- Polson J, Lee WM; American Association for the Study of Liver Disease. AASLD position paper: the management of acute liver failure. Hepatology 2005; 41:1179–1197.
- O’Grady JG, Schalm SW, Williams R. Acute liver failure: redefining the syndromes. Lancet 1993; 342:273–275.
- Ostapowicz G, Fontana RJ, Schiodt FV, et al; US Acute Liver Failure Study Group. Results of a prospective study of acute liver failure at 17 tertiary care centers in the United States. Ann Intern Med 2002; 137:947–954.
- Lee WM, Squires RH Jr, Nyberg SL, Doo E, Hoofnagle JH. Acute liver failure: summary of a workshop. Hepatology 2008; 47:1401–1415.
- Acharya SK, Panda SK, Saxena A, Gupta SD. Acute hepatic failure in India: a perspective from the East. J Gastroenterol Hepatol 2000; 15:473–479.
- Larson AM, Polson J, Fontana RJ, et al; Acute Liver Failure Study Group. Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology 2005; 42:1364–1372.
- Patten CJ, Thomas PE, Guy RL, et al. Cytochrome P450 enzymes involved in acetaminophen activation by rat and human liver microsomes and their kinetics. Chem Res Toxicol 1993; 6:511–518.
- Chen W, Koenigs LL, Thompson SJ, et al. Oxidation of acetaminophen to its toxic quinone imine and nontoxic catechol metabolites by baculovirus-expressed and purified human cytochromes P450 2E1 and 2A6. Chem Res Toxicol 1998; 11:295-301.
- Mitchell JR, Jollow DJ, Potter WZ, Gillette JR, Brodie BB. Acetaminophen-induced hepatic necrosis. IV. Protective role of glutathione. J Pharmacol Exp Ther 1973; 187:211–217.
- Schilling A, Corey R, Leonard M, Eghtesad B. Acetaminophen: old drug, new warnings. Cleve Clin J Med 2010; 77:19–27.
- Lauterburg BH, Corcoran GB, Mitchell JR. Mechanism of action of N-acetylcysteine in the protection against the hepatotoxicity of acetaminophen in rats in vivo. J Clin Invest 1983; 71:980–991.
- Watkins PB, Kaplowitz N, Slattery JT, et al. Aminotransferase elevations in healthy adults receiving 4 grams of acetaminophen daily: a randomized controlled trial. JAMA 2006; 296:87–93.
- Schiødt FV, Rochling FA, Casey DL, Lee WM. Acetaminophen toxicity in an urban county hospital. N Engl J Med 1997; 337:1112–1117.
- Whitcomb DC, Block GD. Association of acetaminophen hepatotoxicity with fasting and ethanol use. JAMA 1994; 272:1845–1850.
- Chalasani N, Fontana RJ, Bonkovsky HL, et al; Drug Induced Liver Injury Network (DILIN). Causes, clinical features, and outcomes from a prospective study of drug-induced liver injury in the United States. Gastroenterology 2008; 135:1924–1934 e1–4
- Reuben A, Koch DG, Lee WM; Acute Liver Failure Study Group. Drug-induced acute liver failure: results of a US multicenter, prospective study. Hepatology 2010; 52:2065–2076.
- Stevens T, Qadri A, Zein NN. Two patients with acute liver injury associated with use of the herbal weight-loss supplement hydroxycut. Ann Intern Med 2005; 142:477–478.
- Bernal W, Lee WM, Wendon J, Larsen FS, Williams R. Acute liver failure: a curable disease by 2024? J Hepatol 2015; 62(suppl 1):S112–S120.
- Schiodt FV, Davern TJ, Shakil AO, McGuire B, Samuel G, Lee WM. Viral hepatitis-related acute liver failure. Am J Gastroenterol 2003; 98:448–453.
- Charlton M, Adjei P, Poterucha J, et al. TT-virus infection in North American blood donors, patients with fulminant hepatic failure, and cryptogenic cirrhosis. Hepatology 1998; 28:839–842.
- Bismuth H, Samuel D, Gugenheim J, et al. Emergency liver transplantation for fulminant hepatitis. Ann Intern Med 1987; 107:337–341.
- Rumack BH, Matthew H. Acetaminophen poisoning and toxicity. Pediatrics 1975; 55:871–876.
- Davern TJ 2nd, James LP, Hinson JA, et al; Acute Liver Failure Study Group. Measurement of serum acetaminophen-protein adducts in patients with acute liver failure. Gastroenterology 2006; 130:687–694.
- Perry HE, Shannon MW. Efficacy of oral versus intravenous N-acetylcysteine in acetaminophen overdose: results of an open-label, clinical trial. J Pediatr 1998; 132:149–152.
- Smilkstein MJ, Knapp GL, Kulig KW, Rumack BH. Efficacy of oral N-acetylcysteine in the treatment of acetaminophen overdose. Analysis of the national multicenter study (1976 to 1985). N Engl J Med 1988; 319:1557–1562.
- Makin AJ, Wendon J, Williams R. A 7-year experience of severe acetaminophen-induced hepatotoxicity (1987-1993). Gastroenterology 1995; 109:1907–1916.
- Tsang SW, Chan HL, Leung NW, et al. Lamivudine treatment for fulminant hepatic failure due to acute exacerbation of chronic hepatitis B infection. Aliment Pharmacol Ther 2001; 15:1737–1744.
- Yu JW, Sun LJ, Yan BZ, Kang P, Zhao YH. Lamivudine treatment is associated with improved survival in fulminant hepatitis B. Liver Int 2011; 31:499–506.
- Garg H, Sarin SK, Kumar M, Garg V, Sharma BC, Kumar A. Tenofovir improves the outcome in patients with spontaneous reactivation of hepatitis B presenting as acute-on-chronic liver failure. Hepatology 2011; 53:774–780.
- Pinna AD, Rakela J, Demetris AJ, Fung JJ. Five cases of fulminant hepatitis due to herpes simplex virus in adults. Dig Dis Sci 2002; 47:750–754.
- Farr RW, Short S, Weissman D. Fulminant hepatitis during herpes simplex virus infection in apparently immunocompetent adults: report of two cases and review of the literature. Clin Infect Dis 1997; 24:1191–1194.
- Czaja AJ, Freese DK; American Association for the Study of Liver Disease. Diagnosis and treatment of autoimmune hepatitis. Hepatology 2002; 36:479–497.
- Roberts EA, Schilsky ML. A practice guideline on Wilson disease. Hepatology 2003; 37:1475–1492.
- Berman DH, Leventhal RI, Gavaler JS, Cadoff EM, Van Thiel DH. Clinical differentiation of fulminant Wilsonian hepatitis from other causes of hepatic failure. Gastroenterology 1991; 100:1129–1134.
- Lee WM, Hynan LS, Rossaro L, et al. Intravenous N-acetylcysteine improves transplant-free survival in early stage non-acetaminophen acute liver failure. Gastroenterology 2009; 137:856–864.
- O’Grady JG, Alexander GJ, Hayllar KM, Williams R. Early indicators of prognosis in fulminant hepatic failure. Gastroenterology 1989; 97:439–445.
- Clemmesen JO, Larsen FS, Kondrup J, Hansen BA, Ott P. Cerebral herniation in patients with acute liver failure is correlated with arterial ammonia concentration. Hepatology 1999; 29:648–653.
- Swain M, Butterworth RF, Blei AT. Ammonia and related amino acids in the pathogenesis of brain edema in acute ischemic liver failure in rats. Hepatology 1992; 15:449–453.
- Haussinger D, Laubenberger J, vom Dahl S, et al. Proton magnetic resonance spectroscopy studies on human brain myo-inositol in hypo-osmolarity and hepatic encephalopathy. Gastroenterology 1994; 107:1475–1480.
- Blei AT, Olafsson S, Webster S, Levy R. Complications of intracranial pressure monitoring in fulminant hepatic failure. Lancet 1993; 341:157–158.
- Cordoba J, Gottstein J, Blei AT. Chronic hyponatremia exacerbates ammonia-induced brain edema in rats after portacaval anastomosis. J Hepatol 1998; 29:589–594.
- Canalese J, Gimson AE, Davis C, Mellon PJ, Davis M, Williams R. Controlled trial of dexamethasone and mannitol for the cerebral oedema of fulminant hepatic failure. Gut 1982; 23:625–629.
- Jalan R, SW OD, Deutz NE, Lee A, Hayes PC. Moderate hypothermia for uncontrolled intracranial hypertension in acute liver failure. Lancet 1999; 354:1164–1168.
- Rose C, Michalak A, Rao KV, Quack G, Kircheis G, Butterworth RF. L-ornithine-L-aspartate lowers plasma and cerebrospinal fluid ammonia and prevents brain edema in rats with acute liver failure. Hepatology 1999; 30:636–640.
- Acharya SK, Bhatia V, Sreenivas V, Khanal S, Panda SK. Efficacy of L-ornithine L-aspartate in acute liver failure: a double-blind, randomized, placebo-controlled study. Gastroenterology 2009; 136:2159–2168.
- Bhatia V, Batra Y, Acharya SK. Prophylactic phenytoin does not improve cerebral edema or survival in acute liver failure—a controlled clinical trial. J Hepatol 2004; 41:89–96.
- Canalese J, Gove CD, Gimson AE, Wilkinson SP, Wardle EN, Williams R. Reticuloendothelial system and hepatocytic function in fulminant hepatic failure. Gut 1982; 23:265–269.
- Rolando N, Harvey F, Brahm J, et al. Prospective study of bacterial infection in acute liver failure: an analysis of fifty patients. Hepatology 1990; 11:49–53.
- Rolando N, Wade JJ, Stangou A, et al. Prospective study comparing the efficacy of prophylactic parenteral antimicrobials, with or without enteral decontamination, in patients with acute liver failure. Liver Transpl Surg 1996; 2:8–13.
- Rolando N, Harvey F, Brahm J, et al. Fungal infection: a common, unrecognised complication of acute liver failure. J Hepatol 1991; 12:1–9.
- Vaquero J, Polson J, Chung C, et al. Infection and the progression of hepatic encephalopathy in acute liver failure. Gastroenterology 2003; 125:755–764.
- Rolando N, Philpott-Howard J, Williams R. Bacterial and fungal infection in acute liver failure. Semin Liver Dis 1996; 16:389–402.
- Rolando N, Wade J, Davalos M, Wendon J, Philpott-Howard J, Williams R. The systemic inflammatory response syndrome in acute liver failure. Hepatology 2000; 32:734–739.
- Rolando N, Gimson A, Wade J, Philpott- Howard J, Casewell M, Williams R. Prospective controlled trial of selective parenteral and enteral antimicrobial regimen in fulminant liver failure. Hepatology 1993; 17:196–201.
- Karvellas CJ, Cavazos J, Battenhouse H, et al; US Acute Liver Failure Study Group. Effects of antimicrobial prophylaxis and blood stream infections in patients with acute liver failure: a retrospective cohort study. Clin Gastroenterol Hepatol 2014; 12:1942–1949.
- Acharya SK, Dasarathy S, Kumer TL, et al. Fulminant hepatitis in a tropical population: clinical course, cause, and early predictors of outcome. Hepatology 1996; 23:1148–1155.
- Cook DJ, Fuller HD, Guyatt GH, et al. Risk factors for gastrointestinal bleeding in critically ill patients. Canadian Critical Care Trials Group. N Engl J Med 1994; 330:377–381.
- MacDougall BR, Williams R. H2-receptor antagonist in the prevention of acute gastrointestinal hemorrhage in fulminant hepatic failure: a controlled trial. Gastroenterology 1978; 74:464–465.
- Stange J, Mitzner SR, Risler T, et al. Molecular adsorbent recycling system (MARS): clinical results of a new membrane-based blood purification system for bioartificial liver support. Artif Organs 1999; 23:319–330.
- Vaid A, Chewich H, Balk EM, Jaber BL. Molecular adsorbent recirculating system as artificial support therapy for liver failure: a meta-analysis. ASAIO J 2012; 58:51–59.
- Khuroo MS, Khuroo MS, Farahat KL. Molecular adsorbent recirculating system for acute and acute-on-chronic liver failure: a meta-analysis. Liver Transpl 2004; 10:1099–1106.
- Kjaergard LL, Liu J, Als-Nielsen B, Gluud C. Artificial and bioartificial support systems for acute and acute-on-chronic liver failure: a systematic review. JAMA 2003; 289:217–222.
- Sommer BG, Sutherland DE, Matas AJ, Simmons RL, Najarian JS. Hepatocellular transplantation for treatment of D-galactosamine-induced acute liver failure in rats. Transplant Proc 1979; 11:578–584.
- Demetriou AA, Reisner A, Sanchez J, Levenson SM, Moscioni AD, Chowdhury JR. Transplantation of microcarrier-attached hepatocytes into 90% partially hepatectomized rats. Hepatology 1988; 8:1006–1009.
- Strom SC, Fisher RA, Thompson MT, et al. Hepatocyte transplantation as a bridge to orthotopic liver transplantation in terminal liver failure. Transplantation 1997; 63:559–569.
- Pauwels A, Mostefa-Kara N, Florent C, Levy VG. Emergency liver transplantation for acute liver failure. Evaluation of London and Clichy criteria. J Hepatol 1993; 17:124–127.
- Anand AC, Nightingale P, Neuberger JM. Early indicators of prognosis in fulminant hepatic failure: an assessment of the King's criteria. J Hepatol 1997; 26:62–68.
- Schmidt LE, Larsen FS. MELD score as a predictor of liver failure and death in patients with acetaminophen-induced liver injury. Hepatology 2007; 45:789–796.
- Bernuau J, Goudeau A, Poynard T, et al. Multivariate analysis of prognostic factors in fulminant hepatitis B. Hepatology 1986; 6:648–651.
- Donaldson BW, Gopinath R, Wanless IR, et al. The role of transjugular liver biopsy in fulminant liver failure: relation to other prognostic indicators. Hepatology 1993; 18:1370–1376.
- Schmidt LE, Dalhoff K. Serum phosphate is an early predictor of outcome in severe acetaminophen-induced hepatotoxicity. Hepatology 2002; 36:659–665.
- Polson J, Lee WM; American Association for the Study of Liver Disease. AASLD position paper: the management of acute liver failure. Hepatology 2005; 41:1179–1197.
- O’Grady JG, Schalm SW, Williams R. Acute liver failure: redefining the syndromes. Lancet 1993; 342:273–275.
- Ostapowicz G, Fontana RJ, Schiodt FV, et al; US Acute Liver Failure Study Group. Results of a prospective study of acute liver failure at 17 tertiary care centers in the United States. Ann Intern Med 2002; 137:947–954.
- Lee WM, Squires RH Jr, Nyberg SL, Doo E, Hoofnagle JH. Acute liver failure: summary of a workshop. Hepatology 2008; 47:1401–1415.
- Acharya SK, Panda SK, Saxena A, Gupta SD. Acute hepatic failure in India: a perspective from the East. J Gastroenterol Hepatol 2000; 15:473–479.
- Larson AM, Polson J, Fontana RJ, et al; Acute Liver Failure Study Group. Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology 2005; 42:1364–1372.
- Patten CJ, Thomas PE, Guy RL, et al. Cytochrome P450 enzymes involved in acetaminophen activation by rat and human liver microsomes and their kinetics. Chem Res Toxicol 1993; 6:511–518.
- Chen W, Koenigs LL, Thompson SJ, et al. Oxidation of acetaminophen to its toxic quinone imine and nontoxic catechol metabolites by baculovirus-expressed and purified human cytochromes P450 2E1 and 2A6. Chem Res Toxicol 1998; 11:295-301.
- Mitchell JR, Jollow DJ, Potter WZ, Gillette JR, Brodie BB. Acetaminophen-induced hepatic necrosis. IV. Protective role of glutathione. J Pharmacol Exp Ther 1973; 187:211–217.
- Schilling A, Corey R, Leonard M, Eghtesad B. Acetaminophen: old drug, new warnings. Cleve Clin J Med 2010; 77:19–27.
- Lauterburg BH, Corcoran GB, Mitchell JR. Mechanism of action of N-acetylcysteine in the protection against the hepatotoxicity of acetaminophen in rats in vivo. J Clin Invest 1983; 71:980–991.
- Watkins PB, Kaplowitz N, Slattery JT, et al. Aminotransferase elevations in healthy adults receiving 4 grams of acetaminophen daily: a randomized controlled trial. JAMA 2006; 296:87–93.
- Schiødt FV, Rochling FA, Casey DL, Lee WM. Acetaminophen toxicity in an urban county hospital. N Engl J Med 1997; 337:1112–1117.
- Whitcomb DC, Block GD. Association of acetaminophen hepatotoxicity with fasting and ethanol use. JAMA 1994; 272:1845–1850.
- Chalasani N, Fontana RJ, Bonkovsky HL, et al; Drug Induced Liver Injury Network (DILIN). Causes, clinical features, and outcomes from a prospective study of drug-induced liver injury in the United States. Gastroenterology 2008; 135:1924–1934 e1–4
- Reuben A, Koch DG, Lee WM; Acute Liver Failure Study Group. Drug-induced acute liver failure: results of a US multicenter, prospective study. Hepatology 2010; 52:2065–2076.
- Stevens T, Qadri A, Zein NN. Two patients with acute liver injury associated with use of the herbal weight-loss supplement hydroxycut. Ann Intern Med 2005; 142:477–478.
- Bernal W, Lee WM, Wendon J, Larsen FS, Williams R. Acute liver failure: a curable disease by 2024? J Hepatol 2015; 62(suppl 1):S112–S120.
- Schiodt FV, Davern TJ, Shakil AO, McGuire B, Samuel G, Lee WM. Viral hepatitis-related acute liver failure. Am J Gastroenterol 2003; 98:448–453.
- Charlton M, Adjei P, Poterucha J, et al. TT-virus infection in North American blood donors, patients with fulminant hepatic failure, and cryptogenic cirrhosis. Hepatology 1998; 28:839–842.
- Bismuth H, Samuel D, Gugenheim J, et al. Emergency liver transplantation for fulminant hepatitis. Ann Intern Med 1987; 107:337–341.
- Rumack BH, Matthew H. Acetaminophen poisoning and toxicity. Pediatrics 1975; 55:871–876.
- Davern TJ 2nd, James LP, Hinson JA, et al; Acute Liver Failure Study Group. Measurement of serum acetaminophen-protein adducts in patients with acute liver failure. Gastroenterology 2006; 130:687–694.
- Perry HE, Shannon MW. Efficacy of oral versus intravenous N-acetylcysteine in acetaminophen overdose: results of an open-label, clinical trial. J Pediatr 1998; 132:149–152.
- Smilkstein MJ, Knapp GL, Kulig KW, Rumack BH. Efficacy of oral N-acetylcysteine in the treatment of acetaminophen overdose. Analysis of the national multicenter study (1976 to 1985). N Engl J Med 1988; 319:1557–1562.
- Makin AJ, Wendon J, Williams R. A 7-year experience of severe acetaminophen-induced hepatotoxicity (1987-1993). Gastroenterology 1995; 109:1907–1916.
- Tsang SW, Chan HL, Leung NW, et al. Lamivudine treatment for fulminant hepatic failure due to acute exacerbation of chronic hepatitis B infection. Aliment Pharmacol Ther 2001; 15:1737–1744.
- Yu JW, Sun LJ, Yan BZ, Kang P, Zhao YH. Lamivudine treatment is associated with improved survival in fulminant hepatitis B. Liver Int 2011; 31:499–506.
- Garg H, Sarin SK, Kumar M, Garg V, Sharma BC, Kumar A. Tenofovir improves the outcome in patients with spontaneous reactivation of hepatitis B presenting as acute-on-chronic liver failure. Hepatology 2011; 53:774–780.
- Pinna AD, Rakela J, Demetris AJ, Fung JJ. Five cases of fulminant hepatitis due to herpes simplex virus in adults. Dig Dis Sci 2002; 47:750–754.
- Farr RW, Short S, Weissman D. Fulminant hepatitis during herpes simplex virus infection in apparently immunocompetent adults: report of two cases and review of the literature. Clin Infect Dis 1997; 24:1191–1194.
- Czaja AJ, Freese DK; American Association for the Study of Liver Disease. Diagnosis and treatment of autoimmune hepatitis. Hepatology 2002; 36:479–497.
- Roberts EA, Schilsky ML. A practice guideline on Wilson disease. Hepatology 2003; 37:1475–1492.
- Berman DH, Leventhal RI, Gavaler JS, Cadoff EM, Van Thiel DH. Clinical differentiation of fulminant Wilsonian hepatitis from other causes of hepatic failure. Gastroenterology 1991; 100:1129–1134.
- Lee WM, Hynan LS, Rossaro L, et al. Intravenous N-acetylcysteine improves transplant-free survival in early stage non-acetaminophen acute liver failure. Gastroenterology 2009; 137:856–864.
- O’Grady JG, Alexander GJ, Hayllar KM, Williams R. Early indicators of prognosis in fulminant hepatic failure. Gastroenterology 1989; 97:439–445.
- Clemmesen JO, Larsen FS, Kondrup J, Hansen BA, Ott P. Cerebral herniation in patients with acute liver failure is correlated with arterial ammonia concentration. Hepatology 1999; 29:648–653.
- Swain M, Butterworth RF, Blei AT. Ammonia and related amino acids in the pathogenesis of brain edema in acute ischemic liver failure in rats. Hepatology 1992; 15:449–453.
- Haussinger D, Laubenberger J, vom Dahl S, et al. Proton magnetic resonance spectroscopy studies on human brain myo-inositol in hypo-osmolarity and hepatic encephalopathy. Gastroenterology 1994; 107:1475–1480.
- Blei AT, Olafsson S, Webster S, Levy R. Complications of intracranial pressure monitoring in fulminant hepatic failure. Lancet 1993; 341:157–158.
- Cordoba J, Gottstein J, Blei AT. Chronic hyponatremia exacerbates ammonia-induced brain edema in rats after portacaval anastomosis. J Hepatol 1998; 29:589–594.
- Canalese J, Gimson AE, Davis C, Mellon PJ, Davis M, Williams R. Controlled trial of dexamethasone and mannitol for the cerebral oedema of fulminant hepatic failure. Gut 1982; 23:625–629.
- Jalan R, SW OD, Deutz NE, Lee A, Hayes PC. Moderate hypothermia for uncontrolled intracranial hypertension in acute liver failure. Lancet 1999; 354:1164–1168.
- Rose C, Michalak A, Rao KV, Quack G, Kircheis G, Butterworth RF. L-ornithine-L-aspartate lowers plasma and cerebrospinal fluid ammonia and prevents brain edema in rats with acute liver failure. Hepatology 1999; 30:636–640.
- Acharya SK, Bhatia V, Sreenivas V, Khanal S, Panda SK. Efficacy of L-ornithine L-aspartate in acute liver failure: a double-blind, randomized, placebo-controlled study. Gastroenterology 2009; 136:2159–2168.
- Bhatia V, Batra Y, Acharya SK. Prophylactic phenytoin does not improve cerebral edema or survival in acute liver failure—a controlled clinical trial. J Hepatol 2004; 41:89–96.
- Canalese J, Gove CD, Gimson AE, Wilkinson SP, Wardle EN, Williams R. Reticuloendothelial system and hepatocytic function in fulminant hepatic failure. Gut 1982; 23:265–269.
- Rolando N, Harvey F, Brahm J, et al. Prospective study of bacterial infection in acute liver failure: an analysis of fifty patients. Hepatology 1990; 11:49–53.
- Rolando N, Wade JJ, Stangou A, et al. Prospective study comparing the efficacy of prophylactic parenteral antimicrobials, with or without enteral decontamination, in patients with acute liver failure. Liver Transpl Surg 1996; 2:8–13.
- Rolando N, Harvey F, Brahm J, et al. Fungal infection: a common, unrecognised complication of acute liver failure. J Hepatol 1991; 12:1–9.
- Vaquero J, Polson J, Chung C, et al. Infection and the progression of hepatic encephalopathy in acute liver failure. Gastroenterology 2003; 125:755–764.
- Rolando N, Philpott-Howard J, Williams R. Bacterial and fungal infection in acute liver failure. Semin Liver Dis 1996; 16:389–402.
- Rolando N, Wade J, Davalos M, Wendon J, Philpott-Howard J, Williams R. The systemic inflammatory response syndrome in acute liver failure. Hepatology 2000; 32:734–739.
- Rolando N, Gimson A, Wade J, Philpott- Howard J, Casewell M, Williams R. Prospective controlled trial of selective parenteral and enteral antimicrobial regimen in fulminant liver failure. Hepatology 1993; 17:196–201.
- Karvellas CJ, Cavazos J, Battenhouse H, et al; US Acute Liver Failure Study Group. Effects of antimicrobial prophylaxis and blood stream infections in patients with acute liver failure: a retrospective cohort study. Clin Gastroenterol Hepatol 2014; 12:1942–1949.
- Acharya SK, Dasarathy S, Kumer TL, et al. Fulminant hepatitis in a tropical population: clinical course, cause, and early predictors of outcome. Hepatology 1996; 23:1148–1155.
- Cook DJ, Fuller HD, Guyatt GH, et al. Risk factors for gastrointestinal bleeding in critically ill patients. Canadian Critical Care Trials Group. N Engl J Med 1994; 330:377–381.
- MacDougall BR, Williams R. H2-receptor antagonist in the prevention of acute gastrointestinal hemorrhage in fulminant hepatic failure: a controlled trial. Gastroenterology 1978; 74:464–465.
- Stange J, Mitzner SR, Risler T, et al. Molecular adsorbent recycling system (MARS): clinical results of a new membrane-based blood purification system for bioartificial liver support. Artif Organs 1999; 23:319–330.
- Vaid A, Chewich H, Balk EM, Jaber BL. Molecular adsorbent recirculating system as artificial support therapy for liver failure: a meta-analysis. ASAIO J 2012; 58:51–59.
- Khuroo MS, Khuroo MS, Farahat KL. Molecular adsorbent recirculating system for acute and acute-on-chronic liver failure: a meta-analysis. Liver Transpl 2004; 10:1099–1106.
- Kjaergard LL, Liu J, Als-Nielsen B, Gluud C. Artificial and bioartificial support systems for acute and acute-on-chronic liver failure: a systematic review. JAMA 2003; 289:217–222.
- Sommer BG, Sutherland DE, Matas AJ, Simmons RL, Najarian JS. Hepatocellular transplantation for treatment of D-galactosamine-induced acute liver failure in rats. Transplant Proc 1979; 11:578–584.
- Demetriou AA, Reisner A, Sanchez J, Levenson SM, Moscioni AD, Chowdhury JR. Transplantation of microcarrier-attached hepatocytes into 90% partially hepatectomized rats. Hepatology 1988; 8:1006–1009.
- Strom SC, Fisher RA, Thompson MT, et al. Hepatocyte transplantation as a bridge to orthotopic liver transplantation in terminal liver failure. Transplantation 1997; 63:559–569.
- Pauwels A, Mostefa-Kara N, Florent C, Levy VG. Emergency liver transplantation for acute liver failure. Evaluation of London and Clichy criteria. J Hepatol 1993; 17:124–127.
- Anand AC, Nightingale P, Neuberger JM. Early indicators of prognosis in fulminant hepatic failure: an assessment of the King's criteria. J Hepatol 1997; 26:62–68.
- Schmidt LE, Larsen FS. MELD score as a predictor of liver failure and death in patients with acetaminophen-induced liver injury. Hepatology 2007; 45:789–796.
- Bernuau J, Goudeau A, Poynard T, et al. Multivariate analysis of prognostic factors in fulminant hepatitis B. Hepatology 1986; 6:648–651.
- Donaldson BW, Gopinath R, Wanless IR, et al. The role of transjugular liver biopsy in fulminant liver failure: relation to other prognostic indicators. Hepatology 1993; 18:1370–1376.
- Schmidt LE, Dalhoff K. Serum phosphate is an early predictor of outcome in severe acetaminophen-induced hepatotoxicity. Hepatology 2002; 36:659–665.
KEY POINTS
- In the United States, the most common cause of acute liver failure is acetaminophen toxicity, followed by viral hepatitis.
- Testing for the cause of acute liver failure needs to start as soon as possible so that specific treatment can be initiated and the patient can be placed on the transplant list if needed.
- Acetylcysteine and either a proton pump inhibitor or a histamine H2 receptor blocker should be given to all patients with acute liver failure. Liver transplant is the cornerstone of therapy in patients not responding to other treatments.
- There are a number of prognostic scores for acute liver failure, but each has limitations.
A tale of two sisters with liver disease
A 25-year-old woman presents to the emergency department with a 7-day history of fatigue and nausea. On presentation she denies having abdominal pain, headache, fever, chills, night sweats, vomiting, diarrhea, melena, hematochezia, or weight loss. She recalls changes in the colors of her eyes and darkening urine over the last few days. Her medical history before this is unremarkable. She takes no prescription, over-the-counter, or herbal medications. She works as a librarian and has no occupational toxic exposures. She is single and has one sister with no prior medical history. She denies recent travel, sick contacts, smoking, recreational drug use, or pets at home.
On physical examination, her vital signs are temperature 37.3°C (99.1°F), heart rate 90 beats per minute, blood pressure 125/80 mm Hg, respiration rate 14 per minute, and oxygen saturation 97% on room air. She has icteric sclera and her skin is jaundiced. Cardiac examination is normal. Lungs are clear to auscultation and percussion bilaterally. Her abdomen is soft with no visceromegaly, masses, or tenderness. Extremities are normal with no edema. She is alert and oriented, but she has mild asterixis of the outstretched hands. The neurologic examination is otherwise unremarkable.
The patient’s basic laboratory values are listed in Table 1. Shortly after admission, she develops changes in her mental status, remaining alert but becoming agitated and oriented to person only. In view of her symptoms and laboratory findings, acute liver failure is suspected.
ACUTE LIVER FAILURE
1. The diagnostic criteria for acute liver failure include all of the following except which one?
- Acute elevation of liver biochemical tests
- Presence of preexisting liver disease
- Coagulopathy, defined by an international normalized ratio (INR) of 1.5 or greater
- Encephalopathy
- Duration of symptoms less than 26 weeks
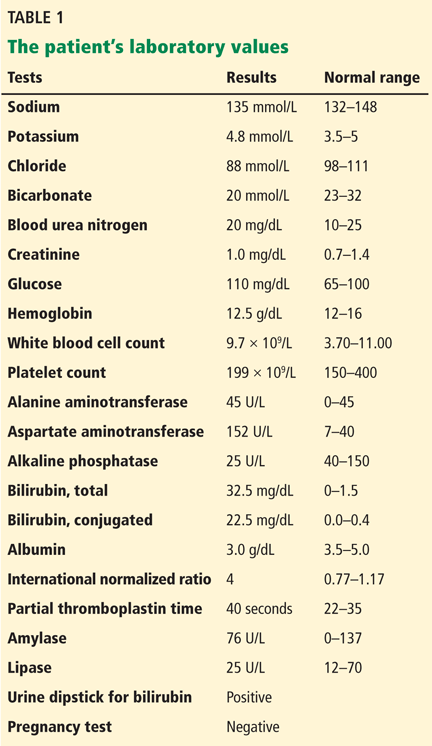
Acute liver failure is defined by acute onset of worsening liver tests, coagulopathy (INR ≥ 1.5), and encephalopathy in patients with no preexisting liver disease and with symptom duration of less than 26 weeks.1 With a few exceptions, a history of preexisting liver disease negates the diagnosis of acute liver failure. Our patient meets the diagnostic criteria for acute liver failure.
Immediate management
Once acute liver failure is identified or suspected, the next step is to transfer the patient to the intensive care unit for close monitoring of mental status. Serial neurologic evaluations permit early detection of cerebral edema, which is considered the most common cause of death in patients with acute liver failure. Additionally, close monitoring of electrolytes and plasma glucose is necessary since these patients are susceptible to electrolyte disturbances and hypoglycemia.
Patients with acute liver failure are at increased risk of infections and should be routinely screened by obtaining urine and blood cultures.
Gastrointestinal bleeding is not uncommon in patients with acute liver failure and is usually due to gastric stress ulceration. Prophylaxis with a histamine 2 receptor antagonist or proton pump inhibitor should be considered in order to prevent gastrointestinal bleeding.
Treatment with N-acetylcysteine is beneficial, not only in patients with acute liver failure due to acetaminophen overdose, but also in those with acute liver failure from other causes.
CASE CONTINUES:
TRANSFER TO THE INTENSIVE CARE UNIT
The patient, now diagnosed with acute liver failure, is transferred to the intensive care unit. Arterial blood gas measurement shows:
- pH 7.38 (reference range 7.35–7.45)
- Pco2 40 mm Hg (36–46)
- Po2 97 mm Hg (85–95)
- Hco3 22 mmol/L (22–26).
A chest radiograph is obtained and is clear. Computed tomography (CT) of the brain reveals no edema. Transcranial Doppler ultrasonography does not show any intracranial fluid collections.
Blood and urine cultures are negative. Her hemoglobin level remains stable, and she does not develop signs of bleeding. She is started on a proton pump inhibitor for stress ulcer prophylaxis and is empirically given intravenous N-acetylcysteine until the cause of acute liver failure can be determined.
CAUSES OF ACUTE LIVER FAILURE
2. Which of the following can cause acute liver failure?
- Acetaminophen overdose
- Viral hepatitis
- Autoimmune hepatitis
- Wilson disease
- Alcoholic hepatitis
Drug-induced liver injury is the most common cause of acute liver failure in the United States,2,3 and of all drugs, acetaminophen overdose is the number-one cause. In acetaminophen-induced liver injury, serum aminotransferase levels are usually elevated to more than 1,000 U/L, while serum bilirubin remains normal in the early stages. Antimicrobial agents, antiepileptic drugs, and herbal supplements have also been implicated in acute liver failure. Our patient has denied taking herbal supplements or medications, including over-the-counter ones.
Acute viral hepatitis can explain the patient’s condition. It is a common cause of acute liver failure in the United States.2 Hepatitis A and E are more common in developing countries. Other viruses such as cytomegalovirus, Epstein-Barr virus, herpes simplex virus type 1 and 2, and varicella zoster virus can also cause acute liver failure. Serum aminotransferase levels may exceed 1,000 U/L in patients with viral hepatitis.
Autoimmune hepatitis is a rare cause of acute liver failure, but it should be considered in the differential diagnosis, particularly in middle-aged women with autoimmune disorders such as hypothyroidism. Autoimmune hepatitis can cause marked elevation in aminotransferase levels (> 1,000 U/L).
Wilson disease is an autosomal-recessive disease in which there is excessive accumulation of copper in the liver and other organs because of an inherited defect in the biliary excretion of copper. Wilson disease can cause acute liver failure and should be excluded in any patient, particularly if under age 40 with acute onset of unexplained hepatic, neurologic, or psychiatric disease.
Alcoholic hepatitis usually occurs in patients with a long-standing history of heavy alcohol use. As a result, most patients with alcoholic hepatitis have manifestations of chronic liver disease due to alcohol use. Therefore, by definition, it is not a cause of acute liver failure. Additionally, in patients with alcoholic hepatitis, the aspartate aminotransferase (AST) level is elevated but less than 300 IU/mL, and the ratio of AST to alanine aminotransferase (ALT) is usually more than 2.
CASE CONTINUES: FURTHER TESTING
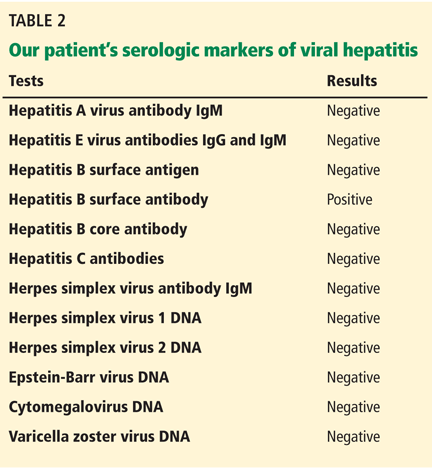
The results of our patient’s serologic tests are shown in Table 2. Other test results:
- Autoimmune markers including antinuclear antibodies, antimitochondrial antibodies, antismooth muscle antibodies, and liver and kidney microsomal antibodies are negative; her immunoglobulin G (IgG) level is normal
- Serum ceruloplasmin 25 mg/dL (normal 21–45)
- Free serum copper 120 µg/dL (normal 8–12)
- Abdominal ultrasonography is unremarkable, with normal liver parenchyma and no intrahepatic or extrahepatic biliary dilatation
- Doppler ultrasonography of the liver shows patent blood vessels.
3. Based on the new data, which of the following statements is correct?
- Hepatitis B is the cause of acute liver failure in this patient
- Herpetic hepatitis cannot be excluded on the basis of the available data
- Wilson disease is most likely the diagnosis, given her elevated free serum copper
- A normal serum ceruloplasmin level is not sufficient to rule out acute liver failure secondary to Wilson disease
Hepatitis B surface antigen and hepatitis B core antibodies were negative in our patient, excluding hepatitis B virus infection. The positive hepatitis B surface antibody indicates prior immunization.
Herpetic hepatitis is an uncommon but important cause of acute liver failure because the mortality rate is high if the patient is not treated early with acyclovir. Fever, elevated aminotransferases, and leukopenia are common with herpetic hepatitis. Fewer than 50% of patients with herpetic hepatitis have vesicular rash.4,5 The value of antibody serologic testing is limited due to high rates of false-positive and false-negative results. The gold standard diagnostic tests are viral load (detection of viral RNA by polymerase chain reaction), viral staining on liver biopsy, or both. In our patient, herpes simplex virus polymerase chain reaction testing was negative, which makes herpetic hepatitis unlikely.
Wilson disease is a genetic condition in which the ability to excrete copper in the bile is impaired, resulting in accumulation of copper in the hepatocytes. Subsequently, copper is released into the bloodstream and eventually into the urine.
However, copper excretion into the bile is impaired in patients with acute liver failure regardless of the etiology. Therefore, elevated free serum copper and 24-hour urine copper levels are not specific for the diagnosis of acute liver failure secondary to Wilson disease. Moreover, Kayser-Fleischer rings, which represent copper deposition in the limbus of the cornea, may not be apparent in the early stages of Wilson disease.
Since it is challenging to diagnose Wilson disease in the context of acute liver failure, Korman et al6 compared patients with acute liver failure secondary to Wilson disease with patients with acute liver failure secondary to other conditions. They found that alkaline phosphatase levels are frequently decreased in patients with acute liver failure secondary to Wilson disease,6 and that a ratio of alkaline phosphatase to total bilirubin of less than 4 is 94% sensitive and 96% specific for the diagnosis.6
Hemolysis is common in acute liver failure due to Wilson disease. This leads to disproportionate elevation of AST compared with ALT, since AST is present in red blood cells. Consequently, the ratio of AST to ALT is usually greater than 2.2, which provides a sensitivity of 94% and a specificity of 86% for the diagnosis.6 These two ratios together provide 100% sensitivity and 100% specificity for the diagnosis of Wilson disease in the context of acute liver failure.6
Ceruloplasmin. Patients with Wilson disease typically have a low ceruloplasmin level. However, because it is an acute-phase reaction protein, ceruloplasmin can be normal or elevated in patients with acute liver failure from Wilson disease.6 Therefore, a normal ceruloplasmin level is not sufficient to rule out acute liver failure secondary to Wilson disease.
CASE CONTINUES: A DEFINITIVE DIAGNOSIS
Our patient undergoes further testing, which reveals the following:
- Her 24-hour urinary excretion of copper is 150 µg (reference value < 30)
- Slit-lamp examination is normal and shows no evidence of Kayser-Fleischer rings
- Her ratio of alkaline phosphatase to total bilirubin is 0.77 based on her initial laboratory results (Table 1)
- Her AST-ALT ratio is 3.4.
The diagnosis in our patient is acute liver failure secondary to Wilson disease.
4. What is the most appropriate next step?
- Liver biopsy
- d-penicillamine by mouth
- Trientine by mouth
- Liver transplant
- Plasmapheresis
Liver biopsy. Accumulation of copper in the liver parenchyma in patients with Wilson disease is sporadic. Therefore, qualitative copper staining on liver biopsy can be falsely negative. Quantitative copper measurement in liver tissue is the gold standard for the diagnosis of Wilson disease. However, the test is time-consuming and is not rapidly available in the context of acute liver failure.
Chelating agents such as d-pencillamine and trientine are used to treat the chronic manifestations of Wilson disease but are not useful for acute liver failure secondary to Wilson disease.
Acute liver failure secondary to Wilson disease is life-threatening, and liver transplant is considered the only definitive life-saving therapy.
Therapeutic plasmapheresis has been reported to be a successful adjunctive therapy to bridge patients with acute liver failure secondary to Wilson disease to transplant.7 However, liver transplant is still the only definitive treatment.
CASE CONTINUES: THE PATIENT’S SISTER SEEKS CARE
The patient undergoes liver transplantation, with no perioperative or postoperative complications.
The patient’s 18-year-old sister is now seeking medical attention in the outpatient clinic, concerned that she may have Wilson disease. She is otherwise healthy and denies any symptoms or complaints.
5. What is the next step for the patient’s sister?
- Reassurance
- Prophylaxis with trientine
- Check liver enzyme levels, serum ceruloplasmin level, and urine copper, and order a slit-lamp examination
- Genetic testing
Wilson disease can be asymptomatic in its early stages and may be diagnosed incidentally during routine blood tests that reveal abnormal liver enzyme levels. All patients with a confirmed family history of Wilson disease should be screened even if they are asymptomatic. The diagnosis of Wilson disease should be established in first-degree relatives before specific treatment for the relatives is prescribed.
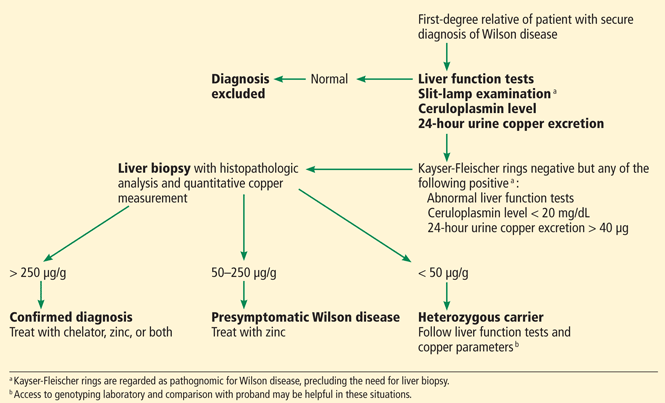
The first step in screening a first-degree relative for Wilson disease is to check liver enzyme levels (specifically aminotransferases, alkaline phosphatase, and bilirubin), serum ceruloplasmin level, and 24-hour urine copper, and order an ophthalmologic slit-lamp examination. If any of these tests is abnormal, liver biopsy should be performed for histopathologic evaluation and quantitative copper measurement. Kayser-Fleischer rings are seen in only 50% of patients with Wilson disease and hepatic involvement, but they are pathognomic. Guidelines8 for screening first-degree relatives of Wilson disease patients are shown in Figure 1.
Genetic analysis. ATP7B, the Wilson disease gene, is located on chromosome 13. At least 300 mutations of the gene have been described,2 and the most common mutation is present in only 15% to 30% of the Wilson disease population.8–10 Routine molecular testing of the ATP7B
CASE CONTINUES: WORKUP OF THE PATIENT’S SISTER
The patient’s sister has no symptoms and her physical examination is normal. Slit-lamp examination reveals no evidence of Kayser-Fleischer rings. Her laboratory values, including complete blood counts, complete metabolic panel, and INR, are within normal ranges. Other test results, however, are abnormal:
- Free serum copper level 27 µg/dL (normal 8–12)
- Serum ceruloplasmin 9.0 mg/dL (normal 20–50)
- 24-hour urinary copper excretion 135 µg (normal < 30).
She undergoes liver biopsy for quantitative copper measurement, and the result is very high at 1,118 µg/g dry weight (reference range 10–35). The diagnosis of Wilson disease is established.
TREATING CHRONIC WILSON DISEASE
6. Which of the following is not an appropriate next step for the patient’s sister?
- Tetrathiomolybdate
- d-penicillamine
- Trientine
- Zinc salts
- Prednisone
The goal of medical treatment of chronic Wilson disease is to improve symptoms and prevent progression of the disease.
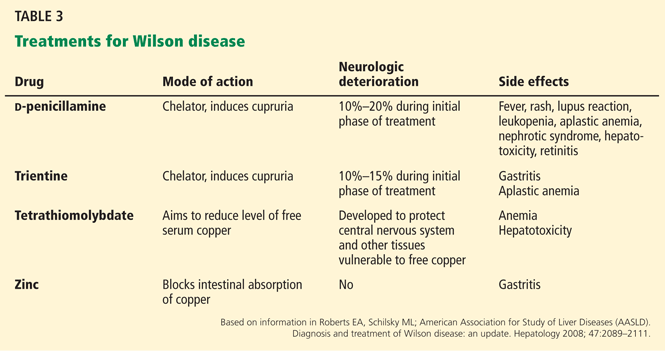
Chelating agents and zinc salts are the most commonly used medicines in the management of Wilson disease. Chelating agents remove copper from tissue, whereas zinc blocks the intestinal absorption of copper and stimulates the synthesis of endogenous chelators such as metallothioneins. Tetrathiomolybdate is an alternative agent developed to interfere with the distribution of excess body copper to susceptible target sites by reducing free serum copper (Table 3). There are no data to support the use of prednisone in the treatment of Wilson disease.
During treatment with chelating agents, 24-hour urinary excretion of copper is routinely monitored to determine the efficacy of therapy and adherence to treatment. Once de-coppering is achieved, as evidenced by a normalization of 24-hour urine copper excretion, the chelating agent can be switched to zinc salts to prevent intestinal absorption of copper.
Clinical and biochemical stabilization is achieved typically within 2 to 6 months of the initial treatment with chelating agents.8 Organ meats, nuts, shellfish, and chocolate are rich in copper and should be avoided.
The patient’s sister is started on trientine 250 mg orally three times daily on an empty stomach at least 1 hour before meals. Treatment is monitored by following 24-hour urine copper measurement. A 24-hour urine copper measurement at 3 months after starting treatment has increased from 54 at baseline to 350 µg, which indicates that the copper is being removed from tissues. The plan is for early substitution of zinc for long-term maintenance once de-coppering is completed.
KEY POINTS
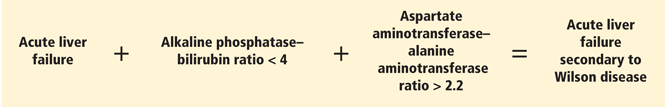
- Acute liver failure is severe acute liver injury characterized by coagulopathy (INR ≥ 1.5) and encephalopathy in a patient with no preexisting liver disease and with duration of symptoms less than 26 weeks.
- Acute liver failure secondary to Wilson disease is uncommon but should be excluded, particularly in young patients.
- The diagnosis of Wilson disease in the setting of acute liver failure is challenging because the serum ceruloplasmin level may be normal in acute liver failure secondary to Wilson disease, and free serum copper and 24-hour urine copper are usually elevated in all acute liver failure patients regardless of the etiology.
- A ratio of alkaline phosphatase to total bilirubin of less than 4 plus an AST-ALT ratio greater than 2.2 in a patient with acute liver failure should be regarded as Wilson disease until proven otherwise (Figure 2).
- Acute liver failure secondary to Wilson disease is usually fatal, and emergency liver transplant is a life-saving procedure.
- Screening of first-degree relatives of Wilson disease patients should include a history and physical examination, liver enzyme tests, complete blood cell count, serum ceruloplasmin level, serum free copper level, slit-lamp examination of the eyes, and 24-hour urinary copper measurement. Genetic tests are supplementary for screening but are not routinely available.
- Lee WM, Larson AM, Stravitz T. AASLD Position Paper: The management of acute liver failure: update 2011. www.aasld.org/sites/default/files/guideline_documents/alfenhanced.pdf. Accessed December 9, 2015.
- Bernal W, Auzinger G, Dhawan A, Wendon J. Acute liver failure. Lancet 2010; 376:190–201.
- Larson AM, Polson J, Fontana RJ, et al; Acute Liver Failure Study Group. Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology 2005; 42:1364–1372.
- Hanouneh IA, Khoriaty R, Zein NN. A 35-year-old Asian man with jaundice and markedly high aminotransferase levels. Cleve Clin J Med 2009; 76:449–456.
- Norvell JP, Blei AT, Jovanovic BD, Levitsky J. Herpes simplex virus hepatitis: an analysis of the published literature and institutional cases. Liver Transpl 2007; 13:1428–1434.
- Korman JD, Volenberg I, Balko J, et al; Pediatric and Adult Acute Liver Failure Study Groups. Screening for Wilson disease in acute liver failure: a comparison of currently available diagnostic tests. Hepatology 2008; 48:1167–1174.
- Morgan SM, Zantek ND. Therapeutic plasma exchange for fulminant hepatic failure secondary to Wilson's disease. J Clin Apher 2012; 27:282–286.
- Roberts EA, Schilsky ML; American Association for Study of Liver Diseases (AASLD). Diagnosis and treatment of Wilson disease: an update. Hepatology 2008; 47:2089–2111.
- Shah AB, Chernov I, Zhang HT, et al. Identification and analysis of mutations in the Wilson disease gene (ATP7B): population frequencies, genotype-phenotype correlation, and functional analyses. Am J Hum Genet 1997; 61:317–328.
- Maier-Dobersberger T, Ferenci P, Polli C, et al. Detection of the His1069Gln mutation in Wilson disease by rapid polymerase chain reaction. Ann Intern Med 1997; 127:21–26.
A 25-year-old woman presents to the emergency department with a 7-day history of fatigue and nausea. On presentation she denies having abdominal pain, headache, fever, chills, night sweats, vomiting, diarrhea, melena, hematochezia, or weight loss. She recalls changes in the colors of her eyes and darkening urine over the last few days. Her medical history before this is unremarkable. She takes no prescription, over-the-counter, or herbal medications. She works as a librarian and has no occupational toxic exposures. She is single and has one sister with no prior medical history. She denies recent travel, sick contacts, smoking, recreational drug use, or pets at home.
On physical examination, her vital signs are temperature 37.3°C (99.1°F), heart rate 90 beats per minute, blood pressure 125/80 mm Hg, respiration rate 14 per minute, and oxygen saturation 97% on room air. She has icteric sclera and her skin is jaundiced. Cardiac examination is normal. Lungs are clear to auscultation and percussion bilaterally. Her abdomen is soft with no visceromegaly, masses, or tenderness. Extremities are normal with no edema. She is alert and oriented, but she has mild asterixis of the outstretched hands. The neurologic examination is otherwise unremarkable.
The patient’s basic laboratory values are listed in Table 1. Shortly after admission, she develops changes in her mental status, remaining alert but becoming agitated and oriented to person only. In view of her symptoms and laboratory findings, acute liver failure is suspected.
ACUTE LIVER FAILURE
1. The diagnostic criteria for acute liver failure include all of the following except which one?
- Acute elevation of liver biochemical tests
- Presence of preexisting liver disease
- Coagulopathy, defined by an international normalized ratio (INR) of 1.5 or greater
- Encephalopathy
- Duration of symptoms less than 26 weeks
Acute liver failure is defined by acute onset of worsening liver tests, coagulopathy (INR ≥ 1.5), and encephalopathy in patients with no preexisting liver disease and with symptom duration of less than 26 weeks.1 With a few exceptions, a history of preexisting liver disease negates the diagnosis of acute liver failure. Our patient meets the diagnostic criteria for acute liver failure.
Immediate management
Once acute liver failure is identified or suspected, the next step is to transfer the patient to the intensive care unit for close monitoring of mental status. Serial neurologic evaluations permit early detection of cerebral edema, which is considered the most common cause of death in patients with acute liver failure. Additionally, close monitoring of electrolytes and plasma glucose is necessary since these patients are susceptible to electrolyte disturbances and hypoglycemia.
Patients with acute liver failure are at increased risk of infections and should be routinely screened by obtaining urine and blood cultures.
Gastrointestinal bleeding is not uncommon in patients with acute liver failure and is usually due to gastric stress ulceration. Prophylaxis with a histamine 2 receptor antagonist or proton pump inhibitor should be considered in order to prevent gastrointestinal bleeding.
Treatment with N-acetylcysteine is beneficial, not only in patients with acute liver failure due to acetaminophen overdose, but also in those with acute liver failure from other causes.
CASE CONTINUES:
TRANSFER TO THE INTENSIVE CARE UNIT
The patient, now diagnosed with acute liver failure, is transferred to the intensive care unit. Arterial blood gas measurement shows:
- pH 7.38 (reference range 7.35–7.45)
- Pco2 40 mm Hg (36–46)
- Po2 97 mm Hg (85–95)
- Hco3 22 mmol/L (22–26).
A chest radiograph is obtained and is clear. Computed tomography (CT) of the brain reveals no edema. Transcranial Doppler ultrasonography does not show any intracranial fluid collections.
Blood and urine cultures are negative. Her hemoglobin level remains stable, and she does not develop signs of bleeding. She is started on a proton pump inhibitor for stress ulcer prophylaxis and is empirically given intravenous N-acetylcysteine until the cause of acute liver failure can be determined.
CAUSES OF ACUTE LIVER FAILURE
2. Which of the following can cause acute liver failure?
- Acetaminophen overdose
- Viral hepatitis
- Autoimmune hepatitis
- Wilson disease
- Alcoholic hepatitis
Drug-induced liver injury is the most common cause of acute liver failure in the United States,2,3 and of all drugs, acetaminophen overdose is the number-one cause. In acetaminophen-induced liver injury, serum aminotransferase levels are usually elevated to more than 1,000 U/L, while serum bilirubin remains normal in the early stages. Antimicrobial agents, antiepileptic drugs, and herbal supplements have also been implicated in acute liver failure. Our patient has denied taking herbal supplements or medications, including over-the-counter ones.
Acute viral hepatitis can explain the patient’s condition. It is a common cause of acute liver failure in the United States.2 Hepatitis A and E are more common in developing countries. Other viruses such as cytomegalovirus, Epstein-Barr virus, herpes simplex virus type 1 and 2, and varicella zoster virus can also cause acute liver failure. Serum aminotransferase levels may exceed 1,000 U/L in patients with viral hepatitis.
Autoimmune hepatitis is a rare cause of acute liver failure, but it should be considered in the differential diagnosis, particularly in middle-aged women with autoimmune disorders such as hypothyroidism. Autoimmune hepatitis can cause marked elevation in aminotransferase levels (> 1,000 U/L).
Wilson disease is an autosomal-recessive disease in which there is excessive accumulation of copper in the liver and other organs because of an inherited defect in the biliary excretion of copper. Wilson disease can cause acute liver failure and should be excluded in any patient, particularly if under age 40 with acute onset of unexplained hepatic, neurologic, or psychiatric disease.
Alcoholic hepatitis usually occurs in patients with a long-standing history of heavy alcohol use. As a result, most patients with alcoholic hepatitis have manifestations of chronic liver disease due to alcohol use. Therefore, by definition, it is not a cause of acute liver failure. Additionally, in patients with alcoholic hepatitis, the aspartate aminotransferase (AST) level is elevated but less than 300 IU/mL, and the ratio of AST to alanine aminotransferase (ALT) is usually more than 2.
CASE CONTINUES: FURTHER TESTING
The results of our patient’s serologic tests are shown in Table 2. Other test results:
- Autoimmune markers including antinuclear antibodies, antimitochondrial antibodies, antismooth muscle antibodies, and liver and kidney microsomal antibodies are negative; her immunoglobulin G (IgG) level is normal
- Serum ceruloplasmin 25 mg/dL (normal 21–45)
- Free serum copper 120 µg/dL (normal 8–12)
- Abdominal ultrasonography is unremarkable, with normal liver parenchyma and no intrahepatic or extrahepatic biliary dilatation
- Doppler ultrasonography of the liver shows patent blood vessels.
3. Based on the new data, which of the following statements is correct?
- Hepatitis B is the cause of acute liver failure in this patient
- Herpetic hepatitis cannot be excluded on the basis of the available data
- Wilson disease is most likely the diagnosis, given her elevated free serum copper
- A normal serum ceruloplasmin level is not sufficient to rule out acute liver failure secondary to Wilson disease
Hepatitis B surface antigen and hepatitis B core antibodies were negative in our patient, excluding hepatitis B virus infection. The positive hepatitis B surface antibody indicates prior immunization.
Herpetic hepatitis is an uncommon but important cause of acute liver failure because the mortality rate is high if the patient is not treated early with acyclovir. Fever, elevated aminotransferases, and leukopenia are common with herpetic hepatitis. Fewer than 50% of patients with herpetic hepatitis have vesicular rash.4,5 The value of antibody serologic testing is limited due to high rates of false-positive and false-negative results. The gold standard diagnostic tests are viral load (detection of viral RNA by polymerase chain reaction), viral staining on liver biopsy, or both. In our patient, herpes simplex virus polymerase chain reaction testing was negative, which makes herpetic hepatitis unlikely.
Wilson disease is a genetic condition in which the ability to excrete copper in the bile is impaired, resulting in accumulation of copper in the hepatocytes. Subsequently, copper is released into the bloodstream and eventually into the urine.
However, copper excretion into the bile is impaired in patients with acute liver failure regardless of the etiology. Therefore, elevated free serum copper and 24-hour urine copper levels are not specific for the diagnosis of acute liver failure secondary to Wilson disease. Moreover, Kayser-Fleischer rings, which represent copper deposition in the limbus of the cornea, may not be apparent in the early stages of Wilson disease.
Since it is challenging to diagnose Wilson disease in the context of acute liver failure, Korman et al6 compared patients with acute liver failure secondary to Wilson disease with patients with acute liver failure secondary to other conditions. They found that alkaline phosphatase levels are frequently decreased in patients with acute liver failure secondary to Wilson disease,6 and that a ratio of alkaline phosphatase to total bilirubin of less than 4 is 94% sensitive and 96% specific for the diagnosis.6
Hemolysis is common in acute liver failure due to Wilson disease. This leads to disproportionate elevation of AST compared with ALT, since AST is present in red blood cells. Consequently, the ratio of AST to ALT is usually greater than 2.2, which provides a sensitivity of 94% and a specificity of 86% for the diagnosis.6 These two ratios together provide 100% sensitivity and 100% specificity for the diagnosis of Wilson disease in the context of acute liver failure.6
Ceruloplasmin. Patients with Wilson disease typically have a low ceruloplasmin level. However, because it is an acute-phase reaction protein, ceruloplasmin can be normal or elevated in patients with acute liver failure from Wilson disease.6 Therefore, a normal ceruloplasmin level is not sufficient to rule out acute liver failure secondary to Wilson disease.
CASE CONTINUES: A DEFINITIVE DIAGNOSIS
Our patient undergoes further testing, which reveals the following:
- Her 24-hour urinary excretion of copper is 150 µg (reference value < 30)
- Slit-lamp examination is normal and shows no evidence of Kayser-Fleischer rings
- Her ratio of alkaline phosphatase to total bilirubin is 0.77 based on her initial laboratory results (Table 1)
- Her AST-ALT ratio is 3.4.
The diagnosis in our patient is acute liver failure secondary to Wilson disease.
4. What is the most appropriate next step?
- Liver biopsy
- d-penicillamine by mouth
- Trientine by mouth
- Liver transplant
- Plasmapheresis
Liver biopsy. Accumulation of copper in the liver parenchyma in patients with Wilson disease is sporadic. Therefore, qualitative copper staining on liver biopsy can be falsely negative. Quantitative copper measurement in liver tissue is the gold standard for the diagnosis of Wilson disease. However, the test is time-consuming and is not rapidly available in the context of acute liver failure.
Chelating agents such as d-pencillamine and trientine are used to treat the chronic manifestations of Wilson disease but are not useful for acute liver failure secondary to Wilson disease.
Acute liver failure secondary to Wilson disease is life-threatening, and liver transplant is considered the only definitive life-saving therapy.
Therapeutic plasmapheresis has been reported to be a successful adjunctive therapy to bridge patients with acute liver failure secondary to Wilson disease to transplant.7 However, liver transplant is still the only definitive treatment.
CASE CONTINUES: THE PATIENT’S SISTER SEEKS CARE
The patient undergoes liver transplantation, with no perioperative or postoperative complications.
The patient’s 18-year-old sister is now seeking medical attention in the outpatient clinic, concerned that she may have Wilson disease. She is otherwise healthy and denies any symptoms or complaints.
5. What is the next step for the patient’s sister?
- Reassurance
- Prophylaxis with trientine
- Check liver enzyme levels, serum ceruloplasmin level, and urine copper, and order a slit-lamp examination
- Genetic testing
Wilson disease can be asymptomatic in its early stages and may be diagnosed incidentally during routine blood tests that reveal abnormal liver enzyme levels. All patients with a confirmed family history of Wilson disease should be screened even if they are asymptomatic. The diagnosis of Wilson disease should be established in first-degree relatives before specific treatment for the relatives is prescribed.
The first step in screening a first-degree relative for Wilson disease is to check liver enzyme levels (specifically aminotransferases, alkaline phosphatase, and bilirubin), serum ceruloplasmin level, and 24-hour urine copper, and order an ophthalmologic slit-lamp examination. If any of these tests is abnormal, liver biopsy should be performed for histopathologic evaluation and quantitative copper measurement. Kayser-Fleischer rings are seen in only 50% of patients with Wilson disease and hepatic involvement, but they are pathognomic. Guidelines8 for screening first-degree relatives of Wilson disease patients are shown in Figure 1.
Genetic analysis. ATP7B, the Wilson disease gene, is located on chromosome 13. At least 300 mutations of the gene have been described,2 and the most common mutation is present in only 15% to 30% of the Wilson disease population.8–10 Routine molecular testing of the ATP7B
CASE CONTINUES: WORKUP OF THE PATIENT’S SISTER
The patient’s sister has no symptoms and her physical examination is normal. Slit-lamp examination reveals no evidence of Kayser-Fleischer rings. Her laboratory values, including complete blood counts, complete metabolic panel, and INR, are within normal ranges. Other test results, however, are abnormal:
- Free serum copper level 27 µg/dL (normal 8–12)
- Serum ceruloplasmin 9.0 mg/dL (normal 20–50)
- 24-hour urinary copper excretion 135 µg (normal < 30).
She undergoes liver biopsy for quantitative copper measurement, and the result is very high at 1,118 µg/g dry weight (reference range 10–35). The diagnosis of Wilson disease is established.
TREATING CHRONIC WILSON DISEASE
6. Which of the following is not an appropriate next step for the patient’s sister?
- Tetrathiomolybdate
- d-penicillamine
- Trientine
- Zinc salts
- Prednisone
The goal of medical treatment of chronic Wilson disease is to improve symptoms and prevent progression of the disease.
Chelating agents and zinc salts are the most commonly used medicines in the management of Wilson disease. Chelating agents remove copper from tissue, whereas zinc blocks the intestinal absorption of copper and stimulates the synthesis of endogenous chelators such as metallothioneins. Tetrathiomolybdate is an alternative agent developed to interfere with the distribution of excess body copper to susceptible target sites by reducing free serum copper (Table 3). There are no data to support the use of prednisone in the treatment of Wilson disease.
During treatment with chelating agents, 24-hour urinary excretion of copper is routinely monitored to determine the efficacy of therapy and adherence to treatment. Once de-coppering is achieved, as evidenced by a normalization of 24-hour urine copper excretion, the chelating agent can be switched to zinc salts to prevent intestinal absorption of copper.
Clinical and biochemical stabilization is achieved typically within 2 to 6 months of the initial treatment with chelating agents.8 Organ meats, nuts, shellfish, and chocolate are rich in copper and should be avoided.
The patient’s sister is started on trientine 250 mg orally three times daily on an empty stomach at least 1 hour before meals. Treatment is monitored by following 24-hour urine copper measurement. A 24-hour urine copper measurement at 3 months after starting treatment has increased from 54 at baseline to 350 µg, which indicates that the copper is being removed from tissues. The plan is for early substitution of zinc for long-term maintenance once de-coppering is completed.
KEY POINTS
- Acute liver failure is severe acute liver injury characterized by coagulopathy (INR ≥ 1.5) and encephalopathy in a patient with no preexisting liver disease and with duration of symptoms less than 26 weeks.
- Acute liver failure secondary to Wilson disease is uncommon but should be excluded, particularly in young patients.
- The diagnosis of Wilson disease in the setting of acute liver failure is challenging because the serum ceruloplasmin level may be normal in acute liver failure secondary to Wilson disease, and free serum copper and 24-hour urine copper are usually elevated in all acute liver failure patients regardless of the etiology.
- A ratio of alkaline phosphatase to total bilirubin of less than 4 plus an AST-ALT ratio greater than 2.2 in a patient with acute liver failure should be regarded as Wilson disease until proven otherwise (Figure 2).
- Acute liver failure secondary to Wilson disease is usually fatal, and emergency liver transplant is a life-saving procedure.
- Screening of first-degree relatives of Wilson disease patients should include a history and physical examination, liver enzyme tests, complete blood cell count, serum ceruloplasmin level, serum free copper level, slit-lamp examination of the eyes, and 24-hour urinary copper measurement. Genetic tests are supplementary for screening but are not routinely available.
A 25-year-old woman presents to the emergency department with a 7-day history of fatigue and nausea. On presentation she denies having abdominal pain, headache, fever, chills, night sweats, vomiting, diarrhea, melena, hematochezia, or weight loss. She recalls changes in the colors of her eyes and darkening urine over the last few days. Her medical history before this is unremarkable. She takes no prescription, over-the-counter, or herbal medications. She works as a librarian and has no occupational toxic exposures. She is single and has one sister with no prior medical history. She denies recent travel, sick contacts, smoking, recreational drug use, or pets at home.
On physical examination, her vital signs are temperature 37.3°C (99.1°F), heart rate 90 beats per minute, blood pressure 125/80 mm Hg, respiration rate 14 per minute, and oxygen saturation 97% on room air. She has icteric sclera and her skin is jaundiced. Cardiac examination is normal. Lungs are clear to auscultation and percussion bilaterally. Her abdomen is soft with no visceromegaly, masses, or tenderness. Extremities are normal with no edema. She is alert and oriented, but she has mild asterixis of the outstretched hands. The neurologic examination is otherwise unremarkable.
The patient’s basic laboratory values are listed in Table 1. Shortly after admission, she develops changes in her mental status, remaining alert but becoming agitated and oriented to person only. In view of her symptoms and laboratory findings, acute liver failure is suspected.
ACUTE LIVER FAILURE
1. The diagnostic criteria for acute liver failure include all of the following except which one?
- Acute elevation of liver biochemical tests
- Presence of preexisting liver disease
- Coagulopathy, defined by an international normalized ratio (INR) of 1.5 or greater
- Encephalopathy
- Duration of symptoms less than 26 weeks
Acute liver failure is defined by acute onset of worsening liver tests, coagulopathy (INR ≥ 1.5), and encephalopathy in patients with no preexisting liver disease and with symptom duration of less than 26 weeks.1 With a few exceptions, a history of preexisting liver disease negates the diagnosis of acute liver failure. Our patient meets the diagnostic criteria for acute liver failure.
Immediate management
Once acute liver failure is identified or suspected, the next step is to transfer the patient to the intensive care unit for close monitoring of mental status. Serial neurologic evaluations permit early detection of cerebral edema, which is considered the most common cause of death in patients with acute liver failure. Additionally, close monitoring of electrolytes and plasma glucose is necessary since these patients are susceptible to electrolyte disturbances and hypoglycemia.
Patients with acute liver failure are at increased risk of infections and should be routinely screened by obtaining urine and blood cultures.
Gastrointestinal bleeding is not uncommon in patients with acute liver failure and is usually due to gastric stress ulceration. Prophylaxis with a histamine 2 receptor antagonist or proton pump inhibitor should be considered in order to prevent gastrointestinal bleeding.
Treatment with N-acetylcysteine is beneficial, not only in patients with acute liver failure due to acetaminophen overdose, but also in those with acute liver failure from other causes.
CASE CONTINUES:
TRANSFER TO THE INTENSIVE CARE UNIT
The patient, now diagnosed with acute liver failure, is transferred to the intensive care unit. Arterial blood gas measurement shows:
- pH 7.38 (reference range 7.35–7.45)
- Pco2 40 mm Hg (36–46)
- Po2 97 mm Hg (85–95)
- Hco3 22 mmol/L (22–26).
A chest radiograph is obtained and is clear. Computed tomography (CT) of the brain reveals no edema. Transcranial Doppler ultrasonography does not show any intracranial fluid collections.
Blood and urine cultures are negative. Her hemoglobin level remains stable, and she does not develop signs of bleeding. She is started on a proton pump inhibitor for stress ulcer prophylaxis and is empirically given intravenous N-acetylcysteine until the cause of acute liver failure can be determined.
CAUSES OF ACUTE LIVER FAILURE
2. Which of the following can cause acute liver failure?
- Acetaminophen overdose
- Viral hepatitis
- Autoimmune hepatitis
- Wilson disease
- Alcoholic hepatitis
Drug-induced liver injury is the most common cause of acute liver failure in the United States,2,3 and of all drugs, acetaminophen overdose is the number-one cause. In acetaminophen-induced liver injury, serum aminotransferase levels are usually elevated to more than 1,000 U/L, while serum bilirubin remains normal in the early stages. Antimicrobial agents, antiepileptic drugs, and herbal supplements have also been implicated in acute liver failure. Our patient has denied taking herbal supplements or medications, including over-the-counter ones.
Acute viral hepatitis can explain the patient’s condition. It is a common cause of acute liver failure in the United States.2 Hepatitis A and E are more common in developing countries. Other viruses such as cytomegalovirus, Epstein-Barr virus, herpes simplex virus type 1 and 2, and varicella zoster virus can also cause acute liver failure. Serum aminotransferase levels may exceed 1,000 U/L in patients with viral hepatitis.
Autoimmune hepatitis is a rare cause of acute liver failure, but it should be considered in the differential diagnosis, particularly in middle-aged women with autoimmune disorders such as hypothyroidism. Autoimmune hepatitis can cause marked elevation in aminotransferase levels (> 1,000 U/L).
Wilson disease is an autosomal-recessive disease in which there is excessive accumulation of copper in the liver and other organs because of an inherited defect in the biliary excretion of copper. Wilson disease can cause acute liver failure and should be excluded in any patient, particularly if under age 40 with acute onset of unexplained hepatic, neurologic, or psychiatric disease.
Alcoholic hepatitis usually occurs in patients with a long-standing history of heavy alcohol use. As a result, most patients with alcoholic hepatitis have manifestations of chronic liver disease due to alcohol use. Therefore, by definition, it is not a cause of acute liver failure. Additionally, in patients with alcoholic hepatitis, the aspartate aminotransferase (AST) level is elevated but less than 300 IU/mL, and the ratio of AST to alanine aminotransferase (ALT) is usually more than 2.
CASE CONTINUES: FURTHER TESTING
The results of our patient’s serologic tests are shown in Table 2. Other test results:
- Autoimmune markers including antinuclear antibodies, antimitochondrial antibodies, antismooth muscle antibodies, and liver and kidney microsomal antibodies are negative; her immunoglobulin G (IgG) level is normal
- Serum ceruloplasmin 25 mg/dL (normal 21–45)
- Free serum copper 120 µg/dL (normal 8–12)
- Abdominal ultrasonography is unremarkable, with normal liver parenchyma and no intrahepatic or extrahepatic biliary dilatation
- Doppler ultrasonography of the liver shows patent blood vessels.
3. Based on the new data, which of the following statements is correct?
- Hepatitis B is the cause of acute liver failure in this patient
- Herpetic hepatitis cannot be excluded on the basis of the available data
- Wilson disease is most likely the diagnosis, given her elevated free serum copper
- A normal serum ceruloplasmin level is not sufficient to rule out acute liver failure secondary to Wilson disease
Hepatitis B surface antigen and hepatitis B core antibodies were negative in our patient, excluding hepatitis B virus infection. The positive hepatitis B surface antibody indicates prior immunization.
Herpetic hepatitis is an uncommon but important cause of acute liver failure because the mortality rate is high if the patient is not treated early with acyclovir. Fever, elevated aminotransferases, and leukopenia are common with herpetic hepatitis. Fewer than 50% of patients with herpetic hepatitis have vesicular rash.4,5 The value of antibody serologic testing is limited due to high rates of false-positive and false-negative results. The gold standard diagnostic tests are viral load (detection of viral RNA by polymerase chain reaction), viral staining on liver biopsy, or both. In our patient, herpes simplex virus polymerase chain reaction testing was negative, which makes herpetic hepatitis unlikely.
Wilson disease is a genetic condition in which the ability to excrete copper in the bile is impaired, resulting in accumulation of copper in the hepatocytes. Subsequently, copper is released into the bloodstream and eventually into the urine.
However, copper excretion into the bile is impaired in patients with acute liver failure regardless of the etiology. Therefore, elevated free serum copper and 24-hour urine copper levels are not specific for the diagnosis of acute liver failure secondary to Wilson disease. Moreover, Kayser-Fleischer rings, which represent copper deposition in the limbus of the cornea, may not be apparent in the early stages of Wilson disease.
Since it is challenging to diagnose Wilson disease in the context of acute liver failure, Korman et al6 compared patients with acute liver failure secondary to Wilson disease with patients with acute liver failure secondary to other conditions. They found that alkaline phosphatase levels are frequently decreased in patients with acute liver failure secondary to Wilson disease,6 and that a ratio of alkaline phosphatase to total bilirubin of less than 4 is 94% sensitive and 96% specific for the diagnosis.6
Hemolysis is common in acute liver failure due to Wilson disease. This leads to disproportionate elevation of AST compared with ALT, since AST is present in red blood cells. Consequently, the ratio of AST to ALT is usually greater than 2.2, which provides a sensitivity of 94% and a specificity of 86% for the diagnosis.6 These two ratios together provide 100% sensitivity and 100% specificity for the diagnosis of Wilson disease in the context of acute liver failure.6
Ceruloplasmin. Patients with Wilson disease typically have a low ceruloplasmin level. However, because it is an acute-phase reaction protein, ceruloplasmin can be normal or elevated in patients with acute liver failure from Wilson disease.6 Therefore, a normal ceruloplasmin level is not sufficient to rule out acute liver failure secondary to Wilson disease.
CASE CONTINUES: A DEFINITIVE DIAGNOSIS
Our patient undergoes further testing, which reveals the following:
- Her 24-hour urinary excretion of copper is 150 µg (reference value < 30)
- Slit-lamp examination is normal and shows no evidence of Kayser-Fleischer rings
- Her ratio of alkaline phosphatase to total bilirubin is 0.77 based on her initial laboratory results (Table 1)
- Her AST-ALT ratio is 3.4.
The diagnosis in our patient is acute liver failure secondary to Wilson disease.
4. What is the most appropriate next step?
- Liver biopsy
- d-penicillamine by mouth
- Trientine by mouth
- Liver transplant
- Plasmapheresis
Liver biopsy. Accumulation of copper in the liver parenchyma in patients with Wilson disease is sporadic. Therefore, qualitative copper staining on liver biopsy can be falsely negative. Quantitative copper measurement in liver tissue is the gold standard for the diagnosis of Wilson disease. However, the test is time-consuming and is not rapidly available in the context of acute liver failure.
Chelating agents such as d-pencillamine and trientine are used to treat the chronic manifestations of Wilson disease but are not useful for acute liver failure secondary to Wilson disease.
Acute liver failure secondary to Wilson disease is life-threatening, and liver transplant is considered the only definitive life-saving therapy.
Therapeutic plasmapheresis has been reported to be a successful adjunctive therapy to bridge patients with acute liver failure secondary to Wilson disease to transplant.7 However, liver transplant is still the only definitive treatment.
CASE CONTINUES: THE PATIENT’S SISTER SEEKS CARE
The patient undergoes liver transplantation, with no perioperative or postoperative complications.
The patient’s 18-year-old sister is now seeking medical attention in the outpatient clinic, concerned that she may have Wilson disease. She is otherwise healthy and denies any symptoms or complaints.
5. What is the next step for the patient’s sister?
- Reassurance
- Prophylaxis with trientine
- Check liver enzyme levels, serum ceruloplasmin level, and urine copper, and order a slit-lamp examination
- Genetic testing
Wilson disease can be asymptomatic in its early stages and may be diagnosed incidentally during routine blood tests that reveal abnormal liver enzyme levels. All patients with a confirmed family history of Wilson disease should be screened even if they are asymptomatic. The diagnosis of Wilson disease should be established in first-degree relatives before specific treatment for the relatives is prescribed.
The first step in screening a first-degree relative for Wilson disease is to check liver enzyme levels (specifically aminotransferases, alkaline phosphatase, and bilirubin), serum ceruloplasmin level, and 24-hour urine copper, and order an ophthalmologic slit-lamp examination. If any of these tests is abnormal, liver biopsy should be performed for histopathologic evaluation and quantitative copper measurement. Kayser-Fleischer rings are seen in only 50% of patients with Wilson disease and hepatic involvement, but they are pathognomic. Guidelines8 for screening first-degree relatives of Wilson disease patients are shown in Figure 1.
Genetic analysis. ATP7B, the Wilson disease gene, is located on chromosome 13. At least 300 mutations of the gene have been described,2 and the most common mutation is present in only 15% to 30% of the Wilson disease population.8–10 Routine molecular testing of the ATP7B
CASE CONTINUES: WORKUP OF THE PATIENT’S SISTER
The patient’s sister has no symptoms and her physical examination is normal. Slit-lamp examination reveals no evidence of Kayser-Fleischer rings. Her laboratory values, including complete blood counts, complete metabolic panel, and INR, are within normal ranges. Other test results, however, are abnormal:
- Free serum copper level 27 µg/dL (normal 8–12)
- Serum ceruloplasmin 9.0 mg/dL (normal 20–50)
- 24-hour urinary copper excretion 135 µg (normal < 30).
She undergoes liver biopsy for quantitative copper measurement, and the result is very high at 1,118 µg/g dry weight (reference range 10–35). The diagnosis of Wilson disease is established.
TREATING CHRONIC WILSON DISEASE
6. Which of the following is not an appropriate next step for the patient’s sister?
- Tetrathiomolybdate
- d-penicillamine
- Trientine
- Zinc salts
- Prednisone
The goal of medical treatment of chronic Wilson disease is to improve symptoms and prevent progression of the disease.
Chelating agents and zinc salts are the most commonly used medicines in the management of Wilson disease. Chelating agents remove copper from tissue, whereas zinc blocks the intestinal absorption of copper and stimulates the synthesis of endogenous chelators such as metallothioneins. Tetrathiomolybdate is an alternative agent developed to interfere with the distribution of excess body copper to susceptible target sites by reducing free serum copper (Table 3). There are no data to support the use of prednisone in the treatment of Wilson disease.
During treatment with chelating agents, 24-hour urinary excretion of copper is routinely monitored to determine the efficacy of therapy and adherence to treatment. Once de-coppering is achieved, as evidenced by a normalization of 24-hour urine copper excretion, the chelating agent can be switched to zinc salts to prevent intestinal absorption of copper.
Clinical and biochemical stabilization is achieved typically within 2 to 6 months of the initial treatment with chelating agents.8 Organ meats, nuts, shellfish, and chocolate are rich in copper and should be avoided.
The patient’s sister is started on trientine 250 mg orally three times daily on an empty stomach at least 1 hour before meals. Treatment is monitored by following 24-hour urine copper measurement. A 24-hour urine copper measurement at 3 months after starting treatment has increased from 54 at baseline to 350 µg, which indicates that the copper is being removed from tissues. The plan is for early substitution of zinc for long-term maintenance once de-coppering is completed.
KEY POINTS
- Acute liver failure is severe acute liver injury characterized by coagulopathy (INR ≥ 1.5) and encephalopathy in a patient with no preexisting liver disease and with duration of symptoms less than 26 weeks.
- Acute liver failure secondary to Wilson disease is uncommon but should be excluded, particularly in young patients.
- The diagnosis of Wilson disease in the setting of acute liver failure is challenging because the serum ceruloplasmin level may be normal in acute liver failure secondary to Wilson disease, and free serum copper and 24-hour urine copper are usually elevated in all acute liver failure patients regardless of the etiology.
- A ratio of alkaline phosphatase to total bilirubin of less than 4 plus an AST-ALT ratio greater than 2.2 in a patient with acute liver failure should be regarded as Wilson disease until proven otherwise (Figure 2).
- Acute liver failure secondary to Wilson disease is usually fatal, and emergency liver transplant is a life-saving procedure.
- Screening of first-degree relatives of Wilson disease patients should include a history and physical examination, liver enzyme tests, complete blood cell count, serum ceruloplasmin level, serum free copper level, slit-lamp examination of the eyes, and 24-hour urinary copper measurement. Genetic tests are supplementary for screening but are not routinely available.
- Lee WM, Larson AM, Stravitz T. AASLD Position Paper: The management of acute liver failure: update 2011. www.aasld.org/sites/default/files/guideline_documents/alfenhanced.pdf. Accessed December 9, 2015.
- Bernal W, Auzinger G, Dhawan A, Wendon J. Acute liver failure. Lancet 2010; 376:190–201.
- Larson AM, Polson J, Fontana RJ, et al; Acute Liver Failure Study Group. Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology 2005; 42:1364–1372.
- Hanouneh IA, Khoriaty R, Zein NN. A 35-year-old Asian man with jaundice and markedly high aminotransferase levels. Cleve Clin J Med 2009; 76:449–456.
- Norvell JP, Blei AT, Jovanovic BD, Levitsky J. Herpes simplex virus hepatitis: an analysis of the published literature and institutional cases. Liver Transpl 2007; 13:1428–1434.
- Korman JD, Volenberg I, Balko J, et al; Pediatric and Adult Acute Liver Failure Study Groups. Screening for Wilson disease in acute liver failure: a comparison of currently available diagnostic tests. Hepatology 2008; 48:1167–1174.
- Morgan SM, Zantek ND. Therapeutic plasma exchange for fulminant hepatic failure secondary to Wilson's disease. J Clin Apher 2012; 27:282–286.
- Roberts EA, Schilsky ML; American Association for Study of Liver Diseases (AASLD). Diagnosis and treatment of Wilson disease: an update. Hepatology 2008; 47:2089–2111.
- Shah AB, Chernov I, Zhang HT, et al. Identification and analysis of mutations in the Wilson disease gene (ATP7B): population frequencies, genotype-phenotype correlation, and functional analyses. Am J Hum Genet 1997; 61:317–328.
- Maier-Dobersberger T, Ferenci P, Polli C, et al. Detection of the His1069Gln mutation in Wilson disease by rapid polymerase chain reaction. Ann Intern Med 1997; 127:21–26.
- Lee WM, Larson AM, Stravitz T. AASLD Position Paper: The management of acute liver failure: update 2011. www.aasld.org/sites/default/files/guideline_documents/alfenhanced.pdf. Accessed December 9, 2015.
- Bernal W, Auzinger G, Dhawan A, Wendon J. Acute liver failure. Lancet 2010; 376:190–201.
- Larson AM, Polson J, Fontana RJ, et al; Acute Liver Failure Study Group. Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology 2005; 42:1364–1372.
- Hanouneh IA, Khoriaty R, Zein NN. A 35-year-old Asian man with jaundice and markedly high aminotransferase levels. Cleve Clin J Med 2009; 76:449–456.
- Norvell JP, Blei AT, Jovanovic BD, Levitsky J. Herpes simplex virus hepatitis: an analysis of the published literature and institutional cases. Liver Transpl 2007; 13:1428–1434.
- Korman JD, Volenberg I, Balko J, et al; Pediatric and Adult Acute Liver Failure Study Groups. Screening for Wilson disease in acute liver failure: a comparison of currently available diagnostic tests. Hepatology 2008; 48:1167–1174.
- Morgan SM, Zantek ND. Therapeutic plasma exchange for fulminant hepatic failure secondary to Wilson's disease. J Clin Apher 2012; 27:282–286.
- Roberts EA, Schilsky ML; American Association for Study of Liver Diseases (AASLD). Diagnosis and treatment of Wilson disease: an update. Hepatology 2008; 47:2089–2111.
- Shah AB, Chernov I, Zhang HT, et al. Identification and analysis of mutations in the Wilson disease gene (ATP7B): population frequencies, genotype-phenotype correlation, and functional analyses. Am J Hum Genet 1997; 61:317–328.
- Maier-Dobersberger T, Ferenci P, Polli C, et al. Detection of the His1069Gln mutation in Wilson disease by rapid polymerase chain reaction. Ann Intern Med 1997; 127:21–26.

Common infectious complications of liver transplant
The immunosuppressed state of liver transplant recipients makes them vulnerable to infections after surgery.1 These infections are directly correlated with the net state of immunosuppression. Higher levels of immunosuppression mean a higher risk of infection, with rates of infection typically highest in the early posttransplant period.
Common infections during this period include operative and perioperative nosocomial bacterial and fungal infections, reactivation of latent infections, and invasive fungal infections such as candidiasis, aspergillosis, and pneumocystosis. Donor-derived infections also must be considered. As time passes and the level of immunosuppression is reduced, liver recipients are less prone to infection.1
The risk of infection can be minimized by appropriate antimicrobial prophylaxis, strategies for safe living after transplant,2 vaccination,3 careful balancing of immunosuppressive therapy,4 and thoughtful donor selection.5 Drug-drug interactions are common and must be carefully considered to minimize the risk.
This review highlights common infectious complications encountered after liver transplant.
INTRA-ABDOMINAL INFECTIONS
Intra-abdominal infections are common in the early postoperative period.6,7
Risk factors include:
- Pretransplant ascites
- Posttransplant dialysis
- Wound infection
- Reoperation8
- Hepatic artery thrombosis
- Roux-en-Y choledochojejunostomy anastomosis.9
Signs that may indicate intra-abdominal infection include fever, abdominal pain, leukocytosis, and elevated liver enzymes. But because of their immunosuppressed state, transplant recipients may not manifest fever as readily as the general population. They should be evaluated for cholangitis, peritonitis, biloma, and intra-abdominal abscess.
Organisms. Intra-abdominal infections are often polymicrobial. Enterococci, Staphylococcus aureus, gram-negative species including Pseudomonas, Klebsiella, and Acinetobacter, and Candida species are the most common pathogens. Strains are often resistant to multiple drugs, especially in patients who received antibiotics in the weeks before transplant.8,10
Liver transplant recipients are also particularly susceptible to Clostridium difficile-associated colitis as a result of immunosuppression and frequent use of antibiotics perioperatively and postoperatively.11 The spectrum of C difficile infection ranges from mild diarrhea to life-threatening colitis, and the course in liver transplant patients tends to be more complicated than in immunocompetent patients.12
Diagnosis. Intra-abdominal infections should be looked for and treated promptly, as they are associated with a higher mortality rate, a greater risk of graft loss, and a higher incidence of retransplant.6,10 Abdominal ultrasonography or computed tomography (CT) can confirm the presence of fluid collections.
Treatment. Infected collections can be treated with percutaneous or surgical drainage and antimicrobial therapy. In the case of biliary tract complications, retransplant or surgical correction of biliary leakage or stenosis decreases the risk of death.6
Suspicion should be high for C difficile-associated colitis in cases of posttransplant diarrhea. C difficile toxin stool assays help confirm the diagnosis.12 Oral metronidazole is recommended in mild to moderate C difficile infection, with oral vancomycin and intravenous metronidazole reserved for severe cases. Colectomy may be necessary in patients with toxic megacolon.
CYTOMEGALOVIRUS INFECTION
Cytomegalovirus is an important opportunistic pathogen in liver transplant recipients.13 It causes a range of manifestations, from infection (viremia with or without symptoms) to cytomegalovirus syndrome (fever, malaise, and cell-line cytopenias) to tissue-invasive disease with end-organ disease.14 Without preventive measures and treatment, cytomegalovirus disease can increase the risk of morbidity, allograft loss and death.15,16

Risk factors for cytomegalovirus infection (Table 1) include:
- Discordant serostatus of the donor and recipient (the risk is highest in seronegative recipients of organs from seropositive donors)
- Higher levels of immunosuppression, especially when antilymphocyte antibodies are used
- Treatment of graft rejection
- Coinfection with other human herpesviruses, such as Epstein-Barr virus.4,17
Preventing cytomegalovirus infection

The strategy to prevent cytomegalovirus infection depends on the serologic status of the donor and recipient and may include antiviral prophylaxis or preemptive treatment (Table 2).18
Prophylaxis involves giving antiviral drugs during the early high-risk period, with the goal of preventing the development of cytomegalovirus viremia. The alternative preemptive strategy emphasizes serial testing for cytomegalovirus viremia, with the goal of intervening with antiviral medications while viremia is at a low level, thus avoiding potential progression to cytomegalovirus disease. Both strategies have pros and cons that should be considered by each transplant center when setting institutional policy.
A prophylactic approach seems very effective at preventing both infection and disease from cytomegalovirus and has been shown to reduce graft rejection and the risk of death.18 It is preferred in cytomegalovirus-negative recipients when the donor was cytomegalovirus-positive—a high-risk situation.19 However, these patients are also at higher risk of late-onset cytomegalovirus disease. Higher cost and potential drug toxicity, mainly neutropenia from ganciclovir-based regimens, are additional considerations.
Preemptive treatment, in contrast, reserves drug treatment for patients who are actually infected with cytomegalovirus, thus resulting in fewer adverse drug events and lower cost; but it requires regular monitoring. Preemptive methods, by definition, cannot prevent infection, and with this strategy tissue-invasive disease not associated with viremia does occasionally occur.20 As such, patients with a clinical presentation that suggests cytomegalovirus but have negative results on blood testing should be considered for tissue biopsy with culture and immunohistochemical stain.
The most commonly used regimens for antiviral prophylaxis and treatment in liver transplant recipients are intravenous ganciclovir and oral valganciclovir.21 Although valganciclovir is the most commonly used agent in this setting because of ease of administration, it has not been approved by the US Food and Drug Administration in liver transplant patients, as it was associated with higher rates of cytomegalovirus tissue-invasive disease.22–24 Additionally, drug-resistant cytomegalovirus strains have been associated with valganciclovir prophylaxis in cytomegalovirus-negative recipients of solid organs from cytomegalovirus-positive donors.25
Prophylaxis typically consists of therapy for 3 months from the time of transplant. In higher-risk patients (donor-positive, recipient-negative), longer courses of prophylaxis have been extrapolated from data in kidney transplant recipients.26 Extension or reinstitution of prophylaxis should also be considered in liver transplant patients receiving treatment for rejection with antilymphocyte therapy.
Routine screening for cytomegalovirus is not recommended while patients are receiving prophylaxis. High-risk patients who are not receiving prophylaxis should be monitored with nucleic acid or pp65 antigenemia testing as part of the preemptive strategy protocol.
Treatment of cytomegalovirus disease
Although no specific threshold has been established, treatment is generally indicated if a patient has a consistent clinical syndrome, evidence of tissue injury, and persistent or increasing viremia.
Treatment involves giving antiviral drugs and also reducing the level of immunosuppression, if possible, until symptoms and viremia have resolved.
The choice of antiviral therapy depends on the severity of disease. Intravenous ganciclovir (5 mg/kg twice daily adjusted for renal impairment) or oral valganciclovir (900 mg twice daily, also renally dose-adjusted when necessary) can be used for mild to moderate disease if no significant gastrointestinal involvement is reported. Intravenous ganciclovir is preferred for patients with more severe disease or gastrointestinal involvement. The minimum duration of treatment is 2 weeks and may need to be prolonged until both symptoms and viremia completely resolve.18
Drug resistance can occur and should be considered in patients who have a history of prolonged ganciclovir or valganciclovir exposure who do not clinically improve or have persistent or rising viremia. In such cases, genotype assays are helpful, and initiation of alternative therapy should be considered. Mutations conferring resistance to ganciclovir are often associated with cross-resistance to cidofovir. Cidofovir can therefore be considered only when genotype assays demonstrate specific mutations conferring an isolated resistance to ganciclovir.27 The addition of foscarnet to the ganciclovir regimen or substitution of foscarnet for ganciclovir are accepted approaches.
Although cytomegalovirus hyperimmunoglobulin has been used in prophylaxis and invasive disease treatment, its role in the management of ganciclovir-resistant cytomegalovirus infections remains controversial.28
EPSTEIN-BARR VIRUS POSTTRANSPLANT LYMPHOPROLIFERATIVE DISEASE
Epstein-Barr virus-associated posttransplant lymphoproliferative disease is a spectrum of disorders ranging from an infectious mononucleosis syndrome to aggressive malignancy with the potential for death and significant morbidity after liver transplant.29 The timeline of risk varies, but the disease is most common in the first year after transplant.
Risk factors for this disease (Table 1) are:
- Primary Epstein-Barr virus infection
- Cytomegalovirus donor-recipient mismatch
- Cytomegalovirus disease
- Higher levels of immunosuppression, especially with antilymphocyte antibodies.30
The likelihood of Epstein-Barr virus playing a contributing role is lower in later-onset posttransplant lymphoproliferative disease. Patients who are older at the time of transplant, who receive highly immunogenic allografts including a liver as a component of a multivisceral transplant, and who receive increased immunosuppression to treat rejection are at even greater risk of late posttransplant lymphoproliferative disease.31 This is in contrast to early posttransplant lymphoproliferative disease, which is seen more commonly in children as a result of primary Epstein-Barr virus infection.
Recognition and diagnosis. Heightened suspicion is required when considering posttransplant lymphoproliferative disease, and careful evaluation of consistent symptoms and allograft dysfunction are required.
Clinically, posttransplant lymphoproliferative disease should be suspected if a liver transplant recipient develops unexplained fever, weight loss, lymphadenopathy, or cell-line cytopenias.30,32 Other signs and symptoms may be related to the organ involved and may include evidence of hepatitis, pneumonitis, and gastrointestinal disease.31
Adjunctive diagnostic testing includes donor and recipient serology to characterize overall risk before transplantation and quantification of Epstein-Barr viral load, but confirmation relies on tissue histopathology.
Treatment focuses on reducing immunosuppression.30,32 Adding antiviral agents does not seem to improve outcome in all cases.33 Depending on clinical response and histologic classification, additional therapies such as anti-CD20 humanized chimeric monoclonal antibodies, surgery, radiation, and conventional chemotherapy may be required.34
Preventive approaches remain controversial. Chemoprophylaxis with an antiviral such as ganciclovir is occasionally used but has not been shown to consistently decrease rates of posttransplant lymphoproliferative disease. These agents may act in an indirect manner, leading to decreased rates of cytomegalovirus infection, a major cofactor for posttransplant lymphoproliferative disease.24
Passive immunoprophylaxis with immunoglobulin targeting cytomegalovirus has shown to decrease rates of non-Hodgkin lymphoma from posttransplant lymphoproliferative disease in renal transplant recipients in the first year after transplant,35 but data are lacking regarding its use in liver transplant recipients. Monitoring of the viral load and subsequent reduction of immunosuppression remain the most efficient measures to date.36
FUNGAL INFECTIONS
Candida species account for more than half of fungal infections in liver transplant recipients.37 However, a change has been noted in the past 20 years, with a decrease in Candida infections accompanied by an increase in Aspergillus infections.38 Endemic mycoses such as coccidioidomycosis, blastomycosis, and histoplasmosis should be considered with the appropriate epidemiologic history or if disease develops early after transplant and the donor came from a highly endemic region.39Cryptococcus may also be encountered.
Diagnosis. One of the most challenging aspects of fungal infection in liver transplant recipients is timely diagnosis. Heightened suspicion and early biopsy for pathological and microbiological confirmation are necessary. Although available noninvasive diagnostic tools often lack specificity, early detection of fungal markers may be of great use in guiding further diagnostic workup or empiric treatment in the critically ill.
Noninvasive tests include galactomannan, cryptococcal antigen, histoplasma antigen, (1-3)-beta-D-glucan assay and various antibody tests. Galactomannan testing has been widely used to aid in the diagnosis of invasive aspergillosis. Similarly, the (1-3)-beta-D-glucan assay is a non–culture-based tool for diagnosing and monitoring the treatment of invasive fungal infections. However, a definite diagnosis cannot be made on the basis of a positive test alone.40 The complementary diagnostic characteristics of combining noninvasive assays have yet to be fully elucidated.41 Cultures and tissue histopathology are also used when possible.
Treatment is based on targeted specific antifungal drug therapy and reduction of immunosuppressive therapy, when possible. The choice of antifungal agent varies with the pathogen, the site of involvement, and the severity of the disease. A focus on potential drug interactions, their management, and therapeutic drug monitoring when using antifungal medications is essential in the posttransplant period. Combination therapy can be considered in some situations to enhance synergy. The following sections discuss in greater detail Candida species, Aspergillus species, and Pneumocystis jirovecii infections.
Candida infections

Candidiasis after liver transplant is typically nosocomial, especially when diagnosed during the first 3 months (Table 3).37
Risk factors for invasive candidiasis include perioperative colonization, prolonged operative time, retransplant, greater transfusion requirements, and postoperative renal failure.37,42,43 Invasive candidiasis is of concern for its effects on morbidity, mortality, and cost of care.43–46
Organisms. The frequency of implicated species, in particular those with a natural resistance to fluconazole, differs in various reports.37,45,46Candida albicans remains the most commonly isolated pathogen; however, non-albicans species including those resistant to fluconazole have been reported more frequently and include Candida glabrata and Candida krusei.47,48
Signs and diagnosis. Invasive candidiasis in liver transplant recipients generally manifests itself in catheter-related blood stream infections, urinary tract infections, or intra-abdominal infections. Diagnosis can be made by isolating Candida from blood cultures, recovering organisms in culture of a normally sterile site, or finding direct microscopic evidence of the fungus on tissue specimens.49
Disseminated candidiasis refers to the involvement of distant anatomic sites. Clinical manifestations may cause vision changes, abdominal pain or skin nodules with findings of candidemia, hepatosplenic abscesses, or retinal exudates on funduscopy.49
Treatment of invasive candidiasis in liver recipients often involves antifungal therapy and reduction of immunosuppression. Broad-spectrum antifungals are initially advocated in an empirical approach to cover fluconazole-resistant strains of the non-albicans subgroups.50 Depending on antifungal susceptibility, treatment can later be adjusted.
Fluconazole remains the agent of choice in most C albicans infections.47 However, attention should be paid to the possibility of resistance in patients who have received fluconazole prophylaxis within the past 30 days. Additional agents used in treatment may include echinocandins, amphotericin, and additional azoles.
Antifungal prophylaxis is recommended in high-risk liver transplant patients, although its optimal duration remains undetermined.44 Antifungal prophylaxis has been associated with decreased incidence of both superficial and invasive candidiasis.51
Aspergillus infection
Aspergillus, the second most common fungal pathogen, has become a more common concern in liver transplant recipients. Aspergillus fumigatus is the most frequently encountered species.38,52
Risk factors. These infections typically occur in the first year, during intense immunosuppression. Retransplant, renal failure, and fulminant hepatic failure are major risk factors.52 In the presence of risk factors and a suggestive clinical setting, invasive aspergillosis should be considered and the diagnosis pursued.
Diagnosis is suggested by positive findings on CT accompanied by lower respiratory tract symptoms, focal lesions on neuroimaging, or demonstration of the fungus on cultures.49 However, Aspergillus is rarely grown in blood culture. The galactomannan antigen is a noninvasive test that can provide supporting evidence for the diagnosis.41,52 False-positive results do occur in the setting of certain antibiotics and cross-reacting fungi.53
Treatment consists of antifungal therapy and immunosuppression reduction.52
Voriconazole is the first-line agent for invasive aspergillosis. Monitoring for potential drug-drug interactions and side effects is required.54,55 Amphotericin B is considered a second-line choice due to toxicity and lack of an oral formulation. In refractory cases, combined antifungal therapy could be considered.52 The duration of treatment is generally a minimum of 12 weeks.
Prophylaxis. Specific prophylaxis against invasive aspergillosis is not currently recommended; however, some authors suggest a prophylactic approach using echinocandins or liposomal amphotericin B in high-risk patients.51,52 Aspergillosis is associated with a considerable increase in mortality in liver transplant recipients, which highlights the importance of timely management.52,56
Pneumocystis jirovecii
P jirovecii remains a common opportunistic pathogen in people with impaired immunity, including transplant and human immunodeficiency virus patients.
Prophylaxis. Widespread adoption of antimicrobial prophylaxis by transplant centers has decreased the rates of P jirovecii infection in liver transplant recipients.57,58 Commonly used prophylactic regimens after liver transplantation include a single-strength trimethoprim-sulfamethoxazole tablet daily or a double-strength tablet three times per week for a minimum of 6 to 12 months after transplant. Atovaquone and dapsone can be used as alternatives in cases of intolerance to trimethoprim-sulfamethoxazole (Table 2).
Inhaled pentamidine is clearly inferior and should be used only when the other medications are contraindicated.59
Signs and diagnosis. P jirovecii pneumonia is characterized by fever, cough, dyspnea, and chest pain. Insidious hypoxemia, abnormal chest examination, and bilateral interstitial pneumonia on chest radiography are common.
CT may be more sensitive than chest radiography.57 Findings suggestive of P jirovecii pneumonia on chest CT are extensive bilateral and symmetrical ground-glass attenuations. Other less-characteristic findings include upper lobar parenchymal opacities and spontaneous pneumothorax.57,60
The serum (1,3)-beta-D-glucan assay derived from major cell-wall components of P jirovecii might be helpful. Studies report a sensitivity for P jirovecii pneumonia as high as 96% and a negative predictive value of 99.8%.61,62
Definitive diagnosis requires identification of the pathogen. Routine expectorated sputum sampling is generally associated with a poor diagnostic yield. Bronchoscopy and bronchoalveolar lavage with silver or fluorescent antibody staining of samples, polymerase chain reaction testing, or both significantly improves diagnosis. Transbronchial or open lung biopsy are often unnecessary.57
Treatment. Trimethoprim-sulfamethoxazole is the first-line agent for treating P jirovecii pneumonia.57 The minimum duration of treatment is 14 days, with extended courses for severe infection.
Intravenous pentamidine or clindamycin plus primaquine are alternatives for patients who cannot tolerate trimethoprim-sulfamethoxazole. The major concern with intravenous pentamidine is renal dysfunction. Hypoglycemia or hyperglycemia, neutropenia, thrombocytopenia, nausea, dysgeusia, and pancreatitis may also occur.63
Atovaquone might also be beneficial in mild to moderate P jirovecii pneumonia. The main side effects include skin rashes, gastrointestinal intolerance, and elevation of transaminases.64
A corticosteroid (40–60 mg of prednisone or its equivalent) may be beneficial in conjunction with antimicrobial therapy in patients with significant hypoxia (partial pressure of arterial oxygen < 70 mm Hg on room air) in decreasing the risk of respiratory failure and need for intubation.
With appropriate and timely antimicrobial prophylaxis, cases of P jirovecii pneumonia should continue to decrease.
TUBERCULOSIS
Development of tuberculosis after transplantation is a catastrophic complication, with mortality rates of up to 30%.65 Most cases of posttransplant tuberculosis represent reactivation of latent disease.66 Screening with tuberculin skin tests or interferon-gamma-release assays is recommended in all liver transplant candidates. Chest radiography before transplant is necessary when assessing a positive screening test.67
The optimal management of latent tuberculosis in these cases remains controversial. Patients at high risk or those with positive screening results on chest radiography warrant treatment for latent tuberculosis infection with isoniazid unless contraindicated.67,68
The ideal time to initiate prophylactic isoniazid therapy is unclear. Some authors suggest delaying it, as it might be associated with poor tolerance and hepatotoxicity.69 Others have found that early isoniazid use was not associated with negative outcomes.70
Risk factors for symptomatic tuberculosis after liver transplant include previous infection with tuberculosis, intensified immunosuppression (especially anti-T-lymphocyte therapies), diabetes mellitus, and other co-infections (Table 1).71
The increased incidence of atypical presentations in recent years makes the diagnosis of active tuberculosis among liver transplant recipients challenging. Sputum smears can be negative due to low mycobacterial burdens, and tuberculin skin testing and interferon-gamma-release assays may be falsely negative due to immunosuppression.67
Treatment of active tuberculosis consists initially of a four-drug regimen using isoniazid, rifampin, pyrazinamide, and ethambutol for 2 months. Adjustments are made in accordance with culture and sensitivity results. Treatment can then be tapered to two drugs (isoniazid and rifampin) for a minimum of 4 additional months. Prolonged treatment may be required in instances of extrapulmonary or disseminated disease.65,72
Tuberculosis treatment can be complicated by hepatotoxicity in liver transplant recipients because of direct drug effects and drug-drug interactions with immunosuppressive agents. Close monitoring for rejection and hepatotoxicity is therefore imperative while liver transplant recipients are receiving antituberculosis therapy. Drug-drug interactions may also be responsible for marked reductions in immunosuppression levels, especially with regimens containing rifampin.71 Substitution of rifabutin for rifampin reduces the effect of drug interactions.66
VIRAL HEPATITIS
Hepatitis B virus
Hepatitis B virus-related end-stage liver disease and hepatocellular carcinoma are common indications for liver transplant in Asia. It is less common in the United States and Europe, accounting for less than 10% of all liver transplant cases. Prognosis is favorable in recipients undergoing liver transplant for hepatitis B virus, with excellent survival rates. Prevention of reinfection is crucial in these patients.
Treatment with combination antiviral agents and hepatitis B immunoglobulin (HBIG) is effective.73 Lamivudine was the first nucleoside analogue found to be effective against hepatitis B virus. Its low cost and relative safety are strong arguments in favor of its continued use in liver transplant recipients.74 In patients without evidence of hepatitis B viral replication at the time of transplant, monotherapy with lamivudine has led to low recurrence rates, and adefovir can be added to control resistant viral strains.75
The frequent emergence of resistance with lamivudine favors newer agents such as entecavir or tenofovir. These nucleoside and nucleotide analogues have a higher barrier to resistance, and thus resistance to them is rare. They are also more efficient, potentially allowing use of an HBIG-sparing protocol.76 However, they are associated with a higher risk of nephrotoxicity and require dose adjustments in renal insufficiency. Data directly comparing entecavir and tenofovir are scarce.
Prophylaxis. Most studies support an individualized approach for prevention of hepatitis B virus reinfection. High-risk patients, ie, those positive for HBe antigen or with high viral loads (> 100,000 copies/mL) are generally treated with both HBIG and antiviral agents.77 Low-risk patients are those with a negative HBe antigen, low hepatitis B virus DNA levels, hepatitis B virus-related acute liver failure, and cirrhosis resulting from coinfection with both hepatitis B and hepatitis D virus.75 In low-risk patients, discontinuation of HBIG after 1 to 2 years of treatment is appropriate, and long-term prophylaxis with antiviral agents alone is an option. However, levels of hepatitis B DNA should be monitored closely.78,79
Hepatitis C virus
Recurrence of hepatitis C virus infection is the rule among patients who are viremic at the time of liver transplant.80,81 Most of these patients will show histologic evidence of recurrent hepatitis within the first year after liver transplant. It is often difficult to distinguish between the histopathological appearance of a recurrent hepatitis C virus infection and acute cellular rejection.
Progression to fibrosis and subsequently cirrhosis and decompensation is highly variable in hepatitis C virus-infected liver transplant recipients. Diabetes, insulin resistance, and possibly hepatitis steatosis have been associated with a rapid progression to advanced fibrosis. The contribution of immunosuppression to the progression of hepatitis C virus remains an area of active study. Some studies point to antilymphocyte immunosuppressive agents as a potential cause.82 Liver biopsy is a useful tool in this situation. It allows monitoring of disease severity and progression and may distinguish recurrent hepatitis C virus disease from other causes of liver enzyme elevation.
The major concern with the recurrence of hepatitis C virus infection after liver transplant is allograft loss. Rates of patient and graft survival are reduced in infected patients compared with hepatitis C virus-negative patients.83,84 Prophylactic antiviral therapy has no current role in the management of hepatitis C virus disease. Those manifesting moderate to severe necroinflammation or mild to moderate fibrosis indicative of progressive disease should be treated.81,85
Sustained viral clearance with antiviral agents confers a graft survival benefit.
The combination of peg-interferon and weight-based ribavirin has been the standard of treatment but may be associated with increased rates of rejection.86,87 The sustained virologic response rates for hepatitis C virus range from 60% in genotypes 4, 5, and 6 after 48 weeks of treatment to 60% to 80% in genotypes 2 and 3 after 24 weeks, but only about 30% in genotype 1.88
Treatment with the newer agents, especially protease inhibitors, in genotype 1 (peg-interferon, ribavirin, and either telaprevir or boceprevir) has been evaluated. Success rates reaching 70% have been achieved.89 Adverse effects can be a major setback. Serious complications include severe anemia, renal dysfunction, increased risk of infection, and death.
Triple therapy should be carefully considered in liver transplant patients with genotype 1 hepatitis C virus.90 Significant drug-drug interactions are reported between hepatitis C virus protease inhibitors and immunosuppression regimens. Additional new oral direct- acting antivirals have been investigated. They bring promising advances in hepatitis C virus treatment and pave the way for interferon-free regimens with pangenotypic activity.
IMMUNIZATION
Immunization can decrease the risk of infectious complications in liver transplant recipients, as well as in close contacts and healthcare professionals.3
Influenza. Pretransplant influenza vaccine and posttransplant annual influenza vaccines are necessary.
Pneumococcal immunization should additionally be provided prior to transplant and repeated every 3 to 5 years thereafter.3,91
A number of other vaccinations should also be completed before transplant, including the hepatitis A and B vaccines and the tetanus/diphtheria/acellular pertussis vaccines. However, these vaccinations have not been shown to be detrimental to patients after transplant.91
Varicella and zoster vaccines should be given before liver transplant—zoster in patients over age 60, and varicella in patients with no immunity. Live vaccines, including varicella and zoster vaccines, are contraindicated after liver transplant.3
Human papillomavirus. The bivalent human papillomavirus vaccine can be given before transplant in females ages 9 to 26; the quadrivalent vaccine is beneficial in those ages 9 to 26 and in women under age 45.3,91
IMMUNOSUPPRESSION CARRIES RISK OF INFECTION
Most liver transplant patients require prolonged immunosuppressive therapy. This comes with an increased risk of new or recurrent infections, potentially causing death and significant morbidity.
Evaluation of existing risk factors, appropriate prophylaxis and immunization, timely diagnosis, and treatment of such infections are therefore essential steps for the successful management of liver transplant recipients.
- Fishman JA. Infection in solid-organ transplant recipients. N Engl J Med 2007; 357:2601–2614.
- Avery RK, Michaels MG; AST Infectious Diseases Community of Practice. Strategies for safe living after solid organ transplantation. Am J Transplant 2013; 13(suppl 4):304–310.
- Danziger-Isakov L, Kumar D; AST Infectious Diseases Community of Practice. Vaccination in solid organ transplantation. Am J Transplant 2013; 13(suppl 4):311–317.
- San Juan R, Aguado JM, Lumbreras C, et al; RESITRA Network, Spain. Incidence, clinical characteristics and risk factors of late infection in solid organ transplant recipients: data from the RESITRA study group. Am J Transplant 2007; 7:964–971.
- Ison MG, Grossi P; AST Infectious Diseases Community of Practice. Donor-derived infections in solid organ transplantation. Am J Transplant 2013; 13(suppl 4):22–30.
- Kim YJ, Kim SI, Wie SH, et al. Infectious complications in living-donor liver transplant recipients: a 9-year single-center experience. Transpl Infect Dis 2008; 10:316–324.
- Arnow PM. Infections following orthotopic liver transplantation. HPB Surg 1991; 3:221–233.
- Reid GE, Grim SA, Sankary H, Benedetti E, Oberholzer J, Clark NM. Early intra-abdominal infections associated with orthotopic liver transplantation. Transplantation 2009; 87:1706–1711.
- Said A, Safdar N, Lucey MR, et al. Infected bilomas in liver transplant recipients, incidence, risk factors and implications for prevention. Am J Transplant 2004; 4:574–582.
- Safdar N, Said A, Lucey MR, et al. Infected bilomas in liver transplant recipients: clinical features, optimal management, and risk factors for mortality. Clin Infect Dis 2004; 39:517–525.
- Niemczyk M, Leszczyniski P, Wyzgał J, Paczek L, Krawczyk M, Luczak M. Infections caused by Clostridium difficile in kidney or liver graft recipients. Ann Transplant 2005; 10:70–74.
- Albright JB, Bonatti H, Mendez J, et al. Early and late onset Clostridium difficile-associated colitis following liver transplantation. Transpl Int 2007; 20:856–866.
- Lee SO, Razonable RR. Current concepts on cytomegalovirus infection after liver transplantation. World J Hepatol 2010; 2:325–336.
- Ljungman P, Griffiths P, Paya C. Definitions of cytomegalovirus infection and disease in transplant recipients. Clin Infect Dis 2002; 34:1094–1097.
- Beam E, Razonable RR. Cytomegalovirus in solid organ transplantation: epidemiology, prevention, and treatment. Curr Infect Dis Rep 2012; 14:633–641.
- Bodro M, Sabé N, Lladó L, et al. Prophylaxis versus preemptive therapy for cytomegalovirus disease in high-risk liver transplant recipients. Liver Transpl 2012; 18:1093–1099.
- Weigand K, Schnitzler P, Schmidt J, et al. Cytomegalovirus infection after liver transplantation incidence, risks, and benefits of prophylaxis. Transplant Proc 2010; 42:2634–2641.
- Razonable RR, Humar A; AST Infectious Diseases Community of Practice. Cytomegalovirus in solid organ transplantation. Am J Transplant 2013; 13(suppl 4):93–106.
- Meije Y, Fortún J, Len Ó, et al; Spanish Network for Research on Infection in Transplantation (RESITRA) and the Spanish Network for Research on Infectious Diseases (REIPI). Prevention strategies for cytomegalovirus disease and long-term outcomes in the high-risk transplant patient (D+/R-): experience from the RESITRA-REIPI cohort. Transpl Infect Dis 2014; 16:387–396.
- Durand CM, Marr KA, Arnold CA, et al. Detection of cytomegalovirus DNA in plasma as an adjunct diagnostic for gastrointestinal tract disease in kidney and liver transplant recipients. Clin Infect Dis 2013; 57:1550–1559.
- Levitsky J, Singh N, Wagener MM, Stosor V, Abecassis M, Ison MG. A survey of CMV prevention strategies after liver transplantation. Am J Transplant 2008; 8:158–161.
- Marcelin JR, Beam E, Razonable RR. Cytomegalovirus infection in liver transplant recipients: updates on clinical management. World J Gastroenterol 2014; 20:10658–10667.
- Kalil AC, Freifeld AG, Lyden ER, Stoner JA. Valganciclovir for cytomegalovirus prevention in solid organ transplant patients: an evidence-based reassessment of safety and efficacy. PLoS One 2009; 4:e5512.
- Kalil AC, Mindru C, Botha JF, et al. Risk of cytomegalovirus disease in high-risk liver transplant recipients on valganciclovir prophylaxis: a systematic review and meta-analysis. Liver Transpl 2012; 18:1440–1447.
- Eid AJ, Arthurs SK, Deziel PJ, Wilhelm MP, Razonable RR. Emergence of drug-resistant cytomegalovirus in the era of valganciclovir prophylaxis: therapeutic implications and outcomes. Clin Transplant 2008; 22:162–170.
- Kumar D, Humar A. Cytomegalovirus prophylaxis: how long is enough? Nat Rev Nephrol 2010; 6:13–14.
- Lurain NS, Chou S. Antiviral drug resistance of human cytomegalovirus. Clin Microbiol Rev 2010; 23:689–712.
- Torres-Madriz G, Boucher HW. Immunocompromised hosts: perspectives in the treatment and prophylaxis of cytomegalovirus disease in solid-organ transplant recipients. Clin Infect Dis 2008; 47:702–711.
- Burra P, Buda A, Livi U, et al. Occurrence of post-transplant lymphoproliferative disorders among over thousand adult recipients: any role for hepatitis C infection? Eur J Gastroenterol Hepatol 2006; 18:1065–1070.
- Jain A, Nalesnik M, Reyes J, et al. Posttransplant lymphoproliferative disorders in liver transplantation: a 20-year experience. Ann Surg 2002; 236:429–437.
- Allen UD, Preiksaitis JK; AST Infectious Diseases Community of Practice. Epstein-Barr virus and posttransplant lymphoproliferative disorder in solid organ transplantation. Am J Transplant 2013; 13(suppl 4):107–120.
- Allen U, Preiksaitis J; AST Infectious Diseases Community of Practice. Epstein-Barr virus and posttransplant lymphoproliferative disorder in solid organ transplant recipients. Am J Transplant 2009; 9(suppl 4):S87–S96.
- Perrine SP, Hermine O, Small T, et al. A phase 1/2 trial of arginine butyrate and ganciclovir in patients with Epstein-Barr virus-associated lymphoid malignancies. Blood 2007; 109:2571–2578.
- Jagadeesh D, Woda BA, Draper J, Evens AM. Post transplant lymphoproliferative disorders: risk, classification, and therapeutic recommendations. Curr Treat Options Oncol 2012; 13:122–136.
- Opelz G, Daniel V, Naujokat C, Fickenscher H, Döhler B. Effect of cytomegalovirus prophylaxis with immunoglobulin or with antiviral drugs on post-transplant non-Hodgkin lymphoma: a multicentre retrospective analysis. Lancet Oncol 2007; 8:212–218.
- Nowalk AJ, Green M. Epstein-Barr virus–associated posttransplant lymphoproliferative disorder: strategies for prevention and cure. Liver Transpl 2010; 16(suppl S2):S54–S59.
- Pappas PG, Silveira FP; AST Infectious Diseases Community of Practice. Candida in solid organ transplant recipients. Am J Transplant 2009; 9(suppl 4):S173–S179.
- Singh N, Wagener MM, Marino IR, Gayowski T. Trends in invasive fungal infections in liver transplant recipients: correlation with evolution in transplantation practices. Transplantation 2002; 73:63–67.
- Miller R, Assi M; AST Infectious Diseases Community of Practice. Endemic fungal infections in solid organ transplantation. Am J Transplant 2013; 13(suppl 4):250–261.
- Fontana C, Gaziano R, Favaro M, Casalinuovo IA, Pistoia E, Di Francesco P. (1-3)-beta-D-glucan vs galactomannan antigen in diagnosing invasive fungal infections (IFIs). Open Microbiol J 2012; 6:70–73.
- Aydogan S, Kustimur S, Kalkancı A. Comparison of glucan and galactomannan tests with real-time PCR for diagnosis of invasive aspergillosis in a neutropenic rat model [Turkish]. Mikrobiyol Bul 2010; 44:441–452.
- Hadley S, Huckabee C, Pappas PG, et al. Outcomes of antifungal prophylaxis in high-risk liver transplant recipients. Transpl Infect Dis 2009; 11:40–48.
- Pappas PG, Kauffman CA, Andes D, et al; Infectious Diseases Society of America. Clinical practice guidelines for the management of candidiasis: 2009 update by the Infectious Diseases Society of America. Clin Infect Dis 2009; 48:503–535.
- Person AK, Kontoyiannis DP, Alexander BD. Fungal infections in transplant and oncology patients. Infect Dis Clin North Am 2010; 24:439–459.
- Van Hal SJ, Marriott DJE, Chen SCA, et al; Australian Candidaemia Study. Candidemia following solid organ transplantation in the era of antifungal prophylaxis: the Australian experience. Transpl Infect Dis 2009; 11:122–127.
- Singh N. Fungal infections in the recipients of solid organ transplantation. Infect Dis Clin North Am 2003; 17:113–134,
- Liu X, Ling Z, Li L, Ruan B. Invasive fungal infections in liver transplantation. Int J Infect Dis 2011; 15:e298–e304.
- Raghuram A, Restrepo A, Safadjou S, et al. Invasive fungal infections following liver transplantation: incidence, risk factors, survival, and impact of fluconazole-resistant Candida parapsilosis (2003-2007). Liver Transpl 2012; 18:1100–1109.
- De Pauw B, Walsh TJ, Donnelly JP, et al; European Organization for Research and Treatment of Cancer/Invasive Fungal Infections Cooperative Group; National Institute of Allergy and Infectious Diseases Mycoses Study Group (EORTC/MSG) Consensus Group. Revised definitions of invasive fungal disease from the European Organization for Research and Treatment of Cancer/Invasive Fungal Infections Cooperative Group and the National Institute of Allergy and Infectious Diseases Mycoses Study Group (EORTC/MSG) Consensus Group. Clin Infect Dis 2008; 46:1813–1821.
- Moreno A, Cervera C, Gavaldá J, et al. Bloodstream infections among transplant recipients: results of a nationwide surveillance in Spain. Am J Transplant 2007; 7:2579–2586.
- Cruciani M, Mengoli C, Malena M, Bosco O, Serpelloni G, Grossi P. Antifungal prophylaxis in liver transplant patients: a systematic review and meta-analysis. Liver Transpl 2006; 12:850–858.
- Singh N, Husain S; AST Infectious Diseases Community of Practice. Invasive aspergillosis in solid organ transplant recipients. Am J Transplant 2009; 9(suppl 4):S180–S191.
- Fortún J, Martín-Dávila P, Alvarez ME, et al. False-positive results of Aspergillus galactomannan antigenemia in liver transplant recipients. Transplantation 2009; 87:256–260.
- Cherian T, Giakoustidis A, Yokoyama S, et al. Treatment of refractory cerebral aspergillosis in a liver transplant recipient with voriconazole: case report and review of the literature. Exp Clin Transplant 2012; 10:482–486.
- Luong ML, Hosseini-Moghaddam SM, Singer LG, et al. Risk factors for voriconazole hepatotoxicity at 12 weeks in lung transplant recipients. Am J Transplant 2012; 12:1929–1935.
- Neofytos D, Fishman JA, Horn D, et al. Epidemiology and outcome of invasive fungal infections in solid organ transplant recipients. Transpl Infect Dis 2010; 12:220–229.
- Martin SI, Fishman JA; AST Infectious Diseases Community of Practice. Pneumocystis pneumonia in solid organ transplant recipients. Am J Transplant 2009; 9(suppl 4):S227–S233.
- Levine SJ, Masur H, Gill VJ, et al. Effect of aerosolized pentamidine prophylaxis on the diagnosis of Pneumocystis carinii pneumonia by induced sputum examination in patients infected with the human immunodeficiency virus. Am Rev Respir Dis 1991; 144:760–764.
- Rodriguez M, Sifri CD, Fishman JA. Failure of low-dose atovaquone prophylaxis against Pneumocystis jiroveci infection in transplant recipients. Clin Infect Dis 2004; 38:e76–e78.
- Crans CA Jr, Boiselle PM. Imaging features of Pneumocystis carinii pneumonia. Crit Rev Diagn Imaging 1999; 40:251–284.
- Onishi A, Sugiyama D, Kogata Y, et al. Diagnostic accuracy of serum 1,3-beta-D-glucan for Pneumocystis jiroveci pneumonia, invasive candidiasis, and invasive aspergillosis: systematic review and meta-analysis. J Clin Microbiol 2012; 50:7–15.
- Held J, Koch MS, Reischl U, Danner T, Serr A. Serum (1→3)-ß-D-glucan measurement as an early indicator of Pneumocystis jirovecii pneumonia and evaluation of its prognostic value. Clin Microbiol Infect 2011; 17:595–602.
- Fishman JA. Prevention of infection caused by Pneumocystis carinii in transplant recipients. Clin Infect Dis 2001; 33:1397–1405.
- Colby C, McAfee S, Sackstein R, Finkelstein D, Fishman J, Spitzer T. A prospective randomized trial comparing the toxicity and safety of atovaquone with trimethoprim/sulfamethoxazole as Pneumocystis carinii pneumonia prophylaxis following autologous peripheral blood stem cell transplantation. Bone Marrow Transplant 1999; 24:897–902.
- Subramanian A, Dorman S; AST Infectious Diseases Community of Practice. Mycobacterium tuberculosis in solid organ transplant recipients. Am J Transplant 2009; 9(suppl 4):S57–S62.
- Subramanian AK, Morris MI; AST Infectious Diseases Community of Practice. Mycobacterium tuberculosis infections in solid organ transplantation. Am J Transplant 2013; 13(suppl 4):68–76.
- Horne DJ, Narita M, Spitters CL, Parimi S, Dodson S, Limaye AP. Challenging issues in tuberculosis in solid organ transplantation. Clin Infect Dis 2013; 57:1473–1482.
- Holty JE, Gould MK, Meinke L, Keeffe EB, Ruoss SJ. Tuberculosis in liver transplant recipients: a systematic review and meta-analysis of individual patient data. Liver Transpl 2009; 15:894–906.
- Jafri SM, Singal AG, Kaul D, Fontana RJ. Detection and management of latent tuberculosis in liver transplant patients. Liver Transpl 2011; 17:306–314.
- Fábrega E, Sampedro B, Cabezas J, et al. Chemoprophylaxis with isoniazid in liver transplant recipients. Liver Transpl 2012; 18:1110–1117.
- Aguado JM, Torre-Cisneros J, Fortún J, et al. Tuberculosis in solid-organ transplant recipients: consensus statement of the group for the study of infection in transplant recipients (GESITRA) of the Spanish Society of Infectious Diseases and Clinical Microbiology. Clin Infect Dis 2009; 48:1276–1284.
- Yehia BR, Blumberg EA. Mycobacterium tuberculosis infection in liver transplantation. Liver Transpl 2010; 16:1129–1135.
- Katz LH, Paul M, Guy DG, Tur-Kaspa R. Prevention of recurrent hepatitis B virus infection after liver transplantation: hepatitis B immunoglobulin, antiviral drugs, or both? Systematic review and meta-analysis. Transpl Infect Dis 2010; 12:292–308.
- Jiang L, Jiang LS, Cheng NS, Yan LN. Current prophylactic strategies against hepatitis B virus recurrence after liver transplantation. World J Gastroenterol 2009; 15:2489–2499.
- Riediger C, Berberat PO, Sauer P, et al. Prophylaxis and treatment of recurrent viral hepatitis after liver transplantation. Nephrol Dial Transplant 2007; 22(suppl 8):viii37–viii46.
- Cholongitas E, Vasiliadis T, Antoniadis N, Goulis I, Papanikolaou V, Akriviadis E. Hepatitis B prophylaxis post liver transplantation with newer nucleos(t)ide analogues after hepatitis B immunoglobulin discontinuation. Transpl Infect Dis 2012; 14:479–487.
- Fox AN, Terrault NA. Individualizing hepatitis B infection prophylaxis in liver transplant recipients. J Hepatol 2011; 55:507–509.
- Fox AN, Terrault NA. The option of HBIG-free prophylaxis against recurrent HBV. J Hepatol 2012; 56:1189–1197.
- Wesdorp DJ, Knoester M, Braat AE, et al. Nucleoside plus nucleotide analogs and cessation of hepatitis B immunoglobulin after liver transplantation in chronic hepatitis B is safe and effective. J Clin Virol 2013; 58:67–73.
- Terrault NA, Berenguer M. Treating hepatitis C infection in liver transplant recipients. Liver Transpl 2006; 12:1192–1204.
- Ciria R, Pleguezuelo M, Khorsandi SE, et al. Strategies to reduce hepatitis C virus recurrence after liver transplantation. World J Hepatol 2013; 5:237–250.
- Issa NC, Fishman JA. Infectious complications of antilymphocyte therapies in solid organ transplantation. Clin Infect Dis 2009; 48:772–786.
- Kalambokis G, Manousou P, Samonakis D, et al. Clinical outcome of HCV-related graft cirrhosis and prognostic value of hepatic venous pressure gradient. Transpl Int 2009; 22:172–181.
- Neumann UP, Berg T, Bahra M, et al. Long-term outcome of liver transplants for chronic hepatitis C: a 10-year follow-up. Transplantation 2004; 77:226–231.
- Wiesner RH, Sorrell M, Villamil F; International Liver Transplantation Society Expert Panel. Report of the first International Liver Transplantation Society expert panel consensus conference on liver transplantation and hepatitis C. Liver Transpl 2003; 9:S1–S9.
- Dinges S, Morard I, Heim M, et al; Swiss Association for the Study of the Liver (SASL 17). Pegylated interferon-alpha2a/ribavirin treatment of recurrent hepatitis C after liver transplantation. Transpl Infect Dis 2009; 11:33–39.
- Veldt BJ, Poterucha JJ, Watt KD, et al. Impact of pegylated interferon and ribavirin treatment on graft survival in liver transplant patients with recurrent hepatitis C infection. Am J Transplant 2008; 8:2426–2433.
- Faisal N, Yoshida EM, Bilodeau M, et al. Protease inhibitor-based triple therapy is highly effective for hepatitis C recurrence after liver transplant: a multicenter experience. Ann Hepatol 2014; 13:525–532.
- Mariño Z, van Bömmel F, Forns X, Berg T. New concepts of sofosbuvir-based treatment regimens in patients with hepatitis C. Gut 2014; 63:207–215.
- Coilly A, Roche B, Dumortier J, et al. Safety and efficacy of protease inhibitors to treat hepatitis C after liver transplantation: a multicenter experience. J Hepatol 2014; 60:78–86.
- Lucey MR, Terrault N, Ojo L, et al. Long-term management of the successful adult liver transplant: 2012 practice guideline by the American Association for the Study of Liver Diseases and the American Society of Transplantation. Liver Transpl 2013; 19:3–26.
The immunosuppressed state of liver transplant recipients makes them vulnerable to infections after surgery.1 These infections are directly correlated with the net state of immunosuppression. Higher levels of immunosuppression mean a higher risk of infection, with rates of infection typically highest in the early posttransplant period.
Common infections during this period include operative and perioperative nosocomial bacterial and fungal infections, reactivation of latent infections, and invasive fungal infections such as candidiasis, aspergillosis, and pneumocystosis. Donor-derived infections also must be considered. As time passes and the level of immunosuppression is reduced, liver recipients are less prone to infection.1
The risk of infection can be minimized by appropriate antimicrobial prophylaxis, strategies for safe living after transplant,2 vaccination,3 careful balancing of immunosuppressive therapy,4 and thoughtful donor selection.5 Drug-drug interactions are common and must be carefully considered to minimize the risk.
This review highlights common infectious complications encountered after liver transplant.
INTRA-ABDOMINAL INFECTIONS
Intra-abdominal infections are common in the early postoperative period.6,7
Risk factors include:
- Pretransplant ascites
- Posttransplant dialysis
- Wound infection
- Reoperation8
- Hepatic artery thrombosis
- Roux-en-Y choledochojejunostomy anastomosis.9
Signs that may indicate intra-abdominal infection include fever, abdominal pain, leukocytosis, and elevated liver enzymes. But because of their immunosuppressed state, transplant recipients may not manifest fever as readily as the general population. They should be evaluated for cholangitis, peritonitis, biloma, and intra-abdominal abscess.
Organisms. Intra-abdominal infections are often polymicrobial. Enterococci, Staphylococcus aureus, gram-negative species including Pseudomonas, Klebsiella, and Acinetobacter, and Candida species are the most common pathogens. Strains are often resistant to multiple drugs, especially in patients who received antibiotics in the weeks before transplant.8,10
Liver transplant recipients are also particularly susceptible to Clostridium difficile-associated colitis as a result of immunosuppression and frequent use of antibiotics perioperatively and postoperatively.11 The spectrum of C difficile infection ranges from mild diarrhea to life-threatening colitis, and the course in liver transplant patients tends to be more complicated than in immunocompetent patients.12
Diagnosis. Intra-abdominal infections should be looked for and treated promptly, as they are associated with a higher mortality rate, a greater risk of graft loss, and a higher incidence of retransplant.6,10 Abdominal ultrasonography or computed tomography (CT) can confirm the presence of fluid collections.
Treatment. Infected collections can be treated with percutaneous or surgical drainage and antimicrobial therapy. In the case of biliary tract complications, retransplant or surgical correction of biliary leakage or stenosis decreases the risk of death.6
Suspicion should be high for C difficile-associated colitis in cases of posttransplant diarrhea. C difficile toxin stool assays help confirm the diagnosis.12 Oral metronidazole is recommended in mild to moderate C difficile infection, with oral vancomycin and intravenous metronidazole reserved for severe cases. Colectomy may be necessary in patients with toxic megacolon.
CYTOMEGALOVIRUS INFECTION
Cytomegalovirus is an important opportunistic pathogen in liver transplant recipients.13 It causes a range of manifestations, from infection (viremia with or without symptoms) to cytomegalovirus syndrome (fever, malaise, and cell-line cytopenias) to tissue-invasive disease with end-organ disease.14 Without preventive measures and treatment, cytomegalovirus disease can increase the risk of morbidity, allograft loss and death.15,16
Risk factors for cytomegalovirus infection (Table 1) include:
- Discordant serostatus of the donor and recipient (the risk is highest in seronegative recipients of organs from seropositive donors)
- Higher levels of immunosuppression, especially when antilymphocyte antibodies are used
- Treatment of graft rejection
- Coinfection with other human herpesviruses, such as Epstein-Barr virus.4,17
Preventing cytomegalovirus infection
The strategy to prevent cytomegalovirus infection depends on the serologic status of the donor and recipient and may include antiviral prophylaxis or preemptive treatment (Table 2).18
Prophylaxis involves giving antiviral drugs during the early high-risk period, with the goal of preventing the development of cytomegalovirus viremia. The alternative preemptive strategy emphasizes serial testing for cytomegalovirus viremia, with the goal of intervening with antiviral medications while viremia is at a low level, thus avoiding potential progression to cytomegalovirus disease. Both strategies have pros and cons that should be considered by each transplant center when setting institutional policy.
A prophylactic approach seems very effective at preventing both infection and disease from cytomegalovirus and has been shown to reduce graft rejection and the risk of death.18 It is preferred in cytomegalovirus-negative recipients when the donor was cytomegalovirus-positive—a high-risk situation.19 However, these patients are also at higher risk of late-onset cytomegalovirus disease. Higher cost and potential drug toxicity, mainly neutropenia from ganciclovir-based regimens, are additional considerations.
Preemptive treatment, in contrast, reserves drug treatment for patients who are actually infected with cytomegalovirus, thus resulting in fewer adverse drug events and lower cost; but it requires regular monitoring. Preemptive methods, by definition, cannot prevent infection, and with this strategy tissue-invasive disease not associated with viremia does occasionally occur.20 As such, patients with a clinical presentation that suggests cytomegalovirus but have negative results on blood testing should be considered for tissue biopsy with culture and immunohistochemical stain.
The most commonly used regimens for antiviral prophylaxis and treatment in liver transplant recipients are intravenous ganciclovir and oral valganciclovir.21 Although valganciclovir is the most commonly used agent in this setting because of ease of administration, it has not been approved by the US Food and Drug Administration in liver transplant patients, as it was associated with higher rates of cytomegalovirus tissue-invasive disease.22–24 Additionally, drug-resistant cytomegalovirus strains have been associated with valganciclovir prophylaxis in cytomegalovirus-negative recipients of solid organs from cytomegalovirus-positive donors.25
Prophylaxis typically consists of therapy for 3 months from the time of transplant. In higher-risk patients (donor-positive, recipient-negative), longer courses of prophylaxis have been extrapolated from data in kidney transplant recipients.26 Extension or reinstitution of prophylaxis should also be considered in liver transplant patients receiving treatment for rejection with antilymphocyte therapy.
Routine screening for cytomegalovirus is not recommended while patients are receiving prophylaxis. High-risk patients who are not receiving prophylaxis should be monitored with nucleic acid or pp65 antigenemia testing as part of the preemptive strategy protocol.
Treatment of cytomegalovirus disease
Although no specific threshold has been established, treatment is generally indicated if a patient has a consistent clinical syndrome, evidence of tissue injury, and persistent or increasing viremia.
Treatment involves giving antiviral drugs and also reducing the level of immunosuppression, if possible, until symptoms and viremia have resolved.
The choice of antiviral therapy depends on the severity of disease. Intravenous ganciclovir (5 mg/kg twice daily adjusted for renal impairment) or oral valganciclovir (900 mg twice daily, also renally dose-adjusted when necessary) can be used for mild to moderate disease if no significant gastrointestinal involvement is reported. Intravenous ganciclovir is preferred for patients with more severe disease or gastrointestinal involvement. The minimum duration of treatment is 2 weeks and may need to be prolonged until both symptoms and viremia completely resolve.18
Drug resistance can occur and should be considered in patients who have a history of prolonged ganciclovir or valganciclovir exposure who do not clinically improve or have persistent or rising viremia. In such cases, genotype assays are helpful, and initiation of alternative therapy should be considered. Mutations conferring resistance to ganciclovir are often associated with cross-resistance to cidofovir. Cidofovir can therefore be considered only when genotype assays demonstrate specific mutations conferring an isolated resistance to ganciclovir.27 The addition of foscarnet to the ganciclovir regimen or substitution of foscarnet for ganciclovir are accepted approaches.
Although cytomegalovirus hyperimmunoglobulin has been used in prophylaxis and invasive disease treatment, its role in the management of ganciclovir-resistant cytomegalovirus infections remains controversial.28
EPSTEIN-BARR VIRUS POSTTRANSPLANT LYMPHOPROLIFERATIVE DISEASE
Epstein-Barr virus-associated posttransplant lymphoproliferative disease is a spectrum of disorders ranging from an infectious mononucleosis syndrome to aggressive malignancy with the potential for death and significant morbidity after liver transplant.29 The timeline of risk varies, but the disease is most common in the first year after transplant.
Risk factors for this disease (Table 1) are:
- Primary Epstein-Barr virus infection
- Cytomegalovirus donor-recipient mismatch
- Cytomegalovirus disease
- Higher levels of immunosuppression, especially with antilymphocyte antibodies.30
The likelihood of Epstein-Barr virus playing a contributing role is lower in later-onset posttransplant lymphoproliferative disease. Patients who are older at the time of transplant, who receive highly immunogenic allografts including a liver as a component of a multivisceral transplant, and who receive increased immunosuppression to treat rejection are at even greater risk of late posttransplant lymphoproliferative disease.31 This is in contrast to early posttransplant lymphoproliferative disease, which is seen more commonly in children as a result of primary Epstein-Barr virus infection.
Recognition and diagnosis. Heightened suspicion is required when considering posttransplant lymphoproliferative disease, and careful evaluation of consistent symptoms and allograft dysfunction are required.
Clinically, posttransplant lymphoproliferative disease should be suspected if a liver transplant recipient develops unexplained fever, weight loss, lymphadenopathy, or cell-line cytopenias.30,32 Other signs and symptoms may be related to the organ involved and may include evidence of hepatitis, pneumonitis, and gastrointestinal disease.31
Adjunctive diagnostic testing includes donor and recipient serology to characterize overall risk before transplantation and quantification of Epstein-Barr viral load, but confirmation relies on tissue histopathology.
Treatment focuses on reducing immunosuppression.30,32 Adding antiviral agents does not seem to improve outcome in all cases.33 Depending on clinical response and histologic classification, additional therapies such as anti-CD20 humanized chimeric monoclonal antibodies, surgery, radiation, and conventional chemotherapy may be required.34
Preventive approaches remain controversial. Chemoprophylaxis with an antiviral such as ganciclovir is occasionally used but has not been shown to consistently decrease rates of posttransplant lymphoproliferative disease. These agents may act in an indirect manner, leading to decreased rates of cytomegalovirus infection, a major cofactor for posttransplant lymphoproliferative disease.24
Passive immunoprophylaxis with immunoglobulin targeting cytomegalovirus has shown to decrease rates of non-Hodgkin lymphoma from posttransplant lymphoproliferative disease in renal transplant recipients in the first year after transplant,35 but data are lacking regarding its use in liver transplant recipients. Monitoring of the viral load and subsequent reduction of immunosuppression remain the most efficient measures to date.36
FUNGAL INFECTIONS
Candida species account for more than half of fungal infections in liver transplant recipients.37 However, a change has been noted in the past 20 years, with a decrease in Candida infections accompanied by an increase in Aspergillus infections.38 Endemic mycoses such as coccidioidomycosis, blastomycosis, and histoplasmosis should be considered with the appropriate epidemiologic history or if disease develops early after transplant and the donor came from a highly endemic region.39Cryptococcus may also be encountered.
Diagnosis. One of the most challenging aspects of fungal infection in liver transplant recipients is timely diagnosis. Heightened suspicion and early biopsy for pathological and microbiological confirmation are necessary. Although available noninvasive diagnostic tools often lack specificity, early detection of fungal markers may be of great use in guiding further diagnostic workup or empiric treatment in the critically ill.
Noninvasive tests include galactomannan, cryptococcal antigen, histoplasma antigen, (1-3)-beta-D-glucan assay and various antibody tests. Galactomannan testing has been widely used to aid in the diagnosis of invasive aspergillosis. Similarly, the (1-3)-beta-D-glucan assay is a non–culture-based tool for diagnosing and monitoring the treatment of invasive fungal infections. However, a definite diagnosis cannot be made on the basis of a positive test alone.40 The complementary diagnostic characteristics of combining noninvasive assays have yet to be fully elucidated.41 Cultures and tissue histopathology are also used when possible.
Treatment is based on targeted specific antifungal drug therapy and reduction of immunosuppressive therapy, when possible. The choice of antifungal agent varies with the pathogen, the site of involvement, and the severity of the disease. A focus on potential drug interactions, their management, and therapeutic drug monitoring when using antifungal medications is essential in the posttransplant period. Combination therapy can be considered in some situations to enhance synergy. The following sections discuss in greater detail Candida species, Aspergillus species, and Pneumocystis jirovecii infections.
Candida infections
Candidiasis after liver transplant is typically nosocomial, especially when diagnosed during the first 3 months (Table 3).37
Risk factors for invasive candidiasis include perioperative colonization, prolonged operative time, retransplant, greater transfusion requirements, and postoperative renal failure.37,42,43 Invasive candidiasis is of concern for its effects on morbidity, mortality, and cost of care.43–46
Organisms. The frequency of implicated species, in particular those with a natural resistance to fluconazole, differs in various reports.37,45,46Candida albicans remains the most commonly isolated pathogen; however, non-albicans species including those resistant to fluconazole have been reported more frequently and include Candida glabrata and Candida krusei.47,48
Signs and diagnosis. Invasive candidiasis in liver transplant recipients generally manifests itself in catheter-related blood stream infections, urinary tract infections, or intra-abdominal infections. Diagnosis can be made by isolating Candida from blood cultures, recovering organisms in culture of a normally sterile site, or finding direct microscopic evidence of the fungus on tissue specimens.49
Disseminated candidiasis refers to the involvement of distant anatomic sites. Clinical manifestations may cause vision changes, abdominal pain or skin nodules with findings of candidemia, hepatosplenic abscesses, or retinal exudates on funduscopy.49
Treatment of invasive candidiasis in liver recipients often involves antifungal therapy and reduction of immunosuppression. Broad-spectrum antifungals are initially advocated in an empirical approach to cover fluconazole-resistant strains of the non-albicans subgroups.50 Depending on antifungal susceptibility, treatment can later be adjusted.
Fluconazole remains the agent of choice in most C albicans infections.47 However, attention should be paid to the possibility of resistance in patients who have received fluconazole prophylaxis within the past 30 days. Additional agents used in treatment may include echinocandins, amphotericin, and additional azoles.
Antifungal prophylaxis is recommended in high-risk liver transplant patients, although its optimal duration remains undetermined.44 Antifungal prophylaxis has been associated with decreased incidence of both superficial and invasive candidiasis.51
Aspergillus infection
Aspergillus, the second most common fungal pathogen, has become a more common concern in liver transplant recipients. Aspergillus fumigatus is the most frequently encountered species.38,52
Risk factors. These infections typically occur in the first year, during intense immunosuppression. Retransplant, renal failure, and fulminant hepatic failure are major risk factors.52 In the presence of risk factors and a suggestive clinical setting, invasive aspergillosis should be considered and the diagnosis pursued.
Diagnosis is suggested by positive findings on CT accompanied by lower respiratory tract symptoms, focal lesions on neuroimaging, or demonstration of the fungus on cultures.49 However, Aspergillus is rarely grown in blood culture. The galactomannan antigen is a noninvasive test that can provide supporting evidence for the diagnosis.41,52 False-positive results do occur in the setting of certain antibiotics and cross-reacting fungi.53
Treatment consists of antifungal therapy and immunosuppression reduction.52
Voriconazole is the first-line agent for invasive aspergillosis. Monitoring for potential drug-drug interactions and side effects is required.54,55 Amphotericin B is considered a second-line choice due to toxicity and lack of an oral formulation. In refractory cases, combined antifungal therapy could be considered.52 The duration of treatment is generally a minimum of 12 weeks.
Prophylaxis. Specific prophylaxis against invasive aspergillosis is not currently recommended; however, some authors suggest a prophylactic approach using echinocandins or liposomal amphotericin B in high-risk patients.51,52 Aspergillosis is associated with a considerable increase in mortality in liver transplant recipients, which highlights the importance of timely management.52,56
Pneumocystis jirovecii
P jirovecii remains a common opportunistic pathogen in people with impaired immunity, including transplant and human immunodeficiency virus patients.
Prophylaxis. Widespread adoption of antimicrobial prophylaxis by transplant centers has decreased the rates of P jirovecii infection in liver transplant recipients.57,58 Commonly used prophylactic regimens after liver transplantation include a single-strength trimethoprim-sulfamethoxazole tablet daily or a double-strength tablet three times per week for a minimum of 6 to 12 months after transplant. Atovaquone and dapsone can be used as alternatives in cases of intolerance to trimethoprim-sulfamethoxazole (Table 2).
Inhaled pentamidine is clearly inferior and should be used only when the other medications are contraindicated.59
Signs and diagnosis. P jirovecii pneumonia is characterized by fever, cough, dyspnea, and chest pain. Insidious hypoxemia, abnormal chest examination, and bilateral interstitial pneumonia on chest radiography are common.
CT may be more sensitive than chest radiography.57 Findings suggestive of P jirovecii pneumonia on chest CT are extensive bilateral and symmetrical ground-glass attenuations. Other less-characteristic findings include upper lobar parenchymal opacities and spontaneous pneumothorax.57,60
The serum (1,3)-beta-D-glucan assay derived from major cell-wall components of P jirovecii might be helpful. Studies report a sensitivity for P jirovecii pneumonia as high as 96% and a negative predictive value of 99.8%.61,62
Definitive diagnosis requires identification of the pathogen. Routine expectorated sputum sampling is generally associated with a poor diagnostic yield. Bronchoscopy and bronchoalveolar lavage with silver or fluorescent antibody staining of samples, polymerase chain reaction testing, or both significantly improves diagnosis. Transbronchial or open lung biopsy are often unnecessary.57
Treatment. Trimethoprim-sulfamethoxazole is the first-line agent for treating P jirovecii pneumonia.57 The minimum duration of treatment is 14 days, with extended courses for severe infection.
Intravenous pentamidine or clindamycin plus primaquine are alternatives for patients who cannot tolerate trimethoprim-sulfamethoxazole. The major concern with intravenous pentamidine is renal dysfunction. Hypoglycemia or hyperglycemia, neutropenia, thrombocytopenia, nausea, dysgeusia, and pancreatitis may also occur.63
Atovaquone might also be beneficial in mild to moderate P jirovecii pneumonia. The main side effects include skin rashes, gastrointestinal intolerance, and elevation of transaminases.64
A corticosteroid (40–60 mg of prednisone or its equivalent) may be beneficial in conjunction with antimicrobial therapy in patients with significant hypoxia (partial pressure of arterial oxygen < 70 mm Hg on room air) in decreasing the risk of respiratory failure and need for intubation.
With appropriate and timely antimicrobial prophylaxis, cases of P jirovecii pneumonia should continue to decrease.
TUBERCULOSIS
Development of tuberculosis after transplantation is a catastrophic complication, with mortality rates of up to 30%.65 Most cases of posttransplant tuberculosis represent reactivation of latent disease.66 Screening with tuberculin skin tests or interferon-gamma-release assays is recommended in all liver transplant candidates. Chest radiography before transplant is necessary when assessing a positive screening test.67
The optimal management of latent tuberculosis in these cases remains controversial. Patients at high risk or those with positive screening results on chest radiography warrant treatment for latent tuberculosis infection with isoniazid unless contraindicated.67,68
The ideal time to initiate prophylactic isoniazid therapy is unclear. Some authors suggest delaying it, as it might be associated with poor tolerance and hepatotoxicity.69 Others have found that early isoniazid use was not associated with negative outcomes.70
Risk factors for symptomatic tuberculosis after liver transplant include previous infection with tuberculosis, intensified immunosuppression (especially anti-T-lymphocyte therapies), diabetes mellitus, and other co-infections (Table 1).71
The increased incidence of atypical presentations in recent years makes the diagnosis of active tuberculosis among liver transplant recipients challenging. Sputum smears can be negative due to low mycobacterial burdens, and tuberculin skin testing and interferon-gamma-release assays may be falsely negative due to immunosuppression.67
Treatment of active tuberculosis consists initially of a four-drug regimen using isoniazid, rifampin, pyrazinamide, and ethambutol for 2 months. Adjustments are made in accordance with culture and sensitivity results. Treatment can then be tapered to two drugs (isoniazid and rifampin) for a minimum of 4 additional months. Prolonged treatment may be required in instances of extrapulmonary or disseminated disease.65,72
Tuberculosis treatment can be complicated by hepatotoxicity in liver transplant recipients because of direct drug effects and drug-drug interactions with immunosuppressive agents. Close monitoring for rejection and hepatotoxicity is therefore imperative while liver transplant recipients are receiving antituberculosis therapy. Drug-drug interactions may also be responsible for marked reductions in immunosuppression levels, especially with regimens containing rifampin.71 Substitution of rifabutin for rifampin reduces the effect of drug interactions.66
VIRAL HEPATITIS
Hepatitis B virus
Hepatitis B virus-related end-stage liver disease and hepatocellular carcinoma are common indications for liver transplant in Asia. It is less common in the United States and Europe, accounting for less than 10% of all liver transplant cases. Prognosis is favorable in recipients undergoing liver transplant for hepatitis B virus, with excellent survival rates. Prevention of reinfection is crucial in these patients.
Treatment with combination antiviral agents and hepatitis B immunoglobulin (HBIG) is effective.73 Lamivudine was the first nucleoside analogue found to be effective against hepatitis B virus. Its low cost and relative safety are strong arguments in favor of its continued use in liver transplant recipients.74 In patients without evidence of hepatitis B viral replication at the time of transplant, monotherapy with lamivudine has led to low recurrence rates, and adefovir can be added to control resistant viral strains.75
The frequent emergence of resistance with lamivudine favors newer agents such as entecavir or tenofovir. These nucleoside and nucleotide analogues have a higher barrier to resistance, and thus resistance to them is rare. They are also more efficient, potentially allowing use of an HBIG-sparing protocol.76 However, they are associated with a higher risk of nephrotoxicity and require dose adjustments in renal insufficiency. Data directly comparing entecavir and tenofovir are scarce.
Prophylaxis. Most studies support an individualized approach for prevention of hepatitis B virus reinfection. High-risk patients, ie, those positive for HBe antigen or with high viral loads (> 100,000 copies/mL) are generally treated with both HBIG and antiviral agents.77 Low-risk patients are those with a negative HBe antigen, low hepatitis B virus DNA levels, hepatitis B virus-related acute liver failure, and cirrhosis resulting from coinfection with both hepatitis B and hepatitis D virus.75 In low-risk patients, discontinuation of HBIG after 1 to 2 years of treatment is appropriate, and long-term prophylaxis with antiviral agents alone is an option. However, levels of hepatitis B DNA should be monitored closely.78,79
Hepatitis C virus
Recurrence of hepatitis C virus infection is the rule among patients who are viremic at the time of liver transplant.80,81 Most of these patients will show histologic evidence of recurrent hepatitis within the first year after liver transplant. It is often difficult to distinguish between the histopathological appearance of a recurrent hepatitis C virus infection and acute cellular rejection.
Progression to fibrosis and subsequently cirrhosis and decompensation is highly variable in hepatitis C virus-infected liver transplant recipients. Diabetes, insulin resistance, and possibly hepatitis steatosis have been associated with a rapid progression to advanced fibrosis. The contribution of immunosuppression to the progression of hepatitis C virus remains an area of active study. Some studies point to antilymphocyte immunosuppressive agents as a potential cause.82 Liver biopsy is a useful tool in this situation. It allows monitoring of disease severity and progression and may distinguish recurrent hepatitis C virus disease from other causes of liver enzyme elevation.
The major concern with the recurrence of hepatitis C virus infection after liver transplant is allograft loss. Rates of patient and graft survival are reduced in infected patients compared with hepatitis C virus-negative patients.83,84 Prophylactic antiviral therapy has no current role in the management of hepatitis C virus disease. Those manifesting moderate to severe necroinflammation or mild to moderate fibrosis indicative of progressive disease should be treated.81,85
Sustained viral clearance with antiviral agents confers a graft survival benefit.
The combination of peg-interferon and weight-based ribavirin has been the standard of treatment but may be associated with increased rates of rejection.86,87 The sustained virologic response rates for hepatitis C virus range from 60% in genotypes 4, 5, and 6 after 48 weeks of treatment to 60% to 80% in genotypes 2 and 3 after 24 weeks, but only about 30% in genotype 1.88
Treatment with the newer agents, especially protease inhibitors, in genotype 1 (peg-interferon, ribavirin, and either telaprevir or boceprevir) has been evaluated. Success rates reaching 70% have been achieved.89 Adverse effects can be a major setback. Serious complications include severe anemia, renal dysfunction, increased risk of infection, and death.
Triple therapy should be carefully considered in liver transplant patients with genotype 1 hepatitis C virus.90 Significant drug-drug interactions are reported between hepatitis C virus protease inhibitors and immunosuppression regimens. Additional new oral direct- acting antivirals have been investigated. They bring promising advances in hepatitis C virus treatment and pave the way for interferon-free regimens with pangenotypic activity.
IMMUNIZATION
Immunization can decrease the risk of infectious complications in liver transplant recipients, as well as in close contacts and healthcare professionals.3
Influenza. Pretransplant influenza vaccine and posttransplant annual influenza vaccines are necessary.
Pneumococcal immunization should additionally be provided prior to transplant and repeated every 3 to 5 years thereafter.3,91
A number of other vaccinations should also be completed before transplant, including the hepatitis A and B vaccines and the tetanus/diphtheria/acellular pertussis vaccines. However, these vaccinations have not been shown to be detrimental to patients after transplant.91
Varicella and zoster vaccines should be given before liver transplant—zoster in patients over age 60, and varicella in patients with no immunity. Live vaccines, including varicella and zoster vaccines, are contraindicated after liver transplant.3
Human papillomavirus. The bivalent human papillomavirus vaccine can be given before transplant in females ages 9 to 26; the quadrivalent vaccine is beneficial in those ages 9 to 26 and in women under age 45.3,91
IMMUNOSUPPRESSION CARRIES RISK OF INFECTION
Most liver transplant patients require prolonged immunosuppressive therapy. This comes with an increased risk of new or recurrent infections, potentially causing death and significant morbidity.
Evaluation of existing risk factors, appropriate prophylaxis and immunization, timely diagnosis, and treatment of such infections are therefore essential steps for the successful management of liver transplant recipients.
The immunosuppressed state of liver transplant recipients makes them vulnerable to infections after surgery.1 These infections are directly correlated with the net state of immunosuppression. Higher levels of immunosuppression mean a higher risk of infection, with rates of infection typically highest in the early posttransplant period.
Common infections during this period include operative and perioperative nosocomial bacterial and fungal infections, reactivation of latent infections, and invasive fungal infections such as candidiasis, aspergillosis, and pneumocystosis. Donor-derived infections also must be considered. As time passes and the level of immunosuppression is reduced, liver recipients are less prone to infection.1
The risk of infection can be minimized by appropriate antimicrobial prophylaxis, strategies for safe living after transplant,2 vaccination,3 careful balancing of immunosuppressive therapy,4 and thoughtful donor selection.5 Drug-drug interactions are common and must be carefully considered to minimize the risk.
This review highlights common infectious complications encountered after liver transplant.
INTRA-ABDOMINAL INFECTIONS
Intra-abdominal infections are common in the early postoperative period.6,7
Risk factors include:
- Pretransplant ascites
- Posttransplant dialysis
- Wound infection
- Reoperation8
- Hepatic artery thrombosis
- Roux-en-Y choledochojejunostomy anastomosis.9
Signs that may indicate intra-abdominal infection include fever, abdominal pain, leukocytosis, and elevated liver enzymes. But because of their immunosuppressed state, transplant recipients may not manifest fever as readily as the general population. They should be evaluated for cholangitis, peritonitis, biloma, and intra-abdominal abscess.
Organisms. Intra-abdominal infections are often polymicrobial. Enterococci, Staphylococcus aureus, gram-negative species including Pseudomonas, Klebsiella, and Acinetobacter, and Candida species are the most common pathogens. Strains are often resistant to multiple drugs, especially in patients who received antibiotics in the weeks before transplant.8,10
Liver transplant recipients are also particularly susceptible to Clostridium difficile-associated colitis as a result of immunosuppression and frequent use of antibiotics perioperatively and postoperatively.11 The spectrum of C difficile infection ranges from mild diarrhea to life-threatening colitis, and the course in liver transplant patients tends to be more complicated than in immunocompetent patients.12
Diagnosis. Intra-abdominal infections should be looked for and treated promptly, as they are associated with a higher mortality rate, a greater risk of graft loss, and a higher incidence of retransplant.6,10 Abdominal ultrasonography or computed tomography (CT) can confirm the presence of fluid collections.
Treatment. Infected collections can be treated with percutaneous or surgical drainage and antimicrobial therapy. In the case of biliary tract complications, retransplant or surgical correction of biliary leakage or stenosis decreases the risk of death.6
Suspicion should be high for C difficile-associated colitis in cases of posttransplant diarrhea. C difficile toxin stool assays help confirm the diagnosis.12 Oral metronidazole is recommended in mild to moderate C difficile infection, with oral vancomycin and intravenous metronidazole reserved for severe cases. Colectomy may be necessary in patients with toxic megacolon.
CYTOMEGALOVIRUS INFECTION
Cytomegalovirus is an important opportunistic pathogen in liver transplant recipients.13 It causes a range of manifestations, from infection (viremia with or without symptoms) to cytomegalovirus syndrome (fever, malaise, and cell-line cytopenias) to tissue-invasive disease with end-organ disease.14 Without preventive measures and treatment, cytomegalovirus disease can increase the risk of morbidity, allograft loss and death.15,16
Risk factors for cytomegalovirus infection (Table 1) include:
- Discordant serostatus of the donor and recipient (the risk is highest in seronegative recipients of organs from seropositive donors)
- Higher levels of immunosuppression, especially when antilymphocyte antibodies are used
- Treatment of graft rejection
- Coinfection with other human herpesviruses, such as Epstein-Barr virus.4,17
Preventing cytomegalovirus infection
The strategy to prevent cytomegalovirus infection depends on the serologic status of the donor and recipient and may include antiviral prophylaxis or preemptive treatment (Table 2).18
Prophylaxis involves giving antiviral drugs during the early high-risk period, with the goal of preventing the development of cytomegalovirus viremia. The alternative preemptive strategy emphasizes serial testing for cytomegalovirus viremia, with the goal of intervening with antiviral medications while viremia is at a low level, thus avoiding potential progression to cytomegalovirus disease. Both strategies have pros and cons that should be considered by each transplant center when setting institutional policy.
A prophylactic approach seems very effective at preventing both infection and disease from cytomegalovirus and has been shown to reduce graft rejection and the risk of death.18 It is preferred in cytomegalovirus-negative recipients when the donor was cytomegalovirus-positive—a high-risk situation.19 However, these patients are also at higher risk of late-onset cytomegalovirus disease. Higher cost and potential drug toxicity, mainly neutropenia from ganciclovir-based regimens, are additional considerations.
Preemptive treatment, in contrast, reserves drug treatment for patients who are actually infected with cytomegalovirus, thus resulting in fewer adverse drug events and lower cost; but it requires regular monitoring. Preemptive methods, by definition, cannot prevent infection, and with this strategy tissue-invasive disease not associated with viremia does occasionally occur.20 As such, patients with a clinical presentation that suggests cytomegalovirus but have negative results on blood testing should be considered for tissue biopsy with culture and immunohistochemical stain.
The most commonly used regimens for antiviral prophylaxis and treatment in liver transplant recipients are intravenous ganciclovir and oral valganciclovir.21 Although valganciclovir is the most commonly used agent in this setting because of ease of administration, it has not been approved by the US Food and Drug Administration in liver transplant patients, as it was associated with higher rates of cytomegalovirus tissue-invasive disease.22–24 Additionally, drug-resistant cytomegalovirus strains have been associated with valganciclovir prophylaxis in cytomegalovirus-negative recipients of solid organs from cytomegalovirus-positive donors.25
Prophylaxis typically consists of therapy for 3 months from the time of transplant. In higher-risk patients (donor-positive, recipient-negative), longer courses of prophylaxis have been extrapolated from data in kidney transplant recipients.26 Extension or reinstitution of prophylaxis should also be considered in liver transplant patients receiving treatment for rejection with antilymphocyte therapy.
Routine screening for cytomegalovirus is not recommended while patients are receiving prophylaxis. High-risk patients who are not receiving prophylaxis should be monitored with nucleic acid or pp65 antigenemia testing as part of the preemptive strategy protocol.
Treatment of cytomegalovirus disease
Although no specific threshold has been established, treatment is generally indicated if a patient has a consistent clinical syndrome, evidence of tissue injury, and persistent or increasing viremia.
Treatment involves giving antiviral drugs and also reducing the level of immunosuppression, if possible, until symptoms and viremia have resolved.
The choice of antiviral therapy depends on the severity of disease. Intravenous ganciclovir (5 mg/kg twice daily adjusted for renal impairment) or oral valganciclovir (900 mg twice daily, also renally dose-adjusted when necessary) can be used for mild to moderate disease if no significant gastrointestinal involvement is reported. Intravenous ganciclovir is preferred for patients with more severe disease or gastrointestinal involvement. The minimum duration of treatment is 2 weeks and may need to be prolonged until both symptoms and viremia completely resolve.18
Drug resistance can occur and should be considered in patients who have a history of prolonged ganciclovir or valganciclovir exposure who do not clinically improve or have persistent or rising viremia. In such cases, genotype assays are helpful, and initiation of alternative therapy should be considered. Mutations conferring resistance to ganciclovir are often associated with cross-resistance to cidofovir. Cidofovir can therefore be considered only when genotype assays demonstrate specific mutations conferring an isolated resistance to ganciclovir.27 The addition of foscarnet to the ganciclovir regimen or substitution of foscarnet for ganciclovir are accepted approaches.
Although cytomegalovirus hyperimmunoglobulin has been used in prophylaxis and invasive disease treatment, its role in the management of ganciclovir-resistant cytomegalovirus infections remains controversial.28
EPSTEIN-BARR VIRUS POSTTRANSPLANT LYMPHOPROLIFERATIVE DISEASE
Epstein-Barr virus-associated posttransplant lymphoproliferative disease is a spectrum of disorders ranging from an infectious mononucleosis syndrome to aggressive malignancy with the potential for death and significant morbidity after liver transplant.29 The timeline of risk varies, but the disease is most common in the first year after transplant.
Risk factors for this disease (Table 1) are:
- Primary Epstein-Barr virus infection
- Cytomegalovirus donor-recipient mismatch
- Cytomegalovirus disease
- Higher levels of immunosuppression, especially with antilymphocyte antibodies.30
The likelihood of Epstein-Barr virus playing a contributing role is lower in later-onset posttransplant lymphoproliferative disease. Patients who are older at the time of transplant, who receive highly immunogenic allografts including a liver as a component of a multivisceral transplant, and who receive increased immunosuppression to treat rejection are at even greater risk of late posttransplant lymphoproliferative disease.31 This is in contrast to early posttransplant lymphoproliferative disease, which is seen more commonly in children as a result of primary Epstein-Barr virus infection.
Recognition and diagnosis. Heightened suspicion is required when considering posttransplant lymphoproliferative disease, and careful evaluation of consistent symptoms and allograft dysfunction are required.
Clinically, posttransplant lymphoproliferative disease should be suspected if a liver transplant recipient develops unexplained fever, weight loss, lymphadenopathy, or cell-line cytopenias.30,32 Other signs and symptoms may be related to the organ involved and may include evidence of hepatitis, pneumonitis, and gastrointestinal disease.31
Adjunctive diagnostic testing includes donor and recipient serology to characterize overall risk before transplantation and quantification of Epstein-Barr viral load, but confirmation relies on tissue histopathology.
Treatment focuses on reducing immunosuppression.30,32 Adding antiviral agents does not seem to improve outcome in all cases.33 Depending on clinical response and histologic classification, additional therapies such as anti-CD20 humanized chimeric monoclonal antibodies, surgery, radiation, and conventional chemotherapy may be required.34
Preventive approaches remain controversial. Chemoprophylaxis with an antiviral such as ganciclovir is occasionally used but has not been shown to consistently decrease rates of posttransplant lymphoproliferative disease. These agents may act in an indirect manner, leading to decreased rates of cytomegalovirus infection, a major cofactor for posttransplant lymphoproliferative disease.24
Passive immunoprophylaxis with immunoglobulin targeting cytomegalovirus has shown to decrease rates of non-Hodgkin lymphoma from posttransplant lymphoproliferative disease in renal transplant recipients in the first year after transplant,35 but data are lacking regarding its use in liver transplant recipients. Monitoring of the viral load and subsequent reduction of immunosuppression remain the most efficient measures to date.36
FUNGAL INFECTIONS
Candida species account for more than half of fungal infections in liver transplant recipients.37 However, a change has been noted in the past 20 years, with a decrease in Candida infections accompanied by an increase in Aspergillus infections.38 Endemic mycoses such as coccidioidomycosis, blastomycosis, and histoplasmosis should be considered with the appropriate epidemiologic history or if disease develops early after transplant and the donor came from a highly endemic region.39Cryptococcus may also be encountered.
Diagnosis. One of the most challenging aspects of fungal infection in liver transplant recipients is timely diagnosis. Heightened suspicion and early biopsy for pathological and microbiological confirmation are necessary. Although available noninvasive diagnostic tools often lack specificity, early detection of fungal markers may be of great use in guiding further diagnostic workup or empiric treatment in the critically ill.
Noninvasive tests include galactomannan, cryptococcal antigen, histoplasma antigen, (1-3)-beta-D-glucan assay and various antibody tests. Galactomannan testing has been widely used to aid in the diagnosis of invasive aspergillosis. Similarly, the (1-3)-beta-D-glucan assay is a non–culture-based tool for diagnosing and monitoring the treatment of invasive fungal infections. However, a definite diagnosis cannot be made on the basis of a positive test alone.40 The complementary diagnostic characteristics of combining noninvasive assays have yet to be fully elucidated.41 Cultures and tissue histopathology are also used when possible.
Treatment is based on targeted specific antifungal drug therapy and reduction of immunosuppressive therapy, when possible. The choice of antifungal agent varies with the pathogen, the site of involvement, and the severity of the disease. A focus on potential drug interactions, their management, and therapeutic drug monitoring when using antifungal medications is essential in the posttransplant period. Combination therapy can be considered in some situations to enhance synergy. The following sections discuss in greater detail Candida species, Aspergillus species, and Pneumocystis jirovecii infections.
Candida infections
Candidiasis after liver transplant is typically nosocomial, especially when diagnosed during the first 3 months (Table 3).37
Risk factors for invasive candidiasis include perioperative colonization, prolonged operative time, retransplant, greater transfusion requirements, and postoperative renal failure.37,42,43 Invasive candidiasis is of concern for its effects on morbidity, mortality, and cost of care.43–46
Organisms. The frequency of implicated species, in particular those with a natural resistance to fluconazole, differs in various reports.37,45,46Candida albicans remains the most commonly isolated pathogen; however, non-albicans species including those resistant to fluconazole have been reported more frequently and include Candida glabrata and Candida krusei.47,48
Signs and diagnosis. Invasive candidiasis in liver transplant recipients generally manifests itself in catheter-related blood stream infections, urinary tract infections, or intra-abdominal infections. Diagnosis can be made by isolating Candida from blood cultures, recovering organisms in culture of a normally sterile site, or finding direct microscopic evidence of the fungus on tissue specimens.49
Disseminated candidiasis refers to the involvement of distant anatomic sites. Clinical manifestations may cause vision changes, abdominal pain or skin nodules with findings of candidemia, hepatosplenic abscesses, or retinal exudates on funduscopy.49
Treatment of invasive candidiasis in liver recipients often involves antifungal therapy and reduction of immunosuppression. Broad-spectrum antifungals are initially advocated in an empirical approach to cover fluconazole-resistant strains of the non-albicans subgroups.50 Depending on antifungal susceptibility, treatment can later be adjusted.
Fluconazole remains the agent of choice in most C albicans infections.47 However, attention should be paid to the possibility of resistance in patients who have received fluconazole prophylaxis within the past 30 days. Additional agents used in treatment may include echinocandins, amphotericin, and additional azoles.
Antifungal prophylaxis is recommended in high-risk liver transplant patients, although its optimal duration remains undetermined.44 Antifungal prophylaxis has been associated with decreased incidence of both superficial and invasive candidiasis.51
Aspergillus infection
Aspergillus, the second most common fungal pathogen, has become a more common concern in liver transplant recipients. Aspergillus fumigatus is the most frequently encountered species.38,52
Risk factors. These infections typically occur in the first year, during intense immunosuppression. Retransplant, renal failure, and fulminant hepatic failure are major risk factors.52 In the presence of risk factors and a suggestive clinical setting, invasive aspergillosis should be considered and the diagnosis pursued.
Diagnosis is suggested by positive findings on CT accompanied by lower respiratory tract symptoms, focal lesions on neuroimaging, or demonstration of the fungus on cultures.49 However, Aspergillus is rarely grown in blood culture. The galactomannan antigen is a noninvasive test that can provide supporting evidence for the diagnosis.41,52 False-positive results do occur in the setting of certain antibiotics and cross-reacting fungi.53
Treatment consists of antifungal therapy and immunosuppression reduction.52
Voriconazole is the first-line agent for invasive aspergillosis. Monitoring for potential drug-drug interactions and side effects is required.54,55 Amphotericin B is considered a second-line choice due to toxicity and lack of an oral formulation. In refractory cases, combined antifungal therapy could be considered.52 The duration of treatment is generally a minimum of 12 weeks.
Prophylaxis. Specific prophylaxis against invasive aspergillosis is not currently recommended; however, some authors suggest a prophylactic approach using echinocandins or liposomal amphotericin B in high-risk patients.51,52 Aspergillosis is associated with a considerable increase in mortality in liver transplant recipients, which highlights the importance of timely management.52,56
Pneumocystis jirovecii
P jirovecii remains a common opportunistic pathogen in people with impaired immunity, including transplant and human immunodeficiency virus patients.
Prophylaxis. Widespread adoption of antimicrobial prophylaxis by transplant centers has decreased the rates of P jirovecii infection in liver transplant recipients.57,58 Commonly used prophylactic regimens after liver transplantation include a single-strength trimethoprim-sulfamethoxazole tablet daily or a double-strength tablet three times per week for a minimum of 6 to 12 months after transplant. Atovaquone and dapsone can be used as alternatives in cases of intolerance to trimethoprim-sulfamethoxazole (Table 2).
Inhaled pentamidine is clearly inferior and should be used only when the other medications are contraindicated.59
Signs and diagnosis. P jirovecii pneumonia is characterized by fever, cough, dyspnea, and chest pain. Insidious hypoxemia, abnormal chest examination, and bilateral interstitial pneumonia on chest radiography are common.
CT may be more sensitive than chest radiography.57 Findings suggestive of P jirovecii pneumonia on chest CT are extensive bilateral and symmetrical ground-glass attenuations. Other less-characteristic findings include upper lobar parenchymal opacities and spontaneous pneumothorax.57,60
The serum (1,3)-beta-D-glucan assay derived from major cell-wall components of P jirovecii might be helpful. Studies report a sensitivity for P jirovecii pneumonia as high as 96% and a negative predictive value of 99.8%.61,62
Definitive diagnosis requires identification of the pathogen. Routine expectorated sputum sampling is generally associated with a poor diagnostic yield. Bronchoscopy and bronchoalveolar lavage with silver or fluorescent antibody staining of samples, polymerase chain reaction testing, or both significantly improves diagnosis. Transbronchial or open lung biopsy are often unnecessary.57
Treatment. Trimethoprim-sulfamethoxazole is the first-line agent for treating P jirovecii pneumonia.57 The minimum duration of treatment is 14 days, with extended courses for severe infection.
Intravenous pentamidine or clindamycin plus primaquine are alternatives for patients who cannot tolerate trimethoprim-sulfamethoxazole. The major concern with intravenous pentamidine is renal dysfunction. Hypoglycemia or hyperglycemia, neutropenia, thrombocytopenia, nausea, dysgeusia, and pancreatitis may also occur.63
Atovaquone might also be beneficial in mild to moderate P jirovecii pneumonia. The main side effects include skin rashes, gastrointestinal intolerance, and elevation of transaminases.64
A corticosteroid (40–60 mg of prednisone or its equivalent) may be beneficial in conjunction with antimicrobial therapy in patients with significant hypoxia (partial pressure of arterial oxygen < 70 mm Hg on room air) in decreasing the risk of respiratory failure and need for intubation.
With appropriate and timely antimicrobial prophylaxis, cases of P jirovecii pneumonia should continue to decrease.
TUBERCULOSIS
Development of tuberculosis after transplantation is a catastrophic complication, with mortality rates of up to 30%.65 Most cases of posttransplant tuberculosis represent reactivation of latent disease.66 Screening with tuberculin skin tests or interferon-gamma-release assays is recommended in all liver transplant candidates. Chest radiography before transplant is necessary when assessing a positive screening test.67
The optimal management of latent tuberculosis in these cases remains controversial. Patients at high risk or those with positive screening results on chest radiography warrant treatment for latent tuberculosis infection with isoniazid unless contraindicated.67,68
The ideal time to initiate prophylactic isoniazid therapy is unclear. Some authors suggest delaying it, as it might be associated with poor tolerance and hepatotoxicity.69 Others have found that early isoniazid use was not associated with negative outcomes.70
Risk factors for symptomatic tuberculosis after liver transplant include previous infection with tuberculosis, intensified immunosuppression (especially anti-T-lymphocyte therapies), diabetes mellitus, and other co-infections (Table 1).71
The increased incidence of atypical presentations in recent years makes the diagnosis of active tuberculosis among liver transplant recipients challenging. Sputum smears can be negative due to low mycobacterial burdens, and tuberculin skin testing and interferon-gamma-release assays may be falsely negative due to immunosuppression.67
Treatment of active tuberculosis consists initially of a four-drug regimen using isoniazid, rifampin, pyrazinamide, and ethambutol for 2 months. Adjustments are made in accordance with culture and sensitivity results. Treatment can then be tapered to two drugs (isoniazid and rifampin) for a minimum of 4 additional months. Prolonged treatment may be required in instances of extrapulmonary or disseminated disease.65,72
Tuberculosis treatment can be complicated by hepatotoxicity in liver transplant recipients because of direct drug effects and drug-drug interactions with immunosuppressive agents. Close monitoring for rejection and hepatotoxicity is therefore imperative while liver transplant recipients are receiving antituberculosis therapy. Drug-drug interactions may also be responsible for marked reductions in immunosuppression levels, especially with regimens containing rifampin.71 Substitution of rifabutin for rifampin reduces the effect of drug interactions.66
VIRAL HEPATITIS
Hepatitis B virus
Hepatitis B virus-related end-stage liver disease and hepatocellular carcinoma are common indications for liver transplant in Asia. It is less common in the United States and Europe, accounting for less than 10% of all liver transplant cases. Prognosis is favorable in recipients undergoing liver transplant for hepatitis B virus, with excellent survival rates. Prevention of reinfection is crucial in these patients.
Treatment with combination antiviral agents and hepatitis B immunoglobulin (HBIG) is effective.73 Lamivudine was the first nucleoside analogue found to be effective against hepatitis B virus. Its low cost and relative safety are strong arguments in favor of its continued use in liver transplant recipients.74 In patients without evidence of hepatitis B viral replication at the time of transplant, monotherapy with lamivudine has led to low recurrence rates, and adefovir can be added to control resistant viral strains.75
The frequent emergence of resistance with lamivudine favors newer agents such as entecavir or tenofovir. These nucleoside and nucleotide analogues have a higher barrier to resistance, and thus resistance to them is rare. They are also more efficient, potentially allowing use of an HBIG-sparing protocol.76 However, they are associated with a higher risk of nephrotoxicity and require dose adjustments in renal insufficiency. Data directly comparing entecavir and tenofovir are scarce.
Prophylaxis. Most studies support an individualized approach for prevention of hepatitis B virus reinfection. High-risk patients, ie, those positive for HBe antigen or with high viral loads (> 100,000 copies/mL) are generally treated with both HBIG and antiviral agents.77 Low-risk patients are those with a negative HBe antigen, low hepatitis B virus DNA levels, hepatitis B virus-related acute liver failure, and cirrhosis resulting from coinfection with both hepatitis B and hepatitis D virus.75 In low-risk patients, discontinuation of HBIG after 1 to 2 years of treatment is appropriate, and long-term prophylaxis with antiviral agents alone is an option. However, levels of hepatitis B DNA should be monitored closely.78,79
Hepatitis C virus
Recurrence of hepatitis C virus infection is the rule among patients who are viremic at the time of liver transplant.80,81 Most of these patients will show histologic evidence of recurrent hepatitis within the first year after liver transplant. It is often difficult to distinguish between the histopathological appearance of a recurrent hepatitis C virus infection and acute cellular rejection.
Progression to fibrosis and subsequently cirrhosis and decompensation is highly variable in hepatitis C virus-infected liver transplant recipients. Diabetes, insulin resistance, and possibly hepatitis steatosis have been associated with a rapid progression to advanced fibrosis. The contribution of immunosuppression to the progression of hepatitis C virus remains an area of active study. Some studies point to antilymphocyte immunosuppressive agents as a potential cause.82 Liver biopsy is a useful tool in this situation. It allows monitoring of disease severity and progression and may distinguish recurrent hepatitis C virus disease from other causes of liver enzyme elevation.
The major concern with the recurrence of hepatitis C virus infection after liver transplant is allograft loss. Rates of patient and graft survival are reduced in infected patients compared with hepatitis C virus-negative patients.83,84 Prophylactic antiviral therapy has no current role in the management of hepatitis C virus disease. Those manifesting moderate to severe necroinflammation or mild to moderate fibrosis indicative of progressive disease should be treated.81,85
Sustained viral clearance with antiviral agents confers a graft survival benefit.
The combination of peg-interferon and weight-based ribavirin has been the standard of treatment but may be associated with increased rates of rejection.86,87 The sustained virologic response rates for hepatitis C virus range from 60% in genotypes 4, 5, and 6 after 48 weeks of treatment to 60% to 80% in genotypes 2 and 3 after 24 weeks, but only about 30% in genotype 1.88
Treatment with the newer agents, especially protease inhibitors, in genotype 1 (peg-interferon, ribavirin, and either telaprevir or boceprevir) has been evaluated. Success rates reaching 70% have been achieved.89 Adverse effects can be a major setback. Serious complications include severe anemia, renal dysfunction, increased risk of infection, and death.
Triple therapy should be carefully considered in liver transplant patients with genotype 1 hepatitis C virus.90 Significant drug-drug interactions are reported between hepatitis C virus protease inhibitors and immunosuppression regimens. Additional new oral direct- acting antivirals have been investigated. They bring promising advances in hepatitis C virus treatment and pave the way for interferon-free regimens with pangenotypic activity.
IMMUNIZATION
Immunization can decrease the risk of infectious complications in liver transplant recipients, as well as in close contacts and healthcare professionals.3
Influenza. Pretransplant influenza vaccine and posttransplant annual influenza vaccines are necessary.
Pneumococcal immunization should additionally be provided prior to transplant and repeated every 3 to 5 years thereafter.3,91
A number of other vaccinations should also be completed before transplant, including the hepatitis A and B vaccines and the tetanus/diphtheria/acellular pertussis vaccines. However, these vaccinations have not been shown to be detrimental to patients after transplant.91
Varicella and zoster vaccines should be given before liver transplant—zoster in patients over age 60, and varicella in patients with no immunity. Live vaccines, including varicella and zoster vaccines, are contraindicated after liver transplant.3
Human papillomavirus. The bivalent human papillomavirus vaccine can be given before transplant in females ages 9 to 26; the quadrivalent vaccine is beneficial in those ages 9 to 26 and in women under age 45.3,91
IMMUNOSUPPRESSION CARRIES RISK OF INFECTION
Most liver transplant patients require prolonged immunosuppressive therapy. This comes with an increased risk of new or recurrent infections, potentially causing death and significant morbidity.
Evaluation of existing risk factors, appropriate prophylaxis and immunization, timely diagnosis, and treatment of such infections are therefore essential steps for the successful management of liver transplant recipients.
- Fishman JA. Infection in solid-organ transplant recipients. N Engl J Med 2007; 357:2601–2614.
- Avery RK, Michaels MG; AST Infectious Diseases Community of Practice. Strategies for safe living after solid organ transplantation. Am J Transplant 2013; 13(suppl 4):304–310.
- Danziger-Isakov L, Kumar D; AST Infectious Diseases Community of Practice. Vaccination in solid organ transplantation. Am J Transplant 2013; 13(suppl 4):311–317.
- San Juan R, Aguado JM, Lumbreras C, et al; RESITRA Network, Spain. Incidence, clinical characteristics and risk factors of late infection in solid organ transplant recipients: data from the RESITRA study group. Am J Transplant 2007; 7:964–971.
- Ison MG, Grossi P; AST Infectious Diseases Community of Practice. Donor-derived infections in solid organ transplantation. Am J Transplant 2013; 13(suppl 4):22–30.
- Kim YJ, Kim SI, Wie SH, et al. Infectious complications in living-donor liver transplant recipients: a 9-year single-center experience. Transpl Infect Dis 2008; 10:316–324.
- Arnow PM. Infections following orthotopic liver transplantation. HPB Surg 1991; 3:221–233.
- Reid GE, Grim SA, Sankary H, Benedetti E, Oberholzer J, Clark NM. Early intra-abdominal infections associated with orthotopic liver transplantation. Transplantation 2009; 87:1706–1711.
- Said A, Safdar N, Lucey MR, et al. Infected bilomas in liver transplant recipients, incidence, risk factors and implications for prevention. Am J Transplant 2004; 4:574–582.
- Safdar N, Said A, Lucey MR, et al. Infected bilomas in liver transplant recipients: clinical features, optimal management, and risk factors for mortality. Clin Infect Dis 2004; 39:517–525.
- Niemczyk M, Leszczyniski P, Wyzgał J, Paczek L, Krawczyk M, Luczak M. Infections caused by Clostridium difficile in kidney or liver graft recipients. Ann Transplant 2005; 10:70–74.
- Albright JB, Bonatti H, Mendez J, et al. Early and late onset Clostridium difficile-associated colitis following liver transplantation. Transpl Int 2007; 20:856–866.
- Lee SO, Razonable RR. Current concepts on cytomegalovirus infection after liver transplantation. World J Hepatol 2010; 2:325–336.
- Ljungman P, Griffiths P, Paya C. Definitions of cytomegalovirus infection and disease in transplant recipients. Clin Infect Dis 2002; 34:1094–1097.
- Beam E, Razonable RR. Cytomegalovirus in solid organ transplantation: epidemiology, prevention, and treatment. Curr Infect Dis Rep 2012; 14:633–641.
- Bodro M, Sabé N, Lladó L, et al. Prophylaxis versus preemptive therapy for cytomegalovirus disease in high-risk liver transplant recipients. Liver Transpl 2012; 18:1093–1099.
- Weigand K, Schnitzler P, Schmidt J, et al. Cytomegalovirus infection after liver transplantation incidence, risks, and benefits of prophylaxis. Transplant Proc 2010; 42:2634–2641.
- Razonable RR, Humar A; AST Infectious Diseases Community of Practice. Cytomegalovirus in solid organ transplantation. Am J Transplant 2013; 13(suppl 4):93–106.
- Meije Y, Fortún J, Len Ó, et al; Spanish Network for Research on Infection in Transplantation (RESITRA) and the Spanish Network for Research on Infectious Diseases (REIPI). Prevention strategies for cytomegalovirus disease and long-term outcomes in the high-risk transplant patient (D+/R-): experience from the RESITRA-REIPI cohort. Transpl Infect Dis 2014; 16:387–396.
- Durand CM, Marr KA, Arnold CA, et al. Detection of cytomegalovirus DNA in plasma as an adjunct diagnostic for gastrointestinal tract disease in kidney and liver transplant recipients. Clin Infect Dis 2013; 57:1550–1559.
- Levitsky J, Singh N, Wagener MM, Stosor V, Abecassis M, Ison MG. A survey of CMV prevention strategies after liver transplantation. Am J Transplant 2008; 8:158–161.
- Marcelin JR, Beam E, Razonable RR. Cytomegalovirus infection in liver transplant recipients: updates on clinical management. World J Gastroenterol 2014; 20:10658–10667.
- Kalil AC, Freifeld AG, Lyden ER, Stoner JA. Valganciclovir for cytomegalovirus prevention in solid organ transplant patients: an evidence-based reassessment of safety and efficacy. PLoS One 2009; 4:e5512.
- Kalil AC, Mindru C, Botha JF, et al. Risk of cytomegalovirus disease in high-risk liver transplant recipients on valganciclovir prophylaxis: a systematic review and meta-analysis. Liver Transpl 2012; 18:1440–1447.
- Eid AJ, Arthurs SK, Deziel PJ, Wilhelm MP, Razonable RR. Emergence of drug-resistant cytomegalovirus in the era of valganciclovir prophylaxis: therapeutic implications and outcomes. Clin Transplant 2008; 22:162–170.
- Kumar D, Humar A. Cytomegalovirus prophylaxis: how long is enough? Nat Rev Nephrol 2010; 6:13–14.
- Lurain NS, Chou S. Antiviral drug resistance of human cytomegalovirus. Clin Microbiol Rev 2010; 23:689–712.
- Torres-Madriz G, Boucher HW. Immunocompromised hosts: perspectives in the treatment and prophylaxis of cytomegalovirus disease in solid-organ transplant recipients. Clin Infect Dis 2008; 47:702–711.
- Burra P, Buda A, Livi U, et al. Occurrence of post-transplant lymphoproliferative disorders among over thousand adult recipients: any role for hepatitis C infection? Eur J Gastroenterol Hepatol 2006; 18:1065–1070.
- Jain A, Nalesnik M, Reyes J, et al. Posttransplant lymphoproliferative disorders in liver transplantation: a 20-year experience. Ann Surg 2002; 236:429–437.
- Allen UD, Preiksaitis JK; AST Infectious Diseases Community of Practice. Epstein-Barr virus and posttransplant lymphoproliferative disorder in solid organ transplantation. Am J Transplant 2013; 13(suppl 4):107–120.
- Allen U, Preiksaitis J; AST Infectious Diseases Community of Practice. Epstein-Barr virus and posttransplant lymphoproliferative disorder in solid organ transplant recipients. Am J Transplant 2009; 9(suppl 4):S87–S96.
- Perrine SP, Hermine O, Small T, et al. A phase 1/2 trial of arginine butyrate and ganciclovir in patients with Epstein-Barr virus-associated lymphoid malignancies. Blood 2007; 109:2571–2578.
- Jagadeesh D, Woda BA, Draper J, Evens AM. Post transplant lymphoproliferative disorders: risk, classification, and therapeutic recommendations. Curr Treat Options Oncol 2012; 13:122–136.
- Opelz G, Daniel V, Naujokat C, Fickenscher H, Döhler B. Effect of cytomegalovirus prophylaxis with immunoglobulin or with antiviral drugs on post-transplant non-Hodgkin lymphoma: a multicentre retrospective analysis. Lancet Oncol 2007; 8:212–218.
- Nowalk AJ, Green M. Epstein-Barr virus–associated posttransplant lymphoproliferative disorder: strategies for prevention and cure. Liver Transpl 2010; 16(suppl S2):S54–S59.
- Pappas PG, Silveira FP; AST Infectious Diseases Community of Practice. Candida in solid organ transplant recipients. Am J Transplant 2009; 9(suppl 4):S173–S179.
- Singh N, Wagener MM, Marino IR, Gayowski T. Trends in invasive fungal infections in liver transplant recipients: correlation with evolution in transplantation practices. Transplantation 2002; 73:63–67.
- Miller R, Assi M; AST Infectious Diseases Community of Practice. Endemic fungal infections in solid organ transplantation. Am J Transplant 2013; 13(suppl 4):250–261.
- Fontana C, Gaziano R, Favaro M, Casalinuovo IA, Pistoia E, Di Francesco P. (1-3)-beta-D-glucan vs galactomannan antigen in diagnosing invasive fungal infections (IFIs). Open Microbiol J 2012; 6:70–73.
- Aydogan S, Kustimur S, Kalkancı A. Comparison of glucan and galactomannan tests with real-time PCR for diagnosis of invasive aspergillosis in a neutropenic rat model [Turkish]. Mikrobiyol Bul 2010; 44:441–452.
- Hadley S, Huckabee C, Pappas PG, et al. Outcomes of antifungal prophylaxis in high-risk liver transplant recipients. Transpl Infect Dis 2009; 11:40–48.
- Pappas PG, Kauffman CA, Andes D, et al; Infectious Diseases Society of America. Clinical practice guidelines for the management of candidiasis: 2009 update by the Infectious Diseases Society of America. Clin Infect Dis 2009; 48:503–535.
- Person AK, Kontoyiannis DP, Alexander BD. Fungal infections in transplant and oncology patients. Infect Dis Clin North Am 2010; 24:439–459.
- Van Hal SJ, Marriott DJE, Chen SCA, et al; Australian Candidaemia Study. Candidemia following solid organ transplantation in the era of antifungal prophylaxis: the Australian experience. Transpl Infect Dis 2009; 11:122–127.
- Singh N. Fungal infections in the recipients of solid organ transplantation. Infect Dis Clin North Am 2003; 17:113–134,
- Liu X, Ling Z, Li L, Ruan B. Invasive fungal infections in liver transplantation. Int J Infect Dis 2011; 15:e298–e304.
- Raghuram A, Restrepo A, Safadjou S, et al. Invasive fungal infections following liver transplantation: incidence, risk factors, survival, and impact of fluconazole-resistant Candida parapsilosis (2003-2007). Liver Transpl 2012; 18:1100–1109.
- De Pauw B, Walsh TJ, Donnelly JP, et al; European Organization for Research and Treatment of Cancer/Invasive Fungal Infections Cooperative Group; National Institute of Allergy and Infectious Diseases Mycoses Study Group (EORTC/MSG) Consensus Group. Revised definitions of invasive fungal disease from the European Organization for Research and Treatment of Cancer/Invasive Fungal Infections Cooperative Group and the National Institute of Allergy and Infectious Diseases Mycoses Study Group (EORTC/MSG) Consensus Group. Clin Infect Dis 2008; 46:1813–1821.
- Moreno A, Cervera C, Gavaldá J, et al. Bloodstream infections among transplant recipients: results of a nationwide surveillance in Spain. Am J Transplant 2007; 7:2579–2586.
- Cruciani M, Mengoli C, Malena M, Bosco O, Serpelloni G, Grossi P. Antifungal prophylaxis in liver transplant patients: a systematic review and meta-analysis. Liver Transpl 2006; 12:850–858.
- Singh N, Husain S; AST Infectious Diseases Community of Practice. Invasive aspergillosis in solid organ transplant recipients. Am J Transplant 2009; 9(suppl 4):S180–S191.
- Fortún J, Martín-Dávila P, Alvarez ME, et al. False-positive results of Aspergillus galactomannan antigenemia in liver transplant recipients. Transplantation 2009; 87:256–260.
- Cherian T, Giakoustidis A, Yokoyama S, et al. Treatment of refractory cerebral aspergillosis in a liver transplant recipient with voriconazole: case report and review of the literature. Exp Clin Transplant 2012; 10:482–486.
- Luong ML, Hosseini-Moghaddam SM, Singer LG, et al. Risk factors for voriconazole hepatotoxicity at 12 weeks in lung transplant recipients. Am J Transplant 2012; 12:1929–1935.
- Neofytos D, Fishman JA, Horn D, et al. Epidemiology and outcome of invasive fungal infections in solid organ transplant recipients. Transpl Infect Dis 2010; 12:220–229.
- Martin SI, Fishman JA; AST Infectious Diseases Community of Practice. Pneumocystis pneumonia in solid organ transplant recipients. Am J Transplant 2009; 9(suppl 4):S227–S233.
- Levine SJ, Masur H, Gill VJ, et al. Effect of aerosolized pentamidine prophylaxis on the diagnosis of Pneumocystis carinii pneumonia by induced sputum examination in patients infected with the human immunodeficiency virus. Am Rev Respir Dis 1991; 144:760–764.
- Rodriguez M, Sifri CD, Fishman JA. Failure of low-dose atovaquone prophylaxis against Pneumocystis jiroveci infection in transplant recipients. Clin Infect Dis 2004; 38:e76–e78.
- Crans CA Jr, Boiselle PM. Imaging features of Pneumocystis carinii pneumonia. Crit Rev Diagn Imaging 1999; 40:251–284.
- Onishi A, Sugiyama D, Kogata Y, et al. Diagnostic accuracy of serum 1,3-beta-D-glucan for Pneumocystis jiroveci pneumonia, invasive candidiasis, and invasive aspergillosis: systematic review and meta-analysis. J Clin Microbiol 2012; 50:7–15.
- Held J, Koch MS, Reischl U, Danner T, Serr A. Serum (1→3)-ß-D-glucan measurement as an early indicator of Pneumocystis jirovecii pneumonia and evaluation of its prognostic value. Clin Microbiol Infect 2011; 17:595–602.
- Fishman JA. Prevention of infection caused by Pneumocystis carinii in transplant recipients. Clin Infect Dis 2001; 33:1397–1405.
- Colby C, McAfee S, Sackstein R, Finkelstein D, Fishman J, Spitzer T. A prospective randomized trial comparing the toxicity and safety of atovaquone with trimethoprim/sulfamethoxazole as Pneumocystis carinii pneumonia prophylaxis following autologous peripheral blood stem cell transplantation. Bone Marrow Transplant 1999; 24:897–902.
- Subramanian A, Dorman S; AST Infectious Diseases Community of Practice. Mycobacterium tuberculosis in solid organ transplant recipients. Am J Transplant 2009; 9(suppl 4):S57–S62.
- Subramanian AK, Morris MI; AST Infectious Diseases Community of Practice. Mycobacterium tuberculosis infections in solid organ transplantation. Am J Transplant 2013; 13(suppl 4):68–76.
- Horne DJ, Narita M, Spitters CL, Parimi S, Dodson S, Limaye AP. Challenging issues in tuberculosis in solid organ transplantation. Clin Infect Dis 2013; 57:1473–1482.
- Holty JE, Gould MK, Meinke L, Keeffe EB, Ruoss SJ. Tuberculosis in liver transplant recipients: a systematic review and meta-analysis of individual patient data. Liver Transpl 2009; 15:894–906.
- Jafri SM, Singal AG, Kaul D, Fontana RJ. Detection and management of latent tuberculosis in liver transplant patients. Liver Transpl 2011; 17:306–314.
- Fábrega E, Sampedro B, Cabezas J, et al. Chemoprophylaxis with isoniazid in liver transplant recipients. Liver Transpl 2012; 18:1110–1117.
- Aguado JM, Torre-Cisneros J, Fortún J, et al. Tuberculosis in solid-organ transplant recipients: consensus statement of the group for the study of infection in transplant recipients (GESITRA) of the Spanish Society of Infectious Diseases and Clinical Microbiology. Clin Infect Dis 2009; 48:1276–1284.
- Yehia BR, Blumberg EA. Mycobacterium tuberculosis infection in liver transplantation. Liver Transpl 2010; 16:1129–1135.
- Katz LH, Paul M, Guy DG, Tur-Kaspa R. Prevention of recurrent hepatitis B virus infection after liver transplantation: hepatitis B immunoglobulin, antiviral drugs, or both? Systematic review and meta-analysis. Transpl Infect Dis 2010; 12:292–308.
- Jiang L, Jiang LS, Cheng NS, Yan LN. Current prophylactic strategies against hepatitis B virus recurrence after liver transplantation. World J Gastroenterol 2009; 15:2489–2499.
- Riediger C, Berberat PO, Sauer P, et al. Prophylaxis and treatment of recurrent viral hepatitis after liver transplantation. Nephrol Dial Transplant 2007; 22(suppl 8):viii37–viii46.
- Cholongitas E, Vasiliadis T, Antoniadis N, Goulis I, Papanikolaou V, Akriviadis E. Hepatitis B prophylaxis post liver transplantation with newer nucleos(t)ide analogues after hepatitis B immunoglobulin discontinuation. Transpl Infect Dis 2012; 14:479–487.
- Fox AN, Terrault NA. Individualizing hepatitis B infection prophylaxis in liver transplant recipients. J Hepatol 2011; 55:507–509.
- Fox AN, Terrault NA. The option of HBIG-free prophylaxis against recurrent HBV. J Hepatol 2012; 56:1189–1197.
- Wesdorp DJ, Knoester M, Braat AE, et al. Nucleoside plus nucleotide analogs and cessation of hepatitis B immunoglobulin after liver transplantation in chronic hepatitis B is safe and effective. J Clin Virol 2013; 58:67–73.
- Terrault NA, Berenguer M. Treating hepatitis C infection in liver transplant recipients. Liver Transpl 2006; 12:1192–1204.
- Ciria R, Pleguezuelo M, Khorsandi SE, et al. Strategies to reduce hepatitis C virus recurrence after liver transplantation. World J Hepatol 2013; 5:237–250.
- Issa NC, Fishman JA. Infectious complications of antilymphocyte therapies in solid organ transplantation. Clin Infect Dis 2009; 48:772–786.
- Kalambokis G, Manousou P, Samonakis D, et al. Clinical outcome of HCV-related graft cirrhosis and prognostic value of hepatic venous pressure gradient. Transpl Int 2009; 22:172–181.
- Neumann UP, Berg T, Bahra M, et al. Long-term outcome of liver transplants for chronic hepatitis C: a 10-year follow-up. Transplantation 2004; 77:226–231.
- Wiesner RH, Sorrell M, Villamil F; International Liver Transplantation Society Expert Panel. Report of the first International Liver Transplantation Society expert panel consensus conference on liver transplantation and hepatitis C. Liver Transpl 2003; 9:S1–S9.
- Dinges S, Morard I, Heim M, et al; Swiss Association for the Study of the Liver (SASL 17). Pegylated interferon-alpha2a/ribavirin treatment of recurrent hepatitis C after liver transplantation. Transpl Infect Dis 2009; 11:33–39.
- Veldt BJ, Poterucha JJ, Watt KD, et al. Impact of pegylated interferon and ribavirin treatment on graft survival in liver transplant patients with recurrent hepatitis C infection. Am J Transplant 2008; 8:2426–2433.
- Faisal N, Yoshida EM, Bilodeau M, et al. Protease inhibitor-based triple therapy is highly effective for hepatitis C recurrence after liver transplant: a multicenter experience. Ann Hepatol 2014; 13:525–532.
- Mariño Z, van Bömmel F, Forns X, Berg T. New concepts of sofosbuvir-based treatment regimens in patients with hepatitis C. Gut 2014; 63:207–215.
- Coilly A, Roche B, Dumortier J, et al. Safety and efficacy of protease inhibitors to treat hepatitis C after liver transplantation: a multicenter experience. J Hepatol 2014; 60:78–86.
- Lucey MR, Terrault N, Ojo L, et al. Long-term management of the successful adult liver transplant: 2012 practice guideline by the American Association for the Study of Liver Diseases and the American Society of Transplantation. Liver Transpl 2013; 19:3–26.
- Fishman JA. Infection in solid-organ transplant recipients. N Engl J Med 2007; 357:2601–2614.
- Avery RK, Michaels MG; AST Infectious Diseases Community of Practice. Strategies for safe living after solid organ transplantation. Am J Transplant 2013; 13(suppl 4):304–310.
- Danziger-Isakov L, Kumar D; AST Infectious Diseases Community of Practice. Vaccination in solid organ transplantation. Am J Transplant 2013; 13(suppl 4):311–317.
- San Juan R, Aguado JM, Lumbreras C, et al; RESITRA Network, Spain. Incidence, clinical characteristics and risk factors of late infection in solid organ transplant recipients: data from the RESITRA study group. Am J Transplant 2007; 7:964–971.
- Ison MG, Grossi P; AST Infectious Diseases Community of Practice. Donor-derived infections in solid organ transplantation. Am J Transplant 2013; 13(suppl 4):22–30.
- Kim YJ, Kim SI, Wie SH, et al. Infectious complications in living-donor liver transplant recipients: a 9-year single-center experience. Transpl Infect Dis 2008; 10:316–324.
- Arnow PM. Infections following orthotopic liver transplantation. HPB Surg 1991; 3:221–233.
- Reid GE, Grim SA, Sankary H, Benedetti E, Oberholzer J, Clark NM. Early intra-abdominal infections associated with orthotopic liver transplantation. Transplantation 2009; 87:1706–1711.
- Said A, Safdar N, Lucey MR, et al. Infected bilomas in liver transplant recipients, incidence, risk factors and implications for prevention. Am J Transplant 2004; 4:574–582.
- Safdar N, Said A, Lucey MR, et al. Infected bilomas in liver transplant recipients: clinical features, optimal management, and risk factors for mortality. Clin Infect Dis 2004; 39:517–525.
- Niemczyk M, Leszczyniski P, Wyzgał J, Paczek L, Krawczyk M, Luczak M. Infections caused by Clostridium difficile in kidney or liver graft recipients. Ann Transplant 2005; 10:70–74.
- Albright JB, Bonatti H, Mendez J, et al. Early and late onset Clostridium difficile-associated colitis following liver transplantation. Transpl Int 2007; 20:856–866.
- Lee SO, Razonable RR. Current concepts on cytomegalovirus infection after liver transplantation. World J Hepatol 2010; 2:325–336.
- Ljungman P, Griffiths P, Paya C. Definitions of cytomegalovirus infection and disease in transplant recipients. Clin Infect Dis 2002; 34:1094–1097.
- Beam E, Razonable RR. Cytomegalovirus in solid organ transplantation: epidemiology, prevention, and treatment. Curr Infect Dis Rep 2012; 14:633–641.
- Bodro M, Sabé N, Lladó L, et al. Prophylaxis versus preemptive therapy for cytomegalovirus disease in high-risk liver transplant recipients. Liver Transpl 2012; 18:1093–1099.
- Weigand K, Schnitzler P, Schmidt J, et al. Cytomegalovirus infection after liver transplantation incidence, risks, and benefits of prophylaxis. Transplant Proc 2010; 42:2634–2641.
- Razonable RR, Humar A; AST Infectious Diseases Community of Practice. Cytomegalovirus in solid organ transplantation. Am J Transplant 2013; 13(suppl 4):93–106.
- Meije Y, Fortún J, Len Ó, et al; Spanish Network for Research on Infection in Transplantation (RESITRA) and the Spanish Network for Research on Infectious Diseases (REIPI). Prevention strategies for cytomegalovirus disease and long-term outcomes in the high-risk transplant patient (D+/R-): experience from the RESITRA-REIPI cohort. Transpl Infect Dis 2014; 16:387–396.
- Durand CM, Marr KA, Arnold CA, et al. Detection of cytomegalovirus DNA in plasma as an adjunct diagnostic for gastrointestinal tract disease in kidney and liver transplant recipients. Clin Infect Dis 2013; 57:1550–1559.
- Levitsky J, Singh N, Wagener MM, Stosor V, Abecassis M, Ison MG. A survey of CMV prevention strategies after liver transplantation. Am J Transplant 2008; 8:158–161.
- Marcelin JR, Beam E, Razonable RR. Cytomegalovirus infection in liver transplant recipients: updates on clinical management. World J Gastroenterol 2014; 20:10658–10667.
- Kalil AC, Freifeld AG, Lyden ER, Stoner JA. Valganciclovir for cytomegalovirus prevention in solid organ transplant patients: an evidence-based reassessment of safety and efficacy. PLoS One 2009; 4:e5512.
- Kalil AC, Mindru C, Botha JF, et al. Risk of cytomegalovirus disease in high-risk liver transplant recipients on valganciclovir prophylaxis: a systematic review and meta-analysis. Liver Transpl 2012; 18:1440–1447.
- Eid AJ, Arthurs SK, Deziel PJ, Wilhelm MP, Razonable RR. Emergence of drug-resistant cytomegalovirus in the era of valganciclovir prophylaxis: therapeutic implications and outcomes. Clin Transplant 2008; 22:162–170.
- Kumar D, Humar A. Cytomegalovirus prophylaxis: how long is enough? Nat Rev Nephrol 2010; 6:13–14.
- Lurain NS, Chou S. Antiviral drug resistance of human cytomegalovirus. Clin Microbiol Rev 2010; 23:689–712.
- Torres-Madriz G, Boucher HW. Immunocompromised hosts: perspectives in the treatment and prophylaxis of cytomegalovirus disease in solid-organ transplant recipients. Clin Infect Dis 2008; 47:702–711.
- Burra P, Buda A, Livi U, et al. Occurrence of post-transplant lymphoproliferative disorders among over thousand adult recipients: any role for hepatitis C infection? Eur J Gastroenterol Hepatol 2006; 18:1065–1070.
- Jain A, Nalesnik M, Reyes J, et al. Posttransplant lymphoproliferative disorders in liver transplantation: a 20-year experience. Ann Surg 2002; 236:429–437.
- Allen UD, Preiksaitis JK; AST Infectious Diseases Community of Practice. Epstein-Barr virus and posttransplant lymphoproliferative disorder in solid organ transplantation. Am J Transplant 2013; 13(suppl 4):107–120.
- Allen U, Preiksaitis J; AST Infectious Diseases Community of Practice. Epstein-Barr virus and posttransplant lymphoproliferative disorder in solid organ transplant recipients. Am J Transplant 2009; 9(suppl 4):S87–S96.
- Perrine SP, Hermine O, Small T, et al. A phase 1/2 trial of arginine butyrate and ganciclovir in patients with Epstein-Barr virus-associated lymphoid malignancies. Blood 2007; 109:2571–2578.
- Jagadeesh D, Woda BA, Draper J, Evens AM. Post transplant lymphoproliferative disorders: risk, classification, and therapeutic recommendations. Curr Treat Options Oncol 2012; 13:122–136.
- Opelz G, Daniel V, Naujokat C, Fickenscher H, Döhler B. Effect of cytomegalovirus prophylaxis with immunoglobulin or with antiviral drugs on post-transplant non-Hodgkin lymphoma: a multicentre retrospective analysis. Lancet Oncol 2007; 8:212–218.
- Nowalk AJ, Green M. Epstein-Barr virus–associated posttransplant lymphoproliferative disorder: strategies for prevention and cure. Liver Transpl 2010; 16(suppl S2):S54–S59.
- Pappas PG, Silveira FP; AST Infectious Diseases Community of Practice. Candida in solid organ transplant recipients. Am J Transplant 2009; 9(suppl 4):S173–S179.
- Singh N, Wagener MM, Marino IR, Gayowski T. Trends in invasive fungal infections in liver transplant recipients: correlation with evolution in transplantation practices. Transplantation 2002; 73:63–67.
- Miller R, Assi M; AST Infectious Diseases Community of Practice. Endemic fungal infections in solid organ transplantation. Am J Transplant 2013; 13(suppl 4):250–261.
- Fontana C, Gaziano R, Favaro M, Casalinuovo IA, Pistoia E, Di Francesco P. (1-3)-beta-D-glucan vs galactomannan antigen in diagnosing invasive fungal infections (IFIs). Open Microbiol J 2012; 6:70–73.
- Aydogan S, Kustimur S, Kalkancı A. Comparison of glucan and galactomannan tests with real-time PCR for diagnosis of invasive aspergillosis in a neutropenic rat model [Turkish]. Mikrobiyol Bul 2010; 44:441–452.
- Hadley S, Huckabee C, Pappas PG, et al. Outcomes of antifungal prophylaxis in high-risk liver transplant recipients. Transpl Infect Dis 2009; 11:40–48.
- Pappas PG, Kauffman CA, Andes D, et al; Infectious Diseases Society of America. Clinical practice guidelines for the management of candidiasis: 2009 update by the Infectious Diseases Society of America. Clin Infect Dis 2009; 48:503–535.
- Person AK, Kontoyiannis DP, Alexander BD. Fungal infections in transplant and oncology patients. Infect Dis Clin North Am 2010; 24:439–459.
- Van Hal SJ, Marriott DJE, Chen SCA, et al; Australian Candidaemia Study. Candidemia following solid organ transplantation in the era of antifungal prophylaxis: the Australian experience. Transpl Infect Dis 2009; 11:122–127.
- Singh N. Fungal infections in the recipients of solid organ transplantation. Infect Dis Clin North Am 2003; 17:113–134,
- Liu X, Ling Z, Li L, Ruan B. Invasive fungal infections in liver transplantation. Int J Infect Dis 2011; 15:e298–e304.
- Raghuram A, Restrepo A, Safadjou S, et al. Invasive fungal infections following liver transplantation: incidence, risk factors, survival, and impact of fluconazole-resistant Candida parapsilosis (2003-2007). Liver Transpl 2012; 18:1100–1109.
- De Pauw B, Walsh TJ, Donnelly JP, et al; European Organization for Research and Treatment of Cancer/Invasive Fungal Infections Cooperative Group; National Institute of Allergy and Infectious Diseases Mycoses Study Group (EORTC/MSG) Consensus Group. Revised definitions of invasive fungal disease from the European Organization for Research and Treatment of Cancer/Invasive Fungal Infections Cooperative Group and the National Institute of Allergy and Infectious Diseases Mycoses Study Group (EORTC/MSG) Consensus Group. Clin Infect Dis 2008; 46:1813–1821.
- Moreno A, Cervera C, Gavaldá J, et al. Bloodstream infections among transplant recipients: results of a nationwide surveillance in Spain. Am J Transplant 2007; 7:2579–2586.
- Cruciani M, Mengoli C, Malena M, Bosco O, Serpelloni G, Grossi P. Antifungal prophylaxis in liver transplant patients: a systematic review and meta-analysis. Liver Transpl 2006; 12:850–858.
- Singh N, Husain S; AST Infectious Diseases Community of Practice. Invasive aspergillosis in solid organ transplant recipients. Am J Transplant 2009; 9(suppl 4):S180–S191.
- Fortún J, Martín-Dávila P, Alvarez ME, et al. False-positive results of Aspergillus galactomannan antigenemia in liver transplant recipients. Transplantation 2009; 87:256–260.
- Cherian T, Giakoustidis A, Yokoyama S, et al. Treatment of refractory cerebral aspergillosis in a liver transplant recipient with voriconazole: case report and review of the literature. Exp Clin Transplant 2012; 10:482–486.
- Luong ML, Hosseini-Moghaddam SM, Singer LG, et al. Risk factors for voriconazole hepatotoxicity at 12 weeks in lung transplant recipients. Am J Transplant 2012; 12:1929–1935.
- Neofytos D, Fishman JA, Horn D, et al. Epidemiology and outcome of invasive fungal infections in solid organ transplant recipients. Transpl Infect Dis 2010; 12:220–229.
- Martin SI, Fishman JA; AST Infectious Diseases Community of Practice. Pneumocystis pneumonia in solid organ transplant recipients. Am J Transplant 2009; 9(suppl 4):S227–S233.
- Levine SJ, Masur H, Gill VJ, et al. Effect of aerosolized pentamidine prophylaxis on the diagnosis of Pneumocystis carinii pneumonia by induced sputum examination in patients infected with the human immunodeficiency virus. Am Rev Respir Dis 1991; 144:760–764.
- Rodriguez M, Sifri CD, Fishman JA. Failure of low-dose atovaquone prophylaxis against Pneumocystis jiroveci infection in transplant recipients. Clin Infect Dis 2004; 38:e76–e78.
- Crans CA Jr, Boiselle PM. Imaging features of Pneumocystis carinii pneumonia. Crit Rev Diagn Imaging 1999; 40:251–284.
- Onishi A, Sugiyama D, Kogata Y, et al. Diagnostic accuracy of serum 1,3-beta-D-glucan for Pneumocystis jiroveci pneumonia, invasive candidiasis, and invasive aspergillosis: systematic review and meta-analysis. J Clin Microbiol 2012; 50:7–15.
- Held J, Koch MS, Reischl U, Danner T, Serr A. Serum (1→3)-ß-D-glucan measurement as an early indicator of Pneumocystis jirovecii pneumonia and evaluation of its prognostic value. Clin Microbiol Infect 2011; 17:595–602.
- Fishman JA. Prevention of infection caused by Pneumocystis carinii in transplant recipients. Clin Infect Dis 2001; 33:1397–1405.
- Colby C, McAfee S, Sackstein R, Finkelstein D, Fishman J, Spitzer T. A prospective randomized trial comparing the toxicity and safety of atovaquone with trimethoprim/sulfamethoxazole as Pneumocystis carinii pneumonia prophylaxis following autologous peripheral blood stem cell transplantation. Bone Marrow Transplant 1999; 24:897–902.
- Subramanian A, Dorman S; AST Infectious Diseases Community of Practice. Mycobacterium tuberculosis in solid organ transplant recipients. Am J Transplant 2009; 9(suppl 4):S57–S62.
- Subramanian AK, Morris MI; AST Infectious Diseases Community of Practice. Mycobacterium tuberculosis infections in solid organ transplantation. Am J Transplant 2013; 13(suppl 4):68–76.
- Horne DJ, Narita M, Spitters CL, Parimi S, Dodson S, Limaye AP. Challenging issues in tuberculosis in solid organ transplantation. Clin Infect Dis 2013; 57:1473–1482.
- Holty JE, Gould MK, Meinke L, Keeffe EB, Ruoss SJ. Tuberculosis in liver transplant recipients: a systematic review and meta-analysis of individual patient data. Liver Transpl 2009; 15:894–906.
- Jafri SM, Singal AG, Kaul D, Fontana RJ. Detection and management of latent tuberculosis in liver transplant patients. Liver Transpl 2011; 17:306–314.
- Fábrega E, Sampedro B, Cabezas J, et al. Chemoprophylaxis with isoniazid in liver transplant recipients. Liver Transpl 2012; 18:1110–1117.
- Aguado JM, Torre-Cisneros J, Fortún J, et al. Tuberculosis in solid-organ transplant recipients: consensus statement of the group for the study of infection in transplant recipients (GESITRA) of the Spanish Society of Infectious Diseases and Clinical Microbiology. Clin Infect Dis 2009; 48:1276–1284.
- Yehia BR, Blumberg EA. Mycobacterium tuberculosis infection in liver transplantation. Liver Transpl 2010; 16:1129–1135.
- Katz LH, Paul M, Guy DG, Tur-Kaspa R. Prevention of recurrent hepatitis B virus infection after liver transplantation: hepatitis B immunoglobulin, antiviral drugs, or both? Systematic review and meta-analysis. Transpl Infect Dis 2010; 12:292–308.
- Jiang L, Jiang LS, Cheng NS, Yan LN. Current prophylactic strategies against hepatitis B virus recurrence after liver transplantation. World J Gastroenterol 2009; 15:2489–2499.
- Riediger C, Berberat PO, Sauer P, et al. Prophylaxis and treatment of recurrent viral hepatitis after liver transplantation. Nephrol Dial Transplant 2007; 22(suppl 8):viii37–viii46.
- Cholongitas E, Vasiliadis T, Antoniadis N, Goulis I, Papanikolaou V, Akriviadis E. Hepatitis B prophylaxis post liver transplantation with newer nucleos(t)ide analogues after hepatitis B immunoglobulin discontinuation. Transpl Infect Dis 2012; 14:479–487.
- Fox AN, Terrault NA. Individualizing hepatitis B infection prophylaxis in liver transplant recipients. J Hepatol 2011; 55:507–509.
- Fox AN, Terrault NA. The option of HBIG-free prophylaxis against recurrent HBV. J Hepatol 2012; 56:1189–1197.
- Wesdorp DJ, Knoester M, Braat AE, et al. Nucleoside plus nucleotide analogs and cessation of hepatitis B immunoglobulin after liver transplantation in chronic hepatitis B is safe and effective. J Clin Virol 2013; 58:67–73.
- Terrault NA, Berenguer M. Treating hepatitis C infection in liver transplant recipients. Liver Transpl 2006; 12:1192–1204.
- Ciria R, Pleguezuelo M, Khorsandi SE, et al. Strategies to reduce hepatitis C virus recurrence after liver transplantation. World J Hepatol 2013; 5:237–250.
- Issa NC, Fishman JA. Infectious complications of antilymphocyte therapies in solid organ transplantation. Clin Infect Dis 2009; 48:772–786.
- Kalambokis G, Manousou P, Samonakis D, et al. Clinical outcome of HCV-related graft cirrhosis and prognostic value of hepatic venous pressure gradient. Transpl Int 2009; 22:172–181.
- Neumann UP, Berg T, Bahra M, et al. Long-term outcome of liver transplants for chronic hepatitis C: a 10-year follow-up. Transplantation 2004; 77:226–231.
- Wiesner RH, Sorrell M, Villamil F; International Liver Transplantation Society Expert Panel. Report of the first International Liver Transplantation Society expert panel consensus conference on liver transplantation and hepatitis C. Liver Transpl 2003; 9:S1–S9.
- Dinges S, Morard I, Heim M, et al; Swiss Association for the Study of the Liver (SASL 17). Pegylated interferon-alpha2a/ribavirin treatment of recurrent hepatitis C after liver transplantation. Transpl Infect Dis 2009; 11:33–39.
- Veldt BJ, Poterucha JJ, Watt KD, et al. Impact of pegylated interferon and ribavirin treatment on graft survival in liver transplant patients with recurrent hepatitis C infection. Am J Transplant 2008; 8:2426–2433.
- Faisal N, Yoshida EM, Bilodeau M, et al. Protease inhibitor-based triple therapy is highly effective for hepatitis C recurrence after liver transplant: a multicenter experience. Ann Hepatol 2014; 13:525–532.
- Mariño Z, van Bömmel F, Forns X, Berg T. New concepts of sofosbuvir-based treatment regimens in patients with hepatitis C. Gut 2014; 63:207–215.
- Coilly A, Roche B, Dumortier J, et al. Safety and efficacy of protease inhibitors to treat hepatitis C after liver transplantation: a multicenter experience. J Hepatol 2014; 60:78–86.
- Lucey MR, Terrault N, Ojo L, et al. Long-term management of the successful adult liver transplant: 2012 practice guideline by the American Association for the Study of Liver Diseases and the American Society of Transplantation. Liver Transpl 2013; 19:3–26.
KEY POINTS
- After liver transplant, the risk of infection and the likely causal organisms vary with the patient’s state of immunosuppression and the time of infection.
- Recurrent or newly acquired infections may jeopardize the survival of the graft and the recipient.
- Because infections with viruses, fungi, and atypical pathogens can alter the prognosis, they need to be prevented and carefully managed.
- An ongoing assessment of each patient’s risk of infection allows the clinician to constantly and efficiently adapt immunosuppressive, prophylactic, and therapeutic strategies.
Genetics and hepatitis C: It’s good to be ‘CC’
What a difference a single nucleotide can make! The human genome contains more than 3 billion base pairs. Yet having a different nucleotide in only one pair can make a big difference in how we respond to a disease or its treatment.
Specifically, in hepatitis C virus infection, people born with the nucleotide cytosine (C) at location rs12979860 in both alleles of the gene that codes for interleukin 28B (the IL28B CC genotype) can count themselves luckier than those born with thymine (T) in this location in one of their alleles (the CT genotype) or both of their alleles (the TT genotype). Those with the CC genotype are more likely to clear the virus spontaneously, and even if the infection persists, it is less likely to progress to liver cancer and more likely to respond to treatment with interferon.
Here, we review the IL28B polymorphism and its implications in treating hepatitis C.
GENETIC POLYMORPHISM AND HUMAN DISEASE
Of the 3 billion base pairs of nucleotides, fewer than 1% differ between individuals, but this 1% is responsible for the diversity of human beings. Differences in genetic sequences among individuals are called genetic polymorphisms. A single-nucleotide polymorphism is a DNA sequence variation that occurs in a single nucleotide in the genome. For example, two sequenced DNA fragments from different individuals, AAGCCTA and AAGCTTA, contain a difference in a single nucleotide.
Genetic variations such as these underlie some of the differences in our susceptibility to disease, the severity of illness we develop, and our response to treatments. Therefore, identifying genetic polymorphisms may shed light on biologic pathways involved in diseases and may uncover new targets for therapy.1
Genome-wide association studies have looked at hundreds of thousands of single-nucleotide polymorphisms to try to identify most of the common genetic differences among people and relate them to common chronic diseases such as coronary artery disease,2 type 2 diabetes,3 stroke,4 breast cancer,5 rheumatoid arthritis,6 Alzheimer disease,7 and, more recently, hepatitis C virus infection.8
HEPATITIS C VIRUS: A MAJOR CAUSE OF LIVER DISEASE
Hepatitis C virus infection is a major cause of chronic liver disease and hepatocellular carcinoma and has become the most common indication for liver transplantation in the United States.9
This virus has six distinct genotypes throughout the world, with multiple subtypes in each genotype. (A genotype is a classification of a virus based on its RNA.9) In this review, we will focus on genotype 1; hence, “hepatitis C virus” will refer to hepatitis C virus genotype 1.
Our knowledge of the biology, pathogenesis, and treatment of hepatitis C has been advancing. Originally, fewer than 50% of patients responded to therapy with the combination of pegylated interferon and ribavirin,10,11 but since 2011 the response rate has increased to approximately 70% with the approval of the protease inhibitors telaprevir and boceprevir, used in combination with pegylated interferon and ribavirin.12–15
Unfortunately, interferon-based treatment is often complicated by side effects such as fatigue, influenza-like symptoms, hematologic abnormalities, and neuropsychiatric symptoms. An accurate way to predict response would help patients make informed decisions about antiviral treatment, taking into account the risk and possible benefit for individual patients.
GENETIC POLYMORPHISM AND HEPATITIS C VIRUS INFECTION
Genome-wide association studies have identified single-nucleotide polymorphisms in the IL28B gene that are associated with differences in response to hepatitis C treatment.8
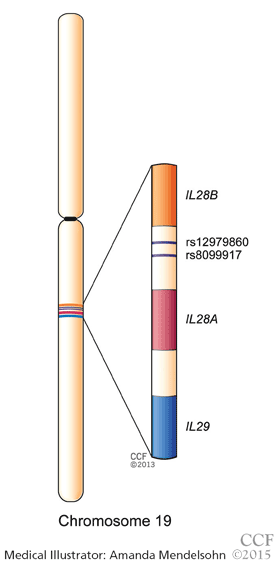
Studying 565,759 polymorphisms in 1,137 patients, researchers at Duke University identified a single-nucleotide polymorphism at location rs12979860 in IL28B (Figure 1) that was strongly associated with response to combination therapy with pegylated interferon and ribavirin.8 The chance of cure with this standard treatment is twice as high in patients who are homozygous for cytosine in this location (the CC genotype) than in those who are heterozygous (CT) or homozygous for thymine in this location (the TT genotype) (Table 1).

Adding one of the new protease inhibitors, telaprevir or boceprevir, to the standard hepatitis C treatment substantially improves the cure rates in all three IL28B genotypes, but especially in people with CT or TT, in whom the response rate almost triples with the addition of one of these drugs. Those with the CC genotype (who are more likely to be cured with pegylated interferon and ribavirin alone) also achieve an increase (although minimal) in cure rates when a protease inhibitor is included in the regimen (TABLE 1).13–15 Thus, it remains unclear if adding a protease inhibitor to pegylated interferon plus ribavirin in patients with the IL28B CC genotype translates into added effectiveness worth the additional cost of the protease inhibitor in previously untreated patients.
Additionally, the effect of the IL28B genotype on telaprevir-based triple therapy has been disputed in more recent studies. In a subgroup analysis of the results of a trial that evaluated telaprevir in the treatment of hepatitis C, researchers found that sustained virologic response rates were significantly higher in the telaprevir group, and this was similar across the different IL28B polymorphisms.16
The favorable IL28B CC genotype is associated with higher rates of rapid virologic response to antiviral therapy.13–15 Of note, almost all patients who achieve a rapid virologic response do well, with a high rate of sustained virologic response even after a shorter duration of therapy (24 vs 48 weeks). Therefore, in addition to predicting response to interferon before starting treatment, the IL28B CC genotype may also identify patients who need only a shorter duration of therapy.
Interestingly, the C allele is much more frequent in white than in African American populations, an important observation that explains the racial difference in response to hepatitis C therapy.8
Two other research groups, from Asia and Australia, performed independent genome-wide association studies that identified different single-nucleotide polymorphisms (eg, rs8099917) in the same IL28B gene as predictors of response to treatment in patients with hepatitis C virus infection.17,18 These findings may be explained by linkage disequilibrium, which means that these single-nucleotide polymorphisms are found more frequently together in the same patient due to their proximity to each other. In this review, we will focus on the rs12979860 polymorphism; hence “IL28B genotype” will refer to the single-nucleotide polymorphism at rs12979860, unless otherwise specified.
The favorable CC genotype is less common in African Americans than in patients of other ethnicities.19 Moreover, although IL28B CC is associated with a better response rate to interferon-based antiviral therapy across all ethnicities, those of African American descent with the CC genotype are less likely to achieve a sustained virologic response than white or Hispanic Americans.8
BIOLOGIC ASSOCIATION: IL28B POLYMORPHISM AND HEPATITIS C
The interferon lambda family consists of three cytokines:
- Interleukin 29 (interferon lambda 1)
- Interleukin 28A (interferon lambda 2)
- Interleukin 28B (interferon lambda 3).
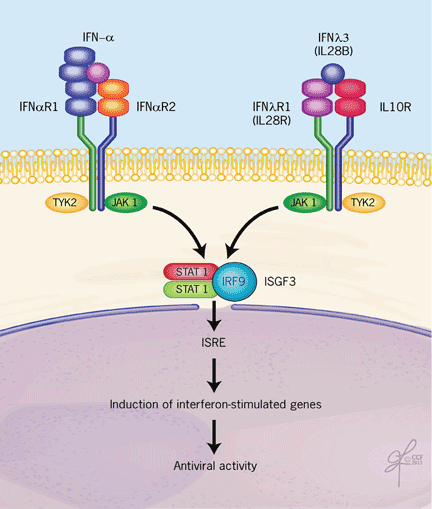
Production of these three molecules can be triggered by viral infection, and they induce antiviral activity through both innate and adaptive immune pathways. They signal through the IL10R-IL28R receptor complex.20–22 This receptor activates the JAK-STAT (Janus kinase-signal transducer and activator of transcription) pathway, which regulates a large number of interferon-stimulated genes, primarily through the interferon-stimulated response element (Figure 2).
A 2013 study found that interferon-stimulated gene expression levels in patients with normal livers were highest in those with the CC genotype, intermediate with CT, and lowest with TT. Interestingly, this pattern was reversed in those with hepatitis C virus infection, indicating a relationship between the IL28B genotype and gene expression before infection.23
The mechanism underlying the association between the IL28B polymorphism and response to hepatitis C treatment is not well understood. The unfavorable TT genotype seems to lead to continuous activation of a subset of interferon-stimulated genes in the presence of intracellular hepatitis C viral RNA. But this level of expression is not sufficient to eliminate the virus from the cells. Instead, it might lead to up-regulation of interferon-inhibitory molecules that suppress JAK-STAT signaling, thereby reducing sensitivity to interferon signaling. Therefore, the hepatocyte not only cannot clear the virus by itself, but also cannot induce strong interferon-stimulated gene expression when interferon is given during therapy.20–22
The recently identified ss469425590 polymorphism, which is located in close proximity to rs12979860 in the IL28B gene, is particularly interesting, as it suggests a possible molecular mechanism. The delta G frameshift variant creates a novel gene called IFNL4, which is transiently activated in response to hepatitis C virus infection.24IFNL4 stimulates STAT1 and STAT2 phosphorylation and induces the expression of interferon-stimulated genes. Increased interferon-stimulated gene expression has been shown to be associated with decreased response to pegylated interferon-ribavirin treatment. These observations suggest that the ss469425590 delta G allele is responsible for the increased activation of interferon-stimulated genes and the lower sustained virologic response rate observed in patients who receive pegylated interferon-ribavirin treatment. It is possible that the activation of interferon-stimulated genes in patients with the ss469425590 delta G/delta G genotype reduces interferon-stimulated gene responsiveness to interferon alpha, which normally activates interferon-stimulated genes and inhibits hepatitis C progression.24
IL28B POLYMORPHISM AND ACUTE HEPATITIS C VIRUS INFECTION
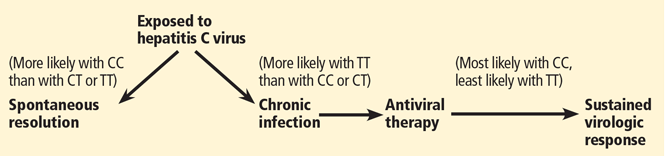
From 70% to 80% of acute hepatitis C virus infections persist and become chronic, while 20% to 30% spontaneously resolve. Epidemiologic, viral, and host factors have been associated with the differences in viral clearance or persistence, and studies have found that a strong host immune response against the virus favors viral clearance. Thus, variation in the genes involved in the immune response may contribute to one’s ability to clear the virus. Consistent with these observations, recent studies have shown that the polymorphism in the IL28 gene region encoding interferon lambda 3 strongly predicts spontaneous resolution of acute hepatitis C virus infection. People who have the IL28B CC genotype are three times more likely to spontaneously clear the virus than those with the CT or TT genotype (Figure 3).24
IL28B POLYMORPHISM AND THE NATURAL HISTORY OF HEPATITIS C
In people in whom hepatitis C virus infection persists, up to 20% develop progressive liver fibrosis and eventually cirrhosis over 10 to 20 years.19,25,26 The speed at which fibrosis develops in these patients is variable and unpredictable.25 The relationship between IL28B polymorphisms and hepatic fibrosis in patients with chronic hepatitis C virus infection has not been clearly established, although a study indicated that in patients with a known date of infection, the IL28B genotype is not associated with progression of hepatic fibrosis.27 Obstacles in this field of study are that it is difficult to determine accurately when the patient contracted the virus, and that serial liver biopsies are needed to investigate the progression of hepatic fibrosis.
Patients with chronic hepatitis C virus infection are also at higher risk of hepatocellular carcinoma compared with the general population.28 An analysis of explanted livers of patients with hepatitis C found that the prevalence of hepatocellular carcinoma in those with the unfavorable TT genotype was significantly higher than with the other genotypes.29 Similarly, an earlier study demonstrated that patients with hepatitis C-associated hepatocellular carcinoma carried the T allele more frequently.30 As with other aspects of IL28B associations with hepatitis C, these findings indicate that the C allele confers a certain degree of protection.
An important implication of these relationships is that they may eventually help identify patients at greater risk, who therefore need earlier intervention.
IL28B POLYMORPHISM AND LIVER TRANSPLANTATION
Hepatitis C virus infection always recurs after liver transplantation, with serious consequences that include cirrhosis and liver failure. Recurrent hepatitis C virus infection has become an important reason for repeat transplantation in the United States.
Results of treatment with pegylated interferon and ribavirin for recurrent hepatitis C after liver transplantation have been disappointing, with response rates lower than 30% and significant side effects.31 Identifying the factors that predict the response to therapy allows for better selection of treatment candidates.
Similar to the way the IL28B genotype predicts response to antiviral therapy in the nontransplant setting, the IL28B genotypes of both the recipient and the donor are strongly and independently associated with response to interferon-based treatment in patients with hepatitis C after liver transplantation. The IL28B CC genotype in either the recipient or the donor is associated with a higher rate of response to pegylated interferon and ribavirin combination therapy after liver transplantation.30,32 For example, the response rate to therapy after liver transplantation reaches 86% in CC-donor and CC-recipient livers, compared with 0% in TT-donor and TT-recipient livers.
Additionally, the IL28B genotype of the recipient may determine the severity of histologic recurrence of hepatitis C, as indicated by progressive hepatic fibrosis. A recipient IL28B TT genotype is associated with more severe histologic recurrence of hepatitis C.33
These data suggest that CC donor livers might be preferentially allocated to patients with hepatitis C virus infection.
IL28B AND OTHER FACTORS IN HEPATITIS C VIRUS INFECTION
Although it is tempting to think that the IL28B polymorphism is the sole predictor of response to antiviral therapy, it is but one of several known factors in the virus and the host.
While IL28B polymorphisms are the most important predictor of sustained virologic response with an interferon-based regimen, a rapid virologic response (undetectable viral load at 4 weeks) had superior predictive value and specificity in one study.34 In fact, for patients with chronic hepatitis C infection who achieved a rapid virologic response with pegylated interferon and ribavirin, the IL28B polymorphism had no effect on the rate of sustained virologic response. However, it did predict a sustained virologic response in the group who did not achieve rapid virologic response.
In a study of patients with acute hepatitis C infection,35 jaundice and the IL28 rs12979860 CC genotype both predicted spontaneous clearance. The best predictor of viral persistence was the combination of the CT or TT genotype plus the absence of jaundice, which had a predictive value of 98%.
IL28B AND THE FUTURE OF HEPATITIS C VIRUS THERAPY
New oral agents were recently approved for treating hepatitis C. As of November 2014, these included simeprevir, sofosbuvir, and ledipasvir.
Simeprevir is a second-generation NS3/4A protease inhibitor approved for use in combination with pegylated interferon and ribavirin. A recent phase 3 trial evaluating simeprevir in patients who had relapsed after prior therapy found sustained virologic response rates to be higher with simeprevir than with placebo, irrespective of IL28B status.36 This finding was similar to that of a trial of telaprevir.16
Sofosbuvir is a nucleotide analogue NS5B polymerase inhibitor that becomes incorporated into the growing RNA, inducing a chain termination event.37 In phase 3 trials,38,39 researchers found an initial rapid decrease in viral load for patients treated with this agent regardless of IL28B status.
In the NEUTRINO trial (Sofosbuvir With Peginterferon Alfa 2a and Ribavirin for 12 Weeks in Treatment-Naive Subjects With Chronic Genotype 1, 4, 5, or 6 HCV Infection),38 which used sofusbuvir in combination with interferon and ribavirin, the rate of sustained virologic response was higher in those with the favorable CC genotype (98%) than with a non-CC genotype (87%).
In COSMOS (A Study of TMC435 in Combination With PSI-7977 [GS7977] in Chronic Hepatitis C Genotype 1-Infected Prior Null Responders to Peginterferon/Ribavirin Therapy or HCV Treatment-Naive Patients),39 which used a combination of simeprevir, sofosbuvir, and ribavirin, the rate of sustained virologic response was higher in those with the CC genotype (100%) than with the TT genotype (83%; Table 1).
These new medications have radically changed the landscape of hepatitis C therapy and have also unlocked the potential for developing completely interferon-free regimens.
Other new interferon-free regimens such as ledipasvir, daclatasvir, and asunaprevir promise high rates of sustained virologic response, which makes the utility of testing for IL28B polymorphisms to predict sustained virologic response very much diminished (Table 1).40,41 However, these new drugs are expected to be expensive, and IL28B polymorphisms may be used to identify candidates who are more likely to respond to pegylated interferon and ribavirin, particularly in resource-poor settings and in developing countries. Additionally, patients who have contraindications to these newer therapies will still likely need an interferon-based regimen, and thus the IL28B polymorphism will still be important in predicting treatment response and prognosis.
IL28B WILL STILL BE RELEVANT IN THE INTERFERON-FREE AGE
The IL28B polymorphism is a strong predictor of spontaneous clearance of hepatitis C virus and responsiveness to interferon-based therapy, and testing for it has demonstrated a great potential to improve patient care. IL28B testing has become available for clinical use and may optimize the outcome of hepatitis C treatment by helping us to select the best treatment for individual patients and minimizing the duration of therapy and the side effects associated with interferon-based antiviral medications.
As newer therapies have shifted toward interferon-free regimens that offer very high sustained virologic response rates, the usefulness of IL28B polymorphism as a clinical test to predict the response rate to antiviral therapy is minimized substantially. It may remain clinically relevant in resource-poor settings and in developing countries, especially in light of the potentially prohibitive costs of the newer regimens, and for patients in whom these treatments are contraindicated. This does not minimize the lesson we learned from the discovery of the IL28B gene and the impact on our understanding of the pathogenesis of hepatitis C virus infection.
- Attia J, Ioannidis JP, Thakkinstian A, et al. How to use an article about genetic association: A: background concepts. JAMA 2009; 301:74–81.
- Samani NJ, Erdmann J, Hall AS, et al; WTCCC and the Cardiogenics Consortium. Genomewide association analysis of coronary artery disease. N Engl J Med 2007; 357:443–453.
- Zeggini E, Weedon MN, Lindgren CM, et al; Wellcome Trust Case Control Consortium (WTCCC). Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science 2007; 316:1336–1341.
- Matarín M, Brown WM, Scholz S, et al. A genome-wide genotyping study in patients with ischaemic stroke: initial analysis and data release. Lancet Neurol 2007; 6:414–420.
- Easton DF, Pooley KA, Dunning AM, et al; AOCS Management Group. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature 2007; 447:1087–1093.
- Plenge RM, Seielstad M, Padyukov L, et al. TRAF1-C5 as a risk locus for rheumatoid arthritis—a genomewide study. N Engl J Med 2007; 357:1199–1209.
- Coon KD, Myers AJ, Craig DW, et al. A high-density whole-genome association study reveals that APOE is the major susceptibility gene for sporadic late-onset Alzheimer’s disease. J Clin Psychiatry 2007; 68:613–618.
- Ge D, Fellay J, Thompson AJ, et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance (letter). Nature 2009; 461:399–401.
- Ali A, Zein NN. Hepatitis C infection: a systemic disease with extrahepatic manifestations. Cleve Clin J Med 2005; 72:1005-1019.
- Hanouneh IA, Feldstein AE, Lopez R, et al. Clinical significance of metabolic syndrome in the setting of chronic hepatitis C virus infection. Clin Gastroenterol Hepatol 2008; 6:584–589.
- Elgouhari HM, Zein CO, Hanouneh I, Feldstein AE, Zein NN. Diabetes mellitus is associated with impaired response to antiviral therapy in chronic hepatitis C infection. Dig Dis Sci 2009; 54:2699–2705.
- Alkhouri N, Zein NN. Protease inhibitors: silver bullets for chronic hepatitis C infection? Cleve Clin J Med 2012; 79:213–222.
- McHutchison JG, Everson GT, Gordon SC, et al; PROVE1 Study Team. Telaprevir with peginterferon and ribavirin for chronic HCV genotype 1 infection. N Engl J Med 2009; 360:1827–1838.
- Jacobson IM, McHutchison JG, Dusheiko G, et al; ADVANCE Study Team. Telaprevir for previously untreated chronic hepatitis C virus infection. N Engl J Med 2011; 364:2405–2416.
- Jacobson IM, Catlett I, Marcellin P, et al. Telaprevir substantially improved SVR rates across all IL28B genotypes in the ADVANCE trial. J Hepatol 2011; 54(suppl 1):S542–S543.
- Pol S, Aerssens J, Zeuzem S, et al. Limited impact of IL28B genotype on response rates in telaprevir-treated patients with prior treatment failure. J Hepatol 2013; 58:883–889.
- Suppiah V, Moldovan M, Ahlenstiel G, et al. IL28B is associated with response to chronic hepatitis C interferon-alpha and ribavirin therapy. Nat Genet 2009; 41:1100–1104.
- Tanaka Y, Nishida N, Sugiyama M, et al. Genome-wide association of IL28B with response to pegylated interferon-alpha and ribavirin therapy for chronic hepatitis C. Nat Genet 2009; 41:1105–1109.
- Thomas DL, Thio CL, Martin MP, et al. Genetic variation in IL28B and spontaneous clearance of hepatitis C virus. Nature 2009; 461:798–801.
- Rehermann B. Hepatitis C virus versus innate and adaptive immune responses: a tale of coevolution and coexistence. J Clin Invest 2009; 119:1745–1754.
- Marcello T, Grakoui A, Barba-Spaeth G, et al. Interferons alpha and lambda inhibit hepatitis C virus replication with distinct signal transduction and gene regulation kinetics. Gastroenterology 2006; 131:1887–1898.
- Doyle SE, Schreckhise H, Khuu-Duong K, et al. Interleukin-29 uses a type 1 interferon-like program to promote antiviral responses in human hepatocytes. Hepatology 2006; 44:896–906.
- Raglow Z, Thoma-Perry C, Gilroy R, Wan YJ. IL28B genotype and the expression of ISGs in normal liver. Liver Int 2013; 33:991–998.
- Prokunina-Olsson L, Muchmore B, Tang W, et al. A variant upstream of IFNL3 (IL28B) creating a new interferon gene IFNL4 is associated with impaired clearance of hepatitis C virus. Nat Genet 2013; 45:164–171.
- Hanouneh IA, Zein NN, Askar M, Lopez R, John B. Interleukin-28B polymorphisms are associated with fibrosing cholestatic hepatitis in recurrent hepatitis C after liver transplantation. Clin Transplant 2012; 26:E335–E336.
- Poynard T, Bedossa P, Opolon P. Natural history of liver fibrosis progression in patients with chronic hepatitis C. The OBSVIRC, METAVIR, CLINIVIR, and DOSVIRC groups. Lancet 1997; 349:825–832.
- Thomas DL, Astemborski J, Rai RM, et al. The natural history of hepatitis C virus infection: host, viral, and environmental factors. JAMA 2000; 284:450–456.
- Bochud PY, Cai T, Overbeck K, et al; Swiss Hepatitis C Cohort Study Group. Genotype 3 is associated with accelerated fibrosis progression in chronic hepatitis C. J Hepatol 2009; 51:655–666.
- Marabita F, Aghemo A, De Nicola S, et al. Genetic variation in the interleukin-28B gene is not associated with fibrosis progression in patients with chronic hepatitis C and known date of infection. Hepatology 2011; 54:1127–1134.
- Fabris C, Falleti E, Cussigh A, et al. IL-28B rs12979860 C/T allele distribution in patients with liver cirrhosis: role in the course of chronic viral hepatitis and the development of HCC. J Hepatol 2011; 54:716–722.
- Eurich D, Boas-Knoop S, Bahra M, et al. Role of IL28B polymorphism in the development of hepatitis C virus-induced hepatocellular carcinoma, graft fibrosis, and posttransplant antiviral therapy. Transplantation 2012; 93:644–649.
- Hanouneh IA, Miller C, Aucejo F, Lopez R, Quinn MK, Zein NN. Recurrent hepatitis C after liver transplantation: on-treatment prediction of response to peginterferon/ribavirin therapy. Liver Transpl 2008; 14:53–58.
- Charlton MR, Thompson A, Veldt BJ, et al. Interleukin-28B polymorphisms are associated with histological recurrence and treatment response following liver transplantation in patients with hepatitis C virus infection. Hepatology 2011; 53:317–324.
- Thompson AJ, Muir AJ, Sulkowski MS, et al. Interleukin-28B polymorphism improves viral kinetics and is the strongest pretreatment predictor of sustained virologic response in genotype 1 hepatitis C virus. Gastroenterology 2010; 139:120–129.e18.
- Beinhardt S, Payer BA, Datz C, et al. A diagnostic score for the prediction of spontaneous resolution of acute hepatitis C virus infection. J Hepatol 2013; 59:972–977.
- Forns X, Lawitz E, Zeuzem S, et al. Simeprevir with peginterferon and ribavirin leads to high rates of SVR in patients with HCV genotype 1 who relapsed after previous therapy: a phase 3 trial. Gastroenterology 2014; 146:1669–1679.e3.
- Sofia MJ, Bao D, Chang W, et al. Discovery of a ß-d-2’-deoxy-2’-ß-fluoro-2’-ß-C-methyluridine nucleotide prodrug (PSI-7977) for the treatment of hepatitis C virus. J Med Chem 2010; 53:7202–7218.
- Lawitz E, Mangia A, Wyles D, et al. Sofosbuvir for previously untreated chronic hepatitis C infection. N Engl J Med 2013; 368:1878–1887.
- Sulkowski MS, Jacobson IM, Ghalib R, et al. Once-daily simeprevir (TMC435) plus sofosbuvir (GS-7977) with or without ribavirin in HCV genotype 1 prior null responders with metavir F0-2: COSMOS study subgroup analysis. 49th EASL, April 2014, London. Oral abstract O7. www.natap.org/2014/EASL/EASL_46.htm. Accesed January 9, 2015.
- Lok AS, Gardiner DF, Lawitz E, et al. Preliminary study of two antiviral agents for hepatitis C genotype 1. N Engl J Med 2012; 366:216–224.
- Afdhal N, Zeuzem S, Kwo P, et al; ION-1 Investigators. Ledipasvir and sofosbuvir for untreated HCV genotype 1 infection. N Engl J Med 2014; 370:1889–1898.
What a difference a single nucleotide can make! The human genome contains more than 3 billion base pairs. Yet having a different nucleotide in only one pair can make a big difference in how we respond to a disease or its treatment.
Specifically, in hepatitis C virus infection, people born with the nucleotide cytosine (C) at location rs12979860 in both alleles of the gene that codes for interleukin 28B (the IL28B CC genotype) can count themselves luckier than those born with thymine (T) in this location in one of their alleles (the CT genotype) or both of their alleles (the TT genotype). Those with the CC genotype are more likely to clear the virus spontaneously, and even if the infection persists, it is less likely to progress to liver cancer and more likely to respond to treatment with interferon.
Here, we review the IL28B polymorphism and its implications in treating hepatitis C.
GENETIC POLYMORPHISM AND HUMAN DISEASE
Of the 3 billion base pairs of nucleotides, fewer than 1% differ between individuals, but this 1% is responsible for the diversity of human beings. Differences in genetic sequences among individuals are called genetic polymorphisms. A single-nucleotide polymorphism is a DNA sequence variation that occurs in a single nucleotide in the genome. For example, two sequenced DNA fragments from different individuals, AAGCCTA and AAGCTTA, contain a difference in a single nucleotide.
Genetic variations such as these underlie some of the differences in our susceptibility to disease, the severity of illness we develop, and our response to treatments. Therefore, identifying genetic polymorphisms may shed light on biologic pathways involved in diseases and may uncover new targets for therapy.1
Genome-wide association studies have looked at hundreds of thousands of single-nucleotide polymorphisms to try to identify most of the common genetic differences among people and relate them to common chronic diseases such as coronary artery disease,2 type 2 diabetes,3 stroke,4 breast cancer,5 rheumatoid arthritis,6 Alzheimer disease,7 and, more recently, hepatitis C virus infection.8
HEPATITIS C VIRUS: A MAJOR CAUSE OF LIVER DISEASE
Hepatitis C virus infection is a major cause of chronic liver disease and hepatocellular carcinoma and has become the most common indication for liver transplantation in the United States.9
This virus has six distinct genotypes throughout the world, with multiple subtypes in each genotype. (A genotype is a classification of a virus based on its RNA.9) In this review, we will focus on genotype 1; hence, “hepatitis C virus” will refer to hepatitis C virus genotype 1.
Our knowledge of the biology, pathogenesis, and treatment of hepatitis C has been advancing. Originally, fewer than 50% of patients responded to therapy with the combination of pegylated interferon and ribavirin,10,11 but since 2011 the response rate has increased to approximately 70% with the approval of the protease inhibitors telaprevir and boceprevir, used in combination with pegylated interferon and ribavirin.12–15
Unfortunately, interferon-based treatment is often complicated by side effects such as fatigue, influenza-like symptoms, hematologic abnormalities, and neuropsychiatric symptoms. An accurate way to predict response would help patients make informed decisions about antiviral treatment, taking into account the risk and possible benefit for individual patients.
GENETIC POLYMORPHISM AND HEPATITIS C VIRUS INFECTION
Genome-wide association studies have identified single-nucleotide polymorphisms in the IL28B gene that are associated with differences in response to hepatitis C treatment.8
Studying 565,759 polymorphisms in 1,137 patients, researchers at Duke University identified a single-nucleotide polymorphism at location rs12979860 in IL28B (Figure 1) that was strongly associated with response to combination therapy with pegylated interferon and ribavirin.8 The chance of cure with this standard treatment is twice as high in patients who are homozygous for cytosine in this location (the CC genotype) than in those who are heterozygous (CT) or homozygous for thymine in this location (the TT genotype) (Table 1).
Adding one of the new protease inhibitors, telaprevir or boceprevir, to the standard hepatitis C treatment substantially improves the cure rates in all three IL28B genotypes, but especially in people with CT or TT, in whom the response rate almost triples with the addition of one of these drugs. Those with the CC genotype (who are more likely to be cured with pegylated interferon and ribavirin alone) also achieve an increase (although minimal) in cure rates when a protease inhibitor is included in the regimen (TABLE 1).13–15 Thus, it remains unclear if adding a protease inhibitor to pegylated interferon plus ribavirin in patients with the IL28B CC genotype translates into added effectiveness worth the additional cost of the protease inhibitor in previously untreated patients.
Additionally, the effect of the IL28B genotype on telaprevir-based triple therapy has been disputed in more recent studies. In a subgroup analysis of the results of a trial that evaluated telaprevir in the treatment of hepatitis C, researchers found that sustained virologic response rates were significantly higher in the telaprevir group, and this was similar across the different IL28B polymorphisms.16
The favorable IL28B CC genotype is associated with higher rates of rapid virologic response to antiviral therapy.13–15 Of note, almost all patients who achieve a rapid virologic response do well, with a high rate of sustained virologic response even after a shorter duration of therapy (24 vs 48 weeks). Therefore, in addition to predicting response to interferon before starting treatment, the IL28B CC genotype may also identify patients who need only a shorter duration of therapy.
Interestingly, the C allele is much more frequent in white than in African American populations, an important observation that explains the racial difference in response to hepatitis C therapy.8
Two other research groups, from Asia and Australia, performed independent genome-wide association studies that identified different single-nucleotide polymorphisms (eg, rs8099917) in the same IL28B gene as predictors of response to treatment in patients with hepatitis C virus infection.17,18 These findings may be explained by linkage disequilibrium, which means that these single-nucleotide polymorphisms are found more frequently together in the same patient due to their proximity to each other. In this review, we will focus on the rs12979860 polymorphism; hence “IL28B genotype” will refer to the single-nucleotide polymorphism at rs12979860, unless otherwise specified.
The favorable CC genotype is less common in African Americans than in patients of other ethnicities.19 Moreover, although IL28B CC is associated with a better response rate to interferon-based antiviral therapy across all ethnicities, those of African American descent with the CC genotype are less likely to achieve a sustained virologic response than white or Hispanic Americans.8
BIOLOGIC ASSOCIATION: IL28B POLYMORPHISM AND HEPATITIS C
The interferon lambda family consists of three cytokines:
- Interleukin 29 (interferon lambda 1)
- Interleukin 28A (interferon lambda 2)
- Interleukin 28B (interferon lambda 3).
Production of these three molecules can be triggered by viral infection, and they induce antiviral activity through both innate and adaptive immune pathways. They signal through the IL10R-IL28R receptor complex.20–22 This receptor activates the JAK-STAT (Janus kinase-signal transducer and activator of transcription) pathway, which regulates a large number of interferon-stimulated genes, primarily through the interferon-stimulated response element (Figure 2).
A 2013 study found that interferon-stimulated gene expression levels in patients with normal livers were highest in those with the CC genotype, intermediate with CT, and lowest with TT. Interestingly, this pattern was reversed in those with hepatitis C virus infection, indicating a relationship between the IL28B genotype and gene expression before infection.23
The mechanism underlying the association between the IL28B polymorphism and response to hepatitis C treatment is not well understood. The unfavorable TT genotype seems to lead to continuous activation of a subset of interferon-stimulated genes in the presence of intracellular hepatitis C viral RNA. But this level of expression is not sufficient to eliminate the virus from the cells. Instead, it might lead to up-regulation of interferon-inhibitory molecules that suppress JAK-STAT signaling, thereby reducing sensitivity to interferon signaling. Therefore, the hepatocyte not only cannot clear the virus by itself, but also cannot induce strong interferon-stimulated gene expression when interferon is given during therapy.20–22
The recently identified ss469425590 polymorphism, which is located in close proximity to rs12979860 in the IL28B gene, is particularly interesting, as it suggests a possible molecular mechanism. The delta G frameshift variant creates a novel gene called IFNL4, which is transiently activated in response to hepatitis C virus infection.24IFNL4 stimulates STAT1 and STAT2 phosphorylation and induces the expression of interferon-stimulated genes. Increased interferon-stimulated gene expression has been shown to be associated with decreased response to pegylated interferon-ribavirin treatment. These observations suggest that the ss469425590 delta G allele is responsible for the increased activation of interferon-stimulated genes and the lower sustained virologic response rate observed in patients who receive pegylated interferon-ribavirin treatment. It is possible that the activation of interferon-stimulated genes in patients with the ss469425590 delta G/delta G genotype reduces interferon-stimulated gene responsiveness to interferon alpha, which normally activates interferon-stimulated genes and inhibits hepatitis C progression.24
IL28B POLYMORPHISM AND ACUTE HEPATITIS C VIRUS INFECTION
From 70% to 80% of acute hepatitis C virus infections persist and become chronic, while 20% to 30% spontaneously resolve. Epidemiologic, viral, and host factors have been associated with the differences in viral clearance or persistence, and studies have found that a strong host immune response against the virus favors viral clearance. Thus, variation in the genes involved in the immune response may contribute to one’s ability to clear the virus. Consistent with these observations, recent studies have shown that the polymorphism in the IL28 gene region encoding interferon lambda 3 strongly predicts spontaneous resolution of acute hepatitis C virus infection. People who have the IL28B CC genotype are three times more likely to spontaneously clear the virus than those with the CT or TT genotype (Figure 3).24
IL28B POLYMORPHISM AND THE NATURAL HISTORY OF HEPATITIS C
In people in whom hepatitis C virus infection persists, up to 20% develop progressive liver fibrosis and eventually cirrhosis over 10 to 20 years.19,25,26 The speed at which fibrosis develops in these patients is variable and unpredictable.25 The relationship between IL28B polymorphisms and hepatic fibrosis in patients with chronic hepatitis C virus infection has not been clearly established, although a study indicated that in patients with a known date of infection, the IL28B genotype is not associated with progression of hepatic fibrosis.27 Obstacles in this field of study are that it is difficult to determine accurately when the patient contracted the virus, and that serial liver biopsies are needed to investigate the progression of hepatic fibrosis.
Patients with chronic hepatitis C virus infection are also at higher risk of hepatocellular carcinoma compared with the general population.28 An analysis of explanted livers of patients with hepatitis C found that the prevalence of hepatocellular carcinoma in those with the unfavorable TT genotype was significantly higher than with the other genotypes.29 Similarly, an earlier study demonstrated that patients with hepatitis C-associated hepatocellular carcinoma carried the T allele more frequently.30 As with other aspects of IL28B associations with hepatitis C, these findings indicate that the C allele confers a certain degree of protection.
An important implication of these relationships is that they may eventually help identify patients at greater risk, who therefore need earlier intervention.
IL28B POLYMORPHISM AND LIVER TRANSPLANTATION
Hepatitis C virus infection always recurs after liver transplantation, with serious consequences that include cirrhosis and liver failure. Recurrent hepatitis C virus infection has become an important reason for repeat transplantation in the United States.
Results of treatment with pegylated interferon and ribavirin for recurrent hepatitis C after liver transplantation have been disappointing, with response rates lower than 30% and significant side effects.31 Identifying the factors that predict the response to therapy allows for better selection of treatment candidates.
Similar to the way the IL28B genotype predicts response to antiviral therapy in the nontransplant setting, the IL28B genotypes of both the recipient and the donor are strongly and independently associated with response to interferon-based treatment in patients with hepatitis C after liver transplantation. The IL28B CC genotype in either the recipient or the donor is associated with a higher rate of response to pegylated interferon and ribavirin combination therapy after liver transplantation.30,32 For example, the response rate to therapy after liver transplantation reaches 86% in CC-donor and CC-recipient livers, compared with 0% in TT-donor and TT-recipient livers.
Additionally, the IL28B genotype of the recipient may determine the severity of histologic recurrence of hepatitis C, as indicated by progressive hepatic fibrosis. A recipient IL28B TT genotype is associated with more severe histologic recurrence of hepatitis C.33
These data suggest that CC donor livers might be preferentially allocated to patients with hepatitis C virus infection.
IL28B AND OTHER FACTORS IN HEPATITIS C VIRUS INFECTION
Although it is tempting to think that the IL28B polymorphism is the sole predictor of response to antiviral therapy, it is but one of several known factors in the virus and the host.
While IL28B polymorphisms are the most important predictor of sustained virologic response with an interferon-based regimen, a rapid virologic response (undetectable viral load at 4 weeks) had superior predictive value and specificity in one study.34 In fact, for patients with chronic hepatitis C infection who achieved a rapid virologic response with pegylated interferon and ribavirin, the IL28B polymorphism had no effect on the rate of sustained virologic response. However, it did predict a sustained virologic response in the group who did not achieve rapid virologic response.
In a study of patients with acute hepatitis C infection,35 jaundice and the IL28 rs12979860 CC genotype both predicted spontaneous clearance. The best predictor of viral persistence was the combination of the CT or TT genotype plus the absence of jaundice, which had a predictive value of 98%.
IL28B AND THE FUTURE OF HEPATITIS C VIRUS THERAPY
New oral agents were recently approved for treating hepatitis C. As of November 2014, these included simeprevir, sofosbuvir, and ledipasvir.
Simeprevir is a second-generation NS3/4A protease inhibitor approved for use in combination with pegylated interferon and ribavirin. A recent phase 3 trial evaluating simeprevir in patients who had relapsed after prior therapy found sustained virologic response rates to be higher with simeprevir than with placebo, irrespective of IL28B status.36 This finding was similar to that of a trial of telaprevir.16
Sofosbuvir is a nucleotide analogue NS5B polymerase inhibitor that becomes incorporated into the growing RNA, inducing a chain termination event.37 In phase 3 trials,38,39 researchers found an initial rapid decrease in viral load for patients treated with this agent regardless of IL28B status.
In the NEUTRINO trial (Sofosbuvir With Peginterferon Alfa 2a and Ribavirin for 12 Weeks in Treatment-Naive Subjects With Chronic Genotype 1, 4, 5, or 6 HCV Infection),38 which used sofusbuvir in combination with interferon and ribavirin, the rate of sustained virologic response was higher in those with the favorable CC genotype (98%) than with a non-CC genotype (87%).
In COSMOS (A Study of TMC435 in Combination With PSI-7977 [GS7977] in Chronic Hepatitis C Genotype 1-Infected Prior Null Responders to Peginterferon/Ribavirin Therapy or HCV Treatment-Naive Patients),39 which used a combination of simeprevir, sofosbuvir, and ribavirin, the rate of sustained virologic response was higher in those with the CC genotype (100%) than with the TT genotype (83%; Table 1).
These new medications have radically changed the landscape of hepatitis C therapy and have also unlocked the potential for developing completely interferon-free regimens.
Other new interferon-free regimens such as ledipasvir, daclatasvir, and asunaprevir promise high rates of sustained virologic response, which makes the utility of testing for IL28B polymorphisms to predict sustained virologic response very much diminished (Table 1).40,41 However, these new drugs are expected to be expensive, and IL28B polymorphisms may be used to identify candidates who are more likely to respond to pegylated interferon and ribavirin, particularly in resource-poor settings and in developing countries. Additionally, patients who have contraindications to these newer therapies will still likely need an interferon-based regimen, and thus the IL28B polymorphism will still be important in predicting treatment response and prognosis.
IL28B WILL STILL BE RELEVANT IN THE INTERFERON-FREE AGE
The IL28B polymorphism is a strong predictor of spontaneous clearance of hepatitis C virus and responsiveness to interferon-based therapy, and testing for it has demonstrated a great potential to improve patient care. IL28B testing has become available for clinical use and may optimize the outcome of hepatitis C treatment by helping us to select the best treatment for individual patients and minimizing the duration of therapy and the side effects associated with interferon-based antiviral medications.
As newer therapies have shifted toward interferon-free regimens that offer very high sustained virologic response rates, the usefulness of IL28B polymorphism as a clinical test to predict the response rate to antiviral therapy is minimized substantially. It may remain clinically relevant in resource-poor settings and in developing countries, especially in light of the potentially prohibitive costs of the newer regimens, and for patients in whom these treatments are contraindicated. This does not minimize the lesson we learned from the discovery of the IL28B gene and the impact on our understanding of the pathogenesis of hepatitis C virus infection.
What a difference a single nucleotide can make! The human genome contains more than 3 billion base pairs. Yet having a different nucleotide in only one pair can make a big difference in how we respond to a disease or its treatment.
Specifically, in hepatitis C virus infection, people born with the nucleotide cytosine (C) at location rs12979860 in both alleles of the gene that codes for interleukin 28B (the IL28B CC genotype) can count themselves luckier than those born with thymine (T) in this location in one of their alleles (the CT genotype) or both of their alleles (the TT genotype). Those with the CC genotype are more likely to clear the virus spontaneously, and even if the infection persists, it is less likely to progress to liver cancer and more likely to respond to treatment with interferon.
Here, we review the IL28B polymorphism and its implications in treating hepatitis C.
GENETIC POLYMORPHISM AND HUMAN DISEASE
Of the 3 billion base pairs of nucleotides, fewer than 1% differ between individuals, but this 1% is responsible for the diversity of human beings. Differences in genetic sequences among individuals are called genetic polymorphisms. A single-nucleotide polymorphism is a DNA sequence variation that occurs in a single nucleotide in the genome. For example, two sequenced DNA fragments from different individuals, AAGCCTA and AAGCTTA, contain a difference in a single nucleotide.
Genetic variations such as these underlie some of the differences in our susceptibility to disease, the severity of illness we develop, and our response to treatments. Therefore, identifying genetic polymorphisms may shed light on biologic pathways involved in diseases and may uncover new targets for therapy.1
Genome-wide association studies have looked at hundreds of thousands of single-nucleotide polymorphisms to try to identify most of the common genetic differences among people and relate them to common chronic diseases such as coronary artery disease,2 type 2 diabetes,3 stroke,4 breast cancer,5 rheumatoid arthritis,6 Alzheimer disease,7 and, more recently, hepatitis C virus infection.8
HEPATITIS C VIRUS: A MAJOR CAUSE OF LIVER DISEASE
Hepatitis C virus infection is a major cause of chronic liver disease and hepatocellular carcinoma and has become the most common indication for liver transplantation in the United States.9
This virus has six distinct genotypes throughout the world, with multiple subtypes in each genotype. (A genotype is a classification of a virus based on its RNA.9) In this review, we will focus on genotype 1; hence, “hepatitis C virus” will refer to hepatitis C virus genotype 1.
Our knowledge of the biology, pathogenesis, and treatment of hepatitis C has been advancing. Originally, fewer than 50% of patients responded to therapy with the combination of pegylated interferon and ribavirin,10,11 but since 2011 the response rate has increased to approximately 70% with the approval of the protease inhibitors telaprevir and boceprevir, used in combination with pegylated interferon and ribavirin.12–15
Unfortunately, interferon-based treatment is often complicated by side effects such as fatigue, influenza-like symptoms, hematologic abnormalities, and neuropsychiatric symptoms. An accurate way to predict response would help patients make informed decisions about antiviral treatment, taking into account the risk and possible benefit for individual patients.
GENETIC POLYMORPHISM AND HEPATITIS C VIRUS INFECTION
Genome-wide association studies have identified single-nucleotide polymorphisms in the IL28B gene that are associated with differences in response to hepatitis C treatment.8
Studying 565,759 polymorphisms in 1,137 patients, researchers at Duke University identified a single-nucleotide polymorphism at location rs12979860 in IL28B (Figure 1) that was strongly associated with response to combination therapy with pegylated interferon and ribavirin.8 The chance of cure with this standard treatment is twice as high in patients who are homozygous for cytosine in this location (the CC genotype) than in those who are heterozygous (CT) or homozygous for thymine in this location (the TT genotype) (Table 1).
Adding one of the new protease inhibitors, telaprevir or boceprevir, to the standard hepatitis C treatment substantially improves the cure rates in all three IL28B genotypes, but especially in people with CT or TT, in whom the response rate almost triples with the addition of one of these drugs. Those with the CC genotype (who are more likely to be cured with pegylated interferon and ribavirin alone) also achieve an increase (although minimal) in cure rates when a protease inhibitor is included in the regimen (TABLE 1).13–15 Thus, it remains unclear if adding a protease inhibitor to pegylated interferon plus ribavirin in patients with the IL28B CC genotype translates into added effectiveness worth the additional cost of the protease inhibitor in previously untreated patients.
Additionally, the effect of the IL28B genotype on telaprevir-based triple therapy has been disputed in more recent studies. In a subgroup analysis of the results of a trial that evaluated telaprevir in the treatment of hepatitis C, researchers found that sustained virologic response rates were significantly higher in the telaprevir group, and this was similar across the different IL28B polymorphisms.16
The favorable IL28B CC genotype is associated with higher rates of rapid virologic response to antiviral therapy.13–15 Of note, almost all patients who achieve a rapid virologic response do well, with a high rate of sustained virologic response even after a shorter duration of therapy (24 vs 48 weeks). Therefore, in addition to predicting response to interferon before starting treatment, the IL28B CC genotype may also identify patients who need only a shorter duration of therapy.
Interestingly, the C allele is much more frequent in white than in African American populations, an important observation that explains the racial difference in response to hepatitis C therapy.8
Two other research groups, from Asia and Australia, performed independent genome-wide association studies that identified different single-nucleotide polymorphisms (eg, rs8099917) in the same IL28B gene as predictors of response to treatment in patients with hepatitis C virus infection.17,18 These findings may be explained by linkage disequilibrium, which means that these single-nucleotide polymorphisms are found more frequently together in the same patient due to their proximity to each other. In this review, we will focus on the rs12979860 polymorphism; hence “IL28B genotype” will refer to the single-nucleotide polymorphism at rs12979860, unless otherwise specified.
The favorable CC genotype is less common in African Americans than in patients of other ethnicities.19 Moreover, although IL28B CC is associated with a better response rate to interferon-based antiviral therapy across all ethnicities, those of African American descent with the CC genotype are less likely to achieve a sustained virologic response than white or Hispanic Americans.8
BIOLOGIC ASSOCIATION: IL28B POLYMORPHISM AND HEPATITIS C
The interferon lambda family consists of three cytokines:
- Interleukin 29 (interferon lambda 1)
- Interleukin 28A (interferon lambda 2)
- Interleukin 28B (interferon lambda 3).
Production of these three molecules can be triggered by viral infection, and they induce antiviral activity through both innate and adaptive immune pathways. They signal through the IL10R-IL28R receptor complex.20–22 This receptor activates the JAK-STAT (Janus kinase-signal transducer and activator of transcription) pathway, which regulates a large number of interferon-stimulated genes, primarily through the interferon-stimulated response element (Figure 2).
A 2013 study found that interferon-stimulated gene expression levels in patients with normal livers were highest in those with the CC genotype, intermediate with CT, and lowest with TT. Interestingly, this pattern was reversed in those with hepatitis C virus infection, indicating a relationship between the IL28B genotype and gene expression before infection.23
The mechanism underlying the association between the IL28B polymorphism and response to hepatitis C treatment is not well understood. The unfavorable TT genotype seems to lead to continuous activation of a subset of interferon-stimulated genes in the presence of intracellular hepatitis C viral RNA. But this level of expression is not sufficient to eliminate the virus from the cells. Instead, it might lead to up-regulation of interferon-inhibitory molecules that suppress JAK-STAT signaling, thereby reducing sensitivity to interferon signaling. Therefore, the hepatocyte not only cannot clear the virus by itself, but also cannot induce strong interferon-stimulated gene expression when interferon is given during therapy.20–22
The recently identified ss469425590 polymorphism, which is located in close proximity to rs12979860 in the IL28B gene, is particularly interesting, as it suggests a possible molecular mechanism. The delta G frameshift variant creates a novel gene called IFNL4, which is transiently activated in response to hepatitis C virus infection.24IFNL4 stimulates STAT1 and STAT2 phosphorylation and induces the expression of interferon-stimulated genes. Increased interferon-stimulated gene expression has been shown to be associated with decreased response to pegylated interferon-ribavirin treatment. These observations suggest that the ss469425590 delta G allele is responsible for the increased activation of interferon-stimulated genes and the lower sustained virologic response rate observed in patients who receive pegylated interferon-ribavirin treatment. It is possible that the activation of interferon-stimulated genes in patients with the ss469425590 delta G/delta G genotype reduces interferon-stimulated gene responsiveness to interferon alpha, which normally activates interferon-stimulated genes and inhibits hepatitis C progression.24
IL28B POLYMORPHISM AND ACUTE HEPATITIS C VIRUS INFECTION
From 70% to 80% of acute hepatitis C virus infections persist and become chronic, while 20% to 30% spontaneously resolve. Epidemiologic, viral, and host factors have been associated with the differences in viral clearance or persistence, and studies have found that a strong host immune response against the virus favors viral clearance. Thus, variation in the genes involved in the immune response may contribute to one’s ability to clear the virus. Consistent with these observations, recent studies have shown that the polymorphism in the IL28 gene region encoding interferon lambda 3 strongly predicts spontaneous resolution of acute hepatitis C virus infection. People who have the IL28B CC genotype are three times more likely to spontaneously clear the virus than those with the CT or TT genotype (Figure 3).24
IL28B POLYMORPHISM AND THE NATURAL HISTORY OF HEPATITIS C
In people in whom hepatitis C virus infection persists, up to 20% develop progressive liver fibrosis and eventually cirrhosis over 10 to 20 years.19,25,26 The speed at which fibrosis develops in these patients is variable and unpredictable.25 The relationship between IL28B polymorphisms and hepatic fibrosis in patients with chronic hepatitis C virus infection has not been clearly established, although a study indicated that in patients with a known date of infection, the IL28B genotype is not associated with progression of hepatic fibrosis.27 Obstacles in this field of study are that it is difficult to determine accurately when the patient contracted the virus, and that serial liver biopsies are needed to investigate the progression of hepatic fibrosis.
Patients with chronic hepatitis C virus infection are also at higher risk of hepatocellular carcinoma compared with the general population.28 An analysis of explanted livers of patients with hepatitis C found that the prevalence of hepatocellular carcinoma in those with the unfavorable TT genotype was significantly higher than with the other genotypes.29 Similarly, an earlier study demonstrated that patients with hepatitis C-associated hepatocellular carcinoma carried the T allele more frequently.30 As with other aspects of IL28B associations with hepatitis C, these findings indicate that the C allele confers a certain degree of protection.
An important implication of these relationships is that they may eventually help identify patients at greater risk, who therefore need earlier intervention.
IL28B POLYMORPHISM AND LIVER TRANSPLANTATION
Hepatitis C virus infection always recurs after liver transplantation, with serious consequences that include cirrhosis and liver failure. Recurrent hepatitis C virus infection has become an important reason for repeat transplantation in the United States.
Results of treatment with pegylated interferon and ribavirin for recurrent hepatitis C after liver transplantation have been disappointing, with response rates lower than 30% and significant side effects.31 Identifying the factors that predict the response to therapy allows for better selection of treatment candidates.
Similar to the way the IL28B genotype predicts response to antiviral therapy in the nontransplant setting, the IL28B genotypes of both the recipient and the donor are strongly and independently associated with response to interferon-based treatment in patients with hepatitis C after liver transplantation. The IL28B CC genotype in either the recipient or the donor is associated with a higher rate of response to pegylated interferon and ribavirin combination therapy after liver transplantation.30,32 For example, the response rate to therapy after liver transplantation reaches 86% in CC-donor and CC-recipient livers, compared with 0% in TT-donor and TT-recipient livers.
Additionally, the IL28B genotype of the recipient may determine the severity of histologic recurrence of hepatitis C, as indicated by progressive hepatic fibrosis. A recipient IL28B TT genotype is associated with more severe histologic recurrence of hepatitis C.33
These data suggest that CC donor livers might be preferentially allocated to patients with hepatitis C virus infection.
IL28B AND OTHER FACTORS IN HEPATITIS C VIRUS INFECTION
Although it is tempting to think that the IL28B polymorphism is the sole predictor of response to antiviral therapy, it is but one of several known factors in the virus and the host.
While IL28B polymorphisms are the most important predictor of sustained virologic response with an interferon-based regimen, a rapid virologic response (undetectable viral load at 4 weeks) had superior predictive value and specificity in one study.34 In fact, for patients with chronic hepatitis C infection who achieved a rapid virologic response with pegylated interferon and ribavirin, the IL28B polymorphism had no effect on the rate of sustained virologic response. However, it did predict a sustained virologic response in the group who did not achieve rapid virologic response.
In a study of patients with acute hepatitis C infection,35 jaundice and the IL28 rs12979860 CC genotype both predicted spontaneous clearance. The best predictor of viral persistence was the combination of the CT or TT genotype plus the absence of jaundice, which had a predictive value of 98%.
IL28B AND THE FUTURE OF HEPATITIS C VIRUS THERAPY
New oral agents were recently approved for treating hepatitis C. As of November 2014, these included simeprevir, sofosbuvir, and ledipasvir.
Simeprevir is a second-generation NS3/4A protease inhibitor approved for use in combination with pegylated interferon and ribavirin. A recent phase 3 trial evaluating simeprevir in patients who had relapsed after prior therapy found sustained virologic response rates to be higher with simeprevir than with placebo, irrespective of IL28B status.36 This finding was similar to that of a trial of telaprevir.16
Sofosbuvir is a nucleotide analogue NS5B polymerase inhibitor that becomes incorporated into the growing RNA, inducing a chain termination event.37 In phase 3 trials,38,39 researchers found an initial rapid decrease in viral load for patients treated with this agent regardless of IL28B status.
In the NEUTRINO trial (Sofosbuvir With Peginterferon Alfa 2a and Ribavirin for 12 Weeks in Treatment-Naive Subjects With Chronic Genotype 1, 4, 5, or 6 HCV Infection),38 which used sofusbuvir in combination with interferon and ribavirin, the rate of sustained virologic response was higher in those with the favorable CC genotype (98%) than with a non-CC genotype (87%).
In COSMOS (A Study of TMC435 in Combination With PSI-7977 [GS7977] in Chronic Hepatitis C Genotype 1-Infected Prior Null Responders to Peginterferon/Ribavirin Therapy or HCV Treatment-Naive Patients),39 which used a combination of simeprevir, sofosbuvir, and ribavirin, the rate of sustained virologic response was higher in those with the CC genotype (100%) than with the TT genotype (83%; Table 1).
These new medications have radically changed the landscape of hepatitis C therapy and have also unlocked the potential for developing completely interferon-free regimens.
Other new interferon-free regimens such as ledipasvir, daclatasvir, and asunaprevir promise high rates of sustained virologic response, which makes the utility of testing for IL28B polymorphisms to predict sustained virologic response very much diminished (Table 1).40,41 However, these new drugs are expected to be expensive, and IL28B polymorphisms may be used to identify candidates who are more likely to respond to pegylated interferon and ribavirin, particularly in resource-poor settings and in developing countries. Additionally, patients who have contraindications to these newer therapies will still likely need an interferon-based regimen, and thus the IL28B polymorphism will still be important in predicting treatment response and prognosis.
IL28B WILL STILL BE RELEVANT IN THE INTERFERON-FREE AGE
The IL28B polymorphism is a strong predictor of spontaneous clearance of hepatitis C virus and responsiveness to interferon-based therapy, and testing for it has demonstrated a great potential to improve patient care. IL28B testing has become available for clinical use and may optimize the outcome of hepatitis C treatment by helping us to select the best treatment for individual patients and minimizing the duration of therapy and the side effects associated with interferon-based antiviral medications.
As newer therapies have shifted toward interferon-free regimens that offer very high sustained virologic response rates, the usefulness of IL28B polymorphism as a clinical test to predict the response rate to antiviral therapy is minimized substantially. It may remain clinically relevant in resource-poor settings and in developing countries, especially in light of the potentially prohibitive costs of the newer regimens, and for patients in whom these treatments are contraindicated. This does not minimize the lesson we learned from the discovery of the IL28B gene and the impact on our understanding of the pathogenesis of hepatitis C virus infection.
- Attia J, Ioannidis JP, Thakkinstian A, et al. How to use an article about genetic association: A: background concepts. JAMA 2009; 301:74–81.
- Samani NJ, Erdmann J, Hall AS, et al; WTCCC and the Cardiogenics Consortium. Genomewide association analysis of coronary artery disease. N Engl J Med 2007; 357:443–453.
- Zeggini E, Weedon MN, Lindgren CM, et al; Wellcome Trust Case Control Consortium (WTCCC). Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science 2007; 316:1336–1341.
- Matarín M, Brown WM, Scholz S, et al. A genome-wide genotyping study in patients with ischaemic stroke: initial analysis and data release. Lancet Neurol 2007; 6:414–420.
- Easton DF, Pooley KA, Dunning AM, et al; AOCS Management Group. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature 2007; 447:1087–1093.
- Plenge RM, Seielstad M, Padyukov L, et al. TRAF1-C5 as a risk locus for rheumatoid arthritis—a genomewide study. N Engl J Med 2007; 357:1199–1209.
- Coon KD, Myers AJ, Craig DW, et al. A high-density whole-genome association study reveals that APOE is the major susceptibility gene for sporadic late-onset Alzheimer’s disease. J Clin Psychiatry 2007; 68:613–618.
- Ge D, Fellay J, Thompson AJ, et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance (letter). Nature 2009; 461:399–401.
- Ali A, Zein NN. Hepatitis C infection: a systemic disease with extrahepatic manifestations. Cleve Clin J Med 2005; 72:1005-1019.
- Hanouneh IA, Feldstein AE, Lopez R, et al. Clinical significance of metabolic syndrome in the setting of chronic hepatitis C virus infection. Clin Gastroenterol Hepatol 2008; 6:584–589.
- Elgouhari HM, Zein CO, Hanouneh I, Feldstein AE, Zein NN. Diabetes mellitus is associated with impaired response to antiviral therapy in chronic hepatitis C infection. Dig Dis Sci 2009; 54:2699–2705.
- Alkhouri N, Zein NN. Protease inhibitors: silver bullets for chronic hepatitis C infection? Cleve Clin J Med 2012; 79:213–222.
- McHutchison JG, Everson GT, Gordon SC, et al; PROVE1 Study Team. Telaprevir with peginterferon and ribavirin for chronic HCV genotype 1 infection. N Engl J Med 2009; 360:1827–1838.
- Jacobson IM, McHutchison JG, Dusheiko G, et al; ADVANCE Study Team. Telaprevir for previously untreated chronic hepatitis C virus infection. N Engl J Med 2011; 364:2405–2416.
- Jacobson IM, Catlett I, Marcellin P, et al. Telaprevir substantially improved SVR rates across all IL28B genotypes in the ADVANCE trial. J Hepatol 2011; 54(suppl 1):S542–S543.
- Pol S, Aerssens J, Zeuzem S, et al. Limited impact of IL28B genotype on response rates in telaprevir-treated patients with prior treatment failure. J Hepatol 2013; 58:883–889.
- Suppiah V, Moldovan M, Ahlenstiel G, et al. IL28B is associated with response to chronic hepatitis C interferon-alpha and ribavirin therapy. Nat Genet 2009; 41:1100–1104.
- Tanaka Y, Nishida N, Sugiyama M, et al. Genome-wide association of IL28B with response to pegylated interferon-alpha and ribavirin therapy for chronic hepatitis C. Nat Genet 2009; 41:1105–1109.
- Thomas DL, Thio CL, Martin MP, et al. Genetic variation in IL28B and spontaneous clearance of hepatitis C virus. Nature 2009; 461:798–801.
- Rehermann B. Hepatitis C virus versus innate and adaptive immune responses: a tale of coevolution and coexistence. J Clin Invest 2009; 119:1745–1754.
- Marcello T, Grakoui A, Barba-Spaeth G, et al. Interferons alpha and lambda inhibit hepatitis C virus replication with distinct signal transduction and gene regulation kinetics. Gastroenterology 2006; 131:1887–1898.
- Doyle SE, Schreckhise H, Khuu-Duong K, et al. Interleukin-29 uses a type 1 interferon-like program to promote antiviral responses in human hepatocytes. Hepatology 2006; 44:896–906.
- Raglow Z, Thoma-Perry C, Gilroy R, Wan YJ. IL28B genotype and the expression of ISGs in normal liver. Liver Int 2013; 33:991–998.
- Prokunina-Olsson L, Muchmore B, Tang W, et al. A variant upstream of IFNL3 (IL28B) creating a new interferon gene IFNL4 is associated with impaired clearance of hepatitis C virus. Nat Genet 2013; 45:164–171.
- Hanouneh IA, Zein NN, Askar M, Lopez R, John B. Interleukin-28B polymorphisms are associated with fibrosing cholestatic hepatitis in recurrent hepatitis C after liver transplantation. Clin Transplant 2012; 26:E335–E336.
- Poynard T, Bedossa P, Opolon P. Natural history of liver fibrosis progression in patients with chronic hepatitis C. The OBSVIRC, METAVIR, CLINIVIR, and DOSVIRC groups. Lancet 1997; 349:825–832.
- Thomas DL, Astemborski J, Rai RM, et al. The natural history of hepatitis C virus infection: host, viral, and environmental factors. JAMA 2000; 284:450–456.
- Bochud PY, Cai T, Overbeck K, et al; Swiss Hepatitis C Cohort Study Group. Genotype 3 is associated with accelerated fibrosis progression in chronic hepatitis C. J Hepatol 2009; 51:655–666.
- Marabita F, Aghemo A, De Nicola S, et al. Genetic variation in the interleukin-28B gene is not associated with fibrosis progression in patients with chronic hepatitis C and known date of infection. Hepatology 2011; 54:1127–1134.
- Fabris C, Falleti E, Cussigh A, et al. IL-28B rs12979860 C/T allele distribution in patients with liver cirrhosis: role in the course of chronic viral hepatitis and the development of HCC. J Hepatol 2011; 54:716–722.
- Eurich D, Boas-Knoop S, Bahra M, et al. Role of IL28B polymorphism in the development of hepatitis C virus-induced hepatocellular carcinoma, graft fibrosis, and posttransplant antiviral therapy. Transplantation 2012; 93:644–649.
- Hanouneh IA, Miller C, Aucejo F, Lopez R, Quinn MK, Zein NN. Recurrent hepatitis C after liver transplantation: on-treatment prediction of response to peginterferon/ribavirin therapy. Liver Transpl 2008; 14:53–58.
- Charlton MR, Thompson A, Veldt BJ, et al. Interleukin-28B polymorphisms are associated with histological recurrence and treatment response following liver transplantation in patients with hepatitis C virus infection. Hepatology 2011; 53:317–324.
- Thompson AJ, Muir AJ, Sulkowski MS, et al. Interleukin-28B polymorphism improves viral kinetics and is the strongest pretreatment predictor of sustained virologic response in genotype 1 hepatitis C virus. Gastroenterology 2010; 139:120–129.e18.
- Beinhardt S, Payer BA, Datz C, et al. A diagnostic score for the prediction of spontaneous resolution of acute hepatitis C virus infection. J Hepatol 2013; 59:972–977.
- Forns X, Lawitz E, Zeuzem S, et al. Simeprevir with peginterferon and ribavirin leads to high rates of SVR in patients with HCV genotype 1 who relapsed after previous therapy: a phase 3 trial. Gastroenterology 2014; 146:1669–1679.e3.
- Sofia MJ, Bao D, Chang W, et al. Discovery of a ß-d-2’-deoxy-2’-ß-fluoro-2’-ß-C-methyluridine nucleotide prodrug (PSI-7977) for the treatment of hepatitis C virus. J Med Chem 2010; 53:7202–7218.
- Lawitz E, Mangia A, Wyles D, et al. Sofosbuvir for previously untreated chronic hepatitis C infection. N Engl J Med 2013; 368:1878–1887.
- Sulkowski MS, Jacobson IM, Ghalib R, et al. Once-daily simeprevir (TMC435) plus sofosbuvir (GS-7977) with or without ribavirin in HCV genotype 1 prior null responders with metavir F0-2: COSMOS study subgroup analysis. 49th EASL, April 2014, London. Oral abstract O7. www.natap.org/2014/EASL/EASL_46.htm. Accesed January 9, 2015.
- Lok AS, Gardiner DF, Lawitz E, et al. Preliminary study of two antiviral agents for hepatitis C genotype 1. N Engl J Med 2012; 366:216–224.
- Afdhal N, Zeuzem S, Kwo P, et al; ION-1 Investigators. Ledipasvir and sofosbuvir for untreated HCV genotype 1 infection. N Engl J Med 2014; 370:1889–1898.
- Attia J, Ioannidis JP, Thakkinstian A, et al. How to use an article about genetic association: A: background concepts. JAMA 2009; 301:74–81.
- Samani NJ, Erdmann J, Hall AS, et al; WTCCC and the Cardiogenics Consortium. Genomewide association analysis of coronary artery disease. N Engl J Med 2007; 357:443–453.
- Zeggini E, Weedon MN, Lindgren CM, et al; Wellcome Trust Case Control Consortium (WTCCC). Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science 2007; 316:1336–1341.
- Matarín M, Brown WM, Scholz S, et al. A genome-wide genotyping study in patients with ischaemic stroke: initial analysis and data release. Lancet Neurol 2007; 6:414–420.
- Easton DF, Pooley KA, Dunning AM, et al; AOCS Management Group. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature 2007; 447:1087–1093.
- Plenge RM, Seielstad M, Padyukov L, et al. TRAF1-C5 as a risk locus for rheumatoid arthritis—a genomewide study. N Engl J Med 2007; 357:1199–1209.
- Coon KD, Myers AJ, Craig DW, et al. A high-density whole-genome association study reveals that APOE is the major susceptibility gene for sporadic late-onset Alzheimer’s disease. J Clin Psychiatry 2007; 68:613–618.
- Ge D, Fellay J, Thompson AJ, et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance (letter). Nature 2009; 461:399–401.
- Ali A, Zein NN. Hepatitis C infection: a systemic disease with extrahepatic manifestations. Cleve Clin J Med 2005; 72:1005-1019.
- Hanouneh IA, Feldstein AE, Lopez R, et al. Clinical significance of metabolic syndrome in the setting of chronic hepatitis C virus infection. Clin Gastroenterol Hepatol 2008; 6:584–589.
- Elgouhari HM, Zein CO, Hanouneh I, Feldstein AE, Zein NN. Diabetes mellitus is associated with impaired response to antiviral therapy in chronic hepatitis C infection. Dig Dis Sci 2009; 54:2699–2705.
- Alkhouri N, Zein NN. Protease inhibitors: silver bullets for chronic hepatitis C infection? Cleve Clin J Med 2012; 79:213–222.
- McHutchison JG, Everson GT, Gordon SC, et al; PROVE1 Study Team. Telaprevir with peginterferon and ribavirin for chronic HCV genotype 1 infection. N Engl J Med 2009; 360:1827–1838.
- Jacobson IM, McHutchison JG, Dusheiko G, et al; ADVANCE Study Team. Telaprevir for previously untreated chronic hepatitis C virus infection. N Engl J Med 2011; 364:2405–2416.
- Jacobson IM, Catlett I, Marcellin P, et al. Telaprevir substantially improved SVR rates across all IL28B genotypes in the ADVANCE trial. J Hepatol 2011; 54(suppl 1):S542–S543.
- Pol S, Aerssens J, Zeuzem S, et al. Limited impact of IL28B genotype on response rates in telaprevir-treated patients with prior treatment failure. J Hepatol 2013; 58:883–889.
- Suppiah V, Moldovan M, Ahlenstiel G, et al. IL28B is associated with response to chronic hepatitis C interferon-alpha and ribavirin therapy. Nat Genet 2009; 41:1100–1104.
- Tanaka Y, Nishida N, Sugiyama M, et al. Genome-wide association of IL28B with response to pegylated interferon-alpha and ribavirin therapy for chronic hepatitis C. Nat Genet 2009; 41:1105–1109.
- Thomas DL, Thio CL, Martin MP, et al. Genetic variation in IL28B and spontaneous clearance of hepatitis C virus. Nature 2009; 461:798–801.
- Rehermann B. Hepatitis C virus versus innate and adaptive immune responses: a tale of coevolution and coexistence. J Clin Invest 2009; 119:1745–1754.
- Marcello T, Grakoui A, Barba-Spaeth G, et al. Interferons alpha and lambda inhibit hepatitis C virus replication with distinct signal transduction and gene regulation kinetics. Gastroenterology 2006; 131:1887–1898.
- Doyle SE, Schreckhise H, Khuu-Duong K, et al. Interleukin-29 uses a type 1 interferon-like program to promote antiviral responses in human hepatocytes. Hepatology 2006; 44:896–906.
- Raglow Z, Thoma-Perry C, Gilroy R, Wan YJ. IL28B genotype and the expression of ISGs in normal liver. Liver Int 2013; 33:991–998.
- Prokunina-Olsson L, Muchmore B, Tang W, et al. A variant upstream of IFNL3 (IL28B) creating a new interferon gene IFNL4 is associated with impaired clearance of hepatitis C virus. Nat Genet 2013; 45:164–171.
- Hanouneh IA, Zein NN, Askar M, Lopez R, John B. Interleukin-28B polymorphisms are associated with fibrosing cholestatic hepatitis in recurrent hepatitis C after liver transplantation. Clin Transplant 2012; 26:E335–E336.
- Poynard T, Bedossa P, Opolon P. Natural history of liver fibrosis progression in patients with chronic hepatitis C. The OBSVIRC, METAVIR, CLINIVIR, and DOSVIRC groups. Lancet 1997; 349:825–832.
- Thomas DL, Astemborski J, Rai RM, et al. The natural history of hepatitis C virus infection: host, viral, and environmental factors. JAMA 2000; 284:450–456.
- Bochud PY, Cai T, Overbeck K, et al; Swiss Hepatitis C Cohort Study Group. Genotype 3 is associated with accelerated fibrosis progression in chronic hepatitis C. J Hepatol 2009; 51:655–666.
- Marabita F, Aghemo A, De Nicola S, et al. Genetic variation in the interleukin-28B gene is not associated with fibrosis progression in patients with chronic hepatitis C and known date of infection. Hepatology 2011; 54:1127–1134.
- Fabris C, Falleti E, Cussigh A, et al. IL-28B rs12979860 C/T allele distribution in patients with liver cirrhosis: role in the course of chronic viral hepatitis and the development of HCC. J Hepatol 2011; 54:716–722.
- Eurich D, Boas-Knoop S, Bahra M, et al. Role of IL28B polymorphism in the development of hepatitis C virus-induced hepatocellular carcinoma, graft fibrosis, and posttransplant antiviral therapy. Transplantation 2012; 93:644–649.
- Hanouneh IA, Miller C, Aucejo F, Lopez R, Quinn MK, Zein NN. Recurrent hepatitis C after liver transplantation: on-treatment prediction of response to peginterferon/ribavirin therapy. Liver Transpl 2008; 14:53–58.
- Charlton MR, Thompson A, Veldt BJ, et al. Interleukin-28B polymorphisms are associated with histological recurrence and treatment response following liver transplantation in patients with hepatitis C virus infection. Hepatology 2011; 53:317–324.
- Thompson AJ, Muir AJ, Sulkowski MS, et al. Interleukin-28B polymorphism improves viral kinetics and is the strongest pretreatment predictor of sustained virologic response in genotype 1 hepatitis C virus. Gastroenterology 2010; 139:120–129.e18.
- Beinhardt S, Payer BA, Datz C, et al. A diagnostic score for the prediction of spontaneous resolution of acute hepatitis C virus infection. J Hepatol 2013; 59:972–977.
- Forns X, Lawitz E, Zeuzem S, et al. Simeprevir with peginterferon and ribavirin leads to high rates of SVR in patients with HCV genotype 1 who relapsed after previous therapy: a phase 3 trial. Gastroenterology 2014; 146:1669–1679.e3.
- Sofia MJ, Bao D, Chang W, et al. Discovery of a ß-d-2’-deoxy-2’-ß-fluoro-2’-ß-C-methyluridine nucleotide prodrug (PSI-7977) for the treatment of hepatitis C virus. J Med Chem 2010; 53:7202–7218.
- Lawitz E, Mangia A, Wyles D, et al. Sofosbuvir for previously untreated chronic hepatitis C infection. N Engl J Med 2013; 368:1878–1887.
- Sulkowski MS, Jacobson IM, Ghalib R, et al. Once-daily simeprevir (TMC435) plus sofosbuvir (GS-7977) with or without ribavirin in HCV genotype 1 prior null responders with metavir F0-2: COSMOS study subgroup analysis. 49th EASL, April 2014, London. Oral abstract O7. www.natap.org/2014/EASL/EASL_46.htm. Accesed January 9, 2015.
- Lok AS, Gardiner DF, Lawitz E, et al. Preliminary study of two antiviral agents for hepatitis C genotype 1. N Engl J Med 2012; 366:216–224.
- Afdhal N, Zeuzem S, Kwo P, et al; ION-1 Investigators. Ledipasvir and sofosbuvir for untreated HCV genotype 1 infection. N Engl J Med 2014; 370:1889–1898.
KEY POINTS
- In IL28B, the rs12979860 location can be occupied by either cytosine (C) or thymine (T). The CC genotype is more favorable than the CT or TT genotype.
- Testing for the IL28B polymorphism is currently available and allows for better outcomes through proper selection of treatment, particularly with interferon-based treatment.
- Although newer therapies have shifted toward regimens that do not use interferon, the IL28B polymorphism remains clinically significant, especially in light of the potentially prohibitive costs of the newer regimens, and for patients in whom these treatments are contraindicated.
A 25-year-old man with very high alkaline phosphatase
A 25-year-old man presented to his primary care physician with generalized malaise. His symptoms started around 2 months earlier with progressive fatigue, nausea, decreased appetite, and weight loss (15 lb in 2 months). He denied having fever, chills, night sweats, abdominal pain, diarrhea, melena, or hematochezia.
His medical history was remarkable only for depression, well controlled with sertraline (Zoloft), which he started taking 3 years ago. He was not taking any other prescribed, over-the-counter, or herbal medications.
He had no family history of cancer or liver disease. He did not smoke and rarely drank alcohol. He had never used recreational drugs. He was sexually active with one female partner, used condoms for protection, and had never been diagnosed with a sexually transmitted disease. He had not traveled recently and had not been exposed to any pet.
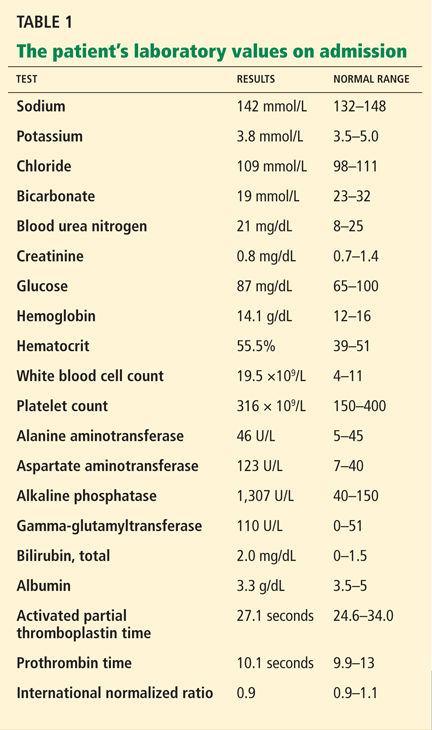
The patient’s laboratory values on admission are shown in Table 1. Of note, his serum alkaline phosphatase level was 1,307 U/L (reference range 40–150 U/L).
LIVER TESTS CAN NARROW THE DIAGNOSIS
The most commonly used laboratory tests of the liver can be classified into those that measure either:
- Liver synthetic function (eg, the serum albumin and bilirubin concentrations and the prothrombin time) or
- Liver damage, as reflected by the serum concentrations of the enzymes alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase, and gamma-glutamyltransferase (GGT).1,2
ALT and AST are normally concentrated in the hepatocytes and thus, when present in the serum in elevated concentrations, are markers of liver cell injury. The serum levels of these enzymes start to increase within a few hours of liver cell injury as they leak out of the cells via the damaged cell membrane. AST is less liver-specific than ALT, since AST levels can be elevated not only in liver injury but also in muscle, cardiac, and red blood cell injury.3,4
Alkaline phosphatase is actually a heterogeneous group of enzymes found mainly in liver and bone cells. Hepatic alkaline phosphatase is concentrated near the biliary canalicular membrane of the hepatocyte. Accordingly, increased levels of hepatic alkaline phosphatase are mainly seen in liver diseases that predominantly affect the biliary system.3
GGT is also concentrated in hepatic biliary epithelial cells, and thus GGT elevation is another marker of hepatobiliary disease. In fact, measuring the GGT level can help to determine whether an isolated elevation of alkaline phosphatase is due to liver injury.2,3
Accordingly, liver diseases can be classified into two broad categories:
- Hepatocellular injury, in which the primary injury occurs to the hepatocytes
- Cholestatic injury, in which the primary injury is to the bile ducts.
In the former, elevated levels of ALT and AST predominate, while in the latter, elevated alkaline phosphatase is the main finding.3
WHAT TEST NEXT FOR OUR PATIENT?
1. What is the next most appropriate diagnostic step for our patient?
- Liver biopsy
- Ultrasonography of the liver
- Computed tomography (CT) of the liver
- Observation
Our patient has an elevated GGT level, which suggests that his elevated alkaline phosphatase is of hepatic rather than bony origin. Moreover, a serum alkaline phosphatase level that is elevated out of proportion to the aminotransferase levels reflects cholestatic liver injury.
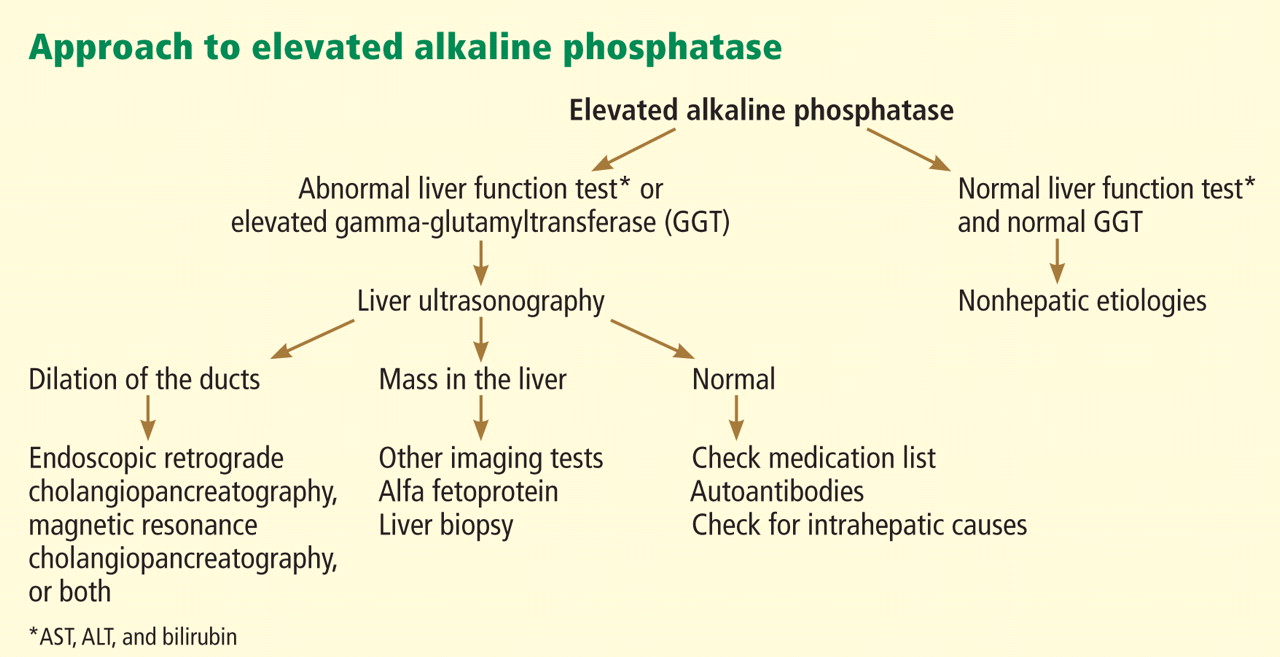
CASE CONTINUED: ULTRASONOGRAPHY IS MOSTLY NORMAL
Ultrasonography of the right upper quadrant revealed that the liver had normal echogenicity and was mildly enlarged. There was no focal hepatic lesion. The gallbladder appeared normal, with no stones or sludge. No dilated intrahepatic or extrahepatic biliary ducts were seen. The common bile duct measured 4 mm. A small amount of ascites not amenable to paracentesis was present.
Thus, in the absence of biliary dilation on ultrasonography, we are dealing with an intrahepatic cholestatic process.
CAUSES OF CHOLESTATIC LIVER DISEASE
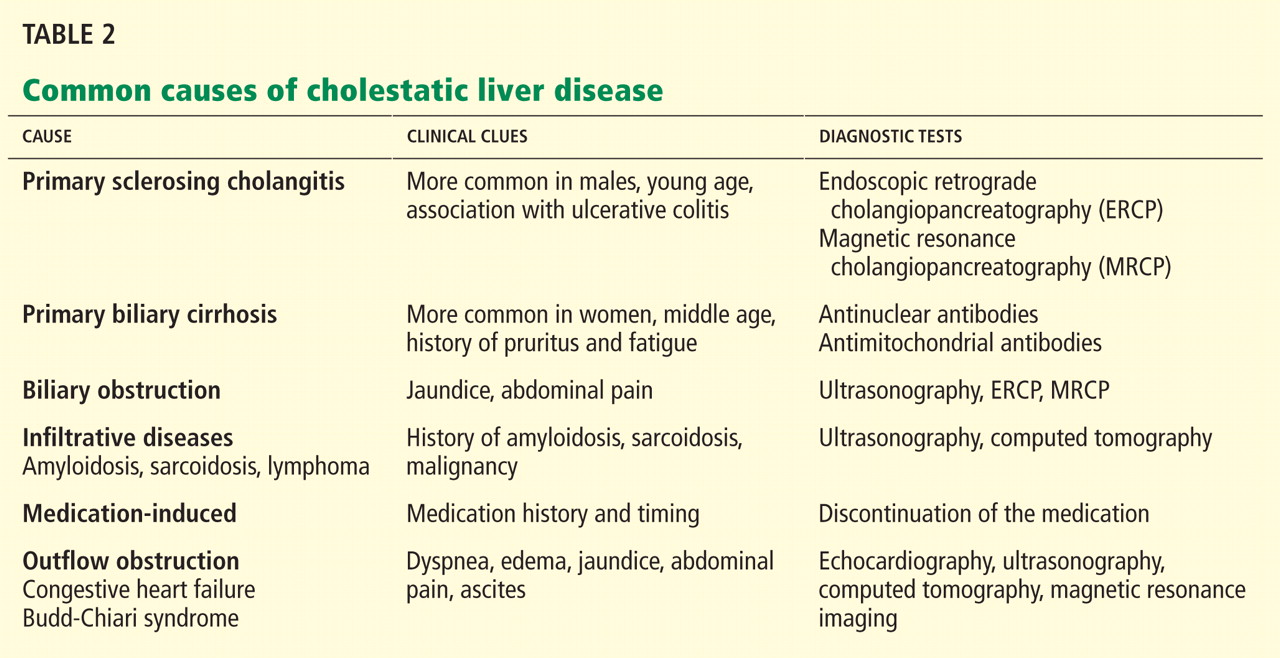
Viral hepatitis
Viral hepatitis most often produces a hepatocellular pattern of injury (ie, AST and ALT elevations predominate). However, in rare cases it can cause a cholestatic pattern of injury.
Our patient subsequently had serologic tests for viral hepatitis, including hepatitis A, B, and C, and the results were negative.
Autoimmune liver disease
The three most common forms of autoimmune liver disease are autoimmune hepatitis, primary biliary cirrhosis, and primary sclerosing cholangitis.
Autoimmune hepatitis is characterized by high serum ALT and AST levels, whereas primary biliary cirrhosis and primary sclerosing cholangitis are associated with predominant elevations of alkaline phosphatase, since they are cholestatic disorders.
Our patient’s alkaline phosphatase level was much higher than his ALT and AST levels, making the latter two diseases more likely.
Primary biliary cirrhosis (and autoimmune hepatitis) are associated with autoantibodies in the serum, such as antinuclear antibody, smooth muscle antibody, and antimitochondrial antibody.
Our patient subsequently was tested for these antibodies, and the results were negative.
Primary sclerosing cholangitis usually affects the extrahepatic biliary system. Thus, if it is present, abnormalities should be seen on imaging.
As mentioned previously, no dilated intrahepatic or extrahepatic biliary ducts were seen on ultrasonography in our patient. Moreover, primary sclerosing cholangitis is associated with inflammatory bowel disease, particularly ulcerative colitis, which our patient did not have.
Drug-induced liver injury
Drug-induced liver injury is a common cause of cholestatic liver disease. However, our patient was not taking any prescribed, over-the-counter, or herbal medications. Additionally, he denied heavy alcohol use.
Infiltrative disorders
Infiltrative disorders such as amyloidosis, sarcoidosis, or lymphoma should be considered in the differential diagnosis of cholestatic liver disease. A clue to a possible infiltrative process is a markedly elevated level of alkaline phosphatase with a mildly increased serum bilirubin concentration, both of which our patient had.
AFTER ULTRASONOGRAPHY, WHAT IS THE NEXT STEP?
2. Which of the following is the next most appropriate diagnostic test for our patient?
- Endoscopic retrograde cholangiopancreatography (ERCP)
- Magnetic resonance cholangiopancreatography (MRCP)
- Liver biopsy
- CT of the abdomen
Figure 1 shows a proposed algorithm for evaluating increased alkaline phosphatase levels.
If there is no biliary duct dilation on ultrasonography, then abnormal levels of alkaline phosphatase most likely represent an intrahepatic pattern of cholestatic liver injury. Therefore, additional imaging with CT or magnetic resonance imaging is of limited diagnostic value. ERCP is used today for therapy rather than diagnosis, so its use is limited to patients known to have dilated biliary ducts on imaging. Liver biopsy, however, can provide useful findings.
Case continued: He undergoes biopsy
Our patient underwent transjugular liver biopsy. During the procedure, transjugular venography showed stenosis in the right, middle, and left hepatic veins and the hepatic portion of the inferior vena cava, consistent with Budd-Chiari syndrome.
The liver biopsy specimen was positive for extensive deposition of slight eosinophilic and amorphous material in a sinusoidal pattern in the liver parenchyma, as well as in the portal tracts, with markedly atrophic hepatocytes. Congo red birefringence confirmed the diagnosis of amyloidosis. The immunohistochemical phenotype was positive for kappa light chains, which is diagnostic for primary-type amyloidosis, also called amyloidosis of light chain composition, or AL.
Bone marrow aspiration and bone marrow biopsy were performed and showed 22% plasma cells, well above the normal range (0–2%), consistent with the diagnosis of multiple myeloma.
BUDD-CHIARI SYNDROME: A CHALLENGING DIAGNOSIS
Budd-Chiari syndrome is a rare condition characterized by obstruction of venous outflow from the liver at a site that may vary from the small hepatic veins up to the inferior vena cava or even the right atrium.5,6 Obstruction of hepatic venous outflow leads to sinusoidal congestion and hypoxic damage of the hepatocytes.7 Hypoxia and necrosis of the hepatocytes result in the release of free radicals. Cirrhosis can eventually occur secondary to ischemic necrosis of hepatocytes and hepatic fibrosis.8
The estimated incidence of this syndrome is 1 in 2.5 million persons per year.7 It is more prevalent in women and young adults.8
Heterogeneous in its causes and manifestations

Budd-Chiari syndrome is also heterogeneous in its manifestations, which depend on the extent of the occlusion, on the acuteness of the obstruction, and on whether venous collateral circulation has developed to decompress the liver sinusoids.9,12,13 Therefore, on the basis of its clinical manifestations, it can be classified as fulminant, acute, subacute, or chronic.12–16
The fulminant form presents with hepatic encephalopathy within 8 weeks after the development of jaundice. The subacute form, which is the most common, has a more insidious onset in which hepatic sinusoids are decompressed by portal and hepatic venous collateral circulation. The patient usually presents with abdominal pain, ascites, hepatomegaly, nausea, vomiting, and mild jaundice. Finally the chronic form presents as complications of cirrhosis.12–16
Imaging plays an important role in diagnosing Budd-Chiari syndrome
Imaging plays an important role in detecting and classifying Budd-Chiari syndrome.
Duplex ultrasonography is useful for detecting this syndrome and has a sensitivity and specificity of 85%.9
CT and magnetic resonance imaging can also help in the diagnosis by showing thrombosis, obstruction, or occlusion in the hepatic vein or the inferior vena cava.5
Venography is the gold standard for diagnosis. However, it should be performed only if noninvasive tests are negative or nondiagnostic and there is a high clinical suspicion of this disease.17 Budd-Chiari syndrome has a characteristic pattern on venography known as “spider web,” which is due to the formation of venous collaterals to bypass the occluded hepatic veins.9
Liver biopsy is not necessarily required to confirm the diagnosis of Budd-Chiari syndrome, but it can help in diagnosing the acute or subacute forms and also in ruling out other causes. Histologic findings can include centrizonal congestion, loss of hepatocytes, hemorrhage, and fibrosis.18,19 Regenerative nodules are found in about 25% of patients.19
TREATING BUDD-CHIARI SYNDROME
The primary goal of treatment is to prevent further extension of the venous thrombosis in the hepatic veins, in their collaterals, and in the intrahepatic and extrahepatic portal venous system. Resolution of hepatic congestion improves liver perfusion and preserves function of the hepatocytes.
Anticoagulation is recommended in the early stages. Heparin therapy should be initiated and subsequently switched to warfarin with the goal of achieving an international normalized ratio of the prothrombin time of 2.0 to 2.5.8,9,19
Thrombolysis is effective in the acute form.20,21 Recanalization, including percutaneous or transhepatic angioplasty of localized segments of the narrowed hepatic veins or inferior vena cava, has long-term patency rates of 80% to 90%.22
If thrombolytic therapy and angioplasty are unsuccessful, a transjugular intrahepatic portosystemic shunt or a surgical procedure (side-to-side portocaval shunt, central splenorenal shunt, or mesocaval shunt) should be considered.9
Liver transplantation is another treatment option in those with fulminant Budd-Chiari syndrome or advanced liver cirrhosis.8
PROGNOSIS HAS IMPROVED
The prognosis of Budd-Chiari syndrome has improved, thanks to both earlier diagnosis and new treatments. The 1-year survival rate, which was about 60% before 1985, has increased to more than 80% in recent cohort studies.19
Studies have shown that the Child-Pugh score, which is based on a combination of serum albumin, bilirubin, prothrombin time, encephalopathy, and ascites, can be considered as an independent prognostic factor. A lower Child-Pugh score and a younger age are associated with a good prognosis.19,23,24 (The Child-Pugh score cannot be applied to our patient because he does not have cirrhosis.)
What happened to our patient?
Our patient was started on anticoagulation for his Budd-Chiari syndrome and on bortezomib (Velcade) and dexamethasone for his multiple myeloma. He achieved remarkable improvement in his liver function tests. Follow-up duplex ultrasonography 1 month after discharge revealed that the stenosis in the hepatic veins had resolved. He is following up with the oncology clinic for management of his multiple myeloma.
- Folwaczny C. Efficient diagnostics for elevated transaminases. [Article in German] MMW Fortschr Med 2007; 149:44–48.
- Moussavian SN, Becker RC, Piepmeyer JL, Mezey E, Bozian RC. Serum gamma-glutamyl transpeptidase and chronic alcoholism. Influence of alcohol ingestion and liver disease. Dig Dis Sci 1985; 30:211–214.
- Aragon G, Younossi ZM. When and how to evaluate mildly elevated liver enzymes in apparently healthy patients. Cleve Clin J Med 2010; 77:195–204.
- Lepper PM, Dufour JF. Elevated transaminases—what to do if everything was done?. [Article in German] Praxis (Bern 1994) 2009; 98:330–334.
- Buzas C, Sparchez Z, Cucuianu A, Manole S, Lupescu I, Acalovschi M. Budd-Chiari syndrome secondary to polycythemia vera. A case report. J Gastrointestin Liver Dis 2009; 18:363–366.
- Valla DC. Primary Budd-Chiari syndrome. J Hepatol 2009; 50:195–203.
- Rautou PE, Moucari R, Cazals-Hatem D, et al. Levels and initial course of serum alanine aminotransferase can predict outcome of patients with Budd-Chiari syndrome. Clin Gastroenterol Hepatol 2009; 7:1230–1235.
- Cura M, Haskal Z, Lopera J. Diagnostic and interventional radiology for Budd-Chiari syndrome. Radiographics 2009; 29:669–681.
- Menon KV, Shah V, Kamath PS. The Budd-Chiari syndrome. N Engl J Med 2004; 350:578–585.
- Darwish Murad S, Plessier A, Hernandez-Guerra M, et al; EN-Vie (European Network for Vascular Disorders of the Liver). Etiology, management, and outcome of the Budd-Chiari syndrome. Ann Intern Med 2009; 151:167–175.
- Valla D, Le MG, Poynard T, Zucman N, Rueff B, Benhamou JP. Risk of hepatic vein thrombosis in relation to recent use of oral contraceptives. A case-control study. Gastroenterology 1986; 90:807–811.
- Bismuth H, Sherlock DJ. Portasystemic shunting versus liver transplantation for the Budd-Chiari syndrome. Ann Surg 1991; 214:581–589.
- Orloff MJ, Daily PO, Orloff SL, Girard B, Orloff MS. A 27-year experience with surgical treatment of Budd-Chiari syndrome. Ann Surg 2000; 232:340–352.
- Dilawari JB, Bambery P, Chawla Y, et al. Hepatic outflow obstruction (Budd-Chiari syndrome). Experience with 177 patients and a review of the literature. Medicine (Baltimore) 1994; 73:21–36.
- Mahmoud AE, Mendoza A, Meshikhes AN, et al. Clinical spectrum, investigations and treatment of Budd-Chiari syndrome. QJM 1996; 89:37–43.
- Klein AS, Cameron JL. Diagnosis and management of the Budd-Chiari syndrome. Am J Surg 1990; 160:128–133.
- Plessier A, Valla DC. Budd-Chiari syndrome. Semin Liver Dis 2008; 28:259–269.
- Cazals-Hatem D, Vilgrain V, Genin P, et al. Arterial and portal circulation and parenchymal changes in Budd-Chiari syndrome: a study in 17 explanted livers. Hepatology 2003; 37:510–519.
- Hoekstra J, Janssen HL. Vascular liver disorders (I): diagnosis, treatment and prognosis of Budd-Chiari syndrome. Neth J Med 2008; 66:334–359.
- Frank JW, Kamath PS, Stanson AW. Budd-Chiari syndrome: early intervention with angioplasty and thrombolytic therapy. Mayo Clin Proc 1994; 69:877–881.
- Raju GS, Felver M, Olin JW, Satti SD. Thrombolysis for acute Budd-Chiari syndrome: case report and literature review. Am J Gastroenterol 1996; 91:1262–1263.
- Fisher NC, McCafferty I, Dolapci M, et al. Managing Budd-Chiari syndrome: a retrospective review of percutaneous hepatic vein angioplasty and surgical shunting. Gut 1999; 44:568–574.
- Zeitoun G, Escolano S, Hadengue A, et al. Outcome of Budd-Chiari syndrome: a multivariate analysis of factors related to survival including surgical portosystemic shunting. Hepatology 1999; 30:84–89.
- Darwish Murad S, Valla DC, de Groen PC, et al. Determinants of survival and the effect of portosystemic shunting in patients with Budd-Chiari syndrome. Hepatology 2004; 39:500–508.
A 25-year-old man presented to his primary care physician with generalized malaise. His symptoms started around 2 months earlier with progressive fatigue, nausea, decreased appetite, and weight loss (15 lb in 2 months). He denied having fever, chills, night sweats, abdominal pain, diarrhea, melena, or hematochezia.
His medical history was remarkable only for depression, well controlled with sertraline (Zoloft), which he started taking 3 years ago. He was not taking any other prescribed, over-the-counter, or herbal medications.
He had no family history of cancer or liver disease. He did not smoke and rarely drank alcohol. He had never used recreational drugs. He was sexually active with one female partner, used condoms for protection, and had never been diagnosed with a sexually transmitted disease. He had not traveled recently and had not been exposed to any pet.
The patient’s laboratory values on admission are shown in Table 1. Of note, his serum alkaline phosphatase level was 1,307 U/L (reference range 40–150 U/L).
LIVER TESTS CAN NARROW THE DIAGNOSIS
The most commonly used laboratory tests of the liver can be classified into those that measure either:
- Liver synthetic function (eg, the serum albumin and bilirubin concentrations and the prothrombin time) or
- Liver damage, as reflected by the serum concentrations of the enzymes alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase, and gamma-glutamyltransferase (GGT).1,2
ALT and AST are normally concentrated in the hepatocytes and thus, when present in the serum in elevated concentrations, are markers of liver cell injury. The serum levels of these enzymes start to increase within a few hours of liver cell injury as they leak out of the cells via the damaged cell membrane. AST is less liver-specific than ALT, since AST levels can be elevated not only in liver injury but also in muscle, cardiac, and red blood cell injury.3,4
Alkaline phosphatase is actually a heterogeneous group of enzymes found mainly in liver and bone cells. Hepatic alkaline phosphatase is concentrated near the biliary canalicular membrane of the hepatocyte. Accordingly, increased levels of hepatic alkaline phosphatase are mainly seen in liver diseases that predominantly affect the biliary system.3
GGT is also concentrated in hepatic biliary epithelial cells, and thus GGT elevation is another marker of hepatobiliary disease. In fact, measuring the GGT level can help to determine whether an isolated elevation of alkaline phosphatase is due to liver injury.2,3
Accordingly, liver diseases can be classified into two broad categories:
- Hepatocellular injury, in which the primary injury occurs to the hepatocytes
- Cholestatic injury, in which the primary injury is to the bile ducts.
In the former, elevated levels of ALT and AST predominate, while in the latter, elevated alkaline phosphatase is the main finding.3
WHAT TEST NEXT FOR OUR PATIENT?
1. What is the next most appropriate diagnostic step for our patient?
- Liver biopsy
- Ultrasonography of the liver
- Computed tomography (CT) of the liver
- Observation
Our patient has an elevated GGT level, which suggests that his elevated alkaline phosphatase is of hepatic rather than bony origin. Moreover, a serum alkaline phosphatase level that is elevated out of proportion to the aminotransferase levels reflects cholestatic liver injury.
CASE CONTINUED: ULTRASONOGRAPHY IS MOSTLY NORMAL
Ultrasonography of the right upper quadrant revealed that the liver had normal echogenicity and was mildly enlarged. There was no focal hepatic lesion. The gallbladder appeared normal, with no stones or sludge. No dilated intrahepatic or extrahepatic biliary ducts were seen. The common bile duct measured 4 mm. A small amount of ascites not amenable to paracentesis was present.
Thus, in the absence of biliary dilation on ultrasonography, we are dealing with an intrahepatic cholestatic process.
CAUSES OF CHOLESTATIC LIVER DISEASE
Viral hepatitis
Viral hepatitis most often produces a hepatocellular pattern of injury (ie, AST and ALT elevations predominate). However, in rare cases it can cause a cholestatic pattern of injury.
Our patient subsequently had serologic tests for viral hepatitis, including hepatitis A, B, and C, and the results were negative.
Autoimmune liver disease
The three most common forms of autoimmune liver disease are autoimmune hepatitis, primary biliary cirrhosis, and primary sclerosing cholangitis.
Autoimmune hepatitis is characterized by high serum ALT and AST levels, whereas primary biliary cirrhosis and primary sclerosing cholangitis are associated with predominant elevations of alkaline phosphatase, since they are cholestatic disorders.
Our patient’s alkaline phosphatase level was much higher than his ALT and AST levels, making the latter two diseases more likely.
Primary biliary cirrhosis (and autoimmune hepatitis) are associated with autoantibodies in the serum, such as antinuclear antibody, smooth muscle antibody, and antimitochondrial antibody.
Our patient subsequently was tested for these antibodies, and the results were negative.
Primary sclerosing cholangitis usually affects the extrahepatic biliary system. Thus, if it is present, abnormalities should be seen on imaging.
As mentioned previously, no dilated intrahepatic or extrahepatic biliary ducts were seen on ultrasonography in our patient. Moreover, primary sclerosing cholangitis is associated with inflammatory bowel disease, particularly ulcerative colitis, which our patient did not have.
Drug-induced liver injury
Drug-induced liver injury is a common cause of cholestatic liver disease. However, our patient was not taking any prescribed, over-the-counter, or herbal medications. Additionally, he denied heavy alcohol use.
Infiltrative disorders
Infiltrative disorders such as amyloidosis, sarcoidosis, or lymphoma should be considered in the differential diagnosis of cholestatic liver disease. A clue to a possible infiltrative process is a markedly elevated level of alkaline phosphatase with a mildly increased serum bilirubin concentration, both of which our patient had.
AFTER ULTRASONOGRAPHY, WHAT IS THE NEXT STEP?
2. Which of the following is the next most appropriate diagnostic test for our patient?
- Endoscopic retrograde cholangiopancreatography (ERCP)
- Magnetic resonance cholangiopancreatography (MRCP)
- Liver biopsy
- CT of the abdomen
Figure 1 shows a proposed algorithm for evaluating increased alkaline phosphatase levels.
If there is no biliary duct dilation on ultrasonography, then abnormal levels of alkaline phosphatase most likely represent an intrahepatic pattern of cholestatic liver injury. Therefore, additional imaging with CT or magnetic resonance imaging is of limited diagnostic value. ERCP is used today for therapy rather than diagnosis, so its use is limited to patients known to have dilated biliary ducts on imaging. Liver biopsy, however, can provide useful findings.
Case continued: He undergoes biopsy
Our patient underwent transjugular liver biopsy. During the procedure, transjugular venography showed stenosis in the right, middle, and left hepatic veins and the hepatic portion of the inferior vena cava, consistent with Budd-Chiari syndrome.
The liver biopsy specimen was positive for extensive deposition of slight eosinophilic and amorphous material in a sinusoidal pattern in the liver parenchyma, as well as in the portal tracts, with markedly atrophic hepatocytes. Congo red birefringence confirmed the diagnosis of amyloidosis. The immunohistochemical phenotype was positive for kappa light chains, which is diagnostic for primary-type amyloidosis, also called amyloidosis of light chain composition, or AL.
Bone marrow aspiration and bone marrow biopsy were performed and showed 22% plasma cells, well above the normal range (0–2%), consistent with the diagnosis of multiple myeloma.
BUDD-CHIARI SYNDROME: A CHALLENGING DIAGNOSIS
Budd-Chiari syndrome is a rare condition characterized by obstruction of venous outflow from the liver at a site that may vary from the small hepatic veins up to the inferior vena cava or even the right atrium.5,6 Obstruction of hepatic venous outflow leads to sinusoidal congestion and hypoxic damage of the hepatocytes.7 Hypoxia and necrosis of the hepatocytes result in the release of free radicals. Cirrhosis can eventually occur secondary to ischemic necrosis of hepatocytes and hepatic fibrosis.8
The estimated incidence of this syndrome is 1 in 2.5 million persons per year.7 It is more prevalent in women and young adults.8
Heterogeneous in its causes and manifestations
Budd-Chiari syndrome is also heterogeneous in its manifestations, which depend on the extent of the occlusion, on the acuteness of the obstruction, and on whether venous collateral circulation has developed to decompress the liver sinusoids.9,12,13 Therefore, on the basis of its clinical manifestations, it can be classified as fulminant, acute, subacute, or chronic.12–16
The fulminant form presents with hepatic encephalopathy within 8 weeks after the development of jaundice. The subacute form, which is the most common, has a more insidious onset in which hepatic sinusoids are decompressed by portal and hepatic venous collateral circulation. The patient usually presents with abdominal pain, ascites, hepatomegaly, nausea, vomiting, and mild jaundice. Finally the chronic form presents as complications of cirrhosis.12–16
Imaging plays an important role in diagnosing Budd-Chiari syndrome
Imaging plays an important role in detecting and classifying Budd-Chiari syndrome.
Duplex ultrasonography is useful for detecting this syndrome and has a sensitivity and specificity of 85%.9
CT and magnetic resonance imaging can also help in the diagnosis by showing thrombosis, obstruction, or occlusion in the hepatic vein or the inferior vena cava.5
Venography is the gold standard for diagnosis. However, it should be performed only if noninvasive tests are negative or nondiagnostic and there is a high clinical suspicion of this disease.17 Budd-Chiari syndrome has a characteristic pattern on venography known as “spider web,” which is due to the formation of venous collaterals to bypass the occluded hepatic veins.9
Liver biopsy is not necessarily required to confirm the diagnosis of Budd-Chiari syndrome, but it can help in diagnosing the acute or subacute forms and also in ruling out other causes. Histologic findings can include centrizonal congestion, loss of hepatocytes, hemorrhage, and fibrosis.18,19 Regenerative nodules are found in about 25% of patients.19
TREATING BUDD-CHIARI SYNDROME
The primary goal of treatment is to prevent further extension of the venous thrombosis in the hepatic veins, in their collaterals, and in the intrahepatic and extrahepatic portal venous system. Resolution of hepatic congestion improves liver perfusion and preserves function of the hepatocytes.
Anticoagulation is recommended in the early stages. Heparin therapy should be initiated and subsequently switched to warfarin with the goal of achieving an international normalized ratio of the prothrombin time of 2.0 to 2.5.8,9,19
Thrombolysis is effective in the acute form.20,21 Recanalization, including percutaneous or transhepatic angioplasty of localized segments of the narrowed hepatic veins or inferior vena cava, has long-term patency rates of 80% to 90%.22
If thrombolytic therapy and angioplasty are unsuccessful, a transjugular intrahepatic portosystemic shunt or a surgical procedure (side-to-side portocaval shunt, central splenorenal shunt, or mesocaval shunt) should be considered.9
Liver transplantation is another treatment option in those with fulminant Budd-Chiari syndrome or advanced liver cirrhosis.8
PROGNOSIS HAS IMPROVED
The prognosis of Budd-Chiari syndrome has improved, thanks to both earlier diagnosis and new treatments. The 1-year survival rate, which was about 60% before 1985, has increased to more than 80% in recent cohort studies.19
Studies have shown that the Child-Pugh score, which is based on a combination of serum albumin, bilirubin, prothrombin time, encephalopathy, and ascites, can be considered as an independent prognostic factor. A lower Child-Pugh score and a younger age are associated with a good prognosis.19,23,24 (The Child-Pugh score cannot be applied to our patient because he does not have cirrhosis.)
What happened to our patient?
Our patient was started on anticoagulation for his Budd-Chiari syndrome and on bortezomib (Velcade) and dexamethasone for his multiple myeloma. He achieved remarkable improvement in his liver function tests. Follow-up duplex ultrasonography 1 month after discharge revealed that the stenosis in the hepatic veins had resolved. He is following up with the oncology clinic for management of his multiple myeloma.
A 25-year-old man presented to his primary care physician with generalized malaise. His symptoms started around 2 months earlier with progressive fatigue, nausea, decreased appetite, and weight loss (15 lb in 2 months). He denied having fever, chills, night sweats, abdominal pain, diarrhea, melena, or hematochezia.
His medical history was remarkable only for depression, well controlled with sertraline (Zoloft), which he started taking 3 years ago. He was not taking any other prescribed, over-the-counter, or herbal medications.
He had no family history of cancer or liver disease. He did not smoke and rarely drank alcohol. He had never used recreational drugs. He was sexually active with one female partner, used condoms for protection, and had never been diagnosed with a sexually transmitted disease. He had not traveled recently and had not been exposed to any pet.
The patient’s laboratory values on admission are shown in Table 1. Of note, his serum alkaline phosphatase level was 1,307 U/L (reference range 40–150 U/L).
LIVER TESTS CAN NARROW THE DIAGNOSIS
The most commonly used laboratory tests of the liver can be classified into those that measure either:
- Liver synthetic function (eg, the serum albumin and bilirubin concentrations and the prothrombin time) or
- Liver damage, as reflected by the serum concentrations of the enzymes alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase, and gamma-glutamyltransferase (GGT).1,2
ALT and AST are normally concentrated in the hepatocytes and thus, when present in the serum in elevated concentrations, are markers of liver cell injury. The serum levels of these enzymes start to increase within a few hours of liver cell injury as they leak out of the cells via the damaged cell membrane. AST is less liver-specific than ALT, since AST levels can be elevated not only in liver injury but also in muscle, cardiac, and red blood cell injury.3,4
Alkaline phosphatase is actually a heterogeneous group of enzymes found mainly in liver and bone cells. Hepatic alkaline phosphatase is concentrated near the biliary canalicular membrane of the hepatocyte. Accordingly, increased levels of hepatic alkaline phosphatase are mainly seen in liver diseases that predominantly affect the biliary system.3
GGT is also concentrated in hepatic biliary epithelial cells, and thus GGT elevation is another marker of hepatobiliary disease. In fact, measuring the GGT level can help to determine whether an isolated elevation of alkaline phosphatase is due to liver injury.2,3
Accordingly, liver diseases can be classified into two broad categories:
- Hepatocellular injury, in which the primary injury occurs to the hepatocytes
- Cholestatic injury, in which the primary injury is to the bile ducts.
In the former, elevated levels of ALT and AST predominate, while in the latter, elevated alkaline phosphatase is the main finding.3
WHAT TEST NEXT FOR OUR PATIENT?
1. What is the next most appropriate diagnostic step for our patient?
- Liver biopsy
- Ultrasonography of the liver
- Computed tomography (CT) of the liver
- Observation
Our patient has an elevated GGT level, which suggests that his elevated alkaline phosphatase is of hepatic rather than bony origin. Moreover, a serum alkaline phosphatase level that is elevated out of proportion to the aminotransferase levels reflects cholestatic liver injury.
CASE CONTINUED: ULTRASONOGRAPHY IS MOSTLY NORMAL
Ultrasonography of the right upper quadrant revealed that the liver had normal echogenicity and was mildly enlarged. There was no focal hepatic lesion. The gallbladder appeared normal, with no stones or sludge. No dilated intrahepatic or extrahepatic biliary ducts were seen. The common bile duct measured 4 mm. A small amount of ascites not amenable to paracentesis was present.
Thus, in the absence of biliary dilation on ultrasonography, we are dealing with an intrahepatic cholestatic process.
CAUSES OF CHOLESTATIC LIVER DISEASE
Viral hepatitis
Viral hepatitis most often produces a hepatocellular pattern of injury (ie, AST and ALT elevations predominate). However, in rare cases it can cause a cholestatic pattern of injury.
Our patient subsequently had serologic tests for viral hepatitis, including hepatitis A, B, and C, and the results were negative.
Autoimmune liver disease
The three most common forms of autoimmune liver disease are autoimmune hepatitis, primary biliary cirrhosis, and primary sclerosing cholangitis.
Autoimmune hepatitis is characterized by high serum ALT and AST levels, whereas primary biliary cirrhosis and primary sclerosing cholangitis are associated with predominant elevations of alkaline phosphatase, since they are cholestatic disorders.
Our patient’s alkaline phosphatase level was much higher than his ALT and AST levels, making the latter two diseases more likely.
Primary biliary cirrhosis (and autoimmune hepatitis) are associated with autoantibodies in the serum, such as antinuclear antibody, smooth muscle antibody, and antimitochondrial antibody.
Our patient subsequently was tested for these antibodies, and the results were negative.
Primary sclerosing cholangitis usually affects the extrahepatic biliary system. Thus, if it is present, abnormalities should be seen on imaging.
As mentioned previously, no dilated intrahepatic or extrahepatic biliary ducts were seen on ultrasonography in our patient. Moreover, primary sclerosing cholangitis is associated with inflammatory bowel disease, particularly ulcerative colitis, which our patient did not have.
Drug-induced liver injury
Drug-induced liver injury is a common cause of cholestatic liver disease. However, our patient was not taking any prescribed, over-the-counter, or herbal medications. Additionally, he denied heavy alcohol use.
Infiltrative disorders
Infiltrative disorders such as amyloidosis, sarcoidosis, or lymphoma should be considered in the differential diagnosis of cholestatic liver disease. A clue to a possible infiltrative process is a markedly elevated level of alkaline phosphatase with a mildly increased serum bilirubin concentration, both of which our patient had.
AFTER ULTRASONOGRAPHY, WHAT IS THE NEXT STEP?
2. Which of the following is the next most appropriate diagnostic test for our patient?
- Endoscopic retrograde cholangiopancreatography (ERCP)
- Magnetic resonance cholangiopancreatography (MRCP)
- Liver biopsy
- CT of the abdomen
Figure 1 shows a proposed algorithm for evaluating increased alkaline phosphatase levels.
If there is no biliary duct dilation on ultrasonography, then abnormal levels of alkaline phosphatase most likely represent an intrahepatic pattern of cholestatic liver injury. Therefore, additional imaging with CT or magnetic resonance imaging is of limited diagnostic value. ERCP is used today for therapy rather than diagnosis, so its use is limited to patients known to have dilated biliary ducts on imaging. Liver biopsy, however, can provide useful findings.
Case continued: He undergoes biopsy
Our patient underwent transjugular liver biopsy. During the procedure, transjugular venography showed stenosis in the right, middle, and left hepatic veins and the hepatic portion of the inferior vena cava, consistent with Budd-Chiari syndrome.
The liver biopsy specimen was positive for extensive deposition of slight eosinophilic and amorphous material in a sinusoidal pattern in the liver parenchyma, as well as in the portal tracts, with markedly atrophic hepatocytes. Congo red birefringence confirmed the diagnosis of amyloidosis. The immunohistochemical phenotype was positive for kappa light chains, which is diagnostic for primary-type amyloidosis, also called amyloidosis of light chain composition, or AL.
Bone marrow aspiration and bone marrow biopsy were performed and showed 22% plasma cells, well above the normal range (0–2%), consistent with the diagnosis of multiple myeloma.
BUDD-CHIARI SYNDROME: A CHALLENGING DIAGNOSIS
Budd-Chiari syndrome is a rare condition characterized by obstruction of venous outflow from the liver at a site that may vary from the small hepatic veins up to the inferior vena cava or even the right atrium.5,6 Obstruction of hepatic venous outflow leads to sinusoidal congestion and hypoxic damage of the hepatocytes.7 Hypoxia and necrosis of the hepatocytes result in the release of free radicals. Cirrhosis can eventually occur secondary to ischemic necrosis of hepatocytes and hepatic fibrosis.8
The estimated incidence of this syndrome is 1 in 2.5 million persons per year.7 It is more prevalent in women and young adults.8
Heterogeneous in its causes and manifestations
Budd-Chiari syndrome is also heterogeneous in its manifestations, which depend on the extent of the occlusion, on the acuteness of the obstruction, and on whether venous collateral circulation has developed to decompress the liver sinusoids.9,12,13 Therefore, on the basis of its clinical manifestations, it can be classified as fulminant, acute, subacute, or chronic.12–16
The fulminant form presents with hepatic encephalopathy within 8 weeks after the development of jaundice. The subacute form, which is the most common, has a more insidious onset in which hepatic sinusoids are decompressed by portal and hepatic venous collateral circulation. The patient usually presents with abdominal pain, ascites, hepatomegaly, nausea, vomiting, and mild jaundice. Finally the chronic form presents as complications of cirrhosis.12–16
Imaging plays an important role in diagnosing Budd-Chiari syndrome
Imaging plays an important role in detecting and classifying Budd-Chiari syndrome.
Duplex ultrasonography is useful for detecting this syndrome and has a sensitivity and specificity of 85%.9
CT and magnetic resonance imaging can also help in the diagnosis by showing thrombosis, obstruction, or occlusion in the hepatic vein or the inferior vena cava.5
Venography is the gold standard for diagnosis. However, it should be performed only if noninvasive tests are negative or nondiagnostic and there is a high clinical suspicion of this disease.17 Budd-Chiari syndrome has a characteristic pattern on venography known as “spider web,” which is due to the formation of venous collaterals to bypass the occluded hepatic veins.9
Liver biopsy is not necessarily required to confirm the diagnosis of Budd-Chiari syndrome, but it can help in diagnosing the acute or subacute forms and also in ruling out other causes. Histologic findings can include centrizonal congestion, loss of hepatocytes, hemorrhage, and fibrosis.18,19 Regenerative nodules are found in about 25% of patients.19
TREATING BUDD-CHIARI SYNDROME
The primary goal of treatment is to prevent further extension of the venous thrombosis in the hepatic veins, in their collaterals, and in the intrahepatic and extrahepatic portal venous system. Resolution of hepatic congestion improves liver perfusion and preserves function of the hepatocytes.
Anticoagulation is recommended in the early stages. Heparin therapy should be initiated and subsequently switched to warfarin with the goal of achieving an international normalized ratio of the prothrombin time of 2.0 to 2.5.8,9,19
Thrombolysis is effective in the acute form.20,21 Recanalization, including percutaneous or transhepatic angioplasty of localized segments of the narrowed hepatic veins or inferior vena cava, has long-term patency rates of 80% to 90%.22
If thrombolytic therapy and angioplasty are unsuccessful, a transjugular intrahepatic portosystemic shunt or a surgical procedure (side-to-side portocaval shunt, central splenorenal shunt, or mesocaval shunt) should be considered.9
Liver transplantation is another treatment option in those with fulminant Budd-Chiari syndrome or advanced liver cirrhosis.8
PROGNOSIS HAS IMPROVED
The prognosis of Budd-Chiari syndrome has improved, thanks to both earlier diagnosis and new treatments. The 1-year survival rate, which was about 60% before 1985, has increased to more than 80% in recent cohort studies.19
Studies have shown that the Child-Pugh score, which is based on a combination of serum albumin, bilirubin, prothrombin time, encephalopathy, and ascites, can be considered as an independent prognostic factor. A lower Child-Pugh score and a younger age are associated with a good prognosis.19,23,24 (The Child-Pugh score cannot be applied to our patient because he does not have cirrhosis.)
What happened to our patient?
Our patient was started on anticoagulation for his Budd-Chiari syndrome and on bortezomib (Velcade) and dexamethasone for his multiple myeloma. He achieved remarkable improvement in his liver function tests. Follow-up duplex ultrasonography 1 month after discharge revealed that the stenosis in the hepatic veins had resolved. He is following up with the oncology clinic for management of his multiple myeloma.
- Folwaczny C. Efficient diagnostics for elevated transaminases. [Article in German] MMW Fortschr Med 2007; 149:44–48.
- Moussavian SN, Becker RC, Piepmeyer JL, Mezey E, Bozian RC. Serum gamma-glutamyl transpeptidase and chronic alcoholism. Influence of alcohol ingestion and liver disease. Dig Dis Sci 1985; 30:211–214.
- Aragon G, Younossi ZM. When and how to evaluate mildly elevated liver enzymes in apparently healthy patients. Cleve Clin J Med 2010; 77:195–204.
- Lepper PM, Dufour JF. Elevated transaminases—what to do if everything was done?. [Article in German] Praxis (Bern 1994) 2009; 98:330–334.
- Buzas C, Sparchez Z, Cucuianu A, Manole S, Lupescu I, Acalovschi M. Budd-Chiari syndrome secondary to polycythemia vera. A case report. J Gastrointestin Liver Dis 2009; 18:363–366.
- Valla DC. Primary Budd-Chiari syndrome. J Hepatol 2009; 50:195–203.
- Rautou PE, Moucari R, Cazals-Hatem D, et al. Levels and initial course of serum alanine aminotransferase can predict outcome of patients with Budd-Chiari syndrome. Clin Gastroenterol Hepatol 2009; 7:1230–1235.
- Cura M, Haskal Z, Lopera J. Diagnostic and interventional radiology for Budd-Chiari syndrome. Radiographics 2009; 29:669–681.
- Menon KV, Shah V, Kamath PS. The Budd-Chiari syndrome. N Engl J Med 2004; 350:578–585.
- Darwish Murad S, Plessier A, Hernandez-Guerra M, et al; EN-Vie (European Network for Vascular Disorders of the Liver). Etiology, management, and outcome of the Budd-Chiari syndrome. Ann Intern Med 2009; 151:167–175.
- Valla D, Le MG, Poynard T, Zucman N, Rueff B, Benhamou JP. Risk of hepatic vein thrombosis in relation to recent use of oral contraceptives. A case-control study. Gastroenterology 1986; 90:807–811.
- Bismuth H, Sherlock DJ. Portasystemic shunting versus liver transplantation for the Budd-Chiari syndrome. Ann Surg 1991; 214:581–589.
- Orloff MJ, Daily PO, Orloff SL, Girard B, Orloff MS. A 27-year experience with surgical treatment of Budd-Chiari syndrome. Ann Surg 2000; 232:340–352.
- Dilawari JB, Bambery P, Chawla Y, et al. Hepatic outflow obstruction (Budd-Chiari syndrome). Experience with 177 patients and a review of the literature. Medicine (Baltimore) 1994; 73:21–36.
- Mahmoud AE, Mendoza A, Meshikhes AN, et al. Clinical spectrum, investigations and treatment of Budd-Chiari syndrome. QJM 1996; 89:37–43.
- Klein AS, Cameron JL. Diagnosis and management of the Budd-Chiari syndrome. Am J Surg 1990; 160:128–133.
- Plessier A, Valla DC. Budd-Chiari syndrome. Semin Liver Dis 2008; 28:259–269.
- Cazals-Hatem D, Vilgrain V, Genin P, et al. Arterial and portal circulation and parenchymal changes in Budd-Chiari syndrome: a study in 17 explanted livers. Hepatology 2003; 37:510–519.
- Hoekstra J, Janssen HL. Vascular liver disorders (I): diagnosis, treatment and prognosis of Budd-Chiari syndrome. Neth J Med 2008; 66:334–359.
- Frank JW, Kamath PS, Stanson AW. Budd-Chiari syndrome: early intervention with angioplasty and thrombolytic therapy. Mayo Clin Proc 1994; 69:877–881.
- Raju GS, Felver M, Olin JW, Satti SD. Thrombolysis for acute Budd-Chiari syndrome: case report and literature review. Am J Gastroenterol 1996; 91:1262–1263.
- Fisher NC, McCafferty I, Dolapci M, et al. Managing Budd-Chiari syndrome: a retrospective review of percutaneous hepatic vein angioplasty and surgical shunting. Gut 1999; 44:568–574.
- Zeitoun G, Escolano S, Hadengue A, et al. Outcome of Budd-Chiari syndrome: a multivariate analysis of factors related to survival including surgical portosystemic shunting. Hepatology 1999; 30:84–89.
- Darwish Murad S, Valla DC, de Groen PC, et al. Determinants of survival and the effect of portosystemic shunting in patients with Budd-Chiari syndrome. Hepatology 2004; 39:500–508.
- Folwaczny C. Efficient diagnostics for elevated transaminases. [Article in German] MMW Fortschr Med 2007; 149:44–48.
- Moussavian SN, Becker RC, Piepmeyer JL, Mezey E, Bozian RC. Serum gamma-glutamyl transpeptidase and chronic alcoholism. Influence of alcohol ingestion and liver disease. Dig Dis Sci 1985; 30:211–214.
- Aragon G, Younossi ZM. When and how to evaluate mildly elevated liver enzymes in apparently healthy patients. Cleve Clin J Med 2010; 77:195–204.
- Lepper PM, Dufour JF. Elevated transaminases—what to do if everything was done?. [Article in German] Praxis (Bern 1994) 2009; 98:330–334.
- Buzas C, Sparchez Z, Cucuianu A, Manole S, Lupescu I, Acalovschi M. Budd-Chiari syndrome secondary to polycythemia vera. A case report. J Gastrointestin Liver Dis 2009; 18:363–366.
- Valla DC. Primary Budd-Chiari syndrome. J Hepatol 2009; 50:195–203.
- Rautou PE, Moucari R, Cazals-Hatem D, et al. Levels and initial course of serum alanine aminotransferase can predict outcome of patients with Budd-Chiari syndrome. Clin Gastroenterol Hepatol 2009; 7:1230–1235.
- Cura M, Haskal Z, Lopera J. Diagnostic and interventional radiology for Budd-Chiari syndrome. Radiographics 2009; 29:669–681.
- Menon KV, Shah V, Kamath PS. The Budd-Chiari syndrome. N Engl J Med 2004; 350:578–585.
- Darwish Murad S, Plessier A, Hernandez-Guerra M, et al; EN-Vie (European Network for Vascular Disorders of the Liver). Etiology, management, and outcome of the Budd-Chiari syndrome. Ann Intern Med 2009; 151:167–175.
- Valla D, Le MG, Poynard T, Zucman N, Rueff B, Benhamou JP. Risk of hepatic vein thrombosis in relation to recent use of oral contraceptives. A case-control study. Gastroenterology 1986; 90:807–811.
- Bismuth H, Sherlock DJ. Portasystemic shunting versus liver transplantation for the Budd-Chiari syndrome. Ann Surg 1991; 214:581–589.
- Orloff MJ, Daily PO, Orloff SL, Girard B, Orloff MS. A 27-year experience with surgical treatment of Budd-Chiari syndrome. Ann Surg 2000; 232:340–352.
- Dilawari JB, Bambery P, Chawla Y, et al. Hepatic outflow obstruction (Budd-Chiari syndrome). Experience with 177 patients and a review of the literature. Medicine (Baltimore) 1994; 73:21–36.
- Mahmoud AE, Mendoza A, Meshikhes AN, et al. Clinical spectrum, investigations and treatment of Budd-Chiari syndrome. QJM 1996; 89:37–43.
- Klein AS, Cameron JL. Diagnosis and management of the Budd-Chiari syndrome. Am J Surg 1990; 160:128–133.
- Plessier A, Valla DC. Budd-Chiari syndrome. Semin Liver Dis 2008; 28:259–269.
- Cazals-Hatem D, Vilgrain V, Genin P, et al. Arterial and portal circulation and parenchymal changes in Budd-Chiari syndrome: a study in 17 explanted livers. Hepatology 2003; 37:510–519.
- Hoekstra J, Janssen HL. Vascular liver disorders (I): diagnosis, treatment and prognosis of Budd-Chiari syndrome. Neth J Med 2008; 66:334–359.
- Frank JW, Kamath PS, Stanson AW. Budd-Chiari syndrome: early intervention with angioplasty and thrombolytic therapy. Mayo Clin Proc 1994; 69:877–881.
- Raju GS, Felver M, Olin JW, Satti SD. Thrombolysis for acute Budd-Chiari syndrome: case report and literature review. Am J Gastroenterol 1996; 91:1262–1263.
- Fisher NC, McCafferty I, Dolapci M, et al. Managing Budd-Chiari syndrome: a retrospective review of percutaneous hepatic vein angioplasty and surgical shunting. Gut 1999; 44:568–574.
- Zeitoun G, Escolano S, Hadengue A, et al. Outcome of Budd-Chiari syndrome: a multivariate analysis of factors related to survival including surgical portosystemic shunting. Hepatology 1999; 30:84–89.
- Darwish Murad S, Valla DC, de Groen PC, et al. Determinants of survival and the effect of portosystemic shunting in patients with Budd-Chiari syndrome. Hepatology 2004; 39:500–508.
A 35-year-old Asian man with jaundice and markedly high aminotransferase levels
A 35-year-old man who was born in Vietnam presents to the emergency department of a local hospital because he has had jaundice for 5 days and fatigue, malaise, and anorexia for 2 weeks. He also has nausea and mild epigastric and right upper quadrant abdominal pain. He denies having fevers, chills, night sweats, vomiting, diarrhea, melena, hematochezia, or weight loss.
His medical history is remarkable only for perinatally acquired hepatitis B virus (HBV) infection, for which he never received antiviral therapy. He does not take any prescribed, over-the-counter, or herbal medications.
He lives in the Midwest region of the United States and works full-time as a physician in private practice. He is married and has two children.
He has not travelled recently. He has no pets at home and has not been exposed to any.
He has never smoked. He drinks alcohol socially but has never used recreational drugs.
In a laboratory evaluation performed a year ago for insurance purposes, his liver function tests—serum albumin, total bilirubin, alkaline phosphatase, alanine aminotransferase, and aspartate aminotransferase levels—were all normal. He was positive for HBV surface antigen and HBV e antigen and negative for antibodies against these antigens.
PHASES OF HBV INFECTION
1. Which of the following best describes the status of HBV infection in this patient before his current symptoms developed?
- Resolved HBV infection
- Chronic inactive HBV infection
- Chronic active HBV infection
- Immune-tolerant chronic HBV infection
The correct answer is immune-tolerant chronic HBV infection.
Resolved infection. In immunocompetent adults, most primary HBV infections are self-limited: people clear the virus and gain lasting immunity (defined as the loss of HBV surface antigen, the development of antibody against surface antigen, no detectable HBV DNA in the serum, and normal alanine and aspartate aminotransferase levels). However, a minority of primary HBV infections persist and become chronic.
Chronic HBV infection is defined as the persistence of HBV surface antigen in the serum for at least 6 months. Patients with chronic HBV infection can be broadly classified as having either inactive disease (the inactive surface antigen carrier state) or chronic active hepatitis B (Figure 1).1–9
Chronic inactive HBV infection. Carriers of inactive HBV infection have low serum levels of HBV DNA (< 2,000 IU/mL), persistently normal aminotransferase levels, and no HBV e antigen; if a liver biopsy is performed, no significant hepatitis is found.
Chronic active HBV infection. Patients with chronic active HBV infection, in contrast, have high serum HBV DNA levels (> 20,000 IU/mL) and persistently or intermittently high aminotransferase levels; they do have HBV e antigen, and a liver biopsy shows moderate or severe necroinflammation.
A small group of patients with chronic active hepatitis B may be negative for e antigen but still have high aminotransferase levels, high HBV DNA levels, and continued necroinflammation in the liver.4 The virus in these patients has a mutation in its precore or core promoter gene that prevents the production of e antigen.
Patients with chronic active HBV infection (whether positive or negative for e antigen) are at a significantly greater risk of progressive liver injury and developing cirrhosis and hepatocellular carcinoma than are inactive carriers of HBV.
Immune-tolerant chronic HBV infection. Patients who acquired HBV at birth (eg, our patient) may have immune-tolerant HBV infection, which is characterized by significant HBV replication manifested by the presence of HBV e antigen and high levels of HBV DNA in the serum. However, these patients have no clinical or pathologic evidence of active liver disease (no symptoms, normal serum alanine aminotransferase levels, and minimal changes on liver biopsy).5 This was obviously the case in our patient, based on his history and laboratory results before his current symptoms developed.
Case continues: Liver function abnormalities
On physical examination, the patient’s temperature is 99.9°F (37.7°C), heart rate 106 per minute, blood pressure 98/54 mm Hg, respiratory rate 18 per minute, and oxygen saturation 100% while breathing ambient air. He is alert and oriented to time, place, and person.
He has icteric sclera, and his skin is jaundiced. His lymph nodes are not palpable. His cardiac examination is normal except for tachycardia. His lungs are clear to auscultation and percussion. He has mild epigastric and right upper quadrant abdominal tenderness with no peritoneal signs, hepatosplenomegaly, or masses.
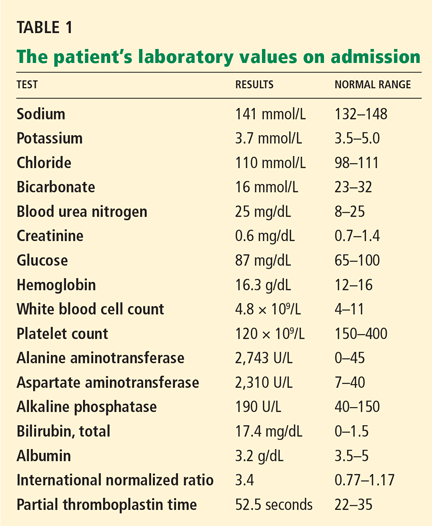
His basic laboratory values on admission are listed in Table 1. His amylase and lipase levels are normal. A urine dipstick test is positive for bilirubin.
WHAT IS THE LEAST LIKELY DIAGNOSIS?
2. Which one of the following is the least likely diagnosis in this patient?
- Reactivation of hepatitis B
- Drug-associated liver injury
- Acute viral hepatitis
- Acute alcoholic hepatitis
- Ischemic hepatitis
The degree and pattern of liver function abnormalities in our patient reflect hepatocellular injury rather than cholestatic liver disease, because his aminotransferase levels are elevated much higher than his alkaline phosphatase level (Table 1). Bilirubin elevation does not help differentiate the two conditions.
The degree and pattern of aminotransferase elevations are also helpful in narrowing the differential diagnosis. Serum aminotransferase levels of more than 1,000 U/L are mainly seen in patients with ischemic, viral, and toxininduced liver injury. Other rare causes of such high levels include Budd-Chiari syndrome, Wilson disease, and autoimmune hepatitis.
Ischemic hepatitis. Our patient has mild hypotension, but it does not seem to have been severe enough or of long enough duration to have caused ischemic hepatitis.
Drug-associated liver injury. Hepatotoxicity associated with drugs (most commonly acetaminophen [Tylenol]), herbal therapy, or mushroom poisoning should be considered in any patient whose aminotransferase levels are this high. However, our patient denies taking any medications (prescribed or over-the-counter), herbal remedies, or illicit drugs.
Acute viral hepatitis can certainly explain the patient’s clinical picture. Infection with hepatitis A virus, hepatitis D virus, hepatitis E virus, cytomegalovirus, Epstein-Barr virus, herpes simplex viruses types 1 and 2, and varicella zoster virus have all been implicated in severe acute hepatitis. Although hepatitis E virus infection is more common in developing countries, it has been reported in the United States.6 It is unlikely that acute hepatitis C virus infection is producing this degree of elevation in aminotransferase levels.
Reactivation of the patient’s chronic HBV infection can also account for his clinical presentation.
Acute alcoholic hepatitis should be suspected clinically if a patient has a history of heavy alcohol use and clinical and laboratory findings that are compatible with the diagnosis. However, the absolute values of serum aspartate aminotransferase and alanine aminotransferase in acute alcoholic hepatitis are almost always less than 500 IU/L (and typically less than 300 IU/L). Our patient’s values are much higher, and he says he does not drink very much. Although people sometimes underestimate their alcohol intake, alcoholic hepatitis is the least likely diagnosis in our patient.
Case continues: The patient is hospitalized
The patient is admitted with a diagnosis of acute hepatitis. Given his history of chronic hepatitis B, he is empirically started on lamivudine (Epivir-HBV).

- Antinuclear antibody negative
- Autoimmune liver disease panel negative
- Serum ceruloplasmin 30 mg/dL (normal range 15–60)
- Alpha fetoprotein 35.1 μg/L (< 10).
Abdominal ultrasonography is performed and reveals a small stone in the gallbladder with no evidence of biliary dilatation; otherwise, the gallbladder appears normal. Doppler ultrasonography shows the liver vessels to be patent; the liver is normal in appearance. The abdomen and pelvis appear to be normal on computed tomography without intravenous contrast.
On the third hospital day, the patient’s blood test results are:
- Aspartate aminotransferase 199 U/L (normal range 7–40)
- Alanine aminotransferase 735 U/L (0–45)
- Total bilirubin 22.9 mg/dL (0–1.5)
- International normalized ratio 6.0 (0.77–1.17)
- White blood cell count 5.1 × 109/L (4–11)
- Hemoglobin 11.7 g/dL (12–16)
- Platelet count 166 × 109/L (150–400)
- Blood and urine cultures negative.
WHAT IS CAUSING HIS ACUTE HEPATITIS?
3. On the basis of the new data, which of the following statements about the cause of acute hepatitis in this patient is the most accurate?
- Herpetic hepatitis is the most likely cause, given his positive test for immunoglobulin M (IgM) against herpes simplex virus
- Hepatitis C cannot be excluded with the available data
- Negative HBV e antigen does not exclude the diagnosis of acute exacerbation of HBV infection
- Hepatocellular carcinoma is the likely diagnosis, given the elevated alpha fetoprotein level
The third answer above is correct: a negative test for hepatitis B e antigen does not exclude the diagnosis of acute exacerbation of HBV infection
Herpetic hepatitis. Although not common, hepatitis due to herpes simplex virus infection should be considered in the differential diagnosis of any patient presenting with severe acute hepatitis, particularly when fever is present. Common features of herpetic hepatitis on presentation include high fever, leukopenia, markedly elevated aminotransferases, and mild cholestasis. Vesicular rash occurs in only less than half of cases of herpetic hepatitis.10
Serologic testing is of limited value because it has high rates of false-positive and false-negative results. The diagnosis can be confirmed only by viral polymerase chain reaction testing or by identifying herpes simplex viral inclusions in the liver biopsy.
However, the death rate is high in this disease, and since herpetic hepatitis is one of the few treatable causes of acute liver failure, parenteral acyclovir (Zovirax) should be considered empirically in patients presenting with acute liver failure. Our patient was started on acyclovir when his tests for IgM against herpes simplex virus came back positive.
Hepatitis C. Antibodies against hepatitis C virus do not develop immediately after this virus is contracted; they may take up to 12 weeks to develop after exposure. For this reason, about 30% to 50% of patients with acute hepatitis C virus infection are negative for these antibodies initially. In those patients, hepatitis C virus RNA in the blood is the most sensitive test to detect acute hepatitis C virus infection.
Our patient has neither antibodies against hepatitis C virus nor hepatitis C virus RNA by polymerase chain reaction testing, which rules out hepatitis C virus infection.
Disappearance of e antigen in HBV infection. The disappearance of HBV e antigen is usually associated with a decrease in serum HBV DNA and remission of liver disease. However, some patients continue to have active liver disease and high levels of HBV DNA despite e antigen seroconversion. This is due to a stop codon mutation in the precore region of the viral genome that decreases or prevents production of HBV e antigen.4 In other words, even though HBV e antigen is a good marker of HBV replication in general, a subgroup of patients with chronic HBV infection are negative for e antigen but still have a high rate of viral replication as evidenced by high serum HBV DNA levels.
Patients with perinatally acquired chronic HBV infection most often have immune-tolerant chronic HBV infection. Among those patients (mostly Asian),5,7 the virus is spontaneously cleared at a rate of approximately 2% to 3% per year,8 most often during the second and third decades of age.
Transition from the immune-tolerant phase to the immune clearance phase is frequently associated with mild transient worsening of the liver function profile.9,11,12 However, in a small percentage of patients, hepatic decompensation and even (rarely) death from hepatic failure may occur secondary to a sudden activation of the immune system as it attempts to clear the virus. This may result in an increase in immune-mediated lysis of infected hepatocytes.
Hepatocellular carcinoma. Exacerbation of hepatitis B may be associated with an elevation of alpha fetoprotein, which may falsely raise concerns about the possibility of hepatocellular carcinoma. However, our patient had abdominal imaging with both ultrasonography and computed tomography, which showed no evidence of hepatocellular carcinoma.
Comment. The most likely cause of the patient’s acute liver failure is an acute exacerbation of hepatitis B. However, herpetic hepatitis should be ruled out by testing for herpes simplex virus by polymerase chain reaction, performing a liver biopsy, or both.
Case continues: His condition worsens
A transjugular liver biopsy shows changes associated with chronic hepatitis B, severe acute hepatitis with extensive confluent and submassive hepatic necrosis, and no intracellular viral inclusions. Subsequently, acyclovir is stopped.
On the 6th hospital day, he develops progressive metabolic acidosis and hypotension, with worsening hypoxemia. A chest radiograph is obtained to look for pneumonia, but it is indeterminate; computed tomography of the chest without contrast medium is likewise unremarkable. Duplex ultrasonography of the four extremities is negative for venous thrombosis.
The patient becomes more lethargic and difficult to arouse. He is transferred to the intensive care unit and intubated. His prothrombin and partial thromboplastin times continue to rise, the prothrombin time reaching values of more than 50 seconds. In addition, progressive renal insufficiency develops.
WHAT IS THE NEXT STEP?
4. Which of the following is the most appropriate next step in the management of this patient?
- Liver transplantation
- HBV immunoglobulin only
- Interferon and a nucleoside analogue
- Liver-assist devices
- Continue supportive care only
Liver transplantation. Since the patient’s severe acute hepatitis is accompanied by coagulopathy and encephalopathy, he meets the definition of having acute liver failure. Liver transplantation remains the only definitive therapy.

HBV immune globulin immunoprophylaxis is indicated in patients with HBV infection undergoing liver transplantation, to prevent recurrence of hepatitis B after the transplant, particularly in those with a high pretransplant viral load.17 The use of pretransplant antiviral therapy and the posttransplant combination of antiviral therapy and HBV immune globulin has reduced the rate of hepatitis B recurrence to less than 10%. However, immune globulin is by no means the best single next step in managing this patient, who clearly needs a new liver.
Interferon, nucleoside analogues. Options for antiviral treatment are interferon alfa and nucleoside analogues. Interferon therapy is contraindicated in patients such as ours, who have decompensated liver disease, because it can exacerbate the disease.18

Liver-assist devices. Because liver allografts are in short supply, there has been a strong interest in developing a device that would provide the same benefits for patients with liver failure as hemodialysis does for patients with renal failure. Trials are under way to determine the efficacy and safety of these devices.20
Case continues: He receives a liver
The patient undergoes liver transplantation. He is given HBV immune globulin during and after the surgery.
Pathologic review. Under the microscope, his old liver has widespread necrosis and hemorrhage as well as inflammatory changes suggesting a chronic viral process. Regenerative nodules are present in the small amount of surviving liver parenchyma, consistent with early cirrhosis. Iron staining shows +3 depositions in areas of hepatic collapse (a nonspecific finding). Periodic acid-Schiff staining after diastase (used to detect alpha-1 antitrypsin deficiency) is negative. Herpetic viral inclusions are not present.
An immunoassay for herpes simplex virus antigen is negative. Immunostaining with antibodies to the HBV core antigen is negative. HBV surface antigen is strongly and diffusely positive in the cytoplasm of 80% to 90% of hepatocytes. The immunohistologic staining pattern is consistent with integration of HBV DNA into the DNA of hepatic tissue.
Postoperative course. Lamivudine is continued after surgery, and the patient is sent home. He has resumed the level of functioning he had before becoming ill.
Comment. The outcome of liver transplantation for hepatitis B has notably improved since HBV immune globulin and nucleoside analogues were introduced. The results of liver transplantation for hepatitis B, particuarly patient and graft survival rates, are now better than those in transplant patients with hepatitis C and similar to those in transplant patients with other types of liver disease.21 The combination of HBV immune globulin and lamivudine has cut the rate of HBV reinfection after liver transplantation to approximately 10% and increased the 5-year survival rate after transplantation to about 80%.17,22
KEY POINTS
- In immunocompetent adults, most primary HBV infections are self-limited.
- Chronic HBV infection is defined as the persistence of HBV surface antigen in the serum for at least 6 months. Patients having chronic HBV infection can be broadly classified as inactive carriers or having chronic active disease.
- Most patients with chronic active HBV infection are positive for HBV e antigen, except patients in whom the virus has a mutation in the precore or core region of its genome that prevents the production of e antigen.
- Patients who carry inactive HBV or who are immune-tolerant require serial measurements of aminotransferase and HBV DNA levels. Treatment can be considered if the patient has a high viral load (> 2,000 IU/mL), elevated aminotransferases, or active disease on liver biopsy.
- Carriers of chronic active HBV (whether positive or negative for HBV e antigen) should be referred to a hepatologist for consideration of liver biopsy and treatment.
- Interferon should not be used in immunocompromised patients or those with decompensated liver disease because it can further exacerbate the liver disease.
- Liver transplantation should be considered in patients with acute liver failure who have a poor prognosis according to the King’s College Hospital criteria.
- Dusheiko G. Hepatitis B. In:Bircher J, Benhamou JP, McIntyre N, Rizzetto M, Rodes J, editors. Oxford Textbook of Clinical Hepatology. 2nd ed. Oxford, UK: Oxford University Press; 1999:876–896.
- Chu CJ, Hussain M, Lok AS. Quantitative serum HBV DNA levels during different stages of chronic hepatitis B infection. Hepatology 2002; 36:1408–1415.
- Pawlotsky JM, Bastie A, Hezode C, et al. Routine detection and quantification of hepatitis B virus DNA in clinical laboratories: performance of three commercial assays. J Virol Methods 2000; 85:11–21.
- Brunetto MR, Giarin MM, Oliveri F, et al. Wild-type and e-antigen-minus hepatitis viruses and course of chronic hepatitis. Proc Natl Acad Sci USA 1991; 88:4186–4190.
- Lok AS, Lai CL. A longitudinal follow-up of asymptomatic hepatitis B surface antigen-positive Chinese children. Hepatology 1988; 5:1130–1133.
- Hsu HY, Chang MH, Hsieh KH, et al. Cellular immune response to HBcAg in mother-to-infant transmission of hepatitis B virus. Hepatology 1992; 15:770–776.
- Chang MH, Hsu HY, Hsu HC, Ni YH, Chen JS, Chen DS. The significance of spontaneous hepatitis B e antigen seroconversion in childhood: with special emphasis on the clearance of hepatitis B e antigen before 3 years of age. Hepatology 1995; 22:1387–1392.
- Ruiz-Moreno M, Otero M, Millan A, et al. Clinical and histological outcome after hepatitis B e antigen to antibody seroconversion in children with chronic hepatitis B. Hepatology 1999; 29:572–575.
- Liaw YF, Chu CM, Su IJ, Huang MJ, Lin DY, Chang-Chien CS. Clinical and histological events preceding hepatitis B e antigen seroconversion in chronic type B hepatitis. Gastroenterology 1983; 84:216–219.
- Norvell JP, Blei AT, Jovanovic BD, Levitsky J. Herpes simplex virus hepatitis: an analysis of the published literature and institutional cases. Liver Transplant 2007; 13:1428–1434,
- Liaw YF, Pao CC, Chu CM, Sheen IS, Huang MJ. Changes of serum hepatitis B virus DNA in two types of clinical events preceding spontaneous hepatitis B e antigen seroconversion in chronic type B hepatitis. Hepatology 1987; 7:1–3.
- Maruyama T, Iino S, Koike K, Yasuda K, Milich DR. Serology of acute exacerbation in chronic hepatitis B virus infection. Gastroenterology 1993; 105:1141–1151.
- O'Grady JG, Alexander GJ, Hayllar KM, Williams R. Early indicators of prognosis in fulminant hepatic failure. Gastroenterology 1989; 97:439–445.
- Shakil AO, Kramer D, Mazariegos GV, Fung JJ, Rakela J. Acute liver failure: clinical features, outcome analysis, and applicability of prognostic criteria. Liver Transplant 2000; 6:163–169.
- Anand AC, Nightingale P, Neuberger JM. Early indicators of prognosis in fulminant hepatic failure: an assessment of the King’s criteria. J Hepatol 1997; 26:62–68.
- Schmidt LE, Dalhoff K. Serum phosphate is an early predictor of outcome in severe acetaminophen-induced hepatotoxicity. Hepatology 2002; 36:659–665.
- Samuel D, Muller R, Alexander G, et al. Liver transplantation in European patients with the hepatitis B surface antigen. N Engl J Med 1993; 329:1842–1847.
- Lok A, McMahon BJ. Chronic hepatitis B. Hepatology 2007; 45:507–539.
- Sorren MF, Belangia EA, Costa J, et al. National Institutes of Health consensus development conference statement: management of hepatitis B. Ann Intern Med 2009; 150:104–110.
- Kjaergard LL, Liu J, Als-Nielsen B, Gluud C. Artificial and bioartificial support systems for acute and acute-on-chronic liver failure: a systematic review. JAMA 2003; 289:217–222.
- Kim WR, Poterucha JJ, Kremers WK, Ishitani MB, Dickson ER. Outcome of liver transplantation for hepatitis B in the United States. Liver Transplant 2004; 10:968–974.
- Terrault NA, Zhou S, Combs C, et al. Prophylaxis in liver transplant recipients using a fixed dosing schedule of hepatitis B immunoglobulin. Hepatology 1996; 24:1327–1333.
A 35-year-old man who was born in Vietnam presents to the emergency department of a local hospital because he has had jaundice for 5 days and fatigue, malaise, and anorexia for 2 weeks. He also has nausea and mild epigastric and right upper quadrant abdominal pain. He denies having fevers, chills, night sweats, vomiting, diarrhea, melena, hematochezia, or weight loss.
His medical history is remarkable only for perinatally acquired hepatitis B virus (HBV) infection, for which he never received antiviral therapy. He does not take any prescribed, over-the-counter, or herbal medications.
He lives in the Midwest region of the United States and works full-time as a physician in private practice. He is married and has two children.
He has not travelled recently. He has no pets at home and has not been exposed to any.
He has never smoked. He drinks alcohol socially but has never used recreational drugs.
In a laboratory evaluation performed a year ago for insurance purposes, his liver function tests—serum albumin, total bilirubin, alkaline phosphatase, alanine aminotransferase, and aspartate aminotransferase levels—were all normal. He was positive for HBV surface antigen and HBV e antigen and negative for antibodies against these antigens.
PHASES OF HBV INFECTION
1. Which of the following best describes the status of HBV infection in this patient before his current symptoms developed?
- Resolved HBV infection
- Chronic inactive HBV infection
- Chronic active HBV infection
- Immune-tolerant chronic HBV infection
The correct answer is immune-tolerant chronic HBV infection.
Resolved infection. In immunocompetent adults, most primary HBV infections are self-limited: people clear the virus and gain lasting immunity (defined as the loss of HBV surface antigen, the development of antibody against surface antigen, no detectable HBV DNA in the serum, and normal alanine and aspartate aminotransferase levels). However, a minority of primary HBV infections persist and become chronic.
Chronic HBV infection is defined as the persistence of HBV surface antigen in the serum for at least 6 months. Patients with chronic HBV infection can be broadly classified as having either inactive disease (the inactive surface antigen carrier state) or chronic active hepatitis B (Figure 1).1–9
Chronic inactive HBV infection. Carriers of inactive HBV infection have low serum levels of HBV DNA (< 2,000 IU/mL), persistently normal aminotransferase levels, and no HBV e antigen; if a liver biopsy is performed, no significant hepatitis is found.
Chronic active HBV infection. Patients with chronic active HBV infection, in contrast, have high serum HBV DNA levels (> 20,000 IU/mL) and persistently or intermittently high aminotransferase levels; they do have HBV e antigen, and a liver biopsy shows moderate or severe necroinflammation.
A small group of patients with chronic active hepatitis B may be negative for e antigen but still have high aminotransferase levels, high HBV DNA levels, and continued necroinflammation in the liver.4 The virus in these patients has a mutation in its precore or core promoter gene that prevents the production of e antigen.
Patients with chronic active HBV infection (whether positive or negative for e antigen) are at a significantly greater risk of progressive liver injury and developing cirrhosis and hepatocellular carcinoma than are inactive carriers of HBV.
Immune-tolerant chronic HBV infection. Patients who acquired HBV at birth (eg, our patient) may have immune-tolerant HBV infection, which is characterized by significant HBV replication manifested by the presence of HBV e antigen and high levels of HBV DNA in the serum. However, these patients have no clinical or pathologic evidence of active liver disease (no symptoms, normal serum alanine aminotransferase levels, and minimal changes on liver biopsy).5 This was obviously the case in our patient, based on his history and laboratory results before his current symptoms developed.
Case continues: Liver function abnormalities
On physical examination, the patient’s temperature is 99.9°F (37.7°C), heart rate 106 per minute, blood pressure 98/54 mm Hg, respiratory rate 18 per minute, and oxygen saturation 100% while breathing ambient air. He is alert and oriented to time, place, and person.
He has icteric sclera, and his skin is jaundiced. His lymph nodes are not palpable. His cardiac examination is normal except for tachycardia. His lungs are clear to auscultation and percussion. He has mild epigastric and right upper quadrant abdominal tenderness with no peritoneal signs, hepatosplenomegaly, or masses.
His basic laboratory values on admission are listed in Table 1. His amylase and lipase levels are normal. A urine dipstick test is positive for bilirubin.
WHAT IS THE LEAST LIKELY DIAGNOSIS?
2. Which one of the following is the least likely diagnosis in this patient?
- Reactivation of hepatitis B
- Drug-associated liver injury
- Acute viral hepatitis
- Acute alcoholic hepatitis
- Ischemic hepatitis
The degree and pattern of liver function abnormalities in our patient reflect hepatocellular injury rather than cholestatic liver disease, because his aminotransferase levels are elevated much higher than his alkaline phosphatase level (Table 1). Bilirubin elevation does not help differentiate the two conditions.
The degree and pattern of aminotransferase elevations are also helpful in narrowing the differential diagnosis. Serum aminotransferase levels of more than 1,000 U/L are mainly seen in patients with ischemic, viral, and toxininduced liver injury. Other rare causes of such high levels include Budd-Chiari syndrome, Wilson disease, and autoimmune hepatitis.
Ischemic hepatitis. Our patient has mild hypotension, but it does not seem to have been severe enough or of long enough duration to have caused ischemic hepatitis.
Drug-associated liver injury. Hepatotoxicity associated with drugs (most commonly acetaminophen [Tylenol]), herbal therapy, or mushroom poisoning should be considered in any patient whose aminotransferase levels are this high. However, our patient denies taking any medications (prescribed or over-the-counter), herbal remedies, or illicit drugs.
Acute viral hepatitis can certainly explain the patient’s clinical picture. Infection with hepatitis A virus, hepatitis D virus, hepatitis E virus, cytomegalovirus, Epstein-Barr virus, herpes simplex viruses types 1 and 2, and varicella zoster virus have all been implicated in severe acute hepatitis. Although hepatitis E virus infection is more common in developing countries, it has been reported in the United States.6 It is unlikely that acute hepatitis C virus infection is producing this degree of elevation in aminotransferase levels.
Reactivation of the patient’s chronic HBV infection can also account for his clinical presentation.
Acute alcoholic hepatitis should be suspected clinically if a patient has a history of heavy alcohol use and clinical and laboratory findings that are compatible with the diagnosis. However, the absolute values of serum aspartate aminotransferase and alanine aminotransferase in acute alcoholic hepatitis are almost always less than 500 IU/L (and typically less than 300 IU/L). Our patient’s values are much higher, and he says he does not drink very much. Although people sometimes underestimate their alcohol intake, alcoholic hepatitis is the least likely diagnosis in our patient.
Case continues: The patient is hospitalized
The patient is admitted with a diagnosis of acute hepatitis. Given his history of chronic hepatitis B, he is empirically started on lamivudine (Epivir-HBV).
- Antinuclear antibody negative
- Autoimmune liver disease panel negative
- Serum ceruloplasmin 30 mg/dL (normal range 15–60)
- Alpha fetoprotein 35.1 μg/L (< 10).
Abdominal ultrasonography is performed and reveals a small stone in the gallbladder with no evidence of biliary dilatation; otherwise, the gallbladder appears normal. Doppler ultrasonography shows the liver vessels to be patent; the liver is normal in appearance. The abdomen and pelvis appear to be normal on computed tomography without intravenous contrast.
On the third hospital day, the patient’s blood test results are:
- Aspartate aminotransferase 199 U/L (normal range 7–40)
- Alanine aminotransferase 735 U/L (0–45)
- Total bilirubin 22.9 mg/dL (0–1.5)
- International normalized ratio 6.0 (0.77–1.17)
- White blood cell count 5.1 × 109/L (4–11)
- Hemoglobin 11.7 g/dL (12–16)
- Platelet count 166 × 109/L (150–400)
- Blood and urine cultures negative.
WHAT IS CAUSING HIS ACUTE HEPATITIS?
3. On the basis of the new data, which of the following statements about the cause of acute hepatitis in this patient is the most accurate?
- Herpetic hepatitis is the most likely cause, given his positive test for immunoglobulin M (IgM) against herpes simplex virus
- Hepatitis C cannot be excluded with the available data
- Negative HBV e antigen does not exclude the diagnosis of acute exacerbation of HBV infection
- Hepatocellular carcinoma is the likely diagnosis, given the elevated alpha fetoprotein level
The third answer above is correct: a negative test for hepatitis B e antigen does not exclude the diagnosis of acute exacerbation of HBV infection
Herpetic hepatitis. Although not common, hepatitis due to herpes simplex virus infection should be considered in the differential diagnosis of any patient presenting with severe acute hepatitis, particularly when fever is present. Common features of herpetic hepatitis on presentation include high fever, leukopenia, markedly elevated aminotransferases, and mild cholestasis. Vesicular rash occurs in only less than half of cases of herpetic hepatitis.10
Serologic testing is of limited value because it has high rates of false-positive and false-negative results. The diagnosis can be confirmed only by viral polymerase chain reaction testing or by identifying herpes simplex viral inclusions in the liver biopsy.
However, the death rate is high in this disease, and since herpetic hepatitis is one of the few treatable causes of acute liver failure, parenteral acyclovir (Zovirax) should be considered empirically in patients presenting with acute liver failure. Our patient was started on acyclovir when his tests for IgM against herpes simplex virus came back positive.
Hepatitis C. Antibodies against hepatitis C virus do not develop immediately after this virus is contracted; they may take up to 12 weeks to develop after exposure. For this reason, about 30% to 50% of patients with acute hepatitis C virus infection are negative for these antibodies initially. In those patients, hepatitis C virus RNA in the blood is the most sensitive test to detect acute hepatitis C virus infection.
Our patient has neither antibodies against hepatitis C virus nor hepatitis C virus RNA by polymerase chain reaction testing, which rules out hepatitis C virus infection.
Disappearance of e antigen in HBV infection. The disappearance of HBV e antigen is usually associated with a decrease in serum HBV DNA and remission of liver disease. However, some patients continue to have active liver disease and high levels of HBV DNA despite e antigen seroconversion. This is due to a stop codon mutation in the precore region of the viral genome that decreases or prevents production of HBV e antigen.4 In other words, even though HBV e antigen is a good marker of HBV replication in general, a subgroup of patients with chronic HBV infection are negative for e antigen but still have a high rate of viral replication as evidenced by high serum HBV DNA levels.
Patients with perinatally acquired chronic HBV infection most often have immune-tolerant chronic HBV infection. Among those patients (mostly Asian),5,7 the virus is spontaneously cleared at a rate of approximately 2% to 3% per year,8 most often during the second and third decades of age.
Transition from the immune-tolerant phase to the immune clearance phase is frequently associated with mild transient worsening of the liver function profile.9,11,12 However, in a small percentage of patients, hepatic decompensation and even (rarely) death from hepatic failure may occur secondary to a sudden activation of the immune system as it attempts to clear the virus. This may result in an increase in immune-mediated lysis of infected hepatocytes.
Hepatocellular carcinoma. Exacerbation of hepatitis B may be associated with an elevation of alpha fetoprotein, which may falsely raise concerns about the possibility of hepatocellular carcinoma. However, our patient had abdominal imaging with both ultrasonography and computed tomography, which showed no evidence of hepatocellular carcinoma.
Comment. The most likely cause of the patient’s acute liver failure is an acute exacerbation of hepatitis B. However, herpetic hepatitis should be ruled out by testing for herpes simplex virus by polymerase chain reaction, performing a liver biopsy, or both.
Case continues: His condition worsens
A transjugular liver biopsy shows changes associated with chronic hepatitis B, severe acute hepatitis with extensive confluent and submassive hepatic necrosis, and no intracellular viral inclusions. Subsequently, acyclovir is stopped.
On the 6th hospital day, he develops progressive metabolic acidosis and hypotension, with worsening hypoxemia. A chest radiograph is obtained to look for pneumonia, but it is indeterminate; computed tomography of the chest without contrast medium is likewise unremarkable. Duplex ultrasonography of the four extremities is negative for venous thrombosis.
The patient becomes more lethargic and difficult to arouse. He is transferred to the intensive care unit and intubated. His prothrombin and partial thromboplastin times continue to rise, the prothrombin time reaching values of more than 50 seconds. In addition, progressive renal insufficiency develops.
WHAT IS THE NEXT STEP?
4. Which of the following is the most appropriate next step in the management of this patient?
- Liver transplantation
- HBV immunoglobulin only
- Interferon and a nucleoside analogue
- Liver-assist devices
- Continue supportive care only
Liver transplantation. Since the patient’s severe acute hepatitis is accompanied by coagulopathy and encephalopathy, he meets the definition of having acute liver failure. Liver transplantation remains the only definitive therapy.
HBV immune globulin immunoprophylaxis is indicated in patients with HBV infection undergoing liver transplantation, to prevent recurrence of hepatitis B after the transplant, particularly in those with a high pretransplant viral load.17 The use of pretransplant antiviral therapy and the posttransplant combination of antiviral therapy and HBV immune globulin has reduced the rate of hepatitis B recurrence to less than 10%. However, immune globulin is by no means the best single next step in managing this patient, who clearly needs a new liver.
Interferon, nucleoside analogues. Options for antiviral treatment are interferon alfa and nucleoside analogues. Interferon therapy is contraindicated in patients such as ours, who have decompensated liver disease, because it can exacerbate the disease.18
Liver-assist devices. Because liver allografts are in short supply, there has been a strong interest in developing a device that would provide the same benefits for patients with liver failure as hemodialysis does for patients with renal failure. Trials are under way to determine the efficacy and safety of these devices.20
Case continues: He receives a liver
The patient undergoes liver transplantation. He is given HBV immune globulin during and after the surgery.
Pathologic review. Under the microscope, his old liver has widespread necrosis and hemorrhage as well as inflammatory changes suggesting a chronic viral process. Regenerative nodules are present in the small amount of surviving liver parenchyma, consistent with early cirrhosis. Iron staining shows +3 depositions in areas of hepatic collapse (a nonspecific finding). Periodic acid-Schiff staining after diastase (used to detect alpha-1 antitrypsin deficiency) is negative. Herpetic viral inclusions are not present.
An immunoassay for herpes simplex virus antigen is negative. Immunostaining with antibodies to the HBV core antigen is negative. HBV surface antigen is strongly and diffusely positive in the cytoplasm of 80% to 90% of hepatocytes. The immunohistologic staining pattern is consistent with integration of HBV DNA into the DNA of hepatic tissue.
Postoperative course. Lamivudine is continued after surgery, and the patient is sent home. He has resumed the level of functioning he had before becoming ill.
Comment. The outcome of liver transplantation for hepatitis B has notably improved since HBV immune globulin and nucleoside analogues were introduced. The results of liver transplantation for hepatitis B, particuarly patient and graft survival rates, are now better than those in transplant patients with hepatitis C and similar to those in transplant patients with other types of liver disease.21 The combination of HBV immune globulin and lamivudine has cut the rate of HBV reinfection after liver transplantation to approximately 10% and increased the 5-year survival rate after transplantation to about 80%.17,22
KEY POINTS
- In immunocompetent adults, most primary HBV infections are self-limited.
- Chronic HBV infection is defined as the persistence of HBV surface antigen in the serum for at least 6 months. Patients having chronic HBV infection can be broadly classified as inactive carriers or having chronic active disease.
- Most patients with chronic active HBV infection are positive for HBV e antigen, except patients in whom the virus has a mutation in the precore or core region of its genome that prevents the production of e antigen.
- Patients who carry inactive HBV or who are immune-tolerant require serial measurements of aminotransferase and HBV DNA levels. Treatment can be considered if the patient has a high viral load (> 2,000 IU/mL), elevated aminotransferases, or active disease on liver biopsy.
- Carriers of chronic active HBV (whether positive or negative for HBV e antigen) should be referred to a hepatologist for consideration of liver biopsy and treatment.
- Interferon should not be used in immunocompromised patients or those with decompensated liver disease because it can further exacerbate the liver disease.
- Liver transplantation should be considered in patients with acute liver failure who have a poor prognosis according to the King’s College Hospital criteria.
A 35-year-old man who was born in Vietnam presents to the emergency department of a local hospital because he has had jaundice for 5 days and fatigue, malaise, and anorexia for 2 weeks. He also has nausea and mild epigastric and right upper quadrant abdominal pain. He denies having fevers, chills, night sweats, vomiting, diarrhea, melena, hematochezia, or weight loss.
His medical history is remarkable only for perinatally acquired hepatitis B virus (HBV) infection, for which he never received antiviral therapy. He does not take any prescribed, over-the-counter, or herbal medications.
He lives in the Midwest region of the United States and works full-time as a physician in private practice. He is married and has two children.
He has not travelled recently. He has no pets at home and has not been exposed to any.
He has never smoked. He drinks alcohol socially but has never used recreational drugs.
In a laboratory evaluation performed a year ago for insurance purposes, his liver function tests—serum albumin, total bilirubin, alkaline phosphatase, alanine aminotransferase, and aspartate aminotransferase levels—were all normal. He was positive for HBV surface antigen and HBV e antigen and negative for antibodies against these antigens.
PHASES OF HBV INFECTION
1. Which of the following best describes the status of HBV infection in this patient before his current symptoms developed?
- Resolved HBV infection
- Chronic inactive HBV infection
- Chronic active HBV infection
- Immune-tolerant chronic HBV infection
The correct answer is immune-tolerant chronic HBV infection.
Resolved infection. In immunocompetent adults, most primary HBV infections are self-limited: people clear the virus and gain lasting immunity (defined as the loss of HBV surface antigen, the development of antibody against surface antigen, no detectable HBV DNA in the serum, and normal alanine and aspartate aminotransferase levels). However, a minority of primary HBV infections persist and become chronic.
Chronic HBV infection is defined as the persistence of HBV surface antigen in the serum for at least 6 months. Patients with chronic HBV infection can be broadly classified as having either inactive disease (the inactive surface antigen carrier state) or chronic active hepatitis B (Figure 1).1–9
Chronic inactive HBV infection. Carriers of inactive HBV infection have low serum levels of HBV DNA (< 2,000 IU/mL), persistently normal aminotransferase levels, and no HBV e antigen; if a liver biopsy is performed, no significant hepatitis is found.
Chronic active HBV infection. Patients with chronic active HBV infection, in contrast, have high serum HBV DNA levels (> 20,000 IU/mL) and persistently or intermittently high aminotransferase levels; they do have HBV e antigen, and a liver biopsy shows moderate or severe necroinflammation.
A small group of patients with chronic active hepatitis B may be negative for e antigen but still have high aminotransferase levels, high HBV DNA levels, and continued necroinflammation in the liver.4 The virus in these patients has a mutation in its precore or core promoter gene that prevents the production of e antigen.
Patients with chronic active HBV infection (whether positive or negative for e antigen) are at a significantly greater risk of progressive liver injury and developing cirrhosis and hepatocellular carcinoma than are inactive carriers of HBV.
Immune-tolerant chronic HBV infection. Patients who acquired HBV at birth (eg, our patient) may have immune-tolerant HBV infection, which is characterized by significant HBV replication manifested by the presence of HBV e antigen and high levels of HBV DNA in the serum. However, these patients have no clinical or pathologic evidence of active liver disease (no symptoms, normal serum alanine aminotransferase levels, and minimal changes on liver biopsy).5 This was obviously the case in our patient, based on his history and laboratory results before his current symptoms developed.
Case continues: Liver function abnormalities
On physical examination, the patient’s temperature is 99.9°F (37.7°C), heart rate 106 per minute, blood pressure 98/54 mm Hg, respiratory rate 18 per minute, and oxygen saturation 100% while breathing ambient air. He is alert and oriented to time, place, and person.
He has icteric sclera, and his skin is jaundiced. His lymph nodes are not palpable. His cardiac examination is normal except for tachycardia. His lungs are clear to auscultation and percussion. He has mild epigastric and right upper quadrant abdominal tenderness with no peritoneal signs, hepatosplenomegaly, or masses.
His basic laboratory values on admission are listed in Table 1. His amylase and lipase levels are normal. A urine dipstick test is positive for bilirubin.
WHAT IS THE LEAST LIKELY DIAGNOSIS?
2. Which one of the following is the least likely diagnosis in this patient?
- Reactivation of hepatitis B
- Drug-associated liver injury
- Acute viral hepatitis
- Acute alcoholic hepatitis
- Ischemic hepatitis
The degree and pattern of liver function abnormalities in our patient reflect hepatocellular injury rather than cholestatic liver disease, because his aminotransferase levels are elevated much higher than his alkaline phosphatase level (Table 1). Bilirubin elevation does not help differentiate the two conditions.
The degree and pattern of aminotransferase elevations are also helpful in narrowing the differential diagnosis. Serum aminotransferase levels of more than 1,000 U/L are mainly seen in patients with ischemic, viral, and toxininduced liver injury. Other rare causes of such high levels include Budd-Chiari syndrome, Wilson disease, and autoimmune hepatitis.
Ischemic hepatitis. Our patient has mild hypotension, but it does not seem to have been severe enough or of long enough duration to have caused ischemic hepatitis.
Drug-associated liver injury. Hepatotoxicity associated with drugs (most commonly acetaminophen [Tylenol]), herbal therapy, or mushroom poisoning should be considered in any patient whose aminotransferase levels are this high. However, our patient denies taking any medications (prescribed or over-the-counter), herbal remedies, or illicit drugs.
Acute viral hepatitis can certainly explain the patient’s clinical picture. Infection with hepatitis A virus, hepatitis D virus, hepatitis E virus, cytomegalovirus, Epstein-Barr virus, herpes simplex viruses types 1 and 2, and varicella zoster virus have all been implicated in severe acute hepatitis. Although hepatitis E virus infection is more common in developing countries, it has been reported in the United States.6 It is unlikely that acute hepatitis C virus infection is producing this degree of elevation in aminotransferase levels.
Reactivation of the patient’s chronic HBV infection can also account for his clinical presentation.
Acute alcoholic hepatitis should be suspected clinically if a patient has a history of heavy alcohol use and clinical and laboratory findings that are compatible with the diagnosis. However, the absolute values of serum aspartate aminotransferase and alanine aminotransferase in acute alcoholic hepatitis are almost always less than 500 IU/L (and typically less than 300 IU/L). Our patient’s values are much higher, and he says he does not drink very much. Although people sometimes underestimate their alcohol intake, alcoholic hepatitis is the least likely diagnosis in our patient.
Case continues: The patient is hospitalized
The patient is admitted with a diagnosis of acute hepatitis. Given his history of chronic hepatitis B, he is empirically started on lamivudine (Epivir-HBV).
- Antinuclear antibody negative
- Autoimmune liver disease panel negative
- Serum ceruloplasmin 30 mg/dL (normal range 15–60)
- Alpha fetoprotein 35.1 μg/L (< 10).
Abdominal ultrasonography is performed and reveals a small stone in the gallbladder with no evidence of biliary dilatation; otherwise, the gallbladder appears normal. Doppler ultrasonography shows the liver vessels to be patent; the liver is normal in appearance. The abdomen and pelvis appear to be normal on computed tomography without intravenous contrast.
On the third hospital day, the patient’s blood test results are:
- Aspartate aminotransferase 199 U/L (normal range 7–40)
- Alanine aminotransferase 735 U/L (0–45)
- Total bilirubin 22.9 mg/dL (0–1.5)
- International normalized ratio 6.0 (0.77–1.17)
- White blood cell count 5.1 × 109/L (4–11)
- Hemoglobin 11.7 g/dL (12–16)
- Platelet count 166 × 109/L (150–400)
- Blood and urine cultures negative.
WHAT IS CAUSING HIS ACUTE HEPATITIS?
3. On the basis of the new data, which of the following statements about the cause of acute hepatitis in this patient is the most accurate?
- Herpetic hepatitis is the most likely cause, given his positive test for immunoglobulin M (IgM) against herpes simplex virus
- Hepatitis C cannot be excluded with the available data
- Negative HBV e antigen does not exclude the diagnosis of acute exacerbation of HBV infection
- Hepatocellular carcinoma is the likely diagnosis, given the elevated alpha fetoprotein level
The third answer above is correct: a negative test for hepatitis B e antigen does not exclude the diagnosis of acute exacerbation of HBV infection
Herpetic hepatitis. Although not common, hepatitis due to herpes simplex virus infection should be considered in the differential diagnosis of any patient presenting with severe acute hepatitis, particularly when fever is present. Common features of herpetic hepatitis on presentation include high fever, leukopenia, markedly elevated aminotransferases, and mild cholestasis. Vesicular rash occurs in only less than half of cases of herpetic hepatitis.10
Serologic testing is of limited value because it has high rates of false-positive and false-negative results. The diagnosis can be confirmed only by viral polymerase chain reaction testing or by identifying herpes simplex viral inclusions in the liver biopsy.
However, the death rate is high in this disease, and since herpetic hepatitis is one of the few treatable causes of acute liver failure, parenteral acyclovir (Zovirax) should be considered empirically in patients presenting with acute liver failure. Our patient was started on acyclovir when his tests for IgM against herpes simplex virus came back positive.
Hepatitis C. Antibodies against hepatitis C virus do not develop immediately after this virus is contracted; they may take up to 12 weeks to develop after exposure. For this reason, about 30% to 50% of patients with acute hepatitis C virus infection are negative for these antibodies initially. In those patients, hepatitis C virus RNA in the blood is the most sensitive test to detect acute hepatitis C virus infection.
Our patient has neither antibodies against hepatitis C virus nor hepatitis C virus RNA by polymerase chain reaction testing, which rules out hepatitis C virus infection.
Disappearance of e antigen in HBV infection. The disappearance of HBV e antigen is usually associated with a decrease in serum HBV DNA and remission of liver disease. However, some patients continue to have active liver disease and high levels of HBV DNA despite e antigen seroconversion. This is due to a stop codon mutation in the precore region of the viral genome that decreases or prevents production of HBV e antigen.4 In other words, even though HBV e antigen is a good marker of HBV replication in general, a subgroup of patients with chronic HBV infection are negative for e antigen but still have a high rate of viral replication as evidenced by high serum HBV DNA levels.
Patients with perinatally acquired chronic HBV infection most often have immune-tolerant chronic HBV infection. Among those patients (mostly Asian),5,7 the virus is spontaneously cleared at a rate of approximately 2% to 3% per year,8 most often during the second and third decades of age.
Transition from the immune-tolerant phase to the immune clearance phase is frequently associated with mild transient worsening of the liver function profile.9,11,12 However, in a small percentage of patients, hepatic decompensation and even (rarely) death from hepatic failure may occur secondary to a sudden activation of the immune system as it attempts to clear the virus. This may result in an increase in immune-mediated lysis of infected hepatocytes.
Hepatocellular carcinoma. Exacerbation of hepatitis B may be associated with an elevation of alpha fetoprotein, which may falsely raise concerns about the possibility of hepatocellular carcinoma. However, our patient had abdominal imaging with both ultrasonography and computed tomography, which showed no evidence of hepatocellular carcinoma.
Comment. The most likely cause of the patient’s acute liver failure is an acute exacerbation of hepatitis B. However, herpetic hepatitis should be ruled out by testing for herpes simplex virus by polymerase chain reaction, performing a liver biopsy, or both.
Case continues: His condition worsens
A transjugular liver biopsy shows changes associated with chronic hepatitis B, severe acute hepatitis with extensive confluent and submassive hepatic necrosis, and no intracellular viral inclusions. Subsequently, acyclovir is stopped.
On the 6th hospital day, he develops progressive metabolic acidosis and hypotension, with worsening hypoxemia. A chest radiograph is obtained to look for pneumonia, but it is indeterminate; computed tomography of the chest without contrast medium is likewise unremarkable. Duplex ultrasonography of the four extremities is negative for venous thrombosis.
The patient becomes more lethargic and difficult to arouse. He is transferred to the intensive care unit and intubated. His prothrombin and partial thromboplastin times continue to rise, the prothrombin time reaching values of more than 50 seconds. In addition, progressive renal insufficiency develops.
WHAT IS THE NEXT STEP?
4. Which of the following is the most appropriate next step in the management of this patient?
- Liver transplantation
- HBV immunoglobulin only
- Interferon and a nucleoside analogue
- Liver-assist devices
- Continue supportive care only
Liver transplantation. Since the patient’s severe acute hepatitis is accompanied by coagulopathy and encephalopathy, he meets the definition of having acute liver failure. Liver transplantation remains the only definitive therapy.
HBV immune globulin immunoprophylaxis is indicated in patients with HBV infection undergoing liver transplantation, to prevent recurrence of hepatitis B after the transplant, particularly in those with a high pretransplant viral load.17 The use of pretransplant antiviral therapy and the posttransplant combination of antiviral therapy and HBV immune globulin has reduced the rate of hepatitis B recurrence to less than 10%. However, immune globulin is by no means the best single next step in managing this patient, who clearly needs a new liver.
Interferon, nucleoside analogues. Options for antiviral treatment are interferon alfa and nucleoside analogues. Interferon therapy is contraindicated in patients such as ours, who have decompensated liver disease, because it can exacerbate the disease.18
Liver-assist devices. Because liver allografts are in short supply, there has been a strong interest in developing a device that would provide the same benefits for patients with liver failure as hemodialysis does for patients with renal failure. Trials are under way to determine the efficacy and safety of these devices.20
Case continues: He receives a liver
The patient undergoes liver transplantation. He is given HBV immune globulin during and after the surgery.
Pathologic review. Under the microscope, his old liver has widespread necrosis and hemorrhage as well as inflammatory changes suggesting a chronic viral process. Regenerative nodules are present in the small amount of surviving liver parenchyma, consistent with early cirrhosis. Iron staining shows +3 depositions in areas of hepatic collapse (a nonspecific finding). Periodic acid-Schiff staining after diastase (used to detect alpha-1 antitrypsin deficiency) is negative. Herpetic viral inclusions are not present.
An immunoassay for herpes simplex virus antigen is negative. Immunostaining with antibodies to the HBV core antigen is negative. HBV surface antigen is strongly and diffusely positive in the cytoplasm of 80% to 90% of hepatocytes. The immunohistologic staining pattern is consistent with integration of HBV DNA into the DNA of hepatic tissue.
Postoperative course. Lamivudine is continued after surgery, and the patient is sent home. He has resumed the level of functioning he had before becoming ill.
Comment. The outcome of liver transplantation for hepatitis B has notably improved since HBV immune globulin and nucleoside analogues were introduced. The results of liver transplantation for hepatitis B, particuarly patient and graft survival rates, are now better than those in transplant patients with hepatitis C and similar to those in transplant patients with other types of liver disease.21 The combination of HBV immune globulin and lamivudine has cut the rate of HBV reinfection after liver transplantation to approximately 10% and increased the 5-year survival rate after transplantation to about 80%.17,22
KEY POINTS
- In immunocompetent adults, most primary HBV infections are self-limited.
- Chronic HBV infection is defined as the persistence of HBV surface antigen in the serum for at least 6 months. Patients having chronic HBV infection can be broadly classified as inactive carriers or having chronic active disease.
- Most patients with chronic active HBV infection are positive for HBV e antigen, except patients in whom the virus has a mutation in the precore or core region of its genome that prevents the production of e antigen.
- Patients who carry inactive HBV or who are immune-tolerant require serial measurements of aminotransferase and HBV DNA levels. Treatment can be considered if the patient has a high viral load (> 2,000 IU/mL), elevated aminotransferases, or active disease on liver biopsy.
- Carriers of chronic active HBV (whether positive or negative for HBV e antigen) should be referred to a hepatologist for consideration of liver biopsy and treatment.
- Interferon should not be used in immunocompromised patients or those with decompensated liver disease because it can further exacerbate the liver disease.
- Liver transplantation should be considered in patients with acute liver failure who have a poor prognosis according to the King’s College Hospital criteria.
- Dusheiko G. Hepatitis B. In:Bircher J, Benhamou JP, McIntyre N, Rizzetto M, Rodes J, editors. Oxford Textbook of Clinical Hepatology. 2nd ed. Oxford, UK: Oxford University Press; 1999:876–896.
- Chu CJ, Hussain M, Lok AS. Quantitative serum HBV DNA levels during different stages of chronic hepatitis B infection. Hepatology 2002; 36:1408–1415.
- Pawlotsky JM, Bastie A, Hezode C, et al. Routine detection and quantification of hepatitis B virus DNA in clinical laboratories: performance of three commercial assays. J Virol Methods 2000; 85:11–21.
- Brunetto MR, Giarin MM, Oliveri F, et al. Wild-type and e-antigen-minus hepatitis viruses and course of chronic hepatitis. Proc Natl Acad Sci USA 1991; 88:4186–4190.
- Lok AS, Lai CL. A longitudinal follow-up of asymptomatic hepatitis B surface antigen-positive Chinese children. Hepatology 1988; 5:1130–1133.
- Hsu HY, Chang MH, Hsieh KH, et al. Cellular immune response to HBcAg in mother-to-infant transmission of hepatitis B virus. Hepatology 1992; 15:770–776.
- Chang MH, Hsu HY, Hsu HC, Ni YH, Chen JS, Chen DS. The significance of spontaneous hepatitis B e antigen seroconversion in childhood: with special emphasis on the clearance of hepatitis B e antigen before 3 years of age. Hepatology 1995; 22:1387–1392.
- Ruiz-Moreno M, Otero M, Millan A, et al. Clinical and histological outcome after hepatitis B e antigen to antibody seroconversion in children with chronic hepatitis B. Hepatology 1999; 29:572–575.
- Liaw YF, Chu CM, Su IJ, Huang MJ, Lin DY, Chang-Chien CS. Clinical and histological events preceding hepatitis B e antigen seroconversion in chronic type B hepatitis. Gastroenterology 1983; 84:216–219.
- Norvell JP, Blei AT, Jovanovic BD, Levitsky J. Herpes simplex virus hepatitis: an analysis of the published literature and institutional cases. Liver Transplant 2007; 13:1428–1434,
- Liaw YF, Pao CC, Chu CM, Sheen IS, Huang MJ. Changes of serum hepatitis B virus DNA in two types of clinical events preceding spontaneous hepatitis B e antigen seroconversion in chronic type B hepatitis. Hepatology 1987; 7:1–3.
- Maruyama T, Iino S, Koike K, Yasuda K, Milich DR. Serology of acute exacerbation in chronic hepatitis B virus infection. Gastroenterology 1993; 105:1141–1151.
- O'Grady JG, Alexander GJ, Hayllar KM, Williams R. Early indicators of prognosis in fulminant hepatic failure. Gastroenterology 1989; 97:439–445.
- Shakil AO, Kramer D, Mazariegos GV, Fung JJ, Rakela J. Acute liver failure: clinical features, outcome analysis, and applicability of prognostic criteria. Liver Transplant 2000; 6:163–169.
- Anand AC, Nightingale P, Neuberger JM. Early indicators of prognosis in fulminant hepatic failure: an assessment of the King’s criteria. J Hepatol 1997; 26:62–68.
- Schmidt LE, Dalhoff K. Serum phosphate is an early predictor of outcome in severe acetaminophen-induced hepatotoxicity. Hepatology 2002; 36:659–665.
- Samuel D, Muller R, Alexander G, et al. Liver transplantation in European patients with the hepatitis B surface antigen. N Engl J Med 1993; 329:1842–1847.
- Lok A, McMahon BJ. Chronic hepatitis B. Hepatology 2007; 45:507–539.
- Sorren MF, Belangia EA, Costa J, et al. National Institutes of Health consensus development conference statement: management of hepatitis B. Ann Intern Med 2009; 150:104–110.
- Kjaergard LL, Liu J, Als-Nielsen B, Gluud C. Artificial and bioartificial support systems for acute and acute-on-chronic liver failure: a systematic review. JAMA 2003; 289:217–222.
- Kim WR, Poterucha JJ, Kremers WK, Ishitani MB, Dickson ER. Outcome of liver transplantation for hepatitis B in the United States. Liver Transplant 2004; 10:968–974.
- Terrault NA, Zhou S, Combs C, et al. Prophylaxis in liver transplant recipients using a fixed dosing schedule of hepatitis B immunoglobulin. Hepatology 1996; 24:1327–1333.
- Dusheiko G. Hepatitis B. In:Bircher J, Benhamou JP, McIntyre N, Rizzetto M, Rodes J, editors. Oxford Textbook of Clinical Hepatology. 2nd ed. Oxford, UK: Oxford University Press; 1999:876–896.
- Chu CJ, Hussain M, Lok AS. Quantitative serum HBV DNA levels during different stages of chronic hepatitis B infection. Hepatology 2002; 36:1408–1415.
- Pawlotsky JM, Bastie A, Hezode C, et al. Routine detection and quantification of hepatitis B virus DNA in clinical laboratories: performance of three commercial assays. J Virol Methods 2000; 85:11–21.
- Brunetto MR, Giarin MM, Oliveri F, et al. Wild-type and e-antigen-minus hepatitis viruses and course of chronic hepatitis. Proc Natl Acad Sci USA 1991; 88:4186–4190.
- Lok AS, Lai CL. A longitudinal follow-up of asymptomatic hepatitis B surface antigen-positive Chinese children. Hepatology 1988; 5:1130–1133.
- Hsu HY, Chang MH, Hsieh KH, et al. Cellular immune response to HBcAg in mother-to-infant transmission of hepatitis B virus. Hepatology 1992; 15:770–776.
- Chang MH, Hsu HY, Hsu HC, Ni YH, Chen JS, Chen DS. The significance of spontaneous hepatitis B e antigen seroconversion in childhood: with special emphasis on the clearance of hepatitis B e antigen before 3 years of age. Hepatology 1995; 22:1387–1392.
- Ruiz-Moreno M, Otero M, Millan A, et al. Clinical and histological outcome after hepatitis B e antigen to antibody seroconversion in children with chronic hepatitis B. Hepatology 1999; 29:572–575.
- Liaw YF, Chu CM, Su IJ, Huang MJ, Lin DY, Chang-Chien CS. Clinical and histological events preceding hepatitis B e antigen seroconversion in chronic type B hepatitis. Gastroenterology 1983; 84:216–219.
- Norvell JP, Blei AT, Jovanovic BD, Levitsky J. Herpes simplex virus hepatitis: an analysis of the published literature and institutional cases. Liver Transplant 2007; 13:1428–1434,
- Liaw YF, Pao CC, Chu CM, Sheen IS, Huang MJ. Changes of serum hepatitis B virus DNA in two types of clinical events preceding spontaneous hepatitis B e antigen seroconversion in chronic type B hepatitis. Hepatology 1987; 7:1–3.
- Maruyama T, Iino S, Koike K, Yasuda K, Milich DR. Serology of acute exacerbation in chronic hepatitis B virus infection. Gastroenterology 1993; 105:1141–1151.
- O'Grady JG, Alexander GJ, Hayllar KM, Williams R. Early indicators of prognosis in fulminant hepatic failure. Gastroenterology 1989; 97:439–445.
- Shakil AO, Kramer D, Mazariegos GV, Fung JJ, Rakela J. Acute liver failure: clinical features, outcome analysis, and applicability of prognostic criteria. Liver Transplant 2000; 6:163–169.
- Anand AC, Nightingale P, Neuberger JM. Early indicators of prognosis in fulminant hepatic failure: an assessment of the King’s criteria. J Hepatol 1997; 26:62–68.
- Schmidt LE, Dalhoff K. Serum phosphate is an early predictor of outcome in severe acetaminophen-induced hepatotoxicity. Hepatology 2002; 36:659–665.
- Samuel D, Muller R, Alexander G, et al. Liver transplantation in European patients with the hepatitis B surface antigen. N Engl J Med 1993; 329:1842–1847.
- Lok A, McMahon BJ. Chronic hepatitis B. Hepatology 2007; 45:507–539.
- Sorren MF, Belangia EA, Costa J, et al. National Institutes of Health consensus development conference statement: management of hepatitis B. Ann Intern Med 2009; 150:104–110.
- Kjaergard LL, Liu J, Als-Nielsen B, Gluud C. Artificial and bioartificial support systems for acute and acute-on-chronic liver failure: a systematic review. JAMA 2003; 289:217–222.
- Kim WR, Poterucha JJ, Kremers WK, Ishitani MB, Dickson ER. Outcome of liver transplantation for hepatitis B in the United States. Liver Transplant 2004; 10:968–974.
- Terrault NA, Zhou S, Combs C, et al. Prophylaxis in liver transplant recipients using a fixed dosing schedule of hepatitis B immunoglobulin. Hepatology 1996; 24:1327–1333.