User login
Gallstones: Watch and wait, or intervene?
The prevalence of gallstones is approximately 10% to 15% of the adult US population.1,2 Most cases are asymptomatic, as gallstones are usually discovered incidentally during routine imaging for other abdominal conditions, and only about 20% of patients with asymptomatic gallstones develop clinically significant complications.2,3
Nevertheless, gallstones carry significant healthcare costs. In 2004, the median inpatient cost for any gallstone-related disease was $11,584, with an overall annual cost of $6.2 billion.4,5
Laparoscopic cholecystectomy is the standard treatment for symptomatic cholelithiasis. For asymptomatic cholelithasis, the usual approach is expectant management (“watch and wait”), but prophylactic cholecystectomy may be an option in certain patients at high risk.
CHEMICAL COMPOSITION
Gallstones can be classified into 2 main categories based on their predominant chemical composition: cholesterol or pigment.
Cholesterol gallstones
About 75% of gallstones are composed of cholesterol.3,4 In the past, this type of stone was thought to be caused by gallbladder inflammation, bile stasis, and absorption of bile salts from damaged mucosa. However, it is now known that cholesterol gallstones are the result of biliary supersaturation caused by cholesterol hypersecretion into the gallbladder, gallbladder hypomotility, accelerated cholesterol nucleation and crystallization, and mucin gel accumulation.
Pigment gallstones
Black pigment gallstones account for 10% to 15% of all gallstones.6 They are caused by chronic hemolysis in association with supersaturation of bile with calcium hydrogen bilirubinate, along with deposition of calcium carbonate, phosphate, and inorganic salts.7
Brown pigment stones, accounting for 5% to 10% of all gallstones,6 are caused by infection in the obstructed bile ducts, where bacteria that produce beta-glucuronidase, phospholipase, and slime contribute to formation of the stone.8,9
RISK FACTORS FOR GALLSTONES

Age. After age 40, the risk increases dramatically, with an incidence 4 times higher for those ages 40 to 69 than in younger people.10
Female sex. Women of reproductive age are 4 times more likely to develop gallstones than men, but this gap narrows after menopause.11 The higher risk is attributed to female sex hormones, pregnancy, and oral contraceptive use. Estrogen decreases secretion of bile salts and increases secretion of cholesterol into the gallbladder, which leads to cholesterol supersaturation. Progesterone acts synergistically by causing hypomobility of the gallbladder, which in turn leads to bile stasis.12,13
Ethnicity. The risk is higher in Mexican Americans and Native Americans than in other ethnic groups.14
Rapid weight loss, such as after bariatric surgery, occurs from decreased caloric intake and promotes bile stasis, while lipolysis increases cholesterol mobilization and secretion into the gallbladder. This creates an environment conducive to bile supersaturation with cholesterol, leading to gallstone formation.
Chronic hemolytic disorders carry an increased risk of developing calcium bilirubinate stones due to increased excretion of bilirubin during hemolysis.
Obesity and diabetes mellitus are both attributed to insulin resistance. Obesity also increases bile stasis and cholesterol saturation.
CLINICAL PRESENTATION OF GALLSTONES (CHOLELITHIASIS)
Most patients with gallstones (cholelithiasis) experience no symptoms. Their gallstones are often discovered incidentally during imaging tests for unrelated or unexplained abdominal symptoms. Most patients with asymptomatic gallstones remain symptom-free, while about 20% develop gallstone-related symptoms.2,3
Abdominal pain is the most common symptom. The phrase biliary colic—suggesting pain that is fluctuating in nature—appears ubiquitously in the medical literature, but it does not correctly characterize the pain associated with gallstones.
Most patients with gallstone symptoms describe a constant and often severe pain in the right upper abdomen, epigastrium, or both, often persisting for 30 to 120 minutes. Symptoms are frequently reported in the epigastrium when only visceral pain fibers are stimulated due to gallbladder distention. This is usually called midline pain; however, pain occurs in the back and right shoulder in up to 60% of patients, with involvement of somatic fibers.15,16 Gallstone pain is not relieved by change of position or passage of stool or gas.
Onset of symptoms more than an hour after eating or in the late evening or at night also very strongly suggests biliary pain. Patients with a history of biliary pain are more likely to experience it again, with a 69% chance of developing recurrent pain within 2 years.17
GALLSTONE-RELATED COMPLICATIONS

Acute gallbladder inflammation (cholecystitis)
Gallbladder inflammation (cholecystitis) is the most common complication, occurring in up to 10% of symptomatic cases. Many patients with acute cholecystitis present with right upper quadrant pain that may be accompanied by anorexia, nausea, or vomiting. Inspiratory arrest on deep palpation of the right upper quadrant (Murphy sign) has a specificity of 79% to 96% for acute cholecystitis.20 Markers of systemic inflammation such as fever, elevated white blood cell count, and elevated C-reactive protein are highly suggestive of acute cholecystitis.20,21
Bile duct stones (choledocholithiasis)
Bile duct stones (choledocholithiasis) are detected in 3.4% to 12% of patients with gallstones.22,23 Most stones in the common bile duct migrate there from the gallbladder via the cystic duct. Less commonly, primary duct stones form in the duct due to biliary stasis. Removing the gallbladder does not completely eliminate the risk of bile duct stones, as stones can remain or recur after surgery.
Bile duct stones can obstruct the common bile duct, which disrupts normal bile flow and leads to jaundice. Other symptoms may include pruritus, right upper quadrant pain, nausea, and vomiting. Serum levels of bilirubin, aspartate aminotransferase, alanine aminotransferase (ALT), and alkaline phosphatase are usually high.24
Acute bacterial infection (cholangitis)
Acute bacterial infection of the biliary system (cholangitis) is usually associated with obstruction of the common bile duct. Common symptoms of acute cholangitis include right upper quadrant pain, fever, and jaundice (Charcot triad), and these are present in about 50% to 75% of cases.21 In severe cases, patients can develop altered mental status and septicemic shock in addition to the Charcot triad, a condition called the Reynold pentad. White blood cell counts and serum levels of C-reactive protein, bilirubin, aminotransferases, and alkaline phosphatase are usually elevated.21
Pancreatitis
Approximately 4% to 8% of patients with gallstones develop inflammation of the pancreas (pancreatitis).25 The diagnosis of acute pancreatitis requires at least 2 of the following:26,27
- Abdominal pain (typically epigastric, often radiating to the back)
- Amylase or lipase levels at least 3 times above the normal limit
- Imaging findings that suggest acute pancreatitis.
Gallstone-related pancreatitis should be considered if the ALT level is greater than 150 U/mL, which has a 97% specificity for gallstone-related pancreatitis.28
ABDOMINAL ULTRASONOGRAPHY FOR DIAGNOSIS
Transabdominal ultrasonography, with a sensitivity of 84% to 89% and a specificity of up to 99%, is the test of choice for detecting gallstones.29 The characteristic findings of acute cholecystitis on ultrasonography include enlargement of the gallbladder, thickening of the gallbladder wall, presence of pericholecystic fluid, and tenderness elicited by the ultrasound probe over the gallbladder (sonographic Murphy sign).
Scintigraphy as a second test
Acute cholecystitis is primarily a clinical diagnosis and typically does not require additional imaging beyond ultrasonography. When there is discordance between clinical and ultrasonographic findings, the most accurate second imaging test is scintigraphy of the biliary tract, usually performed with technetium-labeled hydroxy iminodiacetic acid. Given intravenously, the radionuclide is rapidly taken up by the liver and then secreted into the bile. In acute cholecystitis, the cystic duct is functionally occluded and the isotope does not enter the gallbladder, creating an imaging void compared with a normal appearance.
Scintigraphy is more sensitive than abdominal ultrasonography, with a sensitivity of up to 97% vs 81% to 88%, respectively.29,30 The tests have about equal specificity.
Even though scintigraphy is more sensitive, abdominal ultrasonography is often the initial test for patients with suspected acute cholecystitis because it is more widely available, takes less time, does not involve radiation exposure, and can assess for the presence or absence of gallstones and dilation of the intra- and extrahepatic bile ducts.
Looking for stones in the common bile duct
When acute cholangitis due to choledocholithiasis is suspected, abdominal ultrasonography is a prudent initial test to look for gallstones or biliary dilation suggesting obstruction by stones in the common bile duct. Abdominal ultrasonography has only a 22% to 55% sensitivity for visualizing stones in the common bile duct, but it has a 77% to 87% sensitivity for detecting common bile duct dilation, a surrogate marker of stones.31
The normal bile duct diameter ranges from 3 to 6 mm, although mild dilation is often seen in older patients or after cholecystectomy or Roux-en-Y gastric bypass surgery.32,33 Bile duct dilation of up to 10 mm can be considered normal in patients after cholecystectomy.34 A normal-appearing bile duct on ultrasonography has a negative predictive value of 95% for excluding common bile duct stones.31
Endoscopic ultrasonography (EUS), magnetic resonance cholangiopancreatography (MRCP), and endoscopic retrograde cholangiopancreatography (ERCP) have similar sensitivity (89%–94%, 85%–92%, and 89%–93%, respectively) and specificity (94%–95%, 93%–97%, and 100%, respectively) for detecting common bile duct stones.35–37 EUS is superior to MRCP in detecting stones smaller than 6 mm.38
ERCP should be reserved for managing rather than diagnosing common bile duct stones because of the risk of pancreatitis and perforation. Patients undergoing cholecystectomy who are suspected of having choledocholithiasis may undergo intraoperative cholangiography or laparoscopic common bile duct ultrasonography.
WATCH AND WAIT, OR INTERVENE?
Asymptomatic gallstones

Standard treatment for these patients is expectant management. Cholecystectomy is not recommended for patients with asymptomatic gallstones.47 Nevertheless, some patients may benefit from prophylactic cholecystectomy. We and others48 suggest considering cholecystectomy in the following patients.
Patients with chronic hemolytic anemia (including children with sickle cell anemia and spherocytosis). These patients have a higher risk of developing calcium bilirubinate stones, and cholecystectomy has improved outcomes.49 It should be noted that most of these data come from pediatric populations and have been extrapolated to adults.
Native Americans, who have a higher risk of gallbladder cancer if they have gallstones.2,50
Conversely, calcification of the gallbladder wall (“porcelain gallbladder”) is no longer considered an absolute indication for cholecystectomy. This condition was thought to be associated with a high rate of gallbladder carcinoma, but analyses of larger, more recent data sets found much smaller risks.51,52 Further, cholecystectomy in these patients was found to be associated with high rates of postoperative complications. Thus, prophylactic cholecystectomy is no longer recommended in asymptomatic cases of porcelain gallbladder.
In addition, concomitant cholecystectomy in patients undergoing bariatric surgery is no longer considered the therapeutic standard. Historically, cholecystectomy was performed in these patients because of the increased risk of gallstones associated with rapid weight loss after surgery. However, research now weighs against concomitant cholecystectomy with bariatric surgery and most other abdominal surgeries for asymptomatic gallstones.53
Laparoscopic surgery for symptomatic gallstones

For patients experiencing acute cholecystitis, laparoscopic cholecystectomy within 72 hours is recommended.48 There were safety concerns regarding higher rates of morbidity and conversion from laparoscopic to open cholecystectomy in patients who underwent surgery before the acute cholecystitis episode had settled. However, a large meta-analysis found no significant difference between early and delayed laparoscopic cholecystectomy in bile duct injury or conversion rates.54 Further, early cholecystectomy—defined as within 1 week of symptom onset—has been found to reduce gallstone-related complications, shorten hospital stays, and lower costs.55–57 If the patient cannot undergo surgery, percutaneous cholecystotomy or novel endoscopic gallbladder drainage interventions can be used.
.")
Several variables predict the presence of bile duct stones in patients who have symptoms (Table 4). Based on these predictors, the ASGE classifies the probabilities as low (< 10%), intermediate (10% to 50%), and high (> 50%)31:

- High-risk patients should undergo preoperative ERCP and stone extraction if needed
- Intermediate-risk patients should undergo preoperative imaging with EUS or MRCP or intraoperative bile duct evaluation, depending on the availability, costs, and local expertise.

Patients with associated cholangitis should be given intravenous fluids and broad-spectrum antibiotics. Biliary decompression should be done as early as possible to decrease the risk of morbidity and mortality. For acute cholangitis, ERCP is the treatment of choice.25
Patients with acute gallstone pancreatitis should receive conservative management with intravenous isotonic solutions and pain control, followed by laparoscopic cholecystectomy.48
The timing of laparoscopic cholecystectomy in acute gallstone pancreatitis has been debated. Studies conducted during the era of open cholecystectomy reported similar or worse outcomes if cholecystectomy was done sooner rather than later.
However, in 1999, Uhl et al58 reported that 48 of 77 patients admitted with acute gallstone pancreatitis were able to undergo laparoscopic cholecystectomy during the same admission. Success rates were 85% (30 of 35 patients) in those with mild disease and 62% (8 of 13 patients) in those with severe disease. They concluded laparoscopic cholecystectomy could be safely performed within 7 days in patients with mild disease, whereas in severe disease at least 3 weeks should elapse because of the risk of infection.
In a randomized trial published in 2010, Aboulian et al59 reported that hospital length of stay (the primary end point) was shorter in 25 patients who underwent laparoscopic cholecystectomy early (within 48 hours of admission) than in 25 patients who underwent surgery after abdominal pain had resolved and laboratory enzymes showed a normalizing trend, 3.5 vs 5.8 days (P = .0016). Rates of perioperative complications and need for conversion to open surgery were similar between the 2 groups.
If there is associated cholangitis, patients should also be given broad-spectrum antibiotics and should undergo ERCP within 24 hours of admission.25–27
SUMMARY
Gallstones are common in US adults. Abdominal ultrasonography is the diagnostic imaging test of choice to detect gallbladder stones and assess for findings suggestive of acute cholecystitis and dilation of the common bile duct. Fortunately, most gallstones are asymptomatic and can usually be managed expectantly. In patients who have symptoms or have gallstone complications, laparoscopic cholecystectomy is the standard of care.
- Schirmer BD, Winters KL, Edlich RF. Cholelithiasis and cholecystitis. J Long Term Eff Med Implants 2005; 15(3):329–338. doi:10.1615/JLongTermEffMedImplants.v15.i3.90
- Stinton LM, Shaffer EA. Epidemiology of gallbladder disease: cholelithiasis and cancer. Gut Liver 2012; 6(2):172–187. doi:10.5009/gnl.2012.6.2.172
- Lee JY, Keane MG, Pereira S. Diagnosis and treatment of gallstone disease. Practitioner 2015; 259(1783):15–19.
- Russo MW, Wei JT, Thiny MT, et al. Digestive and liver diseases statistics, 2004. Gastroenterology 2004; 126(5):1448–1453. doi:10.1053/j.gastro.2004.01.025
- Everhart JE, Ruhl CE. Burden of digestive diseases in the United States part I: overall and upper gastrointestinal diseases. Gastroenterology 2009; 136(2):376–386. doi:10.1053/j.gastro.2008.12.015
- Cariati A. Gallstone classification in Western countries. Indian J Surg 2015; 77(suppl 2):376–380. doi.org/10.1007/s12262-013-0847-y
- Carey MC. Pathogenesis of gallstones. Am J Surg 1993; 165(4):410–419. doi:10.1016/S0002-9610(05)80932-8
- Lammert F, Gurusamy K, Ko CW, et al. Gallstones. Nat Rev Dis Primers 2016; 2:16024. doi:10.1038/nrdp.2016.24
- Stewart L, Oesterle AL, Erdan I, Griffiss JM, Way LW. Pathogenesis of pigment gallstones in Western societies: the central role of bacteria. J Gastrointest Surg 2002; 6(6):891–904.
- Barbara L, Sama C, Morselli Labate AM, et al. A population study on the prevalence of gallstone disease: the Sirmione Study. Hepatology 1987; 7(5):913–917. doi:10.1002/hep.1840070520
- Sood S, Winn T, Ibrahim S, et al. Natural history of asymptomatic gallstones: differential behaviour in male and female subjects. Med J Malaysia 2015; 70(6):341–345.
- Maringhini A, Ciambra M, Baccelliere P, et al. Biliary sludge and gallstones in pregnancy: incidence, risk factors, and natural history. Ann Intern Med 1993; 119(2):116–120. doi:10.7326/0003-4819-119-2-199307150-00004
- Etminan M, Delaney JA, Bressler B, Brophy JM. Oral contraceptives and the risk of gallbladder disease: a comparative safety study. CMAJ 2011; 183(8):899–904. doi:10.1503/cmaj.110161
- Everhart JE, Khare M, Hill M, Maurer KR. Prevalence and ethnic differences in gallbladder disease in the United States. Gastroenterology 1999; 117(3):632–639.
- Festi D, Sottili S, Colecchia A, et al. Clinical manifestations of gallstone disease: evidence from the multicenter Italian study on cholelithiasis (MICOL). Hepatology 1999; 30(4):839–846. doi:10.1002/hep.510300401
- Berhane T, Vetrhus M, Hausken T, Olafsson S, Sondenaa K. Pain attacks in non-complicated and complicated gallstone disease have a characteristic pattern and are accompanied by dyspepsia in most patients: the results of a prospective study. Scand J Gastroenterol 2006; 41(1):93–101. doi:10.1080/00365520510023990
- Thistle JL, Cleary PA, Lachin JM, Tyor MP, Hersh T. The natural history of cholelithiasis: the National Cooperative Gallstone Study. Ann Intern Med 1984; 101(2):171–175. doi:10.7326/0003-4819-101-2-171
- Friedman GD. Natural history of asymptomatic and symptomatic gallstones. Am J Surg 1993; 165(4):399–404. doi:0.1016/S0002-9610(05)80930-4
- Friedman GD, Raviola CA, Fireman B. Prognosis of gallstones with mild or no symptoms: 25 years of follow-up in a health maintenance organization. J Clin Epidemiol 1989; 42(2):127–136. doi:10.1016/0895-4356(89)90086-3
- Hirota M, Takada T, Kawarada Y, et al. Diagnostic criteria and severity assessment of acute cholecystitis: Tokyo guidelines. J Hepatobiliary Pancreat Surg 2007; 14(1):78–82. doi:10.1007/s00534-006-1159-4
- Miura F, Takada T, Kawarada Y, et al. Flowcharts for the diagnosis and treatment of acute cholangitis and cholecystitis: Tokyo guidelines. J Hepatobiliary Pancreat Surg 2007; 14(1):27–34. doi:10.1007/s00534-006-1153-x
- Koo KP, Traverso LW. Do preoperative indicators predict the presence of common bile duct stones during laparoscopic cholecystectomy? Am J Surg 1996; 171(5):495–499. doi:10.1016/S0002-9610(97)89611-0
- Collins C, Maguire D, Ireland A, Fitzgerald E, O’Sullivan GC. A prospective study of common bile duct calculi in patients undergoing laparoscopic cholecystectomy: natural history of choledocholithiasis revisited. Ann Surg 2004; 239(1):28–33. doi:10.1097/01.sla.0000103069.00170.9c
- Costi R, Gnocchi A, Di Mario F, Sarli L. Diagnosis and management of choledocholithiasis in the golden age of imaging, endoscopy and laparoscopy. World J Gastroenterol 2014; 20(37):13382–13401. doi:10.3748/wjg.v20.i37.13382
- European Association for the Study of the Liver (EASL). EASL Clinical Practice Guidelines on the prevention, diagnosis and treatment of gallstones. J Hepatol 2016; 65(1):146–181. doi:10.1016/j.jhep.2016.03.005
- Greenberg JA, Hsu J, Bawazeer M, et al. Clinical practice guideline: management of acute pancreatitis. Can J Surg 2016; 59 (2):128–140. doi:10.1503/cjs.015015
- Tenner S, Baillie J, DeWitt J, Vege SS; American College of Gastroenterology. American College of Gastroenterology guideline: management of acute pancreatitis. Am J Gastroenterol 2013; 108(9):1400–1416. doi:10.1038/ajg.2013.218
- Moolla Z, Anderson F, Thomson SR. Use of amylase and alanine transaminase to predict acute gallstone pancreatitis in a population with high HIV prevalence. World J Surg 2013; 37(1):156–161. doi:10.1007/s00268-012-1801-z
- Shea JA, Berlin JA, Escarce JJ, et al. Revised estimates of diagnostic test sensitivity and specificity in suspected biliary tract disease. Arch Intern Med 1994; 154(22):2573–2581. doi:10.1001/archinte.1994.00420220069008
- Kiewiet JJ, Leeuwenburgh MM, Bipat S, et al. A systematic review and meta-analysis of diagnostic performance of imaging in acute cholecystitis. Radiology 2012; 264(3):708–720. doi:10.1148/radiol.12111561
- ASGE Standards of Practice Committee; Maple JT, Ben-Menachem T, Anderson MA, et al. The role of endoscopy in the evaluation of suspected choledocholithiasis. Gastrointest Endosc 2010; 71(1):1–9. doi:10.1016/j.gie.2009.09.041
- Bachar GN, Cohen M, Belenky A, Atar E, Gideon S. Effect of aging on the adult extrahepatic bile duct: a sonographic study. J Ultrasound Med 2003; 22(9):879–885. doi:10.7863/jum.2003.22.9.879
- El-Hayek K, Timratana P, Meranda J, Shimizu H, Eldar S, Chand B. Post Roux-en-Y gastric bypass biliary dilation: natural process or significant entity? J Gastrointest Surg 2012; 16(12):2185–2189. doi:10.1007/s11605-012-2058-4
- Park SM, Kim WS, Bae IH, et al. Common bile duct dilatation after cholecystectomy: a one-year prospective study. J Korean Surg Soc 2012; 83(2):97–101. doi:10.4174/jkss.2012.83.2.97
- Tse F, Liu L, Barkun AN, Armstrong D, Moayyedi P. EUS: a meta-analysis of test performance in suspected choledocholithiasis. Gastrointest Endosc 2008; 67(2):235–244. doi:10.1016/j.gie.2007.09.047
- Verma D, Kapadia A, Eisen GM, Adler DG. EUS vs MRCP for detection of choledocholithiasis. Gastrointest Endosc 2006; 64(2):248–254. doi:10.1016/j.gie.2005.12.038
- Tseng LJ, Jao YT, Mo LR, Lin RC. Over-the-wire US catheter probe as an adjunct to ERCP in the detection of choledocholithiasis. Gastrointest Endosc 2001; 54(6):720–723. doi:10.1067/mge.2001.119255
- Kondo S, Isayama H, Akahane M, et al. Detection of common bile duct stones: comparison between endoscopic ultrasonography, magnetic resonance cholangiography, and helical-computed-tomographic cholangiography. Eur J Radiol 2005; 54(2):271–275. doi:10.1016/j.ejrad.2004.07.007
- Attili AF, De Santis A, Capri R, Repice AM, Maselli S. The natural history of gallstones: the GREPCO experience. The GREPCO Group. Hepatology 1995; 21(3):656–660. doi:10.1016/0270-9139(95)90514-6
- Sakorafas GH, Milingos D, Peros G. Asymptomatic cholelithiasis: is cholecystectomy really needed? A critical reappraisal 15 years after the introduction of laparoscopic cholecystectomy. Dig Dis Sci 2007; 52(5):1313–1325. doi:10.1007/s10620-006-9107-3
- Gracie WA, Ransohoff DF. The natural history of silent gallstones: the innocent gallstone is not a myth. N Engl J Med 1982; 307(13):798–800. doi:10.1056/NEJM198209233071305
- McSherry CK, Ferstenberg H, Calhoun WF, Lahman E, Virshup M. The natural history of diagnosed gallstone disease in symptomatic and asymptomatic patients. Ann Surg 1985; 202(1):59–63. doi:10.1097/00000658-198507000-00009
- Wada K, Wada K, Imamura T. Natural course of asymptomatic gallstone disease. Nihon Rinsho 1993; 51(7):1737–1743. Japanese.
- Halldestam I, Enell EL, Kullman E, Borch K. Development of symptoms and complications in individuals with asymptomatic gallstones. Br J Surg 2004; 91(6):734–738. doi:10.1002/bjs.4547
- Festi D, Reggiani ML, Attili AF, et al. Natural history of gallstone disease: expectant management or active treatment? Results from a population-based cohort study. J Gastroenterol Hepatol 2010; 25(4):719–724. doi:10.1111/j.1440-1746.2009.06146.x
- Shabanzadeh DM, Sorensen LT, Jorgensen T. A prediction rule for risk stratification of incidentally discovered gallstones: results from a large cohort study. Gastroenterology 2016; 150(1):156–167e1. doi:10.1053/j.gastro.2015.09.002
- Overby DW, Apelgren KN, Richardson W, Fanelli R; Society of American Gastrointestinal and Endoscopic Surgeons. SAGES guidelines for the clinical application of laparoscopic biliary tract surgery. Surg Endosc 2010; 24(10):2368–2386. doi:10.1007/s00464-010-1268-7
- Abraham S, Rivero HG, Erlikh IV, Griffith LF, Kondamudi VK. Surgical and nonsurgical management of gallstones. Am Fam Physician 2014; 89(10):795–802.
- Currò G,, Iapichino G, Lorenzini C, Palmeri R, Cucinotta E. Laparoscopic cholecystectomy in children with chronic hemolytic anemia. Is the outcome related to the timing of the procedure? Surg Endosc 2006; 20(2):252–255. doi:10.1007/s00464-005-0318-z
- Hundal R, Shaffer EA. Gallbladder cancer: epidemiology and outcome. Clin Epidemiol 2014; 6:99–109. doi:10.2147/CLEP.S37357
- Chen GL, Akmal Y, DiFronzo AL, Vuong B, O’Connor V. Porcelain gallbladder: no longer an indication for prophylactic cholecystectomy. Am Surg 2015; 81(10):936–940.
- Schnelldorfer T. Porcelain gallbladder: a benign process or concern for malignancy? J Gastrointest Surg 2013; 17(6):1161–1168. doi:10.1007/s11605-013-2170-0
- Warschkow R, Tarantino I, Ukegjini K, et al. Concomitant cholecystectomy during laparoscopic Roux-en-Y gastric bypass in obese patients is not justified: a meta-analysis. Obes Surg 2013; 23(3)3979–408. doi:10.1007/s11695-012-0852-4
- Gurusamy K, Samraj K, Gluud C, Wilson E, Davidson BR. Meta-analysis of randomized controlled trials on the safety and effectiveness of early versus delayed laparoscopic cholecystectomy for acute cholecystitis. Br J Surg 2010; 97(2):141–150. doi:10.1002/bjs.6870
- Papi C, Catarci M, D’Ambrosio L, et al. Timing of cholecystectomy for acute calculous cholecystitis: a meta-analysis. Am J Gastroenterol 2004; 99(1):147–155. doi:10.1046/j.1572-0241.2003.04002.x
- Gurusamy KS, Davidson C, Gluud C, Davidson BR. Early versus delayed laparoscopic cholecystectomy for people with acute cholecystitis. Cochrane Database Syst Rev 2013; 6:CD005440. doi:10.1002/14651858
- Menahem B, Mulliri A, Fohlen A, Guittet L, Alves A, Lubrano J. Delayed laparoscopic cholecystectomy increases the total hospital stay compared to an early laparoscopic cholecystectomy after acute cholecystitis: an updated meta-analysis of randomized controlled trials. HPB (Oxford) 2015; 17(10):857–862. doi:10.1111/hpb.12449
- Uhl W, Müller CA, Krähenbühl L, Schmid SW, Schölzel S, Büchler MW. Acute gallstone pancreatitis: timing of laparoscopic cholecystectomy in mild and severe disease. Surg Endosc 1999; 13(11):1070–1076. doi:10.1007/s004649901175
- Aboulian A, Chan T, Yaghoubian A, et al. Early cholecystectomy safely decreases hospital stay in patients with mild gallstone pancreatitis: a randomized prospective study. Ann Surg 2010(4): 251:615–619. doi:10.1097/SLA.0b013e3181c38f1f
The prevalence of gallstones is approximately 10% to 15% of the adult US population.1,2 Most cases are asymptomatic, as gallstones are usually discovered incidentally during routine imaging for other abdominal conditions, and only about 20% of patients with asymptomatic gallstones develop clinically significant complications.2,3
Nevertheless, gallstones carry significant healthcare costs. In 2004, the median inpatient cost for any gallstone-related disease was $11,584, with an overall annual cost of $6.2 billion.4,5
Laparoscopic cholecystectomy is the standard treatment for symptomatic cholelithiasis. For asymptomatic cholelithasis, the usual approach is expectant management (“watch and wait”), but prophylactic cholecystectomy may be an option in certain patients at high risk.
CHEMICAL COMPOSITION
Gallstones can be classified into 2 main categories based on their predominant chemical composition: cholesterol or pigment.
Cholesterol gallstones
About 75% of gallstones are composed of cholesterol.3,4 In the past, this type of stone was thought to be caused by gallbladder inflammation, bile stasis, and absorption of bile salts from damaged mucosa. However, it is now known that cholesterol gallstones are the result of biliary supersaturation caused by cholesterol hypersecretion into the gallbladder, gallbladder hypomotility, accelerated cholesterol nucleation and crystallization, and mucin gel accumulation.
Pigment gallstones
Black pigment gallstones account for 10% to 15% of all gallstones.6 They are caused by chronic hemolysis in association with supersaturation of bile with calcium hydrogen bilirubinate, along with deposition of calcium carbonate, phosphate, and inorganic salts.7
Brown pigment stones, accounting for 5% to 10% of all gallstones,6 are caused by infection in the obstructed bile ducts, where bacteria that produce beta-glucuronidase, phospholipase, and slime contribute to formation of the stone.8,9
RISK FACTORS FOR GALLSTONES
Age. After age 40, the risk increases dramatically, with an incidence 4 times higher for those ages 40 to 69 than in younger people.10
Female sex. Women of reproductive age are 4 times more likely to develop gallstones than men, but this gap narrows after menopause.11 The higher risk is attributed to female sex hormones, pregnancy, and oral contraceptive use. Estrogen decreases secretion of bile salts and increases secretion of cholesterol into the gallbladder, which leads to cholesterol supersaturation. Progesterone acts synergistically by causing hypomobility of the gallbladder, which in turn leads to bile stasis.12,13
Ethnicity. The risk is higher in Mexican Americans and Native Americans than in other ethnic groups.14
Rapid weight loss, such as after bariatric surgery, occurs from decreased caloric intake and promotes bile stasis, while lipolysis increases cholesterol mobilization and secretion into the gallbladder. This creates an environment conducive to bile supersaturation with cholesterol, leading to gallstone formation.
Chronic hemolytic disorders carry an increased risk of developing calcium bilirubinate stones due to increased excretion of bilirubin during hemolysis.
Obesity and diabetes mellitus are both attributed to insulin resistance. Obesity also increases bile stasis and cholesterol saturation.
CLINICAL PRESENTATION OF GALLSTONES (CHOLELITHIASIS)
Most patients with gallstones (cholelithiasis) experience no symptoms. Their gallstones are often discovered incidentally during imaging tests for unrelated or unexplained abdominal symptoms. Most patients with asymptomatic gallstones remain symptom-free, while about 20% develop gallstone-related symptoms.2,3
Abdominal pain is the most common symptom. The phrase biliary colic—suggesting pain that is fluctuating in nature—appears ubiquitously in the medical literature, but it does not correctly characterize the pain associated with gallstones.
Most patients with gallstone symptoms describe a constant and often severe pain in the right upper abdomen, epigastrium, or both, often persisting for 30 to 120 minutes. Symptoms are frequently reported in the epigastrium when only visceral pain fibers are stimulated due to gallbladder distention. This is usually called midline pain; however, pain occurs in the back and right shoulder in up to 60% of patients, with involvement of somatic fibers.15,16 Gallstone pain is not relieved by change of position or passage of stool or gas.
Onset of symptoms more than an hour after eating or in the late evening or at night also very strongly suggests biliary pain. Patients with a history of biliary pain are more likely to experience it again, with a 69% chance of developing recurrent pain within 2 years.17
GALLSTONE-RELATED COMPLICATIONS
Acute gallbladder inflammation (cholecystitis)
Gallbladder inflammation (cholecystitis) is the most common complication, occurring in up to 10% of symptomatic cases. Many patients with acute cholecystitis present with right upper quadrant pain that may be accompanied by anorexia, nausea, or vomiting. Inspiratory arrest on deep palpation of the right upper quadrant (Murphy sign) has a specificity of 79% to 96% for acute cholecystitis.20 Markers of systemic inflammation such as fever, elevated white blood cell count, and elevated C-reactive protein are highly suggestive of acute cholecystitis.20,21
Bile duct stones (choledocholithiasis)
Bile duct stones (choledocholithiasis) are detected in 3.4% to 12% of patients with gallstones.22,23 Most stones in the common bile duct migrate there from the gallbladder via the cystic duct. Less commonly, primary duct stones form in the duct due to biliary stasis. Removing the gallbladder does not completely eliminate the risk of bile duct stones, as stones can remain or recur after surgery.
Bile duct stones can obstruct the common bile duct, which disrupts normal bile flow and leads to jaundice. Other symptoms may include pruritus, right upper quadrant pain, nausea, and vomiting. Serum levels of bilirubin, aspartate aminotransferase, alanine aminotransferase (ALT), and alkaline phosphatase are usually high.24
Acute bacterial infection (cholangitis)
Acute bacterial infection of the biliary system (cholangitis) is usually associated with obstruction of the common bile duct. Common symptoms of acute cholangitis include right upper quadrant pain, fever, and jaundice (Charcot triad), and these are present in about 50% to 75% of cases.21 In severe cases, patients can develop altered mental status and septicemic shock in addition to the Charcot triad, a condition called the Reynold pentad. White blood cell counts and serum levels of C-reactive protein, bilirubin, aminotransferases, and alkaline phosphatase are usually elevated.21
Pancreatitis
Approximately 4% to 8% of patients with gallstones develop inflammation of the pancreas (pancreatitis).25 The diagnosis of acute pancreatitis requires at least 2 of the following:26,27
- Abdominal pain (typically epigastric, often radiating to the back)
- Amylase or lipase levels at least 3 times above the normal limit
- Imaging findings that suggest acute pancreatitis.
Gallstone-related pancreatitis should be considered if the ALT level is greater than 150 U/mL, which has a 97% specificity for gallstone-related pancreatitis.28
ABDOMINAL ULTRASONOGRAPHY FOR DIAGNOSIS
Transabdominal ultrasonography, with a sensitivity of 84% to 89% and a specificity of up to 99%, is the test of choice for detecting gallstones.29 The characteristic findings of acute cholecystitis on ultrasonography include enlargement of the gallbladder, thickening of the gallbladder wall, presence of pericholecystic fluid, and tenderness elicited by the ultrasound probe over the gallbladder (sonographic Murphy sign).
Scintigraphy as a second test
Acute cholecystitis is primarily a clinical diagnosis and typically does not require additional imaging beyond ultrasonography. When there is discordance between clinical and ultrasonographic findings, the most accurate second imaging test is scintigraphy of the biliary tract, usually performed with technetium-labeled hydroxy iminodiacetic acid. Given intravenously, the radionuclide is rapidly taken up by the liver and then secreted into the bile. In acute cholecystitis, the cystic duct is functionally occluded and the isotope does not enter the gallbladder, creating an imaging void compared with a normal appearance.
Scintigraphy is more sensitive than abdominal ultrasonography, with a sensitivity of up to 97% vs 81% to 88%, respectively.29,30 The tests have about equal specificity.
Even though scintigraphy is more sensitive, abdominal ultrasonography is often the initial test for patients with suspected acute cholecystitis because it is more widely available, takes less time, does not involve radiation exposure, and can assess for the presence or absence of gallstones and dilation of the intra- and extrahepatic bile ducts.
Looking for stones in the common bile duct
When acute cholangitis due to choledocholithiasis is suspected, abdominal ultrasonography is a prudent initial test to look for gallstones or biliary dilation suggesting obstruction by stones in the common bile duct. Abdominal ultrasonography has only a 22% to 55% sensitivity for visualizing stones in the common bile duct, but it has a 77% to 87% sensitivity for detecting common bile duct dilation, a surrogate marker of stones.31
The normal bile duct diameter ranges from 3 to 6 mm, although mild dilation is often seen in older patients or after cholecystectomy or Roux-en-Y gastric bypass surgery.32,33 Bile duct dilation of up to 10 mm can be considered normal in patients after cholecystectomy.34 A normal-appearing bile duct on ultrasonography has a negative predictive value of 95% for excluding common bile duct stones.31
Endoscopic ultrasonography (EUS), magnetic resonance cholangiopancreatography (MRCP), and endoscopic retrograde cholangiopancreatography (ERCP) have similar sensitivity (89%–94%, 85%–92%, and 89%–93%, respectively) and specificity (94%–95%, 93%–97%, and 100%, respectively) for detecting common bile duct stones.35–37 EUS is superior to MRCP in detecting stones smaller than 6 mm.38
ERCP should be reserved for managing rather than diagnosing common bile duct stones because of the risk of pancreatitis and perforation. Patients undergoing cholecystectomy who are suspected of having choledocholithiasis may undergo intraoperative cholangiography or laparoscopic common bile duct ultrasonography.
WATCH AND WAIT, OR INTERVENE?
Asymptomatic gallstones
Standard treatment for these patients is expectant management. Cholecystectomy is not recommended for patients with asymptomatic gallstones.47 Nevertheless, some patients may benefit from prophylactic cholecystectomy. We and others48 suggest considering cholecystectomy in the following patients.
Patients with chronic hemolytic anemia (including children with sickle cell anemia and spherocytosis). These patients have a higher risk of developing calcium bilirubinate stones, and cholecystectomy has improved outcomes.49 It should be noted that most of these data come from pediatric populations and have been extrapolated to adults.
Native Americans, who have a higher risk of gallbladder cancer if they have gallstones.2,50
Conversely, calcification of the gallbladder wall (“porcelain gallbladder”) is no longer considered an absolute indication for cholecystectomy. This condition was thought to be associated with a high rate of gallbladder carcinoma, but analyses of larger, more recent data sets found much smaller risks.51,52 Further, cholecystectomy in these patients was found to be associated with high rates of postoperative complications. Thus, prophylactic cholecystectomy is no longer recommended in asymptomatic cases of porcelain gallbladder.
In addition, concomitant cholecystectomy in patients undergoing bariatric surgery is no longer considered the therapeutic standard. Historically, cholecystectomy was performed in these patients because of the increased risk of gallstones associated with rapid weight loss after surgery. However, research now weighs against concomitant cholecystectomy with bariatric surgery and most other abdominal surgeries for asymptomatic gallstones.53
Laparoscopic surgery for symptomatic gallstones
For patients experiencing acute cholecystitis, laparoscopic cholecystectomy within 72 hours is recommended.48 There were safety concerns regarding higher rates of morbidity and conversion from laparoscopic to open cholecystectomy in patients who underwent surgery before the acute cholecystitis episode had settled. However, a large meta-analysis found no significant difference between early and delayed laparoscopic cholecystectomy in bile duct injury or conversion rates.54 Further, early cholecystectomy—defined as within 1 week of symptom onset—has been found to reduce gallstone-related complications, shorten hospital stays, and lower costs.55–57 If the patient cannot undergo surgery, percutaneous cholecystotomy or novel endoscopic gallbladder drainage interventions can be used.
Several variables predict the presence of bile duct stones in patients who have symptoms (Table 4). Based on these predictors, the ASGE classifies the probabilities as low (< 10%), intermediate (10% to 50%), and high (> 50%)31:
- High-risk patients should undergo preoperative ERCP and stone extraction if needed
- Intermediate-risk patients should undergo preoperative imaging with EUS or MRCP or intraoperative bile duct evaluation, depending on the availability, costs, and local expertise.
Patients with associated cholangitis should be given intravenous fluids and broad-spectrum antibiotics. Biliary decompression should be done as early as possible to decrease the risk of morbidity and mortality. For acute cholangitis, ERCP is the treatment of choice.25
Patients with acute gallstone pancreatitis should receive conservative management with intravenous isotonic solutions and pain control, followed by laparoscopic cholecystectomy.48
The timing of laparoscopic cholecystectomy in acute gallstone pancreatitis has been debated. Studies conducted during the era of open cholecystectomy reported similar or worse outcomes if cholecystectomy was done sooner rather than later.
However, in 1999, Uhl et al58 reported that 48 of 77 patients admitted with acute gallstone pancreatitis were able to undergo laparoscopic cholecystectomy during the same admission. Success rates were 85% (30 of 35 patients) in those with mild disease and 62% (8 of 13 patients) in those with severe disease. They concluded laparoscopic cholecystectomy could be safely performed within 7 days in patients with mild disease, whereas in severe disease at least 3 weeks should elapse because of the risk of infection.
In a randomized trial published in 2010, Aboulian et al59 reported that hospital length of stay (the primary end point) was shorter in 25 patients who underwent laparoscopic cholecystectomy early (within 48 hours of admission) than in 25 patients who underwent surgery after abdominal pain had resolved and laboratory enzymes showed a normalizing trend, 3.5 vs 5.8 days (P = .0016). Rates of perioperative complications and need for conversion to open surgery were similar between the 2 groups.
If there is associated cholangitis, patients should also be given broad-spectrum antibiotics and should undergo ERCP within 24 hours of admission.25–27
SUMMARY
Gallstones are common in US adults. Abdominal ultrasonography is the diagnostic imaging test of choice to detect gallbladder stones and assess for findings suggestive of acute cholecystitis and dilation of the common bile duct. Fortunately, most gallstones are asymptomatic and can usually be managed expectantly. In patients who have symptoms or have gallstone complications, laparoscopic cholecystectomy is the standard of care.
The prevalence of gallstones is approximately 10% to 15% of the adult US population.1,2 Most cases are asymptomatic, as gallstones are usually discovered incidentally during routine imaging for other abdominal conditions, and only about 20% of patients with asymptomatic gallstones develop clinically significant complications.2,3
Nevertheless, gallstones carry significant healthcare costs. In 2004, the median inpatient cost for any gallstone-related disease was $11,584, with an overall annual cost of $6.2 billion.4,5
Laparoscopic cholecystectomy is the standard treatment for symptomatic cholelithiasis. For asymptomatic cholelithasis, the usual approach is expectant management (“watch and wait”), but prophylactic cholecystectomy may be an option in certain patients at high risk.
CHEMICAL COMPOSITION
Gallstones can be classified into 2 main categories based on their predominant chemical composition: cholesterol or pigment.
Cholesterol gallstones
About 75% of gallstones are composed of cholesterol.3,4 In the past, this type of stone was thought to be caused by gallbladder inflammation, bile stasis, and absorption of bile salts from damaged mucosa. However, it is now known that cholesterol gallstones are the result of biliary supersaturation caused by cholesterol hypersecretion into the gallbladder, gallbladder hypomotility, accelerated cholesterol nucleation and crystallization, and mucin gel accumulation.
Pigment gallstones
Black pigment gallstones account for 10% to 15% of all gallstones.6 They are caused by chronic hemolysis in association with supersaturation of bile with calcium hydrogen bilirubinate, along with deposition of calcium carbonate, phosphate, and inorganic salts.7
Brown pigment stones, accounting for 5% to 10% of all gallstones,6 are caused by infection in the obstructed bile ducts, where bacteria that produce beta-glucuronidase, phospholipase, and slime contribute to formation of the stone.8,9
RISK FACTORS FOR GALLSTONES
Age. After age 40, the risk increases dramatically, with an incidence 4 times higher for those ages 40 to 69 than in younger people.10
Female sex. Women of reproductive age are 4 times more likely to develop gallstones than men, but this gap narrows after menopause.11 The higher risk is attributed to female sex hormones, pregnancy, and oral contraceptive use. Estrogen decreases secretion of bile salts and increases secretion of cholesterol into the gallbladder, which leads to cholesterol supersaturation. Progesterone acts synergistically by causing hypomobility of the gallbladder, which in turn leads to bile stasis.12,13
Ethnicity. The risk is higher in Mexican Americans and Native Americans than in other ethnic groups.14
Rapid weight loss, such as after bariatric surgery, occurs from decreased caloric intake and promotes bile stasis, while lipolysis increases cholesterol mobilization and secretion into the gallbladder. This creates an environment conducive to bile supersaturation with cholesterol, leading to gallstone formation.
Chronic hemolytic disorders carry an increased risk of developing calcium bilirubinate stones due to increased excretion of bilirubin during hemolysis.
Obesity and diabetes mellitus are both attributed to insulin resistance. Obesity also increases bile stasis and cholesterol saturation.
CLINICAL PRESENTATION OF GALLSTONES (CHOLELITHIASIS)
Most patients with gallstones (cholelithiasis) experience no symptoms. Their gallstones are often discovered incidentally during imaging tests for unrelated or unexplained abdominal symptoms. Most patients with asymptomatic gallstones remain symptom-free, while about 20% develop gallstone-related symptoms.2,3
Abdominal pain is the most common symptom. The phrase biliary colic—suggesting pain that is fluctuating in nature—appears ubiquitously in the medical literature, but it does not correctly characterize the pain associated with gallstones.
Most patients with gallstone symptoms describe a constant and often severe pain in the right upper abdomen, epigastrium, or both, often persisting for 30 to 120 minutes. Symptoms are frequently reported in the epigastrium when only visceral pain fibers are stimulated due to gallbladder distention. This is usually called midline pain; however, pain occurs in the back and right shoulder in up to 60% of patients, with involvement of somatic fibers.15,16 Gallstone pain is not relieved by change of position or passage of stool or gas.
Onset of symptoms more than an hour after eating or in the late evening or at night also very strongly suggests biliary pain. Patients with a history of biliary pain are more likely to experience it again, with a 69% chance of developing recurrent pain within 2 years.17
GALLSTONE-RELATED COMPLICATIONS
Acute gallbladder inflammation (cholecystitis)
Gallbladder inflammation (cholecystitis) is the most common complication, occurring in up to 10% of symptomatic cases. Many patients with acute cholecystitis present with right upper quadrant pain that may be accompanied by anorexia, nausea, or vomiting. Inspiratory arrest on deep palpation of the right upper quadrant (Murphy sign) has a specificity of 79% to 96% for acute cholecystitis.20 Markers of systemic inflammation such as fever, elevated white blood cell count, and elevated C-reactive protein are highly suggestive of acute cholecystitis.20,21
Bile duct stones (choledocholithiasis)
Bile duct stones (choledocholithiasis) are detected in 3.4% to 12% of patients with gallstones.22,23 Most stones in the common bile duct migrate there from the gallbladder via the cystic duct. Less commonly, primary duct stones form in the duct due to biliary stasis. Removing the gallbladder does not completely eliminate the risk of bile duct stones, as stones can remain or recur after surgery.
Bile duct stones can obstruct the common bile duct, which disrupts normal bile flow and leads to jaundice. Other symptoms may include pruritus, right upper quadrant pain, nausea, and vomiting. Serum levels of bilirubin, aspartate aminotransferase, alanine aminotransferase (ALT), and alkaline phosphatase are usually high.24
Acute bacterial infection (cholangitis)
Acute bacterial infection of the biliary system (cholangitis) is usually associated with obstruction of the common bile duct. Common symptoms of acute cholangitis include right upper quadrant pain, fever, and jaundice (Charcot triad), and these are present in about 50% to 75% of cases.21 In severe cases, patients can develop altered mental status and septicemic shock in addition to the Charcot triad, a condition called the Reynold pentad. White blood cell counts and serum levels of C-reactive protein, bilirubin, aminotransferases, and alkaline phosphatase are usually elevated.21
Pancreatitis
Approximately 4% to 8% of patients with gallstones develop inflammation of the pancreas (pancreatitis).25 The diagnosis of acute pancreatitis requires at least 2 of the following:26,27
- Abdominal pain (typically epigastric, often radiating to the back)
- Amylase or lipase levels at least 3 times above the normal limit
- Imaging findings that suggest acute pancreatitis.
Gallstone-related pancreatitis should be considered if the ALT level is greater than 150 U/mL, which has a 97% specificity for gallstone-related pancreatitis.28
ABDOMINAL ULTRASONOGRAPHY FOR DIAGNOSIS
Transabdominal ultrasonography, with a sensitivity of 84% to 89% and a specificity of up to 99%, is the test of choice for detecting gallstones.29 The characteristic findings of acute cholecystitis on ultrasonography include enlargement of the gallbladder, thickening of the gallbladder wall, presence of pericholecystic fluid, and tenderness elicited by the ultrasound probe over the gallbladder (sonographic Murphy sign).
Scintigraphy as a second test
Acute cholecystitis is primarily a clinical diagnosis and typically does not require additional imaging beyond ultrasonography. When there is discordance between clinical and ultrasonographic findings, the most accurate second imaging test is scintigraphy of the biliary tract, usually performed with technetium-labeled hydroxy iminodiacetic acid. Given intravenously, the radionuclide is rapidly taken up by the liver and then secreted into the bile. In acute cholecystitis, the cystic duct is functionally occluded and the isotope does not enter the gallbladder, creating an imaging void compared with a normal appearance.
Scintigraphy is more sensitive than abdominal ultrasonography, with a sensitivity of up to 97% vs 81% to 88%, respectively.29,30 The tests have about equal specificity.
Even though scintigraphy is more sensitive, abdominal ultrasonography is often the initial test for patients with suspected acute cholecystitis because it is more widely available, takes less time, does not involve radiation exposure, and can assess for the presence or absence of gallstones and dilation of the intra- and extrahepatic bile ducts.
Looking for stones in the common bile duct
When acute cholangitis due to choledocholithiasis is suspected, abdominal ultrasonography is a prudent initial test to look for gallstones or biliary dilation suggesting obstruction by stones in the common bile duct. Abdominal ultrasonography has only a 22% to 55% sensitivity for visualizing stones in the common bile duct, but it has a 77% to 87% sensitivity for detecting common bile duct dilation, a surrogate marker of stones.31
The normal bile duct diameter ranges from 3 to 6 mm, although mild dilation is often seen in older patients or after cholecystectomy or Roux-en-Y gastric bypass surgery.32,33 Bile duct dilation of up to 10 mm can be considered normal in patients after cholecystectomy.34 A normal-appearing bile duct on ultrasonography has a negative predictive value of 95% for excluding common bile duct stones.31
Endoscopic ultrasonography (EUS), magnetic resonance cholangiopancreatography (MRCP), and endoscopic retrograde cholangiopancreatography (ERCP) have similar sensitivity (89%–94%, 85%–92%, and 89%–93%, respectively) and specificity (94%–95%, 93%–97%, and 100%, respectively) for detecting common bile duct stones.35–37 EUS is superior to MRCP in detecting stones smaller than 6 mm.38
ERCP should be reserved for managing rather than diagnosing common bile duct stones because of the risk of pancreatitis and perforation. Patients undergoing cholecystectomy who are suspected of having choledocholithiasis may undergo intraoperative cholangiography or laparoscopic common bile duct ultrasonography.
WATCH AND WAIT, OR INTERVENE?
Asymptomatic gallstones
Standard treatment for these patients is expectant management. Cholecystectomy is not recommended for patients with asymptomatic gallstones.47 Nevertheless, some patients may benefit from prophylactic cholecystectomy. We and others48 suggest considering cholecystectomy in the following patients.
Patients with chronic hemolytic anemia (including children with sickle cell anemia and spherocytosis). These patients have a higher risk of developing calcium bilirubinate stones, and cholecystectomy has improved outcomes.49 It should be noted that most of these data come from pediatric populations and have been extrapolated to adults.
Native Americans, who have a higher risk of gallbladder cancer if they have gallstones.2,50
Conversely, calcification of the gallbladder wall (“porcelain gallbladder”) is no longer considered an absolute indication for cholecystectomy. This condition was thought to be associated with a high rate of gallbladder carcinoma, but analyses of larger, more recent data sets found much smaller risks.51,52 Further, cholecystectomy in these patients was found to be associated with high rates of postoperative complications. Thus, prophylactic cholecystectomy is no longer recommended in asymptomatic cases of porcelain gallbladder.
In addition, concomitant cholecystectomy in patients undergoing bariatric surgery is no longer considered the therapeutic standard. Historically, cholecystectomy was performed in these patients because of the increased risk of gallstones associated with rapid weight loss after surgery. However, research now weighs against concomitant cholecystectomy with bariatric surgery and most other abdominal surgeries for asymptomatic gallstones.53
Laparoscopic surgery for symptomatic gallstones
For patients experiencing acute cholecystitis, laparoscopic cholecystectomy within 72 hours is recommended.48 There were safety concerns regarding higher rates of morbidity and conversion from laparoscopic to open cholecystectomy in patients who underwent surgery before the acute cholecystitis episode had settled. However, a large meta-analysis found no significant difference between early and delayed laparoscopic cholecystectomy in bile duct injury or conversion rates.54 Further, early cholecystectomy—defined as within 1 week of symptom onset—has been found to reduce gallstone-related complications, shorten hospital stays, and lower costs.55–57 If the patient cannot undergo surgery, percutaneous cholecystotomy or novel endoscopic gallbladder drainage interventions can be used.
Several variables predict the presence of bile duct stones in patients who have symptoms (Table 4). Based on these predictors, the ASGE classifies the probabilities as low (< 10%), intermediate (10% to 50%), and high (> 50%)31:
- High-risk patients should undergo preoperative ERCP and stone extraction if needed
- Intermediate-risk patients should undergo preoperative imaging with EUS or MRCP or intraoperative bile duct evaluation, depending on the availability, costs, and local expertise.
Patients with associated cholangitis should be given intravenous fluids and broad-spectrum antibiotics. Biliary decompression should be done as early as possible to decrease the risk of morbidity and mortality. For acute cholangitis, ERCP is the treatment of choice.25
Patients with acute gallstone pancreatitis should receive conservative management with intravenous isotonic solutions and pain control, followed by laparoscopic cholecystectomy.48
The timing of laparoscopic cholecystectomy in acute gallstone pancreatitis has been debated. Studies conducted during the era of open cholecystectomy reported similar or worse outcomes if cholecystectomy was done sooner rather than later.
However, in 1999, Uhl et al58 reported that 48 of 77 patients admitted with acute gallstone pancreatitis were able to undergo laparoscopic cholecystectomy during the same admission. Success rates were 85% (30 of 35 patients) in those with mild disease and 62% (8 of 13 patients) in those with severe disease. They concluded laparoscopic cholecystectomy could be safely performed within 7 days in patients with mild disease, whereas in severe disease at least 3 weeks should elapse because of the risk of infection.
In a randomized trial published in 2010, Aboulian et al59 reported that hospital length of stay (the primary end point) was shorter in 25 patients who underwent laparoscopic cholecystectomy early (within 48 hours of admission) than in 25 patients who underwent surgery after abdominal pain had resolved and laboratory enzymes showed a normalizing trend, 3.5 vs 5.8 days (P = .0016). Rates of perioperative complications and need for conversion to open surgery were similar between the 2 groups.
If there is associated cholangitis, patients should also be given broad-spectrum antibiotics and should undergo ERCP within 24 hours of admission.25–27
SUMMARY
Gallstones are common in US adults. Abdominal ultrasonography is the diagnostic imaging test of choice to detect gallbladder stones and assess for findings suggestive of acute cholecystitis and dilation of the common bile duct. Fortunately, most gallstones are asymptomatic and can usually be managed expectantly. In patients who have symptoms or have gallstone complications, laparoscopic cholecystectomy is the standard of care.
- Schirmer BD, Winters KL, Edlich RF. Cholelithiasis and cholecystitis. J Long Term Eff Med Implants 2005; 15(3):329–338. doi:10.1615/JLongTermEffMedImplants.v15.i3.90
- Stinton LM, Shaffer EA. Epidemiology of gallbladder disease: cholelithiasis and cancer. Gut Liver 2012; 6(2):172–187. doi:10.5009/gnl.2012.6.2.172
- Lee JY, Keane MG, Pereira S. Diagnosis and treatment of gallstone disease. Practitioner 2015; 259(1783):15–19.
- Russo MW, Wei JT, Thiny MT, et al. Digestive and liver diseases statistics, 2004. Gastroenterology 2004; 126(5):1448–1453. doi:10.1053/j.gastro.2004.01.025
- Everhart JE, Ruhl CE. Burden of digestive diseases in the United States part I: overall and upper gastrointestinal diseases. Gastroenterology 2009; 136(2):376–386. doi:10.1053/j.gastro.2008.12.015
- Cariati A. Gallstone classification in Western countries. Indian J Surg 2015; 77(suppl 2):376–380. doi.org/10.1007/s12262-013-0847-y
- Carey MC. Pathogenesis of gallstones. Am J Surg 1993; 165(4):410–419. doi:10.1016/S0002-9610(05)80932-8
- Lammert F, Gurusamy K, Ko CW, et al. Gallstones. Nat Rev Dis Primers 2016; 2:16024. doi:10.1038/nrdp.2016.24
- Stewart L, Oesterle AL, Erdan I, Griffiss JM, Way LW. Pathogenesis of pigment gallstones in Western societies: the central role of bacteria. J Gastrointest Surg 2002; 6(6):891–904.
- Barbara L, Sama C, Morselli Labate AM, et al. A population study on the prevalence of gallstone disease: the Sirmione Study. Hepatology 1987; 7(5):913–917. doi:10.1002/hep.1840070520
- Sood S, Winn T, Ibrahim S, et al. Natural history of asymptomatic gallstones: differential behaviour in male and female subjects. Med J Malaysia 2015; 70(6):341–345.
- Maringhini A, Ciambra M, Baccelliere P, et al. Biliary sludge and gallstones in pregnancy: incidence, risk factors, and natural history. Ann Intern Med 1993; 119(2):116–120. doi:10.7326/0003-4819-119-2-199307150-00004
- Etminan M, Delaney JA, Bressler B, Brophy JM. Oral contraceptives and the risk of gallbladder disease: a comparative safety study. CMAJ 2011; 183(8):899–904. doi:10.1503/cmaj.110161
- Everhart JE, Khare M, Hill M, Maurer KR. Prevalence and ethnic differences in gallbladder disease in the United States. Gastroenterology 1999; 117(3):632–639.
- Festi D, Sottili S, Colecchia A, et al. Clinical manifestations of gallstone disease: evidence from the multicenter Italian study on cholelithiasis (MICOL). Hepatology 1999; 30(4):839–846. doi:10.1002/hep.510300401
- Berhane T, Vetrhus M, Hausken T, Olafsson S, Sondenaa K. Pain attacks in non-complicated and complicated gallstone disease have a characteristic pattern and are accompanied by dyspepsia in most patients: the results of a prospective study. Scand J Gastroenterol 2006; 41(1):93–101. doi:10.1080/00365520510023990
- Thistle JL, Cleary PA, Lachin JM, Tyor MP, Hersh T. The natural history of cholelithiasis: the National Cooperative Gallstone Study. Ann Intern Med 1984; 101(2):171–175. doi:10.7326/0003-4819-101-2-171
- Friedman GD. Natural history of asymptomatic and symptomatic gallstones. Am J Surg 1993; 165(4):399–404. doi:0.1016/S0002-9610(05)80930-4
- Friedman GD, Raviola CA, Fireman B. Prognosis of gallstones with mild or no symptoms: 25 years of follow-up in a health maintenance organization. J Clin Epidemiol 1989; 42(2):127–136. doi:10.1016/0895-4356(89)90086-3
- Hirota M, Takada T, Kawarada Y, et al. Diagnostic criteria and severity assessment of acute cholecystitis: Tokyo guidelines. J Hepatobiliary Pancreat Surg 2007; 14(1):78–82. doi:10.1007/s00534-006-1159-4
- Miura F, Takada T, Kawarada Y, et al. Flowcharts for the diagnosis and treatment of acute cholangitis and cholecystitis: Tokyo guidelines. J Hepatobiliary Pancreat Surg 2007; 14(1):27–34. doi:10.1007/s00534-006-1153-x
- Koo KP, Traverso LW. Do preoperative indicators predict the presence of common bile duct stones during laparoscopic cholecystectomy? Am J Surg 1996; 171(5):495–499. doi:10.1016/S0002-9610(97)89611-0
- Collins C, Maguire D, Ireland A, Fitzgerald E, O’Sullivan GC. A prospective study of common bile duct calculi in patients undergoing laparoscopic cholecystectomy: natural history of choledocholithiasis revisited. Ann Surg 2004; 239(1):28–33. doi:10.1097/01.sla.0000103069.00170.9c
- Costi R, Gnocchi A, Di Mario F, Sarli L. Diagnosis and management of choledocholithiasis in the golden age of imaging, endoscopy and laparoscopy. World J Gastroenterol 2014; 20(37):13382–13401. doi:10.3748/wjg.v20.i37.13382
- European Association for the Study of the Liver (EASL). EASL Clinical Practice Guidelines on the prevention, diagnosis and treatment of gallstones. J Hepatol 2016; 65(1):146–181. doi:10.1016/j.jhep.2016.03.005
- Greenberg JA, Hsu J, Bawazeer M, et al. Clinical practice guideline: management of acute pancreatitis. Can J Surg 2016; 59 (2):128–140. doi:10.1503/cjs.015015
- Tenner S, Baillie J, DeWitt J, Vege SS; American College of Gastroenterology. American College of Gastroenterology guideline: management of acute pancreatitis. Am J Gastroenterol 2013; 108(9):1400–1416. doi:10.1038/ajg.2013.218
- Moolla Z, Anderson F, Thomson SR. Use of amylase and alanine transaminase to predict acute gallstone pancreatitis in a population with high HIV prevalence. World J Surg 2013; 37(1):156–161. doi:10.1007/s00268-012-1801-z
- Shea JA, Berlin JA, Escarce JJ, et al. Revised estimates of diagnostic test sensitivity and specificity in suspected biliary tract disease. Arch Intern Med 1994; 154(22):2573–2581. doi:10.1001/archinte.1994.00420220069008
- Kiewiet JJ, Leeuwenburgh MM, Bipat S, et al. A systematic review and meta-analysis of diagnostic performance of imaging in acute cholecystitis. Radiology 2012; 264(3):708–720. doi:10.1148/radiol.12111561
- ASGE Standards of Practice Committee; Maple JT, Ben-Menachem T, Anderson MA, et al. The role of endoscopy in the evaluation of suspected choledocholithiasis. Gastrointest Endosc 2010; 71(1):1–9. doi:10.1016/j.gie.2009.09.041
- Bachar GN, Cohen M, Belenky A, Atar E, Gideon S. Effect of aging on the adult extrahepatic bile duct: a sonographic study. J Ultrasound Med 2003; 22(9):879–885. doi:10.7863/jum.2003.22.9.879
- El-Hayek K, Timratana P, Meranda J, Shimizu H, Eldar S, Chand B. Post Roux-en-Y gastric bypass biliary dilation: natural process or significant entity? J Gastrointest Surg 2012; 16(12):2185–2189. doi:10.1007/s11605-012-2058-4
- Park SM, Kim WS, Bae IH, et al. Common bile duct dilatation after cholecystectomy: a one-year prospective study. J Korean Surg Soc 2012; 83(2):97–101. doi:10.4174/jkss.2012.83.2.97
- Tse F, Liu L, Barkun AN, Armstrong D, Moayyedi P. EUS: a meta-analysis of test performance in suspected choledocholithiasis. Gastrointest Endosc 2008; 67(2):235–244. doi:10.1016/j.gie.2007.09.047
- Verma D, Kapadia A, Eisen GM, Adler DG. EUS vs MRCP for detection of choledocholithiasis. Gastrointest Endosc 2006; 64(2):248–254. doi:10.1016/j.gie.2005.12.038
- Tseng LJ, Jao YT, Mo LR, Lin RC. Over-the-wire US catheter probe as an adjunct to ERCP in the detection of choledocholithiasis. Gastrointest Endosc 2001; 54(6):720–723. doi:10.1067/mge.2001.119255
- Kondo S, Isayama H, Akahane M, et al. Detection of common bile duct stones: comparison between endoscopic ultrasonography, magnetic resonance cholangiography, and helical-computed-tomographic cholangiography. Eur J Radiol 2005; 54(2):271–275. doi:10.1016/j.ejrad.2004.07.007
- Attili AF, De Santis A, Capri R, Repice AM, Maselli S. The natural history of gallstones: the GREPCO experience. The GREPCO Group. Hepatology 1995; 21(3):656–660. doi:10.1016/0270-9139(95)90514-6
- Sakorafas GH, Milingos D, Peros G. Asymptomatic cholelithiasis: is cholecystectomy really needed? A critical reappraisal 15 years after the introduction of laparoscopic cholecystectomy. Dig Dis Sci 2007; 52(5):1313–1325. doi:10.1007/s10620-006-9107-3
- Gracie WA, Ransohoff DF. The natural history of silent gallstones: the innocent gallstone is not a myth. N Engl J Med 1982; 307(13):798–800. doi:10.1056/NEJM198209233071305
- McSherry CK, Ferstenberg H, Calhoun WF, Lahman E, Virshup M. The natural history of diagnosed gallstone disease in symptomatic and asymptomatic patients. Ann Surg 1985; 202(1):59–63. doi:10.1097/00000658-198507000-00009
- Wada K, Wada K, Imamura T. Natural course of asymptomatic gallstone disease. Nihon Rinsho 1993; 51(7):1737–1743. Japanese.
- Halldestam I, Enell EL, Kullman E, Borch K. Development of symptoms and complications in individuals with asymptomatic gallstones. Br J Surg 2004; 91(6):734–738. doi:10.1002/bjs.4547
- Festi D, Reggiani ML, Attili AF, et al. Natural history of gallstone disease: expectant management or active treatment? Results from a population-based cohort study. J Gastroenterol Hepatol 2010; 25(4):719–724. doi:10.1111/j.1440-1746.2009.06146.x
- Shabanzadeh DM, Sorensen LT, Jorgensen T. A prediction rule for risk stratification of incidentally discovered gallstones: results from a large cohort study. Gastroenterology 2016; 150(1):156–167e1. doi:10.1053/j.gastro.2015.09.002
- Overby DW, Apelgren KN, Richardson W, Fanelli R; Society of American Gastrointestinal and Endoscopic Surgeons. SAGES guidelines for the clinical application of laparoscopic biliary tract surgery. Surg Endosc 2010; 24(10):2368–2386. doi:10.1007/s00464-010-1268-7
- Abraham S, Rivero HG, Erlikh IV, Griffith LF, Kondamudi VK. Surgical and nonsurgical management of gallstones. Am Fam Physician 2014; 89(10):795–802.
- Currò G,, Iapichino G, Lorenzini C, Palmeri R, Cucinotta E. Laparoscopic cholecystectomy in children with chronic hemolytic anemia. Is the outcome related to the timing of the procedure? Surg Endosc 2006; 20(2):252–255. doi:10.1007/s00464-005-0318-z
- Hundal R, Shaffer EA. Gallbladder cancer: epidemiology and outcome. Clin Epidemiol 2014; 6:99–109. doi:10.2147/CLEP.S37357
- Chen GL, Akmal Y, DiFronzo AL, Vuong B, O’Connor V. Porcelain gallbladder: no longer an indication for prophylactic cholecystectomy. Am Surg 2015; 81(10):936–940.
- Schnelldorfer T. Porcelain gallbladder: a benign process or concern for malignancy? J Gastrointest Surg 2013; 17(6):1161–1168. doi:10.1007/s11605-013-2170-0
- Warschkow R, Tarantino I, Ukegjini K, et al. Concomitant cholecystectomy during laparoscopic Roux-en-Y gastric bypass in obese patients is not justified: a meta-analysis. Obes Surg 2013; 23(3)3979–408. doi:10.1007/s11695-012-0852-4
- Gurusamy K, Samraj K, Gluud C, Wilson E, Davidson BR. Meta-analysis of randomized controlled trials on the safety and effectiveness of early versus delayed laparoscopic cholecystectomy for acute cholecystitis. Br J Surg 2010; 97(2):141–150. doi:10.1002/bjs.6870
- Papi C, Catarci M, D’Ambrosio L, et al. Timing of cholecystectomy for acute calculous cholecystitis: a meta-analysis. Am J Gastroenterol 2004; 99(1):147–155. doi:10.1046/j.1572-0241.2003.04002.x
- Gurusamy KS, Davidson C, Gluud C, Davidson BR. Early versus delayed laparoscopic cholecystectomy for people with acute cholecystitis. Cochrane Database Syst Rev 2013; 6:CD005440. doi:10.1002/14651858
- Menahem B, Mulliri A, Fohlen A, Guittet L, Alves A, Lubrano J. Delayed laparoscopic cholecystectomy increases the total hospital stay compared to an early laparoscopic cholecystectomy after acute cholecystitis: an updated meta-analysis of randomized controlled trials. HPB (Oxford) 2015; 17(10):857–862. doi:10.1111/hpb.12449
- Uhl W, Müller CA, Krähenbühl L, Schmid SW, Schölzel S, Büchler MW. Acute gallstone pancreatitis: timing of laparoscopic cholecystectomy in mild and severe disease. Surg Endosc 1999; 13(11):1070–1076. doi:10.1007/s004649901175
- Aboulian A, Chan T, Yaghoubian A, et al. Early cholecystectomy safely decreases hospital stay in patients with mild gallstone pancreatitis: a randomized prospective study. Ann Surg 2010(4): 251:615–619. doi:10.1097/SLA.0b013e3181c38f1f
- Schirmer BD, Winters KL, Edlich RF. Cholelithiasis and cholecystitis. J Long Term Eff Med Implants 2005; 15(3):329–338. doi:10.1615/JLongTermEffMedImplants.v15.i3.90
- Stinton LM, Shaffer EA. Epidemiology of gallbladder disease: cholelithiasis and cancer. Gut Liver 2012; 6(2):172–187. doi:10.5009/gnl.2012.6.2.172
- Lee JY, Keane MG, Pereira S. Diagnosis and treatment of gallstone disease. Practitioner 2015; 259(1783):15–19.
- Russo MW, Wei JT, Thiny MT, et al. Digestive and liver diseases statistics, 2004. Gastroenterology 2004; 126(5):1448–1453. doi:10.1053/j.gastro.2004.01.025
- Everhart JE, Ruhl CE. Burden of digestive diseases in the United States part I: overall and upper gastrointestinal diseases. Gastroenterology 2009; 136(2):376–386. doi:10.1053/j.gastro.2008.12.015
- Cariati A. Gallstone classification in Western countries. Indian J Surg 2015; 77(suppl 2):376–380. doi.org/10.1007/s12262-013-0847-y
- Carey MC. Pathogenesis of gallstones. Am J Surg 1993; 165(4):410–419. doi:10.1016/S0002-9610(05)80932-8
- Lammert F, Gurusamy K, Ko CW, et al. Gallstones. Nat Rev Dis Primers 2016; 2:16024. doi:10.1038/nrdp.2016.24
- Stewart L, Oesterle AL, Erdan I, Griffiss JM, Way LW. Pathogenesis of pigment gallstones in Western societies: the central role of bacteria. J Gastrointest Surg 2002; 6(6):891–904.
- Barbara L, Sama C, Morselli Labate AM, et al. A population study on the prevalence of gallstone disease: the Sirmione Study. Hepatology 1987; 7(5):913–917. doi:10.1002/hep.1840070520
- Sood S, Winn T, Ibrahim S, et al. Natural history of asymptomatic gallstones: differential behaviour in male and female subjects. Med J Malaysia 2015; 70(6):341–345.
- Maringhini A, Ciambra M, Baccelliere P, et al. Biliary sludge and gallstones in pregnancy: incidence, risk factors, and natural history. Ann Intern Med 1993; 119(2):116–120. doi:10.7326/0003-4819-119-2-199307150-00004
- Etminan M, Delaney JA, Bressler B, Brophy JM. Oral contraceptives and the risk of gallbladder disease: a comparative safety study. CMAJ 2011; 183(8):899–904. doi:10.1503/cmaj.110161
- Everhart JE, Khare M, Hill M, Maurer KR. Prevalence and ethnic differences in gallbladder disease in the United States. Gastroenterology 1999; 117(3):632–639.
- Festi D, Sottili S, Colecchia A, et al. Clinical manifestations of gallstone disease: evidence from the multicenter Italian study on cholelithiasis (MICOL). Hepatology 1999; 30(4):839–846. doi:10.1002/hep.510300401
- Berhane T, Vetrhus M, Hausken T, Olafsson S, Sondenaa K. Pain attacks in non-complicated and complicated gallstone disease have a characteristic pattern and are accompanied by dyspepsia in most patients: the results of a prospective study. Scand J Gastroenterol 2006; 41(1):93–101. doi:10.1080/00365520510023990
- Thistle JL, Cleary PA, Lachin JM, Tyor MP, Hersh T. The natural history of cholelithiasis: the National Cooperative Gallstone Study. Ann Intern Med 1984; 101(2):171–175. doi:10.7326/0003-4819-101-2-171
- Friedman GD. Natural history of asymptomatic and symptomatic gallstones. Am J Surg 1993; 165(4):399–404. doi:0.1016/S0002-9610(05)80930-4
- Friedman GD, Raviola CA, Fireman B. Prognosis of gallstones with mild or no symptoms: 25 years of follow-up in a health maintenance organization. J Clin Epidemiol 1989; 42(2):127–136. doi:10.1016/0895-4356(89)90086-3
- Hirota M, Takada T, Kawarada Y, et al. Diagnostic criteria and severity assessment of acute cholecystitis: Tokyo guidelines. J Hepatobiliary Pancreat Surg 2007; 14(1):78–82. doi:10.1007/s00534-006-1159-4
- Miura F, Takada T, Kawarada Y, et al. Flowcharts for the diagnosis and treatment of acute cholangitis and cholecystitis: Tokyo guidelines. J Hepatobiliary Pancreat Surg 2007; 14(1):27–34. doi:10.1007/s00534-006-1153-x
- Koo KP, Traverso LW. Do preoperative indicators predict the presence of common bile duct stones during laparoscopic cholecystectomy? Am J Surg 1996; 171(5):495–499. doi:10.1016/S0002-9610(97)89611-0
- Collins C, Maguire D, Ireland A, Fitzgerald E, O’Sullivan GC. A prospective study of common bile duct calculi in patients undergoing laparoscopic cholecystectomy: natural history of choledocholithiasis revisited. Ann Surg 2004; 239(1):28–33. doi:10.1097/01.sla.0000103069.00170.9c
- Costi R, Gnocchi A, Di Mario F, Sarli L. Diagnosis and management of choledocholithiasis in the golden age of imaging, endoscopy and laparoscopy. World J Gastroenterol 2014; 20(37):13382–13401. doi:10.3748/wjg.v20.i37.13382
- European Association for the Study of the Liver (EASL). EASL Clinical Practice Guidelines on the prevention, diagnosis and treatment of gallstones. J Hepatol 2016; 65(1):146–181. doi:10.1016/j.jhep.2016.03.005
- Greenberg JA, Hsu J, Bawazeer M, et al. Clinical practice guideline: management of acute pancreatitis. Can J Surg 2016; 59 (2):128–140. doi:10.1503/cjs.015015
- Tenner S, Baillie J, DeWitt J, Vege SS; American College of Gastroenterology. American College of Gastroenterology guideline: management of acute pancreatitis. Am J Gastroenterol 2013; 108(9):1400–1416. doi:10.1038/ajg.2013.218
- Moolla Z, Anderson F, Thomson SR. Use of amylase and alanine transaminase to predict acute gallstone pancreatitis in a population with high HIV prevalence. World J Surg 2013; 37(1):156–161. doi:10.1007/s00268-012-1801-z
- Shea JA, Berlin JA, Escarce JJ, et al. Revised estimates of diagnostic test sensitivity and specificity in suspected biliary tract disease. Arch Intern Med 1994; 154(22):2573–2581. doi:10.1001/archinte.1994.00420220069008
- Kiewiet JJ, Leeuwenburgh MM, Bipat S, et al. A systematic review and meta-analysis of diagnostic performance of imaging in acute cholecystitis. Radiology 2012; 264(3):708–720. doi:10.1148/radiol.12111561
- ASGE Standards of Practice Committee; Maple JT, Ben-Menachem T, Anderson MA, et al. The role of endoscopy in the evaluation of suspected choledocholithiasis. Gastrointest Endosc 2010; 71(1):1–9. doi:10.1016/j.gie.2009.09.041
- Bachar GN, Cohen M, Belenky A, Atar E, Gideon S. Effect of aging on the adult extrahepatic bile duct: a sonographic study. J Ultrasound Med 2003; 22(9):879–885. doi:10.7863/jum.2003.22.9.879
- El-Hayek K, Timratana P, Meranda J, Shimizu H, Eldar S, Chand B. Post Roux-en-Y gastric bypass biliary dilation: natural process or significant entity? J Gastrointest Surg 2012; 16(12):2185–2189. doi:10.1007/s11605-012-2058-4
- Park SM, Kim WS, Bae IH, et al. Common bile duct dilatation after cholecystectomy: a one-year prospective study. J Korean Surg Soc 2012; 83(2):97–101. doi:10.4174/jkss.2012.83.2.97
- Tse F, Liu L, Barkun AN, Armstrong D, Moayyedi P. EUS: a meta-analysis of test performance in suspected choledocholithiasis. Gastrointest Endosc 2008; 67(2):235–244. doi:10.1016/j.gie.2007.09.047
- Verma D, Kapadia A, Eisen GM, Adler DG. EUS vs MRCP for detection of choledocholithiasis. Gastrointest Endosc 2006; 64(2):248–254. doi:10.1016/j.gie.2005.12.038
- Tseng LJ, Jao YT, Mo LR, Lin RC. Over-the-wire US catheter probe as an adjunct to ERCP in the detection of choledocholithiasis. Gastrointest Endosc 2001; 54(6):720–723. doi:10.1067/mge.2001.119255
- Kondo S, Isayama H, Akahane M, et al. Detection of common bile duct stones: comparison between endoscopic ultrasonography, magnetic resonance cholangiography, and helical-computed-tomographic cholangiography. Eur J Radiol 2005; 54(2):271–275. doi:10.1016/j.ejrad.2004.07.007
- Attili AF, De Santis A, Capri R, Repice AM, Maselli S. The natural history of gallstones: the GREPCO experience. The GREPCO Group. Hepatology 1995; 21(3):656–660. doi:10.1016/0270-9139(95)90514-6
- Sakorafas GH, Milingos D, Peros G. Asymptomatic cholelithiasis: is cholecystectomy really needed? A critical reappraisal 15 years after the introduction of laparoscopic cholecystectomy. Dig Dis Sci 2007; 52(5):1313–1325. doi:10.1007/s10620-006-9107-3
- Gracie WA, Ransohoff DF. The natural history of silent gallstones: the innocent gallstone is not a myth. N Engl J Med 1982; 307(13):798–800. doi:10.1056/NEJM198209233071305
- McSherry CK, Ferstenberg H, Calhoun WF, Lahman E, Virshup M. The natural history of diagnosed gallstone disease in symptomatic and asymptomatic patients. Ann Surg 1985; 202(1):59–63. doi:10.1097/00000658-198507000-00009
- Wada K, Wada K, Imamura T. Natural course of asymptomatic gallstone disease. Nihon Rinsho 1993; 51(7):1737–1743. Japanese.
- Halldestam I, Enell EL, Kullman E, Borch K. Development of symptoms and complications in individuals with asymptomatic gallstones. Br J Surg 2004; 91(6):734–738. doi:10.1002/bjs.4547
- Festi D, Reggiani ML, Attili AF, et al. Natural history of gallstone disease: expectant management or active treatment? Results from a population-based cohort study. J Gastroenterol Hepatol 2010; 25(4):719–724. doi:10.1111/j.1440-1746.2009.06146.x
- Shabanzadeh DM, Sorensen LT, Jorgensen T. A prediction rule for risk stratification of incidentally discovered gallstones: results from a large cohort study. Gastroenterology 2016; 150(1):156–167e1. doi:10.1053/j.gastro.2015.09.002
- Overby DW, Apelgren KN, Richardson W, Fanelli R; Society of American Gastrointestinal and Endoscopic Surgeons. SAGES guidelines for the clinical application of laparoscopic biliary tract surgery. Surg Endosc 2010; 24(10):2368–2386. doi:10.1007/s00464-010-1268-7
- Abraham S, Rivero HG, Erlikh IV, Griffith LF, Kondamudi VK. Surgical and nonsurgical management of gallstones. Am Fam Physician 2014; 89(10):795–802.
- Currò G,, Iapichino G, Lorenzini C, Palmeri R, Cucinotta E. Laparoscopic cholecystectomy in children with chronic hemolytic anemia. Is the outcome related to the timing of the procedure? Surg Endosc 2006; 20(2):252–255. doi:10.1007/s00464-005-0318-z
- Hundal R, Shaffer EA. Gallbladder cancer: epidemiology and outcome. Clin Epidemiol 2014; 6:99–109. doi:10.2147/CLEP.S37357
- Chen GL, Akmal Y, DiFronzo AL, Vuong B, O’Connor V. Porcelain gallbladder: no longer an indication for prophylactic cholecystectomy. Am Surg 2015; 81(10):936–940.
- Schnelldorfer T. Porcelain gallbladder: a benign process or concern for malignancy? J Gastrointest Surg 2013; 17(6):1161–1168. doi:10.1007/s11605-013-2170-0
- Warschkow R, Tarantino I, Ukegjini K, et al. Concomitant cholecystectomy during laparoscopic Roux-en-Y gastric bypass in obese patients is not justified: a meta-analysis. Obes Surg 2013; 23(3)3979–408. doi:10.1007/s11695-012-0852-4
- Gurusamy K, Samraj K, Gluud C, Wilson E, Davidson BR. Meta-analysis of randomized controlled trials on the safety and effectiveness of early versus delayed laparoscopic cholecystectomy for acute cholecystitis. Br J Surg 2010; 97(2):141–150. doi:10.1002/bjs.6870
- Papi C, Catarci M, D’Ambrosio L, et al. Timing of cholecystectomy for acute calculous cholecystitis: a meta-analysis. Am J Gastroenterol 2004; 99(1):147–155. doi:10.1046/j.1572-0241.2003.04002.x
- Gurusamy KS, Davidson C, Gluud C, Davidson BR. Early versus delayed laparoscopic cholecystectomy for people with acute cholecystitis. Cochrane Database Syst Rev 2013; 6:CD005440. doi:10.1002/14651858
- Menahem B, Mulliri A, Fohlen A, Guittet L, Alves A, Lubrano J. Delayed laparoscopic cholecystectomy increases the total hospital stay compared to an early laparoscopic cholecystectomy after acute cholecystitis: an updated meta-analysis of randomized controlled trials. HPB (Oxford) 2015; 17(10):857–862. doi:10.1111/hpb.12449
- Uhl W, Müller CA, Krähenbühl L, Schmid SW, Schölzel S, Büchler MW. Acute gallstone pancreatitis: timing of laparoscopic cholecystectomy in mild and severe disease. Surg Endosc 1999; 13(11):1070–1076. doi:10.1007/s004649901175
- Aboulian A, Chan T, Yaghoubian A, et al. Early cholecystectomy safely decreases hospital stay in patients with mild gallstone pancreatitis: a randomized prospective study. Ann Surg 2010(4): 251:615–619. doi:10.1097/SLA.0b013e3181c38f1f
KEY POINTS
- Abdominal pain is the primary symptom associated with gallstones.
- Abdominal ultrasonography is the diagnostic test of choice to detect gallstones and assess for findings suggestive of acute cholecystitis and dilation of the common bile duct.
- First-line therapy for asymptomatic gallstones is expectant management.
- First-line therapy for symptomatic gallstones is cholecystectomy.
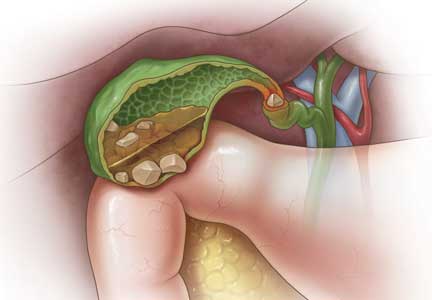
In reply: Wilson disease
In Reply: We thank Dr. Mirrakhimov and colleagues for bringing important questions to our attention.
In terms of the differential diagnosis of cholestatic liver injury, we agree that pathologic processes such choledocholithiasis, cholangitis, primary biliary cirrhosis, and primary sclerosing cholangitis should be generally considered. However, in the case we described, the patient had no abdominal pain or fever, which makes choledocholithiasis or cholangitis very unlikely. Primary biliary cirrhosis and primary sclerosing cholangitis can cause chronic liver disease but should not be considered in the differential diagnosis of acute liver injury (acute hepatitis), such as in the case we described.
We agree that the hemolytic anemia typically seen in patients with Wilson disease is Coombs-negative, and that Coombs testing and a peripheral smear should be performed. Both were negative in our patient.
We also agree with Dr. Mirrakhimov and colleagues that Kayser-Fleischer rings are not necessarily specific for Wilson disease and can be seen in patients with other forms of cholestatic liver disease such as primary biliary cirrhosis. However, Kayser-Fleischer rings are pathognomonic for acute liver failure from Wilson disease. In other words, when Kayser-Fleischer rings are seen in a patient with acute liver failure, the diagnosis is Wilson disease until proven otherwise.
We discussed on page 112 of our article other treatments such as plasmapheresis as adjunctive therapy to bridge patients with acute liver failure secondary to Wilson disease to transplant. However, liver transplant is still the only definitive and potentially curative treatment.
In Reply: We thank Dr. Mirrakhimov and colleagues for bringing important questions to our attention.
In terms of the differential diagnosis of cholestatic liver injury, we agree that pathologic processes such choledocholithiasis, cholangitis, primary biliary cirrhosis, and primary sclerosing cholangitis should be generally considered. However, in the case we described, the patient had no abdominal pain or fever, which makes choledocholithiasis or cholangitis very unlikely. Primary biliary cirrhosis and primary sclerosing cholangitis can cause chronic liver disease but should not be considered in the differential diagnosis of acute liver injury (acute hepatitis), such as in the case we described.
We agree that the hemolytic anemia typically seen in patients with Wilson disease is Coombs-negative, and that Coombs testing and a peripheral smear should be performed. Both were negative in our patient.
We also agree with Dr. Mirrakhimov and colleagues that Kayser-Fleischer rings are not necessarily specific for Wilson disease and can be seen in patients with other forms of cholestatic liver disease such as primary biliary cirrhosis. However, Kayser-Fleischer rings are pathognomonic for acute liver failure from Wilson disease. In other words, when Kayser-Fleischer rings are seen in a patient with acute liver failure, the diagnosis is Wilson disease until proven otherwise.
We discussed on page 112 of our article other treatments such as plasmapheresis as adjunctive therapy to bridge patients with acute liver failure secondary to Wilson disease to transplant. However, liver transplant is still the only definitive and potentially curative treatment.
In Reply: We thank Dr. Mirrakhimov and colleagues for bringing important questions to our attention.
In terms of the differential diagnosis of cholestatic liver injury, we agree that pathologic processes such choledocholithiasis, cholangitis, primary biliary cirrhosis, and primary sclerosing cholangitis should be generally considered. However, in the case we described, the patient had no abdominal pain or fever, which makes choledocholithiasis or cholangitis very unlikely. Primary biliary cirrhosis and primary sclerosing cholangitis can cause chronic liver disease but should not be considered in the differential diagnosis of acute liver injury (acute hepatitis), such as in the case we described.
We agree that the hemolytic anemia typically seen in patients with Wilson disease is Coombs-negative, and that Coombs testing and a peripheral smear should be performed. Both were negative in our patient.
We also agree with Dr. Mirrakhimov and colleagues that Kayser-Fleischer rings are not necessarily specific for Wilson disease and can be seen in patients with other forms of cholestatic liver disease such as primary biliary cirrhosis. However, Kayser-Fleischer rings are pathognomonic for acute liver failure from Wilson disease. In other words, when Kayser-Fleischer rings are seen in a patient with acute liver failure, the diagnosis is Wilson disease until proven otherwise.
We discussed on page 112 of our article other treatments such as plasmapheresis as adjunctive therapy to bridge patients with acute liver failure secondary to Wilson disease to transplant. However, liver transplant is still the only definitive and potentially curative treatment.
A tale of two sisters with liver disease
A 25-year-old woman presents to the emergency department with a 7-day history of fatigue and nausea. On presentation she denies having abdominal pain, headache, fever, chills, night sweats, vomiting, diarrhea, melena, hematochezia, or weight loss. She recalls changes in the colors of her eyes and darkening urine over the last few days. Her medical history before this is unremarkable. She takes no prescription, over-the-counter, or herbal medications. She works as a librarian and has no occupational toxic exposures. She is single and has one sister with no prior medical history. She denies recent travel, sick contacts, smoking, recreational drug use, or pets at home.
On physical examination, her vital signs are temperature 37.3°C (99.1°F), heart rate 90 beats per minute, blood pressure 125/80 mm Hg, respiration rate 14 per minute, and oxygen saturation 97% on room air. She has icteric sclera and her skin is jaundiced. Cardiac examination is normal. Lungs are clear to auscultation and percussion bilaterally. Her abdomen is soft with no visceromegaly, masses, or tenderness. Extremities are normal with no edema. She is alert and oriented, but she has mild asterixis of the outstretched hands. The neurologic examination is otherwise unremarkable.
The patient’s basic laboratory values are listed in Table 1. Shortly after admission, she develops changes in her mental status, remaining alert but becoming agitated and oriented to person only. In view of her symptoms and laboratory findings, acute liver failure is suspected.
ACUTE LIVER FAILURE
1. The diagnostic criteria for acute liver failure include all of the following except which one?
- Acute elevation of liver biochemical tests
- Presence of preexisting liver disease
- Coagulopathy, defined by an international normalized ratio (INR) of 1.5 or greater
- Encephalopathy
- Duration of symptoms less than 26 weeks
Acute liver failure is defined by acute onset of worsening liver tests, coagulopathy (INR ≥ 1.5), and encephalopathy in patients with no preexisting liver disease and with symptom duration of less than 26 weeks.1 With a few exceptions, a history of preexisting liver disease negates the diagnosis of acute liver failure. Our patient meets the diagnostic criteria for acute liver failure.
Immediate management
Once acute liver failure is identified or suspected, the next step is to transfer the patient to the intensive care unit for close monitoring of mental status. Serial neurologic evaluations permit early detection of cerebral edema, which is considered the most common cause of death in patients with acute liver failure. Additionally, close monitoring of electrolytes and plasma glucose is necessary since these patients are susceptible to electrolyte disturbances and hypoglycemia.
Patients with acute liver failure are at increased risk of infections and should be routinely screened by obtaining urine and blood cultures.
Gastrointestinal bleeding is not uncommon in patients with acute liver failure and is usually due to gastric stress ulceration. Prophylaxis with a histamine 2 receptor antagonist or proton pump inhibitor should be considered in order to prevent gastrointestinal bleeding.
Treatment with N-acetylcysteine is beneficial, not only in patients with acute liver failure due to acetaminophen overdose, but also in those with acute liver failure from other causes.
CASE CONTINUES:
TRANSFER TO THE INTENSIVE CARE UNIT
The patient, now diagnosed with acute liver failure, is transferred to the intensive care unit. Arterial blood gas measurement shows:
- pH 7.38 (reference range 7.35–7.45)
- Pco2 40 mm Hg (36–46)
- Po2 97 mm Hg (85–95)
- Hco3 22 mmol/L (22–26).
A chest radiograph is obtained and is clear. Computed tomography (CT) of the brain reveals no edema. Transcranial Doppler ultrasonography does not show any intracranial fluid collections.
Blood and urine cultures are negative. Her hemoglobin level remains stable, and she does not develop signs of bleeding. She is started on a proton pump inhibitor for stress ulcer prophylaxis and is empirically given intravenous N-acetylcysteine until the cause of acute liver failure can be determined.
CAUSES OF ACUTE LIVER FAILURE
2. Which of the following can cause acute liver failure?
- Acetaminophen overdose
- Viral hepatitis
- Autoimmune hepatitis
- Wilson disease
- Alcoholic hepatitis
Drug-induced liver injury is the most common cause of acute liver failure in the United States,2,3 and of all drugs, acetaminophen overdose is the number-one cause. In acetaminophen-induced liver injury, serum aminotransferase levels are usually elevated to more than 1,000 U/L, while serum bilirubin remains normal in the early stages. Antimicrobial agents, antiepileptic drugs, and herbal supplements have also been implicated in acute liver failure. Our patient has denied taking herbal supplements or medications, including over-the-counter ones.
Acute viral hepatitis can explain the patient’s condition. It is a common cause of acute liver failure in the United States.2 Hepatitis A and E are more common in developing countries. Other viruses such as cytomegalovirus, Epstein-Barr virus, herpes simplex virus type 1 and 2, and varicella zoster virus can also cause acute liver failure. Serum aminotransferase levels may exceed 1,000 U/L in patients with viral hepatitis.
Autoimmune hepatitis is a rare cause of acute liver failure, but it should be considered in the differential diagnosis, particularly in middle-aged women with autoimmune disorders such as hypothyroidism. Autoimmune hepatitis can cause marked elevation in aminotransferase levels (> 1,000 U/L).
Wilson disease is an autosomal-recessive disease in which there is excessive accumulation of copper in the liver and other organs because of an inherited defect in the biliary excretion of copper. Wilson disease can cause acute liver failure and should be excluded in any patient, particularly if under age 40 with acute onset of unexplained hepatic, neurologic, or psychiatric disease.
Alcoholic hepatitis usually occurs in patients with a long-standing history of heavy alcohol use. As a result, most patients with alcoholic hepatitis have manifestations of chronic liver disease due to alcohol use. Therefore, by definition, it is not a cause of acute liver failure. Additionally, in patients with alcoholic hepatitis, the aspartate aminotransferase (AST) level is elevated but less than 300 IU/mL, and the ratio of AST to alanine aminotransferase (ALT) is usually more than 2.
CASE CONTINUES: FURTHER TESTING
The results of our patient’s serologic tests are shown in Table 2. Other test results:
- Autoimmune markers including antinuclear antibodies, antimitochondrial antibodies, antismooth muscle antibodies, and liver and kidney microsomal antibodies are negative; her immunoglobulin G (IgG) level is normal
- Serum ceruloplasmin 25 mg/dL (normal 21–45)
- Free serum copper 120 µg/dL (normal 8–12)
- Abdominal ultrasonography is unremarkable, with normal liver parenchyma and no intrahepatic or extrahepatic biliary dilatation
- Doppler ultrasonography of the liver shows patent blood vessels.
3. Based on the new data, which of the following statements is correct?
- Hepatitis B is the cause of acute liver failure in this patient
- Herpetic hepatitis cannot be excluded on the basis of the available data
- Wilson disease is most likely the diagnosis, given her elevated free serum copper
- A normal serum ceruloplasmin level is not sufficient to rule out acute liver failure secondary to Wilson disease
Hepatitis B surface antigen and hepatitis B core antibodies were negative in our patient, excluding hepatitis B virus infection. The positive hepatitis B surface antibody indicates prior immunization.
Herpetic hepatitis is an uncommon but important cause of acute liver failure because the mortality rate is high if the patient is not treated early with acyclovir. Fever, elevated aminotransferases, and leukopenia are common with herpetic hepatitis. Fewer than 50% of patients with herpetic hepatitis have vesicular rash.4,5 The value of antibody serologic testing is limited due to high rates of false-positive and false-negative results. The gold standard diagnostic tests are viral load (detection of viral RNA by polymerase chain reaction), viral staining on liver biopsy, or both. In our patient, herpes simplex virus polymerase chain reaction testing was negative, which makes herpetic hepatitis unlikely.
Wilson disease is a genetic condition in which the ability to excrete copper in the bile is impaired, resulting in accumulation of copper in the hepatocytes. Subsequently, copper is released into the bloodstream and eventually into the urine.
However, copper excretion into the bile is impaired in patients with acute liver failure regardless of the etiology. Therefore, elevated free serum copper and 24-hour urine copper levels are not specific for the diagnosis of acute liver failure secondary to Wilson disease. Moreover, Kayser-Fleischer rings, which represent copper deposition in the limbus of the cornea, may not be apparent in the early stages of Wilson disease.
Since it is challenging to diagnose Wilson disease in the context of acute liver failure, Korman et al6 compared patients with acute liver failure secondary to Wilson disease with patients with acute liver failure secondary to other conditions. They found that alkaline phosphatase levels are frequently decreased in patients with acute liver failure secondary to Wilson disease,6 and that a ratio of alkaline phosphatase to total bilirubin of less than 4 is 94% sensitive and 96% specific for the diagnosis.6
Hemolysis is common in acute liver failure due to Wilson disease. This leads to disproportionate elevation of AST compared with ALT, since AST is present in red blood cells. Consequently, the ratio of AST to ALT is usually greater than 2.2, which provides a sensitivity of 94% and a specificity of 86% for the diagnosis.6 These two ratios together provide 100% sensitivity and 100% specificity for the diagnosis of Wilson disease in the context of acute liver failure.6
Ceruloplasmin. Patients with Wilson disease typically have a low ceruloplasmin level. However, because it is an acute-phase reaction protein, ceruloplasmin can be normal or elevated in patients with acute liver failure from Wilson disease.6 Therefore, a normal ceruloplasmin level is not sufficient to rule out acute liver failure secondary to Wilson disease.
CASE CONTINUES: A DEFINITIVE DIAGNOSIS
Our patient undergoes further testing, which reveals the following:
- Her 24-hour urinary excretion of copper is 150 µg (reference value < 30)
- Slit-lamp examination is normal and shows no evidence of Kayser-Fleischer rings
- Her ratio of alkaline phosphatase to total bilirubin is 0.77 based on her initial laboratory results (Table 1)
- Her AST-ALT ratio is 3.4.
The diagnosis in our patient is acute liver failure secondary to Wilson disease.
4. What is the most appropriate next step?
- Liver biopsy
- d-penicillamine by mouth
- Trientine by mouth
- Liver transplant
- Plasmapheresis
Liver biopsy. Accumulation of copper in the liver parenchyma in patients with Wilson disease is sporadic. Therefore, qualitative copper staining on liver biopsy can be falsely negative. Quantitative copper measurement in liver tissue is the gold standard for the diagnosis of Wilson disease. However, the test is time-consuming and is not rapidly available in the context of acute liver failure.
Chelating agents such as d-pencillamine and trientine are used to treat the chronic manifestations of Wilson disease but are not useful for acute liver failure secondary to Wilson disease.
Acute liver failure secondary to Wilson disease is life-threatening, and liver transplant is considered the only definitive life-saving therapy.
Therapeutic plasmapheresis has been reported to be a successful adjunctive therapy to bridge patients with acute liver failure secondary to Wilson disease to transplant.7 However, liver transplant is still the only definitive treatment.
CASE CONTINUES: THE PATIENT’S SISTER SEEKS CARE
The patient undergoes liver transplantation, with no perioperative or postoperative complications.
The patient’s 18-year-old sister is now seeking medical attention in the outpatient clinic, concerned that she may have Wilson disease. She is otherwise healthy and denies any symptoms or complaints.
5. What is the next step for the patient’s sister?
- Reassurance
- Prophylaxis with trientine
- Check liver enzyme levels, serum ceruloplasmin level, and urine copper, and order a slit-lamp examination
- Genetic testing
Wilson disease can be asymptomatic in its early stages and may be diagnosed incidentally during routine blood tests that reveal abnormal liver enzyme levels. All patients with a confirmed family history of Wilson disease should be screened even if they are asymptomatic. The diagnosis of Wilson disease should be established in first-degree relatives before specific treatment for the relatives is prescribed.
The first step in screening a first-degree relative for Wilson disease is to check liver enzyme levels (specifically aminotransferases, alkaline phosphatase, and bilirubin), serum ceruloplasmin level, and 24-hour urine copper, and order an ophthalmologic slit-lamp examination. If any of these tests is abnormal, liver biopsy should be performed for histopathologic evaluation and quantitative copper measurement. Kayser-Fleischer rings are seen in only 50% of patients with Wilson disease and hepatic involvement, but they are pathognomic. Guidelines8 for screening first-degree relatives of Wilson disease patients are shown in Figure 1.
Genetic analysis. ATP7B, the Wilson disease gene, is located on chromosome 13. At least 300 mutations of the gene have been described,2 and the most common mutation is present in only 15% to 30% of the Wilson disease population.8–10 Routine molecular testing of the ATP7B
CASE CONTINUES: WORKUP OF THE PATIENT’S SISTER
The patient’s sister has no symptoms and her physical examination is normal. Slit-lamp examination reveals no evidence of Kayser-Fleischer rings. Her laboratory values, including complete blood counts, complete metabolic panel, and INR, are within normal ranges. Other test results, however, are abnormal:
- Free serum copper level 27 µg/dL (normal 8–12)
- Serum ceruloplasmin 9.0 mg/dL (normal 20–50)
- 24-hour urinary copper excretion 135 µg (normal < 30).
She undergoes liver biopsy for quantitative copper measurement, and the result is very high at 1,118 µg/g dry weight (reference range 10–35). The diagnosis of Wilson disease is established.
TREATING CHRONIC WILSON DISEASE
6. Which of the following is not an appropriate next step for the patient’s sister?
- Tetrathiomolybdate
- d-penicillamine
- Trientine
- Zinc salts
- Prednisone
The goal of medical treatment of chronic Wilson disease is to improve symptoms and prevent progression of the disease.
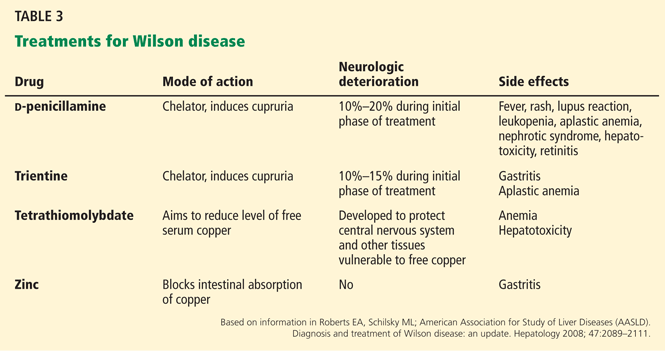
Chelating agents and zinc salts are the most commonly used medicines in the management of Wilson disease. Chelating agents remove copper from tissue, whereas zinc blocks the intestinal absorption of copper and stimulates the synthesis of endogenous chelators such as metallothioneins. Tetrathiomolybdate is an alternative agent developed to interfere with the distribution of excess body copper to susceptible target sites by reducing free serum copper (Table 3). There are no data to support the use of prednisone in the treatment of Wilson disease.
During treatment with chelating agents, 24-hour urinary excretion of copper is routinely monitored to determine the efficacy of therapy and adherence to treatment. Once de-coppering is achieved, as evidenced by a normalization of 24-hour urine copper excretion, the chelating agent can be switched to zinc salts to prevent intestinal absorption of copper.
Clinical and biochemical stabilization is achieved typically within 2 to 6 months of the initial treatment with chelating agents.8 Organ meats, nuts, shellfish, and chocolate are rich in copper and should be avoided.
The patient’s sister is started on trientine 250 mg orally three times daily on an empty stomach at least 1 hour before meals. Treatment is monitored by following 24-hour urine copper measurement. A 24-hour urine copper measurement at 3 months after starting treatment has increased from 54 at baseline to 350 µg, which indicates that the copper is being removed from tissues. The plan is for early substitution of zinc for long-term maintenance once de-coppering is completed.
KEY POINTS
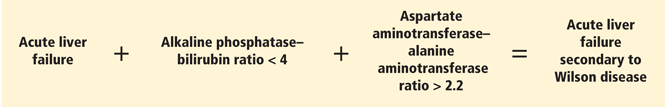
- Acute liver failure is severe acute liver injury characterized by coagulopathy (INR ≥ 1.5) and encephalopathy in a patient with no preexisting liver disease and with duration of symptoms less than 26 weeks.
- Acute liver failure secondary to Wilson disease is uncommon but should be excluded, particularly in young patients.
- The diagnosis of Wilson disease in the setting of acute liver failure is challenging because the serum ceruloplasmin level may be normal in acute liver failure secondary to Wilson disease, and free serum copper and 24-hour urine copper are usually elevated in all acute liver failure patients regardless of the etiology.
- A ratio of alkaline phosphatase to total bilirubin of less than 4 plus an AST-ALT ratio greater than 2.2 in a patient with acute liver failure should be regarded as Wilson disease until proven otherwise (Figure 2).
- Acute liver failure secondary to Wilson disease is usually fatal, and emergency liver transplant is a life-saving procedure.
- Screening of first-degree relatives of Wilson disease patients should include a history and physical examination, liver enzyme tests, complete blood cell count, serum ceruloplasmin level, serum free copper level, slit-lamp examination of the eyes, and 24-hour urinary copper measurement. Genetic tests are supplementary for screening but are not routinely available.
- Lee WM, Larson AM, Stravitz T. AASLD Position Paper: The management of acute liver failure: update 2011. www.aasld.org/sites/default/files/guideline_documents/alfenhanced.pdf. Accessed December 9, 2015.
- Bernal W, Auzinger G, Dhawan A, Wendon J. Acute liver failure. Lancet 2010; 376:190–201.
- Larson AM, Polson J, Fontana RJ, et al; Acute Liver Failure Study Group. Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology 2005; 42:1364–1372.
- Hanouneh IA, Khoriaty R, Zein NN. A 35-year-old Asian man with jaundice and markedly high aminotransferase levels. Cleve Clin J Med 2009; 76:449–456.
- Norvell JP, Blei AT, Jovanovic BD, Levitsky J. Herpes simplex virus hepatitis: an analysis of the published literature and institutional cases. Liver Transpl 2007; 13:1428–1434.
- Korman JD, Volenberg I, Balko J, et al; Pediatric and Adult Acute Liver Failure Study Groups. Screening for Wilson disease in acute liver failure: a comparison of currently available diagnostic tests. Hepatology 2008; 48:1167–1174.
- Morgan SM, Zantek ND. Therapeutic plasma exchange for fulminant hepatic failure secondary to Wilson's disease. J Clin Apher 2012; 27:282–286.
- Roberts EA, Schilsky ML; American Association for Study of Liver Diseases (AASLD). Diagnosis and treatment of Wilson disease: an update. Hepatology 2008; 47:2089–2111.
- Shah AB, Chernov I, Zhang HT, et al. Identification and analysis of mutations in the Wilson disease gene (ATP7B): population frequencies, genotype-phenotype correlation, and functional analyses. Am J Hum Genet 1997; 61:317–328.
- Maier-Dobersberger T, Ferenci P, Polli C, et al. Detection of the His1069Gln mutation in Wilson disease by rapid polymerase chain reaction. Ann Intern Med 1997; 127:21–26.
A 25-year-old woman presents to the emergency department with a 7-day history of fatigue and nausea. On presentation she denies having abdominal pain, headache, fever, chills, night sweats, vomiting, diarrhea, melena, hematochezia, or weight loss. She recalls changes in the colors of her eyes and darkening urine over the last few days. Her medical history before this is unremarkable. She takes no prescription, over-the-counter, or herbal medications. She works as a librarian and has no occupational toxic exposures. She is single and has one sister with no prior medical history. She denies recent travel, sick contacts, smoking, recreational drug use, or pets at home.
On physical examination, her vital signs are temperature 37.3°C (99.1°F), heart rate 90 beats per minute, blood pressure 125/80 mm Hg, respiration rate 14 per minute, and oxygen saturation 97% on room air. She has icteric sclera and her skin is jaundiced. Cardiac examination is normal. Lungs are clear to auscultation and percussion bilaterally. Her abdomen is soft with no visceromegaly, masses, or tenderness. Extremities are normal with no edema. She is alert and oriented, but she has mild asterixis of the outstretched hands. The neurologic examination is otherwise unremarkable.
The patient’s basic laboratory values are listed in Table 1. Shortly after admission, she develops changes in her mental status, remaining alert but becoming agitated and oriented to person only. In view of her symptoms and laboratory findings, acute liver failure is suspected.
ACUTE LIVER FAILURE
1. The diagnostic criteria for acute liver failure include all of the following except which one?
- Acute elevation of liver biochemical tests
- Presence of preexisting liver disease
- Coagulopathy, defined by an international normalized ratio (INR) of 1.5 or greater
- Encephalopathy
- Duration of symptoms less than 26 weeks
Acute liver failure is defined by acute onset of worsening liver tests, coagulopathy (INR ≥ 1.5), and encephalopathy in patients with no preexisting liver disease and with symptom duration of less than 26 weeks.1 With a few exceptions, a history of preexisting liver disease negates the diagnosis of acute liver failure. Our patient meets the diagnostic criteria for acute liver failure.
Immediate management
Once acute liver failure is identified or suspected, the next step is to transfer the patient to the intensive care unit for close monitoring of mental status. Serial neurologic evaluations permit early detection of cerebral edema, which is considered the most common cause of death in patients with acute liver failure. Additionally, close monitoring of electrolytes and plasma glucose is necessary since these patients are susceptible to electrolyte disturbances and hypoglycemia.
Patients with acute liver failure are at increased risk of infections and should be routinely screened by obtaining urine and blood cultures.
Gastrointestinal bleeding is not uncommon in patients with acute liver failure and is usually due to gastric stress ulceration. Prophylaxis with a histamine 2 receptor antagonist or proton pump inhibitor should be considered in order to prevent gastrointestinal bleeding.
Treatment with N-acetylcysteine is beneficial, not only in patients with acute liver failure due to acetaminophen overdose, but also in those with acute liver failure from other causes.
CASE CONTINUES:
TRANSFER TO THE INTENSIVE CARE UNIT
The patient, now diagnosed with acute liver failure, is transferred to the intensive care unit. Arterial blood gas measurement shows:
- pH 7.38 (reference range 7.35–7.45)
- Pco2 40 mm Hg (36–46)
- Po2 97 mm Hg (85–95)
- Hco3 22 mmol/L (22–26).
A chest radiograph is obtained and is clear. Computed tomography (CT) of the brain reveals no edema. Transcranial Doppler ultrasonography does not show any intracranial fluid collections.
Blood and urine cultures are negative. Her hemoglobin level remains stable, and she does not develop signs of bleeding. She is started on a proton pump inhibitor for stress ulcer prophylaxis and is empirically given intravenous N-acetylcysteine until the cause of acute liver failure can be determined.
CAUSES OF ACUTE LIVER FAILURE
2. Which of the following can cause acute liver failure?
- Acetaminophen overdose
- Viral hepatitis
- Autoimmune hepatitis
- Wilson disease
- Alcoholic hepatitis
Drug-induced liver injury is the most common cause of acute liver failure in the United States,2,3 and of all drugs, acetaminophen overdose is the number-one cause. In acetaminophen-induced liver injury, serum aminotransferase levels are usually elevated to more than 1,000 U/L, while serum bilirubin remains normal in the early stages. Antimicrobial agents, antiepileptic drugs, and herbal supplements have also been implicated in acute liver failure. Our patient has denied taking herbal supplements or medications, including over-the-counter ones.
Acute viral hepatitis can explain the patient’s condition. It is a common cause of acute liver failure in the United States.2 Hepatitis A and E are more common in developing countries. Other viruses such as cytomegalovirus, Epstein-Barr virus, herpes simplex virus type 1 and 2, and varicella zoster virus can also cause acute liver failure. Serum aminotransferase levels may exceed 1,000 U/L in patients with viral hepatitis.
Autoimmune hepatitis is a rare cause of acute liver failure, but it should be considered in the differential diagnosis, particularly in middle-aged women with autoimmune disorders such as hypothyroidism. Autoimmune hepatitis can cause marked elevation in aminotransferase levels (> 1,000 U/L).
Wilson disease is an autosomal-recessive disease in which there is excessive accumulation of copper in the liver and other organs because of an inherited defect in the biliary excretion of copper. Wilson disease can cause acute liver failure and should be excluded in any patient, particularly if under age 40 with acute onset of unexplained hepatic, neurologic, or psychiatric disease.
Alcoholic hepatitis usually occurs in patients with a long-standing history of heavy alcohol use. As a result, most patients with alcoholic hepatitis have manifestations of chronic liver disease due to alcohol use. Therefore, by definition, it is not a cause of acute liver failure. Additionally, in patients with alcoholic hepatitis, the aspartate aminotransferase (AST) level is elevated but less than 300 IU/mL, and the ratio of AST to alanine aminotransferase (ALT) is usually more than 2.
CASE CONTINUES: FURTHER TESTING
The results of our patient’s serologic tests are shown in Table 2. Other test results:
- Autoimmune markers including antinuclear antibodies, antimitochondrial antibodies, antismooth muscle antibodies, and liver and kidney microsomal antibodies are negative; her immunoglobulin G (IgG) level is normal
- Serum ceruloplasmin 25 mg/dL (normal 21–45)
- Free serum copper 120 µg/dL (normal 8–12)
- Abdominal ultrasonography is unremarkable, with normal liver parenchyma and no intrahepatic or extrahepatic biliary dilatation
- Doppler ultrasonography of the liver shows patent blood vessels.
3. Based on the new data, which of the following statements is correct?
- Hepatitis B is the cause of acute liver failure in this patient
- Herpetic hepatitis cannot be excluded on the basis of the available data
- Wilson disease is most likely the diagnosis, given her elevated free serum copper
- A normal serum ceruloplasmin level is not sufficient to rule out acute liver failure secondary to Wilson disease
Hepatitis B surface antigen and hepatitis B core antibodies were negative in our patient, excluding hepatitis B virus infection. The positive hepatitis B surface antibody indicates prior immunization.
Herpetic hepatitis is an uncommon but important cause of acute liver failure because the mortality rate is high if the patient is not treated early with acyclovir. Fever, elevated aminotransferases, and leukopenia are common with herpetic hepatitis. Fewer than 50% of patients with herpetic hepatitis have vesicular rash.4,5 The value of antibody serologic testing is limited due to high rates of false-positive and false-negative results. The gold standard diagnostic tests are viral load (detection of viral RNA by polymerase chain reaction), viral staining on liver biopsy, or both. In our patient, herpes simplex virus polymerase chain reaction testing was negative, which makes herpetic hepatitis unlikely.
Wilson disease is a genetic condition in which the ability to excrete copper in the bile is impaired, resulting in accumulation of copper in the hepatocytes. Subsequently, copper is released into the bloodstream and eventually into the urine.
However, copper excretion into the bile is impaired in patients with acute liver failure regardless of the etiology. Therefore, elevated free serum copper and 24-hour urine copper levels are not specific for the diagnosis of acute liver failure secondary to Wilson disease. Moreover, Kayser-Fleischer rings, which represent copper deposition in the limbus of the cornea, may not be apparent in the early stages of Wilson disease.
Since it is challenging to diagnose Wilson disease in the context of acute liver failure, Korman et al6 compared patients with acute liver failure secondary to Wilson disease with patients with acute liver failure secondary to other conditions. They found that alkaline phosphatase levels are frequently decreased in patients with acute liver failure secondary to Wilson disease,6 and that a ratio of alkaline phosphatase to total bilirubin of less than 4 is 94% sensitive and 96% specific for the diagnosis.6
Hemolysis is common in acute liver failure due to Wilson disease. This leads to disproportionate elevation of AST compared with ALT, since AST is present in red blood cells. Consequently, the ratio of AST to ALT is usually greater than 2.2, which provides a sensitivity of 94% and a specificity of 86% for the diagnosis.6 These two ratios together provide 100% sensitivity and 100% specificity for the diagnosis of Wilson disease in the context of acute liver failure.6
Ceruloplasmin. Patients with Wilson disease typically have a low ceruloplasmin level. However, because it is an acute-phase reaction protein, ceruloplasmin can be normal or elevated in patients with acute liver failure from Wilson disease.6 Therefore, a normal ceruloplasmin level is not sufficient to rule out acute liver failure secondary to Wilson disease.
CASE CONTINUES: A DEFINITIVE DIAGNOSIS
Our patient undergoes further testing, which reveals the following:
- Her 24-hour urinary excretion of copper is 150 µg (reference value < 30)
- Slit-lamp examination is normal and shows no evidence of Kayser-Fleischer rings
- Her ratio of alkaline phosphatase to total bilirubin is 0.77 based on her initial laboratory results (Table 1)
- Her AST-ALT ratio is 3.4.
The diagnosis in our patient is acute liver failure secondary to Wilson disease.
4. What is the most appropriate next step?
- Liver biopsy
- d-penicillamine by mouth
- Trientine by mouth
- Liver transplant
- Plasmapheresis
Liver biopsy. Accumulation of copper in the liver parenchyma in patients with Wilson disease is sporadic. Therefore, qualitative copper staining on liver biopsy can be falsely negative. Quantitative copper measurement in liver tissue is the gold standard for the diagnosis of Wilson disease. However, the test is time-consuming and is not rapidly available in the context of acute liver failure.
Chelating agents such as d-pencillamine and trientine are used to treat the chronic manifestations of Wilson disease but are not useful for acute liver failure secondary to Wilson disease.
Acute liver failure secondary to Wilson disease is life-threatening, and liver transplant is considered the only definitive life-saving therapy.
Therapeutic plasmapheresis has been reported to be a successful adjunctive therapy to bridge patients with acute liver failure secondary to Wilson disease to transplant.7 However, liver transplant is still the only definitive treatment.
CASE CONTINUES: THE PATIENT’S SISTER SEEKS CARE
The patient undergoes liver transplantation, with no perioperative or postoperative complications.
The patient’s 18-year-old sister is now seeking medical attention in the outpatient clinic, concerned that she may have Wilson disease. She is otherwise healthy and denies any symptoms or complaints.
5. What is the next step for the patient’s sister?
- Reassurance
- Prophylaxis with trientine
- Check liver enzyme levels, serum ceruloplasmin level, and urine copper, and order a slit-lamp examination
- Genetic testing
Wilson disease can be asymptomatic in its early stages and may be diagnosed incidentally during routine blood tests that reveal abnormal liver enzyme levels. All patients with a confirmed family history of Wilson disease should be screened even if they are asymptomatic. The diagnosis of Wilson disease should be established in first-degree relatives before specific treatment for the relatives is prescribed.
The first step in screening a first-degree relative for Wilson disease is to check liver enzyme levels (specifically aminotransferases, alkaline phosphatase, and bilirubin), serum ceruloplasmin level, and 24-hour urine copper, and order an ophthalmologic slit-lamp examination. If any of these tests is abnormal, liver biopsy should be performed for histopathologic evaluation and quantitative copper measurement. Kayser-Fleischer rings are seen in only 50% of patients with Wilson disease and hepatic involvement, but they are pathognomic. Guidelines8 for screening first-degree relatives of Wilson disease patients are shown in Figure 1.
Genetic analysis. ATP7B, the Wilson disease gene, is located on chromosome 13. At least 300 mutations of the gene have been described,2 and the most common mutation is present in only 15% to 30% of the Wilson disease population.8–10 Routine molecular testing of the ATP7B
CASE CONTINUES: WORKUP OF THE PATIENT’S SISTER
The patient’s sister has no symptoms and her physical examination is normal. Slit-lamp examination reveals no evidence of Kayser-Fleischer rings. Her laboratory values, including complete blood counts, complete metabolic panel, and INR, are within normal ranges. Other test results, however, are abnormal:
- Free serum copper level 27 µg/dL (normal 8–12)
- Serum ceruloplasmin 9.0 mg/dL (normal 20–50)
- 24-hour urinary copper excretion 135 µg (normal < 30).
She undergoes liver biopsy for quantitative copper measurement, and the result is very high at 1,118 µg/g dry weight (reference range 10–35). The diagnosis of Wilson disease is established.
TREATING CHRONIC WILSON DISEASE
6. Which of the following is not an appropriate next step for the patient’s sister?
- Tetrathiomolybdate
- d-penicillamine
- Trientine
- Zinc salts
- Prednisone
The goal of medical treatment of chronic Wilson disease is to improve symptoms and prevent progression of the disease.
Chelating agents and zinc salts are the most commonly used medicines in the management of Wilson disease. Chelating agents remove copper from tissue, whereas zinc blocks the intestinal absorption of copper and stimulates the synthesis of endogenous chelators such as metallothioneins. Tetrathiomolybdate is an alternative agent developed to interfere with the distribution of excess body copper to susceptible target sites by reducing free serum copper (Table 3). There are no data to support the use of prednisone in the treatment of Wilson disease.
During treatment with chelating agents, 24-hour urinary excretion of copper is routinely monitored to determine the efficacy of therapy and adherence to treatment. Once de-coppering is achieved, as evidenced by a normalization of 24-hour urine copper excretion, the chelating agent can be switched to zinc salts to prevent intestinal absorption of copper.
Clinical and biochemical stabilization is achieved typically within 2 to 6 months of the initial treatment with chelating agents.8 Organ meats, nuts, shellfish, and chocolate are rich in copper and should be avoided.
The patient’s sister is started on trientine 250 mg orally three times daily on an empty stomach at least 1 hour before meals. Treatment is monitored by following 24-hour urine copper measurement. A 24-hour urine copper measurement at 3 months after starting treatment has increased from 54 at baseline to 350 µg, which indicates that the copper is being removed from tissues. The plan is for early substitution of zinc for long-term maintenance once de-coppering is completed.
KEY POINTS
- Acute liver failure is severe acute liver injury characterized by coagulopathy (INR ≥ 1.5) and encephalopathy in a patient with no preexisting liver disease and with duration of symptoms less than 26 weeks.
- Acute liver failure secondary to Wilson disease is uncommon but should be excluded, particularly in young patients.
- The diagnosis of Wilson disease in the setting of acute liver failure is challenging because the serum ceruloplasmin level may be normal in acute liver failure secondary to Wilson disease, and free serum copper and 24-hour urine copper are usually elevated in all acute liver failure patients regardless of the etiology.
- A ratio of alkaline phosphatase to total bilirubin of less than 4 plus an AST-ALT ratio greater than 2.2 in a patient with acute liver failure should be regarded as Wilson disease until proven otherwise (Figure 2).
- Acute liver failure secondary to Wilson disease is usually fatal, and emergency liver transplant is a life-saving procedure.
- Screening of first-degree relatives of Wilson disease patients should include a history and physical examination, liver enzyme tests, complete blood cell count, serum ceruloplasmin level, serum free copper level, slit-lamp examination of the eyes, and 24-hour urinary copper measurement. Genetic tests are supplementary for screening but are not routinely available.
A 25-year-old woman presents to the emergency department with a 7-day history of fatigue and nausea. On presentation she denies having abdominal pain, headache, fever, chills, night sweats, vomiting, diarrhea, melena, hematochezia, or weight loss. She recalls changes in the colors of her eyes and darkening urine over the last few days. Her medical history before this is unremarkable. She takes no prescription, over-the-counter, or herbal medications. She works as a librarian and has no occupational toxic exposures. She is single and has one sister with no prior medical history. She denies recent travel, sick contacts, smoking, recreational drug use, or pets at home.
On physical examination, her vital signs are temperature 37.3°C (99.1°F), heart rate 90 beats per minute, blood pressure 125/80 mm Hg, respiration rate 14 per minute, and oxygen saturation 97% on room air. She has icteric sclera and her skin is jaundiced. Cardiac examination is normal. Lungs are clear to auscultation and percussion bilaterally. Her abdomen is soft with no visceromegaly, masses, or tenderness. Extremities are normal with no edema. She is alert and oriented, but she has mild asterixis of the outstretched hands. The neurologic examination is otherwise unremarkable.
The patient’s basic laboratory values are listed in Table 1. Shortly after admission, she develops changes in her mental status, remaining alert but becoming agitated and oriented to person only. In view of her symptoms and laboratory findings, acute liver failure is suspected.
ACUTE LIVER FAILURE
1. The diagnostic criteria for acute liver failure include all of the following except which one?
- Acute elevation of liver biochemical tests
- Presence of preexisting liver disease
- Coagulopathy, defined by an international normalized ratio (INR) of 1.5 or greater
- Encephalopathy
- Duration of symptoms less than 26 weeks
Acute liver failure is defined by acute onset of worsening liver tests, coagulopathy (INR ≥ 1.5), and encephalopathy in patients with no preexisting liver disease and with symptom duration of less than 26 weeks.1 With a few exceptions, a history of preexisting liver disease negates the diagnosis of acute liver failure. Our patient meets the diagnostic criteria for acute liver failure.
Immediate management
Once acute liver failure is identified or suspected, the next step is to transfer the patient to the intensive care unit for close monitoring of mental status. Serial neurologic evaluations permit early detection of cerebral edema, which is considered the most common cause of death in patients with acute liver failure. Additionally, close monitoring of electrolytes and plasma glucose is necessary since these patients are susceptible to electrolyte disturbances and hypoglycemia.
Patients with acute liver failure are at increased risk of infections and should be routinely screened by obtaining urine and blood cultures.
Gastrointestinal bleeding is not uncommon in patients with acute liver failure and is usually due to gastric stress ulceration. Prophylaxis with a histamine 2 receptor antagonist or proton pump inhibitor should be considered in order to prevent gastrointestinal bleeding.
Treatment with N-acetylcysteine is beneficial, not only in patients with acute liver failure due to acetaminophen overdose, but also in those with acute liver failure from other causes.
CASE CONTINUES:
TRANSFER TO THE INTENSIVE CARE UNIT
The patient, now diagnosed with acute liver failure, is transferred to the intensive care unit. Arterial blood gas measurement shows:
- pH 7.38 (reference range 7.35–7.45)
- Pco2 40 mm Hg (36–46)
- Po2 97 mm Hg (85–95)
- Hco3 22 mmol/L (22–26).
A chest radiograph is obtained and is clear. Computed tomography (CT) of the brain reveals no edema. Transcranial Doppler ultrasonography does not show any intracranial fluid collections.
Blood and urine cultures are negative. Her hemoglobin level remains stable, and she does not develop signs of bleeding. She is started on a proton pump inhibitor for stress ulcer prophylaxis and is empirically given intravenous N-acetylcysteine until the cause of acute liver failure can be determined.
CAUSES OF ACUTE LIVER FAILURE
2. Which of the following can cause acute liver failure?
- Acetaminophen overdose
- Viral hepatitis
- Autoimmune hepatitis
- Wilson disease
- Alcoholic hepatitis
Drug-induced liver injury is the most common cause of acute liver failure in the United States,2,3 and of all drugs, acetaminophen overdose is the number-one cause. In acetaminophen-induced liver injury, serum aminotransferase levels are usually elevated to more than 1,000 U/L, while serum bilirubin remains normal in the early stages. Antimicrobial agents, antiepileptic drugs, and herbal supplements have also been implicated in acute liver failure. Our patient has denied taking herbal supplements or medications, including over-the-counter ones.
Acute viral hepatitis can explain the patient’s condition. It is a common cause of acute liver failure in the United States.2 Hepatitis A and E are more common in developing countries. Other viruses such as cytomegalovirus, Epstein-Barr virus, herpes simplex virus type 1 and 2, and varicella zoster virus can also cause acute liver failure. Serum aminotransferase levels may exceed 1,000 U/L in patients with viral hepatitis.
Autoimmune hepatitis is a rare cause of acute liver failure, but it should be considered in the differential diagnosis, particularly in middle-aged women with autoimmune disorders such as hypothyroidism. Autoimmune hepatitis can cause marked elevation in aminotransferase levels (> 1,000 U/L).
Wilson disease is an autosomal-recessive disease in which there is excessive accumulation of copper in the liver and other organs because of an inherited defect in the biliary excretion of copper. Wilson disease can cause acute liver failure and should be excluded in any patient, particularly if under age 40 with acute onset of unexplained hepatic, neurologic, or psychiatric disease.
Alcoholic hepatitis usually occurs in patients with a long-standing history of heavy alcohol use. As a result, most patients with alcoholic hepatitis have manifestations of chronic liver disease due to alcohol use. Therefore, by definition, it is not a cause of acute liver failure. Additionally, in patients with alcoholic hepatitis, the aspartate aminotransferase (AST) level is elevated but less than 300 IU/mL, and the ratio of AST to alanine aminotransferase (ALT) is usually more than 2.
CASE CONTINUES: FURTHER TESTING
The results of our patient’s serologic tests are shown in Table 2. Other test results:
- Autoimmune markers including antinuclear antibodies, antimitochondrial antibodies, antismooth muscle antibodies, and liver and kidney microsomal antibodies are negative; her immunoglobulin G (IgG) level is normal
- Serum ceruloplasmin 25 mg/dL (normal 21–45)
- Free serum copper 120 µg/dL (normal 8–12)
- Abdominal ultrasonography is unremarkable, with normal liver parenchyma and no intrahepatic or extrahepatic biliary dilatation
- Doppler ultrasonography of the liver shows patent blood vessels.
3. Based on the new data, which of the following statements is correct?
- Hepatitis B is the cause of acute liver failure in this patient
- Herpetic hepatitis cannot be excluded on the basis of the available data
- Wilson disease is most likely the diagnosis, given her elevated free serum copper
- A normal serum ceruloplasmin level is not sufficient to rule out acute liver failure secondary to Wilson disease
Hepatitis B surface antigen and hepatitis B core antibodies were negative in our patient, excluding hepatitis B virus infection. The positive hepatitis B surface antibody indicates prior immunization.
Herpetic hepatitis is an uncommon but important cause of acute liver failure because the mortality rate is high if the patient is not treated early with acyclovir. Fever, elevated aminotransferases, and leukopenia are common with herpetic hepatitis. Fewer than 50% of patients with herpetic hepatitis have vesicular rash.4,5 The value of antibody serologic testing is limited due to high rates of false-positive and false-negative results. The gold standard diagnostic tests are viral load (detection of viral RNA by polymerase chain reaction), viral staining on liver biopsy, or both. In our patient, herpes simplex virus polymerase chain reaction testing was negative, which makes herpetic hepatitis unlikely.
Wilson disease is a genetic condition in which the ability to excrete copper in the bile is impaired, resulting in accumulation of copper in the hepatocytes. Subsequently, copper is released into the bloodstream and eventually into the urine.
However, copper excretion into the bile is impaired in patients with acute liver failure regardless of the etiology. Therefore, elevated free serum copper and 24-hour urine copper levels are not specific for the diagnosis of acute liver failure secondary to Wilson disease. Moreover, Kayser-Fleischer rings, which represent copper deposition in the limbus of the cornea, may not be apparent in the early stages of Wilson disease.
Since it is challenging to diagnose Wilson disease in the context of acute liver failure, Korman et al6 compared patients with acute liver failure secondary to Wilson disease with patients with acute liver failure secondary to other conditions. They found that alkaline phosphatase levels are frequently decreased in patients with acute liver failure secondary to Wilson disease,6 and that a ratio of alkaline phosphatase to total bilirubin of less than 4 is 94% sensitive and 96% specific for the diagnosis.6
Hemolysis is common in acute liver failure due to Wilson disease. This leads to disproportionate elevation of AST compared with ALT, since AST is present in red blood cells. Consequently, the ratio of AST to ALT is usually greater than 2.2, which provides a sensitivity of 94% and a specificity of 86% for the diagnosis.6 These two ratios together provide 100% sensitivity and 100% specificity for the diagnosis of Wilson disease in the context of acute liver failure.6
Ceruloplasmin. Patients with Wilson disease typically have a low ceruloplasmin level. However, because it is an acute-phase reaction protein, ceruloplasmin can be normal or elevated in patients with acute liver failure from Wilson disease.6 Therefore, a normal ceruloplasmin level is not sufficient to rule out acute liver failure secondary to Wilson disease.
CASE CONTINUES: A DEFINITIVE DIAGNOSIS
Our patient undergoes further testing, which reveals the following:
- Her 24-hour urinary excretion of copper is 150 µg (reference value < 30)
- Slit-lamp examination is normal and shows no evidence of Kayser-Fleischer rings
- Her ratio of alkaline phosphatase to total bilirubin is 0.77 based on her initial laboratory results (Table 1)
- Her AST-ALT ratio is 3.4.
The diagnosis in our patient is acute liver failure secondary to Wilson disease.
4. What is the most appropriate next step?
- Liver biopsy
- d-penicillamine by mouth
- Trientine by mouth
- Liver transplant
- Plasmapheresis
Liver biopsy. Accumulation of copper in the liver parenchyma in patients with Wilson disease is sporadic. Therefore, qualitative copper staining on liver biopsy can be falsely negative. Quantitative copper measurement in liver tissue is the gold standard for the diagnosis of Wilson disease. However, the test is time-consuming and is not rapidly available in the context of acute liver failure.
Chelating agents such as d-pencillamine and trientine are used to treat the chronic manifestations of Wilson disease but are not useful for acute liver failure secondary to Wilson disease.
Acute liver failure secondary to Wilson disease is life-threatening, and liver transplant is considered the only definitive life-saving therapy.
Therapeutic plasmapheresis has been reported to be a successful adjunctive therapy to bridge patients with acute liver failure secondary to Wilson disease to transplant.7 However, liver transplant is still the only definitive treatment.
CASE CONTINUES: THE PATIENT’S SISTER SEEKS CARE
The patient undergoes liver transplantation, with no perioperative or postoperative complications.
The patient’s 18-year-old sister is now seeking medical attention in the outpatient clinic, concerned that she may have Wilson disease. She is otherwise healthy and denies any symptoms or complaints.
5. What is the next step for the patient’s sister?
- Reassurance
- Prophylaxis with trientine
- Check liver enzyme levels, serum ceruloplasmin level, and urine copper, and order a slit-lamp examination
- Genetic testing
Wilson disease can be asymptomatic in its early stages and may be diagnosed incidentally during routine blood tests that reveal abnormal liver enzyme levels. All patients with a confirmed family history of Wilson disease should be screened even if they are asymptomatic. The diagnosis of Wilson disease should be established in first-degree relatives before specific treatment for the relatives is prescribed.
The first step in screening a first-degree relative for Wilson disease is to check liver enzyme levels (specifically aminotransferases, alkaline phosphatase, and bilirubin), serum ceruloplasmin level, and 24-hour urine copper, and order an ophthalmologic slit-lamp examination. If any of these tests is abnormal, liver biopsy should be performed for histopathologic evaluation and quantitative copper measurement. Kayser-Fleischer rings are seen in only 50% of patients with Wilson disease and hepatic involvement, but they are pathognomic. Guidelines8 for screening first-degree relatives of Wilson disease patients are shown in Figure 1.
Genetic analysis. ATP7B, the Wilson disease gene, is located on chromosome 13. At least 300 mutations of the gene have been described,2 and the most common mutation is present in only 15% to 30% of the Wilson disease population.8–10 Routine molecular testing of the ATP7B
CASE CONTINUES: WORKUP OF THE PATIENT’S SISTER
The patient’s sister has no symptoms and her physical examination is normal. Slit-lamp examination reveals no evidence of Kayser-Fleischer rings. Her laboratory values, including complete blood counts, complete metabolic panel, and INR, are within normal ranges. Other test results, however, are abnormal:
- Free serum copper level 27 µg/dL (normal 8–12)
- Serum ceruloplasmin 9.0 mg/dL (normal 20–50)
- 24-hour urinary copper excretion 135 µg (normal < 30).
She undergoes liver biopsy for quantitative copper measurement, and the result is very high at 1,118 µg/g dry weight (reference range 10–35). The diagnosis of Wilson disease is established.
TREATING CHRONIC WILSON DISEASE
6. Which of the following is not an appropriate next step for the patient’s sister?
- Tetrathiomolybdate
- d-penicillamine
- Trientine
- Zinc salts
- Prednisone
The goal of medical treatment of chronic Wilson disease is to improve symptoms and prevent progression of the disease.
Chelating agents and zinc salts are the most commonly used medicines in the management of Wilson disease. Chelating agents remove copper from tissue, whereas zinc blocks the intestinal absorption of copper and stimulates the synthesis of endogenous chelators such as metallothioneins. Tetrathiomolybdate is an alternative agent developed to interfere with the distribution of excess body copper to susceptible target sites by reducing free serum copper (Table 3). There are no data to support the use of prednisone in the treatment of Wilson disease.
During treatment with chelating agents, 24-hour urinary excretion of copper is routinely monitored to determine the efficacy of therapy and adherence to treatment. Once de-coppering is achieved, as evidenced by a normalization of 24-hour urine copper excretion, the chelating agent can be switched to zinc salts to prevent intestinal absorption of copper.
Clinical and biochemical stabilization is achieved typically within 2 to 6 months of the initial treatment with chelating agents.8 Organ meats, nuts, shellfish, and chocolate are rich in copper and should be avoided.
The patient’s sister is started on trientine 250 mg orally three times daily on an empty stomach at least 1 hour before meals. Treatment is monitored by following 24-hour urine copper measurement. A 24-hour urine copper measurement at 3 months after starting treatment has increased from 54 at baseline to 350 µg, which indicates that the copper is being removed from tissues. The plan is for early substitution of zinc for long-term maintenance once de-coppering is completed.
KEY POINTS
- Acute liver failure is severe acute liver injury characterized by coagulopathy (INR ≥ 1.5) and encephalopathy in a patient with no preexisting liver disease and with duration of symptoms less than 26 weeks.
- Acute liver failure secondary to Wilson disease is uncommon but should be excluded, particularly in young patients.
- The diagnosis of Wilson disease in the setting of acute liver failure is challenging because the serum ceruloplasmin level may be normal in acute liver failure secondary to Wilson disease, and free serum copper and 24-hour urine copper are usually elevated in all acute liver failure patients regardless of the etiology.
- A ratio of alkaline phosphatase to total bilirubin of less than 4 plus an AST-ALT ratio greater than 2.2 in a patient with acute liver failure should be regarded as Wilson disease until proven otherwise (Figure 2).
- Acute liver failure secondary to Wilson disease is usually fatal, and emergency liver transplant is a life-saving procedure.
- Screening of first-degree relatives of Wilson disease patients should include a history and physical examination, liver enzyme tests, complete blood cell count, serum ceruloplasmin level, serum free copper level, slit-lamp examination of the eyes, and 24-hour urinary copper measurement. Genetic tests are supplementary for screening but are not routinely available.
- Lee WM, Larson AM, Stravitz T. AASLD Position Paper: The management of acute liver failure: update 2011. www.aasld.org/sites/default/files/guideline_documents/alfenhanced.pdf. Accessed December 9, 2015.
- Bernal W, Auzinger G, Dhawan A, Wendon J. Acute liver failure. Lancet 2010; 376:190–201.
- Larson AM, Polson J, Fontana RJ, et al; Acute Liver Failure Study Group. Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology 2005; 42:1364–1372.
- Hanouneh IA, Khoriaty R, Zein NN. A 35-year-old Asian man with jaundice and markedly high aminotransferase levels. Cleve Clin J Med 2009; 76:449–456.
- Norvell JP, Blei AT, Jovanovic BD, Levitsky J. Herpes simplex virus hepatitis: an analysis of the published literature and institutional cases. Liver Transpl 2007; 13:1428–1434.
- Korman JD, Volenberg I, Balko J, et al; Pediatric and Adult Acute Liver Failure Study Groups. Screening for Wilson disease in acute liver failure: a comparison of currently available diagnostic tests. Hepatology 2008; 48:1167–1174.
- Morgan SM, Zantek ND. Therapeutic plasma exchange for fulminant hepatic failure secondary to Wilson's disease. J Clin Apher 2012; 27:282–286.
- Roberts EA, Schilsky ML; American Association for Study of Liver Diseases (AASLD). Diagnosis and treatment of Wilson disease: an update. Hepatology 2008; 47:2089–2111.
- Shah AB, Chernov I, Zhang HT, et al. Identification and analysis of mutations in the Wilson disease gene (ATP7B): population frequencies, genotype-phenotype correlation, and functional analyses. Am J Hum Genet 1997; 61:317–328.
- Maier-Dobersberger T, Ferenci P, Polli C, et al. Detection of the His1069Gln mutation in Wilson disease by rapid polymerase chain reaction. Ann Intern Med 1997; 127:21–26.
- Lee WM, Larson AM, Stravitz T. AASLD Position Paper: The management of acute liver failure: update 2011. www.aasld.org/sites/default/files/guideline_documents/alfenhanced.pdf. Accessed December 9, 2015.
- Bernal W, Auzinger G, Dhawan A, Wendon J. Acute liver failure. Lancet 2010; 376:190–201.
- Larson AM, Polson J, Fontana RJ, et al; Acute Liver Failure Study Group. Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology 2005; 42:1364–1372.
- Hanouneh IA, Khoriaty R, Zein NN. A 35-year-old Asian man with jaundice and markedly high aminotransferase levels. Cleve Clin J Med 2009; 76:449–456.
- Norvell JP, Blei AT, Jovanovic BD, Levitsky J. Herpes simplex virus hepatitis: an analysis of the published literature and institutional cases. Liver Transpl 2007; 13:1428–1434.
- Korman JD, Volenberg I, Balko J, et al; Pediatric and Adult Acute Liver Failure Study Groups. Screening for Wilson disease in acute liver failure: a comparison of currently available diagnostic tests. Hepatology 2008; 48:1167–1174.
- Morgan SM, Zantek ND. Therapeutic plasma exchange for fulminant hepatic failure secondary to Wilson's disease. J Clin Apher 2012; 27:282–286.
- Roberts EA, Schilsky ML; American Association for Study of Liver Diseases (AASLD). Diagnosis and treatment of Wilson disease: an update. Hepatology 2008; 47:2089–2111.
- Shah AB, Chernov I, Zhang HT, et al. Identification and analysis of mutations in the Wilson disease gene (ATP7B): population frequencies, genotype-phenotype correlation, and functional analyses. Am J Hum Genet 1997; 61:317–328.
- Maier-Dobersberger T, Ferenci P, Polli C, et al. Detection of the His1069Gln mutation in Wilson disease by rapid polymerase chain reaction. Ann Intern Med 1997; 127:21–26.
