User login
Seeking medical care abroad: A challenge to empathy
On an otherwise pleasant evening during the first week of July 2016, a businessman who was a citizen of the United Arab Emirates visiting Cleveland for medical treatment was falsely accused of links to a terror organization. Officers stormed his hotel with assault rifles and handcuffed and arrested him—all this, apparently, because the man was dressed in traditional Emirati clothing.
This case highlights a level of complexity in providing medical care to foreigners far beyond language interpreting services and outside the borders of the institution where medical care is provided. In the current issue of the Journal, Cawcutt and Wilson1 review their experiences in the care of international patients and the unique challenges associated with it.
FROM THE TEMPLE OF AESCULAPIUS TO CLEVELAND CLINIC
In 2015, patients from more than 100 countries traveled to Cleveland seeking care at Cleveland Clinic. But medical travel was part of the practice of medicine long before major US hospitals became destinations for international patients, and it has been refined over the years.
Ancient cultures had a thriving tradition of patients traveling long distances for the best and most advanced medical treatment.2–4 In ancient Greece, people from all around the Mediterranean came to the city of Epidaurus to be cured in its famous temple of Aesculapius, built as a medical center.
Similarly, early Islamic cultures established a healthcare system that catered to foreigners. A noted example is the Mansuri hospital in Cairo, built in 1248 ce and considered the most advanced hospital of its time. Accommodating nearly 8,000 patients, the Mansuri hospital became a healthcare destination for foreigners regardless of race or religion.2–4
Europe also had a great tradition of providing medical care to foreign patients. Between the 15th and 17th centuries, belief in the healing power of mineral water led to the establishment of spas and the rise of spa towns, particularly in the south of France near mineral springs. The poor sanitary conditions of Europe at the time may have prompted the interest in the healing effect of mineral spas, but wealthy individuals from all over the world traveled to these destinations, creating local prosperity due to medical tourism.2–4
The city of Bath, in England, is a great example. In the 1720s, Bath was a popular destination for those traveling for healthcare. It became the first city in England to build a covered sewage system, ahead of London by several years. It also had paved roads, lights, hotels, and restaurants in much greater numbers than other cities in England, a likely result of prosperity associated with medical tourism.
ALL PATIENTS WANT TO BE TREATED WITH RESPECT AND KINDNESS
While medical knowledge and health delivery models have changed over the years, caring for foreign patients is perhaps as old as medicine itself. The central focus of restoring health is certainly not unique to international patients, but understanding their unique needs is important in order to achieve the best outcomes, something that Cawcutt and Wilson highlight well.1
A number of studies have addressed the question of what patients really want. Responses were surprisingly consistent: they want to be treated with respect and kindness.5,6 In other words, they want empathy, and this is true of all patients regardless of ethnicity or background. Empathy is a tremendous therapeutic force and can narrow what may look like an unbridgeable gap between patient and physician.7,8
EMPATHY REQUIRES EFFECTIVE COMMUNICATION
Empathy, though sometimes innate, requires effective communication and shared experiences. Neither of these two requirements is easily achievable in the care of foreign patients.
Communication is hampered by language barriers, although it can be enhanced significantly by language translating services and the work of certified medical interpreters. These often-invisible heroes should be recognized as essential members of the medical team. Their work requires cultural sensitivity and formal training to avoid miscommunication and medical errors. Codes of ethics for medical interpreters include confidentiality, accuracy in conveying the content and spirit of the message, freedom from personal biases, cultural training, and professional boundaries.9
TOWARD CULTURAL COMPETENCY
Lack of shared experiences between the foreign patient and care provider is an even greater obstacle to overcome in eliminating any empathy deficit. Shared experiences, whether cultural, religious, or social, help us to see the world through the eyes of the patient.
International patients may differ from us in background, ethnicity, religion, dress, expectations, and other areas. Cultural and religious backgrounds often dictate certain behaviors in the event of critical illness or death. Even in routine and less acute medical care, the background of a foreign patient may lead to logistical quandaries such as the need for same-sex caregivers or a private room.
A paradox currently exists in our efforts to meet patients’ need and desire for empathy. While culturally empathic care is necessary to achieve the best medical outcomes, this topic is not yet part of the curriculum for physicians or other healthcare providers in training. A culturally sensitive institution has many business advantages.10 Thorough and focused cultural training of medical staff is essential. Shared experiences can potentially be fashioned through a well-designed cultural competency training program to enhance empathy for foreign patients.
A SERVICE-ORIENTED APPROACH
Besides cultural competency and language training, a service-oriented approach to accommodate the needs of medical travelers and their family members is of paramount importance. Many of the complaints and burdens of medical visitors concern services that are not medical in nature, such as daily living necessities. Transportation, religious services, banking, extended-stay facilities, cell phone service, legal services, shopping, dining, and entertainment are among many other living needs for those receiving medical care abroad. These services are inconsistently provided throughout medical institutions in the United States, which provide care to thousands of international patients annually.
Unique challenges of providing medical care to international patients have direct effects on medical outcomes. A population-based cohort study of US-born and foreign-born adults with lung or colorectal cancer suggested disparities in quality and type of care.11 Foreign-born patients reported lower-quality care and were less likely to receive complex cancer treatments recommended by clinical guidelines. The authors proposed that quality of care and outcomes may be improved with greater emphasis on coordination of care and improving communication. Similar findings were reported in foreign-born patients with breast cancer.12
‘WHAT WOULD YOU THINK TO BE USED THUS?’
Four hundred years ago, in the play Sir Thomas More (a collaboration between several Elizabethan playwrights),13 the title character confronts a mob of anti-immigrant rioters, and in a speech believed to have been written by William Shakespeare (Act 2, Scene 4), asks them to imagine themselves banished to a foreign country and subjected to hostility such as they were meting out:
To be used thus?”
Empathy for foreigners seeking medical care is not merely an act of kindness; rather, it is a central piece of healing. Medical institutions interested in providing healthcare to this unique group of patients should take these principles into account and carefully examine their ability to deliver compassionate care collectively to local and foreign-born patients alike.
- Cawcutt KA, Wilson JW. The benefits and challenges of caring for international patients. Cleve Clin J Med 2016; 83:794–800.
- Health-Tourism.com. The history of medical tourism. Health-Tourism.com. www.health-tourism.com/medical-tourism/history/. Accessed September 21, 2016.
- Chen LH, Hochberg NS, Magill AJ. The pre-travel consultation. US Centers for Disease Control and Prevention. wwwnc.cdc.gov/travel/yellowbook/2016/the-pre-travel-consultation/the-pre-travel-consultation. Accessed September 21, 2016.
- Rogers K. Medical tourism. Encyclopedia Britannica. www.britannica.com/topic/medical-tourism. Accessed September 21, 2016.
- Detsky AS. What do patients really want from healthcare? JAMA 2011; 306:2500–2501.
- Shaywitz D. What do patients really want from healthcare? Forbes Dec 24, 2011. www.forbes.com/sites/davidshaywitz/2011/12/24/what-do-patients-really-want-from-health-care/print/. Accessed September 21, 2016.
- Lee TH. How to spread empathy in healthcare. Harvard Business Review July 17, 2014.
- Friedman R. Understanding empathy: can you feel my pain? New York Times April 24, 2007.
- National Council on Interpreting in Health Care. A national code of ethics for interpreters in healthcare. July 2004. www.ncihc.org/assets/documents/publications/NCIHC%20National%20Code%20of%20Ethics.pdf. Accessed September 21, 2016.
- Minguet L. Creating a culturally sensitive corporation. Harvard Business Review, September 2014.
- Nielsen SS, He Y, Ayanian JZ, Gomez SL, Khan KL, West DW, et al. Quality of cancer care among foreign-born patients with lung or colorectal cancer. Cancer 2010; 116:5497–5506.
- Kouri EM, He Y, Winer EP, Keating NL. Influence of birthplace on breast cancer diagnosis and treatment for Hispanic women. Breast Cancer Res Treat 2009; 121:743–751.
- Dyce A, editor. Sir Thomas More, a play. London: The Shakespeare Society, 1844. https://archive.org/details/sirthomasmorepla00mund. Accessed September 21, 2016.
On an otherwise pleasant evening during the first week of July 2016, a businessman who was a citizen of the United Arab Emirates visiting Cleveland for medical treatment was falsely accused of links to a terror organization. Officers stormed his hotel with assault rifles and handcuffed and arrested him—all this, apparently, because the man was dressed in traditional Emirati clothing.
This case highlights a level of complexity in providing medical care to foreigners far beyond language interpreting services and outside the borders of the institution where medical care is provided. In the current issue of the Journal, Cawcutt and Wilson1 review their experiences in the care of international patients and the unique challenges associated with it.
FROM THE TEMPLE OF AESCULAPIUS TO CLEVELAND CLINIC
In 2015, patients from more than 100 countries traveled to Cleveland seeking care at Cleveland Clinic. But medical travel was part of the practice of medicine long before major US hospitals became destinations for international patients, and it has been refined over the years.
Ancient cultures had a thriving tradition of patients traveling long distances for the best and most advanced medical treatment.2–4 In ancient Greece, people from all around the Mediterranean came to the city of Epidaurus to be cured in its famous temple of Aesculapius, built as a medical center.
Similarly, early Islamic cultures established a healthcare system that catered to foreigners. A noted example is the Mansuri hospital in Cairo, built in 1248 ce and considered the most advanced hospital of its time. Accommodating nearly 8,000 patients, the Mansuri hospital became a healthcare destination for foreigners regardless of race or religion.2–4
Europe also had a great tradition of providing medical care to foreign patients. Between the 15th and 17th centuries, belief in the healing power of mineral water led to the establishment of spas and the rise of spa towns, particularly in the south of France near mineral springs. The poor sanitary conditions of Europe at the time may have prompted the interest in the healing effect of mineral spas, but wealthy individuals from all over the world traveled to these destinations, creating local prosperity due to medical tourism.2–4
The city of Bath, in England, is a great example. In the 1720s, Bath was a popular destination for those traveling for healthcare. It became the first city in England to build a covered sewage system, ahead of London by several years. It also had paved roads, lights, hotels, and restaurants in much greater numbers than other cities in England, a likely result of prosperity associated with medical tourism.
ALL PATIENTS WANT TO BE TREATED WITH RESPECT AND KINDNESS
While medical knowledge and health delivery models have changed over the years, caring for foreign patients is perhaps as old as medicine itself. The central focus of restoring health is certainly not unique to international patients, but understanding their unique needs is important in order to achieve the best outcomes, something that Cawcutt and Wilson highlight well.1
A number of studies have addressed the question of what patients really want. Responses were surprisingly consistent: they want to be treated with respect and kindness.5,6 In other words, they want empathy, and this is true of all patients regardless of ethnicity or background. Empathy is a tremendous therapeutic force and can narrow what may look like an unbridgeable gap between patient and physician.7,8
EMPATHY REQUIRES EFFECTIVE COMMUNICATION
Empathy, though sometimes innate, requires effective communication and shared experiences. Neither of these two requirements is easily achievable in the care of foreign patients.
Communication is hampered by language barriers, although it can be enhanced significantly by language translating services and the work of certified medical interpreters. These often-invisible heroes should be recognized as essential members of the medical team. Their work requires cultural sensitivity and formal training to avoid miscommunication and medical errors. Codes of ethics for medical interpreters include confidentiality, accuracy in conveying the content and spirit of the message, freedom from personal biases, cultural training, and professional boundaries.9
TOWARD CULTURAL COMPETENCY
Lack of shared experiences between the foreign patient and care provider is an even greater obstacle to overcome in eliminating any empathy deficit. Shared experiences, whether cultural, religious, or social, help us to see the world through the eyes of the patient.
International patients may differ from us in background, ethnicity, religion, dress, expectations, and other areas. Cultural and religious backgrounds often dictate certain behaviors in the event of critical illness or death. Even in routine and less acute medical care, the background of a foreign patient may lead to logistical quandaries such as the need for same-sex caregivers or a private room.
A paradox currently exists in our efforts to meet patients’ need and desire for empathy. While culturally empathic care is necessary to achieve the best medical outcomes, this topic is not yet part of the curriculum for physicians or other healthcare providers in training. A culturally sensitive institution has many business advantages.10 Thorough and focused cultural training of medical staff is essential. Shared experiences can potentially be fashioned through a well-designed cultural competency training program to enhance empathy for foreign patients.
A SERVICE-ORIENTED APPROACH
Besides cultural competency and language training, a service-oriented approach to accommodate the needs of medical travelers and their family members is of paramount importance. Many of the complaints and burdens of medical visitors concern services that are not medical in nature, such as daily living necessities. Transportation, religious services, banking, extended-stay facilities, cell phone service, legal services, shopping, dining, and entertainment are among many other living needs for those receiving medical care abroad. These services are inconsistently provided throughout medical institutions in the United States, which provide care to thousands of international patients annually.
Unique challenges of providing medical care to international patients have direct effects on medical outcomes. A population-based cohort study of US-born and foreign-born adults with lung or colorectal cancer suggested disparities in quality and type of care.11 Foreign-born patients reported lower-quality care and were less likely to receive complex cancer treatments recommended by clinical guidelines. The authors proposed that quality of care and outcomes may be improved with greater emphasis on coordination of care and improving communication. Similar findings were reported in foreign-born patients with breast cancer.12
‘WHAT WOULD YOU THINK TO BE USED THUS?’
Four hundred years ago, in the play Sir Thomas More (a collaboration between several Elizabethan playwrights),13 the title character confronts a mob of anti-immigrant rioters, and in a speech believed to have been written by William Shakespeare (Act 2, Scene 4), asks them to imagine themselves banished to a foreign country and subjected to hostility such as they were meting out:
To be used thus?”
Empathy for foreigners seeking medical care is not merely an act of kindness; rather, it is a central piece of healing. Medical institutions interested in providing healthcare to this unique group of patients should take these principles into account and carefully examine their ability to deliver compassionate care collectively to local and foreign-born patients alike.
On an otherwise pleasant evening during the first week of July 2016, a businessman who was a citizen of the United Arab Emirates visiting Cleveland for medical treatment was falsely accused of links to a terror organization. Officers stormed his hotel with assault rifles and handcuffed and arrested him—all this, apparently, because the man was dressed in traditional Emirati clothing.
This case highlights a level of complexity in providing medical care to foreigners far beyond language interpreting services and outside the borders of the institution where medical care is provided. In the current issue of the Journal, Cawcutt and Wilson1 review their experiences in the care of international patients and the unique challenges associated with it.
FROM THE TEMPLE OF AESCULAPIUS TO CLEVELAND CLINIC
In 2015, patients from more than 100 countries traveled to Cleveland seeking care at Cleveland Clinic. But medical travel was part of the practice of medicine long before major US hospitals became destinations for international patients, and it has been refined over the years.
Ancient cultures had a thriving tradition of patients traveling long distances for the best and most advanced medical treatment.2–4 In ancient Greece, people from all around the Mediterranean came to the city of Epidaurus to be cured in its famous temple of Aesculapius, built as a medical center.
Similarly, early Islamic cultures established a healthcare system that catered to foreigners. A noted example is the Mansuri hospital in Cairo, built in 1248 ce and considered the most advanced hospital of its time. Accommodating nearly 8,000 patients, the Mansuri hospital became a healthcare destination for foreigners regardless of race or religion.2–4
Europe also had a great tradition of providing medical care to foreign patients. Between the 15th and 17th centuries, belief in the healing power of mineral water led to the establishment of spas and the rise of spa towns, particularly in the south of France near mineral springs. The poor sanitary conditions of Europe at the time may have prompted the interest in the healing effect of mineral spas, but wealthy individuals from all over the world traveled to these destinations, creating local prosperity due to medical tourism.2–4
The city of Bath, in England, is a great example. In the 1720s, Bath was a popular destination for those traveling for healthcare. It became the first city in England to build a covered sewage system, ahead of London by several years. It also had paved roads, lights, hotels, and restaurants in much greater numbers than other cities in England, a likely result of prosperity associated with medical tourism.
ALL PATIENTS WANT TO BE TREATED WITH RESPECT AND KINDNESS
While medical knowledge and health delivery models have changed over the years, caring for foreign patients is perhaps as old as medicine itself. The central focus of restoring health is certainly not unique to international patients, but understanding their unique needs is important in order to achieve the best outcomes, something that Cawcutt and Wilson highlight well.1
A number of studies have addressed the question of what patients really want. Responses were surprisingly consistent: they want to be treated with respect and kindness.5,6 In other words, they want empathy, and this is true of all patients regardless of ethnicity or background. Empathy is a tremendous therapeutic force and can narrow what may look like an unbridgeable gap between patient and physician.7,8
EMPATHY REQUIRES EFFECTIVE COMMUNICATION
Empathy, though sometimes innate, requires effective communication and shared experiences. Neither of these two requirements is easily achievable in the care of foreign patients.
Communication is hampered by language barriers, although it can be enhanced significantly by language translating services and the work of certified medical interpreters. These often-invisible heroes should be recognized as essential members of the medical team. Their work requires cultural sensitivity and formal training to avoid miscommunication and medical errors. Codes of ethics for medical interpreters include confidentiality, accuracy in conveying the content and spirit of the message, freedom from personal biases, cultural training, and professional boundaries.9
TOWARD CULTURAL COMPETENCY
Lack of shared experiences between the foreign patient and care provider is an even greater obstacle to overcome in eliminating any empathy deficit. Shared experiences, whether cultural, religious, or social, help us to see the world through the eyes of the patient.
International patients may differ from us in background, ethnicity, religion, dress, expectations, and other areas. Cultural and religious backgrounds often dictate certain behaviors in the event of critical illness or death. Even in routine and less acute medical care, the background of a foreign patient may lead to logistical quandaries such as the need for same-sex caregivers or a private room.
A paradox currently exists in our efforts to meet patients’ need and desire for empathy. While culturally empathic care is necessary to achieve the best medical outcomes, this topic is not yet part of the curriculum for physicians or other healthcare providers in training. A culturally sensitive institution has many business advantages.10 Thorough and focused cultural training of medical staff is essential. Shared experiences can potentially be fashioned through a well-designed cultural competency training program to enhance empathy for foreign patients.
A SERVICE-ORIENTED APPROACH
Besides cultural competency and language training, a service-oriented approach to accommodate the needs of medical travelers and their family members is of paramount importance. Many of the complaints and burdens of medical visitors concern services that are not medical in nature, such as daily living necessities. Transportation, religious services, banking, extended-stay facilities, cell phone service, legal services, shopping, dining, and entertainment are among many other living needs for those receiving medical care abroad. These services are inconsistently provided throughout medical institutions in the United States, which provide care to thousands of international patients annually.
Unique challenges of providing medical care to international patients have direct effects on medical outcomes. A population-based cohort study of US-born and foreign-born adults with lung or colorectal cancer suggested disparities in quality and type of care.11 Foreign-born patients reported lower-quality care and were less likely to receive complex cancer treatments recommended by clinical guidelines. The authors proposed that quality of care and outcomes may be improved with greater emphasis on coordination of care and improving communication. Similar findings were reported in foreign-born patients with breast cancer.12
‘WHAT WOULD YOU THINK TO BE USED THUS?’
Four hundred years ago, in the play Sir Thomas More (a collaboration between several Elizabethan playwrights),13 the title character confronts a mob of anti-immigrant rioters, and in a speech believed to have been written by William Shakespeare (Act 2, Scene 4), asks them to imagine themselves banished to a foreign country and subjected to hostility such as they were meting out:
To be used thus?”
Empathy for foreigners seeking medical care is not merely an act of kindness; rather, it is a central piece of healing. Medical institutions interested in providing healthcare to this unique group of patients should take these principles into account and carefully examine their ability to deliver compassionate care collectively to local and foreign-born patients alike.
- Cawcutt KA, Wilson JW. The benefits and challenges of caring for international patients. Cleve Clin J Med 2016; 83:794–800.
- Health-Tourism.com. The history of medical tourism. Health-Tourism.com. www.health-tourism.com/medical-tourism/history/. Accessed September 21, 2016.
- Chen LH, Hochberg NS, Magill AJ. The pre-travel consultation. US Centers for Disease Control and Prevention. wwwnc.cdc.gov/travel/yellowbook/2016/the-pre-travel-consultation/the-pre-travel-consultation. Accessed September 21, 2016.
- Rogers K. Medical tourism. Encyclopedia Britannica. www.britannica.com/topic/medical-tourism. Accessed September 21, 2016.
- Detsky AS. What do patients really want from healthcare? JAMA 2011; 306:2500–2501.
- Shaywitz D. What do patients really want from healthcare? Forbes Dec 24, 2011. www.forbes.com/sites/davidshaywitz/2011/12/24/what-do-patients-really-want-from-health-care/print/. Accessed September 21, 2016.
- Lee TH. How to spread empathy in healthcare. Harvard Business Review July 17, 2014.
- Friedman R. Understanding empathy: can you feel my pain? New York Times April 24, 2007.
- National Council on Interpreting in Health Care. A national code of ethics for interpreters in healthcare. July 2004. www.ncihc.org/assets/documents/publications/NCIHC%20National%20Code%20of%20Ethics.pdf. Accessed September 21, 2016.
- Minguet L. Creating a culturally sensitive corporation. Harvard Business Review, September 2014.
- Nielsen SS, He Y, Ayanian JZ, Gomez SL, Khan KL, West DW, et al. Quality of cancer care among foreign-born patients with lung or colorectal cancer. Cancer 2010; 116:5497–5506.
- Kouri EM, He Y, Winer EP, Keating NL. Influence of birthplace on breast cancer diagnosis and treatment for Hispanic women. Breast Cancer Res Treat 2009; 121:743–751.
- Dyce A, editor. Sir Thomas More, a play. London: The Shakespeare Society, 1844. https://archive.org/details/sirthomasmorepla00mund. Accessed September 21, 2016.
- Cawcutt KA, Wilson JW. The benefits and challenges of caring for international patients. Cleve Clin J Med 2016; 83:794–800.
- Health-Tourism.com. The history of medical tourism. Health-Tourism.com. www.health-tourism.com/medical-tourism/history/. Accessed September 21, 2016.
- Chen LH, Hochberg NS, Magill AJ. The pre-travel consultation. US Centers for Disease Control and Prevention. wwwnc.cdc.gov/travel/yellowbook/2016/the-pre-travel-consultation/the-pre-travel-consultation. Accessed September 21, 2016.
- Rogers K. Medical tourism. Encyclopedia Britannica. www.britannica.com/topic/medical-tourism. Accessed September 21, 2016.
- Detsky AS. What do patients really want from healthcare? JAMA 2011; 306:2500–2501.
- Shaywitz D. What do patients really want from healthcare? Forbes Dec 24, 2011. www.forbes.com/sites/davidshaywitz/2011/12/24/what-do-patients-really-want-from-health-care/print/. Accessed September 21, 2016.
- Lee TH. How to spread empathy in healthcare. Harvard Business Review July 17, 2014.
- Friedman R. Understanding empathy: can you feel my pain? New York Times April 24, 2007.
- National Council on Interpreting in Health Care. A national code of ethics for interpreters in healthcare. July 2004. www.ncihc.org/assets/documents/publications/NCIHC%20National%20Code%20of%20Ethics.pdf. Accessed September 21, 2016.
- Minguet L. Creating a culturally sensitive corporation. Harvard Business Review, September 2014.
- Nielsen SS, He Y, Ayanian JZ, Gomez SL, Khan KL, West DW, et al. Quality of cancer care among foreign-born patients with lung or colorectal cancer. Cancer 2010; 116:5497–5506.
- Kouri EM, He Y, Winer EP, Keating NL. Influence of birthplace on breast cancer diagnosis and treatment for Hispanic women. Breast Cancer Res Treat 2009; 121:743–751.
- Dyce A, editor. Sir Thomas More, a play. London: The Shakespeare Society, 1844. https://archive.org/details/sirthomasmorepla00mund. Accessed September 21, 2016.

In reply: Wilson disease
In Reply: We thank Dr. Mirrakhimov and colleagues for bringing important questions to our attention.
In terms of the differential diagnosis of cholestatic liver injury, we agree that pathologic processes such choledocholithiasis, cholangitis, primary biliary cirrhosis, and primary sclerosing cholangitis should be generally considered. However, in the case we described, the patient had no abdominal pain or fever, which makes choledocholithiasis or cholangitis very unlikely. Primary biliary cirrhosis and primary sclerosing cholangitis can cause chronic liver disease but should not be considered in the differential diagnosis of acute liver injury (acute hepatitis), such as in the case we described.
We agree that the hemolytic anemia typically seen in patients with Wilson disease is Coombs-negative, and that Coombs testing and a peripheral smear should be performed. Both were negative in our patient.
We also agree with Dr. Mirrakhimov and colleagues that Kayser-Fleischer rings are not necessarily specific for Wilson disease and can be seen in patients with other forms of cholestatic liver disease such as primary biliary cirrhosis. However, Kayser-Fleischer rings are pathognomonic for acute liver failure from Wilson disease. In other words, when Kayser-Fleischer rings are seen in a patient with acute liver failure, the diagnosis is Wilson disease until proven otherwise.
We discussed on page 112 of our article other treatments such as plasmapheresis as adjunctive therapy to bridge patients with acute liver failure secondary to Wilson disease to transplant. However, liver transplant is still the only definitive and potentially curative treatment.
In Reply: We thank Dr. Mirrakhimov and colleagues for bringing important questions to our attention.
In terms of the differential diagnosis of cholestatic liver injury, we agree that pathologic processes such choledocholithiasis, cholangitis, primary biliary cirrhosis, and primary sclerosing cholangitis should be generally considered. However, in the case we described, the patient had no abdominal pain or fever, which makes choledocholithiasis or cholangitis very unlikely. Primary biliary cirrhosis and primary sclerosing cholangitis can cause chronic liver disease but should not be considered in the differential diagnosis of acute liver injury (acute hepatitis), such as in the case we described.
We agree that the hemolytic anemia typically seen in patients with Wilson disease is Coombs-negative, and that Coombs testing and a peripheral smear should be performed. Both were negative in our patient.
We also agree with Dr. Mirrakhimov and colleagues that Kayser-Fleischer rings are not necessarily specific for Wilson disease and can be seen in patients with other forms of cholestatic liver disease such as primary biliary cirrhosis. However, Kayser-Fleischer rings are pathognomonic for acute liver failure from Wilson disease. In other words, when Kayser-Fleischer rings are seen in a patient with acute liver failure, the diagnosis is Wilson disease until proven otherwise.
We discussed on page 112 of our article other treatments such as plasmapheresis as adjunctive therapy to bridge patients with acute liver failure secondary to Wilson disease to transplant. However, liver transplant is still the only definitive and potentially curative treatment.
In Reply: We thank Dr. Mirrakhimov and colleagues for bringing important questions to our attention.
In terms of the differential diagnosis of cholestatic liver injury, we agree that pathologic processes such choledocholithiasis, cholangitis, primary biliary cirrhosis, and primary sclerosing cholangitis should be generally considered. However, in the case we described, the patient had no abdominal pain or fever, which makes choledocholithiasis or cholangitis very unlikely. Primary biliary cirrhosis and primary sclerosing cholangitis can cause chronic liver disease but should not be considered in the differential diagnosis of acute liver injury (acute hepatitis), such as in the case we described.
We agree that the hemolytic anemia typically seen in patients with Wilson disease is Coombs-negative, and that Coombs testing and a peripheral smear should be performed. Both were negative in our patient.
We also agree with Dr. Mirrakhimov and colleagues that Kayser-Fleischer rings are not necessarily specific for Wilson disease and can be seen in patients with other forms of cholestatic liver disease such as primary biliary cirrhosis. However, Kayser-Fleischer rings are pathognomonic for acute liver failure from Wilson disease. In other words, when Kayser-Fleischer rings are seen in a patient with acute liver failure, the diagnosis is Wilson disease until proven otherwise.
We discussed on page 112 of our article other treatments such as plasmapheresis as adjunctive therapy to bridge patients with acute liver failure secondary to Wilson disease to transplant. However, liver transplant is still the only definitive and potentially curative treatment.
A tale of two sisters with liver disease
A 25-year-old woman presents to the emergency department with a 7-day history of fatigue and nausea. On presentation she denies having abdominal pain, headache, fever, chills, night sweats, vomiting, diarrhea, melena, hematochezia, or weight loss. She recalls changes in the colors of her eyes and darkening urine over the last few days. Her medical history before this is unremarkable. She takes no prescription, over-the-counter, or herbal medications. She works as a librarian and has no occupational toxic exposures. She is single and has one sister with no prior medical history. She denies recent travel, sick contacts, smoking, recreational drug use, or pets at home.
On physical examination, her vital signs are temperature 37.3°C (99.1°F), heart rate 90 beats per minute, blood pressure 125/80 mm Hg, respiration rate 14 per minute, and oxygen saturation 97% on room air. She has icteric sclera and her skin is jaundiced. Cardiac examination is normal. Lungs are clear to auscultation and percussion bilaterally. Her abdomen is soft with no visceromegaly, masses, or tenderness. Extremities are normal with no edema. She is alert and oriented, but she has mild asterixis of the outstretched hands. The neurologic examination is otherwise unremarkable.
The patient’s basic laboratory values are listed in Table 1. Shortly after admission, she develops changes in her mental status, remaining alert but becoming agitated and oriented to person only. In view of her symptoms and laboratory findings, acute liver failure is suspected.
ACUTE LIVER FAILURE
1. The diagnostic criteria for acute liver failure include all of the following except which one?
- Acute elevation of liver biochemical tests
- Presence of preexisting liver disease
- Coagulopathy, defined by an international normalized ratio (INR) of 1.5 or greater
- Encephalopathy
- Duration of symptoms less than 26 weeks
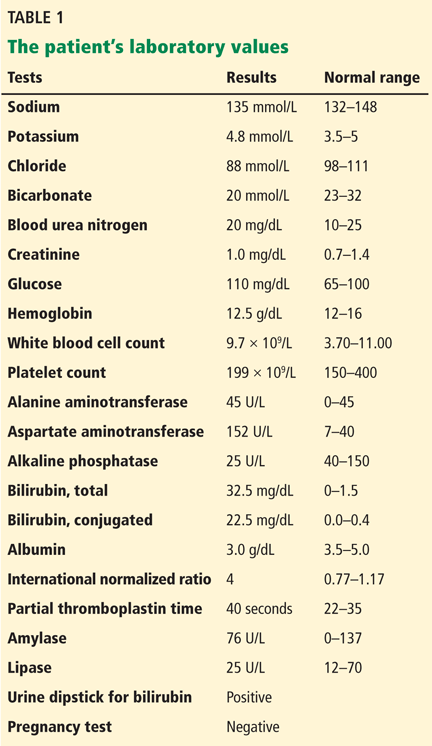
Acute liver failure is defined by acute onset of worsening liver tests, coagulopathy (INR ≥ 1.5), and encephalopathy in patients with no preexisting liver disease and with symptom duration of less than 26 weeks.1 With a few exceptions, a history of preexisting liver disease negates the diagnosis of acute liver failure. Our patient meets the diagnostic criteria for acute liver failure.
Immediate management
Once acute liver failure is identified or suspected, the next step is to transfer the patient to the intensive care unit for close monitoring of mental status. Serial neurologic evaluations permit early detection of cerebral edema, which is considered the most common cause of death in patients with acute liver failure. Additionally, close monitoring of electrolytes and plasma glucose is necessary since these patients are susceptible to electrolyte disturbances and hypoglycemia.
Patients with acute liver failure are at increased risk of infections and should be routinely screened by obtaining urine and blood cultures.
Gastrointestinal bleeding is not uncommon in patients with acute liver failure and is usually due to gastric stress ulceration. Prophylaxis with a histamine 2 receptor antagonist or proton pump inhibitor should be considered in order to prevent gastrointestinal bleeding.
Treatment with N-acetylcysteine is beneficial, not only in patients with acute liver failure due to acetaminophen overdose, but also in those with acute liver failure from other causes.
CASE CONTINUES:
TRANSFER TO THE INTENSIVE CARE UNIT
The patient, now diagnosed with acute liver failure, is transferred to the intensive care unit. Arterial blood gas measurement shows:
- pH 7.38 (reference range 7.35–7.45)
- Pco2 40 mm Hg (36–46)
- Po2 97 mm Hg (85–95)
- Hco3 22 mmol/L (22–26).
A chest radiograph is obtained and is clear. Computed tomography (CT) of the brain reveals no edema. Transcranial Doppler ultrasonography does not show any intracranial fluid collections.
Blood and urine cultures are negative. Her hemoglobin level remains stable, and she does not develop signs of bleeding. She is started on a proton pump inhibitor for stress ulcer prophylaxis and is empirically given intravenous N-acetylcysteine until the cause of acute liver failure can be determined.
CAUSES OF ACUTE LIVER FAILURE
2. Which of the following can cause acute liver failure?
- Acetaminophen overdose
- Viral hepatitis
- Autoimmune hepatitis
- Wilson disease
- Alcoholic hepatitis
Drug-induced liver injury is the most common cause of acute liver failure in the United States,2,3 and of all drugs, acetaminophen overdose is the number-one cause. In acetaminophen-induced liver injury, serum aminotransferase levels are usually elevated to more than 1,000 U/L, while serum bilirubin remains normal in the early stages. Antimicrobial agents, antiepileptic drugs, and herbal supplements have also been implicated in acute liver failure. Our patient has denied taking herbal supplements or medications, including over-the-counter ones.
Acute viral hepatitis can explain the patient’s condition. It is a common cause of acute liver failure in the United States.2 Hepatitis A and E are more common in developing countries. Other viruses such as cytomegalovirus, Epstein-Barr virus, herpes simplex virus type 1 and 2, and varicella zoster virus can also cause acute liver failure. Serum aminotransferase levels may exceed 1,000 U/L in patients with viral hepatitis.
Autoimmune hepatitis is a rare cause of acute liver failure, but it should be considered in the differential diagnosis, particularly in middle-aged women with autoimmune disorders such as hypothyroidism. Autoimmune hepatitis can cause marked elevation in aminotransferase levels (> 1,000 U/L).
Wilson disease is an autosomal-recessive disease in which there is excessive accumulation of copper in the liver and other organs because of an inherited defect in the biliary excretion of copper. Wilson disease can cause acute liver failure and should be excluded in any patient, particularly if under age 40 with acute onset of unexplained hepatic, neurologic, or psychiatric disease.
Alcoholic hepatitis usually occurs in patients with a long-standing history of heavy alcohol use. As a result, most patients with alcoholic hepatitis have manifestations of chronic liver disease due to alcohol use. Therefore, by definition, it is not a cause of acute liver failure. Additionally, in patients with alcoholic hepatitis, the aspartate aminotransferase (AST) level is elevated but less than 300 IU/mL, and the ratio of AST to alanine aminotransferase (ALT) is usually more than 2.
CASE CONTINUES: FURTHER TESTING
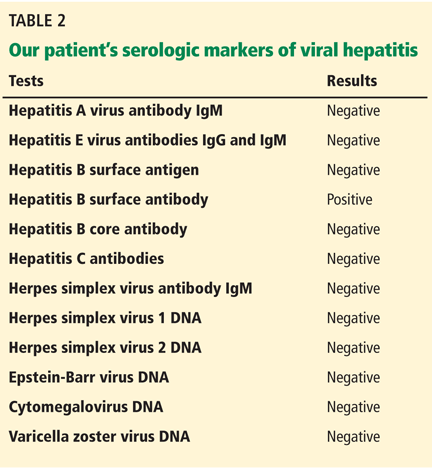
The results of our patient’s serologic tests are shown in Table 2. Other test results:
- Autoimmune markers including antinuclear antibodies, antimitochondrial antibodies, antismooth muscle antibodies, and liver and kidney microsomal antibodies are negative; her immunoglobulin G (IgG) level is normal
- Serum ceruloplasmin 25 mg/dL (normal 21–45)
- Free serum copper 120 µg/dL (normal 8–12)
- Abdominal ultrasonography is unremarkable, with normal liver parenchyma and no intrahepatic or extrahepatic biliary dilatation
- Doppler ultrasonography of the liver shows patent blood vessels.
3. Based on the new data, which of the following statements is correct?
- Hepatitis B is the cause of acute liver failure in this patient
- Herpetic hepatitis cannot be excluded on the basis of the available data
- Wilson disease is most likely the diagnosis, given her elevated free serum copper
- A normal serum ceruloplasmin level is not sufficient to rule out acute liver failure secondary to Wilson disease
Hepatitis B surface antigen and hepatitis B core antibodies were negative in our patient, excluding hepatitis B virus infection. The positive hepatitis B surface antibody indicates prior immunization.
Herpetic hepatitis is an uncommon but important cause of acute liver failure because the mortality rate is high if the patient is not treated early with acyclovir. Fever, elevated aminotransferases, and leukopenia are common with herpetic hepatitis. Fewer than 50% of patients with herpetic hepatitis have vesicular rash.4,5 The value of antibody serologic testing is limited due to high rates of false-positive and false-negative results. The gold standard diagnostic tests are viral load (detection of viral RNA by polymerase chain reaction), viral staining on liver biopsy, or both. In our patient, herpes simplex virus polymerase chain reaction testing was negative, which makes herpetic hepatitis unlikely.
Wilson disease is a genetic condition in which the ability to excrete copper in the bile is impaired, resulting in accumulation of copper in the hepatocytes. Subsequently, copper is released into the bloodstream and eventually into the urine.
However, copper excretion into the bile is impaired in patients with acute liver failure regardless of the etiology. Therefore, elevated free serum copper and 24-hour urine copper levels are not specific for the diagnosis of acute liver failure secondary to Wilson disease. Moreover, Kayser-Fleischer rings, which represent copper deposition in the limbus of the cornea, may not be apparent in the early stages of Wilson disease.
Since it is challenging to diagnose Wilson disease in the context of acute liver failure, Korman et al6 compared patients with acute liver failure secondary to Wilson disease with patients with acute liver failure secondary to other conditions. They found that alkaline phosphatase levels are frequently decreased in patients with acute liver failure secondary to Wilson disease,6 and that a ratio of alkaline phosphatase to total bilirubin of less than 4 is 94% sensitive and 96% specific for the diagnosis.6
Hemolysis is common in acute liver failure due to Wilson disease. This leads to disproportionate elevation of AST compared with ALT, since AST is present in red blood cells. Consequently, the ratio of AST to ALT is usually greater than 2.2, which provides a sensitivity of 94% and a specificity of 86% for the diagnosis.6 These two ratios together provide 100% sensitivity and 100% specificity for the diagnosis of Wilson disease in the context of acute liver failure.6
Ceruloplasmin. Patients with Wilson disease typically have a low ceruloplasmin level. However, because it is an acute-phase reaction protein, ceruloplasmin can be normal or elevated in patients with acute liver failure from Wilson disease.6 Therefore, a normal ceruloplasmin level is not sufficient to rule out acute liver failure secondary to Wilson disease.
CASE CONTINUES: A DEFINITIVE DIAGNOSIS
Our patient undergoes further testing, which reveals the following:
- Her 24-hour urinary excretion of copper is 150 µg (reference value < 30)
- Slit-lamp examination is normal and shows no evidence of Kayser-Fleischer rings
- Her ratio of alkaline phosphatase to total bilirubin is 0.77 based on her initial laboratory results (Table 1)
- Her AST-ALT ratio is 3.4.
The diagnosis in our patient is acute liver failure secondary to Wilson disease.
4. What is the most appropriate next step?
- Liver biopsy
- d-penicillamine by mouth
- Trientine by mouth
- Liver transplant
- Plasmapheresis
Liver biopsy. Accumulation of copper in the liver parenchyma in patients with Wilson disease is sporadic. Therefore, qualitative copper staining on liver biopsy can be falsely negative. Quantitative copper measurement in liver tissue is the gold standard for the diagnosis of Wilson disease. However, the test is time-consuming and is not rapidly available in the context of acute liver failure.
Chelating agents such as d-pencillamine and trientine are used to treat the chronic manifestations of Wilson disease but are not useful for acute liver failure secondary to Wilson disease.
Acute liver failure secondary to Wilson disease is life-threatening, and liver transplant is considered the only definitive life-saving therapy.
Therapeutic plasmapheresis has been reported to be a successful adjunctive therapy to bridge patients with acute liver failure secondary to Wilson disease to transplant.7 However, liver transplant is still the only definitive treatment.
CASE CONTINUES: THE PATIENT’S SISTER SEEKS CARE
The patient undergoes liver transplantation, with no perioperative or postoperative complications.
The patient’s 18-year-old sister is now seeking medical attention in the outpatient clinic, concerned that she may have Wilson disease. She is otherwise healthy and denies any symptoms or complaints.
5. What is the next step for the patient’s sister?
- Reassurance
- Prophylaxis with trientine
- Check liver enzyme levels, serum ceruloplasmin level, and urine copper, and order a slit-lamp examination
- Genetic testing
Wilson disease can be asymptomatic in its early stages and may be diagnosed incidentally during routine blood tests that reveal abnormal liver enzyme levels. All patients with a confirmed family history of Wilson disease should be screened even if they are asymptomatic. The diagnosis of Wilson disease should be established in first-degree relatives before specific treatment for the relatives is prescribed.
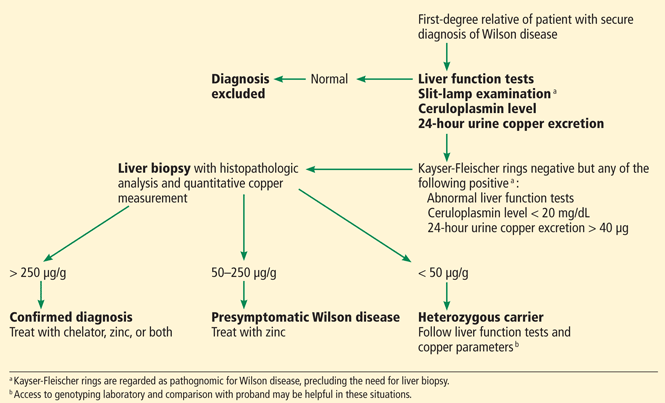
The first step in screening a first-degree relative for Wilson disease is to check liver enzyme levels (specifically aminotransferases, alkaline phosphatase, and bilirubin), serum ceruloplasmin level, and 24-hour urine copper, and order an ophthalmologic slit-lamp examination. If any of these tests is abnormal, liver biopsy should be performed for histopathologic evaluation and quantitative copper measurement. Kayser-Fleischer rings are seen in only 50% of patients with Wilson disease and hepatic involvement, but they are pathognomic. Guidelines8 for screening first-degree relatives of Wilson disease patients are shown in Figure 1.
Genetic analysis. ATP7B, the Wilson disease gene, is located on chromosome 13. At least 300 mutations of the gene have been described,2 and the most common mutation is present in only 15% to 30% of the Wilson disease population.8–10 Routine molecular testing of the ATP7B
CASE CONTINUES: WORKUP OF THE PATIENT’S SISTER
The patient’s sister has no symptoms and her physical examination is normal. Slit-lamp examination reveals no evidence of Kayser-Fleischer rings. Her laboratory values, including complete blood counts, complete metabolic panel, and INR, are within normal ranges. Other test results, however, are abnormal:
- Free serum copper level 27 µg/dL (normal 8–12)
- Serum ceruloplasmin 9.0 mg/dL (normal 20–50)
- 24-hour urinary copper excretion 135 µg (normal < 30).
She undergoes liver biopsy for quantitative copper measurement, and the result is very high at 1,118 µg/g dry weight (reference range 10–35). The diagnosis of Wilson disease is established.
TREATING CHRONIC WILSON DISEASE
6. Which of the following is not an appropriate next step for the patient’s sister?
- Tetrathiomolybdate
- d-penicillamine
- Trientine
- Zinc salts
- Prednisone
The goal of medical treatment of chronic Wilson disease is to improve symptoms and prevent progression of the disease.
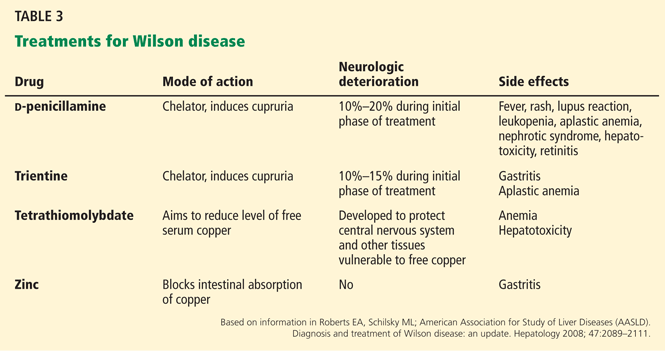
Chelating agents and zinc salts are the most commonly used medicines in the management of Wilson disease. Chelating agents remove copper from tissue, whereas zinc blocks the intestinal absorption of copper and stimulates the synthesis of endogenous chelators such as metallothioneins. Tetrathiomolybdate is an alternative agent developed to interfere with the distribution of excess body copper to susceptible target sites by reducing free serum copper (Table 3). There are no data to support the use of prednisone in the treatment of Wilson disease.
During treatment with chelating agents, 24-hour urinary excretion of copper is routinely monitored to determine the efficacy of therapy and adherence to treatment. Once de-coppering is achieved, as evidenced by a normalization of 24-hour urine copper excretion, the chelating agent can be switched to zinc salts to prevent intestinal absorption of copper.
Clinical and biochemical stabilization is achieved typically within 2 to 6 months of the initial treatment with chelating agents.8 Organ meats, nuts, shellfish, and chocolate are rich in copper and should be avoided.
The patient’s sister is started on trientine 250 mg orally three times daily on an empty stomach at least 1 hour before meals. Treatment is monitored by following 24-hour urine copper measurement. A 24-hour urine copper measurement at 3 months after starting treatment has increased from 54 at baseline to 350 µg, which indicates that the copper is being removed from tissues. The plan is for early substitution of zinc for long-term maintenance once de-coppering is completed.
KEY POINTS
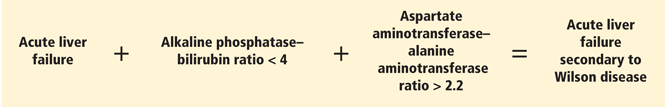
- Acute liver failure is severe acute liver injury characterized by coagulopathy (INR ≥ 1.5) and encephalopathy in a patient with no preexisting liver disease and with duration of symptoms less than 26 weeks.
- Acute liver failure secondary to Wilson disease is uncommon but should be excluded, particularly in young patients.
- The diagnosis of Wilson disease in the setting of acute liver failure is challenging because the serum ceruloplasmin level may be normal in acute liver failure secondary to Wilson disease, and free serum copper and 24-hour urine copper are usually elevated in all acute liver failure patients regardless of the etiology.
- A ratio of alkaline phosphatase to total bilirubin of less than 4 plus an AST-ALT ratio greater than 2.2 in a patient with acute liver failure should be regarded as Wilson disease until proven otherwise (Figure 2).
- Acute liver failure secondary to Wilson disease is usually fatal, and emergency liver transplant is a life-saving procedure.
- Screening of first-degree relatives of Wilson disease patients should include a history and physical examination, liver enzyme tests, complete blood cell count, serum ceruloplasmin level, serum free copper level, slit-lamp examination of the eyes, and 24-hour urinary copper measurement. Genetic tests are supplementary for screening but are not routinely available.
- Lee WM, Larson AM, Stravitz T. AASLD Position Paper: The management of acute liver failure: update 2011. www.aasld.org/sites/default/files/guideline_documents/alfenhanced.pdf. Accessed December 9, 2015.
- Bernal W, Auzinger G, Dhawan A, Wendon J. Acute liver failure. Lancet 2010; 376:190–201.
- Larson AM, Polson J, Fontana RJ, et al; Acute Liver Failure Study Group. Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology 2005; 42:1364–1372.
- Hanouneh IA, Khoriaty R, Zein NN. A 35-year-old Asian man with jaundice and markedly high aminotransferase levels. Cleve Clin J Med 2009; 76:449–456.
- Norvell JP, Blei AT, Jovanovic BD, Levitsky J. Herpes simplex virus hepatitis: an analysis of the published literature and institutional cases. Liver Transpl 2007; 13:1428–1434.
- Korman JD, Volenberg I, Balko J, et al; Pediatric and Adult Acute Liver Failure Study Groups. Screening for Wilson disease in acute liver failure: a comparison of currently available diagnostic tests. Hepatology 2008; 48:1167–1174.
- Morgan SM, Zantek ND. Therapeutic plasma exchange for fulminant hepatic failure secondary to Wilson's disease. J Clin Apher 2012; 27:282–286.
- Roberts EA, Schilsky ML; American Association for Study of Liver Diseases (AASLD). Diagnosis and treatment of Wilson disease: an update. Hepatology 2008; 47:2089–2111.
- Shah AB, Chernov I, Zhang HT, et al. Identification and analysis of mutations in the Wilson disease gene (ATP7B): population frequencies, genotype-phenotype correlation, and functional analyses. Am J Hum Genet 1997; 61:317–328.
- Maier-Dobersberger T, Ferenci P, Polli C, et al. Detection of the His1069Gln mutation in Wilson disease by rapid polymerase chain reaction. Ann Intern Med 1997; 127:21–26.
A 25-year-old woman presents to the emergency department with a 7-day history of fatigue and nausea. On presentation she denies having abdominal pain, headache, fever, chills, night sweats, vomiting, diarrhea, melena, hematochezia, or weight loss. She recalls changes in the colors of her eyes and darkening urine over the last few days. Her medical history before this is unremarkable. She takes no prescription, over-the-counter, or herbal medications. She works as a librarian and has no occupational toxic exposures. She is single and has one sister with no prior medical history. She denies recent travel, sick contacts, smoking, recreational drug use, or pets at home.
On physical examination, her vital signs are temperature 37.3°C (99.1°F), heart rate 90 beats per minute, blood pressure 125/80 mm Hg, respiration rate 14 per minute, and oxygen saturation 97% on room air. She has icteric sclera and her skin is jaundiced. Cardiac examination is normal. Lungs are clear to auscultation and percussion bilaterally. Her abdomen is soft with no visceromegaly, masses, or tenderness. Extremities are normal with no edema. She is alert and oriented, but she has mild asterixis of the outstretched hands. The neurologic examination is otherwise unremarkable.
The patient’s basic laboratory values are listed in Table 1. Shortly after admission, she develops changes in her mental status, remaining alert but becoming agitated and oriented to person only. In view of her symptoms and laboratory findings, acute liver failure is suspected.
ACUTE LIVER FAILURE
1. The diagnostic criteria for acute liver failure include all of the following except which one?
- Acute elevation of liver biochemical tests
- Presence of preexisting liver disease
- Coagulopathy, defined by an international normalized ratio (INR) of 1.5 or greater
- Encephalopathy
- Duration of symptoms less than 26 weeks
Acute liver failure is defined by acute onset of worsening liver tests, coagulopathy (INR ≥ 1.5), and encephalopathy in patients with no preexisting liver disease and with symptom duration of less than 26 weeks.1 With a few exceptions, a history of preexisting liver disease negates the diagnosis of acute liver failure. Our patient meets the diagnostic criteria for acute liver failure.
Immediate management
Once acute liver failure is identified or suspected, the next step is to transfer the patient to the intensive care unit for close monitoring of mental status. Serial neurologic evaluations permit early detection of cerebral edema, which is considered the most common cause of death in patients with acute liver failure. Additionally, close monitoring of electrolytes and plasma glucose is necessary since these patients are susceptible to electrolyte disturbances and hypoglycemia.
Patients with acute liver failure are at increased risk of infections and should be routinely screened by obtaining urine and blood cultures.
Gastrointestinal bleeding is not uncommon in patients with acute liver failure and is usually due to gastric stress ulceration. Prophylaxis with a histamine 2 receptor antagonist or proton pump inhibitor should be considered in order to prevent gastrointestinal bleeding.
Treatment with N-acetylcysteine is beneficial, not only in patients with acute liver failure due to acetaminophen overdose, but also in those with acute liver failure from other causes.
CASE CONTINUES:
TRANSFER TO THE INTENSIVE CARE UNIT
The patient, now diagnosed with acute liver failure, is transferred to the intensive care unit. Arterial blood gas measurement shows:
- pH 7.38 (reference range 7.35–7.45)
- Pco2 40 mm Hg (36–46)
- Po2 97 mm Hg (85–95)
- Hco3 22 mmol/L (22–26).
A chest radiograph is obtained and is clear. Computed tomography (CT) of the brain reveals no edema. Transcranial Doppler ultrasonography does not show any intracranial fluid collections.
Blood and urine cultures are negative. Her hemoglobin level remains stable, and she does not develop signs of bleeding. She is started on a proton pump inhibitor for stress ulcer prophylaxis and is empirically given intravenous N-acetylcysteine until the cause of acute liver failure can be determined.
CAUSES OF ACUTE LIVER FAILURE
2. Which of the following can cause acute liver failure?
- Acetaminophen overdose
- Viral hepatitis
- Autoimmune hepatitis
- Wilson disease
- Alcoholic hepatitis
Drug-induced liver injury is the most common cause of acute liver failure in the United States,2,3 and of all drugs, acetaminophen overdose is the number-one cause. In acetaminophen-induced liver injury, serum aminotransferase levels are usually elevated to more than 1,000 U/L, while serum bilirubin remains normal in the early stages. Antimicrobial agents, antiepileptic drugs, and herbal supplements have also been implicated in acute liver failure. Our patient has denied taking herbal supplements or medications, including over-the-counter ones.
Acute viral hepatitis can explain the patient’s condition. It is a common cause of acute liver failure in the United States.2 Hepatitis A and E are more common in developing countries. Other viruses such as cytomegalovirus, Epstein-Barr virus, herpes simplex virus type 1 and 2, and varicella zoster virus can also cause acute liver failure. Serum aminotransferase levels may exceed 1,000 U/L in patients with viral hepatitis.
Autoimmune hepatitis is a rare cause of acute liver failure, but it should be considered in the differential diagnosis, particularly in middle-aged women with autoimmune disorders such as hypothyroidism. Autoimmune hepatitis can cause marked elevation in aminotransferase levels (> 1,000 U/L).
Wilson disease is an autosomal-recessive disease in which there is excessive accumulation of copper in the liver and other organs because of an inherited defect in the biliary excretion of copper. Wilson disease can cause acute liver failure and should be excluded in any patient, particularly if under age 40 with acute onset of unexplained hepatic, neurologic, or psychiatric disease.
Alcoholic hepatitis usually occurs in patients with a long-standing history of heavy alcohol use. As a result, most patients with alcoholic hepatitis have manifestations of chronic liver disease due to alcohol use. Therefore, by definition, it is not a cause of acute liver failure. Additionally, in patients with alcoholic hepatitis, the aspartate aminotransferase (AST) level is elevated but less than 300 IU/mL, and the ratio of AST to alanine aminotransferase (ALT) is usually more than 2.
CASE CONTINUES: FURTHER TESTING
The results of our patient’s serologic tests are shown in Table 2. Other test results:
- Autoimmune markers including antinuclear antibodies, antimitochondrial antibodies, antismooth muscle antibodies, and liver and kidney microsomal antibodies are negative; her immunoglobulin G (IgG) level is normal
- Serum ceruloplasmin 25 mg/dL (normal 21–45)
- Free serum copper 120 µg/dL (normal 8–12)
- Abdominal ultrasonography is unremarkable, with normal liver parenchyma and no intrahepatic or extrahepatic biliary dilatation
- Doppler ultrasonography of the liver shows patent blood vessels.
3. Based on the new data, which of the following statements is correct?
- Hepatitis B is the cause of acute liver failure in this patient
- Herpetic hepatitis cannot be excluded on the basis of the available data
- Wilson disease is most likely the diagnosis, given her elevated free serum copper
- A normal serum ceruloplasmin level is not sufficient to rule out acute liver failure secondary to Wilson disease
Hepatitis B surface antigen and hepatitis B core antibodies were negative in our patient, excluding hepatitis B virus infection. The positive hepatitis B surface antibody indicates prior immunization.
Herpetic hepatitis is an uncommon but important cause of acute liver failure because the mortality rate is high if the patient is not treated early with acyclovir. Fever, elevated aminotransferases, and leukopenia are common with herpetic hepatitis. Fewer than 50% of patients with herpetic hepatitis have vesicular rash.4,5 The value of antibody serologic testing is limited due to high rates of false-positive and false-negative results. The gold standard diagnostic tests are viral load (detection of viral RNA by polymerase chain reaction), viral staining on liver biopsy, or both. In our patient, herpes simplex virus polymerase chain reaction testing was negative, which makes herpetic hepatitis unlikely.
Wilson disease is a genetic condition in which the ability to excrete copper in the bile is impaired, resulting in accumulation of copper in the hepatocytes. Subsequently, copper is released into the bloodstream and eventually into the urine.
However, copper excretion into the bile is impaired in patients with acute liver failure regardless of the etiology. Therefore, elevated free serum copper and 24-hour urine copper levels are not specific for the diagnosis of acute liver failure secondary to Wilson disease. Moreover, Kayser-Fleischer rings, which represent copper deposition in the limbus of the cornea, may not be apparent in the early stages of Wilson disease.
Since it is challenging to diagnose Wilson disease in the context of acute liver failure, Korman et al6 compared patients with acute liver failure secondary to Wilson disease with patients with acute liver failure secondary to other conditions. They found that alkaline phosphatase levels are frequently decreased in patients with acute liver failure secondary to Wilson disease,6 and that a ratio of alkaline phosphatase to total bilirubin of less than 4 is 94% sensitive and 96% specific for the diagnosis.6
Hemolysis is common in acute liver failure due to Wilson disease. This leads to disproportionate elevation of AST compared with ALT, since AST is present in red blood cells. Consequently, the ratio of AST to ALT is usually greater than 2.2, which provides a sensitivity of 94% and a specificity of 86% for the diagnosis.6 These two ratios together provide 100% sensitivity and 100% specificity for the diagnosis of Wilson disease in the context of acute liver failure.6
Ceruloplasmin. Patients with Wilson disease typically have a low ceruloplasmin level. However, because it is an acute-phase reaction protein, ceruloplasmin can be normal or elevated in patients with acute liver failure from Wilson disease.6 Therefore, a normal ceruloplasmin level is not sufficient to rule out acute liver failure secondary to Wilson disease.
CASE CONTINUES: A DEFINITIVE DIAGNOSIS
Our patient undergoes further testing, which reveals the following:
- Her 24-hour urinary excretion of copper is 150 µg (reference value < 30)
- Slit-lamp examination is normal and shows no evidence of Kayser-Fleischer rings
- Her ratio of alkaline phosphatase to total bilirubin is 0.77 based on her initial laboratory results (Table 1)
- Her AST-ALT ratio is 3.4.
The diagnosis in our patient is acute liver failure secondary to Wilson disease.
4. What is the most appropriate next step?
- Liver biopsy
- d-penicillamine by mouth
- Trientine by mouth
- Liver transplant
- Plasmapheresis
Liver biopsy. Accumulation of copper in the liver parenchyma in patients with Wilson disease is sporadic. Therefore, qualitative copper staining on liver biopsy can be falsely negative. Quantitative copper measurement in liver tissue is the gold standard for the diagnosis of Wilson disease. However, the test is time-consuming and is not rapidly available in the context of acute liver failure.
Chelating agents such as d-pencillamine and trientine are used to treat the chronic manifestations of Wilson disease but are not useful for acute liver failure secondary to Wilson disease.
Acute liver failure secondary to Wilson disease is life-threatening, and liver transplant is considered the only definitive life-saving therapy.
Therapeutic plasmapheresis has been reported to be a successful adjunctive therapy to bridge patients with acute liver failure secondary to Wilson disease to transplant.7 However, liver transplant is still the only definitive treatment.
CASE CONTINUES: THE PATIENT’S SISTER SEEKS CARE
The patient undergoes liver transplantation, with no perioperative or postoperative complications.
The patient’s 18-year-old sister is now seeking medical attention in the outpatient clinic, concerned that she may have Wilson disease. She is otherwise healthy and denies any symptoms or complaints.
5. What is the next step for the patient’s sister?
- Reassurance
- Prophylaxis with trientine
- Check liver enzyme levels, serum ceruloplasmin level, and urine copper, and order a slit-lamp examination
- Genetic testing
Wilson disease can be asymptomatic in its early stages and may be diagnosed incidentally during routine blood tests that reveal abnormal liver enzyme levels. All patients with a confirmed family history of Wilson disease should be screened even if they are asymptomatic. The diagnosis of Wilson disease should be established in first-degree relatives before specific treatment for the relatives is prescribed.
The first step in screening a first-degree relative for Wilson disease is to check liver enzyme levels (specifically aminotransferases, alkaline phosphatase, and bilirubin), serum ceruloplasmin level, and 24-hour urine copper, and order an ophthalmologic slit-lamp examination. If any of these tests is abnormal, liver biopsy should be performed for histopathologic evaluation and quantitative copper measurement. Kayser-Fleischer rings are seen in only 50% of patients with Wilson disease and hepatic involvement, but they are pathognomic. Guidelines8 for screening first-degree relatives of Wilson disease patients are shown in Figure 1.
Genetic analysis. ATP7B, the Wilson disease gene, is located on chromosome 13. At least 300 mutations of the gene have been described,2 and the most common mutation is present in only 15% to 30% of the Wilson disease population.8–10 Routine molecular testing of the ATP7B
CASE CONTINUES: WORKUP OF THE PATIENT’S SISTER
The patient’s sister has no symptoms and her physical examination is normal. Slit-lamp examination reveals no evidence of Kayser-Fleischer rings. Her laboratory values, including complete blood counts, complete metabolic panel, and INR, are within normal ranges. Other test results, however, are abnormal:
- Free serum copper level 27 µg/dL (normal 8–12)
- Serum ceruloplasmin 9.0 mg/dL (normal 20–50)
- 24-hour urinary copper excretion 135 µg (normal < 30).
She undergoes liver biopsy for quantitative copper measurement, and the result is very high at 1,118 µg/g dry weight (reference range 10–35). The diagnosis of Wilson disease is established.
TREATING CHRONIC WILSON DISEASE
6. Which of the following is not an appropriate next step for the patient’s sister?
- Tetrathiomolybdate
- d-penicillamine
- Trientine
- Zinc salts
- Prednisone
The goal of medical treatment of chronic Wilson disease is to improve symptoms and prevent progression of the disease.
Chelating agents and zinc salts are the most commonly used medicines in the management of Wilson disease. Chelating agents remove copper from tissue, whereas zinc blocks the intestinal absorption of copper and stimulates the synthesis of endogenous chelators such as metallothioneins. Tetrathiomolybdate is an alternative agent developed to interfere with the distribution of excess body copper to susceptible target sites by reducing free serum copper (Table 3). There are no data to support the use of prednisone in the treatment of Wilson disease.
During treatment with chelating agents, 24-hour urinary excretion of copper is routinely monitored to determine the efficacy of therapy and adherence to treatment. Once de-coppering is achieved, as evidenced by a normalization of 24-hour urine copper excretion, the chelating agent can be switched to zinc salts to prevent intestinal absorption of copper.
Clinical and biochemical stabilization is achieved typically within 2 to 6 months of the initial treatment with chelating agents.8 Organ meats, nuts, shellfish, and chocolate are rich in copper and should be avoided.
The patient’s sister is started on trientine 250 mg orally three times daily on an empty stomach at least 1 hour before meals. Treatment is monitored by following 24-hour urine copper measurement. A 24-hour urine copper measurement at 3 months after starting treatment has increased from 54 at baseline to 350 µg, which indicates that the copper is being removed from tissues. The plan is for early substitution of zinc for long-term maintenance once de-coppering is completed.
KEY POINTS
- Acute liver failure is severe acute liver injury characterized by coagulopathy (INR ≥ 1.5) and encephalopathy in a patient with no preexisting liver disease and with duration of symptoms less than 26 weeks.
- Acute liver failure secondary to Wilson disease is uncommon but should be excluded, particularly in young patients.
- The diagnosis of Wilson disease in the setting of acute liver failure is challenging because the serum ceruloplasmin level may be normal in acute liver failure secondary to Wilson disease, and free serum copper and 24-hour urine copper are usually elevated in all acute liver failure patients regardless of the etiology.
- A ratio of alkaline phosphatase to total bilirubin of less than 4 plus an AST-ALT ratio greater than 2.2 in a patient with acute liver failure should be regarded as Wilson disease until proven otherwise (Figure 2).
- Acute liver failure secondary to Wilson disease is usually fatal, and emergency liver transplant is a life-saving procedure.
- Screening of first-degree relatives of Wilson disease patients should include a history and physical examination, liver enzyme tests, complete blood cell count, serum ceruloplasmin level, serum free copper level, slit-lamp examination of the eyes, and 24-hour urinary copper measurement. Genetic tests are supplementary for screening but are not routinely available.
A 25-year-old woman presents to the emergency department with a 7-day history of fatigue and nausea. On presentation she denies having abdominal pain, headache, fever, chills, night sweats, vomiting, diarrhea, melena, hematochezia, or weight loss. She recalls changes in the colors of her eyes and darkening urine over the last few days. Her medical history before this is unremarkable. She takes no prescription, over-the-counter, or herbal medications. She works as a librarian and has no occupational toxic exposures. She is single and has one sister with no prior medical history. She denies recent travel, sick contacts, smoking, recreational drug use, or pets at home.
On physical examination, her vital signs are temperature 37.3°C (99.1°F), heart rate 90 beats per minute, blood pressure 125/80 mm Hg, respiration rate 14 per minute, and oxygen saturation 97% on room air. She has icteric sclera and her skin is jaundiced. Cardiac examination is normal. Lungs are clear to auscultation and percussion bilaterally. Her abdomen is soft with no visceromegaly, masses, or tenderness. Extremities are normal with no edema. She is alert and oriented, but she has mild asterixis of the outstretched hands. The neurologic examination is otherwise unremarkable.
The patient’s basic laboratory values are listed in Table 1. Shortly after admission, she develops changes in her mental status, remaining alert but becoming agitated and oriented to person only. In view of her symptoms and laboratory findings, acute liver failure is suspected.
ACUTE LIVER FAILURE
1. The diagnostic criteria for acute liver failure include all of the following except which one?
- Acute elevation of liver biochemical tests
- Presence of preexisting liver disease
- Coagulopathy, defined by an international normalized ratio (INR) of 1.5 or greater
- Encephalopathy
- Duration of symptoms less than 26 weeks
Acute liver failure is defined by acute onset of worsening liver tests, coagulopathy (INR ≥ 1.5), and encephalopathy in patients with no preexisting liver disease and with symptom duration of less than 26 weeks.1 With a few exceptions, a history of preexisting liver disease negates the diagnosis of acute liver failure. Our patient meets the diagnostic criteria for acute liver failure.
Immediate management
Once acute liver failure is identified or suspected, the next step is to transfer the patient to the intensive care unit for close monitoring of mental status. Serial neurologic evaluations permit early detection of cerebral edema, which is considered the most common cause of death in patients with acute liver failure. Additionally, close monitoring of electrolytes and plasma glucose is necessary since these patients are susceptible to electrolyte disturbances and hypoglycemia.
Patients with acute liver failure are at increased risk of infections and should be routinely screened by obtaining urine and blood cultures.
Gastrointestinal bleeding is not uncommon in patients with acute liver failure and is usually due to gastric stress ulceration. Prophylaxis with a histamine 2 receptor antagonist or proton pump inhibitor should be considered in order to prevent gastrointestinal bleeding.
Treatment with N-acetylcysteine is beneficial, not only in patients with acute liver failure due to acetaminophen overdose, but also in those with acute liver failure from other causes.
CASE CONTINUES:
TRANSFER TO THE INTENSIVE CARE UNIT
The patient, now diagnosed with acute liver failure, is transferred to the intensive care unit. Arterial blood gas measurement shows:
- pH 7.38 (reference range 7.35–7.45)
- Pco2 40 mm Hg (36–46)
- Po2 97 mm Hg (85–95)
- Hco3 22 mmol/L (22–26).
A chest radiograph is obtained and is clear. Computed tomography (CT) of the brain reveals no edema. Transcranial Doppler ultrasonography does not show any intracranial fluid collections.
Blood and urine cultures are negative. Her hemoglobin level remains stable, and she does not develop signs of bleeding. She is started on a proton pump inhibitor for stress ulcer prophylaxis and is empirically given intravenous N-acetylcysteine until the cause of acute liver failure can be determined.
CAUSES OF ACUTE LIVER FAILURE
2. Which of the following can cause acute liver failure?
- Acetaminophen overdose
- Viral hepatitis
- Autoimmune hepatitis
- Wilson disease
- Alcoholic hepatitis
Drug-induced liver injury is the most common cause of acute liver failure in the United States,2,3 and of all drugs, acetaminophen overdose is the number-one cause. In acetaminophen-induced liver injury, serum aminotransferase levels are usually elevated to more than 1,000 U/L, while serum bilirubin remains normal in the early stages. Antimicrobial agents, antiepileptic drugs, and herbal supplements have also been implicated in acute liver failure. Our patient has denied taking herbal supplements or medications, including over-the-counter ones.
Acute viral hepatitis can explain the patient’s condition. It is a common cause of acute liver failure in the United States.2 Hepatitis A and E are more common in developing countries. Other viruses such as cytomegalovirus, Epstein-Barr virus, herpes simplex virus type 1 and 2, and varicella zoster virus can also cause acute liver failure. Serum aminotransferase levels may exceed 1,000 U/L in patients with viral hepatitis.
Autoimmune hepatitis is a rare cause of acute liver failure, but it should be considered in the differential diagnosis, particularly in middle-aged women with autoimmune disorders such as hypothyroidism. Autoimmune hepatitis can cause marked elevation in aminotransferase levels (> 1,000 U/L).
Wilson disease is an autosomal-recessive disease in which there is excessive accumulation of copper in the liver and other organs because of an inherited defect in the biliary excretion of copper. Wilson disease can cause acute liver failure and should be excluded in any patient, particularly if under age 40 with acute onset of unexplained hepatic, neurologic, or psychiatric disease.
Alcoholic hepatitis usually occurs in patients with a long-standing history of heavy alcohol use. As a result, most patients with alcoholic hepatitis have manifestations of chronic liver disease due to alcohol use. Therefore, by definition, it is not a cause of acute liver failure. Additionally, in patients with alcoholic hepatitis, the aspartate aminotransferase (AST) level is elevated but less than 300 IU/mL, and the ratio of AST to alanine aminotransferase (ALT) is usually more than 2.
CASE CONTINUES: FURTHER TESTING
The results of our patient’s serologic tests are shown in Table 2. Other test results:
- Autoimmune markers including antinuclear antibodies, antimitochondrial antibodies, antismooth muscle antibodies, and liver and kidney microsomal antibodies are negative; her immunoglobulin G (IgG) level is normal
- Serum ceruloplasmin 25 mg/dL (normal 21–45)
- Free serum copper 120 µg/dL (normal 8–12)
- Abdominal ultrasonography is unremarkable, with normal liver parenchyma and no intrahepatic or extrahepatic biliary dilatation
- Doppler ultrasonography of the liver shows patent blood vessels.
3. Based on the new data, which of the following statements is correct?
- Hepatitis B is the cause of acute liver failure in this patient
- Herpetic hepatitis cannot be excluded on the basis of the available data
- Wilson disease is most likely the diagnosis, given her elevated free serum copper
- A normal serum ceruloplasmin level is not sufficient to rule out acute liver failure secondary to Wilson disease
Hepatitis B surface antigen and hepatitis B core antibodies were negative in our patient, excluding hepatitis B virus infection. The positive hepatitis B surface antibody indicates prior immunization.
Herpetic hepatitis is an uncommon but important cause of acute liver failure because the mortality rate is high if the patient is not treated early with acyclovir. Fever, elevated aminotransferases, and leukopenia are common with herpetic hepatitis. Fewer than 50% of patients with herpetic hepatitis have vesicular rash.4,5 The value of antibody serologic testing is limited due to high rates of false-positive and false-negative results. The gold standard diagnostic tests are viral load (detection of viral RNA by polymerase chain reaction), viral staining on liver biopsy, or both. In our patient, herpes simplex virus polymerase chain reaction testing was negative, which makes herpetic hepatitis unlikely.
Wilson disease is a genetic condition in which the ability to excrete copper in the bile is impaired, resulting in accumulation of copper in the hepatocytes. Subsequently, copper is released into the bloodstream and eventually into the urine.
However, copper excretion into the bile is impaired in patients with acute liver failure regardless of the etiology. Therefore, elevated free serum copper and 24-hour urine copper levels are not specific for the diagnosis of acute liver failure secondary to Wilson disease. Moreover, Kayser-Fleischer rings, which represent copper deposition in the limbus of the cornea, may not be apparent in the early stages of Wilson disease.
Since it is challenging to diagnose Wilson disease in the context of acute liver failure, Korman et al6 compared patients with acute liver failure secondary to Wilson disease with patients with acute liver failure secondary to other conditions. They found that alkaline phosphatase levels are frequently decreased in patients with acute liver failure secondary to Wilson disease,6 and that a ratio of alkaline phosphatase to total bilirubin of less than 4 is 94% sensitive and 96% specific for the diagnosis.6
Hemolysis is common in acute liver failure due to Wilson disease. This leads to disproportionate elevation of AST compared with ALT, since AST is present in red blood cells. Consequently, the ratio of AST to ALT is usually greater than 2.2, which provides a sensitivity of 94% and a specificity of 86% for the diagnosis.6 These two ratios together provide 100% sensitivity and 100% specificity for the diagnosis of Wilson disease in the context of acute liver failure.6
Ceruloplasmin. Patients with Wilson disease typically have a low ceruloplasmin level. However, because it is an acute-phase reaction protein, ceruloplasmin can be normal or elevated in patients with acute liver failure from Wilson disease.6 Therefore, a normal ceruloplasmin level is not sufficient to rule out acute liver failure secondary to Wilson disease.
CASE CONTINUES: A DEFINITIVE DIAGNOSIS
Our patient undergoes further testing, which reveals the following:
- Her 24-hour urinary excretion of copper is 150 µg (reference value < 30)
- Slit-lamp examination is normal and shows no evidence of Kayser-Fleischer rings
- Her ratio of alkaline phosphatase to total bilirubin is 0.77 based on her initial laboratory results (Table 1)
- Her AST-ALT ratio is 3.4.
The diagnosis in our patient is acute liver failure secondary to Wilson disease.
4. What is the most appropriate next step?
- Liver biopsy
- d-penicillamine by mouth
- Trientine by mouth
- Liver transplant
- Plasmapheresis
Liver biopsy. Accumulation of copper in the liver parenchyma in patients with Wilson disease is sporadic. Therefore, qualitative copper staining on liver biopsy can be falsely negative. Quantitative copper measurement in liver tissue is the gold standard for the diagnosis of Wilson disease. However, the test is time-consuming and is not rapidly available in the context of acute liver failure.
Chelating agents such as d-pencillamine and trientine are used to treat the chronic manifestations of Wilson disease but are not useful for acute liver failure secondary to Wilson disease.
Acute liver failure secondary to Wilson disease is life-threatening, and liver transplant is considered the only definitive life-saving therapy.
Therapeutic plasmapheresis has been reported to be a successful adjunctive therapy to bridge patients with acute liver failure secondary to Wilson disease to transplant.7 However, liver transplant is still the only definitive treatment.
CASE CONTINUES: THE PATIENT’S SISTER SEEKS CARE
The patient undergoes liver transplantation, with no perioperative or postoperative complications.
The patient’s 18-year-old sister is now seeking medical attention in the outpatient clinic, concerned that she may have Wilson disease. She is otherwise healthy and denies any symptoms or complaints.
5. What is the next step for the patient’s sister?
- Reassurance
- Prophylaxis with trientine
- Check liver enzyme levels, serum ceruloplasmin level, and urine copper, and order a slit-lamp examination
- Genetic testing
Wilson disease can be asymptomatic in its early stages and may be diagnosed incidentally during routine blood tests that reveal abnormal liver enzyme levels. All patients with a confirmed family history of Wilson disease should be screened even if they are asymptomatic. The diagnosis of Wilson disease should be established in first-degree relatives before specific treatment for the relatives is prescribed.
The first step in screening a first-degree relative for Wilson disease is to check liver enzyme levels (specifically aminotransferases, alkaline phosphatase, and bilirubin), serum ceruloplasmin level, and 24-hour urine copper, and order an ophthalmologic slit-lamp examination. If any of these tests is abnormal, liver biopsy should be performed for histopathologic evaluation and quantitative copper measurement. Kayser-Fleischer rings are seen in only 50% of patients with Wilson disease and hepatic involvement, but they are pathognomic. Guidelines8 for screening first-degree relatives of Wilson disease patients are shown in Figure 1.
Genetic analysis. ATP7B, the Wilson disease gene, is located on chromosome 13. At least 300 mutations of the gene have been described,2 and the most common mutation is present in only 15% to 30% of the Wilson disease population.8–10 Routine molecular testing of the ATP7B
CASE CONTINUES: WORKUP OF THE PATIENT’S SISTER
The patient’s sister has no symptoms and her physical examination is normal. Slit-lamp examination reveals no evidence of Kayser-Fleischer rings. Her laboratory values, including complete blood counts, complete metabolic panel, and INR, are within normal ranges. Other test results, however, are abnormal:
- Free serum copper level 27 µg/dL (normal 8–12)
- Serum ceruloplasmin 9.0 mg/dL (normal 20–50)
- 24-hour urinary copper excretion 135 µg (normal < 30).
She undergoes liver biopsy for quantitative copper measurement, and the result is very high at 1,118 µg/g dry weight (reference range 10–35). The diagnosis of Wilson disease is established.
TREATING CHRONIC WILSON DISEASE
6. Which of the following is not an appropriate next step for the patient’s sister?
- Tetrathiomolybdate
- d-penicillamine
- Trientine
- Zinc salts
- Prednisone
The goal of medical treatment of chronic Wilson disease is to improve symptoms and prevent progression of the disease.
Chelating agents and zinc salts are the most commonly used medicines in the management of Wilson disease. Chelating agents remove copper from tissue, whereas zinc blocks the intestinal absorption of copper and stimulates the synthesis of endogenous chelators such as metallothioneins. Tetrathiomolybdate is an alternative agent developed to interfere with the distribution of excess body copper to susceptible target sites by reducing free serum copper (Table 3). There are no data to support the use of prednisone in the treatment of Wilson disease.
During treatment with chelating agents, 24-hour urinary excretion of copper is routinely monitored to determine the efficacy of therapy and adherence to treatment. Once de-coppering is achieved, as evidenced by a normalization of 24-hour urine copper excretion, the chelating agent can be switched to zinc salts to prevent intestinal absorption of copper.
Clinical and biochemical stabilization is achieved typically within 2 to 6 months of the initial treatment with chelating agents.8 Organ meats, nuts, shellfish, and chocolate are rich in copper and should be avoided.
The patient’s sister is started on trientine 250 mg orally three times daily on an empty stomach at least 1 hour before meals. Treatment is monitored by following 24-hour urine copper measurement. A 24-hour urine copper measurement at 3 months after starting treatment has increased from 54 at baseline to 350 µg, which indicates that the copper is being removed from tissues. The plan is for early substitution of zinc for long-term maintenance once de-coppering is completed.
KEY POINTS
- Acute liver failure is severe acute liver injury characterized by coagulopathy (INR ≥ 1.5) and encephalopathy in a patient with no preexisting liver disease and with duration of symptoms less than 26 weeks.
- Acute liver failure secondary to Wilson disease is uncommon but should be excluded, particularly in young patients.
- The diagnosis of Wilson disease in the setting of acute liver failure is challenging because the serum ceruloplasmin level may be normal in acute liver failure secondary to Wilson disease, and free serum copper and 24-hour urine copper are usually elevated in all acute liver failure patients regardless of the etiology.
- A ratio of alkaline phosphatase to total bilirubin of less than 4 plus an AST-ALT ratio greater than 2.2 in a patient with acute liver failure should be regarded as Wilson disease until proven otherwise (Figure 2).
- Acute liver failure secondary to Wilson disease is usually fatal, and emergency liver transplant is a life-saving procedure.
- Screening of first-degree relatives of Wilson disease patients should include a history and physical examination, liver enzyme tests, complete blood cell count, serum ceruloplasmin level, serum free copper level, slit-lamp examination of the eyes, and 24-hour urinary copper measurement. Genetic tests are supplementary for screening but are not routinely available.
- Lee WM, Larson AM, Stravitz T. AASLD Position Paper: The management of acute liver failure: update 2011. www.aasld.org/sites/default/files/guideline_documents/alfenhanced.pdf. Accessed December 9, 2015.
- Bernal W, Auzinger G, Dhawan A, Wendon J. Acute liver failure. Lancet 2010; 376:190–201.
- Larson AM, Polson J, Fontana RJ, et al; Acute Liver Failure Study Group. Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology 2005; 42:1364–1372.
- Hanouneh IA, Khoriaty R, Zein NN. A 35-year-old Asian man with jaundice and markedly high aminotransferase levels. Cleve Clin J Med 2009; 76:449–456.
- Norvell JP, Blei AT, Jovanovic BD, Levitsky J. Herpes simplex virus hepatitis: an analysis of the published literature and institutional cases. Liver Transpl 2007; 13:1428–1434.
- Korman JD, Volenberg I, Balko J, et al; Pediatric and Adult Acute Liver Failure Study Groups. Screening for Wilson disease in acute liver failure: a comparison of currently available diagnostic tests. Hepatology 2008; 48:1167–1174.
- Morgan SM, Zantek ND. Therapeutic plasma exchange for fulminant hepatic failure secondary to Wilson's disease. J Clin Apher 2012; 27:282–286.
- Roberts EA, Schilsky ML; American Association for Study of Liver Diseases (AASLD). Diagnosis and treatment of Wilson disease: an update. Hepatology 2008; 47:2089–2111.
- Shah AB, Chernov I, Zhang HT, et al. Identification and analysis of mutations in the Wilson disease gene (ATP7B): population frequencies, genotype-phenotype correlation, and functional analyses. Am J Hum Genet 1997; 61:317–328.
- Maier-Dobersberger T, Ferenci P, Polli C, et al. Detection of the His1069Gln mutation in Wilson disease by rapid polymerase chain reaction. Ann Intern Med 1997; 127:21–26.
- Lee WM, Larson AM, Stravitz T. AASLD Position Paper: The management of acute liver failure: update 2011. www.aasld.org/sites/default/files/guideline_documents/alfenhanced.pdf. Accessed December 9, 2015.
- Bernal W, Auzinger G, Dhawan A, Wendon J. Acute liver failure. Lancet 2010; 376:190–201.
- Larson AM, Polson J, Fontana RJ, et al; Acute Liver Failure Study Group. Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology 2005; 42:1364–1372.
- Hanouneh IA, Khoriaty R, Zein NN. A 35-year-old Asian man with jaundice and markedly high aminotransferase levels. Cleve Clin J Med 2009; 76:449–456.
- Norvell JP, Blei AT, Jovanovic BD, Levitsky J. Herpes simplex virus hepatitis: an analysis of the published literature and institutional cases. Liver Transpl 2007; 13:1428–1434.
- Korman JD, Volenberg I, Balko J, et al; Pediatric and Adult Acute Liver Failure Study Groups. Screening for Wilson disease in acute liver failure: a comparison of currently available diagnostic tests. Hepatology 2008; 48:1167–1174.
- Morgan SM, Zantek ND. Therapeutic plasma exchange for fulminant hepatic failure secondary to Wilson's disease. J Clin Apher 2012; 27:282–286.
- Roberts EA, Schilsky ML; American Association for Study of Liver Diseases (AASLD). Diagnosis and treatment of Wilson disease: an update. Hepatology 2008; 47:2089–2111.
- Shah AB, Chernov I, Zhang HT, et al. Identification and analysis of mutations in the Wilson disease gene (ATP7B): population frequencies, genotype-phenotype correlation, and functional analyses. Am J Hum Genet 1997; 61:317–328.
- Maier-Dobersberger T, Ferenci P, Polli C, et al. Detection of the His1069Gln mutation in Wilson disease by rapid polymerase chain reaction. Ann Intern Med 1997; 127:21–26.

Genetics and hepatitis C: It’s good to be ‘CC’
What a difference a single nucleotide can make! The human genome contains more than 3 billion base pairs. Yet having a different nucleotide in only one pair can make a big difference in how we respond to a disease or its treatment.
Specifically, in hepatitis C virus infection, people born with the nucleotide cytosine (C) at location rs12979860 in both alleles of the gene that codes for interleukin 28B (the IL28B CC genotype) can count themselves luckier than those born with thymine (T) in this location in one of their alleles (the CT genotype) or both of their alleles (the TT genotype). Those with the CC genotype are more likely to clear the virus spontaneously, and even if the infection persists, it is less likely to progress to liver cancer and more likely to respond to treatment with interferon.
Here, we review the IL28B polymorphism and its implications in treating hepatitis C.
GENETIC POLYMORPHISM AND HUMAN DISEASE
Of the 3 billion base pairs of nucleotides, fewer than 1% differ between individuals, but this 1% is responsible for the diversity of human beings. Differences in genetic sequences among individuals are called genetic polymorphisms. A single-nucleotide polymorphism is a DNA sequence variation that occurs in a single nucleotide in the genome. For example, two sequenced DNA fragments from different individuals, AAGCCTA and AAGCTTA, contain a difference in a single nucleotide.
Genetic variations such as these underlie some of the differences in our susceptibility to disease, the severity of illness we develop, and our response to treatments. Therefore, identifying genetic polymorphisms may shed light on biologic pathways involved in diseases and may uncover new targets for therapy.1
Genome-wide association studies have looked at hundreds of thousands of single-nucleotide polymorphisms to try to identify most of the common genetic differences among people and relate them to common chronic diseases such as coronary artery disease,2 type 2 diabetes,3 stroke,4 breast cancer,5 rheumatoid arthritis,6 Alzheimer disease,7 and, more recently, hepatitis C virus infection.8
HEPATITIS C VIRUS: A MAJOR CAUSE OF LIVER DISEASE
Hepatitis C virus infection is a major cause of chronic liver disease and hepatocellular carcinoma and has become the most common indication for liver transplantation in the United States.9
This virus has six distinct genotypes throughout the world, with multiple subtypes in each genotype. (A genotype is a classification of a virus based on its RNA.9) In this review, we will focus on genotype 1; hence, “hepatitis C virus” will refer to hepatitis C virus genotype 1.
Our knowledge of the biology, pathogenesis, and treatment of hepatitis C has been advancing. Originally, fewer than 50% of patients responded to therapy with the combination of pegylated interferon and ribavirin,10,11 but since 2011 the response rate has increased to approximately 70% with the approval of the protease inhibitors telaprevir and boceprevir, used in combination with pegylated interferon and ribavirin.12–15
Unfortunately, interferon-based treatment is often complicated by side effects such as fatigue, influenza-like symptoms, hematologic abnormalities, and neuropsychiatric symptoms. An accurate way to predict response would help patients make informed decisions about antiviral treatment, taking into account the risk and possible benefit for individual patients.
GENETIC POLYMORPHISM AND HEPATITIS C VIRUS INFECTION
Genome-wide association studies have identified single-nucleotide polymorphisms in the IL28B gene that are associated with differences in response to hepatitis C treatment.8
Studying 565,759 polymorphisms in 1,137 patients, researchers at Duke University identified a single-nucleotide polymorphism at location rs12979860 in IL28B (Figure 1) that was strongly associated with response to combination therapy with pegylated interferon and ribavirin.8 The chance of cure with this standard treatment is twice as high in patients who are homozygous for cytosine in this location (the CC genotype) than in those who are heterozygous (CT) or homozygous for thymine in this location (the TT genotype) (Table 1).

Adding one of the new protease inhibitors, telaprevir or boceprevir, to the standard hepatitis C treatment substantially improves the cure rates in all three IL28B genotypes, but especially in people with CT or TT, in whom the response rate almost triples with the addition of one of these drugs. Those with the CC genotype (who are more likely to be cured with pegylated interferon and ribavirin alone) also achieve an increase (although minimal) in cure rates when a protease inhibitor is included in the regimen (TABLE 1).13–15 Thus, it remains unclear if adding a protease inhibitor to pegylated interferon plus ribavirin in patients with the IL28B CC genotype translates into added effectiveness worth the additional cost of the protease inhibitor in previously untreated patients.
Additionally, the effect of the IL28B genotype on telaprevir-based triple therapy has been disputed in more recent studies. In a subgroup analysis of the results of a trial that evaluated telaprevir in the treatment of hepatitis C, researchers found that sustained virologic response rates were significantly higher in the telaprevir group, and this was similar across the different IL28B polymorphisms.16
The favorable IL28B CC genotype is associated with higher rates of rapid virologic response to antiviral therapy.13–15 Of note, almost all patients who achieve a rapid virologic response do well, with a high rate of sustained virologic response even after a shorter duration of therapy (24 vs 48 weeks). Therefore, in addition to predicting response to interferon before starting treatment, the IL28B CC genotype may also identify patients who need only a shorter duration of therapy.
Interestingly, the C allele is much more frequent in white than in African American populations, an important observation that explains the racial difference in response to hepatitis C therapy.8
Two other research groups, from Asia and Australia, performed independent genome-wide association studies that identified different single-nucleotide polymorphisms (eg, rs8099917) in the same IL28B gene as predictors of response to treatment in patients with hepatitis C virus infection.17,18 These findings may be explained by linkage disequilibrium, which means that these single-nucleotide polymorphisms are found more frequently together in the same patient due to their proximity to each other. In this review, we will focus on the rs12979860 polymorphism; hence “IL28B genotype” will refer to the single-nucleotide polymorphism at rs12979860, unless otherwise specified.
The favorable CC genotype is less common in African Americans than in patients of other ethnicities.19 Moreover, although IL28B CC is associated with a better response rate to interferon-based antiviral therapy across all ethnicities, those of African American descent with the CC genotype are less likely to achieve a sustained virologic response than white or Hispanic Americans.8
BIOLOGIC ASSOCIATION: IL28B POLYMORPHISM AND HEPATITIS C
The interferon lambda family consists of three cytokines:
- Interleukin 29 (interferon lambda 1)
- Interleukin 28A (interferon lambda 2)
- Interleukin 28B (interferon lambda 3).
Production of these three molecules can be triggered by viral infection, and they induce antiviral activity through both innate and adaptive immune pathways. They signal through the IL10R-IL28R receptor complex.20–22 This receptor activates the JAK-STAT (Janus kinase-signal transducer and activator of transcription) pathway, which regulates a large number of interferon-stimulated genes, primarily through the interferon-stimulated response element (Figure 2).
A 2013 study found that interferon-stimulated gene expression levels in patients with normal livers were highest in those with the CC genotype, intermediate with CT, and lowest with TT. Interestingly, this pattern was reversed in those with hepatitis C virus infection, indicating a relationship between the IL28B genotype and gene expression before infection.23
The mechanism underlying the association between the IL28B polymorphism and response to hepatitis C treatment is not well understood. The unfavorable TT genotype seems to lead to continuous activation of a subset of interferon-stimulated genes in the presence of intracellular hepatitis C viral RNA. But this level of expression is not sufficient to eliminate the virus from the cells. Instead, it might lead to up-regulation of interferon-inhibitory molecules that suppress JAK-STAT signaling, thereby reducing sensitivity to interferon signaling. Therefore, the hepatocyte not only cannot clear the virus by itself, but also cannot induce strong interferon-stimulated gene expression when interferon is given during therapy.20–22
The recently identified ss469425590 polymorphism, which is located in close proximity to rs12979860 in the IL28B gene, is particularly interesting, as it suggests a possible molecular mechanism. The delta G frameshift variant creates a novel gene called IFNL4, which is transiently activated in response to hepatitis C virus infection.24IFNL4 stimulates STAT1 and STAT2 phosphorylation and induces the expression of interferon-stimulated genes. Increased interferon-stimulated gene expression has been shown to be associated with decreased response to pegylated interferon-ribavirin treatment. These observations suggest that the ss469425590 delta G allele is responsible for the increased activation of interferon-stimulated genes and the lower sustained virologic response rate observed in patients who receive pegylated interferon-ribavirin treatment. It is possible that the activation of interferon-stimulated genes in patients with the ss469425590 delta G/delta G genotype reduces interferon-stimulated gene responsiveness to interferon alpha, which normally activates interferon-stimulated genes and inhibits hepatitis C progression.24
IL28B POLYMORPHISM AND ACUTE HEPATITIS C VIRUS INFECTION
From 70% to 80% of acute hepatitis C virus infections persist and become chronic, while 20% to 30% spontaneously resolve. Epidemiologic, viral, and host factors have been associated with the differences in viral clearance or persistence, and studies have found that a strong host immune response against the virus favors viral clearance. Thus, variation in the genes involved in the immune response may contribute to one’s ability to clear the virus. Consistent with these observations, recent studies have shown that the polymorphism in the IL28 gene region encoding interferon lambda 3 strongly predicts spontaneous resolution of acute hepatitis C virus infection. People who have the IL28B CC genotype are three times more likely to spontaneously clear the virus than those with the CT or TT genotype (Figure 3).24
IL28B POLYMORPHISM AND THE NATURAL HISTORY OF HEPATITIS C
In people in whom hepatitis C virus infection persists, up to 20% develop progressive liver fibrosis and eventually cirrhosis over 10 to 20 years.19,25,26 The speed at which fibrosis develops in these patients is variable and unpredictable.25 The relationship between IL28B polymorphisms and hepatic fibrosis in patients with chronic hepatitis C virus infection has not been clearly established, although a study indicated that in patients with a known date of infection, the IL28B genotype is not associated with progression of hepatic fibrosis.27 Obstacles in this field of study are that it is difficult to determine accurately when the patient contracted the virus, and that serial liver biopsies are needed to investigate the progression of hepatic fibrosis.
Patients with chronic hepatitis C virus infection are also at higher risk of hepatocellular carcinoma compared with the general population.28 An analysis of explanted livers of patients with hepatitis C found that the prevalence of hepatocellular carcinoma in those with the unfavorable TT genotype was significantly higher than with the other genotypes.29 Similarly, an earlier study demonstrated that patients with hepatitis C-associated hepatocellular carcinoma carried the T allele more frequently.30 As with other aspects of IL28B associations with hepatitis C, these findings indicate that the C allele confers a certain degree of protection.
An important implication of these relationships is that they may eventually help identify patients at greater risk, who therefore need earlier intervention.
IL28B POLYMORPHISM AND LIVER TRANSPLANTATION
Hepatitis C virus infection always recurs after liver transplantation, with serious consequences that include cirrhosis and liver failure. Recurrent hepatitis C virus infection has become an important reason for repeat transplantation in the United States.
Results of treatment with pegylated interferon and ribavirin for recurrent hepatitis C after liver transplantation have been disappointing, with response rates lower than 30% and significant side effects.31 Identifying the factors that predict the response to therapy allows for better selection of treatment candidates.
Similar to the way the IL28B genotype predicts response to antiviral therapy in the nontransplant setting, the IL28B genotypes of both the recipient and the donor are strongly and independently associated with response to interferon-based treatment in patients with hepatitis C after liver transplantation. The IL28B CC genotype in either the recipient or the donor is associated with a higher rate of response to pegylated interferon and ribavirin combination therapy after liver transplantation.30,32 For example, the response rate to therapy after liver transplantation reaches 86% in CC-donor and CC-recipient livers, compared with 0% in TT-donor and TT-recipient livers.
Additionally, the IL28B genotype of the recipient may determine the severity of histologic recurrence of hepatitis C, as indicated by progressive hepatic fibrosis. A recipient IL28B TT genotype is associated with more severe histologic recurrence of hepatitis C.33
These data suggest that CC donor livers might be preferentially allocated to patients with hepatitis C virus infection.
IL28B AND OTHER FACTORS IN HEPATITIS C VIRUS INFECTION
Although it is tempting to think that the IL28B polymorphism is the sole predictor of response to antiviral therapy, it is but one of several known factors in the virus and the host.
While IL28B polymorphisms are the most important predictor of sustained virologic response with an interferon-based regimen, a rapid virologic response (undetectable viral load at 4 weeks) had superior predictive value and specificity in one study.34 In fact, for patients with chronic hepatitis C infection who achieved a rapid virologic response with pegylated interferon and ribavirin, the IL28B polymorphism had no effect on the rate of sustained virologic response. However, it did predict a sustained virologic response in the group who did not achieve rapid virologic response.
In a study of patients with acute hepatitis C infection,35 jaundice and the IL28 rs12979860 CC genotype both predicted spontaneous clearance. The best predictor of viral persistence was the combination of the CT or TT genotype plus the absence of jaundice, which had a predictive value of 98%.
IL28B AND THE FUTURE OF HEPATITIS C VIRUS THERAPY
New oral agents were recently approved for treating hepatitis C. As of November 2014, these included simeprevir, sofosbuvir, and ledipasvir.
Simeprevir is a second-generation NS3/4A protease inhibitor approved for use in combination with pegylated interferon and ribavirin. A recent phase 3 trial evaluating simeprevir in patients who had relapsed after prior therapy found sustained virologic response rates to be higher with simeprevir than with placebo, irrespective of IL28B status.36 This finding was similar to that of a trial of telaprevir.16
Sofosbuvir is a nucleotide analogue NS5B polymerase inhibitor that becomes incorporated into the growing RNA, inducing a chain termination event.37 In phase 3 trials,38,39 researchers found an initial rapid decrease in viral load for patients treated with this agent regardless of IL28B status.
In the NEUTRINO trial (Sofosbuvir With Peginterferon Alfa 2a and Ribavirin for 12 Weeks in Treatment-Naive Subjects With Chronic Genotype 1, 4, 5, or 6 HCV Infection),38 which used sofusbuvir in combination with interferon and ribavirin, the rate of sustained virologic response was higher in those with the favorable CC genotype (98%) than with a non-CC genotype (87%).
In COSMOS (A Study of TMC435 in Combination With PSI-7977 [GS7977] in Chronic Hepatitis C Genotype 1-Infected Prior Null Responders to Peginterferon/Ribavirin Therapy or HCV Treatment-Naive Patients),39 which used a combination of simeprevir, sofosbuvir, and ribavirin, the rate of sustained virologic response was higher in those with the CC genotype (100%) than with the TT genotype (83%; Table 1).
These new medications have radically changed the landscape of hepatitis C therapy and have also unlocked the potential for developing completely interferon-free regimens.
Other new interferon-free regimens such as ledipasvir, daclatasvir, and asunaprevir promise high rates of sustained virologic response, which makes the utility of testing for IL28B polymorphisms to predict sustained virologic response very much diminished (Table 1).40,41 However, these new drugs are expected to be expensive, and IL28B polymorphisms may be used to identify candidates who are more likely to respond to pegylated interferon and ribavirin, particularly in resource-poor settings and in developing countries. Additionally, patients who have contraindications to these newer therapies will still likely need an interferon-based regimen, and thus the IL28B polymorphism will still be important in predicting treatment response and prognosis.
IL28B WILL STILL BE RELEVANT IN THE INTERFERON-FREE AGE
The IL28B polymorphism is a strong predictor of spontaneous clearance of hepatitis C virus and responsiveness to interferon-based therapy, and testing for it has demonstrated a great potential to improve patient care. IL28B testing has become available for clinical use and may optimize the outcome of hepatitis C treatment by helping us to select the best treatment for individual patients and minimizing the duration of therapy and the side effects associated with interferon-based antiviral medications.
As newer therapies have shifted toward interferon-free regimens that offer very high sustained virologic response rates, the usefulness of IL28B polymorphism as a clinical test to predict the response rate to antiviral therapy is minimized substantially. It may remain clinically relevant in resource-poor settings and in developing countries, especially in light of the potentially prohibitive costs of the newer regimens, and for patients in whom these treatments are contraindicated. This does not minimize the lesson we learned from the discovery of the IL28B gene and the impact on our understanding of the pathogenesis of hepatitis C virus infection.
- Attia J, Ioannidis JP, Thakkinstian A, et al. How to use an article about genetic association: A: background concepts. JAMA 2009; 301:74–81.
- Samani NJ, Erdmann J, Hall AS, et al; WTCCC and the Cardiogenics Consortium. Genomewide association analysis of coronary artery disease. N Engl J Med 2007; 357:443–453.
- Zeggini E, Weedon MN, Lindgren CM, et al; Wellcome Trust Case Control Consortium (WTCCC). Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science 2007; 316:1336–1341.
- Matarín M, Brown WM, Scholz S, et al. A genome-wide genotyping study in patients with ischaemic stroke: initial analysis and data release. Lancet Neurol 2007; 6:414–420.
- Easton DF, Pooley KA, Dunning AM, et al; AOCS Management Group. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature 2007; 447:1087–1093.
- Plenge RM, Seielstad M, Padyukov L, et al. TRAF1-C5 as a risk locus for rheumatoid arthritis—a genomewide study. N Engl J Med 2007; 357:1199–1209.
- Coon KD, Myers AJ, Craig DW, et al. A high-density whole-genome association study reveals that APOE is the major susceptibility gene for sporadic late-onset Alzheimer’s disease. J Clin Psychiatry 2007; 68:613–618.
- Ge D, Fellay J, Thompson AJ, et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance (letter). Nature 2009; 461:399–401.
- Ali A, Zein NN. Hepatitis C infection: a systemic disease with extrahepatic manifestations. Cleve Clin J Med 2005; 72:1005-1019.
- Hanouneh IA, Feldstein AE, Lopez R, et al. Clinical significance of metabolic syndrome in the setting of chronic hepatitis C virus infection. Clin Gastroenterol Hepatol 2008; 6:584–589.
- Elgouhari HM, Zein CO, Hanouneh I, Feldstein AE, Zein NN. Diabetes mellitus is associated with impaired response to antiviral therapy in chronic hepatitis C infection. Dig Dis Sci 2009; 54:2699–2705.
- Alkhouri N, Zein NN. Protease inhibitors: silver bullets for chronic hepatitis C infection? Cleve Clin J Med 2012; 79:213–222.
- McHutchison JG, Everson GT, Gordon SC, et al; PROVE1 Study Team. Telaprevir with peginterferon and ribavirin for chronic HCV genotype 1 infection. N Engl J Med 2009; 360:1827–1838.
- Jacobson IM, McHutchison JG, Dusheiko G, et al; ADVANCE Study Team. Telaprevir for previously untreated chronic hepatitis C virus infection. N Engl J Med 2011; 364:2405–2416.
- Jacobson IM, Catlett I, Marcellin P, et al. Telaprevir substantially improved SVR rates across all IL28B genotypes in the ADVANCE trial. J Hepatol 2011; 54(suppl 1):S542–S543.
- Pol S, Aerssens J, Zeuzem S, et al. Limited impact of IL28B genotype on response rates in telaprevir-treated patients with prior treatment failure. J Hepatol 2013; 58:883–889.
- Suppiah V, Moldovan M, Ahlenstiel G, et al. IL28B is associated with response to chronic hepatitis C interferon-alpha and ribavirin therapy. Nat Genet 2009; 41:1100–1104.
- Tanaka Y, Nishida N, Sugiyama M, et al. Genome-wide association of IL28B with response to pegylated interferon-alpha and ribavirin therapy for chronic hepatitis C. Nat Genet 2009; 41:1105–1109.
- Thomas DL, Thio CL, Martin MP, et al. Genetic variation in IL28B and spontaneous clearance of hepatitis C virus. Nature 2009; 461:798–801.
- Rehermann B. Hepatitis C virus versus innate and adaptive immune responses: a tale of coevolution and coexistence. J Clin Invest 2009; 119:1745–1754.
- Marcello T, Grakoui A, Barba-Spaeth G, et al. Interferons alpha and lambda inhibit hepatitis C virus replication with distinct signal transduction and gene regulation kinetics. Gastroenterology 2006; 131:1887–1898.
- Doyle SE, Schreckhise H, Khuu-Duong K, et al. Interleukin-29 uses a type 1 interferon-like program to promote antiviral responses in human hepatocytes. Hepatology 2006; 44:896–906.
- Raglow Z, Thoma-Perry C, Gilroy R, Wan YJ. IL28B genotype and the expression of ISGs in normal liver. Liver Int 2013; 33:991–998.
- Prokunina-Olsson L, Muchmore B, Tang W, et al. A variant upstream of IFNL3 (IL28B) creating a new interferon gene IFNL4 is associated with impaired clearance of hepatitis C virus. Nat Genet 2013; 45:164–171.
- Hanouneh IA, Zein NN, Askar M, Lopez R, John B. Interleukin-28B polymorphisms are associated with fibrosing cholestatic hepatitis in recurrent hepatitis C after liver transplantation. Clin Transplant 2012; 26:E335–E336.
- Poynard T, Bedossa P, Opolon P. Natural history of liver fibrosis progression in patients with chronic hepatitis C. The OBSVIRC, METAVIR, CLINIVIR, and DOSVIRC groups. Lancet 1997; 349:825–832.
- Thomas DL, Astemborski J, Rai RM, et al. The natural history of hepatitis C virus infection: host, viral, and environmental factors. JAMA 2000; 284:450–456.
- Bochud PY, Cai T, Overbeck K, et al; Swiss Hepatitis C Cohort Study Group. Genotype 3 is associated with accelerated fibrosis progression in chronic hepatitis C. J Hepatol 2009; 51:655–666.
- Marabita F, Aghemo A, De Nicola S, et al. Genetic variation in the interleukin-28B gene is not associated with fibrosis progression in patients with chronic hepatitis C and known date of infection. Hepatology 2011; 54:1127–1134.
- Fabris C, Falleti E, Cussigh A, et al. IL-28B rs12979860 C/T allele distribution in patients with liver cirrhosis: role in the course of chronic viral hepatitis and the development of HCC. J Hepatol 2011; 54:716–722.
- Eurich D, Boas-Knoop S, Bahra M, et al. Role of IL28B polymorphism in the development of hepatitis C virus-induced hepatocellular carcinoma, graft fibrosis, and posttransplant antiviral therapy. Transplantation 2012; 93:644–649.
- Hanouneh IA, Miller C, Aucejo F, Lopez R, Quinn MK, Zein NN. Recurrent hepatitis C after liver transplantation: on-treatment prediction of response to peginterferon/ribavirin therapy. Liver Transpl 2008; 14:53–58.
- Charlton MR, Thompson A, Veldt BJ, et al. Interleukin-28B polymorphisms are associated with histological recurrence and treatment response following liver transplantation in patients with hepatitis C virus infection. Hepatology 2011; 53:317–324.
- Thompson AJ, Muir AJ, Sulkowski MS, et al. Interleukin-28B polymorphism improves viral kinetics and is the strongest pretreatment predictor of sustained virologic response in genotype 1 hepatitis C virus. Gastroenterology 2010; 139:120–129.e18.
- Beinhardt S, Payer BA, Datz C, et al. A diagnostic score for the prediction of spontaneous resolution of acute hepatitis C virus infection. J Hepatol 2013; 59:972–977.
- Forns X, Lawitz E, Zeuzem S, et al. Simeprevir with peginterferon and ribavirin leads to high rates of SVR in patients with HCV genotype 1 who relapsed after previous therapy: a phase 3 trial. Gastroenterology 2014; 146:1669–1679.e3.
- Sofia MJ, Bao D, Chang W, et al. Discovery of a ß-d-2’-deoxy-2’-ß-fluoro-2’-ß-C-methyluridine nucleotide prodrug (PSI-7977) for the treatment of hepatitis C virus. J Med Chem 2010; 53:7202–7218.
- Lawitz E, Mangia A, Wyles D, et al. Sofosbuvir for previously untreated chronic hepatitis C infection. N Engl J Med 2013; 368:1878–1887.
- Sulkowski MS, Jacobson IM, Ghalib R, et al. Once-daily simeprevir (TMC435) plus sofosbuvir (GS-7977) with or without ribavirin in HCV genotype 1 prior null responders with metavir F0-2: COSMOS study subgroup analysis. 49th EASL, April 2014, London. Oral abstract O7. www.natap.org/2014/EASL/EASL_46.htm. Accesed January 9, 2015.
- Lok AS, Gardiner DF, Lawitz E, et al. Preliminary study of two antiviral agents for hepatitis C genotype 1. N Engl J Med 2012; 366:216–224.
- Afdhal N, Zeuzem S, Kwo P, et al; ION-1 Investigators. Ledipasvir and sofosbuvir for untreated HCV genotype 1 infection. N Engl J Med 2014; 370:1889–1898.
What a difference a single nucleotide can make! The human genome contains more than 3 billion base pairs. Yet having a different nucleotide in only one pair can make a big difference in how we respond to a disease or its treatment.
Specifically, in hepatitis C virus infection, people born with the nucleotide cytosine (C) at location rs12979860 in both alleles of the gene that codes for interleukin 28B (the IL28B CC genotype) can count themselves luckier than those born with thymine (T) in this location in one of their alleles (the CT genotype) or both of their alleles (the TT genotype). Those with the CC genotype are more likely to clear the virus spontaneously, and even if the infection persists, it is less likely to progress to liver cancer and more likely to respond to treatment with interferon.
Here, we review the IL28B polymorphism and its implications in treating hepatitis C.
GENETIC POLYMORPHISM AND HUMAN DISEASE
Of the 3 billion base pairs of nucleotides, fewer than 1% differ between individuals, but this 1% is responsible for the diversity of human beings. Differences in genetic sequences among individuals are called genetic polymorphisms. A single-nucleotide polymorphism is a DNA sequence variation that occurs in a single nucleotide in the genome. For example, two sequenced DNA fragments from different individuals, AAGCCTA and AAGCTTA, contain a difference in a single nucleotide.
Genetic variations such as these underlie some of the differences in our susceptibility to disease, the severity of illness we develop, and our response to treatments. Therefore, identifying genetic polymorphisms may shed light on biologic pathways involved in diseases and may uncover new targets for therapy.1
Genome-wide association studies have looked at hundreds of thousands of single-nucleotide polymorphisms to try to identify most of the common genetic differences among people and relate them to common chronic diseases such as coronary artery disease,2 type 2 diabetes,3 stroke,4 breast cancer,5 rheumatoid arthritis,6 Alzheimer disease,7 and, more recently, hepatitis C virus infection.8
HEPATITIS C VIRUS: A MAJOR CAUSE OF LIVER DISEASE
Hepatitis C virus infection is a major cause of chronic liver disease and hepatocellular carcinoma and has become the most common indication for liver transplantation in the United States.9
This virus has six distinct genotypes throughout the world, with multiple subtypes in each genotype. (A genotype is a classification of a virus based on its RNA.9) In this review, we will focus on genotype 1; hence, “hepatitis C virus” will refer to hepatitis C virus genotype 1.
Our knowledge of the biology, pathogenesis, and treatment of hepatitis C has been advancing. Originally, fewer than 50% of patients responded to therapy with the combination of pegylated interferon and ribavirin,10,11 but since 2011 the response rate has increased to approximately 70% with the approval of the protease inhibitors telaprevir and boceprevir, used in combination with pegylated interferon and ribavirin.12–15
Unfortunately, interferon-based treatment is often complicated by side effects such as fatigue, influenza-like symptoms, hematologic abnormalities, and neuropsychiatric symptoms. An accurate way to predict response would help patients make informed decisions about antiviral treatment, taking into account the risk and possible benefit for individual patients.
GENETIC POLYMORPHISM AND HEPATITIS C VIRUS INFECTION
Genome-wide association studies have identified single-nucleotide polymorphisms in the IL28B gene that are associated with differences in response to hepatitis C treatment.8
Studying 565,759 polymorphisms in 1,137 patients, researchers at Duke University identified a single-nucleotide polymorphism at location rs12979860 in IL28B (Figure 1) that was strongly associated with response to combination therapy with pegylated interferon and ribavirin.8 The chance of cure with this standard treatment is twice as high in patients who are homozygous for cytosine in this location (the CC genotype) than in those who are heterozygous (CT) or homozygous for thymine in this location (the TT genotype) (Table 1).
Adding one of the new protease inhibitors, telaprevir or boceprevir, to the standard hepatitis C treatment substantially improves the cure rates in all three IL28B genotypes, but especially in people with CT or TT, in whom the response rate almost triples with the addition of one of these drugs. Those with the CC genotype (who are more likely to be cured with pegylated interferon and ribavirin alone) also achieve an increase (although minimal) in cure rates when a protease inhibitor is included in the regimen (TABLE 1).13–15 Thus, it remains unclear if adding a protease inhibitor to pegylated interferon plus ribavirin in patients with the IL28B CC genotype translates into added effectiveness worth the additional cost of the protease inhibitor in previously untreated patients.
Additionally, the effect of the IL28B genotype on telaprevir-based triple therapy has been disputed in more recent studies. In a subgroup analysis of the results of a trial that evaluated telaprevir in the treatment of hepatitis C, researchers found that sustained virologic response rates were significantly higher in the telaprevir group, and this was similar across the different IL28B polymorphisms.16
The favorable IL28B CC genotype is associated with higher rates of rapid virologic response to antiviral therapy.13–15 Of note, almost all patients who achieve a rapid virologic response do well, with a high rate of sustained virologic response even after a shorter duration of therapy (24 vs 48 weeks). Therefore, in addition to predicting response to interferon before starting treatment, the IL28B CC genotype may also identify patients who need only a shorter duration of therapy.
Interestingly, the C allele is much more frequent in white than in African American populations, an important observation that explains the racial difference in response to hepatitis C therapy.8
Two other research groups, from Asia and Australia, performed independent genome-wide association studies that identified different single-nucleotide polymorphisms (eg, rs8099917) in the same IL28B gene as predictors of response to treatment in patients with hepatitis C virus infection.17,18 These findings may be explained by linkage disequilibrium, which means that these single-nucleotide polymorphisms are found more frequently together in the same patient due to their proximity to each other. In this review, we will focus on the rs12979860 polymorphism; hence “IL28B genotype” will refer to the single-nucleotide polymorphism at rs12979860, unless otherwise specified.
The favorable CC genotype is less common in African Americans than in patients of other ethnicities.19 Moreover, although IL28B CC is associated with a better response rate to interferon-based antiviral therapy across all ethnicities, those of African American descent with the CC genotype are less likely to achieve a sustained virologic response than white or Hispanic Americans.8
BIOLOGIC ASSOCIATION: IL28B POLYMORPHISM AND HEPATITIS C
The interferon lambda family consists of three cytokines:
- Interleukin 29 (interferon lambda 1)
- Interleukin 28A (interferon lambda 2)
- Interleukin 28B (interferon lambda 3).
Production of these three molecules can be triggered by viral infection, and they induce antiviral activity through both innate and adaptive immune pathways. They signal through the IL10R-IL28R receptor complex.20–22 This receptor activates the JAK-STAT (Janus kinase-signal transducer and activator of transcription) pathway, which regulates a large number of interferon-stimulated genes, primarily through the interferon-stimulated response element (Figure 2).
A 2013 study found that interferon-stimulated gene expression levels in patients with normal livers were highest in those with the CC genotype, intermediate with CT, and lowest with TT. Interestingly, this pattern was reversed in those with hepatitis C virus infection, indicating a relationship between the IL28B genotype and gene expression before infection.23
The mechanism underlying the association between the IL28B polymorphism and response to hepatitis C treatment is not well understood. The unfavorable TT genotype seems to lead to continuous activation of a subset of interferon-stimulated genes in the presence of intracellular hepatitis C viral RNA. But this level of expression is not sufficient to eliminate the virus from the cells. Instead, it might lead to up-regulation of interferon-inhibitory molecules that suppress JAK-STAT signaling, thereby reducing sensitivity to interferon signaling. Therefore, the hepatocyte not only cannot clear the virus by itself, but also cannot induce strong interferon-stimulated gene expression when interferon is given during therapy.20–22
The recently identified ss469425590 polymorphism, which is located in close proximity to rs12979860 in the IL28B gene, is particularly interesting, as it suggests a possible molecular mechanism. The delta G frameshift variant creates a novel gene called IFNL4, which is transiently activated in response to hepatitis C virus infection.24IFNL4 stimulates STAT1 and STAT2 phosphorylation and induces the expression of interferon-stimulated genes. Increased interferon-stimulated gene expression has been shown to be associated with decreased response to pegylated interferon-ribavirin treatment. These observations suggest that the ss469425590 delta G allele is responsible for the increased activation of interferon-stimulated genes and the lower sustained virologic response rate observed in patients who receive pegylated interferon-ribavirin treatment. It is possible that the activation of interferon-stimulated genes in patients with the ss469425590 delta G/delta G genotype reduces interferon-stimulated gene responsiveness to interferon alpha, which normally activates interferon-stimulated genes and inhibits hepatitis C progression.24
IL28B POLYMORPHISM AND ACUTE HEPATITIS C VIRUS INFECTION
From 70% to 80% of acute hepatitis C virus infections persist and become chronic, while 20% to 30% spontaneously resolve. Epidemiologic, viral, and host factors have been associated with the differences in viral clearance or persistence, and studies have found that a strong host immune response against the virus favors viral clearance. Thus, variation in the genes involved in the immune response may contribute to one’s ability to clear the virus. Consistent with these observations, recent studies have shown that the polymorphism in the IL28 gene region encoding interferon lambda 3 strongly predicts spontaneous resolution of acute hepatitis C virus infection. People who have the IL28B CC genotype are three times more likely to spontaneously clear the virus than those with the CT or TT genotype (Figure 3).24
IL28B POLYMORPHISM AND THE NATURAL HISTORY OF HEPATITIS C
In people in whom hepatitis C virus infection persists, up to 20% develop progressive liver fibrosis and eventually cirrhosis over 10 to 20 years.19,25,26 The speed at which fibrosis develops in these patients is variable and unpredictable.25 The relationship between IL28B polymorphisms and hepatic fibrosis in patients with chronic hepatitis C virus infection has not been clearly established, although a study indicated that in patients with a known date of infection, the IL28B genotype is not associated with progression of hepatic fibrosis.27 Obstacles in this field of study are that it is difficult to determine accurately when the patient contracted the virus, and that serial liver biopsies are needed to investigate the progression of hepatic fibrosis.
Patients with chronic hepatitis C virus infection are also at higher risk of hepatocellular carcinoma compared with the general population.28 An analysis of explanted livers of patients with hepatitis C found that the prevalence of hepatocellular carcinoma in those with the unfavorable TT genotype was significantly higher than with the other genotypes.29 Similarly, an earlier study demonstrated that patients with hepatitis C-associated hepatocellular carcinoma carried the T allele more frequently.30 As with other aspects of IL28B associations with hepatitis C, these findings indicate that the C allele confers a certain degree of protection.
An important implication of these relationships is that they may eventually help identify patients at greater risk, who therefore need earlier intervention.
IL28B POLYMORPHISM AND LIVER TRANSPLANTATION
Hepatitis C virus infection always recurs after liver transplantation, with serious consequences that include cirrhosis and liver failure. Recurrent hepatitis C virus infection has become an important reason for repeat transplantation in the United States.
Results of treatment with pegylated interferon and ribavirin for recurrent hepatitis C after liver transplantation have been disappointing, with response rates lower than 30% and significant side effects.31 Identifying the factors that predict the response to therapy allows for better selection of treatment candidates.
Similar to the way the IL28B genotype predicts response to antiviral therapy in the nontransplant setting, the IL28B genotypes of both the recipient and the donor are strongly and independently associated with response to interferon-based treatment in patients with hepatitis C after liver transplantation. The IL28B CC genotype in either the recipient or the donor is associated with a higher rate of response to pegylated interferon and ribavirin combination therapy after liver transplantation.30,32 For example, the response rate to therapy after liver transplantation reaches 86% in CC-donor and CC-recipient livers, compared with 0% in TT-donor and TT-recipient livers.
Additionally, the IL28B genotype of the recipient may determine the severity of histologic recurrence of hepatitis C, as indicated by progressive hepatic fibrosis. A recipient IL28B TT genotype is associated with more severe histologic recurrence of hepatitis C.33
These data suggest that CC donor livers might be preferentially allocated to patients with hepatitis C virus infection.
IL28B AND OTHER FACTORS IN HEPATITIS C VIRUS INFECTION
Although it is tempting to think that the IL28B polymorphism is the sole predictor of response to antiviral therapy, it is but one of several known factors in the virus and the host.
While IL28B polymorphisms are the most important predictor of sustained virologic response with an interferon-based regimen, a rapid virologic response (undetectable viral load at 4 weeks) had superior predictive value and specificity in one study.34 In fact, for patients with chronic hepatitis C infection who achieved a rapid virologic response with pegylated interferon and ribavirin, the IL28B polymorphism had no effect on the rate of sustained virologic response. However, it did predict a sustained virologic response in the group who did not achieve rapid virologic response.
In a study of patients with acute hepatitis C infection,35 jaundice and the IL28 rs12979860 CC genotype both predicted spontaneous clearance. The best predictor of viral persistence was the combination of the CT or TT genotype plus the absence of jaundice, which had a predictive value of 98%.
IL28B AND THE FUTURE OF HEPATITIS C VIRUS THERAPY
New oral agents were recently approved for treating hepatitis C. As of November 2014, these included simeprevir, sofosbuvir, and ledipasvir.
Simeprevir is a second-generation NS3/4A protease inhibitor approved for use in combination with pegylated interferon and ribavirin. A recent phase 3 trial evaluating simeprevir in patients who had relapsed after prior therapy found sustained virologic response rates to be higher with simeprevir than with placebo, irrespective of IL28B status.36 This finding was similar to that of a trial of telaprevir.16
Sofosbuvir is a nucleotide analogue NS5B polymerase inhibitor that becomes incorporated into the growing RNA, inducing a chain termination event.37 In phase 3 trials,38,39 researchers found an initial rapid decrease in viral load for patients treated with this agent regardless of IL28B status.
In the NEUTRINO trial (Sofosbuvir With Peginterferon Alfa 2a and Ribavirin for 12 Weeks in Treatment-Naive Subjects With Chronic Genotype 1, 4, 5, or 6 HCV Infection),38 which used sofusbuvir in combination with interferon and ribavirin, the rate of sustained virologic response was higher in those with the favorable CC genotype (98%) than with a non-CC genotype (87%).
In COSMOS (A Study of TMC435 in Combination With PSI-7977 [GS7977] in Chronic Hepatitis C Genotype 1-Infected Prior Null Responders to Peginterferon/Ribavirin Therapy or HCV Treatment-Naive Patients),39 which used a combination of simeprevir, sofosbuvir, and ribavirin, the rate of sustained virologic response was higher in those with the CC genotype (100%) than with the TT genotype (83%; Table 1).
These new medications have radically changed the landscape of hepatitis C therapy and have also unlocked the potential for developing completely interferon-free regimens.
Other new interferon-free regimens such as ledipasvir, daclatasvir, and asunaprevir promise high rates of sustained virologic response, which makes the utility of testing for IL28B polymorphisms to predict sustained virologic response very much diminished (Table 1).40,41 However, these new drugs are expected to be expensive, and IL28B polymorphisms may be used to identify candidates who are more likely to respond to pegylated interferon and ribavirin, particularly in resource-poor settings and in developing countries. Additionally, patients who have contraindications to these newer therapies will still likely need an interferon-based regimen, and thus the IL28B polymorphism will still be important in predicting treatment response and prognosis.
IL28B WILL STILL BE RELEVANT IN THE INTERFERON-FREE AGE
The IL28B polymorphism is a strong predictor of spontaneous clearance of hepatitis C virus and responsiveness to interferon-based therapy, and testing for it has demonstrated a great potential to improve patient care. IL28B testing has become available for clinical use and may optimize the outcome of hepatitis C treatment by helping us to select the best treatment for individual patients and minimizing the duration of therapy and the side effects associated with interferon-based antiviral medications.
As newer therapies have shifted toward interferon-free regimens that offer very high sustained virologic response rates, the usefulness of IL28B polymorphism as a clinical test to predict the response rate to antiviral therapy is minimized substantially. It may remain clinically relevant in resource-poor settings and in developing countries, especially in light of the potentially prohibitive costs of the newer regimens, and for patients in whom these treatments are contraindicated. This does not minimize the lesson we learned from the discovery of the IL28B gene and the impact on our understanding of the pathogenesis of hepatitis C virus infection.
What a difference a single nucleotide can make! The human genome contains more than 3 billion base pairs. Yet having a different nucleotide in only one pair can make a big difference in how we respond to a disease or its treatment.
Specifically, in hepatitis C virus infection, people born with the nucleotide cytosine (C) at location rs12979860 in both alleles of the gene that codes for interleukin 28B (the IL28B CC genotype) can count themselves luckier than those born with thymine (T) in this location in one of their alleles (the CT genotype) or both of their alleles (the TT genotype). Those with the CC genotype are more likely to clear the virus spontaneously, and even if the infection persists, it is less likely to progress to liver cancer and more likely to respond to treatment with interferon.
Here, we review the IL28B polymorphism and its implications in treating hepatitis C.
GENETIC POLYMORPHISM AND HUMAN DISEASE
Of the 3 billion base pairs of nucleotides, fewer than 1% differ between individuals, but this 1% is responsible for the diversity of human beings. Differences in genetic sequences among individuals are called genetic polymorphisms. A single-nucleotide polymorphism is a DNA sequence variation that occurs in a single nucleotide in the genome. For example, two sequenced DNA fragments from different individuals, AAGCCTA and AAGCTTA, contain a difference in a single nucleotide.
Genetic variations such as these underlie some of the differences in our susceptibility to disease, the severity of illness we develop, and our response to treatments. Therefore, identifying genetic polymorphisms may shed light on biologic pathways involved in diseases and may uncover new targets for therapy.1
Genome-wide association studies have looked at hundreds of thousands of single-nucleotide polymorphisms to try to identify most of the common genetic differences among people and relate them to common chronic diseases such as coronary artery disease,2 type 2 diabetes,3 stroke,4 breast cancer,5 rheumatoid arthritis,6 Alzheimer disease,7 and, more recently, hepatitis C virus infection.8
HEPATITIS C VIRUS: A MAJOR CAUSE OF LIVER DISEASE
Hepatitis C virus infection is a major cause of chronic liver disease and hepatocellular carcinoma and has become the most common indication for liver transplantation in the United States.9
This virus has six distinct genotypes throughout the world, with multiple subtypes in each genotype. (A genotype is a classification of a virus based on its RNA.9) In this review, we will focus on genotype 1; hence, “hepatitis C virus” will refer to hepatitis C virus genotype 1.
Our knowledge of the biology, pathogenesis, and treatment of hepatitis C has been advancing. Originally, fewer than 50% of patients responded to therapy with the combination of pegylated interferon and ribavirin,10,11 but since 2011 the response rate has increased to approximately 70% with the approval of the protease inhibitors telaprevir and boceprevir, used in combination with pegylated interferon and ribavirin.12–15
Unfortunately, interferon-based treatment is often complicated by side effects such as fatigue, influenza-like symptoms, hematologic abnormalities, and neuropsychiatric symptoms. An accurate way to predict response would help patients make informed decisions about antiviral treatment, taking into account the risk and possible benefit for individual patients.
GENETIC POLYMORPHISM AND HEPATITIS C VIRUS INFECTION
Genome-wide association studies have identified single-nucleotide polymorphisms in the IL28B gene that are associated with differences in response to hepatitis C treatment.8
Studying 565,759 polymorphisms in 1,137 patients, researchers at Duke University identified a single-nucleotide polymorphism at location rs12979860 in IL28B (Figure 1) that was strongly associated with response to combination therapy with pegylated interferon and ribavirin.8 The chance of cure with this standard treatment is twice as high in patients who are homozygous for cytosine in this location (the CC genotype) than in those who are heterozygous (CT) or homozygous for thymine in this location (the TT genotype) (Table 1).
Adding one of the new protease inhibitors, telaprevir or boceprevir, to the standard hepatitis C treatment substantially improves the cure rates in all three IL28B genotypes, but especially in people with CT or TT, in whom the response rate almost triples with the addition of one of these drugs. Those with the CC genotype (who are more likely to be cured with pegylated interferon and ribavirin alone) also achieve an increase (although minimal) in cure rates when a protease inhibitor is included in the regimen (TABLE 1).13–15 Thus, it remains unclear if adding a protease inhibitor to pegylated interferon plus ribavirin in patients with the IL28B CC genotype translates into added effectiveness worth the additional cost of the protease inhibitor in previously untreated patients.
Additionally, the effect of the IL28B genotype on telaprevir-based triple therapy has been disputed in more recent studies. In a subgroup analysis of the results of a trial that evaluated telaprevir in the treatment of hepatitis C, researchers found that sustained virologic response rates were significantly higher in the telaprevir group, and this was similar across the different IL28B polymorphisms.16
The favorable IL28B CC genotype is associated with higher rates of rapid virologic response to antiviral therapy.13–15 Of note, almost all patients who achieve a rapid virologic response do well, with a high rate of sustained virologic response even after a shorter duration of therapy (24 vs 48 weeks). Therefore, in addition to predicting response to interferon before starting treatment, the IL28B CC genotype may also identify patients who need only a shorter duration of therapy.
Interestingly, the C allele is much more frequent in white than in African American populations, an important observation that explains the racial difference in response to hepatitis C therapy.8
Two other research groups, from Asia and Australia, performed independent genome-wide association studies that identified different single-nucleotide polymorphisms (eg, rs8099917) in the same IL28B gene as predictors of response to treatment in patients with hepatitis C virus infection.17,18 These findings may be explained by linkage disequilibrium, which means that these single-nucleotide polymorphisms are found more frequently together in the same patient due to their proximity to each other. In this review, we will focus on the rs12979860 polymorphism; hence “IL28B genotype” will refer to the single-nucleotide polymorphism at rs12979860, unless otherwise specified.
The favorable CC genotype is less common in African Americans than in patients of other ethnicities.19 Moreover, although IL28B CC is associated with a better response rate to interferon-based antiviral therapy across all ethnicities, those of African American descent with the CC genotype are less likely to achieve a sustained virologic response than white or Hispanic Americans.8
BIOLOGIC ASSOCIATION: IL28B POLYMORPHISM AND HEPATITIS C
The interferon lambda family consists of three cytokines:
- Interleukin 29 (interferon lambda 1)
- Interleukin 28A (interferon lambda 2)
- Interleukin 28B (interferon lambda 3).
Production of these three molecules can be triggered by viral infection, and they induce antiviral activity through both innate and adaptive immune pathways. They signal through the IL10R-IL28R receptor complex.20–22 This receptor activates the JAK-STAT (Janus kinase-signal transducer and activator of transcription) pathway, which regulates a large number of interferon-stimulated genes, primarily through the interferon-stimulated response element (Figure 2).
A 2013 study found that interferon-stimulated gene expression levels in patients with normal livers were highest in those with the CC genotype, intermediate with CT, and lowest with TT. Interestingly, this pattern was reversed in those with hepatitis C virus infection, indicating a relationship between the IL28B genotype and gene expression before infection.23
The mechanism underlying the association between the IL28B polymorphism and response to hepatitis C treatment is not well understood. The unfavorable TT genotype seems to lead to continuous activation of a subset of interferon-stimulated genes in the presence of intracellular hepatitis C viral RNA. But this level of expression is not sufficient to eliminate the virus from the cells. Instead, it might lead to up-regulation of interferon-inhibitory molecules that suppress JAK-STAT signaling, thereby reducing sensitivity to interferon signaling. Therefore, the hepatocyte not only cannot clear the virus by itself, but also cannot induce strong interferon-stimulated gene expression when interferon is given during therapy.20–22
The recently identified ss469425590 polymorphism, which is located in close proximity to rs12979860 in the IL28B gene, is particularly interesting, as it suggests a possible molecular mechanism. The delta G frameshift variant creates a novel gene called IFNL4, which is transiently activated in response to hepatitis C virus infection.24IFNL4 stimulates STAT1 and STAT2 phosphorylation and induces the expression of interferon-stimulated genes. Increased interferon-stimulated gene expression has been shown to be associated with decreased response to pegylated interferon-ribavirin treatment. These observations suggest that the ss469425590 delta G allele is responsible for the increased activation of interferon-stimulated genes and the lower sustained virologic response rate observed in patients who receive pegylated interferon-ribavirin treatment. It is possible that the activation of interferon-stimulated genes in patients with the ss469425590 delta G/delta G genotype reduces interferon-stimulated gene responsiveness to interferon alpha, which normally activates interferon-stimulated genes and inhibits hepatitis C progression.24
IL28B POLYMORPHISM AND ACUTE HEPATITIS C VIRUS INFECTION
From 70% to 80% of acute hepatitis C virus infections persist and become chronic, while 20% to 30% spontaneously resolve. Epidemiologic, viral, and host factors have been associated with the differences in viral clearance or persistence, and studies have found that a strong host immune response against the virus favors viral clearance. Thus, variation in the genes involved in the immune response may contribute to one’s ability to clear the virus. Consistent with these observations, recent studies have shown that the polymorphism in the IL28 gene region encoding interferon lambda 3 strongly predicts spontaneous resolution of acute hepatitis C virus infection. People who have the IL28B CC genotype are three times more likely to spontaneously clear the virus than those with the CT or TT genotype (Figure 3).24
IL28B POLYMORPHISM AND THE NATURAL HISTORY OF HEPATITIS C
In people in whom hepatitis C virus infection persists, up to 20% develop progressive liver fibrosis and eventually cirrhosis over 10 to 20 years.19,25,26 The speed at which fibrosis develops in these patients is variable and unpredictable.25 The relationship between IL28B polymorphisms and hepatic fibrosis in patients with chronic hepatitis C virus infection has not been clearly established, although a study indicated that in patients with a known date of infection, the IL28B genotype is not associated with progression of hepatic fibrosis.27 Obstacles in this field of study are that it is difficult to determine accurately when the patient contracted the virus, and that serial liver biopsies are needed to investigate the progression of hepatic fibrosis.
Patients with chronic hepatitis C virus infection are also at higher risk of hepatocellular carcinoma compared with the general population.28 An analysis of explanted livers of patients with hepatitis C found that the prevalence of hepatocellular carcinoma in those with the unfavorable TT genotype was significantly higher than with the other genotypes.29 Similarly, an earlier study demonstrated that patients with hepatitis C-associated hepatocellular carcinoma carried the T allele more frequently.30 As with other aspects of IL28B associations with hepatitis C, these findings indicate that the C allele confers a certain degree of protection.
An important implication of these relationships is that they may eventually help identify patients at greater risk, who therefore need earlier intervention.
IL28B POLYMORPHISM AND LIVER TRANSPLANTATION
Hepatitis C virus infection always recurs after liver transplantation, with serious consequences that include cirrhosis and liver failure. Recurrent hepatitis C virus infection has become an important reason for repeat transplantation in the United States.
Results of treatment with pegylated interferon and ribavirin for recurrent hepatitis C after liver transplantation have been disappointing, with response rates lower than 30% and significant side effects.31 Identifying the factors that predict the response to therapy allows for better selection of treatment candidates.
Similar to the way the IL28B genotype predicts response to antiviral therapy in the nontransplant setting, the IL28B genotypes of both the recipient and the donor are strongly and independently associated with response to interferon-based treatment in patients with hepatitis C after liver transplantation. The IL28B CC genotype in either the recipient or the donor is associated with a higher rate of response to pegylated interferon and ribavirin combination therapy after liver transplantation.30,32 For example, the response rate to therapy after liver transplantation reaches 86% in CC-donor and CC-recipient livers, compared with 0% in TT-donor and TT-recipient livers.
Additionally, the IL28B genotype of the recipient may determine the severity of histologic recurrence of hepatitis C, as indicated by progressive hepatic fibrosis. A recipient IL28B TT genotype is associated with more severe histologic recurrence of hepatitis C.33
These data suggest that CC donor livers might be preferentially allocated to patients with hepatitis C virus infection.
IL28B AND OTHER FACTORS IN HEPATITIS C VIRUS INFECTION
Although it is tempting to think that the IL28B polymorphism is the sole predictor of response to antiviral therapy, it is but one of several known factors in the virus and the host.
While IL28B polymorphisms are the most important predictor of sustained virologic response with an interferon-based regimen, a rapid virologic response (undetectable viral load at 4 weeks) had superior predictive value and specificity in one study.34 In fact, for patients with chronic hepatitis C infection who achieved a rapid virologic response with pegylated interferon and ribavirin, the IL28B polymorphism had no effect on the rate of sustained virologic response. However, it did predict a sustained virologic response in the group who did not achieve rapid virologic response.
In a study of patients with acute hepatitis C infection,35 jaundice and the IL28 rs12979860 CC genotype both predicted spontaneous clearance. The best predictor of viral persistence was the combination of the CT or TT genotype plus the absence of jaundice, which had a predictive value of 98%.
IL28B AND THE FUTURE OF HEPATITIS C VIRUS THERAPY
New oral agents were recently approved for treating hepatitis C. As of November 2014, these included simeprevir, sofosbuvir, and ledipasvir.
Simeprevir is a second-generation NS3/4A protease inhibitor approved for use in combination with pegylated interferon and ribavirin. A recent phase 3 trial evaluating simeprevir in patients who had relapsed after prior therapy found sustained virologic response rates to be higher with simeprevir than with placebo, irrespective of IL28B status.36 This finding was similar to that of a trial of telaprevir.16
Sofosbuvir is a nucleotide analogue NS5B polymerase inhibitor that becomes incorporated into the growing RNA, inducing a chain termination event.37 In phase 3 trials,38,39 researchers found an initial rapid decrease in viral load for patients treated with this agent regardless of IL28B status.
In the NEUTRINO trial (Sofosbuvir With Peginterferon Alfa 2a and Ribavirin for 12 Weeks in Treatment-Naive Subjects With Chronic Genotype 1, 4, 5, or 6 HCV Infection),38 which used sofusbuvir in combination with interferon and ribavirin, the rate of sustained virologic response was higher in those with the favorable CC genotype (98%) than with a non-CC genotype (87%).
In COSMOS (A Study of TMC435 in Combination With PSI-7977 [GS7977] in Chronic Hepatitis C Genotype 1-Infected Prior Null Responders to Peginterferon/Ribavirin Therapy or HCV Treatment-Naive Patients),39 which used a combination of simeprevir, sofosbuvir, and ribavirin, the rate of sustained virologic response was higher in those with the CC genotype (100%) than with the TT genotype (83%; Table 1).
These new medications have radically changed the landscape of hepatitis C therapy and have also unlocked the potential for developing completely interferon-free regimens.
Other new interferon-free regimens such as ledipasvir, daclatasvir, and asunaprevir promise high rates of sustained virologic response, which makes the utility of testing for IL28B polymorphisms to predict sustained virologic response very much diminished (Table 1).40,41 However, these new drugs are expected to be expensive, and IL28B polymorphisms may be used to identify candidates who are more likely to respond to pegylated interferon and ribavirin, particularly in resource-poor settings and in developing countries. Additionally, patients who have contraindications to these newer therapies will still likely need an interferon-based regimen, and thus the IL28B polymorphism will still be important in predicting treatment response and prognosis.
IL28B WILL STILL BE RELEVANT IN THE INTERFERON-FREE AGE
The IL28B polymorphism is a strong predictor of spontaneous clearance of hepatitis C virus and responsiveness to interferon-based therapy, and testing for it has demonstrated a great potential to improve patient care. IL28B testing has become available for clinical use and may optimize the outcome of hepatitis C treatment by helping us to select the best treatment for individual patients and minimizing the duration of therapy and the side effects associated with interferon-based antiviral medications.
As newer therapies have shifted toward interferon-free regimens that offer very high sustained virologic response rates, the usefulness of IL28B polymorphism as a clinical test to predict the response rate to antiviral therapy is minimized substantially. It may remain clinically relevant in resource-poor settings and in developing countries, especially in light of the potentially prohibitive costs of the newer regimens, and for patients in whom these treatments are contraindicated. This does not minimize the lesson we learned from the discovery of the IL28B gene and the impact on our understanding of the pathogenesis of hepatitis C virus infection.
- Attia J, Ioannidis JP, Thakkinstian A, et al. How to use an article about genetic association: A: background concepts. JAMA 2009; 301:74–81.
- Samani NJ, Erdmann J, Hall AS, et al; WTCCC and the Cardiogenics Consortium. Genomewide association analysis of coronary artery disease. N Engl J Med 2007; 357:443–453.
- Zeggini E, Weedon MN, Lindgren CM, et al; Wellcome Trust Case Control Consortium (WTCCC). Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science 2007; 316:1336–1341.
- Matarín M, Brown WM, Scholz S, et al. A genome-wide genotyping study in patients with ischaemic stroke: initial analysis and data release. Lancet Neurol 2007; 6:414–420.
- Easton DF, Pooley KA, Dunning AM, et al; AOCS Management Group. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature 2007; 447:1087–1093.
- Plenge RM, Seielstad M, Padyukov L, et al. TRAF1-C5 as a risk locus for rheumatoid arthritis—a genomewide study. N Engl J Med 2007; 357:1199–1209.
- Coon KD, Myers AJ, Craig DW, et al. A high-density whole-genome association study reveals that APOE is the major susceptibility gene for sporadic late-onset Alzheimer’s disease. J Clin Psychiatry 2007; 68:613–618.
- Ge D, Fellay J, Thompson AJ, et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance (letter). Nature 2009; 461:399–401.
- Ali A, Zein NN. Hepatitis C infection: a systemic disease with extrahepatic manifestations. Cleve Clin J Med 2005; 72:1005-1019.
- Hanouneh IA, Feldstein AE, Lopez R, et al. Clinical significance of metabolic syndrome in the setting of chronic hepatitis C virus infection. Clin Gastroenterol Hepatol 2008; 6:584–589.
- Elgouhari HM, Zein CO, Hanouneh I, Feldstein AE, Zein NN. Diabetes mellitus is associated with impaired response to antiviral therapy in chronic hepatitis C infection. Dig Dis Sci 2009; 54:2699–2705.
- Alkhouri N, Zein NN. Protease inhibitors: silver bullets for chronic hepatitis C infection? Cleve Clin J Med 2012; 79:213–222.
- McHutchison JG, Everson GT, Gordon SC, et al; PROVE1 Study Team. Telaprevir with peginterferon and ribavirin for chronic HCV genotype 1 infection. N Engl J Med 2009; 360:1827–1838.
- Jacobson IM, McHutchison JG, Dusheiko G, et al; ADVANCE Study Team. Telaprevir for previously untreated chronic hepatitis C virus infection. N Engl J Med 2011; 364:2405–2416.
- Jacobson IM, Catlett I, Marcellin P, et al. Telaprevir substantially improved SVR rates across all IL28B genotypes in the ADVANCE trial. J Hepatol 2011; 54(suppl 1):S542–S543.
- Pol S, Aerssens J, Zeuzem S, et al. Limited impact of IL28B genotype on response rates in telaprevir-treated patients with prior treatment failure. J Hepatol 2013; 58:883–889.
- Suppiah V, Moldovan M, Ahlenstiel G, et al. IL28B is associated with response to chronic hepatitis C interferon-alpha and ribavirin therapy. Nat Genet 2009; 41:1100–1104.
- Tanaka Y, Nishida N, Sugiyama M, et al. Genome-wide association of IL28B with response to pegylated interferon-alpha and ribavirin therapy for chronic hepatitis C. Nat Genet 2009; 41:1105–1109.
- Thomas DL, Thio CL, Martin MP, et al. Genetic variation in IL28B and spontaneous clearance of hepatitis C virus. Nature 2009; 461:798–801.
- Rehermann B. Hepatitis C virus versus innate and adaptive immune responses: a tale of coevolution and coexistence. J Clin Invest 2009; 119:1745–1754.
- Marcello T, Grakoui A, Barba-Spaeth G, et al. Interferons alpha and lambda inhibit hepatitis C virus replication with distinct signal transduction and gene regulation kinetics. Gastroenterology 2006; 131:1887–1898.
- Doyle SE, Schreckhise H, Khuu-Duong K, et al. Interleukin-29 uses a type 1 interferon-like program to promote antiviral responses in human hepatocytes. Hepatology 2006; 44:896–906.
- Raglow Z, Thoma-Perry C, Gilroy R, Wan YJ. IL28B genotype and the expression of ISGs in normal liver. Liver Int 2013; 33:991–998.
- Prokunina-Olsson L, Muchmore B, Tang W, et al. A variant upstream of IFNL3 (IL28B) creating a new interferon gene IFNL4 is associated with impaired clearance of hepatitis C virus. Nat Genet 2013; 45:164–171.
- Hanouneh IA, Zein NN, Askar M, Lopez R, John B. Interleukin-28B polymorphisms are associated with fibrosing cholestatic hepatitis in recurrent hepatitis C after liver transplantation. Clin Transplant 2012; 26:E335–E336.
- Poynard T, Bedossa P, Opolon P. Natural history of liver fibrosis progression in patients with chronic hepatitis C. The OBSVIRC, METAVIR, CLINIVIR, and DOSVIRC groups. Lancet 1997; 349:825–832.
- Thomas DL, Astemborski J, Rai RM, et al. The natural history of hepatitis C virus infection: host, viral, and environmental factors. JAMA 2000; 284:450–456.
- Bochud PY, Cai T, Overbeck K, et al; Swiss Hepatitis C Cohort Study Group. Genotype 3 is associated with accelerated fibrosis progression in chronic hepatitis C. J Hepatol 2009; 51:655–666.
- Marabita F, Aghemo A, De Nicola S, et al. Genetic variation in the interleukin-28B gene is not associated with fibrosis progression in patients with chronic hepatitis C and known date of infection. Hepatology 2011; 54:1127–1134.
- Fabris C, Falleti E, Cussigh A, et al. IL-28B rs12979860 C/T allele distribution in patients with liver cirrhosis: role in the course of chronic viral hepatitis and the development of HCC. J Hepatol 2011; 54:716–722.
- Eurich D, Boas-Knoop S, Bahra M, et al. Role of IL28B polymorphism in the development of hepatitis C virus-induced hepatocellular carcinoma, graft fibrosis, and posttransplant antiviral therapy. Transplantation 2012; 93:644–649.
- Hanouneh IA, Miller C, Aucejo F, Lopez R, Quinn MK, Zein NN. Recurrent hepatitis C after liver transplantation: on-treatment prediction of response to peginterferon/ribavirin therapy. Liver Transpl 2008; 14:53–58.
- Charlton MR, Thompson A, Veldt BJ, et al. Interleukin-28B polymorphisms are associated with histological recurrence and treatment response following liver transplantation in patients with hepatitis C virus infection. Hepatology 2011; 53:317–324.
- Thompson AJ, Muir AJ, Sulkowski MS, et al. Interleukin-28B polymorphism improves viral kinetics and is the strongest pretreatment predictor of sustained virologic response in genotype 1 hepatitis C virus. Gastroenterology 2010; 139:120–129.e18.
- Beinhardt S, Payer BA, Datz C, et al. A diagnostic score for the prediction of spontaneous resolution of acute hepatitis C virus infection. J Hepatol 2013; 59:972–977.
- Forns X, Lawitz E, Zeuzem S, et al. Simeprevir with peginterferon and ribavirin leads to high rates of SVR in patients with HCV genotype 1 who relapsed after previous therapy: a phase 3 trial. Gastroenterology 2014; 146:1669–1679.e3.
- Sofia MJ, Bao D, Chang W, et al. Discovery of a ß-d-2’-deoxy-2’-ß-fluoro-2’-ß-C-methyluridine nucleotide prodrug (PSI-7977) for the treatment of hepatitis C virus. J Med Chem 2010; 53:7202–7218.
- Lawitz E, Mangia A, Wyles D, et al. Sofosbuvir for previously untreated chronic hepatitis C infection. N Engl J Med 2013; 368:1878–1887.
- Sulkowski MS, Jacobson IM, Ghalib R, et al. Once-daily simeprevir (TMC435) plus sofosbuvir (GS-7977) with or without ribavirin in HCV genotype 1 prior null responders with metavir F0-2: COSMOS study subgroup analysis. 49th EASL, April 2014, London. Oral abstract O7. www.natap.org/2014/EASL/EASL_46.htm. Accesed January 9, 2015.
- Lok AS, Gardiner DF, Lawitz E, et al. Preliminary study of two antiviral agents for hepatitis C genotype 1. N Engl J Med 2012; 366:216–224.
- Afdhal N, Zeuzem S, Kwo P, et al; ION-1 Investigators. Ledipasvir and sofosbuvir for untreated HCV genotype 1 infection. N Engl J Med 2014; 370:1889–1898.
- Attia J, Ioannidis JP, Thakkinstian A, et al. How to use an article about genetic association: A: background concepts. JAMA 2009; 301:74–81.
- Samani NJ, Erdmann J, Hall AS, et al; WTCCC and the Cardiogenics Consortium. Genomewide association analysis of coronary artery disease. N Engl J Med 2007; 357:443–453.
- Zeggini E, Weedon MN, Lindgren CM, et al; Wellcome Trust Case Control Consortium (WTCCC). Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science 2007; 316:1336–1341.
- Matarín M, Brown WM, Scholz S, et al. A genome-wide genotyping study in patients with ischaemic stroke: initial analysis and data release. Lancet Neurol 2007; 6:414–420.
- Easton DF, Pooley KA, Dunning AM, et al; AOCS Management Group. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature 2007; 447:1087–1093.
- Plenge RM, Seielstad M, Padyukov L, et al. TRAF1-C5 as a risk locus for rheumatoid arthritis—a genomewide study. N Engl J Med 2007; 357:1199–1209.
- Coon KD, Myers AJ, Craig DW, et al. A high-density whole-genome association study reveals that APOE is the major susceptibility gene for sporadic late-onset Alzheimer’s disease. J Clin Psychiatry 2007; 68:613–618.
- Ge D, Fellay J, Thompson AJ, et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance (letter). Nature 2009; 461:399–401.
- Ali A, Zein NN. Hepatitis C infection: a systemic disease with extrahepatic manifestations. Cleve Clin J Med 2005; 72:1005-1019.
- Hanouneh IA, Feldstein AE, Lopez R, et al. Clinical significance of metabolic syndrome in the setting of chronic hepatitis C virus infection. Clin Gastroenterol Hepatol 2008; 6:584–589.
- Elgouhari HM, Zein CO, Hanouneh I, Feldstein AE, Zein NN. Diabetes mellitus is associated with impaired response to antiviral therapy in chronic hepatitis C infection. Dig Dis Sci 2009; 54:2699–2705.
- Alkhouri N, Zein NN. Protease inhibitors: silver bullets for chronic hepatitis C infection? Cleve Clin J Med 2012; 79:213–222.
- McHutchison JG, Everson GT, Gordon SC, et al; PROVE1 Study Team. Telaprevir with peginterferon and ribavirin for chronic HCV genotype 1 infection. N Engl J Med 2009; 360:1827–1838.
- Jacobson IM, McHutchison JG, Dusheiko G, et al; ADVANCE Study Team. Telaprevir for previously untreated chronic hepatitis C virus infection. N Engl J Med 2011; 364:2405–2416.
- Jacobson IM, Catlett I, Marcellin P, et al. Telaprevir substantially improved SVR rates across all IL28B genotypes in the ADVANCE trial. J Hepatol 2011; 54(suppl 1):S542–S543.
- Pol S, Aerssens J, Zeuzem S, et al. Limited impact of IL28B genotype on response rates in telaprevir-treated patients with prior treatment failure. J Hepatol 2013; 58:883–889.
- Suppiah V, Moldovan M, Ahlenstiel G, et al. IL28B is associated with response to chronic hepatitis C interferon-alpha and ribavirin therapy. Nat Genet 2009; 41:1100–1104.
- Tanaka Y, Nishida N, Sugiyama M, et al. Genome-wide association of IL28B with response to pegylated interferon-alpha and ribavirin therapy for chronic hepatitis C. Nat Genet 2009; 41:1105–1109.
- Thomas DL, Thio CL, Martin MP, et al. Genetic variation in IL28B and spontaneous clearance of hepatitis C virus. Nature 2009; 461:798–801.
- Rehermann B. Hepatitis C virus versus innate and adaptive immune responses: a tale of coevolution and coexistence. J Clin Invest 2009; 119:1745–1754.
- Marcello T, Grakoui A, Barba-Spaeth G, et al. Interferons alpha and lambda inhibit hepatitis C virus replication with distinct signal transduction and gene regulation kinetics. Gastroenterology 2006; 131:1887–1898.
- Doyle SE, Schreckhise H, Khuu-Duong K, et al. Interleukin-29 uses a type 1 interferon-like program to promote antiviral responses in human hepatocytes. Hepatology 2006; 44:896–906.
- Raglow Z, Thoma-Perry C, Gilroy R, Wan YJ. IL28B genotype and the expression of ISGs in normal liver. Liver Int 2013; 33:991–998.
- Prokunina-Olsson L, Muchmore B, Tang W, et al. A variant upstream of IFNL3 (IL28B) creating a new interferon gene IFNL4 is associated with impaired clearance of hepatitis C virus. Nat Genet 2013; 45:164–171.
- Hanouneh IA, Zein NN, Askar M, Lopez R, John B. Interleukin-28B polymorphisms are associated with fibrosing cholestatic hepatitis in recurrent hepatitis C after liver transplantation. Clin Transplant 2012; 26:E335–E336.
- Poynard T, Bedossa P, Opolon P. Natural history of liver fibrosis progression in patients with chronic hepatitis C. The OBSVIRC, METAVIR, CLINIVIR, and DOSVIRC groups. Lancet 1997; 349:825–832.
- Thomas DL, Astemborski J, Rai RM, et al. The natural history of hepatitis C virus infection: host, viral, and environmental factors. JAMA 2000; 284:450–456.
- Bochud PY, Cai T, Overbeck K, et al; Swiss Hepatitis C Cohort Study Group. Genotype 3 is associated with accelerated fibrosis progression in chronic hepatitis C. J Hepatol 2009; 51:655–666.
- Marabita F, Aghemo A, De Nicola S, et al. Genetic variation in the interleukin-28B gene is not associated with fibrosis progression in patients with chronic hepatitis C and known date of infection. Hepatology 2011; 54:1127–1134.
- Fabris C, Falleti E, Cussigh A, et al. IL-28B rs12979860 C/T allele distribution in patients with liver cirrhosis: role in the course of chronic viral hepatitis and the development of HCC. J Hepatol 2011; 54:716–722.
- Eurich D, Boas-Knoop S, Bahra M, et al. Role of IL28B polymorphism in the development of hepatitis C virus-induced hepatocellular carcinoma, graft fibrosis, and posttransplant antiviral therapy. Transplantation 2012; 93:644–649.
- Hanouneh IA, Miller C, Aucejo F, Lopez R, Quinn MK, Zein NN. Recurrent hepatitis C after liver transplantation: on-treatment prediction of response to peginterferon/ribavirin therapy. Liver Transpl 2008; 14:53–58.
- Charlton MR, Thompson A, Veldt BJ, et al. Interleukin-28B polymorphisms are associated with histological recurrence and treatment response following liver transplantation in patients with hepatitis C virus infection. Hepatology 2011; 53:317–324.
- Thompson AJ, Muir AJ, Sulkowski MS, et al. Interleukin-28B polymorphism improves viral kinetics and is the strongest pretreatment predictor of sustained virologic response in genotype 1 hepatitis C virus. Gastroenterology 2010; 139:120–129.e18.
- Beinhardt S, Payer BA, Datz C, et al. A diagnostic score for the prediction of spontaneous resolution of acute hepatitis C virus infection. J Hepatol 2013; 59:972–977.
- Forns X, Lawitz E, Zeuzem S, et al. Simeprevir with peginterferon and ribavirin leads to high rates of SVR in patients with HCV genotype 1 who relapsed after previous therapy: a phase 3 trial. Gastroenterology 2014; 146:1669–1679.e3.
- Sofia MJ, Bao D, Chang W, et al. Discovery of a ß-d-2’-deoxy-2’-ß-fluoro-2’-ß-C-methyluridine nucleotide prodrug (PSI-7977) for the treatment of hepatitis C virus. J Med Chem 2010; 53:7202–7218.
- Lawitz E, Mangia A, Wyles D, et al. Sofosbuvir for previously untreated chronic hepatitis C infection. N Engl J Med 2013; 368:1878–1887.
- Sulkowski MS, Jacobson IM, Ghalib R, et al. Once-daily simeprevir (TMC435) plus sofosbuvir (GS-7977) with or without ribavirin in HCV genotype 1 prior null responders with metavir F0-2: COSMOS study subgroup analysis. 49th EASL, April 2014, London. Oral abstract O7. www.natap.org/2014/EASL/EASL_46.htm. Accesed January 9, 2015.
- Lok AS, Gardiner DF, Lawitz E, et al. Preliminary study of two antiviral agents for hepatitis C genotype 1. N Engl J Med 2012; 366:216–224.
- Afdhal N, Zeuzem S, Kwo P, et al; ION-1 Investigators. Ledipasvir and sofosbuvir for untreated HCV genotype 1 infection. N Engl J Med 2014; 370:1889–1898.
KEY POINTS
- In IL28B, the rs12979860 location can be occupied by either cytosine (C) or thymine (T). The CC genotype is more favorable than the CT or TT genotype.
- Testing for the IL28B polymorphism is currently available and allows for better outcomes through proper selection of treatment, particularly with interferon-based treatment.
- Although newer therapies have shifted toward regimens that do not use interferon, the IL28B polymorphism remains clinically significant, especially in light of the potentially prohibitive costs of the newer regimens, and for patients in whom these treatments are contraindicated.
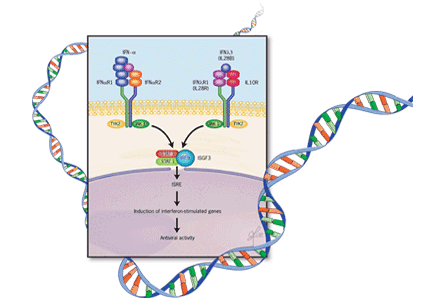
Protease inhibitors: Silver bullets for chronic hepatitis C infection?
The treatment of hepatitis c virus (HCV) infection is on the brink of major changes with the recent approval of the first direct-acting antiviral agents, the protease inhibitors boceprevir (Victrelis) and telaprevir (Incivek).
Both drugs were approved by the US Food and Drug Administration (FDA) Advisory Panel for Chronic Hepatitis C in May 2011 and are believed to significantly improve treatment outcomes for patients with HCV genotype 1 infection.
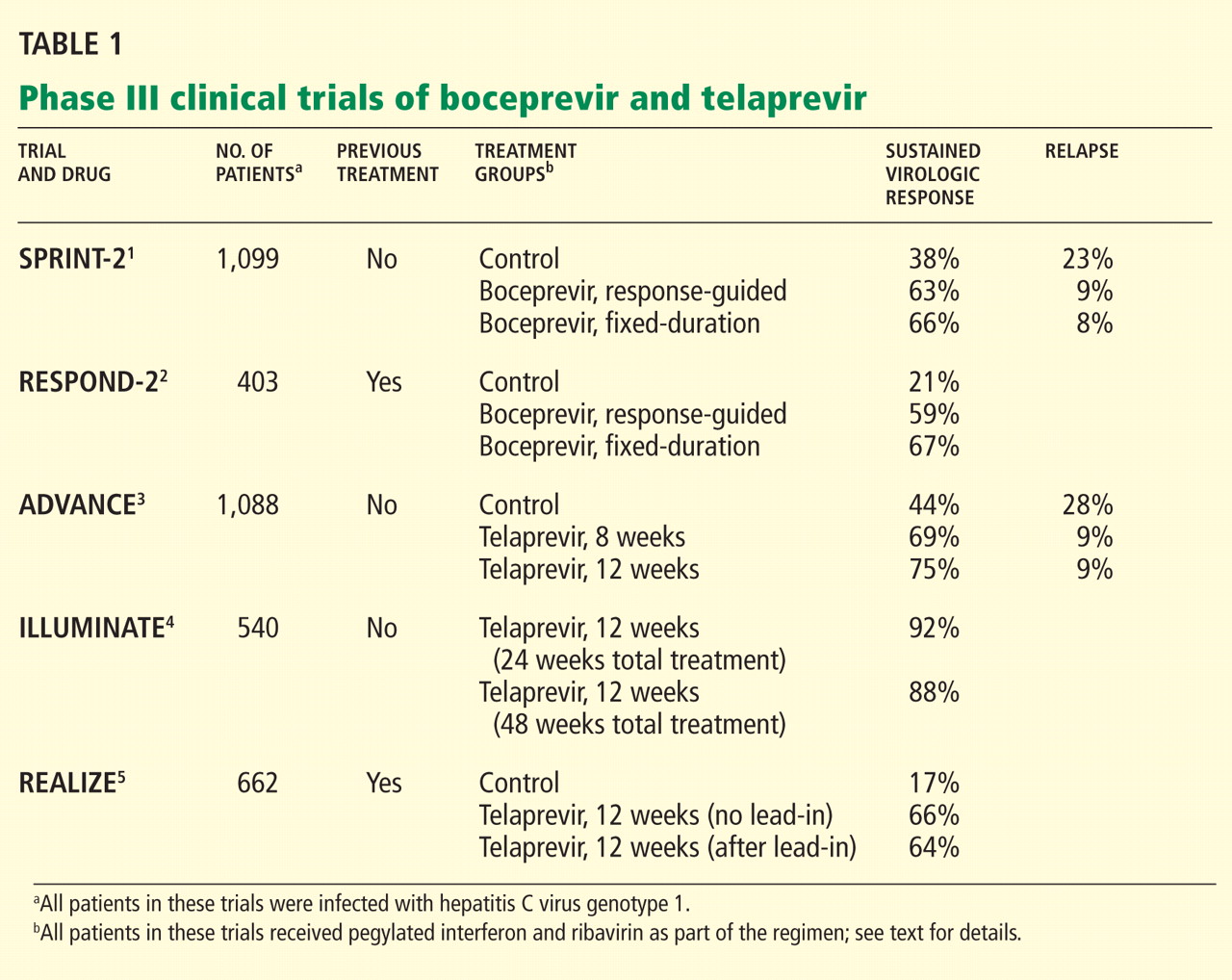
A MAJOR PUBLIC HEALTH PROBLEM
HCV infection is a major public health problem. Nearly 4 million people in the United States are infected.6,7 Most patients with acute HCV infection become chronically infected, and up to 25% eventually develop cirrhosis and its complications, making HCV infection the leading indication for liver transplantation.8–10
Chronic HCV infection has a large global impact, with 180 million people affected across all economic and social groups.11 The highest prevalence of HCV has been reported in Egypt (14%), in part due to the use of inadequately sterilized needles in mass programs to treat endemic schistosomiasis. In developed countries, hepatocellular carcinoma associated with HCV has the fastest growing cancer-related death rate.12
CURRENTLY, FEWER THAN 50% OF PATIENTS ARE CURED
The goal of HCV treatment is to eradicate the virus. However, most infected patients (especially in the United States and Europe) are infected with HCV genotype 1, which is the most difficult genotype to treat.
Successful treatment of HCV is defined as achieving a sustained virologic response—ie, the absence of detectable HCV RNA in the serum 24 weeks after completion of therapy. Once a sustained virologic response is achieved, lifetime “cure” of HCV infection is expected in more than 99% of patients.13
The current standard therapy for HCV, pegylated interferon plus ribavirin for 48 weeks, is effective in only 40% to 50% of patients with genotype 1 infection.14 Therefore, assessing predictors of response before starting treatment can help select patients who are most likely to benefit from therapy.
Viral factors associated with a sustained virologic response include HCV genotypes other than genotype 1 and a low baseline viral load.
Beneficial patient-related factors include younger age, nonblack ethnicity, low body weight (≤ 75 kg), low body mass index, absence of insulin resistance, and absence of advanced fibrosis or cirrhosis.
More recently, a single-nucleotide polymorphism near the interleukin 28B (IL28B) gene, coding for interferon lambda 3, was found to be associated with a twofold difference in the rates of sustained virologic response: patients with the favorable genotype CC were two times more likely to achieve a sustained virologic response than patients with the CT or TT genotypes.15–17
PROTEASE INHIBITORS: MECHANISM OF ACTION
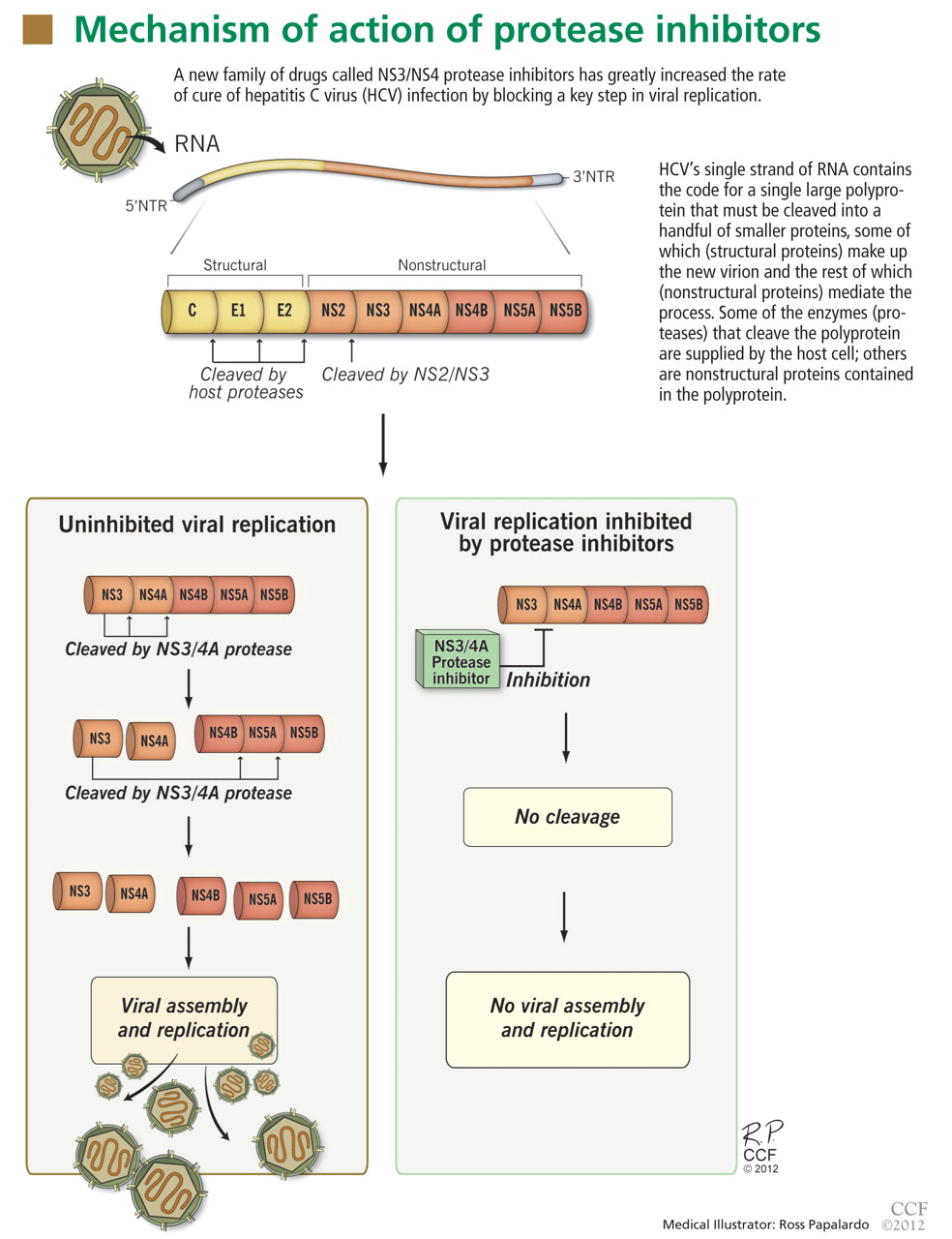
NS3/4A protease inhibitors rely on the principle of end-product inhibition, in which the cleavage product of the protease (a peptide) acts to inhibit the enzyme activity; this is why they are called peptidomimetics. The active site of the NS3/4A protease is a shallow groove composed of three highly conserved amino acid residues, which may explain why protease inhibitors display high antiviral efficacy but pose a low barrier to the development of resistance.20
Protease inhibitors are prone to resistance
The development of viral resistance to protease inhibitors has been a major drawback to their use in patients with chronic HCV infection.21
HCV is a highly variable virus with many genetically distinct but closely related quasispecies circulating in the blood at any given time. Drug-resistant, mutated variants preexist within the patient’s quasispecies, but only in small quantities because of their lesser replication fitness compared with the wild-type virus.22 When direct-acting antiviral therapy is started, the quantity of the wild-type virus decreases and the mutated virus gains replication fitness. Using protease inhibitors as monotherapy selects resistant viral populations rapidly within a few days or weeks.
HCV subtypes 1a and 1b may have different resistance profiles. With genotype 1a, some resistance-associated amino acid substitutions require only one nucleotide change, but with genotype 1b, two nucleotide changes are needed, making resistance less frequent in patients with HCV genotype 1b.23
BOCEPREVIR
Boceprevir is a specific inhibitor of the HCV viral protease NS3/4A.
In phase 3 clinical trials, boceprevir 800 mg three times a day was used with pegylated interferon alfa-2b (PegIntron) 1.5 μg/kg/week and ribavirin (Rebetol) 600 to 1,400 mg daily according to body weight.
Before patients started taking boceprevir, they went through a 4-week lead-in phase, during which they received pegylated interferon and ribavirin. This schedule appeared to reduce the incidence of viral breakthrough in phase 2 trials, and it produced higher rates of sustained virologic response and lower relapse rates compared with triple therapy without a lead-in phase.
Rapid virologic response was defined as undetectable HCV RNA at week 4 of boceprevir therapy (week 8 of the whole regimen).
Boceprevir in previously untreated patients with HCV genotype 1: The SPRINT-2 trial
The Serine Protease Inhibitor Therapy 2 (SPRINT-2) trial1 included more than 1,000 previously untreated adults with HCV genotype 1 infection (938 nonblack patients and 159 black patients; two other nonblack patients did not receive any study drug and were not included in the analysis). In this double-blind trial, patients were randomized into three groups:
- The control group received the standard of care with pegylated interferon and ribavirin for 48 weeks
- The response-guided therapy group received boceprevir plus pegylated interferon and ribavirin for 24 weeks after the 4-week lead-in phase; if HCV RNA was undetectable from week 8 to week 24, treatment was considered complete, but if HCV RNA was detectable at any point from week 8 to week 24, pegylated interferon and ribavirin were continued for a total of 48 weeks.
- The fixed-duration therapy group received boceprevir, pegylated interferon, and ribavirin for 44 weeks after the lead-in period.
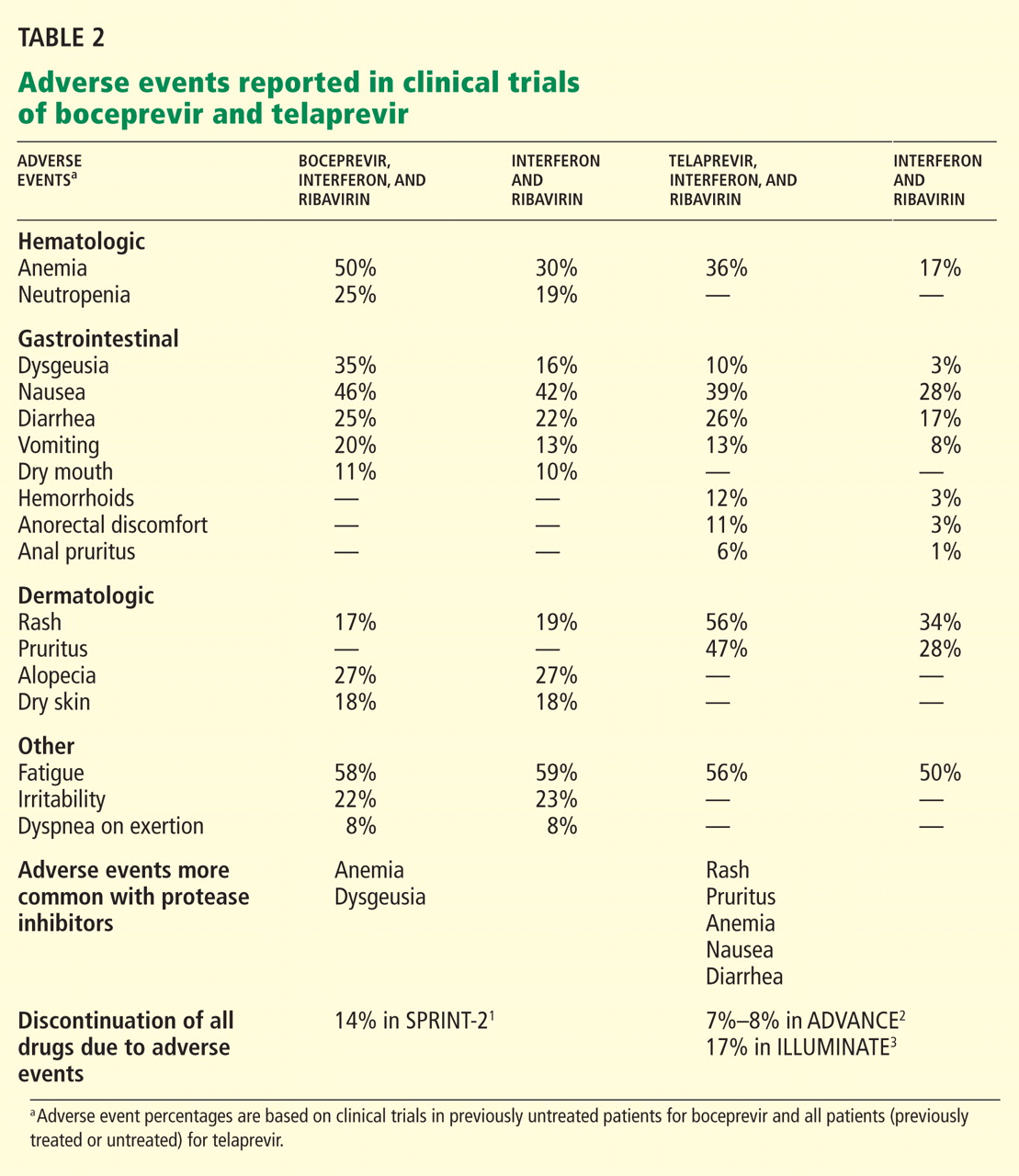
The rate of relapse was 8% and 9% in the boceprevir groups vs 23% in the control group. Patients in the boceprevir groups who had a decrease in HCV RNA of less than 1 log10 during the lead-in phase were found to have a significantly higher rate of boceprevirresistant variants than those who achieved a decrease of HCV RNA of 1 log10 or more.
Boceprevir in previously treated patients with HCV genotype 1: The RESPOND-2 trial
The Retreatment With HCV Serine Protease Inhibitor Boceprevir and PegIntron/Rebetol 2) (RESPOND-2) trial2 was designed to assess the efficacy of combined boceprevir, pegylated interferon, and ribavirin for repeat treatment of patients with HCV genotype 1. These patients had previously undergone standard treatment and had a reduction of 2 log10 or more in HCV RNA after 12 weeks of therapy but with detectable HCV RNA during the therapy period or had had a relapse (defined as undetectable HCV RNA at the end of a previous course of therapy with HCV RNA positivity thereafter). Importantly, null-responders (those who had a reduction of less than 2 log10 in HCV RNA after 12 weeks of therapy) were excluded from this trial.
After a lead-in period of interferon-ribavirin treatment for 4 weeks, 403 patients were assigned to one of three treatment groups:
- Pegylated interferon and ribavirin for 44 weeks (the control group)
- Boceprevir, pegylated interferon, and ribavirin in a response-guided regimen
- Boceprevir, pegylated interferon, and ribavirin for 44 weeks (the fixed-duration group).
Sustained virologic response was achieved in only 21% of patients in the control group. Adding boceprevir increased the rate to 59% in the response-guided therapy group and to 67% in the fixed-duration group. Previous relapsers had better rates than partial responders (69%–75% vs 40%–52%).
Importantly, patients who had a poor response to pegylated interferon and ribavirin during the lead-in phase (defined as having less than a 1-log decrease in the virus before starting boceprevir) had significantly lower rates of sustained virologic response and higher rates of resistance-associated virus variants.
Side effects of boceprevir
Overall, boceprevir is well tolerated. The most common side effects of triple therapy are those usually seen with pegylated interferon and ribavirin, such as flulike symptoms and fatigue (Table 2). However, anemia was more frequent in the boceprevir groups in both SPRINT-2 and RESPOND-2 (45%–50% compared with 20%–29% in the control groups). Erythropoietin was allowed in these studies and was used in about 40% of patients.
The other common side effect associated with boceprevir was dysgeusia (alteration of taste). Dysgeusia was reported by approximately 40% of patients; however, most dysgeusia events were mild to moderate in intensity and did not lead to treatment cessation.
In the SPRINT-2 trial,1 the study drugs had to be discontinued in 12% to 16% of patients in the boceprevir groups because of adverse events, which was similar to the rate (16%) in the control group. Erythropoietin was allowed in this trial, and it was used in 43% of patients in the boceprevir groups compared with 24% in the control group, with discontinuation owing to anemia occurring in 2% and 1% of cases, respectively.
TELAPREVIR
Telaprevir, the other protease NS3/4A inhibitor, has also shown efficacy over current standard therapy in phase 3 clinical trials. It was used in a dose of 750 mg three times a day with pegylated interferon alfa-2a (Pegasys) 180 μg per week and ribavirin (Copegus) 1,000 to 1,200 mg daily according to body weight. A lead-in phase with pegylated interferon and ribavirin was not applied with telaprevir, as it was in the boceprevir trials. Extended rapid virologic response was defined as an undetectable HCV RNA at weeks 4 and 12 of therapy.
Telaprevir in previously untreated patients with HCV genotype 1
The ADVANCE study3 was a double-blind randomized trial assessing the efficacy and safety of telaprevir in combination with pegylated interferon and ribavirin in more than 1,000 previously untreated patients. The three treatment groups received:
- Telaprevir, pegylated interferon, and ribavirin for 8 weeks, followed by pegylated interferon and ribavirin alone for 16 weeks in patients who achieved an extended rapid virologic response (total duration of 24 weeks) or 40 weeks in patients who did not (total duration of 48 weeks)
- Telaprevir, pegylated interferon, and ribavirin for 12 weeks, followed by pegylated interferon-ribavirin alone for 12 (total of 24 weeks) or 36 weeks (total of 48 weeks) according to extended rapid virologic response
- Standard care with pegylated interferon and ribavirin for 48 weeks.
The rate of sustained virologic response was 69% in the group that received telaprevir for 8 weeks and 75% in the group that received it for 12 weeks compared with 44% in the control group (P < .0001 for both) (Table 2). Patients infected with HCV genotype 1b had a higher sustained virologic response rate (79%) than those infected with HCV genotype 1a (71%).
Sustained virologic response rates were lower in black patients and patients with bridging fibrosis or cirrhosis, but were still significantly higher in the telaprevir groups than in the control group. The results of this subset analysis were limited by small numbers of patients in each category.
In total, 57% of those who received telaprevir for 8 weeks and 58% of those who received it for 12 weeks achieved an extended rapid virologic response and were able to cut the duration of their therapy in half (from 48 weeks to 24 weeks).
The relapse rates were 9% in the telaprevir groups and 28% in the control group.
The rate of virologic failure was lower in patients who received triple therapy than in those who received interferon-ribavirin alone (8% in the group that got telaprevir for 12 weeks and 13% in the group that got it for 8 weeks, vs 32% in the control group). The failure rate was also lower in patients with HCV genotype 1b infection than in those with genotype 1a.
The ILLUMINATE study4 (Illustrating the Effects of Combination Therapy With Telaprevir) investigated whether longer duration of treatment than that given in the ADVANCE trial increased the rate of sustained virologic response. Previously untreated patients received telaprevir, interferon, and ribavirin for 12 weeks, and those who achieved an extended rapid virologic response were randomized at week 20 to continue interferonribavirin treatment for 24 or 48 weeks of total treatment.
The sustained virologic response rates in patients who achieved an extended rapid virologic response were 92% in the group that received pegylated interferon and ribavirin for 12 weeks, and 88% in those who received it for 48 weeks. Thus, the results of this study support the use of response-guided therapy for telaprevir-based regimens.
Telaprevir in previously treated patients with HCV genotype 1: The REALIZE trial
In this phase 3 placebo-controlled trial,5 622 patients with prior relapse, partial response, or null response were randomly allocated into one of three groups:
- Telaprevir for 12 weeks plus pegylated interferon and ribavirin for 48 weeks
- Lead-in for 4 weeks followed by 12 weeks of triple therapy and another 32 weeks of pegylated interferon and ribavirin
- Pegylated interferon and ribavirin for 48 weeks (the control group).
The overall sustained virologic response rates were 66% and 64%, respectively, in the telaprevir groups vs 17% in the control group (P < .0001). The sustained virologic response rates in the telaprevir groups were 83% to 88% in prior relapsers, 54% to 59% in partial responders, and 29% to 33% in null-responders. Of note, patients did not benefit from the lead-in phase.
This was the only trial to investigate the response to triple therapy in null-responders, a group in which treatment has been considered hopeless. A response rate of approximately 31% was encouraging, especially if we compare it with the 5% response rate achieved with the current standard of care with pegylated interferon and ribavirin.
Telaprevir side effects
As with boceprevir-based triple therapy, the most common adverse events were related to pegylated interferon (Table 2).
Nearly 50% of patients who receive telaprevir develop a skin rash that is primarily eczematous, can be managed with topical steroids, and usually resolves when telaprevir is discontinued. Severe rashes occurred in 3% to 6% of patients in the ADVANCE trial,3 and three suspected cases of Stevens-Johnson syndrome have been reported to the FDA.
Other side effects that were more frequent with telaprevir included pruritus, nausea, diarrhea, and anemia. On average, the hemoglobin level decreased by an additional 1 g/dL in the telaprevir treatment groups compared with the groups that received only pegylated interferon-ribavirin. Erythropoietin use was not allowed in the phase 3 telaprevir studies, and anemia was managed by ribavirin dose reduction.
In the ADVANCE trial,3 study drugs were discontinued owing to adverse events in 7% to 8% of the patients in the telaprevir groups compared with 4% in the control group. In the ILLUMINATE trial,4 17% of patients had to permanently discontinue all study drugs due to adverse events.
FDA-APPROVED TREATMENT REGIMENS FOR BOCEPREVIR AND TELAPREVIR
For treatment algorithms, see the eFigures that accompany this article online.
Boceprevir in previously untreated patients
- Week 0—Start pegylated interferon and ribavirin
- Week 4—Add boceprevir
- Week 8—Measure HCV RNA
- Week 12—Measure HCV RNA; stop treatment if it is more than 100 IU/mL
- Week 24—Measure HCV RNA; stop treatment if it is detectable
- Week 28—Stop all treatment if HCV RNA was undetectable at weeks 8 and 24
- Week 36—Measure HCV RNA; stop boceprevir
- Week 48—Stop all treatment (eFigure 1).
Boceprevir in previously treated patients
- Week 0—Start pegylated interferon and ribavirin
- Week 4—Add boceprevir
- Week 8—Measure HCV RNA
- Week 12—Measure HCV RNA; stop treatment if it is more than 100 IU/mL
- Week 24—Measure HCV RNA; stop treatment if it is detectable
- Week 36—if HCV RNA was not detectable at week 8, stop all treatment now; if HCV RNA was detectable at week 8, stop boceprevir now but continue pegylated interferon and ribavirin
- Week 48—Stop all treatment (eFigure 2).
Telaprevir in previously untreated patients and prior relapsers
- Week 0—start telaprevir, pegylated interferon, and ribavirin
- Week 4—measure HCV RNA; stop all treatment if it is more than 1,000 IU/mL
- Week 12—Stop telaprevir; measure HCV RNA; stop all treatment if HCV RNA is more than 1,000 IU/mL
- Week 24—Stop pegylated interferon and ribavirin if HCV RNA was undetectable at week 12; measure HCV RNA and stop treatment if it is detectable; otherwise, continue pegylated interferon and ribavirin
- Week 48—Stop all treatment (eFigure 3).
Telaprevir in patients who previously achieved a partial or null response
- Week 0—Start telaprevir, pegylated interferon, and ribavirin
- Week 4—Measure HCV RNA; stop treatment if it is more than 1,000 IU/mL
- Week 12—Measure HCV RNA; stop all treatment if it is more than 1,000 IU/mL; if less than 1,000 IU/mL then stop telaprevir but continue pegylated interferon and ribavirin
- Week 24—Measure HCV RNA; stop treatment if HCV RNA is detectable
- Week 48—Stop all treatment (eFigure 4).
Drug interactions with boceprevir and telaprevir
Both boceprevir and telaprevir inhibit cytochrome P450 3A (CYP3A) and thus are contraindicated in combination with drugs highly dependent on CYP3A for clearance and with drugs for which elevated plasma concentrations are associated with serious adverse events, such as atorvastatin (Lipitor), simvastatin (Zocor), sildenafil (Viagra), midazolam (Versed), and St. John’s wort. Giving potent inducers of CYP3A with boceprevir or telaprevir may lead to lower exposure and loss of efficacy of both protease inhibitors.
EMERGING THERAPIES FOR HCV
Thanks to a better understanding of the biology of HCV infection, the effort to develop new therapeutic agents started to focus on targeting specific steps of the viral life cycle, including attachment, entry into cells, replication, and release.24
Currently, more than 50 clinical trials are evaluating new direct-acting antivirals to treat HCV infection.25 Monoclonal and polyclonal antibodies that target the molecular process involved in HCV attachment and entry are being developed.26 The nonstructural protein NS5B (RNA polymerase) is intimately involved in viral replication and represents a promising target.27 Several nucleosides and nonnucleoside protease inhibitors have already entered clinical trials.
The low fidelity of the HCV replication machinery leads to a very high mutation rate, thus enabling the virus to quickly develop mutations that resist agents targeting viral enzymes.28 Therefore, a novel approach is to target host cofactors that are essential for HCV replication. An intriguing study by Lanford et al29 demonstrated that antagonizing microRNA-122 (the most abundant microRNA in the liver and an essential cofactor for viral RNA replication) by the oligonucleotide SPC3649 caused marked and prolonged reduction of HCV viremia in chronically infected chimpanzees.29
Although we are still in the early stages of drug development, the future holds great promise for newer drugs to improve the sustained virologic response, shorten the duration of treatment, improve tolerability with interferon-sparing regimens, and decrease viral resistance.
FUTURE PERSPECTIVES
With the introduction of the first direct-acting antiviral medications for HCV (boceprevir and telaprevir), 2011 will be marked as the year that changed hepatitis C treatment for the better. Triple therapy with pegylated interferon, ribavirin, and either boceprevir or telaprevir has the potential for increasing the rate of sustained virologic response to around 70% in previously untreated patients and 65% in previously treated patients who are infected with HCV genotype 1. The IL28B polymorphisms appear to play a role in the rate of sustained virologic response achieved with triple therapy, with preliminary data showing a better response rate in patients who have the CC genotype.17
These drugs will add up to $50,000 to the cost of treating hepatitis C virus infection, depending on the drug used and the length of treatment. However, they may be well worth it if they prevent liver failure and the need for transplantation.
Many questions remain, such as how to use these new regimens to treat special patient populations—for example, those with a recurrence of HCV infection after liver transplantation, those co-infected with HCV and human immunodeficiency virus, and those infected with HCV genotypes other than genotype 1.
Other direct-acting antiviral agents that specifically target the replication cycle of HCV are currently in clinical development. In fact, the future has already started with the release of the Interferon-Free Regimen for the Management of HCV (INFORM-1) study results.30 This was the first trial to evaluate an interferon-free regimen for patients with chronic HCV infection using two direct-acting antiviral drugs (the protease inhibitor danoprevir and the polymerase inhibitor RG7128), with promising results.
- Poordad F, McCone J, Bacon BR, et al. Boceprevir for untreated chronic HCV genotype 1 infection. N Engl J Med 2011; 364:1195–1206.
- Bacon BR, Gordon SC, Lawitz E, et al. Boceprevir for previously treated chronic HCV genotype 1 infection. N Engl J Med 2011; 364:1207–1217.
- Jacobson IM, McHutchison JG, Dusheiko G, et al; for the ADVANCE Study Team. Telaprevir for previously untreated chronic hepatitis C virus infection. N Engl J Med 2011; 364:2405–2416.
- Sherman KE, Flamm SL, Afdhal NH, et al; for the ILLUMINATE Study Team. Response-guided telaprevir combination treatment for hepatitis C virus infection. N Engl J Med 2011; 365:1014–1024.
- Zeuzem S, Andreone P, Pol S, et al; for the REALIZE Study Team. Telaprevir for retreatment of HCV infection. N Engl J Med 2011; 364:2417–2428.
- Armstrong GL, Wasley A, Simard EP, McQuillan GM, Kuhnert WL, Alter MJ. The prevalence of hepatitis C virus infection in the United States, 1999 through 2002. Ann Intern Med 2006; 144:705–714.
- Mitchell AE, Colvin HM, Palmer Beasley R. Institute of Medicine recommendations for the prevention and control of hepatitis B and C. Hepatology 2010; 51:729–733.
- Kim WR. The burden of hepatitis C in the United States. Hepatology 2002; 36:S30–S34.
- Marcellin P, Asselah T, Boyer N. Fibrosis and disease progression in hepatitis C. Hepatology 2002; 36:S47–S56.
- Seeff LB. Natural history of chronic hepatitis C. Hepatology 2002; 36:S35–S46.
- Lavanchy D. The global burden of hepatitis C. Liver Int 2009; 29(suppl 1):74–81.
- National Institutes of Health Consensus Development Conference Statement: Management of hepatitis C: 2002—June 10–12, 2002. Hepatology 2002; 36:S3–S20.
- Pearlman BL, Traub N. Sustained virologic response to antiviral therapy for chronic hepatitis C virus infection: a cure and so much more. Clin Infect Dis 2011; 52:889–900.
- Hoofnagle JH, Seeff LB. Peginterferon and ribavirin for chronic hepatitis C. N Engl J Med 2006; 355:2444–2451.
- Ge D, Fellay J, Thompson AJ, et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature 2009; 461:399–401.
- Suppiah V, Moldovan M, Ahlenstiel G, et al. IL28B is associated with response to chronic hepatitis C interferon-alpha and ribavirin therapy. Nat Genet 2009; 41:1100–1104.
- Thompson AJ, Muir AJ, Sulkowski MS, et al. Interleukin-28B polymorphism improves viral kinetics and is the strongest pretreatment predictor of sustained virologic response in genotype 1 hepatitis C virus. Gastroenterology 2010; 139:120–129.e118.
- Nielsen SU, Bassendine MF, Burt AD, Bevitt DJ, Toms GL. Characterization of the genome and structural proteins of hepatitis C virus resolved from infected human liver. J Gen Virol 2004; 85:1497–1507.
- Penin F, Dubuisson J, Rey FA, Moradpour D, Pawlotsky JM. Structural biology of hepatitis C virus. Hepatology 2004; 39:5–19.
- Nelson DR. The role of triple therapy with protease inhibitors in hepatitis C virus genotype 1 naive patients. Liver Int 2011; 31(suppl 1):53–57.
- Pawlotsky JM. Treatment failure and resistance with direct-acting antiviral drugs against hepatitis C virus. Hepatology 2011; 53:1742–1751.
- Monto A, Schooley RT, Lai JC, et al. Lessons from HIV therapy applied to viral hepatitis therapy: summary of a workshop. Am J Gastroenterol 2010; 105:989–1004.
- McCown MF, Rajyaguru S, Kular S, Cammack N, Najera I. GT-1a or GT-1b subtype-specific resistance profiles for hepatitis C virus inhibitors telaprevir and HCV-796. Antimicrob Agents Chemother 2009; 53:2129–2132.
- Cholongitas E, Papatheodoridis GV. Review article: novel therapeutic options for chronic hepatitis C. Aliment Pharmacol Ther 2008; 27:866–884.
- Naggie S, Patel K, McHutchison J. Hepatitis C virus directly acting antivirals: current developments with NS3/4A HCV serine protease inhibitors. J Antimicrob Chemother 2010; 65:2063–2069.
- Mir HM, Birerdinc A, Younossi ZM. Monoclonal and polyclonal antibodies against the HCV envelope proteins. Clin Liver Dis 2009; 13:477–486.
- Birerdinc A, Younossi ZM. Emerging therapies for hepatitis C virus. Expert Opin Emerg Drugs 2010; 15:535–544.
- Khattab MA. Targeting host factors: a novel rationale for the management of hepatitis C virus. World J Gastroenterol 2009; 15:3472–3479.
- Lanford RE, Hildebrandt-Eriksen ES, Petri A, et al. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science 2010; 327:198–201.
- Gane EJ, Roberts SK, Stedman CA, et al. Oral combination therapy with a nucleoside polymerase inhibitor (RG7128) and danoprevir for chronic hepatitis C genotype 1 infection (INFORM-1): a randomised, double-blind, placebo-controlled, dose-escalation trial. Lancet 2010; 376:1467–1475.
The treatment of hepatitis c virus (HCV) infection is on the brink of major changes with the recent approval of the first direct-acting antiviral agents, the protease inhibitors boceprevir (Victrelis) and telaprevir (Incivek).
Both drugs were approved by the US Food and Drug Administration (FDA) Advisory Panel for Chronic Hepatitis C in May 2011 and are believed to significantly improve treatment outcomes for patients with HCV genotype 1 infection.
A MAJOR PUBLIC HEALTH PROBLEM
HCV infection is a major public health problem. Nearly 4 million people in the United States are infected.6,7 Most patients with acute HCV infection become chronically infected, and up to 25% eventually develop cirrhosis and its complications, making HCV infection the leading indication for liver transplantation.8–10
Chronic HCV infection has a large global impact, with 180 million people affected across all economic and social groups.11 The highest prevalence of HCV has been reported in Egypt (14%), in part due to the use of inadequately sterilized needles in mass programs to treat endemic schistosomiasis. In developed countries, hepatocellular carcinoma associated with HCV has the fastest growing cancer-related death rate.12
CURRENTLY, FEWER THAN 50% OF PATIENTS ARE CURED
The goal of HCV treatment is to eradicate the virus. However, most infected patients (especially in the United States and Europe) are infected with HCV genotype 1, which is the most difficult genotype to treat.
Successful treatment of HCV is defined as achieving a sustained virologic response—ie, the absence of detectable HCV RNA in the serum 24 weeks after completion of therapy. Once a sustained virologic response is achieved, lifetime “cure” of HCV infection is expected in more than 99% of patients.13
The current standard therapy for HCV, pegylated interferon plus ribavirin for 48 weeks, is effective in only 40% to 50% of patients with genotype 1 infection.14 Therefore, assessing predictors of response before starting treatment can help select patients who are most likely to benefit from therapy.
Viral factors associated with a sustained virologic response include HCV genotypes other than genotype 1 and a low baseline viral load.
Beneficial patient-related factors include younger age, nonblack ethnicity, low body weight (≤ 75 kg), low body mass index, absence of insulin resistance, and absence of advanced fibrosis or cirrhosis.
More recently, a single-nucleotide polymorphism near the interleukin 28B (IL28B) gene, coding for interferon lambda 3, was found to be associated with a twofold difference in the rates of sustained virologic response: patients with the favorable genotype CC were two times more likely to achieve a sustained virologic response than patients with the CT or TT genotypes.15–17
PROTEASE INHIBITORS: MECHANISM OF ACTION
NS3/4A protease inhibitors rely on the principle of end-product inhibition, in which the cleavage product of the protease (a peptide) acts to inhibit the enzyme activity; this is why they are called peptidomimetics. The active site of the NS3/4A protease is a shallow groove composed of three highly conserved amino acid residues, which may explain why protease inhibitors display high antiviral efficacy but pose a low barrier to the development of resistance.20
Protease inhibitors are prone to resistance
The development of viral resistance to protease inhibitors has been a major drawback to their use in patients with chronic HCV infection.21
HCV is a highly variable virus with many genetically distinct but closely related quasispecies circulating in the blood at any given time. Drug-resistant, mutated variants preexist within the patient’s quasispecies, but only in small quantities because of their lesser replication fitness compared with the wild-type virus.22 When direct-acting antiviral therapy is started, the quantity of the wild-type virus decreases and the mutated virus gains replication fitness. Using protease inhibitors as monotherapy selects resistant viral populations rapidly within a few days or weeks.
HCV subtypes 1a and 1b may have different resistance profiles. With genotype 1a, some resistance-associated amino acid substitutions require only one nucleotide change, but with genotype 1b, two nucleotide changes are needed, making resistance less frequent in patients with HCV genotype 1b.23
BOCEPREVIR
Boceprevir is a specific inhibitor of the HCV viral protease NS3/4A.
In phase 3 clinical trials, boceprevir 800 mg three times a day was used with pegylated interferon alfa-2b (PegIntron) 1.5 μg/kg/week and ribavirin (Rebetol) 600 to 1,400 mg daily according to body weight.
Before patients started taking boceprevir, they went through a 4-week lead-in phase, during which they received pegylated interferon and ribavirin. This schedule appeared to reduce the incidence of viral breakthrough in phase 2 trials, and it produced higher rates of sustained virologic response and lower relapse rates compared with triple therapy without a lead-in phase.
Rapid virologic response was defined as undetectable HCV RNA at week 4 of boceprevir therapy (week 8 of the whole regimen).
Boceprevir in previously untreated patients with HCV genotype 1: The SPRINT-2 trial
The Serine Protease Inhibitor Therapy 2 (SPRINT-2) trial1 included more than 1,000 previously untreated adults with HCV genotype 1 infection (938 nonblack patients and 159 black patients; two other nonblack patients did not receive any study drug and were not included in the analysis). In this double-blind trial, patients were randomized into three groups:
- The control group received the standard of care with pegylated interferon and ribavirin for 48 weeks
- The response-guided therapy group received boceprevir plus pegylated interferon and ribavirin for 24 weeks after the 4-week lead-in phase; if HCV RNA was undetectable from week 8 to week 24, treatment was considered complete, but if HCV RNA was detectable at any point from week 8 to week 24, pegylated interferon and ribavirin were continued for a total of 48 weeks.
- The fixed-duration therapy group received boceprevir, pegylated interferon, and ribavirin for 44 weeks after the lead-in period.
The rate of relapse was 8% and 9% in the boceprevir groups vs 23% in the control group. Patients in the boceprevir groups who had a decrease in HCV RNA of less than 1 log10 during the lead-in phase were found to have a significantly higher rate of boceprevirresistant variants than those who achieved a decrease of HCV RNA of 1 log10 or more.
Boceprevir in previously treated patients with HCV genotype 1: The RESPOND-2 trial
The Retreatment With HCV Serine Protease Inhibitor Boceprevir and PegIntron/Rebetol 2) (RESPOND-2) trial2 was designed to assess the efficacy of combined boceprevir, pegylated interferon, and ribavirin for repeat treatment of patients with HCV genotype 1. These patients had previously undergone standard treatment and had a reduction of 2 log10 or more in HCV RNA after 12 weeks of therapy but with detectable HCV RNA during the therapy period or had had a relapse (defined as undetectable HCV RNA at the end of a previous course of therapy with HCV RNA positivity thereafter). Importantly, null-responders (those who had a reduction of less than 2 log10 in HCV RNA after 12 weeks of therapy) were excluded from this trial.
After a lead-in period of interferon-ribavirin treatment for 4 weeks, 403 patients were assigned to one of three treatment groups:
- Pegylated interferon and ribavirin for 44 weeks (the control group)
- Boceprevir, pegylated interferon, and ribavirin in a response-guided regimen
- Boceprevir, pegylated interferon, and ribavirin for 44 weeks (the fixed-duration group).
Sustained virologic response was achieved in only 21% of patients in the control group. Adding boceprevir increased the rate to 59% in the response-guided therapy group and to 67% in the fixed-duration group. Previous relapsers had better rates than partial responders (69%–75% vs 40%–52%).
Importantly, patients who had a poor response to pegylated interferon and ribavirin during the lead-in phase (defined as having less than a 1-log decrease in the virus before starting boceprevir) had significantly lower rates of sustained virologic response and higher rates of resistance-associated virus variants.
Side effects of boceprevir
Overall, boceprevir is well tolerated. The most common side effects of triple therapy are those usually seen with pegylated interferon and ribavirin, such as flulike symptoms and fatigue (Table 2). However, anemia was more frequent in the boceprevir groups in both SPRINT-2 and RESPOND-2 (45%–50% compared with 20%–29% in the control groups). Erythropoietin was allowed in these studies and was used in about 40% of patients.
The other common side effect associated with boceprevir was dysgeusia (alteration of taste). Dysgeusia was reported by approximately 40% of patients; however, most dysgeusia events were mild to moderate in intensity and did not lead to treatment cessation.
In the SPRINT-2 trial,1 the study drugs had to be discontinued in 12% to 16% of patients in the boceprevir groups because of adverse events, which was similar to the rate (16%) in the control group. Erythropoietin was allowed in this trial, and it was used in 43% of patients in the boceprevir groups compared with 24% in the control group, with discontinuation owing to anemia occurring in 2% and 1% of cases, respectively.
TELAPREVIR
Telaprevir, the other protease NS3/4A inhibitor, has also shown efficacy over current standard therapy in phase 3 clinical trials. It was used in a dose of 750 mg three times a day with pegylated interferon alfa-2a (Pegasys) 180 μg per week and ribavirin (Copegus) 1,000 to 1,200 mg daily according to body weight. A lead-in phase with pegylated interferon and ribavirin was not applied with telaprevir, as it was in the boceprevir trials. Extended rapid virologic response was defined as an undetectable HCV RNA at weeks 4 and 12 of therapy.
Telaprevir in previously untreated patients with HCV genotype 1
The ADVANCE study3 was a double-blind randomized trial assessing the efficacy and safety of telaprevir in combination with pegylated interferon and ribavirin in more than 1,000 previously untreated patients. The three treatment groups received:
- Telaprevir, pegylated interferon, and ribavirin for 8 weeks, followed by pegylated interferon and ribavirin alone for 16 weeks in patients who achieved an extended rapid virologic response (total duration of 24 weeks) or 40 weeks in patients who did not (total duration of 48 weeks)
- Telaprevir, pegylated interferon, and ribavirin for 12 weeks, followed by pegylated interferon-ribavirin alone for 12 (total of 24 weeks) or 36 weeks (total of 48 weeks) according to extended rapid virologic response
- Standard care with pegylated interferon and ribavirin for 48 weeks.
The rate of sustained virologic response was 69% in the group that received telaprevir for 8 weeks and 75% in the group that received it for 12 weeks compared with 44% in the control group (P < .0001 for both) (Table 2). Patients infected with HCV genotype 1b had a higher sustained virologic response rate (79%) than those infected with HCV genotype 1a (71%).
Sustained virologic response rates were lower in black patients and patients with bridging fibrosis or cirrhosis, but were still significantly higher in the telaprevir groups than in the control group. The results of this subset analysis were limited by small numbers of patients in each category.
In total, 57% of those who received telaprevir for 8 weeks and 58% of those who received it for 12 weeks achieved an extended rapid virologic response and were able to cut the duration of their therapy in half (from 48 weeks to 24 weeks).
The relapse rates were 9% in the telaprevir groups and 28% in the control group.
The rate of virologic failure was lower in patients who received triple therapy than in those who received interferon-ribavirin alone (8% in the group that got telaprevir for 12 weeks and 13% in the group that got it for 8 weeks, vs 32% in the control group). The failure rate was also lower in patients with HCV genotype 1b infection than in those with genotype 1a.
The ILLUMINATE study4 (Illustrating the Effects of Combination Therapy With Telaprevir) investigated whether longer duration of treatment than that given in the ADVANCE trial increased the rate of sustained virologic response. Previously untreated patients received telaprevir, interferon, and ribavirin for 12 weeks, and those who achieved an extended rapid virologic response were randomized at week 20 to continue interferonribavirin treatment for 24 or 48 weeks of total treatment.
The sustained virologic response rates in patients who achieved an extended rapid virologic response were 92% in the group that received pegylated interferon and ribavirin for 12 weeks, and 88% in those who received it for 48 weeks. Thus, the results of this study support the use of response-guided therapy for telaprevir-based regimens.
Telaprevir in previously treated patients with HCV genotype 1: The REALIZE trial
In this phase 3 placebo-controlled trial,5 622 patients with prior relapse, partial response, or null response were randomly allocated into one of three groups:
- Telaprevir for 12 weeks plus pegylated interferon and ribavirin for 48 weeks
- Lead-in for 4 weeks followed by 12 weeks of triple therapy and another 32 weeks of pegylated interferon and ribavirin
- Pegylated interferon and ribavirin for 48 weeks (the control group).
The overall sustained virologic response rates were 66% and 64%, respectively, in the telaprevir groups vs 17% in the control group (P < .0001). The sustained virologic response rates in the telaprevir groups were 83% to 88% in prior relapsers, 54% to 59% in partial responders, and 29% to 33% in null-responders. Of note, patients did not benefit from the lead-in phase.
This was the only trial to investigate the response to triple therapy in null-responders, a group in which treatment has been considered hopeless. A response rate of approximately 31% was encouraging, especially if we compare it with the 5% response rate achieved with the current standard of care with pegylated interferon and ribavirin.
Telaprevir side effects
As with boceprevir-based triple therapy, the most common adverse events were related to pegylated interferon (Table 2).
Nearly 50% of patients who receive telaprevir develop a skin rash that is primarily eczematous, can be managed with topical steroids, and usually resolves when telaprevir is discontinued. Severe rashes occurred in 3% to 6% of patients in the ADVANCE trial,3 and three suspected cases of Stevens-Johnson syndrome have been reported to the FDA.
Other side effects that were more frequent with telaprevir included pruritus, nausea, diarrhea, and anemia. On average, the hemoglobin level decreased by an additional 1 g/dL in the telaprevir treatment groups compared with the groups that received only pegylated interferon-ribavirin. Erythropoietin use was not allowed in the phase 3 telaprevir studies, and anemia was managed by ribavirin dose reduction.
In the ADVANCE trial,3 study drugs were discontinued owing to adverse events in 7% to 8% of the patients in the telaprevir groups compared with 4% in the control group. In the ILLUMINATE trial,4 17% of patients had to permanently discontinue all study drugs due to adverse events.
FDA-APPROVED TREATMENT REGIMENS FOR BOCEPREVIR AND TELAPREVIR
For treatment algorithms, see the eFigures that accompany this article online.
Boceprevir in previously untreated patients
- Week 0—Start pegylated interferon and ribavirin
- Week 4—Add boceprevir
- Week 8—Measure HCV RNA
- Week 12—Measure HCV RNA; stop treatment if it is more than 100 IU/mL
- Week 24—Measure HCV RNA; stop treatment if it is detectable
- Week 28—Stop all treatment if HCV RNA was undetectable at weeks 8 and 24
- Week 36—Measure HCV RNA; stop boceprevir
- Week 48—Stop all treatment (eFigure 1).
Boceprevir in previously treated patients
- Week 0—Start pegylated interferon and ribavirin
- Week 4—Add boceprevir
- Week 8—Measure HCV RNA
- Week 12—Measure HCV RNA; stop treatment if it is more than 100 IU/mL
- Week 24—Measure HCV RNA; stop treatment if it is detectable
- Week 36—if HCV RNA was not detectable at week 8, stop all treatment now; if HCV RNA was detectable at week 8, stop boceprevir now but continue pegylated interferon and ribavirin
- Week 48—Stop all treatment (eFigure 2).
Telaprevir in previously untreated patients and prior relapsers
- Week 0—start telaprevir, pegylated interferon, and ribavirin
- Week 4—measure HCV RNA; stop all treatment if it is more than 1,000 IU/mL
- Week 12—Stop telaprevir; measure HCV RNA; stop all treatment if HCV RNA is more than 1,000 IU/mL
- Week 24—Stop pegylated interferon and ribavirin if HCV RNA was undetectable at week 12; measure HCV RNA and stop treatment if it is detectable; otherwise, continue pegylated interferon and ribavirin
- Week 48—Stop all treatment (eFigure 3).
Telaprevir in patients who previously achieved a partial or null response
- Week 0—Start telaprevir, pegylated interferon, and ribavirin
- Week 4—Measure HCV RNA; stop treatment if it is more than 1,000 IU/mL
- Week 12—Measure HCV RNA; stop all treatment if it is more than 1,000 IU/mL; if less than 1,000 IU/mL then stop telaprevir but continue pegylated interferon and ribavirin
- Week 24—Measure HCV RNA; stop treatment if HCV RNA is detectable
- Week 48—Stop all treatment (eFigure 4).
Drug interactions with boceprevir and telaprevir
Both boceprevir and telaprevir inhibit cytochrome P450 3A (CYP3A) and thus are contraindicated in combination with drugs highly dependent on CYP3A for clearance and with drugs for which elevated plasma concentrations are associated with serious adverse events, such as atorvastatin (Lipitor), simvastatin (Zocor), sildenafil (Viagra), midazolam (Versed), and St. John’s wort. Giving potent inducers of CYP3A with boceprevir or telaprevir may lead to lower exposure and loss of efficacy of both protease inhibitors.
EMERGING THERAPIES FOR HCV
Thanks to a better understanding of the biology of HCV infection, the effort to develop new therapeutic agents started to focus on targeting specific steps of the viral life cycle, including attachment, entry into cells, replication, and release.24
Currently, more than 50 clinical trials are evaluating new direct-acting antivirals to treat HCV infection.25 Monoclonal and polyclonal antibodies that target the molecular process involved in HCV attachment and entry are being developed.26 The nonstructural protein NS5B (RNA polymerase) is intimately involved in viral replication and represents a promising target.27 Several nucleosides and nonnucleoside protease inhibitors have already entered clinical trials.
The low fidelity of the HCV replication machinery leads to a very high mutation rate, thus enabling the virus to quickly develop mutations that resist agents targeting viral enzymes.28 Therefore, a novel approach is to target host cofactors that are essential for HCV replication. An intriguing study by Lanford et al29 demonstrated that antagonizing microRNA-122 (the most abundant microRNA in the liver and an essential cofactor for viral RNA replication) by the oligonucleotide SPC3649 caused marked and prolonged reduction of HCV viremia in chronically infected chimpanzees.29
Although we are still in the early stages of drug development, the future holds great promise for newer drugs to improve the sustained virologic response, shorten the duration of treatment, improve tolerability with interferon-sparing regimens, and decrease viral resistance.
FUTURE PERSPECTIVES
With the introduction of the first direct-acting antiviral medications for HCV (boceprevir and telaprevir), 2011 will be marked as the year that changed hepatitis C treatment for the better. Triple therapy with pegylated interferon, ribavirin, and either boceprevir or telaprevir has the potential for increasing the rate of sustained virologic response to around 70% in previously untreated patients and 65% in previously treated patients who are infected with HCV genotype 1. The IL28B polymorphisms appear to play a role in the rate of sustained virologic response achieved with triple therapy, with preliminary data showing a better response rate in patients who have the CC genotype.17
These drugs will add up to $50,000 to the cost of treating hepatitis C virus infection, depending on the drug used and the length of treatment. However, they may be well worth it if they prevent liver failure and the need for transplantation.
Many questions remain, such as how to use these new regimens to treat special patient populations—for example, those with a recurrence of HCV infection after liver transplantation, those co-infected with HCV and human immunodeficiency virus, and those infected with HCV genotypes other than genotype 1.
Other direct-acting antiviral agents that specifically target the replication cycle of HCV are currently in clinical development. In fact, the future has already started with the release of the Interferon-Free Regimen for the Management of HCV (INFORM-1) study results.30 This was the first trial to evaluate an interferon-free regimen for patients with chronic HCV infection using two direct-acting antiviral drugs (the protease inhibitor danoprevir and the polymerase inhibitor RG7128), with promising results.
The treatment of hepatitis c virus (HCV) infection is on the brink of major changes with the recent approval of the first direct-acting antiviral agents, the protease inhibitors boceprevir (Victrelis) and telaprevir (Incivek).
Both drugs were approved by the US Food and Drug Administration (FDA) Advisory Panel for Chronic Hepatitis C in May 2011 and are believed to significantly improve treatment outcomes for patients with HCV genotype 1 infection.
A MAJOR PUBLIC HEALTH PROBLEM
HCV infection is a major public health problem. Nearly 4 million people in the United States are infected.6,7 Most patients with acute HCV infection become chronically infected, and up to 25% eventually develop cirrhosis and its complications, making HCV infection the leading indication for liver transplantation.8–10
Chronic HCV infection has a large global impact, with 180 million people affected across all economic and social groups.11 The highest prevalence of HCV has been reported in Egypt (14%), in part due to the use of inadequately sterilized needles in mass programs to treat endemic schistosomiasis. In developed countries, hepatocellular carcinoma associated with HCV has the fastest growing cancer-related death rate.12
CURRENTLY, FEWER THAN 50% OF PATIENTS ARE CURED
The goal of HCV treatment is to eradicate the virus. However, most infected patients (especially in the United States and Europe) are infected with HCV genotype 1, which is the most difficult genotype to treat.
Successful treatment of HCV is defined as achieving a sustained virologic response—ie, the absence of detectable HCV RNA in the serum 24 weeks after completion of therapy. Once a sustained virologic response is achieved, lifetime “cure” of HCV infection is expected in more than 99% of patients.13
The current standard therapy for HCV, pegylated interferon plus ribavirin for 48 weeks, is effective in only 40% to 50% of patients with genotype 1 infection.14 Therefore, assessing predictors of response before starting treatment can help select patients who are most likely to benefit from therapy.
Viral factors associated with a sustained virologic response include HCV genotypes other than genotype 1 and a low baseline viral load.
Beneficial patient-related factors include younger age, nonblack ethnicity, low body weight (≤ 75 kg), low body mass index, absence of insulin resistance, and absence of advanced fibrosis or cirrhosis.
More recently, a single-nucleotide polymorphism near the interleukin 28B (IL28B) gene, coding for interferon lambda 3, was found to be associated with a twofold difference in the rates of sustained virologic response: patients with the favorable genotype CC were two times more likely to achieve a sustained virologic response than patients with the CT or TT genotypes.15–17
PROTEASE INHIBITORS: MECHANISM OF ACTION
NS3/4A protease inhibitors rely on the principle of end-product inhibition, in which the cleavage product of the protease (a peptide) acts to inhibit the enzyme activity; this is why they are called peptidomimetics. The active site of the NS3/4A protease is a shallow groove composed of three highly conserved amino acid residues, which may explain why protease inhibitors display high antiviral efficacy but pose a low barrier to the development of resistance.20
Protease inhibitors are prone to resistance
The development of viral resistance to protease inhibitors has been a major drawback to their use in patients with chronic HCV infection.21
HCV is a highly variable virus with many genetically distinct but closely related quasispecies circulating in the blood at any given time. Drug-resistant, mutated variants preexist within the patient’s quasispecies, but only in small quantities because of their lesser replication fitness compared with the wild-type virus.22 When direct-acting antiviral therapy is started, the quantity of the wild-type virus decreases and the mutated virus gains replication fitness. Using protease inhibitors as monotherapy selects resistant viral populations rapidly within a few days or weeks.
HCV subtypes 1a and 1b may have different resistance profiles. With genotype 1a, some resistance-associated amino acid substitutions require only one nucleotide change, but with genotype 1b, two nucleotide changes are needed, making resistance less frequent in patients with HCV genotype 1b.23
BOCEPREVIR
Boceprevir is a specific inhibitor of the HCV viral protease NS3/4A.
In phase 3 clinical trials, boceprevir 800 mg three times a day was used with pegylated interferon alfa-2b (PegIntron) 1.5 μg/kg/week and ribavirin (Rebetol) 600 to 1,400 mg daily according to body weight.
Before patients started taking boceprevir, they went through a 4-week lead-in phase, during which they received pegylated interferon and ribavirin. This schedule appeared to reduce the incidence of viral breakthrough in phase 2 trials, and it produced higher rates of sustained virologic response and lower relapse rates compared with triple therapy without a lead-in phase.
Rapid virologic response was defined as undetectable HCV RNA at week 4 of boceprevir therapy (week 8 of the whole regimen).
Boceprevir in previously untreated patients with HCV genotype 1: The SPRINT-2 trial
The Serine Protease Inhibitor Therapy 2 (SPRINT-2) trial1 included more than 1,000 previously untreated adults with HCV genotype 1 infection (938 nonblack patients and 159 black patients; two other nonblack patients did not receive any study drug and were not included in the analysis). In this double-blind trial, patients were randomized into three groups:
- The control group received the standard of care with pegylated interferon and ribavirin for 48 weeks
- The response-guided therapy group received boceprevir plus pegylated interferon and ribavirin for 24 weeks after the 4-week lead-in phase; if HCV RNA was undetectable from week 8 to week 24, treatment was considered complete, but if HCV RNA was detectable at any point from week 8 to week 24, pegylated interferon and ribavirin were continued for a total of 48 weeks.
- The fixed-duration therapy group received boceprevir, pegylated interferon, and ribavirin for 44 weeks after the lead-in period.
The rate of relapse was 8% and 9% in the boceprevir groups vs 23% in the control group. Patients in the boceprevir groups who had a decrease in HCV RNA of less than 1 log10 during the lead-in phase were found to have a significantly higher rate of boceprevirresistant variants than those who achieved a decrease of HCV RNA of 1 log10 or more.
Boceprevir in previously treated patients with HCV genotype 1: The RESPOND-2 trial
The Retreatment With HCV Serine Protease Inhibitor Boceprevir and PegIntron/Rebetol 2) (RESPOND-2) trial2 was designed to assess the efficacy of combined boceprevir, pegylated interferon, and ribavirin for repeat treatment of patients with HCV genotype 1. These patients had previously undergone standard treatment and had a reduction of 2 log10 or more in HCV RNA after 12 weeks of therapy but with detectable HCV RNA during the therapy period or had had a relapse (defined as undetectable HCV RNA at the end of a previous course of therapy with HCV RNA positivity thereafter). Importantly, null-responders (those who had a reduction of less than 2 log10 in HCV RNA after 12 weeks of therapy) were excluded from this trial.
After a lead-in period of interferon-ribavirin treatment for 4 weeks, 403 patients were assigned to one of three treatment groups:
- Pegylated interferon and ribavirin for 44 weeks (the control group)
- Boceprevir, pegylated interferon, and ribavirin in a response-guided regimen
- Boceprevir, pegylated interferon, and ribavirin for 44 weeks (the fixed-duration group).
Sustained virologic response was achieved in only 21% of patients in the control group. Adding boceprevir increased the rate to 59% in the response-guided therapy group and to 67% in the fixed-duration group. Previous relapsers had better rates than partial responders (69%–75% vs 40%–52%).
Importantly, patients who had a poor response to pegylated interferon and ribavirin during the lead-in phase (defined as having less than a 1-log decrease in the virus before starting boceprevir) had significantly lower rates of sustained virologic response and higher rates of resistance-associated virus variants.
Side effects of boceprevir
Overall, boceprevir is well tolerated. The most common side effects of triple therapy are those usually seen with pegylated interferon and ribavirin, such as flulike symptoms and fatigue (Table 2). However, anemia was more frequent in the boceprevir groups in both SPRINT-2 and RESPOND-2 (45%–50% compared with 20%–29% in the control groups). Erythropoietin was allowed in these studies and was used in about 40% of patients.
The other common side effect associated with boceprevir was dysgeusia (alteration of taste). Dysgeusia was reported by approximately 40% of patients; however, most dysgeusia events were mild to moderate in intensity and did not lead to treatment cessation.
In the SPRINT-2 trial,1 the study drugs had to be discontinued in 12% to 16% of patients in the boceprevir groups because of adverse events, which was similar to the rate (16%) in the control group. Erythropoietin was allowed in this trial, and it was used in 43% of patients in the boceprevir groups compared with 24% in the control group, with discontinuation owing to anemia occurring in 2% and 1% of cases, respectively.
TELAPREVIR
Telaprevir, the other protease NS3/4A inhibitor, has also shown efficacy over current standard therapy in phase 3 clinical trials. It was used in a dose of 750 mg three times a day with pegylated interferon alfa-2a (Pegasys) 180 μg per week and ribavirin (Copegus) 1,000 to 1,200 mg daily according to body weight. A lead-in phase with pegylated interferon and ribavirin was not applied with telaprevir, as it was in the boceprevir trials. Extended rapid virologic response was defined as an undetectable HCV RNA at weeks 4 and 12 of therapy.
Telaprevir in previously untreated patients with HCV genotype 1
The ADVANCE study3 was a double-blind randomized trial assessing the efficacy and safety of telaprevir in combination with pegylated interferon and ribavirin in more than 1,000 previously untreated patients. The three treatment groups received:
- Telaprevir, pegylated interferon, and ribavirin for 8 weeks, followed by pegylated interferon and ribavirin alone for 16 weeks in patients who achieved an extended rapid virologic response (total duration of 24 weeks) or 40 weeks in patients who did not (total duration of 48 weeks)
- Telaprevir, pegylated interferon, and ribavirin for 12 weeks, followed by pegylated interferon-ribavirin alone for 12 (total of 24 weeks) or 36 weeks (total of 48 weeks) according to extended rapid virologic response
- Standard care with pegylated interferon and ribavirin for 48 weeks.
The rate of sustained virologic response was 69% in the group that received telaprevir for 8 weeks and 75% in the group that received it for 12 weeks compared with 44% in the control group (P < .0001 for both) (Table 2). Patients infected with HCV genotype 1b had a higher sustained virologic response rate (79%) than those infected with HCV genotype 1a (71%).
Sustained virologic response rates were lower in black patients and patients with bridging fibrosis or cirrhosis, but were still significantly higher in the telaprevir groups than in the control group. The results of this subset analysis were limited by small numbers of patients in each category.
In total, 57% of those who received telaprevir for 8 weeks and 58% of those who received it for 12 weeks achieved an extended rapid virologic response and were able to cut the duration of their therapy in half (from 48 weeks to 24 weeks).
The relapse rates were 9% in the telaprevir groups and 28% in the control group.
The rate of virologic failure was lower in patients who received triple therapy than in those who received interferon-ribavirin alone (8% in the group that got telaprevir for 12 weeks and 13% in the group that got it for 8 weeks, vs 32% in the control group). The failure rate was also lower in patients with HCV genotype 1b infection than in those with genotype 1a.
The ILLUMINATE study4 (Illustrating the Effects of Combination Therapy With Telaprevir) investigated whether longer duration of treatment than that given in the ADVANCE trial increased the rate of sustained virologic response. Previously untreated patients received telaprevir, interferon, and ribavirin for 12 weeks, and those who achieved an extended rapid virologic response were randomized at week 20 to continue interferonribavirin treatment for 24 or 48 weeks of total treatment.
The sustained virologic response rates in patients who achieved an extended rapid virologic response were 92% in the group that received pegylated interferon and ribavirin for 12 weeks, and 88% in those who received it for 48 weeks. Thus, the results of this study support the use of response-guided therapy for telaprevir-based regimens.
Telaprevir in previously treated patients with HCV genotype 1: The REALIZE trial
In this phase 3 placebo-controlled trial,5 622 patients with prior relapse, partial response, or null response were randomly allocated into one of three groups:
- Telaprevir for 12 weeks plus pegylated interferon and ribavirin for 48 weeks
- Lead-in for 4 weeks followed by 12 weeks of triple therapy and another 32 weeks of pegylated interferon and ribavirin
- Pegylated interferon and ribavirin for 48 weeks (the control group).
The overall sustained virologic response rates were 66% and 64%, respectively, in the telaprevir groups vs 17% in the control group (P < .0001). The sustained virologic response rates in the telaprevir groups were 83% to 88% in prior relapsers, 54% to 59% in partial responders, and 29% to 33% in null-responders. Of note, patients did not benefit from the lead-in phase.
This was the only trial to investigate the response to triple therapy in null-responders, a group in which treatment has been considered hopeless. A response rate of approximately 31% was encouraging, especially if we compare it with the 5% response rate achieved with the current standard of care with pegylated interferon and ribavirin.
Telaprevir side effects
As with boceprevir-based triple therapy, the most common adverse events were related to pegylated interferon (Table 2).
Nearly 50% of patients who receive telaprevir develop a skin rash that is primarily eczematous, can be managed with topical steroids, and usually resolves when telaprevir is discontinued. Severe rashes occurred in 3% to 6% of patients in the ADVANCE trial,3 and three suspected cases of Stevens-Johnson syndrome have been reported to the FDA.
Other side effects that were more frequent with telaprevir included pruritus, nausea, diarrhea, and anemia. On average, the hemoglobin level decreased by an additional 1 g/dL in the telaprevir treatment groups compared with the groups that received only pegylated interferon-ribavirin. Erythropoietin use was not allowed in the phase 3 telaprevir studies, and anemia was managed by ribavirin dose reduction.
In the ADVANCE trial,3 study drugs were discontinued owing to adverse events in 7% to 8% of the patients in the telaprevir groups compared with 4% in the control group. In the ILLUMINATE trial,4 17% of patients had to permanently discontinue all study drugs due to adverse events.
FDA-APPROVED TREATMENT REGIMENS FOR BOCEPREVIR AND TELAPREVIR
For treatment algorithms, see the eFigures that accompany this article online.
Boceprevir in previously untreated patients
- Week 0—Start pegylated interferon and ribavirin
- Week 4—Add boceprevir
- Week 8—Measure HCV RNA
- Week 12—Measure HCV RNA; stop treatment if it is more than 100 IU/mL
- Week 24—Measure HCV RNA; stop treatment if it is detectable
- Week 28—Stop all treatment if HCV RNA was undetectable at weeks 8 and 24
- Week 36—Measure HCV RNA; stop boceprevir
- Week 48—Stop all treatment (eFigure 1).
Boceprevir in previously treated patients
- Week 0—Start pegylated interferon and ribavirin
- Week 4—Add boceprevir
- Week 8—Measure HCV RNA
- Week 12—Measure HCV RNA; stop treatment if it is more than 100 IU/mL
- Week 24—Measure HCV RNA; stop treatment if it is detectable
- Week 36—if HCV RNA was not detectable at week 8, stop all treatment now; if HCV RNA was detectable at week 8, stop boceprevir now but continue pegylated interferon and ribavirin
- Week 48—Stop all treatment (eFigure 2).
Telaprevir in previously untreated patients and prior relapsers
- Week 0—start telaprevir, pegylated interferon, and ribavirin
- Week 4—measure HCV RNA; stop all treatment if it is more than 1,000 IU/mL
- Week 12—Stop telaprevir; measure HCV RNA; stop all treatment if HCV RNA is more than 1,000 IU/mL
- Week 24—Stop pegylated interferon and ribavirin if HCV RNA was undetectable at week 12; measure HCV RNA and stop treatment if it is detectable; otherwise, continue pegylated interferon and ribavirin
- Week 48—Stop all treatment (eFigure 3).
Telaprevir in patients who previously achieved a partial or null response
- Week 0—Start telaprevir, pegylated interferon, and ribavirin
- Week 4—Measure HCV RNA; stop treatment if it is more than 1,000 IU/mL
- Week 12—Measure HCV RNA; stop all treatment if it is more than 1,000 IU/mL; if less than 1,000 IU/mL then stop telaprevir but continue pegylated interferon and ribavirin
- Week 24—Measure HCV RNA; stop treatment if HCV RNA is detectable
- Week 48—Stop all treatment (eFigure 4).
Drug interactions with boceprevir and telaprevir
Both boceprevir and telaprevir inhibit cytochrome P450 3A (CYP3A) and thus are contraindicated in combination with drugs highly dependent on CYP3A for clearance and with drugs for which elevated plasma concentrations are associated with serious adverse events, such as atorvastatin (Lipitor), simvastatin (Zocor), sildenafil (Viagra), midazolam (Versed), and St. John’s wort. Giving potent inducers of CYP3A with boceprevir or telaprevir may lead to lower exposure and loss of efficacy of both protease inhibitors.
EMERGING THERAPIES FOR HCV
Thanks to a better understanding of the biology of HCV infection, the effort to develop new therapeutic agents started to focus on targeting specific steps of the viral life cycle, including attachment, entry into cells, replication, and release.24
Currently, more than 50 clinical trials are evaluating new direct-acting antivirals to treat HCV infection.25 Monoclonal and polyclonal antibodies that target the molecular process involved in HCV attachment and entry are being developed.26 The nonstructural protein NS5B (RNA polymerase) is intimately involved in viral replication and represents a promising target.27 Several nucleosides and nonnucleoside protease inhibitors have already entered clinical trials.
The low fidelity of the HCV replication machinery leads to a very high mutation rate, thus enabling the virus to quickly develop mutations that resist agents targeting viral enzymes.28 Therefore, a novel approach is to target host cofactors that are essential for HCV replication. An intriguing study by Lanford et al29 demonstrated that antagonizing microRNA-122 (the most abundant microRNA in the liver and an essential cofactor for viral RNA replication) by the oligonucleotide SPC3649 caused marked and prolonged reduction of HCV viremia in chronically infected chimpanzees.29
Although we are still in the early stages of drug development, the future holds great promise for newer drugs to improve the sustained virologic response, shorten the duration of treatment, improve tolerability with interferon-sparing regimens, and decrease viral resistance.
FUTURE PERSPECTIVES
With the introduction of the first direct-acting antiviral medications for HCV (boceprevir and telaprevir), 2011 will be marked as the year that changed hepatitis C treatment for the better. Triple therapy with pegylated interferon, ribavirin, and either boceprevir or telaprevir has the potential for increasing the rate of sustained virologic response to around 70% in previously untreated patients and 65% in previously treated patients who are infected with HCV genotype 1. The IL28B polymorphisms appear to play a role in the rate of sustained virologic response achieved with triple therapy, with preliminary data showing a better response rate in patients who have the CC genotype.17
These drugs will add up to $50,000 to the cost of treating hepatitis C virus infection, depending on the drug used and the length of treatment. However, they may be well worth it if they prevent liver failure and the need for transplantation.
Many questions remain, such as how to use these new regimens to treat special patient populations—for example, those with a recurrence of HCV infection after liver transplantation, those co-infected with HCV and human immunodeficiency virus, and those infected with HCV genotypes other than genotype 1.
Other direct-acting antiviral agents that specifically target the replication cycle of HCV are currently in clinical development. In fact, the future has already started with the release of the Interferon-Free Regimen for the Management of HCV (INFORM-1) study results.30 This was the first trial to evaluate an interferon-free regimen for patients with chronic HCV infection using two direct-acting antiviral drugs (the protease inhibitor danoprevir and the polymerase inhibitor RG7128), with promising results.
- Poordad F, McCone J, Bacon BR, et al. Boceprevir for untreated chronic HCV genotype 1 infection. N Engl J Med 2011; 364:1195–1206.
- Bacon BR, Gordon SC, Lawitz E, et al. Boceprevir for previously treated chronic HCV genotype 1 infection. N Engl J Med 2011; 364:1207–1217.
- Jacobson IM, McHutchison JG, Dusheiko G, et al; for the ADVANCE Study Team. Telaprevir for previously untreated chronic hepatitis C virus infection. N Engl J Med 2011; 364:2405–2416.
- Sherman KE, Flamm SL, Afdhal NH, et al; for the ILLUMINATE Study Team. Response-guided telaprevir combination treatment for hepatitis C virus infection. N Engl J Med 2011; 365:1014–1024.
- Zeuzem S, Andreone P, Pol S, et al; for the REALIZE Study Team. Telaprevir for retreatment of HCV infection. N Engl J Med 2011; 364:2417–2428.
- Armstrong GL, Wasley A, Simard EP, McQuillan GM, Kuhnert WL, Alter MJ. The prevalence of hepatitis C virus infection in the United States, 1999 through 2002. Ann Intern Med 2006; 144:705–714.
- Mitchell AE, Colvin HM, Palmer Beasley R. Institute of Medicine recommendations for the prevention and control of hepatitis B and C. Hepatology 2010; 51:729–733.
- Kim WR. The burden of hepatitis C in the United States. Hepatology 2002; 36:S30–S34.
- Marcellin P, Asselah T, Boyer N. Fibrosis and disease progression in hepatitis C. Hepatology 2002; 36:S47–S56.
- Seeff LB. Natural history of chronic hepatitis C. Hepatology 2002; 36:S35–S46.
- Lavanchy D. The global burden of hepatitis C. Liver Int 2009; 29(suppl 1):74–81.
- National Institutes of Health Consensus Development Conference Statement: Management of hepatitis C: 2002—June 10–12, 2002. Hepatology 2002; 36:S3–S20.
- Pearlman BL, Traub N. Sustained virologic response to antiviral therapy for chronic hepatitis C virus infection: a cure and so much more. Clin Infect Dis 2011; 52:889–900.
- Hoofnagle JH, Seeff LB. Peginterferon and ribavirin for chronic hepatitis C. N Engl J Med 2006; 355:2444–2451.
- Ge D, Fellay J, Thompson AJ, et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature 2009; 461:399–401.
- Suppiah V, Moldovan M, Ahlenstiel G, et al. IL28B is associated with response to chronic hepatitis C interferon-alpha and ribavirin therapy. Nat Genet 2009; 41:1100–1104.
- Thompson AJ, Muir AJ, Sulkowski MS, et al. Interleukin-28B polymorphism improves viral kinetics and is the strongest pretreatment predictor of sustained virologic response in genotype 1 hepatitis C virus. Gastroenterology 2010; 139:120–129.e118.
- Nielsen SU, Bassendine MF, Burt AD, Bevitt DJ, Toms GL. Characterization of the genome and structural proteins of hepatitis C virus resolved from infected human liver. J Gen Virol 2004; 85:1497–1507.
- Penin F, Dubuisson J, Rey FA, Moradpour D, Pawlotsky JM. Structural biology of hepatitis C virus. Hepatology 2004; 39:5–19.
- Nelson DR. The role of triple therapy with protease inhibitors in hepatitis C virus genotype 1 naive patients. Liver Int 2011; 31(suppl 1):53–57.
- Pawlotsky JM. Treatment failure and resistance with direct-acting antiviral drugs against hepatitis C virus. Hepatology 2011; 53:1742–1751.
- Monto A, Schooley RT, Lai JC, et al. Lessons from HIV therapy applied to viral hepatitis therapy: summary of a workshop. Am J Gastroenterol 2010; 105:989–1004.
- McCown MF, Rajyaguru S, Kular S, Cammack N, Najera I. GT-1a or GT-1b subtype-specific resistance profiles for hepatitis C virus inhibitors telaprevir and HCV-796. Antimicrob Agents Chemother 2009; 53:2129–2132.
- Cholongitas E, Papatheodoridis GV. Review article: novel therapeutic options for chronic hepatitis C. Aliment Pharmacol Ther 2008; 27:866–884.
- Naggie S, Patel K, McHutchison J. Hepatitis C virus directly acting antivirals: current developments with NS3/4A HCV serine protease inhibitors. J Antimicrob Chemother 2010; 65:2063–2069.
- Mir HM, Birerdinc A, Younossi ZM. Monoclonal and polyclonal antibodies against the HCV envelope proteins. Clin Liver Dis 2009; 13:477–486.
- Birerdinc A, Younossi ZM. Emerging therapies for hepatitis C virus. Expert Opin Emerg Drugs 2010; 15:535–544.
- Khattab MA. Targeting host factors: a novel rationale for the management of hepatitis C virus. World J Gastroenterol 2009; 15:3472–3479.
- Lanford RE, Hildebrandt-Eriksen ES, Petri A, et al. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science 2010; 327:198–201.
- Gane EJ, Roberts SK, Stedman CA, et al. Oral combination therapy with a nucleoside polymerase inhibitor (RG7128) and danoprevir for chronic hepatitis C genotype 1 infection (INFORM-1): a randomised, double-blind, placebo-controlled, dose-escalation trial. Lancet 2010; 376:1467–1475.
- Poordad F, McCone J, Bacon BR, et al. Boceprevir for untreated chronic HCV genotype 1 infection. N Engl J Med 2011; 364:1195–1206.
- Bacon BR, Gordon SC, Lawitz E, et al. Boceprevir for previously treated chronic HCV genotype 1 infection. N Engl J Med 2011; 364:1207–1217.
- Jacobson IM, McHutchison JG, Dusheiko G, et al; for the ADVANCE Study Team. Telaprevir for previously untreated chronic hepatitis C virus infection. N Engl J Med 2011; 364:2405–2416.
- Sherman KE, Flamm SL, Afdhal NH, et al; for the ILLUMINATE Study Team. Response-guided telaprevir combination treatment for hepatitis C virus infection. N Engl J Med 2011; 365:1014–1024.
- Zeuzem S, Andreone P, Pol S, et al; for the REALIZE Study Team. Telaprevir for retreatment of HCV infection. N Engl J Med 2011; 364:2417–2428.
- Armstrong GL, Wasley A, Simard EP, McQuillan GM, Kuhnert WL, Alter MJ. The prevalence of hepatitis C virus infection in the United States, 1999 through 2002. Ann Intern Med 2006; 144:705–714.
- Mitchell AE, Colvin HM, Palmer Beasley R. Institute of Medicine recommendations for the prevention and control of hepatitis B and C. Hepatology 2010; 51:729–733.
- Kim WR. The burden of hepatitis C in the United States. Hepatology 2002; 36:S30–S34.
- Marcellin P, Asselah T, Boyer N. Fibrosis and disease progression in hepatitis C. Hepatology 2002; 36:S47–S56.
- Seeff LB. Natural history of chronic hepatitis C. Hepatology 2002; 36:S35–S46.
- Lavanchy D. The global burden of hepatitis C. Liver Int 2009; 29(suppl 1):74–81.
- National Institutes of Health Consensus Development Conference Statement: Management of hepatitis C: 2002—June 10–12, 2002. Hepatology 2002; 36:S3–S20.
- Pearlman BL, Traub N. Sustained virologic response to antiviral therapy for chronic hepatitis C virus infection: a cure and so much more. Clin Infect Dis 2011; 52:889–900.
- Hoofnagle JH, Seeff LB. Peginterferon and ribavirin for chronic hepatitis C. N Engl J Med 2006; 355:2444–2451.
- Ge D, Fellay J, Thompson AJ, et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature 2009; 461:399–401.
- Suppiah V, Moldovan M, Ahlenstiel G, et al. IL28B is associated with response to chronic hepatitis C interferon-alpha and ribavirin therapy. Nat Genet 2009; 41:1100–1104.
- Thompson AJ, Muir AJ, Sulkowski MS, et al. Interleukin-28B polymorphism improves viral kinetics and is the strongest pretreatment predictor of sustained virologic response in genotype 1 hepatitis C virus. Gastroenterology 2010; 139:120–129.e118.
- Nielsen SU, Bassendine MF, Burt AD, Bevitt DJ, Toms GL. Characterization of the genome and structural proteins of hepatitis C virus resolved from infected human liver. J Gen Virol 2004; 85:1497–1507.
- Penin F, Dubuisson J, Rey FA, Moradpour D, Pawlotsky JM. Structural biology of hepatitis C virus. Hepatology 2004; 39:5–19.
- Nelson DR. The role of triple therapy with protease inhibitors in hepatitis C virus genotype 1 naive patients. Liver Int 2011; 31(suppl 1):53–57.
- Pawlotsky JM. Treatment failure and resistance with direct-acting antiviral drugs against hepatitis C virus. Hepatology 2011; 53:1742–1751.
- Monto A, Schooley RT, Lai JC, et al. Lessons from HIV therapy applied to viral hepatitis therapy: summary of a workshop. Am J Gastroenterol 2010; 105:989–1004.
- McCown MF, Rajyaguru S, Kular S, Cammack N, Najera I. GT-1a or GT-1b subtype-specific resistance profiles for hepatitis C virus inhibitors telaprevir and HCV-796. Antimicrob Agents Chemother 2009; 53:2129–2132.
- Cholongitas E, Papatheodoridis GV. Review article: novel therapeutic options for chronic hepatitis C. Aliment Pharmacol Ther 2008; 27:866–884.
- Naggie S, Patel K, McHutchison J. Hepatitis C virus directly acting antivirals: current developments with NS3/4A HCV serine protease inhibitors. J Antimicrob Chemother 2010; 65:2063–2069.
- Mir HM, Birerdinc A, Younossi ZM. Monoclonal and polyclonal antibodies against the HCV envelope proteins. Clin Liver Dis 2009; 13:477–486.
- Birerdinc A, Younossi ZM. Emerging therapies for hepatitis C virus. Expert Opin Emerg Drugs 2010; 15:535–544.
- Khattab MA. Targeting host factors: a novel rationale for the management of hepatitis C virus. World J Gastroenterol 2009; 15:3472–3479.
- Lanford RE, Hildebrandt-Eriksen ES, Petri A, et al. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science 2010; 327:198–201.
- Gane EJ, Roberts SK, Stedman CA, et al. Oral combination therapy with a nucleoside polymerase inhibitor (RG7128) and danoprevir for chronic hepatitis C genotype 1 infection (INFORM-1): a randomised, double-blind, placebo-controlled, dose-escalation trial. Lancet 2010; 376:1467–1475.
KEY POINTS
- Standard care with the combination of pegylated interferon and ribavirin produces a sustained virologic response in about 40% of patients infected with HCV genotype 1, the most prevalent genotype in North America.
- New phase 3 trials showed that the addition of an oral protease inhibitor (boceprevir or telaprevir) increased the sustained virologic response rates to 70% in patients infected with HCV genotype 1.
- Boceprevir and telaprevir must be used in combination with pegylated interferon and ribavirin; they should not be used as monotherapy because of concern about the development of drug-resistant mutations.
- The main side effects of boceprevir were anemia and dysgeusia. Adverse events associated with telaprevir included rash, pruritus, anemia, and diarrhea.
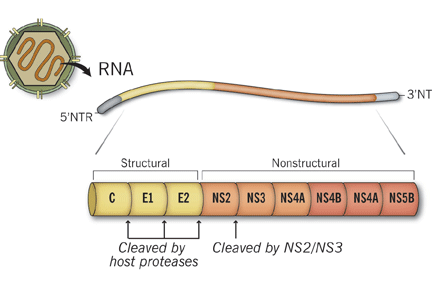
A 25-year-old man with very high alkaline phosphatase
A 25-year-old man presented to his primary care physician with generalized malaise. His symptoms started around 2 months earlier with progressive fatigue, nausea, decreased appetite, and weight loss (15 lb in 2 months). He denied having fever, chills, night sweats, abdominal pain, diarrhea, melena, or hematochezia.
His medical history was remarkable only for depression, well controlled with sertraline (Zoloft), which he started taking 3 years ago. He was not taking any other prescribed, over-the-counter, or herbal medications.
He had no family history of cancer or liver disease. He did not smoke and rarely drank alcohol. He had never used recreational drugs. He was sexually active with one female partner, used condoms for protection, and had never been diagnosed with a sexually transmitted disease. He had not traveled recently and had not been exposed to any pet.
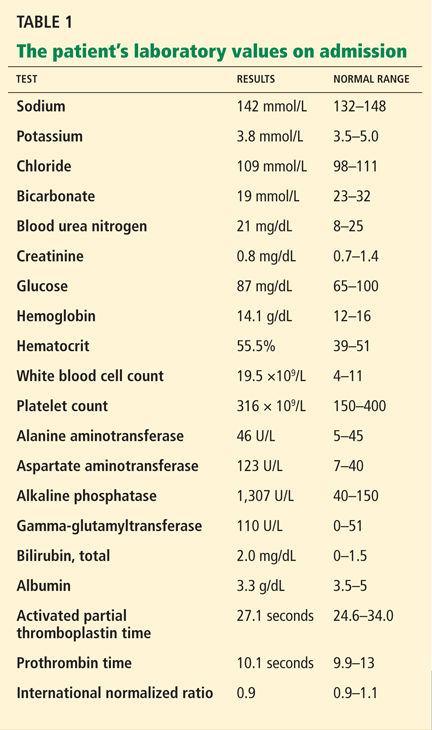
The patient’s laboratory values on admission are shown in Table 1. Of note, his serum alkaline phosphatase level was 1,307 U/L (reference range 40–150 U/L).
LIVER TESTS CAN NARROW THE DIAGNOSIS
The most commonly used laboratory tests of the liver can be classified into those that measure either:
- Liver synthetic function (eg, the serum albumin and bilirubin concentrations and the prothrombin time) or
- Liver damage, as reflected by the serum concentrations of the enzymes alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase, and gamma-glutamyltransferase (GGT).1,2
ALT and AST are normally concentrated in the hepatocytes and thus, when present in the serum in elevated concentrations, are markers of liver cell injury. The serum levels of these enzymes start to increase within a few hours of liver cell injury as they leak out of the cells via the damaged cell membrane. AST is less liver-specific than ALT, since AST levels can be elevated not only in liver injury but also in muscle, cardiac, and red blood cell injury.3,4
Alkaline phosphatase is actually a heterogeneous group of enzymes found mainly in liver and bone cells. Hepatic alkaline phosphatase is concentrated near the biliary canalicular membrane of the hepatocyte. Accordingly, increased levels of hepatic alkaline phosphatase are mainly seen in liver diseases that predominantly affect the biliary system.3
GGT is also concentrated in hepatic biliary epithelial cells, and thus GGT elevation is another marker of hepatobiliary disease. In fact, measuring the GGT level can help to determine whether an isolated elevation of alkaline phosphatase is due to liver injury.2,3
Accordingly, liver diseases can be classified into two broad categories:
- Hepatocellular injury, in which the primary injury occurs to the hepatocytes
- Cholestatic injury, in which the primary injury is to the bile ducts.
In the former, elevated levels of ALT and AST predominate, while in the latter, elevated alkaline phosphatase is the main finding.3
WHAT TEST NEXT FOR OUR PATIENT?
1. What is the next most appropriate diagnostic step for our patient?
- Liver biopsy
- Ultrasonography of the liver
- Computed tomography (CT) of the liver
- Observation
Our patient has an elevated GGT level, which suggests that his elevated alkaline phosphatase is of hepatic rather than bony origin. Moreover, a serum alkaline phosphatase level that is elevated out of proportion to the aminotransferase levels reflects cholestatic liver injury.
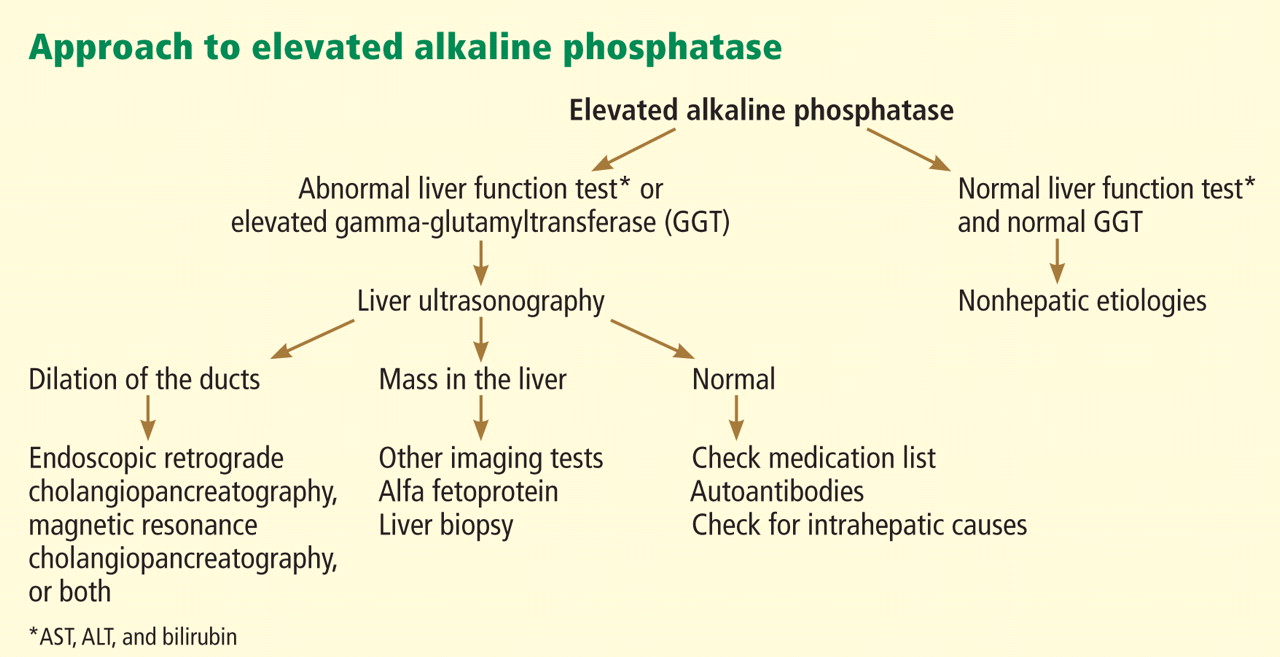
CASE CONTINUED: ULTRASONOGRAPHY IS MOSTLY NORMAL
Ultrasonography of the right upper quadrant revealed that the liver had normal echogenicity and was mildly enlarged. There was no focal hepatic lesion. The gallbladder appeared normal, with no stones or sludge. No dilated intrahepatic or extrahepatic biliary ducts were seen. The common bile duct measured 4 mm. A small amount of ascites not amenable to paracentesis was present.
Thus, in the absence of biliary dilation on ultrasonography, we are dealing with an intrahepatic cholestatic process.
CAUSES OF CHOLESTATIC LIVER DISEASE
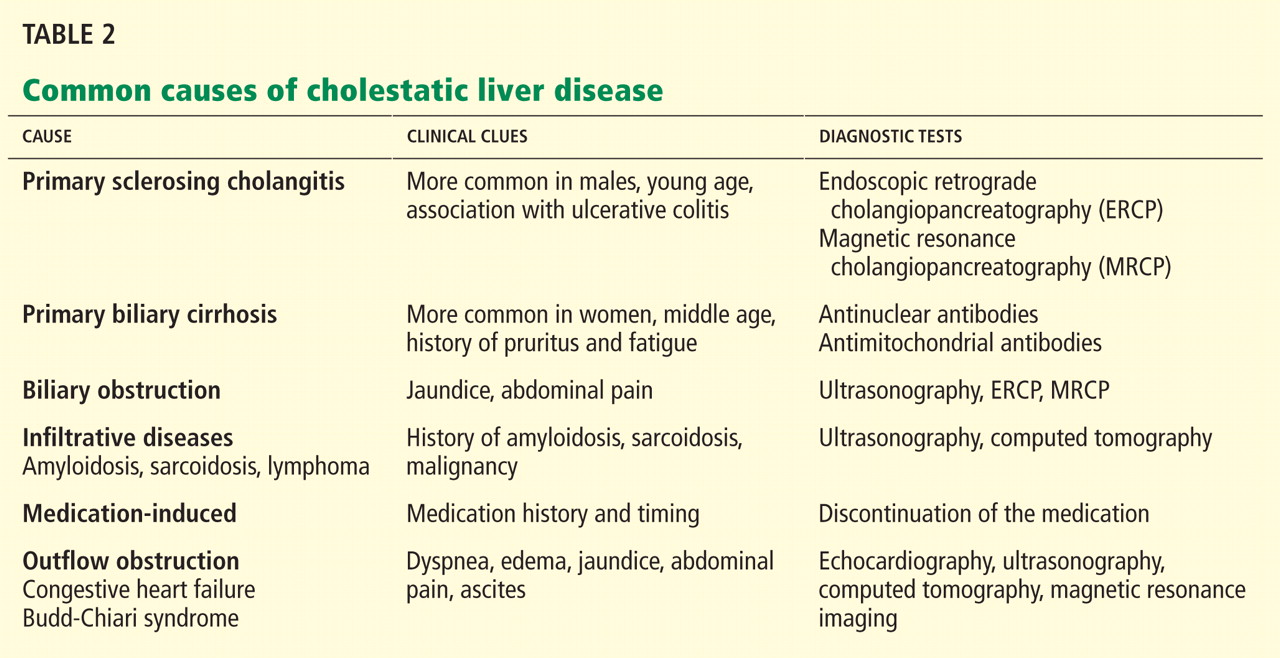
Viral hepatitis
Viral hepatitis most often produces a hepatocellular pattern of injury (ie, AST and ALT elevations predominate). However, in rare cases it can cause a cholestatic pattern of injury.
Our patient subsequently had serologic tests for viral hepatitis, including hepatitis A, B, and C, and the results were negative.
Autoimmune liver disease
The three most common forms of autoimmune liver disease are autoimmune hepatitis, primary biliary cirrhosis, and primary sclerosing cholangitis.
Autoimmune hepatitis is characterized by high serum ALT and AST levels, whereas primary biliary cirrhosis and primary sclerosing cholangitis are associated with predominant elevations of alkaline phosphatase, since they are cholestatic disorders.
Our patient’s alkaline phosphatase level was much higher than his ALT and AST levels, making the latter two diseases more likely.
Primary biliary cirrhosis (and autoimmune hepatitis) are associated with autoantibodies in the serum, such as antinuclear antibody, smooth muscle antibody, and antimitochondrial antibody.
Our patient subsequently was tested for these antibodies, and the results were negative.
Primary sclerosing cholangitis usually affects the extrahepatic biliary system. Thus, if it is present, abnormalities should be seen on imaging.
As mentioned previously, no dilated intrahepatic or extrahepatic biliary ducts were seen on ultrasonography in our patient. Moreover, primary sclerosing cholangitis is associated with inflammatory bowel disease, particularly ulcerative colitis, which our patient did not have.
Drug-induced liver injury
Drug-induced liver injury is a common cause of cholestatic liver disease. However, our patient was not taking any prescribed, over-the-counter, or herbal medications. Additionally, he denied heavy alcohol use.
Infiltrative disorders
Infiltrative disorders such as amyloidosis, sarcoidosis, or lymphoma should be considered in the differential diagnosis of cholestatic liver disease. A clue to a possible infiltrative process is a markedly elevated level of alkaline phosphatase with a mildly increased serum bilirubin concentration, both of which our patient had.
AFTER ULTRASONOGRAPHY, WHAT IS THE NEXT STEP?
2. Which of the following is the next most appropriate diagnostic test for our patient?
- Endoscopic retrograde cholangiopancreatography (ERCP)
- Magnetic resonance cholangiopancreatography (MRCP)
- Liver biopsy
- CT of the abdomen
Figure 1 shows a proposed algorithm for evaluating increased alkaline phosphatase levels.
If there is no biliary duct dilation on ultrasonography, then abnormal levels of alkaline phosphatase most likely represent an intrahepatic pattern of cholestatic liver injury. Therefore, additional imaging with CT or magnetic resonance imaging is of limited diagnostic value. ERCP is used today for therapy rather than diagnosis, so its use is limited to patients known to have dilated biliary ducts on imaging. Liver biopsy, however, can provide useful findings.
Case continued: He undergoes biopsy
Our patient underwent transjugular liver biopsy. During the procedure, transjugular venography showed stenosis in the right, middle, and left hepatic veins and the hepatic portion of the inferior vena cava, consistent with Budd-Chiari syndrome.
The liver biopsy specimen was positive for extensive deposition of slight eosinophilic and amorphous material in a sinusoidal pattern in the liver parenchyma, as well as in the portal tracts, with markedly atrophic hepatocytes. Congo red birefringence confirmed the diagnosis of amyloidosis. The immunohistochemical phenotype was positive for kappa light chains, which is diagnostic for primary-type amyloidosis, also called amyloidosis of light chain composition, or AL.
Bone marrow aspiration and bone marrow biopsy were performed and showed 22% plasma cells, well above the normal range (0–2%), consistent with the diagnosis of multiple myeloma.
BUDD-CHIARI SYNDROME: A CHALLENGING DIAGNOSIS
Budd-Chiari syndrome is a rare condition characterized by obstruction of venous outflow from the liver at a site that may vary from the small hepatic veins up to the inferior vena cava or even the right atrium.5,6 Obstruction of hepatic venous outflow leads to sinusoidal congestion and hypoxic damage of the hepatocytes.7 Hypoxia and necrosis of the hepatocytes result in the release of free radicals. Cirrhosis can eventually occur secondary to ischemic necrosis of hepatocytes and hepatic fibrosis.8
The estimated incidence of this syndrome is 1 in 2.5 million persons per year.7 It is more prevalent in women and young adults.8
Heterogeneous in its causes and manifestations

Budd-Chiari syndrome is also heterogeneous in its manifestations, which depend on the extent of the occlusion, on the acuteness of the obstruction, and on whether venous collateral circulation has developed to decompress the liver sinusoids.9,12,13 Therefore, on the basis of its clinical manifestations, it can be classified as fulminant, acute, subacute, or chronic.12–16
The fulminant form presents with hepatic encephalopathy within 8 weeks after the development of jaundice. The subacute form, which is the most common, has a more insidious onset in which hepatic sinusoids are decompressed by portal and hepatic venous collateral circulation. The patient usually presents with abdominal pain, ascites, hepatomegaly, nausea, vomiting, and mild jaundice. Finally the chronic form presents as complications of cirrhosis.12–16
Imaging plays an important role in diagnosing Budd-Chiari syndrome
Imaging plays an important role in detecting and classifying Budd-Chiari syndrome.
Duplex ultrasonography is useful for detecting this syndrome and has a sensitivity and specificity of 85%.9
CT and magnetic resonance imaging can also help in the diagnosis by showing thrombosis, obstruction, or occlusion in the hepatic vein or the inferior vena cava.5
Venography is the gold standard for diagnosis. However, it should be performed only if noninvasive tests are negative or nondiagnostic and there is a high clinical suspicion of this disease.17 Budd-Chiari syndrome has a characteristic pattern on venography known as “spider web,” which is due to the formation of venous collaterals to bypass the occluded hepatic veins.9
Liver biopsy is not necessarily required to confirm the diagnosis of Budd-Chiari syndrome, but it can help in diagnosing the acute or subacute forms and also in ruling out other causes. Histologic findings can include centrizonal congestion, loss of hepatocytes, hemorrhage, and fibrosis.18,19 Regenerative nodules are found in about 25% of patients.19
TREATING BUDD-CHIARI SYNDROME
The primary goal of treatment is to prevent further extension of the venous thrombosis in the hepatic veins, in their collaterals, and in the intrahepatic and extrahepatic portal venous system. Resolution of hepatic congestion improves liver perfusion and preserves function of the hepatocytes.
Anticoagulation is recommended in the early stages. Heparin therapy should be initiated and subsequently switched to warfarin with the goal of achieving an international normalized ratio of the prothrombin time of 2.0 to 2.5.8,9,19
Thrombolysis is effective in the acute form.20,21 Recanalization, including percutaneous or transhepatic angioplasty of localized segments of the narrowed hepatic veins or inferior vena cava, has long-term patency rates of 80% to 90%.22
If thrombolytic therapy and angioplasty are unsuccessful, a transjugular intrahepatic portosystemic shunt or a surgical procedure (side-to-side portocaval shunt, central splenorenal shunt, or mesocaval shunt) should be considered.9
Liver transplantation is another treatment option in those with fulminant Budd-Chiari syndrome or advanced liver cirrhosis.8
PROGNOSIS HAS IMPROVED
The prognosis of Budd-Chiari syndrome has improved, thanks to both earlier diagnosis and new treatments. The 1-year survival rate, which was about 60% before 1985, has increased to more than 80% in recent cohort studies.19
Studies have shown that the Child-Pugh score, which is based on a combination of serum albumin, bilirubin, prothrombin time, encephalopathy, and ascites, can be considered as an independent prognostic factor. A lower Child-Pugh score and a younger age are associated with a good prognosis.19,23,24 (The Child-Pugh score cannot be applied to our patient because he does not have cirrhosis.)
What happened to our patient?
Our patient was started on anticoagulation for his Budd-Chiari syndrome and on bortezomib (Velcade) and dexamethasone for his multiple myeloma. He achieved remarkable improvement in his liver function tests. Follow-up duplex ultrasonography 1 month after discharge revealed that the stenosis in the hepatic veins had resolved. He is following up with the oncology clinic for management of his multiple myeloma.
- Folwaczny C. Efficient diagnostics for elevated transaminases. [Article in German] MMW Fortschr Med 2007; 149:44–48.
- Moussavian SN, Becker RC, Piepmeyer JL, Mezey E, Bozian RC. Serum gamma-glutamyl transpeptidase and chronic alcoholism. Influence of alcohol ingestion and liver disease. Dig Dis Sci 1985; 30:211–214.
- Aragon G, Younossi ZM. When and how to evaluate mildly elevated liver enzymes in apparently healthy patients. Cleve Clin J Med 2010; 77:195–204.
- Lepper PM, Dufour JF. Elevated transaminases—what to do if everything was done?. [Article in German] Praxis (Bern 1994) 2009; 98:330–334.
- Buzas C, Sparchez Z, Cucuianu A, Manole S, Lupescu I, Acalovschi M. Budd-Chiari syndrome secondary to polycythemia vera. A case report. J Gastrointestin Liver Dis 2009; 18:363–366.
- Valla DC. Primary Budd-Chiari syndrome. J Hepatol 2009; 50:195–203.
- Rautou PE, Moucari R, Cazals-Hatem D, et al. Levels and initial course of serum alanine aminotransferase can predict outcome of patients with Budd-Chiari syndrome. Clin Gastroenterol Hepatol 2009; 7:1230–1235.
- Cura M, Haskal Z, Lopera J. Diagnostic and interventional radiology for Budd-Chiari syndrome. Radiographics 2009; 29:669–681.
- Menon KV, Shah V, Kamath PS. The Budd-Chiari syndrome. N Engl J Med 2004; 350:578–585.
- Darwish Murad S, Plessier A, Hernandez-Guerra M, et al; EN-Vie (European Network for Vascular Disorders of the Liver). Etiology, management, and outcome of the Budd-Chiari syndrome. Ann Intern Med 2009; 151:167–175.
- Valla D, Le MG, Poynard T, Zucman N, Rueff B, Benhamou JP. Risk of hepatic vein thrombosis in relation to recent use of oral contraceptives. A case-control study. Gastroenterology 1986; 90:807–811.
- Bismuth H, Sherlock DJ. Portasystemic shunting versus liver transplantation for the Budd-Chiari syndrome. Ann Surg 1991; 214:581–589.
- Orloff MJ, Daily PO, Orloff SL, Girard B, Orloff MS. A 27-year experience with surgical treatment of Budd-Chiari syndrome. Ann Surg 2000; 232:340–352.
- Dilawari JB, Bambery P, Chawla Y, et al. Hepatic outflow obstruction (Budd-Chiari syndrome). Experience with 177 patients and a review of the literature. Medicine (Baltimore) 1994; 73:21–36.
- Mahmoud AE, Mendoza A, Meshikhes AN, et al. Clinical spectrum, investigations and treatment of Budd-Chiari syndrome. QJM 1996; 89:37–43.
- Klein AS, Cameron JL. Diagnosis and management of the Budd-Chiari syndrome. Am J Surg 1990; 160:128–133.
- Plessier A, Valla DC. Budd-Chiari syndrome. Semin Liver Dis 2008; 28:259–269.
- Cazals-Hatem D, Vilgrain V, Genin P, et al. Arterial and portal circulation and parenchymal changes in Budd-Chiari syndrome: a study in 17 explanted livers. Hepatology 2003; 37:510–519.
- Hoekstra J, Janssen HL. Vascular liver disorders (I): diagnosis, treatment and prognosis of Budd-Chiari syndrome. Neth J Med 2008; 66:334–359.
- Frank JW, Kamath PS, Stanson AW. Budd-Chiari syndrome: early intervention with angioplasty and thrombolytic therapy. Mayo Clin Proc 1994; 69:877–881.
- Raju GS, Felver M, Olin JW, Satti SD. Thrombolysis for acute Budd-Chiari syndrome: case report and literature review. Am J Gastroenterol 1996; 91:1262–1263.
- Fisher NC, McCafferty I, Dolapci M, et al. Managing Budd-Chiari syndrome: a retrospective review of percutaneous hepatic vein angioplasty and surgical shunting. Gut 1999; 44:568–574.
- Zeitoun G, Escolano S, Hadengue A, et al. Outcome of Budd-Chiari syndrome: a multivariate analysis of factors related to survival including surgical portosystemic shunting. Hepatology 1999; 30:84–89.
- Darwish Murad S, Valla DC, de Groen PC, et al. Determinants of survival and the effect of portosystemic shunting in patients with Budd-Chiari syndrome. Hepatology 2004; 39:500–508.
A 25-year-old man presented to his primary care physician with generalized malaise. His symptoms started around 2 months earlier with progressive fatigue, nausea, decreased appetite, and weight loss (15 lb in 2 months). He denied having fever, chills, night sweats, abdominal pain, diarrhea, melena, or hematochezia.
His medical history was remarkable only for depression, well controlled with sertraline (Zoloft), which he started taking 3 years ago. He was not taking any other prescribed, over-the-counter, or herbal medications.
He had no family history of cancer or liver disease. He did not smoke and rarely drank alcohol. He had never used recreational drugs. He was sexually active with one female partner, used condoms for protection, and had never been diagnosed with a sexually transmitted disease. He had not traveled recently and had not been exposed to any pet.
The patient’s laboratory values on admission are shown in Table 1. Of note, his serum alkaline phosphatase level was 1,307 U/L (reference range 40–150 U/L).
LIVER TESTS CAN NARROW THE DIAGNOSIS
The most commonly used laboratory tests of the liver can be classified into those that measure either:
- Liver synthetic function (eg, the serum albumin and bilirubin concentrations and the prothrombin time) or
- Liver damage, as reflected by the serum concentrations of the enzymes alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase, and gamma-glutamyltransferase (GGT).1,2
ALT and AST are normally concentrated in the hepatocytes and thus, when present in the serum in elevated concentrations, are markers of liver cell injury. The serum levels of these enzymes start to increase within a few hours of liver cell injury as they leak out of the cells via the damaged cell membrane. AST is less liver-specific than ALT, since AST levels can be elevated not only in liver injury but also in muscle, cardiac, and red blood cell injury.3,4
Alkaline phosphatase is actually a heterogeneous group of enzymes found mainly in liver and bone cells. Hepatic alkaline phosphatase is concentrated near the biliary canalicular membrane of the hepatocyte. Accordingly, increased levels of hepatic alkaline phosphatase are mainly seen in liver diseases that predominantly affect the biliary system.3
GGT is also concentrated in hepatic biliary epithelial cells, and thus GGT elevation is another marker of hepatobiliary disease. In fact, measuring the GGT level can help to determine whether an isolated elevation of alkaline phosphatase is due to liver injury.2,3
Accordingly, liver diseases can be classified into two broad categories:
- Hepatocellular injury, in which the primary injury occurs to the hepatocytes
- Cholestatic injury, in which the primary injury is to the bile ducts.
In the former, elevated levels of ALT and AST predominate, while in the latter, elevated alkaline phosphatase is the main finding.3
WHAT TEST NEXT FOR OUR PATIENT?
1. What is the next most appropriate diagnostic step for our patient?
- Liver biopsy
- Ultrasonography of the liver
- Computed tomography (CT) of the liver
- Observation
Our patient has an elevated GGT level, which suggests that his elevated alkaline phosphatase is of hepatic rather than bony origin. Moreover, a serum alkaline phosphatase level that is elevated out of proportion to the aminotransferase levels reflects cholestatic liver injury.
CASE CONTINUED: ULTRASONOGRAPHY IS MOSTLY NORMAL
Ultrasonography of the right upper quadrant revealed that the liver had normal echogenicity and was mildly enlarged. There was no focal hepatic lesion. The gallbladder appeared normal, with no stones or sludge. No dilated intrahepatic or extrahepatic biliary ducts were seen. The common bile duct measured 4 mm. A small amount of ascites not amenable to paracentesis was present.
Thus, in the absence of biliary dilation on ultrasonography, we are dealing with an intrahepatic cholestatic process.
CAUSES OF CHOLESTATIC LIVER DISEASE
Viral hepatitis
Viral hepatitis most often produces a hepatocellular pattern of injury (ie, AST and ALT elevations predominate). However, in rare cases it can cause a cholestatic pattern of injury.
Our patient subsequently had serologic tests for viral hepatitis, including hepatitis A, B, and C, and the results were negative.
Autoimmune liver disease
The three most common forms of autoimmune liver disease are autoimmune hepatitis, primary biliary cirrhosis, and primary sclerosing cholangitis.
Autoimmune hepatitis is characterized by high serum ALT and AST levels, whereas primary biliary cirrhosis and primary sclerosing cholangitis are associated with predominant elevations of alkaline phosphatase, since they are cholestatic disorders.
Our patient’s alkaline phosphatase level was much higher than his ALT and AST levels, making the latter two diseases more likely.
Primary biliary cirrhosis (and autoimmune hepatitis) are associated with autoantibodies in the serum, such as antinuclear antibody, smooth muscle antibody, and antimitochondrial antibody.
Our patient subsequently was tested for these antibodies, and the results were negative.
Primary sclerosing cholangitis usually affects the extrahepatic biliary system. Thus, if it is present, abnormalities should be seen on imaging.
As mentioned previously, no dilated intrahepatic or extrahepatic biliary ducts were seen on ultrasonography in our patient. Moreover, primary sclerosing cholangitis is associated with inflammatory bowel disease, particularly ulcerative colitis, which our patient did not have.
Drug-induced liver injury
Drug-induced liver injury is a common cause of cholestatic liver disease. However, our patient was not taking any prescribed, over-the-counter, or herbal medications. Additionally, he denied heavy alcohol use.
Infiltrative disorders
Infiltrative disorders such as amyloidosis, sarcoidosis, or lymphoma should be considered in the differential diagnosis of cholestatic liver disease. A clue to a possible infiltrative process is a markedly elevated level of alkaline phosphatase with a mildly increased serum bilirubin concentration, both of which our patient had.
AFTER ULTRASONOGRAPHY, WHAT IS THE NEXT STEP?
2. Which of the following is the next most appropriate diagnostic test for our patient?
- Endoscopic retrograde cholangiopancreatography (ERCP)
- Magnetic resonance cholangiopancreatography (MRCP)
- Liver biopsy
- CT of the abdomen
Figure 1 shows a proposed algorithm for evaluating increased alkaline phosphatase levels.
If there is no biliary duct dilation on ultrasonography, then abnormal levels of alkaline phosphatase most likely represent an intrahepatic pattern of cholestatic liver injury. Therefore, additional imaging with CT or magnetic resonance imaging is of limited diagnostic value. ERCP is used today for therapy rather than diagnosis, so its use is limited to patients known to have dilated biliary ducts on imaging. Liver biopsy, however, can provide useful findings.
Case continued: He undergoes biopsy
Our patient underwent transjugular liver biopsy. During the procedure, transjugular venography showed stenosis in the right, middle, and left hepatic veins and the hepatic portion of the inferior vena cava, consistent with Budd-Chiari syndrome.
The liver biopsy specimen was positive for extensive deposition of slight eosinophilic and amorphous material in a sinusoidal pattern in the liver parenchyma, as well as in the portal tracts, with markedly atrophic hepatocytes. Congo red birefringence confirmed the diagnosis of amyloidosis. The immunohistochemical phenotype was positive for kappa light chains, which is diagnostic for primary-type amyloidosis, also called amyloidosis of light chain composition, or AL.
Bone marrow aspiration and bone marrow biopsy were performed and showed 22% plasma cells, well above the normal range (0–2%), consistent with the diagnosis of multiple myeloma.
BUDD-CHIARI SYNDROME: A CHALLENGING DIAGNOSIS
Budd-Chiari syndrome is a rare condition characterized by obstruction of venous outflow from the liver at a site that may vary from the small hepatic veins up to the inferior vena cava or even the right atrium.5,6 Obstruction of hepatic venous outflow leads to sinusoidal congestion and hypoxic damage of the hepatocytes.7 Hypoxia and necrosis of the hepatocytes result in the release of free radicals. Cirrhosis can eventually occur secondary to ischemic necrosis of hepatocytes and hepatic fibrosis.8
The estimated incidence of this syndrome is 1 in 2.5 million persons per year.7 It is more prevalent in women and young adults.8
Heterogeneous in its causes and manifestations
Budd-Chiari syndrome is also heterogeneous in its manifestations, which depend on the extent of the occlusion, on the acuteness of the obstruction, and on whether venous collateral circulation has developed to decompress the liver sinusoids.9,12,13 Therefore, on the basis of its clinical manifestations, it can be classified as fulminant, acute, subacute, or chronic.12–16
The fulminant form presents with hepatic encephalopathy within 8 weeks after the development of jaundice. The subacute form, which is the most common, has a more insidious onset in which hepatic sinusoids are decompressed by portal and hepatic venous collateral circulation. The patient usually presents with abdominal pain, ascites, hepatomegaly, nausea, vomiting, and mild jaundice. Finally the chronic form presents as complications of cirrhosis.12–16
Imaging plays an important role in diagnosing Budd-Chiari syndrome
Imaging plays an important role in detecting and classifying Budd-Chiari syndrome.
Duplex ultrasonography is useful for detecting this syndrome and has a sensitivity and specificity of 85%.9
CT and magnetic resonance imaging can also help in the diagnosis by showing thrombosis, obstruction, or occlusion in the hepatic vein or the inferior vena cava.5
Venography is the gold standard for diagnosis. However, it should be performed only if noninvasive tests are negative or nondiagnostic and there is a high clinical suspicion of this disease.17 Budd-Chiari syndrome has a characteristic pattern on venography known as “spider web,” which is due to the formation of venous collaterals to bypass the occluded hepatic veins.9
Liver biopsy is not necessarily required to confirm the diagnosis of Budd-Chiari syndrome, but it can help in diagnosing the acute or subacute forms and also in ruling out other causes. Histologic findings can include centrizonal congestion, loss of hepatocytes, hemorrhage, and fibrosis.18,19 Regenerative nodules are found in about 25% of patients.19
TREATING BUDD-CHIARI SYNDROME
The primary goal of treatment is to prevent further extension of the venous thrombosis in the hepatic veins, in their collaterals, and in the intrahepatic and extrahepatic portal venous system. Resolution of hepatic congestion improves liver perfusion and preserves function of the hepatocytes.
Anticoagulation is recommended in the early stages. Heparin therapy should be initiated and subsequently switched to warfarin with the goal of achieving an international normalized ratio of the prothrombin time of 2.0 to 2.5.8,9,19
Thrombolysis is effective in the acute form.20,21 Recanalization, including percutaneous or transhepatic angioplasty of localized segments of the narrowed hepatic veins or inferior vena cava, has long-term patency rates of 80% to 90%.22
If thrombolytic therapy and angioplasty are unsuccessful, a transjugular intrahepatic portosystemic shunt or a surgical procedure (side-to-side portocaval shunt, central splenorenal shunt, or mesocaval shunt) should be considered.9
Liver transplantation is another treatment option in those with fulminant Budd-Chiari syndrome or advanced liver cirrhosis.8
PROGNOSIS HAS IMPROVED
The prognosis of Budd-Chiari syndrome has improved, thanks to both earlier diagnosis and new treatments. The 1-year survival rate, which was about 60% before 1985, has increased to more than 80% in recent cohort studies.19
Studies have shown that the Child-Pugh score, which is based on a combination of serum albumin, bilirubin, prothrombin time, encephalopathy, and ascites, can be considered as an independent prognostic factor. A lower Child-Pugh score and a younger age are associated with a good prognosis.19,23,24 (The Child-Pugh score cannot be applied to our patient because he does not have cirrhosis.)
What happened to our patient?
Our patient was started on anticoagulation for his Budd-Chiari syndrome and on bortezomib (Velcade) and dexamethasone for his multiple myeloma. He achieved remarkable improvement in his liver function tests. Follow-up duplex ultrasonography 1 month after discharge revealed that the stenosis in the hepatic veins had resolved. He is following up with the oncology clinic for management of his multiple myeloma.
A 25-year-old man presented to his primary care physician with generalized malaise. His symptoms started around 2 months earlier with progressive fatigue, nausea, decreased appetite, and weight loss (15 lb in 2 months). He denied having fever, chills, night sweats, abdominal pain, diarrhea, melena, or hematochezia.
His medical history was remarkable only for depression, well controlled with sertraline (Zoloft), which he started taking 3 years ago. He was not taking any other prescribed, over-the-counter, or herbal medications.
He had no family history of cancer or liver disease. He did not smoke and rarely drank alcohol. He had never used recreational drugs. He was sexually active with one female partner, used condoms for protection, and had never been diagnosed with a sexually transmitted disease. He had not traveled recently and had not been exposed to any pet.
The patient’s laboratory values on admission are shown in Table 1. Of note, his serum alkaline phosphatase level was 1,307 U/L (reference range 40–150 U/L).
LIVER TESTS CAN NARROW THE DIAGNOSIS
The most commonly used laboratory tests of the liver can be classified into those that measure either:
- Liver synthetic function (eg, the serum albumin and bilirubin concentrations and the prothrombin time) or
- Liver damage, as reflected by the serum concentrations of the enzymes alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase, and gamma-glutamyltransferase (GGT).1,2
ALT and AST are normally concentrated in the hepatocytes and thus, when present in the serum in elevated concentrations, are markers of liver cell injury. The serum levels of these enzymes start to increase within a few hours of liver cell injury as they leak out of the cells via the damaged cell membrane. AST is less liver-specific than ALT, since AST levels can be elevated not only in liver injury but also in muscle, cardiac, and red blood cell injury.3,4
Alkaline phosphatase is actually a heterogeneous group of enzymes found mainly in liver and bone cells. Hepatic alkaline phosphatase is concentrated near the biliary canalicular membrane of the hepatocyte. Accordingly, increased levels of hepatic alkaline phosphatase are mainly seen in liver diseases that predominantly affect the biliary system.3
GGT is also concentrated in hepatic biliary epithelial cells, and thus GGT elevation is another marker of hepatobiliary disease. In fact, measuring the GGT level can help to determine whether an isolated elevation of alkaline phosphatase is due to liver injury.2,3
Accordingly, liver diseases can be classified into two broad categories:
- Hepatocellular injury, in which the primary injury occurs to the hepatocytes
- Cholestatic injury, in which the primary injury is to the bile ducts.
In the former, elevated levels of ALT and AST predominate, while in the latter, elevated alkaline phosphatase is the main finding.3
WHAT TEST NEXT FOR OUR PATIENT?
1. What is the next most appropriate diagnostic step for our patient?
- Liver biopsy
- Ultrasonography of the liver
- Computed tomography (CT) of the liver
- Observation
Our patient has an elevated GGT level, which suggests that his elevated alkaline phosphatase is of hepatic rather than bony origin. Moreover, a serum alkaline phosphatase level that is elevated out of proportion to the aminotransferase levels reflects cholestatic liver injury.
CASE CONTINUED: ULTRASONOGRAPHY IS MOSTLY NORMAL
Ultrasonography of the right upper quadrant revealed that the liver had normal echogenicity and was mildly enlarged. There was no focal hepatic lesion. The gallbladder appeared normal, with no stones or sludge. No dilated intrahepatic or extrahepatic biliary ducts were seen. The common bile duct measured 4 mm. A small amount of ascites not amenable to paracentesis was present.
Thus, in the absence of biliary dilation on ultrasonography, we are dealing with an intrahepatic cholestatic process.
CAUSES OF CHOLESTATIC LIVER DISEASE
Viral hepatitis
Viral hepatitis most often produces a hepatocellular pattern of injury (ie, AST and ALT elevations predominate). However, in rare cases it can cause a cholestatic pattern of injury.
Our patient subsequently had serologic tests for viral hepatitis, including hepatitis A, B, and C, and the results were negative.
Autoimmune liver disease
The three most common forms of autoimmune liver disease are autoimmune hepatitis, primary biliary cirrhosis, and primary sclerosing cholangitis.
Autoimmune hepatitis is characterized by high serum ALT and AST levels, whereas primary biliary cirrhosis and primary sclerosing cholangitis are associated with predominant elevations of alkaline phosphatase, since they are cholestatic disorders.
Our patient’s alkaline phosphatase level was much higher than his ALT and AST levels, making the latter two diseases more likely.
Primary biliary cirrhosis (and autoimmune hepatitis) are associated with autoantibodies in the serum, such as antinuclear antibody, smooth muscle antibody, and antimitochondrial antibody.
Our patient subsequently was tested for these antibodies, and the results were negative.
Primary sclerosing cholangitis usually affects the extrahepatic biliary system. Thus, if it is present, abnormalities should be seen on imaging.
As mentioned previously, no dilated intrahepatic or extrahepatic biliary ducts were seen on ultrasonography in our patient. Moreover, primary sclerosing cholangitis is associated with inflammatory bowel disease, particularly ulcerative colitis, which our patient did not have.
Drug-induced liver injury
Drug-induced liver injury is a common cause of cholestatic liver disease. However, our patient was not taking any prescribed, over-the-counter, or herbal medications. Additionally, he denied heavy alcohol use.
Infiltrative disorders
Infiltrative disorders such as amyloidosis, sarcoidosis, or lymphoma should be considered in the differential diagnosis of cholestatic liver disease. A clue to a possible infiltrative process is a markedly elevated level of alkaline phosphatase with a mildly increased serum bilirubin concentration, both of which our patient had.
AFTER ULTRASONOGRAPHY, WHAT IS THE NEXT STEP?
2. Which of the following is the next most appropriate diagnostic test for our patient?
- Endoscopic retrograde cholangiopancreatography (ERCP)
- Magnetic resonance cholangiopancreatography (MRCP)
- Liver biopsy
- CT of the abdomen
Figure 1 shows a proposed algorithm for evaluating increased alkaline phosphatase levels.
If there is no biliary duct dilation on ultrasonography, then abnormal levels of alkaline phosphatase most likely represent an intrahepatic pattern of cholestatic liver injury. Therefore, additional imaging with CT or magnetic resonance imaging is of limited diagnostic value. ERCP is used today for therapy rather than diagnosis, so its use is limited to patients known to have dilated biliary ducts on imaging. Liver biopsy, however, can provide useful findings.
Case continued: He undergoes biopsy
Our patient underwent transjugular liver biopsy. During the procedure, transjugular venography showed stenosis in the right, middle, and left hepatic veins and the hepatic portion of the inferior vena cava, consistent with Budd-Chiari syndrome.
The liver biopsy specimen was positive for extensive deposition of slight eosinophilic and amorphous material in a sinusoidal pattern in the liver parenchyma, as well as in the portal tracts, with markedly atrophic hepatocytes. Congo red birefringence confirmed the diagnosis of amyloidosis. The immunohistochemical phenotype was positive for kappa light chains, which is diagnostic for primary-type amyloidosis, also called amyloidosis of light chain composition, or AL.
Bone marrow aspiration and bone marrow biopsy were performed and showed 22% plasma cells, well above the normal range (0–2%), consistent with the diagnosis of multiple myeloma.
BUDD-CHIARI SYNDROME: A CHALLENGING DIAGNOSIS
Budd-Chiari syndrome is a rare condition characterized by obstruction of venous outflow from the liver at a site that may vary from the small hepatic veins up to the inferior vena cava or even the right atrium.5,6 Obstruction of hepatic venous outflow leads to sinusoidal congestion and hypoxic damage of the hepatocytes.7 Hypoxia and necrosis of the hepatocytes result in the release of free radicals. Cirrhosis can eventually occur secondary to ischemic necrosis of hepatocytes and hepatic fibrosis.8
The estimated incidence of this syndrome is 1 in 2.5 million persons per year.7 It is more prevalent in women and young adults.8
Heterogeneous in its causes and manifestations
Budd-Chiari syndrome is also heterogeneous in its manifestations, which depend on the extent of the occlusion, on the acuteness of the obstruction, and on whether venous collateral circulation has developed to decompress the liver sinusoids.9,12,13 Therefore, on the basis of its clinical manifestations, it can be classified as fulminant, acute, subacute, or chronic.12–16
The fulminant form presents with hepatic encephalopathy within 8 weeks after the development of jaundice. The subacute form, which is the most common, has a more insidious onset in which hepatic sinusoids are decompressed by portal and hepatic venous collateral circulation. The patient usually presents with abdominal pain, ascites, hepatomegaly, nausea, vomiting, and mild jaundice. Finally the chronic form presents as complications of cirrhosis.12–16
Imaging plays an important role in diagnosing Budd-Chiari syndrome
Imaging plays an important role in detecting and classifying Budd-Chiari syndrome.
Duplex ultrasonography is useful for detecting this syndrome and has a sensitivity and specificity of 85%.9
CT and magnetic resonance imaging can also help in the diagnosis by showing thrombosis, obstruction, or occlusion in the hepatic vein or the inferior vena cava.5
Venography is the gold standard for diagnosis. However, it should be performed only if noninvasive tests are negative or nondiagnostic and there is a high clinical suspicion of this disease.17 Budd-Chiari syndrome has a characteristic pattern on venography known as “spider web,” which is due to the formation of venous collaterals to bypass the occluded hepatic veins.9
Liver biopsy is not necessarily required to confirm the diagnosis of Budd-Chiari syndrome, but it can help in diagnosing the acute or subacute forms and also in ruling out other causes. Histologic findings can include centrizonal congestion, loss of hepatocytes, hemorrhage, and fibrosis.18,19 Regenerative nodules are found in about 25% of patients.19
TREATING BUDD-CHIARI SYNDROME
The primary goal of treatment is to prevent further extension of the venous thrombosis in the hepatic veins, in their collaterals, and in the intrahepatic and extrahepatic portal venous system. Resolution of hepatic congestion improves liver perfusion and preserves function of the hepatocytes.
Anticoagulation is recommended in the early stages. Heparin therapy should be initiated and subsequently switched to warfarin with the goal of achieving an international normalized ratio of the prothrombin time of 2.0 to 2.5.8,9,19
Thrombolysis is effective in the acute form.20,21 Recanalization, including percutaneous or transhepatic angioplasty of localized segments of the narrowed hepatic veins or inferior vena cava, has long-term patency rates of 80% to 90%.22
If thrombolytic therapy and angioplasty are unsuccessful, a transjugular intrahepatic portosystemic shunt or a surgical procedure (side-to-side portocaval shunt, central splenorenal shunt, or mesocaval shunt) should be considered.9
Liver transplantation is another treatment option in those with fulminant Budd-Chiari syndrome or advanced liver cirrhosis.8
PROGNOSIS HAS IMPROVED
The prognosis of Budd-Chiari syndrome has improved, thanks to both earlier diagnosis and new treatments. The 1-year survival rate, which was about 60% before 1985, has increased to more than 80% in recent cohort studies.19
Studies have shown that the Child-Pugh score, which is based on a combination of serum albumin, bilirubin, prothrombin time, encephalopathy, and ascites, can be considered as an independent prognostic factor. A lower Child-Pugh score and a younger age are associated with a good prognosis.19,23,24 (The Child-Pugh score cannot be applied to our patient because he does not have cirrhosis.)
What happened to our patient?
Our patient was started on anticoagulation for his Budd-Chiari syndrome and on bortezomib (Velcade) and dexamethasone for his multiple myeloma. He achieved remarkable improvement in his liver function tests. Follow-up duplex ultrasonography 1 month after discharge revealed that the stenosis in the hepatic veins had resolved. He is following up with the oncology clinic for management of his multiple myeloma.
- Folwaczny C. Efficient diagnostics for elevated transaminases. [Article in German] MMW Fortschr Med 2007; 149:44–48.
- Moussavian SN, Becker RC, Piepmeyer JL, Mezey E, Bozian RC. Serum gamma-glutamyl transpeptidase and chronic alcoholism. Influence of alcohol ingestion and liver disease. Dig Dis Sci 1985; 30:211–214.
- Aragon G, Younossi ZM. When and how to evaluate mildly elevated liver enzymes in apparently healthy patients. Cleve Clin J Med 2010; 77:195–204.
- Lepper PM, Dufour JF. Elevated transaminases—what to do if everything was done?. [Article in German] Praxis (Bern 1994) 2009; 98:330–334.
- Buzas C, Sparchez Z, Cucuianu A, Manole S, Lupescu I, Acalovschi M. Budd-Chiari syndrome secondary to polycythemia vera. A case report. J Gastrointestin Liver Dis 2009; 18:363–366.
- Valla DC. Primary Budd-Chiari syndrome. J Hepatol 2009; 50:195–203.
- Rautou PE, Moucari R, Cazals-Hatem D, et al. Levels and initial course of serum alanine aminotransferase can predict outcome of patients with Budd-Chiari syndrome. Clin Gastroenterol Hepatol 2009; 7:1230–1235.
- Cura M, Haskal Z, Lopera J. Diagnostic and interventional radiology for Budd-Chiari syndrome. Radiographics 2009; 29:669–681.
- Menon KV, Shah V, Kamath PS. The Budd-Chiari syndrome. N Engl J Med 2004; 350:578–585.
- Darwish Murad S, Plessier A, Hernandez-Guerra M, et al; EN-Vie (European Network for Vascular Disorders of the Liver). Etiology, management, and outcome of the Budd-Chiari syndrome. Ann Intern Med 2009; 151:167–175.
- Valla D, Le MG, Poynard T, Zucman N, Rueff B, Benhamou JP. Risk of hepatic vein thrombosis in relation to recent use of oral contraceptives. A case-control study. Gastroenterology 1986; 90:807–811.
- Bismuth H, Sherlock DJ. Portasystemic shunting versus liver transplantation for the Budd-Chiari syndrome. Ann Surg 1991; 214:581–589.
- Orloff MJ, Daily PO, Orloff SL, Girard B, Orloff MS. A 27-year experience with surgical treatment of Budd-Chiari syndrome. Ann Surg 2000; 232:340–352.
- Dilawari JB, Bambery P, Chawla Y, et al. Hepatic outflow obstruction (Budd-Chiari syndrome). Experience with 177 patients and a review of the literature. Medicine (Baltimore) 1994; 73:21–36.
- Mahmoud AE, Mendoza A, Meshikhes AN, et al. Clinical spectrum, investigations and treatment of Budd-Chiari syndrome. QJM 1996; 89:37–43.
- Klein AS, Cameron JL. Diagnosis and management of the Budd-Chiari syndrome. Am J Surg 1990; 160:128–133.
- Plessier A, Valla DC. Budd-Chiari syndrome. Semin Liver Dis 2008; 28:259–269.
- Cazals-Hatem D, Vilgrain V, Genin P, et al. Arterial and portal circulation and parenchymal changes in Budd-Chiari syndrome: a study in 17 explanted livers. Hepatology 2003; 37:510–519.
- Hoekstra J, Janssen HL. Vascular liver disorders (I): diagnosis, treatment and prognosis of Budd-Chiari syndrome. Neth J Med 2008; 66:334–359.
- Frank JW, Kamath PS, Stanson AW. Budd-Chiari syndrome: early intervention with angioplasty and thrombolytic therapy. Mayo Clin Proc 1994; 69:877–881.
- Raju GS, Felver M, Olin JW, Satti SD. Thrombolysis for acute Budd-Chiari syndrome: case report and literature review. Am J Gastroenterol 1996; 91:1262–1263.
- Fisher NC, McCafferty I, Dolapci M, et al. Managing Budd-Chiari syndrome: a retrospective review of percutaneous hepatic vein angioplasty and surgical shunting. Gut 1999; 44:568–574.
- Zeitoun G, Escolano S, Hadengue A, et al. Outcome of Budd-Chiari syndrome: a multivariate analysis of factors related to survival including surgical portosystemic shunting. Hepatology 1999; 30:84–89.
- Darwish Murad S, Valla DC, de Groen PC, et al. Determinants of survival and the effect of portosystemic shunting in patients with Budd-Chiari syndrome. Hepatology 2004; 39:500–508.
- Folwaczny C. Efficient diagnostics for elevated transaminases. [Article in German] MMW Fortschr Med 2007; 149:44–48.
- Moussavian SN, Becker RC, Piepmeyer JL, Mezey E, Bozian RC. Serum gamma-glutamyl transpeptidase and chronic alcoholism. Influence of alcohol ingestion and liver disease. Dig Dis Sci 1985; 30:211–214.
- Aragon G, Younossi ZM. When and how to evaluate mildly elevated liver enzymes in apparently healthy patients. Cleve Clin J Med 2010; 77:195–204.
- Lepper PM, Dufour JF. Elevated transaminases—what to do if everything was done?. [Article in German] Praxis (Bern 1994) 2009; 98:330–334.
- Buzas C, Sparchez Z, Cucuianu A, Manole S, Lupescu I, Acalovschi M. Budd-Chiari syndrome secondary to polycythemia vera. A case report. J Gastrointestin Liver Dis 2009; 18:363–366.
- Valla DC. Primary Budd-Chiari syndrome. J Hepatol 2009; 50:195–203.
- Rautou PE, Moucari R, Cazals-Hatem D, et al. Levels and initial course of serum alanine aminotransferase can predict outcome of patients with Budd-Chiari syndrome. Clin Gastroenterol Hepatol 2009; 7:1230–1235.
- Cura M, Haskal Z, Lopera J. Diagnostic and interventional radiology for Budd-Chiari syndrome. Radiographics 2009; 29:669–681.
- Menon KV, Shah V, Kamath PS. The Budd-Chiari syndrome. N Engl J Med 2004; 350:578–585.
- Darwish Murad S, Plessier A, Hernandez-Guerra M, et al; EN-Vie (European Network for Vascular Disorders of the Liver). Etiology, management, and outcome of the Budd-Chiari syndrome. Ann Intern Med 2009; 151:167–175.
- Valla D, Le MG, Poynard T, Zucman N, Rueff B, Benhamou JP. Risk of hepatic vein thrombosis in relation to recent use of oral contraceptives. A case-control study. Gastroenterology 1986; 90:807–811.
- Bismuth H, Sherlock DJ. Portasystemic shunting versus liver transplantation for the Budd-Chiari syndrome. Ann Surg 1991; 214:581–589.
- Orloff MJ, Daily PO, Orloff SL, Girard B, Orloff MS. A 27-year experience with surgical treatment of Budd-Chiari syndrome. Ann Surg 2000; 232:340–352.
- Dilawari JB, Bambery P, Chawla Y, et al. Hepatic outflow obstruction (Budd-Chiari syndrome). Experience with 177 patients and a review of the literature. Medicine (Baltimore) 1994; 73:21–36.
- Mahmoud AE, Mendoza A, Meshikhes AN, et al. Clinical spectrum, investigations and treatment of Budd-Chiari syndrome. QJM 1996; 89:37–43.
- Klein AS, Cameron JL. Diagnosis and management of the Budd-Chiari syndrome. Am J Surg 1990; 160:128–133.
- Plessier A, Valla DC. Budd-Chiari syndrome. Semin Liver Dis 2008; 28:259–269.
- Cazals-Hatem D, Vilgrain V, Genin P, et al. Arterial and portal circulation and parenchymal changes in Budd-Chiari syndrome: a study in 17 explanted livers. Hepatology 2003; 37:510–519.
- Hoekstra J, Janssen HL. Vascular liver disorders (I): diagnosis, treatment and prognosis of Budd-Chiari syndrome. Neth J Med 2008; 66:334–359.
- Frank JW, Kamath PS, Stanson AW. Budd-Chiari syndrome: early intervention with angioplasty and thrombolytic therapy. Mayo Clin Proc 1994; 69:877–881.
- Raju GS, Felver M, Olin JW, Satti SD. Thrombolysis for acute Budd-Chiari syndrome: case report and literature review. Am J Gastroenterol 1996; 91:1262–1263.
- Fisher NC, McCafferty I, Dolapci M, et al. Managing Budd-Chiari syndrome: a retrospective review of percutaneous hepatic vein angioplasty and surgical shunting. Gut 1999; 44:568–574.
- Zeitoun G, Escolano S, Hadengue A, et al. Outcome of Budd-Chiari syndrome: a multivariate analysis of factors related to survival including surgical portosystemic shunting. Hepatology 1999; 30:84–89.
- Darwish Murad S, Valla DC, de Groen PC, et al. Determinants of survival and the effect of portosystemic shunting in patients with Budd-Chiari syndrome. Hepatology 2004; 39:500–508.
Recognizing and treating cutaneous signs of liver disease
Dysfunction in the body’s second largest organ, the liver, often yields changes in the body’s largest organ, the skin. If we can recognize these manifestations early, we are better able to promptly diagnose and treat the underlying liver disease, as well as the skin lesions.
The liver has many jobs: synthesizing proteins such as clotting factors, complements, and albumin; neutralizing toxins; and metabolizing lipids and carbohydrates. Insults to the liver can compromise any of these functions, affecting visceral organs, joints, gastrointestinal tissues, and the skin. Dermatologic signs of specific liver diseases include alopecia and vitiligo associated with autoimmune hepatitis, and xanthelasma in chronic cholestatic liver disease.
This article reviews the important cutaneous manifestations of specific liver diseases. We focus first on skin conditions that may represent liver disease, and then we discuss several major liver diseases and their typical cutaneous manifestations.
JAUNDICE AND HYPERBILIRUBINEMIA
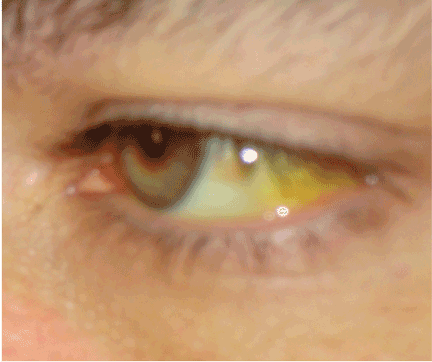
Establishing whether the excess bilirubin is conjugated or unconjugated gives a clue as to whether the cause is prehepatic, intrahepatic, or posthepatic.3–8 One of the liver’s main functions is to conjugate bilirubin into a secretable form. Prehepatic causes of jaundice include hemolysis and ineffective erythropoiesis, both of which lead to higher levels of circulating unconjugated bilirubin.4 Intrahepatic causes of jaundice can lead to both unconjugated and conjugated hyperbilirubinemia.4,8 Posthepatic causes such as bile duct obstruction primarily result in conjugated hyperbilirubinemia.4
PRURITUS AND PRURIGO NODULARIS
Pruritus can be multifactorial or the result of a specific dermatologic or systemic condition.9 A thorough history and physical examination are warranted to rule out hepatic or systemic causes of itching.10
The liver neutralizes toxins and filters bile salts. If its function is impaired, these materials can accumulate in the body, and deposition in the skin causes irritation and itching.11,12 In cholestatic liver disorders such as primary sclerosing cholangitis and obstructive gallstone disease, pruritus tends to be generalized, but worse on the hands and feet.13
Although the severity of pruritus is not directly associated with the level of bile salts and toxic substances, lowering bile salt levels can mitigate symptoms.11
Treatment. Pruritus due to liver disease is particularly resistant to therapy.
In a strategy described by Mela et al for managing pruritus in chronic liver disease,14 the initial treatment is the anion exchange resin cholestyramine (Questran) at a starting dose of 4 g/day, gradually increased to 24 g/day in two doses at mealtimes.
If the pruritus does not respond adequately to cholestyramine or the patient cannot tolerate the drug, then the antituberculosis drug rifampin (Rifadin) can be tried. Rifampin promotes metabolism of endogenous pruritogens and has been effective against cholestatic pruritus when started at 150 mg/day and increased up to 600 mg/day, depending on the clinical response.14
Third-line drug therapies include opioid antagonists such as naltrexone (ReVia) and nalmefene (Revex).14,15
Plasmapheresis can be considered if drug therapy fails.16 Experimental therapies include albumin dialysis using the molecular adsorbent recirculating system (a form of artificial liver support), antioxidant treatment, and bright-light therapy.15 Liver transplantation, when appropriate, also resolves cholestatic pruritus.14
Prurigo nodularis
Prurigo nodularis, distinguished by firm, crusty nodules, is associated with viral infections (eg, hepatitis C, human immunodeficiency virus), bacterial infections, and kidney dysfunction.17,18 The lesions are intensely pruritic and often lead to persistent scratching, excoriation, and, ultimately, diffuse scarring.19
Treatment. Although the exact cause of prurigo nodularis is not known and no cure exists, corticosteroid or antihistamine ointments control the symptoms in most patients with hepatitis.19 Low doses of thalidomide (Thalomid), a tumor necrosis factor antagonist, have also been used safely and effectively.18,19
SUPERFICIAL VASCULAR SIGNS
Spider angiomas
Spider angiomas can occur anywhere on the body, but they occur most often on the face and the trunk.21,23 A key feature is that they disappear when pressure is applied and reappear when pressure is removed.23,24 Biopsy is rarely necessary for diagnosis.
These lesions occur with elevated estrogen levels, such as in cirrhosis, during estrogen therapy, or during pregnancy.25–28 Although spider angiomas are common in pregnant women and in children, adults with spider angiomas deserve a workup for liver dysfunction.29
Given their innocuous nature and asymptomatic course, spider angiomas themselves require no medical treatment.
Bier spots
Bier spots are small, irregularly shaped, hypopigmented patches on the arms and legs. They are likely due to venous stasis associated with functional damage to the small vessels of the skin.30
Since Bier spots are a sign of liver disease, they must be distinguished from true pigmentation disorders. A key distinguishing feature is that Bier spots disappear when pressure is applied. Also, raising the affected limb from a dependent position causes the hypopigmented macules of Bier spots to disappear, which is not the case in true pigmentation disorders.10,30
Paper-money skin
Paper-money skin (or “dollar-paper” markings) describes the condition in which the upper trunk is covered with many randomly scattered, needle-thin superficial capillaries. It often occurs in association with spider angiomas. The name comes from the resemblance the thread-like capillaries have to the finely chopped silk threads in American dollar bills.10,31 The condition is commonly seen in patients with alcoholic cirrhosis and may improve with hemodialysis.31
PALMAR ERYTHEMA
Palmar erythema is a florid, crimson coloration of the palms of the hands and the fingertips. It can occur anywhere on the palm and fingers but is most common on the hypothenar eminence. It can occur in a number of liver conditions but most often with cirrhosis.32 Hepatic compromise, as seen in alcoholic liver disease, disrupts the body’s androgen balance, causing local vasodilation and erythema.32,33 Although the exact mechanism remains unknown, research suggests that prostacyclins and nitric oxide play a role, as both are increased in liver disease.32,33
XANTHELASMA
Xanthelasma—a localized cholesterol deposit beneath the skin and especially beneath the eyelids—is a common manifestation of hypercholesterolemia. Xanthelasma often presents as a painless, yellowish, soft plaque with well-defined borders,34 which may enlarge over the course of weeks.
Several liver diseases can lead to various forms of secondary dyslipoproteinemia.35 The most common dyslipoproteinemias in liver disease are hypertriglyceridemia and low levels of high-density lipoprotein cholesterol, and both of these often accompany fatty liver disease.36 Hypercholesterolemia is a common feature of primary biliary cirrhosis and other forms of cholestatic liver disease.37 Studies suggest that the total plasma cholesterol level is elevated in as many as 50% of patients with compromised liver function.38
Treatment. The underlying hyperlipidemia is treated with cholesterol-lowering drugs. Laser treatment and surgical excision have proven efficacious in treating the lesions.39
OTHER CUTANEOUS FINDINGS IN LIVER DISEASE
Bleeding and bruising. Liver disease can cause hypersplenism and thrombocytopenia, in addition to a decrease in clotting factors. These may present with a myriad of cutaneous symptoms, including purpura, bleeding gums, and easy bruising and bleeding, even from minor trauma.40–42
Hyperpigmentation of the skin may accompany hemochromatosis, alcoholic liver disease, and cirrhosis.43–45
“Terry’s nails,” in which the proximal two-thirds of the nail plate turns powdery white with a ground-glass opacity, may develop in patients with advanced cirrhosis.48
ALCOHOLIC CIRRHOSIS AND THE SKIN
The cutaneous changes associated with alcoholic cirrhosis are more widely recognized than those due to other forms of liver dysfunction. In the United States, approximately 3 million people have alcoholic cirrhosis, the second-leading reason for liver transplantation.49,50
As the body’s main site of alcohol metabolism, the liver is the organ most affected by excessive alcohol intake, which can lead to end-stage liver disease secondary to alcoholic cirrhosis.41,51 The characteristic feature of cirrhosis is advanced fibrous scarring of parenchymal tissue and the formation of regenerative nodules with increased resistance to blood flow throughout the organ.41,52 The insufficient blood flow damages vital structures in the liver and compromises liver function. For example, liver cirrhosis leads to defective hepatic synthesis of clotting factors and results in bleeding disorders.
Cutaneous lesions often accompany alcoholic cirrhosis and have been detected in up to 43% of people with chronic alcoholism.53 Skin changes in alcoholic cirrhosis can be of great diagnostic value. The combined prevalence of spider angiomas, palmar erythema, and Dupuytren contracture in alcoholic cirrhosis was found to be 72%. Paper-money skin and Dupuytren contracture are more distinct lesions for alcoholic cirrhosis.31 Recognizing these skin changes contributes to the diagnosis and staging of liver cirrhosis.51,52
Dupuytren contracture
Dupuytren contracture is characterized by progressive fibrosis and thickening of tendons in the palmar fascia, the connective tissue that lies beneath the skin of the palms.54 Over time, as fibrotic involvement expands across the fascia, rampant stiffness of the joints ensues, sometimes to a point where the fingers cannot fully flex or extend.54
Although the exact cause of Dupuytren contracture is unknown, it appears to be associated with excess alcohol consumption and can be found in patients with alcoholic cirrhosis.54,55 These patients often present with painless stiffness of the fingers, curling of fingers, and loss of motion in involved fingers.54 Surgery in the form of limited fasciectomy has been curative in such patients.54
Disseminated superficial porokeratosis
Porokeratosis is a keratinization disorder of clonal origin that presents as a linear configuration of white scaly papules that coalesce into plaques throughout the body.56 Although it most commonly afflicts fair-skinned people, patients with alcoholic cirrhosis have a much greater susceptibility than the general population.57,58
A recent study58 documented that the lesions completely resolved when liver function improved, thus underlining the relationship between the two conditions. Since immunosuppression has been linked to eruption of the lesion, the fact that both humoral and cell-mediated immune responses are impaired in alcoholic liver disease provides another dimension to the association between porokeratosis and alcoholic cirrhosis.58
These lesions can transform into squamous cell carcinoma.59 The risk of widespread metastases in squamous cell carcinoma highlights the importance of dermatologic consultation in such patients.59
HEPATITIS C AND THE SKIN
Extrahepatic manifestations have been documented in up to 74% of people with hepatitis C virus infection.60 In addition to parasthesias, arthralgias, and myalgias, hepatitis C has a significant association with porphyria cutanea tarda, lichen planus, vitiligo, sialadenitis, urticarial vasculitis, corneal ulcers, xerosis, pruritus, and prurigo nodularis.60–64 Although the primary causative agents of sialadenitis are bacteria, viruses such as hepatitis C have been implicated as a cause of chronic sialadenitis with associated xerostomia.65
Patients with hepatitis C being treated with interferons also present with cutaneous manifestations such as hyperkeratosis and vasculitis.63
Porphyria cutanea tarda
Porphyria cutanea tarda is the most common of the porphyrias, disorders distinguished by deficiencies or defects in one or more of the enzymes responsible for hepatic production of heme.66 If these enzymes are impaired, heme precursors such as porphyrins accumulate.66
Porphyria cutanea tarda results from a deficiency of the hepatic enzyme uroporphyrin decarboxylase. In the absence of this enzyme, shortwave visible light activates uroporphyrin deposited in the skin, resulting in a photochemical reaction that generates reactive oxygen species that lead to the characteristic skin blistering.
Although porphyria cutanea tarda is associated with liver disease in general, recent studies confirm that patients with hepatitis C are at particularly high risk.67 Those with the disorder often present with skin photosensitivity. 68 Many develop blisters on sun-exposed skin, including the dorsal aspects of the hands and forearms and on the neck and face. Chronic porphyria cutanea tarda can lead to scarring, alopecia, and skin ulceration.69 As the blisters heal, keratin-filled milial cysts may develop in the areas of ulceration.
The condition is also commonly associated with melasma-like hyperpigmentation and hypertrichosis in sun-exposed areas of the head and neck. People of Northern European ancestry may be more at risk than the general population because of a presumed genetic susceptibility.70
Treatment. Because many patients with porphyria cutanea tarda have iron overload, they need to restrict foods rich in iron and to avoid alcohol.71,72 Severe cases may necessitate iron removal via phlebotomy or antimalarial therapy. Patients with porphyria cutanea tarda induced by hepatitis C should have their bodily iron stores depleted before starting antiviral therapy.60
Lichen planus
Lichen planopilaris is a subset of lichen planus that causes scaling and atrophy of the scalp and permanent hair loss (Figure 4).73
Interferon-induced vitiligo
Vitiligo is an autoimmune disease in which melanocytes in the skin are destroyed, with resulting depigmentation in affected areas.75 Although it has no specific association with liver disease, it has been linked to treatments for hepatitis C such as interferons.76 Interferon-induced vitiligo often completely resolves when interferon is stopped.77
Typical findings include aggregations of irregularly shaped white patches in a focal or segmental pattern.78 The diagnosis is based on the medical history, physical examination, and sometimes skin biopsy.
HEMOCHROMATOSIS
Hemochromatosis or “bronze diabetes” is a devastating multisystem disease with a relentless course. It is among the most common genetic disorders of metabolism, and results in deposition of iron in tissues and organs throughout the body, including the liver, usually in patients ages 30 to 40.
As iron stores increase in tissues and organs, multiorgan failure and associated complications may ensue. In addition, surplus iron stores can also result in widespread bronze discoloration of skin exposed to the sun. Hemochromatosis also results in loss of body hair, ichthyosiform alterations, and koilonychia.79
Treatments that lower serum iron levels can reverse the cutaneous manifestations of the disorder and minimize the risk of organ failure.
Since the condition is inherited in an autosomal-recessive pattern, family members of patients should consider being screened.80
Hyperpigmentation in hemochromatosis
Hyperpigmentation is an early sign of hemochromatosis, affecting up to 90% of patients. Usually, sun-exposed areas of the body are the most prone and take on a grayish or brownish-bronze hue.81 Cutaneous iron deposits injure vital skin structures, initiating a process that culminates in enhanced melanin production by melanocytes.82 Exposure to ultraviolet light may have synergistic effects with iron, hastening the process of hyperpigmentation. As a result of this synergistic effect, many patients with hemochromatosis notice tanning with minimal sun exposure.
Although organ function can improve immediately with phlebotomy to reduce iron stores, skin hyperpigmentation does not immediately resolve.81,82
Ichthyosiform alterations in hemochromatosis
Ichthyosiform changes, in which the skin takes on the appearance of fish scales,83 can be seen in patients with hemochromatosis.80 Affected areas typically become extremely dry. Treatment includes topical hydrating creams and ointments. Avoiding sunlight is paramount, as sunlight exposure may exacerbate the condition.83
- Morioka D, Togo S, Kumamoto T, et al. Six consecutive cases of successful adult ABO-incompatible living donor liver transplantation: a proposal for grading the severity of antibody-mediated rejection. Transplantation 2008; 85:171–178.
- Bertini G, Rubaltelli FF. Non-invasive bilirubinometry in neonatal jaundice. Semin Neonatol 2002; 7:129–133.
- Clementi M, Di Gianantonio E, Fabris L, et al. Inheritance of hyperbilirubinemia: evidence for a major autosomal recessive gene. Dig Liver Dis 2007; 39:351–355.
- Roche SP, Kobos R. Jaundice in the adult patient. Am Fam Physician 2004; 69:299–304.
- Odemis B, Parlak E, Basar O, Yüksel O, Sahin B. Biliary tract obstruction secondary to malignant lymphoma: experience at a referral center. Dig Dis Sci 2007; 52:2323–2332.
- Caddy GR, Tham TC. Gallstone disease: symptoms, diagnosis and endoscopic management of common bile duct stones. Best Pract Res Clin Gastroenterol 2006; 20:1085–1101.
- Heathcote EJ. Diagnosis and management of cholestatic liver disease. Clin Gastroenterol Hepatol 2007; 5:776–782.
- Faust TW, Reddy KR. Postoperative jaundice. Clin Liver Dis 2004; 8:151–166.
- Maticic M, Poljak M, Lunder T, Rener-Sitar K, Stojanovic L. Lichen planus and other cutaneous manifestations in chronic hepatitis C: pre- and post-interferon-based treatment prevalence vary in a cohort of patients from low hepatitis C virus endemic area. J Eur Acad Dermatol Venereol 2008; 22:779–788.
- Polat M, Oztas P, Ilhan MN, Yalçin B, Alli N. Generalized pruritus: a prospective study concerning etiology. Am J Clin Dermatol 2008; 9:39–44.
- Gaspari R, Avolio AW, Zileri Dal Verme L, et al. Molecular adsorbent recirculating system in liver transplantation: safety and efficacy. Transplant Proc 2006; 38:3544–3551.
- Dasgupta R, Saha I, Pal S, et al. Immunosuppression, hepatotoxicity and depression of antioxidant status by arecoline in albino mice. Toxicology 2006; 227:94–104.
- Mayo MJ, Handem I, Saldana S, Jacobe H, Getachew Y, Rush AJ. Sertraline as a first-line treatment for cholestatic pruritus. Hepatology 2007; 45:666–674.
- Mela M, Mancuso A, Burroughs AK. Review article: pruritus in cholestatic and other liver diseases. Aliment Pharmacol Ther 2003; 17:857–870.
- Montero JL, Pozo JC, Barrera P, et al. Treatment of refractory cholestatic pruritus with molecular adsorbent recirculating system (MARS). Transplant Proc 2006; 38:2511–2513.
- Neff GW, O'Brien CB, Reddy KR, et al. Preliminary observation with dronabinol in patients with intractable pruritus secondary to cholestatic liver disease. Am J Gastroenterol 2002; 97:2117–2119.
- Neri S, Raciti C, D’Angelo G, Ierna D, Bruno CM. Hyde’s prurigo nodularis and chronic HCV hepatitis. J Hepatol 1998; 28:161–164.
- Brown MA, George CR, Dunstan CR, Kalowski S, Corrigan AB. Prurigo nodularis and aluminium overload in maintenance haemodialysis. Lancet 1992; 340:48.
- Stander S, Luger T, Metze D. Treatment of prurigo nodularis with topical capsaicin. J Am Acad Dermatol 2001; 44:471–478.
- Requena L, Sangueza OP. Cutaneous vascular anomalies. Part I. Hamartomas, malformations, and dilation of preexisting vessels. J Am Acad Dermatol 1997; 37:523–549.
- Khasnis A, Gokula RM. Spider nevus. J Postgrad Med 2002; 48:307–309.
- Kaul V, Friedenberg FK, Braitman LE, et al. Development and validation of a model to diagnose cirrhosis in patients with hepatitis C. Am J Gastroenterol 2002; 97:2623–2628.
- Banyai AL. There is more than surface appearance to skin spiders. Chest 1971; 60:48.
- Errickson CV, Matus NR. Skin disorders of pregnancy. Am Fam Physician 1994; 49:605–610.
- Li CP, Lee FY, Hwang SJ, et al. Spider angiomas in patients with liver cirrhosis: role of alcoholism and impaired liver function. Scand J Gastroenterol 1999; 34:520–523.
- Li CP, Lee FY, Hwang SJ, et al. Role of substance P in the pathogenesis of spider angiomas in patients with nonalcoholic liver cirrhosis. Am J Gastroenterol 1999; 94:502–507.
- Sadick NS, Niedt GW. A study of estrogen and progesterone receptors in spider telangiectasias of the lower extremities. J Dermatol Surg Oncol 1990; 16:620–623.
- Henry F, Quatresooz P, Valverde-Lopez JC, Pierard GE. Blood vessel changes during pregnancy: a review. Am J Clin Dermatol 2006; 7:65–69.
- Finn SM, Rowland M, Lawlor F, et al. The significance of cutaneous spider naevi in children. Arch Dis Child 2006; 91:604–605.
- Peyrot I, Boulinguez S, Sparsa A, Le Meur Y, Bonnetblanc JM, Bedane C. Bier’s white spots associated with scleroderma renal crisis. Clin Exp Dermatol 2007; 32:165–167.
- Satoh T, Yokozeki H, Nishioka K. Vascular spiders and paper money skin improved by hemodialysis. Dermatology 2002; 205:73–74.
- Serrao R, Zirwas M, English JC. Palmar erythema. Am J Clin Dermatol 2007; 8:347–356.
- Matsumoto M, Ohki K, Nagai I, Oshibuchi T. Lung traction causes an increase in plasma prostacyclin concentration and decrease in mean arterial blood pressure. Anesth Analg 1992; 75:773–776.
- Otto AI, Horvath I, Feldmann J. Multiple firm, painless erythematous papules with a yellowish hue. Arch Dermatol 2005; 141:1595–1600.
- Gandelman G, Aronow WS, Weiss MB. Resolving hyperlipidemia after liver transplantation in a patient with primary sclerosing cholangitis. Am J Ther 2006; 13:171–174.
- Assy N, Kaita K, Mymin D, Levy C, Rosser B, Minuk G. Fatty infiltration of liver in hyperlipidemic patients. Dig Dis Sci 2000; 45:1929–1934.
- Allocca M, Crosignani A, Gritti A, et al. Hypercholesterolaemia is not associated with early atherosclerotic lesions in primary biliary cirrhosis. Gut 2006; 55:1795–1800.
- Dickson E, Fleming C, Ludwig J. Primary biliary cirrhosis. In:Popper H, Schaffner F, editors. Progress in Liver Diseases. New York: Grune and Stratton; 1978:487.
- Elner VM, Mintz R, Demirci H, Hassan AS. Local corticosteroid treatment of eyelid and orbital xanthogranuloma. Trans Am Ophthalmol Soc 2005; 103:69–73.
- Craxi A, Camma C, Giunta M. Clinical aspects of bleeding complications in cirrhotic patients. Blood Coagul Fibrinolysis 2000; 11( suppl 1):S75–579.
- Kajihara M, Okazaki Y, Kato S, et al. Evaluation of platelet kinetics in patients with liver cirrhosis: similarity to idiopathic thrombocytopenic purpura. J Gastroenterol Hepatol 2007; 22:112–118.
- Levine N. Patient reports six-month history of minimally pruritic purple dots on legs. Non-blanching macules developed over six months. Geriatrics 2006; 61:22.
- Barton JC, Rao SV, Pereira NM, et al. Juvenile hemochromatosis in the southeastern United States: a report of seven cases in two kinships. Blood Cells Mol Dis 2002; 29:104–115.
- Smith AG, Shuster S, Bomford A, Williams R. Plasma immunoreactive beta-melanocyte-stimulating hormone in chronic liver disease and fulminant hepatic failure. J Invest Dermatol 1978; 70:326–327.
- Barton JC, McDonnell SM, Adams PC, et al. Management of hemochromatosis. Hemochromatosis Management Working Group. Ann Intern Med 1998; 129:932–939.
- Bahnsen M, Gluud C, Johnsen SG, et al. Pituitary-testicular function in patients with alcoholic cirrhosis of the liver. Eur J Clin Invest 1981; 11:473–479.
- Kumar N, Aggarwal SR, Anand BS. Comparison of truncal hair distribution in alcoholic liver disease and alcohol-related chronic pancreatitis. J Gastroenterol Hepatol 2001; 16:855–856.
- Holzberg M, Walker HK. Terry's nails: revised definition and new correlations. Lancet 1984; 1:896–899.
- Mandayam S, Jamal MM, Morgan TR. Epidemiology of alcoholic liver disease. Semin Liver Dis 2004; 24:217–232.
- Belle SH, Beringer KC, Detre KM. Liver transplantation in the United States: results from the National Pitt-UNOS Liver Transplant Registry. United Network for Organ Sharing Clin Transpl 1994:19–35.
- Dunn W, Xu R, Schwimmer JB. Modest wine drinking and decreased prevalence of suspected nonalcoholic fatty liver disease. Hepatology 2008; 47:1947–1954.
- Afford SC, Fisher NC, Neil DA, et al. Distinct patterns of chemokine expression are associated with leukocyte recruitment in alcoholic hepatitis and alcoholic cirrhosis. J Pathol 1998; 186:82–89.
- Evstaf’ev VV, Levin MM. Dermatologic pathology in chronic alcoholics. Vestn Dermatol Venerol 1989; 8:72–74.
- Jerosch-Herold C, Shepstone L, Chojnowski AJ, Larson D. Splinting after contracture release for Dupuytren's contracture (SCoRD): protocol of a pragmatic, multi-centre, randomized controlled trial. BMC Musculoskelet Disord 2008; 9:62.
- Houghton S, Holdstock G, Cockerell R, Wright R. Dupuytren’s contracture, chronic liver disease and IgA immune complexes. Liver 1983; 3:220–224.
- Ibbotson SH. Disseminated superficial porokeratosis: what is the association with ultraviolet radiation? Clin Exp Dermatol 1996; 21:48–50.
- Kono T, Kobayashi H, Ishii M, Nishiguchi S, Taniguchi S. Synchronous development of disseminated superficial porokeratosis and hepatitis C virus-related hepatocellular carcinoma. J Am Acad Dermatol 2000; 43:966–968.
- Park BS, Moon SE, Kim JA. Disseminated superficial porokeratosis in a patient with chronic liver disease. J Dermatol 1997; 24:485–487.
- Murata Y, Kumano K, Takai T. Type 2 segmental manifestation of disseminated superficial porokeratosis showing a systematized pattern of involvement and pronounced cancer proneness. Eur J Dermatol 2001; 11:191–194.
- Galossi A, Guarisco R, Bellis L, Puoti C. Extrahepatic manifestations of chronic HCV infection. J Gastrointestin Liver Dis 2007; 16:65–73.
- El-Serag HB, Hampel H, Yeh C, Rabeneck L. Extrahepatic manifestations of hepatitis C among United States male veterans. Hepatology 2002; 36:1439–1445.
- Stefanova-Petrova DV, Tzvetanska AH, Naumova EJ, et al. Chronic hepatitis C virus infection: prevalence of extrahepatic manifestations and association with cryoglobulinemia in Bulgarian patients. World J Gastroenterol 2007; 13:6518–6528.
- Vassilopoulos D, Calabrese LH. Extrahepatic immunological complications of hepatitis C virus infection. AIDS 2005; 19( suppl 3):S123–S127.
- Hsing AW, Zhang M, Rashid A, et al. Hepatitis B and C virus infection and the risk of biliary tract cancer: a population-based study in China. Int J Cancer 2008; 122:1849–1853.
- Madrid C, Courtois B, Duran D. Chronic sialadenitis revealing hepatitis C: a case report. Med Oral 2004; 9:328–332.
- Lançoni G, Ravinal RC, Costa RS, Roselino AM. Mast cells and transforming growth factor-beta expression: a possible relationship in the development of porphyria cutanea tarda skin lesions. Int J Dermatol 2008; 47:575–581.
- Toll A, Celis R, Ozalla MD, Bruguera M, Herrero C, Ercilla MG. The prevalence of HFE C282Y gene mutation is increased in Spanish patients with porphyria cutanea tarda without hepatitis C virus infection. J Eur Acad Dermatol Venereol 2006; 20:1201–1206.
- Badminton MN, Elder GH. Management of acute and cutaneous porphyrias. Int J Clin Pract 2002; 56:272–278.
- Jackson JM, Callen JP. Scarring alopecia and sclerodermatous changes of the scalp in a patient with hepatitis C infection. J Am Acad Dermatol 1998; 39:824–826.
- Mortimore M, Merryweather-Clarke AT, Robson KJ, Powell LW. The haemochromatosis gene: a global perspective and implications for the Asia-Pacific region. J Gastroenterol Hepatol 1999; 14:838–843.
- Shehan JM, Huerter CJ. Porphyria cutanea tarda associated with an acute gastrointestinal bleed: the roles of supplemental iron and blood transfusion. Cutis 2001; 68:147–150.
- Lambrecht RW, Thapar M, Bonkovsky HL. Genetic aspects of porphyria cutanea tarda. Semin Liver Dis 2007; 27:99–108.
- d’Ovidio R, Sgarra C, Conserva A, Angelotti UF, Erriquez R, Foti C. Alterated integrin expression in lichen planopilaris. Head Face Med 2007; 3:11.
- Chuang TY, Stitle L, Brashear R, Lewis C. Hepatitis C virus and lichen planus: a case-control study of 340 patients. J Am Acad Dermatol 1999; 41:787–789.
- Kemp EH, Gavalas NG, Gawkrodger DJ, Weetman AP. Autoantibody responses to melanocytes in the depigmenting skin disease vitiligo. Autoimmun Rev 2007; 6:138–142.
- Tomasiewicz K, Modrzewska R, Semczuk G. Vitiligo associated with pegylated interferon and ribavirin treatment of patients with chronic hepatitis C: a case report. Adv Ther 2006; 23:139–142.
- Simsek H, Savas C, Akkiz H, Telatar H. Interferon-induced vitiligo in a patient with chronic viral hepatitis C infection. Dermatology 1996; 193:65–66.
- Mulekar SV, Al Issa A, Asaad M, Ghwish B, Al Eisa A. Mixed vitiligo. J Cutan Med Surg. 2006; 10:104–107.
- Waalen J, Felitti V, Gelbart T, Ho NJ, Beutler E. Prevalence of hemochromatosis-related symptoms among individuals with mutations in the HFE gene. Mayo Clin Proc 2002; 77:522–530.
- Walker AP, Tucker DC, Hall MA, et al. Results communication and patient education after screening for possible hemochromatosis and iron overload: experience from the HEIRS study of a large ethnically and linguistically diverse group. Genet Med 2007; 9:778–791.
- Stulberg DL, Clark N, Tovey D. Common hyperpigmentation disorders in adults: Part I. Diagnostic approach, cafe au lait macules, diffuse hyperpigmentation, sun exposure, and phototoxic reactions. Am Fam Physician 2003; 68:1955–1960.
- Tsuji T. Experimental hemosiderosis: relationship between skin pigmentation and hemosiderin. Acta Derm Venereol 1980; 60:109–114.
- Oji V, Traupe H. Ichthyoses: differential diagnosis and molecular genetics. Eur J Dermatol 2006; 16:349–359.
Dysfunction in the body’s second largest organ, the liver, often yields changes in the body’s largest organ, the skin. If we can recognize these manifestations early, we are better able to promptly diagnose and treat the underlying liver disease, as well as the skin lesions.
The liver has many jobs: synthesizing proteins such as clotting factors, complements, and albumin; neutralizing toxins; and metabolizing lipids and carbohydrates. Insults to the liver can compromise any of these functions, affecting visceral organs, joints, gastrointestinal tissues, and the skin. Dermatologic signs of specific liver diseases include alopecia and vitiligo associated with autoimmune hepatitis, and xanthelasma in chronic cholestatic liver disease.
This article reviews the important cutaneous manifestations of specific liver diseases. We focus first on skin conditions that may represent liver disease, and then we discuss several major liver diseases and their typical cutaneous manifestations.
JAUNDICE AND HYPERBILIRUBINEMIA
Establishing whether the excess bilirubin is conjugated or unconjugated gives a clue as to whether the cause is prehepatic, intrahepatic, or posthepatic.3–8 One of the liver’s main functions is to conjugate bilirubin into a secretable form. Prehepatic causes of jaundice include hemolysis and ineffective erythropoiesis, both of which lead to higher levels of circulating unconjugated bilirubin.4 Intrahepatic causes of jaundice can lead to both unconjugated and conjugated hyperbilirubinemia.4,8 Posthepatic causes such as bile duct obstruction primarily result in conjugated hyperbilirubinemia.4
PRURITUS AND PRURIGO NODULARIS
Pruritus can be multifactorial or the result of a specific dermatologic or systemic condition.9 A thorough history and physical examination are warranted to rule out hepatic or systemic causes of itching.10
The liver neutralizes toxins and filters bile salts. If its function is impaired, these materials can accumulate in the body, and deposition in the skin causes irritation and itching.11,12 In cholestatic liver disorders such as primary sclerosing cholangitis and obstructive gallstone disease, pruritus tends to be generalized, but worse on the hands and feet.13
Although the severity of pruritus is not directly associated with the level of bile salts and toxic substances, lowering bile salt levels can mitigate symptoms.11
Treatment. Pruritus due to liver disease is particularly resistant to therapy.
In a strategy described by Mela et al for managing pruritus in chronic liver disease,14 the initial treatment is the anion exchange resin cholestyramine (Questran) at a starting dose of 4 g/day, gradually increased to 24 g/day in two doses at mealtimes.
If the pruritus does not respond adequately to cholestyramine or the patient cannot tolerate the drug, then the antituberculosis drug rifampin (Rifadin) can be tried. Rifampin promotes metabolism of endogenous pruritogens and has been effective against cholestatic pruritus when started at 150 mg/day and increased up to 600 mg/day, depending on the clinical response.14
Third-line drug therapies include opioid antagonists such as naltrexone (ReVia) and nalmefene (Revex).14,15
Plasmapheresis can be considered if drug therapy fails.16 Experimental therapies include albumin dialysis using the molecular adsorbent recirculating system (a form of artificial liver support), antioxidant treatment, and bright-light therapy.15 Liver transplantation, when appropriate, also resolves cholestatic pruritus.14
Prurigo nodularis
Prurigo nodularis, distinguished by firm, crusty nodules, is associated with viral infections (eg, hepatitis C, human immunodeficiency virus), bacterial infections, and kidney dysfunction.17,18 The lesions are intensely pruritic and often lead to persistent scratching, excoriation, and, ultimately, diffuse scarring.19
Treatment. Although the exact cause of prurigo nodularis is not known and no cure exists, corticosteroid or antihistamine ointments control the symptoms in most patients with hepatitis.19 Low doses of thalidomide (Thalomid), a tumor necrosis factor antagonist, have also been used safely and effectively.18,19
SUPERFICIAL VASCULAR SIGNS
Spider angiomas
Spider angiomas can occur anywhere on the body, but they occur most often on the face and the trunk.21,23 A key feature is that they disappear when pressure is applied and reappear when pressure is removed.23,24 Biopsy is rarely necessary for diagnosis.
These lesions occur with elevated estrogen levels, such as in cirrhosis, during estrogen therapy, or during pregnancy.25–28 Although spider angiomas are common in pregnant women and in children, adults with spider angiomas deserve a workup for liver dysfunction.29
Given their innocuous nature and asymptomatic course, spider angiomas themselves require no medical treatment.
Bier spots
Bier spots are small, irregularly shaped, hypopigmented patches on the arms and legs. They are likely due to venous stasis associated with functional damage to the small vessels of the skin.30
Since Bier spots are a sign of liver disease, they must be distinguished from true pigmentation disorders. A key distinguishing feature is that Bier spots disappear when pressure is applied. Also, raising the affected limb from a dependent position causes the hypopigmented macules of Bier spots to disappear, which is not the case in true pigmentation disorders.10,30
Paper-money skin
Paper-money skin (or “dollar-paper” markings) describes the condition in which the upper trunk is covered with many randomly scattered, needle-thin superficial capillaries. It often occurs in association with spider angiomas. The name comes from the resemblance the thread-like capillaries have to the finely chopped silk threads in American dollar bills.10,31 The condition is commonly seen in patients with alcoholic cirrhosis and may improve with hemodialysis.31
PALMAR ERYTHEMA
Palmar erythema is a florid, crimson coloration of the palms of the hands and the fingertips. It can occur anywhere on the palm and fingers but is most common on the hypothenar eminence. It can occur in a number of liver conditions but most often with cirrhosis.32 Hepatic compromise, as seen in alcoholic liver disease, disrupts the body’s androgen balance, causing local vasodilation and erythema.32,33 Although the exact mechanism remains unknown, research suggests that prostacyclins and nitric oxide play a role, as both are increased in liver disease.32,33
XANTHELASMA
Xanthelasma—a localized cholesterol deposit beneath the skin and especially beneath the eyelids—is a common manifestation of hypercholesterolemia. Xanthelasma often presents as a painless, yellowish, soft plaque with well-defined borders,34 which may enlarge over the course of weeks.
Several liver diseases can lead to various forms of secondary dyslipoproteinemia.35 The most common dyslipoproteinemias in liver disease are hypertriglyceridemia and low levels of high-density lipoprotein cholesterol, and both of these often accompany fatty liver disease.36 Hypercholesterolemia is a common feature of primary biliary cirrhosis and other forms of cholestatic liver disease.37 Studies suggest that the total plasma cholesterol level is elevated in as many as 50% of patients with compromised liver function.38
Treatment. The underlying hyperlipidemia is treated with cholesterol-lowering drugs. Laser treatment and surgical excision have proven efficacious in treating the lesions.39
OTHER CUTANEOUS FINDINGS IN LIVER DISEASE
Bleeding and bruising. Liver disease can cause hypersplenism and thrombocytopenia, in addition to a decrease in clotting factors. These may present with a myriad of cutaneous symptoms, including purpura, bleeding gums, and easy bruising and bleeding, even from minor trauma.40–42
Hyperpigmentation of the skin may accompany hemochromatosis, alcoholic liver disease, and cirrhosis.43–45
“Terry’s nails,” in which the proximal two-thirds of the nail plate turns powdery white with a ground-glass opacity, may develop in patients with advanced cirrhosis.48
ALCOHOLIC CIRRHOSIS AND THE SKIN
The cutaneous changes associated with alcoholic cirrhosis are more widely recognized than those due to other forms of liver dysfunction. In the United States, approximately 3 million people have alcoholic cirrhosis, the second-leading reason for liver transplantation.49,50
As the body’s main site of alcohol metabolism, the liver is the organ most affected by excessive alcohol intake, which can lead to end-stage liver disease secondary to alcoholic cirrhosis.41,51 The characteristic feature of cirrhosis is advanced fibrous scarring of parenchymal tissue and the formation of regenerative nodules with increased resistance to blood flow throughout the organ.41,52 The insufficient blood flow damages vital structures in the liver and compromises liver function. For example, liver cirrhosis leads to defective hepatic synthesis of clotting factors and results in bleeding disorders.
Cutaneous lesions often accompany alcoholic cirrhosis and have been detected in up to 43% of people with chronic alcoholism.53 Skin changes in alcoholic cirrhosis can be of great diagnostic value. The combined prevalence of spider angiomas, palmar erythema, and Dupuytren contracture in alcoholic cirrhosis was found to be 72%. Paper-money skin and Dupuytren contracture are more distinct lesions for alcoholic cirrhosis.31 Recognizing these skin changes contributes to the diagnosis and staging of liver cirrhosis.51,52
Dupuytren contracture
Dupuytren contracture is characterized by progressive fibrosis and thickening of tendons in the palmar fascia, the connective tissue that lies beneath the skin of the palms.54 Over time, as fibrotic involvement expands across the fascia, rampant stiffness of the joints ensues, sometimes to a point where the fingers cannot fully flex or extend.54
Although the exact cause of Dupuytren contracture is unknown, it appears to be associated with excess alcohol consumption and can be found in patients with alcoholic cirrhosis.54,55 These patients often present with painless stiffness of the fingers, curling of fingers, and loss of motion in involved fingers.54 Surgery in the form of limited fasciectomy has been curative in such patients.54
Disseminated superficial porokeratosis
Porokeratosis is a keratinization disorder of clonal origin that presents as a linear configuration of white scaly papules that coalesce into plaques throughout the body.56 Although it most commonly afflicts fair-skinned people, patients with alcoholic cirrhosis have a much greater susceptibility than the general population.57,58
A recent study58 documented that the lesions completely resolved when liver function improved, thus underlining the relationship between the two conditions. Since immunosuppression has been linked to eruption of the lesion, the fact that both humoral and cell-mediated immune responses are impaired in alcoholic liver disease provides another dimension to the association between porokeratosis and alcoholic cirrhosis.58
These lesions can transform into squamous cell carcinoma.59 The risk of widespread metastases in squamous cell carcinoma highlights the importance of dermatologic consultation in such patients.59
HEPATITIS C AND THE SKIN
Extrahepatic manifestations have been documented in up to 74% of people with hepatitis C virus infection.60 In addition to parasthesias, arthralgias, and myalgias, hepatitis C has a significant association with porphyria cutanea tarda, lichen planus, vitiligo, sialadenitis, urticarial vasculitis, corneal ulcers, xerosis, pruritus, and prurigo nodularis.60–64 Although the primary causative agents of sialadenitis are bacteria, viruses such as hepatitis C have been implicated as a cause of chronic sialadenitis with associated xerostomia.65
Patients with hepatitis C being treated with interferons also present with cutaneous manifestations such as hyperkeratosis and vasculitis.63
Porphyria cutanea tarda
Porphyria cutanea tarda is the most common of the porphyrias, disorders distinguished by deficiencies or defects in one or more of the enzymes responsible for hepatic production of heme.66 If these enzymes are impaired, heme precursors such as porphyrins accumulate.66
Porphyria cutanea tarda results from a deficiency of the hepatic enzyme uroporphyrin decarboxylase. In the absence of this enzyme, shortwave visible light activates uroporphyrin deposited in the skin, resulting in a photochemical reaction that generates reactive oxygen species that lead to the characteristic skin blistering.
Although porphyria cutanea tarda is associated with liver disease in general, recent studies confirm that patients with hepatitis C are at particularly high risk.67 Those with the disorder often present with skin photosensitivity. 68 Many develop blisters on sun-exposed skin, including the dorsal aspects of the hands and forearms and on the neck and face. Chronic porphyria cutanea tarda can lead to scarring, alopecia, and skin ulceration.69 As the blisters heal, keratin-filled milial cysts may develop in the areas of ulceration.
The condition is also commonly associated with melasma-like hyperpigmentation and hypertrichosis in sun-exposed areas of the head and neck. People of Northern European ancestry may be more at risk than the general population because of a presumed genetic susceptibility.70
Treatment. Because many patients with porphyria cutanea tarda have iron overload, they need to restrict foods rich in iron and to avoid alcohol.71,72 Severe cases may necessitate iron removal via phlebotomy or antimalarial therapy. Patients with porphyria cutanea tarda induced by hepatitis C should have their bodily iron stores depleted before starting antiviral therapy.60
Lichen planus
Lichen planopilaris is a subset of lichen planus that causes scaling and atrophy of the scalp and permanent hair loss (Figure 4).73
Interferon-induced vitiligo
Vitiligo is an autoimmune disease in which melanocytes in the skin are destroyed, with resulting depigmentation in affected areas.75 Although it has no specific association with liver disease, it has been linked to treatments for hepatitis C such as interferons.76 Interferon-induced vitiligo often completely resolves when interferon is stopped.77
Typical findings include aggregations of irregularly shaped white patches in a focal or segmental pattern.78 The diagnosis is based on the medical history, physical examination, and sometimes skin biopsy.
HEMOCHROMATOSIS
Hemochromatosis or “bronze diabetes” is a devastating multisystem disease with a relentless course. It is among the most common genetic disorders of metabolism, and results in deposition of iron in tissues and organs throughout the body, including the liver, usually in patients ages 30 to 40.
As iron stores increase in tissues and organs, multiorgan failure and associated complications may ensue. In addition, surplus iron stores can also result in widespread bronze discoloration of skin exposed to the sun. Hemochromatosis also results in loss of body hair, ichthyosiform alterations, and koilonychia.79
Treatments that lower serum iron levels can reverse the cutaneous manifestations of the disorder and minimize the risk of organ failure.
Since the condition is inherited in an autosomal-recessive pattern, family members of patients should consider being screened.80
Hyperpigmentation in hemochromatosis
Hyperpigmentation is an early sign of hemochromatosis, affecting up to 90% of patients. Usually, sun-exposed areas of the body are the most prone and take on a grayish or brownish-bronze hue.81 Cutaneous iron deposits injure vital skin structures, initiating a process that culminates in enhanced melanin production by melanocytes.82 Exposure to ultraviolet light may have synergistic effects with iron, hastening the process of hyperpigmentation. As a result of this synergistic effect, many patients with hemochromatosis notice tanning with minimal sun exposure.
Although organ function can improve immediately with phlebotomy to reduce iron stores, skin hyperpigmentation does not immediately resolve.81,82
Ichthyosiform alterations in hemochromatosis
Ichthyosiform changes, in which the skin takes on the appearance of fish scales,83 can be seen in patients with hemochromatosis.80 Affected areas typically become extremely dry. Treatment includes topical hydrating creams and ointments. Avoiding sunlight is paramount, as sunlight exposure may exacerbate the condition.83
Dysfunction in the body’s second largest organ, the liver, often yields changes in the body’s largest organ, the skin. If we can recognize these manifestations early, we are better able to promptly diagnose and treat the underlying liver disease, as well as the skin lesions.
The liver has many jobs: synthesizing proteins such as clotting factors, complements, and albumin; neutralizing toxins; and metabolizing lipids and carbohydrates. Insults to the liver can compromise any of these functions, affecting visceral organs, joints, gastrointestinal tissues, and the skin. Dermatologic signs of specific liver diseases include alopecia and vitiligo associated with autoimmune hepatitis, and xanthelasma in chronic cholestatic liver disease.
This article reviews the important cutaneous manifestations of specific liver diseases. We focus first on skin conditions that may represent liver disease, and then we discuss several major liver diseases and their typical cutaneous manifestations.
JAUNDICE AND HYPERBILIRUBINEMIA
Establishing whether the excess bilirubin is conjugated or unconjugated gives a clue as to whether the cause is prehepatic, intrahepatic, or posthepatic.3–8 One of the liver’s main functions is to conjugate bilirubin into a secretable form. Prehepatic causes of jaundice include hemolysis and ineffective erythropoiesis, both of which lead to higher levels of circulating unconjugated bilirubin.4 Intrahepatic causes of jaundice can lead to both unconjugated and conjugated hyperbilirubinemia.4,8 Posthepatic causes such as bile duct obstruction primarily result in conjugated hyperbilirubinemia.4
PRURITUS AND PRURIGO NODULARIS
Pruritus can be multifactorial or the result of a specific dermatologic or systemic condition.9 A thorough history and physical examination are warranted to rule out hepatic or systemic causes of itching.10
The liver neutralizes toxins and filters bile salts. If its function is impaired, these materials can accumulate in the body, and deposition in the skin causes irritation and itching.11,12 In cholestatic liver disorders such as primary sclerosing cholangitis and obstructive gallstone disease, pruritus tends to be generalized, but worse on the hands and feet.13
Although the severity of pruritus is not directly associated with the level of bile salts and toxic substances, lowering bile salt levels can mitigate symptoms.11
Treatment. Pruritus due to liver disease is particularly resistant to therapy.
In a strategy described by Mela et al for managing pruritus in chronic liver disease,14 the initial treatment is the anion exchange resin cholestyramine (Questran) at a starting dose of 4 g/day, gradually increased to 24 g/day in two doses at mealtimes.
If the pruritus does not respond adequately to cholestyramine or the patient cannot tolerate the drug, then the antituberculosis drug rifampin (Rifadin) can be tried. Rifampin promotes metabolism of endogenous pruritogens and has been effective against cholestatic pruritus when started at 150 mg/day and increased up to 600 mg/day, depending on the clinical response.14
Third-line drug therapies include opioid antagonists such as naltrexone (ReVia) and nalmefene (Revex).14,15
Plasmapheresis can be considered if drug therapy fails.16 Experimental therapies include albumin dialysis using the molecular adsorbent recirculating system (a form of artificial liver support), antioxidant treatment, and bright-light therapy.15 Liver transplantation, when appropriate, also resolves cholestatic pruritus.14
Prurigo nodularis
Prurigo nodularis, distinguished by firm, crusty nodules, is associated with viral infections (eg, hepatitis C, human immunodeficiency virus), bacterial infections, and kidney dysfunction.17,18 The lesions are intensely pruritic and often lead to persistent scratching, excoriation, and, ultimately, diffuse scarring.19
Treatment. Although the exact cause of prurigo nodularis is not known and no cure exists, corticosteroid or antihistamine ointments control the symptoms in most patients with hepatitis.19 Low doses of thalidomide (Thalomid), a tumor necrosis factor antagonist, have also been used safely and effectively.18,19
SUPERFICIAL VASCULAR SIGNS
Spider angiomas
Spider angiomas can occur anywhere on the body, but they occur most often on the face and the trunk.21,23 A key feature is that they disappear when pressure is applied and reappear when pressure is removed.23,24 Biopsy is rarely necessary for diagnosis.
These lesions occur with elevated estrogen levels, such as in cirrhosis, during estrogen therapy, or during pregnancy.25–28 Although spider angiomas are common in pregnant women and in children, adults with spider angiomas deserve a workup for liver dysfunction.29
Given their innocuous nature and asymptomatic course, spider angiomas themselves require no medical treatment.
Bier spots
Bier spots are small, irregularly shaped, hypopigmented patches on the arms and legs. They are likely due to venous stasis associated with functional damage to the small vessels of the skin.30
Since Bier spots are a sign of liver disease, they must be distinguished from true pigmentation disorders. A key distinguishing feature is that Bier spots disappear when pressure is applied. Also, raising the affected limb from a dependent position causes the hypopigmented macules of Bier spots to disappear, which is not the case in true pigmentation disorders.10,30
Paper-money skin
Paper-money skin (or “dollar-paper” markings) describes the condition in which the upper trunk is covered with many randomly scattered, needle-thin superficial capillaries. It often occurs in association with spider angiomas. The name comes from the resemblance the thread-like capillaries have to the finely chopped silk threads in American dollar bills.10,31 The condition is commonly seen in patients with alcoholic cirrhosis and may improve with hemodialysis.31
PALMAR ERYTHEMA
Palmar erythema is a florid, crimson coloration of the palms of the hands and the fingertips. It can occur anywhere on the palm and fingers but is most common on the hypothenar eminence. It can occur in a number of liver conditions but most often with cirrhosis.32 Hepatic compromise, as seen in alcoholic liver disease, disrupts the body’s androgen balance, causing local vasodilation and erythema.32,33 Although the exact mechanism remains unknown, research suggests that prostacyclins and nitric oxide play a role, as both are increased in liver disease.32,33
XANTHELASMA
Xanthelasma—a localized cholesterol deposit beneath the skin and especially beneath the eyelids—is a common manifestation of hypercholesterolemia. Xanthelasma often presents as a painless, yellowish, soft plaque with well-defined borders,34 which may enlarge over the course of weeks.
Several liver diseases can lead to various forms of secondary dyslipoproteinemia.35 The most common dyslipoproteinemias in liver disease are hypertriglyceridemia and low levels of high-density lipoprotein cholesterol, and both of these often accompany fatty liver disease.36 Hypercholesterolemia is a common feature of primary biliary cirrhosis and other forms of cholestatic liver disease.37 Studies suggest that the total plasma cholesterol level is elevated in as many as 50% of patients with compromised liver function.38
Treatment. The underlying hyperlipidemia is treated with cholesterol-lowering drugs. Laser treatment and surgical excision have proven efficacious in treating the lesions.39
OTHER CUTANEOUS FINDINGS IN LIVER DISEASE
Bleeding and bruising. Liver disease can cause hypersplenism and thrombocytopenia, in addition to a decrease in clotting factors. These may present with a myriad of cutaneous symptoms, including purpura, bleeding gums, and easy bruising and bleeding, even from minor trauma.40–42
Hyperpigmentation of the skin may accompany hemochromatosis, alcoholic liver disease, and cirrhosis.43–45
“Terry’s nails,” in which the proximal two-thirds of the nail plate turns powdery white with a ground-glass opacity, may develop in patients with advanced cirrhosis.48
ALCOHOLIC CIRRHOSIS AND THE SKIN
The cutaneous changes associated with alcoholic cirrhosis are more widely recognized than those due to other forms of liver dysfunction. In the United States, approximately 3 million people have alcoholic cirrhosis, the second-leading reason for liver transplantation.49,50
As the body’s main site of alcohol metabolism, the liver is the organ most affected by excessive alcohol intake, which can lead to end-stage liver disease secondary to alcoholic cirrhosis.41,51 The characteristic feature of cirrhosis is advanced fibrous scarring of parenchymal tissue and the formation of regenerative nodules with increased resistance to blood flow throughout the organ.41,52 The insufficient blood flow damages vital structures in the liver and compromises liver function. For example, liver cirrhosis leads to defective hepatic synthesis of clotting factors and results in bleeding disorders.
Cutaneous lesions often accompany alcoholic cirrhosis and have been detected in up to 43% of people with chronic alcoholism.53 Skin changes in alcoholic cirrhosis can be of great diagnostic value. The combined prevalence of spider angiomas, palmar erythema, and Dupuytren contracture in alcoholic cirrhosis was found to be 72%. Paper-money skin and Dupuytren contracture are more distinct lesions for alcoholic cirrhosis.31 Recognizing these skin changes contributes to the diagnosis and staging of liver cirrhosis.51,52
Dupuytren contracture
Dupuytren contracture is characterized by progressive fibrosis and thickening of tendons in the palmar fascia, the connective tissue that lies beneath the skin of the palms.54 Over time, as fibrotic involvement expands across the fascia, rampant stiffness of the joints ensues, sometimes to a point where the fingers cannot fully flex or extend.54
Although the exact cause of Dupuytren contracture is unknown, it appears to be associated with excess alcohol consumption and can be found in patients with alcoholic cirrhosis.54,55 These patients often present with painless stiffness of the fingers, curling of fingers, and loss of motion in involved fingers.54 Surgery in the form of limited fasciectomy has been curative in such patients.54
Disseminated superficial porokeratosis
Porokeratosis is a keratinization disorder of clonal origin that presents as a linear configuration of white scaly papules that coalesce into plaques throughout the body.56 Although it most commonly afflicts fair-skinned people, patients with alcoholic cirrhosis have a much greater susceptibility than the general population.57,58
A recent study58 documented that the lesions completely resolved when liver function improved, thus underlining the relationship between the two conditions. Since immunosuppression has been linked to eruption of the lesion, the fact that both humoral and cell-mediated immune responses are impaired in alcoholic liver disease provides another dimension to the association between porokeratosis and alcoholic cirrhosis.58
These lesions can transform into squamous cell carcinoma.59 The risk of widespread metastases in squamous cell carcinoma highlights the importance of dermatologic consultation in such patients.59
HEPATITIS C AND THE SKIN
Extrahepatic manifestations have been documented in up to 74% of people with hepatitis C virus infection.60 In addition to parasthesias, arthralgias, and myalgias, hepatitis C has a significant association with porphyria cutanea tarda, lichen planus, vitiligo, sialadenitis, urticarial vasculitis, corneal ulcers, xerosis, pruritus, and prurigo nodularis.60–64 Although the primary causative agents of sialadenitis are bacteria, viruses such as hepatitis C have been implicated as a cause of chronic sialadenitis with associated xerostomia.65
Patients with hepatitis C being treated with interferons also present with cutaneous manifestations such as hyperkeratosis and vasculitis.63
Porphyria cutanea tarda
Porphyria cutanea tarda is the most common of the porphyrias, disorders distinguished by deficiencies or defects in one or more of the enzymes responsible for hepatic production of heme.66 If these enzymes are impaired, heme precursors such as porphyrins accumulate.66
Porphyria cutanea tarda results from a deficiency of the hepatic enzyme uroporphyrin decarboxylase. In the absence of this enzyme, shortwave visible light activates uroporphyrin deposited in the skin, resulting in a photochemical reaction that generates reactive oxygen species that lead to the characteristic skin blistering.
Although porphyria cutanea tarda is associated with liver disease in general, recent studies confirm that patients with hepatitis C are at particularly high risk.67 Those with the disorder often present with skin photosensitivity. 68 Many develop blisters on sun-exposed skin, including the dorsal aspects of the hands and forearms and on the neck and face. Chronic porphyria cutanea tarda can lead to scarring, alopecia, and skin ulceration.69 As the blisters heal, keratin-filled milial cysts may develop in the areas of ulceration.
The condition is also commonly associated with melasma-like hyperpigmentation and hypertrichosis in sun-exposed areas of the head and neck. People of Northern European ancestry may be more at risk than the general population because of a presumed genetic susceptibility.70
Treatment. Because many patients with porphyria cutanea tarda have iron overload, they need to restrict foods rich in iron and to avoid alcohol.71,72 Severe cases may necessitate iron removal via phlebotomy or antimalarial therapy. Patients with porphyria cutanea tarda induced by hepatitis C should have their bodily iron stores depleted before starting antiviral therapy.60
Lichen planus
Lichen planopilaris is a subset of lichen planus that causes scaling and atrophy of the scalp and permanent hair loss (Figure 4).73
Interferon-induced vitiligo
Vitiligo is an autoimmune disease in which melanocytes in the skin are destroyed, with resulting depigmentation in affected areas.75 Although it has no specific association with liver disease, it has been linked to treatments for hepatitis C such as interferons.76 Interferon-induced vitiligo often completely resolves when interferon is stopped.77
Typical findings include aggregations of irregularly shaped white patches in a focal or segmental pattern.78 The diagnosis is based on the medical history, physical examination, and sometimes skin biopsy.
HEMOCHROMATOSIS
Hemochromatosis or “bronze diabetes” is a devastating multisystem disease with a relentless course. It is among the most common genetic disorders of metabolism, and results in deposition of iron in tissues and organs throughout the body, including the liver, usually in patients ages 30 to 40.
As iron stores increase in tissues and organs, multiorgan failure and associated complications may ensue. In addition, surplus iron stores can also result in widespread bronze discoloration of skin exposed to the sun. Hemochromatosis also results in loss of body hair, ichthyosiform alterations, and koilonychia.79
Treatments that lower serum iron levels can reverse the cutaneous manifestations of the disorder and minimize the risk of organ failure.
Since the condition is inherited in an autosomal-recessive pattern, family members of patients should consider being screened.80
Hyperpigmentation in hemochromatosis
Hyperpigmentation is an early sign of hemochromatosis, affecting up to 90% of patients. Usually, sun-exposed areas of the body are the most prone and take on a grayish or brownish-bronze hue.81 Cutaneous iron deposits injure vital skin structures, initiating a process that culminates in enhanced melanin production by melanocytes.82 Exposure to ultraviolet light may have synergistic effects with iron, hastening the process of hyperpigmentation. As a result of this synergistic effect, many patients with hemochromatosis notice tanning with minimal sun exposure.
Although organ function can improve immediately with phlebotomy to reduce iron stores, skin hyperpigmentation does not immediately resolve.81,82
Ichthyosiform alterations in hemochromatosis
Ichthyosiform changes, in which the skin takes on the appearance of fish scales,83 can be seen in patients with hemochromatosis.80 Affected areas typically become extremely dry. Treatment includes topical hydrating creams and ointments. Avoiding sunlight is paramount, as sunlight exposure may exacerbate the condition.83
- Morioka D, Togo S, Kumamoto T, et al. Six consecutive cases of successful adult ABO-incompatible living donor liver transplantation: a proposal for grading the severity of antibody-mediated rejection. Transplantation 2008; 85:171–178.
- Bertini G, Rubaltelli FF. Non-invasive bilirubinometry in neonatal jaundice. Semin Neonatol 2002; 7:129–133.
- Clementi M, Di Gianantonio E, Fabris L, et al. Inheritance of hyperbilirubinemia: evidence for a major autosomal recessive gene. Dig Liver Dis 2007; 39:351–355.
- Roche SP, Kobos R. Jaundice in the adult patient. Am Fam Physician 2004; 69:299–304.
- Odemis B, Parlak E, Basar O, Yüksel O, Sahin B. Biliary tract obstruction secondary to malignant lymphoma: experience at a referral center. Dig Dis Sci 2007; 52:2323–2332.
- Caddy GR, Tham TC. Gallstone disease: symptoms, diagnosis and endoscopic management of common bile duct stones. Best Pract Res Clin Gastroenterol 2006; 20:1085–1101.
- Heathcote EJ. Diagnosis and management of cholestatic liver disease. Clin Gastroenterol Hepatol 2007; 5:776–782.
- Faust TW, Reddy KR. Postoperative jaundice. Clin Liver Dis 2004; 8:151–166.
- Maticic M, Poljak M, Lunder T, Rener-Sitar K, Stojanovic L. Lichen planus and other cutaneous manifestations in chronic hepatitis C: pre- and post-interferon-based treatment prevalence vary in a cohort of patients from low hepatitis C virus endemic area. J Eur Acad Dermatol Venereol 2008; 22:779–788.
- Polat M, Oztas P, Ilhan MN, Yalçin B, Alli N. Generalized pruritus: a prospective study concerning etiology. Am J Clin Dermatol 2008; 9:39–44.
- Gaspari R, Avolio AW, Zileri Dal Verme L, et al. Molecular adsorbent recirculating system in liver transplantation: safety and efficacy. Transplant Proc 2006; 38:3544–3551.
- Dasgupta R, Saha I, Pal S, et al. Immunosuppression, hepatotoxicity and depression of antioxidant status by arecoline in albino mice. Toxicology 2006; 227:94–104.
- Mayo MJ, Handem I, Saldana S, Jacobe H, Getachew Y, Rush AJ. Sertraline as a first-line treatment for cholestatic pruritus. Hepatology 2007; 45:666–674.
- Mela M, Mancuso A, Burroughs AK. Review article: pruritus in cholestatic and other liver diseases. Aliment Pharmacol Ther 2003; 17:857–870.
- Montero JL, Pozo JC, Barrera P, et al. Treatment of refractory cholestatic pruritus with molecular adsorbent recirculating system (MARS). Transplant Proc 2006; 38:2511–2513.
- Neff GW, O'Brien CB, Reddy KR, et al. Preliminary observation with dronabinol in patients with intractable pruritus secondary to cholestatic liver disease. Am J Gastroenterol 2002; 97:2117–2119.
- Neri S, Raciti C, D’Angelo G, Ierna D, Bruno CM. Hyde’s prurigo nodularis and chronic HCV hepatitis. J Hepatol 1998; 28:161–164.
- Brown MA, George CR, Dunstan CR, Kalowski S, Corrigan AB. Prurigo nodularis and aluminium overload in maintenance haemodialysis. Lancet 1992; 340:48.
- Stander S, Luger T, Metze D. Treatment of prurigo nodularis with topical capsaicin. J Am Acad Dermatol 2001; 44:471–478.
- Requena L, Sangueza OP. Cutaneous vascular anomalies. Part I. Hamartomas, malformations, and dilation of preexisting vessels. J Am Acad Dermatol 1997; 37:523–549.
- Khasnis A, Gokula RM. Spider nevus. J Postgrad Med 2002; 48:307–309.
- Kaul V, Friedenberg FK, Braitman LE, et al. Development and validation of a model to diagnose cirrhosis in patients with hepatitis C. Am J Gastroenterol 2002; 97:2623–2628.
- Banyai AL. There is more than surface appearance to skin spiders. Chest 1971; 60:48.
- Errickson CV, Matus NR. Skin disorders of pregnancy. Am Fam Physician 1994; 49:605–610.
- Li CP, Lee FY, Hwang SJ, et al. Spider angiomas in patients with liver cirrhosis: role of alcoholism and impaired liver function. Scand J Gastroenterol 1999; 34:520–523.
- Li CP, Lee FY, Hwang SJ, et al. Role of substance P in the pathogenesis of spider angiomas in patients with nonalcoholic liver cirrhosis. Am J Gastroenterol 1999; 94:502–507.
- Sadick NS, Niedt GW. A study of estrogen and progesterone receptors in spider telangiectasias of the lower extremities. J Dermatol Surg Oncol 1990; 16:620–623.
- Henry F, Quatresooz P, Valverde-Lopez JC, Pierard GE. Blood vessel changes during pregnancy: a review. Am J Clin Dermatol 2006; 7:65–69.
- Finn SM, Rowland M, Lawlor F, et al. The significance of cutaneous spider naevi in children. Arch Dis Child 2006; 91:604–605.
- Peyrot I, Boulinguez S, Sparsa A, Le Meur Y, Bonnetblanc JM, Bedane C. Bier’s white spots associated with scleroderma renal crisis. Clin Exp Dermatol 2007; 32:165–167.
- Satoh T, Yokozeki H, Nishioka K. Vascular spiders and paper money skin improved by hemodialysis. Dermatology 2002; 205:73–74.
- Serrao R, Zirwas M, English JC. Palmar erythema. Am J Clin Dermatol 2007; 8:347–356.
- Matsumoto M, Ohki K, Nagai I, Oshibuchi T. Lung traction causes an increase in plasma prostacyclin concentration and decrease in mean arterial blood pressure. Anesth Analg 1992; 75:773–776.
- Otto AI, Horvath I, Feldmann J. Multiple firm, painless erythematous papules with a yellowish hue. Arch Dermatol 2005; 141:1595–1600.
- Gandelman G, Aronow WS, Weiss MB. Resolving hyperlipidemia after liver transplantation in a patient with primary sclerosing cholangitis. Am J Ther 2006; 13:171–174.
- Assy N, Kaita K, Mymin D, Levy C, Rosser B, Minuk G. Fatty infiltration of liver in hyperlipidemic patients. Dig Dis Sci 2000; 45:1929–1934.
- Allocca M, Crosignani A, Gritti A, et al. Hypercholesterolaemia is not associated with early atherosclerotic lesions in primary biliary cirrhosis. Gut 2006; 55:1795–1800.
- Dickson E, Fleming C, Ludwig J. Primary biliary cirrhosis. In:Popper H, Schaffner F, editors. Progress in Liver Diseases. New York: Grune and Stratton; 1978:487.
- Elner VM, Mintz R, Demirci H, Hassan AS. Local corticosteroid treatment of eyelid and orbital xanthogranuloma. Trans Am Ophthalmol Soc 2005; 103:69–73.
- Craxi A, Camma C, Giunta M. Clinical aspects of bleeding complications in cirrhotic patients. Blood Coagul Fibrinolysis 2000; 11( suppl 1):S75–579.
- Kajihara M, Okazaki Y, Kato S, et al. Evaluation of platelet kinetics in patients with liver cirrhosis: similarity to idiopathic thrombocytopenic purpura. J Gastroenterol Hepatol 2007; 22:112–118.
- Levine N. Patient reports six-month history of minimally pruritic purple dots on legs. Non-blanching macules developed over six months. Geriatrics 2006; 61:22.
- Barton JC, Rao SV, Pereira NM, et al. Juvenile hemochromatosis in the southeastern United States: a report of seven cases in two kinships. Blood Cells Mol Dis 2002; 29:104–115.
- Smith AG, Shuster S, Bomford A, Williams R. Plasma immunoreactive beta-melanocyte-stimulating hormone in chronic liver disease and fulminant hepatic failure. J Invest Dermatol 1978; 70:326–327.
- Barton JC, McDonnell SM, Adams PC, et al. Management of hemochromatosis. Hemochromatosis Management Working Group. Ann Intern Med 1998; 129:932–939.
- Bahnsen M, Gluud C, Johnsen SG, et al. Pituitary-testicular function in patients with alcoholic cirrhosis of the liver. Eur J Clin Invest 1981; 11:473–479.
- Kumar N, Aggarwal SR, Anand BS. Comparison of truncal hair distribution in alcoholic liver disease and alcohol-related chronic pancreatitis. J Gastroenterol Hepatol 2001; 16:855–856.
- Holzberg M, Walker HK. Terry's nails: revised definition and new correlations. Lancet 1984; 1:896–899.
- Mandayam S, Jamal MM, Morgan TR. Epidemiology of alcoholic liver disease. Semin Liver Dis 2004; 24:217–232.
- Belle SH, Beringer KC, Detre KM. Liver transplantation in the United States: results from the National Pitt-UNOS Liver Transplant Registry. United Network for Organ Sharing Clin Transpl 1994:19–35.
- Dunn W, Xu R, Schwimmer JB. Modest wine drinking and decreased prevalence of suspected nonalcoholic fatty liver disease. Hepatology 2008; 47:1947–1954.
- Afford SC, Fisher NC, Neil DA, et al. Distinct patterns of chemokine expression are associated with leukocyte recruitment in alcoholic hepatitis and alcoholic cirrhosis. J Pathol 1998; 186:82–89.
- Evstaf’ev VV, Levin MM. Dermatologic pathology in chronic alcoholics. Vestn Dermatol Venerol 1989; 8:72–74.
- Jerosch-Herold C, Shepstone L, Chojnowski AJ, Larson D. Splinting after contracture release for Dupuytren's contracture (SCoRD): protocol of a pragmatic, multi-centre, randomized controlled trial. BMC Musculoskelet Disord 2008; 9:62.
- Houghton S, Holdstock G, Cockerell R, Wright R. Dupuytren’s contracture, chronic liver disease and IgA immune complexes. Liver 1983; 3:220–224.
- Ibbotson SH. Disseminated superficial porokeratosis: what is the association with ultraviolet radiation? Clin Exp Dermatol 1996; 21:48–50.
- Kono T, Kobayashi H, Ishii M, Nishiguchi S, Taniguchi S. Synchronous development of disseminated superficial porokeratosis and hepatitis C virus-related hepatocellular carcinoma. J Am Acad Dermatol 2000; 43:966–968.
- Park BS, Moon SE, Kim JA. Disseminated superficial porokeratosis in a patient with chronic liver disease. J Dermatol 1997; 24:485–487.
- Murata Y, Kumano K, Takai T. Type 2 segmental manifestation of disseminated superficial porokeratosis showing a systematized pattern of involvement and pronounced cancer proneness. Eur J Dermatol 2001; 11:191–194.
- Galossi A, Guarisco R, Bellis L, Puoti C. Extrahepatic manifestations of chronic HCV infection. J Gastrointestin Liver Dis 2007; 16:65–73.
- El-Serag HB, Hampel H, Yeh C, Rabeneck L. Extrahepatic manifestations of hepatitis C among United States male veterans. Hepatology 2002; 36:1439–1445.
- Stefanova-Petrova DV, Tzvetanska AH, Naumova EJ, et al. Chronic hepatitis C virus infection: prevalence of extrahepatic manifestations and association with cryoglobulinemia in Bulgarian patients. World J Gastroenterol 2007; 13:6518–6528.
- Vassilopoulos D, Calabrese LH. Extrahepatic immunological complications of hepatitis C virus infection. AIDS 2005; 19( suppl 3):S123–S127.
- Hsing AW, Zhang M, Rashid A, et al. Hepatitis B and C virus infection and the risk of biliary tract cancer: a population-based study in China. Int J Cancer 2008; 122:1849–1853.
- Madrid C, Courtois B, Duran D. Chronic sialadenitis revealing hepatitis C: a case report. Med Oral 2004; 9:328–332.
- Lançoni G, Ravinal RC, Costa RS, Roselino AM. Mast cells and transforming growth factor-beta expression: a possible relationship in the development of porphyria cutanea tarda skin lesions. Int J Dermatol 2008; 47:575–581.
- Toll A, Celis R, Ozalla MD, Bruguera M, Herrero C, Ercilla MG. The prevalence of HFE C282Y gene mutation is increased in Spanish patients with porphyria cutanea tarda without hepatitis C virus infection. J Eur Acad Dermatol Venereol 2006; 20:1201–1206.
- Badminton MN, Elder GH. Management of acute and cutaneous porphyrias. Int J Clin Pract 2002; 56:272–278.
- Jackson JM, Callen JP. Scarring alopecia and sclerodermatous changes of the scalp in a patient with hepatitis C infection. J Am Acad Dermatol 1998; 39:824–826.
- Mortimore M, Merryweather-Clarke AT, Robson KJ, Powell LW. The haemochromatosis gene: a global perspective and implications for the Asia-Pacific region. J Gastroenterol Hepatol 1999; 14:838–843.
- Shehan JM, Huerter CJ. Porphyria cutanea tarda associated with an acute gastrointestinal bleed: the roles of supplemental iron and blood transfusion. Cutis 2001; 68:147–150.
- Lambrecht RW, Thapar M, Bonkovsky HL. Genetic aspects of porphyria cutanea tarda. Semin Liver Dis 2007; 27:99–108.
- d’Ovidio R, Sgarra C, Conserva A, Angelotti UF, Erriquez R, Foti C. Alterated integrin expression in lichen planopilaris. Head Face Med 2007; 3:11.
- Chuang TY, Stitle L, Brashear R, Lewis C. Hepatitis C virus and lichen planus: a case-control study of 340 patients. J Am Acad Dermatol 1999; 41:787–789.
- Kemp EH, Gavalas NG, Gawkrodger DJ, Weetman AP. Autoantibody responses to melanocytes in the depigmenting skin disease vitiligo. Autoimmun Rev 2007; 6:138–142.
- Tomasiewicz K, Modrzewska R, Semczuk G. Vitiligo associated with pegylated interferon and ribavirin treatment of patients with chronic hepatitis C: a case report. Adv Ther 2006; 23:139–142.
- Simsek H, Savas C, Akkiz H, Telatar H. Interferon-induced vitiligo in a patient with chronic viral hepatitis C infection. Dermatology 1996; 193:65–66.
- Mulekar SV, Al Issa A, Asaad M, Ghwish B, Al Eisa A. Mixed vitiligo. J Cutan Med Surg. 2006; 10:104–107.
- Waalen J, Felitti V, Gelbart T, Ho NJ, Beutler E. Prevalence of hemochromatosis-related symptoms among individuals with mutations in the HFE gene. Mayo Clin Proc 2002; 77:522–530.
- Walker AP, Tucker DC, Hall MA, et al. Results communication and patient education after screening for possible hemochromatosis and iron overload: experience from the HEIRS study of a large ethnically and linguistically diverse group. Genet Med 2007; 9:778–791.
- Stulberg DL, Clark N, Tovey D. Common hyperpigmentation disorders in adults: Part I. Diagnostic approach, cafe au lait macules, diffuse hyperpigmentation, sun exposure, and phototoxic reactions. Am Fam Physician 2003; 68:1955–1960.
- Tsuji T. Experimental hemosiderosis: relationship between skin pigmentation and hemosiderin. Acta Derm Venereol 1980; 60:109–114.
- Oji V, Traupe H. Ichthyoses: differential diagnosis and molecular genetics. Eur J Dermatol 2006; 16:349–359.
- Morioka D, Togo S, Kumamoto T, et al. Six consecutive cases of successful adult ABO-incompatible living donor liver transplantation: a proposal for grading the severity of antibody-mediated rejection. Transplantation 2008; 85:171–178.
- Bertini G, Rubaltelli FF. Non-invasive bilirubinometry in neonatal jaundice. Semin Neonatol 2002; 7:129–133.
- Clementi M, Di Gianantonio E, Fabris L, et al. Inheritance of hyperbilirubinemia: evidence for a major autosomal recessive gene. Dig Liver Dis 2007; 39:351–355.
- Roche SP, Kobos R. Jaundice in the adult patient. Am Fam Physician 2004; 69:299–304.
- Odemis B, Parlak E, Basar O, Yüksel O, Sahin B. Biliary tract obstruction secondary to malignant lymphoma: experience at a referral center. Dig Dis Sci 2007; 52:2323–2332.
- Caddy GR, Tham TC. Gallstone disease: symptoms, diagnosis and endoscopic management of common bile duct stones. Best Pract Res Clin Gastroenterol 2006; 20:1085–1101.
- Heathcote EJ. Diagnosis and management of cholestatic liver disease. Clin Gastroenterol Hepatol 2007; 5:776–782.
- Faust TW, Reddy KR. Postoperative jaundice. Clin Liver Dis 2004; 8:151–166.
- Maticic M, Poljak M, Lunder T, Rener-Sitar K, Stojanovic L. Lichen planus and other cutaneous manifestations in chronic hepatitis C: pre- and post-interferon-based treatment prevalence vary in a cohort of patients from low hepatitis C virus endemic area. J Eur Acad Dermatol Venereol 2008; 22:779–788.
- Polat M, Oztas P, Ilhan MN, Yalçin B, Alli N. Generalized pruritus: a prospective study concerning etiology. Am J Clin Dermatol 2008; 9:39–44.
- Gaspari R, Avolio AW, Zileri Dal Verme L, et al. Molecular adsorbent recirculating system in liver transplantation: safety and efficacy. Transplant Proc 2006; 38:3544–3551.
- Dasgupta R, Saha I, Pal S, et al. Immunosuppression, hepatotoxicity and depression of antioxidant status by arecoline in albino mice. Toxicology 2006; 227:94–104.
- Mayo MJ, Handem I, Saldana S, Jacobe H, Getachew Y, Rush AJ. Sertraline as a first-line treatment for cholestatic pruritus. Hepatology 2007; 45:666–674.
- Mela M, Mancuso A, Burroughs AK. Review article: pruritus in cholestatic and other liver diseases. Aliment Pharmacol Ther 2003; 17:857–870.
- Montero JL, Pozo JC, Barrera P, et al. Treatment of refractory cholestatic pruritus with molecular adsorbent recirculating system (MARS). Transplant Proc 2006; 38:2511–2513.
- Neff GW, O'Brien CB, Reddy KR, et al. Preliminary observation with dronabinol in patients with intractable pruritus secondary to cholestatic liver disease. Am J Gastroenterol 2002; 97:2117–2119.
- Neri S, Raciti C, D’Angelo G, Ierna D, Bruno CM. Hyde’s prurigo nodularis and chronic HCV hepatitis. J Hepatol 1998; 28:161–164.
- Brown MA, George CR, Dunstan CR, Kalowski S, Corrigan AB. Prurigo nodularis and aluminium overload in maintenance haemodialysis. Lancet 1992; 340:48.
- Stander S, Luger T, Metze D. Treatment of prurigo nodularis with topical capsaicin. J Am Acad Dermatol 2001; 44:471–478.
- Requena L, Sangueza OP. Cutaneous vascular anomalies. Part I. Hamartomas, malformations, and dilation of preexisting vessels. J Am Acad Dermatol 1997; 37:523–549.
- Khasnis A, Gokula RM. Spider nevus. J Postgrad Med 2002; 48:307–309.
- Kaul V, Friedenberg FK, Braitman LE, et al. Development and validation of a model to diagnose cirrhosis in patients with hepatitis C. Am J Gastroenterol 2002; 97:2623–2628.
- Banyai AL. There is more than surface appearance to skin spiders. Chest 1971; 60:48.
- Errickson CV, Matus NR. Skin disorders of pregnancy. Am Fam Physician 1994; 49:605–610.
- Li CP, Lee FY, Hwang SJ, et al. Spider angiomas in patients with liver cirrhosis: role of alcoholism and impaired liver function. Scand J Gastroenterol 1999; 34:520–523.
- Li CP, Lee FY, Hwang SJ, et al. Role of substance P in the pathogenesis of spider angiomas in patients with nonalcoholic liver cirrhosis. Am J Gastroenterol 1999; 94:502–507.
- Sadick NS, Niedt GW. A study of estrogen and progesterone receptors in spider telangiectasias of the lower extremities. J Dermatol Surg Oncol 1990; 16:620–623.
- Henry F, Quatresooz P, Valverde-Lopez JC, Pierard GE. Blood vessel changes during pregnancy: a review. Am J Clin Dermatol 2006; 7:65–69.
- Finn SM, Rowland M, Lawlor F, et al. The significance of cutaneous spider naevi in children. Arch Dis Child 2006; 91:604–605.
- Peyrot I, Boulinguez S, Sparsa A, Le Meur Y, Bonnetblanc JM, Bedane C. Bier’s white spots associated with scleroderma renal crisis. Clin Exp Dermatol 2007; 32:165–167.
- Satoh T, Yokozeki H, Nishioka K. Vascular spiders and paper money skin improved by hemodialysis. Dermatology 2002; 205:73–74.
- Serrao R, Zirwas M, English JC. Palmar erythema. Am J Clin Dermatol 2007; 8:347–356.
- Matsumoto M, Ohki K, Nagai I, Oshibuchi T. Lung traction causes an increase in plasma prostacyclin concentration and decrease in mean arterial blood pressure. Anesth Analg 1992; 75:773–776.
- Otto AI, Horvath I, Feldmann J. Multiple firm, painless erythematous papules with a yellowish hue. Arch Dermatol 2005; 141:1595–1600.
- Gandelman G, Aronow WS, Weiss MB. Resolving hyperlipidemia after liver transplantation in a patient with primary sclerosing cholangitis. Am J Ther 2006; 13:171–174.
- Assy N, Kaita K, Mymin D, Levy C, Rosser B, Minuk G. Fatty infiltration of liver in hyperlipidemic patients. Dig Dis Sci 2000; 45:1929–1934.
- Allocca M, Crosignani A, Gritti A, et al. Hypercholesterolaemia is not associated with early atherosclerotic lesions in primary biliary cirrhosis. Gut 2006; 55:1795–1800.
- Dickson E, Fleming C, Ludwig J. Primary biliary cirrhosis. In:Popper H, Schaffner F, editors. Progress in Liver Diseases. New York: Grune and Stratton; 1978:487.
- Elner VM, Mintz R, Demirci H, Hassan AS. Local corticosteroid treatment of eyelid and orbital xanthogranuloma. Trans Am Ophthalmol Soc 2005; 103:69–73.
- Craxi A, Camma C, Giunta M. Clinical aspects of bleeding complications in cirrhotic patients. Blood Coagul Fibrinolysis 2000; 11( suppl 1):S75–579.
- Kajihara M, Okazaki Y, Kato S, et al. Evaluation of platelet kinetics in patients with liver cirrhosis: similarity to idiopathic thrombocytopenic purpura. J Gastroenterol Hepatol 2007; 22:112–118.
- Levine N. Patient reports six-month history of minimally pruritic purple dots on legs. Non-blanching macules developed over six months. Geriatrics 2006; 61:22.
- Barton JC, Rao SV, Pereira NM, et al. Juvenile hemochromatosis in the southeastern United States: a report of seven cases in two kinships. Blood Cells Mol Dis 2002; 29:104–115.
- Smith AG, Shuster S, Bomford A, Williams R. Plasma immunoreactive beta-melanocyte-stimulating hormone in chronic liver disease and fulminant hepatic failure. J Invest Dermatol 1978; 70:326–327.
- Barton JC, McDonnell SM, Adams PC, et al. Management of hemochromatosis. Hemochromatosis Management Working Group. Ann Intern Med 1998; 129:932–939.
- Bahnsen M, Gluud C, Johnsen SG, et al. Pituitary-testicular function in patients with alcoholic cirrhosis of the liver. Eur J Clin Invest 1981; 11:473–479.
- Kumar N, Aggarwal SR, Anand BS. Comparison of truncal hair distribution in alcoholic liver disease and alcohol-related chronic pancreatitis. J Gastroenterol Hepatol 2001; 16:855–856.
- Holzberg M, Walker HK. Terry's nails: revised definition and new correlations. Lancet 1984; 1:896–899.
- Mandayam S, Jamal MM, Morgan TR. Epidemiology of alcoholic liver disease. Semin Liver Dis 2004; 24:217–232.
- Belle SH, Beringer KC, Detre KM. Liver transplantation in the United States: results from the National Pitt-UNOS Liver Transplant Registry. United Network for Organ Sharing Clin Transpl 1994:19–35.
- Dunn W, Xu R, Schwimmer JB. Modest wine drinking and decreased prevalence of suspected nonalcoholic fatty liver disease. Hepatology 2008; 47:1947–1954.
- Afford SC, Fisher NC, Neil DA, et al. Distinct patterns of chemokine expression are associated with leukocyte recruitment in alcoholic hepatitis and alcoholic cirrhosis. J Pathol 1998; 186:82–89.
- Evstaf’ev VV, Levin MM. Dermatologic pathology in chronic alcoholics. Vestn Dermatol Venerol 1989; 8:72–74.
- Jerosch-Herold C, Shepstone L, Chojnowski AJ, Larson D. Splinting after contracture release for Dupuytren's contracture (SCoRD): protocol of a pragmatic, multi-centre, randomized controlled trial. BMC Musculoskelet Disord 2008; 9:62.
- Houghton S, Holdstock G, Cockerell R, Wright R. Dupuytren’s contracture, chronic liver disease and IgA immune complexes. Liver 1983; 3:220–224.
- Ibbotson SH. Disseminated superficial porokeratosis: what is the association with ultraviolet radiation? Clin Exp Dermatol 1996; 21:48–50.
- Kono T, Kobayashi H, Ishii M, Nishiguchi S, Taniguchi S. Synchronous development of disseminated superficial porokeratosis and hepatitis C virus-related hepatocellular carcinoma. J Am Acad Dermatol 2000; 43:966–968.
- Park BS, Moon SE, Kim JA. Disseminated superficial porokeratosis in a patient with chronic liver disease. J Dermatol 1997; 24:485–487.
- Murata Y, Kumano K, Takai T. Type 2 segmental manifestation of disseminated superficial porokeratosis showing a systematized pattern of involvement and pronounced cancer proneness. Eur J Dermatol 2001; 11:191–194.
- Galossi A, Guarisco R, Bellis L, Puoti C. Extrahepatic manifestations of chronic HCV infection. J Gastrointestin Liver Dis 2007; 16:65–73.
- El-Serag HB, Hampel H, Yeh C, Rabeneck L. Extrahepatic manifestations of hepatitis C among United States male veterans. Hepatology 2002; 36:1439–1445.
- Stefanova-Petrova DV, Tzvetanska AH, Naumova EJ, et al. Chronic hepatitis C virus infection: prevalence of extrahepatic manifestations and association with cryoglobulinemia in Bulgarian patients. World J Gastroenterol 2007; 13:6518–6528.
- Vassilopoulos D, Calabrese LH. Extrahepatic immunological complications of hepatitis C virus infection. AIDS 2005; 19( suppl 3):S123–S127.
- Hsing AW, Zhang M, Rashid A, et al. Hepatitis B and C virus infection and the risk of biliary tract cancer: a population-based study in China. Int J Cancer 2008; 122:1849–1853.
- Madrid C, Courtois B, Duran D. Chronic sialadenitis revealing hepatitis C: a case report. Med Oral 2004; 9:328–332.
- Lançoni G, Ravinal RC, Costa RS, Roselino AM. Mast cells and transforming growth factor-beta expression: a possible relationship in the development of porphyria cutanea tarda skin lesions. Int J Dermatol 2008; 47:575–581.
- Toll A, Celis R, Ozalla MD, Bruguera M, Herrero C, Ercilla MG. The prevalence of HFE C282Y gene mutation is increased in Spanish patients with porphyria cutanea tarda without hepatitis C virus infection. J Eur Acad Dermatol Venereol 2006; 20:1201–1206.
- Badminton MN, Elder GH. Management of acute and cutaneous porphyrias. Int J Clin Pract 2002; 56:272–278.
- Jackson JM, Callen JP. Scarring alopecia and sclerodermatous changes of the scalp in a patient with hepatitis C infection. J Am Acad Dermatol 1998; 39:824–826.
- Mortimore M, Merryweather-Clarke AT, Robson KJ, Powell LW. The haemochromatosis gene: a global perspective and implications for the Asia-Pacific region. J Gastroenterol Hepatol 1999; 14:838–843.
- Shehan JM, Huerter CJ. Porphyria cutanea tarda associated with an acute gastrointestinal bleed: the roles of supplemental iron and blood transfusion. Cutis 2001; 68:147–150.
- Lambrecht RW, Thapar M, Bonkovsky HL. Genetic aspects of porphyria cutanea tarda. Semin Liver Dis 2007; 27:99–108.
- d’Ovidio R, Sgarra C, Conserva A, Angelotti UF, Erriquez R, Foti C. Alterated integrin expression in lichen planopilaris. Head Face Med 2007; 3:11.
- Chuang TY, Stitle L, Brashear R, Lewis C. Hepatitis C virus and lichen planus: a case-control study of 340 patients. J Am Acad Dermatol 1999; 41:787–789.
- Kemp EH, Gavalas NG, Gawkrodger DJ, Weetman AP. Autoantibody responses to melanocytes in the depigmenting skin disease vitiligo. Autoimmun Rev 2007; 6:138–142.
- Tomasiewicz K, Modrzewska R, Semczuk G. Vitiligo associated with pegylated interferon and ribavirin treatment of patients with chronic hepatitis C: a case report. Adv Ther 2006; 23:139–142.
- Simsek H, Savas C, Akkiz H, Telatar H. Interferon-induced vitiligo in a patient with chronic viral hepatitis C infection. Dermatology 1996; 193:65–66.
- Mulekar SV, Al Issa A, Asaad M, Ghwish B, Al Eisa A. Mixed vitiligo. J Cutan Med Surg. 2006; 10:104–107.
- Waalen J, Felitti V, Gelbart T, Ho NJ, Beutler E. Prevalence of hemochromatosis-related symptoms among individuals with mutations in the HFE gene. Mayo Clin Proc 2002; 77:522–530.
- Walker AP, Tucker DC, Hall MA, et al. Results communication and patient education after screening for possible hemochromatosis and iron overload: experience from the HEIRS study of a large ethnically and linguistically diverse group. Genet Med 2007; 9:778–791.
- Stulberg DL, Clark N, Tovey D. Common hyperpigmentation disorders in adults: Part I. Diagnostic approach, cafe au lait macules, diffuse hyperpigmentation, sun exposure, and phototoxic reactions. Am Fam Physician 2003; 68:1955–1960.
- Tsuji T. Experimental hemosiderosis: relationship between skin pigmentation and hemosiderin. Acta Derm Venereol 1980; 60:109–114.
- Oji V, Traupe H. Ichthyoses: differential diagnosis and molecular genetics. Eur J Dermatol 2006; 16:349–359.
KEY POINTS
- Pruritus due to liver disease is particularly resistant to therapy. Cholestyramine (Questran) 4 g/day, gradually increased to 24 g/day, is one option. If the pruritus does not respond or the patient cannot tolerate cholestyramine, rifampin (Rifadin) can be tried.
- Spider angiomas, Bier spots, and “paper-money” skin are all superficial vascular problems that may be related to liver disease.
- Cutaneous lesions often accompany alcoholic cirrhosis and have been detected in up to 43% of people with chronic alcoholism. The combination of spider angiomas, palmar erythema, and Dupuytren contracture is common in alcoholic cirrhosis.
- Although porphyria cutanea tarda is associated with liver disease in general, recent studies show that patients with hepatitis C are at particularly high risk.
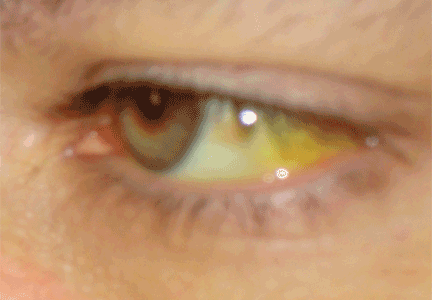
A 35-year-old Asian man with jaundice and markedly high aminotransferase levels
A 35-year-old man who was born in Vietnam presents to the emergency department of a local hospital because he has had jaundice for 5 days and fatigue, malaise, and anorexia for 2 weeks. He also has nausea and mild epigastric and right upper quadrant abdominal pain. He denies having fevers, chills, night sweats, vomiting, diarrhea, melena, hematochezia, or weight loss.
His medical history is remarkable only for perinatally acquired hepatitis B virus (HBV) infection, for which he never received antiviral therapy. He does not take any prescribed, over-the-counter, or herbal medications.
He lives in the Midwest region of the United States and works full-time as a physician in private practice. He is married and has two children.
He has not travelled recently. He has no pets at home and has not been exposed to any.
He has never smoked. He drinks alcohol socially but has never used recreational drugs.
In a laboratory evaluation performed a year ago for insurance purposes, his liver function tests—serum albumin, total bilirubin, alkaline phosphatase, alanine aminotransferase, and aspartate aminotransferase levels—were all normal. He was positive for HBV surface antigen and HBV e antigen and negative for antibodies against these antigens.
PHASES OF HBV INFECTION
1. Which of the following best describes the status of HBV infection in this patient before his current symptoms developed?
- Resolved HBV infection
- Chronic inactive HBV infection
- Chronic active HBV infection
- Immune-tolerant chronic HBV infection
The correct answer is immune-tolerant chronic HBV infection.
Resolved infection. In immunocompetent adults, most primary HBV infections are self-limited: people clear the virus and gain lasting immunity (defined as the loss of HBV surface antigen, the development of antibody against surface antigen, no detectable HBV DNA in the serum, and normal alanine and aspartate aminotransferase levels). However, a minority of primary HBV infections persist and become chronic.
Chronic HBV infection is defined as the persistence of HBV surface antigen in the serum for at least 6 months. Patients with chronic HBV infection can be broadly classified as having either inactive disease (the inactive surface antigen carrier state) or chronic active hepatitis B (Figure 1).1–9
Chronic inactive HBV infection. Carriers of inactive HBV infection have low serum levels of HBV DNA (< 2,000 IU/mL), persistently normal aminotransferase levels, and no HBV e antigen; if a liver biopsy is performed, no significant hepatitis is found.
Chronic active HBV infection. Patients with chronic active HBV infection, in contrast, have high serum HBV DNA levels (> 20,000 IU/mL) and persistently or intermittently high aminotransferase levels; they do have HBV e antigen, and a liver biopsy shows moderate or severe necroinflammation.
A small group of patients with chronic active hepatitis B may be negative for e antigen but still have high aminotransferase levels, high HBV DNA levels, and continued necroinflammation in the liver.4 The virus in these patients has a mutation in its precore or core promoter gene that prevents the production of e antigen.
Patients with chronic active HBV infection (whether positive or negative for e antigen) are at a significantly greater risk of progressive liver injury and developing cirrhosis and hepatocellular carcinoma than are inactive carriers of HBV.
Immune-tolerant chronic HBV infection. Patients who acquired HBV at birth (eg, our patient) may have immune-tolerant HBV infection, which is characterized by significant HBV replication manifested by the presence of HBV e antigen and high levels of HBV DNA in the serum. However, these patients have no clinical or pathologic evidence of active liver disease (no symptoms, normal serum alanine aminotransferase levels, and minimal changes on liver biopsy).5 This was obviously the case in our patient, based on his history and laboratory results before his current symptoms developed.
Case continues: Liver function abnormalities
On physical examination, the patient’s temperature is 99.9°F (37.7°C), heart rate 106 per minute, blood pressure 98/54 mm Hg, respiratory rate 18 per minute, and oxygen saturation 100% while breathing ambient air. He is alert and oriented to time, place, and person.
He has icteric sclera, and his skin is jaundiced. His lymph nodes are not palpable. His cardiac examination is normal except for tachycardia. His lungs are clear to auscultation and percussion. He has mild epigastric and right upper quadrant abdominal tenderness with no peritoneal signs, hepatosplenomegaly, or masses.
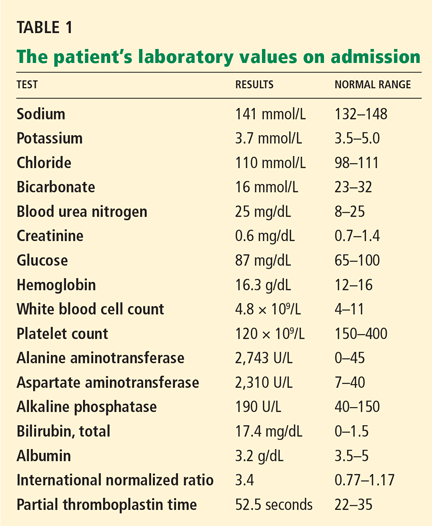
His basic laboratory values on admission are listed in Table 1. His amylase and lipase levels are normal. A urine dipstick test is positive for bilirubin.
WHAT IS THE LEAST LIKELY DIAGNOSIS?
2. Which one of the following is the least likely diagnosis in this patient?
- Reactivation of hepatitis B
- Drug-associated liver injury
- Acute viral hepatitis
- Acute alcoholic hepatitis
- Ischemic hepatitis
The degree and pattern of liver function abnormalities in our patient reflect hepatocellular injury rather than cholestatic liver disease, because his aminotransferase levels are elevated much higher than his alkaline phosphatase level (Table 1). Bilirubin elevation does not help differentiate the two conditions.
The degree and pattern of aminotransferase elevations are also helpful in narrowing the differential diagnosis. Serum aminotransferase levels of more than 1,000 U/L are mainly seen in patients with ischemic, viral, and toxininduced liver injury. Other rare causes of such high levels include Budd-Chiari syndrome, Wilson disease, and autoimmune hepatitis.
Ischemic hepatitis. Our patient has mild hypotension, but it does not seem to have been severe enough or of long enough duration to have caused ischemic hepatitis.
Drug-associated liver injury. Hepatotoxicity associated with drugs (most commonly acetaminophen [Tylenol]), herbal therapy, or mushroom poisoning should be considered in any patient whose aminotransferase levels are this high. However, our patient denies taking any medications (prescribed or over-the-counter), herbal remedies, or illicit drugs.
Acute viral hepatitis can certainly explain the patient’s clinical picture. Infection with hepatitis A virus, hepatitis D virus, hepatitis E virus, cytomegalovirus, Epstein-Barr virus, herpes simplex viruses types 1 and 2, and varicella zoster virus have all been implicated in severe acute hepatitis. Although hepatitis E virus infection is more common in developing countries, it has been reported in the United States.6 It is unlikely that acute hepatitis C virus infection is producing this degree of elevation in aminotransferase levels.
Reactivation of the patient’s chronic HBV infection can also account for his clinical presentation.
Acute alcoholic hepatitis should be suspected clinically if a patient has a history of heavy alcohol use and clinical and laboratory findings that are compatible with the diagnosis. However, the absolute values of serum aspartate aminotransferase and alanine aminotransferase in acute alcoholic hepatitis are almost always less than 500 IU/L (and typically less than 300 IU/L). Our patient’s values are much higher, and he says he does not drink very much. Although people sometimes underestimate their alcohol intake, alcoholic hepatitis is the least likely diagnosis in our patient.
Case continues: The patient is hospitalized
The patient is admitted with a diagnosis of acute hepatitis. Given his history of chronic hepatitis B, he is empirically started on lamivudine (Epivir-HBV).

- Antinuclear antibody negative
- Autoimmune liver disease panel negative
- Serum ceruloplasmin 30 mg/dL (normal range 15–60)
- Alpha fetoprotein 35.1 μg/L (< 10).
Abdominal ultrasonography is performed and reveals a small stone in the gallbladder with no evidence of biliary dilatation; otherwise, the gallbladder appears normal. Doppler ultrasonography shows the liver vessels to be patent; the liver is normal in appearance. The abdomen and pelvis appear to be normal on computed tomography without intravenous contrast.
On the third hospital day, the patient’s blood test results are:
- Aspartate aminotransferase 199 U/L (normal range 7–40)
- Alanine aminotransferase 735 U/L (0–45)
- Total bilirubin 22.9 mg/dL (0–1.5)
- International normalized ratio 6.0 (0.77–1.17)
- White blood cell count 5.1 × 109/L (4–11)
- Hemoglobin 11.7 g/dL (12–16)
- Platelet count 166 × 109/L (150–400)
- Blood and urine cultures negative.
WHAT IS CAUSING HIS ACUTE HEPATITIS?
3. On the basis of the new data, which of the following statements about the cause of acute hepatitis in this patient is the most accurate?
- Herpetic hepatitis is the most likely cause, given his positive test for immunoglobulin M (IgM) against herpes simplex virus
- Hepatitis C cannot be excluded with the available data
- Negative HBV e antigen does not exclude the diagnosis of acute exacerbation of HBV infection
- Hepatocellular carcinoma is the likely diagnosis, given the elevated alpha fetoprotein level
The third answer above is correct: a negative test for hepatitis B e antigen does not exclude the diagnosis of acute exacerbation of HBV infection
Herpetic hepatitis. Although not common, hepatitis due to herpes simplex virus infection should be considered in the differential diagnosis of any patient presenting with severe acute hepatitis, particularly when fever is present. Common features of herpetic hepatitis on presentation include high fever, leukopenia, markedly elevated aminotransferases, and mild cholestasis. Vesicular rash occurs in only less than half of cases of herpetic hepatitis.10
Serologic testing is of limited value because it has high rates of false-positive and false-negative results. The diagnosis can be confirmed only by viral polymerase chain reaction testing or by identifying herpes simplex viral inclusions in the liver biopsy.
However, the death rate is high in this disease, and since herpetic hepatitis is one of the few treatable causes of acute liver failure, parenteral acyclovir (Zovirax) should be considered empirically in patients presenting with acute liver failure. Our patient was started on acyclovir when his tests for IgM against herpes simplex virus came back positive.
Hepatitis C. Antibodies against hepatitis C virus do not develop immediately after this virus is contracted; they may take up to 12 weeks to develop after exposure. For this reason, about 30% to 50% of patients with acute hepatitis C virus infection are negative for these antibodies initially. In those patients, hepatitis C virus RNA in the blood is the most sensitive test to detect acute hepatitis C virus infection.
Our patient has neither antibodies against hepatitis C virus nor hepatitis C virus RNA by polymerase chain reaction testing, which rules out hepatitis C virus infection.
Disappearance of e antigen in HBV infection. The disappearance of HBV e antigen is usually associated with a decrease in serum HBV DNA and remission of liver disease. However, some patients continue to have active liver disease and high levels of HBV DNA despite e antigen seroconversion. This is due to a stop codon mutation in the precore region of the viral genome that decreases or prevents production of HBV e antigen.4 In other words, even though HBV e antigen is a good marker of HBV replication in general, a subgroup of patients with chronic HBV infection are negative for e antigen but still have a high rate of viral replication as evidenced by high serum HBV DNA levels.
Patients with perinatally acquired chronic HBV infection most often have immune-tolerant chronic HBV infection. Among those patients (mostly Asian),5,7 the virus is spontaneously cleared at a rate of approximately 2% to 3% per year,8 most often during the second and third decades of age.
Transition from the immune-tolerant phase to the immune clearance phase is frequently associated with mild transient worsening of the liver function profile.9,11,12 However, in a small percentage of patients, hepatic decompensation and even (rarely) death from hepatic failure may occur secondary to a sudden activation of the immune system as it attempts to clear the virus. This may result in an increase in immune-mediated lysis of infected hepatocytes.
Hepatocellular carcinoma. Exacerbation of hepatitis B may be associated with an elevation of alpha fetoprotein, which may falsely raise concerns about the possibility of hepatocellular carcinoma. However, our patient had abdominal imaging with both ultrasonography and computed tomography, which showed no evidence of hepatocellular carcinoma.
Comment. The most likely cause of the patient’s acute liver failure is an acute exacerbation of hepatitis B. However, herpetic hepatitis should be ruled out by testing for herpes simplex virus by polymerase chain reaction, performing a liver biopsy, or both.
Case continues: His condition worsens
A transjugular liver biopsy shows changes associated with chronic hepatitis B, severe acute hepatitis with extensive confluent and submassive hepatic necrosis, and no intracellular viral inclusions. Subsequently, acyclovir is stopped.
On the 6th hospital day, he develops progressive metabolic acidosis and hypotension, with worsening hypoxemia. A chest radiograph is obtained to look for pneumonia, but it is indeterminate; computed tomography of the chest without contrast medium is likewise unremarkable. Duplex ultrasonography of the four extremities is negative for venous thrombosis.
The patient becomes more lethargic and difficult to arouse. He is transferred to the intensive care unit and intubated. His prothrombin and partial thromboplastin times continue to rise, the prothrombin time reaching values of more than 50 seconds. In addition, progressive renal insufficiency develops.
WHAT IS THE NEXT STEP?
4. Which of the following is the most appropriate next step in the management of this patient?
- Liver transplantation
- HBV immunoglobulin only
- Interferon and a nucleoside analogue
- Liver-assist devices
- Continue supportive care only
Liver transplantation. Since the patient’s severe acute hepatitis is accompanied by coagulopathy and encephalopathy, he meets the definition of having acute liver failure. Liver transplantation remains the only definitive therapy.

HBV immune globulin immunoprophylaxis is indicated in patients with HBV infection undergoing liver transplantation, to prevent recurrence of hepatitis B after the transplant, particularly in those with a high pretransplant viral load.17 The use of pretransplant antiviral therapy and the posttransplant combination of antiviral therapy and HBV immune globulin has reduced the rate of hepatitis B recurrence to less than 10%. However, immune globulin is by no means the best single next step in managing this patient, who clearly needs a new liver.
Interferon, nucleoside analogues. Options for antiviral treatment are interferon alfa and nucleoside analogues. Interferon therapy is contraindicated in patients such as ours, who have decompensated liver disease, because it can exacerbate the disease.18

Liver-assist devices. Because liver allografts are in short supply, there has been a strong interest in developing a device that would provide the same benefits for patients with liver failure as hemodialysis does for patients with renal failure. Trials are under way to determine the efficacy and safety of these devices.20
Case continues: He receives a liver
The patient undergoes liver transplantation. He is given HBV immune globulin during and after the surgery.
Pathologic review. Under the microscope, his old liver has widespread necrosis and hemorrhage as well as inflammatory changes suggesting a chronic viral process. Regenerative nodules are present in the small amount of surviving liver parenchyma, consistent with early cirrhosis. Iron staining shows +3 depositions in areas of hepatic collapse (a nonspecific finding). Periodic acid-Schiff staining after diastase (used to detect alpha-1 antitrypsin deficiency) is negative. Herpetic viral inclusions are not present.
An immunoassay for herpes simplex virus antigen is negative. Immunostaining with antibodies to the HBV core antigen is negative. HBV surface antigen is strongly and diffusely positive in the cytoplasm of 80% to 90% of hepatocytes. The immunohistologic staining pattern is consistent with integration of HBV DNA into the DNA of hepatic tissue.
Postoperative course. Lamivudine is continued after surgery, and the patient is sent home. He has resumed the level of functioning he had before becoming ill.
Comment. The outcome of liver transplantation for hepatitis B has notably improved since HBV immune globulin and nucleoside analogues were introduced. The results of liver transplantation for hepatitis B, particuarly patient and graft survival rates, are now better than those in transplant patients with hepatitis C and similar to those in transplant patients with other types of liver disease.21 The combination of HBV immune globulin and lamivudine has cut the rate of HBV reinfection after liver transplantation to approximately 10% and increased the 5-year survival rate after transplantation to about 80%.17,22
KEY POINTS
- In immunocompetent adults, most primary HBV infections are self-limited.
- Chronic HBV infection is defined as the persistence of HBV surface antigen in the serum for at least 6 months. Patients having chronic HBV infection can be broadly classified as inactive carriers or having chronic active disease.
- Most patients with chronic active HBV infection are positive for HBV e antigen, except patients in whom the virus has a mutation in the precore or core region of its genome that prevents the production of e antigen.
- Patients who carry inactive HBV or who are immune-tolerant require serial measurements of aminotransferase and HBV DNA levels. Treatment can be considered if the patient has a high viral load (> 2,000 IU/mL), elevated aminotransferases, or active disease on liver biopsy.
- Carriers of chronic active HBV (whether positive or negative for HBV e antigen) should be referred to a hepatologist for consideration of liver biopsy and treatment.
- Interferon should not be used in immunocompromised patients or those with decompensated liver disease because it can further exacerbate the liver disease.
- Liver transplantation should be considered in patients with acute liver failure who have a poor prognosis according to the King’s College Hospital criteria.
- Dusheiko G. Hepatitis B. In:Bircher J, Benhamou JP, McIntyre N, Rizzetto M, Rodes J, editors. Oxford Textbook of Clinical Hepatology. 2nd ed. Oxford, UK: Oxford University Press; 1999:876–896.
- Chu CJ, Hussain M, Lok AS. Quantitative serum HBV DNA levels during different stages of chronic hepatitis B infection. Hepatology 2002; 36:1408–1415.
- Pawlotsky JM, Bastie A, Hezode C, et al. Routine detection and quantification of hepatitis B virus DNA in clinical laboratories: performance of three commercial assays. J Virol Methods 2000; 85:11–21.
- Brunetto MR, Giarin MM, Oliveri F, et al. Wild-type and e-antigen-minus hepatitis viruses and course of chronic hepatitis. Proc Natl Acad Sci USA 1991; 88:4186–4190.
- Lok AS, Lai CL. A longitudinal follow-up of asymptomatic hepatitis B surface antigen-positive Chinese children. Hepatology 1988; 5:1130–1133.
- Hsu HY, Chang MH, Hsieh KH, et al. Cellular immune response to HBcAg in mother-to-infant transmission of hepatitis B virus. Hepatology 1992; 15:770–776.
- Chang MH, Hsu HY, Hsu HC, Ni YH, Chen JS, Chen DS. The significance of spontaneous hepatitis B e antigen seroconversion in childhood: with special emphasis on the clearance of hepatitis B e antigen before 3 years of age. Hepatology 1995; 22:1387–1392.
- Ruiz-Moreno M, Otero M, Millan A, et al. Clinical and histological outcome after hepatitis B e antigen to antibody seroconversion in children with chronic hepatitis B. Hepatology 1999; 29:572–575.
- Liaw YF, Chu CM, Su IJ, Huang MJ, Lin DY, Chang-Chien CS. Clinical and histological events preceding hepatitis B e antigen seroconversion in chronic type B hepatitis. Gastroenterology 1983; 84:216–219.
- Norvell JP, Blei AT, Jovanovic BD, Levitsky J. Herpes simplex virus hepatitis: an analysis of the published literature and institutional cases. Liver Transplant 2007; 13:1428–1434,
- Liaw YF, Pao CC, Chu CM, Sheen IS, Huang MJ. Changes of serum hepatitis B virus DNA in two types of clinical events preceding spontaneous hepatitis B e antigen seroconversion in chronic type B hepatitis. Hepatology 1987; 7:1–3.
- Maruyama T, Iino S, Koike K, Yasuda K, Milich DR. Serology of acute exacerbation in chronic hepatitis B virus infection. Gastroenterology 1993; 105:1141–1151.
- O'Grady JG, Alexander GJ, Hayllar KM, Williams R. Early indicators of prognosis in fulminant hepatic failure. Gastroenterology 1989; 97:439–445.
- Shakil AO, Kramer D, Mazariegos GV, Fung JJ, Rakela J. Acute liver failure: clinical features, outcome analysis, and applicability of prognostic criteria. Liver Transplant 2000; 6:163–169.
- Anand AC, Nightingale P, Neuberger JM. Early indicators of prognosis in fulminant hepatic failure: an assessment of the King’s criteria. J Hepatol 1997; 26:62–68.
- Schmidt LE, Dalhoff K. Serum phosphate is an early predictor of outcome in severe acetaminophen-induced hepatotoxicity. Hepatology 2002; 36:659–665.
- Samuel D, Muller R, Alexander G, et al. Liver transplantation in European patients with the hepatitis B surface antigen. N Engl J Med 1993; 329:1842–1847.
- Lok A, McMahon BJ. Chronic hepatitis B. Hepatology 2007; 45:507–539.
- Sorren MF, Belangia EA, Costa J, et al. National Institutes of Health consensus development conference statement: management of hepatitis B. Ann Intern Med 2009; 150:104–110.
- Kjaergard LL, Liu J, Als-Nielsen B, Gluud C. Artificial and bioartificial support systems for acute and acute-on-chronic liver failure: a systematic review. JAMA 2003; 289:217–222.
- Kim WR, Poterucha JJ, Kremers WK, Ishitani MB, Dickson ER. Outcome of liver transplantation for hepatitis B in the United States. Liver Transplant 2004; 10:968–974.
- Terrault NA, Zhou S, Combs C, et al. Prophylaxis in liver transplant recipients using a fixed dosing schedule of hepatitis B immunoglobulin. Hepatology 1996; 24:1327–1333.
A 35-year-old man who was born in Vietnam presents to the emergency department of a local hospital because he has had jaundice for 5 days and fatigue, malaise, and anorexia for 2 weeks. He also has nausea and mild epigastric and right upper quadrant abdominal pain. He denies having fevers, chills, night sweats, vomiting, diarrhea, melena, hematochezia, or weight loss.
His medical history is remarkable only for perinatally acquired hepatitis B virus (HBV) infection, for which he never received antiviral therapy. He does not take any prescribed, over-the-counter, or herbal medications.
He lives in the Midwest region of the United States and works full-time as a physician in private practice. He is married and has two children.
He has not travelled recently. He has no pets at home and has not been exposed to any.
He has never smoked. He drinks alcohol socially but has never used recreational drugs.
In a laboratory evaluation performed a year ago for insurance purposes, his liver function tests—serum albumin, total bilirubin, alkaline phosphatase, alanine aminotransferase, and aspartate aminotransferase levels—were all normal. He was positive for HBV surface antigen and HBV e antigen and negative for antibodies against these antigens.
PHASES OF HBV INFECTION
1. Which of the following best describes the status of HBV infection in this patient before his current symptoms developed?
- Resolved HBV infection
- Chronic inactive HBV infection
- Chronic active HBV infection
- Immune-tolerant chronic HBV infection
The correct answer is immune-tolerant chronic HBV infection.
Resolved infection. In immunocompetent adults, most primary HBV infections are self-limited: people clear the virus and gain lasting immunity (defined as the loss of HBV surface antigen, the development of antibody against surface antigen, no detectable HBV DNA in the serum, and normal alanine and aspartate aminotransferase levels). However, a minority of primary HBV infections persist and become chronic.
Chronic HBV infection is defined as the persistence of HBV surface antigen in the serum for at least 6 months. Patients with chronic HBV infection can be broadly classified as having either inactive disease (the inactive surface antigen carrier state) or chronic active hepatitis B (Figure 1).1–9
Chronic inactive HBV infection. Carriers of inactive HBV infection have low serum levels of HBV DNA (< 2,000 IU/mL), persistently normal aminotransferase levels, and no HBV e antigen; if a liver biopsy is performed, no significant hepatitis is found.
Chronic active HBV infection. Patients with chronic active HBV infection, in contrast, have high serum HBV DNA levels (> 20,000 IU/mL) and persistently or intermittently high aminotransferase levels; they do have HBV e antigen, and a liver biopsy shows moderate or severe necroinflammation.
A small group of patients with chronic active hepatitis B may be negative for e antigen but still have high aminotransferase levels, high HBV DNA levels, and continued necroinflammation in the liver.4 The virus in these patients has a mutation in its precore or core promoter gene that prevents the production of e antigen.
Patients with chronic active HBV infection (whether positive or negative for e antigen) are at a significantly greater risk of progressive liver injury and developing cirrhosis and hepatocellular carcinoma than are inactive carriers of HBV.
Immune-tolerant chronic HBV infection. Patients who acquired HBV at birth (eg, our patient) may have immune-tolerant HBV infection, which is characterized by significant HBV replication manifested by the presence of HBV e antigen and high levels of HBV DNA in the serum. However, these patients have no clinical or pathologic evidence of active liver disease (no symptoms, normal serum alanine aminotransferase levels, and minimal changes on liver biopsy).5 This was obviously the case in our patient, based on his history and laboratory results before his current symptoms developed.
Case continues: Liver function abnormalities
On physical examination, the patient’s temperature is 99.9°F (37.7°C), heart rate 106 per minute, blood pressure 98/54 mm Hg, respiratory rate 18 per minute, and oxygen saturation 100% while breathing ambient air. He is alert and oriented to time, place, and person.
He has icteric sclera, and his skin is jaundiced. His lymph nodes are not palpable. His cardiac examination is normal except for tachycardia. His lungs are clear to auscultation and percussion. He has mild epigastric and right upper quadrant abdominal tenderness with no peritoneal signs, hepatosplenomegaly, or masses.
His basic laboratory values on admission are listed in Table 1. His amylase and lipase levels are normal. A urine dipstick test is positive for bilirubin.
WHAT IS THE LEAST LIKELY DIAGNOSIS?
2. Which one of the following is the least likely diagnosis in this patient?
- Reactivation of hepatitis B
- Drug-associated liver injury
- Acute viral hepatitis
- Acute alcoholic hepatitis
- Ischemic hepatitis
The degree and pattern of liver function abnormalities in our patient reflect hepatocellular injury rather than cholestatic liver disease, because his aminotransferase levels are elevated much higher than his alkaline phosphatase level (Table 1). Bilirubin elevation does not help differentiate the two conditions.
The degree and pattern of aminotransferase elevations are also helpful in narrowing the differential diagnosis. Serum aminotransferase levels of more than 1,000 U/L are mainly seen in patients with ischemic, viral, and toxininduced liver injury. Other rare causes of such high levels include Budd-Chiari syndrome, Wilson disease, and autoimmune hepatitis.
Ischemic hepatitis. Our patient has mild hypotension, but it does not seem to have been severe enough or of long enough duration to have caused ischemic hepatitis.
Drug-associated liver injury. Hepatotoxicity associated with drugs (most commonly acetaminophen [Tylenol]), herbal therapy, or mushroom poisoning should be considered in any patient whose aminotransferase levels are this high. However, our patient denies taking any medications (prescribed or over-the-counter), herbal remedies, or illicit drugs.
Acute viral hepatitis can certainly explain the patient’s clinical picture. Infection with hepatitis A virus, hepatitis D virus, hepatitis E virus, cytomegalovirus, Epstein-Barr virus, herpes simplex viruses types 1 and 2, and varicella zoster virus have all been implicated in severe acute hepatitis. Although hepatitis E virus infection is more common in developing countries, it has been reported in the United States.6 It is unlikely that acute hepatitis C virus infection is producing this degree of elevation in aminotransferase levels.
Reactivation of the patient’s chronic HBV infection can also account for his clinical presentation.
Acute alcoholic hepatitis should be suspected clinically if a patient has a history of heavy alcohol use and clinical and laboratory findings that are compatible with the diagnosis. However, the absolute values of serum aspartate aminotransferase and alanine aminotransferase in acute alcoholic hepatitis are almost always less than 500 IU/L (and typically less than 300 IU/L). Our patient’s values are much higher, and he says he does not drink very much. Although people sometimes underestimate their alcohol intake, alcoholic hepatitis is the least likely diagnosis in our patient.
Case continues: The patient is hospitalized
The patient is admitted with a diagnosis of acute hepatitis. Given his history of chronic hepatitis B, he is empirically started on lamivudine (Epivir-HBV).
- Antinuclear antibody negative
- Autoimmune liver disease panel negative
- Serum ceruloplasmin 30 mg/dL (normal range 15–60)
- Alpha fetoprotein 35.1 μg/L (< 10).
Abdominal ultrasonography is performed and reveals a small stone in the gallbladder with no evidence of biliary dilatation; otherwise, the gallbladder appears normal. Doppler ultrasonography shows the liver vessels to be patent; the liver is normal in appearance. The abdomen and pelvis appear to be normal on computed tomography without intravenous contrast.
On the third hospital day, the patient’s blood test results are:
- Aspartate aminotransferase 199 U/L (normal range 7–40)
- Alanine aminotransferase 735 U/L (0–45)
- Total bilirubin 22.9 mg/dL (0–1.5)
- International normalized ratio 6.0 (0.77–1.17)
- White blood cell count 5.1 × 109/L (4–11)
- Hemoglobin 11.7 g/dL (12–16)
- Platelet count 166 × 109/L (150–400)
- Blood and urine cultures negative.
WHAT IS CAUSING HIS ACUTE HEPATITIS?
3. On the basis of the new data, which of the following statements about the cause of acute hepatitis in this patient is the most accurate?
- Herpetic hepatitis is the most likely cause, given his positive test for immunoglobulin M (IgM) against herpes simplex virus
- Hepatitis C cannot be excluded with the available data
- Negative HBV e antigen does not exclude the diagnosis of acute exacerbation of HBV infection
- Hepatocellular carcinoma is the likely diagnosis, given the elevated alpha fetoprotein level
The third answer above is correct: a negative test for hepatitis B e antigen does not exclude the diagnosis of acute exacerbation of HBV infection
Herpetic hepatitis. Although not common, hepatitis due to herpes simplex virus infection should be considered in the differential diagnosis of any patient presenting with severe acute hepatitis, particularly when fever is present. Common features of herpetic hepatitis on presentation include high fever, leukopenia, markedly elevated aminotransferases, and mild cholestasis. Vesicular rash occurs in only less than half of cases of herpetic hepatitis.10
Serologic testing is of limited value because it has high rates of false-positive and false-negative results. The diagnosis can be confirmed only by viral polymerase chain reaction testing or by identifying herpes simplex viral inclusions in the liver biopsy.
However, the death rate is high in this disease, and since herpetic hepatitis is one of the few treatable causes of acute liver failure, parenteral acyclovir (Zovirax) should be considered empirically in patients presenting with acute liver failure. Our patient was started on acyclovir when his tests for IgM against herpes simplex virus came back positive.
Hepatitis C. Antibodies against hepatitis C virus do not develop immediately after this virus is contracted; they may take up to 12 weeks to develop after exposure. For this reason, about 30% to 50% of patients with acute hepatitis C virus infection are negative for these antibodies initially. In those patients, hepatitis C virus RNA in the blood is the most sensitive test to detect acute hepatitis C virus infection.
Our patient has neither antibodies against hepatitis C virus nor hepatitis C virus RNA by polymerase chain reaction testing, which rules out hepatitis C virus infection.
Disappearance of e antigen in HBV infection. The disappearance of HBV e antigen is usually associated with a decrease in serum HBV DNA and remission of liver disease. However, some patients continue to have active liver disease and high levels of HBV DNA despite e antigen seroconversion. This is due to a stop codon mutation in the precore region of the viral genome that decreases or prevents production of HBV e antigen.4 In other words, even though HBV e antigen is a good marker of HBV replication in general, a subgroup of patients with chronic HBV infection are negative for e antigen but still have a high rate of viral replication as evidenced by high serum HBV DNA levels.
Patients with perinatally acquired chronic HBV infection most often have immune-tolerant chronic HBV infection. Among those patients (mostly Asian),5,7 the virus is spontaneously cleared at a rate of approximately 2% to 3% per year,8 most often during the second and third decades of age.
Transition from the immune-tolerant phase to the immune clearance phase is frequently associated with mild transient worsening of the liver function profile.9,11,12 However, in a small percentage of patients, hepatic decompensation and even (rarely) death from hepatic failure may occur secondary to a sudden activation of the immune system as it attempts to clear the virus. This may result in an increase in immune-mediated lysis of infected hepatocytes.
Hepatocellular carcinoma. Exacerbation of hepatitis B may be associated with an elevation of alpha fetoprotein, which may falsely raise concerns about the possibility of hepatocellular carcinoma. However, our patient had abdominal imaging with both ultrasonography and computed tomography, which showed no evidence of hepatocellular carcinoma.
Comment. The most likely cause of the patient’s acute liver failure is an acute exacerbation of hepatitis B. However, herpetic hepatitis should be ruled out by testing for herpes simplex virus by polymerase chain reaction, performing a liver biopsy, or both.
Case continues: His condition worsens
A transjugular liver biopsy shows changes associated with chronic hepatitis B, severe acute hepatitis with extensive confluent and submassive hepatic necrosis, and no intracellular viral inclusions. Subsequently, acyclovir is stopped.
On the 6th hospital day, he develops progressive metabolic acidosis and hypotension, with worsening hypoxemia. A chest radiograph is obtained to look for pneumonia, but it is indeterminate; computed tomography of the chest without contrast medium is likewise unremarkable. Duplex ultrasonography of the four extremities is negative for venous thrombosis.
The patient becomes more lethargic and difficult to arouse. He is transferred to the intensive care unit and intubated. His prothrombin and partial thromboplastin times continue to rise, the prothrombin time reaching values of more than 50 seconds. In addition, progressive renal insufficiency develops.
WHAT IS THE NEXT STEP?
4. Which of the following is the most appropriate next step in the management of this patient?
- Liver transplantation
- HBV immunoglobulin only
- Interferon and a nucleoside analogue
- Liver-assist devices
- Continue supportive care only
Liver transplantation. Since the patient’s severe acute hepatitis is accompanied by coagulopathy and encephalopathy, he meets the definition of having acute liver failure. Liver transplantation remains the only definitive therapy.
HBV immune globulin immunoprophylaxis is indicated in patients with HBV infection undergoing liver transplantation, to prevent recurrence of hepatitis B after the transplant, particularly in those with a high pretransplant viral load.17 The use of pretransplant antiviral therapy and the posttransplant combination of antiviral therapy and HBV immune globulin has reduced the rate of hepatitis B recurrence to less than 10%. However, immune globulin is by no means the best single next step in managing this patient, who clearly needs a new liver.
Interferon, nucleoside analogues. Options for antiviral treatment are interferon alfa and nucleoside analogues. Interferon therapy is contraindicated in patients such as ours, who have decompensated liver disease, because it can exacerbate the disease.18
Liver-assist devices. Because liver allografts are in short supply, there has been a strong interest in developing a device that would provide the same benefits for patients with liver failure as hemodialysis does for patients with renal failure. Trials are under way to determine the efficacy and safety of these devices.20
Case continues: He receives a liver
The patient undergoes liver transplantation. He is given HBV immune globulin during and after the surgery.
Pathologic review. Under the microscope, his old liver has widespread necrosis and hemorrhage as well as inflammatory changes suggesting a chronic viral process. Regenerative nodules are present in the small amount of surviving liver parenchyma, consistent with early cirrhosis. Iron staining shows +3 depositions in areas of hepatic collapse (a nonspecific finding). Periodic acid-Schiff staining after diastase (used to detect alpha-1 antitrypsin deficiency) is negative. Herpetic viral inclusions are not present.
An immunoassay for herpes simplex virus antigen is negative. Immunostaining with antibodies to the HBV core antigen is negative. HBV surface antigen is strongly and diffusely positive in the cytoplasm of 80% to 90% of hepatocytes. The immunohistologic staining pattern is consistent with integration of HBV DNA into the DNA of hepatic tissue.
Postoperative course. Lamivudine is continued after surgery, and the patient is sent home. He has resumed the level of functioning he had before becoming ill.
Comment. The outcome of liver transplantation for hepatitis B has notably improved since HBV immune globulin and nucleoside analogues were introduced. The results of liver transplantation for hepatitis B, particuarly patient and graft survival rates, are now better than those in transplant patients with hepatitis C and similar to those in transplant patients with other types of liver disease.21 The combination of HBV immune globulin and lamivudine has cut the rate of HBV reinfection after liver transplantation to approximately 10% and increased the 5-year survival rate after transplantation to about 80%.17,22
KEY POINTS
- In immunocompetent adults, most primary HBV infections are self-limited.
- Chronic HBV infection is defined as the persistence of HBV surface antigen in the serum for at least 6 months. Patients having chronic HBV infection can be broadly classified as inactive carriers or having chronic active disease.
- Most patients with chronic active HBV infection are positive for HBV e antigen, except patients in whom the virus has a mutation in the precore or core region of its genome that prevents the production of e antigen.
- Patients who carry inactive HBV or who are immune-tolerant require serial measurements of aminotransferase and HBV DNA levels. Treatment can be considered if the patient has a high viral load (> 2,000 IU/mL), elevated aminotransferases, or active disease on liver biopsy.
- Carriers of chronic active HBV (whether positive or negative for HBV e antigen) should be referred to a hepatologist for consideration of liver biopsy and treatment.
- Interferon should not be used in immunocompromised patients or those with decompensated liver disease because it can further exacerbate the liver disease.
- Liver transplantation should be considered in patients with acute liver failure who have a poor prognosis according to the King’s College Hospital criteria.
A 35-year-old man who was born in Vietnam presents to the emergency department of a local hospital because he has had jaundice for 5 days and fatigue, malaise, and anorexia for 2 weeks. He also has nausea and mild epigastric and right upper quadrant abdominal pain. He denies having fevers, chills, night sweats, vomiting, diarrhea, melena, hematochezia, or weight loss.
His medical history is remarkable only for perinatally acquired hepatitis B virus (HBV) infection, for which he never received antiviral therapy. He does not take any prescribed, over-the-counter, or herbal medications.
He lives in the Midwest region of the United States and works full-time as a physician in private practice. He is married and has two children.
He has not travelled recently. He has no pets at home and has not been exposed to any.
He has never smoked. He drinks alcohol socially but has never used recreational drugs.
In a laboratory evaluation performed a year ago for insurance purposes, his liver function tests—serum albumin, total bilirubin, alkaline phosphatase, alanine aminotransferase, and aspartate aminotransferase levels—were all normal. He was positive for HBV surface antigen and HBV e antigen and negative for antibodies against these antigens.
PHASES OF HBV INFECTION
1. Which of the following best describes the status of HBV infection in this patient before his current symptoms developed?
- Resolved HBV infection
- Chronic inactive HBV infection
- Chronic active HBV infection
- Immune-tolerant chronic HBV infection
The correct answer is immune-tolerant chronic HBV infection.
Resolved infection. In immunocompetent adults, most primary HBV infections are self-limited: people clear the virus and gain lasting immunity (defined as the loss of HBV surface antigen, the development of antibody against surface antigen, no detectable HBV DNA in the serum, and normal alanine and aspartate aminotransferase levels). However, a minority of primary HBV infections persist and become chronic.
Chronic HBV infection is defined as the persistence of HBV surface antigen in the serum for at least 6 months. Patients with chronic HBV infection can be broadly classified as having either inactive disease (the inactive surface antigen carrier state) or chronic active hepatitis B (Figure 1).1–9
Chronic inactive HBV infection. Carriers of inactive HBV infection have low serum levels of HBV DNA (< 2,000 IU/mL), persistently normal aminotransferase levels, and no HBV e antigen; if a liver biopsy is performed, no significant hepatitis is found.
Chronic active HBV infection. Patients with chronic active HBV infection, in contrast, have high serum HBV DNA levels (> 20,000 IU/mL) and persistently or intermittently high aminotransferase levels; they do have HBV e antigen, and a liver biopsy shows moderate or severe necroinflammation.
A small group of patients with chronic active hepatitis B may be negative for e antigen but still have high aminotransferase levels, high HBV DNA levels, and continued necroinflammation in the liver.4 The virus in these patients has a mutation in its precore or core promoter gene that prevents the production of e antigen.
Patients with chronic active HBV infection (whether positive or negative for e antigen) are at a significantly greater risk of progressive liver injury and developing cirrhosis and hepatocellular carcinoma than are inactive carriers of HBV.
Immune-tolerant chronic HBV infection. Patients who acquired HBV at birth (eg, our patient) may have immune-tolerant HBV infection, which is characterized by significant HBV replication manifested by the presence of HBV e antigen and high levels of HBV DNA in the serum. However, these patients have no clinical or pathologic evidence of active liver disease (no symptoms, normal serum alanine aminotransferase levels, and minimal changes on liver biopsy).5 This was obviously the case in our patient, based on his history and laboratory results before his current symptoms developed.
Case continues: Liver function abnormalities
On physical examination, the patient’s temperature is 99.9°F (37.7°C), heart rate 106 per minute, blood pressure 98/54 mm Hg, respiratory rate 18 per minute, and oxygen saturation 100% while breathing ambient air. He is alert and oriented to time, place, and person.
He has icteric sclera, and his skin is jaundiced. His lymph nodes are not palpable. His cardiac examination is normal except for tachycardia. His lungs are clear to auscultation and percussion. He has mild epigastric and right upper quadrant abdominal tenderness with no peritoneal signs, hepatosplenomegaly, or masses.
His basic laboratory values on admission are listed in Table 1. His amylase and lipase levels are normal. A urine dipstick test is positive for bilirubin.
WHAT IS THE LEAST LIKELY DIAGNOSIS?
2. Which one of the following is the least likely diagnosis in this patient?
- Reactivation of hepatitis B
- Drug-associated liver injury
- Acute viral hepatitis
- Acute alcoholic hepatitis
- Ischemic hepatitis
The degree and pattern of liver function abnormalities in our patient reflect hepatocellular injury rather than cholestatic liver disease, because his aminotransferase levels are elevated much higher than his alkaline phosphatase level (Table 1). Bilirubin elevation does not help differentiate the two conditions.
The degree and pattern of aminotransferase elevations are also helpful in narrowing the differential diagnosis. Serum aminotransferase levels of more than 1,000 U/L are mainly seen in patients with ischemic, viral, and toxininduced liver injury. Other rare causes of such high levels include Budd-Chiari syndrome, Wilson disease, and autoimmune hepatitis.
Ischemic hepatitis. Our patient has mild hypotension, but it does not seem to have been severe enough or of long enough duration to have caused ischemic hepatitis.
Drug-associated liver injury. Hepatotoxicity associated with drugs (most commonly acetaminophen [Tylenol]), herbal therapy, or mushroom poisoning should be considered in any patient whose aminotransferase levels are this high. However, our patient denies taking any medications (prescribed or over-the-counter), herbal remedies, or illicit drugs.
Acute viral hepatitis can certainly explain the patient’s clinical picture. Infection with hepatitis A virus, hepatitis D virus, hepatitis E virus, cytomegalovirus, Epstein-Barr virus, herpes simplex viruses types 1 and 2, and varicella zoster virus have all been implicated in severe acute hepatitis. Although hepatitis E virus infection is more common in developing countries, it has been reported in the United States.6 It is unlikely that acute hepatitis C virus infection is producing this degree of elevation in aminotransferase levels.
Reactivation of the patient’s chronic HBV infection can also account for his clinical presentation.
Acute alcoholic hepatitis should be suspected clinically if a patient has a history of heavy alcohol use and clinical and laboratory findings that are compatible with the diagnosis. However, the absolute values of serum aspartate aminotransferase and alanine aminotransferase in acute alcoholic hepatitis are almost always less than 500 IU/L (and typically less than 300 IU/L). Our patient’s values are much higher, and he says he does not drink very much. Although people sometimes underestimate their alcohol intake, alcoholic hepatitis is the least likely diagnosis in our patient.
Case continues: The patient is hospitalized
The patient is admitted with a diagnosis of acute hepatitis. Given his history of chronic hepatitis B, he is empirically started on lamivudine (Epivir-HBV).
- Antinuclear antibody negative
- Autoimmune liver disease panel negative
- Serum ceruloplasmin 30 mg/dL (normal range 15–60)
- Alpha fetoprotein 35.1 μg/L (< 10).
Abdominal ultrasonography is performed and reveals a small stone in the gallbladder with no evidence of biliary dilatation; otherwise, the gallbladder appears normal. Doppler ultrasonography shows the liver vessels to be patent; the liver is normal in appearance. The abdomen and pelvis appear to be normal on computed tomography without intravenous contrast.
On the third hospital day, the patient’s blood test results are:
- Aspartate aminotransferase 199 U/L (normal range 7–40)
- Alanine aminotransferase 735 U/L (0–45)
- Total bilirubin 22.9 mg/dL (0–1.5)
- International normalized ratio 6.0 (0.77–1.17)
- White blood cell count 5.1 × 109/L (4–11)
- Hemoglobin 11.7 g/dL (12–16)
- Platelet count 166 × 109/L (150–400)
- Blood and urine cultures negative.
WHAT IS CAUSING HIS ACUTE HEPATITIS?
3. On the basis of the new data, which of the following statements about the cause of acute hepatitis in this patient is the most accurate?
- Herpetic hepatitis is the most likely cause, given his positive test for immunoglobulin M (IgM) against herpes simplex virus
- Hepatitis C cannot be excluded with the available data
- Negative HBV e antigen does not exclude the diagnosis of acute exacerbation of HBV infection
- Hepatocellular carcinoma is the likely diagnosis, given the elevated alpha fetoprotein level
The third answer above is correct: a negative test for hepatitis B e antigen does not exclude the diagnosis of acute exacerbation of HBV infection
Herpetic hepatitis. Although not common, hepatitis due to herpes simplex virus infection should be considered in the differential diagnosis of any patient presenting with severe acute hepatitis, particularly when fever is present. Common features of herpetic hepatitis on presentation include high fever, leukopenia, markedly elevated aminotransferases, and mild cholestasis. Vesicular rash occurs in only less than half of cases of herpetic hepatitis.10
Serologic testing is of limited value because it has high rates of false-positive and false-negative results. The diagnosis can be confirmed only by viral polymerase chain reaction testing or by identifying herpes simplex viral inclusions in the liver biopsy.
However, the death rate is high in this disease, and since herpetic hepatitis is one of the few treatable causes of acute liver failure, parenteral acyclovir (Zovirax) should be considered empirically in patients presenting with acute liver failure. Our patient was started on acyclovir when his tests for IgM against herpes simplex virus came back positive.
Hepatitis C. Antibodies against hepatitis C virus do not develop immediately after this virus is contracted; they may take up to 12 weeks to develop after exposure. For this reason, about 30% to 50% of patients with acute hepatitis C virus infection are negative for these antibodies initially. In those patients, hepatitis C virus RNA in the blood is the most sensitive test to detect acute hepatitis C virus infection.
Our patient has neither antibodies against hepatitis C virus nor hepatitis C virus RNA by polymerase chain reaction testing, which rules out hepatitis C virus infection.
Disappearance of e antigen in HBV infection. The disappearance of HBV e antigen is usually associated with a decrease in serum HBV DNA and remission of liver disease. However, some patients continue to have active liver disease and high levels of HBV DNA despite e antigen seroconversion. This is due to a stop codon mutation in the precore region of the viral genome that decreases or prevents production of HBV e antigen.4 In other words, even though HBV e antigen is a good marker of HBV replication in general, a subgroup of patients with chronic HBV infection are negative for e antigen but still have a high rate of viral replication as evidenced by high serum HBV DNA levels.
Patients with perinatally acquired chronic HBV infection most often have immune-tolerant chronic HBV infection. Among those patients (mostly Asian),5,7 the virus is spontaneously cleared at a rate of approximately 2% to 3% per year,8 most often during the second and third decades of age.
Transition from the immune-tolerant phase to the immune clearance phase is frequently associated with mild transient worsening of the liver function profile.9,11,12 However, in a small percentage of patients, hepatic decompensation and even (rarely) death from hepatic failure may occur secondary to a sudden activation of the immune system as it attempts to clear the virus. This may result in an increase in immune-mediated lysis of infected hepatocytes.
Hepatocellular carcinoma. Exacerbation of hepatitis B may be associated with an elevation of alpha fetoprotein, which may falsely raise concerns about the possibility of hepatocellular carcinoma. However, our patient had abdominal imaging with both ultrasonography and computed tomography, which showed no evidence of hepatocellular carcinoma.
Comment. The most likely cause of the patient’s acute liver failure is an acute exacerbation of hepatitis B. However, herpetic hepatitis should be ruled out by testing for herpes simplex virus by polymerase chain reaction, performing a liver biopsy, or both.
Case continues: His condition worsens
A transjugular liver biopsy shows changes associated with chronic hepatitis B, severe acute hepatitis with extensive confluent and submassive hepatic necrosis, and no intracellular viral inclusions. Subsequently, acyclovir is stopped.
On the 6th hospital day, he develops progressive metabolic acidosis and hypotension, with worsening hypoxemia. A chest radiograph is obtained to look for pneumonia, but it is indeterminate; computed tomography of the chest without contrast medium is likewise unremarkable. Duplex ultrasonography of the four extremities is negative for venous thrombosis.
The patient becomes more lethargic and difficult to arouse. He is transferred to the intensive care unit and intubated. His prothrombin and partial thromboplastin times continue to rise, the prothrombin time reaching values of more than 50 seconds. In addition, progressive renal insufficiency develops.
WHAT IS THE NEXT STEP?
4. Which of the following is the most appropriate next step in the management of this patient?
- Liver transplantation
- HBV immunoglobulin only
- Interferon and a nucleoside analogue
- Liver-assist devices
- Continue supportive care only
Liver transplantation. Since the patient’s severe acute hepatitis is accompanied by coagulopathy and encephalopathy, he meets the definition of having acute liver failure. Liver transplantation remains the only definitive therapy.
HBV immune globulin immunoprophylaxis is indicated in patients with HBV infection undergoing liver transplantation, to prevent recurrence of hepatitis B after the transplant, particularly in those with a high pretransplant viral load.17 The use of pretransplant antiviral therapy and the posttransplant combination of antiviral therapy and HBV immune globulin has reduced the rate of hepatitis B recurrence to less than 10%. However, immune globulin is by no means the best single next step in managing this patient, who clearly needs a new liver.
Interferon, nucleoside analogues. Options for antiviral treatment are interferon alfa and nucleoside analogues. Interferon therapy is contraindicated in patients such as ours, who have decompensated liver disease, because it can exacerbate the disease.18
Liver-assist devices. Because liver allografts are in short supply, there has been a strong interest in developing a device that would provide the same benefits for patients with liver failure as hemodialysis does for patients with renal failure. Trials are under way to determine the efficacy and safety of these devices.20
Case continues: He receives a liver
The patient undergoes liver transplantation. He is given HBV immune globulin during and after the surgery.
Pathologic review. Under the microscope, his old liver has widespread necrosis and hemorrhage as well as inflammatory changes suggesting a chronic viral process. Regenerative nodules are present in the small amount of surviving liver parenchyma, consistent with early cirrhosis. Iron staining shows +3 depositions in areas of hepatic collapse (a nonspecific finding). Periodic acid-Schiff staining after diastase (used to detect alpha-1 antitrypsin deficiency) is negative. Herpetic viral inclusions are not present.
An immunoassay for herpes simplex virus antigen is negative. Immunostaining with antibodies to the HBV core antigen is negative. HBV surface antigen is strongly and diffusely positive in the cytoplasm of 80% to 90% of hepatocytes. The immunohistologic staining pattern is consistent with integration of HBV DNA into the DNA of hepatic tissue.
Postoperative course. Lamivudine is continued after surgery, and the patient is sent home. He has resumed the level of functioning he had before becoming ill.
Comment. The outcome of liver transplantation for hepatitis B has notably improved since HBV immune globulin and nucleoside analogues were introduced. The results of liver transplantation for hepatitis B, particuarly patient and graft survival rates, are now better than those in transplant patients with hepatitis C and similar to those in transplant patients with other types of liver disease.21 The combination of HBV immune globulin and lamivudine has cut the rate of HBV reinfection after liver transplantation to approximately 10% and increased the 5-year survival rate after transplantation to about 80%.17,22
KEY POINTS
- In immunocompetent adults, most primary HBV infections are self-limited.
- Chronic HBV infection is defined as the persistence of HBV surface antigen in the serum for at least 6 months. Patients having chronic HBV infection can be broadly classified as inactive carriers or having chronic active disease.
- Most patients with chronic active HBV infection are positive for HBV e antigen, except patients in whom the virus has a mutation in the precore or core region of its genome that prevents the production of e antigen.
- Patients who carry inactive HBV or who are immune-tolerant require serial measurements of aminotransferase and HBV DNA levels. Treatment can be considered if the patient has a high viral load (> 2,000 IU/mL), elevated aminotransferases, or active disease on liver biopsy.
- Carriers of chronic active HBV (whether positive or negative for HBV e antigen) should be referred to a hepatologist for consideration of liver biopsy and treatment.
- Interferon should not be used in immunocompromised patients or those with decompensated liver disease because it can further exacerbate the liver disease.
- Liver transplantation should be considered in patients with acute liver failure who have a poor prognosis according to the King’s College Hospital criteria.
- Dusheiko G. Hepatitis B. In:Bircher J, Benhamou JP, McIntyre N, Rizzetto M, Rodes J, editors. Oxford Textbook of Clinical Hepatology. 2nd ed. Oxford, UK: Oxford University Press; 1999:876–896.
- Chu CJ, Hussain M, Lok AS. Quantitative serum HBV DNA levels during different stages of chronic hepatitis B infection. Hepatology 2002; 36:1408–1415.
- Pawlotsky JM, Bastie A, Hezode C, et al. Routine detection and quantification of hepatitis B virus DNA in clinical laboratories: performance of three commercial assays. J Virol Methods 2000; 85:11–21.
- Brunetto MR, Giarin MM, Oliveri F, et al. Wild-type and e-antigen-minus hepatitis viruses and course of chronic hepatitis. Proc Natl Acad Sci USA 1991; 88:4186–4190.
- Lok AS, Lai CL. A longitudinal follow-up of asymptomatic hepatitis B surface antigen-positive Chinese children. Hepatology 1988; 5:1130–1133.
- Hsu HY, Chang MH, Hsieh KH, et al. Cellular immune response to HBcAg in mother-to-infant transmission of hepatitis B virus. Hepatology 1992; 15:770–776.
- Chang MH, Hsu HY, Hsu HC, Ni YH, Chen JS, Chen DS. The significance of spontaneous hepatitis B e antigen seroconversion in childhood: with special emphasis on the clearance of hepatitis B e antigen before 3 years of age. Hepatology 1995; 22:1387–1392.
- Ruiz-Moreno M, Otero M, Millan A, et al. Clinical and histological outcome after hepatitis B e antigen to antibody seroconversion in children with chronic hepatitis B. Hepatology 1999; 29:572–575.
- Liaw YF, Chu CM, Su IJ, Huang MJ, Lin DY, Chang-Chien CS. Clinical and histological events preceding hepatitis B e antigen seroconversion in chronic type B hepatitis. Gastroenterology 1983; 84:216–219.
- Norvell JP, Blei AT, Jovanovic BD, Levitsky J. Herpes simplex virus hepatitis: an analysis of the published literature and institutional cases. Liver Transplant 2007; 13:1428–1434,
- Liaw YF, Pao CC, Chu CM, Sheen IS, Huang MJ. Changes of serum hepatitis B virus DNA in two types of clinical events preceding spontaneous hepatitis B e antigen seroconversion in chronic type B hepatitis. Hepatology 1987; 7:1–3.
- Maruyama T, Iino S, Koike K, Yasuda K, Milich DR. Serology of acute exacerbation in chronic hepatitis B virus infection. Gastroenterology 1993; 105:1141–1151.
- O'Grady JG, Alexander GJ, Hayllar KM, Williams R. Early indicators of prognosis in fulminant hepatic failure. Gastroenterology 1989; 97:439–445.
- Shakil AO, Kramer D, Mazariegos GV, Fung JJ, Rakela J. Acute liver failure: clinical features, outcome analysis, and applicability of prognostic criteria. Liver Transplant 2000; 6:163–169.
- Anand AC, Nightingale P, Neuberger JM. Early indicators of prognosis in fulminant hepatic failure: an assessment of the King’s criteria. J Hepatol 1997; 26:62–68.
- Schmidt LE, Dalhoff K. Serum phosphate is an early predictor of outcome in severe acetaminophen-induced hepatotoxicity. Hepatology 2002; 36:659–665.
- Samuel D, Muller R, Alexander G, et al. Liver transplantation in European patients with the hepatitis B surface antigen. N Engl J Med 1993; 329:1842–1847.
- Lok A, McMahon BJ. Chronic hepatitis B. Hepatology 2007; 45:507–539.
- Sorren MF, Belangia EA, Costa J, et al. National Institutes of Health consensus development conference statement: management of hepatitis B. Ann Intern Med 2009; 150:104–110.
- Kjaergard LL, Liu J, Als-Nielsen B, Gluud C. Artificial and bioartificial support systems for acute and acute-on-chronic liver failure: a systematic review. JAMA 2003; 289:217–222.
- Kim WR, Poterucha JJ, Kremers WK, Ishitani MB, Dickson ER. Outcome of liver transplantation for hepatitis B in the United States. Liver Transplant 2004; 10:968–974.
- Terrault NA, Zhou S, Combs C, et al. Prophylaxis in liver transplant recipients using a fixed dosing schedule of hepatitis B immunoglobulin. Hepatology 1996; 24:1327–1333.
- Dusheiko G. Hepatitis B. In:Bircher J, Benhamou JP, McIntyre N, Rizzetto M, Rodes J, editors. Oxford Textbook of Clinical Hepatology. 2nd ed. Oxford, UK: Oxford University Press; 1999:876–896.
- Chu CJ, Hussain M, Lok AS. Quantitative serum HBV DNA levels during different stages of chronic hepatitis B infection. Hepatology 2002; 36:1408–1415.
- Pawlotsky JM, Bastie A, Hezode C, et al. Routine detection and quantification of hepatitis B virus DNA in clinical laboratories: performance of three commercial assays. J Virol Methods 2000; 85:11–21.
- Brunetto MR, Giarin MM, Oliveri F, et al. Wild-type and e-antigen-minus hepatitis viruses and course of chronic hepatitis. Proc Natl Acad Sci USA 1991; 88:4186–4190.
- Lok AS, Lai CL. A longitudinal follow-up of asymptomatic hepatitis B surface antigen-positive Chinese children. Hepatology 1988; 5:1130–1133.
- Hsu HY, Chang MH, Hsieh KH, et al. Cellular immune response to HBcAg in mother-to-infant transmission of hepatitis B virus. Hepatology 1992; 15:770–776.
- Chang MH, Hsu HY, Hsu HC, Ni YH, Chen JS, Chen DS. The significance of spontaneous hepatitis B e antigen seroconversion in childhood: with special emphasis on the clearance of hepatitis B e antigen before 3 years of age. Hepatology 1995; 22:1387–1392.
- Ruiz-Moreno M, Otero M, Millan A, et al. Clinical and histological outcome after hepatitis B e antigen to antibody seroconversion in children with chronic hepatitis B. Hepatology 1999; 29:572–575.
- Liaw YF, Chu CM, Su IJ, Huang MJ, Lin DY, Chang-Chien CS. Clinical and histological events preceding hepatitis B e antigen seroconversion in chronic type B hepatitis. Gastroenterology 1983; 84:216–219.
- Norvell JP, Blei AT, Jovanovic BD, Levitsky J. Herpes simplex virus hepatitis: an analysis of the published literature and institutional cases. Liver Transplant 2007; 13:1428–1434,
- Liaw YF, Pao CC, Chu CM, Sheen IS, Huang MJ. Changes of serum hepatitis B virus DNA in two types of clinical events preceding spontaneous hepatitis B e antigen seroconversion in chronic type B hepatitis. Hepatology 1987; 7:1–3.
- Maruyama T, Iino S, Koike K, Yasuda K, Milich DR. Serology of acute exacerbation in chronic hepatitis B virus infection. Gastroenterology 1993; 105:1141–1151.
- O'Grady JG, Alexander GJ, Hayllar KM, Williams R. Early indicators of prognosis in fulminant hepatic failure. Gastroenterology 1989; 97:439–445.
- Shakil AO, Kramer D, Mazariegos GV, Fung JJ, Rakela J. Acute liver failure: clinical features, outcome analysis, and applicability of prognostic criteria. Liver Transplant 2000; 6:163–169.
- Anand AC, Nightingale P, Neuberger JM. Early indicators of prognosis in fulminant hepatic failure: an assessment of the King’s criteria. J Hepatol 1997; 26:62–68.
- Schmidt LE, Dalhoff K. Serum phosphate is an early predictor of outcome in severe acetaminophen-induced hepatotoxicity. Hepatology 2002; 36:659–665.
- Samuel D, Muller R, Alexander G, et al. Liver transplantation in European patients with the hepatitis B surface antigen. N Engl J Med 1993; 329:1842–1847.
- Lok A, McMahon BJ. Chronic hepatitis B. Hepatology 2007; 45:507–539.
- Sorren MF, Belangia EA, Costa J, et al. National Institutes of Health consensus development conference statement: management of hepatitis B. Ann Intern Med 2009; 150:104–110.
- Kjaergard LL, Liu J, Als-Nielsen B, Gluud C. Artificial and bioartificial support systems for acute and acute-on-chronic liver failure: a systematic review. JAMA 2003; 289:217–222.
- Kim WR, Poterucha JJ, Kremers WK, Ishitani MB, Dickson ER. Outcome of liver transplantation for hepatitis B in the United States. Liver Transplant 2004; 10:968–974.
- Terrault NA, Zhou S, Combs C, et al. Prophylaxis in liver transplant recipients using a fixed dosing schedule of hepatitis B immunoglobulin. Hepatology 1996; 24:1327–1333.