User login
Enlarging Mass on the Scalp
Enlarging Mass on the Scalp
THE DIAGNOSIS: Malignant Proliferating Trichilemmal Tumor
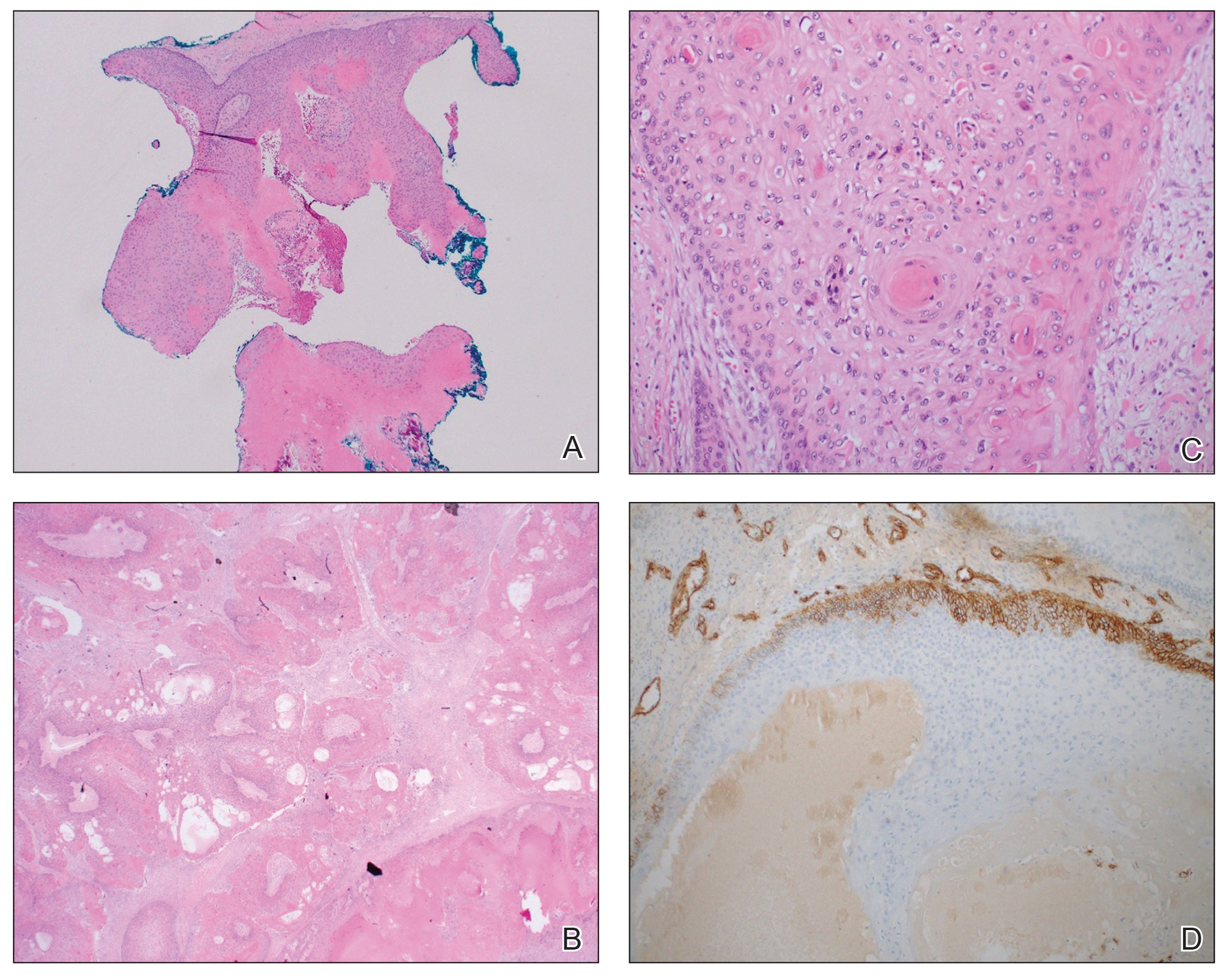
Histologic examination revealed atypical keratinocytes, nuclear pleomorphism, and lobulating epithelial masses with trichilemmal keratinization (Figure). The presence of CD34 positivity, a marker of outer follicular root sheath–derived cells, supported the diagnosis of a malignant proliferating trichilemmal tumor (MPTT). Imaging also revealed signs of bone invasion, further supporting a malignant process. Based on these findings, the patient underwent complete excision of the mass with scalp reconstruction, lymph node dissection, and systemic evaluation for metastases. Final pathology confirmed negative surgical margins and no lymph node involvement. Adjuvant radiation was not required, given the absence of skull invasion or confirmed distant metastasis.
The differential diagnosis for rapidly enlarging scalp tumors can be broad and includes both benign and malignant processes. In this patient, the differential diagnoses included trichilemmal carcinoma, cutaneous squamous cell carcinoma (SCC), sebaceous carcinoma (SC), proliferating trichilemmal tumor (PTT), and MPTT. Due to the notable clinical and histologic overlap among these lesions, definitive diagnosis required histopathologic evaluation in our patient.
Proliferating trichilemmal tumors were first described in 1966 by Wilson-Jones,1 who used the term proliferating epidermoid cysts, noting their distinct histologic features and resemblance to SCC.2 These tumors generally are benign and arise from the isthmus of the outer root sheath of the hair follicle; however, malignant transformation can occur, resulting in a rare entity known as MPTT. This malignant variant was first described in 1983 by Saida et al,3 who emphasized its distinct clinical behavior, including infiltrative growth, high mitotic activity, and potential for local recurrence and metastasis.
A recent literature review identified 60 reported cases of MPTT, with an average patient age of 57 years and a female predominance.4 Clinically, MPTTs often manifest as large (>5 cm) lobulated masses located on sun-exposed, hair-bearing areas of the skin, especially the scalp. These lesions may be flesh-colored to pink and often exhibit ulceration, necrosis, or calcification.5 Typically, MPTTs follow a biphasic course, beginning with a slow-growing phase followed by a period of rapid growth. Due to their aggressive behavior and resemblance to other cutaneous malignancies, accurate differentiation of MPTT from benign PTTs, cutaneous SCCs, SCs, and trichilemmal carcinomas is critical.
Malignant proliferating trichilemmal tumors demonstrate a substantially higher metastatic potential than either benign PTTs or cutaneous SCCs. While cutaneous SCCs carry a metastasis rate of approximately 1.9% to 2.6%, MPTTs carry a considerably higher rate of approximately 25.0%.6 Regional lymphatic spread is the most common route of dissemination, making comprehensive lymph node assessment—both radiographic and clinical—an important component of tumor staging. When lymph node involvement is suspected, surgical dissection may be indicated, along with consideration of adjuvant therapies.
Histopathologically, MPTT is characterized by nuclear atypia, mitotic figures, and lobulated masses of proliferating epithelium showing trichilemmal differentiation and infiltrative growth.4 The presence of CD34 positivity, reflecting outer follicular root sheath differentiation, helps distinguish MPTT from cutaneous SCC and SC, which typically lack this marker.6,7 Immunohistochemistry is therefore a valuable adjunct in differentiating these lesions.
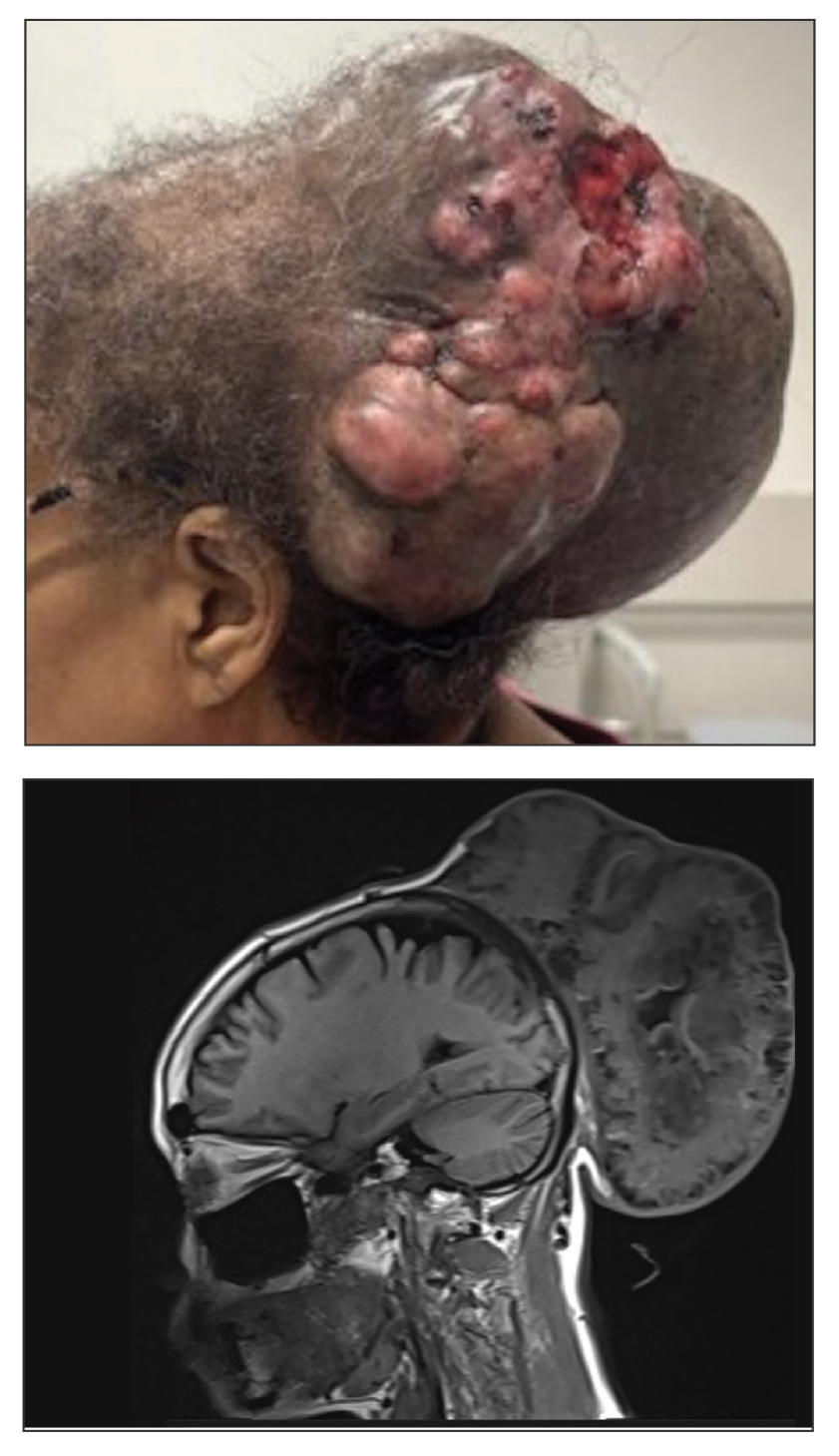
The mainstay of treatment for MPTT is wide local excision with clear margins. Margins of at least 1 cm generally are recommended. Although Mohs micrographic surgery may be used in anatomically sensitive areas, it typically is not preferred due to the potential for skip lesions in MPTT, which may lead to incomplete excision and recurrence.8 In cases with evidence of regional or distant metastasis or when clear margins cannot be achieved confidently, adjunctive treatments such as radiation therapy and systemic chemotherapy may be indicated. Preoperative imaging is used to evaluate for local invasion (skull or bone involvement) and regional lymph node status, which may inform adjuvant therapy postoperatively.
The prognosis for MPTT is variable and depends largely on early recognition, accurate histopathologic diagnosis, complete surgical excision with clear margins, and the presence or absence of metastasis. When the tumor is fully excised with negative margins and no lymph node involvement, the risk for recurrence is substantially reduced; however, MPTT is known for its potential aggressive behavior. Delays in diagnosis or incomplete resection can lead to local recurrence, regional spread, or even distant metastasis. In the literature review discussed previously, the mortality rate of patients with MPTT was 11.7%,4 which is notably higher than that of more common cutaneous malignancies such as cutaneous SCC, which is reported at 1.2%.9
The clinical course of MPTT remains difficult to predict due to its rarity and the limited availability of large-scale studies. Most published data are derived from isolated case reports or small case series, making standardized treatment guidelines challenging. Given this uncertainty, long-term follow-up is strongly recommended to monitor for recurrence or metastatic progression.2
This case highlights the critical role of clinicopathologic correlation in the evaluation of atypical or rapidly growing scalp lesions. The expertise of dermatologists in recognizing atypical presentations, combined with precise histopathologic analysis, including immunohistochemical staining, is vital to ensuring accurate diagnosis and optimal treatment. Early intervention can improve patient outcomes by reducing the risk for local recurrence and metastatic progression as well as the need for more intensive therapies.
- Jones EW. Proliferating epidermoid cysts. Arch Dermatol. 1966;94:11-19.
- Kemaloglu CA, Öztürk M, Aydın B, et al. Malignant proliferating trichilemmal tumor of the scalp: report of 4 cases and a short review of the literature. Case Reports Plast Surg Hand Surg. 2022;9:158-164. doi:10.1080/23320885.2022.2077208
- Saida T, Oohard K, Hori Y, et al. Development of a malignant proliferating trichilemmal cyst in a patient with multiple trichilemmal cysts. Dermatology. 1983;166:203-208. doi:10.1159/000249868
- Abdelhammed MH, Siatecka H, Diwan AH, et al. A rare case of a malignant proliferating trichilemmal tumor: a molecular study harboring potential therapeutic significance and a review of literature. Dermatopathology (Basel). 2024;11:354-363. doi:10.3390 /dermatopathology11040038
- Fronek L, Brahs A, Farsi M, et al. A rare case of trichilemmal carcinoma: histology and management. J Clin Aesthet Dermatol. 2021;14:25-30.
- Osto M, Parry N, Rehman R, et al. Malignant proliferating trichilemmal tumor of the scalp: a systematic review. Am J Dermatopathol. 2021;43:851-866. doi:10.1097/DAD.0000000000001991
- Plaza JA, Mackinnon A, Carrillo L, et al. Role of immunohistochemistry in the diagnosis of sebaceous carcinoma: a clinicopathologic and immunohistochemical study. Am J Dermatopathol. 2015;37:809-821. doi:10.1097/DAD.0000000000000255
- Singh P, Usman A, Motta L, et al. Malignant proliferating trichilemmal tumour. BMJ Case Rep. Published online August 17, 2018. doi:10.1136/bcr-2018-224460
- Ran NA, Granger EE, Brodland DG, et al. Risk factor number and recurrence, metastasis, and disease-related death in cutaneous squamous cell carcinoma. JAMA Dermatol. 2025;161:597-604. doi:10.1001/jamadermatol.2025.0128
THE DIAGNOSIS: Malignant Proliferating Trichilemmal Tumor
Histologic examination revealed atypical keratinocytes, nuclear pleomorphism, and lobulating epithelial masses with trichilemmal keratinization (Figure). The presence of CD34 positivity, a marker of outer follicular root sheath–derived cells, supported the diagnosis of a malignant proliferating trichilemmal tumor (MPTT). Imaging also revealed signs of bone invasion, further supporting a malignant process. Based on these findings, the patient underwent complete excision of the mass with scalp reconstruction, lymph node dissection, and systemic evaluation for metastases. Final pathology confirmed negative surgical margins and no lymph node involvement. Adjuvant radiation was not required, given the absence of skull invasion or confirmed distant metastasis.
The differential diagnosis for rapidly enlarging scalp tumors can be broad and includes both benign and malignant processes. In this patient, the differential diagnoses included trichilemmal carcinoma, cutaneous squamous cell carcinoma (SCC), sebaceous carcinoma (SC), proliferating trichilemmal tumor (PTT), and MPTT. Due to the notable clinical and histologic overlap among these lesions, definitive diagnosis required histopathologic evaluation in our patient.
Proliferating trichilemmal tumors were first described in 1966 by Wilson-Jones,1 who used the term proliferating epidermoid cysts, noting their distinct histologic features and resemblance to SCC.2 These tumors generally are benign and arise from the isthmus of the outer root sheath of the hair follicle; however, malignant transformation can occur, resulting in a rare entity known as MPTT. This malignant variant was first described in 1983 by Saida et al,3 who emphasized its distinct clinical behavior, including infiltrative growth, high mitotic activity, and potential for local recurrence and metastasis.
A recent literature review identified 60 reported cases of MPTT, with an average patient age of 57 years and a female predominance.4 Clinically, MPTTs often manifest as large (>5 cm) lobulated masses located on sun-exposed, hair-bearing areas of the skin, especially the scalp. These lesions may be flesh-colored to pink and often exhibit ulceration, necrosis, or calcification.5 Typically, MPTTs follow a biphasic course, beginning with a slow-growing phase followed by a period of rapid growth. Due to their aggressive behavior and resemblance to other cutaneous malignancies, accurate differentiation of MPTT from benign PTTs, cutaneous SCCs, SCs, and trichilemmal carcinomas is critical.
Malignant proliferating trichilemmal tumors demonstrate a substantially higher metastatic potential than either benign PTTs or cutaneous SCCs. While cutaneous SCCs carry a metastasis rate of approximately 1.9% to 2.6%, MPTTs carry a considerably higher rate of approximately 25.0%.6 Regional lymphatic spread is the most common route of dissemination, making comprehensive lymph node assessment—both radiographic and clinical—an important component of tumor staging. When lymph node involvement is suspected, surgical dissection may be indicated, along with consideration of adjuvant therapies.
Histopathologically, MPTT is characterized by nuclear atypia, mitotic figures, and lobulated masses of proliferating epithelium showing trichilemmal differentiation and infiltrative growth.4 The presence of CD34 positivity, reflecting outer follicular root sheath differentiation, helps distinguish MPTT from cutaneous SCC and SC, which typically lack this marker.6,7 Immunohistochemistry is therefore a valuable adjunct in differentiating these lesions.
The mainstay of treatment for MPTT is wide local excision with clear margins. Margins of at least 1 cm generally are recommended. Although Mohs micrographic surgery may be used in anatomically sensitive areas, it typically is not preferred due to the potential for skip lesions in MPTT, which may lead to incomplete excision and recurrence.8 In cases with evidence of regional or distant metastasis or when clear margins cannot be achieved confidently, adjunctive treatments such as radiation therapy and systemic chemotherapy may be indicated. Preoperative imaging is used to evaluate for local invasion (skull or bone involvement) and regional lymph node status, which may inform adjuvant therapy postoperatively.
The prognosis for MPTT is variable and depends largely on early recognition, accurate histopathologic diagnosis, complete surgical excision with clear margins, and the presence or absence of metastasis. When the tumor is fully excised with negative margins and no lymph node involvement, the risk for recurrence is substantially reduced; however, MPTT is known for its potential aggressive behavior. Delays in diagnosis or incomplete resection can lead to local recurrence, regional spread, or even distant metastasis. In the literature review discussed previously, the mortality rate of patients with MPTT was 11.7%,4 which is notably higher than that of more common cutaneous malignancies such as cutaneous SCC, which is reported at 1.2%.9
The clinical course of MPTT remains difficult to predict due to its rarity and the limited availability of large-scale studies. Most published data are derived from isolated case reports or small case series, making standardized treatment guidelines challenging. Given this uncertainty, long-term follow-up is strongly recommended to monitor for recurrence or metastatic progression.2
This case highlights the critical role of clinicopathologic correlation in the evaluation of atypical or rapidly growing scalp lesions. The expertise of dermatologists in recognizing atypical presentations, combined with precise histopathologic analysis, including immunohistochemical staining, is vital to ensuring accurate diagnosis and optimal treatment. Early intervention can improve patient outcomes by reducing the risk for local recurrence and metastatic progression as well as the need for more intensive therapies.
THE DIAGNOSIS: Malignant Proliferating Trichilemmal Tumor
Histologic examination revealed atypical keratinocytes, nuclear pleomorphism, and lobulating epithelial masses with trichilemmal keratinization (Figure). The presence of CD34 positivity, a marker of outer follicular root sheath–derived cells, supported the diagnosis of a malignant proliferating trichilemmal tumor (MPTT). Imaging also revealed signs of bone invasion, further supporting a malignant process. Based on these findings, the patient underwent complete excision of the mass with scalp reconstruction, lymph node dissection, and systemic evaluation for metastases. Final pathology confirmed negative surgical margins and no lymph node involvement. Adjuvant radiation was not required, given the absence of skull invasion or confirmed distant metastasis.
The differential diagnosis for rapidly enlarging scalp tumors can be broad and includes both benign and malignant processes. In this patient, the differential diagnoses included trichilemmal carcinoma, cutaneous squamous cell carcinoma (SCC), sebaceous carcinoma (SC), proliferating trichilemmal tumor (PTT), and MPTT. Due to the notable clinical and histologic overlap among these lesions, definitive diagnosis required histopathologic evaluation in our patient.
Proliferating trichilemmal tumors were first described in 1966 by Wilson-Jones,1 who used the term proliferating epidermoid cysts, noting their distinct histologic features and resemblance to SCC.2 These tumors generally are benign and arise from the isthmus of the outer root sheath of the hair follicle; however, malignant transformation can occur, resulting in a rare entity known as MPTT. This malignant variant was first described in 1983 by Saida et al,3 who emphasized its distinct clinical behavior, including infiltrative growth, high mitotic activity, and potential for local recurrence and metastasis.
A recent literature review identified 60 reported cases of MPTT, with an average patient age of 57 years and a female predominance.4 Clinically, MPTTs often manifest as large (>5 cm) lobulated masses located on sun-exposed, hair-bearing areas of the skin, especially the scalp. These lesions may be flesh-colored to pink and often exhibit ulceration, necrosis, or calcification.5 Typically, MPTTs follow a biphasic course, beginning with a slow-growing phase followed by a period of rapid growth. Due to their aggressive behavior and resemblance to other cutaneous malignancies, accurate differentiation of MPTT from benign PTTs, cutaneous SCCs, SCs, and trichilemmal carcinomas is critical.
Malignant proliferating trichilemmal tumors demonstrate a substantially higher metastatic potential than either benign PTTs or cutaneous SCCs. While cutaneous SCCs carry a metastasis rate of approximately 1.9% to 2.6%, MPTTs carry a considerably higher rate of approximately 25.0%.6 Regional lymphatic spread is the most common route of dissemination, making comprehensive lymph node assessment—both radiographic and clinical—an important component of tumor staging. When lymph node involvement is suspected, surgical dissection may be indicated, along with consideration of adjuvant therapies.
Histopathologically, MPTT is characterized by nuclear atypia, mitotic figures, and lobulated masses of proliferating epithelium showing trichilemmal differentiation and infiltrative growth.4 The presence of CD34 positivity, reflecting outer follicular root sheath differentiation, helps distinguish MPTT from cutaneous SCC and SC, which typically lack this marker.6,7 Immunohistochemistry is therefore a valuable adjunct in differentiating these lesions.
The mainstay of treatment for MPTT is wide local excision with clear margins. Margins of at least 1 cm generally are recommended. Although Mohs micrographic surgery may be used in anatomically sensitive areas, it typically is not preferred due to the potential for skip lesions in MPTT, which may lead to incomplete excision and recurrence.8 In cases with evidence of regional or distant metastasis or when clear margins cannot be achieved confidently, adjunctive treatments such as radiation therapy and systemic chemotherapy may be indicated. Preoperative imaging is used to evaluate for local invasion (skull or bone involvement) and regional lymph node status, which may inform adjuvant therapy postoperatively.
The prognosis for MPTT is variable and depends largely on early recognition, accurate histopathologic diagnosis, complete surgical excision with clear margins, and the presence or absence of metastasis. When the tumor is fully excised with negative margins and no lymph node involvement, the risk for recurrence is substantially reduced; however, MPTT is known for its potential aggressive behavior. Delays in diagnosis or incomplete resection can lead to local recurrence, regional spread, or even distant metastasis. In the literature review discussed previously, the mortality rate of patients with MPTT was 11.7%,4 which is notably higher than that of more common cutaneous malignancies such as cutaneous SCC, which is reported at 1.2%.9
The clinical course of MPTT remains difficult to predict due to its rarity and the limited availability of large-scale studies. Most published data are derived from isolated case reports or small case series, making standardized treatment guidelines challenging. Given this uncertainty, long-term follow-up is strongly recommended to monitor for recurrence or metastatic progression.2
This case highlights the critical role of clinicopathologic correlation in the evaluation of atypical or rapidly growing scalp lesions. The expertise of dermatologists in recognizing atypical presentations, combined with precise histopathologic analysis, including immunohistochemical staining, is vital to ensuring accurate diagnosis and optimal treatment. Early intervention can improve patient outcomes by reducing the risk for local recurrence and metastatic progression as well as the need for more intensive therapies.
- Jones EW. Proliferating epidermoid cysts. Arch Dermatol. 1966;94:11-19.
- Kemaloglu CA, Öztürk M, Aydın B, et al. Malignant proliferating trichilemmal tumor of the scalp: report of 4 cases and a short review of the literature. Case Reports Plast Surg Hand Surg. 2022;9:158-164. doi:10.1080/23320885.2022.2077208
- Saida T, Oohard K, Hori Y, et al. Development of a malignant proliferating trichilemmal cyst in a patient with multiple trichilemmal cysts. Dermatology. 1983;166:203-208. doi:10.1159/000249868
- Abdelhammed MH, Siatecka H, Diwan AH, et al. A rare case of a malignant proliferating trichilemmal tumor: a molecular study harboring potential therapeutic significance and a review of literature. Dermatopathology (Basel). 2024;11:354-363. doi:10.3390 /dermatopathology11040038
- Fronek L, Brahs A, Farsi M, et al. A rare case of trichilemmal carcinoma: histology and management. J Clin Aesthet Dermatol. 2021;14:25-30.
- Osto M, Parry N, Rehman R, et al. Malignant proliferating trichilemmal tumor of the scalp: a systematic review. Am J Dermatopathol. 2021;43:851-866. doi:10.1097/DAD.0000000000001991
- Plaza JA, Mackinnon A, Carrillo L, et al. Role of immunohistochemistry in the diagnosis of sebaceous carcinoma: a clinicopathologic and immunohistochemical study. Am J Dermatopathol. 2015;37:809-821. doi:10.1097/DAD.0000000000000255
- Singh P, Usman A, Motta L, et al. Malignant proliferating trichilemmal tumour. BMJ Case Rep. Published online August 17, 2018. doi:10.1136/bcr-2018-224460
- Ran NA, Granger EE, Brodland DG, et al. Risk factor number and recurrence, metastasis, and disease-related death in cutaneous squamous cell carcinoma. JAMA Dermatol. 2025;161:597-604. doi:10.1001/jamadermatol.2025.0128
- Jones EW. Proliferating epidermoid cysts. Arch Dermatol. 1966;94:11-19.
- Kemaloglu CA, Öztürk M, Aydın B, et al. Malignant proliferating trichilemmal tumor of the scalp: report of 4 cases and a short review of the literature. Case Reports Plast Surg Hand Surg. 2022;9:158-164. doi:10.1080/23320885.2022.2077208
- Saida T, Oohard K, Hori Y, et al. Development of a malignant proliferating trichilemmal cyst in a patient with multiple trichilemmal cysts. Dermatology. 1983;166:203-208. doi:10.1159/000249868
- Abdelhammed MH, Siatecka H, Diwan AH, et al. A rare case of a malignant proliferating trichilemmal tumor: a molecular study harboring potential therapeutic significance and a review of literature. Dermatopathology (Basel). 2024;11:354-363. doi:10.3390 /dermatopathology11040038
- Fronek L, Brahs A, Farsi M, et al. A rare case of trichilemmal carcinoma: histology and management. J Clin Aesthet Dermatol. 2021;14:25-30.
- Osto M, Parry N, Rehman R, et al. Malignant proliferating trichilemmal tumor of the scalp: a systematic review. Am J Dermatopathol. 2021;43:851-866. doi:10.1097/DAD.0000000000001991
- Plaza JA, Mackinnon A, Carrillo L, et al. Role of immunohistochemistry in the diagnosis of sebaceous carcinoma: a clinicopathologic and immunohistochemical study. Am J Dermatopathol. 2015;37:809-821. doi:10.1097/DAD.0000000000000255
- Singh P, Usman A, Motta L, et al. Malignant proliferating trichilemmal tumour. BMJ Case Rep. Published online August 17, 2018. doi:10.1136/bcr-2018-224460
- Ran NA, Granger EE, Brodland DG, et al. Risk factor number and recurrence, metastasis, and disease-related death in cutaneous squamous cell carcinoma. JAMA Dermatol. 2025;161:597-604. doi:10.1001/jamadermatol.2025.0128
Enlarging Mass on the Scalp
Enlarging Mass on the Scalp
A 61-year-old woman presented to the emergency department with worsening pain and bleeding from a scalp tumor of 16 years’ duration. Initially noted as a small nodule on the left parietal scalp on computed tomography of the head, the mass had grown rapidly in recent years and currently measured 22×10×15 cm. At prior consultations with plastic and general surgery, the patient had declined surgical intervention. At the current presentation, biopsies were performed by plastic surgery, and a dermatopathology consultation was ordered. Histopathology revealed atypical keratinocytes, nuclear pleomorphism, lobulating epithelial masses with trichilemmal keratinization, and CD34 positivity. Subsequent computed tomography and positron emission tomography of the head showed occipital skull erosion and bilateral cervical lymphadenopathy, suggesting metastasis.

Disseminated Papules and Nodules on the Skin and Oral Mucosa in an Infant
The Diagnosis: Congenital Cutaneous Langerhans Cell Histiocytosis
Although the infectious workup was positive for herpes simplex virus type 1 and cytomegalovirus antibodies, serologies for the rest of the TORCH (toxoplasmosis, other agents [syphilis, hepatitis B virus], rubella, cytomegalovirus) group of infections, as well as other bacterial, fungal, and viral infections, were negative. A skin biopsy from the right fifth toe showed a dense infiltrate of CD1a+ histiocytic cells with folded or kidney-shaped nuclei mixed with eosinophils, which was consistent with Langerhans cell histiocytosis (LCH) (Figure 1). Skin lesions were treated with hydrocortisone cream 2.5% and progressively faded over a few weeks.
.")
Langerhans cell histiocytosis is a rare disorder with a variable clinical presentation depending on the sites affected and the extent of involvement. It can involve multiple organ systems, most commonly the skeletal system and the skin. Organ involvement is characterized by histiocyte infiltration. Acute disseminated multisystem disease most commonly is seen in children younger than 3 years.1
Congenital cutaneous LCH presents with variable skin lesions ranging from papules to vesicles, pustules, and ulcers, with onset at birth or in the neonatal period. Various morphologic traits of skin lesions have been described; the most common presentation is multiple red to yellow-brown, crusted papules with accompanying hemorrhage or erosion.1 Other cases have described an eczematous, seborrheic, diffuse eruption or erosive intertrigo. One case of a child with a solitary necrotic nodule on the scalp has been reported.2
Our patient presented with disseminated, nonblanching, purple to dark red papules and nodules of the skin and oral mucosa, as well as nail dystrophy (Figure 2). However, LCH in a neonate can mimic other causes of congenital papulonodular eruptions. Red-brown papules and nodules with or without crusting in a newborn can be mistaken for erythema toxicum neonatorum, transient neonatal pustular melanosis, congenital leukemia cutis, neonatal erythropoiesis, disseminated neonatal hemangiomatosis, infantile acropustulosis, or congenital TORCH infections such as rubella or syphilis. When LCH presents as vesicles or eroded papules or nodules in a newborn, the differential diagnosis includes incontinentia pigmenti and hereditary epidermolysis bullosa.

Langerhans cell histiocytosis may even present with a classic blueberry muffin rash that can lead clinicians to consider cutaneous metastasis from various hematologic malignancies or the more common TORCH infections. Several diagnostic tests can be performed to clarify the diagnosis, including bacterial and viral cultures and stains, serology, immunohistochemistry, flow cytometry, bone marrow aspiration, or skin biopsy.3 Langerhans cell histiocytosis is diagnosed with a combination of histology, immunohistochemistry, and clinical presentation; however, a skin biopsy is crucial. Tissue should be taken from the most easily accessible yet representative lesion. The characteristic appearance of LCH lesions is described as a dense infiltrate of histiocytic cells mixed with numerous eosinophils in the dermis.1 Histiocytes usually have folded nuclei and eosinophilic cytoplasm or kidney-shaped nuclei with prominent nucleoli. Positive CD1a and/or CD207 (Langerin) staining of the cells is required for definitive diagnosis.4 After diagnosis, it is important to obtain baseline laboratory and radiographic studies to determine the extent of systemic involvement.
Treatment of congenital LCH is tailored to the extent of organ involvement. The dermatologic manifestations resolve without medications in many cases. However, true self-resolving LCH can only be diagnosed retrospectively after a full evaluation for other sites of disease. Disseminated disease can be life-threatening and requires more active management. In cases of skin-limited disease, therapies include topical steroids, nitrogen mustard, or imiquimod; surgical resection of isolated lesions; phototherapy; or systemic therapies such as methotrexate, 6-mercaptopurine, vinblastine/vincristine, cladribine, and/or cytarabine. Symptomatic patients initially are treated with methotrexate and 6-mercaptopurine.5 Asymptomatic infants with skin-limited involvement can be managed with topical treatments.
Our patient had skin-limited disease. Abdominal ultrasonography, skeletal survey, and magnetic resonance imaging of the brain revealed no abnormalities. The patient’s family was advised to monitor him for reoccurrence of the skin lesions and to continue close follow-up with hematology and dermatology. Although congenital LCH often is self-resolving, extensive skin involvement increases the risk for internal organ involvement for several years.6 These patients require long-term follow-up for potential musculoskeletal, ophthalmologic, endocrine, hepatic, and/or pulmonary disease.
- Pan Y, Zeng X, Ge J, et al. Congenital self-healing Langerhans cell histiocytosis: clinical and pathological characteristics. Int J Clin Exp Pathol. 2019;12:2275-2278.
- Morren MA, Vanden Broecke K, Vangeebergen L, et al. Diverse cutaneous presentations of Langerhans cell histiocytosis in children: a retrospective cohort study. Pediatr Blood Cancer. 2016;63:486-492. doi:10.1002/pbc.25834
- Krooks J, Minkov M, Weatherall AG. Langerhans cell histiocytosis in children: diagnosis, differential diagnosis, treatment, sequelae, and standardized follow-up. J Am Acad Dermatol. 2018;78:1047-1056. doi:10.1016/j.jaad.2017.05.060
- Haupt R, Minkov M, Astigarraga I, et al. Langerhans cell histiocytosis (LCH): guidelines for diagnosis, clinical work-up, and treatment for patients till the age of 18 years. Pediatr Blood Cancer. 2013;60:175-184. doi:10.1002/pbc.24367
- Allen CE, Ladisch S, McClain KL. How I treat Langerhans cell histiocytosis. Blood. 2015;126:26-35. doi:10.1182/blood-2014-12-569301
- Jezierska M, Stefanowicz J, Romanowicz G, et al. Langerhans cell histiocytosis in children—a disease with many faces. recent advances in pathogenesis, diagnostic examinations and treatment. Postepy Dermatol Alergol. 2018;35:6-17. doi:10.5114/pdia.2017.67095
The Diagnosis: Congenital Cutaneous Langerhans Cell Histiocytosis
Although the infectious workup was positive for herpes simplex virus type 1 and cytomegalovirus antibodies, serologies for the rest of the TORCH (toxoplasmosis, other agents [syphilis, hepatitis B virus], rubella, cytomegalovirus) group of infections, as well as other bacterial, fungal, and viral infections, were negative. A skin biopsy from the right fifth toe showed a dense infiltrate of CD1a+ histiocytic cells with folded or kidney-shaped nuclei mixed with eosinophils, which was consistent with Langerhans cell histiocytosis (LCH) (Figure 1). Skin lesions were treated with hydrocortisone cream 2.5% and progressively faded over a few weeks.
Langerhans cell histiocytosis is a rare disorder with a variable clinical presentation depending on the sites affected and the extent of involvement. It can involve multiple organ systems, most commonly the skeletal system and the skin. Organ involvement is characterized by histiocyte infiltration. Acute disseminated multisystem disease most commonly is seen in children younger than 3 years.1
Congenital cutaneous LCH presents with variable skin lesions ranging from papules to vesicles, pustules, and ulcers, with onset at birth or in the neonatal period. Various morphologic traits of skin lesions have been described; the most common presentation is multiple red to yellow-brown, crusted papules with accompanying hemorrhage or erosion.1 Other cases have described an eczematous, seborrheic, diffuse eruption or erosive intertrigo. One case of a child with a solitary necrotic nodule on the scalp has been reported.2
Our patient presented with disseminated, nonblanching, purple to dark red papules and nodules of the skin and oral mucosa, as well as nail dystrophy (Figure 2). However, LCH in a neonate can mimic other causes of congenital papulonodular eruptions. Red-brown papules and nodules with or without crusting in a newborn can be mistaken for erythema toxicum neonatorum, transient neonatal pustular melanosis, congenital leukemia cutis, neonatal erythropoiesis, disseminated neonatal hemangiomatosis, infantile acropustulosis, or congenital TORCH infections such as rubella or syphilis. When LCH presents as vesicles or eroded papules or nodules in a newborn, the differential diagnosis includes incontinentia pigmenti and hereditary epidermolysis bullosa.
Langerhans cell histiocytosis may even present with a classic blueberry muffin rash that can lead clinicians to consider cutaneous metastasis from various hematologic malignancies or the more common TORCH infections. Several diagnostic tests can be performed to clarify the diagnosis, including bacterial and viral cultures and stains, serology, immunohistochemistry, flow cytometry, bone marrow aspiration, or skin biopsy.3 Langerhans cell histiocytosis is diagnosed with a combination of histology, immunohistochemistry, and clinical presentation; however, a skin biopsy is crucial. Tissue should be taken from the most easily accessible yet representative lesion. The characteristic appearance of LCH lesions is described as a dense infiltrate of histiocytic cells mixed with numerous eosinophils in the dermis.1 Histiocytes usually have folded nuclei and eosinophilic cytoplasm or kidney-shaped nuclei with prominent nucleoli. Positive CD1a and/or CD207 (Langerin) staining of the cells is required for definitive diagnosis.4 After diagnosis, it is important to obtain baseline laboratory and radiographic studies to determine the extent of systemic involvement.
Treatment of congenital LCH is tailored to the extent of organ involvement. The dermatologic manifestations resolve without medications in many cases. However, true self-resolving LCH can only be diagnosed retrospectively after a full evaluation for other sites of disease. Disseminated disease can be life-threatening and requires more active management. In cases of skin-limited disease, therapies include topical steroids, nitrogen mustard, or imiquimod; surgical resection of isolated lesions; phototherapy; or systemic therapies such as methotrexate, 6-mercaptopurine, vinblastine/vincristine, cladribine, and/or cytarabine. Symptomatic patients initially are treated with methotrexate and 6-mercaptopurine.5 Asymptomatic infants with skin-limited involvement can be managed with topical treatments.
Our patient had skin-limited disease. Abdominal ultrasonography, skeletal survey, and magnetic resonance imaging of the brain revealed no abnormalities. The patient’s family was advised to monitor him for reoccurrence of the skin lesions and to continue close follow-up with hematology and dermatology. Although congenital LCH often is self-resolving, extensive skin involvement increases the risk for internal organ involvement for several years.6 These patients require long-term follow-up for potential musculoskeletal, ophthalmologic, endocrine, hepatic, and/or pulmonary disease.
The Diagnosis: Congenital Cutaneous Langerhans Cell Histiocytosis
Although the infectious workup was positive for herpes simplex virus type 1 and cytomegalovirus antibodies, serologies for the rest of the TORCH (toxoplasmosis, other agents [syphilis, hepatitis B virus], rubella, cytomegalovirus) group of infections, as well as other bacterial, fungal, and viral infections, were negative. A skin biopsy from the right fifth toe showed a dense infiltrate of CD1a+ histiocytic cells with folded or kidney-shaped nuclei mixed with eosinophils, which was consistent with Langerhans cell histiocytosis (LCH) (Figure 1). Skin lesions were treated with hydrocortisone cream 2.5% and progressively faded over a few weeks.
Langerhans cell histiocytosis is a rare disorder with a variable clinical presentation depending on the sites affected and the extent of involvement. It can involve multiple organ systems, most commonly the skeletal system and the skin. Organ involvement is characterized by histiocyte infiltration. Acute disseminated multisystem disease most commonly is seen in children younger than 3 years.1
Congenital cutaneous LCH presents with variable skin lesions ranging from papules to vesicles, pustules, and ulcers, with onset at birth or in the neonatal period. Various morphologic traits of skin lesions have been described; the most common presentation is multiple red to yellow-brown, crusted papules with accompanying hemorrhage or erosion.1 Other cases have described an eczematous, seborrheic, diffuse eruption or erosive intertrigo. One case of a child with a solitary necrotic nodule on the scalp has been reported.2
Our patient presented with disseminated, nonblanching, purple to dark red papules and nodules of the skin and oral mucosa, as well as nail dystrophy (Figure 2). However, LCH in a neonate can mimic other causes of congenital papulonodular eruptions. Red-brown papules and nodules with or without crusting in a newborn can be mistaken for erythema toxicum neonatorum, transient neonatal pustular melanosis, congenital leukemia cutis, neonatal erythropoiesis, disseminated neonatal hemangiomatosis, infantile acropustulosis, or congenital TORCH infections such as rubella or syphilis. When LCH presents as vesicles or eroded papules or nodules in a newborn, the differential diagnosis includes incontinentia pigmenti and hereditary epidermolysis bullosa.
Langerhans cell histiocytosis may even present with a classic blueberry muffin rash that can lead clinicians to consider cutaneous metastasis from various hematologic malignancies or the more common TORCH infections. Several diagnostic tests can be performed to clarify the diagnosis, including bacterial and viral cultures and stains, serology, immunohistochemistry, flow cytometry, bone marrow aspiration, or skin biopsy.3 Langerhans cell histiocytosis is diagnosed with a combination of histology, immunohistochemistry, and clinical presentation; however, a skin biopsy is crucial. Tissue should be taken from the most easily accessible yet representative lesion. The characteristic appearance of LCH lesions is described as a dense infiltrate of histiocytic cells mixed with numerous eosinophils in the dermis.1 Histiocytes usually have folded nuclei and eosinophilic cytoplasm or kidney-shaped nuclei with prominent nucleoli. Positive CD1a and/or CD207 (Langerin) staining of the cells is required for definitive diagnosis.4 After diagnosis, it is important to obtain baseline laboratory and radiographic studies to determine the extent of systemic involvement.
Treatment of congenital LCH is tailored to the extent of organ involvement. The dermatologic manifestations resolve without medications in many cases. However, true self-resolving LCH can only be diagnosed retrospectively after a full evaluation for other sites of disease. Disseminated disease can be life-threatening and requires more active management. In cases of skin-limited disease, therapies include topical steroids, nitrogen mustard, or imiquimod; surgical resection of isolated lesions; phototherapy; or systemic therapies such as methotrexate, 6-mercaptopurine, vinblastine/vincristine, cladribine, and/or cytarabine. Symptomatic patients initially are treated with methotrexate and 6-mercaptopurine.5 Asymptomatic infants with skin-limited involvement can be managed with topical treatments.
Our patient had skin-limited disease. Abdominal ultrasonography, skeletal survey, and magnetic resonance imaging of the brain revealed no abnormalities. The patient’s family was advised to monitor him for reoccurrence of the skin lesions and to continue close follow-up with hematology and dermatology. Although congenital LCH often is self-resolving, extensive skin involvement increases the risk for internal organ involvement for several years.6 These patients require long-term follow-up for potential musculoskeletal, ophthalmologic, endocrine, hepatic, and/or pulmonary disease.
- Pan Y, Zeng X, Ge J, et al. Congenital self-healing Langerhans cell histiocytosis: clinical and pathological characteristics. Int J Clin Exp Pathol. 2019;12:2275-2278.
- Morren MA, Vanden Broecke K, Vangeebergen L, et al. Diverse cutaneous presentations of Langerhans cell histiocytosis in children: a retrospective cohort study. Pediatr Blood Cancer. 2016;63:486-492. doi:10.1002/pbc.25834
- Krooks J, Minkov M, Weatherall AG. Langerhans cell histiocytosis in children: diagnosis, differential diagnosis, treatment, sequelae, and standardized follow-up. J Am Acad Dermatol. 2018;78:1047-1056. doi:10.1016/j.jaad.2017.05.060
- Haupt R, Minkov M, Astigarraga I, et al. Langerhans cell histiocytosis (LCH): guidelines for diagnosis, clinical work-up, and treatment for patients till the age of 18 years. Pediatr Blood Cancer. 2013;60:175-184. doi:10.1002/pbc.24367
- Allen CE, Ladisch S, McClain KL. How I treat Langerhans cell histiocytosis. Blood. 2015;126:26-35. doi:10.1182/blood-2014-12-569301
- Jezierska M, Stefanowicz J, Romanowicz G, et al. Langerhans cell histiocytosis in children—a disease with many faces. recent advances in pathogenesis, diagnostic examinations and treatment. Postepy Dermatol Alergol. 2018;35:6-17. doi:10.5114/pdia.2017.67095
- Pan Y, Zeng X, Ge J, et al. Congenital self-healing Langerhans cell histiocytosis: clinical and pathological characteristics. Int J Clin Exp Pathol. 2019;12:2275-2278.
- Morren MA, Vanden Broecke K, Vangeebergen L, et al. Diverse cutaneous presentations of Langerhans cell histiocytosis in children: a retrospective cohort study. Pediatr Blood Cancer. 2016;63:486-492. doi:10.1002/pbc.25834
- Krooks J, Minkov M, Weatherall AG. Langerhans cell histiocytosis in children: diagnosis, differential diagnosis, treatment, sequelae, and standardized follow-up. J Am Acad Dermatol. 2018;78:1047-1056. doi:10.1016/j.jaad.2017.05.060
- Haupt R, Minkov M, Astigarraga I, et al. Langerhans cell histiocytosis (LCH): guidelines for diagnosis, clinical work-up, and treatment for patients till the age of 18 years. Pediatr Blood Cancer. 2013;60:175-184. doi:10.1002/pbc.24367
- Allen CE, Ladisch S, McClain KL. How I treat Langerhans cell histiocytosis. Blood. 2015;126:26-35. doi:10.1182/blood-2014-12-569301
- Jezierska M, Stefanowicz J, Romanowicz G, et al. Langerhans cell histiocytosis in children—a disease with many faces. recent advances in pathogenesis, diagnostic examinations and treatment. Postepy Dermatol Alergol. 2018;35:6-17. doi:10.5114/pdia.2017.67095
A 38-week-old infant boy presented at birth with disseminated, nonblanching, purple to dark red papules and nodules on the skin and oral mucosa. He was born spontaneously after an uncomplicated pregnancy. The mother experienced an episode of oral herpes simplex virus during pregnancy. The infant was otherwise healthy. Laboratory tests including a complete blood cell count and routine serum biochemical analyses were within reference range; however, an infectious workup was positive for herpes simplex virus type 1 and cytomegalovirus antibodies. Ophthalmologic and auditory screenings were normal.

